/










































































































































































































































































































Текст
THE INFRA-RED SPECTRA
OF
COMPLEX MOLECULES
by
L. J. BELLAMY
B. Sc., Ph. D.
Senior Principal Scientific Officer,
Ministry of Supply
LONDON: METHUEN & CO. LTD
NEW YORK: JOHN WILEY & SONS, INC.
Л. БЕЛЛАМИ
ИНФРАКРАСНЫЕ
СПЕКТРЫ
СЛОЖНЫХ МОЛЕКУЛ
ПЕРЕВОД С АНГЛИЙСКОГО
В- (М. АКИМОВА, Ю. А. ПЕНТИНА,
Э. Г. ТЕТЕРИНА
ПОД РЕДАКЦИЕЙ
канд. хим. наук Ю. А. П Е Н Т И Н;А
ИЗДАТЕЛЬСТВО ИНОСТРАННОЙ ЛИТЕРАТУРЫ
МОСКВА 1963
В книге дается богатейший, хорошо систематизи-
рованный справочный материал по инфракрасным спек-
трам поглощения различных классов органических соеди-
нений, а также многих неорганических соединений.
Приведены в качестве иллюстраций 30 спектров различ-
ных соединений и 5 корреляционных диаграмм.
Книга предназначена для химиков, биологов, био-
химиков, медиков и других специалистов различных
квалификаций: научных сотрудников, аспирантов, сту-
дентов, инженеров, техников, лаборантов
Редакция литературы по химии
ПРЕДИСЛОВИЕ
Предлагаемая вниманию читателя книга Л. Беллами
«Инфракрасные спектры сложных молекул» представляет
собой заново переработанное и значительно расширенное
издание книги того же автора, вышедшей в Лондоне
в 1954 г., которая была переведена на русский язык и издана
в Советском Союзе в 1957 г.
Метод инфракрасной спектроскопии является одним
из важнейших современных физических методов иденти-
фикации химических соединений, изучения строения
молекул и количественного анализа смесей; с каждым
годом он находит все более широкое применение в самых
различных областях науки и в первую очередь в химии,
биохимии и биологии. В настоящее время накоплен бога-
тейший экспериментальный материал по инфракрасным
спектрам поглощения химических соединений, и основная
ценность книги Беллами состоит в том, что она содержит
весьма полный обзор хорошо систематизированных спра-
вочных данных об особенностях спектров (характеристи-
ческих или групповых частотах) различных классов орга-
нических соединений, а также многих неорганических
соединений.
Второе издание книги качественно намного превос-
ходит первое. Помимо серьезной переработки текста,
проведенной с учетом новых литературных данных, автор
Дает много совершенно нового материала по целому ряду
вопросов, которые раньше не были освещены. Основываясь
главным образом на своих собственных исследованиях
и привлекая также работы других авторов, Беллами
написал новый раздел (часть V, гл. 23), посвященный вопро-
сам происхождения, смысла и интерпретации смещений
6 П редисловие
характеристических частот. Здесь рассмотрено влияние
на частоты изменений агрегатного состояния, раствори-
телей, водородных связей, напряжения, а также электри-
ческие эффекты (индукционный, сопряжения и мезомерии,
дипольного поля и некоторые другие). Этот раздел пред-
ставляет самостоятельный интерес и немаловажен с точ-
ки зрения основной задачи книги, так как автор обсуждает
возможность использования рассмотренных эффектов для
интерпретации спектров и более правильного отнесения
полос поглощения. Все остальные главы, посвященные рас-
смотрению особенностей спектров и строения соединений
различных классов, а особенно непредельных, ароматиче-
ских, кислород- и азотсодержащих, фосфорорганических
и некоторых других, значительно пополнены новыми кор-
реляционными данными. Всего введено около двадцати
новых разделов; многие главы переписаны заново почти
целиком.
В книге больше внимания уделяется проблеме интен-
сивности поглощения, обсуждаются такие специальные
вопросы, как трансанулярный эффект в больших цикли-
ческих системах, влияние сопряжения с циклопропаном
альдегидов, кетонов и др. Естественно, что объем книги
значительно возрос, а число цитируемых литературных
источников увеличилось почти вдвое по сравнению с первым
изданием. Гораздо лучше отражены в книге труды совет-
ских ученых.
Необходимо подчеркнуть, что, пользуясь книгой Бел-
лами, читатель должен отдавать себе отчет в том, что
почти все приводимые в ней корреляции спектров
и строения соединений получены только эмпирическим
путем; некоторые из них вполне надежны, другие же тре-
буют критического отношения. Во многих случаях даваемая
автором интерпретация тех или иных экспериментальных
данных вызывает серьезные сомнения. Основным недостат-
ком книги Беллами, как и подавляющего большинства опуб-
ликованных корреляционных исследований, является то,
что авторы описываемых работ мало или почти совсем не
пользуются теорией колебательных спектров и расчетными
методами; такие методы в настоящее время хорошо разра-
ботаны в трудах советских ученых Б. И. Степанова,
М. А. Ельяшевича, Л. С. Маянца и др., а также и за рубе-
П редисловие
7
жом, например Е. Вильсоном и др *. Наиболее объективные
и достоверные данные по отнесению частот могут быть
получены лишь с применением теоретических расчетов,
и последние работы некоторых советских исследователей
показали, что при этом могут быть достигнуты серьезные
успехи даже в случае интерпретации колебательных
спектров весьма сложных молекул.
При обсуждении вопросов связи строения молекул
и спектров Беллами часто пользуется представлениями
о резонансе канонических структур, хотя как раз сами
колебательные спектры являются ярким опровержением
сосуществования таких «резонирующих» структур. Не
являются убедительными и некоторые другие рассуждения
автора. Заслуживает, например, весьма критического отно-
шения большая часть материала главы 23, где многие
попытки автора объяснить причины экспериментально
наблюдаемых смещений характеристических частот недо-
статочно обоснованы, а иногда встречают серьезные воз-
ражения.
Указанные недостатки не снижают, однако, ценности
книги как прекрасного справочного пособия по инфракрас-
ным спектрам, которое является пока в своем роде един-
ственным в литературе.
При переводе книги сделаны некоторые исправления
по сравнению с переводом первого издания, касающиеся
принятой в спектроскопии терминологии. Здесь важно
отметить следующее: деформационным колебаниям груп-
пы
СН2 (которые в пропане или в любой молекуле, если
говорить о локальной симметрии группы, относятся
к типам симметрии В2, А2) соответствуют англий-
ские термины (в том же порядке) bending, rocking, wagging
* Обобщение этих работ дано в следующих основных моногра-
фиях: Волькенштейн М. В., Ельяшевич М. А., Степа-
нов Б. И., Колебания молекул, Гостехиздат, М.—Л., 1949; Ма-
я н ц Л. С., Теория и расчет колебаний молекул, Издательство
АН СССР, Москва, I960; Ель я ш е в и ч М. А.,Атомная и молеку-
лярная спектроскопия, Госфизматпздат, Москва, 1962; Виль-
сон Е., Цешиус Дж., Кросс П., Теория колебательных спек-
тров молекул, Издатинлит, Москва, 1960.
8
Предисловие
и twisting. Для этих колебаний в настоящее время в рус-
ской литературе установились соответственно следующие
адекватные термины, принятые и при переводе данной книги:
ножничное (или просто деформационное 6), маятниковое,
веерное и крутильное деформационное. В первом издании
название колебания В2 (rocking) было переведено как «кру-
тильное».
Несомненно, что в связи с расширением применений
методов инфракрасной спектроскопии в различных обла-
стях науки круг читателей, для которых интересна книга
Беллами, сильно возрос, и она найдет еще более широкий
спрос, чем первое ее издание. Книгу можно рекомендовать
всем специалистам самой различной квалификации, кото-
рые используют или собираются использовать в своей работе
методы инфракрасной спектроскопии.
Ю. Пентин
ПРЕДИСЛОВИЕ АВТОРА
КО ВТОРОМУ ИЗДАНИЮ
Читатель, как я надеюсь, увидит из текста, что выход
в свет второго издания настоящей книги вскоре после
первого вполне оправдан. Хотя большое количество мате-
риала было заменено результатами последующих исследо-
ваний, общий объем второго издания книги значительно
увеличился. Некоторым указанием на объем проведенной
работы служит тот факт, что после отбора в книгу было
включено более 700 новых литературных источников.
Я стремился отметить все работы по данной тематике,
основываясь на том, что читателю нетрудно будет исключить
те из них, которые не представляют для него интереса.
В основном теория колебательно-вращательных спект-
ров небольших молекул здесь не рассматривается, так как
она хорошо освещена в других превосходных работах.
Однако я не смог устоять против соблазна включить в конце
книги главу о причинах и значении смещений групповых
частот в больших молекулах. В этом направлении за по-
следние несколько лет были достигнуты большие успехи, а
главное было положено наконец начало рациональному
упорядочению большого эмпирического материала, касаю-
щегося групповых частот в больших молекулах. Многие
проблемы в данной области еще подлежат разрешению,
а некоторые из излагаемых идей, как увидит читатель,
находятся в значительной степени в стадии формирования.
Тем не менее найденные спектральные закономерности уже
частично используются в структурном анализе. Кроме того,
для установления соответствующих корреляций весьма
важно знать основные факторы, обусловливающие странные
особенности некоторых групповых частот. При написании
последней главы я пытался быть по возможности объектив-
ным, но читателю следует рассматривать отдельные ее раз-
10
Предисловие автора ко второму изданию
делы критически, как это и принято делать, если автор
излагает результаты своих исследований.
Серьезной проблемой при подготовке второго издания
явился выбор подходящего способа выражения интенсив-
ности. Нет никаких сомнений, что в дальнейшем при рас-
познавании специфических групп в молекулах абсолютные
интенсивности будут использоваться в такой же степени,
как и абсолютные частоты, а в некоторых случаях первые
дадут больше сведений. В настоящее время применяются
качественно различные способы выражения интенсивности,
и потому часто нельзя оценить надежность представленных
.данных. В связи с этим я не стал касаться этого вопроса,
а, как и в первом издании, включил имеющиеся данные по
интенсивности вместе с данными по соответствующим
групповым частотам.
Задача по просмотру очень большого объема литературы
была облегчена тем, что многие авторы любезно предоста-
вили мне оттиски своих статей. Всем им я выражаю свою
глубокую благодарность. В надежде на то, что эти авторы,
а также и другие, согласятся и в будущем оказывать подоб-
ную помощь, я помещаю ниже свой адрес. Во всех книгах
типа данной книги авторы должны учитывать результаты
работ своих предшественников; поэтому при подготовке
обоих изданий я считал необходимым включить ряд опуб-
ликованных корреляционных графиков и таблиц и поль-
зовался материалом книг Рендалла, Фаулера, Фьюзона
и Денгла, а также Барнса, Гора, Лиддела и Уильямса.
Значительную помощь оказал мне прекрасный обзор Джонса
и Сендорфи. Я благодарен также правлению Химического
общества за разрешение воспроизвести ряд иллюстраций,
опубликованных в издаваемом им журнале.
Е. R. D. Е. Ministry of
Supply, Waltham Abbey,
Essex.
Л. Дж. Б.,
июль 1957
Глава 1
ВВЕДЕНИЕ
Инфракрасная спектроскопия находит все большее
применение в промышленности и органической химии для
распознавания и количественного и структурного анализа
неизвестных соединений. Почти все имеющиеся на про-
мышленных предприятиях спектрометры применяются
именно для этих целей. В то же время физики-теоретики
используют спектры также для получения данных по меха-
нике простых молекул. Такие исследования исключительно
полезны для целей анализа, так как дают возможность опре-
делить групповые движения в молекуле, связанные с раз-
личными характеристическими частотами, и позволяют,
таким образом, в какой-то мере оценить смещения частот
при изменении внутримолекулярного окружения группы.
Однако эти исследования обычно ограничиваются изучением
простых молекул, у которых частоты, связанные с различ-
ными структурными элементами, отличаются от соответ-
ствующих частот, найденных для более сложных веществ.
Поэтому при интерпретации спектров следует опи-
раться на имеющиеся эмпирические данные о связи между
инфракрасными полосами поглощения и структурными
элементами молекул. Для этого необходимо ознакомиться
с большим числом опубликованных работ, что представляет
собой нелегкую задачу. Время от времени в литературе
публиковались сводные таблицы, обобщавшие такие сведе-
ния; эти таблицы при правильном использовании могут быть
очень полезны. Однако, как указывают сами авторы, не все
корреляции имеют общее значение, так как в некоторых
случаях они выведены на основании изучения ограниченной
группы соединений; поэтому неосмотрительное применение
их может привести к ошибочным результатам. Кроме того,
нецелесообразно для данной специфической группы, напри-
12
Гл. 1. Введение
мер карбонила, указывать широкий интервал частот, охва-
тывающий все известные случаи взаимодействия, так как
такая корреляция не представляет какой-либо ценности.
Исследование же отдельных полос поглощения различных
карбонилсодержащих групп в пределах такого интервала
часто позволяет выяснить природу этих структурных групп.
Настоящая работа представляет собой попытку дать
критический обзор данных по инфракрасным спектрам,
на которых основываются спектрально-структурные корре-
ляции, с указанием исследованных в каждом случае сое-
динений и известных факторов, которые могут влиять на
частоты и интенсивности характеристических полос. Особое
внимание уделялось публикациям, посвященным исследо-
ваниям серий соединений, содержащих одну общую струк-
турную группу. Хотя в книге рассматривается большое
число работ, посвященных фундаментальным исследова-
ниям отдельных молекул, попытки дать полную библиогра-
фию таких работ не делалось, так как поглощение струк-
турными группами очень простых соединений часто не
является типичным для таких же групп в больших моле-
кулах. Для удобства пользования оригинальными работами
принятые в них наименования химических соединений
сохранены и в данной книге, хотя иногда номенклатура
отличается от принятой в Англии*.
Многие из обсуждаемых корреляций, особенно в области
высоких частот, дают очень ценные и точные сведения о
строении соединений, поскольку по положению и интенсив-
ности полосы поглощения можно судить о наличии опреде-
ленной группы, а также о ее внутримолекулярном окруже-
нии. Другие корреляции, особенно относящиеся к коле-
баниям атомов скелета, показывают значительные изме-
нения частот в зависимости от структурных изменений.
Такими корреляциями следует пользоваться с большой
осторожностью; они неприменимы к соединениям, строение
которых значительно отличается от строения соединений,
использованных при установлении корреляции. Тем не менее
они могут дать указания на возможные структуры, а отсут-
ствие каких-либо полос в соответствующей области сви-
* Этот принцип в наименовании химических соединений в ос-
новном сохранен при переводе книги.— Прим. ред.
Гл. 1. Введение
13
детельствует об отсутствии в молекуле данной группы.
В любом случае спектроскопист укажет, очевидно, не только
ту группировку, в отношении которой у него не возникает
сомнений, но и рассмотрит структуры, которыемогут присут-
ствовать, хотя их с уверенностью нельзя идентифицировать.
При таких исследованиях очень полезно располагать све-
дениями о достоверности тех или иных опубликованных
данных по идентификации структур. Можно надеяться, что
в этом отношении некоторую помощь окажет данная книга.
Содержание этой книги строго ограничено • эмпириче-
ской интерпретацией инфракрасных спектров, и следует
отметить, что до настоящего времени не было опубликовано
ни одной подобной монографии. В книге не описаны опе-
рации, сопутствующие спектроскопическим измерениям,
как, например, приготовление образцов, конструирование
кювет, качественный анализ, а также не рассмотрено соот-
ветствующее оборудование. Этим вопросам посвящено боль-
шое число уже опубликованных статей, а также некоторые
работы, готовящиеся к печати. Кроме того, очень немногие
исследователи имеют в своем распоряжении приборы более
чем одного типа, а характеристики и особенности своего
прибора каждый сможет изучить практически гораздо быст-
рее и подробнее, чем при использовании общего обзора
по этому вопросу.
Когда имеются эмпирические данные о большом числе
различных структурных групп, материал можно распо-
ложить любым способом; автор следовал общепринятой
в периодически публикуемых корреляционных таблицах
системе [1—8]. Данные сгруппированы в соответствии
с подразделением типов связей на четыре основные группы:
I — углерод-углеродные и углерод-водородные связи;
II — углерод-кислородные и кислород-водородные связи;
III—углерод-азотные и азот-водородные связи; IV—:
связи между другими элементами (неорганические струк-
туры). Однако такое разделение строго не выдерживается.
Материал, относящийся к амидам, например, неудобно
распределять между частями II и III, он рассматривается
в части III, т. е. после обсуждения карбонильных частот
в части II. Или, например, глава по нитросоединениям и
родственным структурам включена в часть IV, т. е. после
обсуждения углерод-азотных связей.
14
Гл. 1. Введение
Что касается выбора единиц, то следует отметить, что
автор везде пользуется волновыми числами. Волновые
числа, а не длины волн применяются автором, поскольку
ими рекомендует пользоваться Британский объединенный
комитет; кроме того, по мнению автора, шкала волновых
чисел является единственно подходящей для корреляцион-
ных целей. Применение этой шкалы значительно облегчает
идентификацию обертонов и составных частот, а главное
позволяет непосредственно сравнивать инфракрасные спект-
ры со спектрами комбинационного рассеяния. По-видимому,
наиболее веским аргументом, который можно привести
в пользу шкалы длин волн, является простота конструк-
ции приборов с линейной шкалой отсчета длин волн. Для
удобства пользования такими приборами в конце книги
даны таблицы перевода одних единиц в другие.
В начале каждой главы кратко перечислены корреля-
ции, рассматриваемые далее подробно, и дана таблица,
включающая соответствующие частоты. В конце настоя-
щей главы приведены сводные корреляционные диаграммы,
обобщающие данные о поглощении для различных струк-
турных групп (диаг. 1—5). Диаграммы помещены с целью
дать сведения о структурных группах, которые могут
мешать при использовании тех или иных корреляций.
Прежде чем пользоваться приведенными значениями жела-
тельно во избежание ошибок ознакомиться с оригиналь-
ными работами. Автор пытался дать наиболее полные све-
дения, поэтому он приводит много предположительных
и даже несколько сомнительных корреляций, которые экс-
периментально полностью не подтверждены. Такие корре-
ляции можно применять лишь после того, как детально
выяснена их надежность. Приближенное указание интен-
сивности полос дается в таблицах и на графиках символа-
ми; с.— сильная, ср.—средняя, сл.— слабая и пер.—
переменная.
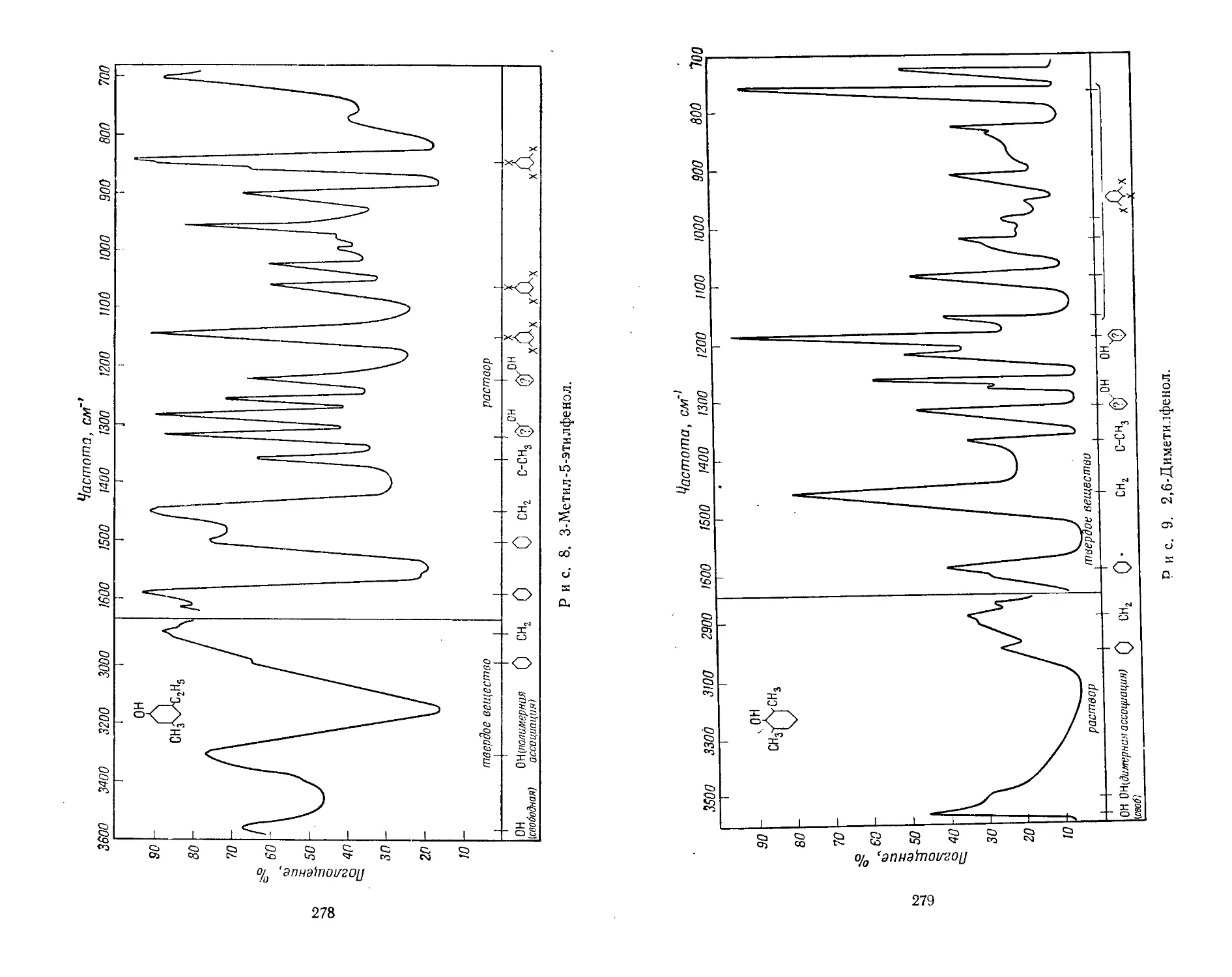
В конце каждой части книги приведены спектральные
кривые, иллюстрирующие некоторые рассмотренные в тек-
сте корреляции. При этом, как уже упоминалось выше,
для полноты сведений иногда использовались сомнительные
данные. Некоторые из них отмечены на спектрах вопроси-
тельным знаком. Приведенные кривые особенно полезны для
тех, кто только начинает знакомиться с данной областью
Гл. 1. Введение
15
науки. Данные спектры были получены на двухлучевом
спектрометре 21В (фирма «Perkin—Elmer») с призмой из
каменной соли. В отдельных случаях спектры были сняты
на однолучевом приборе D209 (фирма «Hilger»).
ЛИТЕРАТУР А
1. Barnes, Liddel, Williams, Ind. Eng. Chem. (Anal.),
15, 659 (1943).
2. Thompson, J. Chem. Soc., 1948 , 328.
3. Williams, Rev. Sci. Instruments, 19, 135 (1948).
4. Barnes, Gore, Stafford, Williams, Analyt. Chem.,
20, 402 (1948).
5. С о 1 t h u p, J. Opt. Soc. Amer., 40, 397 (1950).
6. «The Spectroscopic Panel of the Hydrocarbon Research Group»,
J. Inst. Petroleum, 37, 109 (1951).
7. Barnes, Gore, Liddel, Williams, Infra-red Spectro-
scopy, Reinhold Pub. Corp., New York, 1944.
8. Randall, Fowler, Fuson, Dangl, Infra-red Deter-
mination of Organic Structures, Van Nostrand, New York,
1949.
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1. Чуланове кий В. М., Введение в молекулярный спек-
тральный анализ, Гостехиздат, М.—Л., 1951.
2. Волькенштейн М. В., Ельяшевич М. А,, Сте-
панов Б. И., Колебания молекул, т. II, гл. 29 и 31,
Гостехиздат, М.—Л., 1949.
3. «Рассеяние света и инфракрасная спектроскопия», библиогра-
фический указатель 1928—1940, Издательство АН СССР,
Москва, 1961.
4. Hershenson Н., Infra-red Spectra, Index for 1945—1957,
London, 1959,
Сн,
СН
сн=сн
сн=снг
CH=CR
RC=CR’
CHU
СН ароматич
COOH
ОН
с-0
NH,
3/50
3500
3250
Частота, слГ’
ЗООО 2750
кэ
Ja
. Беллами
=NH
CONH:
CONH (открытая цепь)
CONH (лактамы)
NHjRCOOH
NHJCIRCOOH
NHjRCOO’X*
Пиридин и т n
Азиды
C=N
л,р-Ненасыщенные нитрилы
NHj
CN’ OCN" ит.п
Р-0»
SH
SkH
полимеры
связанна*
своМндя
серия полос
ср
Ср
оч.с
СР; мономер
свобод, одинмостил,
пер пер
свободно я, 2 полосы, си
I ->
сяйобяоц 1П0МХ0. CD
с полосы, с
хелаты, ел
2500
2250
2000
димер, ил., широком
NH*
CN" ОСИ' ц т.п
Р-ОН
SH
StH
а,^-ненасыщенные нитрилы
CeN-
чаще ел
сн,
CM,
CN
CH=CH
CH=CHt
C=CHt
C№CR
RC=CR’
CHO
CH ароматич
COOH
OH
C^O
NHt
NH
=NH
CONNt
CONH (открытая цепь)
CONH (лактамы)
NHjRGOOH
NH^CIRCOOH
NHjRCOO’X4
Пиридин и т.п
Азиды
1. Корреляционная диаграмма для области 3750 — 2000 слг1. Валентные колебания водородных связей
и колебания тройных связей.
Частота. см~'
2000 ____1900 1800 ПОР_______________1600_____________1500
Аллены -сн=снг ^с=снг С=€ С=С (сопряженная) Ароматические системы (все типы замещения, исключая унозонные ниже) — п — (пора- а о симметричные тризамещенныв) — — (тризомещенные с рядам расположенными заместителями) — ~ (сопряженные) — и— щсстипы замещения) C=N C=N (сопряженная или в цикле) N=N СОННг(2лл СО см. в диаг 3) CONH (только открытые цепи) nh2 NH NH* NH*C1R COO’ c-no2 0—NO, N-NO, -0-N=0 Пиридин и т.п Пиримидин и т.п Трополаны каждый то t ср замещения харантери.зу ср. ется определенным спекп пер прр пер с. ср Ср | t ср . . ср - ср ср Аллены -сн=снг >с=сн2 С=С С=С (сопряженная) Ароматические системы (все типы замещения, иск л ю чоя укизонныр ниже) —,,— (пара-и асимметричные тризомещб.нные) — (тпризпмещенныесрядом расположенными заместителями) — и— (сопряженные) — (все типы замещения) C=N C=N (сопряженная или в цикле) N^N СОNHJдля СО см вдиаг 3) CONH (только открытые цепи) NH? NH NHj NH*CIR COO’ C-NO2 O-NOj n-no2 -0-N=0 Пиридин и m П Пиримидин и т.п. Трополоны
пром ср' пер пер
пер
пер связанная, с. сЬдбоднз. ср оч ел '1 с связанная, с свибодная, с
ел ел, с с. _ ср ср. Ср , ср . ср.
2. Корреляционная диаграмма для области 2000 — 1500 см \ Колебания групп с двойными связями
и другие.
Частота, см‘
Насыщенные кетоны и кислоты ft, ^-Ненасыщенные кетоны Арилкртоны ft>3_ а.',^'-Ненасыщенныр и диарилкетс а- Гологенкетоны а-.а'-Дигалогеннетоны Кетоны в виде хелатов Кетоны г 6-членным циклом Кетоны с 5- членным циклом Кртоны ( 4-членным циклом Насыщенные альдегиды a,(i-Ненасыщенные альдегиды а.р-а'.р’-Ненасыщенные альдегиды Альдегиды в виде хелатов ft,^-Ненасыщенные кислоты ft-Галогенкислоты Ароматические кислоты Кислоты с внутримолекулярной водород Ионизированные кислоты Насыщенные сложные эфиры, лактоны сЬ ft, ^-Ненасыщенные и ароматические слс Виниловые и а-гологенза.мещенныр ело. Салицилаты и антранилаты Сложные эфиры в виде * слотов Лактоны с 5-членным циклом ft. р-Ненасыщенные лактоны с 5 щ рнныр Тиоловыр сложные, эфиры Сэлогенангидриды кислот Хлоркарбонаты Ангидриды (открытая цепы ны ной связью - и 7- членным жные эфиры мныр эфиры циклом Л/М <1 г циклами фазност MJU ! бОСАГ' •ни i — 1 W J 350 1 300 Г 550 / 500 Насыщенные кетоны а кислоты a..ft-Ненасыщенные кетоны Арилкетоны а>&~ ^'.^'-Ненасыщенные и диарилкетоны а.- Гзлогенкетоны а, а'-Дигалогенкепфны Кетоны в виде хелатов Кетоны с 6-членным Кетоны с 5- членным Кетоны с 4-членным Насыщенные альдегиды ^.^-Ненасыщенные альдегиды а,р- а'^'-Ненасыщенные альдегиды Альдегиды в виде хелатов а, ^-Ненасыщенные кислоты сс-Галогенкислоты Ароматические кислоты Кислоты евнутрималекулярной водородной Ионизированные кислоты связью Насыщенные сложные эфиры, лактоны я.^Венасыщенные и ароматические Гл^е Виниловые и а.-галогензамещенныё сложные Салицилаты и антранилаты зфиры Сложные эфиры в виде хелатов Лактоны с 5-членным циклом а,^-Ненасыщенныелактоны с 5-членным Тиоловые сложные эфиры циклом Галогенангидриды кислот Хлоркарбонаты
Ангидриды <циклические* Алкильные перекиси Ароматические перекиси Первичные амиды (СО) Вторичные омидыид-лактамы (СО) Третичные амиды (СО) у-Лактамы Лактамы разность разност) ра Чем-' 25 см' ’ Кость 25см~' юнденси- ненок эованные -poke 21 1 < онденсирован ные циклы денси- некой иные сован лы адр ’вободндя свозе зободная сеязс Ъенси ные лы иная иная Ангидриды (открытая цепы Ангидриды (циклические) Алкильные перекиси Ароматические перекиси Персичные амиды (СО) Вторичные амиды и 8-лактамы (СО) Третичные амиды (СО) у-Лантамы р-Лахтамы
3. Корреляционная диаграмма для области 1900-1500 см >. Колебания карбонильной группы (все полосы
С ЦЛ ЬН FJPI,
Частота. сяГ’
1500 то 1300 1200 1100 1000 900 800 ТОО ООО
ПН>С (СН^С . (СН8)>С Сн2 (сн,ц циклогексан Циклобутин Циклопропан Г.Н^Н-1ццС) - СН=Сн- {транс» -СН=Сн7 X=Uh, -С=СН Ароматические соединения 1 монозамещенные) - {1,2 замещенные) - •• - (1.3-зомещенные! - <• - (1,4-замещвнные! - - - 4.2,3-залющенные/ -••-(( 2,4-замещенные! - - - (1.3.5-замещенные) RCHQ С^СНО Я COR CgH$COR COOK ГИ) Формиаты Ацетаты Фенолацетаты Пропионаты и т п Акрилаты и т п Бензоаты и т.п Эпоксиды Алифатические перекиси Ароматические перекиси Ангидриды (открытаяцвпы — <• — (циклические) CONHj сн2осн8 Ароматические простые эфиры Ср ср- ср ср с пер А сс ffi £ ср £ с. & СЛ сл СЛ 3L. СЛ. СЛ. £Е Ср . а с. <?Р ср Ср “* а. с сл сн8с <СН3|гС 1СНа),с снг <СНг)4 циклогексан Циклобутан Циклопропан -СН^СН-щисГ -СМ=СН- (транс) -сн=снг >С=СНг >С=СН- Аромати ческие соединения (монозамещенные) - v -(1,2-замещенные) - я— Ц,3-замещенные) - Ц 4-замещенные) -11,2.3-замещенные) - “-(1.2.4-замещенные) — •• —(/. 3,5-замещенные) ясно с6н5сно RCOR CgHjCOR СООН гх0 Формиаты Ацетаты Фенолацетаты Пропионаты а т. п Акрилаты и т п Бензоаты и п< п Эпоксиды Алифатические перекиси Ароматические перекиси Ангидриды (открытая цель) — " —- щиялические/ CONHt CHjOCNs Ароматические простые эфиры
г • ср сл. 1 полоса, сл, 1 'н
пер. 2ПОЛОСЫ.СЛ. а с. 1
пер СЛ, СЛ сл. .)палосо,сл
пер пер 2ПОЛОСЫ.СЛ. с
СЛ сл. сл_1полоса ил. ср с. *“
пер .2ПОЛОСЫ, СЛ.
пер ил. Ср 1 полоса, ел. Ср сл.
ср сл
Ф ср ср ср ЕЕ. ер •
с с.. с, с
с с ЙР _
a
с
с с.
с.
—а—
4. Корреляционная диаграмма для области 1500—650 сл-1. Колебания групп с простыми связями и
другие (I).
Частота г см ’ 1300 BOO ПОР 1000 SOO S00 7ПП т
CHjOH снон 5С-0Н Фенолы C6Hs-NH2 CfcH5-NH- C6HS-N< Пиридин Пиримидин Р=0 Р-0—С(алкил) Р-О-С(арил) Р- P=S P-F <RO)aPCg C-F C-Cl -CF-CF3 C-nOj o-nd2,n-no2 C-№0, N-N=0 no; S.ICHj), Si{CM3)2 Si(CH3) S|“C6H5 Si“O~Si (открытая цепь) Sl-O“Si(e<jUKrte) co; so; no; nh4* PO4.HPO4.Hjpq, Силикаты N-C=S c-s- -S0- ) -5 Of -S03H -SOjCI.SOjO-.SOjN- c c. — <? OOj ССЫ, c. CH20H СНОН SO-OH <' Фенолы CsHrNHx CsH5-NH~ CSHS-N< Пиридин Пиримидин P=0 Р-Ъ-Ъ(алкил) P~-0-C(apuM P-CSH5 p^-s P-F iROIjPig C-Cl -CF-CFj C-NOj 0-N02, N-NOt C-N=O, N-N=0 no; SilCHji, Si(CHslj Si(CH3) Si-CbH5 Si- 0 -Sv (открытая цепь) Si-O-Si (в цикле) co; SO4 N0£ nh4* PO4.HPO4.MjPq! Силикаты N-C=S C-S- -so- -so2- -SO3H -SOjCl, SOjO-.SOjN-
c.
c c. c
c.
0 £
c
C6QC
’’дная, с связан -<вя, c cn3 c,hs алкил a арил, c. c nep.
ou.c
cp
cp t СЛ
nep c tN 1. 04
cp.
q
оч.с . c c
04 c
СЛ
04 c
—
c
4.
5. Корреляционная диаграмма для области 1500-650 См~\ Колебания групп с простыми связями и
другие (II).
Глава 2
АЛКАНЫ
1. ВВЕДЕНИЕ
Изучение характеристических полос поглощения для
различных типов углеводородных структур привело к уста-
новлению ряда надежных соотношений между положением
полос и типами присутствующих групп. Наиболее изучены
корреляции, касающиеся валентных и деформационных
колебаний С—Н, а для некоторых типов разветвленных
структур найдены также корреляции, относящиеся к коле-
баниям С—С скелета. Данные о поглощении, соответствую-
щем валентным колебаниям С—Н, особенно полезны при
распознавании насыщенных и ненасыщенных соединений;
располагая же сведениями о деформационных колебаниях,
можно получить указание о примерном количественном
соотношении групп СН3, СН2 и СН в углеводородах.
Данные о симметричных деформационных колебаниях
группы СН3 используются также в структурных исследо-
ваниях для распознавания как связи С—СН3, так и раз-
ветвленных структур; идентификация последних может
быть проверена по частотам колебаний скелета.
В этой главе рассматриваются также корреляции для
группы —(СН2)4— и для насыщенных циклических угле-
водородов. Обсуждаемые корреляции приведены в табл. 1.
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С - Н
а. Положение полос. Инфракрасные полосы поглоще-
ния, соответствующие валентным колебаниям С—Н для
органических веществ, находятся в области около 3000 см~'.
Это было установлено еще в 1905 г. Кобленцем [11, а
в 1929 г. Бонино [21 указал, что характеристические поло-
сы колебаний С — Н ароматических и алифатических сое-
динений находятся в данной области в различных местах.
Таблица 1
Частота,
Валентные колебания С—Н
сн3 2962 и 2872J-10 (с.)
сн2 2926 и 2853^-10 (с.)
СН (третичный) 2890±10 (сл )
Деформационные колебания С—Н
С—СН3 (антисимметричные) 1450^20 (ср.)
-СН2- 1465±20 (ср.)
С—СН3 (симметричные) 1380—1370 (с.)
—С—(СН3)2 1385-1380 и 1370-1365 (с., интенсивности при- близительно равны)
—С—(СН3)3 1395 — 1385 (ср.), 1365 (с.)
—СН— Около 1340 (сл.)
Колебания скелета
(СН3)3—С—R 1250±5 (с.), 1250—1200 (с.), около 415
(СН3)2—С 1170±5 (с), 1170-1140 (с.), около 800
-(СН2)4- 750—720 (с.)
—(СН2)4—0— (или другая функцио- 742—734 (с.) (к углево-
нальная группа) дородам неприменимо)
Циклические системы
Циклопропан 1020—1000 (ср.)
Циклобутан (предположительные зна- 920-866 или 1000—960
чения) (ср-)
Циклопентан » нет характеристич. ко- лебаний
Циклогексан » 1005—925 и 1055—1000 (ср-)
24
Гл. 2. Алканы
25
Фокс и Мартин [3—6] изучили природу этих полос и уста-
новили связь их положения с различными типами строе-
ния. В ряде работ, опубликованных в период с 1937 по
1940 г., этим авторам удалось показать, что в случае угле-
водородов положение максимума поглощения, соответ-
ствующего валентным колебаниям С — Н, почти цели-
ком определяется природой самой связи и фактически не
зависит от других особенностей строения. Так, группам
СН3, СН2 и СН насыщенных соединений соответствуют
близко расположенные максимумы поглощения; двойная
связь ароматических соединений, образованная углеродом,
к которому присоединен атом водорода, вызывает смеще-
ние полосы С — Н в сторону больших частот, тогда как
при наличии одних лишь ненасыщенных алифатических
соединений наблюдается дальнейшее и еще большее ее сме-
щение. При использовании призмы из хлористого натрия
с ее сравнительно низкой дисперсией удается только рас-
познать главные классы соединений, но и это представляет-
ся ценным. Расмуссен и Браттен [7], например, исполь-
зовали указанную спектральную область для обнаруже-
ния ненасыщенности в олефинах. Между тем дисперсия
призмы из фтористого лития уже достаточна для дифферен-
цирования различных типов групп с СН-связями основ-
ных классов соединений, так что могут быть отдельно иден-
тифицированы группы СН3, СН2 и СН. Максимумы погло-
щения, соответствующие колебаниям в ненасыщенных и
ароматических соединениях, рассматриваются в гл. 3 и 5,
а в настоящей главе обсуждаются лишь полосы, которые
обусловлены валентными колебаниями С — Н, когда все
связи данного атома углерода являются насыщенными.
Группа СН3. Фокс и Мартин [6] исследовали большое
число углеводородов, содержащих метильные группы, и
нашли, что во всех случаях имеются две сильные полосы—
при 2962 и 2872 сл/'1, соответствующие антисимметричным
и симметричным валентным колебаниям. Для всех исследо-
вавшихся соединений эти значения менялись не более чем
на + 10 сл/'1. Кроме того, в некоторых случаях были заме/
чены две более слабые полосы с промежуточными часто-
тами. Положение обеих главных полос не выходило за ука-
занные пределы при наличии по соседству с метильной
группой двойной связи, но в этих случаях полоса с мень-
26 Часть!
шей частотой колебаний СН3 (2872 слГ1) расщеплялась
в результате резонансного эффекта на две. Среднее значе-
ние частоты для этой пары полос оставалось, однако,
с точностью до нескольких обратных сантиметров равным
2872 см1.
Группа СН2. Эта группа дает начало двум характери-
стическим полосам при 2926 и 2853 см', которые также
соответствуют антисимметричным и симметричным валент-
ным колебаниям [4, 6]. Как и в рассмотренном выше случае,
средние значения частот оставались постоянными в пределах
т? 10 слГ1, причем это относится не только к углеводородам
алифатического ряда, но также и к колебаниям СН2 для
таких соединений, как бензиловый спирт и дифенилметан.
Для соединений, содержащих группу— СН2— С=С,
наблюдался резонансный эффект расщепления, аналогич-
ный наблюдаемому для группы СН3 (см. выше). Средняя
частота и в этом случае близка к нормальному значению.
Группа СН. Интенсивность полосы поглощения группы
/7СН очень мала по сравнению с группами СН2 и СН3;
однако Фокс и Мартин [61 полагают, что слабая полоса,
которая может быть замечена при 2890 слГ ' у таких соеди-
нений, как трифенилметан, является характеристичной.
Эти данные имеют, однако, очень ограниченную ценность.
Когда группа СН непосредственно связана с кислородом,
интенсивность полосы значительно возрастает, но смеще-
ние ее, обусловленное присутствием атома кислорода,
усложняет картину, так что Позевский и Коггешелл [501
не сумели, например, идентифицировать у кислородсодер-
жащих веществ какого-либо специфичного поглощения
связанного с этой группой.
Все рассмотренные выше корреляции были позднее
полностью подтверждены другими исследователями [8, 9],
и не известно ни одного случая отклонения от них для
насыщенных углеводородов. Однако изменения силового
поля в непосредственной близости от группы СН могут
влиять на частоту валентных колебаний, и именно по этой
I
причине частота =С — Н отлична от частоты —С—Н.
Аналогичным образом замещение такими атомами, как
Гл. 2. Алканы
27
галогены, с присоединением их к углероду группы СН или
введение групп СН2 в напряженный цикл должно привести
к изменению частот валентных колебаний, и эти корреляции
уже не будут пригодны.
Область 2850—2500 см 2. Для этой области не было
предложено никаких корреляций, но известно, что в ней
лежат полосы поглощения многих углеводородов. Никаких
объяснений происхождения этих полос нет. Они обычно
очень слабы в случае углеводородов, но такие соединения,
как диоксан, тетр а гидрофуран и циклогексанон, имеют
несколько более интенсивные полосы, которые легко можно
спутать с поглощением, обусловленным валентными коле-
баниями S — Н, поскольку последнее наблюдается в этой
же области и сравнимо по интенсивности.
б. Факторы, влияющие на характеристичность частоты
валентных колебаний С—Н. Как было указано выше, кор-
реляции, выведенные для частот валентных колебаний
С — Н, непосредственно применимы только к ненапряжен-
ным углеводородам, в .которых рассматриваемые группы
связаны с другими углеродными атомами. Влияние заме-
стителей другого рода исследовано еще недостаточно глу-
боко, чтобы можно было сделать сколько-нибудь точные
выводы относительно степени смещения полосы в каждом
случае. Однако смещение может быть весьма значитель-
ным; например, метилдейтерпд и моногалогенпроизводные
метана (исключая фтористый метил) поглощают вблизи
2970 и 3060 см 1, причем разности частот соответствующих
полос составляют 87 и 86 слГ1. Когда метильная группа
присоединена к азоту, полоса 2872 см'1 смещается обычно
в сторону больших частот [10], тогда как присоединение
к кислороду вызывает обычно смещение в сторону боль-
ших частот полосы 2960 см'1. Кроме того, в интервале
2832—2815 см'1 появляется новая полоса, которая весьма
характеристична для метоксильной группы (1131. Если
метильная группа находится по соседству с карбониль-
ной, как в ацетатах, то происходит также некоторое сме-
щение всех полос.
Позевский.и Коггешелл [50] провели недавно исследо-
вание для того, чтобы выяснить, можно ли применить
представление о характеристичности частот валентных
28
Часть I
колебаний С — Н к соединениям, содержащим серу и кис-
лород. Они исследовали 24 серусодержащих и 45 кислород-
содержащих соединений, используя высокую дисперсию
призмы из фтористого лития. Полученные ими результаты
приводятся ниже.
2962 см~1 (колебания СН3). Правильность отнесения
полосы 2962 см1 к антисимметричным валентным колеба-
ниям метильной группы была подтверждена тем фактом,
что относительная интенсивность этой полосы очень сильно
возрастает у соединений с высокой степенью разветвления
цепи. Исключая первые члены ряда, которые часто пока-
зывают аномалии, авторы нашли, что введение атома серы
мало влияет на эту полосу. У таких соединений полоса
находится в интервале 2969—2960 сл/-1; исключением
являются этилсульфиды, у которых она лежит между 2980
и 2970 см-1. Кислородсодержащие вещества претерпевают
большие изменения, и для метил-н-бутирата могут быть
разрешены две полосы при 2970 и 2952 сл/-1, соответствую-
щие двум различно расположенным метильным группам.
У большинства соединений, содержащих кислород, эта
полоса лежит в области 2972—2960 сл/-1, но при наличии
связи С2Н5— О частота возрастает до 2990—2980 сл/-1,
а при наличии изопропиловой, втор-бутиловой и трет-бу-
тиловой групп —до 2978—2973 сл/-1.
Было найдено, что у соединений с неразветвленной
цепью легко разрешаются две полосы, а у многих развет-
вленных соединений имеется в этой области одна-един-
ственная полоса. Таким образом, отсутствие отдельной
полосы, присущей группе СН2, не означает еще отсутствия
этой группы, тогда как наличие в этой области двух полос
служит достаточно обоснованным доказательством наличия
как группы СН3, так и группы СН2.
Интересной особенностью спектров является то, что
у веществ, содержащих кислород и серу, коэффициент пога-
шения полос валентных колебаний С — Н в этой области
заметно больше найденного для углеводородов Фоксом и
Мартином.
2930 сл/-1 (колебания СН3 и СН2). Все исследованные
соединения поглощают в области 2948—2922 сл/-1 [50].
В некоторых случаях могут быть разрешены отдельные
полосы, обусловленные наличием групп CHS и CHai
Гл. 2. Алканы
29
• 2890—2850 см'1 (колебания СН3 и СНг). Было найдено,
что наличие в этой области полос колебаний групп СН3
и СН2 хорошо согласуется с корреляцией, установленной
Фоксом и Мартином [61, а введение в молекулу кислорода
или серы не приводит к заметному изменению частот.
Среднее значение частоты колебаний метильной группы
равно 2880 см'1 для веществ, содержащих кислород, и
2875 см'1 для веществ, содержащих серу. Колебания
СН2 для обоих типов соединений проявляются при среднем
значении частоты 2861 см'1.
Эти корреляции для кислородсодержащих соединений
полностью подтвердили Нол ин и Джонс [79], которые изу-
чали различные частично дейтерированные диэтилкетоны.
Так, например, С2Н5СОС2Н5 имеет полосы при 2977, 2936,
2883 и 2902 см'1. Три первые полосы отсутствуют в спект-
рах CD3CH2COCH2CD3 и, следовательно, должны быть
связаны с группами СН3. Присутствие только одной полосы
валентных колебаний СН2 в спектрах этих соединений обус-
ловлено, вероятно, значительными изменениями интен-
сивности, которые вызваны тем, что группа СН2 связана
с карбонилом, причем вторая, очень слабая полоса, по-ви-
димому, появляется при 2955 см'1.
Еще одним фактором, который может привести к сме-
щению полос валентных колебаний С — Н, является на-
пряжение цикла. Частоты валентных колебаний С — Н
ненапряженных циклов имеют обычные значения. Плайлер
и Аквиста [41] показали, что у 19 производных циклопен-
тана смещение полос СН2 от нормального значения неве-
лико . Однако в случае меньших циклов влияние размеров
цикла на длины связей становится заметным и имеет место
смещение полос. Робертс и Чемберс [51] показали, что
при уменьшении размеров цикла частоты полос СН2 увели-
чиваются, а интенсивности уменьшаются. Из этого они
заключили, что распределение электронной плотности по
связям С — Н является функцией размера кольца. Эти
авторы исследовали полосы СН2 трех-, четырех-, пяти-
и шестичленных циклов, содержащих один атом хлора или
брома. Они нашли, что для хлор- и бромзамещенных шести-
членных циклов частоты имеют нормальные значения;
Для замещенных пятичленных циклов значения частот
очень близки к нормальным, а при дальнейшем уменьше-
30
Ч а с in 1> 1
нии кольца они сильно увеличиваются, так что для
бромциклопропана частоты колебаний СН2 равны 3077
и 2985 сл/-1, в то время как для бромциклогексана они
равны 2924 и 2841 слг1. Для циклобутана [80] соответ-
ствующее увеличение частоты валентных колебаний С — Н
является не настолько большим, чтобы ее значение пре-
высило 3000 см'1, но производные циклопропана уже можно
легко идентифицировать по частотам этих колебаний [62, 73],
если отсутствуют ненасыщенные связи. Частота валент-
ных колебаний метилена высока также у стероидов и три-
терпенов, которые содержат мостиковые структуры, вклю-
чающие циклопропановое кольцо; для их идентификации
было использовано поглощение в области 3060^-3040 см 1
[74]. Таким же образом можно обнаруживать эпоксигруп-
пы [114], при условии, что они неполностью алкилированы,
по поглощению в интервале 3056—2990 слг-1.
Высокое значение частот валентных колебаний С—Н
для упомянутых трехчленных колец отражает их хорошо
известный «ненасыщенный» характер. В связи с этим бы-
вает затруднительно отличить их по поглощению от аро-
матических и олефиновых соединений.
Остается отметить еще два второстепенных фактора,
которые могут обусловить небольшие смещения полос.
Фокс и Мартин [4] нашли, что малые смещения происходят
в результате изменения физического состояния вещества.
В этих случаях смещения всегда невелики и происходят
в одном и том же направлении. Переход от парообразного
состояния к раствору сопровождается уменьшением частоты
примерно на 7 сл/-1, а при переходе от раствора к чистой
жидкости частота уменьшается еще на такую же величину.
Эти данные получены для раствора в четыреххлористом
углероде. Другим фактором, который может привести
к небольшим изменениям характеристических частот,
является возникновение водородной связи. Предполагалось,
что на валентные колебания С — Н этот фактор не влияет,
однако Сазерленд [12] недавно показал, что имеется уже
достаточно много доказательств противного. Например,
установлено [81, 82], что в случае хлороформа может
возникнуть водородная связь с атомами кислорода, а нитро-
форм CH(NO2)3 является сильным донором протонов и его
можно титровать щелочью. Тем не менее изменения частот,
Гл. 2. Алканы
31
которых можно при этом ожидать, должны быть очень
малыми по сравнению с изменениями, обусловленными
межмолекулярными силами, так что появление водород-
ной связи не имеет сколько-нибудь большого значения
при исследовании характеристических частот.
в. Факторы, влияющие на интенсивность. Роуз [13]
в 1938 г., изучая обертоны колебаний С — Н, заметил,
что интенсивность поглощения различных структурных
групп молекулы в значительной степени остается постоянной,
что было подтверждено Фоксом и Мартином [6], исследо-
вавшими основные частоты. Используя в качестве меры
интенсивности площадь полос поглощения, они нашли,
что для парафинов с длинной цепью интегральные интен-
сивности полос СН2 и СН3 непосредственно связаны с чис-
лом этих групп, присутствующих в соединении, и интен-
сивность полосы СН2 при каждом увеличении длины цепи
на одну группу увеличивается на постоянную величину.
В случае разветвленных цепей это правило выполняется
несколько хуже, однако если при оценке интегральных
интенсивностей принимаются во внимание все полосы СН3,
то оно выполняется удовлетворительно. Для 2,3-диметил-
бутана, 2,2,3-триметилбутана и 2,3,4-триметилбутана соот-
ношение числа групп СН2 было найдено равным 4,42 : 5 : 5,21
вместо 4:5:5.
Если по соседству с группой СН3 находится двойная
связь, то при измерениях, ограничивающихся нормаль-
ными частотами, из-за расщепления полос этот метод
непригоден, однако опять-таки определение интегрального
поглощения по всем полосам метильных групп дает доволь-
но хорошие результаты. Для бутена-1, цис- и транс-буче-
нов-2 и триметилэтилена авторы указанной работы полу-
чили соотношение числа метильных групп в молекулах,
равное 1 : 1,76 : 1,89 : 3,50 вместо 1:2:2: 3.
Подобное же постоянство интенсивностей поглощения
было найдено для групп =СН и =СН2. Те же авторы при-
водят следующие значения относительных интенсивностей,
определенные как суммарное интегральное поглощение по
всем полосам, связанным с соответствующими группами:
^СН 410, =СН2 1800, ^СН 120, >СН2 2400, ^>С — СН3
32
Часть I
2230,
СН — СН3 3800. Из этих данных видно, что интен-
сивность поглощения групп СН2 и СН3 уменьшается, когда
они располагаются у двойной связи, и что интенсивность
поглощения групп СН сравнительно мала.
В какой-то степени аналогичное исследование было
проведено Френсисом [9], хотя для получения значений
интенсивностей, характерных для таких структурных эле-
ментов, какими являются группы СН3, СН2 и СН, им
использовались полосы 2900, 1460 и 1375 см'1. Френсису
удалось показать, что для 12 алифатических углеводородов
рассчитанные интенсивности поглощения каждой из этих
полос в общем близки к наблюдаемым значениям и что
только для очень сложных веществ с разветвленной цепью
имеются заметные отличия от аддитивных значений, полу-
ченных при рассмотрении углеводорода как суммы невза-
имодействующих групп СН3, —СН2— и —СН—. В одной
из последующих работ [72] он предложил использовать для
определения метиленовых групп парафинов интегральную
интенсивность полосы между 790 и 685 см'1. Радикалы цик-
лопентил и циклогексил можно определять отдельно, произ-
водя оценку интенсивностей полос при 2957 и 2926 см'1
после вычета накладывающегося поглощения метильной и
метиленовой групп.
Таким образом, количественные измерения в области
3000 см'1 позволяют определить относительное содержание
в углеводороде групп СН3, СН2 и СН. Хастингс и др. [61]
применили аналогичный метод, включая также измерения
в области 800—700 см'1, для определения метильных и мети-
леновых групп в парафинах и метиленовых групп в заме-
щенных нафталина в сложных смесях.
Однако, как уже отмечалось, Позевский и Коггешелл
[50] нашли, что коэффициент погашения для групп СН2 и
СН3 в области 2890—2850 см'1 не остается тем же самым
в случае кислородсодержащих соединений, так что рассмат-
риваемые закономерности без дополнительных фундамен-
тальных исследований не могут быть применены для сое-
динений, не являющихся углеводородами. Это было под-
тверждено Френсисом [71], который обнаружил заметное
уменьшение интегральной интенсивности поглощения мети-
льных и метиленовых групп, связанных с атомом кислорода
Гл. 2. Алканы
33
или карбонильной группой. Мироне и Фаббри [83] также
заметили, чго общая интенсивность полосы СН2 для вто-
ричных и третичных спиртов ниже, чем для соответствую-
щих первичных соединений. Эго вызвано, по-видимому,
увеличением числа групп СН2, связанных с атомом кисло-
рода.
Как показал Роуз [13], аналогичные определения воз-
можны в области обертонов; Хиббард и Кливе [14] исполь-
зовали для этой цели область вторых обертонов; Туот и
Барчевиц [15] нашли, чго интенсивности полос СН3 и СН2
в этой области являются функцией числа присутствующих
групп, и использовали этот факт для анализов. Готье [52]
применил аналогичный метод для определения числа раз-
ветвлений в жидких углеводородах, исследуя область
8000—5J00 слГ1. Эванс, Хиббард и Пауэлл 153] также
интенсивно изучали эту область и показали, что она может
быть удобной для выяснения строения больших молекул
с молекулярным весом вплоть до 5J0. Они пришли к выводу,
что можно определить относительное содержание различ-
ных групп с точностью до 0,40 группы (или большей) в пара-
финах, а в нафтенах и ароматических соединениях с точ-
ностью, равной (или большей) соответственно 1,2 и 0,9 груп-
пы. Обзор характеристических частот и анализ области
обертонов был сделан в работах Кея [84] и других авто-
ров [85].
Эга область имеет для исследований такого рода много
преимуществ, поскольку может быть использована обычная
оптика и имеется возможность применения высокочувстви-
тельных приемников.
3. ДЕФОРМАЦИОННЫЕ КОЛЕБАНИЯ ГРУПП СН3 —С и СН2
а. Положение полос. Уже давно при изучении инфра-
красных спектров было установлено, что группы СН3
и СН2 обнаруживают поглощение при 1460 см *, обусло-
вленное деформационными колебаниями [16—18]. По-
скольку впоследствии в лаборатории Американского неф-
тяного института (АНИ) было получено большое число
спектров углеводородов и проведен полный математический
анализ спектров многих простых углеводородов, это погло-
щение было интерпретировано весьма точно. В настоящее
3 Л. Беллами
34
Часть 1
время общепринято, что поглощение, соответствующее де-
формационным колебаниям СН2, очень близко к 1465 см'1,
а поглощение при 1450 слг1 соответствует антисимметрич-
ным деформационным колебаниям СН3. Изменение этих
значений более чем на 10—20 см'1 наблюдается исключи-
тельно редко и разве только в тех случаях, когда присут-
ствуют сильно электроотрицательные атомы, вызывающие
изменение распределения заряда на углеродном атоме.
Данные, на которых основывается такое отнесение,
первоначально были получены при изучении спектров срав-
нительно большого числа простых углеводородов; эти
данные были затем подвергнуты различными исследователя-
ми математической обработке. Правильность отнесения
полос поглощения групп СН3 и СН2 в эту область подтвер-
ждается спектрами АНИ; кроме того, сведения о спектрах
комбинационного рассеяния также свидетельствуют о пра-
вильности отнесения частот в этой области к деформацион-
ным колебаниям групп СН2 и СН3 [19, 34]. В случае груп-
пы СНз деформационные колебания атомов водорода, свя-
занных с углеродом, обусловливают появление двух полос:
антисимметричные колебания дают полосу 1460 см'1, тогда
как симметричные обусловливают появление полосы при
1375 см'1. Соответствующая полоса деформационных коле-
।
।
баний —С—Н определена менее точно. Это объясняется
I
прежде всего ее низкой интенсивностью по сравнению
с интенсивностью полос СН2 и СН3, и, кроме того, следует
отметить, что соединений, содержащих эту группу, изучено
меньше, чем соединений, содержащих группы СН2 и СН3.
Такая слабая полоса появляется у углеводородов при
1340 см'1 [10, 63, 69], но она обычно недостаточно интен-
сивна, чтобы иметь большое значение для структурного
анализа, хотя в определенных случаях, когда указанная
группа соединена с атомом кислорода или азота, происходит
значительное увеличение интенсивности.
Полосы в области 1460 см'1, обусловленные группами
СН2 и СН3, лежат слишком близко одна от другой, и потому
обе эти группы нелегко различить; кроме того, в зависимости
от изменения состояния могут происходить небольшие сме-
щения полос, что вносит дополнительную трудность. Однако
Г л. 2. Алканы
35
полная интенсивность поглощения в области 1460 см'1
прямо пропорциональна числу таких групп, что и было
использовано Френсисом [9], располагавшим также дан-
ными о других полосах, для расчета относительного содер-
жания групп СН3, СН2 и СН, присутствующих в молекуле.
Симметричные деформационные колебания водородных
атомов метильной группы связаны с появлением полосы
поглощения в интервале 1385—1370 см'1, которая исклю-
чительно стабильна по своему положению, если только эта
группа присоединена к другому атому углерода. Эта корре-
ляция имеет большое значение особенно потому, что проис-
ходящее в определенных условиях расщепление полосы
позволяет идентифицировать системы с разветвленными
цепями. Она была установлена в результате интенсивного
изучения углеводородов во время второй мировой войны
[16, 17, 38, 54] и была доказана Расмуссеном [28] в 1948 г.
Как и в случае полос поглощения деформационных колеба-
ний метиленовых групп, указанная корреляция выведена
на основании данных о многочисленных спектрах углеводо-
родов, которые были проанализированы математически,
а также на основании данных, полученных из спектров АНИ,
которые эту закономерность подтверждают [60, 64].
Интенсивность упомянутой полосы, приходящаяся на
каждую группу СН3, значительно больше, чем интенсив-
ность полос, соответствующих деформационным колебаниям
метиленовой группы и антисимметричным деформацион-
ным колебаниям метильной группы, а полная интенсив-
ность прямо пропорциональна числу присутствующих групп.
Френсис [9] показал, что даже тогда, когда имеет место
расщепление полосы, как в случае разветвленных цепей,
интегральное поглощение в области 1380 см'1 связано
с числом групп СН3 и в сочетании с данными из других
областей спектра может использоваться для определения
числа присутствующих метильных групп. Эти данные о по-
глощении были также использованы Фрименом [65] при
определении степени разветвленности жирных кислот.
Лютер и Червоный [86] провели обширное исследование
интегральных интенсивностей полос поглощения СН для
нормальных парафинов состава до Сзг и для многих веществ
с разветвленной цепью. Они приводят коэффициенты инте-
гральной интенсивности поглощения для ножничных и веер-
3*
36
Часть t
ных деформационных колебаний СН2 и для антисимметрич-
ных и симметричных деформационных колебаний СН3.
Поскольку теперь имеется больше данных об изменениях
интенсивностей в зависимости от структурных изменений,
подобные исследования очень полезны для корреляционных
работ.
б. Факторы, влияющие на положение и интенсивность
полос деформационных колебаний СН2 и СН3. Как и в слу-
чае частот валентных колебаний С—Н, изменение агрегат-
ного состояния может вызвать изменение этих частот на
величину до 10 см"1. Ричардс и Томпсон [21], например,
обнаружили смещения такого порядка при исследовании
ряда углеводородов в жидком и твердом состояниях. Однако
эти смещения сопровождались и другими изменениями
спектра. Было замечено, что в спектрах твердых веществ
полосы становятся резче и во многих случаях раздваи-
ваются. Этот эффект был подтвержден Сазерлендом [12],
который нашел, что ниже точки перехода твердые пара-
фины имеют при 1460 см"1 двойную полосу поглощения,
а выше этой точки и для жидкости имеется одна полоса.
Это происходит в результате взаимодействия между дефор-
мационными колебаниями СН2. Аналогичные данные сооб-
щались Шеппардом и Симпсоном [64] для многих других
углеводородов.
Изменения частот деформационных колебаний СН2
вызываются также введением в непосредственной близости
электроотрицательных групп. Так, например, в случае
ненасыщенных веществ частота деформационных колеба-
ний СН2 в группе винильного типа = СН2 смещена до
1420—1410 см"1, и этот факт имеет значение для иденти-
фикации двойной связи такого типа (гл. 3). В алленах
[22] частота СН2 уменьшается еще больше —до 1389 см"1.
Влияние ненасыщенной группировки простирается только
на атом углерода, участвующий в образовании двойной
связи, а частоты соседних групп СН2 изменяются сравни-
тельно мало. Когда же двойная связь находится в цикличе-
ской системе, иногда происходят небольшие смещения полос
в сторону меньших частот и для соседних метиленовых
групп. Например, у многих стеринов полоса деформацион-
ных колебаний СН2 в группировке —С = С—СН2 — нахо-
Гл. 2. Алканы
37
-дится при 1438 см'1 [66]. Изменения, обусловленные заме-
щением атомами галогенов, опять-таки касаются лишь
деформационных колебаний С — Н при том атоме угле-
рода, к которому присоединен галоген. В случае фтори-
стого метилена [23] частота увеличивается до 1508 см'1,
тогда как у хлористого метилена [24] она равна 1429 см'1;
антисимметричным деформационным колебаниям фтористого
метила соответствует полоса поглощения при 1471 см'1,
а йодистого метила [25] — при 1441 см'1. Такие смещения
наглядно проиллюстрированы Брауном и Шеппардом [26]
на примере спектров некоторых бромпроизводных углево-
дородов; 1,2-дибромэтан поглощает при 1435 см'1, а бро-
мистый н-бутил и бромистый н-пропил поглощают при
1435 см'1 и, кроме того, при 1460 см'1; 1,4-дибромбутан
имеет две частоты деформационных колебаний СН2: 1433
и 1440 см'1. Смещения имеют также место, когда рассмат-
риваемая группа присоединена к другим атомам, таким,
как азот или сера, однако их величина неопределенна и
в какой-то степени зависит от природы других замести-
телей у этих атомов.
Интенсивность полосы деформационных колебаний мети-
леновой группы, прилегающей к карбонилу, сильно воз-
растает, и полоса смещается в сторону меньших частот,
аналогично тому, как это происходит в случае винильног!
группировки. Ацетон, например, поглощает при 1431 см'1,
а уксусная кислота — при 1418 см'1. Френсис [71] отнес
к деформационным колебаниям метиленовой группы, при-
лежащей к карбонилу, частоту 1410 см'1, и это значение
было подтверждено для многих соединений. Так, Хаджи
и Шеппард [20] наблюдали для кислот с группировкой
— СН2СООН, сильную полосу при этой частоте, которой
нет у других кислот и которая отличается от полосы
1420 см'1 самой карбоксильной! группы (гл. 10). Резуль-
таты изучения кетонов СН3— СН2— СО — СН2—СН3,
СН3— CD2— СО— CD2-CH3 и CD3— СН2— СО —СН2—CD:i
показали [67, 79], что деформационным колебаниям СН2
соответствует частота 1415 см'1, а симметричным дефор-
мационным колебаниям СН3— 1380 см'1. Было изучено
также очень много стероидов и найдено, что их активные
метиленовые группы, прилежащие к карбонилу в 3-м или
17-м положении иди в боковой цепи, поглощают в области
38
Часть I
1418—1408 сл'1. Между тем карбонильная группа в поло-
жениях 4, 6, 7, 11 или 12 оказывает, по-видимому, несколько
меньшее влияние на прилежащие группы СН2, которые
поглощают при 1434 ел/'1, т. е. при частоте, которая близка
к частоте колебаний метиленовой группы в цикле, активи-
рованной двойной связью [66, 67, 87]. Эти данные были
подтверждены результатами исследования дейтерирован-
ных соединений.
В стероидацетатах влияние группы СО на симметричные
колебания СН3 незначительно, и хотя для 3- и 17-ацетатов
обнаружены две полосы при 1375 и 1365 см"1, это обусло-
влено, вероятно, пространственными эффектами, анало-
гичными тем, которые приводят к расщеплению полосы
валентных колебаний С—О (гл. 11), а не влиянием карбо-
нильной группы. Однако когда метильная группа присое-
динена непосредственно к группе СО, влияние последней,
конечно, более ярко выражено [66], и к симметричным дефор-
мационным колебаниям СН3 стеринов с группой СОСН3
в боковой цепи отнесена частота 1356 см"1.
В напряженных циклических системах также может
происходить небольшое перераспределение электронной
плотности в метиленовых группах и, следовательно, изме-
нение характеристической частоты. Частоты СН2 окиси
этилена, имида и сульфида равны соответственно 1500,
1475 и 1471 см"1 [27]. Легко заметить, что этот эффект
невелик, а при увеличении цикла и уменьшении его напря-
женности частоты СН2 приближаются к нормальному зна-
чению.
У шестичленных стероидных колец эта частота на-
ходится между 1456 и 1448 см"1, т. е. она немного меньше,
чем частота метиленовых групп в боковой цепи (1468—1466
ел'1), так что в благоприятных случаях можно различить
эти два типа групп [66, 67]. Если циклопропановые кольца
включены в большие системы, такие, например, как три-
терпены, то частоты деформационных колебаний СН недо-
статочно характерны для их непосредственной идентифика-
ции [74]. Однако положение и тип таких колец могут
быть найдены путем изучения изменений частот деформа-
ционных колебаний СН3 и СН2, происходящих при размы-
кании колец под действием хлористого водорода или хло-
ристого дейтерия [88].
Гл. 2. Алканы
39
Смещения полос поглощения симметричных колебаний
СН3, обусловленные изменением агрегатного состояния,
изучались Ричардсом и Томпсоном [21 ], которые нашли, что
такие смещения сравнительно невелики и редко превышают
10 см'1. Изменение характера заместителей при углеродном
атоме, к которому присоединена метильная группа, также
мало влияет на положение полосы, за исключением тех
случаев, когда присоединяется вторая метильная группа.
Это иллюстрируется следующими значениями частот, отне-
сенных к симметричным колебаниям СН3 в молекулах,
которые подвергались математическому анализу [29J:
ацетальдегид 1370, метилизоцианат 1377, метилацетилен
1379, уксусная кислота 1381 и метилцианпд 1396 см'1.
Когда к одному и тому же углеродному атому присоединены
две метильные группы, происходит резонансное расщепле-
ние и появляются две полосы [17,54,64]. Относительные
интенсивности этих полос обычно прямо указывают на сте-
пень разветвленности цепи. Наиболее простым веществом,
для которого наблюдается такой эффект, является этан
[301, поглощающий при 1374 и 1379 см'1. При наличии изо-
пропиловой группировки (СН3)2—СН — или (СН3)2—CR —
расщепление больше; полосы находятся на равном расстоя-
нии по обе стороны от среднего значения в области 1385—
1350 см 1 и имеют примерно одинаковую интенсивность.
Впервые этот эффект был замечен Томпсоном и Торкингто-
ном [17], которые обнаружили дублетность полос СН3 для
ряда 2,2-диметилзамещенных углеводородов. В случае
третичной бутиловой группировки (СН3)3—С— происходит
еще большее расщепление, и полосы находятся при 1395
и 1365 см'1, причем интенсивность второй полосы при-
мерно в два раза больше первой. Данные о положении
и относительной интенсивности полос в этой области позво-
ляют, таким образом, судить о наличии или отсутствии
в молекуле различных типов разветвлений. Эти корреляции
очень подробно рассмотрены Шеппардом и Симпсоном [641,
Мак-Мурри и Торнтоном [60]. Соботка и Стинлер [311
использовали их, изучая спектры этой области при иден-
тификации нормальной, изо- и нео-форм пальмитиновой
и стеариновой кислот. Следует, однако, напомнить, что
интенсивность поглощения в этой области зависит и от
наличия метильных групп в других частях молекулы, так
40
Часть I
как каждая такая группа дает здесь свое поглощение. Поэто-
му для идентификации изопропиловой и третичной бутило-
вой группировок должно быть найдено также поглощение,
соответствующее колебаниям скелета и характерное для
того или иного типа разветвления цепи. Влияние строения
молекулы на частоту симметричных деформационных коле-
баний СН3 можно проиллюстрировать также на примере
ряда стероидов [66]. Было найдено, что большое число
стеринов, у которых имеются две группы СН3, располо-
женные между кольцами, поглощает при 1384 и 1378 см'1,
тогда как 13 стероидов, у которых имеется лишь одна такая
группа, дают только по одной из этих полос. Положение
этих полос может, однако, зависеть в какой-то степени от
стереохимических факторов. Характеристические частоты
для метильной и метиленовой групп, расположенных в сте-
роидах в различном окружении, были рассмотрены Джон-
сом и Херлингом [111].
4. ДЕФОРМАЦИОННЫЕ КОЛЕБАНИЯ МЕТИЛЬНЫХ ГРУПП,
ПРИСОЕДИНЕННЫХ К НЕУГЛЕРОДНЫМ АТОМАМ
Численное значение частоты симметричных деформа-
ционных колебаний СН3 в том случае, когда метильная
группа присоединена не к углероду, а к атому другого эле-
мента, в основном определяется электроотрицательностью
последнего и его положением в периодической системе[89,
93]. Величина смещений, обусловливаемая влиянием неугле-
родных атомов, может быть очень значительной. Фтористый,
хлористый, бромистый и йодистый метилы поглощают соот-
ветственно при 1475, 1355, 1305 и 1255 см'1 [29]. Измене-
ние частоты этих колебаний можно проиллюстрировать
на примерах метанола и диметилового эфира (соответствен-
но 1456 и 1466 см1, группа СН3—-О), диметилсульфида
(1323 см'1, группа СН3— S) и метиламина (1418 см'х, группа
СН3— N). Силиконы с группой СН3— Si поглощают по-
стоянно при 1250 см'1, а фосфорорганическиесоединения, со-
держащие связь СН3—Р, могут поглощать вблизи 1280 см'1.
Если метильная группа соединена с ароматическим коль-
цом, появляется очень специфичная полоса маятниковых
колебаний вблизи 1042 см'1 [91]. Эту корреляцию следует
применять с осторожностью, так как л«етсг-производные
Гл. 2. Алканы
41
имеют в этой области полосу колебаний кольца, а у орто-
производных могут возникать взаимодействия между
группами.
Беллами и Уильямс [89] показали, что в любом сое-
динении типа СН3Х отдельные колебания по связям СН
непосредственно связаны друг с другом и валентными коле-
баниями НX. Этот вопрос более полно обсуждается в гл. 23.
5. КОЛЕБАНИЯ УГЛЕРОДНОГО СКЕЛЕТА
а. Изопропиловая и яг/ге/и-бутиловая структуры. Коле-
бания скелета обычно очень чувствительны к изменениям
строения окружающих групп, что приводит к значитель-
ному смещению частот. Однако это не относится к таким
СНзх СН3СНзх
разветвленным структурам, как /СК и рСН—С,
сн3/ \с СН3Х
относительно которых можно ожидать, что они имеют
присущие им характеристические частоты. Симпсон и
Сазерленд [32, 33] вычислили ожидаемые для этих группи-
ровок частоты v2 и v3, приняв за основу сравнительно про-
стое силовое поле. Они показали, что в случае 2,2-диметил-
замещенных парафинов (CH3)3CR нельзя ожидать боль-
шого изменения каждой из этих частот при изменении мо-
лекулярного веса группы R от 15 (метил, СН3) до 99 (геп-
тил, С7Н15). Можно предположить, что в этом ряду про-
исходит медленное уменьшение частоты v2 с максималь-
ным снижением на 37 см'1 и что частота v3 уменьшается на
97 см1.
Эти авторы установили, что в ряду парафинов до нона-
нов включительно каждый раз, когда имеется 2,2-диметиль-
ная группировка, появляются сильные полосы поглоще-
ния при 1250 и 1200 см'1. Частота первой из них почти
постоянна и меняется по ряду не более чем на величину
в несколько обратных сантиметров, тогда как частота вто-
рой, совпадающая у неопентана с первой, уменьшается
по ряду до 1200 см'1. Постоянство положения полосы
1250 см 1 не является неожиданным, так как эта полоса
соответствует перпендикулярным колебаниям v2 группы
(СН3)3С, которые зависят от изменений остальной угле-
водородной цепи гораздо меньше, чем параллельные
42
Часть I
колебания v3. Колебания С—С скелета можно иногда отли-
чить от веерных деформационных колебаний СН2 путем
сравнения спектров углеводородов и соответствующих
соединений с полярными заместителями. Полосы СН2 во
втором случае становятся часто более интенсивными, и
таким способом было получено дополнительное подтвержде-
ние указанного выше отнесения колебаний скелета [92].
Аналогичные расчеты тех же авторов для скелета изо-
пропилового типа показали, что частоты v2 и v3 составляют
приблизительно 1170 и 1145 c.i!1, что было подтверждено
экспериментально. Эти авторы нашли, что по ряду от
2-метилпропана до 2-метилнонана одна из частот остается
постоянной в интервале 1170—1167 см'1, тогда как дру-
гая постепенно уменьшается от 1170 см'1 для первого
члена до 1142 см'1 для последнего. Когда у центрального
углерода группировки (СН3)2С^ свободного водородного
атома нет, в этой области спектра имеется только одна
полоса при 1195 см'1 [54]. В некоторых случаях наблю-
далась вторая полоса около 1210 см'1, однако положение
ее было менее постоянным [64].
Эти корреляции полностью подтверждаются также рабо-
той Расмуссена [28]. Не соглашаясь с интерпретацией упо-
мянутых полос, он приводит экспериментальные данные,
полученные для большого ряда разветвленных парафинов,
которые подтверждают, что указанные полосы могут быть
связаны с этими различными типами разветвленных
структур. Эти корреляции справедливы, по-видимому, для
многих производных углеводородов, если только атом
кислорода не присоединен у ответвления цепи. Группа
С\ /С
поглощает примерно при 900 см'1 (гл. 7).
с/ \о
Симпсон и Сазерленд указали, что, помимо этих корре-
ляций, может быть предсказана также третья частота коле-
баний скелета (v4) для каждой из рассматриваемых развет-
вленных структур. Для mpem-бутиловых соединений она
соответствует 415 см'1, а для изопропиловых соединений —
около 850 см'1. Эти частоты представляются, однако,
менее важными, чем другие. Относительно первой из них
имеется очень мало сведений, вторая же для ряда упомя-
Гл 2. Алканы
43
нутых 2-метилсоединений меняется в интервале 840 —
793 ел1.
Найдено также значительное число корреляций для
ряда других углеводородных структур. Они были устано-
влены Шеппардом и Симпсоном [64], а также Мак-Мурри
и Торнтоном [60] на ссновании спектров АНИ и касаются
веерных, маятниковых и крутильных деформационных
колебаний СН2, валентных колебаний С—С и колебаний
для различного типа замещений метильной группой. По-
скольку эти корреляции не являются общепринятыми, они
здесь не рассматриваются, и познакомиться с ними можно
по оригинальным работам. Во многих случаях эти работы
содержат подробные сведения о ряде соединений, при ис-
следованиях которых была выведена та или иная корреля-
ция, а также некоторые данные об интенсивности рассмат-
риваемых полос.
б. Парафиновые цепи. Как уже упоминалось выше,
корреляции для колебаний скелета в случае открытой цепи
охватывают широкую область частот и обычно не являются
надежными. Частоты валентных колебаний простой связи
в этиловой и н-пропиловой группах приведены Кольрау-
шем [34] и Шеппардом и Симпсоном [641; они включены
также в сводную таблицу частот Колтупа [10] и таблицу,
выпущенную спектроскопической группой АНИ [55].
Однако эти полосы часто настолько слабы и расположены
в таком широком интервале частот, что при изучении слож-
ных молекул не имеют большого практического значения.
Известна между тем одна очень важная для практики кор-
реляция, касающаяся колебаний в открытой цепи. Она
характерна для длинных цепей типа — (СН2)П—(и > 4),
которые дают сильную полосу поглощения вблизи 720 см"1.
Эта полоса относится к маятниковым колебаниям групп СН2
и, строго говоря, не связана с колебаниями скелета, однако
ее удобно рассмотреть здесь, так как появление ее, по-ви-
Димому, обусловлено не единичным структурным элемен-
том, а группировкой в целом.
Туот и Леконт [35] исследовали в 1943 г. ряд спир-
тов и заметили, ’ что когда в цепи имеется подряд по
меньшей мере четыре метиленовые группы, появляется
Полоса вблизи 720 см'1. К этому же случаю Томпсон и Тор-
44 Часть!
кингтон [171 относят дублет 721 и 732 см'1 политена.
Шеппард и Сазерленд [36], исследуя полностью дейтери-
рованные парафины, показали, что эта полоса обусловлена
маятниковыми колебаниями групп СН2. Сазерленд и Джонс
[371, используя поляризованное излучение, также пока-
зали, что рассматриваемые колебания перпендикулярны
к углеводородной цепи. В первой из этих работ для полос
таких колебаний в углеводородах указывается интервал
760—720 см'1.
Таким образом, ясно, что наличие ряда непосредствен-
но связанных групп — СН2— приводит к появлению в этой
области полосы поглощения, однако что касается числа
таких групп, необходимых для появления полосы, то его
точно определить, по-видимому, нельзя. Обычно таким
числом считается четыре, и неизвестно ни одного углево-
дорода, содержащего четыре или более непосредственно
связанных групп СН2, в спектре которого не было бы этой
полосы. С другой стороны, она появляется у некоторых сое-
динений, имеющих только три связанные группы СН2.
Предполагается, что при уменьшении числа групп проис-
ходит постепенное смещение полосы в сторону больших
частот [38, 601. Поэтому указанная корреляция, безуслов-
но, может использоваться для доказательства отсутствия
группы — (СН2)4—, при наличии же полосы последняя
должна интерпретироваться с осторожностью.
Виберли и Бассетт [39] предложили недавно более точ-
ную корреляцию для самой «-бутиловой группы. Было
найдено, что для ряда, состоящего из 18 н-бутилпроизвод-
ных, у которых бутиловая группа присоединена к кислороду,
азоту или какой-либо другой функциональной группе, зна-
чение частоты всегда находится в интервале 742—734 см'1.
Авторы отмечают, что эта корреляция справедлива только
для указанных соединений, но не годится для углеводо-
родов. В этой связи они ссылаются на Смита, который сооб-
щил им, что из 700 углеводородных спектров АНИ 220
имеют полосы в области 750—720 см'1. Из них 34 имеют
полосы в интервале 742—734 см'1, причем только шесть
относятся к соединениям, содержащим бутиловую группу,
а 17 — к соединениям, содержащим пропиловую группу.
Шесть соединений с «-бутиловой группой поглощают в обла-
сти от 728 до 730 см'1, тогда как остальные дают полосу
Гл. 2 Алканы
45
поглощения выше 742 сл'1. Эти данные подтверждаются
Мак-Мурри и Торнтоном [60] и свидетельствуют о том,
что полосы в указанной области могут использоваться для
целей идентификации структуры лишь с большой осторож-
ностью. Эта полоса рассматривалась также Бомштейном [93].
Важным доказательством того, что полоса в области
750—720 сл'1 относится к маятниковым колебаниям метиле-
новой группы, являются данные об изменении полосы при
изменении агрегатного состояния вещества. Как и в случае
ножничных деформационных колебаний СН2, в спектрах
твердых веществ полоса всегда является двойной, а в слу-
чае раствора или жидкого состояния — простой [12, 68].
Этого свойства достаточно для распознавания данной полосы
среди других полос указанной спектральной области,
имеющих другое происхождение и не обладающих такой
особенностью. Это явление было подробно изучено мно-
гими исследователями [77, 94—100], которые применяли
поляризованное излучение, изучали зависимость спектра
от температуры и т. д. В настоящее время установлено,
что расщепление этой полосы в кристаллических веществах
происходит в результате взаимодействия между соседними
молекулами. Изучение интенсивности подобных полос
может, таким образом, служить удобным способом для опре-
деления относительного содержания кристаллического и
аморфного веществ. Исследуя спектральную область
1300—1200 см'1, можно получить также некоторое под-
тверждение наличия длинной цепи метиленовых групп.
Браун, Шеппард и Симпсон [40] нашли для ряда парафи-
нов с длинной цепью слабые полосы около 1300 и 1240 см'1,
которые они отнесли к веерным деформационным колеба-
ниям СН2. Интерпретация этой области осложняется взаи-.
модействием валентных колебаний С — С с веерными дефор-
мационными колебаниями СН2; однако, как показали
исследования, полоса при 1300 см'1 появляется у всех
парафинов с четным числом углеродных атомов в цепи
длиной от четырех до четырнадцати атомов, а полоса
1240 см'1— у всех парафинов с длиной цепи, составляю-
щей шесть и более углеродных атомов. Ряд исследователей
подробно изучали эту область [75, 100—104, 112]; поэтому
могут быть расшифрованы и определены различные частоты
многих простых углеводородов. Число полос увеличивается
46
Часть i
с длиной цепи и уменьшается с введением заместителей,
приводящим к снижению количества смежных метиленовых
групп. Однако у нормальных углеводородов эти полосы
очень слабы, и только у соединений с полярной группой
в конце цепи они становятся настолько интенсивными, что
могут быть без труда идентифицированы. Имеется по край-
ней мере возможность использования этой области для
определения длины цепей; некоторые успехи в этом напра-
влении были достигнуты для ряда жирных кислот (гл. 10).
в. Циклические системы. Насыщенные циклические сое-
динения образуют такую группу, для которой следует ожи-
дать наличия характеристических частот, обусловленных
деформационными колебаниями кольца. Этим соединениям
посвящен ряд опубликованных работ [41, 51, 54]. Здесь
будут рассмотрены корреляции для индивидуальных цикли-
ческих систем, однако следует иметь в виду, что для сое-
динений с относительно большими циклами приводимые
корреляции не имеют большого практического значения.
Наиболее характеристичны для циклических систем
частоты пульсационных колебаний, которые, к сожалению,
неактивны в инфракрасных спектрах, но легко идентифици-
руются в спектрах комбинационного рассеяния [105].
Циклопропан. Бартлесон, Берк и Ланкелма [42] в
1946 г. заметили, что производные циклопропана погло-
щают в области частот 1026 и 866 слг1. Затем были опуб-
ликованы данные Дерфера, Пикетта и Бурда [43] по ис-
следованию 14 различных производных циклопропана.
Они нашли, что сильная полоса при 866 см~1 присутствует
не всегда, но во всех случаях наблюдается полоса при
1020—1000 сл-1. Единственным соединением, которое не
подчиняется этому правилу, является сам циклопропан,
поглощающий несколько в стороне от этой области.
В ЗООспектрах других углеводородов эта полоса обнаружи-
валась так редко, что ее появление можно считать слу-
чайным. Поэтому указанные авторы считают эту полосу
типичной для циклопропановой структуры и предпола-
гают, что она относится к деформационным колебаниям
кольца. Такая полоса наблюдалась и в спектрах соедине-
ний, исследовавшихся Бартлесоном и др. [42], а Мерри-
сон [44] установил, что она характерна также для спектра
Гл. 2. Алканы
47
винилциклопропана [45]. Это же подтвердил Слейби [58], ко-
торый обнаружил данную полосу также у дициклопропила.
В последующих работах [59, 73] он приводит результаты
исследования большого числа производных циклопропана.
Было установлено, что 34 соединения с циклопропановой
структурой поглощают в интервале 1048—1017 см'1, из
них 29 соединений поглощают при 1021 ± 5 см'1. Наличие
этой полосы для 9 циклопропановых соединений подтвер-
дили также Виберли и Бане [62], а Бридсон-Джонс и др.
[69, 70] наблюдали ее еще у семи соединений. Расположение
этой полосы в очень узком интервале частот характерно
не только для простых структур; она имеется, напри-
мер, в спектре нзо-холестана [46], у которого циклопропа-
новое кольцо составляет часть системы, представляющей
собой шестичленный цикл с мостиком, а также в спектрах
карена [47] и карена-3, у которых одна из связей цикло-
пропанового кольца является одновременно связью шести-
членного цикла. Наличие аналогичной полосы в спектре
артенола (тритерпеноид) было использовано Бартоном
[46] как доказательство присутствия циклопропанового
кольца. Однако, как указал Коул [74], при использовании
этой корреляции в случае кислородсодержащих соединений
необходимо соблюдать крайнюю осторожность. Наблюдая
у некоторых тритерпенов, содержащих циклопропановые
кольца, полосу вблизи 1010 см'1, он не мог считать ее
достаточно характеристичной без дополнительных данных
по валентным колебаниям СН. Эта корреляция подтвер-
ждается также работой Шеппарда [54] по нафтеновым угле-
водородам.
Циклобутан. Положение характеристических полос
циклобутана не так четко определено, как для циклопро-
пана, отчасти из-за того, что они гораздо менее интенсив-
ны. Вильсон [48] отнес полосы циклобутана при 920
и 903 см'1 к маятниковым колебаниям СН2, а Дерфер и др.
[43] согласились с этим, обнаружив, что семь замещенных
Циклобутана поглощают в узком интервале 920 — 910 см'1.
Они между тем указали, что если это отнесение к колеба-
ниям СН2 правильно, то нельзя ожидать появления таких
полос у полностью замещенных соединений. Позднее Рейд
и Сак [49] изучили восемь полностью замещенных цикло-
бутанов. Образцы исследовались в твердом состоянии,
48
Часть
причем все они поглощали в интервале от 888 до 868 см'1
и не поглощали при 920—910 см'1. Меррисон [44] уста-
новил, что ни метиленциклобутан, ни октафторциклобутан
не поглощают в интервале 920—910 см'1, но все цикло-
бутаны, спектры которых были получены АНИ, погло-
щают в интервале 1000—960 см1. Значимость последней
корреляции несколько уменьшается из-за того, что многие
производные циклопентана, как наблюдалось, также по-
глощают в этой области. Шеппард [54] придерживается
мнения, что для циклобутановой структуры характерна
полоса при 910 см'1, но не сообщает, какие соединения
он исследовал. Полоса, имеющая примерно такую же ча-
стоту, наблюдалась в спектрах ряда производных цикло-
бутана, описанных Робертсом и Чемберсом [51, 56] и
Робертсом и Симмонсом [57].
Циклопентан. Как уже указывалось, Меррисон [44]
наблюдал в спектрах производных циклопентана при-
сутствие полосы, среднее значение частоты которой равно
977 см1. Однако изменения этой частоты от соединения
к соединению слишком велики, чтобы считать эту корреля-
цию надежной. Шеппард [54] рассматривает полосы около
930 и 890 см'1 как характеристические, но не указывает,
какие соединения им исследовались.
Циклогексан. Меррисон [44] заметил, что, за исключе-
нием двух, все опубликованные спектры производных цик-
логексана имеют полосы в интервалах 1005—952 и 1055—
1000 см'1. Эго относится к 50 опубликованным спектрам,
а оба соединения, являющиеся исключениями (циклогек-
сан и |3-гаммексан), имеют лишь одну из этих полос. Кроме
того, Меррисон приводит еще 9 исследованных им веществ,
которые поглощают в указанных областях. Эти полосы,
однако, не обнаружены у производных циклогексанона,
для которых введение карбонильной группы должно при-
водить к изменению всех характеристических частот дефор-
мационных колебаний цикла. Шеппард [54] указывает
для циклогексанов частоты около 1260 и 890 см'1, но
опять-таки не приводит никаких других данных.
г. Поглощение в области низких частот. Углеводороды
дают ряд слабых полос поглощения в области ниже 650 см'1,
обусловливаемых колебаниями скелета. Они обычно очень
Гл. 2. Алканы
19
слабы, и поэтому их можно изучать только при относитель-
но толстых пленках. Плайлер [1061 привел данные, касаю-
щиеся интервала 650—400 см"1 цля ряда веществ, а Бо-
релло и Мусса [107] предположили, что в этой области
имеется полоса, относящаяся к характеристическим коле-
баниям метильной группы, которая может быть исполь-
зована для обнаружения групп СН3 в полиэтилене.
Поглощение в области низких частот изучал Гейтс
[108], а позже Донно [109, ПО] обнаружил некоторые инте-
ресные различия между полосами в этой области для сое-
динений с прямой и разветвленной цепью и рассмотрел
вопрос о их применении для аналитических целей.
ЛИТЕРАТУРА
1. Coblentz, Investigations of Infra-red Spectra, Part 1, Car-
negie Institute, Washington, 1905.
2. Bonino, Trans. Faraday Soc., 25, 876 (1929).
3. Fox, Martin, Proc. Roy. Soc., A162, 419 (1937).
4. Fox, Martin, Proc. Roy. Soc., A167, 257 (1938).
5. Fox, Martin, J. Chem. Soc., 1939, 318.
6. Fox, Martin, Proc. Roy. Soc., A175, 208, 234 (1940).
7. Rasmussen, Brattain, J. Chem. Phys., 15, 120, 131,
135 (1947).
8. Saier, Coggeshall, Analyt. Chem., 20, 812 (1948).
9. Francis, J. Chem. Phys., 18, 861 (1950).
10. С о I t h u p, J. Opt. Soc. Amer., 40, 397 (1950).
11. J о n e s, M cK ay, S i n с 1 a i r, J. Amer. Chem. Soc., 74,
2575 (1952).
12. Sutherland, Discuss. Faraday Soc., 9, 274 (1950).
13. Rose, J. Rec. Nat. Bur. Stand., 20, 129 (1938).
14. H i b b a r d, Cleaves, Analyt. Chem., 21, 486 (1949).
15. T u о t, В a r c h e w i t z, Bull. Soc. Chim., 1950, 851.
16. В a r n e s, Gore, Liddel, Van Zandt Williams,
Infra-red spectroscopy, Reinhold Pub. Corp., New York, 1944.
17. Thompson, Torkington, Proc. Roy. Soc., A184,
3 (1945).
18. Thompson, Torkington, Trans. Faraday Soc., 41,
246 (1945).
19. Crawford, Avery, Linnet t, J. Chem. Phys., 6, 682
(1938).
20. H a d 1 i, S h e p p a r d, Proc. Roy. Soc., A216, 247 (1953).
21. Richards, Thompson, Proc. Roy. Soc., A195,
1 (1948).
22. Гер цбер г, Колебательные и вращательные спектры много-
атомных молекул, Издатинлит, М., 1949, стр. 363.
Л. Беллами
50
Часть I
23. Pitzer, J. Chem. Phys., 12, 310 (1944).
24. Гер цберг, Колебательные и вращательные спектры много-
атомных молекул, Издатинлит, М., 1949, стр. 341.
25. Герцберг, Колебательные и вращательные спектры много-
атомных молекул, Издатинлит, М., 1949, стр. 339.
26. Brown, Sheppard, Discuss. Faraday Soc., 9, 144 (1950).
27. T h о m p s о п, C a v e, Trans. Faraday Soc., 47, 946, 951 (1951).
28. Rasmussen, J. Chem. Phys., 16, 712 (1948).
29. Randall, Fowler, Fuson, Da ng 1, Infra-red deter-
mination of organic structures, Van Nostrand, New York,
1949.
30. Герцберг, Колебательные и вращательные спектры много-
атомных молекул, Издатинлит, М., 1949, стр. 370.
31. Sobotka, S t у и 1 е г, J. Amer. Chem. Soc., 72, 5139 (1950).
32. Sutherland, Simpson, J. Chem. Phys., 15, 153 (1947).
33. Simpson, Sutherland, Proc. Roy. Soc., A199,169 (1949)-
34. К о л ь p а у ш, Спектры комбинационного рассеяния, Издатин-
лит, М., 1952.
35. Т и о t, L е с о m t е, С. R. Acad. sci. Paris, 216, 339 (1943).
36. Sheppard, Sutherland, Nature, 159, 739 (1947).
37. Sutherland, Vallence Jones, Nature, 160, 567 (1947).
38. Fellgett, Harris, Simpson, Sutherland,
Thompson, Whiffen, Willis, Inst. Petroleum
Reports, XI. 1946.
39. Wiberley, Bassett, Analyt. Chem., 22, 841 (1950).
40. Brown, Sheppard, Simpson, Discuss. Faraday Soc.,
9, 261 (1950).
41. Plyler, A c q u i s t a, J. Res. Nut. Bur. Stand., 43, 37 (1949).
42. Bartleson, Burk, L a n k e 1 m a, J. Amer. Chem Soc.,
68, 2513 (1946).
43. D e r f e r, Pickett, В о о r d, J. Amer. Chem. Soc., 71,
2482 (1949).
44. Marrison, J. Chem. Soc., 1951, 1614.
45. Van Vol kenb urgh, Greenlee, Derfer,
В о о r d, J. Amer. Chem. Soc., 71, 3595 (1949).
46. Barton, J. Chem Soc., 1951, 1444.
47. Pliva, Herout, Coll. Trav. Chim. Tchecosl., 15, 160 (1950).
48. Wilson, J. Chem. Phus., 11, 369 (1943).
49. Reid, Sack, J. Amer. Chem. Soc., 73, 1985 (1951).
50. Pozefsky, Coggesha 11,Analyt. Chem., 23, 1611 (1951).
51. Roberts, Chambers, J. Amer. Chem. Soc., 73, 5030
(1951).
52. G a u t h i e r, C. R. Acad. sci. Paris, 231, 837 (1950).
53. E v a n s, Hibbard, Powell, Analyt. Chem., 23, 1604
(1951).
54. Sheppard, J. Inst. Pet., 37, 95 (1951).
55. «The Spectroscopic Panel of the Hydrocarbon Research Group»,
J. Inst. Pet., 37, 109 (1951).
56. Roberts, Chambers, J. Amer. Chem. Soc., 73, 5034
(1951).
57. R о b e r t s, S i m m о n s, J. Amer. Chem. Soc., 73 5487 (1951).
Гл. 2. Алканы
51
58. S 1 а b е у, J. Amer. Chem. Soc., 74, 4930 (1952).
59. S 1 a bey, J. Amer. Chem. Soc., 74, 4928 (1952).
60. McMurry, Thornton, Analyt. Chem., 24, 318 (1952).
61. Hastings, Watson, Williams, Anderson,
Analyt. Chem., 24, 611 (1952).
62. W i b e r 1 e y, Bunce, Analyt. Chem., 24, 623 (1952).
63. P i t z e r, Kilpatrick, Chem. Reviews, 39, 435 (1946).
64. Sheppard, Simpsom, Quarterly Reviews, 7, 19 (1953).
65. Freeman, J. Amer. Chem. Soc., 75, 1859 (1953).
66. J о n e s, С о 1 e, J. Amer. Chem. Soc., 74, 5648 (1952).
67. J о n e s, С о 1 e, N о 1 i n, J. Amer. Chem. Soc., 74, 5662 (1952).
68. Robert, F a v r e, C. R. Acad. Sci. (Paris), 234, 2270 (1952).
69. В r i d s о n-J ones, Buckley, Cross, Driver,
J Chem. Soc., 1951, 2999.
70. В r i d s о n-J ones, В u с к 1 e y, J. Chem. Soc., 1951, 3009.
71. Francis, J. Chem. Phys., 19, 942 (1951).
72. Francis, Analyt. Chem., 25, 1466 (1953).
73. S la bey, J. Amer. Chem. Soc., 76, 3604 (1954).
74. С о 1 e, J. Chem. Soc., 1954, 3807, 3810.
75. Tschamler, J. Chem. Phys., 22, 1845 (1954).
76. Keller, S a n d e m a n, J. Polimer. Sci., 15, 133 (1955).
77. T о b i n, C a r r a n o, J. Polymer. Sci., 24, 93 (1957).
78. Stein, Sutherland, J. Polimer. Sci., 12, 370 (1953).
79. N о 1 i n, J о n e s, J. Amer. Chem. Soc., 75, 5626 (1953).
80. Rathgens, Freeman, Gwinn, Pitzer, J. Amer.
Chem. Soc., 75, 5634 (1953).
81. L о r d, N о 1 i n, S t i d h a m, J. Amer. Chem. Soc., 77,
1365 (1955).
82. Huggins, Pimentel, Schooler y, J. Chem. Phys.,
23, 1244 (1955).
83. M i г о n e, F a b b r i, Gazz. chim., 84, 187 (1954).
84. Kaye, .^Spectrochim. Acta, 6, 257 (1954).
85. Lauer, Rosenbau m., App. Spectroscopy, 6, 29 (1952).
86. Luther, Czerwany, Z. Phys. Chem., 6, 286 (1956).
87. J о n e s, N о 1 i n, R о b e r t s, J. Amer. Chem. Soc., 77, 6331
(1955).
88. В a r t о n, P a g e, W a r n h о f f, J. Chem. Soc., 1954, 2715.
89. В e 1 1 a m y, Williams, J. Chem. Soc., 1956, 2753.
90. Sheppard, Trans. Faraday Soc., 51, 1465 (1955).
91. R a n d 1 e, W h i f f e n, J. Chem. Soc., 1955, 3497.
92. S h e p p a r d, S i m p s о n, J. Chem. Phys., 23, 582 (1955).
93. В о m s t e i n, Analyt. Chem., 25, 512 (1953).
94. К e 1 1 e r, S a n d e m a n, J. Polymer. Sci., 13, 511 (1954).
95. S t e i n, S u t h e r 1 a n d, J. Chem. Phys., 22, 1993 (1954).
96. К r i m m, J. Chem. Phys., 22, 567 (1954).
97. К r i m m, Phys. Rev., 94, 1426 (1954).
98. Новак, Жур. техн, физ., 24, 18 (1954).
99. Никитин, Покровский, Доклады АН СССР, 95,
109 (1954).
100. К г i m m, L iang, Sutherland, J. Chem. Phys., 25, 549
(1956).
4*
52
Часть J
101. Primas, Gunthard, Helv. Chim. Acta, 36, 1791 (1953);
39, 1182 (1956).
102. Brown, Sheppard, Simpson, Phil. Trans., A247,
35 (1954).
103. Brown, Sheppard, Simpson, J. Phys. Radium,
15, 593 (1954).
104. Мидзусима С., Строение молекул и внутреннее враще-
ние, Издатинлит, М., 1957, стр. 119.
105. Батуев Н. И., Изв. АН СССР, Отд. хим. наук, 1, 3 (1947).
106. Plyler, J. Opt. Soc. Amer., 37, 746 (1947).
107. В о г e 1 1 о, Mussa, J. Polymer. Sci., 13, 402 (1954).
108. Gates, J. Chem. Phys., 17, 393 (1949).
109. D о n n e a u d, C. R. Acad. sci. (Paris), 239, 1480 (1954).
110. D о n n e a u d, Rev. Inst. Franc. Petrole, 10, 1525 (1955).
111. Jones, H e r 1 i n g, J. Org. Chem., 19, 1252 (1954).
112. В r i n i, Bull. soc. Chim. France, 1955, 996.
113. Henbest, Meakins, Nicholls, Wagland,
J. Chem. Soc., 1957, 1462.
114. Henbest, Meakins, Nicholls, Taylor, J. Chem.
Soc., 1957, 1459.
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1. ВолькенштейнМ. В,, ЕльяшевичМ. А., Степа-
нов Б. И., Колебания молекул, т. I и II, гл. 14 и 30, Гос-
техиздат, М.—Л., 1949.
2. Иогансен А. В., Инфракрасные спектры углеводородов,
«Физико-химические свойства индивидуальных углеводоро-
дов», Гостоптехиздат, М.—Л., вып. 6, 1957.
3. Кеслер X., Пентин Ю. А., Трещева Е. Г., Татев-
с к и й В. М., Оптика и спектроскопия, 7, 301 (1959).
Глава з
АЛКЕНЫ
1. ВВЕДЕНИЕ
Инфракрасные полосы поглощения, соответствующие
различным типам двойной связи, изучены довольно хоро-
шо, так что о наличии и типе ненасыщенных связей в неиз-
вестном соединении можно судить с достаточной опреде-
ленностью, хотя на положение полосы в некоторой степени
влияет число заместителей при двойной связи. Это обус-
ловлено тем, что наиболее пригодны для целей идентифика-
ции характеристические частоты валентных и деформацион-
ных колебаний С—Н, и, например, у веществ типа
R1R2C=CHR3 число таких характеристических частот умень-
шено. Ненасыщенность в соединениях типа RIR2C=CR3R4 оп-
ределяется с большим трудом, так как у них остается един-
ственная характеристическая частота валентных колеба-
ний С=С, а интенсивность этой полосы значительно пони-
жена вследствие симметричности групп относительно
связи. Весьма подробный обзор основных работ, посвящен-
ных простым молекулам такого рода, сделан Шеппардом
и Симпсоном [47]. Можно рекомендовать также обзоры
Леконта и Нава [80], Груздева [81] и О’Коннора [82].
Многие из характеристических частот двойных связей
находятся в области частот валентных колебаний С—О и
С—С, так что наличие полосы поглощения с характерным
значением частоты еще нельзя считать доказательством
присутствия двойной связи определенного типа. Однако
при отсутствии такой полосы можно с уверенностью гово-
рить об отсутствии этой группы.
С другой стороны, исследование всех интервалов харак-
терных частот поглощения обычно позволяет с большой
определенностью идентифицировать двойную связь и ее
тип по крайней мере для частично замещенных соединений.
Сравнительное постоянство относительно молекулярного
54
Часть I
веса интенсивности характеристических полос валентных
и внеплоскостных деформационных колебаний СН дало
возможность разработать количественные методы анализа
сложных углеводородов и полимеров. Некоторые исследо-
ватели [83] предположили, что между частотами колеба-
ний С=С и СН существует статистическая зависимость.
Это предположение может быть справедливым в случае
некоторых близких по строению углеводородов, однако
широко им нельзя руководствоваться, так как валентные
колебания С=С более чувствительны к изменениям массы,
чем деформационные колебания.
Структурно-частотные корреляции приведены в табл. 2.
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С = С
а. Положение и интенсивность полосы. В противопо-
ложность интенсивному поглощению С = О валентным коле-
баниям С=С несопряженных соединений соответствует
лишь слабая полоса поглощения инфракрасного спектра.
Положение пика поглощения в какой-то мере зависит от
природы заместителей при обоих углеродных атомах, но
главным образом как на частоту, так и на интенсивность
оказывают влияние симметрия группировки, сопряжение
и замещение фтором. Каждый из этих факторов рассмат-
ривается ниже.
В случае соединений, содержащих только одну изоли-
рованную двойную связь, пик поглощения обычно нахо-
дится в интервале 1680—1620 см'1, причем подавляющее
большинство соединений поглощает в области 1660—
1640 см'1. Барнс и др. [1 ] перечислили около 20 соединений,
обнаруживающих поглощение в области 1660—1640 ел’1;
валентные колебания С=С были отнесены к данной области
также в других многочисленных фундаментальных иссле-
дованиях малых молекул углеводородов. Так, например,
Шеппард [2] обнаружил для бутена-1 полосу при 1645 слг'1,
которую он отнес к этим колебаниям, а Томпсон и Торкинг-
тон [3] указывают на наличие полосы около 1650 слг’1 у
ряда галоидных аллилов и аллилового спирта. Ряд примеров-
соединений, для которых колебания С=С также отнесены
к этой области, приводится в работе Рендалла и др. [4],
Таблица 2
Частота, см1
Валентные колебания С=С
Несопряженная связь .............
Связь, сопряженная с фенилом . .
Связь, сопряженная с С = О или С=С
Валентные и деформационные
колебания С—Н
—СН=СН— (транс)
валентные ......................
внеплоскостные деформационные
плоскостные деформационные . .
—СН=СН— (цис)
валентные ......................
внеплоскостные деформационные
(корреляция ненадежная) . . . .
—СН=СН2 (винил)
валентные ......................
валентные......................
внеплоскостные деформационные
внеплоскостные деформацион-
ные СН2........................
возможный обертон предыдущих
колебаний.....................
плоскостные деформационные СН2
плоскостные деформационные . . .
CR1R2=CH2
валентные ......................
внеплоскостные деформационные
возможный обертон предыдущих
колебаний .....................
плоскостные деформационные СН2
CR1R2=cHR3
валентные .....................
внеплоскостные деформационные
1680—1620 (интенсивность
сильно меняется)
Около 1625 (повышенная
интенсивность)
Около 1600 (повышенная
интенсивность)
3040-3010 (ср.)
970—960 (с.)
1310—1295 (с,—сл.)
3040—3010 (ср.)
Около 690
3040-3010 (ср.)
3095-3075 (ср.)
995—985 (с.)
915—905 (с.)
1856—1800 (ср.)
1420—1410 (с.)
1300—1290 (с,—сл.)
3095—3075 (ср )
895—885 (с.)
1800—1750 (ср.)
1420—1410 (с.)
3040—3010 (ср.)
840—790 (с.)
56
Часть I
например изобутилен с полосой при 1637 сл/’1, пропилен
и бутен-2 с полосами при 1647 см~г.
Эту корреляцию изучали в дальнейшем Шеппард и
Сазерленд [5], которые, основываясь на результатах иссле-
дования углеводородов, сделали предположение о том,
что двойные связи типов R1R2C=CH2 и R1R2C=CHR3 можно
распознавать по положению максимума поглощения, и
привели для них интервалы соответственно 1655—1645 смг1
и 1680—1670 сл/’1. Указанные типы связей можно распо-
знавать также по интенсивности поглощения в данной обла-
сти, так как концевая двойная связь (первый из приведен-
ных типов) дает значительно более интенсивную полосу,
чем связь, находящаяся в цепи (второй тип). Эти сведения
подтвердили Томпсон и Уиффен [6], которые указали, что
они согласуются также с данными о спектрах комбина-
ционного рассеяния аналогичных соединений [7]. Ими же
руководствовались Томпсон и Торкингтон [8] при отне-
сении полос 1645 и 1690 сл/’1 полимеризованного 2,3-ди-
метилбутадиена к двум различным типам двойных связей,
к связи типа R1R2C=CR3R4 не отнесено никакой характе-
ристической полосы, однако на основании изучения спект-
ров комбинационного рассеяния можно сделать вывод, что
этой связи также соответствует поглощение в области 1690—
1680 сл/’1 [47, 58]. Интенсивность этой полосы в инфракрас-
ном спектре всегда мала, а часто полоса вообще не обна-
руживается. Такое отнесение подтверждается данными
Мак-Мурри и Торнтона [58], анализировавших спектры
АНИ, а также обобщениями результатов исследований
простых олефиновых соединений, которые были проведены
Шеппардом [55] и Шеппардом и Симпсоном [47]. Послед-
ние указывают следующие средние значения частот коле-
баний С=С для изучавшихся простых олефинов:
CHR = СН2 1643 CM~i
CHR1 = CHR2 (транс) 1673 »
CHR1-CHR2 (цис) 1657
CR1R2=CH2 1653 »
CR1R2 = CHR3 1670
CR1R2 = CR3R4 1670
Такое подразделение частот бывает удобным при изуче-
нии сравнительно простых молекул углеводородов. Однако
Гл. 3. Алкены
57
оно неприемлемо для соединений с олефиновой связью,
содержащих кислород, и для других соединений с поляр-
ными группами, так как в этом случае происходит неко-
торое возмущение колебаний С=С. Виниловые соединения
с кислородным атомом или карбонильной группой при
двойной связи поглощают между 1652 и 1611 см'1, а соот-
ветствующие производные винилидена — между 1670 и
1632 см'1 [74]. Аналогичным образом небольшие изме-
нения частот колебаний С=С происходят даже при незна-
чительных изменениях напряжения в различных стероид-
ных кольцах. Джонс и др. [9] изучали много таких ве-
ществ и нашли, что положение полосы поглощения С=С
зависит от положения двойной связи в циклической систе-
ме. Так, например, десять различных А5-ненасыщенных
стероидов поглощают в области 1672—1664 см'1, тогда как
ряд А1]-стероидов поглощает между 1628 и 1624 см'1.
Были также найдены соответствующие корреляции для
других положений двойной связи, и это позволило авторам
заключить, что частота максимума зависит лишь от поло-
жения двойной связи и почти не зависит от других струк-
турных изменений. У всех указанных стероидов поглоще-
ние одинаково малоинтенсивно. Ни у одного из этих несо-
пряженных соединений (всего 29) полоса поглощения не
лежит вне области 1680—1620 см'1. Хенбест и др. [77, 108]
и Коул и Торнтон [109] получили аналогичные данные для
стероидов и тритерпенов с г{ис-двойными связями. Изуче-
ние влияния значительных напряжений кольца было пред-
принято Лордом и Уокером [75]. Как показали опыты, при
уменьшении размера кольца и увеличении напряжения уве-
личивается частота валентных колебаний С—Н и умень-
шается частота колебаний —С=С—. Частота колебаний
С=С у циклогептена равна 1651 см'1, а у циклобутена —
1566 см'1. Присоединение второго кольца приводит к уве-
личению напряжения, поэтому пятичленные кольца би-
цикло- [2,2,1 ]-гептадиена-2 поглощают почти как и ци-
клобутен при 1568 см'1. Поглощение похожей на бицикли-
ческие соединения мостиковой системы с шестичленным
кольцом мало отличается от поглощения циклопентена.
Справедливость вывода о том, что конденсация колец дей-
ствительно приводит к увеличению напряжения, была
подтверждена изучением [106] реакционной способности.
58
Часть 1
С другой стороны, замещение метильной группой у угле-
рода с олефиновой связью приводит, по-видимому, к умень-
шению напряжения и к повышению частоты колебаний
С=С (например, у 1-метилциклогептена до 1673 см-1).
Причины и значение этого явления обсуждались Лордом
и Миллером [84]. В случае экзоциклических двойных свя-
зей напряжение действует в противоположном направлении,
т. е. при его увеличении частота растет. Группировки
С=СН2 при четырехчленных, пятичленных и шестичлен-
ных циклических системах поглощают соответственно при
1678, 1657 и 1651 см-1 [84]. Сопряжение у 1,2-диметилен-
циклопентана понижает эту частоту до 1626 см-1 [НО],
однако интересно отметить, что у 1,2-диметиленциклобу-
тана этого явления не наблюдается и он поглощает при
той же частоте, что и 1-метиленциклобутан [111]. Влияние
напряжения цикла на интенсивность поглощения С=С
подробно не изучалось, однако заслуживает внимания тот
факт, что некоторые мостиковые соединения с циклогепте-
новыми кольцами, изученные Хенбестом и др. [108],
у которых, как показывает высокая частота колебаний
—СН, существует значительное напряжение, не обнару-
живают какого-либо поглощения С=С.
Таким образом, поглощение С=С обычно приводит
к появлению слабой полосы в области 1680—1620 сл'1.
Для простых углеводородов и стероидов [85, 108] в этой
области установлен ряд корреляций, дающих дополнитель-
ные ценные сведения, однако при применении их для
сложных молекул необходимо соблюдать осторожность.
При распознавании типа двойной связи, особенно в сим-
метричных соединениях с несопряженной двойной связью,
у которых полоса С=С может отсутствовать, очень важно
изучить другие спектральные области колебаний СН, а
также относительные интенсивности всех полос, связанных
с наличием структуры, включающей двойную связь. В сом-
нительных случаях могут быть изучены изменения, сопро-
вождающие гидрогенизацию, бромирование или другое
химическое воздействие на двойную связь. Подобный спо-
соб, предложенный Леонардом и Гашем [78], полезен
при идентификации аф-ненасыщенных третичных аминов.
Под действием хлорной кислоты происходит частичная
Гл. 3. Алкены
59
эпимеризация этих соединений в структуру /СН—C=N6 ,
Z I Х
что приводит к смещению поглощения двойной связи на
20—50 иГ1 в сторону более высоких частот. Однако Р,у-не-
насыщенные амины не дают такого эффекта и продолжают
поглощать в исходном интервале 1665—1640 смг1.
Как показано ниже, интенсивность поглощения С=С
значительно изменяется в зависимости от симметрии груп-
пировки и от таких факторов, как сопряжение и т. п.
Детальных исследований абсолютных интенсивностей не
производилось, однако Джонс и Сандорфи [86] составили
обзор по величинам интенсивностей спектров АНИ, а
Девисон и Бейтс [74] приводят в своей работе данные о
коэффициентах погашения для многих кислородсодержа-
щих олефинов.
б. Влияние симметрии. Исходя из теоретических пред-
ставлений, можно ожидать, что у соединений, которые
имеют транс-конфигурацию относительно двойной связи,
совпадающей с центром симметрии, валентные колебания
С=С в инфракрасной области будут отсутствовать. Это
объясняется тем фактом, что поглощение в инфракрасной
области происходит только при таких колебаниях, кото-
рые сопровождаются изменением дипольного момента, а
при валентных колебаниях С=С молекулы с транс-сим-
метрией заметного изменения дипольного момента не про-
исходит. С другой стороны, в случае соединений, имею-
щих /gzc-конфигурацию относительно двойной связи, при
колебаниях С=С происходит некоторое изменение диполь-
ного момента. Оба этих типа колебаний активны в спект-
рах комбинационного рассеяния [47, 55]. Поэтому следует
ожидать, что интенсивность полосы двойной связи С=С
в инфракрасном спектре будет уменьшаться при переме-
щении данной связи от конца цепи к центру молекулы,
которая становится при этом более симметричной. Этим
легко объясняется повышенная интенсивность поглощения
концевой двойной связи R1R2C=CH2 по сравнению с погло-
щением этой связи в соединениях типа R1R2C=CHR3.
fcc Известны многочисленные примеры, иллюстрирующие
этот эффект. Так, валентные колебания С=С этилена и
60
Часть 1
транс-дихлорэтилена неактивны в инфракрасном спектре,
но дают интенсивные полосы соответственно при 1623 и
1577 см'1 в спектрах комбинационного рассеяния [10].
С другой стороны, цис-дихлорэтилен [11] дает сильную
полосу поглощения в инфракрасном спектре при 1590 см'1.
Флетт [12] не обнаружил полос поглощения С=С у фу-
маровой кислоты, что опять-таки связано с наличием центра
симметрии.
Имеется много работ, в которых сообщается об умень-
шении интенсивности полосы С=С при возрастании сим-
метрии молекул. Клетц и Самнер [13] обратили внимание
на уменьшение интенсивности этой полосы в ряду триме-
тилпентенов, где симметрия постепенно увеличивается,
и выдвинули предположение о том, что сравнение интенсив-
ностей может дать указание на тип имеющейся связи. Так,
2,4,4-триметилпентен-1 с двойной связью в конце цепи
=СН2 обнаруживает самое сильное поглощение в этом
ряду. Если произвольно принять интенсивность этого
поглощения за единицу, то значение оптической плотности
для 2,4,4-триметилпентена-2 получается равным только
0,35. Это же значение получено и для 3,4,4-триметилпенте-
на-2, также имеющего структуру R1R2C=CHR3. При даль-
нейшем увеличении симметрии происходит уменьшение
интенсивности до значения 0,14 для 2,3,4-триметилпенте-
на с двойной связью типа R1R2C=CR3R1.
Шрив и др. [14] не обнаружили полос валентных коле-
баний С=С у транс-конфигураций ненасыщенных жирных
кислот и спиртов с длинной цепью, когда двойная связь
расположена в середине цепи. С другой стороны, у соот-
ветствующих цис-соединений полосы двойных связей
иногда можно обнаружить. В противоположность этому
Д10-ундеценовая кислота и ее метиловый эфир дают
сильное поглощение С=С, обусловленное концевой ви-
нильной группировкой.
Данные об интенсивности поглощения связи С=С ука-
зывают, таким образом, на природу присутствующей двой-
ной связи, и в тех случаях, когда она известна, могут
быть использованы при исследовании цис-транс-пзоме-
рии. Типичным примером таких исследований является
работа Бернштейна и Полинга [15], изучавших цис- и
транс-1,2-дихлорпропен-1 и их дейтеропроизводные. После
Г л. 3. Алкены
61
разделения двух изомеров можно было легко идентифи-
цировать fjuc-соединение по сравнительно интенсивным
полосам при 1606 см'1 (D) и 1614 сл/*1 (Н) и транс-соеди-
нение по полосам низкой интенсивности при 1605 см'1
(D) и 1615 сл/*1 (Н). Дополнительные сведения о цис- или
транс-строении соединений можно получить также, уста-
новив полное число полос спектра. Это число значительно
ниже у более симметричного изомера; в приведенном слу-
чае число полос поглощения в спектре транс-соединения
примерно в два раза меньше, чем в спектре /щс-соединения.
Существует также общее, но не всегда соблюдающееся пра-
вило, что частота поглощения С=С у ^ас-изомеров несколь-
ко ниже, чем у соответствующих транс-изомеров [47, 112—
114]. Для его иллюстрации представляют определенный
интерес небольшие изменения частот у различных изоме-
ров соединения СН3СН=СН—СН=СН—СООСН3, иссле-
дованных Алланом, Микинсом и Уайтингом [87]. транс-
транс-Соермнение поглощает при 1642 и 1614 см'1, а
//пс-гщс-соединение—при 1623 и 1587 см'1, тогда как
смешанные изомеры дают промежуточные значения. Это
явление было, в частности, использовано для распознава-
ния цис- и транс-пропенилбензолов [73].
в. Влияние сопряжения. Сопряжение в алифатических
соединениях. При сопряжении двух алифатических связей
С=С происходит расщепление поглощения на две полосы
[16, 17]. Это объясняется частично механическим взаимо-
действием, а частично тем, что меняется характер нормаль-
ных колебаний, и для двух идентичных связей возникают
формы колебаний в фазе и не в фазе. Тем не менее у про-
стых сопряженных систем одна полоса сильнее другой и
имеет обычно частоту примерно на 30 см'1 меньше частоты
соответствующего несопряженного соединения. Барнс и
др. [1] приводят ряд соединений, дающих в результате
наличия системы сопряженных связей С=С полосы погло-
щения около 1600 см'1; другими примерами служат 1,3-бу-
тадиен (1597 см'1) [18] и винилацетилен (1600 см'1) [2].
Во всех случаях при этом в инфракрасной области наблю-
дается сильное увеличение интенсивности поглощения.
Наиболее полное исследование влияния сопряженных
связей друг на друга выполнено Блоутом, Филдсом и Карп-
62
Часть 1
ласом [19]. Ими изучался ряд сопряженных систем с длин-
ной цепью, включающий полиены с двумя, тремя, четырьмя
и пятью сопряженными связями С=С. Как и ожидали
эти авторы, спектр поглощения в области 1650—1600 слг1
является сложным, но они заметили, что диен имеет здесь
две двойные полосы, триен — три и тетраен — четыре. У всех
соединений этого ряда наиболее сильная полоса наблю-
дается вблизи 1650 слТ1. Они исследовали также полиено-
вые азиды и полиеновые альдегиды, содержащие по две,
четыре, шесть, восемь и десять сопряженных двойных
связей. В этих случаях имеет место сопряжение различных
типов ненасыщенных связей, и отнесение частот было более
трудным, однако оказалось, что полосы колебаний С=С
постепенно смещаются в сторону меньших частот при
увеличении числа сопряженных связей. Авторы указали,
что их результаты не могут быть точно интерпретированы
без математической обработки, однако они подтвердили об-
щее положение о том, что сопряжение приводит к смеще-
нию главной полосы поглощения С=С в сторону мень-
ших частот. Наличие этого смещения и величина его
зависят от многих факторов, которые еще не вполне
изучены.
Сопряжение с ацетиленовой связью изучалось Алланом
и др. [87], при этом наблюдались сходные явления, если
не считать того, что, конечно, не происходит расщепления
полосы колебаний — С = С —. Интенсивность поглощения
также увеличивается, но этот эффект проявляется меньше,
чем при сопряжении с карбонильной группой. Так,
два соединения СН3— С = С — CH = СН — СООСН3 и
СН3 — СН = СН—С = С — СООСН3 можно легко отли-
чить по различным соотношениям интенсивностей поглоще-
ния групп С = С и С = С, обусловленным различным их
расположением относительно карбонильной группы. Влия-
ние сопряжения у олефинов было рассмотрено Скрокко и
Салветти [92]; некоторые примеры приведены в статьях
Сёренсена И. и Сёренсена Н. [115], а также в статье Воль-
мана и Мангардта [116].
Джонс и др. [9] исследовали небольшое число сопряжен-
ных диенов ряда стеринов, причем оказалось, что все эти
соединения дают два максимума в области валентных коле-
баний С=С. Например, Д35-стерин поглощает при 1618 и
Гл. 3. Алкены
63
1578 см'1. Однако когда в молекуле присутствуют также
сложноэфирные группы, имеют место заметные эффекты
взаимодействия; два Д3’5-эфира поглощают при 1670 и
1639 см'1, хотя сложноэфирная группа находится не близко
к двойным связям. Следует отметить, что расщепление
поглощения С = С является обычным явлением как для
виниловых эфиров, так и для акрилатов [74]. По-видимому,
это связано с появлением обертонов или с явлением поворот-
ной изомерии, и таким образом появление более чем одной
полосы поглощения С = С не означает, что обязательно
присутствуют две или несколько олефиновых групп. Влия-
ние сопряжения у стероидов и тритерпенов было также изу-
чено Хенбестом и др. [108] и Коулом и Торнтоном [109].
Сопряжение с ароматическим кольцом. При сопряжении
двойной связи с ароматическим кольцом смещение полосы
С = С обычно бывает меньшим, чем при сопряжении али-
фатических связей. Это не является неожиданным, так
как в ароматической системе двойная связь слабее, чем
в алифатической. Поэтому поглощение наблюдается вблизи
1625 см'1.
Барнс и др. [1] приводят примеры соединений с та-
ким смещением. Одним из типичных примеров служит
коричная кислота, у которой главная полоса поглощения
С = С расположена вблизи 1626 см'1. Поскольку для не-
сопряженных связей С = С весь интервал частот равен
.1680—1620 см'1, поглощение около 1625 см'1 не обязатель-
но указывает на сопряжение с арильной группой. Однако
на основании относительной интенсивности этой полосы и
интенсивности полосы ароматического кольца, которая
также становится при сопряжении больше, часто оказы-
вается возможным сделать вывод о том, имеется ли сопря-
жение такого типа. При сопряжении с ароматическим
кольцом появляется также еще третья полоса колебаний
С = С, расположенная около 1590 см'1, которая может зна-
чительно облегчить интерпретацию (см. гл. 5).
а, ^-Ненасыщенные кетоны и другие соединения.
Сопряжение двойной связи с карбонильной группой при-
водит к смещению полосы С = С в сторону меньших частот.
Это смещение аналогично тому, которое происходит в ре-
зультате обычного сопряжения пары двойных связей, но
меньше его по величине. Так, Расмуссен и др. [20] отно-
64
Часть I
сят к колебаниям С — С полосу поглощения изофорона при
1639 см'1 и указывают, что по другим неопубликованным
данным о кетонах с таким сопряжением полосы С = С
находятся в интервале 1647 —1621 см'1. Они установили
также, что ацетилацетонацетат дает сильное поглощение
при 1633 см'1, а Блоут и др. [19] обнаружили у кротонового
альдегида и 2,4-гексадиеналя в растворах очень сильные
полосы в области частот С = С соответственно при 1638
и 1642 см'1. Для этих соединений в твердом состоянии часто-
ты примерно на 10 см'1 больше. Исследование спектров
комбинационного рассеяния таких систем дало в основном
аналогичные результаты (21 ]. Однако, как и в случае сопря-
женных диенов, величина смещения может, сильно зави-
сеть от других факторов, например от наличия циклических
систем. Так, у ненасыщенных кетостероидов частота в не-
которых случаях уменьшается даже до 1588 см'1, причем
ее значение почти полностью определяется расположением
ненасыщенных связей в системе. Джонс и др. [9], а также
Фёрчготт и др. [22] исследовали большое число таких
соединений и установили зависимость обнаруженных
частот от положения ненасыщенных связей; например,
Д16,го.кетоны поглощают при 1592 — 1588 см'1, тогда как
Д4,з-кетоны поглощают при 1617 см'1. Частота С = С
лишь немного уменьшается в ряду а, ^-ненасыщенных
кислот. Фримен [37] приводит для таких колебаний в ряду
2-гексеновых кислот интервал частот 1653 —1631 см'1.
В настоящее время опубликованы величины молекулярных
коэффициентов погашения для ряда соединений подобного
типа [74, 87].
Точное положение полос поглощения С = С сложных
молекул, как видно, может быть надежно предсказано
только в немногих случаях; так, например, детально иссле-
дованы модельные соединения стероидных систем.
г. Влияние замещения фтором. Торкингтон и Томпсон
[23] обратили внимание на заметное увеличение частоты
валентных колебаний С = С, которое имеет место, когда
у одного из атомов углерода, образующих двойную связь,
происходит замещение атомом фтора. Полоса поглощения
С = С 1626 см'1, характерная для этилена, при замещении
одним атомом фтора смещается лишь до 1650 — 1645 см'1, но
Гл. 3. Алкены 65
ее частота возрастает до 1730 см 1 для СН2 — CF2 п
CC12 = CF2. Приведенная частота этилена взята, разумеется,
из данных о спектрах комбинационного рассеяния, тогда
как остальные соединения обладают достаточной асиммет-
рией для того, чтобы эти колебания были активны в инфра-
красном спектре. Причины таких смещений неизвестны,
но последние указывают, по-видимому, на укорочение двой-
ной связи под влиянием атомов фтора. Эти данные подтвер-
ждены Хатчером и Постом 164], а также другими исследо-
вателями [65, 66]; известны случаи еще большего смещения
частоты при дальнейшем замещении водородов этилена атс-
мами фтора. Эджелл [24] установил, что частота валентных
колебаний С = С полностью фторированного пропена равна
1798 см1. Парк, Лайкан и Лачер [25] исследовали спектр
трифторэтилена и обнаружили в нем сильную полосу около
1780 см'1. Эти авторы не указали, к какой структурной груп-
пе можно отнести данную полосу, но примечательно, что
такой полосы нет у соответствующих насыщенных соеди-
нений, а именно у дихлорида CF2C1 — CFC1H и у анало-
гичного дибромида. Важно отметить, что перфторбутен-1
также поглощает при 1792 см 1 [88], а у перфторбутена-2
частота снижена до 1733 см'1. Другие сходные случаи
рассмотрены Брайсом и др. [88] и Вейбленом [118]. Хас-
зельдин [67] составил таблицу частот С — С для различных
замещенных этилена и показал, что когда атом фтора не
присоединен непосредственно к углероду, образующему
двойную связь, как, например, в случае группировки
—"СН = СН — CF3, то его влияние практически ничтожно.
3. ВАЛЕНТНЫЕ КОЛЕБАНИЯ = СН
. Наиболее надежным способом обнаружения двойных
связей по инфракрасным спектрам является исследование
области 3000 см'1 с использованием призмы из фтористого
лития, обладающей высокой дисперсией. В этой области
проявляются валентные колебания С — Н, частоты кото-
рых фактически никак не зависят от строения молекулы
в целом, но зависят от валентного состояния углеродного
атома и ближайшего окружения. Можно поэтому распоз-
нать характеристические частоты валентных колебаний
С — Н для групп =СН2 и =CRH, которые легко отли-
5 Л. Беллами
66
Часть I
чимы от колебаний — СН2 и — СН3, имеющих меньшие зна-
чения частот. Фокс и Мартин [26] показали, что частоты
валентных колебаний С — Н ряда углеводородов, содержа-
щих группу = СН2, находятся в интервале 3092—3077 см'1.
Все они, кроме этилена, имеют вторую полосу в обла-
сти 3025—3012 см'1, которая характерна также еще для
пяти других ненасыщенных углеводородов и приписывается
колебаниям = СН —. При помощи этих полос можно поэто-
му дифференцировать структуры — СН = СН2 (обе
полосы), ^)С = СН2 (только полоса 3079 см1) и ^)С = СН—
(только полоса 3019 см'1). Шеппард и Симпсон [47], напри-
мер, зарегистрировали полосу валентных колебаний СН
для большого числа замещенных этилена. Полоса погло-
щения = СН2 наблюдалась ими в интервале 3095—3075 см'1
13 примеров), а полоса поглощения СН—в ин-
тервале 3030—3000 см'1(16 примеров). Нормальные колеба-
ния СН насыщенных структур имеют частоты ниже 3000 см1
и не накладываются на рассматриваемые колебания. Одна-
ко в некоторых случаях у углеводородов с большим моле-
кулярным весом полоса поглощения метильной группы
может маскировать близкую по частоте полосу = СН.
Эти данные были подтверждены для углеводородов Сон-
дерсом и Смитом [63] и применялись Сейером и Коггешел-
лом [27] для количественного анализа сложных смесей
углеводородов. Был успешно проведен анализ смеси изо-
меров 2,4,4-триметилпентена-1 и 2,4,4-триметилпентена-2
при использовании полосы =СН2 первого из них
вблизи 3090 см'1, а смеси н-октана, октена-2 и октена-1
при применении полос 3067, 3012 и 2857 см'1. Валентные
колебания = С — Н ароматических колец, проявляющиеся
около 3030 см'1, также использовались для количественных
исследований, например для анализа смесей бензола и н-геп-
тана и определения бензилового спирта в н-октаноле и ме-
тилэтилкетоне.
Известны некоторые другие примеры анализов такого
типа [27], причем во всех случаях полученные результаты
совпадали с точностью до 1% с теоретическими значениями.
Основываясь на указанных полосах поглощения, можно
исследовать кинетику таких реакций, как полимеризация
стирола [90].
Гл. 3. Алкены
67
Различными авторами [9, 28, 108, 109] была проведена
идентификация типов двойных связей у стер инов и тритер-
пенов, для чего использовалась область высоких частот.
Было установлено, что у насыщенных стероидов первый
максимум поглощения в этой области расположен при
2970 см"1, а стероиды, содержащие этиленовую двойную
связь, имеют полосы в области 3040—3030 см"1. В некото-
рых случаях плохая растворимость веществ делает затруд-
нительным наблюдение пиков поглощения растворов, однако
у значительного числа ненасыщенных стероидов полосы
поглощения были найдены в соответствующей области, при-
чем они, по-видимому, мало смещаются с изменением струк-
туры колец или при сопряжении с кетонной группой. Винил-
тестостерон также имеет слабую полосу около 3085 см"1,
обусловленную присутствием концевой винильной группы,
тогда как четыре этинилпроизводных поглощают при
3310 см"1 из-за наличия группы С = С — Н. Микинс [87]
недавно обратил внимание на то, что частота валентных
колебаний СН некоторых винилиденовых эфиров равна
3120 см1. Эта частота примерно на 40 см'1 выше обычного
значения, и если окажется, что такая корреляция имеет
общий характер, то ее можно будет использовать для
диагностических целей.
Таким образом, ясно, что при изучении этой области
могут быть получены ценные данные, а при большей доступ-
ности призм из фтористого лития применение данной
спектральной области будет, вероятно, значительно рас-
ширено. Между прочим, следует напомнить, что все эти
корреляции были выведены для углеводородов, и присоеди-
нение новых групп, например атомов галогенов, к атому
углерода, к которому присоединен атом водорода, должно
приводить к изменению частот. Так, ряд насыщенных гало-
генпроизводных углеводородов поглощает в области 3090—
ЗОЮ см 1, что обусловлено смещением полос СН под влия-
нием атома галогена. Например, цис- и /прпнс-дихлорэти-
лены поглощают при 3085 см"1, бромистый метил и йоди-
стый метил — при 3058 см"1 и хлористый метилен — при
3049 см"1. Фокс и Мартин [26] подчеркивают, что даже
в случае нормальных углеводородов необходимо основы^
ваться на исследовании молекул, близких по строению
канализируемым. Изменения полос поглощения = СН при
5*
68
Часть I
переходе от алифатических систем к ароматическим уже
отмечались и будут более подробно обсуждаться в разделе,
посвященном циклическим системам. При интерпретации
частот в этой области необходимо учитывать возможность
появления полос в интервале 3090—3000 см'1, обусловлен-
ных как ненасыщенными системами, так и галогенпроиз-
водными предельных углеводородов, и наличие двойной
связи того или иного типа следует подтверждать данными,
относящимися к какой-либо другой части спектра.
В области 8000—5000 см'1 появляются первые обертоны
валентных колебаний С — Н, которые можно использовать,
как это делал Роуз [29], для идентификации различных
структурных элементов,
4. ВНЕПЛОСКОСТНЫЕ ДЕФОРМАЦИОННЫЕ
КОЛЕБАНИЯ =СН
Одним из наиболее тщательно изученных методов опре-
деления углерод-углеродных двойных связей и дифферен-
цирования их различных типов является исследование
внеплоскостных деформационных колебаний атомов водо-
рода, связанных с углеродом. Эти колебания дают характе-
ристические полосы поглощения в области 1000—800 см'1,
которые почти совершенно не зависят от строения молекулы
в целом и неоднократно применялись в качественных и коли-
чественных исследованиях.
Ламберт и Леконт [30, 31] впервые установили, что эти
полосы обусловлены наличием ненасыщенных связей, но
более точные корреляции были установлены позднее Томп-
соном и Торкингтоном [8], Гором и Джонсоном [32],
Расмуссеном и Браттеном [16, 33] и Шеппардом [55].
Детальное отнесение различных форм колебаний, дающих
эти характеристические полосы, было выполнено Шеппар-
дом и Сазерлендом [34] и Шеппардом и Симпсоном [47],
а Торкингтон [45] дал математическую трактовку связи
наблюдаемых смещений частот с электроотрицательностью
других заместителей при атоме углерода.
а. Дизамещенные этилена (— CH = СН —, транс).
Этиленовые двойные связи в присутствии заместителей
в транс-положении дают полосу при 990—965 см'1, интен-
Гл. 3. Алкены
6
сивность которой изменяется от средней до высокой., Эта
полоса, как было показано Килпатриком и Питцеррм 135],
обусловлена деформационными колебаниями атомов ,водо-
рода при двойной связи, выходящих из плоскости данной
связи. Поэтому изменение массы любого из заместителей
при атомах углерода очень мало влияет на положение
полосы, а интенсивность изменяется обратно пропорцио-
нально молекулярному весу. Это объясняется дем,, что аб-
солютная интенсивность полосы на каждую .двойную
связь есть величина постоянная, а количество двойных
связей на единицу объема вещества при увеличении длины
цепи уменьшается. Расмуссен и Браттен [33] показалц,
что указанная полоса поглощения появляется только при
транс-конфигурации молекул, что служит ценным .ука-
занием при выяснении вопросов цис- и транс-изомерии.
Существует так много примеров этого специфичного
поглощения, что здесь можно отметить лишь немногие щз
них; некоторые примеры будут приведены в качестве иллю-
страции использования данного поглощения для анали-
зов. Следует между тем указать, что, кроме галогенсодер-
жащих соединений, которые являются, по-видимому., исклю-
чением, не было обнаружено еще ни одного случая каких-
либо нарушений указанной корреляции, хотя было иссле-
довано большое число спектров углеводородов, полученных
АНИ. Эта корреляция была выведена для углеводородов,
но она, по-видимому, хорошо выполняется также для мно-
жества других соединений, например для кислот, аромати-
ческих соединений, полимеров и др. ,
Типичные случаи появления полос поглощения-в. обла-
сти 980—965 см 1 у ряда производных олефинов и этилеца
описаны в работах Шеппарда и Сазерленда [34], Шеппарда
и Симпсона [47] и Шеппарда [55], а Хемптон [36] приводит
дополнительные данные для других углеводородов. Почди
во всех этих случаях значение частоты близко к 965 см'1-.
Все высокомолекулярные жирные кислоты и спирты
с транс-конфигурацией относительно двойной связи погло!-
щают. при 965 см 1 [38], но если двойная связь располо-
жена в а,|3-положении к карбонильной группе, происходи^
небольшое смещение поглощения в сторону более высоки^
частот. Кротоновая, коричная, о-кумаровая- и акриловая
кислоты [12] поглощают в интервале980—974 см'1; известны
70
Часть I
примеры поглощения в этой области многими другими ве-
ществами [70, 71, 87, 93, 112]. Это поглощение является
общим для всех типов карбонильных групп, включая амиды
и другие аналогичные вещества. Замещение у а-углеродного
атома на группу CN, ОН или OR не оказывает влияния на
частоту поглощения [76, 87, 93], однако у некоторых метил-
производных этилена частота достигает 978 см'1. Следо-
вательно, присутствие у олефина атома хлора в a-поло-
жении непосредственно по рассматриваемой полосе может
быть не обнаружено, но у таких веществ находят новую
характеристическую полосу неизвестного происхождения
вблизи 1250 см1 [76, 87]. Замещение галогеном непо-
средственно у двойной связи оказывает более заметное
влияние. Хасзельдин [67] относит к деформационным коле-
баниям СН у — СН=СНВг и — СН = СНС1 частоту
935 см'1-, это подтверждает Китсон [76]. транс-1,2-Диал-
килвиниловые эфиры также поглощают приблизительно на
30 см 1 ниже нормального значения частоты [93].
Влияние сопряжения на частоту рассматриваемого
поглощения было изучено подробно, особенно влияние
многократного сопряжения, которое имеет место у расти-
тельных масел и других соединений. Подробный обзор
данных по этому вопросу с полной библиографией был
сделан О’Коннором [82]. Эффект сопряжения опять-таки
невелик, но обычно наблюдается смещение полосы в сто-
рону больших частот, особенно у длинных сопряженных
цепей. Аллан и др. [87] описали много соединений, у кото-
рых происходит постепенное увеличение частоты с удли-
нением сопряженной цепи; если эта цепь сопряжена, кроме
того, с кислотными и сложноэфирными группами, то ча-
стота достигает предельной величины около 1000 см'1.
Были сделаны некоторые попытки [82, 94, 107] подразде-
лить интервал 1000—965 см 1 на участки, характерные для
различных пространственных форм жирных кислот с длин-
ной цепью. Так, транс-транс-соединения поглощают вблизи
980 см'1, цис-пгранс-соедшеняя— вблизи 984 см'1, транс-
транс-транс-соединения — вблизи 995 см 1 и т. д. Такой
подход может быть полезным в отношении ряда жирных
кислот, но его нельзя неосмотрительно применять к дру-
гим соединениям. Например, Чепман и Тейлор [96] ука-
зывают, что в случае p-каротина с одиннадцатью сопряжен-
Гл. 3. Алкены
71
ными двойными связями деформационные колебания СН,
по существу, имеют постоянную частоту 968 с.и1.
Сопряжение с ароматическими кольцами, как у стиль-
бенов [39, 97], не влияет на положение полосы 965 см'1,
но при наличии сопряжения с ацетиленовыми связями [87]
эта полоса слегка смещается в сторону низких частот. Ин-
тенсивность данного поглощения у различных замещенных
стильбена изучал Орр [119], который обнаружил значи-
тельные изменения ширины полосы, зависящие, вероятно,
от стерических эффектов.
Коэффициент погашения рассматриваемой полосы изме-
рялся многими исследователями, и хотя трудно сравнивать
значения, полученные на различных приборах, из этих
измерений видно, что он имеет довольно постоянное значе-
ние. Это также является подтверждением вывода, что
полоса поглощения при 965 см 1 обусловлена структурой
—СН = СН— (транс). Поскольку данная полоса находится
в той спектральной области, в которой обнаруживаются
также многие другие колебания, то ее отсутствие можно
рассматривать как доказательство отсутствия связи ука-
занного типа; с другой стороны, если эта полоса име-
ется, необходимо искать другие подтверждения наличия
двойной связи путем измерения интенсивности и путем
использования других спектральных областей, например
областей валентных колебаний С — Н и С = С.
Андерсон и Сейфрид [40] выразили величину коэффи-
циента погашения полосы при 967 см1, обусловленной эти-
леновой связью с транс-конфигурацией, через коэффи-
циент поглощения функциональной группы. Полученная
ими величина хорошо согласуется с величиной, приведенной
Хемптоном [36] и с получающейся из спектров АНИ.
Последняя величина (вычисленная при учете полного моле-
кулярного веса соединений) меняется от 132 до 141, тогда
как для элаидиновой кислоты и родственных соединений,
по данным Шрива и др. [38], значение коэффициента равно
140—156. Изменения коэффициента от соединения к соеди-
нению довольно заметны, но по сравнению с изменениями
этих коэффициентов для других веществ их можно считать
небольшими; например, Шрив приводит значения коэффи-
циентов для метилолеата и триолеина, равные соответ-
ственно 12,2 и 74,4.
7‘2 Часть /
Сравнительно постоянное отношение интенсивности этой
полосы к количеству двойных связей использовалось мно-
гими исследователями для количественного анализа. В ряду
жирных кислот интенсивность этой полосы приблизительно
аддитивна даже у веществ с сопряженными связями [94, 98],
однако Аллан и др. [87] отметили заметные отклонения
в случае сопряжения с COOR или с ацетиленовыми
связями.
В уже упоминавшейся работе Шрива была сделана
попытка разработать метод определения транс-октадеце-
новых кислот, сложных эфиров и спиртов в сложных сме-
сях. Аналогичные методы применяются для определения
количества транс-олефинов в смазочных маслах [41, 68,
95] и в бензине [42]. Особенно полезно пользоваться этими
методами при количественном и качественном изучении
реакций полимеризации. Объектами исследований явились
низкокипящие полимеры бутадиена и стирола [36, 431,
природный и синтетический каучуки [44, 60, 69], изуча-
лась также вулканизация природного каучука [59]. В слу-
чае терпенов [46] и стеринов (48, 61] изучение поглощения
при 965 слг1 вместе с исследованием деформационных коле-
баний СН при двойных связях других типов также дало
ценные сведения о строении соединений. Все эти работы
свидетельствуют о постоянстве рассматриваемой частоты
для широкого круга различных соединений.
Доказательство того, что эта полоса специфична для
транс-конфигурации при этиленовой связи, основывается,
конечно, на факте отсутствия такой полосы у соответ-
ствующих соединений цис-ряда [16]. Часто бывает трудно
получить цис-соединение совершенно свободным от
транс-изомера, но во многих случаях полоса при 965 слг1
практически исчезает. В других случаях интенсивность
полосы при 965 слг1 предельно мала, например у олеи-
новой кислоты, так что часто изомеры различают по по-
глощению в этой области. Примерами применения данного
метода для определения изомеров служат исследования
цис- и транс-фенилбутадиена [49], полиуретанов [50],
высокомолекулярных ненасыщенных енолов [51] и изо-
меров хлористого кротонила [52]. Однако при использова-
нии этого метода необходима осторожность. Например,
если группа — СН = С(СН3) — сопряжена с карбонильной
Гл- 3. Алкены
7
группой, то при наличии /пране-конфигурации она может
также давать поглощение в области 1000—980 см 1 [80].
б. Дизамещенные этилена (цис). Из-за того что в чистом
виде цис-нзомер можно получить лишь в редких случаях,
о внеплоскостных деформационных колебаниях атомов водо-
рода в г^ис-дизамещенных этилена [34] получено гораздо
меньше сведений. Несколько спектров г^ис-соединений описа-
но в работах Американского нефтяного института (АНИ)
и в некоторых других работах [47, 55]. Эти данные позво-
ляют предположить, что пик поглощения, обусловленный
данной формой колебаний, лежит около 690 см'1, причем
его положение гораздо менее постоянно, чем положение соот-
ветствующей полосы транс-формы. Так, г^нс-пентен-2 погло-
щает при 698 см'1, а цис-4-метилпентен-2 — при 719 см'1.
Шеппард и Симпсон [47] приводят для ряда простых про-
изводных этилена интервал 728—675 см'1, но даже при
таком широком интервале отнесение полос в некоторых
случаях сомнительно. Указанное расположение полосы
поглощения подтверждено Многими исследователями [76,
79, 87, 99, 112, 120]. У углеводородов эта полоса обычно
лежит вблизи 730 см'1, однако она намного чувствительнее
к изменениям структуры прилегающих групп, нежели соот-
ветствующая полоса при 965 см'1 у транс-форм. Замещение
хлором [76], метилом [76, 79, 99] или кислородсодержащей
группой [87] в a-положении вызывает заметное смещение
в сторону больших частот. Так, цис- 1-хлорбутен-2 [76]
поглощает при 769 см'1 и цис-1,4-дихлорбутен-2 при
787 см'1, а смещение в присутствии других упомянутых
заместителей имеет величину такого же порядка, как
и в присутствии хлора.
Сопряжение двойных связей также вызывает повышение
частоты, величина которого зависит от того, имеется ли
сопряжение с двух сторон и сколько двойных связей сопря-
жено [79]; известно смещение частоты до 780 см'1 [100, 101].
С другой стороны, сопряжение с ацетиленовой связью вызы-
вает лишь небольшое смещение в сторону более низких
частот; в этих случаях было найдено довольно постоянное
значение частоты, равное 720 см'1 [87]. Большое влияние
оказывает на частоту сопряжение с карбонильной груп-
пой. Так, группа — СН = СН — COOR (цис) поглощает
74
Часть I
вблизи 820 см"1, причем постоянство данной частоты делает
ее весьма полезной для анализа, особенно если дальнейшее
сопряжение не оказывает значительного воздействия. Ин-
тенсивность указанного поглощения много слабее, чем
в случае транс-соединений, но она значительно повышается
при сопряжении.
У таких сложных соединений, как стероиды, тритерпены
и др., щ/с-олефины рассматриваемого типа дают серию из
нескольких полос в области 800—650 см"1. При изучении
данной области можно получить сведения о расположении
заместителей в циклах [77, 108, 109], однако обычно не
удается обнаружить полосу, отвечающую поглощению
— СН = СН — (цис)-, кроме того, значительное искажение
картины вызывает введение заместителей, приводящих
к появлению сопряженных систем. Поэтому при идентифи-
кации tjuc-олефинов необходимо соблюдать осторожность
и не следует забывать, что в этой области поглощают также
соединения другой структуры, в том числе и некоторые
транс-олефины. Например, транс-фенилпропенилкарбинол
[54] и хлористый транс-кротонил [53] поглощают в обла-
сти 690 см"1.
в. Монозамещенные этилена. Двойная связь винилового
типа дает две сильные полосы около 990 и 910 см"1. Первая
из них связана с деформационными колебаниями водорода
в группе —СН = С
[34] и отсутствует у несимметрич-
ных дизамещенных этилена. Вторая полоса обусловлена
внеплоскостными деформационными колебаниями атомов
водорода в группе = СН2 и характерна как для монозаме-
щенных, так и для несимметричных дизамещенных этилена.
Как и в случае связи —СН = СН— , указанные частоты
довольно постоянны. Отнесение их к рассматриваемому
типу связи подтверждается многими данными, в том числе
спектрами АНИ. Например, Андерсон и Сейфрид [40] при-
водят 12 виниловых соединений, поглощающих при 995+2
и 910±2 см"1, а Хемптон [36] упоминает еще о 9 веществах,
в спектрах которых (полученных АНИ) интервал второй
полосы составляет 912—909 см"1. Мак-Мурри и Торнтон
[58] отмечают 15 таких соединений, а Шеппард и Сазер-
ленд [34] и Шеппард и Симпсон [47] также сообщают о ряде
Гл. 3. Алкены
75
виниловых соединений, поглощающих в интервалах частот
995—985 и 915—905 см'1. Однако при некоторых условиях
происходят и небольшие смещения. Так, если в молекуле
RCH = СН2 все атомы водорода у а-углерода группы R
замещены, то частота смещается в интервал 1005—995 см'1,
а если один из заместителей при этом углероде, не обра-
зующем ненасыщенных связей, является функциональной
группой, то полоса 910 см'1 смещается в интервал 930—
918 см'1 [54, 55]. Например, линалоол поглощает при 925
и 997 см'1. Эти явления были довольно подробно изучены
рядом исследователей [74, 76, 93, 102], и влияние на обе
полосы различных заместителей теперь хорошо известно.
Присоединение к углеродному атому, находящемуся в a-по-
ложении относительно двойной связи, таких полярных
групп, как СН2С1 [76],CH2CN [76],CH,OR [74, 93], CH2SR
[62] и СН2ОН, оказывает небольшое влияние, причем
более высокая частота остается без изменения, а более
низкая повышается на 12 см'1. В случае группы CH2OCOR
это влияние более заметно [74], так как смещаются обе
частоты, значения которых становятся равными 985
и 932 см'1. Немного большее влияние оказывает присоеди-
нение карбонильной группы непосредственно у двойной
связи. При этом более высокая частота равняется 982 см1,
а более низкая в зависимости от природы карбонильной
группы находится в интервале 965—950 см'1 [74, 93].
Однако наиболее заметные смещения происходят при
непосредственном присоединении атома кислорода, как
это имеет место у виниловых эфиров и подобных соедине-
ний. Частота 990 см'1 понижается до 962 см1 у простых
виниловых эфиров и до 948 см'1 у сложных виниловых
эфиров [74, 89, 93]. Положение более низкочастотной по-
лосы (910 см'1) у этих соединений менее определенно. Де-
висон [74] сначала предположил, что эта полоса у простых
виниловых эфиров лежит при 942 см'1 и у виниловых слож-
ных эфиров — при 870 см'1. Однако Микинс [89] обнару-
жил эту полосу у простых виниловых эфиров вблизи 810 см'1,
что было подтверждено более подробными исследования-
ми Филпоттса [93], а также тем фактом, что полоса 890 см'1
для соединений СН2= CR2 смещается приблизительно
в два раза больше по сравнению со смещением полосы обыч-
ного винилового эфира, если обе группы R представляют
76
Часть I
собой алкоксигруппы [93]. С другой стороны, у винилни-
тратов полосы лежат при 965 и 940 см 1 [103], хотя можно
было бы ожидать аналогии с кислородсодержащими соеди-
нениями.
Замещение галогенами, если оно происходит у одного
из атомов углерода при двойной связи, также приводит
к некоторому усложнению картины, но возможные случаи
здесь ограничиваются фтористым, хлористым и иодистым
винилом. Эти вещества изучались Томпсоном и Торкинг-
тоном [56], которые обнаружили ожидаемое понижение
частоты 910 см'1 алкилзамещенных до частот соответствен-
но 860, 895 и 902 см'1.
Обе рассматриваемые здесь полосы имеют постоянную
интенсивность, если учитывать молекулярный вес, что ха-
рактерно для полос внеплоскостных деформационных
колебаний СН при двойной связи. Для количественных ис-
следований используется обычно полоса при 910 см1,
так как она более удобна для анализа полимеров и углево-
дородов на содержание олефинов. Андерсон [40] нашел,
что значение интенсивности полосы при 910 см'1 при учете
молекулярного веса постоянно в пределах примерно 7%.
Из спектров АНИ, приведенных Хемптоном [36], следует,
что значение коэффициента погашения находится в преде-
лах 143—153 (также при учете молекулярного веса). Бо-
лее детальные исследования провел Иогансен [121], кото-
рый показал, что интегральная интенсивность упомянутой
полосы почти постоянна, хотя ее ширина заметно изменяет-
ся. Изменения наблюдаемых коэффициентов погашения
в зависимости от присутствия различных полярных заме-
стителей исследовались Девисоном [74].
Использование этих полос в качественном и количе-
ственном анализе рассматривается в работах, которые упо-
минались при обсуждении связи — СН = СН—, так как
прежде всего в этих работах ставилась задача обнаруже-
ния и количественного определения различных типов
двойных связей в смесях [36, 38, 40—46]. Данные корре-
ляции при некотором расширении интервалов частот, по-
видимому, применимы также и к неуглеводородам.
Помимо упомянутой пары характеристических полос,
виниловые соединения почти всегда дают полосу средней
интенсивности в области 1850—1800 см'1, которая, воз-
Гл. 3. Алкены 77
можно, представляет собой обертон, связанный с полосами
910 и 990 см'1. Это предположение подтверждается тем,
что в случае двойной связи типа RJR2C = СН2 с частотой
внеплоскостных деформационных колебаний водорода ниже
900 см'1 полоса, которая, возможно, соответствует обер-
тону, также наблюдается при более низкой частоте (около
1800—1750 см'1). С другой стороны, интенсивность этого
«обертона» часто бывает значительно выше, чем можно
было бы ожидать. Таким образом, происхождение этой
полосы не вполне ясно, однако во многих случаях она слу-
жит доказательством наличия группы = СН2.
г. Несимметричные дизамещенные этилена (RLR2C=СН2).
Полоса внеплоскостных деформационных колебаний СН
у соединений типа R1R3C = СН2 имеет частоту 890 см'1.
Эта частота постоянна в такой же степени, как и частоты
в случае уже рассмотренных типов двойных связей. На-
пример, Шеппард и Симпсон [47] приводят для семи раз-
личных соединений с этой группой интервал 892—887 см'1.
Влияние полярных групп на частоту по существу является
таким же, как и у виниловых соединений. Группы Cl, CN,
OR и ОН, находящиеся у а-углеродного атома, обычно вы-
зывают небольшое смещение до 900 см'1 [74, 76, 93]. Однако
это происходит не всегда; например, 2,3-дихлорбутадиен-1,3
дает обычную полосу поглощения при 892 слГ1 [105]. Кар-
бонильная группа, присоединенная непосредственно у двой-
ной связи, вызывает увеличение частоты до 930—945 см'1
[74, 76, 93, 104]. Такие же частоты получаются, если не-
посредственно у двойной связи находится нитрогруппа
[103]. Присоединение кислорода у двойной связи приводит,
как и в рассмотренном выше случае, к значительному сме-
щению частоты, а именно до 795 см'1 [89, 93], причем у ди-
этоксисоединений частота снижается даже до 710 см'1
[93]. Сопряжение с олефиновой связью иногда приводит
к небольшому повышению частоты [104]; большее влияние
оказывает сопряжение с ацетиленовой связью (908 см'1)
или присоединение нитрильной группы непосредственно
к.двойной связи (934 см'1). Как и в рассмотренных выше слу-
чаях, при замещении атомами галогенов полоса смещается
в сторону меньших частот. Томпсон и Торкингтон [56]
сообщают о смещениях полосы у бромистого, хлористого
78
Часть 1
и фтористого винилидена соответственно на 5, 11 и 71 см1.
Интенсивность рассматриваемой полосы несколько менее
постоянна; Андерсон [40] сообщает об отклонениях от сред-
него значения в пределах до 30%. Тем не менее анализы пу-
тем измерения интенсивности поглощения при этой ча-
стоте во многих случаях могут выполняться с достаточной
точностью. Типичные примеры использования поглоще-
ния для анализов можно найти в цитированной литературе
[36, 38, 40—46].
Так же, как и в случае виниловых соединений, имеется
полоса в области 1800—1750 см которая может быть обер-
тоном частоты 890 с.и1 и часто является ценным подтверж-
дением присутствия несимметричной этиленовой связи.
д. Тризамещенные этилена (R4R2C = CHR3). У три-
замещенных этилена характеристичность полосы внеплос-
костных деформационных колебаний СН сильно уменьше-
на, и частота в большей степени зависит от природы заме-
стителей. Это объясняется, вероятно, тем, что заместители
R1, R2 и R3 могут вызывать гораздо большие изменения
силовых постоянных связей, чем заместители в моно-
и дизамещенных этилена. Интервал частот, характерный
для этой структуры, обычно составляет 840—800 см
хотя сообщалось о нескольких соединениях, которые по-
глощают даже при 790 см1. Как и в предыдущих случаях,
при отнесении указанных частот принимались в расчет
спектры АНИ [58], а также данные работ Шеппарда и Са-
зерленда [34], Томпсона и Торкингтона [3] и других.
Недостаточное постоянство частоты этой полосы делает ее
менее пригодной для аналитических целей по сравнению
с полосами, характерными для двойных связей других ти-
пов, но тем не менее она была с успехом использована
для качественной идентификации углеводородов и количе-
ственного анализа олефиновых смесей. В частности, эту
полосу применяли Ружичка и др. [57], которые по наличию
или отсутствию поглощения в области 840—800 см 1
распознавали сесквитерпены, содержащие' группу
R1R2C=CHR3, и полностью замещенные сесквитерпены типа
R1R2C=CR3R4, у которых не может быть полосы деформаци-
онных колебаний водорода при двойной связи. Хиршман [72]
нашел у большого ряда ненасыщенных стеринов эту полосу
Гл. 3. Алкены
79
при 800 см'1, а также обнаружил вторую полосу прибли-
зительно при 812 см'1, которая появляется, по-видимому,
только у Л5 - 30-ацетоксисоединений. Влияние замещения
галогеном при двойной связи полностью еще не изучено,
но предполагается, что замещение приводит к уменьшению
частоты; неопрен, имеющий структуру R1CIC = CHR2,
поглощает при 826 см'1. Однако эту корреляцию нельзя
считать вполне надежной, и ее нужно применять с большой
осторожностью. Для некоторых иронов Леконт и Нав
[80] отнесли полосу в области 1000—980 см'1 к группе
С = СН— (транс); она может вызываться валентными ко-
СН3
лебаниями СН3 — С. Примерно такая же корреляция была
предложена Гунцлером и др. [117] для полностью заме-
щенных этиленов, содержащих в качестве заместителя ме-
тильную группу. В этих случаях появляется довольно по-
стоянная полоса поглощения вблизи 1155 см'1.
5. ПЛОСКОСТНЫЕ ДЕФОРМАЦИОННЫЕ КОЛЕБАНИЯ =СН
Полосы деформационных колебаний водородных атомов
в плоскости двойной связи мало использовались в аналити-
ческих целях, и данный тип колебаний тщательно не изу-
чался. Это объясняется тем, что условия симметрии при-
водят в некоторых случаях либо к тому, что указанные
колебания становятся запрещенными в ИК-спектре, либо
к уменьшению интенсивности соответствующей полосы.
Тем не менее частоты этих колебаний определены с такой
же точностью, как и частоты внеплоскостных деформа-
ционных колебаний. В случае таких типов двойных свя-
зей, которые дают сильные полосы, обнаружение частот
плоскостных колебаний может служить ценным подтверж-
дением наличия двойной связи, несмотря на то что эти
полосы обычно находятся в области, связанной также
с валентными колебаниями С — С и деформационными
колебаниями — СН у насыщенных групп.
а. Дизамещенные этилена (транс). Плоскостные дефор-
мационные колебания vt атомов водорода в случае симмет-
ричных замещенных этилена должны быть активны только
80
Часть I
в спектре комбинационного рассеяния и являются запре-
щенными в инфракрасном спектре. Шеппард и Сазерленд
[34] показали, что в действительности для ряда подобного
типа углеводородов как в спектре комбинационного рассея-
ния, так и в инфракрасном спектре появляется интенсивная
полоса в области 1310—1290 см'1. Авторы отнесли эту по-
лосу к плоскостным деформационным колебаниям vt и v2
двух различных классов симметрии. Полоса в этой области
инфракрасного спектра наблюдается у всех углеводородов
такого типа, однако интенсивность ее, по-видимому, пере-
менна и иногда очень мала [34, 47, 55].
б. Дизамещенные этилена (цис). Дизамещенные эти-
лена с ^ис-конфигурацией дают сильную линию в спектре
комбинационного рассеяния вблизи 1260 см'1, которая от-
носится к колебаниям vt [47, 55]. Такая полоса по усло-
виям симметрии является допустимой также в инфракрас-
ном спектре, однако в этой области инфракрасного спект-
ра обнаружить поглощение не всегда удается. Между тем
во многих случаях найдена полоса средней интенсивности
вблизи 1405 см'1, которая также приписывается плоскост-
ным деформационным колебаниям СН [47].
в. Монозамещенные этилена. Как и в случае внеплоскост-
ных деформационных колебаний атомов водорода, соответ-
ствующие плоскостные колебания у соединений винилового
типа приводят к появлению двух полос. Первая полоса
с частотой Vi обусловлена деформационными колебаниями
атомов водорода в группе =СН2, тогда как полоса v2
обусловлена главным образом группой —СН = С —. Обе
полосы находятся как в инфракрасном спектре, так и
в спектре комбинационного рассеяния в интервалах 1420—
1410 и 1300—1290 см'1. Полоса с большей частотой отне-
сена к деформационным колебаниям =СН2 [34, 47, 55]
по аналогии со структурой КЧ^2С = СН2, у которой она
также обнаруживается, а соответствующая полоса деформа-
ционных колебаний СН2 насыщенных углеводородов нахо-
дится вблизи 1450 см'1. Полоса с меньшей частотой также
лежит в ожидаемой области частот, причем аналогичная
полоса имеется и у симметричных транс-дизамещенных
этилена.
Гл. 3. Алкены
81
Полоса с большей частотой, около 1415 см'1, в инфра-
красном спектре имеет обычно высокую интенсивность
и очень постоянное положение, так что она может быть
успешно использована для анализов. Замещение группами
CH2OR и CH2OCOR почти или совсем не влияет на эту
частоту, но в случае акрилатов и метилакрилатов она слег-
ка понижается до 1405 см1 [74]. Полоса с меньшей часто-
той также имеет сравнительно постоянное положение,
но интенсивность ее, по-видимому, более переменна.
г. Несимметричные дизамещенные этилена. Согласно
изложенному выше, следует ожидать, что структура этого
типа дает сильную полосу в интервале 1420—1410 см'1,
обусловленную плоскостными деформационными колеба-
ниями = СН2, как в спектре комбинационного рассеяния,
так и в инфракрасном спектре. Такая полоса была найдена
экспериментально, причем ее положение было, как и ожи-
далось, постоянно в пределах нескольких обратных санти-
метров [34, 47, 55].
д. Тризамещенные этилена. Плоскостным деформацион-
ным колебаниям единственного атома водорода этой струк-
туры соответствует сильная линия спектра комбинацион-
ного рассеяния в области 1390—1375 см'1. Хотя в инфра-
красном спектре такая полоса также разрешена, интен-
сивность ее в большинстве случаев мала, а поэтому и зна-
чение для структурных анализов невелико.
ЛИТЕРАТУРА
1. Barnes, Gore, Liddel, Williams., Infra-red
spectroscopy, Reinhold, New York, 1944.
2. Sheppard, J. Chem. Phys., 17, 74 (1949).
3. Thompson, Torkington, Trans. Faraday Soc., 42,
432 (1946).
4. Randall, Fowler, Fuson, D a n g 1, Infra-red deter-
mination of organic structures, Van Nostrand Co., New York,
1949.
5. Sheppard, Sutherland, J. Chem. Soc., 1947, 1540.
6. T h о m p s о n, W h i f f e n, J. Chem. Soc., 1948, 1412.
7. H i b b e n, The Raman effect and its chemical applications,
Reinhold, New York, 1939, p. 166.
c
° Л. Беллами
82
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
31.
35.
36.
37.
38.
Част I
Thompson, Torkington, Trans. Faraday Soc., 41,
246 (1945).
Jones, Humphries, Packard, Dobriner,
J. Amer. Chem. Soc., 72, 86 (1950).
Г ерцберг, Колебательные и вращательные спектры много-
атомных молекул, Издатинлит, М., 1949, стр. 350.
Герцберг, Колебательные и вращательные спектры много-
атомных молекул, Издатинлит, М., 1949, стр. 353.
Flett, J. Chem. Soc., 1951, 962.
К 1 е t z, Sumner, J. Chem. Soc., 1948, 1456.
Shreve, Heether, Knight, Swern, Analyt. Chem.,
22, 1498 (1950).
Bernstein, Fowling, J. Amer. Chem. Soc., 73, 1843
(1951).
Rasmussen, Brattai n, J. Chem. Phys., 15, 120
(1947).
Bradacs, Kahovec, Z. physikal. Chem , B48, 63 (1940).
Rasmussen, Tunnicliff, Brattai n, J. Chem.
Phys., 11, 432 (1943).
Blout, Fields, Karplus, J. Amer. Chem. Soc., 70,
194, (1948).
Rasmussen, Tunnicliff, Brattai n, J. Amer. Chem.
Soc., 71, 1068 (1949).
Kohlrausch, Pongrat z, Z. Chem. Phys., B27, 176
(1934).
Furchgott, Rosenkrantz, Harris, Shorr, J.
Biol. Chem., 163, 375 (1946).
Torkington, Thompson, Trans. Faraday Soc., 41,
236 (1945).
Edge 11, J. Amer. Chem. Soc., 70, 2816 (1948).
Park, L у c a n, Lacher, J. Amer. Chem. Soc., 73, 711
(1951).
Fox, Martin, Proc. Roy. Soc., A175, 208 (1940).
Saier, Coggeshell, Analyt. Chem., 20, 812 (1948).
Jones, Williams, Whalen, Dobriner, J. Amer
Chem. Soc., 70, 2024 (1948).
Rose, J. Res. Nat. Bur. Stand., 20, 129 (1938).
Lambert, Lecomte, Compt. rend. Acad. sci. Paris, 206,
1007 (1938).
Lambert, Lecomte, Ann. phys., 10, 503 (1938).
Gore, Johnson, Phys. Rev., 68, 283 (1945).
Rasmussen, Brattai n, J. Chem. Phys., 15, 131, 135
(1947).
Sheppard, Sutherland, Proc Roy. Soc., A196, 195
(1949).
Kilpatrick, Pitzer, J. Res. Nat. Bur. Stand., 38, 191
(1947).
Hampton, Analyt. Chem., 21, 923 (1949).
Freeman, J. Amer. Chem. Soc., 75, 1859 (1953).
Shrexe, Heether, Knight, Swern, Analyt. Chem .
22, 1261 (1950).
Гл. 3. Алкены
83
39. Thompson, V a g о, С о г f i е 1 d, О г г, J. Chem. Soc.,
1950, 214.
40. Anderson, Seyfried, Analyt. Chem., 20, 998 (1948).
41. Fred, P u t s c h e r, Analyt. Chem., 21, 900 (1949).
42. J о h n s t о n, A p p 1 e b v, Baker, Analyt. Chem., 20,
805 (1948).
43. T r e u m a n n, Wall, Analyt. Chem., 21, 1161 (1949).
•14. Dinsmore, Smith, Analyt. Chem., 20, 11 (1948).
45. Torkington, Proc. Roy. Soc., A206, 17 (1951).
46. Bateman, Cunneen, Fabian, Koch, J. Chem.
Soc., 1950, 936.
47. S h e p p a r d, S i m p s о n, Quart. Rev., 6, 1 (1952).
48. J о n e s, J. Amer. Chem. Soc., 72, 5332 (1950).
49. Grum m it, Christoph, J. Amer. Chem. Soc., 73, 3479
(1951).
50. Marvel, Young, J. Amer. Chem. Soc., 73, 1066 (1951).
51. С г о m b i e, Harper, J. Chem. Soc., 1950, 1707, 1714.
52. Fl a t c h, Nesbitt, J. Amer. Chem. Soc., 72, 727 (1950).
53. P h i 1 p о t t s, T h a i n, Nature, 166, 1028 (1950).
54. В a r n a r d, Bateman, Harding, Koch, Shep-
pard, Sutherland, J. Chem. Soc., 1950, 915.
55. Sheppard, J. Inst. Pet., 37, 95 (1951).
56. Thompson, Torkington, Proc. Roy. Soc., A184, 21
(1945).
57. Meyer, J e g e r, Ru’icka, Helv. chim. acta, 33, 672 (1950);
Dietrich, J e g e r, ibid., 33, 71 1 (1950); Koller, H i e-
stand, Dietrich, Jeger, ibid., 33, 1050 (1950).
58. McMurry, Thornton, Analyt. Chem., 24, 318 (1952).
59. Sheppard, Sutherland, Trans. Faraday Soc., 41,
261 (1945).
60. Salomon, Van der Schee, Ketelaar, Van Eyk,
Discuss. Faraday Soc., 9, 291 (1950).
61. Bladon, Fabian, Henbest, Koch, Wood, J. Chem.
Soc., 1951, 2402.
62. T a r b e 1 1, McCall, J. Amer. Chem. Soc., 74, 48 (1952).
63. S a u n d e r s, Smith, J. Applied Phys., 20, 953 (1949).
64. Hatcher, Yost, J. Chem. Phys., 5, 992 (1937).
65. Morin o, Kuchitsu, Shimanouchi, J. Chem. Phys.,
20, 726 (1952).
66. Nielsen, Liang, Smith, J. Chem. Phys., 20, 1090 (1952).
67. Haszeldine, Nature, 168, 1028 (1951).
68. P u t s c h e r, Analyt. Chem., 24, 1551 (1952).
69. Richardson, Sacher, J. Polymer. Sci., 10, 353 (1953).
70. С г о m b i e, J. Chem. Soc., 1952, 2997, 4338.
71. Sinclair, McKay, Myers, Jones, J. Amer. Chem.
Soc., 74, 2578 (1952).
72. H i r s c h m a n n, J. Amer. Chem. Soc., 74, 5357 (1952).
73. M i x e r, Fl e c k, W i n s t e i n, Y о u n g, J. Amer. Chem.
Soc., 75, 4098 (1953).
74. D a v i s о n, Bates, J. Chem. Soc., 1953, 2607.
75. L о r cl Walker, J Amer. Chem. Soc., 76, 2518 (1954).
6*
84
Часть 1
76. К i t s о n, Analyt. Chem., 25, 1470 (1953).
77. Henbest, Meakins, Wood, J. Chem. Soc., 1954, 800.
78. L e о n a r d, G a s h, J. Amer. Chem. Soc., 76, 2781 (1954).
79. Oroshnik, Mebane, J. Amer. Chem. Soc., 76, 5719
(1954).
80. Lecomte, Naves, J. Chim. Phys., 1956, 462.
81. Груздев П. Ф., ЖФХ, 28 507 (1954).
82. O’Connor, J. Amer. Oil Chem. Soc., 33, 1 (1956).
83. Werner, Lark, J. Chem. Soc., 1954 1152.
84. Lord, Miller, Applied Spectroscopy, 10, 115 (1956).
85. J о n e s, H e r 1 i n g, J. Org. Chem., 19, 1252 (1954).
86. Джонс P. H., Сандорфи К., Применение спектроско-
пии в химии, Издатинлит, Москва, 1959, стр. 315.
87. Allan, М е a k i n s, Whiting, J. Chem. Soc.,
1955, 1874.
88. Brice, Lazerte, Hals, Petersen, J. Amer. Chem.
Soc., 75, 2698 (1953).
89. M e a к i n s, J. Chem. Soc., 1953, 4170.
90. Slowinski, Emil, Claver, J. Polymer. Sci., 17,
269 (1955).
91. Bellamy, J. Chem. Soc., 1955, 4221.
92. Scrocco, Salvetti, Bull. Sci. della Fac. Chim. Bolog-
na, 12, 1 (1954).
93. Philpotts, частное сообщение.
94. Ahlers, Brett, McTaggart, J. Appl. Chem., 3,
433 (1953).
95. Francis, Analyt. Chem., 28, 1171 (1956).
96. Chapman, Taylor, Nature, 174, 1611 (1954).
97. DeTar, Carpin o, J. Amer. Chem. Soc., 78, 475
(1956).
98. Bickford, DuPre, Mack, O’C о n n о r, J. Amer.
Oil. Chem. Soc., 30, 376 (1953).
99. Hall, M i к о s, Analyt. Chem., 21, 422 (1949).
100. Kuhn, Inhoffen, Staab, Otting, Chem. Ber.,
86, 965 (1953).
101. Zechmeister, Experimentia, 10, 9 (1954).
102. Kirmann, Chancel, Bull. Soc. Chim. Fr.. 1954,
1338.
103. Brown, J. Amer. Chem. Soc., 77, 6341 (1955).
104. В r u g e 1, Angew. Chem., 68, 441 (1956).
105. Sheppard, Szasz, Trans. Faraday Soc., 49, 358
(1953).
106. Traynham, Sehnert, J. Amer. Chem. Soc., 78, 4024
(1956).
107. Jackson, Paschke, Tolberg, Boyd, Wheeler
J. Amer. Oil Chem. Soc., 29, 229 (1952).
108. Henbest, Meakins, Nicholls, Wilson,
J. Chem. Soc., 1957, 997.
109. Cole, Thornton, J. Chem. Soc., 1957, 1332.
110. Blomquist, Wolinsky, Meinwald, Longo-
n'e, J. Amer. Chem. Soc., 78, 6057 (1956).
Гл. 3. Алкены
85
111. В I о m q u i s t, V e r d о 1, J. Amer. Chem. Soc., 77, 1806
(1955).
112. Heilman n, Gaudemaris, Arnaud, Bull. Soc.
Chim. Fr., 1957, 112, 119.
113. Gambon i, Theus, Sehin z, Helv. Chim. Acta, 38,
255 (1955).
114. Theus, Surber, Colombi, Sehin z, Helv.
Chim. Acta, 38, 239 (1955).
115. Sorensen J. S., Sorensen N. A., Acta Chem.
Scand., 8., 1741, 1763 (1954).
116. Bohlmann, Mannhardt, Chem. Ber., 88, 429, 1336
(1955).
117. Gunzler, Kienitz, Neuhaus, Naturwiss., 43, 299
(1956).
118. Weiblen, Fluorine Chemistry, Ed. Simons, Vol. 2, Academic
Press, New York, 1954, p. 449.
119. О r r, Spectrochim. Acta, 8, 218 (1956).
120. Jones, Mansfield, Whiting, J. Chem. Soc., 1956,
4073.
121. Иогансен А. В., Изв. АН СССР, сер. физ., 18, 708
(1954).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1. Прилежаева Е. Н., Сыркин Я. К-, В о л ь-
кен штейн М. В., ЖФХ, 14, 1936 (1940).
2. И о г а н с е и А. В., Инфракрасные спектры углеводородов,
«Физико-химические свойства индивидуальных углеводо-
родов», Гостоптехиздат, М.—Л., вып. 6, 1957.
3. П е н т и н Ю. А., Панченко Ю. Н., Т р е щ е-
в а Е. Г., Шарипов 3., Оптика и спектроскопия, 10,
55 (1961).
Глава 4
АЛКИНЫ И АЛЛЕНЫ
I. ВВЕДЕНИЕ
Возможность определения ацетиленовых связей у неиз-
вестных веществ зависит во многом от степени замещения
при тройной связи. Монозамещенные ацетилена могут быть
легко идентифицированы по очень характерным валентным
колебаниям зС — Н, а также по колебаниям С = С,
которым соответствует сильная полоса, если тройная связь
расположена в конце цепи. У дизамещенных веществ пер-
вая из этих полос, конечно, отсутствует, тогда как вторая
имеет переменную интенсивность в зависимости от поло-
жения тройной связи. У симметричных структур полоса
поглощения С = С часто бывает слишком слабой, чтобы
ее можно было обнаружить, и таким образом не имеется на-
дежного способа для распознавания такого типа связи.
С другой стороны, если полоса валентных колебаний
С- С присутствует, то ее. положение является весьма ха-
рактеристичным, а при исследовании обычных смещений,
обусловленных сопряжением или наличием ароматического
заместителя, также можно получить ценные дополнитель-
ные сведения.
Единственной иной возможностью получить данные
о монозамещенных ацетилена является исследование об-
ласти деформационных и составных частот sCH. Полоса
этих деформационных колебаний появляется вблизи 650 см'1,
а составная частота наблюдалась в некоторых случаях
при 1250 см'1. Однако в настоящее время имеется еще не-
достаточно данных, чтобы можно было безошибочно иден-
тифицировать каждую из этих полос.
Шеппард и Симпсон [20] опубликовали обзор полос по-
глощения, найденных у простых алкинов, в том числе на-
блюдавшихся у этих веществ частот колебаний С = С
и sC — Н. Подробное изучение производных ацетилена
Гл. 4. Алкины и аллены
87
с сопряженными связями было проведено Алланом, Микин-
сом и Уайтингом [25].
Алленовые вещества мало распространены и сравни-
тельно плохо изучены. Они ведут себя так, как если бы их
молекулы включали не две двойные связи, а связи, близкие
к тройной и простой; характеристические полосы появ-
ляются вблизи 1950 и 1060 сл"1, что соответствует валент-
ным колебаниям структуры С==С ~ С. Интересно отме-
тить, что поглощение в области 2000 сл"1 наблюдается
и у других аналогичных структур [1], например
у HN = С = О (2274 сл"1), О = С = О (2349 сл'1),
HN -N-N (2140 сл"1), О = N=N (2223 сл"1), HN =C=S
(1963 сл"1) и О = С = S (2050 сл"1).
Рассматриваемые корреляции приведены в табл. 3.
Таблица 3
Частота, см-1
Ацетиленовые производные
Валентные колебания =СН .... Валентные колебания С С, 3300 (с.)
монозамещенные производные . . Валентные колебания С=С, 2140—2100 (с.)
дизамещенные производные . . . Другие обсуждаемые полосы погло- 2260—2190 (различные ин тенсивности)
щения Алленовые производные 1250, 650
Валентные колебания С = С=С . . . 1950 и 1060 (ср )
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ s СН
О работах Фокса и Мартина [2] и других авторов, по-
священных изучению валентных колебаний С — Н, уже
говорилось при рассмотрении алканов и алкенов. Основы-
ваясь на результатах этих работ, можно было ожидать.
88
Часть I
что где-то около 3000 см'1 должна появляться полоса по-
глощения, которая соответствует валентным колебаниям
водородного атома, связанного с атомом углерода при
тройной связи у монозамещенного ацетилена. Кроме того,
следовало ожидать, что эта полоса довольно интенсивна
и мало зависит от строения остальной части молекулы, тем
более что в данном случае невозможно присоединение
каких-либо других заместителей к тому же углеродному
атому, у которого находится атом водорода. Эти предполо-
жения были подтверждены на практике; Рендалл и др.
[31 приводят математически обработанные данные для ряда
молекул, у которых поглощение, соответствующее валент-
ным колебаниям = СН, обнаруживается в интервале
3390—3290 см1, а Шеппард [4] приводит значения 3300 см'1
для бутина-1 и 3305 см 1 для винилацетилена (спектры ком-
бинационного рассеяния). Уотиз и сотрудники [5, 6] изу-
чили 12 монозамещенных ацетилена и установили, что эта
полоса появляется у них приблизительно при 3270 см'1.
Однако они отметили, что из-за низкой дисперсии камен-
ной соли в этой области полученные ими значения частот
на 20—40 см'1 ниже тех, которые были получены для не-
которых из этих соединений по спектрам комбинационного
рассеяния; поэтому для приведения найденных ими значе-
ний в соответствие с другими данными должна быть вве-
дена такая поправка. Интересно отметить, что полоса ва-
лентных колебаний СН у HCN [7] тоже появляется при
3311 см'1; это свидетельствует о том, что на указанную
полосу поглощения не оказывает заметного влияния при-
рода другого атома, соединенного с углеродом тройной
связью.
Таким образом, путем изучения указанной области
частот можно легко отличить монозамещенные ацетилена
от соединений с двойной связью и предельных соеди-
нений.
Единственными другими группами, которые могут, по-
видимому, поглощать в этой области, являются группы
ОН и NH, образующие водородную связь. Эти группы лег-
ко можно отличить по их широким полосам поглощения,
а подтверждение наличия ацетиленовой структуры может
быть в любом случае получено при рассмотрении области
валентных колебаний С = С.
Гл. 4. Алкины и аллены
89
3. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С=С
а. Положение полосы. Путем детального исследо-
вания простых молекул (3, 4, 8, 20] было установле-
но, что валентные колебания С = С обусловливают по-
явление поглощения вблизи 2100 см'1. Положение и интен-
сивность этой полосы поглощения на примере двадцати
восьми различным образом замещенных ацетиленов изу-
чали Уотиз и сотрудники [5, 6]. Их данные соответствуют
полученным ранее из спектров комбинационного рассея-
ния [8] и показывают, что положение полосы зависит от
числа заместителей. У соединений ряда НС s CR полоса
всегда появляется в интервале 2140—2100 см'1, а в случае
ряда RC = CR1 она находится в интервале 2260—2190 см'1.
Данные интервалы относятся к несопряженным системам,
у которых R является предельным или любым алифатиче-
ским радикалом или галогеном. Эти результаты были под-
тверждены данными [6], полученными для другой серии
из 16 производных ацетилена, хотя в некоторых случаях
в зависимости от заместителя у а-углеродного атома наб-
людались небольшие отклонения частот от указанных
интервалов. Например, было найдено, что соединения
С4Н9СНХ — С ~~ СН, где X означает ОН и Вг, обнаружи-
вают поглощение соответственно при 2080 и 2090 см'1.
Колебания же С = С у CF3C s СН имеют нормальную
частоту [26]; не влияет на частоту замещение хлором в а-
положении у дизамещенных ацетилена. В последнем случае
наличие замещения хлором можно легко установить, так
как группа —С ССН2С1 имеет характеристическую ча-
стоту вблизи 1250 см'1, аналогичную частоте соответствую-
щих замещенных олефинов [25]. Следовательно, присоеди-
нение полярной группы не всегда приводит к смещению
частоты, и даже СН3С s С — С1 дает полосу в нормальном
положении [33], несмотря на то что хлор присоединен
непосредственно к атому при тройной связи. Аналогич-
ным образом замещение на фенильную группу в соединении
типа R — С ~ С — СН2— X приводит к понижению
частоты от 2260 (насыщенные заместители) до 2210 см'1
(ароматические заместители), причем эта величина не вы-
ходит за пределы интервала, указанного для дизамещенных
производных. С указанной корреляцией согласуются
90
Часть
данные Шеппарда и Симпсона [20] для 17 различных произ-
водных ацетилена, а также данные, полученные недавно
Бломквистом [21] для колебаний CsC у циклодецина
и циклононина.
б. Влияние сопряжения. В последнее время был прояв-
лен значительный интерес к химии сопряженных произ-
водных ацетилена, и уже получены некоторые данные по
инфракрасным спектрам этих соединений [25, 27—31, 35].
Сопряжение с ацетиленовыми группами обычно приводит,
подобно сопряжению с олефиновыми группами, к некото-
рому увеличению интенсивности поглощения и к неболь-
шому его смещению в сторону более низких частот. Однако
эти изменения значительно меньше тех, которые происхо-
дят у олефинов. Сопряжение с карбонильными группами во
многих случаях не вызывает заметного изменения частоты
[25, 37—39], но для некоторых соединений, содержащих
карбонил в a-положении, наблюдается смещение частоты
до 2170 см"1.
Аналогичным образом у большого ряда сопряженных
кислот и эфиров, изученных Алланом и др. [25], главная
частота поглощения С<С лежит в области 2260—2235 см"1,
т. е. у верхнего предела обычного интервала частот. Уотиз
и Миллер [5] нашли, что полоса валентных колебаний
С = С у С6НИС ~ CCN лежит при 2120 см"1, т. е. ниже
обычного интервала частот для дизамещенных ацетилена,
а диацетилен [9] и винилацетилен [4] поглощают при 2024
и 2099 см"1 соответственно. Такого смещения полос не на-
блюдается у а,Р-а'Р'- сопряженных алкинов, у которых
отдельные эффекты сопряжения взаимно компенсируются.
Например, дицианоацетилен [22] поглощает при 2267 см"1.
Это явление наблюдалось также у сопряженных кислот
и эфиров R(—Cs С—)n COOR, где п > 1. В некоторых
случаях наблюдаются три полосы, хотя присутствуют
только две тройные связи. Например, нонадиин-1,4 и три-
декадиин-5,8 дают три полосы в области 2000 см"1 [34].
Двойная полоса колебаний CsC найдена у фторопроиз-
водных, например у трифторпропина [10] и перфтордиме-
тилацетилена [32].
Происхождение этих дополнительных полос неизвест-
но. Некоторые авторы объясняют их механическими взаи-
Гл. 4. Алкины и аллены
91
модействиями, другие считают их обертонами или состав-
ными полосами. Уотиз и Миллер [5] наблюдали интересную
особенность, касающуюся рассматриваемой корреляции,
которая состоит в том, что монозамещенные соединения
дают только одну полосу валентных колебаний С = С,
а дизамещенные — две полосы или больше. Эти дополни-
тельные полосы наблюдались только в тех случаях, когда
основная полоса была достаточно интенсивна, но их появ-
ление согласуется с полученными ранее данными из спект-
ров комбинационного рассеяния [8]. Общее правило для
растворов таково, что полоса с более высокой частотой зна-
чительно интенсивнее других, однако наблюдались и об-
ратные соотношения, когда при определении относитель-
ных интенсивностей образцы применялись в виде табле-
ток или взвесей.
Расщепление полос в этой области наблюдается у три-
фторпропина [101 и перфторбутина-2, которые, кроме глав-
ной полосы при 2128 см'1, дают еще полосу при 1960 слГ1.
в. Интенсивность. Интенсивность основной полосы ва-
лентных колебаний С zs С крайне непостоянна и непосред-
ственно зависит от положения тройной связи в молекуле.
Уотиз и Миллер [5] нашли, что у соединений с тройной
связью, расположенной в конце цепи, полоса является
интенсивной, а с удалением тройной связи от конца цепи
интенсивность этой полосы постепенно уменьшается. Это
происходит в связи с появлением в молекуле псевдоцентра
симметрии; валентные колебания тройной связи становятся
симметричными относительно него и, следовательно, не-
активными в инфракрасном спектре. Точно такое же яв-
ление обнаруживается и в случае связи С = С, о чем доста-
точно подробно говорилось в гл. 3. Поэтому для такого
соединения, как С3Н7—С = С — (СН2)2С1, основная по-
лоса С s С не обнаруживается, и, следовательно, отсут-
ствие полосы поглощения в интервале, соответствующем
тройной связи, нельзя считать доказательством отсутствия
такой связи. Напротив, в спектрах комбинационного рас-
сеяния эта полоса, конечно, всегда бывает сильной.
Сопряжение обычно приводит к некоторому увеличению
интенсивности, однако для того, чтобы у дизамещенных
веществ интенсивность полосы была средней, необходимо
92
Часть 1
сопряжение по крайней мере четырех групп С = С [27].
Например, полоса поглощения для СН3(С = С)3СН3 при
2222 сл"1 имеет слабую интенсивность, тогда как полоса
при 2237 см"1 для СН3(С s С)4СН3 имеет среднюю интенсив-
ность. При сопряжении с карбонильными группами также
происходит увеличение интенсивности; Аллан и др. [25]
приводят для ряда таких соединений значения коэффициен-
тов погашения.
4. ДЕФОРМАЦИОННЫЕ КОЛЕБАНИЯ =СН
Полосы поглощения этих колебаний систематически
не изучались, если не считать нескольких веществ, для
которых была проведена математическая обработка спек-
тральных данных. По аналогии с соединениями с двойной
связью деформационные колебания = С — Н должны давать
полосы, положение которых довольно постоянно, однако
интервал 700—600 см~\ в который они попадают, лежит да-
леко за пределами прозрачности каменной соли, поэтому
эти полосы были изучены лишь для небольшого числа ве-
ществ. Тем не менее известно, что для ряда простых моно-
замещенных алкинов полосы, отвечающие колебаниям
= СН, появляются в интервале642—615 сл"1 [20]. В ряде
случаев обнаруживалась сильная составная полоса в ин-
тервале 1300—1200 сл"1, которая была отнесена к этим
колебаниям [4, 11, 12, 20]. Однако в данной области погло-
щают очень многие вещества различных типов, и поэтому
обнаружение такой полосы не может иметь большого зна-
чения для структурной диагностики до тех пор, пока в ре-
зультате последующих исследований указанный интервал
не будет подразделен на более узкие участки, отвечающие
различным типам структур.
5. АЛЛЕНОВЫЕ СТРУКТУРЫ
Алленовая структура С = С = С в органической хи-
мии встречается редко, поэтому исследованы спектры лишь
немногих веществ, содержащих такую группу. Однако
известно, что обычной полосы валентных колебаний С = С
вблизи 1600 сл"1 у таких веществ не обнаруживается,
но что сильная взаимосвязь этих колебаний приводит
к появлению двух полос, близких к полосам колебаний
Гл. 4. Алкины и аллены
93
тройной и простой связей. Простейшим соединением рас-
сматриваемого типа является аллен; он был исследован
Линнеттом и Эйвери [13], а также Томпсоном и Гаррисом
[14], которые отнесли к указанным колебаниям две полосы
вблизи 1965 и 1070 см'1*. С другой стороны, полоса валент-
ных колебаний — С — Н соответствует больше полосам
структур с двойной, а не с тройной связью. Расмуссен и Брат-
тен [15] исследовали бутадиен-1,2 и нашли, что он также
дает полосы поглощения при 1970 и 1064 см'1. Уотиз [16]
сообщает, что гептадиен-1,2 поглощает при 1950 см'1;
эту полосу он использовал для определения гептадиена-1,2
в смеси с гептином-1 и гептином-2.
Как уже отмечалось, такое поведение алленов сходно
с поведением многих различных типов соединений, содер-
жащих структурный элемент = С = ; например, кетены дают
полосы при 2160 и ИЗО см'1, а дифенилкетен [17, 18]
имеет полосу при 2130 см'1. Поэтому, несмотря на то что
структуры алленового типа можно идентифицировать
по инфракрасным спектрам довольно надежно, степень пе-
рекрывания полос такова, что только по одним этим поло-
сам нельзя различить, например, аллены и изоцианаты;
пользуясь инфракрасным спектром, можно только иденти-
фицировать скелет X = С = X по появлению полосы по-
глощения средней интенсивности около 1950 см'1.
Обзор алленовых структур, спектры которых были под-
вергнуты математическому анализу, был сделан Шеппардом
и Симпсоном [20]. Уотиз и Келмер [ 19] исследовали один-
надцать различных алленов, семь из которых содержали
группы COOH, CONH2 или COOR, непосредственно свя-
занные с концевой алленовой связью. В этих семи случаях
наблюдался дублет при 1950 и 1930 см'1, тогда как в четы-
рех других, когда такие заместители отсутствовали или
алленовая группа находилась не в конце цепи, была об-
наружена только одна полоса поглощения в данной обла-
сти. На этом основании можно в какой-то степени разли-
чать между собой алленовые соединения. Сопряженные
аллены встречаются очень редко; у немногих исследованных
* Очевидно, что первая из них просто относится к антисимме-
тричным, а вторая — к симметричным колебаниям алленовой струк-
туры и сравнение с полосами тройной, двойной и ординарной связи
неуместно. — Прим. ред.
94
Часть 1
соединений [23, 24], в частности у магнамицина, харак-
теристическая полоса оказалась смещенной до 1900 см'1.
Как и у алкинов, у этих соединений смещение меньше,
если имеется сопряжение с обеих сторон алленовой группы;
полоса появляется тогда вблизи 1930 см'1.
О валентных колебаниях = С — Н этих соединений
имеется мало сведений, и поэтому область 3000 см'1 не
может быть использована для структурного анализа. Рас-
муссен и Браттен [15], например, применявшие призму
из каменной соли, не смогли идентифицировать какую-
либо полосу = С— Ну бутадиена-1,2, которая была бы
отлична от главных полос СН в интервале 3030—2940 см"1.
ЛИТЕРАТУРА
1. Herzberg, Reid, Discuss. Faraday Soc., 9, 92 (1950).
2. F о x, Martin, Proc. Roy. Soc., A175, 208 (1940).
3. Randall, Fowler, Fuson, Da ngl, Infra-red
Determination of Organic Structures, Van Nostrand, New
York, 1949.
4. S h e p p a r d, J. Chem. Phys., 17, 74 (1949).
5, W о t i z, M i i i e r, J. Amer. Chem. Soc., 71, 3441 (1949).
6. W о t i z, Miller, Palchak, J. Amer. Chem. Soc., 72,
5055 (1950).
7. Герцберг, Спектры и строение двухатомных молекул,
Издатиплит, М., 1949, стр. 301.
8. Н i b b е n, The Raman Effect and its Chemical Application,
Reinhold New York., 1939, p. 200.
9. Герцберг, Колебательные и вращательные спектры много-
атомных молекул, Издатиплит, М., 1949, стр. 348.
10. Н е n n е, N a g е г, J. Amer. Chem. Soc., 73, 1042 (1951).
11. W u, Vibrational Spectra and Structure of Polyatomic Mole-
cules, Edwards, Ann. Arbor (Michigan, USA), 1946,
p. 176.
12. C r a w f о r d, J. Chem. Phys., 8, 526 (1940).
13. L i n n e t t, Avery, J. Chem. Phys., 6, 686 (1938); Герц-
берг, Колебательные и вращательные спектры многоатом-
ных молекул, Издатинлит, М., 1949, стр. 363.
14. Thompson, Harris, Trans. Faraday Soc., 40, 295 (1944).
15. R a s m u s s e n, В r a t t a i n, J. Chem. Phys., 15, 120—
135 (1947).
16. W о t i z, J. Amer. Chem. Soc., 73, 693 (1951).
17. W h i f f e n, Thompson, J. Chem. Soc., 1946, 1005.
18. Drayton, Thompson, J. Chem. Soc., 1948, 1416.
19. W о t i z, С e 1 m e r, j. Amer. Chem. Soc., 74, 1860 (1952).
20 Sheppard, Simpson, Quarterly Reviews, 6, 1 (1952).
Гл. 4. Алкины и аллены
95
21. Blomquist, Burge, Huang Liu, Bohrer,
S u с г у, Kleis, J. Amer. Chem. Soc., 73, 5510 (1951).
22. M i 1 1 e r, Hannan, J. Chem. Phys., 21 110 (1953).
23. Oroshnik, Mebane, Karma s, J. Amer. Chem. Soc.,
75, 1050 (1953).
о '
e
r, S
M
1 о
a к
mon
24. C e 1 m e
25. Allan,
1874.
H a s z e
Cook,
Bohl
Bohl
30. S c h u
e о r g i e f f,
76, 5494 (1954).
enne, Finnegan,
avidson, Bernst
ensler, Mahadeven,
Soc., 78, 163 (1956).
Sorensen J. S., So r e n s
8, 1741, 1763 (1954).
ohlmann, Mannhard
h e u s, Surber, С о 1 о m b i,
s, J. Amer. Chem. Soc., 75, 1372 (1953).
Whit' ' ~ ~ -----
i n g, J.
Chem.
Soc.,
1955,
26.
27.
28.
29.
31.
32.
33.
34.
35.
36.
37.
n
G
H
D
G
В
T
1 d i
Jone
m a n n,
m a n n,
bach,
Lee
W h
e,
s,
Chem.
V i e h e,
T r a u t s
C a v e,
J. Chem.
J. Chem.
1755 (1955).
Ber., 88, 1017,
2150.
2883.
1952,
1952,
d h a m,
i t i n g,
Ber.. 88,
Chem.
c h о 1 d, Annalen, 594, 67 (1955).
В 1 a к i e, J. Amer. Chem. Soc.,
Soc.,
Soc.,
1245 (1955).
J. Amer. Chem. Soc., 71, 298 (1949).
e i n,
Can. J. Chem., 33, 1226 (1955).
Casella, J. Amer. Chem.
e n N. A., Acta Chem. Scand.,
Acta, 38, 239 (1955).
38. Ga mbon i, T h e u s.
t, Chem. Ber., 88, 133u (1955).
S c h i n z, Helv. Chim.
S c h i n z, Helv. Chim. Acta, 38,
255 (1955).
39. G г о b, Fischer, Helv. Chim. Acta. 38, 1794 (1955).
Глава 5
АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
I. ВВЕДЕНИЕ
Соединения ароматического ряда дают большое число
очень резких характеристических полос, поэтому их мож-
но идентифицировать методом инфракрасной спектроскопии
вполне надежно; более того, изменения в некоторых обла-
стях спектра, являющиеся результатом замещения, мало
зависят от природы заместителей, поэтому обычно бывает
возможным определить также степень и тип замещения.
У таких сложных молекул, как бензол, возможно боль-
шое число различных форм нормальных колебаний. Ре-
зультаты изучения инфракрасных спектров свидетельствуют
о плоском строении бензольного кольца [1]; нормальные
колебания его изучались целым рядом исследователей,
что позволило идентифицировать многие из них [2—8].
Таким образом, происхождение многих полос, используе-
мых в структурно-аналитических работах, известно, и часто
можно оценить изменения, являющиеся, по-видимому, след-
ствием структурных модификаций. Изучение корреляций,
связывающих изменения спектров с различными типами
замещения, было начато Леконтом [9—11], опубликовав-
шим целый ряд работ в период между 1937 и 1946 гг.,
а затем продолжено другими исследователями.
Структуры ароматического типа лучше всего обнаружи-
ваются по наличию полос валентных колебаний = С—Н
при 3030 см'1 и колебаний С = С в области 1600—1500 см'1.
На полосы поглощения, находящиеся в этих областях,
очень мало влияет характер замещения, хотя изучение
области колебаний С = С также может в некоторых слу-
чаях пролить свет на природу замещения и наличие сопря-
жения с двойными связями кольца.
Если наличие ароматического кольца установлено, то
присутствие и относительное расположение заместителей
Гл. 5. Ароматические соединения
97
можно установить по поглощению в интервалах 2000—1660,
1250—1000 и 1000—650 см"1. Первый из этих интервалов
является наиболее важным, и, исследуя его, можно полу-
чить вполне определенные данные о типе замещения. В об-
ласти низких частот и в меньшей степени в области 1250—
1000 см"1 следует затем искать подтверждение этих дан-
ных. В некоторых случаях наличие групп, поглощающих
или дающих обертоны в интервале 2000—1660 см1, затруд-
няет интерпретацию; кроме того, на все три области ока-
зывают глубокое влияние сильные электроноакцепторные
или электронодонорные группы, например нитрогруппа.
В этом случае интерпретация спектра становится менее
точной.
Обсуждаемые корреляции приведены в табл. 4.
Таблица 4
Частота, с.и-1
Валентные колебания =С—Н
Область 2000—1660
Плоскостные колебания скелета С = С
3030 (резкое поглощение)
Спектр поглощения специ-
фичен для каждого типа
замещения (см. рис. 6)
Вблизи 1600 (пер.), 1500
(пер.), 1580 (ср., сопря-
женные кольца) и 1450
(ср.)
Внеплоскостные деформационные
колебания СН
Пять соседних незамещенных ато-
мов водорода
Четыре то же
Три » »
Два » »
Один незамещенный атом водорода
Область 1225—950 сл!-1
1,2-, 1,4- и 1,2,4-замещение
770—730 (оч.с.) и 710—690(с.)
770—735 (оч. с.)
810—750 (оч. с.)
860—800 (оч. с.)
900—860 (ср.)
1225—1175, 1125—1090 и две
полосы при 1070—1000 (все
полосы сл.)
7 Л. Беллами
98
Часть I
Продолжение
Частота, см 1
Моно-, 1,3-, 1,2,3- и 1,3,5-замеще- 1175—1125, 1110—1070 (кро-
ние ме 1,3,5-замещения) и одна полоса при 1070—1000 (все полосы сл.)
1,2-, 1,2,3- и 1,2,4-замещение 1000—960 (сл.)
Трополоны 1630—1600, 1570—1535 (пер.)
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ =СН
Результаты исследований валентных колебаний СН
предельных и непредельных соединений уже были рас-
смотрены достаточно подробно (гл. 3). Выводы относи-
тельно стабильности положения этих полос относятся
и к ароматическим соединениям. Фокс и Мартин [12—14],
используя высокую дисперсию дифракционной решетки,
исследовали область 3000 см 1 в спектрах многих аромати-
ческих веществ, включая полициклические соединения,
такие, как нафталин и хинолин. Они нашли, что валент-
ные колебания СН ароматических соединений дают полосы
вблизи 3038 см"1, число которых обычно равно трем. Мно-
гие монозамещенные ароматических соединений, растворен-
ные в четыреххлористом углероде, дают характерный трип-
лет вблизи 3058 см"1, однако иногда количество линий бы-
вает больше трех, и, в частности, очень сложную картину
могут давать полициклические системы. Однако почти во
всех случаях одна из полос в этой области значительно
интенсивнее других. Эту область подробно изучили в усло-
виях высокого разрешения Жозьен и Леба [61] на примере
монозамещенных ароматических соединений. Они показа-
ли, что в этих случаях сложные системы полос просто объ-
ясняются наличием одной основной полосы и ряда более
слабых полос, представляющих составные частоты колеба-
ний, лежащих в области 1600—1400 см~г. Основная часто-
та С — Н лежит в интервале от 3079 (нитробензол) до
3030 см'1 (толуол).
Гл. 5. Ароматические соединения
99
Данные исследователи рассмотрели также возможную
связь указанных частот с электроноакцепторной или элек-
тронодонорной способностью заместителей [62, 68], а так-
же с такими свойствами, как дипольный момент, электрон-
ная плотность, рассчитанная по молекулярным схемам,
и реакционная способность. Например, интересно отметить,
что существует приблизительно линейная зависимость
между частотой валентных колебаний С — Ни относитель-
ным содержанием метазамещенного вещества, полученного
нитрованием, но замещение галогенами представляет при
этом исключение.
Валентные колебания СН в полициклических системах
были изучены Фоксом и Мартином [14] и Фьюзоном и Жо-
зьен [63], которые особое внимание обратили на замещен-
ные бензантрацена. Была рассмотрена возможная зави-
симость между частотами колебаний СН и канцерогенной
активностью этого соединения.
Итак, на практике идентификация поглощения СН
у ароматических соединений не представляет затрудне-
ний. Обычно оно обнаруживается при работе с призмой из
каменной соли в виде резкой и относительно слабой полосы
вблизи 3030 см'1 рядом с главными более интенсивными
полосами поглощения СН2 и СН3 несколько ниже 3000 см'1.
Интенсивность этого поглощения меньше интенсивности
колебаний насыщенных групп СН2, однако она растет про-
порционально количеству ароматических групп.
Изменения в агрегатном состоянии вызывают смеще-
ния порядка 10—15 см'1, но если только не используется
призма из фтористого лития, эти смещения в рассматривае-
мой области длин волн мало заметны.
Полосы валентных колебаний СН ароматических соеди-
нений успешно использовались Сейером и Коггешеллом
[15] в количественном анализе для определений содержа-
ния бензилового спирта в н-октаноле и метилэтилкетоне,
бензола в н-гексане, этилбензола в смеси с н-октаном, окте-
ном-2 и карбитолом* и составных частей других смесей.
Поглощение в области выше 3000 см 1 может быть так-
же вызвано валентными колебаниями СН при двойных свя-
зях различных типов, а равно и колебаниями групп СНХ,
где X — галоген. Алифатические двойные связи иденти-
* Этиловый эфир диэтиленгликоля. — Прим. ред.
7*
100
Часть I
фицируют обычно по положению полос, обнаруживаемых
при применении призмы из фтористого лития, а также по
характеристическим полосам, которые они дают в других
областях спектра. Поглощение СН галогензамещенных
групп не так легко идентифицировать, однако ароматиче-
ское строение обычно подтверждается данными, получае-
мыми при исследовании области 1600—1500 см Помехи,
обусловленные водородными связями групп ОН и NH,
обычно исключаются применением разбавленных раство-
ров, но в ряде случаев, когда имеются внутримолекуляр-
ные связи, область ароматических валентных колебаний
= СН может быть замаскирована.
Рассматриваемые валентные колебания имеют оберто-
ны в области от 8000 до 5000 см \ которые также являются
характеристическими для ароматических соединений [66],
и Роуз [16]использовал эту область для анализа аромати-
ческих компонентов в углеводородах.
3. ПОГЛОЩЕНИЕ В ОБЛАСТИ 2000—1650 иг1
Изучение спектров поглощения замещенных аромати-
ческих соединений в интервале 2000—1650 см'1 привело
к разработке одного из лучших и наиболее селективных
методов идентификации типа замещения в бензольном
кольце. Этот метод был предложен Юнгом, Дюваллом
и Райтом [17], которые показали, что каждый из различных
типов замещения дает весьма характерную картину спект-
ра поглощения в данной области, не зависящую в определен-
ных пределах от природы заместителей.
Ароматические соединения по сравнению с большин-
ством других соединений имеют в указанной области бо-
лее сложный спектр поглощения, который включает целую
серию обертонов и составных частот. Обертоны и состав-
ные частоты относительно слабы по сравнению с основны-
ми частотами, поэтому для изучения этой области приме-
няют кюветы с достаточно большой толщиной слоя (0,1 мм
для жидкостей и 1 мм для 10%-ных растворов твердых
веществ). В последнем случае желательно применение двух-
лучевой методики для исключения слабого поглощения,
которое дает в этой области растворитель — четыреххло-
ристый углерод.
Гл. 5. Ароматические соединения
101
Уиффену [64] недавно удалось показать, что все полосы
в области 2000—1650 см"1 могут быть с достаточной точно-
стью отнесены к результирующим составным полосам основ-
ных частот внеплоскостных колебаний СН, лежащих между
1000 и 700 см'1. Это обстоятельство делает понятными ха-
рактерные изменения в рассматриваемой области спектра,
происходящие при изменении положения заместителей,
и объясняет, почему общий вид спектра в этом случае более
важен, чем абсолютные значения частот. Становится также
понятной меньшая сложность спектра у более симметрич-
ных структур, которые имеют меньшее количество основ-
ных частот внеплоскостных колебаний СН.
Юнг и др. [17] сообщают, что они использовали рассмат-
риваемую спектральную область в течение ряда лет для
характеристики замещения в бензольном кольце и полу-
чили удовлетворительные результаты при исследовании
большого числа разнообразных веществ. После опубли-
кования цитированной работы мы успешно идентифициро-
вали в нашей лаборатории широкий ряд ароматических
соединений, используя данную область спектра; при этом мы
не встречали каких-либо неожиданностей и затруднений
за немногими исключениями, когда при наличии некото-
рых заместителей получалась необычная картина. Этот
факт был уже отмечен упомянутыми авторами и обсуж-
дается ниже.
Существенное различие между методикой анализа
в этой спектральной области и обычной методикой инфра-
красной спектроскопии заключается в том, что в первом
случае интенсивность и число полос играют относитель-
но большую роль, чем точные длины волн. Поэтому
желательно, чтобы исследователи имели для рассматривае-
мой области комплект стандартных спектров (эталонов),
используемых для сравнения. Такие спектры приведены
в работеЮнга и др. [17], однако предпочтительнее получать
их на том же приборе, который используется для исследо-
ваний; в частности рекомендуется получить типичные
спектры толуола, ксилола и т. д.
Такие спектры являются настолько характерными, что
небольшие изменения интенсивности и положения полос
не вызывают серьезных затруднений при распознавании
типа замещения. Так, например, монозамещенные дают
102
Часть I
обычно серию из четырех полос, интенсивность которых
постепенно уменьшается в направлении больших длин
волн.
В случае хлорбензола и бромбензола интенсивность
первой из полос немного меньше, чем интенсивность второй;
у фенола же четвертая полоса сильнее второй и третьей
и все полосы несколько смещены в сторону больших длин
волн. Тем не менее эти спектры достаточно заметно отли-
чаются от спектров, получающихся в случае других типов
замещения, и настолько сходны со стандартным спектром,
что позволяют легко установить, что соединения отно-
сятся к монозамещенным ароматических веществ.
Дизамещенные, тризамещенные и тетразамещенные
имеют еще более характерные спектры. Юнг и др. [17]
отмечают, что иногда у этих веществ происходит более за-
метное расщепление полос на дублеты, чем это бывает
обычно, но серьезных затруднений в идентификации при
этом не возникает. Были получены также образцы спектров
для пентазамещенных и гексазамещенных, но их следует
применять с большой осторожностью, поскольку с увели-
чением числа заместителей спектр становится менее харак-
терным; кроме того, сами авторы не считают, что ими было
изучено достаточное число однотипных соединений, чтобы
считать эти данные правильными во всех случаях. Уиф-
фен [64] показал, что 1,2,3,4- и 1,2,3,5-тетразамещенные
ароматических соединений имеют только две частоты вне-
плоскостных колебаний СН и что можно получить не более
трех суммарных полос (а -'-а, а b и b + Ь), из которых
две попадают в интервал 2000—1650 см'1. В первом слу-
чае они могут быть легко различимы, но во втором случае
они расположены вблизи друг от друга и, перекрываясь,
появляются иногда в виде одной полосы. У 1,2,4,5-заме-
щенных и пентазамещенных ароматических соединений
возможна только одна основная частота СН. Она лежит
обычно вблизи 870 см'1, поэтому следует ожидать одну
сильную полосу вблизи 1740 см'1.
Использованию этих типичных спектров могут мешать
следующие причины: появление в этой же области спектра
какой-либо основной полосы или какого-либо обертона
других колебаний и наличие некоторых заместителей, зна-
чительно изменяющих исходные основные частоты.
Гл. 5. Ароматические соединения
103
Однако другие полосы основных тонов и обертонов
в этой области спектра распознать нетрудно, так как они
легко выделяются как дополнительные полосы, накладываю-
щиеся на основную картину спектра. Например, у стирола
появляется полоса при 1818 см~', которая является обер-
тоном колебаний двойной связи при 910 см1; она накла-
дывается на обычный спектр монозамещенного соедине-
ния, полосы которого лежат по обе стороны от нее.
Если присутствует карбонильная группа, то при требуе-
мой толщине слоя кюветы интенсивность основной полосы
группы СО обычно такова, что маскируется значительная
часть рассматриваемой области спектра и уменьшается
возможность идентификации.
Фтор, группа R —О— или нитрогруппа у монозаме-
щенных вызывают значительные изменения общей карти-
ны спектра. В первых двух случаях спектр очень близок
к нормальному, но имеет дополнительную полосу несколь-
ко выше 2000 см1, которая вносит некоторую путаницу.
Ее можно считать также суммарной полосой [64] и при
анализе не принимать во внимание. Спектр нитробензола
заметно отличается от спектров других монозамещенных,
так как у этого соединения основные частоты внеплоскост-
ных колебаний СН, как будет показано позже, сами зна-
чительно смещены. Существует общее правило, что если
спектр соединения отличается от нормального спектра дан-
ной конфигурации, то он не будет похожим и на спектры
других конфигураций, и поэтому только в очень редких слу-
чаях исследование этой области спектра не дает каких-
либо полезных сведений. С последующим замещением
в большинстве случаев спектры становятся нормальными;
например, нитротолуолы и нитроксилолы дают нормаль-
ные или близкие к нормальным спектры. Однако мы уста-
новили, что при большом числе нитрогрупп в некоторых
случаях могут получаться нетипичные спектры.
Существует мало данных о применимости таких типич-
ных спектров поглощения для целей анализа в случае
нафтеновых или полициклических углеводородов, но можно
предположить, что в результате суммарного эффекта двух
или нескольких колец, замещенных различным образом,
рассматриваемая область спектра окажется непригодной
Для идентификации. Спектры АНИ для различных заме-
104
Часть I
щенных нафталина нельзя классифицировать, основываясь
на поглощении в данной области, так как они, как и спек-
тры, полученные ранее, значительно отличаются от любых
стандартных спектров. Это было подтверждено Фьюзоном
и Жозьен [63] при изучении замещенных бензантраценов.
Хотя они не считали данную область спектра пригодной
для определения типа замещения, однако указали на ряд
закономерностей, которые могут быть полезными для клас-
сификации этих соединений.
4. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С=С
Фундаментальные исследования колебаний бензольного
кольца, упомянутые выше [2—7], показали, что характе-
ристические валентные колебания полуторных углерод-
углеродных связей скелета приводят к появлению группы
из четырех полос между 1650 и 1450 слЕ из них две полосы,
лежащие около 1600 и 1500 см'1, весьма характерны для
самого ароматического кольца. Вместе с полосой валент-
ных колебаний С — Н вблизи 3030 см'1 они позволяют лег-
ко распознавать ароматическую структуру. На положение
этих полос в некоторой степени влияет природа замещаю-
щих групп, но в гораздо большей степени их положение
зависит от распределения этих групп относительно кольца,
так что в некоторых случаях можно получить определен-
ные указания на тип замещения. Однако, как будет пока-
зано ниже, в результате частичного перекрывания интер-
валов частот, характерных для различных типов замеще-
ния, количество случаев, когда такая идентификация воз-
можна, ограничено.
До последнего времени в литературе не было опубли-
ковано данных о положении этих полос для какого-либо
достаточно большого ряда соединений, однако Барнс и др.
[18] составили таблицу частот двух главных полос для неко-
торых моно- и дизамещенных веществ, а в настоящее время
серия углеводородных спектров АНИ пополнена спектра-
ми многих ароматических веществ. Последние служат хоро-
шей иллюстрацией относительных смещений полос при
изменении массы и относительного расположения заме-
стителей в кольце; однако поскольку в серии АНИ содер-
жатся в основном спектры только для одного типа замеще-
Гл. 5. Ароматические соединения
105
ния, то они не дают представления о всех интервалах ча-
стот поглощения для каждого возможного типа замещения
при наличии в качестве заместителей различных функцио-
нальных групп. Рендалл и др. [19] указали приблизитель-
ные интервалы частот: 1613—1600 и 1504—-1493 сл"1 для
более чем 50 исследованных ими ароматических соедине-
ний, тогда как Колтуп [20] установил более точные корре-
ляции, используя данные о большом числе ароматических
соединений, исследованных в лабораториях фирмы «Ameri-
can Cyanamide Company». Кеннон и Сазерленд [42] также
обсуждали соответствующие корреляции для ароматических
соединений и опубликовали спектры более ста таких соеди-
нений различных типов.
Интерпретации частот в этой области весьма способ-
ствовали работы Рендла и Уиффена [65], суммировавших
большое количество опубликованного материала о различ-
ных замещенных ароматических соединений. Эти авторы
отнесли четыре полосы области 1650—1450 см1 к колеба-
ниям скелета С — С, что согласуется с ранее опубликован-
ными данными Леконта [66]. Одна из этих полос, вблизи
1580 сл'1, часто бывает слишком слабой, тогда как другая,
около 1450 сл"1, расположена возле полосы деформацион-
ных колебаний группы СН2 и потому часто маскируется.
Такое отнесение частот было полностью подтверждено
более подробными исследованиями монозамещенных арома-
тических соединений, проведенными Жозьен и Леба [61],
и, таким образом, эти корреляции можно считать точно уста-
новленными. Однако, как будет показано ниже, интенсив-
ность данных полос изменчива, так что в некоторых случаях
они появляются лишь как слабые перегибы на крыльях
Других полос, и это создает трудности в их распознавании.
а. Полоса в области 1600СЛГ'1. Колтуп [20] показал, что
эта полоса появляется у большинства ароматических соеди-
нений в интервале 1625—1575 сл"1. У паразамещенных
наблюдается небольшое смещение в сторону более высоких
частот (1650—1585 сл"1); в меньшей степени это относится
также и к несимметричным тризамещенным ароматических
соединений. Напротив, у веществ с тремя соседними заме-
стителями смещение происходит в сторону низких частот
(1610—1560 сл'1).
106 Часть!
Указанные интервалы полностью согласуются с нашими
данными, со спектрами АНИ и работами Уиффена [65, 67]
и Жозьен [61]. У подавляющего большинства монозаме-
щенных полоса лежит в пределах интервала 1600 + 5 см-1,
причем у таких соединений, как хлорбензол, бромбензол
и тиофенол, она лежит ближе к нижней границе этого ин-
тервала, а у нитробензола она соответствует значению, пре-
вышающему среднее. В связи с этим можно предположить,
что положение полосы определяется в какой-то степени элек-
троотрицательностью замещающих групп, однако это
только один из многих действующих факторов, так как
дальнейшее введение электроотрицательных групп не при-
водит к дальнейшему уменьшению частоты. Кроме того,
эти смещения значительно меньше, чем те, которые могут
быть получены путем изменения положения заместителей.
Таким образом, положение данной полосы дает мало све-
дений о природе заместителей или совсем их не дает. Только
при благоприятных обстоятельствах, когда полоса лежит
заметно выше или ниже 1600 см1, мы имеем надежные ука-
зания на тип замещения. Однако соответствующие смеще-
ния претерпевает также и полоса при 1500 см'1 (см. ниже),
так что о типе замещения можно судить на основании поло-
жения обеих полос, что иногда бывает полезным для под-
тверждения данных, получаемых при исследовании других
областей спектра. Кеннон и Сазерленд [42] показали, что
эта корреляция справедлива не только для более простых
веществ, но и для широкого ряда полициклических арома-
тических соединений.
б. Область частот 1600—1560 см-1. В литературе
имеются противоречивые данные о положении и происхож-
дении полосы в этой области частот. Барнс и др. [18] по-
казали, что эта вторая полоса в отдельных случаях распо-
лагается близко к первой, но они никак ее не интерпрети-
ровали. Согласно Колтупу [20], данная полоса имеет
среднюю интенсивность и в общем случае находится
в интервале, указанном выше, а Рендалл и др. [19] счи-
тают, что она находится в интервале 1587—1575 сл"1 и ука-
зывает на сопряжение двойной связи с ароматическим
кольцом. Расмуссен, Танниклифф и Браттен [21] предпо-
ложили, что поглощение при 1600 и 1585 сл"1 служит при-
Гл. 5. Ароматические соединения
107
знаком присутствия группы бензоила, а Кеннон и Сазер-
ленд [42] отметили раздвоение полосы при 1600 смГ1 у ди-
фенила, производных нафталина и фенантрена и других
соединений.
Такая путаница возникла, вероятно, по двум причинам:
во-первых, потому, что эта полоса лежит в области погло-
щения атмосферных паров воды, в которой обнаружение
слабых полос на однолучевом приборе является затрудни-
тельным, и, во-вторых, из-за различий в толщинах пленок,
исследуемых в разных лабораториях. При исследовании
образца в виде тончайшей пленки или в виде тонкого слоя
взвеси в нуйоле мы обнаружили, пользуясь двухлучевым
прибором, что при отсутствии сопряжения кольца эта вто-
рая полоса выглядит как слабый выступ, прилегающий
к главной полосе при 1600 сл/-1. В некоторых случаях она
так слаба, что ее практически не удается обнаружить;
однако, используя более толстые слои, интенсивность этой
полосы можно увеличить. Во всех случаях, когда исследо-
вались достаточно толстые пленки, полоса находилась
в положении, указанном Колтуном. В некоторых случаях
относительные интенсивности полос при 1600 и 1580 сл/-1
изменяются и, например, у орто-дихлорбензола и у неко-
торых ксиленолов более слабая полоса находится со сто-
роны более коротких волн. Однако это не противоречит ука-
занным закономерностям, так как интервалы, в которых
появляются эти полосы, перекрываются. Жозьен и Леба
[61] и Рендл и Уиффен [65] подтвердили присутствие сла-
бой дополнительной полосы основных частот С — С.
Если непосредственно к кольцу присоединена карбо-
нильная или какая-нибудь другая ненасыщенная группа,
то интенсивность второй полосы при 1580 сл/' значительно
увеличивается и она становится более заметной в спектре.
Поэтому и было сделано предположение, что такая полоса
может служить для обнаружения сопряжения с кольцом.
Во всех случаях, изученных нами, усиление интенсивности
этой полосы было достаточным для того, чтобы можно было
при некоторых экспериментальных навыках установить
наличие сопряжения. Таким образом, противоречия в ли-
тературе устраняются, если считать, что полоса при
1580 сл/ 1 соответствует нормальным колебаниям аромати-
ческого кольца, но она является всегда слабой и трудно
108
Часть I
обнаруживаемой при отсутствии сопряжения с внешней
группой; при наличии сопряжения она превращается
в полосу средней интенсивности, которую легко различить.
Однако при идентификации такого сопряжения следует
учитывать толщину использованной пленки и относитель-
ную интенсивность этой полосы по сравнению с полосой
при 1600 сл"1.
в. Полоса в интервале 1525—1475 см'1. Эта полоса
в зависимости от положения заместителей относительно
кольца ведет себя таким же образом, как и полоса
при 1600 сл'1. В согласии с данными Колтупа и со спект-
рами АНИ мы нашли, что полоса всегда появляется в ин-
тервале 1525—1475 сл"1 и обычно лежит ближе к 1500 см'1,
за исключением случаев небольшого смещения в сторону
более высоких частоту пара-замещенных и несимметричных
тризамещенных соединений и в сторону более низких ча-
стот у тризамещенных, заместители которых расположены
по-соседству. Эти данные согласуются с результатами работ
Уиффена [65] и Жозьен [61].
г. Полоса при 1450 см'1. Предположение о существова-
нии четвертой полосы колебаний скелета С — Св области
1450 см'1 было сделано Леконтом [66] и полностью под-
тверждено последующими исследователями [61, 65]. Для
ряда монозамещенных ароматических соединений, изучен-
ных Жозьен и Леба [61], четвертая полоса находилась
в интервале 1470—1439 сл'1 и имела умеренную или высо-
кую интенсивность. Однако в некоторых случаях, напри-
мер у анилина и нитробензола, ее интенсивность была срав-
нительно слабой. Эта полоса часто перекрывается сильной
полосой деформационных колебаний СН2, что уменьшает
возможности ее использования для целей идентификации,
однако она может быть легко применена для таких соеди-
нений, как галогенпроизводные бензола и близкие к ним
вещества, а также для соединений с конденсированными
циклами [63].
д. Полициклические соединения. Теоретически можно
ожидать, что главные полосы ароматического кольца при
1600 и 1500 сл"1 имеются также и у полициклических
Гл. 5. Ароматические соединения
109
веществ, хотя из-за фиксирования связей у таких веществ
и из-за того, что у них не все связи имеют одинаковую дли-
ну [22], можно предположить, что интервалы частот этих
полос более широки. Это подтверждается исследованиями
Колтупа 120], который указал на смещение полосы при
1600 сл'1 в сторону более коротких длин волн (1650—
1600 сл"1) и на расширение интервала частот, характерных
для второй полосы, до 1630—1575 сл'1 у а- и Р-замещенных
нафталина. Третья и четвертая полосы всегда наблюдаются
в интервале 1525—1450 сл1. Мы не располагаем доста-
точным количеством данных, которые позволили бы судить
о точных границах этих интервалов, но практически мы
никогда не испытывали затруднений при распознавании
ароматического характера как полициклических соедине-
ний, так и замещенных хинолина и других аналогичных
веществ. Эти полосы можно также без труда различить
в спектрах ряда многоядерных углеводородов, изученных
Орром и Томпсоном [23], производных стильбена, исследо-
ванных Томпсоном, Ваго, Корфилдом и Орром [24], заме-
щенных нафталина [60, 69], а также в спектрах многоядер-
ных углеводородов, полученных Кенноном и Сазерлендом
[42] и Фьюзоном и Жозьен [63]. Поглощение в интервале
1640—1600 сл"1 успешно использовалось также для рас-
познавания фенильной группы в алкалоидах [25] и в аро-
матических стероидах [26, 71].
е. Интенсивность полос поглощения ароматических
соединений в области 1600—1450 см'1. Интенсивность по-
лос поглощения в области 1600—1500 сл"1, обусловленных
ароматическими структурами, меняется в очень широких
пределах. У несопряженных структур полосы обычно бы-
вают слабыми и часто имеют вид выступов на других поло-
сах. Полоса при 1500 сл'1 немного более интенсивна,
чем полоса при 1600 сл'1. Сопряжение кольца с двойной
связью, например с группами СО, С = С, NO2 и т. д.,
в большинстве случаев вызывает очень заметное увеличение
интенсивности всех полос, включая полосу в интервале
1600—1560 сл"1, интенсивность которой уже обсуждалась
выше. В некоторых случаях увеличение интенсивности пер-
вых двух полос, по-видимому, сопровождается уменьше-
нием интенсивности третьей полосы. У меркаптобензтиазола,
110
Часть I
например, полоса при 1500 см'1 легко обнаруживается
в случае несопряженной тиокетоформы и является предель-
но слабой у производных сопряженной меркаптановой
формы. Это не является, однако, общим правилом, и у боль-
шинства сопряженных ароматических веществ полоса при
1500 см 1 распознается без затруднений, хотя ее интенсив-
ность бывает часто несколько пониженной. Интенсивность
полосы вблизи 1450 см 1 вообще довольно постоянна и из-
меняется заметно только в присутствии некоторых замести-
телей.
Интенсивность этих полос, по-видимому, зависит от
относительного положения заместителей, так как измене-
ния в их положении могут вызывать заметные изменения
спектра. В некоторых случаях полоса при 1600 см'1 ста-
новится такой сильной, что ее интенсивность превышает
интенсивность полосы карбонильной группы, с которой
сопряжено кольцо. Это происходит, например, в спект-
рах п-оксиацетофенона и о-оксибензальдегида. В некоторых
случаях, когда полоса поглощения карбонила под влия-
нием сильной водородной связи может сместиться в об-
ласть 1600 см \ иногда возникают затруднения в разре-
шении обеих полос и в определении степени смещения
полосы карбонила.
ж. Идентификация ароматических структур в присут-
ствии других групп. Три полосы в области 1600—1500 см \
характерные для ароматической структуры, можно в по-
давляющем большинстве случаев распознать без затруд-
нений, если при этом пользоваться пленками образца соот-
ветствующей толщины. Эти полосы наряду с данными из
области 3000 см'1 обычно достаточны для надежной иден-
тификации ароматической структуры. Однако гетероцикли-
ческие ароматические соединения, такие, как пиридин
и пиримидины, дают довольно сходную группу полос в об-
ласти 1600 см 1 [27]. Это относится также к циклоокта-
тетраену, спектр которого напоминает спектр стирола,
содержащего обычную связь С = С, сопряженную с коль-
цом [28].
Идентификация ароматических соединений может быть
также осложнена из-за присутствия групп СО — NH,
С = С, NO2 и NH2. Их полосы поглощения иногда пере-
Гл. 5. Ароматические соединения
111
крываютту или иную из важных областей. Полоса обычной
связи С = С около 1640 см'1 лежит почти рядом с полосой
фенила при 1600 сл'1, и когда связь С = С сопряжена
с кольцом, то ее полоса часто попадает в область погло-
щения ароматической группы. Однако вторая полоса С = С,
появляющаяся в результате резонансного расщепления
двух колебаний С = С неароматических соединений, имеет
частоту заметно более высокую, чем полоса ароматических
соединений при 1500 сл1. Трудности встречаются также
при идентификации полосы в области 1600 сл-1 у некото-
рых ароматических р-дикетонов и у других структур, у ко-
торых в эту же область смещена полоса поглощения карбо-
нила. Затруднено также распознавание некоторых хино-
нов и сходных с ним веществ, у которых имеются фиксиро-
ванные двойные связи, так как полоса, характерная для
ароматической структуры, появляется у них возле самых
границ обычного для нее интервала частот.
В общем при такой идентификации встречается мало за-
труднений, и рассмотренные корреляции могут приме-
няться к спектрам образцов, исследуемых как в растворе,
так и в виде пасты с парафиновым маслом, так как смеще-
ния полос в зависимости от агрегатного состояния вещества
очень малы [29] и находятся в пределах описанных выше
интервалов.
5. ВНЕПЛОСКОСТНЫЕ ДЕФОРМАЦИОННЫЕ
КОЛЕБАНИЯ СН
Внеплоскостные деформационные колебания незамещен-
ных водородных атомов кольца вызывают появление силь-
ных полос в области 1000—650 см'1. Частоты этих полос
определяются в основном положением, а не природой заме-
стителей, и с некоторыми ограничениями они служат пре-
восходным средством для распознавания типа замещения.
Вопрос о числе и положении таких полос у ароматических
соединений с различным замещением рассматривался Ренд-
лом и Уиффеном [65]. Однако хотя положение всех полос
отличается устойчивостью, интенсивность большинства из
них мала, поэтому внимание исследователей было обраще-
но на одну или две наиболее сильные полосы. Очень высо-
кая интенсивность этих полос делает их особенно удобными
112
Часть I
при количественных определениях; в литературе приво-
дятся многие примеры их применения для определения
относительных количеств орто-, мета- и лара-замещенных
изомеров и некоторых тризамещенных веществ. Например,
Уиффен и Томпсон [31] разработали метод количественного
анализа крезола, основанный на использовании этих полос,
а также основы количественного анализа смесей ксилено-
лов. Эти исследования были продолжены Фриделом, Пейр-
сом и МакТоверном [38], которые предложили метод ана-
лиза фенолов, крезолов, ксиленолов и этилфенолов. Ме-
тод анализа четырехкомпонентной смеси хлорбензола
с о-, м- и н-хлорбромбензолами описан Фергусоном и Леван-
том [39], метод анализа смесей трет-бутилфенолов —
Хейлсом [40], а смеси из пяти ароматических соединений
состава С1о — Перри [41].
Вопрос о зависимости появления рассматриваемых по-
лос и их положения в спектре от орто-, мета- и пара-
замещения был рассмотрен Барнсом и др. [18] и более
широко изучен в работах Уиффена, Торкингтона и Томп-
сона [30], а также Уиффена и Томпсона ]31], которые опре-
делили положение полос для ряда моно- и дизамещенных
веществ и для некоторых три- и тетразамещенных. Позд-
нее их данные были дополнены данными Ричардса и Томп-
сона [32] по замещенным фенола, Томпсона, Ваго, Корфил-
да и Орра [24] по замещенным стильбена, Орра и Томпсо-
на [23] по многоядерным углеводородам, Камада [52]
и Кеннона и Сазерленда [42], исследовавших большое чис-
ло ароматических веществ.
Эти данные были включены в различные таблицы [20,
33, 34], причем предложенные границы интервалов по-
степенно расширялись с увеличением числа и ~тт''’^ванных
соединений. Сведения о положении полос в зс^исимости
от различных типов замещения обобщил Колтуп [20], кото-
рый положил в основу классификации число водородных
атомов, связанных с углеродами кольца и расположенных
рядом; эти данные будут рассмотрены ниже. Наконец,
Коул и Томпсон [35] обосновали эмпирические корреля-
ции математической обработкой спектров и рассчитали
ожидаемые основные частоты внеплоскостных колебаний
СН для различных типов замещения, используя простое
силовое поле только с двумя силовыми постоянными.
Гл. 5. Ароматические соединения
113
Их результаты находятся в хорошем согласии с эксперимен-
тальными данными, которые они получили для значитель-
ного ряда галогенопроизводных бензола. Найденные кор-
реляции применимы даже для о-, м- и п-дидейтеробензолов,
хотя значения характеристических частот у этих соедине-
ний примерно на 20 см'1 выше обычных значений [53].
Таким образом, корреляции, выведенные для ди- и три-
замещенных, вполне обоснованы, так как они получены на
основании очень большого количества литературных дан-
ных. При применении рассмотренных корреляций к тетра-
и пентазамещенным нафталина [69] и полициклическим
соединениям наблюдаются некоторые отклонения. Так, для
циклопентенофенантренов [72] и для бензантраценов [63]
указанные корреляции полос с учетом числа соседних
водородных атомов в каждом кольце еще сохраняются,
однако у них в этих же областях появляются и дополни-
тельные полосы, что и можно было ожидать, принимая
во внимание сложную структуру данных соединений. В по-
добных случаях для анализа является более существенным
отсутствие какой-либо полосы, а не ее присутствие.
Заметные отклонения от обычных значений частот могут
проявляться, если в кольце в качестве заместителей имеется
несколько сильно полярных групп [54], например СО,
CF з или NO2; такие особые случаи будут рассмотрены ниже.
а. Пять соседних незамещенных атомов водорода. Мо-
нозамещенные ароматических соединений. Уиффен и Томп-
сон [31 ] первые предположили, что поглощение, соответ-
ствующее внеплоскостным деформационным колебаниям
СН для монозамещенных бензола, лежит в интервале
760—740 сл"к. который Колтуп [20] несколько расширил
до 770—730 см'1. Этот последний интервал согласуется
с результатами, полученными в нашей лаборатории, а так-
же со многими опубликованными данными [58, 61, 65].
Иногда в случаях сильного взаимодействия заместителя
с кольцом надежное отнесение этой частоты оказывается
невозможным. Например, бензойная кислота очень сильно
поглощает при 714 и слабо — при 806 см"1 [68], при этом
неясно, какая из этих двух полос поглощения связана
с колебаниями, дающими в обычных условиях указанное
характеристическое поглощение. Однако в большинстве
8 Л. Беллами
114
Ч а с т ь I
случаев полоса, отвечающая характеристическому погло-
щению СН, будучи самой сильной в рассматриваемой области
спектра, легко распознается, хотя ее положение внутри
указанного интервала заметно меняется даже при неболь-
ших изменениях в структуре молекулы. Например, при
переходе от фенилгидразина к хлористоводородному фе-
нилгидразину смещение полосы составляет 15 см'1. Верх-
няя часть интервала частот этой полосы перекрывается
интервалом, характерным для полос орто-дизамещенных
веществ, поэтому для отличия монозамещенных веществ
от opmo-дизамещенных нельзя пользоваться только одним
этим интервалом. Однако Уиффен [36, 65] показал, что мо-
нозамещенные вещества сильно поглощают также вблизи
690 см'1, где opmo-замещенные практически не поглощают;
таким образом, можно различить данные вещества. Эта по-
следняя корреляция подтверждена Колтупом [20], Рен-
даллом и др. [19], Жозьен и Леба [61] и Кенноном и Са-
зерлендом [42]. Указанная полоса у монозамещенных нахо-
дится почти всегда в пределах 700 + 10 см'1, а Рендалл
и другие считают ее весьма специфичной для фенильной
группы. Ее следует всегда изучать одновременно с полосой
750 см'1, так как при 700 см'1 поглощают также мета-
замещенные и некоторые тризамещенные, хотя для них
интервал частот несколько шире. Отсутствие полосы в ин-
тервале 700 ± 10 см 1 служит доказательством отсутствия
монозамещенного, но обратное положение не обязательно
верно, и подтверждение наличия монозамещенного следует
искать в области 750 см'1.
В некоторых очень немногих случаях полоса при 750 см'1
расщепляется на несколько более слабых полос в том же
интервале частот. Это явление наблюдается у zjzzc-форм
фенилпропенилкарбинола и стирилметилкарбинола [37],
хотя их mptzHc-формы имеют только одну нормальную по
положению и интенсивности полосу. Причины данного
явления неизвестны, но оно может быть связано со стери-
ческими затруднениями в ^ис-соединениях.
б. Четыре соседних незамещенных атома водорода.
О/?/ЯО-Дизамещенные ароматические соединения. Как было
упомянуто выше, opmo-дизамещенные поглощают приблизи-
тельно в той же области частот, в которой поглощают
Гл. 5. Ароматические соединения
115
и монозамещенные производные; соответствующая сильная
полоса лежит в интервале 770—735 см'1. Уменьшение чис-
ла соседних незамещенных атомов водорода от пяти до че-
тырех не оказывает значительного влияния на частоту.
Эта корреляция справедлива также и для таких цикличе-
ских соединений, как фталевый ангидрид, для нафталинов,
хинолинов и соединений с большим числом колец. Однако,
как показал Колтуп, для этих веществ следует ожидать
несколько более широкого интервала частот. В соответ-
ствии со сказанным Орр и Томпсон 123] наблюдали сильную
полосу вблизи 750 см1 у значительного числа канцероген-
ных углеводородов, содержащих 1,2-дизамещенные много-
ядерные структуры, хотя во многих случаях наблюдались
отклонения порядка + 10 см'1. Кеннон и Сазерленд [42]
отнесли к этим же колебаниям полосу при 750 см 1 у 9,10-
дигидроантрацена и полосу при 725 см~1 у самого антра-
цена. Большое количество замещенных нафталина было изу-
чено Ценцелем и Хаджи [69], Вернером и др. [60, 70].
Во всех случаях, когда в одном из колец не имеется заме-
стителей, появляется сильная полоса, соответствующая
opmo-замещению, и в то же время обычно бывает возмож-
ным проследить полосы, отвечающие колебаниям остав-
шихся незамещенными атомов водорода другого кольца,
которые характерны для тризамещенных или тетразаме-
щенных бензола. Однако если заместители присутствуют
в двух кольцах, то эти полосы идентифицировать труднее,
и в некоторых случаях использование рассматриваемых
корреляций может привести к ошибочным выводам. Полосы
в интервале 746—738 см'1 были обнаружены у соответствую-
щих замещенных бензантрацена [63], но, как было указа-
но выше, появление дополнительных полос понижает
возможность применения этой корреляции для полицикли-
ческих соединений.
в. Три соседних незамещенных атома водорода. Заме-
щение в 1,3- и 1,2,3-положениях. Ароматические кольца
с тремя соседними атомами водорода обнаруживают погло-
щение в интервале 810—750 см 1 одновременно с другим по-
глощением средней интенсивности в области 725—680 см'1.
Колтуп [20] приводит для веществ с тремя соседними заме-
стителями несколько более узкие интервалы частот обеих,
8*
116
Часть I
полос, чем для мета-дизамещенных, однако это может быть
связано с тем, что было изучено меньшее количество три-
замещенных соединений. Частичное перекрывание этих
двух интервалов частот с интервалами частот монозаме-
щенных может в некоторых случаях затруднять исследова-
ние, но поскольку первая полоса расположена обычно выше
770 см1, а положение второй является более переменным,
чем у монозамещенных, то полосы этих двух типов веществ
могут только случайно находиться в пределах одного и того
же узкого интервала частот.
Главную полосу в интервале 810—750 см'1 можно без
труда обнаружить также в спектрах замещенных нафтали-
на, содержащих три соседних незамещенных атома водо-
рода [69, 70]. Например, спектр 1,5-диметилнафталина
из серии АНИ имеет сильную полосу при 790 см'1, являю-
щуюся в этой области самой интенсивной. Монозамещенные
нафталина с заместителем в положении 1 тоже дают эту
полосу и, кроме того, полосу в интервале 770—735 см х,
обусловленную четырьмя незамещенными атомами водо-
рода второго кольца. Однако при последующем замещении
эта область спектра бывает у нафталинов часто слишком
сложна и тип замещения не удается идентифицировать.
г. Два соседних незамещенных атома водорода. Заме-
щение в 1,4-, 1,2,4- и 1,2,3,4-положениях. С последующим
уменьшением числа незамещенных соседних атомов водо-
рода в кольце частота внеплоскостных колебаний СН сме-
щается еще дальше в сторону более высоких частот и силь-
ная полоса появляется в интервале 860—800 см'1. Обзор
данных по этой полосе недавно сделали Рендл и Уиффен
[65] и Бомстейн [55]. В интервале 750—700 см'1 сильная
полоса не появляется, но иногда присутствует слабая по-
лоса. Тем не менее в тех случаях, когда главная полоса
попадает в область 810—800 см'1, отличить исследуемое
замещение от жета-замещения можно только по присут-
ствию или отсутствию второй полосы средней или сильной
интенсивности в интервале 750—700 см1. Аналогичное
поглощение показывают спектры монозамещенных нафта-
лина [70], у которых, кроме этой полосы, имеется погло-
щение, обусловленное другим кольцом. Отличить друг
от друга все три возможных типа замещения, при которых
Гл. 5. Ароматические соединения 117
поглощение происходит в интервале 860—800 см1, можно
лучше всего посредством изучения области 2000—1600 см'1,
хотя несимметричные тризамещенные можно также отли-
чить по полосе средней интенсивности, обусловленной неза-
мещенным атомом водорода, который расположен между
двумя заместителями (см. ниже). Число 1,2,3,4-тетразаме-
щенных, о которых сообщается в литературе или которые
доступны для исследования, мало [65], но в изученных слу-
чаях корреляция хорошо выполняется. Например, 1,2,3,4-
тетраметилбензол поглощает при 804 с.и1. Полосу в этой
области дают также неостероиды, у которых кольцо В
является ароматическим [71], и соответствующим образом
замещенные бензантрацена [68].
д. Атом водорода, расположенный между двумя заме-
стителями. Замещение в 1,3-, 1,2,4-, 1,3,5-, 1,2,3,5-,
1,2,4,5-положениях; пентазамещенные ароматических
веществ. Когда число незамещенных соседних атомов водо-
рода уменьшается до одного, то полоса снова смещается
в сторону более высоких частот; при этом полоса поглощения
СН появляется в области 900—860 с.и1. Уменьшение числа
поглощающих групп отражается и на интенсивности полос,
так что в отличие от очень сильных полос, наблюдавшихся
в случаях, описанных выше, эта полоса имеет среднюю ин-
тенсивность. У 1,3,5-тризамещенных одна сильная полоса
лежит в интервале 865—810 сл'1, а вторая полоса— между
675 и 730 сл’1. Например, 1,3,5-триметилбензол имеет
полосы поглощения при 881 (сл.), 835 (с.) и 687 (с.) см'1
(спектр 354 из серии АНИ). Последняя из этих полос по-
зволяет отличить замещенные такого типа от пара-заме,-
щенных и сходных с ними веществ, поглощающих вблизи
830 сл'1. Мак-Коли и др. [57] нашли, что у симметричных
триалкилбензолов эта полоса расположена между 874
и 835 сл'1, и рассмотрели вопрос о влиянии разветвления
цепи заместителей на частоту. Количество исследованных
тетра- и пентазамещенных невелико. Мы зарегистрировали
почти тридцать таких веществ, спектры которых согла-
суются с обсуждаемыми корреляциями, но пока не будет
исследовано большее число спектров, их следует применять
осмотрительно. В спектрах многоядерных углеводородов
с соответствующим замещением можно заметить полосу
118
Часть I
средней интенсивности в интервале 900—800 см'1, но из-за
сложной картины спектра в данной области идентификацию
по этой полосе нельзя считать надежной.
е. Влияние замещающих групп. Как уже было сказано,
положение частот СН определяется почти полностью поло-
жением заместителей и не зависит от их природы. Однако
это не совсем верно, так как электроноакцепторные или
электронодонорные свойства заместителей оказывают боль-
шое влияние на точное положение полос поглощения в пре-
делах указанных интервалов частот. В крайних случаях,
например при наличии нескольких нитрогрупп, это влия-
ние приводит к смещению поглощения вообще за пределы
ожидаемого интервала. Пытаясь оценить величину и на-
правление смещения полос, мы исследовали 26 из 29 воз-
можных моно-, ди- и тризамещенных бензола, содержа-
щих метильные группы и нитрогруппы или же только ни-
трогруппы. Результаты показывают, что замещение нитро-
группами приводит к сдвигу полос в сторону более высоких
частот и что величина смещения зависит от степени заме-
щения. При замещении несколькими нитрогруппами полосы
достигают некоторого положения, далее которого они при
последующем замещении этими же группами не смещаются.
Например, частота деформационных колебаний СН, когда
атом водорода находится между двумя заместителями у со-
единений, содержащих три нитрогруппы и больше, очень
близка к 920 см'1. Подобные специальные исследования по-
зволяют иногда распознавать у таких веществ вид замеще-
ния при условии, что известны какие-либо другие данные,
такие, как число нитрогрупп, так как в противном случае
бывает трудно отличить, например, пара-замещенные с од-
ной нитрогруппой от мета-замещенного с двумя нитро-
группами. Поэтому когда присутствует несколько нитро-
групп или когда сильная электроноакцепторная группа
расположена в пара-положении к сильной электронодонор-
ной группе, как, например, у пара-аминобензойной кислоты,
при интерпретации этой спектральной области нужна край-
няя осторожность. В таких случаях часто появляются слож-
ные полосы, интерпретация которых затруднена. Марго-
шее и Фассел [58] привели несколько подобных примеров.
Однако соединения с большим числом заместителей, вызы-
Гл. 5. Ароматические соединения
119
вающих затруднения при интерпретации, редки; поэтому
характер замещения, отвечающего рассматриваемой спек-
тральной области, как правило, достаточно ясен, и если
при этом воспользоваться данными, относящимися к об-
ласти 2000—1600 см \ то можно произвести надежную
идентификацию типа замещения. Как уже указывалось,
точность рассмотренных корреляций уменьшается с увели-
чением степени замещения, так что идентификация тетра-
и пентазамещенных гораздо менее надежна, чем, напри-
мер, идентификация монозамещенных.
Некоторые исследователи изучали возможную связь
этих частот с изменениями электронной плотности вокруг
кольца, происходящими при введении в него заместителей.
Например, Беллами [59] рассмотрел вопрос о зависимости
упомянутых выше полос поглощения от значений постоян-
ной Гаммета о для заместителей, которая определяет элек-
тронодонорные и электроноакцепторные свойства этих
заместителей. Если не считать галогензамещенных, кото-
рые ведут себя так, как если бы величина и была у них
равна нулю, то можно найти связь между величинами о и
частотами для трех, двух соседних или одного изолирован-
ного атомов водорода, связанных с углеродом кольца; эта
связь оказывается иногда полезной для предсказания частот
у неизвестных соединений. Аномальное поведение галогенов
можно объяснить, пользуясь современными данными, полу-
ченными методом ядерного магнитного резонанса, которые
показывают, что при замещении галогенами электронная
плотность углеродного кольца заметно не изменяется [73].
Аддитивность величин постоянной Гаммета о сказывает-
ся на приблизительной аддитивности величин смещения
частот. Например, было замечено, что отклонение частоты
от главного значения для naptz-дизамещенного соединения
представляет собой приблизительно сумму смещений для
отдельно взятых монозамещенных веществ [58]. Кросс
и др. [74] также рассматривали этот вопрос и выдвинули
объяснение причин смещения частот у нитросоединений
и других веществ с точки зрения теории молекулярных
орбит. Однако нет удовлетворительного объяснения того
факта, что во всех случаях при наличии как электроноак-
цепторных, так и электронодонорных заместителей частоты
б СН больше, чем они бывают у метилзамещенных.
120
Часть I
6. ХАРАКТЕРИСТИЧЕСКОЕ ПОГЛОЩЕНИЕ В ОБЛАСТИ
ЧАСТОТ 1225 — 950 см~1
В этой области частот для всех ароматических соедине-
ний возникает серия сравнительно слабых полос, положе-
ние которых меняется в зависимости от характера распре-
деления заместителей. Из-за малой интенсивности они
обычно менее пригодны для аналитических целей, чем
описанные выше полосы поглощения с более низкими часто-
тами. Однако при изучении этих полос можно найти под-
тверждение данных, полученных при исследовании других
частей спектра и касающихся как наличия заместителей,
так и типа замещения ароматических соединений. Рендл
и Уиффен [65, 67, 75] изучили эти полосы достаточно
подробно и показали, что они относятся главным образом
к плоскостным деформационным колебаниям С — Н, хотя
в некоторых случаях частоты в этой области чувствитель-
ны к массе заместителей.
Барнс и др. [18] заметили появление характеристиче-
ских полос в данной области спектров ароматических соеди-
нений и установили следующие корреляции: монозамещен-
ные 1075—1065, орто-замещенные 1125—1085, лета-заме-
щенные 1170—1140 и пара-замещенные 1120—1090 слг1.
В согласии с этими данными находится таблица Томпсона
[38], хотя в ней несколько расширены интервалы частот
и указана еще одна полоса пара-замещенных в интервале
1225—1175 см"1. Эти данные были значительно дополнены
Колтупом [20], который исследовал тризамещенные, а так-
же показал, что в случае определенных типов замещения
может появляться до шести характеристических полос. По-
скольку рассматриваемые полосы в значительной степени
перекрывают друг друга, ни одна отдельно взятая полоса
без одновременного рассмотрения других полос не дает воз-
можности распознать структуру. Наши данные находятся
в согласии с данными Колтупа, а также и Уиффена [65].
Мы будем классифицировать различные полосы поглоще-
ния в этой области следующим образом.
а. Замещение в 1,2-, 1,4-и 1,2,4-положениях. Соедине-
ния с таким замещением дают слабые полосы поглощения
в интервалах 1225—1175 и 1125—1090 см 1 наряду с двумя
слабыми дополнительными полосами в интервале 1070—
Гл. 5. Ароматические соединения
121
1000 щи'1. Эти полосы очень резки и, несмотря на их малую
интенсивность, распознаются обычно без больших затруд-
нений. 1,2,4-Замещенные можно отличить от других типов
соединений по присутствию дополнительной полосы в интер-
вале 1175—1125 сл'1.
б. Монозамещенные; замещение в 1,3-, 1,2,3- и 1,3,5-
положениях. Соединения с таким замещением дают слабое
поглощение в области 1175—1125 сл'1, как и 1,2,4-заме-
щенные, упомянутые выше. Кроме того, монозамещенные,
мета- и 1,2,3-замещенные поглощают в области 1110—
1070 сл'1, а все четыре типа дают еще одну полосу в интер-
вале 1070—1000 сл'1.
в. Замещение в 1,2-, 1,2,3- и 1,2,4-положениях. При
таком замещении обычно обнаруживается слабое поглоще-
ние в интервале 1000—960 сл'1.
Таким образом, на основании данных о количестве по-
лос и их положении можно дифференцировать главные ука-
занные здесь классы соединений, но идентификация отдель-
ного типа замещения внутри класса часто зависит от опре-
деления одной-единственной слабой полосы. Поскольку
эти полосы появляются в основной области спектра, свя-
занной с валентными колебаниями С — С и С — О, то та-
кая идентификация конечно очень ненадежна. Поэтому
полученные сведения могут быть использованы только
для подтверждения фактов, установленных на основании
данных о поглощении в каких-либо других областях
спектра.
Что касается стабильности положения рассматриваемых
полос в зависимости от природы заместителей, то, как по-
казали наши опыты, сильно ориентирующие группы влияют
на эти полосы меньше, чем на полосы в области более низ-
ких частот. Например, все полосы динитросоединений по-
падают в ожидаемые интервалы, и только дальнейшее за-
мещение нитрогруппами приводит к нарушениям. Однако
Кросс и Фассел [76] показали, что одна полоса в этой об-
ласти все же чувствительна по отношению к электроотри-
цательности замещающей группы.
Пинхас и Семуэль [56] полагают, что трифенилметиль-
ная группа дает характеристическую полосу вблизи
1185 см'1, но из-за появления в этой же области полосы СН
122
Часть I
многих других ароматических соединений применимость
данной корреляции ограничена.
Из-за сложности данной области спектра ароматиче-
ских соединений, имеющих более трех заместителей, при-
менение ее для анализа подобных соединений не рассма-
тривалось.
7. ДРУГИЕ АРОМАТИЧЕСКИЕ СИСТЕМЫ
Трополоны и родственные им соединения, содержащие
три сопряженные двойные связи в семичленном кольце,
имеют много свойств, присущих ароматическим соедине-
ниям. При изучении их инфракрасных спектров некоторые
исследователи [44—51] отметили следующие три характе-
ристические области поглощения: 1624— 1605, 1570— 1538
и 1280—1250 см'1, считая, что первые две области отно-
сятся к валентным колебаниям С = С. Однако Николс
и Тарбелл показали, что полоса в области 1570—1538 см'1
отсутствует у ряда бензотрополонов [47] и что в спектрах
колхицинов, не имеющих гидроксильной группы, отсут-
ствует полоса при 1275 сл-1 [48]. Таким образом, пригод-
ность этих корреляций сомнительна, поскольку первую
полосу трудно отличить от обычной полосы поглощения
фенила, а две другие, по-видимому, не специфичны.
Изучение ароматических соединений С5Н5 и С7Н7 пока-
зало, что их спектры в основном сходны со спектром бензола
[77]. Некоторый ароматический характер тиофена [78]
и аналогичных ему соединений также отражается на часто-
тах валентных колебаний СН и на общем виде их спектров.
ЛИТЕРАТУРА
1. Ingold, Proc. Roy. Soc., A169, 149 (1938); Angus,
Bailey, Ingold, Wilson, J. Chem. Soc., 1936,
912; Wilson, ibid., 1936, 1210; Bailey, Ingold,
Pool, Wilson, ibid., 1946, 222.
2. Wilson, Phys. Review, 45, 706 (1934).
3. Langset h, Lord, Danske Vidensk Selskab. Math. Fys.,
16, 6 (1938).
Гл. 5. Ароматические соединения
123
4. A ngus, Bailey, НА 1 е, Ingold, L е с к i е, Rai-
sin, Thomps o^n, Wilson, J. Chem. Soc., 1936,
966, 971.
5. Bailey, Hale, Ingold, Thompson, J. Chen'.
Soc., 1936, 931.
6. Герцберг, Колебательные и вращательные спектры много-
атомных молекул, Издатинлит, М., 1949, стр. 396.
7. Р i t z е г, Scott, J. Amer. Chem. Soc., 65, 803 (1943),
8. Plyler, Discuss. Faraday Soc., 9, 100 (1950).
9. L e с о m t e, Compt. rend. Acad. sci. Paris, 204, 1186 (1937);
206, 1568 (1938); 208, 1636 (1939).
10. L e с о m t e, J. Phys. Radium, 8, 489 (1937); 9, 13 (1938); 9,
512 (1938); 10, 423 (1939).
11. Delay, Lecomte, J. Phys. Radium, 7, 33 (1946).
12. F о x, Martin, J. Chem. Soc., 1939, 318.
13. F о x, Martin, Proc. Roy. Soc., A175, 208 (1940).
14. Fox, Martin, Proc. Roy. Soc., A167, 257 (1938).
15. S a i e г, С о g g e s h a 1 1, Analyt. Chem., 20, 812 (1948).
16. Rose, J. Res. Nat. Bur. Stand., 20, 129 (1938).
17. Y о u n g, Duvall, Wright, Analyt. Chem., 23, 709
(1951).
18. Barnes, Gore, Liddel, Van Zandt Williams,
Infra-red Spectroscopy, Reinhold, 1944.
19. Randall, Fowler, Fuson, Dangl, The Infra-red
Determination of Organic Structure, Van Nostrand,
1949.
20. С о 1 t h u p, J. Opt. Soc. Amer., 40, 397 (1950).
21. Rasmussen, Tunnicliff, Brattain, J. Amer
Chem. Soc., 71, 1068 (1949).
22. Badger, Quarterly Reviews, 5, 147 (1951).
23. Orr, Thompson, J. Chem. Soc., 1950, 218.
24. Thompson, Vago, Corfield, Orr, J. Chem. Soc.,
1950, 214.
25. Marion, Ramsay, Jones, J. Amer. Chem. Soc., 73,
305 (1951).
26. Jones, Humphries, Packard, Dobriner,
J. Amer. Chem. Soc., 72, 86 (1950).
27. Brownlie, J. Chem Soc., 1950, 3062.
28. Lippincott, Lord, McDonald, J. Amer. Chem.
Soc., 73, 3370 (1951).
29. Richards, Thompson, Proc. Roy. Soc., A195, 1 (1948).
124
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
Часть I
Whiffen, Torkington, Thompson, Trans.
Faraday Soc., 41, 200 (1945).
Whiffen, Thompson, J. Chem. Soc., 1945, 268.
Richards, Thompson, J. Chem. Soc., 1947, 1260.
Thompson, J. Chem. Soc., 1948, 328.
Van Zandt Williams, Rev. Sci. Instr., 19, 135 (1948).
Cole, Thompson, Trans. Faraday Soc., 46, 103 (1950).
Whiffen, Thesis, Oxford, 1946.
Philpotts, Thain, Nature, 166,
Friedel, Peirce, McGovern,
1028 (1950).
Analyt. Chem., 22,
418 (1950).
Ferguson, Levant, Analyt. Chem., 23, 1510 (1951).
Hales, Analyst, 75, 146 (1950).
Perry, Analyt. Chem., 23, 495 (1951).
Cannon, Sutherland, Spectrochimica Acta, 4, 373
(1951).
Laurer, McCaulay, Analyt. Chem., 23, 1875 (1951).
Koch, J. Chem. Soc., 1951, 512.
A u 1 i n-E rdtman, Theorell, Acta Chem. Scand.,
4, 1490 (1950).
Bartel s-K e i t h, Johnson, Chem. and Ind., 1950,
677.
Nicholls, Tarbell, J. Amer. Chem. Soc., 74, 4935
(1952).
Nicholls, T a г b e 1 1, J. Amer. Chem. Soc., 75, 1104 (1953).
Doering, Knox, J. Amer. Chem. Soc., 74, 5683 (1952).
Doering, Knox, J. Amer. Chem. Soc., 75, 297 (1953).
Doering, Hiskey, J. Amer. Chem. Soc., 74 5688 (1952).
К a m a d a, Japan Analyst, 1, 141 (1952).
Tiers G., Tiers J., J. Chem. Phys., 20, 761 (1952).
R a n d 1 e, W h i f f e n, J. Chem. Soc., 1952, 4153.
Bomstei n, Analyt. Chem., 25, 512 (1953).
Pinchas, Samuel, J. Chem. Soc., 1954, 863.
McCaulay, Lien, Launer, J. Amer. Chem. Soc., 76,
2364 (1954).
Margoshes, Fassel, Spectrochimica Acta, 7, 14 (1955).
Bellamy, J. Chem. Soc., 1955, 2818.
Ferguson, Werner, J. Chem. Soc., 1954, 3645.
Josie n, Lebas, Bull. Soc. Chim. Fr., 1956, 53, 57, 62.
Josie n, Lebas, Compt. Rend. Acad. Sci. Paris, 240, 181
(1955).
Fuson, Josie n, J. Amer. Chem. Soc., 78, 3049 (1956).
Whiffen, Spectrochim. Acta, 7, 253 (1955).
Гл. 5. Ароматические соединения
125
65. Randle, Whiffen, Molecular Spectroscopy, Inst, of
Petroleum, 1955.
66. D e p a i g n e-D e 1 a y, Lecomte, J. Phys. Radium, 7,
38 (1946).
67. Whiffen, J. Chem. Soc., 1956, 1350.
68. Bellamy, J. Chem. Soc., 1955, 4221.
69. Cencelj, Hadzi, Spectrochim. Acta, 7, 274 (1955).
70. W e r n e r, Kennard, Rayson, Austral. J. Chem., 8,
346 (1955).
71. Scheer, Nes, Smeetzer, J. Amer. Chem. Soc., 77,
3300 (1955).
72. Dannenberg, Schiedt, Steidle, Z. Naturforsch.,
8B, 269 (1953).
73. Corio, Dailey, J. Amer. Chem. Soc., 78, 3043 (1956).
74. Kross, Fassel. Margoshes, J. Amer. Chem. Soc.,
78, 1332 (1956).
75. Randle, Whiffen, Trans. Faraday Soc., 52, 9 (1956)
76. Kross, Fassel, J. Amer. Chem. Soc., 77, 5858 (1955)
77. N e 1 s о n, F a t e 1 e y, Lippincott, J. Amer. Chem
Soc., 78, 4870 (1956).
78. Hildago, Compt. rend. Acad. Sci. Paris, 239, 253 (1954)
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1. Иогансен А. В., Инфракрасные спектры углеводородов,
«Физико-химические свойства индивидуальных углеводоро-
дов», Гостоптехиздат, Москва — Ленинград, вып. 6, 1957.
2. Джонс Н., Сандорфи К., Применение спектроскопии
в химии, Издатинлит, М., 1959, стр. 315.
Рис. 1. 2,3-Диметилбутан,
Поглощение, */в Поглощение,
Частота, см'’
Рис. 2. 2,2-Диметилбутан,
Частота, см
Рис. 4. Стирол.
Поглощением 0/о Поглощение, %
Частота, см'
Рис. 6. Замещенные бензола, область 2000—1600 см х.
Частота, см"'
1700 1600 1500 1400 1300 1200 1100 1000 900 800 700
Рис. 7. п-Оксибеизальдегид.
(уо 'эпна'тосгои
132
ЧАСТЬ
II
КОЛЕБАНИЯ
УГЛЕРОД-КИСЛОРОДНЫХ
И КИСЛОРОД-ВОДОРОДНЫХ
СВЯЗЕЙ
Глава 6
СПИРТЫ И ФЕНОЛЫ
1. ВВЕДЕНИЕ
Полоса поглощения валентных колебаний О — Н в ин-
фракрасном спектре известна очень давно и изучена наи-
более детально. Еще в 1895 г. Ашкинасс [1] и Ранзохов [2]
впервые отметили, что полоса, появляющаяся вблизи
3300 см~1, связана, по-видимому, с гидроксильной группой,
после чего Кобленц [3] и Венигер [4, 5] привели дополни-
тельные сведения об этой полосе. Особенно интенсивно
поглощение ОН стало изучаться начиная с 1933 г. в связи
с работами Вулфа, Хендрикса, Хилберта и Лиддела [6—9],
которые показали, что инфракрасная спектроскопия
является удобным и простым методом изучения водородной
связи, так как при образовании последней увеличивается
длина связи ОН и полоса поглощения смещается в сторону
более низких частот. Эти авторы изучали главным обра-
зом область обертонов, но Фокс и Мартин [10, 11 ] получили
по существу сходные результаты, исследуя основные ча-
стоты ОН; Беджер и Бауэр [12] показали, что величина сме-
щения основной частоты, обусловленного образованием
водородной связи, может быть использована для оценки
прочности водородной связи, так как эта величина состав-
ляет примерно 35 см 1 на каждую килокалорию.
Очень полные обзоры по спектроскопическому исследо-
ванию водородной связи были сделаны рядом авторов [12—
16, 94—96], поэтому здесь будет показано только, какое
значение имеют данные о водородной связи при интерпре-
тации инфракрасных спектров спиртов. В соответствии
с этим даны ссылки лишь на те статьи, где приводится
большой экспериментальный материал.
Гидроксильная группа сильно полярна и потому взаи-
модействует с любыми другими молекулами, которые
136
Часть ! I
в какой-то степени поляризованы. Поэтому поглощение не-
связанной группы ОН можно наблюдать только в парах или
в разбавленных растворах с неполярными растворителями
(хотя имеются и некоторые исключения, а именно для соеди-
нений, у которых пространственные затруднения мешают
образованию связей или уменьшают силу связи). Однако
даже в растворе сероуглерода при очень сильном разбав-
лении обнаруживаются некоторые изменения частоты
колебаний группы ОН спиртов, обусловленные ассоциацией
с молекулами растворителя [78]. Изменение агрегатного
состояния, как, например, переход из жидкого в кристал-
лическое состояние, также вызывает смещение полосы
в сторону более высоких частот из-за того, что в кристалли-
ческом состоянии увеличивается возможность ориентации
П7, 18].
Аналогичным образом, изменения температуры также
должны заметно влиять на положение полос поглоще-
ния ОН [19, 20]. Эти явления очень затрудняют установ-
ление определенных интервалов частот для различных
типов связи. Тем не менее возможно, как будет показано
ниже, провести различие между межмолекулярными, внут-
римолекулярными и хелатными связями и установить,
например, является ли межмолекулярная ассоциация ди-
мерной или полимерной. Приблизительные интервалы ча-
стот поглощения, относящиеся к различным типам водород-
ной связи, указаны в табл. 5, но следует учитывать, что
они установлены лишь ориентировочно, и при оценке ти-
пов связи нужно принимать во внимание также физическое
состояние образца, а в некоторых случаях даже темпера-
туру, при которой проводились наблюдения.
Можно ожидать также наличия полос поглощения ва-
лентных колебаний С — О и деформационных колеба-
ний ОН в области более низких частот спектра. Как будет
показано, многие спирты дают две относящиеся к этим
колебаниям полосы поглощения, для которых характерны
довольно широкие интервалы частот. Обе полосы чувст-
вительны к изменениям характера водородной связи и
не представляют большого значения при интерпретации
спектров.
Рассматриваемые корреляции приведены в табл. 5.
Таблица 5
Частота, см-1
Валентные колебания ОН
Несвязанная группа ОН...........
Межмолекулярные водородные связи3
1. Соединения с одним мостиком
2. Полимеры ..................
Внутримолекулярная водородная
связь. Соединения с одним мости-
ком6 ...........................
Хелатные соединения. Сильные внут-
римолекулярные связи6 . . . . .
Деформационные колебания ОН
и валентные колебания С—Ов
Первичные спирты................
Вторичные спирты ...............
Третичные спирты ...............
Фенолы..........................
3650—3590 (пер.), узкая
полоса
3550—3450 (пер.), узкая по-
лоса
3400—3200 (с.), широкая
полоса
3570—3450 лоса (пер.), узкая по-
3200—2500 (сл.), рокая полоса очень ши-
Вблизи 1050 (с.), 1350—1260
(с.)
Вблизи 1100 (с.), 1350—1260
(с.)
Вблизи 1150 (с.), 1410—1310
(с.)
Вблизи 1200 (с.), 1410—1310
(с-)
а Мостики разрываются при разбавлении растворов.
6 Мостики ие изменяются при разбавлении растворов.
в Для несвязанных групп частоты понижаются. Значение данных для ана-
лиза ограничено.
137
138
Часть II
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ НЕСВЯЗАННОЙ ГРУППЫ ОН
Обычно для частот валентных колебаний ОН несвязан-
ной гидроксильной группы приводят интервал 3700—
3500 сл 1 [22—25]. Верхней границей частот таких колеба-
ний может служить полоса воды при 3760 см'1 [26, 27].
Однако этот случай безусловно особый, так как оба атома
водорода присоединены к одному и тому же атому кисло-
рода, а более характерным следует, по-видимому, считать
поглощение при 3590 см'1 [28], которое дает перекись водо-
рода. Из-за того, что трудно отличить поглощение свобод-
ного гидроксила от поглощения гидроксила, образующего
очень слабую водородную связь с растворителями и дру-
гими веществами, установить нижнюю границу нелегко.
Димеры, у которых группа ОН слабо связана одним водо-
родным мостиком, поглощают вблизи 3500 см'1 [29]; это
значение часто рассматривают как нижний предел интер-
вала частот поглощения свободной группы ОН.
Однако весь интервал 3700—3500 см'1 значительно
больше, чем можно было бы ожидать на основании теории
и при сравнении с аналогичными валентными колебания-
ми С — Н. В последнем случае частота определяется почти
целиком валентным состоянием (насыщенностью) углерод-
ного атома и присутствием или отсутствием в а-положении
электроноакцепторных групп. Поэтому для группы ОН
следовало бы ожидать даже большего постоянства поло-
жения полосы. Значительная ширина приводимого интер-
вала частот объясняется, вероятно, низкой дисперсией
призмы из хлористого натрия, которая очень широко ис-
пользовалась в более ранних исследованиях и которая не
позволяет наблюдать описываемые частоты с большой точ-
ностью. В последующих работах показано, что частоты
валентных колебаний свободной группы ОН, измеренные
с применением призмы из фтористого лития, лежат в бо-
лее узком интервале, который не превышает 3650—3590 см'1,
этот интервал с некоторыми оговорками приведен в табл. 5.
Фокс и Мартин [10] , например, нашли, что полоса погло-
щения свободной группы ОН у значительной части алифа-
тических и ароматических спиртов лежит в области 3636—
3618 см'1. Кун [74] провел также точные измерения интер-
валов частот несвязанной группы ОН для 35 спиртов
Гл 6. Спирты и фенолы
139
и фенолов. Во всех случаях, исключая метанол, поглощение
было обнаружено в интервале 3644—3605 см1. В пределах
какого-нибудь одного класса соединений общий интервал
частот еще меньше; например, полоса несвязанной группы
ОН у большого числа 1,2-диолов [97, 132] лежит в пределах
3630 ± 5 см'1. Фенолы также образуют общую группу
с а, p-ненасыщенными спиртами и поглощают вблизи ниж-
ней границы общего интервала [74, 98]. Влияние аромати-
ческого кольца, проявляющееся в небольшом понижении
частоты колебаний свободной группы ОН, было изучено
Ингрехемом и др. [75], которые нашли, что у 25 замещен-
ных фенола эта полоса лежит в интервале 3616—3588 см'1,
а у 40 замещенных пирокатехина — в интервале 3615—
3592 см1. Эти исследователи установили существование
зависимости между частотами валентных колебаний ОН
замещенных фенола и значениями постоянной Гаммета
о замещенных ароматических соединений, а Гулден [98]
показал, что эти частоты связаны также с величинами рКа.
Хансбергер и др. [76] заметили закономерное падение ча-
стоты ОН в ряду: метанол (3645), фенол (3628), а- или р-
нафтол (3618) и оксифенантрен (3605) см'1. Соседняя кар-
бонильная группа оказывает значительно большее влияние
на частоту, чем двойная связь в а,р-положении. Например,
кислоты в виде мономеров поглощают вблизи 3520 см'1,
тогда как замещение фтором в a-положении очень мало
влияет на значение частоты колебаний свободной группы
ОН, хотя и уменьшает тенденцию к образованию водород-
ной связи [79, 80]. Батуев и Матвеева [30] показали, что
в случае спектров комбинационного рассеяния можно
по характеристическим полосам при 3632, 3622 и 3615 см'1
различить первичные, вторичные и третичные спирты, но
по инфракрасным спектрам такое распознавание можно
провести только в случае простых спиртов. Коул и Джеф-
ферис [132] сообщают о неопубликованных данных, со-
гласно которым первичные спирты поглощают вблизи
3642 см"1 (с коэффициентом погашения е = 70), вторич-
ные—вблизи 3629 см 1 (е = 60—50) и третичные — вблизи
3618 см'1 (е = 45). Эти исследования были продолжены Ане-
том и Бейвином [133], которые получили такие же данные
Для ряда простых спиртов; однако они наблюдали также
падение частоты до 3545 см'1, как, например, в случае
140
Часть II
третичных спиртов, у которых электронное облако похоже
на облако л-электронов двойных связей и у которых предпо-
лагается наличие стерических эффектов. Возможно по-
этому, что в этих случаях происходит образование слабой
водородной связи с л-облаком, сходное с ассоциацией не-
которых доноров протонов с бензолом. Однако аномалия
наблюдается и у бензилового спирта, поглощающего при
3614 см-1, поэтому указанные авторы считают, что было бы
неблагоразумно на основании их данных о частотах простых
спиртов делать широкие обобщения. Тем не менее у такого
ряда изученных соединений, как стероиды и тритерпены,
отмеченные различия существуют; Олсоп и др. [134] по-
казали, что у тритерпенов можно различать также аксиаль-
ные и экваториальные [30] гидроксильные группы, погло-
щающие соответственно при 3629—3630 и 3637—3639 см-1.
Все эти значения частот относятся к поглощению несвя-
занных ОН для веществ в неполярных растворителях, и при
изменении полярности растворителя или температуры и дав-
ления они слегка изменяются. Изменения частот в связи
с изменениями полярности растворителя связаны с разли-
чиями диэлектрической постоянной окружающей среды,
хотя в некоторых случаях, например у фенола, они могут
быть связаны с образованием молекулярных комплексов
[99]. Изменения с температурой [100] и давлением [101]
невелики и возникают в результате изменения расстояний
между отдельными молекулами.
Частоты несвязанного гидроксила группы N — ОН
оксимов аналогичны частотам гидроксилов спиртов и лежат
в пределах 3650—3500 сл-1 1136—138].
3. КОЛЕБАНИЯ СВЯЗАННОЙ ГРУППЫ ОН (ОБЩИЕ
СВЕДЕНИЯ)
Фрейманн и Хейлман [33], исследуя область обертонов,
различали три типа водородных связей с участием гидро-
ксильных групп: межмолекулярную связь между двумя
или несколькими молекулами, внутримолекулярную связь
и хелатную связь, в случае которой могут быть резонанс-
ные структуры. Системы со связями этих типов подробно
изучались многими исследователями, и в настоящее время
можно определить тип связи в каждом отдельном случае.
Гл. б. Спирты и фенолы
141
Частота поглощения связанной группы ОН является мерой
прочности водородной связи. Например, у структуры
С — ОН...О— она почти такая же, как и у структуры
С —ОН ... О = С—, если только нет возможности резо-
нанса, при котором происходит стабилизация, вызывающая
образование намного более прочной водородной связи
и значительное смещение полос в сторону более низких
частот. Например, полоса поглощения у диацетонового
спирта при 3484 см'1 почти не отличается от полосы погло-
щения ОН димерных спиртов, тогда как ацетилацетон по-
глощает при 2700 см'1 [34]. У обоих этих веществ группа
ОН образует водородную связь с карбонильной группой,
но у последнего вещества происходит резонансная стабили-
зация, вследствие чего атомы кислорода приближены
друг к другу. Можно показать, что у хелатных соединений
этого типа частота колебаний карбонила, которая служит
также мерой прочности водородной связи, является линей-
ной функцией степени ненасыщенности двойной связи в
а,р-положении [76, 102], поскольку именно этим фактором
определяется расстояние О ... О.
Проводились исследования [77, 84, 103, 104, 135]
зависимости частот связанной группы ОН от расстояния
между двумя атомами, соединенными водородной связью.
Лорд и Меррифилд [84] показали, что у кислот исходных
соединений, образующих водородную связь, существует
приблизительно линейная зависимость между смещением
частоты ОН и расстоянием О ... О. Эта зависимость была
более подробно изучена Накамото и др. [103] и Пименте-
лом [104, 135]. Для связей X — H...Y существуют различ-
ные зависимости, но для соединений в пределах одного
типа связи имеется простая линейная зависимость, если
только три участвующих в водородной связи атома лежат
на одной прямой. Это привело к заключению, что откло-
нение от линейной зависимости может быть использовано
как мера угла изгиба водородной связи.
Прочность водородной (или дейтериевой) связи может
служить в качестве характеристики относительной прото-
ноакцепторной способности оснований. Так частота валент-
ных колебаний OD дейтерометанола в замещенных пири-
дина является прямой функцией основных свойств этих
соединений [105].
142
Часть 11
4. МЕЖМОЛЕКУЛЯРНЫЕ ВОДОРОДНЫЕ СВЯЗИ
Межмолекулярные водородные связи в случае отсут-
ствия резонансных структур приводят к появлению широ-
ких полос поглощения в интервале 3450—3200 см'1. Интен-
сивность этих полос обычно значительно выше интенсив-
ности полос колебаний несвязанной группы ОН, а их боль-
шая ширина объясняется, по-видимому, тем, что спирт
образует различные полимерные формы с водородными свя-
зями различной прочности, так что наблюдаемая широкая
полоса составляется из ряда более узких полос. Разное
положение этих полос в пределах указанного интервала
обусловлено только неодинаковой прочностью связей и яв-
ляется функцией физического состояния образца, его кон-
центрации и природы растворителя, если вещество нахо-
дится в растворе, температуры [106], а также типа образую-
щейся связи. Однако сравнение различных типов спиртов
при сходных условиях показывает, что у димеров с одним
мостиком, у которых из-за пространственных затруднений
не могут образоваться полимерные формы, возникают лишь
слабые водородные связи [29, 35, 36], так что они погло-
щают обычно вблизи 3500 см'1; нормальные же спирты,
у которых образуется очень мало димеров [19], поглощают
в интервале 3400—3200 см'1. Кун [74] установил, что ди-
меризованные спирты поглощают в интервале 3525—
3472 см1, а полимеризованные — в интервале 3341—
3338 см1. Марринен и Манн [107, 108] сообщают, что по-
лимерные ассоциаты в целлюлозе поглощают в интервале
3347—3324 см'1, а димеры с простыми мостиками — при
3404 см'1. Они разработали изящный метод определения
относительного содержания кристаллического и аморф-
ного вещества в целлюлозе, основанный на различиях ско-
рости дейтерообмена в группах ОН. От природы группы
R в соединении R — ОН заметно зависит интенсивность
полосы поглощения; соответствующие данные могут иногда
использоваться для выяснения структуры молекулы. Надо
отметить, что количественный анализ смесей на основе погло-
щения связанной группы ОН осуществить гораздо труднее.
а. Комплексы с одним водородным мостиком. Проч-
ность водородной связи между спиртами и растворителями
может быть изучена в очень разбавленных растворах, в ко-
Гл. 6. Спирты и фенолы
143
торых димеризация самих спиртов исключается. Работы
по изучению такого взаимодействия спиртов и раствори-
теля были выполнены многими исследователями, в том чис-
ле Мекке [38], который изучал поглощение ОН растворен-
ного фенола, применяя в опытах 18 различных растворите-
лей. Его результаты согласуются с данными предыдущих
исследователей, которые показали, что при переходе от
таких индифферентных растворителей, как циклогексан
илиСС14, к полярным растворителям наблюдается расшире-
ние полос, сопровождающееся уменьшением интенсивно-
сти. По силе влияния растворители располагаются в сле-
дующем порядке: циклогексан < сероуглерод < хлорбен-
зол < бензол. Интересно отметить, что при более высоких
концентрациях, когда преобладает эффект полимеризации
спирта, этот порядок изменяется, а также начинают играть
роль такие факторы, как диэлектрическая постоянная и раз-
мер молекулы растворителя. Если судить по частоте колеба-
ний группы ОН, при больших концентрациях ассоциация
молекул увеличивается в зависимости от растворителя
в таком порядке: бензол < хлороформ < сероуглерод [35].
Комплексы с одним мостиком типа С—О—Н ... О —
и С —О— Н ... О = С— обнаруживают поглощение при
одних и тех же частотах, а именно вблизи 3510 см'1. Ког-
гешелл и Сейер [31] нашли такую полосу у разбавленных
растворов этилового спирта и ацетона в четыреххлористом
углероде, а также у растворов этилового спирта и диоксана
в том же растворителе. Кун [74] указывает, что для колеба-
ний группы ОН в системе ROH ... О(С2Н5)2, где R —
алкильная группа, значения частот составляют 3492—
3482 см'1. Взаимодействие с другими растворителями, на-
пример с аминами, влияет аналогичным образом [39].
При использовании бензола в качестве растворителя
Мекке [38] наблюдал в области обертонов интересное яв-
ление, а именно: частичное расщепление полосы поглоще-
ния. Он объясняет это тем, что молекулы растворителя
могут располагаться относительно связи ОН двумя раз-
личными способами.
Комплексы с одним мостиком, не считая тех, которые
получаются при взаимодействии неодинаковых молекул,
образуются только в особых случаях, когда полной
полимеризации препятствуют пространственные факторы.
144
Часть II
Типичными соединениями, для которых наблюдаются стери-
ческие эффекты, являются opmo-замещенные фенола, изу-
чавшиеся многими исследователями [18, 31, 35, 36, 40, 41,
74, 139]. На основании химических данных уже давно из-
вестно, что opmo-замещенные фенола отличаются от других
типов замещенных, а 2,6-замещенные фенола с большими
заместителями вообще не участвуют в обычных реакциях
фенола и не растворяются в воде. Это объясняется простран-
ственными затруднениями, и можно показать, что у 2,6-ди-
замещенных фенола с большими замещающими группами
водородная связь не образуется, а у opmo-замещенных фе-
нола с заместителями, размеры которых больше размеров
метильной группы, она значительно ослаблена. Сере и Кит-
чен [40] опубликовали данные для обширного ряда фенолов
различного типа, исследованных в твердом и жидком состоя-
ниях и в растворах; они показали, что полоса поглощения
ОН появляется, например-, у 2,6-ди-трет-бутилфенола
в положении, которое характерно для несвязанной группы
ОН, и что при переходе от раствора к жидкому или твердому
состоянию заметного смещения полосы не происходит. Для
производных фенола, у которых имеются некоторые про-
странственные затруднения, наблюдается меньшее измене-
ние длины волны пика ОН по сравнению с несвязанной
группой ОН, чем у производных фенола, отличающихся
отсутствием пространственных затруднений. Как показал
Коггешелл [41], это обусловлено тем, что в первом случае
образование связей приводит преимущественно к димери-
зации, а не к полимеризации. Однако Хантер [95] показал,
что возможно другое объяснение неполной ассоциации. Это
объяснение состоит в невозможности достижения группой
ОН полной копланарности с кольцом, что приводит к умень-
шению ее кислотности и протонодонорных свойств.
Образование димеров с одним мостиком наблюдается так-
же у алифатических спиртов с разветвленной цепью, у кото-
рых полимерная ассоциация невозможна из-за стерических
препятствий, связанных с разветвлением цепей. Смит
и Крейц [29] наблюдали это явление у ряда таких спиртов
и отнесли частоту 3500 см'1 к водородному мостику, кото-
рый образуют эти спирты. Они также отметили, что в этих
случаях димер дает такое же поглощение несвязанной
группы ОН, как и мономер. Аналогичное явление наблю-
Гл. 6. Спирты и фенолы
145
дается и у mpmz-амилового спирта, поглощение которого
в жидком состоянии из-за пространственных затруднений
соответствует поглощению свободной группы ОН; погло-
щение связанной группы ОН происходит при более высо-
кой частоте, чем у нормальных спиртов. Подобные случаи
имеют место у углеводов, алюминиевых мыл и аналогич-
ных веществ [46, 47, 81].
б. Полимерная ассоциация. Наиболее обычный случай
образования водородных связей наблюдается при полимер-
ной ассоциации, когда молекулы соединяются в длинные
цепи при помощи относительно прочных водородных свя-
зей. При отсутствии хелатных образований область по-
глощения обычно лежит между 3400 и 3200 см'1, но точное
положение полосы зависит от природы растворителя и от
того, в каком состоянии — твердом или жидком — нахо-
дится исследуемое вещество. В связи с этим интересно отме-
тить, что полосы поглощения ОН в случае быстро охлаж-
денных расплавленных пленок обычно лежат гораздо бли-
нке к полосам, характерным для концентрированных раство-
ров, чем к полосам веществ в кристаллическом состоянии.
Это соответствует тому взгляду, что у вещества в кристалли-
ческом состоянии молекулы ориентированы таким образом,
что водородная связь обладает максимальной прочностью.
Интервал частот этого типа поглощения группы ОН очень
хорошо установлен на основании многочисленных экспери-
ментальных данных и не нуждается в подробном рассмот-
рении; типичные полосы такого поглощения имеются в полу-
ченных АНИ спектрах различных простых спиртов. В ряду
спиртов от метанола до деканола, взятых в виде концентри-
рованных растворов в неполярных растворителях, полоса
поглощения связанной группы ОН появляется в интервале
3370—3344 см'1. Кун [74] также дает перечень значитель-
ного числа спиртов и диолов, исследованных в виде концен-
трированных растворов, у которых полосы ОН лежат в пре-
делах интервалов, приведенных в табл. 5.
Путем исследования относительных интенсивностей
полос поглощения свободной и связанной групп ОН при
различных концентрациях можно определять константы
Диссоциации спиртов в случае их полимерных ассоциатов
[140] и спиртов, связанных с карбонильной группой [141].
Ю Л. Беллами
146
Часть II
5. ВНУТРИМОЛЕКУЛЯРНЫЕ ВОДОРОДНЫЕ СВЯЗИ
В большинстве случаев внутримолекулярная водород-
ная связь претерпевает резонансную стабилизацию, а это
приводит к образованию очень сильной связи и, следова-
тельно, к большому смещению частоты колебаний ОН. Эти
случаи будут рассмотрены в разделе, посвященном хелат-
ным соединениям. При отсутствии резонансной стабилиза-
ции возникает внутримолекулярная связь в виде одного
водородного мостика, похожая на водородную связь димер-
ных образований, рассмотренную выше. Поэтому такая
водородная связь является слабой; ей соответствует отно-
сительно слабая полоса поглощения в интервале 3570—
3450 см'. Существенное различие между двумя типами свя-
зей заключается в том, что межмолекулярные водородные
связи разрываются при разбавлении растворов неполяр-
ными растворителями, тогда как внутримолекулярные свя-
зи не зависят от концентрации раствора.
а. Комплексы с одним мостиком. Типичными примерами
внутримолекулярных комплексов с одним мостиком являют-
ся такие циклические соединения, как 2-оксициклогекса-
ноны [109], фенолы [7, 42], нитроспирты [ПО], 1,2-хлор-
гидрины [142] и циклические 1,2-диолы [97, 132]. Для по-
следнего типа соединений, как показал Кун [97], изучение
водородной связи может дать сведения об их вероятной
конфигурации. Наиболее стабильными мостиковыми струк-
турами такого типа являются, как и следовало ожидать,
структуры, представляющие шестичленные кольца [111].
Интересное явление наблюдается у opmo-замещенных фено-
ла, у которых возникает водородная связь между группой
ОН и другим заместителем. Полинг [43] показал, что связь
С—О у фенолов имеет в какой-то степени характер двойной
связи, поэтому водородный атом должен лежать в плоскости
кольца. Таким образом, фенол может иметь две равноценные
конфигурации I и II:
О-Н Н-0
6 6
I и
Гл. 6. Спирты и фенолы
147
С галогеном или другой протоноакцепторной группой
в орто-положении возможны цис- и транс-конфигурации
III и IV:
которые неравноценны, тогда как 2,6-дихлорфенол дает
снова равноценные структуры V и VI:
Таким образом, следует ожидать, что фенол и 2,6-дихлорфе-
нол дадут одну полосу поглощения ОН, а о-хлорфенол даст
две полосы, так как у рнс-формы может образовываться
водородная связь, а у транс-формы она не образуется. Это
было подтверждено экспериментально. Оказалось, что
в области обертонов фенол поглощает при 7050 см'1, 2,6-
дихлорфенол — при 6890 см'1, а о-хлорфенол дает обе эти
полосы поглощения, причем вторая полоса намного сильнее
первой [44]. Эго указывает на значительное преобладание
zjuc-формы, вследствие стабилизирующего влияния водород-
ной связи. У жидкого о-хлорфенола максимум поглощения
транс-формы появляется при 6620 см'1. Этот максимум
соответствует межмолекулярной связи, которая образуется
при димеризации [45]; сравнимые данные были получены
и в основной области спектра [112, 113]. Аналогичные ре-
зультаты дало исследование о-метоксифенола, фенилпро-
изводных и о-цианофенола [20]; кроме того, некоторые
замещенные виниловых спиртов тоже дают пару полос,
соответствующих двум конфигурациям [48]. Для пирока-
техинов [75] обнаружен такой же эффект; их внутримоле-
10*
148
Часть II
кулярной связи с одним мостиком соответствует полоса
поглощения в интервале 3575—3535 см1.
Явление, сходное с орто-эффектом, имеющим место при
образовании межмолекулярной связи у орто-замещенных
фенола, может также наблюдаться в случае внутримолеку-
лярной связи у замещенных бис-фенолалканов [41, 82].
Внутримолекулярная связь между двумя фенольными гид-
роксилами появляется в одной из конфигураций, но вторая,
транс-конфигурация, также стабилизирована, так как сво-
бодное вращение заторможено из-за наличия большой заме-
щающей группы в орто-положении. Таким образом, цис-
форма дает полосу поглощения, характерную для внутри-
молекулярной водородной связи, тогда как транс-форма
дает полосу поглощения, характерную для свободной груп-
пы ОН, если только условия не допускают образования
межмолекулярной связи.
б. Хелатные соединения. В ранних работах Хилберта,
Вулфа, Хендрикса и Лиддела [6—9] и других исследовате-
лей было найдено, что водородные связи у орто-замещенных
фенола, у которых в качестве заместителей в орто-положе-
нии присутствуют карбонильные группы или нитрогруппы,
более прочны, чем у соответствующих мета- и нара-заме-
щенных или других производных фенола. В области оберто-
нов у орто-производных полностью исчезала полоса погло-
щения несвязанной группы ОН вблизи 7000 сл-1, и это
рассматривалось как доказательство присутствия прочной
водородной связи. Поскольку водородная связь является
внутримолекулярной, то это происходит и в растворе. Подоб-
ное явление наблюдалось и в случае ацетилацетона, который
вместе с другими енольными Р-дикетонами повторно иссле-
довался в области основных частот Расмуссеном и др. [34].
Было найдено, что у этих соединений поглощению ОН соот-
ветствует полоса вблизи 2700 см'1 и что полоса поглощения
карбонила также смещается в большей степени, чем это
бывает при наличии обычной водородной связи. Упомяну-
тые авторы установили сходство между частотами ОН этих
соединений и димеров жирных кислот, которые также ха-
рактеризуются необычными свойствами. Полинг [13] объяс-
нил прочную связь у димеров жирных кислот наличием ре-
зонансной ионной структуры наряду с нормальной кова-
Гл. 6. Спирты и фенолы
149
лентной структурой; в результате связь ОН ослабляется
под влиянием зарядов и образуется сильная водородная
связь.
Расмуссен и др. [34] полагают, что сходные рассуждения
могут быть применены к енольным формам 0-дикетонов;
последние включают следующие резонансные структуры:
О—Н.....О
I II
R'C = CR"— CR’"
VIH
+О-Н.....О’
r'-C-CR''=CR!"
IX
Этим объясняется также исчезновение полосы в области
обертонов при 7000 см 1 у соединений, изученных Вулфом
и др., так как все соединения, у которых наблюдалось такое
ОН О
I II
явление, включали структуру R — С = С — С —. Действи-
тельно, уменьшение соответствующей основной частоты до
2700 см 1 приводит к исчезновению обертона в области
7000 см'1 и появлению его вблизи 5000 см'1. Для описания
этого явления Расмуссен и др. ввели термин «хелатное со-
пряжение». Аналогичные данные в отношении частот ОН
приведены Мартином [49], который показал, что салици-
ловый альдегид и о-оксиацетофенон дают широкие слабые
полосы поглощения ОН, занимающие область от 3500 до
2900 см 1 и сравнимые с полосой салициловой кислоты,
но гораздо меньшей интенсивности. Однако о-оксибензило-
вый спирт, у которого не может быть ионной структуры,
дает одну узкую полосу при 3600 см 1 (свободная группа
ОН) и другую полосу при 3436 см'1 (внутримолекулярная
связь с одним водородным мостиком). Хансбергер [50]
показал, что явление хелатного сопряжения характерно для
оксинафтальдегидов и аналогичных веществ; было описано
также много других примеров этого явления^ ацетилаце-
тонов [114, 115, 143], оксиантрахин нов [51, 83, 93, 116,
144], оксифлавонов [117], оксиинданонов [118] и других
соединений этого типа [119, 120]. Частота колебаний ОН
при 2600 см'1 была также обнаружена у соединений, у ко-
торых гидроксильная группа образует хелатную связь
150
Часть II
и структура которых включает гетероциклический атом
азота [121]. Для многих соединений такого типа сообщалось
об отсутствии полосы поглощения ОН в области основных
частот. Это объясняется главным образом перекрыванием
слабой полосы ОН сильными полосами поглощения валент-
ных колебаний СН вблизи 3000 см'1. В случае 5,5-диметил-
циклогександиона-1,3 хелатное сопряжение возникает ско-
рее всего при межмолекулярной ассоциации, а не при вну-
тримолекулярной. Это соединение содержит р-кетоенольную
структуру и дает полосу поглощения валентных колебаний
ОН при 2632 см'1. Стерические препятствия не допускают
образования у него внутримолекулярной водородной связи,
поэтому Расмуссен и др. [34] предполагают, что такое по-
глощение наблюдается в результате образования димера,
структура которого также обладает способностью к резо-
нансной стабилизации.
Нижнюю границу частот, при которых появляется поло-
са ОН этого типа, установить крайне трудно, так как поло-
сы поглощения относительно слабы и очень широки. Однако
Рейд и Руби [52] обнаружили размытую полосу поглощения
ОН у 6-фенил-4-окси-5,6-дигидропропирона при 2500 сл’1;
они, кроме того, отмечают, что по неопубликованным дан-
ным тетроновые кислоты имеют полосы поглощения ОН
с частотой, близкой к 2500 см'1. Беллами и Бичер [53]
и Даш и Смит [51] наблюдали полосы поглощения ОН фос-
форорганических кислот при таких же низких частотах,
хотя эти соединения, возможно, димерны, как и карбоновые
кислоты. Поглощение при 2440 см'1 наблюдалось также у не-
которых оксисоединений в растворах [55, 56], но хотя Горди
[56] и считает, что оно является слабым поглощением ОН,
это остается неясным, так как такая полоса может быть обер-
тоном деформационных колебаний СН хлороформа [57]*.
Однако во всех этих случаях характерная форма и положе-
ние полос поглощения прочно связанных групп ОН доста-
точно специфичны для распознавания структур данного
типа. При идентификации бывают затруднения в тех слу-
чаях, когда поглощение ОН совпадает с сильным поглоще-
нием СН вблизи 3000 см'1. Так, Амстуц и др. [58] сообщают
* Хлороформ использовался в качестве растворителя.—
Прим. ред.
Гл 6. Спирты и фенолы
151
об отсутствии полосы поглощения ОН у 1-нитрозонафтола-2,
а о-оксидифенилсульфоксид также не дает отчетливой по-
лосы, а лишь показывает возрастающее поглощение между
3250 и 2900 см х. В подобных случаях дополнительные дан-
ные можно иногда получить путем дейтерирования, приво-
дящего к появлению полосы OD при более низких частотах.
Однако дейтерирование необходимо проводить осторожно,
так как замещение водородного атома групп СН приведет
к появлению полосы поглощения CD примерно в том же ме-
сте, где находится полоса поглощения группы OD. Самая
прочная водородная связь, о которой когда-либо сообща-
лось, образуется тогда, когда водород близок к симметрич-
ному положению относительно других атомов. Например,
для диметил глиоксима никеля путем изучения дейтеропро-
изводных [77] было установлено, что очень слабая полоса
вблизи 1775 см"1 отвечает частоте ОН.
6. ИНТЕНСИВНОСТЬ И ФОРМА ПОЛОС ВАЛЕНТНЫХ
КОЛЕБАНИЙ ОН
Форма полосы поглощения ОН и ее относительная интен-
сивность могут иногда служить указанием на тип структуры.
Этот вопрос еще полностью не изучен, но выполнено боль-
шое число работ по выяснению причин характерного уве-
личения интенсивности и ширины полос поглощения гидро-
ксильных групп, связанных водородной связью. Большую
ширину этих полос в спектрах жидкостей приписывали обыч-
но тому, что молекулы образуют различные по прочности
водородные связи, так что вид полосы должен был бы слу-
жить указанием на распределение водородных связей по
прочности [10, 57]. Однако, как указал Клец [59], это объяс-
нение нельзя применить к кристаллам, так как у них все
водородные связи должны быть одинаковы. В спектрах кри-
сталлических фенолов полоса поглощения ОН уже, чем
в спектрах жидких фенолов, но все-таки шире, чем для фено-
лов в разбавленных растворах. Можно сделать другое пред-
положение, согласно которому ширина этих полос обуслов-
лена комбинированием с рядом низкочастотных колебаний,
включая частоты деформационных колебаний [12, 60,611.
С этим согласуется наблюдаемое увеличение ширины
полосы с увеличением прочности водородной^ связи и значи-
152
Часть II
тельное ее уменьшение при замещении водорода дейтерием.
Что касается кристаллов, то для них, согласно Мекке и
Уббелоде [62], одним из факторов, влияющих на уширение
полос поглощения ОН, является силовое поле в кристалле.
Вопрос о причине увеличения интенсивности полос связан-
ных групп ОН с теоретической точки зрения был рассмот-
рен Цубомура [122, 123], а зависимость интенсивности от
температуры была исследована Хьюзом и др. [100], а также
Финчем и Липпинкоттом [106].
Интенсивность полос поглощения несвязанной группы
ОН подчиняется закону Бера, и для каждого отдельного
класса родственных веществ, например фенолов [51],
коэффициент погашения является в основном постоянной
величиной. Однако он значительно меняется от одного клас-
са соединений к другому. Эти данные использовались при
определении цис- и транс-форм фенолов, испытывающих
пространственные затруднения. Можно также оценивать
содержание свободной группы ОН в алифатических соеди-
нениях по измерениям поглощения в области 3610 см'1
[29]. У соединений с одним водородным мостиком полоса
поглощения ОН остается довольно узкой [38], а изучение
некоторых групп веществ показало, что по крайней мере
в пределах отдельного класса веществ коэффициент погаше-
ния можно считать постоянным, т. е. закон Бера выполняет-
ся [29, 35]. Димеры с одним мостиком дают полосы погло-
щения, характерные как для свободной, так и для связан-
ной группы ОН.
Барроу [124] и Браун и Роджерс [145] изучали абсо-
лютные интенсивности полос поглощения ОН для различ-
ных типов спиртов. В зависимости от природы заместителей
интенсивность может меняться даже в два раза. Например,
фенол поглощает намного сильнее, чем трет-бутанол. Браун
и Роджерс [145] связывают это с индукционным эффектом
заместителей; они показали, что интенсивность при раство-
рении в четыреххлористом углероде повышается с увели-
чением электроотрицательности заместителей. В раствори-
телях, в которых имеет место ассоциация, интенсивность
сильно возрастает [124], так что интенсивность поглоще-
ния гидроксильной группы метанола в эфире приблизи-
тельно в восемь раз, а в триэтиламине — в двенадцать раз
больше, чем в четыреххлористом углероде.
Гл. 6. Спирты и фенолы
153
При наличии прочной межмолекулярной связи наблю-
даются широкие и очень сильные полосы. Интенсивность
их изменяется в зависимости от типа вещества, но в пределах
данного класса веществ она опять-таки остается примерно
постоянной. Фридел [35] нашел, что средняя интенсивность
полос поглощения для десяти фенолов с различными заме-
стителями в мета- и пара-положениях является постоянной
в пределах 2,1 %. Однако в случае хелатных соединений
интенсивность значительно уменьшается, и пол оса становится
очень широкой и слабой. В этом случае интенсивность зна-
чительно меньше, чем, например, интенсивность полос
поглощения связанной группы ОН у димерных кислот, а по-
лоса является такой широкой, что измерение коэффициента
погашения нецелесообразно.
Такие изменения частоты и интенсивности поглощения
гидроксила важны прежде всего с точки зрения количест-
венного анализа, и любой анализ, базирующийся на иссле-
довании полосы группы ОН, должен, несомненно, прово-
диться при очень точном соблюдении стандартных условий.
В то же время сам факт наличия таких изменений полос
может иногда быть успешно использован. Коггешелл и Сейер
[31] применяли данные об интенсивности при изучении сте-
пени полимеризации, хотя Френсис [63], обсуждая интен-
сивность полос связанных групп ОН, подверг сомнению их
метод подхода к интерпретации результатов измерений.
Факт образования внутримолекулярной водородной
связи у о-бромпроизводных фенола успешно использовал
Симард и др. [64] при анализе смесей фенолов, крезолов
и ксиленолов; измерения интенсивности полос поглощения
свободной группы ОН в разбавленных растворах до и после
бромирования позволили им определить соединения, орто-
положения которых заняты метильными группами. У этих
последних соединений частота ОН остается, конечно, без
изменений, тогда как у всех других дизамещенных проис-
ходит образование внутримолекулярной водородной связи
и появляется вторая полоса ОН.
При количественном изучении спиртов и фенолов лучше
всего пользоваться разбавленными растворами, поскольку
в этом случае другие водородные связи, кроме внутри-
молекулярной, не образуются. Иногда можно провести на-
дежные анализы, основываясь на поглощении связанной
154
Часть //
группы ОН, если стандартизировать условия и пользоваться
одними и теми же растворителями. При этом, однако,
коэффициенты погашения должны соответствовать иссле-
дуемым или очень близким по строению молекулам, изу-
ченным при тех же условиях, и должны определяться инте-
гральные интенсивности поглощения.
При определении структур, например при изучении свя-
занных групп ОН у поливинилового спирта [65] и углево-
дов [46], регистрировалось появление или исчезновение
полос поглощения ОН у монокристаллов, исследуемых
в поляризованном излучении. Как и в случае наличия сво-
бодных групп ОН метод дейтерирования весьма полезен
при идентификации связанных групп и изучении процессов
образования водородной связи; для этих целей его применя-
ли Роуэн и Плайлер [66], Шеппард [116], Сазерленд [125]
и многие другие исследователи.
7. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С — О И ДЕФОРМАЦИОННЫЕ
КОЛЕБАНИЯ О-Н
Оба эти вида колебаний должны приводить к появлению
сильных полос поглощения, характеризующих спирты. Как
будет показано ниже, большинство спиртов дает в низко-
частотной области две полосы, которые чувствительны к из-
менениям характера водородных связей и которые при раз-
бавлении раствора сдвигаются в сторону более низких ча-
стот. К сожалению, из-за взаимодействия колебаний было
много путаницы в вопросе о том, какая из этих полос отно-
сится к первому и какая — ко второму виду колебаний.
Недавние исследования позволили получить некоторые
сведения, однако полностью этот вопрос еще не выяснен.
Первую, обычно более интенсивную из этих полос, заме-
тил в 1910 г. Венигер [4], который обнаружил сильную
полосу в спектрах жидких первичных спиртов вблизи
1050, вторичных спиртов — вблизи 1100 и третичных спир-
тов— вблизи 1150 см1. Эти данные были подтверждены
Леконтом [67, 85], который покааал, что перечисленные
полосы связаны с гидроксильной группой, так как при
исследовании большого ряда жирных спиртов наблюдалось
систематическое уменьшение интенсивности этих полос
по мере увеличения длины цепи и уменьшения относитель-
Гл. 6. Спирты и фенолы
155
ного содержания группы ОН. Леконт также отметил, что
полосы поглощения чувствительны к структурным изме-
нениям, поскольку наблюдалось заметное уменьшение
интенсивности поглощения при 1100 сл'1 у некоторых ви-
нилкарбинолов по сравнению с предельными вторичными
спиртами. Спектр фенола был найден слишком сложным для
проведения каких-либо надежных отнесений.
Цейсе и Цуцуи [861 недавно провели систематическое
исследование зависимости положения полос поглощения
спиртов от строения молекул. Они установили, что у насы-
щенных спиртов с неразветвленной цепью полоса появляется
в относительно узких интервалах частот: 1075—1010 см'1
для первичных и 1119—1105 см 1 для вторичных спиртов
и вблизи 1140 сл'1 для третичных спиртов. Эти данные
в основном подтвердили Стюарт и Сазерленд [125], изу-
чавшие жидкие спирты. Однако у всех спиртов с развет-
влением цепи в a-положении, у аф-ненасыщенных и али-
циклических спиртов наблюдается смещение полос за пре-
делы этих интервалов, а в величине и направлении этих
смещений имеется ряд странных аномалий. Для цепей, раз-
ветвленных у а-углеродного атома, частота во всех трех слу-
чаях уменьшается на 10—15сл-1. Если, однако, имеется два
таких разветвления цепи, то получается обратный эффект;
так ди-/пре/п-бутиловый спирт поглощает при 1163 сл’1,
т. е. в пределах обычного для таких спиртов интервала. Не-
насыщенность в аф-положении приводит к значительному
уменьшениючастоты; например, у вторичных аф-ненасыщен-
ных спиртов полоса находится в интервале 1074—1012 сл'1.
Найдено, что смещение в случае ароматической ненасыщен-
ности больше, чем в случае алкенильной, несмотря на то
что кратность связи в первом случае меньше, чем во втором.
Хотя изучено мало спектров соответствующих первичных
и третичных спиртов, но и у них, по-видимому, происходит
смещение полос в сторону низких частот, причем алиловый
спирт опять-таки поглощает при более высокой частоте
(1030 сл'1), чем бензиловый (1010 сл’1). У алициклических
вторичных спиртов частота постепенно уменьшается с уве-
личением размеров кольца в ряду от циклобутанола
(1090 сл’1) до циклогептанола (1025 сл’1), так что большее
смещение частоты наблюдается у тех спиртов, у которых
имеет место меньшее напряжение цикла. У растворенных
156
Часть II
стеринов полоса лежит в интервале 1075—1000 см'1. В ре-
зультате многочисленных тщательных исследований найде-
но, что существует корреляция положения полосы в этом
интервале со стереохимическим расположением гидроксиль-
ной группы при атоме С3 [87—90]; таким образом, в настоя-
щее время можно, используя это свойство, получать ценные
сведения о структуре. Аналогичным путем можно различать
аксиальные и экваториальные гидроксильные группы в поло-
жении 3 у тритерпенов [134]. Примерно такие же данные
получены для деканолов [91], хотя у них различия в зави-
симости от структурных особенностей молекулы выражены
менее отчетливо.
Значительное перекрывание интервалов частот, харак-
терных для различных типов спиртов, с частотами других
функциональных групп делает идентификацию типа присут-
ствующей структуры по результатам исследования только
этой области очень затруднительной, но если имеются допол-
нительные данные, то исследование этой области часто
может дать очень ценные сведения.
Вторая полоса спиртов обычно лежит в области 1300 см'1
[69], но ее положение значительно изменяется в зависимо-
сти от строения молекулы, и она не подвергалась система-
тическому изучению. Обе указанные полосы поглощения
чувствительны к изменениям состояния веществ, что сви-
детельствует о влиянии на них водородной связи; поэтому
по вопросу о том, какую из них следует отнести к деформа-
ционным колебаниям ОН, имеются весьма противоречивые
данные. В случае метилового спирта некоторые авторы
относят к деформационным колебаниям ОН полосу при
1109 см'1 [70], тогда как другие относят к этим колебаниям
полосу при 1340 слГ1 [71, 26]. Кроме того, метанол дает
сильную полосу при 1С34 см'1, которую связывают с колег
баниями С — О [72, 73]. Что касается фенола, то, несмотря
на исследования его различных состояний и изучения
с применением дейтерирования [32, 68], остались сомнения,
следует ли к деформационным колебаниям ОН отнести
полосу вблизи 1200 или вблизи 1350 см \ так как обе эти
полосы поглощения чувствительны к изменениям в характе-
ре водородных связей. В этом последнем случае полосы
находятся ближе друг к другу и по аналогии с карбоновыми
кислотами [37] можно ожидать взаимосвязи соответствую-
Гл. 6. Спирты и фенолы
157
щих колебаний. Учитывая такую взаимосвязь, Куратани
[92], изучавший пентахлорфенол с применением дейтери-
рования, отнес обе полосы (при 1280 и 1195 см1) к де-
формационным колебаниям ОН. Вопрос об идентификации
этих полос у замещенного фенола рассматривается также
в статьях Мекке и Росми [126], Дейвиса и Джонса [127].
Большинство исследователей считает, что поглощение ряда
стеринов в интервале 1075—1000 см"1 обусловлено валент-
ными колебаниями С — О [87—89], но другие относят его
к деформационным колебаниям ОН [90]. Положение далее
усложняется появлением у некоторых спиртов дополни-
тельных полос, чувствительных к дейтерированию, которые
могут быть обусловлены поворотной изомерией [146].
Чтобы преодолеть эти трудности Сазерленд и его сотруд-
ники [21, 125, 128, 129], а также Куайнен и Виберли [130,
131 ] провели недавно большую работу по изучению дейтеро-
производных спиртов. Эти две группы авторов приводят
различные данные по отнесению полос, и потому получен-
ные результаты нельзя считать окончательными, однако, по-
Видимому, полоса алифатических спиртов вблизи 1100 см'1
связана в основном с валентными колебаниями С—О, тогда
как простой или двойной пик в области 1400—1300 см"1
отвечает плоскостным деформационным колебаниям ОН.
Последние, как указывалось, вероятно, взаимосвязаны с де-
формационными колебаниями соседних групп СН2, что при-
водит к смешанным колебаниям, и потому изменения спектра
не будут соответствовать ожидаемым в результате дейтери-
рования. Братож и др. [114] провели аналогичное отнесе-
ние полос для производных ацетил ацетона, причем к дефор-
мационным колебаниям ОН они отнесли полосу при
1435 см1, а к валентным колебаниям С—О, частота которых
повышена из-за резонанса,— полосу при 1284 слГ1.
У фенолов и близких к ним соединений положение совсем
иное, так как валентным колебаниям С—О в результате
влияния ароматического кольца соответствует поглощение
при более высоких частотах. Таким образом, имеется воз-
можность взаимодействия между колебаниями ОН и С—О
или между колебаниями С—О и ароматического кольца.
Шеппард [116] отнес большую из двух главных чувстви-
тельных к дейтерированию частот оксихинолинов к валент-
ным колебаниям С—О. Мекке и Росми [126] сделали ана-
158
Часть 11
логичное отнесение в случае фенола; они считают, что поло-
са 1180 сл 1 соответствует в основном колебаниям ОН.
По-видимому, эти колебания взаимосвязаны главным обра-
зом с колебаниями ароматического цикла, имеющими
частоту 1310 сл1, хотя во взаимодействии могут участвовать
и другие колебания ароматического цикла.
Из этих исследований ясно, что большинство спиртов
дают две полосы, которые независимо от их происхождения
достаточно характерны для того, чтобы по ним можно было
судить о структуре, если бы при этом имелось достаточно
данных, связывающих частоты с различными структурными
изменениями. Однако в настоящее время число таких дан-
ных ограничено; по этой причине, а также из-за изменений
спектров между 1450 и 600 см 1 в зависимости от состояния
вещества эта область имеет ограниченное применение для
структурного анализа.
Следует добавить, что все спирты в жидком состоянии
дают очень широкие полосы в интервале 750—650 сл’1,
возникающие в результате внеплоскостных деформацион-
ных колебаний связанных групп ОН. Эти полосы не нашли
применения в корреляционных исследованиях.
ЛИТЕРАТУРА
1. A s с h k i n a s s, Wied. Ann., 55, 401 (1895).
2. Ransohoff, Dissert. Berlin, 1896.
3. Coblentz, Investigations of Infra-red Spectra, Part I,
Carnegie Institute, 1905, p. 35.
4. W e n i g e r, Phys. Review, 31, 388 (1910).
5. W e n i g e r, J. Opt. Soc. Amer., 7, 517 (1923).
6. L 1 d d e 1, Wulf, J. Amer. Chem. Soc., 55, 3574 (1933).
7. W u 1 f, L i d d e 1, J. Amer. Chem. Soc., 57, 1464 (1935).
8. Hilbert, Wulf, Hendricks, Liddel, Nature,
135, 147 (1935).
9. Hilbert, Wulf, Hendricks, Liddel, J. Amer.
Chem. Soc., 58, 548, 1991 (1936).
10. Fox, Martin, Proc. Roy. Soc., A162, 419 (1937); ibid.,
A175, 208 (1941).
11. Fo x, Martin, Trans. Faraday Soc., 36, 897 (1940).
12 Badger, Bauer, J. Chem. Phys., 5, 839 (1937).
13. Pauling, The Nature of the Chemical Bond, Oxford Univer-
sity Press, 1950, p, 316.
14. Wright, Ind. Eng. Chem. (Anal. Ed), 13, 1 (1941).
Гл. 6. Спирты и фенолы
159
15. Н u n t е г, Price, Martin, Report of a Symposium on the
Hydrogen Bond, Institute of Chemistry Monograph, 1950.
16. H u g g i n s, J. Org. Chem., 1, 407 (1936).
17. К 1 e t z, Price, J. Chem. Soc., 1947, 644.
18. R i c h a r d s, Thompson, J. Chem. Soc., 1947, 1260.
19. H о f f m a n n, Z. physikal. Chem., B53, 179 (1943).
20. Liittke, Mecke, Z. Elektrochem., 53, 241 (1949).
21. Ramsay, Sutherland, Discuss. Faraday Soc., 9, 274
(1950).
22. Thompson, J. Chem. Soc., 1948, 328.
23. С о I t h u p, J. Opt. Soc. Amer., 40, 397 (1950).
24. Barnes, Gore, Liddel, Van Zandt Williams.,
Infra-red Spectroscopy, Reinhold 1944.
25. Randall, Fowler, Fuson, Dang 1, Infra-red Deter-
mination of Organic Structure, Van Nostrand, 1949.
26. Герцберг, Колебательные и вращательные спектры много-
атомных молекул, Издатинлит, М., 1949, стр. 304.
27. С г о s s, Burnham, Leighton, J. Amer. Chem. Soc.,
59, 1134 (1937).
28. G i g u e r e, J. Chem. Phys., 18, 88 (1950).
29. Smith, Creitz, J. Res. Nat. Bur. Stand., 46, 145 (1951).
30. Батуев, Матвеева, Изв. АН СССР, Отдел, хим. наук,
1951, 448.
31. Coggeshall, Saie г, J. Amer. Chem. Soc., 73, 5414 (1951).
32. Williams, H о f s t a d t e r, Herman, J. Chem. Phys.,
7, 802 (1939).
33. F г e у m a n n, H e i 1 m a n n, Compt. rend. Acad. sci. Paris,
219, 415 (1944).
34. Rasmussen, Tunnicliff, Brattai n, J. Amer.
Chem. Soc., 71, 1068 (1949).
35. Friedel, J. Amer. Chem. Soc., 73, 2881 (1951).
36. Coggeshall, J. Amer. Chem. Soc., 69, 1620 (1947).
37. Ha dz i, Sheppard, Proc. Roy. Soc., A216, 247 (1953).
38. Mecke, Discuss. Faraday Soc., 9, 161 (1950).
39. Baker, Davies, Gaunt, J. Chem. Soc., 1949, 24.
40. S e a r s, Kitchen, J. Amer. Chem. Soc., 71, 4110 (1949).
41. Coggeshall, J. Amer. Chem. Soc., 72, 2836 (1950).
42. Liittke, Mecke, Z. physikal. Chem., 56, 196 (1950).
43. Pauling, J. Amer. Chem. Soc., 58, 94 (1936).
44. W u 1 f, Liddel, Hendricks, J. Amer. Chem. Soc.,
58, 2287 (1936).
45. E г г e г a, M о 1 1 e t, J. Phys. Radium, 6, 281 (1935).
46. Thompson, Nicholson, Short, Discuss. Faraday
Soc., 9, 222 (1950).
47. Brown, Holliday, Trotter,J. Chem. Soc., 1951,
1532.
48. Buswell, Rodebush, Whitney, J. Amer. Chem. Soc.,
89, 770 (1947).
49. Martin, Nature, 166, 474 (1950).
50. Hunsberger, J. Amer. Chem. Soc., 72, 5626 (1950).
51. Flett, J. Chem. Soc., 1948, 1441.
160
Часть II
52. Reid, Ruby, J. Amer. Chem. Soc., 73, 1054 (1951).
53. Bellamy, Beecher, J. Chem. Soc., 1952, 1701.
54. Daasch, Smith, Analyt. Chem., 23, 853 (1951).
55. Buswell, R о d e b u s h, Roy, J. Amer. Chem. Soc., 60.
2528 (1938).
56. Gordy, J. Chem. Phys., 7, 163 (1939).
57. Sutherland, Trans. Faraday Soc., 36, 889 (1940).
58. Amstutz, Hunsberger, Chessick, J. Amer. Chem.
Soc., 73, 1220 (1951).
59. К 1 e t z, Discuss. Faraday Soc., 9, 211 (1950).
60. Davies, Sutherland, J. Chem. Phys., 6, 755, 762 (1938).
61. D a v i e s, J. Chem. Phys., 8, 577 (1940); Discuss. Faraday Soc.,
9, 212 (1950).
62. Davies, Discuss. Faraday Soc., 9, 213 (1950).
63. Francis, J. Chem. Phys., 19, 505 (1951).
64. Simard, Hasegawa, Bandaruk, Headington,
Analyt. Chem., 23, 1384 (1951).
65. Glatt, Webber, Seaman, Ellis, J. Chem. Phys., 18,
413 (1950).
66. Rowen, Plyler, J. Res. Nat. Bur. Stand., 44, 313 (1950).
67. L e с о m t e, Traite de chimie organique, Masson et Cie, Paris,
2, 143 (1936).
68. В r a t t a i n, J. Chem. Phys., 6, 298 (1938).
69. L e с о m t e, Compt. rend. Acad. Sci. Paris, 180, 825 (1925).
70. Davies, J. Chem. Phys., 16, 267 (1948).
71. Noether, J. Chem. Phys., 10, 693 (1942).
72. Borden, Barker, J. Chem. Phys., 6, 553 (1938).
73. Barker, Bosschieter, J. Chem. Phys., 6, 563 (1938).
74. Kuhn, J. Amer. Chem. Soc., 74, 2492 (1952).
75. Ingraham, Corse, Bailey, Stitt, J. Amer. Chem.
Soc., 74, 2297 (1952).
76. Hunsberger, Ketcham, Gutowsky, J. Amer.
Chem. Soc., 74, 4839 (1952).
77. Rundle, Parasol, J. Chem. Phys., 20, 1487 (1952).
78. S t u a r t, J. Chem. Phys., 21, 1115 (1953).
79. Henne, Francis, J. Amer. Chem. Soc., 75, 991 (1953).
80. Haszeldine, J. Chem. Soc., 1953, 1757.
81. Harple, Wiberley, Bauer, Analyt. Chem., 24, 635
(1952).
82. Amberlang, Binder, J. Amer. Chem. Soc., 75, 947 (1953).
83. Josien, Fuson, Lebas, Gregory, J. Chem. Phys.,
10, 331 (1953).
84. Lord, Merrifield, J. Chem. Phys., 10, 166 (1953).
85. Tuot, Lecomte, Bull. Soc. Chim. France, 10, 542 (1943).
86. Zeiss, T s u t s u i, J. Amer. Chem. Soc., 75, 897 (1953).
87. Rosenkrantz, Milhorat, Farber, J. Biol. Chem.,
195, 509 (1952).
88. Cole, Jones, Dobriner, J. Amer. Chem. Soc., 74, 5571
(1952).
89. Rosenkrantz, Zablow, J. Amer. Chem. Soc., 75,
903, (1953). :
Гл. 6. Спирты и фенолы
161
90. Furst, Kuhn, S с о t о n i, G u n t li a r d, Helv. Chim.
Acta, 35, 951 (1952).
91. Dauben, Hoerger, Freeman, J. Amer. Chem. Soc.,
74, 5206 (1952).
92. К u r a t a n i, J. Chem. Soc. Japan, 73, 758 (1952).
93 Hergert, К u r t h, J. Amer. Chem. Soc., 75, 1622 (1953).
94. W h e 1 a n d, Resonance on Organic Chemistry, Wiley,
New York, 1955, p. 47.
95. Hunter, Progress in Stereochem., Vol. I, Butterworth,
London, 1954, p. 224.
96. Ferguson, Electronic Theories of Organic Chemistry,
Prentice Hall, New York, 1952.
97. Kuhn, J. Amer. Chem. Soc., 76, 4323 (1954).
98. Goul den, Spectrochim. Acta, 6, 129 (1954).
99. J о s i e n, D i z a b c, Compt. Rend. Acad. Sci. Paris, 243,
44 (1956).
100. Hughes, Martin, Coggeshall, J. Chem. Phys.,
24, 489 (1956).
101. Fishman, Drickamer, J. Chem. Phys., 24, 548 (1956).
102. Bellamy, Beecher, J. Chem. Soc., 1954, 4487.
103. Nakamoto, Margoshes, Rundle, J. Amer. Chem.
Soc., 77, 6480 (1955).
104. Pimentel, Seder holm, J. Chem. Phys., 24 639 (1956).
105. Tamres, Searles, Leigh ly, Mohr in an, J. Amer.
Chem. Soc., 76, 3983 (1954).
106. Finch, Lippincott, J. Chem. Phys., 24, 908 (1956).
107. Marrinan, Mann, J. App. Chem., 4, 204 (1954).
108. Marrinan, Mann, Trans. Faraday Soc., 52, 481, 487, 492
(1956).
109. Huitric, Kumler J. Amer. Chem. Soc., 78, 1147 (1956).
110. Urbanski, Bull. Acad. Polon. Sci., Cl. Ill, 4, 87 (1956).
111. Hoyer, Chem. Ber., 89, 146 (1956).
112. Rossmy, Liittke, Mecke, J. Chem. Phys., 21, 1606
(1953).
113. К e u s s 1 e r, Rossmy, Z. Electrochem., 60, 136 (1956).
114. В r a t о z, H a dz 1, Rossmy, Trans. Faraday Soc., 52,
464 (1956).
115. E i s t e r t, Reiss, Chem. Ber., 88, 92 (1955).
116. H a d z i, S h e p p a r d, Trans. Faraday Soc., 50, 911 (1954).
117. S h a w, Simpson, J. Chem. Soc., 1955, 655.
118. Farmer, Hayes, Thomson, J. Chem. Soc., 1956,
3600.
119. Musso, Chem. Ber., 88, 1915 (1955).
120. Duncanson, J. Chem. Soc., 1953, 1207.
121. Branch, Nature, 177, 671 (1956).
122. Tsubomura, J. Chem. Phys., 24, 927 (1956).
123. Tsubomura, J. Chem. Phys., 23, 2130 (1955).
124. В a г г о w, J. Phys. Chem., 59, 1129 (1955).
125. Stuart, Sutherland, J. Chem. Phys., 24, 559 (1956).
126. Mecke, Rossmy, Z. Electrochem., 59, 866 (1955).
127. Davies, Jones, J. Chem Soc., 1954, 120.
11 Л. Беллами
162
Часть 11
128. Stuart, Sutherland, J. Phys. Radium, 15, 321 (1954).
129. К r i m m, Liang, Sutherland, J. Chem. Phys., 24,
778 (1956).
130. Quinan, Wiberley, J. Chem. Phys., 21, 1896 (1953).
131. Quinan, Wiberley, Analyt. Chem., 26, 1762 (1954).
132. Cole, Jefferies, J. Chem. Soc., 1956, 4391.
133. A n e t, Bavin, Can. J. Chem., 34, 1756 (1956).
134. Allsop, Cole, White, W ill i x, J. Chem. Soc., 1956,
4868.
135. Huggins, Pimentel, J. Phys. Chem., 60, 1615 (1956)
136. Palm, W e r b i n, Can. J. Chem., 31, 1004 (1953).
137. Califano, Liittke, Z. Phys. Chem., 5, 240 (1955).
138. Califano, Liittke, Z. Phys. Chem., 6 , 83 (1956).
139. Bowman, Stevens, Baldwin, J. Amer. Chem. Soc.,
79, 87 (1957).
140. Ens, Murray, Can. J. Chem., 35, 170 (1957).
141. Widom, Philippe, Hobbs, J. Amer. Chem. Soc., 79,
1383 (1957).
142. Nickson, J. Amer. Chem. Soc., 79, 243 (1957).
143. Mecke, F u n c k, Z. Electrochem., 60, 1124 (1956).
144. Шигорин, Докунихин, ДАН СССР, 100, 323 (1955).
145. Brown, Rogers, J. Amer. Chem. Soc., 79, 577 (1957).
146. Ma cl ou, Henry, Compt. Rend. Acad. Sci. Paris, 224, 1494
(1957).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Батуев M. И., ДАН СССР, 52, 5 (1946); 53, 4 (1946); 21, 4 (1947).
Степанов Б. И., ФЖХ, 19, 507 (1945); 20, 407 (1946).
Шубин А. А., Изв. АН СССР (сер. физ.), 14, 442 (1950).
Шигорин Д. Н., ЖФХ, 23, 505 (1949); 24, 924, 932, 955 (1950);
Изв. АН СССР (сер. физ.), 24, 4 (1950); 27, 596 (1953).
Соколов Н. Д., Успехи физ. наук, 57, 2 (1955).
Шигорин Д. Н., Ш е м я к и н М. М., Щукина Л. А.,
Колосов М. Н., Менделевии Ф. А., ДАН СССР,
108, 673 (1956).
Глава 1
ПРОСТЫЕ ЭФИРЫ, ПЕРЕКИСИ И ОЗОНИДЫ
1. ВВЕДЕНИЕ
Единственной характерной особенностью спектров про-
стых эфиров, на основании которой их можно идентифици-
ровать в инфракрасной области, является очень интен-
сивная полоса поглощения, обусловленная валентными коле-
баниями по ординарной связи С—О. Было уже показано,
что у спиртов, т. е. у структур, содержащих группу СН2ОН,
эта полоса появляется вблизи 1050 см \ но она очень чув-
ствительна к изменениям строения прилежащей части
молекулы, так что даже такое разветвление цепи, как у вто-
ричных спиртов, вызывает смещение полосы до 1100 см'1.
Поэтому не следует ожидать, что для простых эфиров поло-
жение этой полосы будет постоянным, тем более, что в этом
случае будет проявляться взаимодействие между коле-
баниями С—О и О—R и изменение частоты будет обуслов-
ливаться природой заместителей, расположенных по обе
стороны указанных групп. Однако корреляции для отдель-
ных структур, как и для спиртов, возможны. Можно, напри-
мер, ожидать, что группа СН2—О—СН2 поглощает в значи-
тельно бол ее узком интервале, чем интервал 1150—1000 см'1,
который был первоначально указан Леконтом [1 ] как общий
для предельных простых эфиров. Хотя полосу такой струк-
туры обычно легко распознать по ее очень высокой интен-
сивности, появление ее следует рассматривать лишь как
указание на возможное присутствие такой структуры.
Это объясняется отчасти тем, что до настоящего времени
простые эфиры с разветвленными цепями и другие типы
эфиров еще недостаточно хорошо изучены и не исследовано
влияние структурных изменений на рассматриваемую
частоту; трудности возникают также в связи с тем, что эта
полоса перекрывается, например, сильными полосами вто-
ричных и третичных спиртов, перекисей и структурных
групп Р—О—С и Si—О—С.
11*
164
Часть 11
Примером соединений, у которых структурные измене-
ния вызывают значительное изменение частоты колебаний
группы С — О, являются соединения, содержащие группу
= С — О —. В случае фенола полоса смещается в сторону
высоких частот до 1259 см1, и такой же эффект наблюдает-
ся у сложных эфиров и кислот. Поэтому не удивительно,
что ароматические и непредельные простые эфиры также
имеют сильные полосы поглощения С—О вблизи 1250 см1.
Тот факт, что полоса в этой области обнаруживается у мно-
гих веществ, является дополнительной иллюстрацией труд-
ностей, встречающихся при использовании данной области
Для идентификации структур. В лучшем случае сильная
полоса вблизи 1250 см 1 является указанием на наличие
группы С—О с ненасыщенным углеродным атомом, а для
того, чтобы установить, входит ли эта группа в молекулу
сложного эфира, спирта или какого-либо другого соедине-
ния, нужно исследовать остальную часть спектра.
Другое затруднение возникает в результате того, что
на длину связи С — О влияют также положение и природа
заместителей в ароматическом кольце. Колтуп [2] нашел,
что частота валентных колебаний С — О для лета-заме-
щенных ароматических простых эфиров выше нормальной,
и в его таблицах для этих колебаний приводится значение
частоты 1300 см~1. Он указал также на присутствие полосы
средней интенсивности в интервале 1070—1000 см~1 в случае
моноарилэфиров типа СН2—О—С6Н5. Мы так же, как и дру-
гие авторы 130], в ряде случаев наблюдали эту полосу, воз-
никновение которой связано, возможно, с колебаниями
остатка молекулы СН2—О—. Однако в этой области появ-
ляется так много полос средней интенсивности, соответствую-
щих углерод-углеродным колебаниям и другим колебаниям
скелета, что эти данные нельзя использовать для анализов.
По поглощению циклические простые эфиры несколько
отличаются от эфиров с открытой цепью, поскольку кольцо
может иметь собственные колебания, которые, хотя и не
являются колебаниями С—О как таковыми, тем не менее
достаточно характеристичны для идентификации гетеро-
цикла. Для установления соответствующих корреляций
изучались соединения, содержащие эпоксигруппы, и раз-
личные типы циклических структур сахаров. Таким путем
можно, например, по характеристическим частотам, отно-
Г-л. 1. Простые эфиры, перекиси и озониды
165
сящимся к тетрагидропирановому и тетрагидрофурановому
кольцам, различить а- и |3-аномеры и производные фуранозы
[32—35].
К колебаниям перекисной цепочки —О—О— предполо-
жительно относят поглощение при 890—830 см~у, но надеж-
ность такого вывода вызывает некоторые сомнения, так как
найдено, что в этой области поглощают также другие струн-
ец /С
туры, например, скелет . Перекиси, содержащие
CZ О
группы —СО—О—О—, могут быть идентифицированы
также по характеристическим колебаниям карбонила (гл. 9).
Никаких удовлетворительных корреляций для озонидов
пока еще не установлено.
Рассматриваемые корреляции приведены в табл. 6.
Таблица 6
Частота, сл»-1
Диалкилэфиры —СН2—О—СН2—
Ароматические и другие простые эфи-
ры с группой =С—0— .......
Циклические эфиры
1. Эпсксиды ..................
2. Соединения с большими циклами
Алкильные перекиси..............
1150—1060 (оч. с.)
1270—1230 (оч. с.)
Вблизи 1250 (с.); вблизи 890,
транс; вблизи 830, цис
(ср.)
1140—1070 (оч. с.)
890—820 (оч. сл )
2. АЛИФАТИЧЕСКИЕ ПРОСТЫЕ ЭФИРЫ
Спектры ряда низших алифатических эфиров приведены
Барнсом и др. [3]. Точное положение полос С—О—С неука-
зано, но из спектров можно видеть, что все эти соединения
поглощают приблизительно в интервале 1150—1060 см1.
Эти же авторы опубликовали спектры аллилэтилэфира и ди-
аллилэфира, в которых полоса С—О—С занимает прибли-
зительно такое же положение, как и в спектрах насыщенных
166
Часть II
эфиров. Этот факт, по-видимому, свидетельствует о том,
что структура С = С — С — О —С не оказывает заметного
влияния на колебания С — О, что согласуется с данными
Леконта [1], согласно которым частоты валентных колеба-
ний С — О виниловых спиртов имеют по существу нормаль-
ные значения.
С другой стороны, сравнивая эфиры со спиртами, можно
ожидать, что разветвление цепи у углеродного атома, осу-
ществляющего эфирную связь, будет приводить к значитель-
ному изменению частоты колебаний С — О. В литературе
об этом нет почти никаких сведений, однако следует отме-
тить, что в спектре диизопропилового эфира, приведенном
Барнсом и др. [3], имеется триплет в области 1100—
1070 с.и1, тогда как диизоамилэфир [4], у которого развет-
вление начинается на большем расстоянии от связи С — О,
дает только одну сильную полосу при 1111 см Ч Чамлер
и Лейтнер [30, 36] приводят для предельных эфиров интер-
вал 1150—1080 см'1 и указывают, что спектры эфиров,
содержащих группу С — О — С — О — С, имеют в этом
же интервале интенсивный дублет. Бергманн и Пинхас
[37] изучили 18 кеталей и ацеталей, многие из которых
являются циклическими диоксаланами, и идентифицировали
серию из четырех полос в области 1200—1000 см'1, которую
они считают специфичной для группы С — О — С — О — С
и относят к различным взаимодействующим колебаниям
С — О. Однако Лагранж и Мастагли [38], рассмотрев дан-
ные корреляции, указали, что большинство из этих полос
появляется также у производных диоксана и что они не
всегда присутствуют в спектрах диоксаланов.
Кроме данных по указанным соединениям, в литературе
имеется очень мало сведений об инфракрасных спектрах
простых эфиров. Из соединений, обычно встречающихся
в промышленности, исследовались различные полигликоли,
моноэфиры и диэфиры этиленгликоля и диэтиленгликоля
и замещенные феноксиуксусных сложных эфиров. Мы ис-
следовали ди-, три- и различные полиэтиленгликоли, а так-
же метил-, этил- и бутилцеллозольвы и карбитолы и обна-
ружили, что все они дают очень широкие сильные полосы
приблизительно в пределах указанного интервала. Частота,
соответствующая полосе С — Оу феноксиуксусных слож-
ных эфиров, также является обычной, но при 1250 см'1
Гл. 7. Простые эфиры, перекиси и озониды
167
появляется еще полоса С — О сложноэфирной группировки.
Также не наблюдается отклонений от обычных значений
частот С — О у 0-хлорэтилэфира. Влияние замещения
галогеном у а-углеродного атома не изучалось. Девисон
[39] исследовал смещения полос спектра, происходящие при
кристаллизации полиэтиленгликоля, и сделал предполо-
жение, что две полосы С — О — С, появляющиеся при
1120 и 1061 сл"1, как и в случае диметилового эфира, соот-
ветствуют антисимметричным и симметричным валентным
колебаниям. Простые эфиры полиэтиленгликолей изуча-
лись также Чаковским и др. [40]; кроме того, были выпол-
нены исследования полиоксиметилена [41] и окиси поли-
пропилена [42].
На основании сказанного был предположительно при-
нят для этих соединений интервал частот 1150—1060 см~1.
Однако следует помнить, что это предположение осно-
вывается на спектральных данных ограниченного количе-
ства соединений, и вполне возможно, что интервал придет-
ся расширить, чтобы охватить другие типы алифатических
эфиров, например соединения, которые содержат группу
— СН — О — СН2— и т. д. Что касается интенсивности
полосы поглощения С — О — С, то она должна быть мень-
ше у высокомолекулярных веществ с небольшим числом
эфирных связей. У алкилацеталей смежная с кислородом
группа СН2, по-видимому, дает характеристическую полосу
вблизи 2825 сл"1 [57].
3. ДИАРИЛЭФИРЫ И АРИЛАЛКИЛЭФИРЫ
Данные, относящиеся к спектрам ароматических эфиров,
так же как и алифатических эфиров, приведены в работе
Барнса и др. [3]. Все эти спектры имеют сильную полосу
приблизительно в области 1270—1230 см-1; сравнение орто-,
мета- и пара-изомеров между собой показывает, в согласии с
данными Колтупа [2], что у мета-замещенных веществ эта по-
лоса имеет несколько большую частоту, чем у других веществ.
Работа Леконта [1! также подтверждает эти данные.
Кроме обычных ароматических эфиров, Леконт исследовал
соединения ряда СбЩСЦСНг^ОСсНа, где п имеет значе-
ния от 1 до 10. У всех этих соединении сильная полоса
вблизи 1250 сл!'1 не изменяет своего положения. Имеется
168
Часть 11
и вторая полоса в области 1150—1060 см'1, которая, веро-
ятно, обусловлена колебаниями СН2 — О —. Чамлер и
Лейтнер [30] также наблюдали обе эти полосы в спектрах
арилалкилэфиров; дифенилэфиры были изучены Кимото
[43]. Наши данные о спектрах ряда ароматических эфиров
согласуются с приведенными корреляциями. Общее коли-
чество исследованных веществ, как и в случае алифатиче-
ских эфиров, еще относительно невелико, и вполне возмож-
но, что частоты некоторых соединений, например нитро-
арилэфиров и других, выходят за указанные пределы
значений. Большая часть из немногих имеющихся в лите-
ратуре данных относится к диарилэфирам, а применение
указанных корреляций к виниловым спиртам ненадежно.
Например, частоты поглощения цис- и /лранс-бутиленилбу-
тилэфиров в этой области заметно отличаются от частот
арилэфиров, и наиболее интенсивная полоса у них появ-
ляется в интервале 1200—1100 см'1 [11]. Для немногих
изучавшихся веществ такого типа [30, 44] полоса колеба-
ний С — О наблюдалась в пределах интервала 1240—
1150 см'1. Имеются также некоторые доказательства того,
что на нее не влияет сопряжение двойных связей.
При использовании этих данных не следует забывать,
что фенолы, сложные эфиры, кислоты и другие соединения,
содержащие группу — С — О —, могут также поглощать
вблизи 1250 см'1.
4. ЦИКЛИЧЕСКИЕ ПРОСТЫЕ ЭФИРЫ
а. Эпоксиды. Исследователи, изучавшие окись этилена
[5—7, 45], отнесли к эпоксигруппе полосы при 865, 1165
и 1265 см'1. В связи с вопросами окисления растительных
масел и углеводородов эта группа представляет значитель-
ный интерес, и потому было сделано много попыток выяс-
нить возможность определения ее характеристических
полос поглощения. С целью определения последних в при-
веденных спектральных областях Филд, Коул и Вудфорд
[8] исследовали восемь довольно сложных эпоксидов,
а именно окись изобутилена, окись стирола и т. д. Хотя
они установили, что все перечисленные соединения дают
полосы в каждой из этих областей, они пришли к выводу,
что только полосу при 1250 см'1 можно с достаточной уве-
Гл. 7. Простые эфиры, перекиси и озониды
169
ревностью отнести к эпоксигруппе. Данная полоса у изучен-
ных веществ всегда находится в интервале 1260—1240 см'1.
Идентификация остальных полос, по мнению указанных
авторов, затруднительна из-за многих колебаний С — С
и маятниковых колебаний С — Н в этих молекулах.
Шрив и др. [9] также изучали этот вопрос и получили
спектры нескольких простых эпоксидов, а также эпокси-
производных некоторых 1,2-олефинов с длинной цепью.
Их результаты согласуются с результатами Филда и др.
[8], поскольку во всех случаях была найдена полоса в ин-
тервале 1260—1250 см'1. Следует отметить, что у более
простых соединений эта полоса сравнительно интенсивна,
а у соединений, у которых молярная концентрация эпокси-
групп значительно ниже, например у 1,2-эпокситетрадека-
на, она имеет среднюю или слабую интенсивность. У окиси
а-метилстирола эта полоса отсутствует, но Паттерсон [46]
обнаружил ее у 25 других эпоксидов. Гюнтард и др. [47]
также отметили отсутствие этой полосы у стероидэпоксидов.
Однако Шрив и др. [9] не считают, что эта полоса, относя-
щаяся, по их мнению, наряду с полосами при 1430 и 1136 см'1
к валентным колебаниям С — О, является всегда наиболее
характерной для эпоксидов. У всех исследованных соеди-
нений этого класса они нашли также полосы вблизи 910
и 830 см1, хотя вторая из них у 3,4-эпоксибутена-1, вероят-
но, под влиянием сопряженной двойной связи была смещена
к 818 см1. Кроме того, изучение эпоксипроизводных жир-
ных кислот, сложных эфиров и спиртов с одной ненасыщен-
ной связью показало, что спектры транс-эпоксидов качест-
венно сходны со спектрами предельных соединений, но име-
ют новую полосу вблизи 890 см'1, в то время как все цис-
эпоксиды дают полосу около 830 см'1. Они предположи-
тельно отнесли эти полосы к колебаниям кольца эпокси-
группы цис- и транс-соединений. Эти низкочастотные поло-
сы поглощения изучали также Паттерсон [46] и Гюнтард
и др. [47]. У соединений, изученных Паттерсоном, были
найдены две полосы в интервалах 950—863 и 864—786 см'1,
расположенные, однако, чаще всего вблизи 910 и 830 см'1,
что хорошо согласуется с ранее опубликованными данными.
С другой стороны, у стероидэпоксидов Гюнтард и др. нашли,
что полосы эпоксигруппы у одних соединений лежат между
900 и 800 см'1, а у других — между 1050 и 1035 см'1.
170
Часть II
Пока не будет получено достаточное число спектров,
этими данными следует пользоваться с большой осторож-
ностью, так как многие другие группы также поглощают
при 830 и 890 см'1, и применять эти корреляции к соеди-
нениям, которые не вполне сходны с описанными, можно
лишь весьма предположительно.
Туот и Баршевиц [12] сделали предположение о том, что
поглощение в области обертонов при 7042 см'1 может быть
характеристичным для эпоксидов, однако эта область иссле-
довалась у очень немногих веществ и требует дальнейшего
изучения.
б. Соединения с четырех-, пяти- и шестичленными
кольцами. У больших колец, напряжение которых меньше,
частота валентных колебаний группы С — О — С должна
быть близка к нормальному значению. Шрив и др. [9]
привели спектры тетрагидропирана и диоксана, в которых
имеется по нескольку полос в интервале 1124—1030 см'1.
Полоса 1100 см 1 тетрагидропирана была отнесена к коле-
баниям С — О [31 ]. Чамлер и др. [30, 48] исследовали
несколько сходных соединений и нашли, что группа
С—О—С—О—С приводит к появлению дублета в том же
интервале частот. Изучение диоксанов и диоксаланов под-
твердило образование сложной структуры полос, однако
число характеристических полос в обоих этих случаях опре-
делить неудалось [37, 38]. У непредельных циклических сое-
динений с группой = С — О — С = полоса смещается в сто-
рону больших частот. Тетрагидрофуран, имеющий пяти-
членный цикл, поглощает при 1076 см'1; Барроу и Серлс
[311 нашли, что пять родственных ему соединений также
поглощают в интервале 1100—1075 см'1. У других гетеро-
циклических систем, например у азоксимов и оксадиазолов,
обнаружена сильная полоса при 1030 см'1, которая отсут-
ствует у фуразанов [10] и, вероятно, обусловлена связью
С—О. В случае четырехчленных циклов влияние напряже-
ния становится более заметным, и полоса поглощения
С—О у окиси триметилена появляется в области 980—
970 см'1 [31]. Это косвенно подтверждает правильность
интервала 890—830 см'1, предложенного для эпоксидов,
у которых поглощения следует ожидать при более низких
частотах.
Гл. 7. Простые эфиры, перекиси и озониды
171
Однако имеется слишком мало материала для того, что-
бы можно было установить более точные корреляции. Можно
сделать только обобщение, что эфирная связь в соединениях
этого типа с большими кольцами, по-видимому, поглощает
примерно при той же частоте, что и в случае соединений
с открытой цепью. Джонс и Сандорфи [491 составили по ли-
тературным данным полезную сводку о ряде циклических
простых эфиров.
5. ПЕРЕКИСИ
Возможность определения перекисных и гидроперекис-
ных структур методом инфракрасной спектроскопии пред-
ставляет практический интерес в связи с выяснением меха-
низма окислительных процессов в углеводородах. Теорети-
чески этот вопрос обсуждался Шеппардом [13], который
указал, что наличие каких-либо достаточно характеристиче-
ских полос колебаний О—Оу перекисей типа R—О—О—R'
является маловероятным, так как эти колебания довольно
симметричны и не связаны с большим изменением диполь-
ного момента; поэтому следует ожидать, что соответствую-
щие полосы в инфракрасном спектре будут слабыми. Более
того, массы и силовые постоянные для группы О—О на-
столько близки к соответствующим величинам для групп
С—О и С—С, что появление достаточно характеристической
полосы весьма сомнительно. У гидроперекисей, у которых
группа О—О находится в конце цепи, вероятность этого
несколько больше, но даже если это и так, колебания О—О
будут вносить вклад в колебания скелета всей молекулы
и поэтому частота будет сильно зависеть от природы заме-
щающих групп. Однако Шеппард высказал предположение,
что для некоторых групп заместителей корреляции- могут
быть установлены аналогично тому, как они были установ-
лены, в частности, для первичных, вторичных и третичных
спиртов, но при этом необходимо изучить большой ряд сое-
динений различных типов.
Область, в которой можно ожидать каких-либо колеба-
ний скелета О—О, легко определить из спектра перекиси
водорода, который изучался различными исследователями
[14, 151. Жигер [14] показал, что положение полосы D2O2
существенно не отличается от установленного для Н2О2,
т. е. близко к 877 см~\ и потому эту полосу можно вполне
172
Часть 11
обоснованно приписать валентным колебаниям О—О. Более
того, такое отнесение соответствует длине связи, которая
получена из электронографических данных.
Эти исследования были продолжены Лидбитером [16],
который получил инфракрасный спектр и спектр комбина-
ционного рассеяния перекиси, образующейся в этиловом
эфире, а также спектры комбинационного рассеяния соот-
ветствующих а-монооксиперекисей и а-диоксиперекисей
и перекиси бензоила. Во всех случаях он наблюдал доста-
точно интенсивную полосу в интервале 883—881 см"1.
В противоположность толкованиям Шеппарда он отмечает,
что при переходе от Н2О2 к более сложным молекулам
частота колебаний О—О изменяется в меньшей мере, чем
частота колебаний С—С углеводородов. Однако количество
изученных веществ весьма ограничено, и в отношении
чистоты по крайней мере одного из них — перекиси бен-
зоила— имеются сомнения, так как Шрив и др. [17]
не обнаружили полосы при 883 см"1 в инфракрасном
спектре этого соединения, что соответствует и нашим
наблюдениям.
В других работах было показано, что алифатические
вещества такого типа поглощают в области 870 см"1, но
для них, как и следовало ожидать, характерный интервал
частот сравнительно широк. Минков [18, 50] изучил 30 чи-
стых перекисей, гидроперекисей и перкислот и во всех
случаях обнаружил полосу в интервале 890—830 см"1.
Предположительно приписав эту полосу колебаниям груп-
пы О—О, он указал, что интенсивность ее невелика и что она
может быть замаскирована или спутана с некоторыми поло-
сами сравнимой интенсивности, соответствующими колеба-
ниям скелета больших молекул. Были изучены только
алифатические соединения и в основном метил-, этил- и бу-
тилпроизводные. Однако, несмотря на низкую интенсив-
ность полосы при 813 см"1, Релей и др. [20] использовали
ее для определения перекиси ди-трет-бутила, содержащей-
ся в mpem-бутиловом спирте.
Правильность такого отнесения была подтверждена ре-
зультатами систематических исследований рядов соедине-
ний, содержащих связь О—О [17, 19, 51—53], а также дан-
ными наблюдений отдельных соединений [54—56]. Однако,
как и в ранее рассмотренных случаях, полоса была слабой
Гл. 7. Простые эфиры, перекиси и озониды
173
и идентификация ее представлялась трудной. Это можно
показать на примере работы Уильямса и Мошера [51],
которые сравнивали спектры гидроперекисей алкилов со
спектрами соответствующих спиртов. Хотя в рассматривае-
мой области между ними были найдены заметные различия,
многие спирты все же дают здесь собственное поглощение.
Как упоминалось выше, эта корреляция не пригодна для
перекиси бензоила, которая сильно поглощает вблизи
1000 см'1. Шрив и др. 117] указывают, что перекиси фталои-
ла, n-хлорбензоила и бензальдегида имеют примерно такую
же полосу поглощения и авторы полагают, что эта полоса
является характерной для колебаний О—О ароматических
перекисей. Мы исследовали пербензойную кислоту и обна-
ружили полосы поглощения вблизи 1030 и 880 см'1.
Таким образом, полоса поглощения, соответствующая
колебаниям группы О—О алифатических соединений, нахо-
дится где-то в интервале 890—820 см'1, а для ароматических
перекисей, возможно, вблизи 1000 см'1. Полосы поглощения
соответствующих перекисей и гидроперекисей должны нахо-
диться в пределах указанного интервала, но несколько отли-
чаться по своему положению. Однако ввиду упоминавших-
ся затруднений едва ли имеет смысл пытаться подразде-
лить этот интервал на более узкие участки. Было бы очень
рискованно отнести, например, полосу вблизи 870 см'1
у неизвестного соединения к перекисной связи, если только
нет дополнительных надежных доказательств такой струк-
туры. Это стало особенно ясно после того, как Филпотте
С\ /С
и Тейн [21] показали, что группа также дает силь-
(У \о
ную полосу поглощения в области 920—800 см'1. Такое
заключение было сделано на основании сравнения спектров
нескольких типов третичных перекисей, гидроперекисей
и спиртов, которое показало, что полоса, первоначально
отнесенная к группе О—О, находится и в спектрах спиртов.
Мы со своей стороны также заметили сильное поглощение
при 901 см'1 у mpem-бутилата алюминия, отсутствующее
у изопропилата алюминия. Эго не ставит под вопрос пра-
вильность отнесения колебаний О—О, но свидетельствует
о необходимости особого внимания при использовании
этой корреляции.
i?4
Часть II
Существует также ряд других возможностей для распо-
знавания перекисных и близких структур. Например, весь-
ма характеристична частота перекисного карбонила (гл. 9).
В то же время можно ожидать, что типичные частоты группы
ОН, связанной с кислородом, у перкислот и гидроперекисей
значительно отличаются от таковых у карбоновых кислот
и спиртов, позволяя тем самым идентифицировать их.
В этом направлении проводилось много работ, но чтобы
можно было прийти к определенным выводам, необходимы
дальнейшие подробные исследования. Вероятно, наиболее
успешно удастся использовать частоту валентных колеба-
ний ОН гидроперекисей. Если учесть электроотрицатель-
ности, то следует ожидать, что эта частота значительно
ниже, чем у спиртов; Карякин и др. [52] на основании ис-
следования растворов очень ограниченного числа веществ
считают ее равной 3450 см'1. Некоторым косвенным под-
тверждением этого следует считать тот факт, что в жидкой
фазе частота валентных колебаний ОН перекисных соеди-
нений значительно ниже, чем у соответствующих спиртов
[51J. Эти рассуждения не применимы, однако, к перки-
слотам, которые образуют системы с внутримолекулярными
связями [53, 56] и у которых колебания ОН лежат вблизи
3280 см'1.
Был сделан ряд других попыток отнесения деформаци-
онных колебаний ОН и валентных колебаний С—О [19, 52,
53], однако сравнение со спиртами [51 ] показало, что полу-
ченные корреляции менее удачны. У перкислот частота де-
формационных колебаний ОН появляется, как и у карбо-
новых кислот, вблизи 950 см'1, хотя интенсивность погло-
щения перкислот значительно слабее [53]. Были получены
некоторые другие корреляции для гидроперекисей [51]
и близких соединений, таких, как перкарбэнагы и сложные
эфиры перкислот* [50], но для их подтверждения необхо-
димы более детальные исследования.
6. ОЗОНИДЫ
Впервые методы инфракрасной спектроскопии для
исследования образования, устойчивости и разложения
озонидов были применены Бринером и его сотрудниками
* Имеются в виду ацилалкилперекиси.—Прим. ред.
Гл. 7. Простые эфиры, перекиси и озониды 175
[22—24]. При предварительном исследовании они пропу-
скали озон через растворы ненасыщенных соединений в че-
тыреххлористом углероде и наблюдали в спектрах измене-
ния, соответствующие различным степеням озонолиза.
Вначале был сделан вывод о поглощении озонидами в обла-
сти частот карбонила, однако последующие работы этих
авторов [25, 26], а также результаты, полученные Криге
и др. [27], показали, что данное поглощение обусловлено
продуктами разложения. В последней из цитированных ра-
бот было изучено около 18 озонидов различных типов и по-
казано, что у большинства из них после озонолиза появ-
ляется полоса умеренной интенсивности в интервале 1064—
1042 см1. Эта полоса, по-видимому, соответствует валент-
ным колебаниям С—О озонидов, что было подтверждено
Бринером и Дальвиком [25, 2б]; Гарвин и Шуберт [28]
также наблюдали озонидное поглощение в этой области.
Значение данной корреляции, однако, ограничено тем, что
поблизости лежат полосы поглощения валентных колеба-
ний С—О других типов.
Были предприняты также некоторые исследования дей-
ствия озона на эластомеры, а именно на натуральный кау-
чук, неопрен и др. [8]. В этих случаях не было получено
никаких указаний на присутствие озонидов, но были обна-
ружены продукты разложения, которые характеризовались
полосами поглощения ОН и карбонила.
ЛИТЕРАТУРА
1. L е с о m t е, Traite de chimie organique, ed. Grignard et Baud,
Masson et Cie, Paris, 2, 143 (1936).
2. Col th up, J. Opt. Soc. Amer., 40, 397 (1950).
3. Barnes, Gore, Liddel, Van Zandt, Williams,
Infra-red Spectroscopy, Reinhold, 1944.
4. Randall, Fowler, Fuson, Dangl, Infra-red Deter-
mination of Organic Structures, Van Nostrand, 1949.
5. Герцберг, Колебательные и вращательные спектры много-
атомных молекул, Издатинлит, М, 1949, стр. 365.
6. Bonner, J. Chem. Phys., 5, 704 (1937).
7. Thompson, Cave, Trans. Faraday Soc., 47, 946 (1951).
8. F i e 1 d, С о 1 e, Woodford, J. Chem. Phys., 18, 1298
(1950).
9. Shreve, Heether, Knight, Swern, Analyt. Chem ,
23, 277 (1951).
176
Часть 11
10. М i 1 о п е, В о г е 1 1 о, Gazz. Chim. Italia, 81, 368 (1951).
11, И а 1 1, Philpotts, Stern, Thain, J. Chem. Soc..
1951 3341.
12. T u о t, В a r c h e w i t z, Bull. Soc. chim. (France), 1950, 851.
13. Sheppard, Discuss. Faraday Soc., 9, 322 (1950).
14. Giguere, J. Chem. Phys., 18, 88 (1950).
15. Taylor, J. Chem. Phys., 18, 898 (1950).
16. L e a d b e a t e r, Compt. rend. Acad. sci. Paris, 230, 829 (1950).
17. Shreve, Heether, Knight, Swern, Analyt. Chem.,
23, 282 (1951).
18. M i n к о f f, Discuss. Faraday Soc., 9, 320 (1950).
19. Whitcomb, Moorhead, Sha rp, Symposium on Mole-
cular Structure and Spectrography, Ohio, 1950.
20. Raley, Rust, V a u g h a n, J. Amer. Chem. Soc., 70, 1336
(1948).
21. P h i 1 p о t t s, Thain, Analyt. Chem., 24, 638 (1952).
22. В r i n e r, S u s z, D a 1 1 w i g k, Arch. Sci. Phys. Nat., 4,
199 (1951).
23. S u s z, Da 1 1 wigk, В r i n e r, Arch. Sci. Phys. Nat., 4,
202 (1951).
24. В r i n e r, Susz, Da 11 wigk, Compt. rend. Acad. Sci.
Paris 944 1Q49
25. Br i ner, D’a 1 1 w i g k, Helv. Chim. Acta. 39, 1446, 1826
(1956).
26. В r i n e r, Dall wigk, Compt. Rend. Acad. Sci. Paris, 243,
630 (1956).
27. Cr i eg ее, К er cko w, Z i n к e, Chem. Ber., 88, 1878 (1955)
28. Garvin, Schubert, J. Phys. Chem., 60, 807 (1956).
29. Allison, Stanley, Analyt. Chem., 29, 630 (1952).
30. Tschamler, Leutner, Monatsh., 83, 1502 (1952).
31. Barrow, Searles, J. Amer. Chem. Soc., 75, 1175 (1953).
32. Barker, Bourne, Stacey, WhiHen, J. Chem. Soc.,
1954, 171.
33. Barker, Bourne, Stephens, Whiffen, J. Chem.
Soc., 1954, 3468, 4211.
34. Barker, Stephens, J. Chem. Soc., 1954, 4550.
35. W h i s t 1 e r, House, Analyt. Chem., 25, 1463 (1953).
36. Tschamler, Spectrochim. Acta, 6, 95 (1953).
37. Bergmann, Pinchas, Rec. trav. chim., 71, 161 (1952).
38. Lagrange, Mastagli, Compt. Rend. Acad. Sci. Paris,
241, 1947 (1955).
39. Davison, J. Chem. Soc., 1955, 3270.
40. Chakhovskoy, Martin, Van Neche 1, Bull. Soc.
Chim. Beiges, 65, 453 (1956).
41. Philpotts, Evans, Sheppard, Trans. Faraday Soc.
51, 1051 (1955).
42. Price, Osgan, J. Amer. Chem. Soc., 78, 4787 (1956).
43. К i m о t o, J. Pharm. Soc. Japan, 75, 763 (1955).
44. M e a к i n s, J. Chem. Soc., 1953, 4170.
45. Lord, Nolin, J. Chem. Phys., 24, 656 (1956).
46. Patterson, Analyt. Chem., 26, 823 (1954).
Гл. 7. Простые эфиры, перекиси и озониды 177
47. Giinthard, Heusser, Furst, Helv. Chim. Acta, 36,
1900 (1953).
48. Tschamler, V о e t t e r, Monatsh., 83, 303, 1228 (1952).
49. Джонс, Сандорфи, Применение спектроскопии в химии,
Издатинлит, М., 1959, стр. 353.
50. М i n к о f f, Proc. Roy. Soc., A224, 176 (1954).
51. W i 1 1 i a m.s, Mosher, Analyt. Chem., 27, 517 (1955).
52. Карякин, Никитин, Иванов, ЖФХ, 27, 1856 (1953).
53. Swern, Witnauer, Eddy, Parker, J. Amer. Chem.
Soc, 77, 5537 (1955).
54. Criegee, Pauli g, Chem. Ber., 88, 712 (1955).
55. Milos, Mageli, J. Amer. Chem. Soc., 75, 5970 (1953).
56. Giguere, Olmos, Can J. Chem., 30, 821 (1952).
57. В r u g e I, Oster, Angew. Chem , 68, 441 (19561.
12 л. Беллами
Глава 8
ГАЛОГЕНАНГИДРИДЫ КИСЛОТ, КАРБОНАТЫ,
АНГИДРИДЫ КИСЛОТ И КАРБОНИЛЫ МЕТАЛЛОВ
1. ВВЕДЕНИЕ
Ни один из классов соединений, рассматриваемых в этой
главе, не изучался систематически, однако небольшие груп-
пы соединений были исследованы, и полученные результаты
оказались достаточно надежны для установления соответ-
ствующих корреляций.
Рассматриваемые корреляции приведены в табл. 7.
Таблица У
Частота, см-1
Колебания С=О
(все полосы сильные)
Галогенангидриды кислот ..........
Карбонаты.........................
Ангидриды кислот (открытая цепь)
Циклические ангидриды (пятичленное
кольцо)...........................
Перекиси жирного ряда ............
Перекиси ароматического ряда . . .
Колебания С—О
Ангидриды кислот (открытая цепь)
Циклические ангидриды кислот . .
Колебания С = О
1815—17703
1780—1740
1850—1800 и 1790—17400
1870—1820 и 1800—17505
1820—1810 и 1800—1780
1805—1780 и 1785—1755
1170—1050 (с.)
1300—1200 (с.)
Вблизи 2050 и 2040 (ср.)
а При наличии сопряжения полоса лежит в нижней части этого интервала.
0 Сопряжение понижает частоту в каждом случае на 20 см-1.
Гл. 8. Галогенангидриды кислот, карбонаты и др.
179
Все интервалы частот колебаний карбонила являются
довольно широкими, но это объясняется ограниченным
количеством имеющихся данных; установление более
точных корреляций требует изучения большего числа ве-
ществ.
2. ГАЛОГЕНАНГИДРИДЫ КИСЛОТ
У сложных эфиров, кислот и кетонов замещение хлором
или бромом в a-положении по отношению к карбонильной
группе вызывает смещение частоты колебаний С=О в сто-
рону более высоких значений. При непосредственном при-
соединении галогена к карбонильной группе, как и следо-
вало ожидать, имеет место значительно большее смещение
в том же направлении. Газообразный фосген поглощает при
1828 см'1, а хлористый ацетил [1, 2] и бромистый ацетил —
соответственно при 1802 и 1812 см'1. Увеличение размеров
радикала не оказывает значительного влияния на частоту;
так, хлористый фенилацетил поглощает при 1802 см'1
[1, 2], тогда как, по нашим данным, значение частоты для
хлористого капроила составляет 1790 см'1. Аналогичные
результаты получены при исследовании спектров комбина-
ционного рассеяния хлористого оксалила (1776 см'1)
и хлористого пропионила (1786 см'1).
Следовало ожидать, что у галогенангидридов сопряже-
ние галогена с двойной связью в аф-положении или с арома-
тическим радикалом также должно привести к уменьшению
частоты СО; это действительно наблюдается для хлористого
бензоила, который поглощает при 1773 см'1 [1, 21. Однако
данное соединение дает вторую полосу СО при 1736 см'1,
происхождение которой неизвестно. В литературе имеются
сведения об инфракрасных спектрах очень немногих фтор-
ангидридов кислот; из-за высокой электроотрицательности
фтора они поглощают при значительно более высоких
частотах. Например, соединение COF2 [18] поглощает при
1928 см'1, причем эта частота одна из самых высоких
из известных частот карбонила, пары же фтористого аце-
тила поглощают при 1872 см'1. Замещение галогенами в
a-положении также приводит к повышению частоты карбо-
нила; так, фтористый трифторацетил поглощает при
1901 см'1. При меньшем замещении галогенами—у хло-
ристого хлорацетила и других аналогичных соединений,
12*
180
Ч а с т ь II
изученных Мидзусимой и др. [19],— возможность поворот-
ной изомерии приводит к появлению двух карбонильных
частот. В общем случае одна из этой пары частот выше, чем
карбонильная частота хлористого ацетила, а другая —
ниже [20]. Более высокая частота отвечает изомеру, у кото-
рого атом хлора расположен в пространстве ближе к атому
кислорода, тогда как меньшая соответствует структуре,
в которой два атома хлора расположены близко друг к другу.
В последнем случае влияние внутреннего поля по всей
вероятности приводит к уменьшению эффективной электро-
отрицательности атомов галогена, в результате чего проис-
ходит уменьшение частоты. Частоты ряда галогенангидри-
дов жирных кислот и их галогенпроизводных были рассмот-
рены Беллами и Уильямсом [20], а фторпроизводныегалоген-
ангидридов также изучались Хасзельдином [17], Хастедом
и Альбрехтом [16] и Вибленом [21].
Приведенные выше значения частот, кроме тех, о которых
было специально оговорено, получены при исследовании
растворов или жидких веществ. В газообразном состоянии
наблюдалось увеличение частоты приблизительно на 20 см'1
[3], указывающее на то, что в жидкости имеют место какие-
то формы ассоциации молекул.
3. КАРБОНАТЫ И ХЛОРФОРМИАТЫ
Данные об органических карбонатах и хлорформиатах
приведены лишь в статье Хейлса, Джонса и Кинестона [41]
и в работе, где сообщаются подробные сведения о корреля-
циях, относящихся к этиленкарбонату [42]. Естественно
было ожидать, что влияние двух атомов кислорода у кар-
бонатов приведет к повышению частоты поглощения кар-
бонильной группы по сравнению с частотой для сложных
эфиров и что аналогичное повышение будет иметь место
у хлорформиатов.
Эго было подтверждено экспериментально. Диацилкар-
бонаты ROCOOR поглощают в интервале 1760—1740 см'1
[41 ], причем точное значение частоты отчасти определяется
стереоизомерной формой. Дифенилкарбонат поглощает
выше, а именно при 1775 см'1, что типично для виниловых
сложных эфиров. Замещение галогенами в радикалах R
еще больше повышает частоту колебаний карбонила, и
Гл. 8 Галогенангидриды кислот, карбонаты и др.
181
CCI3OCOOCCI3, например, поглощает при 1832 см'1.
У циклических карбонатов на остальные эффекты наклады-
вается влияние напряжения кольца, и у изученных [41,42]
систем с пятичленным кольцом частоты колебаний карбо-
нила попадают в интервал 183С—1800 см'1. Эти значения
изменяются с изменением агрегатного состояния; в качестве
примера можно привести этиленкарбонат [42], который
в парах поглощает при 1870 см'1, в растворах в неполяр-
ных растворителях — в интервале 1830—1820 см'1 и в жид-
ком состоянии — при 1805 см'1. В некоторых случаях
появляется вторая полоса поглощения вблизи 1780 см'1,
которая, как полагают, отвечает обертону.
Как циклические карбонаты, так и карбонаты с открытой
цепью дают, кроме того, сильные полосы валентных коле-
баний С—О [41]. У соединений с открытой цепью их поло-
жение приблизительно соответствует положению аналогич-
ных полос сложных эфиров, но у систем с пятичленным
кольцом наблюдается некоторое смещение под влиянием
напряжения кольца.
Хлорформиаты поглощают при несколько более высоких
частотах, чем карбонаты. Их полоса С=О обычно лежит
между 1780 и 1770 см'1. Она тоже смещается в сторону более
высоких частот при замещении галогенами в а-положении,
появляясь у производных, содержащих группу СС13, при
1806 см'1.
4. АНГИДРИДЫ КИСЛОТ
Систематического изучения спектров поглощения ангид-
ридов не проводилось, и в отдельных работах имеются
данные по исследованию лишь одного-двух веществ. Эти
данные трудно сравнивать между собой, так как изучавшие-
ся вещества часто находились в различных физических
состояниях. Однако установлено, что все ангидриды дают
две карбонильные полосы поглощения. Их положение
в спектре и относительно друг друга, а также интенсивность
зависят от того, сопряжена ли карбонильная группа и вхо-
дит ли она в состав напряженного пятичленного кольца.
Рендалл и др. [1] обратили внимание на тот факт, что ука-
занные две полосы всегда находятся приблизительно на оди-
наковом расстоянии друг от друга (около 60 сл'1); это слу-
жит, несмотря на имеющиеся исключения, очень полезным
182
Часть II
ориентиром для их идентификации. Вещества, молекулы
которых не являются напряженными, дают следующие
типичные значения частот:
Уксусный ангидрид [1]
Фенилуксусный ангидрид
Капроновый ангидрид [4]
Глутаровый ангидрид [6]
1824 и 1748 сл-1
1808 и 1745 »
1825 и 1763 »
1802 и 1761 »
аф-Сопряжение приводит к понижению этих значений
на 20—40 см'1. Кротоновый ангидрид 14] поглощает вблизи
1780 и 1725 сма у бензоилбензоата эти полосы, по нашим
данным, появляются при 1789 и 1727 см'1. Из приведенных
значений видно, что расстояние между двумя полосами,
несмотря на изменения отдельных частот, довольно постоян-
но. Факторы, которые определяют взаимное положение этих
полос, были рассмотрены Куком [23].
У циклических соединений, у которых группы СО участ-
вуют в образовании пятичленного кольца, влияние напря-
жения кольца приводит к смещению полосы в сторону более
высоких частот, которое аналогично соответствующему
смещению у циклических кетонов и лактонов с пятичлен-
ными кольцами. Янтарный ангидрид [1], например, погло-
щает при 1865 и 1782 см'1. Однако сопряжение вызывает
некоторое понижение этих значений у циклических веществ,
так что отличить а,(3-ненасыщенные циклические ангидриды
от насыщенных ангидридов с открытой цепью на основе
только карбонильных полос поглощения не всегда возмож-
но. Типичные значения частот для ненасыщенных цикличе-
ских соединений составляют 1845 и 1775 см'1 у фталевого
ангидрида и 1848 и 1790 см'1 у малеинового ангидрида [4].
Ангидрид нафталин-1,2-дикарбоновой кислоты и некоторые
его производные ]7] поглощают при 1848—1845 и 1783—
1779 см'1. В случае циклических ангидридов с шестичлен-
ным кольцом, конденсированным с ароматическими систе-
мами, было зарегистрировано значительно меньшее расстоя-
ние (34 см'1) между двумя частотами, которые сами оказа-
лись ниже обычных величин (1770 и 1736 см'1) [22]. Другие
данные об отдельных ангидридах приведены в работах
Уокера [24], Ку [25], Сторка и Бреслова [26] и Хохштейна
и др. [27]. В последней даны сведения об ангидриде 3-меток-
си-6-метилпиромеллитовой кислоты с двумя ангидридными
Гл 8. Галогенангидриды кислот, карбонаты и др. 183
системами при одном и том же ароматическом кольце, бла-
годаря чему появляются четыре карбонильных частоты.
Замещение фтором в a-положении приводит к обычному для
карбонильных полос смещению обеих частот вверх. Так
перфторуксусный ангидрид поглощает при 1884 и 1818 см'1
[28].
Обнаружение ангидридами и перекисями (см. ниже)
двух карбонильных частот представляет собой любопытную
загадку, на которую до сих пор еше нет точного ответа.
Обычным и наиболее вероятным объяснением является то,
что при взаимодействии колебаний двух карбонильных
групп происходит резонансное расщепление. Однако при
этом объяснении остается неясным, почему низшая из двух
карбонильных частот во многих случаях имеет приблизи-
тельно то же значение, что и частота соответствующих слож-
ных эфиров и лактонов, и почему в отдельных случаях у ан-
гидридов жирных кислот с длинной цепью, растворенных
в полиэтилене, появляется только одна полоса поглощения,
имеющая меньшую частоту (1757 сл"1) со слабыми плечами
при еще более низких частотах [29].
Изменение интенсивности также происходит путем, ко-
торый нельзя предугадать. У большинства ангидридов,
в отличие от перекисей, полоса с более высокой частотой
интенсивнее, чем полоса с низшей частотой [15]. Однако
в некоторых случаях, например у фталевого ангидрида [4]
и у некоторых замещенных ангидрида нафталин-1,2-дикар-
боновой кислоты 17], полоса с низшей частотой сильнее.
Для выяснения этого явления нужны дальнейшие иссле-
дования.
Кроме полосы поглощения карбонила, ангидриды дают
также сильные полосы, обусловленные валентными коле-
баниями С—О—С. Эти полосы можно обычно идентифици-
ровать по их высокой интенсивности, но, как и в случае
простых эфиров, интервал частот, в пределах которого они
появляются, довольно широк. Колтуп [5] приводит следую-
щие интервалы: 1175—1045 см'1 для веществ с открытой
цепью и 1310 —1210 см'1 для циклических веществ
с напряженным кольцом. Эти полосы поглощения представ-
ляются полезными для получения негативных доказательств,
так как отсутствие ангидрида вполне доказывается отсут-
ствием сильных полос в каждой из указанных областей.
184
Часть II
Напротив, появление таких полос еще не является доказа-
тельством присутствия ангидрида, так как сильные полосы
в рассматриваемых областях могут давать многие другие
типы соединений.
5. ПЕРЕКИСИ
Полосы поглощения группы СО у перекисей, содержащих
структуру СО—О—О—СО, очень похожи на такие же поло-
сы ангидридов. Во всех случаях у перекисей появляются
полосы, частоты которых сравнимы с частотами соответ-
ствующих полос ангидридов, но расстояние между двумя
полосами перекисей составляет немногим более 30 слг1,
тогда как соответствующая величина для ангидридов равна
60 см'1.
Девисон [15] исследовал ряд жирных, ароматических
и несимметричных (смешанных) перекисей и сравнил полу-
ченные значения частот. Семь жирных перекисей обнару-
жили поглощение в интервалах 1820—1811 и 1796—1785c.it1
со средним расстоянием между полосами в 25 см'1. Введе-
ние двух ароматических групп вызывает понижение обеих
частот, как этого и следовало ожидать, хотя расстояние
между ними остается приблизительно таким же.
Для одиннадцати ароматических перекисей были найде-
ны полосы поглощения в интервалах 1805—1780 и 1783—
1758 см'1. У несимметричных веществ, содержащих одну
арильную и одну алкильную группы, среднее расстояние
между полосами групп СО больше нормального, что обу-
словлено сопряжением арильной группы с ближайшей кар-
бонильной группой. Однако этот эффект недостаточен для
того, чтобы расстояние между полосами достигло такой же
величины, как и у ангидридов.
Небольшое число перкислот и их сложных эфиров было
исследовано Девисоном [15] и Сверном и др. [30]; кроме
того, несколько спектров получил Минков [31], который,
однако, не сделал никаких выводов о карбонильных часто-
тах поглощения. Эти соединения дают только одну полосу
поглощения карбонила во всем интервале 1790—1740 см'1.
Девисон нашел, что частота варьирует гораздо больше, чем
согласно данным Сверна и др., которые у значительного
числа пер кислот наблюдал и поглощение при 1747—1748 см' ’.
Гл. 8. Галогенангидриды кислот, карбонаты и др. 185
Частота этого поглощения заметно не изменяется при пере-
ходе от твердого вещества к раствору, что подтверждает
существование у этих кислот образований с внутримолеку-
лярными связями [32].
6. КАРБОНИЛЫ МЕТАЛЛОВ
Спектры карбонилов металлов интересны прежде всего
тем, что они помогают выяснить природу углерод-кислород-
ной и металл-углеродной связей. Они почти не имеют прак-
тического применения, тем не менее результаты немногих
исследований в этой области будут кратко рассмотрены
ниже.
Окись углерода поглощает при 2143 см1, т. е. в области,
связываемой обычно с колебаниями по тройной связи [8],
и в настоящее время является общепринятым представле-
ние о том, что это соединение существует по крайней мере
частично в виде структуры с тройной связью. Однако в отно-
шении карбонилов металлов существуют серьезные разно-
гласия по поводу того, имеют ли они структуру X—С=О
или Х=С—О. Кроуфор [9, 10] исследовал карбонил никеля
и показал, что он дает полосы поглощения при 2039 и
2050 сл"1. Ряд других карбонилов, в том числе Fe(CO)5,
Fe3(CO)12 и Fe2(CO)9, был изучен Шелайном [11, 12] и Ше-
лайном и Питцером [13]. Каждое из этих соединений дает
аналогичную карбонилу никеля пару полос, a Fe3(CO)12
и Fe2(CO)9 дают, кроме того, полосу вблизи 1830 сл"1.
Для карбонилов марганца, рения [33, 34] и хрома [35]
обнаруживается поглощение только в области 2000 сл"1,
а для дикобальтоктакарбонила и Со4(СО)12 [36, 37] — до-
полнительное поглощение вблизи 1850 сл"1.
Появление полос поглощения в области частот колеба-
ний тройных связей рассматривается Шелайном как дока-
зательство существования в карбонилах металлов струк-
туры —С^ О; однако этот довод не является достаточно убе-
дительным, так как многие структуры типа Х=С=Х также
дают характеристические полосы ближе к области колебаний
обычных тройных связей, чем к области колебаний двой-
ных связей. Имеется дополнительное доказательство нали-
чия у этих веществ структуры —С^О, так как силовые
постоянные, вычисленные по частотам колебаний металд-
186
Часть II
углеродных связей [38], соответствуют тем значениям, кото-
рые получены для алкилметаллов, структура которых вклю-
чает ординарную металл-углеродную связь. Кинг и Лип-
пинкотт [38] установили, что частоты валентных колебаний
металл-углеродных связей у карбонилов железа и никеля
равны соответственно 410 и 382 см'1.
Большой интерес представляет появление полос погло-
щения вблизи 1830 см'1 у некоторых карбонилов железа
и кобальта, поскольку, как показывают рентгенографиче-
ские данные, структура этих соединений состоит из колец
с мостиками, у которых одни карбонильные группы явля-
ются, по существу, кетонными, а остальные присоединены по
периметру в виде групп —С=О. Согласно рентгенографиче-
ским данным [14] угол
С=О у Fe2(CO)9 составляет 87°.
Это говорит о том, что степень напряжения здесь даже боль-
ше, чем у структур четырехчленных лактонов и лактамов;
поэтому частота поглощения полосы при 1828 см'1 не яв-
ляется в этом случае неожиданной. Fe3(CO)12 также погло-
щает при 1833 см'1 и, очевидно, имеет аналогичный валент-
ный угол. В случае мостиковых систем карбонила кобальта
доказательство мостиковой структуры может быть получено
путем замещения карбонильных групп, образующих мо-
стики, ацетиленовыми связями [39]. Кобальт образует,
кроме того, интересное соединение — гидрокарбонил
НСо(СО)4. Последний не содержит гидроксильной группы,
а водород связан с образованием мостика [37, 40].
ЛИТЕРАТУРА
1. Randall, Fowler, Fuson, Dangl, Infra-red De-
termination of Organic Structures, Van Nostrand, 1949.
2. Rasmussen, Brattai n, J. Amer. Chem. Soc., 71, 1073
(1949).
3. Hartwell, Richards, Thompson, J. Chem. Soc.,
1948, 1436.
4. Barnes, Gore, Liddel, Van Zandt Williams,
Infrared Spectroscopy, Reinhold, 1944.
5. С о 1 t h u p, J. Opt. Soc. Amer., 40, 397 (1950).
6. Wasserman, Zimmerman, J. Amer. Chem. Soc., 72,
5787 (1950).
7. Modest, Szmus zk о v i c z, J. Amer. Chem. Soc., 72,
577 (1950).
Гл. 8. Галогенангидриды кислот, карбонаты и др. 187
8. Герцберг, Спектры и строение двухатомных молекул,
Издатинлит, М., 1949, стр. 91.
9. Crawford, Cross, J Chem. Phys., 6, 525 (1938).
10. С r a w f о r d, Horwitz, J. Chem. Phys., 16, 147 (1948).
ll. Sheline, J. Amer. Chem. Soc., 72, 5761 (1950).
12. S h e 1 i n e, J. Amer. Chem. Soc., 73, 1615 (1951).
13. S h e 1 i n e, P i t z e r, J. Amer. Chem. Soc., 72, 1107 (1950).
14. P о w e 1 1, E w e n s, J. Chem. Soc., 1939, 286.
15. Davison, J. Chem. Soc., 1951, 2456.
16. Husted, Ahlbrecht, J. Amer. Chem. Soc., 75, 1605 (1953).
17. H a s z e 1 d i n e, Nature, 168, 1028 (1951).
18. Nielsen, Burke, Woltz, Jones, J. Chem. Phys.,
20, 596 (1952).
19. Nakagawa, Ichishima, Kurtani, Miyazawa,
Shimanouchi, Mizushima, J. Chem. Phys., 20,
1720 (1952).
20. В e 1 1 a m y, Williams, J. Chem. Soc., в печати.
21. W i e b 1 e n, Fluorine Chemistry, ed. Simons, v. 2, Academic
Press, New York, 1954, p. 449.
22. Brown, Todd, J. Chem. Soc., 1954, 1280.
23. Cooke, Chem. and Ind., 1955, 142.
24. W a 1 k e r, J. Amer. Chem. Soc., 75, 3387, 3390 (1953).
25. Koo, J. Amer. Chem. Soc., 75, 720 (1953).
26. Stork, Breslow, J. Amer. Chem. Soc., 75, 3291 (1953).
27. Hochstein, Conover, R eg n a, Pasternack,
Gordon, Woodward, J. Amer. Chem. Soc., 75, 5455
(1953).
28. Fuson, J о s i e n, Jones, Lawson, J. Chem. Soc.,
20, 1627 (1952).
29. R u g g, Smith, В а с о n, J. Polymer. Sci., 13, 535 (1954).
30. S w e r n, W i t n a u e r, E d d у, P a r k e r, J. Amer. Chem.
Soc., 77, 5537 (1955).
31. Minkoff, Proc. Roy. Soc., A224, 176 (1954).
32. Giguere, Olmos, Can. J. Chem-., 30, 821 (1952).
33. Brimm, Lynch, Sens y, J. Amer. Chem. Soc., 76, 3831
(1954).
34. С о t t о n, L i e h r, W i 1 k i n s о n, J. Inorg. Nuclear Chem.,
2, 141 (1956).
35. S h u f 1 e r, Sternberg, Fr i e d e 1, J. Amer. Chem. Soc.,
78, 2867 (1956).
36. Cable, Nyholm, Sheline, J. Amer. Chem. Soc.,
76, 3373 (1954).
37. Friedel, W e n d e r, S h u f 1 e r, Sternberg, J.Amer.
Chem Soc., 77, 3951 (1955).
38. King, L i p p i n с о t t, J. Amer. Chem. Soc., 79, 4192 (1956).
39. Greenfield, Sternberg, Friedel, Wotiz,
Markby, Wender, J. Amer. Chem. Soc., 78, 120 (1956).
40. E d g e 1 1. Magee, G a 1 1 u p, J. Amer. Chem. Soc., 78,
4185 (1956).
41. Hales, Jones, Kynaston, J. Chem. Soc., 1957, 618
42. Angell, Trans. Faraday Soc., 52, 1178 (1956).
Глава 9
АЛЬДЕГИДЫ И КЕТОНЫ
1. ВВЕДЕНИЕ
Полоса поглощения в инфракрасной области спектра,
обусловленная валентными колебаниями С—О, по-видимо-
му, более изучена, чем какая-либо другая полоса, и полу-
чено много сведений относительно факторов, которые
влияют на ее частоту и интенсивность. Как у кетонов, так
и у альдегидов частота карбонильного поглощения опреде-
ляется почти полностью строением ближайших групп, окру-
жающих С=О, а строение остальной части молекулы не имеет
большого значения, если только отсутствуют хелатные или
сходные с ними связи. Полоса карбонильного поглощения
смещается, таким образом, от нормального положения
у а,р-ненасыщенных соединений и у карбонильных соеди-
нений с сильно электроотрицательными заместителями при
а-атоме углерода, тогда как у циклических кетонов величи-
на сдвига полосы и его направление зависят от степени
напряженности кольца. В некоторых случаях могут проис-
ходить значительные изменения частоты вследствие образо-
вания хелатов (внутрикомплексные связи), а также вслед-
ствие пространственных затруднений. Однако в каждом
из этих случаев величина ожидаемого сдвига полосы извест-
на, и новый характерный интервал частот бывает сравни-
тельно узок. Поэтому при изучении положения и интенсив-
ности одной этой полосы часто можно установить наличие
карбонильной группы и получить значительное количество
данных о ее окружении.
Опубликован обзор, специально посвященный этому
вопросу [82], а Джонс и Сандорфи [83] и Джонс и Герлинг
[84] составили очень полезные таблицы характеристиче-
ских карбонильных частот. Рядом авторов [83, 85—87]
были рассмотрены факторы, вызывающие смещения частоты
С=О; они довольно подробно описаны в гл. 23. Следует
Гл. 9. Альдегиды и кетоны
189
отметить, что был предложен ряд полезных корреляций,
связывающих смещения карбонильной полосы с такими
физическими свойствами, как длина связи [89], потенциал
полуволны [90], электронная плотность [91], электроотри-
цательность [88], мезомерный эффект заместителей [87],
а в отдельных случаях реакционная способность [90, 92]
и дипольный момент связи С = О [93].
Имеет место частичное перекрывание интервалов частот,
характерных для карбонильных групп разного типа, но
в некоторых случаях это затруднение может быть устранено
исследованием интенсивности поглощения. Кроме упомяну-
тых выше факторов, влияющих на изменение частоты, не-
большие сдвиги могут вызывать также изменения агрегат-
ного состояния и образование Водородной связи. Эти фак-
торы будут обсуждаться в разделе, посвященном нормаль-
ным простым кетонам, но приведенные там сведения в рав-
ной степени относятся и к другим типам кетонов.
Альдегиды были изучены менее подробно, однако имеется
достаточно доказательств того, что смещения полосы при-
изменениях в окружении группы С=О аналогичны смеще-
ниям в случае кетонов. При идентификации альдегидов
обычно для подтверждения используются данные о частоте
валентных колебаний С—Н альдегидной группы. Благодаря
сильному влиянию карбонильного кислорода эта частота
фактически не зависит от строения всей молекулы и поэтому
является весьма характеристичной.
Интервалы частот, характерные для различных типов
альдегидов и кетонов, представлены в табл. 8
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С=О В СЛУЧАЕ
КЕТОНОВ С ОТКРЫТОЙ ЦЕПЬЮ
а. Положение полосы поглощения. Когда карбонильная
группа расположена между двумя метиленовыми группами,
мы встречаемся с простейшим случаем невозмущенных ва-
лентных колебаний С=О. Венигер [1] в 1910 г. впервые
установил, что альдегиды и кетоны всегда имеют сильную
полосу поглощения вблизи 1680 слГ1, которая, возможно,
связана с карбонильной группировкой, а в 1944 г. Барнс
и др. [2] опубликовали сообщениео десяти кетонах, у которых
Таблица 8
Частота, см-1
Кетонная карбонильная
а б группа ’
Насыщенные кетоны с открытой цепью
-СН2-СО-СН2 1725—1705
а,р-Ненасыщенные кетоны
—СН=СН-СО— 1685—1665
а,р-а',Р'-Ненасыщенные кетоны
—СН=СН—СО—СН=СН— 1670—1663
Арилкетоны8 С6Н5—СО— 1700—1680
Диарилкетоныв С6Н5—СО—С6Н6 1670—1660
а-Галогензамещенные кетоны
—СВг—СО и т. п. 1745-1725
а,а'-Дигалоген замещенные кетоны
—СВг—СО—СВг и т. п. 1765—1745
а-Дикетоны —СО—СО— 1730—1710
Р-Дикетоны (енольные)
-СО-СН2-СО- 1640—1540
а,Р-Ненасыщенные [J-окси- или амино-
кетоны —СО—С = С 1640—1540
ОН или NH2
1-Кето-2-оксиарилкетоны или 1-кето-2-
аминоарилкетоны
О О 1 1655-1635»
ХОН или NH2
у-Дикетоны —СО—СН2—СН2—СО— 1725—1705
-СО-СН2-О-СО— 1745—1725
Кетоны с шести- и семичленным коль-
цом 1725—1705
Кетоны с пятичленным кольцом .... 1750—1740
Кетоны с четырехчленным кольцом . . Вблизи 1775
Хиноны
две группы СО в одном кольце . . . 1690—1660 ’
две группы СО в двух кольцах . . . 1655—1635
Тропслоны Вблизи 1600
Продолжение таблицы 8
Частота, см-1
Алкилкетоны 1 частоты не относятся 1325—1215 (с.)
Арилкетоны ) к колебаниям С=О Альдегид ыа> б Колебания СО Насыщенные алифатические альдегиды а,р-Ненасыщенные альдегиды а,Р-у,б-Ненасыщенные альдегиды . . . Арилальдегиды8 а,Р-Ненасыщенные-Р-сксиальдегиды . . Валентные колебания С—Н 1225—1075 (с.) 1740—1720 1705—1680 1680—1660 1715-1695 1670—1645
Все типы соединений Деформационные колебания С—Н 2900—2700 (сл.) Обычно имеются две по- лосы, одна из которых находится вблизи 2720 c.W1
Все типы Другие колебания 975—780 (ср.)
Алифатические альдегиды 1440—1325 (с.)
Ароматические альдегиды 1415—1350 (с.) 1320—1260 » 1230—1160 »
а Характеристические сдвиги обычно обладают аддитивностью, так что
например, полоса колебаний СО а-бром-а.ф-иенасыщенного алифатического
кетоиа должна быть смещена относительно нормального положения в резуль-
тате увеличения частоты из-за наличия брома и уменьшения частоты из-за
сопряжения.
б Все указанные значения относятся к разбавленным растворам. В конден-
сированной фазе и в растворителях различной полярности происходят значитель-
ные сдвиги полос. Все полосы карбонильного поглощения сильные.
в Положение полосы изменяется также в зависимости от природы и поло-
жения заместителей в цикле.
192
Часть II
полосы поглощения карбонильной группы находятся
в интервале 1725—1690 сл’1, и отметили, что сопряжение
приводит к смещению полосы в сторону низких частот.
Затем многими исследователями было показано, что полоса
карбонильного поглощения такого рода простых кетонов
в растворе всегда лежит внутри узкого интервала 1725—
1706 см'1, если только при этом не возникает водородной
связи или других препятствующих этому явлений.
Так, например, Томпсоном и Торкингтоном [3] были
обнаружены полосы поглощения у восьми таких веществ
вблизи 1710 см'1, а последующие данные, подтверждающие
правильность отнесения этой области поглощения, были
приведены Расмуссеном, Танниклиффом и Браттеном [4],
Бонино и Скрокко [51, Кромвеллом и др. [6]. Жозьен и со-
трудники [90, 94] и Джонс и сотрудники [7—11, 62, 84]
опубликовали серию статей о частотах карбонильного погло-
щения стеринов и показали, что в пределах класса этих сое-
динений возможны дальнейшие уточнения. Например, им
удалось отличить стерины с карбонильной группой в боковой
цепи при атоме углерода С20 от других стеринов, у которых
карбонильная группа расположена в шестичленном кольце.
Как будет показано ниже, карбонильные группы в нена-
пряженных насыщенных циклах поглощают в том же общем
интервале частот 1720—1706 см'1, но было найдено, что
в случае стеринов, у которых карбонил находится в цепи
при атоме С20 (свыше тридцати примеров), полосы поглоще-
ния попадают в более узкий интервал 1710—1706 см'1,
тогда как карбонильные группы в цикле поглощают при не-
сколько более высоких частотах. Указанными исследовате-
лями было тщательно изучено несколько сотен стеринов,
содержащих карбонильные группы обоих типов, и многое
сделано для выяснения типов структур, приводящих к сме-
щениям полос. Такого рода сравнения, при которых рас-
сматриваются только малые различия, должны проводить-
ся при условиях, идентичных условиям получения исход-
ных данных. Следует, например, применять один и тот же
растворитель. Было обнаружено, что некоторые 3-кетосоеди-
нения, растворенные в сероуглероде, поглощают при 1719—
1715 см'1, а в хлороформе при 1702—1705 см'1 [62]. Это
почти точно равно разнице частот поглощения карбониль-
ных групп при атомах С20 и С3. Поглощение карбонильных
Гл. 9. Альдегиды и кетоны
193
групп при различном их положении в тритерпеновом
скелете изучали Коул и Торнтон [95].
Приведенные выше корреляции относятся поэтому толь-
ко к спектрам веществ в неполярных растворителях, и име-
ются всегда отклонения от них, если образцы исследуются
в твердом или жидком состоянии. Хартвелл, Ричардс и
Томпсон [12] изучали влияние физического состояния на
примере группы карбонильных соединений и обнаружили,
что семь нормальных кетонов с одной группой С=О погло-
щают в жидком состоянии в области 1747—1714 см"1. Для
соединений в парообразном состоянии карбонильные частоты
поглощения оказались заметно отличными от частот, полу-
ченных для растворов. Ацетон, например, в парообразном
состоянии поглощает при 1742 см"1, в то время как в случае
раствора полоса поглощения лежит между 1728—1718 см'1
в зависимости от растворителя. Аналогичным образом диде-
цилкетон поглощает при 1740 см'1 в парообразном состоя-
нии и в интервале 1724—1717 см'1 — в растворе. Возмож-
но, таким образом, что в конденсированной фазе существует
некая форма дипольной ассоциации [100, 101], в результате
чего происходит сдвиг поглощения в направлении более
высоких частот на величину порядка 20 см'1. Поэтому,
насколько это возможно, измерения частот поглощения ке-
тонов следует производить для растворов.
Небольшие изменения карбонильной частоты, наблю-
дающиеся при применении различных растворителей, по-
дробно изучались рядом исследователей [96—99]. Причины
этих смещений не вполне ясны, но, по-видимому, между рас-
творителем и растворенным веществом имеет место какой-то
вид электростатического взаимодействия. В тех случаях,
когда растворитель может образовывать водородные связи
с карбонильной группой, обнаруживается небольшое сме-
щение полосы, которое весьма незначительно по сравнению
с эффектом взаимодействия, сопровождающегося резонанс-
ной стабилизацией. Расмуссен и др. [4] обнаружили,
например, что полоса поглощения карбонила диацетонового
спирта находится при 1712 см'1, несмотря на то что частоты
поглощения ОН указывают на наличие сильной водородной
связи, а Гров и Уиллис [13] оценивают влияние водородных
связей на некоторые карбонильные группы величиной
смещения не более 10 см'1.
13 Л. Беллами
194
Чаете 11
б. Влияние сопряжения олефиновых связей. Сопряже-
ние карбонильной группы со связью С=С приводит к умень-
шению частоты колебаний С=О на величину, зависящую от
природы двойной связи. Первоначально сведения об этом
эффекте были получены на основании изучения спектров
комбинационного рассеяния [14], а затем были полностью
подтверждены данными об инфракрасных спектрах погло-
щения.
Алифатическая связь С—С при сопряжении с карбониль-
ной группой приводит к понижению частоты колебаний при-
мерно на 40 сл”1, т. е. до 1685—1665 см-1. Так, например,
изофорон поглощает при 1672 сл'1 [4]. Тернер и Войтл [15]
измерили недавно с целью сравнения частоту колебаний СО
для 1-ацетил-2-метил-А 1-циклогексена и для вещества А2,
в котором карбонил не сопряжен с двойной связью; полу-
ченные ими значения соответственно равны 1686 и 1709 сл*1.
Аналогичные смещения Мекке и Ноак [102] продемонстри-
ровали, сравнивая кетоны с а,|3- или р,у-двойной связью,
а дальнейшим подтверждением этих результатов служит
работа Фасона и сотрудников [90]. Хейльман и сотрудники
[103] показали, что величина смещения полосы не зависит
от типа олефиновой связи. Однако в случае цис-транс-
изомерии карбонильная частота гщс-изомера обычно на 10—
20 сл*1 выше. В ряду стеринов карбонильная группа, сопря-
женная с а,р-непредельной связью, независимо от того,
находится ли она в боковой цепи [7] или в шестичленном
цикле [9], поглощает в одном и том же интервале час-
тот [84].
Сопряжение с у,ё-двойной связью оказывает лишь не-
большое влияние на положение рассматриваемой полосы
поглощения, которая смещается в сторону низких частот
в пределах указанного интервала, и для трех стеринов этого
типа Джонс и др. [9] установили интервал поглощения
1669—1663 сл*1. Это было подтверждено Кромвеллом и др.
[6], которые отметили, что любой дополнительный сдвиг
полосы при подобном сопряжении является незначительным.
Это не является неожиданным, так как, согласно Блоуту,
Филдсу и Карпласу [16] частота а,|3-ненасыщенной альде-
гидной карбонильной группы уменьшается не более чем
на несколько обратных сантиметров даже при увеличении
длины сопряженной цепи до семи двойных связей.
Г л. 9. Альдегиды и кетоны
195
Аналогичным образом а,Р- а',Р'-сопряжение не приво-
дит к сколько-нибудь заметному дополнительному эффекту,
кроме смещения частоты поглощения по направлению к низ-
кочастотной границе указанного интервала. Джонс и др.
[7] сообщили о пяти соединениях с частотой поглощения
а,р-а',0'-ненасыщенной карбонильной группы в интервале
1666—1663 сл-1.
В противоположность этому диарилкетоны показывают
большее смещение полосы, чем кетоны с одним ароматическим
радикалом, и поглощают в том же интервале, что и соответ-
ствующие олефины. При исследовании нескольких соедине-
ний [104—106], характеризующихся сопряжением с аце-
тиленовыми связями, было отмечено, что смещения имеют,
по-видимому, тот же порядок величины и то же направле-
ние, как и в случае олефинов.
Эти смещения полос примерно в той же мере проявляются
й у кетонов, у которых полоса уже сдвинута по какой-либо
иной причине. Так, 2-бром-З-кетостероиды поглощают вбли-
зи 1735 см 1 вследствие влияния галогена, а А1-2-бром-3-
кетостероиды поглощают при 1697 см'1 [7]; таким образом,
частота уменьшается на 40 см'1, т. е. на величину того же
порядка, как и при обычном а,р-сопряжении. Подобные
эффекты обнаруживаются также в случае полос поглоще-
ния карбонила, частота которых повышается из-за наличия
пятичленного цикла. Понятно поэтому, что корреляции,
указанные для этого типа соединений, применимы только
тогда, когда отсутствуют другие факторы, также способные
влиять на частоту поглощения. Однако в общем случае
отдельные сдвиги полос, вызываемые различными фактора-
ми, по-видимому, аддитивны, и потому все-таки можно
произвести оценку положения полосы карбонильного погло-
щения.
Рассматриваемая корреляция неприменима к а, р-нена-
сыщенным кетонам (ароматическим и жирным), в которых
находится также гидроксильная группа или аминогруппа
при Р-атоме углерода. В этих соединениях имеет место внут-
реннее комплексообразование, которое оказывает очень
большое влияние на частоту карбонильного поглощения.
Такие соединения представляют поэтому особый случай
и будут рассмотрены отдельно в разделе, посвященном р-ди-
кетонам, которым они родственны.
13*
196
Часть II
в. Влияние ароматического сопряжения. Когда аромати-
ческий радикал непосредственно присоединен к атому угле-
рода карбонильной группы, частота карбонила изменяется
меньше, чем в случае сопряжения с этиленовой двойной
связью, и полоса поглощения находится в интервале 1700—
1680 см'1. Томпсон и Торкингтон [3] рассмотрели три таких
случая. Ацетофенон, например, поглощает при 1686 см'1
в отличие от изофорона, поглощающего при 1672 см'1. Одна-
ко когда к карбонилу непосредственно присоединены два
ароматических радикала, наблюдается дальнейшее умень-
шение частоты поглощения до 1670—1660 см'1. Хансбергер
[17] сделал предположение, что меньшее влияние аромати-
ческого ядра может объясняться тем, что степень двоесвя-
занности его связей меньше, чем в случае двойных алифати-
ческих связей С=С. Бензальацетофенон, у которого с груп-
пой СО сопряжены и алифатическая двойная связь и арома-
тическое ядро, поглощает при 1667 см'1. Как и в случае
других типов кетонов, изменения состояния могут сильно
влиять на эту частоту поглощения, и все указанные выше
величины относятся к поглощению в разбавленных раство-
рах. Так, например, Хартвелл, Ричардс и Томпсон [12]
нашли, что в конденсированной фазе фен ил метил кетон
поглощает при 1707 см'1.
Уже было отмечено, что замещение группами ОН или
NH2 в орто-положении относительно карбонильной группы
представляет особый случай образования внутримолеку-
лярной водородной связи (см. ниже). Другие заместители
в кольце, по-видимому, тоже способны в некоторой степени
влиять на частоту колебаний. Например, наличие в орто-
положениях алкильных групп приводит к увеличению
частоты в результате уменьшения степени копланарности
карбонильной группы с циклом [107]. Ацетомезитилен
в жидком состоянии поглощает при 1701 см'1, т. е. частота
на 9 см'1 выше, чем у ацетофенона. Аналогичные резуль-
таты были получены для некоторых бензоцикланонов.
Кроме того, карбонильные частоты замещенных ацетофено-
нов значительно различаются в зависимости от электро-
фильных и нуклеофильных свойств замещающих групп.
Соловей и Фрейе [18] показали, что происходят большие
изменения в твердой фазе, а при более детальных исследова-
ниях растворов ацетофенонов и бензофенонов [90, 169]
Г л. 9. Альдегиды и кетоны
197
было обнаружено, что карбонильные частоты могут быть
непосредственно связаны со значениями постоянной Гам-
мета а для заместителей. Величина а, получаемая в резуль-
тате кинетических исследований, является мерой электрон-
нодонорных или акцепторных свойств замещающих групп.
В случае бензофенонов была установлена зависимость сме-
щения частот от полярографического потенциала полуволны
[90], а в случае ацетофенонов было показано, что смещения
являются функцией вычисленного порядка карбонильной
связи [108].
Как и в рассмотренных ранее случаях, влияние арома-
тического кольца в a-положении должно аддитивно скла-
дываться с эффектом какой-либо другой структуры, которая
также может влиять на частоту колебаний С=О. Гутше,
например [19], привел данные о ряде трициклических кето-
нов, содержащих одно ароматическое кольцо. Было найдено,
что частота поглощения группы С=О, находящейся в шести-
членном цикле с арильной группой в a-положении, равна
1695—1686 см'1, т. е. имеет такое же значение, как и у сход-
ных веществ с открытой цепью. Однако частота поглощения
группы С=О в пятичленном цикле увеличена до 1715—
1706 см'1, причем влияние напряжения пятичленного цикла
в какой-то мере уменьшено вследствие сопряжения с арома-
тическим кольцом.
г. Влияние сопряжения с циклопропаном. Известно,
что в результате высокой электронной плотности в центре
кольца циклопропан по своим свойствам аналогичен соеди-
нению с двойной олефиновой связью. Влияние циклопропа-
нового кольца, присоединенного к карбонильной группе,
мало исследовано, но некоторые данные об алкилциклопро-
пилкетонах приведены в работах Виберлея и Бунсе [109]
и Кромвелла [170], где указывается, что карбонильные ча-
стоты этих веществ в жидком состоянии имеют пониженное
значение (1704—1686 см'1). Примеры такого влияния коль-
ца приведены также Фасоном и др. [90, НО, 111], причем
особый интерес представляют такие соединения, как г-сте-
рины, в частности г-холестанон-6.
Влияние других напряженных циклических систем ме-
нее изучено. В нескольких случаях было обнаружено, что
«сопряжение» с эпоксидным кольцом не приводит к изме-
198
Часть II
нению карбонильной частоты, но Кромвелл и Гудсон [112]
наблюдали понижение этой частоты у некоторых этилен-
иминов с а-карбонильной группой. Они рассмотрели также
условия, при которых у таких систем может возникать
эффект сопряжения. Однако уже указывалось [90], что со-
поставление частот, полученных для твердого состояния,
с частотами жидкостей может приводить к ошибочным
выводам, поэтому желательно получить подтверждение пра-
вильности данных указанных авторов.
д. Влияние галогенов в a-положении. Давно известно,
что замещение галогеном в непосредственной близости
от карбонильной группы приводит к сдвигу полосы погло-
щения карбонила в сторону более высоких частот. Этот
эффект особенно заметно выражен у хлорангидридов кислот,
где хлор непосредственно связан с карбонильной группой,
но он также имеет место и в том случае, когда галоген нахо-
дится при а-атоме углерода.
Это хорошо известное явление широко изучалось путем
исследования спектров комбинационного рассеяния [20].
Сказанное относится ко всем типам карбонильного погло-
щения. Хамптон [21] и Хартвелл, Ричардс и Томпсон [12]
наблюдали подобный эффект у сложных эфиров, причем
последние авторы отнесли его за счет усиления двойной свя-
зи карбонильной группы. Влияние замещения галогеном
наиболее широко исследовалось в случае кетонных карбо-
нилов в стероидах Вудвардом [22], Джерасси и Шольцом
[23], Диксоном и Пейджем [113] и особенно Джонсом и др.
[9, 10, 63, 83, 119]. У циклических систем этого ряда отдель-
ные циклы жестко закреплены в форме кресла или ванны,
так что они являются особенно удобными объектами иссле-
дования стереохимических эффектов, связанных с различ-
ным конфигурационным расположением галогенов.
Было обнаружено, что обычно замещение в а-положении
атомом брома приводит к повышению частоты колебаний
С=О примернэ на 20 см'1. Так, например, несопряженные
3-кетостероиды поглощают при 1719—1715 см'1, a 2- или
4-бром-З-кетостероиды поглощают вблизи 1735 см'1. Анало-
гично Д]-3-кетостероиды вследствие сопряжения поглощают
при 1684—1680 см'1, а для AJ-2- или -4-бром-З-кетостерои-
дов частота полосы поглощения повышается до 1697 см'1.
Гл. 9. Альдегиды и кетоны
199
Смещение полосы СО увеличивается при замещении
галогенами по обе стороны от карбонильной группы, так
что 2,4-дибром-З-кетоны поглощают вблизи 1760 см'1,
причем второй инкремент повышения частоты равен перво-
му. Между тем замещение двумя атомами брома при одном
и том же а-атоме углерода дает очень немногим больший
эффект, чем замещение одним атомом. Действительно, как
2-бромхолестанон-З, так и 2,2-дибромохолестанон-З погло-
щают при 1739—1735 см"1. Это можно объяснить, если
учесть, что указанный выше эффект возникает лишь в слу-
чае, когда связь С—Вг является копланарной с кетонной
группой и увеличена доля полярной структуры II [63].
Таким образом, замещение бромом в а-экваториальном
положении в кольце, имеющем креслообразную конфигу-
рацию, вызывает повышение частоты колебаний соседней
группы СО на 20 сл”1. С другой стороны, замещение в ак-
сиальном положении не приводит к какому-либо изменению
частоты [63]. Аналогичным образом частота колебаний
карбонильной группы при атоме С20 повышается на 20 сл”1,
когда бромом замещается водород при атоме C2i, но при
замещении у атома С17 в a-положении частота колебаний
не меняется.
Корей и сотрудники [115—118], Сандрис и Оуриссон
[119] и Инаяма [120] исследовали такие простейшие си-
стемы, как а-галогензамещенные циклогексаноны и цикло-
пентаноны. Результаты этих работ весьма близки к тем
данным, которые были получены для ряда стеринов, и таким
образом являются дополнительным их подтверждением.
В то же время они менее убедительны, так как в случае
указанных систем конверсия форм кресла и ванны происхо-
дит сравнительно легко, и, действительно, последние ис-
следования методом измерения дипольных моментов [121]
поставили их под сомнение. Положение о том, что взаимо-
действие карбонила и галогена имеет место лишь при их
экваториальном, но не при аксиальном положении, было
2 00
Часть II
использовано при попытке оценить степень согнутости
циклопентанонового кольца [122]. Аналогичные эффекты
были замечены также в циклогептаноновом ряду [25, 70].
Учитывая стереоспецифичность указанного эффекта
взаимодействия, Джонс и Сандорфи [83], Беллами и Уиль-
ямс [93, 123] независимо друг от друга выдвинули предпо-
ложение, что механизм его связан главным образом с влия-
ниями пространственного поля, а не с индукционными
силами, действующими по связям. При этом возможность
внутреннего вращения а-галогенкетонов вокруг связей
С — С должна приводить к существованию двух поворот-
ных изомеров, имеющих различные карбонильные частоты.
В случаях хлорацетона [124] и 1,3-дихлорацетона [125]
наличие эффекта поворотной изомерии уже было установ-
лено, а дальнейшие исследования такого рода соединений
и ю-хлорацетофенонов показало, что у жидких моно-
и дигалогенкетонов и их растворов наблюдаются две четкие
карбонильные частоты. Большая из этих частот относится
к более полярной форме (I), у которой атом галогена нахо-
дится ближе к карбонильному кислороду, а более низкая
частота, которая близка к частоте незамещенного кетона,
соответствует форме (II).
Относительные интенсивности этих полос, как это обычно
бывает при наличии поворотной изомерии, сильно зависят
от полярности растворителя, так что в полярных раство-
рителях и в самой жидкости интенсивность полосы с мень-
шей частотой сильно понижена. С другой стороны, в твер-
дом состоянии существует только одна форма молекул,
и наблюдается только одна карбонильная частота. В неко-
торых случаях только одна форма устойчива также в парах.
Учитывая зависимость эффекта раздвоения частоты С = О
от пространственных факторов, легко объяснить тот факт,
что появление второго атома галогена в а-поло>кении
Г л. 9. Альдегиды и кетоны
201
незначительно увеличивает карбонильную частоту, так как
в цис-положении к кислороду может находиться только один
из этих двух атомов. Дальнейшие сведения об этих эффек-
тах даны в гл. 23. Величина смещения карбонильной ча-
стоты в случаях хлор- и бромзамещенных примерно оди-
накова, тогда как для фторзамещенных следует ожидать
большего смещения. Так, например, 1,1,1-трифтораце-
тон [1761 поглощает при 1780 см'1, т. е. на 40 см'1 выше,
чем ацетон в паровой фазе. Об аналогичных результатах
для а-фторкетонов сообщает также Хазельдин [68]. Подоб-
ные пространственные эффекты наблюдались для а-гало-
гензамещенных сложных эфиров, галогенпроизводных кис-
лот, альдегидов и, по-видимому, амидов [93].
О влиянии других полярных групп в a-положении из-
вестно очень мало. Окси-группа в a-положении обычно
понижает карбонильную частоту, очевидно в результате
образования водородной связи [13, 127].
3. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С==О У ДИКЕТОНОВ
а. а-Дикетоны. В результате исследований спектров
комбинационного рассеяния установлено, что между сосед-
ними карбонильными группами существует очень слабое
взаимодействие и что повышение частоты колебаний в ре-
зультате этого взаимодействия не превышает 5—15 см'1.
Кольрауш и Понграц [27] установили, что в спектре ком-
бинационного рассеяния бензила полоса смещается в сто-
рону высоких частот на расстояние, большее лишь на 5 см'1
по сравнению с полосой ацетофенона. Томпсон же [28] ука-
зал, что глиоксаль поглощает при 1730 см'1 в отличие от
формальдегида, поглощающего при 1745 см'1, т. е. сдвиг
полосы происходит в противоположном направлении. При-
чина появления в области карбонильного поглощения
у бензила и диацетила только по одной полосе объяснена
Расмуссеном и др. [4], которые предположили, что эти
соединения существуют в виде транс-формы, так что в
инфракрасном спектре в каждом случае активно только
антисимметричное колебание. Частоты в спектрах комби-
национного рассеяния отличаются очень мало, и это лиш-
ний раз доказывает, что взаимодействие между карбониль-
ными группами невелико.
202
Часть II
Из стероидов Джонс и др. [9] изучили некоторые 11,
12-дикетоны в растворе и обнаружили полосы поглощения
карбонила при 1726 см'1. Это означает, что повышение
частоты поглощения за пределы интервала 1716—1706 см'1,
который включает полосы поглощения индивидуальных
11- и 12-монокетонов, очень невелико. Барнс и Пинкней
[69], а также и Леонард и др. [70, 127] исследовали многие
а-дикетоны и наблюдали только очень слабые эффекты
взаимодействия.
Поэтому у а-дикетонов обычно следует ожидать лишь
небольшого повышения частоты поглощения. Исключением
являются а-кетометиловый эфир пировиноградной кисло-
ты [29], у которого и эфирная и кетонная карбонильные
группы поглощают при 1748 см'1, и пировиноградная кис-
лота, у которой обе группы поглощают при 1745 см'1.
Исключения возможны также для некоторых цикличес-
ких систем, когда карбонильные группы не могут зани-
мать транс-положения. В этих случаях может иметь место
дипольное взаимодействие, аналогичное тому, которое
характерно для а-галогенкетонов. Алдер и др. [128] наблю-
дали для ряда таких соединений повышенные значения кар-
бонильной частоты.
б. Р-Дикетоны. Расмуссен и др. [4] описали эффект
внутреннего комплексообразования для некоторых р-дике-
тонов, приводящий к очень значительному сдвигу карбо-
нильной полосы поглощения. Химическими методами и с
помощью спектров комбинационного рассеяния доказано,
что и ацетилацетон, и дибензоилметан существуют главным
образом в виде моноенольной формы, и ни один из них не
имеет полосы поглощения СО, соответствующей обычному
сопряженному кетону. Вместо нее наблюдалась очень
широкая и в сто раз более интенсивная полоса в области
1639—1538 см'1. Указанные исследователи полагают, что
это поглощение вызывается карбонильной группой с двой-
ной связью, ослабленной вследствие резонанса структур:
<?н....9 +он.....р-
I || и || I
R—C=CR'—С—R" R—С—CR'=C—R"
Гл. 9. Альдегиды и кетоны 203
Этот резонансный эффект описан как «внутреннее комплек-
сообразование с сопряжением» в отличие от обычного эф-
фекта образования водородной связи, как в случае диаце-
тонового спирта. То же явление может всегда иметь место
при наличии структуры типа
Х-Н Y
I II
С = С —С
где X и Y ведут себя, как доноры или акцепторы электро-
нов; исследованы многие подобные соединения. Наблю-
давшиеся во всех случаях очень большие изменения спектра
в области поглощения ОН подтверждают ту точку зрения,
что образующиеся здесь связи гораздо сильнее, чем обычные
водородные мостики. Аналогичные эффекты внутреннего
комплексообразования в сложных эфирах описаны также
Расмуссеном и Браттеном [30]. Было показано, что вели-
чина смещения карбонильной частоты является функцией
степени двоесвязности аф-связи [64, 129]. В случае еноль-
ной формы дикетонов эта связь обычно имеет характер
двойной олефиновой связи и обнаруживается частота,
близкая к 1600 см'1; если же ненасыщенность в аф-поло-
жении связана с ароматической системой, то наблюдаются
несколько меньшие смещения. Примеры енольных р-дике-
тонов, образующих хелатные связи, обсуждались также
Шигориным [130], Парком, Брауном и Лачером [131],
которым удалось показать, что при замещении обоих ато-
мов водорода центральной метиленовой группы возмож-
ность енолизации предотвращается и наблюдается обычная
карбонильная частота. Это явление обнаружено также
в случае халконов [67, 71], в замещенных ароматических
системах, содержащих группы — СО — СН2 — СО —
[69], и даже у тетроновых кислот [143] и Р-трикетонов,
у которых при центральном атоме углерода остается един-
ственный атом водорода [72].
Резонансные системы такого типа могут быть получены
также при димеризации. Например, 5,5-диметилциклогек-
сандион-1,3 [4] поглощает при 1700 и при 1605 слГ1. Пер-
вая из этих полос относится к обычной карбонильной груп-
пе, а вторая соответствует сопряженной внутрикомплекс-
ной форме, полученной при димеризации. Однако в этом
204
Часть II
случае межмолекулярные связи при разбавлении неполяр-
ными растворителями разрываются и снова наблюдаются
обычные частоты, не относящиеся к хелатам [129, 132].
Обнаружены также соответствующие изменения в области
валентных колебаний ОН.
в. а,р-Ненасыщенные 0-оксикетоны. В случае аромати-
ческих соединений одна из двойных связей кольца может
иметь тот же характер, что и двойная связь в «^-ненасы-
щенной группировке, и потому 1-кето-2-оксисоединения
также обнаруживают аномальные частоты карбонильного
поглощения. Горди [31] указал, что для о-оксиацетофенона
карбонильное поглощение наблюдается при 1639—1613 см'1,
а Хансбергер [17], исследовавший многие производные
нафталина, обнаружил сдвиги полос поглощения вслед-
ствие этого эффекта на 50—60 см'1. Например, а- и р-аце-
тонафтоны поглощают при частоте 1685 см'1, соответствую-
щей поглощению обычного а,p-а',6'-ненасыщенного аро-
матического кетона. Однако и 1-окси-2-ацетонафтон, и
2-окси-1-ацетонафтон вследствие внутреннего комплексо-
образования поглощают при 1625 см'1. В этой связи осо-
бенно интересно отметить, что З-окси-2-ацетонафтон погло-
щает при 1657 см'1. Здесь имеет место значительно мень-
шее смещение, что обусловлено уменьшенной! кратностью
двойной связи 2,3 в результате фиксации связей. В литера-
туре имеются сведения об альдегидах, обнаруживающих
такие же свойства. Сдвиг полос для этих соединений не так
велик, как для истинных Р-дикетонов, но тем не менее он
хорошо заметен. Аналогичные результаты были получены
для производных фенантрена, содержащих гидроксильную
группу при Сщ и группу СООСН3 при С9 [64]. Для этих
соединений наблюдается смещение порядка 75 см'1; срав-
нение со смещением для метилированного фенантрилового
сложного эфира или кетона указывает на то, что 9,10-двой-
ная связь фенантрена, по-видимому, ближе по характеру
к олефиновой связи, чем 1,2-двойная связь нафталина.
Понижение частоты происходит не только вследствие уси-
ления арильного сопряжения, поскольку частоты колеба-
ний СО в группах СНО, СОСН3 и СООСН3, находящихся
при атоме С9 фенантрена, всего на 2 см'1 меньше, чем у соот-
ветствующих производных нафталина, когда отсутствуют-
Гл. 9. Альдегиды и кетоны
205
группы ОН. Результаты исследования смещений частот
карбонильных групп при замещении в орто-положении
окси-группой были использованы Хансбергером и др. [1331
для оценки сравнительной степени двоесвязанности раз-
личных связей цикла в индане.
Указанное смещение возможно также в том случае,
когда карбонильная группа является частью циклической
системы, но имеется а,|3-ненасыщенность и группа ОН на-
ходится при р-атоме углерода. Примерами таких соедине-
ний являются 1-оксиантрахиноны, антроны и оксантроны,
которые изучались Флеттом [32], а также Хаджи и
Шеппардом [134], и оксихиноны, изучавшиеся Жозьен
и др. [73].
Как антрахинон, так и 2-оксиантрахинон поглощают
в интервале 1673—1676 слГ1, в то время как 1-оксиантра-
хинон имеет две полосы поглощения СО при 1680—1675 см'1
и при 1630—1622 см1, соответствующие свободной! и свя-
занной карбонильным группам. В случае двух гидроксиль-
ных групп, находящихся в ^-положениях, наблюдается
только одна полоса при 1639—1623 см'1, а в предельном
случае 1,4,5,8-тетраоксиантрахинона частота карбониль-
ного поглощения уменьшается до 1595 см1.
Аналогичным образом у 1-оксиантрона частота карбо-
нильного поглощения уменьшается на 20 см'1, а у 4-окси-
антрона — на 10 см'1. Причина уменьшения частоты во
втором случае не ясна, но, как будет показано ниже, из-
вестны многочисленные примеры подобного явления при
4-ам инозамещении.
Хотя, казалось бы, эти эффекты вообще удовлетвори-
тельно объясняются внутренним комплексообразованием
с сопряжением, имеются еще некоторые особенности, кото-
рые не совсем понятны и требуют дальнейшего изучения.
Кроме аномальных отклонений, отмеченных в отношении
влияния 4-замещения в антрахинонах, Вудвард и др. [33]
обнаружили заметные смещения полос карбонильного по-
глощения для Р-дикетоенольных простых эфиров, у кото-
рых замещение препятствует и образованию обычной водо-
родной связи, и внутреннему комплексообразованию. Ссы-
лаясь на исследования димедонметилового эфира, 3- и 4-ме-
токситолухинонов и их комплексов с некоторыми Дионами,
указанные авторы считают, что для таких систем сильное
206
Часть 11
поглощение при 1660 и 1625 см"1 является характеристи-
ческим. Интересные аномалии наблюдаются также в случае
флавонов и флаванонов [71, 115]. 5-Оксифлаванон пока-
зывает обычное уменьшение карбонильной частоты на
40 см'1, обусловленное образованием шестичленного хе-
латного кольца (I). В то же время для 5-оксифлавона, не-
смотря на аналогию структуры (II), не наблюдается ника-
кого смещения частоты. Замещение флавонов оксигруппой
в положении 3 приводит опять-таки к уменьшению частоты
примерно на 30 см'1 (III), хотя, казалось бы, образую-
щийся при этом пятичленный хелатный цикл должен быть
менее устойчив, чем шестичленная циклическая система.
В случае 3,5-диоксифлавонов не наблюдается никакого
смещения частоты, что кажется еще более удивительным,
однако удовлетворительного объяснения этому пока еще
не найдено.
г. а,Р-Ненасыщенные Р-аминокетоны. Можно было бы
ожидать, что соединения этого типа так же, как и оксисое-
динения, рассмотренные выше, образуют внутрикомплекс-
ные соединения (хелаты). Однако не ясно, справедлива ли
такая аналогия между указанными соединениями, так как
они могут также рассматриваться как винилоги амидов,
на которые они в некоторых отношениях походят; поэтому
атом азота может оказывать влияние на частоты карбониль-
ного поглощения независимо от каких-либо эффектов внут-
реннего комплексообразования. Это наблюдается, например,
в случае у-хинолонов.
В согласии с данным предположением Кромвелл и др.
[6, 35] обнаружили у р-аминоненасыщенных кетонов сдви-
ги карбонильного поглощения порядка 20—80 см"1, хотя
а-амино-а,Р-ненасыщенные кетоны дают обычное поглоще-
Гл. 9. Альдегиды и кетоны
207
ние. Оказалось, что у полностью замещенных аминов также
происходит значительное изменение частоты поглощения,
но не такое большое, как у соединений, которые могут
образовывать водородные связи. На этом основании указан-
ные авторы пришли к выводу, что у этих замещенных ами-
нов имеет место резонанс структур
— N — О" — N —
II I I II
R -СО—С = С— R—С=С—С—
Аналогичные явления были обнаружены Флеттом [32]
для аминоантрахинонов. Он показал, что в этих структу-
рах водородная связь может играть лишь небольшую роль
даже в случае незамещенных аминов. Так, например, ча-
стота валентных колебаний у 1 -аминоантрахинона не очень
значительно отличается от частоты колебаний у 2-нафтил-
амина, а частота колебаний NH у 1-метиламиноантрахинона
всего на 100 см'1 меньше частоты, обычной для вторичных
аминов. Это находится в большом противоречии с поведе-
нием соответствующих оксисоединений, у которых в обыч-
ном интервале частот валентных колебаний ОН нет никаких
полос. Более того, Флетт обнаружил, что карбонильное
поглощение у 2-аминоантрахинона смещено почти в такой
же степени, как и у 1-аминосоединений, на основании чего
он заключил, что могут иметь место резонансные структуры
двух типов:
Аналогичные изменения происходят и у 4-аминозаме-
щенных соединений, которые не могут образовать внутри-
молекулярных водородных связей таким путем, как
1 -аминосоединения.
208
Часть 1J
Таким образом, несмотря на большое сходство этой
системы с «.[^-ненасыщенными кетонами, содержащими
Р-гидроксильные группы, она имеет и свои специфические
особенности, так что требуется осторожность при интерпре-
тации частот карбонильного поглощения «^-ненасыщен-
ных соединений с Р- или даже у-аминозамещением.
д. Внутрикомплексные соединения Р-дикетонов с метал-
лами. р-Дикетоны легко связываются со многими метал-
лами, образуя такие внутрикомплексные соединения, как
ацетилацетонаты металлов. Эти соединения изучались
Леконтом [36, 66] и Морганом [37]. В спектрах 11 ацетил-
ацетонатов Леконт обнаружил сильную полосу поглоще-
ния в интервале 1562—1550 см"1 и предположил, что она
относится к карбонильной группе, ослабленной резонансом
между связями С—О—М и С=О...М. Этот сдвиг полосы
поглощения подобен сдвигу, имеющемуся в спектрах иони-
зированных жирных кислот, которые можно в некоторой
степени сопоставить с упомянутыми соединениями [38, 39].
У всех этих веществ имеется вторая полоса поглощения
вблизи 1515 см"\ отнесенная к связи С = С. Эти данные были
подтверждены Беллами и Бранчем [136], которые иссле-
довали ряд внутрикомплексных соединений меди с различ-
ными р-дикетонами, а также хелаты этих лигандов с дру-
гими металлами. Характерные частоты в этих случаях
лежат, как правило, в интервалах 1608—1524 см"1 и
1390—1309 сл/"1; однако у салицилового альдегида смеще-
ние значительно меньше, что обусловлено меньшей сте-
пенью двоесвязности, и у соединений этого ряда карбо-
нильная частота является функцией устойчивости хелата.
Бендер и Фигуэрас [137] отметили также смещение частоты,
связанное с образованием енолят-иона у ацетилацетона,
к которому добавлялся этоксид натрия; при этом карбо-
нильная частота имела значение 1604 сл/"1. Очень близкое
значение характерно также для глицината цинка [138].
Для карбонильной частоты комплексов кетонов с треххло-
ристым алюминием и треххлористой сурьмой первоначально
было установлено значение, примерно равное 1550 сл/"1
[139, 140]. Такая полоса действительно имеется по данным
Джевелла и Спура [141] у бензоильных соединений. Тем
не менее происхождение ее теперь неясно, так как недавно
Гл. 9. Альдегиды и кетоны
209
Саз и др. [177] на основании результатов своих исследова-
ний предположили, что полоса карбонильного поглощения
у таких соединений лежит значительно выше, т. е. отвечает
частоте около 2390 см'1 вследствие преобладания форм
типа [ Хел3Со]+AICI7.
е. у- и 6-Дикетоны и сходные вещества. Карбонильные
группы у и 5-дикетонов слишком удалены одна от другой,
чтобы непосредственно взаимодействовать. Следует, однако,
отметить, что когда одна из карбонильных групп является
частью кислотной или сложноэфирной структуры, то может
происходить лактонизация. При этом карбонильная полоса
поглощения с частотой, характерной для кетонов, исчезает,
а полоса с частотой, характерной для кислот или сложных
эфиров, замещается полосой, соответствующей лактону.
Часто эти две формы существуют совместно и потому обна-
руживаются все три частоты поглощения. Путем неболь-
ших изменений внешних условий можно получить ту или
иную из этих форм. Мандей [40], например, полагает, что
пенициллиновая кислота может быть полностью переведена
из кето-формы в форму лактона при растирании с нуйолом.
Гров и Уиллис [13] изучили многие другие случаи лакто-
низации.
Был обнаружен еще один случай взаимодействия у-дике-
тонов, а именно соединений со структурой СО—С—О—СО—;
здесь, по-видимому, происходит взаимодействие между
карбонильными группами, отличающееся от химической
перегруппировки при образовании лактона.
Джонс и др. [9, 62] отметили большое число 21-ацетокси-
20-кетостероидов, у которых частоты С=О, как кетонной,
так и эфирной карбонильных групп, увеличены вследствие
эффекта взаимодействия соответственно на 20 и 10 см'1.
Происхождение этого эффекта непонятно, но он наблю-
дается как при полярном, так и при экваториальном поло-
жении ацетокси-группы в 12-ацетокси-11 -кетостероидах
[113]. С другой стороны, он, по-видимому, не должен иметь
места в 17-ацетокси-16-кетозамещенных, когда кето-группа
является частью пятичленного цикла. Это привело Беллами
и Уильямса [142] к выводу о том, что взаимодействие имеет
Дипольный характер, а смещение частоты обусловлено силь-
ным сближением кислородных атомов карбонильных групп.
14 Л. Беллами
210
Часть 11
4. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С = О в АЛИФАТИЧЕСКИХ
ЦИКЛИЧЕСКИХ СИСТЕМАХ
а. Семичленные циклы. Карбонильные группы, входя-
щие в алифатические семичленные циклы, имеют обычные
частоты колебаний, очень близкие к частотам, характерным
для соединений с открытой цепью. Это было показано Шел-
лом и его сотрудниками при исследовании циклических
кетонов в связи с изучением химии пенициллина [41],
а затем подтверждено другими исследователями. Скотт
и Тарбелл [25] указывают, что циклогептанон поглощает
при 1699 см'1, тогда как 2-хлорциклогептанон вследствие
влияния атома галогена поглощает при 1715 см'1. Тетра-
гидроколхицин, который, по-видимому, представляет собой
семичленный циклический кетон с «-заместителями, притя-
гивающими электроны, также поглощает при 1710 сл"1.
Изменения этого карбонильного поглощения под влия-
нием сопряжения и т. п. также очень сходны с соответст-
вующими эффектами в случае соединений с открытой
цепью. Гутше [43] показал, что 2- и 3-фенилциклогептаноны
в жидком состоянии поглощают при 1690 сл"1, а Скотт
и Тарбелл [25] приводят для поглощения 2,3-бензоцикло-
гептанона значение частоты 1683 сл'1 и для дважды сопря-
женного 2,6,6-триметилциклогептадиенона-2,4 значение
1661 сл"1.
Имеются также многочисленные данные по спектрам
комбинационного рассеяния циклогептанонов [44, 45],
которые подтверждают, что частоты колебаний карбониль-
ной группы этих соединений по существу такие же, как
для кетонов с открытой цепью.
б. Шестичленные циклы. Как и следовало ожидать,
частоты полос поглощения карбонильной группы ненапря-
женных шестичленных циклов такие же, как у соединений
с открытой цепью. Уиффен и Томпсон [46] нашли значение
частоты, соответствующей карбонильному поглощению
циклогексанона, равным 1710 см1, а общий характерный
интервал частот карбонила был установлен для этих соеди-
нений Шеллом с сотрудниками [41].
Очень большое число стероидов, содержащих карбо-
нильные группы в шести- и пятичленных циклах, изучили
Гл. 9. Альдегиды и кетоны
211
Джонс и др. [7—11 ], которые обнаружили, что первые ведут
себя совершенно аналогично соединениям с открытой цепью.
Имеются очень малые, но вполне заметные различия частот
колебаний карбонильных групп С=О, расположенных
в различных шестичленных стеринных циклах, но ни в одном
случае частота не выходит за пределы общего интервала
1720—1706 см'1, если только нет сопряжения или каких-
либо других эффектов.
Все факторы, такие, как а-галогенирование, аф-сопря-
жение ит. п., которые влияют на частоту колебаний обыч-
ных карбонильных групп, точно в такой же мере влияют
на частоты карбонильных групп шестичленного цикла.
Например, Джонс и др. [9] обнаружили, что все 29 иссле-
дованных а,р-ненасыщенных шестичленных циклических
кетонов поглощают в области 1684—1674 см'1, а пять
соединений с а,р-а',р'-сопряжением поглощают в сбласти
1663—1660 смх. Большое количество работ, как уже указы-
валось, выполнено по исследованию а-галогензамещенных
циклогексанонов [115—121].
Введение в цикл гетероатома дает заметный эффект.
Обычные лактоны и лактамы рассматриваются ниже в соот-
ветствующей главе. Пироны и тиопироны имеют необычные
карбонильные частоты, и по реакционной способности они
значительно отличаются от обычных кетонов. Тарбелл
и Хоффманн [144] нашли у 1,4-тиопирона очень сильную
карбонильную полосу с частотой около 1639 см'1 и вторую
сильную полосу при 1574 см'1, что объясняется возросшим
резонансом, связанным с наличием у атома серы свободной
пары электронов; правильность такого объяснения под-
тверждается данными о сульфонах. Для последних при
наличии а,р-а',р'-непредельных связей обнаруживаются
обычные карбонильные частоты (1657 см'1).
Присоединение новых циклов, такого типа, как в мости-
ковых циклических структурах*, обычно приводит к уве-
личению напряженности в цикле, в связи с чем карбониль-
ная частота повышается [171, 172]. В то же время, когда
циклопентадиеноны конденсируются с ароматическими систе-
мами, частота С=О, как заметили Жозьен и Фасон [145],
* Имеются в виду мостики типа водородных или внутриком-
плексиых связей.— Прим. ред.
14*
212
Часть 11
закономерно уменьшается с увеличением числа конденси-
руемых циклов. Это обусловлено, по-видимому, возраста-
нием возможности резонанса структур.
в. Пятичленные циклы. Хартвелл, Ричардс и Томпсон
[12] установили, что циклопентанон в жидком состоянии
поглощает при 1772 слГ1, а Уиффен и Томпсон [46] нашли
значение частоты в случае раствора равным 1740 см'1.
Позднее результаты работ Джонса [9] по стеринам пол-
ностью подтвердили вывод о том, что частоты карбонила
пятичленных циклических соединений несколько повышены.
Этот автор отмечает 22 соединения такого типа, содержащие
карбонильную группу, которые в растворе поглощают
в области 1749—1745 см1. На этом основании ему удалось
дифференцировать карбонильные группы в пяти- и шести-
членных циклах. Подобное же увеличение частоты погло-
щения сверх значения частот для соответствующих слож-
ных эфиров обнаружено у пятичленных циклических лак-
тонов.
Эго усиление связи С=О с увеличением напряженности
цикла, по-видимому, является частным случаем более
общего явления, которое подробно обсуждалось теорети-
чески [47]. В основе его лежат, по-видимому, изменения
характера sp-гибридизации валентных о-орбит углерода,
приводящие к тому, что все связи, непосредственно отхо-
дящие от напряженного цикла, становятся сильнее, а все
связи самого цикла — слабее.
Для пятичленных циклических кетонов изменения кар-
бонильной частоты в результате сопряжения имеют тот же
порядок величины, как в случае ненапряженных циклов.
Так, например, аф-ненасыщенные группы СО пятичлен-
ных циклов поглощают [9, 90] при 1716 см'1; уменьшение
частоты на 38 см'1 такое же, как у нормальных аф-ненасы-
щенных кетонов. Дальнейшее сопряжение не дает, однако,
заметного эффекта, и тетрациклон поглощает при 1713 см'1
[146]. В то же время между эндо- и экзо-сопряжением
имеется некоторое отличие [90], и соединения второго типа
поглощают при несколько более низкой частоте (1690 см'1).
Джонс [9] показал, что|3,у-ненасыщенность не оказывает вли-
яния на полосу карбонильного поглощения и что два иссле-
довавшихся Ли-17-кетостероида поглощают при 1754 см'1.
Гл. 9. Альдегиды и кетоны
213
Замещение галогеном в a-положении повышает карбониль-
ную частоту несколько меньше, чем в случае циклогекса-
нона, и величина смещения отчасти зависит от степени
изогнутости циклопентанонового кольца 1119, 122].
г. Четырехчленные циклы. Четырехчленные циклы
с карбонилом широко не исследовались, за исключением
особого случая р-лактамов, но, согласно теории, частота
полосы поглощения в этом случае должна быть еще выше,
чем для пятичленных циклов. Это подтверждается немного-
численными имеющимися данными; так, например, изве-
стно, что циклобутанон в растворе [46] поглощает при
1775 см1.
д. Большие циклические системы. Зависимость карбо-
нильной частоты от размеров цикла изучалась многими
исследователями, а недавно эти работы пополнились также
исследованиями интенсивности полос. Фрейе и Франкен-
берг [147] показали, что для шести-, семи- и восьмичленных
циклов имеют место очень малые отличия карбонильных
частот и что последние не связаны с реакционной способ-
ностью карбонила. Прелог [148] и Гюнтард [149, 173]
подтвердили этот вывод на ряде кетонов (СН2)П—С=О
при п от 3 до 14 и обсудили изменения частоты, дипольного
момента и интенсивности в зависимости от размеров цикла.
Частота повышается при возрастании п до 5, а затем пре-
терпевает лишь очень незначительные изменения. В то же
время коэффициент поглощения закономерно падает с рос-
том п до 9, а затем становится примерно постоянным, полу-
ширина же полосы, наоборот, сначала растет, а затем падает
с увеличением размеров кольца.
Что касается карбонильной частоты, то аналогичные
результаты были получены и для очень больших цикли-
ческих систем, образующихся из бензоилцикланонов
[107, 150].
е. Трансанулярный эффект в больших циклических
системах. Для некоторых больших циклических систем,
содержащих атом азота, было найдено аномальное значе-
ние карбонильной частоты, хотя азот был отделен в этих
системах от карбонильной группы несколькими группами
^->СН2—. Этот эффект наблюдается только при совершенно
214
Часть II
определенных размерах и конфигурации циклов. Впервые
он был обнаружен для некоторых алкалоидных производ-
ных, дающих пониженную относительно ожидаемой карбо-
нильную частоту [151], а затем полностью подтвержден
результатами обширных исследований Леонарда и др.
[152—157]. Для систем типа (I), где R равно СН3 или С2Н3,
частота С=О была найдена равной примерно 1665 см'1
при п = 3 или п = 2 и 3, но при п = 2 или 4 или для
циклов из 11, 13, 15, 17, 19 или 23 членов эта частота равна
около 1700 см'1.
ЛСНг) — с = о
I
Х(сн,)п—снон
I
Происхождение этого эффекта становится понятным, если
формулу девятичленной циклической системы записать
в виде II. Действительно, становится очевидным, что непо-
средственная близость атома азота с его свободной электрон-
ной парой приводит к прямому взаимодействию с карбо-
нильной группой через внутримолекулярное пространство.
Близость азота и карбонильной группы в таких системах
подтверждается образованием ими с хлорной кислотой
мостиковых циклических производных и тем фактом, что
при увеличении размеров радикала R и связанных с этим
пространственных эффектов карбонильная частота растет.
Эти результаты представляют очень большой интерес для
объяснения смещений частот. Другие примеры такого
эффекта отмечались также Марионом и др. [158, 159].
5. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С = О В АРОМАТИЧЕСКИХ
ЦИКЛИЧЕСКИХ СИСТЕМАХ
а. Хиноны. Инфракрасные спектры хинонов имеют ряд
интересных особенностей. n-Бензохинон имеет в растворе
две карбонильные частоты (1669 см'1 и 1656 см'1), хотя
полоса с меньшей частотой довольно слабая [91]. Анало-
Гл. 9. Альдегиды и кетоны
215
гичная картина наблюдается и у многих других хинонов,
у которых с хиноновым циклом конденсирован только один
другой цикл; если же хиноновый цикл находится в центре,
например у 9,10-антрахинона, то наблюдается лишь одна
полоса. Точно также и некоторые замещенные п-бензо-
хиноны имеют две частоты, а другие только одну [160, 161 ].
Эти факты многократно обсуждались, и еще окончательно
не решен вопрос о том, представляет ли вторая полоса
частоту взаимосвязанных колебаний С=С или обе частоты
соответствуют несвязанным колебаниям отдельных групп
С=О. В большинстве случаев, однако, главная полоса
наблюдается в ожидаемом для а,|3-а',|3'-непредельных
кетонов интервале частот, хотя положение полосы и зависит
от природы и числа соседних циклов и электронных свойств
заместителей [174]. В случае полициклических хинонов
карбонильные частоты могут быть поставлены в соответст-
вие с рассчитанной электронной плотностью у атомов
углерода карбонильных групп [91].
Ряд антрахинонов с различными заместителями в цикле
был исследован Флеттом [32]. Незамещенный антрахинон
поглощает при 1681 см'1, т. е. при частоте, немного большей
обычной частоты для а,р-а',р'-диарилкетона. Замещение
различными группами (за исключением гидроксильной
и амино-групп, которые могут образовывать внутримоле-
кулярные водородные связи) приводит к изменению частоты
поглощения в пределах от 1692 (1,8-дихлорантрахинон)
до 1675 см'1 (1 -метоксиантрахинон). Аналогичные неболь-
шие изменения были обнаружены в случае антронов и оксан-
тронов. Таким образом, кроме случая сопряженных цепей
или циклов, замещение, по-видимому, не оказывает замет-
ного влияния на частоту карбонильного поглощения поли-
циклических хинонов, пока отсутствуют хелатные группы,
хотя природа и число циклов влияют на частоту карбонила.
Нельзя точно предсказать, в какой степени изменяется
частота. Так, например, в растворах бензохинон поглощает
при 1664 см'1, антрахинон — при 1681 см'1, антрон — при
1653 см'1 и оксантрон — при 1676 см'1. Жозьен и Фюзон
[48] наблюдали полосу карбонильного поглощения пирен-
хинона при 1639 см'1 и предположили, что такой большой
сдвиг полосы может быть связан с размерами сильно сопря-
женной молекулы. Поглощение хризенхинона при 1658 см'1
216
Ч а с т ь II
ближе к нормальному, и обычно с ростом числа циклов
наблюдается повышение частоты колебаний карбонила,
если только добавочные циклы не располагаются под углом
к циклу, содержащему карбонильную группу [73]. Поли-
циклические хиноны были также изучены Хаджи и Шеп-
пардом [49]. Эти авторы находят, что частоты полос карбо-
нильного поглощения в случае, когда два хиноноидных
карбонила расположены в различных циклах, ниже, чем
когда они находятся в одном и том же цикле. Соединения
первого типа поглощают в интервале 1655—1635 см'1 [49],
а соединения второго типа — в интервале 1680—1660 см'1
[49, 65]. Правильность последнего интервала была под-
тверждена Жозьен и Фюзоном [65, 73, 74, 90]. Спектры
полициклических хинонов рассматривались также Берг-
маном и Пинхасом [75, 81].
о-Хиноны привлекали пока меньшее внимание, хотя ряд
из них был исследован Жозьен и Фюзоном [90] и Оттингом
и Стайджером [162]. Карбонильные частоты этих соедине-
ний, по-видимому, не сильно отличаются от частот «-хино-
нов, но во второй из указанных работ для некоторых соеди-
нений в твердом состоянии приводятся несколько более
низкие значения. В случае дифенохинонов резонанс обусло-
вливает понижение карбонильных частот [90 ] и соединения
этого типа поглощают при —1640 см'1.
б. Трополоны. Кроме изучения ароматических шести-
членных циклов, большая работа была проведена по иссле-
дованию частот карбонильного поглощения семичленных
циклов, имеющих в состоянии резонанса три двойные связи
[25, 34, 42, 50, 57, 76—78, 163]. Типичным соединением
этого рода является трополон, который поглощает при
1615 см'1. Такая частота поглощения могла бы быть обу-
словлена в частности водородной связью с гидроксильной
группой, которая также имеется в молекуле, но прочность
водородной связи в этом случае меньше, чем у Р-дикетонов,
и валентные колебания ОН проявляются при 3100 см'1.
Уменьшение частоты полосы карбонильного поглощения
должно поэтому происходить по какой-либо другой при-
чине, и Кох [50] считает, что это явление связано с разме-
рами цикла. Он предполагает, что если у пятичленного
цикла происходит усиление связи С—О и ослабление связей
Гл. 9. Альдегиды и кетоны
217
С—С цикла, то у ароматического семичленного цикла дол-
жен наблюдаться обратный эффект: ослабление связи С=О
и усиление связей цикла. Трополоны могут также образо-
вывать внутрикомплексные соединения с медью, у которых
частота карбонильного поглощения уменьшается еще
больше, а именно до 1590 и 1595 см'1. Несколько удиви-
тельным является тот факт, что при наличии в орто-поло-
жении карбоксильной группы карбонильная частота ока-
зывается еще ниже [114] и циклические кетоны такого типа
поглощают при 1580—1570 см1.
Многие аналогичные случаи известны для ряда колхи-
цинов, у которых предполагается существование структур
подобного типа. Скотт и Тарбелл [25] для шести производ-
ных колхицина нашли сильную полосу 1620—1612 см'1,
которую они отнесли к карбонильной группе. Это в какой-то
мере подтверждается тем, что у тетрагидроколхицина ука-
занная полоса исчезает и заменяется полосой при 1710 см'1,
которая соответствует нормальной частоте колебаний кар-
бонила семичленного насыщенного цикла. Подобная полоса
вблизи 1610 см1 была обнаружена Никольсом и Тарбеллом
[42], а также Фабианом, Делароффом, Пойрером и Легран-
дом [1651 у ряда бензотрополонов и у колхицинов [79].
6. ИНТЕНСИВНОСТЬ КАРБОНИЛЬНОГО ПОГЛОЩЕНИЯ
Вследствие высокой интенсивности карбонильного погло-
щения измерение его особенно удобно при количественных
исследованиях, и не удивительно, что выполнено много
работ по определению молекулярных коэффициентов пога-
шения карбонильных соединений. Хамптон и Невел [21]
показали, что коэффициенты погашения карбонильных полос
поглощения сложных эфиров значительно меняются, при-
чем ход этих изменений в какой-то мере аналогичен ходу
изменения частот поглощения в результате сопряжения
и т, п. Сложные эфиры весьма сходного строения, погло-
щающие при примерно одинаковых частотах, имеют также
сравнимые молекулярные коэффициенты погашения.
Более того, пределы изменений интенсивности карбониль-
ного поглощения сложных эфиров очень сильно отличаются
от пределов интенсивности карбонильного поглощения
кетонов, так что на этом основании возможна их. грубая
218
Часть II
дифференциация. Например, для кетонных карбонильных
групп стероидов характерна интенсивность поглощения
в пределах 350—1350 (в единицах молекулярного коэффи-
циента погашения), тогда как для сложных эфиров харак-
терен интервал значений 350—770 [55]. На этом основании
Марион, Рамсай и Джене [55] смсгли ди44ерсьиирсЕать
карбонильное поглощение сложных эфиров и кетонов в ряду
22 алкалоидов, используя как характеристические интен-
сивности полос, так и характеристические частоты полос
поглощения.
Совсем недавно Кросс и Рольф [56] измерили молеку-
лярные коэффициенты погашения широкого класса карбо-
нильных соединений. Их результаты подтверждают вывод
о том, что те же факторы, которые влияют на частоту,
соответствующую карбонильному поглощению, влияют
также и на его характеристическую интенсивность, но что
для данного класса соединений интенсивности постоянны
в той же мере, что и частоты полос поглощения. Так,
например, молекулярный коэффициент погашения семи
диалкил кетонов, поглощающих винтервале 1724—1715С.Ш1,
колеблется от 174 до 199. Четыре циклических кетона
с шестичленными кольцами, поглощающие при 1721 —
1716 см'1, имеют коэффициент погашения от 301 до 334,
а для двух диарилкетонов, поглощающих в области 1665—
1664 см1, значение коэффициента погашения находится
в пределах от 404 до 412. Что касается двух насыщенных
алифатических альдегидов, поглощающих в области
1736—1735 см1, то они имеют молекулярные коэффициенты
погашения, равные 130 и 148.
Таким образом, изучение интенсивности карбонильных
полос поглощения представляет собой еще один способ
характеристики карбонильных групп, а совместное исполь-
зование данных об интенсивности и положении полос погло-
щения позволяет легко дифференцировать различные типы
карбонильных групп. Действительно, в отношении рас-
познавания шестичленных циклов и открытых цепей метод,
основанный на определении интенсивности поглощения,
даже более селективен и, несмотря на совпадение полос
карбонильного поглощения, позволяет выяснить тип при-
сутствующей группировки. Этот метод был успешно при-
менен Джонсом и др. [10, 83] для идентификации карбо-
Гл. 9. Альдегиды и кетоны
219
нильных групп, находящихся в цикле или в боковой цепи
стеринов.
Метод определения карбонильного поглощения по
интенсивности полос является сравнительно новым, и хотя
его пригодность вполне очевидна, он был широко исполь-
зован только для стероидных кетонов, которые исследова-
лись Джонсом, Рамсаем, Дейром и Добрайнером [10].
Эти авторы определяли интегральное поглощение, а не
молекулярные коэффициенты погашения в максимумах
полос, и таким путем они получили более удовлетворитель-
ные значения интенсивности. Ими была изучена интенсив-
ность карбонильного поглощения 55 различных кетостерои-
дов и обнаружено, что интегральная интенсивность изме-
няется в пределах от 1,2 до 4,1. Интенсивность поглощения
карбонильных групп кетонов различных типов может, сле-
довательно, изменяться максимально в 4 раза, что нахо-
дится в согласии с ранними исследованиями Ричардса
и Бартона [58]. Однако в этих пределах интенсивность
поглощения какого-либо отдельного специфического типа
карбонильных соединений остается постоянной. Так, напри-
мер, интенсивность карбонильного поглощения стеринов
не претерпевает заметных изменений при различных поло-
жениях заместителей в кольцах, но сильно отличается от
интенсивности поглощения соединений с карбонильной
группой, расположенной в боковой цепи. Аналогичным
образом обнаружены систематические изменения интенсив-
ностей карбонильных полос поглощения в тех случаях,
когда карбонил сопряжен или у соседней метиленовой
группы имеется атом галогена. Обзор данных об изменениях
молярного коэффициента погашения в зависимости от типа
карбонильной группы в стероидах был сделан Джонсом
и Сандорфи [83]. Чтобы проиллюстрировать указанное
положение, ниже приведены типичные значения для неко-
торых из этих классов соединений (в единицах интенсив-
ности):
а-Бромкетоны............................. 1,2—1,99
20-Кетостероиды (СО в боковой цепи) 1,79
Насыщенные циклические кетосте-
роиды ........................... 2,16—2,74
аф-Сопряженные кетостероиды
(кольцо)....................... 3,4—4,1
220
Часть II
Ричардс и Бартон обнаружили также, что у ди- и поликар-
бонильных стероидов интенсивности поглощения С=О
аддитивны, так что можно определять число карбониль-
ных групп, входящих в неизвестное соединение, даже в
случае перекрытия полос, если только известны типы сое-
динений.
Таким образом, определение интенсивности полосы слу-
жит существенным дополнением к ее характеристике по
частоте, соответствующей поглощению, и знание частоты
и интенсивности позволяет обычно однозначно идентифи-
цировать данный частный тип карбонильной структуры.
Точное определение интенсивности поглощения в растворах
представляет исключительно трудную задачу, и возмож-
ность такого определения многократно обсуждалась. Тем
не менее для целей идентификации различия интенсивностей
достаточно велики и ошибки измерений не могут играть
большой роли и снижать надежность выводов. Для успеш-
ного проведения исследований необходимо располагать до-
статочно большим количеством основных данных об измене-
ниях интенсивности карбонильной полосы в зависимости
от строения молекулы. При этом можно получить очень
полезные сведения, и это подтверждается недавними
исследованиями Френсиса [80], Бюрера и Гюнтарда [149]
и Барроу [24]. Особенно интересной в этом отношении
является работа Барроу, в которой на большом ряде раз-
личных карбонилсодержащих соединений ему удалось уста-
новить соотношение между интенсивностью карбонильной
полосы и энергией резонанса системы.
7. ХАРАКТЕРИСТИЧЕСКИЕ ЧАСТОТЫ ПОЛОС
ПОГЛОЩЕНИЯ КЕТОНОВ, НЕ ОБУСЛОВЛЕННЫХ ВАЛЕНТ-
НЫМИ КОЛЕБАНИЯМИ С = О
Присутствие в молекуле карбонильной группы часто
приводит к появлению, помимо полосы, характерной для
карбонильного поглощения, полосы средней интенсивности
в области частот валентных колебаний ординарных связей.
Хаджи и Шеппард [49] для полициклических хинонов,
например, наблюдали сильную полосу поглощения в интер-
вале 1350—1200 см'1, которая отсутствует у соответствую-
щих углеводородов. Они предположили, что эта полоса
Гл. 9. Альдегиды и кетоны
221
может быть обусловлена каким-то движением карбонильной
-группы, связанной с остальной частью молекулы. Несом-
ненно, что у большинства кетонов полоса поглощения в этой
области имеется, и Колтуп [59] указал для нее следующие
широкие пределы частот: для алифатических кетонов интер-
вал 1325—1215 см'1 и для ароматических кетонов интервал
1225—1075 см1. Однако эти интервалы частот настолько
широки и имеется так много других колебаний, обусловли-
вающих появление полос поглощения при сравнимых
частотах, что эти данные о поглощении не нашли приме-
нения при определении кетонов; для ограниченной же
группы очень родственных соединений изучение этой
области может дать полезные сведения. Указанные выше
и некоторые другие полосы поглощения карбонильной
группы в области низших частот рассматривались Джон-
сом и др. [52, 531.
Кетоны могут быть идентифицированы также по полосам
деформационных колебаний группы С=О. Группировка
—СО—СН3 дает три полосы, связанные с этими колеба-
ниями [51]: около 600, 500 и 400 см'1. Если, однако, вблизи
карбонильной группы имеется разветвление цепи, то про-
исходят значительные изменения Спектра, так что в случае,
например, метилизопропилкетона указанные полосы не
могут быть идентифицированы. Поэтому данная корреляция
представляет главным образом академический интерес.
8. ПОГЛОЩЕНИЕ ВАЛЕНТНЫХ КОЛЕБАНИЙ
С=О У АЛЬДЕГИДОВ
Относительно альдегидов имеется гораздо меньше све-
дений, чем относительно кетонов, но есть указания на то,
что в обоих случаях на частоту поглощения влияют почти
одни и те же факторы.
У нормальных несопряженных алкилальдегидов полоса
карбонильного поглощения характеризуется несколько
большей частотой, чем у соответствующих кетонов, и интер-
вал поглощения составляет примерно 1740—1720 см'1.
Как и у кетонов, значение этой частоты, получаемое при
изучении вещества в газовой фазе, заметно выше, и Харт-
велл и др. [12], например, приводят значение частоты
поглощения 1752 см'1 для ацетальдегида и 1757 см'1.для
222
Часть 11
пропионового альдегида. Аналогичным образом имеется
лишь очень небольшое взаимодействие между двумя сосед-
ними альдегидными группами, и глиоксаль поглощает
почти при такой же частоте, что и формальдегид [28].
У ненасыщенных альдегидов с двойной связью в
а,р-положении полоса поглощения карбонила сдвигается
на величину того же порядка, что и для кетонов, и они
поглощают в интервале 1705—1685 см'1. Эффект удлинения
сопряженной цепи хорошо изучен Блоутом и др. [16].
Для ряда полиеновых альдегидов было обнаружено, что
первый член ряда — кротоновый альдегид, поглощает в
растворе при 1685 см'1, в то время как остальные члены
ряда, содержащие от двух до семи сопряженных двойных
связей, поглощают в интервале 1677—1664 см1. Авторы
пришли поэтому к заключению, что влияние второй сопря-
женной у,д-связи мало, а эффект дальнейшего сопряжения
совершенно незначителен. Точно так же в ряду а-фурилаль-
дегидов, содержащих длинные полиеновые цепи и фуриль-
ный остаток, не наблюдается сколько-нибудь заметного
смещения полосы карбонильного поглощения при переходе
от первого члена ряда к последующему. Аналогичные
результаты были получены Скрокко и Сальветти [54],
которые обсудили их также с теоретической точки зрения.
Ароматические циклы в сопряжении с альдегидными
группами мало влияют на полосу карбонильного поглоще-
ния, и ароматические альдегиды в растворе поглощают
в интервале 1710—1695 см1. Бензальдегид поглощает при
1704 см'1, а а- и р-нафтальдегиды [17] поглощают соответ-
ственно при 1700 и 1702 см'1. В этом случае замещение
в цикле группами, сильно притягивающими или отталки-
вающими электроны, также оказывает заметное влияние
на полосу карбонильного поглощения.
Хотя замещение галогеном в a-положении широко
не изучалось, однако есть все основания предполагать, что
здесь будут иметь место те же эффекты внутренних полей,
как и в соответствующих кетонах, и карбонильная частота
будет повышена. Например, у хлораля в растворе карбо-
нильная полоса имеет частоту 1762 см'1. Точно так же
поворотная изомерия моно- и ди-а-галогенальдегидов при-
водит, по-видимому, к появлению двух карбонильных
частот.
Гл. 9. Альдегиды и кетоны
223
Обычным является также образование р-окси-а,^-непре-
дельными альдегидами хелатных структур. Хансбергер [17]
обнаружил подобные эффекты для 1-окси-2-нафтальдегида
и для 2-окси-1-нафтальдегида. У З-окси-2-нафтальдегида
этот эффект меньше, что, по мнению автора, объясняется
в какой-то степени фиксацией связей в нафталине.
Метоксипроизводные этого ряда имеют нормальные кар-
бонильные частоты и внутрикомплексных соединений не
образуют.
Указанные выше эффекты наблюдались также в случае
альдегидов инданового ряда [133] и альдегидов родствен-
ных камфоре [26]. С другой стороны, как и у а-дикетонов,
взаимодействия соседних карбонильных групп не наблю-
дается. Косгров и др. [166] исследовали ряд соединений,
таких как фенилглиоксаль и подобные кетоальдегиды,
у которых обе карбонильные частоты имеют по существу
обычные значения.
Интенсивность карбонильного поглощения альдегидов
систематически не изучалась, но Кросс и Рольф [56] при-
вели некоторые значения коэффициентов погашения аль-
дегидов, которые можно сравнить со значениями для соот-
ветствующих кетонов. Пропиональ и гептаналь (1735
и 1736 см1) имеют молекулярные коэффициенты погашения
130 и 148, а диалкилкетоны — примерно 180. Для кротоно-
вого альдегида коэффициент погашения равен 234, а для
бензальдегида 324. Последнее значение сравнимо со значе-
нием коэффициента ацетофенона 310. Из имеющихся
ограниченных данных следует, по-видимому, что интенсив-
ность карбонильных полос поглощения альдегидов должна
меняться с изменением структуры почти так же, как в слу-
чае кетонов.
В спиртовом растворе наблюдается отчетливое уменьше-
ние интенсивности карбонильного поглощения альдегидов
вследствие реакции RCHO + ROH -> RCH(OH)OR.
Ашдаун и Клетц [60] сообщили о многих таких случаях
и показали, что наблюдается также соответствующее умень-
шение интенсивности полос поглощения валентных и дефор-
мационных колебаний С—Н альдегидов и в то же время
появляется новая полоса вблизи 1020—1110 см1, которая
может быть отнесена к связи С—О—.
224
Часть II
9. ПОГЛОЩЕНИЕ ВАЛЕНТНЫХ КОЛЕБАНИЙ
С—Н у АЛЬДЕГИДОВ
Высокая характеристичность валентных колебаний С—Н
уже обсуждалась в предыдущих разделах. Следует ожидать,
что альдегиды будут иметь в той же области характеристи-
ческое поглощение, вызываемое валентными колебаниями
атома водорода, присоединенного к карбонильной группе.
Частота этих колебаний должна, очевидно, отличаться
от частоты колебаний СН в метиленовой группе. Это под-
тверждается имеющимися данными, хотя следует отметить,
что число исследованных соединений очень ограниченно.
Колтуп [59] приводит для частот этих колебаний интервал
2900—2700 см'1, а Позевский и Когешелл [61] недавно
обнаружили у многих альдегидов две полосы поглощения
в данной области. Они находятся вблизи 2720 и 2820 см'1.
Возникновение двух полос поглощения связано, по-види-
мому, с появлением, помимо фундаментальной частоты,
обертона или составной частоты. Однако не ясно, какая
из них соответствует фундаментальной частоте. Тем не
менее появление полосы поглощения вблизи 2720 см'1
вместе с наличием полосы поглощения С=О является
надежным доказательством присутствия группы СНО.
Пинхас [167, 175] наблюдал эту полосу для ряда альдеги-
дов. Он заметил, однако, что иногда она смещается в сто-
рону больших частот, особенно у opmo-замещенных бензаль-
дегида, и предположил, что это может быть обусловлено
образованием водородной связи. Последнее, однако, едва ли
возможно, так как частота при этом должна была бы сме-
щаться в противоположном направлении. Тем не менее
в цитированных работах содержится большое количество
полезных сведений о положении обеих полос поглощения
С—Н альдегидов.
10. ДРУГИЕ ХАРАКТЕРИСТИЧЕСКИЕ КОЛЕБАНИЯ
АЛЬДЕГИДОВ
Деформационные колебания группы СНО значительно
труднее надежно идентифицировать, но можно предполо-
жить, что им соответствует полоса поглощения около
900 см'1. Поскольку интенсивность невелика, то полосу труд-
Гл. 9. Альдегиды и кетоны
225
но идентифицировать среди многих других полос в данной
области, и она поэтому не особенно удобна для анализа.
Колтуп 159] приводит для этих колебаний интервал 975—
825 см \ что отражает значительное изменение в положении
полосы, однако Ашдаун и Клетц [60] приводят значение
780 см 1 для «-бутиральдегида и 795 и 905 см 1 для изобу-
тиральдегида.
Как и в случае кетонов, присутствие карбонильной
группы вызывает изменение колебаний некоторых сосед-
них связей С—С, так что появляются дополнительные
полосы поглощения в области валентных колебаний угле-
род-углеродных связей. Эти полосы имеют среднюю интен-
сивность и могут быть использованы лишь в качестве не
очень надежного подтверждения наличия альдегидной
группы. Колтуп [59] указывает следующие интервалы
частот поглощения: 1440—1325 см'1 для алифатических
альдегидов, 1415—1350, 1320—1260 и 1230—1160 см'1
для ароматических альдегидов. Все немногие исследован-
ные нами альдегиды также поглощают в этих интервалах,
но мы обнаружили, кроме того, заметные смещения полос
поглощения внутри этих интервалов для ароматических
соединений, отличающихся только относительным рас-
положением второго заместителя в кольце. Имеется, оче-
видно, много различных факторов, определяющих значе-
ния этих частот, и до тех пор, пока они не будут полностью
изучены, нецелесообразно пытаться применять данную
область для идентификации альдегидов.
ЛИТЕРАТУРА
1. Wen iger, Phys. Rev., 31, 388 (1910).
2. Barnes, Gore, Liddel, Van Zandt Will ia ms,
Infra-red Spectroscopy (Reinhold, 1944).
3. Thompson, Torkington, J. Chem. Soc., 1945, 640.
4. Rasmussen, Tunnicliff, Brattai n, J. Amer.
Chem Soc., 71, 1068 (1949).
5. Bonino, Scrocco, Atti accad. Naz. Lincei, 6. 421 (1949).
6. Cromwell, Miller, Johnson, Frank, Wallace,
J. Amer. Chem. Soc., 71, 3337 (1949).
7. Jones, Van Zandt Williams, Whalen, Do-
brine r, J. Amer. Chem. Soc., 70, 2024 (1948).
8. Jones, Humphries, Dobriner, J. Amer. Chem. Soc.,
71, 241 (1949).
15 Л. Беллами
226
Часть 11
9. Jones, Humphries, Dobriner, J. Amer. Chem.
Soc., 72, 956 (1950).
10. J о n e s, Ramsay, К e i r, D о b r i n e r, J. Amer. Chem.
Soc., 74, 80 (1952).
11. Jones, Dobriner, Vitamins and Hormones, Vol. 7 (N. Y.
Academic Press, 1949), p. 294.
12. Hartwell, Richards, Thompson, J. Chem. Soc.,
1948, 1436.
13. G г о v e, Willis, J. Chem. Soc., 1951, 877.
14. Kohlrausch, Pongrat z, Z. Physik. Chem., B27, 176,
1934.
15. T u r n e г, V о i t 1 e, J. Amer. Chem. Soc., 73, 1403 (1951).
16. В 1 о u t, Fields, Karp lus, J. Amer. Chem. Soc., 70,
194 (1948).
17. H u n s b e r g e r, J. Amer. Chem. Soc., 72, 5626 (1950).
18. Soloway, F r i e s s, J. Amer. Chem. Soc., 73, 5000 (1951).
19. G u t s c h e, J. Amer. Chem. Soc., 73, 786 (1951).
20. Cheng, Z. physikal Chem., B24, 293 (1934).
21. Hampton, Newell, Analyt. Chem., 21, 914 (1949).
22. W о о d w a r d, J. Amer. Chem. Soc., 63, 1123 (1941).
23. Djerassi, Scholz, J. Amer. Chem. Soc., 70, 1911
(1948).
24. В a г г о w, J. Chem. Phys., 21, 2008 (1953).
25. S с о t t, Tar bell, J. Amer. Chem. Soc., 72, 240 (1950).
26. Bonino, Miron e, Gazz., 89, 1058 (1954).
27. Kohlrausch, Pongratz, Ber., 67, 976 (1934).
28. Thompson, Trans. Faraday Soc., 36, 988 (1940).
29. Randall, Fowler, Fuson, Dangl, Infra-red De-
termination of Organic Structures (Van Nostrand, 1949).
30. Rasmussen, Brattai n, J. Amer. Chem. Soc., 71, 1073
(1949).
31. G о r d y, J. Chem. Phys., 8, 516 (1940).
32. Flett, J. Chem. Soc., 1948, 1441.
33. Woodward, Stork, Wineman, Nelson, Both-
n e r-B у, в печати; Woodward, Kovach, J. Amer.
Chem. Soc., 72, 1009 (1950).
34. A u 1 i n-E rdtman, Theorell, Acta Chem. Scand., 4,
1490 (1950).
35. Cromwell, Barker, Wankel, Vanderhorst,
Olson, A n g 1 i n, J. Amer. Chem. Soc., 73, 1044 (1951).
36. L e с о m t e, Discuss. Faraday Soc., 9, 125 (1950).
37. M о r g a n, U. S. Atomic Commission, AECD 12659, 16 (1949).
38. L e с о m t e, Rev. Optique, 28, 253 (1949).
39. Duval, Lecumte, Douville, Ann. Physique, 17, 5
(1942).
40. Munday, Nature, 163, 443 (1949).
41. The Chemistry of Penicillin (Princeton University Press, 1949),
p. 404.
42. N i c h о 1 1 s, T a r b e 1 1, J. Amer. Chem. Soc., 74, 4935 (1952).
43. G u t s c h e, J. Amer. Chem. Soc., 71, 3513 (1949).
44. В i g u a r d, Bull. Soc. Chem., 7 (5), 894 (1940).
Гл. 9. АльдегидьЦи кетоны
227
45. G о d с h о t, С a n q u i 1, Compt. rend. Acad. Sci. (Paris),
208, 1065 (1939).
46. W h i f f e n, Thompson,!. Chem. Soc., 1946, 1005.
47. Coulson, Moffitt, Phil. Mag., 40, 1 (1949).
48. J о s 1 e n, F и s о n, J. Amer. Chem. Soc., 73, 478 (1951).
49. H a d-i i, S h e p p a r d, J. Amer. Chem. Soc., 73, 5460 (1951).
50. К о c h, J. Chem. Soc., 1951, 512.
51. Lecomte, Josien, Lascombe, Bull. Soc. Chim.
France, 1956, 163.
52. J о n e s, H e r I i n g, К a t z e n e 1 1 e n b о g e n, J. Amer.
Chem. Soc., 77, 651 (1955).
53. J о n e s, N о 1 i n, R о b e r t s, J. Amer. Chem. Soc., 77, 6331
(1955).
54. Scrocco, Sal vet t i, Boll. Sci. Fac. Chim. Ind. Bologna,
12, 93 (1954).
55. Marion, Ramsay, Jones, J. Amer. Chem. Soc., 73,
305 (1951).
56. Cross, Rolfe, Trans. Faraday Soc., 47, 354 (1951).
57. Bartels-Keith, Johnson, Chem. and Ind., 1950, 677.
58. Richards, Burton, Trans. Faraday Soc., 45, 874 (1949).
59. С о 1 t h и p, J. Opt. Soc. Amer., 40, 379 (1950).
60. Ashdown, К 1 e t z, J. Chem. Soc., 1948, 1454.
61. Pozefsky, Coggeshall, Analyt. Chem., 23. 1611 (1951),
62. Jones, Humphries, Herling, Dobriner, J.
Amer. Chem. Soc., 74, 2820 (1952).
63. Jones, Ramsay, Herling, Dobriner, J. Amer.
Chem. Soc., 74, 2828 (1952).
64. Hunsberger, Ketcham, Gutowsky, J. Amer.
Chem. Soc., 74, 4839 (1952).
65. Josien, Fuson, Compt. rend. Acad. Sci. (Paris), 234, 1680
(1952).
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
Duval, Freymann, Lecomte, Bull. Soc. Chim.
(France), 1952, 106.
Bella my, Spicer, Strickland, J. Chem. Soc.,
1952, 4653.
Haszeldine, Nature, 168, 1028 (1951).
Barnes, Pinkney, J. Amer. Chem. Soc., 75, 479 (1953).
Leonard, Robinson, J. Amer. Chem. Soc , 75, 2143
(1953).
Hergert, Kurth, J. Amer. Chem. Soc., 75, 1622 (1953).
Birch, J. Chem. Soc., 1951, 3026.
Josien, Fuson, Lebas, Gregory, J. Chem. Phys.,
21, 331 (1953).
Josien, Fuson, Bull. Soc. Chim. (France), 1952, 389.
Bergmann, Pinchas, Bull. Res. Council Israel, 1, 87
(1952).
Doering, Knox, J. Amer. Chem. Soc., 74, 5683 (1952).
Doering, Knox, J. Amer. Chem. Soc., 75, 297 (1953).
Doering, Hiskey, J. Amer. Chem. Soc., 74, 5688 (1952).
Nicholls, Tarbell, J. Amer. Chem. Soc., 75, 1104
(1953).
15*
228
Часть 11
80. Francis, J. Chem. Phys., 19, 942 (1951).
81. В e r g m a n n, Pinchas, J. Chim. Phys., 49, 537 (1952).
82. Lecomte, Bull. Soc. Chim. France, 1955, 1026.
83. Д ж о н с, С а н д о p ф и, Применение спектроскопии в хи-
мии, Издатинлит, М., 1959, стр. 374.
84. J о n е s, Н е г 1 i n g, J Org. к.hem., 19, 1252 (1954).
85. L о г d, Miller, Applied Sperro-. py, 10, 115 (1956).
86. Ш и г о p и н, ДАН СССР, 96, 769 (1954).
87. В е 1 1 a m у, J. Chem. Soc., 1955, 4221.
88. К a g а г i s е, J. Amer. Chem. Soc., 77, 1377 (1955).
89. Margoshes, Fillwalk, Fassel, Rundle,
J. Chem., Phys., 22, 381 (1954).
90. Fuson, Josie n, Shelton, J. Amer. Chem. Soc., 76,
2526 (1954).
91. J о s i e n, D e s c h a m p s, J. Chim. Phys., 52, 213 (1955).
92. Scrocco, Libert i, Ricerca. Sci., 24, 1687 (1954).
93. Bellamy, Williams, J. Chem. Soc., 1957, в печати.
94. Lascombe, Josie n, Bull. Soc. Chim. France, 1955, 1227.
95. Cole, T h о r n t о n, J. Chem. Soc., 1956, 1007.
96. Josie n, Lascombe, J. Chim. Phys., 52, 162 (1955).
97. Josie n, Lascombe, Compt. Rend. Acad. Sci. (Paris),
238, 2414 (1954).
98. Bayliss, Cole, Little, Aust. J. Chem., 8, 26 (1955).
99. Чулановский, ДАН СССР, 101, 457 (1955).
100. Josie n, Lascombe, Lecomte, Mathieu, Compt.
Rend. Acad. Sci. (Paris), 240, 1982 (1955).
101. W h e 1 a n d, Resonance in Organic Chemistry, Wiley, New
York, 1955, p. 52.
102. Mecke, Noack, Angew. Chem., 18, 150 (1956).
103. Heilman n, De Gaudemaris, Arnaud, Compt.
Rend. Acad. Sci. (Paris), 240, 1995 (1955).
104. Franzen, Chem. Ber., 88, 717 (1955).
105. Theus, Surber, Colombi, Sehin z, Helv. Chim.
Acta, 38, 239 (1955).
106. Gambon i, Theus, Sehin z, Helv. Chim. Acta, 38,
255 (1955).
107. Schubert, Sweeney, J. Amer. Chem. Soc., 77, 4172
(1955).
108. Tanaka, Nagakura, Kobayashi, J. Chem. Phys.,
24, 311 (1956).
109. Wiberley, Bunce, Analyt. Chem., 24, 623 (1952).
110. F u s о n, J б s i e n, C a r y, J. Amer. Chem. Soc., 73, 4445
(1951).
111. J о s i e n, Fuson, Bull. Soc. Chim. (France), 1952, 389.
112. Cromwell, Hudson, J. Amer. Chem. Soc., 75, 872 (1953).
113. D i c k s о n, P a g e, J. Chem. Soc., 1955, 447.
114. J ones, J. Amer. Chem. Soc., 75, 4§39 (1953).
115. Corey, J. Amer. Chem. See., 75, 2301, 3297 (1953); 76, 175
(1954).
116. Corey, Experimentia, 9, 329 (1953).
117. С о г e у, В u r k e, J. Amer. Chem. Soc., 77, 5418 (1955).
Гл. 9. Альдегиды и кетоны
229
118. С о г е у, Т о р i е, W о z n i а к, J. Amer. Chem. Soc., 77,
5415 (1955).
119. S a n d г i s, О u r i s s о n, Bull. Soc. Chim. (France), 1956,
958.
120. I n a у a m a, Pharm. Bull. Japan, 4, 198 (1956).
121. К u m m 1 e r, H u i t r i c, J. Amer. Chem. Soc., 78, 3369
(1956).
122. Brutcher, Roberts, Barr, Pearson, J. Amer.
Chem. Soc., 78, 1507 (1956).
123. Bellamy, Thomas, Williams, J. Chem. Soc.,
1956, 3704.
124. Mizushima, Shimanouchi, Miyazawa,
Ichishima, Kuratani, Nakagawa, Shido,
J. Chem. Phys., 21, 815 (1953).
125. Daasch, Kagarise, J. Amer. Chem. Soc., 77, 6156 (1955).
126. Fuson, House, Melby, J. Amer. Chem. Soc., 75, 5952
(1953).
127. Leonard, L e i t e п e n, M о t t u s, J. Amer. Chem. Soc.,
75, 3300 (1953).
128. Alder, Schafer, Esser, Kriger, Reubke, Ann.,
593, 23 (1955).
129. В ellamy, Beecher, J. Chem. Soc., 1954, 4487.
130. Ш и г о p и н, ЖФХ, 28, 584 (1954).
131. P a г к. Brown, Lacher, J. Amer. Chem. Soc., 75, 4753
(1953).
132. Fistert, Reiss, Chem. Ber., 87, 92 (1954).
133. Hunsberger, Lednicer, Gu tow sky, Bunker,
T u n s о i g, J. Amer. Chem. Soc., 77, 2466 (1955).
134. Hadji, Sheppard, Trans. Faraday Soc., 50, 911 (1954).
135. Shaw, S i m p s о n, J. Chem. Soc., 1955, 655.
136. Bellamy, В r a n c h, J. Chem. Soc., 1954, 4491.
137. Bender, Figueras, J. Amer. Chem. Soc., 75, 6304 (1953).
138. Sweeney, Curran, Quagliano, J. Amer. Chem.
Soc., 77, 5508 (1955).
139. Susz, Cooke, Helv. (Jhirn. Acta, 37, 1273 (1954).
140. Cooke, Susz, Herschmann, Helv. Chim. Acta, 37,
1280 (1954).
141. J e w e 1 1, Spur, Ohio State Symposium, 1956, cf. Spectro-
chim. Acta, 8, 305 (1956).
142. Bellamy, Williams, J. Chem. Soc., 1957, 861.
143. Duncanson, J. Chem. Soc., 1953, 1207.
144. Tarbell, Hoffman, J. Amer. Chem. Soc., 76, 2451 (1954).
145. J о s i e n, F u s о n, J. Phys. Radium, 15, 654 (1954).
146. Jones, Sandorfy, Trucker, J. Phys. Radium, 15,
320 (1954).
147. Freis s, Frankenberg, J. Amer. Chem. Soc., 74, 2679
(1952).
148. P r e 1 о g, J. Chem. Soc., 1950, 420.
149. В u r er, GQnthard, Helv. Chim. Acta, 39, 356 (1956).
150. Schubert, Sweeney, Latourette, J. Amer.
Chem. Soc., 76, 5462 (1954),
230
Часть II
151. Anet, Bailey, Robinson, Chem. and Ind., 1953, 944.
152. Leonard Fox, Oki, Chiavarelli.J. Amer. Chem.
Soc , 76, 630 (1954).
153. Leonard, Oki, J. Amer. Chem. Soc., 76, 3463 (1954).
154. Leonard, Fox, Oki, J. Amer. Chem. Soc., 76, 5708 (1954).
155. Leonard, Oki, Chiavarelli.J. Amer. Chem. Soc.,
77, 6234 (1955).
156. Leonard, Oki, Brader, Boaz, J. Amer. Chem. Soc.,
77, 6237 (1955).
157. Leonard, О к i, J. Amer. Chem. Soc., 77 , 6241, 6245 (1955).
158. Mottus, Schwarz, Marion, Can. J. Chem., 31, 1144
(1953).
159. Anet, Marion, Can. J. Chem., 32, 452 (1954).
160. В archewitz, Tatibouet, Souchay, Compt.
Rend. Acad. Sci. (Paris), 236, 1652 (1953).
161. Yates, Ardas, Fieser, J. Amer. Chem. Soc., 78, 650
(1956).
162. Otting, Staiger, Chem. Ber., 88, 828 (1955).
163. Kuratani, Tsuboi, Shimanouchi, Bull. Chem.
Soc. Japan, 25, 250 (1952).
164. Tarbell, Smith, Boekelheide, J. Amer. Chem.
Soc., 76, 2470 (1954).
165. Fabian, Delaroff, Poirier, Legrand, Bull.
Soc. Chim. France, 1955, 1455.
166. Cosgrove, Daniels, Whitehead, Goulden,
J. Chem. Soc., 1952, 4821.
167. P i n c h a s, Analyt. Chem., 27, 2 (1955).
168. Heilman n, Gaudemaris, Arnaud, Bull. Soc.
Chim. France, 1957, 112, 119.
169. Jones, Forbes, Mueller, Can. J. Chem., 35, 504 (1957).
170. Mo hr ba cher, Cromwell, J Amer. Chem. Soc., 79,
401 (1957).
171. Allen, Davis, Stewart, Van Allen, J. Org.
Chem., 20, 306 (1955).
172. Allen, Van Allen, J. Org Chem., 20, 323 (1955).
173. BQrer, Giinthard, Chimica, 11, 96 (1957).
174. Josien, Deschamps, J. Chim. Phys., 54, 885 (1957).
175. P i n c h a s, Analyt. Chem., 29, 334 (1957).
176. Whiffen, частное сообщение.
177. Susz, Wuhmann, Helv. Chim. Acta, 40, 722, 971 (1957).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Шигорин Д. Н., Сыркин я. К-, ДАН СССР, 70, 1033
(1950).
Карякин, А. В., Никитин В. А., Иванов К. И.,
ЖФХ, 27, 1856, 1867 (1953).
Ней л а ид О. Я., В а н а г Г. Я., Успехи Химии, 28, 436
(1959).
Глава 10
КАРБОНОВЫЕ КИСЛОТЫ
1. ВВЕДЕНИЕ
Карбоновые кислоты обычно существуют в виде димеров
с очень прочными водородными мостиками между карбо-
нильной и гидроксильной группами двух молекул. Такого
рода ассоциация молекул в какой-то мере сохраняется даже
в парообразном состоянии и в разбавленных растворах
(для некоторых растворителей), так что частота колебаний
карбонила значительно изменена по сравнению с нормаль-
ной частотой. Поэтому такие вещества при исследовании
их методом инфракрасной спектроскопии обычно приме-
няются в твердом или жидком состоянии. При этом в какой-
то степени маскируются различия, обусловленные измене-
ниями структуры молекулы непосредственно вблизи кар-
бонильной группы, так что различия между а,р-ненасыщен-
ными или а-галогензамещенными кислотами не всегда
бывают так отчетливо выражены, как в случае кетонов.
Тем не менее те же самые факторы, которые влияют на
карбонильные частоты кетонов, обычно действуют в слу-
чае карбоновых кислот и смещение полос происходит
в одном и том же направлении. Карбонильные полосы
поглощения карбоновых кислот лежат почти в той же
спектральной области, что и у кетонов и альдегидов, однако
кислоты могут быть идентифицированы путем изучения
других областей спектра.
Поскольку водородная связь в этих кислотах является
чрезвычайно сильной, то и частоты валентных колебаний
ОН настолько отличаются от нормальных значений, что
становятся характеристичными, поэтому изучение соот-
ветствующей спектральной области позволяет установить
присутствие карбоновых кислот.
Имеются также другие области спектра, при исследо-
вании которых можно получить некоторые данные, хотя
232
Часть II
и менее надежные, чем те, о которых упоминалось выше.
Такими областями являются области вблизи 1400, 1250
и 920 см'1. Происхождение полос здесь неясно. Первые две
полосы относятся различными авторами к валентным коле-
баниям ординарной связи С—О—. Третья полоса, по-види-
мому, возникает в результате деформационных колеба-
ний ОН.
Данные об этих спектральных областях можно успешно
использовать для идентификации карбоновых кислот, осо-
бенно если при этом сравнивать полученную интенсивность
карбонильного поглощения с его интенсивностью у из-
вестных кислот. Кроме того, при изучении частоты С=О
можно иногда получить некоторые данные о непо-
средственном окружении группы СООН.
Таблица 9
Частота, сл-1
Валентные колебания свободной группы
ОН карбоксила ...................
Валентные колебания связанной груп-
пы ОН карбоксила...................
Колебания С=О:
Насыщенные алифатические кислоты
а-Г алогензамещенные алифатические
кислоты .........................
аф-Ненасыщенные кислоты..........
Ароматические кислоты ...........
Кислоты, имеющие внутреннюю водо-
родную связь ....................
Валентные колебания С—О или дефор-
мационные колебания ОН...........
Деформационные (внеплоскостные) коле-
бания ОН ..........................
,0
-Cf
хо-
Прогрессия полос твердых жирных кис-
лот ...............................
3560—3500 (ср.)
2700—2500 (сл.)
1725-1700 (с.)
1740—1720 »
1715—1690 »
1700—1680 »
1670—1650 »
1440—1395 (сл.)
1320-1211 (с.)
950— 900 (перем.)
1610—1550 (с.)
1420—1300 »
1350—1180 (сл.)
Гл. 10. Карбоновые кислоты
233
Присутствие карбоновой кислоты легко установить
в результате исследования соли или водного раствора
кислоты, в котором происходит диссоциация. Диссоциация
кислот приводит к тому, что оба атома кислорода, связан-
ные с углеродом, становятся равноценными, причем исче-
зает полоса карбонильного поглощения и появляются две
новые полосы вблизи 1550 и 1400 см1; эти полосы возни-
кают в результате симметричных и антисимметричных коле-
баний группы СОО‘. При добавлении-минеральных кислот
к водным растворам, содержащим диссоциированную кар-
боновую кислоту, степень диссоциации последней умень-
шается и снова появляется полоса карбонильного погло-
щения.
Таким образом, идентификация карбоновых кислот
на основании изучения инфракрасных спектров обычно
достаточно надежна, а иногда, как будет показано ниже,
можно получить некоторые дополнительные данные, касаю-
щиеся окружения карбоксильной группы.
Эти корреляции приведены в табл. 9 и подробно рас-
смотрены ниже.
Особый случай представляют аминокислоты, образую-
щие биполярные ионы; они будут рассмотрены ниже в раз-
деле, посвященном азотуглеродным соединениям.
Все приводимые данные относятся к образцам, иссле-
дованным в твердом или жидком состоянии.
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ ОН КАРБОКСИЛЬНОЙ
ГРУППЫ
Частоты валентных колебаний ОН кислот были изучены
довольно подробно в связи с наблюдавшимися для них
аномалиями, состоящими в том, что кислоты в твердом
состоянии не обнаруживают полос поглощения ни свобод-
ной, ни связанной группы ОН.
Некоторые жирные кислоты были исследованы в виде
паров при различных температурах [1, 2, 52, 53] и в виде
растворов в четыреххлористом углероде и других раство-
рителях [3—8, 54—56]. Результаты исследований показы-
вают, что если полоса валентных колебаний ОН мономеров
расположена вблизи 3550 слг1, то для димеров имеется
широкая область поглощения со многими подмаксимумами
234
Часть II
между 3000 и 2500 см'1. Аналогичные изменения происхо-
дят и в области поглощения карбонила. Димеры являются
довольно прочными, и даже в относительно разбавленном
растворе в четыреххлористом углероде и в виде паров при
низких температурах кислоты имеют полосы, соответст-
вующие как мономерам, так и димерам.
Полосы мономеров детально исследовались при этих
условиях Гулденом [54], который указывает для них
интервал от частоты 3504 см'1, соответствующей полосе
фторуксусной кислоты, наиболее сильной из исследован-
ных кислот, до 3545 см'1, соответствующей полосе наиболее
слабой п- А-диметиламинобензойной кислоты. Эта зави-
симость частоты от силы кислоты отражает роль индукцион-
ных эффектов заместителей при карбоксильной группе,
которые влияют как на частоту, так и на величину рКа.
Значения частот колебаний ОН и величин рКа связаны
для отдельных рядов алифатических, ароматических
и Р,у-непредельных кислот простой линейной зависи-
мостью. Были также приближенно измерены коэффи-
циенты погашения некоторых полос мономеров, причем
было найдено, что величина их составляет примерно лишь
одну пятую от соответствующего значения для фенолов.
Измерение относительной интенсивности полос мономерной
и димерной форм при различных концентрациях исполь-
зовалось также для оценки констант равновесия [57].
У жидких и твердых кислот полосы мономеров отсут-
ствуют и, как показали Флетт [9] и Шрив и др. [10], в
этих агрегатных состояниях кислоты существуют пол-
ностью в виде димеров или высших полимеров. При изу-
чении инфракрасных спектров этих кислот лучше всего
брать их в указанных состояниях, когда не возникает
осложнений из-за присутствия мономера; по этой причине
большинство данных, касающихся частот карбоновых ки-
слот, получено при изучении твердых или жидких веществ.
Исследования очень большого ряда кислот [9, 10, 55,
56, 58, 89, 91] показали, что поглощение, связанное с коле-
баниями ОН, проявляется в виде широкой полосы с рядом
более мелких пиков в интервале 3000—2500 см'1. В боль-
шинстве случаев главный пик наблюдается около 3000 см'1,
а главная побочная полоса — около 2650 см'1. Хотя обычно
эти полосы перекрываются в какой-то степени с полосами
Гл. 10. Карбоновые кислоты
235
СН, тем не менее, общая картина весьма характерна. Диме-
ризацией хорошо можно объяснить сильное смещение ча-
стоты ОН, но только ею нельзя объяснить появления сател-
литов и большой ширины поглощения. Эти вопросы много-
кратно обсуждались в литературе. Высказывалось пред-
положение, что фундаментальной частоте валентных коле-
баний ОН сопутствует ряд дополнительных частот, обу-
словленных взаимосвязью этих колебаний с более низко-
частотными колебаниями димеров, которые сопровождаются
растяжением по водородной связи [3, 59—63]. Аналогичное,
предположение было выдвинуто с квантовомеханических
позиций Батуевым [61] и Степановым [62, 63]. Согласно
другой точке зрения, а именно: Фасона и Жозьен [58],
широкая полоса рассматривается как результат наложения
ряда отдельных пиков, обусловленных водородными свя-
зями различной силы. Данная проблема была недавно
детально обсуждена Братозом, Хаджи и Шеппардом [55, 56],
которые указали, что трудности возникают при обоих этих
объяснениях и в частности в связи с отсутствием данных
о температурной зависимости полос ОН и данных по дей-
терированным кислотам. Они утверждают, что все наблю-
даемые в этой области частот пики можно удовлетвори-
тельно объяснить, если принять, что они представляют
собой составные полосы низкочастотных колебаний, вклю-
чая колебания группы СООН, и что интенсивность повы-
шается за счет резонанса Ферми с основными колебаниями
ОН. Так, например, главную побочную полосу обычных
кислот при 2700 см'1 можно считать составной полосой,
обусловленной колебаниями С—О и О—Н, соответственно
около 1420 см'1 и 1300 см'1. У дейтерированных кислот эта
полоса смещается до 2100 см'1, что соответствует суммиро-
ванию совпадающих по величине частот колебаний Ви
и Ag, равных 1050 см'1. Побочные полосы, наблюдаемые
при частотах выше основной частоты, также могут быть
удовлетворительно объяснены как суммарные частоты
одного из указанных выше колебаний и основного колеба-
ния карбонильной группы. Однако и при таком рассмотре-
нии очень большая ширина главной полосы поглощения ОН
пока еще не находит объяснения.
Полинг [11] полагает, что повышенная прочность водо-
родной связи в димерах жирных кислот обусловлена сидь-
236
Часть II
ним влиянием ионной резонансной структуры
О' . . . +НО.
RC( )CR,
ХОН+ . . . -о7
и на этом основании ему удалось объяснить также большее
смещение полос ОН у таких димеров по сравнению со спир-
тами, у которых водородная связь не стабилизируется
влиянием зарядов.
Поскольку при 2700—2500 см"1 поглощают лишь немно-
гие другие соединения, этот интервал особенно удобно
использовать для идентификации карбоновых кислот, хотя
характерные для них полосы бывают часто слабыми, что
может вызвать затруднения при идентификации кислот
с большим молекулярным весом. Расстояние между этими
полосами и полосами валентных колебаний СН при исполь-
зовании призмы из каменной соли достаточно велико для
того, чтобы их нельзя было спутать. Таким образом, погло-
щение в области 2700—2500 см"1 обычно служит хорошим
доказательством присутствия димеров карбоновых кислот,
но их идентификацию никогда не следует проводить без
учета данных по другим областям спектра, и особенно
области поглощения карбонила. Поглощение в интервале
2700—2500 см 1 служит только признаком наличия группы
ОН с сильной водородной связью, и хотя такие прочные
водородные связи необычны для других соединений, кроме
карбоновых кислот, они все-таки в некоторых случаях
образуются. При исключительных обстоятельствах воз;
можно также появление при еще более низких частотах
полос поглощения ОН у кислот, образующих внутримоле-
кулярные связи. Например, у основной калиевой соли
малеиновой кислоты [64] атом водорода, по-видимому,
расположен симметрично между двумя атомами кислорода.
У этого соединения в области частот выше 1600 см"1 не
наблюдается ни одной сильной полосы, которая сколько-
нибудь менялась бы при дейтерировании.
Примерами других веществ со сравнительно прочными
водородными связями являются енольные формы Р-дике-
тонов и сходные с ними вещества [12], которые образуют
прочные хелатные кольца. Аналогично обычные полосы ОН
не наблюдаются ни у о-оксидифенил сульфоксида, ни у
Гл. 10. Карбоновые кислоты
237
а-нитрозо-р-нафтола, что объясняется образованием внут-
рикомплексных соединений (хелатов) [13]. Мы нашли
также, что в этой области дают полосы поглощения ОН
сульфоновые и фосфорорганические кислоты [141. Карбо-
нильное поглощение р-дикетонов смещено, однако, в сто-
рону значительно более низких частот, чем те, которые
характерны для карбоновых кислот, так что затруднения
не возникают, если только при этом учитываются и коле-
бания С=О и О—Н. Кроме того, полоса поглощения хелат-
ной оксигруппы указанного типа не имеет той структуры,
которая так характерна для карбоновых кислот.
Маловероятно, что рассматриваемые полосы можно-
перепутать с полосами поглощения группы SH или SiH,
также находящимися в этой области [38], так как последние
являются узкими и легко отличимы от довольно широких
полос кислот.
Для окончательной проверки можно провести дейтеро-
водородный обмен, при котором полоса ОН претерпевает
определенную степень смещения [39, 55, 56]. Например,
полоса валентных ОН-колебаний у CD3COOH находится
при 3125 см1, а полоса OD-колебаний у CD3COOD —
при 2299 см1.
Характеристические полосы группы ОН могут исполь-
зоваться также для получения сведений о кристаллической
структуре твердых кислот. Куратани [15] исследовал
в области 3570—2850 см'1 инфракрасный дихроизм у корич-
ной, адипиновой и других кислот, кристаллизующихся
в виде игл, и нашел, что поглощение интенсивнее тогда,
когда электрический вектор поляризованного излучения
параллелен длинной оси игл, откуда следует, что связь
О—Н‘’‘О параллельна этой оси.
3. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С = О
а. Влияние физического состояния. Физическое состоя-
ние исследуемых карбоновых кислот непосредственно
влияет на частоту колебаний карбонильной группы. Как
уже упоминалось выше, рядом исследователей были изучены
некоторые простейшие кислоты в парообразном состоянии
[1, 2], когда в зависимости от температуры соотношение
мономерной и димерной форм меняется. Хартвелл, Ричардс
238
Часть 11
и Томпсон [16] исследовали таким путем ряд кислот и срав-
нили свои результаты с данными, полученным при изучении
жидкостей. Они нашли, что для паров уксусной кислоты,
являющейся типичным примером, при 20° С частоты, соот-
ветствующие полосам поглощения, равны 1735 и 1785 см'1,
а при 60° С—1735 и 1790 см'1, но во втором случае полоса
с более высокой частотой значительно более интенсивна,
что указывает на увеличение доли мономера. В жидком
состоянии уксусная кислота поглощает при 1717 см'1.
Для разбавленных растворов в таких неполярных рас-
творителях, как четыреххлористый углерод, наблюдаются
две карбонильные частоты, соответствующие мономеру
и димеру. Путем исследования изменений интенсивности
этих полос при разбавлении, точно так же как и полос
поглощения ОН, можно получить данные о константах
равновесия [65]. Значение частоты мономера в разбавлен-
ных растворах обычно несколько меньше, чем в парах.
Например у трифторуксусной кислоты в парах мономер
поглощает при 1820 см'1, а в четыреххлористом углероде
частота уменьшается до 1810 см'1 [66]. У нефторирован-
ных предельных кислот, растворенных в четыреххлористом
углероде, частота мономера в среднем имеэт значение
1760 см'1 [17].
В некоторых растворителях, таких как диоксан, наблю-
дается только одна карбонильная частота. Вначале это
объясняли существованием кислот в таких растворах в виде
свободных мономеров [9]. Однако частоты, наблюдаемые
в диэксане и простых эфирах, обычно на 20 см 1 ниже
частоты мономера в чегыреххлористом углероде [67],
так чго, по-видимому, здесь имеет место сильный эффект
взаимодействия с растворителем. Эго был-о подтверждено
Френкелем и др. [68], которые обнаружили зависимость
частоты от полярности растворителя. С такими соедине-
ниями, как пиридин, образуются комплексы состава 1 : 1
и 1 : 2, что приводит к появлению карбонильной полосы
мономера и полосы карбоксилат-иона [68].
Однако основное количество данных о карбонильных
частотах карбоновых кислот получено при изучении твер-
дой или жидкой фазы и растворов с концентрацией, доста-
точной для сохранения водородной связи. Поэтому для
структурноаналитических целей интерес представляют
Гл. 10. Карбоновые кислоты
239
именно частоты димеризованных кислот. Изменения кри-
сталлической формы, влияющие на водородную связь,
приводят к заметным изменениям и низкочастотной области
спектров таких веществ, но карбонильные частоты меняются
при этом незначительно. Типичным примером является
коричная кислота. Леконт и Ги [18] приводят значения
1702, 1707 и 1709 см"1 для колебаний С=О трех различных
форм г^нс-коричной кислоты и значение 1699 слГ1 для обеих
форм транс-коричной кислоты.
В то же время при плавлении карбонильная частота
увеличивается на 30 см-1, что обусловлено разрывом силь-
ных водородных связей в кристаллах и возникновением
более слабой ассоциации [67, 89].
В некоторых случаях заметные изменения наблюдаются
также при исследовании кислот в прессованных таблетках,
что объясняется взаимодействием с галогенидами щелоч-
ных металлов [78].
Влияние на карбонильные частоты структурных изме-
нений в непосредственной близости от карбоксильной группы
не было так хорошо изучено, как в случае кетонов; тем
не менее некоторое количество данных по этому вопросу
имеется.
Барнс и др. [19] привели значения частот коле-
баний С=О для ряда кислот, а Леконт [20] на примере
небольшого числа соединений рассмотрел зависимость
частоты карбонильной группы от структуры молекулы.
Такая зависимость изучалась затем Жиллеттом [21] и
Флеттом [9, 22]; в то же время рядом других исследователей
[23—25] были получены данные по отдельным группам
карбоновых кислот. Найденные результаты обсуждаются
ниже.
б. Насыщенные алифатические кислоты. Насыщенные
•одноосновные жирные кислоты, не содержащие электро-
фильных заместителей, в твердом или жидком состоянии
поглощают в интервале 1725—1705 см'1. Это согласуется
как с данными Флетта [9], исследовавшего около 60 кар-
боновых кислот, так и с данными других уже упоминав-
шихся авторов, а также и с нашими данными. Синклер,
Мак-Кей и Джонс [42] установили, что полоса карбониль-
ного поглощения у насыщенных жирных кислот (Сц—C2i)
240
Часть /
в растворе находится при 1708 см'1-, такое же значение
получается для ненасыщенных жирных кислот, у которых
ненасыщенная связь не находится в а,р-положении [43].
Фримен [44] приводит значение 1712 ± 6 см'1 для этой
полосы у 27 кислот с разветвленными цепями, исследован-
ными в растворе. У соединений в твердом состоянии частота
на несколько см'1 ниже. Данные закономерности были
подтверждены также последующими работами [56, 67,
89, 90]. Тиоуксусная кислота [27] поглощает немного
ниже указанного интервала, а именно при 1696 см'1.
в. Влияние галогенов, находящихся в a-положении. Как
и в случае кетонов, замещение галогенами или цианогруп-
пами в a-положении приводит к смещению карбонильного
поглощения в сторону более высоких частот. Например,
а-хлорпропионовая кислота поглощает при 1730 см'1, тогда
как Р-хлорпропионовая кислота — при 1710 см'1 [9].
У 2-бромстеариновой кислоты смещение меньше, и полоса
лежит при 1716 см'1, тогда как у стеариновой кислоты она
находится при 1708 см'1 [43]. У мономеров в растворах
происходит подобное, хотя и меньшее по величине смещение
полосы. Аналогичным образом, пары хлоруксусной кислоты
поглощают при 1794 см'1, а уксусной кислоты — при
1785 см'1. Такую же закономерность наблюдал Жиллетт
[21] для метилуксусной и галогенуксусных кислот. Он
нашел, что частота колебаний карбонильной группы в ряду
монохлоруксусная кислота — трихлоруксусная кислота
постепенно возрастает. Исследования частот поглощения
других типов карбонильных групп показывают, что при
введении второго и третьего атомов галогена в a-положе-
ние смещения полос значительно меньше, чем при первом
замещении [70] и аналогичного эффекта можно ожидать
в случае кислот. По аналогии с замещенными кетонами
и сложными эфирами [71] у моно- и ди-а-галогензамещен-
ных кислот возможно наличие поворотной изомерии, при-
водящей к расщеплению карбонильных частот. В этой
связи интересно отметить, что в спектре жидкой дихлорук-
сусной кислоты, как показали Братож и др. [56], наблю-
даются две карбонильные полосы, хотя, судя по полосам
поглощения ОН, никаких мономеров в этом случае нет.
Однако в случае кислот обнаружение и изучение поворот-
Гл. 10. Карбоновые кислоты
241
ной изомерии сильно затруднено из-за наличия в растворах
равновесия мономер димер; дело осложняется также
тем фактом, что кислоты сами по себе высоко полярны,
и концентрации менее полярных форм молекул в жидком
состоянии могут быть очень малы. В общем случае смеще-
ние полос в результате замещения галогеном в а-положении
составляет величину порядка 20 см'1, так что можно было бы
для этого класса соединений в твердом и жидком состояниях
указать интервал 1740—1720 см'1. Кислоты, содержащие
фтор в a-положении, поглощают даже при более высоких
частотах, а именно при 1790—1770 см'1 [46, 47, 66, 72].
г. Влияние сопряжения. В случае кетонов ненасыщен-
ная связь в аф-положении оказывает большее влияние
на длину связи С=О, чем арильный радикал и это объяс-
няли тем, что двойные связи кольца имеют несколько
менее выраженный двоесвязный характер. Однако для
кислот иногда наблюдается обратное явление, и арильный
радикал оказывает большее влияние, чем ненасыщенная
связь в аф-положении.
У ряда аф-ненасыщенных кислот, исследованных Флет-
том [9], частота колебаний С=О составляет 1710—1700 см'1,
за исключением коричной кислоты, у которой имеется аро-
матическое у,6-сопряжение и частота меньше 1700 см'1;
иными словами, здесь наблюдается небольшое смещение
в сторону нижней границы обычного интервала карбониль-
ных частот. С другой стороны, при замещении на аромати-
ческий радикал значение частоты колебаний С=О нахо-
дится в пределах от 1700 до 1680 см'1, и оно выше только
для производных бензойной кислоты, содержащих сильные
электрофильные нитрогруппы.
Поскольку, однако, многие предельные кислоты
являются жидкими, то отмеченные отличия могут быть
обусловлены, по крайней мере частично, различием фаз,
а не какими-либо иными причинами. Кроме того, известны
и многие аф-непредельные кислоты, которые поглощают
в интервале 1680—1700 см1. Для ряда таких кислот Фри-
мен [48] указывает интервал 1700—1692 см'1; аналогичные
значения приводились и другими исследователями [73, 74].
Таким образом ясно, что сопряжение, как с ароматическим
циклом, так и с олефиновой двойной связью, приводит
16 Л. Беллами
242
Часть II
к понижению частоты, но сомнительно, чтобы эти два типа
соединений можно было различать по карбонильным час-
тотам.
Дальнейшее сопряжение с а,|3-двойной связью в оле-
финовых соединениях не влияет заметным образом на кар-
бонильную частоту. При сопряжении с ацетиленовой связью
наблюдается несколько большее смещение, часто до значе-
ний несколько ниже 1680 см'1, но при дальнейшем сопря-
жении (трех и более ацетиленовых связей) происходит
небольшое увеличение частоты по сравнению с минималь-
ным значением. Это явление рассматривалось Алланом
и др. [74] в связи с вопросом о кислотности таких соеди-
нений.
д. Влияние образования хелатных структур. Некото-
рые кислоты способны к образованию внутримолекулярных
водородных связей; при этом значение карбонильной
частоты у них уменьшается. Из девяти кислот, у которых,
как отметил Флетт [9], карбонильные частоты меньше
1680 см'1, семь относятся как раз к такому типу. Простей-
шим примером является фумаровая кислота, которая погло-
щает при 1680 см'1 в отличие от малеиновой кислоты, погло-
щающей при частоте 1705см'1 [9]. о-Оксибензойные кислоты
(81 могут образовывать внутрикомплексные соединения,
аналогичные тем, которые образуются о-оксиацетофенона-
ми и о-оксибензальдегидами, с соответственно большим изме-
нением карбонильной частоты. Салициловая кислота погло-
щает при 1655 см'1, и такое смещение полосы сравнимо
с тем, которое происходит у |3-окси-а,|3-ненасыш.енных
кетонов. Как и следовало ожидать, такой же эффект наблю-
дается и у [3-амино-а,|3-ненасыщенных кислот. З-Амино-
2-нафтойная кислота поглощает при 1665 см'1’, Флетт 19]
и Муссо 175] приводят также ряд других аналогичных
случаев. Мы также наблюдали подобное явление у М-фенил-
антраниловой кислоты, у которой поглощение карбонила
обнаруживается при 1660 см'1.
При рассмотрении частот колебаний карбонила окси-,
амино- и кетокислот необходимо принимать во внимание
возможность замыкания колец с образованием лактонов,
лактамов или лактолов, в результате чего характеристи-
ческая карбонильная полоса карбоновой кислоты исчезает.
Гл. 10. Карбоновые кислоты
243
Как было показано [28], пенициллиновая кислота отно-
сится как раз к таким кислотам; она существует как сво-
бодная кислота только при строго определенных условиях.
Ацетофенон-орто-карбонова я кислота поглощает при
1732 см'1. Это поглощение соответствует скорее пятичлен-
ному лактольному кольцу, чем ароматической кислоте.
о-Формилбензойная кислота (поглощение при 1738 см'1)
также существует скорее всего в виде лактола, а не свобод-
ной кислоты [17]. Известны также и другие случаи. Для
некоторых соединений, например для бензил-орто-карбо-
новой кислоты, известны обе формы. Кетоформа поглощает
при 1698 и 1683 см'1, что соответствует ароматическому
кетону и ароматической кислоте. Вместе с тем лактольная
форма поглощает при 1692 и 1745 см'1, что соответствует
ароматическому кетону и карбонилу пятичленного лактоль-
ного кольца.
е. Дикарбоновые кислоты. Флетт [9] и Шонманн [26]
изучили целый ряд дикарбоновых кислот. Согласно полу-
ченным данным, щавелевая кислота очень интенсивно по-
глощает в области 1710—1690 слг1, а у малоновой кислоты
появляются две полосы при 1740 и 1710 см'1. Эффект взаимо-
действия карбонилов уменьшается в случае янтарной
кислоты, которая, кроме главного карбонильного поглоще-
ния при 1700 см'1, дает лишь слабую полосу при 1780 см'1.
Высшие члены ряда дикислот, такие, как адипиновая кис-
лота, обнаруживают только одну полосу вблизи 1700 см’1,
что было подтверждено на примерах других высших жир-
ных кислот Коришем и Девисоном [67]. Для фталевой
кислоты также известна одна полоса СО при 1695 см'1,
а для терефталевой кислоты — полоса при 1690 см'1.
Следовательно, можно считать, что дикарбоновые кислоты
имеют обычные карбонильные частоты, если не принимать
во внимание первые члены ряда этих кислот.
Дикарбоновые кислоты исследовались также Шоттом
и Розенбергом [76, 77], особенно в отношении стереоизо-
мерии их а,а'-дизамещенных. По-видимому, в результате
различий кристаллической структуры карбонильные
частоты оптически активных и рацемических форм не-
сколько отличаются, что оказывается достаточным для
идентификации каждой из этих форм.
16*
244
Часть II
ж. Интенсивность поглощения. Интенсивность карбо-
нильного поглощения карбоновых кислот больше, чем
у кетонов. Кросс и Рольф [29] приводят значения молеку-
лярного коэффициента погашения, равные 502—564 для
олеиновой, стеариновой и пальмитиновой кислот, в то время
как для нормальных насыщенных кетонов коэффициент
равен в среднем 200. Определения этих коэффициентов
проводились для растворов веществ в гексане, но из при-
веденных значений карбонильных частот очевидно, что
концентрации веществ были достаточны для образования
димеров.
Интенсивность карбонильных полос определялась также
Флеттом [9]. Полученные им результаты указывают на зна-
чительные различия в интенсивности при переходе от
одного класса соединений к другому. По-видимому, кис-
лоты сходны в этом отношении с кетонами, каждый тип
которых имеет собственный интервал частот и характери-
стическую интенсивность поглощения. Однако для того
чтобы установить, так ли обстоит дело в случае кислот,
необходимо провести ряд исследований, и только тогда для
идентификации соединений можно будет использовать
абсолютные значения интенсивностей. Тем не менее, если
при исследовании неизвестного соединения у него пред-
полагается наличие карбоксильной группы, то можно ожи-
дать, что интенсивность его карбонильной полосы будет
близка к тому значению, которое характерно для известной
карбоновой кислоты с такой же структурой вблизи группы
соон.
Характеристическая интенсивность карбонильного
поглощения карбоксильной группы окисленной целлюлозы
была использована Форциати, Роуэном и Плайлером [30]
при определении в целлюлозе карбоксильных групп, но при
этом в качестве стандарта применялся окисленный образец
с известным содержанием карбоксильных групп.
4. ДРУГИЕ ХАРАКТЕРИСТИЧЕСКИЕ КОЛЕБАНИЯ
КАРБОНОВЫХ КИСЛОТ
а. Поглощение около 1400 см'1. Флетт [9] наблюдал
полосу вблизи 1400 см'1 в спектрах 45 из 60 исследованных
кислот. Поэтому ее можно считать менее полезной для
Гл. 10. Карбоновые кислоты
245
идентификации по сравнению с теми двумя полосами,
которые рассматривались выше. Данной полосе свойственна
малая интенсивность, и этим, возможно, объясняется тот
факт, что она в некоторых случаях не наблюдается. Такая
полоса была обнаружена нами также у десяти кислот, не
упоминавшихся Флеттом. Интервал частот, при которых
может появляться эта полоса, составляет, по-видимому,
1440—1395 см"1. Имеющиеся данные позволяют сде-
лать вывод о том, что структурные изменения вызывают
смещение этой полосы в направлении, противоположном
смещению полосы колебаний С=О. Так, например, по дан-
ным Флетта [9], из шести кислот, поглощающих выше
1430 см1, четыре являются ненасыщенными или имеют
хелатную структуру, а из восьми кислот, поглощающих
ниже 1415 см"1, четыре содержат сильные электрофильные
заместители. Хаджи и Шеппард [49] также обнаружили
полосу поглощения в этом интервале частот у 15 карбоновых
кислот. В числе их была исследована диацетиленкарбоновая
и трихлоруксусная кислоты, а так как ни связь CsC,
ни структура СС1з не могут иметь основной полосы вблизи
1400 см"1, то очевидно, что такая полоса относится к кар^
боксильной группе. Пытаясь установить тип колебаний,
указанные авторы сравнили свои спектры со спектрами
соответствующих дейтеропроизводных и показали, что эта
полоса вместе с полосой вблизи 1300 см"1 возникает в резуль-
тате колебаний С—О, которые так тесно связаны с плоско-
стными деформационными колебаниями ОН, что у исходных
кислот ни одну из этих полос нельзя отнести к определен-
ному типу колебаний. У дейтерированных веществ взаимо^
действие сильно уменьшается и можно более отчетливо раз-
личить отдельные полосы поглощения. В случае дикарбо-
новых кислот соответствующая полоса при 1435 см"1 была
приписана колебаниям только лишь димеризованных кар-
боксильных групп, так как при плавлении кислот, когда
образование водородных связей более беспорядочно, она
исчезает [67, 89]. Возможно, что сообщения об отсутствии
такой полосы у некоторых кислот объясняется этой причи-
ной. Френсис [50] и Синклер и др. [42, 43] недавно пока-
зали, что все насыщенные жирные кислоты и сложные
эфиры, у которых к карбонильной группе присоединена
метиленовая группа, поглощают при 1410 см"1, тогда как
246
Часть II
у а,р-ненасыщенных кислот никаких сходных полос не
наблюдается. Авторы отнесли поэтому такую полосу погло-
щения к деформационным колебаниям СН2, частота кото-
рых изменяется под влиянием прилежащей группы СО.
Эта полоса, конечно, совершенно отлична от полосы погло-
щения карбоксильной группы, рассмотренной выше, и
появляется наряду с последней у кислот, содержащих
группировку —СН2СООН [49, 67]. Интенсивность полосы
выше, чем в случае обычной группы СН2 и на основании
интенсивности можно у низших жирных кислот установить
наличие разветвления в a-положении. При длине цепи
кислоты менее, чем С14, полоса 1410 см'1 сильнее, чем
полоса деформационных колебаний метиленовой группы при
1475 см'1, если только нет а-разветвления, когда группи-
ровка СН2СООН преобразуется.
б. Поглощение около 1250 см'1. Полоса вблизи 1250 см'1
появляется у всех кислот, исследованных Флеттом [9],
а Шрив и др. [10] заметили у большого числа жирных кис-
лот с длинной цепью присутствие дублета между 1280
и 1250 см'1, который, по их мнению, можно считать харак-
терным для таких соединений. Полученные результаты
указывают на то, что положение данной полосы сильно
изменяется при переходе от одного соединения к другому
и что она может появляться где-либо в пределах интервала
1320—1210 см *. Ее обычно удавалось обнаружить, по-
скольку она является самой сильной полосой спектра между
1600 и 700 см'1. Так, например, Флетт [9] приводит 18 кис-
лот, поглощающих выше 1280 см'1, из которых 14 являются
ароматическими, а две — ненасыщенными; из десяти кис-
лот, поглощающих ниже 1240 см'1, шесть являются произ-
водными уксусной кислоты, а другие, как, например,
а-хлорпропионовая кислота, содержат электроотрицатель-
ные заместители.
С другой стороны, все другие исследователи, изучавшие
насыщенные жирные кислоты, а также кислоты, в которых
двойные связи были достаточно удалены от карбоксильной
группы [42—44], нашли, что такая полоса поглощения
лежит очень близко к 1290 см'1, а Хаджи и Шеппард [49]
приводят для нее сравнительно узкий интервал 1300 4-
ф- 15 см'1. Фримен [44] указывает, что в спектрах ряда
Гл. 10. Карбоновые кислоты
247
жирных кислот этой полосе неизменно сопутствует вторая
полоса поглощения при более низких частотах. Если раз-
ветвление цепи начинается не ближе 6-атома углерода (от
карбоксила), то как частоты 1285 ± 5 слг1 и 1235+5 см'1,
так и относительные интенсивности этих полос остаются
почти постоянными для всех кислот. С приближением
места разветвления цепи к карбоксильной группе наблю-
дается явное изменение частот и интенсивностей этих полос,
так что в некоторых случаях интенсивность второй полосы
становится больше интенсивности первой. Гертин и др. [79],
а также Кориш и Чепмен [89] продолжили эти исследова-
ния и для низших членов ряда жирных кислот и получили,
по существу, такие же результаты. На основании этих
данных можно, по-видимому, удовлетворительно объяс-
нить значительное несоответствие между широкими интер-
валами частот, приводимыми одной группой авторов,
и узкими интервалами, которые указываются другими
авторами. По-видимому, все кислоты дают поглощение
в области 1300 см'1, ново многих случаях оно сопровож-
дается второй полосой при более низких частотах, которая
может быть интенсивнее первой. Поэтому для идентифика-
ции карбоксильной группы по наиболее сильной полосе
в этой области требуется изучить гораздо более широкий
интервал частот, а не ограничиваться областью вблизи
1300 см'1. Полоса, соответствующая более высокой частоте,
очевидно, возникает в результате комбинирования плоско-
стных деформационных колебаний ОН и С—О [49]; про-
исхождение же второй полосы менее понятно. Шрив и др.
[10] относят найденную ими вблизи 1200 см"1 полосу
к валентным колебаниям С—О и указывают, что сложные
эфиры и подобные им вещества поглощают примерно при
такой же частоте [31]. К этим же колебаниям Куратани
[51] относит полосу у трихлоруксусной кислоты, основы-
ваясь на том, что она сохраняется у ацетат-иона. Однако,
учитывая данные Хаджи и Шеппарда об эффектах взаимо-
действия, следует отметить, что сделанные предположения
нуждаются в дальнейшей проверке.
О’Коннор и др. [24] сообщают также о полосе в интер-
вале 1170—1150 см1, имеющейся в спектрах большого
числа жирных кислот с четным числом атомов углерода,
которая отсутствует в спектрах соответствующих сложных
248
Ч а с т ь II
эфиров. Однако более низкая частота этой полосы свидетель-
ствует, возможно, о том, что последняя обусловлена дру-
гими колебаниями и непосредственно не связана с рассмат-
риваемыми полосами.
в. Деформационные колебания ОН карбоксильной
группы. Девис и Сазерленд [3] предположили, что внепло-
скостные деформационные колебания гидроксила карбок-
сильной группы у низших жирных кислот могут прояв-
ляться вблизи 935 см1. Шрив и др. [10] обнаружили
сильную полосу поглощения в интервале 939—926 см1
в спектрах ряда жирных кислот с длинной цепью, которая
исчезает при этерификации и может быть, по их мнению,
отнесена к колебаниям гидроксила.
Флетт [9] наблюдал эту полосу у 43 из 60 кислот, но он
не обнаружил какой-либо определенной связи между их
появлением или исчезновением, с одной стороны, и частотой
или структурными факторами — с другой. Интенсивность
этой полосы меняется в широких пределах, а в некоторых
случаях она бывает очень слабой. Этим можно объяснить
тот факт, что иногда ее трудно заметить. О полосе в области
953—844 см1 у всех членов ряда кислот С8—Ci8 с четным
числом углеродных атомов сообщают также О’Коннор
и др. [24] и Синклер и др. [42]; однако данная полоса
отсутствует у соответствующих этиловых или метиловых
сложных эфиров. Хаджи и Шеппард нашли полосу при
935 ± 15 см 1 у всех 15 исследованных ими кислот. Они
отнесли ее к внеплоскостным деформационным колебаниям
ОН и показали, что при дейтерировании она, как и следо-
вало ожидать, смещается до 675 ± 15 см1.
Таким образом, эта полоса, по-видимому, достаточно
характерна для карбоновых кислот и возникает в резуль-
тате деформационных колебаний ОН. Если это так, то сле-
дует ожидать, что она заметно сместится при изменении
физического состояния вещества, так как физическое со-
стояние может оказывать влияние на водородную связь.
Мы наблюдали в некоторых случаях значительное смещение
этой полосы при переходе от твердого состояния к раство-
ру; иногда такое смещение достигает большой величины,
и можно считать, что полоса исчезла. Боннер и Хофстадтер
[1] полагают, что данная полоса относится только к диме-
Гл. 10. Карбоновые кислоты
249
рам и имеет связь со структурой О—Н--О. Однако значи-
тельное изменение частоты происходит также при переходе
от жидкости (расплава) к пасте кристаллического веще-
ства с нуйолом. В предельном случае двух полиморфных
форм стеариновой кислоты при переходе от стабильной
модификации к нестабильной нами было найдено смещение,
равное 48 см1, что подтверждается также данными Син-
клера и др. [42]. Вследствие очень непостоянной интен-
сивности эта полоса не особенно удобна для идентификации
кислот, хотя и может быть использована как дополни-
тельное доказательство их присутствия. Она встречается в
интервале частот 940—900 см-1, а у соединений, исследуе-
мых в твердом состоянии, эта полоса обычно широка и плохо
очерчена. В случае разбавленных растворов, в которых
кислоты существуют только в виде мономеров, полоса
совсем не появляется [49].
г. Прогрессия полос жирных кислот. Жирные кислоты
с длинной цепью, исследовавшиеся в кристаллическом
состоянии, обнаруживают серию полос, определенным
образом расположенных в области 1350—1180 см1. Джонс
и др. [41] указывают, что число этих полос дает сведения
относительно длины углеродной цепи. Например, для лау-
риновой кислоты (С12) известны только три равномерно рас-
положенные полосы в этой области, причем низшей из них
соответствует частота 1195 см1. Если углеродная цепь
увеличивается, то число таких полос растет; для генэйко-
зановой кислоты (C2i), например, известно Девять полос,
из которых низшая лежит при 1184 см1. В ряду от С18
до С21 при увеличении углеродной цепи на один углеродный
атом число полос увеличивается на одну полосу. Эфиры
жирных кислот, а также парафины дают аналогичные
полосы, хотя их интенсивность в случае парафинов очень
сильно ослаблена. Эти полосы можно отнести поэтому
к одной из форм деформационных колебаний (маятниковым
или крутильным) групп СН2 в цепи. Наличие таких полос
связано прежде всего с транс-расположением метиленовых
групп, как например в вытянутой цепи н-парафина.
Поэтому любое нарушение такого расположения меняет
и картину спектра в данной области. Кориш и Девисон
[67] предположили, что исчезновение этих полос у рас-
250
Часть II
плавленных кислот и появление сплошного поглощения
обусловлено незакономерным изгибом цепей (СН2)П. Изме-
нения характера поглощения в рассматриваемой области,
сопровождающие изменения агрегатного состояния, были
полностью подтверждены и другими исследователями
[80, 81, 89—92]. Разветвление цепи или присутствие двой-
ных связей также приводят к изменению вида прогрессии
полос. У тетрабромстеариновой кислоты характерного
ряда полос не наблюдается, но у 2-бромстеариновой кис-
лоты отчетливо заметны узкие полосы. Таким образом,
в определенных случаях изучение этой области может дать
полезные сведения. Можно, например, дифференцировать
насыщенные жирные кислоты с различной длиной цепи,
тогда как другими способами соединения этого класса
почти невозможно отличить друг от друга. Мейклежон и
др. [91] показали, что имеются также существенные разли-
чия между спектрами солей жирных кислот в этой области,
что с успехом может использоваться для целей идентифи-
кации. Они также предположили, что изучение этих полос
поглощения окажется полезным при определении длины
цепи.
5. ИОНИЗОВАННАЯ КОРБОКСИЛЬНАЯ
ГРУППА; СОЛИ
Леконт и его сотрудники провели обширные исследова-
ния инфракрасных спектров солей органических кислот
и изучили почти тысячу таких веществ [32—35]. Когда
происходит диссоциация с образованием группы СОО“,
становится возможным резонанс между двумя связями
С—О. Поэтому характеристическая полоса карбонильного
поглощения исчезает и вместо нее появляются две полосы
в области 1610—1550 смТ1 и 1400—1300 см \ которые соот-
ветствуют антисимметричным и симметричным колебаниям
группы СОО-.
Первая из этих полос весьма характерна, поскольку
она имеет более постоянную частоту, тогда как в широком
интервале 1400—1300 смг1 появляются полосы, связанные
со многими другими колебаниями скелета. Данные Леконта
были подтверждены другими исследователями, изучав-
шими, в частности, пенициллин [45]. Как будет показано
ниже, некоторое сходство обнаруживают аминокислоты,
Гл. 10. Карбоновые кислоты 251
у которых благодаря наличию биполярных ионов становится
возможным резонанс. Данные спектров комбинационного
рассеяния [40] для ряда карбоновых кислот также указы-
вают на то, что образование солей приводит к исчезновению
полосы поглощения С=О и к появлению вместо нее полосы
вблизи 1430 см'1. В этом случае ионизованную карбок-
сильную группу можно идентифицировать по частоте сим-
метричных колебаний, что отвечает теории, согласно кото-
рой соответствующая полоса должна быть сильной в спектре
комбинационного рассеяния и слабой в инфракрасном
спектре. Для полосы антисимметричных колебаний спра-
ведливо обратное, и, согласно Эрлиху [80], отношение
интенсивностей полос полимерных кислот в инфракрасном
спектре составляет примерно 7,6 : 1.
В случае твердого состояния обе эти частоты несколько
изменяются в зависимости от природы иона металла,
а также от природы той группы, к которой присоединена
ионизованная карбоксильная группа. Кейгарайз [83]
показал, что для одно- и двухвалентных элементов имеет
место линейная зависимость частоты антисимметричных
валентных колебаний группы СОО“ соли от величины
электроотрицательности элемента; такую же зависимость
отметил и Стимпсон [84]. Изменение природы соседней
замещающей группы также дает заметный эффект, так что
имеются, например, вполне четкие отличия характеристи-
ческих частот формиат-, ацетат- и оксалат-ионов [85].
Аналогичным образом в случае замещенных бензойной
кислоты частота карбоксилат-иона меняется в зависимости
от природы ароматических заместителей [84]. Указанные
изменения лучше можно изучить, определяя разность
частот между двумя полосами поглощения карбоксилат-
иона, так как эта разность может быть измерена точнее,
чем положение каждой из полос в отдельности. В случае
трифторацетата натрия влияние группы CF3 сказывается
особенно сильно, так что частоты СОО~ имеют значения
1680 см'1 и 1457 см'1 [86]. Первая из них близка к обычному
интервалу частот карбонильной группы неионизованных
кислот, но ее нужно сравнивать с частотой 1825 см'1,
характерной для неассоциированной кислоты.
Образование водородной связи с ионизованной карбок-
сильной группой может привести к некоторому уменьше-
252
Ч а.с т ь II
нию частоты антисимметричных колебаний. В случае соли
CF3COONa-2CF3COOH, например, полоса поглощения
(асимм) связанного карбоксилат-иона лежит при 1625 см'1
[86], в то время как в случае трифторуксусной кислоты
в пиридиновом растворе она лежит при 1667 см'1 [67].
Для динатрийэтилендиаминтетрауксусной кислоты частота
асимметричных колебаний карбоксилат-иона имеет значе-
ние 1637 см'1, т. е. она выше частоты тетранатриевой соли
(1597 см'1). Это также может объясняться влиянием водо-
родной связи [85], но в этом частном случае могут иметь
значение и другие факторы.
Обычно спектры солей жирных кислот в твердом состоя-
нии имеют более четкие различия, чем спектры исходных
кислот; Чайльдерс и Стразерс [88] использовали это явле-
ние для анализов смесей кислот различного типа.
Колебания карбонильной группы диссоциированных
в растворе кислот изучались Гором, Барнсом и Петерсе-
ном [36], которые применяли растворы в обычной и тяже-
лой воде. Чтобы можно было исследовать всю спектральную
область, они пользовались кюветой из хлористого серебра.
Было найдено, что полосы поглощения группы СОСУ аце-
тата натрия, являющегося типичным примером, находятся
около 1560 и 1410 см1. Используя тяжелую воду, которая
позволяет избежать перекрывания области колебаний кар-
бонила полосами обычной воды, и добавив в раствор DC1,
можно наблюдать по появлению сильной полосы при
1730 см'1 образование недиссоциированной кислоты, а сле-
довательно, и карбонильной группы. При добавлении NaOD
происходит обратное явление. Характерное изменение кар-
бонильной частоты при переходе недиссоциированной кис-
лоты в диссоциированную весьма типично для карбоновых
кислот и позволяет легко доказать наличие этих веществ.
Этот метод был применен в работе Кросса, Ричардса
и Уиллиса [37]. При изучении окисленных полиэтиленов эти
авторы оценивали долю карбонильного поглощения, при-
ходящуюся на карбоксильную группу, путем сравнения
интенсивностей полос при 1550 и 1720 см'1 до и после обра-
зования соли. Следует отметить, что эти полосы солей кар-
боновых кислот очень похожи на полосы нитрогруппы,
электронная структура которой напоминает ионизиро-
ванную карбонильную группу; такие полосы возникают
Гл. 10. Карбоновые кислоты
253
в результате одинаковых форм колебаний. Однако на прак-
тике при дифференциации этих групп затруднений почти
не встречается.
ЛИТЕРАТУРА
1. Bonner, Hofstadter, J. Chem. Phys., 6, 531 (1938).
2. Herman, Hofstadter, ibid., p. 534.
3. Davies, Sutherland, ibid., p. 755.
4. Davies, J. Chem. Phys., 8, 577 (1940).
5. Bus well, Rodebush, Roy, J. Amer. Chem. Soc., 60,
2239 (1938).
6. Klotz, G r u e n, J. Phys, and Colloid Chem., 52, 961 (1948).
7. McCutcheon, Crawford, Welsh, Oil and Soap,
18, 9 (1941).
8. Martin, Nature, 166, 474 (1950).
9. Flett, J. Chem. Soc., 1951, 962.
10. S h r e v e, H e e t h e г, К n i g h t, S w er n, Analyt. Chem.,
22, 1498 (1950).
11. П а у л и н г, Природа химической связи, Госхимиздат, М.—Л.,
1947.
12. Rasmussen, Tunnicliff, Brattai n, J. Amer.
Chem. Soc., 71, 1068 (1949).
13. Amstutz, Hunsberger, Chessick, J. Amer.
Chem. Soc., 73, 1220 (1951).
14. Bellamy, В e e c h e r, J. Chem. Soc., 1952, 1701.
15. К u r a t a n i, J. Chem. Soc. Japan, 71, 401 (1950).
16. Hartwell, Richards, Thompson, J. Chem. Soc.,
1948, 1436.
17. G г о v e, Willis, J. Chem. Soc., 1951, 877.
18. Lecomte, Guy, Compt. rend. Acad. Sci. Paris, 227, 54 (1948).
19. В a r n e s, Gore, Liddel, Williams, Infra-red Spec-
troscopy, Reinhold, 1944.
20. Lecomte, Traite de Chemie Organique, Ed. Grignard et Baud,
Masson et Cie., Paris, 1936, vol. 2, p. 143.
21. Gillette, J. Amer. Chem. Soc., 58, 1143 (1936).
22. Flett, Trans. Faraday Soc., 44, 767 (1948).
'23. Bartlett, Rylander, J. Amer. Chem. Soc., 73, 4275
(1951).
24. O’C onnor, Field, Singleton, J. Amer. Oil Chem.
Soc., 28, 154 (1951).
25. Rao, D a u b e r t, J. Amer. Chem. Soc., 70. 1102 (1948).
26. Schonmann, Helv. Phys. Acta, 16, 343 (1943).
27. Sheppard, Trans. Faraday Soc., 45, 693 (1949).
28. Munday, Nature, 163, 443 (1949).
29. Cross, Rolfe, Trans. Faraday Soc., 47, 354 (1951).
30. Forziati, Rowen, Plyler, J. Res. Nat. Bur. Stand.,
46, 288 (1951).
31. Thompson, Torkington, J. Chem. Soc.. 1945, 640
•32. Lecomte, Rev. Optique, 28, 353 (1949).
254
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
Часть II
Douville, Ann. Physique, 17, 5
Lecomte, Bull. Soc. chim., 9,
Leccmte, Rev. trav. chim., 69,
t e r s e n,. Analyt. Chem., 21, 382
Willis, Discuss, Faraday Soc., 9,
Duval, Lecomte,
(1942).
Douville, Duval,
548 (1942).
Duval, Ger d i ng,
391 (1950).
Gore, Barnes, Pe
(1949).
Cross, Richards,
235 (1950).
С о 1 t h u p, J. Opt. Soc. Amer., 40, 397 (1950).
Herman, Hofstadter, J. Chem. Phys., 7, 460 (1939).
E d s a 1 1, J. Chem. Phys., 5, 508 (1937).
Jones, McKay, Sinclair, J. Amer. Chem. Soc., 74,
2575 (1952).
Sinclair, McKay, Jones, J. Amer. Chem. Soc., 74,
2570 (1952).
Sinclair, McKay, Myers, Jones, J. Amer. Chem.
Soc., 74, 2578 (1952).
Freeman, J. Amer. Chem. Soc., 74, 2523 (1952).
The Chemistry of Penicillin (Princeton University Press, 1949),
p. 382.
Haszei dine, Nature, 168, 1028 (1951).
Husted, Ahlbrecht, J. Amer. Chem. Soc., 75, 1607
(1953).
Freeman, J. Amer. Chem. Soc., 75, 1859 (1953).
Hadzi, Sheppard, Proc. Roy. Soc., A216, 247 (1953).
Francis, J. Chem. Phys., 19, 942 (1951).
Kuratani, J. Chem. Soc. Japan, 73, 758 (1952).
Fuson, Josie n, Jones, Lawson, J. Chem. Phys.,
20, 1627 (1952).
Josie n, Fuson, Compt. Rend. Acad. Sci. (Paris), 235, 1025
(1952).
G о u 1 d e n, Spectrochim. Acta., 6, 129 (1954).
Bratoz, Hadzi, Sheppard,
Youqoslavie, 1, 71 (1953).
Bull. Sci. Acad. RPF.
Spectrochim. Acta, 8,
Bratoz, Hadzi, Sheppard,
' 249 (1956).
Harris, Hobbs, J. Amer. Chem. Soc., 76, 1419 (1954).
Fuson, J о s i e n, J. Opt. Soc. Amer., 43, 1102 (1953).
Davies, E v a n s, J. Chem Phys., 20, 342 (1952).
Чулановский, Симонова, ДАН СССР, 68, 1033
(1949).
Батуев, Изв. АН СССР, отд. хим. наук, 1950, 402; Изв.
АН СССР, сер. физ., 14, 429 (1950).
Степанов, ЖФХ, 19, 507 (1945); 20, 907 (1946).
Степанов, Nature, 157, 808 (1956).
Cardwell, Dunitz, О г g е 1, J. Chem Soc., 1953, 3740.
Barrow, Yerger, J. Amer. Chem. Soc., 1954, 5428.
Josie n, Fuson, Lawson, Jones, Compt. Rend. Acad.
Sci. Paris, 234, 1163 (1952).
Гл. 10. Карбоновые кислоты
255
67. Coris h, Davison, J. Chem. Soc., 1955, 6005.
68. Fraenkel, Belford, Yankwich, J. Amer. Chem.
Soc., 76, 15 (1954).
69. Barrow, J. Amer. Chem. Soc., 78, 5802 (1956).
70. Bellamy, Thomas, Williams, J. Chem. Soc.,
1955, 3704.
71. J о s i e n, Callas, Compt. Rend. Acad. Sci. (Paris), 240,
1641 (1955).
72. H a s z e 1 d i n e, J. Chem. Soc., 1954, 4026.
73. Harran d, Tuerna 1-V a t r a n, Ann. Phys., 10, 5 (1955).
74. Allan, Meakins, Whiting, J. Chem. Soc., 1955, 1874.
75. M u s s o, Chem. Ber., 88, 1915 (1955).
76. Schott e, Rosenberg, Arkiv. Kemi., 8, 551 (1956).
77. Rosenberg, Schotte, Acta. Chem. Scand., 8, 867 (1954).
78. Farmer, Chem. and Ind., 1955, 586.
79. Guertin, Wiberley, Bauer, Goldenson, Ana-
lyt. Chem., 28, 1553 (1956).
80. Neuilly, Compt. Rend. Acad. Sci. (Paris), 238, 65 (1954).
81. R i g a u x, Compt. Rend. Acad. Sci. (Paris), 238, 63, 783 (1954).
82. Ehrlich, J. Amer. Chem. Soc., 76, 5263 (1954).
83. К a g a г i s e, J. Phys. Chem., 59, 271 (1955).
84. Stimpson, J. Chem. Phys., 22, 1942 (1954).
85. I t o, Berstein, Can. J. Chem., 34, 170 (1956).
86. Klemperer, Pimentel, J. Chem. Phys., 22, 1399 (1954).
87. Chapman, J. Chem. Soc., 1955, 1766.
88. Childers, Struthers, Analyt. Chem , 27, 737 (1955).
89. Coris h, Chapman, J. Chem. Soc., 1957, 1746.
90. Wensel, Schiedt, Breusch, Z. Naturforsch, 12B, 71
(1957).
91. M e i к 1 e j о h n, Meyer, A г о n о v i c, Schuette,
Meloch, Analyt. Chem., 29, 329 (1957).
92. V о v S у d о w, Acta Chem. Scand., 9, 1119 (1955).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Пентин Ю. А., Трубников И. С., Шу шер и-
на Н. П., Левина Р. Я-, ЖОХ, 31,2092 (1961)
Глава 11
СЛОЖНЫЕ ЭФИРЫ И ЛАКТОНЫ
1. ВВЕДЕНИЕ
Сложные эфиры имеют две характеристические полосы
поглощения, обусловленные наличием групп С=О и С—О—.
Карбонильная частота в этом случае заметно больше частоты
обычных кетонов, что объясняется наличием по соседству
атома кислорода; поэтому обычно можно дифференцировать
эти два типа связей. Однако интервалы частот, характерные
для таких полос, несколько перекрываются, например
в случае ненасыщенных сложных эфиров, у которых частота
СО понижена, и таких кетонов, как а-галогензамещенные,
у которых частота С=О больше нормального значения.
Поэтому необходимо принимать во внимание интенсивность
полосы С=О, а также полосу ординарной связи С—О,
которая у сложных эфиров очень интенсивна и обычно
легко отличается от более слабых полос С—С кетонов,
появляющихся в одной и той же спектральной области.
Аналогичное увеличение интенсивности карбонильной
полосы под влиянием соседнего атома кислорода происхо-
дит, конечно, и в случае кислот, однако кислоты обычно
исследуются в таком состоянии, в котором они ассоцииро-
ваны, что приводит на практике к компенсирующему сме-
щению полосы в сторону меньших частот. Тем не менее
некоторое перекрывание интервалов карбонильных частот,
которые характерны для сложных эфиров и кислот, все же
имеется. Полосы поглощения валентных колебаний С—О
также находятся у них в одной и той же области, но, как
было показано выше (гл. 10), идентификация кислот по
области колебаний ОН или при использовании способа
перевода кислот в соли не встречает обычно затруднений,
а кроме того, имеются заметные различия интенсивностей
карбонильных полос поглощения этих двух классов соеди-
нений.
Гл. 11. Сложные эфиры и лактоны
257
Поведение карбонильной полосы поглощения сложных
эфиров очень сходно с поведением кетонной карбонильной
полосы, если иметь в виду изменения частоты, обусловлен-
ные изменениями внутримолекулярного окружения и, как
будет показано, смещения полосы имеют в обоих случаях
Таблица 10:'
Частота, СЛ4-1
Валентные колебания С = О
Нормальные насыщенные сложные эфиры а,р-Ненасыщенные сложные эфиры и 1750—1735
арилпроизводные сложных эфиров . . 1730—1717
Винилпроизводные сложных эфиров . . Сложные эфиры с электроотрицатель- 1770—1800
ным заместителем в а-положении . . 1770—1745
Сложные а-кетоэфиры 1755-1740
Сложные Р-кетоэфиры (енольные) . . . Около 1650
Салицилаты и антранилаты Сложные кетоэфиры с карбонильной 1690—1670
группой в у, 6 и т. д. положениях 1750—1735
6-Лактоны 1750-1735
у-Лактоны, насыщенные 1780—1760
у-Лактоны, аф-ненасыщенные .... 1760-1740
у-Лактоны, р,у-ненасыщенные ..... Около 1800
Р-Лактоны » 1820
Сложные тиоэфиры всех типов .... » 1675
Валентные колебания С — О
Формиаты 1200—1180
Ацетаты 1250—1230
Феиолацетаты Около 1205
Пропионаты и высшие гомологи . . . 1200—1150
Акрилаты, фумараты, малеаты .... 1300—1200 1180-1130
Бензоаты и фталаты 1310-1250 1150-1100
а Все приведенные полосы имеют высокую интенсивность.
И Л. Беллами
258
Часть II
один и тот же порядок величины. Это относится также и к
циклическим системам, для которых изменения частоты
С=О циклических кетонов в зависимости от размеров
цикла совершенно аналогичны соответствующим измене-
ниям в случае лактонов. Поскольку различные структурные
факторы, вызывающие смещения полосы, подробно обсуж-
дались при рассмотрении альдегидов и кетонов в гл. 9,
то здесь они будут рассматриваться менее детально.
Как и в случае кетонов, в этот раздел не включены
сложные эфиры, в которых к карбонильной группе присое-
динен атом азота, так как они подробно описаны в гл. 12.
Полоса поглощения ординарной связи С—О находится
в той же области, в которой появляются полосы валентных
колебаний С—О ненасыщенных простых эфиров и спиртов;
эта полоса не имеет существенного значения, так как
частота ее менее постоянна, чем частота карбонильной
полосы. В некоторых случаях были установлены более
узкие и специфичные интервалы частот для отдельных
классов сложных эфиров, например ацетатов, бутиратов,
бензоатов и т. д.; кроме того, некоторые сложные эфиры
дают две отдельные полосы поглощения в этой области,
что значительно облегчает идентификацию этих соединений.
Что касается ацетатов, то интенсивное изучение полос
С—О для большого числа стеринацетатов позволило Джонсу
с сотрудниками [1] сделать выводы о стереохимическом
строении Сз-ацетоксистеринов, а именно об относительном
расположении атома водорода при пятом атоме углерода.
Соответствующие корреляции приведены в табл. 10.
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С = О В СЛОЖНЫХ
ЭФИРАХ
а. Насыщенные сложные эфиры. Томпсон и Торкингтон
[2] исследовали в 1945 г. карбонильные частоты 36 слож-
ных эфиров, в число которых входили формиаты, ацетаты,
бутираты и др. В случае формиатов, для которых можно
было ожидать аномальных значений частот, поскольку они
являются первыми членами ряда, карбонильная полоса
появляется в интервале 1724—1722 см'1. Во всех остальных
случаях эта полоса находится вблизи 1740 см1. Все эти
данные были получены для жидких образцов, а позднее
Гл. 11. Сложные эфиры и лактоны
259
Хартвелл, Ричардс и Томпсон [3] пополнили их, исследо-
вав те же сложные эфиры в газообразном состоянии. Они
установили, что для всех соединений частота увеличивается
примерно на 20—30 см'1 по сравнению со значением ее для
жидкой фазы. Относительное постоянство частот карбо-
нильных групп сохраняется также в спектрах паров. Этими
исследователями изучались также спектры метилацетата
в девяти различных растворителях, причем было найдено,
что рассматриваемая полоса находится в интервале 1756—
1738 см'1. Этот интервал достаточно близок к среднему зна-
чению частоты для жидкого состояния, что свидетельствует
о малом изменении частот при переходе от жидкости
К раствору.
Образование водородных связей также дает лишь неболь-
шой эффект, если только не происходит резонансной стаби-
лизации. Сирлес и др. [41] показали, что в метаноловом
растворе смещение карбонильной полосы составляет около
8 см х, т. е. такое же, как в случае кетонов. Лактоны
несколько более нуклеофильны, что приводит к смещениям
до 15 см'1, зависящим также от размеров цикла. Хенбест
и Ловелл [68] обратили внимание на некоторые необычные
случаи, когда водородные связи образуются преимуще-
ственно с кислородными атомами спиртов и при этом карбо-
нильная частота становится примерно на 10 см'1 выше обыч-
ного значения.
Все эти данные были подтверждены многими другимш
исследователями. Хамптон и Невелл [4] привели значения
карбонильных частот для девятнадцати сложных эфиров-
различного типа. Они заметили, что частота сильно меняет-
ся при усложнении молекулы; это главным образом относит-
ся к уже известным случаям, когда происходит замещение
галогеном в a-положении или имеет место а,|3-ненасыщен-
ность. Что же касается значения данной частоты, например
1738 слг1 для этилпальмитата, то оно, по существу, такое же
как для более простых насыщенных веществ. Эти иссле-
дователи определили также значение молекулярного коэф-
фициента погашения для полос сложных эфиров. При пере-
ходе от одной группы к другой значение этой величины
Довольно сильно меняется, но для отдельной данной группы
соединений оно почти постоянно. Значения, приведенные,
например, для низших насыщенных сложных эфиров рас-
17*
260
Часть II
сматриваемого типа, изменяются в пределах от 569 до 610.
Андерсон и Сейфрид [5] также получили данные о положе-
нии и интенсивности полос поглощения карбонила слож-
ных эфиров, которые согласуются с указанными.
Обсуждаемая закономерность остается справедливой
и в случае более сложных молекул. Расмуссен и Браттен
[6] установили, что карбонильные частоты метилпивалата,
этилдифенилацетата и метилсаркоцината близки к 1739 см 1;
это указывает на то, что а-метил-, арил- или аминозамести-
тели не влияют или мало влияют на связь С = О. В случае
этилцианоацетата частота увеличивается, однако, до
1751 см1, что отражает влияние групп С = N. Шрив и др. [7]
и Синклер и др. [33, 34] исследовали метиловые эфиры
жирных кислот с длинной цепью и нашли, что карбониль-
ная полоса находится у них в интервале 1748—1739 см'1,
тогда как ряд триглицеридов поглощает [7] в пределах
от 1751 до 1748 см'1. О'Коннор и др. [8] приводят анало-
гичные данные для метиловых и этиловых эфиров жирных
кислот.
Наиболее обширное исследование карбонильных полос
сложных эфиров провели Джонс и его сотрудники [9—11,
28, 42, 43], изучившие карбонильные частоты свыше 100
.стер ин ацетатов, пропионатов и т. п. За некоторыми исклю-
чениями все насыщенные вещества поглощают в интервале
1742—1735 см'1 независимо от того, где расположена
по отношению к стероидным кольцам эфирная группировка;
однако имеются указания, что для каждой группы ана-
логичным образом замещенных веществ характерен более
узкий интервал частот.
Что касается интенсивностей этих карбонильных полос,
то систематическое изучение их еще не проводилось. Можно,
«эднако, ожидать, что в этом случае применимы выводы,
«сделанные для кетонов, согласно которым группы соедине-
ний различного структурного типа, классифицируемые
«по частотам, имеют различные значения коэффициента
.погашения, но примерно постоянные значения в пределах
.каждой группы [12].
Указания на это даются в работах Хамптона и Невелла
П4] и Андерсона и Сейфрида [5]. Джонс и др. [12] показали,
что пять стероидарилацетатов имеют сравнимые интенсив-
ности карбонильных полос и что они в 1,5—2,5 раза больше,
Гл. И. Сложные эфиры и лактоны
261
чем интенсивности в случае насыщенных кетонов. Стал
и Пессен [35] нашли, что постепенное уменьшение интен-
сивности карбонильной полосы алкилсебацинатов при
последовательном увеличении длины цепи настолько
закономерно, что имеется возможность дифференцировать
таким образом различные соединения этого ряда. Кросс
и Рольф [13] также определили коэффициенты погашения
для ряда сложных эфиров. Значения коэффициентов для
насыщенных веществ сравнительно близки, тогда как зна-
чение, найденное для одного аф-ненасыщенного эфира,
заметно отличается. Молекулярные коэффициенты пога-
шения были использованы для идентификации сложно-
эфирных группировок в алкалоидах Марионом, Рамсаем
и Джонсом [27]. Дополнительные данные об интенсивности,
относящиеся к небольшому числу сложных эфиров, были
приведены также Барроу [44] и Френсисом [45]. Эти авторы
подтвердили вывод о том, что интенсивность карбонильной
полосы меняется главным образом в связи с изменением
функции карбонила, причем Барроу предполагает, что
данный факт отражает различия в энергиях резонанса раз-
личных систем.
б. Влияние сопряжения. При наличии а,(3-ненасыщен-
ности следует ожидать понижения карбонильной частоты,
что и было подтверждено в случае сложных эфиров многими
исследователями [2—4, 6, 10, 46, 47]. Между тем интенсив-
ность карбонильной полосы снижается вследствие наличия
ненасыщенной связи в а,0-положении в меньшей степени,
чем в случае кетонов, а величина смещения полосы, как
при замещении ароматическим радикалом, так и радикалом
с простой двойной связью, одинакова. Поэтому если при
исследовании кетонов можно отличить эти два типа ради-
калов, то при исследовании сложных эфиров это сделать
невозможно. Метилметакрилат поглощает при 1718 см'1,
пропилметакрилат [13] — при 1721 см'1, а для 11 стероид-
ных бензоатов [10] такая полоса поглощения найдена
в интервале 1724—1719 см'1. Прямым подтверждением того,
что уменьшение частоты обусловлено влиянием двойной
связи, является тот факт, что эти же стероидные бензоаты
после гидрогенизации до гекса гидробензоатов поглощают
при 1739—1735 см'1 [10]. Таким образом, частота умень-
262
Ч а с т ь II
шается примерно на 20 еж-1. Сопряжение другой двойной
связи в у,6-положении оказывает очень небольшое влияние;
этиловый эфир коричной кислоты [4], например, поглощает
при 1717 см'1, а метилбензоат поглощает при 1724 см'1
[6]. Метиловые эфиры нафталиновых и фенантреновых кар-
боновых кислот также поглощают при 1724 см'1 [301.
Эги значения получены при изучении жидких веществ
и растворов, а для веществ в газообразном состоянии часто-
ты, по-видимому, на 20—30 см'1 выше [3].
Частоты претерпевают также изменения в зависимости
от других изменений строения. Например, о-, м- и и-нитро-
бензоаты поглощают при 1733 см'1 [6], что отражает замет-
ное уменьшение влияния арила. Такие соединения, как
этил-|3,|3-диэтоксиакрилат (1736 см'1) [6], диаллилмалеат
(1738 см'1) [4] и диэтил-1,2-дицианоэтандикарбоксилат
(1755 см'1) [46], также обнаруживают необычно высокие
карбонильные частоты. Это обусловлено, по-видимому,
эффектами полей, аналогичными тем, которые действуют
в а-галогензамещенных соединениях.
Сопряжение с ацетиленовыми связями приводит к боль-
шему смещению, и соединения этого типа поглощают
в интервале 1720—1708 см'1.
в. Виниловые эфиры. Характер изменения частоты
сложных эфиров из-за наличия структуры СО — О—С= С
не похож на изменение у кетонов. Соединения этого типа
показывают заметное увеличение карбонильной частоты
независимо от того, является ли двойная связь обычной
или она входит в ароматическое ядро. Барнс и др. [14]
отметили это для винилацетата, который поглощает при
1776 см'1. Хартвелл и др. [3] наблюдали такое же явление
для фенилацетата, а Джонс и др. [28] —для стероидных
фенольных эфиров. Расмуссен и Браттен [6] указывают
на еще большее смещение частоты у о-нитробензоилацетата
(1786 см'1), обусловленное, вероятнее всего, влиянием
нитрогруппы. Другие примеры приводят Гров и Уиллис
[15] и Уэлш [16]; как показал опыт, для некоторых соеди-
нений, например для фталидилиденуксусной кислоты, сме-
щение может быть еще большим и карбонильная частота
равна 1800 см'1. Беллами высказал следующее предположе-
ние [48]: поскольку у винилацетата нет энергии резонанса.
Гл. 11. Сложные эфиры и лактоны
263
то обычные мезомерные переходы к таким формам, как
о-
— C+=OR в этих случаях не происходят, и частота кар-
бонильного поглощения исключительно определяется силь-
ной электроотрицательностью атома кислорода.
г. Влияние атомов галогена в a-положении. Влияние
замещения сложных эфиров атомами галогена в а-положении
совершенно аналогично рассмотренному выше влиянию
в случае кетонов. Введение в a-положение группы CF3
значительно увеличивает карбонильную частоту [36—38,
49, 50J, и такие соединения поглощают около 1790 сл-1.
В то же время изучение соединений только с одним или
двумя атомами галогена в a-положении первоначально
привело к противоречивым результатам. Хаупчейн и др.
[25] нашли, что у моно-а-фторзамещенных сложных эфиров
карбонильная частота в среднем имеет значение 1786 м ’,
как у трифторзамещенных соединений. Однако Бендер [49]
получил для жидких монозамещенных значения в интервале
1776—1749 см'1. Мак-Би и Кристмен [51], изучая растворы
моно-, ди- и трифторзамещенных показали, что иногда
возможно появление двух карбонильных частот. Этот факт
был объяснен Жозьен и Каласом [52], которые получили
дополнительные данные и показали, что здесь имеет место
эффект поворотной изомерии. Причины, вызывающие сме-
щения карбонильных частот, обсуждались Беллами и Уиль-
ямсом [53], которые предположили, что здесь имеет место
механизм дипольного взаимодействия. Отсюда следует, что
на частоту влияет значительным образом лишь атом гало-
гена, находящийся ближе в пространстве к карбонильному
кислороду. Этим объясняется тот факт, что у моногалоген -
замещенных смещение такое же как у тригалогензамещен-
ных. Иллюстрацией могут служить значения частот хлор-
уксусных эфиров [52]. Метилтрихлорацетат поглощает при
1776 см'1, а метилацетат при 1750 см'1. Моно- и дихлор-
замещенные имеют по две полосы поглощения с этими же
частотами. Полоса с более высокой частотой соответствует,
таким образом, поворотному изомеру, у которого атом хлора
находится близко к карбонильному атому кислорода,
а полоса с более низкой частотой — изомеру, у которого
264
Часть II
атом хлора удален от карбонильного кислорода. Если
соединения дают эффект поворотной изомерии такого рода,
то всегда следует ожидать появления двух полос поглоще-
ния. Однако не всегда оказывается возможным наблюдать
обе эти полосы в спектрах жидкостей, так как более поляр-
ная форма (которой соответствует большая частота) в сильно
полярной жидкости стабилизируется и ее концентрация
возрастает.
В отдельных случаях наблюдалось смещение частоты
карбонильного поглощения сложных эфиров при наличии
группировки —СО — О — СН2—CF3, несмотря на то что
атомы галогена казалось бы удалены от карбонильного
кислорода [50]. Такого эффекта не обнаружено в случае
P-замещения фтором по другую сторону карбонильной
группы, и соединения типа CF3CH2CO— О — R дают обыч-
ную для сложных эфиров частоту. Увеличение частоты
в первом случае не вполне понятно; оно может быть свя-
зано с /{uc-строением сложноэфирной группы, которое
позволяет атомам фтора приблизиться к кислороду карбо-
нила. Аналогичные эффекты взаимодействия, по-видимому,
имеют место и в случае таких заместителей, как а-нитрил-
группа, но они еще недостаточно хорошо изучены. Необычно
высокая частота 1,1-диацетоксипропана 1761 см'1 также
была объяснена эффектами поля [54].
3. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С = О В СЛОЖНЫХ ЭФИРАХ
ДВУХОСНОВНЫХ КИСЛОТ И КЕТОЭФИРАХ
а. а-Диэфиры и а-кетоэфиры. Как и в случае а-дикето-
нов, две соседние группы СО, по-видимому, очень слабо
взаимодействуют между собой в этих соединениях. Дибу-
тилоксалат поглощает при 1746 см'1 [4], а метиловый эфир
пировиноградной кислоты —при 1748 см1 [17]. Гров и Уил-
лис [15] сообщают еще о двух аналогичных случаях, когда
эффект взаимодействия настолько мал, что его едва удается
заметить.
б. p-Кетоэфиры и родственные соединения. Образование
сопряженных внутримолекулярных (хелатных) связей
было уже довольно подробно описано при рассмотрении
дикетонов (гл. 9); аналогичное явление наблюдается и в слу-
чае соответствующих сложных эфиров. Такие эфиры изуча-
Гл. 11. Сложные эфиры и лактоны 265’
лись Расмуссеном и Браттеном [6]. Этил-а,а-диметилацето-
ацетат не может енолизоваться, и в растворе он обнару-
живает обычное поглощение при 1718 и 1742 сж’1, обусло-
вленное наличием кетонной и эфирной карбонильных
групп. Как этил-а-метилацетоацетат, так и этилацетоацетат
дают еще одну полосу при 1650 см1, которую относят
к эфирной карбонильной группе, соединенной внутримо-
лекулярной связью с гидроксильной группой енола. Мети-
ловый эфир также дает полосу поглощения при 1632 см
которую относят [30] к связи С=С. С другой стороны,
поскольку для диэтилдиацетилсукцината наблюдаются
только обычные кетонная и эфирная полоса, то нельзя
для него предположить возможность енолизации. [J-Диэфир,.
диэтилмалонат, также характеризуется обычной карбо-
нильной частотой [2], хотя при растворении его в четырех-
хлористом углероде наблюдается некоторое расщепление
полосы на дублет [46].
В ароматическом ряду эфирная полоса поглощения
обнаружена для н-бутилсалицилата при 1675 см-1 [6],
а для метилсалицилата при 1684 см~г [4], что опять-таки
объясняют образованием сопряженных внутримолекуляр-
ных связей. Смещение полосы в этом случае меньше, чем
у енолизованных хелатов [J-дикетонов или алкилф-кето-
эфиров, что указывает на меньшую силу внутримолекуляр-
ной связи. Это отражает, вероятно, и менее выраженный
двоесвязный характер связей ароматического ядра. Влия-
ние внутримолекулярных связей ясно обнаружилось при.
исследовании ацетилированного н-бутилсалицилата, для
которого частота оказалась равной 1723 см'1, т. е. обычной
для ароматических сложных эфиров. Ацетатная полоса
также является обычной; она соответствует 1770 см С
указывая на строение типа винилового эфира.
У таких соединений, как метил-10-окси-9-фенантрен-
карбоксилат [30], увеличение степени фиксирования двой-
ных связей колец приводит к образованию более сильных
внутримолекулярных связей, так что смещение полосы
возрастает примерно до 75 см-1. Как и в случае соответст-
вующих кетонов, величина смещения карбонильной частоты
сложных эфиров в хелатных комплексах использовалась
для определения степени двоесвязности, например в ряду
индана [55].
266
Часть II
Образование внутримолекулярных связей наблюдается
также для циклических |3-кетоэфиров, таких, как произ-
водные кетоэфиров циклогексанона и циклопентанона,
которые способны к енолизации. Ряд таких соединений был
изучен Леонардом и др. [31], а также Беллами и Бичером
[56]. Не способные к енолизации кетоэфиры циклогекса-
нона дают полосы поглощения около 1735 и 1718 см'1,
соответствующие эфирной и кетонной карбонильным груп-
пам. Однако такие соединения, как этилциклогексанон-2-
карбоксилат, помимо этих полос, дают также две другие
при 1656 и 1618 см'1, которые обусловлены, должно быть,
хелатной структурой енольной формы. Леонард и др. [31]
относят первую из этих новых полос поглощения к связан-
ному карбонилу (хелатная связь), а вторую — к двойной
связи. Аналогичные полосы наблюдаются и для производ-
ных циклопентанона, но интенсивность их в этом случае
сильно понижена.
По аналогии с кетонами следует ожидать также образо-
вания внутримолекулярных водородных связей в |3-амино-
«ф-ненасыщенных эфирах [6], что и было обнаружено
экспериментально в случае метил-Ы-метилантранилата.
Последний поглощает при 1685 см'1 и это указывает на
образование примерно таких же внутримолекулярных
связей, как и в случае салицилата. Удаление водородного
атома, участвующего в образовании внутримолекулярной
связи, приводит к появлению вновь эфирной карбонильной
полосы 1730 см'1.
б. у- и б-диэфиры и кетоэфиры. Сколько-нибудь замет-
ного взаимодействия карбонилов у у- и б-диэфиров не
происходит. Диэтиловый эфир янтарной кислоты [17] пог-
лощает при 1733 см'1, а фталаты поглощают обычно около
1730 см'1. Последнее значение является для ароматических
эфиров несколько завышенным, что может быть обусловлено
влиянием электронов цикла на две карбонильные группы.
Эфирные группы, даже расположенные на большем рас-
стоянии одна от другой, не взаимодействуют. Диэтиловый
эфир адипиновой кислоты ведет себя обычным образом
и поглощает при 1739 см'1.
При рассмотрении у- и б-кетоэфиров и подобных веществ
нельзя не учесть возможности образования лактолов.
Гл. 11. Сложные эфиры и лактоны
267
Значения частот эфиров будут при этом, по-видимому,
те же, что и частот соответствующих пяти- или шестичлен-
ных циклических лактонов. Характеристические частоты
последних будут рассмотрены ниже. Использование данных
о числе и положении карбонильных полос для идентифи-
кации лактольных структур продемонстрировали на ряде
примеров Гров и Уиллис [15].
В гл. 9 уже отмечались эффекты взаимодействия, обна-
руженные для структуры —СО—С — О — СО—, например
в случае 21-ацетокси-20-кетостероидов [10, 54]. Влияние,
оказываемое при этом на поглощение эфирного карбонила,
меньше, чем на кетонную карбонильную полосу, и смеще-
ние достигает величины порядка 4-10 см1.
4. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С = О В ЛАКТОНАХ,
ЛАКТОЛАХ и т. п.
а. Шестичленные кольца (б-лактоны). Полоса поглоще-
ния эфирной карбонильной группы б-лактонов характери-
зуется той же частотой, что и полоса соединений с открытой
цепью. Это аналогично случаю циклических кетонов и
связано с отсутствием напряжения в цикле такого размера.
К этому заключению пришли Расмуссен и Браттен 118]
на основании неопубликованной работы; ими приведено
также [6] значение частоты для б-валеролактона (1738 см ').
Джонс и др. [10, 42] подтвердили правильность такой
корреляции для ряда стероидных б-лактонов, а Гров
и Уиллис [15] установили, что она справедлива для ряда
шестичленных циклических лактолов. Ненасыщенные
циклы этого типа широко не изучались, но на основании
сравнения с поведением у-лактонов следует ожидать, что
вследствие а,(3-сопряжения, а-электроотрицательных заме-
стителей или ненасыщенности типа той, которая характерна
для виниловых эфиров, изменение карбонильной частоты
будет таким же, как и в случае эфиров с открытой цепью.
Эта корреляция неприменима к лактонам с двумя кар-
бонильными группами в одном и том же шестичленном
цикле. Вассерман и Циммерман [23] исследовали мезолак-
тид и бензилид, у которых карбонильные полосы находятся
в интервале 1767—1757 с.и-1, а Рендалл и др. [17] при-
водят для дегидроуксусной кислоты значение 1721 сл/-1.
268
Часть II
Для некоторых соединений, по-видимому, имеют место-
сложные эффекты взаимодействия, вследствие чего невоз-
можна интерпретация смещений полос.
Еще одной причиной, которая должна вызывать смеще-
ние частоты, является напряжение цикла, возникающее-
при конденсации лактона с какой-либо другой циклической
системой. Оно имеет место у некоторых б-лактонов спиро-
типа в ряду стероидов, так как поглощение наблюдается
в интервале 1793—1786 см'1 [42]. Эта частота даже выше,
чем в случае соответствующих спиро-у-лактонов (1781 —
1777 см'1), для которых повышение частоты вполне нор-
мально. Уилдер и Уинстон [57] приводят значения карбо-
нильных частот, близкие к 1764 см'1, для трициклических
лактонов, которые по другим данным должны быть система-
ми шестичленных циклов. Величина смещения зависит, ко-
нечно, от степени дополнительно создаваемого напряжения.
б. Пятичленные кольца. Проводились исследования
у-лактонов, и в настоящее время известны примеры смеще-
ний частот в зависимости от структурных изменений, кото-
рые аналогичны смещениям для эфиров с открытой цепью.
Уиффен и Томпсон [19] указали в 1946 г., что интервал
частот для у-лактонов составляет 1800—1740 см'1, а также
отметили, что у |3,у-ненасыщенных веществ полоса погло-
щения лежит ближе к 1800 см'1. Это подтвердили Ричардс
и Томпсон [20, 21J, которые сообщили, что четыре пол-
ностью насыщенных у-лактона поглощают при 1740 см'1.
Однако последующие работы показали, что в действитель-
ности карбонильная частота у-лактонов выше этого значе-
ния [6, 10, 15, 18], и что происходит такое же смещение
полосы, как в случае пятичленных циклических кетонов.
Расмуссен [6] нашел значение частоты для у-валеролактона,
равное 1770 см'1, а Джонс для стероидных у-лактонов нашел
частоты в пределах 1780—1778 см'1. Эти значения были
подтверждены также Гровом и Уиллисом [15], Вудвардом
и Ковачем [22], Марионом и др. [27]. Они согласуются
также со значениями, полученными для а,|3- и 0,у-ненасы-
щенных у-лактонов, так как смещения частот в этих слу-
чаях имеют порядок величины, которого можно ожидать,
если принять, что нормальное значение частоты примерно
равно 1770 см'1. Было сделано предположение, что для
Гл. 11. Сложные эфиры и лактоны
269
вф-ненасыщенных у-лактонов полоса поглощения сдви-
нута в сторону меньших частот примерно на 20 см'1 по
сравнению с полосой для насыщенных соединений и лежит
при 1750 см'1. Это было подтверждено Гровом и Уиллисом
[15], которые изучали ароматические лактолы, такие, как
ацетофенон-о-карбоновая кислота, лактол бензил-о-кар-
•боновой кислоты, фталида, и ряд аналогичных соединений.
При наличии ненасыщенной связи в а,у-положении отно-
сительно карбонильной группы пятичленного циклического
лактона образуется структура типа винилового эфира,
и частота возрастает приблизительно до 1800 см'1. Это
можно проиллюстрировать на примере Р,у-ангеликового
лактона, который поглощает при 1799 см'1, а также 3-мети-
ленфталида и фталидилиденуксусной кислоты [15], погло-
щающих соответственно при 1780 и 1800 см'1. Вудвард
и Ковач [22] считают характерной для енольных лактонов,
имеющих такую структуру, частоту 1792 см'1.
В тех случаях, когда одна из двойных связей является
экзоциклической, возможно наличие одновременно обоих
типов сопряжения. У протоанемонина и его гомопроизвод-
ных имеется система а,0-сопряженного у-лактона, которая
также содержит структуру винилового эфира, обусловлен-
ную наличием экзоциклической двойной связи при у-атоме
углерода. Такие соединения поглощают в жидком состоянии
при 1776 см'1 [58]. В противоположном случае [59], когда
цикл включает группировку винилового эфира, а сопря-
женная аф-непредельная связь является экзоциклической,
соответствующая частота равна 1750 см'1.
Еще один случай смещения частоты у-лактонов был
отмечен для у-ацетокси-у-валеролактона, у которого лак-
тонная карбонильная частота имеет значение 1797 см'1
(6]. Этот эффект непонятен, однако Брюгель и сотрудники
(60] сообщили о ряде аналогичных случаев, когда значе-
ние частоты у-лактонов с электроотрицательными замести-
телями в у-положении равно примерно 1800 см'1.
Иногда могут образовываться также внутрикомплексные
соединения. К таким соединениям Расмуссен и Браттен
[6] относят а-ацетил-у-бутиролактон, который имеет 0-кето-
эфирную структуру. Для неенолизованной формы полосы
появляются при 1773 см'1 (пятичленный циклический лак-
тон) и 1718 см'1 (кетон), но имеется также полоса при
270
Часть II
1656 см1, которую авторы относят к связанной карбониль-
ной группе (хелатная связь) и которая аналогична полосе,
наблюдаемой в случае соединений с открытой цепью.
Замещение галогеном в a-положении также повышает
карбонильные частоты. Полоса поглощения перфторбути-
ролактона в результате влияния атомов фтора находится
при 1873 см'1 [29].
Можно ожидать, что еще одним фактором, влияющим
на карбонильную частоту, является напряжение цикла,
возникающее в конденсированных циклических системах
и при образовании водородных связей. Однако в отличие от
б-лактонов, три- и тетрациклические у-лактоны поглощают
обычно около 1770 см'1 [61], что справедливо также и для
стероидных у-лактонов спиро-типа [42]. С другой стороны,
образование водородных связей, по-видимому, оказывает
большее влияние на у- и б-лактоны, чем на соответствую-
щие сложные эфиры, и в этих случаях смещение составляет
до 15 см'1 [41].
в. Четырехчленные кольца. Такие соединения почты
не изучались и редко встречаются на практике. Спектр,
комбинационного рассеяния ^-бутиролактона был получен
Тауфеном и Муреем [32] и имеет полосу при 1818 см'1.
Инфракрасный спектр поглощения |3-пропионолактона.
исследовался Сирльсом и др. [41]. Это соединение будучи
растворено в четыреххлористом углероде поглощает при
1841 см'1. Приведенные данные подтверждают предположе-
ние о том, что увеличенное напряжение цикла должно при-
водить к дальнейшему смещению полосы в сторону боль-
ших частот.
5. ТИОЛОВЫЕ СЛОЖНЫЕ ЭФИРЫ
О тиоловых эфирах имеется очень ограниченное коли-
чество данных. Расмуссен и Браттен [18] изучали ряд слож-
ных тиоловых эфиров с открытой цепью. Как у насыщенных,
так и у а-амино и а,|3-ненасыщенных соединений карбониль-
ная полоса находится при 1675 см'1. Поэтому можно счи-
тать, что структурные факторы, вызывающие смещения
карбонильной полосы для обычных эфиров, в случае тио-
ловых эфиров роли не играют. Об этом еще свидетельствует
Гл. 11. Сложные эфиры и лактсны 2/Г
постоянство значения карбонильной частоты фториро-
ванных тиоловых эфиров (1680 слГ1), которое можно объяс-
нить сравнительно большой поляризуемостью атома
серы [25, 29].
Было показано [24], что в спектре ненасыщенного
у-тиолактона также имеется полоса поглощения около.
1670 см'1.
6. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С—О
а. Общие сведения. Валентным колебаниям С—О в кис-
лотах, спиртах, простых и сложных эфирах соответствуют
очень сильные полосы поглощения в области 1300—1000 см"1.
Будучи одной из форм колебаний скелета, они имеют
гораздо менее постоянную частоту, чем соответствующие
колебания карбонила, и очень чувствительны к изменениям
массы и природы прилегающих групп. Несмотря на то что
полосы обладают очень большой интенсивностью, их часто
бывает трудно распознать, так как они находятся в области
спектра, где обычно расположены многие другие сильные
полосы. Более того, нельзя ожидать, что валентные коле-
бания С—О как таковые дадут характеристичную полосу,
так как они несомненно сильно зависят от движения дру-
гих атомов. Фаулер и Смит [62] исследовали ряд сложных
эфиров и пришли к выводу, что в указанной области нельзя
однозначно идентифицировать ни одну полосу. Тем не менее,
изучение наиболее интенсивных полос в этой области,
которые, вероятнее всего, связаны с колебаниями кисло-
рода, может дать полезные сведения.
Формиаты, ацетаты, бутираты и другие простейшие слож-
ные эфиры были довольно детально изучены Томпсоном
и Торкингтоном [2], которые показали, что частота самой
сильной полосы около 1200 см' была сравнительно постоян-
ной в пределах каждой группы. Однако всегда наблюда-
лась еще одна сильная полоса в области 1200—1000 см'1,
которая смещалась в разной степени в зависимости от
различной природы спиртового остатка. Первую из полос
эти авторы предположительно отнесли к связи С—О при
карбонильной группе, и ее интенсивность детально иссле-
довалась Френсисом [45] и Расселом и Томпсоном [63],
а вторую, менее постоянную, полосу — к связи С—О
272
Часть II
спиртового остатка. В последующих работах было уста-
новлено, что аф-ненасыщенные сложные эфиры дают две
сильные полосы в области 1300—1100 см'1, и некоторые
исследователи сумели использовать эти частоты С—О при
изучении пространственных эффектов в молекулах стерои-
дов. Поскольку эти корреляции изменяются при переходе
от одного класса соединений к другому, то они должны
быть установлены для каждого класса в отдельности.
б. Формиаты. Метиловый эфир муравьиной кислоты дает
в области 1250—1050 см1 самую сильную полосу при
1214 см'1; частота такой полосы у этилового эфира умень-
шается до 1195 см'1, а у высших гомологов до 1185 см'1
[2, 631. Если отбросить первый член ряда, то для этой полосы
можно, по-видимому, указать интервал 1200—1180 см'1,
но частота будет изменяться под влиянием сопряжения
у фенолформиатов и подобных веществ. Степень такого
влияния неизвестна, так что эту корреляцию и все остальные
данные, касающиеся валентных колебаний С—О, нельзя
считать вполне надежными. Джонс и др. [1] подтвердили
наличие полосы 1180 см'1 для двух стеринформиатов. Фор-
миаты дают также вторую полосу поглощения гораздо
меньшей интенсивности в интервале 1161—1151 см'1 [63].
в. Ацетаты. Ацетаты изучены гораздо лучше других
эфиров. Томпсон и Торкингтон [2] обнаружили у восьми
низших ацетатов, кончая бутилацетатом, характеристич-
ную полосу примерно при 1245 см'1, причем последняя
не изменяется и у разветвленных изомеров. Джонс и др.
[1, 65] недавно определяли положение этой полосы свыше
чем у ста 3-ацетостероидов и получили очень интересные
результаты. Прежде всего следует отметить, что у всех
изученных ацетатов обнаруживается очень сильная полоса
в данной области. Во многих случаях имеется одна полоса,
лежащая в интервале 1247—1236 см'1, но в ряде случаев
обнаруживается расщепление на три компоненты, причем
полосы находятся в интервале 1250—1200 см'1. При сопо-
ставлении этих данных с известным строением молекул
стеринов было обнаружено, что спектры с одной такой
полосой дают все соединения, у которых ацетатная группа
при Сз и атом водорода при С5 находятся в транс-положении
Гл. 11 Сложные эфиры и лактоны
273
а также соединения, у которых при С5 нет атома водорода,
а имеется А5-двойная связь. Триплет полос найден во всех
случаях, когда наблюдается /{uc-конфигурация. Каждая
из транс- и щ/с-форм имеет, конечно, два стероизомера,
но последние легко дифференцируются осаждением спирта
дигитонином, что и определяет конфигурацию при С3.
Этим методом можно установить относительное расположе-
ние в пространстве оксигруппы при третьем атоме С и водо-
родного атома при пятом атоме С для любого данного сте-
роида. Из-за того что спектр с одной полосой наблюдается
также для Аа-стеринов, затруднений не возникает, так как
наличие двойной связи спектроскопически может быть легко
установлено. Метод неприменим, однако, к соединениям, у
которых при С5 находится не водород, а какой-то другой
атом или группа. Аналогичные результаты были также
получены другими исследователями при изучении как сте-
ринацетатов [39, 64], так и деканолацетатов [40].
Авторы предполагают, что причина указанных разли-
чий, возможно, состоит в том, что /{uc-соединения, дающие
несколько полос, могут существовать в виде смеси поворот-
ных изомеров, возникающих в результате заторможенного
вращения вокруг связи С—О ацетатной группы. Однако
данные Олсопа и др. [66] ставят это предположение под
сомнение, так как ничего подобного не наблюдается у три-
терпеноид-3-ацетатов, у которых и экваториальное, и акси-
альное положение заместителей приводит к появлению
одной и той же полосы при 1243 см'1. Это может быть отне-
сено за счет наличия в молекулах гем-диметильной группы
при С4, но авторы указывают, что сама по себе такая группа
не должна была бы препятствовать возможности появления
поворотной изомерии. В случае растворов простых ацетатов
имеет также место расщепление полосы 1250 см'1 [63].
Джонс и др. [1] исследовали три стероидных ацетата,
у которых цикл I является ароматическим. Последнее
обстоятельство, как и следовало ожидать,’приводит к сме-
щению полосы валентных колебаний С—О, и самая сильная
полоса этой области находится уже при 1205 см'1. Положе-
ние полосы поглощения С—О в указанных и предельных
стероидных ацетатах меняется обратно тому, как это имеет
место в случае карбонильной полосы поглощения, причем
эги полосы связаны простой линейной зависимостью [65].
Л. Беллами
274
Часть II
Вторая полоса поглощения, характерная для ацетатов,
лежит в интервале 1060—1000 см'1, но ее гораздо труднее
идентифицировать из-за очень слабой интенсивности. Она
может быть, однако, достаточно надежно определена в
спектрах стероидных и тритерпеноидных ацетатов и так же,
как полоса 1250 см'1, является полезной для установления
пространственной конфигурации. Так, Розенкранц и
Скрогстром [64], а также Джонс и Герлинг [65] показали,
что точное положение данной полосы в указанном интер-
вале может служить для идентификации аксиальной и эква-
ториальной конформаций. Эта полоса по своим особенно-
стям, по-видимому, очень сходна с соответствующей полосой
ОН оксистероидных соединений, лежащей в этом же интер-
вале частот. Те же данные, позволяющие дифференцировать
конформации, могут быть получены и в случае тритерпе-
нового ряда [66], хотя интересно отметить, что направле-
ние смещения полосы при переходе от аксиальной к эква-
ториальной конформации в этом случае, по-видимому, про-
тивоположно тому, которое имеет место в стеринах.
г. Пропионаты, бутираты и высшие гомологи. Томпсон
и Торкингтон [2] приводят следующие средние значения
частот валентных колебаний С—О для ряда исследованных
ими эфиров: 1190 см 1 для пропионатов, 1190 см'1 для нор-
мальных бутиратов, 1200см'1 для изобутиратов и 1195 см'1
для изовалератов.
Частоты, найденные для соединений в пределах каждой
группы, сохраняют примерно постоянное значение. Кен-
далл и др. [67] приводят близкие значения характеристи-
ческих частот для высших членов ряда, а именно: 1175 см'1
для адипатов, 1174 см'1 для рицинолеатов, 1168 см'1 для
2-этилгексоатов, 1163 см'1 для лауринатов, 1172 см'1 для
олеатов, 1172 см 1 для себацинатов, 1174 см'1 для стеара-
тов, 1183 см'1 для цитратов, 1280 и 1120 см'1 для бензоатов.
Если не считать последнего случая, то различия частот,
по-видимому, слишком малы, чтобы иметь значение для
идентификации отдельных эфиров, однако такая полоса
надежно коррелируется со сложноэфирной группировкой
вообще. Шрив и др. [7] рассмотрели положение полос
колебаний С—О метиловых эфиров жирных кислот с длин-
ной цепью. Они отметили три полссы около 1250, 1205
Гл. 11. Сложные эфиры и лактоны 275
и 1175 см'1, которые отнесли к связям С—О; эти данные
были подтверждены Синклером и др. [33]. Полоса вблизи
1175 см'1 является во всех спектрах самой сильной, так
что это отнесение согласуется с указанными ранее корре-
ляциями.
Примерно аналогичная картина наблюдается в случае
триглицеридов, но в данном случае по обе стороны от силь-
ной полосы около 1163 см'1 расположены слабые полосы
1250 и 1110 см'1 [7]. Таким образом, возможно, что у слож-
ных эфиров выше пропионатов сильная полоса находится
между 1250 и 1160 см'1, если только при этом нет, конечно,
никаких других особенностей строения, нарушающих эту
закономерность, таких, например, как кратные связи
в а.6-положении относительно связи С—О— или группы
С=О.
д. а,р-Ненасыщенные сложные эфиры. Опубликовано
лишь небольшое количество спектров сложных эфиров
такого типа, и систематических исследований данного
класса соединений не проводилось. Колтуп [26] указывает,
что все акрилаты, фумараты, малеаты, бензоаты и фталаты
имеют две сильные полосы примерно в интервалах 1310—
1250 см-1 и 1200—1100 см-1, хотя точных данных о положе-
нии полос автор не приводит. Мы можем подтвердить, что
как бензоаты, так и фталаты дают две сильные и обычно
легко распознаваемые полосы в интервалах 1300—1250 см'1
и 1150—1100 см'1, которые часто могут быть очень полезны
для идентификации этих групп соединений. Джонс и др. [1]
нашли сильную полосу при 1270 см'1 для пяти стероидных
бензоатов, что также подтверждает указанное соотношение.
ЛИТЕРАТУРА
I. Jones, Humphries, Herling, Dobriner, J. Amer
Chem. Soc., 73, 3215 (1951).
2. Thompson, Torkington, J. Chem. Soc., 1945, 640.
3. Hartwell, Richards, Thompson, J. Chem. Soc.,
1948, 1436.
4. Hampton, Newell, Analyt. Chem., 21, 914 (1949).
5. Anderson, Seyfried, Analyt. Chem., 20, 998 (1948).
6. Rasmussen, Brattai n, J. Amer. Chem. Soc., 71, 1073
(1949).
7. Shreve, H e e t her, Knight, Swern, Analyt. Chem ,
22, 1498 (1950).
18*
276
Часть 11
8. O’C onno г, Field, Singleton, J. Am. Oil Chem. Soc.,
28, 154 (1951).
9. Jones, Humphries, Dobriner, J. Amer. Chem.
Soc., 71, 241 (1949).
10. J о n e s, H u m p h г i e s, D о b г i n e r, J. Amer. Chem. Soc.,
72, 956 (1950).
II. Jones, Dobriner, Vitamins and Hormones, N. Y. Acade-
mic Press, 1949, 294.
12. Jones, Ramsay, Keir, Dobriner, J. Amer. Chem.
Soc., 74, 80 (1952).
13. Cross, Rolfe, Trans. Faraday Soc., 47, 354 (1951).
14. Barnes, Gore, Liddel, Williams, Infra-red Spectro-
scopy, Reinhold, 1944.
15. Grove, Willis, J. Chem. Soc., 1951, 877.
16. Walsh, Trans. Faraday Soc., 43, 75 (1947).
17. Randall, Fowler, Fuson, Dangl, Infra-red Deter-
mination of Organic Structures. Van Nostrand, 1949.
18. Rasmussen, Brattai n, The Chemistry of Penicillin,
Princeton Univ. Press, 1949, p. 404.
19. Whiffen, Thompson, J. Chem. Soc., 1946, 1005.
20. Richards, Thompson, С. P. S. Reports, 442, 511.
21. Richards, Thompson, The Chemistry of Penicillin,
Princeton Univ. Press, 1949, p. 386.
22. Woodward,Kovach,J. Amer. Chem. Soc., 72, 1019 (1950).
23. W a s s e г m a n, Zimmerman, J. Amer. Chem. Soc., 72,
5787 (1950).
24. H u r d , К г e u z, J. Amer. Chem. Soc., 72, 5543 (1950).
25. H auptsch ein, Stoke s, Nodiff, J. Amer. Chem. Soc.,
74, 4005 (1952).
26. С о 1 t h u p, J. Opt. Soc. Amer., 40, 397 (1950).
27. Marion, Ramsay, Jones, J Amer. Chem. Soc., 73,
305 (1951).
28. Jones, Humphries, Herling, Dobriner, J
Amer. Chem. Soc., 74, 2820 (1952).
29. Hauptschein, Stokes, Grosse, J Amer. Chem. Soc.,
74, 1974 (1952).
30. Hunsberger, Ketch am, Gutow sky, J. Amer. Chem.
Soc., 74, 4839 (1952).
31. Leonard, Gutowsky, Middleton, Peterson,
J. Amer. Chem. Soc., 74, 4070 (1952).
32. T a u f e n, Murray, J. Amer. Chem. Soc., 67, 754 (1945).
33. Sinclair, McKay, Jones, J. Amer. Chem. Soc., 74,
2570 (1952).
34. Sinclair, McKav, Myers, Jones. J. Amer. Chem.
Soc., 74, 2578 (1952).'
35. Stahl, Pessen, J. Airier. Chem. Soc., 74, 5487 (1952).
36. H a s z e 1 d i n e, Nature, 168, 1028 (1951).
37. Husted, Ahlbrecht, J. Amer. Chem. Soc., 75, 1607
(1953).
38. Rappaport Hauptschein, O’Brien, Filler,
J. Amer. Chem. Soc., 75, 2695 (1953).
Гл. 11. Сложные эфиры и лактоны
277
39. Furst, Kuhn, S со to п i, Giinthard, Helv. Chim.
Acta, 35, 951 (1952).
40. Dauben, Haerger, Freeman, J. Amer. Chem. Soc.,
74, 5206 (1953).
41. Searles, Tamres, Barrow, J. Amer. Chem. Soc., 75,
71 (1953).
42. Jones, H e r 1 i n g, J. Org. Chem., 19, 1252 (1954).
43. Джонс, Сандорфи, Применение спектроскопии в
химии, Издатинлит, М., 1959, стр. 365.
44? Barrow, J. Chem. Phys., 21, 2008 (1953).
45. Francis, J. Chem. Phys., 19, 942 (1951).
46. Felton, Orr, J. Chem. Soc., 1955, 2170.
47. A 1 1 a n, M e a k i n s, Whiting, J. Chem. Soc., 1955, 1874,
48. Bellamy, J. Chem. Soc., 1955, 4221.
49. Bender, J. Amer. Chem. Soc., 75, 5986 (1954).
50. Filler, J. Amer. Chem. Soc., 75, 1376 (1954).
51. McBee, Christman, J. Amer. Chem. Soc., 77, 755 (1955).
52. Josie n, Calas, Compt. Rend. Acad. Sci. (Paris), 240, 1641
(1955).
.53. Bellamy, Williams, J. Chem. Soc., 1957, 4294.
54. Bellamy, Williams, J Chem. Soc., 1957, 861.
55. Hunsberger, Le d nicer, G utowsky, Bunker,
T u n s о i g, J. Amer. Chem. Soc., 77, 2466 (1955).
56. Bellamy, Beecher, J. Chem. Soc., 1954, 4487.
57. Welder, Winston, J. Amer. Chem. Soc., 76, 5598 (1955).
58. Grundmann, Kober, J. Amer. Chem. Soc., 76, 2332
(1955).
59. Briigel, Dury, Stengel, Suter, Angew. Chem., 68,
440 (1956).
60. Briigel, Stengal, Reicheneder, Suter, Angew.
Chem., 68, 441 (1956).
61 В er son, J. Amer. Chem. Soc., 76, 4975 H954).
62. F о w 1 e r, Smith, J. Opt. Soc. Amer., 43, 1054 (1953).
63. Russel, Thompson, J. Chem. Soc., 1955, 479
64. Rosen krantz, Skrogstrom, J. Amer. Chem. Soc ,
77, 2237 (1955).
65. Jones, Herling, J. Amer Chem. Soc., 78, 1 152 (1956).
66. Allsop, Cole, White, Willis, J. Chem. Soc., 1956,
4868.
67. К e n d a 1 1, H ampton, Ha usdorff, Pristera, App
Spectroscopy, 7, 179 (1953).
68. Henbest. Lovell, J. Chem. Soc., 1957, 1965.
гр
Рис. 9. 2,6-Диметилфенол.
Поглощение, % Поглощение,
Частота, см~'
1900 1800 1700 1600 1500 1400 1300 12ПП
90
80
70
60
50
40
30
20
10
Р и с, 10. Ангидрид фенилуксусной кислоты.
Рис, 11. п-Метоксиацетофенон,
Частота, см'1
Рис. 13. Перекись беизоила.
Рис. 15. N-Фенилантраниловая кислота.
ЧАСТЬ
III
КОЛЕБАНИЯ
УГЛЕРОД-АЗОТНЫХ
И АЗОТ-ВОДОРОДНЫХ
СВЯЗЕЙ
Г л а в a 12
АМИДЫ, БЕЛКИ И ПОЛИПЕПТИДЫ
1. ВВЕДЕНИЕ
Теоретически первичные и вторичные амиды могут су-
ществовать в кетонной или енольной форме R —СО — NH —
или R — С(ОН) = N—, каждая из которых, вероятно, ста-
билизируется в результате резонанса с биполярной фор-
/° z/0H+
мой R — С< или R —Cxz
X'NH'— XN2-
Хантш [1] предположил существование енольной формы
у некоторых простых амидов на основании данных по
спектрам поглощения в ультрафиолетовой области; резуль-
таты рентгенографических измерений [2] указывают на то,
что в твердом состоянии эти соединения находятся в форме
циклических полимеров и что существование енольной фор-
мы маловероятно. Эта точка зрения в дальнейшем подт-
верждена такими химическими данными, как факт сущест-
вования 2-оксипиридинов целиком в форме лактамов, нес-
мотря на потенциальную возможность существования лак-
тимной формы, которая могла бы стабилизироваться вслед-
ствие резонанса цикла [70]. Аналогичный вывод следует
из результатов измерения дипольных моментов рассматри-
ваемых соединений [71—73, 106], а данные анализа ин-
фракрасных спектров поглощения как амидов, так и окси-
пиридинов полностью согласуются с предположением о ке-
тонном строении этих соединений.
Поэтому можно предположить, что все амиды имеют
обусловленную карбонильной группой полосу поглоще-
ния, частота которой в некоторой степени изменяется под
влиянием биполярной резонансной структуры. В меньшей
мере на частоту влияет природа группы R, примыкающей
к карбонильной группе. Эта полоса поглощения называет-
ся полосой амид-1; она является общей для всех типов
19 Л. Беллами
290
Часть III
амидов, включая циклические формы. Частота этой полосы
заметно подверждена влиянию водородной связи, вследствие
чего возможны значительные смещения при переходе от
твердого состояния к раствору. Это же относится (и даже
в большей степени) к другим характеристическим полосам
амидов; поэтому при работе с амидами и другими подоб-
ными веществами особое внимание должно быть обращено
на агрегатное состояние, в котором исследуется вещество.
Кроме полосы поглощения карбонильной группы, пер-
вичные и вторичные амиды имеют характеристические по-
лосы, обусловленные колебаниями NH. Эти полосы в сово-
купности с первой полосой обычно вполне характеризуют
амидную группировку. У первичных амидов имеются две
формы деформационных колебаний NH, соответствующие
антисимметричным и симметричным колебаниям водород-
ных атомов, в то время как у вторичных амидов обычно
имеется только одиночная полоса, положение которой зави-
сит от того, существует ли данное соединение в цис- или
транс-форме. Большинство вторичных амидов характери-
зуется полосами, соответствущими наличию в растворе
обеих форм, хотя почти у всех этих соединений преобла-
дающей является транс-форма [74]. Образование водород-
ной связи опять-таки приводит к значительным смещениям
частот в спектре вещества в твердом состоянии, а для кон-
центрированных растворов часто можно наблюдать полосы
колебаний одновременно как свободных, так и связанных
групп NH. Положение полос, относящихся к колебаниям
связанных групп NH, меняется также в зависимости от
природы образующихся водородных связей; имеются, на-
пример, различия между частотами колебаний связанных
групп NH вторичных амидов в цис- и /нранс-формах. В спек-
трах многих вторичных амидов вторая полоса поглоще-
ния NH находится при меньших частотах, а у более слож-
ных веществ, таких, как полипептиды и белки, в этой обла-
сти имеется несколько полос поглощения. Например, дике-
топиперазин в твердом состоянии имеет пять полос, причем
все они, по-видимому, связаны с валентными колебаниями
NH. Интерпретация этих сложных полос рассматривается
в разделе, посвященном полипептидам и белкам. Следует,
однако, заметить, что имеются отчетливые различия в пове-
дении ОН-и NH-групп при изменении состояния вещества.
Г л. 12. Амиды, белки и полипептиды
291
Например, при повышении концентрации N-этилацетамида
и других аналогичных веществ наблюдается постепенное
изменение частоты валентных колебаний NH, в отличие от
скачкообразного изменения частоты валентных колебаний
ОН [75, 76]. Эго обстоятельство привело некоторых иссле-
дователей к предположению, что механизм взаимодействия
различных групп может быть различным. Выдвинуто,
например, предположение, что у амидов может осущест-
вляться перенос протона [77], однако остается неясным,
почему это не приводит к существованию кетоенольных
форм. Другим возможным объяснением является предпо-
ложение, сделанное Кенноном [76, 78] о том, что у неко-
торых вторичных амидов взаимодействие биполярных групп
'OCN+ может играть значительно большую роль, чем водо-
родные связи.
Полагают, что деформационные колебания NH2 в пер-
вичных амидах проявляются в области 1600 слг1; этим
колебаниям обычно приписывается полоса поглощения, ко-
торая находится у всех соединений такого типа (полоса
амид-II). Однако вторичные амиды также имеют сильную
характеристическую полосу при несколько более низких
частотах, природа которой являлась предметом многочис-
ленных дискуссий. Ее относят либо к деформационным ко-
лебаниям NH, либо к валентным колебаниям С — N, либо
к сложному колебанию, включающему колебания обоих
этих типов. До настоящего времени не удалось удовлетво-
рительно объяснить все наблюдаемые явления, но какой
бы ни была природа этой полосы, факт ее систематического
появления у всех нециклических вторичных амидов может
быть успешно использован при расшифровке спектров.
У вторичных амидов эта полоса сопровождается более сла-
бой полосой в области 1300 слг1 (полоса амид-Ш), кото-
рая имеется и у циклических соединений и у соединений
с открытой цепью. У первичных амидов она находится
около 1400 см"1 и относится к валентным колебаниям
С — N.
У лактамов с небольшими кольцами происходит измене-
ние частоты колебаний группы С=О амидов в связи с изме-
нением напряжения кольца; частота изменяется также при
конденсации лактамового кольца с другой циклической
структурой, при которой атом азота теряет присоединен-
19*
292
Часть HI
ный атом водорода. Имеются также различия в валентных
колебаниях NH циклических соединений, в которых груп-
па NH связана водородной связью; это объясняется тем,
что в этих соединениях амидная группа, очевидно, нахо-
дится в ^«с-положении.
Кроме того, нахождение других замещающих функцио-
нальных групп в непосредственной близости от амидной
группировки часто приводит к заметным изменениям в спек-
тре. Например, спектры соединений, содержащих струк-
турную группу СО — NH — СО, настолько отличаются
от спектров нормальных амидов, что их можно дифферен-
цировать. Уретаны и анилиды более сходны с нормальными
амидами, но у замещенных мочевины интерпретация обла-
сти 1600 слг1 становится чрезвычайно затруднительной из-
за сложности полос поглощения NH. Однако, согласно
имеющимся данным, полоса карбонильного поглощения
амида имеет обычную частоту. Амидокислоты рассматри-
ваются в гл. 13 вследствие их сходства с аминокисло-
тами.
Другие полосы поглощения вторичных амидов, находя-
щиеся в области более низких частот, имеют меньшее ана-
литическое значение. К числу таких полос относится поло-
са поглощения с частотой примерно 700 слг1, соответствую-
щая внеплоскостным деформационным колебаниям NH
[79]. Эга полоса чрезвычайно широка в спектрах твердых
веществ и концентрированных растворов. По предложе-
нию Мидзусима и др. [80] ее называют полосой амид-V.
Имеются также еще более низкочастотные полосы: полоса
амид-1 V и полоса амид-VI. Эти полосы относятся по суще-
ству к скелетным колебаниям и более подробно рассмот-
рены Миядзава [81].
Указанные корреляции представлены в табл. 11.
2 ВАЛЕНТНЫЕ КОЛЕБАНИЯ NH
а. Первичные амиды. В разбавленных растворах простые
первичные амиды имеют две полосы поглощения, связан-
ные с валентными колебаниями свободных групп NH; поло-
сы располагаются вблизи 3500 и 3400 слг1, как и у обычных
аминов. Ацетамид [3], например, в разбавленном растворе
в хлороформе поглощает при 3538 и 3420 слг1.
Таблица 11
Частота, см-1
Валентные колебания NH
Первичные амиды
свободная группа NH Вблизи 3500 и 3400 (ср.)
связанная группа NH » 3350 и 3180 »
Вторичные амиды
свободная группа NH транс- . . 3460- 3400 >
свободная группа NH цис- .... 3440- 3420 »
связанная группа NH транс 3320- 3270 »
в » » цис 3180- 3140 »
в в в цис и транс 3100- 3070 (сл.)
Полоса поглощения СО
Полоса амид-1
Первичные амиды
твердые Вблизи 1650 1
разбавленные растворы » 1690J
Вторичные амиды
твердые 1680—1630 1
разбавленные растворы 1700—1670 J (С.)
Третичные амиды
твердые и разбавленные растворы 1670—1630 »
Циклические амиды
а) большие кольца (раствор) . . . Вблизи 1680 »
б) у-лактамы
1) моноциклические > 1700 л
2) полициклические 1750—1700
в) р-лактамы
1) моноциклические (раствор) 1760-1730 (с.)
2) полициклические, с тиазолн-
диновыми кольцами (раствор) 1780-1770 в
294
Часть III
Продолжение табл. И
Частота, cai-1
Деформационные колебания NH2
Полоса амид-Н
Только первичные амиды
твердые............................. 1650—1620 (с.)
раствор ....................... 1620—1590 »
Только вторичные нециклические
амиды
твердые 1570—1515 (с.)
раствор 1550—1510 »
Полоса а.адЗ-Ш
Только вторичные амиды
Вблизи 1290 (ср.)
Деформационные колебания NH
Полоса амид-V
Только связанные вторичные амиды
Другие корреляции
Полосы амид-lV и амид-VI
Только вторичные амиды...........
Первичные амиды .................
Вблизи 720
широкая
(ср.)
Вблизи 620 и 600
1420*—1400 (ср.)
Некоторые авторы [4—9, 82] исследовали эти полосы
поглощения в спектрах твердых веществ и растворов. Наи-
более детальным является исследование Клеверли [83],
который определил частоты и интенсивности полос погло-
щения, обусловленных валентными колебаниями связан-
ных и свободных групп NH, для широкого круга амидов,
растворенных в хлороформе. Длина и природа боковых
алкильных цепей мало влияют на частоты этих полос погло-
щения, которые практически всегда остаются постоянными.
Г л. 12. Амиды, белки и полипептиды
265
В спектрах растворов средней концентрации наблюдает-
ся довольно сложная картина, так как проявляются обе
полосы поглощения свободной и связанной групп.
N-октанамид, например, в хлороформе дает полосы погло-
щения, соответствующие свободным группам NH2, 3530
и 3415 см1, но дополнительно поглощает и при 3498, 3345,
3300 и 3182 см'1. Из этого следует, что одновременно осу-
ществляются различные типы ассоциации. В спектре твер-
дого вещества картина вновь упрощается и наблюдаются
две более широкие полосы поглощения NH вблизи 3350 и
3180 см1.
б. Вторичные амиды. При использовании спектрометров
с невысокой разрешающей способностью в спектрах сильно
разбавленных растворов простых вторичных амидов обна-
ружена только одна полоса поглощения в области 3460—
3420 см'1 [4—7]. Эта частота много меньше частоты, соот-
ветствующей колебаниям О — Н, так что в этом случае
имеется надежное доказательство кетонного строения. При
высоком разрешении эта полоса во многих случаях рас-
щепляется на две компоненты. Такие амиды, как у-бути-
ролактам и 6-валеролактам, которые могут существовать
только в цис-форме, дают только одну полосу поглощения
в области 3440—3420 см1, тогда как такие вещества, как
бензанилид, который имеет транс-конфигурацию, дают одну
полосу в области несколько более высоких частот (3460—
3440 см'1). Двойные полосы поглощения, характерные для
большинства вторичных амидов, должны быть поэтому отне-
сены к цис- и транс-поворотным изомерам [74]. Благодаря
этому можно определить относительные концентрации изо-
меров, сравнивая относительные интенсивности полос. У
нормальных вторичных амидов, таких, как N-метилацет-
амид, преобладающей является транс-форма, содержание
которой 95%, но у соединений с пространственными затруд-
нениями, таких, как N-mpcm-бутилфенилацетамид, при-
мерно на 70% осуществляется цис-форма. Частота полосы
поглощения веществ в твердом состоянии определяется глав-
ным образом типом имеющейся водородной связи, и в неко-
торых случаях, особенно у дипептидов, полипептидов и бел-
ков, наблюдаются две полосы или, в некоторых случаях,
Сложная полоса поглощения. У вторичных амидов с откры-
296
Часть III
той цепью, находящихся в твердом состоянии, главная
полоса NH имеет частоту около 3270 см'1 [4, 6, 7, 88].
В растворах проявляются обе полосы свободных и связан-
ных групп NH, и частота последней очень сильно зависит
от природы растворителя и выбранной концентрации рас-
твора [75, 76, 82]. Для.твердых веществ эта полоса нахо-
дится около 3280 см'1, тогда как у раствора N-этилацет-
амида в четыреххлористом углероде она наблюдается в за-
висимости от концентрации между 3372 и 3309 см'1. Это
явление обычно рассматривается как доказательство изме-
нения длины полимерной цепочки [75,76].
Отнесение этих полос к колебаниям связанной группы
NH подтверждено исследованием дейтерированных соеди-
нений, а также простых аминов при условиях, когда обра-
зуется водородная связь NH- • О = С. При замене в груп-
пе NH водорода на атом D при обработке D2O полоса
3280 см'1 у многих простых вторичных амидов смещается до
2450 см'1, т. е. до положения, ожидаемого для соответству-
ющих колебаний ND [5, 9]. При удовлетворительном раз-
решении эта полоса также может быть расщеплена на дуб-
лет, соответствующий цис- и транс-формам. Кроме того,
дифениламин при условиях, которые допускают образова-
ние межмолекулярной связи NH- • О=С, поглощает вбли-
зи 3320 см'1 [4]. В разбавленном растворе дифениламин по-
глощает при 3430 см'1, а в твердом состоянии вместо этой
полосы появляется более широкая полоса при 3380 см'1
вследствие образования водородных связей NH - — N. Одна-
ко в растворах, содержащих NN-диэтилацетамид, значе-
ние частоты поглощения уменьшается до 3320 см'1, которое
мало отличается от значения частоты, наблюдаемой у вто-
ричных амидов в твердом состоянии, и указывает на зна-
чительно более сильную связь NH'O=C.
Мидзусима и др. [84] провели сравнительное изучение
протоноакцепторных и донорных свойств амидов, сопо-
ставляя их с аналогичными свойствами фенола и анилина,
и пришли к заключению, что по протонодонорным свой-
ствам амиды примерно соответствуют спиртам и являются
более сильными донорами, чем амины. '
Кроме сильного поглощения при 3280 см'1, у многих
вторичных амидов наблюдалась более слабая полоса при
3080 см'1. Она также связана с колебаниями NH и, подоб-
Гл. 12. Амиды, белки и полипептиды
297
но полосе 3280 см'1, исчезает в разбавленном растворе
и заменяется полосой поглощения 3420 см'1. Эта полоса
представляет особый интерес в случае полипептидов и бел-
ков, в спектрах которых она появляется почти всегда;
более подробные сведения будут даны в следующем раз-
деле. Необходимо также отметить, что такая полоса имеет-
ся и в спектрах многих простых вторичных амидов. Обе
эти полосы наблюдаются также при соответствующих час*
тотах у тиоамидных соединений [85].
Циклические лактамы и родственные им соединения
аналогичны по своему поведению в' разбавленных раство-
рах [6, 10, 16] соединениям с открытой цепью и поглощают
около 3420 см'1. Однако в конденсированной фазе полоса
вблизи 3280 с.и1 не появляется, а вместо нее возникает
другая полоса при 3175 см'1. В спектре раствора эта поло-
са имеет частоту 3220 см'1 независимо от концентрации
раствора [75]. Это доказывает, что в таких случаях име-
ются только димеры. Описываемые вещества имеют так-
же полосу 3080 см'1, которая обычно более интенсивна,
чем у амидов с открытой цепью. На основании различий
в частотах колебаний групп NH этих двух типов амидов
в конденсированной фазе Дармон и Сазерленд [6, 10] пред-
положили, что поглощение при 3280 см'1 обусловлено ами-
дами, которые образуют водородную связь и находятся
в транс-форме I.
ЧМ-Н.....
O = (L^ N-H.....О
I
Поглощение же при 3175 см'1 возникает при цис-конфигу-
рациях II или III.
О Н
С—N-
III
298
Ч а с т ь III
Тот факт, что полоса 3175 см"1 наблюдается только
у циклических лактамов, в случае которых должна иметь
место гщс-конфигурация, подтверждает эту точку зрения,
особенно в связи с наличием в настоящее время убедитель-
ных доказательств, полученных из исследований инфракрас-
ных спектров и измерений дипольных моментов и свиде-
тельствующих о том, что амиды с открытой цепью сущест-
вуют преимущественно в виде транс-изомеров [73, 74,
86, 87].
Одно время предполагалось, что полоса 3080 см"1 возни-
кает в результате образования мостиков с цас-конфигура-
циями [6, 7], но в последующих работах было показано,
что в этом случае действуют другие факторы, и данная точ-
ка зрения большинством исследователей теперь не разде-
ляется [10—12, 90]. При попытках установить наличие
в спектрах полосы 3080 см"1 нужно иметь в виду, что аро-
матические соединения, в том числе анилиды, обычно по-
глощают в этой области вследствие валентных колебаний
водородных атомов кольца.
3. ПОЛОСА АМИД-1 (КАРБОНИЛЬНОЕ ПОГЛОЩЕНИЕ)
а. Общие сведения. В твердом состоянии все амиды име-
ют сильную полосу поглощения вблизи 1640 слГ1. Нет
никакого сомнения, что у NN-дизамещенных веществ она
соответствует карбонильному поглощению, так как в этом
случае енольные формы не могут образоваться. Это погло-
щение соответствует заметно меньшей частоте, чем карбо-
нильное поглощение обычных кетонов, что обусловлено
возникновением резонансной ионной формы. Частота погло-
щения еще более понижается вследствие заметных эффек-
тов ассоциации молекул в твердом состоянии; соответ-
ствующие вещества в парах поглощают при значительно
более высоких частотах. Формамид, например,-поглсщает
[89] в парах при 1740 см"1, а в разбавленных растворах
в хлороформе частота поглощения уменьшается до 1709
см"1. Аналогичным образом N-метил- [90] и N-этилацет-
амиды [75] в парах поглощают в области 1720—1715 см"1,
и частота поглощения уменьшается примерно до 1700 см"1
в разбавленных растворах и до 1650 см"1 в жидких образ-
цах. Приведенные данные показывают, что у веществ,
Гл. 12. Амиды, белки и полипептиды
299
находящихся в виде паров, вклад ионной формы сравни-
тельно невелик. Для NN-дизамещенных амидов в твердом
состоянии, у которых не может быть образования водород-
ных связей, характерно тем не менее понижение частоты,
обусловленное, вероятно, тем, что некоторую роль играют
эффекты сильной ассоциации биполярных ионов [76].
В случае простых или N-монозамещенных амидов эту по-
лосу можно было бы интерпретировать как полосу валент-
ных колебаний C=N енольной формы, так как группа
C=N также поглощает в этой области. Однако можно
представить много убедительных доказательств, что эта
полоса относится к карбонильной группе. Так, например,
смещения этой полосы, наблюдающиеся при замещении
электрофильными группами при атоме азота, происходят
в том направлении, которого следовало бы ожидать для
полосы, соответствующей карбонильной группе, и в про-
тивоположном тому, которого можно было бы ожидать для
колебаний C=N [4, 9]. Подобно этому, значительные сдви-
ги полосы, сопровождающие изменения состояния, легче
объяснить образованием или нарушением водородных свя-
зей с карбонильной группой. Кроме того, если полосу
амид-1 отнести к группе C=N енольной формы, то не оста-
ется никаких других групп, к которым можно было бы
отнести полосу амид-11 первичных и вторичных амидов.
Этот довод, а также то, что карбонильная группа
NN-дизамещенных амидов поглощает при сравнител ьно мень-
ших частотах, убедительно свидетельствуют в пользу ке-
тонной структуры амидной группы. С отнесением данной
полосы к группе С=О теперь согласны все исследователи,
работающие в этой области, хотя принято считать, что
указанная полоса в какой-то небольшой мере обусловлена
валентными колебаниями С — N.
Точное положение полосы амид-1 определяется в зави-
симости от присутствия или отсутствия каких-либо заме-
стителей при атоме азота и от их электроотрицательности;
положение полосы изменяется при введении амидной груп-
пы в напряженный цикл, при наличии в некоторых слу-
чаях атомов галогенов в a-положении и особенно в зави-
симости от того, в каком физическом состоянии исследуется
образец. Все эти случаи рассмотрены ниже. Что касается
структур — СО — NH — СО — и О — СО — NH —, то
300
Часть III
они. описаны в разделе, посвященном специальным видам
структур (стр. 316—319).
б. Первичные амиды. R — СО — NH2. Простые первич-
ные амиды, исследованные в твердом состоянии, поглощают
вблизи 1650 см'1. Точные интервалы характерных частот
не могут быть указаны, так как число изученных образ-
цов невелико. В связи с исследованиями белков и пеницил-
лина основное внимание привлекали вторичные амиды.
Однако рассматриваемые корреляции для первичных ами-
дов были подтверждены в ряде работ [4, 5, 9, 25, 291. Аце-
тамид является исключением, поскольку он поглощает
в твердом состоянии при 1694 см'1 [5], хотя рентгенографи-
ческие данные определенно указывают на ассоциацию мо-
лекул в этом состоянии. Полоса амид-1 претерпевает зна-
чительные изменения при изменении состояния вещества,
сопровождаемого разрывом водородной связи [4, 5, 13—
15, 25]; она смещается также в зависимости от полярности
используемого растворителя [4]. Так, например, все пять
незамещенных амидов, исследованных Ричардсом и Томп-
соном [4], в разбавленном растворе в диоксане поглощают
вблизи 1690 см'1 в отличие от поглощения при 1650 см'1,
характерного для соответствующих твердых веществ. В слу-
чае концентрированных растворов частота полосы погло-
щения имеет некоторое среднее значение в зависимости от
разбавления. Так, например, гексамид в твердом состоя-
нии поглощает при 1655 см'1, в концентрированных раство-
рах— при 1668 см'1 и в разбавленном растворе (в хло-
роформе) — при 1680 см'1. Значения частоты полосы погло-
щения при растворении вещества в диоксане и в метаноле
[4], равные соответственно 1692 и 1672 см'1, указывают
на степень смещения полосы в зависимости от изменения
типа применяемого растворителя. Все амиды общей фор-
мулы CH3(CH2)nCONH2 [91], где п=1—10, в виде раство-
ров в хлороформе поглощают всегда при 1679 см'1. Влия-
ние замещения атомом галогена в a-положении на частоту
полосы поглощения подробно не изучено, но, по данным
Хасзельдина [65], у полностью фторированных амидов на-
блюдается сдвиг частоты поглощения к более высоким
частотам, а трихлорацетамид [91 ] в растворе в хлороформе
поглощает при 1732 с.м"1,
Гл. 12. Амиды, белки и полипептиды
301
в. Вторичные амиды (N-монозамещенные амиды, откры-
тая цепь). Вторичные амиды до последнего времени под-
вергались исследованию главным образом только в твер-
дом состоянии. Рендалл и другие [5] составили таблицу
частот 76 таких соединений, которые были ими исследованы
главным образом с связи с изучением пенициллина, и мно-
гие результаты приведены в монографии «Химия пени-
циллина» [9]. У простых N-монозамещенных амидов [4,
5, 9], находящихся в твердом состоянии, полоса амид-1
расположена очень близко к 1640 см1. Ричардс и Томпсон
[4], однако, установили, что электрофильные заместители
при атоме азота могут вызвать повышение указанной часто-
ты до 1680 см'1. Это было подтверждено Лачером и др. [66]
и Гирером [92], которые исследовали также N-хлориро-
ванные соединения, поглощающие при более высоких час-
тотах. Рендалл и др. [5] обнаружили тот же эффект у очень
сложных амидов с молекулами, имеющими такие размеры
и форму, при которых должна уменьшаться тенденция
к полной ориентации внутри кристалла. В случае разбав-
ленных растворов в инертных растворителях частота
карбонильного поглощения повышается примерно до
1680С.И1 для простых монозамещенных амидов идо 1700 см'1
для анилидов и подобных им веществ [4, 5, 9]. С другой
стороны, как и следовало ожидать, в случае очень слож-
ных и, следовательно, слабо ориентированных амидов не
наблюдается сколько-нибудь заметных изменений частоты
поглощения вследствие изменения состояния. Пенициллин,
например, поглощает при 1681—1667 см'1 в разбавленном
растворе и при 1667 см'1 в твердом состоянии [9]. Отсут-
ствие какого-либо смещения полосы, подобного смещению
у простых амидов, при первоначальной идентификации
амидной группы в пеницилине являлось одним из главных
затруднений; однако затем удалось показать, что другие
комплексные амиды, заведомо содержащие связь СО—NH—,
ведут себя аналогично. Следующая трудность в случае
сложных амидов возникает в связи с тем, что у различ-
ных кристаллических форм иногда наблюдаются раз-
личные частоты карбонильного поглощения. Например,
амидная полоса натрийбензилпенициллина в кристалли-
ческом состоянии находится около 1700 с.и-1, а в аморфном
состоянии —около 1667 см'1 [9], тогда как в случае
302
Часть ll 1
свободной кислоты происходит дальнейшее смещение до
1650 см'1.
Принимая во внимание все эти сложные эффекты, невоз-
можно установить точные соотношения между полосой
амид-1 и структурой групп, окружающих карбонильную
группу. Можно констатировать, что все исследованные ней-
тральные амиды в твердом состоянии имеют эту полосу
в пределах 1680—1630 см'1, но, за исключением некото-
рых простых амидов, даже на основании точных данных
о положении полосы СО нельзя с уверенностью делать
какие-либо выводы относительно природы внутримолеку-
лярного окружения амидной группы. Например, можно
было ожидать, что частота полосы амид-1 соединения
С6Н5 — СО — NH — СН3 из-за сопряжения с аромати-
ческим ядром должна быть меньше, чем у соединения
СвН5СН2—СО—NH — С2Н5. В действительности же между
ними нет никакого различия 15], и оба эти соединения по-
глощают при1649 см'1. Это, вероятно, происходит потому,
что изменения, обусловленные сопряжением, компенсируют-
ся или маскируются эффектами кристаллической упаковки.
Влияние сопряжения на частоту карбонильного поглощения
амидов с открытой цепью, по-видимому, не изучено, а эффект
замещения атомом галогена в a-положении исследован не-
которыми авторами, и полученные ими результаты рас-
смотрены Беллами и Уильямсом [93]. В отличие от кето-
нов с атомами галогенов в a-положении введение одного
атома галогена в a-положение в случае амидов не вызы-
вает существенного изменения частоты карбонильного по-
глощения [94]. Это обусловлено тем, что атом галогена
находится в гош-положении по отношению к карбониль-
ному кислороду. Однако у ди- и тризамещенных соедине-
ний становится возможным большее приближение атома
галогена к атому кислорода, и поглощение наблюдается
при более высоких частотах 188, 91,95]. Алкилтрифторметил-
амиды типа CF3CONHR, например, в твердом состоянии
поглощают в области 1698—1718 см'1.
Указанная для твердых веществ область частот погло-
щения характерна только для нейтральных продуктов,
так как у амидов, содержащих карбоксильную группу,
обнаружены отклонения, и они обычно имеют полосу
амид-1 при значительно меньших частотах. Например,
Гл. 12. Амиды, белки и полипептиды
303
этиловый эфир гиппуровой кислоты поглощает в обычной
области, а именно при 1650 см'1, свободная же кислота
[5] имеет полосу амид-1 при 1597 см'1. Рендалл и др. [5]
установили, что из многих амидокислот этого типа, кото-
рые были ими исследованы, только а-амидокислоты погло-
щают ниже 1618 см'1, в то время как иным образом заме-
щенные кислоты поглощают в интервале 1680—1618 см'1,
причем подавляющее большинство из них поглощает вбли-
зи нижней границы этого интервала.
Ряд более простых монозамещенных амидов был иссле-
дован в последнее время в виде паров и растворов в раз-
личных растворителях. Как указывалось выше, частота
карбонильного поглощения в спектрах паров гораздо бли-
же к частоте поглощения кетонов, что подтверждает очень
малый вклад ионной формы при этих условиях. В растворе
частота поглощения зависит от природы растворителя и вы-
бранной концентрации раствора. Например, карбонильное
поглощение растворов N-этилацетамида в различных рас-
творителях лежит в области частот 1687—1663 см'1 даже
при таких концентрациях, когда исключается влияние
водородной связи [751. При более высоких концентрациях
частота поглощения изменяется непрерывно при измене-
нии концентрации раствора, вследствие изменения сил
межмолекулярного взаимодействия 176, 90]. Влияние этих
эффектов ограничивает возможность использования точных
значений частоты поглощения для получения данных о струк-
туре исследуемого соединения.
г. Третичные амиды. NN'-Дизамещенные амиды не мо-
гут образовывать водородных связей, а поэтому их карбо-
нильная полоса поглощения мало зависит от изменения
агрегатного состояния или от сложности молекулы. Поло-
са обычно находится вблизи 1650 см'1, если только при
атоме азота нет замещающей фенильной группы, когда час-
тота поглощения повышается до 1690 см'1. Как и в случае
ацетанилидов, последнее происходит вследствие конкурен-
ции цикла за неподеленную пару электронов атома азота.
В результате вклад ионнсгй формы амидов уменьшается,
и частота карбонильного поглощения повышается. Анало-
гичным эффектом может объясняться высокое значение
частоты поглощения N-нитрозоамидов, растворы которых
304
Часть 111
поглощают около 1740 см'1 [961, хотя в этом случае возмож-
ны и другие объяснения. Гирер [92] отметил, что обратный
эффект наблюдается у диметилмочевины, где ионный харак-
тер карбонильной связи усиливается наличием второго
атома азота, так что частота поглощения соединений в твер-
дом состоянии уменьшается до 1610 см'1. Постоянство этой
частоты в случае растворов характерно, конечно, только
для неполярных растворителей; в тех же растворителях,
в которых может образовываться водородная связь, наб-
людается заметное уменьшение карбонильной частоты. На-
пример, NN-диэтилацетамид поглощает при 1647 см'1
в растворе в диоксане, но эта же полоса находится при
1615 см'1 в случае раствора в метаноле [4]. Рендалл и др.
[51 на основании исследования 11 веществ этого типа уста-
новили интервал 1667—1631 см'1, что согласуется со зна-
чениями частот, указанными Ричардсом и Томпсоном [4].
Как и в случае вторичных амидов, эта корреляция не всегда
остается справедливой, если где-либо в другой части моле-
кулы имеются карбоксильные группы. Например, /-N-фен-
ацетилпролин в твердом состоянии поглощает при 1590 см'1.
В случае других веществ, таких, как N-фенацил-М-фенил-
глицин, полоса карбонильного поглощения появляется
у верхней границы интервала частот, обычного для слож-
ных эфиров, и очень близка к нижней границе интервала
для свободных кислот [5]. Летау и Гроппом [88] было
показано, что, как и в случае вторичных амидов, заметное
изменение частоты поглощения наблюдается при введении
более одного атома галогена в a-положение. В этом случае
у всех изученных дигалогензамещенных в растворах наблю-
дается по две карбонильные полосы поглощения, что свя-
зано, как предполагают,с наличием поворотной изомерии.
д. Лактамы. Карбонильное поглощение лактамов являет-
ся нормальным для ненапряженных циклов из шести или
более углеродных атомов, но полоса смещается в напра-
влении более высоких частот в случае меньших циклов
из-за наличия в них напряжения. Это напоминает явле-
ния, имеющие место в случае лактонов, если не считать того,
что лактамы могут существовать в виде систем конденсиро-
ванных колец с третичным атомом азота, что связано с до-
полнительными изменениями.
Гл. 12. Амиды, белки и полипептиды
305
Кольца с шестью и более членами. Такие соединения
мало исследованы, но полученные результаты показывают,
что карбонильное поглощение амидных групп, включенных
в большие циклические системы, очень сходно с поглоще-
нием нормальных амидов [9]. Эдвардс и др. [971 считают,
что из шестичленных циклических систем вторичные лак-
тамы в виде растворов в хлороформе поглощают при
1665см1, а третичные соединения поглощают о кол о 1637 см ‘.
Конденсация дополнительного кольца не приводит в этих
случаях к заметному изменению частоты. Теми же авторами
было сделано интересное наблюдение, что отсутствие
аф-насыщенностп у этих соединений не уменьшает частоты
карбонильного поглощения, а по-видимому, слегка ее
повышает, хотя положение усложняется вследствие появ-
ления дополнительных полос поглощения при более низ-
ких частотах. Тем не менее этот эффект не является не-
ожиданным, так как в случае неплоских колец возможна
структура, при которой не может быть достигнута копла-
нарность связей С — N и С — С^= с карбонильной связью.
Дальнейшее сопряжение обнаружено у таких систем,
как пироллидоны и хинолоны, которые были исследованы
несколькими группами авторов [98—101]. Спектры этих
соединений в общем более сложны, но имеется ясное ука-
зание на то, что не только 2-окси-, но и 4-оксипиридины су-
ществуют в форме лактамов. Аналогичные эффекты на-
блюдаются у пиримидинов. Имеются указания на совпадение
частоты карбонильного поглощения 2- и 4-оксисоединений
с частотой поглощения лактамов, что отсутствует у соот-
ветствующих метоксипроизводных [102, 103].
Ангидриды некоторых аминокислот, содержащие по две
вторичные амидные связи в одном и том же шестичленном
кольце, изученные Брокманном и Муссо [107], обладают,
по-видимому, обычными свойствами.
Что касается более сложных циклических систем, то
частота карбонильного поглощения у них практически
не меняется и для растворов равна примерно 1670 см~1.
Так, различными авторами были исследованы семи- [85,
104], восьми- [104] и девятичленные [18] циклические сис-
темы. Шидтом [105] были изучены еще более сложные сис-
темы, относительно которых им было дополнительно отме-
чено, что кольца с девятью и более членами характеризуют-
20 л. Беллами
306 Часть 111
ся также полосой амид-11, хотя последняя отсутствует
в спектрах более простых циклических соединений. Это
наблюдение имеет важное значение, так как недавние изме-
рения дипольных моментов показали, что в циклических
системах этого рода преобладает транс-форма [114].
Пятичленные кольца. Изучено ограниченное число про-
стых у-лактамов, тогда как по гетероциклическим соедине-
ниям, например оксазалонам и др., имеется большее число
данных.
у-Бутиролактам [5] в жидком состоянии и в растворе
[75, 85] поглощает при 1700 см'1, а в парах — при 1754 см'1
[85], в то время как результаты исследования ряда поли-
циклических у-лактамов, проведенного в связи с изуче-
нием пенициллина, показывают, что полоса карбонильной
группы расположена в интервале 1750—1700 см"1 и в боль-
шинстве случаев ближе к границе, соответствующей низким
частотам [9, 64].
Четырехчленные кольца. В связи с тем интересом, кото-
рый вызвал пенициллин, изучено большое число (3-лакта-
мов разного типа. Английскими и американскими исследо-
вателями изучено 17 моноциклических (3-лактамов [9],
причем было найдено, что полоса карбонильного поглоще-
ния у них лежит в интервале 1760—1730 см'1 (в растворе).
Для образцов, исследованных в твердом состоянии, преде-
лы области поглощения несколько шире, но смещения при
переходе из одного состояния в другое обычно невелики.
У р-лактамов, конденсированных с тиазолидиновыми коль-
цами, частота поглощения значительно больше, и для трех
изученных образцов врастворезначение ее ограниченоузким
интервалом 1780—1770 см'1. В твердом же состоянии час-
тота поглощения меньше только на несколько обратных
сантиметров. Таким образом, это значение сравнимо со зна-
чением 1779 см'1, которое относят к Р-лактамной груп-
пировке пенициллина, причем интересно отметить, что ча-
стота уменьшается до 1739 см'1 в дезтиопенициллине,
в котором кольцо Р-лактама сохранено, но уже не кон-
денсировано с тиазолидиновым циклом.
Значительное количество р-лактамов изучено позднее
Шиханом и его сотрудниками [19—24] в связи с исследова-
ниями проблем синтеза соединений. Их результаты в основ-
ном подтверждают данные прежних исследователей, однако
Гл. 12. Амиды, белки и полипептиды
307
эти авторы отметили, что замещение двух групп COOR при
углероде, находящемся в a-положении, амидным азотом
приводит у неконденсированных р-лактамов к смещению
полосы в сторону более высоких частот, а именно до
1770 см'1. Очевидно, таким образом, что конденсирование
колец не является единственным фактором, вызывающим
сдвиг полосы. Кроме того, установлено, что для конденсиро-
ванных тиазолидиновых р-лактамов происходит еще боль-
шее смещение полосы СО (до 1800 см'1), вследствие окисле-
ния атома серы тиазолидинового кольца с образованием
сульфона. Тот факт, что изменение в группировке, столь
далекой от карбонильной группы, приводит к такому боль-
шому смещению, указывает на необходимость учета ряда
факторов при интерпретации частот СО в этой области.
Следует подчеркнуть также, что корреляции для конденси-
рованных р-лактамов установлены только на основании дан-
ных о структурах р-лактам — тиазолидин и не обязатель-
но применимы для других типов систем с конденсирован-
ными кольцами.
е. Интенсивность полосы амид-1. В подавляющем боль-
шинстве случаев амиды подвергались исследованию в твер-
дом состоянии, а поэтому относительно факторов, которые
могут оказывать влияние на интенсивность карбонильного
поглощения, имеется мало сведений. Ричардс и Бартон
[26] измерили коэффициенты погашения полос карбониль-
ного поглощения для нескольких простых амидов и нашли,
что они имеют довольно постоянные значения. Ленорман
[27, 29] установил, однако, что интенсивности значительно
изменяются, и указал, что у N-этилбензамида полоса СО
при 1659 см'1 заметно слабее, чем полоса амид-11 при
1550 см'1. Аналогичные отклонения от нормального соот-
ношения интенсивностей имеют место в случае N-метилбен-
замида. В ряду ацетамид, хлорацетамид, дихлорацетамид,
трихлорацетамид такие отклонения для интенсивностей
полос амид-1 и амид-II постепенно увеличиваются [28].
Неясно, однако, является ли это результатом изменения
абсолютной интенсивности полосы поглощения СО или из-
менения интенсивности полосы амид-11, с которой сравни-
вается поглощение СО. Ленорман [27] указал также, что
интенсивность полосы СО может меняться при изменениях
20*
308
Часть 111
кислотности растворов. Джонсом и Клеверли [91] прове-
дены некоторые исследования интенсивности поглощения
N-алкиламидов и получены вполне устойчивые значения
коэффициентов погашения, равные 600—800. Однако изме-
нения полярного характера карбонильной связи, которые
сопровождают изменения физического состояния образца
или замену растворителя, должны оказывать большое
влияние на измеряемые величины, а поэтому исследования
этого рода связаны с очень большими трудностями. Работы
в этой области желательно продолжить, учитывая их боль-
шое значение для исследований полимеров.
4. ПОЛОСА АМИД-П
Первичные и вторичные амиды имеют в области 1600—
1500 см'1 вторую сильную полосу, которая отсутствует
в спектрах третичных амидов, а также в спектрах цикли-
ческих лактамов при нормальных условиях. Происхожде-
ние этой полосы точно не установлено, неясно также, обу-
словлена ли она в обоих случаях (первичных и вторичных
амидов) одними и теми же причинами. Тем не менее эту
полосу обычно рассматривают как полосу амид-П.
а. Первичные амиды. RCONH2. Все первичные амиды
имеют вторую менее интенсивную полосу, расположенную
очень близко к основной полосе карбонильного поглоще-
ния. Обычно ее интенсивность составляет примерно от У2
до 1/з интенсивности полосы СО. Эта полоса расположена
в интервале 1650—1620 см'1, т. е. при более низких часто-
тах, чем полоса СО (твердые вещества) [4, 5, 9]. Рендалл
и др. [5] сообщают, что в редких случаях эта полоса сдви-
нута относительно полосы карбонильного поглощения в сто-
рону высоких частот, но это не подтверждается данными
этих же авторов о спектрах простых амидов. При такой
интерпретации трудно было бы установить, происходит
ли смещение полос или изменение интенсивностей. Иногда
в спектрах веществ, исследуемых в твердом состоянии, обе
полосы располагаются настолько близко одна к другой,
что наблюдается только одиночная полоса. Однако вслед-
ствие того, что эти две полосы смещаются в противополож-
ных направлениях при изменении состояния вещества, мо-
Гл. 12. Амиды, белки и полипептиды
309
жет быть установлено наличие обеих этих полос. Напри-
мер, фенацетамид в твердом состоянии имеет одну широкую
полосу при 1645 сл/-1, которая в спектре разбавленного
раствора разрешается в виде двух полос при 1678 и
1580 см'1 [5].
Результаты, полученные в этой области различными ис-
следователями [4, 5, 9], показывают, что в случае твердых
веществ полоса находится в интервале 1650—1620 см'1
и смещается в интервал 1620—1590 сл/'1 в случае разбавлен-
ных растворов. Все первичные амиды CH3(CH2)riCONH2
в виде растворов в хлороформе поглощают при 1590—
1588 см1, коэффициент погашения емакс равен 180—210 для
всех соединений с п от 1 до 10 [91]. Природа радикала R
в структуре RCONH2 существенно не влияет на эту час-
тоту, и цианацетамид, например, поглощает при 1620 см'1
(твердое вещество). В результате разрыва водородных свя-
зей происходит смещение полосы в таком направлении,
которое дает основание предположить, что она связана
с деформационными колебаниями. Большинство исследо-
вателей считает, что это поглощение, подобно поглоще-
нию простых аминов в этой области, обусловлено дефор-
мационными колебаниями NH2. Если принять это поло-
жение как вероятное, то следует помнить, что здесь возмо-
жно также какое-то взаимодействие с колебаниями С — N,
которое предполагалось, например, для вторичных амидов.
Это колебание может быть сложным, но, очевидно, преобла-
дающими в нем являются деформационные колебания NH.
б. Вторичные амиды. RCONHR. Вопрос об идентифи-
кации полосы поглощения амид-U, имеющейся у всех
вторичных амидов в области 1550 см'1 (твердые вещества),
является предметом многочисленных дискуссий. Многие
аргументы, выдвинутые для объяснения этой полосы, очень
подробно обсуждать здесь не представляется возможным,
но ввиду важности отнесения этой полосы при исследова-
ниях белков ниже рассматриваются некоторые из основ-
ных точек зрения.
в. Происхождение полосы амид-П у вторичных амидов.
Принято считать, что эта полоса обусловлена деформацион-
ными -колебаниями- NH- и,- таким-образом, она-по-своему-
310
Часть 111
происхождению аналогична полосе амид-П незамещенных
амидов. Эта точка зрения подтверждается тем, что подоб-
ная полоса отсутствует у третичных амидов [4, 9] и осла-
бляется при дейтерировании исследуемых соединений [9,
10, 581; направление сдвига полосы при изменениях состо-
яния вещества и данные исследования поляризации также
говорят о ее связи с плоскостными деформационными коле-
баниями NH [35]. Однако это простое объяснение нельзя
считать полностью удовлетворительным по ряду причин.
Например, не объяснимо отсутствие этой полосы у цик-
лических лактамов, содержащих менее чем девятичленные
кольца. Возможно, что само по себе это не является очень
серьезным возражением, так как измерениями дипольных
моментов было показано, что более простые циклические
системы этого типа существуют в цпс-конфигурации, тогда
как более сложные системы имеют транс-конфигурацию
[114]. Соединения, находящиеся в ассоциированном состоя-
нии, для которых было сделано большинство этих измере-
ний, характеризуются частотой валентных колебаний NH
в цнс-форме, примерно на 100 см"1 меньшей, чем у транс-
формы, а поэтому могла бы ожидаться заметно более высо-
кая частота деформационных колебаний. Для соединений,
присутствующих в цис-форме, можно было бы ожидать, что
полоса амид-П располагается очень близко к полосе амид-1
и может совпадать с ней. Тем не менее, остаются другие
возражения против этой точки зрения, такие, как отсут-
ствие полосы в спектрах комбинационного рассеяния и не-
однозначность результатов исследования дейтерированных
соединений, поскольку изменения в спектрах при этом
наблюдаются и в области 1300 слг1, где можно было бы
ожидать полосу поглощения ND, и в других областях
спектра.
Наиболее распространенное альтернативное объяснение
состоит в том, что эта полоса возникает в результате ва-
лентных колебаний С — N, когда связь С — N имеет в зна-
чительной степени характер двойной связи из-за резонанса
с карбонильной группой [5, 28]. Эта точка зрения частично
подтверждается в результатах исследования дейтерирован-
ных соединений и согласуется с результатами рентгеногра-
фических работ [34, 671, но имеет другие недостатки, заклю-
чающиеся в том, что при этом не объясняются отсутствие
Гл. 12. Амиды, белки и полипептиды 311
полосы у третичных амидов и данные поляризационных
измерений.
Третья точка зрения высказана Ленорманом [27—31],
развившим видоизмененную теорию двух молекулярных
форм. Согласно этой теории, и полоса амид-1 и полоса
амид-П обусловлены колебаниями карбонильной группы.
Первая полоса соответствует обычной кето-форме, а вторая—
полярной форме, при которой молекулы ассоциируются
в димеры. Этой теории противоречат данные Гирера [92]
о том, что полоса амид-П сохраняется в поглощении паров,
когда имеются только неассоциирсванные молекулы.
Эти разноречивые точки зрения были примирены, по
крайней мере частично, Фрейзером и Прайсом [59, 108],
которые указали, что рассмотрение какого-либо отдельного
движения пары атомов системы — СО — NH — изолиро-
ванно от всей системы является чрезмерным упрощением.
Двоесвязный характер и распределение зарядов по связям
С=О и С—N сами изменяются при колебании, влияя тем
самым на другие колебания группы. Взаимодействие между
этими связями приводит к асимметричному колебанию OCN
(полоса амид-1) и симметричному колебанию, которое в свою
очередь взаимодействует с деформационными колебаниями
NH. В соответствии с этим, авторы относят полосу амид-П
к сложному колебанию, которое лучше всего может быть
представлено как комбинация колебаний OCN и NH в раз-
ных фазах, причем большую роль играет угловое смещение
атома водорода. Соответствующему синфазному колеба-
нию, которое носит главным образом характер валентного
колебания С — N, приписывают полосу амид-ПI (см. ниже)
с частотой около 1290 см Эта точка зрения, включающая
в рассмотрение сложные колебания, получила почти всеоб-
щее признание, хотя Мидзусима и др. [80] отдают предпо-
чтение иным корреляциям и рассматривают полосу амид-П
как соответствующую скорее колебаниям С — N, а не NH.
При таком истолковании они являются последователями
Бехера и др. [111, 112], которые аналогичным образом
интерпретируют результаты исследования дейтерирования
диметил- н метиленмочевины. Гирер [92] предпочитает
корреляцию, связывающую полосу амид-П со смешанными
колебаниями NH и N—СН3 в N-метилацетамиде, но такая
Корреляция встречает ряд серьезных возражений [90, 109].
312
Часть III
Корреляция полосы со смешанными колебаниями дает
возможность значительно лучше, чем раньше, объяснить
результаты работ с дейтерированными соединениями, и пра-
вильность этого объяснения почти не вызывает сомнений.
Тем не менее некоторые незначительные противоречия
остаются независимо от принятой точки зрения относи-
тельно природы смешанных колебаний. Например, Абботт
и Амброз [109] указали на трудности объяснения резуль-
татов исследования дейтерирования ацетанилида в соот-
ветствии со схемой Фрейзера и Прайса, в то же время Мид-
зусима и др. [80] также указали на некоторые противоречия
в их интерпретации результатов работ с дейтерированными
соединениями. Если принять, что оба колебания С — N
и N — Н обусловливают полосы амид-11 иамид-Ш, то воп-
рос о том, какое из колебаний играет большую роль в тон
или иной полосе поглощения, является уже академическим.
Однако, что касается корреляции полосы амид-11, то дан-
ные Мекке и др. [85, ПО] по тиоамидам определенно под-
тверждают точку зрения, развитую Фрейзером и Прайсом.
У тиоамидов полоса амид-П примерно при 1550 с.и1 не
только обладает большой интенсивностью, но обнаружена
также и в случае циклических лактамов с малыми кольцами;
при дейтерировании соединений полоса смещается почти
точно на теоретически предсказанную величину, что позво-
ляет отнести ее к колебаниям только группы NH. С дру-
гой стороны, полоса амид-1 II всегда находится вблизи
1290 см1. В связи с этим нельзя считать необоснованным
предположение, что у нормальных амидов, имеющих в зна-
чительной мере двоесвязный характер C=N, полоса, соот-
ветствующая деформационным колебаниям NH, может быть
сильно смещена от 1550 с.и1, и ее положение восстанавли-
вается при взаимодействии колебаний с колебаниями OCN.
г. Область частот полосы амид-П. Положение поло-
сы поглощения амид-П N-монозамещенных амидов изуча-
лось рядом исследователей. Спектры многих веществ полу-
чили Ричардс и Томпсон [4], а Рендалл и др. [5] сооб-
щили подробности относительно положения полосы для
72 монозамещенных амидов. Опубликованы также данные
некоторых других исследователей, получивших согласу-
ющиеся результаты [9]. Эти исследователи установили
Гл. 12. Амиды, белки и полипептиды
313
область частот 1570—1515 слг1 для веществ в твердом сос-
тоянии, большинство которых поглощает вблизи среднего
значения 1540 см'1. Ричардс и Томпсон [9] показали, что
электрофильные заместители при атоме азота в некоторых
случаях приводят к небольшому сдвигу полосы в напра-
влении больших длин волн, но в спектре N-хлорметилацет-
амида полоса амид-П, по-видимому, отсутствует [92]. Одна-
ко, за исключением этой работы, не предпринималось
никаких попыток установить связь между положением поло-
сы и природой окружения амидной группы. Это объясняет-
ся частично тем, что изменения, сопутствующие растворе-
нию или изменению состояния, чрезвычайно велики и долж-
ны маскировать незначительные различия, вызванные
указанной причиной. Наличие где-либо в другой части моле-
кулы карбоксильных групп не оказывает столь заметного
влияния на полосу поглощения амид-П, как на полосу
амид-1, и направление происходящего при этом сдвига
полосы непостоянно. Например, гиппуровая кислота и ме-
тилфенацетурат поглощают при 1555 см'1 (в твердом состоя-
нии), в то время как этилгиппурат и фенацетуровая кисло-
та поглощают при 1530 см'1 [5].
В случае растворов полоса амид-П сдвигается в напра-
влении меньших частот в пределах интервала 1650—
1510 см'1; так, например, метилфенацетурат поглощает при
1555 см 1 в твердом состоянии, при 1535 см'1 в расплаве
и при 1517 см'1 в разбавленном растворе [5]. Это смещение
в сторону меньших частот при разрыве водородных связей
имеет направление, противоположное тому, которое харак-
терно для полос поглощения, обусловленных валентными
колебаниями, а именно полос ОН и NH вблизи 3300 см'1;
однако такого направления сдвига и следует ожидать в слу-
чае изменений деформационных частот. Как и в случае
полосы амид-1, величина смещения при переходе от твердого
состояния к разбавленному раствору зависит от сложности
молекулы. Для сложных веществ, в которых геометрия мо-
лекулы такова, что водородные связи в кристаллической
решетке не образуются, смещения, наблюдаемые при рас-
творении, невелики. Частота полосы 1515 см'1 твердого
бензилпенициллина, например при его растворении, замет-
но не изменяется. Типичными в отношении сдвига полос
поглощения при изменении состояния вещества являются
314
Часть III
N-метилацетамид (1565 см 1 в жидком состоянии, 1534 см"1
в растворе, 1490 см"1 в парах [87, 90—92]) и а-хлор-N-
метилацетамид [94] (1565см1 в твердом состоянии, 1550 см"1
в жидком состоянии, 1515 слг1 в парах). Однако вооб-
ще опубликовано чрезвычайно мало данных о характери-
стических частотах этого поглощения в разбавленных раст-
ворах или в парах.
Как было показано ранее, у циклических лактамов
и сходных с ними веществ такая полоса поглощения отсут-
ствует [9, 28], но наблюдается у соответствующих тиолак-
тамов. Частота поглощения претерпевает изменения, анало-
гичные описанным выше. У тиовалеролактама, например,
полоса амид-П смещается при переходе от твердого состо-
яния к парам от 1572 до 1513 см"1 [85].
5. ДРУГИЕ КОРРЕЛЯЦИИ
а. Первичные амиды. Рендалл и др. [5] заметили у всех
исследованных ими незамещенных амидов полосу в обла-
сти 1418—1399 см"1, которая отсутствует в спектрах N-заме-
щенных амидов. Они отнесли эту полосу к валентным коле-
баниям С — N и считают, что это подтверждает их точку
зрения об аналогичном происхождении полосы амид-11.
Однако число исследованных ими простых амидов очень
ограниченно, а другими исследователями эта корреляция
не обсуждалась.
Поскольку поглощение в этой области не является спе-
цифичным, то при работах по идентификации веществ эта
корреляция имеет ограниченное значение. Мы заметили
слабое поглощение в этой области у многих азотсодержа-
щих соединений, не являющихся амидами, например у ди-
фениламина; следовательно, правильность первоначально-
го отнесения не подтверждается, так как нет оснований по-
лагать, что связь С—N в этих соединениях имеет в какой
то мере характер двойной связи.
б. Полоса амид-П1. Эта полоса поглощения проявляется
у вторичных амидов [80, 90, 108] в области 1305—1200 слг1
и обычно заметно слабее, чем полосы амид-1 или амид-П.
Полоса амид-1II почти определенно связана со смешан-
ными колебаниями, включающими колебания OCN и N — Н;
Гл. 12. Амиды, белки и полипептиды
315
этот вопрос* был уже обсужден выше в связи с корреляцией
полосы амид-П. У обычных амидов полоса амид-1 II чув-
ствительна к дейтерированию соединений, что указывает
на связь со смешанными колебаниями, но у тиоамидов ее
частота практически не меняется, а поэтому ее относят пол-
ностью к колебанию С — N [85]. Изменения состояния ве-
щества приводят к заметным изменениям частоты полосы
поглощения обычных амидов, но, как и следовало ожидать,
этот эффект заметно ослаблен у тиопроизводных. Эта полоса
только недавно была определенно идентифицирована как
характеристическая полоса поглощения вторичных амидов
и подробно еще не изучена.
в. Скелетные колебания. Низкочастотные полосы погло-
щения, названные Мидзусимой и др. [80] полосами амид-IV
и амид-VI, проявляются у вторичных амидов соответствен-
но около 620 см'1 и около 600 см'1 [80, 81]. Эти полосы
поглощения связаны со скелетными колебаниями и имеют
ограниченное значение для характеристики соединений.
г. Внеплоскостные деформационные колебания NH. Ха-
рактерной для белков и вторичных амидов является раз-
мытая полоса поглощения с максимумом около 700 см'1.
При дейтерировании соединений интенсивность такой по-
лосы уменьшается и вблизи 530 см'1 появляется новая поло-
са поглощения. Это уменьшение частоты в 1,36 раза почти
точно такое, какого можно бы ожидать для чисто дефор-
мационного колебания NH, которое, таким образом, и обу-
словливает появление этой полосы поглощения [79]. При
растворении вещества интенсивность полосы уменьшается
и у вторичных амидов при сильном разбавлении в конце
концов полностью исчезает. Ясно поэтому, что поглощение
связано с ассоциированной формой этих соединений и
совершенно аналогично полосам поглощения тиоамидов
[85], претерпевающим такие же изменения. Частота коле-
баний у свободной NH-группы определена не вполне точно.
В разбавленном растворе N-метилацетамида к этому коле-
банию отнесена полоса 648 см'1 [80], а вообще эта частота
лежит за нижним пределом рабочей области призмы из
каменной соли.
316
Часть 111
6. АМИДЫ, содержащие ОСОБЫЕ СТРУКТУРЫ
а. Соединения, содержащие группу СО — NH — СО.
Группа СО — NH — СО обычно не встречается в веществах
с открытой цепью, но находится во многих циклических
соединениях, например в хидантоине, сукцинимиде, пури-
нах и родственных им веществах. Возможность таутоме-
рии у некоторых урацилов и пиримидинов рассматривается
в гл. 16. Остальные вещества дают одну полосу поглоще-
ния связанной амидной группы NH в области 3200 см"1,
и из исследований ограниченного числа веществ [5] следует,
что их поглощение, по-видимому, сходно с поглощением
циклических лактамов, которое имеет место при меньших
частотах, чем для соединений с открытой цепью.
Карбонильное поглощение этих веществ [51 проявляет-
ся в виде двух отдельных полос в интервалах 1790—
1720 и 1710—1670 см'1. Первая из них отнесена к карбо-
нильной группе в положении 4, поскольку она наблюдается
у соединений, у которых группа СО в положении 2 заме-
щена группой С = S. Вторая полоса могла бы быть обуслов-
лена колебаниями С = N, но ее относят к карбонилу в поло-
жении 2, так как она появляется у соединений, в кото-
рых группа NH замещена группой N — СН3.
Замена NH на NCH3 не приводит к сколько-нибудь за-
метному смещению карбонильной полосы, и, поскольку
спектры твердого вещества и раствора очень сходны, то пред-
полагается, что в твердом состоянии водородные связи
в значительном количестве не образуются [5]. Однако
при таком заключении трудно объяснить малые значения
частот валентной полосы поглощения NH для веществ
в твердом состоянии и в разбавленных растворах и
желательно провести дальнейшие исследования в этой
области.
У циклических соединений, содержащих три карбониль-
ные группы, те группы, которые находятся в непосредствен-
ной близости от группы NH, ведут себя, как указано выше,
в то время как третья группа дает обычную полосу погло-
щения СО. Спектры шести триалкилпирролидинтрионов,
исследованных Скиннером и Перкинсом [32], имеют поло-
сы поглощения при 1786, 1724 и 1667 см"1. Поглощение
при 1724 см'1 относится к группе СО в положении 3,
Гл. 12. Амиды, белки и полипептиды
317
б. Соединения, содержащие группу R — NH —СО — OR.
У соединений этого типа карбонильное поглощение, соот-
ветствующее полосе амид-1, вероятно, должно иметь, как
и в случае обычных амидов, меныпую частоту в связи
с наличием группы NH. Однако остаток С — О — R должен
влиять противоположным образом, так же как и для слож-
ных эфиров, у которых полоса карбонильного поглощения
соответствует более высоким частотам, чем у кетонов. Это
и наблюдается в действительности, по крайней мере для
уретанов, исследованных Рендаллом и др. [5], и карба-
матов, которые были изучены нами. Полоса амид-1 ряда
уретанов находится в интервале 1736—1700 см"1 [5], тогда
как у небольшого ряда различных замещенных карбаматов
эта полоса появляется в области 1710—1690 см~1. Полоса
амид-П у соединений с группой NH имеет нормальные час-
тоты, и это обстоятельство можно рассматривать как неко-
торое подтверждение отнесения полосы амид-П к деформа-
ционным колебаниям NH, а не к связи OCN. В последнем
случае можно было бы ожидать большого смещения полосы
при замене С — N— С— на С — N — О—. Сам уретан по-
глощает при 1618 сл/-1,что соответствует полосе поглощения
амид-П незамещенного амида, тогда как NN-дизамещенные
соединения не имеют полосы в этой области. Простые
N-монозамещенные уретаны [5] и карбаматы в твердом состо-
янии дают полосу поглощения валентных колебаний NH
в области 3300—3250 см-1, которая является обычной для
амидов с открытой цепью.
Та же группа имеется в таких циклических системах,
как оксазолоны, которые были изучены Гомпером и Хер-
лингеромП 15] и сопоставлены с соответствующими тиокето-
соединениями. Частота карбонильного поглощения пони-
жается в этом случае примерно до 1750 сл/’1, а частота поло-
сы амид-Ш повышается до 1380 см~1.
в. Анилиды. С6Н5 — NHCOR. Замещение ароматическим
радикалом при амидном азоте не оказывает сколько-ни-
будь значительного влияния на спектр, за исключением
небольшого повышения частоты полосы амид-1, обуслов-
ленной наличием карбонильной группы. Ричардс и Томп-
сон [4] нашли, что полоса СО у ряда анилидов в разбавлен-
ном растворе расположена при 1700 сл/’1; это значение
318
Часть 111
возрастает до 1715слг1 для соединений, у которых замести-
тели в кольце имеют повышенные электроноакцепторные
свойства. Полоса амид-П должна, по-видимому, значи-
тельно меньше зависеть от такого рода изменений, и могли
быть замечены только небольшие сдвиги ее в сторону мень-
ших частот. Практически аналогичные результаты полу-
чены Гирером [92] для ряда ацетанилидов с различными
циклическими заместителями. Частота поглощения СО
у ариламидов в твердом состоянии несколько выше, чем
у алкиламидов. Спектры большого числа анилидов жир-
ных кислот получены Дийкстра и Де Жонжом [116].
Полосы поглощения, соответствующие валентным коле-
баниям NH, являются обычными для амидов с открытой
цепью и находятся вблизи 3270 см'1 (твердое вещество),
но идентификация какого-либо возможного дополнитель-
ного поглощения в области 3070 см'1 в этом случае затруд-
нена, так как в эту же область попадают полосы поглоще-
ния СН ароматического ядра.
г. Замещенные производные мочевины — NHCONH —
и т. д. Спектры производных мочевины исследовались Том-
псоном, Николсоном и Шортом [25], Рендгллом и др. [5],
Бойвином и Бойвин [117, 118] и Бехером [111, 112], но
полностью интерпретировать полученные ими результаты
не удается, так как спектры часто являются сложными,
и весьма возможно существование различных биполярных
и енольных форм. У полностью замещенных мочевины
имеется полоса поглощения СО вблизи 1660 см'1, и в этой
же области наблюдается полоса поглощения у моно- и диза-
мещенных веществ, которую с полным основанием можно
также объяснить наличием группы С = О. Полосы поглоще-
ния NH, однако, более сложны. Как у ацетил-, так и у
фенацетилмочевины обнаружено поглощение при 1634 см'1,
которое можно было бы отнести к деформационным колеба-
ниям NH2 [5], но нет никаких указаний на какую-либо
соответствующую полосу при более низких частотах, появ-
ления которой естественно ожидать при замещении в груп-
пе NH. Для установления надежных корреляций соответ-
ствующих амидных полос поглощения имеют значение
результаты исследования дейтерированных производных
диметилмочевины и метиленмочевины [111, 112], которые
Г л. 12. Амиды, белки и полипептиды
319
могут быть сопоставлены с имеющимися данными о произ-
водных.тиомочевины [85, ПО]. Тем не менее интерпрета-
ция столь сложных спектров остается весьма затруднитель-
ной. Характеристические частоты колебаний группы
NH—СО — N— ациклических системах были обсуждены
Гомпером и др.
д. Соединения, содержащие группу R — СО — NH — N —.
Наличие дополнительного атома азота, по-видимому, мало
сказывается на спектроскопических свойствах этих соеди-
нений, и спектр, например, диформил гидразина не очень
сильно отличается от спектров других вторичных амидов
[80]. Однако в таких циклических системах, как пиразо-
лоны, значительно труднее идентифицировать характери-
стические частоты колебаний амидов из-за сложности на-
блюдающихся полос поглощения [119, 120].
7. ПОЛИПЕПТИДЫ И БЕЛКИ
а. Общие сведения. Методы инфракрасной спектроско-
пии нашли применение уже в ранних исследованиях строе-
ния белков и подобных веществ [13—15, 36—40]. Пока-
зано, что белки дают обычную картину полос поглощения,
включающую полосы при 3300 слГ1; последние обуслов-
лены группами NH, участвующими в образовании водород-
ной связи. Имеются также полосы амид-1 и амид-П в об-
ласти 1600—1500 см'1; остальная же часть спектра опреде-
ляется в основном природой компонентов аминокислот.
Кроме того, некоторыми исследователями замечена слабая
полоса поглощения вблизи 3080 см'1, предположительно
интерпретированная ими как дополнительное поглощение
группы NH [40]. В последние годы исследования стали
проводиться в больших масштабах благодаря развитию
методов изучения поляризованного излучения, которые
позволяют изучать внутреннюю ориентацию различных
группировок. Кроме того, достигнуты успехи в приготов-
лении простых модельных полипептидов и в очистке самих
белков, в связи с чем работы в этой области начинают да-
вать существенные результаты, обзор которых дан Сазер-
лендом [121] и Эллиоттом [122].
320
Часть III
Проведены также исследования простых пептидов,
содержащих лишь небольшое число аминокислотных остат-
ков. В случае простейших членов ряда преобладает харак-
теристический спектр пептидов, обусловленный присут-
ствующими биполярными ионами. По этой причине такие
соединения рассмотрены отдельно, но данные об этих сое-
динениях должны учитываться при обсуждении характери-
стических частот для более высокомолекулярных поли-
пептидов.
б. Простые пептиды. Спектры простых пептидов изу-
чены рядом исследователей [25, 41, 123—127]. В дипеп-
тидах, например в глицилглицине, влияние структуры би-
полярных ионов наибольшее и спектр более сложен, чем
спектры высших членов ряда. Из рентгенографических ис-
следований известно, что это соединение существует в виде
биполярного иона с открытой цепью [34]. При 3300, 3080,
1630, 1575, 1540 и 1240 см'1 у глицилглицина имеются поло-
сы поглощения NH, на что указывают смещения этих полос
при дейтерировании [25, 41]. Первые две полосы соответ-
ствуют валентным колебаниям NH, тогда как полоса
1540 см'1 является обычной полосой амид-П, которая может
быть обусловлена деформационными колебаниями NH. По-
лосы 1630 и 1575 см'1 аналогичны полосам амид-1 и амид-П
аминокислот и могут быть связаны с группировкой NH+.
Карбонильная группа пептидной цепи дает обычное погло-
щение при 1655 слг1, и, кроме того, имеются еще сильные
полосы при 1608 и 1400 см'1, которые, по-видимому, соот-
ветствуют группе СОО~. Подобная полоса при 1400 см'1
замечена Эдселломидр. [42] в спектре комбинационного рас-
сеяния. Указанными исследователями в этой области обна-
ружена в инфракрасном спектре слабая полоса вблизи
1680 см1, которую они предположительно отнесли к коле-
баниям СО неионизованной карбоксильной группы. Однако
имеется некоторое расхождение в данных, так как Эдселл
и др. [42] обнаружили эту полосу в спектре комбинацион-
ного рассеяния при 1740 см'1. Отнесение ее подтверждает-
ся спектрами низших полиглицинов. При рассмотрении
ряда этих соединений вплоть до гекса- и полиглицинов
можно отметить постепенное уменьшение влияния биполяр-
ной ионной структуры, а поэтому в конце концов полоса
Гл. 12. Амиды, белки и полипептиды
321
1400 см'1 (которая из двух полос СОО~ является наибо-
лее сильной) исчезает, тогда как полоса 1680 см'1 остается.
Томпсон и др. [25] сообщают, что последняя усиливается
у высших членов ряда, в то время как Блоут и Линслей
[41] утверждают, что ее интенсивность уменьшается. Одна-
ко различие здесь, по-видимому, только в выражениях
одной и той же мысли; дело в том, что если полоса действи-
тельно обусловлена неионизованной группой СООН, то ее
абсолютная интенсивность уменьшается с увеличением дли-
ны цепи и молекулярного веса соединения, тогда как ее
относительная интенсивность на группу СООН повышает-
ся вследствие увеличения относительного содержания
неионизованных карбоксильных групп.
Ни один из полиглицинов не поглощает выше 3330 см'1,
что указывает на способность всех групп NH образовы-
вать в кристаллическом состоянии водородные связи. При
увеличении молекулярного веса интенсивность полосы
3300 см 1 закономерно повышается относительно интенсив-
ности поглощения СН2, причем имеется тенденция прибли-
жения отношения интенсивностей к предельному значению,
по мере того как число групп СН2 приближается к числу
амидных связей [41]. Это дает основание отнести полосу
к валентным колебаниям связанной группы NH.
С другой стороны, в случае полосы 3080 см'1 такие
явления не наблюдались, хотя ее связь с группой NH была
установлена в результате исследования продуктов дейте-
рирования, в спектре которых указанная полоса исчезает
и заменяется двумя полосами J41] при 2530 и 2480 см'1.
Остальные характеристические полосы поглощения высших
членов ряда пептидов с увеличением длины цепочки
и уменьшением влияния биполярных ионов все больше
принимают вид, типичный для спектров белков. Все поли-
глицины поглощают при 1015 1 10 см'1, и это поглощение
можно считать характеристическим для диглициновой струк-
туры [41], особенно в связи с тем, что оно не обнаружено
у глицилаланина или у каких-либо иных полипептидов,
у которых два глициновых остатка непосредственно не свя-
заны [127]. Спектры поглощения полипептидов с амино-
кислотными остатками какого-либо одного типа прибли-
жаются к спектрам белков, если число аминокислотных
остатков становится равным трем или более [109], и дают
21 л. Беллами
322
Часть 111
типичные полосы поглощения амидов, такие, как амид-
ные полосы II, III и V, корреляции для которых были
обсуждены выше |127]. Полипролин и полисаркозин |124]
ведут себя, конечно, несколько иначе, так как у них отсут-
ствуют связи NH.
Смешанные полипептиды также изучены обеими груп-
пами исследователей |25, 41]. В общем картина оказалась
подобной наблюдавшейся для полиглицинов, но были за-
мечены некоторые интересные отличия. В частности, мно-
гие глицинлейцинпептиды, помимо полос поглощения при
3300 и 3080 см'1, дают полосы при 3400 и 3520 см1.
Эти данные ясно указывают на присутствие несвязанных
группировок NH2 и позволяют пре;положить, что кристал-
лическая упаковка этих веществ такова, что ни одна из кон-
цевых групп NH2 не может участвовать в образовании водо-
родных связей. Интересные результаты получены также
в исследованиях происходящих изменений при использо-
вании для синтезов различных оптических форм амино-
кислот. Когда имеется более одного асимметричного атома
углерода, наблюдаются явные изменения в спектре веще-
ства. Так, спектр с(/-глицилаланина идентичен спектру
соответствующего /d-соединения, но заметно отличается от
спектров dd- или //-соединений, которые в свою очередь
идентичны друг другу. Аналогично, спектр ///-триаланина
идентичен спектру ddd-формы, но резко отличается от спек-
тра смешанного изомера 1123]. Эти отличия относятся глав-
ным образом к полосам поглощения валентных колебаний
NH и полосам амид-1 и амид-П и связаны с различиями
в пространственном строении соединений. Эллиотт |126|
сообщил также о некоторых различиях между активной
и мезо-формами таких полипептидов, как полиглицин, по-
лиаланин и т. п. Эти вещества существуют явно скорее в
свернутой a-форме, чем в |3-форме, но для первого из них
это выражено еще не очень сильно, и наблюдается сдвиг
полосы карбонильного поглощения примерно на 9 см1.
Ряд простых пептидов был исследован в виде растворов
в тяжелой воде Ленорманом и др. [62, 128], которые про-
следили изменения полос карбонильного поглощения и по-
глощения NH2 в зависимости от изменений pH раствора.
Подобное исследование сложных молекул было проведено
Эрлихом и Сазерлендом [129, 130]. Они установили, что
Гл. 12. Амиды, белки и полипептиды
323
полоса поглощения 1550 см'1 представляет наложение по-
лосы поглощения СОО и полосы амид-П. Вследствие этого
изменения степени ионизации, которые происходят в про-
цессе денатурирования таких веществ, должны влиять на это
поглощение независимо от каких-либо происходящих при
этом изменений конфигурационных состояний молекул.
Изучены также ацетилированные соединения, не содер-
жащие группы NH2 или СООН [25, 43, 131, 132]. В этих
случаях спектры в области 3080 и 1600 см'1 сходны со
спектрами полиамидов. Изучая такие соединения, как
ацетилглицин-Ы-метиламид в виде разбавленных растворов
в четыреххлористом углероде, Мидзусима и его сотрудни-
ки |8, 43, 63, 68, всегда наблюдали помимо полосы свобод-
ной группы NH при 3450 см 1 присутствие полосы вблизи
3330 см'1. Кроме того, более подробные исследования [44,
132, 137] показывают, что межмолекулярное и внутримоле-
кулярное взаимодействия могут вызвать поглощение с од-
ной и той же частотой. Таким образом, интерпретация
структуры соединения на основе данных о поглощении
в этой области спектра весьма затруднительна. Предпола-
гается, что в растворе в четыреххлористом углероде все
молекулы ацетилпролин-М-метиламида присутствуют в свер-
нутой форме, а в хлороформе, где тенденция к образованию
внутримолекулярных водородных связей уменьшается, мо-
жет осуществляться вторая конфигурация, в случае кото-
рой может происходить межмолекулярное взаимодействие.
Тенденция к образованию межмолекулярных связей еще
более повышается в случае ацетилпиперидина и-карбоновой
кислоты N-метиламида ] 132|, у которого стерические эф-
фекты затрудняют образование внутримолекулярных водо-
родных связей. В разбавленном растворе в четыреххлори-,
стом углероде это вещество поглощает при 3390 см'1, что
обусловлено образованием внутримолекулярных связей,
а также при 3367 см-1 в связи с наличием межмолекулярных
связей. Эти данные свидетельствуют о том, что нельзя с уве-
ренностью отнести полосы поглощения белков при 3280 см'1
полностью к тра«с-межмолекулярным связям.
в. Полоса валентных колебаний NH вблизи 3330 см \
Как показали исследования с дейтерированными соединения-,
ми и исследования с применением метода поляризации,полоса,
21*
324
Ч а с т ь III
расположенная вблизи 3330 см'1, обусловлена связан-
ной группой NH. Как и в случае простых пептидов, имеют-
ся разногласия в вопросе о том, обусловлена ли эта поло-
са поглощения целиком образованием водородных связей
транс-типа между цепочками молекул или образованием
внутримолекулярных связей или обеими этими причинами.
Показано, что поли-у-бензил-а-глутамат в разбавленном
растворе в хлороформе не поглощает при 3450 см'1, что гово-
рит о способности всех групп NH образовывать между собой
связи внутри молекулы. Напротив, поликарбобензокси-
о!/-лизин, который имеет группы NH и СО и в главной, и в
боковой цепях, поглощает в этих условиях при 3450 см'1,
что указывает на отсутствие связи между полипептидными
цепями [11, 49, 51]. Свертывание полипептидных цепей
в растворе может быть не таким, как в твердом состоянии,
но и в данном случае можно обоснованно считать, что при
свернутой a-форме пептидных цепей водородные связи яв-
ляются внутримолекулярными. С другой стороны, Шиго-
рин и др. [133—135] на основании изучения поликапролакта-
мов и сходных веществ относят полосу поглощения 3300 см1
целиком к внутримолекулярному взаимодействию, свя-
зывая более слабое поглощение вблизи 3200 см'1 с межмо-
лекулярными ассоциациями. Так же как и в случае про-
стых пептидов, поглощение около 3300 см'1, вероятно, обу-
словлено ассоциациями обоих типов. Так, например, и по-
ли-у-бензил-/-глутамат [136] и поли-а-/-глутамовая кисло-
та [ 137] могут быть получены в виде двух различных модифи-
каций, которые отличаются друг от друга ориентацией
группы NH. Одной из них, вероятно, является свернутая
a-форма с внутримолекулярными связями, а другой —
открытая или произвольная форма. В обоих случаях погло-
щение наблюдается около 3300 си'1. Аналогичным образом
найлон и другие синтетические полимеры с длинными цепя-
ми, у которых внутримолекулярные связи невозможны, так-
же поглощают в этой области. В связи с этим интерпрета-
ция изменений интенсивности этой полосы, которыми со-
провождается денатурирование [45], чрезвычайно сложна.
Как отмечалось выше, поглощение при 3300 см'1 сопро-
вождается более слабым поглощением около 3200 слг1 [50].
Это поглощение, согласно Шигорину и др. [133,135], соответ-
ствует только внутримолекулярным связям, но эти данные
Гл. 12. Амиды, белки и полипептиды
325
представляются не окончательными в связи с причинами,
рассмотренными выше, а также в связи с тем, что в этой
же области поглощают и а- и |3-кератины [135].
г. Полоса валентных колебаний NH вблизи 3080 см1.
Первоначально считали, что эта полоса обусловлена вну-
тримолекулярными водородными связями групп NH, суще-
ствующими помимо связей между цепочками, которым соот-
ветствует полоса поглощения при 3330 см 1 [6, 7]. Однако
в последней работе указывается на то, что здесь имеют
значение другие факторы. На основании того, что интен-
сивность этой полосы у найлона постепенно уменьшается
при повышении температуры, а частота полосы при этом не
меняется, можно вывести заключение, что эта полоса, не-
смотря на ее меньшую частоту, соответствует более слабым
связям, чем полоса 3330 см'1. Относительно природы этой
полосы было выдвинуто много различных предположений.
Например, считали, что она связана с взаимодействиями
в кристаллическом состоянии [12], что она возникает вслед-
ствие «закручивания» межмолекулярных связей в ^wc-поло-
жение [134], а также что она возникает у цис- и транс-
изомеров, что эта частота является составной из сЛювной
частоты колебаний NH и низкочастотных колебаний ассо-
циатов или что эта полоса вместе с полосой 3300 см'1 соот-
ветствует нормальным колебаниям NH последовательно
связанных осцилляторов. В результате обсуждения этой
проблемы в последнее время указанную полосу поглоще-
ния, в соответствии с прежним предположением Бедже-
ра [82], рассматривают как полосу, соответствующую обер-
тону полосы амид-П.
Очевидно, что в настоящее время возникновение полосы
поглощения в этой области просто объяснить невозможно,
особенно потому, что некоторые более простые соединения,
такие, как дикетопиперазин, которые, казалось бы, могли
служить удобными объектами исследования, в действитель-
ности являются более сложными и дают несколько полос
в рассматриваемой области спектра.
д. Полосы амид-1 и амид-П. У всех полипептидов и бел-
ков имеются полосы амид-1 и амид-П. Большинство этих
веществ обычно поглощает в областях 1650 и 1550 см'1,
326
Часть III
но встречаются и небольшие отличия, которые могут в ка-
кой-то мере характеризовать изменения в образовании
водородных связей. Например, циклический пептид грами-
цидин [47] поглощает при 1610 и 1513 см'1. Влияние изме-
нений типа водородных связей на частоты этих полос погло-
щения очень хорошо выявляется при исследованиях свер-
нутых и вытянутых форм этих веществ наряду с изучением
дихроизма и анализом рентгенографических данных [46,
52 , 54]. Поли-/-глутамбензиловый эфир обнаруживает амид-
ное поглощение СО в виде одной полосы при 1658 см'1,
а рентгенографическое исследование показывает, что это
соединение существует исключительно в виде а-формы.
Однако при синтезе этого соединения из веществ с низким
молекулярным весом оно получается в произвольной или
в какой-либо развернутой форме; полоса амид-1 находится
при этом около 1630 см~1 [136]. Исследования, проведенные
с растворами, подтверждают наличие этих форм [113]. Ме-
тиловый эфир имеет, однако, две полосы поглощения с час-
тотами 1658 и 1628 см1, последняя из которых не поляри-
зована. В случае производного муравьиной кислоты, кото-
рое, как полагают, существует в виде 0-формы, имеется
только одна полоса при 1629 см'1. Аналогичным образом
две полосы амид-П 1527 и 1550 слг1 соответствуют 0- и
а-формам.
Хард и др. [69] наблюдали у синтетических поли-
пептидов различия в частотах для полос карбонильного
поглощения, которые они приписывают существованию
а- и 0-форм, образующихся в результате применения раз-
личных методов приготовления образцов, а Кримм [138]
связывает это с различиями в углах, образованных водород-
ными связями О...Н — N —. Этот вопрос обсуждался также
Кэнноном [78], который предпочитает относить эти разли-
чия в поглощении за счет различной степени взаимодей-
ствия диполей 'OCN*.
Таким образом, исследуя положение этих полос, мож-
но получить некоторые сведения о молекулярной форме,
но следует учесть, что в твердом состоянии на поглоще-
ние могут влиять также и другие факторы. Например, при
повышении температуры найлона происходят изменения
частот СО и NH, которые очень похожи на уже описанные
изменения при переходе из a-формы в 0-форму. Доказано,
Гл. 12. Амиды, белки и полипептиды
327
однако, что найлон сохраняет при этих условиях вытяну-
тую р-форму [48]. Аналогичным образом полоса поглоще-
ния хитина в области 1625 см'1 была предположительно от-
несена к амидной группе СО, которая связана водородной
связью с группой ОН [55]. Интерпретация полосы амид-П
также сопряжена со значительными трудностями. Эта
полоса претерпевает, например, явные изменения при дей-
терировании соединений и при изменениях их конфигура-
ции (128, 139, 140]; решение вопроса о происхождении
этой полосы затрудняется из-за наличия в той же области
спектра полосы поглощения, обусловленной колебаниями
ионизованной карбоксильной группы. У многих белков
полоса амид-П частично перекрывается полосой поглоще-
ния ионизованной карбоксильной группы, так что измене-
ния в этой области спектра, сопутствующие изменениям со-
стояния вещества, могут быть обусловлены целиком изме-
нениями группы СОО' при удалении от изоэлектрической
точки [128—130, 137]. Интерпретация частот поглощения
в этой области требует поэтому осторожности.
Ряд важных сведений о свойствах белков может быть
также получен при изучении области обертонов, тем более,
что при этом можно работать в присутствии жидкой воды.
Полосы поглощения в этой области также проявляют дихро-
изм, но следует иметь в виду, что он не обязательно дол-
жен быть того же направления, что и дихроизм полосы
основных колебаний, тем более что многие из полос погло-
щения в этой области спектра соответствуют составным
частотам от нескольких тонов. Так, полоса около 4600 см 1
в спектрах многих белков была первоначально интерпрети-
рована, как соответствующая карбонильному поглоще-
нию [53], поскольку она смещается при изменениях спи-
ральной формы соединения и обнаружена у полиметилме-
такрилата, но отсутствует в спектре полиэтилена. Одна-
ко, как было недавно показано Хехтом и Вудом [141],
эта полоса соответствует составной частоте 2\СО плюс
частота 1250 см'1. Те же исследователи выяснили проис-
хождение ряда других полос поглощения этого типа,
расположенных в высокочастотной области спектра. Ряд
корреляций для этой области спектра рассмотрен Фрейзе-
ром [142], который обсудил также результаты, полученные
при исследовании дихроизма.
328
Ч а с т ь III
8. ДИХРОИЗМ В ИНФРАКРАСНОЙ ОБЛАСТИ СПЕКТРА
ПРИ ИССЛЕДОВАНИЯХ БЕЛКОВ
В последние годы методы исследования в поляризован-
ном инфракрасном излучении получили широкое примене-
ние для изучения ориентации специфических группировок
в кристалле или в ориентированной пленке. Метод состоит
в пропускании поляризованного излучения через образец
в двух взаимно перпендикулярных направлениях и наблю-
дении изменений интенсивности определенных полос
поглощения. Таким путем можно определить ориентацию
отдельных групп молекулы в образце как целом. Этот
метод был применен в 1947 г. Глеттом и др. при изучении
белков [56, 57] и затем широко использован другими иссле-
дователями в этой области, причем доказано, что при его
помощи можно провести дифференциацию свернутой (а)
и вытянутой (р) форм белков и полипептидов [46, 49—
51, 59, 61, 125]. Детальные обзоры техники эксперимента
недавно опубликованы Эллиоттом [122] и Бемфордом
и др. [17].
Амброз, Эллиотт и Темпл [46] изучали полосы поглоще-
ния, соответствующие валентным колебаниям NH для
ориентированных полиэфиров глутаминовой кислоты, а- и
Р-кератинов и ориентированных миозина и тропомиозина. Во
всех случаях, когда предполагали,что вещество находится
в виде свернутой a-формы, обе полосы валентных коле-
баний NH (3310 и 3060 см1) проявляли параллельный ди-
хроизм, тогда как у кератина птичьего пера, который су-
ществует в виде p-формы, он перпендикулярен. Аналогич-
ную картину наблюдали Амброз и Хенби [51] у поли-
эфира глутаминовой кислоты. Это исключает возможность
проявления дихроизма группами NH боковых цепей, так
как в данном веществе они отсутствуют. Продолжением этих
исследований явилось изучение валентных колебаний С = О
и деформационных колебаний NH [49 , 52], которое пока-
зало, что, кроме изменений частот, при изменении молеку-
лярной формы происходят также изменения направления
ориентации групп NH. У полиэфира глутаминовой кисло-
ты, например, в a-форме проявляется сильный параллель-
ный дихроизм. В случае (3-формы не найдено никакой ори-
ентации, но дихроизм полосы поглощения СО при измене-
Гл. 12. Амиды, белки и полипептиды
329
нии формы таких сополимерных веществ полностью меняет-
ся от параллельного до перпендикулярного.
Для деформационных колебаний NH наблюдаются те же
особенности, но в этом случае a-форма имеет перпендику-
лярный дихроизм, а р-форма — параллельный дихроизм.
Подобные результаты можно также получить при исследо-
вании полосы 4810 см1, которая соответствует составной
частоте валентных колебаний NH, и полосы амид-П [56,
141]. Эта методика исследования применена также при изу-
чении вторичных амидов в кристаллическом состоянии
[33, 105].
Дармон и Сазерленд [6] подвергли критике эту интер-
претацию результатов, при которой для а-кератина и по-
добных соединений предполагается только простая форма
водородной связи, как чрезмерно упрощенную. Они ука-
зали, что вряд ли можно придавать большое значение чис-
ленным величинам дихроических отношений, учитывая то
расхождение, которое имеется между малыми величинами
отношений, найденных для полос NH в а-кератине и най-
лоне, и большой величиной отношения, которую следова-
ло бы ожидать [6]
На необходимость соблюдения осторожности при интер-
претации результатов поляризационных исследований ука-
зывали в недавней работе Фрейзер и Прайс [59]. Они
установили, что момент перехода для карбонильного валент-
ного колебания, обусловливающего полосу поглощения
амид-1, смещен от направления связи СО вследствие взаи-
модействия с колебанием С — N и соответствующего орби-
тального эффекта. Они вычислили дихроизм ориентирован-
ного полипептида, в котором цепочки имеют конфигура-
цию, установленную Полингом и Корей [60] (3,7-спираль),
и показали, что если допустить отклонение момента пере-
хода СО на 20° по направлению к NO, то величина дихро-
изма окажется очень близкой к полученной эксперимен-
тально [54]. Поэтому возражения против наличия
3,7-спирали, основанные на данных поляризационных иссле-
дований [61], не являются убедительными.
ЛИТЕРАТУРА
1. Hantzsch, Вег., 64, 661 (1931).
2. Senti, Harker, J. Amer. Chem. Soc., 62, 2008 (1940),
330
Часть III
3. Davies, Discuss. Faraday Soc., 9, 325 (1950).
4. Richards, Thompson, J. Chem. Soc., 1947, 1248.
5. Randall, Fowler, Fuson, Dangl, Infra-red Deter-
mination of Organic Structures, Van Nostrand, 1949.
6. Darmon, Sutherland, Nature, 164, 440 (1949).
7. Astbury, Dalgliesh, Darmon, Sutherland,
Nature, 162, 596 (1948).
8. Mizushima, Shimanouchi, Tsuboi, Nature,
166, 406 (1950).
9. The Chemistry of Penicillin, Princeton University Press, 1949,
p. 390.
10. Darmon, Discuss. Faraday Soc., 9, 325 (1950).
11. Elliot, Ambrose, Nature, 165, 921 (1950).
12. Sutherland, Discuss. Faraday Soc., 9, 274 (1950).
13. В u s w e 1 1, R о d e b u s h, Roy, J. Amer. Chem. Soc., 60,
2444 (1938).
14. В u s w e 1 1, D owning, R о d e b u s h, J. Amer. Chem.
Soc., 62, 2759 (1940).
15. В u s w e 1 1, G о r e, J. Phys. Chem., 46, 575 (1942).
16. Tsuboi, Bull. Chem. Soc. Japan, 22, 215 (1949).
17. Bamford, Brown, Elliott, Hanby, Trotter,
Proc. Roy. Soc., 141B, 49 (1953).
18. W i t к о p, P a t r i c k, R о s e n b 1 u m, J. Amer. Chem. Soc.,
73, 2641 (1951).
19. Sheehan, Bose, J. Amer. Chem. Soc., 72, 5158 (1950).
20. Sheehan, Bose, J. Amer. Chem. Soc., 73, 1761 (1951).
21. Sheehan, Hill, В u h 1 e, J. Amer. Chem. Soc., 73, 4373
(1951).
22. Sheehan, Corey, J. Amer. Chem. Soc., 73, 4756 (1951).
23. Sheehan, Laubach, J. Amer. Chem. Soc., 73, 4752 (1951).
24. Sheehan, Ryan, J. Amer. Chem. Soc., 73, 4367 (1951).
25. Thompson, N icholson, Short, Discuss. Faraday Soc.,
9, 222 (1950).
26. R i c h a r d s, В u r t о n, Trans. Faraday Soc., 45, 874 (1949).
27. Lenormant, Discuss. Faraday Soc., 9, 319 (1950).
28. Lenormant, Bull. Soc. chim., 15, 33 (1948).
29. Lenormant, Ann. Chim., 5, 459 (1950).
30. Lenorm ant, Chouteau, J. Physiol., A203, 41 (1949).
31. Chouteau, Lenormant, Compt. rend. Acad. Sci.
(Paris), 232, 1479 (1951).
32. Skinner, Perkins, J. Amer. Chem. Soc., 72, 5569 (1950).
33. Sandeman, Proc. Roy. Soc., A232, 105 (1955).
34. Hughes, Moore, J. Amer. Chem. Soc., 71, 2618 (1949).
35. E 1 1 i о t, Ambrose, Temple, J. Chem. Phys., 16,
877 (1948).
36. S t a i г, С о b 1 e n t z, J. Res. Nat. Bur. Stand., 15, 295 (1935).
37. Wright, J. Biol. Chem., 127, 137 (1939).
38. Bath, Ellis, J. Phys. Chem., 45, 204 (1941).
39. L e n о r m a n t, Compt. rend. Acad. Sci. (Paris), 221, 58 (1945).
40. Darmon, Sutherland, J. Amer. Chem. Soc., 69, 2074
(1947).
Гл. 12. Амиды, белки и полипептиды
331
41. В 1 о u t, L i n s 1 e у, J. Amer. Chem. Soc., 74, 1946 (1952).
42. Edsall, Otvos, Rich, J. Amer. Chem. Soc., 72, 474
(1950).
43. Mizushima, Sh imanouchi, Tsuboi, Sugita,
Kato, К о n d o, J. Amer. Chem. Soc., 73, 1330 (1951).
44. Mizushima, Shimanouchi, Tsuboi, Souda,
J. Amer. Chem. Soc., 74, 270 (1952).
45 U z m a n, В 1 о u t, Nature, 166, 862 (1950).
46. Ambrose, Elliot, Temple, Nature, 163, 859 (1949).
47. Klotz, Griswold, G r u e n, J. Amer. Chem. Soc., 71,
1615 (1949).
48. Sutherland, Tanner, Ohio Symposium on Molecular
Spectroscopy, 1951.
49. Elliot, Ambrose, Discuss. Faraday Soc., 9, 324 (1950).
50. Holliday, Discuss. Faraday Soc., 9, 325 (1950).
51. Ambrose, Hanby, Nature, 163, 483 (1949).
52. Hanby, Waley, Watson, J. Chem. Soc., 1950, 3239.
53. Ambrose, Elliot, Temple, Proc. Roy. Soc., A206, 192
(1951).
54. Ambrose, Elliot, Proc. Roy. Soc., A205, 47 (1951).
55. Darmon, Rudall, Discuss. Faraday Soc., 9, 251 (1950).
56. Glatt, Ellis, J. Chem. Phys., 16, 551 (1948).
57. Glatt, J. Chem. Phys., 15, 880 (1947).
58. Pauling, Corey, Branson, Proc. U. S. Nat. Acad.
Sci., 37, 205 (1951).
59. Fraser, Price, Nature, 170, 490 (1952).
60. Pauling, Corey, Proc. U. S. Nat. Acad. Sci., 37, 235 (1951).
61. Bamford, Brown, Elliot, Hanby, Trotter,
Nature, 169, 357 (1952).
62. Lenormant, Chouteau, Compt. rend. Acad. Sci.
(Paris), 234, 2057 (1952).
63. Mizushima, Shimanouchi, Tsuboi, Sugita,
Kurosaki, Mataga, Souda, J. Amer. Chem. Soc.,
74, 4639 (1952).
64. Wasserman, Precopio, Liu, J. Amer. Chem. Soc.,
74, 4093 (1952).
65. Haszeldine, Nature, 168, 1028 (1951).
66. Lacher, Olsen, Park, J. Amer. Chem. Soc., 74, 5578
(1952).
67. Yakel, Hughes, J. Amer. Chem. Soc., 74, 6302 (1952).
68. Mizushima, Shimanouchi, Tsuboi, Sugita,
Kurosaki, Matoga, Souda, J. Amer. Chem. Soc.,
75, 1863 (1953).
69. H u r d, В a u e г, К 1 о t z, J. Amer. Chem. Soc., 75, 624 (1953).
70. W h e 1 a n d, Resonance in Organic Chemistry, Wiley, New
York, 1956, p. 410.
71. Smith, Electric Dipole Moments, Butterworths, London, 1955,
p. 241.
72. Bates, Hobbs, J. Amer. Chem. Soc., 73, 2151 (1951).
73. Worsham, Hobbs, J. Amer. Chem. Soc., 76, 206 (1954).
74. Russell, Thompson, Spectrochim. Acta, 8, 138 (1956).
332
Часть III
75. Klemperer, Crony n, Maki, Pimentel, J. Amer.
Chem. Soc., 76, 5846 (1954).
76. С a n о n n, Mikrochem. Acta, 1955, 555.
77. Osh ida, О о s h i к a, M i у a s a к a, J. Phys. Soc. Japan,
10, 849 (1955).
78. Cannon, J. Chem. Phys., 24, 491 (1956).
79. Kessler, Sutherland, J. Chem. Phys., 21, 570 (1953).
80. Miyazaw a, Shimanouchi, Mizushima, J. Chem.
Phys., 24, 408 11956).
81. M i у a z a w a, J. Chem. Soc. Japan, 77, 321, 619 (1956).
82. Badger, Rubakava, Proc. Nat. Acad. Sci. U. S., 40,
12 (1954).
83. Клеверли, цитируется no книге «Применение спектро-
скопии в химии», Издатинлит, 1959, стр. 413—414.
84. Mizushima, Tsuboi, Shimanouchi, Tsud а,
Spectrochim. Acta, 7, 100 (1955).
Mecke, Mecke, Chem. Ber., 89, 343 (1956).
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
Мидзусима С., Строение молекул и внутреннее вращение,
М., 1957, стр. 143.
Mizushima, Shimanouchi, Nagakura, К u-
ratani, Tsuboi, Baba, Fujioka, J. Amer. Chem.
Soc., 72, 3490 (1950).
Letaw, Gropp, J. Chem. Phys., 21, 1621 (1953).
Evans, J. Chem. Phys., 22, 1228 (1954).
Davies, Evans, Lumley Jones, Trans. Faraday Soc.,
51, 761 (1955).
Джонс, Клеверли, см. Применение спектроскопии
в химии, Издатинлит, М., 1959, стр. 419—420.
G i е г е г, Z. Naturforsch., 8В, 644, 654 (1953).
Bellamy, Williams, J. Chem. Soc., 1957, 4294.
Mizushima, Shimanouchi, Ichishima,
Miyazawa, Nakaga wa, Araki, J. Amer. Chem.
Soc., 78, 2038 (1956).
Robson, Reinhart, J. Amer. Chem. Soc., 77, 498 (1955).
White, J. Amer. Chem. Soc., 77, 6008 (1955).
Edwards, Singh, Can. J. Chem., 32, 683 (1954).
Ramirez, Paul, J. Amer. Chem. Soc., 77, 1035 (1955).
Шейнкер Ю. H-, Резников В. M., ДАН СССР,
102, 109 (1955).
Шейнкер Ю. Н., Померанцев Ю. И., ЖФХ, 30,
79 (1956).
Gibson, Kynaston, Lindsey, J. Chem. Soc., 1955,
4340.
Brown, Short, J. Chem. Soc., 1953, 168.
Tanner, Spectrochim. Acta, 8, 9 (1956).
В r fl g e 1, частное сообщение.
S c h i e d t, Angew. Chem., 66, 609 (1954).
Kotera, Shibata, Sorve, J. Amer. Chem. Soc., 77,
6187 (1955).
Brockman n, M u s s o, Chem. Ber., 89, 241 (1956).
Frazer, Price, Proc. Roy. Soc., 141B, 66 (1953).
Гл. 12. Амиды, белки и полипептиды
333
109. Abbott, Ambrose, Proc. Roy. Soc., 234A, 247 (1956).
110. Mecke, Mecke, Luttringhaus, Zeit. Naturforsch.,
10B, 367 (1955).
111. Becher, Griffel, Naturwiss., 43, 467 (1956).
112. Becher, Chem. Ber., 89, 1593 (1956).
113. Doty, Holtzer, Bradbury, Blout, J. Amer. Chem.
Soc., 76, 4493 (1954).
114. H u i s g e n, Walz, Chem. Ber., 89, 2616 (1956).
115. Gompper, Herlinger, Chem. Ber., 89, 2825 (1956).
116. Dijkstra, De Jonge, Rec. trav. chim., 75, 1173 (1956).
117. Boivin, Boivin, Can. J. Chem., 32, 561 (1954).
118. Boivin, Bridges, Boivin, Can. J. Chem., 32 , 242
(1954).
119. Gargon, Boivin, Paquin, Can.J.Chem.,31, 1025 (1953).
120. Gargon, Boivin, McDonald, Yaffe, Can. J.
Chem., 32, 823 (1954).
121. Sutherland, Advances in Protein Chemistry, 7, 291 (1952).
122. Elliott, J. App. Chem., 8, 341 (1956).
123. Ellenbogen, J. Amer. Chem. Soc., 78, 363, 366, 369 (1956).
124. Berger, Kurtz, Katchalski, J. Amer. Chem. Soc.,
76, 5552 (1954).
125. Elliott, Malcolm, Trans. Faraday Soc., 52, 528 (1956).
126. E 1 1 i о t t, Proc. Roy. Soc., A221, 104 (1954).
127. Asai, Tsuboi, Shimanouchi, Mizushima,
J. Phys. Chem., 59, 322 (1954).
128. Lenormant, Blout, Nature, 172, 770 (1953).
129. Ehrlich, Sutherland, Nature, 172, 671 (1953).
130. Ehrlich, Sutherland, J. Amer. Chem. Soc., 76, 5268
(1954).
131. Mizushima, Tsuboi, Shimanouchi, Sugita,
Yoshimoto, J. Amer. Chem. Soc., 76, 2479 (1954).
132. Mizushima, Tsuboi, Shimanouchi, Asai,
J. Amer. Chem. Soc., 76, 6003 (1954).
133. Михайлов H. В., Шигорин Д. H., Макарьева
С. П., ДАН СССР, 87, 1009 (1952).
134. Шигорин Д. Н., Михайлов Н. В., Макарьева
С. П., ДАН СССР, 94, 711 (1954).
135. Шигорин Д. Н., Михайлов Н. В., Клюева О. А.,
ЖФХ, 30, 1591 (1956).
136. Blout, A s a d о и г i а п, J. Amer.Chem. Soc., 78, 955 (1955).
137. Blout, I d e 1 s о n, J. Amer. Chem. Soc., 78, 497 (1956).
138. К r i m m, J. Chem. Phys., 23, 1371 (1955).
139. Lenormant, Blout, Bull. Soc. Chim. Fr., 1954, 859.
140. Lenormant, Trans. Faraday Soc., 52, 549 (1956).
141. Hecht, Wood, Proc. Roy. Soc., A235, 174 (1956).
142. Frazer, J. Chem. Phys., 24, 89 (1956).
143. Cannon, Spectrochim. Acta, в печати.
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Чибисов А, к., Пентин Ю. А. ЖОХ, 31, 11, 359 (1961).
Глава 13
АМИНОКИСЛОТЫ, ИХ ХЛОРИСТОВОДОРОДНЫЕ
И ДРУГИЕ СОЛИ И АМИДОКИСЛОТЫ
1. ВВЕДЕНИЕ
При исследованиях спектров комбинационного рас-
сеяния аминокислот, проведенных Эдселлом [1, 2], полу-
чены первые оптические доказательства биполярной струк-
туры этих веществ, и в результате более поздних работ
[3—5] эти данные полностью подтверждены. Первые иссле-
дования инфракрасных спектров проведены Фрейманном,
Фрейманн и Румпфом [6], изучавшими область обертонов
валентных колебаний NH. Эти авторы доказали также на-
личие в нейтральных аминокислотах четвертичного атома
азота. Дальнейшие сведения об основных частотах были
получены рядом других авторов, главным образом при
исследовании области 3000 см1 [7—10]. Среди первых
исследователей в этой области можно назвать Райта [11,
12], который впервые установил, что спектр рацемата цис-
тина в твердом состоянии отличается от спектра любого
чистого оптического изомера. Этот факт полностью под-
твержден более поздними исследователями на других ами-
нокислотах [13, 14]; так же дело, по-видимому, обстоит
и в случае мезовинной кислоты [15]. Кегелом и др. [32]
изучено около 50 пар d- и Z-изомеров аминокислот. Как
и в случае полипептидов, исследованных Элленбогеном [33],
индивидуальные d- и /-изомеры имеют идентичные спек-
тры, которые, однако, могут заметно отличаться от спек-
тра смешанной формы. Но когда имеются два асимметрич-
ных атома углерода, как в обычной или в алло-формах, то
становятся возможными различия в спектрах оптических
изомеров. Аналогичные результаты получены Брокман-
ном и Муссо [34] и Эрхартом и Хеем [35].
В связи с изучением продуктов гидролиза белков, а
также в связи с работами, проведенными для решения проб-
лемы синтеза пенициллина, интерес к аминокислотам в пос-
Гл. 13. Аминокислоты и их хлористоводородные соли 335
леднее время значительно возрос. Несколькими группами
исследователей [17—23, 33, 34] описано теперь большое
количество спектров аминокислот и установлен ряд кор-
реляций, при помощи которых аминокислоты и их гидро-
хлориды могут быть идентифицированы. Эти корреляции
следующие:
а) Область частот валентных колебаний NH. В обычной
для таких колебаний области 3500—3300 см'1 никаких по-
лос поглощения нет, а вместо этого появляется полоса
поглощения вблизи 3070оГ1, обусловленная группой NH,.
Эта корреляция остается, по-видимому, справедливой так-
же для гидрохлоридов, но нарушается в случае N-заме-
щенных аминокислот, например саркозина или пролина,
которые содержат только группу NHJ. При образовании
соли вновь появляется нейтральная группа NH2 и обнару-
живаются обычные полосы валентных колебаний NH, если
только не образуются внутрикомплексные (хелатные) свя-
зи, как это имеет место в случае глицинатов металлов.
б) У аминокислот всех типов и их солей имеется погло-
щение, соответствующее ионизованной карбонильной груп-
пе. У соответствующих гидрохлоридов эта полоса исчезает
и заменяется типичной полосой карбонильного поглоще-
ния. У гидрохлоридов а-аминокислот полоса карбониль-
ного поглощения под влиянием группы NH, смещается
примерно на 20 см'1 в сторону более высоких частот.
в) У большинства аминокислот есть две характеристиче-
ские полосы в области 3000—2000 см1, тогда как у их гидро-
хлоридов имеются почти непрерывные ряды полос между
3030 и 2500 см'1.
г) Кроме полосы поглощения ионизованной карбоксиль-
ной группы, в области 1600—1500 см'1 имеются еще две
полосы, из которых по крайней мере одна должна быть
связана с группой NH*. Как и в случае соответствующих
валентных колебаний, у аминокислот с группой NH+ эти
полосы отсутствуют. Указанные две полосы имеются также
у гидрохлоридов аминокислот, содержащих группу NH+.
д) Наконец, многие аминокислоты поглощают вблизи
1300 см'1, а многие гидрохлориды — вблизи 2000 см'1,
но происхождение этих полос поглощения неизвестно.
Эти корреляции являются надежной основой для иден-
тификации аминокислот как класса соединений. При идеи-
336
Часть III
тификации их следует учитывать также многие другие дан-
ные, особенно данные об изменениях частот колебаний
NH, С/ и С=О в гидрохлоридах и солях. В этом отно-
ХГ
шении ценными являются также исследования растворов
в воде и в тяжелой воде [24, 30]. Для специальных це-
лей можно, кроме того, использовать в качестве раствори-
теля треххлористую сурьму [36].
Присутствие серы или других функциональных групп,
по-видимому, не нарушает этих корреляций [13, 18, 32].
Для дикарбоновых моноаминокислот обнаружена, как
и следовало ожидать, кроме полос поглощения, обуслов-
ленных ионизованным карбоксилом и NH* [13], полоса
карбонильного поглощения, а у орнитина [22] и у других
диаминокарбоновых кислот, кроме поглощения, типичного
для аминокислот, имеются полосы поглощения, обуслов-
ленные свободной группой NH2; однако такие сложные
соединения еще недостаточно изучены. Эти соединения,по-
видимому, независимо от числа атомов углерода, разде-
ляющих группы NH2 и СООН, существуют в виде биполяр-
ных ионов. Рендалл и др. [17] считают е-аминокапроновую
кислоту типичной в этом отношении аминокислотой, а Гей-
манн и Гюнтард сделали аналогичные выводы относительно
других аминокислот с далеко отстоящими функциональны-
ми группами [29]. Ленорман [13] показал также наличие
полосы поглощения NH* у НН2(СН2)иСООН и родствен-
ных соединений. Аналогичные результаты получены Дес-
пасом и др. [37].
В случае ароматических кислот наличие полос погло-
щения ароматического цикла в интервале 1600—1500 см~1
значительно усложняет спектр, однако р-фенилаланин,
тирозин и подобные им вещества имеют почти обычные
для аминокислот полосы поглощения [13, 17].
С другой стороны, для антраниловой кислоты обычные
карбоксильная и аминная полосы поглощения несколько
видоизменены вследствие наличия внутренней водородной
связи, и эта кислота ведет себя не как типичная ами-
нокислота. Отсутствие в этом случае биполярной фор-
мы, возможно, объясняется образованием водородной
связи.
Гл 13. Аминокислоты и их хлористоводородные соли 337
Замещение водорода при атоме азота первичных амино-
кислот алкильными радикалами приводит к образованию
таких соединений, как саркозин, которые содержат не бо-
лее одной группы NH+. У этих соединений по-прежнему
имеется полоса поглощения, обусловленная ионизованным
карбоксилом, но для колебаний NHJ адекватные корре-
ляции не установлены, потому что число доступных соеди-
нений такого рода ограниченно.
N-Ацилзамещение приводит к образованию соединений,
которые сочетают свойства, характерные для аминокислот
и амидов. Их спектры имеют ряд отличительных черт, при-
чем некоторые из них характерны для аминокислот, так
что эти соединения удобнее рассматривать здесь, а не
вместе с амидами. Однако, как и в случае амидов, поло-
жение используемых для анализа полос поглощения этих
соединений при изменении состояния вещества может менять-
ся; поэтому важно, чтобы сравнения проводились только
между веществами, исследованными в одинаковых состоя-
ниях. Различия в спектрах сложных эфиров в твердом
и жидком состоянии использованы Рендаллом и др. [17]
для их идентификации. Эти соединения не существуют в ви-
де биполярных ионов и имеют поэтому полосы поглоще-
ния NH и СО, а также полосы поглощения амид-1 и амид-П.
Подобно аминокислотам они поглощают также в интервале
3000—2000 см'1. Дипептиды и сходные с ними вещества,
также содержащие группу NH+, были рассмотрены отдель-
но в гл. 12.
Обсуждаемые корреляции приведены в табл. 12. Во
всех случаях указанные интервалы частот поглощения отно-
сятся к веществам в твердом состоянии, в каком они обы-
чно исследуются.
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ ГРУППЫ NH+
В обычной области валентных колебаний группы NH,
а именно в интервале 3500—3300 см'1, не поглощают ни
аминокислоты, ни гидрохлориды [10—12, 17—19]. Вместо
этого появляется лишь одна полоса в интервале 3130—
3030 см \ Фрейманн и Фрейманн [10], а также Томпсон
и др. [19] сообщают о наблюдавшейся у ряда аминокислот
полосе поглощения вблизи 3070 см'1, которую они отнесли
22 Л. Беллами
Таблица 12
Частота, см~'-
Колебания NHJ
(NH2 аминокислот и их гидрохлори-
дов. Не относится к солям)
Частоты валентных колебаний NH3
Частоты деформационных колеба-
ний МЩ
аминокислотная полоса I . . . .
аминокислотная полоса II . . . .
Колебания NH
Амидокислоты.....................
Частоты колебаний СООН
Ионный карбоксил (все типы амино-
кислот и солей, за исключением
гидрохлоридов) .................
Обычный кислотный карбонил (неио-
низованный). Гидрохлориды
а-аминокислоты..................
а-амидокислоты.................
Р-, у- и низшие аминокислоты
Область 3000—2000 см~ 1
Аминокислоты.....................
»
Гидрохлориды аминокислот
3130—3030 (ср.)
1660—1610 (сл.) (у гидро-
хлоридов уменьшается до
1590)
1550—1485 (ср.)
3390—3260 (ср.) (у сложных
эфиров частота меньше) л
1600—1560 (с.)
1754—1720 (с.)
1724—1695 (с.)
1730—1700 (с.)
2760—2530 (сл.) 1 Имеются
2140—2080 (сл.) J в боль-
шинстве
случаев,
но не
всегда
Ряды почти непрерывных
полос поглощения,
3030—2500 (сл.)
Гл. 13. Аминокислоты и их хлористоводородные соли 339
Продолжение табл. 12
Частота, см-1
Амидокислоты 2640—2360 (сл.) 4 Имеются 1945—1835(сл.) J в боль- шинстве случаев, но не
всегда.
Другие корреляции (аминокислоты)
Большинство гидрохлоридов амино-
кислот . ........................
Большинство аминокислот и их про-
изводных ........................
Другие корреляции (амидокислоты)
Полоса поглощения амид-1 (СО)
а-кислоты .....................
другие кислоты ................
Полоса амид-П ...................
Вблизи 2000 (сл.)
Вблизи 1300 (ср.)
1620—1600
1650—1620 (с.)
1570—1500 (с.)
к валентным колебаниям группы NH*; Рендалл и др. [17]
указанную корреляцию не обсуждали, но Фасон и др.
[ 18] исследовали те же соединения вновь и нашли, что все
13 исследовавшихся Рендаллом аминокислот поглощают
в интервале 3130—3030 сл'1. Они [18] привели данные еще
о пяти аминокислотах, поглощающих в этом же интервале
частот. Накоцец, Кегел и др. [32] также обнаружили эту
полосу поглощения у многих исследованных ими амино-
кислот различных типов. Эта полоса почти наверное обу-
словлена антисимметричными валентными колебаниями
NH*, тогда как полоса, соответствующая симметричным
колебаниям, находится среди полос в интервале 3000—
2000 см-1.
Относительно полос гидрохлоридов сведений имеется
немного, но можно ожидать, что они дают аналогичные
22*
340
Часть III
полосы поглощения. В той же области спектра поглощают
гидрохлориды простых аминов: бутиламина [37] и метил-
амина [38]. Томпсон и др. [19] обнаружили полосу погло-
щения 3100 см'1 у гидрохлорида глицинового эфира, в то
время как фенилглициновая соль натрия с незаряженной
группой NH2 дает обычную аминную полосу поглощения
вблизи 3370 см'1. Гор и др. [24] подтвердили наличие поло-
сы поглощения вблизи 3000 см'1 у растворов глицина в тя-
желой воде, содержащей DC1. С другой стороны, при рас-
смотрении спектров гидрохлоридов аминокислот, опубли-
кованных Рендаллом и др. [17], можно сделать заключе-
ние, что семь соединений, содержащих группу NH*, и три
соединения, содержащих группу NH*, не поглощают в этой
области, в то время как пять других соединений с группой
NH* поглощают в пределах 3145—3049 см1. Это свидетель-
ствует о необходимости дальнейшей работы в этом напра-
влении, так как если бы было подтверждено, что гидро-
хлориды аминокислот не поглощают в этой области, то бы-
ло бы обоснованным сомнение в правильности отнесения
этого поглощения к группе NH*. Однако поскольку все
исследованные до настоящего времени нейтральные ами-
нокислоты определенно поглощают в этой области, данная
корреляция вполне может быть использована для их иден-
тификации. Полосы валентных колебаний NH* наблю-
даются также у координационных соединений, таких как
аммины кобальта. Эти соединения были изучены рядом
исследователей [39—45]. Однако в этих случаях заряд,
сосредоточенный на атоме азота, существенно меньше и
частоты антисимметричных и симметричных колебаний
равны соответственно примерно 3300 и 3150 см'1.
Эту корреляцию следует, конечно, применять только
для аминокислот, которые могут содержать группу NH*.
N-Замещенные аминокислоты, такие, как N-фенилглицин,
саркосин или пролин, имеют только группу NH*, которая
поглощает, по-видимому, при меньших частотах. Ограни-
ченное число изученных соединений такого рода не позво-
ляет установить какой-либо корреляции для последней
структуры, но результаты исследований гидрохлоридов
вторичных аминов [38, 46] показывают, что поглощение,
соответствующее валентным колебаниям NH*, можно ожи-
дать где-то вблизи 2700 см'1. В случае гидрохлоридов
Гл. 13. Аминокислоты и их хлористоводородные соли 341
нескольких третичных аминов, исследованных в твердом со-
стоянии Лордом и Меррифилдом [31], отмечено поглощение,
соответствующее группе NH+, при столь низкой частоте,
как 2425 см'1.
3. ДЕФОРМАЦИОННЫЕ КОЛЕБАНИЯ NH|
АМИНОКИСЛОТНЫЕ ПОЛОСЫ ПОГЛОЩЕНИЯ I И И
Как указано в табл. 12, у всех аминокислот, которые
могут содержать группу NH*, и их гидрохлоридов, кроме
полосы поглощения ионного карбоксила, имеются по край-
ней мере одна, а вероятно, две характеристические полосы
поглощения в области 1600—1500 см'1 [13, 17—19]. Первая
из них, в интервале 1660—1610 см'1, бывает обычно сла-
бой, а в некоторых случаях выглядит только как выступ
на главной полосе поглощения ионного карбоксила. Вто-
рая характеристическая полоса обычно более интенсивна,
но ее абсолютная интенсивность, по-видимому, изменяется
[17]. Эта полоса находится в интервале 1550—1485 см'1.
Причины возникновения этих полос неясны, но отсутствие
их у солей и N-замещенных аминокислот [13, 17] и нали-
чие у гидрохлоридов указывает на то, что они, вероятно,
связаны с деформационными колебаниями NHg. Ленорман
[13] относит вторую полосу к деформационным колеба-
ниям NH*, но не дает никаких объяснений относительно
первой полосы, тогда как Рендалл и др. [17] относят пер-
вую полосу поглощения к группе NH* и не рассматривают
вторую.
Было выдвинуто предположение, что эти частоты обу-
словлены симметричными и антисимметричными дефор-
мационными колебаниями NH+, причем оба эти колебания
должны быть активны в инфракрасном спектре; такое пред-
положение представляется весьма вероятным. Однако в слу-
чае гидрохлоридов первичных аминов [38], а также в слу-
чае сложных аминов [41, 44, 47—49] полоса, соответствую-
щая колебаниям группы NH* претерпевает значительно
более сильное расщепление, и исследования, проведенные с
дейтерированными соединениями [39], показывают, что
вторая полоса поглощения находится вблизи 1300 см'1.
Аминокислотная полоса поглощения I. Что касается
первой из рассматриваемых полос поглощения, то она
342
Ч а с т ь III
имеется у всех моноаминомонокарбоновых кислот, иссле-
дованных Рендаллом и др. [17], а также еще у пяти
кислот, исследованных Фасоном и др. [18]; в боль-
шинстве случаев она попадает в узкий интервал частот
1640—1610 слг1. Была, однако, указана и более широкая
область поглощения, чтобы охватить также такие соедине-
ния, как сР-а-аминокапроновая кислота [17], поглощаю-
щая при 1658 см'1, и несколько других аминокислот, у ко-
торых Ленорман обнаружил поглощение в интервале 1640—
1600 см'1. Данные Ленормана [13] весьма сходны с дан-
ными Рендалла и др. [17], но он не смог обнаружить эту
полосу во всех без исключения случаях. Аналогично Томп-
сон, Николсон и Шорт [19] указывают на эту полосу только
в некоторых случаях, а Клотц и Груен [20] не упоминают
о ней при рассмотрении полученных ими спектров. Эта
полоса наблюдается лишь в виде исключения в спектрах
только некоторых соединений из 50 аминокислот, иссле-
дованных Кегелом и др. [32], несмотря на то что мно-
гие из этих веществ те же, что и изученные Рендаллом
и Ленорманом. Это может объясняться малой интенсив-
ностью полосы поглощения в некоторых случаях и раз-
ной толщиной применявшихся пленок, но возможно также,
что это связано с различиями исследовавшихся кристалли-
ческих форм. Из данных Ленормана очевидна очень боль-
шая зависимость этой полосы поглощения, как и полосы
поглощения ионной карбоксильной группы, от изменений
оптической формы молекул, и связанные с этим сдвиги
полосы могут оказаться достаточными, чтобы замаски-
ровать более слабое поглощение, обусловленное группой
NH+. Брокманн и Муссо [34] отметили также появление
этой полосы у смешанных изомеров, хотя она не обнаружена
у индивидуальных d- и /-изомеров. Эти различия авторы
связывают с различием в порядке построения водородных
мостиков в кристаллах.
Поэтому хотя представляется чрезвычайно вероятным
наличие поглощения в этой области частот у всех амино-
кислот, у которых может образоваться группа NHJ, тем не
менее нельзя не учитывать, что это поглощение может быть
очень слабым или может маскироваться поглощением ион-
ной карбоксильной группы, если последнему соответствует
высокая частота колебаний.
Гл. 13. Аминокислоты и их хлористоводородные соли
343
Положение полосы поглощения, обусловленной дефор-
мационными колебаниями NHJ для таких кислот, как сар-
косин, нельзя достаточно надежно предсказать на осно-
вании имеющихся данных. (//-Пролин [13] поглощает при
1653 см1, а саркозин [17] при 1645 см~\ ноу N-фен ил глицина
в этой области нет никаких полос поглощения.
У гидрохлоридов, содержащих группу NHJ, обнару-
жено аналогичное поглощение. Соединения такого типа
изучены в значительно меньшем количестве, однако сле-
дует отметить, что данные Рендалла и др. [17], которые
указывают для исследованных ими соединений интервал
частот 1610—1590 с.и-1, в основном согласуются с дан-
ными Ленормана [13].
Обнаружение этого поглощения в данном случае сильно
упрощается ввиду отсутствия в той же области полосы
поглощения ионного карбоксила.
Аминокислотная полоса поглощения II. Относительно
наличия этой полосы, которая обычно значительно интен-
сивнее первой, имеются гораздо более согласованные дан-
ные. Ее присутствие в указанной области отмечено для всех
кислот и гидрохлоридов, содержащих группу NH+, кото-
рые изучались Рендаллом и др. [17], Фасоном и др. [18],
Томпсоном и др. [19], Клотцом и Груеном [20] и другими
исследователями [22, 32, 34]. Эта полоса поглощения от-
сутствует в спектрах солей [13], чем подтверждается отне-
сение ее к деформационным колебаниям NH*. Как указано
выше, интенсивность ее обычно велика, хотя в некоторой
степени она может изменяться. Эта полоса с частотой око-
ло 1515 см~х обнаружена Кегелом и др. [32] в спектрах
почти всех 50 исследованных ими соединений. Однако
она отсутствует в спектре изовалина и расщепляется на
дублет у некоторых других аминокислот, таких как
а,р-диаминопропионовая и аф-диаминомасляная кислоты.
У оксикислот полоса поглощения имеет интересную особен-
ность: она смещается постепенно в область более высоких
частот по мере последовательного приближения группы ОН
к группе NH+. Частота полосы поглощения гексагомосери-
на равна 1504 см~х и увеличивается до 1522 см'1 в случае
пентагомосерина. В спектре самого гомосерина она прояв-
ляется при 1538 см'1 и исчезает в случае серина, вероятно,
вследствие наложения [34] полосы поглощения ООО-
344
Ч а с т ь III
при 1600 см'1. Данная полоса поглощения отсутствует
у аминокислот с группой NH+, таких как саркозин, про-
лин и соединение CH3NH (СН2)юСООН [13], но у гидро-
хлоридов вторичных аминов наблюдается полоса погло-
щения около 1600 см'1, так что она может совпадать с по-
глощением ионизованной карбоксильной группы. Как уже
отмечалось, число исследованных гидрохлоридов гораздо
меньше, но во всех изученных случаях эта полоса имеется.
Исчезновение полосы при образовании соли позволяет
легко убедиться в правильности идентификации, прово-
димой с использованием этой полосы.
4. КОЛЕБАНИЯ СООН
а. Ионизованная карбоксильная группа. Положение
полос поглощения ионизованной карбоксильной группы
хорошо установлено (гл. 10) в результате работ Леконта
[25] и проведенных Эдселлом [1—5] исследований спек-
тров комбинационного рассеяния как солей обычных кис-
лот, так и аминокислот. Две полосы поглощения, соответ-
ствующие антисимметричным и симметричным колебаниям,
находятся около 1550 и 1410 см'1. Идентификация первой
из них в инфракрасных спектрах поглощения обычно прохо-
дит легче, в области же частот второй полосы имеются мно-
гие другие полосы поглощения.
Спектры аминокислот соответствуют указанным дан-
ным: нейтральные кислоты и их соли имеют полосу по-
глощения вблизи 1600 см'1, которая может быть отнесена
к ионизованной карбоксильной группе [13, 17—22, 32,
34, 50].
Все исследователи, изучавшие эту область, соглашаются
с таким отнесением полосы, которое подтверждается полу-
ченными ими данными о спектрах большого числа ами-
нокислот. Томпсон и др. [19] указали для нее интервал
частот 1600—1590 см'1, который, несомненно, является
слишком узким, если учесть результаты, полученные дру-
гими исследователями. Фасон и др. [18], так же как и
Рендалл [17], указывают интервал 1600—1560 см'1, кото-
рый достаточно широк, чтобы охватить полосы поглоще-
ния всех исследованных кислот, за исключением саркози-
на [17], поглощающего при 1616 см'1. Все значения частот
Гл. 13. Аминокислоты и их хлористоводородные соли 345
поглощения, приведенные Ленорманом [13], также попа-
дают в этот интервал, хотя, как уже отмечалось, из извест-
ного спектра NH2(CH2)10COOH (для этого соединения
значения частот карбоксильного поглощения не приве-
дены) следует, что частота колебаний СООН у этого соеди-
нения выше 1650 см'1. Последнее соединение, возможно,
является исключением, поскольку длина его цепи значи-
тельно больше, чем обычная, и может привести к иной
кристаллической упаковке.
Рассматриваемая полоса поглощения исчезает при обра-
зовании четвертичного гидрохлорида, при котором иони-
зации карбоксильной группы не происходит. Это подтвер-
ждено как Рендаллом и др. [17], так и Ленорманом [13].
Может показаться, что в спектрах некоторых гидрохлори-
дов, приведенных Клотцем и Груеном [20], эта полоса
остается, но на самом деле наблюдаемая здесь полоса
поглощения является аминокислотной полосой I, обуслов-
ленной группой NH+, которая авторами не исследовалась.
Эта полоса при отсутствии поглощения ионного карбок-
сила видна более отчетливо. Изменения в спектре, проис-
ходящие при переходе карбоксила из ионной формы в не-
диссоциированную, хорошо продемонстрированы Гором
и др. [24], изучавшими растворы глицина в обычной и тя-
желой воде. У нейтрального раствора наблюдаются полосы
поглощения ионизованной карбоксильной группы (1610
и 1400 см1), которые заменяются полосой карбонильного
поглощения при 1740 см'1 после добавления DC1. При
добавлении NaOD первоначальные полосы поглощения
ионной группы вновь появляются примерно при тех же
частотах (1590 и 1400 см'1).
Ленорман [28] также исследовал растворы некоторых
аминокислот в тяжелой воде и показал, что поглощение
аланина в нейтральном растворе наблюдается при 1618 см'1,
а в щелочном растворе — при 1570 см'1. В отличие
от аланина Р-аланин в обоих случаях поглощает
при 1618 см'1.
Эта корреляция применима к аминокислотам всех ти-
пов, независимо от того, имеет ли аминогруппа структуру
NH+ или NH+. Аналогичный эффект обнаружен у пиридин-
бетаина [26], хотя в этом случае полоса поглощения имеет
часто!у 1652 см1.
34fl
Ч а с m ь III
У некристаллических образцов иногда можно обнару-
живать полосы поглощения как ионизованной, так и неио-
низованной группы [21], что может явиться причиной
ошибок при идентификации. Однако этого обычно не наб-
людается.
Вторую полосу поглощения, обусловленную ионной
карбоксильной группой, труднее различить в инфракрасном
спектре, где ее интенсивность заметно меньше, чем интен-
сивность полосы 1600 слг1, тогда как во многих спектрах
комбинационного рассеяния она отчетливо видна [5].
Хотя многие аминокислоты, несомненно, поглощают в этой
области, трудно доказать, что какая-либо отдельная поло-
са обусловлена указанной группой. Фасон и др. [18] обна-
ружили у ряда ароматических цистеинов полосы поглоще-
ния вблизи 1400 и 1406 слг-1, одну из которых, согласно
их предположению, можно рассматривать как поглощение
ионизованной карбонильной группы; Кегел и др. [32]
также относят к этому поглощению полосу 1408 слг-1, кото-
рая проявляется в спектрах подавляющего большинства
исследованных ими аминокислот. Эрлих и Сазерленд [50]
также относят полосу 1410 слг-1 в спектре N-метиламино-
янтарной кислоты к симметричному колебанию СОО-. Учи-
тывая, однако, малую интенсивность полосы в инфракрас-
ном спектре, при идентификации ее следует быть очень
осторожным.
б. Поглощение неионизованной карбоксильной группы.
Обычная полоса карбонильного поглощения группы СООН
появляется в ряду аминокислот только у гидрохлоридов,
если не считать, правда, дикарбоновых моноаминокислот,
у которых имеются и ионная, и недиссоциированная фор-
мы карбоксила.
Рендалл и др. [17] приводят интервал необычно высо-
ких частот, соответствующих этому поглощению (1754—
1724 слг-1), который частично перекрывается интервалом
частот поглощения обычных сложных эфиров. Однако
Эдселл [5] на основании спектров комбинационного рас-
сеяния установил, что аномально высокие частоты погло-
щения в интервале 1750—1740 слг-1 наблюдаются только
для а-аминокислот, а остальные аминокислоты имеют обыч-
ные полосы поглощения в интервале 1730—1700 слг-1.
Гл. 13. Аминокислоты и их хлористоводородные соли 347
На основании спектральных данных Рендалла и др. [17]
и Ленормана [13] можно сделать вывод, что это верно
также и для инфракрасных спектров поглощения. Подав-
ляющее большинство соединений, исследованных, напри-
мер, Рендаллом и др. [17], является а-аминокислотами,
поглощающими в интервале высоких частот, тогда как
гидрохлорид 6-амино-«-валерьяновой кислоты поглощает
при 1701 см'1.
Повышение у а-аминокислот частоты поглощения при-
мерно на 20 см 1 неудивительно и объясняется, очевидно,
влиянием группировки NH+СГна длину связи С=О сосед-
ней карбонильной группы. Аналогичный эффект дает в слу-
чае кислот и кетонов а-галогензамещение, тогда как в
P-положении галоген достаточно удален от карбонильной
группы и не влияет на частоту ее колебаний.
Эта корреляция должна быть применима и для гидро-
хлоридов кислот, содержащих группу NH+, но, как и в
предыдущих случаях, сведений об этом мало. Гидрохлорид
саркосина опять-таки проявляет несколько отличные свой-
ства, поскольку его полоса карбонильного поглощения
находится при 1757 см'1, тогда как пролин и гидрохлорид
N-фен ил глицина поглощают при 1730 см'1. Гидрохлорид
гистидина поглощает при 1706 см'1, а дигидрохлорид орни-
тина [13] поглощает при 1739 см'1. Таким образом, постоль-
ку поскольку это допускают имеющиеся ограниченные
данные, можно, по-видимому, предполагать, что указан-
ная корреляция справедлива и для этого типа соединений.
5. ОБЛАСТЬ 3000—2000 см'1
Томпсон и др. [19] обратили внимание на наличие в спек-
трах простых аминокислот слабой, но отчетливой полосы
поглощения вблизи 2100 см'1, которая отсутствует в спек-
трах натриевой солифенилглицина и п-аминофенилуксусной
кислоты. Это наводит на мысль о связи данной полосы
с наличием биполярной ионной структуры. Еще раньше
Рендалл и др. [17] заметили в спектрах большинства, хотя
и не всех, аминокислот две полосы поглощения в этой
области. Тем, что гидрохлориды обычно поглощают в этой
области, подтверждается предположение о возможной зави-
348
Ч а с т ь III
симости появления этой полосы поглощения от наличия
группы NH* Указанные полосы поглощения имеют боль-
шое аналитическое значение, так как они расположены
в спектральной области, сравнительно свободной от дру-
гих полос поглощения, но они появляются не во всех
случаях.
Эти исследования были продолжены Фасоном и др.
[18], которые обнаружили по одной или по две полосы
поглощения в интервале 2760—2530 см 1 для 10 соединений
из 13, исследованных Рендаллом и др. [17], и, кроме этого,
еще для 5 исследованных ими соединений. Другая полоса
поглощения найдена ими в интервале 2140—2080 см'1 для
11 соединений из 13, исследованных Рендаллом и др. [17],
и для 3 из 5 соединений, исследованных ими самими. Ке-
гел и др. [32] обнаружили полосу 2128 слГ1 у всех иссле-
дованных ими а-аминокислот, включая многие соединения,
у которых имеется разветвление при углеродном атоме
в a-положении. Они отметили также поглощение при
2564 сл-1 во всех случаях, кроме аминокапроновой кислоты.
Однако, когда в состав молекулы вводятся другие поляр-
ные группы, положение становится более сложным. У орни-
тина, например, полоса 2128 см'1 отсутствует. Она появляе-
тся в этом месте спектра у гексагомосерина и постепенно
смещается в низкочастотную область при уменьшении дли-
ны цепи. У серина она появляется при 2033 см'1. Эти свой-
ства полосы сходны со свойствами поглощения, обуслов-
ленного деформационными колебаниями NH* соединений
этого типа, так что естественно относить эту полосу к ва-
лентным колебаниям NH+. Аномальным при этом является
поглощение аспарагиновой кислоты (2062 и 1905 см'1)
и аминоадипиновой кислоты (1923 см'1). Поэтому хотя
и ясно, что в этой области спектра должно обнаруживаться
характеристическое поглощение, связанное с валентными
колебаниями NH+, в случае каких-либо соединений более
сложных, чем простые аминокислоты, пользоваться этой
корреляцией нужно осторожно.
Гидрохлориды аминокислот наряду с другими типами
соединений имеют почти непрерывный ряд полос погло-
щения средней интенсивности, расположенных между 3030
и 2500 см 1 [17].
Гл. 13. Аминокислоты и их хлористоводородные соли 349
6. ДРУГИЕ КОРРЕЛЯЦИИ (АМИНОКИСЛОТЫ)
Рендалл и др. [17] заметали наличие у гидрохло-
ридов аминокислот, содержащих группу NH+, полос по-
глощения вблизи 2000 с.и’1, которых нет у соединений
с группой NH+, но никакого объяснения природы этих
полос предложено не было и никаких других данных об
этой полосе больше не имеется.
Кроме того, Клотц и Груен [20] отметили всегда при-
сутствующую в спектрах исследованных ими аминокислот
полосу поглощения вблизи 1300 см'1, а также полосу погло-
щения средней интенсивности в интервале 1335—1300 см'1,
которая имеется почти во всех опубликованных спек-
трах аминокислот, охватывающих эту область частот [12,
17—19, 22].
Кегел и др. [32] обнаружили полосы при 1370
и 1316 см'1 у всех простейших аминокислот, исследо-
ванных ими, и интересно отметить, что к симметричному
деформационному колебанию NH+ у сложных аминов была
отнесена полоса примерно такой же частоты [39].
Жозьен и Фасон [27] заметили интенсивную полосу
поглощения в интервале 1232—1215 см'1, имеющуюся
в спектрах аминокислот, гидрохлоридов аминокислот и ди-
карбоновых аминокислот. Наличие этой полосы поглоще-
ния в тех случаях, когда группа СООН не ионизована,
дает основание предположить ее связь с валентными коле-
баниями С — О или с деформационными колебаниями С—ОН
карбоксильной группы, причем следует напомнить, что
и многие карбоновые кислоты поглощают вблизи 1250 см'1
{гл. 10). Это, таким образом, вторая область частот,
при исследовании которой можно получить данные, под-
тверждающие наличие группы СООН. Деспас и др. [37]
также связывают полосы в интервалах 1250—1100 см 1
и 1000—900 см'1 с поглощением NH+, но трудно сказать,
какое именно колебание группы могло бы обусловить это
поглощение, так как маятниковые колебания приводят
к поглощению при более низких частотах.
Другие возможные корреляции относятся к области
880 см'1, в которой находятся полосы поглощения многих
аминокислот. Фасон и др. [18] предполагают, что поглоще-
ние в этой области возможно связано е деформационными
350
Ч а с т ь III
колебаниями ионной карбоксильной группы, но по анало-
гии со сложными аминами более вероятно, что оно отно-
сится к маятниковым колебаниям группы NH+.
7. СОЛИ АМИНОКИСЛОТ
Аминокислоты могут, очевидно, давать основные и кис-
лыесоли. Частоты колебаний, характерные для гидрохлори-
дов, уже рассмотрены выше. Образование соли по карбок-
сильной группе аминокислот с длинными цепочками лик-
видирует биполярный характер молекулы, и аминогруппа
дает при этом обычное поглощение NH2 первичного амина.
Однако в случае а-аминокислот могут возникнуть различ-
ные ситуации, так как со многими металлами аминогруппа
может координироваться, давая на связь неспаренные
электроны. Соединения этого типа имеют большое биоло-
гическое значение и изучены многими исследователями
[51—54]. Соединениями этого типа являются глицинаты
металлов, но их спектры сложны из-за наличия дополни-
тельных полос поглощения кристаллизационной воды. Тем
не менее у них обнаруживаются типичные полосы валент-
ных колебаний NH2 вблизи 3250 и 3130 см1. Эти частоты
выше, чем соответствующие частоты гидрохлоридов, что
согласуется с представлением о донорно-акцепторной свя-
зи с участием свободной пары электронов атома азота.
Полосы поглощения ионизованной карбонильной группы
проявляются при обычных частотах. Это обстоятельство
было интерпретировано как свидетельство чисто ионной
связи между атомом кислорода и атомом металла, хотя
при этом подразумевалась в какой-то мере эквивалентность
двух атомов кислорода карбоксильной группы, что трудно
согласовать с плоской квадратной конфигурацией, харак-
терной для многих внутрикомплексных соединений, напри-
мер таких, как соединения меди. Розенберг [54] провел
исследование спектров внутрикомплексных соединений гли-
цина и лейцина при низких температурах, при которых по-
лосы становятся резче. У негидратированных соединений
лейцина полосы валентных колебаний NH2 проявляются
около 3350 и 3250 см~1, но поглощение СОО" остается вбли-
зи 1600 см~*. Можно также различать цис- и транс-изомеры
глициновых комплексов платины, используя небольшие раз-
личия в частотах полос поглощения NH2 в этих двух случаях.
Гл. 13. Аминокислоты, и их хлористоводородные соли 351
8. АМИДОКИСЛОТЫ
а. Колебания NH. Фасон и др. [18] проанализировали
наряду со своими данными результаты, полученные Рендал-
лом и др. [17], и показали, что валентным колебаниям NH
у твердых веществ соответствует частота 3390—3260 см'1.
В этом интервале находятся значения частот колебаний
15 а-амидокислот, исследованных Рендаллом и др., и 10
других образцов, исследованных Фасоном и др. При обра-
зовании сложных эфиров, у которых возможность возник-
новения водородных связей меньше, эта частота повышается
и ее значение поэтому приближается к нормальному. Май-
хил и Шлеппингхоф [16] также обнаружили поглощение
при частоте 3350 сл-1 у ряда N-ацетиламинокислот.
б. Область 3000—2000 см'1. Подобно аминокислотам,
амидокислоты во многих случаях имеют в этой области широ-
кие и довольно слабые полосы поглощения. Проведенный
Фасоном и др. [18]анализ опубликованных данных показы-
вает, что полоса поглощения в интервале 2640—2360 см'1
наблюдается для 23 соединений из 26, а другая полоса в
интервале 1945—1835 слг-1 наблюдается для 16 из 26 соеди-
нений. Эти полосы являются иногда сложными и, вероятно,
составными; они наблюдаются не всегда, что уменьшает их
значение. При интерпретации спектров неизвестных веществ
надо помнить, что в этой области, кроме аминокислот и их
гидрохлоридов, могут поглощать также и амидокислоты.
Майхил и др. [16] также рассмотрели вопрос о наличии
в спектре всех исследованных ими соединений полосы
поглощения в интервале 2600—2500 см'1. Это, вероятно,
характеристическое поглощение карбоновых кислот, соот-
ветствующее поглощению вблизи 2650 см'1, которое наб-
людается у димеров карбоновых кислот (гл. 10).
в. Поглощение, обусловленное карбоксильной груп-
пой. Для амидокислот в твердом состоянии обнаружены
обычные карбоксильные полосы поглощения [17, 18] в ин-
тервале частот 1724—1695 см'1. Исключение составляют
гиппуровая кислота, поглощающая при 1740 см'1, и
N-хлорацетил-Ш-серин, который поглощает при 1730 см'1.
Рендалл и др. 117] предполагают, что значение частоты
352
Ч а с т ь III
этой полосы поглощения больше 1695 см'1 для а-кислот
и меньше для других кислот, но число исследованных ими
амидокислот, не являющихся а-кислотами, невелико. Неко-
торые из таких кислот поглощают при 1695 см'1, а другие,
например е-бензамидокапроновая кислота, поглощают при
1698 см'1. Сомнительно поэтому, можно ли проводить раз-
личие между полосами поглощения в данной области.
Растворение веществ приводит к сдвигу полосы в сторону
более высоких частот на 30—60 см'1 [18] в случае разбав-
ленных растворов.
г. Полосы поглощения амид-1 и амид II. Рендалл и др.
[17] считают, что для а-амидокислот характерно расположе-
ние полосы поглощения амид-1 при 1620 см'1 или при более
низкой частоте, тогда как у других кислот эта полоса
появляется при более высоких частотах. В общем эта
точка зрения хорошо согласуется с полученными ими
результатами (интервал частот 1620—1600 см'1 в 12 слу-
чаях из 15), а также с данными Фасона и др. [18] и Май-
хила и Шлеппингхофа [16]. Однако эта корреляция не
всегда оправдывается. Фасон и др. обнаружили, что
dZ-ацетилцистин поглощает при 1587 см'1, а dZ-фенацетил-Р-
фенилаланин [17] — при 1665 см'1. В растворе эта полоса
поглощения опять-таки заметно смещается в сторону более
высоких частот (1700—1680 см'1).
Полоса поглощения амид'-П находится в обычном интер-
вале 1570—1500 см'1 для 25 соединений, изучавшихся дву-
мя основными группами исследователей [17, 18]. Смеще-
ния полосы при растворении вещества заметно меньше,
чем в первом случае, и имеют противоположное направле-
ние. Найденный интервал частот составляет примерно 1530—
1510 см'1, и данные Фасона и др. [18] находятся в согласии
с этими значениями. Майхил и др. [16] не рассматривают
вопрос об этой полосе поглощения в спектрах исследован-
ных ими соединений.
д. Другие корреляции. Наличие у многих амидокислот,
а также у гидрохлоридов аминокислот полосы поглощения,
соответствующей частоте 1225 см'1 или близкой к этому
значению, было предложено считать еще одним возмож-
ным критерием при идентификации данных соединений
Гл. 13. Аминокислоты и их хлористоводородные соли 353
[18, 271. Это, по-видимому, полоса поглощения группы
СООН, обусловленная валентными колебаниями С — О или
деформационными колебаниями ОН, и ее значение для це-
лей идентификации соединений обсуждается в разделе,
посвященном карбоновым кислотам.
ЛИТЕРАТУРА
1. Е
2. Е
3. Е
4. Е
6. F
d s а 1 1, J. Chem. Phys., 4, 1 (1936).
d s а 1 1, J. Chem. Phys., 5, 225, 508 (1937).
d s a 1 1, Scheinberg, J. Chem. Phys., 8, 520 (1940).
d s a 1 1, J. Amer. Chem. Soc., 65, 1767 (1943).
d s a 1 1, О tv о s, Rich, J. Amer. Chem. Soc., 72, 474
(1950).
reymann, Freymann, Rumpf, J. Phys. Radium,
7, 30 (1936).
7. Heintz, Compt. rend. Acad.
8. Lenormant, Compt. rend
(1946).
9. Duval, Lecomte, Bull.
Sci. (Paris.), 201, 1478 (1935).
Acad. Sci. (Paris), 222, 1432
Soc. Chim. France, 10, 187
(1943).
10. Freymann, Freymann, Proc. Indian Acad. Sci.,
8, 301 (1938).
11. Wright, J. Biol. Chem., 120, 641 (1937).
12. Wright, J. Biol. Chem., 127, 137 (1939).
13. L e n о r m a n t, J. Chem. Phys., 43, 327 (1946).
14. Sutherland, Discuss. Faraday Soc., 9, 319 (1950).
15. Lecomte, неопубликованная работа в [13].
16. Micheel, Schleppinghoff, Chem. Ber., 88, 763
(1955).
17. Randall, Fowler, Fuson, Dangl, Infra-red De-
termination of Organic Structures, Van Nostrand, 1949.
18. F u s о n, J о s i e n, Powell, J. Amer. Chem. Soc., 74,
1 (1952).
19. Thompson, Nicholson, Short, Discuss, Faraday
Soc., 9, 222 (1950).
20. К 1 о t z, G r u e n, J. Phys. Colloid. [Chem., 52, 961 (1948).
21. The Chemistry of Penicillin, Princeton University Press, 1949,
p. 407.
22. Larsson, Acta Chem. Scand., 4, 27 (1950).
23. К u r a t a n i, J. Chem. Soc. Japan, 70, 453 (1949).
24. Gore, Barnes, Petersen, Analyt. Chem., 21, 382
(1949).
25. D u v a 1, Lee omte, Douville, Annals de Physique,
17, 5 (1942).
26. R a s m u s s e n, В r a t t a i n, J. Amer. Chem. Soc., 71,
1073 (1949).
27, J о s i e n, Fuson, Compt. rend. Acad. Sci. (Paris.), 232
2016 (1951).
23 Л. Беллами
354
Часть Hi
28. Lenorman
(1952).
29. G a u m a n n,,
'30. Lenorman
M.e r
G r e
1 1 u m,
о g e n,
t, Compt. rend. Acad. Sci. (Paris), 234, 1959
33. E
Giinthard, Helv. chim. Acta, 35, 53 (1952).
t, J. Chim. Phys,, 49, 635 (1952).
r i f i e 1 d, J. Chem. Phys., 21, 166 (1953).
e n s t e i n, W 1 n i. t z,
J. Amer. Chem. Soc., 77, 5708 (1955).
J. Amer. Chem. Soc., 78, 363, 366, 369
Birnbaum,
34. В
35. E
36. L
37. D
38. В
,31. Lord,
32. Ko e g e 1,
M с C a
1 1 e n b
(1956).
г о с к m
r h a r t,
a c h e r,
58, 206
e s p a s ,
1953, 1105.
ellanato, Barcelo,
M u s s o, Chem. Ber., 89, 241 (1956).
Chem. Ber., '
К i a n p
a n n,
Hey,
Croy,
(1954).
К h a 1 a d j i, V
о
e
89, 2124 (1956).
u r, P
ark, J. Phys. Chem.,
r g о z,
Anales.
Bull. Soc. Chim, Fr.,
Fiz. Quim., 52B, 469
(1956).
39. Powell, Scheppard, J. Chem. Soc., 1956, 3108.
40. Chat t, Duncanson, Venanzi, J. Chem. Soc.,
1956, 2712.
41. Hill, Rosenberg, J. Chem. Phys., 24, 1219 (1956).
42. Beattie, Tyrrell, J. Chem. Soc., 1956, 2849.
43. Caglioti, Silvestrom, Sartori, Scrocco,
Ricerca. Sci., 26, 1743 (1956).
44. P e n t 1 a n d, Lane, Quagliano, J. Amer. Chem.
Soc., 78, 887 (1956).
45. Fujita, Nakamoto, Kobayashi, J. Amer. Chem.
Soc., 78, 3295 (1956).
46. H e а с о c k, Marion, Can. J. Chem., 34, 1782 (1956).
47. Merritt, Wiberley, J. Phys. Chem., 59, 55 (1955).
48. Barrow, Kreuger, Basolo, J. Inorg. Nuclear
Chem., 2, 340 (1956).
49. Mizushima, N a к a g a w a, Quagliano, J. Chem.
Phys., 25, 1367 (1956).
50. Ehrlich, Sutherland, J. Amer. Chem. Soc., 76,
5268 (1954).
51. Sweeney, Curran, Quagliano, J. Amer. Chem.
Soc., 77, 5508 (1955).
52. Sen, Mizushima, Curran, Quagliano, J. Amer.
Chem. Soc., 77, 211 (1955).
53. Svatos, Curran, Quagliano,
Soc., 77, 6159 (1955).
54. Rosenberg, Acta Chem. Scand., 10, 840 (1956).
J. Amer. Chem
Глава 14
АМИНЫ И ИМИНЫ
1. ВВЕДЕНИЕ
Первичные амины можно идентифицировать по наличию
в области частот валентных колебаний NH двух полос
поглощения, обусловленных симметричными и антисимме-
тричными колебаниями водородных атомов. В некоторых
случаях в этой области наблюдается третья полоса погло-
щения, обусловленная образованием водородной связи.
-Последняя всегда приводит к смещению полос поглощения
в направлении более низких частот, но в данном случае
связи значительно слабее, чем в случае групп ОН, и по-
этому полосы поглощения резче, а их смещение в указан-
ном направлении меньше. К сожалению, в этой области
•имеется наложение колебаний ОН и NH, в связи с чем
дифференциация их не всегда возможна. Изменения частот
валентных колебаний NH наблюдаются также у аминов
и гидрохлоридов, у которых аминогруппа несет поло-
жительный заряд (NHg). Эти случаи рассмотрены
отдельно.
У первичных аминов имеется также полоса' поглощения
вблизи 1650 см1, обусловленная деформационными коле-
баниями NH2, и подобно частоте валентных колебаний
частота, соответствующая этому поглощению, изменяется
под влиянием водородной связи. Удобным средством рас-
познавания этих полос поглощения является метод дейте-
рирования. Кроме указанных полос поглощения, у арома-
тических аминов часто можно идентифицировать характе-
ристическую полосу поглощения, обусловленную, по-ви-
димому, валентными колебаниями С — N. Имеется также
полоса поглощения в низкочастотной области спектра,
соответствующая внешним деформационным колебаниям
-группы NH2, но положение этой полосы в зависимости
23*
356
Часть III
от строения прилежащих к группе частей молекулы опре-
деляется неоднозначно.
У вторичных аминов имеется только одна полоса погло-
щения в интервале 3500—3200 см'1, обусловленная валент-
ными колебаниями NH, положение которой также несколь-
ко меняется под влиянием водородной связи; имины погло-
щают практически в той же области. Полосы деформацион-
ных колебаний NH у этих соединений слишком слабы, что-
бы их можно было использовать для целей идентификации
соединений, хотя в ограниченном числе случаев у соответ-
ствующих полос наблюдается повышенная интенсивность
поглощения. Так, например, деформационными колебания-
ми NH, возможно, обусловлена полоса поглощения амид-П,
наблюдающаяся у всех вторичных амидов, что, однако, еще
не доказано (гл. 12). В то время как в случае первичных
ароматических аминов связи С — N могут быть идентифици-
рованы, у алифатических соединений соответствующие
полосы поглощения слабее и положение их менее
постоянно.
Третичные амины чрезвычайно трудно идентифициро-
вать спектроскопическим способом. В некоторых случаях у
ароматических соединений можно идентифицировать полосу
валентных колебаний С — N, но для алифатических соеди-
нений удовлетворительной корреляции нет. Обсуждаются
также возможности идентификации группы СН3 — N.
Указанные корреляции представлены в табл. 13.
Таблица 13
Частота, си-1
Полосы поглощения валентных
колебаний NH
Первичные амины. Две полосы. Под
влиянием водородной связи или
в твердом состоянии частота умень-
шается ..........................
Вторичные амины. Одна полоса
Имины. Одна полоса ..............
3500—3300 (ср.)
3500—3300 »
3400—3300 »
Гл. 14. Амины и имины
357
Продолжение табл. 13
Частота, см-1
Частсты деформационных колебаний NH Первичные амины 1650—1590 (с.—ср.)
Вторичные амины 1650—1550 (оч. сл.)
Колебания С—N Ароматические амины первичные 1340—1250 (с.)
вторичные 1350—1280 »
третичные 1360—1310 »
Алифатические амины 1220—1020 (ср, —сл.) Вблизи 1410 (сл.)
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ NH
а. Общие сведения. У первичных аминов полосу погло-
щения вблизи 3500 см 1 впервые заметил Кобленц [11, а тот
факт, что эта полоса поглощения появляется почти всегда
и связана с валентными колебаниями NH, установлен Бел-
лом [2—5] и Эллисом [6—7], которые исследовали боль-
шое число соединений и показали, что такое поглощение
характерно для всех первичных и вторичных аминов, но не
наблюдается для третичных соединений. Разрешающая
способность приборов, имевшихся в распоряжении этих
исследователей, была, однако, недостаточна для определе-
ния точных значений характеристических частот погло-
щения различных соединений. Позднее было опубликовано
много работ по изучению отдельных аминов, а в некоторых
случаях также эффектов образования водородной связи.
Однако детальных исследований групп соединений с це-
лью установления корреляций между точным значением
частоты поглощения и структурой молекулы в целом не
проводилось, за исключением амидов и аминокислот. Более
того, возможность использования для этой цели разроз-
ненных литературных данных ограничена тем обстоятель-
ством, что только очень небольшая часть их получена с при-
менением призм из фторидов лития или кальция, а разре-
358
Часть HI
шение, даваемое в этой области каменной солью, недостаточ-
но, чтобы обнаружить малые различия частот поглощения,
которые, очевидно, имеются у различных классов аминов.
б. Первичные амины. У первичных аминов в разбавлен-
ных растворах в неполярном растворителе имеются две
полосы поглощения в интервале 3500—3300 см'1. Первая
из них, обусловленная антисимметричными валентными
колебаниями, обычно находится вблизи 3500 см'1, а вторая,
возникающая вследствие соответствующих симметричных
колебаний, находится вблизи 3400 см'1. Положение обеих
полос несколько меняется при изменении полярности рас-
творителя, а в концентрированных растворах, в которых
может происходить ассоциация молекул, эти изменения
проявляются сильнее. Образование внутримолекулярных
связей также приводит к уменьшению частот этих полос,
что будет показано ниже при обсуждении эффектов водо-
родной связи.
Эта корреляция частот сделана главным образом на
основании данных о таких простых соединениях, как метил-
амин [8—9, 461 и анилин [10—12, 47—48], которые очень
хорошо изучены. Первое из них поглощает при 3470
и 3360 см'1, а второе — при 3481 и 3395 см'1.
Калифано и Моккиа [47] исследовали на приборе с вы-
соким разрешением обе полосы валентных колебаний NH
у ряда замещенных анилинов. Они обнаружили, что каж-
дая из частот этих колебаний находится в прямой зависи-
мости от реакционной способности соединения, определяе-
мой значением постоянной Гаммета о. Эти результаты нахо-
дятся в согласии с ранее выдвинутыми предположениями
Ричардса [14] и Флетта [15], которые пытались установить
связь между частотами колебаний NH и основностью ами-
нов. Фасон и др. [13] показали, однако, что хотя эта связь,
по-видимому, и существует у сходных соединений, но, по
крайней мере у вторичных аминов, другие изменения час-
тот, обусловленные структурными особенностями, такими,
как в случае дифениламина и пиррола, достаточно велики,
чтобы замаскировать смещения частот, вызванные первой
причиной. Альтернативное рассмотрение этой проблемы
дано Сирлесом и др. [49], которые определяли основность
соединений по инфракрасным спектрам поглощения, изме-
Г л. 14. Амины и имины
359
ряя смещения полос OD дейтерометанола в различных ами-
нах. Аналогичные исследования проведены с аминоантра-
хинонами [161, нафтиламинами [171 и аминопиридинами
[50—52]. Хотя исследования были выполнены на прибо-
рах с небольшим разрешением, полученные данные хорошо
согласуются с приведенными интервалами частот для этих
колебаний.
Имеется также большое количество данных, полученных
при исследованиях на приборах с высокой разрешающей
силой координационных соединений металлов, в которых
первичные амины выступают в качестве доноров электро-
нов. Такие соединения, как (L-am-PtCl2), где L — какой-
либо лиганд, агп — первичный или вторичный амин,
были изучены в виде растворов Четтом, Дункансоном
и Венанзи [53, 54].
Обзор результатов, полученных при исследованиях на
приборах с высокой разрешающей силой, опубликован не-
давно Беллами и Уильямсом [55]. Они отметили, что час-
тоты как симметричных, так и антисимметричных колеба-
ний зависят главным образом от некоторых силовых постоян-
ных, причем значения частот должны быть непосредственно
связаны друг с другом. Они нашли, что для всех 64 пер-
вичных аминов, для которых имеются надежные данные,
выполняется равенство vCIIMM = 345,53 + 0,876 тантисимм
со среднеквадратичной ошибкой 4,8 см"1. Это соотноше-
ние имеет большое значение для идентификации частот
колебаний свободной группы NH2, особенно в связи
с тем, что оно выполняется1 также и для координационных
соединений, у которых наблюдается значительное (иногда
на 200 см'1) понижение частот колебаний. Они указали,
однако, что это равенство, вероятно, не будет выполняться
в тех случаях, когда в соединении одна группа NH являет-
ся связанной, а другая — свободной, как это имеет место
у димерных комплексов, описанных Четтом и др. [54], или
у таких соединений, как антраниловая кислота. Однако
у некоторых твердых координационных соединений равен-
ство между связями NH восстанавливается, и полученное
соотношение вновь выполняется. Таким образом, про-
верка применимости указанного соотношения может слу-
жить чувствительным методом определения неэквивалент-
ности связей NH у первичных дминор.
360
Часть III
Изучение соотношения интенсивностей этих полос
было проведено только в некоторых исследованиях; можно
указать, например, на работу Калифано и Моккиа [861,
довольно детально исследовавших ряд ароматических сое-
динений. Интересное наблюдение сделано Вампири [56],
показавшим, что интегральное поглощение первичных ами-
нов в области 3600—3300 см'1 приблизительно вдвое боль-
ше, чем соответствующее поглощение вторичных аминов.
Имеется очень мало данных о частотах поглощения групп
NH2, присоединенных не к атому углерода, а к другим
атомам, хотя гидразин, представляющий собой в этом смы-
сле специфическое соединение, был исследован рядом авто-
ров. В последнем исследовании, проведенном Жигером
и Лиу [18], указаны полосы поглощения паров при 3350,
3325 и 3314 см'1, что также согласуется с указанной
областью частот. Шеллом и др. [571 исследован также
1,1 -диметилгидразин.
в. Вторичные амины и имины. В разбавленном растворе
вторичные амины дают только одну валентную полосу
поглощения NH, однако при высоких концентрациях, когда
происходит образование водородной связи, иногда появ-
ляется вторая полоса поглощения в области более низких
частот.
Как и в предыдущем случае, данные, на которых осно-
вана соответствующая корреляция, довольно значительно
отличаются друг от друга. Фасон и др. [13] точно опреде-
лили частоты колебаний свободных групп NH индола,
пиррола, карбазола и дифениламина, равные соответст-
венно 3491, 3496, 3483 и 3433 с.Щ1. Ричардс и Бартон [33]
подтвердили правильность последней из этих частот и ука-
зали, что частота поглощения метил- и этиланилинов равна
3430 см'1. Марион, Рамсай и Джонс [34] приводят для ря-
да вторичных аминов, относящихся к алкалоидам, интервал
частот 3480—3440 см'1 и указывают, что эта полоса сохра-
няется у пиперидина и сходных соединений, но интенсив-
ность ее сильно уменьшена и в некоторых случаях у рас-
творов в хлороформе не может быть обнаружена. Хикок
и Марион [58] наблюдали аналогичный эффект уменьше-
ния интенсивности у растворов пирролидина и пиперазина
в хлороформе. Они указывают для ряда оснований этого
Гл. 14. Амины и имины
361
типа область частот поглощения 3380—3205 слт1, но эти
данные относятся к свободным основаниям, которые могут
быть ассоциированы. Уиткоп [35, 41, 42] и Майрон и Вам-
пири [45] исследовали также ряд производных индсла
и обнаружили поглощение в интервале 3472—3378 см'1.
У этиленимина [20] частота колебаний NH равна 3341 слг"1.
Рендалл и др. [19] указывают для частот колебаний свобод-
ных групп NH интервал 3500—3050 см'1, но большинство
спектров, на которых основывались эти авторы, получено
для твердых веществ и сомнительно, чтобы частота погло-
щения свободной группы NH была значительно меньше
3300 см'1.
Наиболее детальное исследование вторичных аминов
проведено Расселом и Томпсоном [59], которые определяли
как частоту, так и интенсивность этой полосы в спектрах
широкого круга соединений. Обе эти характеристики поло-
сы оказались очень чувствительны к природе окружающей
структуры. У алифатических вторичных аминов частота
поглощения уменьшается до 3310—3350 см'1 с одновре-
менным уменьшением интенсивности поглощения. У алкил-
арильных аминов эта частота резко повышается примерно
до 3450 см'1 и интенсивность возрастает в 50 раз. У пир-
рола, индола и других аналогичных соединений частота
достигает 3490 см'1, и интенсивность вновь увеличивается.
По крайней мере у этого ряда соединений, измерения интен-
сивности полосы, по-видимому, имеют большое значение
для качественного анализа. Заметные изменения частоты
и интенсивности полос поглощения NH обнаружены Бар-
ром н Хасзельдином [60] у соединений с атомами фтора
в виде заместителей при а-атоме углерода. Частота коле-
баний NH б«с-2,2,2-трифтордиметиламина, например, рав-
на 3460 см'1, а коэффициент погашения примерно в 50 раз
больше, чем у самого диметиламина. Спектры вторичных
ариламинов изучены также Салимовым и Татевским [74].
Для иминогруппы =NH характерен, по-видимому, не-
сколько более узкий интервал частот, соответствующих
поглощению свободной группы NH; это следует из того фак-
та, что группировка, включающая связь NH, почти всегда
имеет строение — C=NH. Колтуп [21] приводит для ими-
нов такого строения интервал частот поглощения 3400—
3300 см'1, но вообще в литературе имеется очень мало
332
Часть 111
данных об этих соединениях. Пиккард и Полли [61 1 приводят
для ряда кетиминов аналогичного строения частоту около
3200 см но это значение относится, вероятно, к образцам,
исследованным в твердом состоянии. Рендалл и др. [19]
и Либер, Леверинг и Паттерсон [221 получили спектры
большого числа замещенных гуанидинов, но интерпрета-
ция этих спектров осложнена наличием в соединениях дру-
гих групп NH. Кроме того, Браунли [23] и Шорт и Томп-
сон [24] исследовали большое количество аминопиримиди-
нов; но, хотя многие из этих соединений существуют, по-
видимому, в форме имидов, трудно установить какие-либо
корреляции из-за сложности явлений таутомерии.
3. ВОДОРОДНЫЕ СВЯЗИ АМИНОВ
При соответствующих условиях у большинства аминов
можно наблюдать эффекты образования межмолекулярной
или внутримолекулярной водородной связи (или их обеих).
Поскольку имеется ряд подробных обзоров по этому вопро-
су [25, 26, 62], то, как и в случае гидроксильной группы,
здесь не будут упоминаться все опубликованные работы
и тем более исследования в области обертонов. Однако
ниже приводятся сведения об условиях, при которых можно
ожидать сдвигов полос поглощения, а также о возможной
величине этих сдвигов.
а. Межмолекулярные водородные связи. У аминов в кон-
денсированной фазе водородная связь вызывает лишь не-
большое уменьшение частоты поглощения, которое обычно
значительно меньше 100 см'1. Этот факт вызвал дискуссии
относительно того, действительно ли это смещение обуслов-
лено водородной связью; изменение частоты того же поряд-
ка можно с успехом объяснить изменениями состояния ве-
щества, поскольку в твердом состоянии возможны взаи-
модействия колебаний атомов водорода соседних молекул.
Однако исследования, проведенные Фасоном и др. [13]
с растворами на приборе с высокой разрешающей силой,
показали, что у пиррола и родственных соединений действи-
тельно образуются межмолекулярные связи того типа,
какой еще раньше предполагали Горди [27] и Томпсон
и Гаррис [28]. Фасон и др. [13] наблюдали при переходе
Г л. 14. Амины и имины
363
от жидкостей или сильно концентрированных растворов
к сильно разбавленным растворам типичные сдвиги полос
поглощения в область более низких частот, составлявшие
приблизительно 80—90 см 1 для пиррола и индола, 70 см'1
для карбазола и 30слг-1для дифениламина. Точная величи-
на смещения в каждом отдельном случае зависит, конеч-
но, от растворителя и концентрации вещества; сдвиг поло-
сы поглощения связанной группы несколько меньше в бо-
лее разбавленном растворе, тогда как положение полосы
поглощения свободной группы NH слегка меняется в зави-
симости от полярности растворителя [13]. Изменения поло-
жения и интенсивности полосы NH, которые имеют место
в пирроле в различных растворителях при разных кон-
центрациях, изучены несколькими группами исследовате-
лей [63—68]. Хотя никакого окончательного вывода по
этому поводу сделать нельзя, представляется по крайней
мере весьма вероятным, что у этого соединения при высо-
ких концентрациях осуществляется скорее чистая водород-
ная связь, а не ассоциация диполей. Однако в случае ани-
лина Беллами и Уильямсом [55] показано, что полученное
ими соотношение между частотами колебаний NH выпол-
няется в случае жидкого образца, что свидетельствует об
отсутствии у него водородных связей. Это подтверждается
также наличием последовательного смещения полосы по-
глощения в область более низких частот по мере увеличе-
ния концентрации растворов, в то время как у пиррола
при этих условиях появляется вторая полоса поглощения.
Группы NH могут также взаимодействовать со спиртами
]29] и с кетонными группами [30], хотя сдвиги полос при
этом опять-таки малы. Сазерленд [30] показал, что ассо-
циация с кетонными группами приводит обычно к погло-
щению в интервале 3320—3240 см'1, а с атомами азота —
к поглощению в пределах 3300 — 3150 см'1. В связи с труд-
ностью определения сколько-нибудь точных интервалов
частот колебаний свободных групп NH в аминах область
поглощения 3400—3100 см'1 для ассоциированных групп
NH этого типа можно считать установленной лишь ориенти-
ровочно. Нельзя упускать из виду возможность одновре-
менного появления полос поглощения, соответствующих
как свободным, так и связанным группам NH. Так, напри-
мер, Флетт заметил, что у твердых р-нафтиламина и 2-амино-
364
Часть Hl
антрахинона имеются три максимума поглощения в обла-
сти частот колебаний NH [161, то же самое мы нашли для
о- и ж-хлоранилинов, /г-фенилендиамина, и-нитроанилина
и у других аминов. Для пиррола и других вторичных аминов
при соответствующих концентрациях обнаружены две
полосы поглощения [13]. В последнем случае имеется опас-
ность ошибочно принять эти полосы за две полосы поглоще-
ния свободных групп NH первичных аминов, однако этого
можно избежать, проводя исследования при различных
концентрациях. Во всяком случае, надежным указанием на
наличие водородной связи является обычно наличие более
широкой низкочастотной полосы поглощения.
б. Внутримолекулярные связи. В спиртах простейший
случай внутримолекулярной связи наблюдается у сопря-
женных систем, содержащих группу С— С = С, т. е.
II I
О ОН
таких, как салициловая кислота и ацетилацетон. Подобная
сопряженная система также благоприятствует образованию
внутренних связей у аминов. Однако в таких случаях
наблюдаемые сдвиги полос поглощения NH обычно неве-
лики, что указывает на значительно более слабую связь,
хотя смещения карбонильного поглощения гораздо больше,
чем можно было бы при этом ожидать. Например, в случае
метил-Ы-метилантранилата частота карбонильного погло-
щения 1685 см-1 указывает на наличие сильной водородной
связи, в то время как частота поглощения NH 3361 см-1
незначительно отличается от ее обычного значения при меж-
молекулярной связи [31]. Тот факт, что значение частоты
карбонильного поглощения у метил-М,М-диметилантра-
нилата снова характерно для обычного сложного эфира
(1730 см-1), показывает все же, что образование внутрикомп-
лексных (хелатных) связей в указанных выше случаях имеет
место. ХатвейиФлетт [17] получили аналогичные результа-
ты для ряда нитронафтиламинов. У 2-нитро-1-нафтиламина
в разбавленном растворе частота антисимметричных коле-
баний NH повышается от 3486 см-1 (частота а-нафтиламина)
до 3528 см \ тогда как частота симметричных колебаний NH
в то же время понижается от 3412 до 3378 см Ч Несколь-
ко меньшее понижение частоты наблюдалось в случае
Г л. 14. Амины и имины 365
1-нитро-2-нафтиламина. В обоих этих случаях изменения
частоты колебаний N = O вблизи 1340 см1 подтверждают
образование хелатов. Между тем связь оказывается слабой
или вообще отсутствует в случаях З-нитро-2-нафтиламина
и 8-нитро-1-нафтиламина, у которых частоты колебаний NH
остаются фактически такими же, как у самих нафтиламинов.
У первого из указанных соединений причиной этого являет-
ся менее выраженный двоесвязный характер 2,3-двойной
связи нафталинового ядра, тогда как у второго соединения
отсутствие сопряженной двойной связи, очевидно, препят-
ствует образованию внутримолекулярной связи, несмотря
на то что в этом соединении амино- и нитрогруппы факти-
чески расположены ближе одна к другой, чем в 1-нитро-
2-аминосоединениях. Хелатные связи образуются также
в случае 1-аминоакридина и 8-аминохинолина [40] и у не-
которых аминоазосоединений [75], но изменения частот
поглощения при этом невелики.
Кромвелл и др. [32] нашли, что аналогичные эффекты
образования хелатов имеют место для р-амино-аф-ненасы-
щенных кетонов, но в этом случае наблюдается значительное
изменение частоты колебаний NH, а поэтому у исследован-
ных соединений не удалось обнаружить никаких полос NH;
был сделан вывод о том, что они совпали с полосами погло-
щения валентных колебаний СН вблизи 3000 см1. Трудно-
сти возникают также в связи с тем, что у четырех соедине-
ний, у которых аминогруппа полностью замещена и не
может образовывать водородных связей, наблюдается,
правда, меньший, но все же заметный сдвиг карбонильной
полосы поглощения. Было высказано предположение о том,
что в этих случаях некоторая часть молекул в основном
состоянии имеет строение
— \ 4- /
О
I II
R-C=C-С—R
Возможно при этом, что именно такая резонансная форма
в большей степени, чем водородная связь, обусловливает
те небольшие изменения частот колебаний NH, которые
наблюдались в указанных выше случаях; эти изменения
были значительно меньше тех, каких следовало бы ожидать,
исходя из величин сдвигов полос карбонильного поглощения.
366
Часть 111
Флетт [16] предложил такое же объяснение для не-
больших изменений частот колебаний NH, наблюдавшихся
у 1-амино- и 1,4-диаминоантрахинонов; в этом случае
указанное предположение подтверждается тем, что у 2-ами-
ноантрахинонов также наблюдается большое смещение кар-
бонильного поглощения.
Однако в этих случаях возможно, что отнесение частот
колебаний С = О ненадежно, так как в той же области
должны быть и полосы деформационных колебаний NH2.
4. ДЕФОРМАЦИОННЫЕ КОЛЕБАНИЯ NH
а. Первичные амины. Наши знания о деформационных
колебаниях NH весьма ограниченны, что недавно отмеча-
лось в обзоре Сазерленда [30]. Общепринято, что у про-
стых аминов частота внутренних деформационных колеба-
ний NH2 составляет 1650—1590 слг1, но этот интервал уста-
новлен в результате исследований очень небольшого числа
соединений, а именно метиламина [8, 9, 46], анилина [10]—
[12], гидразина [18], замещенных гидразина [36] и неко-
торых. аминов с длинными цепями [69]. Имеется, однако,
значительно больше данных о простых амидах, у которых
частоты деформационных колебаний NH2 находятся в этой
же области и которые хорошо изучены многими авторами
(см. гл. 12). Эти данные косвенным образом свидетельствуют
о правильности такой корреляции для аминов, однако неко-
торые авторы высказывают сомнение в отношении того,
является ли вообще соответствующая полоса поглощения
вторичных амидов связанной с деформационными колебания-
ми NH. Работы с дейтерированными соединениями подтвер-
ждают точку зрения, что поглощение в указанной области
у первичных амидов и аминов обусловлено деформацион-
ными колебаниями NH, хотя с ними связаны также некото-
рые полосы поглощения в более длинноволновой части
спектра. Однако полоса поглощения в интервале 1650—
1590 слг1 имеется почти во всех спектрах первичных аминов,
сведения о которых нам удалось найти в литературе, а также
у значительного числа таких веществ, исследованных нами.
Исключения составляют; большей частью некоторые аро-
матические вещества, у которых полосы поглощения дефор-
мационных колебаний NH маскируются поглощением,
Гл. 14. Амины и имины
337
обусловленным колебаниями ароматического цикла с часто-
той около 1600 см'1. Так, у о-хлоранилина имеется только
одна сильная полоса поглощения при 1613 см'1, тогда как
-мета-замещенное соединение сильно поглощает при 1613 и
.1597 см'1. Известно еще небольшое число соединений, пред-
ставляющих исключение. Например, появление у боль-
-шинства аминозамещенных пиримидинов полосы поглоще-
ния при 1650 см'1 рассматривается как доказательство
существования их скорее в амино-, чем в имино-форме,
но у 2-метил-4,6-диметокси-5-аминопиримидина, который
должен быть первичным амином, в этой области нет ника-
кой полосы поглощения [24, 37]. Указанная корреляция,
по-видимому, вполне справедлива также для тиоамидных
структур. Например, тиооксамид поглощает при 1600 слг1.
Ввиду отсутствия более точных данных невозможно
никак связать смещения этой полосы в пределах данного
интервала частот со структурными изменениями. Посколь-
ку данная полоса поглощения обусловлена деформацион-
ными колебаниями, то при образовании водородных связей
она смещается в сторону более высоких частот [37], однако
эти смещения чаще всего недостаточно велики для того,
чтобы вывести полосу поглощения за пределы указанного
интервала частот. В этом отношении особенно детально
исследованы первичные амиды, и в типичном случае дефор-
мационных колебаний NH у гексамида частота полосы по-
глощения, равная в случае твердого состояния 1635 см'1,
уменьшается до 1595 см'1 в сильно разбавленном раство-
ре [38].
Первичные амины должны также давать поглощение
при больших длинах волн, обусловленное внешними дефор-
мационными колебаниями группы NH2, но этот вопрос
мало изучен. Большинство первичных аминов имеет широ-
кие полосы поглощения в интервале 900—650 см'1, частота
и форма которых различна в зависимости от степени обра-
зования водородных связей; это дало основание предполо-
жить, что данные полосы связаны с указанными выше коле-
баниями. Хотя в случае некоторых простых соединений
[8, 9, 46] отнесение полос с этими колебаниями было прове-
дено, однако в нашем распоряжении нет данных, которые
могли бы быть успешно использованы в аналитической
работе.
368
Часть 111
б. Вторичные амины. Полосы поглощения деформацион-
ных колебаний NH имеют в случае первичных аминов сред-
нюю или высокую интенсивность, у вторичных же алифа-
тических аминов они обычно очень слабы и часто могут
быть не замечены при использовании пленок вещества
обычной толщины. В случае вторичных ароматических
аминов положение в какой-то степени осложняется нали-
чием в той же спектральной области полос поглощения, обус-
ловленных валентными колебаниями С — С в цикле, кото-
рые в некоторых случаях, когда атом азота присоединен
непосредственно к циклу, становятся более интенсивными
[31]. Этим объясняется наличие противоречивых данных
о характеристических частотах полос поглощения анили-
довых структур. Так, например, Уиткоп и Патрик [39]
считают полосу поглощения при 1613 слГ1 типичной для
структурного элемента С6Н5—N—С~, где атом С не
участвует в образовании двойной связи. В спектрах боль-
шого числа производных индола, которые они привели для
иллюстрации этой точки зрения, в данной области имеется
только одна полоса поглощения, обусловленная, по-види-
мому, ароматическим ядром, тогда как в спектре метилке-
тола, имеющего двойную связь при С, интенсивность этой
полосы заметно ослаблена. Однако теми же авторами была
предложена [35, 41, 42] и вторая корреляция, согласно
которой сильную полосу поглощения в интервале 1639—
1600 см 1 они считают «характеристической для анилиновых
структур вообще и для индоленинов в частности» . Струк-
тура С6Н5 — N = С —, которую они относят к этой полосе
поглощения, содержит, конечно, связь C = N, и ей также
может соответствовать полоса поглощения в данной обла-
сти; или, если эта связь сопряжена, то возможно взаимодей-
ствие ее колебаний с колебаниями ароматического цикла.
Рендалл и др. [19] также рассматривали «анилиновую»
структуру и характерные для нее корреляции, но эти корре-
ляции, по-видимому, тождественны с теми, которые извест-
ны для обычной ароматической системы; только в случае
анилиновой структуры происходит усиление третьей полосы
поглощения при 1610—1590 см1, которая в обычных слу-
чаях слаба или отсутствует. Этот эффект сходен с тем,
который наблюдается при сопряжении кольца (гл. 4).
Гл. 14. Амины и имины
369
Однако хотя у некоторых из ароматических соединений,
исследованных в виде тонких пленок, в этой области не
обнаружено никаких дополнительных полос поглощения,
у других, например у фенил-0-нафтиламина и метил-М-ме-
тилантранилата [31], наблюдается некоторое отличие от
дизамещенных аминов. Хаджи и Скрбляк [70] недавно
вновь исследовали этот вопрос, используя метод дейте-
рирования соединений. Полученные ими результаты ясно
показывают, что у ароматических вторичных аминов дей-
ствительно имеется слабое дополнительное поглощение
в области 1510 см'1. Оно обусловлено деформационными
колебаниями NH, взаимодействующими с низкочастотными
валентными колебаниями С — N. Однако у некоторых ве-
ществ, таких, как метиланилин, оно перекрывается погло-
щением, обусловленным колебаниями кольца, и его трудно
идентифицировать. У тиоамидов полоса хорошо видна и
имеет значительно большую интенсивность [71—73].
У алифатических вторичных аминов эта полоса слишком
слаба, чтобы ее можно было легко определить. У цикличе-
ских оснований, например, в этой области обнаружено толь-
ко обычное поглощение, характерное для ароматических
соединений. Положение изменяется только при образова-
нии соли [58]. Аналогичным образом Барр и Хасзельдин
[60] не смогли идентифицировать эту полосу в спектрах
диметил- или диэтиламинов, хотя у некоторых фторопро-
изводных была прослежена полоса поглощения около
1500 см'1. Имины, содержащие структурную группу
—C = NH, сходны с другими вторичными амидами в том
отношении, что полосы поглощения деформационных коле-
баний NH у них очень слабы, но, конечно, они имеют в той
же спектральной области и полосы поглощения, соответ-
ствующие концевой связи C = N.
5. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С N
а. Ароматические амины. Корреляции для ароматиче-
ских аминов приведены Колтупом [21]. Сильные полосы
поглощения появляются у таких аминов в следующих интер-
валах:
1340—1250 CAi’i у первичных аминов
1350—1280 » » вторичных »
1360—1310 » » третичных »
24 Л. Беллами
370
Часть Ill
Эти корреляции установлены в результате исследований,
проведенных Колтуном (неопубликованная работа), и ни-
каких других сведений о них в литературе не приведено.
Однако наличие полосы поглощения примерно в одной и
той же спектральной области во всех трех случаях пока-
зывает, что она должна соответствовать валентным коле-
баниям С—N, частота которых в большей мере опреде-
ляется ароматическим характером структуры при атоме
углерода. Такие же выводы сделаны Барнсом и др. 144],
которые сформулировали эту корреляцию в более общем
виде.
Данные Колту па полностью подтверждаются данными
о спектрах примерно сорока ароматических аминов различ-
ных типов, исследованных нами, а также результатами,
полученными при изучении спектров большого числа про-
изводных анилина, содержащих фосфор и имеющих струк-
туру Р — NH — Аг [43]. При различных типах замеще-
ния в цикле рассматриваемая полоса, по-видимому, несколь-
ко смещается в пределах указанного интервала частот, но
для оценки величины этих эффектов не имеется еще доста-
точного количества данных. Интенсивность таких полос
поглощения изменяется от довольно большой величины
до средней. Это также может быть связано с какими-либо
различиями в замещении или другими особенностями
строения.
Либер и др. [22] обнаружили эти полосы поглощения
также у 13 тетразолов различного типа. Недавно Хаджи
[70] исследовал влияние дейтерирования соединений на это
поглощение в спектрах производных анилина. Полоса
вблизи 1260 см'1 смещается примерно до 1350 см1. Это
позволяет отнести ее к валентным колебаниям С — N,
частота которых у соединений, содержащих водород, пони-
жена вследствие связи с деформационными колебаниями NH.
б. Алифатические амины. Колтуп [21] показал также,
что алифатические амины имеют полосы поглощения в ин-
тервале 1220—1020 см'1, которые, по-видимому, соответ-
ствуют валентным колебаниям несопряженной связи С — N;
они часто имеют малую интенсивность. По этой причине,
а также вследствие широкого интервала частот, в который
они попадают наряду с полосами многих других типов
Гл. 14. Амины и имины
371
колебаний скелета, сведения об этих полосах не имеют
большого значения, если не считать того, что в некоторых
случаях их можно использовать для подтверждения других
данных. У метиламина эта полоса проявляется [46] при
1044 см1.
В случае ненасыщенных третичных аминов можно разли-
чать а,0- и р,у-ненасыщенные связи, наблюдая при обра-
зовании соли за изменениями частот поглощения в области
1600—1700 см'1 [87].
6. ГРУППА СН3—N
Можно было бы ожидать, что деформационные колеба-
ния группы СНз, связанной с атомом азота, будут иметь
характеристическую частоту, подобно тому как и колеба-
ния в случае группировки СНз — С, полоса поглощения кото-
рой находится при 1375 см'1. Однако замена заместителей
при атоме азота может оказать большее влияние на частоту
колебаний. У простых молекул частота этих колебаний
лежит в интервале 1460—1430 см'. Исследование спектров
ряда более сложных молекул, содержащих данную группу,
показывает, что хотя у многих из этих соединений имеется
полоса поглощения вблизи 1430 см'1, но у некоторых
других, если даже она и есть, ее нельзя отличить
от главной полосы, т. е. от поглощения деформационных
колебаний СН.
Рассматриваемая полоса поглощения мало интенсивна, и
применение этой корреляции ограничивается еще тем обстоя-
тельством, что значительное число других азотсодержа-
щих соединений также имеет слабые полосы поглощения
в интервале 1430—1400 см'1. У диметиламиносоединений,
таких, как диметиламин и окись бис-диметиламинофтор-
фосфина, в отличие от изопропилпроизводных расщепле-
ния этой полосы поглощения на две компоненты равной
интенсивности не наблюдается. Поэтому можно вывести
заключение, что, несмотря на постоянство частоты дефор-
мационных колебаний группы СН3 в случаях присоедине-
ния ее к атомам углерода, кремния и многих других эле-
ментов, полосы соответствующих колебаний в группе МСНз
слишком слабы и непостоянны по частоте, и поэтому эта
корреляция не может быть успешно применена.
24*
372
Часть Hl
7. ГРУППЫ NH+ NHt и NH*
Производные аминов с положительно заряженными
атомами азота обычно наблюдаются в солях, координацион-
ных соединениях, включая аммониевые, и биполярных
структурах. Последние обсуждались в предыдущей главе
и здесь не рассматриваются.
а. Поглощение NH+. Поглощение, обусловленное нали-
чием этой группы, обнаружено у сложных аминов и гид-
рохлоридов первичных аминов. Первые из этих соединений
изучены многими исследователями [53, 54, 76—83]. Заряд,
сосредоточенный на атоме азота, в этом случае заметно
меньше, чем в случае гидрохлоридов, вследствие чего
и частоты соответствующего поглощения ближе к частотам
поглощения обычных аминов. Несколько таких соедине-
ний, исследованных в растворе, характеризуются частотами
валентных колебаний вблизи 3380 и 3280 слг"1. Однако боль-
шинство исследований выполнено с твердыми веществами,
у которых вследствие эффектов межмолекулярного взаимо-
действия эти частоты понижены до 3359—3150 слг"1, при-
чем иногда наблюдаются сложные полосы. Частота антисим-
метричных деформационных колебаний, как и в случае пер-
вичных аминов, равна примерно 1600 сл/"1, частота сим-
метричных колебаний лежит около 1300 слг"1, а частота
маятниковых колебаний NH3 составляет примерно 800 слг"1.
Эти данные подтверждаются при исследовании дейтериро-
ванных соединений.
Гидрохлориды простых первичных аминов менее иссле-
дованы, но, по-видимому, они подобны гидрохлоридам ами-
нокислот. Гидрохлорид метиламина, например, поглощает
при 3075 и 2972 слг"1, а полоса поглощения, соответствую-
щая деформационным колебаниям NH 184], находится при
1617 см Г У гидрохлорида гидроксиламина связи NH
взаимодействуют сильнее, и полосы поглощения [84] на-
ходятся при 2955 и 2667 слг"1.
б. Поглощение NH*. Четтом и др. [53] изучено в обла-
сти высоких частот большое число координационных соеди-
нений, в том числе производные анилина, метиламин и дру-
гие амины. В разбавленных растворах эти соединения ведут
Гл. 14. Амины и имины
373
себя подобно обычным первичным аминам, для которых
выполняется соотношение Беллами и Уильямса [55],
но влияние отдачи электрона сказывается на значительном
уменьшении частот валентных колебаний NH. Эти частоты
примерно на 200 см"1 меньше, чем те же частоты колебаний
у анилина. Образование водородной связи в концентрирован-
ных растворах и в твердых веществах приводит к даль-
нейшему уменьшению этих частот. Деформационные колеба-
ния NH2 у соединений! этого типа исследованы Мидзусимой
и др. [815; частоты этих колебаний более близки к обычным
значениям.
Гидрохлориды вторичных оснований изучены Хикоком
и Марионом [58], которые обнаружили целые серии полос
поглощения в интервале частот 2800—2000 см"1. Это явле-
ние особенно заметно у ароматических соединений, у кото-
рых, помимо других полос поглощения в области меньших
частот, воспроизводятся три или четыре полосы в интервале
частот 2780—2600 см"1. При дейтерировании соединений
эти полосы ослабляются, что указывает на их связь с коле-
баниями NH. Из 17 изученных солей у 15 наблюдается
полоса поглощения в интервале 2760—2690 см"1-, все соли
поглощают в интервале 1620—1560 см 1 (деформационные
колебания NH2) и около 800 см"1 (маятниковые колеба-
ния NH2). Барселло и др. [84] обнаружили аналогичную
картину в случае гидрохлорида диметиламина.
в. Поглощение NH*. У гидрохлорида триметиламина
полоса валентных колебаний NH+ [84] обнаружена при
2735 см'1. У солей пиридина, индолена и оснований Шиффа
поглощение, обусловленное колебаниями группы С = NH+,
находится в интервале 2500—2325 см"1. Однако когда около
группы NH+ нет никакого другого гетероатома, то появ-
ляется вторая полоса поглощения в интервале частот 2200—
1800 см"1. Использование этого эффекта для целей каче-
ственного анализа рассмотрено Уиткопом [85].
ЛИТЕРАТУРА
1. С о b 1 е n t z, Investigations of Infra-red Spectra, Carnegie
Institute, 1905.
2. В e 1 1, J. Am. Chem. Soc., 47, 2192 (1925).
3. Bel 1, J. Am. Chem. Soc., 47, 3039 (1925).
374
Часть 111
4. В е 1 1, J. Am. Chem. Soc., 48, 813 (1926).
5. В е 1 1, J. Am. Chem. Soc., 48, 818 (1926).
6. Е 1 И s, J. Am. Chem. Soc., 49, 347 (1927).
7. Е 1 1 i s, J. Am. Chem. Soc.. 50, 685 (1928).
8. Cleaves, Plyler, J. Chem. Phys., 7, 563 (1939).
9. В a i 1 e y, Carson, Daly, Proc. Roy. Soc., A 173, 339
(1939).
10. Buswell, Downing, Rodebush, J. Am. Chem.
Soc., 61, 3252 (1939).
11. Gordy, J. Am. Chem. Soc., 59, 464 (1937).
12. Williams, Hofstadter, Herman, J. Chem. Phys.,
7, 802 (1939).
13. Fuson, Josie n, Powell, Utterback, J. Chem.
Phys., 20, 145 (1952).
14. Richards, Trans. Faraday Soc., 44, 40 (1948).
15. Flett, Trans. Faraday Soc., 44, 767 (1948)
16. Flett, J. Chem. Soc., 1948, 1441.
17. H a t h w a y, Flett, Trans. Faraday Soc., 45, 818 (1949).
18. Giguere, Liu, J. Chem. Phys., 20, 136 (1952).
19. Randall, Fowler, Fuson, Dangl, Infra-red De-
termination of Organic Structures, Van Nostrand, 1949.
20. Hoff ma n, Evans, G 1 о с к 1 e r, J. Amer. Chem. Soc.,
73, 3028 (1951).
21. С о 1 t h u p, J. Opt. Soc. Amer., 40, 397 (1950).
22. Lieber, Levering, Patterson, Analyt. Chem.,
23, 1594 (1951).
23. Brownl ie, J. Chem. Soc., 1950, 3062.
24. Short, Thompson, J. Chem. Soc., 1952, 168.
25. П а у л и и г Л., Природа химической связи, Госхимиздат,
М,—Л., 1947.
26. Hunter, Price, Martin, Report on the Symposium on
the Hydrogen Bond, Institute of Chemistry, 1950.
27. Gordy, J. Chem. Soc., 7, 167 (1939).
28. T h о m p s о n, Harris, J. Chem. Soc., 1944, 301.
29. В a к e r, Davies, Gaunt, J. Chem. Soc., 1949, 24.
30. Sutherland, Discuss. Faraday Soc., 9, 274 (1950).
31. Rasmussen, Brattai n, J. Amer. Chem. Soc., 71,
1073 (1949).
32. Cromwell, Miller, Johnson, Frank, Wal-
lace, J. Amer. Chem. Soc., 71, 3337 (1949).
33. Richards, Burton, Trans. Faraday Soc., 45, 874
(1949).
34. Marion, Ramsay, Jones, J. Amer. Chem. Soc., 73,
305 (1951).
35. W i t к о p, J. Amer. Chem. Soc., 72, 614 (1950).
36. Axford, J a n z, Russell, J. Chem. Phys., 19, 704
(1951).
37. Thompson, Nicholson, Short, Discuss. Faraday
Soc., 9, 222 (1950).
38. Richards, Thompson, J. Chem. Soc., 1947, 1248.
39. W i t к о p, Patrick, J. Amer. Chem. Soc., 73, 713 (1951).
Гл. 14. Амины и имины
375
40. Short, J. Chem. Soc., 1952, 4584.
41. Witkop, Patrick, J. Amer. Chem. Soc., 73, 1558 (1951).
42. W i t к о p, Patrick, J. Amer. Chem. Soc., 73, 2188 (1951).
43. В e 1 1 a m y, Beecher, J. Chem. Soc., 1952, 1701.
44. Barnes, Gore, Stafford, V. Zandt Willi-
ams, Analyt. Chem., 20, 402 (1948).
45. Mirone, V a m p i r i, Atti accad. Nazi. Lincei Rend.
Classe Sci. fis. mat. e. Nat., 12, 405 (1952).
46. В a г с e I 1 о, В e 1 1 a n a t o, Spectrochim. Acta, 8, 27 (1956).
47. С a 1 i f a n o, Moccia, Gazz. Chim., 86, 1014 (1956).
48. R i c h t e r i n g, Z. Phys. Chem., 9, 393 (1956).
49. S e a r 1 e s, T a m r e s, В I о c k, Q u a r t e r m a n, J. Amer.
Chem. Soc., 78, 4917 (1956).
50. A n g у a 1, Werner, J. Chem. Soc., 1952, 2911.
51. Ш и г о p и н Д. И., Данюшевский Я. Л-, Гольд-
фарб Я- Л., Изв. АН СССР, ОХН, № 1, 120 (1956).
52. Costa, Blasina, Sartori, Z. Phys. Chem., 7, 123
(1956).
53. Chat t. Duncanson, Venanzi, J. Chem. Soc.
1955, 4461.
54. C h a t t, Duncanson, Venanzi, J. Chem. Soc., 1956,
2712.
55. Bellamy, Williams, Spectrochim. Acta, 9, 341 (1957).
56. V a m p i r i, Gazz. Chim., 84, 1087 (1954).
57. Shull, Wood, Aston, Rank, J. Chem. Phys., 22,
1191 (1954).
58. H e а с о c k, Marion, Can. J. Chem., 34, 1782 (1956).
59. Russel, Thompson, J. Chem. ,Soc., 1955, 483.
60. Barr, Haszeldine, J. Chem. Soc., 1955, 4169.
61. Pickard, Polly. J. Amer. Chem. Soc., 76, 5169 (1954).
62. Hunter, Progress in Stereochemistry, Vol. 1, Butterworth,
London, 1954, p. 224.
63. J о s i e n, Fuson, J. Chem. Phys., 22, 1169 (1954).
64. F u s о n, J о s i e n, J. Phys. Radium, 15, 652 (1954).
65. Josie n, Fuson, J. Chem. Phys., 22, 1264 (1954).
66. Mirone, Fabbri, Gazz. Chim., 86, 1079 (1956).
67. T u о m i к о s к i, Mikrochem. Acta, 1955, 505.
68. T u о m i к о s к i, J. Phys. Radium, 16, 347 (1955).
69. Despas, Khaladji, Vergoz, Bull. Soc. Chim. Fr..
1953, 1105.
70. H a d z i, S к r b I j a к , J. Chem. Soc., 1957, 843.
71. H a d z i, J. Chem. Soc., 1957, 847.
72. Mecke, Mecke, Chem. Ber., 89, 343 (1956).
73. Mecke, Mecke, Luttinghaus, Zeit. Naturforsch..
10B, 367 (1955).
74. Салимов M. A., T а т e в с к и й В. М., ДАН СССР,
112, 890 (1957).
75. Багр атишви ли Г. Д., ДАН СССР, 96, 753 (1954).
76. Powell, Sheppard, J. Chem. Soc., 1956, 3108.
77. D u v a 1, Duval, Lecomte, Compt. Rend. Acad. Sci
(Paris), 224, 1632 (1947).
376
Часть 111
78. Beattie, Tyrell, J. Chem. Soc., 1956, 2849.
79. H i 1 1, Rosenberg, J. Chem. Phys., 24, 1219 (1956)
80. M i z us h i m a, Na kagawa, Q u a g 1 i a n o, J. Chem.
Phys., 25, 1367 (1956).
81. Mizushima, Nakagawa, Sweeney, J. Chem.
Phys., 25, 1006 (1956).
82. Svatos, Curran, Quagliano, J. Amer. Chem.
Soc., 77, 6159 (1955).
83. Barrow, Kreuger, Basolo, J. Inorg. Nuclear
Chem., 2, 340 (1956).
84. В e 1 1 a n a t о, В a r c e 1 1 o, Anales. real Soc. Espan. fis.
y. quim. Madrid, 52B, 469 (1956).
85. W i t к о p, Experimentia, 10, 420 (1954).
86. Califano, Moccia, Gazz. Chim., 87, 58 (1957).
87. Leonard, Gash, J. Amer. Chem. Soc., 76, 2781 (1954).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Н„
5,
Изв. АН СССР,
Ярославский Н. Г.,
184 (1941).
Н. Г., ЖФХ, 22, 256 (1948).
Григоровский А. М., Ярослав-
ДАН СССР, 67, 697 (1949).
Л., Кондакова М. С., Ш
н А.
физ.,
В С К и й
н и
сер.
ела
якин А. В.,
скин Н. Г.,
ь д ф а р б Я-
Д. Н., Изв. АН СССР, ОХН, № 3, 336 (1956).
Шей и кер Ю.Н., Куш кин Б.В.,Постовск
ЖФХ, 31, 214 (1957).
Домбровская У., Пентин Ю. А.,
с к и й Я , Татевский В М.,
ЖФХ, 32, 135 (1958).
Яро
Кар
Гол
и й И. я.,
До
м б ров-
Кочетков Н. К-,
T e
P
e
и г о p и н
Глава 15
СОЕДИНЕНИЯ С НЕНАСЫЩЕННЫМИ СВЯЗЯМИ
ПРИ АТОМАХ АЗОТА
I. ВВЕДЕНИЕ
Наблюдаются большие расхождения в имеющихся дан-
ных о ненасыщенных азотсодержащих соединениях. Поло-
жение полосы валентных колебаний C^N нитрилов тща-
тельно исследовано и точно определено; исследовано также
влияние сопряжения или замещения атомами галогенов.
Аналогичным образом азиды можно без труда определить
по характеристическому поглощению N=N. Число иссле-
дованных в этом случае соединений еще сравнительно неве-
лико, но положение полосы поглощения вблизи 2140 см'1
почти не оставляет сомнений в том, что она обусловлена
колебаниями тройной связи. Интересно отметить, что эта
полоса поглощения ясно видна и у неорганических азидов.
Колебания двойных связей С = N и N = N изучены
меньше. Приблизительное положение соответствующих
полос поглощения известно, но мало что известно о факто-
рах, влияющих на их положение и интенсивность. У сопря-
женных циклических соединений частота колебаний С = N
изменяется гораздо больше, чем частота соответствующих
колебаний С = С, и потому при интерпретации спектров
поглощения соединений, содержащих такие структуры,
требуется большая осторожность. Указанные корреляции
представлены в табл. 14.
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ—С = N
а. Общие сведения. В случае нитрилов следует ожидать
появления полос поглощения в области частот колебаний
тройных связей 2300—2000 слГ1, но до последнего времени
исследованы только некоторые соединения этого типа.
Рейтц и Сабати [1, 2] и Рейтц и Скрабел [31 исследовали,
однако, спектры комбинационного рассеяния мономерных
378
Часть III
Таблица 14
Частота, еле-1
Валентные колебания С N
Насыщенные алкилпитрилы . . . .
а.Р'Арнлнитрилы ................
аф-Ненасыщенные алкилнитрилы
Изоцианаты.......................
Валентные колебания С N
Соединения с открытой цепью . . .
аф-Ненасыщенные соединения с
открытой цепью..................
Сопряженные циклические соедине-
ния ...........................
Валентные колебания N = N
— N = С = N —
Валентные колебания N = N (азиды)/
2260—2240 (с.)
2240—2220 (»)
2235-2215 (»)
2275—2240 (»)
1690—1640 (перем.)
1660—1630 »
1660—1480 »
1630—1575 »
2155—2130 (с.)
1340—1180 (сл.)
2160—2120 (с.)
нитрилов и нашли следующие значения частот: 2245 см'1
для алифатических нитрилов; 2229 см'1 для ароматических
нитрилов и 2220 см'1 для сопряженных соединении.
Последующие исследования показали, что эти значения
приблизительно верны также и для инфракрасных спектров
поглощения. Снайдером и Элилом [4] в 1948 г. опублико-
ваны спектры поглощения двух нитрилов, один из которых
является сопряженным, а другой — несопряженным, и,
хотя частоты полос поглощения авторами не указаны, из
спектров видно, что сопряженное соединение поглощает
при частоте примерно на 30 см'1 меньшей, чем частота
поглощения несопряженного соединения. Марвел и др.
[5, 6] изучили мономеры и димеры ацетиловых и бензоило-
вых нитрилов и приводят частоты: 2235 см1 для мономера
и 2256 см 1 для димера, у которого сопряжения с группой
СО нет. Исследован также димерный 2-цнано-1,3-бутадиен.
Он содержит две группы CN, из которых только одна соп-
ряжена. Для этого соединения наблюдаются два пика при
2220 и 2243 см'1.
Гл. 15. Соединения с ненасыщенными связями при атомах азота 379
Данные об этой полосе поглощения получены также при
исследовании фосфорилцианидов [7], которые поглощают
при 2232 см 1, и синильной кислоты. Последняя изучалась
в виде смеси, содержащей изотопы С12 и С13 [8]. Частоты
колебаний С == N оказались равными соответственно 2213
и 2157 см1, причем изотопическое смещение отличается
от теоретического не более чем на 1 см'1.
Более подробное исследование нитрилов, подтверждаю-
щее и дополняющее все эти данные, проведено Китсоном
и Гриффитом [9], которые изучали спектры примерно 70 раз-
личных нитрилов с целью установления зависимости наб-
людаемых сдвигов полос поглощения от строения молекул.
Эти авторы нашли, что у 34 насыщенных моно- и динит-
рилов (за исключением малононитрила) частоты колебаний
С s N имеют значения в пределах 2260—2240 см'1 и этот же
интервал включает частоты еще 17 ненасыщенных, но
не сопряженных нитрилов. Двенадцать других нитрилов,
у которых связь С = N сопряжена с двойной связью (за
исключением фумаронитрила), имеют сильную полосу по-
глощения в области 2232—2218 см'1, а у четырех соедине-
ний с нитрильными группами обоих типов имеются обе эти
полосы поглощения. Подобным же образом те ароматические
соединения, у которых группа С ss N присоединена непо-
средственно к циклу (12 соединений), поглощают в интер-
вале 2240—2221 см'1, тогда как у двух других соединений,
у которых группа С е= N отделена от ароматического ядра
метиленовыми группами, обнаружены обычные частоты
поглощения, равные 2254 и 2248 см'1. Как и следовало
ожидать, природа других заместителей кольца в какой-то
мере влияет на частоту поглощения, но указанный выше
интервал уже включает частоты поглощения соединений
как с заместителями, отталкивающими электроны, так и
с заместителями, притягивающими их. Эти данные были
затем подтверждены в рабэге Фелтона и Орра [35] и Скин-
нера и Томпсона [36], которые также исследовали большое
число нитрилов и получили аналогичные результаты.
Из сказанного ясно, что группа С = N может быть
идентифицирована по инфракрасному спектру, если только
соответствующая ей полоса поглощения достаточно интен-
сивна. Некоторое количество дополнительных данных мож-
но также получить и о влиянии сопряжения, причем поведе-
380
Часть III
ние групп С = N оказывается в этом отношении весьма
сходным с поведением карбонильной группы, о которой
имеется больше сведений. Имеются, однако, и явные раз-
личия в поведении этих двух групп. Так, например, если
наличие атомов фтора в a-положении повышает частоту
колебаний нитрильной группы, как и в случае соединений,
содержащих карбонильные группы, то присоединение ато-
мов хлора понижает эту частоту. Перфторалкилнитрилы
поглощают при 2275ц 5 см у CF2= CFCN частота погло-
щения (2255 см"1) также повышена по сравнению с винил-
цианидом [37]. Однако цианогенхлорид поглощает при
2214 см'1, в отличие от повышенной частоты колебаний
карбонильной группы фосгена. У арилнитрилов влияние
ароматического цикла усиливается при наличии группы
n-N(CH3)2 [38], что приводит к несколько пониженным
частотам поглощения. Аналогичное явление наблюдается
в случае ацетофенонов.
Что касается малононитрила и фумаронитрила, упомя-
нутых выше как исключения, то в обоих этих случаях
следует ожидать взаимодействия между группами С = N;
аналогичные эффекты наблюдаются и в случае колебаний
С = О у соответствующих карбоновых кислот. Вследствие
этого рассмотренные случаи несущественны с точки зрения
общей применимости указанных корреляций. Еще одним
исключением является цианистый диазогуанидин, кото-
рый, поданным Либераидр. [13], поглощает при 2126 см'1.
Такой сдвиг полосы поглощения гораздо больше того,
которого можно было бы ожидать при сопряжении с
—N = N—.
Приведенные выше корреляции неприменимы, конечно,
к изонитрилам. Число исследованных соединений этого
класса очень невелико, но, по-видимому, можно сказать,
что их тройная связь Cs N обусловливает появление
полосы поглощения в интервале 2180—2120 см 1 [11].
Мак-Брайд и Бичелл [29] в своей'работе сопоставили данные
о спектрах поглощения некоторых нитрилов и изонитри-
лов. Нитрилы поглощают в указанной выше области, при-
чем значение частоты несколько зависит от природы заме-
стителей, тогда как алкилизонитрилы поглощают при
2183—2144 см'1, а ароматические производные — вблизи
2145 см'1. Авторы наблюдали также в спектрах изонитри-
Гл. 15. Соединения с ненасыщенными связями при атомах азота 381
лов новую полосу поглощения вблизи 1592 см"1, которая
отсутствует у нитрилов. Однако у изометилцианида, иссле^
дованного Уильямсом [39], в этой области нет никаких полос
поглощения. Частота поглощения, обусловленного коле-
баниями N = С, в этом случае равна 2166 см"1.
б. Интенсивность полос поглощения С N. Китсон и
Гриффит [9] изучили также интенсивность полосы погло-
щения нитрилов. Они нашли, что интенсивность полосы
в зависимости от строения нитрила довольно сильно колеб-
лется, меняясь от очень высокой до столь слабой, что полосу
не удается обнаружить. Эта полоса поглощения интенсивна
обычно у соединений, содержащих только атомы С, Н и N,
причем интенсивность полосы у этих соединений является
функцией числа нитрильных групп в молекуле, а потому
величину интенсивности в расчете на каждую группу С s N
можно считать во всех случаях практически одинаковой.
Сопряжение также оказывает на интенсивность некоторое
влияние. Кросс и Ролф [12] отмечают, что полоса погло-
щения группы С s N бензонитрила (2250 слГ1) вдвое ин-
тенсивнее, чем в двух других случаях, когда эта группа
не сопряжена с бензольным кольцом.
Введение в молекулу кислородсодержащей группы при-
водит к значительному уменьшению интенсивности полосы
поглощения С s= N, причем влияние этой группы больше
тогда, когда она присоединена к тому же атому углерода,
что и нитрил. У циангидрина ацетона, например, интен-
сивность полосы поглощения С = N составляет примерно
одну треть обычной величины, а у dl- и лгсзо-диацетилциан-
гидрина нитрильная полоса слаба, но заметна. У соответ-
ствующих ацетоксисоединений она может быть вообще
неразличима.
Это исследование наглядно показывает возможности
и пределы использования метода инфракрасной спектров
метрии для изучения этой специфической группы. Когда
в спектре имеется соответствующая полоса поглощения,
группа может быть точно идентифицирована, а также
могут быть получены некоторые данные о сопряжении. Ис-
пользуя весьма сходные соединения в качестве эталонов,
можно таким путем определить содержание групп С = N.
Магат и др. [10] определили группу Cs N в полимерах,
382
Часть 111
подобных найлону, используя в качестве стандарта интен-
сивности адипонитрил.
С другой стороны, отсутствие в рассматриваемой спект-
ральной области интенсивной полосы поглощения не может
служить доказательством отсутствия групп С == N, если
только не известно, что в молекуле отсутствует кислород.
Количественное определение групп С N в неизвестных
веществах нельзя производить при помощи измерений ин-
тенсивности, поскольку произвольный выбор в качестве
стандарта интенсивности соединения, недостаточно близ-
кого по строению к исследуемому веществу, может при-
вести к большим ошибкам.
Интенсивность, а также полуширина полос поглощения
С N очень сильно меняются в зависимости от структуры
соединения. Эти изменения значительно больше, чем
соответствующие изменения частот полос, вследствие чего
их можно успешно использовать при идентификации соеди-
нений. Например, Фелтон и Орр [35] отметили, что интен-
сивность полос поглощения нитрильной группы в таком
соединении, как 3-метилбутен-2-нитрил, более чем в 10 раз
превышает интенсивность поглощения ацетонитрила. Наи-
более детальные исследования этого вопроса выполнены
Скиннером и Томпсоном [36] и Сенси и Галло [40]. У ал-
килцианидов интенсивность поглощения равна 2,0-1СГ8
— 3,2-10’s, тогда как у всех ароматических нитрилов она
значительно больше, что частично зависит от природы
и положения заместителей в цикле, и связана, вероятно,
с изменениями энергии резонанса, а,£-Ненасыщенные сое-
динения характеризуются частотами поглощения, анало-
гичными частотам ароматических нитрилов, но интенсив-
ности их полос поглощения гораздо меньше. Введение в мо-
лекулу полярных групп в a-положении также сопровож-
дается заметными изменениями интенсивности полос погло-
щения С s N.
3. ИЗОЦИАНАТЫ И КАРБОДИИМИДЫ
а. Изоцианаты. Этот класс соединений изучен Дэвисо-
ном [42] и Хойером [41 ]. Дэвисон исследовал 10 изоцианатов
и обнаружил у них интенсивную полосу поглощения
2269± 6 см1. Она не изменяется при изменениях состояния
Гл. 16. Соединения с ненасыщенными связями при атомах азота 383
вещества и при наличии сопряжения. Интенсивность этой
полосы поглощения в случае двух ароматических изоциа-
натов более чем в 100 раз превышает интенсивность соот-
ветствующей, полосы алкилцианидов, которые поглощают
в той же области спектра. Таким образом, это обстоятельство
может быть использовано для идентификации рассматривае-
мых соединений. Эги данные полностью подтверждены Хойе-
ром 141]. Он исследовал более 40 изоцианатов и отметил,
что и алкильные и арильные производные поглощают в обла-
сти 2274—2242 см1, причем большинство из них погло-
щает около 2270 см'1. Не отмечено никакого четкого раз-
личия в частоте между сопряженными и несопряженными
соединениями, и подтверждена высокая интенсивность этой
полосы поглощения. У 1-циано-З-изоцианатопропана, на-
пример, поглощение N = С = О при 2272 см'1 легко можно
отличить от заметно более слабой полосы поглощения нит-
рильной группы, находящейся при несколько меньшей
частоте. Фенилизоцианат представляет собой исключение
и дает дублет при 2278 и 2260 см'1. Полоса поглощения
изоцианатов 2270 см'1 соответствует антисимметричным
валентным колебаниям группы N = С = О, а менее интен-
сивная полоса, соответствующая симметричным колеба-
ниям, теоретически должна находиться около 1350 см1.
Эйстером и Жиллеттом 125] эта полоса обнаружена у ме-
тилизоцианата при 1377 см'1. Если бы идентификация этой
полосы могла быть проведена вполне надежно, то можно
было бы гораздо проще различать изоцианаты и нитрилы.
Но, к сожалению, эта вторая полоса слишком малоинтен-
сивна, чтобы ее можно было зарегистрировать сколько-
нибудь надежно [42], к тому же она маскируется полосами
поглощения метильной и метиленовой групп, которые нахо-
дятся в этой области спектра.
б. Карбодиимиды. Соединения этого класса, содержащие
группу R—N=C=N—R, мало изучены. Очень ограничен-
ное число образцов исследовано Кхорана [43, 45], кото-
рый отметил поглощение при 2150 см'1. Более детальное
исследование частот и интенсивности поглощения выполнено
Микинсом и Моссом [44]. Этими авторами изучены 9 кар-
бодиимидов, и у всех обнаружено очень интенсивное по-
глощение в области 2152—2128 см'1, сопровождающееся
384
Часть 111
у трех веществ значительно менее интенсивной полосой при
меньших частотах. Между алкильными и арильными про-
изводными имеется небольшое различие в частоте или ин-
тенсивности поглощения. Интенсивность полосы опре-
делялась различными методами, и коэффициент погаше-
ния еа оказался порядка 1400. Это примерно в 2,5 раза
больше, чем у обычного карбонильного поглощения кетонов.
4. ВАЛЕНТНЫЕ КОЛЕБАНИЯ -C=N-
а. Общие сведения. Проведена большая работа по опре-
делению положения полосы поглощения валентных коле-
баний — С—N— у различных классов соединений; деталь-
ный обзор имеющихся данных сделан Фабианом, Легра-
ндом и Пуарье [46]. Тем не менее при идентификации этой
полосы возникают затруднения из-за значительных изме-
нений ее интенсивности при изменениях структуры соеди-
нения. Кроме того, имеющиеся сведения об эффектах
сопряжения в циклических системах противоречивы и не-
определенны. Частично это обусловлено тем, что у многих
исследованных соединений имеет место сопряжение связи
C=N со связями С=С, а частоты колебаний этих связей
столь близки, что, по крайней мере в случае циклических
систем, сомнительно, чтобы могли быть обнаружены коле-
бания, присущие каждой группе в отдельности. По этой
причине Рендалл и др. [14] предпочитают идентифицировать
группы полос в интервале 1600—1500 см~1 как характери-
стические для структуры тиазолов в целом, а не определять
происхождение отдельных полос. Ценный перечень ориги-
нальных работ, посвященных исследованию частот коле-
баний гетероциклических соединений этого типа, приведен
Джонсом и Сандорфи [47]. У пиридина и подобных ему
ароматических соединений полос поглощения, характерных
для C=N, конечно, не имеется; системы этого типа рассмот-
рены отдельно в разделе, посвященном гетероциклическим
ароматическим соединениям (гл. 16).
Вторая трудность при идентификации полос поглощения
C=N обусловлена тем, что многие работы по изучению этой
группы были проведены в связи с исследованиями пени-
циллина, а поэтому изучавшиеся соединения часто содер-
жали амидные группы, аминогруппы и ароматические
Гл. 15. Соединения с ненасыщенными связями при атомах азота 385
циклические системы, которые могли давать поглощение
в той же спектральной области, что и группа C=N.
Как будет видно из последующего обсуждения, полосы
поглощения С = N у систем с открытой цепью или у не-
сопряженных циклических систем наблюдаются в интер-
вале 1690—1640 см'1, а влияние сопряжения обычно неве-
лико. В случае сопряженных циклических систем положе-
ние значительно менее определенно, и полосы поглощения
C—N относят к интервалу частот 1660—1480 см'1. Для
ациклических соединений и циклических веществ без
внутреннего сопряжения полоса поглощения С = N отне-
сена Барнсом и др. [15] к 1650 см'1, и правильность этого
отнесения полностью подтверждена данными более позд-
них работ, особенно результатами исследований пеницил-
лина, которые полностью не опубликованы, но суммированы
Томпсоном, Браттеном, Рендаллом и Расмуссеном [16].
На основании данных, полученных для оксазинов, оксазо-
линов, оксимов и иминов, эти авторы выводят заключение,
что полосы поглощения С = N лежат в области 6,0 ц
(1667 слГ1). Эта корреляция подтверждена также Фабианом
и др. [46], которые считают характерным для несопряжен-
ных соединений типа R — СН = N — R поглощение в ин-
тервале частот 1674—1665 см'1. В некоторых случаях
наблюдается небольшое понижение частоты, когда один
из углеводородных радикалов имеет разветвленную цепь.
Для оксимов эта корреляция также выполняется довольно
хорошо. Ацетоксим, например, поглощает при 1675 см'1,
оксим циклогексанона — при 1669 см'1, а оксим циклопен-
танона [12] — при 1684 см'1. Небольшое повышение ча-
стоты при переходе от шестичленного цикла к пятичлен-
ному аналогично повышению частоты колебаний С = О
у циклопентанона по сравнению с циклогексаноном, что
можно объяснить напряжением цикла. Формальдоксим
и оксим ацетона были довольно детально исследованы Кали-
фано и Люттке [48], а Палм и Вербин [49] изучали вопрос
о геометрических изомерах подобных соединений. Несо-
пряженные оксимы в твердом состоянии поглощают около
1640 см'1 независимо от геометрической формы. Однако
на эту частоту влияет межмолекулярная ассоциация,
и у разбавленного раствора частоты поглощения несколько
выше. Фабиан и др. [46], приняв также во внимание
25 Л. Беллами
386
Часть III
данные по спектрам комбинационного рассеяния, указывают
для оксимов в растворе интервал частот 1684—1652 см"1.
Имины проявляют, по-видимому, аналогичные свой-
ства. Для несопряженных соединений Пиккард и Полли [50]
указали интервал частот поглощения 1640—1633 см-1.
Однако эти данные относятся, вероятно, к образцам в кон-
денсированной фазе, и в случае растворов частоты погло-
щения, по-видимому, несколько выше и попадают в интер-
вал частот 1680—1660 с.и1. Для исследованных в твердом
виде соединений с пятичленным циклом, содержащих экзо-
циклическую связь = NH, Бегалей и Элвидж [51] ука-
зывают частоту поглощения 1664 слГ1, что, вероятно,
несколько выше обычного значения. Когда к атому углерода
связи С = N присоединены одна или несколько групп NH,
как это имеет место в случае гуанидинов и родственных им
веществ, идентифицировать сколько-нибудь надежно свя-
зи С = N становится труднее. В общем, можно отметить,
что полосы поглощения проявляются в этих случаях при
частотах, несколько выше обычных. Для соединений этого
типа Либер, Леверинг и Паттерсон [13] приводят интервал
частот 1689—1657 см"1, а Пиккард и Полли отмечают,
что частоты сильно меняются в пределах 1718—1590 см"1.
Поглощение, соответствующее несопряженным связям
С = N в оксазолонах, также приходится на область 1683—
1668 см 1 [171. Таким образом, в тех случаях, когда связь
С = N является несопряженной и на атоме азота нет заря-
да, полосу валентных колебаний для ненапряженных сое-
динений всех типов можно ожидать в области 1680—1650 см"1
и при несколько меньших значениях частот для веществ
в твердом состоянии. Имеются, однако, и отклонения от
этого общего правила, особенно у гуанидинов и у соеди-
нений с напряженными циклами. У пиразолонов, напри-
мер [52], частота валентных колебаний С = N может быть
повышена до 1700 см"1. Однако, когда атом азота связи
С = N при замещении приобретает полярный характер,
характеристическая частота сильно меняется. Для ряда
соединений этого типа Гулден [30] привел интервал частот
поглощения 1659—1510 см"1; значительные изменения
в частотах колебаний С = N обнаружены также у хелат-
ных соединений металлов с некоторыми оксимами [311.
Направление сдвига полосы поглощения в таких случаях
Гл. 15. Соединения с ненасыщенными связями при атомах азота 387
частично зависит от природы заряда на атоме азота. У сое-
динений с группой N+= С, например, частоты несколько
повышены по сравнению с обычными значениями. Леонард
и др. [53, 54] использовали это обстоятельство, применив
его для дифференциации а,|3-ненасыщенных третичных ами-
нов, прослеживая повышение частот колебаний С = N+(^
при образовании солей, способных существовать в раз-
личных таутомерных формах.
Влияние сопряжения на эту частоту вполне аналогич-
но влиянию на частоту С == С, хотя происходящие изме-
нения несколько меньше. Ацетилазин [19], имеющий две
сопряженные связи С = N, поглощает при 1664 слГ1,
но в случае азина кротонового альдегида [19] интерпрета-
ция полос поглощения более сложна, так как наряду с дву-
мя связями С = С он имеет также две связи С = N. Однако
и в этом случае наиболее сильная полоса поглощения на-
ходится при 1650 см \ ct-Фурилазин поглощает при 1635 см'1.
Когда сопряженную систему образуют ароматические цик-
лы, эффект сопряжения опять-таки невелик. Частота коле-
баний С = N для растворов лежит в пределах [45] 1660—
1630 см'1; несколько меньшие значения характерны для
соединений в твердом и в ассоциированном состояниях,
как это имеет место в случае ароматических альдоксимов.
Наличие второго ароматического кольца с другой стороны
от связи С — N приводит к уплотнению частот у низко-
частотного края указанного интервала. Влияние сопряже-
ния на поглощение С = N у оксазолонов также было под-
робно изучено в связи с исследованиями пенициллина.
Группой американских исследователей [17] установлено,
что несопряженные оксазолоны поглощают в интервале
1683—1668 см'1 и что частота поглощения при сопряжении
с ароматическими радикалами понижается только до
1657—1641 см'1. Аналогичные результаты получены груп-
пой английских исследователей [18], которые обнаружили,
что даже у оксазолонов, у которых связь С = N с одной
стороны сопряжена с ароматическим ядром, а с другой
стороны — со связью С = С, полоса поглощения С = N
расположена в интервале 1672—1634 см \ несмотря на то
что частоты других колебаний цикла значительно при этом
меняются.
25*
388
Часть II
б. Сопряженные циклические системы. Как указывалось
выше, у сопряженных циклических систем полосы погло-
щения связей С = N нельзя рассматривать изолированно,
так как эти связи взаимодействуют с другими двойными
связями гораздо сильнее, чем в случае ациклических сое-
динений. Кроме того, у таких систем могут возникать боль-
шие сдвиги полос поглощения как связи С = С, так и связи
С = N, а поэтому дифференцировать их бывает - трудно
и лучше рассматривать имеющуюся группу полос погло-
щения в целом как специфическую для данного структур-
ного элемента.
В качестве типичного примера можно привести тиазолы.
Рендалл и др. [14] исследовали семь образцов соединений
этого типа, причем в интервалах 1634—1570 и 1538—
1493 см'1 были обнаружены полосы поглощения, которые
авторы считают типичными для тиазольной структуры.
Аналогично этому бензтиазолы имеют две полосы поглоще-
ния в интервалах 1647—1513 и 1529—1473 см'1. В таких
случаях в колебаниях участвует также ароматическое
кольцо, и для аналитических целей систему следует рас-
сматривать как единое целое. Это относится в какой-то
мере также к триазинам и сходным с ними соединениям,
исследованным Рузенсом [20]. У тиазолинов, исследован-
ных Оттингом и Дровертом [55], частота поглощения С = N
в случае несопряженных соединений достигает приблизи-
тельно 1640 сл1 1 и падает примерно до 1610 см'1 (твердое
вещество) при наличии ароматического сопряжения. Одна-
ко сопряжение со связью С = С приводит к появлению
сложных полос поглощения. Обнаружено до 4 полос погло-
щения, среди которых полосы примерно 1640 и 1570 см'1
относятся соответственно к антисимметричным и симме-
тричным валентным колебаниям системы — С = С — C=N.
Вторым классом сопряженных циклических систем,
содержащих связь С = N, являются соединения, у кото-
рых сопряжение приводит к полному исчезновению соот-
ветствующих полос поглощения. Либер и др. [13] сооб-
щили о таких явлениях для многих тетразолов, у которых
не удалось обнаружить полос поглощения С = N и N = N,
хотя у исследованного ими простого триазола при 1626 см'1
наблюдалась сильная полоса поглощения, которую они
отнесли к связи С = N.
Гл. 15. Соединения с ненасыщенными связями при атомах азота 389
Ясно поэтому, что корреляции, предлагаемые для связи
С = N, можно применить к сопряженным циклическим
системам только после исследования достаточно большого
числа сходных веществ, а в настоящее время эти корреля-
ции нельзя с уверенностью применять для идентификации
группы С = N в неизвестных веществах.
в. Интенсивность полос поглощения C=N. В зави-
симости от природы заместителей интенсивность полос
поглощения С = N— сильно меняется. Кросс и Ролф [121,
отметили, что у оксимов поглощение чрезвычайно слабое,
но даже его интенсивность меняется в два раза у несопря-
женных соединений. С другой стороны, Гулден [30] отме-
тил, что у иминотиоэфиров интенсивность поглощения
— С == N— сравнительно велика, а у Ы,Ы'-диметилбенз-
амида — чрезвычайно велика [56]. Фабиан и Легранд
[56] сравнили коэффициенты погашения полос поглощения
— С = N — у различных соединений. У такого соедине-
ния, как N-пропилиденпропиламин, значение е в растворе
в четыреххлористом углероде достигает 140. Замена одной
или обоих алкильных групп ароматическими циклами при-
водит к повышению этой величины почти до 180. В то же
время у таких оксимов, как оксим циклогексанона, значе-
ние е при тех же условиях составляет 20 и уменьшается
в случае оксима ацетофенона до величины меньшей 8,5.
Замещение атомом серы при атоме углерода двойной
связи повышает коэффициент погашения до 218 (при нали-
чии сопряжения £=270), в то время как замещение атомом
азота приводит к еще большему эффекту. Для различных
замещенных бензамидинов значение е меняется в пределах
365—880. Присоединение атома кислорода к атому углерода
при двойной связи также дает значительный эффект; метил-
N-фенилбензимидат, например, характеризуется значе-
нием е = 670.
Хотя значения коэффициентов погашения были опре-
делены не на основании точных измерений интегральных
интенсивностей полос поглощения, уже сам порядок полу-
ченных величин, очевидно, настолько сильно меняется,
что определенные таким путем интенсивности полос могут
дать ценные сведения относительно ближайшего окружения
связи — С = N —.
390
Ч а с т ь III
5. ВАЛЕНТНЫЕ КОЛЕБАНИЯ —N = N-
Относительно полос поглощения в инфракрасной обла-
сти спектра, обусловленных связью — N = N —, имеется
мало сведений. По аналогии с поглощением групп — С = N —
и С = С можно было бы ожидать появления этой полосы
поглощения в области 1600 см'1, но она должна быть, по-ви-
димому, слабой в случае несопряженных систем и даже от-
сутствовать в случае симметричных структур.
Герцберг [21] указывает на сильную полосу поглощения
в спектре комбинационного рассеяния азометана при
1575 см'1, которую он относит к валентным колебаниям
— N = N —, а Либер, Леверинг и Паттерсон [13] обна-
ружили у четырех азосоединений полосы поглощения
в интервале 1631—1613 см'1. Однако идентификацию полос
поглощения данными авторами нельзя считать надежной,
так как у двух из этих соединений в указанном интервале
имеются по две полосы поглощения, а поскольку в сое-
динениях присутствует также группа С = N, то дифферен-
циация этих полос затруднительна. Указанные авторы
приводят также спектры многих тетразолов; в этих спект-
рах не имеется полос поглощения, которые можно было бы
отнести к связи — N = N —, хотя присутствие последней
не вызывает сомнений. Лефевр и др. [60] исследовали также
частоты колебаний связи — N — N в цикле у формазанов.
Нами исследовано небольшое число ароматических диазо-
соединений, но ни в одном случае не удалось наблюдать
сколько-нибудь отчетливых полос поглощения в ожидаемом
интервале частот, за исключением полос, характерных для
самих сопряженных ароматических систем. Поскольку все
эти соединения в какой-то мере симметричны, то возможно,
что полосы поглощения — N = N — либо слишком слабы,
чтобы их можно было заметить, либо замаскированы более
интенсивными полосами поглощения связей С = С цикла.
С той же трудностью столкнулись Лефевр и др. [34, 57],
исследовавшие большое количество ароматических диазо-
соединений. У всех соединений они обнаружили полосы
поглощения при 1406 + 14 и 1577 i 8 см'1; хотя последняя
полоса была отнесена ими к связи — N = N —, авторы
указывают, что она вполне может быть обусловлена ко-
лебаниями кольца. Возможность наложения полос этих
Гл. 15. Соединения с ненасыщенными связями при атомах азота 391
колебаний, очевидно, ограничивает пригодность корреляции,
предложенной для связи — N = N —, которая в против-
ном случае могла бы найти широкое применение при иссле-
довании азокрасителей. Такая же трудность встретилась
в работе Долинского и Джонса [58], которым не удалось
обнаружить в спектрах поглощения ароматических азосо-
единений полосы 1577 и 1408 см'1. Ими сделано, однако,
интересное заключение об аномальном поведении о-окси-
и аминоазосоединений, отличающем их от п-замещенных
соединений. На основании полученных данных авторы
пришли к заключению, что указанные соединения содер-
жат биполярные ионные структуры с группой — N= NH—.
Хаджи [59] продолжил эти исследования, используя дей-
терированные соединения. Он сделал вывод, что при этом
образуются сложные смеси таутомеров, включая гидра-
зоны, тогда как такие соединения, как 4-фенилазо-1 -наф-
тол и о-метилпроизводное 1- и 4-фенилазофенола присут-
ствуют в форме истинных — N = N — соединений. Он,
однако, не смог определить какие-либо характеристиче-
ские частоты колебаний — N = N — ни в одном из этих
соединений.
Малая интенсивность полосы поглощения — N = N —
и то обстоятельство, что она находится, вероятно, в обла-
сти 1600 см'1, где наблюдается поглощение ароматических
соединений, препятствуют, таким образом, установлению
каких-либо приемлемых корреляций для этой группы.
У такого азоксисоединения, как гексафторазоксиметан,
поглощение, обусловленное группой^ С — N = N — С Х
z 4 х
о
[61], наблюдается для колебаний N = N при 1570 см'1 и
для колебаний N —> О при 1270 см'1.
Возможно также, что существует характеристическая
частота колебаний скелета — С — N = N — С —. Тетлоу
[23] показал, что как у цис-, так и у /п/аис-азобензолов
имеется полоса поглощения при 927 см'1, которая отсут-
ствует у гидразобензола, и что у этой полосы обнару-
жен фильтр-эффект Христиансена только в случае транс-
формы, что может указывать на ее связь с колебаниями
скелета в направлении максимальной поляризуемости.
392
Ч а с т ь III
6. ДИАЗОГРУППА
Диазометан представляет линейную молекулу следую-
щего строения: СН2 = N = N и поглощает при 2101 см'1.
Отнесение этой полосы поглощения к валентным колебаниям
подтверждается подробнейшими исследованиями двух диа-
зоцианидов [32, 33]. Недавно Арони и др. [62] и Ветсел
и др. [63] изучили соли диазония. Этими группами иссле-
дователей изучено в общей сложности более 50 солей арил-
диазония. По данным Арони и др. [62], частота характери-
стических колебаний—CN* равна 2261 ±20 см1. Ветсел
[63] и др. подтверждают эти данные, несколько расширяя
интервал частот. Последняя группа авторов исследовала
влияние на эту частоту колебаний как анионов, так и ка-
тиона диазония. Замена аниона приводит лишь к незна-
чительным смещениям полосы, но замещения в цикле груп-
пами с сильными электроноакцепторными свойствами ока-
зывают сильное влияние на частоту поглощения. Диазо-
ниевые соли n-нитроанилина, например, поглощают при
2294 см'1, тогда как соответствующие п-диэтиламинопро-
изводные поглощают при 2151 см'1. В предельном случае
диазофенола частота уменьшается до 2110 см'1. Однако
Лефевр и др. [64] показали, что и диазофенолы и диазо-
нафтолы существуют фактически в форме диазидов хинона,
а поэтому уменьшение частоты из-за возросшего резонанса
вполне понятно. Область частот поглощения для соединений
этого типа в зависимости от природы и места замещения
других циклических групп составляет 2173—2014 см'1.
Высокая частота этой полосы поглощения типична для
структур X = X = X, и поэтому ее не всегда удается
легко отличить от полосы изоцианатов и других аналогич-
ных соединений. Однако обнаружение этой полосы в спектре
природного азосерина привело недавно к успешному синтезу
[65] этого вещества.
7. АЗИДЫ
Аз иды могут быть легко идентифицированы по сильной
полосе поглощения, соответствующей антисимметричным
валентным колебаниям N = N = N, которая всегда наблю-
дается в спектрах вблизи 2130 см'1. Соответствующие сим-
Гл. 15. Соединения с ненасыщенными связями при атомах азота 393
метричные колебания имеют значительно меньшую частоту.
Эта полоса поглощения не только намного слабее, но и
гораздо менее постоянна по частоте, а поэтому находит
сравнительно меньшее применение для аналитических
целей.
Спектр азотистоводородной кислоты исследован Эйсте-
ром [24], который отнес полосу поглощения при 2141 см'1
к антисимметричным колебаниям N = N — N, а полосу
при 1269 см'1— к соответствующим симметричным коле-
баниям. Хорошо изучен также метилазид [25], у которого
соответствующие полосы находятся при 2141 и 1351 см'1.
Систематические исследования азидов проведены Шейн-
кером и Сыркиным [26], которые изучили спектры ком-
бинационного рассеяния азида натрия и еще шести соеди-
нений. Во всех случаях эти авторы обнаружили сильную
полосу в интервале 2167—2080 см 1 и вторую, более слабую
полосу в интервале 1343—1177 см1. В согласии с данными
более ранних работ эти полосы отнесены соответственно
к антисимметричным и симметричным колебаниям. Сле-
дует, однако, заметить, что некоторые неорганические азиды
имеют простую линейную структуру, так что частота
симметричных колебаний неактивна в инфракрасном спектре
поглощения. У кристаллов азида аммония, например,
наблюдается только полоса 2050 см'1, а полоса, соответ-
ствующая симметричным колебаниям, отсутствует [66].
Дальнейшее подтверждение этих результатов получено
в работе Бойера [27], а также Либера, Леверинга и Паттер-
сона [13]. Бойер исследовал инфракрасные спектры погло-
щения продуктов взаимодействия азотистоводородной кис-
лоты и соединений с сопряженными связями. Всего было
изучено 10 азидов и установлено, что все они сильно погло-
щают вблизи 2128 см'1. Частота поглощения оказалась не
очень чувствительной к изменениям структуры молекулы;
обнаружено, что 1 -фенил-1 -азидо-2-нитроэтан, например,
поглощает при той же частоте, что и триазоацетон.
Либер и др. [13] исследовали повторно азид натрия и
еще пять ранее не исследованных веществ. Они сообщили
о наличии во всех случаях сравнительно сильной полосы
поглощения в интервале 2151—2128 см'1 и второй, более
слабой и менее постоянной по частоте полосы поглощения
близи 1282 см'1. Из данных этой работы, по-видимому,
394
Ч а с т ь III
следует, что Шейнкер и Сыркин [26] указали для частот
антисимметричных колебаний более широкий интервал
частот, чем это обычно наблюдается, и что это поглощение
практически можно ожидать в интервале частот 2160—
2120 см1. Эти данные по существу подобны данным для
диазо-, изоциановых и других аналогичных групп.
Спектры комбинационного рассеяния неорганических
азидов исследовали Каховец и Кольрауш [28]. Полученные
ими результаты согласуются с данными исследования
инфракрасных спектров, поскольку они показывают, что
частоты, характерные для различных катионов, заклю-
чаются в пределах узкого интервала.
ЛИТЕРАТУРА
1. Reitz, Sabathy, Monatsh., 71, 100 (1938).
2. Reitz, Sabathy, Monatsh., 71, 131 (1938).
3. R e i t z, S k r a b e 1, Monatsh., 70, 398 (1937).
4. S n у d e r, E 1 i e 1, J. Amer. Chem. Soc., 70, 1857 (1948).
5. Marvel, Brace, Miller, Johnson, J. Amer.
Chem. Soc., 71, 34 (1949).
6. Marvel, Brace, J. Amer. Chem. Soc., 71, 37 (1949).
7. Holmstedt, Larsson, Acta Chem. Scand., 5, 1179
(1951).
8. Richardson, Brigh t-W i 1 s о n, J. Chem. Phys., 18,
155 (1950).
9. К i t s о n, Griffith, Analyt. Chem., 24, 334 (1952).
10. Magat, Chandler, Faris, Reith, Salisbury,
J. Amer. Chem. Soc., 73, 1031 (1951).
11. Mellon, Analytical Spectroscopy, Wiley, 1950.
12 Cross, Rolfe, Trans. Faraday Soc., 47, 354 (1951).
13. Lieber, Levering, Patterson, Analyt. Chem.,
23, 1594 (1951).
14. Randall, Fowler, Fuson, Dangl, Infra-red De-
termination of Organic Structures, Van Nosfrand, 1949.
15. Barnes, Gore, Liddel, Van Zandt Willi-
a m s, Infra-red Spectroscopy, Reinhold, 1944.
16. Thompson, Brattai n, Randall, Rasmus-
sen, The Chemistry of Penicillin, Princeton University
Press, 1949, p. 382.
17. Rasmussen, Brattai n, The Chemistry of Penicillin,
Princeton University Press, 1949, p. 400.
18. Thompson, The Chemistry of Penicillin, Princeton Univer-
sity Press, 1949, p. 387.
19. В 1 о u t, F i e 1 d s, К a r p 1 u s, J. Amer. Chem. Soc., 70,
194 (1948).
Гл. 15. Соединения с ненасыщенными связями при атомах азота 395
20. Roosens, Bull. Soc. Chim. (Belg.), 59, 377 (1950).
21. Г e рцбер г Г., Колебательные и вращательные спектры
многоатомных молекул, Издатинлит, М., 1949, стр. 386.
22. Crawford, Fletcher, Ramsay, J. Chem. Phys.,
19, 406 (1951).
23. T e t 1 о w, Research, 3, 187 (1950).
24. E у s t e r, J. Chem. Phys., 8, 135 (1940).
25. Eyster, Gillette, J. Chem. Phys., 8, 369 (1940).
26. Ill e й н к e p IO. H., Сыркин Я. K-, Изв. АН СССР
сер. физ., 14, 478 (1950).
27. Boyer, J. Amer. Chem. Soc., 73, 5248 (1951).
28 Kahovec, Kohlrausch, Mh. Chem., 77, 180 (1947).
29. McBride, Beachell, J. Amer. Chem. Soc., 74, 5247
(1952).
30. G о u 1 d e n, J. Chem., Soc., 1953, 997.
31. D u у c k a e r t s, Bull. Soc. Roy. Sci. Liege, 21, 196
(1952).
32. Sheppard, Sutherland, J. Chem. Soc., 1947, 453.
33. Anderson, Le Fevre, Savage, J. Chem. Soc.,
1947, 443.
34. Le Fevre, O’ Dwyer, Werner, Chem. and Ind.,
1953 , 378.
35. F e 1 t о n, Orr, J. Chem. Soc., 1955, 2170.
36. Skinner, Thompson, J. Chem. Soc., 1955, 487.
37. W e i b 1 e n, Fluorine Chemistry, Vol. 2, Academic Press,
New York, 1954.
38. L i p p e r t, Vogel, Zeit. Phys. Chem., 9. 133 (1956).
39. Williams, J. Chem. Phys., 25, 656 (1956).
40. S e n s i, Gallo, Gazz. Chim., 85, 224, 235 (1955).
41. Hoyer, Chem. Ber., 89, 2677 (1956).
42. Davison, J. Chem. Soc., 1953, 3712.
43. К h о r a n a, Can. J. Chem., 32, 261 (1954).
44. M e a k i n s, Moss, J. Chem. Soc., 1957, 993.
45. К h о r a n a, Chem. Reviews, 53, 145 (1953).
46. Fabian, Legrand, Poirier, Bull. Soc. Chim. Fr.,
1956, 1499.
47. Джонс H., Сандорфи K-, Применение спектро-
скопии в химии, Издатинлит, М., 1959, стр. 429.
48. Califano, Liittke, Zeit. Phys. Chem., 6, 83 (1956).
49. Palm, W e r b i n, Can. J. Chem., 31, 1004 (1953).
50. Pickard, Polly, J. Amer. Chem. Soc., 76, 5169 (1954).
51. В a g u 1 e у, E 1 v i d g e, J. Chem. Soc., 1957, 709.
52. Gagon, Boivin, McDonald, Yaffe, Can. J.
Chem., 32, 823 (1954).
53. Leonard, Gash, J. Amer. Chem. Soc., 76, 2781 (1951).
54. Leonard, Thomas, Gash, J. Amer. Chem. Soc., 77,
1552 (1955).
55. Otting, Drawer t, Chem. Ber., 88, 1469 (1955).
56. Fabian, Legrand, Bull. Soc. Chim. Fr., 1956, 1461.
57. Le Fevre, O’Dwyer, Werner, Austral. J. Chem.,
6, 341 (1953).
396
Часть III
58. D о 1 i n s k y, Jones, J. Assoc. Off. Agric. Chem., 37, 197
(1954).
59. Н adzi, J. Chem. Soc., 1956, 2143.
60. L e Fevre, Sousa, Werner, Austral. J. Chem., 9, 151 (1956).
61. J ander, Haszeldine, J. Chem. Soc., 1954, 919.
62. А roney, Le Fevre, Werner, J. Chem. Soc., 1955, 276.
63. Whetsei, Hawkins, Johnson, J. Amer. Chem. Soc., 78, 3360 (1956).
64. L e Fevre, Sousa, Werner, J. Chem. Soc., 1954, 4686.
65. F usari, Frohardt, Ryder, Haskell, J о ha-
nessen, Elder, Bartz, J. Amer. Chem. Soc., 76, 2878 (1954).
66. D о w s, Whittle, Pimentel, J. Chem. Phys., 23, 1475 (1955).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Шигорин Д. Н., Сыркин Я. К., ЖФХ, 23, 24 (1949).
Шигорин Д. Н., ЖФХ, 25, 798 (1951); 29, 1033 (1955).
Глава 16
ГЕТЕРОЦИКЛИЧЕСКИЕ АРОМАТИЧЕСКИЕ
СОЕДИНЕНИЯ
1. ВВЕДЕНИЕ
Колебания колец бензола очень сходны с колебаниями
колец пиридина и хинолина, деформационные же колебания
связей водорода у них значительно отличаются. Однако
внеплоскостные деформационные колебания связей водо-
рода у пиридина и хинолина, по-видимому, аналогичны
соответствующим колебаниям бензольных соединений с лиш-
ним заместителем. Так например, а-монозамещенный пири-
дин ведет себя ц-этом отношении подобно орто-дизамещен-
ному бензолу. Поскольку исследовано не очень большое
число пиридинов или хинолинов, то ни один из этих об-
щих выводов нельзя применять без соответствующих огово-
рок или без подтверждения их правильности на аналогич-
ных по строению соединениях.
Пиримидины и пурины изучались гораздо шире в связи
с тем, что они представляют интерес как соединения, важ-
ные с биологической точки зрения. Однако исследовались
главным образом окси- и аминопроизводные, которые
могут существовать более чем в одной форме. Во всяком
случае, до тех пор пока не будут получены дополнитель-
ные данные, свидетельствующие о существовании в этих
соединениях гетероцикла ароматического типа, нельзя на
основании имеющихся в настоящее время сведений сделать
общие выводы.
Число данных о других типах пиримидинов более огра-
ниченно; однако они все же показывают, что эти соедине-
ния также сходны с бензолом в отношении валентных
колебаний С — Н и колебаний кольца, но, как и сле-
довало ожидать, сильно отличаются от него по частотам
деформационных колебаний связей водорода.
Указанные корреляции представлены в табл. 15.
398
Часть III
Таблица 15
Частота, см~1
Пиридины и хинолины
Валентные колебания СН . . . .
Валентные колебания С = С и
C = N
То же
Колебания кольца; деформацион-
ные колебания СН............
То же
То же
То же
Пиримидины
Валентные колебания СН . . . .
Валентные колебания С = С, С =N
Колебания кольца; деформаци- Г
онные колебания СН . . . . 1
Вблизи 3020 (с.)
1660—1590 (иногда имеется
вторая полоса с более низкой
частотой) (ср.)
Вблизи 1500
Вблизи 1200 (с.)
1100—1000 (с.)
900-650 (с.)
Вблизи 710 (с.) (дополнитель-
ная полоса к полосе, указан-
ной выше) (с.)
3060—3010 (с.)
1580—1520 (ср.)
1000—960 »
825—775 »
2. ПИРИДИНЫ И ХИНОЛИНЫ
а. Общие сведения. О характеристических полосах
поглощения ароматических гетероциклических систем
имеется мало данных. Клайн и Туркевич [1], а также Кор-
син и др. [21] показали, что колебания кольца пиридина
очень сходны с соответствующими колебаниями бензола,
но что деформационные колебания связей водорода
у этих соединений сильно различаются. Когда полосы,
соответствующие деформационным колебаниям, удается
обнаружить, то в большинстве случаев можно заметить,
что при переходе от бензола к пиридину наблюдается сдвиг
полосы поглощения в область более низких частот. Более
или менее детально исследованы также полностью или ча-
стично дейтерированные соединения [23]. Кеннон и Сазер-
ленд [2] рассмотрели корреляции, которые можно устано-
Гл. 16. Гетероциклические ароматические соединения 399
вить на основании полученных ими данных о спектрах и
у-пиколинов и 2,6-лутидина; спектр а-пиколина исследован
Фрейзером и Гловацким [3] и включен в серию спектров
Американского нефтяного института (АНИ). Спектры пи-
колинов рассмотрены также Робертсом и Шварком [4].
Данные по отдельным участкам спектров замещенных про-
изводных пиридина получены Марионом, Рамсаем и Джон-
сом [5] при исследованиях алкалоидов, Коулсоном, Хейл-
сом и Херингтоном [6] в связи с работой по определению
воды и Деншемом, Ленгстоном и Гоухом [8] в работе по
определению пиридина. Хинолины изучены плохо; имеются
данные АНИ по спектру изохинолина, а Хеннаном и др. [7]
опубликованы спектры пяти тиолов хинолина. Частоты
внеплоскостных деформационных колебаний СН у заме-
щенных пиридинов рассмотрены Шиндо и Икекава [26],
а валентные колебания СН — Таллентом и Зиверсом [27].
Довольно большое сходство колебаний кольца произ-
водных пиридина и бензола, которое стало очевидно из
этих исследований, подтверждает в какой-то мере предпо-
ложительно выдвинутые корреляции, обсуждаемые ниже.
Но при любом использовании этих корреляций нельзя забы-
вать, что они основываются лишь на очень ограниченном
количестве данных, главным образом по алкилпроизводным.
б. Валентные колебания СН. Имеющиеся немногие
данные подтверждают предположение о том, что валент-
ные колебания С — Ну пиридинов и хинолинов, по суще-
ству, подобны таким же колебаниям у бензола. Пиридин
и пиколины имеют полосы поглощения СН в интервале
3070—3020 см 1 [1, 4—6], которые при более высоком
разрешении разделяются на ряд отдельных полос [25, 27].
Их положение и число меняются при наличии замещающих
групп, но частоты полос в каждом таком случае оказы-
ваются очень близки к частотам соответствующих бензоль-
ных гомологов. Марион и др. [5] обнаружили у анабазина
полосу поглощения вблизи 3030 см'1, которая отсутствует
в спектре пиперидина и которая относится к валентным
колебаниям С — Н пиридинового цикла. Мы нашли, что
у производных хинолина также имеется несколько полос
поглощения вблизи 3030 см'1, но их и следовало ожидать
в связи с наличием углеродного цикла. Как и для бензола,
400
Часть III
в этой области обнаружены также другие, более слабые,
полосы поглощения, однако о них имеется мало сведений.
Для пиридина, например, обнаружена слабая полоса погло-
щения с частотой 3420 см1, которая находится в области
валентных колебаний NH [6]. Многие гетероциклические
соединения поглощают в области более низких частот
в интервале 2830—2790 слг1 [27]. Это поглощение отсут-
ствует в случае производных бензола и может быть исполь-
зовано для идентификации некоторых веществ. Следует,
однако, помнить, что многие кислородсодержащие соеди-
нения, такие, как диизопропиловый эфир, также погло-
щают примерно в этой области спектра.
в. Колебания С = С и С = N. Взаимодействия между
колебаниями С = С и С = N пиридинового цикла сходны,
по-видимому, с аналогичными взаимодействиями колебаний
бензола и приводят к появлению двух полос поглощения,
находящихся одна от другой на расстоянии 100 слг1 в об-
ласти несколько меньших частот, чем в случае бензола.
У многих соединений полосе поглощения с более высокой
частотой сопутствует еще одна полоса с меньшей частотой
[2]. Она не связана, по-видимому, с внешним сопряжением
цикла, как это имеет место в случае бензола, но вид этой
полосы нередко весьма сходен с видом полос поглощения
такого рода структур, так что при исследованиях гетеро-
циклических соединений легко можно допустить ошибку,
спутав полосы поглощения производных пиридина с по-
лосами сопряженных ароматических соединений.
Сам пиридин поглощает при 1580, 1570 и 1485 см~1.
В спектрах пиколинов и 2,6-лутидинов имеются полосы
поглощения в интервале 1600—1590 слг1 [2] и вблизи
1500 слг-1, а различные алкалоиды, содержащие пиридино-
вые или хинолиновые циклы, также поглощают между 1600
и 1560 слг-1 [5]. Анабазин, например, поглощает при 1592
и 1576 слг-1, но эти полосы поглощения отсутствуют в спект-
ре пиперидина. Положение этих полос поглощения у ряда
алкилпиридинов определено также Шиндо и Икекава [26]
и Куком и Черчем [41], которые обсудили изменения отно-
сительной интенсивности полос при изменениях положения
замещающих групп. Эти данные, однако, мало что дают для
целей качественного анализа.
Гл. 16. Гетероциклические ароматические соединения 401
В случае хинолина и изохинолина возможны более •
сложные взаимодействия; сам изохинолин имеет три полосы
поглощения между 1600 и 1500 слг1, тогда как многие ди-
метил хинол ины, исследованные нами, имеют в этой области
четыре полосы. Спектр поглощения в общем сходен со
спектром пиридина и отличается в большинстве случаев
тем, что в нем имеет место наложение одной или большего
числа полос. У 8-оксихинолина главные полосы поглощения
расположены при 1577 и 1495 см'1 и гораздо более слабые
полосы — при 1563 и 1504 см'1. Тиолы хинолина, рассмот-
ренные Хеннаном и др. [7], обычно имеют сравнительно
простую пару полос поглощения вблизи 1600 см'1, но во
всех этих случаях, за исключением одного, существует
возможность таутомерии с тиокетонной формой. Интересно
отметить, что для одного соединения, представляющего
исключение, в котором тиольная группа замещена, наблю-
дается более сложный спектр поглощения, состоящий из
четырех сильных полос между 1600 и 1500 см'1.
г. Колебания кольца и деформационные колебания
С — Н. Отнесение частот в случае замещенных пиридина
и хинолина. У замещенных производных пиридина обна-
ружен целый ряд полос поглощения, которые могут ока-
заться полезными при идентификации этого типа струк-
туры. Кеннон и Сазерленд [2] установили, что у пиридина
и исследованных ими производных имеется сильная полоса
поглощения вблизи 1200 см'1 и еще одна полоса между
1100 и 1000 см'1. В спектре 2-метилпиридина, согласно
данным АНИ, также имеются две полосы поглощения
при 1240 и 1043 см'1, причем вторая полоса особенно интен-
сивна. Она связана либо с колебаниями кольца, либо
с деформационными колебаниями связей водорода, так
что вряд ли эта корреляция применима для ряда хиноли-
нов. Ни у одного из исследованных нами замещенных хино-
линов не наблюдалось в этих интервалах частот сильной
полосы поглощения..
Остается еще область характеристических частот
между 900 и 700 см'1, включающая частоты деформационных
колебаний СН. В случае замещенных бензола показано,
что наиболее интенсивная полоса поглощения в этом интер-
вале связана с внеплоскостными колебаниями незамещен-
26 л* Беллами
402
Часть III
ных атомов водорода в цикле, и главным фактором, опре-
деляющим частоту этих колебаний, является число таких
свободных атомов водорода, находящихся по соседству
друг с другом. Аналогичный эффект можно ожидать в слу-
чае производных пиридина, так что спектр поглощения пи-
ридина с пятью свободными атомами водорода должен был
бы походить в этой области частот на спектр поглощения
замещенного бензола, тогда как у-пиколин, содержащий две
пары свободных атомов водорода, должен в этом отноше-
нии походить на n-дизамещенный бензол. Насколько можно
судить по имеющимся ограниченным данным, это предпо-
ложение подтверждается на практике. Так, например,
поглощение пиридина [2] при 750 см"1, а-пиколина |3] при
755c.tr1, Р-пиколина при 790 см"1 и у-пиколина при 800 см"1
сходно с картиной поглощения соответственно моно-,
орто-, мета- и пара-замещенных бензола. Эта корреляция
с небольшими отклонениями [26] выполняется также для
8 этилпиридинов с различными заместителями и 19 заме-
щенных метилпиридинов. Аналогичные результаты получены
Куком и др. [41 ], которые также отметили характеристиче-
ское поглощение в области 2080—1650 см"1, подобное
поглощению бензольных систем.
В спектрах хинолиновых соединений полоса поглощения
710 см 1 не появляется. С другой стороны, корреляция,
относящаяся к внеплоскостным деформационным колеба-
ниям СН, если рассматривать каждый цикл отдельно,
хорошо выполняется. Так, например, и 2,6- и 2,7-диметил-
хинолины имеют только по два соседних свободных атома
водорода в каждом из двух циклов. Оба соединения очень
сильно поглощают соответственно при 831 и 835 см"1 (ана-
логично пара-замещенным ароматическим соединениям).
Кроме того, у обоих соединений в интервале 900—850 см"1
имеется вторая, более слабая полоса поглощения, которую,
возможно, следует отнести к колебаниям еще одного, от-
дельно расположенного в цикле атома водорода.
2,3- и 2,4-Диметилхинолины содержат четыре свобод-
ных атома водорода в углеродном цикле и только по одному
в гетероцикле. У этих соединений в рассматриваемой обла-
сти имеются очень интенсивные полосы поглощения соот-
ветственно при 755 и 758 см"1 (аналогично ор/по-замещен-
ным). Оба соединения поглощают также в интервале
Гл. 16. Гетероциклические ароматические соединения 403
900—850 см1. Аналогичным образом изохинолин (спектр
АНИ) имеет в этой области очень сильную полосу погло-
щения при 745 см'1, которая может быть обусловлена коле-
баниями четырех атомов водорода в углеродном цикле.
Возможно, что вторая сильная полоса поглощения при
829 см 1 в этом случае связана с наличием двух соседних
незамещенных атомов водорода гетероцикла, а полоса при
864 см'1—с наличием еще одного незамещенного атома
водорода. На этом основании можно было предположить,
что 8-оксихинолин окажется сходным с мета-замещенным
ароматическим соединением, содержащим по три соседних
незамещенных атома водорода в каждом цикле. Хотя спектр
поглощения в этой области частот более сложен, чем в слу-
чае алкилпроизводных, тем не менее наиболее интенсивная
полоса поглощения, как и следовало ожидать, находится
при 780 см'1. У многих хиназолинов и хиназолонов [20]
также имеется полоса поглощения в интервале 780—740 см-1.
Удивительно, что эти корреляции так хорошо приме-
нимы для ряда производных хинолина, однако хорошее
соответствие спектров поглощения рассмотренных шести
соединений с ожидаемыми спектрами нельзя считать слу-
чайным. Не следует забывать при этом, что, как и в случае
ароматических соединений, замещение сильно полярными
группировками существенно изменяет эти частоты колеба-
ний СН.
д. Окси- и аминопиридины и хинолины. Строение таких
соединений, как 2-оксипиридины, представляет интерес
с той точки зрения, что у них возможны таутомерные пири-
доновые структуры. Результаты измерения дипольных мо-
ментов свидетельствуют в пользу предположения о том,
чтопиридоновая форма является исключительно благоприят-
ной. Эта точка зрения полностью подтверждается исследо-
ваниями инфракрасных спектров поглощения, проведен-
ными рядом авторов [28—31]. В этой форме существует не
только 2-окси-, но также и 4-оксипиридин и многие другие
родственные производные хинолина.
В то же время в случае аминопиридинов очень мало
или вообще нет доказательств образования иминов. 2-Ами-
нопиридин [32] в разбавленном растворе поглощает при
3500 и 3410 см'1, и, следовательно, в этом отношении подо-
26*
404
Часть 111
бен анилину; аналогичным образом ведет себя и 4-амино-
пиридин [33]. Беллами и Уильямсом [34] показано, что
эти частоты и соответствующие частоты других гетероцик-
лических аминов [35] подчиняются соотношению, устанав-
ливающему связь между антисимметричными и симметрич-
ными частотами колебаний у первичных аминов, а поэтому
не приходится сомневаться в подобии структур рассматри-
ваемых соединений.
3. ПИРИМИДИНЫ И ПУРИНЫ
а. Общие сведения. Вследствие биологического значе-
ния пиримидинов как компонентов нуклеиновых кислот их
исследованию посвящено очень много работ. Однако из-за
наличия большого числа таутомерных форм, которые можно
приписать некоторым замещенным пиримидинам (в том
числе таким соединениям, в которых циклы совершенно не
носят ароматического характера), еще невозможно пока
установить удовлетворительные корреляции для иденти-
фикации этого специфического класса соединений, и основ-
ное внимание в настоящее время направлено главным обра-
зом на выяснение реально существующих структур.
Впервые спектральные данные о пиримидинах были
опубликованы Блоутом и Филдсом [9—11], которые при-
вели спектры многих нуклеиновых кислот, пуринов и пи-
римидинов и показали, что урацил, по-видимому, суще-
ствует в виде кето-формы. Эти данные подтверждены Лаче-
ром, Кемпионом и Парком [12], которые исследовали
спектры поглощения урацила, 5-хлорурацила и тимина
в области обертонов. Блоут и Меллорс [13] также выяснили
возможности применения метода инфракрасной спектро-
скопии для исследования образцов тканей. Браунли, Са-
зерленд и Тодд [14] использовали метод инфракрасной
спектроскопии, чтобы показать наличие водородных связей
у некоторых гликозидаминопиримидинов, но они изучали
лишь область карбонильного поглощения. Данные о спект-
рах многих пиримидинов и пуринов собраны также Рен-
даллом и др. [15], а спектры их растворов в трихлориде
сурьмы приведены Лачером и др. [36].
Очень большое число замещенных пиримидинов изу-
чено в последнее время многими исследователями [16—18,
Гл. 16. Гетероциклические ароматические соединения 405
22, 37, 38]. Основные результаты, полученные этими авто-
рами, обсуждены ниже, но, как уже указывалось, нали-
чие прототропных изменений не позволяет в настоящее
время сформулировать никаких точных корреляций.
б. Валентные колебания СН. По аналогии с пиридином
и бензолом можно ожидать, что валентным колебаниям СН
у пиримидина и его производных должны соответствовать
полосы поглощения вблизи 3050 см~г. Шорт и Томпсон [18]
обнаружили в этой области группу полос поглощения для
самого пиримидина и наблюдали поглощение в этой области
также у большинства замещенных пиримидинов. Однако
у тризамещенных пиримидинов только с одним свободным
атомом водорода в цикле эти полосы поглощения очень
слабы, и поглощения нельзя ожидать для тетразамещенных
соединений. Браунли [16] также отметил наличие полос
поглощения в интервале 3650 — 3100 см1 у девяти триза-
мещенных пиримидинов и отсутствие такого поглощения
у тетразамещенных соединений.
в. Колебания С — С и С = N. У полностью аромати-
ческих пиримидиновых систем можно ожидать поглощения
в области 1600—1500 см~г, обусловленного эффектами
взаимодействия двойных связей циклов. У самого пири-
мидина [16—18] имеются две полосы поглощения, а именно
при 1570 и 1610 [18] или 1650 слг1 [15], возникновение
которых, вероятно, можно объяснить указанной выше при-
чиной. Браунли [16] сообщает о наличии полос поглоще-
ния в интервалах 1640—1620 и 1580—1560 слг1 в спектрах
всех исследованных им 24 замещенных пиримидинов, за
исключением двух образцов, имеющих только одну полосу
поглощения при 1590 слг1. Такие же выводы сделаны для
ряда пиримидинов Блоутом и Филдсом [11]. Не возникает
сомнений, что полоса поглощения с более низкой частотой
обусловлена колебаниями цикла; что же касается первой
полосы, то в правильности такого отнесения нет уверен-
ности, так как большинство соединений, исследованных
Браунли, содержит аминогруппу, частоты деформационных
колебаний которой вполне могут находиться в интервале
1650—1600 см’1. В связи с этим следует указать, что в при-
веденных Шортом и Томпсоном [18] спектрах для ряда
406
Часть III
замещенных пиримидинов, которые не содержат групп ОН
или NH2, имеется только одна полоса поглощения в интер-
вале 1580—1520 смТаннер [38] и Монтгомери [37] обнару-
жили полосу около 1650 слГ1 у аминопиримидинов, но
высказали предположение, что она вполне может соответ-
ствовать деформационным колебаниям группы NH2. Окси-
пиримидины также не являются подходящими объектами
для установления корреляций, касающихся таких колеба-
ний кольца, так как они могут существовать в виде тауто-
мерной кетоформы; в этом случае полосы поглощения двой-
ных связей цикла, возможно, имеют иные частоты, чем
в случае полностью ароматических систем. Поскольку ис-
следованные до настоящего времени пиримидины являются
главным образом либо амино-, либо оксипроизводными,
то корреляция для колебаний двойных связей кольца
пока не может быть установлена.
г. Колебания кольца. Шорт и Томпсон [18] обратили
внимание на тот факт, что все исследованные ими хлор-
и этоксизамещенные пиримидины поглощают около 990
и 810 слГ1. Первую полосу поглощения относят к колеба-
ниям кольца и предполагают, что вторая также обусло-
влена этими колебаниями, хотя нельзя исключить возмож-
ности, что она связана с деформационными колебаниями
связей СН. Полосы поглощения, соответствующие примерно
этим частотам, имеются у большинства из 39 амино- и окси-
производных, спектры которых приведены в работе [18].
Браунли [1б] нашел у всех исследованных им пиримидинов
полосу поглощения в интервале 1000—960 см~1, которую
он по не вполне ясным причинам относит к ножничным коле-
баниям С — NH2. Во всех случаях он обнаружил также
полосу поглощения в интервале 825—775 с.и-1, которую
отнес к деформационным колебаниям связей СН в случае
ди- и тризамещенных пиримидинов и к колебаниям скелета
в случае тетразамещенных соединений.
Постоянное присутствие этих полос в спектрах погло-
щения столь различных замещенных соединений, вероят-
но, не является случайным, но соответствующие корреля-
ции не могут, по-видимому, иметь большого значения до
тех пор, пока окончательно не будет установлено строе-
ние амино- и оксипиримидинов, и только после этого можно
Гл. 16. Гетероциклические ароматические соединения 407
будет объяснить причины отсутствия этих полос поглоще-
ния в отдельных случаях.
д. Окси- и аминопиримидины. Выше указано, что по
вопросу о строении амино- и оксипиримидинов, для которых
можно допустить существование таутомерных форм, еще
не имеется единой точки зрения, и доказательства, полу-
ченные с помощью инфракрасной спектроскопии, сами по
себе не являются бесспорными. Так, например, в области
3000 см1 происходит наложение полос поглощения, харак-
терных для связанных групп ОН и NH, что приводит
к определенным трудностям, тогда как в области 1600 см'1
могут появиться полосы, обусловленные деформационными
колебаниями связей NH, колебаниями карбонила и двой-
ных связей кольца.
Этот вопрос нет возможности рассмотреть здесь подроб-
но, но взгляды различных авторов можно резюмировать
следующим образом. Браунли [16] интерпретирует данные,
которые он получил методом инфракрасной спектроскопии,
как подтверждение существования этих соединений в имино-
и енольной формах, тогда как Томпсон, Николсон и Шорт
[17] отдают предпочтение кетонной структуре в случае
оксипроизводных и иминоструктуре — в случае амино-
производных. Обзор всех имеющихся данных сделан Шортом
и Томпсоном [18], которые считают, что для 2- и 4-оксипи-
римидинов, а, возможно, также и для 2,4-диоксипроизводных
полученные данные свидетельствуют в пользу кетонной
структуры, а что касается 4,6-диоксипиримидина,то он может
содержать одну кетонную и одну енольную группу. Таннер
[38J исследовал многие ди- и триоксипиримидины, и его
данные в общем подтверждают предположение, что наличие
замещения в положении 4 приводит к кетонным структурам,
тогда как присутствие оксигрупп в положении 5 или 6 ста-
билизирует имевшуюся первоначальную структуру. Пред-
полагается, что аминогруппы обычно не дают таутомерных
форм. Шорт и Томпсон [18], однако, указывают, что в от-
дельных случаях электронные эффекты различных замести-
телей могут привести к образованию таутомерных форм.Ука-
занные выводы в дальнейшем подтверждены результатами,
полученными Брауном и Шортом [22], которые исследовали
ультрафиолетовую и инфракрасную области спектра.
408
Часть III
4. ДРУГИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СТРУКТУРЫ
Ряд хиназолинов, хиназолонов и хиназолиндионов иссле-
дован Калбертсоном и др. [20]. Эти соединения имеют по
три полосы поглощения, обозначаемые как хиназолиновые
полосы I, II и III соответственно при 1628—1618, 1581 —
1566 и 1517—1478 см'1. Все полосы узкие, относительно
интенсивные и обусловлены, очевидно, колебаниями циклов.
Более детально эти соединения не охарактеризованы. Что
касается триазинов, т. е. таких соединений, как циану-
ровая кислота, меламин и т. п., то здесь возникают те же
трудности, что и при интерпретации спектров таутомерных
пиримидинов, причем о строении указанных соединений
известно значительно меньше. Рузенс [19] опубликовал
спектры поглощения циануровой кислоты, хлористого циа-
нурата, а также меламина и его гидрохлорида. Мы также
исследовали эти соединения, а кроме того, аммелин, тиоам-
мелин и аммелид, но имеющихся данных еще недостаточно,
чтобы установить какие-либо корреляции или получить
сведения о строении этих веществ. Возникают трудности
при интерпретации спектров в области 1700—1500 см 1
в случае аммелина и тиоаммелина. Как аммелин, так
и аммелид интенсивно поглощают при 1715 см-1, и это слу-
жит убедительным доказательством существования их в ви-
де кето-формы. Однако тиоаммелин поглощает также интен-
сивно при 1695 см'1, и в этом случае полоса поглощения
может быть обусловлена колебаниями кольца или пред-
ставляет собой смещенную полосу колебаний NH. Поэто-
му для этой группы соединений в настоящее время не уста-
новлено никаких надежных корреляций.
Оттинг [39] недавно исследовал 1,3,4-триазол и тет-
разол и показал, что оба эти соединения имеют в значитель-
ной мере характер ароматических соединений. Итоидр. [40]
изучали диазины и рассмотрели различия между частотами
поглощения пиразина, пиримидина и пиридазина. Последние
два соединения, например, поглощают при 1565 см1,ay
пиразина нет полос поглощения с частотой выше 1490 см1,
если не считать полосы валентных колебаний СН при
3054 см'1. Семичленные гетероциклические системы арома-
тического типа изучены также Димротом и Ленке [24], а
Финнегеном и др. [25] приведены спектры ряда 5-аминотет-
разолов, но никаких корреляций при этом не обсуждалось.
Гл. 16. Гетероциклические ароматические соединения 409
ЛИТЕРАТУРА
1. Kline, Tur kevitch, J. Chem. Phys., 12, 300 (1944).
2. Cannon, Sutherland, Spectrochimica Acta, 4, 373
(1951).
3. F r e i s e r,
(1948).
G 1 о w а с к i,
J. Amer. Chem. Soc., 70, 2575
4. Roberts, S z w a r c, J. Chem. Phys., 16, 981 (1948).
5. Marion, Ramsay, Jones, J. Amer. Chem. Soc., 73,
305 (1951).
6. Coulson, Hales, Herington, J. Chem. Soc., 1951,
2125.
7. Hannan, Lieblich, Renfrew, J. Amer. Chem.
Soc., 71, 3733 (1949).
8. Densham, Langston, Gough, J. Chem. Soc., 1952,
2433.
9. В 1 о u t, Fields, Science, 107, 252 (1948).
10. В 1 о u t, Fields, J. Biol. Chem., 178, 335 (1949).
11. В 1 о u t, Fields, J. Amer. Chem. Soc., 72, 479
(1950).
12. Lacher, Campion, Park, Science, 110, 300 (1949).
13. В 1 о u t, Mellors, Science, 110, 137 (1949).
14. В г о w n 1 i e, Sutherland, Todd, J. Chem. Soc.,
1948, 2265.
15.
16.
Randall, Fowler, Fuson,
termination of Organic Structures,
Brownlie, J. Chem. Soc., 1950,
D a n g 1, Infra-red
Van Nostrand, 1949.
3062.
De-
17. Thompson, Nicholson, Short, Discuss. Faraday
Soc., 9, 222 (1950).
18. S h о r t, Thompson, J. Chem. Soc., 1952, 168.
19. R о о s e n s, Bull. Soc. Chim. (Belg.), 59, 377 (1950).
20. Culbertson, Decius, Christensen, J. Amer.
Chem. Soc., 74, 4834 (1952).
21. С о r r s i n, Fox, Lord, J. Chem. Phys., 21, 1170
(1953).
22. Brown, Short, J. Chem. Soc., 1953, 331.
23. Anderson, Bak, Brodersen, Rastru p-A n d e r-
s e n, J. Chem. Phys., 23, 1047 (1955).
24. D i m г о t h, L e n к e, Chem. Ber., 89, 2608 (1956).
25. F i n n e g a n, Henry, Olsen, J. Amer., Chem. Soc.,
77, 4420 (1955).
410
Часть III
26. Shindo, I к e к a w a, Pharm. Bull. Japan, 4, 192 (1956).
27. T a 1 1 e n t, Siewers, Analyt. Chem., 28, 953 (1956).
28. Gibson, Kynaston, Lindsey, J. Chem. Soc., 1955,
4340.
29/Ramirez, Paul, J. Amer. Chem. Soc., 77, 1035
(1955).
30. Ш e й н к e p Ю. H., Резников В. M., ДАН СССР,
102, 109 (1955).
31. Ш е й н к е р Ю. Н., Померанцев Ю. И., ЖФХ, 30,
79 (1956).
32. Ш и г о р и и Д. Н., Данюшевский Я. Л., Г о л ь д -
фар б Я. Л., Изв. АН СССР, ОХН, № 1, 120 (1956).
33. Costa, Blasina, Sartori, Z. Phys. Chem., 7, 123
(1956).
34. Bellamy, Williams, Spectrochim. Acta, 9, 341 (1957).
35. Angyal, Werner, J. Chem. Soc., 1952, 2911.
36. L a c h e r, Bitner, Emery, S e f e 1, Park, J. Phys.
Chem., 59, 615 (1955).
37. M о n t g о m e r y, J. Amer. Chem. Soc., 78, 1928 (1956).
38. Tanner, Spectrochim. Acta, 8, 9, (1956).
39. Otting, Chem. Ber., 89, 2887 (1956).
40. Ito, Shimada, Kuraishi, Mizushima,
J. Chem. Phys., 25, 597 (1956).
41. Cook, Church, J. Phys. Chem., 61, 458 (1957),
Частота, см"'
3200 3000 2800 2600 2400 2200 1760 1600 1500 1400 1300 1200 1100 1000 000 800
% ‘эпна1по1/гоц
Рис. 16. dZ-Метионин.
411
Рис. 18. Гиппуровая кислота.
Sitz
Рис. 20. 1-п-Хлорфенил-3,3-диметилмочевина.
Частота, смГ
1700 1600 1500 1400 1300 1200 1100 1000 300
Рис. 19. Фенил ацетамид.
Частота, см"’
3400 3200 3000 1700 1600 1500 1400 1300 1200 1100 1000 300 800
Частота, см
1600 1500 MOO 1300 1200 1100 1000 000 800
90
80
70
°. 60
| 50
3
1«0
t
30
20
10
Рис. 21. 2-Нитродифеииламии.
Рис. 22. Дициандиамид,
ЧАСТЬ
IV
СВЯЗИ МЕЖДУ ДРУГИМИ
ЭЛЕМЕНТАМИ.
НЕОРГАНИЧЕСКИЕ
СТРУКТУРЫ
Глава 17
НИТРО- И НИТРОЗОСОЕДИНЕНИЯ, НИТРАТЫ
И НИТРИТЫ
1. введение
В этой главе рассматриваются соединения, содержащие
группировки R — NO2) R — NO и NO" а также окиси
аминов. У таких соединений, как оксимы, у которых связь
NO имеет характер простой одинарной связи, обуслов-
ленная ею частота колебаний переменна [34], а сами колеба-
ния, вероятно, связаны с другими колебаниями. В связи
с указанными особенностями этих колебаний они здесь
не рассматриваются.
Группу R — NO2 содержат ковалентные нитросоедине-
ния, ковалентные нитраты и нитрамины. Соединения каж-
дого из этих трех классов дают по две чрезвычайно сильные
полосы поглощения в интервалах 1650—1500 и 1350—
1250 слГ1, соответствующие антисимметричным и симмет-
ричным валентным колебаниям группы NO2. Частоты этих
полос зависят от природы группы R, а поэтому часто можно
дифференцировать соединения указанных трех основных
классов, несмотря на то что наблюдается перекрывание
характерных для них интервалов частот. Кроме того,
имеются некоторые сведения о наличии характеристиче-
ского поглощения в области более низких частот, обуслов-
ленного колебаниями скелета или деформационными колеба-
ниями.
Валентные колебания в основном не зависят от измене-
ний состояния вещества, за исключением тех нескольких
случаев, когда группа NO2 может связываться водородной
связью. Происходящее при этом изменение частоты никог-
да не бывает таким большим, чтобы полосы поглощения ока-
зались вне пределов указанных интервалов частот. Однако
что касается области более низких частот, то здесь при
изменении состояния вещества или кристаллической
формы часто происходят значительные изменения частоты,
422
4 a с'т ь IV
а потому имеющиеся данные нельзя считать вполне на-
дежными.
В случае соединений, содержащих группу R — N = О,
следовало бы ожидать появления лишь одной полосы
в области 1600 сл'1, но у нитритов она почти всегда дублет-
на в результате сосуществования цис - и транс-форм. Изу-
чая относительные интенсивности этих двух полос, часто
можно получить некоторые сведения о природе углеводо-
родного остатка R в R — О — N = О. Для соединений,
у которых нитрозогруппа присоединена непосредственно
к атому углерода или азота, положение полосы поглощения
N = О труднее определить, в частности, вследствие лег-
кости димеризации некоторых соединений этого класса.
Тем не менее в настоящее время приближенные значения
частот определены достаточно хорошо. Для нитритов, кроме
указанных полос, имеются еще две полосы в более низко-
частотной области; для нитрозосоедйнений такие полосы
не обнаружены.
Ионизованные нитраты имеют сильную полосу с цент-
ром вблизи 1380 сл’1 и полосу средней интенсивности
около 800 сл’1, которые весьма характеристичны.
Рассматриваемые корреляции приведены в табл. 16.
2. НИТРОСОЕДИНЕНИЯ
а. Группа С — NO2. В 1944 г. Барнс и др. [1] обнару-
жили для группы NO2 полосы поглощения в областях
1550 и 1340 сл’1, правильность этой корреляции подтверж-
дена данными по спектрам комбинационного рассеяния [2]
и результатами исследования простых нитропарафинов
[3, 4]. Затем данные были пополнены Колтупом [51, кото-
рый показал, что частоты поглощения у соединений, со-
держащих группу С — NO2, и соединений, содержащих
группу О — NO2, различны. В последние годы эта корреля-
ция подтверждена детальными исследованиями влияния
окружения группы CNO2 на ее колебания, так что роль
сопряжения, замещения атомом галогена в а-положении
и других изменений окружения группы уже выяснена.
Первоначальные исследования низших нитропарафинов
[3, 4] показали, что нитрометан поглощает при 1580 и
1375 сл’1, тогда как полосы поглощения, соответствующие
Гл. 17. Нитро- и нитрозосоединения, нитраты и нитриты 423
Таблица 16
Частота, см-1
R —ЫО2 а) Валентные колебания — с—no2 — о—no2 — N—NO2 б) Колебания в низкочастот- ной области R—N=O а) Валентные колебания N=O —О—N=O 1570—1500; 1370—1300 (эти дан- ные относятся к ароматическим соединениям; у алифатических соединений частоты иногда выше). 1650—1600; 1300—1250 1630—1550; 1300-1250 800—500 1681—1610 (две полосы
О II Z 1 и /|\ 1600—1500 (с.)
Z Z II о 1500—1430 (с.)
Окиси аминов б) Другие характеристические колебания NO; 1300—1200 (с.) 1410—1340 и 860—800
антисимметричным колебаниям, у высших членов ряда
имеют несколько меньшие частоты. Эти данные полностью
подтверждены более поздними исследованиями. Ранняя
работа Смита и др. [4] повторена Хасзельдином [24],
дополнительно исследовавшим некоторые другие соедине-
ния. Интервалы частот поглощения для простых алкильных
производных составляют 1567—1550 см'1 (антисимметрич-
ные колебания) и 1379—1368слг-1(симметричныеколебания).
Данные значения частот относятся, по-видимому, к образ-
цам, исследованным в жидком состоянии, но у этих соеди-
нений при растворении наблюдается сравнительно неболь-
шое изменение частот. Браун [25] и Корнблюм, Ангнейд
и Смайли [26] исследовали примерно по 35 нитрона-
424
Часть IV
рафинов и получили практически одинаковые результаты.
Первичные и вторичные соединения RCH2NO2 и R jR 2CHNO2
поглощают в интервалах 1565—1545 и 1383—1360 см'1.
В общем первичные соединения поглощают при несколько
более высоких частотах, чем другие соединения, но смеще-
ния полос, указанные Брауном (8 слг1 для антисимметрич-
ных колебаний и 6 см'1 для симметричных ^колебаний),
не имеют существенного значения, так как при этом частоты
не выходят за пределы всего интервала частот, которого
можно ожидать для этих соединений. В то же время у тре-
тичных нитросоединений R3CNO2 наблюдается заметное
уменьшение обеих частот колебаний до 1545—1530 и 1358—
1342 см'1. Вследствие этого их можно отличить от первич-
ных и вторичных нитросоединений. Поскольку кратные свя-
зи, участвующие в этих колебаниях, не входят непосред-
ственно в кольцо, то влияние напряжения цикла в случае
циклических нитросоединений очень мало. Нитроциклогек-
сан и нитроциклопентан, например, ведут себя как типич-
ные вторичные алкильные нитросоединения и поглощают
около нижних пределов указанных интервалов [25, 26].
У 1-метил-2-нитроциклопропана наблюдается заметное
уменьшение [25] обеих частот поглощения (1538 и 1357 см'1),
но оно, вероятно, вызвано эффектом сопряжения циклопро-
панового цикла, а не влиянием напряжения цикла.
Сопряжение нитрогруппы с этиленовой двойной связью
приводит к уменьшению обеих частот поглощения подобно
тому, как это имеет место в случае карбонильного погло-
щения. Шехтер и Шеппард [27] обнаружили, например,
что 2-метил-1-нитропропен поглощает при 1515 и 1350 см'1,
тогда как 2-метил-3-нитропропен поглощает при 1555 и
1366 см'1. Эти данные подтверждены также Брауном [25],
который установил области поглощения моноалкилнитро-
этиленов (1524 ± 4 и 1353 ± 6 слг1) и несколько более
низкочастотные области (1515 ± 4 и 1346 ± 9 см'1) для
ди- и триалкилнитроэтиленов.
Замещение атомом галогена в a-положении оказывает
заметное влияние на частоту поглощения нитрогруппы, ана-
логичное эффекту, который наблюдается в случае карбо-
нильного поглощения. Однако в этом случае частота анти-
симметричного колебания значительноувеличивается и в то
же время примерно в такой же мере уменьшается частот”
Гл. 17. Нитро- и нитрозосоединения, нитраты и нитриты 425
симметричного колебания. В предельных случаях это при-
водит к появлению этих полос в области частот, характер-
ной обычно для поглощения групп — О — NO2. Этот эф-
фект, разумеется, более заметен в случае полностью гало-
гензамещенных нитрометанов. Несколькими исследовате-
лями [8, 24, 28] изучен хлорпикрин, и показано, что он
поглощает при 1625 и 1311 см'1. Интересно отметить, что
в случае соответствующего трифторпроизводного смещение
полос поглощения несколько меньше [28, 29] (1620 и
1315 слГ1), тогда как можно было бы ожидать обратного
эффекта. У трибромпроизводного [28] сдвиг также меньше
(1606 и 1311 сл!-1). Влияние моно- и дизамещения атомами
галогенов аналогично. Ряд соединений этого типа изучен
Брауном [25] и Хасзельдином [24]. Из полученных ими
данных следует, что соединения, содержащие один атом хло-
ра в а-положении, поглощают при 1575 ± 5 и 1348 ± 6 см'1,
а наличие двух атомов хлора в a-положениях приводит
к смещению полос поглощения до 1587 ± 10 и 1332 ± 5 см'1.
Данных о нитросоединениях с другими электроотрицатель-
ными группами в a-положении получено немного, но, по-
видимому, для них должен наблюдаться аналогичный эф-
фект. У соединений, содержащих, например, гел«-динитро-
группу [25], частоты полос поглощения уменьшаются до
1580 и 1330 см'1. Хасзельдин отметил аналогичный эффект
для одного замещенного соединения с группой СООС2Н5
в а-положении.
Ароматические нитросоединения изучены многими иссле-
дователями, но, несмотря на то что некоторые соединения
были исследованы неоднократно, имеются разногласия
относительно средних значений частот поглощения этих
веществ. Так, Франсел [6] приводит частоты полос погло-
щения около 1530 и 1360 см'1, и эти данные подтверждаются
результатами работы Лотропа и др. [7]. Браун [25] опуб-
ликовал значения частот для 12 соединений, поглощающих
в интервалах 1527 ± 16 и 1348 ± 11 см'1, Рендл и Уиф-
фен [30] приводят следующие средние значения частот по-
глощения (23 соединения): 1518 и 1349 см'1, а Кросс и Фас-
сел [31] (34 соединения) — 1523 и 1344 см'1. Последние
исследователи показали также, что полосы поглощения
этих соединений очень мало смещаются при изменении со-
стояния от твердого вещества до его раствора в неполяр-
426
Часть IV
ном растворителе. Различия между средними значениями
частот поглощения, полученными различными авторами,
отражают фактически только тот факт, что частоты колеба-
ний нитрогруппы изменяются в зависимости от природы
других заместителей в кольце. Таким образом, приведен-
ные средние значения частот зависят в значительной мере
от типа исследованных соединений. У /г-динитробензола,
например, частота антисимметричных колебаний равна
1560 см'1, в то же время у /г-нитрофеноксида натрия [31]
она составляет 1501 см'1. Аналогичным образом замена
метиловой группы в нитротолуоле электронодонорной груп-
пой N(CH3)3 приводит к изменению частот колебаний груп-
пы NO2 [32] от 1527 и 1350 до 1506 и 1332 см'1. Френк,
Хормен и Шайбе [33] недавно сопоставили наблюдавшиеся
значения частот антисимметричных колебаний для обшир-
ного круга заместителей. В своих измерениях они исполь-
зовали метод прессования таблеток, в результате чего могли
иметь место некоторые изменения частот по сравнению с обыч-
ными значениями для вещества в твердом состоянии (как
это отмечено в случае карбоновых кислот), но относитель-
ные величины смещения частот, вероятно, при этом не за-
трагиваются, и данные этих исследователей хорошо согла-
суются с результатами, полученными другими авторами.
Для частот колебаний обычной плоской нитрогруппы в аро-
матических производных указан интервал 1548—1520 см'1.
Эти значения согласуются с ожидаемыми величинами;
они несколько выше, чем частоты колебаний в случае соот-
ветствующих соединений с сопряженными этиленовыми груп-
пами. Присутствие в молекуле сильно электроотрицательной
группы в napa-положенни или большой группы в орто-поло-
жении, приводящее к стремлению нитрогруппы выйти из
плоскости кольца, влечет за собой повышение частоты коле-
баний группы до 1565—1540 см-1, тогда как наличие электро-
нодонорных групп в орто- или /гара-положениях вызывает
уменьшение частоты до 1525—1490 см'1. Авторы не рас-
сматривают частоты соответствующих симметричных колеба-
ний, но и они, вероятно, смещаются от среднего значения
около 1350 см'1 примерно на ту же величину и в том же на-
правлении, что и частоты антисимметричных колебаний.
При наличии в соединении двух или более нитрогрупп влия-
ние, оказываемое на них заместителями, различно в зави-
Гл. 17. Нитро- и нитрозосоединения, нитраты и нитриты 427
симости от положения групп, что приводит к появлению
нескольких различных частот колебаний групп. У 1,3-бис-
метиламино-2,4,6-тринитробензола [33], например, наблю-
дается поглощение при 1554 и 1538 см"1, а также слабое
поглощение при 1508 и 1493 см"1.
В нашей лаборатории также были исследованы вещества,
у которых обе полосы поглощения нитрогруппы прояв-
ляются в виде дублетов. Это явление наблюдается в тех
случаях, когда одна нитрогруппа остается в плоскости
кольца, а другая под влиянием стерических эффектов вы-
ходит из этой плоскости. При этом происходит понижение
степени сопряжения ароматического цикла и появляется
новая более высокочастотная полоса поглощения. В слу-
чае полинитроароматических соединений, у которых может
образовываться водородная связь, также возможно появ-
ление в спектрах сложных полос. Однако смещение полос,
обусловленное этой причиной, сравнительно мало и несу-
щественно изменяет указанные области частот погло-
щения.
Другая возможная корреляция для алифатических нит-
росоединений относится к частотам колебаний СН2 и СН3
соседних метильных или метиленовых групп. Корнблюмом
и др. [26] полоса 1379 см"1 отмечена у всех исследованных
ими первичных нитросоединений, в том числе и тогда, когда
нет метильной группы. Эта полоса поглощения появляется
дополнительно к полосе самой ЫО2-группы и почти навер-
няка обусловлена деформационными колебаниями СН2
возмущенной метиленовой группы. По своему характеру
она подобна характеристической частоте колебаний груп-
пы СН2СООН в случае кислот. У вторичных алифатических
нитросоединений в той же области спектра наблюдается
несколько полос поглощения в дополнение к поглощению
самой нитрогруппы. Некоторые наблюдения в этой области
были сделаны Брауном [25]. Он обнаружил, что у простых
нитроалканов полосы поглощения, соответствующие сим-
метричным валентным колебаниям NO2 и антисимметрич-
ным деформационным колебаниям метильной группы, на-
лагаются при 1379 см"1. Однако в тех случаях, когда ме-
тильная группа присоединена к тому же атому углерода,
что и нитрогруппа, появляются две полосы поглощения
при 1395 и 1370 см"1. Аналогичным образом структурная
428
Часть IV
группа (CH3)2CNO2 характеризуется сильными полосами
поглощения при 1397, 1374 и 1351 слГ1.
б. Ковалентные нитраты — О —NO2. Данные по спек-
трам комбинационного рассеяния [3] показывают, что в слу-
чае ковалентных нитратов две полосы антисимметричных
и симметричных колебаний NO2 находятся на большем рас-
стоянии друг от друга и появляются при 1640 и 1260 сл"1.
Опубликованных данных по инфракрасным спектрам нитра-
тов имеется мало; в литературе приведены лишь некоторые
данные о нитроцеллюлозе [91 и нитроглицерине [10, 23].
Нами зарегистрированы спектры восьми соединений, в част-
ности изопропилнитрата, этиленгликольдинитрата и пента-
эритриттетранитрата, которые содержат рассматриваемую
группировку. Значения частоты антисимметричных колеба-
ний для этих соединений находятся в интервале 1650—
1610 сл"1, а симметричных колебаний — в интервале 1300—
1250 сл"1. Это согласуется с прежними исследованиями
Леконтом и Матье [35] простых нитропарафинов; для ча-
стоты антисимметричных валентных колебаний у некоторых
нитратов енольных эфиров Камлер [36] приводит значение
1656 сл"1. Наиболее обширные исследования в этой об-
ласти выполнены Брауном [25], который исследовал 21 ни-
трат сложных эфиров и определил интервалы характеристи-
ческих валентных колебаний 1639 ± 13 и 1279 л 7 сл"1.
в. Ковалентные нитрамины — N — NO2. Полоса анти-
симметричных колебаний NO2 характеризуется для нитр-
аминов почти такой же частотой, как и для соединений
с группировкой С — NO2, тогда как полоса симметричных
колебаний смещена в сторону меньших частот. Однако
в этом случае частота, по-видимому, в большей степени
зависит от строения прилежащих групп. Это. вероятно,
связано с тем, что электронная структура атома азота,
к которому присоединена нитрогруппа, подвержена боль-
шим изменениям. Либер и др. [11] исследовали 17 N-нитро-
соединений различного типа. У 14 из них значения частоты
симметричных колебаний NO2 находятся в интервале 1315—
1260 сл"1. При более высоких частотах поглощают либо
кислоты, либо соли, что связано, вероятно, с наличием
группировки N = N+= О.;. С другой стороны, для частоты
Гл. 17. Нитро- и нитрозосоединения, нитраты и нитриты 429
антисимметричных колебаний найден более широкий ин-
тервал, и ее значение, по-видимому, больше зависит от при-
роды заместителей. Нитрогуанидин и родственные ему соеди-
нения с алкильными заместителями поглощают в интервале
1634—1605 сл'1, тогда как арилгуанидины и нитромочевина
поглощают в интервале 1587—1575 сл'1. Полинитрамины
и соли 5-нитроаминотетразола поглощают при 1563—
1547 сл'1. Для частоты антисимметричных колебаний
у нитрогаунидинов Камлер [37] также привел значения
в интервале 1655—1620 сл'1. Салямоном и Ярославским
[38] на основании изучения области обертонов показано,
что нитрамины присутствуют в форме RNHNO2, а не в дру-
гой возможной форме RN = NOOH; в той же лаборатории
проведено исследование спектров комбинационного рассея-
ния солей нитраминов, которое показало, что у этих соеди-
нений частота симметричных валентных колебаний повы-
шается до 1400 сл'1 [39].
Мы исследовали десять полинитраминов различного
типа и установили, что для них полосы поглощения NO2
находятся в интервалах 1587—-1530 и 1292—1260 сл'1.
Эти интервалы, очевидно, характерны для поглощения
всех нитраминов вообще, за исключением нитрогуаниди-
нов и мочевин, которые могут существовать в прототроп-
ных формах и в этих случаях поглощают при более высоких
частотах.
г. Интенсивность полос валентных колебаний группы
R — NO2. Во всех описанных выше случаях обе полосы
поглощения валентных колебаний группы NO2 очень ин-
тенсивны. Однако абсолютные значения интенсивности раз-
ных соединений очень различны, и при переходе от моно-
нитроароматических соединений к ди- и полинитросоедине-
ниям интенсивность возрастает нелинейно относительно
молярной концентрации групп NO2. Это относится даже
к сходным соединениям; так, например, у диэтилен-
гликольдинитрата коэффициент погашения, рассчитанный
на одну нитрогруппу, примерно на 80% выше, чем у нитро-
глицерина [10]. Поэтому интенсивность этих полос погло-
щения не может быть использована для определения числа
групп NO2 в неизвестных соединениях. В общем полоса
поглощения, соответствующая антисимметричным колеба-
430
Ч а с т ь IV
ниям, заметно интенсивнее, чем полоса, соответствующая
симметричным колебаниям. В этом отношении положение
сходно с тем, которое наблюдается для ионизованной карбо-
ксильной группы, изоэлектронной нитрогруппе. Однако
это правило не без исключений, и у некоторых веществ обе
полосы имеют сравнимые интенсивности. В тех случаях,
когда наличие в соединении нитрогруппы приводит к стери-
ческим затруднениям, наблюдаются заметные изменения
формы обеих полос поглощения нитрогруппы. У о-нитро-
толуола, например, полуширина полосы поглощения, соот-
ветствующей симметричным колебаниям нитрогруппы, бо-
лее чем в 2 раза меньше, чем у /г-нитротолуола [40]. Эти
данные могут быть очень полезны для целей качественного
анализа, хотя интегральная абсолютная интенсивность
обеих полос изменяется очень мало.
д. Поглощение группы RNO2 в области низких частот.
Можно предположить, что полосы поглощения в низкоча-
стотной области обусловлены колебаниями скелета и дефор-
мационными колебаниями группировки RNO2. В случае
алкилнитритов Тарт [12, 13, 22] обнаружил полосу почти
с постоянной частотой 800 см'1, которую он отнес к валент-
ным колебаниям N — О. Однако на основании полученных
нами спектров алкилнитритов можно вывести заключение,
что такая полоса имеется лишь в нескольких случаях и что
она значительно смещается при изменении состояния ве-
щества. В случае нитраминов мы заметили появление по-
лосы средней интенсивности в области 790—770 см'1;
полоса в этом интервале имеется также в спектрах всех
исследованных Либером и др. [11] N-нитросоединений,
кроме двух. Эти данные, возможно, и заслуживают внима-
ния, но указанная корреляция является весьма предполо-
жительной, и ее можно использовать лишь для подтвержде-
ния правильности других, более надежных данных.
В случае алифатических нитросоединений к поглоще-
нию, обусловленному колебаниями С — N, Хасзельди-
ном [24] была предположительно отнесена полоса погло-
щения большой или средней интенсивности, наблюдающаяся
в интервале 920—830 см'1. Рендл и Уиффен [30] также пред-
полагают, что колебанию С — N у ароматических нитро-
соединений может соответствовать полоса поглощения,
Гл. 17. Нитро- и нитрозосоединения, нитраты и нитриты 431
обычно проявляющаяся около 850 см'1. Эта гипотеза была
затем обсуждена Брауном [251 и Кроссом и Фасселом [311.
Браун отметил, что порядок связи С — N, а также частота
колебаний по этой связи, по-видимому, сильно изменяются
при переходе от соединения одного типа к соединению ино-
го типа; а Кросс и Фассел [311 попытались идентифициро-
вать полосу поглощения С — N при исследовании дейте-
рированного нитробензола. Имеются только две полосы,
отличные от поглощения, присущего кольцу и группе
NO2, и практически не меняющие своего положения. Это
полоса 850 слГ1 (в случае иара-замещенных соединений
частично перекрывающаяся полосами, соответствующими
внеплоскостным колебаниям СН) и полоса 1300 см'1. Авто-
ры считают наиболее вероятным, что из этих двух полос
вторая связана с колебаниями С — N. Однако при реше-
нии задачи о типе замещения цикла по полосам деформа-
ционных колебаний СН, лежащих в этой же области спек-
тра, не следует забывать, что у ароматических нитросоеди-
нений часто наблюдается сильное поглощение в низкоча-
стотной области спектра около 850 см'1 и иногда около
750 см'1.
3. ВАЛЕНТНЫЕ КОЛЕБАНИЯ R — N = O
У таких соединений, как хлористый и бромистый ни-
трозил [14], частота N = О лежит около 1800 см'1, но это
значение выше обычного, так как длина связи N = О
под влиянием атома галогена уменьшается. В случае нитри-
тов результаты ряда исследований свидетельствуют о том,
что частота валентных колебаний N = О лежит около
1660 см'1. У нитрозосоединений частоты колебаний N = О
довольно различны, но обычно им соответствует поглоще-
ние в области 1600—1500 см'1. Нитрозамины поглощают
при еще более низких частотах, обычно ниже 1500 см'1.
а. Нитриты — О — N = О. Тарт [12, 13, 22] показал,
что у 15 изученных им нитритов колебаниям N = О соот-
ветствуют две полосы в интервалах 1681—1653 и 1625—
1613 см'1. Эти полосы приписываются соответственно
транс- и /{wc-формам нитрита. Частоты постепенно умень-
шаются при увеличении размеров присоединенной группы,
432
Ч а с т ь IV
причем наивысшие их значения у метилнитрита состав-
ляют 1681 и 1625 см'1, а наинизшие у амилнитрита —1653
и 1613 см'1. Указанные интервалы частот подтверждены
аналогичными исследованиями Хасзельдина и др. [41, 421,
проведенными на небольшом числе ал к ил нитритов. В слу-
чае 2,2,2-трифторэтилнитрита полоса поглощения сме-
щается до 1736 (транс-форма) и 1695 см'1 (цис-форма).
Этот сдвиг весьма значителен, если учесть удаленность
места присоединения атомов фтора по цепи от связи N = О.
Эффект вызван, вероятно, особенностями пространствен-
ного строения молекулы и подобен в какой-то мере тем
эффектам, которые наблюдаются в случае некоторых р-фто-
рированных сложных эфиров.
Данные об относительной интенсивности этих полос по-
зволяют просто распознавать тип замещения, что представ-
ляет интерес, так как соотношение цис- и транс-форм у раз-
ных нитритов различно. Так, например, отношение коэф-
фициентов погашения для полос транс- и цис-форм ме-
тилнитрита равно 1 : 1, а для этилнитрита оно становится
равным 2 : 3 и достигает постоянного значения 3 : 3,5 для
высших первичных нитритов. Однако в случае вторичных
нитритов это отношение равно 6 : 10, а в случае третич-
ных нитритов, у которых содержание цис-формы сильно
уменьшается, оно составляет 40 : 1.
Интенсивности полос в этой спектральной области весь-
ма значительны, и с такой интенсивностью, какую имеют
нитритные полосы, приходится встречаться редко.
б. Нитрозосоединения > С — NO. Отнесение полосы
валентных колебаний — N = О у нитрозосоединений встре-
чает затруднения, которые обусловлены главным образом
тем обстоятельством, что первичные и вторичные нитрозо-
соединения легко переходят в оксимы, а третичные соеди-
нения легко димеризуются. Томпсон, Николсон и Шорт
[15] первоначально предположили, что этим колебаниям
соответствует полоса поглощения в области 1400—1300 см'1,
в то время как Браунли [16] на основании данных, полу-
ченных при изучении некоторых нитрозопиримиденов, от-
нес ее к 1650 см'1. В последнее время эта проблема интен-
сивно изучалась Люттке [43—45] и Тартом [46], и теперь
вопрос стал несколько проясняться.
Гл. /7. Нитро- и нитрозосоединения, нитраты и нитриты 433
Люттке проследил изменения во времени в спектрах
первичных и вторичных нитрозосоединений при их испаре-
нии. У нитрозометана и нитрозоциклогексана, например,
имеются полосы поглощения при 1564 и 1558 см'1, которые
могут быть отнесены к колебаниям —N = О. При стоя-
нии образцов интенсивность этих полос быстро уменьшается
и появляются новые полосы, так что в сравнительно корот-
кое время спектры заменяются соответственно спектрами
формальдоксима и оксима циклогексанона. В растворе эти
соединения присутствуют преимущественно в виде димеров,
и нитрозометан, например, в этой форме поглощает около
1290 см'1.
У третичных нитрозосоединений R’R2R3CN = О исклю-
чена возможность образования оксимов. У этих соедине-
ний в твердом состоянии или в растворе также обычно на-
блюдается димеризация, но в спектрах разбавленного рас-
твора и паров часто можно выделить частоты колебаний,
относящиеся к мономеру. Однако многие соединения этого
типа, изученные Тартом [461 и Люттке [43], содержат один
или более атомов галогенов. (R—галоген). Это приводит
к дальнейшим усложнениям, поскольку в таком случае
могут проявляться эффекты поля, подобные тем, которые
обнаружены у а-галогензамещенных кетонов и которые
зависят от конфигурации молекулы. У моно- и ди-а-галоген-
замещенных соединений наблюдаются поэтому дублетные
полосы поглощения N = О, что связано, как это подтвер-
ждено, с поворотной изомерией. Этот эффект приводит также
к повышению частоты поглощения, как и в случае соответ-
ствующих карбонильных соединений. Нитрозоциклогексан,
например, поглощает при 1558 см'1, а 1,4-дихлор-1,4-
динитрозоциклогексан [43] — при 1570 см'1. Поглощение
N = О вообще очень сильно меняется в зависимости от
природы окружения группы. Его частота уменьшается до
1495 см'1 в случае 2,4,6-триметилнитрозобензола и повы-
шается до 1620 см'1 в случае CClgNO. Интересно отметить,
что частота поглощения у последнего соединения выше,
чем у соответствующего трифторпроизводного [29, 47],
на основании чего можно предположить, что в этом случае
играют роль не только индукционный эффект и эффекты
поля, но также и явление мезомерии с участием структур,
включающих атомы галогена, связанные двойной связью.
28 л. Беллами
434
Часть IV
В связи с этим важным может быть и тот факт, что в отли-
чие от карбонильной группы в аналогичных условиях ча-
стота колебаний группы N = О постоянно повышается по
мере присоединения все большего числа атомов галогенов
в а-положение.
В общем ароматические нитрозосоединения поглощают
около 1500 см'1, третичные алифатические соединения при
более высоких частотах — около 1550 см'1, а а-галоген-
замещенные соединения поглощают при еще более высоких
частотах —до 1620 см'1. В последнем случае полосы по-
глощения часто имеют дублетный характер вследствие
явлений поворотной изомерии. Таким образом, благодаря
этим исследованиям характеристическая частота колеба-
ний -^С — N = О, по-видимому, установлена довольно
хорошо. Накамото и Рендлом [48] выдвинуто альтернатив-
ное предположение об отнесении к колебаниям этой груп-
пы частоты 1380—1340 слГ1, однако эти авторы не иссле-
довали область 1600—1500 см'1, которая представляется
более вероятной для этой частоты с точки зрения рассмо-
тренных выше работ. Косвенным подтверждением правиль-
ности отнесения этих колебаний к более высокочастотной
области служат результаты, полученные Беллами и Уильям-
сом [49] при сравнении частот колебаний карбонильной
группы и группы N = О у соединений с аналогичным заме-
щением. Это сопоставление позволяет оценить значение
частоты колебаний группы на основании известных частот
для кетонов и приводит к значению в области 1600 см'1.
в. Нитрозамины — N — N = О. В первоначальных
работах Ирла и др. [17] сделано предположение, что погло-
щение N = О у N-нитрозосоединений находится около
1400 см'1. Это предположение подтверждено данными по
спектрам комбинационного рассеяния [18], но Либером
и др. [11] выдвинуто альтернативное предположение о том,
что в случае исследованных ими нитрозогуанидинов и сход-
ных соединений это поглощение проявляется в интервале
1600—1500 см'1. Несмотря на то что число исследованных
соединений этого типа остается сравнительно небольшим,
вопрос о месте характеристического поглощения N = О
теперь проясняется.
Гл. 17. Нитро- и нитрозосоединения, нитраты и нитриты 435
Небольшое число этих соединений в различных состоя-
ниях исследовано Хасзельдином и др. [41, 42, 50] и Тартом
[46, 50], и если в прежних исследованиях [41] возникали
сомнения относительно возможности димеризации в жид-
ком состоянии, то теперь этот вопрос удовлетворительно
разрешен [50]. Все данные говорят за то, что у диалкилни-
трозаминов в мономерном состоянии в парах частота колеба-
ний N = О равна примерно 1490 с.и-1. В случае раствора
в четыреххлористом углероде она падает до 1450 см'1
и еще немного уменьшается в случае жидкого состояния.
Полосы поглощения, соответствующие димерам, прояв-
ляются около 1310 см'1. Имеющихся данных все же недо-
статочно, чтобы получить какие-либо точные интервалы
частот полос поглощения, но у нормальных алкилнитроз-
аминов эти частоты обычно на 10 см'1 меньше, чем указан-
ные значения. Нет никаких данных об а-фторзамещенных
соединениях, но интересно отметить, что N-нитрозо-бнс-
2,2,2-трифторэтиламин в парах [42] характеризуется по-
глощением N = О при 1550 см'1. Это свидетельствует о
весьма значительном сдвиге частоты колебаний, если учесть,
что атомы фтора так далеко находятся от связи N = О.
Такое смещение может быть обусловлено, однако, тем, что
вследствие определенной геометрической конфигурации мо-
лекулы группы оказываются расположенными недалеко
одна от другой, несмотря на большое число промежуточ-
ных связей. Повышенные частоты поглощения — N = О
отмечены также в случае амидов с присоединенной к атому
азота нитрозогруппой. Уайт [51] исследовал ряд соедине-
ний этого типа и отнес поглощение N = О к интервалу ча-
стот 1527—1515 см'1. Для этих веществ характерны также
повышенные значения частот карбонильного поглощения,
соответствующего скорее сложным эфирам, чем амидам.
Вследствие этого вполне возможно, что у ряда этих
веществ имеет место значительное дипольное взаимодей-
ствие.
Аналогичным явлением могут быть обусловлены и более
высокие частоты, отмеченные Либером и др. [11] в случае
нитрозогуанидинов, однако имеется слишком мало данных
о свойствах этих соединений, чтобы можно было сделать
какие-либо однозначные выводы.
28*
436
Часть JV
г. Другие характеристические полосы поглощения групп
R — N = О. В низкочастотной области спектра имеются
некоторые полосы поглощения, обусловленные главным
образом скелетными колебаниями, но иногда и они могут
быть полезны для подтверждения выводов, сделанных на
основании рассмотрения каких-либо иных данных. Тарт
[12, 13, 22], например, высказал ряд предположений отно-
сительно поглощения алкилнитритов, которые могут быть
полезны в этих целях. У всех исследованных им 15 нитри-
тов обнаружены полссы поглощения в интервале 814—
751 слг"1, которые этот автор относит к основным колеба-
ниям vN O; имеются также еще две полосы в интервалах
691—617 и 625—565 см'1, соответствующие деформацион-
ным колебаниям — О — N = 0 цис- и транс-форм. Не-
смотря на то что для колебаний скелета характерен до-
вольно широкий интервал частот, эти корреляции могут
успешно применяться для подтверждения наличия нитрит-
ных групп, поскольку интенсивность обеих полсс довольно
высока. Помимо этого, были определены составные полосы
с частотами 2300—2250 C4«^1(6ONO + vN=;0), 2500 см 1 (vN_0+
VN=O) и полоса, соответствующая первому обертону колеба-
ний vN=0, при 3300—3200 см'1. Аналогичные пслссы погло-
щения, соответствующие валентным колебаниям N — N
и деформационным колебаниям N — N = О, он идентифи-
цировал также в случае нитрэзсаминсв [46]. Эти полосы
наблюдаются соответственно около 1050 и 660 см'1. Вто-
рая полсса, таким образом, не сильно отличается от полос,
соответствующих колебанию О — N = О. Наличие пер-
вой полосы поглощения у некоторых соединений подтверж-
дено также Хасзельдином [41, 42|.
Никаких других удобных полсс поглощения в случае
соединений, содержащих i руппу — С— N = О, не иденти-
фицирсвано, хотя к валентному колебанию С — N относят
сильно размытую полосу около 1100 см'1. .
д. Окиси аминов. Окиси аминов также содержат группы
N — О, присоединенные кратной связью, и поэтому их удоб-
но рассмотреть в этой же главе. Коста с сотрудниками [52—
54] исследовал поглощение, обуслсвленное колебаниями
этой группы, у ряда N-окисей пиридина и различным
Гл. 17. Нитро- и нитрозосоединения, нитраты и нитриты 437
образом замещенных производных соединений. Сама окись
пиридина в растворах в неполярных растворителях по-
глощает при 1270 см'1, но частота поглощения, по-види-
мому, очень чувствительна к электрическим свойствам за-
мещающих групп. Заместители с сильно выраженными
электронодонсрными свойствами вызывают заметное умень-
шение этой частоты, и окись п-метокси-Ы-пиридина [521 по-
глощает при 1238 см'1. Еще более эффективными в этом
отношении оказываются /г-окси- и аминогруппы [53]
и соединения, содержащие аминогруппу, поглощают, по-
видимому, около 1200 см'1. Обратный эффект наблюдается
в случае заместителей с сильно выраженными электроноак-
цепторными свойствами, и у /г-нитропроизводного соедине-
ния [52] поглощение N — О наблюдается при 1304 см'1. Ме-
тис-Ноел, Вольф и Галле [55] исследовали несколько оки-
сей третичных аминов. У соединений этого типа полоса
поглощения N—>0 отнесена к области 970—950 см'1.
Эго очень большое смещение по сравнению с соответствую-
щим поглощением у пиридинового ряда, но в тех случаях,
когда кратность связи становится минимальной, можно
ожидать сдвига полосы поглощения в более низкочастот-
ную область спектра.
Уайли и Слеймекером [58] исследованы также и сопо-
ставлены с N-окисями пиридина N-окиси пиримидина.
У соединений обоих типов наблюдается поглощение N —» О
в области 1300—1255 си-1. Интересным наблюдением, сде-
ланным в этой работе, является обнаружение значительных
различий между спектрами N-окисей пиколинов. Окиси
а- и р-пиколина сходны с окисью пиридина и поглощают
между 1264 и 1260 см'1, а окись у-пиколина поглощает
при 1290 см'1. Вещества, родственные окислам аминов,
встречаются в ряду азоксисоединений, содержащих связь
О
t
— N = N —. Уиткоп и Киссмен [56] исследовали несколько
таких соединений и обнаружили у них обычную полосу
поглощения в области 1310—1250 см'1. Авторы относят эту
полосу к колебаниям по связи N —>0, но осторожно отме-
чают, что в той же области должно обнаруживаться поглоще-
ние, соответствующее валентным колебаниям С — N арома-
тических соединений. Джендер и Хасзельдин [57] исследо-
438
Часть IV
вали гексафтсразоксиметан, который поглощает при 1282
и 1256 см'1. Учитывая тесную аналогию в свойствах с оки-
сями аминов, можно, вероятно, отнести эти полосы к по-
глощению N—»О азоксисоединений.
4. ИОНИЗОВАННЫЕ НИТРАТЫ
Нитрат-ион имеет четыре основные частоты колебаний,
причем все они при определенных условиях могут наблю-
даться в инфракрасном спектре. Однако только двум из
них соответствуют достаточно интенсивные полосы погло-
щения, которыми можно пользоваться в целях идентифика-
ции соединений. Эти полосы соответствуют колебаниям v3
и v2n имеют частоты 13S0 и 800 см'1.
Сильные полосы вблизи этих частот найдены у некото-
рых неорганических нитратов, например у нитратов аммо-
ния, таллия, бария и свинца [19, 20]; кроме того, полоса
1390 сл-1 наблюдалась также для многих веществ в спек-
трах комбинационного рассеяния [21]. Интервалы частот
для полсс органических нитратов составляют 1410—1340
и 860—800 см'1 [5], что подтверждается также спектрами
таких соединений, как, например, нитрат гуанидина, кото-
рые изучены Либером и др. [И], а также данными о неко
торых соединениях, исследованных нами. Полоса с боль-
шей частотой всегда значительно сильнее, чем другая по-
лоса, которая редко имеет интенсивность выше средней.
При изучении спектров комбинационного рассеяния
растворов неорганических веществ показано [21], что поло-
са колебаний v3 при высоких концентрациях расщепляется
на две полосы, которые находятся в интервале от 1500 см'1
(Th) до 1315 см'1 (А1). Это заставляет предположить, что
при таких концентрациях изменяется форма ионной нитрат-
ной группы. Однако если концентрации не очень высоки,
то полосы не выходят за пределы интервала 1410—1345 см'1;
в случае органических нитратов эта корреляция не нару-
шается при изменении концентрации.
ЛИТЕРАТУРА
1 Barnes, Gore, Liddel, Williams, Infra-red Spect-
roscopy, Reinhold, 1944.
2. H i b b e n, The Raman Effect and its Chemical Applicati-
ons, Reinhold, 1939.
Гл. 17. Нитро- и нитрозоссединения, нитраты и нитриты 439
3. N i е 1 s е и, Smith, Ind. Eng. Chem. (Anal.), 15, 609 (1943).
4. S m i t h, Pan, Nielsen, J. Chem. Phys., 18, 706 (1950).
5. Col thup, J. Opt. Soc. Amer., 40, 397 (1950).
6. F r a n c e 1, J. Amer. Chem. Soc., 74, 1265 (1952).
7. Lothrop, Handrick, Hainer, J. Amer. Chem.
Soc., 73, 3581 (1951).
8. Randall, Fowler, Fuson, Dangl, Infra-red
Determination of Organic Structures, Van Nostrand, 1949.
9. H и к и т и н В. H., ЖФХ, 23, 786 (1949).
10. Р inchas, Analyt. Chem., 23, 201 (1951).
11. Lieber, Levering, Patterson, Analyt. Chem.,
23, 1594 (1951).
12. Tarte, Bull. Soc.’Chim. (Belg.), 60 227 (1951).
13. Tarte, Bull. Soc. Chim. (Belg.), 60, 240 (1951).
14. В u г n s, Bernstein, J. Chem. Phys., 18, 1669 (1950).
15. Thompson, Nicholson, Short, Discuss. Faraday
Soc., 9, 222 (1950).
16. В г о w n 1 i e, J. Chem. Soc., 1951, 3062.
17. E a r 1, L e Fevre, P u 1 f о r d, Walsh, J. Chem. Soc.,
1951, 2207.
18. В a r r e d o, G о u b e a u, Z. anorg. Chem., 251, 2 (1943).
19. N e w m a n, Halford, J. Chem. Phys., 18, 1276 (1950).
20. Newman, Halford, J. Chem. Phys., 18, 1291 (1950).
21. Mathieu, Lounsbury, Discuss. Faraday Soc., 9, 196
(1950).
22. Tarte, J. Chem. Phys., 20, 1570 (1952).
23. P r i s t e r a, Analyt. Chem., 25, 844 (1953).
24. H a s z e 1 d i n e, J. Chem. Soc., 1953, 2525.
25. В г о w n, J. Amer. Chem. Soc., 77, 6341 (1955).
26. Kornblum, Ungnade, Smiley, J. Org. Chem., 21,
377 (1956).
27. Shechter, Sheppard, J. Amer. Chem. Soc., 76, 3617
(1954).
28. Mason, Dunder dale, J. Chem. Soc., 1956, 754.
29. J a n d e r, H a s z e 1 d i n e, J. Chem. Soc., 1954, 912.
30. Randle, Whiffen, J. Chem. Soc., 1952, 4153.
31. Kross, Fassel, J. Amer. Chem. Soc., 78, 4225 (1956).
32. L i p p e r t, Vogel, Z. Phys. Chem., 9, 133 (1956).
33. Fra nk, H о r m a n n, S c h e i b e, Chem. Ber., 90, 330
(1957).
34. Pal m, W e r b i n, Can. J. Chem., 31, 1004 (1953).
35. L e с о m t e, Mathieu, J. Chim. Phys., 39, 57 (1942,.
36. Kumler, J. Amer. Chem. Soc., 75, 4346 (1953).
37. К u m 1 e r, J. Amer. Chem. Soc., 76, 814 (1954).
38. Салямон Г. С., Ярославский Н. Г., Сб. статей по
общей химии, 2, 1325 (1953).
39. Салямон Г. С., Бобович Я. С., Сб. статей по общей
химии, 2, 1332 (1953).
40. Conduit, частное сообщение.
41. Haszeldine, dander, J. Chem. Soc., 1954, 691.
42. Haszeldine, Mattinson, J. Chem. Soc., 1955, 4172.
440
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
Ч а с т ь IV
L ii t t к е, Zeit. Electrochem., 61, 302 (.1957).
L ii t t к е, J. Phys. Radium, 15, 633 (1954).
Schindler, Liittke, Hol leek, Chem. Ber., 90,
157 (1957).
T a r t e, Bull. Soc. Chim Beiges, 63, 525 (1954).
Mason, Dunderdale, J. Chem. Soc., 1956, 759.
Nakamoto, Rundle, J. Amer. Chem. Soc., 78, 1113 (1956).
Bellamy, Williams, J. Chem. Soc., 1957 , 863.
Haszeldine, dander, Tarte, J. Chem. Phys., 23,
979 (1955).
White, J. Amer. Chem. Soc., 77, 6008 (1955).
Costa, В 1 a s i n a, Z. Phys. Chem., 4, 24 (1955).
Costa, Blasina, Sartori, Z. Phys. Chem., 7, 123 (1956).
Sartori, Costa, Blasina, Gazz. Chim., 85, 1085
(1955).
M a t h i s-N о e 1, Wolf, G a 1 1 a i s, Compt. Rend. Acad.
Sci. (Paris), 242, 1873 (1956).
Witkop, Kissman, J. Amer. Chem. Soc., 75, 1975
(1953).
Jander, Haszeldine, J. Chem. Soc., 1954, 919.
Wiley, Slaymaker, J. Amer. Chem. Soc., 79, 2233
(1957).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Ill о p ы г и н
Словохот
П. П., ЖФХ, 22, 897 (1948).
ова Н. А., ЖФХ, 25, 768 (1951).
Глава 18
ФОСФОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
1. ВВЕДЕНИЕ
Инфракрасные спектры фссфорсрганических соедине-
ний стали изучать только в течение последних нескольких
лет. Однако широкое применение этих соединений в хо-
зяйстве и промышленности в качестве инсектицидов, доба-
вок к маслам, пластификаторов и т. п., а также возможность
получить сведения о метаболизме фосфатов в нуклеиновых
кислотах, лецитинах и аналогичных продуктах стимули-
ровали проведение исследований в широком масштабе,
и в настоящее время уже установлен ряд корреляций по
таким спектрам. Эти данные собраны в виде корреляцион-
ных таблиц для связей фосфора в неорганических соеди-
нениях [22J и для фссфорсрганических соединений [23]
Корбриджем.
Предложенные корреляции приведены в табл. 17.
Следует, однако, отметить, что не все они одинаково
хорошо обоснованы; многие из них установлены в резуль-
тате исследований большого числа соединений, другие же
являются лишь очень приближенными корреляциями
и основаны на данных исследований ограниченного числа
соединений или недостаточно типичных соединений.
Эти корреляции обсуждаются ниже; даны также неко-
торые указания на предполагаемую степень их надеж-
ности.
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ Р = О
| Мейрик и Томпсон [1] первые исследовали инфракрас-
ные спектры и спектры комбинационного рассеяния фос-
форорганических соединений. В результате изучения пяти
фосфонатов они предположили, что полоса средней интен-
сивности, появляющаяся во всех случаях в области 1260—
1250 см'1, обусловлена валентными колебаниями Р = О.
Такое отнесение подтверждено данными о спектрах
Таблица 17
Частота, сл-1
Фосфор-кислородные связи
а) Р = О (свободная)........
Р = О (при водородной связи)
б) Р — О — С (в ароматических
соединениях) ..............
в) Р — О — С (в алифатических
соединениях) ...............
г) р_0—С2Н5 ................
Д) Р-О- СН;,................
е) Р— О — Р ................
ж) Р-ОН.....................
Фосфор-водородные связи Р—Н
Фосфор-углеродные связи
а) р-с0н5..................
б) Р — Алкил................
Р-СН3 .....................
Фосфор-серные связи
P=S........................
Фосфор-азотные связи
а) Р —NH2 иР-NH-............
б) Р —N—....................
Фосфор-галогенные связи
а) Р—С1.....................
б) Р —F.....................
Ионные фосфатные группы
Арилзамещенные..............
Алкилзамещенные ............
1350—1250 (иногда 1175) (с.)
1250—1150 (оч. с.)
1240—1190 (с.)
1050—990 (оч. с.) (иногда ниже)
1170—1150 (сл.) (имеется так-
же в случае Р — О — Аг)
1 180± 10 (сл.)
970—940
2700—2560 (широкая и нерез-
кая полоса)
2440—2350 (ср.)
1450—1435 (ср.)
Надежных корреляций нет,
1320—1280
840—600 (оч. сл.)
Обычные значения для групп
NH2 и NH’
Около 715 (сомнительно)
580—440 (с.)
885—810 (с.)
1090—1040
1180—1150 (с.) и около 1080
Гл. 18. Фосфорорганические соединения
443
комбинационного рассеяния РОС13 [2] (в котором полоса
колебаний Р = О находится при 1295 см'1) и других фос-
фитов и фссфонатов [3].
Эти исследования продолжили и развили Гор [4] и Даш
и Смит [5], которые независимо друг от друга изучили зна-
чительное количество фссфонатов и фосфатов. Гору [4]
удалось показать, что отнесение полосы в области 1250 см'1
к этим колебаниям достаточно хорошо согласуется с теорией,
так как применение правила Горди дает в зависимости от
выбора длины связи Р — О значения частоты в пределах
интервала 1300—1100 см'1. Кроме того, он исследовал зна-
чительное число фосфатов и сравнил их с соответствующими
тиофосфатами. Во всех случаях основное отличие состояло
в том, что в спектрах фосфатов имелась полоса в области
1300—1250 см'1. Эти данные полностью подтверждены Да-
шем и Смитом [5], которые также показали, что значение
частоты этих колебаний определяется в значительной сте-
пени числом электроотрицательных заместителей у атома
фосфора. У соединений с тремя электроотрицательными за-
местителями, например фосфатов или фторфосфонатов, по-
лоса Р = О находится приблизительно в интервале 1310—
1275 см'1, тогда как у соединений с двумя электроотрица-
тельными заместителями, например у кислых фссфонатов
и алкилфосфонатов, она лежит в области 1275—1250 см'1.
Найдено, что фосфинаты и подобные им вещества погло-
щают около 1240 см'1, в предельных же случаях трифенил-
и триметилфссфиноксидов частоты уменьшаются соответ-
ственно до 1190 и 1176 см'1. Последние два соединения пред-
ставляют интерес также потому, что на примере их можно
проиллюстрировать явное отсутствие влияния на частоту
Р = О сопряжения ароматических колец. Вообще связь
этой полосы с группой Р = О была установлена сравнением
соответствующих фссфитов с подобными соединениями трех-
валентного фосфора, у которых данной группы нет.
Правильность этой корреляции подтверждена Беллами
и Бичером [6—81, изучавшими главным образом фосфаты
и фосфонаты, а также работами Холмштедта и Ларссона
[9], Бергманна, Литтауера и Пинхаса [10], Харвеем
и Мейхудом [24], Марсеном [25], Эмелеусом и др. [26,
27] и Томасом [28]. Изучены также спектры нескольких
природных соединений фосфора [18—21].
444
Часть IV
Эту корреляцию можно поэтому считать окончательно
установленной; мы также исследовали спектры свыше
100 соединений, содержащих группу Р = О, и обнаружили
для них полосу поглощения в области 1300—1250 см"1.
Этот интервал частот и указан на стр. 20, хотя в неко-
торых случаях полосы поглощения могут наблюдаться
и вне этой области спектра. Как отмечалось выше, частота
валентных колебаний зависит главным образом от электро-
отрицательности замещающих групп, а поэтому при наличии
сильно электроотрицательных заместителей эта частота
резко увеличивается. Трифторфосфиноксид, например,
поглощает при 1404 см"1 [29]. Наличие заместителей,
обладающих минимальной электроотрицательностью, та-
ких, как атомы углерода или водорода, присоединенных не-
посредственно к атому фосфора, может привести к тому,
что частота колебаний будет меньше, чем нижний предел
указанного интервала частот. Томас [28] исследовал это
поглощение у 250 фосфорорганических соединений и при-
вел интервал частот 135С—1175 см"1 (в отсутствие водород-
ной связи), что указывает на наличие ожидаемых крайних
случаев поглощения.
Беллом и др. [30] установлено полуколичественное со-
отношение между частотой этого поглощения и электро-
отрицательностью заместителей. Ими показано, что для
галогензамещенных фссфиноксидов соблюдается линейная
зависимость между суммой электроотрицательностей ато-
мов галогенов и частотой фосфорильного поглощения. На-
личие этой зависимссти использовано для вычисления
значений эффективной электроотрицательности других
групп заместителей, причем полученные значения, по-ви-
димому, имеют довольно общее значение. В некоторых слу-
чаях, например в случае групп OR, значение эффективной
электроотрицательности, вычисленное таким путем, ока-
зывается весьма близким к тому, которое можно ожидать
для кислородсодержащих заместителей, но значения элек-
троотрицательности для таких заместителей, как ОН
и NH2, оказываются аномально малыми, по-видимому,
вследствие образования этими группами водородных свя-
зей. Недавно полученные результаты [24] показывают,
что в случае соединений, у которых к атому фосфора при-
соединен атом азота, значения эффективной электроотри-
Гл. 18. Фосфорорганические соединения
445
цательности азотсодержащих заместителей также оказы-
ваются меньше, чем можно было бы предполагать. Частота
поглощения Р = О, например, при переходе от (С2Н6О)3РО
к [N(C2H 6)2]3РО уменьшается на 57 си’1. По-видимому,
в этих случаях неподеленная пара электронов атома азота
оказывает каким-то образом влияние на частоту колебаний,
помимо прямого индукционного эффекта.
Полоса Р — О смещается на 50—80 см'1, если молекула
содержит ОН- или NH-группы, с которыми Р = О-группа
может образовать водородную связь [5, 7, 8, 26, 27]. Связь
особенно сильна в случае соединений, содержащих группу
ОН, на что указывает сопутствующее ее образованию боль-
шое смещение полосы валентных колебаний О — Н (см. ни-
же). Помимо смещения в сторону меньших частот, значи-
тельно возрастает интенсивность полосы Р = О. Когдс.
карбонильная группа связана водородной связью, то
увеличение ее интенсивности не всегда наблюдается;
исключение составляют соединения с группировкой
— СО — С =С(ОН) — , у которых такое увеличение объяс-
няется наличием резонирующих структур. В случае груп-
// 0
пы Р 7 наблюдаемое увеличение интенсивности, вероят-
\ОН
но, также обусловлено наличием аналогичных резонансных
структур.
Такой тип образования водородной связи, по-видимому,
встречается только в окси- и аминссоединениях, и частота
Р — О у водородсодержащих фссфонатов, например, имеет
обычное значение. В самом деле, хотя данные о теплоте сме-
шения и другие данные показывают, что многие типы фос-
форе рганических соединений ассоциированы [11], однако
Р = О-группа не принимает участия в ассоциации посред-
ством водородной связи, так как ее частота в инфракрас-
ном спектре остается сравнительно постоянной. Из стери-
ческих соображений следует, что образование внутримоле-
кулярной связи с участием оксигрупп затруднено, и более
вероятной в этих случаях представляется ассоциация путем
образования димеров, как это имеет место у карбоновых кис-
лот. Однако ассоциация при этом чрезвычайно сильна,
и в тех немногих случаях, когда были исследованы раз-
бавленные растворы и применялись неполярные раствори-
446
Часть IV
тели, смещение полосы Р = О все же наблюдалось, но
часто при этом была заметна также вторая полоса погло-
щения, соответствующая колебаниям несвязанной группы
Р = О [7]. Аминосоединения также характеризуются меж-
молекулярными связями, и в разбавленных растворах ча-
стоты NH и Р = О имеют обычные для них значения. При
использовании данной корреляции удалось показать, что
диалкилфосфит, содержащий водород, существует только
в форме фосфонага.
/0Н zo
Р—OR . Р—Н
is™
О наличии сильных полос поглощения Р = О и Р —Н
в спектрах ряда соединений такого типа сообщили Мейрик
и Томпсон [1]; аналогичные сведения приведены Беллами
и Бичером [6]. Отсутствие полос поглощения Р —ОН и тот
факт, что полоса поглощения Р = О соответствует несвя-
занной фосфорильной группе, указывают, что лишь очень
небольшая часть вещества существует в виде водородсодер-
жащего фосфита.
Полоса поглощения группы Р = О имеет довольно силь-
ную интенсивность, но иногда она меняется в зависимости
от природы присоединенных группировок. Сравнение абсо-
лютных интенсивностей для различных классов веществ
осложняется тем, что данная полоса очень часто появляется
в виде дублета. Гор [4] первоначально предположил, что
вторая из этих полос у этилфосфатов обусловлена колеба-
ниями О — С2Н5, однако наличие дублетного характера
полосы у многих полностью ароматических фосфатов и фос-
фонатов свидетельствует о том, что эта полоса связана с са-
мими колебаниями Р = О. Вопрос о природе дублетного
характера полосы окончательно еще не решен, но были вы-
двинуты предположения о взаимодействии группы с близ-
лежащими группами СН [25, 28], возможно также, что здесь
сказывается явление поворотной изомерии.
3. ВАЛЕНТНЫЕ КОЛЕБАНИЯ Р —О—С
а. Колебания Р—О—С (арил). Подобно тому как
частоты колебаний связей С — О — С (алкил ) и С — О — С
Гл. 18. Фосфорорганические соединения
447
(арил) значительно различаются, имеются заметные раз-
личия и между частотами колебаний аналогичных типов
связей Р — О — С. При исследовании фосфорсодержащих
соединений наблюдалось большее постоянство частот, одна-
ко следует отметить, что большинство изученных до настоя-
щего времени соединений очень сходны друг с другом по
строению.
Различие между обоими типами соединений с группами
Р — О — С наглядно продемонстрировано при исследова-
нии ряда таких родственных соединений, как трифенил-,
дифенилэтил-, фенилдиэтил-, триэтилфссфаты. При одних
и тех же концентрациях этих веществ и при использовании
одной и той же кюветы было обнаружено постепенное ослаб-
ление и затем полное исчезновение сильной полосы около
1200 см'1; при этом для полосы 1030 см'1, отсутствующей
у трифенилфосфата, в указанном ряду соединений наблю-
далось увеличение интенсивности [6].
На основании этих и других данных [6, 7] к Р — О — С
(ароматический цикл) отнесена полоса поглощения в об-
ласти 1200 см'1 и показано, что такая полоса имеется у всех
исследованных до настоящего времени ароматических фос-
фатов, фосфонатов и фосфитов, а также у некоторых арил-
тиофосфатов и аналогичных веществ, тогда как у полностью
алифатических соединений такой полосы нет.
Установлено, что интервал, в котором обнаруживалась
данная полоса, составляет 1240—1190 см'1, причем у пара-
нитрозамещенных соединений, например у некоторых ин-
сектицидных препаратов, мы наблюдали эту полосу вблизи
1240 см'1.
У некоторых других соединений этого типа, изученных
Томасом [28], частота поглощения иногда уменьшалась
даже до 1180 см'1.
Найдена также вторая полоса поглощения Р — О — С
(арил) [6], которая не так интенсивна и лежит около
1030 см'1. Она менее полезна для целей идентификации,
так как попадает в область частот, соответствующую погло-
щению алифатических эфиров, а также некоторых типов
колебаний ароматического кольца. Однако интенсивность,
присущая этой полосе поглощения, настолько меньше ин-
тенсивности соответствующей полосы Р-—О — С (алкил),
что последнюю легко можно отличить.
448
Часть IV
б. Колебания Р —О — С (алкил). В результате описан-
ных выше исследований найдено также, что частота полосы
поглощения Р — О — С (алкил) равна примерно 1030 слг1;
такое же соотношение установлено еще раньше Дашем
и Смитом [5].
В настоящее время известны спектры очень большого
ряда алкилфосфатов и аналогичных веществ [1,4—10];
оказалось, что во всех случаях имеется очень интен-
сивная полоса поглощения в области 1050—995 см-1, при-
чем подавляющее большинство соединений поглощает
при 1030 см'1. Бергманн и др. [10] сообщают, что у изо-
прэпилфссфата эта полоса отсутствует, а возникновение
полосы при 995 см'1 объясняют иной причиной. Однако
описанные Мейриком и Томпсоном [1], Беллами и Бичером
[6, 7| спектры других веществ с разветвленной цепью нахо-
дятся в согласии с указанной корреляцией, и нет основа-
ний предполагать, что вещества с разветвленной цепью
представляют какое-либо исключение.
Томас [28] недавно исследовал около ЗОЭ фосфороргани-
ческих соединений и отметил, что у метиловых и этиловых
эфиров это поглощение наблюдается в области 1050—
1000 см'1. Однако у соединений с более длинными углево-
дородными цепями алкильных радикалов частота погло-
щения несколько меньше и достигает в предельных слу-
чаях 950 см'1 при условии, что в молекулу не входит атом
фтора или другие электроотрицательные заместители. Эта
корреляция применима, конечно, только к колебаниям
Р—О — С фоефатэв, причем изменение валентного со-
стояния и переход к трзхвалентному фосфору приводят
к смещению полосы поглощения в более низкочастотную
область спектра.
Некоторые исследователи пытались идентифицировать
это поглощение, связывая рассмотренную ниже полосу по-
глощения вблизи 1150 см'1 с независимыми валентными
колебаниями отдельных частей этой группировки, т. е.
связями Р — О — и С — О — Так, Томас [28] и Мак-
Айвор и др. [31] относят полосу 1030 см'1 к колебанию
Р — О — (С), а более высокочастотную полосу к колеба-
нию С —О — (Р). С другой стороны, Марсен [25] связы-
вает колебание С — О — (Р) с поглощением в области
1030 см 1 и относит другое колебание к меньшим частотам.
Гл. 18. Фосфорорганические соединения 449
Если возможна какая-либо дифференциация между этими
двумя колебаниями, то представляется более правильным
отнесение полосы 1030 см'1 к колебанию С — О — (Р),
ввиду близкой аналогии с алифатическими сложными эфи-
рами и спиртами. Кроме того, при переходе к арилфосфатам
эта полоса смещается, занимая практически то же положе-
ние, что и в случае ароматических сложных эфиров. Од-
нако, по-видимому, столь глубокую дифференциацию от-
дельных полос поглощения вряд ли можно считать надеж-
ной, так как массы атомов близки и колебания могут сильно
взаимодействовать. Могут также проявляться, например,
симметричные и антисимметричные валентные колебания
О
и
— О — Р — О— , приводящие к появлению разных полос
поглощения. Бергманн и др. [10] предположили, что появ-
ление полосы 1030 см 1 объясняется именно этой причи-
ной. Однозначное решение вопроса о природе этих полос
поглощения поэтому затруднительно, что ни в коем случае
не влияет на возможность использования их для решения
различных аналитических задач. Никаких детальных иссле-
дований интенсивности этой полосы не проводилось, но
в спектрах триалкилфосфатов она обычно имеет очень боль-
шую интенсивность. Однако следует отметить, что многие
другие скелетные колебания могут вызывать поглощение
в той же области спектра, хотя маловероятно, чтобы это
поглощение было сравнимой интенсивности. Помимо таких
известных примеров веществ со связью С — О— , как слож-
ные эфиры и спирты, сильное поглощение в области 1000—
980 см'1 отмечено Харвеем и Мейхудом [24] у таких соеди-
нений, как (CH3)2NPOC12.
в. Колебания Р — О — С2Н5. По наблюдению Даша
и Смита [5] этилфосфаты и подобные им вещества, содержа-
щие группу Р — О — С2Н5, все обладают узкой, но слабой
по интенсивности полосой поглощения при 1160 см'1. Мы
подтвердили эти данные для многих других соединений
и отметили также, что аналогичная, но более интенсивная
полоса появляется и при наличии группы Р — О — Аг.
У всех 70 изученных нами соединений, содержащих
какую-либо из этих групп, полоса появляется в интервале
29 л. Беллами
450
Часть IV
1156—1163 см”1, причем единственным исключением
является этилфосфородихлоридит, у которого полоса на-
ходится при 1143 см”1. Таким образом, присутствие по-
лосы в этом интервале частот указывает на наличие одной
из этих групп; в зависимости от того, появляется ли другая
полоса при 1200 или при 1030 см-1, можно установить, ка-
кая именно группа присутствует. Томас [28] недавно иссле-
довал это поглощение у более широкого круга алифатиче-
ских соединений и показал, что рассматриваемая корреля-
ция с несколько расширенным интервалом частот может
быть применена и к структурным группам — СН2 — О — Р.
Из 196 соединений, содержащих такую структуру, 185 по-
глощают в области 1170—1140 см'1, тогда как 65 из 69 слож-
ных эфиров с разветвленными цепями, содержащими группу
^>СН — О — Р—.поглощают в интервале частот 1150—
ИЗО см”1. Эта полоса поглощения представляется, следо-
вательно, достаточно характеристичной для самой группы
С — О — Р, что служит основанием для отнесения ее
к колебанию С — О — (Р). Однако, как предполагалось
выше, в той мере, в какой вообще возможна какая-либо
дифференциация связей С — О — и —О — Р—, более есте-
ственным представляется сопоставление более высокоча-
стотной полосы поглощения с колебанием [91] по связи
Р — О — по аналогии с соответствующей полосой 1250 см"1
у сложных эфиров и более низкочастотной полосы
с остатком С—О — по аналогии со сложными эфирами
и спиртами.
г. Колебания Р — О — СН3. Группа Р — О—СН3
дает слабую, но вполне отчетливую полосу поглощения при
1190 см"1. Это показано Дашем и Смитом [5] и подтверждено
на ряде примеров в нашей лаборатории. Частоты полос по-
глощения Р — О — СН3 и Р — О — С2Н5 обычных алкил-
фосфатов достаточно постоянны, но при введении в молекулу
атома галогена или других заместителей они заметно из-
меняются. Томас [28], например, привел более широкую
область для поглощения СН3—О — Р (1190—1170 см*"1).
Тем не менее и эти пределы оказываются еще удовлетвори-
тельными для определения этой группировки у алкилфосфа-
тов, если учитывать при этом наличие полосы 1030 см”1.
Гл. 18. Фосфорорганические соединения
451
4. ПИРОФОСФАТЫ
Различными группами исследователей выполнена боль-
шая работа с целью идентификации поглощения, характер-
ного для связи Р — О—Р пирофосфатов. Бергманн и др.
[10] исследовали три пирофосфата и сравнили их спектры
со спектрами исходных фосфатов. Они нашли, что в каж-
дом случае пирофосфат имеет на одну полосу больше, чем
фосфат, и что эта полоса лежит в области 970—930 см'1.
Они указали также, что, по данным Даша и Смита [5],
у н-бутилпирофосфата также имеется полоса при 950 см'1.
Поэтому они отнесли поглощение в этой области к антисим-
метричным колебаниям Р — О — Р.
С другой стороны, Холмштедт и Ларссон [9] ориенти-
ровочно предположили, что колебания Р — О пирофос-
фатной группы проявляются в области 710 см'1. Корбрид-
жем и Лоувом [32] изучено поглощение в этой области
у ряда органических соединений. У соединений этого клас-
са ими обнаружено поглощение Р — О — Р около 900 см'1,
а также вблизи 700 см'1, что подтверждает, таким образом,
обе предложенные корреляции. Однако многие из получен-
ных ими данных относятся к полипирофосфатам, и предпо-
лагается, что полоса поглощения -— 900 см 1 в случае фос-
форорганических пирофосфатов характеризуется более вы-
сокой частотой. Харвей и Мейхуд [24] на основании иссле-
дования 14 соединений того же типа пришли к заключению,
что для этого поглощения характерна область 950—910 см'1,
а Мак-Айвор и др. [31] подтвердили эти корреляции, ис-
следовав ряд других соединений и указав в качестве верх-
него предела частоты поглощения 970 см 1. Аналогичные
данные привели Томас [28] и Симон и др. [33]. Таким обра-
зом, имеются, по-видимому, достаточные основания для
отнесения поглощения Р — О — Р у пирофосфатов к этой
области частот. У тионопирофосфатов, однако, характери-
стическая полоса поглощения смещена в область более
низких частот [31]. Теми же исследователями изучена
и область 700 см'1, но не получено никаких удобных кор-
реляций, по-видимому, из-за множества пиков поглощения,
которые обычно имеют место в этой области спектра.
В некоторых особых случаях соответствующая полоса
поглощения может быть довольно полезна, однако следует
29*
452
Часть IV
иметь в виду, что она находится в области спектра, где по-
глощают многие другие фосфорорганические соединения.
Со стороны более высоких частот такое поглощение частично
перекрывается полосой Р — О — С (алкил) и полосой в об-
ласти 980 см-1, рассмотренной ниже в разделе 12. Хар-
вей и Мейхуд [24] предполагают, что отличить это погло-
щение от полос Р — О — С , по-видимому, можно, измеряя
полуширину полос, так как полоса поглощения пирофос-
фатов заметно шире, чем другие полосы в этой области.
Тем не менее идентификация этой группы в неизвестном
соединении остается проблемой, представляющей значитель-
ные затруднения.
5. ВАЛЕНТНЫЕ КОЛЕБАНИЯ Р—ОН
Известно, что валентные колебания группы ОН сравни-
тельно мало зависят от природы присоединенной к кисло-
роду группы. Однако в фосфорных соединениях кислотного
О
характера, содержащих группу РТ7 , эффект образова-
ли
ния водородной связи даже больше, чем в случае карбоно-
вых кислот, а поэтому полоса колебаний ОН имеет вполне
характеристическую частоту. Даш и Смит [5] и Беллами
и Бичер [7, 8] не обнаружили обычной полосы ОН ни у од-
ной из исследованных ими кислот; вместо них эти авторы
наблюдали широкую нерезкую полосу поглощения в обла-
сти 2700—2560 слг \ В случае растворов с неполярными рас-
творителями эта полоса сохраняется. Она, очевидно, свя-
зана с группой ОН, так как исчезает при образовании
соли, причем значение частоты колебаний Р = О в этом
случае также подтверждает предположение о наличии очень
сильной водородной связи. Аналогичные результаты полу-
чены также по синтетическим кефалинам [8, 12] и кислым
нафтилфосфатам [13]. Подобные же свойства обнаружены
у неорганических фосфорных кислот и родственных им
соединений [22], а Томас [28] подтвердил, что тиофосфор-
ные кислоты также поглощают в этой области спектра,
хотя отметил при этом, что у соединений, содержащих
\ S
группу , эта частота имеет более высокое значе-
/ ХОН
Гл. 18. Фосфорорганические соединения
453
ние вследствие меньших сил ассоциации. С другой
стероны, у таких кислот, как трифторметилфосфонат
натрия, полоса поглощения наблюдается при более
низких частотах [26, 27] (около 2350 с’.и ') в связи с уси-
лением водородной связи из-за наличия фторметилза-
мещения.
Кроме того, фосфорорганические кислоты дают широкие
нерезкие полосы поглощения в области 2500—1600 см"1,
которые еще не достаточно исследованы. Эти полосы погло-
щения обычно являются слабыми и широкими, и во многих
случаях их трудно обнаружить. Томас [28] отнес полосу
поглощения 1650 evr1 к деформационному колебанию ОН,
однако это значение частоты представляется слишком
высоким по сравнению с частотами, характерными для
карбоновых кислот и фенолов.
Для полосы деформационных колебаний Р — ОН не
установлена точная корреляция, и вряд ли ей может соот-
ветствовать очень узкий интервал частот. Однако в некото-
рых случаях эта полоса находится, по-видимому, вблизи
1030 см"1. Так, например, кислый дифенилфос4ат погло-
щает при 1030 см""1, тогда как ни у серебряной соли, ни
у трифенилфосфата такого поглощения не обнаружено.
Аналогичные данные приведены Корбриджем и Лоувом [22],
тогда как Эмелеусом и др. [26, 27] частота деформационных
колебаний Р — ОН у ряда фторированных соединений отне-
сена примерно к 940 см1. Эта корреляция представляется
правдоподобной, так как соответствующая частота в слу-
чае димеров карбоновых кислот имеет значение около
900 см Однако в некоторых случаях идентификация этой
полосы поглощения затрудняется в связи с возможностью
присутствия в той же области других полос поглощения,
особенно полосы Р — О — С. Действительно, Томас [28]
в соответствии с предложенными им ранее керреляциями
отдает предпочтение отнесению этой полосы к валентным
колебаниям Р—О(Н). Было бы неразумным пытаться
идентифицировать группы Р — ОН по поглощению
в низкочастотной области спектра, но в тех случаях,
когда отыскивается поглощение Р — О — С (алкил),
следует иметь в виду, что в области примерно 1030 см'1
можно наблюдать поглощение, обусловленное и груп-
пами Р — ОН.
454
Часть IV
6. ВАЛЕНТНЫЕ КОЛЕБАНИЯ Р—Н
Различными исследователями [1, 5—7, 27, 28, 32]
изучено небольшое число неорганических и органических
соединений, содержащих связи Р — Н, и у всех у них в об-
ласти 2440—2350 см'1 обнаружены резкие полосы погло-
щения средней интенсивности. Следовало ожидать, что
полосы валентных колебаний Р — Н должны появляться
как раз в этой области; кроме того, оказалось, что сам
фосфин поглощает при 2327 и 2421 см'1 [14]. Таким обра-
зом, справедливость указанного отнесения можно считать
полностью доказанной. Почти во всех случаях найдено,
что полоса является узкой, а тот факт, что у соответствую-
щих диалкилфосфонатов полоса Р = О появляется почти
точно в ожидаемой области, позволяет предположить, что
группа Р — Н не может заметно участвовать в образова-
нии водородной связи.
Полный интервал 2440—2350 см'1, в котором появляется
полоса поглощения Р — Н (исключение составляет фосфин),
по литературным данным, по-видимому, шире, чем можно
было ожидать для простых валентных колебаний этого
типа, и, согласно предположению Томаса [28], вероятно,
имеется возможность различать фосфонаты (2450—2400 см'1)
и фосфинаты (2350—2325 см'1) по положению этой полосы
поглощения. Деформационное колебание Р — Н не иден-
тифицировано ни в одном случае, кроме спектра самого
фосфина.
7. ФОСФОР-УГЛЕРОДНЫЕ СВЯЗИ
а. Ароматические соединения. Значительное количе-
ство фосфорсодержащих ароматических соединений с груп-
пой Р — Аг изучено Дашем и Смитом [5], причем во всех
случаях найдены резкие полосы средней интенсивности
в интервалах 1450—1435 и 1005 —995 см'1. Эти полосы от-
несены к связи Р — С6Н5, но авторы указывают, что с та-
ким же успехом они могут быть обусловлены активирован-
ными колебаниями кольца. Это заключение, по-видимому,
справедливо для полосы с меньшей частотой, поскольку
в этой же области нами была обнаружена довольно слабая
полоса поглощения для многих соединений, содержащих
группу Р—О—СвН5, а также было показано, что в зави-
Г л. 18. Фосфорорганические соединения
455
симости от природы заместителей в ароматическом кольце
эта полоса смещается так, как если бы она была обуслов-
лена колебаниями кольца. Полосы 1440 см"1, однако, не
обнаружено в каком-либо из этих спектров, и нет основа-
ний думать, что она недостаточно характерна для группы
Р—С0Н5. Эта корреляция подтверждена также Корбрид-
жем [23] на основании его неопубликованных данных. Вбли-
зи от этой частоты поглощают также соединения, содержа-
щие группу Si — С0Н5 (гл. 20), но это не существенно, так
как обе эти группы находятся вместе весьма редко.
б. Алифатические соединения. Пригодных корреляций
для Р — С-связи в алифатических соединениях не уста-
новлено, и поскольку полосы валентных колебаний Р — С
у таких соединений, как триметилфосфиноксид [15], ле-
жат, по-видимому, в области 750—650 см"1, где поглощают
также многие другие фосфорорганические соединения,
то сомнительно, чтобы какие-либо корреляции могли быть
вообще установлены.
В случае соединений со связью Р — СН3 характеристи-
ческая полоса поглощения должна быть обусловлена анти-
симметричными деформационными колебаниями СН3. Най-
дено, что эта полоса появляется [22, 24, 28, 31] в области
1320—1280 см"1 и в большинстве случаев является полосой
сильной или средней интенсивности. У соединений, содер-
жащих пятивалентный фосфор, эта полоса обычно наблю-
дается в более высокочастотной части указанного интервала,
тогда как у соединений трехвалентного фосфора [28] она
проявляется ближе к 1285 см"1. Эта полоса, несмотря на то
что она проявляется в узком интервале частот, не является,
однако, совершенно специфичной для группы, так как
вблизи этого интервала поглощают также группы N(CH3)2,
входящие часто в состав фосфорорганических инсектицидов.
Выдвинуто предположение, что дифференциация этих двух
полос может оказаться возможной при измерении их полу-
ширины, так как полуширина полосы Р — СН3 заметно
меньше [24, 28].
8. СВЯЗИ ФОСФОРА С СЕРОЙ
Предполагалось, что корреляции, относящиеся к поло-
сам Р = S и Р — S— инфракрасных спектров, могут быть
456
Часть IV
успешно использованы при изучении фосфорорганических
инсектицидов, так как вместе с данными о соответству-
ющих фосфоркислородных связях они позволяют получить
сведения о сложной изомеризации, которая происходит
при нагревании тиофосфатов. Однако, хотя в этой области
выполнена значительная работа, оказалось, что соответст-
вующее характеристическое поглощение очень мало интен-
сивно и поэтому имеет ограниченное значение.
а. Валентные колебания Р S. Соединения PSC13
и PSBr3 поглощают при 753 и 718 см"1, а у соответствую-
щих оксихлоридов данные полосы отсутствуют. Этот факт
позволил сделать предположение о наличии полосы Р = S
в области 750 см'1, которое затем было подтверждено Го-
ром, вычислившим на основании правила Горди значение
752 см'1. Даш и Смит [5] и Гор [4] исследовали значитель-
ное число тиофосфатов различных типов и сравнили полу-
ченные спектры со спектрами соответствующих фосфатов.
Все результаты свидетельствуют о том, что полоса погло-
щения Р = S лежит в области 750—600 см'1, однако на-
блюдаемая интенсивность очень переменна. В немногих
отдельных случаях полоса Р = S очень интенсивна, но
у подавляющего большинства соединений она сравни-
тельно слабая, а иногда и Вообще не может быть обнаруже-
на. Для триметил тиофосфата, например, не наблюдалось
заметной полосы в области 800—600 см'1. Эти общие дан-
ные подтверждены Корбриджем [23], но в то же время
Мак-Айвор и др. [31] и Томас [28] предполагают, что область
поглощения лежит при несколько более высоких частотах.
Мак-Айвор и др. обнаружили поглощение средней интен-
сивности в области 840—780 см'1 у ряда исследованных
ими соединений, содержащих связь Р = S. У некоторых
соединений, у которых к атому фосфора присоединены
также SR, SH или атомы хлора, появляется дополнитель-
ная интенсивная полоса поглощения около 650 см'1. Изу-
чавшие эти. соединения исследователи не смогли провести
отнесения полосы поглощения Р = S, но считают более
вероятным отнесение ее к области 800 см"1. Более опре-
деленное разделение соединений этого типа на различные
классы проведено Томасом. Он указывает, что соединения
типа (RO)3PS поглощают в интервале 845—800 см'1,
ГлЯ 18.Ч Фосфорорганические соединения
457
(RO)2RP = S —в интервале 805—775c.tr1, (RO)R1R2PS —
в интервале 790—770 см'1 и фосфинсульф иды — в интер-
вале 770—750 см'1. Это изменение частоты поглощения
хорошо согласуется с ходом изменения частоты Р = О,
чем объясняется, например, считавшееся прежде аномаль-
ным поведение триметилтиофосфата, который поглощает
при 815 см'1. С другой стороны, при этом не ясно, почему,
например, РОС13 поглощает в более высокочастотной об-
ласти, чем соединения типа PO(OR)3, а частота поглощения
PSC13 753 см'1 заметно меньше, чем частота поглощения
его аналога с заместителями OR. Тем не менее рассмотрен-
ные представления дают некоторые основания для предпо-
ложительных корреляций частот Р = S, хотя наличие
в этой же области спектра во многих случаях ряда допол-
нительных полос поглощения снижает значение этих кор-
реляций.
б. Валентные колебания Р — S—. Положение каких-
либо полос Р — S— известно недостаточно точно и, ве-
роятно, не является постоянным. Гор заметил, однако,
что полоса 1200 см'1, характерная для группировки
Р — О—С6Н5, не наблюдается в случае группировки
Р—S—С0Н5. Если полоса поглощения P = S лежит
около 700 см'1, то колебания группы с одинарной связью
должны, по-видимому, иметь более низкие значения частот,
находящиеся за пределами обычного рабочего интервала
призмы из каменной соли. Томас [28] и Мак-Айвор и др.
[31] предполагают, что это поглощение зависит частично
от природы других заместителей и проявляется в интервале
575—510 см'1.
9. СВЯЗИ’ ФОСФОРА С АЗОТОМ
а. Колебания Р — NH2 и Р — NH—. Исследовано
лишь ограниченное число фосфорамидатов и родственных
им веществ [7, 9, 32]. Валентные и деформационные колеба-
ния NH очень мало зависят от наличия атома фосфора,
и соответствующая им полоса появляется почти в том же
месте, как и у обычных аминов. Так, при исследовании
в твердом состоянии соединения с группой Р — NH2 дают
две и иногда три полосы валентных колебаний NH в области
458
Часть IV
3330—3150 см"1. Разбавленные растворы, в случае кото-
рых водородные связи нарушены, поглощают около 3400 слГ1.
Аналогичным образом и соединения с группой Р — NH —
дают в этой области обычное поглощение. У соединений
с группой Р — NH2 обнаружены также полосы поглощения
деформационных колебаний в области 1570—1550 см"1;
исключением является структура типа биполярного иона
[7], как, например, у кислого бензилфосфорамидата.
У анилидофосфорных соединений характеристичные по-
лосы валентных колебаний в группировке Аг — NH —
наблюдаются в обычной области 1280 см1, где их можно
спутать с полосами колебаний Р О [7].
б. Валентные колебания Р — N. Ряд исследователей
пытался идентифицировать характеристическую частоту
валентных колебаний Р — N. Холмштедт и Ларссон [91
выдвинули предположение, что колебание может прояв-
ляться в спектре около 775 см"1; это предположение было
подтверждено Харвеем и Мейхудом [24] и Корбриджем
[23], которые обнаружили соответствующее поглощение
в области 750—680 см"1 в тех случаях, когда имеется груп-
па Р — N. Ларсеном [34] рассмотрен также случай, когда
/С
имеется структурная группировка Р — , которой,
по его мнению, обусловливается поглощение в области
820—780 см"1. При любой корреляции частот для связи
Р — N соответствующая полоса должна быть, по-видимо-
му, чувствительна к изменению масс заместителей и вслед-
ствие этого должна претерпевать значительные смещения
даже при небольших изменениях в структуре молекулы.
Корреляция для этих колебаний не представляет поэтому
большой ценности, а в ряде случаев оказалось вообще не-
возможным идентифицировать рассматриваемую полосу
поглощения. Это подтверждено Марсеном [25] и отнесено
им за счет наложения других полос, таких, как полоса,
соответствующая валентным колебаниям С — Р, а Томас
[28] отметил также ряд веществ, у которых в указанной
области не наблюдается поглощения, обусловленного нали-
чием связи Р — N —; эти данные согласуются с получен-
ными нами результатами.
Гл. 18. Фосфорорганические соединения
459
В случае циклических структур, когда появляется связь
Р = N, в области более высоких частот обнаружено погло-
щение, которое может быть связано с колебаниями этой
группы. У фосфонитрилхлоридов к этому поглощению
Дашем [34] отнесены полосы в области 1300—1200 см1;
у солей фосфонитриловых кислот также обнаружены [22]
интенсивные полосы поглощения в области 1300—1100 см'.
В связи с отсутствием какого-либо специфического по-
глощения Р — N были предприняты попытки найти часто-
ты, характеристические для больших остатков, таких,
как Р — N — СН3 и Р — N(CH3)2. Действительно, при-
соединение метильной группы к связи N — Р приводит
к появлению характеристической полосы деформационных
колебаний СН3 [24, 25, 28] в области 1325—1290 с.и1.
Этот интервал перекрывается областью частот, соответ-
ствующих поглощению Р — СН3, но эти две полосы можно
различить, измеряя полуширину полос [24,28]. Рассмотрен
также ряд других частот, характерных, как установлено,
для структурной группы N(CH3)2 [24, 25]. Большинству
из них соответствуют полосы средней интенсивности, кото-
рые очень трудно надежно идентифицировать. Следует,
однако, отметить, что наличие диметиламиновой группы,
присоединенной непосредственно к атому фосфора, при-
водит, по-видимому, к появлению сильного поглощения
в области 1010—990 см 1 [24, 25, 28], где оно может быть
спутано с поглощением Р — О — С (алкил).
10. СВЯЗИ ФОСФОРА С ГАЛОГЕНАМИ
а. Колебания Р — С1.’ Треххлористый фосфор [16]
имеет две полосы поглощения (при 511 и 488 см'1), которые
соответствуют валентным колебаниям Р — С1; для каждого
из соединений РОС13 и PSC13 также обнаружены две полосы
поглощения в этой области спектра. Даш и Смит [5] иссле-
довали семь других соединений со связью Р — С1, в молеку-
лах которых, за исключением одного соединения, содержа-
лось более чем по одному атому хлора. Полосы наблюдались
в интервалах частот 572—500 и 500—433 см'1. Окись ди-
фенилхлорфосфина, как и следовало ожидать, имеет только
одну полосу при 521 см'1. Эти данные подтверждены
4(0
Часть IV
в последующих работах Корбриджа [23], Томаса [28] и Мак-
Айвора и др. [31]. Все эти исследователи обнаружили ин-
тенсивную двойную полосу, обычно находящуюся в области
580—440 см'1. Мак-Айвор и др. [31] рассмотрели эту кор-
реляцию более детально и разбили всю область на интер-
валы, соответствующие соединениям различных классов.
Так, у соединений трехвалентного фосфора этот дублет на-
ходится около 500 см-1, смещаясь в высокочастотную об-
ласть (до 540 см'1) в случае фосфатов и в низкочастотную
область (примерно до 475 см'1) в случае тионофосфатов.
Однако эти исследователи отметили также, что положение
полосы зависит и от таких факторов, как число заместите-
лей — галогенов и наличие связей С — Р, а поэтому ука-
занные более детальные корреляции следует использовать
с осторожностью.
б. Колебания Р — F. Положение пиков полос погло-
щения Р — F изучали также Даш и Смит [5]. Они нашли,
что, как и в случае связей С — F, имеется интенсивная
полоса поглощения, положение которой в спектре, оче-
видно, меняется от 900 до 800 см'1. Исследованы шесть
соединений этого типа, причем некоторые из них содержали
более одной связи Р — F и давали несколько пиков. Все
эти соединения дают в указанной области по крайней мере
одну интенсивную полосу, и на основании своих наблюде-
ний указанные авторы предположительно отнесли колеба-
ния Р — F соединений пятивалентного фосфора к области
900—850 см'1, а соединений трехвалентного фосфора —
к. 900—750 см'1. У PF3 первая основная полоса наблю-
дается при 890 см'1 [ 16]. Эта корреляция была подтверждена
Ксрбриджем, который указал также общие пределы погло-
щения (£00—720 с.и-1). Более детальное исследование вы-
полнено Томасом [28], который изучил 64 соединения этого
типа. Полученные им результаты показывают, что эти
довольно широкие пределы области поглощения для всех
соединений, кроме самих PF3 и POF3, практически могут
быть сужены до 885—810 см'1.
в. Связи Р — Вг и Р — I. Связи фосфора с бромом
и иодом мало изучены. Частоты валентных колебаний
Р — Вг в РВг3 [16] равны 400 и 380 см'1.
Гл. 18. Фосфорорганические соединения
461
11. ИОННАЯ ФОСФАТНАЯ ГРУППА
У неорганических фосфатов типа М;РО4' найдена [17]
интенсивная полоса в области 1050—1000 см'1, а в некото-
рых случаях имеются также указания [10, 14] на появление
второй полосы поглощения в области 980 см'1. У кислых
фосфатов наблюдается отчетливое и возрастающее смещение
поглощения в сторону больших частот. Многие соединения
типа НРО( поглощают в области 1070—1050 см'1, а типа
Н2РО;— в области 1090—1030 см'1 [17]. В обоих случаях
наблюдается также более слабая полоса поглощения в об-
ласти 1000—900 см'1 (см. гл. 21). Эти первоначально уста-
новленные корреляции были затем подтверждены и развиты
в результате детальных исследований неорганических фос-
фатов, выполненных Корбриджем и Лоувом [22, 23, 32].
Благодаря этим работам теперь имеются корреляции для
ортофосфатов (одно-, двух- и трехосновных), фосфор-
фторидатов, метилфосфонатов, одноосновных фосфорамида-
тов, гипофосфатов, пирофосфатов и т. п., полученные, как
правило, на основании исследования по крайней мере
восьми солей каждого класса соединений. Эти данные сум-
мированы в очень удобной форме в виде корреляционных
диаграмм [23], относящихся исключительно к солям фос-
форных оксикислот, и находят широкое применение в ана-
литической работе.
У фосфорорганических кислот положение полос еще
более неопределенно, так как электроотрицательность ор-
ганических заместителей может оказывать непосредствен-
ное влияние на длины связей резонансного гибрида, проис-
,О
хождение которого обусловливается структурой Ру
В настоящее время нет достаточных данных для установле-
ния определенной корреляции, однако у шести солей аро-
матических кислот типа
.О
(RO)2P\
ХО
обнаружены силь-
ные полосы поглощения в области 1090—1040 см'1, в кото-
рой соответствующие триарилфосфаты не поглощают [7, 8].
Изучались также четыре соли монозамещенных аромати-
ческих кислот [(RO)PO3]2~ [7, 8], которые поглощают
462
Ч а с т ь IV
в области 1100—1050 см'1. Почти для всех этих солей
полосы поглощения наблюдались вблизи верхней границы
указанного интервала, но число исследованных соедине-
ний слишком мало, чтобы можно было решить вопрос о том,
происходят ли какие-либо смещения частот, подобные тем,
которые наблюдаются у соответствующих неорганических
соединений.
При наличии алифатических заместителей область
1050 см'1 не может быть исследована, так как связь
Р—О —С (алифатический радикал) сама по себе дает здесь
сильное поглощение, но для шести солей алифатических
кислот (RO)2PO“ найдено, что они поглощают в области
1185—1156 см'1, где ни соответствующие арилпроизводные,
ни соответствующие алкилфосфаты не поглощают [8].
Эта область поглощения также отнесена к ионной фосфат-
ной группе. В более поздней работе Марсен [25] отнес
эту полосу к антисимметричным валентным колебаниям РО2,
тогда как поглощение, соответствующее симметричному
колебанию, проявляется вблизи поглощения Р — О — С
(алкил), т. е. около 1080 см'1. Эмелеус и др. [26, 27] иссле-
довали также ряд ионных солей, у которых к атому фос-
фора присоединена группа CF3. У этих веществ основное
поглощение, соответствующее группе ^РО2, обнаружено
вблизи 1250 см'1, что указывает на зависимость частоты
этих колебаний от величины электроотрицательности за-
местителей. Эти предварительные данные позволяют, таким
образом, предположить, что полоса поглощения ионной
фосфатной группы солей фосфорорганических кислот рас-
положена где-либо в пределах 1250—1040 см'1 и что ее
истинное положение определяется больше природой орга-
нических заместителей, чем величиной отрицательного
заряда ионной группировки.
У многих ароматических фосфатных ионных соедине-
ний найдены также полосы поглощения в области 920—
880 см'1, которые сходны с аналогичными полосами неорга-
нических веществ [8]. Однако они часто малоинтенсивны,
и хотя предполагалось, что эти полосы также связаны
с колебаниями ионной фосфатной группы [7], детального
сравнения этой области спектра для ароматических и али-
фатических производных не проводилось.
Гл, 18. Фосфорорганические соединения
463
12. ДРУГИЕ КОРРЕЛЯЦИИ ДЛЯ ФОСФОРОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
Из многочисленных опубликованных в литературе спект-
ров следует, что для подавляющего большинства фосфор-
органических соединений обнаружена очень интенсивная
полоса поглощения около 980 сж-1 [1, 4—6, 8—10]. Был
выдвинут ряд предположений, объясняющих ее происхож-
дение, но, поскольку эта полоса появляется у соединений
различных типов, отнесение ее к определенной структур-
ной группировке молекулы затруднительно. Холмштедт
и Ларссон [9], например, на основании данных об ограни-
ченном числе соединений приписали ее валентным С — N-
। /N
колебаниям структуры Р — С < , но это объяснение
XN
не является единственно возможным, так как такая полоса
имеется у большинства фосфатов, фосфонатов и фосфони-
тов. Бергманн и др. [10] полагают, что эта полоса у
алкилфосфатов и пирофосфатов соответствует симметричным
валентным колебаниям трех групп Р — OR, а потому ана-
логична полосе поглощения 980 см"1 ионного фосфатного
радикала. Однако, как и в случае полосы 1030 см"1 (кото-
рую они относят к соответствующим антисимметричным
колебаниям), трудно объяснить, почему для фосфатов, фос-
фонатов и фосфонитов, как и для соответствующих кислот
с органическими заместителями, частота должна оставаться
неизменной.
Происхождение этой полосы обсуждалось также Бел-
лами и Бичером [8], которые весьма предположительно от-
несли ее к валентным колебаниям Р — О соединений пяти-
валентного фосфора. Указанные авторы отмечают между тем,
что если их предположение справедливо, то можно сделать
вывод о связи полосы 1030 см"1 с колебаниями О — С,
а полосы 980 см"1 с колебаниями Р — О группировки
Р — О — С. Некоторые подтверждения правильности этой
точки зрения получены Марсеном [25], который отнес по-
следнюю полосу к антисимметричным валентным колеба-
ниям Р — О, но очевидно, что, прежде чем можно будет
сделать окончательные выводы, требуется провести допол-
нительные исследования. Это особенно ясно в связи
464
Часть IV
с исследованием, выполненным Томасом [28], результаты
которого показывают, что в некоторых случаях полосы, со-
ответствующие колебаниям? — О — С, можно ожидать при
частотах меньше 990 см'1.
Однако исключительно высокая интенсивность полосы
поглощения делает ее особенно пригодной для целей иденти-
фикации, а наличие сильной полосы около 980 см'1 часто
служит подтверждением данных о присутствии фосфорорга-
нического соединения, полученных при исследовании дру-
гих областей спектра.
литература
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
Meyrick, Thompson, J. Chem. Soc., 1950, 225.
Yost, Anderson, J. Chem. Phys., 2, 624 (1934).
Арбузов Б. А., Батуев M. И., Виноградова В. С.,
ДАН СССР, 54, 599 (1946).
Gore, Discuss. Faraday Soc., N 9, 138 (1950).
Daasch, Smith, Analyt. Chem., 23, 853 (1951), N R.L.
Report, 3657.
Bellamy, Beecher, J. Chem.' Soc., 1952, 475.
Bellamy, Beecher, J. Chem. Soc., 1952, 1701.
Bellamy, Beecher, J. Chem. Soc., 1953, 728.
Holmstedt, Larsson, Acta Chem. Scand., 5, 1179
(1951).
Bergmann, Littaue
1952, 847.
Kosolapoff, M с C u 1
73, 5392 (1951).
Baer, Maurukas, Rt
r, P i n c h a s, J. Chem. Soc.,
lough, J. Amer. Chem. Soc.,
i s s e 1 1, J. Amer. Chem. Soc.,
74, 152 (1952).
Friedman, Seligman, J. Amer. Chem. Soc., 73, 5292
(1951).
14. Г e p ц б e p г Г., Колебательные и вращательные спектры
многоатомных молекул., Издатинлит, М., 1949, стр. 326
15. Daasch, Smith, J. Chem. Phys., 19, 22 (1951).
16. Герцберг Г., Колебательные и вращательные спектры
многоатомных молекул,. Издатинлит, М., 1949, стр. 182.
17. М i 1 1 е г, Wilkins, Analyt. Chem., 24, 1253 (1952).
18. Н a n a h a n, J а у к о, J. Amer. Chem. Soc., 74, 5070 (1952).
19. M i s I о w, J. Amer. Chem. Soc., 75, 5155 (1952).
20. Baer, J. Amer. Chem. Soc., 75, 621 (1953).
21. Marinetti, Berry, Rouse r, Stotz, J. Amer.
Chem. Soc., 75, 313 (1953).
22. Corbridge, Lowe, J. Chem. Soc., 1954, 4555.
23. С о r b r i d g e, J., Appl. Chem., 6, 456 (1956).
24. Harvey, Mayhood, Can. J. Chem., 33, 1552 (1955).
25. M a a r s e n, Thesis, University of Amsterdam, 1956.
Г л. 18. Фосфорорганические соединения
465
26. Е in е I е u s, Haszeldine, Paul, J. Chem. Soc., 1955,
563.
27. Benet t, Emeleus, Haszeldine, J. Chem. Soc ,
1954, 3598.
28. Thomas, J. Appl. Chem., 1957, 198, частное сообщение.
29. G u t о w s к у, Liehr, J. Chem. Phys., 20, 1652 (1952).
30. Bell, Heisler, Tannebaum, Goldenson,
J. Amer. Chem. Soc., 76, 5185 (1954).
31. Me Ivor, Grant, Hubley, Can. J. Chem., 39,
1611 (1956).
32. Cor br i dge, Lowe, J. Chem. Soc., 1954, 493.
33. Simon, S t о 1 z e r, Chem. Ber., 89, 2253 (1956).
34. L a a r s e n, Acta Chem. Scand., 6, 1470 (1952).
35. Daasch, J. Amer. Chem. Soc., 40, 397 (1954).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Попов E. M., Кабачннк М. И., М а я н ц Л. С., Успехи
химии, 30, 846 (1961).
30 л. Беллами
Глава 19
ГАЛОГЕНПРОИЗВОДНЫЕ ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
1. ВВЕДЕНИЕ
Для определения галогенов в органических веществах
нет необходимости пользоваться методами инфракрасной
спектроскопии, поскольку галогены гораздо проще опре-
делить другими методами. Иногда, хотя и редко, прихо-
дится прибегать и к этим методам, если количества иссле-
дуемых веществ очень малы или если вещества очень дороги.
Тем не менее желательно располагать сведениями о положе-
нии полос поглощения углерод — галоген и о влиянии
замещения этого типа на другие области спектра.
Единственной характеристической полосой поглоще-
ния галогенпроизводных в области спектра, исследуемой
при помощи призмы из каменной соли, является полоса
валентных колебаний С — X. Эти колебания соответствуют
колебаниям скелета, содержащего одинарные связи, и,
следовательно, их частота значительно меняется в резуль-
тате взаимодействия с колебаниями соседних групп. Это
наблюдается в случае легких галогенов — фтора и хлора,
массы которых не сильно отличаются от массы углеродного
скелета. В случае бромпроизводных органических соедине-
ний эффект взаимодействия меньше, и поэтому полоса ва-
лентных колебаний С — Вг имеет более постоянную частоту.
При изучении жидких галоидных алкилов и их растворов
обнаружено несколько частот валентных колебаний С — X.
Это обусловлено сосуществованием различных поворотных
изомеров с отличающимися фундаментальными частотами.
В случае простых галоидных алкилов таких форм только
две: конфигурации транс и гош, но у более сложных соеди-
нений, таких, как 1,2-дихлорпропан или галогениды с длин-
ной цепью, число возможных изомеров возрастает. Вы-
полненено очень большое число работ по исследованию
Гл. 19 Галогенпроизводные органических соединений 467
отдельных молекул такого типа, в которых имеется много
данных относительно валентных колебаний С— X. Об-
ширный обзор этих данных сделан Мидзусима [12], деталь-
ное изучение этого вопроса провели недавно Браун и Шеп-
пард [13, 14]. В этих работах установлено, что в твердом
состоянии остается лишь одна поворотно-изомерная форма;
обычно это транс-изомер, имеющий более высокую ча-
стоту валентных колебаний С — X. Аналогичные резуль-
таты получены по частотам валентных колебаний С — X
аксиальной и экваториальной форм хлорированных цикло-
гексанов [15] и стеринов [16], которые весьма полезны для
целей диагностики.
При замещении фтором наблюдается наибольший эффект
взаимодействия, и полоса валентных колебаний С — F
появляется где-либо в интервале 1400—1000 см'1 в зави-
симости от природы соединения и степени фторирования.
Например, при большом содержании фтора в углеводоро-
дах в этом интервале появляется целый ряд очень интен-
сивных полос поглощения. Интенсивность полос С — F
исключительно высока, и фтороуглероды можно уже рас-
познать только по одному этому признаку. Замещение фто-
ром приводит к очень значительным смещениям в сторону
больших частот полос валентных колебаний прилежащих
групп СН, С = С и С = О; подробные сведения приводятся
в соответствующих главах, где рассматриваются данные
группы. Таким образом, точные корреляции можно уста-
новить только для некоторых форм колебаний и при усло-
вии постоянного внутримолекулярного окружения данной
группы. Одна из таких корреляций предлагается, например,
для группировки CF — CF3.
Замещение хлором также приводит к эффектам взаимо-
действия групп, но они сравнительно слабее; влияние хлора
на соседнюю связь С = С, например, невелико. Для полосы
валентных колебаний С — С1 у соединений только с одной
такой связью характерен поэтому узкий интервал частот,
но для сложных хлорированных продуктов более чем
с одним атомом хлора этот интервал значительно шире.
Положение полосы валентных колебаний С — Вг вообще
более постоянно, но бывают и случайные аномалии,
например в случае бронированных стеринов, у которых
частоты необычно высоки.
30*
468
Часть IV
Полосы поглощения С — I изучались только на примере
небольшого числа соединений. Они появляются в длинно-
волновой области около 500 см"1.
Рассматриваемые корреляции приведены в табл. 18.
Таблица 18
Частота, см-1
Валентные колебания
углерод — галоген
С—F
С— С1
С —Вг
С —I
Группировка CF — CF3
1400—1000 (оч. с.)
800—600 (с.)
) 600—500 (с.)
1 Около 500 (с.)
i
745—730 (оч. с.)
; 1365—1325 (ср. с.)
2. СВЯЗИ С—F
В случаеТпростых молекул присутствие одного атома
фтора приводит обычно к появлению довольно интенсив-
ной полосы поглощения в области 1100—1000 см'1. Хорошо
изучено [1, 2, 4] значительное количество простых фтори-
рованных соединений и найдено, что при увеличении числа
атомов фтора частота полосы поглощения возрастает,
а полоса расщепляется на два пика, обусловленных сим-
метричными и антисимметричными колебаниями. В слу-
чае больших молекул, содержащих много атомов фтора,
очень интенсивная полоса поглощения наблюдается в ин-
тервале 1400—1000 см"1. Соединения этого типа широко
изучались; опубликовано значительное число работ, од-
нако общие корреляции не установлены.
Типичный ряд соединений, у которых наблюдается за-
кономерное смещение полосы в указанной выше области,
представляют фторопроизводные метана, описанные Томп-
соном и Темплом [3]. Эти авторы приводят следующие
данные для полос валентных колебаний CF:
Гл. 19. Галогенпроизводные органических соединений 469
Соединение cci2fh CC13F CC12F2 CC1F3 Частота, см 1 1072 1102 1155 и 1095 1210 и 1102
У несколько более сложных молекул, например у 1,1,1-
трифторэтана, в результате взаимодействия групп появ-
ляется несколько пиков, так что это соединение имеет
полосы при 1290, 1278, 1266, 1230 и 1135 см’1, которые
окончательно интерпретировать не удается.
Спектры полностью фторированных углеводородов очень
сложны, и в интервале 1350—1100 см’1 наблюдается целый
ряд очень сильных полос, большинство которых тем или
иным образом связано, по-видимому, с валентными колеба-
ниями С — F. Спектры ряда таких веществ приведены
Томпсоном и Темплом [5]; опубликовано также 'значи-
тельное число данных АНИ. Фторированные углеводороды
легко узнать по исключительно высокой интенсивности
полос поглощения в указанной области и по отсутствию
заметного поглощения выше 1350 саг1. Между тем никаких
точных корреляций не установлено, и если учесть имею-
щиеся данные о сильных взаимодействиях в таких молеку-
лах, то едва ли можно рассчитывать, что какие-либо про-
стые корреляции будут установлены в дальнейшем. Воз-
можно, что удастся это сделать, если рассматривать большие
молекулы, в которых строение групп, расположенных
вокруг данной связи, остается постоянным. Барнс и др.
[6] предположили, что для группы = CF2 характеристиче-
скими являются полосы при 1340 и 1200 см’1, что подтверж-
дается данными о спектрах многих простых фторированных
соединений, содержащих эту группу. Однако значение
этой корреляции снижается тем, что при указанных часто-
тах появляются полосы у многих фторопроизводных насы-
щенных углеводородов.
3. СВЯЗИ С—С1
Положение характеристических полос хлорированных
соединений в общем более постоянно, чем у фторированных,
и полоса валентных колебаний С — С1 появляется в более
470
Часть IV
узком, но все еще довольно широком интервале частот.
Взаимодействие этих колебаний и перекрывание характери-
стическими полосами таких соседних групп, как С = О
или С = С, у них также менее заметны. Как и в случае
фторпроизводных, изучено очень много простых хлор-
содержащих соединений [1, 7, 12—14]. Мы не можем по-
дробно рассмотреть все опубликованные работы, но вообще
у соединений, содержащих только один атом хлора, основ-
ная полоса валентных колебаний С — С1 появляется в ин-
тервале 750—700 см'1. Эта полоса соответствует более устой-
чивой транс-конфигурации; в жидких хлористых алкилах
и их растворах наблюдается также вторая полоса, обуслов-
ленная аош-изомером, которая находится около 650 см'1.
В случае циклических структур, например циклогексановых,
замещение хлором может происходить в аксиальном или
экваториальном положениях, и каждая из конформаций,
как и в предыдущем случае, имеет свою характеристиче-
скую частоту. Связь этих двух случаев обсуждалась Шеп-
пардом [17]. У производных циклогексана полосы С — С1
находятся при 742 см'1 (экваториальная) и 688 см'1 (ак-
сиальная) [15]; аналогичные данные получены и для стеро-
идов. Однако в последнем случае имеются и некоторые
отличия, связанные с зависимостью положения характе-
ристических полос от положения заместителя в стероидной
системе [16]. Частоты валентных колебаний экваториальной
группы С — С1, например у 2-, 3- и 7-хлорзамещенных сте-
роидов, находятся соответственно при 755, 782—750
и 749 см'1, а соответствующие частоты аксиальных форм
равны 693, 730—617 и 588 см'1. Присоединение несколь-
ких атомов хлора к одному и тому же атому углерода при-
водит обычно к повышению частоты колебаний С—С1.
В случае четыреххлористого углерода, например, очень
сильная полоса имеется при 797 см'1. Когда в молекуле при-
сутствует также фтор, спектр интерпретировать трудно,
но, согласно имеющимся данным, эффекты взаимодействия
приводят при этом к повышению частоты. Аналогичным
образом в случае фосгена, дихлорэтилена и хлорирован-
ных ароматических соединений взаимодействия связей при-
водят к повышению частоты колебаний С — С1 примерно
до 845 см 1 [7]. В случае хлоропрена и некоторых других
сложных соединений полоса С — О появляется ниже
Гл. 19. Галогенпроизводные органических соединений 471
650 см'1 [8]. У многих сложных соединений, содержащих
хлор, имеется довольно интенсивная полоса в интервале
750—700 см'1, обусловленная колебаниями С — С1, одна-
ко эта корреляция мало пригодна для целей идентифика-
ции соединений, так как интервал частот, характерный
для этой группы, довольно широк. Для соединений, у ко-
торых имеется несколько атомов хлора и основная полоса
поэтому очень интенсивна, наблюдается обычно полоса
средней! интенсивности в интервале 1510—1470 слг1, яв-
ляющаяся первым обертоном.
4. СВЯЗИ С-Вг
Исследование простых молекул показывает, что полоса
поглощения С — Вг появляется в области 600—500 см'1,
но, как и в случае хлорпроизводных, у жидких бромистых
алкилов и растворов наблюдается две полосы; в случае
циклических структур частоты также зависят от ориента-
ции заместителей. Спектры ряда бромистых алкилов —
от этил-до mpem-бутилбромида и различных ароматических
и бензилбромвдов — получены Мортимером, Блодгеттом
п Даниелсом [9], которые привели в таблице пары характе-
ристических частот. Главная, транс-полоса наблюдается
в интервале 689—645 см'1, что указывает на более слабые
эффекты взаимодействия для бромсодержащих соединений
по сравнению с соединениями, содержащими легкие гало-
гены. Однако интервал остается еще широким, и полосы
С — Вг идентифицировать трудно, если только не присут-
ствует несколько атомов брома, когда полосы поглощения
С — Вг можно узнать по их высокой интенсивности.
Отнесение полосы поглощения около 650 си-1 к транс-
форме в общем было подтверждено Брауном и Шеппардом
[13, 14], которые идентифицировали также частоту колеба-
ний С—Вг гош-формы, имеющую значение около 560 см'1.
Изучено также некоторое число соединений ряда стеринов
[16] и показано, что при экваториальном замещении появля-
ется полоса в области 750—700 см'1, а при аксиальном — в
области 690—590 см'1. Для аксиального замещения в неко-
торых дибромстероидах характерен даже более широкий
интервал, так как они поглощают около 550 см'1. Причины
такого расширения границ и аномально высокие значения
472
Часть IV
частот экваториальных форм по сравнению с бромистыми
алкилами пока непонятны.
5. СВЯЗИ С—I
Изучено лишь очень немного простых иодсодержащих
соединений. У пентаметилен иодила и н-пропилиодида [14],
а также у других простых молекул [1] обнаружена полоса
С — I транс-формы около 600 см"1. Полоса гош-формы на-
блюдается у жидкостей при более низких частотах (около
503 см"1). Исследован спектр одного иодзамещенного сте-
рина [16], имеющего полосу при 672 см"1. Опять-таки при-
чина такого смещения по сравнению с производными нор-
мальных алканов неизвестна.
6. ДРУГИЕ КОРРЕЛЯЦИИ
Выше было высказано предположение, что из-за силь-
ного взаимодействия колебаний С — С и С — F для послед-
них могла бы иметь какое-то значение лишь корреляция,
относящаяся к большим структурным группировкам, в ко-
торых указанное взаимодействие постоянно. Действительно,
несколько таких корреляций установлено.
Так, например, в случае производных CF3CeH5 частоты
антисимметричных и симметричных колебаний группы CF3
имеют постоянное значение и не сильно зависят от природы
и положения других заместителей в ароматическом кольце.
Эти значения следующие: 1321 ± 9 см"1 (симметричные),
1179 ± 7 и 1140 ± 9 см"1 (антисимметричные) [11], причем
расщепление частоты антисимметричных колебаний обуслов-
лено ароматическим кольцом. Эта корреляция весьма по-
лезна для распознавания группировки CF3 — арил.
Вторая менее важная корреляция такого рода отно-
сится к валентным колебаниям С — F в группе CF3, при-
соединенной к фторметиленовой группе. Структура
CF3CF2— X имеет характеристическую частоту в интервале
1365—1325 см"1; соответствующая полоса достаточно интен-
сивна, если X — галоген или карбонильная группа [18, 19].
Группировка CF3CF также имеет характеристическую ча-
стоту [10], связанную, по-видимому, с деформационными
колебаниями CF3 и лежащую в интервале 745—730 см-1.
Гл. 19. Галогенпроизводные органических соединений 47$
ЛИТЕРАТУРА
1 .
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
Герцберг Г., Колебательные и вращательные спектры1
многоатомных молекул, Издатинлит, М., 1949.
Torkington, ~ "
236 (1945).
h
h
h
a
Chem., 20, 402
Randall,
T h
о
m p
son, Trans. Faraday
Soc., 41
1422.
1428.
1432.
Analyt.
J., Chem. Soc.,
J. Chem. Soc.,
Chem. Soc.,
Willi
1 e,
1 e,
1 e,
T
T
T
r e,
(1948).
Fowler,
termination of Organic
Thompson, Torki
(1945).
Mortimer, Blodgett, D
Soc., 69, 822 (1947).
Bellamy, Branch, Nature,
Randle, Whiffen, J. Chem. Soc., 1955, 1'311.
Мидзусима С., Строение молекул и внутреннее вращение,
Издатинлит, М., 1957.
T
T
T
В
о
о
о
r
m p s
m p s
m p s
n e s,
о
о
0
n,
n,
n,
G о
e
e
e
m
m
m ,
Stafford,
P
P
P
J.
1948,
1948,
1948,
a m s,
Fuson,
Structures,
n g t о n,
D a n g 1, Infra-red De-
Van Nostrand, 1949.
Proc. Roy. Soc., A184, 21.
a n i e 1 s, J. Amer. Chem..
173, 633 (1954).
Brown, Sheppard, Trans. Faraday Soc., 50, 1164 (1954).
Brown, Sheppard, Proc. Roy. Soc., A231, 555 (1955).
Larnaudie, Compt. Rend. Acad. Sci. (Paris), 235, 154
(1952); 236, 909 (1953).
Barton, Page, Shoppe e, J. Chem. Soc., 1956, 331.
Sheppard, Chem. and Ind., 1957, 192.
Hauptschein, Stokes, Nodiff, J. Amer. Chem..
Soc., 74 , 4005 (1952).
Tiers, Brown, Reid, J. Amer. Chem. Soc., 75, 5978
(1953).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Волькенштейн M. В., ЕльяшевичМ. А., Степа-
не в Б. И., Колебания молекул, т. I, гл. 14, Гостехиздат,
М,—Л., 1949.
Пентин Ю. А., Татевский В. М., Вестник МГУ, № 3,
63, 73 (1955); ДАН СССР, 108, 290 (1956).
Поздышев В. А..Пентин Ю.А., Татевский В.М.,
Оптика и спектроскопия, 3, 211 (1957).
Глава 20
КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
1. ВВЕДЕНИЕ
Инфракрасные полосы поглощения, обусловленные
группами, включающими атомы кремния, примерно в пять
раз более интенсивны, чем полосы соответствующих угле-
родных связей. Причины такого соотношения интенсивно-
стей рассматривались Райтом и Хантером [1]. Высокая
интенсивность полос сильно облегчает проведение анали-
тических исследований, и инфракрасные спектры погло-
щения широко использовались при изучении и описании
кремнийорганических полимеров и родственных соедине-
ний. Помимо Райта и Хантера [1], кремнийсодержащие
соединения подробно изучались Юнгом, Серве, Кюрри
и Хантером [2], Ричардсом и Томпсоном [3] и Кларком,
Гордоном, Юнгом и Хантером [4], которые установили ряд
полезных корреляций. Первые две группы авторов иссле-
довали растворы в сероуглероде и четыреххлористом угле-
роде, а Ричардс и Томпсон работали главным образом
с тонкими пленками жидкостей. Сравнения спектров рас-
творов и пленок жидкостей показывает, что смещения глав-
ных полос невелики, если только не происходит образова-
ния водородных связей. Это же подтвердили Симон и Мак-
Магон [18], сравнивая спектры некоторых алкилсиланов
и силоксанов в газообразном, жидком и твердом состоя-
ниях. Следует, однако, иметь в виду, что эти соединения,
согласно Хайду [5], могут существовать в различных кри-
сталлических формах. Можно поэтому ожидать, что при
полиморфных превращениях вид спектра поглощения су-
щественно изменяется. По этой причине предпочтительно,
если это возможно, использовать при исследованиях рас-
творы, тем более что для многих соединений в растворах
установленные корреляции исключительно постоянны,
т. е. частоты практически не меняются.
Гл. 20. Кремнийорганические соединения 475
Кроме упоминавшихся выше корреляционных работ, про-
водились фундаментальные исследования силана [6, 7],
тетраметилсилана [8, 9], метил- [19, 23, 24] и галогенси-
ланов [10, 11, 20, 21, 23], а также кварца [12, 13, 17].
Ниже не приводится никаких сведений о группе Si = О.
В области 6,5 р,, где можно было бы ожидать поглощения
этой группы, ни одним из исследователей не обнаружено
никаких полос; кроме того, имеется достаточно много хими-
ческих данных о том, что в кремнийорганических соедине-
ниях такая группа не встречается. Частоты, предложенные
в качестве характеристических для отдельных группиро-
вок, включающих кремний, приведены в табл. 19, а данные,
на которых основываются корреляции, обсуждаются
в тексте.
Таблица 19
Частота3, слс-1
Si (СН3)3 1250 , 841, 756 — 754
Si (СН3)2 1259, 814—800
Si (СН3) 1259, около 800
Si —С6Н5 1429, 1130—1090
Si—0 — Si в цикле
тримеры 1020—1010
тетрамеры 1090—1080
большие кольца 1080—1050
Si—0 — С и Si—0 — Si (в от-
крытой цепи) 1090—1020
Si—Н 2300—2100
а Все указанные полосы поглощения очень интенсивны.
2. СВЯЗИ Si—С
Валентные колебания Si — С проявляются в спектраль-
ной области 900—700 слГ1 и в большой степени зависят от
природы замещающих групп. Так, например, валентным
476
Часть IV
колебаниям Si — С тетраметилсилана соответствует полоса
860 см'1, тогда как тетраэтилсилан имеет полосу при
733 см'1. Поэтому только полосы, относящиеся к такой
связи в одних и тех же группировках, например таких,
как Si—СН3, Si—СЙН5 и т. д., имеют, по-видимому,
постоянное положение. Можно ожидать, что маятниковым
колебаниям этих групп должны соответствовать характери-
стические полосы поглощения при более высоких частотах.
а. Валентные Si—СН3-колебания.—51(СНз)з- У ряда
соединений с открытой цепью, исследованных Райтом и Хан-
тером [1] (от гексаметилдисилоксана до октадекаметилок-
тасилоксана), имеется сильная полоса при 841 см'1, интен-
сивность которой при увеличении длины цепи уменьшается
и которая неизменно отсутствует у соответствующих цикли-
ческих соединений (от гексаметилциклотрисилоксана до
гексадекаметилциклооктасилоксана). У тех же открытых
соединений имеется вторая полоса при 756—754 см'1.
интенсивность которой также меняется при изменении
длины цепи. Указанные полосы, несомненно, связаны
с колебаниями группы Si(CH3)3 на конце открытой цепи
и, по-видимому, достаточно характеристичны для иденти-
фикации этой группировки. Эти данные подтверждены Ри-
чардсом и Томпсоном [3] на ряде соединений. В то же время
12 арилметилсиланов, исследованных в растворах Клар-
ком и др. [4], имеют полосы поглощения около 840 см'1.
У триметилхлорсилана обнаружена первая из указанных
полос при 857 сж-1, отнесенная к маятниковым колебаниям
СН3 [19].
— 5ЦСНз)г— Все соединения, изучавшиеся Райтом
и Хантером [1], у которых имеются группы —Si(CH3)2—.
поглощают в области 800 см'1. Эта частота отнесена к маят-
никовым колебаниям метильной группы, так же как в слу-
чае метилхлорсиланов, у которых соответствующая поло-
са находится в области 850—800 см'1 в зависимости от
числа присутствующих метильных групп [19]. Это погло-
щение, по-видимому, весьма характеристично для указан-
ной группировки, тем более что оно отсутствует в случае
димера, у которого имеются лишь группировки Si(CH3)s.
Ричардс и Томпсон [3] подтвердили эти данные и пополнили
их данными об алкоксисиланах, у которых при наличии
Гл. 20. Кремнийорганические соединения
477
двух метильных групп, непосредственно присоединенных
к атому кремния, эта полоса обнаружена в спектре. С дру-
гой стороны, Юнг и др. [2] не наблюдали этой полосы
в спектрах циклических диметилтримеров и тетрамеров,
хотя предыдущими авторами она у этих соединений была
зарегистрирована. В случае циклических полимеров эта
полоса появляется в интервале 814—802 см'1; у соединений
с открытой цепью и метилсиланов она имеет практически
постоянную частоту 800 сл-1. Ричардс и Томпсон [3] обра-
тили также внимание на вторую полосу, около 700 слг1,
которая имеется как у соединений с открытой цепью, так
и у циклических соединений. Они полагают, что эта полоса
также может быть связана с группировкой —ЗЦСНзЬ— •
Однако данная корреляция имеет, по-видимому, меньшее
значение, чем предыдущая, так как очень близко к этой
области расположены полосы в спектрах этил- и фенилза-
мещенных соединений, приведенных Юнгом и др. [2].
— Si(CHs)— В случае соединений, содержащих груп-
пировку Si(CH3)R1R2R3, Ричардс и Томпсон [3] обнару-
жили, что полоса появляется обычно вблизи 800 см'1,
но положение ее в большей степени зависит от природы
групп R, чем в случае соединений более чем с одной метиль-
ной группой. В соответствии с этим Юнг и др. [2] нашли,
что два изомерных циклических фенилметилтримера по-
глощают при 791 и 788 см'1. Когда к атому кремния при-
соединены две метильные группы, всегда можно ожидать
появления полосы около 800 см'1, и ее отсутствие служит
доказательством отсутствия такой группировки. С другой
стороны, наличие такой полосы может быть связано также
с группой, содержащей лишь один метильный радикал.
б. Деформационные колебания Si — СН3. Все соедине-
ния, исследованные Райтом и Хантером, имеют сильную
полосу при 1258 см'1. Аналогичная полоса при 1250 см'1
наблюдается для тетраметилсилана [8, 9], и указанные
авторы относят ее к маятниковым колебаниям метильной
группы. Ричардс и Томпсон разделяют эту точку зрения,
однако такое отнесение встречает и ряд возражений. Юнг,
Келер и Мак-Кини [8] приписывают ее симметричным
деформационным колебаниям, а Ренк и др. [9] — колеба-
ниям у-СН. Последующие исследования таких соедине-
478
Часть IV
ний, как метилсиланы, по-видимому, подтверждают отне-
сение этой полосы к симметричным деформационным
колебаниям метильной группы; маятниковым же колеба-
ниям соответствует, по-видимому, полоса около 800 см'1.
Во всяком случае, характеристичность полосы 1258 см
для силиконов не вызывает сомнений, и при подходящих
условиях при ее помощи можно легко распознавать ме-
тильную группу. Райт и Хантер [1] указывают, что у ряда
исследованных ими соединений с открытой цепью имеется
вторая полоса при 1250 см1, интенсивность которой умень-
шается при увеличении длины цепи. Эта полоса отсутствует
у циклических соединений, и поэтому авторы относят ее
к концевым группам Si(CH3)3. Два исследованных ими по-
лимера с разветвленной цепью также имеют эту полосу по-
глощения. Юнг и др. [2] сообщают, что у всех исследован-
ных ими циклических соединений, содержащих метильные
группы, всегда имелась полоса поглощения при 1259 см'1.
Эти соединения не имеют, конечно, второй полосы при
1250 см1, так как у них нет концевых групп. Указанные
авторы заметили, однако, у этилзамещенных соединений
также наличие полосы 1241 см'1, которая, как они
полагают, обусловлена деформационными колебаниями
— CH2Si— . В случае присутствия группировки Si — CHS
очень существенно точно определить длины волн полос
в этой спектральной области, для чего необходимо поль-
зоваться растворами. Об этом свидетельствуют данные, по-
лученные Ричардсом и Томпсоном в результате исследова-
ния веществ в жидком состоянии; эти авторы обнаружили
у метилполимеров с открытой цепью и циклических метил-
полимеров лишь одну полосу в интервале 1265—1260 см'1.
Поскольку Райт и Хантер [11 изучали те же соединения,
то имеющиеся различия данных обусловлены, по-видимому,
изменениями физического состояния образцов. Кларк и др.
[4] не приводят точных значений частот этих полос погло-
щения у исследованных ими ароматических триметилсила-
нов, но сообщают, что во всех спектрах обнаружена полоса
около 1256 см'1.
Эта корреляция в какой-то степени справедлива для
силоксимегилсиланов, изученных Ричардсом и Томпсоном
[3]. Для 10 соединений, исследованных в жидком состоя-
нии, у которых метильная группа непосредственно при-
Гл. 20.Кремнийорганические соединения
479"
соединена к кремнию, эти авторы нашли сильные полосы
в интервале 1269—1256 см'1, тогда как у соединений,
содержащих этильные группы, частота уменьшается до*
1250—1239 см'1. Какая связь существует между положе-
нием полосы метилсиликонов и эффектами упаковки в жид-
ком состоянии и в какой степени смещение полосы может
быть обусловлено непосредственным влиянием алкокси-
заместителей, нельзя определить до тех пор, пока эти соеди-
нения не будут исследованы в растворе. Кэй и Танненбаум
[23] исследовали возможность идентификации группы;
— Si— С2Н5 по полосам поглощения при более низ-
ких частотах. Для систем — Si— СН2— R они приводят
частоты 1281 (R = Н), 1235—1220 (R = СН3) и 1220 —
1163 см 1 (R > СН3). Однако Харвей и др. [25] отме-
тили, что хотя такие полосы поглощения действительно'
наблюдаются в этой области для ряда от метил- до гексил-
фенилсилана, аналогичные же полосы обнаружены также-
у циклогексилфенилсилана и изопропилфенилсилана, по-
этому указанная корреляция имеет лишь ограниченное зна-
чение.
в. Колебания Si — С6Н5. Юнг и др. [2] изучили семь-
R R
циклических силиконов типа (SiO)3 или (SiO)4, у которых.
R \
непосредственно к атому кремния присоединены неза-
мещенные ароматические радикалы. У всех силиконов,
в растворах найдены две полосы: при 1429 см'1 и вблизи
1110 см'1, которые могут быть характеристичными для связи1
Si — С6Н5; в то же время обычные частоты колебаний моно-
замещенных ароматических циклов при 740—735 и 700 см'1
остаются по существу без изменений. Указанные авторы,
считают полосу 1429 см'1 характеристичной для связи:
кремний—фенил, но следует заметить, что колебания СН
метильной и этильной групп, присоединенных к кремнию,
имеют частоту 1412 см'1, так что и в этом случае при исполь-
зовании данной корреляции предпочтительнее проводить-
измерения с растворами. Ричардс и Томпсон [3] нашли зна-
чения частот для шести соединений с такой связью, рав-
4?0
Часть IV
ные 1430—1425 см 1, хотя исследовались твердые вещества;
это также подтверждает правильность рассматриваемой
корреляции. Последняя была подтверждена и работой
Харвея и др. [25], которые нашли у значительного числа
фенилалкилсиланов поглощение при 1431 см'1.
Юнг и др. [2] сообщают, что вторая полоса имеет менее
постоянную длину волны, но во всех приведенных спектрах
•ее частота очень близка к 1124 сж-1. В случае тетрамеров
она перекрывается сильной полосой поглощения Si — О
с частотой 1090—1080 слг"1, в случае же тримеров, у кото-
рых полоса Si — О смещается до 1020—1010 см'1, ее легко
различить. Связь этой полосы с колебаниями Si—С6Н5
подтверждается тем, что когда к одному и тому же атому
кремния присоединено больше одного фенильного радикала,
полоса дублетна. Аналогичную сильную полосу в области
1123—1110 см'1 можно наблюдать в спектрах четырех аро-
матических силиконов, исследованных Ричардсом и Томп-
соном [3]. Все они имеют еще более сильную полосу при
меньшей частоте, которую можно отнести к связи Si—О.
В случае трифенилоксисилана и дифенилдиоксисилана
эффекты взаимодействия или образования водородной свя-
зи приводят, по-видимому, к слиянию полос Si—О nSi—С6Н5
в простую или дублетную полосу около 1120—1111 см'1.
Хотя для арилтриметилсиланов [4] точных значений ча-
стот не приведено, они, по-видимому, имеют полосы в не-
сколько более широких интервалах: 1430—1390 и 1135—
1090 см"1. Значения, относящиеся к высокочастотной по-
лосе, были определены при исследовании пленок жидкостей.
Ни а-, ни Р-нафтилтрихлорсиланы не дают ни одной из этих
полос [14]. Однако для фенилалкилсиланов, исследован-
ных Харвеем и др. [25], полоса при 1116 см'1 является
характеристической, что опять-таки подтверждает ее отне-
сение к связи Si—С6Н5.
3. ВАЛЕНТНЫЕ КОЛЕБАНИЯ Si—О
В спектре отражения кристаллического кварца имеются
две сильные полосы валентных колебаний Si —О (при 1179
и 1109 см'1) [13], так что примерно в этой же области мож-
но было бы ожидать полосы поглощения у кремнийоргани-
ческих соединений со связью Si — О. Такие полосы в дей-
Гл. 20-Кремнийорганические соединения
481
ствительности и наблюдались. Циклические соединения
и соединения с открытой цепью обсуждаются ниже отдель-
но, поскольку между ними имеются некоторые различия.
а. Циклические силоксаны. Райт и Хантер [1] привели
спектры шести циклических соединений от гексаметилцик-
лотрисилоксана до гексадекаметилциклооктасилоксана.
У тримера полоса Si —О появляется при 1018 см'1, у ос-
тальных же соединений она лежит в интервале 1076—
1056 см'1, причем с увеличением размеров кольца частота
закономерно уменьшается в небольшой степени. Ричардс
и Томпсон [3] подтвердили эти данные для пяти первых
членов ряда, а, кроме того, исследовали еще два цикличе-
ских ароматических силоксана [Si(C6H5)201s и [Si(C6H5)2O]4.
Первое из этих соединений имеет сильную полосу при
1015 см'1, которая близка по частоте к полосе тримера ме-
тилового ряда, тогда как для тетрамера самая сильная
полоса в этой области расположена около 1100 см'1.
Юнг и др. 121 изучали тримеры и тетрамеры, содержа-
щие различные заместители у атома кремния. Они иссле-
довали циклические тримеры и тетрамеры [общая формула
(SiRгРгО)„] диметил-, диэтил-, дифенил-, метилфенил-
п этилфенилпроизводных, а также смешанный дифенилте-
траэтилтример. Эти авторы установили, что положение
полосы поглощения Si — О — Si мало меняется в зависи-
мости от природы заместителей, тогда как между часто-
тами тримеров и тетрамеров имеется значительное разли-
чие. Они обнаружили, что все тримеры дают полосу погло-
щения Si—О — Si в узком интервале 1020—1010 см'1,
а все тетрамеры поглощают в интервале 1093—1081 см'1.
Эти значения частот метилтримеров и тетрамеров согла-
суются в пределах ошибок эксперимента со значениями,
полученными в ранних работах. Расхождение примерно
в 20 см'1 между значением, полученным указанными авто-
рами для дифенилтетрамера, и значением, приведенным
Ричардсом и Томпсоном, возможно, обусловлено различ-
ным физическим состоянием объектов или взаимодействием
колебаний Si — О и Si — С(;Н5 для вещества в жидком со-
стоянии. Юнг и др. [2] указывают, что упомянутое выше
четкое различие между тримерами и тетрамерами можно
использовать для дифференциации этих форм циклических
31 Л. Беллами
482
Часть IV
силиконовых полимеров, обычно получаемых при гидро-
лизе веществ типа R2SiCl2.
Таким образом, имеется значительное количество дан-
ных, свидетельствующих о правильности отнесения
к колебаниям эфирной связи Si — О полос в интервале
1020—1010 см'1 в случае циклических тримеров и в интер-
вале 1090—1080 см'1 в случае циклических тетрамеров;
кроме того, установлено, что положение этих полос мало
зависит от природы замещающих групп. В случае больших
циклов частота уменьшается до 1050 см'1. Барри и др. [261
получены также спектры некоторых органосилсесквиоксаце-
нов, но отнесение частот подробно не обсуждалось.
б. Полимерные и мономерные силоксаны с открытой
цепью. Для ряда полимерных силоксанов с открытой цепью
от гексаметилдисилоксана до октадекаметил октасилоксана
Райт и Хантер [1 ] нашли полосу поглощения Si — О в пре-
делах интервала 1055—1024 см'1 и также наблюдали не-
большое уменьшение частоты при увеличении длины цепи
на одну группу. Для двух полимеров с разветвленной цепью
они приводят несколько более высокие значения (1056—
и 1068 см'1). Эти данные для первых пяти членов ряда
подтверждены Ричардсом и Томпсоном, которые исследо-
вали также два полимерных фенилзамещенных силанола,
причем самые сильные полосы обнаружены у них в этой
области при 1078—1076 см'1. Таким образом, оказывается,
что поглощение Si — О — Si у соединений с открытой цепью
наблюдается в той же области, что и у циклических соеди-
нений, но, по-видимому, ближе к нижнему пределу ука-
занного интервала частот.
Ричардс и Томпсон исследовали также 10 алкоксисила-
нов, которые имеют очень интенсивную полосу в интервале
1090—1050 см'1. Трифенилоксисилан и дифенилдиоксиси-
лан поглощают при несколько более высокой частоте
(~1100 см'1), но это обусловлено, вероятно, эффектами вза-
имодействия связей или образованием водородной связи. Эти
полосы, по-видимому, относятся к группе Si — О — С.
Необходимо отметить, что полоса поглощения в данном
случае имеет несколько более высокую частоту, чем обыч-
ные полосы поглощения Si — О — Si для соединений с от-
крытой- цепью. Однако мы не имеем достаточных данных,
Гл. 20-Кремнийорганические соединения
483
чтобы судить о том, является ли это различие существен-
ным (т. е. обусловлено ли оно различными типами связи).
В настоящее время можно только сказать, что соединения
с открытой цепью имеют очень сильную полосу поглоще-
ния Si — О в интервале 1090—1020 см'1, что подтверждено
работами Крешкова и др. [27, 28], в которых были иссле-
дованы некоторые простые кремнийорганические эфиры.
Следует отметить, что полоса Si — О — Si находится
в одной и той же области с полосами С —О— С и Р— О— С;
это свидетельствует о том, что изменение масс атомов
в этом случае, по-видимому, не влияет на частоту коле-
баний.
Помимо полосы валентных колебаний Si — О, можно
ожидать в области 500—300 см'1 появления второй полосы
поглощения, обусловленной деформационными колебания-
ми связей Si — О, которые соответствуют полосе 482 см'1
в спектре отражения кварца [13]. Райт и Хантер [1]
используя призму из бромистого калия, исследовали в этой
области несколько соединений. Во всех случаях они нашли
очень сильную полосу в области 450—435 см'1 (у нижней
границы рабочего интервала призмы), но дальнейшие ис-
следования этих колебаний не проводились.
4. ВАЛЕНТНЫЕ КОЛЕБАНИЯ Si - Н
Отнесение частоты валентных колебаний Si — Н опи-
рается на исследования ряда алкил- и галогензамещенных
силанов, поэтому соответствующий интервал частот срав-
нительно широк. У силана частота валентных колебаний
Si — Н имеет значение 2187 см'1 [6, 7], а Ричардс и Томп-
сон [3] сообщают, что у трихлорсилана, дихлорэтилсилана
и хлордиэтилсилана имеются сильные полосы валентных
колебаний Si — Н вблизи 2200 см'1. В последнее время
для трихлорсилана получено [15] значение частоты
2274 см 1 в случае газообразного состояния и 2258 см'1
для жидкости. Столь высокие значения частот отражают
влияние трех атомов хлора, а у моногалогенсиланов обе
частоты валентных колебаний SiH3 почти перекрываются
в области 2205—2195 см'1. Если к атому кремния присоеди-
нены алкильные группы, то частота Si — Н заметно пони-
жается. Вест и Рочестер [291 полагают, что диалкилсиланы
31*
484
Часть IV
(которые поглощают между 2096 и 2083 см 1) можно отли-
чить от диарилсиланов (2096—2111 см'1), однако их частоты
различаются весьма мало. Харвей и др. [25] сообщили да-
лее, что ряд алкиларилсиланов поглощает при постоянной
частоте 2128 см'1. Таким образом, можно считать, что по-
лосы Si — Н лежат около 2100 см'1, причем частота повы-
шается при присоединении атома галогена.
5. ВАЛЕНТНЫЕ КОЛЕБАНИЯ Si-С!
В отличие от только что рассмотренной группировки
следует ожидать, что частота поглощения Si — Cl сильно
зависит от природы групп, присоединенных к атому
кремния.
Исследования с целью установления частоты колеба-
ний по этой связи не проводились, но Ричардс и Томпсон
[3] обнаружили у трех упомянутых выше хлорсиланов
сильную полосу около 810 см'1, которая, как полагают
эти авторы, относится к связи Si—Cl; SiCl4 поглощает
при 800 см'1 [ 16].
6. ВАЛЕНТНЫЕ КОЛЕБАНИЯ Si — ОН
Как и следовало ожидать, атом кремния не влияет на
валентные колебания О — Н, если не считать случаев,
когда образуется водородная связь. Ричардс и Томпсон [3]
исследовали четыре силанола, которые в твердом состоя-
нии имеют интенсивную полосу поглощения около 3250 см'1.
В случае разбавленных растворов в четыреххлористом уг-
лероде вместо этой полосы появляется обычно полоса по-
глощения «свободной» группы ОН при 3690 см'1. Полосы
деформационных колебаний ОН у этих соединений не иден-
тифицированы. Авторы заметили во всех случаях сильную
полосу в области 880—830 см'1, которая отсутствует у цик-
лических дифенилсилоксанов, но они считают, что на ос-
новании имеющихся данных нельзя ее с уверенностью
отнести к деформационным колебаниям. Указанное выше
положение полосы валентных колебаний свободной груп-
пировки Si — ОН подтверждено в случае силанола [30]
разбавленный раствор которого поглощает при 3676 сл'1.
Гл. 20. Кремнийорганические соединения
485
7. ВЛИЯНИЕ СВЯЗИ Si - С НА КОЛЕБАНИЯ СН
Возможное влияние связи Si — С на колебания группы
СН, присоединенной к атому углерода при Si, рассматри-
валось Райтом и Хантером [1] и Юнгом и др. [2] Райт
отмечает, что у всех исследованных ими полимеров мети-
лового ряда полоса валентных колебаний СН имеет точно
такую же частоту, что и в случае метильной группы углево-
дородов, но что интенсивность этой полосы составляет от 1/3
до х/4 величины, соответствующей группировке С — СН3.
Деформационным колебаниям связей СН соответствует
немного меньшая частота, а именно 1412 слГ1 у всех соеди-
нений, исследовавшихся в растворах. Юнг и др. [2] под-
твердили эти данные, рассматривая метил- и этилполимеры,
а также высказали свои соображения по поводу неизмен-
ности положения обеих полос и их относительной интен-
сивности для веществ, находящихся в виде растворов.
У ароматических соединений, изучавшихся Юнгом
[2], частоты валентных колебаний связей С = С кольца
имеют обычные значения 1595 и 1493 ел-1; полосы внеплос-
костных деформационных колебаний связей СН в длинно-
волновой области также не отличаются от обычных. Осталь-
ные полосы подробно не изучены, но, помимо указанных
полос при 1429 и 1124 см'1, относящихся к связи Si — С6Н 5,
Юнг и др. [2] отмечают, что у всех изученных ими аромати-
ческих соединений неизменно наблюдались полосы 1190,
1031 и 996 см'1, которые, возможно, связаны с ароматиче-
ским кольцом. Полосы, близкие по частотам к указанным,
наблюдаются также еще у четырех ароматических соеди-
нений, исследованных Ричардсом и Томпсоном [3].
Л И Т Е Р А Т У Р А
I. Wright, Hunter, J. Amer. Chem. Soc., 69, 803 (1947).
2. Young, Serva is, Currie, Hunter, J. Amer
Chem. Soc., 70, 3758 (1948).
3. Richards, Thompson, J. Chem. Soc , 1949, 124.
4. Clark, Gordon, Young, Hunter, J. Amer. Chem.
Soc., 73, 3798 (1951).
5. Hyde, Frevel, Nutting, Petrie, Purcell,
J. Amer. Chem. Soc., 69, 488 (1947).
6. Steward, Nielsen, J. Chem. Phvs., 2, 712 (1934); Phys
Rev., 47, 828 (1935).
486
Часть IV
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
Т i n d а 1, Strale у, Nielsen, Proc. Nat. Acad. Sci
U. S., 27, 208 (1941).
Young, Koehler, McKinney, J. Amer. Chem. Soc.,
69, 1410 (1947).
Rank, Saksena, Shull, Discuss. Faraday Soc., 9, 187
(1950).
Bailey, Hale, Thompson, Proc. Roy. Soc., A167,
555 (1938).
Scott, Frisch, J. Amer. Chem. Soc., 73, 2599 (1951).
Barnes, Gore, Li d del, Williams, Infra-red
Spectroscopy, Reinhold, 1944.
Coblentz, Investigations of Infra-red Spectra, Carnegie
Inst of Washington, 1905.
Gilkey, Tyler, J. Amer. Chem. Soc., 73, 4982 (1951).
Gibian, McKinney, J. Amer. Chem. Soc., 73, 1431
(1951).
Герцберг Г., Колебательные и вращательные спектры
многоатомных молекул, Издатинлит, М., 1949, стр. 186.
Simon, McMahon, J. Chem. Phys., 21, 23 (1953).
Simon, McMahon, J. Chem. Phys., 20, 905 (1952).
Smith, J. Chem. Phys., 21, 1997 (1953).
Newman, О ' L о а п e, P о I o, Wi 1 so n, J. Chem. Phys.,
25, 855 (1956).
Andersen, Bak, Acta Chem. Scand., 8, 738 (1954).
Mayo, Optiz, Peake, J. Chem. Phys., 23, 1344 (1955).
Kaye, Tannenbaum, J. Org. Chem., 18, 1750 (1953).
Cerato, Lauer, Beachell, J. Chem. Phys., 22, 1
(1954).
Harvey, Nebergall, Peake, J. Amer. Chem. Soc.,
76, 4555 (1954).
Barry, Daudt, Domicone, G i key, J. Amer. Chem.
Soc., 77, 4248 (1955).
Крешков, Михайленко, Якимович, Ж. анал.
хим., 9, 208 (1954).
Крешков, Михайленко, Якимович, ЖФХ,
28, 538 (1954)
West, Rochest'er, J. Org. Chem., 18, 303 (1953).
Kakudo, Kasai, Watase, J. Chem. Phys., 21, 1894
(1953).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Касаточкин В. И., Ш о с т а к о в с к и й М. С., 3 и л ь -
бербранд О. И., Кочкин Д. А., Изв. АН СССР,
сер. физ., № 6, 726 (1954).
Лазарев А.Н., Воронков М.Г., Тенишева Т.Ф.,
Оптика и спектроскопия, 5, 365 (1958).
Глава 21
НЕОРГАНИЧЕСКИЕ ИОНЫ
1. ВВЕДЕНИЕ
До недавнего времени исследования колебательных спект-
ров неорганических веществ велись главным образом при
помощи метода комбинационного рассеяния света. В неко-
торых ранних работах изучалась инфракрасная область
спектров для ряда веществ и применялся метод отражения
[1]; позднее большое число этих же веществ Леконт и его
сотрудники [2, 3] изучали при помощи обычного метода
абсорбционной инфракрасной спектроскопии. Однако при-
менявшаяся этими исследователями аппаратура не давала
возможности получать спектры с достаточно высоким раз-
решением. Основные трудности возникали в связи с тем,
что не всегда удавалось приготовить образцы неорганиче-
ских веществ в подходящем для исследований виде Если
размеры частиц вещества не достаточно малы, то потери
на рассеяние света очень значительны, особенно если изу-
чается область высоких частот, и в . пектре наблюдается
лишь ряд широких размытых полос поглощения.
Недавно такие исследования возобновились, поскольку
было обнаружено, что четкие спектры, сравнимые со спект-
рами органических соединений, могут быть получены при
использовании взвесей в парафиновом масле или тонких
слоев порошков, если размер частиц достаточно мал. Так
например, удалось детально исследовать отдельные не-
органические соли, а также некоторые группы родствен-
ных им соединений. Дюваль, Леконт и Морандат [4] изу-
чили ряд нитритов и хлоритов и доказали, что эти ионы
имеют нелинейное строение; Дюваль и Леконт 15] приме-
нили также метод инфракрасных спектров поглощения
для определения степени полимеризации ортоборатов
и метаборатов и для изучения реакций в твердой фазе [10].
Внедрение техники прессования таблеток из бромистого
488
Частя IV
калия и появление соответствующего оборудования для
тонкого измельчения образцов также стимулировало рабо-
ты в этом направлении.
Наибольший интерес представляет недавно опублико-
ванная работа Миллера и Уилкинса, в которой описан
ряд корреляций, пригодных для аналитических целей и иден-
тификации соединений [6]. Эти авторы исследовали и опуб-
ликовали спектры 159 неорганических солей и составили
таблицы характерных частот для большого числа различ-
ных ионных групп. Наиболее важные из них рассмотрены
ниже; для получения более подробных сведений следует
обратиться к оригинальной работе. Хант, Вишерд и Бон-
хем [7] показали пригодность инфракрасных спектров по-
глощения для идентификации сложных горных пород
и минералов и, исследовав также спектры 15 неорганиче-
ских соединений, установили некоторые корреляции. Эти
авторы предложили также простой и удобный способ
приготовления порошкообразных образцов с размером
частиц до 5 ц (седиментационный метод).
Метод инфракрасных спектров поглощения не позво-
ляет провести полный анализ сложных смесей, за исклю-
чением небольшого числа минералов, для которых можно
получить полные спектральные данные; однако, как пока-
зали Миллер и Уилкинс, этот метод очень полезен в неорга-
ническом анализе, особенно если его сочетать с эмиссион-
ным анализом и рентгенографическими исследованиями.
Эгот метод успешно также использован для проведения
ряда количественных анализов различных пород и минера-
лов [12], а детальные исследования силикатных пород
выявили различия между породами различных типов, до-
статочные для того, чтобы можно было применить метод
инфракрасных спектров для идентификации соединений.
Рассматриваемые корреляции приведены в табл. 20.
2. КАРБОНАТЫ И БИКАРБОНАТЫ
Хант и др. [7], а также Миллер и Уилкинс [6] исследо-
вали инфракрасные спектры 12 различных карбонатов
и, кроме того, ряд основных карбонатов, бикарбонатов и
аналогичных веществ, а Луисферт и Побегуин [И] изучали
различные кристаллические формы карбоната кальция.
Гл. 21. Неорганические ионы
489
Таблица 20
Частота, cm-i
Карбонаты (СО|_) . , . Сульфаты (SO^“) . . . 1450—1410 (оч. 1130—1080 (оч. с.), 880—860 (ср. ) с.), 680—610 (ср. — сл.)
Нитраты (NO3) .... 1380—1350 (оч. с.), 840—815 (ср.)
Нитриты (NOj) .... 1250—1230 (оч. с.), 840—800 (сл.)
Аммоний (NHJ) .... Цианиды, тиоцианаты, цианаты и комплекс- ные ионы Фосфаты (РО^~, HPOJ-, Н2РО;) Силикаты (всех типов) 3300—3030 (оч. 2200—2000 (с.) 1100—1000 (с.) 1100—900 (с.) с.), 1430—1390 (с.)
Во всех случаях обычные карбонаты характеризуются
сильной полосой в области 1450—1410 см 1 и полосой сред-
ней интенсивности в области 880—860 см'1. Однако в от-
ношении второй полосы карбонат свинца представляет,
по-видимому, исключение, так как он имеет лишь очень
слабую полосу поглощения с частотой 840 см'1. Хант и др.
[7] указали на связь положения этой второй полосы с мас-
сой катиона и нашли логарифмическую зависимость сме-
щений в сторону меньших частот от увеличения атомного
веса. Для карбонатов, изученных Миллером и Уилкинсом,
это правило также применимо (за исключением карбоната
лития). У карбоната кальция эти полосы проявляются при
876 и 1430 см'1, и первая из них использовалась для опре-
деления карбоната в фосфатах [13, 14]. Отнесение частот
колебаний индивидуальных карбонатов проведено Луис-
фертом [15] и Дециусом [16]. С использованием методов
инфракрасной спектрометрии выполнены также исследо-
вания природы костных карбонатов [17].
Кроме того, многие карбонаты обнаруживают полосы
поглощения в области 750—700 см'1. Не все из них погло-
щают в этой области, но если такое поглощение имеется,
то наблюдаемые полосы часто можно использовать для идеи-
490
Часть IV
тификации минералов. Так, например, кальцит и доломит
имеют полосы, частоты которых отличаются одна от другой
на 17 слг1 17].
Спектры основных карбонатов и бикарбонатов значи-
тельно отличаются друг от друга. Основные карбонаты
имеют более сложные спектры, а полоса поглощения при
1430 см 1 почти всегда бывает двойной. Кроме того, число
полос в интервале 1110—700 см 1 в большинстве случаев
увеличивается от двух до пяти, а также появляется полоса
при 3300 см'1, соответствующая связанной группе ОН.
Спектры различных основных карбонатов тоже значитель-
но отличаются друг от друга, что во многих случаях позво-
ляет идентифицировать индивидуальные соединения.
Изучено лишь очень ограниченное число бикарбонатов
[6], но установлено, что в области 1431 см'1 они не погло-
щают. Вместо этого по обе стороны от этой полосы наблю-
даются полосы, довольно далеко отстоящие одна от дру-
гой. Однако в настоящее время определенных корреляций
не установлено.
3. СУЛЬФАТЫ
Миллер и Уилкинс [6] исследовали 10 неорганических
сульфатов, а Хант и др. [71 — два других сульфата. Во
всех случаях обнаружена очень сильная полоса в области
ИЗО—1080 см'1, которой в большинстве случаев сопут-
ствует значительно более слабая полоса в области 680—
610 см'1. Для дигидрата сульфата кальция найдена силь-
ная полоса поглощения с частотой 1140 см'1, а после на-
гревания при 170° в течение 12 час в этой области обнару-
жена группа из трех полос. Эти результаты свидетель-
ствуют о влиянии на такие спектры степени гидратации,
а также симметрии кристалла, которые, таким образом,
должны приниматься во внимание при оценке спектров.
В отличие от карбонатов между наблюдаемыми часто-
тами полос поглощения и свойствами положительного иона
не имеется, по-видимому, непосредственной связи, хотя
спектры сульфатов настолько различны, что обычно можно
идентифицировать каждое из этих соединений в отдель-
ности. Леконт и др. [18] исследовали также сульфат-ион
и пришли, по-существу, к аналогичным выводам. Они от-
Гл. 21. Неорганические ионы
491
метили, в частности [191, что полоса около 610 слГ1 яв-
ляется простой в случае Mg, К или Мп, но дублетна в слу-
чае Са, Си, Be или Ва.
Подобно бикарбонатам, бисульфаты еще мало изучены,
однако на основании результатов исследования трех образ-
цов [6] можно сделать вывод, что у них нет интенсивной
полосы поглощения при 1110 см'1. Имеется указание-на то,
что эта пслсса расщепляется на две полосы средней интен-
сивности. Сульфиты в этой области не поглощают.
4. НИТРАТЫ И НИТРИТЫ
Все 10 неорганических нитратов, изученных Миллером
и Уилкинсом [6], обнаруживают характеристические по-
лосы поглощения в интервалах 1380—1350 см'1 (оч. с.)
и 840—815'с.и1 (ср.). Никаких корреляций, связывающих
положение этих полос с природой положительного иона,
в данном случае установить не удалось. Указанные выше
корреляции подтверждены для ряда органических нитра-
тов, например нитрата гуанидина, у которого группа имеет
ионный характер. Такие соединения поглощают в той же
области, хотя интервал характерных частот в этих случаях
иногда несколько шире (гл. 17). Нитриты также имеют
полосу от слабой до средней интенсивности в области 840—
815 см'1, но их легко отличить от нитратов по наличию силь-
ного поглощения [5, 6] в области 1250—1230 см'1. Отнесе-
ние частот колебаний для нитрита натрия выполнено
Тартом [201; Хафель [41] рассмотрел спектры некоторых
кристаллических нитратов.
5. ИОН АММОНИЯ
Изучены 18 различных солей аммония [6]. Во всех слу-
чаях найдены две характеристические полосы поглощения.
Одна из них соответствует валентным колебаниям NH
и появляется в области 3200 см'1, тогда как другая, также
интенсивная полоса, находится в области 1430—1390 см'1.
Число и положение полос в области валентных колеба-
ний NH отражает возможности образования водородных
связей внутри кристалла, а в тех случаях, когда обнару-
492
Часть IV
живаются мультиплетные пики, это может также указы-
вать на образование нескольких водородных связей раз-
личной прочности. У перхлората аммония частота валент-
ных колебаний NH равна 3300 см'1, чго указывает либо
на наличие слабых водородных связей, либо на полное
их отсутствие; это хорошо согласуется с тем, что перхло-
рат аммония не присоединяет воду и не образует гидрата.
Для большинства солей аммония наблюдается та или иная
степень образования водородных связей, причем полосы
NH в подавляющем большинстве случаев лежат в области
3200—3100 см'1. Нитрат аммония представляет крайний
случай, когда образуются очень сильные связи, и он имеет
три полосы поглощения с частотами 3160, 3060 и 3030 см'1 [6].
Спектр азида аммония был изучен Пиментелом и др. [21],
а Уолдрон и Хорниг [22] исследовали некоторые кристал-
логидраты солей аммония. Полоса поглощения с частотой
1410 см 1 довольно интенсивна и резко очерчена, так что
при использовании ее вместе с полосами валентных колеба-
ний этот ион можно легко идентифицировать.
6. ЦИАНИДЫ
Число исследованных простых цианидов очень невели-
ко, но хорошее соответствие их спектров со спектрами орга-
нических нитрилов и изоцианидов указывает на то, что
колебания С = N не сильно различаются и в неорганиче-
ских ионах. Поэтому весьма вероятно, что и другие про-
стые цианиды также можно идентифицировать по погло-
щению в области 2200—2000 см'1. Цианистые натрий и ка-
лий дают сильную полосу в области 2080—2070 см'1.
У расплавов эта полоса смещается до 2250 см'1; обсуждался
вопрос о том [23], не связано ли это с наличием изомерной
формы — изоцианида. Ферро- и феррицианиды калия и ци-
аниды кобальта, которые также поглощают в интервале
2100—2000 см'х, изучены рядом исследователей [6,24—27].
Исследованы также инфракрасные спектры водных рас-
творов цианидных комплексов серебра, меди и золота
[28—30].
Тиоцианаты (всего исследовано шесть) поглощают в об-
ласти 2090—2020 см'1, а цианат серебра [6] имеет сильную
полосу при 2170 см'1
Гл. 21. Неорганические ионы
493
Таким образом, можно считать, что область 2200—
2000 см1 является характерной для различных неоргани-
ческих соединений, содержащих группировку — С = N.
7. ФОСФАТЫ
Из результатов работ по комбинационному рассеянию
света следовало, что фосфат-ион поглощает при 1080
и 980 см1 [8]. Однако при исследовании методом инфра-
красной спектроскопии трехосновных фосфатов во многих
случаях была обнаружена только одна очень сильная и ши-
рокая полоса. Для девяти ортофосфатов, изучавшихся
Миллером и Уилкинсом [6], найдено, что она лежит в об-
ласти 1030—1000 слГ1.
Позднее несколькими группами исследователей, особен-
но Корбриджем [31, 32], Леконтом [33, 34] и Цубои [42],
изучены основные частоты колебаний фосфат-иона всех
типов, в том числе метафосфатов, пирофосфатов, гипофос-
фитов и т. д. Полученные ими результаты представлены
Корбриджем [32] в виде корреляционной диаграммы для
фосфорных оксикислот, и теперь имеется достаточно дан-
ных, чтобы можно было различить большинство из этих
типов соединений. Эти полосы поглощения уже рассмотре-
ны в гл. 18, к которой мы и отсылаем читателя. Буесом
и Герке [35] опубликованы спектры высокополимеров фос-
фатов, находящихся в расплавленном, стеклообразном
и кристаллическом состояниях.
8. СИЛИКАТЫ
Хант и др. [7] исследовали 15 различных силикатов. Для
них найдены очень интенсивные полосы поглощения в обла-
сти 1100—ЭООс.ч Как число, так и положение этих полос
значительно изменяются при переходе от одного минерала
к другому, но все вещества, в которых содержатся сход-
ные силикат-ионы, дают сходные спектры. Тремолит
(OH)2Ca2Mg5(Si4O11)12H актинолит(ОН)2Са2(М£ре) 6(Si40n)12,
например, имеют очень сходные спектры с несколькими
интенсивными полосами в этой области, тогда как горн-
бленд Ca(MgFe)3Si40i2, CaMg2(AlFe)2Si3Oi2 имеет толь-
ко одну полосу при 1010 см1. Показано, что эти раз-
личия достаточны, чтобы их можно было использовать
494
Часть IV
при анализе минеральных руд, причем различные классы
силикатов могут быть идентифицированы по их характерным
спектрам поглощения ]9]. Выполнены также исследования
спектров таких веществ, как стекловидный фарфор [36],
и отдельных кристаллических силикатов, таких, как афвил-
лит [371.
9. ДРУГИЕ КОРРЕЛЯЦИИ
Миллер и Уилкинс [7] исследовали также ряд сульфи-
тов, хроматов, боратов, силикатов, броматов, йодатов,
ванадатов, манганатов и т. д. и получили предваритель-
ные сведения об их характеристических частотах. Резуль-
таты этих работ здесь детально не рассматриваются, так
как во многих случаях исследовалось лишь по три или
четыре соединения определенного типа, и хотя имеются осно-
вания ожидать, что и для других соединений должны быть
обнаружены аналогичные спектры, корреляции не могут
пока считаться установленными. В некоторых случаях
корреляции подтверждаются большим числом данных,
но при этом имеются исключения. Так, у пяти из шести
сульфитов обнаружена сильная полоса поглощения в об-
ласти 960—910 см-1, но у сульфита аммония нет полос по-
глощения с частотой, меньшей 1105 см \
Выполнены некоторые исследования эфиров борной
кислоты |38, 39, 43| и самой борной кислоты [40]. В послед-
нем случае частота валентных колебаний В — О состав-
ляет примерно 1400 см 1, а частота деформационных колеба-
ний около 810 см \ но обе они изменяются при изменении
структуры соединения. У простых боратов связь В — О
проявляется [43] около 1340 ± 10 см1, что позволяет
думать о наличии в какой-то мере характера двойной связи.
Изучены также частоты деформационных колебаний
ионов ХО4 и отмечены изменения, возникающие при заме-
не X [19]. Имеются также данные о некоторых неоргани-
ческих перекисях и надперекисях [44].
ЛИТЕРАТУРА
1. Шефер, Матосе и, Инфракрасные спектры, ОНТИ,
М.-Л., 1935.
2. Lecomte, Anal. Chim. Acta, 2, 727 (1948).
3. Lecomte, Cahiers Phys., 17, 1 (1943).
Гл. 21. Неорганические ионы
495
4. Duval, Lecomte, Mor an dat, Bull. Soc. chim.
(France), 18, 745 (1951).
5. Duval, Lecomte, Compt. Rend. Acad. Sci. (Paris), 232,
2306 (1951).
6. M i 1 1 e r, Wilkins, Analyt. Chem., 24, 1253 (1952).
7. Hunt, Wisherd, В о n h a m, Analyt. Chem., 22, 1478 (1950).
8. Герцберг Г., Колебательные и вращательные спектры
многоатомных молекул, Издатинлит, М., 1949.
9. L a u n е г, Amer. Mineralogist, 37, 764 (1952).
10. Duval, Lecomte, Compt. Rend. Acad. Sci. (Paris),
234, 2445 (1952).
II. Louisfert, Pobequin, Compt. Rend. Acad. Sci.
(Paris) 235, 287 (1952).
12. H и n t, Turner, Analyt. Chem., 25, 1169 (1953).
13. Pobequin, Lecomte, Compt. Rend. Acad. Sci. (Paris),
236, 1544 (1953).
14. Pobequin, J. Phys. Radium, 15, 410 (1954).
15. Louisfert, Compt. Rend. Acad. Sci. (Paris), 241, 940
(1955).
16. D e c i и s, J. Chem. Phys., 22, 1956 (1954).
17. Underwood, Toribara, Neuman, J. Amer. Chem.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
Soc., 77, 317 (1955).
Pascal, Duval, Lecomte, Pa caul t, Compt. Rend.
Acad. Sci. (Paris), 233, 118 (1951).
Duval, Lecomte, Compt. Rend. Acad. Sci. (Paris),
239, 249 (1954).
T a r t e, Ann. Soc. Sci. Bruxelles, 70, 244 (1956).
Dows, Whittle, Pimentel, J. Chem. Phys., 23,
1475 (1955).
Waldron, H о r n i g, J. Amer. Chem. Soc., 75, 6079
(1953).
Hoyer, Angew. Chem., 68, 440 (1956).
Bonino, Fabbri, Atti Accad. Naz. Lincei. Rend. Classe.
Sci. Fis. mat. Nat., 21, 246 (1956).
Bonino, Fabbri, Atti Accad Naz. Lincei. Rend. Classe.
Sci. Fis. mat. Nat., 19, 386 (1955); 20, 414 (1956).
Bonino Salvetti, Atti Accad. Naz. Lincei. Rend.
Classe.
m s c h
(1954).
J о
J о
P e
С о
С о
E
n
n
n
r
r
Sci.
w i 1
Fis. mat. Nat., 20, 150 (1956).
1 e r,
Compt. Rend. Acad. Sci. (Paris), 238, 1414
e s,
e s,
n e
b r i
b r i
D и v a
240,
D и v a
В и e s,
А в г у
637 (1954).
n n e
man,
J.
P e
J. Chem. Phys., 22, J135 (1954)'
man,'
1 d g e,
‘ d g e,
L e c i
(1955).
L e с о m
Gehrke,
с т и н и к,
Chem. Phys., 22, 965 (1954).
24, 293 (1956).
Chem. Soc., 1954, 493, 4555.
J. Chem. Phys
J. -
Jones,
Lowe,
J. Appl. Chem., 6, 456 (1956).
66
i e, Compt. Rend. Acad. Sci. (Paris),
t e, Mikrochim. Acta, 1956, 454.
Z. Anorg. Chem., 288, 307 (1956).
Сеткина, Федорова, ЖФХ, 28,
496
Часть IV
37. Р е t с h, Sheppard, М е g a w, Acta cryst., 9, 29 (1956).
38. S t е е 1 е, De с i us, J. Chem. Phys., 25, 1184 (1956).
39. D u v a 1, Lecomte, J. Opt. Soc. Amer., 44, 261 (1954).
40. Bethell, Sheppard, Trans. Faraday Soc., 51, 9 (1955).
41. Hafele, Z. Physik., 148, 262 (1957).
42. Tsuboi, J. Amer. Chem. Soc., 79, 1351 (1957).
43. Werner, O’Brien, Austral. J. Chem., 8, 355 (1955).
44. В r a m e, Cohen, Margrave, M e 1 о c h e, J. Inorg.
Nuclear Chem., 4, 90 (1957).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Ахманова M. В., Успехи хим., 28, 312 (1959),
А 1 1 р г е s s J. G., II a m Ыу A. N., Austral. J. Chem., 12, 569
(1959).
Lawson К. Е., Infra-red Absorption of Inorganic Substances,
Reinhold P. C., New York, 1961.
Г л а в а 22
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ
СЕРУ
1. ВВЕДЕНИЕ
В литературе приводится значительное количество кор-
реляций для полос поглощения групп, включающих атомы
серы, однако лишь некоторые из них находят широкое при-
менение в структурном анализе. Связи серы с водородом
или кислородом изучены не так подробно, как связи ОН
и СО, но соответствующие им полосы поглощения хорошо
определены и обычно распознаются без особого труда.
Полоса S — Н находится в области частот, при которых по-
глощают лишь немногие другие соединения, тогда как
полоса S — О определяется обычно по ее высокой интен-
сивности и характерной сложной структуре. Поскольку
известно, что в большинстве случаев для связи серы с кис-
лородом имеется более одной характеристической полосы,
идентификация этих связей облегчается. Показано [37],
что в случае группировки XSO2Y частоты антисимметричных
и симметричных валентных колебаний сера — кислород
связаны простой линейной зависимостью, так что, исполь-
зуя эту закономерность, можно проводить проверку иден-
тификации указанной группы.
С другой стороны, связи С — S, S — S и т. п. не так
легко идентифицировать, поскольку соответствующие им
полосы поглощения исключительно малоинтенсивны и по-
ложение полос очень переменно. Обычно наблюдаются од-
новременно обе эти особенности. Однако иногда некоторые
ценные сведения об этих связях можно получить, а по-
этому предложенные корреляции здесь рассматриваются
Все обсуждаемые корреляции приведены в табл. 21.
2. ВАЛЕНТНЫЕ КОЛЕБАНИЯ SH
Положение полосы валентных колебаний SH изучено
в некоторых ранних работах. Белл {1, 2] показал, что ряд
32 Л, Беллами
498
Часть IV
Таблица 21
Частота, см-1
- S -H — S —S — -C-S — c=s 2600—2550 (сл.) 500—400 (сл.) 700—600 (сл.) 1200—1050 (с.) (У тиомочевины до 1400 слг1)
— N — C=S -s=o 1500—1470 (с.) 1060—1040 (с) (Для твердого состояния на 10—20 с.и"1 ниже)
— SO--ОН — SO—OR —RO —SO —OR —so2— — SO2C1 — SO2 —N — -o-so2- — O-SO2 —O — SO3H и SO3 — Около 1090 (с.) 1125—1135 (с.) 1215—1150 (с.) 1160—1140 (с.) и 1300—1350 (с.) Несколько выше, чем для R — SO2 — R 1180—1140 (с.) и 1350—1300 (с.) 1200—1145 (с.) и 1420—1330 (с.) 1230—1150 (с.) и 1440—1350 (с.) 1210—1150 (с.) и 1060—1030 (с.), около 650 (с.)
P=S См. гл. 18
простых меркаптанов, а именно: пропил-, бутил- и изо-
амилмеркаптаны, имеют четкую, но довольно слабую полосу
поглощения в интервале 2650—2550 см1. Ему удалось
также установить, что полосы тиофенола и тпо-/г-крезола
около 2550 см1 отсутствуют у соответствующих сульфидов.
Эти данные, относящиеся к колебаниям SH, подтверждены
Эллисом [3], который нашел первый обертон указанных
колебаний в области 5000 см-1, и Уильямсом [41, показав-
шим, что и другие меркаптаны, помимо исследованных Бел-
лом, поглощают около 2630 см
Результаты этих ранних работ полностью подтверждены
последующими исследованиями. Троттер и Томпсон [5],
а также Шеппард [6] изучили большое число простых мер-
каптанов и сравнили полученные данные с данными о соот-
Гл. 22. Органические соединения, содержащие серу 499
ветствующих сульфидах. Основное различие между этими
соединениями состоит в следующем: в случае сульфидов
наблюдается исчезновение полосы SH, которая по данным
Троттера и Томпсона имеет частоту около 2575 см 1 и для
которой Шеппард установил интервал частот 2590—2560 слг1.
Согласно Рендаллу и др. [7], исследовавшим несколько
меркаптанов, интервал частот составляет 2688—2560 см”1,
но он включает, очевидно, и полосу антисимметричных
колебаний SH сероводорода с частотой 2688 см”1. Эта моле-
кула представляет исключение, и органические меркап-
таны, по-видимому, поглощают при частотах, не превышаю-
щих 2600 см'1. Поэтому в табл. 21 приводится интервал
2600—2550 см”1. Такая корреляция подтверждена несколь-
кими работами, опубликованными в последнее время. Хейнс
и др. [38] приводят три тиола, которые поглощают при
2564 см”1; несколько аналогичных примеров указывают
также Вагнер, Бехер и Коттенхан [39]. Абсолютным исклю-
чением является, по-видимому, 1,2-димеркаптоэтан, кото-
рый в жидком состоянии, как указывают Свинэй и др. [40],
поглощает при 2350 см”1. Даже тиоуксусная кислота [8]
и дитиоуксусная кислота [41] поглощают при более высо-
ких частотах (соответственно при 2550 и 2481 см”1), что
указывает на отсутствие заметной ассоциации их молекул,
и действительно, для группы SH не характерна возмож-
ность образования сильных водородных связей, как это
имеет место в случае групп ОН и NH. Частота полосы SH
очень мало изменяется при переходе от жидкости к раз-
бавленному раствору, а поэтому образование межмолеку-
лярных водородных связей, если и происходит, то в очень
небольшой степени. С другой стороны, малые смещения
полос для растворов в некоторых основаниях свидетель-
ствуют об образовании слабой водородной связи S — Н • • • N.
Горди и Стенфорд [9] исследовали частоты валентных
колебаний SH тиофенола, бутил- и бензилмеркаптанов
и тиоуксусной кислоты, растворенных в различных раство-
рителях. Когда в качестве растворителей применяются
простые эфиры и подобные им соединения, частота остается
без изменений, однако при растворении в анилине и изо-
пропиловом эфире наблюдается некоторое ее уменьшение,
а в случае пиридина, а-пиколина и дибензиламина обна-
ружено большое изменение частоты (на величину до 80 см”1).
32*
500
Часть IV
Аналогичные результаты получены Вагнером и др. [39],
которые показали, что тиофенол способен образовывать
с сульфоксидами водородные связи и при этом частота
валентных колебаний SH может смещаться на 100 см1.
В случае ароматических сульфоксидов величина смеще-
ния несколько меньше. Эти исследователи доказали также,
что тиосалициловая кислота (2515 см'1) и ее этиловый
эфир (2542 см'1) могут образовывать хелатные соедине-
ния. Жозьен и др. [42] также опубликовали работу, пока-
зывающую, что тиофенол образует водородные связи;
аналогичные данные получены также для дитиофосфор-
ной кислоты [43]. Плант и др. [69] не обнаружили обра-
зования водородных связей группами SH в жидких ве-
ществах.
Полосы SH не всегда интенсивны, и хотя обычно они
хорошо определяются, но при применении разбавленных
растворов или очень тонких слоев исследуемых образцов
их часто трудно обнаружить. Они маскируются также
в случае соединений, содержащих группу СООН, которая
поглощает в этой области. Между тем, при соответствую-
щих условиях наличие полосы в этой области свидетель-
ствует о присутствии меркаптогруппы. Исследование этой
спектральной области проводилось, например, с целью
выяснения, существуют ли некоторые тиолы хинолина
в меркапто- или тиокетоформе [10]. Отсутствие полосы
поглощения SH в спектре меркаптобензтиазола является,
например, одним из самых веских доказательств того, что
это вещество существует при обычных условиях в виде тио-
кетона.
3. ВАЛЕНТНЫЕ КОЛЕБАНИЯ S-S
Полоса поглощения, соответствующая связи — S — S —,
может быть использована для аналитических целей, по-
видимому, в очень редких случаях. Эта полоса лежит в об-
ласти 500—400 см'1 и имеет очень слабую интенсивность.
Ее можно определить только потому, что соответствующая
линия комбинационного рассеяния очень интенсивна. Вен-
катесварен [11] отнес линию 470 см 1 спектра комбина-
ционного рассеяния жидкой серы к валентным колеба-
ниями — S — S —, а проведенные исследования инфра-
Гл. 22. Органические соединения, содержащие серу
501
красных спектров и спектров комбинационного рассеяния
органических дисульфидов показали, что и в случае этих
соединений связи S — S соответствует полоса поглощения
в той же области.
Троттер и Томпсон [51 отнесли к валентным колеба-
ниям S — S диметил- и диэтил дисульфидов слабые полосы
с частотой 517 см'1, а у ди-н-пропилсульфида они заме-
тили вблизи 500 см'1 лишь очень слабую полосу. Анало-
гичным образом Шеппард [6] отнес к этим колебаниям
в дициклогексилдисульфиде полосу 510 см'1, но он не мог
обнаружить соответствующих полос у ди-н-бутил- и ди-
mpem-бутилдисульфидов. В случае ароматических соеди-
нений полоса, по-видимому, обнаруживается легче, и Ци-
мерман и Уиллис [12], исследовав 15 ароматических дисуль-
фидов, тиолсульфонатов и т. п., приводят для связи S— S
интервал 490—430 см'1; в то же время они отмечают отсут-
ствие такой полосы у соответствующих соединений, не имею-
щих связи S — S. Согласно данным указанных авторов,
эти полосы едва заметны и с трудом подвергаются иден-
тификации, а в случае дифенил дисульфона никакой полосы,
соответствующей колебаниям S — S, не обнаружено. Это
предположительно объясняется тем, что данная молекула
имеет центр симметрии, и если все молекулы вещества
существуют в виде транс-формы, то следует ожидать, что
колебания S — S будут неактивны в инфракрасном спектре.
Цимерман и Уиллис приводят также для колебаний S — S
в цистине значение частоты, равное 454 см~1, что же ка-
сается меркаптанцистеина, то оказалось, что он не имеет
соответствующей полосы поглощения.
Таким образом, наличие этой полосы в области 500—
400 см'1 достаточно хорошо установлено; однако учитывая
малую энергию источников излучения в этой области
спектра и большое количество рассеянного излучения,
использовать указанную полосу (если только ее интенсив-
ность не очень высока) можно лишь с большой осмотри-
тельностью. Поэтому такая корреляция может быть полезна
только в немногих случаях, когда происходят лишь неболь-
шие изменения в характере связей S — S и в данной спект-
ральной области нет какой-либо другой полосы поглоще-
ния, что позволяет применять слой образца относительно
большой толщины.
502
Часть IV
4. ВАЛЕНТНЫЕ КОЛЕБАНИЯ С — S
Валентным колебаниям С — S обычно соответствует
в инфракрасном спектре слабая полоса поглощения в обла-
сти 700—600 см1. Эта корреляция относится ко всем типам
соединений с такой связью; полоса в этой области обнару-
жена у меркаптанов, сульфидов, сульфоксидов, сульфонов
и т. д. Частота колебаний, по-видимому, зависит от харак-
тера связи, образуемой с участием серы, но условия, при
которых происходят ее изменения, подробно не изучены.
Малая интенсивность этой полосы и непостоянство ее поло-
жения весьма ограничивают ее использование в аналити-
ческих исследованиях, тем более что в этой же области
имеется еще ряд полос скелетных колебаний. Поэтому
идентификация связи С — S требует большой осторожности,
и наличие этой связи в исследуемом соединении должно
быть подтверждено данными о спектрах комбинационного
рассеяния, в которых колебаниям С — S соответствует
интенсивная линия. Для аналитических целей полоса
в области 700—600 см1 может быть использована, по-види-
мому, лишь в очень редких случаях.
Первые исследования соединений, содержащих связь
С — S, проведены Троттером и Томпсоном [5], которые
описали спектры поглощения в инфракрасной области
для семи простых меркаптанов от метил- до mpem-бутил-
меркаптана и сопоставили полученные ими результаты
с опубликованными данными по спектрам комбинационного
рассеяния [11, 13—15]. Образцы исследовались в газооб-
разном и жидком состояниях, причем было замечено, что
при сравнительно небольших изменениях строения проис-
ходит значительное смещение полосы С — S. Частота
С — S для метилмеркаптана, например, оказалась равной
705 см-1, тогда как у этилмеркаптана она составляет
660 см'1. Для некоторых высших гомологов в этой области
найдено несколько полос, что затрудняет определение
полосы С — S. Исследованы также три сульфида и три
дисульфида, причем установлено, что полоса С — S лежит
в интервале 705—587 см"1. У четвертого дисульфида,
в котором связь С — S сопряжена с ароматическим коль-
цом, частота С — S оказалась несколько меньшей, а именно
570 см'1. На основании этой работы и более раннего исследо-
вания этилендисульфида Томпсоном и Дюпре [16] Томпсон
Гл. 22. Органические соединения, содержащие серу
503
[17] сделал вывод, что группировке С — S — С должен соот-
ветствовать интервал частот, приблизительно 700—600 см'1.
Эта работа была продолжена затем Шеппардом [6], кото-
рый сравнил инфракрасные спектры и спектры комбина-
ционного рассеяния большого количества меркаптанов,
сульфидов и дисульфидов. Он заметил закономерное пони-
жение частоты при переходе от первичных к вторичным
и затем к третичным соединениям со связью С — S и под-
разделил область 700—600 см 1 следующим образом:
Первичные
Вторичные
Третичные
Соединение
CH3S-
RCH2 — S —
R1R2CH — S —
R1R2R3C — S —
Частота, см 1
705—685
660—630
630—600
600—570
Циклогексиловые производные не подчиняются этой
классификации, хотя пентаметиленсульфид поглощает при
частоте, характерной для первичных сульфидов (654 см'1).
Поэтому такое подразделение интервала частот справед-
ливо только для насыщенных ациклических соединений
и может с успехом использоваться лишь для алкилмеркап-
танов, сульфидов и дисульфидов, аналогичных тем, кото-
рые исследованы и данные о которых послужили основа-
нием для указанной классификации. Шеппард наблюдал
также закономерное изменение частоты по ряду меркап-
тан — сульфид — дисульфид. Во всех случаях полоса
смещается в сторону меньших частот, но величина смеще-
ния составляет примерно 10 см'1 и не умаляет значения
приведенного выше подразделения. Изучено также некото-
рое количество соединений, у которых связь С — S сопря-
жена с двойной связью, причем установлено, что для них
наблюдается смещение полосы в сторону меньших частот
порядка 60 см'1. Сульфид карбонила является, по-види-
мому, исключением, но данные об этом соединении нена-
дежны. Замечено, что сопряжение сопровождается повыше-
нием интенсивности полосы поглощения С — S; так, напри-
мер, дициклогексен ил сульфид дает сильную полосу
634 см'1, тогда как у циклогексилциклогексенилсульфида
эта полоса имеет среднюю интенсивность. Однако число
исследованных соединений еще слишком мало, чтобы можно
было делать какие-либо определенные выводы.
504
Часть IV
Шеппард обратил также внимание на большую бли-
зость между значениями частот колебаний С — S и С — С1.
Из данных о спектрах комбинационного рассеяния извест-
но, что частоты С — С1 закономерно изменяются по ряду
производных следующим образом: метил > этил ~ про-
пил > втор-пропил > трет-бутил ~ аллил ~ бензил,
и для соответствующих соединений со связью С — S
не только наблюдается такая же закономерность в изме-
нении частот, но и почти те же значения частоты. Если
предположить, что эффект уменьшения частоты у соеди-
нений со связью С — С1 соответствует уменьшению сило-
вой постоянной, то следует сделать вывод, что небольшое
отличие масс атомов С1 и S не должно иметь большого зна-
чения и для соединений, содержащих серу, характерен тот
же порядок уменьшения частоты.
Отнесение полос С — S обсуждали также Цимерман
и Уиллис [12], которые провели большую работу по иссле-
дованию ароматических дисульфидов и тиолсульфонатов.
Они встретили при идентификации слабых полос С — S
те же самые затруднения, какие встречались и в более
ранних исследованиях, тем более что в области 700 см'1
проявляются колебания ароматического кольца. Для
всех ароматических соединений, которые были исследов-
ны, полоса колебаний С — S лежит у верхнего предела
указанного выше интервала, составляющего 702—673 см'1.
Для некоторых соединений точное отнесение частоты колеба-
ний С — S невозможно, но и для них также обнаружена
полоса в этой области. В тех случаях, когда фенил непо-
средственно присоединен к атому серы, полоса С — S,
по-видимому, лежит ближе к верхней границе интервала
частот. Цимерман и Уиллис приводят также значения
частот С — S для нескольких аминокислот, содержащих серу,
которые находятся в интервале 700—600 см'1.
Эта корреляция в дальнейшем подтверждена исследо-
ваниями таких отдельных молекул, как дитиоуксусная
кислота [41]—частота 581 см'1, 1,2-димеркаптоэтан
[40] — частота 735 см'1, а также различных сульфонов
и сульфоксисоединений, однако непостоянство положения
и интенсивности полосы ограничивает применимость корре-
ляции, исключая лишь особые случаи.
Гл. 22. Органические соединения, содержащие серу 505
5. ВАЛЕНТНЫЕ КОЛЕБАНИЯ C=S
Индентификация полосы поглощения С = S встретила
определенные затруднения. В случае сероуглерода к валент-
ным колебаниям С = S отнесены полосы с частотами 1522
и 650 см'1 [19], тогда как у карбонилсульфида значение
этой частоты составляет 859 см'1 [20]. В этих соединениях,
однако, углерод образует две двойные связи, а поэтому
приведенные данные не дают никаких указаний о вероят-
ном положении полосы С = S для насыщенных тиоэфиров
и подобных им соединений.
Первоначально Колтуп [18] и Томпсон и др. [21] пред-
положительно указали для этого поглощения интервал
частот 1400—1300 см'1, однако Шеппарду [6] не удалось на
основании спектров ряда исследованных им простых алкил-
тиокетонов установить какую-либо пригодную корреляцию,
что подтвердилось и в лаборатории автора данной моно-
графии. Такая неопределенность может быть объяснена,
очевидно, тем, что в качестве первых объектов исследова-
ния выбирались простые тиокетоны, которые, как теперь
известно, во многих случаях существуют в виде димеров,
т. е. тиокетонная структура исчезает. Успеха в этом направ-
лении можно ожидать от исследований больших молекул,
что и подтверждено работами Мекке [44, 45] и последую-
щими работами. Мекке и др. [44] сопоставили полосы
поглощения карбонила и тиокарбонила в спектрах более
семидесяти соединений различных типов. Предварительные
расчеты показали, что отношение vco : vcs равняется при-
мерно 1,5, а частота С = S находится в интервале примерно
1200—1050 см'1. Это подтверждено экспериментально
сравнением спектров отдельных пар. Как и в случае кар-
бонильной группы, полоса поглощения С = S чувстви-
тельна к природе окружающей структуры, но эффекты
влияния различных заместителей не всегда одинаковы.
Например, атом галогена, присоединенный к атому угле-
рода в тиоэфире, не так сильно повышает частоту С = S,
как атом кислорода, тогда как в случае карбонилсодержа-
щих соединений наблюдается обратное явление. Отноше-
ние карбонильной и тиокарбонильной частот меняется
поэтому в интервале 1,6—1,14. Для различных типов
тиокетонов указанные авторы нашли следующие общие
506
Ч а с т ь IV
интервалы частот: C12CS 1121, (RS)2CS 1058—1053,
(RO)2CS 1212—1234, > С = С — С = S 1156—1143,
(С6Н5)2С = S 1226—1219 см'1, а также производные
тиомочевины, для которых характерен наиболее широкий
интервал 1400—ИЗО см'1. Естественно, что число изучен-
ных соединений различных классов неодинаково, поэтому
приведенные данные по мере дальнейших исследований
новых веществ могут нуждаться в дополнении. Производ-
ные тиомочевины интересны в том отношении, что они могут
облегчить отнесение полос поглощения амидов; тиоамиды
отдельно исследованы Мекке [45]. Для ряда соединений
этого типа найдено, что частота С = S тиолактамов лежит
около 1120 см'1 и мало зависит от изменения агрегатного
состояния. Тиокапролактам, например, поглощает при
1121 см'1 в парах, при 1117 см'1 в растворе в хлороформе
и при 1113 см 1 в твердом состоянии. Производные тиомо-
чевины вообще поглощают при более высокой частоте —
около 1205 см'1. Как в случае тиолактамов, так и в послед-
нем случае имеется полоса поглощения С — N (амид-Ш)
около 1300 см'х.
Доказательства, подтверждающие эту корреляцию, име-
ются еще в нескольких работах. Хаджи [46] исследовал
тиобензанилид и родственные ему соединения, у которых
частота С = S лежит около 1000 см'1, а Джонс и др. [48]
исследовали этилентиокарбонат (С = S 1074 слГ1), 2,4-ди-
оксидитиобензойную кислоту (1125 см'1) и другие ана-
логичные вещества. Эти работы подтвердили также,
что у производных тиомочевины и родственных ей соеди-
нений частота весьма непостоянна. Сама тиомочевина погло-
щает при 1408 см1, тиокарбанилид — при 1344 см'1, а дитио-
оксамид— при 1425 см'1. Марвел и др. [49] опубликовали
данные по нескольким тиокетонам и относят к колебаниям
дитиоэфиров полосы поглощения в интервале 1195—1170 см'1.
Несколько тиокетонов с а-трифторметильной группой изу-
чены Хасзельдином и Киддом [47], причем, как и следо-
вало ожидать, частота С = S у этих соединений выше, чем
у исходных.
У соединений, отличных от тиоамидов и производных
мочевины, колебаниям С = S соответствует в общем случае
интенсивная полоса в интервале 1234—1053 см'1, причем
Гл. 22. Органические соединения, содержащие серу
507
точное значение частоты зависит от природы заместителей.
В случае производных тиомочевины, как уже указывалось,
частота обычно выше, но во всяком случае лежит где-то
в интервале 1400—1150 см1. Столь широкий интервал ука-
зывает, по-видимому, на возможность различной степени
взаимодействия колебаний.
6. ТИОУРЕИДЫ — N—C=S и -NH-C=S
I I I
Рендалл и др. [7] обратили внимание на сильную поло-
су, которая появляется в области 1613—1471 смт1 в спект-
рах 18 исследованных ими соединений с группировкой
R — N — С = S или R — NH — С = S. Авторы пола-
гают, что указанная полоса обусловлена колебаниями
С — N, причем эта связь имеет меньшую против обычной
длину из-за влияния связи С = S; они считают, что приве-
денное значение частоты слишком велико, чтобы его можно
было приписать самой связи С = S.
В случае вторичных амидов это, вероятно, полоса
амид-П, которую Мекке [45] отнес к деформационным коле-
баниям NH. В то же время сильная полоса поглощения
наблюдается в этой области даже у М,М-диалкилпроизвод-
ных, например у дитиокарбаматов. Мы обнаружили силь-
ную полосу в интервале 1490—1470 см'1 у веществ, выде-
ляемых из соответствующих ксантатов, а последующие иссле-
дования Манна [22] и Четта, Дункансона и Венанзи [50]
подтвердили этот факт. В последней работе эта полоса
поглощения, для которой авторы указывают более широ-
кий интервал, 1542—1480 см'1, относится к валентным коле-
баниям по связи С — N, укороченной из-за наличия кано-
нической структуры:
X C---NR
Эти авторы исследовали также изменения частоты в зави-
симости от валентности и направленности связей элемен-
та X. Наибольшая частота наблюдается в случае раство-
ров бивалентных плоских комплексов Pt, Ni, Pd или Си,
508
Часть IV
а в случае бивалентных тетраэдрических комплексов имеет-
ся, по-видимому, тенденция к тому, что частота погло-
щения оказывается несколько ниже. Эта тенденция еще
более ярко выражена для октаэдрических комплексов
таких трехвалентных элементов, как кобальт.
Интересно, что полоса С — N, частота которой у обычных
тиоамидов равна приблизительно 1300 см'1, сильно сме-
щена у таких N.N-диалкилсоединений в сторону высоких
частот (см. выше), однако вполне возможно, что это обус-
ловлено влиянием второго атома серы и значительной
ролью показанной выше ионной формы даже в таких сое-
динениях, как дитиодисульфиды. Следует указать, что
применять эту корреляцию вне пределов ограниченного
круга соединений, на изучении которых она основана,
необходимо с осторожностью.
7. ВАЛЕНТНЫЕ КОЛЕБАНИЯ S=O
О
II
а. Сульфоксиды.S.Числоописанныхвлитературеинфра-
красных спектров сульфоксидов сравнительно невелико, но
проведена довольно большая работа с целью определения,
по крайней мере приблизительно, положения главной
полосы, соответствующей этой группе. Шрибер [231 при
изучении ряда сульфонов исследовал также инфракрасные
спектры дифенил сульфоксида, ди-н-бутилсульфоксида
и хлористого тионила. Он обнаружил полосу около 1190 см'1
и, основываясь на данных о сульфонах, которые поглощают
при частоте, близкой по значению, предположил, что эта
частота (1190 см'1) также связана с колебаниями S = О.
Позднеебыло проведено более подробное исследование ин-
фракрасных спектров сульфоксидов Бернардом, Фабианом
и Кохом [24], которые изучили 7 сульфоксидов, причем
степень чистоты 5 из них была очень высокой. Во всех
случаях эти авторы наблюдали очень сильную полосу около
1050 см'1, которую они отнесли к колебаниям S = О по ана-
логии с моноокисью серы [25], имеющей полосу валентных
колебаний S = О с частотой 1124 см'1.
Установлено, что эта полоса не только очень интенсивна,
но характеризуется также и довольно постоянным значе-
Гл. 22. Органические соединения, содержащие серу
509
нием частоты в случае разбавленных растворов. Это связано
с низкой частотой колебаний прилегающих групп С — S,
в результате чего взаимодействие колебаний является мало-
вероятным. Так, например, и циклогексилметилсульфоксид,
и фенилметилсульфоксид в разбавленном растворе в четы-
реххлористом углероде интенсивно поглощают при 1055 см'1,
и даже у таких ненасыщенных соединений, как диаллил-
сульфоксид (1047 см'1), частота заметно не уменьшается.
Правда, в одном случае (фенилвинилсульфоксид) Прайс
и Джиллис [51 ] обнаружили небольшое увеличение частоты
по сравнению с насыщенными соединениями. Постоянством
частоты сульфоксиды отличаются, по-видимому, от кар-
бонильных соединений, у которых частота сильно зависит
от сопряжения с двойной связью. С другой стороны, при
переходе от твердого состояния к разбавленным растворам
происходит смещение полосы на 10—20 см'1 в сторону
больших частот, так что рассматриваемая группа в какой-
то мере способна, по-видимому, в случае твердого состоя-
ния вещества участвовать в образовании водородной связи.
Так, например, фенилметилсульфоксид поглощает в твер-
дом состоянии при 1035 см1; аналогичные смещения
полос наблюдаются и у других исследованных соединений.
Образование у этих соединений водородных связей не
является неожиданным, поскольку, как показал Моффит
[26], сульфоксидная связь носит характер двойной связи.
Однако при таком отнесении трудно объяснить, почему
у хлористого тионила наблюдаемая частота S = О имеет
высокое значение. Шрибер [23] при исследовании инфра-
красного спектра получил значение частоты S = О, равное
1238 см'1, а соответствующая частота в спектре комбина-
ционного рассеяния [27] равна 1229 см'1. Фтористый тио-
нил поглощает при еще более высокой частоте (1330 см'1)
[52]. Бернард и др. [24] указывают, что хлор, непосред-
ственно присоединенный к атому серы, оказывает, как и сле-
довало ожидать, заметное влияние на частоту колебаний
сульфоксидной связи, так как приводит к уменьшению коли-
чества групп RRS+ — О’ по сравнению с RRS = O. Бернард
отмечает аналогию с поведением карбонильной группы,
в случае которой Хартвелл, Ричардс и Томпсон [28] предло-
жили подобное объяснение изменения частоты от 1718 см'1
для (СН3)2СО до 1803 см'1 для С12СО.
510
Часть IV
В дальнейшем правильность отнесения полосы 1050 см 1
была подтверждена также Цимерманом и Уиллисом [12],
повторно исследовавшими два органических сульфоксида,
о которых сообщал Шрибер [23]. Они нашли, что полоса
1190 слГ1 обладает довольно малой интенсивностью по
сравнению с очень интенсивной полосой вблизи 1050 см'1,
имеющейся у обоих соединений. Так, дифенилсульфоксид
дает сильную полосу при 1053 см'1 (в твердом состоянии—
1042 см'1), а ди-н-бутилсульфоксид — сильную полосу
при 1039 см'1 (в твердом состоянии — 1019 см'1). Эти авторы
исследовали также четыре сульфоксида, относительно кото-
рых предполагалось, что они содержат группировку
—SO — SO —. Однако ни для одного из них не было обна-
ружено сильной полосы при 1050 см'1, а у соответствующих
сульфидов наблюдались полосы только в области 1100—
1000 см'1. Все эти соединения имеют интенсивные полосы
около 1150 и 1340 см'1, которые характерны для сульфонов;
поэтому авторы пришли к выводу, что указанные соедине-
ния существуют в виде тиолсульфонатов со структурой
—S — SO2-.
Амстуц, Хансбергер и Чессик [29] также приводят
некоторые данные об инфракрасных спектрах двух сульф-
оксидов. Наибольший интерес представляют данные об
о-оксидифенилсульфоксиде, у которого в твердом состоя-
нии авторы обнаружили слабую полосу при 1034 см'1
и сильную полосу при 994 см'1. Они объясняют это обра-
зованием водородной связи и, исходя из общих соображе-
ний, указывают, что сульфоксиды должны обладать боль-
шей способностью к образованию такой связи, чем суль-
фоны. Смещение полосы, согласно их данным, больше по
сравнению с тем, которое наблюдалось раньше при переходе
от твердого состояния к раствору, и возможно, что в этом
случае происходило образование внутримолекулярных
водородных связей, подобное тому, которое имеет место
у о-оксиацетофенона. Для карбонильных соединений, у кото-
рых группа СО отделена от 0-гидроксильной группы, сопря-
женной двойной связью, наблюдаются очень большие
смещения полосы СО. Они гораздо больше, чем те, которые
происходят при образовании обычных водородных связей.
Возможно, что смещение полосы для о-оксидифенилсульф-
оксида следует объяснять аналогичным образом, хотя
Гл. 22. Органические соединения, содержащие серу 511
величина его не столь велика, как в случае карбонильных
соединений.
Учитывая сказанное выше, можно предположить, что
для большинства обычных сульфоксидов в случае раство-
ров должна появляться сильная полоса в интервале 1060—
1040 см'1, а в случае твердого состояния частота полосы
должна быть меньше на 10—20 см'1. Следует, однако, пом-
нить, что до настоящего времени изучено небольшое число
соединений, и новые исследования несомненно расширят
круг соединений, у которых частота S = О не будет укла-
дываться в пределы указанного интервала. Колтуп [18]
приводит для сульфоксидов интервал 1090—1000 см1;
этот интервал шире, чем обычно наблюдаемый на практике,
но он, по-видимому, примерно определяет область частот,
соответствующих данной группе. Частота должна быть,
однако, значительно выше у таких соединений, как галоид-
ные тионилы и ковалентные сульфиты, у которых к атому
серы присоединены сильно электроотрицательные атомы.
б. Ковалентные сульфиты— SO—О— и— О — SO — О—.
Сульфиновые кислоты RSOOH и их сложные эфиры
исследованы Детонн и Хаджи [53, 54], а несколько диал-
килсульфитов (RO)2S = О изучены Симоном, Кригсманом
и Дутцем [55]. В случае сульфиновых кислот в растворах
сильная полоса находится около 1090 см'1; частота ее
очень мало меняется при дейтерировании, а у соответ-
ствующих сложных эфиров она несколько выше (1126—
1136 см'1). Сравнение с карбоновыми кислотами и их слож-
ными эфирами подтверждает отнесение этой частоты к коле-
баниям S = О. Частоту такого порядка можно ожидать
и из сравнения с сульфоксидами. Когда к атому серы при-
соединяется еще один атом кислорода, как вдиалкилсуль-
фитах, можно ожидать дальнейшего повышения частоты;
у нескольких таких соединений, которые были исследованы
[55], действительно имеется сильная полоса около 1200 см'1,
относимая к колебаниям S = О.
Такое отнесение основывается на данных по очень
небольшому числу соединений, но хорошо согласуется
с общими представлениями и косвенно подтверждается
данными по соответствующим циклическим соединениям.
Де ла Мер и др. [56] исследовали ряд циклических сульфи-
12
Часть IV
тов в твердом состоянии и обнаружили сильные полосы
поглощения в интервале 1213—1215 см'1 у 1,2-производ-
ных (пятичленный цикл) и в интервале 1195—1208 см'1
у 1,3-производных (шестичленный цикл). Эти интервалы
установлены при изучении довольно ограниченного числа
соединений, и очевидно, что для этих групповых частот
должен быть характерен более широкий интервал частот.
Самант и Эмерсон [571 подтвердили эти данные, хотя и име-
ются некоторые различия в указываемых значениях частот,
обусловленные, по-видимому, тем, что эти исследователи
работали с растворами. Они приводят для поглощения цик-
лических сульфитов интервал 1210—1220 см'1, причем им не
удалось заметить никаких различий между пяти- и шести-
членными циклическими системами. Эти авторы изучали
также зависимость частоты S = О от электроотрицатель-
ности заместителей, используя «групповые электроотрица-
тельности», предложенные Кейгарайзом, но никаких зако-
номерностей им обнаружить не удалось. Это связано,
по-видимому, с различиями в геометрии сульфоксидов и кар-
бонилсодержащих соединений, от которой зависит электро-
отрицательность групп; в последнем случае группировка
плоская, и электроотрицательность может включать эле-
мент мезомерного сопряжения, тогда как в случае сульфо-
ксидов этот эффект вполне может отсутствовать.
в. Сульфоны — SO2 —. Основное исследование харак-
теристических полос поглощения сульфонов в инфракрас-
ном спектре проведено Шрибером [23], который изучил
спектры растворов 13 сульфонов и сравнил их со спек-
трами соответствующих сульфидов. Он нашел, что у сульфо-
нов имеются две полосы, которых нет у сульфидов. Этим
полосам соответствуют частоты 1160—1120 и 1350—1300 см'1.
Полосы очень интенсивны, и их легко распознать, поскольку
при твердом состоянии вещества они расщепляются, образуя
группу сильных полос с очень близкими частотами. Их отне-
сение к валентным колебаниям S = О подтверждается на
примере двуокиси серы [30], у которой обнаружены полосы
при 1151 и 1361 см'1. Шрибер нашел для исследовавшихся
им соединений интервалы 1159—1128 и 1352—1313 см'1.
Он предложил принять для колебаний S = О сульфонов
интервалы 1160—1120 и 1350—1300 см'1. Как и для суль-
Гл. 22. Органические соединения, содержащие серу
513
фоксидов, сопряжение не оказывает заметного влияния на
частоты; действительно, у большинства изученных арома-
тических сульфонов низкочастотная полоса обнаружена
у верхней границы указанного интервала, а именно при
1150 см'1. Шрибер применял в своей работе только рас-
творы и зависимость частот от физического состояния не
изучал.
Бернард, Фабиан и Кох [24] сравнили спектры семи
сульфонов, находившихся в твердом состоянии, со спек-
трами растворов и нашли, что, как ив случае сульфоксидов,
частоты, при которых поглощают твердые вещества, на 10—
20 см'1 ниже, чем частоты для растворов в четыреххлористом
углероде. Данные этих авторов дополняют данные Шри-
бера; как и следовало ожидать, все исследованные ими со-
единения имеют полосы поглощения в интервалах 1160—
1120 и 1350—1300 см'1. Наблюдения показали, что низко-
частотная полоса смещается в результате изменения агре-
гатного состояния или образования водородной связи
несколько меньше, чем высокочастотная. Так, например,
циклогексилметилсульфон поглощает в растворе при
1144 и 1321 см'1, а в жидком состоянии—при 1138 и 1309 см1.
Указанные авторы исследовали также область 500 см'1,
пытаясь обнаружить третью полосу, соответствующую этой
группе и аналогичную полосе двуокиси серы при 520 см'1.
Им не удалось, однако, установить никаких корреляций,
и они предположили, что поскольку в этой области нахо-
дятся также полосы связей С—S, то имеет место влияние этих
связей на частоты рассматриваемой группы. В этом они
расходятся с Колтупом [18], который приписывает суль-
фонам, помимо полос в интервалах 1170—1110 и 1350—
1290 см'1, полосы в области 600—520 см'1. Первые два
интервала мало отличаются от интервалов, приведенных
Шрибером. Подтверждение того, что полосы в интервале
600—500 см 1 могут относиться к деформационным колеба-
ниям SO2, можно, однако, найти в работе Симона и др.
[55, 60].
Амстуц, Хансбергер и Чессик [29] также исследовали
ряд сульфонов, но изучали при этом только полосу 1160—
1120 см'1. Они заметили ряд интересных изменений в этой
области, связанных с образованием водородной связи.
Три сульфона, не содержащих гидроксильной группы,
33 л. Беллами
5 14
Часть IV
поглощают, как было найдено, в области 1151—1146 см'1,
и четвертый с n-оксифенильной группировкой также погло-
щает при 1154 см'1 (1151 см'1 в растворе). С другой стороны,
два о-оксифенилсульфона поглощают в твердом состоянии
при 1138 и ИЗО см'1, а в растворе опять-таки при 1145
и 1148 см'1. Образование водородной 'связи наблюдается
в случае о-метокси-п-оксифенилсульфона, который погло-
щает при 1148 см1 в растворен при 1133 см'1 в твердом со-
стоянии. В работе Прайса и др. [51, 58] также сообщается,
что сульфоны поглощают около 1310 и 1140 см'1, и подтвер-
ждается, что в этом случае сопряжение также не влияет на
частоты колебаний — SO2 —•. Был сделан вывод, что у этих
соединений эффект сопряжения отсутствует. На основании
измерений интенсивностей в инфракрасных спектрах и дан-
ных по дипольным моментам это оспаривалось Барроу и др.
[59]; однако эти авторы сами приводят для частоты симме-
тричных колебаний интервал 1154—1141 см'1, что подтвер-
ждает во всяком случае неизменность частоты при сопря-
жении.
Хотя Цимерман и Уиллис [12] указывают, что дисуль-
фоксиды являются, по-видимому, тиолсульфонатами, дан-
ные о дисульфоксидах нельзя использовать при проверке
правильности корреляции для группы SO2, так как выводы
Цимермана и Уиллиса о строении этих соединений были
сделаны на основании изучения только инфракрасных
спектров. Тем не менее интересно отметить, что четыре
ароматических соединения, которые были исследованы
в твердом состоянии, поглощают в узких интервалах
1154—1144 и 1342—1333 см'1. Так же, как Бернард и др.,
указанные авторы не обнаружили никакой полосы в обла-
сти 520 см'1, которую можно было бы отнести к группе SO2.
В связи с изучением строения сульфонов были опубли-
кованы инфракрасные спектры различных сульфонов также
и другими авторами. Так, например, Росс и Рате [31] при-
водят в своей работе инфракрасные спектры 1-циано-
2-бензилсульфонилциклогексана и соответствующего
2-фенилпроизводного. Они приводят также спектры соот-
ветствующих меркаптопроизводных. Сравнение этих спек-
тров со спектрами сульфонов показывает, что главным отли-
чием последних является в обоих случаях наличие у них
двух полос около 1150 и 1320 см'1. Коп, Моррисон и Филдс
Гл. 22. Органические соединения, содержащие серу 515
[32] также описывают спектры ряда сульфонов, в том числе
карбонилфенилсульфона и а-метилаллилфенилсульфона.
Авторы не обсуждают корреляций, но спектры показывают
наличие сильных полос в указанных интервалах. Спектры
еще двух сульфонов опубликованы Россом, Буши и Роли-
хом [35].
Таким образом, рассматриваемые корреляции можно
считать хорошо установленными, а вследствие очень высо-
кой интенсивности и часто наблюдаемого расщепления
этих полос последние могут быть легко идентифицированы.
У подавляющего большинства изученных веществ первая
полоса в случае растворов находится в узком интервале
1160—1140 см 1 и лишь немного смещается при твердом
состоянии. Однако это можно отчасти объяснить тем, что
многие исследовавшиеся вещества являются ароматически-
ми. Эффект образования водородной связи может приводить
к несколько большему смещению полосы, но оно все-таки
сравнительно невелико (меньше, чем для сульфоксидов).
Таким образом, сульфоны имеют, по-видимому, меньшую
тенденцию к образованию водородной связи, что согла-
суется и с теорией [29]. В работе Беллами и Уильямса [37]
показано, что частоты антисимметричных и симметричных
колебаний группы SO2 связаны линейной зависимостью,
и это можно успешно использовать для проверки правиль-
ности идентификации группы SO2.
г. Сульфонилхлориды. Можно ожидать, что присоеди-
нение атома галогена непосредственно к атому серы в суль-
фоне приведет к смещению обеих характеристических полос
в сторону больших частот. О таких соединениях опубли-
ковано очень мало сведений, однако Шрибер [23] заметил,
что полосы бензолсульфонилхлорида в растворе появля-
ются при 1185 и 1340 см1, а мы обнаружили, что п-толуол-
сульфонилхлорид в твердом состоянии имеет полосы 1166
и 1366 см1. Кроме того, имеется некоторое количество слу-
чайных данных, которые можно найти в работах по иссле-
дованию единичных соединений или небольших групп
[60, 61]. Например, Симон и др. [60] нашли в спектре
метансульфонилхлорида полосы 1370 и 1175 см'1.
д. Сульфонамиды. Для обычных амидов наличие тре-
тичного атома азота с двумя свободными электронами, кото-
33*
516
Ч а с т ь I V
рый непосредственно присоединен к карбонильной группе,
приводит к удлинению связи С — О и смещению полосы
в сторону меньших частот. Смещение имеет одинаковую
величину для первичных, вторичных и третичных амидов,
и следовало бы ожидать, что примерно такой же эффект
будет и в случае сульфонамидов. Количество опубликован-
ных данных по сульфонамидам сравнительно невелико, но
ясно, что смещение полос в сторону меньших частот обычно
не наблюдается. Действительно, в некоторых случаях поло-
сы SO2 выходят за пределы интервала характерных ча-
стот, но они при этом имеют более высокие частоты, чем
обычно.
Адамс и Тьепкема [331 опубликовали инфракрасные
спектры 16 М,М'-дизамещенных сульфонамидов и отме-
тили, что во всех случаях сильные полосы, которые они
относят к группе—SO2—, лежат в интервале 1180—
ПбОслГ1. Шрибер [23], кроме того, указал, что эти спектры
имеют также сильную полосу в интервале 1360—1330 см1,
соответствующую второй сульфоновой полосе. Системати-
ческого изучения первичных и вторичных сульфонамидов
не проводилось. Барнс и др. [34] приводят спектры о-,
м- и «-сульфаниламидов, причем во всех этих спектрах
имеются сильные полосы около 1300 и 1150 см\ а Шрибер
сообщает о трех сульфонамидах, которые поглощают при
1167—1162 и 1358—1346 см-1.
Последующие исследования Адамса [631 подтвердили
его же более ранние данные; наиболее обширное исследо-
вание этих соединений провели Бакстер, Цимерман-Крейг
и Уиллис [64], которые изучили спектры 25 первичных,
вторичных и третичных сульфонамидов в разбавленных
растворах и в твердом состоянии. Для всех трех типов со-
единений частоты SO2 одинаковы и находятся в случае рас-
творов в интервалах 1370—1333 и 1178—1159 см~г. Как
и в случае сульфонов, частота симметричных колебаний
при переходе к твердому состоянию не меняется, тогда как
частота антисимметричных колебаний уменьшается на 10—
20 см-1. Частоты валентных колебаний NH2-rpynn у пер-
вичных сульфонамидов очень близки к частотам первичных
аминов, но в случае вторичных соединений валентные коле-
бания NH проявляются около 3390 см-1, что примерно
на 40 см'1 ниже, чем у соответствующих аминов.
Гл. 22. Органические соединения, содержащие серу
517
8. КОВАЛЕНТНЫЕ СУЛЬФАТЫ И СУЛЬФОНАТЫ
Следует ожидать, что присоединение к группе SO2
атома кислорода приведет к некоторому смещению полосы
SO2 в сторону больших частот. Это находится в соответ-
ствии с тем фактом, что у сложных эфиров частота колеба-
ний СО имеет несколько большее значение, чем у кетонов.
Когда в ковалентном сульфате имеется два таких атома
кислорода, следует ожидать еще большего смещения поло-
сы. Количество имеющихся данных очень мало, но, по-
видимому, такие смещения действительно имеют место.
Шрибер [231 нашел, что два н-толуолсульфоната в раство-
рах поглощают при 1375—1370 и 1185 см'1, а диметил-
и диэтилсульфаты поглощают соответственно при 1193
и 1187 см'1. Пять сложных эфиров п-толуолсульфоновой
кислоты поглощают в интервалах 1375—1350 и 1192—
1170 см'1 [65]; очень близкие значения приводятся для
сульфонов [66], у которых группировка — SO2 — О —
содержится в шестичленной циклической системе. Симон
и др. [68] нашли у нескольких сложных эфиров алкил-
сульфоновых кислот поглощение около 1350 и 1176 см'1.
В согласии со сказанным выше, Колтуп [18] приводит для
ковалентных сульфонатов интервалы частот 1420—1330
и 1200—1145 см 1, а для ковалентных сульфатов 1440—1350
и 1230—1150 см1.
9. СУЛЬФОНОВЫЕ кислоты И СОЛИ
Систематических исследований органических соедине-
ний этого типа не проводилось. Колтуп [18] предложил
интервалы 1260—1150, 1080—1010 и 700—600 см'1. Шри-
бер [23] сообщает, что три сульфоновые кислоты погло-
щают между 1182 и 1170 см'1, но не комментирует никаких
других полос. Хасзельдин и Кидд [67] исследовали 12 суль-
фоновых кислот и солей и указали, что сильные полосы
поглощения наблюдаются в интервалах 1190—1170 и 1064—
1040 см'1, причем между самими кислотами и их солями раз-
ница очень небольшая. Хотя эти интервалы уже, чем при-
веденные Колтупом, однако они относятся и к меньшему
кругу алкил- и фторалкилсульфоновых кислот, а поэтому
приведенные выше более широкие интервалы, по-види-
мому, найдут большее применение.
518
Часть IV
литература
1. Bell, Вег., В60, 1749 (1927).
2. Bell, Вег., В61, 1918 (1928).
3. Е 1 1 t s, J. Amer. Chem. Soc., 50, 2113 (1928).
4. Williams, Phys. Rev., 54, 504 (1938).
5. Trotter, Thompson, J. Chem. Soc., 1946, 481.
6. Sheppard, Trans. Faraday Soc., 46, 429 (1950).
7. Randall, Fowler, Fuson, Dangl, Infra-red Deter-
mination of Organic Structures, Van Nostrand, 1949.
8. Sheppard, Trans. Faraday Soc., 45, 693 (1949).
9. Gordy, Stanford, J. Amer. Chem. Soc., 62, 497 (1940).
10. Hannan, Lieblich, Renfrew, J. Amer. Chem. Soc.,
71, 3733 (1949).
11. Venkateswaren, Indian J. Physics, 5, 219 (1930).
12. С у m e r m a n, Willis, J. Chem. Soc., 1951, 1332.
13. Kohl r a usch, Dadieu, Pongrat z, Wien. Ber.,
141, Ila, 276 (1932).
14. Корре r, Seka, Kohlrausch, Wien. Ber., 141,
Ila, 465 (1932).
15. Kopp 1, Kohlrausch, Wien. Ber., 142, 1 lb, 477 (1933).
16. T h о m p s о n, Dupre, Trans. Faraday Soc., 36, 805 (1940).
17. T h о m p s о n, J. Chem. Soc., 1948, 328.
18. С о 1 t h u p, J. Opt. Soc. Amer., 40, 397 (1950).
19. Г e p ц б e p г Г., Колебательные и вращательные спектры
многоатомных молекул, Издатинлит, М., 1949, стр. 299.
20. Bailey, Cassie, Proc. Roy. Soc., A135, 375 (1932).
21. Thompson, Nicholson, Short, Discuss. Faraday
Soc., 9, 222 (1950).
22. Mann, Trans. Inst. Rubber Ind., 27, 232 (1951).
23. S c h r i e b e r, Analyt. Chem., 21, 1168 (1949).
24. Barnard, Fabian, Koch, J. Chem. Soc., 1949, 2442.
25. Herzberg, Molecular Spectra and Molecular Structure, Vol. 1,
Van Nostrand, 1950, p. 573.
26. M о f f i t, Proc. Roy. Soc., A200, 409 (1950).
27. Cabannes, Rousset, Ann. Physique, 19, 229 (1933).
28. Hartwell, Richards, Thompson, J. Chem. Soc.,
1948, 1436.
29. Amstutz, Hunsberger, Chessick, J. Amer. Chem.
Soc., 73, 1220 (1951).
30. Герцберг Г., Колебательные и вращательные спектры
многоатомных молекул, Издатинлит, М., 1949, стр. 308.
31. R о s s, Raths, J. Amer. Chem. Soc., 73, 129 (1951).
32. С о p e, Morrison, Field, J. Amer. Chem. Soc., 72,
59 (1950).
33. Adams, Tjepkema, J. Amer. Chem. Soc., 70,4204 (1948).
34. Barnes, Gore, Liddel, Williams, Infra-red
Spectroscopy, Reinhold, 1944, p. 87.
35. R о s s, Bushey, R о 1 i h, J. Amer. Chem. Soc., 73, 540
(1951).
36. Flett, J. Chem. Soc., 1953, 347.
Гл. 22 Органические соединения, содержащие серу
519
37.
38.
39.
40.
41.
42.
Bellamy, Williams, J. Chem. Soc., 1957, 863.
Haines, Helm, Bailey, Ball, J. Phys. Chem., 58,
270 (1954).
Wagner, Becher, KoHenhahn, Chem. Ber., 89,
1708 (1956).
Sweeney, Mizushima, Quagliano, J. Amer.
Chem. Soc., 77, 6521 (1955).
Mecke, Spiesecke, Chem. Ber., 89, 1110 (1956).
Josie n, Dizabo, Saumagne, Bull. Soc. Chim.
France, 1957, 423.
43. Meneefe, Alford, Scott, J. Chem. Phys., 25, 370 (1956).
44. Mecke, Mecke, Liittringhaus, Z. Naturforsch.,
105B, 367 (1955).
45. Mecke, Mecke, Chem. Ber., 89, 343 (1956).
46. Hadzi, J. Chem. Soc., 1957, 847.
47. Haszeldine, Kidd, J. Chem. Soc., 1955, 3871.
48. Jones, Ky naston, Hales, J. Chem. Soc., 1957, 614.
49. M a r v e 1, R a d z i t z к у, В r a d e r, J. Amer. Chem.
Soc., 77, 5997 (1955).
50. Chatt, Duncanson, Venanzi, Suomen. Kem., 29B,
75 (1956).
51. P r i c e, Gillis, J. Amer. Chem. Soc., 75, 4750 (1953).
52. Haszeldine, Kidd, J. Chem. Soc., 1955, 2901.
53. Deton i, Hadzi, J. Chem. Soc., 1955, 3163.
54. D e t о n i, Hadzi, Bull. Sci. Yougoslavie, 2, 44 (1955).
55. Simon, Kriegsmann, Dutz, Chem. Ber., 89, 2390 (1956).
56. De la Mere, К 1 у n e, Millen, Pritchard,
Watson, J. Chem. Soc., 1956, 1813.
57. Samant, Emerson, J Amer. Chem. Soc., 78, 454 (1956).
58. Price, Morita, J. Amer. Chem. Soc., 75, 4747 (1953).
59. Rogers, Barrow, Bordwell, J. Amer. Chem. Soc.,
78, 1790 (1956).
60. S i m о n, Kriegsmann, Dutz, Chem. Ber., 89, 1883 (1956).
61. Ham, Hambly, Aust. J. Chem., 6, 33 (1953).
62. Dudley, Cady, Eggers, J. Amer. Chem. Soc., 78,
290 (1956).
63. Adams, Cosgrove, J. Amer. Chem. Soc., 76, 3584 (1954).
64. Baxter, Cymerma n-C r a i g, Willis, J. Chem. Soc.,
1955, 669.
65. T i p s о n, J. Amer. Chem. Soc., 74, 1354 (1952).
66. Philbin, Stuart, Timoney, Wheeler, J. Chem.
Soc., 1956, 4414.
67. H a s z e 1 d i n e, Kidd, J. Chem. Soc., 1954, 4228.
68. Simon, Kriegsmann, Dutz, Chem.В er.,84, 2378 (1956).
69. P 1 a n t, T a r b e 1 1, W h i t e m a n, J. Amer. Chem. Soc.,
77, 1572 (1955).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Юрьев Ю. К-, Пентин Ю. А., Ревенко О. М., Нефте-
химия, 1, 163, 742 (1961).
Поглощение, % Поглощение, %
Р и’с. 24. Изопропилнитрат.
Частота, см'
Рис. 26. Дифенилфосфорная кислота.
Рис. 28. Перфтордиметилциклогексан.
ЬЭ
Частота, см
Рис. 30. Меркаптобензтиазол.
ЧАСТЬ
V
ПРИЧИНЫ
ИЗМЕНЕНИЯ ЧАСТОТ КОЛЕБАНИЙ
ГРУПП АТОМОВ
И ЗНАЧЕНИЕ ЭТИХ ИЗМЕНЕНИЙ
34 Л. Беллами
Глава 23
ПРИЧИНЫ ИЗМЕНЕНИЯ ЧАСТОТ КОЛЕБАНИЙ
ГРУПП АТОМОВ И ЗНАЧЕНИЕ ЭТИХ ИЗМЕНЕНИЙ
1. ВВЕДЕНИЕ
Концепция характеристичности частот колебаний групп
атомов является вообще чрезмерно упрощенной. В этом
приближении частота колебаний вдоль связи АВ опре-
деляется упругостью связи, характеризуемой силовой
постоянной, и массами А и В. В случае сложных молекул
частота изменяется под действием ряда других факторов,
влияние которых и определяет точное положение соответ-
ствующей полосы поглощения. Некоторые из этих факто-
ров связаны с внешними воздействиями на колеблю-
щуюся группу; сюда относятся эффекты изменения агре-
гатного состояния вещества или его кристаллической формы,
влияние растворителей и наличие или отсутствие водород-
ной связи. Другая группа факторов носит характер вну-
тренних воздействий и включает изменения геометрии моле-
кулы или массы замещающих групп, наличие механической
связи между различными колебаниями молекулы, эффекты
стерических затруднений и влияния электрических полей
замещающих групп, действующих вдоль связей или в неко-
торых случаях во всем внутреннем объеме молекулы.
Учитывая столь большое число возможных переменных
параметров, с первого взгляда трудно даже понять, почему
вообще возможно существование характеристических частот
колебаний групп атомов; но следует помнить, что при неко-
торых благоприятных обстоятельствах действие многих
из рассмотренных факторов может быть сведено к мини-
муму. Например, можно показать, что частота колебаний
карбонильной группы почти совершенно не зависит от массы
заместителей во всех случаях, когда их атомный вес
больше 12, а высокое значение силовой постоянной этой
связи по сравнению с силовыми постоянными соседних
связей обеспечивает отсутствие механической связи
34*
532
Часть V
колебаний. Поэтому основная частота колебаний остается
по существу постоянной, и наблюдаются лишь небольшие
различия в частотах под влиянием пространственных затруд-
нений или под воздействием электрических эффектов заме-
стителей. Эти небольшие различия приводят, однако, к важ-
ным отличиям между частотами карбонильного поглощения
кетонов, сложных эфиров, хлоридов карбоновых кислот
ит. п., которые имеют очень большое значение в анали-
тической работе. Вообще колебания по сложным связям
и колебания с участием атомов водорода являются наиме-
нее чувствительными к изменениям масс заместителей и к
эффектам механической связи колебаний, вследствие чего
колебания по связям этого типа отличаются наиболее харак-
теристическими частотами. С другой стороны, в случае таких
связей, как одинарная связь углерод — углерод, полосы
поглощения появляются в такой широкой области, что не
представляют никакой ценности для целей идентификации.
Несмотря на то что имеется значительное количество
переменных параметров, которые определяют положение
индивидуальной полосы поглощения, часто можно создать
такие условия, при которых один какой-либо параметр
будет практически отделен от других и может быть исследо-
ван отдельно. Например, при изотопическом обмене одного
из атомов изменяются массы колеблющихся атомов, но не
меняется химическая природа связи; одно и то же веще-
ство может быть исследовано в растворах в различных
растворителях или в разных состояниях; влияние напря-
жения может быть изучено путем сравнения аналогичных
циклических систем различных размеров. Исследования
этого рода позволяют лучше понять, при каких условиях
какой-либо из рассмотренных факторов оказывает сильное
влияние на данную частоту колебаний, и при некоторых
благоприятных обстоятельствах может быть предсказано
ожидаемое направление, а иногда даже и примерная вели-
чина смещения полосы, которое вызывается данным изме-
нением окружения группы. При этом не предполагается,
что все действующие факторы известны или измерены. Ниже
отдельно рассматриваются различные факторы, и можно
видеть, что наши знания о них неравноценны. Довольно
хорошо исследованы условия, при которых могут наблю-
даться значительные смещения полос поглощения, обу-
Гл. 23. Причины изменения частот колебаний групп атомов 533
словленные изменением масс атомов, участвующих в коле-
баниях, эффектами связи колебаний и водородной связью,
но по вопросу о влиянии различных растворителей име-
ются только отрывочные данные. Имеющиеся представле-
ния о влиянии электрических факторов и особенно о вну-
тримолекулярных дипольных взаимодействиях также нахо-
дятся еще в стадии формирования, а потому проводимое
ниже обсуждение этих проблем следует рассматривать как
выражающее в какой-то мере точку зрения автора. Многие,
казалось бы, аномальные значения частот могут быть
удовлетворительно объяснены на основе уже известных
представлений, но ряд явлений, таких, как высокая частота
валентных колебаний С = С у тетрафторэтилена (активных
в спектре комбинационного рассеяния), остаются без какой-
либо разумной интерпретации, и ясно, что для надлежаще-
го истолкования необходимо дальнейшее развитие теории.
Несмотря на эти ограничения, исследования природы
смещения полос поглощения, характерных для колебаний
различных групп, позволили получить ряд корреляций
между положением различных полос поглощения в отдель-
ном спектре, которые полезны при качественном анализе.
Обнаружен также ряд количественных соотношений между
смещениями полос поглощения и другими физическими
свойствами, такими, как реакционная способность, кислот-
ность и основность, длина связи и т. д. Эти соотношения
могут быть успешно использованы для ряда дальнейших
исследований и последовательно обсуждаются ниже.
2. ЭФФЕКТЫ ИЗМЕНЕНИЯ СОСТОЯНИЯ ВЕЩЕСТВА
Хотя некоторые вещества, например фтористый водо-
род, и характеризуются наличием ассоциации молекул
даже в парах при малых давлениях, однако такие веще-
ства являются редкими исключениями. Поэтому при иссле-
дованиях колебаний молекул в парах их можно рассма-
тривать как колебания практически свободных молекул.
В то же время в жидкости колеблющаяся группа окру-
жена другими молекулами, которые могут влиять на частоту
ее колебаний либо в связи с наличием диэлектрических
свойств среды, либо вследствие ассоциации молекул.
У карбоновых кислот димеризация с образованием сильных
534
Часть V
водородных связей приводит к очень сильному уменьшению
частот колебаний карбонильной и гидроксильных групп
соответственно на 50 и 500 слГ1 по сравнению с частотами
колебаний в случае пара. Однако ассоциация молекул не
всегда приводит обязательно к образованию специфи-
ческого молекулярного комплекса. Например, у- жидкого
ацетона силы притяжения между отдельными диполями
\с = 6, вероятно, способствуют образованию непрочных
ассоциатов типа = О \С = О, в которых отдель-
ные молекулы связаны в цепи электростатическими силами.
При этом каждый положительный заряд индуцирует неболь-
шой дополнительный отрицательный заряд на атоме кис-
лорода соседней молекулы и полярность связи С = О
повышается. Это означает, что возрастает роль резонансной
структуры типа ^>С—О, и, следовательно, длина связи
увеличивается, а частота колебаний карбонильной группы
уменьшается. Возможно, что именно этой причиной и обу-
словлено в основном смещение полосы поглощения ацетона
при переходе от пара (vc01742 слг1) к жидкости (vc0 1718 сл/'1).
Несколько иное положение наблюдается при переходе от
жидкого состояния к твердому кристаллическому состоя-
нию. Происходящие при этом из-за дальнейшего усиления
межмолекулярного взаимодействия смещения частот обычно
невелики, за исключением случая, когда взаимодействие
осуществляется с участием водородных связей. Однако
вследствие возросшего порядка системы в спектре часто
исчезают некоторые полосы. Иногда, наоборот, появляются
дополнительные полосы поглощения. Первый из этих эффек-
тов обусловлен явлением поворотной изомерии, так как раз-
личные поворотные изомеры существуют у вещества в жид-
ком и газообразном состоянии, но не в ориентированном кри-
сталле, вследствие чего в спектре последнего уже не появ-
ляется полос поглощения, соответствующих менее стабиль-
ному изомеру*. Например, у жидкого со-хлорацетофенона
* Поворотный изомер, остающийся в кристалле, не обязатель"
но является самым стабильным в жидкости и газе. Возможно также
образование, кристаллической модификации более чем с одним
поворотным^изомером.— Прим. ред.
Гл. 23. Причины изменения частот колебаний групп атомов 535
наблюдаются две полосы поглощения карбонильной группы
примерно одинаковой интенсивности, а в спектре кристалла
остается только одна из этих полос. И наоборот, значитель-
ная часть полос поглощения, имеющих аналитическое зна-
чение и наблюдающихся в спектрах кристаллических жир-
ных кислот, обусловлена главным образом полностью транс-
расположением метиленовых групп. В спектрах жидких
образцов этих прогрессий полос не обнаружено*.
Появление дополнительных полос поглощения в спект-
рах твердых веществ обусловлено различными эффектами.
В жестких кристаллических структурах осуществляются
значительные межмолекулярные взаимодействия, вслед-
ствие чего колебания каждой группы атомов зависят от
свойств элементарной ячейки. При соответствующих усло-
виях проявляются синфазные колебания и колебания не
в фазе одинаковых групп атомов у различных молекул,
приводящие к расщеплению соответствующей простой
полосы на два компонента. Особенно чувствительна к это-
му эффекту полоса поглощения около 720 см~\ соответ-
ствующая маятниковым колебаниям — (СН2)„—. Расщеп-
ление этой полосы в спектре кристаллического полиэти-
лена используется для простого определения степени кри-
сталличности вещества. Аналогичным образом такие изме-
нения в кристаллической решетке, какие имеют место при
переходе от одной полиморфной формы к другой, также
могут привести к значительным изменениям спектра вслед-
ствие тех изменений, которые происходят в ближайшем
окружении колеблющихся групп. Этот эффект особенно
сильно сказывается в низкочастотной области спектра, где
нередко различия между кристаллическими формами одного
и того же вещества сказываются настолько сильно, что их
можно использовать для определения отдельных компо-
нентов смеси. Различного рода эффекты влияния кристал-
лической решетки наблюдаются также в некоторых случаях
при исследовании образцов, запрессованных в виде табле-
ток с галогенидами щелочных металлов. Показано, что
изменения в наблюдающихся спектрах зависят от степени
* В жидкой фазе в результате беспорядочной ориентировки
метиленовых групп в молекулах вместо прогрессии узких полос
появляется очень широкая размытая полоса поглощения.—
Прим. ред.
536
Часть V
измельчения смеси и что в случае мелкодисперсных образ-
цов обнаруживаются взаимодействия между колебаниями
молекул образца и решетки галогенида щелочного металла.
При этом могут быть получены спектры, заметно отличаю-
щиеся от спектров того же вещества, исследованного в виде
взвеси в парафиновом масле. Подобные явления наблюда-
лись у карбогидратов [2], гидроксилсодержащих соедине-
ний и кислот [3, 4] и у других веществ и были обсуждены
Фармером [3] и Баркером [5].
Таким образом, факторы, определяющие изменения
в спектрах при изменениях состояния вещества, в общих
чертах известны, но уровень наших знаний в настоящее
время еще недостаточен, чтобы объяснить или предсказать
все наблюдающиеся эффекты- Как правило, наличие моле-
кулярной ассоциации приводит к уменьшению частот
валентных колебаний и увеличению частот деформационных
колебаний, и, за исключением соединений с сильными водо-
родными связями, смещения полос, обусловленные этой
причиной, обычно невелики. Например, для частот колеба-
ний карбонильной группы весь интервал изменений частот
при переходе от пара к твердому веществу редко превышает
25 см"1, хотя в исключительных случаях при наличии резо-
нансных структур он может достигать 100 см"1. Труднее
предсказать величины изменения частот, возникающие
вследствие эффектов кристаллической упаковки. Эффекты
расщепления полос поглощения в кристаллах сопряжены
только с небольшими изменениями частот, но, как указы-
валось выше, многие низкочастотные колебания, по-види-
мому, очень чувствительны к ближайшему окружению
группы в кристалле, и иногда наблюдаются значительные
изменения этих колебаний.
3. ВЛИЯНИЕ РАСТВОРИТЕЛЕЙ
Изменения частот, которые наблюдаются при переходе
от одного растворителя к другому, обычно малы, если отсут-
ствует водородная связь. Эти эффекты не вполне ясны
и не очень детально изучены. Наблюдающиеся изменения
объясняю.. । обычно подобно тому, как объяснялись выше
изменения частот, происходящие при изменении состояния
вещества. Например, при замене одного растворителя
Гл. 23 П ричины изменения частот колебаний групп атомов 537
другим изменяется диэлектрическая проницаемость среды,
в которой находится колеблющаяся молекула. Это сообра-
жение привело к многочисленным попыткам сопоставить
изменения частот при смене растворителя с диэлектрической
проницаемостью или показателем преломления раствори-
теля [6—13]. Кирквуд [6] и Бауэр и Мегет [7] приводят
следующее выражение, связывающее частоты колебаний
простого диполя в парах (v0) и в растворе (vs) с диэлектри-
ческой проницаемостью растворителя D:
vc—vs К (Г - -1)
v,, ~ (2Р + 1) ’
где К — константа. Исследования этих авторов выполнены,
в основном, с неполярными растворителями, а в более позд-
них работах Жозьен и др. [8—11 ] и Бейлисса, Коула и Лит-
тла [12] было показано, что хотя это выражение прибли-
женно выполняется для большинства неполярных раство-
рителей, однако для полярных растворителей, включая даже
в ряде случаев бензол, оно не выполняется. Несколько луч-
шее совпадение с экспериментальными данными получено
[11, 12] при замене D квадратом показателя преломления,
но поскольку смещения полос при переходе от одного непо-
лярного растворителя к другому сравнительно невелики,
то ясно, что для адекватного описания имеющихся данных
необходимо включить в рассмотрение ряд дополнительных
факторов. Эта точка зрения подтверждается тем обстоятель-
ством, что в случае более концентрированных растворов,
когда в спектре появляются полосы, соответствующие вза-
имодействию между молекулами растворенного вещества,
наблюдаемая частота колебаний в некоторых случаях не
зависит от диэлектрической проницаемости растворителя
[11], хотя можно было бы ожидать, что она особенно чув-
ствительна к влиянию растворителя.
Главной причиной отклонений от соотношения Кирк-
вуда, Бауэра и Мегета должны быть явления ассоциации
молекул растворенного вещества с молекулами раствори-
теля. Один предельный случай представляют вещества, обла-
дающие очень большой способностью образовывать ассо-
циаты, например соляная кислота в эфире, которая обра-
зует комплекс с соотношением компонентов 1 : 1, а другой
предельный случай представляют растворители с доста-
538
Часть V
точно произвольной ориентацией диполей, вследствие чего
наблюдается значительное разнообразие во взаимном рас-
положении молекул растворенного вещества и раствори-
теля, не являющемся, однако, совершенно произвольным.
Бауэр и Мегет [7] установили, что использование в приве-
денном выше выражении значения статической диэлектри-
ческой проницаемости вполне обосновано только в том
случае, если компоненты электронной и ориентационной
поляризации следуют за любыми мгновенными изменениями
ориентации диполей растворителя, вызванными тепловым
движением, колебанием и колебательными переходами.
Поскольку же время релаксации ориентации диполей во
много раз больше [98], чем период колебания молекулы, то
это условие в случае растворов не выполняется.
В свете этих проблем Бейлиссом и др. [12] было пред-
принято исследование изменений частот и интенсивности
полос, соответствующих колебаниям С = О, С — Си С — Н.
При смене растворителя полосы, соответствующие коле-
баниям С — С, претерпевают очень малые смещения, смеще-
ния полос С = О и С — Н порядка 10—20 см-1, а полосы,
соответствующие обертонам колебаний С = О, смещаются
на величины, почти вдвое большие, чем смещения полос,
соответствующих основным частотам. Полученные авторами
этого исследования результаты в общем хорошо согласу-
ются с развитыми ими теоретическими представлениями, но
результаты, относящиеся к колебаниям С — Н, труднее
поддаются такой интерпретации.
У некоторых веществ, представляющих исключение, при
увеличении полярности растворителя наблюдается не пони-
жение, а повышение частоты валентных колебаний. Таким
необычным соединением является нитрозилхлорид, иссле-
дованный в наших лабораториях. Это вещество в парах пог-
глощает при 1800 сл/-1, а в растворе в четыреххлористом уг-
лероде частота поглощения увеличивается до 1814 см
При повышении полярности растворителя наблюдается
дальнейшее смещение полосы поглощения в более высоко-
частотную область. Частота поглощения принимает следую-
щие значения в случае раствора: в бензоле 1836, в диок-
сане 1859, в ацетонитриле 1874 и в нитрометане 1881 сл/-1.
При этом нельзя предположить образование комплексов,
так как в смесях с четыреххлористым углеродом и ацето-
Гл. 23. Причины изменения частот колебаний групп атомов 539
нитрилом наблюдаемое смещение полосы поглощения в сто-
рону более высоких частот увеличивается при увеличении
концентрации более полярного растворителя. В этом слу-
чае должно происходить какое-либо дипольное взаимодей-
ствие с растворителем, при котором наличие более поляр-
ных растворителей приводит к повышению роли ионной
канонической формы (О = N)* — СГ и тем самым к при-
ближению частоты колебаний N = О к частоте колебаний
радикала (N s? О)+.
До сих пор нет никакой теории, которая могла бы объяс-
нить все эти экспериментальные данные. Однако в некото-
рых отношениях поставленная проблема аналогична той,
которая возникает при сопоставлении дипольных моментов
молекул, измеренных с использованием различных раство-
рителей. При этом полуэмпирическое рассмотрение вопро-
са позволяет получить некоторые достаточно обоснованные
соотношения, с помощью которых обычно удается с боль-
шой точностью предсказать те изменения, которые наблю-
даются при переходе от одного растворителя к другому
[14—16]. Эти соотношения содержат произвольные члены,
отражающие влияние структурных факторов, и весьма
вероятно, что аналогичный подход к рассмотрению сме-
щений полос поглощения окажется достаточно плодотвор-
ным. В этой связи следует, вероятно, заметить, что диполь-
ный момент нитрозилхлорида также изменяется аномально,
подобно изменению частот колебаний при переходе от пара
к жидкости, т. е. в направлении, обратном обычному.
4. ВОДОРОДНАЯ СВЯЗЬ
Водородная связь является, очевидно, особым случаем
рассматривавшейся выше ассоциации молекул. Однако
она изучена гораздо детальнее, чем другие виды ассоциа-
ции, и ее следует обсудить отдельно, так как она является
единственным примером, когда удовлетворительные коли-
чественные данные о строении и свойствах могут быть полу-
чены непосредственно из смещений полос, наблюдающихся
при изменении состояния вещества. Величины смещений
полос, которые соответствуют различным типам водород-
ной связи, уже приведены в соответствующих главах кни-
ги, посвященных отдельным группам атомов, и нет необ-
540
Часть И
ходимости вновь останавливаться на этом вопросе. Основ-
ные изменения наблюдаются у частот валентных и дефор-
мационных колебаний связей X — Н, влияние же на про-
тоноакцепторную группу обычно невелико, если только не
наблюдается резонансной стабилизации. Однако в тех слу-
чаях, когда имеет место механическое взаимодействие коле-
бания X — Нс каким-либо другим колебанием, как это
наблюдается в случае деформационных колебаний ОН
и валентных колебаний СО у карбоновых кислот [17], то
изменения в характере водородной связи естественно вызы-
вают изменения частот полос поглощения и в других обла-
стях спектра.
Первые попытки получения количественных данных из
величин изменений частот валентных колебаний X — Н
при ассоциации сделаны в работе Беджера [18], который
указал, что изменения частот колебаний связанных гидро-
ксильных групп являются хорошим критерием для оценки
величин силовых постоянных образующихся водородных
связей. Изменения частот связаны также с расстоянием
О ... О и с энергией водородной связи. Была получена фор-
мула, с помощью которой можно прямо вычислить энергии
таких связей, имея экспериментальные данные об измене-
ниях частот.
В последующих работах Лорда и Меррифилда [19]
показано, что в случае органических кислот, ацетилгли-
цина, льда и других веществ, у которых осуществляются
сильные связи ОН ... О, изменения частот колебаний ОН
обратно пропорциональны расстояниям между атомами
кислорода. Эго соотношение успешно применено для диф-
ференциации геометрических изомеров. Полученные таким
образом расстояния О — Н ... О у циклогександиола-1,2,
например, были использованы для определения вероятных
конфигураций этого соединения [20, 21]. При использова-
нии такого соотношения в его первоначальном виде следу-
ет, однако, соблюдать осторожность, так как могут возни-
кать отклонения от него в тех случаях, когда три атома,
участвующие в образовании водородной связи, не лежат на
одной прямой. Эго обстоятельство выяснилось благодаря
дальнейшим исследованиям Накамото, Маргошеса и Ранд-
ла [22] и Пиментела и Седерхолма [23], выполненным на
широком круге различных соединений, способных образо-
Гл. 23. Причины изменения частот колебаний групп атомов 541
вывать водородные связи. Эти исследователи подтвердили,
что для всех связей ОН ... О (кроме случая наибольших
расстояний О ... О) изменение частоты колебаний ОН
является линейной функцией расстояния между атомами
кислорода, при условии, что связи лежат на одной прямой.
Это соотношение выражается формулой
Av = 4,43-103 (2,84 - 7?),
где 7? — расстояние О ... О. Когда атомы кислорода нахо*
дятся на большом расстоянии друг от друга, водородные
связи очень слабы и можно ожидать, что форма указанного
соотношения изменится и будет приближаться к асимптоте.
В более детальных исследованиях этого вопроса Липпин-
коттом и Шредером [100] предложена потенциальная функ-
ция, которая, по-видимому, достаточно хорошо описывает
систему О — Н ... О. Некоторые уточнения функции пред-
ложены Уэлшем [101].
Аналогичные соотношения получены [22] для ОН ... N,
ОН ... С1 и других типов водородных связей, но наклон
графиков, полученных из этих соотношений, весьма разли-
чен, а разброс экспериментальных точек заметно больше.
Соотношения этого типа рассмотрены также Кенноном [24]
в связи с исследованиями ассоциации вторичных амидов,
у которых изменения частот колебаний NH очень малы по
сравнению со значительными изменениями частот колеба-
ний карбонильной группы. На основании полученных дан-
ных Кеннон высказал мнение, что в этих случаях осуще-
ствляется не водородная связь какого-то типа, а имеет место
дипольное взаимодействие. Однако не вполне ясно, на-
сколько сильно это предположение отличается от более
обычных представлений, так как водородные связи в любом
случае должны в некоторой мере иметь характер диполь-
ного взаимодействия.
Другим удобным применением изменений частоты коле-
баний X — Н является определение с их помощью теплот
смешения веществ, образующих водородные связи. Тем-
ресом и др. [25], например, показано, что изменение часто-
ты валентных колебаний OD в случае растворов D-метано-
ла в органических основаниях связано линейной зависи-
мостью с теплотой смешения, а также зависит от основности
растворителя.
542
Ч а с т ь V
5. ЭФФЕКТ МАСС И ВЛИЯНИЕ ВЗАИМОСВЯЗИ
КОЛЕБАНИЙ
Эффект масс в чистом виде трудно исследовать, не ис-
пользуя метод изотопического обмена, так как замена одного
элемента другим обязательно приводит к изменению и про-
чих факторов, обычно электронного характера. Замещения
в колеблющейся группе могут также изменить ее воспри-
имчивость к эффектам механической связи колебаний и при-
вести таким образом к большим изменениям частот колеба-
ний, чем можно было бы ожидать для взятого в отдельности
эффекта масс. Однако в некоторых случаях вероятные эф-
фекты простых изменений масс, не участвующих в связан-
ных колебаниях, могут быть предсказаны на основании
общей теории. Согласно этой теории, наименее чувствитель-
ными к изменению масс заместителей должны быть коле-
бания групп с участием атомов водорода или групп с крат-
ными связями, только одним концом присоединенных к ос-
тальной части молекулы. В случае валентных и деформа-
ционных колебаний групп X — Н все движение почти пол-
ностью локализуется на атоме водорода, а более тяжелый
атом X вообще почти не движется, так что в первом при-
ближении такие колебания являются не чувствительными
к изменению массы любых заместителей при атоме X.
В случае групп с кратными связями, таких, как карбониль-
ная группа, у которой в колебании участвуют атомы почти
равных масс, картина сложнее, но и подобные случаи рас-
смотрены в ряде теоретических работ. Случай с карбониль-
ной связью обсужден Холфордом [26], который показал,
что в молекулах типа СОХ2 масса атома X может меняться
от 12 до сколь угодно большой величины, вызывая при этом
изменения частоты колебаний карбонильной группы не
более чем на 25 см"1. Изменения же частоты, обусловлен-
ные изменением масс заместителей при атоме X, вообще
незначительны по сравнению с рассмотренным эффектом.
При массе атома X, несколько меньшей 12, появляются
смешанные колебания, которые рассматриваются ниже.
Однако в тех случаях, когда заместителями являются водо-
род и дейтерий, изменение частоты валентных колебаний
С — X выходит за пределы ожидаемого при наличии вза-
Гл. 23. Причины изменения частот колебаний групп атомов 543
имодействия колебаний, а поэтому разница частот колеба-
ний карбонильной группы формальдегида и формальдеги-
да-Н2 обусловлена, по-видимому, действительно эффектом
масс. Однако в практической работе, проявляя должную
осторожность в случае альдегидов, частоту карбонильных
колебаний можно считать не зависящей от масс замести-
телей. Рассмотрение Уиффеном [27] ряда соединений
X —С = N, а Лордом и Миллером [28] ацетиленовой
связи привело к аналогичному выводу.
Именно по этим причинам из всех корреляций, рассмот-
ренных в предыдущих главах, наиболее важными и полез-
ными являются те из них, которые относятся либо к коле-
баниям с участием атомов водорода в таких группах, как
ОН, СН2, NH2 и фенильное кольцо, либо к валентным коле-
баниям таких групп, как С = О, Р = О, NO2, SO2, и т. п.
Когда группа с кратной связью замещена с обеих сторон, за-
висимость частоты колебаний от масс заместителей заметно
больше, и хотя эта частота в какой-то мере стабилизиро-
вана значительной силовой постоянной, для поглощения, со-
ответствующего таким связям, как ^)С = С^ и ^>С = N—,
характерен обычно более широкий интервал частот.
В случае колебаний групп с простой связью, не включаю-
щих атомов водорода, зависимость частоты колебаний от
эффекта масс и механического взаимодействия повышает-
ся до такой степени, что удобные корреляции могут быть
получены только при рассмотрении достаточно больших
структурных элементов молекул, в которых эффект масс
практически постоянен. Например, для валентных колеба-
ний группы С — Ос простой связью характерен широкий
интервал частот, но в нем могут быть выделены частоты,
характерные для специфичных групп эфиров, первичных,
вторичных и третичных спиртов и т. д., т. е. для каждого
класса соединений необходимы свои корреляции. Анало-
гично и для группы CF3 характерны постоянные частоты
колебаний только в том случае, пока ее ближайшее окру-
жение остается постоянным. Например, когда группа при-
соединена к фенильному кольцу [29], то проявляются опре-
деленные характерные частоты колебаний; то же наблю-
дается и в случае, когда эта группа присоединена к группе
CF2 [30], но эти характерные частоты различны и в этих
544
Часть V
К&ух. случаях и не могут наблюдаться у каких-либо других
соединений.
Эффекты, обусловленные механическим взаимодействи-
ем колебаний различных групп, очень хорошо рассмотрены
в статье Лорда и Миллера [28]. Связь колебаний осуще-
ствляется в том случае, когда две колеблющиеся с доста-
точно высокими и практически равными частотами группы
находятся вблизи друг друга и при условии, что они обла-
дают одинаковой симметрией. Необходимостью выполнения
этих условий объясняется тот факт, что кратные связи отно-
сительно свободны от таких эффектов, поскольку наличие
у одного и того же атома более одной кратной связи яв-
ляется необычным. Если же это имеет место, как в случае
аллена или двуокиси углерода, то возникающее взаимодей-
ствие колебаний чрезвычайно велико и частоты антисим-
метричного и симметричного валентных колебаний отлича-
ются очень сильно. Характер изменения эффектов связи
колебаний хорошо иллюстрируется на примере рас-
смотренных выше молекул типа Х2СО. Когда X—водород
или дейтерий, частоты валентных колебаний С — X слиш-
ком велики, чтобы эти колебания могли взаимодействовать
с колебаниями карбонильной группы, имеющими частоту
около 1700 см'1, но когда масса X повышается, то и частота
соответствующих колебаний приближается к указанному
значению. В тех случаях, когда масса X близка.к 6, вза-
имодействие колебаний становится настолько сильным, что
ни связь С — X, ни связь С О не могут быть идентифи-
цированы отдельно и в спектре наблюдаются две далеко
отстоящие полосы поглощения. При дальнейшем возраста-
нии массы X до 12 частота колебаний С — X становится
достаточно малой по сравнению с частотой карбонильной
группы, чтобы устранить взаимодействие этих колебаний,
и последние опять можно идентифицировать.
Явление взаимодействия колебаний, характерных для
связен, включающих атомы водорода, очень удобно иссле-
довать, используя метод дейтерирования,' так как при этом
массы атомов сильно меняются, а химический характер
связи остается прежним. Если колебания по связи X — Н
и X — D не взаимодействуют с другими колебаниями, то
отношение их частот равно корню из двух. Поэтому если
дейтерирование приводит к такому сдвигу полосы погло-
Г л. 23. Причины изменения частот колебаний групп атомов 545
щения, что ее частота изменяется в указанном отношении,
то эта полоса может быть определенно идентифицирована,
как относящаяся к несвязанному колебанию ХН. Если же
отношение частот не равно]/ 2, то это показывает, что либо
колебание X — Н, либо колебание X — D взаимодейст-
вует с другим колебанием. Это явление может быть нагляд-
но продемонстрировано на примере частот колебаний моле-
кул HCN, DCN и TCN [77]. Наивысшими частотами у соот-
ветствующих молекул являются 3312, 2629 и 2460 сл"1.
Эти частоты соответствуют колебаниям, включающим глав-
ным образом валентные колебания С — Н (D или Т), но
поскольку отношение этих величин не равно]/2 и ]/3, то
очевидно, что имеет место взаимодействие колебаний. Ана-
логично в случае колебаний, связанных в основном с груп-
пой С = N, соответствующие частоты для указанного ряда
соединений равны 2096, 1928 и 1724 сл"1, и существенное
изменение значений по сравнению, например, со значением
2267 сл"1 для ацетонитрила также показывает, что у дей-
терированных и тритиированных соединений имеет место
значительное механическое взаимодействие колебаний.
При рассмотрении возможных причин аномальных зна-
чений частот колебаний групп атомов всегда следует иметь
в виду возможность взаимодействия колебаний, но это не
мешает установлению удобных в аналитической работе
корреляций для многих таких колебаний. Например, хоро-
шо известные полосы поглощения амид-П и амид-III явля-
ются высоко характеристическими для вторичных амидов
е открытой цепью, но обе они обязаны своим происхожде-
нием связанным колебаниям с участием деформационных
колебаний NH и валентных колебаний С — N [31 ]. При
постоянном окружении RCONHR степень взаимодействия
колебаний остается практически постоянной, а поэтому
частоты характеристичны. Аналогичным образом связаны
валентные колебания С — О и деформационные колеба-
ния ОН карбоновых кислот [17], но это не препятствует
получению полезных корреляций для частот колебаний
группы RCOOH при ее постоянном окружении. Однако
необходимо помнить, что такого рода корреляции относятся
не к индивидуальным колебаниям, сопоставляемым с от-
дельными связями, а поэтому изменения в характере еодо-
Зэ Л. Беллами
546
Часть V
родной связи или дейтерирование в случае кислот приве-
дет к изменению не только частот колебаний ОН, но также
и частот валентных колебаний С — О. По той же причине
дейтерирование вторичных амидов приводит к изменению
частот полос поглощения амид-II и амид-Ш, тогда как
у тиоамидов при этом изменяется частота только полосы
амид-П [32]. Последнее объясняется тем, что в случае тио-
амидов наблюдаются меньшие мезомерные эффекты и час-
тота колебаний С — N не достигает столь высокого зна-
чения, чтобы вступить во взаимодействие с деформацион-
ными колебаниями NH. Поэтому при дейтерировании этих
соединений полоса поглощения С — N остается неизменной,
а частота деформационных колебаний NH уменьшается
в ]'2 раз.
6. ЭФФЕКТЫ НАПРЯЖЕНИЯ
Геометрия расположения заместителей при атоме угле-
рода, находящегося в данном валентном состоянии, опре-
деляется гибридизацией его электронных орбит. Напри-
мер, насыщенный 5р3-атом углерода имеет тетраэдрическую
систему валентностей, а олефиновый 5р2-атом углерода —
плоскую тригональную конфигурацию. Любые изменения
в валентных углах, вызванные стерическими напряжения-
ми, приводят к некоторому перераспределению вкладов
s- и р-орбит в отдельные связи. s-Орбиты являются сфери-
ческими и, вообще говоря, меньше, чем р-орбиты, обладаю-
щие цилиндрической симметрией, так что изменения в их
соотношении при гибридизации приводят к изменениям
длин отдельных связей и вызывают изменения частот коле-
баний. Поскольку олефиновый атом углерода имеет только
одну s-орбиту и две р-орбиты, способные гибридизоваться,
то усиление s-характера одной связи должно компенсиро-
ваться усилением р-характера другой связи. Простым при-
мером, иллюстрирующим это положение, служит цикло-
пропан, у которого валентные углы углерода с естествен-
ной тетраэдрической направленностью связей уменьшены
вследствие напряжения цикла. Это означает, что несколько
увеличивается кратность связей С — С, а у связей С — Н
больше проявляется s-характер, вследствие чего они ста-
новятся более короткими и частоты их колебаний повыша-
ются до 3030 см1.
Гл. 23. Причины изменения частот колебаний групп атомов 547
Эффекты напряжения этого рода проявляются наиболее
отчетливо в случае колебаний, включающих двойные связи.
В настоящее время имеются многочисленные данные о пове-
дении связей \с = и /С = О при наличии различ-
ного типа напряжений; этот вопрос был уже подробно рас-
смотрен Лордом и Миллером [28]. Когда группа =
является частью шестичленной циклической системы, ва-
лентные углы, а следовательно, и частоты колебаний имеют
обычныезначения. Циклогексен [33] поглощает при 1646 см'1.
Это положение остается в силе также и в случае больших
циклических систем, у которых вледствие изгибания цикла
достигается такая конфигурация, при которой напряжение
уменьшается и валентные углы приближаются к их обыч-
ным значениям. Однако при уменьшении размеров цикла
напряжение возрастает и частота колебаний по связи
\с = С<^ уменьшается. Циклопентен поглощает при
1611 см'1, а циклобутен при 1566 см'1. По не вполне ясным
причинам присоединение к циклу второго цикла, по-види-
мому, увеличивает напряжение в системе, а поэтому часто-
та колебаний по связи С = С еще более уменьшается. Этот
эффект примерно равен эффекту уменьшения цикла на один
атом углерода. Так, например, частота полосы поглощения,
соответствующей колебаниям по двойной связи у системы
с конденсированными насыщенными шестичленными цик-
лами, бицикло-2,2,1-октена-2 [33], равна 1614 см'1, т. е.
близка к частоте полосы поглощения циклопентена. Анало-
гичные эффекты наблюдаются в случае бицикло-2,2,1-
гептена-2, но у соединений типа бицикло-2,2,1-гептен-5-
ил-2 возникает напряжение, оказывающее заметное влия-
ние на интенсивность полосы поглощения С = С и умень-
шающее ее до такой степени, что поглощение не может быть
обнаружено [38].
Подобные же эффекты напряжения обнаружены у более
сложных систем. Аналогичныезначения имеют частоты коле-
баний А2 стероидов и тритерпенов, у которых эффекты
напряжения одинаковы, но поглощение, обусловленное
колебаниями А9<11’-связей стеролов, лежит при более низ-
ких частотах [39] (1643—1648 см'1), чем соответствующее
35*
548
Ч а с т ь V
поглощение у тритерпенов (1660 сл-1), вследствие различий
в размерах соседних циклов. Влияние присоединения новых
циклов на напряжение олефиновых связей находит также
отражение в их реакционной способности, которая изме-
няется аналогичным образом [34].
Как и следовало ожидать, частота валентных колебаний
СН повышается при понижении частоты колебаний С = С
и у циклобутена [28] достигает значения 3060 сл-1 (в слу-
чае циклогексена 3017сл-1). Изменения частот внеплоскост-
ных деформационных колебаний СН изучены, однако, менее
детально. У стероидов и циклогексенов трудно выделить
какую-либо отдельную полосу поглощения, соответствую-
щую деформационному колебанию цис-СН = СН, но наблю-
дается ряд полос поглощения, характерных для данной
системы в целом [41]. У ряда циклогексенов эта картина
поглощения проще, чем у стероидов, и, по-видимому, при
увеличении напряжения вследствие конденсирования цик-
лов она смещается в область более высоких частот. Однако
в случае циклопентена и ряда бицикл о гептенов наблюдает-
ся обратный эффект, и поглощение обнаруживается при
более низких частотах, чем в случае самого циклогексена
[38]. Если имеются экзоциклические метиленовые группы
(С = СН2), то напряжение распределяется по различным
связям, и поэтому частота колебаний увеличивается при
уменьшении размеров циклов. Это является следствием ис-
кажения простых связей С — Си частичного приобретения
ими характера p-связей. По этой же причине в олефиновой
связи принимает большее участие s-орбита атома углерода
цикла, и длина связи уменьшается. Метиленциклобутан
[28] поглощает при 1678 см"1, а метиленциклогексан [35] —
при 1651 см г. Влияние сопряжения еще сказывается у
1,2-диметиленциклопентана [36], который поглощает при
меньшей частоте (1626 см 1), чем 1-метиленциклопентан
[351 (1657 см1), но как 1-метилен-, так и 1,2-диметиленцик.-
лобутаны поглощают при 1650 см1, на основании чего мож-
но предполагать, что две экзоциклические двойные связи
в этом случае не могут достичь полной копланарности.
Изменения карбонильного поглощения циклических
систем, по существу, аналогичны тем, которые наблюда-
ются для колебаний экзоциклических метиленовых групп;
детальные сведения о различных изменениях частот даны
Гл. 23. Причины изменения частот колебаний групп атомов 549
в соответствующих предыдущих главах. В случае циклов,
содержащих более шести членов, имеется возможность
некоторой внутренней перестройки, приводящей к уменьше-
нию напряжения в системах; они поглощают при обычных
частотах, частоты же колебаний у всех пятичленных цикли-
ческих систем, таких, как лактоны, лактамы и ангидриды,
выше, чем у соответствующих соединений с открытыми
цепями. У четырехчленных циклических систем этот эффект
еще больше, а у некоторых неорганических карбонильных
соединений, содержащих очень сильно напряженные мос-
тики, частоты карбонильного поглощения становятся
выше 1800 см~1. Холфорд [26] теоретически рассмотрел
изменения частот колебаний карбонильной группы
при изменениях угла между связями; результаты в общем
хорошо согласуются с этими экспериментальными дан-
ными.
Частоты колебаний карбонильной группы также чув-
ствительны к эффектам конденсирования циклов. В первых
исследованиях спектра пенициллина было отмечено, что
Р-лактамы, конденсированные с пятичленными цикличе-
скими системами, поглощают при более высоких частотах,
чем изолированные ^-лактамы. Аналогичные эффекты на-
блюдаются у циклических кетонов; Алленом и др. [38,
39] исследованы шести-, семи- и восьмичленные цикличе-
ские системы с карбонильными мостиками, и у всех этих
соединений наличие дополнительного напряжения находит
отражение в более высоких значениях частот колебаний кар-
бонильных групп.
Эффекты напряжения, возникающие вследствие стери-
ческих препятствий, изучены менее полно, но, по-видимо-
му, основные изменения при этом относятся скорее
к ширине полосы поглощения, а не к ее частоте. Орр [42]
исследовал mpawc-opmo-замещенные стильбены с замести-
телями, достаточно большими для того, чтобы могло про-
исходить искажение углов; и хотя частота деформацион-
ных колебаний СН оставалась неизменной, ширина полосы
возрастала в зависимости от степени напряжения. Анало-
гичные эффекты мы наблюдали для полос, соответствующих
валентным колебаниям NO2 некоторых орто-замещенных
нитросоединений. Однако в случае ароматических соеди-
нений значительные стерические затруднения могут при-
550
Часть V
вести к изменениям частоты и интенсивности полос вслед-
ствие разворота нитрогрупп и выхода их из плоскости
цикла, когда сопряжение отсутствует. В некоторых случаях
заниженные значения частот валентных колебаний ОН
у ненасыщенных спиртов объяснялись [43] эффектами сте-
рических затруднений, обусловленными наличием л-элек-
тронов, но возможно, что в этих случаях осуществляется
водородная связь с облаком л-электронов.
7. ЭЛЕКТРИЧЕСКИЕ ЭФФЕКТЫ
Название «электрические эффекты» является весьма
неопределенным, однако трудно найти иной термин, доста-
точно хорошо объединяющий и выражающий различные
индукционные, мезомерные эффекты и эффекты диполь-
ного взаимодействия, имеющие место в локальном окруже-
нии колеблющейся группы. Все эти эффекты изменяют,
по-существу, состояние гибридизации орбит атомов, на
которые они действуют, и с этой точки зрения простран-
ственные эффекты, рассмотренные выше, вероятно, могли
бы быть с таким же успехом включены в этот раздел. Раз-
граничение эффектов было проведено, однако, специально
для того, чтобы подчеркнуть строго локальную природу
эффектов, которые рассматриваются в этом разделе.
Индукционный и мезомерный эффекты и эффект поля —
это основные факторы, обусловливающие небольшие, но
очень важные различия, которые наблюдаются между
частотами колебаний карбонильных групп кетонов,
эфиров и хлоридов карбоновых кислот. Эти эффекты могут
быть успешно исследованы только в случае таких групп,
которые нечувствительны к эффектам масс или связи коле-
баний, и особенно важно, чтобы при любых сопоставле-
ниях между различными молекулами были выдержаны
аналогичные условия ассоциации и т. п. Например, в слу-
чае растворов в четыреххлористом углероде частоты карбо-
нильного поглощения кислот меньше, чем у соответствую-
щих кетонов, но это обусловлено тем, что кислоты в этих
условиях ассоциированы в виде димеров. Для паров в дей-
ствительности наблюдается обратное соотношение, и замена
группы OR на ОН также повышает частоту карбонильного
поглощения.
Гл. 23. Причины изменения частот колебаний групп атомов 551
В идеальном случае на основе количественных иссле-
дований этих эффектов в отдельности можно было бы пред-
сказывать направление и величины изменений частот дан-
ного колебания, соответствующих специфическим изме-
нениям заместителей. До такого положения еще очень дале-
ко, а на деле наше понимание роли этих эффектов в измене-
ниях частот колебаний в основном относится пока к области
предположений. Кроме того, эти эффекты иногда действуют
в противоположных направлениях; влияние одного из них
ведет к понижению частоты, а влияние другого — к ее по-
вышению, так что наблюдающиеся в итоге направление
и величина изменения частоты определяются суммарным
действием таких эффектов. Этот факт хорошо иллюстри-
руется на примере различного изменения частот валентных
колебаний ОН и NH2 при изменении их заместителей.
Так, в ряду СНзОН > СвН5ОН > СН3СООН частота
колебаний ОН постепенно уменьшается, тогда как частоты
валентных колебаний NH2 у аналогичного ряда аминов
повышаются: CH3NH2 < C0H5NH2 < CH3CONH2. В обоих
случаях возрастающий индукционный эффект, который
сопровождает возрастающую степень ненасыщенности замес-
тителей в этих рядах, должен был бы приводить к увели-
чению полярности связей X — Н и уменьшению частот
валентных колебаний. Однако ненасыщенность влияет
также и на восприимчивость групп ОН и NH2 к мезомерному
эффекту. Как атом кислорода, так и атом азота имеют непо-
деленныепары электронов, обладающие известной степенью
подвижности. Поэтому в случае кислот и амидов можно
ожидать, что будут осуществляться такие канонические
•формы, как
-О
1
СН3—с = он+
и
-о
I
СН3—C=NHJ
У этих структур частоты валентных колебаний X—Н вы-
ше, точно так же как в случае соответствующих колебаний
•СН2 винильных групп частоты выше, чем у групп СН2
предельных соединений. Поэтому, казалось бы, что боль-
552
Чисть V
шая подвижность неподеленной пары электронов атома
азота должна приводить к существенной роли мезомерного
эффекта у амидов, влияние которого превосходит влияние
индукционного эффекта и приводит к более высоким зна-
чениям частот валентных колебаний NH2, чем в случае
аминов. Однако у кислот подвижность неподеленной пары
электронов атома кислорода значительно меньше, а поэтому
превалирует индукционный эффект и частота колебаний
ОН меньше, чем у спиртов. Совершенно аналогичное явле-
ние наблюдается для кислот и амидов у карбонильных час-
тот, которые имеют в первом случае большее, а во втором—
меньшее значение, чем в случае кетонов. Это явление было
объяснено Томпсоном и др. [44] подобным же образом.
а. Индукционные эффекты. Теперь в общем установ-
лено, что почти все ковалентные связи имеют в некоторой
мере полярный характер, и только в исключительных слу-
чаях, например у молекулы азота или центральной связи
С — С этана, наблюдаются чисто ковалентные связи.
Колебания по этим связям в инфракрасном спектре неактив-
ны, а поэтому практически мы всегда имеем дело с коле-
баниями по связям, которые являются в какой-то мере
полярными. Если изменяется электроотрицательность
заместителя, участвующего в связи, по которой происходит
колебание, то полярность связи, а следовательно, и частота
колебаний изменяются. Это явление обусловлено индук-
ционным эффектом; он действует только вдоль связи, не
зависит от геометрии молекулы и прежде всего определяется
электроотрицательностями замещающих атомов или эффек-
тивными электроотрицательностями замещающих групп.
Простейшая схема действия индукционного эффекта
может быть прослежена при рассмотрении изменений час-
тоты карбонильной полосы, которые наблюдаются при
переходе от ацетона к ацетилхлориду. У ацетона карбо-
нильная связь имеет в некоторой степени полярный харак-
тер и атом кислорода несет какой-то отрицательный заряд.
Это означает, что электронное облако, образующее связь,
смещено относительно геометрического центра связи
в направлении атома кислорода. Если теперь одну из
метильных групп заменить значительно более электроотри-
цательным заместителем, например хлором, то возросшая
Гл. 23. Причины изменения частот колебаний групп атомов 55.3
сила притяжения электрона сместит электронное облако
назад, несколько ближе к геометрическому центру связи.
Следовательно, полярность карбонильной связи умень-
шается и частота колебаний возрастает.
Рис. 31.
Поэтому можно ожидать, что изменения частоты, свя-
занные с индукционными эффектами, могут быть сопостав-
лены главным образом с электроотрицательностями замес-
тителей, и в тех случаях, когда проявляется только индук-
ционный эффект, это сопоставление может быть проведено
количественно.
Такая возможность может быть проверена на примере
простых молекул ХН„ при прямом сравнении частот валент-
ных колебаний X — Н (исправленных, как полагается, на
незначительные эффекты масс) и значений электроотрица-
тельностей элементов X. На рис. 31 показан результат
такого сравнения. Точки, соответствующие значениям,
полученным для одновалетных элементов, лежат на прямой
линии, отражающей простую линейную зависимость частот
554
Часть V
от значений электроотрицательностей по Полингу, а точки,
соответствующие значениям для поливалентных элементов,
лежат на подобных прямых, смещенных на ту или иную
величину, в зависимости от валентного состояния элемента.
Это показывает, что действительно имеется тесная коли-
чественная связь между частотами колебаний и электро-
отрицательностями, но имеются также некоторые допол-
нительные факторы, которые играют роль в случае эле-
ментов с валентностью больше единицы. Последнее не
совсем понятно, но следует помнить, что сами колебания
X — Н в этих случаях являются не простыми; они дают
частоты антисимметричных и симметричных колебаний,
и, возможно, поэтому необходимо учитывать также эффекты
изменения углов между связями.
Более простой случай представляет собой изменение
частот валентных колебаний ОН карбоновых кислот
RCOOH. Давно отмечено, что относительная легкость иони-
зации атома водорода, характеризуемая для кислоты вели-
чиной pf(a, является мерой индукционного эффекта груп-
пы R. У кислот жирного ряда, у которых проявляются
только индукционные силы, должно быть, таким образом,
прямое соотношение между частотами валентных колеба-
ний ОН мономеров и значениями рК.а-' Гульден [45] пока-
зал, что простая линейная зависимость такого типа дей-
ствительно существует. У ароматических кислот проявля-
ются как индукционные, так и мезомерные эффекты, и зна-
чения рКа с частотой колебаний ОН связывает другое
линейное соотношение; различие в наклоне обусловлено
действием дополнительного эффекта.
Соотношение между электроотрицательностью и часто-
тами колебаний групп изучено также у виниловых соеди-
нений типа СН2 — CXY, для которых частоты деформа-
ционных колебаний СН2 рассматривались в зависимости
от сумм электроотрицательностей заместителей [46]. В слу-
чае таких заместителей, как алкильные группы или атомы
галогенов, для которых по химическим соображениям
нельзя ожидать заметных мезомерных эффектов, была
найдена линейная зависимость. Для таких заместителей,
как атомы кислорода или азота, у которых, вероятно,
заметен мезомерный эффект, такое соотношение, как и сле-
довало ожидать, не выполняется. Чтобы проявлялись мезо-
Гл. 23. Причины изменения частот колебаний групп атомов 555
мерные эффекты, требуется выполнение условия копланар-
ности рассматриваемых связей, так как иначе не могут
образоваться подходящие молекулярные орбиты. У неплос-
ких структур, таких, как пирамидальные фосфаты и сульф-
оксиды, возможность проявления этого эффекта соответ-
ственно сильно уменьшена, и этим объясняется относи-
тельная независимость частот колебаний групп Р = О
и S = О от эффектов сопряжения. Поэтому индукцион-
ный эффект является основным фактором, обусловливаю-
щим изменения этих частот при замене заместителей. Бел-
лом и др. (47] показано, что у галогенидов фосфорила
частота колебаний Р = О является линейной функцией
суммы электроотрицательностей атомов галогенов. Им уда-
лось также для групп атомов других заместителей полу-
чить значения электроотрицательностей, которые, по-види-
мому, могут успешно применяться для этого ряда соедине-
ний, хотя для заместителей ОН и NH2 имеются некоторые
неточности, обусловленные, по-видимому, наложением
эффектов водородной связи. Кейгарайз [48] провел анало-
гичную работу с частотами карбонильного поглощения
и получил другие значения «электроотрицательностей
групп». Интересно отметить, что эти значения явно отли-
чаются от значений, полученных Беллом и др., по-види-
мому, вследствие того, что плоская карбонильная струк-
тура в большей степени допускает мезомерные эффекты.
В случае таких заместителей, как, например, группа OR
у эфиров, при получении значений «электроотрицатель-
ностей групп» на основании сопоставления частот карбо-
нильного поглощения должны учитываться одновременно
и индукционный и мезомерный эффекты. В то же время
у соответствующих триалкилфосфатов мезомерный эффект
сильно уменьшен, поскольку все связи Р — О не могут
лежать в одной плоскости с фосфорильной группой. Это
замечание показывает, что при использовании значений
электроотрицательностей групп, полученных таким путем,
следует соблюдать осторожность. Во всяком случае, едва
ли при этом можно получить что-либо большее, чем весьма
приблизительные соотношения, так как сами значения
электроотрицательностей элементов далеки от точных вели-
чин и меняются при изменении рассматриваемого состоя-
ния гибридизации [49].
556
Часть V
Количественный подход к исследованию индукционных
эффектов, не зависящих от величин электроотрицатель-
ностей, предложен Беллами и Уильямсом [1] и развит на
основе их исследований природы эффектов поля. Проведение
этих исследований было связано с предшествующей рабо-
той Смита, Ри, Мэйджи и Эйринга [51] по количественным
соотношениям между индукционными силами и диполь-
ными моментами связей. Этими авторами разработан полу-
классический метод, который позволяет вычислять оста-
точные суммарные заряды отдельных атомов небольших
молекул в предположении, что вдоль связей действуют толь-
ко индукционные силы. Значения, полученные таким обра-
зом, могут быть выражены в величинах дипольных момен-
тов отдельных связей, и показано, что у ряда метилгалоге-
нидов при векторном сложении их моментов связей полу-
чаются значения дипольных моментов молекул, которые
находятся в блестящем согласии с экспериментальными дан-
ными. Этот метод развит Беллами и Уильямсом для вычис-
ления дипольных моментов карбонильных связей таких
соединений, у которых мезомерные эффекты малы или совсем
отсутствуют. Они также получили значения дипольных
моментов молекул, хорошо согласующиеся с эксперимен-
тальными результатами, и продолжили это исследование
с целью показать, что полученные таким образом значе-
ния дипольных моментов карбонильной связи находятся
в простой линейной зависимости от наблюдающихся частот
колебаний. Полученные результаты иллюстрируются ниж-
ней прямой на рис. 32. Верхняя прямая относится к спе-
циальному случаю соединений CF3СОХ, у которых эффек-
ты поля превалируют над индукционными эффектами,
и будет обсуждаться ниже в соответствующем разделе.
Поэтому представляется вполне надежно установлен-
ным, что в тех случаях, когда действуют только индук-
ционные эффекты, изменения в частотах колебаний групп
атомов являются мерой силы действия этих эффектов. Дей-
ствительно, колебательные частоты, по-видимому, значи-
тельно более чувствительны к этим эффектам, чем любые
другие возможные параметры, но необходимо соблюдать
очень большую осторожность в использовании этого поло-
жения, так как наши знания об условиях, в которых прояв-
ляются только индукционные эффекты, являются еще
Гл. 23. Причины изменения частот колебаний групп атомов 557
очень неполными. Например, невозможно однозначно
объяснить данные наблюдений, показывающих, что частота
колебаний С = С бром- и иодацетиленов примерно на
100 см"1 меньше [28], чем у алкил- или хлорзамещенных
соединений. В этих случаях играют, по-видимому, неко-
торую роль геометрические размеры заместителя, что,
вероятно, имеет место также у некоторых 1,2-галогидри-
нов, исследованных Никсоном [52]. Можно было бы пред-
положить, что сила образующейся водородной связи между
Р и с. 32. Воспроизводится по J. Chem. Soc., 1957,
4294.
соседними атомом галогена и гидроксильной группой
является функцией электроотрицательностей атомов гало-
генов, так как расстояния между центрами ядер атомов
кислорода и галогена остаются одинаковыми. В действи-
тельности верно обратное, и наиболее сильные связи обра-
зуются иодом, обладающим наименьшей электроотрица-
тельностью в ряду галогенов. Сказанное позволяет пред-
положить, что в этом случае более важными оказываются
величины атомных объемов и что именно расстоянием
между протоном и внешними валентными электронами
атома галогена определяется сила образующейся связи.
б. Сопряжение и мезомерия. Электроны, образующие
л-облака кратных связей, могут в какой-то мере поляризо-
ваться, т. е. обладают некоторой степенью подвижности.
У такого соединения, как бутадиен-1,3, у которого все
атомы углерода лежат в одной плоскости, это приводит
558
Часть V
к образованию молекулярных орбит, которые охватывают
все атомы углерода, в результате чего центральная оди-
нарная связь С — С приобретает в некоторой мере харак-
тер двойной связи, тогда как кратные связи становятся
несколько слабее. Система, как целое, выигрывает за счет
энергии резонанса и в результате имеет более высокую
стабильность. Это и есть простое сопряжение, причем
легко видеть, что небольшое увеличение длины двойных
связей, которое при этом происходит, должно привести
к уменьшению частот их колебаний. В предыдущих главах
это иллюстрировалось множеством примеров, и нет необ-
ходимости их продолжать.
Сходные эффекты наблюдаются также, когда к кратной
связи присоединен атом элемента, имеющий легко поляри-
зуемые электроны в виде неподеленных пар. У амидов, эфи-
ров и хлоридов карбоновых кислот частота колебаний
карбонильной группы зависит не только от электроотрица-
тельностей заместителей, но также от вклада таких кано-
нических форм, как
о- о- о-
I + I + I +
R —c = nr2, r —c = or, R—С=С1 >
которые во всех случаях стремятся уменьшить частоту
колебаний в результате увеличения длины связи С — О.
Это так называемый мезомерный эффект, и мера его дей-
ствия зависит от наличия и способности неподеленной
пары электронов к образованию кратной связи, что-
определяется поляризуемостями элементов, рассмат-
риваемых в конкретном окружении. Обычно при нали-
чии заместителей — галогенов вклад канонических форм
указанного типа невелик, а поэтому индукционный эффект
превалирует над мезомерным эффектом и частота колеба-
ний карбонильной группы высока. В случае легче поляри-
зуемого атома кислорода этот эффект больше, и у атомов
азота он наиболее заметен. В последнем случае связь
С — N сильно укорочена, и частота колебаний карбониль-
ной группы меньше, чем у кетонов, несмотря на более высо-
кую электроотрицательность атома азота. В особых слу-
чаях (винилацетат) обнаружено отсутствие у системы энер-
гии резонанса, что практически означает отсутствие мезомер-
Гл. 23. Причины изменения частот колебаний групп атомов 559
ного эффекта и отсутствие какого-либо вклада таких резо-
О
нансных форм, как СН3 — С = О+ — СН = СН2. Это
может быть обусловлено тем, что как карбонильная, так
и винильная группы стараются оттянуть к себе неподелен-
ную пару электронов атома кислорода. Если эти эффекты
уравновешиваются, то резонанс отсутствует, а частота
колебаний карбонильной группы при этом определяется
только значением эффективной электроотрицательности
группы OR. Поэтому важно отметить, что частоты колеба-
ний карбонильной группы у виниловых эфиров значитель-
но выше, чем у эфиров жирного ряда, и приближаются
к значениям для хлоридов карбоновых кислот. Таким обра-
зом, мезомерные эффекты следует предполагать у таких
систем, у которых атом с имеющимися у него поляризуе-
мыми электронами присоединен непосредственно к группе
с кратной связью, и все три атома лежат в одной плоскости.
Так, у сф-а'|3'-ненасыщенных карбонильных соединений
обе двойные связи играют роль в понижении частоты коле-
баний карбонильной группы, но в случае неплоских систем,
таких, как диаллилсульфоксид, частота колебаний S = О
не изменяется. Аналогично у ацетофенонов с большими
заместителями в орто-положении карбонильная группа
выходит из плоскости цикла и частота колебаний группы
повышается.
Мезомерные эффекты не могут быть изолированы от
индукционных эффектов тех же заместителей, а поэтому
всегда необходимо рассматривать их комбинированное
действие. Влияние результирующего эффекта имеет очень
большое значение не только в отношении изменения частот
поглощения в инфракрасной области спектра, но также
и вследствие того, что от него зависят многие другие важ-
ные физические свойства. Изменения степени кислотности
и основности, величин дипольных моментов, длин связей
и в соответствующих случаях скоростей реакций — все-
это определяется прежде всего результирующим эффектом,
обусловленным действием индукционного (/) и мезомер»
ного (/И) эффектов. Предпринято много различных попыток
количественного исследования этих эффектов, но сколько»
нибудь точное их исследование возможно только в отдел ь-
560
Часть V
ных случаях. Вполне возможно, что в некоторых случаях
смещения полос поглощения в инфракрасной области
спектра могут служить мерой действия этих эффектов, но
при этом необходимо прежде всего доказать, что колеба-
ния, использованные для этой цели, действительно коли-
чественно зависят только от /- и М-эффектов и не зависят
ни от каких иных факторов.
Доказательства, что колебания, частоты которых под-
ходят для этой цели, действительно имеются, даны в много-
численных исследованиях, в которых показана прямая
связь изменений частот колебаний групп атомов с другими
физическими свойствами, зависящими от /- и М-эффектов.
О связи частот с кислотностью соединений уже говорилось
[45]; аналогичные данные имеются относительно связи
частот валентных колебаний NH первичных аминов с их
основностью [53, 54]. Частоты колебаний карбонильной
группы были сопоставлены с полярографическими потен-
циалами полуволны [55], окислительно-восстановитель-
ными потенциалами [56], длинами связей [57] и констан-
тами нестойкости внутрикомплексных соединений [58];
были установлены также соотношения между изменениями
частот колебаний карбонильной группы, а также частот коле-
баний СН2 и изменениями рефракции, которые являются
важной характеристикой /- и ТИ-эффектов [46]. Однако
наиболее важным физическим свойством, которое связано
с этими эффектами, является реакционная способность;
были предприняты многочисленные исследования с целью
получения соотношений между изменениями частот колеба-
ний групп и изменениями реакционной способности.
В этом вопросе особенно важно четко разграничивать
типы реакций, скорости которых определяются исключи-
тельно /- и М-эффектами и которые можно, по-видимому,
как-то связать со сдвигами частот, и реакций, скорости
которых зависят от других факторов. Индукционный и мезо-
мерный эффекты можно рассматривать как статические
факторы, которыми определяется реакционная способность,
а у некоторых веществ только ими и определяются ско-
рости реакций. В некоторых других случаях действуют
также динамические факторы. Например, распределение
электронной плотности в молекуле может быть сильно
возмущено приближением воздействующего реагента, что
Г л. 23. Причины изменения частот колебаний групп атомов 561
вносит новый элемент, который не может быть выявлен
спектроскопическим исследованием исходной молекулы в ее
основном состоянии. Эти динамические факторы были
названы электромерными эффектами и очень подробно
обсуждены Ингольдом [59, 60]. Таким образом, наличия
каких-либо соотношений между реакционной способ-
ностью и частотой колебаний групп атомов можно ожидать
только в случае таких реакций, когда динамические эффекты
рассмотренного типа не играют сколько-нибудь заметной
роли. Такие реакции обычно имеют место у ароматического
ряда соединений, особенно в случае участия в них замес-
тителей в циклах. Очень широко исследовал реакции
этого типа Гаммет [61 ]; его работа была продолжена Джаф-
фе [62] и Тафтом [63, 64]. Гаммету удалось показать, что
каждая отдельная группа, находящаяся в данном положе-
нии в ароматическом цикле, дает один и тот же эффект,
влияющий на реакционную способность соединения, который
зависит от индукционных и мезомерных свойств этой группы.
Величина этого эффекта может быть выражена в виде посто-
янной сг, значение которой служит мерой способности замес-
тителя притягивать или отдавать электроны. Постоянные а
применимы в широкой области реакций различных типов;
они являются величинами аддитивными, а поэтому реак-
ционная способность дважды замещенного соединения может
быть предсказана, если известна сумма значений <т для
замещающих групп.
Величина постоянной Гаммета <т является удобной
мерой /- и 7И-эффектов индивидуальных групп в аромати-
ческих системах; были предприняты многочисленные иссле-
дования с целью установления возможных соотношений
между этими величинами и изменениями частот колебаний
групп. Линейные зависимости значений <т и сдвигов частот
колебаний установлены для валентных колебаний NH2
анилинов [53, 65], валентных колебаний ОН фенолов [66],
колебаний карбонильной группы ацетофенонов и подобных
им соединений [55, 68—70], а также внеплоскостных
деформационных колебаний СН у ароматических циклов
[71]. Сходимость получающихся результатов хорошо иллю-
стрируется последним примером, данные для которого
воспроизведены на рис. 33. Рассматриваемым колебаниям
соответствуют хорошо изученные интенсивные полосы
36 Л. Беллами
562
Ч а с т ь И
поглощения ароматических соединений, положение кото-
рых характеризует тип замещения и определяется в свою
очередь числом смежных свободных (незамещенных) ато-
мов водорода в цикле. Известно, что эти частоты нечув-
ствительны к эффекту масс, и нетрудно видеть, что, за
исключением заместителей галогенов, наблюдается очень
хорошая сходимость результатов. Все галогены в каче-
стве заместителей ведут себя так, как если бы значения о
были у них равны нулю, и все точки для них отложены на
графике в соответствии с этим. Указанное выше согласует-
ся с данными измерений ядерного магнитного резонанса,
полученными Корио и Дэйли [72], которые обнаружили,
что атомы хлора и брома в качестве заместителей не изме-
няют электронную плотность у ароматических групп СН,
из чего авторы заключили, что в реакционной способности
таких соединений важную роль играют динамические
электромерные эффекты. Закономерность, иллюстрируемая
рис. 31, также была использована для получения значений о
непосредственно из величин наблюдающихся частот коле-
баний; например, значение о для mpem-бутильной группы
получено из соответствующих частот поглощения в спектре
1,3,5-три-трет-бутилбензола. Полученные таким образом
значения могут быть затем использованы для предсказа-
ния частот поглощения других соединений, содержащих
эту группу, и совпадение с экспериментальными данными
бывает обычно порядка нескольких сантиметров в минус
первой степени.
Леба и Жозьен [73] провели весьма сходные исследова-
ния частот валентных колебаний СН и обнаружили, что,
за исключением, как и прежде, заместителей — галогенов,
наблюдающаяся частота колебаний может быть прямо
коррелирована с выраженным в процентах выходом мета-
нитросоединений, получающихся при нитровании моно-
замещенного соединения.
При рассмотрении общих проблем использования этих
данных для оценки специфических свойств соединений
необходимо обращать внимание на вопрос применимости
для этой цели выбранной частоты колебаний группы. В иде-
альном случае она должна быть особенно чувствительна
к 7- и М-эффектам и нечувствительна ко всем другим эффек-
там. В некоторых из рассмотренных выше примеров весь
Гл. 23. Причины изменения частот колебаний групп атсмов 563
интервал сдвигов частот сравнительно невелик, а поэтому
их нельзя рассматривать в качестве достаточно точной меры
изменений I- и М-эффектов. У других веществ, таких, как
Рис. 33. Колебания S-CH:
ф 3 атома водорода, расположенных в цикле рядом;
А 3 атома водорода, расположенных в цикле (гало-
гепзамещенные) рядом; Q 2 атома водорода, располо-
женных в цикле рядом; х изолиоованный атом водорода
в цикле; Д изолированный атом водорода в цикле (гало-
геиза-мещеиные). Воспроизводится по J.Chem. Soc.,
1955, 2818.
ряд метил галогенидов, изменения частот валентных ноле-
баний СН v4 при переходе от метилфторида к метилиодиду
составляют около 50 см'1. Если они происходят в высоко-
частотной области спектра, их можно измерять с большой
точностью; поэтому они служат чувствительной относи-
тельной мерой индукционных эффектов атомов галогенов.
36*
564
Часть V
Весь интервал изменений длин связей СН у соединений
этого ряда составляет всего 0,001 Л; это само по себе
указывает на чувствительность частот к электрическим
эффектам.
Проведенное выше обсуждение ограничивалось рас-
смотрением возможности использования сдвигов частот
колебаний групп для оценки индукционного и мезомер-
ного эффектов и физических свойств, которые также свя-
заны с ними. Некоторые предварительные исследования
по сходным вопросам были выполнены также значительно
более сложным методом измерения интенсивностей полос.
В некоторых случаях изменения интенсивности, которыми
сопровождаются небольшие изменения в распределении
электронной плотности в колеблющейся группе, несравнен-
нобольше, чем соответствующие изменения частот. Посколь-
ку изменения интенсивности зависят от тех же факторов,
то измерение этого параметра представляет собой альтер-
нативный и часто более предпочтительный метод исследо-
вания. В некоторых случаях оба метода дают, по-видимому,
сходные результаты. Значения постоянных Гаммета о были
сопоставлены, например, с интенсивностями полос погло-
щения, соответствующих валентным колебаниям NH2 [75]
и ОН [76]. У других веществ изменение интенсивности
поглощения следует иному закону, чем изменение частот,
и между ними нет никакой связи. Так, интенсивность кар-
бонильного поглощения у замещенных ацетофенонов при
изменении заместителей меняется лишь весьма незначи-
тельно, и эти изменения не могут быть сопоставлены со
значениями постоянных Гаммета о [68]. Аналогичным обра-
зом Барроу [78] нашел, что для достаточно большого ряда
различных соединений, содержащих карбонильную группу,
изменение интенсивности поглощения не сопоставимо со
сдвигами частот, но является функцией энергии резонанса.
Последняя, по-видимому, никак не связана с индукцион-
ными эффектами, и возможно, что изменения интенсив-
ности карбонильного поглощения зависят в первую оче-
редь от мезомерных эффектов. Ясно, что имеется обширное
поле деятельности для дальнейшей работы в этой области,
и результаты, получаемые при совместном изучении интен-
сивностей и частот поглощения, вероятно, являются глав-
ным источником надежды на успех в этой работе.
Гл. 23 Причины изменения частот колебаний групп атомов 565
в. Эффекты поля диполей. До недавнего времени счи-
талось, что индукционный и мезомерный эффекты являются,
по-видимому, единственными факторами электрического
характера, влияющими на частоты колебаний групп
в ненапряженных системах, но более детальное исследова-
ние некоторых аномальных значений частот колебаний при-
вело к необходимости рассмотрения третьего эффекта.
Джонс и др. [79, 80] отметили, что у а-галогензамещенных
кетостероидов экваториальное замещение приводит к повы-
шению частоты колебаний карбонильной группы примерно
на 25 сл1-1, аксиальное же замещение не дает подобных
эффектов. Аналогичные случаи отмечены Пейджем [811;
Корей и др. обнаружили этот эффекту а-галогензамещенных
циклогексанонов [82—85]. У этих соединений можно ожи-
дать очень малые мезомерные эффекты, а индукционный
эффект не зависит от геометрии молекулы, так что полу-
ченные результаты не могут быть адекватно описаны влия-
нием /- и М-эффектов. Положение еще более осложняется,
если учесть, что введение второго атома галогена при атоме
углерода в a-положение влияет очень мало или вообще не
влияет на частоту колебаний карбонильной группы, но
когда этот атом вводится в а'-положение, то наблюдается
дальнейшее повышение частоты колебаний. Эти данные
привели Джонса и др. [68, 86] и Беллами и Уильямса
[1, 50, 87, 88] к предположению, что в подобных случаях
могут иметь место эффекты поля и что частота колебаний
подвержена влиянию электрических полей диполей, дей-
ствующих в межмолекулярном пространстве, а не вдоль
связей, как это обычно принято считать.
Эта концепция разработана Беллами и Уильямсом [1]
с точки зрения простой электростатической теории. Они
предполагают, что близость отрицательно заряженного атома
галогена к легко поляризуемому отрицательно заряжен-
ному атому кислорода карбонильной группы вызывает
взаимное индуцирование противоположных зарядов. В ре-
зультате этого отрицательные заряды атомов как хлора,
так и кислорода уменьшаются и связи С — С1 и С = О
становятся менее полярными. Частоты колебаний по этим
связям, таким образом, возрастают,что подтверждается
и опытом. Этот эффект, по-существу, аналогичен обсуж-
денным выше электростатическим эффектам, наблюдаю-
566
Часть V
щимся в случае жидкого ацетона, у которого сами диполи
С = О ориентируются преимущественно таким образом,
что разноименные заряды оказываются рядом. Однако
в более жестком внутримолекулярном окружении не всегда
возможно, чтобы сами диполи связей могли так ориенти-
роваться, и можно ожидать, что сдвиги частот колебаний
в том или ином направлении осуществляются в зависимости
от того, между какими зарядами происходят взаимодей-
ствия — между одноименными или разноименными.
Эта гипотеза отчасти базируется на результатах экспе-
риментальных исследований а-галогензамещенных соеди-
нений, содержащих карбонильную группу и, частично, на
результатах приближенных вычислений, которые показы-
вают, что порядок наблюдающихся сдвигов частот колеба-
ний имеет такую величину, какую можно было бы ожидать
в случае, если бы эти сдвиги были обусловлены подобными
электростатическими взаимодействиями. Быть может, наи-
более важной чертой этой гипотезы является то, что она
представляет несложную единообразную схему, которая
дает возможность объяснить и ряд других аномальных зна-
чений частот колебаний.
Экспериментальные исследования эффектов поля сосре-
доточены в первую очередь на а-галогензамещенных соеди-
нениях, содержащих карбонильную группу; кроме стери-
нов и циклогексанонов, исследовано также значительное
количество кетонов с открытой цепью и родственных им
соединений. Эти соединения в жидком состоянии способны
давать эффект поворотной изомерии, а поэтому такое соеди-
нение, как хлорацетон, существует в виде смеси структур
I и II:
i п
Вследствие этого в случае жидкости или раствора прояв-
ляются две частоты колебаний карбонильной группы, отно-
сящиеся к отдельным изомерам, и, прослеживая измене-
Гл. 23. Причины изменения частот колебаний групп атомов 567
ния интенсивностей соответствующих полос поглощения
при изменениях температуры и полярности растворителей,
можно установить, какая частота соответствует более
полярной структуре. В случае некоторых веществ, напри-
мер самого хлорацетона, полученные результаты подтверж-
дены измерениями дипольных моментов [89]. У всех иссле-
дованных соединений, у которых возможно существование
различных поворотных изомеров такого типа, более высо-
кая частота колебаний карбонильной группы была при-
писана более полярной структуре, подобной I [1, 50],
тогда как меньшая частота, соответствующая таким фор-
мам, как II, обычно близка по величине к частоте, харак-
терной для исходных кетонов, что находится в согласии
с предложенным механизмом. Эта теория дает также удов-
летворительное объяснение того факта, что наличие второго
атома галогена в а-положении не приводит к дальнейшему
повышению частоты колебаний, так как в цнс-положении
относительно атома кислорода карбонильной группы может
находиться не более одного из двух атомов галогена
в а-положении. Однако когда атомы галогена находятся
в цепи с обеих сторон от карбонильной группы, то каждый
из них может давать свои эффекты поля. Это подтверждено
на примере 1,3-дихлорацетона, молекулы которого могут
находиться в изомерных формах III, IV и V, приведенных
ниже. У этого соединения обнаружены в жидком состоя-
нии три частоты колебаний карбонильной группы, которые
III vco 1755 cjm-1
V V(2Q 17 28 CJW-1
могут быть скоррелированы при исследованиях температур-
ной зависимости интенсивности полос поглощения.
Аналогичные результаты получены в наших лаборато-
риях для а-галогензамещенных альдегидов и хлоридов
кислот; эффект поля также может сказываться во взаимном
влиянии атома карбонильного кислорода ацетофенона
и атома хлора или нитрогруппы в орто-положении [50, 68).
568
Часть V
Имеющиеся в литературе данные показывают, что эти
эффекты имеют место также у кислот, эфиров и амидов.
В случае эфиров это подтверждается данными, которые
получили Жозьен и Калас [90] при исследовании раство-
ров метилацетата и его хлорпроизводных в четыреххлори-
стом углероде. В этих условиях метилацетат поглощает
при 1750 слг1. Такую же полосу поглощения имеют как
моно-, так и дихлорпроизводные. Эта частота соответ-
ствует поворотным изомерам, у которых атомы хлора уда-
лены от атома карбонильного кислорода. Однако у обоих
производных обнаружено также дополнительное поглоще-
ние при 1775 сл-1, обусловленное теми изомерами, у кото-
рых имеет место эффект поля. В случае метилтрихлораце-
тата, как и следовало ожидать, наблюдается только одна
полоса поглощения при 1770 слТ1.
Интересно, что галогензамещенные амиды ведут себя
несколько аномально по сравнению с соответствующими
кетонами; у них введение атома галогена в а-положении
обычно не приводит к сколько-нибудь заметному повыше-
нию частоты до тех пор, пока при том же атоме углерода
не появляется второй атом галогена, который и вызывает
ожидаемое повышение частоты. Данные по такого рода
соединениям приведены Летау и Трупом [91] и обсуждены
Беллами и Уильямсом [1]. Последние установили, что
такие свойства не являются неожиданными, так как в соот-
ветствии с простыми электростатическими представлениями
действие обычного мезомерного эффекта у амидов приводит
к тому, что у атома азота появляется некоторый формаль-
ный положительный заряд. Электростатическое отталкива-
ние между отрицательно заряженными атомами галогена
и кислорода усилено, таким образом, электростатическим
притяжением между атомами хлора и азота. По этой при-
чине следует ожидать значительного увеличения относи-
тельной устойчивости аош-формы VI. Это предположение
О
Н ||
I /9
cz nr2
н/ \ci
VI
Гл. 23. Причины изменения частот колебаний групп атомов 569
полностью подтверждено результатами недавних измерений
дипольного момента метилхлорацетамида, которые показы-
вают, что в неполярных растворителях присутствует почти
исключительно эта форма [99].
Развита также количественная сторона теории эффектов
поля. В предыдущем разделе был рассмотрен метод вычис-
ления дипольных моментов карбонильной группы, и ниж-
ний график на рис. 32 иллюстрирует полученное соотно-
шение между частотами ее колебаний и дипольными момен-
тами связи для соединений, у которых действуют только
индукционные эффекты вдоль связей. Если провести анало-
гичное рассмотрение для трифторметилкарбонильных соеди-
нений, то получается график, приведенный на верхней
части рис. 32, причем смещение прямой от исходного поло-
жения связано с наложением эффектов поля, которые не
были приняты во внимание при этих вычислениях. Это сме-
щение, составляющее примерно 0,2 дебая, является, таким
образом, мерой изменения дипольного момента связи
С = О под влиянием эффекта поля группы CF3. Имеется
возможность провести отдельный расчет вероятной вели-
чины этого эффекта поля, рассматривая атомы фтора и кис-
лорода как точечные заряды, используя значения диполь-
ных моментов, вычисленные отдельно для связей С — F
и С = О, и выбирая разумные значения для продольной
поляризуемости связи. Такой расчет дает величину
0,19 дебая для данного рассматриваемого ряда соединений.
Столь прекрасное совпадение результатов почти наверняка
носит случайный характер, так как при расчете делается
множество приближений, однако полученный результат
свидетельствует о том, что наблюдаемые эффекты поля по
порядку величины совпадают с теми, которые могут быть
предсказаны в соответствии с теорией простых электроста-
тических взаимодействий.
В связи с этим представляется вероятным, что эффекты
поля являются важнейшей причиной сдвига частот коле-
баний С = О у а-галогензамещенных соединений, содер-
жащих карбонильную группу. Однако это не означает, что
индукционные эффекты полностью отсутствуют. Экспери-
менты показывают, что атомы галогенов, находящиеся
в гош-положении относительно атома кислорода, не вызы-
вают сколько-нибудь заметного изменения частоты; это
570
Часть V
может быть обусловлено также компенсацией индукцион-
ных эффектов небольшими остаточными эффектами поля,
которые у таких изомеров действуют, по-видимому, в про-
тивоположном направлении. Указанное может быть причи-
ной того обстоятельства, что наблюдается некоторый раз-
брос значений частот колебаний карбонильной группы
у таких соединений, как этилтрифторацетат, который погло-
щает при частоте на 9 см~1 большей, чем дифторзамещенное
соединение [93], и метил трихлорацетат, который погло-
щает при частоте, на ослГ1 меньшей, чем соответствующее
дихлорпроизводное [90].
У соединений других типов эффекты поля широко не
изучены, хотя можно ожидать, что они должны прояв-
ляться у всех молекул, у которых имеются два близко рас-
положенных в пространстве заряженных атома с легко
поляризуемыми электронами. Выдвинуто, например, пред-
положение, что аномальные значения частот колебаний
карбонильных групп в случае 21-ацетокси-20-кетосте-
роидов могут быть обусловлены взаимодействиями тако-
го типа между двумя атомами кислорода карбонильных
групп [87].
Внутримолекулярные эффекты поля, которые имеют,
по-видимому, несколько иной характер, осуществляются
у некоторых циклических аминоацилоинов и родственных
им соединений, имеющих аномально низкие значения час-
тот колебаний карбонильных групп [94]. Большая работа
по исследованию этого класса соединений выполнена Лео-
нардом и его сотрудниками, и соответствующая литература
указана в гл. 9. Эти соединения представляют собой коль-
цевые циклические системы типа VII и VIII, конфигу-
рация которых такова, что атом азота подходит очень близко
Гл. 23. Причины изменения частот колебаний групп атомов 571
к атому углерода карбонильной группы. При этом имеет
место взаимодействие либо обусловленное простым эффектом
поля двух разноименных зарядов, когда происходит умень-
шение частоты колебаний, либо (и это более вероятно),
обусловленное наличием резонансных форм Vila и
VIII а. Любой механизм должен приводить к увеличе-
нию полярности связи С = О и уменьшению частоты
колебаний.
В результате для соединений этого типа характерны низ-
кие значения частот колебаний карбонильной группы, близ-
кие к значениям частот амидов, а не кетонов. Близость атома
азота к атому углерода карбонильной группы подтверж-
дается тем фактом, что при образовании соли с хлорной
кислотой получаются мостиковые циклические соединения
с гидроксильными группами. Можно также заставить
взаимодействующие атомы отойти друг от друга, либо изме-
няя размеры цикла, либо вводя громоздкие заместители
при атоме азота. При этом частота колебаний карбониль-
ной группы возвращается к ее обычному значению, харак-
терному для больших циклических кетонов, и проявляется
около 1720 сл/"1.
г. Применения. Представленные выше данные дают доста-
точно веские основания считать, что исследование смеще-
ний частот колебаний групп, выбранных надлежащим обра-
зом, может открыть путь к новым применениям методов
инфракрасной спектрометрии для прослеживания измене-
ний физических свойств и электрических эффектов, кото-
рыми вызываются эти смещения, хотя для полного исполь-
зования таких возможностей почти наверняка необходимо
дополнять исследования частот полос поглощения изуче-
нием интенсивностей соответствующих полос. Однако, как
неоднократно указывалось, подобная работа требует вели-
чайшей осторожности как в выборе подходящих частот
колебаний, так и в применении полученных данных
к какой-либо конкретной проблеме.
Несмотря на все эти ограничения, известно уже много
случаев, подобных указанным в предыдущих разделах,
когда данные измерений частот могут быть использованы
для подтверждения результатов, получаемых другими физи-
ческими методами. Исследования в этой области позво-
572
Часть V
ляют также установить некоторые полезные корреляции,
связывающие положение двух или более полос в пределах
одного и того же спектра. В качестве примеров такого
применения метода могут служить частоты валентных
колебаний SO2 и NH2, которые практически свободны от
эффектов масс и взаимодействия колебаний. Обе эти груп-
пы имеют антисимметричное и симметричное валентные
колебания, точные значения частот которых зависят от
электрических свойств заместителей. Таким образом, в каж-
дом отдельном случае смещения частот антисимметричных
Рис. 34. (Воспроизводится по J. Chem. Soc.,
1957, 863).
и симметричных колебаний зависят от общего фактора,
и они могут быть поэтому прямо сопоставлены друг
с другом.
На рис. 34 показана зависимость этих двух частот
друг от друга для ряда различных замещений группы SO2.
Нетрудно видеть, что имеется прямая линейная зависимость
между этими частотами, и это очень удобно для целей кор-
реляций полос, так как предварительная идентификация
одной из них может быть подтверждена при точном опреде-
лении положения другой полосы [88]. Аналогичные свой-
ства характерны для двух валентных колебаний NH2
Гл. 23. Причины изменения частот колебаний групп атомов 573
первичных аминов, частоты которых связаны друг с дру-
гом [94] соотношением
'Vchmm ~ 345,5 4~ 0,876 v^hthchmm*
Выполнимость этих соотношений зависит от эквива-
лентности природы двух рассматриваемых полос и их зави-
симости от одних и тех же электрических факторов. Если
же эта эквивалентность нарушается образованием водо-
родной связи или другими эффектами ассоциации, которые
влияют на одну связь больше, чем на другую, то соотноше-
ние перестает выполняться. Это положение подтверждено
в случае первичных аминов, у которых один атом водорода
группы NH2 связан, а другой свободен, и это обстоятельство
косвенно может служить в качестве чувствительного крите-
рия для определения ассоциаций такого типа.
Еще одно соотношение такого рода, связывающее частоты
валентных колебаний карбонильной группы и группы
С — О, было получено в случае стероидных ацетатов [95].
Обычная чувствительность связи С — О к эффекту масс
уменьшена в этих условиях до предела вследствие постоян-
ства окружения группы, и частота ее колебаний, подобно
частоте колебаний карбонильной группы, закономерно
изменяется при изменениях электрических свойств замес-
тителей. При этом существует простая линейная зависи-
мость обеих частот колебаний, имеющая важное значение
для целей идентификации соединений.
В качестве примера, показывающего необходимость
соблюдения осторожности при исследованиях такого рода,
можно указать на результаты, полученные при попытке
найти функциональную зависимость между частотами
антисимметричных и симметричных валентных колебаний
нитрогруппы. В этом случае не обнаружено никакой общей
монотонной зависимости. Причины этого рассмотрены Брау-
ном [96], который установил, что если антисимметричные
колебания свободны от эффектов взаимодействия колеба-
ний, то дело обстоит, вероятно, иначе в случае симметрич-
ных колебаний, частота которых близка к частоте колеба-
ний С — N и которые имеют ту же симметрию. Симметрич-
ные колебания являются, следовательно, объектом, на кото-
рый воздействует дополнительный переменный фактор,
в результате чего частота этих колебаний не связана
574
Часть V
линейной зависимостью с частотой антисимметричных
колебаний.
Могут быть найдены также соотношения, связывающие
целый ряд полос поглощения в одном спектре, как это имеет
место в случае шести фундаментальных колебаний метиль-
ной группы. Можно показать, что каждое из валентных,
деформационных и веерных колебаний СН в ряду метил-
галогенидов зависит от электроотрицательностей атомов
галогенов, причем это верно даже в случае валентных коле-
баний С— X, если правильно учтен эффект масс. Эта корре-
ляция сама по себе не представляет очень большой ценности
из-за отсутствия точных значений электроотрицательности.
Однако частоты валентных колебаний Н — Ху галоген-
содержащих кислот, исправленные с учетом эффекта масс,
также закономерно изменяются при изменении электроотри-
цательности (рис. 31), и при нанесении значений этих час-
тот колебаний НХ как функций от частот колебаний СН3Х
Рис. 35. (Воспроизводится по J. Chem. Soc., 1956, 2753).
неопределенности в значениях электроотрицательностей
могут быть компенсированы. Характер совпадения полу-
ченных результатов иллюстрируется графиками рис. 35,
на которых нанесены данные для пяти видов колебаний СН,
причем для одного из них приведен график, рассчитанный
с учетом эффекта взаимодействия колебаний с колебаниями
С — X, частота которых исправлена с учетом эффектов масс.
Гл. 23. Причины изменения частот колебаний групп атомов 575
Рассмотрение данных рис. 31 показывает, что, кроме
значений электроотрицательностей, имеются и другие фак-
торы, оказывающие влияние на частоты валентных колеба-
ний X — Н поливалентных элементов. Если предположить,
что влияние этих факторов одинаково для ряда соединений
СНз — Х(Н)„, то из данных рис. 35 можно, зная, например,
частоты валентных колебаний ОН воды, найти шесть фун-
даментальных частот колебаний метильной группы в случае
метилового спирта. Показано, что результаты, полученные
таким образом, довольно хорошо согласуются с данными
для достаточно широкого круга соединений, содержащих
метильную группу, с различными заместителями [74],
и эти данные находят важное применение для проверки
правильности отнесения частот колебаний.
Проводились также исследования по сопоставлению
частот колебаний различных групп с одними и теми же
заместителями. Например, закономерные изменения частот
колебаний карбонильных групп у соединений RCOCHS
при изменениях природы радикала R были сопоставлены
непосредственно со смещениями частот валентных колеба-
ний N - = О у соединений R — N = О с тем же рядом замес-
тителей R. При этом можно было видеть вполне отчетливые
линейные соотношения [88, 97], если только обе сравни-
ваемые группы в одинаковой мере подвержены влиянию
электрических эффектов. Однако, как и следовало ожидать,
расхождения наблюдаются в тех случаях, когда проявля-
ются эффекты поля,так как при этом различия в межатомных
расстояниях и атомных поляризуемостях приводят к раз-
личной степени взаимодействия.
Еще одной областью, в которой изучение причин сме-
щений частот колебаний вполне может найти широкое при-
менение, является более детальное исследование эффектов
внутримолекулярного поля в связи с межмолекулярными
эффектами, которыми сопровождаются изменения агрегат-
ного состояния вещества.
Механизмы взаимодействия, по крайней мере в некото-
рых случаях, представляются весьма сходными, и изуче-
ние внутримолекулярных сил, которые действуют при
известных значениях углов и вдоль известных расстояний,
может открыть путь к лучшему пониманию более сложных
вопросов межмолекулярного взаимодействия.
576
Часть И
ЛИТЕРАТУРА
1. В е 1 1 a m у, Williams, J. Chem. Soc., 1957, 4294.
2. Barker, Bourne, Neely, Wh i f fen, Chem. and
Ind., 1954, 1418.
3. Farmer, Chem. and Ind., 1955, 87.
4. Farmer, Spectrochim. Acta, 8, 374 (1956).
5. Barker, J. Phys. Chem., 61, 450 (1957).
6. Kirkwood, cited by We s t and Edwards, J Chem.
Phys., 5, 14 (1937).
7. В a u e r, M a g a t, J. Phys. Radium, 9, 319 (1938).
8. Josien, Sourisseau, Castin el, Bull. Soc. Chim.
France, 1955, 1539.
9. Josien, Spurisseau, Bull. Soc. Chim. France, 1955, 178.
10. J о s i e n, S a u m a g n e, Bull. Soc. Chim. France, 1956, 937.
11. J о s i e n, F u s о n, J. Chem. Phys., 22, 1169 (1954).
12. В а у 1 i s s, С о 1 e, Little, Austral. J. Chem., 8, 26 (1955).
13. H i г о t a, Bull. Chem. Soc., Japan, 27, 295 (1954).
14. Buckingham, Le Fevre, J. Chem. Soc , 1952, 132.
15. L e F ё v r e, L e Fevre, Austral. J. Chem., 7, 33 (1954).
16. Smith, Electric Dipole Moments, Butterworths, London,
1955, pp. 159 et seq.
17. H a d z i, Sheppard, Proc. Roy. Soc., A216, 247 (1953).
18. Badger, J. Chem. Phys., 8, 288 (1940).
19. L о r d, Merrifield, J. Chem. Phys., 21, 166 (1953).
20. Kuhn, J. Amer. Chem. Soc., 74, 2492 (1952).
21. Джонс H., Сандорфи K-> Применение спектроскопии
в химии, Издатинлит, М., 1959, стр. 342.
22. N a k a m о t о, М а г g о s h е s, Rundle, J. Amer. Chem.
Soc., 77, 6480 (1955).
23. Pimentel, Sederholm, J. Chem. Phys., 24, 639
(1956).
24. Cannon, Mikrochem. Acta, 1955, 555.
25. Tam res, Searles, Leigh 1 y, Mohr m a n, J. Amer.
Chem. Soc., 76, 3984 (1954).
26. Halford, J. Chem. Phys., 24, 830 (1956).
27. W h i f f e n, Chem. and Ind., 1957, 193.
28. Lord, M ill er, Applied Spectroscopy, 10, 115 (1956).
29. Randle, W h i f f e n, J. Chem. Soc., 1955, 1311.
30. В e 1 1 a m y, Branch, Nature, 173, 634 (1954).
31. Frazer, Price, Nature, 170, 490 (1952).
32. Mecke, Mecke, Chem. Ber., 89, 343 (1956).
Гл. 23. Причины изменения частот колебаний групп атомов 577
33.
34.
35.
А
36.
В
37.
В
№
1 о
J.
i о
Walker, J. Amer. Chem. Soc., 76, 2518 (1954).
hnert, J. Amer. Chem. Soc., 78, 4024
Татевск ий В. М., Вестник МГУ,
ist, Wolinsky, Meinwald, Longone,
Chem. Soc., 78, 6057 (1956).
ist, V e r d о 1,
J. Amer. Chem.
Soc., 77, 1806
Lord,
Traynham, Se
(1956).
к и ш и и П. А.,
2, сер. физ.-мат., 103 (1951).
m q u
Amer.
m q u
(1955).
Henbest,
J.
Jon
J. .
С о 1 <
Hen
О r r,
A n e
H a r
1948, 1436.
G о u I d e n, Spectrochim. Acta, 6, 129 (1954).
Bellamy, J.
Bell, H ei si
J. Amer. Chem.
К a g a r i s e,
49. Skinner, P
Bellamy,
Smith, Re
73, 2263 (1951).
Nickson, J.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
50.
51.
52.
Meakins,
Soc., 1957, 997.
Humphries,
Amer. Chem. Soc., 72, 86
Thornton, J. Chem. Soc., 1957,
Wood, J. Chem.
Nicholls,
Wilson,
Chem.
e s,
Packard,
(1950).
Dobriner,
1332.
Soc., 1954, 800.
e,
ibest, Meakins,
, Spectrochim. Acta, 8, 218 (1956).
: t, Bavin, Can. J. Chem., 34, 1756 (1956).
(well, Richards, Thompson, J. Chem. Soc.,
Chem. Soc., 1955, 4221.
Tannebaum, Goldenson,
Soc., 76, 5185 (1954)
Amer. Chem. Soc., 77, 1377 (1955).
e r,
J.
г i t с h а г d, Trans. Faraday Soc., 49, 1254 (1953).
Williams,
e,
J. Chem. Soc., 1956, 3704.
Magee, Eyring, J. Amer. Chem. Soc.,
Amer. Chem. Soc., 79, 243 (1957).
53. Flett, Trans. Faraday Soc., 44, 767 (1948).
54. Richards, Trans. Faraday Soc., 44, 40 (1948).
55. Fuson, Josien, Shelton, J. Amer. Chem. Soc., 76,
* 2526 (1954).
56. Josien, Fuson, Lebas, Gregory, J. Chem. Phys.,
21, 331 (1953).
57. Fillwalk, Fassel, Rundle, J. Chem. Phys., 22,
381 (1954).
58. Bellamy, Branch, J. Chem. Soc., 1954, 4491.
59. Ingold, Chem. Reviews, 15, 225 (1934).
60.. Ingold, Structure and Mechanism in Organic Chemistry,
Bell, London, 1954.
37 Л. Беллами
578
Часть V
61. Hammett, Physical
New York, 1940.
Organic Chemistry, McGraw Hill,
62. Jaffe, Chem. Reviews, 53, 191 (1953).
63. Taft, J. Amer. Chem. Soc., 75, 4231 (1953).
64. Taft, Steric Effects in Organic Chemistry, Wiley, New York,
1956.
65. Califano, Moccia, Gazz. Chim., 86, 1014 (1956).
66. Ingraham, Corse, Bailey, Stitt, J. Amer. Chem.
Soc., 74, 2297 (1952).
67. Forbes, Mueller, Can. J. Chem., 35, 488 (1957).
68. Jones, Forbes, Mueller, Can. J. Chem., 35, 504
(1957).
69. Scrocco, Libert i, Ricerca Sci., 24, 1687 (1954).
70. Davison, J. Chem. Soc., 1951, 2456.
71. Bellamy, J. Chem. Soc., 1955, 2818.
72. С о r i o, Dailey, J. Amer. Chem. Soc., 78, 3043 (1956).
73. Lebas, Josie n, Bull. Soc. Chim. France, 1956, 62.
74. В e 1 1 a m y, W i 1 1 i a m s, J. Chem. Soc., 1956, 2753.
75. С a 1 i f a n o, Moccia, Gazz. Chim., 87, 58 (1957).
76. Stone, Thompson, Spectrochim. Acta, 10, 17 (1957).
77. Staats, Morgan, Goldstein, J. Chem. Phys., 25,
582 (1956).
78. В a г г о w, J. Chem. Phys., 21, 2008 (1953).
79. Jones, Ramsey, Herl i ng, Dobriner, J. Amer.
Chem. Soc., 74, 2828 (1952).
80. Jones, J. Amer. Chem. Soc., 75, 4839 (1953).
81. Dickson, Page, J. Chem. Soc., 1955, 447.
82. Corey, J. Amer. Chem. Soc., 75, 2301 (1953).
83. С о г e у, T о p i e, W о z n i a k, J . Amer. Chem. Soc., 77,
5415 (1955).
84. Corey, Burke, J. Amer. Chem. Soc., 77, 5418 (1955).
85. I n a у a m a, Pharm. Bull. Japan, 4, 198 (1956).
86. Джонс H., Санд орфи K-, Применение спектроЬкопии
в химии, Издатинлит, М., 1959, стр. 386.
87. Bellamy, Wil/ i a m s, J. Chem. Soc., 1957, 861.
88. В e 1 1 a m y, W 1 i m s, J. Chem. Soc., 1957, 863.
89. Nakagawa, chishim a, Kuratani, Miy-
azawa, Shimanouchi, Mizushima, J. Chem.
Phys., 20, 170 (1952).
90. Josie n, Calas, Compt. Rend. Acad. Sci. (Paris), 247,
1641 (1955).
Гл. 23. Причины изменения частот колебаний групп атомов 579
91. L е t a w, Groop, J. Chem. Phys., 21, 1621 (1953).
92. В е n d е г, J. Amer. Chem. Soc., 75, 5986 (1953).
93. Leonard, Fo x, О к i, C h i a v a r e 1 1 i, J. Amer. Chem.
Soc., 76, 630 (1954).
94. Bellamy, Williams, Spectrochim. Acta, 9, 341 (1957).
95. Джонс H., Сандорфи K-, Применение спектроскопии
в химии, Издатинлит, М., 1959, стр. 388—390.
96. Brown, J. Amer. Chem. Soc., 77, 6341 (1955).
97. Bellamy, Chem. and Ind., 1957, 26.
98. W h i f f e n, Quart. Reviews, Chem. Soc., 4, 131 (1950).
99. Mizushima, Shimanouchi, Ichishima, Miya-
zawa, Nakawaga, Arkai, J. Amer. Chem. Soc.,
78, 2038 (1956).
100. Lippincott, Schroeder, J. Chem. Phys., 23, 1099
(1955).
101. Welsh, J. Chem. Phys., 26, 710 (1957).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
Волькен штейн М. В., Строение и физические свойства
молекул, Изд. АН СССР, М.—Л., 1955, гл. 10.
Мидзусима С., Строение молекул и внутреннее вращение,
Издатинлит, М., 1957.
Чулановский В. М., сб. Молекулярная спектроскопия.
Изд. ЛГУ, Л., 1960, стр. 3,
37*
00
ТАБЛИЦА ОБРАТНЫХ ВЕЛИЧИН
(для пересчета длин волн в волновые числа)
О 1 2 3 4 5 6 7 8 9123
4 5 6
7 8 9а
1,0
1,1
1,2
1,3
1,4
1,5
1,6
1,7
1,8
1,9
2,0
2,1
2,2
2,3
2,4
2,5
2,6
2,7
2,8
2,9
1,0000
0,9091
0,8333
0,7692
0,7143
0,6667'
0,6250
0,5882
0,5556
0,5263
0,9901
0,9009
0,8264
0,7634
0,7092
0,6623
0,6211
0,5848
0,5525
0,5236
0,9804
0,8929
0,8197
0,7576
0,7042
0,6579
0,6173
0,5814
0,5495
0,5208
0,5000
0,4762
0,4545
0,4348
0,4167
0,4000
0,3846
0,3704
0,3571
0,3448
0,4975
0,4739
0,4525
0,4329
0,4149
0,3984
0,3831
0,3690
0,3559
0,3436
0,4950
0,4717
0,4505
0,4310
0,4132
0,3968
0,3817
0,3676
0,3546
0,3425
0,9709
0,8850
0,8130
0,7519
0,6993
0,6536
0,6135
0,5780
0,5464
0,5181
0,4926
0,4695
0,4484
0,4292
0,4115
0,3953
0,3802
0,3663
0,3534
0,3413
0,9615
0,8772
0,8065
0,7463
0,6944
0,6494
0,6098
0,5747
0,5435
0,5155
0,4902
0,4673
0,4464
0,4274
0,4098
0,3937
0,3788
0,3650
0,3521
0,3401
0,9524
0,8696
0,8000
0,7407
0,6897
0,6452
0,6061
0,5714
0,5405
0,5128
0,4878
0,4651
0,4444
0,4255
0,4082
0,3922
0,3774
0,3636
0,3509
0,3390
0,9434
0,8621
0,7937
0,7353
0,6849
0,6410
0,6024
0,5682
0,5376
0,5102
0,4854
0,4630
0,4425
0,4237
0,4065
0,3906
0,3759
0,3623
0,3497
0,3378
0,9346
0,8547
0,7874
0,7299
0,6803
0,6369
0,5988
0,5650
0,5348
0,5076
0,9259 0,9174
0,8475 0,8403
0,7813 0,7752
0,72460,7194
0,6757
0,6329
0,5952
0,6711
0,6289
0,5917
0,5618
0,5319
0,5051
0,5587
0,5291
0,5025
3,0
3,1
3,2
3,3
3,4
3,5
3,6
3,7
3,8
3,9
0,3333
0,3226
0,3125
0,3030
0,2941
0,2857
0,2778
0,2703
0,2632
0,2564
4,0 0,2500
4,1 0,2439
4,2 0,2381
4,3
4,4
4,5
4,6
4,7
4,8
4,9
5,0
5,1
5,2
5,3
5,4
0,2326
0,2273
0,2222
0,2174
0,2128
0,2083
0,2041
0,2000
0,1961
0,1923
0,1887
0,1852
0,3322
0,3215
0,3115
0,3021
0,2933
0,2849
0,2770
0,2695
0,2625
0,2558
0,2494
0,2433
0,2375
0,2320
0,2268
0,2217
0,2169
0,2123
0,2079
0,2037
0,1996
0,1957
0,1919
0,1883
0,1848
0,3311
0,3205
0,3106
0,3012
0,2924
0,2841
0,2762
0,2688
0,2618
0,2551
0,2488
0,2427
0,2370
0,2315
0,2262
0,2212
0,2165
0,2119
0,2075
0,2033
0,1992
0,1953
0,1916
0,1880
0,1845
0,3300
0,3195
0,3096
0,3003
0,2915
0,2833
0,2755
0,2681
0,2611
0,2545
0,2481
0,2421
0,2364
0,2309
0,2257
0,2208
0,2160
0,2114
0,2070
0,2028
0,1988
0,1949
0,1912
0,1876
0,1842
0,3289
0,3185
0,3086
0,2994
0,2907
0,2825
0,2747
0,2674
0,2604
0,2538
0,2475
0,2415
0,2358
0,2304
0,2252
0,2203
0,2155
0,2110
0,2066
0,2024
0,1984
0,1946
0,1908
0,1873
0,1838
0,3279
0,3175
0,3077
0,2985
0,2899
0,2817
0,2740
0,2667
0,2597
0,2532
0,2469
0,2410
0,2353
0,2299
0,2247
0,2198
0,2151
0,2105
0,2062
0,2020
0,1980
0,1942
0,1905
0,1869
0,1835
0,4831
0,4608
0,4405
0,4219
0,4049
0,3891
0,3745
0,3610
0,3484
0,3367
0,3268
0,3165
0,3067
0,2976
0,2890
0,2809
0,2732
0,2660
0,2591
0,2525
0,2463
0,2404
0,2347
0,2294
0,2242
0,2193
0,2146
0,2101
0,2058
0,2016
0,1976
0,1938
0,1901
0,1866
0,1832
0,4808
0,4587
0,4386
0,4202
0,4032
0,3876
0,3731
0,3597
0,3472
0,3356
0,4785
0,4566
0,4367
0,4184
0,4016
0,3861
0,3717
0,3584
0,3460
0,3344
9 18 27
8 15 23
6 13 19
5 11 16
5 10 14
4 8 13
4 7 11
3 7 10
3 6 9
3 5 8
2 5 7
2 4 7
2 4 6
2 4 5
2 3 5
2 3 5
1 3 4
1 3 4
1 2 4
1 2 3
36 45 55
30 38 45
26 32 38
22 27 33
19 24 29
17 21 25
15 18 22
13 16 20
12 15 18
11 13 16
10 12 14
9 11 13
8 10 12
7 9 11
7 8 10
6 8 9
6 7 8
5 7 8
5 6 7
5 6 7
64 73 82
53 61 68
45 51 58
38 44 49
33 38 43
29 33 38
26 29 33
23 26 30
20 23 26
18 21 24
17 19 21
15 17 20
14 16 18
13 14 16
12 13 15
11 12 14
10 11 13
9 11 12
9 10 11
8 9 10
0,3257
0,3155
0,3058
0,2967
0,2882
0,2801
0,2725
0,2653
0,2584
0,2519
0,3247
0,3145
0,3049
0,2959
0,2874
0,2793
0,2717
0,2646
0,2577
0,2513
0,3236
0,3135
0,3040
0,2950
0,2865
0,2786
0,2710
0,2639
0,2571
0,2506
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
2
1
1
1
0,2457
0,2398
0,2342
0,2288
0,2237
0,2188
0,2141
0,2096
0,2053
0,2012
0,1972
0,1934
0,1898
0,1862
0,1828
0,2445
0,2387
0,2331
0,2278
0,2227
0,2451
0,2392
0,2336
0,2283
0,2232
0,2183 0,2179|
0,2137
0,2092
0,2049
0,2008
0,1969
0,1931
0,1894
0,1859
0,1825
0,2132
0,2088
0,2045
0,2004
0,1965
0,1927
0,189С
0,1855
0,1821
1
1
1
1
1
0
0
О
О
О
О
О
О
О
О
1
1
I
1
1
1
1
1
1
1
1
1
1
1
1
3
3
3
3
3
2
2
2
2
2
2
2
2
2
2
1
1
1
1
1
1
1
1
1
1
4 5 6
4 5 6
4 5 6
4 4 5
3 4 5
3 4 5
3 4 5
3 4 4
3 3 4
3 3 4
2 3 4
2 3 3
2 3 3
2 3 3
2 3 3
2 2 3
2 2 3
2 2 3
2 2 3
2 2 2
2 2 2
2 2 2
1 2 2
1 2 2
1 2 2
7 9 10
7 8 9
7 8 9
6 7 8
6 7 8
6 6 7
5 6 7
5 6 6
5 5 6
4 5 6
4 5 5
4 5 5
4 4 5
4 4 5
4 4 5
3 4 4
3 4 4
3 4 4
3 3 4
3 3 4
3 3 4
3 3 3
3 3 3
3 3 3
2 3 3
Продолжение
to
5,5
5,6
5,7
5,8
5,9
6,0
6,1
6,2
6,3
6,4
6,5
6,6
6,7
6,8
6,9
7,0
7,1
7,2
7,3
7,4
7,5
7,6
7,7
7,8
7,9
Cn
00
co
8,0
8,1
8,2
8,3
8,4
8,5
8,6
8,7
8,8
8,9
9,0
9,1
9,2
9,3
9,4
9,5
9,6
9,7
9,8
9,9
О l 2 3 4 5 6 7 8 9
_________________________________________________________I_______________________________I_______________________________________
2 3 4
5 6 7 8 9®
0,1818
0,1786
0,1754
0,1724
0,1695
0,1667
0,1639
0,1613
0,1587
0,1563
0,1538
0,1515
0,1493
0,1471
0,1449
0,1429
0,1408
0,1389
0,1370
0,1351
0,1333
0,1316
0,1299
0,1282
0,1266
0,1250
0,1235
0,1220
0,1205
0,1190
0,1176
0,1163
0,1149
0,1136
0,1124
0,1111
0,1099
0,1087
0,1075
0,1064
0,1053
0,1042
0,1031
0,1020
0,1010
0,1815
0,1783
0,1751
0,1721
0,1692
0,1664
0,1637
0,1610
0,1585
0,1560
0,1536
0,1513
0,1490
0,1468
0,1447
0,1427
0,1406
0,1 387
0,1368
0,1350
0,1332
0,1812
0,1779
0,1748
0,1718
0,1689
0,1661
0,1634
0,1608
0,1582
0,1558
0,1534
0,1511
0,1488
0,1466
0,1445
0,1425
0,1404
0,1385
0,1366
0,1348
0,1330
0,1808
0,1776
0,1745
0,1715
0,1686
0,1658
0,1631
0,1605
0,1580
0,1555
0,1531
0,1508
0,1486
0,1464
0,1443
0,1422
0,1403
0,1383
0,1364
0,1346'
0,1328
0,1805
0,1773
0,1742
0,1712
0,1684
0,1656
0,1629
0,1603
0,1577
0,1553
0,1529
0,1506
0,1484
0,1462
0,1441
0,1420
0,1401
0,1381
0,1362
0,1344
0,1326
0,1802
0,1770
0,1739
0,1709
0,1681
0,1653
0,1626
0,1600
0,1575
0,1550
0,1527
0,1504
0,1481
0,1460
0,1439
0,1418
0,1399
0,1379
0,1361
0,1342
0,1325
0,1799
0,1767
0,1736
0,1706
0,1678
0,1650
0,1623
0,1597
0,1572
0,1548
0,1524
0,1502
0,1479
0,1458
0,1437
0,1416
0,1397
0,1377
0,1359
0,1340
0,1323
0,1795
0,1764
0,1733
0,1704
0,1675
0,1792
0,1761
0,1730
0,1701
0,1672
0,1789
0,1757
0,1727
0,1698
0,1669
0,1645
0,1618
0,1592
0,1543
0,1520
0,1497
0.1475
0,1647
0,1621
0,1595
0,1570 0,1567
0,1546
0,1522
0,1499
0,1477
0,14560,1453
0,1435Ю, 1433
0,14140,1412
0,1395
0,1376
0,1357
0,1339
0,1321
0,1642
0,1616
0,1590
0,1565
0,1541
0,1517
0,1495
0,1473
0,1451
0,14311
0,1410
0,1391
0,1372
0,1353
0,1335!
0,1393
0,1374
0,1355
0,1337
0,1319 0,1318!
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
1
1
1
1
1
1
1
1
0
0
0
0
0
0
0
0
0
0
0
0
0
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
I
2 2
2 2
2 2
1 2
1 2
1 2
1 2
1 2
1 1
1 1
1 1
1 1
1 1
1 1
1 1
1 1
1 1
1 1
1 1
1 1
1 1
2 3 3
2 3 3
2 2 3
2 2 3
2 2 3
2 2 3
2 2 2
2 2 2
2 2 2
2 2 2
2 2 2
2 2 2
2 2 2
2 2 2
1 2 2
1 2 2
1 2 2
1 2 2
1 2 2
1 1 2
1 1 2
0,1314
0,1297
0,1280
0,1264
0,1248
0,1233
0,1218
0,1203
0,1189
0,1175
0,1161
0,1148
0,1135
0,1122
0,1110
0,1098
0,1086
0,1074
0,1063
0,1052
0,1041
0,1030
0,1019
0,1009
0,1312
0,1295
0,1279
0,1263
0,1247
0,1232
0,1217
0,1202
0,1188
0,1174
0,1160
0,1147
0,1134
0,1121
0,1109
0,1096
0,1085
0,1073
0,1062
0,1050
0,1040
0,1029
0,1018
0,1008
0,1311
0,1294
0,1277
0,1261
0,1245
0,1230
0,1215
0,1200
0,1186
0,1172
0,1159
0,1145
0,1133'
0,1120
0,1107
0,1095
0,1083
0,1072
0,1060
0,1049
0,1038
0,1028
0,1017
0,1007
0,1309
0,1292
0,1276
0,1259
0,1244
0,1229
0,1214
0,1199
0,1185
0,1171
0,1157
0,1144
0,1131
0,1119
0,1106
0,1094
0,1082
0,1071
0,1059
0,1048
0,1037
0,1027
0,1016
0,1006
0,1307
0,1290
0,1274
0,1258
0,1242
0,1227
0,1212
0,1198
0,1183
0,1170
0,1156
0,1143
0,1130
0,1117
0,1105
0,1093
0,1081
0,1070
0,1058
0,1047
0,1036
0,1026
0,1015
0,1005
0,1305
0,1289
0,1272
0,1256
0,1241
0,1225
0,1211
0,1196
0,1182
0,1168
0,1155
0,1142
0,1129
0,1116
0,1104
0,1092
0,1080
0,1068
0,1057
0.1046
0,1035
0,1025
0,1014
0,1004
0,1304
0,1287
0,1271
0,1255
0,1239
0,1224
0,1209
0,1195
0,1181
0,1167
0,1153
0,1140
0,1127
0,1115
0,1103
0,1091
0,1079
0,1067
0,1056
0,1045
0,1034
0,1024
0,1013
0,1003
0,1302
0,1285
0,1269
0,1253
0,1238
0,1222
0,1208
0,1193
0,1179
0,1166
0,1152
0,1139
0,1126
0,1114
0,1101
0,1089
0,1078
0,1066
0,1055
0,1044
0,1033
0,1022
0,1012
0,1002
0,1300
0,1284
0,1267
0,1252
0,1236
0,1221
0,1206
0,1192
0,1178
0,1164
0,1151
0,1138
0,1125
0,1112
0,1100
0,1088
0,1076
0,1065
0,1054
0,1043
0,1032
0,1021
0,1011
0,tooli
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0 1
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
о 0
1 1 1
1 1 1
1 1 1
1 1 1
1 1 I
1 1 1
1 1 1
1 1 1
1 1. 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
0 1 1
0 1 1
0 1 1
0 1 1
0 1 1
0 1 1
0 1 1
0 1 1
0 1 1
1 I 2
1 I 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 I 1
1 1 1
1 1 1
1 1 1
I 1 1
1 1 1
1 I 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
1 1 1
а В последних колонках 1 — 9 даются увеличенные в 10 000 раз поправки, вычитаемые при учете тысячных долей.
СОДЕРЖАНИЕ
Предисловие ........................................... 5
Предисловие автора ко второму изданию.................. 9
Глава 1. Введение .................................. 11
Литература......................................... 15
Часть 1
КОЛЕБАНИЯ УГЛЕРОД-УГЛЕРОДНЫХ
И УГЛЕРОД-ВОДОРОДНЫХ СВЯЗЕЙ
Глава 2. Алканы....................................... 23
1. Введение ...................................... 23
2. Валентные колебания С — Н...................... 23
3. Деформационные колебания групп СН3— С и СН2 33
4. Деформационные колебания метильных групп, при-
соединенных к неуглеродным атомам................. 40
5. Колебания углеродного скелета.................. 41
Литература........................................ 49
Глава 3. Алкены....................................... 53
1. Введение....................................... 53
2. Валентные колебания С = С...................... 54
3. Валентные колебания = СН....................... 65
4. Внеплоскостные деформационные колебания — СН 68
5. Плоскостные деформационные колебания =СН ... 7g
Литература........................................ 81
Глава 4. Алкины и аллены.............................. 86
1. Введение....................................... 86
2. Валентные колебания =СН........................ 87
3. Валентные колебания С = С ..................... 89
4. Деформационные колебания =СН .................. 92
5. Алленовые структуры............................ 92
Ли тература...................................... 94
Содержание 585
Глава 5. Ароматические соединения................... 96
1. Введение........................................ 96
2. Валентные колебания СН....................... 98
3. Поглощение в области 2000—1650 см~1............ 100
4. Валентные колебания С= С..................... 104
5. Внеплоскостные деформационные колебания СН . . 111
6. Характеристическое поглощение в области частот
1225—950 см1...................................... 120
7. Другие ароматические системы .................. 122
Литература.................................... . 122
Часть II
КОЛЕБАНИЯ УГЛЕРОД-КИСЛОРОДНЫХ
И КИСЛОРОД-ВОДОРОДНЫХ СВЯЗЕЙ
Глава 6. Спирты и фенолы ............................. 135
1. Введение ...................................... 135
2. Валентные колебания несвязанной группы ОН . . 138
3. Колебания связанной группы ОН (общие сведения) 140
4. Межмолекулярные водородные связи............... 142
5. Внутримолекулярные водородные связи ........... 146
6. Интенсивность и форма полос валентных колебаний
ОН ............................................... 151
7. Валентные колебания С—О н деформационные коле-
бания О — Н ...................................... 154
Литература........................................ 158
Глава 7. Простые эфиры, перекиси и озониды .... 163
1. Введение....................................... 163
2. Алифатические простые эфиры ................... 165
3. Диарилэфиры и арилалкилэфиры .............. 167
4. Циклические простые эфиры ................ 168
5. Перекиси ...................................... 171
6. Озониды........................................ 174
Литература........................................ 175
Глава 8. Галогенангидриды кислот, карбонаты, ангид-
риды кислот и карбонилы металлов............... . . . 178
1. Введение ...............................
2. Галогенангидриды кислот........................ 179
3. Карбонаты и хлорформиаты....................... 180
4. Ангидриды кислот............................... 181
586
Содержание
5. Перекиси ...................................... 184
6. Карбонилы металлов........................... 185
Литература.......................................... 186
Глава 9. Альдегиды и кетоны......................... 188
1. Введение..................................... 188
2. Валентные колебания С = О в случае кетонов с от-
крытой цепью ...................................... 189
3. Валентные колебания С — О у дикетонов........ 201
4. Валентные колебания С = О в алифатических цик-
лических системах ................................. 210
5. Валентные колебания С = О в ароматических цик-
лических системах............................... 214
6. Интенсивность карбонильного поглощения .... 217
7. Характеристические частоты полос поглощения
кетоиов, не обусловленных валентными колебаниями
С = О........................................... 220
8. Поглощение валентных колебаний С — О у аль-
дегидов .......................................... 221
9. Поглощение валентных колебаний С — Н у аль-
дегидов ............................................ 224
10. Другие характеристические колебания альдегидов 224
Литература...................................... 225
Глава 10. Карбоновые кислоты........................ 231
1. Введение..................................... 231
2. Валентные колебания ОН карбоксильной группы . 233
3. Валентные колебания С^О...................... 237
4. Другие характеристические колебания карбоновых
кислот.......................................... 244
5. Ионизованная карбоксильная группа; соли .... 250
Литература...................................... 253
Глава 11. Сложные эфиры и лактоны ...................... 256
1. Введение..................................... 256
2. Валентные колебания С = О в сложных эфирах . . 258
3. Валентные колебания С=О в сложных эфирах двух-
основных кислот и кетоэфирах .................. . 264
4. Валентные колебания С = О в лактонах, лактолах и
т. п.............................................. 267
5. Тиоловые сложные эфиры........................... 270
6. Валентные колебания С — О........................ 271
Литература...................................... . 275
Содержание 587
Часть III
КОЛЕБАНИЯ УГЛЕРОД-АЗОТНЫХ
И АЗОТ-ВОДОРОДНЫХ СВЯЗЕЙ
Глава 12. Амиды, белки и полипептиды.................. 289
1. Введение....................................... 289
2. Валентные колебания NH .................. 292
3. Полоса амид-1 (карбонильное поглощение)........ 298
4. Полоса амид-П ................................. 308
5. Другие корреляции)............................. 314
6. Амиды, содержащие особые структуры............. 316
7. Полипептиды и белки............................ 319
8. Дихроизм в инфракрасной области спектра при иссле-
дованиях белков................................... 328
Литература........................................ 329
Глава 13. Аминокислоты, их хлористоводородные и другие
соли и амидокислоты ............................. 334
1. Введение....................................... 334
2. Валентные колебания группы NHj;................ 337
3. Деформационные колебания NH3. Аминокислотные
полосы поглощения I и II ..................... 341
4. Колебания СООН ................................ 344
5. Область 3000—2000 слг1 ........................ 347
6. Другие корреляции (аминокислоты)............... 349
7. Соли аминокислот............................... 350
8. Амидо кислоты ................................. 351
Литература........................................ 353
Глава 14. Амины и имины............................... 355
1. Введение....................................... 355
2. Валентные колебания NH......................... 357
3. Водородные связи аминов........................ 362
4. Деформационные колебания NH................. 366
5. Валентные колебания С — N................. 369
6. Группа СН3 — N ................................ 371
7. Группы NH3, NH2 и NH+.......................... 372
Литература........................................ 373
Глава 15. Соединения с ненасыщенными связями при
атомах азота ..................................... 377
1. Введение....................................... 377
2. Валентные колебания —С == N.................... 377
3. Изоцианаты и карбодиимиды ..................... 382
588
Содержание
4. Валентные колебания —С = N —............... 384
5. Валентные колебания —N —- N —............... 390
6. Диазогруппа..................................... 392
7. Азиды........................................... 392
Литература........................................ 394
Глава 16. Гетероциклические ароматические соединения 397
1. Введение........................................ 397
2. Пиридины и хинолины........................... 398
3. Пиримидины и пурины........................... 404
4. Другие гетероциклические структуры.............. 408
Литература......................................... 409
Часть IV
СВЯЗИ МЕЖДУ ДРУГИМИ ЭЛЕМЕНТАМИ.
НЕОРГАНИЧЕСКИЕ СТРУКТУРЫ
Глава 17. Нитро- и нитрозосоединения, нитраты и нитриты 421
1. Введение........................................ 421
2. Нитросоединения................................ 422
3. Валентные колебания R — N — О ................. 431
4. Ионизованные нитраты........................... 438
Литература......................................... 438
Глава 18. Фосфорорганические соединения................ 441
1. Введение....................................... 441
2. Валентные колебания Р = О...................... 441
3. Валентные колебания Р — О — С.................. 446
4. Пирофосфаты.................................... 451
5. Валентные колебания Р — ОН..................... 452
6. Валентные колебания Р — Н ..................... 454
7. Фосфор-углеродные связи........................ 454
8. Связи фосфора с серой.......................... 455
9. Связи фосфора с азотом......................... 457
10. Связи фосфора с галогенами..................... 459
И. Ионная фосфатная группа........................ 461
12. Другие корреляции для фосфорорганических соеди-
нений............................................. 463
Литература......................................... 464
Глава 19. Галогенпроизводные органических соединений 466
1. Введение........................................ 466
2. Связи С — F.................................... 468
3. Связи С — С1................................... 469
Содержание
589
4. Связи С — Вг ................................... 471
5. Связи С — I..................................... 472
6. Другие корреляции............................... 472
Литература......................................... 473
Глава 20. Кремнийорганические соединения............... 474
1. Введение........................................ 474
2. Связи Si — С ................................... 475
3. Валентные колебания Si — О..................... 480
4. Валентные колебания Si — Н..................... 483
5. Валентные колебания Si — Cl.................. 484
6. Валентные колебания Si — ОН..................... 484
7. Влияние связи Si — С на колебания СН............ 485
Литература......................................... 485
Глава 21. Неорганические ионы ......................... 487
1. Введение ..................................... 487
2. Карбонаты и бикарбонаты....................... 488
3. Сульфаты...................................... 490
4. Нитраты и нитриты............................. 491
5. Ион аммония .................................. 491
6. Цианиды....................................... 492
7. Фосфаты....................................... 493
8. Силикаты ..................................... 493
9. Другие корреляции............................. 494
Литература....................................... 494
Глава 22. Органические соединения, содержащие серу 497
1. Введение....................................... 497
2. Валентные колебания SH......................... 497
3. Валентные колебания S — S ..................... 500
4. Валентные колебания С — S ..................... 502
5. Валентные колебания С = S...................... 505
6. Тиоуреиды —N — С — S и — NH — С = S . . . . 507
7. Валентные колебания S = O ..................... 508
8. Ковалентные сульфаты и сульфонаты.............. 517
9. Сульфоновые кислоты и соли..................... 517
Литература........................................ 518
Часть V
Глава 23. Причины изменения частот колебаний групп
атомов и значение этих изменений.................. 531
1. Введение.......................................... 531
590 Содержание
2. Эффекты изменения состояния вещества ............ 533
3. Влияние растворителей............................ 536
4. Водородная связь................................. 539
5. Эффект масс и влияние взаимосвязи колебаний . . 542
6. Эффекты напряжения............................... 546
7. Электрические эффекты............................ 550
Литература.......................................... 576
Таблица обратных величин................................ 580
Л. Беллами
ИНФРАКРАСНЫЕ СПЕКТРЫ
СЛОЖНЫХ МОЛЕКУЛ
Редакторы: Н. А. Оганджанова
и В. В. Арнольдов
Переплет художника
В. И. Те ле пн е в а
Художественный редактор
Е. И. Подмарькова
Технический редактор М. А. Бел е в а
Корректор И. П. Максимова
Сдано в производство 13/V 1963 г.
Подписано к печати 14/Х 1963 г.
Бумага 84 х 1 081/32=9,2 бум. л.
30,3 печ. л.
Уч.-изд. л 30,0. Изд. № 3/1028
Цена 2 р. 30 к. Зак. 847
* * *
ИЗДАТЕЛЬСТВО
ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва, 1-й Рижский пер. 2
* * *
Московская типография № •
Мосгорсовнархоза
Москва, Трехпрудный пер., 9