/















































































































































































Автор: Калви Т.Н. Уильямс Н.Е.
Теги: лечение хирургия медицина фармакология анестезиология
ISBN: 978-5-9518-0216-3
Год: 2007




Текст
medlit.ucoz.ru
Фармакология
для анестезиолога
книга первая
Т.Н. Калви
Н.Е. Уильямс
Перевод с английского под редакцией
докт. мед. наук, профессора В.М. Мизикова,
канд. мед. наук А.М. Цейтлина
Издательство БИНОМ
МОСКВА
2007
УДК 616-089.5
ББК 54.5
К17
medlit.ucoz.ru
Перевод с англ:. А. Анваер, И. Никитенкова
Калви Т.Н., Уильямс Н.Е.
К17 фармакология для анестезиолога/ Пер. с англ. - М.: «Издательство БИНОМ», 2007,- 176 с., ил.
ISBN 978-5-9518-0216-3
Книга представляет собой первую часть британского руководства по фармакологии для
анестезиологов. Прошедшая уже несколько переизданий, она впервые переведена на русский язык.
В книге освещены вопросы фармакокинетики и фармакодинамики, действие ингаляционных,
неингаляционных и местных анестетиков, миорелаксантов и их антагонистов, а также препаратов,
влияющих на гемостаз. Ее отличает полнота и детальность изложения материала.
Книга предназначена для анестезиологов-реаниматологов.
ISBN 978-5-9518-0216-3
ISBN 0-632-05605-3 (англ.)
© Издательство БИНОМ, 2007
ПРЕДИСЛОВИ^О^НИЮ
НА РУССКОМ ЯЗЫКЕ
Существует точка зрения, что сегодня книги не
пишет только ленивый. И действительно, на при-
лавках специализированных отечественных магази-
нов стало немало книг по анестезиологии. Первый
ажиотажный спрос на них после многолетнего ин-
формационного голода уже удовлетворен. Для тех
специалистов, кто читает преимущественно на
русском, появилось много откровений... Для тех же,
кто и раньше читал по-английски, некоторые из них
становятся источником своеобразного «дежавю»:
«Где-то я это уже видел, но явно не в отечественном
издании...».
Увы, фрагментированные переводы-подстроч-
ники для некоторых отечественных авторов стали
основой «своих» монографий. Но действительно хо-
роших переводов полновесных оригинальных
изданий немного. По клинической фармакологии —
тем более. А по клинической фармакологии для анес-
тезиологов — фактически нет.
Книга с названием «Фармакология для анес-
тезиолога» хорошо известна зарубежным специ-
алистам. Прошедшая несколько переизданий, она
впервые переведена на русский язык. Ее отличает
полнота и детальность представления материала:
наряду с подробным изложением основных прин-
ципов и механизмов фармакологии, дающих
цельное понимание действия и поведения (фарма-
кодинамики и фармакокинетики) лекарственных
средств, применяемых в анестезиологии, она содер-
жит глубокие сведения об их клиническом приме-
нении в реальной обстановке современного анесте-
зиологического обеспечения.
Хороший профессиональный перевод — труд-
ная задача, требующая времени. Видимо, именно
этим - стремлением сделать его таковым - и объяс-
няется то обстоятельство, что сегодня в свет выхо-
дит лишь первая часть книги. Но очень важно, что
она уже выходит: у нас есть время на ее вниматель-
ное прочтение, а там, глядишь, подоспеет и вторая!
Эта книга - блестящая основа знаний для тех,
кто начинает свой путь в специальности, и свое-
временная помощь для тех анестезиологов, кто
реально хочет улучшить и систематизировать свои
представления о влиянии и взаимодействии т.н. «анес-
тезиологических» препаратов. Это действительно
«Теория и практика фармакологии для анестезио-
логов».
Докт. мед. наук, профессор В.М. Мизиков
СОДЕРЖАНИЕ
ПРЕДИСЛОВИЕ......................................................................8
СПИСОК СОКРАЩЕНИЙ................................................................9
Глава 1. ВСАСЫВАНИЕ, РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ......................................................................11
Транспорт лекарственных препаратов через клеточные мембраны...................12
Зависимость фармакологического эффекта от концентрации лекарственного
препарата в плазме............................................................15
Пути введения лекарственных препаратов........................................15
Распределение.................................................................19
Метаболизм лекарственных препаратов...........................................22
Выведение (экскреция).........................................................29
Глава 2. ФАРМАКОКИНЕТИКА........................................................32
Объем распределения...........................................................32
Клиренс.......................................................................33
Практическое значение фармакокинетических констант VhCL.......................34
Конечный период полувыведения.................................................36
Контекст-чувствительный период полувыведения..................................37
Измерение объема распределения, клиренса и конечного периода полувыведения
с помощью камерных моделей....................................................38
Камерные модели и фармакологическое действие..................................42
Другие фармакокинетические модели.............................................42
Приложение ....................................................................47
Глава 3. ФАРМАКОДИНАМИКА........................................................48
Зависимость между дозой лекарственного препарата и его эффектом...............48
Основные механизмы действия лекарственных препаратов..........................50
Эффекты, обусловленные физическими или химическими свойствами препарата.......50
Подавление активности ферментов, регулирующих метаболические процессы.........50
Воздействие на ионные каналы и транспортные белки клеточных мембран...........52
Воздействие на рецепторы......................................................52
Количественные аспекты взаимодействия лекарственных препаратов с рецепторами..62
Антагонисты лекарственных препаратов..........................................63
Частичные агонисты............................................................66
Обратные агонисты.............................................................67
Десенситизация................................................................68
Приложения....................................................................69
Глава 4. НЕИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ.............................................73
Барбитураты...................................................................74
Тиопентал.....................................................................76
Метогекситал..................................................................80
Пропофол......................................................................83
Этомидат......................................................................84
Кетамин.......................................................................86
Побочное действие неингаляционных анестетиков.................................87
Тотальная внутривенная анестезия..............................................89
СОДЕРЖАНИЕ
7
Глава 5. ИНГАЛЯЦИОННЫЕ AHECTEffifttfJ It U.GO£ fU................................91
Механизм действия..............................................................91
Анатомические мишени...........................................................95
Мощность.......................................................................95
Скорость наступления анестезии.................................................96
Отдельные препараты...........................................................100
Закись азота..................................................................100
Галотан.......................................................................103
Энфлуран......................................................................106
Изофлуран..................................................................... 108
Севофлуран....................................................................110
Десфлуран.....................................................................111
Циклопропан....................................................................ИЗ
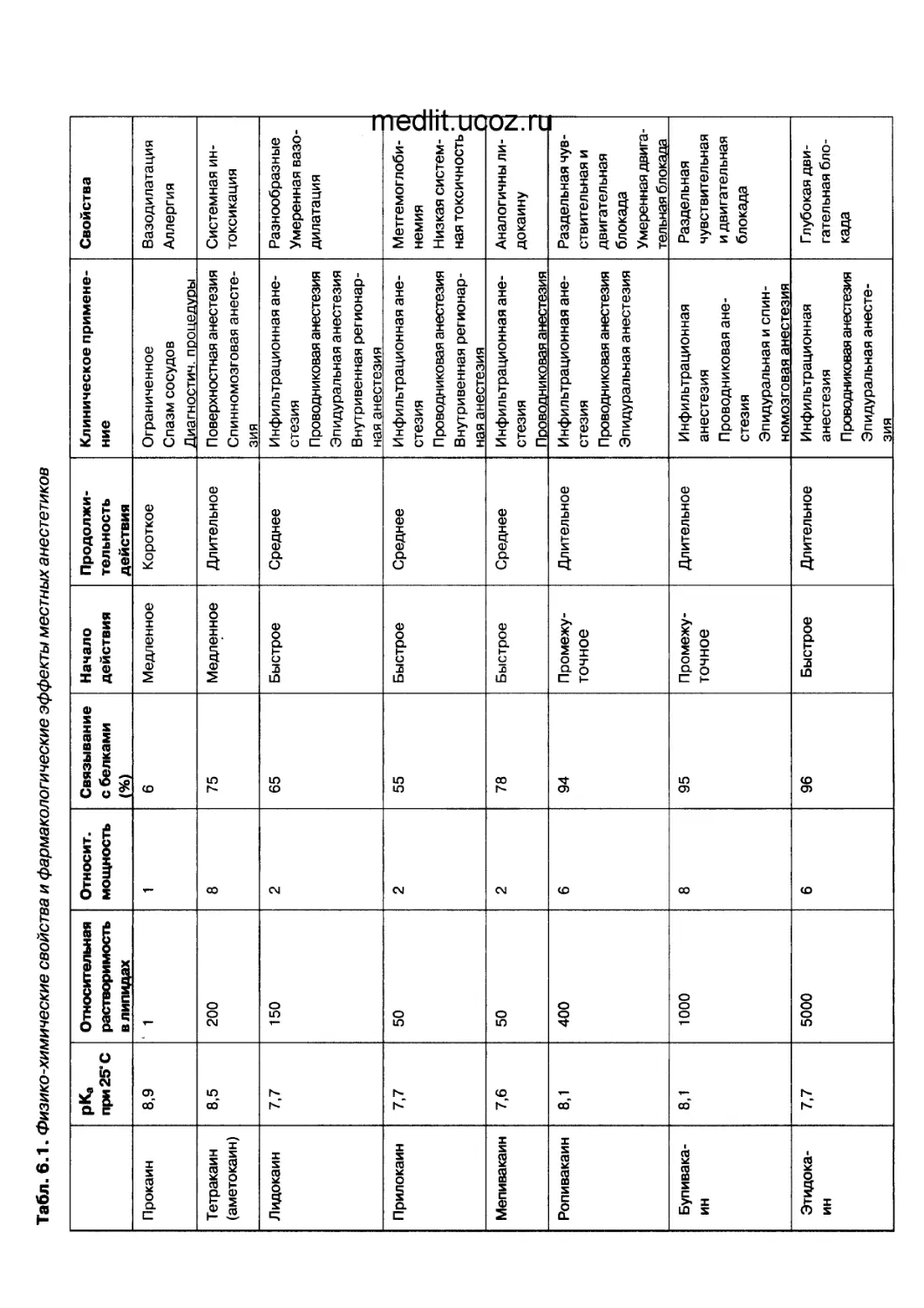
Глава 6. МЕСТНЫЕ АНЕСТЕТИКИ.....................................................115
Историческая справка..........................................................115
Строение и функция нервных волокон............................................116
Молекулярное строение натриевых и калиевых каналов............................117
Механизм действия.............................................................118
Препараты местных анестетиков.................................................. 120
Химическое строение и физико-химические свойства..............................121
Всасывание местных анестетиков в системный кровоток...........................124
Связывание с белками плазмы и проникновение через плаценту....................125
Фармакокинетика...............................................................125
Метаболизм и элиминация.......................................................125
Отдельные эффекты.............................................................127
Побочное действие местных анестетиков.........................................127
Клиническое применение........................................................129
Глава 7. ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ
НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС.......................................................136
Строение нервно-мышечного синапса.............................................136
Синтез и хранение ацетилхолина................................................136
Нервно-мышечная передача......................................................139
Лекарственные препараты, влияющие на нервно-мышечную передачу.................141
Лекарственные препараты, влияющие на высвобождение ацетилхолина...............141
Лекарственные препараты, влияющие на действие ацетилхолина....................142
Деполяризующие миорелаксанты..................................................145
Недеполяризующие миорелаксанты................................................148
Лекарственные препараты, влияющие на гидролиз ацетилхолина....................155
Лекарственные препараты, влияющие на электромеханическое сопряжение...........158
Практические рекомендации.....................................................158
Восстановление нервно-мышечной проводимости...................................159
Мониторинг нервно-мышечной блокады............................................159
Глава 8. АНТИКОАГУЛЯНТЫ, АНТИАГРЕГАНТЫ И ТРОМБОЛИТИКИ...........................161
Механизмы гемостаза...........................................................161
Расстройства гемостаза........................................................162
Антикоагулянты................................................................164
Антиагреганты.................................................................169
Тромболитики (фибринолитики)..................................................173
Антифибринолитические средства................................................175
Другие лекарственные средства, влияющие на гемостаз...........................176
ПРЕДИСЛОВИЕ .
Книга с названием «Фармакология для анесте-
зиолога» могла представлять собой просто описание
лекарственных препаратов, используемых в анесте-
зиологии. Но авторы добились качественно иного ре-
зультата. Благодаря их подходу читатель не только
станет более компетентным анестезиологом, но и по-
лучит особое удовлетворение от своей работы,
более точно понимая нюансы применения лекарст-
венных препаратов.
Анестезиология занимает уникальное место сре-
ди всех медицинских специальностей. Лекарст-
венные препараты, применяемые в анестезиологии,
вызывают глубокое угнетение различных физио-
логических механизмов. У врача любой другой спе-
циальности не возникает необходимости приме-
нять лекарственные средства таким образом.
Анестезиолог должен уметь управлять продолжи-
тельностью действия препаратов и при необходи-
мости быстро устранить их эффект. Часто это тре-
буется выполнить на фоне сопутствующих заболе-
ваний и применения других лекарств. Чтобы быть
готовым к разрешению таких проблем, врач должен
учитывать все факторы, влияющие на эффект ле-
карственного препарата, его элиминацию, а также
взаимодействие с другими препаратами. Эти зна-
ния необходимы анестезиологу больше, чем
представителю любой другой медицинской спе-
циальности. Авторы достойно справились с труд-
ной задачей описания фундаментальных принципов
фармакологии. Подробному описанию этих прин-
ципов посвящена треть книги. Такой подход дол-
жен избавить преподавателя от муки общения с уче-
никами, которые знают, какое воздействие
оказывает тот или иной лекарственный препарат,
но молчанием и каменным выражением лица реа-
гируют на вопрос: «Почему это происходит?»
В разделах, посвященных отдельным лекарст-
венным препаратам, тоже уделено особое внимание
механизмам действия, что придает жизни отдель-
ным темам, часто излагаемым с губительной ску-
кой. Врач, читающий эту книгу, приобретет не только
колоссальный объем бесценной информации, но
и определенный подход к клиническим ситуациям,
который послужит благополучию больных и чув-
ству удовлетворенности от своей работы.
Джексон Риз (Jackson Rees), MB, ChB
СПИСОК СОКРАЩЕНИИ
-----------------medlit.ucoz.ru
АВ — атриовентрикулярный
АД — артериальное давление
АДГ — антидиуретический гормон
АТФ — аденозинтрифосфат
АМФ — аденозинмонофосфат
АПФ — ангиотензин-превращающий фермент
АХЭ — ацетилхолинэстераза
АЧТВ — активированное частичное тромбо-
пластиновое время
в/в — внутривенно(ный)
в/м — внутримышечно(ный)
ВОЗ — Всемирная организация здравоохране-
ния
ВЧД — внутричерепное давление
ГМФ — гуанозинмомнофосфат
ГТФ — гуанозинтрифосфат
ДАГ 1,2-диацилглицерил
ДВС — диссеминированное внутрисосудистое
свертывание
ДЗЛА — давление заклинивания легочной ар-
терии
ДНК — дезоксирибонуклеиновая кислота
ЖКТ — желудочно-кишечный тракт
ИБС — ишемическая болезнь сердца
ИВЛ — искусственная вентиляция легких
ИМФ — инозитолмонофосфат
ИФ3 — инозитол-1,4,5-трифосфат
КоА — кофермент А
КТ — компьютерная томография
ЛСС — легочное сосудистое сопротивление
МАК — минимальная альвеолярная концен-
трация
МАО — моноаминокисдаза
МНО — международное нормализованное от-
ношение
МОД — минутный объем дыхания
МПКП — миниатюрный потенциал концевой
пластинки
НАД — никотинамидадениндинуклеотид
НАДФ — никотинамидадениндинуклеотид-
фосфат
НПВС — нестероидные противовоспалитель-
ные средства
ОПН — острая почечная недостаточность
ОПСС — общее сосудистое периферическое
сопротивление
ОЦК — объем циркулирующей крови
ПВ — протромбиновое время
п/к — подкожно(ный)
ПФК — площадь под фармакокинетической
кривой
РДСВ — респираторный дистресс-синдром
взрослых
РНК — рибонуклеиновая кислота
СКФ — скорость клубочковой фильтрации
ТЭЛА — тромбоэмболия легочной артерии
УЗИ — ультразвуковое исследование
ХОЗ Л — хронические обструктивные заболе-
вания легких
ХПН — хроническая почечная недостаточ-
ность
цАМФ — циклический аденозинмонофосфат
цГМФ — циклический гуанозинмонофосфат
ЦНС — центральная нервная система
ЦОГ — циклооксигеназа
ЦСЖ — цереброспинальная жидкость
ЧДД — частота дыхательных движений
ЧСС — частота сердечных сокращений
ЭДТА — этилендиаминтетрауксусная кислота
ЭКГ — электрокардиография (электрокардио-
грамма, электрокардиографический)
ЭхоКГ — эхокардиография
ЭЭГ — электроэнцефалография
FiO2 — фракционная концентрация кислорода
во вдыхаемой смеси
IgA — иммуноглобулины класса А
IgE — иммуноглобулины класса Е
IgG — иммуноглобулины класса G
IgM — иммуноглобулины класса М
k<i — константа диссоциации
LD50 — медиана летальной дозы
NMDA N — метил D-аспартат
10;Глава
medlit.ucoz.ru
РаСО2 — парциальное давление углекислого рКа — показатель кислотной диссоциации (от-
газа в артериальной крови РАСО2 — парциальное давление углекислого газа в альвеолах РаО2 — парциальное давление кислорода в ар- териальной крови РСО2 — парциальное давление углекислого газа pH — водородный показатель (pH = -1g [Н+]) рицательный десятичный логарифм константы диссоциации) РО2 — парциальное давление кислорода Rh — резус SaO2 — насыщение гемоглобина кислородом в артериальной крови Т1/2 — период полувыведения (полужизни)
ВСАСЫВАНИЕ,
РАСП РЕДЕЛ ЕН tfTW,C0Z ru
ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ
Лекарственными препаратами называют веще-
ства, которые модифицируют биологические реак-
ции. Для осуществления фармакологического
эффекта лекарственный препарат должен прони-
кать через клеточные мембраны. Скорость транс-
порта препарата через мембрану зависит от струк-
туры и физико-химических свойств мембран.
Эти мембраны имеют толщину около 10 нм и со-
стоят из бимолекулярного фосфолипидного слоя
и белка (рис. 1.1). Липидный слой имеет свойства
жидкости, и молекулы свободно перемещаются
в боковой плоскости мембраны. Внешние (перифе-
рические) белки располагаются на внутренней или
внешней поверхности клеточной мембраны и могут
перемешаться при изменениях pH или концентра-
ции ионов. Внутренние (интегральные) белки
часто пронизывают всю толщину плазматической
мембраны и могут образовывать кольца вокруг
мелких пор или ионных каналов диаметром при-
близительно 0.5 им (см. рис. 1.1). В клетках эндо-
телия капилляров поры значительно крупнее
(4-5 нм), и обычно имеются межклеточные про-
странства. Как периферические, гак и интеграль-
ные белки могут выполнять функцию ферментов
и рецепторов и способствуют активному транспор-
ту лекарственных препаратов.
Приблизительно на 5-10% клеточная мембрана
состоит из углеводов, которые обычно располага-
ются на внешней поверхности мембраны в виде
гликолипидов или гликопротеинов. Полагают, что
эти соединения отвечают за иммунологические
свойства клеток и играют важную роль в распозна-
вании молекул. Кроме того, многие мембраны со-
держат неорганические вещества, такие как ионы
кальция, связанные с отрицательно заряженными
группами фосфолипидов и холестерина.
Липидная плазматическая мембрана является
превосходным электрическим изолятором. Таким
образом, по обеим сторонам клеточной мембраны
существует разница потенциалов, которая облегча-
ет либо затрудняет транспорт заряженных молекул
по ионным каналам.
Рис. 1.1. Клеточная мембрана
12
Глава 1
ТРАНСПОРТ ЛЕКАРСТВЕННьРЙ6^^- ucWa+h+
ПРЕПАРАТОВ ЧЕРЕЗ КЛЕТОЧНЫЕ
МЕМБРАНЫ
Лекарственные препараты проникают через
клеточные мембраны пассивно или активно — с по-
мощью мембранных переносчиков.
ПАССИВНЫЙ ТРАНСПОРТ
В большинстве случаев лекарственные препара-
ты проникают через клеточную мембрану путем
пассивной диффузии молекул по градиенту кон-
центрации (из зоны высокой концентрации в зону
низкой); хаотичное движение молекул обусловле-
но тепловой энергией. Скорость транспорта прямо
пропорциональна концентрационному градиенту и
растворимости лекарственного препарата в двой-
ном липидном слое клеточной мембраны. Раство-
римость в липидах определяется коэффициентом
распределения между липидной и водной фазами.
Полярные молекулы (например, четвертичные
амины) удерживаются в водной фазе водородны-
ми связями, и их коэффициент распределения
можно считать равным нулю. Лекарственные
препараты с высоким коэффициентом распреде-
ления между липидами и водой (например, диазепам,
фентанил) легко растворяются в фосфолипидном
слое мембраны и быстро проникают в клетку для
оказания фармакологического действия. Напро-
тив, лекарственные препараты с низким коэффи-
циентом распределения (например, морфин) про-
никают в клетку медленнее и начинают действо-
вать с некоторой задержкой. В меньшей степени
проникновение через мембрану зависит от размера
молекул: полярные вещества с молекулярной мас-
сой более 100-200 Да не способны проникать
сквозь клеточные мембраны. Напротив, некото-
рые мелкие полярные молекулы способны посту-
пать в клетку через ионные каналы и поры, образо-
ванные интегральными белками мембран. Эти мо-
лекулы могут также проникать через мелкие меж-
клеточные и околоклеточные каналы (особенно
в фенестрированных эпителиальных мембранах).
Проницаемость сосудистого эндотелия выше, чем
других тканей, и большинство ионизированных
соединений легко проникает через капиллярные
мембраны.
Большинство лекарственных препаратов являют-
ся слабыми кислотами или слабыми основаниями,
и поэтому в физиологических условиях они могут
существовать как в ионизированной, так и в неио-
низированной форме. Ионизацию или диссоциа-
цию можно представить следующим образом:
для кислот
ВН+ В + Н+ для оснований
В кислой среде слабые кислоты и основания
в основном присутствуют в виде частиц АН и ВН+,
а в щелочной среде — в виде частиц А и В. Неиони-
зированные формы АН и В растворимы в липидах
и поэтому легко проникают через клеточные мем-
браны; в то же время для ионизированных форм А-
и ВН+ клеточные мембраны практически непрони-
цаемы. Поскольку доля ионизированной формы
лекарственного препарата зависит от pH, разница
концентраций Н+ по обеим сторонам мембраны
обеспечивает диффузионный градиент для пассив-
ного транспорта неионизированной фракции.
Рассмотрим слабокислое вещество, диссоции-
рующее следующим образом:
АН А - + Н+
Используя уравнение Гендерсона-Хассель-
бальха, диссоциацию можно представить следую-
щим образом:
рК.-рН - log([АН]/[А ]),
где [АН] и [А] - концентрации неионизиро-
ванной и ионизированной форм, а рХа (отрицатель-
ный логарифм константы диссоциации) равен зна-
чению pH, при котором [АН] = [А ]. Если рХа = 6,
то при pH 2 (например, в желудке) почти 100%
лекарственного препарата представлено формой
АН (рис. 1.2). Эта неионизированная форма легко
проникает в плазму (pH 7,4), где приблизительно
96% вещества переходит в ионизированную форму
А, обеспечивая концентрационный градиент для
дальнейшей диффузии формы АН. Последующий
перенос лекарственного препарата в другие среды
организма также будет зависеть от относительного
градиента pH. При pH 8 (интерстициальная
жидкость или защелоченная моча) концентрация
АН будет меньше, чем при pH 7,4. Таким образом,
создается градиент pH для пассивной диффузии
АН через эпителий почечных канальцев с после-
дующей ионизацией до А' и выведением из орга-
низма (рис. 1.2). Напротив, если pH мочи состав-
ляет 7 или меньше, концентрация АН в моче выше,
чем в плазме, и экскретированное лекарственное
вещество начнет диффундировать из мочи обратно
в плазму.
Аналогичным образом от градииента pH также
зависит неионная диффузия слабых оснований:
В + Н+<=>ВН+
Используя уравнение Гендерсона-Хассель-
бальха, диссоциацию слабого основания можно
представить следующим образом:
ВСАСЫВАНИЕ, РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
13
к medlit.ucoz.ru
Клеточная Клеточная
Рис. 1.2. Неионная диффузия слабой кислоты АН (рКа = 6). Только неионизированная форма АН может проникать
через клеточные мембраны; диффузионный градиент зависит от разницы pH в различных камерах или тканях. Числа
в скобках — доля (в процентах) лекарственного препарата, находящегося в форме АН и А при pH 2; 7,4 и 8
pKa-pH = log([BH+]/[B]),
где [ВН+] и [В] — концентрации ионизиро-
ванной и неионизированной форм соответственно,
а рКа — это значение pH, при котором [ВН+] = [В].
Если рКа препарата со свойствами основания равен 7,
то при pH 2 (например, в желудке), почти 100% ве-
щества будет находиться в ионизированной форме
ВН+ (рис. 1.3). При pH 5,5 (например, в тонком ки-
шечнике) только 3% лекарственного средства будет
представлено неионизированной формой В, спо-
собной проникать через клеточную мембрану. Хотя
эффективный градиент pH не способствует не-
ионной диффузии слабых оснований из просвета
тонкого кишечника (pH 5,5) в плазму (pH 7,4),
постоянный кровоток в капиллярах кишечника
обеспечивает небольшой концентрационный гра-
диент для всасывания лекарственного препарата.
Напротив, при pH 7,4 (то есть, в плазме) слабые
основания в основном представлены неионизиро-
ванными частицами В. В этих условиях возникает
значительный концентрационный градиент, облег-
чающий их диффузию в желудок (pH 2) и кислую
мочу (pH 5). После в/в инъекции опиоидных
анальгетиков фентанила или феноперидина пер-
воначальное снижение их концентрации в плаз-
ме через 30-40 мин сменяется вторичным пиком.
Эти слабые основания сразу после введения выво-
дятся из плазмы в желудок вследствие значитель-
ного концентрационного градиента, облегчающего
диффузию. Последующее всасывание этих опиои-
дов из тонкой кишки обеспечивает вторичное по-
вышение их концентрации в плазме. Слабые осно-
вания легко переходят из плазмы (pH 7,4) в мочу
(pH 5) в виде неионизированных частиц В, где пре-
вращаются в ионизированную форму ВН+ и быстро
выводятся из организма (см. рис. 1.3).
Теоретически, изменяя pH мочи, можно увели-
чить долю слабых кислот (рКа = 3,0-7,5) и слабых
оснований (рКа = 7,5-10,5), присутствующих в
моче в ионизированной форме, тем самым усили-
вая их выведение при лекарственных отравлениях.
Однако на практике форсированный щелочной или
кислотный диурез находит весьма ограниченное
применение; метод эффективен только в тех случа-
ях, когда токсические вещества не связаны с белка-
ми (1), находятся исключительно во внеклеточной
жидкости (2) и в основном выводятся с мочой в не-
измененном виде (3). К сожалению, многие ле-
карственные препараты, обладающие свойствами
кислоты или основания, подвержены активному
метаболизму и имеют большой объем распределе-
ния, что свидетельствует о значительной секвестра-
ции в тканях. В таких условиях лишь небольшое
количество лекарственных препаратов выводится
в неизменном виде с подкисленной или защелочен-
ной мочой; кроме того, количество препарата, спо-
собного диффундировать из плазмы, относительно
невелико. Форсированный диурез — потенциально
опасная процедура, поскольку требует введения
большого количества жидкости и петлевых диуре-
тиков или маннитола для стимуляции диуреза.
Возможные осложнения форсированного диуреза
(особенно у пожилых) — отек легких и головного
мозга. Тем не менее щелочной форсированный
диурез иногда используют при отравлении салицила-
тами и фенобарбиталом. Кислотный форсированный
диурез может применяться при отравлениях амфе-
таминами, фенфлурамином и фенциклидином.
14
Глава 1
Рис. 1.3. Неионная диффузия слабого основания В (рКа = 7). Только неионизированная форма В может проникать
через клеточные мембраны; диффузионный градиент зависит от разницы pH в различных камерах и тканях. Числа
в скобках — доля (в процентах) лекарственного препарата, находящегося в форме В и ВН+ при pH 2; 7,4 и 5
ОБЛЕГЧЕННАЯ ДИФФУЗИЯ И АКТИВНЫЙ
ТРАНСПОРТ
Облегченная диффузия — это трансмембран-
ный транспорт с участием переносчиков, не тре-
бующий затрат энергии. Многие физиологически
активные молекулы поступают в клетку по концен-
трационному градиенту, но с большей скоростью,
чем можно ожидать, исходя из их размеров и рас-
творимости в липидах. Облегченная диффузия
обеспечивает всасывание некоторых простых саха-
ров, кортикостероидов, аминокислот и пиримиди-
нов из тонкого кишечника и их последующее про-
никновение через клеточные мембраны (например,
в мышцах и эритроцитах). Вероятно, облегченная
диффузия осуществляется путем связывания этих
веществ со специфическими участками (сайтами)
белков и последующими конформационными
(аллостерическими) изменениями, облегчающими
их перемещение на противоположную сторону
мембраны.
Напротив, активный транспорт требует затрат
клеточной или метаболической энергии и может
осуществляться против концентрационного гради-
ента. В некоторых случаях метаболическая энергия
непосредственно генерируется в процессе гидроли-
за АТФ (первичный активный транспорт). Однако
чаще энергия обеспечивается активным транспор-
том Na+ или зависит от электрохимического гради-
ента, создаваемого натриевым насосом —
Ма+/К+-зависимой АТФазой (вторичный актив-
ный транспорт). Принято считать, что лекарствен-
ный препарат или субстрат вначале связывается
с интегральным белком-переносчиком (например,
ионным каналом или Na+/K*-зависимой АТФазой).
Затем комплекс белок-лекарственный препарат
проникает в клетку, где лекарство высвобождается,
а белок-переносчик возвращается на наружную по-
верхность клеточной мембраны.
Системы активного транспорта характеризуются
насыщаемостью и специфичностью. Они могут ин-
гибироваться некоторыми лекарственными препа-
ратами (см. главу 4). Системы активного транспорта
играют важную роль в переносе лекарств через кле-
точные мембраны многих органов и тканей, вклю-
чая тонкий кишечник, проксимальные почечные
канальцы, желчные протоки и сосудистое сплете-
ние боковых желудочков головного мозга (табл. 1.1
и 1.2). Они часто транспортируют через мембраны
эндогенные вещества. Например, повторный захват
норадреналина окончаниями симпатических нер-
вов зависит от активного транспорта Na+.
Таблица 1.1. Некоторые лекарственные препара-
ты, элиминация которых происходит путем активно-
го транспорта в проксимальных почечных канальцах
Лекарственные препа- раты со свойствами кислоты Лекарственные препа- раты со свойствами основания
Пенициллины Дофамин
Цефалоспорины Г истамин
Салицилаты Морфин
Тиазидные диуретики Неостигмин
Фуросемид Лидокаин
Хлорпропамид Хинидин
Метотрексат Тиамин
ВСАСЫВАНИЕ, РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
15
Таблица 1.2. Некоторые лекарственные
ты, секретируемые печеночными клетками в желч-
ные протоки
Лекарственные препа- раты со свойствами кислоты Лекарственные препа- раты со свойствами основания
Амоксициллин Векуроний
Ампициллин Панкуроний
Цефалоридин Алкуроний
Сульфобромофталеин Тубокурарин
Пробенецид Гликопирролат
Рентгеноконтрастные вещества Мепензолат
Рифампицин
ЗАВИСИМОСТЬ
ФАРМАКОЛОГИЧЕСКОГО
ЭФФЕКТА ОТ КОНЦЕНТРАЦИИ
ЛЕКАРСТВЕННОГО ПРЕПАРАТА В
ПЛАЗМЕ
Чаще всего невозможно определить эффектив-
ную концентрацию лекарственного препарата в тка-
нях человека, но можно измерить его концентра-
цию в плазме. Основными факторами, определяю-
щими концентрацию лекарственного препарата
в плазме, являются всасывание, распределение,
метаболизм и экскреция. Они могут влиять на кон-
npmedlit.iMBQ^afUo лекарственного препарата
в ткани-ми-
шени, изменяя силу и продолжительность эффекта
(рис. 1.4).
Термин «биодоступность» означает, какая доля
лекарственного препарата достигает ткани-мише-
ни после введения. На практике биодоступность
обычно оценивают, сравнивая концентрации ле-
карственного препарата в плазме после внутривен-
ной инъекции и при использовании других путей
введения, предусматривающих всасывание. Для та-
кого сравнения требуется фармакокинетический
анализ повременных изменений концентрации ле-
карственного препарата в плазме (глава 2).
ПУТИ ВВЕДЕНИЯ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
Лекарственные препараты можно вводить
внутрь, п/к, в/м, в/в, путем местной инфильтрации
или аппликации тканей, а также ингаляционно.
ПРИЕМ ВНУТРЬ
Для больного прием лекарственных препаратов
внутрь очевидно является наиболее удобным и при-
емлемым. Тем не менее довольно часто больные
не соблюдают предписанный режим лечения. Неко-
торые лекарственные препараты быстро распада-
ются в кислой среде желудка (например, бензилпе-
нициллин, эритромицин), другие могут раздражать
слизистую желудка и вызывать тошноту, рвоту или
желудочное кровотечение (например, салицилаты,
фенилбутазон, концентрированные растворы
Внутрь
В/м
Растворение
Подкожно
сасывание
Белки
плазмы
Местная
Ингаляция
Местная
инфильтрация аппликация
Ткань-мишень
Вода
плазмы
В/в
Распределение Л
в тканях
Метаболические изменения
Экскреция
Экскреция
Желчь Моча
Моча
Рис. 1.4. Связь между всасыванием, распределением, метаболизмом, экскрецией и концентрацией лекарственных
препаратов в ткани-мишени
16
Глава 1
большинства солей). Этих осложнений
бежать, если использовать таблетки, которые по-
крыты специальной оболочкой (препятствующей
их растворению в кислом желудочном содержи-
мом), или же лекарственные формы с медленно вы-
свобождающимся активным веществом.
После приема внутрь максимальная концентра-
ция лекарственного препарата в плазме или фарма-
кологический эффект достигаются лишь через
30-120 мин. Достижение адекватной концентрации
препарата в плазме зависит от растворимости (1),
всасываемости (2) и отсутствия выраженного эф-
фекта первичного прохождения через стенку кишки
или печень (3).
РАСТВОРЕНИЕ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ ПРИ ПРИЕМЕ ВНУТРЬ
Необходимым условием для всасывания лекар-
ственного препарата является растворение приня-
той внутрь таблетки или капсулы. Растворение
обычно происходит в желудке и зависит от кислот-
ности желудочного сока. Скорость растворения, а
также скорость и характер опорожнения желудка
влияют на количество растворенного лекарствен-
ного препарата, достигшего проксимальной части
тонкой кишки (где в основном происходит всасы-
вание).
Растворимость таблеток и капсул зависит от
многих факторов, включая размеры частиц, хими-
ческую формулу, состав внутренних инертных на-
полнителей и наружных оболочек. Оригинальные
и генерические формы одного и того же лекарствен-
ного препарата могут иметь разную растворимость,
поэтому их концентрация в плазме после приема
внутрь может отличаться. В свое время путем кли-
нических наблюдений были выявлены различия в
мощности между таблетками дигоксина разных
производителей; причиной этого явилась как раз
неодинаковая растворимость препаратов. Таблетки
фенилгидантоина (фенитоина) также стали прояв-
лять токсические свойства после того, как наполни-
тель сульфат кальция заменили лактозой. Препа-
рат стал быстрее растворяться, что привело к уско-
рению и повышению всасывания, и, соответственно,
увеличению концентрации в плазме.
Удобнее и безопаснее использовать высвобож-
дающиеся с постоянной скоростью лекарственные
препараты; их биодоступность выше, а разброс
плазменной концентрации меньше. К другим пре-
имуществам относится снижение выраженности
побочных эффектов, а также уменьшение дозы, час-
тоты приема и стоимости лечения.
Постепенно высвобождающиеся лекарственные
препараты для приема внутрь обычно состоят из
и®@2оЭДфйных распадающихся полимеров; фикси-
рованные дозы таких препаратов высвобождаются
через равные промежутки времени. Некоторые ле-
карственные формы предназначены для медленно-
го непрерывного высвобождения; они могут быть
осмотически активными или включать ионообмен-
ные смолы, что позволяет лекарственному вещест-
ву высвобождаться в водной среде с соответствую-
щей концентрацией ионов и pH. Таким способом
можно вводить многие лекарственные препараты,
включая некоторые опиоидные анальгетики, несте-
роидные противовоспалительные средства, брон-
ходилататоры, гипотензивные и антиаритмические
препараты, а также соли калия.
ВСАСЫВАНИЕ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ
Всасывание лекарственных препаратов в желуд-
ке и тонкой кишке зависит от способности прони-
кать через липидные клеточные мембраны. Следо-
вательно, скорость и степень всасывания лекарст-
венного препарата в основном определяются его
физико-химическими свойствами. Неионизиро-
ванные вещества (например, этанол) и вещества
с низким молекулярным весом (например, моче-
вина) легко преодолевают клеточные мембраны
путем пассивной диффузии и быстро всасываются
в ЖКТ. Препараты, являющиеся слабыми кислота-
ми (например, аспирин), в кислой среде преимуще-
ственно не ионизированы и хорошо растворимы
в липидах; поэтому они лучше всасываются в же-
лудке, чем в тонкой кишке. Напротив, лекарствен-
ные препараты, обладающие основными свойства-
ми (пропранолол, большинство бензодиазепинов)
меньше ионизированы в щелочной среде; поэтому
они преимущественно всасываются в двенадцати-
перстной кишке (pH 5-6). Сильные основания
(четвертичные амины) всегда ионизированы в рас-
творе и поэтому плохо всасываются в ЖКТ.
В реальности на зону всасывания лекарствен-
ных препаратов влияют многие факторы. Площадь
поверхности слизистой верхних отделов тонкой
кишки больше, чем желудка, и большинство лекар-
ственных препаратов (обладающих как кислот-
ными, так и основными свойствами) преимущест-
венно всасывается в двенадцатиперстной кишке.
Тем не менее неионизированные соединения и ле-
карственные препараты со свойствами кислоты
в значительной степени всасываются в желудке, что
может быстро повышать их концентрацию в плазме
после приема внутрь.
Соединения, влияющие на моторику желудка,
могут также влиять на растворимость и скорость
ВСАСЫВАНИЕ, РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
17
(но не степень) всасывания лекарственньрсщепа| ПОДКОЖНОЕ И ВНУТРИМЫШЕЧНОЕ
ратов. В частности, препараты, снижаюпще^ери^-^ВтЗДЕНИЕ
стальтику и замедляющие опорожнение желудка
(например, атропин и морфин), уменьшают ско-
рость всасывания лекарств. Взаимодействие лекар-
ственных препаратов (например, тетрациклинов
и препаратов железа, или холестирамина и дигок-
сина) может влиять на степень всасывания и изме-
нять общую биодоступность. Всасывание лекарст-
венных препаратов может замедляться при заболе-
ваниях ЖКТ, особенно целиакии, болезни Крона,
механической желтухе или обширной резекции
тонкой кишки.
Хотя в желудке и тонкой кишке большинство
лекарственных препаратов всасывается путем пас-
сивной диффузии, иногда всасывание происходит
при участии мембранных переносчиков. Так, лево-
дофа всасывается с помощью переносчика, обычно
отвечающего за транспорт аминокислот, а фторура-
цил — при участии переносчика пиримидиновых
оснований.
ЭФФЕКТ ПЕРВОГО ПРОХОЖДЕНИЯ
Чтобы оказать фармакологическое действие, ле-
карственные препараты после приема внутрь и вса-
сывания должны пройти через слизистую оболочку
тонкой кишки и печень. Некоторые лекарства под-
вергаются метаболическим превращениям в ки-
шечной стенке (хлорпромазин, дофамин, изопроте-
ренол) или печени (лидокаин, петидин), прежде
чем попасть в системное кровообращение (эффект
первого прохождения). В таких случаях прием
препарата внутрь не всегда позволяет добиться аде-
кватной концентрации в плазме или воспроизводи-
мого фармакологического эффекта. Эффект перво-
го прохождения через печень больше выражен у ле-
карств с высоким коэффициентом печеночной
экстракции (концентрация препарата в печеночной
вене составляет менее 50% концентрации в ворот-
ной вене). В таких случаях печеночный клиренс
препарата в основном ограничен печеночным
кровотоком, а не активностью метаболических фер-
ментов. Препараты, снижающие печеночный кро-
воток (например, пропранолол), влияют на величи-
ну эффекта первого прохождения через печень.
Большинство лекарств, принимаемых внутрь,
имеют низкий коэффициент печеночной экстрак-
ции и значимость эффекта первого прохождения
через печень у них невелика, поэтому их печеноч-
ный клиренс больше зависит от активности фер-
ментов, чем от печеночного кровотока. Иногда во
избежание эффекта первого прохождения лекарст-
венные препараты назначают под язык или рек-
тально.
Некоторые препараты, которые не достигают
адекватной концентрации в плазме или не оказыва-
ют фармакологического действия после приема
внутрь, целесообразно вводить подкожно (п/к) или
внутримышечно (в/м). Особенно это касается пре-
паратов, которые распадаются в ЖКТ (бензилпе-
нициллин, полипептидные гормоны), плохо или
непредсказуемо всасываются (аминогликозиды,
многие четвертичные амины), или в значительной
мере подвержены эффекту первого прохождения
через печень (опиоидные анальгетики). Иногда
препараты назначают в/м, если при приеме внутрь
они плохо переносятся (например, соли железа)
или больные не способны полноценно выполнять
предписания (например, при шизофрении).
Всасывание лекарственных препаратов при вве-
дении п/к или в/м обычно не зависит от константы
диссоциации или pH, но часто определяется регио-
нарным кровотоком. При в/м введении препараты
действуют быстрее и короче, чем при п/к, что объ-
ясняется различиями в перфузии мышц и подкож-
ной клетчатки. Иногда плохорастворимые препа-
раты или лекарственные комплексы вводят п/к,
чтобы замедлить всасывание и увеличить продол-
жительность действия (например, инсулин или пе-
нициллин). В таких случаях высвобождение лекар-
ственного вещества из комплекса и его последую-
щее всасывание определяют продолжительность
действия.
Для заместительной гормональной терапии
используются подкожные имплантаты. По-види-
мому, популярность этой лекарственной формы
будет возрастать (например, для инсулина и других
полипептидных гормонов). Разрабатываются сис-
темы регулируемого высвобождения, способные
ускорять поступление лекарственного средства по
требованию (например, с помощью внешнего маг-
нитного поля, ультразвука или ферментов).
ВНУТРИВЕННОЕ ВВЕДЕНИЕ
Лекарственные средства вводят в/в, если тре-
буется быстрое или немедленное начало действия.
Такой способ введения обеспечивает надежный и
воспроизводимый эффект, позволяет использовать
дозы, точно соответствующие желаемому резуль-
тату, а также свести к минимуму индивидуальные
различия в реакции на лекарственный препарат.
Большинство препаратов можно вводить в/в струй-
но; но некоторые (например, аминофиллин) нужно
вводить медленно во избежание сердечных ослож-
нений, обусловленных нарастанием концентрации
18
Глава 1
препарата в плазме. Раздражающие вещЩ@(}д4-. (JCJOZ3.FtlIcllb- Разработаны защечные формы нитро-
дятся в/в, чтобы не допустить местного поврежде-
ния тканей и сосудов. После в/в введения некото-
рых лекарственных препаратов (диазепам) может
развиться местная реакция в виде поверхностного
тромбофлебита. Неизвестно, связано ли это ослож-
нение с pH раствора или с какими-либо другими
факторами. После введения препаратов, высвобож-
дающих гистамин из тучных клеток (например, ту-
бокурарин, петидин), может развиться локальная
вазодилатация, проявляющаяся покраснением,
припухлостью и отеком окружающих тканей.
Разработка инфузионных насосов для доставки
лекарственных препаратов особенно важна для ле-
чения боли. Некоторые из этих приборов снабжены
электронными устройствами, обеспечивающими
струйное введение лекарств «по требованию». Грави-
тационные методы и баллонные резервуары — это
механические приспособления, позволяющие дос-
таточно точно регулировать введение лекарствен-
ных препаратов. Портативные шприцевые насосы,
работающие на батарейках, используются для не-
прерывного введения опиоидов, особенно при лече-
нии на дому стойких болевых синдромов, вызван-
ных злокачественными заболеваниями.
Желательно, чтобы системы доставки обеспечи-
вали селективное распределение лекарственных
препаратов. Это позволит избежать попадания ле-
карственных препаратов в зоны быстрой инактива-
ции или наиболее чувствительные к токсическому
воздействию. Разрабатываются системы микрочас-
тиц-переносчиков (липосомы, эритроциты, микро-
сферы), которые можно широко использовать для
лечения инфекционных и онкологических заболе-
ваний. Точно так же соединения лекарственных
препаратов с антителами могут быть полезны при
лечении злокачественных опухолей, а более широ-
кое использование «пролекарств» повышает специ-
фичность доставки.
ДРУГИЕ ПУТИ ВВЕДЕНИЯ
Лекарственные формы препаратов для местного
применения (например, антибиотики, кортикосте-
роиды, различные местные анестетики) наносят на
слизистые носоглотки, гортани, мочеполового
тракта, на конъюнктиву. Слизистая хорошо васку-
ляризована, всасывание лекарственных препаратов
с нее происходит быстро, поэтому, например, мест-
ные анестетики могут вызывать системные токси-
ческие реакции.
Иногда лекарственные препараты наносят на сли-
зистые, чтобы быстрее достигнуть системного дей-
ствия или избежать эффекта первого прохождения
глицерина, гиосцина и прохлорперазина; эти таб-
летки закладываются между десной и верхней
губой. Для лечения послеоперационной боли при-
меняются пропитанные опиоидами «леденцы».
Некоторые гипоталамические и гипофизарные
полипептидные гормоны, которые разрушаются
в ЖКТ, вводят через слизистую носа. Интрана-
зально можно вводить опиоиды, кортикостероиды,
антигистаминные препараты, пропранолол и вита-
мин В12- Этот метод введения обеспечивает быстрое
и полное всасывание лекарственного препарата
через слизистую носа в ЦСЖ и сосуды мозга, по-
скольку подслизистое пространство носовой по-
лости непосредственно сообщается с субарахнои-
дальным пространством области обонятельных
долей головного мозга. После интраназального
введения концентрация лекарственных препара-
тов в ЦСЖ может быть выше, чем в плазме.
Большинство лекарственных препаратов, осо-
бенно полярных веществ, плохо всасывается через
неповрежденную кожу. Основным барьером для
всасывания является роговой слой эпидермиса, в том
числе межклеточные липиды этого слоя. Тем не ме-
нее некоторые сильнодействующие лекарственные
препараты, обладающие высокой растворимостью
в липидах (например, нитроглицерин, гиосцин,
этинил эстрад иол, фентанил), всасываются через
кожу и могут оказывать системное действие. В та-
ких случаях роговой слой кожи служит резервуа-
ром жирорастворимых препаратов еще в течение
нескольких дней после их отмены. На проникнове-
ние через кожу могут влиять формообразующие ве-
щества (так называемые основы) и различные уси-
лители всасывания (например, диметилсульфок-
сид, диметилацетамид, N-метилпирролидон).
Местные анестетики (смесь лидокаина с при-
локаином, тетракаин) применяются для обезбо-
ливания при венепункции. Чтобы обеспечить
анальгезию, эти препараты после нанесения на
кожу необходимо закрыть повязкой по крайней
мере на 45 мин.
Местные анестетики можно вводить непосред-
ственно в ткани (инфильтрационная анестезия),
так что область инъекции отграничивает зону их
действия. При этом местные анестетики комбини-
руют с вазоконстрикторами, чтобы уменьшить вса-
сывание в системный кровоток и увеличить про-
должительность анестезии. Для обеспечения про-
водниковой анестезии местные анестетики
вводятся в различные участки около нервов или
нервных сплетений. Кроме того, местные анестети-
ки, анальгетики и даже антибиотики можно вво-
дить интратекально.
ВСАСЫВАНИЕ, РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
19
medlit.
Поглощение и распределение ингаляционных
анестетиков зависит от их проникновения в легоч-
ные капилляры через альвеолы. На скорость этого
процесса влияют многие факторы, включая кон-
центрацию анестетика во вдыхаемой смеси, адекват-
ность легочной вентиляции, растворимость в липи-
дах и коэффициент распределения кровь/газ.
Кортикостероиды и некоторые бронходилата-
торы могут назначать путем ингаляции, чтобы
обеспечить местное воздействие на респиратор-
ные бронхиолы и избежать системных эффектов.
На распределение лекарственного препарата влия-
ет размер частиц аэрозоля. Обычно частицы диа-
метром больше 10 мкм оседают в верхних дыхатель-
ных путях, частицы диаметром 2—10 мкм — в брон-
хиолах, а частицы менее 2 мкм достигают альвеол.
РАСПРЕДЕЛЕНИЕ
После введения и всасывания лекарственные
препараты попадают в плазму, и часть молекул свя-
зывается с белками. Затем они проникают в другие
ткани и органы, и по окончании распределения кон-
центрации препаратов в плазме и внеклеточной
жидкости выравниваются.
Распределение различных лекарственных препа-
ратов в организме очень сильно различается. Распре-
деление оценивают, проводя исследования на жи-
вотных или применяя фармакокинетические мето-
ды у человека. Некоторые препараты (варфарин,
толбутамид) в значительной степени связываются
с белками и преимущественно остаются в плазме.
Ионизированные соединения (литий, большинст-
во четвертичных аминов) плохо проникают через
клеточные мембраны, и их распределение в основ-
ном ограничено внеклеточной жидкостью. Эти пре-
параты плохо проникают в ткани, то есть харак-
теризуются низким объемом распределения (9-20 л
у взрослых). Напротив, жирорастворимые препа-
раты с относительно низким молекулярным весом
хорошо проникают в ткани. Например, этанол, мо-
чевина и некоторые сульфаниламиды равномерно
распределяются в водной среде организма, и их
объем распределения равен общему объему воды в
организме (30-45 л у взрослых). Другие препараты
проникают через клеточные мембраны и активно
связываются с тканевыми белками или секвести-
руются в жировой ткани (например, морфин, тио-
пентал, дигоксин); в таких случаях объем распре-
деления превышает общий объем воды организма
(>30-45 л).
После в/в введения некоторые лекарствен-
ные препараты вначале попадают в хорошо крово-
снабжаемые ткани, а затем по мере уменьшения
ucoz.ru
концентрации в плазме постепенно перераспреде-
ляются в другие структуры. Приблизительно 25%
введенного тиопентала и метогекситала вначале
поступает в головной мозг благодаря мощному моз-
говому кровотоку и хорошей растворимости барби-
туратов в липидах. По мере снижения концентра-
ции в плазме тиопентал и метогекситал поступают
в хуже кровоснабжаемые ткани с повышенным
сродством к этим соединениям (например, в мы-
шечную и жировую). Вследствие этого барбитура-
ты быстро перераспределяются из головного мозга
в мышцы, а потом в подкожно-жировую клетчатку.
Перераспределение является главной причиной
короткого действия этих препаратов, хотя они мо-
гут задерживаться в организме в течение 24 ч.
Некоторые лекарственные препараты обладают
способностью накапливаться в определенных тка-
нях или органах: например, сульфабромфталеин —
в печени, гуанетидин — в постганглионарных сим-
патических окончаниях, йод — в щитовидной же-
лезе, тетрациклины — в растущих зубах и костях.
Концентрация лекарственных препаратов в этих
тканях может значительно превышать их концен-
трацию в плазме. Хорошо проникающие в ткани
и накапливающиеся в клетках лекарственные пре-
параты имеют очень большой объем распределе-
ния, который обычно превышает общий объем
воды в организме (например, фенотиазины, три-
циклические антидепрессанты).
ГЕМАТОЭНЦЕФАЛИЧЕСКИЙ БАРЬЕР
Многие лекарственные препараты, несмотря
на хорошее распределение в различных тканях, не
могут проникнуть в ЦНС. В капиллярах головного
мозга клетки эндотелия соединены друг с другом
посредством непрерывных плотных контактов,
ограничивающих пассивную диффузию, а пиноци-
тозные пузырьки обычно отсутствуют. Окружаю-
щая капилляры базальная мембрана плотно приле-
гает к периферическим отросткам астроцитов
(клетки нейроглии, которые играют важную роль
в питании нейронов). Чтобы попасть из капилля-
ров в головной мозг, большинству препаратов надо
пройти через эндотелий, базальную мембрану и пе-
риферические отростки астроцитов путем простой
диффузии или фильтрации. Некоторые препараты
не способны преодолеть эти структуры, обозначае-
мые собирательным термином «гематоэнцефали-
ческий барьер».
Кроме этого морфологического барьера сущест-
вует также метаболический или ферментативный
гематоэнцефалический барьер, который преимуще-
ственно создается периферическими отростками
астроцитов. Многие потенциально нейротоксичные
20
Глава 1
вещества (например, свободные жирные к
аммиак) могут без труда преодолевать капилляр-
ный эндотелий, но подвергаются расщеплению,
прежде чем достигают ЦНС. В эндотелии капилля-
ров также присутствуют моноаминоксидаза и холи-
нэстераза, и некоторые лекарственные препараты
(например, норадреналин, дофамин, местные ане-
стетики эфирного типа, 5-гидрокситриптамин) при
переходе через гематоэнцефалический барьер мета-
болизируются.
Гематоэнцефалический барьер — это не просто
пассивная и неизменная структура, а динамическая
мембранная поверхность между кровью и голов-
ным мозгом. Структура и функция этой мембраны
зависят от трофических факторов, выделяемых аст-
роцитами. Гематоэнцефалический барьер форми-
руется в течение первого триместра беременности,
но к моменту рождения остается еще незрелым и
может пропускать слаборастворимые в липидах ле-
карственные препараты и эндогенные вещества.
Некоторые метаболически активные вещества и
гормоны (глюкоза, инсулин, L-аминокислоты,
L-тироксин, трансферрин) проникают через гема-
тоэнцефалический барьер посредством эндоцитоза
или с помощью переносчиков. Кроме того, низко-
молекулярные жирорастворимые лекарственные
препараты (общие и местные анестетики, опиоиды)
легко проникают через гематоэнцефалический
барьер в ЦНС. Если препараты активно связывают-
ся с белками плазмы (толбутамид, варфарин), то ге-
матоэнцефалический барьер способны преодоле-
вать только их свободные фракции, и концентрация
этих препаратов в головном мозге составляет 1-2%
от их общей концентрации в плазме. Высокоиони-
зированные препараты (например, четвертичные
амины) не проникают через гематоэнцефаличе-
ский барьер; следовательно, миорелаксанты не ока-
зывают влияния на головной мозг. Связывающиеся
с белками красители (синька Эванса) и препараты
с большим молекулярным весом (циклоспорин)
плохо проникают через гематоэнцефалический
барьер. Некоторые лекарства (бензилпенициллин)
могут попадать в головной мозг при воспалении
(например, бактериальном менингите), когда про-
ницаемость гематоэнцефалического барьера повы-
шается. Нормальная непроницаемость гематоэнце-
фалического барьера может нарушаться при вос-
палении, отеке мозга, острой или хронической
внутричерепной гипертензии.
В некоторых участках головного мозга (напри-
мер, zona postrema, срединное возвышение, шишко-
видная железа и сосудистые сплетения желудочков
мозга) гематоэнцефалический барьер неполноце-
нен или полностью отсутствует. В этих участках
лекарственных препаратов и обмен эн-
догенных веществ ничем не ограничены. В сосуди-
стом сплетении лекарственные препараты могут
свободно проникать из капилляров в ЦСЖ через
относительно проницаемый эпителий сосудистого
сплетения. Эпендима, выстилающая мозговые
желудочки, также не ограничивает диффузию
большинства лекарственных препаратов. Нейро-
пептиды и некоторые ионизированные соединения
(бензилпенициллин, пробенецид) могут активно
секретироваться в противоположном направлении,
то есть из желудочков мозга в капилляры.
ПЛАЦЕНТА
На поздних сроках беременности происходят
структурные изменения плаценты, заключающие-
ся в постепенном исчезновении цитотрофобласта
и хориональной соединительной ткани из ворсин
плаценты. К моменту родов материнская кровь
и кровь плода разъединены одним слоем хориона
(синцитиотрофобластом), который непрерывно
связан с эндотелиальными клетками фетальных
капилляров. Следовательно, плацентарный барьер
состоит из сосудисто-синцитиальной оболочки,
которая с функциональной точки зрения является
типичной липидной мембраной. Через плаценту
легко проникает большинство низкомолекулярных
жирорастворимых лекарственных препаратов;
скорость их поступления в кровь плода зависит от
плацентарного кровотока, площади диффузии и эф-
фективного диффузионного градиента. Напротив,
полярные молекулы с высоким молекулярным
весом плохо проникают через плацентарный барь-
ер. Почти все лекарственные препараты, способные
преодолевать гематоэнцефалический барьер и воз-
действовать на ЦНС, преодолевают и плацентарный
барьер, а их элиминация из тканей плода затрудне-
на и замедлена.
Известно, что во время беременности некоторые
лекарственные препараты, хорошо проникающие
через плаценту, могут вызывать аномалии у плода.
К ним относятся цитотоксические препараты,
антагонисты фолиевой кислоты, фенитоин, эстро-
гены, прогестагены, аминогликозидные антибио-
тики, тетрациклины, антитиреоидные препараты.
Ингаляционные анестетики, в/в барбитураты,
местные анестетики и многие анальгетики (вклю-
чая морфин и петидин) могут проникать из плазмы
матери в кровь плода и вызывать осложнения
в родах. Преодолевать плацентарный барьер и вы-
зывать у плода брадикардию и гипогликемию могут
p-адреноблокаторы (например, пропранолол). Если
на поздних сроках беременности для лечения эк-
лампсии или преэклампсии применяется диазепам,
ВСАСЫВАНИЕ, РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
21
он легко проникает через плаценту и не
расщепляется в организме плода; накопле0иДМ>р-^
ганизме плода некоторых активных метаболитов
диазепама, таких как десметилдиазепам или окса-
зепам, может привести к артериальной гипотонии
и гипотермии у новорожденного. Напротив, иони-
зированные соединения (например, миорелаксанты)
плохо проникают через плаценту, и их введениие
матери редко вызывает осложнения у плода на
поздних сроках беременности (а также у новорож-
денного во время лактации).
СВЯЗЫВАНИЕ С БЕЛКАМИ
Связывание с белками — один из способов быст-
рого распределения лекарственных препаратов из
области всасывания к ткани-мишени. Связывание
с белками критически важно для транспортировки
лекарственных средств с высокой жирораствори-
мостью, поскольку они плохо растворяются в плаз-
ме. Когда кровь поступает в ткань, то концентрация
любого свободного (не связанного с белком) препа-
рата в плазме падает, что приводит к диссоциации
комплексов белок-препарат. Таким образом гене-
рируется постоянный концентрационный гради-
ент, обеспечивающий диффузию лекарств из плазмы
в ткани. Концентрацию свободной фракции лекар-
ственного препарата в плазме можно определить,
измерив его концентрацию в слюне или ЦСЖ, а так-
же in vitro — с помощью равновесного диализа, ульт-
рацентрифугирования или ультрафильтрации.
Главным белком, участвующим в связывании
лекарственных препаратов, является альбумин.
Этот белок имеет ряд участков связывания со срод-
ством к различным лекарственным препаратам.
Альбумин в основном связывает нейтральные или
кислые вещества, в том числе салицилаты, фенил-
бутазон, индометацин, толбутамид, карбеноксолон
и пероральные антикоагулянты. Альбумин также
связывает некоторые лекарственные препараты со
свойствами основания и физиологические субстра-
ты (билирубин, жирные кислоты, триптофан).
Глобулины связывают многие препараты со ще-
лочными свойствами (например, хлорпромазин,
бупивакаин, опиоиды). Чаще всего в роли белка-пе-
реносчика оснований выступает кислый агглико-
протеин. Глобулины плазмы играют важную роль в
связывании минералов, витаминов и гормонов.
Гидрокортизон (кортизол) в плазме чаще всего пе-
реносится специализированным глобулином
(транскортином), имеющим высокое сродство к кор-
тикостероидам. Некоторые лекарственные препа-
раты (тубокурарин, панкуроний) связываются как
с альбумином, так и с иммуноглобулином (IgG).
Резистентность к миорелаксантам, которая иногда
.ОДМеяается при заболеваниях печени, обычно обу-
1сЯовяенА4ювышенным связыванием этих препара-
тов глобулинами плазмы. Препараты, в значитель-
ной степени связывающиеся с белками, могут
конкурировать и вытеснять друг друга из соедине-
ний с альбумином.
Печеночный клиренс многих лекарственных
препаратов ограничивается не связыванием с бел-
ками плазмы, а печеночным кровотоком. Диссоциа-
ция комплекса белок-препарат происходит в тече-
ние микро- или миллисекунд, тогда как перфузия
печени занимает несколько секунд. Таким образом,
снижение печеночного клиренса возможно только
том случае, если препарат в значительной степени
связан с белками, а способность печени к метабо-
лизму или экскреции препарата снижена. Малове-
роятно, чтобы связь с белками нарушила элимина-
цию препаратов почечными клубочками и каналь-
цами. Через проксимальные почечные канальцы
выделяются только несвязанные препараты, но по-
следующее снижение концентрации препаратов
в плазме немедленно приводит к диссоциации
комплексов белок-препарат для поддержания рав-
новесия. Многие связанные с белками препараты
полностью выводятся через почки за один пассаж
(например, бензилпенициллин).
Связывание лекарственных препаратов с белка-
ми плазмы нарушается при патологических состоя-
ниях, вызывающих гипоальбуминемию (цирроз пе-
чени, нефротический синдром, травмы, ожоги).
При этом концентрация свободной фракции препа-
рата увеличивается и может привести к токсическо-
му эффекту (например, при лечении фенитоином
или преднизолоном). Значительные изменения
особенно вероятны, если препараты применяют
в больших дозах или в/в. В таких случаях связыва-
ние с альбумином или другими белками быстро
достигает насыщения, что приводит к непропор-
циональному повышению концентрации свобод-
ной фракции препарата. В хорошо кровоснабжае-
мые ткани и органы (головной мозг, миокард,
печень) могут поступить непропорционально вы-
сокие дозы лекарственных препаратов, что чревато
передозировкой. Аналогичные случаи передози-
ровки возникают у пожилых, а также при дисфунк-
ции почек и уремии, что может быть обусловлено
нарушением сродства лекарственных средств к аль-
бумину. При некоторых заболеваниях (инфаркт
миокарда, ревматоидный артрит, болезнь Крона,
почечная недостаточность, злокачественные ново-
образования), а также после хирургических вмеша-
тельств может повыситься концентрация кислого
ai-гликопротеина плазмы, что приводит к увеличе-
нию степени связывания препаратов со свойствами
22
Глава 1
основания (например, пропранолол, хл
зин); соответственно, концентрация свободной
фракции этих препаратов снижается.
МЕТАБОЛИЗМ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ
Чаще всего действие лекарственного препарата
заканчивается благодаря его метаболическим пре-
вращениям в печени. В результате метаболизма
концентрация активного препарата в плазме сни-
жается, что способствует его диффузии из тканей
(в том числе из ткани-мишени) в кровь. Основное
предназначение печеночного метаболизма — пре-
вращение жирорастворимых лекарственных препа-
ратов в водорастворимые (полярные) соединения,
которые могут фильтроваться в почечных клубоч-
ках или секретироваться с мочой или желчью.
Большинство препаратов метаболизируется в пе-
чени, но некоторые (например, сукцинилхолин,
мивакурий, эсмолол, ремифентанил) гидролизуют-
ся эстеразами плазмы. Многие лекарства частично
или полностью метаболизируются в других тканях,
например, в кишке (хлорпромазин, изопреналин),
почках (мидазолам, дофамин) или легких (ангио-
тензин I, прилокаин). Тем не менее именно печень
играет главную роль в метаболизме лекарств.
После приема внутрь препараты поступают
в кровь воротной вены, а затем могут в значительной
мере удаляться из печеночных синусоидов и подвер-
гаться метаболизму в печени, прежде чем попасть
в системный кровоток. Этот феномен (эффект пер-
вого прохождения) — важная причина неэффектив-
ности принятых внутрь препаратов у некоторых
больных. После приема внутрь значительная доля
лидокаина, пропранолола и большинства опиоидов
переходит из воротной вены в печеночные синусои-
ды; для всех них характерен выраженный эффект
первого прохождения, и клиренс в основном зависит
от печеночного кровотока. Напротив, элиминация
других препаратов (варфарина, толбутамида) опре-
деляется не печеночным кровотоком, а метаболиче-
ской активностью печени.
Как правило, метаболизм лекарственных препа-
ратов обычно снижает их биологическую актив-
ность. Большинство метаболитов менее активны,
чем исходные вещества. Кроме того, способность
метаболитов проникать к рецепторам в ткани-ми-
шени ограничена вследствие более высокой поляр-
ности и меньшей растворимости в жирах. С другой
стороны, некоторые препараты, наоборот, относи-
тельно неактивны в исходной форме, и для прояв-
ления или усиления фармакологического эффекта
требуются метаболические превращения (табл. 1.3).
а 1.3. Лекарственные препараты, кото-
рым для оказания фармакологического действия
требуются метаболические превращения
Лекарственный препа- рат Активный метаболит
Пронтозил красный Сульфаниламид
Хлоралгидрат Трихлорэтанол
Циклофосфамид Фосфорамидный азоти- стый иприт
Преднизон Преднизолон
Кортизон Г идрокортизон
Метилдофа Метилнорадреналин
Прогуанил Циклогуанил
Эналаприл Эналаприлат
Иногда фармакологический эффект продуктов ме-
таболизма значительно отличается от спектра дей-
ствия препаратов-предшественников (петидин,
атракурий). Некоторые антибиотики (ампицил-
лин, хлорамфеникол) назначают внутрь в виде эфи-
ра, потому что так они лучше всасываются, после
чего гидролизуются до активных компонентов.
Иногда метаболизм приводит к образованию со-
единений, обладающих токсическим действием.
Например, парацетамол частично превращается
в JV-ацетил-п-бензохинонимин; если этот высоко-
активный метаболит не подвергается быстрой
конъюгации в печени, то он алкилирует макромоле-
кулы гепатоцитов и вызывает их некроз. Метаболи-
ты галотана тоже обладают способностью кова-
лентно связываться с макромолекулами, вызывая
повреждение гепатоцитов. Существуют веские до-
казательства, что расщепление галотана до реак-
тивных промежуточных метаболитов играет важ-
ную роль в развитии так называемого «галотаново-
го гепатита».
Выделяют две фазы метаболизма лекарствен-
ных средств. Реакции первой фазы состоят в окис-
лении, восстановлении или гидролизе лекарствен-
ных препаратов (таблица 1.4). В ходе второй фазы
образуется ковалентная связь между лекарствен-
ным препаратом (или продуктом реакции первой
фазы) и эндогенным соединением (глюкуроновая
кислота, сульфат, ацетат, глутатион). Реакции пер-
вой и второй фазы повышают растворимость ле-
карств или их метаболитов в воде, ускоряя их эли-
минацию. Метаболизм некоторых препаратов (на-
пример, салицилат натрия) почти полностью
протекает в ходе второй фазы.
ВСАСЫВАНИЕ, РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
23
ению, восстановлению и гидролизу ле-
Таблица 1.4. Типичные реакции первой фазыуЯаивааяимеуММТ^
царственных препаратов IIIcUIll.UUUZT. I U
Реакция Локализация реакции Фермент Пример
Окисление Эндоплазматический ре- тикулум гепатоцитов Цитохром Р-450 Галотан -»трифторуксус- ная кислота Тиопентал -> пентобар- битал
Митохондрии Моноаминоксидаза Дофамин -> дигидрокси- фенилацетальдегид
Цитоплазма гепатоцитов Алкогольдегидрогеназа Этанол -> ацетальдегид
Восстановление Эндоплазматический ре- тикулум гепатоцитов Цитохром Р-450 Пронтозил -> сульфани- ламид
Цитоплазма гепатоцитов Алкогольдегидрогенаэа Хлоралгидрат -»трихлор- этанол
Г идролиз Эндоплазматический ре- тикулум гепатоцитов Эстераза Петидин -> петидиновая кислота
Плазма Холинэстераза Сукцинилхолин -> сукци- нат + холин
Нервно-мышечный си- напс Ацетилхолинэстераза Ацетилхолин -► ацетат + холин
РЕАКЦИИ ПЕРВОЙ ФАЗЫ
Большинство реакций первой фазы и конъюга-
ция с глюкуроновой кислотой осуществляются в
гладком эндоплазматическом ретикулуме гепато-
цитов (рис. 1.5). В ходе ультра центрифуги рован ня
ткани печени эндоплазматический ретикулум отде-
ляется от других органелл клетки и разрушается,
а обломки его мембран образуют мелкие гранулы —
так называемые микросомы. Неспецифические
Рис. 1.5. Электронная микрофотография участка пече-
ночной клетки мыши с изображением митохондрий
(М),эндоплазматического ретикулума (ER) и окружаю-
щей ядро клетки (N) ядерной мембраны; (х 30 000)
24
Глава 1
ферментные системы эндоплазматическое
лума, которые называют цитохромом Р-450 или
«системой оксидаз смешанного типа», катализиру-
ют большинство окислительных и восстановитель-
ных реакций, а также некоторые реакции гидролиза.
Цитохром Р-450 представляет собой суперсе-
мейство генетически родственных гем-тиолатных
белков, которые являются конечным звеном в сис-
теме оксидаз смешанного типа. Его название проис-
ходит от способности в восстановленном состоя-
нии связывать СО и образовывать комплексы,
которые максимально поглощают световые волны
длиной 450 нм.
В ходе окисления окисленная форма цитохрома
Р-450 соединяется с лекарственным препаратом
или другим субстратом. Затем образовавшийся
комплекс восстанавливается под воздействием вто-
рого мембранного фермента (цитохром Р-450-ре-
дуктазы), что влечет за собой перенос электрона
с НАДФ-Н. После этого происходит реакция с мо-
лекулярным кислородом и присоединение второго
электрона от НАДФ-Н. В итоге один атом кислоро-
да высвобождается в виде Н2О, а второй переходит
к субстрату. Затем окисленный метаболит высво-
бождается, и цитохром Р-450 регенерирует в окис-
ленную форму (рис. 1.6).
Ферменты цитохрома Р-450 могут также участ-
вовать в восстановительном метаболизме некото-
рых препаратов (галотан, пронтозил, хлорамфени-
кол; табл. 1.4). Этот процесс зависит от способности
препаратов акцептировать электроны непосредст-
венно от комплекса «восстановленный цитохром
Р-450-лекарство» (рис. 1.6); он усиливается при ги-
поксии.
У человека идентифицировано множество изо-
ферментов цитохрома Р-450. Обычно они классифи-
цируются на семейства и подсемейства по сходству
аминокислотных последовательностей (табл. 1.5).
Члены каждого семейства (CYP 1, CYP 2 и т.д.)
имеют 40% и более общих аминокислотных после-
довательностей, а члены каждого подсемейства
( CYP 1 A, CYP 1В и т.д.)—более 55% общих аминокис-
лотных последовательностей. У млекопитающих су-
ществует 14 различных семейств; шесть из них
(CYP 7, CYP 11, CYP17, CYP19, CYP 21 и CYP 27)
осуществляют исключительно синтез стероидных
гормонов, желчных кислот и холестерина и не уча-
ствуют в метаболизме лекарственных препаратов.
Семейства CYP 1, CYP 2 и CYP 3 составляют 70%
всего печеночного цитохрома Р-450 и отвечают за
большинство реакций первой фазы у человека
(табл. 1.5). Кроме того, изоферменты цитохрома
Р-450 присутствуют не только в печени, но и в тон-
кой кишке, поджелудочной железе, головном моз-
, надпочечниках, почках, костном мозге,
тучных клетках, коже, яичниках и яичках. Цереб-
ральные изоферменты играют важную роль в регу-
ляции уровня некоторых стероидных гормонов, оп-
ределяющих особенности настроения и сна.
Изоферменты обладают различной, но перекры-
вающейся специфичностью к лекарственным суб-
стратам и расщепляют лекарственные препараты
с разной скоростью. Они также различаются своей
восприимчивостью к возбуждающим и угнетающим
стимулам. Кроме того, некоторые из них (например,
CYP 2С18 и CYP 2D6) подвержены генетическому
полиморфизму, и различаются по выраженности
действия в разных органах.
Окисленный цитохром Р-450
+ лекарственный
препарат
(Окисленный цитохром Р-450) -
лекарственный препарат
+ электрон
(от НАДФ-Н цитохром-
Р-450-редуктазы)
(Восстановленный цитохром Р-450) -
лекарственный препарат
+ О2
(Восстановленный цитохром Р-450) -
лекарственный препарат
О2
+ электрон
(Восстановленный цитохром Р-450) -
------------лекарственный препарат
О2 (активн. кисл.)
Окисленный лекарственный препарат + Н2О
Рис. 1.6. Система оксидаз смешанного типа
(цитохром Р-450)
ВСАСЫВАНИЕ. РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
25
Таблица 1.5. Основные изоферменты Шка
Изофермент Субстраты И нгибиторы Биологические свойства
CYP 1А1 Теофиллин Пропофол NO Широко распространен вне пече- ни Индуцируется полициклическими углеводородами Выраженные индивидуальные различия индуцированной экс- прессии
CYP 1А2 Эритромицин Кофеин Парацетамол Теофиллин R-варфарин Галоперидол Ропивакаин Трициклические анти- депрессанты Эстрогены Ондансетрон Такрин Циметидин Фурафиллин Присутствует только в печени Индуцируется фенобарбиталом, фенитоином, омепразолом, куре- нием сигарет и физическими на- грузками Выраженная изменчивость (?), обусловленная генетическим полиморфизмом
CYP2A6 Кумарин Значительные индивидуальные различия концентрации в печени (?) Существует неактивный вариант Индуцируется пиразолом и дру- гими углеводородами
CYP 2В6 Циклофосфамид Основная форма цитохрома Р-450 Индуцируется барбитуратами Выраженная индивидуальная из- менчивость, вызванная индукци- ей или структурной мутацией
CYP 2C8 Диазепам Гексобарбитал Трициклические анти- депрессанты Паклитаксель Циметидин Индуцируется фенобарбиталом и рифампицином
CYP2C9/10 Толбутамид Фенитоин Диклофенак S-варфарин Ибупрофен Мефенамовая кислота Омепразол Сульфафеназол Сульфинпиразон Индивидуальные особенности экспрессии в печени Не чувствителен к агентам, инду- цирующим ферментативную ак- тивность Алкилируется тиениловой кисло- той Метаболиты -»гепатит
26
Глава 1
medlit.ucoz.ru
Таблица 1.5. (продолжение)
CYP 2С19 Диазепам Трициклические анти- депрессанты Пропранолол Мефенитоин Сульфафеназол Индуцируется фенобарбита- лом(?); подвержен генетическо- му полиморфизму (аутосом- но-рецессивный тип наследова- ния)
CYP 2D6 Кодеин Декстрометорфан Трициклические анти- депрессанты р-адреноблокаторы Флекаинид Флуфеназин Циметидин Хинидин Метадон Присутствует в печени, почках и кишечнике Участвует в метаболизме многих препаратов Генетический полиморфизм (ау- тосомно-рецессивный тип насле- дования, С22) Нарушение метаболизма обу- словлено одним из четырех гене- тических вариантов (2D6A, 2D6B, 2D6C, 2D6D)
CYP2E1 Парацетамол Этанол Ингаляционные ане- стетики Трихлорэтилен Дисульфирам Диэтилкарбамат Присутствует в печени, почках и лейкоцитах Индуцируется голоданием, ожи- рением, этанолом.изониазидом и бензеном Превращает проканцерогены в активные соединения
CYP ЗАЗ/4 Нифедипин Мидазолам Лидокаин Ропивакаин Фентанил Альфентанил Г идрокортизон Г ранисетрон Циклоспорин Эритромицин Этинилэстрадиол Лозартан Терфенадин Циметидин Т ролеандомицин Пропофол Кетоконазол Гестоден Сок грейпфрута Почти идентичные изоферменты Р-450 в печени Индуцируется кортикостероида- ми, макролидами, фенобарбита- лом Обеспечивает метаболизм мно- гих экзогенных и эндогенных со- единений
CYP ЗА5 Кофеин Дилтиазем Тестостерон Т ролеандомицин Присуствует в печени плода и подростков, и лишь у 25% взрос- лых; экспрессируется в плаценте и почках
CYP4B1
Индуцируется дексаметазоном
Экспрессируется в легких и дру-
гих внепеченочных эпителиаль-
ных тканях (?); опосредует
<о-окисление
ВСАСЫВАНИЕ, РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
27
Изофермент 2Е1 обеспечивает
и расщепление фторированных ингаляционных
анестетиков. Скорость дефторирования анестети-
ков (оценивается по скорости образования фтора)
убывает в следующем порядке: метоксифлуран >
севофлуран > энфлуран > изофлуран > десфлуран.
Другие изоферменты цитохрома Р-450 (1А2,
2С9/10 и 2D6) также могут участвовать в метабо-
лизме метоксифлурана. Индукция этанолом или фе-
нобарбиталом ускоряет дефторирование; CYP 2Е1
может также индуцироваться голодом, ожирением,
сахарным диабетом, изониазидом, кетоновыми тела-
ми и изопропиловым спиртом. CYP 2Е1 обладает
спсобностью превращать проканцерогены в актив-
ные соединения (табл. 1.5).
Хотя большинство окислительных и востанови-
тельных реакций катализируется изоформами
цитохрома Р-450, некоторые препараты (дофамин,
тирамин) окисляются митохондриальным фермен-
том моноаминоксидазой. Под воздействием алко-
гол ьдегидрогеназы, присутствующей в цитоплазме
печеночных клеток, окисляется этанол и восстанав-
ливается хлоралгидрат (табл. 1.4).
Гидролиз обеспечивает метаболизм эфиров и ами-
дов. Гидролиз препаратов может происходить в эн-
доплазматическом ретикулуме и осуществляться
микросомальными ферментами (например, гидро-
лиз петидина до петидиновой кислоты). Точно так
же гидролиз может проходить в плазме (гидро-
лиз сукцинилхолина и прокаина холинэстера-
зой), в нервно-мышечных синапсах (гидролиз аце-
тилхолина ацетилхолинэстеразой) или цитоплазме
гепатоцитов (гидролиз лидокаина и прилокаина
амидазами).
РЕАКЦИИ ВТОРОЙ ФАЗЫ
Реакции второй фазы представляют собой обра-
зование ковалентной связи между некоторыми эн-
догенными соединениями (глюкуроновой кислотой,
сульфатом, ацетатом, глициновой или метиловой
группы аминокислоты) с окисленными, восстанов-
ленными или гидролизованными продуктами ре-
акций первой фазы. Некоторые относительно по-
лярные молекулы могут метаболизироваться толь-
ко в ходе реакций второй фазы. Наиболее важной
реакцией является конъюгация с глюкуроновой ки-
слотой.
Конъюгация препаратов с глюкуроновой кислотой
в основном осуществляется ферментами эндоплаз-
матического ретикулума печени. Микросомальный
фермент глюкуронил-трансфераза катализирует
перенос остатка глюкуроновой кислоты от УДФ-
глюкуронида на неконъюгированный субстрат.
Этот процесс обеспечивает конъюгацию эндогенных
иО^^нрцрй (билирубин, тироксин), а также многих
препаратов (хлорамфеникол, морфин, салициловая
кислота, кортикостероиды, сульфаниламиды).
Конъюгация с глюкуроновой кислотой обычно при-
водит к образованию метаболитов со свойствами
относительно сильной кислоты (с низким рКа),
которые хорошо растворимы в воде.
Конъюгация с сульфатом происходит в стенке
кишки или цитоплазме гепатоцитов. Отвечающие
за эти реакции ферменты участвуют в синтезе
конъюгированных с сульфатом полисахаридов
(например, гепарина). Образование сульфатных
соединений может быть конечной стадией мета-
болизма хлорамфеникола, изопреналина, норад-
реналина, парацетамола и некоторых кортикосте-
роидов.
Ацетилирование препаратов происходит в раз-
личных тканях, в том числе в селезенке, легких
и печени. В печени конъюгация преимущественно
происходит не в гепатоцитах, а в купферовских
клетках, и заключается в переносе ацетильных
групп коэнзима А на неконъюгированный препа-
рат. У человека скорость и степень ацетилирования
регулируется генетически. Изониазид, многие
сульфаниламиды, гидралазин и фенелзин частично
метаболизируются путем ацетилирования.
Конъюгация с глицином происходит в цито-
плазме гепатоцитов. Часть сульфобромофталеина
конъюгируется с глицином, после чего выделяется
с желчью.
Ферменты, участвующие в реакциях метилиро-
вания, присутствуют в цитоплазме многих тканей.
Метилирование играет важную роль в метаболизме
катехоламинов (адреналин и норадреналин); оно
катализируется ферментом катехол-О-метил-
трансферазой.
Одни препараты повышают активность микро-
сомальных ферментов, в то время как другие угне-
тают их. Индукторы и ингибиторы метаболизма
могут быть причиной индивидуальных различий
в реакции на препараты (включая толерантность),
а также влиять на межлекарственные взаимодей-
ствия. Эти вопросы подробно обсуждаются в по-
следующих главах книги.
ИНДИВИДУАЛЬНЫЕ РАЗЛИЧИЯ
В МЕТАБОЛИЗМЕ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ
Когда некоторые лекарственные препараты
назначают разным больным в одной и той же дозе,
их концентрация в плазме может различаться в 10
и более раз. Этот феномен отчасти обусловлен ин-
дивидуальными особенностями лекарственного
метаболизма, которые являются важной причиной
28
Глава 1
изменчивости лекарственных реакции,
и характер метаболизма лекарственных препаратов
регулируются генетическими факторами. Некото-
рые виды метаболизма подвержены полиморфизму
(например, ацетилирование лекарственных препа-
ратов, гидролиз эфиров и некоторые реакции гид-
роксилирования и деалкилирования). Факторы ок-
ружающей среды (диета, курение сигарет, алкоголь,
инсектициды и другие индукторы и ингибиторы
лекарственного метаболизма) играют не столь важ-
ную роль.
В разных возрастных группах метаболизм пре-
паратов (особенно в печени) протекает по-разному.
У новорожденных метаболизм снижен (особой не-
зрелостью отличаются некоторые изоформы цито-
хрома Р-450 и глюкуронилтрансфераза). У пожи-
лых метаболизм также изменен; при этом повыша-
ется роль факторов окружающей среды.
На метаболизм и клиренс лекарственных препа-
ратов могут непредсказуемо влиять различные за-
болевания. При тяжелых заболеваниях печени
(цирроз или гепатит) нарушается выведение препа-
ратов, которые подвергаются интенсивному мета-
болизму. Снижение клиренса может привести к ку-
муляции препаратов, при этом снижается выведе-
ние их метаболитов с мочой. Заболевания печени
могут также усиливать и пролонгировать действие
лекарственных препаратов, которые метаболизи-
руются холинэстеразой плазмы. Любое снижение
сердечного выброса (при блокаде сердца, инфаркте
миокарда или артериальной гипертонии) может
уменьшить выведение лекарственных препаратов,
если их клиренс зависит от печеночного кровотока.
Обычно заболевания почек оказывают незначи-
тельное влияние или совсем не влияют на метабо-
лизм препаратов, хотя полярные метаболиты могут
накапливаться в плазме и оказывать токсическое
действие. Так, норпетидин (деметилированный ме-
таболит петидина) в норме быстро выводится с мо-
чой. При почечной недостаточности его экскреция
нарушается, иногда с развитием возбуждения и су-
дорог.
Реакция на лекарственные препараты при забо-
леваниях печени, почек и сердца может значитель-
но изменяться.
ВЫВЕДЕНИЕ (ЭКСКРЕЦИЯ)
Основной путь экскреции лекарственных пре-
паратов —с мочой или желчью. В небольшом коли-
честве некоторые препараты выводятся со слюной
или грудным молоком.
Важную роль, определяющую маршрут экскре-
ции (с мочой или желчью), играет молекулярный
ата и его метаболитов. Большинство низ-
комолекулярных соединений и их метаболитов вы-
деляется с мочой. Напротив, препараты с высоким
молекулярным весом (у человека приблизительно
400-500 Да) преимущественно выводятся с жел-
чью. Так желчная экскреция играет важную роль
в выведении некоторых миорелаксантов, антибак-
териальных средств, многих стероидных соедине-
ний (табл. 1.2).
Выведение с мочой зависит от трех различных
процессов, происходящих в разных участках неф-
рона (рис. 1.7):
1. Клубочковая фильтрация
2. Секреция в проксимальных канальцах
3. Диффузия в дистальных канальцах.
КЛУБОЧКОВАЯ ФИЛЬТРАЦИЯ
В результате клубочковой фильтрации с мочой
выделяются некоторые плохо растворимые в жи-
рах препараты и их метаболиты. В клубочках спо-
собна фильтроваться только свободная (не связан-
ная с белками) фракция препарата. Клубочковая
перфузия занимает значительно больше времени,
чем диссоциация комплекса препарат-белок, поэто-
му в клубочках может фильтроваться значительное
количество препаратов, связанных с белками.
СЕКРЕЦИЯ В ПРОКСИМАЛЬНЫХ
КАНАЛЬЦАХ
Активная секреция препаратов в проксималь-
ных канальцах почек приводит к их быстрому выве-
дению из организма. Секреция в проксимальных
канальцах осуществляется с помощью переносчи-
ка, требует затрат энергии и может происходить
против значительного концентрационного гради-
ента. Этим путем выводятся многие препараты и их
метаболиты (табл. 1.1). Секреция препаратов, обла-
дающих свойствами кислоты или основания, осу-
ществляется двумя различными системами актив-
ного транспорта. Обе системы расположены на оп-
ределенных участках клеток канальцевого
эпителия и требуют затрат энергии. Препараты со
свойствами кислоты конкурируют друг с другом за
эти участки. Препараты со свойствами основания
могут препятствовать выведению других основа-
ний или катионов. Кислоты обычно не конкуриру-
ют с основаниями и не препятствуют их секреции.
Иногда конкурентное угнетение канальцевого
транспорта кислот или оснований имеет практиче-
ское значение (например, подавление секреции пе-
нициллина пробенецидом; угнетение транспорта
уратов тиазидными диуретиками).
В ходе секреции в клетки канальцевого эпите-
лия из плазмы проникают только несвязанные пре-
ВСАСЫВАНИЕ. РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
29
параты. Вместе с тем не доказано, что6ы1дд£СШ1. USfitianFAJuipoBaTb обратно в плазму; при этом
н не лекарствен кого препарата белками или эритро-
цитами ограничивало канальцевую секрецию,
некоторые препараты, в значительной степени свя-
занные с белками плазмы (фенол красный, некото-
рые пенициллины), полностью выделяются за одно
прохождение с кровью через почки. Этот феномен
может объясняться тем. что диссоциация комплек-
са препарат-белок происходит значительно быст-
рее, чем перфузия почечных канальцев.
ДИФФУЗИЯ В ДИСТАЛЬНЫХ КАНАЛЬЦАХ
В дистальных почечных канальцах реабсорбция
и элиминация кислот и оснований частично осуще-
ствляется путем неионной диффузии. В этом сег-
менте нефрона создается значительный градиент
Н’ между плазмой и мочой с кислой (нормальной)
реакцией. Большинство препаратов со свойствами
кислоты выделяется преимущественно с мочой со
щелочной реакцией, где они представляют собой не
способные к диффузии анионы. Напротив, в моче
с кислой реакцией они трансформируются в неио-
низированные молекулы, которые могут легко
их элиминация может значительно замедляться.
Например, слабая кислота пробенецид активно сек-
ретируется в проксимальных почечных канальцах
как анион, то есть RCOO ; в кислой среде (то есть
в дистальных канальцах)она частично превращает-
ся в неионизировапное соединение RCOOH и ак-
тивно реабсорб и ру ется.
Следовательно, экскреция пробеницида происхо-
дит достаточно медленно, и период нолуныведения
(Ti 9 составляет 6-12 ч.
Препараты со свойствами основания (напри-
мер, вторичные и третичные амины) выделяются
преимущественно с мочой с кислой реакцией; в этих
условиях они свободно диффундируют из плазмы
в мочу, где и задерживаются в виде катионов. Это обес-
печивает градиент для диффузии неионизирован-
ных препаратов из плазмы в мочу. Многие препа-
раты со свойствами основания хорошо растворимы
в жирах, активно связываются с белками плазмы
и неспособны в значительном количестве выво-
диться путем клубочковой фильтрации или каналь-
цевой секреции. Единственным способом выведения
КЛУБОЧЕК
Фильтрация лекарственных препаратов, не связан-
ных с белками
ПРОКСИМАЛЬНЫЙ ПОЧЕЧНЫЙ КАНАЛЕЦ
Активная секреция слабых кислот (анионов) и слабых
оснований(катионов)
ДИСТАЛЬНЫЙ ПОЧЕЧНЫЙ КАНАЛЕЦ
Пассивная реабс орбция или экскреция ненонизиро-
ванных форм слабых кислот и оснований
Рис. 1.7. Почечная экскреция лекарственных препаратов, включающая клубочковую фильтрацию, секрецию
в проксимальных канальцах и реабсорбцию или экскрецию в дистальных канальцах. В дистальных канальцах
слабые кислоты и основания могут реабсорбироваться или выделяться с мочой в зависимости от рК, и градиента
pH между плазмой и мочой
30
Глава 1
таких препаратов является диффузия их н
рованной фракции из относительно щелочной
плазмы в кислую мочу (рис. 1.3).
Изменения pH мочи влияют на экскрецию сла-
бых кислот и оснований, что иногда используется
для ускорения элиминации препаратов при передо-
зировке и отравлении.
ВЫВЕДЕНИЕ С ЖЕЛЧЬЮ
Выведение лекарственных средств и их метабо-
литов с желчью обычно играет менее важную роль,
чем экскреция с мочой. Тем не менее после приема
внутрь или парентерального введения почти все
препараты или их метаболиты обнаруживаются
в желчи (часто в следовых количествах). Выведе-
ние с желчью является основным путем выведения
веществ с молекулярным весом более 400-500 Да.
Ионизированные или частично ионизирован-
ные препараты и их метаболиты обычно выводятся
из гепатоцитов путем активного транспорта. Высо-
комолекулярные анионы (включая глюкурониды
и сульфаты) и катионы (в том числе четвертич-
ные амины) активно переносятся из гепатоцитов
в желчные протоки разными транспортными систе-
мами, зависящими от Na+/K+ АТФазы. Желчная
секреция относительно неспецифична, насыщаема
и может конкурентно или неконкурентно угнетать-
ся другими препаратами; так, анионы конкурируют
между собой за выведение через желчные протоки,
а препараты со свойствами основания препятст-
вуют выведению других оснований или катионов.
Во многих отношениях желчная секреция анионов
и катионов напоминает механизм активного транс-
порта в проксимальных канальцах нефрона, что
объясняет высокую концентрацию некоторых пре-
паратов в желчи (иногда более чем в сто раз выше,
чем в плазме).
Этот феномен может иметь практическое значе-
ние. Визуализация желчных протоков в ходе рент-
генологических исследований зависит от активной
секреции рентгеноконтрастных препаратов и их
концентрации в желчи. Эффективность ампицил-
лина и некоторых других антибиотиков в лечении
кишечных инфекций обусловлена их способностью
накапливаться в желчи в высокой концентрации.
Многие миорелаксанты частично выводятся с жел-
чью, присутствуя в ней в высокой концентрации.
Моночетвертичные соединения (например, веку-
роний, тубокурарин) выводятся активнее, чем
бис-четвертичные (например, панкуроний, диме-
тил тубокурарин), и этим отчасти объясняется раз-
личная продолжительность их действия.
Многие препараты, выводящиеся с желчью в ви-
де конъюгатов с глюкуроновой кислотой, гидроли-
тонкой кишке под воздействием микро-
флоры, которая выделяет фермент гиалуронидазу.
После гидролиза неизмененные препараты по-
вторно всасываются, метаболизируются в печени
и вновь выводятся с желчью в виде конъюгатов
с глюкуроновой кислотой (рис. 1.8). Этот «кишеч-
но-печеночный кругооборот» может происходить
многократно, прежде чем препарат окончательно
выводится из организма, часто сочетается со значи-
тельным эффектом первого прохождения и дли-
тельным Т1/2- Кишечно-печеночный кругооборот
характерен для многих стероидных соединений
(пероральные контрацептивы, эстрогены). Анти-
биотики широкого спектра действия угнетают или
полностью уничтожают рост кишечной микрофло-
ры, поэтому конъюгаты препаратов с глюкуроно-
вой кислотой не гидролизуются в тонкой кишке
и их элиминация повышается. Считают, что этот
механизм может быть причиной неэффективности
оральной контрацепции при приеме антибиотиков
широкого спектра действия.
Следовые количества многих препаратов, выво-
дящихся с желчью в неизменном виде, могут непо-
средственно переходить из системы печеночной
артерии во внутрипеченочные желчные протоки
через перибилиарные сплетения. На этот процесс
оказывает влияние гормон секретин, который так-
же обладает желчегонным действием.
ВЫВЕДЕНИЕ СО СЛЮНОЙ И ГРУДНЫМ
МОЛОКОМ
Небольшое количество многих лекарственных
препаратов в неизмененном виде выделяется со
слюной и грудным молоком. Несвязанные с белка-
ми, жирорастворимые низкомолекулярные препа-
раты легко проникают в слюну и молоко, где их
концентрация может быть такой же, как и в плазме.
Поскольку pH слюны и грудного молока немного
ниже, чем в плазме, то концентрация слабых кислот
в этих биологических жидкостях будет немного
ниже, а слабых оснований — немного выше, чем
в плазме. Некоторые ионы (например, хлориды
и йодиды) могут активно секретироваться в слюну
и грудное молоко. Выведение препаратов со слю-
ной и грудным молоком редко имеет практическое
значение. Из-за выведения следовых количеств не-
которых препаратов с молоком (например, опиои-
дов, большинства гипнотиков и транквилизаторов)
грудное вскармливание становится нежелатель-
ным, если мать постоянно принимает эти препараты.
Миорелаксанты и их антагонисты выделяются
в слюну и молоко в незначительном количестве.
ВСАСЫВАНИЕ, РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
31
medlit.ucoz.ru
КИШКА
Неизмененный
препарат
Гидролиз под воздействием
бактериальной флоры
Конъюгированный
препарат
ВОРОТНАЯ
ВЕНА
ПЕЧЕНЬ
Неизмененный Печеночная СИСТЕМНОЕ
препарат вена”----* КРОВООБРАЩЕНИЕ
t
I
I
▼ I
Неизмененный » Конъюгированный
препарат препарат
Секреция
ЖЕЛЧЬ
Конъюгированный
препарат
Рис. 1.8. Кишечно-печеночный кругооборот лекарственных препаратов. Лишь небольшое количество всосавшегося
препарата и его метаболита (показано пунктирными стрелками) избегает рециркуляции и поступает в системный
кровоток
Список литературы
Bates, I.P. (1985) Permeability of the blood-brain barrier.
Trends in Pharmacological Sciences 6, 447-450.
Birkett, O., Mackenzie, P.L, Veronese, M.E. & Miners. J.O. (1993).
In vitro approaches can predict human drug metabolism. Trends in
Pharmacological Sciences 14, 292-294.
Chang, G.W.M. & Cam, P.C.A. (1999). Тне physiological and pharma-
cological roles of cytochrome P450 isoenzymes. Anaesthesia 54,42-50.
Cholerton, S., Daly, A.K. & Idle, J.R. (1992). The role of individual
human cytochromes P450 in drug metabolism and clinical response.
Trends in Pharmacological Sciences 13,434-439.
Fehrenbach, A. (1987). Drugs in breast milk. Britishjoumal of Pharma-
ceutical Practice 9,288-290.
George, C.F. (1981). Drug metabolism by the gastrointestinal mucosa.
Clinical Pharmacokinetics 6, 259-274.
Gonzalez, FJ. (1992). Human cytochromes P450: problems and
prospects. Trends in Pharmacological Sciences 13,346-352.
Guengerich, F.P. (1989). Characterization of human microsomal
cytochrome P-450 enzymes. Annual Review of Pharmacology and
Toxicology 29,241-264.
Kharasch, E.D. & Thummel, K.E. (1993). Identification of
cytochrome P450 2E1 as the predominant enzyme catalysing human
liver microsomal defluorination of sevoflurane, isoflurane and
methoxyflurane. Anesthesiology 79,795-807.
Nelson, D.R, Kamataki, T., Waxman, D. J. et al. (1993). The P-450 super-
family. update on new sequences, gene mapping, accession numbers,
ear- ly trivial names of enzymes and nomenclature. DNA Cell Biology 12,
1-51.
Okey, A.B. (1990). Enzyme induction in the cytochrome P-450
system. Pharmacology and Therapeutics 45, 241-298.
Pardridge, W.M. (1988). Recent advances in blood-brain barrier
transport. Annual Review of Pharmacology and Toxicology 28,25-39.
Reynolds, F. (1979). Drug transfer across the placenta. In: Placenta
Transfer (edsG. Chamberlain & A. Wilkinson), pp. 166-181. Pitman
Medical, Tunbridge Wells.
Routledge, P.A. (1986). The plasma protein binding of basic drugs.
British Journal of Clinical Pharmacology 22, 499-506.
Schanker, L.S. (1962). Passage of drugs across body membranes.
Pharmacological Reviews 14,501-530.
Singer, S.J. & Nicolson, G.L. (1972). The fluid mosaic model of the
structure of cell membranes. Science 175, 720-731.
Somogyi, A. (1987). New insights into the renal secretion of drugs.
Trends in Pharmacological Sciences 8, 354-357.
Somogyi, A. (1996). Renal transport of drugs: specificity and
molecular mechanisms. Clinical and Experimental Pharmacology and
Physiology 23, 986-989.
Stoughton, R.B. (1989). Percutaneous absorption of drugs. Annual
Review of Pharmacology and Toxicology 29, 55-69.
Tucker, G.T. (1979). Drug metabolism. British Journal of Anaes-
thesia 51,603-618.
Warburg, E. (1922). Carbonic acid compounds and hydrogen
ion activities in blood and salt solutions: a contribution to the theory
of L.J. Henderson and KA. Hasselbalch. Biochemical Journal 16,
153-340.
ФАРМАКОКИНЙГЙ1Ж2 u
2
Понятие «фармакокинетика» претерпело зна-
чительную эволюцию. Раньше под ним подразуме-
вали измерение и математический анализ концен-
траций лекарственных препаратов и их метаболи-
тов в организме. В настоящее время это понятие
включает изучение и количественную оценку про-
цессов всасывания, распределения, метаболизма,
элиминации. Иными словами, фармакокинетика —
это то, «что организм делает с лекарством».
С 1960-х гг. для измерения концентрации ле-
карственных препаратов и их метаболитов в крови
и моче используются многие передовые аналитиче-
ские методики, включая жидкостную хроматогра-
фию высокого разрешения, масс-спектрометрию
и радиоиммунный анализ. Динамика изменения
концентрации препарата во времени позволяет
рассчитать фармакокинетические константы, ха-
рактеризующие поведение лекарства в организме.
Эти константы используют для определения нагру-
зочной дозы и скорости инфузии, обеспечивающих
постоянную концентрацию препарата в плазме,
а также для прогнозирования скорости и степени
кумуляции. Кроме того, фармакокинетические
константы позволяют определить, на сколько необ-
ходимо снизить дозу препарата при заболеваниях
почек и печени, а также оценить влияние других хи-
мических веществ и заболеваний на поведение ле-
карства в организме.
Двумя важнейшими фармакокинетическими
константами являются объем распределения (V) и
клиренс (CL). Объем распределения — это кажу-
щийся объем, доступный для распределения препа-
рата в организме. Клиренс отражает способность
организма к элиминации препарата. Еще одним
важным фармакокинетическим параметром явля-
ется период полувыведения Т1/2 — время, за которое
концентрация вещества в плазме снижается вдвое.
Объем распределения и клиренс связаны с Т1/2
следующим образом:
Ti/2~V/CL=kx V/CL,
где к — константа (1п2; 0,693).
Таким образом, Tt/2 зависит от первичных
констант V и CL. Увеличение Ti/2 может отражать
повышение объема распределения и/или снижение
клиренса; соответственно, уменьшение Т]/2 — умень-
шение объема распределения и/или повышение
клиренса. По величине Т]/2 не всегда можно судить
об элиминации препарата.
Объем распределения
Объем распределения равен отношению общего
содержания препарата в организме к его концентра-
ции в плазме (или сыворотке). Объем распределения
зависит от коэффициента распределения препара-
та между липидной и водной фазами, регионарного
тканевого кровотока и степени связывания лекар-
ства с белками плазмы и тканей. Объем распреде-
ления, хотя и не отражает никакого реального
анатомического или физиологического простран-
ства, имеет существенное практическое значение.
Это объем, необходимый для равномерного распре-
деления вещества в концентрации, равной его кон-
центрации в плазме.
Объем распределения различных лекарствен-
ных препаратов у взрослых может колебаться от
5 до 2000 л. Если объем распределения препарата
относительно низок (5-20 л), то он либо распреде-
ляется преимущественно в плазме и внеклеточной
жидкости (например, миорелаксанты и другие четвер-
тичные амины), либо активно связывается с белка-
ми плазмы (например, варфарин и фенитоин).
Напротив, если объем распределения превышает
общее содержание воды в организме (45 л), то пре-
парат активно связывается с компонентами тканей,
где его концентрация может оказаться выше, чем
в плазме. Многие препараты активно связываются
с компонентами тканей и характеризуются отно-
сительно высоким объемом распределения; к ним
ФАРМАКОКИНЕТИКА
33
относятся дигоксин, фентанил, бупивакаин, ооль-
шинство фенотиазинов и антидепрессантов. На объ-
ем распределения влияют факторы, от которых зави-
сит соотношение водных пространств организма,
в том числе возраст, беременность, некоторые заболе-
вания (почечная и сердечная недостаточность).
Объем распределения может определять такти-
ку лечения лекарственных отравлений. Для полно-
го выведения из организма препарата с большим
объемом распределения требуется много часов или
даже дней, и форсированный диурез неэффективен
для лечения отравления этим лекарством. Напро-
тив, форсированный диурез показан при отравле-
нии препаратами с относительно малым объемом
распределения (если только они не подвержены ак-
тивному метаболизму и не связываются с белками
плазмы). Теоретически можно предположить, что
вытеснение препаратов из связи с белками плазмы
может привести к непропорциональному измене-
нию объема распределения и концентрации свобод-
ного препарата в тканях (относительно первона-
чального объема распределения).
КЛИРЕНС
Клиренс представляет собой объем крови или
плазмы, который полностью очищается от лекарст-
венного препарата за единицу времени. Клиренс —
теоретическое понятие, поскольку в реальности
препарат не может быть полностью удален из доста-
точно больших объемов плазмы. Клиренс обычно
измеряется в мл/мин, его можно представить как
сумму нескольких путей элиминации препарата
различными органами:
CL = CLr + CLh + CLX,
где CLr — почечный клиренс, CLH — печеночный
клиренс, и С Lx - клиренс, обеспечиваемый други-
ми органами. Клиренс можно также определить
как отношение скорости элиминации лекарствен-
ного препарата (выражаемой в мг/мин) к концен-
трации в цельной крови или плазме (выражае-
мой в мг/мл). Поскольку клиренс — величина
постоянная, скорость элиминации лекарственного
препарата обычно прямо пропорциональна его кон-
центрации в плазме.
Во многих случаях почечный клиренс лекарст-
венных препаратов измеряется напрямую класси-
ческими способами или вычисляется делением об-
щего количества лекарства, выведенного с мочой,
на площадь под фармакокинетической кривой
(ПФК), описывающей зависимость концентрации
препарата от времени. Следовательно, можно от-
дельно определить почечный (CLr) и внепочечный
U.CQZ^rU ГЧ ГЛ ГТ
(CLer) клиренс, где CLEr = CL - CLR. Эти показате-
ли можно использовать для оценки относительно-
го вклада почек и печени в элиминацию препарата.
Если общий клиренс обеспечивается в основ-
ном за счет почечной экскреции (например, CLR >
0,7 х CL), то при почечной недостаточности или во
время трансплантации почки высок риск кумуля-
ции этого препарата. Напротив, если CLr < 0,3 х CL,
то состояние почек не оказывает особого влияния
на элиминацию препарата; в этих случаях клиренс
зависит от метаболизма или экскреции с желчью.
Дисфункция печени оказывает влияние на метабо-
лизм препарата в печени и его экскрецию с желчью,
но степень этого влияния оценить трудно. В то же
время элиминация большинства лекарственных
средств зависит от печени (где они либо подверга-
ются метаболизму, либо экскретируются с жел-
чью). В стационарном состоянии элиминацию
лекарственных препаратов печенью можно выра-
зить коэффициентом экстракции (ER):
ER = (Са - Cv)/Ca = 1 - Cv/Ca,
где Са — концентрация препарата в смешан-
ной крови из воротной вены и печеночной артерии,
a Cv — концентрация препарата в крови, оттекаю-
щей от печени. Коэффициент экстракции — это
интегративный показатель способности пече-
ни удалять лекарственный препарат из печеноч-
ных капилляров путем метаболизма и экскреции
с желчью.
Наиболее распространенная модель печеноч-
ного клиренса допускает, что концентрация сво-
бодного препарата в крови, оттекающей от вены,
и в жидкой фазе гепатоцитов одинакова. При та-
ких условиях выведение препарата печенью зави-
сит от следующих факторов:
1. Печеночного кровотока,
2. Относительной доли свободного препарата в кро-
ви,
3. Активности ферментов, обеспечивающих ме-
таболизм препарата.
Печеночный клиренс можно представить в виде
произведения печеночного кровотока (Q) и коэф-
фициента экстракции (ER), то есть,
CLH = QxER
Его также можно выразить через печеночный
кровоток и так называемый «внутренний кли-
ренс» (CLintrinsic )'•
CLh = Qx (CLintrinsic x 0/(0. + (CLintrinsic x 0),
где CLintrinsic — скорость очищения печеночной
воды от препарата (мл/мин), a f — фракция сво-
бодного лекарства в крови. Внутренний клиренс
не зависит от кровотока и отражает максимальную
34
Глава 2
способность печени к необратимой элими^^^Ц^ф ЦСОЁ эрцр условиях печеночный клиренс определя-
карств путем их метаболизма или экскреции с жел- ется преимущественно печеночным кровотоком,
чью. Значения CLintrinsic для разных препаратов Эту разновидность элиминации называют нерест-
отличаются. В ферментативной кинетике CLintrinsic
характеризуется отношением Vmax/Km (см. ниже раз-
дел «Нелинейная фармакокинетика»).
Если внутренний клиренс (CL) значительно
ниже печеночного кровотока (Q), тогда
Q + (CLintrinsIC х 0 * Q
и
CLi! ® CLintrinsIC х t
В этих условиях печеночный клиренс зависит
только от внутреннего клиренса (то есть от активно-
сти печеночных ферментов) и свободной фракции
препарата в крови. Эту разновидность элиминации
называют рестриктивной (синоним: элиминация,
ограниченная емкостью ферментативных систем).
Она характерна для фенитоина, толбутамида,
теофиллина, варфарина, а также для большинства
барбитуратов и бензодиазепинов. Эти препараты
подвергаются незначительному эффекту первого
прохождения после приема внутрь и отличаются
низким коэффициентом печеночной экстракции
(< 0,3). Их клиренс относительно низок, он не зави-
сит от изменений печеночного кровотока, но на него
значительно влияет активность печеночных фер-
ментов. Ферментативные нарушения могут инду-
цироваться другими лекарствами, факторами
окружающей среды, а также возрастом, погрешно-
стями питания или заболеваниями, что сущест-
венно влияет как на общий клиренс, так и на вели-
чину Ti/2 вышеназванных препаратов. Поскольку
CLH « CLintrinsic х f, печеночный клиренс зависит
также от свободной фракции препарата в крови
и может меняться при связывании лекарств с бел-
ками. Клиренс препаратов, незначительно связы-
ваемых с белками (< 20-30%), не зависит от до-
ли связанного препарата (элиминация, ограничен-
ная емкостью ферментативных систем и нечувст-
вительная к связыванию препарата с белками).
Напротив, клиренс и конечная величина Т|/2 лекар-
ственных средств, которые в значительной степени
связываются с белками, зависят от доли связанного
препарата (элиминация, ограниченная емкостью
ферментативных систем и чувствительная к связы-
ванию препарата с белками).
Если внутренний клиренс (CLintrinsic) высок по
сравнению с печеночным кровотоком, тогда спра-
ведливы следующие допущения:
(Q + (CLintrinsic х 0 ~ (CLintrinsic х 0
и уравнение
CLh « Q х (CLintrinsic х 0/(0, + (CLintrinsic х 0)
можно сократить до CLH « Q.
риктивной (синоним: элиминация, ограниченная
кровотоком). Нерестриктивная элиминация опре-
деляет печеночный клиренс некоторых 0-адре-
ноблокаторов, трициклических антидепрессантов,
опиоидов и лидокаина. Эти препараты подверга-
ются значительному эффекту первого прохожде-
ния через печень после приема внутрь и отличают-
ся высоким коэффициентом печеночной экстрак-
ции (> 0,7). Их клиренс относительно высок, он
определяется величиной печеночного.кровотока
(21 мл/кг/мин у взрослых). Величина Т]/2 зависит
от печеночного кровотока и не зависит от активности
печеночных ферментов или связывания лекарств
с белками крови.
Печеночную элиминацию многих препаратов
нельзя однозначно классифицировать как «рест-
риктивную» или «ограниченную кровотоком».
Элиминация таких препаратов определяется как
внутренним клиренсом (то есть активностью пече-
ночных ферментов и/или экскрецией с желчью),
так и печеночным кровотоком — в зависимости от
условий, при которых элиминация оценивается.
Тем не менее концепция рестриктивной и нерест-
риктивной элиминации обеспечивает физиоло-
гический подход к оценке печеночного клиренса
лекарственных препаратов и иллюстрирует не-
предсказуемость взаимоотношений между связы-
ванием препаратов с белками плазмы и их элимина-
цией. Если печеночный клиренс полностью или
частично зависит от внутреннего клиренса, значи-
тельное связывание препарата с белками может
уменьшить концентрацию его свободной фракции
и ограничить элиминацию. Напротив, если клиренс
зависит от печеночного кровотока, связывание
с белками не оказывает значимого влияния на эли-
минацию препарата.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ
ФАРМАКОКИНЕТИЧЕСКИХ
КОНСТАНТУ И CL
Значения объема распределения и клиренса
можно использовать для расчета дозы лекарствен-
ного препарата, в том числе при почечной и пече-
ночной недостаточности. Если известна концентра-
ция препарата в плазме, необходимая для достиже-
ния желаемого эффекта, то дозу для назначения
внутрь или в/м можно рассчитать по формуле:
доза (мг) = (СР х I х CL)/F
ФАРМАКОКИНЕТИКА
35
где Ср — концентрация лекарственного препарата
в плазме (мг/мл), I — интервал между приемами
препарата (мин), CL — клиренс (мл/мин), a F —
доля препарата, поступающая в системный крово-
ток (то есть, та доля всосавшегося препарата, кото-
рая не подверглась эффекту первого прохождения).
К сожалению, для многих препаратов значения F
точно не известны, и расчетная доза может оказать-
ся относительно неточной.
Напротив, при внутривенном введении вполне
реально обеспечить точную и постоянную концен-
трацию препарата в плазме, применяя сочетание
нагрузочной дозы и поддерживающей инфузии.
Требуемая нагрузочная доза (мг) рассчитывается
по формуле Ср х V, а скорость инфузии (мг/мин) —
по формуле Ср х CL, где СР — необходимая концен-
трация препарата в стационарном состоянии
(мг/мл), V — объем распределения в стационарном
состоянии (мл), a CL — клиренс (мл/мин). Если
лекарственные препараты вводят в/в в течение
длительного периода времени (например, опиоиды,
неингаляционные анестетики, недеполяризую-
щие миорелаксанты, антибиотики), можно добиться
их воспроизводимой и постоянной концентрации
в плазме (рис. 2.1). К сожалению, значения V и CL
получают экспериментальным путем, что сопряже-
но с риском ошибок; кроме того, V и CL для одного
и того же препарата у разных людей могут значи-
тельно отличаться. Нагрузочная доза может вы-
звать преходящее чрезмерное повышение концен-
трации препарата, что иногда заставляет изменить
схему введения (например, трансформировать
струйное введение нагрузочной дозы в инфузию
продолжительностью несколько минут).
В последние годы для достижения воспроиз-
водимых и постоянных концентраций лекарст-
венных препаратов в плазме применяют подход,
основанный на использовании соответствующих
камерных моделей (см. ниже). Вначале препарат
вводят струйно для заполнения центральной каме-
ры до требуемой концентрации; затем переходят
к непрерывной инфузии препарата (экспоненци-
ально снижая скорость) для компенсации элими-
нации препарата из центральной камеры и его пе-
ремещения в одну или несколько периферических
камер. Для достижения постоянной концентрации
в крови некоторых препаратов (например, пропо-
фола, альфентанила) и поддержания ее в пределах
терапевтического диапазона применяют управ-
ляемые компьютером программированные инфу-
зионные насосы.
Рис. 2.1. Поддержание постоянной сывороточной кон-
центрации (С) основано на следующих уравнениях:
Нагрузочная доза (стрелка) = объем распределения хСр;
Скорость инфузии (заштрихованный прямоугольник) =
клиренс х Ср
Фармакокинетические константы могут так-
же использоваться для прогнозирования влия-
ния печеночной и почечной дисфункции на сыво-
роточную концентрацию препаратов. Хроническая
почечная недостаточность приводит к сниже-
нию клиренса креатинина и уменьшению почеч-
ной элиминации многих препаратов. Особенно
сильно снижается клиренс препаратов, которые
почти полностью выводятся почками в неизме-
ненном виде (например, аминогликозидные ан-
тибиотики, азопропазон, хлорпропамид и дигок-
син). В таких случаях можно рассчитать дозу пре-
парата, необходимую для достижения заданной
концентрации в плазме при сниженном почечном
клиренсе.
Хроническая почечная недостаточность умень-
шает клиренс и пролонгирует действие некоторых
миорелаксантов (особенно первых поколений).
Клинически показано, что почечная недостаточ-
ность увеличивает продолжительность действия
алкурония, галламина, панкурония и тубокурари-
на. В то же время продолжительность действия ат-
ракурия, цмс-атракурия, мивакурия и векурония не
меняется. Эти клинические наблюдения объясня-
ются влиянием почечной дисфункции на клиренс
препаратов (табл. 2.1).
36
Глава 2
Hedoc£Q&Gllit
Таблица 2.1. Влияние почечной
сти на клиренс недеполяризующих миорелаксантов
у взрослых
Миорелаксант Клиренс (мл/мин)
Нормальная функция почек Почечная не- достаточность
Атракуриий 300-400 340-470
Г алламин 50-100 5-25
Мивакуриий* 2100-8400 2100-8400
Панкуроний 75-125 20-50
Рокуроний 170-420 100-320
Тубокурарин 80-120 50-70
Векуроний 150-350 130-500
* Приведены значения для двух наиболее активных изо-
меров (цис-транс мивакурия и транс-транс мивакурия)
Время (кратные Ti/2 величины)
Рис. 2.2. Кумуляция лекарственных препаратов при
введении (стрелки) с интервалами, равными Т)/2
КОНЕЧНЫЙ ПЕРИОД
ПОЛУВЫВЕДЕНИЯ
Период полувыведения Т|/2 — это время, за кото-
рое сывороточная концентрация вещества сни-
жается вдвое. В рамках однокамерной модели
(см. ниже) определить Т1/2 очень просто. Однако
для многих лекарственных средств приходится
использовать многокамерную модель, так как
динамика их сывороточной концентрации описы-
вается несколькими экспоненциальными функ-
циями. В таких случаях рассчитывают несколько
значений Ti/2. Наиболее важен конечный период по-
лувыведения, представляющий собой период полу-
выведения в фазу элиминации препарата.
Величину Ti/2 можно представить следующим
образом:
Ti/2« 0,693 х —
Таким образом, период полувыведения можно
считать производной константой, зависящей от
первичных фармакокинетических констант V и CL.
Если одну и ту же дозу препарата принимают
внутрь через промежутки времени, равные конеч-
ному Т1/2, то его сывороточная концентрация посте-
пенно увеличивается, пока через 4-5 Тi/2 не достиг-
нет стационарного уровня (рис. 2.2). Латентного
периода до достижения стационарной концентрации
можно избежать применением нагрузочной дозы,
равной двойной разовой. При инфузии стационар-
ная концентрация препарата тоже достигается
через 4-5 конечных Ti/2 (рис. 2.3). Стационарным
Время (кратные Ti/2 величины)
Рис. 2.3.Кумуляция лекарственных препаратов при
длительной внутривенной инфузии (заштрихованный
прямоугольник).
называют такое состояние, когда скорость инфузии
препарата (скорость поступления) равна скорости
его элиминации (произведение клиренса на сыво-
роточную концентрацию препарата). Чтобы дос-
тичь стационарного состояния, интервалы при
дробном введении препарата (или приеме внутрь)
должны быть приблизительно равны Ti/2: это
обеспечивает приемлемый компромисс между до-
пустимым снижением сывороточной концентра-
ции препарата и необходимой частотой введения.
ФАРМАКОКИНЕТИКА
37
„„„ medlit.ucoz.ru
Таблица 2.2. Период полувыведения из плазмы и продолжительность действия миорелаксантов у взрос-
лых пациентов с нормальной функцией почек и печени
Миорелаксант Ttzj(MMH) Продолжительность действия
Сукцинилхолин 3-5 Короткая
Мивакурий* 2-8 Короткая
Атракурий 18-22 Короткая
Цис-Атракурий 18-22 Короткая
Векуроний 36-72 Короткая
Галламин 80-220 Средняя
Рокуроний 70-140 Средняя
Панкуроний 110-150 Длительная
Алкуроний 180-220 Длительная
Тубокурарин 150-230 Длительная
* Значения двух наиболее активных изомеров (цис-транс мивакурия и транс-транс мивакурия).
В прошлом по величине конечного Т| 2 часто судили
о продолжительности действия препарата.
Действительно, в некоторых случаях (напри-
мер, при использовании миорелаксантов) сущест-
вует определенная корреляция между конечным
Т| г и продолжительностью действия препарата
(табл. 2.2). Такой взаимозависимости нс просле-
живается при использовании других лекарств
(например, опиоидов), поскольку для осуществле-
ния действия они должны преодолеть гематоэнце-
фалический барьер. Если препараты вызывают не-
обратимый эффект (например, многие ингибиторы
ацетилхолинэстсразы и моноаминоксидазы, неко-
торые цитостатические препараты, феноксибенза-
мин), то их продолжительность действия может
составлять несколько дней и даже недель, и она
не зависит от сывороточной концентрации препа-
рата или Ти. Если к окончанию эффекта приводит
перераспределение препаратов в тканях (напри-
мер, в/в барбитураты, бензодиазепины и некото-
рые опиоиды), то о продолжительности их дейст-
вия тоже нельзя судить, основываясь на конечном
Т|7. По мерс усовершенствования аналитических
методов определения препаратов в плазме оказа-
лось, что конечный ТРЗ некоторых анестетиков (на-
пример, пропофола) значительно выше, чем счита-
ли раньше.
КОНТЕКСТ-ЧУВСТВИТЕЛЬНЫЙ
ПЕРИОД ПОЛУВЫВЕДЕНИЯ
При внутривенной инфузии конечный Т)2 не
позволяет судить о продолжительности действия
лекарственного препарата, поскольку не отражает
процессов распределения и перераспределения.
При длительной инфузии, в ходе которой поддер-
живается стационарная концентрация, целесооб-
разно оценивать такой показатель, как «кон-
текст-чувствительный Тг2». Контекст-чувстви-
тельный Т|/г — время после окончания инфузии,
необходимое для снижения концентрации вещест-
ва в плазме вдвое; под «контекстом» здесь пони-
мают продолжительность инфузии. Даже после
продолжительной инфузии контекст-чувствитель-
ный Т|,2 всегда меньше соответствующего конеч-
ного T|rj. С помощью компьютерного моделирова-
ния показано, что контекст-чувствительный
некоторых лекарственных препаратов практически
не зависит от длительности инфузии: например Т)/2
ремифентанила при увеличении продолжитель-
ности инфузии с 1 до 8 ч повышается с 2 до 5 мин.
На контекст-чувствительный других препара-
тов (альфентанил, мидазолам и пропофол) дли-
тельность инфузии оказывает умеренное влияние:
например, при увеличении ее с I до 8 ч Т1/2 пропофо-
ла повышается в 3 раза (с 12 до 38 мин). Следо-
вательно, пробуждение после инфузии этих препара-
тов наступает относительно быстро. Напротив, Tw
фентанила при увеличении продолжительности
38
Г лава 2
Таблица 2.3. Контекст-чувствительный пе/£Х^13$в1А&£1^£№которых общих анестетиков
Продолжитель- ность инфузии (часы) Ремифентанил Альфентанил Мидазолам Пропофол Тиопентал Фентанил
1 2 36 34 12 78 24
2 2 53 45 16 100 50
3 3 55 60 18 120 108
4 3 56 63 25 138 175
5 3 57 67 30 157 218
6 3 58 70 32 167 250
7 3 59 72 35 172 268
8 4 60 75 38 — 180 280
инфузии с 1 до 8 ч возрастает почти и 12 рал (с 24
до 280 мин), поскольку значительная его доля по-
сле завершения инфузии быстро волн ратае гея в
плазму из периферических тканей (табл. 2.3). Эти
компьютерные модели показывают, что после пре-
кращения инфузии действие фентанила может
продолжаться несколько часов. По сравнению с
фентанилом остаточные эффекты ремифентанила
(в меньшей степени альфентанила, пропофола и
мидазолама)относительно скоротечны независимо
от продолжительности инфузии.
Хотя концепция контекст-чуветвитсяьного Тр?
не является фармакокинетической константой
и не имеет фармакокинетического значения, она
служит полезным практическим индикатором.
Контекст-чувствительный Тц позволяет предска-
зать вероятную длительность действия препарата и
быстроту пробуждения после инфузии различной
продолжительности, что представляет значитель-
ную ценность при проведении тотальной внутри-
венной анестезии.
ИЗМЕРЕНИЕ ОБЪЕМА
РАСПРЕДЕЛЕНИЯ,КЛИРЕНСА
И КОНЕЧНОГО ПЕРИОДА
ПОЛУВЫВЕДЕНИЯ С ПОМОЩЬЮ
КАМЕРНЫХ МОДЕЛЕЙ
Объем распределения и клиренс лекарственных
препаратов можно определить по снижению их
сывороточной концентрации, используя камерные
и бес камерные математические модели. При камер-
ном анализе значения объема распределения и кли-
ренса определяют, исходя из параметров подходя-
щей фармакокинетической модели.
В прошлом (фармакокинетические характери-
стики многих лекарственных препаратов описыва-
лись на примере простой однокамерной открытой
модели. Эта модель отличается математической
четкостью и простотой, но в настоящее время из-за
серьезных теоретических и практических ограни-
чений она практически не используется в отноше-
нии большинства препаратов, применяемых во вре-
мя анестезии. Для характеристики вводимых в/в
препаратов почти всегда используют двух- и трех-
камерные открытые модели.
ДВУХКАМЕРНАЯ ОТКРЫТАЯ МОДЕЛЬ
После в/в введения сывороточная концентра-
ция большинства препаратов быстро снижается
вследствие их распределения в организме. Этот
период обычно называют (фазой распределения; ее
продолжительность и скорость снижения сыворо-
точной концентрации в основном зависят от физи-
ко-химических свойств препара га ((х-тенно от мо-
лекулярного веса и растворимости в липидах).
За фазой распределения следует период медлен-
ного снижения концентрации (конечный период,
(фаза элиминации). Она отражает элиминацию
препарата, обусловленную метаболизмом и экскре-
цией.
Графически (фармакокинетику препарата удоб-
но представлять в полулогарифмической системе
координат, когда по оси ординат Y откладывают ло-
гарифм сывороточной концентрации, а по оси абс-
цисс X время после введения. Так. после в/в
струйного введения отмечается типичное двух-
фазное снижение сывороточной концентрации
опиоида фенопиридина (рис. 2.4). Первоначаль-
на* резкое падение концентрации, обусловленное
распределением, сменяется фазой более медленного
ФАРМАКОКИНЕТИКА
39
Рис. 2.4. Биэкспоненциальное сни-
жение концентрации [нг/мл] фе-
ноперидина в плазме после его внут-
ривенного введения. Ось абсцисс
(ось X) отражает время в минутах;
ось ординат (ось Y) — логарифмы
концентрации феноперидина в плаз-
ме; отмеченные точки (черные круж-
ки) соответствуют концентрации ле-
карства в плазме в различные момен-
ты времени. Точка В соответствует
начальной концентрации медленной
фазы экспоненциального уклона,
экстраполированного на нулевую
точку времени. Экстраполирован-
ные значения вычитаются из экспе-
риментально полученных данных
и дают остаточные значения (х);
проведенная через эти точки линия
регрессии, полученная по методу
наименьших квадратов, соответству-
ет фазе быстрого выведения.
снижения вследствие элиминации. Выявляется
прямая линейная зависимость логарифма сыворо-
точной концентрации от времени в фазе элимина-
ции. Динамику сывороточной концентрации пре-
парата можно описать двумя экспоненциальными
функциями (рис. 2.4). Для описания применяют
экстраполяцию, в ходе которой прямую, соединяю-
щую значения логарифма сывороточной концен-
трации в фазу элиминации, продолжают до пере-
сечения с осью Y; точку пересечения обозначают
как В. Вычитание экстраполированных величин
из исходных реальных значений дает последова-
тельность остатков (здесь остаток — разница между
реальным и экстраполированным значением), ко-
торая отражает фазу распределения. Начальная
фаза (фаза а, фаза распределения) отображется
регрессионной прямой, которая пересекает ось Y
в точке А. Обе фазы имеют характерные углы на-
клона (а и р) и периоды полувыведения Tja и Т’ р.
Точки пересечения с осью ординат (А и В) соответ-
ствуют доле участия Т|а и Т|рв снижении сыворо-
точной концентрации фенопиридина после струй-
ного введения..
Числовые значения углов наклона кривых а и Р
рассчитываются по формулам:
a = In 2/T^a
P = ln2/Ti₽
Сывороточная концентрация препарата СР в мо-
мент времени t выражается суммой двух экспонен-
циальных компонентов:
СР = А • е *at+ В • е-₽1
где А и В - точки пересечения графиком оси орди-
нат, а и Р - производные константы скорости,
а е = 2,718 (см. Приложение). Значения А, В, а и р
можно получить графическими методами или
рассчитать с помощью компьютерных программ.
Сывороточную концентрацию фенопиридина (СР)
в момент времени t вычисляют по формуле:
СР = 30e-0276t + 12е-°019t,
где СР измеряется в нг/мл, а t — в минутах.
Биэкспоненциальное снижение сывороточной
концентрации фенопиридина интерпретируют в
рамках открытой двухкамерной фармакокинетиче-
ской модели (рис. 2.5). Модель состоит из относи-
тельно небольшой центральной камеры, в которую
лекарственный препарат поступает и из которой
элиминируется, и периферической камеры.
Скорость элиминации определяется константой
скорости kt0. Объем распределения перифериче-
ской камеры обычно больше, чем у центральной;
двунаправленное перемещение препарата между
двумя камерами определяется константами скоро-
стей ki2 и k2t (рис. 2.5). Центральная и перифериче-
ская камеры не имеют непосредственного физиоло-
гического эквивалента, их параметры и константы
определяются только поведением лекарства в орга-
низме.
Двухкамерная модель является теоретической
концепцией, которая объясняет наблюдаемое
биэкспоненциальное снижение сывороточной
концентрации лекарственного препарата (рис. 2.4).
40
Глава 2
Введение
лекарственного
препарата
medlit.ucoz.ru
♦
Центральная
камера
1
Периферическая
камера
2
Рис. 2.5. Двухкамерная
открытая модель, состоя-
щая из центральной каме-
ры 1 и периферической ка-
меры 2. Константы к12 и к21
определяют скорость пе-
ремещения лекарственно-
го препарата между цен-
тральной и перифериче-
ской камерами; к10 —
константа скорости эли-
минации
k2l
кю
Элиминация
лекарственного
препарата
В отношении фенопиридина (и многих других
препаратов) центральная камера, вероятно, соот-
ветствует крови, интерстициальной жидкости и внут-
риклеточной жидкости хорошо кровоснабжаемых
органов (головного мозга, сердца, легких, печени
и почек). Периферическая камера соответствует
менее васкуляризованным органам и тканям (ске-
летные мышцы, жировая ткань, кожа и соедини-
тельная ткань). В фазе распределения сывороточ-
ная концентрация фенопиридина быстро падает
при его перемещении из центральной камеры в пе-
риферическую (рис. 2.5). После достижения рас-
пределительного равновесия между двумя камера-
ми выведение препарата из организма зависит
только от элиминации, скорость которой определя-
ется константой кю.
Двухкамерная модель основана на допущении,
что распределение и элиминация являются экспо-
ненциальными процессами первого порядка (то
есть их скорость прямо пропорциональна количе-
ству препарата в каждой камере). Отсюда можно
вывести дифференциальные уравнения скорости
изменения содержания препарата в каждой камере
(т. е. dXi/dt и dX2/dt, где Xi и Х2 - количество пре-
парата в каждой камере в момент времени t). Реше-
ние этих дифференциальных уравнений с замеще-
нием Xi и Х2 концентрациями препарата позволяет
вывести математическую зависимость, связываю-
щую концентрацию препарата в центральной (Ci)
и периферической (С2) камерах со временем.
Для центральной камеры
Ci = А • е + В • е_Р\
где А, а, В и р - константы, а е = 2,718. Это выраже-
ние идентично формуле биэкспоненциального сни-
жения сывороточной концентрации фенопиридина
после в/в инъекции. Следовательно, значения А, а,
Вир можно получить путем измерения сывороточ-
ной концентрации препарата, а затем по этим кон-
стантам можно определить другие параметры
двухкамерной открытой модели, например, пло-
щадь под фармакокинетической кривой «концен-
трация-время» (ПФК), клиренс (CL), объем рас-
пределения (V), а также константы скорости кю, kt2,
и к21, используя следующие уравнения:
_ АВ
ПФК= — + -
а р
Доза Доза
ПФК
А В
— + —
а р
, Ар+Ва
к2,'“Х7Г
ар
кю'ь
к12 = а + р - (к21 + кю)
Доза Доза
агеа рхПФК АВ
Р(-+т
а р
ФАРМАКОКИНЕТИКА
41
Рис. 2.6. Трехкамерная
открытая модель, со-
стоящая из центральной
камеры 1 и перифериче-
ских камер 2 и 3. Введе-
ние и элиминация ле-
карственного препарата
осуществляются через
центральную камеру.
Константы к12, к21, к13 и к31
определяют перемеще-
ние лекарственного пре-
парата между централь-
ной и периферическими
камерами; к10 — констан-
та скорости элиминации
medlit.ucoz.ru
Введение
лекарственного
препарата
Периферическая
камера
2
▼
Центральная
камера
1
Периферическая
камера
3
Элиминация
лекарственного
препарата
Доза ki2-bk2i
Vss' А+ВХ—
С помощью компьютерных программ были рас-
считаны следующие параметры двухкамерной мо-
дели фенопиридина после его в/в инъекции:
А = 30 нг/мл
а = 0,276 мин4
'1/а - 2,5 мин
В = 12 нг/мл
0 = 0, 019 мин-1
Т^₽= 37,0 мин
ПФК = 725 (нг/мл) мин
CL = 2511 мл/мин
k2i = 0,090 мин-1
кю = 0,057 мин4
ki2= 0,147 мин-1
Varea=1341
Vss =1171
Как показано выше, можно рассчитать два зна-
чения объема распределения (Varea и Vss • Varea),
которые выражают отношение между общим коли-
чеством фенопиридина в организме и его концен-
трацией в центральной камере в фазе элиминации
(т. е. когда устанавливается распределительное рав-
новесие). При быстрой элиминации и высоком кли-
ренсе лекарственного препарата Varea может превы-
шать истинный объем распределения. Объем рас-
пределения в стационарном состоянии (VSs) не
зависит от скорости элиминации и поэтому точнее
отражает истинный объем распределения.
ТРЕХКАМЕРНАЯ МОДЕЛЬ
Хотя фармакокинетика многих в/в препаратов
соответствует характеристикам двухкамерной от-
крытой модели распределения, иногда для интер-
претации данных лучше использовать более слож-
ные модели. Процесс снижения концентрации мно-
гих опиоидов, а также некоторых миорелаксантов
и в/в анестетиков можно разделить на три экспо-
ненциальных составляющих. Тогда сывороточная
концентрация препарата Ср в момент времени t оп-
ределяется триэкспоненциальным уравнением:
СР = А • е -“‘+ В e~₽t + С • е~*
Числовые значения для точек пересечения (А, В
и С) и трех наклонных составляющих (а, 0 и у)
определяют графически или с помощью компью-
терных программ.
Снижение сывороточной концентрации препа-
рата интерпретируют согласно характеристикам
трехкамерной открытой модели, когда препарат
поступает в центральную камеру и из нее же элими-
нируется (рис. 2.6). Рассчитывают также парамет-
ры модели (клиренс, объем распределения, объемы
камер и константы скорости, определяющие пере-
мещение препаратов между камерами). В некото-
рых случаях снижение сывороточной концентра-
ции препарата лучше интерпретировать с помощью
такой трехкамерной модели, где элиминация про-
исходит из одной из периферических камер.
Иногда снижение сывороточной концентрации
препарата соответствует как двух-, так и трехкамер-
ной модели. Наиболее подходящую модель выбира-
ют следующим образом: с помощью компьютерной
программы полулогарифмическую кривую зави-
симости сывороточной концентрации препарата
42
Глава 2
от времени разделяют на две и три экспон
ных составляющих. В каждом случае определяют
разницу между измеренными и расчетными значе-
ниями. Если применение триэкспоненциального
уравнения не приводит к значительному снижению
суммы остатков, то целесообразно использовать
биэкспоненциальное, соответствующее двухкамер-
ной модели. Хотя это и не доказывает абсолютную
достоверность двухкамерной модели, можно пола-
гать, что она является оптимальной для интерпре-
тации фармакокинетических характеристик.
На практике двух- и трехкамерные модели мало
отличаются. Различия между экспериментальны-
ми и компьютерными значениями часто значитель-
но меньше, чем величина аналитической ошибки
при измерении сывороточной концентрации
препарата. Сроки забора крови также влияют на
выбор фармакокинетической модели, а неправиль-
ное время забора крови может привести к ошибоч-
ной интерпретации начальной или конечной фазы
экспоненциального снижения концентрации.
Следовательно, технические, аналитические фак-
торы, а также факторы времени могут играть важ-
ную роль в выборе фармакокинетической модели.
КАМЕРНЫЕ МОДЕЛИ
И ФАРМАКОЛОГИЧЕСКОЕ
ДЕЙСТВИЕ
Если препарат вызывает обратимую реакцию
в ткани-мишени, между его сывороточной концентра-
цией и фармакологическим эффектом существует тес-
ная корреляция. Если ткань- мишень находится в цен-
тральной камере, то существует взаимосвязь меж-
ду количеством лекарства в этой камере и фарма-
кологическим эффектом. Ранее считали, что посколь-
ку сывороточная концентрация тубокурарина (и его
количество в центральной камере) тесно коррели-
рует со степенью нервно-мышечной блокады, то дей-
ствие тубокурарина осуществляется именно в цен-
тральной камере. Однако позднее было показано,
что для лучшей интерпретации фармакокинетики и
фармакодинамики миорелаксантов к фармакоки-
нетической модели следует добавить отдельную
«эффекторную камеру». После начала инфузии не-
деполяризующих миорелаксантов наблюдается
короткий латентный период между подъемом их
сывороточной концентрации и развитием нерв-
но-мышечной блокады, а после прекращения инфу-
зии падение концентрации миорелаксанта несколь-
ко предшествует восстановлению нервно-мышеч-
ной проводимости. Этот феномен гистерезиса или
временного сдвига равновесия можно объяснить, до-
бавив к центральной камере фармакокинетической
ЦОР^^ГУополнительную эффекторную
камеру с
определенными константами скорости.
ДРУГИЕ ФАРМАКОКИНЕТИЧЕСКИЕ
МОДЕЛИ
Распределение и элиминацию некоторых препа-
ратов, включая тиопентал, лидокаин и ингаляци-
онные анестетики, можно описать в рамках физио-
логических перфузионных моделей. Эти модели
интерпретируют распределение препаратов в пре-
делах анатомических или физиологических про-
странств. В рамках этих пространств определяют
объемы распределения, перфузионные характери-
стики и коэффициенты распределения. Поступле-
ние препарата в пределы какого-либо пространства
может быть ограничено либо кровотоком, либо транс-
мембранным транспортом.
В рамках физиологической перфузионной мо-
дели можно описать фармакокинетику тиопентала.
Концентрация препарата в крови, скелетных мыш-
цах и подкожно-жировой клетчатке в различные
моменты времени после введения соответствует
сравнительно простой модели (включающей цен-
тральную камеру — кровь и шесть тканевых камер).
Было высказано предположение, что тиопентал
преимущественно выводится из головного мозга
в нежировые ткани (например, мышцы), а подкож-
ный жир играет лишь вторичную роль. Впоследст-
вии в эту модель были добавлены дополнительные
камеры, отражающие метаболизм, а также связыва-
ние с белками плазмы и компонентами тканей.
Физиологические перфузионные модели имеют
ряд очевидных преимуществ. Их можно использо-
вать для прогнозирования концентрации препара-
тов в ткани-мишени. Они также позволяют точно
описать процессы распределения и элиминации,
учитывать местные и общие физиологические из-
менения во время анестезии (например, изменения
сердечного выброса, регионарного кровотока,
функции почек). Иногда такие модели помогают
понять причину различий в кинетике лекарствен-
ного препарата у одного и того же человека в разное
время. К сожалению, эти методы связаны с необхо-
димостью детального анализа значительного мас-
сива данных, и изъятие необходимых образцов тка-
ней в условиях общей анестезии может оказаться
нежелательным или неэтичным.
БЕСКАМЕРНЫЕ МЕТОДЫ
Затруднения при использовании камерных мо-
делей привели к разработке бескамерных методов
ФАРМАКОКИНЕТИКА
43
фармакокинетического анализа. Эти мет
висят от допущений конкретных фармакокинети-
ческих моделей, но тем не менее позволяют вычис-
лить клиренс (CL) и объем распределения в стацио-
нарном состоянии (Vss)- Конечный Т|/2 можно
оценить с помощью нелинейной регрессии методом
наименьших квадратов (метод наименьших квад-
ратов — статистический прием, с помощью которо-
го неизвестные параметры модели оцениваются
путем минимизации суммы квадратов отклонений
измеренных значений от теоретических). При не-
обходимости полученные данные можно оценить
(обычно с помощью величин, обратных квадрату
сывороточной концентрации препарата).
Клиренс можно рассчитать по формуле:
гт Доза
ПФК
где ПФК — площадь под фармакокинетической кривой
между значениями t = 0 и t = <» (бесконечность). ПФК
можно определить по «правилу трапеции», то есть
путем измерения и сложения площадей всех тра-
пеций между точками, обозначающими время оче-
редного забора крови и соответствующую ему сы-
вороточную концентрацию препарата в плазме
(рис. 2.7.). Конечную, площадь — ту долю ПФК,
которая приходится на время между последним за-
бором крови и t = оо, можно вычислить по формуле:
Конечная площадь = (последняя измеренная сы-
вороточная концентрация х конечный Т1/2)/0,693
Среднее время существования (СВС, среднее
время, в течение которого молекула лекарства суще-
ствует в организме) можно вычислить по формуле:
ПФКМ
СВС~ ПФК
где ПФКМ - площадь под кривой первого момента
(т. е. полная площадь под фармакокинетической
кривой, экстраполированная на время от нуля до
бесконечности). Тогда V в стационарном состоя-
нии (VSs) можно представить в виде:
Vss = CL х СВС.
НЕЛИНЕЙНАЯ ФАРМАКОКИНЕТИКА
Фармакокинетические модели подразумевают,
что распределение и элиминация препарата явля-
ются процессами первого порядка. Это означает,
что скорость перемещения препарата из одной ка-
меры в другую всегда пропорциональна его кон-
центрации. Однако так бывает не всегда. Многие
физиологические процессы, опосредующие рас-
пределение и элиминацию, зависят от активного
транспорта и потенциально насыщаемы (т. е. имеют
ucoz.ru
Рис. 2.7. Определение площади под фармакокинетиче-
ской кривой (ПФК) по правилу трапеций
максимальную конечную транспортную емкость).
Точно так же насыщаемы многие химические реак-
ции, от которых зависит метаболизм препаратов:
при высокой концентрации субстрата их скорость
достигает максимума. В этих условиях транспорт
и метаболизм препарата осуществляются с высо-
кой, но постоянной скоростью, которая не зависит
от его концентрации. Математически это можно
выразить уравнением dX/dt = - к, где X — количест-
во лекарственного препарата в организме или ка-
мере в момент времени t, к — константа скорости.
Кинетика такого типа называется нелинейной или
кинетикой насыщения; она характерна для препа-
ратов с элиминацией, ограниченной емкостью фер-
ментативных систем. Метаболические реакции,
для которых характерно насыщение, протекают в
соответствии с кинетикой Михаэлиса-Ментена:
после насыщения метаболизм трансформируется
из процесса первого порядка (dX/dt = - kX1 = - kX)
в метаболические процессы нулевого порядка
(dX/dt = - kX° = - к), скорость которых постоянна
и не зависит от концентрации лекарства.
Эти отношения можно вывести из уравнения Ми-
хаэлиса-Ментена для ферментативных реакций:
vmax хСр
V кт+ср
где v — скорость элиминации препарата; Vmax — макси-
мальная скорость элиминации; Кт— константа срод-
ства (константа Михаэлиса) и Ср — сывороточная
44
Глава 2
концентрация препарата. Если Ср намног^^^Ц^ UtJOZ5.1 рщивая зависимости концентрации от вре-
Кт, тогда v ® (Vmax/Km) х Ср; поскольку Vmax и Кт — мени приобретает колоколообразную форму,
константы, то v ~ С, и скорость элиминации соот- отличаясь от моноэкспоненциальной и биэкспо-
ветствует кинетике первого порядка. Напротив, ненциальной кривой.
если Ср значительно превышает Кт, тогда v » (Vmax х
Ср)/Ср, то есть v « Vmax; таким образом, при высокой
сывороточной концентрации скорость элиминации
становится постоянной, что соответствует кинети-
ке нулевого порядка. Константа Михаэлиса Кт от-
ражает степень сродства лекарственного препарата
к ферменту, трансмембранному переносчику или
транспортной системе и соответствует такой сыво-
роточной концентрации, при которой скорость эли-
минации препарата равняется половине макси-
мальной скорости (т. е. v/Vmax = 0,5).
На практике феномен нелинейной кинетики
встречается относительно редко, поскольку на
уровне эффективной сывороточной концентрации
препарата емкость транспортных систем и метабо-
лических реакций обычно далека от насыщения.
Тем не менее иногда (особенно если элиминация
происходит преимущественно путем печеночного
метаболизма) кинетика нулевого порядка наблю-
дается in vivo. В таких случаях повышение дозы
и сывороточной концентрации препарата может
привести к удлинению Т1/2 и непропорциональному
увеличению ПФК. (Напротив, если распределение
и элиминация соответствуют кинетике первого по-
рядка, то все фармакокинетические параметры по
определению не зависят от дозы.) Нелинейная
кинетика характерна для элиминации этанола, са-
лицилатов и фенитоина из-за насыщения метабо-
лических систем печени. При лечении фенитоином
насыщение может наступить уже при достижении
субтерапевтической или терапевтической концен-
трации (40-80 мкмоль/л), и последующее введение
препарата вызывает непропорциональное повыше-
ние концентрации. Нелинейная кинетика также
отмечается при использовании больших или по-
вторных доз тиопентала; при этом конечный Т1/2
и продолжительность действия увеличиваются
(вероятно, из-за печеночного метаболизма нулево-
го порядка). Нелинейная фармакокинетика часто
характеризует элиминацию при передозировке
лекарств; однако остается неясным, отражает ли это
насыщение печеночных метаболических систем,
токсическое воздействие препарата или то и дру-
гое одновременно. Если элиминация осуществля-
ется через насыщаемые процессы (например, при
лечении фенитоином или при передозировке),
то по мере снижения сывороточной концентра-
ции кинетика нулевого порядка трансформирует-
ся в процесс первого порядка. При этом во время
элиминации Ti/2 укорачивается, и полулогарифми-
ЗНАЧЕНИЕ И ОГРАНИЧЕНИЯ
ФАРМАКОКИНЕТИЧЕСКОГО
АНАЛИЗА
Клиническое применение анестетиков преиму-
щественно основано на их фундаментальных био-
логических свойствах и фармакодинамических эф-
фектах, то есть, на их внутренней способности вли-
ять на клеточные процессы. Напротив,
фармакокинетика не имеет столь важного практи-
ческого значения. Тем не менее фармакокинетиче-
ские модели и константы могут использоваться для
разработки соответствующих схем применения
внутривенных анестетиков. В некоторых случаях
автоматический компьютерный инфузионный на-
сос может программироваться на соответствую-
щую фармакокинетическую модель и индивиду-
альные характеристики больного, чтобы предопре-
делить концентрацию лекарственного препарата в
плазме. В последнее время для мониторинга лекар-
ственных реакций и управления инфузионными
системами по принципу «закрытой петли» стали
использовать приборы, действующие по механизму
«обратной связи». К сожалению, попытки примене-
ния фармакокинетически обоснованных режимов
лечения в клинической практике вызывают ряд за-
труднений.
Во-первых, фармакокинетические свойства ле-
карственных препаратов, чьи распределение и эли-
минация согласуются с двухкамерной или трехка-
мерной открытой моделью, часто анализируются по-
сле быстрого внутривенного болюсного введения.
При этом точное измерение многих кинетических
параметров может оказаться очень трудной зада-
чей. Определение клиренса (CL) и объема распре-
деления (V) в основном зависит от площади ниже
кривой «концентрация-время» между t = 0 и t = °°
(ПФК). При использовании двухкамерной модели
ПФК = А/а+ В/p. Поскольку величина А обычно
велика, а величина а относительно мала, любые
ошибки при определении этих констант могут
значительно повлиять на ПФК. После струйного
введения лекарственных препаратов показатели
Айа могут оказаться весьма неточными, так как:
1) они больше зависят от вычисления, а не изме-
рения концентраций;
2) обычно определяются на основании относи-
тельно небольшого количества проб;
ФАРМАКОКИНЕТИКА
45
3) концентрация лекарственного nplelftQ(aHit.
в плазме быстро снижается в фазе распределения,
и небольшие нарушения срока или методики забора
крови могут привести к серьезным ошибкам. Сле-
довательно, фармакокинетические параметры, осно-
ванные на измерении концентрации лекарства
в плазме после в/в струйного введения, могут ока-
заться относительно неточными и недостоверными.
Объем распределения и клиренс определяются
точнее, если лекарственные препараты вводятся в
виде в/в инфузии. Фармакокинетические констан-
ты, применимые к двух- или трехкамерным откры-
тым моделям, можно математически рассчитать по
постинфузионным показателям концентрации ле-
карственных препаратов. При непрерывной инфу-
зии быстрого распределения или перераспределе-
ния лекарств больше не происходит (в зависимости
от продолжительности инфузии), и быстрое сниже-
ние концентрации в плазме в фазу а (фаза быстрого
выведения) истощается.
Кроме того, значение фармакокинетики зависит
от воспроизводимости и измеримости зависимости
фармакологических эффектов лекарственных пре-
паратов от их концентрации в плазме. Фармакоки-
нетический анализ имеет небольшое практическое
значение, если активность лекарственного препа-
рата нельзя прогнозировать, исходя из его концен-
трации в плазме. К сожалению, эта зависимость
между концентрацией в плазме и биологическими
эффектами лекарственных препаратов бывает не-
предсказуемой. В некоторых случаях концентра-
ция в плазме не отражает концентрацию в биофазе
или зоне действия лекарственного препарата, и от-
ношение между ними может носить сложный ха-
рактер. Действие многих лекарственных препара-
тов не полностью обратимо (например, неостигми-
на); другие образуют активные метаболиты со
сходной фармакологической активностью (диаце-
тилморфин, морфин, диазепам и тиопентал); а на-
чало и продолжительность действия некоторых ле-
карств не зависят от их фармакокинетических
свойств (некоторые опиоиды и местные анестети-
ки). Во всех этих случаях фармакокинетические
методы мало пригодны для точного прогнозирова-
ния активности лекарственных препаратов.
Использование многих анестетиков в хиральной
или рацемической форме (глава 3) часто основыва-
ется на результатах фармакокинетического анализа.
В большинстве случаев концентрации этих препара-
тов в плазме определялись не стереоселективными
методами. Поскольку соотношение стереоизомеров
в оригинальной рацемической смеси может быстро
меняться в организме, фармакокинетическая ин-
терпретация полученных кинетических констант
LKJjQtZr. Бккзаться неточной и вводить в заблужде-
ние. Недопустимо распространять на энантиомеры
фармакокинетические данные, полученные без
использования стереоселективных методов.
ПОПУЛЯЦИОННАЯ
ФАРМАКОКИНЕТИКА
Популяционная фармакокинетика — метод,
обладающий значительной прогностической цен-
ностью. При расчете констант и параметров, отра-
жающих распределение и элиминацию препарата в
целой популяции, учитываются индивидуальные
различия. Критериям популяционного анализа
обычно не соответствуют ни анализ данных, полу-
ченных от многих людей, ни суммарная статистика
отдельных фармакокинетических исследований.
Последние исследования основываются на «нели-
нейном моделировании смешанных эффектов»;
при этом делается попытка с помощью статистиче-
ских параметров создать единую фармакокинети-
ческую модель, объединяющую присущие отдель-
ным индивидуумам различия (независимо от при-
чины этих различий). Собираются показатели всех
исследуемых индивидуумов. Достоверность анали-
за ограничена данными многочисленных участни-
ков исследования, а также несколькими допуще-
ниями (например, что форма профиля сывороточ-
ной концентрации препарата у каждого индивиду-
ума характерна для популяции в целом).
В рамках этого подхода считают, что различия
между фармакокинетическими константами инди-
видуумов и популяционными показателями обу-
словлены различиями измеримых характеристик
или «постоянных эффектов» (например, возраста,
веса, клиренса креатинина, печеночного клиренса).
Все необъяснимые причины вариабельности опре-
деляются как «случайные эффекты». Насколько
это возможно, популяционная фармакокинетика
пытается объяснить все фармакокинетические раз-
личия измеримыми характеристиками или посто-
янными эффектами.
БИОДОСТУПНОСТЬ
Под биодоступностью препарата понимают его
долю, поступающую в системный кровоток после
приема внутрь. Биодоступность можно определить
как скорость и степень всасывания, после чего пре-
парат становится доступным в ткани-мишени.
Общее количество препарата в системном кровото-
ке обычно имеет большее значение, чем скорость
всасывания; и биодоступность можно определить
с помощью ПФК, которая легко измеряется по пра-
46
Глава 2
вилу трапеций (рис. 2.7). Считается, что п
дении биодоступность равна 100%. Следовательно,
абсолютную биодоступность (%) можно опреде-
лить как
ПФК„ после_ приема_ внутрь
----------------------------------------хЮО
ПФК 0_ю после_ внутривенного_ введения
Абсолютную биодоступность можно также из-
мерить по общей экскреции неизмененного препа-
рата с мочой после приема внутрь и в/в введения.
Некоторые препараты обладают высокой био-
доступностью при приеме внутрь (например, диазе-
пам, дигоксин, фенитоин и варфарин). Эти соеди-
нения устойчивы в ЖКТ, хорошо всасываются
в тонкой кишке и не подвергаются значительному
метаболизму в кишечной стенке или печени до попа-
дания в системный кровоток. Напротив, биодоступ-
ность многих других препаратов при приеме внутрь
низка, что может быть вызвано рядом факторов.
Во-первых, некоторые препараты нестабильны
или расщепляются в ЖКТ (бензилпенициллин,
гепарин, многие полипептидные гормоны). Во-вто-
рых, из-за физико-химических факторов (напри-
мер, в результате плохой растворимости в жирах
и высокого молекулярного веса) может быть затруд-
нено всасывание препаратов. В ЖКТ плохо всасы-
ваются неостигмин, гликопирролат и аминогли-
козидные антибиотики. И, наконец, препараты, зна-
чительно подверженные эффекту первого
прохождения, обладают низкой системной биодос-
тупностью вследствие интенсивного метаболизма
в кишечной стенке или печени (например, морфин,
пропранолол и лидокаин). Значительный эффект
первого прохождения снижает поступление неиз-
мененного препарата в системный кровоток.
Разница в биодоступности препаратов при
приеме внутрь имеет несколько причин. На раство-
римость капсул и таблеток могут влиять многочис-
ленные фармацевтические факторы. При этом осо-
бенности изготовления одних и тех же препаратов
различными производителями могут существенно
влиять на их растворимость, тем самым приводя
к разбросу сывороточных концентраций после
приема внутрь. Такие колебания растворимости
после приема внутрь чаще всего наблюдаются у от-
носительно нерастворимых препаратов, различия в
биодоступности которых могут иметь высокую
клиническую значимость. Индивидуальные особе-
ности печеночного кровотока и эффекта первого
прохождения также могут влиять на биодоступ-
ность. Если печеночный клиренс ограничен крово-
током, то любое уменьшение печеночного кровото-
ка может привести к увеличению биодоступности.
Препараты, стимулирующие или угнетающие пече-
ночные микросомальные ферменты, также могут
pimedW.uoazbru биодоступность (например.
циметидин
может повышать биодоступность пропранолола).
Физиологические изменения (преклонный воз-
раст) и патологические состояния (цирроз печени)
могут нарушать метаболизм препаратов и повы-
шать их биодоступность.
Список литературы
Benet, L.Z. & Galeazzi, R.L. (1979). Non-compartmental determi-
nation of the steady-state volume of distribu tion.Journal ofPharmace-
utical Sciences 68, 1071-1074.
Gabrielsson, J. & Weiner, D. (1994). Pharmacokinetic/ Pharmaco-
dynamic Data Analysis: Concepts and Applications. Swedish
Pharmaceutical Press, Stockholm.
Gibaldi, M. & Perrier, D. (1982). Pharmacokinetics, 2nd edn. Marcel
Dekker, NewYork.
Hunter, J.M., Jones, R.S. & Utting, J.E. (1984) Comparison of
vecuronium, atracurium and tubocurarine in normal patients and in
patients with no renal function. British Journal of Anaesthesia 56,
941-951.
Mather, L.E. (1983). Clinical pharmacokinetics of fentanyl and its
newer derivatives. Clinical Pharmacokinetics 8, 422-445.
Milne, L, Williams, N.E., Calvey, T.N., Murray, G.R. & Chan, K. (1980).
Plasma concentration and metabolism of phenoperidine in man.
British Journal of Anaesthesia 52, 537-540.
Price, H.L., Kovnat, PJ., Safer,J.N., Conner, E.H. & Price, M.L. (1960).
The uptake of thiopental by body tissues and its relation to the dura-
tion of narcosis. ClinicalPharmacology and Therapeutics 1,16-22.
Prys-Roberts, C. & Hug, C.C. (eds). (1984). Pharmacokinetics
ofAnaesthesia. Blackwell Scientific Publications, Oxford.
Riegelman, S., Loo, J.C.K & Rowland, M. (1968). Shortcomings in
pharmacokinetic analysis by conceiving the body to exhibit
properties of a single compartment. Journal of Pharmaceutical
Sciences 57,117-123.
Rowland, M. & Tozer, T.N. (1995). Clinical Pharmacokinetics:
Concepts and Applications, 3rd edn. Williams & Wilkins, Baltimore.
Schuttler, J., Kloos, S., Schwilden, H. & Stoeckel, H. (1988). TIVA
with propofol and alfentanil by computer assisted infusions.
Anaesthesia 43 (Suppl.), 2-7.
Schwilden, H. (1981). A general method for calculating the dosage
scheme in linear pharmacokinetics. European Journal of Clinical
Pharmacology 20,379 383.
Shafer, S.L. & Varvel, J.R. (1991). Pharmacokinetics, pharmaco-
dynamics and rational opioid selection. Anesthesiology 74,53-63.
Sheiner, L.B., Stanski, D.R., Vozeh, S., Miller, R.D. & Нат, J. (1979).
Simultaneous modeling of pharmacokinetics and pharmaco-
dynamics: application to d-tubocurarine. Clinical Pharmacology and
Therapeutics 215,358-371.
Stanski, D.R. & Watkins, W.D. (1982). Drug Disposition in
Anesthesia. Grune & Stratton, NewYork.
Stanski, D.R. & Maitre, P.O. (1990). Population pharmacokinetics
and pharmacodynamics of thiopental: the effect of age revisited.
Anesthesiology 72, 412-422.
Tozer, T.N. (1981). Concepts basic to pharmacokinetics. Pharmaco-
logy and Therapeutics 12,109-131.
Wilkinson, G.R. (1987). Clearance approaches in pharmacology.
Pharmacological Reviews 39,1-47.
Wright, P.M.C. (1998). Population based pharmacokinetic analysis:
why do we need it; what is it; and what has it told us about
anaesthetics? British Journal of Anaesthesia 80, 488-501.
ФАРМАКОКИНЕТИКА
47
Приложение medllt.UCQ^Ukx
dt
ЭКСПОНЕНЦИАЛЬНАЯ ЗАВИСИМОСТЬ
Изменения концентрации лекарственных пре-
паратов в плазме в режиме реального времени
обычно выражаются математическими уравнения-
ми, содержащими один или более экспоненциаль-
ных членов. В математическом выражении
103 = 1000
103 — экспоненциальный член; число 10 надо воз-
вести в третью степень, чтобы получить 1000. Урав-
нения такого типа удобно записывать с помощью
логарифмов чисел. Логарифм — это степень, в кото-
рую надо возвести основание, чтобы получить нуж-
ное число. Следовательно, логарифм 1000 по осно-
ванию 10 равен 3.
Поскольку 10а х 10ь = 10а+ь;
10а / 10ь = 10аЬ,
то умножение чисел можно выполнить путем сло-
жения логарифмов, а деление - путем их вычита-
ния; после чего проводится антилогарифмическое
преобразование. С этой целью обычно используют-
ся десятичные логарифмы, то есть логарифмы
чисел по основанию 10 (обозначаемые logic или 1g).
Натуральные логарифмы (logs или In) — это ло-
гарифмы, где в качестве основания используется ма-
тематическая константа е = 2,718. В численном вы-
ражении константу е можно представить в виде сум-
мы бесконечно сходящейся последовательности:
1 1
Р = 1 + 1 ----1------|-
2x1 3x2x1 -
ххх ххххх
= 1 + х -ь---+--------ь...
2x1 3x2x1
(сходится при всех конечных значениях х).
Константу е можно также представить в виде ир-
рационального числа, равного сумме ряда:
е = (1+7)п (п —> х)
Важность константы е обусловлена экспоненци-
альной зависимостью, при которой скорость изме-
нения переменной пропорциональна ее величине.
Экспоненциальную зависимость математически
можно выразить с помощью натуральных логариф-
мов. Многие процессы, связанные с всасыванием,
распределением и элиминацией лекарственных
препаратов, можно представить в виде экспоненци-
альной зависимости их концентрации от времени.
При этом снижение или повышение концентрации
лекарственного препарата прямо пропорционально
где dX/dt—скорость уменьшения или увеличения
переменной X за бесконечно малый промежуток
времени t, к - константа, а X - значение переменной
в момент времени t.
Интегрируя это выражение в пределах от t = 0 до
t = оо, получаем:
Х=Х0 • ekt (для экспоненциального увеличения);
Х=Х0 • в'к1(для экспоненциального снижения),
где X - значение X в любой момент времени t; Хо —
значение X в начальный момент времени; е — осно-
вание натуральных логарифмов (2,718), а к — кон-
станта. При этом X представляется как экспоненци-
альная функция времени.
Рассмотрим уравнение для экспоненциального
уменьшения концентрации
Х=Х0 • ekt
Вычисляя натуральный логарифм, получаем
1пХ = 1пХ0 - kt
ln(X/X0) = -kt
Следовательно, k = - ln(X/X0)/t; в этом уравне-
нии к может рассматриваться как константа скоро-
сти и выражает пропорциональные изменения пе-
ременной X в единицу времени. Скорость измене-
ния экспоненциальной зависимости можно также
представить как период полувыведения или кон-
станту времени.
k = [-ln(X/X0)]/t
t = [ln(X/X0)]/(-k)
Если (Х/Хо) = ’/г, то t выражает время, в течение
которого X уменьшается вдвое от исходного значе-
ния; следовательно,
t = [1п(*/2)]/(-к) = 1п2/к=0,693/к
Такое значение t выражает “полужизнь” экспо-
ненциально изменяющейся переменной.
Если
Х/Хо = 1/2,718 = 1/е,
то t представляет время, необходимое для уменьше-
ния X до 1/е (37%) от исходного значения. Таким
образом,
t = [ln( 1/е)]/(-к) = [1п(е1 )]/(-к) = (-1пе)/(-к) = 1Д
В этом случае t выражает константу времени
экспоненциальной зависимости, значение которой
обратно константе скорости к.
ее величине, или, записав математически:
ФАРМАКОДИН/ШШ2 u 3
Лекарственные препараты вызывают множе-
ство различных эффектов. Обычно в их основе
лежит изменение клеточной функции под воздей-
ствием веществ с относительно простой молеку-
лярной структурой. В некоторых случаях (напри-
мер, при использовании ингаляционных анестети-
ков, местных анестетиков и миорелаксантов)
имеется достаточно близкое соответствие между
химическим строением или физико-химическими
свойствами лекарственных препаратов и их эффек-
том. В других случаях сходные по химическому
строению лекарственные препараты (например,
промазин и прометазин), оказывают совершенно
разное фармакологическое действие.
ЗАВИСИМОСТЬ МЕЖДУ ДОЗОЙ
ЛЕКАРСТВЕННОГО ПРЕПАРАТА
И ЕГО ЭФФЕКТОМ
Зависимость эффекта лекарственного препара-
та от его дозы (концентрации) описывается кривой
доза-эффект. Концентрацию лекарственного пре-
парата можно измерить в плазме лабораторных жи-
вотных и человека, но концентрация в точке прило-
жения действия обычно неизвестна. Следовательно,
in vivo трудно с точностью определить зависимость
эффекта препарата.от его концентрации. В прошлом
для измерения эффекта лекарственных средств
обычно использовались изолированные тканевые
препараты (например, прямая мышца живота
лягушки, препарат диафрагмального нерва и диа-
фрагмы крысы, подвздошная кишка морской свин-
ки). В раствор, омывающий тканевой препарат,
добавляли до определенной концентрации лекар-
ственное средство, после чего измеряли эффект.
Иногда изучались кумулятивные реакции (между
повторными введениями лекарственного средства
тканевые препараты не промывались). Многие
наблюдаемые и измеримые эффекты препаратов-
агонистов первоначально были изучены именно
в таких экспериментальных условиях. В последние
годы стали широко использоваться более совер-
шенные методы каскадного непрерывного омыва-
ния образцов тканей растворами препаратов
различной концентрации, а также прямое измере-
ние эффекта на клеточном и субклеточном уров-
нях in vitro и в тканевых культурах. Для этих целей
иногда применяются человеческие ткани, удален-
ные во время хирургических операций (в виде изо-
лированных препаратов или тканевых культур).
In vitro зависимость между дозой препарата и его
эффектом отображают кривой доза-эффект. В обыч-
ных координатах эта кривая чаще всего имеет фор-
му гиперболы. Для построения такой кривой необ-
ходим слишком широкий диапазон концентраций,
поэтому на практике ее часто представляют в полу-
логарифмических координатах (по горизонталь-
ной оси откладывают логарифм концентрации);
в этом случае она приобретает S-образную форму
(рис. 3.1). Такой метод представления имеет ряд
преимуществ: так, зависимость на участке от 20 до
80% максимума носит линейный характер, что по-
зволяет одновременно сравнивать и оценивать
лекарственные препараты различной мощности.
В полулогарифмических координатах у лекарст-
венных препаратов с одинаковым механизмом дей-
ствия кривые доза-эффект параллельны. Как пока-
зано на рис. 3.1, при повышении дозы препарата
эффект нарастает — вначале выражение, потом
в меньшей степени, пока наконец не достигает мак-
симума, выходя на плато. В полулогарифмических
координатах кривая доза-эффект мощных лекарст-
венных препаратов (например, фентанила) смеще-
на влево, к вертикальной оси. Напротив, кривая до-
за-эффект лекарственных препаратов меньшей
мощности (например, морфина) смещена вправо.
Зависимость между концентрацией лекарствен-
ного препарата и его эффектом можно представить
также с помощью кривой Хилла. Эта кривая была
впервые использована А.В. Хиллом около ста лет
тому назад при изучении зависимости SO2 от РО2.
Аргументом является логарифм дозы (концентра-
ФАРМАКОДИНАМИКА
49
Рис.3.1. Зависимость
«доза-эффект».
(а). В линейных коор-
динатах кривая имеет
форму гиперболы.
(б). В полулогариф-
мических координатах
кривая принимает S- об-
разную форму, причем
на большем ее протяже-
нии зависимость носит
линейный характер
ции) лекарственного препарата, в то время как
функция представлена следующим выражением:
Emax — Е
где Emax — максимальный эффект, а Е — эффекты,
соответствующие различным дозам (концентраци-
ям) лекарственного препарата (рис. 3.2).
Для многих лекарственных препаратов зависи-
мость доза-эффект имеет гиперболическую форму
(или S-образную в полулогарифмических коорди-
натах; рис 3.1); в этом случае график Хилла пред-
ставляет собой прямую линию с тангенсом угла на-
клона, равным +1 (коэффициент Хилла). Но суще-
ствуют и другие типы зависимости: так, форма
кривой доза-эффект для агонистов никотиновых,
глутаминовых и ГАМК-рецепторов чаще является
скорее сигмовидной, нежели гиперболической. В
некоторых экспериментальных моделях кривая до-
за-эффект в полулогарифмических координатах
двухфазна (например, при воздействии адреналина
и норадреналина на сердце кролика) или имеет ко-
локолообразную форму (воздействие гистамина на
гладкую мускулатуру легочной артерии морской
свинки; рис. 3.3). В таких случаях кривая Хилла не-
линейна, или же коэффициент Хилла значительно
отличается от единицы.
У интактных животных и человека зависимость
доза-эффект более сложна и может изменяться под
влиянием механизмов гомеостаза и фармакокине-
тических процессов (например, в ходе образования
активных метаболитов или связывания препаратов
с белками плазмы). Следовательно, эффект препара-
та зависит от его концентрации в плазме или в точке
приложения действия. Соответственно, наклон
кривой концентрация-эффект может отражать
терапевтическую широту. Если кривая доза-эф-
фект отличается значительной крутизной (напри-
мер, у фентанила), токсический эффект может
наступить при сравнительно небольшом увеличе-
нии дозы. Напротив, если наклон кривой более по-
логий (например, у диазепама), то увеличение
дозы, как правило, менее опасно.
Зависимость вида «доза-эффект» отражает ре-
акцию на постепенное увеличение дозы лекарст-
венного средства в тканевом препарате, а не ответ
типа «все или ничего». Раньше реакции типа «все
или ничего» широко использовали в эксперимен-
тальной фармакологии для определения медианы
эффективной дозы (ED50) и медианы летальной
дозы (LD50), с тем чтобы оценить терапевтическую
Рис. 3.2. Кривая Хилла отражает зависимость между ло-
гарифмом дозы препарата и логарифмом величины
ЕДЕ^-Е), где Етм — максимальный эффект, а Е —эффек-
ты, соответствующие различной дозе (концентрации) ле-
карственного препарата. В данном случае коэффициент
Хилла равен +1,0
50
Глава 3
Рис. 3.3. Атипичные
кривые доза-эффект
(представлены в по-
лулогарифмических
координатах).
(а) двухфазная кри-
вая;
(б) колоколообраз-
ная кривая
широту препарата. В последние годы эти тесты счи-
таются неинформативными и от их применения
практически отказались.
ОСНОВНЫЕ МЕХАНИЗМЫ
ДЕЙСТВИЯ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ
Существует четыре основных механизма дейст-
вия лекарственных препаратов:
1. Эффекты, обусловленные физическими или
химическими свойствами препарата.
2. Подавление активности ферментов, регули-
рующих метаболические процессы.
3. Воздействие на ионные каналы или транс-
портные белки клеточных мембран.
4. Воздействие на рецепторы — функционально
активные макромолекулярные компоненты клеток
(чаще всего белки).
ЭФФЕКТЫ, ОБУСЛОВЛЕННЫЕ
ФИЗИЧЕСКИМИ ИЛИ
ХИМИЧЕСКИМИ СВОЙСТВАМИ
ПРЕПАРАТА
Простые антациды нейтрализуют кислый желу-
дочный сок, оказывая лишь незначительное воз-
действие на pH и кислотно-щелочное состояние.
Большинство кислот и оснований вызывают изме-
нения pH крови и мочи, обусловленные увеличени-
ем или уменьшением концентрации ионов водоро-
да в биологических жидкостях.
Эффекты комплексобразующих лекарственных
препаратов (пеницилламин, кальциево-динатрие-
вая соль ЭДТА, кобальтовая соль ЭДТА, димерка-
прол, дефероксамин) также обусловлены их хими-
ческими свойствами. Эти препараты связываются
с ионами некоторых металлов, образуя водораство-
римые хелатные соединения, которые постепенно
удаляются из организма с мочой или иными путя-
ми. Комплексобразующие препараты применяют-
ся для лечения отравлений тяжелыми металлами,
а также при некоторых заболеваниях (таблица 3.1).
Действие других лекарств, включая некоторые
анестетики, может быть обусловлено их физиче-
скими свойствами. Согласно классическим концеп-
циям, местные и ингаляционные анестетики вызы-
вают неспецифические физические изменения ли-
пидных или белковых компонентов мембран
нейронов, оказывая опосредованный эффект на их
проницаемость и ионный транспорт. Полученные
в последнее время данные позволяют предполо-
жить, что местные и ингаляционные анестетики
оказывают более специфическое воздействие на
мембраны нейронов. Местные анестетики взаимо-
действуют с аминокислотными остатками в натрие-
вых каналах, так что проницаемость мембран для
натрия снижается ниже порогового уровня, необ-
ходимого для проведения возбуждения (глава 6).
Ингаляционные анестетики также относительно
избирательно влияют на мембранные белки или ре-
цепторы в нейронах головного мозга (глава 5).
ПОДАВЛЕНИЕ АКТИВНОСТИ
ФЕРМЕНТОВ, РЕГУЛИРУЮЩИХ
МЕТАБОЛИЧЕСКИЕ ПРОЦЕССЫ
Действие многих лекарственных препаратов
обусловлено угнетением активности ферментов, ре-
гулирующих различные метаболические процессы
(таблица 3.2). Такие препараты по своему химиче-
скому строению часто напоминают естественные
ФАРМАКОДИНАМИКА
51
Таблица 3.1. Комплексобразующие лека,
Комплексобразующий препарат Токсичное вещество Показания к применению
Дефероксамин Железо Алюминий Отравление железом Передозировка железа Перегрузка алюминием
Кобальтовая соль ЭДТА Цианид Отравление цианидами
Димеркапрол Мышьяк Золото Свинец Ртуть Отравление тяжелыми металлами Отравление мышьяком
Пеницилламин Медь Свинец Ртуть Цинк Отравление тяжелыми металлами Болезнь Вильсона Ревматоидный артрит Цистинурия
Кальциево-динатриевая соль ЭДТА Свинец Отравление тяжелыми металлами
субстраты, метаболизирующиеся под воздействи-
ем этих ферментов. Например, аллопуринол (инги-
битор ксантиноксидазы) является близким анало-
гом ксантина и гипоксантина; в норме под воздейст-
вием ксантиноксидазы эти пуриновые основания
превращаются в мочевую кислоту.
Лекарственные препараты могут вызывать об-
ратимое или необратимое угнетение ферментов.
Обратимые ингибиторы ферментов (например, эд-
рофоний, эноксимон и аллопуринол) конкурируют
с естественными субстратами, которые метаболи-
зируются теми же ферментами, и их действие обыч-
но обусловлено образованием обратимого фер-
мент-ингибиторного комплекса:
Фермент + ингибитор о фермент-ингибитор-
ный комплекс.
Концентрация конкурентного ингибитора в плаз-
ме или тканях влияет на степень угнетения фермен-
та. По мере того как конкурентные ингибиторы
элиминируются из организма, активность фермен-
та постепенно восстанавливается. Поскольку дей-
ствие таких ингибиторов не зависит от формирова-
ния стабильных химических связей, их эффекты
относительно кратковременные и, вероятно, отра-
жают присутствие лекарственных препаратов или
их активных метаболитов в среде, непосредственно
окружающей фермент.
Хотя неостигмин, пиридостигмин и физостиг-
мин обычно относят к обратимым ингибиторам
ацетилхолинэстеразы и холинэстеразы, это спра-
ведливо лишь для первой фазы их действия. Во вто-
рой фазе образуются ковалентные химические свя-
зи между МН2СО-группами этих ингибиторов,
с одной стороны, и эфирными участками ацетилхо-
линэстеразы и холинэстеразы — с другой. Кова-
лентные связи медленно гидролизуются, после чего
активность ферментов постепенно восстанавлива-
ется (глава 7).
Необратимое угнетение активности ферментов
обычно обусловлено образованием стабильного хи-
мического комплекса между лекарственным препа-
ратом и ферментом. В таких случаях регенерация
фермента часто невозможна; активность фермента
восстанавливается только спустя время, необходи-
мое для его синтеза de novo в достаточном количе-
стве. Необратимое угнетение ферментов вызывают
фосфорорганические инсектициды, метотрексат и
большинство мощных ингибиторов моноаминок-
сидазы. Эти препараты обладают чрезвычайно дли-
тельной продолжительностью действия, и их эф-
фекты сохраняются спустя дни и недели после эли-
минации из организма. Аспирин оказывает
аналогичное действие на циклооксигеназу тромбо-
цитов (циклооксигеназу-1), вызывая необратимое
ацетилирование фермента на протяжении всей
жизни тромбоцита (7-9 дней).
В некоторых случаях под воздействием фер-
ментов лекарственные препараты превращаются
в активные метаболиты. Метилдофа в перифериче-
ских и центральных симпатических нейронах пре-
вращается в метилдофамин и метилнорадреналин;
эти метилированные метаболиты могут замещать
физиологический нейромедиатор (норадреналин)
в окончаниях симпатических нервов. Цитотоксиче-
52
Глава 3
Таблица 3.2. Лекарственные препараты, USgb ид ИОННЫЕ
которых частично или полностью обусловлено угне-
тением активности ферментов
Лекарственный препарат Угнетаемый фермент
Аллопуринол Ксантиноксидаза
Аминофиллин Эноксимон Милринон Фосфодиэсте раза
Ацетилсалициловая ки- слота (и другие НПВС) Циклооксигеназа (простагландинсинтетаза)
Каптоприл Эналаприл Лизиноприл Ангиотензин-превра- щающий фермент
Бенсеразид ДОФА-декарбоксилаза
Хлорфенилаланин Три птофан гидро ксил аза
Дисульфирам Альдегиддегидрогеназа
Метотрексат Триметоприм Пириметамин Дигидрофолатредуктаза
Метилдофа ДОФА-декарбоксилаза
Эдрофоний Неостигмин Пиридостигмин Органические фосфаты Ацетилхолинэстераза Холинэстераза плазмы (псевдохолинэстераза)
Бензилпенициллин Транспептидаза бакте- риальной стенки
Моклобемид Фенелзин Транилципромин Селигилин Моноаминоксиадаза
Сул ьфан илам иды Фолатсинтетаза
В некоторых случаях угнетение ферментов происхо-
дит под воздействием метаболитов лекарственных пре-
паратов.
ский препарат фторурацил метаболизируется до
ложного промежуточного нуклеотида, который не
способен превращаться в тимидиловую кислоту
под действием тимидилатсинтетазы, что приводит
к угнетению синтеза ДНК.
КАНАЛЫ И ТРАНСПОРТНЫЕ
БЕЛКИ КЛЕТОЧНЫХ МЕМБРАН
Помимо ферментов и рецепторов, лекарствен-
ные препараты могут непосредственно воздейст-
вовать на натриевые, калиевые или кальциевые ка-
налы. Например, местные анестетики вызывают
блокаду потенциалзависмых натриевых каналов
в мембранах нейронов (глава 6). Пероральные ги-
погликемические препараты блокируют АТФ-за-
висимые калиевые каналы в р-клетках поджелу-
дочной железы, вызывая их деполяризацию и сек-
рецию инсулина (глава 16). Диазоксид и тиазидные
диуретики открывают калиевые каналы в глад-
ких мышцах сосудов, вызывая гиперполяризацию,
вазодилатацию и снижение АД; некоторые анти-
ангинальные препараты оказывают аналогичное
действие на калиевые каналы в клетках мио-
карда (например, никорандил). Антагонисты
кальция воздействуют на кальциевые каналы
L-типа в сердце (верапамил) или гладких мышцах
сосудов (дигидропиридины).
ВОЗДЕЙСТВИЕ НА РЕЦЕПТОРЫ
Многие лекарственные препараты осуществляют
свое действие, связываясь с макромолекулярными
компонентами клеток, которые называются рецеп-
торами. Эти компоненты обычно представляют собой
белки или гликопротеины. Функция большинства
рецепторов состоит во взаимодействии с эндоген-
ными гормонами и медиаторами. Рецепторы связы-
ваются с эндогенными (гормоны, факторы роста,
медиаторы) или экзогенными лигандами (лекарст-
венные препараты), после чего передают поступаю-
щий от них сигнал внутрь клетки-мишени.
Концепция рецепторов была пред ложена Дж. Лэнг-
ли, чтобы объяснить явную специфичность дей-
ствия отдельных лекарственных препаратов
(например, воздействие пилокарпина и атропина
на секрецию слюнных желез, а никотина и препара-
тов кураре — на нервно-мышечную передачу). Важ-
ная роль в развитии концепции хеморецепторов
тканей также принадлежит немецкому врачу Пау-
лю Эрлиху. Взгляды Эрлиха сформировались под
влиянием работ Лэнгли, а также собственных
исследований, посвященных перекрестной ре-
зистентности к антитрипаносомным препаратам
с аналогичной химической структурой. Впоследст-
вии А. Дж. Кларк и Дж.Х. Гэддам разработали мате-
матические и количественные аспекты взаимодей-
ствия лекарственных препаратов с рецепторами.
В последующих работах, проводившихся в
1930-1960-е гг., природа и свойства лекарственных
ФАРМ АКОДИ НАМ И КА
53
рецепторов в основном объяснялись вза^(®С№1
шением их структуры и функции. В исследованиях
того времени анализировались реакции изолиро-
ванных тканей на воздействие лекарственных пре-
паратов со схожей химической структурой (напри-
мер, реакция гладкой мышцы на воздействие эфи-
ров холина). Несмотря на существенное значение,
эти работы представляли лишь косвенный подход к
изучению структуры и свойств рецепторов.
Начиная с 1965 г., стало возможным выделять
рецепторы из тканей, а также изучать их свойства в
препаратах клеточных мембран in vitro. В последнее
время для исследования структуры, функции и ор-
ганизации рецепторов применяются методы моле-
кулярной биологии. Интактные рецепторы выде-
ляют из клеточных мембран, помещают в раствор
и очищают путем аффинной хроматографии (или по-
добными методами), а затем определяют их амино-
кислотную последовательность. После этого стано-
вится возможным определить последовательность
оснований соответствующей рецептору матричной
РНК (мРНК) и, используя технологию рекомби-
нантной ДНК, получить комплементарную ДНК
(кДНК). Затем с помощью транскрипции кДНК
получают мРНК и вводят ее в клеточную культуру
in vitro (например, в ооциты лягушки или гибрид-
ные клетки нейробластомы-глиомы), что приводит
к экспрессии рецепторов и позволяет изучать их
свойства (экспрессионное клонирование). В неко-
торых случаях клеточные рецепторы воссоздаются
с помощью собственных систем транскрипции, что
позволяет изучить молекулярные механизмы, ле-
жащие в основе их физиологических и биохимиче-
ских эффектов. Подобным образом, используя биб-
лиотеки кДНК, удалось клонировать различные
типы и подтипы рецепторов и экспрессировать их в
клеточных культурах. Тем не менее физиологиче-
ские функции некоторых подтипов рецепторов не-
известны. В отдельных случаях были идентифици-
рованы так называемые «рецепторы-сироты» (ре-
цепторы с неизвестными эндогенными лигандами).
Для изучения распределения и гетерогенности
рецепторов в тканях млекопитающих и человека
широко используются меченные радиоактивными
изотопами соединения с высоким сродством к спе-
цифическим рецепторам (радиолиганды). Напри-
мер, некоторые нейротоксины змеиного яда (такие,
как а-бунгаротоксин или токсин а-кобра) избира-
тельно связываются с никотиновыми рецепторами
двигательной концевой пластинки. Меченые ней-
ротоксины применяются для выделения холиноре-
цепторов из различных синапсов. М-холинорецеп-
торы в сердце, тонкой кишке, симпатических ганг-
лиях и головном мозге также идентифицированы
UCOMiUwo меченных изотопами антагонистов
(3Н-хиннуклидинилбензилат, 3Н-М-метилскопо-
ламин, 3Н-пирензепин).
С помощью меченых антагонистов дофамина
(3Н-галоперидол, 3Н-спиперон, 3Н-флупентиксол)
в различных участках ЦНС были идентифицирова-
ны дофаминовые рецепторы (черная субстанция,
полосатое тело, лимбическая система, хеморецепто-
ры триггерных зон и гипофиз). Эти рецепторы
встречаются и в гладких мышцах сосудов (почечных
и мезентериальных), и в симпатических ганглиях.
Радиолиганды, как агонисты (3Н-гидроксибен-
зилизопреналин), так и антагонисты (1251-йодоциа-
нопиндолол или 3Н-дигидроальпренолол), позво-
ляют маркировать и идентифицировать а- и р-адре-
норецепторы. Адренорецепторы очень широко
представлены в организме. Относительно часто
встречаются внесинаптические адренорецепторы,
расположенные в участках, отдаленных от адренер-
гических окончаний (например, а2-рецепторы
тромбоцитов, р2-рецепторы гладких мышц сосу-
дов). Плотность адренорецепторов меняется при
различных патологических состояниях (бронхи-
альная астма, сердечная недостаточность, тирео-
токсикоз). При высокой концентрации катехола-
минов количество и плотность адренорецепторов
снижается (down-regulation). Отчасти этот фено-
мен обусловлен секвестрацией (интернализацией)
рецепторов внутри клеток, хотя может изменяться
и сродство рецепторов. Напротив, любое снижение
концентрации адреналина и норадреналина в кро-
ви, вызванное как медикаментозным воздействием,
так и симпатической денервацией, приводит к уве-
личению количества рецепторов (up-regulation).
Подобные изменения плотности рецепторов, веро-
ятно, характерны для большинства нейромедиато-
ров, действующих в синапсах и нейроэффекторных
соединениях.
В некоторых случаях увеличение количества ре-
цепторов может быть одной из причин феномена
суперчувствительности (т. е. избыточной, но, по
сути, нормальной реакции на введение агонистов).
После периферической денервации или поврежде-
ния скелетной мышцы количество рецепторов уве-
личивается (up-regulation), что приводит к усиле-
нию ответной реакции на ацетилхолин и деполяри-
зующие миорелаксанты (глава 7). Хирургическая
или фармакологическая симпатическая денерва-
ция также усиливает периферические реакции на
норадреналин (и, в меньшей степени, на адрена-
лин). В этом случае усиление реакции в основном
обусловлено угнетением обратного синаптическо-
го захвата катехоламинов; повышение количества
адренорецепторов в результате up-regulation имеет
меньшее значение.
54
Глава 3
Многие рецепторные системы характе{ПВ£££Ш1. ЫСО^рГЫ
определенной чувствительностью, специфично-
стью и насыщаемостью. Большинство рецепторов
высокочувствительно к природным агонистам, так
что даже низкая концентрация агониста оказывает
выраженное действие. Следовательно, для оказа-
ния фармакологического эффекта может потребо-
ваться значительное усиление первоначального
стимула. Кроме того, многие рецепторные системы
проявляют высокую селективность, взаимодейст-
вуя только с несколькими химически схожими со-
единениями, которые по структуре часто идентич-
ны природным нейромедиаторам или гормонам.
Наконец, лекарственные рецепторы обладают огра-
ниченной емкостью связывания. Количество ре-
цепторов определенного типа в клетке обычно по-
стоянно, но может меняться под воздействием фи-
зиологических и фармакологических факторов.
Следовательно, связывание многих радиолигандов
и лекарственных препаратов с рецепторами харак-
теризуется конкурентностью и насыщаемостью.
Лекарственные рецепторы имеют высокое сродст-
во и малую емкость по отношению к специфиче-
ским агонистам и антагонистам; напротив, неспе-
цифическое связывание отличается низким сродст-
вом и высокой емкостью.
Большинство обнаруженных и изученных ре-
цепторов располагается на наружной поверхности
клеток. Важным исключением являются рецепто-
ры стероидных гормонов, которые находятся в ци-
топлазме или на ядерной мембране клеток-мише-
ней. Активация этих рецепторов приводит к изме-
нению активности мРНК и опосредованно влияет
на синтез рибосомальных белков.
Обратимое связывание агониста с рецептором —
это первое действие, запускающее последователь-
ность биофизических и биохимических реакций
(например, транспорт ионов, активация фермента и
синтез белка), приводящих к фармакологическому
эффекту. Эти реакции можно представить следую-
щим образом:
Лекарственный препарат + рецептор
Комплекс лекарственный препарат-рецептор
Активированный лекар- Лекарственный
ственным препаратом ->
А к препарат+рецептор
рецепторный комплекс г г
Биофизические или биохимические изменения
аимодеиствии с рецепторами медиаторы,
гормоны и лекарственные препараты способны за-
пускать следующие молекулярные механизмы:
1. Непосредственное изменение проницаемости
мембран для ионов.
2. Повышение или угнетение синтеза внутри-
клеточных метаболитов.
3. Повышение активности тирозинкиназы (ау-
тофосфорилирование ).
4. Модификация транскрипции ДНК.
ИЗМЕНЕНИЕ ПРОНИЦАЕМОСТИ МЕМБРАН
ДЛЯ ИОНОВ
Иногда активация рецепторов приводит к не-
посредственному воздействию на ионные каналы
в синаптических мембранах, что вызывает селек-
тивное или неселективное повышению проницае-
мости для ионов. В этом случае рецептор и ионный
канал представляют собой единый макромолеку-
лярный комплекс (т. е. рецептор содержит внутри
себя ионный канал). Обычно такое повышение
проницаемости для ионов является быстрым и крат-
ковременным; максимальная реакция развивается
в течение микросекунд и продолжается лишь не-
сколько миллисекунд. Таким способом действуют
некоторые нейромедиаторы (ацетилхолин, ГАМК,
глутамин и глицин), повышая проницаемость пост-
синаптических мембран для ионов.
УСИЛЕНИЕ ИЛИ УГНЕТЕНИЕ СИНТЕЗА
ВНУТРИКЛЕТОЧНЫХ МЕТАБОЛИТОВ
Чаще всего активация рецепторов не приводит к
непосредственным изменениям проницаемости
мембран для ионов или другим немедленным эф-
фектам. Многие нейромедиаторы и лекарственные
препараты являются очень мощными агентами,
причем их эффект обеспечивается амплификацией
начального стимула. В таких случаях активация ре-
цептора приводит к увеличению или уменьшению
активности специфических ферментов, сопряжен-
ных с мембранами, что, в свою очередь, приводит к
накоплению или исчезновению внутриклеточных
метаболитов. Эти метаболиты, представляющие со-
бой связующее звено между активацией рецепто-
ров и эффектом лекарственного препарата, называ-
ют вторыми посредниками.
Ниже приведены четыре наиболее важные сис-
темы вторых посредников:
1. Циклический аденозинмонофосфат (цАМФ).
2. Циклический гуанозинмонофосфат (цГМФ).
3. Оксид азота (NO).
4. Инозитолфосфаты и диацилглицерин.
Эффект
ФАРМАКОДИНАМИКА
55
Агонист + рецептор
Комплекс агонист-рецептор
medlit.ucoz.ru
Агонист + рецептор
ь
1
Комплекс агонист-рецептор
Активация рецептора + G-белок (Gs или Gj)
Активация (Gs)
Угнетение (Gj)
»
Аденилатциклаза Фосфодиэстераза
АТФ -----------» цАМФ ——-------—»АМФ
Активация протеинкиназы А
Фосфорилирование белка
о
Изменение проницаемости мембраны для ионов
Деполяризация или гиперполяризация
Повышенное поступление ионов кальция
Синтез белка
Активация ДНК
Рис. 3.4. Синтез цАМФ с участием аденилатциклазы
Циклический аденозинмонофосфат (цАМФ)
Циклический аденозинмонофосфат синтези-
руется из АТФ под действием фермента адени-
латциклазы (рис. 3.4). Ферментная система аде-
нилатциклазы представляет собой семейство
тканеспецифических изоферментов с различными
регуляторными функциями. Многие лекарственные
препараты и гормоны связываются с рецепторами
клеточной мембраны, что изменяет активность аде-
нилатциклазы, таким образом регулируя синтез
цАМФ. Рецепторные системы опосредованно сопря-
жены с аденилатциклазой через регуляторные G-бел-
ки (белки, связывающие ГТФ), которые могут сти-
мулировать (Gs) или подавлять (Gi) активность
фермента. Когда регуляторным белком является
Gs, агонист рецептора активирует аденилатциклазу
и повышает синтез цАМФ; если же регуляторным
Активация рецептора + G-белок (Gs или Gj)
Активация (Gs)
или угнетение (Gi)
аден илатциклазы
а-ГДФ-р-у 4-----------.
Обмен ГДФ/ГТФ; ’
диссоциация р-у
субъединиц
г
<----а-ГТФ
ГТФаза
а-ГДФ + Р-у ..........
Рис. 3.5. Роль регуляторных G-белков в активации (Gs)
или угнетении (GJ аденилатциклазы. Регуляторные бел-
ки Gs и G, состоят из трех субъединиц (а, р и у); в неактив-
ном состоянии а-субъединица связана с ГДФ. Активиро-
ванные рецепторы связывают Gs или G(, приводя к замене
ГДФ на ГТФ (то есть ГДФ высвобождается из связи
с а-субъединицей и замещается ГТФ); происходит также
диссоциация комплекса, состоящего из р-у-субъединиц.
Затем комплекс а-субъединица-ГТФ соединяется с аде-
нилатциклазой, в результате чего происходит активация
(Gs) или угнетение (G.) фермента. Впоследствии ком-
плекс а-субъединица-ГТФ расщепляется собственной
ГТФазой с образованием комплекса а-субъедини-
ца-ГДФ, который вновь соединяется с Р- и у-субъедини-
цами с восстановлением неактивного G-белка. В некото-
рых ферментных системах комплекс из Р-у-субъединиц
выполняет сигнальную функцию
белком является Gr агонист рецептора угнетает
фермент и снижает синтез цАМФ. G-белки могут
вызывать значительную амплификацию сигнала
после связывания лиганда с рецептором, поскольку
Gs (или Gj) может повторно активироваться един-
ственным комплексом агонист-рецептор. Следова-
тельно, многие лекарственные препараты, чье дейст-
вие опосредуется цАМФ, обладают очень большой
мощностью. Регуляторные G-белки имеют гетеро-
тримерную структуру: они состоят из трех различ-
ных субъединиц — а, р и у. В неактивном состоянии
субъединица а связана с ГД Ф (а-ГДФ-р-у). При ак-
тивизации рецептора происходит диссоциация
комплекса р-у-субъединиц, а-субъединица связыва-
ется с ГТФ (в обмен на ГДФ), отделяется от рецеп-
тора и соединяется с аденилатциклазой (рис. 3.5).
56
Глава 3
После активации или угнетения d>e[>.Mjnedht.u(r0ZHHai3.3 Системы рецепторов и вторые по-
плекс а-субъединица- 1ТФ быстро гидролизуется и
вновь связывается с Р-у-субъединицами с образова-
нием неактивного G-белка (а-ГДФ-р-у).
G-белки играют важную роль в деятельности
многих других клеточных систем; например, регу-
ляторный белок Go регулируетопосредованную ре-
цепторами активность фосфолипазы С и расщепле-
ние фосфоинозитидов (см. ниже). Все рецепторы,
эффект которых опосредуется G-белками (метабо-
тропные рецепторы), имеют одинаковую молеку-
лярную структуру, состоящую из одной длинной
полипептидной цени с семью трансмембранными
доменами.
В некоторых системах ГТФ-связывающие бел-
ки (G| или Go) непосредственно регулируют от-
крывание и закрывание ионных каналов; например,
в агадрснорсцепторах, Mr и Мгхолинорецепто-
рах, а также в опиатных рецепторах комплекс
0-у-субъединиц открывает калиевые каналы, что
приводит к гиперполяризации и ингибирующим
эффектам. G-белки также участвуют в реализации
феномена десенситизации и тесно связаны с белка-
ми Ras/Raf, которые опосредуют экспрессию ге-
нов и деление клеток. На регуляторные G-белки
влияют некоторые бактериальные экзотоксины;
например, холерный токсин активирует Gs (путем
АДФ-рибозилирования его а-субъединицы), а ток-
син бациллы коклюша подавляет опосредованную
рецепторами активацию G, и G,,.
Эффекты многих лекарственных препаратов и эн-
догенных соединений опосредуются рецепторами,
сопряженными с регуляторным белком Gs и адени-
латциклазой; следовательно, активация рецептора
приводит к повышению активности фермента и
увеличению синтеза цАМФ. Все Р-алренергические
эффекты катехоламинов обусловлены повышени-
ем внутриклеточной концентрации цАМФ. Роль
цАМФ была открыта Сазерлендом и Роллом, изу-
чавшими влияние адреналина на гликогенолиз в
изолированных гепатоцитах. Позднее выяснилось,
что повышение внутриклеточной концентрации
цАМФ опосредует эффекты многих других рецеп-
торов, включая аденозиновые А2-рецепторы, гиста-
миновые Нгрецепторы, вазопрессиновые Vrpeuen-
торы, дофаминовые Dp и Ds-рецепторы и некоторые
5-ГТ- и простаноидные рецепторы (табл. 3.3).
Напротив, эффекты других лекарственных пре-
паратов и гормонов .зависят от активации рецепто-
ров. связанных с регуляторным белком G,; при этом
подавляется активность аденилатциклазы и снижа-
ется синтез цАМФ. Этим механизмом частично
опосредовано действие опиоидов на ц-. к- и &-ре-
цепторы, а также воздействие норадреналина на
средники
Тип рецептора Главный эффек- торный путь
Рецепторы АКТГ Gs. А:АЦ. Т цАМФ
Аденозиновые рецепторы А|.Аз А? Gt. 1:АЦ, 1 цАМФ Gs. А;АЦ, t цАМФ
Адренорецепторы 0.2 Р> Gq. А:ФЛС, ? ИФ3 Gt. 1.АЦ, 1 цАМФ Gs. А;АЦ. Т цАМФ
Ангиотензиновые рецеп- торы АТ, АТг Go. А ФЛС. Т ИФэ G|, 1:АЦ, 1 цАМФ
Дофаминовые рецепторы d,.d5 D2. Од Gs. А:AC, t цАМФ Gi. 1:АС, 4 цАМФ
Эндотелиновые рецепторы ЕТа.ЕТв Gq, АФЛС. t ИФ3
Глюкагоновые рецепторы Gs, А:АЦ, t цАМФ
Рецепторы глутаминовой кислоты mGlu,, mGlus mGluz-i, mGlug, mGlu; Gq, АФЛС. t ИФз Gt. 1;АЦ, 4 цАМФ
Рецепторы ГАМК0 Gt, Г.АЦ, 4 цАМФ
Г истаминовые рецепторы н, Н2 Go, АФЛС, Т ИФз Gs. А:АЦ, t цАМФ
Г идрокситриптаминовые (5-НТ)рецепторы 5-НТ, 5-НТ2 5-НТд. 5-НТе, 5-НТ7 Gt. 1:АЦ, 4 цАМФ Go. А ФЛС, Т ИФз Gs. А:АЦ, t цАМФ
М-холинорецепторы М,. М3. М5 М2, Мд Go. АФЛС, Т ИФз Gt. 1:АЦ, 4 цАМФ
Опиатные рецепторы д, к,6 Gt. 1:АЦ, 4 цАМФ
Вазопрессиновые рецеп- торы V, v2 Gq, А ФЛС, Т ИФз Gs. А;АЦ, t цАМФ
А - активация; АЦ аденилатциклаза; АКТГ - адре-
нокортикотропный гормон; цАМФ - циклический аде-
нозинмонофосфат; ГАМК - у-аминомасляная кислота;
G|, Gs, Go — G-белки; 1 - подавление; ИФ, - инози-
тол-).4.5-трнфосфат, ФЛС - фосфолипаза С.
ФАРМАКОДИНАМИКА
57
Таблица 3.4. Характеристика
Семейство изоферментов Сродство Распределение в тканях Селективные ингибиторы
цАМФ цГМФ
1 Са2+-кальмоду- лин-зависимые * Высокое Головной мозг Сердце Печень Почки Фенотиазины Винпоцетин
II цГМФ-стиму- лируемые Низкое Низкое Сердце Кора надпочечников
III цГМФ-ингиби- руемые Высокое Высокое Сердце Кровеносные сосуды Дыхательные пути Тромбоциты Пимобендан Милринон Эноксимон Пероксимон
IV цАМФ-специ- фичные Высокое Низкое Головной мозг Сердце Почки Клетки воспалитель- ного инфильтрата Ролипрам Денбуфиллин
V цГМФ-специ- фичные 4 4 Половой член Тромбоциты Силденафил Дипиридамол Запринаст
VI цГМФ-чувст- вительные ★ 4 Сетчатка глаза
* Различные изоферменты могут иметь разное сродство к субстрату
а2-рецепторы. Кроме того, некоторые эффекты аце-
тилхолина, аденозина, дофамина, глутаминовой
кислоты и соматостатина обусловлены ослаблени-
ем активности аденилатциклазы (таблица 3.3).
В некоторых случаях противоположные или до-
полняющие друг друга физиологические реакции
могут зависеть от активации или подавления
аденилатциклазы; примером являются симпати-
ческие и парасимпатические влияния на ЧСС и со-
кратимость миокарда. Повышение симпатического
тонуса приводит к активации p-адренорецепторов
и, в свою очередь, к возрастанию синтеза цАМФ.
Напротив, повышение тонуса блуждающего нерва
активирует М2-холинорецепторы миокарда, ослаб-
ляя синтез цАМФ.
Внутриклеточный цАМФ быстро связывается с
регуляторными (R) субъединицами протеинкина-
зы А; затем активируются свободные каталитиче-
ские (С) субъединицы фермента, что вызывает
фосфорилирование мембранных белков (рис. 3.4).
Эти изменения, в свою очередь, приводят к другим
эффектам (фосфорилирование ионных каналов;
изменение проницаемости для кальция или калия,
вызывающее деполяризацию или гиперполяриза-
цию; синтез белка; активация ДНК).
После активации аденилатциклазы белком Gs
повышенная внутриклеточная концентрация
цАМФ быстро возвращается к исходному уровню
под действием фосфодиэстеразы (ФДЭ). ФДЭ ка-
тализирует превращение цАМФ в АМФ (рис. 3.4).
Другой изофермент ФДЭ превращает родственный
циклический нуклеотид цГМФ в ГМФ. В клетках
млекопитающих обнаружено несколько семейств
изоферментов фосфодиэстеразы, которые отлича-
ются по специфичности, распределению в тканях и
чувствительности к ингибиторам (таблица 3.4).
Важную роль в гидролизе цАМФ играет ФДЭ III и,
в меньшей степени, ФДЭ IV.
Ингибиторы фосфодиэстеразы пролонгируют эф-
фекты цАМФ, что имитирует длительную стимуля-
цию рецепторов, сопряженных с белком Gs (т.е. по-
вышение внутриклеточной концентрации цАМФ).
58
Глава 3
medlit. щшнГтЫп
Некоторые лекарственные препараты о
свое действие, обратимо угнетая один или несколь-
ко изоферментов ФДЭ. Многие метилксантины
(аминофиллин, теофиллин, теобромин и кофеин)
неселективно подавляют активность изофермен-
тов ФДЭ во многих тканях, оказывая положитель-
ный инотропный эффект, вызывая вазо- и брон-
ходилатацию, угнетая агрегацию тромбоцитов и ли-
полиз. Некоторые другие препараты, сходные по
строению с цАМФ (например, эноксимон, перокси-
мон, милринон), селективно подавляют активность
изофермента ФДЭ III, который присутствует пре-
имущественно в сердце и гладких мышцах сосудов
(таблица 3.4). Действие этих препаратов в основ-
ном ограничено сердечно-сосудистой системой, и их
используют как инотропные средства. Ингибито-
ры ФДЭ IV (например, ролипрам) оказывают про-
тивовоспалительное и бронхорасширяющее дейст-
вие. Ингибиторы ФДЭ V (например, запринаст
и дипиридамол) в значительной степени подавля-
ют агрегацию тромбоцитов (таблица 3.4).
Циклический гуанозинмонофосфат (цГМФ)
Циклический нуклеотид цГМФ является вто-
рым посредником, передающим сигналы некото-
рых гормонов и нейромедиаторов. Он синтезирует-
ся из ГТФ при участии фермента гуанилатциклазы,
существующей в двух различных формах. Первая
форма гуанилатциклазы является компонентом не-
которых клеточных мембран (сопряженная с мем-
браной гуанилатциклаза); другая присутствует в
цитоплазме в растворимой форме и активируется
оксидом азота (NO).
Воздействие предсердного натрийуретического
белка (ПНБ) на клетки почек и гладкие мышцы
сосудов опосредовано сопряженной с мембраной
гуанилатциклазой и повышением синтеза цГМФ.
ПНБ (и аналогичные пептиды) связываются с дву-
мя видами рецепторов (ПНБА и ПНБВ), обладаю-
щими внутренней гуанилатциклазной активностью.
Активация фермента зависит не от связи с G-белка-
ми, а от реакции аутофосфорилирования (см. ниже).
Большинство эффектов NO опосредовано цито-
плазматической (растворимой) формой гуанилат-
циклазы, которая стимулирует образование цГМФ
в клетке. Последующая активация цГМФ-зависи-
мых протеинкиназ приводит к фосфорилированию
мембранных белков, снижает поступление кальция
в клетку и в дальнейшем оказывает влияние на ак-
тивность нейронов и кровеносные сосуды.
Метаболизм цГМФ зависит от активности изо-
ферментов ФДЭ; некоторые изоферменты (напри-
мер, ФДЭ I и ФДЭ III) обладают высоким сродст-
вом к цГМФ (таблица 3.4). Подавление активности
ФДЭ V силденафилом пролонгирует и усиливает
олового члена, которая зависит от синте-
за цГМФ in situ. У некоторых людей сильденафил
может вызвать расстройства зрения, что объясняет-
ся его неспецифическим воздействием на ФДЭ VI.
Оксид азота (NO)
Физиологическая роль NO была открыта во вре-
мя исследований эндотелиального релаксирующе-
го фактора (endothelium derived relaxing factor,
EDRF), которые показали, что вызываемая ацетил-
холином вазодилатация зависит от фактора, обра-
зующегося в сосудистом эндотелии. Впоследствии
было доказано, что активность этого фактора за-
висит от синтеза NO эндотелиальными клетками,
и что эндотелиальный релаксирующий фактор —
это и есть NO.
В некоторых рецепторных системах NO являет-
ся внутри- и межклеточным посредником. Актива-
ция М3-холинорецепторов в кровеносных сосудах
повышает синтез NO в клетках эндотелия; за-
тем NO диффундирует в гладкомышечные клетки
и стимулирует продукцию цГМФ, что вызывает ва-
зодилатацию. Кроме того, NO высвобождается из
нейронов и играет важную роль в развитии воспа-
лительных и иммунных реакций.
В большинстве клеток NO синтезируется из
L-аргинина под действием синтазы оксида азота
(NOS). Существует три различные изоформы NOS.
Нейрональные синтазы оксида азота (nNOS;
NOS-I) присутствуют в большинстве перифериче-
ских и центральных нейронов. Эндотелиальные
NOS (eNOS; NOS-III) локализуются в сосудистом
эндотелии и некоторых других тканях (например,
в клетках миокарда, остеобластах и тромбоцитах).
Нейрональные и эндотелиальные NOS являются
кальцийзависимыми конститутивными (постоян-
но присутствующими) ферментами. Индуцируе-
мая NOS (iNOS; NOS II) — это кальцийнезависи-
мая неконстутивная изоформа, которая появляется
в клетках воспалительного инфильтрата и иммун-
ной системы при некоторых патологических со-
стояниях; активность iNOS значительно выше, чем
nNOS и eNOS. Все три изоформы представляют со-
бой гемсодержащие белки (напоминающие строе-
нием цитохром Р-450) и имеют связующие участки
для L-аргинина, Са2+-кальмодулина, флавиновых
нуклеотидов и тетрагидробиоптерина.
Нейрональная NOS опосредует синтез NO во мно-
гих периферических и центральных нейронах.
В периферической нервной системе NO высвобо-
ждается из окончаний неадренергических- нехоли-
нергических нервов в адвентиции кровеносных
сосудов, модулируя нейрональную регуляцию
функции гладких мышц во многих органах и тка-
нях (например, в желудке, тонкой кишке и матке).
ФАРМ АКОДИ НАМ И КА
59
В половом члене при высвобождении NOR36kdUt
чаний неадренергических-нехолинергических нер-
вов увеличивается содержание цГМФ, что приво-
дит к расслаблению гладких мышц кавернозных
тел и эрекции. В ЦНС при активации глутамино-
вых рецепторов NMDA-типа повышается внутри-
клеточная концентрация кальция и увеличивается
его связывание с кальмодулином, что стимулирует
синтез NO и цГМФ. NO диффундирует в соседние
клетки и стимулирует синтез цГМФ, а также участ-
вует в ретроградной нейротрансмиссии и регуля-
ции пластичности синапсов.
иССййдБЬЬлиальная NOS обеспечивает синтез NO
в сосудистом эндотелии. Затем NO проникает из
эндотелия в гладкомышечные клетки, где связыва-
ется с гемом, содержащимся в растворимой (цито-
плазматической) гуанилатциклазе; происходит ак-
тивация фермента и стимуляция синтеза цГМФ.
После этого активируются цГМФ-зависимые про-
теинкиназы, вызывая фосфорилирование мем-
бранных белков, снижение поступления кальция
в клетку и расслабление гладких мышц сосудов
(рис. 3.6). NO также взаимодействует с внутри-
клеточными сульфгидрильными (SH-) группами
Эндотелиальная клетка
Кровь
L-аргинин
+ 0г NOS
L-гидроксиаргинин
+ 0? NOS
▼
NO ♦
Глутатион-S-
-трансфераза
Нитроглицерин
Нитропруссид натрия
Рис. 3.6. Синтез, вы-
свобождение и эф-
фекты NO в гладких
мышцах сосудов.
SH - сульфгидриль-
ные группы
Гладкомышечная клетка
▼ ♦SH'
NO -------------——►S-нитрозотиолы
Растворимая гуанилатциклаза *-------------
Активация
ГТФ .......................—фцГМФ
Протеинкиназы
▼
Фосфорилирование белков
♦
Расслабление гладких мышц сосудов
60
Глава 3
белков; образующиеся
аминокислот и
S-нитрозотиоловые соединения являются мощны-
ми вазодилататорами. Ацетилхолин, брадикинин
и увеличение действующей на эндотелий силы
сдвига повышают синтез NO в эндотелиальных
клетках и вызывают цГМФ-зависимую релакса-
цию гладких мышц сосудов. В сочетании с проста-
циклином NO действует как ингибитор агрегации и
адгезии тромбоцитов. NO может оказывать отрица-
тельный инотропный эффект. Постоянное образо-
вание NO эндотелиальными клетками — важный
фактор, определяющий базальный сосудистый то-
нус и регуляцию периферического сосудистого
сопротивления.
Лекарственные препараты, превращающиеся
в процессе метаболизма в NO, вызывают расслабле-
ние гладких мышц сосудов (рис. 3.6). Нитроглице-
рин и нитропруссид натрия хорошо растворяются
в липидах и поэтому легко проникают из кровенос-
ного русла в эндотелий и гладкомышечные клетки
сосудов. В этих клетках под воздействием глутати-
OH-S-трансферазы и ферментных систем цитохро-
ма Р-450 они расщепляются до NO и нитрозотио-
лов, в результате чего активируется растворимая
гуанилатциклаза и стимулируется синтез цГМФ.
NO также участвует в регуляции легочного кро-
вотока. Ингаляция низких концентраций NO
(30-50 частей NO на миллион частей вдыхаемой
смеси) успешно применяется для лечения легочной
гипертензии и респираторного дистресс-синдрома
взрослых. NO может угнетать высвобождение эн-
дотелина — вызывающего вазоконстрикцию поли-
пептида, синтезируемого в гладких сосудистых
мышцах. В кровеносных сосудах NO быстро расще-
пляется, и его Ti/2 составляет 5-7 секунд. Следова-
тельно, после ингаляции NO быстро удаляется из
организма, не достигая значительной концентра-
ции в системном кровотоке.
Индуцируемая NOS является кальцийнезави-
симой изоформой, которая экспрессируется многи-
ми иммунными клетками и клетками воспалитель-
ного инфильтрата, а также эндотелием и микроглией.
Синтез iNOS индуцируют бактериальные липоса-
хариды, эндотоксины, у-интерферон, TNF-a, ин-
терлейкин- 1р и другие цитокины. При лихорадке
различного генеза в результате воздействия iNOS
часто повышается экскреция нитратов. Подобные
изменения происходят при бактериальном сепсисе
и эндотоксемии, и NO является основной причиной
отсроченной артериальной гипотонии при шоке.
При язвенном колите увеличивается синтез NO
в клетках воспалительного инфильтрата. NO мо-
жет быть агрессивным оксидантом и важным
компонентом цитотоксичности, опосредуемой
анными макрофагами. Индукцию iNOS
подавляют многие кортикоостероиды, что является
важной составляющей их противовоспалительного
действия (глава 16).
Инозитолфосфаты и диацилглицерин
Фосфоинозитиды — это группа мембранных
фосфолипидов. Во многих клетках активация ре-
цепторов и гидролиз мембранных фосфоинозити-
дов приводит к высвобождению кальция из внутри-
клеточных и внеклеточных депо.
Связывание рецептора агонистами активирует
фермент фосфолипазу С (ФЛС). Активация этого
фермента, как и аденилатциклазы, опосредуется
регуляторным Go-белком (рис. 3.7). Фосфолипаза
С гидролизует мембранный фосфолипид фосфати-
дилинозитол 4,5-дифосфат (ФИФ2) до диацилгли-
церина (ДАГ) и инозитол-1,4,5-трифосфата (ИФ3).
Оба продукта этой реакции являются вторыми по-
средниками. ДАГ активирует протеинкиназу С,
вызывая фосфорилирование многих внутрикле-
точных белков. ИФ3 связывается с внутриклеточ-
ными рецепторами эндоплазматического ретику-
лума, что приводит к открытию кальциевых кана-
лов и высвобождению кальция в цитоплазму.
Действие многих лекарственных препаратов и ней-
ромедиаторов зависит от рецепторных механизмов,
которые запускаются при активации ФЛС, гидро-
лизе ФИФ2 до ИФ3 и ДАГ и высвобождении из эн-
доплазматического ретикулума кальция, который
впоследствии связывается с кальмодулином или
тропонином (таблица 3.3).
Связывание кальция с кальмодулином приво-
дит к конформационным изменениям белков и мо-
жет модифицировать активность многих фермен-
тов, что приводит к синтезу белков, изменениям
ионной проницаемости, выделению секрета из же-
лез и сокращению гладких мышц (рис. 3.7). Анало-
гичную роль в сердечных и скелетных мышцах иг-
рает тропонин С.
Хотя цАМФ, цГМФ, ИФ3, ДАГ и кальций обыч-
но считаются независимыми внутриклеточными
посредниками, есть убедительные доказательства
того, что их эффекты взаимосвязаны. Например,
активация единственного рецептора или подтипа
рецептора может опосредоваться множеством
G-белков (или различными субъединицами одного
G-белка), которые регулируют активность не-
скольких вторых посредников. Кроме того, цАМФ
может повышать внутриклеточную концентрацию
кальция и оказывать влияние на различные эффек-
ты, связанные с изменением его концентрации.
Фосфорилирование многих ферментов и белков
с помощью цАМФ или цГМФ часто приводит к
значительному повышению чувствительности этих
ФАРМАКОДИНАМИКА
61
Рис. 3.7. Фосфатиди-
линозитоловый путь.
В скелетных мышцах
и миокарде высвобо-
жденные ионы каль-
ция связываются с
тропонином С.
ДАГ — диацилглице-
рин; ЭР — эндоплазма-
тический ретикулум;
ИФ3 — инозитол-1,4,5-
-трифосфат; ФИФ2 —
фосфатид ил инози-
тол-4,5-дифосфат
medlit.ucoz.ru
Агонист + рецептор
Комплекс агонист-рецептор
Активация рецептора + G-белок (Gq)
Активация
Фосфолипаза С
ФИФ2 ИФ3
ДАГ
Мобилизация ионов кальция
из ЭР и внеклеточной жидкости
Активация протеинкиназы
+ кальмодулин
Са2+-кальмодулиновый
комплекс
Фосфорилирование белков
Синтез белков
Секреция желез
Мышечное сокращение
циклических нуклотидов к кальцию. Кальций счи-
тают универсальным вторым посредником, отве-
чающим за регуляцию многих видов клеточной ак-
тивности.
ПОВЫШЕНИЕ АКТИВНОСТИ
ТИРОЗИНКИНАЗЫ
(АУТОФОСФОРИЛИРОВАНИЕ)
Связывание лиганда с рецепторами на поверх-
ности клеточной мембраны может привести к со-
единению расположенных рядом парных рецепто-
ров, результатом чего является внутриклеточное
фосфорилирование их собственных тирозиновых
остатков (аутофосфорилирование). Инсулиновые
рецепторы состоят из двух внеклеточных а-субъе-
диниц и двух внутриклеточных 0-субъединиц, ко-
торые соединены между собой дисульфидными
связями (S-S), образуя тетрамерную молекулу
0-а-а-0. Связывание инсулина двумя а-субъеди-
ницами приводит к усилению тирозинкиназной
активности в 0-единицах и последующему фосфори-
лированию аминокислотных остатков других внут-
риклеточных белков.
С подобными рецепторами взаимодействуют
многие цитокины и интерлейкины, а также пред-
сердный натрийуретический белок (ПНБ) и анало-
гичные пептиды. Внутриклеточные сегменты ре-
цепторов ПНБаиПНБв обладают гуанилатциклаз-
ной активностью.
МОДИФИКАЦИЯ ТРАНСКРИПЦИИ ДНК
Стероидные гормоны и их синтетические анало-
ги оказывают свое действие, связываясь с внутри-
клеточными рецепторами и косвенно влияя на син-
тез белков. Стероидные рецепторы представляют
собой несвязанные с другими клеточными компо-
нентами цитоплазматические или ядерные белки,
присутствующие в большинстве клеток. Плотность
62
Глава 3
этих рецепторов в разных типах клеток мЛж«¥ЭнаА
чительно отличаться. Все стероидные гормоны хо-
рошо растворяются в липидах и проникают через
клеточные мембраны внутрь клетки, где связыва-
ются со свободными стероидными рецепторами в ци-
топлазме, формируя активированные комплексы.
Рецепторы отделяются от других белков, переме-
щаются из цитоплазмы в ядро и связываются с оп-
ределенными последовательностями ДНК на регу-
ляторных участках генов-мишеней. Транскрипция
генов либо подавляется, либо индуцируется, что
приводит к изменениям мРНК и синтеза белка в ри-
босомах.
Эффекты многих стероидов зависят от их влия-
ния на синтез белка. Так, противовоспалительное
действие кортикостероидов в основном обусловле-
но их связыванием с внутриклеточными стероид-
ными рецепторами, что приводит к изменениям
в синтезе ДНК и РНК (глава 16).
КОЛИЧЕСТВЕННЫЕ АСПЕКТЫ
ВЗАИМОДЕЙСТВИЯ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
С РЕЦЕПТОРАМИ
Классическая теория взаимодействия веществ
с рецепторами (оккупационная теория) была раз-
работана А. Дж. Кларком в 1930-е гг. Согласно этой
теории, каждая молекула лекарственного средства
обратимо связывается с рецептором, образуя ком-
плекс препарат-рецептор. Доля занятых препара-
том рецепторов зависит от концентрации свободно-
го (несвязанного) лекарственного препарата [D]
и константы диссоциации комплекса препарат-ре-
цептор KD (Приложение I):
[D]
Kd+[D]
Если общее количество рецепторов остается по-
стоянным, это математическое выражение можно
представить графически в виде гиперболы. По мере
увеличения концентрации лекарственного препа-
рата доля занятых рецепторов растет, пока при вы-
соких концентрациях занятыми не оказываются
почти 100% рецецторов (рис. 3.8). Допуская, что
фармакологический эффект прямо пропорциона-
лен доле связанных рецепторов, зависимость меж-
ду дозой лекарственного препарата и эффектом
также можно представить гиперболической кри-
вой. Выше в этой главе уже был охарактеризован
такой тип зависимости (рис. 3.1), когда коэффици-
ент Хилла равен +1 (рис. 3.2). До 1950-х гг. господ-
ствовала классическая оккупационная теория,
согласно которой фармакологический эффект
ucoz.ru
Рис. 3.8. Гиперболическая зависимость между концен-
трацией лекарственного препарата и долей занятых ре-
цепторов. Предполагается, что каждая молекула агониста
связывается с рецептором на некооперативной основе
находится в прямой линейной зависимости от ко-
личества рецепторов, связанных с агонистом.
В 1956 г. Стефенсон выявил очевидные недос-
татки оккупационной теории и ввел ряд важных из-
менений. Он обосновал, что максимальный эффект
при введении агониста можно получить уже при
связывании небольшой доли рецепторов, и что во
многих зонах имеются «резервные рецепторы»
(сверх количества, необходимого для генерации
максимального эффекта). Иными словами, чтобы
вызвать максимальный эффект, лекарственным
препаратам достаточно связаться лишь с относи-
тельно небольшой долей от популяции рецепторов.
Наличие резервных рецепторов гарнтирует адек-
ватные фармакологические эффекты при относи-
тельно низкой концентрации препаратов или ме-
диаторов. Считают, что число резервных рецепто-
ров особенно велико в гладких мышцах, в то время
как в других тканях их меньше. Полагают также,
что запасные рецепторы имеются в нервно-мышеч-
ных синапсах. Для индукции максимального мы-
шечного сокращения при непрямой стимуляции
может потребоваться 25% общего количества ре-
цепторов, а для длительного тетанического сокра-
щения — 50%. Следовательно, недеполяризующие
миорелаксанты, чтобы оказать значимый клиниче-
ский эффект, должны связаться с большим количе-
ством рецепторов.
Стефенсон также обосновал, что фармакологи-
ческий эффект не находится в линейной зависимо-
сти от доли занятых рецепторов, как это можно
ФАРМ АКОДИ НАМ И КА
63
предположить из окупационной теории:
могут вызывать одинаковый эффект, даже если за-
нимают разные доли популяции рецепторов.
В настоящее время ясно, что оккупационная
теория представляет собой упрощенный подход,
а многие фармакологические эффекты сложны и не
имеют линейной зависимости от связывания пре-
парата с рецепторами. Кривая доза-эффект не все-
гда имеет форму гиперболы: она может быть, на-
пример, сигмовидной, а коэффициент Хилла —
представлять неинтегральную величину больше
единицы. Такой феномен имеет место при введении
препаратов, действующих на никотиновые, глута-
миновые и ГАМК-рецепторы. Считается, что его
возникновение связано с наличием множества свя-
зывающих участков (сайтов) на одном макромоле-
кулярном комплексе. Связывание молекулы пре-
парата одним рецепторным сайтом может привести
к конформационным (аллостерическим) измене-
ниям и увеличению или уменьшению сродства ос-
тальных участков к этому препарату. Эта концеп-
ция (концепция кооперативности) была выдвинута
в середине 1960-х гг. Моно, Уайменом и Шанже,
чтобы объяснить аллостерические свойства фер-
ментов. Известно, что именно так взаимодействуют
со своим субстратом (кислородом) некоторые бел-
ковые молекулы (гемоглобин). Связывание гемо-
глобина с одной молекулой кислорода влияет на его
сродство к другим молекулам кислорода. Другие
вещества (например, 2,3-дифосфоглицерат) также
могут связываться с гемоглобином и значительно
снижать его сродство к кислороду.
Похожий феномен возникает в нервно-мышеч-
ных синапсах. При изучении эффектов ацетилхо-
лина и других агонистов на н-холинорецепторы
нервно-мышечного синапса установлено, что кри-
вая концентрация-эффект имеет сигмовид-
ную форму, а кривая Хилла — характерный
наклон (коэффициент Хилла > 1,5). Это объясня-
ется наличием двух участков связывания, рас-
положенных в a-у и а-8 субъединицах н-холино-
рецептора. Связывание ацетилхолина с одним
участком изменяет сродство второго, а для откры-
тия ионного канала требуется одновременное свя-
зывание с обоими участками. Коффициент Хилла
может отражать повышение вероятности откры-
вания ионного канала при одновременном связы-
вании обоих участков (положительная коопера-
тивность).
В некоторых экспериментальных моделях кри-
вая доза-эффект в полулогарифмических коорди-
натах двухфазна (например, при воздействии адре-
налина и норадреналина на сердце кролика) или
имеет колоколообразную форму (воздействие
U®«U на гладкие мышцы легочной артерии
морской свинки; рис. 3.3). Форму обеих кривых
можно объяснить тем, что агонисты действуют на
две популяции рецепторов, опосредующие проти-
воположные реакции (например, активацию или
угнетение сократительной способности гладких
мышц сосудов). Природу столь сложной зависимо-
сти доза-эффект позволяют выявить селективные
антагонисты.
АНТАГОНИСТЫ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ
Лекарственные препараты-антагонисты предот-
вращают или уменьшают фармакологический эф-
фект агонистов. Выделяют следующие типы анта-
гонистов:
1. Конкурентные антагонисты
(а) обратимые
(б) необратимые
2. Неконкурентные антагонисты
Конкурентные антагонисты конкурируют с аго-
нистами за одни и те же участки связывания;
чаще всего такой антагонизм является обратимым.
Напротив, неконкурентные антагонисты не конку-
рируют с агонистами за связывание с рецепторами.
КОНКУРЕНТНЫЕ АНТАГОНИСТЫ
Взаимодействие лекарственного препарата с ре-
цептором включает два компонента: связывание
и активацию. Способность лекарственного препа-
рата связываться с рецептором, то есть вступать с ним
в химическое взаимодействие, называется сродст-
вом. Способность лекарственного препарата после
связывания с рецептором вызывать его активацию
называется внутренней активностью. Когда агони-
сты связываются с рецепторами и индуцируют
фармакологический эффект, одновременно прояв-
ляется их сродство к рецепторам и внутренняя ак-
тивность.
Напротив, конкурентный антагонист связыва-
ется с рецептором (чаще всего обратимо), но не
вызывает эффекта, то есть он обладает сродством к
рецепторам, но лишен внутренней активности.
Следовательно, конкурентный антагонист не акти-
вирует рецептор и не индуцирует биохимические
и биофизические изменения, приводящие к фарма-
кологическому эффекту. Большинство конкурент-
ных антагонистов можно полностью вытеснить из
связи с рецепторами достаточно большими дозами
агонистов. Для конкурентных антагонистов харак-
терен параллельный сдвиг вправо кривой доза-эф-
фект (представленной в полулогарифмических
64
Глава 3
Рис. 3.9.Влияние обратимых конкурентных антаго-
нистов на кривую доза-эффект: (а) без антагониста;
(б) в присутствии антагониста. Для кривой доза-эффект
(представленной в полулогарифмических координатах)
характерен параллельный сдвиг вправо; при этом наклон
кривой и максимальный эффект агониста остаются
прежними
координатах); при этом наклон кривой и максималь-
ный эффект агониста остаются прежними (рис. 3.9).
Конкурентные антагонисты широко применя-
ются в анестезиологии (таблица 3.5). Конкурент-
ные антагонисты можно охарактеризовать дозовым
соотношением — т. е. величиной, на которую нужно
умножить дозу агониста, чтобы вызвать эквива-
лентный эффект в присутствии антагониста:
[А]
Дозовое соотношение - 1 = ——
Kd(A)
где [А] - молярная концентрация антагониста,
a Kj(A) - константа диссоциации комплекса анта-
гонист-рецептор, которая аналогична константе
диссоциации комплекса препарат-рецептор (K,i)
и является обратным отражением сродства антаго-
ниста к рецепторам. Это уравнение известно как
уравнение Шилда; если на ось абсцисс нанести зна-
чения log[ А], а на ось ординат — log[ дозовое соотно-
шение - 1], то получается прямая линия зависимо-
сти, по которой легко определить значение Ка(А).
Кривую Шилда, т.е. кривую log[A] - log[flO3O-
вое соотношение -1] можно использовать для срав-
нения мощности конкурентных антагонистов по ве-
личине их рА2 — отрицательного логарифма моляр-
ной дозы антагониста, при которой дозовое соотно-
шение равно 2. В этом случае Kd(A)= [А]; рА2=
-log[K<i(A)]. Значения рА2 некоторых м-холинобло-
каторов, полученные в эксперименте на изолиро-
medlit.uaQz^s.s. Обратимые конкурентные
антагони-
сты, применяемые в анестезиологии
Лекарственный пре- парат-антагонист Эндогенный лиганд
Атропин Гликопирролат Г иосцин Ацетилхолин (конкурен- ция за м-холинорецеп- торы)
Алкуроний Атракурий Цис-Атракурий Галлам ин Панкуроний Рокуроний Тубокурарин Векуроний Ацетилхолин (конкурен- ция за н-холинорецеп- торы нервно-мышечно- го синапса)
Триметафан Тубокурарин Ацетилхолин (конкуренция за м-холинорецепторы вегетативных ганглиев)
Атенолол Эсмолол Пропранолол Адреналин и норадре- налин (конкуренция за Р-адре- норецепторы)
Прохлорперазин Домперидон Метоклопрамид Дофамин
Хлорфенамин* Прометазин Алимемазин| Циметидин Ранитидин Г истамин
Ондансетрон Г ранисетрон Трописетрон Доласетрон 5-гидрокситриптамин (конкуренция за 5-НТз-рецепторы)
Налоксон Налтрексон Эндорфины
Флумазенил Эндогенные бензодиа- зепины
*Хлорфенамин (хлорфенирамин).
ТАлимемазин (тримепразин)
ванных тканевых препаратах, где агонистом был
ацетилхолин, приведены в таблице 3.6. Определе-
ние рА2 — это удобный способ сравнения мощно-
сти обратимых конкурентных антагонистов; чем
препарат активнее, тем выше его рА2. Эти значения
ФАРМАКОДИНАМИКА
65
Таблица 3.6. Значения рА2 некоторых лек ^ЙЙ^пп^Цювами, вначале необратимые антагонисты
ных препаратов, блокирующих м-холинорецепторы
(препарат подвздошной кишки морской свинки)
могут вызывать эффект, характерный для обрати-
мых. При повышении концентрации и увеличении
Лекарственный препарат Значение рА2
Г иосцин 9,5
Гликопирролат 9,5
Атропин 9,0
Прометазин 7,7
Хлорпромазин 7,5
Амитриптилин 6,7
Петидин 5,3
Соталол 5,1
Пропранолол 5,0
Имипрамин 3,4
не зависят от свойств и относительной мощности
агонистов, которые конкурируют с антагонистами
за одни и те же популяции рецепторов.
Некоторые конкурентные антагонисты конку-
рируют с агонистами за связи с рецепторами, но
очень медленно высвобождаются из таких связей;
в результате общее количество рецепторов, доступ-
ных агонистам, уменьшается. Такие конкурентные
антагонисты называются необратимыми. Они ока-
зывают свое действие, образуя стабильные химиче-
ские связи с рецептором, а также нарушая про-
странственное расположение или вызывая дефор-
мацию молекулы рецептора. При неконкурентном
антагонизме наблюдается не только непараллель-
ное (с уменьшением наклона) смещение вправо
кривой доза-эффект, но и уменьшение максималь-
ного эффекта агониста. Необратимым антагони-
стам обычно свойственна значительная продолжи-
тельность действия, и их эффекты не зависят от
концентрации в плазме. Примерами необратимого
антагонизма являются воздействие феноксибенза-
мина на а-адренорецепторы и воздействие а-бунга-
ротоксина на н-холинорецепторы нервно-мышеч-
ного синапса.
Несмотря на эти очевидные различия, точно
разграничить обратимые и необратимые антагони-
сты не всегда возможно. Как уже упоминалось,
многие ткани содержат резервные рецепторы, что
позволяет получить максимальный эффект при
относительно низкой доле занятых рецепторов.
При этом необратимое снижение общего количест-
ва рецепторов первоначально может не вызывать
снижения максимального эффекта, и кривая до-
за-эффект будет смещаться вправо параллельно.
экспозиции проявляются отчетливые признаки не-
обратимого антагонизма. Иногда препарат действу-
ет двухфазно: вначале — как обратимый антаго-
нист, потом — как необратимый.
НЕКОНКУРЕНТНЫЕ АНТАГОНИСТЫ
Лекарственный антагонизм, который не зависит
от конкуренции между агонистами и антагониста-
ми за связь с рецепторами, называют неконкурент-
ным антагонизмом. Он может обуславливаться
конформационными (аллостерическими) измене-
ниями в рецепторах; например, тахикардия, инду-
цированная галламином, отчасти развивается из-за
уменьшения сродства сердечных м-холинорецеп-
торов к ацетилхолину.
Кроме того, неконкурентный антагонизм может
возникать при непосредственном химическом со-
единении агониста и антагониста; при этом дейст-
вие агониста полностью угнетается или ослабевает.
Например, действие ионов некоторых металлов мо-
жет нейтрализоваться различными комплексобра-
зующими средствами (таблица 3.1). Такой тип хи-
мического антагонизма широко используется при
лечении отравлений тяжелыми металлами. Анти-
коагулянтный эффект гепарина подавляется про-
тамином: кислые сульфатные группы гепарина
нейтрализуются щелочными остатками аргинина.
К другому виду неконкурентного антагонизма
относят функциональный антагонизм (физиологи-
ческий антагонизм): эти термины иногда использу-
ют для описания воздействия лекарств на две неза-
висимые рецепторные системы, которые в норме
реализуют противоположные ответные реакции.
Так, гистамин вызывает сокращение гладких мышц
бронхов, а адреналин расслабляет их; таким обра-
зом, адреналин можно считать функциональным
антагонистом гистамина. В некоторых случаях
такой антагонизм наблюдается в физиологических
условиях (например, противоположные эффекты
симпатической и парасимпатической нервных
систем).
И, наконец, фармакокинетические взаимодей-
ствия, которые приводят к уменьшению концентра-
ции лекарственных препаратов в плазме и сниже-
нию их активности в результате нарушения всасы-
вания, распределения, метаболизма или
выведения, могут считаться формой неконкурент-
ного антагонизма (глава 9).
66
Глава 3
medlit.ucoz.ru
Рис. 3.10. (а) Кривая доза-эффект для полного агониста на фоне постоянной концентрации частичного агониста.
Сплошная линия — реакция на полный агонист; пунктирная линия - реакция на полный агонист в присутствии час-
тичного агониста. Если концентрация полного агониста низка, то внутренняя активность обоих препаратов складыва-
ется, и в результате частичный агонист усиливает эффект. При более высокой концентрации полного агониста эффект
снижается, поскольку частичный агонист (с внутренней активностью < 1) конкурирует с полным агонистом (внутрен-
няя активность = 1) за связывание с рецепторами. При очень высокой концентрации полного агониста частичный аго-
нист полностью вытесняется из связи с рецепторами, и максимальный эффект не меняется, (б) Кривая доза-эффект
для частичного агониста на фоне различных концентраций полного агониста (кривые А-Д). При низкой концентра-
ции полного агониста (А и Б) частичный агонист оказывает дополнительный эффект вследствие сложения внутрен-
них активностей; при более высоких концентрациях полного агониста (Г и Д) частичный агонист оказывает антагони-
стическое действие, поскольку конкурирует с полным агонистом за связывание с рецепторами. Когда эффект опреде-
ленной концентрации полного агониста равен максимальному эффекту частичного агониста (В), какие-либо
изменения эффекта отсутствуют или незначительны
ЧАСТИЧНЫЕ АГОНИСТЫ
В 1954 г. Ариенс продемонстрировал, что неко-
торые лекарственные препараты в зависимости от
условий обладают свойствами как агонистов, так
и антагонистов. Концепция внутренней активности
изначально была разработана именно для объясне-
ния этого феномена. Как выше отмечено, агонисты
обладают двумя свойствами: сродством (способно-
стью связываться с рецептором) и внутренней
активностью (способностью после связывания
с рецептором вызывать его активацию).
Конкурентные антагонисты имеют различное
сродство к рецепторам (которое отражают значе-
ния рАг), но не обладают внутренней активностью
или обладают ей в малой степени. Частичные аго-
нисты также обладают сродством к рецепторам
и конкурируют за связи с ними как с полными
агонистами, так и с конкурентными антагонистами.
В то же время частичные агонисты обладают неко-
торой внутренней активностью, которая намного
выше, чем у конкурентных антагонистов (ноль),
но ниже, чем у полных агонистов (единица). Следо-
вательно, частичные агонисты могут проявлять
свойства как агонистов, так и антагонистов в за-
Таблица 3.7. Частичные агонисты опиатных д- и
к-рецепторов
Лекарствен- ный препарат Внутренняя активность в отно- шении опиатных рецепторов
ц-рецепторы к-рецепторы
Налорфин +
Левалорфан +
Пентазоцин 44-
Циклазоцин ++
Буторфанол -Н-+
Налбуфин ++
Бупренорфин 4-4-
+ - слабая; ++ - умеренная; +++ - выраженная
висимости от условий, в которых они применяются
(рис. 3.10). В низких дозах (концентрациях) час-
тичные агонисты обычно вызывают эффекты
агониста: если фоновая концентрация полного аго-
ниста невысока, то внутренняя активность обоих
ФАРМАКОДИНАМИКА
67
препаратов складывается, и происходит Uf^gpWHblE АГОНИСТЫ
эффекта. При более высокой концентрации полно-
го агониста эффект снижается, потому что в этом
случае частичный агонист действует как конку-
рентный антагонист. Когда эффект определенной
концентрации полного агониста равен максималь-
ному эффекту частичного агониста, какие-либо из-
менения эффекта отсутствуют или незначительны
(рис. 3.10).
Активность частичного агониста зависит от экс-
прессии и плотности рецепторов, а также от эффек-
тивности клеточных механизмов передачи сигнала,
которые могут различаться в различных тканях.
Лекарственные препараты, являющиеся частич-
ными агонистами в одной ткани или системе, мо-
гут быть полными агонистами или антагонистами
в другой.
Известны две группы частичных агонистов,
имеющие клиническое значение для человека.
Частичными агонистами Р-адренорецепторов яв-
ляются Р-адреноблокаторы; это их свойство иногда
называют внутренней симпатомиметической ак-
тивностью. Пропранолол не обладает внутренней
симпатомиметической активностью и вызывает
только блокаду Р-адренорецепторов. Напротив,
пиндолол и окспренолол харакризуются значи-
тельной внутренней симпатомиметической актив-
ностью и могут оказывать симпатомиметическое
действие (таблица 13.4). Другие частичные агони-
сты Р-адренорецепторов (например, ксамотерол)
используют в качестве инотропных средств при
сердечной недостаточности и в критических со-
стояниях. К сожалениию, при высоком симпатиче-
ском тонусе и повышенной концентрации норадре-
налина ксамотерол может действовать как Р-адре-
ноблокатор; в этих случаях сердечный выброс
снижается, и клиническое состояние пациента
может ухудшиться. Внутренняя симпатомиметиче-
ская активность других p-адреноблокаторов может
играть важную роль при лечении пограничной сер-
дечной недостаточности.
Некоторые синтетические наркотические
анальгетики являются частичными агонистами
опиатных рецепторов (таблица 3.7) и в зависимо-
сти от клинической ситуации могут проявлять себя
как агонисты, так и как антагонисты. Частичные
агонисты опиатных рецепторов не усиливают
анальгетический эффект полных агонистов (мор-
фина, петидина, фентанила), а, напротив, иногда
оказывают антагонистическое действие.
Обратные агонисты — это лекарственные пре-
параты, имеющие сродство к рецепторам, но обла-
дающие отрицательной внутренней активностью
(противоположно направленной относительно
активности агонистов). Обратные агонисты кон-
курируют с агонистами, частичными агонистами
и конкурентными антагонистами за связывание
с рецепторами; результат зависит от относительно-
го сродства к этим рецепторам. Эффект обратного
агониста обычно противоположен эффекту агони-
ста. Например, простые эфиры Р-карболинов
(Р-ССМ, Р-ССЕ и р-ССР) являются конкурентны-
ми антагонистами бензодиазепинов и могут уст-
ранять их противосудорожное, анксиолитическое
и седативное действие. Введенные в отсутствие
бензодиазепинов, Р-ССМ и Р-ССЕ вызывают воз-
буждение, снижают судорожный порог, провоциру-
ют судороги. Конкурентный антагонист бензодиа-
зепинов флумазенил может вызывать беспокойст-
во и провоцировать судороги (глава 12); эти
эффекты могут быть обусловлены обратной агони-
стической активностью.
Согласно современным концепциям, механизм
действия обратных агонистов состоит в угнетении
спонтанной конститутивной (не обусловленной
связыванием с рецепторами) активности избы-
точно экспрессированных популяций рецепторов.
По классическим представлениям, рецепторы пре-
бывают в пассивном состоянии, пока не активиру-
ются агонистом. Однако в настоящее время доказа-
но, что эта концепция верна только для нормально-
го количества рецепторов. При избыточной
экспрессии некоторые рецепторы начинают про-
являть базальную (конститутивную) активность
и стимулируют эффекторные системы без связыва-
ния с агонистами. Обратные агонисты уменьшают
или полностью прекращают такую спонтанную
активность и, следовательно, оказывают эффект,
противоположный агонистам. В других условиях
обратные агонисты могут действовать как конку-
рентные антагонисты.
Избыточная экспрессия Р-адренорецепторов
может привести к активации аденилатциклазы
и синтезу цАМФ без связывания с агонистами. В этом
случае p-адреноблокаторы начинают оказывать эф-
фект обратных агонистов: при сердечной недоста-
точности они могут уменьшать ЧСС в покое и ис-
пользуются для лечения некоторых тахиаритмий.
68
Глава 3
ДЕСЕНСИТИЗАЦИЯ
Десенситизацией называется уменьшение эф-
фекта агониста при его длительном или повторном
воздействии. Обычно этот эффект наблюдают in
vitro (т. е., в изолированной ткани или культуре
клеток).
Острая десенситизация обычно быстро развива-
ется и легко обратима. Впервые она была описана
35 лет назад Кацем и Теслефом на примере воздей-
ствия агонистов на двигательную концевую пла-
стинку и скелетную мышцу. У человека такая де-
сенситизация иногда развивается при введении
сукцинилхолина в высоких дозах или многократно;
этот феномен более известен как тахифилаксия или
быстро развивающаяся толерантность (глава 10).
Такой тип острой десенситизации можно объяс-
нить превращением активированных комплексов
препарат-рецептор в неактивированные, десенси-
тизированные формы: хотя рецептор сохраняет
способность связываться с агонистом, после этого
связывания эффект не наступает: ионные каналы
не открываются, двигательная концевая пластинка
не деполяризуется. Многие агонисты и некоторые
антагонисты обладают преимущественным сродст-
вом к десенситизированным рецепторам (метафи-
лические антагонисты).
Острая десенситизация может также развивать-
ся в Р-адренорецепторах; в этом случае они, сохра-
няя способность связывать агонисты, не могут
активировать G-белки или аденилатциклазу. Пола-
гают, что такие изменения обусловлены фосфори-
лированием отдельных аминокислотных остатков
в рецепторе.
Хроническая десенситизация обычно развива-
ется медленнее и не так легко обратима. Она может
быть обусловлена утратой или секвестрацией ре-
цепторов с поверхности эффекторных клеток в ре-
зультате эндоцитоза (интернализации), их необра-
тимыми конформационными изменениями или де-
струкцией. Длительное повышение концентрации
гормона или медиатора может вызвать снижение
количества и плотности соответствующих рецеп-
торов (down-regulation), а также их деструкцию;
такой механизм десенситизации описан для холи-
но- и адренорецепторов. Так, утрата рецепторов
может произойти, если эффекторные клетки под-
вергаются воздействию чрезмерной концентрации
агониста (например, длительное использование
02-адреномиметиков при лечении бронхиальной
астмы). Хроническая десенситизация, обусловлен-
ная утратой рецепторов, может развиваться при ау-
тоиммунных процессах (тяжелая миастения).
med lit. изшзькыитературы
Barnes, PJ. & Adcock, I. (1993). Anti-inflammatory actions of
steroids: molecular mechanisms. Trends in Pharmacological Sciences
14,436-441.
Beavo, J.A. & Reifsnyder, D.H. (1990). Primary sequence of cyclic
nucleotide phosphodiesterase isozymes and the design of selective
inhibitors. Trends in Pharmacological Sciences 11, 150-155.
Bennett, B.M., McDonald, B.J, Nigam, R. & Simon, W.C. (1994).
Biotransformation of organic nitrates and vascular smooth muscle
function. Trends in Pharmacological Sciences 15, 245-249.
Berridge, M. (1993). Inositol trisphosphate and calcium
signalling. Nature 361, 315-325.
Berridge, M. (1997). Elementary and global aspects of
calcium signalling. Journal of Physiology 499, 291-306.
Boughton-Smith, N.K. (1994). Pathological and therapeutic
implications for nitric oxide in inflammatory bowel disease.
Journal of the Royal Society of Medicine 87, 312-314.
Butler, A.R., Flitney, F.W. & Williams, D.L.H. (1995). NO,
nitrosonium ions, nitroxide ions, nitrosothiols and iron-nitrosyls
in biology: a chemist’s perspective. Trends in PhamacologicalSciences
16,18-22.
Changeux,J.-P., Giraudat,J. & Dennis, M. (1987). The nicotinic
acetylcholine receptor: molecular architecture of a ligand-regulated
ion channel. Trends in Pharmacological Sciences#, 459-465.
Cooper, J.R., Bloom, F.E. & Roth, R.H. (1978). The Biocherrii-
cdBaiisofNemxgiharnaaJf^y.'ird edn. Oxf. Univer. Press, New York.
Ehrlich, B.E., Kaftan, E., Bezprozvannaya, S. & Bezprozvanny, I. (1994).
The pharmacology of intracellular Ca2+-release channels. Trends
in Pharmacological sciences 15,145-149.
Gilman, A.G. (1987) G proteins: transducers of receptor-
generated signals. Annual Review of Biochemistry 56, 615-649.
Graziano, M.P. & Gilman, A.G. (1987). Guanine nucleotide-
binding regulatory proteins: mediators of transmembrane
signaling. Trends in Pharmacological Sciences 8,478-481.
Gudermann, T., Kalkbrenner, F. & Schultz, G. (1996). Diversity
and selectivity of receptor-G protein signalling. Annual Reoiew of
Pharmacology and Toxicology 36,429-459.
Kenakin, T.P., Bond, R. A. & Bonner, T.I. (1992) Definition of
pharmacological receptors. Pharmacological Reviews 44, 351-362.
Milligan, G. (1993). Mechanisms of multifunctional signalling by
G-protein-linked receptors. Trends in Pharmacological Sciences 14,
239-244.
Milligan, G., Bond, R.A. & Lee, M. (1995). Inverseagonism: pharmacolo-
gical curiosity or potential therapeutic strategy? Trends in Pharmacolo-
gical Sciences 16, 10-13.
Monod, J., Wyman, J. & Changeux, J.-P. (1965). On the nature
of allosteric transitions: a plausible model. Journal of Molecular
Biology 12,88-118
Nicholson, CB., Challiss, R.AJ. & Shahid, M. (1991). Differential
modulation of tissue function and therapeutic potential of selective
inhibitors of cyclic nucleotide phosphodiesterase isoenzymes. Trends
in Pharmacological Sciences 12,19-27.
Putney, J.W. (1987). Calcium-mobilizing receptors. Trends in Pharmacolo-
gical Sciences 8,481 -486.
ФАРМАКОДИНАМИКА
69
яаейУй.исодкЩ
Palmer, RM J., Ferrige, A.G. & Moncada, S. (1987).
accounts for the biological activity of endothelium-derived
relaxing factor. Nature 327, 524-526.
Quinn, A.C., Petros, AJ. & Vallance, P. (1995). Nitric oxide:
an endogenous gas. British Journal of Anaesthesia 74, 443-451.
Schwinn, D. A. (1993) Adrenoceptors as models for G protein-
coupled receptors: structure, function and regulation. British
Journal of Anaesthesia 71,77-85.
Stephenson, R. P. (1956). A modification of receptor theory. British
Journal of Pharmacology 11,379-393.
Sutherland, E. W. & Rail, T. W. (1960) The relation of adenosine-
s’,5’-phosphate and phosphorylase to the actions of catechola-
mines and other hormones. Pharmacological Reviews 12,265-299.
Taussig, R. & Gilman, A.G. (1995). Mammalian membrane-bound
adenylyl cyclases. Journal of Biological Chemistry 270,1-4
Приложение I
КИНЕТИКА ВЗАИМОДЕЙСТВИЯ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ С
РЕЦЕПТОРАМИ
Классический анализ лекарственных эффектов
основан на допущении, что каждая молекула препа-
рата обратимо связывается с рецептором, образуя
комплекс препарат-рецептор, то есть:
kl
D + R<->DR,
k2
где D — количество свободных (несвязанных) мо-
лекул лекарственного препарата; R — количество
свободных (незанятых) рецепторов, DR — количе-
ство занятых участков связывания в рецепторах
сайтов; ki — константа скорости ассоциации, к2 —
константа скорости диссоциации. В состоянии рав-
новесия значения констант ассоциации и диссоциа-
ции равны; если D, R и DR выражены в молярных
концентрациях, то
kt [D][R] = k2 [DR];
k2/kt - [D][R]/[DR] = Kd,
где Ка — равновесная константа диссоциации, отра-
жающая сродство лекарственного препарата к ре-
цептору. При высоком значении Kd сродство
низкое; а при низком Kd — наоборот, высокое. В чис-
ленном выражении Kd представляет собой концен-
трацию лекарства, необходимую для связывания
половины рецепторов в состоянии равновесия.
Если Rt— общее количество рецепторов,
Rt - [R] + [DR];
[R] = [Rt] - [DR];
поэтому
Kd = [D][R]/[DR] = {[D]([Rt] - [DR])}/[DR];
и
[D]([Rt] - [DR]) - Kd [DR];
[D][Rt] - [D][DR] - Kd [DR];
и
[D][Rt] = Kd [DR] + [D][DR] = [DR](Kd + [D]);
следовательно
[DR]/[Rt] = [D]/(Kd + [D]);
и
f=[D]/(Kd + [D]),
поскольку доля связанных рецепторов (f) равна:
f=[DR]/[Rt];
Это уравнение (уравнение Лэнгмюра или Хил-
ла-Лэнгмюра) было впервые выведено А.В. Хиллом
в 1909 году; подобное соотношение было использо-
вано Лэнгмюром для описания абсорбции газов ме-
таллическими поверхностями.
Приложение II
ИЗОМЕРИЯ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
Изомеры — это соединения, содержащие в своем
составе одни и те же элементы в одинаковом коли-
честве (т. е. имеющие одинаковый химический со-
став, одинаковую молекулярную формулу и одина-
ковый молекулярный вес), но различающиеся про-
странственным расположением атомов или групп
атомов. Физические, химические и биологические
свойства изомеров могут практически совпадать
или, напротив, абсолютно различаться. Выделяют
два типа изомеров — структурные изомеры и сте-
реоизомеры.
Структурная изомерия
Структурные изомеры имеют одинаковую мо-
лекулярную формулу, но различное химическое
строение, так как входящие в их состав атомы со-
единены в разном порядке. Хорошо известными
примерами структурных изомеров являются изо-
флуран и энфлуран. Оба препарата имеют одинако-
вую химическую формулу (СзНгСШбО), но их ато-
мы по-разному присоединены к общему углеродно-
му скелету (рис. 5.6).
Эффект структурных изомеров может быть схо-
жим (например, у изофлурана и энфлурана), или,
напротив, их эффекты и применение могут совер-
шенно различаться (прометазин и промазин).
Обычно структурные изомеры легко отличить,
поскольку они являются абсолютно различными
лекарственными препаратами с разными междуна-
родными названиями.
Для таутомерии или динамической изомерии
характерно равновесное состояние двух неста-
бильных структурных изомеров; один изомер мо-
жет обратимо и быстро превращаться в другой при
70
Глава 3
(HanpnJ90,^fiHjt-
изменениях окружающей среды
Таутомерии подвержены некоторые анестетики,
включая тиопентал и метогекситал. В щелочном
растворе (pH 10,5) тиопентал натрия ионизиро-
ван и растворим в воде; после введения в плазму
(pH 7,4) препарат становится неионизированным
и подвергается таутомерии, поэтому его жирорас-
творимость повышается. Подобные изменения
происходят и с метогекситалом (глава 4).
Стереоизомерия
Стереоизомерами называются два или более
вещества с одинаковой молекулярной формулой
и химическим строением, но различной конфигура-
цией: их атомы или химические группы занимают
различное положение в пространстве. Стереоизо-
меры можно разделить на две основные группы:
1. Энантиомеры.
2. Диастереомеры.
Энантиомеры
Энантиомеры (оптические изомеры, зеркаль-
ные изомеры) — это пары изомеров, не совмещае-
мые в пространстве при попытке наложения одно-
го на другой и представляющие собой зеркальное
отражение друг друга. Чтобы обладать свойством
стереоизомерии, молекула должна иметь хираль-
ный центр, который чаще всего представляет со-
бой асимметричный атом углерода, то есть атом,
связанный с четырьмя разными заместительными
группами или другими атомами (хиральностью на-
зывают отсутствие симметрии относительно пра-
вой и левой стороны). Хиральные лекарственные
препараты имеют зеркальные изомеры, которые не
совпадают при наложении друг на друга: существу-
ют правая (R, rectus) и левая (S, sinister, левая) фор-
мы изомеров, то есть R- и S-изомеры. Эти изомеры
отличаются пространственной ориентацией замес-
тительных групп вокруг хирального центра. Кроме
того, энантиомеры также обладают оптической ак-
тивностью и могут вращать плоскость поляризо-
ванного света вправо [(+) или (</)-форма)] или вле-
во [(-) или (/)-форма]. В зависимости от характери-
стик изомеры могут обозначать как R(+), R(-), S(+)
и S(-). Равновесные смеси двух форм изомеров (ра-
цемические смеси), обозначаемые (+/-) или (dl), не
обладают оптической активностью.
Диастереомеры
Диастереомеры - это пары стереоизомеров, не
являющиеся зеркальными отражениями друг друга
(т. е. энантиомерами). Физические и химические
свойства диастереомеров отличаются: у них разные
точки плавления или растворимость, и они по-раз-
ному участвуют в химических реакциях. Некоторые
еры метогекситала, трамадола, атраку-
рия и мивакурия являются диастереомерами.
Различия между стереоизомерами
С точки зрения стереоизомерии препараты
можно разделить на четыре основные группы (таб-
лица 3.8):
1. Ахиральные препараты (не являющиеся сте-
реоизомерами).
2. Препараты, представляющие собой отдель-
ный стереоизомер.
3. Препараты являющиеся смесью двух форм
стереоизомеров.
4. Препараты, являющиеся смесью трех и более
стереоизомеров.
Отдельные стереоизомеры могут различаться
фармакодинамической активностью и фармакоки-
нетическими свойствами. Чаще всего это касается
лекарственных препаратов, представляющих собой
хиральные смеси двух энантиомеров. Хотя R- и S-
изомеры имеют одинаковое строение, их простран-
ственная конфигурация различна. Следовательно,
два энантиомера могут образовывать различные
трехмерные отношения в асимметричном окруже-
нии рецепторов и ферментов, почти полностью со-
стоящих из L-аминокислот со стереоселективными
свойствами. В таких условиях R- и S-изомеры
по-разному взаимодействуют с участками связыва-
ния рецепторов.
Энантиомеры часто различаются мощностью:
например, 8(+)-изофлуран немного мощнее своего
R(-) энантиомера, в то время как S(+)-кетамин в
3-4 раза мощнее R(-)-KeTaMHHa. Иногда один из
стереоизомеров практически лишен активности:
например, J-атропин, ^/-норадреналин и «/-адрена-
лин приблизительно в 50-100 раз слабее своих
/-энантиомеров. Различия в мощности часто выра-
жаются стереоспецифическим индексом (эудисми-
ческое отношение), который представляет собой
отношение активности более мощного изомера (эу-
томера) к активности менее мощного (дистомера).
Стереоспецифический индекс может составлять
100 и выше; величина индекса может зависеть как
от сродства к рецепторам, так и от внутренней ак-
тивности энантиомеров. Кроме того, они могут от-
личаться по характеру влияния на рецепторы: один
энантиомер может быть агонистом, в то время как
другой проявляет свойства частичного агониста
или конкурентного антагониста.
Отдельные стереоизомеры также различаются
метаболизмом и фармакокинетическими свойст-
вами. В некоторых случаях один энантиомер мета-
болизируется быстрее, чем его антипод. Так, R(+)-
пропранолол, R(-)-пpилoкaин и 8(+)-кетамин
ФАРМАКОДИНАМИКА
71
Таблица 3.8. Ахиральные и хиральные анестезиологии. Некоторые препараты
применяются в виде отдельного изомера, большинство из них являются рацемическими смесями двух
энантиомеров
Ахиральные Хиральные
Один изомер Два изомера Более двух изомеров
Пропофол Этомидат Тиопентал Мивакурий
Закись азота Ропивакаин Метогекситал Атракурий
Тетракаин Левобупивакаин Кетамин
Лидокаин Тубокурарин Галотан
Галламин цис-Атракурий Энфлуран
Фентанил Панкуроний Изофлуран
Альфентанил Векуроний Десфлуран
Ремифентанил Морфин Прилокаин
Петидин Г иосцин Бупивакаин
Неостигмин Атропин
Эдрофоний Трамадол
Дофамин Добутамин
Г ликопиррола
метаболизируются быстрее своих энантиомеров и
отличаются повышенным внутренним печеноч-
ным клиренсом. Парадоксально, что метаболизм
5(+)-кетамина подавляется К(-)-кетамином, чем
можно объяснить относительно длительное дейст-
вие рацемической смеси. Подобные метаболиче-
ские взаимодействия характерны и для других хи-
ральных препаратов (например, левометорфана).
Некоторые неактивные энантиомеры подверже-
ны непрямому метаболическому превращению в
свои антиподы. Так в кишке и в печени R(-)-n6y-
профен и К(-)-фенопрофен превращаются в актив-
ные S (+)-энантиомеры. 5(+)-ибупрофен прибли-
зительно в 160 раз активнее своего энантиомера и в
настоящее время применяется в клинической прак-
тике.
Различия в метаболизме и выведении R- и S-энан-
тиомеров некоторых хоральных лекарств могут
влиять на их фармакокинетику, поэтому многие
препараты-энантиомеры имеют разныеТ| кли-
ренс и объем распределения (глава 2).
Практические аспекты
Многие препараты представляют собой хираль-
ные смеси двух и более изомеров. Иногда свойства
энантиомеров дополняют друг друга, вносят важ-
ный вклад в фармакологические характеристики
препарата (например, трамадол. добутамин). Понятно,
что такие препараты целесообразно использовать в
виде хиральных смесей.
Во многих других случаях фармакодинамика
или фармакокинетика отдельных энантиомеров от-
личается друг от друга (например, кетамин, бу п ива-
каин и атракурий). Существование таких препара-
тов целесообразно нс в виде смеси, а в виде отдель-
ного энантиомера с более благоприятными
свойствами. Достигнутый в последние годы про-
гресс в химических технологиях упростил разделе-
ние и синтез отдельных стереоизомеров (например,
путем асимметричного химического синтеза или
хиральной инверсии одного из энантиомеров).
Кроме того, во многих странах организации, регла-
ментирующие применение лекарственных препа-
ратов, поощряют внедрение в клиническую практи-
ку лекарств, представляющих собой отдельный сте-
реоизомер. Начиная с 1990-х гг. отдельными
энантиомерами является большинство новых ле-
карственных препаратов; кроме того, некоторые су-
ществовавшие ранее рацемические смеси тоже за-
менены отдельными энантиомерами (атракурий,
бупивакаин). Вероятно, в дальнейшем эта тенден-
ция сохранится.
medlit.ucoz.ru
НЕИНГАЛЯЦИОННЫЕ
АНЕСТЕТИКИ
Неингаляционные анестетики — это лекарст-
венные препараты, которые вызывают потерю
сознания в течение времени, за которое кровь из
места в/в инъекции попадает в головной мозг
(20-30 с при инъекции в вену руки)1. В конце
XIX — начале XX века многие лекарственные сред-
ства (некоторые опиаты, хлоралгидрат, брометол,
хлороформ и эфир, а также существовавшие в то
время препараты барбитуровой кислоты) пытались
вводить в/в для быстрого выключения сознания.
К сожалению, анестезия при этом развивалась мед-
ленно, продолжалась долго, часто развивались по-
бочные токсические эффекты.
Поворотным пунктом явилось появление бар-
битуратов, обладавших быстрым началом и корот-
кой продолжительностью гипнотического дейст-
вия. Гексобарбитал был впервые применен в Герма-
нии в 1932 г. Веезе (Weese) и Шарпффом (Scharpff).
Благодаря быстрому наступлению гипнотического
действия, препарат оказался приемлемым для ин-
дукции анестезии. Вскоре гексобарбитал был вы-
теснен тиопенталом, который в США независимо
друг от друга исследовали Ланди (Lundy) и Уотерс
(Waters). В декабре 1941 г. в результате катастрофы
в Пёрл Харборе стало широко известно о том, что
тиопентал способен вызывать опасные для жизни
осложнения (особенно при использовании в боль-
ших дозах в качестве единственного анестетика)2.
В последующие годы тиопентал стал широко при-
меняться для внутривенной индукции анестезии.
Хотя подобными свойствами обладают многие тио-
барбитураты, примечателен тот факт, что именно
тиопентал выдержал испытание временем. В Вели-
кобритании, помимо тиопентала, из барбитуратов
применяют метогекситал, который появился в ане-
стезиологической практике в 1959 г.
В1941 г. Ганс Селье (Hans Selye) установил спо-
собность ряда стероидных гормонов вызывать об-
ратимое угнетение сознания у животных. Некото-
рые стероиды стали использоваться в качестве
анестетиков, хотя гормональные эффекты препят-
ствовали их широкому применению в этой области.
В конце 1950-х гг. в анестезиологической практике
появился гидроксидион, который обладал мощной
гипнотической активностью, но при этом не оказы-
вал гормонального действия. Из-за плохой раство-
римости он мог применяться только в виде в/в ин-
фузии в большом объеме физиологического рас-
твора, и эффект его развивался медленно. Позже
были синтезированы полимеризованные и более
концентрированные формулы препарата; к сожале-
нию, они часто вызывали тромбофлебит, и исполь-
зование гидроксидиона было прекращено.
Альтезин (смесь стероидов альфаксолона и аль-
фадолона) стал применяться в 1972 г. Несмотря
на многие преимущества, частые анафилактиче-
ские и анафилактоидные реакции (особенно брон-
хоспазм и выраженную артериальную гипотонию)
привели к тому, что в 1984 г. альтезин был запре-
щен. Возможно, эти побочные эффекты были обу-
словлены полиэтоксилированным касторовым
маслом (кремофор EL), использовавшимся для
улучшения растворимости стероидов. Минаксолон
(водорастворимый стероидный препарат) прохо-
дил клинические испытания в 1979 г. Однако из-за
часто наблюдавшегося двигательного возбужде-
1 Строго говоря, это не совсем так. Понятие «анестетик» подразумевает, наряду с гипнотическим эффектом (способностью
выключать сознание), наличие анальгетических свойств. Применительно к рассматриваемым средствам анальгетический
эффект доказан лишь в отношении кетамина. Однако в западной литературе понятие «анестетик» относят и к гипнотическим
препаратам, применяемым для выключения сознания в процессе индукции и поддержания анестезии (прим. ред.).
2 Имеется в виду анестезия тиопенталом при вмешательствах у раненых военнослужащих ВМФ США, когда нередкие
осложнения (остановка дыхания и кровообращения на фоне кровопотери) приводили к летальным исходам. Это дало
основание Halford F.J. высказаться так: «Тиопентал ... является идеальным препаратом для эвтаназии» (Anesthesiology,
1943, 4,67) (прим. ред.).
НЕИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
73
medlit.ucoz.ru
ния, а также развития опухолей у эксперименталь-
ных животных дальнейшие исследования были
прекращены.
В настоящее время в Великобритании стероид-
ные анестетики не применяются. Однако 5р-пре-
гнанолон, забракованный ранее из-за низкой рас-
творимости в воде, недавно был выпущен в виде
эмульсии и прошел клинические испытания. Он
является хорошим средством для индукции анесте-
зии, предсказуемо отключая сознание, и не вызыва-
ет боль при инъекции. Клинически значимые изме-
нения кровообращения и возбуждение не развива-
ются.
Евгенол по своему химическому составу близок
к феноксиуксусной кислоте и является одним из
основных компонентов гвоздичного масла. Произ-
водное евгенола — пропанидид, использовался в ка-
честве средства для индукции анестезии около два-
дцати лет и был запрещен в 1983 г. Пропанидид яв-
ляется плохо растворимым в воде эфиром.
Содержащийся в нем кремофор EL иногда вызывал
бронхоспазм и тяжелую артериальную гипотонию
во время индукции анестезии. Кроме того, наблю-
дались различные проявления возбуждения (на-
пример, икота, непроизвольные мышечные движе-
ния, учащение дыхания, судороги).
В качестве анестетиков предлагались различ-
ные производные циклогексамина. Один из таких
препаратов, фенциклидин, использовали для ин-
дукции анестезии в 1950-е гг., но вскоре от него от-
казались из-за тяжелых психотомиметических ре-
акций. Сходный по строению с фенциклидином
препарат кетамин стали применять в 1970-е гг.
Хотя препарат не вызывает потери сознания в тече-
ние времени, за которое кровь из места в/в инъек-
ции попадает в головной мозг, его все же использу-
ют для индукции анестезии. Кетамин играет опре-
деленную роль в анестезиологии, хотя его
применение ограничено рядом недостатков1.
Этомидат (производное имидазола), исполь-
зуемый в анестезиологии с 1974 г., обладает гипно-
тическим действием, но практически лишен
анальгетических свойств. Хотя этомидат достаточ-
но безопасен, он имеет несколько недостатков, ог-
раничивающих его применение.
Пропофол является химически инертным про-
изводным фенола2. Впервые пропофол был приме-
нен в смеси с кремофором EL в 1977 г., а с 1985 г. вы-
пускается в виде водной эмульсии. Быстрое и ясное
пробуждение, характерное для пропофола, несо-
мненно, способствует его нынешней популярности.
Пропофол широко используется для индукции ане-
стезии (особенно в амбулаторной практике), а так-
же для поддержания анестезии посредством в/в ин-
фузии.
Все неингаляционные анестетики применяют
в виде водных или водно-масляных растворов
(эмульсий), которые легко смешиваются с плазмой.
Кроме того, в плазме при pH 7,4 определенная часть
препарата должна находиться в неионизированной
форме и быть жирорастворимой, чтобы проникать
через гематоэнцефалический барьер и вызывать
быструю утрату сознания. Эти противоречивые
физико-химические требования обычно разреша-
ются с помощью защелачивания растворов или
применения жирорастворимых препаратов в фор-
ме смешиваемых с водой масел или эмульсий.
Вследствие этого в препараты анестетиков часто
добавляют основания, буферные вещества, а также
вещества, способствующие растворению.
В данной главе будут рассмотрены следующие
лекарственные препараты.
1. Барбитураты.
2. Пропофол.
3. Этомидат.
4. Кетамин.
Хотя для индукции анестезии иногда применя-
ют бензодиазепины, опиоиды и нейролептики, в те-
рапевтических дозах они не вызывают быстрого
выключения сознания и обычно в этом контексте не
рассматриваются.
1 Кетамин, в отличие от большинства неингаляционных анестетиков, обладает доказанными анальгетическими и симпато-
миметическими свойствами. Применение ограничено его галлюциногенными и психотомиметическими эффектами, при-
сущими производным фенциклидина (прим. ред.).
2 Анальгетической активностью не обладает (см. сноску 1).
74
Глава 4
БАРБИТУРАТЫ
medlit.
ХИМИЧЕСКИЕ СВОЙСТВА
Барбитураты являются производными барбиту-
ровой кислоты, которая была впервые синтезиро-
вана в 1863 г. путем конденсации мочевины и мало-
новой кислоты.
иСОЗмГ&Арбитураты (тиопентал) обычно растворя-
ются в липидах лучше, чем оксибарбитураты (пен-
тобарбитал), и поэтому быстрее преодолевают ге-
матоэнцефалический барьер. Многие барбитураты,
используемые в качестве седативных, снотворных
или противосудорожных средств, в пятом положе-
нии гетероцикла содержат две алкильные группы
длиной от 2 до 5 атомов углерода, ^метилирование
в первом или третьем положении гетероцикла (Ni
или N3) приводит к появлению соединений, кото-
рые могут вызывать возбуждение или судороги (на-
пример, метогекситал). Напротив, замещение ал-
кильных групп в пятом положении гетероцикла
ароматическими или гетероциклическими группа-
ми дает соединения, обладающие противосудорож-
ным действием (например, фенобарбитал).
Барбитураты довольно слабо растворяются в
воде, но могут растворяться в щелочных растворах.
Растворимость барбитуратов в водных растворах
зависит от динамического соотношения двух изо-
меров — енольной и кетоновой формы (таутоме-
рия), а также от присутствия в растворе в виде сла-
бых кислот.
Кетоновая форма
Барбитуровая кислота является биологически
инертным соединением, но ее производные с кисло-
родом (оксибарбитураты) или серой (тиобарбиту-
раты) во втором положении гетероцикла обладают
гипнотической активностью.
Енольная форма
Переход кетоновой формы в енольную зависит
от pH раствора и легко происходит в щелочной сре-
де. Растворимость натриевых солей барбитуровых
НЕИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
75
„ medlit.ucoz.ru
кислот определяется енольной формой и ее после- ных каналов.
дующей ионизацией в щелочном растворе.
Константы диссоциации большинства барбиту-
ратов находятся в диапазоне pH 7,3-8,0; следова-
тельно, in vivo степень ионизации этих препаратов
относительно постоянна. Напротив, различные
барбитураты заметно отличаются друг от друга рас-
творимостью в липидах, способностью связываться
с белками плазмы и интенсивностью метаболизма.
Тиобарбитураты хорошо растворяются в липидах,
активно связываются с белками плазмы (60-90%)
и полностью распадаются под действием печеноч-
ных ферментов. Одни оксибарбитураты (например,
пентобарбитал) растворяются в липидах в 20 раз
слабее, умеренно связываются с белками плазмы
(30-50%) и в основном элиминируются путем пе-
ченочного метаболизма. Другие оксибарбитураты
(например, фенобарбитал) растворяются в липи-
дах в 200 раз слабее, слабо связываются с белками
плазмы (< 20%) и почти полностью экскретируют-
ся с мочой в неизмененном виде. Вследствие это-
го фенобарбитал обладает относительно длитель-
ным Т1/2-
МЕХАНИЗМ ДЕЙСТВИЯ
Хотя барбитураты оказывают угнетающее дейст-
вие на функцию многих тканей и органов, наиболее
чувствительной к их эффектам является ЦНС. Ран-
ние экспериментальные исследования показали, что
эффект барбитуратов обусловлен угнетением рети-
кулярной системы. Барбитураты оказывают влия-
ние на соматосенсорные и акустические корковые
потенциалы, угнетают ЭЭГ и реакцию активации
при стимуляции ретикулярной системы. Считалось,
что эффекты барбитуратов в основном обусловлены
влиянием на синаптическую активность.
Действие барбитуратов обусловлено влиянием
на ГАМК-зависимые хлорные каналы в нейронах
головного мозга. ГАМК (гамма-аминомасляная ки-
слота) является главным тормозным нейромедиа-
тором ЦНС и опосредует пре- и постсинаптическое
торможение в 20-40% всех синапсов. После связы-
вания ГАМК с рецептором хлорные каналы откры-
ваются, ионы хлора проникают в нейрон, что приво-
дит к гиперполяризации и снижению возбудимости
нейронов (торможение).
Существует два подтипа ГАМК-рецепторов:
ГАМКд и ГАМКб. Рецепторы ГАМКа представля-
ют собой макромолекулярный комплекс, включаю-
щий в себя участки связывания с бензодиазепина-
ми, барбитуратами, стероидными анестетиками, а
также некоторыми группами проконвульсантов
(например, пикротоксином).
Действие барбитуратов заключается в удлине-
нии вспышек открывания ГАМК-зависимых хлор-
Этот эффект опосредуется
Р-субъединицей рецептора ГАМКа. В отличие от
барбитуратов, бензодиазепины повышают частоту
вспышек открывания хлорных каналов и имеют
большее сродство к а- и у-субъединицам рецептора.
Барбитураты также препятствуют взаимодействию
радиомеченых аналогов пикротоксина с участками
связывания хлорных каналов.
Барбитураты оказывают слабое воздействие на
анионселективные глициновые каналы, которые
опосредуют торможение активности нейронов. На-
ряду с большинством других общих анестетиков,
они также могут влиять на медиаторчувствитель-
ные катионные каналы, опосредующие активность
нейронов. Другими словами, они угнетают воздей-
ствие различных возбуждающих нейромедиаторов
(глутамат, ацетилхолин, 5-гидрокситриптамин
[5-НТ]) на участки связывания рецепторов. С по-
мощью экспериментальных методов изучения
свойств ионных каналов — локальной фиксации
мембранного потенциала (изоляция фрагмента
клеточной мембраны с помощью специальной пи-
петки) и плоского липидного бислоя (встраивание
изолированного мембранного белка в изолирован-
ную липидную мембрану) — доказано непосредст-
венное воздействие барбитуратов на натриевые ка-
налы. Кроме того, барбитураты оказывают различное
влияние на калиевые каналы; это может быть акти-
вация или блокада в зависимости от подтипа кана-
ла, вида экспериментального животного и участка
мембраны. Все эти факты позволяют сделать вы-
вод, что эффекты барбитуратов определяются сло-
жением реакций различных ионных каналов в раз-
ных участках ЦНС. Поскольку тиопентал и мето-
гекситал являются хиральными соединениями
(глава 3), важную роль могут играть различия меж-
ду оптическими изомерами в сродстве к рецепто-
рам и внутренней активности.
ПРИМЕНЕНИЕ
Барбитураты в течение многих лет использова-
лись как седативные и снотоворные средства, а та-
кие препараты, как амобарбитал, бутобарбитал и се-
кобарбитал применяются в Великобритании до сих
пор. Хотя в настоящее время они относятся к кон-
тролируемым лекарственным средствам, их про-
должают использовать как снотворные. У некото-
рых пациентов с длительной и трудно излечимой
бессонницей барбитураты оказывают более надеж-
ный и предсказуемый эффект, чем бензодиазепи-
ны. Тем не менее они имеют много недостатков.
Как и другие снотворные, они ухудшают способность
к суждению и замедляют реакцию. Пациентов, при-
нимающих барбитураты и ведущих активную
жизнь, необходимо предупреждать о недопустимости
76
Глава 4
управления автомобилем, работы на вь
опасными механизмами. Все барбитураты могут
нарушать психомоторные реакции и вызывать син-
дром отмены на следующий день после приема.
Барбитураты вызывают дозозависимую депрессию
дыхания. Классическим симптомом случайного
или преднамеренного отравления барбитуратами
является тяжелое угнетение дыхания. Барбитура-
ты могут взаимодействовать с другими препарата-
ми, угнетающими активность ЦНС, включая эта-
нол, и особенно опасны при легочных заболеваниях
(хронический бронхит или бронхиальная астма).
Барбитураты индуцируют активность печеночных
ферментов, что усиливает метаболизм других ле-
карственных препаратов (непрямые антикоагулян-
ты, противосудорожные средства и пероральные
контрацептивы). Они могут провоцировать острый
приступ порфирии у генетически предрасположен-
ных пациентов. Элиминация барбитуратов нару-
шается при заболеваниях печени и почек. Они пло-
хо переносятся пожилыми пациентами, вызывая
сонливость, дезориентацию, спутанность сознания,
нечеткость речи, ухудшение памяти и неустойчи-
вость походки; в свою очередь, эти осложнения мо-
гут привести к падению и переломам костей.
При продолжительном или постоянном приеме
барбитуратов в результате индукции ферментов
или снижения чувствительности нейронов может
развиться толерантность. Барбитураты могут про-
тиводействовать эффекту анальгетиков. Они спо-
собны вызывать физическую и психическую зави-
симость.
Некоторые барбитураты эффективны при дли-
тельном лечении эпилепсии: в качестве противосу-
дорожных средств применяют фенобарбитал и при-
мидон. Для общей анестезии в настоящее время ис-
пользуют только два барбитурата: тиопентал и
метогекситал.
ТИОПЕНТАЛ
Общие сведения
Тиопентал (5-этил-5’-(1-метилбутил)-2-тио-
барбитуровая кислота) является тиоаналогом пен-
тобарбитала и представляет собой рацемическую
смесь двух стереоизомеров (рис. 4.1). Тиопентал
выпускают в виде натриевой соли с 6% карбонатом
натрия; это светло-желтый порошок с горьким вку-
сом, хорошо растворимый в воде.
Перед употреблением тиопентал растворяют
в воде или физиологическом растворе, при этом
образуется 2,5% щелочной раствор с pH около 10,5.
В растворе карбонат натрия отщепляет свободные
гидроксильные ионы, что позволяет предотвратить
RdGCUit. и€Ма^<Шйствие с атмосферным СО
2 и последую-
щее выпадение в осадок нерастворимой свободной
кислоты (R-S-H). Растворы тиопентала натрия ос-
таются стабильными при комнатной температуре
до двух недель (и дольше при температуре 4°С), но
при помутнении должны немедленно уничтожать-
ся. Обычно растворы тиопентала натрия использу-
ют в течение 48 ч после приготовления.
Благодаря высокому pH, 2,5% растворы тиопен-
тала обычно обладают бактериостатическими свой-
ствами, но несовместимы со многими лекарства-
ми-основаниями. Тиопентал нельзя смешивать с
окисляющими агентами, более кислыми лекарст-
венными средствами, а также с препаратами, кото-
рые применяют в виде сульфатов, хлоридов и гид-
рохлоридов.
Центральная нервная система
Воздействие тиопентала на ЦНС зависит от
дозы и скорости введения. Быстрое выключение
сознания после стандартной дозы для индукции
анестезии (3-5 мг/кг) обусловлено двумя фактора-
ми. Во-первых, головной мозг очень хорошо крово-
снабжается, получая в норме около 25% сердечного
выброса. Во-вторых, тиопентал отлично растворя-
ется в липидах (коэффициент распределения мас-
ло: вода равен 500-700), и более 90% находящегося
в мозговых капиллярах препарата легко преодоле-
вает гематоэнцефалический барьер.
Сознание выключается плавно, фаза возбужде-
ния развивается в редких случях. Утрате сознания
часто предшествуют один-два глубоких вдоха,
фаза быстрых движений глаз с соответствующими
изменениями на ЭЭГ. Характерны высокочастот-
ные (преимущественно 20-30 Гц) волны различ-
ной амплитуды, которые по мере углубления ане-
стезии сменяются медленными волнами. Высоко-
частотная реакция ЭЭГ отражает селективное
угнетение тормозных нейронов ретикулярной фор-
мации, что соответствует стадии возбуждения по
Гведелу и может проявляться повышенными реф-
лекторными реакциями на хирургическое раздра-
жение, усилением активности блуждающего нерва,
ларингоспазмом и гипералгезией (пониженным по-
рогом восприятия болевых стимулов). Такие эф-
фекты могут возникать при введении малых доз
тиопентала и при пробуждении после тиопентало-
вой анестезии.
По мере углубления анестезии, обусловленного
возрастающим поглощением тиопентала головным
мозгом, снижается реактивность коры, и на ЭЭГ до-
минируют низкоамплитудные волны. При после-
дующем увеличении дозы проявляются стволовые
эффекты тиопентала. Угнетение дыхания обуслов-
НЕИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
77
Тиопентал
Рис. 4.1.
Структурные форму-
лы неингаляционных
анестетиков. Хираль-
ные атомы углерода
отмечены звездочкой
Метогекситал
Пропофол
Этомидат
Кетамин
лено непосредственным воздействием на дыхатель-
ный центр и его связи с варолиевым мостом; чувст-
вительность к СО2 снижается пропорционально
глубине анестезии. Как следствие, повышается
РаСО2, снижается pH и развивается апноэ. При глу-
бокой барбитуровой анестезии важную роль в под-
держании дыхания играет гипоксическая стимуля-
ция дыхательного центра, опосредованная аорталь-
ными и каротидными хеморецепторами. В отличие
от опиоидов, барбитураты влияют главным обра-
зом не на частоту, а на глубину дыхания. Влияние
тиопентала на дыхание зависит от степени хирурги-
ческой стимуляции и сочетанного использования
других препаратов, вызывающих угнетение ЦНС.
К тиопенталу может развиваться так называе-
мая острая толерантность. Феномен состоит в том,
что у человека после использования высоких доз
тиопентала сознание восстанавливается при сыво-
роточной концентрации тиопентала более высокой,
чем в случае применения меньших доз. Иными сло-
вами, сывороточная концентрация тиопентала,
при которой происходит пробуждение, находится
78
Глава 4
в прямой зависимости от дозы препаратаПЙ€^сн1л- ивОЗсиГи
является ли этот феномен следствием снижения
чувствительности ЦНС к тиопенталу (т. е. фармако-
динамическим эффектом) или объясняется нерав-
номерным распределением (гистерезисом, т. е. фар-
макокинетическим эффектом). Кроме того, важно
следующее: если одну и ту же дозу препарата вводят
с разной скоростью, то сознание быстрее восстано-
вится в том случае, когда скорость введения выше.
Таким образом, концепция развития острой толе-
рантности к тиопенталу пока не находит четкого
объяснения.
Тиопентал и другие барбитураты угнетают ме-
таболизм мозга и снижают потребление кислорода
мозгом. Барбитураты уменьшают мозговой крово-
ток, внутричерепной объем крови и давление спин-
номозговой жидкости — возможно, благодаря угне-
тению продукции СОг. Эти свойства тиопентала
используются при защите мозга («мозговая реани-
мация») и для снижения ВЧД. Если тиопентал вы-
зывает выраженное угнетение дыхания (не предот-
вращенное своевременно начатой ИВЛ), то разви-
вается гиперкапния и ВЧД, напротив, может
повышаться.
Кровообращение
Стандартные дозы тиопентала для индукции
анестезии в той или иной степени снижают АД даже
в отсутствие патологических заболеваний и состоя-
ний. Тяжелое угнетение кровообращения может
возникнуть при гиповолемии, сердечно-сосуди-
стых заболеваниях (включая артериальную гипер-
тонию), а также при приеме препаратов, влияющих
на активность симпатической нервной системы
(вазодилататоры, (З-адреноблокаторы). В этих слу-
чаях нарушается способность системы кровообра-
щения компенсировать гемодинамические эффекты
тиопентала, и дозу тиопентала для индукции анесте-
зии следует уменьшить. Снижение АД преимущест-
венно обусловлено уменьшением ударного объема и
сердечного выброса; компенсаторная реакция состо-
ит в рефлекторном повышении ОПСС.
Уменьшение сердечного выброса обусловлено
несколькими факторами. Тиопентал угнетает сосу-
додвигательный центр и гипоталамические ядра,
регулирующие силу сердечных сокращений. Кроме
того, нарушается нервная передача в ганглиях и со-
кратимость гладких мышц сосудов, что приводит
к венодилатации и периферическому депонирова-
нию крови. Высокие дозы тиопентала могут непо-
средственно угнетать сократимость миокарда, ве-
роятно, из-за местноанестезирующего (мембрано-
стабилизирующего) действия.
Во время анестезии тиопенталом снижаются
скорость клубочковой фильтрации, плазмоток че-
рез почки, экскреция электролитов и воды. Эти эф-
фекты отчасти можно объяснить снижением по-
чечного кровотока, но главной причиной является
повышенное высвобождение АДГ (антидиуретиче-
ского гормона) задней долей гипофиза. Секреция
АДГ зависит от активности гипоталамических ядер
и целостности гипофизарного тракта. Подавление
тормозящего влияния на гипоталамические ядра
(особенно супраоптическое ядро) приводит к по-
вышению секреции АДГ. Вследствие этого диурез
во время анестезии тиопенталом составляет около
0,1 мл/мин (приблизительно 10% от нормы). При
уремии связывание тиопентала с белками снижает-
ся, что приводит к усилению его эффектов.
Печень
Не существует никаких доказательств, что
стандартные дозы тиопентала для индукции могут
быть причиной нарушения функции печени. Тем
не менее в редких случаях после использования
тиопентала развивается желтуха и определяются
патологические результаты печеночных проб.
Считают, что это может быть обусловлено угне-
тением дыхания (вызывающим гипоксию и ги-
перкапнию), а также применением тиопентала на
фоне заболеваний (печени и внепеченочных),
которые сами по себе оказывают неблагоприятное
воздействие на печеночный кровоток и функцию
гепатоцитов.
Как и другие барбитураты, тиопентал активирует
различные изоферменты цитохрома Р-450. Тиопен-
тал провоцирует острый приступ перемежающейся
порфирии, поэтому он и другие барбитураты абсо-
лютно противопоказаны при этом заболевании.
При хронических нарушениях функции печени
эффекты тиопентала пролонгируются и пробужде-
ние замедляется. При заболеваниях печени связы-
вание тиопентала с белками плазмы снижается
из-за угнетения синтеза альбумина.
Фармакокинетика
Высокая скорость наступления эффекта обу-
словлена быстрым поступлением в головной мозг
неионизированной и не связанной с белками фрак-
ции тиопентала. В норме (при pH 7,4) 75-80% тио-
пентала связывается с альбумином, а 61% свобод-
ного препарата находится в неионизированной
форме. Следовательно, изменение pH внеклеточ-
ной жидкости и доли свободной фракции может
влиять на поступление тиопентала в головной мозг.
НЕИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
79
В эксперименте при pH 6,8 концентрация т
ла в плазме уменьшается на 40% (возможно, из-за
повышенного потребления его неионизированной
формы тканями). Напротив, алкалоз (например, при
гипервентиляции) может повышать концентрацию
и долю ионизированного тиопентала в плазме,
уменьшая эффективность введенной дозы. In vitro
высокие дозы НПВС (индометацин, аспирин) вы-
тесняют тиопентал из связи с альбумином. Клиниче-
ское значение этого феномена до конца не ясно.
Высокая концентрация тиопентала достигается
в головном мозге и других хорошо кровоснабжае-
мых тканях в течение 1 мин после в/в введения. Бы-
строе пробуждение после однократного введения
обусловлено перераспределением тиопентала из
головного мозга в менее васкуляризованные ткани
(преимущественно в мышцы и кожу). Для насыще-
ния этих тканей требуется 15-30 мин, концентра-
ция тиопентала в крови быстро падает, и анестетик
диффундирует в кровь из головного мозга. Для на-
сыщения слабо васкуляризованных тканей (напри-
мер, жировой) может потребоваться несколько ча-
сов. Концентрацию тиопентала в крови, скелетных
мышцах и жировой ткани в различные моменты
времени после введения можно определить с помо-
щью физиологической перфузионной модели,
включающей центральную камеру — кровь и шесть
тканевых камер (глава 2). В соответствии с этой мо-
делью тиопентал из головного мозга перераспреде-
ляется преимущественно в ткани, лишенные жира
(например, в мышцы), а подкожно-жировая клет-
чатка играет лишь незначительную роль в заверше-
нии его анестетического эффекта.
Снижение сывороточной концентрации тио-
пентала после в/в введения индукционной дозы
можно разделить на три экспоненциальных состав-
ляющих. Начальная фаза (начальный Тр2» 2-4 мин)
отражает быстрое перераспределение препарата в
хорошо кровоснабжаемые ткани (головной мозг,
печень, почки). Затем наступает фаза медленного
перераспределения тиопентала в кожу и мышцы
(Ti/2» 45-60 мин). Наконец, элиминация анестети-
ка из организма отражается окончательным сниже-
нием его концентрации в плазме (конечный Т1/2 »
5-10 ч). Объем, распределения тиопентала в ста-
ционарном состоянии (Vss) немного превышает
общий объем воды в организме (1-4 л/кг), а кли-
ренс составляет 10—20% печеночного кровотока
(около 1-5 мл/мин/кг). У новорожденных и детей
младшего возраста клиренс выше, чем у взрослых.
При ожирении и у пожилых, напротив, клиренс
значительно снижен.
Поскольку тиопентал хорошо растворим в ли-
пидах, он активно метаболизируется в печени
но, в других тканях). С мочой выводятся
только следы тиопентала (в норме менее 1%). Хотя
метаболизм играет весьма ограниченную роль в
прекращении эффектов тиопентала, он полностью
отвечает за окончательную элиминацию анестети-
ка из организма. Метаболизм тиопентала осуще-
ствляется относительно медленно (6-15% в час).
Основной путь инактивации — окисление до тио-
пенталкарбоновой кислоты. Другие пути метабо-
лизма включают десульфурацию до оксибарбиту-
рата пентобарбитала с Т1/2 20-50 ч, а также гидро-
ксилирование. Если используют большие дозы
тиопентала (обычно с целью защиты мозга), в ре-
зультате насыщения печеночных ферментных сис-
тем его метаболизм становится нелинейным (кине-
тика нулевого порядка). При этом очень высока
концентрация тиопентала в плазме, что значитель-
но замедляет восстановление сознания.
Побочные эффекты
Тиопентал, несомненно, является надежным и
безопасным средством для индукции анестезии —
настолько, насколько соблюдаются все меры пре-
досторожности при его использовании. В первую
очередь должно быть под рукой все необходимое
оборудование для ИВЛ и оксигенации. Тиопентал
вызывает снижение тонуса гладких мышц ЖКТ и
угнетение гортанных рефлексов, что сопряжено с
риском аспирации желудочного содержимого. При
гиповолемии, у пожилых, у больных в тяжелом со-
стоянии доза тиопентала для индукции анестезии
должна быть не стандартной (4-5 мг/кг), а значи-
тельно ниже.
При острой перемежающейся и вариегатной
порфирии тиопентал усиливает синтез цитохрома
Р-450 из гема, тем самым индуцируя активность
аминолевулинатсинтазы. Этот фермент играет
ключевую роль в синтезе порфирина печенью и
другими тканями. Стимуляция синтеза порфоби-
линогена и других порфиринов может вызывать
прогрессирующую демиелинизацию и нейропа-
тию, слабость произвольной мускулатуры, парали-
чи, боли в животе и психические осложнения. Моча
обычно содержит порфобилиноген и уропорфирин,
и при контакте с дневным светом в течение несколь-
ких часов краснеет. Тиопентал не оказывает небла-
гоприятного влияния при некоторых видах заболе-
вания (эритропоэтическая порфирия, поздняя
кожная порфирия) и не всегда вызывает обостре-
ние даже в случае острой перемежающейся порфи-
рии. Тем не менее приступ острой порфирии может
оказаться смертельным, и при любых формах забо-
левания барбитураты противопоказаны.
80
Глава 4
medht.ucozju
Тиопентал может вызывать преходящую кра- вен. Попадание тиопентала в артерию тоже быстро
пивницу или эритему. Крапивница обусловлена
высвобождением гистамина тучными клетками и
может сопровождаться повышением концентрации
гистамина в плазме. Выраженность эффекта зави-
сит от дозы и скорости введения. Истинные реак-
ции гиперчувствительности (анафилактические
реакции, в которые вовлечены иммуноглобулины
или Т-лимфоциты) развиваются редко, с частотой
1:14 000-1:20 000. Они проявляются бронхоспаз-
мом, артериальной гипотонией, генерализованны-
ми отеками, периферическим сосудистым коллап-
сом и сопровождаются значительным риском ле-
тальности.
Относительно медленная элиминация тиопен-
тала (конечный Ti/2 5-10 ч) требует наблюдения за
больным на протяжении всего ближайшего после-
операционного периода. Не рекомендуется управ-
лять автомобилем или работать с опасными меха-
низмами в течение 24 ч после анестезии, а также
принимать алкоголь и седативные лекарственные
препараты.
В/в инъекция тиопентала обычно безболезнен-
на, в то время как непреднамеренное внесосудистое
введение может привести к серьезным побочным
эффектам. Попадание тиопентала под кожу может
вызывать местные осложнения в диапазоне от не-
значительной боли до обширного некроза тканей.
Такие осложнения реже встречаются при использо-
вании разведенных растворов (2,5%), чем концен-
трированных (5% раствор, который широко ис-
пользовался одно время). Эти эффекты, вероятно,
обусловлены местным раздражением тканей при
выпадении в осадок тиопентала, нерастворимого
при pH внеклеточной жидкости. Маловероятно,
что повреждение тканей вызывается щелочной ре-
акцией раствора тиопентала, поскольку оно реже
развивается при использовании метогекситала
(1%) с более высоким значением pH. Для лечения
этих осложнений используют местные инъекции
гиалуронидазы или снимающие раздражение мест-
ные средства.
Внутриартериальное введение тиопентала сразу
вызывает сильную стреляющую боль, сопровож-
дающуюся артериальным спазмом с побледнением
конечности, нарастающим цианозом и исчезнове-
нием пульса. Выключение сознания замедляется.
При использовании разбавленных растворов часто-
та и тяжесть осложнений уменьшаются. При отсут-
ствии лечения может развиться гангрена конечности.
Сразу после в/в введения тиопентала вследст-
вие резкого изменения pH образуются кристаллы,
что не имеет особого значения благодаря быстрому
разбавлению препарата кровью из коллатеральных
приводит к выпадению кристаллов, которые с то-
ком крови переносятся в более мелкие сосуды и осе-
дают в мелких артериолах. Первоначальный сосу-
дистый спазм усугубляется химическим эндарте-
риитом, который стремительно поражает
эндотелий. Кроме того, происходит окклюзия сосу-
дов кристаллами тиопентала, агрегатами эритроци-
тов и тромбоцитов, в результате чего развивается
артериальный тромбоз и гангрена. Важным факто-
ром патогенеза этого осложнения является попада-
ние тиопентала в артериальное русло, где препарат
нейтрализуется, но не разбавляется.
При попадании тиопентала в артерию необходи-
мо принять срочные меры. Если игла (или катетер)
все еще находится в артерии, через нее необходимо
ввести вазодилататор (например, папаверин, прока-
ин, антагонист кальция). Раскрытию коллатераль-
ных сосудов способствует симпатическая блокада
(осуществляемая путем блокады плечевого сплете-
ния подмышечным доступом или блокады звездча-
того ганглия). Целесообразно применение гепарина.
Фармакологические характеристики тиопента-
ла приведены в таблицах 4.1 и 4.2.
МЕТОГЕКСИТАЛ
Общие сведения
Метогекситал является 1-метил-5-ал-
лил-5’-(1-метил-2-пентинил)-2-барбитуровой ки-
слотой. Существует четыре оптически активных
изомера этого соединения. В клинической практи-
ке используется рацемическая смесь двух из них:
a-d-метогекситала и a-Z-метогекситала (рис. 4.1).
Метогекситал выпускают в виде натриевой соли
с 6% карбонатом натрия; это белый порошок, хоро-
шо растворимый в воде. Перед употреблением ме-
тогекситал растворяют в воде, физиологическом
растворе или растворе глюкозы; при этом образует-
ся 1% щелочной раствор с pH около 11. Водные рас-
творы метогекситала при комнатной температуре
могут храниться в течение 6 недель; если препарат
растворяют в физиологическом растворе или рас-
творе глюкозы, его надо использовать в течение
24 ч. Благодаря высокому pH, растворы метогекси-
тала оказывают бактериостатический эффект и не-
совместимы с растворами многих препаратов-осно-
ваний.
Стандартная доза метогекситала для индукции
анестезии составляет 1-2 мг/кг, то есть он прибли-
зительно в три раза мощнее тиопентала. Быстрое
начало действия обусловлено высокой растворимо-
стью в липидах (коэффициент распределения мас-
ло/вода равен 300) и обильным кровоснабжением
НЕИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
81
Таблица 4.1. Фармакодинамические свой нестетиков
Анестетик Начало действия Пробуждение Кровообращение Другие эффекты
Тиопентал Быстрое Относительно бы- строе; полное пробуждение за- медлено Ш свФ Осложнения при внесо- судистом введении Артериальный тромбоз Ларинго- и бронхоспазм Индукция ферментов П вчд
Метогекситал Быстрое Быстрое ад; СВ^ чссТ Боль в месте инъекции Возбуждение при индук- ции анестезии Непроизвольные мы- шечные движения Кашель и икота
Пропофол Быстрое Быстрое АДФ СВ^ Боль в месте инъекции Угнетение ферментов Замедленное пробужде- ние после продолжи- тельного применения
Этоми дат Быстрое Умеренно быстрое Минимальные Боль в месте инъекции Возбуждение Угнетение коры надпо- чечников
Кетамин Медленное Медленное АДТ свТ ЧССТ Анальгезия Твчд Психотомиметические эффекты
СВ - сердечный выброс; ЧСС - частота сердечных сокращений; ВЧД-внутричерепное давление
головного мозга, который в норме получает до 25%
сердечного выброса. Во многих отношениях, вклю-
чая воздействие на ЦНС, кровообращение, функ-
цию печени и почек, а также способность индуциро-
вать активность печеночных ферментов, провоци-
ровать приступ острой перемежающейся порфирии
и нежелательные системные реакции метогекси-
тал мало отличается от тиопентала (таблица 4.1).
Тем не менее между этими анестетиками существу-
ет несколько важных различий.
1. При непреднамеренном введении метогекси-
тала под кожу или в артерию опасность поврежде-
ния тканей меньше, чем у тиопентала. Эксперимен-
тальные исследования позволяют утверждать, что
при попадании в артерию такой же эффект, как тио-
пентал, вызывают высокие концентрации метогек-
ситала (5% раствор). Таким образом, основным
фактором, определяющим осложнение, является
концентрация (или, наоборот, степень разведения)
лекарственного препарата; другие свойства барбиту-
ратов играют меньшую роль. При инъекции в мелкую
вену 1% раствор метогекситала чаще вызывает
боль, чем 2,5% раствор тиопентала; объяснения это-
му парадоксу пока не найдено.
2. Метогекситал вызывает возбуждение во вре-
мя индукции анестезии значительно чаще, чем тио-
пентал. Непроизвольные движения конечностей,
тремор, повышенный мышечный тонус, кашель и
икота развиваются почти у 80% больных. Частота
этих осложнений зависит от дозы и скорости введе-
ния препарата, ее можно снизить соответствующей
премедикацией. Метогекситал может провоциро-
вать судорожную активность при исходных нару-
шениях на ЭЭГ или сопутствующей парциальной
форме эпилепсии; эти свойства позволяют исполь-
зовать метогекситал для верификации эпилептиче-
ского очага в ходе нейрохирургических вмеша-
тельств. Возможность применения метогекситала
при сопутствующей эпилепсии остается противо-
речивым вопросом; некоторые специалисты счита-
ют это безопасным.
82
Глава 4
Таблица 4.2. Фармакокинетические св ых анестетиков
Анестетик Конечный Т1/2 (мин) Клиренс (мл/мин/кг) Vd (л/кг) Метаболиты
Тиопентал 300-600 1,4-5,7 1,0-4,0 Тиопентал-карбоновая кислота Г идрокситиопентал Пентобарбитал
Метогекситал 90-250 10,0-13,0 1,2-2,2 Г идроксиметогекситал Гидроксиметогекситала глюкуронид
Пропофол 300-700 21,4-28,6 3,3-5,7 2,6-диизопропилфенола глюкуронид 2,6-диизопропилхинола глюкуронид
Этомидат 60-90 10,0-24,3 2,2-4,3 Этанол 1-(а-метилбензил)-имидазол-5-карбо- новая кислота
Кетамин 150-200 17,1-20,0 2,9-3,1 Норкетамин Г идроксиноркетамин Гидроксикетамина глюкуронид Г идроксиноркетамина глюкуронид
3. Метогекситал может вызывать тахикардию и
понижает АД, но в меньшей степени, чем тиопен-
тал; поэтому многие анестезиологи считают, что
при шоке для индукции анестезии предпочтитель-
но использовать именно метогекситал.
4. Метогекситал чаще, чем тиопентал, вызывает
реакции гиперчувствительности. Однако, в отли-
чие от тиопентала, в большинстве случаев наблюда-
ются только периорбитальный отек и отек лица, но
ни об одном смертельном исходе не сообщалось.
5. Восстановление сознания после анестезии ме-
тогекситалом наступает значительно быстрее, чем
после эквивалентных доз тиопентала. После много-
кратного струйного введения или инфузии риск ку-
муляции метогекситала ниже, чем у тиопентала.
Эти различия обусловлены фармакокинетиче-
скими особенностями анестетиков (таблица 4.2).
После в/в введения индукционной дозы концен-
трация метогекситала в плазме быстро снижается.
В этом снижении можно выделить три экспоненци-
альные составляющие: фаза быстрого перераспре-
деления (начальный Т/г «5-6 мин); фаза медленно-
го перераспределения (Тй « 60 мин); фаза элимина-
ции (конечный Т./2« 1,5-4 ч).
Метогекситал и тиопентал связываются белка-
ми плазмы приблизительно на 80%; их объемы рас-
пределения одинаковы. Более короткая продолжи-
тельность действия метогекситала обусловлена
главным образом быстрым печеночным клиренсом.
Метаболизм метогекситала осуществляется путем
гидроксилирования до гидроксиметогекситала,
который обладает некоторой гипнотической актив-
ностью. В неизмененном виде с желчью и мочой выде-
ляются только следовые количества метогекситала.
Фармакологические характеристики метогек-
ситала приведены в таблицах 4.1 и 4.2.
ПРИМЕНЕНИЕ ТИОПЕНТАЛА И
МЕТОГЕКСИТАЛА
Тиопентал широко применяется для индукции
анестезии. Метогекситал был популярен в стомато-
логической практике, но после появления пропо-
фола его роль при амбулаторных вмешательствах
значительно уменьшилась.
Тиопентал является препаратом выбора при ле-
чении эпилептического статуса, который не купи-
руется обычными противосудорожными препара-
тами, особенно когда возможно проведение ИВЛ в
условиях миорелаксации. В этих условиях тиопен-
тал начинает действовать быстрее, чем диазепам,
что является его значительным преимуществом.
Тиопентал и другие барбитураты применяются
для защиты головного мозга от гипоксии при ин-
сульте и нейрохирургических операциях. Гемоди-
намические эффекты барбитуратов могут приво-
дить к снижению мозгового кровотока. Однако уг-
нетение метаболизма уменьшает отек головного
мозга и снижает внутричерепное давление (ВЧД),
так что перфузия головного мозга улучшается. Для
такого лечения требуется обеспечение проходимо-
сти дыхательных путей, ИВЛ, мониторинг крово-
обращения и ВЧД. Использование барбитуратов
для защиты мозга является сложной и противоре-
чивой проблемой.
НЕИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
83
ПРОПОФОЛ
Общие сведения
Пропофол (2,6-диизопропилфенол) является
химически инертным производным фенола, обла-
дающим свойствами общего анестетика. Пропофол
практически нерастворим в воде, но хорошо рас-
творим в жирах, поэтому применяется в виде 1%
(10 мг/мл) изотонической эмульсии в 10% соевом
масле, 2,25% глицерине и 1,2% очищенных яичных
фосфолипидах. Также выпускается 2% раствор про-
пофола, удобный для длительных инфузий. На ран-
них этапах клинических исследований формула
препарата содержала способствующий растворению
кремофор EL (полиэтоксилированную касторовую
кислоту), от которого отказались из-за высокого
риска анафилактоидных реакций. Пропофол пред-
ставляет собой ахиральное соединение (рис. 4.1).
Механизм действия
Основной механизм действия пропофола, как и
у барбитуратов, состоит во влиянии на ГАМК-зави-
симые ионные каналы в ЦНС. В частности, пропо-
фол активирует р-субъединицы ГАМК-рецептора,
которые относительно нечувствительны к самой
ГАМК (на его анестетические эффекты не влияет
антагонист бензодиазепинов флумазенил).
Механизм действия пропофола недавно был
изучен с помощью метода локальной фиксации
мембранного потенциала (изоляция фрагмента
клеточной мембраны с помощью специальной пи-
петки) в рекомбинантных рецепторах, полученных
из тканей млекопитающих — ооцитов лягушки
Xenopus laevis и изоформ рецепторов мушек
Drosophila. Согласно результатам этих исследова-
ний, пропофол регулирует глициновые рецепторы.
Другие исследования продемонстрировали, что
пропофол может уменьшать время открывания на-
триевых каналов в мембране нейрона. Следует от-
метить, что влияние на натриевые каналы может
быть вторичным по отношению к изменениям, про-
исходящим в ГАМК-рецепторах.
ЦНС
Стандартная доза пропофола для индукции ане-
стезии составляет 1,5-2,5 мг/кг. Высокая скорость
наступления эффекта обусловлена быстрым посту-
плением в головной мозг жирорастворимого препа-
рата. Индукция анестезии вызывает снижение час-
тоты и увеличение амплитуды волн на ЭЭГ. По
мере углубления анестезии начинают доминиро-
вать р и 5-волны. Спонтанные движения, которые
иногда наблюдаются во время индукции анестезии,
совпадают с фазой 5-активности. Пропофол снижа-
medlit.ucoz.ru „
ет мозговой кровоток и потребление кислорода
головным мозгом. Хотя цереброваскулярное со-
противление значительно повышается, ВЧД и це-
ребральное перфузионное давление снижаются.
После в/в струйного введения концентрация про-
пофола в плазме быстро уменьшается вследствие
перераспределения в организме и захвата перифе-
рическими тканями. По мере снижения концентра-
ции пропофол диффундирует из ЦНС в кровь.
Если для индукции анестезии пропофол вводили
в/в струйно одномоментно (а не в виде инфузии),
то пробуждение и полное восстановление сознания
наступают очень быстро.
Пропофол вызывает дозозависимое угнетение
дыхания. Возможна остановка дыхания, которая
обычно бывает преходящей, но пролонгируется
при использовании высокой дозы (у верхней грани-
цы рекомендованного диапазона) или одновремен-
ном использовании других препаратов, угнетаю-
щих дыхание.
При использовании пропофола чрезвычайно
редко развивается послеоперационная тошнота и
рвота, особенно в отсутствие других анестетиков.
Это свойство пропофола, несомненно, способству-
ет его популярности в качестве препарата для ин-
дукции анестезии при коротких и амбулаторных
хирургических вмешательствах.
Кровообращение
Пропофол часто вызывает значительное сниже-
ние АД (на 20-30%). Выраженность артериальной
гипотонии зависит от дозы и скорости введения и
обычно максимальна в течение 5-10 мин. Падение
АД обусловлено в первую очередь снижением
ОПСС и обычно не сопровождается тахикардией.
Считают, что пропофол действует как антагонист
кальция или способствует высвобождению NO.
Сердечный выброс тоже может снижаться посред-
ством такого же механизма, как у тиопентала.
Прочие системные эффекты
Пропофол редко вызывает побочные эффекты
со стороны дыхательных путей (кашель, ларин-
госпазм) и поэтому часто является препаратом вы-
бора для индукции анестезии при установке ларин-
геальной маски. Пропофол умеренно угнетает не-
которые изоферменты системы цитохрома Р-450
и пролонгирует действие фентанила, альфентанила
и пропранолола. Какого-либо существенного влия-
ния на почки он не оказывает. Пропофол не влияет
на активность ферментов, участвующих в синтезе
порфирина, и поэтому не противопоказан при
порфирии. Пропофол может угнетать хемотаксис
лейкоцитов.
84
Глава 4
medlit.ucoiefu
Фармакокинетика
После в/в инъекции снижение концентрации
пропофола в плазме носит би- или триэкспоненци-
альный характер. Начальное снижение концентра-
ции происходит очень быстро (Тш 1-3 мин) и отра-
жает почти немедленное распределение препарата
из крови в ткани. Значения фармакокинетических
констант после инъекции пропофола отличаются
большим разбросом. Раньше считали, что конеч-
ный Т1/2 пропофола составляет 1-5 ч, и объем рас-
пределения примерно в 10 раз превышает общий
объем воды в организме. Клиренс пропофола пре-
вышает печеночный кровоток, что указывает на
возможность значительного внепеченочного мета-
болизма (возможно, в легких). Это предположение
получило подтверждение, когда из мочи пациента
во время беспеченочного этапа трансплантации пе-
чени было выделено значительное количество ме-
таболита глюкуронида.
Однако исследование с многократным изъятием
проб крови показало, что конечный Тш пропофола
такой же, как у тиопентала (5-12 ч). Элиминация
пропофола в значительной степени зависит от ме-
таболизма в печени и чувствительна к изменениям
печеночного кровотока (но не к активности фер-
ментов или связыванию с белками).
Пропофол в значительной степени (на 97-98%)
связывается с белками плазмы. Приблизительно
40% выделяется через почки в виде конъюгатов
глюкуроновой кислоты. Остальная доля превраща-
ется в 2,6-диизопропилхинол и выделяется с мочой
как конъюгаты с глюкуронидами и сульфатами.
Менее 1% пропофола экскретируется из организма
в неизмененном виде.
Побочные эффекты
При анестезии пропофолом (как при индукции,
так и во время пробуждения), иногда возникает
возбуждение, в том числе миоклония, опистотонус
и непроизвольные движения в конечностях. Кроме
того, существует риск провокации судорожного
припадка при сопутствующей эпилепсии.
Способность легко преодолевать плацентарный
барьер и оказывать угнетающее воздействие на
плод ограничивает применение пропофола при ке-
саревом сечении.
Боль от инъекции обычно возникает при введе-
нии пропофола в мелкие вены тыла кисти, она мо-
жет ощущаться в области предплечья или плеча.
Механизм боли неясен; обычно она преходящая, и
осложнения в виде тромбофлебита встречаются
крайне редко. Боль возникает реже, если пропофол
вводят в крупные вены локтевой ямки или если к
анестетику добавляется небольшое количество ли-
докаина (5-10 мг).
начальная пропись пропофола часто вы-
зывала реакции гиперчувствительности, обуслов-
ленные кремофором EL. Вместе с тем известно о
30 случаях аллергических или анафилактоидных
реакций уже после применения современной про-
писи пропофола. Однако никаких прямых иммуно-
логических подтверждений аллергенности самого
пропофола получено не было, и виновниками ал-
лергических реакций могли являться другие одно-
временно используемые препараты, в частности
миорелаксанты.
После введения пропофола иногда наблюдается
выраженная брадикардия, для устранения которой
могут потребоваться м-холиноблокаторы (напри-
мер, атропин). Сообщалось о неожиданных леталь-
ных исходах у детей на фоне длительной седации
пропофолом в палатах интенсивной терапии.
Смерть наступала при явлениях нарастающего ме-
таболического ацидоза, брадикардии и прогресси-
рующей сердечной недостаточности. Считается,
что причиной смерти являлся сам пропофол и ли-
пидные составляющие растворителя, но точная
этиология остается неясной.
Терапевтический диапазон пропофола уже, чем
у этомидата, но шире, чем у барбитуратов.
Основным достоинством пропофола является
быстрое пробуждение и полное восстановление
сознания после индукции анестезии; кроме того,
после длительной инфузии или многократных вве-
дений не наблюдается значительной кумуляции.
Тем не менее конечный Т1/2 пропофола относитель-
но высок, поэтому его применение в виде длитель-
ной инфузии или многократных струйных введе-
ний требует осторожности.
Фармакологические свойства пропофола обоб-
щены в таблицах 4.1 и 4.2.
ЭТОМИДАТ
Общие сведения
Этомидат (К-(+)-1-(а-метилбензил)-имида-
зол-5-этилкарбоксилат сульфат) является заме-
щенным производным имидазола и эфиром. Хотя
сульфатная соль этомидата легко растворяется
в воде, пропись препарата содержит пропиленгли-
коль, что повышает стабильность раствора и умень-
шает местное раздражающее действие. Этомидат
обычно применяют в виде 0,2% раствора (2 мг/мл)
в 35% пропиленгликоле. В препарате содержится
только один оптический изомер этомидата (рис. 4.1).
НЕИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
85
medlit.
Механизм действия
Механизм действия этомидата на мембраны
нейронов и/или нейромедиаторы в ЦНС до конца
не ясен. Недавно стало известно, что этомидат ока-
зывает регулирующее действие на отдельный алло-
стерический участок хлорного канала (изоформа
(3-субъединицы), связанного с ГАМК-рецептором.
Подобные селективные эффекты присущи проти-
восудорожному препарату лорекрезолу, который
по химическому строению напоминает этомидат и в
экспериментальных условиях препятствует дейст-
вию пикротоксина.
Системные эффекты
Стандартная доза для индукции анестезии
(0,3 мг/кг) быстро выключает сознание; продолжи-
тельность действия зависит от дозы, а тенденция к
кумуляции практически отсутствует даже при мно-
гократных струйных введениях. Изменения на ЭЭГ
в начале анестезии напоминают эффекты барбиту-
ратов, а непроизвольные мышечные движения не
связаны с аномальной электрической активностью
коры головного мозга. Наиболее важным достоин-
ством этомидата является относительно широкий
терапевтический диапазон: соотношение между
анестетической и летальной дозой равно 30 (у в/в
барбитуратов оно колеблется от 5 до 10). Этомидат
не оказывает значимого влияния на кровообраще-
ние, обычно вызывая небольшое снижение ОПСС
и АД; кровоток в большинстве органов не меняется
или слегка возрастает. Сократительная способ-
ность миокарда, потребление кислорода и коронар-
ный кровоток обычно не меняются. Этомидат
уменьшает мозговой кровоток и ВЧД, а в гипноти-
ческих дозах угнетает дыхание.
Анафилактические и анафилактоидные реак-
ции развиваются редко. Иногда во время индукции
анестезии появляется сыпь, обусловленная высво-
бождением гистамина из тучных клеток. Тяжелые
реакции (генерализованная эритема, артериальная
гипотония и бронхоспазм) возникали при сочетан-
ном использовании сукцинилхолина и алкурония;
считается, что причинным фактором являются
именно миорелаксанты. Некоторые специалисты
считают этомидат наиболее безопасным препаратом
для индукции анестезии и средством выбора при вы-
соком риске реакций гиперчувствительности.
Фармакокинетика
После внутривенной инъекции концентрация
этомидата в плазме снижается биэкспоненциально.
Первоначальное снижение концентрации (началь-
ный Т1/2 2-5 мин) отражает распределение препара-
ucoz.ru
та в хорошо кровоснабжаемые ткани. Последующее
замедленное снижение концентрации обусловлено
элиминацией препарата (конечный Т1/2« 68-75 мин).
Объем распределения незначительно превышает
общий объем воды в организме, а клиренс составля-
ет 50-80% печеночного кровотока (таблица 4.2).
Относительно высокая степень экстракции ане-
стетика соответствует печеночному клиренсу, ог-
раниченному кровотоком. Этомидат в значитель-
ной степени связывается с альбумином плазмы
(на 70-80%), поэтому его эффект усиливается у по-
жилых и при гипоальбуминемии.
Элиминация этомидата в основном происходит
путем метаболизма, в моче и желчи обнаруживают-
ся лишь его следы (1-2%). Препарат метаболизиру-
ется путем гидролиза эфира до этанола и производ-
ного карбоновой кислоты [1-(а-метилбензил)-ими-
дазол-5-карбоновая кислота]. Метаболизм этомида-
та в основном зависит от неспецифических эстераз,
хотя гидролиз может также осуществляться под
действием холинэстеразы. Этомидат может также
подавлять активность холинэстеразы, конкурируя
с другими субстратами.
Побочные эффекты
Несмотря на преимущества, этомидат вызывает
несколько нежелательных эффектов, ограничи-
вающих его применение. Боль при инъекции возни-
кает у 25-50% пациентов. Содержащийся в пропи-
си препарата пропиленгликоль может вызывать ге-
молиз, хотя и не имеющий клинического значения.
Этомидат часто вызывает возбуждение, особенно
непроизвольные движения в конечностях и повы-
шенный мышечный тонус. Эти эффекты можно
предотвратить предварительным введением бензо-
диазепинов или опиоидов (например, фентанила
или альфентанила). Послеоперационная тошнота и
рвота при индукции анестезии этомидатом разви-
ваются чаще, чем при использовании других неин-
галяционных анестетиков.
Длительная в/в инфузия этомидата может при-
вести к угнетению функции коры надпочечников.
Препарат может нарушать синтез глюкокортикои-
дов и минералкортикоидов. Этот эффект опосреду-
ется высоким сродством этомидата к изоформам
цитохрома Р-450, что приводит к угнетению синте-
за гидроксикортизона. Результаты эксперимен-
тальных исследований показывают, что длитель-
ная инфузия этомидата повышает активность ами-
нолевулинсинтазы. Следовательно, этомидат не
рекомендуется применять при острой интермитти-
рующей порфирии.
Фармакологические свойства этомидата приве-
дены в таблицах 4.1 и 4.2.
86
Глава 4
кетамин medlit
Общие сведения
Кетамин [(2-хлорфенил)-2-(метиламино)-цик-
логексанона гидрохлорид] по химическому строе-
нию сходен с циклогексамином и фенциклидином.
Кетамина гидрохлорид хорошо растворяется в
воде, при этом образуется раствор с низким pH
(3,5-5,5). Кетамин выпускается в трех разных кон-
центрациях (1%, 5% и 10% — 10, 50 и 100 мг/мл со-
ответственно) для введения в/в (рекомендуемая
доза 1-4,5 мг/кг) или в/м (рекомендуемая доза
5-10 мг/кг). Кетамин представляет собой рацеми-
ческую смесь S(+) и R(-) энантиомеров (рис. 4.1).
Механизм действия
Электрофизиологические исследования пока-
зали, что кетамин воздействует преимущественно
на проекционные таламокортикальные пути, ока-
зывая лишь минимальное воздействие на ретику-
лярную формацию, лимбическую систему и боль-
шинство таламических ядер. Кетамин и другие ана-
логи фенциклидина являются антагонистами
возбуждающего нейромедиатора глутамата (подоб-
ное действие могут оказывать закись азота и ксе-
нон). Молекулярной мишёнью кетамина является
NMDA-рецептор. Антагонизм между кетамином и
NMDA носит неконкурентный характер, поэтому
предполагают, что препарат может блокировать
ионные каналы в рецепторе. В ЦНС глутамат вы-
свобождается нервными окончаниями кортикост-
риарной зоны и играет важную роль центрального
нейромедиатора; действие кетамина на этом уровне
вызывает «диссоциативную» анестезию. Опосредо-
ванная глутаматом нервная передача в задних рогах
спинного мозга играет важную роль в трансмиссии
ноцицептивных импульсов по спиноталамическим
проводящим путям; действием кетамина на этом
уровне объясняется его анальгетический эффект.
ЦНС
Кетамин отличается от других неингаляцион-
ных анестетиков почти полным отсутствием гипно-
тического эффекта. Он вызывает диссоциативную
(диссоциированную) анестезию, характеризую-
щуюся дозозависимой антероградной амнезией и
выраженной анальгезией. По сравнению с другими
неингаляционными анестетиками, кетамин начи-
нает действовать медленно — приблизительно че-
рез 90 с после в/в инъекции. При введении в/м для
развития эффекта может потребоваться до 8 мин.
Бывает трудно с точностью определить начало дей-
ствия: больной в течение нескольких минут может
смотреть перед собой в пространство, не закрывая
UGQZBEldpeMH индукции анестезии часто развива-
ются повышенный мышечный тонус и непроиз-
вольные движения конечностей (в том числе
клонико-тонического характера). Миорелаксация
часто недостаточная; напротив, может повышаться
тонус жевательных мышц, приводящий к обструк-
ции дыхательных путей. Характер дыхания практи-
чески не изменяется (хотя частота дыхания может
немного повыситься) и сохраняется реакция на
изменения РаСО2. Кашель и ларингоспазм возни-
кают крайне редко; наоборот, в эксперименте ус-
тановлено, что кетамин действует как антагонист
эффектов гистамина, ацетилхолина и 5-гидро-
кситриптамина на гладкие мышцы бронхов. Кетамин
можно без опаски применять при бронхиальной ас-
тме. Кетамин обычно не подавляет рефлекторные
реакции, но иногда вызывает некоторое угнетение
гортанных рефлексов.
Продолжительность действия кетамина (как и
большинства неингаляционных анестетиков) зави-
сит от дозы. Стандартные дозы для в/в введения
действуют в течение 10-20 мин, хотя точное время
определить трудно. Кетамин вызывает характер-
ные изменения на ЭЭГ: a-ритм подавляется и заме-
щается 0- и 5-волнами. Эти изменения могут сохра-
няться на протяжении всего периода анальгезии.
Кровообращение
В отличие от большинства других анестетиков,
кетамин неизменно увеличивает ЧСС, сердечный
выброс и концентрацию норадреналина в плазме.
На 20-40% повышается диастолическое и систо-
лическое АД, а также легочное сосудистое сопро-
тивление. Такие изменения развиваются в течение
5 мин после введения и продолжаются 10-20 мин;
отчасти они обусловлены непосредственным воз-
действием кетамина на проводящие пути ЦНС.
Экспериментально доказано, что положительное
инотропное действие кетамина обусловлено акти-
вацией сердечных 0-адренорецепторов, а на денер-
вированный миокард кетамин оказывает угнетаю-
щее действие. Кетамин повышает мозговой крово-
ток, потребление кислорода мозгом и ВЧД.
Побочные эффекты
В послеоперационном периоде нередко возни-
кают тошнота и рвота. В первые сутки после приме-
нения кетамина высок риск острых психических
нарушений — ярких устрашающих сновидений, ил-
люзий, галлюцинаций и делирия. Эти осложнения
могут быть очень неприятными для больного; пола-
гают, что они обусловлены нарушениями воспри-
ятия и обработки сенсорной информации (особен-
но зрительной и слуховой). Риск этих осложнений
НЕИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
87
iiPfiCjlit.UGQiriJ
можно значительно снизить с помощью
тельного введения опиоидов, безодиазепинов или
дроперидола (который в чистом виде может сам вы-
зывать психотомиметические эффекты). Психиче-
ские нарушения чаще развиваются у женщин и го-
раздо реже у детей, особенно если послеоперацион-
ный период протекает гладко.
Фармакокинетика
При отсутствии нарушений кислотно-основного
состояния (при pH 7,4) 45% кетамина находится
в неионизированной форме. После в/в введения
кетамин быстро распределяется в тканях и легко
преодолевает гематоэнцефалический (а также пла-
центарный) барьер. Снижение концентрации ке-
тамина в плазме носит биэкспоненциальный харак-
тер: после первоначального быстрого падения кон-
центрации (начальный Ti/2 « 10-20 мин) следует
фаза замедленного выведения (конечный Т1/2 «
150-200 мин), отражающая элиминацию (которая
обеспечивается в основном метаболизмом в печени).
Объем распределения в 2-3 раза превышает об-
щий объем воды в организме, а клиренс приблизи-
тельно равен печеночному кровотоку. Относительно
высокая степень экстракции анестетика соответ-
ствует печеночному Клиренсу, ограниченному
кровотоком, поэтому препараты, уменьшающие
печеночный кровоток (например, ингаляционные
анестетики, Р-адреноблокаторы, циметидин), могут
снижать клиренс кетамина и увеличивать его ко-
нечный Ti/2. После введения в/м кетамин быстро
всасывается; максимальная концентрация в плазме
достигается в течение 30 мин.
Элиминация кетамина зависит от системы окси-
даз гладкого эндоплазматического ретикулума.
Главный метаболит, норкетамин, обладает некото-
рой гипнотической активностью, которая слабее,
чем у кетамина. В процессе дальнейшего метабо-
лизма кетамин и норкетамин превращаются в гид-
роксилированные производные, которые образуют
конъюгаты с глюкуроновой кислотой и элиминиру-
ются из организма (табл. 4.2).
Применение
Применение кетамина ограничено его неблаго-
приятным влиянием на ЦНС и кровообращение.
Кетамин не применяют при заболеваниях сердца,
травмах, психических заболеваниях. Тем не менее
кетамин имеет свои показания. Его применяют в/м
у детей, когда по тем или иным причинам трудно ка-
тетеризировать периферическую вену, а также при
многократных анестезиях. Кетамин используют
при катетеризации сердца, при многократных
перевязках у ожоговых больных, при различных
вмешательствах (лучевая терапия, стер-
нальная пункция). Для седации и премедикации
у маленьких детей кетамин можно назначить
внутрь или ректально. В этих случаях он имеет низ-
кую биодоступность и подвергается эффекту пер-
вого прохождения через печень.
У взрослых показания к применению кетамина
противоречивее и определены менее четко. Его ис-
пользуют для обезболивания при многократных
перевязках у ожоговых больных, у пожилых с высо-
ким риском периоперационных осложнений, при
некоторых экстренных ситуациях (например, обез-
боливание на месте дорожно-транспортного проис-
шествия). В некоторых странах кетамин вводят
интратекально и эпидурально для послеоперацион-
ного обезболивания или лечения хронической боли
(в РФ интратекальное и эпидуральное введение ке-
тамина не разрешено— прим. ред.).
Существуют значительные фармакологические
различия между двумя энантиомерами кетамина.
S(+) кетамин приблизительно в три раза мощнее
своего R(-) антипода, гораздо реже вызывает ост-
рые психические нарушения после пробуждения (в
том числе делирий), обеспечивает более надежную
интраоперационную амнезию. Более того, объем
распределения, клиренс и интенсивность метабо-
лизма S(+)-изомера выше. Эти различия отражают
различное сродство изомеров к хиральным рецеп-
торам и ферментам. В настоящее время признано,
что кетамин имел бы гораздо больше преимуществ,
если бы применялся в виде 8(+)-изомера, а не раце-
мической смеси. Фармакологические свойства ке-
тамина представлены в таблицах 4.1 и 4.2.
ПОБОЧНОЕ ДЕЙСТВИЕ
НЕИНГАЛЯЦИОННЫХ
АНЕСТЕТИКОВ
Общие сведения
Первое подтвержденное сообщение о побочной
реакции на неингаляционный анестетик появилось
в 1952 г. Этим препаратом был тиопентал. После
появления небарбитуровых анестетиков число со-
общений о реакциях гиперчувствительности нача-
ло стремительно расти. Из-за побочных эффектов
было запрещено применение двух препаратов —
пропанидида и альтезина. Прописи обоих препара-
тов содержали кремофор EL (полиэтоксилирован-
ное касторовое масло), использовавшийся для
улучшения растворимости активного компонента.
Кремофор EL является смесью жирных кислот с
молекулярным весом приблизительно 3200 и вхо-
дит в пропись некоторых витаминов и противо-
88
Глава 4
Таблица 4.3. Реакциигиперчувствительно^у'в^'^
использовании неингаляционных анестетиков
Анестетик Частота реакций
Тиопентал от 1:14 000 до 1:20 000
Метогекситал от 1:1600 до 1:7000
Пропофол от 1:80 000 до 1:100 000
Этомидат от 1:50 000 до 1:450 000
Кетамин 2 случая
грибковых препаратов. Хотя ранние исследования
не обнаружили иммуногенных или анафилактоид-
ных свойств кремофора EL, в дальнейшем подтвер-
дилось, что он играет важную роль в развитии по-
бочных реакций на неингаляционные анестетики.
С другой стороны, его сурфактантные свойства мо-
гут усиливать иммуногенный потенциал пропани-
дида и альфаксолона (активного стероидного ком-
понента альтезина).
В настоящее время частота интраоперационных
анафилактических и анафилактоидных реакций
составляет 1: 6000. Считают, что причиной 80% из
них могли быть миорелаксанты. Частота реакций
на неингаляционные анестетики приведена в таб-
лице 4.3. Тяжесть реакций гиперчувствительности
оценить трудно. Например, гиперчувствитель-
ность к тиопенталу, которая обычно редко вызыва-
ет тяжелые последствия, в 10 случаях послужила
причиной смерти. С другой стороны, обо многих
реакциях гиперчувствительности не сообщается
(особенно, если они не имели серьезных последст-
вий).
Некоторые побочные эффекты (например, арте-
риальная гипотония) могут быть обусловлены пря-
мым воздействием анестетика на миокард или со-
суды, или же чрезмерной вазовагальной реакцией
на венепункцию. Неправильное положение или за-
купорка интубационной трубки может имитиро-
вать бронхоспазм. Анестезия неизбежно сопровож-
дается полипрагмазией, и вклад каждого препарата
(в том числе миорелаксантов и коллоидных раство-
ров) в генез реакции бывает трудно определить.
Своевременное выявление реакции и немедленно
начатое лечение улучшают прогноз.
Симптомы анафилактоидных и анафилактиче-
ских реакций на неингаляционные анестетики
включают эритему, бронхоспазм, артериальную ги-
потонию, периферический сосудистый коллапс,
крапивницу, отеки и боль в животе. Течение реак-
ции может быть очень разным: в тяжелых случаях
нескольких секунд после инъекции раз-
вивается бронхоспазм, быстро приводящий к циа-
нозу. Пульс не пальпируется, АД не определяется
(хотя на ЭКГ возможно увеличение ЧСС). Затем
развивается местный или генерализованный отек.
Иногда реакция наступает не сразу, а через 10-90 мин,
симптомы относительно доброкачественны, причи-
на остается неясной. Многие симптомы напомина-
ют фармакологические эффекты гистамина или
других медиаторов, включая брадикинин, 5-гидро-
кситриптамин, лейкотриены, простагландины и ге-
парин.
Выделяют четыре типа реакций:
1. Реакции гиперчувствительности немедленно-
го типа (I типа). Развиваются после сенсибилиза-
ции к лекарственному препарату при повторном
контакте с ним. Реакции предшествует образование
антител IgE.
2. Цитотоксические реакции (II типа). В ходе та-
ких реакций антитела IgG или IgM связываются с
антигеном на поверхности клеток, запуская класси-
ческий каскад комплемента.
3. Анафилактоидные реакции развиваются при
первом контакте с препаратом. Обусловлены акти-
вацией альтернативного пути комплемента.
4. Непосредственное воздействие на циркули-
рующие в крови базофилы и тучные клетки с после-
дующим высвобождением гистамина. Тяжесть ре-
акции зависит от дозы и скорости введения препа-
рата. Может развиться при первом контакте с
препаратом. Симптомы обычно нетяжелые.
ЛЕЧЕНИЕ
Необходимо точно установить диагноз реакции
гиперчувствительности и обеспечить проходи-
мость дыхательных путей. Бронхоспазм является
наиболее опасным для жизни осложнением, и ле-
чение в первую очередь должно быть направлено
на профилактику тяжелой гипоксии. Обычно по-
казана интубация трахеи и ИВЛ с FiO2 1,0. Препа-
ратом выбора является адреналин в концентрации
100 мкг/мл, который вводят в/в (в РФ доступен
0,1% адреналин, что соответствует концентрации
1000 мкг/мл; перед в/в введением этот раствор сле-
дует разбавить в 10 раз — прим.ред.). Помимо брон-
хорасширяющего действия адреналин предотвраща-
ет дальнейшее высвобождение гистамина и повы-
шает ОПСС. Если состояние тяжелое и возможен
полноценный мониторинг кровообращения, то
адреналин вводят в/в струйно медленно дробны-
ми дозами по 300-500 мкг, пока не будет получен
желаемый эффект. Если бронхоспазм не столь тя-
желый, дозы адреналина могут быть меньше
(50-100 мкг). Отек голосовой щели или подсвязоч-
НЕИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
89
ного пространства, вызванный пропотеванШОсЦЦ. иС^Эйы.Шределение концентрации IgE
кости в ткани глотки и гортани, может привести
к обструкции дыхательных путей. Для профилак-
тики и лечения этого осложнения (а также кра-
пивницы) применяются Hi-блокаторы; препара-
том выбора является хлорфенамин (10 мг в/в).
Необходимо также ввести гидрокортизона ге-
мисукцинат (до 200 мг в/в). Однако его максималь-
ный эффект достигается только через 1 -6 ч, поэтому
может сохраняться артериальная гипотония и брон-
хоспазм. При упорном бронхоспазме может помочь
аминофиллин в/в или сальбутамол через распы-
литель ингаляционных расворов (небулайзер).
Для увеличения ОЦК рекомендуются инфузии кол-
лоидных и кристаллоидных растворов, поскольку до
1-2 л ОЦК может депонироваться в тканях.
ПРОФИЛАКТИКА
1. Скорость введения и доза анестетика должны
быть тщательно подобраны. Тяжесть многих реак-
ций уменьшается при медленном в/в введении
средних доз препарата.
2. При аллергии в анамнезе значительно повы-
шен риск реакции I типа, особенно, если в крови по-
вышено содержание IgE. В таких случаях повтор-
ное введение препаратй-аллергена чрезвычайно
опасно. Следует применять другие анестетики или
методики анестезии (например, местную или ре-
гионарную).
3. Если общая анестезия все же необходима, в пре-
медикацию включают кортикостероиды, а также
Hi-блокатор (хлорфенамин) и Н2-блокатор (рани-
тидин), которые помогают предотвратить сердеч-
но-сосудистые эффекты гистамина.
4. Исходя из медицинских и юридических сооб-
ражений чрезвычайно важно дифференцировать
истинную реакцию гиперчувствительности от
других состояний. Последние могут быть обуслов-
лены чрезмерным фармакологическим эффектом
препарата, межлекарственными взаимодействия-
ми и механической обструкцией дыхательных пу-
тей. В пробе крови, полученной после развития ре-
акции радиоиммунологическим методом, можно
определить концентрацию гистамина. Примерно в
течение 6 ч после развития реакции гиперчувстви-
тельности в крови сохраняется повышенное содер-
жание триптазы — протеолитического фермента,
высвобождающегося из гранул тучных клеток
(но не базофилов) после их активации. Диагности-
ческое значение также имеет обнаружение в моче
основного метаболита гистамина — М-метилгиста-
мина. Позже для установления причины реакции
гиперчувствительности применяют более специ-
фические исследования, в том числе внутрикожные
в крови, а для
выявления специфических антител — пробы с ра-
диоаллергосорбентом.
ТОТАЛЬНАЯ ВНУТРИВЕННАЯ
АНЕСТЕЗИЯ
Тотальной внутривенной анестезией называет-
ся общая анестезия, при которой все препараты
вводят внутривенно. Неингаляционные анестети-
ки выключают сознание, опиоиды предотвращают
боль, а миорелаксанты облегчают ИВЛ. В послед-
ние годы интерес к тотальной внутривенной ане-
стезии возрос, поскольку, с одной стороны, она не
загрязняет окружающую среду, с другой стороны,
появились препараты с благоприятными фармако-
кинетическими свойствами (пропофол, альфента-
нил, ремифентанил, атракурий). Метод непрерыв-
ной инфузии имеет значительные практические
преимущества, в том числе отсутствие значимой де-
прессии кровообращения, быстрое пробуждение
и исключение осложнений, сопряженных с ингаля-
ционными анестетиками. К сожалению, эта методи-
ка относительно дорога, поскольку для ее осущест-
вления требуется высокоточная управляемая ин-
фузия препаратов ультракороткого действия,
обеспечивающих быстрое пробуждение. Кроме
того, требуются точные фармакокинетические
данные в отношении используемых препаратов,
собранные в определенной популяции пациентов
и должным образом проанализированные. В свою
очередь, на фармакокинетику и активность неинга-
ляционных анестетиков оказывает влияние соче-
танное применение других препаратов, а также со-
путствующие заболевания системы кровообраще-
ния, почек и печени. Существуют значительные
индивидуальные различия в метаболизме и элими-
нации применяемых в анестезиологии препаратов;
некоторые из этих различий обусловлены генети-
ческими факторами. Непреднамеренное восстанов-
ление сознания и сновидения во время операции
являются еще одной проблемой тотальной внутри-
венной анестезии. Несмотря на очевидные экологи-
ческие преимущества, требуются дополнительные
сведения о лекарственных взаимодействиях и бо-
лее точные способы оценки адекватности анесте-
зии, прежде чем эта методика получит более широ-
кое распространение. Была разработана компьюте-
ризованная и основанная на фармакокинетических
моделях система инфузии, управляемой по целевой
концентрации (target controlled infusion). Инфу-
зия, управляемая по целевой концентрации, позво-
ляет поддерживать заданную сывороточную кон-
центрацию пропофола и изменять ее в зависимости
от интраоперационной ситуации.
90
Глава 4
Список литературы
Belelli, D., Pistis, M., Peters, J.A. & Lambert, J.L. (1999). General
anaesthetic action at transmitter-gated inhibitory amino acid
receptors. Trends in Pharmacological Sciences 20, 496-502.
Bevan, J.C. (1993). Propofol related convulsions. Canadian Journal of
Anaesthesia 40, 805-809.
Briggs, L.P., Clarke, R.S.J., Dundee, J.W. et al. (1981). Use of
di-isopropylphenol as main agent for short procedures. British
Journal of Anaesthesia 53,1197-1202.
Christiensen, J.H., Andreasen, F. & Janssen, J.A. (1982).
Pharmacokinetics and pharmacodynamics of thiopentone: a
comparison between young and elderly patients. Anaesthesia 37,
398-404.
Cook, D.J., Carton, E.G. & Housmans, P.R. (1991). Mechanism of
the positive inotropic effect of ketamine in isolated ferret
ventricular papillary muscle. Anesthesiology 74, 880-888.
Doenicke, A. (1974). Etomidate: a new intravenous hypnotic.
Acta Anaesthesiologica Belgica 25,307-315.
Dolin, SJ„ Smith, M.B., Soar, J. & Morris, P. (1992). Does glycine
antagonism underly the excitatory side effects of methohexitone
and Propofol? British Journal of Anaesthesia 68, 523-526.
Domino, E.E., Chodoff, P. & Corrsen, G. (1965). Pharmacologic
effects of CI 581, a new dissociative anesthetic in man. Clinical
Pharmacology and Therapeutics 6 ,279-290.
Dundee, J.W.,Prrce, H.L. & Dripps, R.D. (1956). Acute tole-
rance to thiopentone in man. British Journal of Anaesthesia 28,
344-352.
Dundee, J.W. & Wyant, G.M. (1988). Intravenous Anaesthesia.
Churchill Livingstone, Edinburgh.
Firestone, L.L., Quinlan, J J. & Homanics, G.E. (1995). The
role of gamma-amino butyric acid type-A receptor subtypes in the
pharmacology of general anaesthesia. Current Opinion in
Anesthesiology 8,311 -314.
Fisher, M. (1995). Treatment of acute anaphylaxis. British
Medical Journal 311, 731-733.
Frenkel, C., Duch, D.S. & Urban, B.W. (1993). Effects of IV anaes-
thetics on human brain sodium channels. British Journal of
Anaesthesia 71, 15-24.
medlit.uffiQZury., Stanski, D.R. & Burch, P.G. (1983). Pharma-
cokinetics of methohexital and thiopental in surgical patients.
Anesthesiology 59, 215-219.
Laroche, D., Vergnaud, M.C., Sillard, B. era/. (1991). Bioche-
mical markers of anaphylactoid reactions to drugs: compari-
son of plasma histamine and try ptase. Anesthesiology 75,945-949.
Lindgren, L. (1994). Anaesthetic activity and side effects of
propofol. Current Opinion in Anaesthesiology!, 321-325.
Lundy, J.S. (1935). Intravenous anesthesia: preliminary report
of the use of two new thiobarbiturates. Proceeding? of Staff Meeting?
of the Mayo Clinic 10, 534-543.
Nebauer, A.E., Doenicke, A., Hoernecke, R. etaz(1992). Does
etomidate cause haemolysis? BritishJoumalofAnaesthesia 69,58-60.
Parke, TJ., Stevens, J.E., Arce, A.S.C. etal. (1992). Metabolic
acidosis and fatal myocardial failure after propofol infusion in
children: five case reports. BritishMedicaljoumal305,613-615.
Petros, AJ., Bogle, R.G. & Pearson, J.D. (1993). Propofol stimu-
lates nitric oxide release from porcine aortic endothelial cells.
British Joumalof Pharmacology 109,6-7.
Powell, H., Morgan, M. & Sear, J.W. (1992). Pregnanolone: a
new steroid intravenous anaesthetic: dose finding study.
Anaesthesia 47, 287-290.
Selye, H. (1942). Studies concerning the correlation between
anesthetic potency, hormonal activity and chemical structure among
steroid compounds. Anesthesia and Analgesia 21,41-47.
Trotter, C. & Serpell, M.G. (1992). Neurological sequelae in
children after prolonged propofol infusion. Anaesthesia SI,
340-342.
Veroli, P., O'Reilly, B., Bertrand, F. etal. (1992). Extrahe-
patic metabolism of propofol in man during the anhepatic phase
of orthoptic liver transplantation. British Journal of Anaesthesia
68,183-186.
Wann, K.T. (1993). Neuronal sodium and potassium channels:
structure and function. British Journal of Anaesthesia! t, 2-14.
White, P.F., Ham, J., Way, W.L. & Trevor, AJ. (1980).
Pharmacology of ketamine isomers in surgical patients.
Anesthesiology 52, 231-239.
Wyant, G.M. & Chang, C.A. (1959). Sodium methohexital: a clinical
study. Canadian Anaesthetists' Society Journal 6, 40-50.
ИНГАЛЯЦИОННЫЕ ucoz u
АНЕСТЕТИКИ
5
Хотя ингаляционные анестетики произвели ре-
волюцию в хирургии, медицинское сообщество
поначалу отнеслось к ним достаточно скептически
и признало далеко не сразу. Способность закиси
азота притуплять восприятие и угнетать возмож-
ность произвольных движений была описана Джо-
зефом Пристли еще в 1772 г., однако впервые этот
газ был использован для ингаляционной анальге-
зии и анестезии только в XIX веке. Этиловый эфир
и хлороформ, которые начали применять в 1840-е
годы, получили признание гораздо быстрее. В1934 г.
в клинической практике появились трихлорэтилен
и циклопропан. Приблизительно двадцать лет
спустя был синтезирован галотан, и после его все-
общего признания были разработаны и внедрены
в практику другие фторзамещенные анестетики
в следующем порядке: метоксифлуран, энфлуран,
изофлуран, севофлуран и десфлуран.
Общая анестезия — это состояние обратимой
утраты восприятия, сопровождающееся отключе-
нием сознания, болевой чувствительности, а также
той или иной степенью миорелаксации. На заре
анестезиологии все эти цели достигались примене-
нием одного-единственного анестетика (так назы-
ваемая «моноанестезия»). Около пятидесяти лет
тому назад были разработаны методики «сбаланси-
рованной» анестезии, где для обеспечения каждого
компонента анестезии применяли отдельный
препарат. Такой подход снижает риск осложнений
и ускоряет восстановление жизненно важных
функций после операции.
Ингаляционные анестетики предназначены
прежде всего для выключения сознания, но вместе
с тем они могут оказывать значительный обезболи-
вающий эффект (например, закись азота) или вы-
зывать миорелаксацию (например, севофлуран).
Ингаляционные анестетики хранят при комнатной
температуре как сжиженные газы (в баллонах под
давлением) или как летучие жидкости (в гермети-
чески закупоренных флаконах).
МЕХАНИЗМ ДЕЙСТВИЯ
Молекулярные механизмы действия ингаляци-
онных анестетиков неизвестны. Действие всех ин-
галяционных анестетиков развивается относитель-
но быстро и носит легкообратимый характер, по-
этому представляется сомнительным, чтобы их
эффект зависел от образования ковалентных свя-
зей в ЦНС. Более вероятно, что действие ингаляци-
онных анестетиков зависит от их физичебких
свойств или от низкоэнергетических межмолеку-
лярных взаимодействий (например, сил Ван дер
Ваальса, диполь-дипольных взаимодействий, ион-
ных или водородных связей).
Большинство ингаляционных анестетиков име-
ет сходные физико-химические и биологические
свойства. Обычно это простые алифатические
углеводороды (алканы) или эфиры, содержащие
1-4 атома углерода, с температурой кипения ниже
90°С. Ингаляционные анестетики различаются по
мощности (синонимы: активность, сила действия).
Мощность прямо пропорциональна их раствори-
мости в органических растворителях (рис. 5.1).
Действие анестетиков носит аддитивный характер:
при использовании одновременно нескольких пре-
паратов их дозы, выраженные в МАК (минималь-
ная альвеолярная концентрация; см. ниже), можно
складывать. Избыточное давление модифицирует
или устраняет эффекты анестетиков. Многие про-
стые и относительно инертные химические соеди-
нения могут в определенных условиях приобретать
свойства ингаляционных анестетиков, в том числе
многие газы, углеводороды, галогензамещенные
углеводороды, эфиры, галогензамещенные эфи-
ры и органические растворители. Большинство
этих веществ нельзя использовать в клинической
практике из-за выраженных побочных эффектов
или токсичности. Некоторые вещества приобре-
тают свойства анестетика только при высоком
парциальном давлении: например, в случае с азо-
том это давление должно составлять около 30 атм.
92
Г лава 5
Рис. 5.1. Зависимость между коэффициентом распределения масло/газ при 37°С и МАК (%) различных ингаляцион-
ных анестетиков
Следовательно, азот может проявить себя как ане-
стетик только в гипербарических условиях (напри-
мер, при глубоководных погружениях).
Много лет назад было установлено, что ингаля-
ционные анестетики реализуют свое действие, вли-
яя на синаптическую передачу в головном и спин-
ном мозге. Толщина клеточной мембраны нейро-
нов в ЦНС составляет около 10 нм. Эта мембрана
состоит из бимолекулярного слоя фосфолипидов,
в который включены внешние и интегральные бел-
ки (см. рис. 1.1). Некоторые из этих белков являют-
ся рецепторами или ферментами; другие пробода-
ют липидную мембрану и содержат в себе селектив-
ные ионные каналы, осуществляющие транспорт
специфичных для каждого канала ионов (напри-
мер, Na+, К+, Са2+ или С1). Открытие и закрытие ка-
налов приводит к изменениям ионных токов, кото-
рые определяют активность нейрона. Фосфолипи-
ды, непосредственно окружающие ионные каналы
(пограничные липиды), могут модифицировать
функциональное состояние этих каналов, влияя
на конформационные изменения, от которых зави-
сит перенос ионов (то же относится и к функциям
рецепторов или ферментов). Если ингаляционные
анестетики вызывают анестезию, воздействуя на
синаптические мембраны, то они могут оказывать
преимущественное влияние либо на фосфолипид-
ные, либо на белковые компоненты мембран.
ЛИПИДНЫЕ ТЕОРИИ АНЕСТЕЗИИ
В 1899-1901 годах Мейер и Овертон независи-
мо друг от друга установили, что мощность многих
анестетиков и гипнотиков прямо пропорциональна
их растворимости в оливковом масле. С тех пор
многие гипотезы основывались на корреляции ме-
жду мощностью анестетика и его растворимостью
в жирах, оцениваемой по коэффициенту распреде-
ления масло/газ при температуре 37°С (рис. 5.1).
Согласно большинству ранних теорий, ингаляци-
онные анестетики оказывали свое действие, взаи-
модействуя с фосфолипидами мембраны нейрона.
Считалось, что ингаляционные анестетики рас-
творяются в фосфолипидах мембран, изменяя их
физические свойства (например, текучесть, объем,
поверхностное натяжение или боковое поверхно-
стное давление), что влияет на степень упорядо-
ченности внутри мембраны. Предполагали, что
изменение физических свойств липидов, особенно
ИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
93
пограничных, оказывает вторичное возцр^дд^ . иСС^?ТОоящее время эти гипотезы не пользуются
на мембранные белки, изменяя их конформацию общим признанием. На основании недавно полу-
и, следовательно, активность. Эти изменения сни-
жают или полностью ингибируют высвобождение
нейротрансмиттеров, или же так увеличивают объ-
ем и толщину липидного слоя, что ионные каналы
теряют способность эффективно функциониро-
вать. Согласно этим концепциям, считается, что
главной мишенью действия анестетиков являются
фосфолипиды; изменение их физико-химических
свойств вторично влияет на ферменты, рецепторы и
ионные каналы. Тем не менее трудно понять, каким
образом изменение физических свойств мембран-
ных фосфолипидов может вызвать состояние ане-
стезии, так как применяемые в клинической прак-
тике концентрации ингаляционных анестетиков
практически не влияют на изолированные липид-
ные бислои. Существенное воздействие на липид-
ные мембраны оказывают значительно более высо-
кие концентрации ингаляционных анестетиков.
Была выдвинута и другая подобная гипотеза
с предположением, что ингаляционные анестетики
действуют на фосфолипиды инертно, а влияние их
заключается в физической модификации соотно-
шения между давлением и объемом в мембране ней-
рона. Теория основана на феномене «инверсии ане-
стезии давлением»; во многих экспериментах, вы-
полненных на светящихся бактериях, головастиках
и мышах, было показано, что анестезия может быть
модифицирована или устранена при повышении
давления в окружающей среде. Подобный феномен
наблюдали и у людей при проведении анестезии
альтезином в барокамере под повышенным давле-
нием. Для объяснения этого феномена в 1970-е
годы была выдвинута гипотеза «критического дав-
ления», суть которой заключается в том, что анесте-
тики и давление действуют посредством одного и
того же механизма на одни и те же точки приложе-
ния. Согласно этой гипотезе, состояние анестезии
развивается тогда, когда увеличение объема непо-
лярных компонентов нейронной мембраны превы-
шает некий критический уровень; при восстановле-
нии исходного объема (путем изменений темпера-
туры или давления) наступает реверсия анестезии.
Были предложены модификации этой гипотезы,
например, «гипотеза среднего избыточного объ-
ема» и «гипотеза многоузлового расширения»,
в которых предполагалось, что анестезия и инвер-
сия анестезии опосредуются воздействием не на
один участок мембраны, а на различные молеку-
лярные структуры мембраны, и характеристики их
могут отличаться при использовании различных
анестетиков. Согласно этой гипотезе, каждый ане-
стетик вызывает уникальную картину депрессии и
активации ЦНС.
ченных экспериментальных данных можно ут-
верждать, что прессорная инверсия анестезии явля-
ется высокоспецифичной, селективной и может
быть опосредована воздействием на чувствитель-
ные к давлению глициновые каналы, которые иг-
рают важную роль в модуляции ингибирующего
тонуса в ЦНС. Следовательно, представляется ма-
ловероятным, что анестезия и давление действуют
посредством одного и того же механизма. Устране-
ние анестезии давлением может иметь опреде-
ленное отношение к неврологическому синдрому
(тремор, судороги, приступы кратковременного
сна), развивающемуся у многих эксперименталь-
ных животных и у человека при давлении окружаю-
щей среды выше 30 атмосфер.
БЕЛКОВЫЕ ТЕОРИИ АНЕСТЕЗИИ
До 1980 г. белки не рассматривались в качестве
мишеней действия ингаляционных анестетиков,
так как представлялось маловероятным, чтобы их
жесткая структура могла одинаково модифициро-
ваться под действием молекул различных ингаля-
ционных анестетиков. Тем не менее недавние ис-
следования показали, что ингаляционные анесте-
тики могут взаимодействовать с рецепторными
белками, вызывая конформационные изменения их
структуры, влияющие на функцию ионных каналов
и ферментов.
В последние годы для изучения воздействия ин-
галяционных анестетиков на рецепторы и фермен-
ты использовали свободные от липидов препараты
очищенных и растворенных белков. Многие раство-
ренные белки и ферментные комплексы резистент-
ны к клиническим концентрациям ингаляционных
анестетиков; к ним относятся гликолитические
ферменты, глутаминовые рецепторы, большинство
потенциал-зависимых ионных каналов и систем ак-
тивного транспорта. Фосфатидил-инозитоловая
система, циклические нуклеотиды и регуляторные
G-белки также относительно устойчивы к дейст-
вию ингаляционных анестетиков. Некоторые бел-
ки (например, гемоглобин, альбумин) связывают
ингаляционные анестетики и претерпевают обра-
тимые конформационные изменения без како-
го-либо нарушения физиологических функций.
Вместе с тем функциональные свойства некото-
рых белков под влиянием клинически значимых
концентраций ингаляционных анестетиков под-
вергаются селективной модификации. Так, в виде
свободного от липидов растворенного соединения
была приготовлена люцифераза (фермент, присут-
ствующий в клетках некоторых бактерий и жуков-
94
Глава 5
светляков); проведенные исследования
n)edlit.u<?sg;jjy5,
. Некоторые факторы, влияющие на
тесную корреляцию между концентрациями инга- МАК ингаляционных анестетиков
ляционного анестетика, которые подавляют люци-
феразу, и концентрациями, которые индуцируют
общую анестезию. Связывание и подавлен не актив-
ности люциферазы согласуется также с феноменом
отсечения (т.е. повышения мощности анестетика в
ряду гомологичных спиртов по мере удлинения их
цепи до достижения определенного молекулярно-
го веса, после чего мощность остается постоянной
несмотря на дальнейшее увеличение длины цепи).
Феномен отсечения может быть обусловлен связы-
ванием спиртов неполярными участками ограни-
ченной емкости на внутренней или наружной сто-
роне мембраны, где они контактируют с волной фа-
зой.
Ингаляционные анестетики также модифици-
руют функциональные свойства других белков,
включая ГАМК, глициновые и никотиновые ре-
цепторы. Установлено, что ингаляционные ане-
стетики могут связываться определенными «кар-
манами» внутри ГАМК- и глициновых рецепторов.
Была предложена модель действия анестетика,
в которой связывающий карман формируется
вторым и третьим трансмембранными доменами
(ТМ2 и ТМЗ) субъединиц ГАМК и глицинового ре-
цепторов и располагается приблизительно на рас-
стоянии 3 нм от наружной поверхности мембраны
(т. е. на одной трети глубины мембраны). В этом
кармане расположены две аминокислоты (серин
19’ и аланин 40 ), которые находятся рядом друг
с другом, ио принадлежат к ТМ2 и ТМЗ соответст-
венно. Связывающий карман обладает фиксиро-
ванным молекулярным объемом, что может объяс-
нить существование феномена отсечения. В рабо-
тах с мутантными рецепторами была подтверждена
роль этих двух аминокислотных остатков в модуля-
ции эффектов ингаляционных анестетиков.
В некоторых клинических исследованиях были
сделаны попытки определить возможную роль
рецепторов или белков ионных каналов в дейст-
вии ингаляционных анестетиков. В этих исследо-
ваниях изучали воздействие агонистов или анта-
гонистов различных рецепторов на мощность
ингаляционных анестетиков. Влияние оценивали
по тому, насколько изменялись значения МАК от-
дельных анестетиков. Выявленные изменения
МАК ингаляционных анестетиков позволяют ут-
верждать, что рецепторы или белки ионных кана-
лов могут играть роль в возникновении анестезии.
Например, такие агонисты а2-адренорецепторов,
как клонидин, азепексол и дексмедетомидин,
уменьшают МАК большинства ингаляционных
анестетиков (табл. 5.1) и могут использоваться для
Фактор L Воздействие на МАК
Физиологические и метаболические факторы
Возраст
Дети т
Пожилые 4-
Температура тела
Гипертермия (>40’С) ?
Гипотермия (<30'С) 44
Артериальная гипотония 4-
Функция щитовидной железы
Гипертиреоз ?
Гипотиреоз Ji
Лекарственные препараты
Катехоламины
Увеличение активности
Снижение активности 4-
аг-адреномиметики ±_
Седативные препараты и транквилизаторы и
Опиоиды
Острое опьянение 4
Хроническое потребление Т
Амфетамины
Острое опьянение TTt
Хроническое потребление 4
Этанол
Острое опьянение 4
Хроническое потребление Т
[ Препараты лития 4 1
4 или Т — изменение от 0 до 30%; 44 ii.ui ТТ — изменение
от 30 до 60%; ТТТ — увеличение > 60%
снижения дозы этих анестетиков. Эти эффекты
можно устранить или предотвратить введением ан-
тагонистов аг-адренорецепторов (например, тола-
залином, йохимбином). Результаты исследований
позволяют утверждать, что аг-адренорецепторы иг-
рают важную роль в индукции анестезии.
Вышеупомянутые исследования позволяют
предположить, что ингаляционные анестетики дей-
ствуют на один или несколько белков в мембране
нейронов, вызывая конформационные изменения
их структуры. Клинически значимые концентра-
ИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
95
ции ингаляционных анестетиков воздейс|ПЁ$®€11Й
рецепторы, белки или воротные лиганды ионных
каналов в ЦНС, что и приводит к постсинаптиче-
скому торможению активности нейронов.
Тем не менее надо отчетливо понимать, что та-
кая интерпретация не полностью согласуется с дру-
гими экспериментальными и клиническими дан-
ными. Большинство препаратов, действующих на
рецепторы или ферменты, относительно мощны,
специфичны, стереоселективны и могут конку-
рентно или неконкурентно подавляться другими
лекарственными препаратами; ингаляционные
анестетики не обладают ни одним из перечислен-
ных свойств. Кроме того, очень трудно понять, на-
пример, каким образом инертный газ ксенон или
даже относительно инертная закись азота вызывает
анестезию, взаимодействуя с белками или рецеп-
торными системами.
АНАТОМИЧЕСКИЕ МИШЕНИ
Анализ вызванных во время анестезии корко-
вых потенциалов показывает, что ингаляционные
анестетики могут подавлять возбудимость нейро-
нов и проведение по ним импульсов на многих
уровнях между периферической нервной системой
и корой головного мозга. В основном изменяется
проведение по афферентным путям, хотя также
оказывается влияние на ретикулярную активирую-
щую систему, базальные ганглии, мозжечок, цен-
тры продолговатого мозга и двигательные пути
спинного мозга. Неврологические проявления
анестезии являются результатом множественного
действия на синапсы в различных участках ЦНС
в зависимости от свойств используемого средства;
маловероятно, чтобы эффект анестетиков зависел
целиком и полностью исключительно от их угне-
тающего действия на афферентные пути. Получен
ряд данных о том, что клетки коры головного мозга
более чувствительны к действию анестетиков, чем
клетки активирующей ретикулярной формации,
несмотря на присутствие многочисленных синап-
сов в последней. Моносинаптическая передача
может быть более чувствительна к тормозящему
действию ингаляционных анестетиков.
МОЩНОСТЬ
Ингаляционные анестетики значительно разли-
чаются между собой по мощности, т.е. по той альве-
олярной концентрации, в которой они способны
вызвать состояние анестезии. Закись азота — отно-
сительно слабый анестетик, который даже в кон-
центрации 80% (в смеси с 20% кислородом) может
не в состоянии обеспечить адекватную
анестезию. Мощность других препаратов выше: для
поддержания анестезии вполне достаточно 15%
циклопропана, 5-10% этилового эфира или дес-
флурана, 1-4% галотана, энфлурана, изофлурана и
севофлурана. Для оценки мощности ингаляцион-
ных анестетиков используют минимальную альве-
олярную концентрацию (МАК). Одна МАК соот-
ветствует такой минимальной альвеолярной кон-
центрации ингаляционного анестетика, при
которой у 50% больных отсутствует двигательная
реакция на разрез кожи. Отметим, что под альвео-
лярной концентрацией понимают концентрацию
в конечной порции выдыхаемой дыхательной сме-
си, а измерения проводят в стационарном состоя-
нии и при атмосферном давлении. Мощность инга-
ляционных анестетиков в основном зависит от их
растворимости в жирах. МАК принято выражать
в объемных процентах (%). Если по оси абсцисс
отложить МАК различных анестетиков, а по оси ор-
динат — растворимость в жирах (выраженную
коэффициентами распределения масло/газ при
температуре 37°С), то получится линейная зависи-
мость (рис. 5.1).
Зависимость между мощностью анестетика и его
жирорастворимостью была независимо друг от дру-
га установлена Мейером и Овертоном в 1899-1901 гг.:
они показали, что мощность многих анестетиков
и гипнотиков зависит от их растворимости в олив-
ковом масле (по отношению к их растворимости
в воде). Оливковое масло представляет собой смесь
многих природных жиров; в настоящее время из-
вестно, что растворимость ингаляционных анесте-
тиков в очищенных растворителях с большей по-
лярностью (например, октанол, лецитин) лучше
коррелирует с их мощностью и может дать более
точное представление о молекулярных мишенях
воздействия этих препаратов. Кроме того, эти ре-
зультаты позволяют предположить, что идеальные
анестетики являются амфипатическими соедине-
ниями и обладают как гидрофобными (неполярными),
так и гидрофильными (полярными) свойствами.
На МАК ингаляционных анестетиков влияют
различные физиологические и фармакологические
факторы (табл. 5.1). МАК наиболее высока у детей
младше 3 лет, после чего постепенно снижается
(рис. 5.2). При использовании одновременно двух
ингаляционных анестетиков их дозы, выраженные
в МАК, можно складывать (рис. 5.3). Смесь 0,5 МАК
закиси азота (52%) и 0,5 МАК галотана (0,37%)
оказывает такой же эффект, как 1,0 МАК любого
из этих средств или 1,0 МАК любого другого ин-
галяционного анестетика. Аддитивность инга-
ляционных анестетиков была использована для
определения МАК закиси азота (104%). Феномен
96
Глава 5
Рис. 5.2. Влияние возраста на МАК некоторых ингаля-
ционных анестетиков; во всех случаях МАК с возрастом
уменьшается
аддитивности позволяет с достаточным основа-
нием утверждать, что все ингаляционные анесте-
тики обладают одинаковым механизмом действия.
На практике можно наблюдать небольшие отклоне-
ния от точной аддитивности (например, при комби-
нации закиси азота и изофлурана). Обычно такие
отклонения объясняют индивидуальными особен-
ностями больного.
Помимо способности предотвращать двигатель-
ную реакцию в ответ на различные стимулы (разрез
кожи, интубацию трахеи) мощность анестетика
можно оценить и по другим параметрам. Например,
определяют МАК, при которой 50% больных пере-
стают выполнять словесные инструкции (МАК-
бодрствования) или теряют память на происходя-
щие события (МАКамнезии); эти концентрации
меньше 1 МАК.
СКОРОСТЬ НАСТУПЛЕНИЯ
АНЕСТЕЗИИ
Общая анестезия наступает, когда концентра-
ция ингаляционного анестетика в ЦНС (выражен-
ная его парциальным давлением, или напряжени-
ем) становится достаточной для того, чтобы вы-
звать выключение сознания. Поглощение и
выведение ингаляционных анестетиков можно
представить в виде определенной последователь-
ности экспоненциальных процессов. Во время ин-
галяционной индукции анестезии устанавливается
Рис. 5.3. Графическое отображение альвеолярных кон-
центраций галотана (ось X) и закиси азота (ось У). Отме-
чены МАК галотана (0,75%) и МАК закиси азота (104%);
эти точки соединены прямой. Каждая точка на прямой
соответствует эквиэффективной комбинации двух ане-
стетиков (в сумме составляющей 1 МАК). Следователь-
но, мощности отдельных ингаляционных анестетиков
аддитивны (если выражаются в единицах МАК)
определенная последовательность диффузионных
градиентов, возникающих:
1. между концентрацией анестетика во вдыхае-
мой смеси (Fi) и его альвеолярной концентрацией
(парциальным давлением, напряжением, FA);
2. между напряжением анестетика в альвеолах и
напряжением его в крови легочных капилляров;
3. между напряжением анестетика в крови ле-
гочных капилляров и напряжением в крови сосудов
головного мозга;
4. между напряжением анестетика в крови сосудов
головного мозга и его напряжением в веществе ЦНС.
На практике скорость наступления анестезии
в основном зависит от того, как быстро Fi возраста-
ет и становится такой же, как FA. Эта зависимость
отражена на рис. 5.4. Быстрое нарастание альвео-
лярной концентрации приводит к возникновению
выраженного диффузионного градиента между
альвеолами и кровью легочных капилляров; следо-
вательно, напряжение анестетика в крови легочных
капилляров тоже нарастает быстро, генерируя все
последующие диффузионные градиенты. По дости-
жении равновесия напряжение анестетика в альве-
олах становится приблизительно таким же, как на-
пряжение в головном мозге.
97
ИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
Рис. 5.4. Зависимость
между и родолжитель-
ностьюанестезии и ско-
ростью увеличения со-
отношения FyF,.
При использовании
анестетиков с низкой
растворимостью в кро-
ви F, нарастает быстрее
После отключения иодачи анестетика во время
пробуждения эти изменения повторяются в обрат-
ном порядке. Fi падает до нуля, и возникает после-
довательность диффузионных градиентов между
парциальным давлением анестетика в ИНС и вы-
дыхаемой смесью. Быстрота пробуждения в основ-
ном зависит от скорости, с которой снижается Fa,
создавая выраженный диффузионный градиент ме-
жду кровью легочных капилляров и альвеолами.
Следовательно, факторы, которые влияют на ско-
рость наступления ингаляционной анестезии, в той
же степени (но в противоположном направлении)
влияют и на пробуждение.
КОЭФФИЦИЕНТ РАСПРЕДЕЛЕНИЯ
КРОВЬ/ГАЗ
Мощность ингаляционных анестетиков пря-
мо пропорциональна их жирорастворимости;
иными словами, МАК анестетика прямо пропор-
циональна коэффициенту распределения мас-
ло/газ (рис. 5.1). Напротив, скорость наступления
анестезии определяется другим физическим свой-
ством, а именно растворимостью в крови. Раствори-
мость в крови выражается коэффициентом распре-
деления кровь/газ при температуре 37’ С. Коэффи-
циент распределения кровь/ газ представляет
собой отношение концентраций анестетика в крови
и газе при температуре 37°С при условии, когда эти
две фазы имеют одинаковый объем и находятся в
стационарном состоянии (т. е. парциальное давле-
ние анестетика в них одинаково). Например, для се-
вофлурана коэффицент распределения кровь/газ
при 37*С составляет 0,65; это значит, что 1 мл крови
содержит 0,65 от того количества севофлурана, ко-
торое находится в 1 мл альвеолярного газа, несмот-
ря на одинаковое парциальное давление анестетика
в обеих фазах. Коэффициент распределения
кровь/газ колеблется от 0.42 (десфлуран) до 12
(этиловый эфир и метоксифлуран); он численно
равен коэффициенту растворимости Оствальда (X)
в крови при температуре 37° С.
Ингаляционные анестетики с низким коэффи-
циентом распределения кровь/газ плохо растворя-
ются в крови; во время индукции анестезии их Fa
быстро возрастает, достигая F( (рис. 5.4), а диффу-
зия в легочные капилляры обеспечивает резкий
подъем парциального давления в крови. Препарат
быстро диффундирует в ЦНС. Такая кинетика
обеспечивает высокую скорость наступления ане-
стезии.
Напротив, анестетики с высоким коэффициен-
том распределения кровь/газ хорошо растворяют-
ся в крови. Хотя они быстро удаляются из альвеол в
|>езультатс растворения в крови легочных капилля-
|юв, их парциальное давление в альвеолах и сосудах
остается относительно низким (рис. 5.4). Так как
диффузионный градиент между кровью мозговых
сосудов и тканями ЦНС остается сравнительно низ-
ким, анестезия наступает медлен но. Колебания соста-
ва крови (например, изменения конценграции сво-
бодных жирных кислот, альбумина и гемоглобина)
могут повлиять на коэффициент распределе-
ния кровь/газ. Все ингаляционные анестетики, при-
меняющиеся в настоящее время в Великобритании и
России, имеют низкий коэффициент распределения
98
Глава 5
кровь/газ, плохо растворяются в крови 1^0йхШтрационный ЭФФЕКТ
индуцируют анестезию.
На скорость индукции анестезии могут влиять
и другие факторы. Например, изофлуран и десфлу-
ран обладают едким эфирным запахом, что вызывает
раздражение верхних дыхательных путей и может за-
медлить индукцию анестезии. При использовании
галотана значительное количество препарата (около
25%) претерпевает биотрансформацию; образовав-
шиеся метаболиты, обладающие седативными свой-
ствами (например, ионы брома), могут замедлить
пробуждение после длительной анестезии. По срав-
нению с галотаном, пробуждение после севофлура-
новой анестезии происходит намного быстрее.
ЛЕГОЧНАЯ ВЕНТИЛЯЦИЯ
Гипервентиляция ускоряет прирост Ед ингаляци-
онного анестетика, что повышает диффузионный
градиент между кровью мозговых сосудов и веще-
ством ЦНС, ускоряя наступление анестезии. Напро-
тив, снижение легочной вентиляции уменьшает
прирост Fa анестетика, замедляя наступление ане-
стезии. Влияние легочной вентиляции на скорость
увеличения Fa/Fi особенно наглядно проявляется
при индукции анестезии препаратами, хорошо рас-
творимыми в крови (т. е. с высоким коэффициен-
том распределения кровь/газ), потому что они по-
глощаются кровью в значительной степени; в этой
ситуации удвоение альвеолярной вентиляции при-
водит к удвоению поглощения анестетика кровью.
Функциональная остаточная емкость (ФОЕ)
легких также влияет на FA ингаляционного анесте-
тика. ФОЕ представляет собой своеобразный бу-
фер между F[ и Fa. Уменьшение ФОЕ повышает
скорость возрастания FA; наоборот, увеличение
ФОЕ снижает скорость возрастания FA. Наруше-
ние вентиляционно-перфузионных отношений
тоже влияет на FA и поглощение анестетика кровью
в конце выдоха (эффект мертвого пространства),
либо из-за уменьшения переноса анестетика в ка-
пилляры (эффект шунтирования).
КОНЦЕНТРАЦИЯ АНЕСТЕТИКА ВО
ВДЫХАЕМОЙ СМЕСИ (F,)
У величение Ft повышает альвеолярную концен-
трацию и ускоряет индукцию анестезии. Для уве-
личения скорости индукции ингаляционный ане-
стетик используют в более высокой концентрации,
чем при поддержании анестезии. Этот феномен
особенно ярко проявляется при ингаляционной ин-
дукции анестезии, проводимой по методике одного
вдоха: после максимального выдоха больной вды-
хает смесь, содержащую 7% севофлурана (либо 4%
галотана), и задерживает дыхание насколько воз-
можно, вслед за чем начинает нормально дышать.
Сознание обычно выключается через 1-2 мин.
Концентрационный эффект возникает только
при использовании закиси азота — единственного
современного ингаляционного анестетика, исполь-
зуемого в достаточно высокой концентрации. Увели-
чение концентрации закиси азота во вдыхаемой
смеси ускоряет скорость увеличения ее концентра-
ции в крови. У взрослых при ингаляции смеси, со-
держащей 60% закиси азота, поглощение анестети-
ка в первые несколько минут превышает 1 л/мин.
ЭФФЕКТ ВТОРОГО ГАЗА
Если два ингаляционных анестетика вводят од-
новременно, то поглощение больших объемов од-
ного анестетика может привести к увеличению
альвеолярной концентрации другого, что ускоряет
индукцию анестезии. Например, сочетание гало-
тана с закисью азота ускоряет прирост FA галотана.
Концентрационный эффект и эффект второго
газа могут немного ускорить индукцию анестезии в
первые несколько минут ингаляции.
СЕРДЕЧНЫЙ ВЫБРОС
Изменения сердечного выброса влияют на ско-
рость прохождения крови по легочным капиллярам
во время индукции анестезии. Хотя низкий сердеч-
ный выброс снижает поглощение анестетика, он ус-
коряет темп увеличения FA и, соответственно, ско-
рость наступления анестезии.
Этот эффект играет значительную роль только
при использовании анестетиков с высоким коэф-
фициентом распределения кровь/газ. По достиже-
нии полного равновесия (когда напряжение анесте-
тика в альвеолах и в крови легочных капилляров
становится одинаковым) диффузия прекращается
и поглощения анестетика кровью не происходит.
На практике полное равновесие маловероятно, так
как большинство анестетиков в той или иной степе-
ни выводится из организма путем метаболизма или
экскреции. Следовательно, ингаляционную анесте-
зию можно рассматривать как состояние частично-
го равновесия.
ДЫХАТЕЛЬНЫЙ КОНТУР
Поглощение анестетика различными компо-
нентами дыхательного контура может изменять
его Fi и Fa. Все галогензамещенные анестетики час-
тично растворяются в резине, а некоторые из них
вступают в реакции с натронной известью; следова-
тельно, концентрация анестетика на выходе из испа-
рителя и его Fi может быть различной. В прошлом
высвобождение из резиновых шлангов анестетика,
поглощенного во время предыдущей анестезии,
могло вызвать осложнения. Появление одноразо-
вых пластиковых шлангов устранило эту проблему.
ИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
99
medht.ucoz.ru ,
состав реверсивного дыхательного контура стетиками, хотя было проведено несколько эпи-
входят поглотители углекислого газа, такие как на-
тронная известь и известь с добавкой гидроксида
бария. Некоторые галогензамещенные анестетики
могут поглощаться этими поглотителями или всту-
пать в реакцию с ними, особенно если поглотитель
сухой и температура его высока. В этой ситуации
применение ингаляционных анестетиков, содержа-
щих CHF2-rpynny (например, энфлурана, изофлу-
рана и десфлурана) может привести к образованию
СО (угарного газа) и превращению гемоглобина в
карбоксигемоглобин. Эта реакция не происходит,
если в контуре установлен увлажнитель или если
поглотитель не пересушен, т. е. содержит достаточ-
ное количество воды (6-19%).
Дыхательный контур может опосредовать влия-
ние альвеолярной вентиляции на поглощение инга-
ляционных анестетиков. Если контур нереверсив-
ный, то поглощение анестетика зависит преимуще-
ственно от альвеолярной вентиляции, поскольку F|
задается значением, выставленным на испарителе.
При использовании реверсивного контура ситуа-
ция меняется: Fj зависит не только от значения,
заданного на испарителе (оно определяет исключи-
тельно концентрацию в потоке свежего газа), но
и от концентрации в выдыхаемой смеси. Если по-
глощение анестетика снижается (например, вслед-
ствие гиповентиляции), то выдыхаемая концентра-
ция увеличивается, что в сочетании с неизменной
концентрацией в потоке свежего газа приводит
к повышению Fi, увеличению альвеолярно-арте-
риального градиента и поглощения анестетика.
Этот эффект компенсирует уменьшение альвео-
лярной вентиляции. Изменения сердечного выбро-
са влияют на Fa анестетика сильнее, чем альвеоляр-
ная вентиляция (МОД), потому что поглощение
анестетика кровью оказывает большее воздействие
на остаточную концентрацию анестетика в дыха-
тельном контуре.
ВЛИЯНИЕ ИНГАЛЯЦИОННЫХ
АНЕСТЕТИКОВ НА
МЕДИЦИНСКИЙ ПЕРСОНАЛ
Медицинский персонал подвергается длитель-
ному влиянию следовых концентраций ингаляци-
онных анестетиков, присутствующих в воздухе
операционных. В крови, моче и воздухе, выдыхаемом
сотрудниками, в течение нескольких дней после ра-
боты в операционной можно обнаружить в неболь-
шой концентрации ингаляционные анестетики и их
метаболиты. Точно неизвестно, насколько опасен
профессиональный контакт с ингаляционными ане-
демиологических исследований, в которых пред-
принимались попытки оценить риск такого кон-
такта у женщин-анестезиологов. По результатам од-
ного исследования было высказано
предположение, что у женщин-анестезиологов
может быть незначительно повышен риск выки-
дыша; доказательств в пользу токсического дейст-
вия ингаляционных анестетиков получено не было.
В ходе проспективно- го исследования, проводимого
в течение десяти лет (1977-1986 гг.), у жен-
щин-врачей всех специальностей в возрасте < 40 лет
не удалось обнаружить связи между частотой выки-
дышей и такими показателями, как медицинская
специальность, проведенное в операционной время,
наличие системы эвакуации медицинских газов в
операционной. Нет убедительных данных в пользу
того, что проведение общей анестезии беременной
женщиной-анестезиологом увеличивает риск вро-
жденных аномалий у ее ребенка.
В других исследованиях у анестезиологов, дол-
гое время подвергавшихся воздействию следовых
концентраций галогензамещенных анестетиков,
были обнаружены нарушения функциональных пе-
ченочных проб; в некоторых случаях выявляли ге-
патит, который разрешался после прекращения
контакта с ингаляционными анестетиками. Выяв-
лено подострое и хроническое токсическое воздей-
ствие закиси азота (см. ниже).
Хотя вред, наносимый следовыми количествами
ингаляционных анестетиков, представляется чрез-
вычайно малым, многие медицинские сотрудники
могут подвергаться длительному воздействию этих
препаратов долгие годы. В развитых странах в по-
следние годы начали широко применять мониторы,
позволяющие определять содержания анестети-
ков в воздухе операционных, а также системы эва-
куации медицинских газов. Эти системы, наряду
с методикой низкопоточной анестезии, сводят
к минимуму риск осложнений, обусловленный
длительным профессиональным контактом с инга-
ляционными анестетиками.
МЕТАБОЛИЗМ ИНГАЛЯЦИОННЫХ
АНЕСТЕТИКОВ
С 1960-х гг. известно, что большинство ингаля-
ционных анестетиков в той или иной степени пре-
терпевает биотрансформацию. Метаболизм проис-
ходит главным образом в печени, а его интенсив-
ность зависит от химического строения анестетика.
Все современные галогензамещенные анестетики
содержат больше чем одну углеродно-галогеновую
100
Глава 5
Wdlit.UGQZTU,
связь (C-F, С-С1 или С-Вг), которые по
метаболизму в разной степени. Связь между угле-
родом и фтором отличается высокой стабильно-
стью, следовательно, трифторметильные группы
(Ср3) в галотане, изофлуране, десфлуране и се-
вофлуране чрезвычайно устойчивы и практически
не подвергаются метаболизму. Напротив, связи
С-С1 и С-Вг в метоксифлуране и галотане менее
стабильны и подвергаются биотрансформации.
Если принять во внимание это рассуждение,
то становится ясно, почему степень биотрансфор-
мации уменьшается в следующей последователь-
ности: метоксифлуран (65%) > галотан (20%) > сево-
флуран (3%) > энфлуран (2%) > изофлуран (0,2%) >
десфлуран (0,02%). Углеродно-галогеновые связи
разрушаются под действием окислительных фер-
ментов, в результате этих реакций высвобожда-
ются реакционно-способные метаболиты и ионы
галогенов (например, F, Вг или СГ). Метокси-
флуран, энфлуран и севофлуран высвобождают ио-
ны фтора. В эксперименте у животных выявлено,
что концентрация ионов фтора в плазме свыше
40 мкмоль/л способна стать причиной обратимой
нефропатии. Четко установлено, что в клиниче-
ских условиях повышение концентрации фторидов
в плазме, обусловленное применением севофлурана,
ни в одном случае не привело к дисфункции почек.
Ингаляционные анестетики претерпевают био-
трансформацию под действием ферментов пече-
ночного комплекса цитохрома Р-450. Одна из изо-
форм этой ферментной системы (CYP 2Е1) отвеча-
ет за дефторирование наиболее распространенных
ингаляционных анестетиков, включая энфлуран,
изофлуран, десфлуран и севофлуран. CYP 2Е1 при-
сутствует также в клетках эпителия почечных ка-
нальцев, где она может принимать участие в дефто-
рировании некоторых анестетиков. Индукция фер-
ментов этанолом или изониазидом (но не
барбитуратами или фенитоином) повышает ско-
рость дефторирования; индукторы активности
CYP 2Е1 также включают голодание, ожирение и
сахарный диабет. Метаболизм галотана также про-
исходит при участии этой ферментной системы,
хотя его дефторирование происходит главным об-
разом в условиях гипоксии.
Токсичность ингаляционных анестетиков обу-
словлена их метаболитами. Препараты, которые пре-
терпевают значительную биотрансформацию под
воздействием ферментных систем печени (напри-
мер, галотан и метоксифлуран) оказывают выра-
женное токсическое действие на печень и почки.
Эти проблемы были сведены к минимуму с появлени-
ем новых ингаляционных анестетиков, метаболизм
которых незначителен и не претерпевает особых
й под воздействием большинства индук-
торов ферментативной активности. Эти анестетики
являются относительно мощными и обладают низ-
кой растворимостью в крови и тканях, что ограничи-
вает их способность к накоплению в организме. С по-
явлением новых фторзамещенных ингаляционных
анестетиков риск побочных эффектов, обусловлен-
ных их метаболизмом, существенно снизился.
ОТДЕЛЬНЫЕ ПРЕПАРАТЫ
ЗАКИСЬ АЗОТА
В 1772 г. Джозеф Пристли синтезировал закись
азота и описал клинические проявления ее воздей-
ствия на организм. Двадцать лет спустя Хэмфри
Дэви предположил, что благодаря способности уст-
ранять боль закись азота целесообразно использо-
вать во время хирургических операций. К сожале-
нию, эти свойства закиси азота как анестетика и
анальгетика игнорировались долгие годы. В1844 г. Го-
раций Уэллс, американский стоматолог, впервые
использовал закись азота при экстракции зуба.
Однако первая клиническая демонстрация прошла
неудачно, и новый метод скомпрометировал себя.
Низкая мощность закиси азота и необходимость до-
полнительной оксигенации были осознаны только
в 1870-е годы. С тех пор ее используют как адъю-
вант и газ-носитель для более мощных анестетиков.
Закись азота также применяют для анальгезии, осо-
бенно в акушерстве в виде смеси равных объемов
закиси азота и кислорода (Энтонокс).
Общие сведения
При комнатной температуре и атмосферном
давлении закись азота представляет собой газ без
цвета и запаха, кипящий при температуре - 88° С,
с удельной плотностью 1,54 (тяжелее воздуха).
Закись азота не воспламеняется, но может поддер-
живать горение. Закись азота синтезируют нагрева-
нием нитрата аммония до температуры 250° С; если
не контролировать температуру достаточно точно,
то образуются более окисленные оксиды азота.
Загрязнение медицинской закиси азота этими
примесями может вызвать опасные для жизни ос-
ложнения. После синтеза сжатый газ охлаждают
до - 40°С и хранят в сжиженном состоянии в сталь-
ных баллонах.
Растворимость закиси азота в крови и жире
мала: коэффициенты распределения кровь/газ и
масло/газ составляют 0,46 и 1,4 соответственно.
Индукция анестезии и пробуждение
Индукция анестезии и пробуждение при ис-
пользовании закиси азота наступают очень быстро:
альвеолярная концентрация быстро уравновешива-
ется с концентрацией во вдыхаемой смеси (рис. 5.4).
ИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
101
„ , medlit.ucoz.ru
Во время индукции могут возникать возбуждение и 65: 35%, т.
эйфория. В XIX веке анестетик называли «веселя-
щим газом» (так как вдыхавшие его люди станови-
лись веселыми и громогласными). В отсутствие пре-
медикации анестезия чистой закисью азота в кисло-
роде вызывает странные причудливые видения.
Несмотря на низкий коэффициент распределе-
ния кровь/газ, растворимость закиси азота в крови
в 15-20 раз больше, чем у кислорода и азота. Закись
азота проникает через биологические мембраны пу-
тем диффузии в 15 раз быстрее кислорода и в 25 раз
быстрее азота. Вследствие этого закись азота диф-
фундирует из альвеол в легочные капилляры быст-
рее, чем кислород, и поэтому во время индукции
альвеолярная концентрация кислорода может вре-
менно возрасти. Противоположная картина разви-
вается после отключения анестетика, когда быст-
рая диффузия закиси азота из крови легочных ка-
пилляров в альвеолы может вызывать преходящую
гипоксию («диффузионную гипоксию»). Закись
азота диффундирует через ткани быстрее других
анестетиков: у взрослых пациентов скорость ее чрес-
кожной диффузии составляет около 10 мл/мин.
Так как скорость диффузии закиси азота через
мембраны значительно выше, чем у кислорода и
азота, то она поступает в замкнутые воздухосодер-
жащие пространства быстрее, чем кислород или
азот могут их покинуть. К таким полостям относят-
ся манжетки интубационных трубок, просвет ки-
шечника, пневмоторакс, воздушные эмболы; закись
азота вызывает увеличение их объема пропорцио-
нально своей альвеолярной концентрации. Во вре-
мя длительных операций на брюшной полости
вздутие кишок может отрицательно повлиять на
ход операции и затруднить ушивание операционной
раны. Дыхательная смесь с 75% содержанием заки-
си азота удваивает объем пневмоторакса за 10 мин,
а утраивает—за 30-45 мин. Закись азота может вы-
звать увеличение давления внутри жестких, нерас-
тягивающихся полостей (например, в барабанной
полости, в придаточных пазухах носа, в глазу).
Мощность
Закись азота представляет собой хороший
анальгетик, но слабый анестетик. Жирораствори-
мость закиси азота невысока: ее МАК при атмо-
сферном давлении составляет 104%'. Низкая мощ-
ность ограничивает ее применение в качестве ане-
стетика. В концентрации выше 70% заксь азота не
следует применять из-за риска гипоксии. Смесь
закиси азота и кислорода (обычно в соотношении
. е. около 2:1) используют в качестве га-
за-носителя для более мощных ингаляционных
анестетиков или в сочетании с другими препарата-
ми (например, внутривенными анестетиками или
опиоидами).
Анальгезия
Закись азота является мощным анальгетиком.
В концентрации 20-50% она оказывает такой же
обезболивающий эффект, как стандартные разовые
дозы морфина или петидина. Смесь закиси азота и
кислорода, содержащая равные объемы этих газов
(Энтонокс) применяется в акушерстве для обезбо-
ливания родов. Закись азота используют также для
обезболивания малых хирургических вмеша-
тельств (например, при перевязке небольших ожо-
гов, поверхностных ран). Механизмы анальгетиче-
ского эффекта неизвестны; возможно, закись азота
влияет на опиатные рецепторы, поскольку анальге-
тический эффект частично снимается налоксоном.
Дыхание
В анестетических концентрациях закись азота
увеличивает ЧДД, что компенсирует снижение ды-
хательного объема, так что МОД и РаСО2 сущест-
венно не меняются. Как и другие анестетики, закись
азота подавляет вентиляторную реакцию на гипер-
капнию и гипоксемию. Закись азота уменьшает
ФОЕ и повышает частоту периоперационных ате-
лектазов. Закись азота ингибирует активность
мукоцилиарного эпителия и подвижность нейтро-
филов.
Кровообращение
Закись азота оказывает незначительное отрица-
тельное инотропное действие in vitro, но in vivo этот
эффект не наблюдается, потому что ему противо-
действует симпатикотонический эффект препара-
та. Более того, эта индуцированная симпатическая
стимуляция может уменьшать выраженность кар-
диодепрессивного действия других ингаляцион-
ных анестетиков. В концентрации 70% закись азота
не оказывает значимого влияния на гемодинамику
даже у тяжелых больных (например, при коронар-
ном шунтировании). ЧСС обычно не изменяется,
но ОПСС вследствие симпатической стимуляции
может немного возрасти. Экстрасистолы и эктопи-
ческие ритмы возникают редко. Нет никаких дан-
ных о том, что закись азота влияет на чувствитель-
ность к эндо- или экзогенным катехоламинам.
Соответственно, закись азота не противопоказана
даже при тяжелых заболеваниях сердца.
МАК закиси азота можно определить с помощью гипербарической методики, а также используя феномен аддитивности
доз двух ингаляционных анестетиков: мощность комбинации 0,5 МАК галотана и 0,5 МАК закиси азота равна мощности
1 МАК любого из этих анестетиков.
102
Глава 5
Рис. 5.5. Метаболические реакции, на которые влияет закись азота. Активная форма витамина В12 (коб[1]аламин)
является кофактором метионинсинтазы и активирует этот фермент. Закись азота окисляет ион кобальта в витамине
В12, переводя его из активной одновалентной формы в неактивную двухвалентную форму (коб[П]аламин). Угнетение
активности метионинсинтазы нарушает синтез метионина и тетрагидрофолиевой кислоты, что влияет на синтез ДНК
и белков
Закись азота увеличивает мозговой кровоток
и потенцирует вазодилатацию мозговых сосудов,
обусловленную галогензамещенными ингаляцион-
ными анестетиками. Соответственно, закись азота
нецелесообразно применять при риске внутриче-
репной гипертензии. Если такого риска нет, то этот
эффект выражен слабо и не имеет практической
значимости.
Метаболизм и токсическое действие
Закись азота практически не подвергается мета-
болизму, что отличает ее от многих других ингаля-
ционных анестетиков. Чрезвычайно малые количе-
ства закиси азота превращаются в азот в ходе окис-
ления витамина Bi2, а также под воздействием
бактериальной флоры кишечника.
Закись азота может нарушать синтез метионина,
дезокситимидина и ДНК. Закись азота окисляет
атом кобальта в молекуле витамина В12, превращая
одновалентную форму [кобаламин(1)] в двухва-
лентную форму (кобаламин(П)]. Так как витамин
В12 является кофактором метионинсинтазы, то
окисление В(2 ингибирует активность этого фер-
мента. Ингибирование активности метионинсинта-
зы нарушает синтез метионина и тетрагидрофо-
лиевой кислоты, что в свою очередь, препятствует
образованию дезокситимидина, тимидина и ДНК
(рис. 5.5). Кроме того, закись азота непосредствен-
но инактивирует метионинсинтазу, возможно,
вследствие образования свободных радикалов.
Контакт с закисью азота достаточно быстро на-
чинает влиять на метаболизм витамина В12 и актив-
ность метионинсинтазы: у крыс синтез тимидина
нарушается уже через 1 ч при дыхании смесью с со-
держанием закиси азота 50%. У человека контакт с
закисью азота в течение 2-24 ч может вызвать нару-
шение синтеза дезокситимидина и мегалобластную
трансформацию в костном мозге; тяжесть этих из-
менений прямо пропорциональна длительности
анестезии. Более длительный контакт с закисью
азота (например, в течение 4-6 сут) приводит к аг-
ранулоцитозу, хотя некоторые больные относи-
тельно устойчивы к действию анестетика. Концен-
трация фолиевой кислоты в крови возрастает, ме-
тионина — снижается. После прекращения контакта
с закисью азота костный мозг постепенно возвра-
щается в норму приблизительно в течение одной
недели. Выздоровление можно ускорить введением
лейковорина (синонимы: фолинат кальция, каль-
циевая соль 5-формил-тетрагидрофолиевой кисло-
ты), который является альтернативным источни-
ком тетрагидрофолиевой кислоты и в течение не-
скольких часов нормализует синтез тимидина.
Метионин менее эффективен даже в больших до-
зах. Хотя витамин Bi2 способствует выздоровле-
нию, его эффект развивается достаточно долго: для
образования новой молекулы метионинсинта-
зы требуется 2-3 сут. Перемежающееся или повтор-
ное воздействие закиси азота в течение короткого
ИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
103
Закись азота
Галотан
0 I
Н—-С—С'—F
8г F
medlit.ucoz.ru
угроза здоровью персонала невелика. Напротив,
длительный контакт с высокими концентрациями
закиси азота в помещениях, не оснащенных систе-
мами эвакуации (например, в стоматологических
кабинетах), может вызвать неврологические нару-
шения, напоминающие подострую комбинирован-
ную дегенерацию спинного мозга. Эти нарушения
отражают хронический дефицит витамина Bj2, вы-
званный закисью азота.
Энфлуран
F F Н
Н—С—О—С—С—-F
I I I
f f а
Изофлуран
F И F
Н—С—О—С—С— F
F а F
Десфлуран
Севофлуран
F Н F
Н—С-—О “С—С—F
F F F
Рис. 5.6. Структурные формулы ингаляционных ане-
стетиков. Хиральные атомы углерода отмечены звездоч-
кой
У животных закись азота проявляет умеренный
тератогенный эффект, если ее вводить несколько
дней в концентрации > 50%. Лейковорин предот-
вращает возникновение тератогенного эффекта,
что доказывает его связь с нарушением синтеза
ДНК. Хотя очень большое количество женщин
(больных и медицинских работников) подверга-
лось влиянию закиси азота на ранних сроках бере-
менности, не существует убедительных данных за
то, что этот препарат может оказать неблагоприят-
ное воздействие на плод. Тем не менее целесобраз-
но избегать контакта с закисью азота в первые 6 нед
беременности или же параллельно назначать лей-
коворин.
ГАЛОТАН
Галотан представляет собой фторзамещенный
углеводород, синтезированный Чарльзом Саклин-
гом в 1950-1955 гг. Фармакологические свойства
были исследованы Джеймсом Равентосом, а осо-
бенности галотановой анестезии у человека были
впервые описаны Майклом Джонстоном в 1958 г.
Галотан представлял собой выдающееся достиже-
ние своего времени и не менее двадцати лет оста-
вался ингаляционным анестетиком выбора. В Ве-
ликобритании его стали значительно реже исполь-
зовать у взрослых с 1985 г., главным образом из-за
очень редкого осложнения — «галотанового гепати-
промежутка времени может не дать достаточного
времени для восстановления активности фермента.
Закись азота оказывает незначительное воздей-
та», а также из-за появления новых фторзамещен-
ных ингаляционных анестетиков. Во многих стра-
нах мира галотан до сих пор является наиболее рас-
пространенным ингаляционным анестетиком
ствие на витамин BJ2 и метионинсинтазу или вооб-
ще его не оказывает при концентрации газа во вды-
хаемой смеси < 450 частиц на миллион независимо
от продолжительности контакта. Концентрация
закиси азота в воздухе операционных, не снабженных
системой эвакуации медицинских газов, составляет
200-400 частиц на миллион; у медицинских сотруд-
ников, работающих в таких операционных, концен-
трация метионина в сыворотке находится в преде-
лах нормы. Концентрация закиси азота в воздухе
операционных, снабженных системой эвакуации ме-
дицинских газов, составляет около 50 частей на мил-
лион. Следовательно, в условиях операционных
кроме того, он продолжает играть важную роль в
детской анестезиологии.
Общие сведения
Галотан (2-бром-2-хлор-1,1,1 -трифторэтан)
представляет собой бесцветную летучую жидкость
с температурой кипения 50,2°С. Хранят его в герме-
тичных флаконах темного стекла, потому что он
разрушается на свету. В качестве консерванта до-
бавляют тимол (0,01%), с тем чтобы предотвратить
высвобождение свободного брома. Смесь галотана
с кислородом или воздухом не воспламеняется и не
взрывоопасна. Коэффициент распределения
кровь/газ относительно низок (2,4); следовательно,
104
Глава 5
индукция анестезии и пробуждение прои
носительно быстро, а глубиной анестезии легко
управлять. Коэффициент распределения масло/газ
при температуре 37,0°С равен 224, МАК составляет
0,75%; это означает, что галотан хорошо растворя-
ется в липидах и является мощным анестетиком.
Для индукции анестезии используют концентра-
цию 2-4%, для поддержания — 0,5-1,5%. Галотан —
плохой анальгетик: в некоторых случаях он даже
оказывает некоторое «антианальгетическое» дей-
ствие, снижая болевой порог.
Молекула галотана обладает оптической актив-
ностью, так как содержит асимметричный атом уг-
лерода и может существовать в виде двух энантио-
меров с различной оптической активностью; на
практике галотан применяют в виде рацемической
смеси.
Дыхание
Галотан угнетает дыхание: ЧДД увеличивается,
в то время как дыхательный объем и МОД снижа-
ются. Может нарастать РаСО2. В низкой концен-
трации (< 1 МАК) галотан частично подавляет вен-
тиляторную реакцию на гиперкапнию и гипоксию
вследствие угнетающего действия на перифериче-
ские сосудистые хеморецепторы. В высокой концен-
трации (1-2 МАК) препарат вызывает более глубо-
кую депрессию дыхания в результате угнетения про-
водящих путей в стволе головного мозга. Сенсорная
и хирургическая стимуляция частично нивелирует
депрессию дыхания. Снижая холинергический то-
нус, галотан вызывает бронходилатацию. Галотан
подавляет индуцированную гистамином бронхо-
констрикцию и поэтому может быть полезен при
сопутствующей бронхиальной астме.
Галотан не раздражает дыхательные пути и не
увеличивает секрецию слюнных желез, а также же-
лез слизистой трахеи и бронхов. В концентрациях,
применяемых для индукции анестезии, подавляет
рефлексы гортани и глотки; влияние на продукцию
слизи и мукоцилиарный клиренс может быть раз-
личным.
Кровообращение
Галотан усиливает тонус блуждающего нерва,
угнетает синусовый узел, замедляет АВ-проведе-
ние. Он часто вызывает синусовую брадикардию и
АВ-узловые ритмы. Уменьшаются также сократи-
мость миокарда и сердечный выброс, хотя при дли-
тельной анестезии они нормализуются. Кроме того,
галотан снижает постнагрузку, уменьшает потреб-
ление кислорода миокардом и повышает устойчи-
вость миокарда к ишемии. Как следствие, галотан
вызывает депрессию сегмента ST реже, чем другие
фторзамещенные анестетики. Снижение АД обу-
словлено в основном угнетением сократимости
; оно прямо пропорционально концентра-
ции анестетика, поэтому галотан можно применять
для управляемой интраоперационной гипотонии.
Действует галотан и на периферическое крово-
обращение, хотя это влияние в различных сосуди-
стых бассейнах проявляется по-разному. Отмечают
небольшое снижение ОПСС (около 5% при 1 МАК).
Галотан повышает кровоток в коже и головном моз-
ге, снижает ЛСС и подавляет гипоксическую вазо-
констрикцию. Напротив, кровоток в кишечнике,
печени и почках уменьшается. Галотан практиче-
ски не оказывает прямого воздействия на коронар-
ный кровоток. Нарушается также ауторегуляция
кровообращения, поэтому мозговой кровоток
может стать зависимым от сердечного выброса.
Галотан изменяет реакцию мозговых сосудов на из-
менения РаСО2. Кроме того, галотан снижает кон-
центрацию дофамина, норадреналина и адреналина
в плазме.
Во время анестезии галотаном часто возникают
относительно доброкачественные аритмии (напри-
мер, брадикардия, АВ-узловые ритмы). Иногда раз-
вивается бигеминия или политопная желудочковая
экстрасистолия, которые могут трансформировать-
ся в желудочковую тахикардию и фибрилляцию
желудочков. Провоцирующие факторы аритмий
включают увеличение концентрации циркулирую-
щих катехоламинов (вследствие усиления секре-
ции эндогенного адреналина или при введении
Р-адреномиметиков), изменения кислотно-щелоч-
ного и электролитного баланса. В ходе галотановой
анестезии целесообразно полностью отказаться от
введения адреналина. Если по ходу операции все же
возникает необходимость в адреналине для полно-
ценного местного гемостаза, то его используют в
концентрации < 10 мкг/мл (> 1:100 000) и вводят не
более 100 мкг в течение десятиминутного интерва-
ла (эту дозу можно удвоить, если адреналин сочета-
ют с 0,5% лидокаином).
Аритмии во время галотановой анестезии часто
разрешаются после устранения провоцирующих
факторов без применения антиаритмических пре-
паратов. Для лечения аритмий, индуцированных
адреналином, иногда требуется использовать Р-ад-
реноблокаторы или лидокаин.
Мышцы
Галотан угнетает сократимость гладких мышц
(сосудов, ЖКТ, мочевого пузыря и матки), а также
скелетных мышц и миокарда. Эквипотенциальные
дозы галотана, энфлурана и изофлурана вызывают
одинаковую степень расслабления матки; тем не
менее низкие концентрации галотана (0,5 МАК)
иногда используют для выключения сознания при
кесаревом сечении.
ИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
105
г . medlit.ucoz.ru
I алотан потенцирует действие недеполяризую- Этот относительно распространенный тип дис-
щих миорелаксантов в зависимости от дозы, но в
меньшей степени, чем фторзамещенные эфиры.
Объяснения этому эффекту ингаляционных ане-
стетиков пока нет, хотя известно, что в его реализа-
ции участвуют как центральные, так и перифериче-
ские механизмы. Галогензамещенные анестетики
подавляют активность ЦНС, угнетают пресинапти-
ческое высвобождение ацетилхолина и могут вы-
звать десенситизацию постсинаптических холино-
рецепторов. При использовании галотана для адек-
ватной миорелаксации требуются меньшие дозы
миорелаксантов, а длительность их действия может
увеличиться. Элиминация галотана быстро устра-
няет потенцирование нервно-мышечной блокады.
Метаболизм
Приблизительно 20-25% галотана под воздей-
ствием CYP 2Е1 подвергается метаболизму до
трифторуксусной кислоты, хлора и брома. Эти ме-
таболиты медленно выделяются с мочой: после
длительной анестезии ионы брома можно обнару-
жить в организме спустя еще несколько недель, и их
концентрация может быть достаточно высокой,
чтобы вызвать значительное угнетение сознания в
послеоперационном периоде. Если в печени актив-
но протекают окислительные процессы, то дефто-
рирование галотана крайне незначительно или во-
обще не происходит, так что ионы фтора не образу-
ются. Напротив, в условиях гипоксии в печени
начинает преобладать восстановительный метабо-
лизм галотана, и при этом образуются ионы фтора и
другие восстановленные метаболиты (хлортрифтор-
этан и хлордифторэтилен), которые в конъюгиро-
ванной форме выделяются с мочой. Следовательно,
в зависимости от напряжения кислорода в гепато-
цитах метаболизм галотана может быть либо окис-
лительным, либо восстановительным.
Гепатотоксичность (галотановый гепатит)
Послеоперационная желтуха и смерть после
контакта с галотаном были впервые описаны вско-
ре после его появления в клинической практике
в 1958 г. Доказано, что галотан в чрезвычайно ред-
ких случаях способен вызывать тяжелый некроз пе-
чени. Это осложнение встречается настолько редко,
что его очень трудно дифференцировать от вирус-
ного гепатита или послеоперационной желтухи
другой этиологии.
Галотан может оказывать на печень воздействие
двоякого рода. Во-первых, анестетик может вызы-
вать легкое преходящее повреждение печени в пер-
вые трое суток после операции приблизительно
у 25% больных; отмечаются характерные изменения
печеночных проб, особенно аминотрансфераз, но прак-
тически отсутствуют клинические проявления.
функции печени, вероятно, вызывается умеренной
гипоксией, особенно в центральных отделах пече-
ночных долек, и обычно возникает на фоне восста-
новительного метаболизма галотана. Во-вторых,
в очень редких случаях галотан вызывает молние-
носный некроз печени, характеризующийся высо-
кой летальностью (50-80%); это поражение обычно
развивается в течение 5 сут после операции и сопро-
вождается резким повышением уровня ами-
нотрансфераз. Длительность воздействия не играет
решающей роли, и во многих случаях некроз печени
развивается после сравнительно короткой анесте-
зии. Частота галотанового некроза печени состав-
ляет в среднем 1:10 000 (1:7 000 -1:35 000). Повтор-
ный контакт с галотаном повышает риск некроза
печени и увеличивает обусловленную им леталь-
ность; риск наиболее высок, когда промежуток
между галотановыми анестезиями не превышает 28
сут.
Риск некроза печени выше при ожирении, а также
при воздействии индукторов печеночных фермен-
тов. Перед применением галотана анестезиолог
должен придерживаться следующих рекомендаций:
1. Собрать подробный анестезиологический
анамнез.
2. Не применять галотан, если после предшест-
вующего его использования прошло менее 3 мес.
3. Не применять галотан, если после предшест-
вующего его использования развилась желтуха или
лихорадка.
Следует подчеркнуть, что полностью исклю-
чить риск галотанового гепатита можно только от-
казавшись от применения галотана.
В последние годы были выяснены иммунологи-
ческие механизмы развития галотанового гепатита.
Один из окисленных метаболитов галотана, три-
фторацетилхлорид, ковалентно связывается с ли-
зиновыми остатками различных белков печени,
включая изоферменты цитохрома Р-450. У предрас-
положенных больных трифторацетилированные
белки запускают иммунный ответ с образованием
антител к гепатоцитам. Антитела к трифторацетил-
лизиновым остаткам можно обнаружить в сыворот-
ке 70% больных с галотановым гепатитом в течение
1-5 лет. По неясной причине лишь у небольшой
части популяции этот антиген запускает иммунный
ответ.
Галотан метаболизируется до трифторуксусной
кислоты под действием изофермента CYP 2Е1, ин-
гибитором которого является дисульфирам. Было
высказано предположение о том, что дисульфи-
рам может уменьшить образование этого метаболи-
та, тем самым снижая риск галотанового гепатита.
106
Глава 5
med I it
Другие фторзамещенные анестетики тоже образу-
ют в процессе биотрансформации трифторуксус-
ную кислоту, но не вызывают гепатита, вероятно, в
силу своего менее интенсивного метаболизма.
ЭНФЛУРАН
Энфлуран — это фторзамещенный этилметило-
вый эфир, синтезированный в 1963 г. Р. Терреллом.
В последние 25 лет его широко используют в Вели-
кобритании и других развитых странах.
Общие сведения
Энфлуран (2-хлор-1,1,2-трифторацетилди-
фторметиловый эфир) представляет собой струк-
турный изомер изофлурана, молекула его облада-
ет оптической активностью и является хиральной.
В практике применяется рацемическая смесь его
энантиомеров. Энфлуран представляет собой бес-
цветную летучую жидкость с температурой кипе-
ния 56,5°С, обладающую относительно приятным
запахом. Коэффициент распределения кровь/газ
составляет 1,8; он ниже, чем у галотана, поэтому
индукция анестезии и пробуждение наступают бы-
стрее, а глубину анестезии контролировать легче.
Коэффициент распределения масло/газ при 37°С
составляет 98, МАК — 1,63%. Для индукции анесте-
зии применяют концентрацию 5%, для поддержа-
ния —1-2%. Энфлуран обладает анальгетическими
свойствами при ингаляции в субанестетической
концентрации (около 0,8%), и его можно применять
при перевязках ожогов.
Дыхание
Энфлуран угнетает дыхание сильнее, чем дру-
гие современные фторзамещенные анестетики.
Дыхательный объем и МОД уменьшаются, ЧДД
возрастает. При самостоятельном дыхании энфлу-
ран повышает РаСО2, подавляет вентиляторную
реакцию на гипоксию и гиперкапнию, а также
гипоксическую вазоконстрикцию. Хирургические
стимулы полностью или частично устраняют де-
прессию дыхания.
Энфлуран не раздражает дыхательные пути и рас-
ширяет бронхи, хотя и в меньшей степени, чем гало-
тан. Он не вызывает бронхо- и ларингоспазма и об-
ратимо увеличивает продукцию слизи в дыхатель-
ных путях.
Кровообращение
Энфлуран угнетает сократимость миокарда и сни-
жает ОПСС, что вызывает снижение АД и сердеч-
ного выброса. ЧСС обычно увеличивается. Относи-
тельно небольшие изменения концентрации анесте-
тика во вдыхаемой смеси оказывают значительное
воздействие на функцию сердца: энфлуран облада-
ет низкой терапевтической широтой, меньшей, чем
большинство других современных фторзамещен-
ных анестетиков.
.ucoz.ru
Низкие дозы (0,5 МАК) практически не влияют
на ударный объем и сердечный выброс, но АД не-
много снижается вследствие уменьшения ОПСС;
ЧСС обычно не изменяется. Более высокие дозы
(1,5 МАК) значительно снижают сократимость
миокарда, что приводит к уменьшению ударного
объема на 50% и увеличению ЧСС; в результате сер-
дечный выброс снижается приблизительно на 30%.
Некоторые антагонисты кальция (например, вера-
памил и дилтиазем) и p-адреноблокаторы потенци-
руют отрицательное инотропное действие энфлу-
рана. Считают, что кардиодепрессивное действие
энфлурана обусловлено влиянием на кальциевые
каналы мембран кардиомиоцитов.
Энфлуран не оказывает значимого влияния на
АВ-проводимость, но на фоне введения р-адреноб-
локаторов или верапамила может возрасти время
АВ-проведения и даже возникнуть аритмии, напри-
мер, выскальзывающие АВ-ритмы. Энфлуран зна-
чительно слабее, чем галотан, сенсибилизирует
миокард к действию катехоламинов, поэтому риск
желудочковых аритмий при подкожном или мест-
ном применении адреналина невелик (глава 4).
Эти отличия между галотаном и энфлураном обу-
словлены разницей в их химическом строении.
Энфлуран независимо от дозы вызывает сниже-
ние ОПСС в среднем на 25%. Энфлуран также вы-
зывает небольшое уменьшение сопротивления ко-
ронарных артерий; напротив, кровоток в органах
брюшной полости и почках снижается. Энфлуран
увеличивает мозговой кровоток, а также нарушает
его ауторегуляцию, что чревато ишемией мозга при
снижении АД.
В высокой дозе (1,5 МАК) энфлуран может вы-
зывать выраженное снижение АД в результате уг-
нетения сократимости миокарда и уменьшения
ОПСС. Следовательно, энфлуран можно использо-
вать для управляемой интраоперационной гипото-
нии.
ЦНС
В высокой концентрации (приблизительно 3%)
энфлуран вызывает судорожную активность на
ЭЭГ, особенно на фоне гипокапнии, обусловленной
гипервентиляцией. У детей могут развиваться па-
роксизмы генерализованной электрической актив-
ности, напоминающие большие эпилептические
припадки. У взрослых чаще наблюдают очаговую
судорожную активность. Судорожную активность
на ЭЭГ можно уменьшить или устранить снижени-
ем концентрации энфлурана и увеличением РаСО2,
однако, появившись, она может сохраняться на
протяжении нескольких дней и недель после ане-
стезии. В некоторых случаях изменения на ЭЭГ
сочетаются с судорожными подергиваниями или
ИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
107
Табл. 5.2. Физические свойства совре
анестетиков
Температура кипения при атмосферном давлении (’С) Давление пара при 20’С (кПа) МАК в ки- слороде (%) МАК в смеси с 70% заки- сью азота(%) Коэффициент распределе- ния масло/газ Коэффициент распределе- ния кровь/газ
Закись азота -88 5200 104 36 1.4 0,46
Г алотан 50,2 32,1 0,75 0,26 224 2,4
Энфлуран 56,5 23,3 1,63 0,57 98 1,8
Изофлуран 48,5 32.5 1.17 0,41 98 1.4
Севофлуран 58,5 22,7 1.8 0,62 80 0,65
Десфлуран 23,5 89,2 6,6 2.3 29 0,42
клонико-тоническими судорогами мимических
мышц и мышц конечностей. Стойкие неврологиче-
ские осложнения не описаны. Хотя нет данных за
то, что судорожная активность чаше возникает при
сопутствующей эпилепсии, у пациентов с этим за-
болеванием энфлуран лучше не применять.
Мышцы
Энфлуран вызывает расслабление скелетных
мышц и усиливает действие недеполяризующих
миорелаксантов. В этом отношении он мощнее га-
лотана. Этот (|м*номен обусловлен центральным
действием энфлурана, а также периферическим
влиянием на скелетные мышцы. Потенцирование
миорелаксации быстро исчезает после прекраще-
ния подачи энфлурана.
Энфлуран вызывает расслабление матки: не-
смотря на это, его используют в небольших концен-
трациях для выключения сознания при кесаревом
сечении. Он вызывает расслабление мышц глаза
и значительно (на 30-40%) снижает внутриглазное
давление. В этом отношении энфлуран несколько
слабее галотана и изофлурана.
Метаболизм и токсичность
Около 2% поступившего в организм энфлурана
подвергается метаболизму в печени. Метаболизм
энфлурана зависит от активности изофермента
CYP 2Е1, котдрый индуцируется изониазидом,
этанолом, голоданием, ожирением и сахарным диа-
бетом. Основными метаболитами энфлурана явля-
ются двуокись углерода, ди||порметокси-дифтор-
уксусная кислота, неорганический фтор и хлор.
Известно, что высокая концентрация фтора в плаз-
ме (> 40 мкмоль/л) вызывает обратимую нефро-
патию и резистентную к АДГ полиурию. После эн-
флурановой анестезии пиковые концентрации
фтора в плазме регистрируются в течение 4-8 ч и
обычно не достигают 20 мкмоль/л. 'Гак как энфлу-
ран относительно малорастворим, он быстро выво-
дится из организма, и концентрация фтора в плазме
обычно снижается в течение 1-2 ч, но иногда для
этого может потребоваться несколько дней. После
длительной энфлурановой анестезии может на не-
сколько часов снижаться чувствительность почеч-
ных канальцев к действию АДГ. Хотя вероятность
необратимого повреждения почек чрезвычайно
мала, энфлуран лучше не применять при сопутст-
вующих заболеваниях почек, особенно если пред-
стоит длительная операция.
Поражение печени, обусловленное токсиче-
ским действием или гиперчувствительностью к эн-
флурану, крайне нехарактерно. Даже длительная
анестезия вызывает незначительные изменения
функции печени. Было несколько сообщений о по-
слеоперационной желтухе после анестезии эн-
флураном. но такие случаи чрезвычайно редки.
Тем не менее надо помнить, что метаболиты эн-
флурана ковалентно связываются с печеночными
ферментами и могут реагировать с антителами
больных с галотановым гепатитом. Следователь-
но, есть вероятность того, что существует пере-
крестная сенситизация между галотаном и энфлу-
раном.
ИЗОФЛУРАН
Изофлуран. также как и его структурный ана-
лог энфлуран. является фторзамешенным этилме-
тиловым эфиром, синтезированным Р. Терреллом
в 1960-е гг. В настоящее время изофлуран широко
применяется в Великобритании и большинстве
развитых стран.
108
Глава 5
medlituSfig,/JJ
Общие сведения
Изофлуран (1-хлор-2,2,2-трифторэтилдифтор-
метиловый эфир) представляет собой бесцветную
летучую жидкость с температурой кипения 48,5°С.
В смеси с кислородом или воздухом не воспламеня-
ется. При комнатной температуре это соединение
очень стойкое, не разлагается на свету и не реагиру-
ет с достаточно увлажненной натронной известью.
Коэффициент распределения масло/газ при 37 ° С
составляет 98, МАК для возраста 40 лет равен 1,17%
(рис. 5.2); изофлуран мощнее энфлурана, хотя не
отличается от него растворимостью в липидах (см.
табл. 5.2 и 5.3).
По сравнению с галотаном и энфлураном, коэф-
фициент распределения кровь/газ у изофлурана ни-
же (1 ,4). Следовательно, индукция анестезии и про-
буждение при использовании изофлурана наступа-
ют быстрее, а глубину анестезии контролировать
легче. К сожалению, изофлуран обладает неприят-
ным эфирным запахом, что может вызвать неко-
торые трудности (например, задержку дыхания)
во время индукции. Для индукции требуется кон-
центрация 2-4%, а для поддержания — 1-2%.
Изофлуран обладает анальгетическими свойства-
ми в субанестетических дозах (0,5%), поэтому его
используют, например, при перевязке ожогов.
Изофлуран является структурным изомером
энфлурана, молекула его имеет хиральный центр.
В практике применяется в виде рацемической сме-
си; в исследованиях in vivo и in vitro было показано,
что энантиомеры R(-) и S(+) обладают стереоспе-
цифичностью и оказывают разное биологическое
действие (глава 3).
Дыхание
Изофлуран вызывает депрессию дыхания — слабее,
чем энфлуран, но сильнее, чем галотан. Как следст-
вие возрастает ЧДД и РаСО2, снижается дыхатель-
ный объем, угнетается вентиляторная реакция на
гиперкапнию. Вентиляторная реакция на гипоксию
и гипоксическая вазоконстрикция угнетаются при-
близительно в той же степени, что и при примене-
нии галотана. Хирургическая стимуляция полно-
стью или частично устраняет депрессию дыхания.
Изофлуран обладает несколько резким эфир-
ным запахом; он раздражает верхние дыхательные
пути, но не вызывает бронхоконстрикции неизме-
ненных бронхов. Изофлуран является слабым
бронходилататором и вызывает обратимое угнете-
ние продукции слизи в дыхательных путях.
Кровообращение
В отличие от энфлурана и галотана, изофлуран
оказывает весьма слабое непосредственное воздей-
ствие на сердце. В концентрации 1-1,5 МАК он
лишь незначительно уменьшает сократительную
сть миокарда и ударный объем. Изофлу-
ран вызывает периферическую вазодилатацию и
снижает АД, но активность барорецепторов угнета-
ет лишь в небольшой степени. Вследствие этого
возрастает рефлекторная симпатическая актив-
ность, приводящая к тахикардии, позволяющей
поддерживать сердечный выброс на уровне выше
исходного. У молодых тахикардия может быть вы-
раженной, с ЧСС > 100 мин*1. У пожилых актив-
ность барорецепторов низка, поэтому увеличения
ЧСС часто не происходит или оно незначительно; в
этом случае сердечный выброс может снизиться.
В целом, изофлуран не влияет на АВ-проводи-
мость или ЧСС, хотя может потенцировать замед-
ление АВ-проведения, вызванное р-адреноблока-
торами или антагонистами кальция. Кроме того,
изофлуран не вызывает значительной сенсибили-
зации миокарда к катехоламинам.
Изофлуран снижает ОПСС, являясь мощным
вазодилататором, особенно в высоких концентра-
циях. В большинстве сосудистых бассейнов он под-
держивает или усиливает кровоток (например, в
печеночных и коронарных артериях), что улучшает
оксигенацию тканей.
Применение изофлурана при ИБС вызывает
много споров. Изофлуран является мощным коро-
нарным вазодилататором, и было высказано пред-
положение, что он может вызвать феномен обкра-
дывания — неблагоприятное перераспределение
кровотока из плохо кровоснабжаемых участков
миокарда в хорошо кровоснабжаемые. Исследова-
ния, проведенные на животных со стенозом коро-
нарных артерий, показали, что изофлуран снижает
кровоток в стенозированном сегменте, но увеличи-
вает его в нормальных сосудах. Несмотря на эти ре-
зультаты, изофлуран практически не влияет на час-
тоту ишемических эпизодов и летальность при ко-
ронарном шунтировании.
На миокардиальный кровоток влияют различ-
ные факторы. Ишемию могут вызывать артериаль-
ная гипотония и тахикардия, особенно если отно-
шение АДсреднее/ЧСС < 1. Это сочетание не так
редко встречается при изофлурановой анестезии
вследствие характерного снижения ОПСС и реф-
лекторного увеличения ЧСС. Следовательно, изо-
флуран нецелесообразно применять при тяжелой
ИБС, особенно в сочетании с недостаточностью
ЛЖ. У больных группы риска следует тщательно
поддерживать нормальное АД и не допускать тахи-
кардии.
ЦНС
В низкой концентрации (< 1 МАК) изофлуран
не вызывает увеличения МК или повышения ВЧД,
в то время как в более высокой может привести
Табл. 5.3. Свойства современных ингаляционных анестетиков
Свойства Закись азота Галотан Энфлуран Изофлуран Севофлуран Десфлуран
Наступление и пре- кращение действия Очень быстрое Относительно медленное Быстрое Быстрое Очень быстрое Очень быстрое
Анальгетический эффект Практически отсутствует Умеренный Умеренный Умеренный Умеренный
Дыхание Не раздражает дыха- тельные пути ЧДДТ ДО! РаСОг (норма) Поступает в воздухо- с о держащие полости Не раздражает дыхатель- ные пути чад Г дои РаСОг t Угнетает дыхание слабее др инталяц. анест. Не раздражает дыха- тельные пути ЧДДТ ДО»! РаСОг ТТ Угнетает дыхам. силь- нее др. ингал. анест Слегка раздражает ды- хательные пути ЧДДТ ДО!! РаСОг t Не раздражает дыхательные пути чад t ДО!! РаСОг Т Едкий, сильно раздражает дыхательные пути чад г ДО!! РаСОг t
Кровообращение Не оказывает или оказывает минималь- ное воздействие Сенсибилизация миокарда к катехола- минам Т/! ЧСС!! АД»! Сердечный выброс !! ОПСС! Сенсибилизация миокар- да к катехоламинам ТТТ ЧСС Г АД!! Сердечный выброс ! ОПСС! Сенсибилизация миокарда к катехола- минам Т ЧССТТ АД- Сердечный выброс ! ОПСС !! Сенсибилизац миокар- да к катехоламинам Т Синдром обкрадывания коронарных артерий'’ чсс Г/! АД!! Сердечный выброс ! (слегка) ОПСС! Сенсибилизация миокарда к катехоламинам t чсст АДА! Сердечный выброс » __ ОПСС!! 3 Сенсибилизация миокарП) да к катехоламинам " О
Влияние на ЭЭГ Отсутствует Снижение вольтажа Картина -вспышка-по- давление- ЭЭГ-картина, анало- гичная очаговому припадку или боль- шому судорожному припадку ♦ мышечные подергивания Снижение вольтажа Картина .вспышка-по- давление» Снижение вольтажа Картина «вспышка-подавле- ние» Снижение вольтажа Q О Картина -вспышка-подар^ ленив» L_j С
Мозговой кровоток ♦ ттт ♦ ♦ t
Усиление действия недеполяризующих миорелаксантов Нет Умеренное Выраженное Выраженное Выраженное Выраженное
Влияние на матку Отсутствует Умеренное расслабление Умеренное расслаб- ление Умеренное расслабле- ние Умеренное расслабление Умеренное расслабление
Метаболизм (%) Минимальный 15-25 2 0,2 э ч 0.02
Образование фтори- дов Нет Незначительное Значительное Незначительное Значительное Незначительное
Токсичность и гипер- чувствительность Инактивация витамина В„ Нейропения Поражение печени (редко) Поражение печени (исключительно редко) Нефротоксичность? Нет Нефротоксичность (толыто в эксперименте. в клинических условиях не обнаружена) Нет
Т или ! — минимальные изменения; ТТ или !! — умеренные изменения. ТТ? или !!! выраженные изменения: Т/Х отсутствие изменений.
110
Глава 5
dtnedJit. ucozjjjp
к выраженному расширению сосудов г
мозга. Изофлуран не вызывает значительного на-
рушения ауторегуляции мозгового кровотока, а
также реакции мозговых сосудов на изменение
СО2, что позволяет широко использовать его при
нейрохирургических вмешательствах. Изофлуран
подавляет биоэлектрическую активность коры го-
ловного мозга (вплоть до появления изолинии) и не
вызывает появления судорожной активности
Мышцы
Изофлуран расслабляет скелетные мышцы и уси-
ливает действие недеполяризующих миорелаксан-
тов. В этом отношении он приблизительно равен по
силе энфлурану, десфлурану и севофлурану. Этот
феномен вызывается как центральным действием,
так и периферическим влиянием на скелетные
мышцы. Потенцирование быстро исчезает после
отключения изофлурана. При анестезии изофлура-
ном дозу недеполяризующих миорелаксантов
обычно уменьшают. Он вызывает расслабление
матки; тем не менее в низкой концентрации
(0,5-0,75%) его применяют для выключения созна-
ния во время кесарева сечения.
Метаболизм и токсичность
Изофлуран элиминируется из организма преиму-
щественно в неизмененном виде; приблизительно
0,2% подвергается метаболизму под действием изо-
фермента CYP 2Е1. Основные метаболиты —
трифторуксусная кислота, ионы фтора и неболь-
шое количество других фторированных соедине-
ний. Максимальная концентрация неорганическо-
го фтора составляет 5 мкмоль/л и даже в присутст-
вии индукторов печеночных ферментов никогда
не достигает уровня, способного вызвать поврежде-
ние почек.
СЕВОФЛУРАН
Севофлуран был синтезирован еще в 1970-е го-
ды, но появился в клинической практике значи-
тельно позже, потому что вызывал токсические
эффекты у экспериментальных животных. Вначале
его начали использовать в Японии, в 1990-е годы—
в Великобритании (особенно у детей), США, За-
падной Европе. (С 2004 г. севофлуран разрешен к
применению в Российской Федерации — прим.ред.)
Общие сведения
Севофлуран (1-трифторметил-2,2,2-трифтор-
этилмонофторметиловый эфир) представляет со-
бой бесцветную летучую жидкость с температурой
кипения 58,5° С. Давление пара при температуре
20°С равно170 мм рт. ст. (22,7 кПа). Севофлуран
не воспламеняется на воздухе и стабилен при ком-
натной температуре.
имость севофлурана в крови невелика
(коэффициент распределения кровь/газ 0,65) и лишь
немногим выше, чем у закиси азота и десфлурана.
Поскольку растворимость в других тканях тоже
низка, то индукция анестезии и пробуждение про-
исходят очень быстро, а глубиной анестезии легко
управлять. По сравнению с изофлураном, севофлу-
ран хуже поглощается пластиковыми и резиновы-
ми компонентами дыхательного контура, что явля-
ется значительным преимуществом при низкопо-
точной анестезии.
Коэффициент распределения масло/газ при
37° С равен 80. МАК для больного сорока лет в сме-
си с кислородом равна 1,80% (рис. 5.2). Для индук-
ции анестезии используют концентрацию 5-7%,
а для поддержания—0,5-3%. При использовании в
субанестетических концентрациях севофлуран
проявляет анальгетические свойства.
В отличие от других фторзамещенных анестети-
ков, севофлуран подвергается частичному гидро-
лизу при длительном контакте с водой или с силь-
ными основаниями, включая поглотители углеки-
слого газа — натронную известь, известь с добавкой
гидроксида бария. Метаболическая деградация
севофлурана при контакте с поглотителями угле-
кислого газа — это процесс, зависящий от темпера-
туры и приводящий к образованию пяти продуктов
(соединения А, В, С, D и Е); в условиях, которые
встречаются в клинической практике, образуются
только соединения А и В.
Соединение А (пентафторизопропенилфторме-
тиловый эфир, PIFE) при введении подопытным
крысам в течение 3 ч в концентрации 50 частиц на
миллион или в течение 1 ч в концентрации 200 час-
тиц на миллион вызывает некроз почечных каналь-
цев. Эти концентрации значительно (до 8 раз) пре-
вышают те, которые могут возникнуть в дыхатель-
ном контуре в клинической практике. Причины
повышенной концентрации соединения А: высокая
температура дыхательной смеси; длительное под-
держание высокой концентрации севофлурана;
низкопоточная анестезия (поток свежего газа < 1 л);
применение извести с добавкой гидроксида бария
(температура увеличивается выше, чем при исполь-
зовании натронной извести). (Следует отметить,
что известь с добавкой гидроксида бария в РФ в на-
стоящее время не используется — прим, ред.)
При взаимодействии с поглотителями углеки-
слого газа также образуется соединение В (пента-
фторметоксипропилфторметиловый эфир, PMFE).
При использовании реверсивного контура соеди-
нение В можно обнаружить у 70% больных, хотя
в очень низкой, следовой концентрации (1,5 части-
цы на миллион).
ИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ 111
Севофлуран является химическим
изофлурана, а его молекула содержит семь атомов
фтора. В отличие от других фторированных инга-
ляционных анестетиков он не имеет хирального
центра и не проявляет поэтому оптической актив-
ности.
Дыхание
Севофлуран угнетает дыхание примерно в той
же степени, что другие ингаляционные анестетики.
Он учащает дыхание, снижает дыхательный объем
и МОД, вызывает увеличение РаСО2. Угнетение
дыхания устраняется или уменьшается под воздей-
ствием хирургических стимулов. Севофлуран об-
ладает приятным запахом и не раздражает дыха-
тельные пути. У детей широко применяется для ин-
дукции анестезии.
Кровообращение
Севофлуран практически не оказывает прямого
угнетающего действия на сердце, хотя снижение со-
кратительной способности миокарда иногда отме-
чается. ЧСС, как правило, не меняется, и тахикар-
дия нехарактерна; сердечный выброс сохраняется
на исходном уровне. Севофлуран не оказывает
влияния на синусовый узел и АВ-проводимость,
исключительно редко бывая причиной аритмий.
Вместе с тем севофлуран может удлинить время
проведения в АВ-узле, если оно уже было увеличе-
но с помощью p-адреноблокаторов или антагони-
стов кальция. Подобно другим фторзамещенным
эфирам, севофлуран не вызывает значительной
сенсибилизации миокарда к действию адреналина
или других катехоламинов.
Севофлуран снижает АД, преимущественно за
счет снижения ОПСС. Кровоток в большинстве пе-
риферических бассейнов поддерживается на ис-
ходном уровне или возрастает (например, в органах
брюшной полости и в почках). Сопротивление моз-
говых сосудов также снижается, и мозговой
кровоток может незначительно возрасти, особенно
при высоких концентрациях. Севофлуран также
увеличивает коронарный кровоток; следует под-
черкнуть, что он не вызывает феномен обкрадыва-
ния коронарных артерий.
Мышцы
Севофлуран вызывает миорелаксацию и потен-
цирует действие недеполяризующих миорелаксан-
тов. Его действие на нервно-мышечные синапсы
аналогично эквипотенциальным концентрациям
изофлурана. Этот феномен осуществляется как за
счет центрального, так и за счет периферического
действия севофлурана; потенцирование быстро
исчезает после отключения подачи севофлурана.
Следовательно, при использовании севофлурана
необходимо снижать дозы недеполяризующих
антов. Севофлуран, как и другие ингаля-
ционные анестетики, может инициировать злока-
чественную гипертермию (было не менее четырех
сообщений о таких случаях). Севофлуран вызывает
также расслабление гладкой мускулатуры матки,
хотя в низкой концентрации его можно использо-
вать при плановом кесаревом сечении.
ЦНС
Севофлуран подавляет электрическую актив-
ность коры головного мозга, что отражается на кар-
тине ЭЭГ, и не вызывает появления судорожной ак-
тивности коры ни при нормокапнии, ни при гипер-
капнии. В высокой концентрации (> 2 МАК) может
повыситься внутричерепное давление; потребле-
ние кислорода мозгом снижается приблизительно
на 50%. При индукции анестезии у детей могут на-
блюдаться беспокойство, возбуждение и кашель, но
частота этих осложнений ниже, чем при использо-
вании других ингаляционных анестетиков.
Метаболизм и токсичность
Севофлуран подвергается относительно ак-
тивному метаболизму в печени (приблизительно
на 3%). Главные продукты метаболизма — углекис-
лый газ, неорганические фториды и гексафторизо-
пропанол. Метаболизм происходит при участии
изофермента CYP 2Е1, активность которого инду-
цируют этанол, изониазид и, возможно, фенобарби-
тал. Эти индукторы стимулируют метаболизм се-
вофлурана.
Во время анестезии и в раннем послеоперацион-
ном периоде концентрация фторидов в плазме мо-
жет достигать достаточно высокого уровня (при-
близительно 20-50 мкмоль/л). Уровень фторидов
обычно бывает максимальным в течение 1-2 ч и
возвращается к норме через 24-48 ч. Основным
фторорганическим метаболитом севофлурана яв-
ляется гексафторизопропанол, который быстро
конъюгируется в печени с образованием глюкуро-
нида с относительно длительным Tt/2 (около 55 ч).
Образование фторидов можно уменьшить приме-
нением дисульфирама (селективного ингибитора
CYP2E1).
ДЕСФЛУРАН
Хотя десфлуран был синтезирован еще в 1960-е гг.,
его первые клинические исследования были прове-
дены только в 1987 г. Благодаря очень низкому
коэффициенту распределения кровь/газ (0,42),
десфлуран позволяет легко регулировать глубину
анестезии. К сожалению, десфлуран не лишен ряда
недостатков, и в настоящее время он находит лишь
ограниченное применение в клинике.
Общие сведения
Десфлуран (1-фтор-2,2,2-трифторэтилдифтор-
метиловый эфир) представляет собой бесцветную,
112
Глава 5
легко испаряющуюся жидкость с температур1э@(Ё1б1Ш. ЦООйлШинов. Десфлуран снижает АД вследствие
ния 23,5° С и давлением паров при 20°С 669 мм рт. ст.
(89,2 кПа). Этот анестетик невозможно использо-
вать с помощью испарителей стандартной конст-
рукции: требуются специальные термостатируе-
мые испарители, работающие под повышенным
давлением. Десфлуран не воспламеняется, он чрез-
вычайно стоек при комнатной температуре, не раз-
лагается на свету и не вступает в реакцию с поглоти-
телями углекислого газа. Коэффициент распреде-
ления кровь/газ у десфлурана ниже, чем у других
галогензамещенных анестетиков (0,42). Следова-
тельно, индукция анестезии и пробуждение долж-
ны происходить быстрее, чем при использовании
других галогензамещенных анестетиков, а глуби-
ной анестезии легче управлять. Коэффициент рас-
пределения масло/газ при температуре 37°С равен
29, а МАК — 6,6%. Следовательно, его мощность
ниже, чем у других галогензамещенных анестети-
ков. Для индукции требуется концентрация 6-9%,
а для поддержания — 3-5%. В субанестетических
концентрациях проявляет анальгетические свойства.
По химическому строению десфлуран очень
близок изофлурану; единственная разница заклю-
чается в замене единственного атома хлора в моле-
куле изофлурана на атом фтора (рис. 5.3). Молеку-
ла десфлурана содержит асимметричный атом уг-
лерода; в клинической практике используют
рацемическую смесь стереоизомеров.
Дыхание
Десфлуран угнетает дыхание в меньшей степе-
ни, чем изофлуран и энфлуран. При самостоятель-
ном дыхании он вызывает увеличение ЧДД, РаСО2,
снижение дыхательного объема и МОД, угнетает
вентиляторную реакцию на гиперкапнию. Хирур-
гическая стимуляция устраняет депрессию дыха-
ния. Десфлуран обладает более едким и неприят-
ным эфирным запахом, чем изофлуран или любой
другой ингаляционный анестетик. Он оказывает
раздражающее влияние на дыхательные пути, вы-
зывает обильное слюнотечение, задержку дыхания,
кашель и ларингоспазм, что замедляет скорость ин-
дукции анестезии и может привести к гипоксемии.
По этим причинам десфлуран противопоказан для
индукции анестезии; его используют для поддер-
жания.
Кровообращение
По своему действию на кровообращение десфлу-
ран напоминает изофлуран. Он практически не ока-
зывает прямого действия на сердце; снижение со-
кратительной способности миокарда происходит
только в редких случаях. Десфлуран не влияет на
АВ-проводимость, синусовый узел и не сенсибили-
зирует сердце к действию адреналина и других
уменьшения ОПСС, при этом происходит рефлек-
торное увеличение ЧСС, что позволяет поддер-
живать сердечный выброс на исходном уровне.
Учащение ЧСС обусловлено повышением секре-
ции катехоламинов.
Десфлуран является мощным периферическим
вазодилататором, он снижает АД путем снижения
ОПСС. Как следствие, кровоток в большинстве пе-
риферических сосудистых бассейнов поддержива-
ется на исходном уровне или возрастает (например,
в органах брюшной полости и в почках). Препарат
снижает сопротивление сосудов головного мозга
и может увеличивать мозговой кровоток. Десфлу-
ран повышает коронарный кровоток.
Мышцы
Десфлуран расслабляет скелетные мышцы и уси-
ливает действие недеполяризующих миорелак-
сантов. В этом отношении он мощнее галотана
и приблизительно равен по силе действия другим
фторзамещенным эфирам. Миорелаксация обу-
словлена центральными и периферическими
эффектами десфлурана. Потенцирование быстро
исчезает после прекращения подачи десфлурана.
Десфлуран расслабляет матку, не отличаясь в этом
отношении по силе действия от галотана, энфлура-
на и изофлурана.
Метаболизм и токсичность
Десфлуран содержит шесть относительно ус-
тойчивых связей С-F, и метаболизму подвергается
лишь мизерная доля препарата — 0,02%. Десфлуран
вызывает небольшое, но достоверное повышение
концентрации трифторуксусной кислоты в плазме
(около 10-20% от уровня, возникающего после
применения изофлурана). Отмечается также не-
большой и незначимый подъем концентрации не-
органических и органических фторидов в плазме.
Метаболизм дефлурана обеспечивается изофер-
ментом CYP 2Е1. Индукция этого фермента эта-
нолом или изониазидом усиливает образование
фторидов.
Так как десфлуран практически полностью
элиминируется в неизмененном виде, вероятность
токсического повреждения почек или печени
чрезвычайно мала. Нет никаких данных в пользу
клеточной или органной токсичности десфлурана.
В опытах на животных, получавших индукторы
ферментной активности, многократная и длитель-
ная ингаляция десфлурана не вызывала никаких
значимых гистологических и гистохимических из-
менений.
ИНГАЛЯЦИОННЫЕ АНЕСТЕТИКИ
113
medlit.uffi^GA,
ЦИКЛОПРОПАН
Циклопропан представляет собой газ без цвета и
запаха. Синтезирован в 1882 г., применяется в кли-
нике с 1934 г. Его хранят в металлических баллонах
в сжиженном виде под давлением 5 атм. У цикло-
пропана низкий (близкий к закиси азота) коэффи-
циент распределения кровь/газ (0,5), поэтому ин-
дукция анестезии и пробуждение происходят чрез-
вычайно быстро. Он вызывает более сильную
депрессию дыхания, чем другие ингаляционные
анестетики, поэтому при отсутствии ИВЛ может
развиться гиперкапния. Циклопропан не раздража-
ет дыхательные пути, хотя может вызывать ларин-
го- и бронхоспазм, особенно при бронхиальной аст-
ме, когда тонус бронхов исходно повышен. Во время
анестезии обычно происходит небольшое, но гене-
рализованное повышение симпатического тонуса,
что приводит к незначительному увеличению АД.
Циклопропан является мощным миорелаксантом
и может потенцировать действие недеполяризую-
щих миорелаксантов. О биотрансформации цикло-
пропана в организме человека известно немного,
хотя экспериментальные данные позволяют утвер-
ждать, что он частично метаболизируется до СО2.
Выраженных токсических эффектов циклопропан
не вызывает.
Циклопропан может быть причиной двух опас-
ных осложнений. Во-первых, он повышает возбу-
димость миокарда и может провоцировать различ-
ные аритмии, особенно во время индукции анестезии.
На фоне циклопропана желудочковые экстрасис-
толы или желудочковая тахикардия могут транс-
формироваться в фибрилляцию желудочков. Риск
фибрилляции желудочков выше при введении
Р-адреномиметиков, высоком уровне эндогенных
катехоламинов (например, при гиперкапнии), при
использовании холиноблокаторов. Сочетание цик-
лопропана с симпатомиметиками чрезвычайно
опасно. Во-вторых, циклопропан взрывоопасен
и может воспламеняться. Из-за этих осложнений
циклопропан представляет сегодня преимущест-
венно исторический интерес.
ЭТИЛОВЫЙ ЭФИР
Этиловый эфир был введен в практику Уилья-
мом Мортоном в 1846 г. и стал первым ингаляцион-
ным анестетиком, который с успехом использовал-
ся для обезболивания хирургических операций.
Этиловый эфир представляет собой летучую вос-
пламеняющуюся жидкость. Он хорошо растворим в
крови (коэффициент распределения кровь/газ = 12,0),
поэтому индукция анестезии и пробуждение явля-
ются довольно медленными, и для полной элими-
рганизма может потребоваться много ча-
сов.
Этиловый эфир повышает симпатический то-
нус, поэтому ЧДД, АД и ЧСС сохраняются на ис-
ходном уровне либо слегка возрастают. Напротив,
на денервированное сердце этиловый эфир оказы-
вает угнетающее действие. Одним из достоинств
этилового эфира является отсутствие повреждаю-
щего действия на кровообращение; он не провоци-
рует аритмии и не повышает чувствительность
сердца к катехоламинам.
Этиловый эфир оказывает выраженное раздра-
жающее действие на дыхательные пути и значи-
тельно повышает секрецию в них, что, как правило,
требует премедикации атропином или сходным
препаратом. Этиловый эфир потенцирует эффект
недеполяризующих миорелаксантов и вызывает
некоторую релаксацию гладкой мускулатуры мат-
ки. Высока частота послеоперационной тошноты и
рвоты.
О метаболизме этилового эфира в организме че-
ловека известно очень мало, хотя он не обладает
значительной токсичностью. Около 6% дозы, вве-
денной экспериментальным животным, выделяет-
ся из организма в виде ряда соединений, включая
этанол, уксусный альдегид и уксусную кислоту.
Главные недостатки этилового эфира включают
медленную индукцию анестезии, медленное пробу-
ждение, раздражающее действие на дыхательные
пути и способность воспламеняться. Тем не менее
этот анестетик широко применялся в клинической
практике вплоть до появления галотана в 1958 г. В на-
стоящее время этиловый эфир практически не ис-
пользуют для общей анестезии. В редких случаях
его применяют для лечения тяжелых приступов
бронхиальной астмы: он усиливает бронхорею, что
повышает эффективность бронхиального лаважа.
Список литературы
Belelli, D., Pistis, М., Peters, J.А. & Lambert, JJ. (1999). General
anaesthetic action at transmitter-gated inhibitory amino acid
receptors. Trends in Pharmacological Sciences 20, 496-502.
Carpenter, R.L., Eger, E.I.I.I, Johnson, B.H., Unadkat, J.D. & Sheiner,
L.B. (1986). Pharmacokinetics of inhaled anesthetics in humans:
measurements during and after the simultaneous administration of
enflurane, halothane, isoflurane, methoxyflurane, and nitrous oxide.
Anesthesia and Analgesia 65,575-582.
Daniels, S. & Smith, E.B. (1993). Effects of general anaesthetics on li-
gand-gated ion channels. BritishJournal of Anaesthesia! i, 59-64.
Eger, E.I. II. (ed.) (1974). Anesthetic Uptake and Action. Baltimore:
Williams & Wilkins.
Eger, E.I. II. (1994). New inhaled anesthetics. Anesthesiology 80,
906-922.
114
Глава 5
Eger, E.I. IL, Saidman, L.J. & Bandstater, B. (1965).
medlit.ucoz.ru
Minimumalveo- Mapleson, W.W. (1996). Effect of age on MAC inhumans:ameta-
lar concentxation: a standard of anesthetic potency. Anesthesiology
26,756-763.
Franks, N.P. & Lieb, W.R. (1994). Molecular and cellular mecha-
nisms of general anaesthesia. Nature 367, 607-614.
Halsey, MJ. (1987). Drug interactions in anaesthesia. Britishjour-
nal ofAnaesthesia 59, 112-123.
Hatch, D.J. (1999). New inhalation agents in paediatric anaes-
thesia. British Journal of Anaesthesia 83, 42-49.
Hayashi, Y. & Maze, M. (1993). Alpha2 adrenoceptor agonists
and anaesthesia. Britishjoumcd of Anaesthesia 71,108-118.
Hayashi, Y., Sumikawa, K., Tashiro, C., Yamatodani, A. & Yoshiya, I.
(1988). Arrhythmogenic threshold of epinephrine during sevoflu-
rane, enflurane, and isoflurane anaesthesia in dogs. Anesthe-
siology 59, 145-147.
Jones, R.M., Cashman, J.N., Eger, E.I. II, Damask, M.C. & Johnson,
B.H. (1990). Kinetics and potency ofdesflurane (1-653) in volun-
teers. Anesthesie and Analgesia 70,3-7.
Kenna, J.G., Satoh, H., Christ, D.D. & Pohl, L.R. (1988). Meta-
bolic basis for a drug hypersensitivity: antibodies in sera from
patients with halothane hepatitis recognize liver neoantigens that
contain the trifluoroacetyl group derived from halothane. Journal
of Pharmacology and Experimental Therapeutics 24,5, 1103-1109.
Kharasch, E.D., Hankins, D., Mautz, D. & Thummel, K.E.(1996).
Identification of the enzyme responsible for oxidative halothane
metabolism: implications for prevention of halothane hepatitis.
analysis. British Journal of Anaesthesia 76, 179-185.
Murat, L, Dubois, M.C. & Piat, V. (1995). Sevoflurane. Armales
Francoises д' Anesthesie et de Reanimation 14,489-501.
Nunn, J.F. (1987) Clinical aspects of the interaction between
nitrous oxide and vitamin В12. BritishJoumalofAnaesthesa59,3-13.
Nunn, J.F., Chanarin, L, Tanner, A.G. & Owen, E.R.T.C. (1986).
Megaloblastic bone marrow changes after repeated nitrous
oxide anaesthesia. British Journal of Anaesthesia 58,1469-1470.
Pocock, G. & Richards, C.D. (1993). Excitatory and inhibitory
synaptic mechanisms in anaesthesia. Britishjoumal ofAnaesthesia 71,
134-147.
Priebe, HJ. (1989). Isoflurane and coronary hemodynamics.
Anesthesiology 71, 960-976.
Samer, J.B., Levine, M„ Davis, P.., Lerman, J., Cook, D.R. & Motoya-
ma, E.K. (1995). Clinical characteristics of sevoflurane in children:
a comparison with halothane. Anesthesiology 82,38-46.
Smith, L„ Nathanson, M. & White, P.F. (1996). Sevoflurane: a long-
awaited volatile anaesthetic. British Journal ofAnaesthesia 76,
435-445.
Stoetling, R.K. & Eger, E.I. II. (1969). An additional explana-
tion for the second gas effect: a concentrating effect. Anesthe-
siology 30, 273-277.
Terrell, R.C. (1984). Physical and chemical properties of anaesthe-
tic agents. British Journal of Anaesthesia 56, 3S-7S.
Lancet 347, 1367-1371.
medlit.ucoz.ru
МЕСТНЫЕ АНЕСТЕТИКИ
6
Местные анестетики — это лекарственные средства,
которые обратимо угнетают возникновение и про-
ведение импульсов в периферических нервах
и нервных окончаниях, что приводит к потере
чувствительности в отграниченных участках тела.
В Великобритании в клинической практике ис-
пользуется четыре местных анестетика (лидокаин,
прилокаин, бупивакаин и ропивакаин). Многие пре-
параты, применяемые для других целей, обладают
свойствами местных анестетиков (например,
хлорпромазин, пропранолол). С другой стороны,
местные анестетики могут применяться в качестве
антиаритмических и противосудорожных средств.
ИСТОРИЧЕСКАЯ СПРАВКА
Давно известно, что некоторые встречающиеся
в природе вещества могут вызывать местные или
генерализованные изменения чувствительности
и мышечной силы. В XVIII веке капитан Джеймс
Кук во время путешествия в Южных Морях попро-
бовал рыбу иглобрюха и подробно описал ее воз-
действие на нервную систему; теперь мы знаем, что
эти эффекты вызваны отравлением тетродотокси-
ном. Тетродотоксин — это яд, который содержится
в организме осьминога, саламандры, некоторых
тритонов и лягушек, а также японского иглобрюха
(рыба фугу, или Spheroides spengleri). Аналогично
действуют сакситоксин и некоторые подобные ему
яды, которые вырабатываются динофлагеллятами —
одноклеточными жгутиковыми водорослями — и на-
капливаются в тканях моллюсков и ракообразных
(которые этими'микроорганизмами питаются).
Механизм действия тетродотоксина и сакситокси-
на в значительной степени аналогичен механизму
действия местных анестетиков. Вместе с тем, тетро-
дотоксин и сакситоксин являются относительно
полярными соединениями, поэтому они плохо про-
никают через клеточную мембрану и, несмотря на
свою чрезвычайно высокую мощность, не могут
применяться для местной анестезии.
Природный алкалоид кокаин был первым мест-
ным анестетиком, примененным в клинической
практике. Это вещество получают из листьев кус-
тарника (Erythroxylon coca), произрастающего
у подножий Анд. Перуанские индейцы в течение
многих веков жевали эти листья: в полученном экс-
тракте содержался кокаин, который высоко ценили
за психостимулирующее действие и способность
вызывать эйфорию. Кроме того, было хорошо из-
вестно, что при жевании листьев немеет слизистая
щек. Альберт Ниман в 1860 г. впервые выделил чис-
тый кокаин. Попробовав новое вещество на вкус,
он заметил, что при этом немеет язык. Фармаколо-
гические свойства кокаина были изучены фон Ан-
репом в 1870-1880 гг. В клиническую практику пре-
парат был внедрен в 1884 г. Фрейдом и Коллером.
Зигмунд Фрейд пытался лечить кокаином своего
больного морфинизмом коллегу, но лишь превра-
тил его из морфинового наркомана в кокаинового.
Сам Фрейд принимал кокаин в течение 10 лет (в это
время он писал книгу «Интерпретация сновиде-
ний»), Карл Коллер начал использовать кокаин для
обезболивания роговицы у животных и быстро
оценил его свойства. Коллер внедрил кокаин в кли-
ническую практику для поверхностной анестезии
в офтальмологии, но вскоре этот препарат стали
применять для инфильтрационной, проводнико-
вой и спинномозговой анестезии.
Высокий потенциал развития патологического
пристрастия к кокаину осознали только к 1890 г.,
после чего начался поиск новых, безопасных лекар-
ственных препаратов. Первый синтетический мест-
ный анестетик прокаин стал применяться Эйнгор-
ном в 1905 г. Впоследствии изучались многие мест-
ные анестетики эфирного типа; большинство из
них давно не применяются и в настоящее время
представляют лишь исторический интерес. Тетра-
каин (аметокаин) до сих пор используется для по-
верхностной анестезии.
Важной вехой в развитии местной анестезии
стал синтез лидокаина (осуществленный в 1943 г.
116
Глава 6
medlit.ucoz.ru
Лофгреном) и его появление и клиннчес
тике. Этот аминоацнламид стал прототипом мест-
ных анестетиков амидного типа (в том числе мепи-
вакаииа, бупивакаина. прилокаина, ропивакаина).
Во многих пранах (но не в Великобритании) приме-
няется очень мощный анестетик этидокаин. В по-
следние годы некоторые местные анестетики амид-
ного типа стали выпускаться в виде единственного
стереоизомера (ронивакаин. левобупивакаин), что
обеспечивает значительные преимущества. Суще-
ствуют экспериментальные л ипосома.лы1ые| лекар-
ственные формы мест ных анестетиков, которые по-
зволяют увеличить продолжительность действия и
уменьшить системную токсичность.
СТРОЕНИЕ И ФУНКЦИЯ НЕРВНЫХ
ВОЛОКОН
Периферические нервы представляют собой
пучки нервных волокон аксонов и дендритов.
Нервиги» волокно окружено эндоневрием — тонкой
соединительнотканной оболочкой, которая состо-
ит в основном из продольно ориентированных мо-
лекул коллагена. Пучки нервных волокон окруже-
ны периневрием. Снаружи нервы покрыты рыхлой
соединительнотканной оболочкой эпиневрием.
В эпиневрии проходят кровеносные и лимфатиче-
ские сосуды. Каждое нервное волокно окутано мие-
линовой оболочкой, представленной цитоплазмой
шванновской клетки. Немиелиновые волокна
обычно объединяются в группы, покрытые оболоч-
кой из одной шванновской клетки (длиной до 0,5 мм);
в местах соединений мембраны соседних шваннов-
ских клеток контактируют друг с другом. Напро-
тив, на каждое миелиновое волокно спиралью на-
мотана отдельная шванновская клетка (рис. 6.1).
ЯШ
Z
Рис. 6.1, Схематичное изображение поперечного сечения
(а) миелинизированного и (б) немиелннизированноп)
нервных волокон; ЯШ — ядро шванновской клетки; а
аксон или дендрит; М - миелиновая оболочка
[дельными шванновскими клетками мие-
линовая оболочка отсутствует, и образуются гак
называемые перехваты Ранвье. Расстояние между
перехватами зависит от размеров шванновских
клеток и диаметра нерва. В крупных миелинизиро-
ванных нервах расстояние между перехватами
может достигать 1 2 мм.
Нервные волокна состоят из сердцевины (цито-
плазмы). заключенной в клеточную мембрану.
Цитоплазма содержит митохондрии, микрока-
нальцы и нейрофиламенты, необходимые для нор-
мального питания и метаболизма. Клеточная мем-
брана представляет собой типичную фосфолипид-
ную мембрану и содержит интегральные белки
(рис. 1.1). Некоторые из них имеют поры или ион-
ные каналы, которые играют важную роль в функ-
ции нейронов.
В состоянии покоя по обе стороны клеточной мем-
браны существует разность потенциалов 60 90 мВ:
внутренняя сторона заряжена отрицательно, а на-
ружная — положительно. Эта разность потенциа-
лов (потенциал покоя) в основном отражает изби-
рательную проницаемость клеточной мембраны
для К . В состоянии покоя мембрана непроницаема
для Na’, а К' может медленно диффундировать из
нейроплазмы. Этому процессу противодействует
отрицательный заряд внутриклеточных белков,
которые препятствуют диффузии К’. Равновесие
между этими двумя силами представляет мембран-
ный потенциал покоя, который зависит от отноше-
ния К,’/К(Г (рис. 6.2).
Во время активации нервной клетки потенциал
покоя претерпевает характерные изменения.
В начальную фазу медленной деполяризации за-
ряд клетки становится менее отрицательным.
Когда достигается пороговый потенциал (-50 мВ),
происходит преходящая быстрая деполяризация
приблизительно до + 25 мВ. После этого потенциал
покоя восстанавливается (фаза реполяризации).
Эти изменения называются потенциалом действия
и происходят в течение 1-2 мс (рис. 6.3).
Способность генерировать потенциал действия
обусловлена присутствием в мембране потенциал-
зависимых натриевых каналов. Во время возбужде-
ния натриевые каналы открываются, и Na’ быстро
диффундирует через клеточную мембрану, вызы-
вая преходящую реверсию мембранного потенциа-
ла. В то же время медленно открываются калиевые
каналы, и К проникает через мембрану, вызывая ее
реполяризацию. Во время рефрактерного периода
ионный баланс нормализуется с помощью Na’/K’
АТФазы.
I
Липосомы что везикулы диаметром от 50 нм до 10 мкм; они состоят из полной фазы, (круженной двойным фосфолипид-
ным слоем. Лекарственные препараты могут находиться как в водной, так и в фосфолипидной фазе (прим. ред.).
МЕСТНЫЕ АНЕСТЕТИКИ
117
medlit.ucoz.ru
Белок
Рис. 6.2. Механизм возникновения
мембранного потенциала покоя.
Фермент Ма*/К+-АТФаза обеспе-
чивает высокую концентрацию К+
внутри клетки и высокую концен-
трацию Na+ во внеклеточной жид-
кости. В состоянии покоя клеточ-
ная мембрана практически непро-
ницаема для Na+, в то время как К*
может пассивно диффундировать
из цитоплазмы во внеклеточную
жидкость. Отрицательный заряд
внутриклеточных белков противо-
действует стремлению К+ поки-
нуть нервную клетку, что обеспе-
чивает поддержание мембранного
потенциала покоя
Рис. 6.3. Изменения мембранного потенциала ( — ),
проницаемости клеточной мембраны для Na ( — ) и
К ( — ) во время потенциала действия нерва
В мембране нейрона также присутствует Са2+,
который может влиять на функцию потенциал-
чувствительных натриевых каналов. В экспери-
ментальных условиях при повышении местной
концентрации кальция пороговый потенциал, не-
обходимый для открытия натриевых каналов,
снижается, хотя потенциал покоя не меняется.
Активация и деполяризация немиелиновых
волокон вызывает появление в клеточной мембра-
не местного электрического тока, что уменьшает
мембранный потенциал в прилегающем участке
нерва. Потенциалзависимые натриевые каналы ак-
тивируются, и импульсы распространяются вдоль
нервного волокна. Ретроградное проведение не-
возможно ввиду быстрой инактивации натриевых
каналов по ходу распространения импульса.
В миелиновых волокнах потенциал действия
распространяется от одного перехвата Ранвье к дру-
гому; поскольку расстояние между перехватами
составляет 1-2 мм, проведение импульса в миели-
новых волокнах происходит намного быстрее,
чем в немиелиновых: скорость может составлять до
120 м/сек.
МОЛЕКУЛЯРНОЕ СТРОЕНИЕ
НАТРИЕВЫХ И КАЛИЕВЫХ
КАНАЛОВ
В последнее время с помощью биохимических,
биофизических и молекулярно-биологических
методов удалось установить точную структуру на-
триевых и калиевых каналов в мембране нейрона.
Потенциалчувствительные натриевые каналы
являются интегральными белками, которые прони-
зывают клеточную мембрану нейрона, формируя
трансмембранную пору (рис. 1.1). Натриевые кана-
лы состоят из трех субъединиц (a, Pi и р2); самая
крупная субъединица (а-субъединица; молекуляр-
ная масса — 260 кДа) является длинной полипеп-
тидной цепью, состоящей из четырех гидрофобных
118
Глава 6
гомологичных доменов (I-IV). Эти четы
соединены между собой внутриклеточными мости-
ками (рис. 6.4).
Каждый домен содержит шесть трансмембран-
ных сегментов (S1-S6). Сегмент S4, несущий поло-
жительный заряд, является потенциалчувстви-
тельной (воротной) структурой, которая запускает
процесс открывания канала в ответ на изменение
мембранного потенциала. Трансмембранные сег-
менты S5 и S6, а также короткая внутримембранная
петля между ними формируют стенки поры в цен-
тре почти симметричного четырехугольника, обра-
зованного четырьмя доменами а-субъединицы
(рис. 6.4). Короткий внутриклеточный мостик меж-
ду доменами III и IV является воротами инактива-
ции. Этот мостик закрывает внутреннее устье на-
триевого канала через 1 -2 мс после его открывания.
Калиевые каналы представлены большой гете-
рогенной группой мембранных белков; известно, по
меньшей мере, 14 типов каналов, выполняющих
различные физиологические функции. Некоторые
калиевые каналы являются потенциалзависимы-
ми; другие реагируют на нейромедиаторы, внутри-
клеточные ионы кальция или АТФ. Они могут
инактивироваться или не инактивироваться после
деполяризации. Поскольку потенциал равновесия
для К+ равен -100 мВ, все открытые калиевые кана-
Ц£«>О?и!ткЬают реполяризацию и снижают возбуди-
мость нейрональной мембраны.
Своим строением калиевые каналы во многом
напоминают натриевые каналы. Основное отличие
заключается в том, что обйчно калиевые каналы
состоят из четырех субъединиц; каждая субъедини-
ца эквивалентна отдельному домену в натриевом
канале (рис. 6.4). Калиевые каналы также содержат
потенциалчувствительную структуру в сегменте S4.
Некоторые каналы медленно инактивируются,
поскольку запираются терминальными участками
каждой субъединицы.
МЕХАНИЗМ ДЕЙСТВИЯ
Большинство местных анестетиков являются
третичными аминовыми основаниями (В), которые
применяют в виде растворов водорастворимых со-
лей, в основном гидрохлоридов (В.НС1) с pH 4-5,5.
При попадании во внеклеточную жидкость (pH 7,4)
третичное основание отщепляется от соляной ки-
слоты:
В.НС1 + нсо; В + Н2СОз + сг
Следовательно, местные анестетики присутст-
вуют в тканях как в неионизированной (В), так
и в ионизированной (ВН+) форме, а их соотноше-
ние зависит от разницы между константой диссо-
Рис. 6.4. (а) Строение а-субъеди-
ницы потенциалчувствительного
натриевого канала; а-субъединица
является длинной полипептидной
цепью, состоящей из четырех гидро-
фобных гомологичных доменов
(I-IV). Эти четыре домена форми-
руют почти симметричный четы-
рехугольник, в центре которого
находится трансмембранная пора,
(б) Схема строения одного из
гидрофобных доменов. Каждый
участок состоит из шести транс-
мембранных сегментов (S1-S6).
Сегмент S4, несущий положитель-
ный заряд, является потенциалчув-
ствительной (воротной) структу-
рой. Трансмембранные сегменты S5
и S6, а также короткая внутримем-
бранная петля между ними фор-
мируют стенки поры. Внутрикле-
точные мостики соединяют S1 и S6
с соседними участками. Считают,
что местные анестетики блокируют
натриевый канал вблизи остатков
тирозина и фенилаланина в сегмен-
те S6 домена IV
No*
МЕСТНЫЕ АНЕСТЕТИКИ
119
циации местного анестетика и pH среды!^0.$и1Ш
неионизированная фракция местного анестетика
(В) растворяется в липидах и может проникать
через оболочки нервов и мембрану нейрона в ци-
топлазму, после чего она соединяется с ионами Н+
и взаимодействует с натриевыми каналами уже
в виде катиона: В + Н+ = ВН+.
В ионизированной форме ВН местный анесте-
тик поступает из цитоплазмы в натриевый канал
с его внутренней стороны и запирает его изнутри.
Считают, что местные анестетики взаимодейству-
ют с остатками фенилаланина (в положении 1764)
и тирозина (в положении 1771), находящимися
в сегменте S6 домена IV (рис. 6.4 и 6.5). Вероятно,
местные анестетики блокируют натриевые каналы
неспецифично, поскольку все третичные основа-
ния с рКа« 7,5-9 в той или иной степени проявляют
свойства местных анестетиков. Так как необходи-
.ucoz.ru
в.на
pH 7.4
Рис. 6.5. Механизм и точка приложения действия мест-
ных анестетиков в натриевых каналах.
мым условием для достижения точки-мишени явля-
ется диффузия местного анестетика (в форме ВН+)
через открытые натриевые каналы, выраженность
блокады нерва может зависеть от частоты его сти-
муляции (частотозависимость действия местных
анестетиков).
Убедительное доказательство в пользу этого
достаточно сложного механизма действия (рис. 6.5)
получено в эксперименте, при котором лидокаин и
его ЛГ-этилированное четвертичное производное
наносились на разные стороны клеточной мембра-
ны. Лидокаин вызывает местную анестезию, когда
его наносят как с внутренней, так и с наружной сто-
роны мембраны. Напротив, четвертичное произ-
водное лидокаина действует только с внутренней
поверхности клеточной мембраны. Из этого экспе-
римента следует, что местные анестетики могут
оказывать свое воздействие на натриевые каналы
только со стороны цитоплазмы нейрона; иными
словами, рецептор, с которым взаимодействует ме-
стный анестетик, находится в области внутрикле-
точного конца натриевого канала.
Хотя местные анестетики преимущественно
блокируют натриевые каналы и предотвращают де-
поляризацию, они могут воздействовать и на калие-
вые каналы. Тем не менее местные анестетики не
изменяют потенциал покоя нервных клеток, если
только не применяются в очень высокой концен-
трации. Они также не влияют на пороговый потен-
циал, необходимый для распространения импульса,
хотя деполяризация и реполяризация угнетаются,
Местные анестетики применяют в виде растворов гидро-
хлоридов (В.НС1) с pH 4-5. При попадании во внеклеточ-
ную жидкость (слабощелочная среда с pH = 7,4) третичное
основание отщепляется от соляной кислоты. Только не-
ионизированная форма В может диффундировать
сквозь клеточную мембрану, после чего в цитоплазме
к ней присоединяется Н+. В форме ВН+ анестетик дости-
гает точки приложения действия в открытом натриевом
канале и блокирует его (БК). Неионизированная форма
местного анестетика также может непосредственно диф-
фундировать к натриевому каналу через клеточную мем-
брану и присоединять Н ’ непосредственно в просвете ка-
нала; это вызывает блокаду канала за счет «мембранного
расширения» (МР), т. е. набухания липопротеиновой
мембраны. Тетродотоксин и сакситоксин блокируют
натриевый канал с наружной стороны мембраны, закры-
вая наружную пору
а проведение замедляется. Аналогичные эффекты
развиваются и в других возбудимых тканях; напри-
мер, под действием местных анестетиков уменьша-
ется скорость деполяризации (фаза 0) в миокарде
желудочков (глава 15).
Некоторые местные анестетики (например, бен-
зокаин) присутствуют в организме только в виде
неионизированных третичных оснований, и по-
этому должны иметь другой механизм действия.
Считается, что такие препараты вызывают блокаду
проведения путем «мембранного расширения»
(т. е., вызывая набухание липопротеинового матрик-
са натриевого канала). В определенной степени
таким же способом действуют и другие местные
1
Константа диссоциации, или значение рХ„ определяется модифицированным уравнением Гендерсона-Хассельбальха:
рК0 - pH + log10[BH */ В] и представляет собой то значение pH, при котором концентрации ионизированной (ВН) и не-
ионизированной (В) фракции основания равны между собой. Константу диссоциации можно использовать для вычис-
ления доли двух форм, присутствующих в растворе при различном pH.
120
Глава 6
анестетики, которые частично присутствуют'шкле-
точной мембране в виде незаряженных оснований
(рис. 6.5). Высокую практическую значимость име-
ет доля анестетика, присутствующая в неионизиро-
ванной форме (В), которая способна проникать
через соединительную ткань и клеточную мембра-
ну. Местные анестетики относительно неактивны
в тканях с кислой средой (например, в области
гнойного абсцесса). Вероятно, это вызвано сниже-
нием доли неионизированной фракции, а также бы-
стрым вымыванием анестетика вследствие обиль-
ной васкуляризации.
Факторы, усиливающие превращение свободно-
го основания (В) в активную форму (ВН+) в цито-
плазме, могут повышать диффузионный градиент
и поступление ВН в натриевые каналы, что ускоря-
ет наступление анестезии и углубляет ее. В экспе-
риментальных условиях СО2 быстро диффунди-
рует через клеточную мембрану и снижает внут-
риклеточный pH, способствуя превращению (В)
в активную форму (ВН+). Хотя насыщенные СО2
растворы местных анестетиков применялись для
ускорения развития анестезии и усиления интен-
сивности блокады, их клинические преимущества
вызывают сомнения. СО2 легко диффундирует
через клеточную мембрану, но быстро нейтрализу-
ется благодаря буферным свойствам внутрикле-
точных белков; кроме того, такие растворы неста-
бильны, местные анестетики могут выпадать в оса-
док, а добавленные к раствору вазоконстрикторы
легко гидролизуются. По этим причинам насыщен-
ные СО2 растворы местных анестетиков не получи-
ли широкого распространения в клинической прак-
тике. Другие средства (например, бикарбонат
натрия и различные декстраны) также добавляют-
ся к растворам местных анестетиков для повыше-
ния концентрации неионизированной фракции
(В), что увеличивает интенсивность и продолжи-
тельность анестезии.
ДЕЙСТВИЕ НА РАЗЛИЧНЫЕ СЕНСОРНЫЕ
МОДАЛЬНОСТИ
После введения анестетика сначала исчезает бо-
левая чувствительность, затем температурная, так-
тильная, проприоцептивная, и в последнюю оче-
редь развивается парез. Эти различия могут быть
обусловлены диаметром нервных волокон, прово-
дящих импульсы разной модальности: тонкие во-
локна более чувствительны к местным анестети-
кам, чем толстые. Безмиелиновые волокна имеют
относительно большую площадь контакта с мест-
ным анестетиком из-за отсутствия миелиновой
оболочки. Напротив, миелиновые волокна имеют
Шйисительно малую площадь поверхности и чувст-
вительны к действию местных анестетиков только
в области перехватов Ранвье. Кроме того, для пре-
рывания проведения импульса по миелинизиро-
ванному нерву требуется блокада 2-3 последова-
тельных перехватов Ранвье. Поскольку расстояние
между перехватами удлиняется по мере увеличе-
ния диаметра нервного волокна, толстые миелино-
вые волокна менее чувствительны к местным ане-
стетикам, чем тонкие миелиновые, которые, в свою
очередь, менее чувствительны, чем немиелиновые
волокна. После введения местного анестетика
вначале наступает блокада тонких немиелиновых
волокон группы С (болевая чувствительность) и тон-
ких миелиновых волокон группы А8 (болевая и тем-
пературная чувствительность), затем толстых
миелиновых волокон групп Ар и Ау (тактильная
и проприоцептивная чувствительность), и нако-
нец — толстых миелиновых волокон группы Аа
(управление скелетными мышцами).
Тем не менее эта концепция не может адекватно
объяснить несколько клинических наблюдений.
Например, миелиновые волокна группы А8, про-
водящие ощущение первой (быстрой) боли, блоки-
руются местными анестетиками раньше, чем неко-
торые немиелиновые С-волокна. Этот феномен
может отражать особенности анатомического рас-
пределения в нервном стволе волокон разных
групп и их доступность для местных анестетиков.
Различное воздействие местных анестетиков на
чувствительные и двигательные функции объясня-
ется также физико-химическими факторами. Так,
благодаря своей высокой жирорастворимости бу-
пивакаин и ропивакаин (в низкой концентрации)
легко блокируют тонкие немиелиновые волокна
группы С. С другой стороны, эти же анестетики
из-за относительно высокой константы диссоциа-
ции рХа (из-за чего невелика неионизированная
фракция препарата, способная проникать через
оболочки и мембраны) не могут быстро и эффек-
тивно блокировать толстые миелиновые Аа-волок-
на и вызвать паралич. Иными словами, бупивакаин
и ропивакаин обладают оптимальными физико-хи-
мическими свойствами для раздельной чувстви-
тельной и двигательной блокады.
ПРЕПАРАТЫ МЕСТНЫХ
АНЕСТЕТИКОВ
Большинство местных анестетиков — это осно-
вания, практически нерастворимые в воде. Хорошо
растворимые в воде соли местных анестетиков
(гидрохлориды) обычно разводят в 0,9% NaCl,
МЕСТНЫЕ АНЕСТЕТИКИ
121
medlit.ucoz.ru
в результате чего образуется раствор с низким
pH 4,0-5,5. Препараты, к которым добавлен адре-
налин, содержат восстанавливающий агент (напри-
мер, метабисульфит натрия, чтобы предотвратить
окисление и повысить стабильность вазокон-
стриктора в растворе. Некоторые препараты
(особенно во флаконах для многоразового исполь-
зования) содержат консервант и фунгицид (напри-
мер, метил «-гидроксибензоат). Большинство рас-
творов местных анестетиков очень устойчивы и име-
ют срок годности более двух лет.
Многие местные анестетики вызывают вазоди-
латацию и поэтому быстро всасываются из зоны
инъекции. Для повышения мощности и увеличения
продолжительности действия к местным анестети-
кам часто добавляют вазоконстрикторы. Вазокон-
стрикторы также уменьшают системную токсич-
ность анестетиков и увеличивают их терапевтиче-
ский индекс за счет замедления всасывания
(которое в основном зависит от местного кровото-
ка). Однако эффективность вазоконстрикторов
очень непостоянна. При инфильтрационной и про-
водниковой анестезии вазоконстрикторы в боль-
шинстве случаев удлиняют и усиливают анестезию;
с другой стороны, они могут не оказывать значимо-
го воздействия на продолжительность эпидураль-
ной или спинномозговой анестезии.
Чаще всего в качестве вазоконстриктора приме-
няют адреналин. Его добавляют к растворам мест-
ных анестетиков в концентрации от 12,5 мкг/мл
(Г.80 ООО) до 3,3 мкг/мл (Г. 300 000). Раньше в каче-
стве вазоконстриктора применяли норадреналин
(1:80 000), но в настоящее время из-за выраженного
прессорного эффекта он не используется. Некото-
рые растворы прилокаина (3%), предназначенные
для стоматологической практики, содержат вазо-
констриктор фелипрессин (0,03 МЕ/мл). Фели-
прессин — это некатехоламиновый вазоконстрик-
тор, по своему химическому строению напоминаю-
щий вазопрессин — гормон задней доли гипофиза.
Этот синтетический октапептид влияет исклю-
чительно на периферические кровеносные сосуды
и не оказывает воздействия на сердце. Фелипрес-
син вызывает менее выраженную вазоконстрик-
цию, чем адреналин, но его можно применять при
ИБС или при других состояниях, когда противопо-
казаны катехоламины. В качестве вазоконстрикто-
ров использовались и другие препараты (например,
фенилэфрин), но оказалось, что адреналин гораздо
эффективнее, чем фенилэфрин и норадреналин,
замедляет всасывание большинства местных ане-
стетиков.
ХИМИЧЕСКОЕ СТРОЕНИЕ И
ФИЗИКО-ХИМИЧЕСКИЕ
СВОЙСТВА
Все местные анестетики имеют сходное хи-
мическое строение. Они состоят из гидрофобной
(липофильной) ароматической группы (Ri),
промежуточной эфирной (-СО.О-) или амидной
(-NH.CO-) цепи и гидрофильной вторичной или
третичной аминогруппы (R2). В зависимости от
типа промежуточной цепи местные анестетики де-
лят на эфиры и амиды (рис. 6.6). Кроме того, тип
промежуточной цепи влияет на продолжитель-
ность действия местных анестетиков.
К местным анестетикам группы эфиров относят:
1. Кокаин
2. Прокаин
3. Хлорпрокаин
4. Тетракаин (аметокаин).
Местные анестетики группы амидов включают:
1. Лидокаин
2. Прилокаин
3. Мепивакаин
4. Бупивакаин
5. Ропивакаин
6. Этидокаин
Между этими двумя группами местных анесте-
тиков существуют важные различия. Эфиры отно-
сительно нестабильны в растворах и в организме
быстро гидролизуются холинэстеразой и некото-
рыми другими эстеразами. Одним из продуктов
гидролиза является парааминобензойная кислота,
которая иногда вызывает аллергические реакции.
Напротив, амиды относительно стабильны в рас-
творе и медленно расщепляются амидазами печени.
Кроме того, реакции гиперчувствительности к ане-
стетикам группы амидов развиваются крайне ред-
ко. Единственным анестетиком группы эфиров, ко-
торый в настоящее время применяется в Велико-
британии, является тетракаин (аметокаин).
Химическое строение местных анестетиков оп-
ределяет их физико-химические характеристики и
клинические свойства. Основные различия мест-
ных анестетиков заключаются в (а) растворимости
в липидах, (б) связывании с белками тканей и (в)
константе диссоциации рКа (таблица 6.1).
Существует тесная корреляция между раство-
римостью анестетика в липидах и его мощностью, осо-
бенно in vitro. Она отражает способность проникать
через периневральные ткани и клеточные мем-
браны нейронов, с тем чтобы достичь точки прило-
жения действия в аксоплазме. Например, в боль-
шинстве клинических ситуаций бупивакаин прибли-
122
Глава 6
Рис. 6.6. Химиче-
ское строение мест-
ных анестетиков.
Хиральные атомы
углерода (асиммет-
ричные центры) отме-
чены звездочками.
Прилокаин, мепива-
каин, бупивакаин
и этидокаин обычно
применяют в виде ра-
цемических смесей
R- и S-стереоизо-
меров. Ропивакаин
является 8(-)-энан-
тиомером
medlit.ucoz.ru
Липофильная ароматическая ’Промежуточная | Гидрофильная
Прокаин
Тетракаин
(аметокаин)
Лидокаин
Прилокаин
Этидокаин
Мепивакаин
Ропивакаин
Бупивакаин
группа • цепь I аминогруппа
/тл! i №
ын2—(( ) H-CX>.0.CHj.CH2-|-N
1 < X...
' ' I I CjHj
Табл. 6.1. Физико-химические свойства и фармакологические эффекты местных анестетиков
рКа при 25° С Относительная растворимость влипидах Относит, мощность Связывание с белками (%) Начало действия Продолжи- тельность действия Клиническое примене- ние Свойства
Прокаин 8,9 ' 1 1 6 Медленное Короткое Ограниченное Спазм сосудов Диагностич. пооиедуоы Вазодилатация Аллергия
Тетракаин (аметокаин) 8,5 200 8 75 Медленное Длительное Поверхностная анестезия Спинномозговая анесте- зия Системная ин- токсикация
Лидокаин 7,7 150 2 65 Быстрое Среднее Инфильтрационная ане- стезия Проводниковая анестезия Эпидуральная анестезия Внутривенная регионар- ная анестезия Разнообразные Умеренная вазо- дилатация
Прилокаин 7,7 50 2 55 Быстрое Среднее Инфильтрационная ане- стезия Проводниковая анестезия Внутривенная регионар- ная анестезия Метгемоглоби- q немия С Низкая систем- — ная токсичность Г
Мепивакаин 7,6 50 2 78 Быстрое Среднее Инфильтрационная ане- стезия Пооводниковая анестезия Аналогичны ли- С |\ докаину
Ропивакаин 8,1 400 6 94 Промежу- точное Длительное Инфильтрационная ане- стезия Проводниковая анестезия Эпидуральная анестезия Раздельная чув- ствительная и двигательная блокада Умеренная двига- тельная блокада
Бупивака- ин 8,1 1000 8 95 Промежу- точное Длительное Инфильтрационная анестезия Проводниковая ане- стезия Эпидуральная и спин- номозговая анестезия Раздельная чувствительная и двигательная блокада
Этидока- ин 7,7 5000 6 96 Быстрое Длительное И нфил ьтрацион ная анестезия Проводниковая анестезия Эпидуральная анесте- зия Глубокая дви- гательная бло- када
124
Г лава 6
зительно в четыре раза мощнее лидокаина
их разной растворимости в липидах (табл. 6.1).
Связывание с белками тканей влияет на продол-
жительность действия местных анестетиков.
Например, действие прокаина (слабо связывается
с белками тканей) в большинстве случаев непро-
должительно; напротив, бупивакаин и ропивакаин
в значительной степени связываются с белками и
обладают большей продолжительностью действия.
Лидокаин, мепивакаин и прилокаин умеренно свя-
зываются белками и имеют среднюю продолжи-
тельность действия (таблица 6.1).
Константа диссоциации (рКа) местных анестети-
ков является наиболее важным фактором, влияю-
щим на скорость наступления эффекта. От рХа за-
висит доля неионизированной формы местного
анестетика, которая способна проникать через тка-
невые барьеры в точку приложения действия
(см. выше). Низкие значения рХа коррелируют с бы-
стрым развитием блокады, поскольку при pH 7,4
значительная доля местного анестетика находится
в неионизированной форме. Напротив, при высо-
ких значениях рКа блокада развивается медленнее,
поскольку при pH 7,4 в неионизированной форме
оснований находится меньшая доля анестетика.
Например, у лидокаина и прилокаина рКа со -
ставляет 7,7; это означает, что 33 % местного ане-
стетика при pH 7,4 находится в растворе в не-
ионизированной форме В и способно проникать
через оболочки нервов (расчет проведен в соответ-
ствии с формулой, приведенной в разделе «Меха-
низм действия»). Напротив, бупивакаин и ропива-
каин имеют рХа = 8,1; при pH 7,4 только 17% этих
препаратов находится в растворе в неионизирован-
ной форме В и способно к диффузии. Именно этим
объясняется более быстрое действие лидокаина,
прилокаина и этидокаина по сравнению с бупи-
вакаином и ропивакаином (таблица 6.1). Тем не
менее, на скорость развития блокады и продолжи-
тельность латентного периода могут влиять и дру-
гие факторы (например, доза и конечная концентра-
ция анестетика в тканях).
Большинство анестетиков группы эфиров (про-
каин, хлорпрокаин и тетракаин) не обладают свой-
ствами стереоизомерии, а большинство анестети-
ков группы амидов (за исключением лидокаина),
напротив, имеют хиральный центр. В клинической
практике некоторые хиральные анестетики приме-
няются в виде рацемических смесей, а другие (на-
пример, S-ропивакаин и S-бупивакаин) — в виде
чистых энантиомеров. Хотя S- и R-энантиомеры
имеют примерно одинаковую анестетическую актив-
ность, (5)-энантиомеры обладают явным преимуще-
ством в некоторых других важных отношениях: на-
UQ^^efUnp
одолжительность их действия дольше
(они вызывают усиленную вазоконстрикцию),
риск кардиотоксических эффектов ниже, двига-
тельная блокада слабее и менее продолжительна.
Энантиомеры также отличаются метаболизмом и
фармакокинетическими свойствами.
ВСАСЫВАНИЕ МЕСТНЫХ
АНЕСТЕТИКОВ В СИСТЕМНЫЙ
КРОВОТОК
Значительная доля местных анестетиков после
инъекции поступает в системный кровоток. Коли-
чество всосавшегося анестетика и его пиковая кон-
центрация в плазме зависят от дозы препарата
и присутствия в растворе вазоконстриктора (осо-
бенно при инфильтрационной и проводниковой
анестезии). Имеет значение также методика анесте-
зии: так, концентрация анестетика в плазме выше
после межреберной и каудальной анестезии, чем
после эпидуральной анестезии или блокады плече-
вого сплетения. После введения 100 мг лидокаина
взрослому человеку пиковая концентрация в ве-
нозной крови может составлять 1,5 мкг/мл (межре-
берная блокада), 1,2 мкг/мл (каудальная и пара-
цервикальная блокада), 1,0 мкг/мл (эпидуральная
анестезия), 0,6 мкг/мл (блокада плечевого сплете-
ния) и 0,4 мкг/мл (спинномозговая анестезия).
Следовательно, одна и та же доза местного анесте-
тика может быть сопряжена с различным риском
токсического воздействия в зависимости от мето-
дики анестезии. Такой разброс концентраций в ос-
новном зависит от степени васкуляризации тканей,
хотя определенную роль играют и другие факторы
(например, захват анестетика липидами тканей).
Строгое соблюдение предписаний относительно
допустимой дозы анестетика нивелирует потенци-
альные различия системной токсичности, обуслов-
ленные методикой анестезии.
Многие местные анестетики активно всасыва-
ются через слизистые. Скорость всасывания тесно
коррелирует с площадью обработанной анестети-
ком поверхности (например, скорость всасывания
очень высока после распыления местного анестети-
ка в дыхательных путях). Кроме того, на скорость
всасывания влияет присущее местным анестетикам
воздействие на тонус гладких мышц сосудов. Так,
кокаин блокирует обратный захват катехоламинов
нейронами и подавляет активность моноаминокси-
дазы; это приводит к вазоконстрикции и замедляет
всасывание. Большинство других местных анесте-
тиков оказывает двухфазное действие на гладкую
мускулатуру сосудов. В клинических концентрациях
МЕСТНЫЕ АНЕСТЕТИКИ
125
все они вызывают ту или иную степень вазЬцияата1-1
ции (в порядке убывания: прокаин > прилокаин >
лидокаин > мепивакаин > бупивакаин > ропива-
кин). Прокаин, хлорпрокаин и родственные им ане-
стетики группы эфиров являются мощными вазо-
дилататорами и быстро всасываются после инъек-
ции; они быстро расщепляются в тканях и плазме
под действием эстераз, особенно псевдохолинэсте-
разы. Прокаин быстро инактивируется фермента-
ми слизистых и поэтому практически не вызывает
поверхностной анестезии.
(Б)-изомеры прилокаина, мепивакаина и бупи-
вакаина вызывают менее выраженную вазодилата-
цию, чем (К)-энантиомеры; следовательно,
(5)-формы хиральных местных анестетиков дейст-
вуют дольше, чем их оптические антиподы.
СВЯЗЫВАНИЕ С БЕЛКАМИ
ПЛАЗМЫ И ПРОНИКНОВЕНИЕ
ЧЕРЕЗ ПЛАЦЕНТУ
Связывание местных анестетиков с белками
плазмы влияет на их фармакокинетику и фармако-
динамику. Анестетики группы эфиров связывают-
ся с белками плазмы цезначительно (< 5-10%).
Напротив, анестетики группы амидов активно свя-
зываются с белками, преимущественно с кислым
а]-гликопротеидом (в порядке убывания: бупива-
каин > ропивакаин > мепивакаин > лидокаин > при-
локаин). Степень связывания с белками плазмы
колеблется от 55% до 95%; обычно это связывание
обратимое и не ограничивает захват местных ане-
стетиков большинством тканей и органов. На свя-
зывание анестетиков с белками плазмы значитель-
но влияют физиологические и патологические из-
менения концентрации кислого аг гликопротеида.
Уровень этого белка повышается при онкологиче-
ских заболеваниях, хирургических вмешательст-
вах, травмах, инфаркте миокарда; в этих случаях
связывание местных анестетиков с белками плазмы
возрастает, а концентрация свободной (несвязан-
ной) фракции уменьшается.
Связывание с белками плазмы влияет на про-
никновение местных анестетиков через плацентар-
ный барьер. Если местный анестетик интенсивно
связывается с белками, то отношение его концен-
траций в пупочной вене и материнской крови будет
низким. Так, для бупивакаина и ропивакаина это
отношение равно 0,2, а для лидокаина и прилокаи-
на — 0,5. Эти показатели не всегда отражают отно-
сительную безопасность применения местных ане-
стетиков во время беременности и родов, поскольку
не учитывают различное связывание анестетика с
ЦеМкГМ- гликопротеидом в крови плода и мате-
ри. Тем не менее считается, что бупивакаин и ропи-
вакаин в меньшей степени проникают через плацен-
ту, чем лидокаин и прилокаин, что имеет очевидные
преимущества во время беременности и родов.
ФАРМАКОКИНЕТИКА
После всасывания анестетика из зоны инъекции
его концентрация в плазме зависит от скорости рас-
пределения в тканях и элиминации. Замедленное
всасывание местного анестетика может препятст-
вовать точному определению его фармакокинети-
ческих параметров. После в/в введения концентра-
ция местного анестетика в плазме снижается по би-
экспоненциальному типу. Сначала следует фаза
быстрого распределения (TJ/2 «1-3 мин), обуслов-
ленная быстрым захватом анестетика хорошо
кровоснабжаемыми органами (легкими, печенью,
почками и скелетными мышцами). Затем концен-
трация снижается медленнее; эта фаза отражает ме-
таболизм и экскрецию препарата.
Конечный Т1/2 большинства анестетиков группы
эфиров относительно короток (приблизительно
10 мин) из-за быстрого гидролиза под действием
псевдохолинэстеразы. Напротив, конечный Т1/2
анестетиков группы амидов колеблется от 100 мин
(лидокаин) до 200 мин (бупивакаин). Их объем
распределения намного превышает общий объем
воды в организме, в то время как клиренс приблизи-
тельно соответствует печеночному кровотоку
(табл. 6.2). Патологические состояния могут изме-
нять фармакокинетические свойства препаратов:
сердечно-сосудистые заболевания и цирроз печени
снижают клиренс, объем распределения и конеч-
ный Т]/2 местных анестетиков. У новорожденных
клиренс местных анестетиков понижен, а Тш, на-
оборот, повышен.
МЕТАБОЛИЗМ И ЭЛИМИНАЦИЯ
Метаболизм местных анестетиков определяется
их химическим строением: эфиры и амиды метабо-
лизируются разными способами и в разных тканях.
Анестетики группы эфиров расщепляются псев-
дохолинэстеразой, а также другими эстеразами.
Только небольшая доля анестетиков группы эфи-
ра элиминируется с мочой в неизмененном виде.
Например, прокаин активно гидролизуется с обра-
зованием парааминобензойной кислоты и диэти-
ламиноэтанола; последний под действием ал-
когольдегидрогеназы в дальнейшем превращается
в диэтилглицин. Аналогично метаболизируется
большинство других анестетиков группы эфиров
126
Глава 6
Табл. 6.2. Фармакокинетика и
Т1/2 (мин) Клиренс (л/мин) Объем распределения (л) Метаболиты
Кокаин 48 2,2 140 Норкокаин Экгонин Бензоилированные производные
Прокаин 8 4,2 48 Парааминобензойная кислота Диэтиламиноэтанол
Тетракаин (аметокаин) 15 3,3 70 Бутиламинобензойная кислота Диметиламиноэтанол
Лидокаин 100 1,0 91 Моноэтилглицинксилидид /У-ЭТИЛГЛИЦИН 2,6-ксилидин 4- гидрокси - 2,6- кси л и дин
Прилокаин 100 2,4 191 о-Толуидин /У-пропиламин
Мепивакаин 115 0,8 84 3-гидроксимепивакаин (и глюку- ронид) 4-гидроксимепивакаин
Ропивакаин 110 0,5 47 3-гидроксиропивакаин (и глюку- ронид) 4-гидроксиропивакаин
Бупивакаин 200 0,6 73 Пипеколиновая кислота Пипеколилксилидид
Этидокаин 160 1,1 134 Дезалкилированные метаболиты 2,6-ксилидиды
(хлорпрокаин и тетракаин). Кокаин, напротив, от-
носительно устойчив к гидролитическому действию
холинэстеразы. Он подвергается активному метабо-
лизму в печени, и только следовые количества
(приблизительно 1%) выделяются в неизмененном
виде. Исследования с радиомеченным кокаином по-
казали, что он превращается в норкокаин, экгонин и
их бензоилированные аналоги. Некоторые из них
оказывают стимулирующее действие на ЦНС.
В отличие от большинства анестетиков-эфиров,
анестетики группы амидов активно расщепляются
ферментами печени (в основном амидазами цито-
плазмы и гладкого эндоплазматического ретику-
лума).
Лидокаин почти полностью метаболизируется
в печени, и только менее 10% выделяется в неизме-
ненном виде с мочой. Вначале лидокаин дезалкили-
руется до ацетальдегида и моноэтилглицинкси-
лидида, который затем подвергается гидролизу
до У-этилглицина и 2,6-ксилидина. Последнее
соединение впоследствии превращается в 4-гидро-
кси-2,6-ксилидин и выделяется с мочой. Некоторые
метаболиты лидокаина имеют активность местно-
го анестетика, другие обладают проконвульсивным
действием (моноэтилглицинксилидид). Глицин-
ксилидид (метаболит, образующийся в неболь-
шом количестве) угнетает ЦНС, имеет сравни-
тельно большой Ti/2, и для его элиминации может
потребоваться несколько дней. Клиренс лидокаина
составляет приблизительно 70-80% нормального
печеночного кровотока, и после приема внутрь в
системный кровоток попадают только следовые ко-
личества лидокаина (т. е. резко выражен эффект
первого прохождения).
Прилокаин разрушается быстрее других анесте-
тиков группы амидов. Хотя его основной метабо-
лизм осуществляется в печени, он также расщепля-
ется в почках и легких. Основными метаболитами
являются 7V-пропил амин и о-толуидин; последний
может вызывать метгемоглобинемию и цианоз, что
иногда случается спустя 5-6 ч после введения боль-
ших доз прилокаина. Метгемоглобинемия обычно
разрешается самостоятельно, но иногда требу-
ет применения восстанавливающих препаратов
(аскорбиновой кислоты или метиленовой синьки).
Прилокаин применяют в виде рацемической смеси
двух стереоизомеров; R-прилокаин метаболизиру-
ется быстрее S-прилокаина и повышает концентра-
цию о-толуидина в крови, что приводит к увеличе-
нию уровня метгемоглобина.
МЕСТНЫЕ АНЕСТЕТИКИ
127
Мепивакаин быстро расщепляется в
образованием не менее трех гидроксилированных
метаболитов; большая часть мепивакаина выделя-
ется с мочой в виде 3-гидроксимепивакаина или
4-гидроксимепивакаина.
Бупивакаин отличается сравнительно низким
печеночным клиренсом и метаболизируется в пече-
ни медленно. Только 6% выделяется с мочой в неиз-
мененном виде. Препарат в основном гидролизуется
до пипеколиновой кислоты, но около 5% превраща-
ется в пипеколилксилидид (дезалкилированный
метаболит). Оба метаболита выводятся с мочой.
Печеночный клиренс ропивакаина также отно-
сительно низок (приблизительно 30% печеночного
кровотока). Метаболизм ропивакаина преимуще-
ственно осуществляется в печени, и только 1-2%
препарата выводится с мочой в неизмененном виде.
Ропивакаин в основном превращается в 3-гидро-
ксиропивакаин, который затем конъюгируется и
удаляется с мочой. Также образуются небольшие
количества 4-гидроксиропивакаина и других мета-
болитов, которые также выводятся с мочой.
Этидокаин частично расщепляется до дезалки-
лированных производных под действием печеноч-
ных оксидаз, после чего гидролизуется с образова-
нием 2,6-ксилидина и гидроксилированных произ-
водных и экскретируется с мочой.
ОТДЕЛЬНЫЕ ЭФФЕКТЫ
КОКАИН
Кокаин вызывает тахикардию и аритмии вслед-
ствие центрального и периферического действия.
В ЦНС кокаин повышает активность нейронов
в симпатических центрах гипоталамуса и продол-
говатого мозга, а на периферии вызывает вазокон-
стрикцию, расширение зрачков и другие симпато-
миметические эффекты. Оба эффекта обусловле-
ны подавлением обратного захвата катехоламинов
в окончаниях центральных и периферических нер-
вов. Этот механизм обеспечивает удаление катехо-
ламинов из внеклеточной жидкости (глава 13).
Следовательно, эффекты эндогенных и экзогенных
катехоламинов усиливаются. Кокаин вызывает за-
висимость и патологическое пристрастие.
ПРОКАИН
Прокаин уменьшает возбудимость миокарда
предсердий и желудочков, и раньше он использо-
вался в качестве антиаритмического средства.
К сожалению, он быстро гидролизуется псевдохо-
линэстеразой плазмы. Прокаинамид, близкий
аналог прокаина, не расщепляется холинэстеразой
it. UCG&aRUi большей
продолжительностью дейст-
вия; в недавнем прошлом этот препарат широко
применяли для лечения желудочковых и наджелу-
дочковых аритмий (глава 15). Прокаин является
мощным вазодилататором и используется для ле-
чения сосудистого спазма после непреднамерен-
ных внутриартериальных инъекций, травм и хи-
рургических вмешательств. Другие местные ане-
стетики вызывают вазодилатацию в меньшей
степени. Прокаин также использовали для кардио-
плегии во время хирургических операций с искус-
ственным кровообращением.
ЛИДОКАИН
Воздействие лидокаина и других местных ане-
стетиков на натриевые каналы миокарда и проводя-
щей системы сердца аналогично их воздействию
на периферические нервы (см. выше). Блокада
натриевых каналов дозозависимо замедляет медлен-
ную диастолическую деполяризацию кардиомиоци-
тов. Лидокаин широко применяется для лечения
желудочковых аритмий при инфаркте миокарда
и во время общей анестезии (глава 15).
БУПИВАКАИН
На натриевые каналы сердца бупивакаин оказы-
вает такое же действие, как и лидокаин. Однако он
также влияет на кальциевые и калиевые каналы
и сильнее других местных анестетиков связывается
тканями миокарда. Поэтому в большой дозе и высо-
кой концентрации бупивакаин оказывает кардио-
токсичное действие.
ПОБОЧНОЕ ДЕЙСТВИЕ МЕСТНЫХ
АНЕСТЕТИКОВ
ПОБОЧНОЕ ДЕЙСТВИЕ, ОБУСЛОВЛЕННОЕ
ПЕРЕДОЗИРОВКОЙ
Местные анестетики и их метаболиты являются
слабыми основаниями и относительно легко пре-
одолевают гематоэнцефалический барьер. Тяжелое
и иногда фатальное токсическое воздействие на
ЦНС может наблюдаться при значительной пере-
дозировке местных анестетиков; токсическое воз-
действие на сердечно-сосудистую систему случает-
ся, когда большие дозы анестетиков непреднаме-
ренно попадают в системный кровоток и достигают
сердца.
ЦНС
Для местных анестетиков характерно двухфаз-
ное воздействие на ЦНС: если их вводить не слиш-
ком быстро, то вначале появляются ранние симпто-
мы, потом — поздние, более тяжелые. Низкие дозы
128
Глава 6
лидокаина (2-4 мг/кг) обладают противос^доРШ-.
ным эффектом и используются для лечения эпи-
лептического статуса, но после введения больших
доз развивается возбуждение ЦНС. При увеличе-
нии концентрации местного анестетика в плазме
наблюдаются онемение языка и полости рта, голо-
вокружение, нарушения зрения, невнятная речь,
мышечные подергивания и тремор, беспокойство и
спутанность речи. Если концентрация местного
анестетика в плазме превышает критический уро-
вень (9 мкг/мл для лидокаина и 2 мкг/мл для бупи-
вакаина), то может развиться большой судорожный
припадок. Судорожный порог (критический уро-
вень, по достижении которого возникают судороги)
изменяется под влиянием ацидоза, гипоксии и дру-
гих лекарственных препаратов, воздействующих на
ЦНС. Некоторые местные анестетики (например,
прокаин) практически не оказывают проконвуль-
сантного действия. Возбуждающие эффекты мест-
ных анестетиков, вероятно, обусловлены селектив-
ным угнетением тормозящих корковых проводя-
щих путей; они сменяются симптомами угнетения
активности коры больших полушарий и продолго-
ватого мозга (комой и апноэ).
Лечение судорог состоит в поддержании адекват-
ной вентиляции и оксигенации, а также в примене-
нии противосудорожных препаратов. Диазепам
(10-20 мг в/в; при необходимости повторить) обычно
не усиливает депрессию ЦНС, развивающуюся при
передозировке местного анестетика. Альтернативным
препаратом является тиопентал (150-250 мг в/в).
Случайное введение больших объемов местного
анестетика в ЦСЖ во время эпидуральной анесте-
зии или паравертебральной блокады может привес-
ти к «тотальной спинальной» анестезии. Развиваю-
щаяся двигательная блокада и угнетение продолго-
ватого мозга приводят к полному параличу дыхания,
а вегетативная блокада вызывает артериальную ги-
потонию. Проводят ИВЛ и поддерживают кровооб-
ращение, в том числе с помощью вазопрессоров.
СЕРДЕЧНО-СОСУДИСТАЯ СИСТЕМА
Передозировка местных анестетиков может вы-
зывать глубокую артериальную гипотонию, бради-
кардию, брадиаритмию и даже остановку сердца;
обычно сердечно-.сосудистые осложнения развива-
ются после появления неврологических симпто-
мов. Наиболее выраженный кардиотоксический
эффект наблюдается при высоких концентрациях
бупивакаина в крови. Помимо блокады натриевых
каналов, бупивакаин воздействует на кальциевые и
калиевые каналы в кардиомиоцитах; кроме того, по
сравнению с другими местными анестетиками, он
связывается с миокардом в большей степени.
иССЖуГААакаин и другие местные анестетики оказы-
вают непосредственное воздействие на проводящую
систему сердца и влияют на центры продолговатого
мозга. Подавляется сократимость и проводимость
миокарда с расширением комплексов QRS и откло-
нением сегмента ST. Высокие концентрации бупива-
каина предрасполагают к развитию феномена reentry
(повторный вход волны возбуждения) и желудоч-
ковым аритмиям, которые усугубляются гипоксией,
ацидозом и гиперкалиемией. Аритмии и брадикар-
дию можно устранить атропином (1,2-1,8 мг), а для
увеличения О ЦК необходима инфузия коллоидных
или кристаллоидных растворов.
В Великобритании зарегистрировано пять
смертельных исходов, обусловленных кардиоток-
сичным действием бупивакаина после проведе-
ния внутривенной регионарной анестезии по Биру.
В США отмечено несколько случаев материнской
смерти после случайного в/в введения бупивакаи-
на во время эпидуральной анестезии по поводу ке-
сарева сечения. В настоящее время считается, что
(З)-энантиомеры местных анестетиков уменьшают
риск кардиодепрессии и кардиотоксичности; так,
ропивакаин (S-изомер) и левобупивакаин (S-бупи-
вакаин) имеют значительные преимущества по
сравнению с рацемическим бупивакаином.
Если для обезболивания родов выполняется
парацервикальная блокада, из-за быстрого всасы-
вания местных анестетиков у плода могут разви-
ваться брадикардия и другие симптомы дистресса.
ПОБОЧНОЕ ДЕЙСТВИЕ, ОБУСЛОВЛЕННОЕ
МЕТОДИКОЙ ПРИМЕНЕНИЯ
Множественная межреберная блокада вызыва-
ет дыхательную недостаточность вследствие неиз-
бежного паралича некоторых двигательных нервов.
Во время спинномозговой анестезии в результа-
те симпатической блокады часто развивается арте-
риальная гипотония различной выраженности.
Эпидуральная анестезия во время родов усугубля-
ет артериальную гипотонию, обусловленную сдав-
лением нижней полой вены. Профилактические
мероприятия (например, предварительная ин-
фузия коллоидных или кристаллоидных раство-
ров) обычно сводят эти осложнения к минимуму.
Может потребоваться симптоматическое лечение
(эфедрин в/в).
ПОБОЧНОЕ ДЕЙСТВИЕ, ОБУСЛОВЛЕННОЕ
ДОБАВЛЕНИЕМ ВАЗОКОНСТРИКТОРА
Побочные эффекты вазоконстрикторов обычно
развиваются при их случайном попадании в сис-
темный кровоток. Добавление симпатомиметиков
к раствору местного анестетика всегда сопряжено
МЕСТНЫЕ АНЕСТЕТИКИ
129
с риском аритмий и артериальной гипертоййЮбИЙ
сами местные анестетики в определенной степени
защищают сердце от этих осложнений.
Вазоконстрикторы противопоказаны при бло-
каде пальцевых нервов, поскольку резко развиваю-
щаяся ишемия может привести к гангрене. Раство-
ры местных анестетиков с добавленным вазоконст-
риктором нельзя вводить в/в.
ОТДЕЛЬНЫЕ ПОБОЧНЫЕ ЭФФЕКТЫ
Аллергические реакции на современные мест-
ные анестетики развиваются очень редко. Чаще
всего наблюдаются кожные реакции при повтор-
ном использовании анестетиков группы эфиров;
анафилактические реакции возникают значитель-
но реже. Вероятно, в роли гаптена выступает мета-
болит — парааминобензойная кислота, которая ин-
дуцирует иммунологическую реакцию. Аллергиче-
ские реакции на амидные анестетики встречаются
еще реже. Не доказано существование перекрест-
ной сенсибилизации между эфирными и амидными
анестетиками. Аллергические реакции, возникаю-
щие при использовании многодозовых флаконов
с местными анестетиками, могут вызываться кон-
сервантами.
Прокаин и другие анестетики группы эфиров
гидролизуются с образованием парааминобензой-
ной кислоты, поэтому они являются антагонистами
сульфаниламидных препаратов. Следовательно,
одновременное применение местных анестетиков
группы эфиров и сульфаниламидов (например,
ко-тримоксазола) нежелательно.
Высокие дозы прилокаина (например, более
600 мг эпидурально) могут вызывать метгемогло-
бинемию вследствие накопления основного мета-
болита — о-толуидина и его гидроксилированных
производных. Особому риску подвергается плод,
поскольку его эритроциты не содержат достаточно-
го количества метгемоглобинредуктазы (фермента,
восстанавливающего метгемоглобин в гемогло-
бин). При необходимости немедленно применяют
метиленовую синьку (5 мг/кг), являющуюся эф-
фективным антидотом для матери и ребенка.
КЛИНИЧЕСКОЕ ПРИМЕНЕНИЕ
ПОВЕРХНОСТНАЯ АНЕСТЕЗИЯ
Местные анестетики можно использовать для
анестезии кожи, глаз, слизистых носа, рта, уха, тра-
хеобронхиального дерева и мочеполовых путей.
Препараты наносят непосредствено на кожу или
слизистые. Наиболее эффективными поверхност-
ными анестетиками являются кокаин, тетракаин
UGOsZoRUhn). лидокаин и прилокаин. Они харак-
теризуются быстрым началом (5-10 мин) и сред-
ней продолжительностью действия (30-60 мин).
Мощный вазоконстрикторный эффект кокаина де-
лает его использование особенно целесообразным
в тех случаях, когда требуется сочетание местной
анестезии и уменьшения кровоточивости тканей.
В настоящее время кокаин в основном применяют
для поверхностной анестезии при операциях в по-
лости носа. Используют водные растворы и пасты,
содержащие 4-10% кокаин, к которым иногда до-
бавляют адреналин в разведении 1:1000. Раньше
кокаин применяли для поверхностной анестезии
глаз, но его обезвоживающий эффект часто приво-
дил к изъязвлению роговицы, и на смену ему при-
шли другие препараты (например, тетракаин, обла-
дающий прекрасным поверхностным действием).
Лидокаин выпускают в виде водных растворов
(2-4%) или водорастворимых гелей, мазей, кремов
и спреев (2-10%). Эти препараты используют в ка-
честве глазных капель и для анестезии трахео-
бронхиального дерева перед интубацией трахеи
или бронхоскопическими исследованиями. Они
также применяются для анестезии уретры во вре-
мя диагностических урологических вмеша-
тельств. Поверхность слизистых обрабатывают
спреями перед ушиванием небольших ран или по-
сле эпизиотомии, а также перед выполнением ин-
фильтрационной анестезии в стоматологии. В хо-
рошо васкуляризированных зонах происходит по-
вышенное всасывание местных анестетиков через
слизистые оболочки, что в редких случаях может
приводить к тяжелым осложнениям.
Бензокаин (неионизированный эфир) входит в со-
став ушных капель для облегчения боли при среднем
отите, а также в состав различных мазей, кремов,
гелей и спреев, используемых в качестве симптома-
тических средств при мышечных судорогах, зуде
и болезненных трещинах.
Всасывание местных анестетиков через непо-
врежденную кожу обычно происходит медленно
и непредсказуемо, поэтому для достижения эф-
фекта требуется высокая концентрация анестети-
ка (например, 20% бензокаин или 40% лидокаин).
В последние годы (особенно у детей) для поверхно-
стной анестезии перед венепункцией широко ис-
пользуется эвтектическая смесь местных анестети-
ков (EMLA; eutectic mixture of local anaesthetics).
Кристаллические третичные основания лидокаина
и прилокаина плавятся и при температуре > 16°С
образуют масляный раствор (эвтектическая смесь).
Концентрация местного анестетика в капле смеси,
представляющей собой эмульсию «масло в воде»,
достаточно высока (приблизительно 80%) и обес-
130
Глава 6
печивает хорошую поверхностную анестези
тическая смесь содержит равные доли (по 25 мг/мл)
третичных оснований лидокаина и прилокаина,
имеет вид эмульсии и наносится на кожу как крем.
Препарат вызывает преходящее побледнение кожи
и эритему. Для хорошего контакта с кожей и разви-
тия анестезии обычно требуется наложение давя-
щей повязки не менее чем на 60 мин.
Для обеспечения поверхностной анестезии пе-
ред венепункцией также применяется 4% гель тет-
ракаина (аметокаина). Анестезия развивается не-
много быстрее, чем при использовании EMLA, про-
должается дольше (4-6 ч) и вызывает умеренную
вазодилатацию.
ИНФИЛЬТРАЦИОННАЯ АНЕСТЕЗИЯ
Инфильтрационную анестезию часто применя-
ется в стоматологической практике и при неболь-
ших хирургических вмешательствах. Непосредст-
венной точкой приложения являются немиелини-
зированные нервные окончания. Как правило,
используют анестетики группы амидов средней
продолжительности действия (лидокаин, прило-
каин и мепивакаин). Эти местные анестетики на-
чинают действовать почти сразу после подслизи-
стого или подкожного введения и обеспечивают
адекватное обезболивание более чем в 90% слу-
чаев. Продолжительность инфильтрационной
анестезии различна. Прокаин действует непродол-
жительно (15-30 мин), в то время как лидокаин
и прилокаин имеют среднюю продолжительность
действия (70-140 мин). Наиболее длительную ане-
стезию обеспечивают бупивакаин и ропивакаин
(приблизительно 200 мин). Добавление адренали-
на (1:200 000,5 мкг/мл) повышает качество и про-
должительность анестезии. Остаточная анестезия
сохраняется дольше после подкожного введения
(4-7 ч), чем после введения под слизистую оболоч-
ку (1-3 ч); вероятно, это объясняется различным
кровоснабжением и скоростью всасывания.
ПРОВОДНИКОВАЯ АНЕСТЕЗИЯ
Проводниковой анестезией называют введение
местного анестетика непосредственно в область нер-
ва или сплетения — как небольшого и относительно
поверхностно расположенного (например, локтево-
го, лучевого или межреберного нерва), так и крупно-
го, глубоко расположенного нерва или сплетения с
обширным дерматомным распределением (напри-
мер, седалищного нерва или плечевого сплетения).
В настоящее время для проводниковой анесте-
зии используют только анестетики группы амидов.
Лидокаин и прилокаин обладают средней продол-
кЮбЗйьйЬктью действия, а бупивакаин и ропивака-
ин вызывают длительную анестезию. Для провод-
никовой анестезии часто применяют растворы, со-
держащие вазоконстрикторы, и продолжительность
анестезии главным образом определяется общей
дозой, а не объемом или концентрацией препарата.
Если анестетики группы амидов применяют для
блокады мелких нервов, то их действие развивается
относительно быстро (в течение 3-6 мин). Продол-
жительность анестезии различна: лидокаин и при-
локаин действуют 1-2 ч (средняя продолжитель-
ность действия), в то время как бупивакаин и ропи-
вакаин — 2-6 ч (длительное действие). Действие
пролонгируется повышением дозы анестетика или
добавлением вазоконстриктора. При блокаде мел-
ких нервов адреналин в разведении 1:200 000 про-
дляет действие лидокаина с 1-2 до 4-5 ч. Смеси
местных анестетиков (например, прилокаина и бу-
пивакаина или лидокаина и бупивакаина) обычно
используют для того, чтобы обеспечить сочетание
быстрого начала и высокой продолжительности
действия.
При блокаде крупных нервных стволов скорость
наступления анестезии может колебаться в широ-
ких пределах; она в основном зависит от анатомиче-
ских факторов, ограничивающих поступление ме-
стных анестетиков в зону действия. При блокаде
плечевого сплетения или седалищного нерва рас-
творы местных анестетиков иногда могут непред-
намеренно оказаться введенными снаружи от окру-
жающих нервные стволы фасций и соединительной
ткани. Стимуляция нерва позволяет определить
точное место инъекции. Местные анестетики долж-
ны проникнуть через соединительнотканный
барьер и миелиновые оболочки нервных стволов;
кроме того, они захватываются окружающей жи-
ровой тканью и мышцами, поэтому скорость раз-
вития анестезии может быть различной. Обычно
лидокаин и прилокаин начинают действовать бы-
стрее (примерно через 14 мин), чем бупивакаин
(через 23 мин). Действие лидокаина и прилокаина
продолжается 3-4 ч, бупивакаина или ропивакаи-
на — до 10 ч. Стойкие парестезии после блокады
нервов обусловлены скорее механической травмой,
нежели действием препарата.
Прокаин является идеальным диагностическим
препаратом, поскольку оказывает относительно
короткое (15-45 мин) и локализованное действие
(благодаря низкой диффузионной способности).
При хронической боли он может применяться для
блокады соматических или вегетативных нервов
и позволяет лучше оценить целесообразность ней-
ролитической блокады.
МЕСТНЫЕ АНЕСТЕТИКИ
131
ЭПИДУРАЛЬНАЯ АНЕСТЕЗИЯ medlit.
Введение раствора местного анестетика в про-
странство между твердой мозговой оболочкой и над-
костницей, выстилающей внутреннюю поверхность
позвоночного канала, широко применяется как
часть сбалансированной анестезии, а также для
обезболивания родов и устранения боли в после-
операционном периоде. Эпидуральную анестезию
обычно выполняют на уровне поясничного и груд-
ного отдела позвоночника. Эпидуральное про-
странство заполнено жировой тканью, лимфатиче-
скими и кровеносными сосудами (преимуществен-
но эпидуральным венозным сплетением). После
введения в эпидуральное пространство раствор ме-
стного анестетика распространяется во всех на-
правлениях и вызывает анальгезию, блокируя про-
ведение импульсов по интрадурально расположен-
ным нервным корешкам. Местные анестетики
приникают через рукава нервных корешков, обра-
зованные твердой мозговой оболочкой, чуть про-
ксимальнее спинномозгового узла. В этом участке
твердая мозговая оболочка очень тонкая и непо-
средственно переходит в эпиневрий спинномозго-
вых нервов. Некоторые ворсинки паутинной обо-
лочки также частично или полностью проникают в
эпидуральное пространство через отверстия в твер-
дой мозговой оболочке, и для попадания в субарах-
ноидальное пространство местным анестетикам
достаточно преодолеть лишь эту тонкую оболочку.
После выполнения эпидуральной блокады в нерв-
ных корешках (которые расположены интраду-
рально) достигается высокая концентрация мест-
ного анестетика, и дерматомное распределение
анальгезии происходит по проводниковому типу.
У молодых пациентов местный анестетик может
распространяться в паравертебральные простран-
ства, и некоторое его количество захватывается
спинным мозгом. Получены данные, что воздейст-
вие местного анестетика на спинной мозг сохраня-
ется еще некоторое время после восстановления
проводимости в нервных корешках.
Эпидуральную анестезию можно рассматривать
как множественную блокаду мелких нервов. Дейст-
вие наступает немного позже (возможно, из-за уве-
личения расстояния, которое надо преодолеть ане-
стетику), но продолжительность такая же, как при
блокаде локтевого нерва или при межреберной бло-
каде. Интенсивность и зона распределения эпиду-
ральной анестезии зависят от объема, концентра-
ции и общей дозы местного анестетика. У величение
объема способствует распространению раствора в эпи-
дуральном пространстве и увеличивает зону рас-
пределения блокады. Повышение концентрации
ускоряет наступление анестезии, углубляет ее и вы-
иСО^еГЦвигательную блокаду. Увеличение общей
дозы повышает продолжительность анестезии.
Другими важными факторами являются место инъ-
екции, скорость введения и положение пациента.
Положение на боку углубляет чувствительную и дви-
гательную блокаду с нижерасположенной стороны.
Более обширное распространение местного
анестетика отмечается у рожениц, поскольку из-за
сдавления нижней полой вены эпидуральное
венозное сплетение набухает и объем эпидурально-
го пространства уменьшается. Следовательно, при
выполнении эпидуральной анестезии у рожениц дозу
местного анестетика нужно уменьшить. При атеро-
склерозе и у пожилых всасывание анестетика из
эпидурального пространства нарушается, что так-
же может потребовать уменьшения дозы.
Эпидуральная анестезия выше уровня Т10 мо-
жет вызывать выраженную артериальную гипото-
нию со снижением систолического АД более чем
на 30 мм рт. ст. Это обусловлено блокадой симпати-
ческих вазоконстрикторных проводящих путей
спинного мозга и вегетативных узлов. Расширение
сосудов в кишечнике, печени и почках приводит
к депонированию крови в емкостных сосудах и зна-
чительному снижению венозного возврата. Артери-
альную гипотонию можно предотвратить или зна-
чительно уменьшить предварительной инфузией
кристаллоидных или коллоидных растворов, а так-
же эфедрином (10-15 мг в/в).
В Великобритании для эпидуральной анестезии
применяют только бупивакаин (0,25-0,75%) или
ропивакаин (0,75-1,0%). Чтобы исключить попада-
ние анестетика в системный кровоток или в суб-
арахноидальное пространство, перед введением
полной дозы обычно вводят тест-дозу. Если необ-
ходима длительная блокада, то устанавливают ка-
тетер, через который анестетик можно вводить раз-
ными способами — струйно медленно по необходи-
мости, как непрерывную инфузию, с помощью
управляемых больным инфузионных насосов.
Бупивакаин начинает действовать достаточно
медленно (через 10-20 мин), эффект сохраняется
несколько часов; высокая растворимость в липидах
и значительное связывание с белками замедляет его
всасывание в системный кровоток. По тем же при-
чинам только небольшое количество бупивакаина
проникает через плацентарный барьер и попадает
в системное кровообращение плода. Низкие концен-
трации (0,25%) бупивакаина обеспечивают аналь-
гезию с минимально выраженным двигательным
блоком, тогда как высокие концентрации (0,5-0,75%)
оказывают более длительное действие с умеренной
двигательной блокадой (табл. 6.3). Применяются
также и промежуточные концентрации (0,375%).
132
Глава 6
инические эффекты бупивакаина и ропи-
Табл. 6.3. Концентрация.
вакаина при эпидуральной, аудальной и спинномозговой анестезии (у взрослых}
Концентрация Удель- ный вес1 Разовая доза Начало дейст- вия(мин) Продолжительность действия (мин) Клинические эф- фекты
Доза (мг) Объем (мл)
Эпидуральная анестезия (на поясничном уровне)
Бупивакаин (2.5 мг/мл, водный раствор) 1003 15-40 6-16 15-25 60-120 Анальгезия Нормальная мы- шечная сила
Бупивакаин (3.75 мг/мл. вод- ный раствор) 1004 23-60 6-16 15-25 90-150 Анальгезия Нормальная мы- шечная сила
Бупивакаин (5 мг/мл, водный раствор) 1006 30-80 6-16 15-25 120-180 Анальгезия Умеренная двига- тельная блокада
Бупивакаин (7,5 мг/мл. водный раствор)2 1008 45-120 6-16 15-25 180-240 Анальгезия Умеренная двига- тельная блокада
Ропивакаин (2 мг/мл, водный раствор) 1002 20-40 10-20 10-20 30-90 Анальгезия Нормальная мы- шечная сила
Ропивакаин (7,5 мг/мл, водный раствор) 1007 .112-150 15-20 10-20 180-300 Анальгезия Умеренная двига- тельная блокада
Ропивакаин (10 мг/мл. водный раствор)2 1010 150-200 15-20 10-20 240-360 Анальгезия Полная двигатель- ная блокада
Каудальная анестезия
Бупивакаин (5 мг/мл, водный раствор) 1006 100-150 20-30 10-20 120-180 Анальгезия Умеренная двига- тельная блокада
Спинномозговая анестезия
Бупивакаин (5 мг/мл, водный раствор) 1006 10-20 2-4 5-15 140-200 Спинномозговая анестезия Умеренная двига- тельная блокада
Бупивакаин (5 мг/мл в 8% глю- козе) 1026 10-20 2 4 5-15 120-180 Спинномозговая анестезия Полная двигатель- ная блокада
1 средний удельный вес ЦСЖ при 20°С равен 1007
г запрещен к применению в акушерстве
Иногда к раствору бупивакаина добавляют адре-
налин (1:200 ООО 1.300 000; 3,3-5 мкг/мл). Хотя ад-
реналин в этом случае не удлиняет анестезию,
вазоконстрикция приводиткснижению пиковой
концентрации бупивакаина в плазме, а вызванная
адреналином тахикардия служит ранним индика-
тором непреднамеренного попадания раствора
местного анестетика в кровь.
МЕСТНЫЕ АНЕСТЕТИКИ
133
п „ medlit.ucoz.ru
В последние годы для эпидуральной анестезии ная олок<
часто применяют ропивакаин (0,75-1,0%). Харак-
тер чувствительной блокады после эпидурального
введения эквивалентных доз бупивакаина и ропи-
вакаина идентичен. Напротив, двигательная блока-
ная олокада обычно выполняется бупивакаином
(0,5%; 20-30 мл) и обеспечивает дополнительную
анальгезию после геморроидэктомии и операций
на промежности у взрослых. Медленное развитие
блокады и частые неудачи, вероятно, обусловлены
да при использовании ропивакаина развивается
медленнее, длится меньше, выражена слабее, хотя и
усиливается при увеличении дозы. Ропивакаин
имеет ряд преимуществ перед бупивакаином: он по-
зволяет лучше отграничить чувствительную
блокаду от двигательной, он менее кардиотокси-
чен, имеет более короткий Т1/2 и быстрый клиренс
(табл. 6.2).
При эпидуральном введении лидокаина может
развиваться тахифилаксия (т. е. быстрое разви-
тие толерантности), что очень редко наблюдается
при использовании бупивакаина или ропивакаина.
При развитии тахифилаксии повторные одинако-
вые дозы местного анестетика приводят к прогрес-
сивному снижению глубины блокады, и для ее под-
держания на прежнем уровне требуется повышение
дозы анестетика. Возможно, этот феномен объяс-
няется локальными изменениями pH при введении
в эпидуральное пространство значительного объема
раствора местного анестетика с относительно низ-
ким pH 4-5. Поскольку объем и буферная емкость
тканей эпидурального пространства ограничены,
растворы местных анестетиков понижают их эф-
фективное значение pH, тем самым уменьшая долю
свободных оснований, способных диффундировать
в нервные корешки. При этом происходит прогрес-
сивное снижение распространенности и продолжи-
тельности сенсорной блокады и анальгезии.
В эпидуральное пространство иногда вводят
другие препараты: опиоиды (фентанил, морфин, диа-
морфин), аг-адреномиметики (клонидин, дексме-
детомидин), бензодиазепины (мидазолам). Их при-
меняют в чистом виде или сочетают с местными
анестетиками. (В РФ в эпидуральное пространство
из этих препаратов разрешено вводить только мор-
фин — прим. ред.).
Каудальная анестезия является разновидно-
стью эпидуральной и обычно предназначается для
блокады нервных корешков на уровне нижних по-
ясничных и крестцовых сегментов спинного мозга.
Структуры конского хвоста, расположенного в кре-
стцовом канале, блокируются при введении мест-
ных анестетиков в крестцовую щель (промежност-
ная анестезия). Анестетикам не нужно преодоле-
вать твердую мозговую оболочку (которая
заканчивается у нижнего края второго крестцового
позвонка). Ниже этого уровня в крестцовом канале
находится крупное венозное сплетение (а также
крестцовые нервы, терминальная нить и жир). Каудаль-
активным всасыванием анестетика в крестцовом
и внутреннем позвоночном венозных сплетениях,
а также потерями препарата через многочисленные
крестцовые отверстия. На зону распространения
блокады влияют положение больного, доза и ско-
рость инъекции.
СПИННОМОЗГОВАЯ АНЕСТЕЗИЯ
Введение растворов местных анестетиков непо-
средственно в цереброспинальную жидкость
(ЦСЖ) вызывает спинномозговую (спинальную)
анестезию. Места соединения передних и задних
спинномозговых корешков со спинным мозгом не-
миелинизированы, и попадающие в субарахнои-
дальное пространство местные анестетики быстро
захватываются нервными корешками, спинномоз-
говыми узлами и спинным мозгом. Вследствие это-
го мощность местного анестетика при субарахнои-
дальном введении в 10-15 раз больше, чем при эпи-
дуральном, а двигательная блокада выражена
сильнее (табл. 6.3). Спинномозговая анестезия
развивается быстрее эпидуральной, потому что для
достижения ткани-мишени местным анестетикам
не нужно преодолевать различные барьеры. Так как
спинномозговая анестезия требует малых доз ане-
стетиков, обычно она короче эпидуральной. Интен-
сивность и зона распространения анестезии зависят
от дозы местного анестетика, скорости введения, по-
ложения пациента и удельного веса раствора (по
сравнению с удельным весом ЦСЖ, т. е. 1007).
Увеличение дозы местного анестетика обычно
повышает качество и глубину анестезии, пролонги-
рует ее и может расширять зону анальгезии. Влия-
ние положения тела больного на анестезию зависит
от удельного веса раствора местного анестетика.
Гипобарические растворы (0,25 и 0,5% водный
раствор бупивакаина с удельным весом 1003-1006)
в ЦСЖ поднимаются вверх. Обычно они действуют
дольше гипербарических растворов. Их распро-
странение не зависит от положения тела пациента,
и их эффекты непредсказуемы. Они не во всех слу-
чаях позволяют обеспечить качественную блокаду.
Изобарические растворы местных анестетиков бо-
лее физиологичны, но их распространение в ЦСЖ
также не зависит от положения пациента. К сожале-
нию, эффекты изобарических растворов тоже слабо
предсказуемы.
Чаще всего для спинномозговой анестезии при-
меняют гипербарические растворы местных анесте-
тиков. Их распространение в ЦСЖ определяется
134
Глава 6
силой тяжести и положением тела пацие
гипербарический раствор вводят между позвонками
L2-L3 или L3-L4, то уровень блокады зависит от
положения тела больного: в положении лежа — это
Т4-Т7, в положении сидя (если больной еще ка-
кое-то время сидит после инъекции) — Т7-Т10.
Если анестетик вводят между позвонками L4-L5,
и пациент продолжает сидеть, то развивается «сед-
ловидная» блокада, которую применяют при опера-
циях на промежности. Распространение гиперба-
рических растворов зависит также от возраста, рос-
та, кривизны позвоночника и емкости
субарахноидального пространства. Так, сдавление
нижней полой вены (например, при беременности
или внутрибрюшных опухолях) приводит к набу-
ханию позвоночного венозного сплетения и умень-
шает объем субарахноидального пространства, что
увеличивает зону распространения местного ане-
стетика и глубину анальгезии.
В Великобритании «тяжелый» бупивакаин
(0,5% бупивакаин в 8% растворе глюкозы; удель-
ный вес 1026) является единственным гипербари-
ческим раствором, применяющимся для спинно-
мозговой анестезии. Средние дозы (10-20 мг, т. е.
2-4 мл) начинают действовать через 5-15 мин,
и анестезия продолжается окрло 2-3 ч (табл. 6.3);
продолжительность действия низких доз (и гипер-
барического раствора лидокаина) меньше. Добав-
ление вазоконстриктора (например, адреналина в раз-
ведении 1:200 000) немного пролонгирует блокаду.
Бупивакаин и глюкоза не метаболизируются в ЦСЖ,
но захватываются спинным мозгом или всасывают-
ся в ветвях спинномозговых артерий, образующих
сосудистую сеть в мягкой мозговой оболочке.
Спинномозговая анестезия выше уровня Т10
вызывает значительную артериальную гипотонию
из-за блокады симпатических сосудосуживающих
путей спинного мозга и вегетативных узлов. Расши-
рение сосудов кишечника, печени почек приводит к
депонированию крови в емкостных сосудах и зна-
чительному снижению венозного возврата. Артери-
альную гипотонию можно предотвратить или зна-
чительно уменьшить предварительным перелива-
нием инфузионных растворов, а также эфедрином
(10-15 мг в/в).
Различные сенсорные модальности по-разному
реагируют на спинномозговую блокаду: зона аналь-
гезии обычно обширнее зоны анестезии. Уровень
двигательной блокады также находится примерно
на два дерматома ниже уровня чувствительной
блокады. Хотя применение ропивакаина (и лево-
бупивакаина) позволяет уменьшить степень и про-
должительность двигательной блокады, в настоя-
щее время их не применяют для спинномозговой
анестезии.
логические осложнения спинномозговой
анестезии встречаются относительно редко; обыч-
но они обусловлены утечкой ЦСЖ, механическими
повреждениями, инфекцией или сопутствующими
заболеваниями. Чем выше концентрация местного
анестетика, тем больше риск арахноидита и синдро-
ма конского хвоста.
ВНУТРИВЕННАЯ РЕГИОНАРНАЯ АНЕСТЕЗИЯ
Прокаин (1% прокаин в 0,9% NaCl) раньше при-
меняли в виде в/в инфузии для усиления действия
общих анестетиков, для обезболивания перевязок
при ожогах, для лечения послеоперационной боли.
Действие прокаина при в/в инфузии довольно
непредсказуемо, часто развиваются побочные
эффекты. В настоящее время для в/в анестезии
он практически не применяется.
Для обезболивания небольших хирургических
вмешательств может использоваться внутривен-
ная регионарная анестезия. Впервые этот метод был
описан в 1908 г. Августом Биром, но широкое рас-
пространение получил лишь в 1970-е гг. Конечность
(чаще руку) предварительно обескровливают, при-
подняв и плотно перебинтовав эластическим бин-
том Эсмарха; после этого накладывают турникет
(жгут), который раздувают до давления, превы-
шающего систолическое на 100 мм рт. ст. Местный
анестетик медленно (в течение 2-3 мин) вводится
в вену конечности. Парестезии и анальгезия разви-
ваются практически сразу, а полная чувствитель-
ная блокада наступает в течение 10 мин. Качество
и продолжительность анальгезии зависят от дозы
и области введения местного анестетика, эффек-
тивности обескровливания, продолжительности
ишемии конечности перед инъекцией. Влияние
также оказывают местный ацидоз и вазодилата-
ция в результате накопления СО2. Точкой прило-
жения действия, вероятно, являются немиелинизи-
рованные нервные окончания, к которым местный
анестетик попадает путем ретроградного распро-
странения по сосудистому руслу. Проводимость
нервов обычно не страдает, хотя в результате пре-
и постсинаптических эффектов в нервно-мышеч-
ных соединениях через 15-20 мин после инъекции
наступает миорелаксация.
Большинство современных местных анестети-
ков обеспечивает эффективную регионарную ане-
стезию. В зависимости от анестетика период оста-
точной анальгезии может составлять 30-350 мин.
Через 20-30 мин возникает вызывает дискомфорт
и боль от турникета; их можно частично избежать,
используя двухкамерный пневматический тур-
никет. Тем не менее внутривенную регионарную
анестезию обычно не применяют, если операция
МЕСТНЫЕ АНЕСТЕТИКИ
135
длится дольше 1 ч. Повреждения нервов встречают-
ся крайне редко и обычно обусловлены длитель-
ным пребыванием турникета.
Местные анестетики, в значительной степени
связывающиеся с белками плазмы и тканей (бупи-
вакаин, ропивакаин), не следует применять для
внутривенной регионарной анестезии: достоверно
известно о пяти смертельных исходах при исполь-
зовании этой методики, которые были вызваны
кардиотоксическим действием бупивакаина.
Чаще всего для внутривенной регионарной ане-
стезии руки применяют лидокаин или прилокаин
(200 мг, т. е. 40 мл 0,5% раствора). Для обезболива-
ния нижней конечности применяют более высокие
дозы анестетиков, и результаты в этом случае менее
удовлетворительны. Системная интоксикация
обычно обусловлена случайным опустошением
манжетки турникета. Если турникет снимают не
ранее чем через 15-20 мин после инъекции, то веро-
ятность достижения высокой концентрации лидо-
каина или прилокаина в плазме мала, однако лег-
кие симптомы интоксикации (парестезии и шум
в ушах) наблюдаются достаточно часто.
ПРОЧИЕ ПОКАЗАНИЯ К ПРИМЕНЕНИЮ
МЕСТНЫХ АНЕСТЕТИКОВ
В прошлом кокаин входил в состав некоторых
анальгетических микстур для приема внутрь при
неизлечимых злокачественных новообразованиях.
Препарат угнетал моторику желудка и вызывал
эйфорию. Однако возбуждающее действие (наряду
с юридическими ограничениями) привело к тому,
что кокаин больше не применяют с этой целью.
Прокаин образует малорастворимые конъюгаты
с некоторыми лекарственными препаратами (напри-
мер, пенициллином), что замедляет их высвобож-
дение и уменьшает боль от инъекций. Лидокаин
также используют для уменьшения боли при в/в
или в/м введении лекарственных препаратов
(пропофол, амоксициллин).
Дибукаин (нуперкаин, цинхокаин) избирательно
подавляет активность нормальной холинэстеразы
плазмы (псевдохолинэстеразы); напротив, атипич-
ные формы фермента устойчивы к нему. Вследствие
этого дибукаин широко используют для изучения
генетических вариантов этого фермента.
Литература
Brockway, М., Bannister, J., McKeown, D. & Wildsmith, J. A.W.
(1990). Double-blind comparison of extradural bupivacaine and
ropivacaine. British Journal of Anaesthesia 64, 388.
Bromage, P.R. (1975). Mechanism of action of extradural
analgesia. British Journal of Anaesthesia 47,199-212.
Eyres, R.L & McDougall, R J. (1999). Local and
regional anaesthesia in children. British Journal of Anaesthe- sia
83, 65-77.
Catterall, W.A. (1993). Structure and function of voltage-gated
ion channels. Trends in Neurosciences 16,500-506.
Covino, B.G. (1986). Pharmacology of local anaesthetic agents.
British Journal of Anaesthesia 58, 701-716.
Critchley, L.A.H., Short, T.G. & Gin, T. (1994). Hypotension
during subarachnoid anaesthesia: haemodynamic analysis of three
treatments. British Journal of Anaesthesia 72,151-155.
Denny, N.M. & Selander, D.E. (1998). Continuous spinal anaesthe-
sia. British Journal of Anaesthesia 81,590-597.
Duncan, L & Wildsmith, J.A.W. (1995). Liposomal local anaesthe-
tics. British Journal of Anaesthesia 75, 260-261.
Gristwood, R., Bardsley, H„ Baker, H. & Dickens, J. (1994).
Reduced cardiotoxicity of levobupivacaine compared racemic
bupivacaine (Marcain): new clinical experience. Expert
Opinion in Investigational Drugs 3, 1209-1212.
Hille, B. (1992). Ionic Channels of Excitable Membranes.
Sinauer Associates, Sunderland, Massachussetts.
Lyons, G, Columb, M„ Wilson, R.C. & Johnson, R.V. (1998).
Epidural pain relief in labour: potencies of levobupivacaine and
racemic bupivacaine. British Journal of Anaesthesia 81, 899-901.
McCrae, A.F., Jozwiak, H. & McCrae, J.H. (1995). Com-
parison of ropivacaine and bupivacaine in extradural analgesia for
the relief of pain in labour. British Journal of Anaesthesia 74,
261-265.
McCrae, A.F. & Wildsmith, J.A.W. (1993). Prevention and
treatment of hypotension during central neural Ь1оск. British
Journal of Anaesthesia 70,672-800.
Molodecka, J., Stenhouse, C., Jones, J.M. & Tomlinson, J A.
(1994). Comparison of percutaneous anaesthesia for venous
cannulation after topical application of either amethocaine or
EMLA cream. British journal of Anaesthesia 72, 174-176.
Morrison, L.M.M., Emanuelsson, B.M., McClure, J.H. et aL
(1994). Efficacy and kinetics of extradural ropivacaine:
comparison with bupivacaine. British Journal of Anaesthesia 72,
164-169.
Narahashi, T., Yamada, M. & Frazier, D.T. (1969). Cationic
forms of local anesthetics Ыоск action potentials from inside the
nerve membrane. Nature 223, 748-749.
Ragsdale, D.R., McPhee, J.C., Scheuer, T. & Catterall, W.A. (1994).
Molecular determinants of state dependent Ыоск of Na+ channels
by local anesthetics. Science 265,1724-1728.
Reynolds, F. (1987). Adverse effects of local anaesthetics. British
Journal of Anaesthesia 59, 78-95.
Scott, D.B. (1986). Toxic effects of local anaesthetic agents on the
central nervous system. British Journal of Anaesthesia 58, 732-735.
Strichartz, G.R. (ed.) (1987). Local anesthetics. Handbook of
Experimental Pharmacology 81,1-19. Heidelberg: Springer-Verlag.
Tucker, G.T. (1986). Pharmacokinetics of local anaesthetics.
British Journal of Anaesthesia 58,717-731.
Wann, K.T. (1993). Neuronal sodium and potassium channels:
Structure and function. British Journal of Anaesthesia 71,2-14.
Л ЕКАРСТВ ЕН Н Йй"1 ucoz u
ПРЕПАРАТЫ,
ВОЗДЕЙСТВУЮЩИЕ НА
НЕРВНО-МЫШЕЧНЫЙ СИНАПС
СТРОЕНИЕ
НЕРВНО-МЫШЕЧНОГО СИНАПСА
Тела соматических мотонейронов расположены
в передних рогах спинного мозга. Их аксоны де-
лятся на множество конечных ветвей, каждая из ко-
торых иннервирует одну мышечную клетку
(волокно); благодаря этому один мотонейрон
может иннервировать от 10 до 150 мышечных
клеток. Это так называемая двигательная единица.
В области окончания аксона миелиновая оболочка
исчезает, аксон ветвится и образует обширную сеть
над специализированной поверхностью мембраны
мышечной клетки — двигательной концевой пла-
стинкой. Нервно-мышечный синапс начинается
у немиелинизированного нервного окончания
дистальнее последнего перехвата Ранвье.
Большинство мышечных волокон имеет един-
ственную двигательную концевую пластинку.
Обычно эта пластинка располагается посредине
волокна. Чаще всего концевая пластинка образует
возвышение на поверхности волокна; такой
нервно-мышечный синапс называют дисковидным
(еп plaque). Эти мышечные волокна иннервируют-
ся быстро проводящими аксонами типа Аа, они
быстро сокращаются и расслабляются.
Некоторые мышечные волокна обильно иннер-
вируются, так что каждое волокно имеет несколь-
ко двигательных концевых пластинок; такой нерв-
но-мышечный синапс называют гроздевидным
(еп grappe). Эти мышечные волокна иннервируют-
ся медленно проводящими аксонами типа Ау. Такая
иннервация характерна для глазодвигательных,
гортанных и некоторых мимических мышц, где
окончания двигательных нервов напоминают вино-
градную гроздь. Эти мышечные волокна, иннерви-
руемые в нескольких точках, не способны распро-
странять потенциал действия, и для развития мы-
шечного сокращения требуется несколько
импульсов. В результате этого сокращение и после-
дующее расслабление происходят медленно и ино-
гда описываются как контрактура.
Клеточное строение двигательной концевой
пластинки можно рассмотреть на электронной
микрофотографии (рис. 7.1). Конечная ветвь аксо-
на, окруженная отростками шванновских клеток,
располагается в углублении мембраны мышечной
клетки и содержит многочисленные митохондрии и
синаптические пузырьки. Многие из этих пузырь-
ков связаны с особыми участками мембраны аксо-
на, где происходит высвобождение нейромедиато-
ра. Пузырьки синтезируются в клетках передних
рогов спинного мозга и переносятся к конечным
ветвям двигательного нерва с помощью микротубу-
лярной транспортной системы. Мембрана аксона
отделена от постсинаптической мембраны щелью
шириной около 50 нм; в щели находится базаль-
ная пластинка толщиной около 20 нм, состоящая
преимущественно из мукополисахаридов. Холино-
рецепторы обычно расположены компактными
группами на гребнях субсинаптических складок
постсинаптической мембраны; дистальнее, в углуб-
лениях складок, содержится гидролитический фер-
мент АХЭ (ацетилхолинэстераза). Холинорецепто-
ры и АХЭ имеются и на пресинаптической мембране.
СИНТЕЗ И ХРАНЕНИЕ
АЦЕТИЛХОЛИНА
Нервно-мышечная передача зависит от синтеза,
хранения и высвобождения ацетилхолина в конеч-
ных ветвях двигательных нервов. К двигательной
концевой пластинке часть холина попадает из
плазмы, а часть образуется в результате гидролиза
ацетилхолина. Из внеклеточной жидкости холин
поступает в цитоплазму нейрона с помощью высо-
коаффинной транспортной системы, присутст-
вующей во всех холинергических нервах (рис. 7.2).
Как правило, именно транспорт холина ограничи-
вает скорость синтеза ацетилхолина холинацетил-
трансферазой:
Холин + ацетил-КоА —> ацетилхолин + КоА.
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС
137
Рис. 7.1. Электронная микрофотография части нервно мышечногосннапса. Конечная ветвь аксона (А) лежит в глубо-
кой выемке мембраны мышечного волокна. В ней находится множество митохондрии и синаптических пузырьков
(везикул). Наружная поверхность аксона покрыта цитоплазматическими отростками шванновских клеток (S).
Ниже аксона видны многочисленные складки мембраны мышечною волокна (отмечены стрелками). Между нер-
вом и мышцей (М) находится базальная пластинка (х 30 000)
Синтез ацетилхолина стимулируется его высво-
бождением; обычно запасы нейромедиатора в нерв-
ном окончании не истощаются ни при дефиците хо-
лина, ни при высокочастотной стимуляции нерва.
I (риблизительно 50% ацетилхолина, синтезиро-
ванного в цитоплазме конечных ветвей двигатель-
ных нервов, переносится в мелкие синаптиче-
ские пузырьки (диаметром около50 нм) и хранится
в виде мультимолекулярных пакетов, которые еще
называют квантами. Часть оставшегося ацетилхо-
лина проникает через мембрану аксона и в значи-
тельном количестве может присутствовать в шван-
новских клетках и цитоплазме мышечного волокна,
хотя его функция здесь неизвестна. Каждый пузы-
рек (везикула) содержит 10 000 12 000 молекул
ацетилхолина, а также ионы кальция. ЛТФ и цро-
тогликан (который присутствует также в электри-
ческих органах некоторых видов рыб). Кроме того,
в окончаниях двигательных нервов обнаружены не-
многочисленные крупные пузырьки с плотным
ядром. Эти пузырьки содержат нейромедиатор
кальцнтонинонодобный пептид, который высвобо-
ждается в синаптическую щель при высокочастот-
ной стимуляции. Функция калыштоииноподоб-
ного пептида не вполне ясна; возможно, он вовлечен
всинтез холннорецепторов и регуляцию их числен-
ности.
Около 1% содержащих ацетилхолин пузырьков
расположено в области преспнаптической мембраны
и представляет собой «легкодоступные» запасы ме-
диатора. Остальная часть равномерно распределя-
ется в цитоплазме конечных ветвей двигательных
138
Г лава 7
Рис. 7.2. Схема нерв-
но-мышечной передачи.
АХ ацетилхолин;
АХЭ ацетилхолинэ-
стераза;
КЛМ — кальмодулин;
ПКП потенциал кон-
цевой пластинки;
МИД — мышечный по-
тенциал действия;
ПК — протеинкиназа;
СР саркоплазматиче-
ский ретикулум
ПКП
СР-Са2*
I
I*-----— МПД------------* Т-тубулярная система
I
Тропонин С-Са2‘
Миазнн актиновый комплекс
* Мышечное сокращение
нервов, где связывается с микротрубочками и акти-
новыми нитями (филаментами), формируя боль-
шие резервные запасы ацетилхолина.
Интегральные белки мембраны аксонов играют
важную роль в «складировании» пузырьков в уча-
стках высвобождения ацетилхолина, а также в фор-
мировании выходных пор, через которые ацетилхо-
лин поступает в синаптическую щель. Эти же белки
участвуют в преобразовании пузырьков. Синап-
сии I это фосфопротеин, образующий оболочку
пузырьков и обеспечивающий их прикрепление
к внутриклеточным структурам. При распростра-
нении потенциала действия открываются кальцие-
вые каналы мембраны аксона, ионы кальция посту*
нают в ци топлазму и связываются с кальмодулином.
Вслед за этим активируется протеинкиназа и <|юс-
форилирует остатки серина в хвостовом участке
синапсина I. что приводит к высвобождению пу-
зырьков из пресинаптической мембраны (рис. 7.2).
Ионы кальция также связываются с другим бел-
ком, синаптотагмином, который, взаимодействуя с
пузырьками, облегчает их слияние со складирую-
щими белками (синтаксинами) в мембране аксона.
Это позволяет содержимому пузырька поступать в
щель нервно-мышечного синапса. Другие инте-
гральные белки (синаптобревин и синаптофизины)
также hi рают важную роль в хранении и последую-
щем высвобождении ацетилхолина.
ХОЛИНОРЕЦЕПТОРЫ
За последние тридцать лет произошел значи-
тельный прогресс в выделении и очисткехолиноре-
цепторов, а молекулярные технологии позволили
точно выяснить их струкуру. Чрезвычайно богатый
источник хол инорецепторов — электрические орга-
ны некоторых рыб, например, южноамериканского
пресноводного угря или гигантского электрическо-
го ската (эмбриологически электрические органы
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС
139
medlit.ucoz.ru
происходят из той же закладки, что и мышцы, од-
нако на долю мембраны, несущей холинорецепто-
ры, приходится значительно большая площадь).
Эти рецепторы необратимо связываются с а-нейро-
токсинами некоторых змей, например, тайваньско-
го полосатого крайта Bungarus multicinctus и ин-
дийской кобры Naja naja; это дало возможность
использовать их в качестве маркеров для выявле-
ния холинорецепторов. Холинорецепторы стало
возможно выделять и очищать путем аффинной
хроматографии. Кроме того, с помощью молеку-
лярных технологий можно определить последова-
тельность аминокислотных остатков холинорецеп-
торов и имплантировать их в искусственные мем-
браны и мембраны ооцитов амфибий.
Холинорецепторы нервно-мышечного синапса
являются интегральными мембранными белками
с молекулярным весом около 250 кДа. У взрослых
рецептор состоит из пяти субъединиц (две а-субъе-
диницы, р, е, 5 субъединицы), которые пронизыва-
ют постсинаптическую мембрану и окружают ион-
ный канал. У плода место в-субъединицы занимает
у-субъединица. Считают, что именно е-субъедини-
ца обеспечивает кратковременность пребывания
канала в открытом состоянии и высокую проводи-
мость для ионов. Каждая а-субъединица имеет мо-
лекулярный вес около 40 кДа и содержит участок
связывания ацетилхолина вблизи определенных
аминокислотных остатков (в положениях цистеин
192 и 193, тирозин 93 и 190, триптамин 149). На ак-
тивацию холинорецепторов влияет фосфолипидный
состав окружающих участков постсинаптической
мембраны. Взаимодействие ацетилхолина с участ-
ком связывания на а-субъединице вызывает
конформационные изменения, приводящие к от-
крыванию ионного канала; вероятность этого
значительно повышается, если с ацетилхолином
связываются оба участка (положительный коопе-
ративный эффект). Пресинаптические холиноре-
цепторы имеют несколько иную молекулярную
структуру.
НЕРВНО-МЫШЕЧНАЯ ПЕРЕДАЧА
В отсутствие нервных импульсов постсинапти-
ческие рецепторы проявляют спонтанную электри-
ческую активность. Она проявляется отдельными,
возникающими через неодинаковые промежутки
времени миниатюрными потенциалами концевой
пластинки (МПКП) амплитудой 0,5-1,5 мВ. Счи-
тается, что каждый МПКП отражает высвобожде-
ние одного кванта ацетилхолина, происходящего
вследствие случайного слияния синаптического
пузырька с пресинаптической мембраной.
Когда конечной ветви аксона достигает потен-
циал действия и она деполяризуется, в синаптиче-
скую щель выделяется содержимое 100-200 пу-
зырьков. Высвобождение ацетилхолина квантами
можно описать с помощью распределения Пуассона:
среднее количество квантов, высвобождаемое при
каждом нервном импульсе (т), выражается уравне-
нием m = пр (где п — количество квантов, способ-
ных к немедленному высвобождению, р — вероят-
ность высвобождения кванта при деполяризации
конечной ветви аксона). Ацетилхолин высвобож-
дается в большем количестве, чем это необходимо
для деполяризации концевой пластинки; иными
словами, существует значительный резерв для
обеспечения нервно-мышечной передачи.
Высвобождаемый ацетилхолин диффундирует
через синаптическую щель и соединяется с а-субъ-
единицами рецепторов, окружающих ионный ка-
нал постсинаптической мембраны. Открывание
ионного канала — это процесс, протекающий по
принципу «все или ничего», занимающий очень
мало времени (5-10 мсек) и сопровождающийся
неспецифическим повышением концентрации для
ионов Na+, К+, Са2+. В эксперименте показано, что
ацетилхолин вызывает перенос 10 000 ионов Na+
через одну ионную пору за каждую миллисекун-
ду пребывания канала в открытом состоянии.
Эти ионные изменения приводят к появлению мно-
жественных токов в отдельных каналах, каждый из
которых имеет амплитуду около 4 пА (4 х 10'*2 А).
Из отдельных токов складывается ток концевой
пластинки, который деполяризует постсинаптиче-
скую мембрану. Возникает небольшой локализо-
ванный потенциал концевой пластинки; когда он
достигает критической амплитуды (т. е. изменяется
приблизительно на 10-15 мВ), открываются потен-
циалзависимые натриевые каналы.
Мышечное волокно деполяризируется, и потен-
циал действия распространяется вдоль его поверх-
ности по системе Т-трубочек (поперечных трубо-
чек); Т-трубочки представляют собой инвагинации
сарколеммы — клеточной мембраны мышечного
волокна. Деполяризация системы Т-трубочек при-
водит к высвобождению Са2+ из саркоплазматиче-
ского ретикулума, возможно, вследствие стимуля-
ции синтеза второго посредника ИФ3. Высвободив-
шиеся ионы кальция связываются с тропонином С
и вызывают конформационные изменения других
тропонинов и тропомиозина. Одновременно акти-
вируется АТФаза миозина, и АТФ гидролизуется
с образованием АДФ, что обеспечивает высвобож-
дение большого количества внутриклеточной энер-
гии. При этом между актином и миозином образу-
ются поперечные мостики и развивается мышечное
140
Глава?
Рис. 7.3. Участки свя-
зывания ацетилхолин-
эстеразы (АХЭ) с аце-
тилхолином
сокращение (рис. 7.2). Последовательность физио-
логических и биохимических изменений в проме-
жутке между деполяризацией системы Т-трубочек
и мышечным сокращением получила название
электромеханического сопряжения (или сопряже-
ния возбуждения с сокращением).
Ацетилхолинэстераза (АХЭ) быстро прекраща-
ет действие ацетилхолина. В молекуле АХЭ имеет-
ся два участка связывания с ацетилхолином, анион-
ный участок и активный центр, которые находятся
на расстоянии около 0,5 нм друг от друга (рис. 7.3.).
Анионный участок в основном представлен отри-
цательно заряженной глутаматной группой, он свя-
зывается с четвертичным атомом азота в молекуле
ацетилхолина. Активный центр представлен гидро-
ксильной группой серина и имидазольной груп-
пой гистидина. Существование водородной связи
между гидроксильной и имидазольной группами
позволяет серину образовать ковалентную связь
с ацетильной группой ацетилхолина. После высво-
бождения холина АХЭ ацетилируется, затем в ре-
зультате быстрого гидролиза происходит регенера-
ция фермента с образованием уксусной кислоты.
Такие реакции протекают очень быстро, и Т|/2 аце-
тилированной АХЭ не превышает 40 мксек. Быст-
рая инактивация ацетилхолина необходима для
предотвращения повторного возбуждения двига-
тельной концевой пластинки.
АХЭ содержится во всех холинергических си-
напсах, и, вероятно, там же синтезируется. Она так-
же обнаруживается в эритроцитах (где ее функция
неизвестна) и во многих участках ЦНС. Напротив,
бутирилхолинэстераза (синонимы: псевдохолин-
эстераза, холинэстераза плазмы) синтезируется в пе-
чени и обнаруживается в плазме, коже, кишечнике
и других тканях. Хотя псевдохолинэстераза может
расщеплять ацетилхолин, она выполняет и более
важную функцию гидролиза других эфиров холина
(например, пропионилхолина, бутирилхолина),
присутствующих в стенке кишечника. В анестезио-
логической практике псевдохолинэстераза играет
важную роль в гидролизе некоторых миорелаксан-
тов (суксаметоний, мивакурий), на которые не дей-
ствует ацетилхолинэстераза. Подобно АХЭ, в псев-
дохолинэстеразе также имеется два участка связы-
вания, кроме того, она так же связывается с эфирами
холина своим активным центром. В отличие от
АХЭ, на анионном участке связывания псевдохо-
линэстеразы образуется биполярная связь.
Высвободившийся из конечной ветви двига-
тельного нерва ацетилхолин воздействует главным
образом на постсинаптические рецепторы. Тем не
менее обнаружены пресинаптические холиноре-
цепторы, располагающиеся в конечных ветвях дви-
гательных нервов и в наиболее дистальных пере-
хватах Ранвье. Эти рецепторы играют важную роль
в обеспечении высвобождения нейромедиатора
при высокочастотной стимуляции нерва; их блока-
да недеполяризующими миорелаксантами вызы-
вает феномен затухания (см. ниже). Стимуляция
холинорецепторов, расположенных на наиболее
дистальном перехвате Ранвье, индуцирует ретро-
градное или антидромное возбуждение двигатель-
ного нерва. Так, инъекция ацетилхолина в подко-
ленную артерию животных не только активизирует
соответствующие мышечные группы, но и вызыва-
ет ретроградно направленный потенциал действия.
Эти антидромные потенциалы регистрируются
на передних спинномозговых корешках электро-
физиологическими методами. Подобные эффекты
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС
141
могут вызываться повторной стимуляци
ингибиторами АХЭ или суксаметонием. Следова-
тельно, лекарственные препараты, воздействую-
щие на нервно-мышечный синапс, действуют также
на двигательные нервы.
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ,
ВЛИЯЮЩИЕ НА
НЕРВНО-МЫШЕЧНУЮ ПЕРЕДАЧУ
Лекарственные препараты могут влиять на че-
тыре фазы нервно-мышечной передачи:
1. Высвобождение ацетилхолина
2. Действие ацетилхолина
3. Гидролиз ацетилхолина
4. Электромеханическое сопряжение.
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ,
ВЛИЯЮЩИЕ НА
ВЫСВОБОЖДЕНИЕ
АЦЕТИЛХОЛИНА
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ,
ВЛИЯЮЩИЕ НА ТРАНСПОРТ КАЛЬЦИЯ
Высвобождение ацетилхолина в конечных вет-
вях двигательных нервов зависит от транспорта ио-
нов кальция в аксоплазму. Следовательно, можно
ожидать, что на высвобождение нейромедиатора
влияют изменения концентрации внеклеточного
Са2+. На практике гипо- и гиперкальциемия, оказы-
вающие значительное воздействие на возбуди-
мость тканей и сократимость миокарда, обычно не
влияют на высвобождение ацетилхолина (за ис-
ключением экспериментальных условий).
Тем не менее лекарственные препараты, подав-
ляющие транспорт ионов кальция, могут нарушать
нервно-мышечную передачу и увеличивать про-
должительность действия недеполяризующих
миорелаксантов. Этот феномен в основном вызыва-
ется аминогликозидами и полимиксином, но ино-
гда развивается и под действием других антибиоти-
ков (колистин, тетрациклины, линкомицин).
Классическое нарушение нервно-мышечной
проводимости, описано при длительном лечении
туберкулеза стрептомицином. Миорелаксация, ре-
зистентная к ингибитору АХЭ неостигмину, может
развиваться после абдоминальных операций, в ходе
которых в брюшную полость вводили большие
дозы аминогликозидов. Для устранения нерв-
но-мышечной блокады, индуцированной антибио-
тиками, применяют соли кальция (например, каль-
ция глюконат); ингибиторы АХЭ менее эффективны.
В современной анестезиологической практике эта
diTKB. ЬКрОЗЕейЫвозникает редко. Антибиотики могут на-
рушать нервно-мышечное проведение при тяжелой
миастении.
Ионы магния конкурируют с ионами кальция за
транспорт в конечных ветвях двигательных нервов,
что снижает высвобождение ацетилхолина. Суль-
фат магния (вводимый парентерально) и маг-
ний-содержащие антациды для приема внутрь
уменьшают высвобождение ацетилхолина и усили-
вают действие миорелаксантов, особенно на фоне
почечной недостаточности.
МЕСТНЫЕ И ОБЩИЕ АНЕСТЕТИКИ
Большинство местных анестетиков могут
уменьшать высвобождение ацетилхолина из конеч-
ных ветвей двигательных нервов. После систем-
ного введения или всасывания они нарушают
проведение импульсов по немиелинизированным
нервным окончаниям; кроме того, блокада прово-
димости крупных и мелких нервных стволов пре-
пятствует передаче нервных импульсов от клеток
передних рогов спинного мозга.
Ингаляционные анестетики также подавляют
высвобождение ацетилхолина. Они вызывают де-
прессию ЦНС, угнетают генерацию нервных им-
пульсов и оказывают прямое воздействие на мем-
браны мышечных волокон. Таким образом, ингаля-
ционные анестетики могут оказывать выраженное
влияние на глубину нервно-мышечной блокады
(глава 6). Оказывая влияние на кровообращение,
ингаляционные анестетики изменяют фармакоки-
нетические свойства миорелаксантов.
НЕЙРОТОКСИНЫ
Некоторые нейротоксины препятствуют высво-
бождению ацетилхолина. Яд паука «черная вдова»
содержит а-латротоксин, который влияет на высво-
бождение ацетилхолина во всех холинергических
нервах. В нервно-мышечном синапсе а-латротоксин
вначале вызывает выраженный экзоцитоз синапти-
ческих пузырьков, которые необратимо сливаются
с мембраной аксона. Синаптические пузырьки дез-
организуются и утрачивают способность накапли-
вать вновь синтезированный ацетилхолин. В этом
процессе участвует синаптотагмин, который обес-
печивает слияние синаптических пузырьков со
складирующими белками в конечной ветви нерва.
Токсин ботулизма также поражает все холинерги-
ческие нервы. Этот токсин является цинксодержа-
щей эндопептидазой, расщепляющей синаптобре-
вин и препятствующей высвобождению ацетилхо-
лина из синаптических пузырьков; содержание
нейромедиатора в конечной ветви двигательного
нерва при этом не изменяется.
142
Глава 7
ГУАНИДИН И АМИНОПИРИДИН
Гуанидин и аминопиридин увеличивают про-
должительность нейронального потенциала дейст-
вия и способствуют поступлению Са2+ в клетку, что
стимулирует высвобождение ацетилхолина. Меха-
низмы их действия немного различаются: гуанидин
тормозит инактивацию натриевых каналов, в то
время как аминопиридин скорее инактивирует
калиевые каналы, тем самым препятствуя реполя-
ризации. Оба препарата применяют для стимуля-
ции высвобождения ацетилхолина при ботулизме
и синдроме Итона-Ламберта (редкое расстройство
нервно-мышечной передачи, обычно возникающее
при мелкоклеточном раке бронхов). В Болгарии
аминопиридин используется для восстановления
нервно-мышечной проводимости после недеполя-
ризующей блокады. К сожалению, аминопиридин
и гуанидин легко преодолевают гематоэнцефаличе-
ский барьер и вызывают судороги, что резко огра-
ничивает их клиническое применение.
ДРУГИЕ ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ
Теофиллин и другие производные ксантина мо-
гут усиливать высвобождение ацетилхолина либо в
результате блокады аденозиновых рецепторов,
либо вследствие подавления активности фосфо-
диэстеразы и накопления цАМФ. Этот же меха-
низм объясняет улучшение состояния пациентов с
тяжелой миастенией при лечении симпатомимети-
ками (например, эфедрином).
Везамикол — новый препарат, применяющийся
при экспериментальных исследованиях нерв-
но-мышечного синапса. Этот третичный амин бло-
кирует нервно-мышечную передачу при приеме
внутрь. Предполагаемый механизм действия — уг-
нетение поступления вновь синтезированного аце-
тилхолина в синаптические пузырьки.
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ,
ВЛИЯЮЩИЕ НА ДЕЙСТВИЕ
АЦЕТИЛХОЛИНА
Лекарственные препараты могут изменять дей-
ствие ацетилхолина, связываясь с постсинаптиче-
скими никотиновыми холинорецепторами. Такие
препараты называются миорелаксантами. Их делят
на две группы:
1. Деполяризующие миорелаксанты
2. Недеполяризующие миорелаксанты.
Как следует из названий, деполяризующие мио-
релаксанты вызывают деполяризацию двигатель-
ной концевой пластинки непосредственно перед
развитием нервно-мышечной блокады. Напротив,
medlit. иООйнКНгризующие миорелаксанты
не вызывают
деполяризацию, за исключением эксперименталь-
ных исследований на эмбриональных мышечных
клетках и культурах мышечных клеток.
МЕХАНИЗМ ДЕЙСТВИЯ
Деполяризующая блокада развивается вследст-
вие распространения деполяризации с двигатель-
ной концевой пластинки на мембраны прилежащих
мышечных волокон. Внутриартериальное введение
малых доз ацетилхолина вблизи двигательной кон-
цевой пластинки в норме вызывает кратковремен-
ное сокращение скелетной мышцы; последующая
стимуляция двигательного нерва не приводит к яв-
ному изменению сократительной реакции. При вве-
дении больших доз ацетилхолина или пролонги-
ровании его действия ингибиторами АХЭ после
первого мышечного сокращения развивается нерв-
но-мышечная блокада, заметно снижающая макси-
мальную сократительную реакцию на стимуляцию
двигательного нерва. Подобные эффекты могут вы-
зываться и другими препаратами, по структуре
напоминающими ацетилхолин, но медленнее уда-
ляемыми из двигательной концевой пластинки
(суксаметоний, декаметоний).
Физиологическая деполяризация двигательной
концевой пластинки ацетилхолином активирует
потенциалзависимые натриевые каналы в приле-
жащих мышечных волокнах, что приводит к воз-
никновению и распространению потенциала дейст-
вия в мышце. Напротив, индуцированная стойкая
деполяризация быстро приводит к генерации мест-
ных электрических токов в прилежащих мышеч-
ных волокнах, что вызывает конформационные
изменения в натриевых каналах и повышение про-
ницаемости мембраны для ионов калия. Этот про-
цесс развивается в течение нескольких миллисе-
кунд и приводит к образованию зоны электриче-
ской невозбудимости вокруг двигательной
концевой пластинки. Зона невозбудимости распро-
страняется на 1-2 мм вдоль мембраны мышечного
волокна и не способна проводить импульсы, хотя
остальная часть мышцы сохраняет нормальные
электрофизиологические свойства.
Напротив, недеполяризующая блокада преиму-
щественно обусловлена конкуренцией миорелак-
санта и ацетилхолина за связь с холинорецептора-
ми на постсинаптической мембране. Такие препа-
раты иногда называют «конкурентными»
миорелаксантами, хотя трудно представить, чтобы
все их эффекты являлись следствием конкурент-
ного антагонизма с ацетилхолином. Обратимое
связывание недеполяризующих миорелаксантов
с постсинаптическими холинорецепторами никак
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС
143
Таблица 7.1.
Сравнительная характеристЛ^
и деполяризующих миорелаксантов
Деполяризующий миорелаксант Недеполяризующий миорелаксант
Преходящие мышечные фасцику- ляции, предшествующие блокаде Возникают Отсутствуют
Затухание нервно-мышечной передачи Отсутствует Характерно
Посттетаническое усилениие Отсутствует (как правило) Характерно
Введение ингибиторов АХЭ на фоне миорелаксантов Усиление степени блокады Уменьшение степени блокады
Введение недеполяризующих миорелаксантов Антагонистическое действие Синергизм
Тахифилаксия и двойной блок Возможны Отсутствуют
не влияет на проводимость импульса или проницае-
мость мембран для ионов. Из-за уменьшения коли-
чества свободных холинорецепторов постепенно ос-
лабевает вызываемая ацетилхолином деполяриза-
ция. Амплитуда потенциала двигательной кон-
цевой пластинки постепенно уменьшается, так что
в конечном итоге перестает генерироваться и распро-
страняться мышечный потенциал действия; иными
словами, развивается нервно-мышечная блокада.
Нормальная нервно-мышечная проводимость вос-
станавливается после адекватного перераспределе-
ния и/или элиминации миорелаксанта, или после
того как к постсинаптическим холинорепторам
начнет поступать достаточное количество ацетил-
холина.
В норме при стимуляции нерва ацетилхолин
высвобождается в большем количестве, чем необ-
ходимо для возбуждения потенциала двигательной
концевой пластинки и последующего мышечного
потенциала действия. Следовательно, нервно-мы-
шечная блокада развивается только после связыва-
ния антагониста со значительной долей рецепторов.
Экспериментально доказано, что недеполяризую-
щие миорелаксанты должны блокировать 80%
рецепторов, прежде чем появляются первые призна-
ки угнетения нервно-мышечной передачи. Для раз-
вития полной нервно-мышечной блокады необхо-
димо блокировать 90-95% рецепторов.
Помимо воздействия на постсинаптические
холинорецепторы, недеполяризующие миорелак-
санты связываются с пресинаптическими рецепто-
рами, расположенными на конечных ветвях двига-
тельных нервов. Эти рецепторы стимулируются
ацетилхолином и обеспечивают высвобождение
ацетилхолина в синаптическую щель в ответ на
высокочастотную стимуляцию нерва. Вследствие
этого при повторной стимуляции нерва на фоне
частичной нервно-мышечной блокады количество
высвобождаемого ацетилхолина уменьшается, что
и проявляется затуханием потенциала действия.
Кроме того, недеполяризующие миорелаксанты фи-
зически блокируют ионные каналы на постсинапти-
ческих, а возможно, и пресинаптических мембранах.
ХАРАКТЕРНЫЕ ОСОБЕННОСТИ
НЕРВНО-МЫШЕЧНОЙ БЛОКАДЫ
Деполяризующая и недеполяризующая блока-
ды имеют харкатерные различия. Классически они
описаны на примере суксаметония и тубокурарина
(табл. 7.1).
1. Развитию деполяризующей блокады предше-
ствуют мышечные подергивания (фасцикуляции).
Эти некоординированные сокращения обусловле-
ны повторяющейся стимуляцией, им соответствует
повышенная электромиографическая активность
скелетных мышц. Фасцикуляции объясняют раз-
дражением пресинаптического нервного оконча-
ния с последующим антидромным возбуждением
аксона и двигательной единицы. Рефлекторные
реакции, опосредованные холинорецепторами мы-
шечных веретен, играют вторичную роль.
Напротив, при недеполяризующей блокаде мы-
шечные фасцикуляции отсутствуют, поскольку
пресинаптические рецепторы связываются с анта-
гонистами ацетилхолина. Недеполяризующие
миорелаксанты уменьшают индуцируемые сукса-
метонием фасцикуляции, так как предотвращают
пресинаптическую деполяризацию и развитие ло-
кальных аксональных рефлексов, что уменьшает
частоту сокращений двигательных единиц. Следо-
вательно, предварительное введение недеполяри-
зующих миорелкасантов может предотвратить
фасцикуляции и мышечную боль, вызываемые сук-
саметонием.
144
Глава 7
medlit.ucoz.ru
Рис. 7.4. Потенциалы действия скелетной мышцы, вызы-
ваемые непрямой стимуляцией двигательного нерва на
фоне недеполяризующей блокады. Во всех трех случаях
стимуляция проводилась в режиме серии из четырех им-
пульсов (TOF-режим)
2. Повторяющаяся или длительная стимуляция
двигательного нерва на фоне введения деполя-
ризующего миорелаксанта не вызывает феномена
затухания — постепенного снижения мышечно-
го потенциала действйя и амплитуды мышечного
сокращения. Напротив, для недеполяризующих
миорелаксантов феномен затухания характерен
(рис. 7.4).
При деполяризующей блокаде в ходе тетанизи-
рующей стимуляции сохраняется высвобождение
ацетилхолина, поскольку влияние деполяризации
на пресинаптические рецепторы допускает выброс
нейромедиатора при высокой частоте проведения
импульсов. Изменения, наблюдаемые при частич-
ной блокаде, отражают суммацию электрической
активности в мышечных волокнах с полностью вос-
становленной проводимостью и волокнах, все еще
рефрактерных к действию ацетилхолина.
При повторяющейся стимуляции двигательно-
го нерва на фоне недеполяризующей блокады коли-
чество высвобождающихся квантов ацетилхолина
постепенно снижается. Это может быть обусловле-
но блокадой пресинаптических холинорецепторов.
Когда действие миорелаксанта распространяется
и на постсинаптические рецепторы, «защитный
механизм» истощается, и мышечный ответ затухает
(рис. 7.4).
3. Феноменом посттетанического усиления (сино-
ним: посттетаническое облегчение) называют воз-
растание амплитуды мышечного ответа на одиноч-
ный стимул, возникающее после тетанического
сокращения. При деполяризующей блокаде постте-
танического усиления не наблюдается. Напротив,
для недеполяризующей блокады посттетаническое
усиление характерно и регистрируется на механо-
грамме и электромиограмме (рис. 7.5).
Тетанизирующая стимуляция двигательного
нерва приводит к постепенному уменьшению коли-
чества высвобождаемого ацетилхолина, приходя-
щегося на каждый одиночный стимул. После этого
синтез и мобилизация нейромедиатора быстро уве-
личиваются. По завершении тетанизирующей сти-
муляции количество высвобождаемого ацетилхо-
лина, приходящееся на каждый одиночный стимул,
увеличивается. Благодаря механизму безопасности
синаптической передачи, в нормальных условиях
и во время деполяризующей блокады это не влияет
на нервно-мышечную функцию и не отражается на
электромиограмме.
Б В
Рис. 7.5. Посттетаническое усиление на фоне частичной недеполяризующей блокады. Смешанные потенциалы дейст-
вия скелетной мышцы, вызванные непрямой стимуляцией двигательного нерва (А). Индуцированное тетаническое
сокращение (его форма и амплитуда обусловлена артефактом от наносимого тетанизирующего стимула) (Б).
Посттетаническое усиление после возвращения к начальной частоте стимуляции (В)
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС 145
medlit.ucoz.ru
При недеполяризующей блокаде посттетаниче-
ское усиление развивается за счет облегчения нерв-
но-мышечной передачи, в результате преходящего
увеличения высвобождения ацетилхолина, вытес-
няющего недеполяризующие миорелаксанты из
участков связывания на постсинаптических рецеп-
торах. На фоне глубокой нервно-мышечной блокады
феномен посттетанического усиления используют
для мониторинга нервно-мышечной проводимости.
4. Ингибиторы АХЭ увеличивают степень и про-
должительность деполяризующей блокады. При этом
действие суксаметония удлиняется в результате
угнетения псевдохолинэстеразы. Декаметоний не
расщепляется псевдохолинэстеразой, и ингибитор
АХЭ вызывает накопление ацетилхолина в синап-
тической щели, что также усиливает деполяризую-
щую блокаду.
Напротив, во время недеполяризующей блока-
ды ингибиторы АХЭ вызывают накопление ацетил-
холина у двигательной концевой пластинки, что
приводит к постепенному вытеснению недеполя-
ризующего миорелаксанта из связи с пост- и преси-
наптическими рецепторами и восстанавлению
нервно-мышечной проводимости.
5. Разные недеполяризующие миорелаксанты
усиливают действие друг друга, углубляя и пролон-
гируя блокаду.
Напротив, недеполяризующие миорелаксанты
ослабляют или устраняют действие деполяризую-
щих миорелаксантов. Недеполяризующие миоре-
лаксанты конкурентно связываются с постсинап-
тическими холинорецепторами. Следовательно,
при деполяризующей блокаде они уменьшают де-
поляризацию двигательной концевой пластинки
и могут восстанавливать нормальную нервно-мы-
шечную передачу. Из-за плохо предсказуемого
эффекта с этой целью в клинической практике они
не применяются.
6. При введении деполяризующих миорелак-
сантов может развиваться тахифилаксия и так на-
зываемое двухфазное действие. Недеполяризующие
миорелаксанты таких осложнений не вызывают.
Тахифилаксия обычно проявляется снижением ре-
акции на ранее эффективные дозы миорелаксанта.
Тахифилаксия-может предшествовать постепенному
развитию изменений, характерных для недеполя-
ризующей блокады (вторая фаза действия деполя-
ризующего миорелаксанта). В анестезиологиче-
ской практике такие явления чаще всего встречают-
ся при использовании больших доз или длительной
инфузии суксаметония, а также при замедленной
элиминации препарата вследствие ферментопатии
(врожденный аномальный тип псевдохолинэстеразы,
при котором активность фермента резко снижена)
или на фоне действия других лекарственных препа-
ратов. Причины тахифилаксии и двухфазного дей-
ствия при использовании деполяризующих
миорелаксантов не вполне ясны. Возможно, это
обусловлено длительным присутствием деполяри-
зующего миорелаксанта на двигательной концевой
пластинке, что приводит к десенситизации рецеп-
торов или блокаде ионных каналов.
ДЕПОЛЯРИЗУЮЩИЕ
МИОРЕЛАКСАНТЫ
Деполяризующие миорелаксанты имеют сход-
ное химическое строение. Это тонкие удлиненные
гибкие молекулы с метильными и этильными груп-
пами, присоединенными к четвертичным аммоние-
вым группам, расположенным на расстоянии
1,2-1,4 нм друг от друга. Раньше в клинической
практике применяли суксаметоний и декаметоний;
в настоящее время для миорелаксации используют
только суксаметоний.
Суксаметоний
Суксаметоний — это четвертичный аммониевый
эфир (рис. 7.6), состоящий из двух молекул ацетил-
холина, соединенных своими нечетвертичными
концами. Способность суксаметония вызывать
деполяризующую блокаду впервые была отмечена
в 1949-1951 гг., хотя его фармакологические свой-
ства исследовались на животных еще в 1906 г.
После в/в введения суксаметония глубокая мио-
релаксация развивается в течение 1 мин вслед за от-
четливыми мышечными фасцикуляциями. Продол-
жительность нервно-мышечной блокады после обыч-
ной дозы (1,0-1,5 мг/кг) составляет 4-6 мин; в течение
этого времени концентрация миорелаксанта в плаз-
ме быстро снижается. Суксаметоний вызывает ряд
нежелательных побочных эффектов (табл. 7.2).
1. Мышечные фасцикуляции. Они могут быть
слабыми (затрагивают только мелкие мышцы кис-
тей и мимические мышцы), средневыраженными
(включают более крупные мышцы конечностей)
или сильными и генерализованными. Средневыра-
женные и сильные фасцикуляции облегчают веноз-
ный возврат и увеличивают сердечный выброс;
АД и внутричерепное давление повышаются.
Фасцикуляции мышц передней брюшной стенки
увеличивают давление в желудке.
2. Миалгия. Послеоперационные мышечные
боли являются частым осложнением суксаметония
и в основном возникают в подреберьях, туловище и
плечевом поясе. Чаще отмечаются у женщин, при
ранней мобилизации после операции и у пациентов,
не привыкших к физическим нагрузкам. Частота
146
Глава 7
Рис. 7.6. Структурные фор-
мулы ацетилхолина, суксаме-
тония и декаметония
medlit.ucoz.ru
Ацетилхолин
сн3
сн3—-со—о—сн2—сн2—N—сн3
сн3
Суксаметоний
Декаметоний
сн3
сн2—со—о—сн2—сн2—N—сн3
СН3
Сн3
СН2—СО—О—СНг—-СН2—N—сн3
сн3
сн3 сн3
СН3—hl—(СН2)|о—hl±-СН
сн3 сн3
3
послеоперационной миалгии колеблется от 6% до
60%; значительно повышается уровень креатинки-
назы, а в некоторых случаях развивается миоглоби-
нурия. Эти симптомы указывают на то, что миалгия
обусловлена повреждением мышц в результате
фасцикуляций. Тем не менее, согласно ряду иссле-
дований, частота послеоперационной миалгии не
находится в прямой зависимости от мышечных
фасцикуляций.
Таблица 7.2. Нежелательные побочные эффекты
суксаметония
Мышечные фасцикуляции
Послеоперационная миалгия
Гиперкалиемия
Тахифилаксия и двухфазное действие
Повышение внутриглазного давления
Злокачественная гипертермия
Парасимпатомиметические эффекты
Брадикардия
Увеличение секреции мокроты
в дыхательных путях
Повышение давления в желудке
Повышение тонуса матки
Длительная нервно-мышечная блокада при за-
медлении метаболической деградации
3. Гиперкалиемия. Деполяризация двигатель-
ной концевой пластинки и последующие мышеч-
ные сокращения способствуют выходу К+ во вне-
клеточную жидкость. В норме уровень калия в плаз-
ме повышается примерно на 0,5 ммоль/л, однако
это редко имеет клиническую значимость. Напро-
тив, при тяжелых ожогах и некоторых неврологиче-
ских нарушениях (например, при травме спинно-
го мозга) развивается гораздо более значительное
повышение уровня К+ в плазме (на несколько
ммоль/л). При неврологических нарушениях особен-
но выраженное увеличение концентрации калия
регистрируется в венозной крови, оттекающей от
парализованных или поврежденных конечностей.
От момента неврологического повреждения, сопро-
вождающегося обширными параличами, до раз-
вития этого осложнения проходит определенный
латентный период. Считается, что патологическая
гиперкалиемия служит проявлением денервацион-
ной гиперчувствительности. В денервированных
или сильно поврежденных мышцах (а также у пло-
да) постсинаптические холинорецепторы распола-
гаются не только в области двигательной концевой
пластинки, но и по всей поверхности мышечного во-
локна. Повышенное высвобождение калия из мы-
шечных клеток обусловлено обширной зоной депо-
ляризации мышечной мембраны суксаметонием.
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС
147
medlit
Описаны многочисленные случаи остановки
сердца после введения суксаметония при обшир-
ных ожогах и некоторых неврологических наруше-
ниях (особенно в период с 20-х по 60-е сут после
возникновения паралича). Хотя явные признаки
повреждения или денервации мышц могут отсутст-
вовать, главной причиной остановки сердца, веро-
ятно, является гиперкалиемия. Способствующие
факторы включают гиповолемию, ацидоз и нару-
шение равновесия между симпатической и пара-
симпатической нервной системой. При исходной
гиперкалиемии (например, в случае почечной не-
достаточности или тяжелого ацидоза) применение
сукцинилхолина сопряжено с особенно высоким
риском гиперкалиемической остановки сердца.
4. Тахифилаксия и двухфазное действие. Эти
феномены обусловлены десенситизацией рецепто-
ров или блокадой ионных каналов.
5. Повышение внутриглазного давления. Сукса-
метоний вызывает повышение внутриглазного дав-
ления, которое может сохраняться в течение не-
скольких минут. Следовательно, миорелаксант
следует с осторожностью применять при прони-
кающих ранениях глаз и открытых офтальмологи-
ческих операциях. Глазодвигательные мышцы
обильно иннервируются (каждое волокно имеет
несколько двигательных концевых пластинок) от-
носительно медленно проводящими аксонами Ау.
Особенностями иннервации глазодвигательные
мышцы напоминают произвольные мышцы птиц, в
которых суксаметоний вызывает медленное стой-
кое сокращение (контрактуру). Видимо, подобные
эффекты развиваются и в глазодвигательных мыш-
цах человека.
6. Злокачественная гипертермия — это наслед-
ственное заболевание, поражающее систему регу-
ляции содержания кальция в мембране мышечных
волокон. Суксаметоний является провоцирующим
фактором злокачественной гипертермии у больных
с наследственной предрасположенностью. Введе-
ние суксаметония вместо миорелаксации приводит
к генерализованной мышечной ригидности. В тече-
ние нескольких минут повышается температура
тела. При этом заболевании суксаметоний вызыва-
ет высвобождение Са2+ из саркоплазматического
ретикулума, что приводит к стойкому сокращению
и последующему повреждению мышц.
7. Парасимпатические эффекты. Своим химиче-
ским строением суксаметоний напоминает ацетил-
холин и оказывает некоторое воздействие на муска-
риновые холинорецепторы (м-холинорецепторы).
Высокая однократная доза или повторное введение
суксаметония может привести к брадикардии или
атриовентрикулярному узловому ритму (особенно
ucoz.ru _ ,,
у детей). Эти эффекты усиливаются дигоксином
или другими лекарственными препаратами, повы-
шающими парасимпатическую активность (Р-адре-
ноблокаторы). Нередко наблюдаются и другие
эффекты, обусловленные влиянием на м-холиноре-
цепторы (например, усиление бронхиальной секре-
ции и слюноотделение), особенно при повторном
введении суксаметония. Повышается мышечный
тонус желудка и давление внутри него, хотя этот
эффект отчасти обуславливается фасцикуляциями
мышц передней брюшной стенки. Повторные дозы
суксаметония заметно повышают тонус матки во
время кесарева сечения. Все эти парасимпатиче-
ские эффекты можно предотвратить или ослабить
предварительным введением атропина.
8. Длительное апноэ. Иногда замедленный ме-
таболизм суксаметония может быть причиной
длительной мышечной слабости и апноэ в после-
операционном периоде.
Преходящее действие однократной дозы сукса-
метония обусловлено его быстрым гидролизом под
действием псевдохолинэстеразы. Расщепление
миорелаксанта происходит в два этапа. Вначале
образуется сукцинилмонохолин со слабой миоре-
лаксирующей активностью (примерно 5% мощно-
сти суксаметония), который впоследствии распа-
дается до янтарной кислоты и холина. Причины
продленного действия суксаметония включают
снижение активности или низкий уровень псевдо-
холинэстеразы, а также сочетанное применение ле-
карственных препаратов, угнетающих активность
псевдохолинэстеразы.
Псевдохолинэстераза синтезируется в печени;
при тяжелой дисфункции печени с выраженной ги-
поальбуминемией уровень псевдохолинэстеразы
значительно снижается, что может привести к неко-
торому увеличению продолжительности действия
суксаметония. При генетических нарушениях, при-
водящих к серьезному снижению активности псев-
дохолинэстеразы, увеличение продолжительности
действия суксаметония может быть более значи-
тельным.
Лекарственные препараты, подавляющие ак-
тивность псевдохолинэстеразы, увеличивают про-
должительность действия суксаметония. Первона-
чально с этой целью использовались тетрагидро-
аминакрин и гексафторений, хотя их действие
ненадежно и непредсказуемо. Более значимые ле-
карственные взаимодействия (пролонгирование
нервно-мышечной блокады, тахифилаксия и повы-
шенный риск двухфазного действия) возникают
при одновременном использовании суксаметония
с субстратами или ингибиторами псевдохолинэсте-
разы. К ним относятся эдрофоний, неостигмин,
148
Глава 7
medlit.ucoz.ru
фосфорорганические соединения, алкилирующие
вещества, триметафан, питоцин и местные анесте-
тики эфирного типа.
Декаметоний
Декаметоний впервые использовали в клиниче-
ской практике в 1949 г., после того как были прове-
дены исследования курареподобной активности
ряда бисчетвертичных соединений. Первоначально
считалось, что препарат меньше угнетает дыхание,
поскольку не действует на диафрагму; однако вско-
ре стало ясно, что доза препарата, обеспечивающая
эффективную миорелаксацию, приводит к парали-
чу дыхания.
Декаметоний характеризуется большей ста-
бильностью гемодинамики, отсутствием или низ-
ким высвобождением гистамина, а также меньшей
частотой мышечных фасцикуляций и послеопера-
ционной миалгии по сравнению с суксаметонием.
Декаметоний широко применялся в экспери-
ментальных исследованиях, но в клинической прак-
тике в Великобритании не используется с 1958 г.
НЕДЕПОЛЯРИЗУЮЩИЕ
МИОРЕЛАКСАНТЫ
Все недеполяризующие миорелаксанты содер-
жат четвертичные аммониевые группы и имеют
громоздкую молекулярную структуру. Они оказы-
вают сходное действие на нервно-мышечный си-
напс, но значительно отличаются по фармакокине-
тическим свойствам и побочным эффектам (влия-
ние на кровообращение, выброс гистамина).
Характеристики недеполяризующих миорелаксан-
тов представлены в табл. 7.3.
Кураре
Классический недеполяризующий миорелак-
сант кураре — это собирательный термин, обозна-
чающий различные яды, которыми южноамерикан-
ские индейцы в течение многих столетий смазыва-
ли наконечники стрел и дротиков для охоты на
диких животных. Строго говоря, под кураре пони-
мают содержащие несколько различных алкалои-
дов неочищенные экстракты некоторых видов рас-
тений Chondrodendron и Strychnos. Приготовление
кураре было окутано мистикой и сопровождалось
ритуалами, а образцы яда, впервые попавшие в ци-
вилизованные страны, классифицировались по
контейнерам для транспортировки: горшочный ку-
раре доставлялся в глиняных сосудах, трубочный —
в стеблях бамбука, а калабаш-кураре — в тыквах.
Научный интерес к кураре возник в 1856 г. благода-
ря наблюдениям Клода Бернара. Он правильно
определил местом действия кураре нервно-мышеч-
ный синапс, поскольку яд не влиял на нервную про-
водимость и не подавлял мышечный ответ на
прямую стимуляцию.
В 1932 г. началось клиническое применение ку-
раре; его очищенные фракции использовались для
лечения столбняка и спастических состояний.
Позднее препарат стал применяться для купиро-
вания судорог от пентилентетразола, применяв-
шегося для лечения психических заболеваний.
В качестве миорелаксанта во время анестезии
тубокурарин был впервые применен в Канаде
Гриффитом и Джонсоном в 1942 г, а в Англии Гре-
ем и Гальтоном в 1945 г.
Продолжительность действия тубокурарина дос-
таточна велика и относительно непредсказуема. Он вы-
зывает выраженное высвобождение гистамина, что
провоцирует артериальную гипотонию и реакции
гиперчувствительности. Появление более безопас-
ных миорелаксантов и высокая стоимость произ-
водства стали причиной исчезновения тубокурари-
на из клинической практики в Великобритании.
Галламин
Первый синтетический миорелаксант галламин
был внедрен в клиническую практику Бове (Bovet)
в 1947 г. и в течение многих лет оставался единст-
венной альтернативой тубокурарину. По сравне-
нию с тубокурарином галламин обладает более ко-
роткой продолжительностью действия и вызывает
менее выраженное высвобождение гистамина.
Однако у него есть два существенных недостатка:
1. Способность вызывать тахикардию в сочета-
нии с артериальной гипертонией.
2. Выраженная зависимость от почечной элими-
нации, что исключает его применение при почеч-
ной недостаточности и гиповолемии.
В настоящее время галламин редко использует-
ся в клинической практике.
Алкуроний
Алкуроний — полусинтетическое производ-
ное алкалоида кураре токсиферина; впервые был
применен в Великобритании в 1962 г. Алкуроний
обладает относительно длительной продолжи-
тельностью действия и в значительной степени
выделяется через почки. Кроме того, алкуроний
часто вызывает реакции гиперчувствительности.
Эти факторы в сочетании со сложностью промыш-
ленного производства привели к прекращению ис-
пользования алкурония в Великобритании.
Панкуроний
Панкуроний — бисчетвертичный аммониевый
стероид; применяется в Великобритании с 1967 г.
Препарат обладает длительным действием (особен-
но у пожилых) и вызывает те же гемодинамические
Таблица 7.3. Фармакологические характеристики недеполяризующих миорелаксантов
Начало действия Высвобо- ждение гистамина Гемодина- мические эффекты Продолж. действия Связыва- ние с белками плазмы (%) Объем рапре- деления (мл/кг) Клиренс (мл/мин/кг) т,, (мин) Элиминация
Тубокура- рин Медленное Часто Артериаль- ная гипотония Высокая 30-50 290 1.5 190 Метаболизму не подвергается; элиминация с мочой (70%) и желчью (30%)
Алкуроний Промежу- точное Часто Артериаль- ная гипото- ния Высокая < 20 300 1.5 200 Метаболизму не подвергается; элиминация с мочой (70%) и желчью (30%)
Галламин Быстрое Редко Тахикардия Средняя <20 230 1.2 150 Метаболизму не подвергается; элиминация с мочой (100%)
Панкуроний Промежу- точное Редко Тахикардия Высокая 20-60 270 1.5 130 Метаболизм (30%) до трех деацетилирован- ных метаболитов; элиминация (70%) в неиз- мененном виде (в основном с мочой)
Векуроний Промежу- точное Редко Отсутствуют Короткая < 20 230 4,0 60 Метаболизм (20%) до деацетилированных соединений; элиминация (80%) в неизменен- ном виде (в основном с желчью)
Атракурий Промежу- точное Редко Отсутствуют Короткая <20 160 5.5 20 Метаболизм (95%) до лаудонозина (по меха- низму Хофмана), четвертичного акрилата, ки- слоты и спирта; элиминация (5%) в неизме- ненном виде
Мивакурий цис-цис цис-транс гране-iране Промежу- точное Редко Отсутствуют Короткая < 20 320 210 210 5.5 90 90 55 2 2 Метаболизм (95%) псевдохолинэстеразой до четвертичной кислоты и спирта; элимина- ция (5%) в неизмененном виде
Рокуроний Быстрое Редко Отсутствуют Средняя <20 210 4.5 85 Элиминация с желчью (55%) и мочой (35%); может расщепляться до деацетилированных метаболитов
цис-Атраку- рий Промежу- точное Редко Отсутствуют Короткая <20 140 5.2 25 Метаболизм по мех«энизму Хофмана и неспе- цифическими эстеразами плазмы |
Для объема распределения, клиренса и Т)/? приведены медианы значений, полученных у пациентов без дисфункции печени и почек
medlit.ucoz.ru
150
Глава?
medlit.ucoz.ru
эффекты, что и галламин. Панкуроний провоциру-
ет тахиаритмии, особенно на фоне лечения трицик-
лическими антидепрессантами; это позволяет
предположить, что панкуроний оказывает сумма-
ционный или потенцирующий эффект на обратный
захват катехоламинов. Панкуроний также угнетает
псевдохолинэстеразу. В последние годы его приме-
няют все реже.
Векуроний
Векуроний — моночетвертичный аналог панку-
рония; впервые был применен в 1980 г. Векуроний
относительно нестабилен в водном растворе и вы-
пускается в виде лиофилизированного порошка, ко-
торый растворяют непосредственно перед исполь-
зованием. Он не влияет на сердечно-сосудистую
систему и редко высвобождает гистамин, поэтому
считается миорелаксантом выбора при атопии или
реакциях гиперчувствительности в анамнезе.
Атракурий
Атракурий относится к бензилизохинолинам.
В Великобритании его начали применять в 1982 г.
Благодаря особенностям химического строения ат-
ракурий частично спонтанно расщепляется in
vivo (путем спонтанной деградации, она же элими-
нация Хофмана). Он также метаболизируется эсте-
разами легких и плазмы до моночетвертичного
спирта и кислоты. Гемодинамические побочные
эффекты наблюдаются редко, высвобождение гис-
тамина — умеренное. В настоящее время атракурий
широко применяется в практике.
Мивакурий
Мивакурий стал применяться в Великобрита-
нии с 1993 г. и завоевал популярность при амбула-
торных и других непродолжительных плановых
операциях, требующих интубации трахеи и ИВ Л.
Мивакурий является бензилизохинолиновым
эфиром. По строению он напоминает атракурий,
но более подвержен расщеплению псевдохолин-
эстеразой, нежели элиминации Хофмана. Миваку-
рий является хиральной смесью трех стереоизоме-
ров (цис-транс 36%, транс-транс 58%, цис-цис 6%).
Цис-цис-пзомер обладает лишь десятой долей мощ-
ности двух других изомеров, но не подвергается
ферментативному расщеплению, что отражается
фармакокинетическими данными (табл. 7.3).
По сравнению с атракурием, мивакурий начина-
ет действовать медленнее, но продолжительность
его действия короче. Эти эффекты носят дозоза-
висимый характер: при увеличении дозы препарат
начинает действовать быстрее, но действует доль-
ше. Мивакурий лишен побочных гемодинамиче-
ских эффектов. Как и атракурий, обладает слабым
гистамин-высвобождающим действием.
Рокуроний
Рокуроний — это аммониевый стероид, при-
меняющийся в анестезиологической практике с
1995 г. Структурный аналог векурония, но обладает
лишь одной восьмой мощности соединения-прото-
типа. Считается, что его низкая мощность обуслав-
ливает быстрое начало действия (60-90 сек), хотя
продолжительность действия такая же, как у экви-
валентных доз атракурия и векурония. Рокуроний
может вызывать слабые ваголитические (т. е. анти-
холинергические) эффекты.
цмс-Атракурий
Применяющийся в Великобритании с 1997 г.
цис-Атракурий является одним из 10 стереоизо-
меров атракурия. По фармакокинетическим свой-
ствам этот миорелаксант аналогичен атракурию
и в значительной степени элиминируется путем
гидролиза эфирной связи и по механизму Хофмана.
По сравнению с атракурием, цмс-атракурий в 3-5
раз мощнее (если сравнивать дозы, угнетающие
электромиографическую реакцию на 95%), но на-
чинает действовать медленнее. Гемодинамические
эффекты и высвобождение гистамина незначитель-
ны даже при относительно высоких дозах.
Пипекуроний
Пипекуроний — миорелаксант с медленным на-
чалом и большой продолжительностью действия,
многие годы использующийся в странах Восточной
Европы. Пипекуроний имеет стероидную структу-
ру, обладает такой же мощностью, что и векуроний,
но лишен ваголитического побочного действия.
Доксакурий
Доксакурий применяется в США с 1992 г. Это наи-
более мощный из существующих миорелаксантов,
не имеющий, впрочем, каких-либо преимуществ
перед другими современными миорелаксантами.
Скорость развития нервно-мышечной блокады и ее
продолжительность достаточно непредсказуемы.
СРАВНИТЕЛЬНЫЕ ХАРАКТЕРИСТИКИ
НЕДЕПОЛЯРИЗУЮЩИХ
МИОРЕЛАКСАНТОВ
Влияние на кровообращение
Большинство недеполяризующих миорелаксан-
тов вызывают снижение АД непосредственно после
введения. Это может быть следствием высвобожде-
ния гистамина, блокады вегетативных ганглиев или
снижения венозного возврата из-за утраты мышеч-
ного тонуса.
Тубокурарин обычно снижает артериальное дав-
ление на 20-30 мм рт. ст. и даже больше. Рефлектор-
ная тахикардия развивается редко; напротив, ЧСС
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС 151
medlit.ucoz.ru
обычно слегка уменьшается. Причинами артери-
альной гипотонии являются высвобождение гиста-
мина и блокада симпатических ганглиев. От-
сутствие рефлекторной тахикардии указывает,
что ключевую роль в снижении АД играет ганглио-
нарная блокада. Алкуроний вызывает аналогич-
ные эффекты, хотя значительное высвобождение
гистамина происходит реже.
Напротив, галламин и панкуроний часто вызы-
вают тахикардию. Галламин обычно увеличивает
ЧСС на 20-30 ударов в минуту; при этом АД не
изменяется или повышается. Галламин вызывает кон-
формационные изменения в М2-холинорецепторах
сердца, снижая их сродство (аффинность) к аце-
тилхолину. Атропин, действие которого частично
обусловлено блокадой постсинаптических Мз-хо-
линорецепторов, может усугублять вызванную гал-
ламином тахикардию. Кроме того, галламин может
увеличивать высвобождение норадреналина в ко-
нечных ветвях симпатических нервов сердца.
Панкуроний увеличивает ЧСС, АД и венозный
возврат, хотя в меньшей степени, чем галламин.
Панкуроний оказывает сложное воздействие на ве-
гетативную иннервацию сердца: он подавляет об-
ратный захват катехоламина, а также оказывает
м-холиноблокирующий эффект, косвенно повы-
шающий высвобождение норадреналина.
Современные недеполяризующие миорелаксан-
ты обычно не влияют на сердечно-сосудистую сис-
тему. На фоне их действия могут развиваться бра-
дикардия и артериальная гипотония, но это, как
правило, обусловлено одновременным применени-
ем других лекарственных препаратов, повышаю-
щих тонус блуждающего нерва. Применение атра-
курия во время галотановой анестезии может при-
вести к тяжелой брадикардии.
Высвобождение гистамина
Как и другие лекарственные препараты-основа-
ния (опиоидные анальгетики, триметафан), миоре-
лаксанты способны высвобождать гистамин и дру-
гие биологически активные вещества из тучных
клеток. Тубокурарин и атракурий часто вызывают
местные реакции, состоящие в появлении красных
пятен и волдырей, отеке и вазодилатации. Предпо-
лагают, что такие реакции обусловлены высвобож-
дением гистамина из тучных клеток прилежащей
сосудистой стенки вследствие высокой локальной
концентрации миорелаксанта. Эти местные реак-
ции редко перерастают в системные.
Иногда миорелаксанты вызывают и генерализо-
ванные реакции гиперчувствительности, приводя-
щие к опасному для жизни бронхоспазму и артери-
альной гипотонии. Приблизительно в 65% случаев
это реакции гиперчувствительности I типа; в анам-
незе, как правило, есть указания на атопию и пред-
шествующий контакт с препаратом. В сыворотке
выявляют специфические антитела IgE, а внутри-
кожная проба с миорелаксантом дает положитель-
ный результат.
В остальных случаях вероятной причиной ос-
ложнений является анафилактоидная реакция.
Иногда она опосредуется прямым воздействием на
циркулирующие базофилы и зависит от дозы и ско-
рости введения миорелаксанта. Другим механиз-
мом является активация альтернативного пути
комплемента белком проперидином с последую-
щим высвобождением гистамина. Анафилактоид-
ная реакция развивается без предшествующего
контакта с препаратом и ее клинические проявле-
ния обычно не так тяжелы, как при истинной ана-
филактической реакции.
Тубокурарин, алкуроний и суксаметоний вызы-
вают как анафилактические, так и анафилактоид-
ные реакции. Атракурий, мивакурий и даже аммо-
ниевые стероиды, не вызывающие значительного
высвобождения гистамина, также могут провоци-
ровать эти реакции. Полипрагмазия в современной
анестезиологической практике часто затрудняет
выявление антигена. Лечение реакций гиперчувст-
вительности рассматривается в главе 4.
Распределение и фармакокинетика
Поскольку все недеполяризующие миорелак-
санты являются четвертичными аммониевыми ос-
нованиями, они плохо всасываются в кишечнике.
После в/в инъекции миорелаксанты преимущест-
венно распределяются во внеклеточной жидкости,
частично связываясь с белками плазмы. Степень
связывания миорелаксантов с белками интенсивно
изучали in vitro, например, с помощью плазмафере-
за, равновесного диализа, ультрафильтрации.
Большинство исследований свидетельствуют о том,
что 30-50% тубокурарина и 20-60% панкурония
связывается с IgG и альбумином, и лишь неболь-
шая часть остальных миорелаксантов связывается
с белками плазмы.
Различия в связывании миорелаксантов с бел-
ками плазмы вряд ли имеют особую практичес-
кую значимость. При повышении концентрации IgG
в плазме (обширные ожоги и заболевания печени)
развивается резистентность к тубокурарину и пан-
куронию, но неясно, насколько этот феномен обу-
словлен именно изменением связывания миоре-
лаксантов с белками. В прошлом предпринимались
попытки найти корреляцию между эффективной
дозой различных миорелаксантов и концентрацией
плазменных белков. Установлена положительная
зависимость между эффективной дозой тубокура-
рина и концентрацией IgG в плазме, а также между
152
Глава?
дозой алкурония и концентрацией ал
Эффективная доза панкурония не зависела ни от
одной белковой фракции плазмы.
Благодаря своему химическому строению неде-
поляризующие миорелаксанты практически не
проникают через гематоэнцефалический и плацен-
тарный барьер. Тем не менее небольшие концентра-
ции миорелаксантов (обычно < 5% материнской)
можно обнаружить в крови плода и пуповинной
крови. Применение недеполяризующих миорелак-
сантов во время кесарева сечения может быть при-
чиной апноэ плода лишь в очень редких случаях.
Вместе с тем, многократное введение миорелаксан-
тов при длительной ИВЛ у беременных вполне
может быть причиной депрессии дыхания у ново-
рожденного. Галламин проникает через плаценту
в большей степени, чем другие миорелаксанты, и его
применение в акушерстве противопоказано.
После в/в инъекции концентрация недеполяри-
зующего миорелаксанта в плазме быстро снижает-
ся в течение 2-10 мин, что в основном вызвано
поступлением препарата в клетки печени и почек.
В небольших концентрациях миорелаксанты лока-
лизуются в области двигательных концевых пла-
стинок, и происходит их неспецифическое связыва-
ние с анионными участками синаптической щели
и на базальной мембране мышечных волокон. Затем
наступает замедленная фаза экспоненциального
снижения концентрации, которая отражает экс-
крецию миорелаксанта через почки или с желчью,
а также метаболизм препарата.
Распределение большинства миорелаксантов
изучалось в рамках двух- и трехкамерных моделей.
Полученные константы были использованы для
расчета фармакокинетических параметров. Кроме
того, эти параметры можно рассчитать и другими
способами (глава 2). Объем распределения боль-
шинства миорелаксантов приблизительно равен
объему внеклеточной жидкости (200-400 мл/кг)
или слегка превышает его. Клиренс миорелаксантов
колеблется между 1,2 и 5,5 мл/мин/кг (за исключе-
нием мивакурия), отражая их конечный Тш и соот-
ветствуя относительной продолжительности дей-
ствия (таблица 7.3). Во многих случаях существует
четкая корреляция между сывороточной концен-
трацией миорелаксанта в равновесном состоянии
и глубиной нервно-мышечной блокады.
Заболевания почек и печени влияют на фарма-
кокинетику недеполяризующих миорелаксантов.
При почечной недостаточности из-за снижения
клиренса Ti/2 большинства недеполяризующих
миорелаксантов увеличивается. Однако Т1/2 и кли-
ренс атракурия и векурония существенно не меня-
ются. При заболеваниях печени изменениям под-
фармакокинетические характеристики
тех миорелаксантов, которые в основном элимини-
руются с желчью (например, векурония).
Метаболизм и элиминация
Большинство миорелаксантов частично вы-
деляется с мочой в неизмененном виде и подверга-
ется незначительному метаболизму ферментными
системами печени. Все миорелаксанты поступают
в гепатоциты, и некоторые из них в значительной
степени выделяются с желчью (в зависимости от
количества четвертичных центров и молекулярно-
го веса).
Тубокурарин практически не подвергается ме-
таболизму в печени или других тканях. В норме он
элиминируется в неизмененном виде с мочой, хотя,
являясь моночетвертичным соединением с молекуляр-
ным весом более 400 Да, в небольшом количестве
присутствует в желчи. При почечной недостаточно-
сти доля выделяемого с желчью тубокурарина зна-
чительно возрастает. Сходный по строению миоре-
лаксант алкуроний точно так же не подвергается
метаболизму, а выделяется в неизмененном виде с
мочой и желчью.
Галламин практически полностью выделяется
в неизмененном виде с мочой. Только следовые
количества галламина выделяются с желчью,
поскольку он является низкомолекулярным со-
единением и содержит более одной четвертичной
группы. Вследствие этого при почечной недоста-
точности клиренс галламина значительно снижает-
ся, а Т|/2 увеличивается в 10-20 раз.
Основная доля панкурония выводится с мочой
в неизмененном виде, но 15-40% его подвергается
метаболизму в печени с образованием трех де-
ацетилированных соединений (3-гидроксипан-
курония, 17-гидроксипанкурония и 3,17-дигид-
роксипанкурония). Один из этих метаболитов,
3-гидроксипанкуроний, обладает активностью не-
деполяризующего миорелаксанта (составляющей
половину от мощности панкурония). В течение
некоторого времени он использовался как миоре-
лаксант (дакуроний). С желчью выделяются
лишь следовые количества панкурония.
Напротив, векуроний, как моночетвертичное
соединение, в основном элиминируется из организ-
ма путем активной секреции в желчь. Векуроний
также выделяется с мочой и в небольшом количест-
ве деацетилируется в печени. Рокуроний экскрети-
руется с желчью (приблизительно 55%) и мочой
(35%), а также в небольшом количестве образует
деацетилированные метаболиты. Цирроз печени
может повлиять на начало и продолжительность
действия этих миорелаксантов; их действие слегка
удлиняется при почечной недостаточности.
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС
153
Атракурий является уникальным миоретЛ^Ш'^4$ffStT|4cmeiiлении образуется меньше лаудано-
том: его элиминация происходит двумя путями —
гидролизом эфирной связи эстеразами и спонтан-
ной деградацией (отщеплением N-алкиловой груп-
пы). При pH 7,4 и температуре 37° С происходит
спонтанная химическая реакция (элиминация
Хофмана), и миорелаксант быстро расщепляется
с образованием третичного амина лауданозина.
Поскольку атракурий является бисчетвертичным
эфиром, он также метаболизируется эстеразами
легких и плазмы до моночетвертичногоспирта
и моночетвертичной кислоты. Хотя лауданозин
является биологически активным веществом
(антагонист глицина), его накопление в организме
при заболеваниях почек или печени маловероятно.
Большим преимуществом атракурия является
спонтанное восстановление нервно-мышечной
проводимости, поэтому он считается препаратом
выбора при почечной и печеночной недостаточно-
сти. Днс-атракурий также элиминируется гидроли-
зом эфирной связи и спонтанной деградацией.
Хотя цме-атракурий в 3-5 раз активнее атракурия,
зина.
Два главных изомера мивакурия быстро расще-
пляются псевдохолинэстеразой, что отражается
их высоким клиренсом (90 мл/мин/кг). При цир-
розе печени синтез псевдохолинэстеразы снижает-
ся, и клиренс значительно замедляется. Генетиче-
ские вариации фермента также приводят к заметно-
му удлинению действия мивакурия. Клиренс
цис-цис изомера снижается при почечной недоста-
точности.
pH внеклеточной жидкости
Эффекты тубокурарина усиливаются и пролон-
гируются при респираторном ацидозе, но ослабева-
ют при респираторном алкалозе. Это объясняется
влиянием pH на физико-химические свойства
тубокурарина, молекула которого содержит одну
третичную и одну четвертичную аминогруппу
(рис. 7.7). В кислой среде ионизация третичной
аминогруппы (рК,“ 8) усиливает действие тубоку-
рарина. Сходное влияние на ионизацию оказывают
Тубокурарин
Алкуроний
Рис. 7.7. Структур-
ные формулы неко-
торых недеполяри-
зующих миорелак-
сантов
Галламин
О —
I -О—CMjCH/ClC/Sh
154
Глава?
Векуроний
Рис. 7.7. (продолжение)
Атракурий
Мивакурий
Рокуроний
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС
155
метаболический ацидоз и
Осложняющим фактором в этих условиях является
перемещение К+ между клетками и внеклеточной
жидкостью. Действие галламина усиливается при
алкалозе и ослабляется при ацидозе.
На другие миорелаксанты колебания pH в фи-
зиологических пределах обычно не влияют. Любое
возникшее изменение в действии миорелаксантов
скорее всего будет обусловлено электролитными
нарушениями, развившимися вследствие колеба-
ний pH.
МИОРЕЛАКСАНТЫ БУДУЩЕГО
Поиски совершенного миорелаксанта продол-
жаются. Идеальный миорелаксант должен быть не-
деполяризующим и создавать оптимальные усло-
вия для интубации трахеи через 1 мин после введе-
ния. Тотальная миорелаксация должна
продолжаться 5-10 мин, а полное спонтанное
восстановление нервно-мышечной проводимости
(вызванный мышечный ответ при одиночной сти-
муляции не менее 95% от исходного; Т4/Т1 > 0,7
при стимуляции в TOF-режиме) — наступать в те-
чение 15-20 мин после введения препарата. В слу-
чае медленного восстановления нервно-мышечной
проводимости блокада должна устраняться инги-
битором АХЭ. Идеальный миорелаксант не должен
вызывать высвобождения гистамина, значимо вли-
ять на гемодинамику и взаимодействовать с други-
ми лекарственными препаратами. Эффективность
миорелаксанта не должна зависеть от состояния пе-
чени и почек, а также колебаний pH.
Исследования показали, что быстрое развитие
недеполяризующей блокады достигается только
высокими дозами миорелаксантов. При использо-
вании мощного миорелаксанта такого преимущест-
ва можно добиться только ценой удлинения блока-
ды; для менее мощных миорелаксантов значение
имеют стоимость препарата и побочные эффекты.
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ,
ВЛИЯЮЩИЕ НА ГИДРОЛИЗ
АЦЕТИЛХОЛИНА
Ингибиторы АХЭ (синоним: антихолинэсте-
разные средства) препятствуют гидролизу ацетил-
холина, угнетая активность ацетилхолинэстеразы.
В результате происходит накопление ацетилхо-
лина во всех холинергических синапсах. Все инги-
биторы АХЭ воздействуют на нервно-мышечную
и вегетативную нервную систему, а некоторые из
них (физостигмин, большинство фосфорорганиче-
ских соединений) оказывают влияние и на ЦНС.
тва также подавляют активность псевдо-
холинэстеразы и увеличивают продолжительность
действия суксаметония.
Выделяют три основные группы ингибиторов
АХЭ:
1. Эдрофоний
2. Карбаматы
3. Фосфорорганические соединения
ЭДРОФОНИЙ
Эдрофоний является фенолсодержащим чет-
вертичным аммониевым основанием простой хи-
мической структуры (рис. 7.8). Четвертичная груп-
па взаимодействует с анионным участком фермен-
та АХЭ (рис. 7.3); кроме того, образуется быстро
обратимая водородная связь с активным центром.
Эдрофоний оказывает прямое воздействие на кон-
цевые ветви двигательного нерва. После в/в введе-
ния эдрофония возникают мышечные фасцикуля-
ции, часто сопровождаемые эффектами стимуляции
м-холинорецепторов (брадикардия, увеличение
секреции желез, повышение тонуса гладких
мышц). Являясь четвертичным аммониевым осно-
ванием, эдрофоний не проникает через гематоэнце-
фалический и плацентарный барьеры. Эдрофоний
быстро распределяется во внеклеточной жидкости,
но также поступает в печеночные и почечные клет-
ки. Концентрация препарата в плазме снижается
по биэкспоненциальному типу, и Т и не превышает
2 мин; затем снижение концентрации замедляется
(конечный Ti/2« 25-45 мин). Угнетение АХЭ быст-
ро прекращается по мере снижения концентрации
эдрофония в плазме, поэтому действие препарата
носит преходящий характер. Эдрофоний элимини-
руется путем конъюгации с глюкуронидом, а также
выделяется с мочой в неизмененном виде.
Эдрофоний (2-10 мг в/в) применяется для под-
тверждения диагноза миастении, а также для диф-
ференциальной диагностики миастенического и
холинергического криза при верифицированном
диагнозе миастении. В анестезиологии он иногда
используется для верификации второй фазы дейст-
вия суксаметония или декаметония.
В последние годы эдрофоний стали использо-
вать для устранения остаточного действия векуро-
ния, атракурия и мивакурия. Во избежание рекура-
ризации доза эдрофония должна быть относитель-
но высока (0,5-1,0 мг/кг).
КАРБАМАТЫ
Карбаматы представляют собой третичные ами-
ны или четвертичные аммониевые основания, но
при физиологических значениях pH они сущест-
вуют в виде ионов. Пространственная структура
156
Глава?
молекулы карбаматов сходна с таковой и вегетативные функции. Кроме того,
лина, поэтому они конкурируют с ацетилхолином
за связывание с АХЭ: аминогруппы связываются с
анионными участками АХЭ, а карбамоильные
группы (рис. 7.8) — с остатком серина активного
центра. Часть молекулы быстро диссоциирует, ос-
тавляя карбамоилированную АХЭ. Таким образом,
карбамоилированная АХЭ напоминает ацетилиро-
ванную АХЭ, образующуюся при взаимодействии
АХЭ с ацетилхолином (рис. 7.3). Следует учесть
выраженные фармакокинетические различия: гид-
ролиз карбамоилированной АХЭ (Ti/з — приблизи-
тельно 30 мин) происходит в 107— 108 раз медленнее
гидролиза ацетилированной АХЭ (Т]/2 = 42 мксек).
Именно карбамоилирование является причиной
угнетения активности АХЭ и накопления ацетил-
холина. К реактивации фермента приводит отщеп-
ление карбамоильной группы. Возможно, благода-
ря химическому строению, напоминающему аце-
тилхолин, карбаматы (особенно неостигмин)
оказывают прямое воздействие на нервно-мышеч-
они угнетают псевдохолинэстеразу плазмы и про-
лонгируют эффекты суксаметония.
Неостигмин и пиридостигмин оказывают сход-
ное воздействие на нервно-мышечное проведение и
вегетативную нервную систему. Поскольку оба ле-
карственных препарата являются четвертичными
аммониевыми основаниями, они плохо проникают
через гематоэнцефалический и плацентарный
барьеры.
Неостигмин в 4-5 раз мощнее пиридостигмина,
поскольку его четвертичная группа находится вне
ароматического кольца (рис. 7.8). Оба лекарствен-
ных препарата плохо всасываются в ЖКТ, и их
мощность после приема внутрь составляет только
10-20% от мощности при парентеральном введе-
нии. Неостигмин всасывается быстрее пиридостиг-
мина и в большей степени подвергается эффекту
первого прохождения через печень. После в/в вве-
дения неостигмин начинает действовать быстрее
пиридостигмина, и его максимальный эффект раз-
Эдрофоний
Неостигмин
Пиридостигмин
Физостигмин
Рис. 7.8. Струк-
турные формулы
ингибиторов АХЭ
СН3 СН3
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС
157
medlit.ucoz.ru
вивается через 5-7 мин. Продолжительность дейст-
вия неостигмина и его конечный Т|/2 (30-45 мин)
короче, чем у пиридостигмина. Достаточно дли-
тельное действие неостигмина и пиридостигмина,
продолжающееся после их элиминации из плазмы
и обычно предотвращающее рекураризацию, сви-
детельствует о низкой скорости гидролиза ингиби-
рованной карбоилированной АХЭ в синаптической
щели. Оба лекарственных препарата частично под-
вергаются метаболизму в печени, а частично выде-
ляются в неизмененном виде с мочой. Пиридостиг-
мин оказывает менее выраженное побочное дейст-
вие на вегетативную нервную систему, чем
неостигмин, и одно время использовался для устра-
нения остаточного действия недеполяризующих
миорелаксантов.
Неостигмин и пиридостигмин часто применяют
для длительного лечения миастении, обычно на-
значая внутрь. К их вегетативным побочным эф-
фектам может развиваться толерантность, поэто-
му не всегда возникает необходимость в использо-
вании м-холиноблокаторов. Неостигмин также
применяют для лечения наджелудочковой тахи-
кардии и стимуляции гладкой мускулатуры моче-
вого пузыря и кишечника, особенно в послеопера-
ционном периоде.
Дистигмин состоит из двух молекул пиридо-
стигмина, нечетвертичные концы которых связаны
метиленовой цепочкой. Дистигмин обладает отно-
сительно большой продолжительностью действия
и применяется для лечения послеоперационного
пареза кишечника или задержки мочи.
Третичный амин физостигмин (эзерин) — это
алкалоид, получаемый из высушенных зрелых
семян физостигмы ядовитой (Phy so stigma vene-
nosum), называемых калабарскими бобами, ореха-
ми эзере, судными орехами и пр., когда-то исполь-
зовавшийся племенами Западной Африки для ша-
манского суда. Препарат хорошо всасывается из
ЖКТ, проникает через клеточные мембраны и лег-
ко преодолевает гематоэнцефалический и плацен-
тарный барьеры. Физостигмин применяют при от-
равлении м-холиноблокаторами (атропин, трицик-
лические антидепрессанты). Глазные капли с
физостигмином являются популярным мистиче-
ским препаратом, применяемым для лечения за-
крытоугольной глаукомы.
ФОСФОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Фосфорорганические соединения угнетают
АХЭ путем фосфорилирования ее активного цен-
тра (рис. 7.3) и образования очень стабильного
комплекса, устойчивого к гидролизу и реактива-
ции. В некоторых случаях при фосфорилировании
происходят структурные изменения АХЭ, после
которых реактивация фермента становится невоз-
можной. Следовательно, восстановление активно-
сти АХЭ в основном зависит от ее синтеза de novo,
а фосфорорганические соединения являются необ-
ратимыми ингибиторами фермента. Большинство
этих соединений также являются мощными инги-
биторами псевдохолинэстеразы.
К классическим представителям фосфорорга-
нических соединений относятся диизопропил-
фторфосфат и тетраэтилпирофосфат. Многие их
аналоги широко используются в качестве инсекти-
цидов, а летучие и жирорастворимые соединения,
синтезированные во время Второй мировой войны,
относятся к химическому оружию. Фосфороргани-
ческие соединения быстро всасываются в легких
и через неповрежденную кожу, вызывая различные
токсические эффекты, обусловленные воздействи-
ем на н-холинорецепторы (мышечная слабость,
паралич и артериальная гипотония) и стимуляцией
периферических м-холинорецепторов (повышение
тонуса гладкой мускулатуры, усиление слюноотде-
ления и бронхиальной секреции). Кроме того,
вследствие стимуляции центральных м-холиноре-
цепторов возникает возбуждение ЦНС (тремор, су-
дороги), которое затем сменяется угнетением с раз-
витием комы и остановкой дыхания.
Для лечения отравлений фосфорорганически-
ми соединениями применяются реактиваторы АХЭ
(пралидоксим, облидоксим), которые обеспечи-
вают гидролиз фосфорилированного фермента.
Могут потребоваться повторные дозы атропина
(2-4 мг), противосудорожные средства и ИВЛ.
Для профилактики отравлений фосфорорганиче-
скими соединениями применяются карбаматы.
Длительное воздействие фосфорорганических со-
единений вызывает тяжелый полиневрит.
Фосфорорганическое соединение эхотиофат
содержит четвертичную аммониевую группу и
взаимодействует с анионным участком АХЭ;
затем фосфорилирует активный центр, вызывая
его стабильное и длительное угнетение. Эхотиофат
в настоящее время в Великобритании не применя-
ется; раньше его использовали в виде глазных ка-
пель как мистическое средство для лечения закры-
тоугольной глаукомы. Эхотиофат ингибирует псев-
дохолинэстеразу, поэтому его отменяют задолго до
хирургической операции, чтобы избежать длитель-
ного апноэ после применения суксаметония.
158
Глава?
ЛЕКАРСТВЕННЫЕ ПРЕПАРаАэ?, 1 ^н№,?лаНз
ВЛИЯЮЩИЕ НА
ЭЛЕКТРОМЕХАНИЧЕСКОЕ
СОПРЯЖЕНИЕ
Сокращение скелетной мышцы зависит от вы-
свобождения Са2+ из саркоплазматического рети-
кулума (через рианодиновые рецепторы) и его по-
следующего связывания с тропонином С. Связыва-
ние Са2+ вызывает конформационные изменения
тропонин-тропомиозинового комплекса, в резуль-
тате которых происходит активация миозиновой
АТФазы и мышечное сокращение (электромехани-
ческое сопряжение). У генетически предрасполо-
женных лиц деполяризующие миорелаксанты и га-
логенсодержащие анестетики вызывают чрезмер-
ное высвобождение Са2+ из саркоплазматического
ретикулума и опасную для жизни злокачественную
гипертермию. Основные симптомы: быстрое повы-
шение температуры тела, мышечная ригидность,
судороги, тахикардия и смешанный респиратор-
но-метаболический ацидоз.
При подозрении на злокачественную гипер-
термию необходимо немедленно начать лечение.
Следует устранить действие, предполагаемого про-
воцирующего фактора (например, отключить пода-
чу ингаляционного анестетика) и как можно быст-
рее закончить хирургическую операцию. Проводят
активное охлаждение, поддерживают кровообра-
щение и дыхание, определяют газовый состав арте-
риальной крови: при метаболическом ацидозе вво-
дят бикарбонат натрия. Раньше применялись боль-
шие дозы прокаина и кортикостероидов, но их
эффективность сомнительна. В настоящее время
препаратом выбора при злокачественной гипертер-
мии является натриевая соль дантролена. Дан-
тролен подавляет высвобождение Са2+ из сарко-
плазматического ретикулума скелетных мышц,
предотвращая электромеханическое сопряже-
ние и аномальное теплообразование. Препарат для
в/в введения (натриевая соль дантролена в смеси с
маннитолом и гидроксидом натрия) выпускается в
виде порошка, растворимого в воде. Начальную
дозу (1 мг/кг) при необходимости можно вводить
повторно каждые 5-10 мин до максимальной
дозы 10 мг/кг.
Дантролен также применяется внутрь для лече-
ния спастических состояний при хронических нев-
рологических заболеваниях (острые нарушения
мозгового кровообращения, рассеянный склероз,
травма спинного мозга, детский церебральный па-
ралич). В таких случаях нередко развиваются по-
бочные эффекты со стороны ЦНС (головокруже-
юсть, повышенная утомляемость) и, реже,
печени. Дозу дантролена нужно увеличивать посте-
пенно до достижения оптимального эффекта.
ПРАКТИЧЕСКИЕ РЕКОМЕНДАЦИИ
ВЫБОР МИОРЕЛАКСАНТА
Суксаметоний остается миорелаксантом выбо-
ра для быстрой интубации трахеи, а также при крат-
ковременных вмешательствах (например, при
электросудорожной терапии). В прошлом суксаме-
тоний применяли и во время длительных хирурги-
ческих операций, вводя многократно струйно или
путем непрерывной инфузии. В настоящее время
такой подход используют редко, чтобы избежать
развития второй фазы действия. Общая доза сукса-
метония, при превышении которой может развить-
ся это осложнение, зависит от методики анестезии:
при сочетании закиси азота и опиоидов она состав-
ляет 7-8 мг/кг, а при ингаляционной анестезии
доза еще ниже.
Атропин обязательно должен предварять введе-
ние суксаметония при повторном применении мио-
релаксанта, а также на фоне действия 0-адренобло-
каторов; в остальных случаях использование атро-
пина остается на усмотрение анестезиолога. Часто
перед введением суксаметония назначаются не-
большие дозы недеполяризующих миорелаксантов
(атракурий 5 мг, панкуроний 1 мг), с тем чтобы
уменьшить фасцикуляции и послеоперационную
миалгию, а также ослабить стойкие сокращения
глазодвигательных мышц при проникающих ране-
ниях глазного яблока; в таком случае дозу суксаме-
тония следует увеличить на 50%.
Противопоказания к применению суксаметония:
1. Обширные ожоги; некоторые неврологиче-
ские состояния и травмы, приводящие к параличам
2. Гиперкалиемия (при почечной недостаточно-
сти)
3. Миастения и миотония
4. Выраженные изменения активности псевдо-
холинэстеразы
5. Подозрение на злокачественную гипертер-
мию в семейном анамнезе.
Точно неизвестно, насколько безопасно примене-
ние суксаметония у детей. У них суксаметоний доста-
точно часто вызывает спазм жевательных мышц,
который иногда бывает предшественником злока-
чественной гипертермии. У детей с невыявленной
миопатией применение суксаметония сопряжено с
риском гиперкалиемической остановки сердца.
Многие годы выбор недеполяризующего миоре-
лаксанта в основном зависел от предполагаемой
длительности операции, и только во вторую оче-
ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА НЕРВНО-МЫШЕЧНЫЙ СИНАПС
159
, ттт medlit.ucQz.nj
редь — от состояния больного. Широкая популяр- ^Ггооы
ность атракурия обусловлена способностью препа-
рата к спонтанной деградации при температуре
тела и отсутствием значимых побочных эффектов.
Атракурий является миорелаксантом выбора при
дисфункции почек и печени, у больных в тяжелом
состоянии и преклонного возраста. Векуроний об-
ладает относительно короткой продолжительно-
стью действия и имеет некоторые преимущества
перед атракурием при повышенном риске реакций
гиперчувствительности, а также в случаях, когда
особенно важна стабильность гемодинамики. Веку-
роний выделяется в основном с желчью и не накап-
ливается при почечной недостаточности. Атраку-
рий и векуроний — миорелаксанты выбора при со-
путствующей миастении; у больных с этим
заболеванием другие препараты могут вызывать
длительное апноэ. Атракурий и векуроний вводят
в/в струйно (в том числе многократно), а также
в виде непрерывной инфузии. В последние годы
они широко применяются при длительной ИВЛ
(например, в случае лечения столбняка или эпи-
лептического статуса); они не кумулируются, что
обеспечивает легкую управляемость миорелаксацией.
Альтернативным препаратом является панку-
роний, хотя он может вызывать тахикардию, осо-
бенно в сочетании с инотропными лекарственными
средствами. Ганглиоблокирующее действие тубо-
курарина способствует развитию пареза кишечни-
ка во время продленной ИВЛ.
Панкуроний имеет преимущества перед други-
ми миорелаксантами при гиповолемии, миваку-
рий — при амбулаторных и краткосрочных хирур-
гических вмешательствах, рокуроний — при необ-
ходимости быстрой индукции нервно-мышечной
блокады.
ВОССТАНОВЛЕНИЕ
НЕРВНО-МЫШЕЧНОЙ
ПРОВОДИМОСТИ
Для устранения остаточного действия неде-
поляризующих миорелаксантов чаще всего ис-
пользуют неостигмин (0,07 мг/кг, максимальная
доза 5 мг). Одновременно для предотвращения
м-холиномиметических эффектов неостигмина вво-
дят атропин (0,02 мг/кг, максимальная доза 1,2 мг).
Секреторная активность лучше подавляется, если
атропин вводить за 5 мин до неостигмина. После
применения этих препаратов необходимо поддер-
живать адекватную оксигенацию и эффективную
легочную вентиляцию, поскольку гипоксия и ги-
перкапния повышают риск сердечных аритмий.
свести к минимуму возможность рекура-
ризации, ингибиторы АХЭ следует вводить не ра-
нее чем через 20-30 мин после инъекции полной
дозы недеполяризующих миорелаксантов, а еще
лучше после некоторого восстановления мышечно-
го тонуса. Клинически степень восстановления
нервно-мышечной проводимости оценивается
по дыхательной активности и мышечной силе.
Признаки восстановления нервно-мышечной про-
водимости: адекватный дыхательный объем, появ-
ление кашлевого рефлекса, отсутствие втягивания
яремной и надключичных ямок на вдохе, восста-
новление тонуса нижней челюсти, способность вы-
совывать язык и удерживать голову в приподнятом
положении не менее 5 секунд. Эти признаки тесно
коррелируют с электрическими и механическими
признаками активности скелетных мышц при не-
прямой стимуляции: их появление означает, что
нервно-мышечная проводимость восстановилась
по меньшей мере на 75% (т. е. амплитуда последнего
ответа на стимуляцию в TOF-режиме составляет не
менее 75% от первого). Эти симптомы трудно оце-
нить, не пробуждая пациента, поэтому оценка с по-
мощью стимулятора нервов является методом вы-
бора для оценки остаточной кураризации.
Доза ингибитора АХЭ зависит от степени оста-
точной миорелаксации. Последняя, в свою очередь,
определяется фармакокинетическими свойствами
и дозой миорелаксанта, а также временными ха-
рактеристиками его введения. При использовании
современных миорелаксантов (атракурий, миваку-
рий) с новыми механизмами элиминации (расщеп-
ление эстеразами, спонтанная деградация) часто
удается значительно снизить дозу неостигмина или
вообще обойтись без него. Длительное стимули-
рующее влияние неостигмина на м-холинорецепто-
ры усиливает послеоперационную боль, повышает
частоту послеоперационной тошноты и рвоты,
является причиной брадикардии. Чтобы свести к ми-
нимуму эти эффекты, следует использовать м-хо-
линоблокаторы длительного действия (например,
гликопирролат).
МОНИТОРИНГ
НЕРВНО-МЫШЕЧНОЙ БЛОКАДЫ
Действие миорелаксантов может значительно
изменяться в зависимости от возраста, температу-
ры тела, pH плазмы и внеклеточной жидкости,
электролитных нарушений, эффектов других ле-
карственных препаратов и сопутствующих заболе-
ваний. Следовательно, реакцию на миорелаксанты
и антагонисты АХЭ предсказать трудно. Кроме
того, действие многих препаратов в фазу восста-
новления нервно-мышечной проводимости быстро
160
Глава?
л. medlit.
видоизменяется. Эти факторы делают мониторинг
нервно-мышечной блокады желательным, а в неко-
торых случаях просто необходимым.
Чаще всего мониторинг заключается в непря-
мой супрамаксимальной стимуляции локтевого
нерва и регистрации сложного мышечного потен-
циала действия или механического сокращения
мышцы, приводящей большой палец. Для пре-
дотвращения повторных электрических разрядов
в мышце стимуляция часто проводится короткими
(0,1-0,2 мсек.) импульсами с прямоугольной фор-
мой волны. В условиях общей анестезии можно про-
водить тетанизирующую стимуляцию (> 50 Гц),
но чаще используется стимуляция серией из четы-
рех импульсов (частота 2 Гц, продолжительность
стимуляции 2 сек); этот метод стимуляции еще на-
зывают TOF-режимом (от train-of-four). Степень
нервно-мышечной блокады оценивают по амплиту-
де первого ответа (ТД в сравнении с исходным, по-
лученным до введения миорелаксанта (То). Вероят-
но, отношение Tt/T0 наиболее точно отражает со-
стояние постсинаптической нервно-мышечной
блокады. Чаще нервно-мышечное проведение
оценивают по амплитуде (или по наличию, от-
сутствию, восстановлению) четвертого ответа
(Т4) в сравнении с первым ответом (ТД во время
TOF-стимуляции (т. е., по отношению T4/Tt).
Такой подход позволяет обойтись без контрольно-
го стимула, хотя скорее отражает пресинаптиче-
ские эффекты миорелаксантов, чем их постсинап-
тическое действие. Эти различия практически не
имеют значения, поскольку обычно соотношения
Т1/То и T4/Ti тесно коррелируют между собой.
Список литературы
Andersen, J.L., Marshall, H.W. & Askings, R.W. (1984) A
randomized trial of intravenous and intracoronary streptokinase in
patients with acute myocardial infarction. Circulation 70, 606-618.
Aster, R.H. (1995) Heparin-induced thrombocytopenia and
thrombosis. New EnglandJournal of Medicine 332,1374-1376.
Barnett, D.B. (1988) Myocardial ischaemia: progress in drug
therapy. British Journal of Anaesthesia 61,11-23.
Bullingham, A. & Strunin, L. (1995) Prevention of postoperative
thromboembolism. British Journal of Anaesthesia 75,622-630.
Collins, R., Peto, R., Baigent, C. & Sleight, P. (1997) Aspirin,
heparin and thrombolytic therapy in suspected acute myocardial
infarction. New England Journal of Medicine 336,847-860.
Davie, E.W., Fujikawa, K. & Kisiel, W. (1991) The coagulation
cascade: initiation, maintenance and regulation. Biochemistry 30,
10363-10370.
Diener, H., Cunha, L., Forbes, C., Sivenius, J., Smets, P. &
Lowenthal, A. (1996) European Stroke Prevention Study 2.
Dipyridamole and acetylsalicylic acid in the secondary prevention
of stroke. Journal of Neurological Science 143,1-14.
ucoz.ru
Fears, R. (1990) Biochemical pharmacology and therapeutic aspects
of thrombolytic agents. Pharmacological Reviews 42,201-224.
Furie, B. & Furie, B.C. (1992) Molecular and cellular biology of
blood coagulation. New England Journal of Medicine 326, 800-806.
Gallus, A.S. (1979) Antiplatelet drugs: clinical pharmacology and
therapeutic use. Drugs 18, 439-477.
Haagensen, R. & Steen, P.A. (1988) Perioperative myocardial
infarction. British Journal of Anaesthesia 61, 24-37.
Hirsh, J. (1991) Heparin. New England Journal of Medicine 324,
1565-1574.
Hirsh, J. (1991) Oral anticoagulant drugs. New England Journal of
Medicine 324,1865-1873.
Hirsh, J. & Levine, M.N. (1992) Low molecular weight heparin.
Blood 79,1-17.
Kroll, M.H. & Schafer, A.L. (1989) Biochemical mechanisms of
platelet activation. Blood 74, 1181-1195.
Mannuccio, M. (1998) Hemostatic drugs. New England Journal of
Medicine 333, 245-253.
Nichols, AJ., Ruffolo, R.R., Huffman, W.F., Poste, G. & Samanen, J.
(1992) Development of GPIIb/IIIa antagonists as antithrombotic
drugs. Trends in Pharmacological Sciences 13,413-417.
Patrono, C. (1994) Aspirin as an antiplatelet drug. New England
Journal of Medicine 330,1287-1294.
Raskob, G.E., Carter, C.J. & Hull, R.D. (1989) Heparin therapy for
venous thrombosis and pulmonary embolism. Blood Reviews 2,
251-258.
Salzman, E.W. (1992) Low-molecular-weight heparin and other
new antithrombotic drugs. New England Journal of Medicine 326,
1017-1019.
Taparelli, C., Metternich, R„ Ehrhardt, C. & Cook, N.S. (1993)
Synthetic low molecular weight inhibitors: molecular design and
pharmacological profile. Trends in Pharmacological Sciences 14,
366-376.
Tyrrell, D.J., Kilfeather, S. & Page, C.P. (1995) Therapeutic uses of
heparin beyond its traditional role as an anticoagulant. Trends in
Pharmacological Sciences 16,198-204.
Ware, J.A. & Heisted, D.D. (1993) Platelet-endothelium
interactions. New England Journal of Medicine 328,628-635.
medlit.ucoz.ru
АНТИКОАГУЛЯНТЫ, Я
АНТ И АГР ЕГ АНТ Ы И
ТРОМБОЛИТИКИ
МЕХАНИЗМЫ ГЕМОСТАЗА
Гемостаз — это сложный биологический про-
цесс, направленный на предотвращение потери
крови при повреждении кровеносных сосудов.
Сразу после повреждения происходит спазм арте-
риол, и спустя короткое время тромбоциты склеи-
ваются с коллагеном поврежденной сосудистой
стенки (адгезия) и друг с другом (агрегация). Этот
процесс зависит от высвобождения из тромбоцитов
таких медиаторов, как АДФ, 5-гидрокситриптамин
(серотонин), тромбоксан А2, фактор активации
тромбоцитов. Растворенные в крови факторы свер-
тывания крови активйзируются липопротеином,
высвобождаемым из сосудистой стенки (тканевой
фактор), и фосфолипидами, высвобождаемыми
из тромбоцитов. Под воздействием активирован-
ных факторов VII, X, V и Са2+ неактивный глико-
протеин протромбин превращается в активный
протеолитический фермент тромбин. Тромбин
воздействует на фибриноген, в результате чего от-
щепляются фибринопептиды А и В и образуются
мономеры фибрина. Мономеры фибрина связыва-
ются друг с другом поперечными связями, в ре-
зультате чего образуется нерастворимый фибри-
новый тромб. Тромбин также активирует другие
факторы свертывания и стимулирует агрегацию
тромбоцитов.
Тромбин также влияет на эндотелиальные и глад-
комышечные клетки, лейкоциты, сердце (автома-
тизм и реполяризацию) и активность нейронов.
Кроме того, он индуцирует пролиферацию фибро-
бластов при пневмосклерозе и системной склеро-
дермии.
При контакте крови с чужеродной поверхно-
стью ферментативные процессы происходят в дру-
гой последовательности. В каскад протеолитиче-
ских реакций вовлекается более дюжины фермен-
тов. Факторы свертывания представляют собой
проферменты, которые превращаются в активные
ферменты в результате воздействия продуктов
предшествующих протеолитических реакций.
Затем активные ферменты катализируют следую-
щую стадию процесса.
Выделяют два механизма коагуляционного ге-
мостаза: внешний (запускается повреждением тка-
ней) и внутренний (все активирующие факторы
присутствуют в плазме). Оба механизма приводят
к образованию протромбин-активирующего ком-
плекса (фактор Ха). Конечный этап свертывания
одинаков для обоих механизмов (рис. 8.1). Для аде-
кватного гемостаза необходима сохранность обоих
механизмов.
В клинической практике для оценки коагуляци-
онного гемостаза и мониторинга эффекта антикоа-
гулянтов применяют два лабораторных показателя.
1. Протромбиновое время (ПВ). В этом исследо-
вании тканевой тромбопластин (растворенный
в растворе NaCl экстракт головного мозга, содержа-
щий тканевой фактор и фосфолипиды) добавляет-
ся в рекальцинированную плазму. В норме сгусток
образуется в течение 12-15 сек. Международное
нормализованное отношение (МНО) в норме рав-
но единице и представляет собой отношение ПВ
больного к ПВ стандартной плазмы. МНО широко
используется для мониторинга эффекта непря-
мых антикоагулянтов и позволяет стандартизиро-
вать значения ПВ, полученные в различных лабо-
раториях.
2. Активированное частичное тромбопластино-
вое время (АЧТВ). В этом анализе Са2+, отрица-
тельно заряженные фосфолипиды и взвесь мелких
частиц (например, каолина, или силиката алюми-
ния) добавляются к плазме, в которую предвари-
тельно был добавлен цитрат или комплексобразую-
щее вещество для связывания кальция. В норме
сгусток образуется в течение 25-35 секунд. На это
время могут влиять особенности методики и при-
меняемые реагенты. АЧТВ используют для мони-
торинга эффекта гепарина.
162
Глава 8
Рис. 8.1. Внеш-
ний и внутрен-
ний механизмы
свертывания.
Активирован-
ные формы фак-
торов свертыва-
ния обозначены
буквой “а". ТФ -
тканевой фактор
medlit.ucoz.ru
ВНЕШНИЙ МЕХАНИЗМ ВНУТРЕННИЙ МЕХАНИЗМ
Повреждение тканей Контакт с поверхностью
V» - Vto
ТФ
Фосфолипиды
Са2+
ХО
Калликреин
Высокомолекулярный кининоген
Ml -* Х!Ю
I
XI - Xia
IX- «о
VIII- villa
Фосфолипиды
,Са2+
V-> Va
Фосфолипиды
Са2+
Протромбин ---------^Тромбин
Фибриноген -----1----»
Фибрин
РАССТРОЙСТВА ГЕМОСТАЗА
НАРУШЕНИЯ ПИТАНИЯ
Витамин К является нормальной составной ча-
стью пищевого рациона и играет существенную
роль в синтезе определенных факторов свертыва-
ния в печени. Он был обнаружен в ходе экспери-
ментальных исследований на цыплятах, когда было
установлено, что дефицит растворимого в эфире
вещества вызывает склонность к кровотечениям.
Впоследствии выяснилось, что существует по
меньшей мере две природных формы витамина К.
Один из них (витамин Къ или фитоменадион)
выделен из растений и может применяться у чело-
века. Другой тип (витамин К2, или менахинон) син-
тезируется грамотрицательными бактериями
тонкой кишки. Обе формы витамина К растворя-
ются в жирах, а степень их всасывания в кишечнике
зависит от присутствия солей желчных кислот.
Дефицит витамина К возникает при его недоста-
точном потреблении е пищей, пониженном синтезе
в кишечнике, нарушении всасывания или утилиза-
ции в печени. Дефицит витамина К нарушает син-
тез в печени протромбина, а также факторов VII, IX
и X (так называемые витамин К-зависимые фак-
торы). В ходе биосинтеза остатки глутаминовой
кислоты в белках-предшественниках превращают-
ся в у-карбоксиглутаматные группы, необходимые
для связывания белков с Са2+ и их сопряжения с
фосфолипидами (рис. 8.2). Этот процесс зависит
от витамина К, который одновременно превращает-
ся из восстановленной формы (гидрохинона) в эпок-
сид (витамин К-2,3-эпоксид). При дефиците вита-
мина К не происходит у-карбоксилирования, вита-
мин К-зависимые факторы остаются неактивными
и возникает склонность к кровотечениям.
Дефицит витамина К возникает при геморраги-
ческой болезни новорожденных, нарушениях вса-
сывания в кишечнике, механической желтухе,
желчных свищах и циррозе печени. Фитоменадион
(1 мг в/м) является средством выбора для профи-
лактики и лечения дефицита витамина К у новоро-
жденных. Более высокие дозы (10-20 мг в сутки
в течение 3 дней перед операцией) восстанавлива-
ют нормальный уровень протромбина при желтухе.
Некоторые прописи фитоменадиона содержат кре-
мофор EL (полиэтоксилированное касторовое мас-
ло) и могут вызывать анафилактоидные реакции
при введении в/в. Если желтуха обусловлена тяже-
лым повреждением гепатоцитов, может потребо-
ваться инфузия свежезамороженной плазмы.
Витамин К3 (менадион) является синтетиче-
ским препаратом, который может превращаться
в водорастворимое соединение (менадиола натрия
дифосфат). Он назначается внутрь для профилак-
тики дефицита витамина К при нарушениях всасы-
вания в кишечнике. Иногда при дефиците глюко-
зо-6-фосфатдегидрогеназы менадиола натрия
дифосфат может вызывать гемолиз.
АНТИКОАГУЛЯНТЫ, АНТИАГРЕГАНТЫ И ТРОМБОЛИТИКИ
163
Витамин К (гидрохинон)
(восстановленная форма)
Витамин К (эпоксид)
(окисленная форма)
Факторы предшественники
(протромбин, VII, IX, X)
НАД
medlit.ucoz.ru
НАДН
Эпоксид-
редуктаза
Подавляется непрямыми
антикоагулянтами
Циркулирующие факторы
(протромбин, VII, IX, X)
Рис. 8.2. Синтез актив-
ных факторов сверты-
вания (протромбин,
VII, IX и X) из неактив-
ных предшественни-
ков. Непрямые анти-
коагулянты подавляют
активность эпоксидре-
дуктазы, которая отве-
чает за восстановление
биологически актив-
ной формы витамина К
(т. е. восстановленного
гидрохинона).
НАД — никотинами-
дадениндинуклеотид
R-глутаминовая кислота
R-у-карбоксиглутаминовая кислота
Связывание Са2+
Связывание фосфолипида
НАСЛЕДСТВЕННЫЕ КОАГУЛОПАТИИ
Гемофилия является наиболее известным при-
мером сцепленной с полом наследственной коагу-
лопатии, хотя встречается относительно редко.
Выделяют два основных типа гемофилии. Гемо-
филия А (классическая гемофилия) характеризу-
ется дефицитом фактора VIII (антигемофильного
глобулина), гемофилия В (болезнь Кристмаса) —
дефицитом фактора IX (фактор Кристмаса).
Болезнь фон Виллебранда также является
наследственной коагулопатией, наследуется по ау-
тосомно-доминантному типу, встречается у мужчин
и женщин и обусловливается дефицитом фактора
фон Виллебранда, синтезирующегося в эндотелии
сосудов и способствующего адгезии тромбоцитов
к субэндотелиальному слою сосудистой стенки.
Кроме того, фактор фон Виллебранда служит пере-
носчиком циркулирующего фактора VIII. Дефект
гемостаза при болезни фон Виллебранда обуслов-
лен увеличением времени кровотечения и сниже-
нием активности фактора VIII.
При необходимости хирургического вмеша-
тельства у пациентов с наследственной коагулопа-
тией в периоперационном периоде вводят концен-
трат соответствующего фактора.
ТРОМБОЗ
Тромбоз — это измененное состояние гемостаза,
приводящее к образованию внутрисосудистого
тромба. Основные факторы тромбообразования
были установлены Рудольфом Вирховом более
100 лет назад; к ним относятся повреждение сосу-
дистой стенки, нарушение кровотока (стаз) и повы-
шенная свертываемость крови (триада Вирхова).
Тромбы могут образовываться:
1) в артериях (белые тромбы), что приводит
к ишемии жизненно важных тканей и органов;
2) в венах (красные тромбы), в этом случае высо-
ка вероятность эмболии легочной артерии;
3) в камерах сердца, что может привести к эмбо-
лии сосудов большого круга кровообращения.
Тромбоэмболии являются частой причиной
осложнений и летального исхода, особенно в по-
слеоперационном периоде. Выделяют несколько
предотвратимых и курабельных факторов
риска повреждения сосудистой стенки: курение,
гиперхолестеринемия и артериальная гипертони-
ческая болезнь. Для профилактики венозного ста-
за, вызванного длительной операцией и послеопе-
рационной иммобилизацией, придают больному
164
Глава 8
оптмальное положение на операционн
переливают инфузионные растворы, используют
перемежающуюся пневматическую компрессию
ног и эластичные чулки, проводят физиотерапию.
Тромбофилия (повышенная свертываемость
крови) способствует тромбообразованию, особен-
но в сочетании с дополнительными факторами
риска (сосудистый стаз). Тромбофилия может
быть обусловлена патологическими изменениями
со стороны факторов свертывания, тромбоцитов,
системы фибринолиза и физиологических ингиби-
торов гемостаза. Увеличение концентрации неак-
тивных факторов свертывания не ускоряет образо-
вание фибрина, но повышение уровня фактора VIII
и фибриногена увеличивает риск ИБС. Угнетение
фибринолиза является фактором риска венозного
тромбоза и развивается при дефиците или анома-
лиях активаторов плазминогена, аномалиях фиб-
риногена и плазминогена.
Существует множество физиологических инги-
биторов тромбоцитов и факторов свертывания,
в том числе гепарин, гепаран-сульфат, простацик-
лин, NO, аденозин, тканевой активатор плазмино-
гена (тАП), аг-макроглобулин, at-антитрипсин, ин-
гибитор С1 эстеразы, антитромбин III, протеин С,
протеин S и кофактор гепарина II. Врожденный
дефицит любых эндогенных ингибиторов сверты-
вания проявляется склонностью к тромбозам, т. е.
тромбофилией. При одной из форм наследственной
тромбофилии — лейденской мутации — аномаль-
ный фактор V резистентен к действию активиро-
ванного протеина С. Парадоксально, но дефицит
фактора XII, участвующего во внутреннем меха-
низме свертывания, также предрасполагает к тром-
бофилии.
У большинства больных тромбофилией первый
венозный тромбоз случается в возрасте 20-30 лет
и его причину можно выявить примерно в 50% слу-
чаев. При необходимости операции у нелеченых
больных показан короткий курс антикоагулянтной
терапии. В день операции назначают концентрат
антитромбина III или СЗП.
Комбинированные пероральные контрацепти-
вы увеличивают риск тромбоэмболических ослож-
нений в послеоперационном периоде, поскольку
содержащиеся в них эстрогены повышают свер-
тываемость крови и увеличивают концентрацию
некоторых факторов свертывания (фибриноген,
факторы VII и X). Может снижаться уровень анти-
тромбина III. Сведения о влиянии контрацептивов
на адгезию и агрегацию тромбоцитов носят проти-
воречивый характер. Повышенная свертываемость
может сохраняться еще 2-3 мес после отмены эст-
рогенов. Чем меньше содержание эстрогена в перо-
ОЯОЙУ!,- контрацептиве, тем ниже риск тромбоэм-
болических осложнений. Даже комбинированные
пероральные контрацептивы, содержащие препа-
раты прогестерона третьего поколения (дезогест-
рел или гестоден), повышают риск тромбоэмболии,
особенно когда их принимают женщины с лейден-
ской мутацией гена фактора V — наиболее распро-
страненной наследственной тромбофилией. Счита-
ется, что пероральные контрацептивы необходимо
отменить за 4-6 недель до больших плановых опе-
раций, а также любых операций на ногах. При дру-
гих операциях в периоперационном периоде исполь-
зуют низкомолекулярные гепарины и эластичные
чулки. Некоторые специалисты считают отмену
комбинированных пероральных контрацептивов
или профилактическое применение антикоагулян-
тов необоснованным, если нет других факторов
риска. Отмена пероральных контрацептивов повы-
шает риск нераспознанной беременности, которую
необходимо исключить до операции.
В следующих разделах этой главы рассматрива-
ются лекарственные препараты, предназначенные
для профилактики и лечения тромбоэмболических
осложнений. Их можно разделить на три группы:
1. Препараты, препятствующие свертыванию
крови (антикоагулянты)
2. Препараты, подавляющие агрегацию тромбо-
цитов (антиагреганты)
3. Препараты, растворяющие патологические
тромбы (тромболитики или фибринолитики).
АНТИКОАГУЛЯНТЫ
В клинической практике применяется два типа
антикоагулянтов:
1. Гепарин
2. Непрямые антикоагулянты
ГЕПАРИН
Гепарин, как следует из самого названия, явля-
ется естественным антикоагулянтом, который
впервые был обнаружен в печени, хотя присутству-
ет в высокой концентрации также в легких и слизи-
стой кишечника. Гепарин находится в секреторных
гранулах тучных клеток вместе с гистамином и раз-
личными протеолитическими ферментами. Физио-
логическая роль гепарина неясна. При анафилак-
тическом шоке небольшое количество гепарина
вместе с гистамином выбрасывается из базофилов
и тучных клеток эндотелия, что несколько угнетает
свертываемость крови. Помимо антикоагулянтного
эффекта, гепарин и другие природные гепариноиды
(гепаран-сульфат, хондроитин-4-сульфат) выпол-
няют и иные физиологические функции. Гепарин
АНТИКОАГУЛЯНТЫ, АНТИАГРЕГАНТЫ И ТРОМБОЛИТИКИ
165
medlit.ucoz.ru
угнетает миграцию иммунных клеток при воспали-
тельных заболеваниях и метастазирующих опухо-
лях, а также оказывает антипролиферативное дей-
ствие на гладкомышечные клетки и фибробласты,
опосредующие механизм атеросклероза и бронхи-
альной астмы.
Природный гепарин представляет собой группу
мукополисахаридов с молекулярным весом 1—40 кДа
(средний молекулярный вес 12 кДа). Гепарин со-
стоит из чередующихся остатков глюкуроновой
кислоты и сульфатированных остатков ацетилглю-
козамина, т. е. является гликозаминогликаном.
Благодаря многочисленным ионизированным
сульфатным группам гепарин при физиологиче-
ских значениях pH имеет сильный отрицательный
заряд. Антикоагулянтная активность гепарина за-
висит от особой пентасахаридной последователь-
ности, которая имеется приблизительно в 30% его
молекул.
Гепарин ускоряет взаимодействие антитромби-
на III (эндогенного антитромбина, представляюще-
го собой ингибитор сериновых протеаз) со многими
активированными факторами свертывания (тром-
бином, факторами Vila, IXa, Xia, ХПа и каллик-
реином). Антитромбин III образует стабильный
комплекс с этими факторами, что нейтрализует их
активность. Гепарин значительно повышает ско-
рость этой реакции, оказывая немедленное антикоа-
гулянтное действие in vivo и in vitro. Аналогичным
образом угнетается и фактор Ха (активированный
фактор Стюарта), стимулирующий превращение
протромбина в тромбин.
Гепарин воздействует на свертываемость крови
и с помощью других механизмов. В высоких дозах
он подавляет агрегацию тромбоцитов и увеличива-
ет время кровотечения. Гепарин также активирует
липопротеинлипазу в тканях и сосудистом эндоте-
лии, гидролизуя триглицериды плазмы до глице-
рина и свободных жирных кислот и просветляя
хилезную плазму. Высокая концентрация жирных
кислот в плазме препятствует связыванию с белка-
ми плазмы некоторых лекарственных препаратов
(например, пропранолола и фенитоина), что под-
тверждается анализами крови, полученными через
катетер, периодически промываемый раствором ге-
парина.
Из-за полярности и высокого молекулярного
веса гепарин плохо проникает через клеточные
мембраны, плацентарный и гематоэнцефалический
барьеры, а также неэффективен при приеме внутрь
или под язык. После однократного в/в введения
действие гепарина продолжается 4-6 ч, хотя его
Ti/2 обычно зависит от дозы. Гепарин расщепляется
в печени под действием гепариназы, и его метабо-
литы выводятся с мочой. При почечной недостаточ-
ности или циррозе печени продолжительность
действия гепарина увеличивается.
Гепарин обычно производят из бычьих легких
или слизистой свиного кишечника. Поскольку эти
препараты различаются мощностью, они стандар-
тизируются путем биологических анализов и их
активность обычно измеряют в Ед/мл. В клиниче-
ской практике используются препараты, содержа-
щие 5000 или 25000 Ед натриевой соли гепарина
в одном миллилитре; с профилактической целью
также применяют кальциевую соль гепарина.
Наиболее значимым побочным эффектом ге-
парина является кровотечение, которое обычно
останавливается после его отмены и введения ле-
карственных препаратов-оснований (например,
протамина). Действие гепарина зависит от ионизи-
рованных сульфатных групп, соединенных с остат-
ками глюкозамина; их нейтрализация протамином
быстро устраняет эффекты препарата. Иногда гепа-
рин вызывает гиперчувствительность, особенно
при аллергических реакциях в анамнезе. При дли-
тельном лечении гепарином довольно часто разви-
вается тромбоцитопения. Считается, что в ее основе
лежит цитолитическая реакция (тип гиперчувст-
вительности II), опосредованная антителами IgG
и IgM. Тромбоцитопения может сочетаться с тром-
бозов. Следовательно, если лечение гепарином
продолжается более 5 суток, необходим монито-
ринг уровня тромбоцитов в крови. Иногда тромбо-
цитопения развивается уже после первой дозы
гепарина, что объясняется анафилактоидной реак-
цией с вовлечением альтернативного пути актива-
ции комплемента. При фракционном или постоян-
ном введении гепарина иногда наблюдается посте-
пенное снижение активности антитромбина III, что
повышает риск тромбоза. Длительное лечение гепа-
рином может привести к алопеции и остеопорозу.
Низкомолекулярные гепарины
С помощью гель-фильтрационной хроматогра-
фии стандартный гепарин можно разделить на
фракции и получить гепарины с низким молеку-
лярным весом (1-10 кДа; средний молекулярный
вес 5 кДа). Низкомолекулярные гепарины не спо-
собны связывать тромбин (фактор Па) и антитром-
бин III одновременно. Обычно они вызывают анти-
коагулянтное действие, нейтрализуя фактор Ха;
оказывают менее выраженное действие на фак-
торы IXa, Xia и ХПа и совсем не оказывают прямого
угнетающего действия на тромбин. Низкомолеку-
лярные гепарины (цертопарин, дальтепарин, энок-
сапарин и тинзапарин) обладают большей биодос-
тупностью и оказывают более продолжительное
166
Глава 8
действие по сравнению со стандартным ге
Они меньше влияют на тромбоциты и реже вызыва-
ют геморрагические осложнения. Низкомолеку-
лярные гепарины практически не влияют на АЧТВ,
поэтому их применение обычно не требует монито-
ринга.
Другие антикоагулянты
Данапароид представляет собой смесь низкомо-
лекулярных гепариноидов — гепарансульфата, дер-
матансульфата и хондроитинсульфата, которые
нейтрализуют фактор Ха. Данапароид используют
для профилактики тромбоза глубоких вен ног,
а также при гепариновой тромбоцитопении.
Лепирудин — это генетически модифицирован-
ный препарат, полученный из дрожжевых клеток,
являющийся прямым ингибитором тромбина.
Действие лепирудина не зависит от антитромби-
на III, поэтому он может применяться при гепари-
новой тромбоцитопении.
Применение
Гепарин применяют для профилактики и лече-
ния тромбоза глубоких вен, ТЭЛ А и инфаркта мио-
карда. Гепарин используют для профилактики
тромбозов во время операций на сердце и крупных
сосудах, а также при проведении гемодиализа.
Низкие дозы гепарина часто назначают для профи-
лактики тромбоэмболических осложнений после
различных хирургических вмешательств, особенно
у больных группы повышенного риска. Факторы
риска включают большие операции с длительной
иммобилизацией в послеоперационном периоде,
ожирение, сердечную недостаточность, венозный
стаз в ногах, тромбоэмболические осложнения
в анамнезе. Противопоказания к применению гепа-
рина: кровотечение, недавно перенесенные офталь-
мологические и нейрохирургические операции,
язвенная болезнь желудка и двенадцатиперст-
ной кишки, варикозное расширение вен пищевода,
артериальная гипертония с диастолическим АД
выше 110 мм рт. ст., повышенная чувствительность
к гепарину.
Контроль за лечением проводят, измеряя АЧТВ
или тромбиновое время (время, которое требуется
для свертывания плазмы при добавлении тромби-
на). Высокие дозы влияют и на ПВ, но в меньшей
степени.
Схемы применения
Стандартная схема лечения тромбоза глу-
боких вен и ТЭДА
Гепарин следует назначать в/в и лучше в виде
длительной инфузии. После нагрузочной дозы
5000 ЕД начинают инфузию со скоростью
Дозу следует корректировать, так
чтобы значения АЧТВ были в 1,5—2,5 раза выше
нормы. В большинстве случаев, требующих дли-
тельной антикоагулянтной терапии, одновременно
с гепарином назначают непрямые антикоагулянты.
Для достижениия полного эффекта непрямых ан-
тикоагулянтов обычно требуется 3-4 дня, после
чего гепарин отменяют.
Альтернативная схема лечения тромбоза
глубоких вен и ТЭЛА (применяется, если гепа-
рин по каким-либо причинам нельзя вводить в/в,
а также в ситуации, когда непрямые антикоа-
гулянты противопоказаны, например, во время
беременности)
В таких случаях концентрированные препара-
ты гепарина (25 000 ЕД/мл) вводят п/к в передне-
боковую стенку живота рядом с гребнем подвздош-
ной кости или в бедро. Начальная доза составляет
10 000-20 000 ЕД каждые 12 ч с ежедневным кон-
тролем АЧТВ. Через 5 дней гепаринотерапии необ-
ходимо регулярно определять уровень тромбоци-
тов в крови.
Профилактическое применение у хирургиче-
ских больных группы повышенного риска
Гепарин вводят п/к в дозе 5000 ЕД за 2 ч до опе-
рации, после чего каждые 8-12 ч в течение 7 дней
или до того, как больной начнет ходить. Для п/к
применения лучше использовать не натриевую,
а кальциевую соль гепарина, поскольку последняя
реже вызывает постинъекцинные гематомы. Обыч-
но лечение малыми дозами гепарина не требует мо-
ниторинга АЧТВ.
В последние годы для профилактики венозного
тромбоза широко применяют низкомолекулярные
гепарины. Они не менее безопасны и эффективны,
чем гепарин, и снижают риск ТЭЛА после общехи-
рургических и ортопедических операций. Низко-
молекулярные гепарины обладают большей био-
доступностью, мощностью и продолжительностью
действия, чем гепарин, и с профилактической це-
лью назначаются 1-2 раза в сутки. Доза зависит от
вида низкомолекулярного гепарина и риска тром-
боэмболических осложнений. Низкомолекуляр-
ные гепарины также используются для лечения
тромбоза глубоких вен и ТЭЛА.
ТЭЛА является одной из основных причин
смерти в послеоперационном периоде (7% всех
смертельных исходов). Отказ от профилактического
применения антикоагулянтов у больных группы
повышенного риска влечет за собой серьезные
медицинские и юридические последствия. Вместе
с тем необходимо соблюдать разумный баланс.
Обязательно следует учитывать противопоказания
к применению гепарина. Особое внимание требуется
АНТИКОАГУЛЯНТЫ, АНТИАГРЕГАНТЫ И ТРОМБОЛИТИКИ
167
больным, одновременно получающим Н
препараты этой группы удлиняют время кровоте-
чения. Противопоказано одновременное примене-
ние гепарина и кеторолака в послеоперационном
периоде. Остальные НПВС оказывают дозозависи-
мое действие. На фоне применения антикоагулян-
тов следует четко соблюдать сроки проведения
эпидуральной и спинномозговой блокады. Соот-
ветствующую пункцию и катетеризацию, необхо-
димую для выполнения этих блокад, рекомендует-
ся проводить либо перед введением гепарина, либо
не ранее чем через 4-6 ч после его последнего введе-
ния. Более длительный промежуток (10-12 ч) тре-
буется при использовании низкомолекулярных
гепаринов.
Применение во время хирургических опера-
ций на сердце и крупных сосудах
Гепарин необходимо ввести до начала искусст-
венного кровообращения. Стандартная доза со-
ставляет 300 ЕД/кг, ее вводят за 4 мин до канюля-
ции. В дальнейшем может потребоваться дробное
введение небольших доз гепарина (50-100 ед/кг)
на каждый час искусственного кровообращения.
Лабораторный мониторинг осуществляют, изме-
ряя активированное время свертывания в лабора-
тории, находящейся при операционной; это время
должно в три раза превышать контрольное зна-
чение. Гепарин вводят перед пережатием аорты
во время операций на периферических сосудах.
Первоначальная доза составляет 150 ЕД/кг; для
мониторинга эффекта и подбора последующих доз
тоже рекомендуется определять активированное
время свертывания.
Применение при ДВС-синдроме
Гепарин также может применяться для лечения
ДВС-синдрома (синдрома диссеминированного
внутрисосудистого свертывания), который вы-
зывается массивным поступлением в кровоток
тромбопластина. Это происходит при акушерских
осложнениях, тяжелых травмах, инфекциях, опу-
холях и печеночной недостаточности. Повсемест-
ное образование тромбов приводит к избыточному
потреблению факторов свертывания и высвобож-
дению продуктов распада фибрина; кровь теряет
способность к свертыванию и развивается гемор-
рагический диатез. Гепарин усиливает активность
антитромбина III и нейтрализует активированные
факторы свертывания, что позволяет остановить их
потребление. Иногда гепарин, напротив, усиливает
склонность к кровотечениям. В таких случаях
применяют ингибиторы фибринолиза (см. ниже),
а для устранения активности гепарина — про-
тамин. Еще важнее немедленно перелить тром-
боцитарный концентрат, криопреципитат (богатый
се фибриногеном препарат крови) и СЗП. Хотя свер-
тываемость крови при ДВС-синдроме достаточно
легко контролировать гепарином, летальный исход
часто наступает от респираторного дистресс-син-
дрома взрослых.
Протамин
Протамин — это основной белок, быстро нейтра-
лизующий действие гепарина. Для снижения риска
аллергических реакций его всегда вводят в/в мед-
ленно (в виде протамина сульфата). Доза зависит
от времени, прошедшего с момента введения гепа-
рина, но не должна превышать 1 мг на 80-100 Ед
гепарина (максимум 50 мг). Передозировка может
привести к обострению кровоточивости, поскольку
протамин сам является слабым антикоагулянтом,
подавляющим образование и активность тромбо-
пластина.
НЕПРЯМЫЕ АНТИКОАГУЛЯНТЫ
Непрямые антикоагулянты были открыты слу-
чайно, после того как у коров, накормленных
испорченным клеверным силосом, начались кро-
вотечения. Причиной оказалось снижение уровня
протромбина в плазме, которое можно пре-
дотвратить добавлением в корм люцерны (богатой
витамином К). Впоследствии были установлены
содержавшиеся в силосе вещества, вызывающие
кровоточивость. Ими оказались бисгидроксику-
марин (дикумарол) и его производные. Дикумарол
был впервые применен в качестве антикоагулянта
в 1941 г. Натриевая соль варфарина является ана-
логом дикумарола, первоначально она использова-
лась в качестве крысиного яда и считалась слишком
токсичной для человека. В настоящее время варфа-
рин является эталонным непрямым антикоагулянтом.
Производные индандиона (например, фениндион)
тоже являются непрямыми антикоагулянтами, но
они часто вызывают реакции гиперчувствительно-
сти и в настоящее время практически не применя-
ются.
Механизм действия
Непрямые антикоагулянты подавляют актив-
ность витамин К-зависимых факторов свертыва-
ния (протромбин, VII, IX и X факторы). Карбокси-
лирование остатков глутаминовой кислоты в бел-
ках-предшественниках сопряжено с окислением
витамина К, который при этом превращается из
восстановленного гидрохинона в эпоксид (рис. 8.2).
Регенерация гидрохинона необходима для не-
прерывного синтеза факторов свертывания, она
зависит от НАДН и фермента эпоксидредуктазы.
Непрямые антикоагулянты подавляют актив-
ность фермента, вызывают накопление эпоксида
168
Глава 8
medlit.ucoz.ru
витамина К и истощают внутриклеточные запасы
восстановленного витамина. Синтез витамин К-за-
висимых факторов нарушается, что оказывает ан-
тикоагулянтное действие.
Непрямые антикоагулянты не оказывают дейст-
вия на уже синтезированные факторы свертывания
и поэтому неэффективны in vitro. In vivo их эффек-
ты начинают проявляться только после того, как ра-
нее синтезированные факторы будут израсходова-
ны и удалены из плазмы. Значение Т| 2 факторов
свертывания колеблется от 5 ч (фактор VII) до 60 ч
(протромбин). Следовательно, непрямые антикоа-
гулянты начинают действовать не раньше чем через
12 ч после введения, а для достижения максималь-
ного эффекта требуется 48-72 ч.
Варфарин
Натриевая соль варфарина является стандарт-
ным непрямым аникоагулянтом. После приема
внутрь варфарин быстро всасывается в ЖКТ и дос-
тигает пиковой концентрации в плазме в течение
1 ч. Варфарин практически полностью связывается
с белками плазмы (95-98%), преимущественно с
альбумином. Это частично ограничивает способ-
ность варфарина проникать через клеточные
мембраны, и объем его распределения (0,1 л/кг)
соответствует ОЦК. Тем не менее, являясь жиро-
растворимым соединением, варфарин легко про-
никает через плацентарный барьер. Лечение вар-
фарином во время беременности может вызывать
врожденные пороки развития (хондродисплазию),
а также кровотечения и внутриутробную гибель
плода, особенно после тридцать шестой недели
беременности. Клиренс варфарина не зависит от
печеночного кровотока, Ti/2 составляет 35-40 ч.
Препарат почти полностью метаболизируется в пе-
чени и выводится с мочой в виде неактивных конъ-
югатов с глюкуроновой кислотой.
Варфарин — хиральное соединение. Используе-
мый в клинике препарат представляет собой раце-
мическую смесь оптических изомеров, которые
значительно различаются мощностью и скоростью
метаболизма, а также влиянием на синтез факторов
свертывания.
Индивидуальные различия в чувствительно-
сти к варфарину
Существуют выраженные индивидуальные раз-
личия в чувствительности к варфарину. Для оцен-
ки эффекта варфарина и подбора дозы необходимо
проводить мониторинг МНО и протромбинового
времени (ПВ).
Выделяют три главных фактора, определяю-
щих индивидуальные различия в чувствительности
к варфарину:
1. Биодоступность витамина К и синтез факто-
ров свертывания.
2. Генетические факторы.
3. Взаимодействие с другими лекарственными
препаратами.
Биодоступность витамина К
Любой фактор, ограничивающий биодоступ-
ность витамина К, усиливает действие непрямых
антикоагулянтов. При заболеваниях печени чувст-
вительность к непрямым антикоагулянтам повы-
шается вследствие нарушения синтеза факторов
свертывания. Повышенное поступление витамина
К с жирами, маслами и энтеральным питанием тре-
бует увеличения дозы непрямых антикоагулянтов.
Играет роль скорость катаболизма витамин К-за-
висимых факторов: лихорадка и гипертиреоз
усиливают действие непрямых антикоагулянтов,
а гипотиреоз, наоборот, ослабляет.
Генетические факторы
Иногда индивидуальные различия в чувст-
вительности к непрямым антикоагулянтам обу-
словлены генетическими факторами. Например,
существуют различные варианты ингибиторов
факторов свертывания с разной функциональной
активностью, а также резистентные формы диафо-
разы (эпоксидредуктазы). Врожденная недостаточ-
ность антитромбина III может быть причиной тя-
желых осложнений при лечении варфарином.
Взаимодействие с другими лекарственными
препаратами
Непрямые антикоагулянты вступают во взаимо-
действие со многими лекарственными преапрата-
ми, что может привести к серьезным последствиям.
Аспирин и большинство НПВС усиливают дейст-
вие непрямых антикоагулянтов. Даже одна доза
аспирина угнетает агрегацию тромбоцитов, увели-
чивает время кровотечения и нарушает гемостаз,
а более высокие дозы подавляют синтез протромби-
на и снижают его концентрацию в плазме. Некото-
рые НПВС усиливают антикоагулянтное действие
варфарина, вытесняя препарат из связи с белками и
угнетая его метаболизм.
Некоторые другие лекарственные препараты
также усиливают и пролонгируют эффект непря-
мых антикоагулянтов, тем самым повышая вероят-
ность кровотечения. В большинстве случаев это
происходит в результате вытеснения варфарина из
связи с белками и угнетения метаболизирующих
его ферментов. К таким лекарственным препаратам
относятся циметидин, хлорамфеникол, дисуль-
фирам, омепразол, метронидазол и кетоконазол.
Этанол увеличивает продолжительность действия
варфарина и снижает его клиренс, особенно при со-
путствующей дисфункции печени.
АНТИКОАГУЛЯНТЫ, АНТИАГРЕГАНТЫ И ТРОМБОЛИТИКИ
169
medlit.ucoz.ru
Некоторые лекарственные препараты (напри-
мер, холестирамин и сукральфат) ухудшают всасы-
вание варфарина в ЖКТ. Холестирамин, кроме
того, угнетает всасывание витамина К. Лекарствен-
ные препараты, индуцирующие микросомальные
ферменты печени (барбитураты, дихлоралфеназон,
рифампицин, пероральные контрацептивы), уси-
ливают метаболическую деградацию варфарина,
что требует увеличения его дозы. После отмены
этих препаратов активность антикоагулянта повы-
шается, и во избежание опасного или даже смер-
тельного кровотечения необходимо снизить дозу
варфарина.
Применение
Непрямые антикоагулянты применяются для
профилактики и лечения тромбоза глубоких вен,
ТЭЛА, транзиторных ишемических атак, а также
при устойчивой к лечению мерцательной аритмии.
Непрямые антикоагулянты показаны также для
профилактики пристеночных тромбозов при
протезированных клапанах сердца и сосудистых
трансплантатах. Перед назначением непрямых
антикоагулянтов необходимо определить исходное
ПВ и МНО. Вначале варфарин назначают в дозе
10 мг в сутки на протяжении трех дней; эта началь-
ная доза должна быть меньше у пожилых, при низ-
ком индексе массы тела, при заболеваниях печени,
сердечной недостаточности, а также при исходно
удлиненном ПВ. Поддерживающие суточные дозы
обычно составляют 3-9 мг. Варфарин нужно
принимать в одно и то же время каждый день.
Цель лечения: увеличение ПВ до такой степени,
чтобы поддерживать МНО на уровне 2-3,5. При ле-
чении тромбоза глубоких вен, ТЭЛА и транзитор-
ных ишемических атак МНО следует поддержи-
вать на уровне 2,5. При рецидивирующем венозном
тромбозе или ТЭЛА, после наложения артериаль-
ных шунтов и протезирования сердечных клапанов
целевой уровень МНО составляет 3,5. Обычно до-
пускается отклонение МНО в пределах 0,5 единиц
от целевого значения. Подбор поддерживающей
дозы варфарина определяется целевым и реаль-
ным значением МНО. МНО следует определять
на второй и третий день лечения, а затем через день
до его стабилизации. Как уже упоминалось, для
быстрого достижения лечебного эффекта одновре-
менно с непрямыми антикоагулянтами применяют
гепарин.
Если больному, получающему варфарин, показа-
на операция, то вначале следует определить МНО.
Незначительное увеличение МНО (менее 1,5)
допустимо и даже желательно для профилактики
послеоперационных венозных тромбозов. Малые
операции без риска массивного кровотечения
можно проводить при значениях МНО менее 2,5.
Перед большими операциями варфарин отменяют
за несколько дней (наиболее распространенная ре-
комендация — за четыре дня), чтобы МНО сни-
зилось до безопасного уровня — менее 1,5. Даже
тогда, когда прием варфарина в периоперационном
периоде принципиально возможен, это часто быва-
ет неудобным из-за самой операции (сдвигается
время приема) или взаимодействия с приме-
няемыми в ходе ее лекарственными препаратами.
В таких случаях варфарин отменяют и переходят
к п/к введению или в/в инфузии нефракциони-
рованного или низкомолекулярного гепарина до
увеличения АЧТВ в 1,5-2,5 раза. В экстренных слу-
чаях под контролем ПВ, МНО и АЧТВ переливают
СЗП, что позволяет эффективно, быстро и управ-
ляемо устранить действие антикоагулянтов. Вита-
мин Ki (фитоменадион) не применяют, поскольку
в последующие несколько недель это может значи-
тельно затруднить действие непрямых антикоагу-
лянтов.
Абсолютная или относительная передозировка
варфарина может вызвать сильное кровотечение.
Источником кровотечения могут быть ЖКТ, лег-
кие, ЦНС, кожа. В этом случае показано перелива-
ние СЗП, концентрата протромбинового комплекса,
концентратов различных факторов свертывания.
Кроме того, назначают 2,5-10 мг фитоменадиона
в/в медленно.
АНТИАГРЕГАНТЫ
Тромбоциты играют важную роль в образовании
артериального тромба при атеросклерозе, когда
сосудистая стенка поражена атеромой. Тромбоци-
ты могут активно участвовать в процессе атероге-
неза. Неясно, играют ли тромбоциты какую-либо
роль в венозном тромбозе. Антиагреганты назна-
чают для профилактики и лечения тромбоэмболий
в артериальном русле. Показания: профилактика
тромбозов после кардиохирургических операций
и ангиопластики; профилактика и лечение ише-
мии головного мозга и инфаркта миокарда; пре-
дотвращение тромбообразования при проведении
гемодиализа и использовании мембранного окси-
генатора. Антиагреганты используют для про-
филактики сосудистых нарушений при пересадке
органов: они предотвращают иммунологически опос-
редованное повреждение эндотелия, которое
является причиной ускоренного развития атеро-
склероза коронарных артерий после пересадки
сердца и окклюзии мелких артерий в пересажен-
ной почке.
170
Глава 8
R medlit.ucqz.ru
В настоящее время изучены многие детали адге- Во многих клинических исследованиях оце-
зии и агрегации тромбоцитов. После повреждения
сосуда тромбоциты слипаются с коллагеном и дру-
гими белками субэндотелиального слоя с помощью
белков адгезии (например, фактора фон Виллеб-
ранда). Из гранул тромбоцитов высвобождаются
различные медиаторы (тромбоксан А2,5-гидрокси-
триптамин, АДФ, 0-тромбоглобулин). Последую-
щая активация зависит от связывания АДФ с ре-
цепторами тромбоцитов и экспрессии гликопро-
теидных рецепторов ПЬ/Ша. Гликопротеидные
рецепторы ПЬ/Ша перекрестно связываются с фиб-
риногеном, что вызывает агрегацию тромбоцитов
(рис. 8.3). Экспозиция тромбоцитарных фосфоли-
пидов приводит к образованию тромба и пре-
вращению фибриногена в фибрин. Антиагреганты
нарушают эти процессы, какой-либо один из них
или несколько, в зависимости от препарата.
АСПИРИН
Аспирин необратимо угнетает активность цик-
лооксигеназы-1 в тромбоцитах, ацетилируя сери-
новые остатки вблизи активного центра фермента.
Это нарушает синтез циклических эндопероксидов
из арахидоновой кислоты, в результате чего умень-
шается образование тромбоксана А2, что в свою оче-
редь ингибирует высвобождение АДФ и агрегацию
тромбоцитов. Аспирин также угнетает активность
циклооксигеназы-1 в сосудистом эндотелии, что
ингибирует синтез циклических эндопероксидов
и простагландина 12 и может противодействовать
влиянию препарата на тромбоциты. Обычно влия-
ние аспирина на циклооксигеназу тромбоцитов
преобладает; происходит это по трем причинам.
Во-первых, синтез простациклина в поврежденном
эндотелии протекает с относительно постоянной
скоростью и менее чувствителен к угнетению.
Во-вторых, циклооксигеназа тромбоцитов необра-
тимо ацетилируется аспирином, после чего тром-
боциты (в отличие от сосудистого эндотелия) не
могут синтезировать новый фермент и остаются
неактивными в течение всего своего срока жизни
(7-9 суток). В-третьих, тромбоциты подвергаются
воздействию аспирина еще до поступления в сис-
темный кровоток,- в крови воротной вены, в то
время как на эндотелий артерий влияет лишь доля
препарата, оставшаяся после первого прохождения
через печень и метаболического превращения в са-
лициловую кислоту. В результате суммарный эф-
фект аспирина состоит в ингибировании агрегации
тромбоцитов. Для полного угнетения активности
циклооксигеназы-1 в тромбоцитах достаточно
относительно невысокой дозы аспирина — 150 мг
в сутки.
нивались отдаленные результаты применения раз-
личных доз аспирина (от 75 до 325 мг в сутки) для
первичной и вторичной профилактики инфаркта
миокарда и церебральной ишемии. Аспирин снижа-
ет частоту транзиторных ишемических атак и риск
инсульта у мужчин. Аспирин также снижает риск
инфаркта миокарда при нестабильной стенокар-
дии.
Есть данные, что применение аспирина в пе-
риоперационном периоде может снизить частоту
тромбоэмболических осложнений после некото-
рых хирургических вмешательств, особенно у мужчин
после протезирования тазобедренного сустава.
Другие НПВС могут оказывать аналогичное
дей- ствие, но их обычно не применяют в качестве
антиагрегантов. Применение любых НПВС в пе-
риоперационном периоде может привести к тяже-
лому кровотечению во время хирургического
вмешательства и после него. Иногда при этом выяв-
ляют тромбоцитопению или тромбоцитопатию, что
свидетельствует о возможной реакции гиперчувст-
вительности. Из-за высокого риска периопераци-
онного кровотечения НПВС необходимо отменять
перед подавляющим большинством больших опе-
раций, трансуретральной простатэктомией и опе-
рациями на сетчатке глаза.
ДИПИРИДАМОЛ
Дипиридамол является вазодилататором, кото-
рый первоначально использовался для лечения
стенокардии. С этой целью он больше не применя-
ется, но иногда используется как антиагрегант.
Дипиридамол угнетает агрегацию тромбоцитов,
увеличивая внутриклеточную концентрацию
цАМФ посредством двух различных механизмов.
Дипиридамол ингибирует фосфодиэстеразу V типа,
что приводит к увеличению концентрации цАМФ,
которое вызывает секвестрацию Са2+ в цитозоле
и подавляет активность фосфолипазы. Кроме того,
дипиридамол также тормозит обратный захват аде-
нозина эритроцитами; аденозин плазмы действует
на А2-рецепторы и стимулирует аденилатциклазу
тромбоцитов. Вследствие этого подавляется
АДФ-индуцированная агрегация тромбоцитов
(рис. 8.3).
Дипиридамол назначают в сочетании с непря-
мыми антикоагулянтами для профилактики тром-
бообразования на протезированных клапанах
сердца. Он усиливает действие непрямых анти-
коагулянтов, что чревато риском повышенной
кровоточивости и требует мониторинга ПВ. Кроме
того, дипиридамол применяется для профилак-
тики транзиторных ишемических атак и инсульта.
АНТИКОАГУЛЯНТЫ, АНТИАГРЕГАНТЫ И ТРОМБОЛИТИКИ
171
medlit.ucoz.ru
Повреждение сосуда
Обнажение субэндотелиального слоя
ФфВ
Коллаген
, Тромбин
Адгезия тромбоцитов
I
Синтез и высвобождение тромбоцитарных факторов
Рис. 8.3. Влияние ан-
тиагрегантов на акти-
вацию и агрегацию
тромбоцитов. Назва-
ния лекарственных
препаратов выделены
курсивом.
А К - арахидоновая
кислота, ЦЭ — цикли-
ческие эндоперекси-
ды, ФфВ — фактор
фон Виллебранда
4 Дипиридамол
i Эпопрастенол
АК
±НПВС | Циклооксигеназа-1
ЦЭ
Щазоксибен |
Тромбоксан Аг 4 цАМФ АДФ
| 4—-—'^Тиклопидин
4 Клопидогрель
Активация тромбоцитов
Экспрессия гликопротеидных рецепторов ПЬ/Ша
• I Абцихсимаб
Связывание с фибриногеном 4 Тирофибан
4 Эптифибатид
Агрегация тромбоцитов
Протромбин
Тромбин
Препараты дипиридамола длительного действия
снижают частоту повторных инсультов на 15-20%
и оказывают аддитивное аспирину антиагрегантное
действие. Дипиридамол назначают внутрь в дозе
300-600 мг/сут в 3-4 приема перед едой; препараты
длительного действия — по 200 мг 2 раза в сутки.
Дипиридамол вызывает ортостатическую гипото-
нию и пульсирующую головную боль, особенно при
сопутствующей мигрени.
тиклопидин
Тиклопидин необратимо блокирует аденозин-
дифосфатные рецепторы тромбоцитов, что предот-
вращает АДФ-зависимую активацию гликопроте-
идных рецепторов Ilb/IIIa (рис. 8.3). Тиклопидин
активен только in vivo и практически не оказывает
действия in vitro1, под действием цитохрома Р-450
он превращается в активный метаболит. Угнете-
ние функции тромбоцитов происходит спустя не-
сколько дней, а максимальный эффект наступает на
5-8 сутки. Тиклопидин может вызывать нейтропе-
нию, агранулоцитоз и тромбоцитопению, поэтому
в ходе лечения каждые 2 недели необходимо прово-
дить общий анализ крови. Тиклопидин применяют
для первичной и вторичной профилактики инсуль-
та и инфаркта миокарда; он особенно полезен при
противопоказаниях к аспирину. Тиклопидин также
используют для профилактики осложнений после
коронарной ангиопластики и стентирования, а так-
же тромбообразования в аппарате искусственного
кровообращения.
172
Глава 8
КЛОПИДОГРЕЛЬ
Клопидогрель по химическому строению и ме-
ханизму действия близок к тиклопидину. Он нару-
шает связывание фибриногена рецепторами и угне-
тает агрегацию тромбоцитов. Поскольку действие
клопидогреля необратимо, функция тромбоци-
тов угнетается на весь срок их жизни (7-9 суток).
Клопидогрель также подавляет стимулирующее
действие других медиаторов (например, АДФ) на
агрегацию тромбоцитов. В условиях in vitro клопи-
догрель неактивен; как и тиклопидин, in vivo он пре-
вращается в активный метаболит под действием
цитохрома Р-450. Клопидогрель не уступает по эф-
фективности аспирину в профилактике осложне-
ний и рецидивов инфаркта миокарда и ишемическо-
го инсульта. В отличие от тиклопидина, клопидог-
рель не вызывает нейтропении и тромбоцитопении.
Тем не менее тиклопидин и клопидогрель являются
очень дорогой альтернативой аспирину.
АБЦИКСИМАБ
Абциксимаб является F(ab) фрагментом гиб-
ридных моноклональных антител к гликопротеид-
ным рецепторам ПЬ/Ша. Гликопротеидный рецеп-
тор ПЬ/Ша, представляющий собой интегральный
белок клеточной мембраны тромбоцитов, служит
рецептором для фибриногена и фактора фон Вил-
лебранда; он необходим для связывания тромбоци-
тов с чужеродными поверхностями и друг с другом.
Абциксимаб угнетает агрегацию тромбоцитов,
препятствуя их связыванию с фибриногеном, фак-
тором фон Виллебранда и другими адгезивными
молекулами. Абциксимаб также связывается с витро-
нектиновыми рецепторами на тромбоцитах и эндо-
телиальных клетках; витронектин представляет собой
гликопротеид, задействованный в процессах свер-
тываемости, адгезии и пролиферации. Абциксимаб
применяют в сочетании с гепарином и аспи-
рином для профилактики ишемических осложнений
во время коронарной ангиопластики. Абциксимаб
обладает антигенными свойствами, его повторное
применение может быть менее эффективным, вы-
звать аллергические реакции и послужить причи-
ной тяжелой тромбоцитопении.
ЭПОПРОСТЕНОЛ
Эпопростенол (PGI2) является природным про-
стагландином, вырабатываемым в интиме крове-
носных сосудов. Это очень мощный ингибитор аг-
регации тромбоцитов, угнетающий ее путем повы-
шения внутриклеточной концентрации цАМФ.
Кроме того, эпопростенол является мощным ва-
зодилататором. Для эпопростенола Т1Д составляет все-
го 2-3 мин; при pH 7,4 он подвергается быстрому
medlit.ucoz.ru
спонтанному гидролизу и превращается в 6-ке-
топростагландин FIa. Эпопростенол применяют в виде
длительной в/в инфузии в дозе 5 нг/кг/мин. Эпо-
простенол может заменять гепарин при гемодиали-
зе. Побочные эффекты включают покраснение
кожи, головную боль и артериальную гипотонию;
при увеличении дозы могут возникнуть брадикар-
дия, бледность и потливость.
ДРУГИЕ АНТИАГРЕГАНТЫ
Тирофибан является непептидным конкурент-
ным антагонистом гликопротеидных рецепторов
ПЬ/Ша. Его применяют вместе с аспирином и гепа-
рином при нестабильной стенокардии и инфаркте
миокарда без патологического зубца Q.
Эптифибатид обладает аналогичным действием
и также применяется вместе с аспирином и гепари-
ном при нестабильной стенокардии и инфаркте
миокарда без патологического зубца Q.
Дазоксибен in vitro селективно угнетает синтез
тромбоксана А2, что приводит к увеличению синте-
за простагландина 12. Тем не менее in vivo дазокси-
бен заметно уменьшает агрегацию тромбоцитов
только в сочетании с аспирином.
Сульфинпиразон (урикозурическое средство)
нарушает агрегацию тромбоцитов, вызывая об-
ратимое угнетение циклооксигеназы-1. Первона-
чальное предположение, что длительное лечение
сульфинпиразоном снижает частоту повторных
инфарктов миокарда, не подтвердилось.
НИЗКОМОЛЕКУЛЯРНЫЕ ДЕКСТРАНЫ
Декстраны, которые иногда используются как
инфузионные растворы для восполнения ОЦК, яв-
ляются полисахаридами с молекулярным весом
10-50 кДа, состоящими из остатков глюкозы.
Молекулы декстранов представляют собой раз-
ветвленные цепи. Декстраны образуются в резуль-
тате ферментации сахарозы бактерией Leuconostoc
mesenteroides. Метаболическая деградация декстра-
нов в организме протекает медленно.
Декстран-40 выпускается в виде 10% раствора
на основе 0,9% NaCl или 5% глюкозы. Соединение
содержит молекулы со средним молекулярным ве-
сом 40 кДа. Осмотическое давление раствора чуть
выше, чем у плазмы.
Декстран-70 выпускается в виде 6% раствора на
основе 0,9% NaCl или 5% глюкозы. Соединение
содержит молекулы со средним молекулярным ве-
сом 70 кДа. Осмотическое давление раствора такое
же, как у плазмы.
При добавлении в кровь in vitro декстраны не
оказывают влияния на функцию тромбоцитов.
Однако их в/в инфузия может удлинять время
АНТИКОАГУЛЯНТЫ, АНТИАГРЕГАНТЫ И ТРОМБОЛИТИКИ
173
medlit.u
кровотечения, нарушать полимеризацию фибрина
и угнетать функцию тромбоцитов.
Декстраны применяют не только в качестве кро-
везаменителей для поддержания ОЦК при гипово-
лемическом шоке, но и для профилактики тромбо-
эмболических осложнений после операции. Более
того, инфузия декстрана-40 может уменьшить вяз-
кость крови и внутрисосудистую агглютинацию
эритроцитов, что улучшает микроциркуляцию.
Другие коллоидные кровезаменители (гидрокси-
этилированные крахмалы и растворы желатина)
также угнетают агрегацию тромбоцитов.
Декстраны могут вызывать различные осложне-
ния. Перегрузка жидкостью особенно опасна при
сопутствующей сердечной или почечной недоста-
точности. Возможны анафилактоидные реакции,
которые проявляются крапивницей, бронхоспаз-
мом или артериальной гипотонией; в отсутствие
сознания они протекают тяжелее. Декстраны с бо-
лее высоким молекулярным весом индуцируют аг-
регацию эритроцитов с формированием монетных
столбиков и повышают СОЭ. Кроме того, они за-
трудняют определение группы крови, резус-факто-
ра и проведение пробы на индивидуальную совмес-
тимость, а также искажают результаты некоторых
биохимических анализов. Из-за риска осложнений
декстраны редко назначаются для профилактики
тромбоэмболических осложнений.
трЬмболитики
(ФИБРИНОЛИТИКИ)
В процессе свертывания крови также активиру-
ется и фибринолиз. Пусковым фактором является
повреждение тканей, в результате чего плазмино-
ген (неактивный р-глобулин плазмы) превращает-
ся в плазмин (протеолитический фермент), расще-
пляющий фибрин до продуктов, которые, в свою
очередь, растворяют тромб (рис. 8.4). Плазмин —
это относительно неспецифический фермент, ко-
торый помимо фибрина расщепляет фибриноген
и другие факторы свертывания.
Основным физиологическим активатором пре-
вращения плазминогена в плазмин является ткане-
вой активатор плазминогена. Этот протеолитиче-
ский фермент высвобождается из эндотелия повре-
жденных сосудов и фагоцитов или образуется из
проактиватора в тканях под действием фактора
ХПа (активированный фактор Хагемана). Ткане-
вой активатор плазминогена наиболее эффективен
на этапе связывания плазминогена с фибрином;
следовательно, образование и действие плазмина
ограничено тромбом. Попадающий в циркулирую-
щую кровь плазмин немедленно нейтрализуется
антиплазминами.
Активаторы плазминогена присутствуют также
в секретах различных желез (моче, молоке, слезах
и поте), что препятствует отложению фибрина
Связанный с фибрином
Циркулирующий
плазминоген
плазминоген
Тканевой активатор
f плазминогена
f Анистреплаза
^Алтеплаза
^Ретеплаза
Продукты деградации
фибриногена
Продукты деградации
фибрина
Рис. 8.4. Влияние
тромболитиков и анти-
фибринолитических
средств на фибрино-
лиз. Названия лекар-
ственных препаратов
выделены курсивом
174
Глава 8
„ , medlit
в протоках. Урокиназа была впервые обнаружена
в человеческой моче в 1885 г. и позднее выделена
из культуры почечных клеток. Были идентифи-
цированы экзогенные активаторы плазминогена
(например, стрептокиназа). Некоторые активато-
ры плазминогена широко используются в лече-
нии ТЭЛА и острого инфаркта миокарда. Уроки-
наза применяется реже, в основном для местного
лечения.
Стрептокиназа
Этот лишенный ферментативных свойств белок
с молекулярным весом 47 кДа впервые был выде-
лен в 1945 г. из р-гемолитических стрептококков
группы С. Стрептокиназа образует стабильный
комплекс с плазминогеном (как со свободным, так
и со связанным с фибрином), в результате чего об-
разуется плазмин. Т]/2 плазмина в крови составляет
приблизительно 40-80 мин. Стрептокиназа обла-
дает антигенными свойствами, и ее первоначальная
доза предназначена для нейтрализации антистреп-
тококковых антител, образовавшихся после пред-
шествующего лечения этим препаратом или пере-
несенной стрептококковой инфекции. Повторное
введение стрептокиназы возможно только через
6 месяцев. Будучи антигеном, препарат может вы-
зывать лихорадку и аллергические реакции, в том
числе тяжелые. Частота этих побочных эффектов
уменьшается при медленном введении и профилак-
тическом назначении кортикостероидов. Для лече-
ния тяжелых аллергических реакций могут потре-
боваться адреналин, Нг и Н2-блокаторы.
Стрептокиназа играет важную роль в лечении
тромбозов глубоких вен, острой ТЭЛА и артериаль-
ной тромбоэмболии. Стандартная схема лечения
заключается в отмене антикоагулянтов и медленном
введении нагрузочной дозы 600 000 ME стрептоки-
назы в периферическую вену в течение 30-60 мин,
затем проводят инфузию в дозе 100 000 МЕ/ч
продолжительностью до 72 ч. До начала и во время ин-
фузии стрептокиназы обязательно проводят анализ
свертывающей системы крови, включая тромбино-
вое время, ПВ, гематокрит и количество тромбоци-
тов. После окончания тромболитической терапии,
когда тромбиновое время снижается до значения,
которое превышает норму менее чем в два раза, для
профилактики повторного тромбоза назначают ин-
фузию гепарина, а при необходимости — непрямые
антикоагулянты.
Однако чаще стрептокиназа применяется для
лечения инфаркта миокарда в первые 6 ч от начала
заболевания. Считается, что формирование очага
инфаркта обычно завершается в течение 4-6 ч,
icoz.ru
особенно после появления патологического зубца
0,на ЭКГ, и тромболизис по истечении этого срока
чаще всего оказывается неэффективным. При ост-
ром инфаркте миокарда 1500000 ME стрептокина-
зы разводят в 0,9% NaCl или 5% глюкозе и вводят
в/в в течение 60 мин. Несмотря на относительную
простоту, в/в инфузия стрептокиназы достаточно
опасна. Внутривенная тромболитическая терапия
неизбежно вызывает системный фибринолиз и по-
вышает риск кровотечений. Присутствие свободно-
го плазмина в кровотоке приводит к потреблению
различных факторов свертывания (главным обра-
зом, фибриногена и факторов V и XII).
При инфаркте миокарда стрептокиназу также
можно вводить непосредственно в коронарную ар-
терию. В идеале такое лечение следует начинать в
течение 6 ч от начала заболевания. Тромб визуали-
зируют с помощью селективной коронарографии,
выполняемой через плечевую или бедренную арте-
рию. Затем через катетер, установленный в коронар-
ной артерии, вводят нагрузочную дозу 250 000 ME,
после чего начинают инфузию в дозе 4000 МЕ/мин
в течение 75 мин или до восстановления проходи-
мости сосуда. Впоследствие также необходимо ле-
чение гепарином для профилактики повторного
тромбоза пораженной артерии. Внутрикоронарная
инфузия стрептокиназы — требующая высокой ква-
лификации и сопряженная со значительным риском
осложнений манипуляция, ее можно выполнять
только в условиях регионального кардиологиче-
ского центра или специализированного отделения.
Есть данные, что после внутрикоронарного введе-
ния стрептокиназы реканализация окклюзирован-
ной артерии происходит реже, чем после внутри-
венной инфузии.
Анистреплаза
Анистреплаза является ацилированным (ани-
зоилированным) комплексом стрептокиназы и
лиз-плазминогена человека. После в/в инъекции
анистреплаза в основном связывается с фибрином,
и анизоильная группа, защищающая активный
центр, гидролизуется. Образующийся активатор-
ный комплекс превращает связанный с фибрином
плазминоген в плазмин. Т1/2 деацилирования в кро-
ви и тромбе составляет приблизительно 40-60 мин.
Считается, что в кровеносном русле комплекс отно-
сительно неактивен и активируется только в местах
свежего тромбоза. Следовательно, анистреплаза
преимущественно влияет на фибринолиз в тромбе,
хотя вызывает и системный фибринолиз.
Анистреплазу вводят в/в медленно: 30 единиц
в течение 4-5 минут. Продолжительность действия
АНТИКОАГУЛЯНТЫ, АНТИАГРЕГАНТЫ И ТРОМБОЛИТИКИ
175
medlit.ucoz.ru
составляет 4-6 ч. Препарат применяют в первые 6 ч
после начала инфаркта миокарда. Анистреплаза
обладает антигенными свойствами.
Алтеплаза
Алтеплаза — рекомбинантный тканевой актива-
тор плазминогена. Она представляет собой одиноч-
ную цепь природного белка и не имеет антиген-
ных свойств. Препарат обладает значительно более
низким сродством к циркулирующему плазмино-
гену, чем к плазминогену, связанному с фибрином,
и в кровотоке относительно неактивен даже в высо-
кой концентрации. Алтеплаза активируется только
в комплексе с фибрином и вызывает превраще-
ние связанного с фибрином плазминогена в плаз-
мин. Следовательно, фибринолиз происходит только
в тромбе.
Алтеплаза подвергается интенсивному метабо-
лизму в печени. Препарат применяют в первые 6 ч
от начала острого инфаркта миокарда: полную дозу
(до 100 мг) вводят в течение 90 мин. Пока не доказа-
но, что алтеплаза и анистреплаза имеют явные пре-
имущества перед стрептокиназой.
Ретеплаза
Ретеплаза — это сходный с алтеплазой реком-
бинантный активатор плазминогена, не имеющий
антигенных свойств. Ретеплаза преимущественно
активирует плазминоген, связанный с фибрином.
По сравнению с алтеплазой, Ti/2 ретеплазы длиннее
и она дольше циркулирует в сосудистом русле.
Урокиназа
Урокиназа - это протеолитический фермент,
который впервые был выделен из человеческой
мочи. В настоящее время урокиназу получают из
культуры почечных клеток эмбриона. У человека
урокиназа не проявляет антигенных свойств. Своим
действием она напоминает стрептокиназу. Эндо-
генный плазминоген превращается в плазмин,
вызывая фибринолиз. Урокиназа имеет короткий
Ti/2 (10-20 мин) и быстро удаляется из сосудистого
русла. Хотя урокиназу можно применять системно
(при тромбоэмболических осложнениях), она дей-
ствует относительно неселективно и быстро исто-
щает запасы циркулирующего в крови плазминоге-
на. В связи с этим урокиназу чаще используют для
местного лечения тромбозов. Препарат может вво-
дить в артериовенозную гемодиализную фистулу и
венозные катетеры, а также использовать для оро-
шения передней камеры глаза или стекловидного
тела в дозе 5000-25 000 ME. При местном лечении
нет необходимости проводить мониторинг сверты-
вающей системы крови.
Станозолол
Станозолол является анаболическим стероид-
ным гормоном с относительно низкой андрогенной
активностью. Он усиливает эндогенный фибрино-
лиз, повышая синтез и высвобождение активатора
плазминогена из сосудистой стенки. Станозолол
применяют для лечения болезни Бехчета, профи-
лактики приступов наследственного ангионевро-
тического отека и лечения слипчивого арахноидита
после неудачных операций на позвоночнике.
АНТИФИБРИНОЛИТИЧЕСКИЕ
СРЕДСТВА
Антифибринолитические средства подавляют
превращение плазминогена в плазмин или угнета-
ют действие самого плазмина, ингибируя фибрино-
лиз. Они применяются для лечения состояний,
обусловленных повышенной фибринолитической
активностью (кровотечения в акушерстве, опера-
ции на предстательной железе), и лечения кровоте-
чений у больных гемофилией. Антифибринолити-
ческие средства также являются антидотами при
передозировке тромболитиков.
ТРАНЕКСАМОВАЯ КИСЛОТА
Транексамовая кислота является мощным ин-
гибитором активации плазминогена, предотвраща-
ет образование плазмина и угнетает фибринолиз.
В высоких концентрациях препарат является
неконкурентным ингибитором плазмина. Транек-
самовую кислоту назначают внутрь или в/в для
профилактики и лечения кровотечений при ме-
норрагиях и операциях на предстательной железе,
а также в хирургической стоматологии, в том числе
при удалении зубов у больных гемофилией. Кроме
того, транексамовую кислоту применяют для ле-
чения наследственного ангионевротического отека
и при ДВС-синдроме.
АПРОТИНИН
Апротинин — это ингибитор сериновых протеи-
наз, влияющий на многие протеолитические фер-
менты, в том числе калликреин, кинины плазмы,
трипсины и плазмин. Антифибринолитическое
действие апротинина обусловлено угнетением
плазмина и его активатора. Апротинин применяет-
ся для лечения тяжелых кровотечений, обуслов-
ленных гиперплазминемическими состояниями,
особенно при злокачественных заболеваниях или
осложнениях тромболитической терапии. С недав-
него времени апротинин с успехом используется
176
Глава 8
для уменьшения кровопотери при карди
ческих операциях с искусственным кровообращением
и других обширных операциях. Поскольку апроти-
нин подавляет активность трипсина и химотрипси-
на, он также применяется для лечения острого пан-
креатита. Апротинин вводят путем медленной в/в
инфузии; он может вызывать местные тромбофле-
биты и иногда тяжелые реакции гиперчувствитель-
ности (эритему, крапивницу, бронхоспазм).
ДРУГИЕ ЛЕКАРСТВЕННЫЕ
СРЕДСТВА, ВЛИЯЮЩИЕ НА
ГЕМОСТАЗ
АНКРОД
Анкрод — выделенный из яда малайской гадюки
протеолитический фермент, который расщепляет
фибрин, уменьшая уровень фибриногена в крови.
При этом образуются микроскопические эмболы,
которые быстро удаляются из кровотока. Запасы
фибриногена истощаются, что приводит к сниже-
нию свертываемости крови. Действие препарата ха-
рактеризуется как доброкачественный и управляе-
мый ДВС-синдром. Анкрод иногда применяют для
профилактики и лечения тромбоза глубоких вен.
Анкрод назначают в/м или в/в; во избежание
быстрого высвобождения продуктов деградации
фибрина его нужно вводить медленно. Эффект оце-
нивают по размеру сгустка или уровню фибри-
ногена в плазме. При передозировке назначают
специфический антидот (который обладает анти-
генными свойствами) или переливают препарат кро-
ви с высоким содержанием фибриногена (напри-
мер, криопреципитат). В Великобритании анкрод
назначают только в особых случаях.
ЭТАМЗИЛАТ
Этамзилат является гемостатическим средст-
вом, нормализующим проницаемость капилляров
и функцию тромбоцитов. Препарат назначают
внутрь или парентерально для лечения меноррагий
и предотвращения капиллярных кровотечений во
время операций. У новорожденных этамзилат ис-
пользуют для профилактики и лечения перивен-
трикулярных кровоизлияний. После системного
введения уменьшается время кровотечения, хотя
уровень тромбоцитов и факторов свертывания не
изменяются.
Побочные эффекты встречаются относительно
редко и включают головную боль, тошноту и сыпь;
после в/в введения возможно снижение АД.
тедеи£й^₽Ессин
Десмопрессин — это синтетический аналог вазо-
прессина, не вызывающий вазоконстрикции. Он ис-
пользуется для улучшения гемостаза при легкой
форме гемофилии А и некоторых формах болезни
фон Виллебранда. Десмопрессин индуцирует вы-
свобождение необходимых факторов свертывания.
Десмопрессин укорачивает время кровотечения
при различных тромбоцитопатиях (например, при
уремии, после приема аспирина), а также позволяет
уменьшить кровопотерю при кардиохирургиче-
ских операциях.
Список литературы
Ali, Н.Н. & Savarese, JJ. (1976) Monitoring of neuromuscular
function. Anesthesiology 45, 216-249.
Bevan, D.R. (1994) Newer neuromuscular blocking agents.
Pharmacology and Toxicology 74,3-9.
Bevan, D.R. (1994) Succinylcholine. Canadian Journal of Anaes-
thesia 41,465-468.
Bowman, W.C. (1993) New developments at the neuromuscular
Junction. In: Proceedings of the J.B. Stenlake Symposium, University
of Strathclyde, Glasgow, 15-22 September 1993.
Caldwell, J. (1995) New muscle relaxants. Current Opinion in
Anesthesiology 8, 356-361.
Calvey, T.N. (1984) Assessment neuromuscular blockade by elec-
tromyography. Journal of the Royal Society of Medicine 77,56-59.
Changeux, J.-P., Giraudat, J. & Dennis, M. (1987) The nicotinic
acetylcholine receptor: molecular architecture of a ligand-regulated
ion channel. Trends in Pharmacological Sciences 8,459-465.
Cooper, R., Mirakhur, R.K., Clarke, R.SJ. & Boules, Z. (1992)
Comparison of intubating conditions after administration of Org
9426 (rocuronium) and suxamethonium. British Journal of
Anaesthesia 69,269-273.
Drachman, D.B., Adams, R.N., Josifek, L.F. et al. (1981)
Antibody-mediated mechanisms of Ach receptor loss in myasthenia
gravis: clinical relevance. Annals of the New York Academy of
Sciences 337, 175-188.
Fleming, N.W. & Lewis, B.K. (1994) Cholinesterase inhibitors do
not prolong neuromuscular block produced by mivacurium. British
Journal of Anaesthesia 73,241-243.
Gronert, G.A., Mott, J. & Lee, J. (1988) Aetiology of malignant
hyperthermia. British Journal of Anaesthesia 60, 253-267.
Lambert, JJ., Durant, N.N. & Henderson, E.G. (1983)
Drug-induced modification of ionic conductance at the
neuromuscular junction. Annual Review of Pharmacology and
Toxicology 23, 505-539.
Lenzen, C., Roewer, N., Wappler, F. et al. (1993) Accelerated
contractures after administration of ryanodine to skeletal muscle of
malignant hyperthermia susceptible patients. British Journal of
Anaesthesia 71, 242-246.
Lu, Z. & Smith, D.O. (1991) Adenosine 5’triphosphate increases
acetylcholine channel opening time in rat skeletal muscle. Journal of
Physiology 436,45-46.
177
Глава 8
medlit.ucoz.ru
O’Sullivan, Е.Р., Williams, N.E. & Calvey, T.N. (1988) Differential
effects of neuromuscular blocking agents on suxamethonium-
induced fasciculations and myalgia. British Journal of Anaesthesia
60,367-371.
Paton, W.D.M. & Waud, D.R. (1967) The margin of safety of
neuromuscular transmission. Journal of Physiology 191, 59-90.
Popper, P. & Miceyvych, P.E. (1989) Localisation of calcium
gene-related peptide and its receptors in striated muscle. Brain
Research 496,180-186.
Quik, M., Collier, B., Audhya, T. & Goldstein, G. (1990)
Thymopoietin inhibits function and ligand binding to nicotinic
receptors at the neuromuscular junction. Journal of Pharmacology
and Experimental Therapeutics 254,1113-1119.
Rosenberg, H. & Gronert, G.A. (1992) Intractable cardiac arrest in
children given succinylcholine. Anesthesiology 77, 1054.
Sudhof, T.C. & Jahn, R. (1991) Proteins of synaptic vesicles involved
in exocytosis and membrane recycling. Neurone 6,665-667.
Tolmie, J.D., Joyce, Т.Н. & Mitchell, G.D. (1967) Succinylcholine
danger in the burned patient. Anesthesiology 28,467-470.
Whittaker, V.P. (1986) The storage and release of acetylcholine.
Trends in Pharmacological Sciences 7,312-315.
Williams, N.E., Webb, S.N. & Calvey, T.N. (1980) Differential
effects of myoneural blocking drugs on neuromuscular transmission.
British Journal of Anaesthesia 52,1111-1115.
Wong, S.F. & Chung, F. (2000) Succinylcholine associated
postoperative myalgia. Anaesthesia 55,144-152.
Zaimis, E. (ed.) (1976) Neuromuscular junction. Handbook of
Experimental Pharmacology, Vol. 42. Berlin: Springer-Verlag.
medlit.ucoz.ru
Научное издание
Калви Т.Н., Уильямс Н.Е.
ФАРМАКОЛОГИЯ ДЛЯ АНЕСТЕЗИОЛОГА
Зав. редакцией к.б.н. Е.В. Мосткова
Оформление СО. Мясникова
Компьютерная верстка и корректура: издательство БИНОМ
ISBN 978-5-9518-0216-3
Подписано в печать 09.08.2007
Формат 84x1 08/1 6. Печ. л. 11,5
Бумага офсетная. Печать офсетная. Тираж 4000. Заказ 5469
ООО "Издательство БИНОМ", 2007
103473, Москва, ул. Краснопролетарская, 16
При участии ООО «ЭМПРЕЗА»
Отпечатано в ОАО «ИПК «Ульяновский Дом печати»
432980, г. Ульяновск, ул. Гончарова, 14
978-5-9518-02'
6-3
785951
802163