/





























































































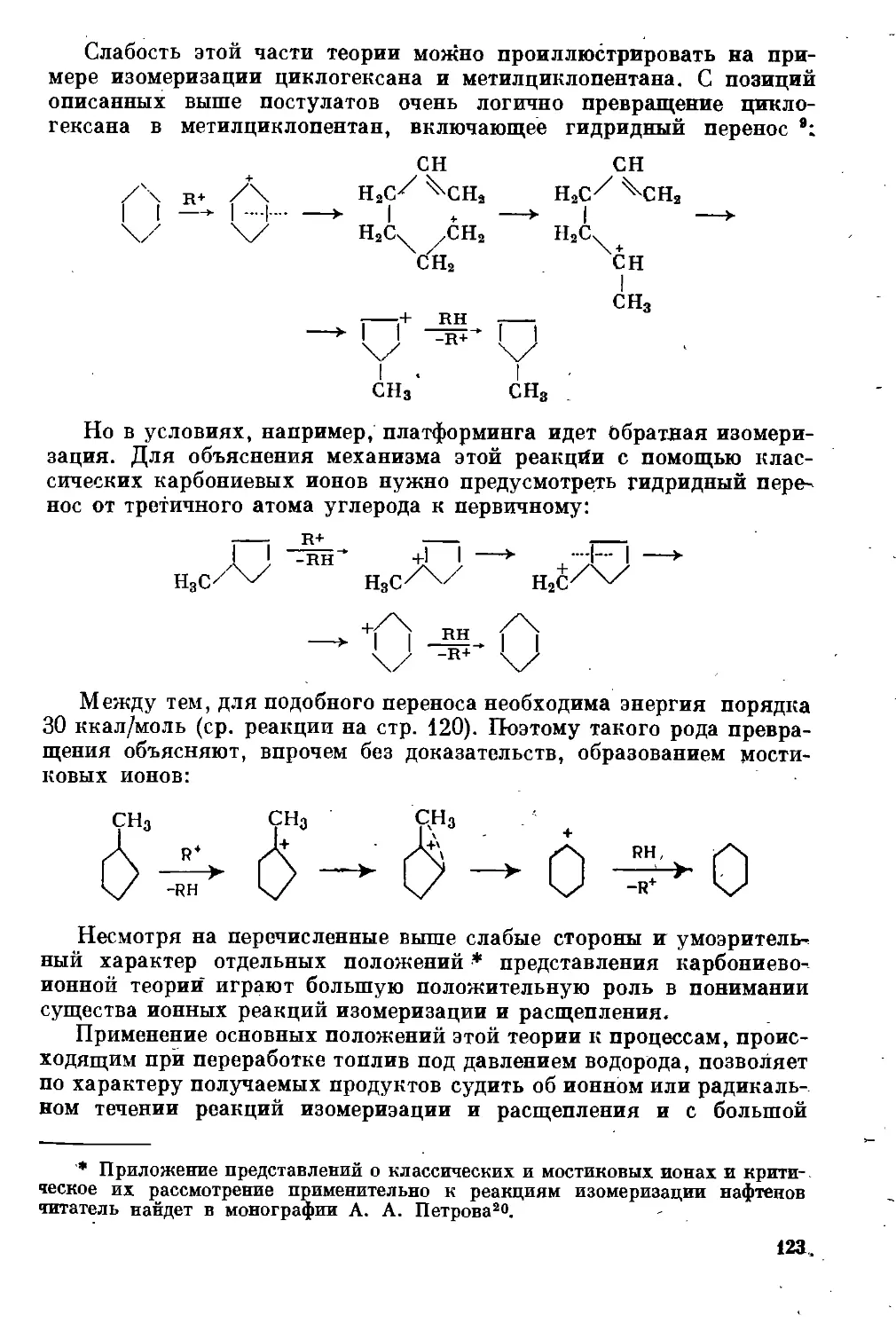






















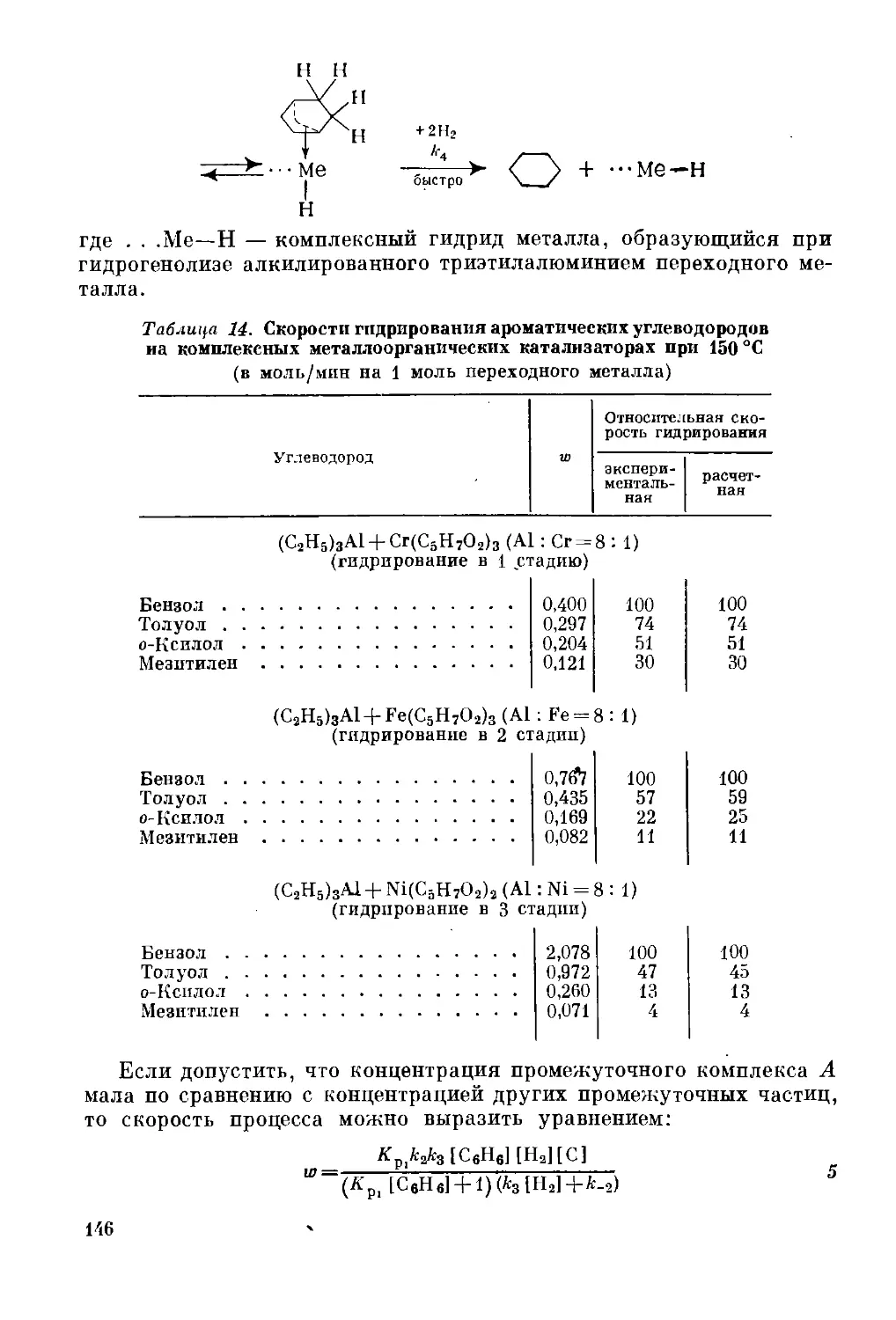



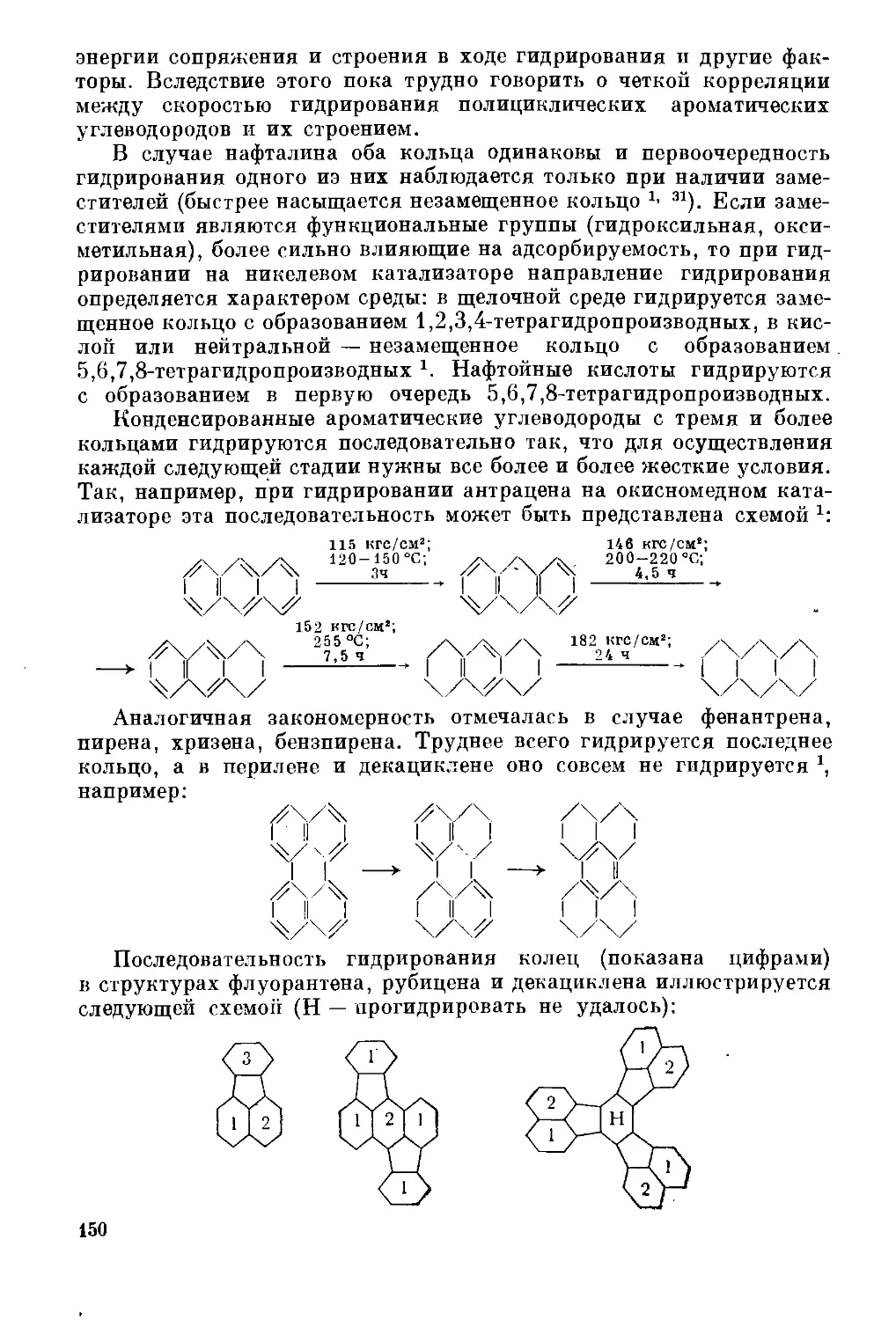




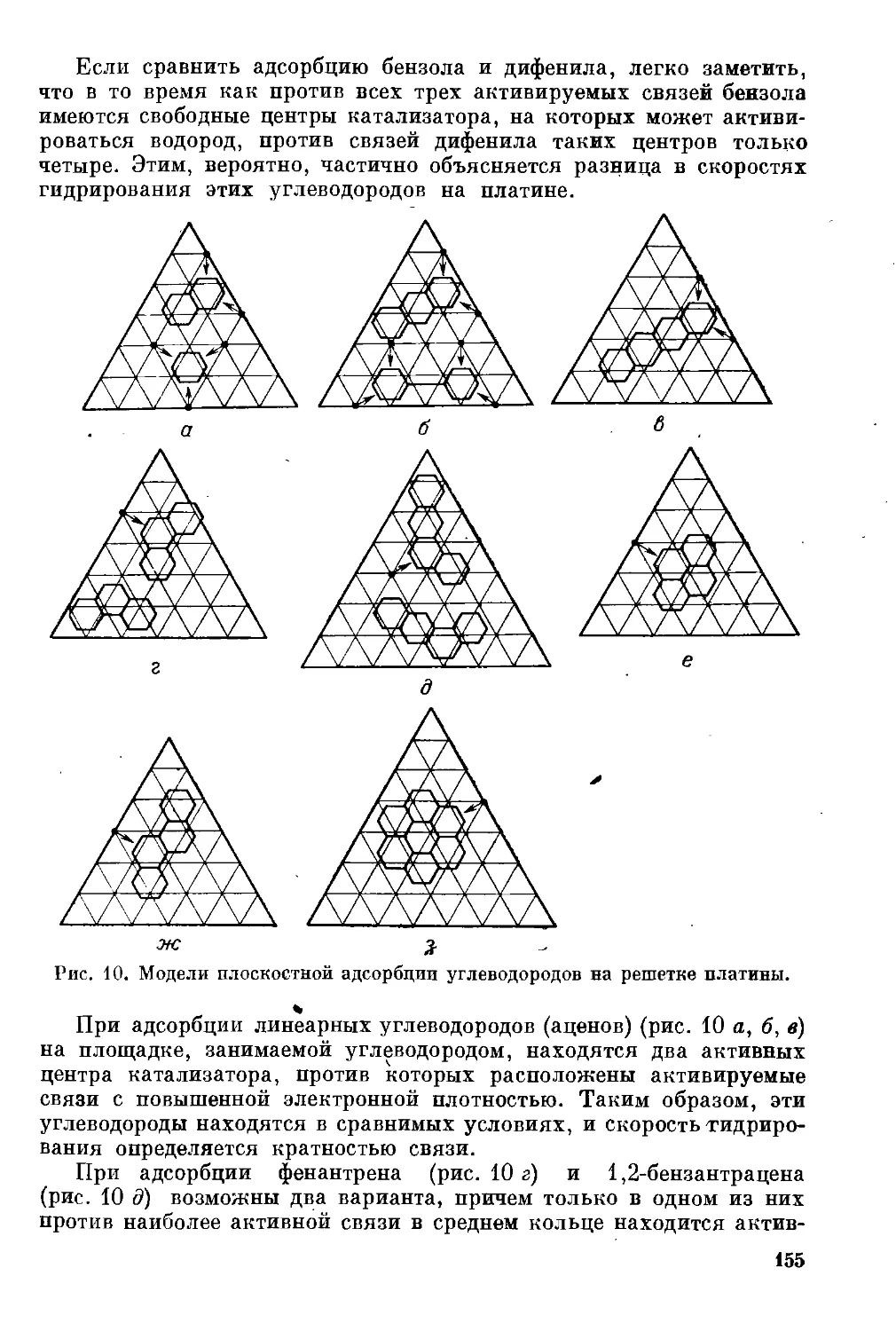














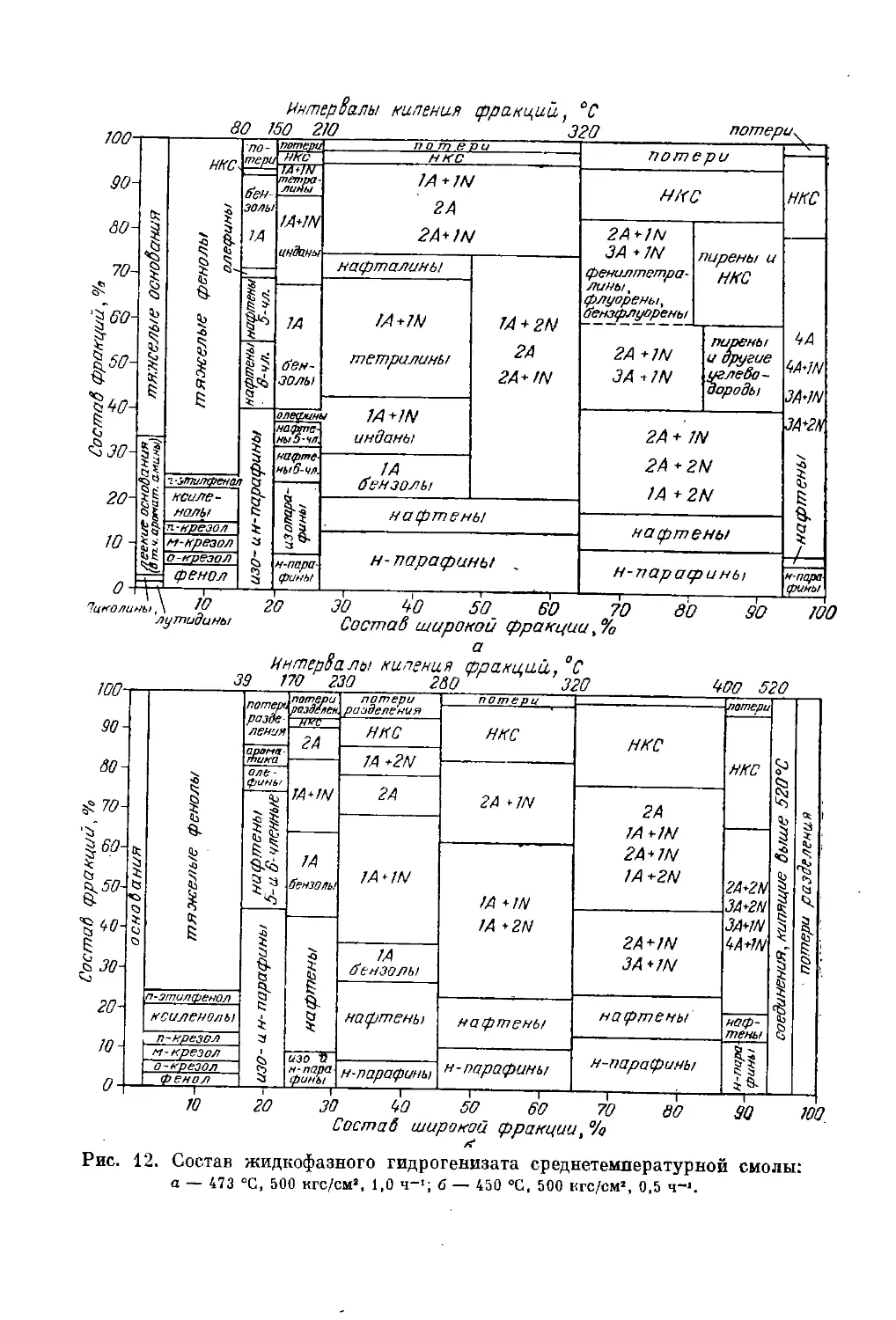





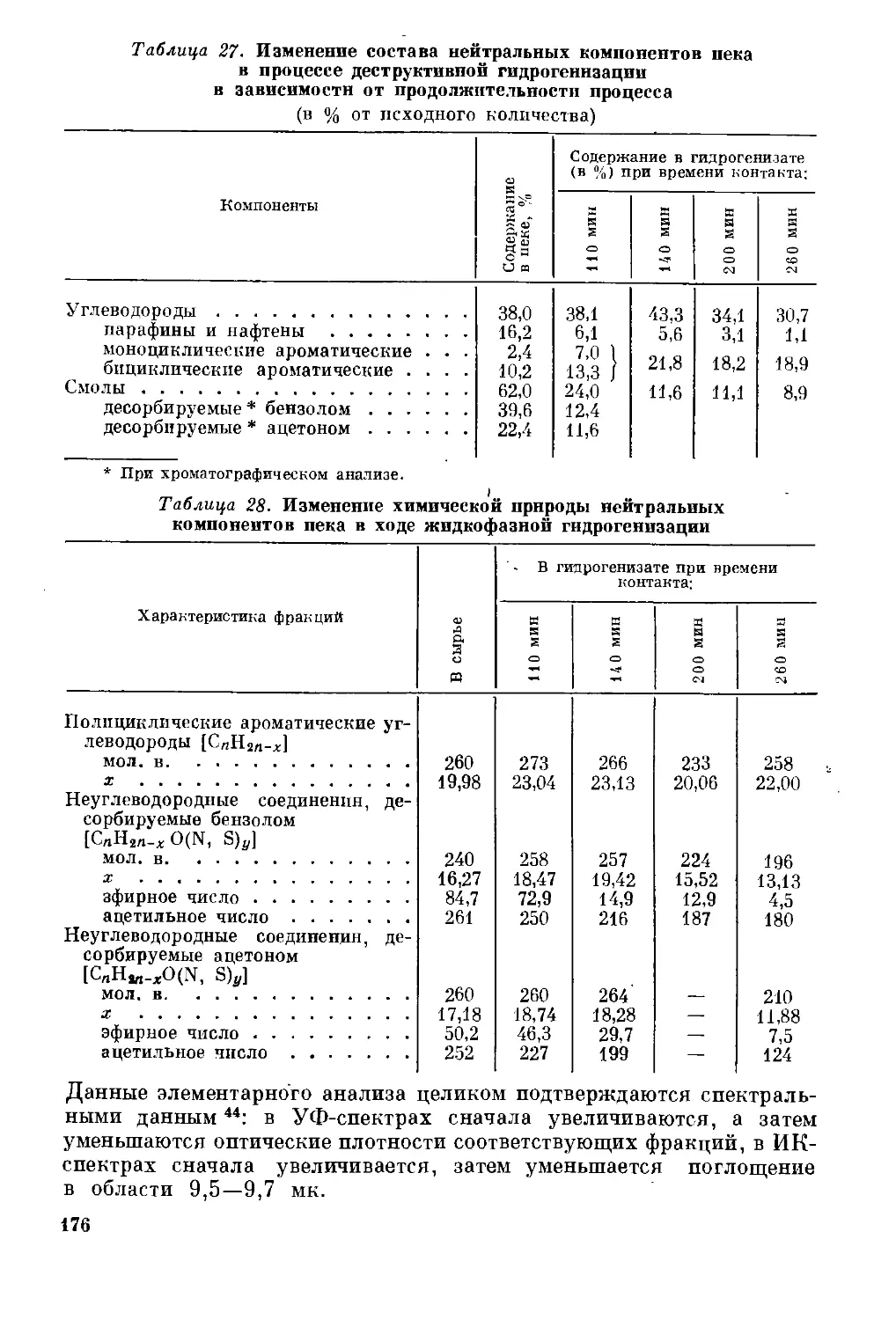

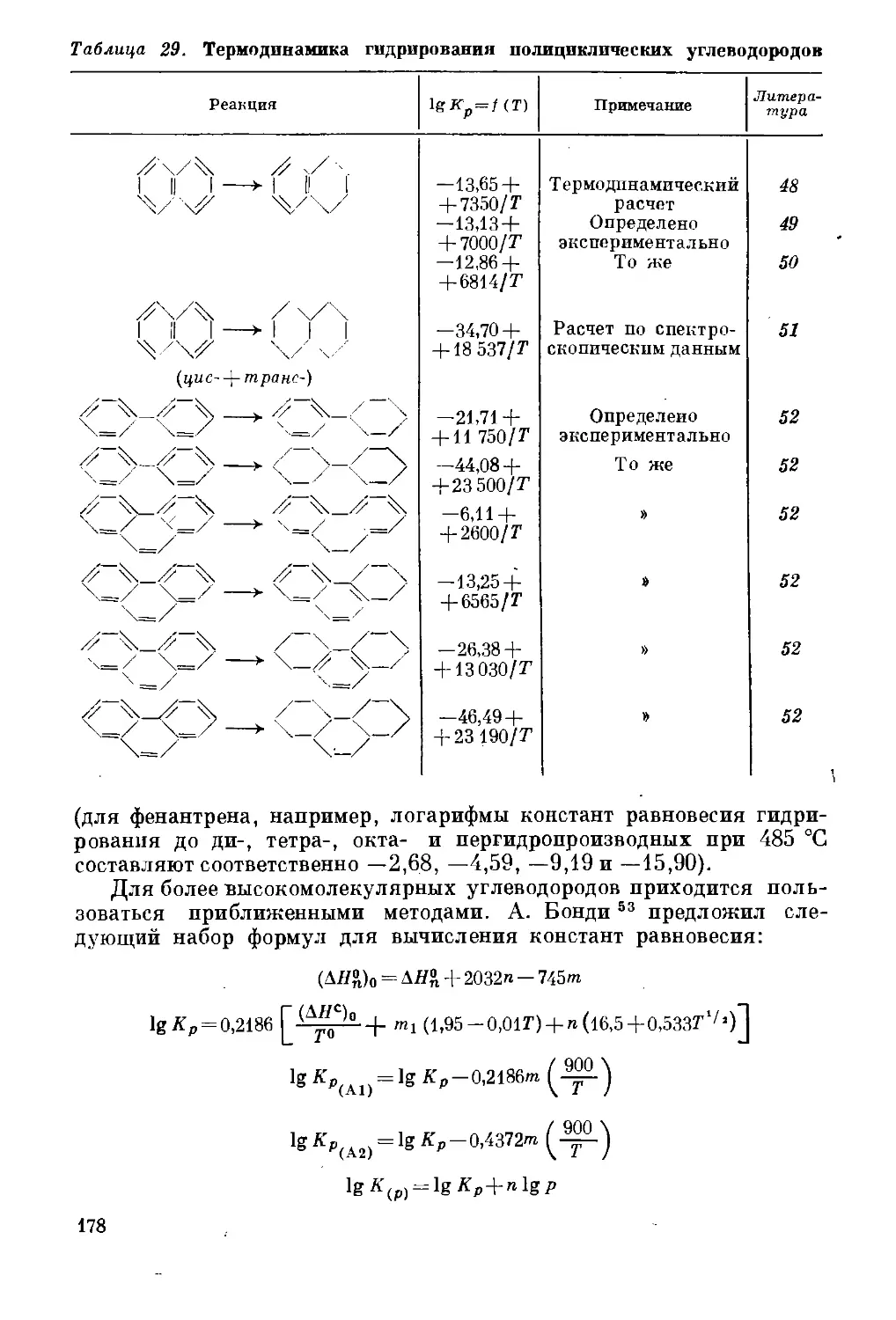












































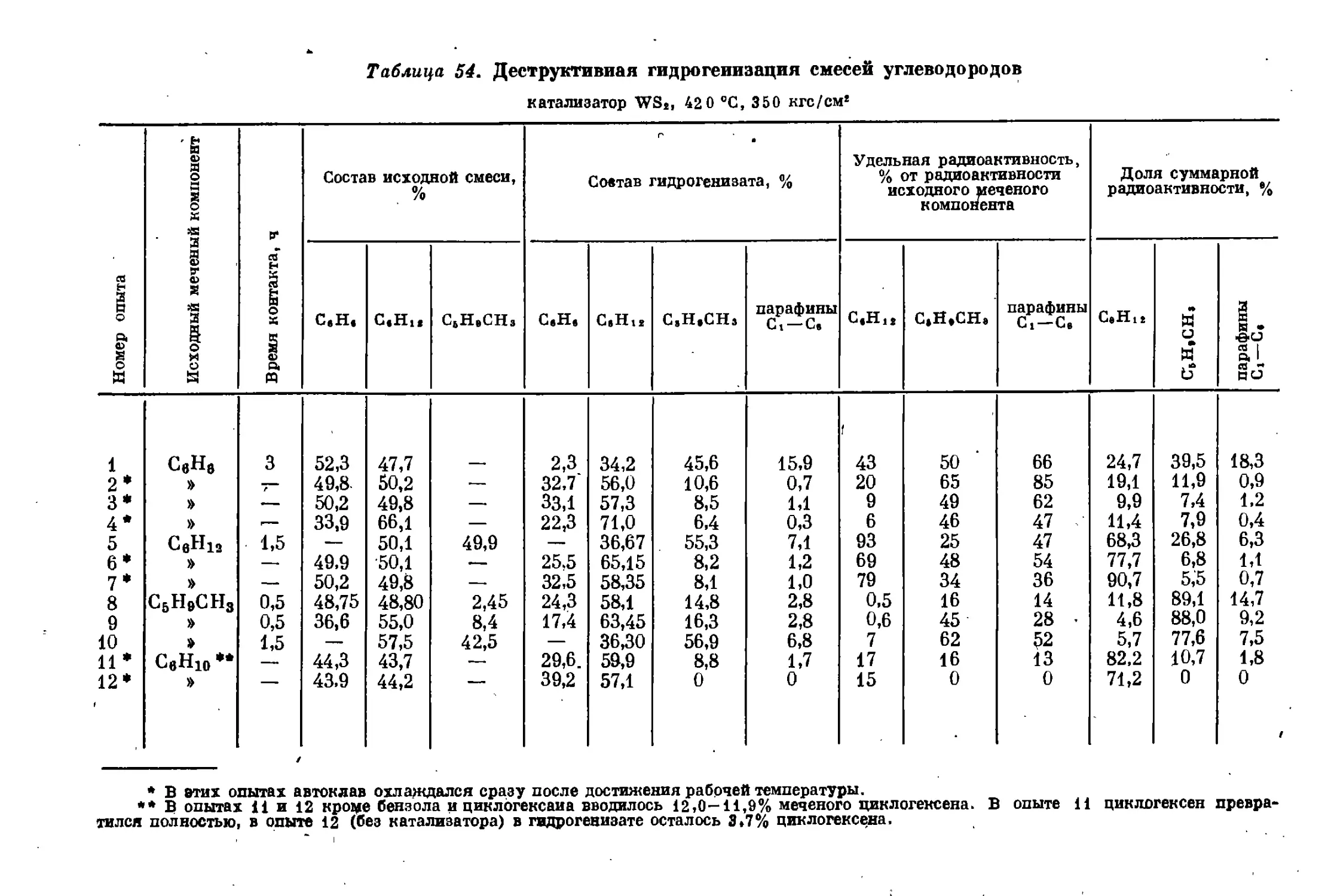



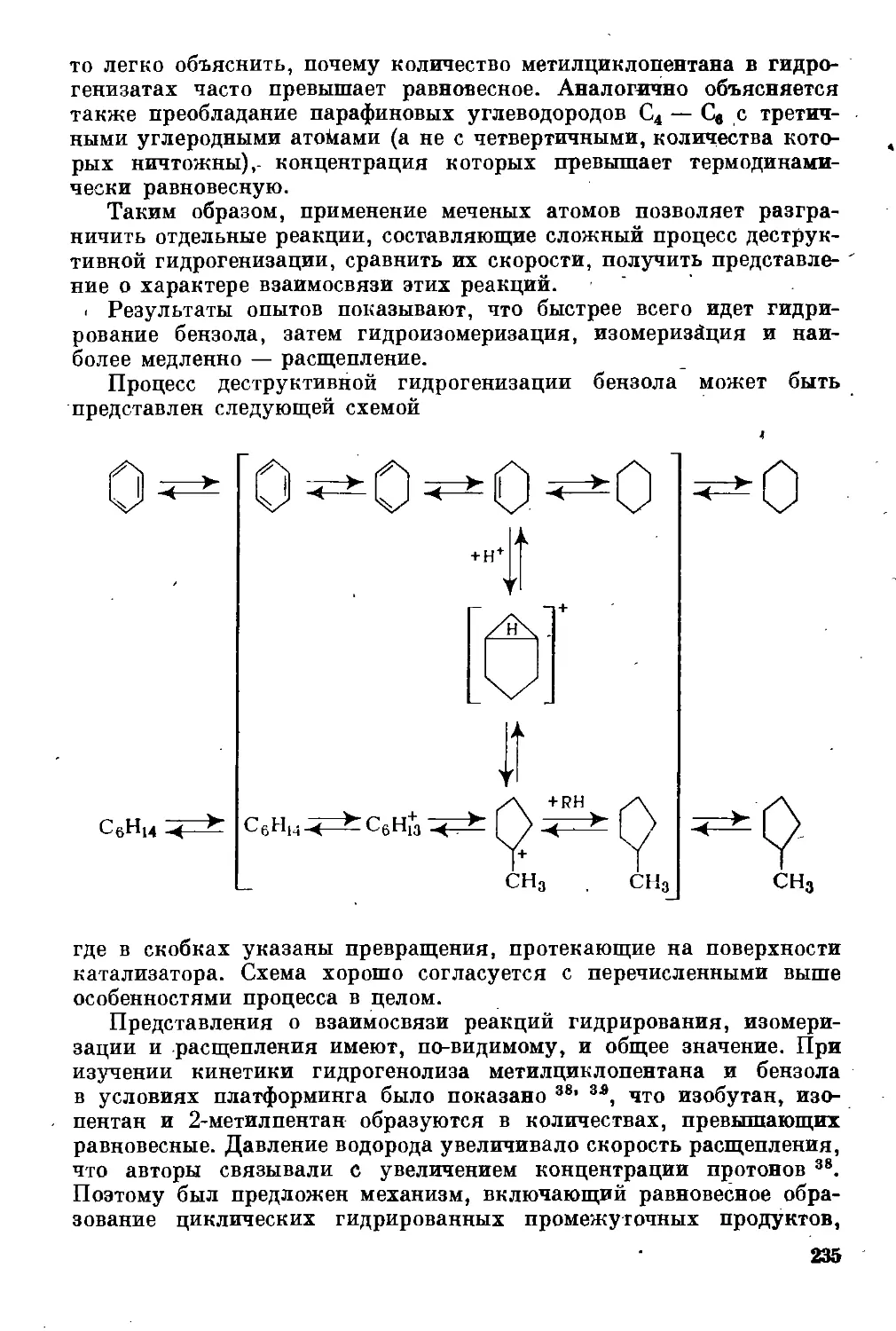














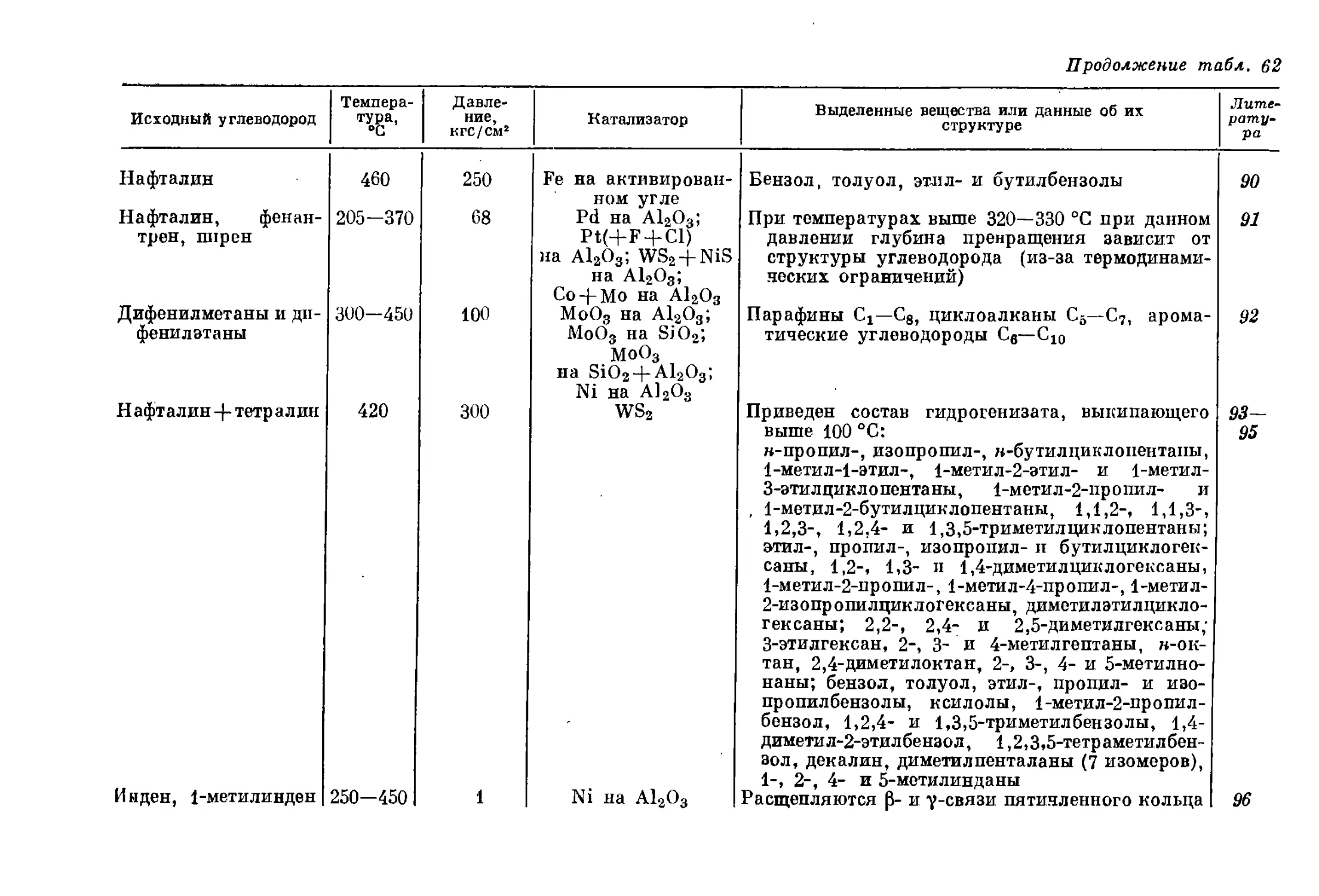



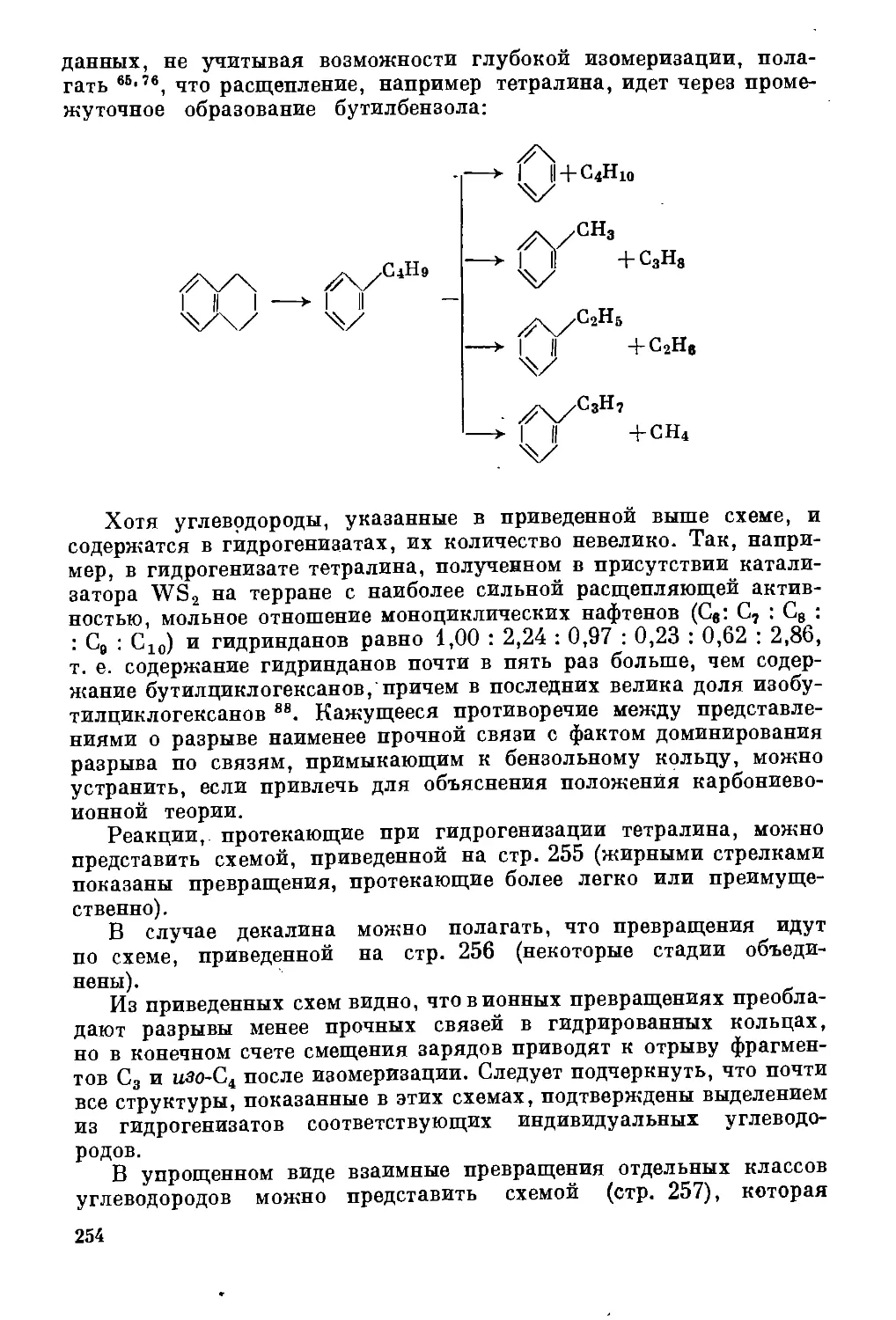










































































Текст
И. В. Калечиц
ХИМИЯ
гидрогенизационных
процессов
в переработке
топлив
МОСКВА
ИЗДАТЕЛЬСТВО «ХИМИЯ»
1973
УДК 662.75.092 : 54
К 17
К 17 Калечпц И. В.
Химия гидрогенизационных процессов в пере-
работке топлив. М., «Химия», 1973.
336 стр.; 79 табл.; 24 рпс.; список литературы 974
ссылки.
В книге впервые в литературе обобщены резуль-
таты многочисленных исследований химии п меха-
низма основных гидрогенизационных процессов, игра-
ющих важную роль в нефтепереработке и нефтехимии.
Даны основные закономерности гидрирования орга-
нических соединений, рассмотрены механизм, кинетика
п катализаторы процессов деструктивной гидроге-
низации, гидрокрекинга, гидроочистки и демети-
лирования.
Книга предназначена для широкого круга хи-
миков-органиков — научных работников, инжене-
ров нефтеперерабатывающей и нефтехимической про-
мышленности. Она представляет интерес для студен-
тов старших курсов химических и нефтяных вузов
и аспирантов, специализирующихся в химии гетеро-
генных процессов.
3147-150
050(01)-73
105-73
39
Редактор М. Н. Пастушенко
Технический редактор В. М. Скитина
Художник А. Я. Михайлов
Корректор Р. А. Шкиперова
Т-14909. Сдановнаб. 3/V 1973 г. Поди. впеч. 10/IX 1973 г. Формат бумаги 60 X ЭО'/ц.
Бумага тип. № 2. Усл. печ. л. 21. Уч.-изд. л. 22.5'2. Тираж 2200 экз.
Зак. К. 271. Изд. № 184.
Цена 2 р. 41 к.
Издательство «Химия». 107076, Москва, Стромынка, 23.
Типография № 6 «Союзполиграфпрома» при Государственном комитете Совета Министров
СССР по делам издательств, полиграфии и книжной торговли.
196006, г. Ленинград, Московский пр., 91.
© Издательство «Химия», 1973 г.
СОДЕРЖАНИЕ
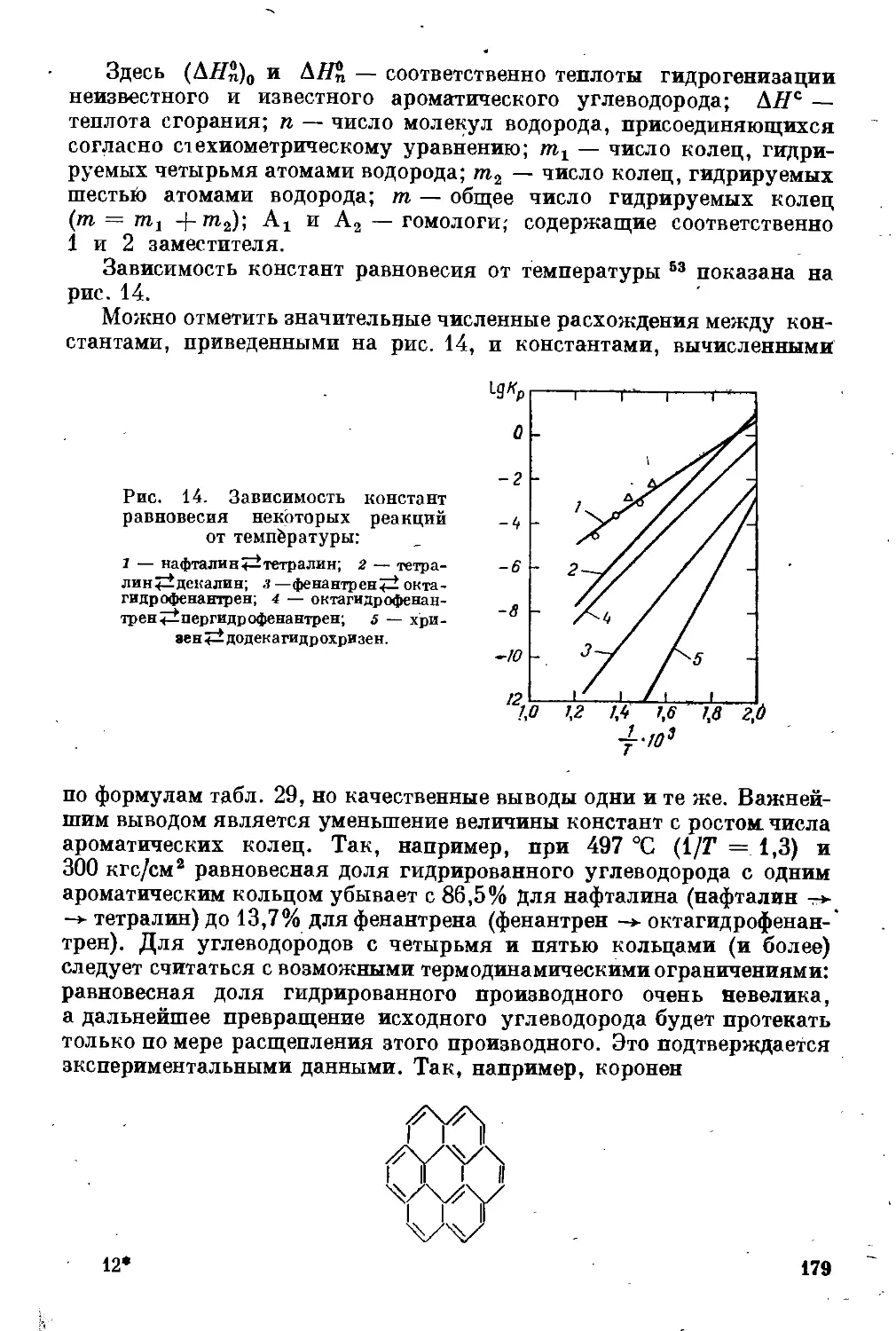
Введение ........................................................ 5
Глава 1. Современное состояние применения гидрогенизационных
процессов в топливной промышленности и тенденции их
дальнейшего развития ......................................... 7
Литература ..................................................... 91
Глава 2. Некоторые теоретические положения протекания ионных
и радикальных реакций ...................................... 111
Литература .............................. 128
Глава 3. Процессы низкотемпературной гидрогенизации ........... 130
Гидрирование бензола ....................................... 131
Гидрирование гомологов бензола ............................. 138
Гидрирование полициклических ароматических углеводородов . . 148
Гидрирование гетероциклических соединений ароматического ха-
рактера ................................................ 159
Литература ...... '....................... 161
Глава 4. Жидкофазная высокотемпературная гидрогенизация ... 163
Состав сырья и продуктов жидкофазной гидрогенизации. Общая
схема процесса ......................................... 163
Превращения нейтральных соединений ......................... 174
Превращения полициклических ароматических/ углеводородов . . 177
Превращения сложных эфиров ................................. 182
Превращения простых эфиров ................................. 185
Превращения спиртов и карбонилсодержащих соединений .... 192
Превращения фенолов и других кислых соединений......... 194
Превращения азотсодержащих соединений ...................... 208
Литература .................................................... 220
Глава 5. Парофазная гидрогенизация........................ 225
Гидрогенизация бензола .................................... 225
Гидрогенизация неароматических углеводородов ............... 236
Гидрогенизация гомологов бензола и циклогексана ............ 240
Гидрогенизация бициклических углеводородов ................ 246
Катализаторы парофазной гидрогенизации ..................... 261
Литература .................... . ................ 274
1* 3
Глава 6. Процессы гидроочистки ......................... 278
Сернистые соединения, входящие в состав нефтей, и их реакционная
способность ......................................... 278
Механизм превращений сернистых соединений в условиях гидро-
очистки ............................................. 283
Кинетика промышленных процессов гидроочистки. Селективность
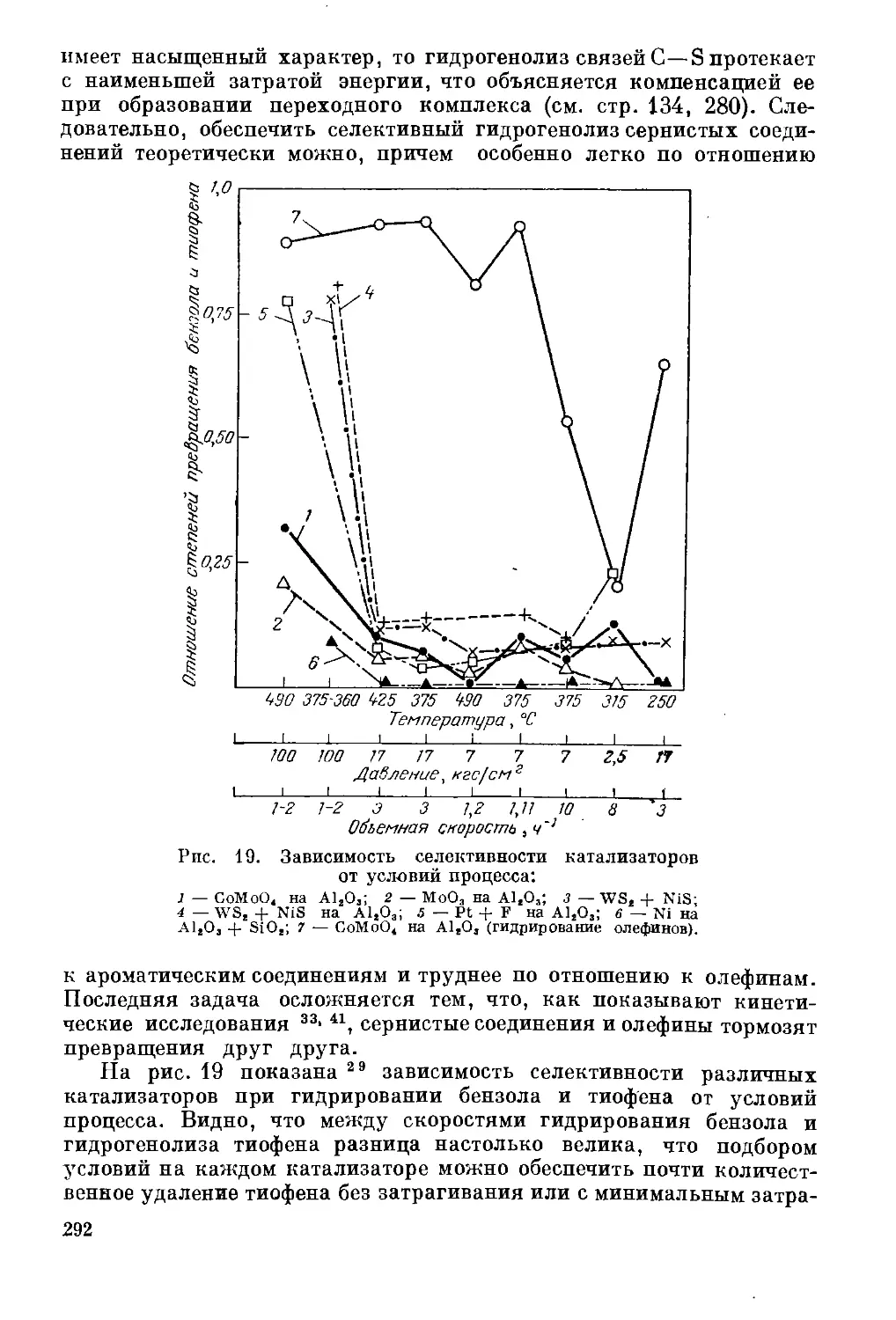
гидрогенолиза сернистых соединений .................. 291
Катализаторы гидроочистки , ....................... 299
Некоторые проблемы дальнейшего развития процессов гидро-
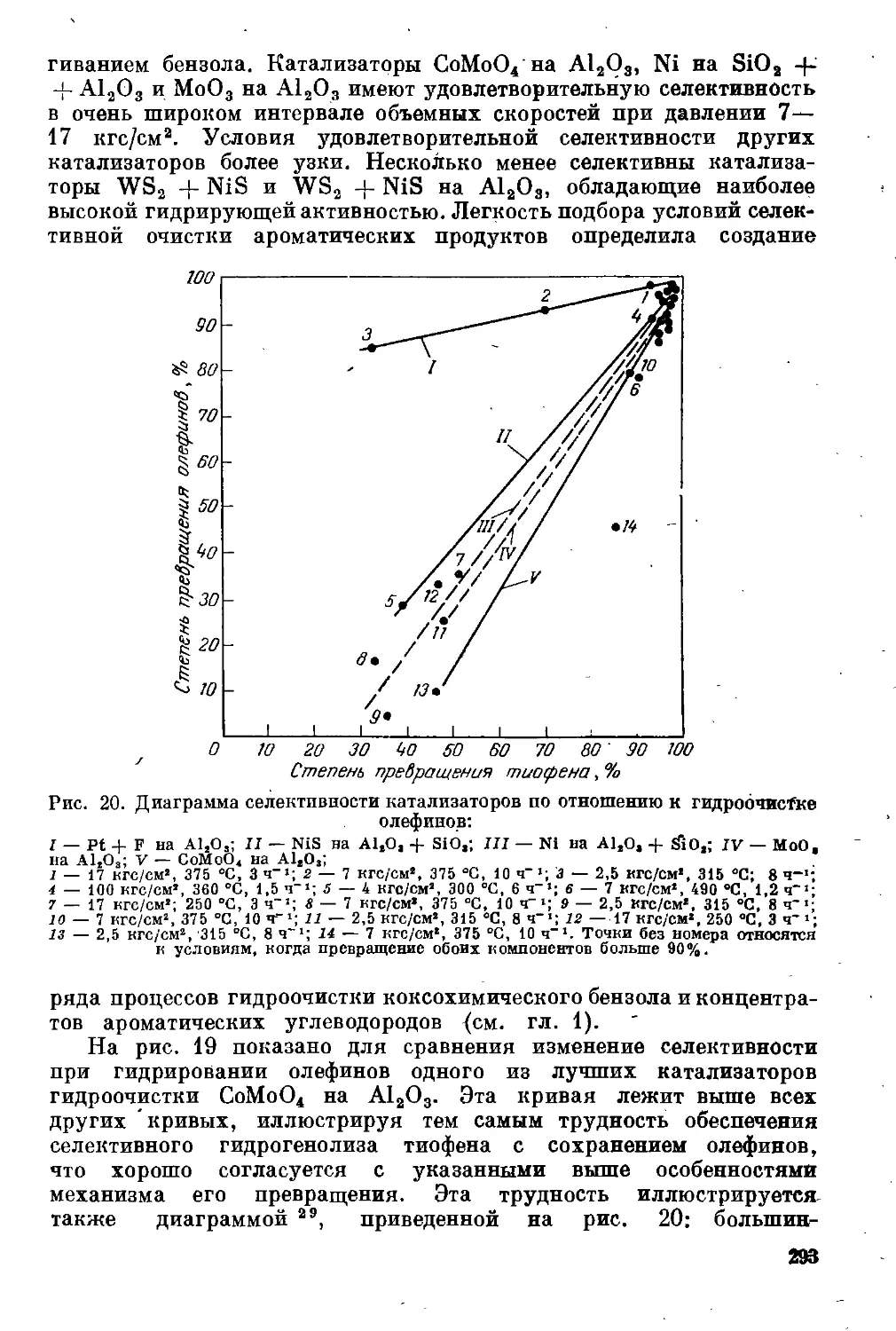
очистки ............................................ 301
Литература ............................. 303
Глава 7. Процессы гидрокрекинга ........................ 306
Литература ..............................................324
Глава 8. Процессы деметилирования ...................... 327
Литература ............................................. 334
Заключение ............................................ 335
ВВЕДЕНИЕ
Технологические процессы, в которых под давлением водорода
осуществляются химические преобразования органических молекул,
играют важнейшую роль в нефтеперерабатывающей и нефтехимиче-
ской промышленности и по масштабам применения занимают первое
место в мире среди каталитических химических процессов.
В самом деле, уже сейчас в мире ежегодно добывается и перера-
батывается более 2 млрд, т нефти и получаются сотни миллионов
тонн угольных и сланцевых смол. Их^чистка от сернистых, азоти-
стых, металлосодержащих соединений и других примесей, превра-
щение в высококачественные моторные, реактивные и котельные
топлива, а также полупродукты для химической переработки невоз-
можны без процессов гидрогенизации. Процессы гидроочистки, гид-
рокрекинга, гидрирования и другие процессы, осуществляемые под
давлением водорода, в настоящее время определяют технический
уровень нефтеперерабатывающей и нефтехимической промышлен-
ности. Уже строятся и проектируются заводы, в которых вся сырая
нефть или все ее погоны так или иначе облагораживаются при по-
мощи процессов гидрогенизации. С развитием методов гидродесуль-
фуризации тяжелых нефтяных продуктов — вакуумных дистилля-
тов, деасфальтизатов и мазутов — уже в ближайшее десятилетие
суммарная мощность гидрогенизационных процессов и процессов
риформинга и изомеризации, также осуществляемых под давлением
водорода, приблизится к миллиарду тонн в год.
Гидрогенизационные процессы непрерывно видоизменяются и со-
вершенствуются; к ним предъявляются все более и более жесткие
требования.
Исследования механизма и кинетики гидрогенизации ведутся
широким фронтом как в СССР, так и за рубежом наряду с разработ-
кой новых технологических процессов. Однако, если работы по тех-
нологии гидрогенизационных процессов обобщаются в обзорных
статьях и монографиях достаточно регулярно и широкий круг чи-
тателей хорошо информирован о достижениях в этой области, много-
численные работы по механизму, кинетике и катализу гидрогениза-
ционных -процессов практически не обобщаются, им обычно посвя-
щаются лишь короткие главы в обзорных монографиях.
Настоящая монография является попыткой восполнить этот
пробел и свести воедино результаты изучения химии и механизма
важнейших процессов гидрогенизации.
5
Чтобы не отрывать основное содержание монографии от тенденций
развития и разработки новых технологических процессов, в первой
главе в конспективной форме даются аннотированные результаты
таких исследований и формулируются основные направления тех-
нического прогресса в этой области. Во второй главе кратко изла-
гаются основные фундаментальные положения механизма радикаль-
ных и ионных реакций, а также теории катализа, необходимые для
интерпретации материала последующих глав.
При рубрикации основного материала монографии — результа-
тов изучения превращений различных углеводородов и их функцио-
нальных производных в условиях гидрогенизационных процессов —
встретились естественные трудности. Действительно, материал
можно было располагать по группам процессов, по классам веществ,
по виду катализаторов, по типу превращений (ионные и радикаль-
ные). Каждый вид рубрикации имел свои преимущества и недостатки.
Автор избрал рубрикацию по группам гидрогенизационных про-
цессов, выделив жидкофазные и парофазные процессы гидрогениза-
ции, а также специфические процессы низкотемпературной гидро-
генизации, гидроочистки, гидрокрекинга и деметилирования. Это
позволило подчеркнуть и охарактеризовать особенности каждой
группы процессов, но, естественно, затруднило сопоставление осо-
бенностей превращений отдельных классов углеводородов и их про-
изводных, а также особенностей ускорения реакций различными ката-
лизаторами. В необходимых случаях такие сопоставления сделаны,
хотя это и нарушает принцип рубрикации, в других случаях чита-
тель найдет ссылки на предыдущие или последующие разделы, в ко-
торых изложен аналогичный материал, но для условий другого
процесса.
Автор выражает надежду, что его труд окажется полезным для
научных работников, инженеров, техников и рабочих, которым пред-
стоит в ближайшие годы управлять процессами гидрогенизации
или создавать новые, еще более эффективные их модификации.
Трудность рубрикации материала, его разрозненность по много-
численным, часто трудно доступным публикациям и, наконец, то,
что данная монография является первой попыткой обобщения такого
материала за последние десятилетия, могут привести к отдельным
недостаткам и неточностям. Автор будет благодарен всем читателям,
которые их укажут.
И. Калечиц.
ГЛАВА 1
СОВРЕМЕННОЕ СОСТОЯНИЕ ПРИМЕНЕНИЯ
ГИДРОГЕНИЗАЦИОННЫХ ПРОЦЕССОВ
В ТОПЛИВНОЙ ПРОМЫШЛЕННОСТИ
И ТЕНДЕНЦИИ ИХ ДАЛЬНЕЙШЕГО РАЗВИТИЯ
Гидрогенизационные процессы в производстве топлив утверди-
лись не сразу, они прошли большой и сложный путь, их развитие
иногда даже было противоречиво.
Этот путь можно схематично разбить на несколько периодов:
1) до середины 30-х годов — период поисковых работ;
2) середина 30-х годов — конец второй мировой войны — пе-
риод промышленного оформления процессов деструктивной гидро-
генизации главным образом смол различного происхождения и углей
и период их наибольшего развития;
3) послевоенный период — период отказа от старой технологии
гидрогенизации углей и смол вследствие невозможности конкури-
ровать с производством топлив из нефти и одновременно период
все большего и большего внедрения гидрогенизационных процессов
в нефтепереработку на основе новых, более эффективных техноло-
гических схем и коренных усовершенствований катализаторов.
В настоящее время процессы гидрогенизации прочно и широко
вошли в нефтеперерабатывающую промышленность, в переработку
с их помощью вовлекаются все более тяжелые погоны нефти. Это
является общей тенденцией, и можно говорить о начале в 70-х годах
четвертого периода, в котором все виды топлив, включая котельные,
а также почти все масла будут облагораживаться при помощи этих
процессов. Все это относится в первую очередь к переработке нефти,
однако остается актуальной и возможность получения моторных то-
плив и масел из ненефтяного сырья, т. е. гидрогенизация может,
но уже на новой технической основе, вернуться к своим истокам.
Исторически сложилось так, что гидрогенизация топлив возникла
и развивалась как метод получения искусственного жидкого топлива
главным образом из ненефтяного сырья — сланцевых и угольных
смол, а также-каменных углей. Это объясняется тем, что в предвоен-
ный период нефти нехватало, а эксплуатируемые сейчас крупней-
шие нефтеносные районы (Ближний Восток, Северная Африка, По-
волжье, Западная Сибирь и др.) еще не были открыты.
Таким образом, как это ни парадоксально, гидрогенизационные
процессы начали развиваться с использованием самого неблаго-
приятного сырья.
В самом деле, очевидно, что для получения бензина — смеси
жидких углеводородов, выкипающих до 150—180 °C и не содер-
жащих неуглеводородных примесей, — самым подходящим сырьем
7
были бы более высококипящие фракции нефти. Задача состояла бы
только в понижении молекулярного веса и устранении неуглеводо-
родных смолистых примесей. Доля последних невелика, и, хотя она
растет с повышением температур кипения, всегда меньше, чем
в сланцевых и угольных полукоксовых. Следовательно, последние
являются менее подходящим сырьем; соответственно, еще более
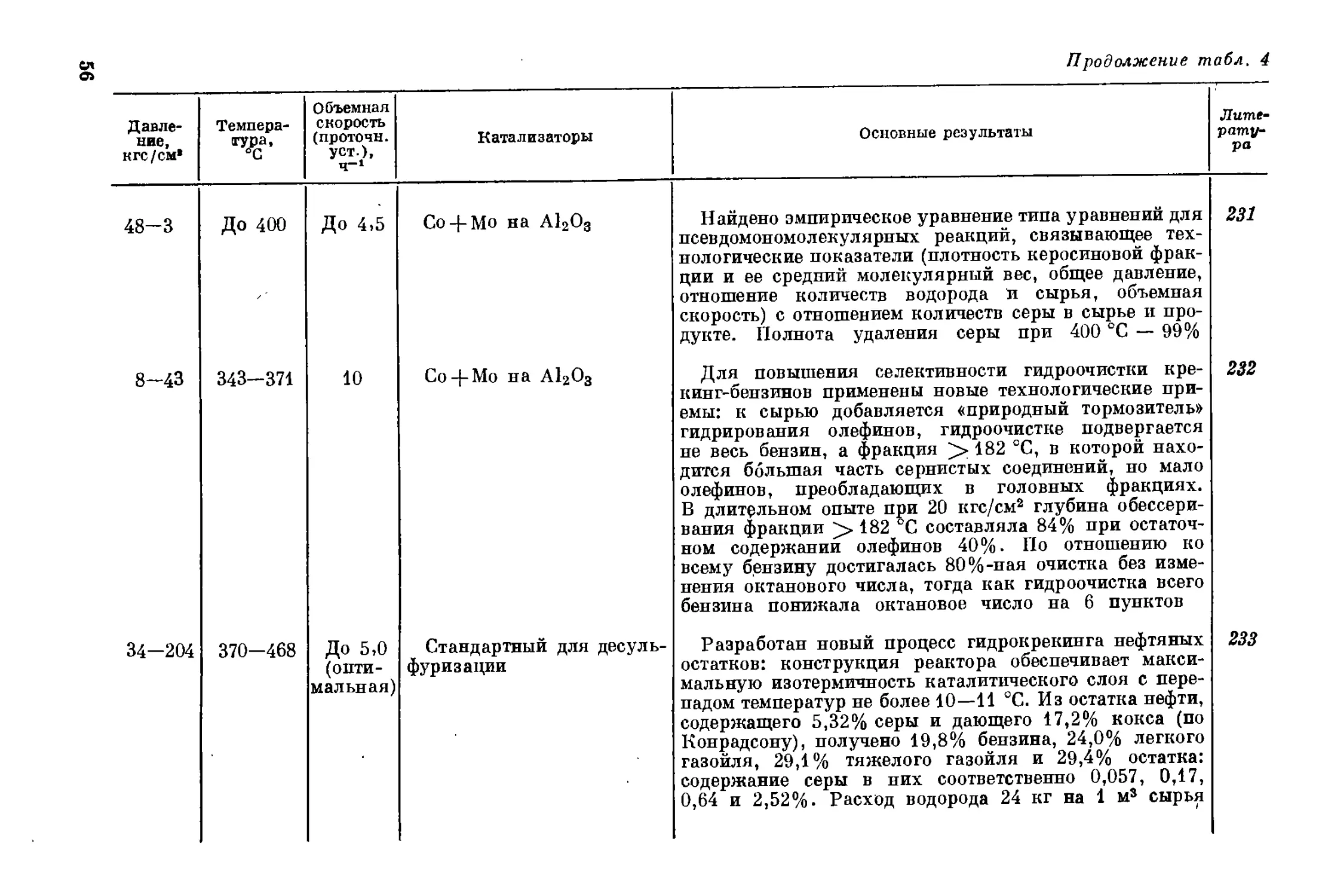
трудным сырьем будет уголь, содержащий не только большие коли-
чества кислорода, серы и азота, но и минеральные включения. Позд-
нее было установлено х, что важным показателем относительной
трудности гидрирования сырья является отношение в нем водорода
и углерода — чем меньше это отношение, тем более ароматизировано
сырье и тем труднее оно перерабатывается гидрогенизационными ме-
тодами. Это отношение составляет1- 2 в бензине 1,95—2,12, в ва-
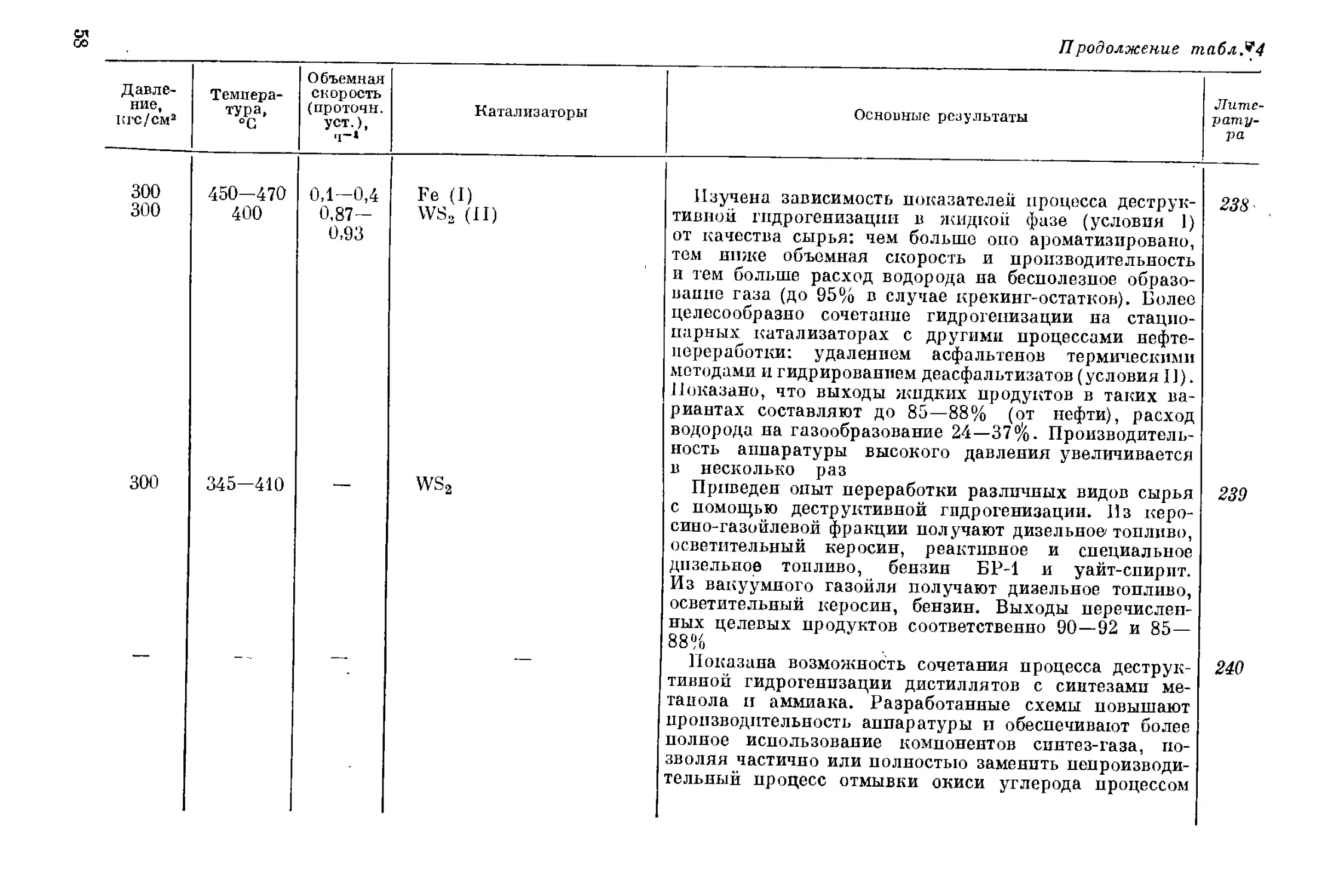
куумном газойле кувейтской нефти 1,89, в остатке вакуумной пере-
гонки той же нефти 1,49, в сланцевой смоле 1,62, в обогащенном угле
0,78-0,8.
Однако ограниченность запасов нефти и полное отсутствие ее
на огромных территориях делало задачу получения моторндгх топлив
из каменных углей весьма заманчивой. Мировые запасы нефти
в 1925 г. оценивались только в 795 млн. т и высказывались серьез-
ные опасения о возможности исчерпания их через несколько десяти-
летий.
В период поисковых работ (до середины тридцатых годов) были
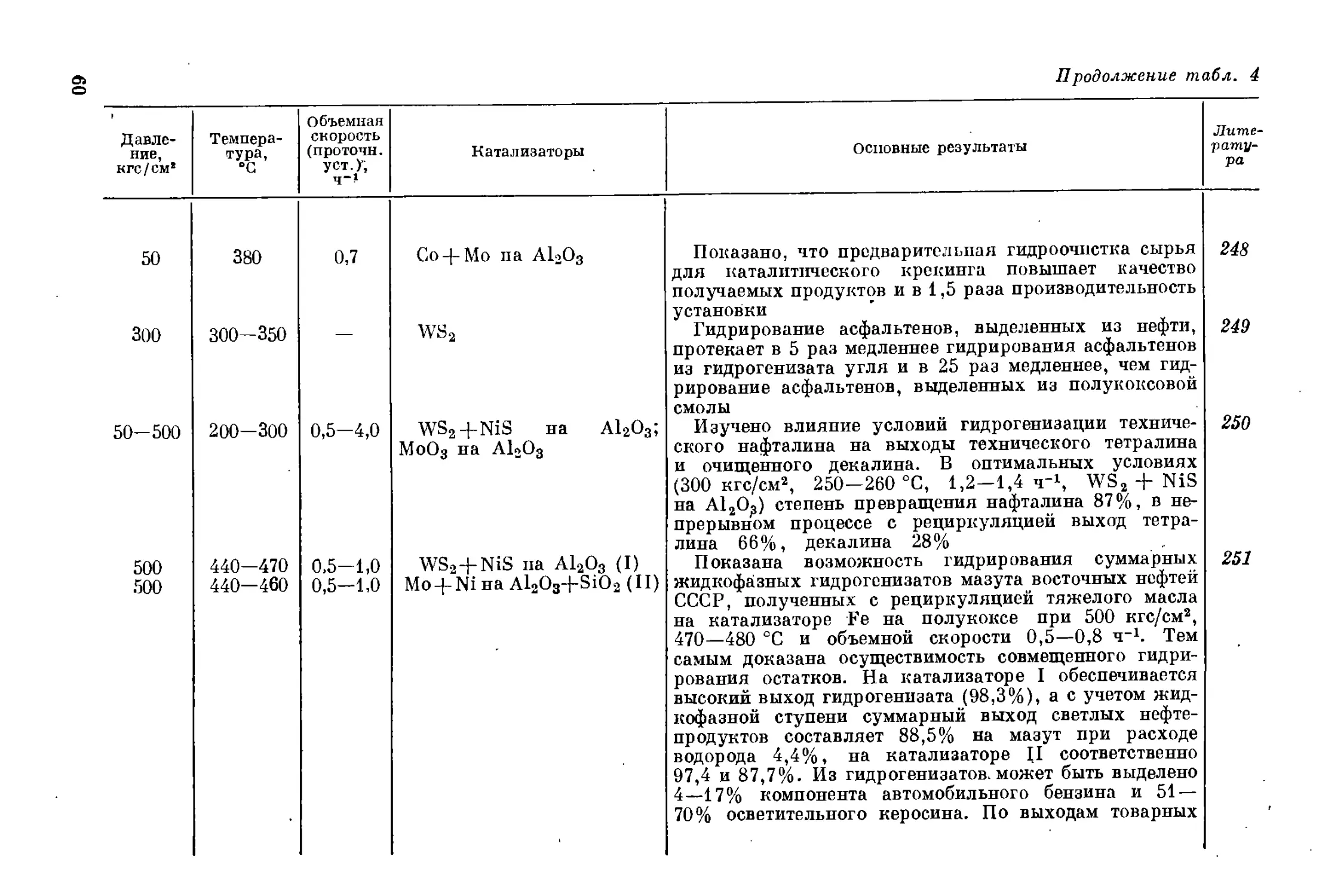
проведены многочисленные опыты по гидрогенизации различных ви-
дов сырья и индивидуальных веществ, моделирующих те или иные
группы компонентов сырья, что подготовило переход к промышлен-
ным испытаниям: были выяснены примерные требования к сырью,
подобраны стабильные катализаторы, определены целесообразные
условия и ступени процессов. Работы этого периода широко известны
и неоднократно обобщались, в том числе в очень подробных
монографиях А. В. Лозового и М. К. Дьяковой 3, А. Д. Петрова 4,
И. Б. Рапопорта J. Следует отметить, что существенный вклад в раз-
витие процессов деструктивной гидрогенизации топлив был сделан
русскими, а позднее советскими учеными 3- 5.
Во втором периоде были созданы промышленные процессы полу-
чения искусственного жидкого топлива путем гидрогенизации углей
и смол J- 6- ’. Основной процесс превращения углей или тяжелых
смол (главным образом в бензин) гидрированием под высоким давле-
нием (200—700 кгс/см2) складывается, как известно, из трех ступе-
ней: жидкофазного гидрирования, парофазного рафинирования (фор-
гидрирование) и расщепления (бензинирование). В жидкофазной сту-
пени применялись давление 700 кгс/см2 и дешевый железный ката-
лизатор, выводимый из цикла вместе с золой и продуктами уплот-
нения. Применением столь высокого давления компенсировалась
низкая активность катализатора, а благодаря его дешевизне реша-
лась проблема отделения минеральной части. На этой стадии за
счет циркуляции тяжелых фракций гидрогенизата сырье превраща-
лось в жидкий продукт — широкую фракцию с температурой конца
8
кипения 325 °C, не содержавшую твердых компонентов или соедине-
ний, склонных к конденсации. Однако в ней присутствовало значи-
тельное количество различных соединений, которые, особенно азот-
содержащие, представляли опасность для катализаторов расщепле-
ния. Превращение этих соединений в углеводороды и, следовательно,
подготовка к расщеплению составляла задачу следующей ступени—
парофазного рафинирования, в которой применяли высокоактивный
стационарный катализатор. Затем рафинированная широкая фрак-
ция превращалась в бензин на стационарном катализаторе расще-
пления.
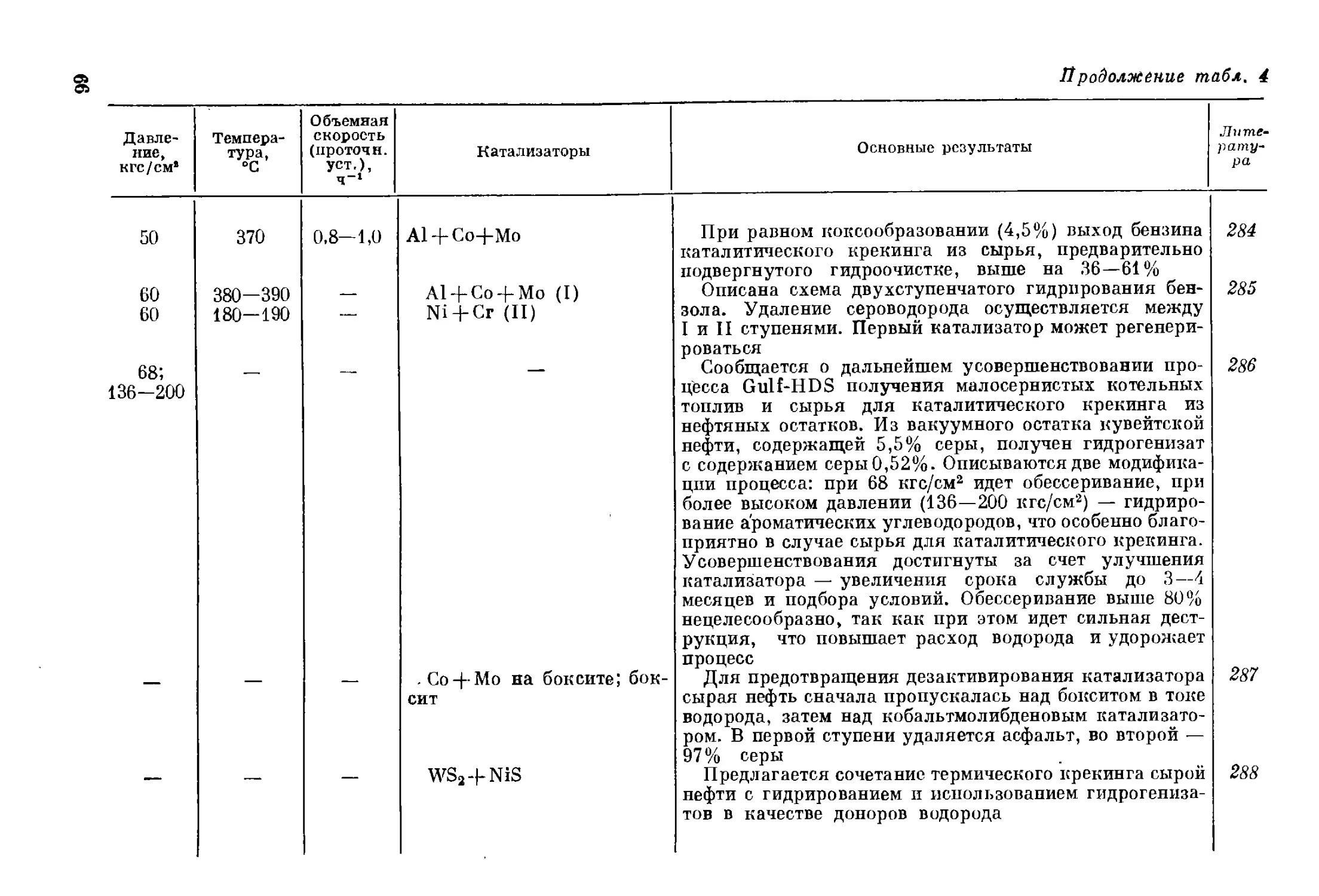
Разделение процесса на три ступени позволило предотвратить
отравление активных, но дорогих катализаторов и уменьшило об-
разование побочных газообразных продуктов, а следовательно,
и расход водорода. Такая многоступенчатая схема давала возмож-
ность перерабатывать практически любое сырье, но большое число
ступеней крайне осложняло и удорожало процесс. Более того, для
получения высококачественных бензинов требовалось введение еще
четвертой ступени — гидроформинга продукта бензинирования —
для превращения нафтеновых углеводородов в ароматические.
Только в случае особо высококачественного сырья — подукок-
совых смол из бурых углей с очень малым содержанием золы и
влаги — осуществлялась одноступенчатая гидрогенизация сразу на ста-
ционарных катализаторах (так называемые процессы ТТН и МТН4- *).
Всего было построено и работало 15 гидрогенизационных заводов*- *• ®,
а их общая мощность оценивалась приблизительно в 6 млн. т сырья
в год Ч
В послевоенные годы было открыто много новых месторождений
нефти, запасы которой уже в пятидесятых годах превосходили при-
мерно в 40 раз уровень мировой добычи, хотя он и возрос в несколько
раз по сравнению с довоенным 10.
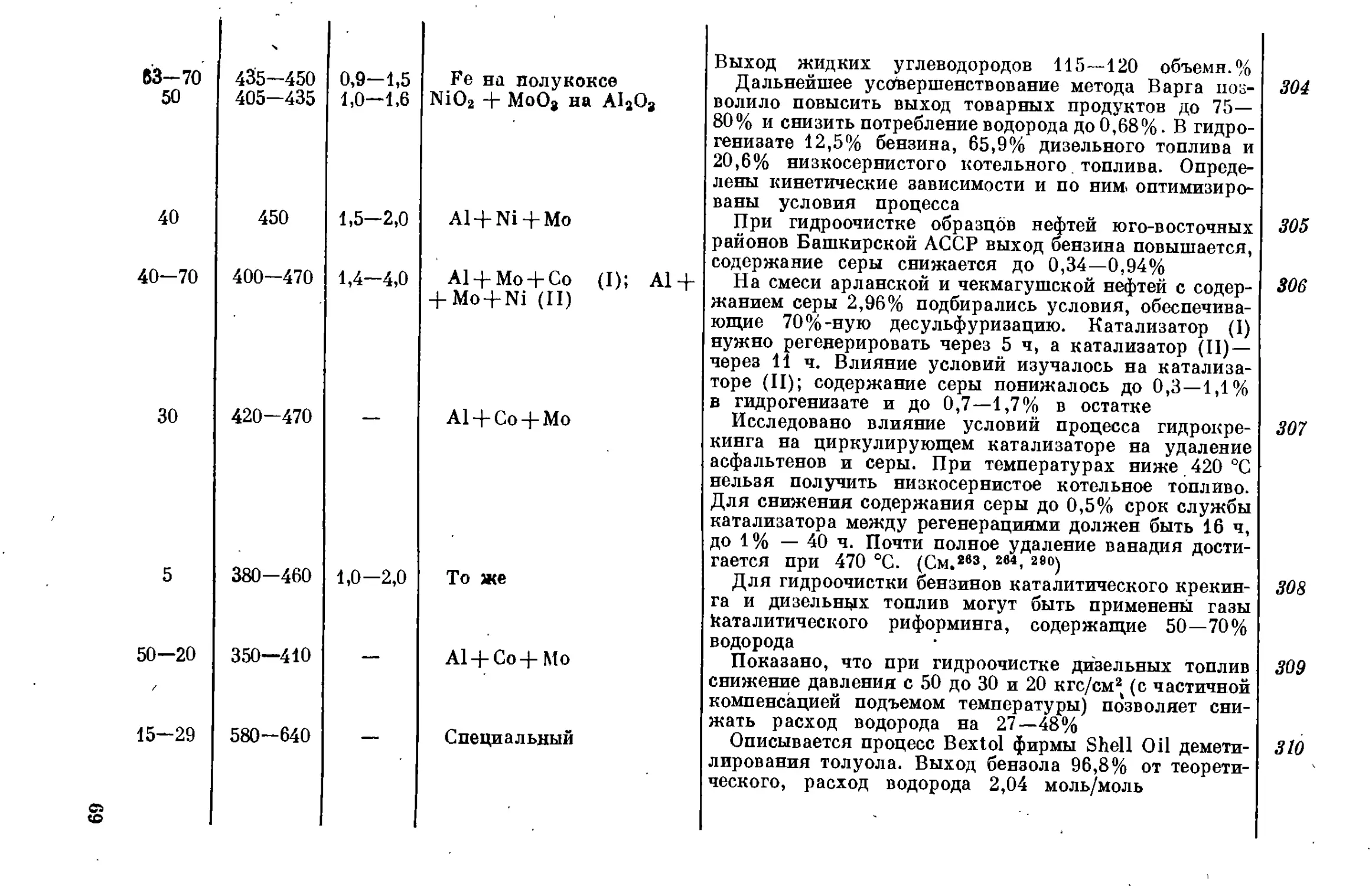
При непрерывном росте запасов и добычи нефти проблема полу-
чения искусственного жидкого топлива потеряла свою остроту,
а дорогостоящий бензин, получаемый гидрогенизацией, не мог кон-
курировать с нефтяным бензином. Стоимость бензина, получаемого
гидрированием угля, составляет 270—316% стоимости бензина из
нефти, а стоимость бензина из сланцевой смолы (с включением стадий
гидрогенизации) — 110—141% стоимости бензина из нефти и. 12.
Эти обстоятельства в значительной степени уменьшили интерес
к гидрогенизации угля, и исследования в области гидрогенизации
были направлены на переработку нефтяных и сланцевых про-
дуктов.
В послевоенный период методы гидрогенизации начинают про-
никать в "переработку нефти. Этому способствовал ряд факторов.
Так, прогресс двигателестроения требовал моторных топлив и масел
все более высокого качества. Первостепенное значение приобрела
необходимость снижения содержания или даже полного удаления
сернистых соединений из бензинов, реактивных и дизельных топлив,
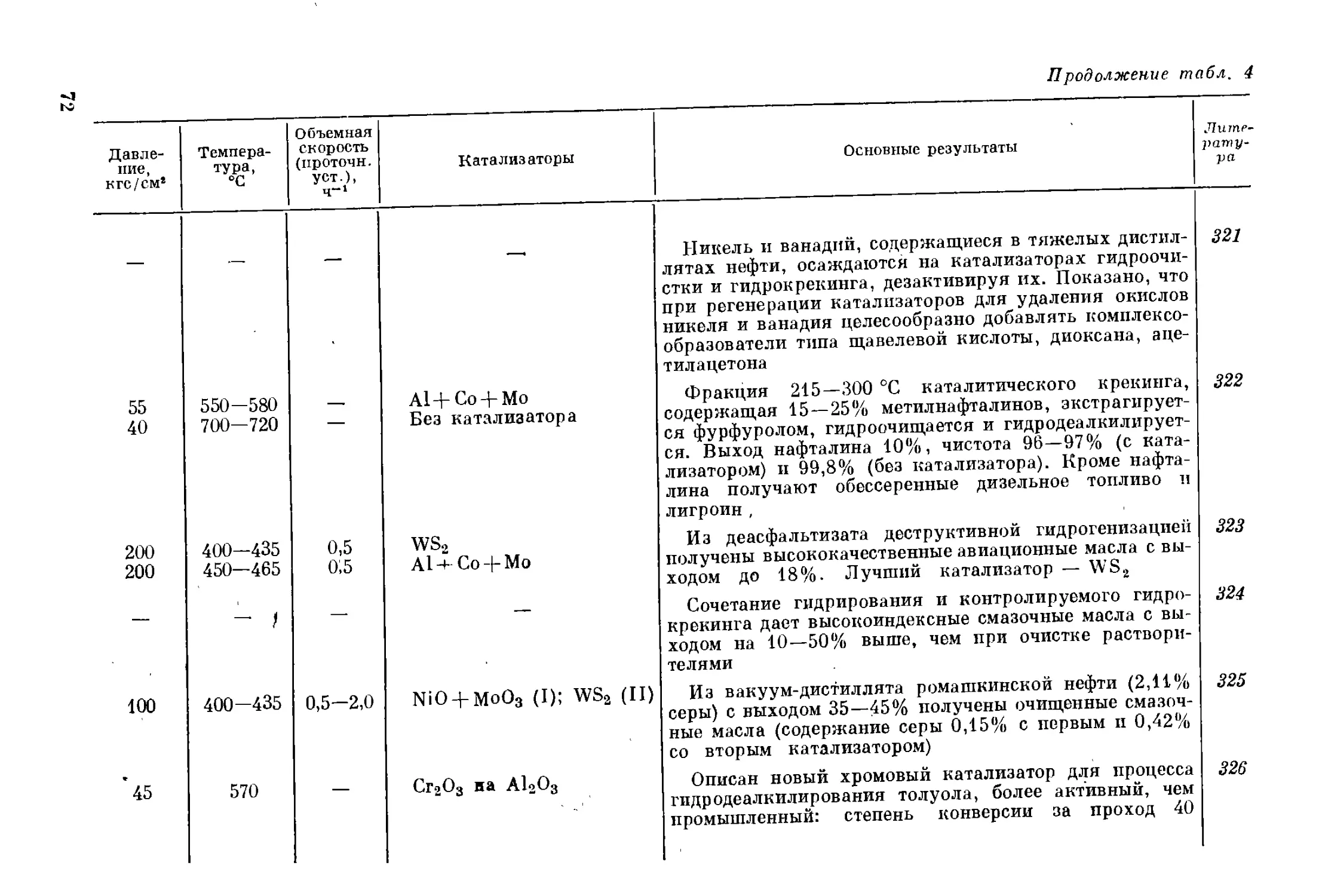
масел.
9
Известно, что сернистые соединения в моторных топливах вызы-
вают повышенный расход топлива, быстрый износ моторов и, как
следствие, более частые ремонты. Так, по данным13, повышение
содержания серы в бензине с 0,033 до 0,15% снижает мощность мотора
на 10,5% и увеличивает удельный расход топлива на 12, 2%. При
этом число капитальных ремонтов двигателей увеличивается в 2,05,
а средних — в 2,13 раза, вызывая необходимость увеличивать на
1,7% парк грузовых автомобилей для компенсации простоев
при ремонте двигателей. На 1000 т израсходованного бензина зто
приносит такие убытки: от перерасхода топлива — 1320 руб., от
повышения числа ремонтов и расхода запасных частей — 4678 руб.,
от необходимости производства дополнительных автомобилей —
8990 руб. (всего — 14 988 руб.). Аналогичные убытки приносит ис-
пользование сернистого дизельного топлива: дополнительные экс-
плуатационные затраты на 1000 т топлива составляют 4540 руб.,
а дополнительные капитальные затраты — 7610 руб. 13.
Однако прирост запасов нефти и рост ее добычи все время шли
преимущественно за счет нефтей с высоким содержанием серы. Нефти
наиболее важных нефтедобывающих районов — Венесуэлы, Ближ-
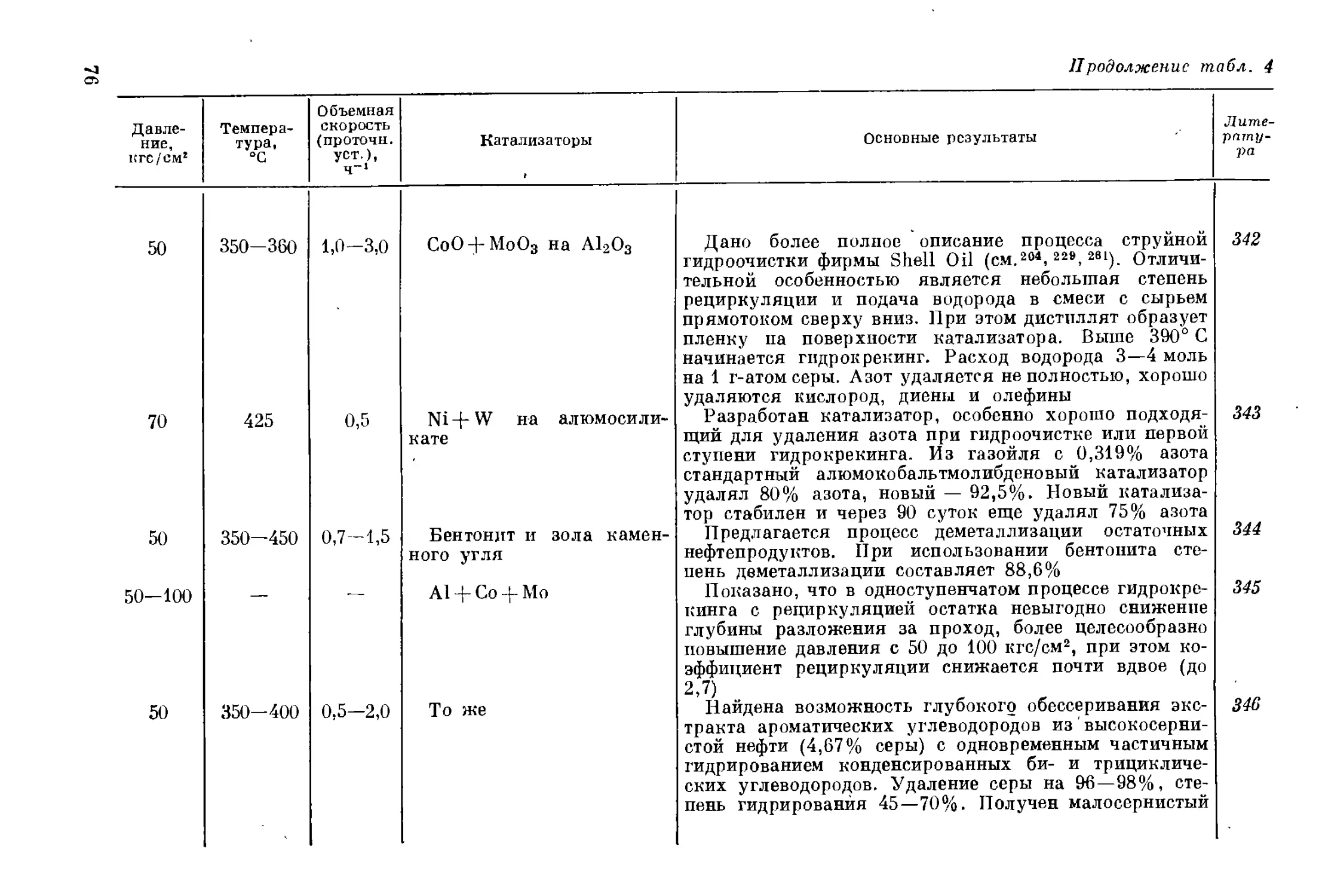
него Востока, ряда районов США и др. — относятся к сернистым
и высокосернистым типам нефтей. Если в 1929 г. сернистые (содержа-
ние серы 0,5—1,9%) и высокосернистые (содержание серы 1,9%)
нефти составляли лишь 25% мировой добычи, то в настоящее время
их количество возросло 14 до 75%. Увеличилась доля сернистых и вы-
сокосернистых нефтей и в СССР.
Важным фактором является также диспропорция между масшта-
бами потребления бензина и других легких дистиллятов и содержа-
нием их в нефтях: прямая перегонка нефти дает их слишком мало,
нужна деструкция тяжелых углеводородов до более легких. В прош-
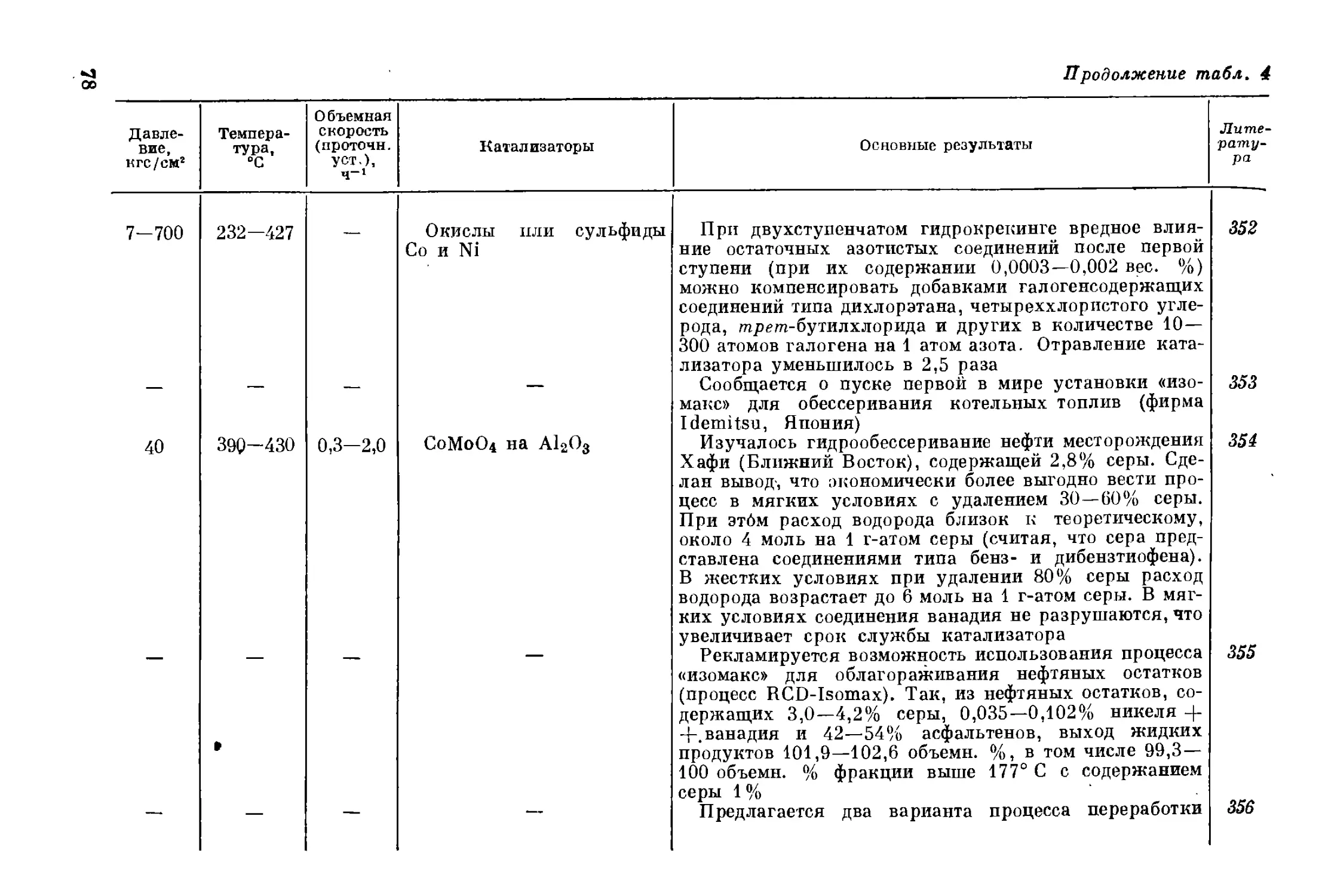
лом эта причина вызвала к жизни сначала термический, а затем ка-
талитический крекинг. Эти процессы и сейчас играют важную роль
в переработке нефти, но их возможности ограничены из-за низкого
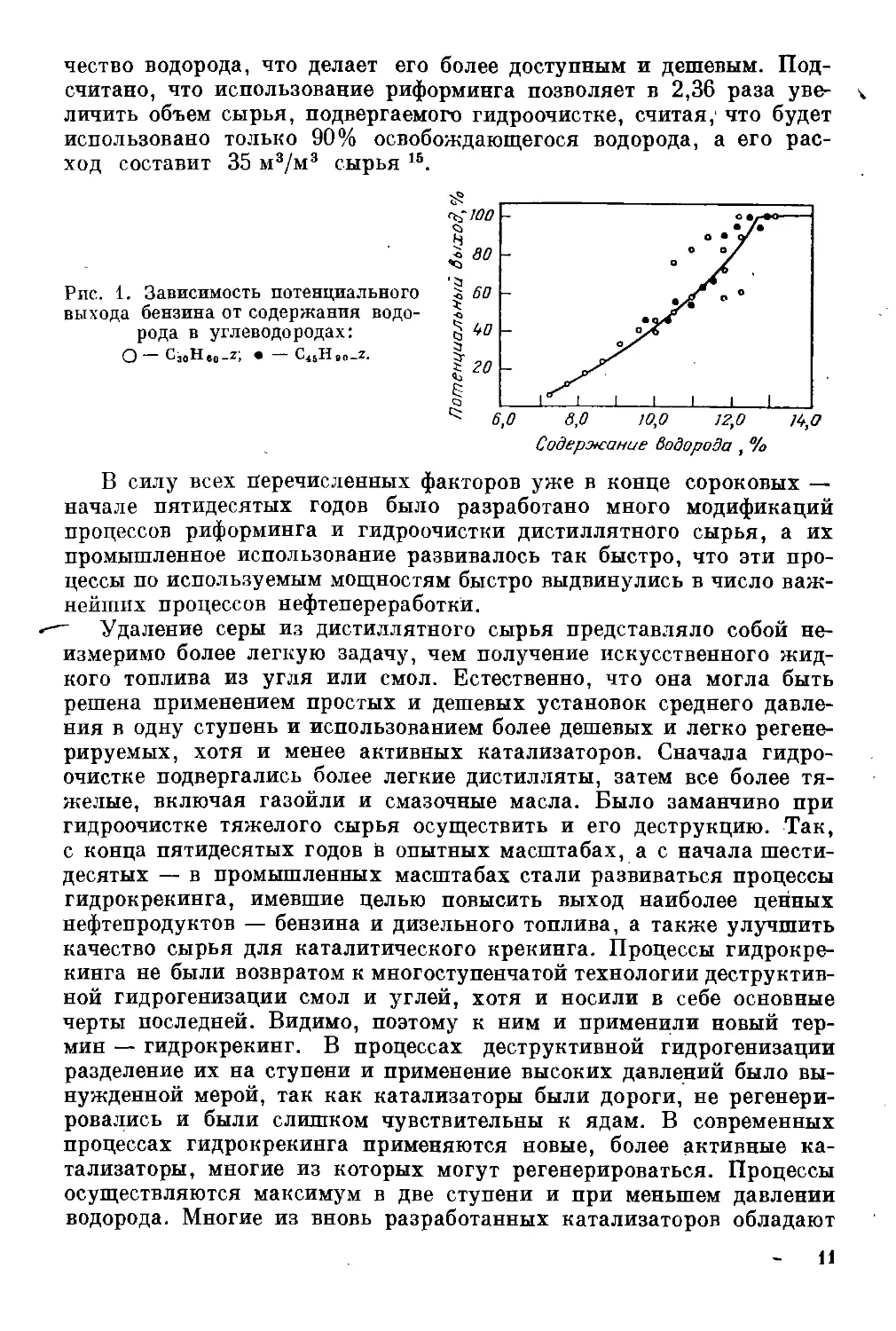
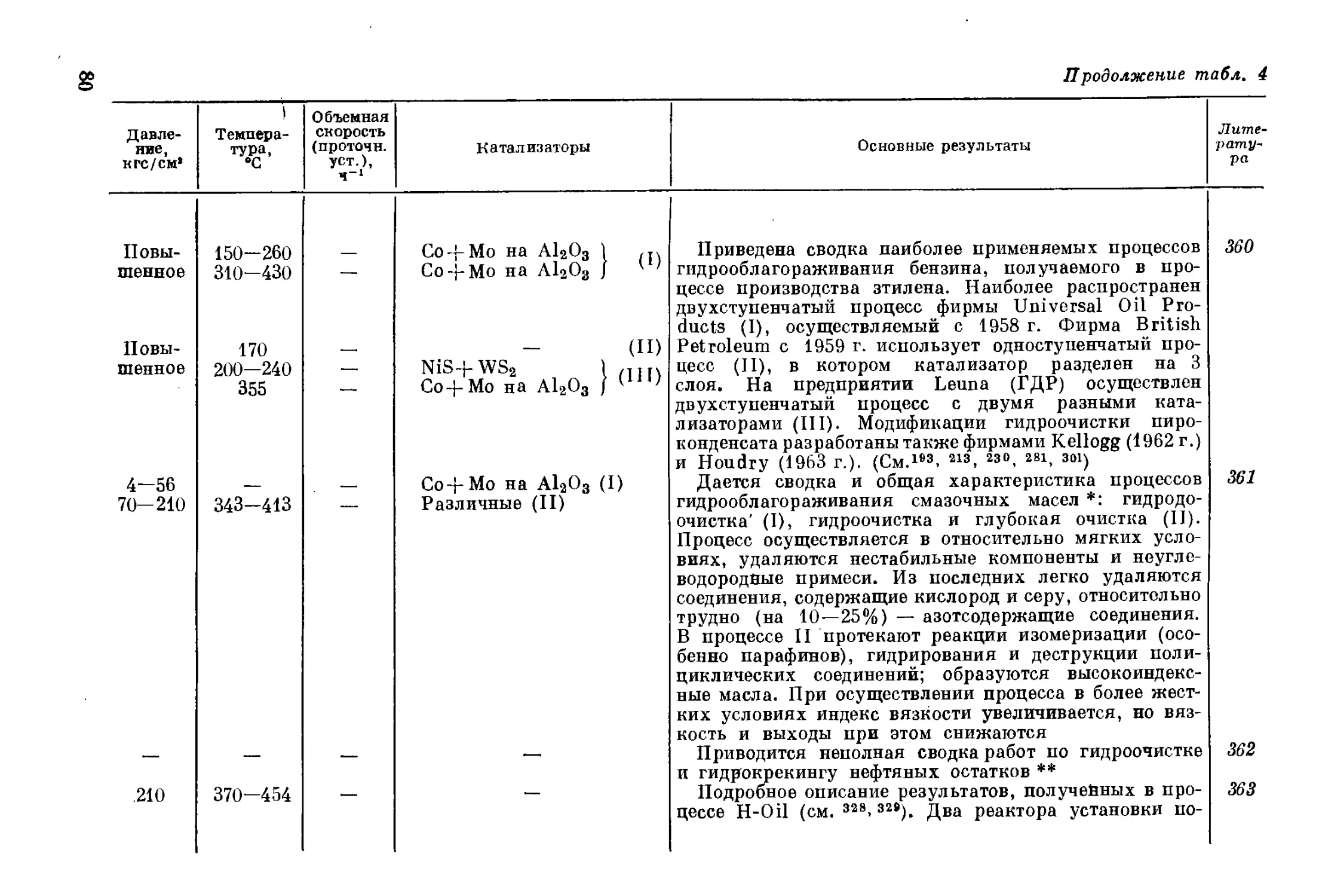
содержания водорода. Хиндс 2 подсчитал потенциальный выход бен-
зина как функцию содержания водорода в сырье в случае так назы-
ваемого идеального катализа, когда водород совсем не участвует
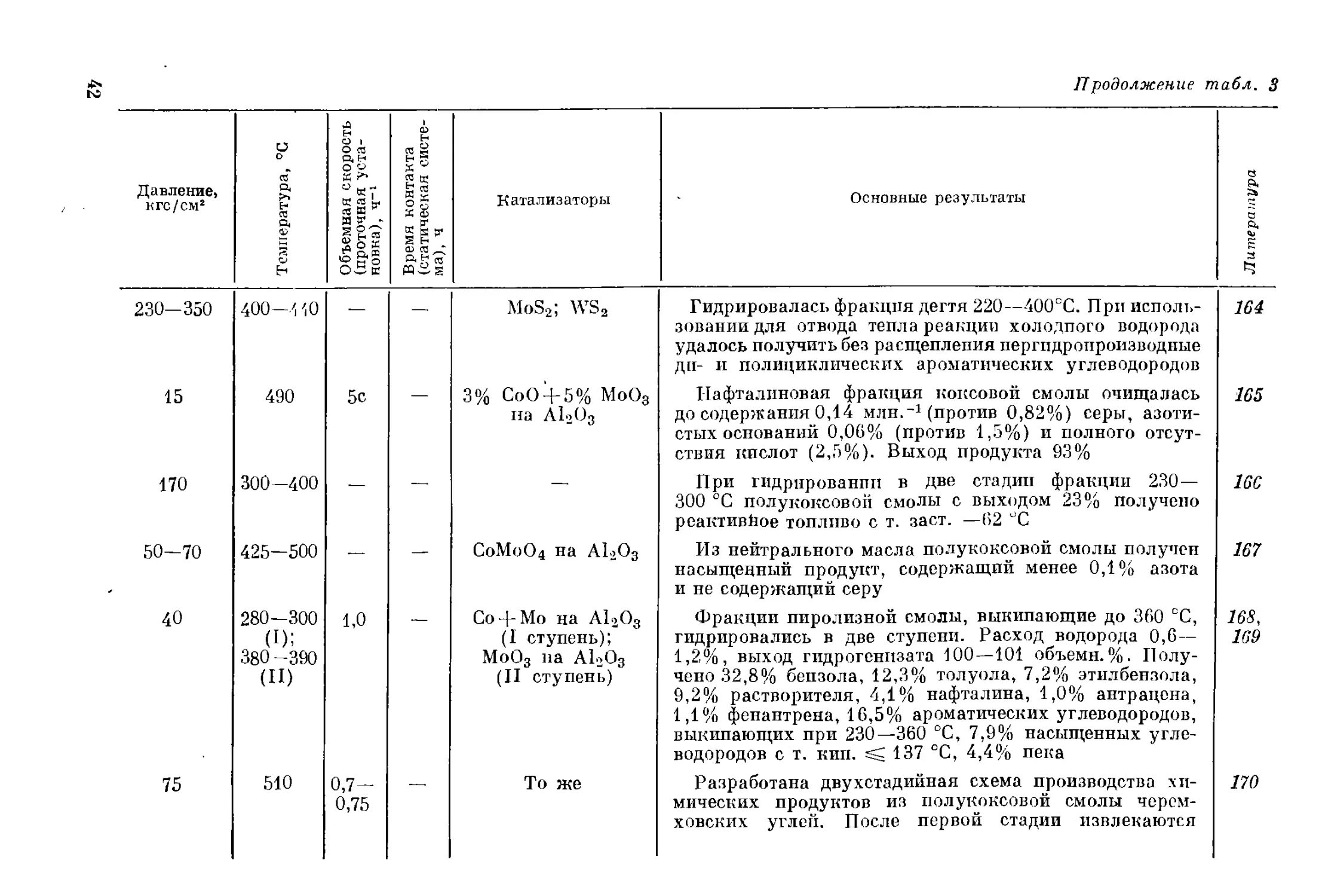
в образовании нежелательных продуктов (рис. 1). Если учесть, что
содержание водорода в тяжелом сырье обычно равно 12%, теоретиче-
ский выход бензина составит не более 75—80%. Фактические выходы
из-за газообразования существенно ниже. Следовательно, для
повышения выходов ценных дистиллятных продуктов в переработке
нефти неизбежно применение гидрогенизационных процессов.
Использованию гидрогенизационных процессов в нефтеперера-
ботке способствовало также повышение требований к детонационной
стойкости бензинов, вследствие чего быстро распространились про-
цессы каталитического риформинга, в результате которых нафтено-
вые и частично парафиновые углеводороды превращаются в детона-
ционностопкие ароматические углеводороды. При этом за счет де-
гидрирования и дегидроциклизации высвобождается большое коли-
10
чество водорода, что делает его более доступным и дешевым. Под-
считано, что использование риформинга позволяет в 2,36 раза уве-
личить объем сырья, подвергаемого гидроочистке, считая, что будет
использовано только 90% освобождающегося водорода, а его рас-
ход составит 35 м3/м3 сырья 15.
Рис. 1. Зависимость потенциального
выхода бензина от содержания водо-
рода в углеводородах:
О CjoHeo-Z', • C^Hgo-Z.
В силу всех перечисленных факторов уже в конце сороковых —
начале пятидесятых годов было разработано много модификаций
процессов риформинга и гидроочистки дистиллятного сырья, а их
промышленное использование развивалось так быстро, что эти про-
цессы по используемым мощностям быстро выдвинулись в число важ-
нейших процессов нефтепереработки.
Удаление серы из дистиллятного сырья представляло собой не-
измеримо более легкую задачу, чем получение искусственного жид-
кого топлива из угля или смол. Естественно, что она могла быть
решена применением простых и дешевых установок среднего давле-
ния в одну ступень и использованием более дешевых и легко регене-
рируемых, хотя и менее активных катализаторов. Сначала гидро-
очистке подвергались более легкие дистилляты, затем все более тя-
желые, включая газойли и смазочные масла. Было заманчиво при
гидроочистке тяжелого сырья осуществить и его деструкцию. Так,
с конца пятидесятых годов в опытных масштабах, а с начала шести-
десятых — в промышленных масштабах стали развиваться процессы
гидрокрекинга, имевшие целью повысить выход наиболее ценных
нефтепродуктов — бензина и дизельного топлива, а также улучшить
качество сырья для каталитического крекинга. Процессы гидрокре-
кинга не были возвратом к многоступенчатой технологии деструктив-
ной гидрогенизации смол и углей, хотя и носили в себе основные
черты последней. Видимо, поэтому к ним и применили новый тер-
мин — гидрокрекинг. В процессах деструктивной гидрогенизации
разделение их на ступени и применение высоких давлений было вы-
нужденной мерой, так как катализаторы были дороги, не регенери-
ровались и были слишком чувствительны к ядам. В современных
процессах гидрокрекинга применяются новые, более активные ка-
тализаторы, многие из которых могут регенерироваться. Процессы
осуществляются максимум в две ступени и при меньшем давлении
водорода. Многие из вновь разработанных катализаторов обладают
И
исключительно высокой изомеризующей и расщепляющей актив-
ностью.
Гидрирующий катализатор должен быть селективным, т. е. он
должен ускорять гидрирование би- и полициклических ароматиче-
ских углеводородов, но быть умеренно активным по отношению к цен-
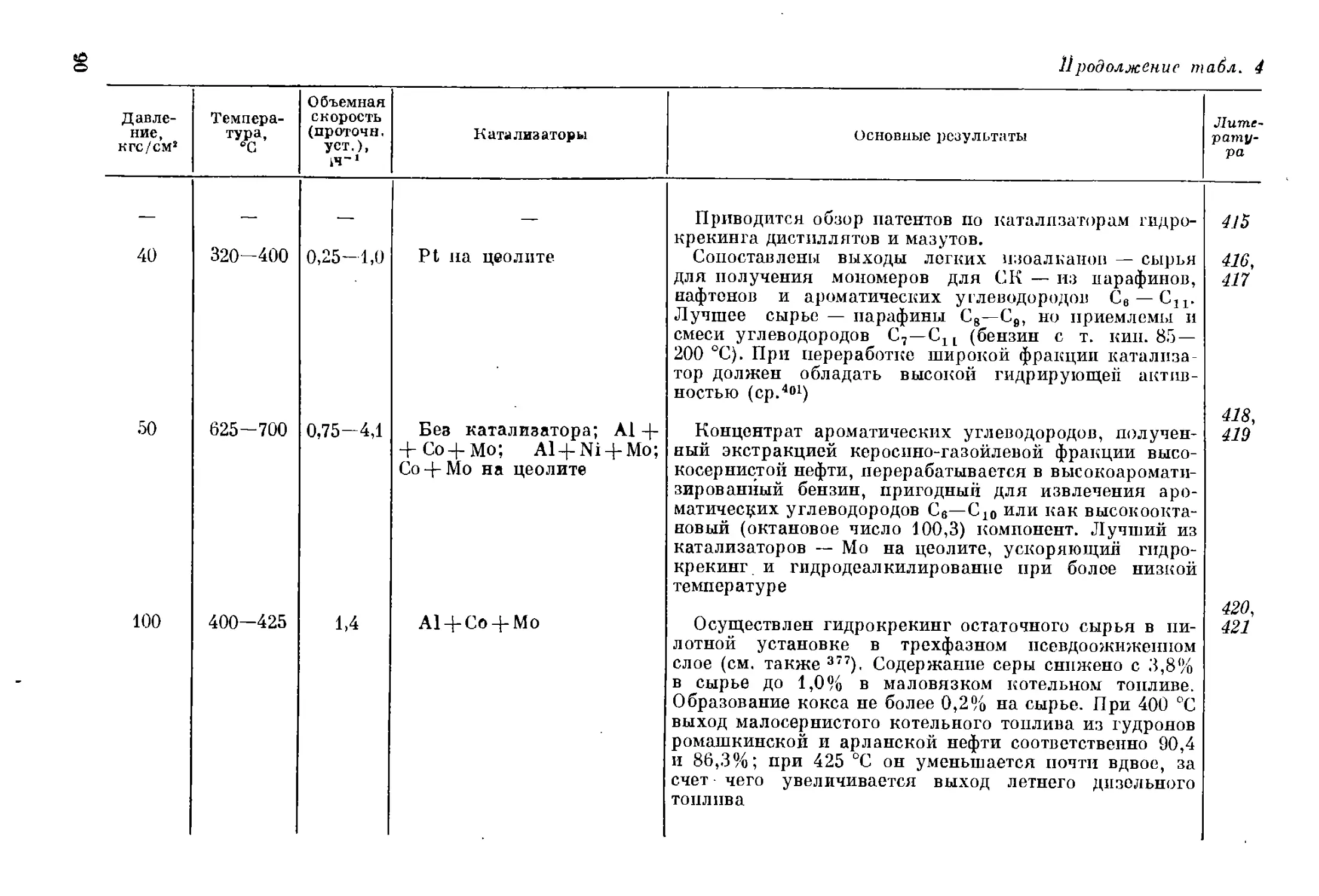
ным моноциклическим ароматическим углеводородам. В продуктах
гидрокрекинга содержание парафиновых углеводородов изострое-
ния выше, чем должно быть по термодинамическому равновесию 1в.
Это является следствием того, что расщеплению сырья предшествует
его глубокая изомеризация на катализаторах гидрокрекинга. Новые
катализаторы гидрокрекинга позволили уменьшить удельные капи-
таловложения при сооружении установок16 в среднем на 20%.
Внесено много технологических и инженерных усовершенствований:
применяются большие реакторы диаметром до 4,5 м, улучшены их
конструкции, удешевлена аппаратура за счет применения биметаллов,
упрощены отделения дистилляции и выделения 1в. Единичные мощ-
ности установок выросли до 12,7 тыс. м3 В'сутки, т. е. —4,5 млн. т
в год 1в. Было разработано несколько модификаций гидрокрекинга,
из которых наиболее распространенными стали процессы «изомакс»,
разработанный фирмами UOP и Chevron, и «юникрекинг», разрабо-
танный фирмами Union Oil и Esso. Суммарная мощность установок
гидрокрекинга в настоящее время быстро растет. Если в 1960 г.
она составляла 16 только 159 м3 в сутки, то к началу 1970 г. — более
180 тыс. м3 в сутки 17. Очень быстро развиваются и другие процессы
гидрогенизации.
Статистические данные по суммарной мощности трех наиболее
важных групп процессов нефтепереработки (каталитический кре-
кинг, каталитический риформинг и гидрогенизационные процессы)
на 1966 г., а также прогнозы по увеличению этих мощностей на 1975 г.
приведены 18 в табл. 1.
Таблица 1. Мощности важнейших каталитических процессов
переработки нефти (млн. т в год)
Зоны Каталитичес кий крекинг Гидрогениза- ционные про- цессы Каталитический риформинг
1966 г. 1975 г. 1966 г. 1975 г. 1966 г. 1975 г.
США и Канада 165.0 187,5 141,0 199,0 87,0 113,6
Западная Европа 22,3 30,9 43,0 129,0 42,0 92,5
Азия и Океания 12,8 21,7 17,3 43,9 13.7 32,4
Латинская Америка 21,1 31,2 7,1 25,2 6,4 21,0
Всего 221,2 271,3 208,4 397,1 149,1 259,5
Предполагают, что гидрогенизационные процессы, из которых
7/8 составляют процессы гидроочистки, выйдут на первое место среди
важнейших каталитических процессов переработки нефти. Однако
12
в будущем следует ожидать еще большего прогресса. Дело в том, что
приведенные выше прогнозы учитывают только гидроочистку дистил-
лятных продуктов и гидрокрекинг. Цроцессы гидроочистки нефтяных
остатков и сырой нефти только начинают разрабатываться. Есть
все основания полагать, что они будут развиваться еще более бурно,
поскольку загрязнение атмосферы становится все более острой со-
циальной проблемой; и она может быть решена только при условии
создания процессов получения малосернистого котельного топлива.
По статистическим данным, в США 18 из 23 млн. т SO2, выбро-
шенного в атмосферу в 1963 г., 41% обусловлен сжиганием угля на
крупных электростанциях и 19% — сжиганием угля на прочих
промышленных установках. Сжигание котельных топлив дает отно-
сительно меньшие выбросы SO2: 2,8% за счет сжигания на электро-
станциях и 13,1 % — на других промышленных объектах. Остальные
выбросы относятся к различным другим технологическим процессам.
Таким образом, хотя использование котельных топлив дает только
15,9% суммарных выбросов SO2, оно представляет едва ли не глав-
ную опасность, так как котельные расположены в густонаселенных
районах.
По данным службы здравоохранения США, предельно допустимая средне-
суточная концентрация SO2 в воздухе20 составляет 0,1 млн'1 (в СССР 0,05 мг/м3).
Считают10, что такая норма может быть обеспечена, если котельные топлива
будут содержать не более 1% серы, а во многих случаях и значительно меньше
(до 0,3%)21.
Возможны три пути предотвращения загрязнения воздуха про-
дуктами горения сернистых котельных топлив: 1) замена их не-
сернистым или малосернистым топливом (природный газ, дистилля-
ты высокого качества); 2) удаление SO2 из дымовых газов или из
газов конверсии сернистого топлива перед их сжиганием; 3) десульфу-
ризация остаточных котельных топлив. Первый путь ограничен
недостатком несернистых топлив или значительно большей стоимостью
дистиллятных. Второй — применим только для крупных котельных
установок и, видимо, будет осуществляться на электростанциях,
потребляющих сернистые угли или мазуты. Этот путь еще требует
разработки и проверки в крупных масштабах. Для относительно
небольших промышленных котельных установок, составляющих
основную массу потребителей тяжелых топлий, применим только
третий путь — гидрообессеривание нефтяных остатков. Он, являясь
универсальным, привлекает наибольший интерес.
Если учесть, что три четверти всего количества добываемой .
нефти приходится на сернистые и высокосернистые нефти 14 и что
почти половина добываемой нефти используется как котельное то-
пливо (особенно в Западной Европе и Японии), то становится ясным,
что при добыче нефти более 2 млрд, т в год производственные мощ-
ности процессов гидроочистки и гидрокрекинга мазутов составят
несколько сот млн. т в год.
Все сказанное выше доказывает исключительную важность гид-,
рогенизационных процессов в нефтепереработке и еще большую
13
их значимость в будущем, когда ни один нефтепродукт не минует
той или иной стадии гидрогенизационного облагораживания. Первый
такой завод уже построен, он будет перерабатывать сернистую нефть
Ближнего Востока; его крупнейшая в мире водородная установка
будет давать в сутки 4 млн. м3 водорода 22.
Хотя гидрогенизационные процессы в переработке нефти оттес-
нили на второй план процессы гидрогенизации смол и полностью
вытеснили процессы гидрогенизации угля, эти области продолжают
привлекать исследователей. Это объясняется, во-первых, требова-
ниями перспективы. Запасы углей на несколько порядков больше
запасов нефти. И хотя опасность истощения последних отодвинута
на много десятилетий, а возможно, и на столетие, в будущем сланцы
и уголь вероятно станут основным поставщиком углеводородного
сырья. Кроме того, многие страны или крупные экономические рай-
оны некоторых стран не имеют собственных запасов нефти.
Во-вторых, метод гидрогенизации и в условиях развития нефте-
переработки сохраняет свое значение как практически единственный
способ переработки различных смол, образующихся в качестве по-
бочных продуктов коксования, полукоксования и газификации углей
п сланцев. С ростом производства металлургического кокса и орга-
низацией дальнего газоснабжения городов количество этих смол бу-
дет возрастать. Без гидрогенизации невозможно их квалифицирован-
ное использование и выделение из них ценных химических продуктов.
В-третьих, заводы, гидрирующие различное ненефтяное сырье,
сохранились и работают в ряде стран 23"28. Технология этих про-
цессов постоянно совершенствуется, что требует непрерывных на-
учных исследований и опытных разработок.
Все вышеизложенное показывает огромную роль гидрогениза-
ционных процессов, блестящие перспективы их развития и объяс-
няет возрастающий интерес ученых многих стран к проблемам гид-
рогенизации.
Общее число публикаций, начиная с 1949—1950 гг., т. е. с пе-
риода, когда были написаны обобщающие монографии советских
ученых 1’3>4> составляет более 1500, кроме того, имеется бойее 1000
патентов. Даже аннотированное перечисление всех этих работ за-
няло бы очень большой объем. Между тем, поскольку эти работы
не обобщались в отечественной научной литературе, их систематиче-
ское рассмотрение представляет большой интерес, позволяя оха-
рактеризовать этапы развития, современное состояние и тенденции
развития процессов гидрогенизации.
Важнейшие результаты и выводы работ двух последних десяти-
летий в области гидрогенизации угля, угольных и сланцевых смол,
нефти, нефтепродуктов и другого углеводородного сырья приведены
в табл. 2—4*.
* В данных таблицах сопоставляются в хронологическом порядке в основ-
ном только публикации, касающиеся разработки новых технологических схем,
усовершенствований технологии гидрогенизации и приложения ее к новым
видам сырья, а также работы, имеющие непосредственно' отношение к технологии.
14
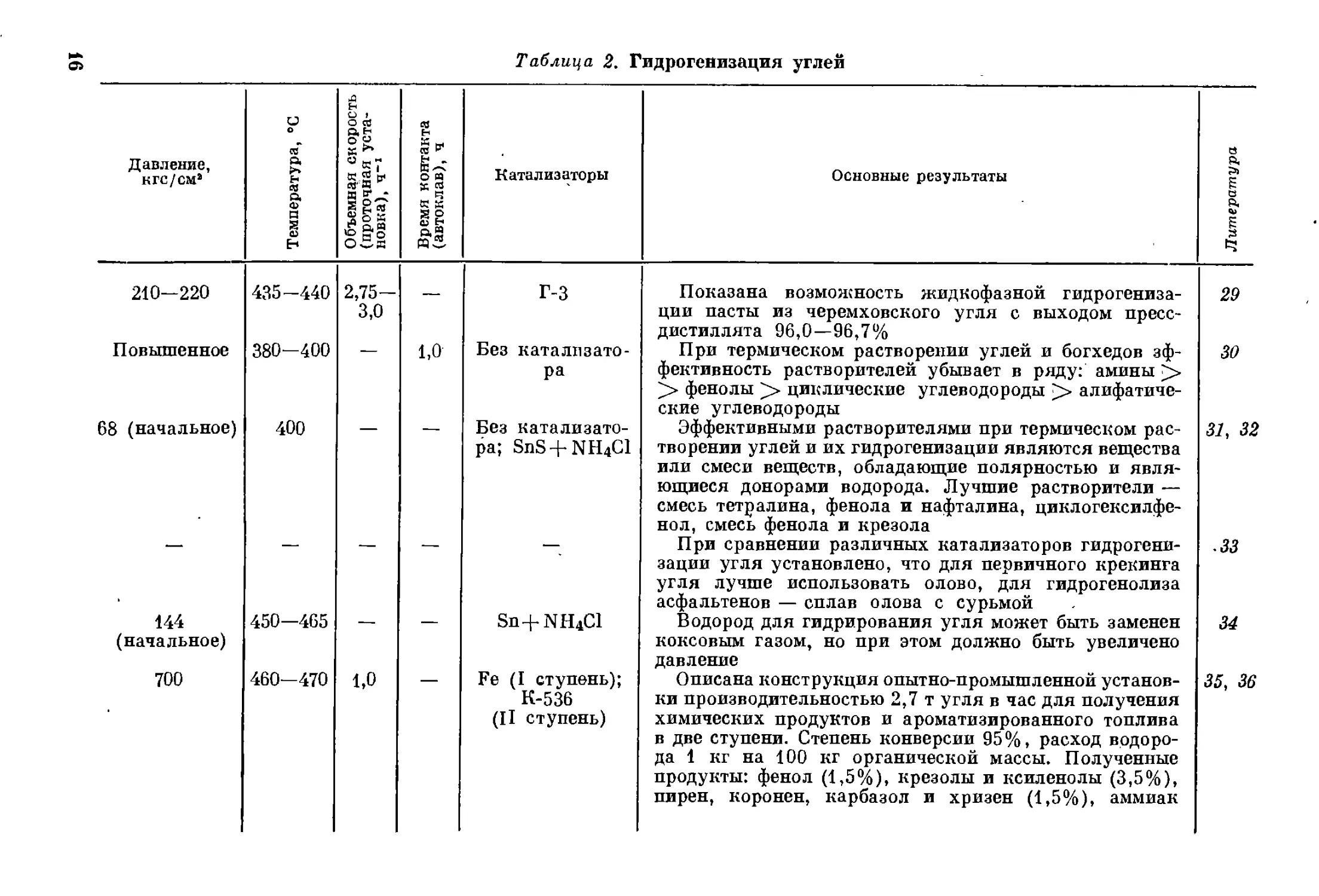
Как видно из данных, приведенных в табл. 2, в последнее время
были значительно развиты теоретические основы гидрогенизации
углей. Еще в довоенный период были определены основные (Законо-
мерности, связывающие относительную легкость ожижения углей
с их элементарным и петрографическим составом4. В настоящее
время можно предсказывать выход жидких продуктов 81. Углуб-
лены представления о влиянии давления на процесс гидрогениза-
ции 47> 6°, 61 и о промежуточных ступенях этого процесса 381 39, 44, 63.
На основании результатов многочисленных работ твердые горючие
ископаемые по легкости их превращения были расположены 96
в следующий ряд:
Торф > Молодые бурые угли > Старые бурыё угли >
> Каменные угли > Антрацит
Легкость ожижения каменных углей убывает в ряду:
Длиннопламенные > Газовые > Паровично-жирные > Коксовые > Тощие
Сапропелитовые угли ожижаются легче гумусовых, горючие
сланцы — легче углей.
В области технологии процессов гидрогенизации наряду с опро-
бованием новых видов сырья и катализаторов выполнялись иссле-
дования по получению из угля различных химических продуктов.
Первая группа работ характеризует непрекращающийся интерес
к углю как к возможному источнику жидкого топлива в ряде
стран, вторая группа — большую перспективность развития хими-
ческой промышленности на основе продуктов углехимии. Гидро-
генизация угля может дать многие вещества, являющиеся
сейчас дефицитными: низшие фенолы и азотистые основания,
полициклические соединения и др. В середине пятидесятых -
годов в США был- разработан проект завода гидрогенизации ,угля
с вариантом углубленной переработки 97, предусматривавшим вы-
пуск, 7,2% фенолов, 50,8% индивидуальных ароматических углево-
дородов, а бензина — только 26,7% (42% по старому варианту).
Однако процесс оказался нерентабельным. По имеющимся в печати
отрывочным данным, неудача объясняется малым выходом низших
фенолов ®8; опыты продолжаются, но в малых масштабах".
Неудачи с осуществлением «обычного» гидрирования угля в пасте
побудили искать другие пути переработки угля, хотя продолжа-
лись и поиски усовершенствований «пастового» процесса 76> 78, 821 8®.
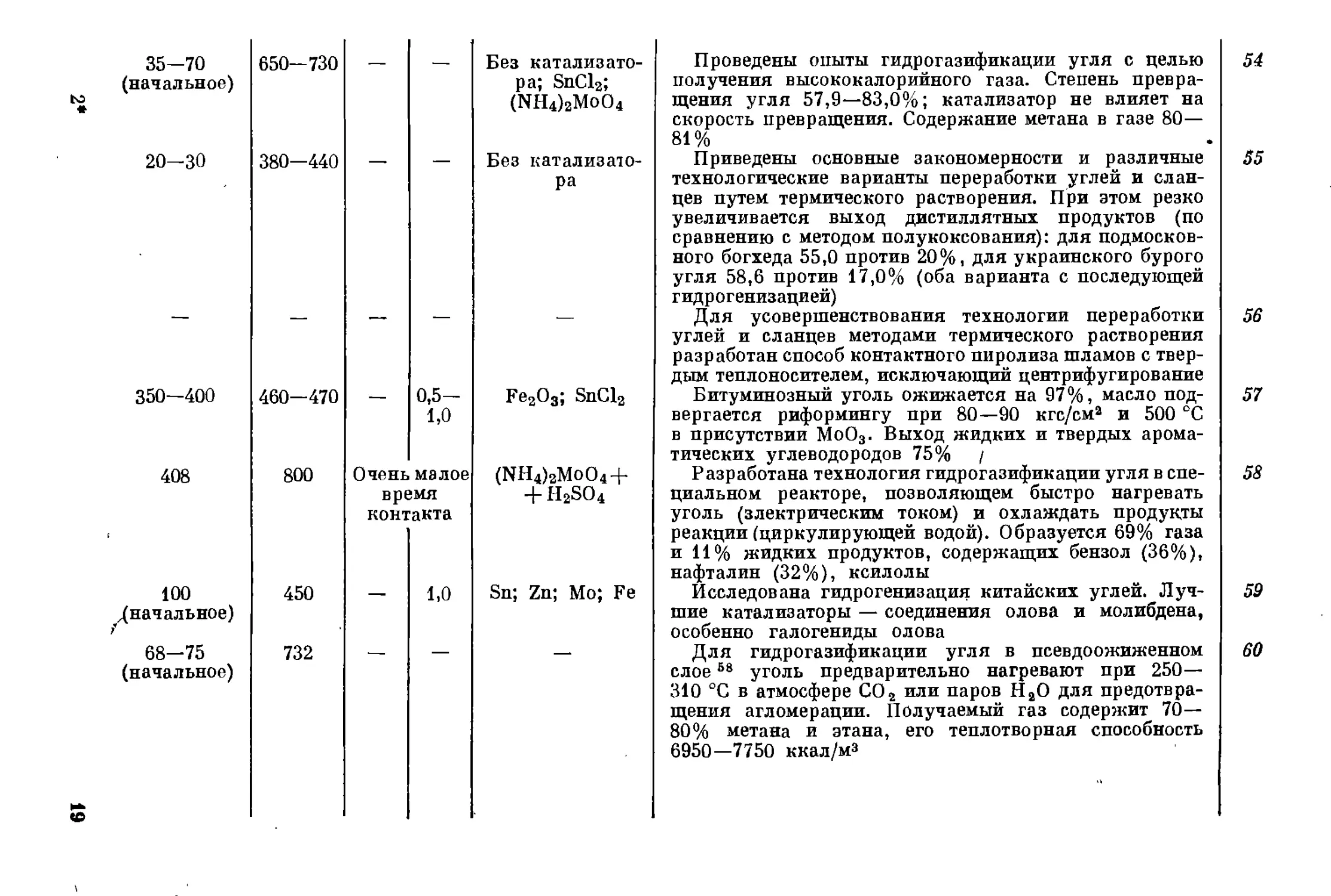
Особенно много работ проведено в США по гидрогазификации угля *.
Их целью был поиск дешевого метода превращения твердого топлива
в газообразное и частично — в жидкое. Но уже й 1964 г. был сделан
вывод о неэкономичности процесса гидрогазификации угля 86, хотя
работы и продолжались 8в- ®°. Бесперспективным оказалось исполь-
зование облучения 77 и атомарного водорода 81.
* См. ссылки 54, 58, 60, 65, 66, 69, 72, 74, 76, 80, 84.
15
о
Таблица 2. Гидрогенизация углей
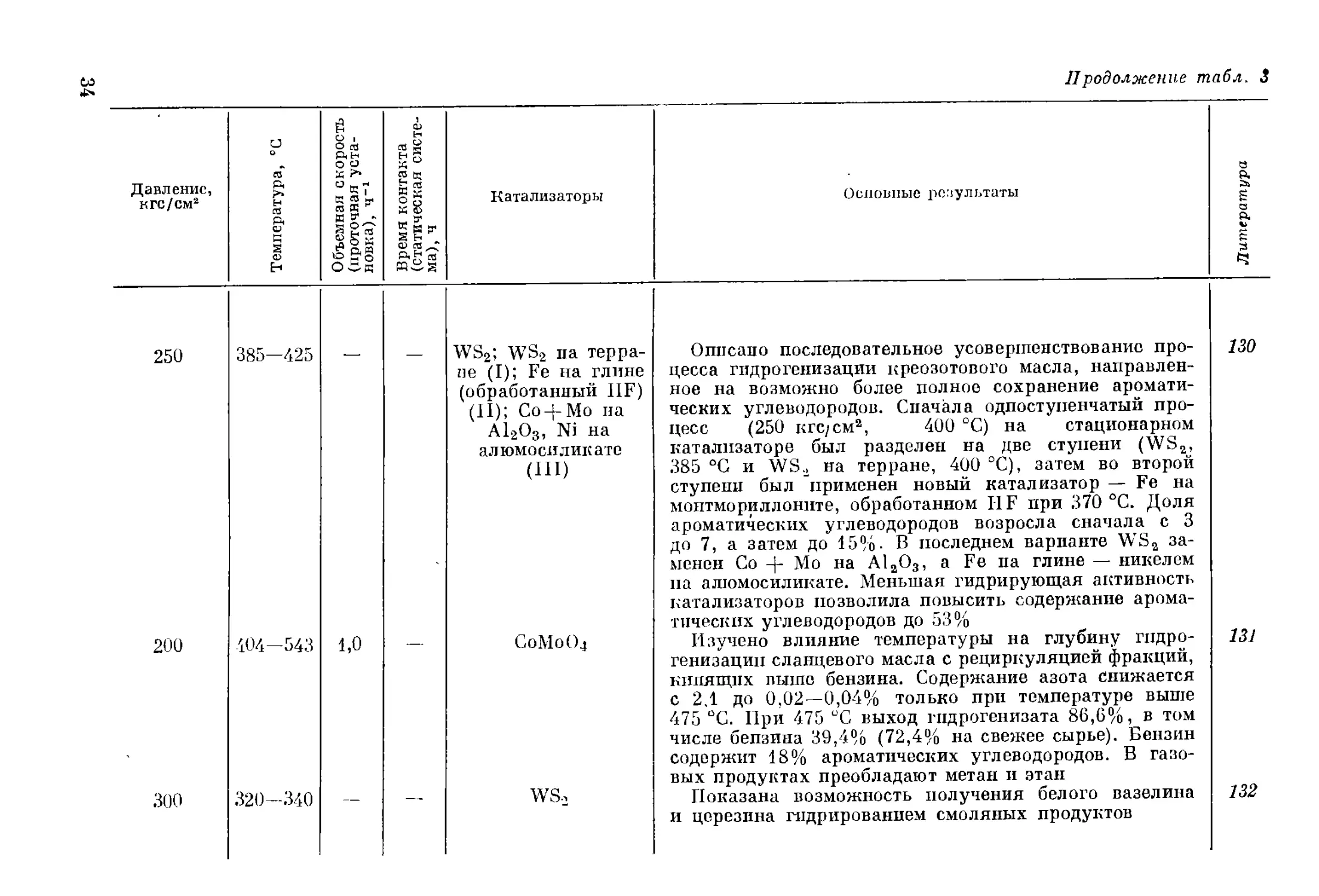
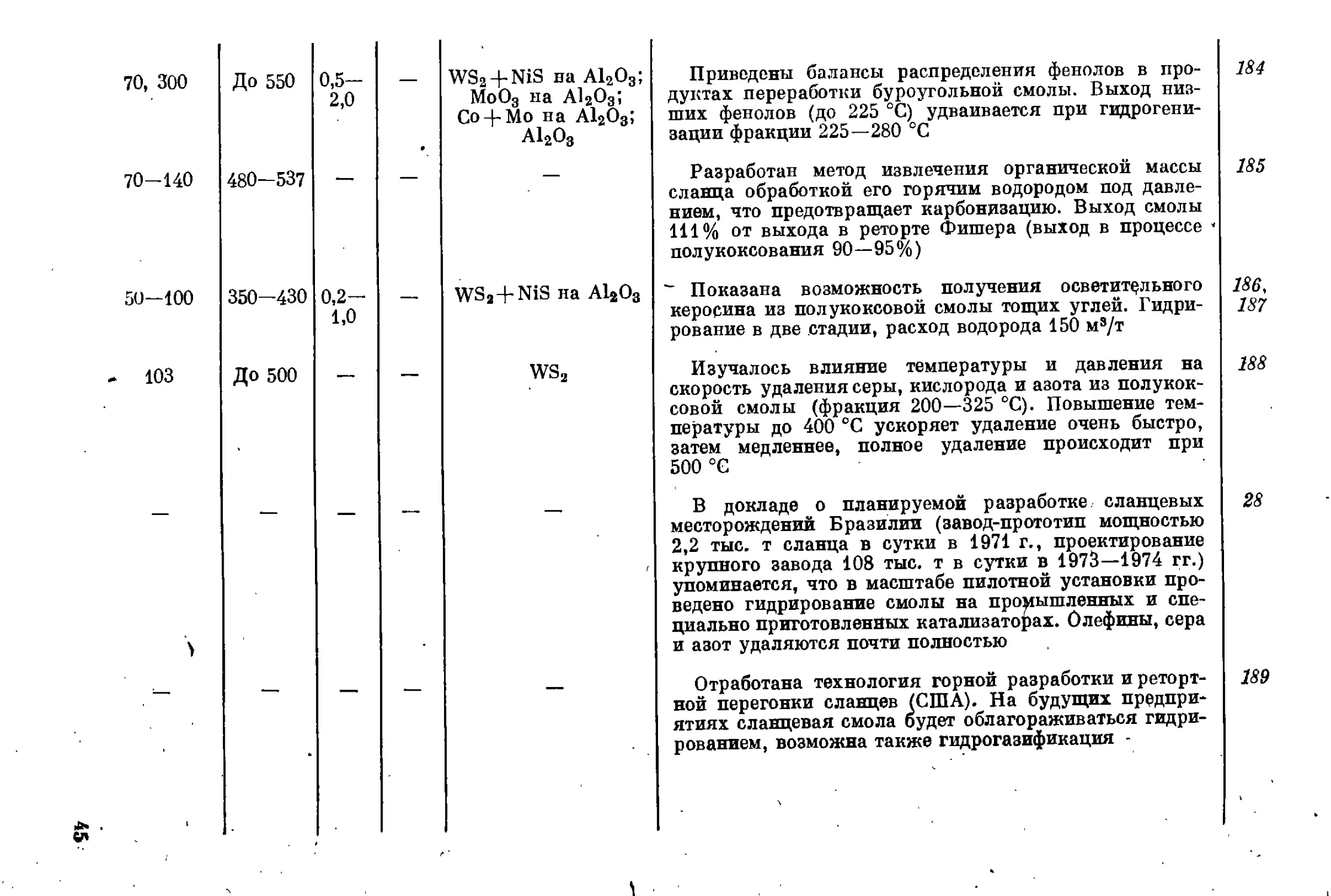
Давление, кгс/см2 Температура, °C Объемная скорость (проточная уста- новка), Ч”1 Время контакта (автоклав), ч Катализаторы Основные результаты Литература
210—220 435-440 2,75— 3,0 — г-з Показана возмоя:ность жидкофазной гидрогениза- ции пасты из черемховского угля с выходом пресс- дистиллята 96,0—96,7% 29
Повышенное 380—400 1,0 Без катализато- ра При термическом растворении углей и богхедов эф- фективность растворителей убывает в ряду: амины >> > фенолы циклические углеводороды >> алифатиче- ские углеводороды 30
68 (начальное) 400 Без катализато- ра; SnS + NH4Cl Эффективными растворителями при термическом рас- творении углей и их гидрогенизации являются вещества или смеси веществ, обладающие полярностью и явля- ющиеся донорами водорода. Лучшие растворители — смесь тетралина, фенола и нафталина, циклогексилфе- нол, смесь фенола и крезола При сравнении различных катализаторов гидрогени- зации угля установлено, что для первичного крекинга угля лучше использовать олово, для гидрогенолиза асфальтенов — сплав олова с сурьмой 31, 32
.33
144 (начальное) 450-465 — — Sn+NH4C1 Водород для гидрирования угля может быть заменен коксовым газом, но при этом должно быть увеличено давление 34
700 460—470 1,0 Fe (I ступень); К-536 (II ступень) Описана конструкция опытно-промышленной установ- ки производительностью 2,7 т угля в час для получения химических продуктов и ароматизированного топлива в две ступени. Степень конверсии 95%, расход водоро- да 1 кг на 100 кг органической массы. Полученные продукты: фенол (1,5%), крезолы и ксиленолы (3,5%), пирен, коронен, карбазол и хризен (1,5%), аммиак 35, 36
to
co * ё cj 70—180 (начальное) 450—475 — 2,5 —
N5
70-250 СО 450 — 1.0 61 соединение я их комбинации
68 СО 68 -U 700 600 460—470 — — Без катализато- ра
10—700 400—470 — 0,25— 1,5 —
70 (начальное) 450 — 1,0 Fe; Ni; Sn; Mo; Cr; Zn; Pb; Cu
85-170 (начальное) 400-440 — 0,5- 3,0 SnS + NH4Cl
4 240 460 0,4- 0,9 — Sn; Ni; Fe; Zn
/\ЧкХ 100 [ Ar^X^^Sk, ’ S . ховмшенное ; fa Р \\ /# ? js \ 460 400 0,5— 1,1 — Fe; Ni; Mo; Sn SnS+NH4Cl; Mo
>>*£- J
(1,5%), сера (1%), бутаны (7,5%), пропан (8,5%),
этан (6%), метан (4%), легкий бензин (17%), бензин
(42%)
Показано, что повторной гидрогенизацией среднего
масла, полученного при гидрогенизации угля, или фе-
нолов, выделенных из него, можно увеличить выход
низших фенолов
Наиболее активны соединения олова в комбинации
с соединениями галоидов. Предполагается, что галоид
ускоряет деструкцию угля и асфальтенов, а олово —
превращение последних в масло
Проведены поисковые опыты по гидрогазификации
угля в псевдоожиженном состоянии. Выход газа 25%,
масла 25%
Описан опыт пуска (на нефтепродуктах) и первого
года работы (на угле) установки (см. 33. 36). В трех
пробегах переработано более 1000 т угля
Разработана методика оценки гидрируемости углей
и показана зависимость ожижения от степени метамор-
физма и давления
Показано, что более эффективна пропитка угля рас-
твором катализатора, чем добавка последнего. При
пропитке Мо лучше Sn и Ni; сульфаты лучше в случае
Со и Fe, хлориды — в случае Sn и Ni
Изучалась кинетика ожижения угля. Показано, что
асфальтены являются промежуточным продуктом
(уголь -► асфальтены -* масло)
В проточной установке эффективность катализаторов
изменяется в рядах: Sn > Ni Fe > Zn (по коли-
честву нерастворимого остатка) и Ni>Sn>> Zn > Fe
(по остаточному содержанию асфальтенов)
Подтверждены выводы работы 43. Активность изме-
няется в ряду: Mo > Sn > Ni > Fe
Найдена прямая пропорциональность между скоро-
стью гидрогенизации и Давлением (ср. 42) : к = аР -j-b
37
38, 39
40
41
42
43
44
45
46
47
сю
Давление, кгс/см2 Температура, СС Объемная скорость (проточная уста- новка), ч4 Время контакта (автоклав), ч Катализаторы
540—700 465-474 1,0 — Sn, Fe (I сту- пень); К-536 (II ступень)
540—700 465-474 1,0 — Sn, Fe (I сту- пень); К-536 (II ступень)
370-1470 420 — 1-2 FeSO4
300-700 420—460 — — Fe
35—280 400 — — Без катализато- ра; Sn; Мо FegOg! MoOgj WO3; Na2S; оксалат олова; 1-2, FeSOi
100 (начальное) 340—450
Прпдолжение тпбл. 2
Основные результаты
Литерат ура
Приводятся уточненные данные по двухступенчатой
гидрогенизации угля. В жидкой фазе оксалат олова
заменен сульфатом железа (из экономических сообра-
жений). На 1000 кг органической массы угля и 59,2 кг
водорода получено в жидкофазной ступени: 579,5 кг
бензина, нафты и среднего масла, 111,2 кг тяжелого
масла, 19,3 кг сероводорода, 8,1 кг аммиака, 155,2 кг
газов, 49,1 кг нерастворимого остатка
Катализатор К-536 был улучшен заменой алюмосили-
катного носителя фильтролем, обработанным фтори-
стым водородом. Получен более ароматизированный
бензин с октановым числом 77—80
Показано, что применением сверхвысоких давлений
можно ожижать и метаморфизованные угли. Повыше-
ние давления понижает выход асфальтенов
На основе экспериментальных данных выведены фор-
мулы, позволяющие по составу угля предсказывать
выход продуктов в промышленных опытах
Выводы работы 47 распространены на случай гидро-
генизации угля при атмосферном давлении
Изучено ожижение венгерских углей в жидкой фазе
в автоклавах. При высоких температурах лучшие ре-
зультаты были получены на катализаторах МоО3,
WO3, Fe3O3 (МоО3 > WO3 > Fe2O3), при низких —на
Sn + I3
48
49
50
51
52
53
35-70 (начальное) 650— 730
to « 20-30 380-440
Без катализато-
ра; SnCl2;
(NH4)2MoO4
Без катализато-
ра
350—400 460—470 — 0,5- 1,0
408 800 Очень вр( КОН7 малое !МЯ акта
100 ^начальное) 450 — 1,0
68-75 (начальное) 732 — —
<0
Fe2O3; SnCl2
(NH4)2MoO4 +
+ H2SO4
Sn; Zn; Mo; Fe
Проведены опыты гидрогазификации угля с целью
получения высококалорийного газа. Степень превра-
щения угля 57,9—83,0%; катализатор не влияет на
скорость превращения. Содержание метана в газе 80—
81%
Приведены основные закономерности и различные
технологические варианты переработки углей и слан-
цев путем термического растворения. При этом резко
увеличивается выход дистиллятных продуктов (по
сравнению с методом полукоксования): для подмосков-
ного богхеда 55,0 против 20%, для украинского бурого
угля 58,6 против 17,0% (оба варианта с последующей
гидрогенизацией)
Для усовершенствования технологии переработки
углей и сланцев методами термического растворения
разработан способ контактного пиролиза шламов с твер-
дым теплоносителем, исключающий центрифугирование
Битуминозный уголь ожижается на 97%, масло под-
вергается риформингу при 80—90 кгс/сма и 500 °C
в присутствии МоО3. Выход жидких и твердых арома-
тических углеводородов 75% /
Разработана технология гидрогазификации угля в спе-
циальном реакторе, позволяющем быстро нагревать
уголь (электрическим током) и охлаждать продукты
реакции (циркулирующей водой). Образуется 69% газа
и 11% жидких продуктов, содержащих бензол (36%),
нафталин (32%), ксилолы
Исследована гидрогенизация китайских углей. Луч-
шие катализаторы — соединения олова и молибдена,
особенно галогениды олова
Для гидрогазификации угля в псевдоожиженном
слое58 уголь предварительно нагревают при 250—
310 °C в атмосфере С02 или паров II2О для предотвра-
щения агломерации. Получаемый газ содержит 70—
80% метана и этана, его теплотворная способность
6950—7750 ккал/м3
54
55
56
57
58
59
60
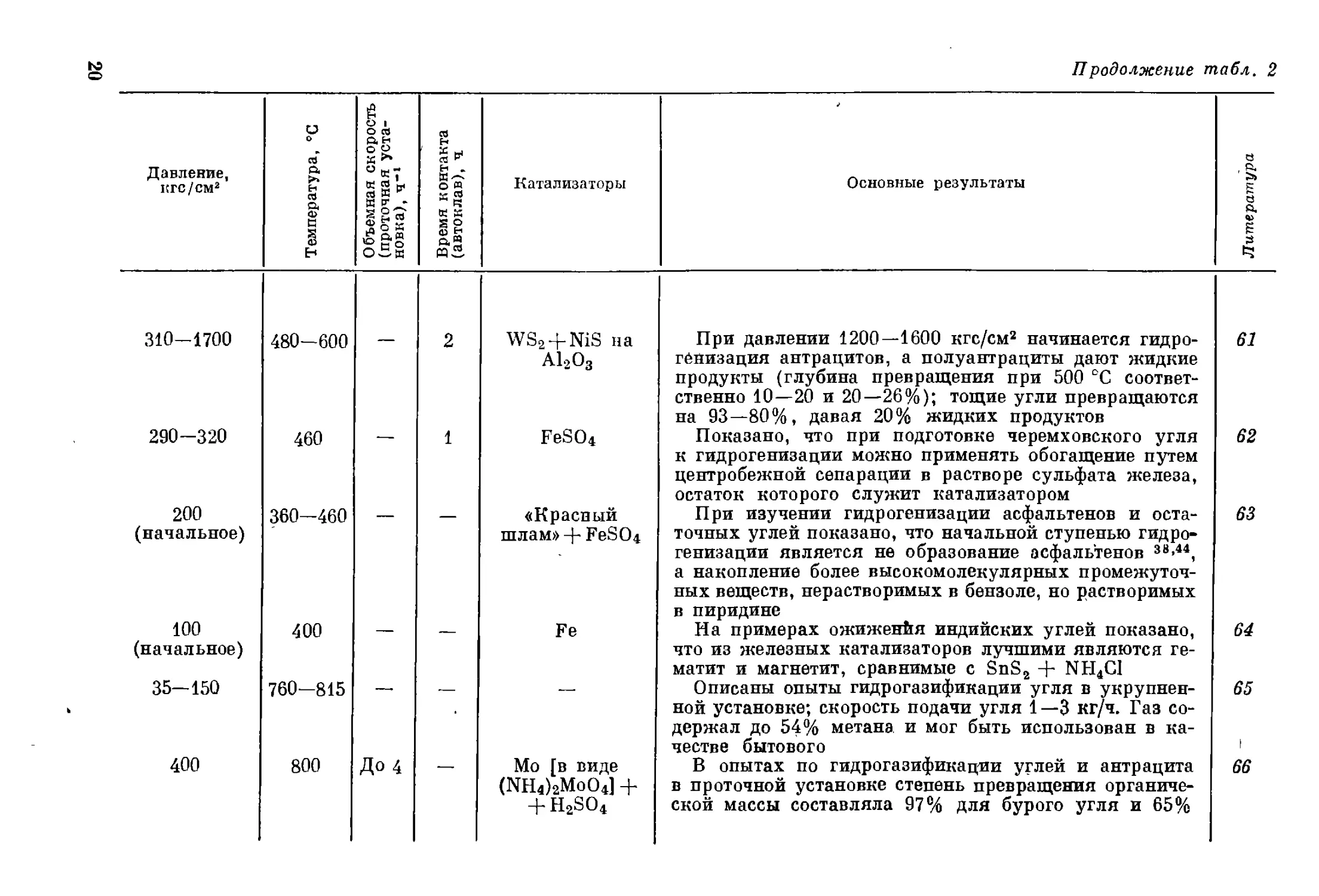
Давление, кгс/см2 Температура, °C Объемная скорость (проточная уста- новка), Ч"1 Время контакта (автоклав), ч Катализаторы
310-1700 480—600 — 2 WS2 + NiS на А12О3
290-320 460 — 1 FeSO4
200 (начальное) 360—460 — «Красный шлам» + FeSO4
100 (начальное) 400 — — Fe
35—150 760-815 — — —
400 800 До 4 — Мо [в виде (NH4)2MoO4] + + H2SO4
Продолжение табл. 2
Основные результаты
При давлении 1200—1600 кгс/см2 начинается гидро-
генизация антрацитов, а полуантрациты дают жидкие
продукты (глубина превращения при 500 °C соответ-
ственно 10—20 и 20—26%); тощие угли превращаются
на 93—80%, давая 20% жидких продуктов
Показано, что при подготовке черемховского угля
к гидрогенизации можно применять обогащение путем
центробежной сепарации в растворе сульфата железа,
остаток которого служит катализатором
При изучении гидрогенизации асфальтенов и оста-
точных углей показано, что начальной ступенью гидро-
генизации является не образование асфальтенов 38-44,
а накопление более высокомолекулярных промежуточ-
ных веществ, нерастворимых в бензоле, но растворимых
в пиридине
На примерах ожиженйя индийских углей показано,
что из железных катализаторов лучшими являются ге-
матит и магнетит, сравнимые с SnS2 + NH4C1
Описаны опыты гидрогазификации угля в укрупнен-
ной установке; скорость подачи угля 1 —3 кг/ч. Газ со-
держал до 54% метана и мог быть использован в ка-
честве бытового
В опытах по гидрогазификации углей и антрацита
в проточной установке степень превращения органиче-
ской массы составляла 97% для бурого угля и 65%
Литература
61
62
63
64
65
66
545 230 46—480 420—480 0,45— 1,6 кг/л 0,2- 1,0 — (NH4)2MoO4 SnS + CH3I (лучший)
— —
700 450-480 — — Оксалат олова
— — — — —
34; 68; 136 400; 450; 538 и выше — o,U- 14,5 мин Без катализато- ра
— —- — — —
50-150 470-480 — 5- 20 мин Fe
Й
для антрацита. Выходы масла от 32 до 0% соответ-
ственно; основной продукт — газ, содержащий 82—
92% метана и 15—18% этана
Степень превращения органической массы угля 98—
99%, максимальный выход широкой фракции (до 325 °C)
47%.- Катализатор (0,01%) вносили пропиткой
Проведены опыты гидрогенизации двух типов углей
в пилотной установке. Исследовано влияние парамет-
ров процесса и найдены условия получения котельного
топлива. Найдено, что при времени контакта 0,45 ч
активность катализаторов изменяется в рядах: Sn >>
> Ni = Ее Zn (по превращению угля) и Sn > Fe >
Г> Ni > Zn (по превращению асфальтенов). Однако
при времени контакта 0,8 ч различия между катализа-
торами незначительны
Разработан реактор для гидрогенизации угольной
пыли в псевдоожиженном слое
.Проведены опыты гидрогенизации японских углей
на установке производительностью 50 кг/сутки
Проведены опыты гидрокрекинга лигнина в раство-
рах фенола, циклогексана и тетралина. Гидрогенизаты
обогащаются ароматическими углеводородами; из кис-
лых компонентов выделены о- и n-крезолы и другие
орто- и пара-производные фенола
Изучено влияние условий на процесс получения го-
рючих газов и кокса из битуминозного угля. Рекомен-
дуемые условия: 900 °C и время контакта 1 мин
Изучено влияние предварительного нагрева пасты,
размера и скорости седиментации частиц в пасте, а
также вязкости пасты на гидрогенолиз угля
Разработан процесс каталитического пиролиза рас-
пыленных топлив под давлением водорода со специаль-
ным методом смешивания сырья и катализатора без
образования пасты. Степень превращения органиче-
ской массы углей и сланцев 91—97%
67
68
69
70
71
72
73
74
Давление, кгс/см2 Температура, °C Объемная скорость (проточная уста- новка), ч"1 Время контакта (автоклав), ч Катализаторы
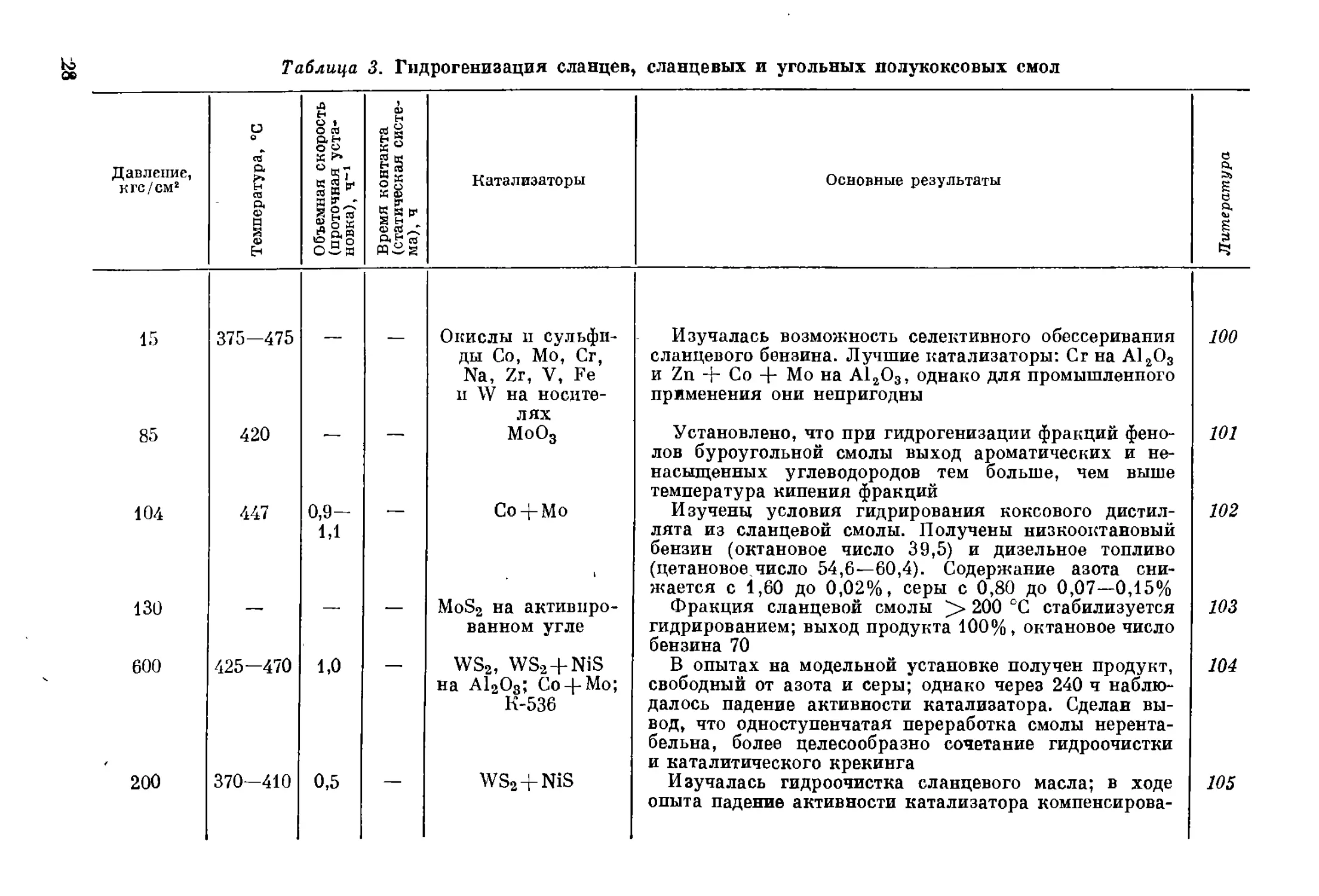
410 480—800 — До 15 мин Мо
410 800 — — Мо
410 400 — 15- 60 мин —
300 440 и 460 6 кг/ч — Байеровская масса
П родолжение табл. 2
Основные результаты
Литература
Изучено влияние времени пребывания угля в зоне
реакции на выход жидких и газообразных продуктов
при применении полунепрерывной аппаратуры для
быстрого нагрева и охлаждения. Уже за 40 с образуется
4,5% масла и 38% газа. С увеличением времени кон-
такта до 15 мин выход газа (93% метана, 7% этана)
вырастает до 80% без увеличения выхода масла. Де-
лается вывод о двухстадийности процесса — сначала
карбонизация, затем медленное превращение карбони-
зированного остатка в газ без образования масла
С целого подбора сырья для получения высококало-
рийного газа из углей испытана гидрогазификация
углей различных марок, полукокса и антрацита. Мак-
симальный выход газа (94%) получен из полукокса
(состав газа: 82—92% СН4, 8—15% С2Нв, 1—3% С3Нв).
Выход масла выше всего из лигиита. Масла выкипают
до 300 °C и содержат менее 4% асфальтенов. Из полу-
кокса и антрацита масло не образуется, из антрацита
получен коронен с небольшим выходом
Сравнивалась гидрогенизация облученного (10’ рад)
и необлученного угля. Установлено, что радиация
либо не влияет на выход газа и масла, либо немного
снижает его (на 4—10%); наибольшее снижение выхода
в случае битуминозного угля
В длительных пробегах на опытной установке подо-
браны оптимальные условия гидрогенизации япон-
76
77
78
До 170 До 925 — — —
300 445-465 0,6— 0,7 кг/л/ч — —
— — — — —
34-410 , 480— 1000 — От 1 ДО 15 мин ( Мо
ского угля. Степень конверсии 97,2%, выход легкого
масла, выкипающего до 300 °C, 71,9%, выход газа
22,1 %
Изучен состав продуктов гидрогенизации итальян-
ских углей. В бензиновой фракции обнаружены спирты,
альдегиды и кетоны, в среднем масле — фенантрен,
карбазол, пирен, метил- и диметилпирены, коронен
и бензперилен
В полупроточном реакторе изучалась гидрогазифика-
ция различных углей в присутствии водорода, водя-
ного пара и их смеси
Показано, что атомарный водород, полученный в элек-
трическом разряде, полностью восстанавливает уголь
и даже коронен до смеси метана, этилена и ацетилена.
Парафины, фенантрен и пирен образуют также высоко-
молекулярные остатки, богатые гидроароматическими
соединениями. В присутствии воды процесс ускоряется,
образуются окислы углерода
Изучены массоперенос при размешивании пасты
и влияние размешивания на процесс гидрогенизации
угля. На размешивание не влияет отношение водород :
: паста, но линейная скорость водорода должна быть
пропорциональна диаметру реактора
Параметр растворимости 6 = \U/v, где U — энер-
гия испарения, а и — мольный объем, предложено
разделить на полярную (X) и неполярную (т) составля- <
ющие, причем б2 = X2 + т2. Предложена формула рас-
чета выхода растворимой фракции
Дальнейшие эксперименты по гидрогазификации
углей при высоких температурах и малом времени кон-
такта. Выход газа (при 800 °C, 410 кгс/см2 и времени
контакта 1 мин) 43%, жидких продуктов 9% (при
15 мин — 68 и 6% соответственно). Нецелесообразно
увеличение скорости водорода, так как растет коли-
чество масла и содержание в нем асфальтенов. Замена
Мо на Fe, а также снижение доли Мо до 0,1 и 0,01%
неэффективны.
79
80
81
82
83
84
Давление, кгс/см* Температура, 'С Объемная скорость (проточная уста- новка), Ч”1 Время контакта (автоклав), ч Катализаторы
55-410 700—800 — До . 15 мин —
3-17-68 800— 1200 — До 15 мин Мо
— — — — —
— — — — Mo + Zn+Cr
Продолжение табл. 2
Основные результаты
Литература
С целью повышения экономичности высокотемператур-
ной гидрогазификации угля изучалась возможность
получения ценных побочных продуктов, в данном слу-
чае — коронена, наивысший выход которого достигнут
из антрацита (0,5—0,7% на ОМУ). Коронен 80%-ной
чистоты, в виде кристаллов
В полунепрерывной установке изучалась возмож-
ность гидрогазификации высокобитуминозного угля при
пониженных давлениях. При 800 °C и 17 кгс/см2 выход
газа составлял 56,5 м3/т при времени контакта не-
сколько секунд и 23 м3/т при времени контакта 15 мин.
Повышение давления в 4 раза увеличивало выход газа
до 56,5 и 198 м3/т соответственно. Однако даже при
3 кгс/см2 может быть превращено в газ 40—60% угля,
но при’ этом температуру следует поднять до 1200 °C
Исследован состав углеводородов гидрогенизата угля;
обнаружены н-алканы С8—С18 и 2-метилалканы С1о —
С18
Среднее масло, полученное гидрогенизацией угля,
переработано в бензин. Выход бензина 91—98% (на
масло) и 39—52% (на уголь). Часть бензина дополни-
тельно должна подвергаться гидрокрекингу
85
86
87
88
/ 70, 120 425 — — Sn; Zn; Ni
N5 СП — — — — —
Первое упоминание о разработке фирмой Hydrocar- bon Research процесса H-Coul 89
Сообщается о продолжающихся опытах Bureau of Mines (США) по гидрогенизации угля как при высоких (210 кгс/см2), так и при низких (70 кгс/см2) давлениях. Степень превращения 81—92% 90
Приводятся данные о применении процесса H-Coul для переработки битуминозных и суббитуминозных углей 91 .
Приведена предпроектная проработка завода по переработке угля. Головным процессом является H-Coul; гидрогенизат (средние фракции) экстраги- руется, рафинат гидроочшцается, далее риформинг, экстракция ароматических соединений, пиролиз. Мощ- ность завода 32 тыс. т угля в сутки или 8,4 млн. т/год (ОМУ). Продукция (в тыс. т/год): этилен — 508, про- пилен — 127, бутадиен — 254, бутены — 51, бензол — 7, толуол — 8,8, ксилолы — 13,5, ароматические углеводороды (Св) — 70, нафталин — 227, фенолы — 74,5, сера — 160, аммиак — 91. Срок окупаемости 4,5 года 92
Обработаны данные опытов термического растворения (см. “) и гидрогенизации углей. Показано, что превра- щение нерастворимой в бензоле части угля или выход экстракта являются функцией полярной части пара- метра растворимости X (см. и), дающей максимум при X = 9,5. Параметр X для бензола 9,2, для фенола 9,9, для тетралина 9,4. 93
Давление, кгс/см* Температура, °C Объемная скорость (проточная уста- новка), ч_| Время контакта (автоклав), ч Катализаторы If ' ,
200—300 425-500 Сульфиды Ni; Со и Мо на А12Од
— — — — —
Продолжение табл. 2
Основные результаты
Литератур*
| Сообщается, что фирмой Consolidation Coul по 94
правительственному заказу разработан и опробован
в масштабе пилотной установки (4,5 кг/ч) процесс ги-
дрогенизации угля, включающий экстракцию фрак-
циями гидрогенизата, отделение экстракта от остатка,
гидрирование экстракта и коксование остатка в псевдо-
ожиженном слое. Гидрирование осуществляется в реак-
торе с трехфазным псевдоожиженным слоем, подобно
процессу H-Oil гидрокрекинга нефтяных остатков
(см. табл. 4). Время пребывания жидкой фазы в реак-
торе 4 ч, степень превращения 85%, выход продукта
С4 — 400 °C 55—57% (на сухой уголь), остальное —
газы и цолукокс. Приведен расчет завода мощностью
9,5 тыс. м3/сутки
На опытной установке процесса Н-Coul (см. 89. 9|) pj
гидрировался (пробег 300 ч) австралийский бурый
уголь, содержавший 23,2 вес. % кислорода. Выходы
продуктов (вес. % на сухой уголь); СО + СО2 — 8,7,
Cj—С3 — 6,6, дистиллят, выкипающий от т. кип. бу-
танов до 524 °C — 48,8, остаток )> 524 °C — 8,3, вода
16,4, аммиак — 0,4, сероводород — 0,7, зола 8,3, не-
превращенный уголь 6,5. Степень превращения 93%,
суммарный выход жидких продуктов 0,62 м3/т сухого
угля. Полный набор процессов для предлагаемого за-
вода включает гидроочистку, гидрокрекинг и рифор-
минг гидрогенизата после процесса H-Coul. Дан рас-
чет завода мощностью 7950 м3/сутки
Лучшие перспективы имеют новые процессы, разработанные
в США фирмами Consolidation Coul и Hydrocarbon Research (про-
цесс H-Coul). В первом из них проблема отделения минеральной части
угля решена введением экстракции растворителем — переносчиком
водорода, которым служит тяжелая фракция следующей ступени —
гидрирования. Заметим, что теоретические основы экстракции твер-
дых горючих ископаемых были разработаны в СССР в так называ-
емом процессе термического растворения б5. Ступень гидрирования
решена совсем по-иному, чем в старом процессе гидрогенизации, —
использованы реактор с псевдоожиженным слоем, возможность вво-
дить активный катализатор и выводить отработанный для регенера-
ции, защита поверхности катализатора от отложений соли и метал-
лов при помощи импульсных струй жидкости 94. В процессе H-Coul
также применен реактор с псевдоожиженным слоем, позволяющий
отдельно выводить пары, жидкие продукты, катализатор и золу 95.
Оба процесса пока не реализованы в промышленности. Несом-
ненно, что чисто топливное направление переработки угля в этих
процессах будет иметь большие трудности, чем топливно-химическое.
Характерно, что в одном из вариантов использования процесса
H-Coul 92 рекламируется выделение из гидрогенизата ценных ком-
понентов (фенолов) с последующей переработкой гидрогенизата
процессами риформинга и пиролиза на ароматические и олефиновые
углеводороды. Таким образом, из данных табл. 2 можно сделать
вывод, что промышленного осуществления процесса гидрогенизации
углей пока нет, но в этом направлении достигнут значительный про-
гресс и накоплен большой опыт работы.
Еще большее число работ опубликовано по гидрогенизации слан-
цев и смол (см. табл. 3).
Из приведенных в табл. 3 данных видно, что наибольший интерес
исследователей вызывают сланцевые смолы, а наименьший — кок-
совые. Это находится в прямой зависимости от легкости гидрирова-
ния смол, убывающей в ряду: сланцевые > угольные полукоксо-
вые угольные коксовые 9в. Как и при гидрировании углей, наи-
большее внимание привлекает получение не столько топливных,
сколько химических продуктов, особенно фенолов *, а также низ-
ших ароматических углеводородов **. Это понятно, так как перера-
ботка смол дороже, чем переработка нефти, и поэтому желательно
получение более ценных, чем топливо, продуктов. Был разработан
ряд принципиальных технологических схем переработки сланцевых и
угольных смол на химические продукты и топлива ***. В этих схемах
помимо технологических приемов, позволяющих сохранять ценные
фенолы и ароматические углеводороды, применялись и специально
разработанные катализаторы 28> 190> 18s. Была осуществлена гидро-
генизация смолы, получаемой при пиролизе нефтепродуктов 198> 169.
* См. ссылки 106, 116, 117, 126, 134, 136, 137, 140, 142, 155, 156,170,
172, 173, 176, 181, 184 и др.
♦* См. ссылки 119, 130, 133, 140, 158, 160, 161, 178, 182.
*** См. ссылки 26, 126, 127, 140, 141, 155, 170, 182.
27
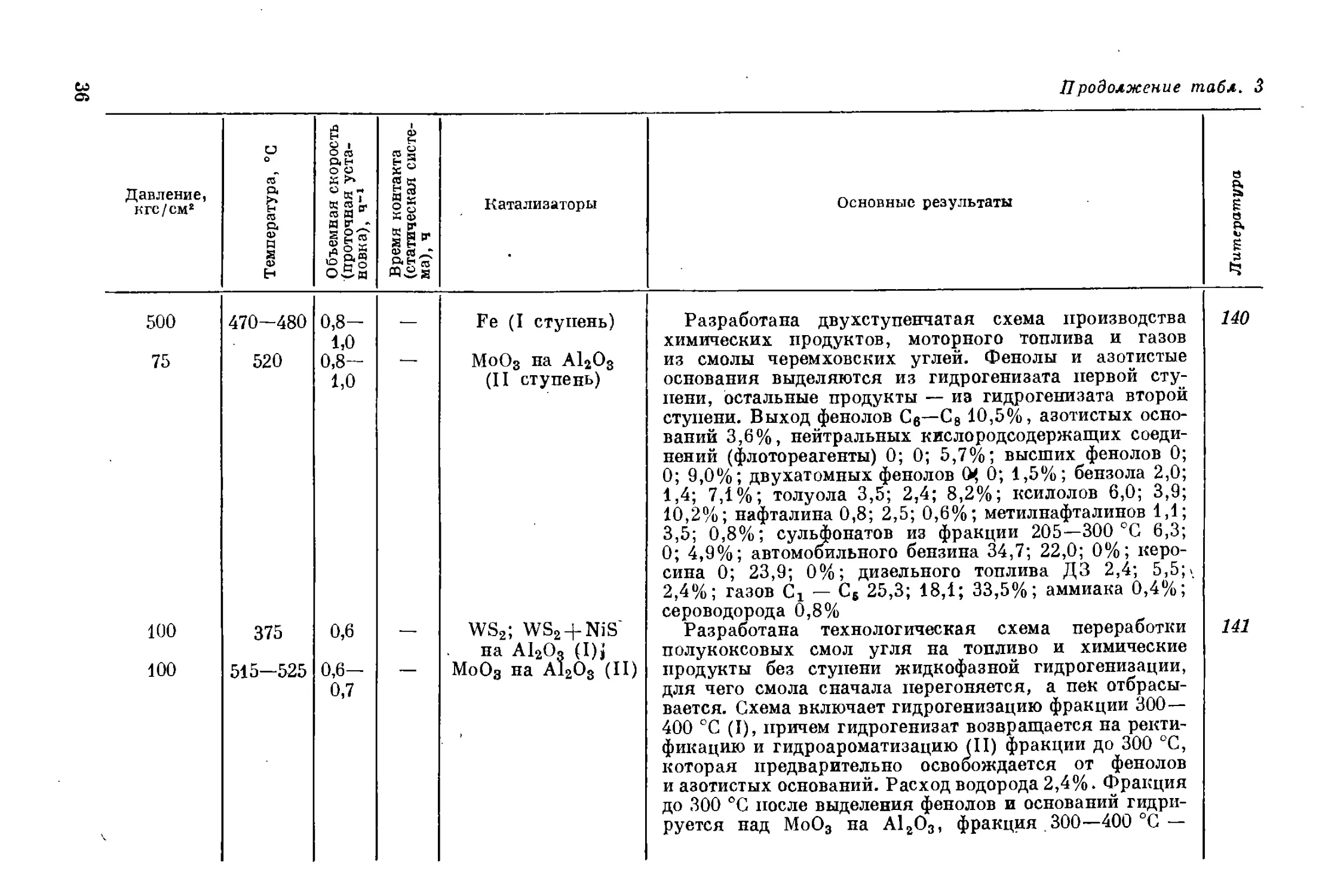
Таблица 3. Гидрогенизация сланцев, сланцевых и угольных полукоксовых смол
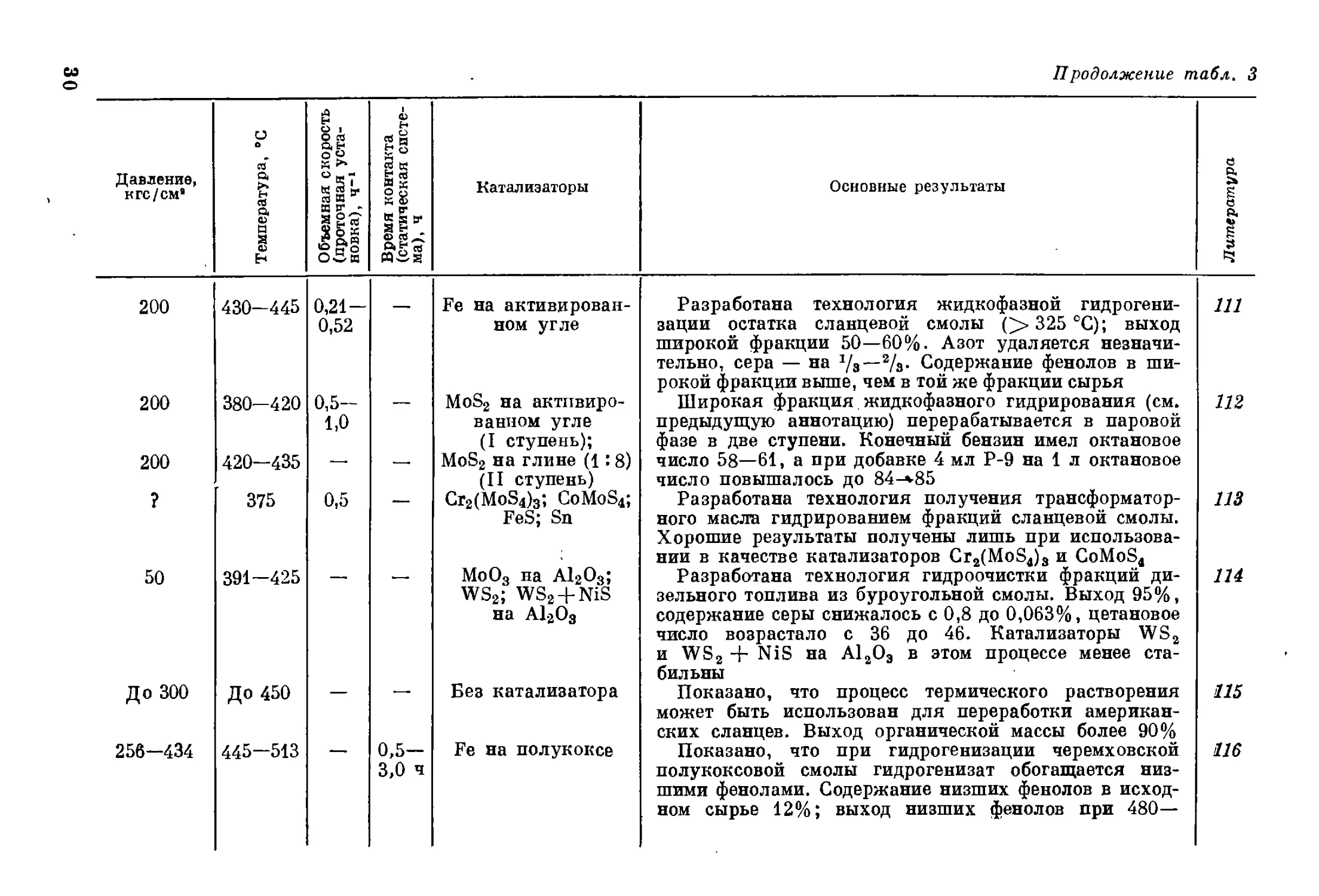
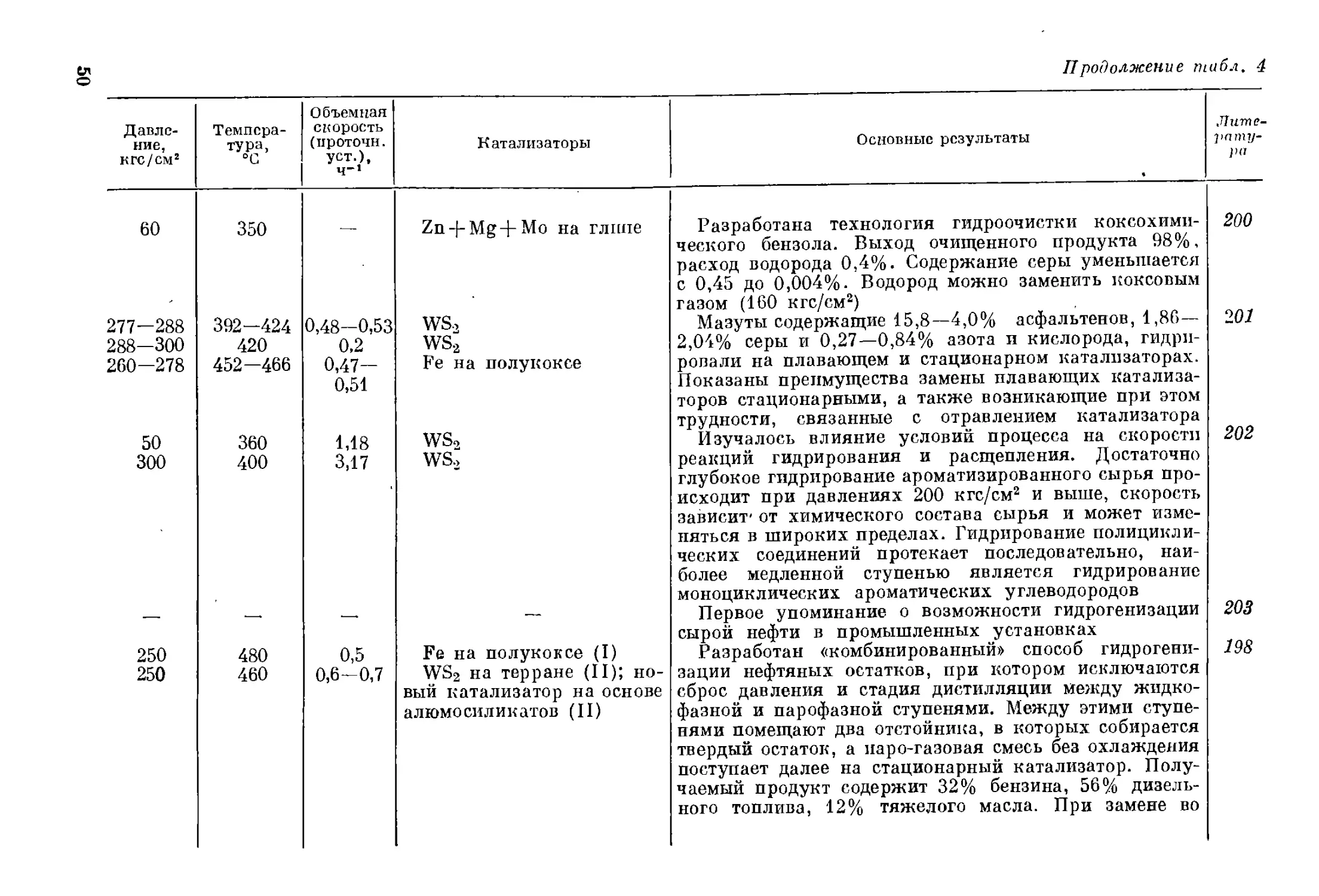
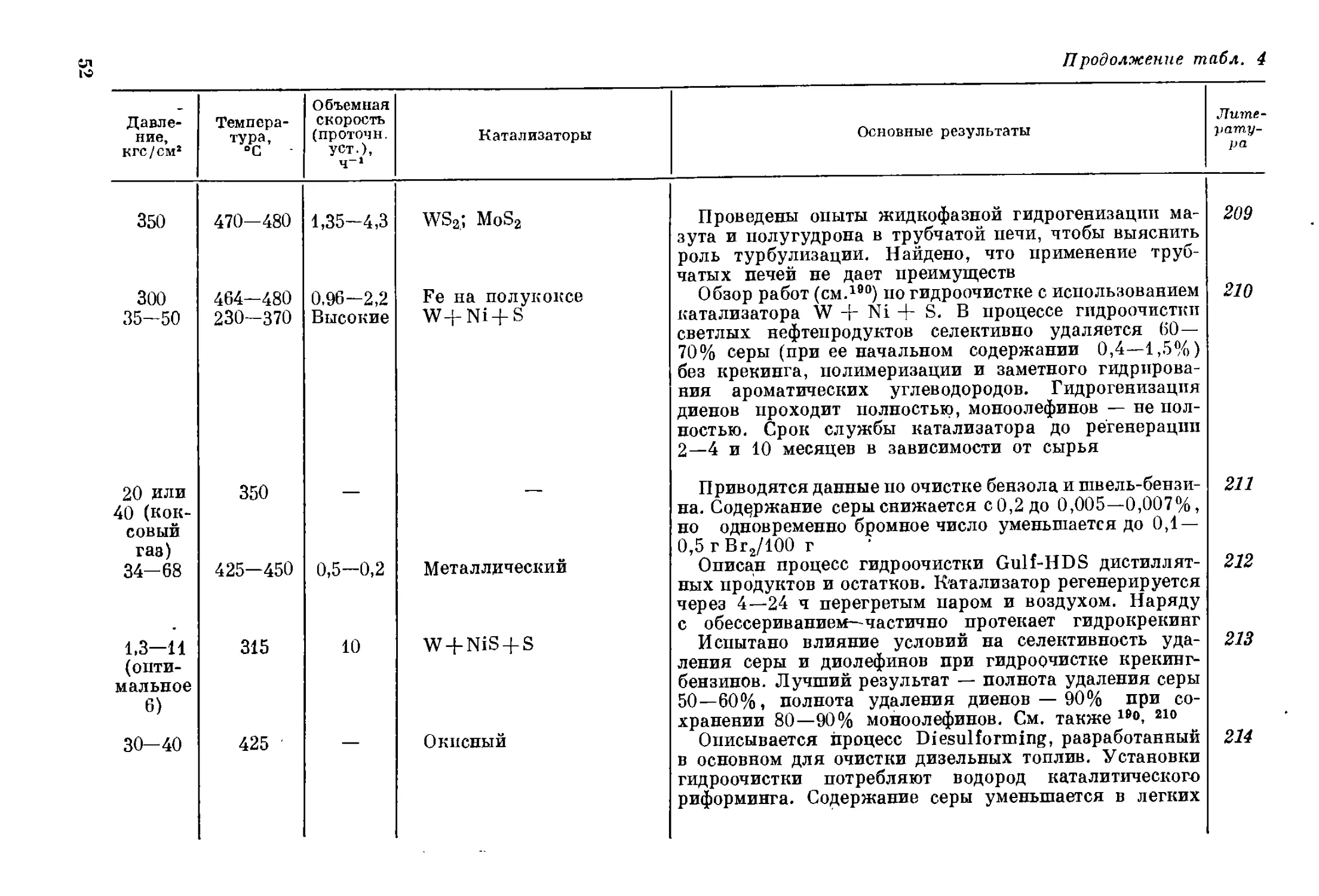
Давление, кгс/см2 Температура, °C Объемная скорость (проточная уста- новка), Ч"1 Время контакта (статическая систе- ма), ч Катализаторы Основные результаты Литература
15 375—475 — — Окислы п сульфи- ды Со, Мо, Сг, Na, Zr, V, Fe и W на носите- лях Изучалась возможность селективного обессеривания сланцевого бензина. Лучшие катализаторы: Сг на А1гО3 и Zn + Со + Мо на А1гО3, однако для промышленного применения они непригодны 100
85 420 — МоО3 Установлено, что при гидрогенизации фракций фено- лов буроугольной смолы выход ароматических и не- насыщенных углеводородов тем больше, чем выше температура кипения фракций 101
104 447 0,9- 1,1 Со -J- Мо Изучены условия гидрирования коксового дистил- лята из сланцевой смолы. Получены низкооктановый бензин (октановое число 39,5) и дизельное топливо (цетановое.число 54,6—60,4). Содержание азота сни- жается с 1,60 до 0,02%, серы с 0,80 до 0,07—0,15% 102
130 — — — MoS2 на активиро- ванном угле Фракция сланцевой смолы 200 °C стабилизуется гидрированием; выход продукта 100%, октановое число 103
600 425—470 1,0 — WS2, WS2 + NiS на А12О3; Со-|-Мо; К-536 В опытах на модельной установке получен продукт, свободный от азота и серы; однако через 240 ч наблю- далось падение активности катализатора. Сделан вы- вод, что одноступенчатая переработка смолы нерента- бельна, более целесообразно сочетание гидроочистки и каталитического крекинга 104
200 370—410 0,5 WS2+NiS Изучалась гидроочистка сланцевого масла; в ходе опыта падение активности катализатора компенсирова- 105
30 (начальное) 460-480 — — . WS2+V2S8; А12О3; МоО3; Fe2OE
200 392-450 0,32— 0,46 — Cr2O3; MoS3; MoS3 на глине; Ёе(ОН)2 на активированном угле
50
300
480—540
400
МоО3 на А12О3
(I ступень); WS2
(II ступень)
/ 300 300—500 — WS2; WS2+NiS на А12О3
200 375 0,16— 0,57 — Cr2(MoS4)3 в 25 %-ном этиленгликоле
лось повышением температуры на 0,5 °C в день. Содер-
жание серы понижалось с 0,90 до 0,10%, азота с 2,0
до 0,70%, а при максимальной температуре — соответ-
ственно до 0,05 и 0,10%
Наиболее высокий выход низших фенолов , (55—
57%) получается при гидрогенизации фракции высших
фенолов буроугольной смолы над катализатором
ws2 + v2s5
Отбензиненная сланцевая смола гидрировалась в про-
точной установке с плавающими катализаторами. Най-
дены условия, при которых предотвращается коксова-
ние и осаждение катализатора. Лучший катализатор —
MoSs, но более перспективен по экономическим сообра-
жениям Fe(OH)2 на активированном угле. Содержание
азота уменьшается с 1,23 до 0,64 (на Мо) и 0,79%
(на Fe)
Разработана двухступенчатая схема переработки
остатка (>200 ?С) перегонки продукта гидрирования
(ступень бензинирования) буроугольной смолы. Выход
бензина 45%, содержание в нем ароматических углево-
дородов 35%. Отбензиненный гидрогениаат первой
ступени гидрируется, давая реактивное топливо с т.
заст. —61 °C. Основными затруднениями на первой
ступени были отравление катализатора азотистыми со-
единениями и отложение кокса, которые были устра-
нены отмывкой циркуляционного газа и конструктив-
ным усовершенствованием реактора
Смесь 85% буроугольного дегтя и 15% легкого масла
гидрировалась до бензина. Максимальный выход бен-
зина 85% при 500 °C. Для получения бензина лучшим
катализатором является WS2, а для парафинистых
масел — WS2 + NiS на A12OS
Изучена гидрогенизация смолы подземного пиролиза
шведских сланцев. Выход бензина 32%, октановое число
66; дизельное топливо содержит 42% ароматических
углеводородов, при хроматографическом отделении ко-
торых его цетановое число повышается с 1Q до 55
106
107
108
109
110
co
о
Давление, к гс/см" Температура, ’С Объемная скорость (проточная уста- новка), ч-1 Время контакта (статическая систе- ма), ч Катализаторы
200 430-445 0,21 — 0,52 — Fe на активирован- ном угле
200 380-420 0,5- 1,0 — MoS2 на активиро- ванном угле (I ступень);
200 420-435 — — MoS2 на глине (1:8) (II ступень)
? 375 0,5 — Cr2(MoSi)3; CoMoS4; FeS; Sn
50 391-425 — MoO3 на A12O3; WS2; WS2 + NiS на A12O3
До 300 До 450 — Без катализатора
256-434 445—513 — 0,5- 3,0 ч Fe на полукоксе
Продолжение табл. 3
Основные результаты
Разработана технология жидкофазной гидрогени-
зации остатка сланцевой смолы (> 325 °C); выход
широкой фракции 50—60%. Азот удаляется незначи-
тельно, сера — на */а—2/3. Содержание фенолов в ши-
рокой фракции выше, чем в той же фракции сырья
Широкая фракция жидкофазного гидрирования (см.
предыдущую аннотацию) перерабатывается в паровой
фазе в две ступени. Конечный бензин имел октановое
число 58—61, а при добавке 4 мл Р-9 на 1 л октановое
число повышалось до 84-»85
Разработана технология получения трансформатор-
ного масла гидрированием фракций сланцевой смолы.
Хорошие результаты получены лишь при использова-
нии в качестве катализаторов Cr2(MoS4)3 и CoMoS4
Разработана технология гидроочистки фракций ди-
зельного топлива из буроугольной смолы. Выход 95%,
содержание серы снижалось с 0,8 до 0,063%, цетановое
число возрастало с 36 до 46. Катализаторы WS2
и WS2 + NiS на А12ОЭ в этом процессе менее ста-
бильны
Показано, что процесс термического растворения
может быть использован для переработки американ-
ских сланцев. Выход органической массы более 90%
Показано, что при гидрогенизации черемховской
полукоксовой смолы гидрогенизат обогащается низ-
шими фенолами. Содержание низших фенолов в исход-
ном сырье 12%; выход низших фенолов при 480—
111
112
113
114
115
116
150 375-450 —
40 408-493 —
50-57 485-527 0,5- 0,75
100 350-360 (I) 400—420 (Н) „ 1,2
25, 80 380-490 —
00
WS2; Fe на полу-
коксе
MoO3 на А12О3
То же
WS2+NiS на А12О3
(I реактор);
WS2 (II реактор)
Со + Мо на А12О3
117
490 °C и 300—340 кгс/см2 17,5%
Гидрировались кислотные и нейтральные компо-
ненты генераторной сланцевой смолы (> 325 °C). Пер-
вые склонны к коксообразованию, но дают большее
количество низших фенолов, вторые легче расщепля-
ются, дают большое количество соединений кислотного
характера
Гидрогенизация буроугольного дегтя при среднем
давлении может быть осуществлена в узких температур-
ных пределах: снижение температуры ухудшает рафи-
нирование, при повышении ее осаждается кокс. Однако
глубина гидрирования недостаточна. Для уменьшения
отложения кокса деготь смешивают с гидрированным
шпиндельным маслом. Возможна двухступенчатая
схема с повторной гидрогенизацией фракций до 350 °C.
Суммарный выход дизельного топлива 98%, цетановое
число дизельного топлива 36
Показана возможность предотвращения коксообра-
зования при гидрировании фракций буроугольных
смол путем установки форполимеризатора перед реак-
тором и использования фракций с более низкой темпе-
ратурой конца кипения (до 210 °C). В промышленных
условиях из легкого масла получено 84,29% гидро-
генизата. Бензин, полученный при 440 , 490 , 510
и 520 °C, содержал соответственно 26, 45, 51 и 53%
ароматических углеводородов; октановые числа бен-
зина соответственно 63, 71, 74, 76
Разработана технология гидроочистки фракций слан-
цевой смолы и продукта термического растворения
сланцев. Содержание серы понижается с 0,87 до 0,020—
0,04 (в бензинах) и 0,09—0,18% (в дизельном топливе).
Схема основана на применении двух реакторов; в реак-
торе II количество катализатора в 4 раза больше, чем
в реакторе I
Показана возможность гидроочистки сланцевого бен-
зина. Содержание серы понижается до 0,05%; выход
бензина 96% и выше. Однако из-за гидрирования оле-
финов октановое число бензина снижается
118
110
120
121
co
ьо
Давление, кгс/см’ Температура, °C i . Объемная скорость К(проточная уста- новка), ч-1 Время контакта (статическая систе- ма), ч Катализаторы
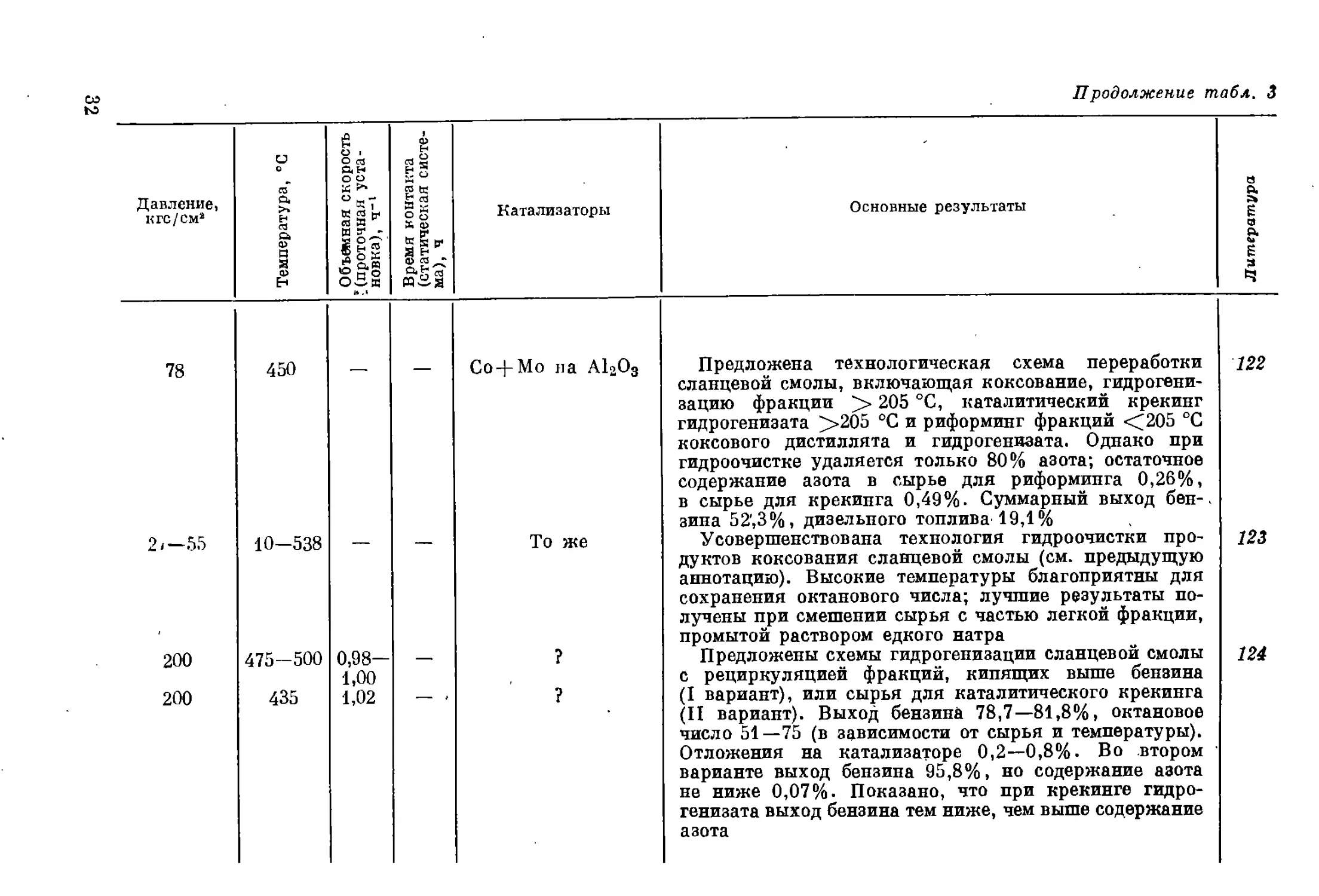
78 450 — — Co-J-Mo па А1гО3
2/—55 10-538 — — То же
200 475-500 0,98- 1,00 — ?
200 435 1,02 ?
Продолжение табл. 3
Основные результаты
Предложена технологическая схема переработки
сланцевой смолы, включающая коксование, гидрогени-
зацию фракции > 205 °C, каталитический крекинг
гидрогенизата >205 °C и риформинг фракций <205 °C
коксового дистиллята и гидрогенизата. Однако при
гидроочистке удаляется только 80% азота; остаточное
содержание азота в сырье для риформинга 0,26%,
в сырье для крекинга 0,49%. Суммарный выход бен-
зина 52',3%, дизельного топлива 19,1%
Усовершенствована технология гидроочистки про-
дуктов коксования сланцевой смолы (см. предыдущую
аннотацию). Высокие температуры благоприятны для
сохранения октанового числа; лучшие результаты по-
лучены при смешении сырья с частью легкой фракции,
промытой раствором едкого натра
Предложены схемы гидрогенизации сланцевой смолы
с рециркуляцией фракций, кипящих выше бензина
(I вариант), или сырья для каталитического крекинга
(II вариант). Выход бензина 78,7—81,8%, октановое
число 51—75 (в зависимости от сырья и температуры).
Отложения на катализаторе 0,2—0,8%. Во втором
варианте выход бензина 95,8%, но содержание азота
не ниже 0,07%. Показано, что при крекинге гидро-
генизата выход бензина тем ниже, чем выше содержание
азота
122
123
124
325
3 Заказ 271
50
100
100
100
350-400 — WS2 + NiS на А12О3
380-425 0,5 — ?
350— 360; 400 1,2- 1,5 — WS2 + NiS на А12О3
400-410 400 2,3 0,8 0,25 Никелевая руда (I) WS2 (II)
V ,
I
27
Описана технология гидрогенизационной перера-
ботки испанской сланцевой смолы; 50% сырья пере-
гоняется выше 350 °C и содержит 1% асфальтенов,
0,06% серы, 0,7% азота, 1,7% кислорода. Полнота
удаления серы, азота и кислорода 95—99%. Выход
гидрогенизата 98%; из него получают бензин, дизель-
ное топливо, парафин, смазочные масла
Описана технологическая схема переработки сланце-
вой смолы, включающая перегонку, термический кре-
кинг, коксование, гидроочистку, риформинг и поли-
меризацию. При гидроочистке содержание азота по-
нижается с 0,34 до 0,02%, но дизельное топливо имеет
высокую температуру застывания (0 °C)
Разработано несколько вариантов переработки сланца
и продуктов его термического растворения или полу-
коксования на моторное топливо, химические продукты
и газ; все варианты включают гидроочистку при пони-
женном давлении. Выход бензина на стадии гидроочи-
стки 95—98,2%, содержание серы понижается до 0,017—
0,042%
Показано, что органическая масса сланца гидри-
руется легче, чем сланцевая смола, подвергавшаяся
термическому воздействию. Принципиальная схема
включает жидкофазную гидрогенизацию с высокой
объемной скоростью (I), термоконтактную перегонку
шлама и гидростабилизацию широкой фракции (II).
Чисто топливный вариант дает 20,9% бензина, 41,1%
дизельного топлива, 23,9% газа, 5,9% полукокса;
топливно-химический — 16,6% бензина, 37,3% дизель-
ного топлива, 5,8% фенолов, 5,7% нейтральных кис-
лородсодержащих соединений, 22,1 % газа, 5,9% полу-
кокса. Разработанная схема характеризуется большей
производительностью аппаратуры (в 5—6 раз), чем
обычная схема гидрогенизации угля и сланцев под
давлением 300—700 кгс/сма
125
126,
127
128,
129
Давление,
кгс/см2
Катализаторы
Продолжение табл.
Основные результаты
S
о
а.
S
ч
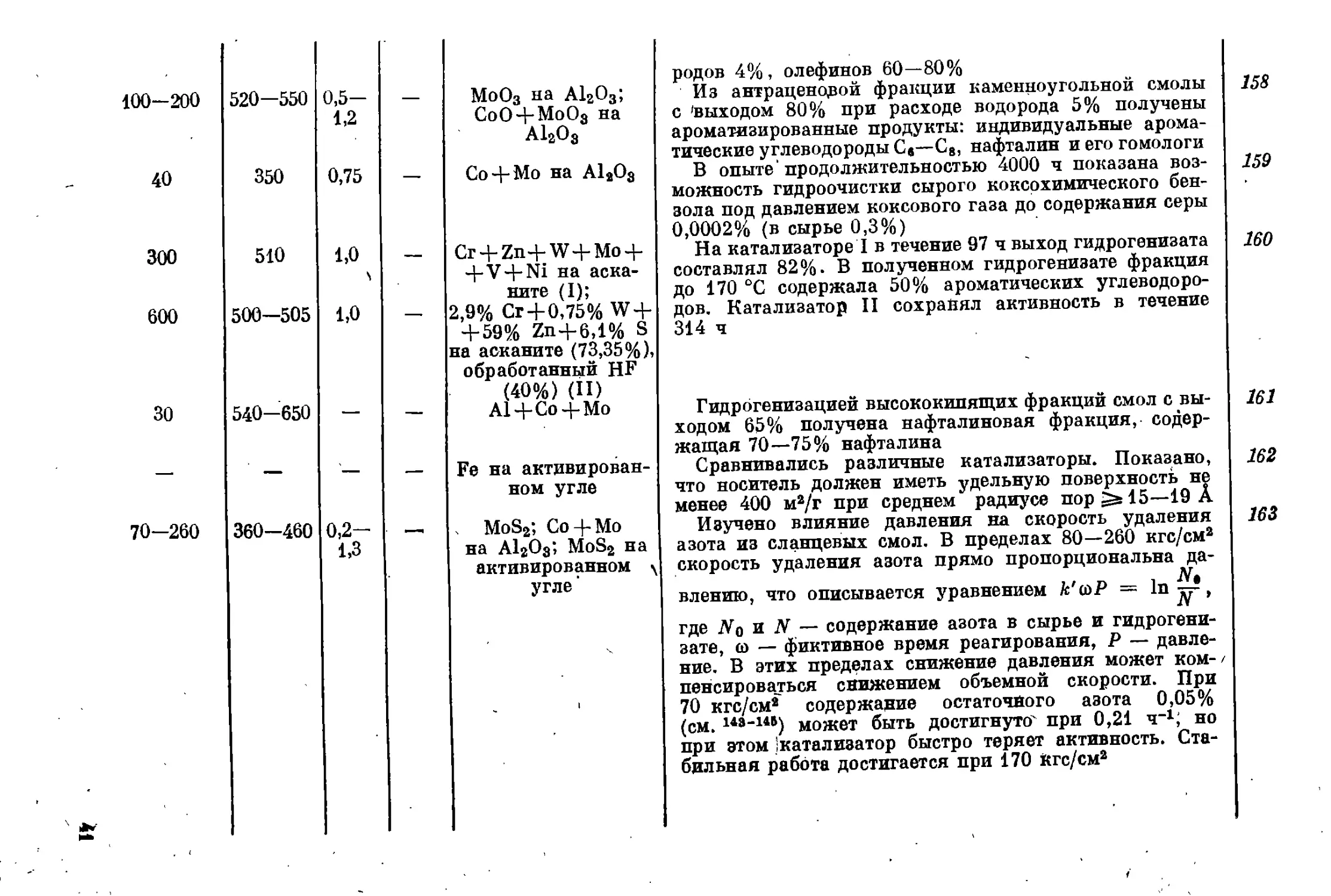
Оппсапо последовательное усовершенствование про-
цесса гидрогенизации креозотового масла, направлен-
ное на возможно более полное сохранение аромати-
ческих углеводородов. Сначала одноступенчатый про-
цесс (250 кгс/см2, 400 °C) на стационарном
катализаторе был разделен на две ступени (WS2,
385 °C и WS2 на терране, 400 °C), затем во второй
ступени был применен новый катализатор — Fe на
монтмориллоните, обработанном И F при 370 °C. Доля
ароматических углеводородов возросла сначала с 3
до 7, а затем до 15% В последнем варианте WS2 за-
менен Со -f- Мо на А12О3, a Fe па глине — никелем
па алюмосиликате. Меньшая гидрирующая активность
катализаторов позволила повысить содержание арома-
тических углеводородов до 53%
Изучено влияние температуры на глубину гидро-
генизации сланцевого масла с рециркуляцией фракций,
кипящих выше бензина. Содержание азота снижается
с 2.1 до 0,02—0,04% только при температуре выше
475 °C. При 475 °C выход гидрогенизата 86,6%, в том
числе бепзина 39,4% (72,4% на свежее сырье). Бензин
содержит 18% ароматических углеводородов. В газо-
вых продуктах преобладают метан и этан
Показана возможность получения белого вазелина
и церезина гидрированием смоляных продуктов
130
131
132
550—600 CO * 493-508 0,45— 0,5 Cr
' ? 300 — — MoS2; NiO
260 375—510 0,5— 2,0 — MoS2
240 460 2—4 Fe на активирован- ном угле
150 (начальное) 425 — 1,0 Fe
50 340- 350; 410—4z0 — __ MoO3 на А12О3 и новый катализа- тор
w
Разработан новый стабильный катализатор, отли-
чающийся хорошей селективностью, сохранения моно-
циклических ароматических углеводородов. Выход бен-
зина из широкой фракции черемховской смолы 60,53%
(68,2% с С5). Октановое число бензина 81,3, а с добавкой
1 мл Р-9 на 1 л — 86,8. Процесс пригоден и для нефтя-
ного сырья, в этом случае выход бензина 62,44% (68,4%
с С5), октановое число 85,2, а с добавкой Р-9 — 91,1
При низкотемпературном гидрировании фракций
нейтральных кислородсодержащих соединений слан-
цевой смолы образуется до 30% фенолов (на MoS2)
или до 60% (на NiO)
Проведен длительный (740 ч) пробег пилотной уста-
новки, показана возможность понижения содержания
азота с 1,01—1,08 до 0,04—0,06%. Выход жидких
продуктов до 98,1%, расход водорода 2,5%
Гидрировались три фракции фенолов из фушуньской
сланцевой смолы с преобладанием соответственно алкил-
фенолов, инданолов и нафтолов. Для первых более
характерно превращение в низшие фенолы, чем восста-
новление, для последних — наоборот. Инданолы за-
нимают промежуточное положение
При гидрогенизации кислотных и нейтральных ком-
понентов сланцевой смолы выход фенолов повышается
при снижении температуры и давления
Разработан новый катализатор для выходной ступени
процесса гидрогенизации среднего масла буроугольной
смолы при среднем давлении. Регенерация МоО3 на
А12О3 ведется в реакторе, коррозия под действием SO2
устраняется экстракцией катализатора перед регене-
рацией (см. также 11В, 122)
Установлено, что одной из причин дезактивирования
стационарных катализаторов при гидрогенизации чеш-
ских буроугольных смол является содержание в смо-
лах мышьяка. Мышьяк удаляется обработкой сырья
паровой фазы отработанным катализатором
133
134
135
136
137
138
139
Давление, кгс/см! Температура, °C Объемная скорость (проточная уста- новка), Ч~1 Время контакта (статическая систе- ма), ч Катализаторы
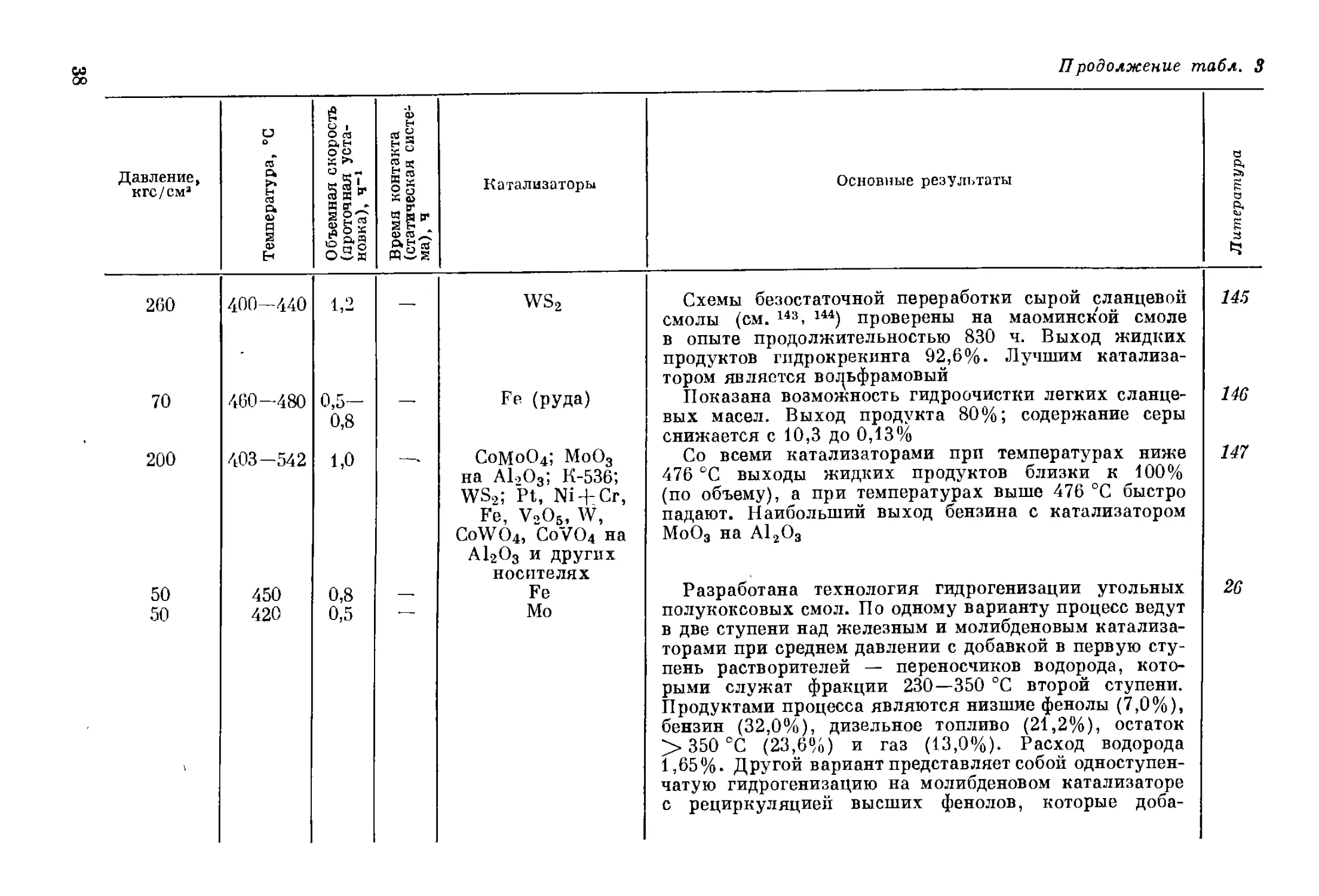
500 470-480 0,8— 1,0 — Fe (I ступень)
75 520 0,8- 1,0 МоО3 на А12О3 (II ступень)
100 375 0,6 — WS2; WS2-|-NiS на А12О? (I)j МоО3 на А12О3 (II)
100 515—525 0,6- 0,7
Иродолжение табл. 3
Основные результаты
Литература
Разработана двухступенчатая схема производства
химических продуктов, моторного топлива и газов
из смолы черемховских углей. Фенолы и азотистые
основания выделяются из гидрогенизата первой сту-
пени, остальные продукты — из гидрогенизата второй
ступени. Выход фенолов Cg—С8 10,5%, азотистых осно-
ваний 3,6%, нейтральных кислородсодержащих соеди-
нений (флотореагенты) 0; 0; 5,7%; высших фенолов 0;
0; 9,0%; двухатомных фенолов О, 0; 1,5%; бензола 2,0;
1,4; 7,1%; толуола 3,5; 2,4; 8,2%; ксилолов 6,0; 3,9;
10,2%; нафталина 0,8; 2,5; 0,6%; метилнафталинов 1,1;
3,5; 0,8%; сульфонатов из фракции 205—300 °C 6,3;
0; 4,9%; автомобильного бензина 34,7; 22,0; 0%; керо-
сина 0; 23,9; 0%; дизельного топлива ДЗ 2,4; 5,5;>.
2,4%; газов Сг — С6 25,3; 18,1; 33,5%; аммиака 0,4%;
сероводорода 0,8%
Разработана технологическая схема переработки
полукоксовых смол угля на топливо и химические
продукты без ступени жидкофазной гидрогенизации,
для чего смола сначала перегоняется, а пек отбрасы-
вается. Схема включает гидрогенизацию фракции 300—
400 °C (I), причем гидрогенизат возвращается на ректи-
фикацию и гидроароматизацию (II) фракции до 300 °C,
которая предварительно освобождается от фенолов
и азотистых оснований. Расход водорода 2,4%. Фракция
до 300 °C после выделения фенолов и оснований гидри-
руется над МоО3 на А12ОЭ, фракция 300—400 °C —
140
141
100-140 (начальное) 420-500 — 3 Fe на полукоксе
260 300—460 0,5— 3,0 — M0S2 i
70—260 360-460 0,35— 1,2 — WS2; MoSa; WS2 + NiS на A12Os; Co 4-Mo на A12O3;; MoS2 на активирог ванном угле
w
над WSa. Суммарный выход фенолов 14,4%, азотистых
оснований 1,1%, ароматических углеводородов 13,4%,
автомобильного бензина 28,4%, газов 10,2%, пека
30,0%
Показано, что с повышением температуры выкипания
гидрируемых фракций фенолов из черемховской полу-
коксовой смолы выход низших фенолов снижается.
Из фракций фенолов 100—125, 100—150 и 100—160 °C
(при 200 мм рт. ст.) получается соответственно 70,3,
50,5 и 49,0% фенол-крезольной фракции
Изучено влияние параметров процесса, выбраны
условия для двух режимов: гидроочистка (260 кгс/см2,
400 °C, объемная скорость 1,2 ч-1) и высокотемператур-
ное расщепление (260 кгс/см2, 440—445 °C, объемная
скорость 1,0—1,2 ч”1). Изучен механизм отравления
катализатора. Для предотвращения отравления необ-
ходимо разделение процесса на две ступени с обяза-
тельной очисткой в первой ступени до содержания азота
0,05%, для чего следует ступенчато повышать темпера-
туру от 400 до 460 °C. Обе ступени проверены в дли-
тельных О 700 ч) опытах. При суммарном расходе
водорода 3,5% общий выход товарных продуктов
(автомобильный бензин, дизельное топливо с т. заст.
—10 или —20 °C, авиационный керосин, веретенное
масло и др.) составил 87,8%
В процессах гидроочистки и гидрокрекинга фушунь-
ской сланцевой смолы (см. предыдущую аннотацию)
испытаны различные катализаторы. По активности их
можно расположить в следующий ряд: WS2 MoS, >
> WS2 -Н NiS на А12О8 > СоМоО4 на А12О8 > MoS2
на активированном угле. На активных катализаторах
можно .снижать давление, компенсируя зто уменьше-
нием объемной скорости. Общий выход жидких про-
дуктов до 90,9%. Температура застывания понижается
с повышением глубины расщепления, поэтому высокое
качество товарных продуктов обеспечивается при вы-
ходе фракции до 325 *С 83%
142
143
144
Давление» кгс/сма Температура, °C Объемная скорость (проточная уста- новка), ч-1 Время контакта (статическая систе- ма), ч Катализаторы
260 400-440 1,2 — ws2
70 460-480 0,5— 0,8 — Fe. (руда)
200 403 -542 1,0 СоМоО4; МоО3 на А1-,О3; К-536; WS,; Pt, Ni + Cr, Fe, V2O5, W, C0W04, СоУОд на A12O3 и других носителях
50 450 0,8 — Fe
50 420 0,5 Мо
Продолжение табл. 3
Основные результаты
е
е.
Схемы безостаточной переработки сырой сланцевой
смолы (см. 143, 144) проверены на маоминск'ой смоле
в опыте продолжительностью 830 ч. Выход жидких
продуктов гидрокрекинга 92,6%. Лучшим катализа-
тором является вольфрамовый
Показана возможность гидроочистки легких сланце-
вых масел. Выход продукта 80%; содержание серы
снижается с 10,3 до 0,13%
Со всеми катализаторами при температурах ниже
476 °C выходы жидких продуктов близки к 100%
(по объему), а при температурах выше 476 °C быстро
падают. Наибольший выход бензина с катализатором
МоО3 на А12О3
145
146
147
Разработана технология гидрогенизации угольных
полукоксовых смол. По одному варианту процесс ведут
в две ступени над железным и молибденовым катализа-
торами при среднем давлении с добавкой в первую сту-
пень растворителей — переносчиков водорода, кото-
рыми служат фракции 230—350 °C второй ступени.
Продуктами процесса являются низшие фенолы (7,0%),
бензин (32,0%), дизельное топливо (21,2%), остаток
> 350 °C (23,6%) и газ (13,0%). Расход водорода
1,65%. Другой вариант представляет собой одноступен-
чатую гидрогенизацию на молибденовом катализаторе
с рециркуляцией высших фенолов, которые доба-
26
300 341, 370 — — WS,
82—376 705 — 2,5 —
70-160 (начальное) 445-500 — 0,5— 3,0 Соединения Ее, So, Мо, Zn на носите- лях
260 360 —400 1,2 — WS2; MoS2; WS2 + N1S на А12Оа; Со-|-Мо на А12О3; '
100 (начальное) 400 — 1,0 MoS2 на активиро- ванном угле
70 380—460 0,8- 1,0 — 5871
вляются к разбавителю. По этой схеме выход низших
фенолов 12,1, легкого дизельного топлива 65,2, остатка
8,7, газа 13,6%
Снижение температуры при гидрогенизации чешских
буроугольных дегтей дает возможность в первой сту-
пени превращать двухатомные фенолы в одноатомные,
а во второй ступени — восстанавливать последние
в ароматические углеводороды. Выход ароматических
углеводородов повышается на 10% по сравнению
с обычной технологией. Из гидрогенизата выделяется
20,5% масел и 7,5% твердого парафина
Предложена технология исчерпывающей гидрогази-
фикации сланца для получения высококалорийного
газа, состоящего почти нацело из метана и пригодного
для дальнейшего газоснабжения
Испытано 33 катализатора для гидрирования в жид-
кой фазе высших фенолов из черемховской смолы.
Лучшими оказались SnCl2, SnCl2 на глине и Fe на
полукоксе, пропитанный серной кислотой. Лучший
выход низших фенолов составил 34,2—34,9% на исход-
ное сырье (фракция 125—180 °C при 20 мм рт. ст.)
Выяснялась возможность оценки катализаторов ги-
дроочистки сланцевдй смолы от соединений азота в про-
точной установке по опытам гидрогенолиза хинолина
в автоклавах. Показано, что порядки активностей в ос-
новном совпадают, образуя ряд WS2^MoS2>WS2+
+NiS на А12О3>Со-(-Мо на А12О3 (> MoS2 на активиро-
ванном угле, т. е. опыты с хинолином могут служить
простейшим критерием оценки активности катализато-
ров
Разработан катализатор для гидроочистки фракций
угольной смолы до 350—370 °C. Остатки коксуются,
коксовые дистилляты поступают на гидрогенизацию.
Из сырья выделяют фенолы. Выходы гидрогенизатов
83,65% (460 °C) — 92,34% (420 °C). Катализатор ста-
билен. Содержание серы уменьшается с 0,244 до 0,071 —
0,118%, содержание фенолов и оснований меняется
незначительно
148
14»
150
151
152
Давление, кгс/см2 Температура, °C 1 Объемная скорость 1 (проточная уста- новка), Ч“1 Время контакта (статическая систе- ма), ч Катализаторы
50-275 400-500 — — Re; RegS?
200 418-440 — — —
300 300 460-485 380-450 — — Fe на активирован- ном угле (I) WS2; WS2-|-NiS на А12О3 (II)
50 300—400 1,0 — WS2; Со + Мо на А12О3
40 380 1,5- 3,0 — 1% СоО+1% Р2О5 + + 10% МоО3 на А12О3 (88%)
Продолжение табл. 3
Основные результаты
Е
з
Сообщается об испытаниях в малых проточных уста-
новках рениевых катализаторов для гидрогенизации
буроугольной смолы. Катализаторы на кислотных
носителях оказались подходящими для расщепления,
давая бензин с октановым числом 73. При среднем
давлении бензин получается только из предварительно
очищенных фракций смолы
Показано, что качество сырья, получаемого из слан-
цевой смолы и используемого для крекинга, зависит
от остаточного содержания азота. При содержании
азота 0,14% и более сырье неудовлетворительное,
при 0,10% и менее — не отличается от прямогонного
легкого газойля
Предложена схема двухступенчатой переработки
сланцевой смолы для получения моторного топлива
и химических продуктов. Гидрогенизат первой сту-
пени, выкипающий до 325 °C, освобождается от фенолов
и азотистых оснований, после чего гидрируется во вто-
рой ступени
Смесь фенолов (73%) и нейтральных веществ (27%)
гидрировались в виде 10—20%-ного раствора в бензине.
Выход очищенных фенолов 82,5%, нейтральные веще-
ства восстанавливаются на 75%
Из бензиновой фракции полукоксовой смолы с вы-
ходом 97% получен гидрогенизат, освобожденный
от серы и кислорода на 90—95%, а от азота только
на 30%. Степень превращения ароматических углеводо-
153
154
155
156
157
100—200 40 520—550 350 0,5- 1,2 0,75 — МоО3 на А12О3; СоО + МоО3 на А1аО3 Со + Мо на A1SO3
300 510 1,0 — Cr + Zn+W+Mo + +V + NI на аска- ните (I);
600 500—505 1,0 2,9% Сг+0,75% W+ + 59% Zn+6,1% S на асканите (73,35%), обработанный HF
30 540-650 — — (40%) (II) AI+Co + Mo
— — — — Fe на активирован- ном угле
70-260 360-460 0,2- 1,3 MoS2; Со + Мо на А13О3; MoS2 на активированном \ угле'
родов 4%, олефинов 60—80%
Из антраценовой фракции каменноугольной смолы
с 'выходом 80% при расходе водорода 5% получены
ароматизированные продукты: индивидуальные арома-
тические углеводороды С»—С8, нафталин и его гомологи
В опыте'продолжительностью 4000 ч показана воз-
можность гидроочистки сырого коксохимического бен-
зола под давлением коксового газа до содержания серы
0,0002% (в сырье 0,3%)
На катализаторе I в течение 97 ч выход гидрогенизата
составлял 82%. В полученном гидрогенизате фракция
до 170 °C содержала 50% ароматических углеводоро-
дов. Катализатор II сохранял активность в течение
314 ч
75S
160
Гидрогенизацией высококипящих фракций смол с вы-
ходом 65% получена нафталиновая фракция, содер-
жащая 70—75% нафталина
Сравнивались различные катализаторы. Показано,
что носитель должен иметь удельную поверхность не
менее 400 м2/г при среднем радиусе пор ^15—19 А
Изучено влияние давления на скорость удаления
азота из сланцевых смол. В пределах 80—260 кгс/см2
скорость удаления азота прямо пропорциональна да-
влению, что описывается уравнением к'ц>Р = 1п^,
где Л'о и Л' — содержание азота в сырье и гидрогени-
зате, со — фиктивное время реагирования, Р — давле-
ние. В этих пределах снижение давления может ком- /
пенсироваться снижением объемной скорости. При
70 кгс/см2 содержание остаточного азота 0,05%
(см. 14э-1«б) может быть достигнуто' при 0,21 ч-1; но
при этом ;катализатор быстро теряет активность. Ста-
бильная работа достигается при 170 Кгс/см2
161
162
163
Давление, кгс/см2 Температура, °C Объемная скорость (проточная уста- новка), Ч“1 Время контакта ; (статическая систе- ма), ч Катализаторы
230-350 400—\ W — — MoS2; WS2
15 490 5с — 3% СоО + 5% MoOg на AloOg
170 300—400 — — —
50-70 425-500 — — С0М0О4 на А1,О3
40 280—300 (I); 380 -390 (II) 1,0 — Со + Мо на AloOg (I ступень); Мо03 па А12О3 (II ступень)
75 510 0,7— 0,75 — То же
Продолжение табл. 3
Основные результаты
Гидрировалась фракция дегтя 220—400'С. При исполь-
зовании для отвода тепла реакции холодного водорода
удалось получить без расщеплепия пергпдропроизводпые
ди- и полициклических ароматических углеводородов
Нафталиновая фракция коксовой смолы очищалась
до содержания 0,14 млн.-1 (против 0,82%) серы, азоти-
стых оснований 0,06% (против 1,5%) и полного отсут-
ствия кислот (2,5%). Выход продукта 93%
При гидрпрованпп в две стадии фракции 230 —
300 °C полукоксовой смолы с выходом 23% получено
реактивйое топливо с т. заст. —62 °C
Из нейтрального масла полукоксовой смолы получен
насыщенный продукт, содержащий менее 0,1% азота
и не содержащий серу
Фракции пиролизной смолы, выкипающие до 360 СС,
гидрировались в две ступени. Расход водорода 0,6—
1,2%, выход гидрогенизата 100—101 объемн.%. Полу-
чено 32,8% бепзола, 12,3% толуола, 7,2% этилбензола,
9,2% растворителя, 4,1% нафталина, 1,0% антрацена,
1,1% фенантрена, 16,5% ароматических углеводородов,
выкипающих при 230—360 °C, 7,9% насыщенных угле-
водородов с т. кип. 137 °C, 4,4% пека
Разработана двухстадийная схема производства хи-
мических продуктов из полукоксовой смолы черем-
ховских углей. После первой стадии извлекаются
164
165
166
167
168,
169
170
500 475-500 — 1,5 —
70 450-600 — — TiO2 на каолините, обработанный HF (I) А12О3, обработан- ный HF (II)
— — . — — WS2; MoS2
125 430 — — Co + Mo на A12O3
320 460 — 0,5 Fe(OH)2 на активи- рованном угле
200 — — — C0M0O4; Zn(CrO2)2
50 420—700 0,5— 1,6 — Co + Mo на Al2Og
л* fid 67—200 480 2,0 — To же
фенолы. Суммарный выход ароматических углеводоро-
дов, нафталина, растворителей и фенолов 62—66%,
газообразных парафинов 33—37%, смол 5,7—6,0%
Изучалась гидрогенизация асфа ль тенистого пека из
смолы битуминозного угля. Выход фракции фенолов
<' 250 °C не выше 5%
Смеси фенолов С7—С8 подвергались деалкилирова-
нию. Лучший выход фенола (13%) получен на катали-
заторе II
При гидрогенизации антраценового масла получено
реактивное топливо с высокой теплотворной способ-
ностью
При гидроочистке сырой сланцевой смолы удается
удалить 93% азота при расходе водорода 2,0%
Из пирокатехина и его гомологов, выделенных из
полукоксовой смолы, наибольший выход фенолов полу-
чен при использовании ароматических растворите-
лей
Гидрокрекинг сырой полукоксовой смолы дает 31 —
59% бензина, который после экстракции щелочью
и кислотой содержит менее 0,01% S и 0,01 % N
Из пековых дистиллятов при рециркуляции среднего
масла получается до 11,2% легких ароматических
углеводородов и до 25,7% нафталина
Установлено, что повышением давления можно пред-
отвратить дезактивацию катализатора, так как скорость
отложения кокса обратно пропорциональна давлению
в третьей степени
171
172,
173
174
175
176
177
178
179
Давление, КГС/СМ2j Температура, °C Объемная скорость (проточная уста- новка), Ч“* Время контакта (статическая сясте- ма), ч Катализаторы
140-385 ООО-760 — — —
500 70 — АЬО3 + 0,3% К, О
— — — — —
' — — — — МоО34-СоО на SiOg-f- AI2O3
— — — — С0М004
Продолжение тдбл. 3
Основные результаты
о
Й.
S
<3
а.
S
а
В лабораторном полупроточном реакторе осущест-
влена гидрогазификация сланца. При времени контакта
5 мин выход СН4 и С2Нв составляет более 90% (считая
на органическую массу сланца)
180
Каталитическое деалкилирование технических кре-
золов в фенол затрудняется отложением кокса на ката-
лизаторе, что может быть уменьшено добавкой щелочи
Описан процесс получения ароматических углеводо-
родов Св—С8 из коксового легкого масла (процесс
«литол»). В процессе осуществляются: полимеризация
стиролов (в форполимеризаторе), гидрогенолиз сер-
нистых соединений, гидрокрекинг, дегидрирование цик-
лоолефинов. Получаемый бензол содержит 0,5 млн.-1
тиофена
Описана модификация катализатора для гидрирова-
ния полукоксовой смолы. Оптимальное содержание
MoOjlO—12%
Выход жидких продуктов при гидрогенизации сырой
сланцевой смолы составил 89%, а при гидрогенизации
коксового дистиллята 97%. Полученные продукты
требуют дальнейшей переработки
181
182,
183
8S
70, 300 До 550 0,5- 2,0 — WS24-NiS на А12О3; МоО3 на А12О3; Со + Мо на А12О3; А12О3
70-140 480—537 — — —
50-100 350-430 0,2- 1,0 — WS2+NiS на А12О3
- 103 До 500 — — ws3
Приведены балансы распределения фенолов в про-
дуктах переработки буроугольной смолы. Выход низ-
ших фенолов (до 225 °C) удваивается при гидрогени-
зации фракции 225—280 °C
Разработан метод извлечения органической массы
сланца обработкой его горячим водородом под давле-
нием, что предотвращает карбонизацию. Выход смолы
111% от выхода в реторте Фишера (выход в процессе *
полукоксования 90—95%)
~ Показана возможность получения осветительного
керосина из полукоксовой смолы тощих углей. Гидри-
рование в две стадии, расход водорода 150 м3/т
Изучалось влияние температуры и давления на
скорость удаления серы, кислорода и азота из полукок-
совой смолы (фракция 200—325 °C). Повышение тем-
пературы до 400 °C ускоряет удаление очень быстро,
затем медленнее, полное удаление происходит при
500 °C
В докладе о планируемой разработке сланцевых
месторождений Бразилии (завод-прототип мощностью
2,2 тыс. т сланца в сутки в 1971 г., проектирование
крупного завода 108 тыс. т в сутки в 1973—1974 гг.)
упоминается, что в масштабе пилотной установки про-
ведено гидрирование смолы на промышленных и спе-
циально приготовленных катализаторах. Олефины, сера
и азот удаляются почти полностью
Отработана технология горной разработки и реторт-
ной перегонки сланцев (США). На будущих предпри-
ятиях сланцевая смола будет облагораживаться гидри-
рованием, возможна также гидрогазификация -
184
185
186,
187
188
28
189
Методы гидрогенизации использованы и для получения и очистки
нафталина 16В.
Большое внимание уделено получению наиболее ценных топлив,
в частности реактивного топлива. Эта задача достаточно сложна
при переработке сланцевых смол, которые содержат много углево-
дородов с прямыми цепями. Между тем для обеспечения необходимой
низкой температуры застывания (—60 °C) должна быть осуществлена
глубокая изомеризация этих углеводородов, которая затрудняется
в присутствии азотистых соединений. Только после тщательного
изучения этих взаимозависимостей удалось предотвращать отравление
катализаторов изомеризации и расщепления 143> 179 и получать реак-
тивное топливо удовлетворительного качества 144 166- 186- 187. Следует
отметить еще одно интересное направление — исчерпывающее гид-
рирование ароматизированной фракции каменноугольных смол с
целью получения полициклических нафтеновых углеводородов, от-
личающихся высокой теплотворной способностью и, следовательно,
представляющих интерес как топливо для ракетных двигателей 164- 174.
Совершенствовалась и технология гидрогенизационной перера-
ботки смол. Здесь, как и в случае гидрогенизации углей, наблюдается
отход от традиционной трехступенчатой технологии деструктивной
гидрогенизации и стремление всемерно упростить технологические
схемы путем сокращения числа ступеней * и снижения давления **.
Выяснены зависимости между давлением и скоростью основных реак-
ций процесса 163- 1Г9. Практически можно легко ориентироваться в вы-
боре давления, с тем чтобы найти разумный компромисс между удо-
рожанием процесса из-за применения более сложной аппаратуры
высокого давления и обеспечением нужных скоростей реакций и пред-
отвращением отравления катализаторов. Возможность защиты ката-
лизаторов при переработке сланцевых смол позволила сократить
или полностью устранить самую неэффективную стадию традици-
онной технологии — жидкофазное гидрирование с плавающим ката-
лизатором, заменив ее гидрированием на активных стационарных
катализаторах ***.
Применение стационарных катализаторов позволяет снизить тем-
пературу процесса и уменьшить расход водорода. Однако при этом
снижается выход химических продуктов, так как именно в жидкофаз-
ном процессе в присутствии малоактивных плавающих катализа-
торов реакции восстановления кислород- и азотсодержащих функ-
циональных групп протекают с умеренной скоростью, сравнимой
со скоростью расщепления сырья с образованием ценных легких
продуктов. Очевидно, что выбор между более или менее активными
катализаторами должен решаться в каждом отдельном случае в за-
висимости от целей процесса и характера сырья. Применительно
к технологическим целям, изложенным выше, подбор катализаторов
* См. ссылки 126, 135, 140, 141, 170, 182.
** См. ссылки 108, 118, 128, 129, 138, 141, 153.
*** См. ссылки 122, 125, 126, 143-145, 152.
46
был направлен на обеспечение возможно более полного сохранения
ценных ароматических углеводородов. Для этого понижали гидри-
рующую активность катализаторов по отношению к этим углеводо-
родам при сохранении или даже повышении расщепляющей актив-
ности и способности гидрировать ди- и полициклические угле-
водороды. Применение новых, более селективных катализаторов
позволяет обеспечить более высокий выход ароматических угле-
водородов 130> 1331 1в0, 183.
Если ароматические углеводороды во многих случаях удается
селективно сохранить, то задача селективного гидрирования с сохра-
нением олефиновых связей еще не решена "> 121. Между тем возмож-
ность получения чистых высших а-олефинов, содержащихся в сланце-
вых смолах в большом количестве, представляет большой
интерес для нефтехимии, например для синтеза поверхностно-
активных веществ. Другой трудностью гидроочистки сланцевых
продуктов является необходимость большой полноты удаления
азота 104> 108’ 120, 122’ 1241 131, даже небольшое количество которого
резко понижает эффективность каталитического крекинга очищенных
продуктов 124> 1М. Для очистки от азота требуется применение
наиболее активных катализаторов 127> 153 или высоких темпера-
тур 131-
Продолжают разрабатываться упрощенные технологические ва-
рианты переработки сланцев: термическое растворение 12в’ 127 и гид-
рогазификация 14 9> 18 °> 18 9.
Общим выводом из рассмотрения результатов работ по гидроге-
низации сланцев и смол является то, что достигнут значительный
прогресс в совершенствовании технологии, в изучении химических
и кинетических закономерностей. Практически проверены и подго-
товлены к внедрению не только топливно-химические, но и химико-
топливные схемы переработки, и их реализация зависит только от
экономической конъюнктуры. По-видимому, из числа стран, исполь-
зующих гидрогенизацию смол, в ближайшие годы наибольшие темпы
развития этих процессов будут в Бразилии 28. '
В табл. 4 сопоставляются работы по гидрогенизации нефти и неф-
тепродуктов.
Как указывалось выше (см. стр. 12) и как видно из данных, при-
веденных в табл. 4, наибольшее развитие в последние годы получили
процессы гидроочистки, применяемые в настоящее время для удале-
ния серы не только из бензинов и прямогонных дизельных топлив,
но и для очистки масляных фракций, нефтяных остатков, сырой
нефти, т. е. практически любых видов сырья, полупродуктов и про-
дуктов нефтепереработки.
Имеется много различных фирменных модификаций процессов
гидроочистки дистиллятных продуктов. Они очень близки и разли-
чаются только деталями приготовления катализаторов и некоторым
варьированием условий процесса. Практически все катализаторы
гидроочистки представляют собой йлюмокобальтмолибденовые или
алюмоникельмолибденовые композиции 43 °. Эти катализаторы хотя
47
00
Таблица 4. Гидрогенизация нефти, продуктов переработки нефти и другого углеводородного сырья
Давле- ние, кгс/см! Темпера- тура, °C Объемная скорость (проточи, уст.), ч-1 Катализаторы Основные результаты Лите- ратур ра
50 343 10,0 W-f-Ni + S (40%W, 25%Ni) Приведены результаты гидроочистки различных неф- тепродуктов: легкий крекинг-бензин — содержание се- ры уменьшается с 0,065 до 0,0013%, бромное число с 56 до 5 г Вг2/100 г; тяжелый газойль — соответственно с 0,26 до 0,002%, с 75 до 8,4; бензин соответственно с 0,51 до 0,008%, ароматизированный дистиллят с 0,08 до 0,003%, с 28 до 0,5. Расщепление практиче- ски не происходит, ароматические углеводороды не затрагиваются, обессеривание протекает несколько быстрее гидрирования олефинов, сохранить которые, однако, не удается 190
15 400 0,4 Со-(-Мо на боксите (I); Zn-|-Mo на боксите (II) При гидроочистке сырой нефти более активен ката- лизатор I: содержание серы снижается с 2,08 до 0,17%, тогда как в случае катализатора II — лишь до 0,32% 191
50 370 1,0 Состав не известен (не- чувствительный к сере)’ Содержание серы в циркулирующем масле каталити- ческого' крекинга уменьшалось от 1,42 до 0,15%. При этом происходило заметное гидрирование аромати- ческих колец (число ароматических атомов на молекулу при нейзменяющемся молекулярном весе 208—209 уменьшается с 11,5 до 8,8, неароматических — возра- стает с 3,8 до 6,9), протекающее за счет бициклических ароматических углеводородов. Для полного насыще- ния ароматических углеводородов необходимо давле- ние 200 кгс/см2 192
10 50 415-425 413 — МоО3 на А12О3 W-j-Ni-1-S Гидроочистке подвергался продукт адсорбционного разделения легкого крекинг-бензина, содержавшего ароматические углеводороды, олефины и 3,4—4,5% серы. Остаточное содержание серы 0,05%, водород расходуется на обессеривание и насыщение олефинов в отношении 3:1, ароматические углеводороды не за- трагиваются 193
4 Заказ 271
20-25 300-450 — 1
1,5-2,0 450-480 —
50 375 —
550 480 0,7
300 327 —
600 460 0,8-1,0
275 - 460. 0,45
50-100 340-380 0,4—0,9
МоО3 на А12О3 (I)
То же (II)
Co-f-Mo
WS2; WS24-NiS на А120
Fe
WS2 на терране
WS2 на терране
Новый катализатор в
основе алюмосиликатов
MoS3 на глнне (1 : 3)
з!
ta
Катализатор дегидрирования, содержащий 60—80 г
МоО3 на 1 л носителя, может быть применен для гидро-
очистки. При режиме I удаляются олефины, сера, азот
и кислород, а ароматические углеводороды сохраня-
ются; при режиме II — сохраняются также и олефины
Проведены опыты гидроочистки тридекана, к ко-
торому добавлялись сернистые соединення до содер-
жания серы 1,4%. Октадецилмеркаптан, диоктилсуль-
фид, бензилоктилсульфид и фенилдецилсульфид вос-
станавливаются почти полностью (до 0,01—0,02% S),
несколько больше остается бензтиофена (0,05% S).
Ароматические кольца гидрируются незначительно
Описан первый вариант переработки нефтяных остат-
ков (выкипающих выше 360 °C) по трехступенчатому
«угольно-смоляному варианту». В первой ступени осу-
ществлялась циркуляция гидрогенизата, кипящего
выше 360 °C; выход бензина 13,2%, широкой фрак-
ции 74,5%. Во второй ступени выход жидкого гидро-
генизата по широкой фракции 97,4%, в третьей —
95,3%. Общий выход бензина 82% (октановое число 64,
с добавкой 0,04% ТЭС — 78,5), выход газов (С2—С4)
8,1%
Найдено, что более целесообразно объединение двух
парофаэных ступеней в одну и переработка .суммар-
ного жидкофазного гидрогенизата (процесс Kombiver-
f ahren), который содержит 48% фракций, кипящих выше
360 °C. За один проход образуется до 50% бензина.
При рециркуляции фракций выше 180 °C произво-
дительность установки повышается на 27%
Суммарный жидкофазный гидрогениэат (см. преды-
дущую аннотацию) перерабатывался над более эффек-
тивным катализатором при меньшем давлении; окта-
новое число бензина с 0,04% ТЭС равно 71—72
Показана возможность гидроочистки коксохимиче-
ского бензола; содержание серы снижается с 0,249
до 0,0028—0,0046%, фактических смол с 14 до ,1 —
7 мг/100 мл
194
195
196
197,
198
197,
198
199
ел
ф
Давле- ние, кгс/см2 Темпера- тура, °C Объемная спорость (проточи, уст.), Ч-1 Катализаторы
60 350 — Zn-f-Mg+Mo на глине
277-288 392-424 0,48-0,53 WS3
288—300 420 0.2 ws2
260-278 452-466 0,47- 0,51 Fe на полукоксе
50 360 1,18 WS,
300 400 3,17 ws.>
. . —
250 480 0,5 Fe на полукоксе (I)
250 460 0,6-0,7 WS2 на терране (II); но- вый катализатор на основе алюмосиликатов (II)
П родолжение табл. 4
Основные результаты
Лите-
1>а ту-
ра
Разработана технология гидроочистки коксохими-
ческого бензола. Выход очищенного продукта 98%,
расход водорода 0,4%. Содержание серы уменьшается
с 0,45 до 0,004%. Водород можно заменить коксовым
газом (160 кгс/см2)
Мазуты содержащие 15,8—4,0% асфальтенов, 1,86—
2,04% серы и 0,27—0,84% азота и кислорода, гидри-
ровали на плавающем и стационарном катализаторах.
Показаны преимущества замены плавающих катализа-
торов стационарными, а также возникающие при этом
трудности, связанные с отравлением катализатора
Изучалось влияние условий процесса на скорости
реакций гидрирования и расщепления. Достаточно
глубокое гидрирование ароматизированного сырья про-
исходит при давлениях 200 кгс/см2 и выше, скорость
зависит' от химического состава сырья и может изме-
няться в широких пределах. Гидрирование полицикли-
ческих соединений протекает последовательно, наи-
более медленной ступенью является гидрирование
моноциклических ароматических углеводородов
Первое упоминание о возможности гидрогенизации
сырой нефти в промышленных установках
Разработан «комбинированный» способ гидрогени-
зации нефтяных остатков, при котором исключаются
сброс давления и стадия дистилляции между жидко-
фазной и парофазной ступенями. Между этими ступе-
нями помещают два отстойника, в которых собирается
твердый остаток, а наро-газовая смесь без охлаждения
поступает далее на стационарный катализатор. Полу-
чаемый продукт содержит 32% бензина, 56% дизель-
ного топлива, 12% тяжелого масла. При замене во
200
201
202
203
198
50 2,0
20-50 350-390 — Со + Мо на AI2O3
35 320-350 —
50-70 400—460 —
6,8—68 415-427 0,5—5,0 Со + Мо на AloOg (обра- ботанный фтором)
27 13,5 400 480 0,25 0,8 V2Os (I) V2OS (II)
втором реакторе катализатора WS2 на терране «но-
вым» катализатором содержание бензина повышается
до 42%, а содержание дизельного топлива и тяжелого
масла составляет соответственно 51 и 7%. Одновременно
повышается качество бензинов. При бензинировании
газойля над катализатором WS2 на терране бензин
содержит 8% ароматических углеводородов и имеет
октановое число 76 (с 0,04% ТЭС), а над новым ката-
лизатором — соответственно 17,5% и 83
Осуществлена гидроочистка прямогонного газойля
и сырой нефти. Содержание серы снижается соответ-
ственно с 0,75 до 0,04—0,22% и с 1,50 до 0,35—0,48%
Разработана технология гидроочистки нефтепродук-
тов, отличающаяся от других методов тем, что созда-
ются условия для сохранения части сырья в жидкой
фазе. Жидкость стекает по большому числу слоев
катализатора, циркулирующий водород просачивается
через нее. Улучшение условий контакта водорода
с сырьем позволяет снизить количество циркулиру-
ющего газа и тем самым удешевить процесс на 15—
20%
Сообщается об очистке сырого бензола под давлением
коксового газа
- Обосновывается нецелесообразность повышения кон-
центрации водорода, растворенного в сырье, за счет
повышения давления. Более эффективна добавка жид-
ких переносчиков водорода, например тетралина;
в реакцию входит 84% водорода из тетралина и только
15% водорода из газовой фазы
Разработана технология гидроочистки продуктов,
выкипающих до 360 °C, водородом, выделяющимся
при дегидрировании нафтенов. Выходы продуктов
96—99,7%, содержание серы снижается с 0,081 —
1,43 до 0,00007-0,36%
Изучалась гидроочистка бензинов, содержащих оле-
фины; в мягких условиях (режим I) обессеривание
сопровождается гидрированием, в жестких (режим II) —
олефины частично сохраняются
198
204
205
206
207
208
Давле- ние, кгс/см2 Темпера- тура, °C Объемная скорость (проточи, уст.), ч-1 Катализаторы
350 470-480 1,35-4,3 WS2; MoS2
300 464—480 0,96-2,2 Fe на полукоксе
35—50 230—370 Высокие W+Ni + S
20 или 40 (кок- совый газ) 350 — —
34-68 425-450 0,5-0,2 Металлический
1,3-11 (опти- мальное 6) 315 10 W + NiS + S
30-40 425 — Окисный
Продолжение табл.
Основные результаты
Лите
нату-
ра.
Проведены опыты жидкофазной гидрогенизации ма-
зута и полугудрона в трубчатой печи, чтобы выяснить
роль турбулизации. Найдено, что применение труб-
чатых печей не дает преимуществ
Обзор работ (см.190) по гидроочистке с использованием
катализатора W + Ni + S. В процессе гидроочистки
светлых нефтепродуктов селективно удаляется 60—
70% серы (при ее начальном содержании 0,4—1,5%)
без крекинга, полимеризации и заметного гидрирова-
ния ароматических углеводородов. Гидрогенизация
диенов проходит полностью, моноолефинов — не пол-
ностью. Срок службы катализатора до регенерации
2—4 и 10 месяцев в зависимости от сырья
Приводятся данные по очистке бензола и швель-бензи-
на. Содержание серы снижается с 0,2 до 0,005—0,007%,
но одновременно бромное число уменьшается до 0,1 —
0,5 г Вг2/100 г
Описан процесс гидроочистки Gulf-HDS дистиллят-
ных продуктов и остатков. Катализатор регенерируется
через 4—24 ч перегретым паром и воздухом. Наряду
с обессериванием—частично протекает гидрокрекинг
Испытано влияние условий на селективность уда-
ления серы и диолефинов при гидроочистке крекинг-
бензинов. Лучший результат — полнота удаления серы
50—60%, полнота удаления диенов — 90% при со-
хранении 80—90% моноолефинов. См. также 19°, 210
Описывается процесс Diesulforming, разработанный
в основном для очистки дизельных топлив. Установки
гидроочистки потребляют водород каталитического
риформинга. Содержание серы уменьшается в легких
209
210
211
212
213
214
20—300 400—460 — Fe на активированном угле
70 450 0,6 То же (оптимальные условия)
148-
зго*
9
(парци-
альное
водоро-
да)
200—300
390
370—410
280-380
0,4-0,9
Ni
Со-)-Мо на А12О3
20-40 380-400
Со-|-Мо на А12О3
сл
со
* Время контакта (стационарная установка) 0,5 я.
дизельных топливах с 1,32 до 0,21%, в тяжелых ди-
зельных топливах — с 1,77 до 0,37%, в котельных то-
пливах — с 2,2 до 0,35 и с 1,02 до 0,105%. Выходы
топлив соответственно 97,3, 98,6, 98,4 и 96,2%
Разработан процесс деструктивной гидрогенизации
венгерской смолистой нефти, содержащей 14,9% ас-
фальтенов. Найдено, что расщепление ускоряется
в первую очередь повышением температуры; продукты
расщепления не успевают гидрироваться растворен-
ным водородом даже при 300 кгс/см2, но в присутствии
доноров водорода (тетралин или фракция гидрогени-
зата) образуют очень мало кокса, выносимого из реак-
тора вместе с катализатором. В оптимальных условиях
образуется 70% перегоняющихся продуктов (против
20% из сырой нефти) и только 0,2% кокса; одновре-
менно удаляется половина (из 3,5%) серы
Проведены опыты по гидрокрекингу высокоаромати-
зированной нефти. Выходы бензина 48,9—56,9%, со-
держание серы уменьшается с 4,57 до 0,29 и с 2,37
до 0,15%. Отношение количеств сырья и катализатора
1:1
Разработан процесс автогидроочистки керосиновых
и газойлевых фракций. Длительность Цикла соот-
ветственно 600 и 200 ч, полнота удаления серы 90—
95 и 60—80%. Отложение кокса на катализаторе 5—
7%, катализатор легко регенерируется
Разработан процесс гидрирования бензола до цикло-
гексана с незначительной изомеризацией. Степень пре-
вращения бензола до 95,5—97,0%, чистота поруча-
ющегося циклогексана 93—97%
При гидроочистке керосиновых дистиллятов восточ-
ных нефтей СССР срок службы катализатора более
2000 ч, этот срок сокращается при понижении давле-
ния водорода (см. 218, 21в) ।
215,
216
217
218,
219
220
221
Давле- ние, кгс/см* Темпера- тура, °C Объемная скорость (проточи, уст.), Ч“* Катализаторы
€-30 378—455 0,5-1,2 МоО3 па А12О3; Сг2О3 на А12О3; NiO на А12О3
350 470 2,5 Fe (I)
250 390—400 2,0 WS2 (II)
50 700- 900 — —
Продолжение табл. 4
Основные результаты
Лите-
рату-
ра
Разработана технология гидрогенизации нефтей и
нефтяных остатков в диспергированном состоянии над
псевдоожиженным катализатором при невысоком да-
влении. Основная идея заключается в облегчении
контакта сырья с катализатором, с тем чтобы ускорить
гидрирование; трудно гидрируемые высокомолекуляр-
ные продукты образуют кокс, который выжигается
при регенерации катализатора. Расход водорода 1,04—
1,36% на сырье. При гидрогенизации 50%-ного мазута
выход автомобильного бензина 30%, дизельного топ-
лива 58%, газа 8,5%, кокса 4,8%. При возврате в цикл
фракций >240 °C выходы соответственно 21,0, 61,0,
14,5 и 6,0. Расход водорода возрастает до 2,5%
Разработана технологическая схема двухступенча-
той гидрогенизации тяжелого остатка с высокими
объемными скоростями. Отравление стационарного
катализатора во второй ступени предотвращается
выводом ~25% гидрогенизата с первой ступени в ва-
куумпуЮ колонну, в которой отделяется шлам, а ва-
куум-дистиллят присоединяется к паро-газовым про-
дуктам с первой ступени, поступающим из сепаратора
во второй реактор. Расход водорода 3,5%. Общий
выход моторных топлив 79,5%, шлама 7,8%, газов
(включая потери) 16,2%
Описаны опыты по гидрогенизации нефтепродуктов
в псевдоожиженном слое кокса. Получается газ с теп-
лотворной способностью 5150—6770 ккал/м3. Из лег-
ких дистиллятов образуется только 3% жидких про-
дуктов. Переработка газойля дает 82,1% газа, 11,9%
бензола и 3,9% нафталина; переработка сырой нефти
дает 78,9% газа, 8,6% жидких продуктов и 2,4% кокса;
222,
223
224
225
300-600 440 1 WS2; WS2+NiS2naAl2O3; WS2 на терране
20-50 380-425 1,5 Co4~Mo на А12О3
— — — СоМоО4 на А12О3
41-52 366-383 — Со Мо на А12О3
3—13 204-288 • 3,0-18,0 WS2 + NiS
3
226
227
переработка котельного топлива дает 68,5% газа,
0,1% жидких продуктов и 4,2% кокса
Гидрогенизация сырой нефти, содержащей 1,64%
серы, 0,03% золы и 2,24% асфальтенов, приводит
к отравлению всех исследованных катализаторов.
Несколько стабильнее WS2 + NiS на А12О3: при
давлении 600 кгс/см2 его активность сохранялась
в течение 600 ч
Разработан активный регенерируемый катализатор
и найдены оптимальные технологические режимы
гидроочистки прямогонных дистиллятов (до 350 °C),
газойлей каталитического крекинга, керосина терми-
ческого крекинга, газойлей коксования. Содержание
серы уменьшается с 0,57—1,92 до 0,03—0,10%. Одно-
временно происходит изменение группового состава
сырья, существенно улучшающее качество продукта
и делающее его пригодным для дальнейшей переработки.
Увеличивается количество парафино-нафтеновых и лег-
ких ароматических углеводородов (соответственно с
44,7 до 52,7 и с 21,7 до 33,1%) и уменьшается доля
тяжелых ароматических углеводородов и смол (соот-
ветственно с 27,5 до 13,3 и с 6,1 до 0,9%). Выходы жид-
ких продуктов во всех случаях 97—98%
Показано, что гидрогенизация сырья для каталити-
ческого крекинга в мягких условиях улучшает пока-
затели последнего: коксоотложепие уменьшается на
30%, содержание серы и металлов — на 90%; пол-
ностью устраняется образование сероводорода
Сообщаются дополнительные данные о процессе гид-
роочистки с противотоком сырья (см. 2М). Расход
водорода 3,1 м3 на 1 кг удаленной серы. Сера удаля-
ется, по промышленным данным, на 80—90%
Установлено, что отношение констант скоростей
гидрирования диенов и олефинов тем больше, чем
ниже температура: 66 при 288 и 103 при 204 °C. Однако
селективность повышается со снижением температуры
медленнее, чем падает скорость гидрирования диенов,
которая уменьшается в этом интервале температур
в 10 раз
228
229
230
Давле- ние, кгс/см* Темпера- тура, Объемная скорость (проточи, уст.), ч-' Катализаторы
48-3 До 400 До 4,5 Со + Мо ня AI2O3
8-43 343-371 10 Со-|-Мо на А12О3
34-204 370-468 До 5,0 (опти- мальная) Стандартный для десуль- фуризации
Продолжение табл. 4
Основные результаты
Лите-
рату-
ра
Найдено эмпирическое уравнение типа уравнений для
псевдомономолекулярных реакций, связывающее тех-
нологические показатели (плотность керосиновой фрак-
ции и ее средний молекулярный вес, общее давление,
отношение количеств водорода И сырья, объемная
скорость) с отношением количеств серы в сырье и про-
дукте. Полнота удаления серы при 400 °C — 99%
Для повышения селективности гидроочистки кре-
кинг-бензинов применены новые технологические при-
емы: к сырью добавляется «природный тормозитель»
гидрирования олефинов, гидроочистке подвергается
не весь бензин, а фракция )> 182 °C, в которой нахо-
дится большая часть сернистых соединений, но мало
олефинов, преобладающих в головных фракциях.
В длительном опыте при 20 кгс/см2 глубина обессери-
вания фракции > 182 °C составляла 84% при остаточ-
ном содержании олефинов 40%. По отношению ко
всему бензину достигалась 80%-ная очистка без изме-
нения октанового числа, тогда как гидроочистка всего
бензина понижала октановое число на 6 пунктов
Разработан новый процесс гидрокрекинга нефтяных
остатков: конструкция реактора обеспечивает макси-
мальную изотермичность каталитического слоя с пере-
падом температур не более 10—11 °C. Из остатка нефти,
содержащего 5,32% серы и дающего 17,2% кокса (по
Конрадсону), получено 19,8% бензина, 24,0% легкого
газойля, 29,1% тяжелого газойля и 29,4% остатка:
содержание серы в них соответственно 0,057, 0,17,
0,64 и 2,52%. Расход водорода 24 кг на 1 м3 сырья
231
232
233
I
27—55 385-440 0,5-2,0 CoMoOi
88 432 0,8 —
100 443-485 0,2-0,44 C0M004 \
s 238 435-438 1,0 2% NiO на Al2Os4-SiOa; 10% NiS на Al2Og“j-SiOaJ Co + Mo на AlgOg \
f . 1 X '
234
Проведены опыты деструктивной гидрогенизации
асфальта, отделяемого на установках деасфальтиза-
ции. Катализатор регенерируется через 500 ч. Степень
превращения сырья —60%. Выход газа 2,3%, бензина
12,1%, фракции дизельного топлива 16,4%, газойля
(343-537 °C) 31%, остатка >537 °C 45,2%
Процесс гидрогенизации нефти с высоким содержа-
нием асфальтенов 216, 216 проведен на пилотной уста-
новке производительностью 10 т/сутки. Получено 6,2%
газа, 14,9% бензина, 62,9% газойля, 9,4% тяжелого
газойля и 21,5% вакуумного остатка. Расход водо-
рода 0,7%
Для производства высококалорийного газа и легких
ароматических углеводородов из нефтяных остатков
предлагается сочетание двух процессов: гидрокрекинга
и гидрогазификации. Выход газа 90,2—90,6%, жид-
ких продуктов 9,4—9,8%. Газ почти нацело состоит
из метана, в жидких продуктах содержится 90,1 —
94,6% бензола, а также толуол, ксилолы и нафталин.
В опытах с индивидуальными углеводородами замет-
ное отложение кокса наблюдалось только при гидро-
газификации нафталина
Описан процесс гидрокрекинга нефтяных остатков
в присутствии разбавителя (большей частью бутана
в количестве 35% от массы остатка). Особенностями'
процесса являются низкий расход водорода и малый
выход газа, высокие выходы сырья для каталитиче-
ского крекинга, отсутствие регенерации катализатора.
Расход водорода (в порядке перечисленных катализа-
торов): 5,5, 7,7 и 10,8 кг/м3; Выход бензина 13,4,14,7
и 20,3%, выход газойля 63,0, 66,8 и 65,5%, выход
остатка'28,2, 22,3 и 18,2%; выход газа 1,5, 1,8 и 1,9%,
баланс бутана — 2,2,—0,2 и -j-1,1%
235
236
237
Давле- ние, Ш'С/СМ2 Темпера- тура, °C Объемная скорость (проточн. уст.), q-i Катализаторы
300 300 450-470 400 04-0,4 0,87— 0,93 Fe (I) WS2 (II)
300 345-410 — ws2
— — —
Продолжение таблУ4
Основные результаты
Лите-
рату-
ра
Изучена зависимость показателей процесса деструк-
тивной гидрогенизации в жидкой фазе (условия 1)
от качества сырья: чем больше оно ароматизировано,
том ниже объемная скорость и производительность
и тем больше расход водорода на бесполезное образо-
вание газа (до 95% в случае крекинг-остатков). Более
целесообразно сочетание гидрогенизации на стацио-
нарных катализаторах с другими процессами нефте-
переработки: удалением асфальтенов термическими
методами и гидрированием деасфальтизатов (условия IJ).
Показано, что выходы жидких продуктов в таких ва-
риантах составляют до 85—88% (от нефти), расход
водорода на газообразование 24—37%. Производитель-
ность аппаратуры высокого давления увеличивается
в несколько раз
Приведен опыт переработки различных видов сырья
с помощью деструктивной гидрогенизации. Из керо-
сино-газойлевой фракции получают дизельное' топливо,
осветительный керосин, реактивное и специальное
дизельное топливо, бензин БР-1 и уайт-спирит.
Из вакуумного газойля получают дизельное топливо,
осветительный керосин, бензин. Выходы перечислен-
ных целевых продуктов соответственно 90—92 и 85—
88 %
Показана возможность сочетания процесса деструк-
тивной гидрогенизации дистиллятов с синтезами ме-
танола и аммиака. Разработанные схемы повышают
производительность аппаратуры и обеспечивают более
полное использование компонентов синтез-газа, по-
зволяя частично или полностью заменить непроизводи-
тельный процесс отмывки окиси углерода процессом
238
239
240
зоо 320-420 1,0-2,0 WS2+NiS на A12O3’, WS2
10-15 400-425 0,6-1,0 Co + Mo на A12O3
— — — To же
50—300 320—400 — »
40 425 0,5 . Co + Mo на A12O3
-- 450 — —
tn <0 • (
синтеза метанола. Блок синтеза метанола в этих усло-
виях может работать автотермически
Показана возможность гидрирования петролатума
с целью получения сырья для производства вазелино-
вого масла и для дальнейшего окисления
Предлагается подготовка сырой нефти для гидриро-
вания путем фильтрования ее через слой глины при
425—475 °C. Обессмоленная таким образом нефть
может гидрироваться при давлении 100 кгс/см2. Даль-
нейшее сочетание гидрирования с каталитическим кре-
кингом дает общий выход светлых нефтепродуктов
84,6%
Установлены параметры автогидроочистки керосина
и дизельного топлива. Содержание серы снижается
соответственно с 0,14—0,37 до 0,01 и с 0,84 до 0,20%
Изучалась динамика отложения кокса в процессе
гидроочистки дизельных топлив. При 300 кгс/см2
и 450 °C кокс может быть удален водородом на 80%.
Кокс удаляется тем легче, чем выше давление и ниже
температура гидроочистки
Разработана технология получения низкозастыва-
ющих масел из сернистого сырья (фракция 200—500 °C
каталитического крекинга мазута) с последующей де-
парафинизацией гидрогенизатов
Гидроочисткой сернистых нефтепродуктов можно
получать турбинное, трансформаторное, веретенное,
вазелиновое и другие масла
Проведены опыты термического крекинга в присут-
ствии тетралина. Выход кокса понижается с 17 до 2%.
Другие разбавители (декалин, гептан, бензол, нафта-
лин) дают худшие результаты. Предполагают, что таким
путем можно будет комбинировать крекинг и гидрогени-
зацию, проводя их при давлениях менее 34 кгс/см2.
Преимуществом метода является гидрирование дистил-
лятов (в том числе нафталина, образовавшегося из
тетралина), а не суммарного остатка, требующего
применения высокого давления
241
242
243
244
245
246
247
О
Давле- ние, кгс/см! Темпера- тура, °C Объемная скорость (проточи, уст. У, Ч“> Катализаторы
50 380 0,7 Со-|-Мо па А12О3
300 300-350 — WS2
50-500 200-300 0,5-4,0 WS2-]-NiS на А12О3; МоО3 на А12О3
500 440-470 0,5-1,0 WS2 + NiS па А12О3 (I)
500 440-460 0,5-1,0 Mo-]-Ni на Al2O3+SiO2 (II)
Продолжение табл. 4
Основные результаты
Лите-
рату-
ра
Показано, что предварительная гидроочистка сырья
для каталитического крекинга повышает качество
получаемых продуктов и в 1,5 раза производительность
установки
Гидрирование асфальтенов, выделенных из нефти,
протекает в 5 раз медленнее гидрирования асфальтенов
из гидрогенизата угля и в 25 раз медленнее, чем гид-
рирование асфальтенов, выделенных из полукоксовой
смолы
Изучено влияние условий гидрогенизации техниче-
ского нафталина на выходы технического тетралина
и очищенного декалина. В оптимальных условиях
(300 кгс/см2, 250—260 °C, 1,2—1,4 ч*1, WS2 + NiS
на А120р) степень превращения нафталина 87%, в не-
прерывном процессе с рециркуляцией выход тетра-
лина 66%, декалина 28%
Показана возможность гидрирования суммарных
жидкофазных гидрогенизатов мазута восточных нефтей
СССР, полученных с рециркуляцией тяжелого масла
на катализаторе Fe на полукоксе при 500 кгс/см2,
470—480 °C и объемной скорости 0,5—0,8 ч-1. Тем
самым доказана осуществимость совмещенного гидри-
рования остатков. На катализаторе I обеспечивается
высокий выход гидрогенизата (98,3%), а с учетом жид-
кофазной ступени суммарный выход светлых нефте-
продуктов составляет 88,5% на мазут при расходе
водорода 4,4%, на катализаторе Ц соответственно
97,4 и 87,7%. Из гидрогенизатов. может быть выделено
4—17% компонента автомобильного бензина и 51 —
70% осветительного керосина. По выходам товарных
248
249
250
251
30 30 430-475 450 0,8—2,0 1,0 МоО3 на A12O3 Та же
30 500-540 2,0-5,0 »
280-300 360-370 1,0 Стационарные катализа-
400-405 1,2 торы 1
370-435 0,5
500 470-483 0,5-0,6 Суспендированный ката- лизатор 1
35 340-380 0,5-3,0 WS2-f-NiS на А12О3
25-300 150-350 0,5-3,0 То же
продуктов и глубине превращения предпочтительнее
применение катализатора II
Гидрогенизация асфальта ромашкинской нефти, раз-
бавленного бензином, в движущемся слое катализатора
дает 66—80% жидких продуктов и 7,6—12,1% газа.
Сера удаляется на 69—97%
При гидрогенизации нефтяных остатков в рециркули-
рующем потоке катализатора разбавление свежего
сырья фракцией 180—300 °C гидрогенизата (1 : 1)
повышает выход жидких продуктов с 84,8 до 90,3%,
уменьшает количество газа с 8,2 до 6,1% и кокса с 7,0
до 5,3%
Показана возможность гидрогенизации туймавин-
ской нефти под невысоким давлением. Выход жидких
продуктов 85—92%, остатка выше 400 °C — не более
5% (СМ. 222, 223)
Описан опыт работы заводских агрегатов при гидри-
ровании различных типов нефтяного сырья: керосино-
газойлевой фракции, вакуум-дистиллята и жидкофаз-
ных нефтяных гидрогенизатов. Высокая степень
очистки обеспечивает хорошее качество продуктов —
бензина, осветительного керосина, дизельного топлива
Описан опыт работы заводского агрегата при жидко-
фазном гидрировании нефтяных остатков. Выход жид-
ких полупродуктов стабильно составлял 86—89%.
В продуктах содержится до 0,17—0,47% коронена,
иногда вызывающего осложнения из-за забивки труб
холодильников
Гидроочистка парафинового сырья существенно по-
вышает его качество как сырья для получения алкил-
арилсульфонатов
Изучено влияние условий на глубину гидроочистки
бензина БР-1. В оптимальных условиях выход высоко-
качественного растворителя 98%; показано, что гидро-
очистка дешевле сернокислотной очистки
252
253
254
255
256 >
251
258
Давле- ние, кгс/см* Темпера- тура, °C Объемная скорость (проточи, уст.), Я-1 Катализаторы
70 440 0,9 Мо на полукоксе; Fe на активированном угле
70 420—440 — Fe на полукоксе; Мо03 на А12О3
43 340-345 — Со + Мо на А12О3
33-130 205-370 — —
30 430-450 1,0-2,0 МоО3 на А12О3; NiO на А12О3
Продолжение табл. 4
Основные результаты
Лите*
рату-
ра
Показана возможность гидрокрекинга нефтяных остат-
ков в присутствии разбавителя — прямогонных фрак-
ций полукоксовой смолы (200—300% и 200—240 °C).
Выход фракций: 10,6—8,0% фракции до 130 °C, 54,4—
39,4% фракции 130—270“ С, 16,2—13,5% фракции
270—360 “С, 11,8—28,2% фракции 350—500 °C (пер-
вые цифры относятся к молибденовому, вторые —
к железному катализаторам)
Технология гидрогенизации при среднем давлении
(70 кгс/см2) с добавкой переносчика водорода
(см.206, 216 > 216, 235) проверена в промышленном мас-
штабе на венгерской и туймазпнской нефтях, а также
на буроугольной и сланцевой смолах. Выходы мотор-
ных топлив соответственно 79, 82, 87 и 71%
Метод,гидроочистки с противотоком сырья (см. 204, 229)
применен для гидроочистки газойля термического кре-
кинга нефтяных остатков. Содержание серы умень-
шается с 0,70 до 0,08%, бромное число с 30,0 до 1,2 г
Вг2/100 г; азот удаляется незначительно (с 0,615 до
0,345 мг/кг)
Сообщается о новом низкотемпературном процессе
гидрокрекинга — изокрекинге; степень превращения
сырья 50—70% при циркуляции всех фракций, кипя-
щих выше бензина
В процессе деструктивной гидрогенизации нефтей и
нефтяных остатков в диспергированном состоянии
(см.222, 223) испытаны катализаторы, приготовленные
различным образом. Молибденовые катализаторы не-
сколько активнее обычных никелевых, но по специаль-
ной методике был приготовлен механически прочный
259
260
261
26
263,
264
33—100 455 До 300 с CoMoCU на А12О3
— — — —
70 420-440 1,1 10 927
55 405 МоО3 на А12О3
100 440 0,5 Fe; МоОэ на А12О3; Сг2034-Мо03 на А12О3
30 452-481 — МоО3 на А13О3
г
никелевый катализатор, не уступающий по активности
промышленному молибденовому. Увеличение доли МоО3
до 10% повышает активность катализатора; дальней-
шее увеличение не оказывает влияния. Оптимальное
содержание NiO 10—12%, при более высоком содер-
жании NiO активность падает
Показано, что гидрогазификация нефти и нефте-
продуктов более эффективна при предварительном
пропускании сырья над кобальтмолибденовым катали-
затором с последующим пиролизом при 816 °C (см.
также 236)
Сообщается о новых модификациях гидрокрекинга —
процессах «юникрекинг» и «ломаке». Первый разрабо-
тан для переработки тяжелых сернистых фракций, вто-
рой — для легких и тяжелых газойлей
На основе опытов на пилотных установках проведен
пробег промышленной установки DHD, переоборудо-
ванной под метод Варга. Сырье — венгерская нефть и
туймазинский мазут (отношение сырье : разбавитель
равно 4:1). Два реактора с плавающим и стационар-
ным катализаторами работали по совмещенной схеме
(см.198). Из нефти получено 75,3% жидких продуктов
при расходе водорода 1,8%; из мазута получено 22%
бензина, 60% дизельного топлива и 11% остатка при
расходе водорода 2,4%
Деструктивная гидрогенизация мазута ромашкин-
ской нефти на порошкообразных катализаторах. Луч-
шим катализатором является хромовый, снижающий
содержание асфальтенов с 5,9 до 0,7% и полностью
обессеривающий фракцию до 325 °C. Длительность
пробега 50 ч
Гидрокрекинг сырой аравийской нефти, содержащей
3,23% серы и 12,9% асфальтенов. Полнота удаления
серы до 90%. Процесс улучшается при добавке разба-
вителя
265
266
267
268
269
Давле- ние, кгс/см* Темпера- тура, “С Объемная скорость (проточи, уст.), ч-« Катализаторы
35 340-380 0,5—3,0 WS24-NiS на А12О3
40-50 250-270 0,4 Ni-f-Mo на А1гОа
— — — —
100 400 — Co-f-Mo на А1гО3
30—100 400 — То же
30-150 350-450 — —
Продолжение табл. 4
Основные результаты
Лите-
рату-
ра
Осуществлена гидроочистка жидких парафинов, полу-
ченных карбамидной депарафинизацией, от серы и смо-
листых примесей. Сульфирование полученного про-
дукта дает кислый гудрон, который может быть исполь-
зован в производстве моющих средств и для окисления
Гидрированием в две ступени ферганских бензинов
получены очищенные растворители: бензин БР-1, уайт-
спирит и др.
Расчетами показано, что в целях уменьшения рас-
хода водорода при гидроочистке можно использовать
циркуляционные газы: например, циркуляционный газ
со стадии гидроочистки летнего дизельного топлива
можно направлять на гидроочистку зимнего дизельного
топлива, а циркуляционный газ этой стадии — на
гидроочистку бензина
Гидрогенизационное облагораживание дистиллятов
контактного коксования уменьшает содержание в них
конденсированных ароматических углеводородов и ас-
фальтенов с 39—45% до 22—33% и облегчает последую-
щий каталитический крекинг. Выход бензина увели-
чивается, снижая на треть отложения кокса. Комбини-
рование коксования, гидрогенизации и каталитического
крекинга дает выход светлых нефтепродуктов 80,5%
Показано, что при облагораживании сернистых
газойлей термического и каталитического крекинга
лучшие результаты достигаются при смешении сырья
с дизельным топливом (1 : 1)
Описывается Процесс Французского Института нефти
для гидроочистки сырой нефти и дистиллятов. Циркуля-
ционный газ очищается от H2S только в случае сырья,
содержащего 3—4% серы. Отложение кокса на катали-
270
271
272
273
274
275
Сг2О3 на А12О3
100 — — Ni или СоМоОа
20* 500-600 — Мо
50 350-425 1,0 Со+Мо на А12О3
40-50 325-350 1,0 А1 + Со + Мо; А14-Мо
40-50 / 300-325 1,0—1,5 А1+Со 4-Мо
40 380 0,5-2,0 То же
* Время контакта (стационарная установка) 0,3-4 с.
заторе предотвращается добавкой легких дистиллятов.
При гидроочистке кувейтской нефти содержание серы
снижено с 2,4 до 0,75%.
Разработан процесс производства нафталина из цир-
куляционных газойлей каталитического крекинга.
На первой ступени в мягких условиях неароматическая
часть гидрокрекируется в бензин, а остаток, обогащен-
ный бициклическими ароматическими углеводородами,
деалкилируется в жестких условиях во второй ступени,
давая нафталин и дополнительное количество бензина
Сообщается о разработке процесса облагораживания
котельных топлив H-Oil
В лабораторных условиях осуществлено деалкилиро-
вание метилнафталиновой фракции. Наряду с нафтали-
ном получено 6—15% продуктов деструкции нафталина
Изучалась возможность гидрообессеривания сырой
нефти (2,81% серы) с целью получения мазутов высо-
кого качества. Обессеривание на 40—68% без замет-
ного крекинга. Активность катализаторов сначала
быстро падала, затем оставалась на уровне 30%
Осуществлена гидроочистка сырого парафина из
высокосернистых нефтей с температурой конца кипения
480 °C и содержанием масла 5:0,8%; расход водорода
0,15%. Срок службы катализатора без регенерации
более 1000 ч
Без сообщения условий гидрирования указывается,
что при гидрогенизации пироконденсата (выход гидро-
генизата 100%, расход водорода 0,64%) получается
47% бензола, 18% толуола, 10% ароматических угле-
водородов С8 и 11% растворителя
Доочистка масел глинами может быть заменена гидро-
доочисткой в указанных условиях с расходом водорода
0,2-0,3 %
Показано, что при гидроочистке дизельных топлив
водород может содержать окись углерода и двуокись
углерода (6,0 и 3,3% соответственно), но это увеличи-
вает расход водорода
276
277
278
279
280
281
282
283
Давле- ние» кгс/см* Темпера- тура. “С Объемная скорость (проточи, уст.), ч-1 Катализаторы
50 370 0,8—1,0 А1 + Со+Мо
60 380—390 — Al + Co + Mo (I)
60 180—190 — Ni + Сг (II)
68; 136-200 —
— — — .Со + Мо на боксите; бок- сит
— — — WS2+NiS
Продолжение табл. 4
Основные результаты
При равном коксообразовании (4,5%) выход бензина
каталитического крекинга из сырья, предварительно
подвергнутого гидроочистке, выше на 36—61%
Описана схема двухступенчатого гидрирования бен-
зола. Удаление сероводорода осуществляется между
I и II ступенями. Первый катализатор может регенери-
роваться
Сообщается о дальнейшем усовершенствовании про-
цесса Gulf-HDS получения малосернистых котельных
топлив и сырья для каталитического крекинга из
нефтяных остатков. Из вакуумного остатка кувейтской
нефти, содержащей 5,5% серы, получен гидрогенизат
с содержанием серы 0,52%. Описываются две модифика-
ции процесса: при 68 кгс/см2 идет обессеривание, при
более высоком давлении (136—200 кгс/см2) — гидриро-
вание ароматических углеводородов, что особенно благо-
приятно в случае сырья для каталитического крекинга.
Усовершенствования достигнуты за счет улучшения
катализатора — увеличения срока службы до 3—4
месяцев и подбора условий. Обессеривание выше 80%
нецелесообразно, так как при этом идет сильная дест-
рукция, что повышает расход водорода и удорожает
процесс
Для предотвращения дезактивирования катализатора
сырая нефть сначала пропускалась над бокситом в токе
водорода, затем над кобальтмолибденовым катализато-
ром. В первой ступени удаляется асфальт, во второй —
97% серы
Предлагается сочетание термического крекинга сырой
нефти с гидрированием и использованием гидрогениза-
тов в качестве доноров водорода
Лите-
р а ту-
ра
284
285
286
287
288
°* 50 ' 30 370 420 1,0 Со0 + Мо03 на А12О3 Co-J-Mo па А12О3
250 340-355 0,4 WS2 + NiS па А2О3
37,5 375 — А1+Со + Мо
100 520—600 — МоО3 (14%) на А12О3
50 400—450 0,5-2,0 Al + Co+Mo; WS2+NiS на А12О3; АП-56; Pt на А12О3 4“ SiO2
— — — —
о> 50 380 Al + Co + Mo (I); Al + 4-Ni + Mo (II)
Описан процесс гидроочистки фирмы Shell OIL
Для удаления серы достаточно давление 50 кгс/см2,
для удаления азота 100—175 кгс/см2. Срок службы
катализатора 3 года. Катализатор регенерируют азо-
том, содержащим 1 % кислорода. В типичном примере
содержание серы понижается с 1,40 до 0,09%
Показана возможность получения малосернистых
котельных топлив гидрогенизацией туймазинской неф-
ти, содержащей 4,35% серы, в циркулирующем потоке
катализатора (см.222, 223, 882-284, 293, seij При регене-
рации катализатора через 16 ч остаточное содержание
серы составляет 0,5%, через 40 ч — 1,0%
Бензол, содержащий 0,1—0,2% серы (в виде тио-
фена), гидрируется с образованием 98,9—99,0%-ного
циклогексана
Осуществлена гидроочистка кумола, полученного
алкилированием бензола в присутствии фосфорной
кислоты. Содержание серы понижено до 0,007%,
деалкилирование протекает не более чем на 2%
Изучено влияние условий и состава сырья на образо-
вание нафталина в ходе высокотемпературной гидро-
генизации. Выход нафталина из различных техниче-
ских продуктов 20—60%; выход тем выше, чем выше
содержание ароматических углеводородов в сырье.
В отсутствие катализатора температура процесса долж-
на быть выше на 100—150 °C.
Осуществлена гидроизомеризация товарного серни-
стого парафина с целью получения высокоиндексных
смазочных масел. Платиновые катализаторы менее при-
годны из-за чувствительности к сере (А П-56) и интен-
сивного расщепления сырья (Pt на А12ОЭ + SiO2)
Осуществлена промышленная гидроочистка бензина
БР-1 с выходом 98,5% (см.258)
Новый катализатор гидроочистки (II) по сравнению
с промышленным катализатором (I) более стабилен,
активен и дешев. При степени обессеривания 80%
объемная скорость вдвое больше
289
290
291
292
293
294
295
296
Давле- ние, кгс/см* Темпера- тура, °C Объемная скорость (проточи, уст.), Ч”1 Катализаторы
84 274-343 — NiS на Al2O3-|-SiO2
— — — —
35-105 '200-425 — —
54 ' — — —
— — — —
— — — —
Продолжение табл, 4
Основные результаты
Лите-
рату-
ра
Из газойлей различных типов (температура конца
кипения до 450 °C) с помощью «изокрекинга» получают
низкозастывающее реактивное топливо. В приведенном
примере из тяжелого газойля (219—452 °C) получено
до 40% авиационного керосина. Общий выход про-
дуктов С. и выше — 117 объемн. % расход водорода
1,2%
Предлагается новый процесс висбрекинга (отноше-
ние мазут : разбавитель от 2:1 до 5:1) с добавкой
донора водорода, которым служит предварительно
гидрированная фракция 371 —482 °C. Расход водорода
на гидрирование циркулирующего разбавителя 27 м3/м3
(см.288)
Сообщается об универсализации процесса «юникре-
кинг», в котором может быть переработано различное,
в том числе утяжеленное, сырье. Процесс осуществляет-
ся в одну или две ступени в зависимости от вида сырья.
Сера и азот удаляются полностью. Срок службы ката-
лизатора без регенерации 6 месяцев
Сообщается о разработке процесса гидрирования лег-
кого пироконденсата. Катализатор регенерируемый
Без приведения условий и сведений о катализаторе
сообщается об осуществлении в промышленном мас-
штабе процесса деметилирования толуола (процесс
«детол» фирмы Houdry). Выход бензола 81,6%, тяжелых
остатков — 1,2%, остальное — газ. Чистота, бензола
99,95%, расход водорода 2,2%
Преимущества процессов «изокрекинг» и «ломаке»
соединены в процессе «изомакс», при помощи которого
можно получать бензин, реактивное и дизельное топ-
ливо из различного сырья, включая вакуумные газойли.
297
298,
299
300
301
302
303
s
83—70 50 435-450 405-435 0,9-1,5 1,0-1,6 Fe на полукоксе NiO2 + MoOg на AI2Oa
40 450 1,5-2,0 Al + Ni + Mo
40-70 400-470 1,4—4,0 Al + Mo + Co (I); A1 + + Mo + Ni (II)
30 420-470 — Al + Co + Mo
5 380-460 1,0-2,0 To же
50—20 / 350—410 — Al + Co+Mo
15-29 580-640 — Специальный
Выход жидких углеводородов 115—120 объемн.%
Дальнейшее усовершенствование метода Варга поз-
волило повысить выход товарных продуктов до 75—
80% и снизить потребление водорода до 0,68%. В гидро-
генизате 12,5% бензина, 65,9% дизельного топлива и
20,6% низкосернистого котельного топлива. Опреде-
лены кинетические зависимости и по ним, оптимизиро-
ваны условия процесса
При гидроочистке образцов нефтей юго-восточных
районов Башкирской АССР выход бензина повышается,
содержание серы снижается до 0,34—0,94%
На смеси арланской и чекмагушской нефтей с содер-
жанием серы 2,96% подбирались условия, обеспечива-
ющие 70%-ную десульфуризацию. Катализатор (I)
нужно регенерировать через 5 ч, а катализатор (II) —
через 11 ч. Влияние условий изучалось на катализа-
торе (II); содержание серы понижалось до 0,3—1,1%
в гидрогенизате и до 0,7—1,7% в остатке
Исследовано влияние условий процесса гидрокре-
кинга на циркулирующем катализаторе на удаление
асфальтенов и серы. При температурах ниже 420 °C
нельзя получить низкосернистое котельное топливо.
Для снижения содержания серы до 0,5% срок службы
катализатора между регенерациями должен быть 16 ч,
до 1% — 40 ч. Почти полное удаление ванадия дости-
гается при 470 °C. (См,!вз, 2М, 2в0)
Для гидроочистки бензинов каталитического крекин-
га и дизельных топлив могут быть применены газы
каталитического риформинга, содержащие 50—70%
водорода
Показано, что при гидроочистке дизельных топлив
снижение давления с 50 до 30 и 20 кгс/см2 (с частичной
компенсацией подъемом температуры) позволяет сни-
жать расход водорода на 27—48%
Описывается процесс Bextol фирмы Shell Oil демети-
лирования толуола. Выход бензола 96,8% от теорети-
ческого, расход водорода 2,04 моль/моль
304
305
306
307
308
309
310
о
Продолжение табл. 4
Давле- ние, кгс/см2 Темпера- тура, °C Объемная скорость (проточи, уст.), ч-1 Катализаторы Основные результаты Лите- рату- ра
— — — СоО + МоО3 на носителе с добавками Na2O Сообщается об осуществлении деалкилирования гомологов нафталина в промышленном масштабе. Рас- ход водорода 0,43 м3 на 1 кг нафталина 311
5-50 350—450 0,5—5,0 Активированный уголь на А1о03 и алюмосиликате Предлагается двухступенчатый процесс переработки остатков п асфальтенпстых сырых нефтей: термоката- литическая коагуляция асфальтенов, затем гидрообес- серивание. Первая ступень при содержании асфальте- нов 1—2% осуществляется на стационарном катализа- торе, при более высоком содержании асфальтенов — на взвешенном, который отфильтровывается вместе с асфальтенами. Повышение температуры облегчает выпадение асфальтенов и приближает процесс к вис- брекингу 312
35—70 400-450 Изучены скорости гидродесульфуризации в процессе Gulf-HD/S (см.286) и факторы, влияющие на нее. Из вакуумного остатка кувейтской нефти с 5,45% серы получены гпдрогенизаты, содержащие от 1,2 до 3,8% серы. Теоретический расход водорода равен 2 моль на 1 г-атом серы, однако фактический расход выше п тем больше, чем глубже обессеривание. Разработан новый катализатор, дающий аналогичные результаты При более низкой температуре 313
63-70 444 (Сред- няя) 1,0 Fe па полукоксе Описаны результаты второго длительного пробега установки процесса гидрокрекинга Варга, в ходе кото- рого было переработано 42 т отбензиненной смеси туй- 314, 315
55 405- 435 1,1 NiO -(- МоО3 на А12О3 мазинской и ромашкинской нефтей плотностью 0,904 г/см3, содержащей 1,05% серы, 1,9% асфальте- нов, 5,4% кокса. Расход водорода 0,0—0,8%, образо- вание кокса 0,5—1,0%, выход жидких продуктов 96,4 %, удаление серы 60—70%
— — —• Статья посвящена химии и механизму процесса 316
— —
Высокое — — Ni + W на алюмосили- кате
50—250
190
275—450
440—460
WS2, МоО3 или металл
VIII группы периодической
системы на фторированном
алюмосиликате
Сульфиды металлов VI
и VIII групп периодиче-
ской системы на А12О3 или
глине
гидрокрекинга «изомакс». Процесс двухступенчатый,
в первой ступени удаляются азот, кислород, сера и
металлы, во второй — идет собственно гидрокрекинг.
Наиболее трудно идет удаление азота
Разработан новый катализатор гидрокрекинга, кото-
рый устойчив к соединениям, содержащим серу и азот.
Катализатор состоит из небольшого количества благо-
родного металла на специальной подложке. Действие
серы и азота можно компенсировать повышением тем-
пературы: при содержании 0,1% азота температуру
повышают на 70, а при содержании 0,5% серы —
только на 13 °C
Описан новый вариант процесса гидрокрекинга Ну-С,
в котором перерабатывались как прямогонные, так и
вторичные газойли с температурой конца кипения до
570 °C. Газойль и водород проходят снизу вверх через
псевдоожиженный слой катализатора. Превращение
пропорционально давлению в степени 1,4. Отложение
кокса незначительное
Описаны дальнейшие разработки процесса гидрокре-
кинга в ФРГ (см.187, 198). Процесс осуществляется в
одну или две стадии в широком интервале условий в за-
висимости от качества сырья. В двухступенчатом про-
цессе давление на обеих ступенях одинаковое —
100 кгс/см2. Разработаны новые катализаторы. Катали-
затор WS2 на терране заменен сульфидом железа,
а также Ni на алюмосиликате. В качестве носителей
испытаны А12О3 и Fe2O3, активированные HF, в каче-
стве гидрирующих агентов — металлы VI и VIII групп
периодической системы
Осуществлен гидрокрекинг вакуум-дистиллятов муха-
новской и ромашкинской нефтей. При расходе водорода
320—430 м3/т получено 20—25% легких продуктов,
а также смазочные масла и парафин. Подбором катали-
заторов процесс может быть направлен на производ-
ство бензина, авиационного керосина или дизельного
топлива
317
318
319
. 320
Давле- ние, кгс/см® Темпера- тура, °C Объемная скорость (проточи, уст.), 4-i Катализаторы
— — —
55 550 -580 А1+ Со + Мо
40 700—720 Беа катализатора
200 400-435 0,5 ws2
200 450-465 о;5 Al -+• Со-|-Мо
— - f — —
100 400-435 0,5-2,0 NiO + MoO3 (I); WS2 (II)
'45 570 — Сг2О3 на А12О3
Продолжение табл. 4
Основные результаты
Лите-
рату-
ра
Никель и ванадий, содержащиеся в тяжелых дистил-
лятах нефти, осаждаются на катализаторах гидроочи-
стки и гидрокрекинга, дезактивируя их. Показано, что
при регенерации катализаторов для удаления окислов
никеля и ванадия целесообразно добавлять комплексо-
образователи типа щавелевой кислоты, диоксана, аце-
тилацетона
Фракция 215—300 °C каталитического крекинга,
содержащая 15—25% метилнафталинов, экстрагирует-
ся фурфуролом, гидроочищается и гидродеалкилирует-
ся. Выход нафталина 10%, чистота 96—97% (с ката-
лизатором) и 99,8% (без катализатора). Кроме нафта-
лина получают обессеренные дизельное топливо п
лигроин,
Из деасфальтизата деструктивной гидрогенизацией
получены высококачественные авиационные масла с вы-
ходом до 18%. Лучший катализатор — WS2
Сочетание гидрирования и контролируемого гидро-
крекинга дает высокоиндексные смазочные масла с вы-
ходом на 10—50% выше, чем при очистке раствори-
телями
Из вакуум-дистиллята ромашкинской нефти (2,11%
серы) с выходом 35—45% получены очищенные смазоч-
ные масла (содержание серы 0,15% с первым п 0,42%
со вторым катализатором)
Описан новый хромовый катализатор для процесса
гидродеалкилирования толуола, более активный, чем
промышленный: степень конверсии за проход 40
321
322
323
324
325
326
70 480 0,5 Pt на A12O3; Pt на фто- рированном A12O3; C0M004 на A12O3; NiO на A12O3
f 430-445 —
— — — Многофункциональный
30-150 350-450. — CoMoO4 на Al2Oa
35-70 540-650 — ?
30—300 300- 460 1,0 NiO + MoO8 на А12Оэ
против 22%, длительность работы в зоне температур
590—610 °C — 500 против 300 ч. Катализатор регене-
рируется смесью азот—воздух
Продукты конденсации терфенилов (полифенилы,
трифенилены и др.), образующиеся при использовании
органических теплоносителей в ядерном реакторе, реге-
нерируются при помощи гидрокрекинга
Сообщается о переработке различных видов тяже*
лого сырья на установке H-Oil. Процесс H-Oil и его
модификация Ну-С позволяют превращать остатки
в более легкие продукты (степень превращения 95%)
Разработан новый процесс гидрокрекинга Hy-G
(фирмы Houdry и Gulf), перерабатывающий широкий
ассортимент тяжелого сырья, вплоть до деасфальтиза-
тов. Катализатор регенерируется через 3—8 месяцев
без выгрузки. Из тяжелого вакуумного газойля можно
получить (в объемн. %): 1) 5,2% углеводородов Cs,
14,1% углеводородов С4, 112,3% бензина или 2) 1,3%
углеводородов С3, 3,2% углеводородов 64, 23,4% бен-
зина, 87,7% печного топлива
Предложен процесс обессеривания сырых нефтей.
В промышленной установке достигнуто 53%-ное обес-
серивание иракской нефти (начальное содержание серы
2,08%). Расход катализатора 3,5 м3/кг
Сообщается об условиях и опыте работы процесса
деметилирования толуола — «детол». Катализатор
очень стабилен и не менялся в течение четырех лет.
Чистота бензола 99,95%, содержание тиофена менее
1 млн*1. (См.302)
| Гидроочистка применена ддя повышения качества
не только парафинов, но и микрокристаллических
восков: содержание серы в восках понижено с 0,45
до 0,02%. Оптимальные условия процесса: 360 РС
и 300 кгс/см2
327
328,
329
330,
331
332
333
334
Давле- ние, кгс/см1 Темпера- тура. °C Объемная скорость (проточи. уст.), ч—1 Катализаторы
— — — —
50—150 380- 425 1,0 Co-j-Mo на А12О3
50 375-400 0,1-1,5 —
Продолжение табл. 4
Основные результаты
Лите-
рату-
ра
Описана модификация процесса гидрокрекинга фир-
мы BASF (DHC-Verfahren) применительно к различ-
ным видам сырья. Катализаторы не содержат благород-
ных металлов. Дизельное топливо можно получать
в одну ступень, бензин и реактивное топливо — лучше
в две. Прп парциальном давлении водорода НО кгс/см2
и циркуляции тяжелых фракций вакуум-дистилляты
перерабатывали с объемной скоростью 0,35—1,1 ч-1.
Выход целевых продуктов 90,3% (ливийская нефть)
и 94,1% (кувейтская). Цетановые числа дизельных
топлив порядка 50, содержание серы не более 0,01%.
(См.187. 1»8, 818)
Показано, что гидрокрекинг арланского вакуумного
дистиллята (3,4% серы) дает гидрогенизаты с содержа-
нием серы 0,08—0,45%. Выход бензина 0,3—4,0%,
дизельйого топлива 28,5—56,1%, остатка 71,2—39,9%.
Расход водорода 1%. Катализатор служит 3 месяца без
снижения активности. При опытном пробеге на про-
мышленной установке выход остатка с 0,1% серы
составил 55,9%. Для более глубокого расщепления
нужны две ступени. Во второй ступени применяется
катализатор NiS на алюмосиликате, удовлетворительно
работающий при достижении в первой ступени содер-
жания азота 0,01%. В бензиновом варианте выход
бензина 55%, дизельного топлива 27,4%, остатка 9,0%;
в дизельнотопливном — соответственно 32,0, 51 0
и 10,2%
Разработана схема гидрирования с катализатором,
взвешенным в жидкой фазе. Из вакуумного газойля
получено до 11,1% бензина, 23,6—49,4% дизелыюгб
топлива и 39,7 — 76,4% остатка, в котором содержа-
лось только 0,11—0,32% серы
335
336
336
tin
20 340-360 5,0—50,0 Al + Co + Mo
270 300—325 1 0,5—0,6 NiS(40%) + WSa(60%)
50 320 1.0 NiS + WS2 на A12O3
— — — —
Показано, что удается гидроочищать бензины вто-
ричного происхождения, лишь незначительно затра-
гивая олефины. В одном из опытов содержание серы
снижалось с 0,41 до 0,12%, иодное число — с 90 до
70 мг 12/100 г
В промышленном масштабе осуществлено глубокое
гидрирование бензола до циклогексана; степень кон-
версии бензола 99%, чистота циклогексана 99,38%
Осуществлено гидрирование нафталиновой фракции
до тетралина и декалина. Степень конверсии 93—95%
Сообщается о возможности использования процесса
«изомакс» для переработки остатков
Разработан процесс гидрокрекинга BASF-IFP [фирма
BASF (см.ззБ) и Французский Институт нефти]. Особен-
ность процесса—возможность производить дизельное
и печное топливо. В одноступенчатом процессе (или в
цервой ступени двухступенчатого варианта) в качестве
катализатора применяются окислы Ni или Со и окис-
лы W или Мо, нанесенные на кристаллические алюмоси-
ликаты. Во второй ступени—платиновый или палладие-
вый катализаторы. Сырье для второй ступени должно
содержать менее 0,001% азота и 0,1% серы. Дизельное
топливо может быть получено из любого сырья, даже
из деасфальтизата. В одном из опытов выходы в одно-
ступенчатом процессе составили: 2,8% H2S + NH3,
1,02% Cj + С2, 3,79% С8 + С4, 5,88% легкого бен-
зина, 13,65% лигроина, 65,36% дизельного топлива,
10,0% печного топлива. В двухступенчатом варианте:
2,75% H2S + NH3, 1,45% Ci + С2, 12,20% С3 + С4,
22,0% легкого бензина (октановое число 82), 64,90%
тяжелого бензина (октановое число 58)
Сообщаются более подробные сведения о процессе
гидрокрекинга Hy-G. Может быть переработан любой
тип газойля (кроме смолистого) в бензин, лигроин,
авиационный керосин или печное топливо. Приведены
расходные коэффициенты, катализатор служит 2 года,
цикл между регенерациями — 6 месяцев. (См.830, 881)
337
338
338
339
340
341
Давле- ние, кгс/см2 Темпера- тура, °C Объемная скорость (проточи, уст.), 4-i Катализаторы
50 350-360 1,0-3,0 СоО-|-МоО3 на А12О3
70 425 0,5 Ni+W на алюмосили- кате
50 350-450 0,7-1,5 Бентонит и зола камен- ного угля
50—100 — — А1 + Со + Мо
50 350—400 0,5—2,0 То же
Лродолжение табл. 4
Основные результаты
Дано более полное описание процесса струйной
гидроочистки фирмы Shell Oil (см.204, 22в,2в1). Отличи-
тельной особенностью является небольшая степень
рециркуляции и подача водорода в смеси с сырьем
прямотоком сверху вниз. При этом дистиллят образует
пленку па поверхности катализатора. Выше 390° С
начинается гидрокрекинг. Расход водорода 3—4 моль
на 1 г-атом серы. Азот удаляется неполностью, хорошо
удаляются кислород, диены и олефины
Разработан катализатор, особенно хорошо подходя-
щий для удаления азота при гидроочистке или первой
ступени гидрокрекинга. Из газойля с 0,319% азота
стандартный алюмокобальтмолибденовый катализатор
удалял 80% азота, новый — 92,5%. Новый катализа-
тор стабилен и через 90 суток еще удалял 75% азота
Предлагается процесс деметаллизации остаточных
нефтепродуктов. При использовании бентонита сте-
пень деметаллизации составляет 88,6%
Показано, что в одноступенчатом процессе гидрокре-
кинга с рециркуляцией остатка невыгодно снижение
глубины разложения за проход, более целесообразно
повышение давления с 50 до 100 кгс/см2, при этом ко-
эффициент рециркуляции снижается почти вдвое (до
2,7)
Найдена возможность глубокого обессеривания экс-
тракта ароматических углеводородов из высокосерни-
стой нефти (4,67% серы) с одновременным частичным
гидрированием конденсированных би- и трицикличе-
ских углеводородов. Удаление серы на 96 —98%, сте-
пень гидрирования 45—70%. Получен малосернистый
Лите-
рату-
ра
342
343
344
345
346
300 370—420 0,4-1,3 WS2; WS2 + NiS на А12О3 Ni
— — — Благородный металл
25-90 ’ 700- 1100 — Беа катализатора
Повы- шенное 400-480 1 —
продукт, содержащий 60% моноциклических аромати-
ческих углеводородов. Селективность катализатора
сохраняется в течение 1000 ч
На примере мазута из ромашкинской нефти показана
возможность получения смазочных масел гидрирова-
нием. Лучший катализатор WS2. С меньшей произво-
дительностью можно работать и при 150 кгс/см2
Фирмой British Petroleum предложено разделить
процесс гидрирования бензола до циклогексана на
3 ступени: гидроочистка, гидрирование основной массы
(до 95%) с использованием газов риформинга и дегид-
рирование. Предложен новый катализатор; утвержда-
ется, что новая технология снижает себестоимость.
Чистота циклогексана 99,99%
Рекламируется процесс НА-84 гидрирования бен-
зола фирм' Sinclair Research и Engelhard Industry.
Новый катализатор RD-260 работал 2 года со 100%-ным
превращением бензола и полным отсутствием изомери-
зации циклогексана в метилциклопентан
Описан процесс МНС—термическое деалкилирование,
разработанный фирмой Mitsubishi. Сырьем служит
пироконденсат производства этилена. Особенностями
процесса являются возможность содержания в сырье
до 30% неароматических углеводородов, низкие тре-
бования к чистоте водорода, специальная конструкция
реактора. Неароматические углеводороды гидрокре-
кируются до метана. Чистота получаемого бензола
99,96—99,99%
При помощи гидрокрекинга может быть получен
бытовой сжиженный газ. В двух вариантах — умерен-
ном и жестком (цифры в скобках) —'из лигроина, со-
держащего 11,5% ароматических углеводородов, полу-
чено: 0,9 (8,7)% Сг—С2, 14,8 (61,4)% С3, 30,0 (14,4)%
С4, 34,1 (0,0)% бензина. Выход углеводородов С8—С4
растет с увеличением степени превращения и достигает
максимума при 85—95%
347
348
349
350
351
00
Давле- вие, кгс/см2 Темпера- тура, °C Объемная скорость (проточи, уст.), ч-1 Катализаторы
7-700 232—427 — Окислы или сульфиды Со и Ni
— — —
40 39Q-430 0,3—2,0 С0М004 на А^Оз
— » — —
— — — —
Продолжение табл. 4
Основные результаты
Лите-
рату-
ра
При двухступенчатом гидрокрекинге вредное влия-
ние остаточных азотистых соединений после первой
ступени (при их содержании 0,0003—0,002 вес. %)
можно компенсировать добавками галогенсодержащих
соединений типа дихлорэтана, четыреххлористого угле-
рода, mpem-бутилхлорида и других в количестве 10—
300 атомов галогена на 1 атом азота. Отравление ката-
лизатора уменьшилось в 2,5 раза
Сообщается о пуске первой в мире установки «изо-
макс» для обессеривания котельных топлив (фирма
Idemitsu, Япония)
Изучалось гидрообессеривание нефти месторождения
Хафи (Ближний Восток), содержащей 2,8% серы. Сде-
лан вывод, что экономически более выгодно вести про-
цесс в мягких условиях с удалением 30—60% серы.
При этбм расход водорода близок к теоретическому,
около 4 моль на 1 г-атом серы (считая, что сера пред-
ставлена соединениями типа бенз- и дибензтиофена).
В жестких условиях при удалении 80% серы расход
водорода возрастает до 6 моль на 1 г-атом серы. В мяг-
ких условиях соединения ванадия не разрушаются, что
увеличивает срок службы катализатора
Рекламируется возможность использования процесса
«изомакс» для облагораживания нефтяных остатков
(процесс RCD-Isomax). Так, из нефтяных остатков, со-
держащих 3,0—4,2% серы, 0,035—0,102% никеля 4-
+.ванадия и 42—54% асфальтенов, выход жидких
продуктов 101,9—102,6 объемн. %, в том числе 99,3—
100 объемн. % фракции выше 177° С с содержанием
серы 1 %
Предлагается два варианта процесса переработки
352
353
354
355
356
нефтяных остатков: RCD-Isomax (максимальное обес-
серивание при незначительном крекинге) и BOC-Iso-
max (максимальный гидрокрекинг со значительным
обессериванием). По каждому из вариантов пущены
промышленные установки. Так, из вакуумного гудрона
с 5,07% серы получено в процессе RCD-Isomax 64,0%
дистиллятов с 0,24% серы и 32,2% остатка с 2,03%
серы (обессеривание на 84%), в процессе BOC-Iso-
max — 81,3% дистиллятов с 1,64% серы и 10,4% остат-
ка с 4,93% серы (обессеривание на 65%).
Фирмой Esso Research предлагается три варианта
получения малосернистого котельного топлива: 1) ва-
куумная перегонка, гидроочистка дистиллята и сме-
шение гидроочищенного продукта с вакуумным остат-
ком (содержание остаточной серы 1—2%); 2) то же -f-
+ деасфальтизация и добавка деасфальтизата к ди-
стилляту для обессеривания (содержание остаточной
серы 0,5—1,0%); 3) прямая гидроочистка мазута до со-
держания серы 0,3—1,8% на новых металлоустойчивых
катализаторах со сроком службы до 2 месяцев
В качестве одного из упрощенных вариантов полу-
чения малосерпистых котельных топлив (см. 21) пред-
лагается легкий термический крекинг нефтяных остат-
ков, содержащих менее 2,2% серы, вакуумная перегон-
ка, гидроочистка дистиллята и смешение с остатком
перегонки
Изучен процесс легкого гидрокрекинга ромашкинской
пефтц в присутствии допоров водорода. Содержание
серы снижается с 1,62 до 1,0%, увеличивается выход
газойля и котельного топлива
Обзор процессов гидроочистки дистиллятного сырья.
Дальнейший прогресс может быть достигнут только
разработкой новых катализаторов, применение кото-
рых позволило бы снизить давление и температуру,
а также облагораживать дистилляты и другими путями,
помимо удаления серы. •
357
358
359
8
Давле- ние, кгс/см* 1 Темпера- тура, “С Объемная скорость (проточи, уст.), ч-1 Катализаторы
Повы- шенное 150—260 310-430 — Co-j-Mo на AI2O3 1 Co-j-Mo на А12О3 J '
Повы- шенное 170 200—240 355 (II) NiS+WS2 1 ,1]]} Со+Мо на А12О3 / (1"'
4-56 70—210 343-413 — Со+Мо на А12О3 (I) Различные (II)
,210 370-454
Продолжение табл. 4
Основные результаты
Лите-
ратур
ра
Приведена сводка наиболее применяемых процессов
гидрооблагораживания бензина, получаемого в про-
цессе производства этилена. Наиболее распространен
двухступенчатый процесс фирмы Universal Oil Pro-
ducts (I), осуществляемый с 1958 г. Фирма British
Petroleum с 1959 г. использует одноступенчатый про-
цесс (II), в котором катализатор разделен на 3
слоя. На предприятии Leuna (ГДР) осуществлен
двухступенчатый процесс с двумя разными ката-
лизаторами (III). Модификации гидроочистки пиро-
конденсата разработаны также фирмами Kellogg (1962 г.)
и Houdry (1963 г.). (См.163, 213, 23°, 281, 361)
Дается сводка и общая характеристика процессов
гидрооблагораживания смазочных масел *: гидродо-
очистка' (I), гидроочистка и глубокая очистка (II).
Процесс осуществляется в относительно мягких усло-
виях, удаляются нестабильные компоненты и неугле-
водородные примеси. Из последних легко удаляются
соединения, содержащие кислород и серу, относительно
трудно (на 10—25%) — азотсодержащие соединения.
В процессе II протекают реакции изомеризации (осо-
бенно парафинов), гидрирования и деструкции поли-
циклических соединений; образуются высокоиндекс-
ные масла. При осуществлении процесса в более жест-
ких условиях индекс вязкости увеличивается, но вяз-
кость и выходы при этом снижаются
Приводится неполная сводка работ по гидроочистке
и гидрокрекингу нефтяных остатков **
Подробное описание результатов, полученных в про-
цессе H-Oil (см. 328, 326). Два реактора установки по-
360
361
362
363
6 Заказ 271
40 70-150 1,0 зволяют добавлять и выводить катализатор. При гид- рокрекинге остатков катализатор добавлялся 1 раз в сутки, при обессеривании масел — 1 раз в несколько суток. Продолжительность жизни катализатора от 39— 40 м3/кг до 9,3 м3/кг. Обессеривание остатков самых различных нефтей достигалось на 75—80% Испытан новый катализатор гидростабилизации пи- роконденсата (см.зв“) 364
Pd сульфидированный
А1 + Со + Мо
50 *** 200 Промывка растворителями закоксованного катализа- тора гидрокрекинга снижает на 60% содержание кокса и облегчает последующую регенерацию катализатора 365
—4U 380-400 0,8-1,2 Al + Co + Mo; Al + Ni + + Мо В промышленных условиях гидроочистки дизельного топлива сравнивались два катализатора. Через 20 ме- сяцев алюмоникельмолибденовый катализатор после регенерации не отличался по активности от свежего, а алюмокобальтмолибденовый — несколько снизил ак- тивность (см. 2вв) 366
Ni или Pd на цеолитах Разработана технологическая схема сочетания про- цессов гидрокрекинга ,и дегидрирования, позволяющая перерабатывать прямогонные тяжелые бензины в вы- сококачественные автомобильные бензины 367
100 425 1,0 Различные ! Сравнивались катализаторы второй ступени двухсту- пенчатого гидрокрекинга вакуумных дистиллятов в мо- торные топлива. Испытаны железные, никедевые, пла- тиновые, хромовые, молибденовые и другие окисные и сульфидные катализаторы на различных носителях. Лучшими оказались Ni и Pt на алюмосиликатах, глав- ным образом никель в частично осерненной форме. Определены нормы очистки сырья от азота в первой ступени; эти нормы тем жестче, чем ниже давление во- дорода на второй ступени 368
50 400—440 1,0-1,5 Fe на алюмосиликате Проведен опытный пробег установки гидроочистки дизельного топлива. Из сырья с. 0,8—0,9% серы полу- чено товарное дизельное топливо с 0,19—0,28% серы 369
* См. также ССЫЛКЖ 243, 232, 323—326, 330, 147.
** См. также ССЫЛКЖ ITS, 2ТТ, 239, 290, 304, 307, 323, 329, 332, 339, 340, 364—363.
•“ Время контакта (стационарная установка) 4 я.
Давле- ние, кгс/см2 Темпера- тура, °C Объемная скорость (проточи, уст.), Ч-1 Катализаторы
50 350-425 0,5-5,0 А1 + Со + Мо
100 Al + Со + Мо; Al + Ni-|-
+ Со
50—150 300—385 0,42— Al - Со - Мо
1,09
100 380—420 0,24- Pt на алюмосиликате
0,94
50 380-390 — А1 + Со + Мо
50 400-460 1,0—1,3 WS2
20 350-465 0,5 А1+ Со + Мо; WS2
250 400—440 0,5-1,6 WS2 + NiS на А12О3
50—250 425 1,0 Со + Мо на Л12О3
Продолже.ние табл. 4
Основные результаты
Лите-
рату-
ра
Показано, что продукт экстракции керосино-газой-
левой фракции высокосернистой нефти обводненным
пиридином может быть гидроочищен. После этого он
становится пригодным либо для каталитического кре-
кинга, либо для высокотемпературной гидрогенизации
с получением ароматических углеводородов С6- С10
Гидрированием в одну или две ступени из фракций
360—500 °C сернистой нефти получены различные
типы депарафинированных моторных масел, успешно
прошедших моторные испытания
Гидроочисткой тяжелого вакуумного дистиллята в пер-
вой ступени и гидрокрекингом — во второй ступени
можно получить 60% масел, в том числе высокоиндекс-
ных, и 40% топлив, в том числе 30% зимнего дизельного
топлива
Технические парафины и гачи в одно- или двухсту-
пенчатом процессе могут быть переработаны в высоко-
индексные смазочные масла
Вязкие масла (авиационные, трансформаторные и ма-
сла для прокатных станов) получают смешением
продуктов гидрирования и гидрокрекинга деасфальти-
рованных гудронов
Глубоким гидрированием фракций сырой нефти,
выкипающих при 300 —400 °C, и фракций продуктов
каталитического крекинга, выкипающих при 200—
500 °C, получены специальные нафтеновые и компрес-
сорные масла
При переработке вакуумного газойля из смеси сер-
нистых нефтей, сЬдержащих 2,16% серы, получены
гидрогенизаты с 0,06—0,26% серы (в зависимости от
давления). Выход тяжелой фракции, выкипающей выше
370
371
372
373
374
375
376
ст> • 100 400—450 1,0-3,0 A14-Ni + Mo
100 400-450 1,0-3,0 То же
50—140 70-140 430-480 340-430 1,0 0,5-1,5 Алюмосиликат Окись металла на А]
100 380—410 0,5—1,0 ?
2 Од
350 °C, равен 38,3—50,9%. При возвращении этой фрак-
ции в цикл выход моторных топлив до 90%, в том числе
81—82% малосернистого дизельного топлива
Гидрокрекинг в трехфазном псевдоожиженном слое
катализатора разработан на холодной модели (см.ззв)
и проверен на пилбтной установке с дистиллятным
сырьем и мазутом арланской нефти. Глубина расщепле-
ния и обессеривания значительно больше, чем в не-
подвижном слое катализатора (см.382). Из мазута с
4,11% серы получено 4,9% бензина, 51,2% дизельного
топлива и 38,5% остатка >360° С, содержащего 0,84%
серы
Гидрокрекинг в циркулирующем потоке катализа-
тора проверен на пилотной установке мощностью
10—35 кг/ч на обессоленной отбензиненной нефти и
вакуумном дистилляте. Расход водорода 0,99—1,12%;
выход гидрогенизата 89,2—91,0%; содержание серы
снижается с 1,70—2,01 до 0,25—0,31%. Остаток
>330 °C с 0,52% серы может быть получен с выходом
35,9% на нефть. (См.2вз,2И, 2!1», 3°7)
В схеме Варга катализатор FeSO4 на полукоксе
заменен природным алюмосиликатом с 7% FeSO4.
В первой ступени удаляется 30% серы, во второй —
89,5% оставшейся. Из отбензиненной ромашкинской
нефти получено 41,35% фракции до 280 °C ((0,0%
серы, 26,1% фракции 280—360 °C (0,12% серы), 19,35%
фракции 360—445 °C (0,18% серы), 2,7% фракции
445—520 °C (0,17% серы), 6,3% фракции >520 °C
Разработан катализатор, устойчивый в условиях
гидроочистки остатков. При очистке до 1 % серы он
работает 1500 ч. При 300 кгс/см2 срок службы увели-
чиваетвяв 2—3 раза. Регенерация его, однако, затруд-
нена, так как он адсорбирует из сырья до1—3% никеля
и 1,5—5,0% ванадия. Рекомендуется для переработки
деасфальтизатов
377
378
379
380
Давле- ние, нгс/см2 Темпера- тура, °C Объемная скорость (проточи, уст.), Ц-1 Катализаторы
— — — —
100 400—450 1,0 WS24~NiS на А13О3 (фто- рированный)
— — — —
40—60 49—249 — —
150—300 360-440 — А1+ Ni + Mo и А]-|-Со -|~ + Мо
Продолжение табл. 4
Основные результаты
Лите-
рату-
ра
Сообщается о работе пилотной установки (в Канаде)
для гидрокрекинга тяжелых остатков, пеков и битумов.
В установке использован принцип процесса Kombi-
verfahren (см.1’8)
С целью подбора условий одноступенчатого гидро-
крекинга вакуумных газойлей до средних дистиллятов
испытаны различные катализаторы. Разработан новый
катализатор (фторированный), на котором при степени
превращения 53,5—69,5% образуется 76—78% фрак-
ций >150° С. Катализатор работает 1800 ч, после чего
в сырье нужно добавлять фтор (в виде фтортолуола)
Описан опыт работы самой крупной в мире установки
гидрокрекинга. Установка состоит из двух блоков
типа «изомакс»: первый — одноступенчатый, перера-
батывающий деасфальтизат на сырье для каталитиче-
ского кфекинга: второй — двухступенчатый, перераба-
тывающий газойли каталитического крекинга на бен-
зин и реактивное топливо. Комбинирование с катали-
тическим крекингом существенно улучшило показатели
и производительность второго блока
Описан двухступенчатый процесс гидростабилизации
пиробензинов (фирма Kellogg). В первой ступени диены
и ацетиленовые углеводороды селективно гидрируются
до моноолефинов; продукт первой ступени может быть
добавлен в товарный бензин. Во второй ступени гид-
рируются моноолефины и разрушаются сернистые сое-
динения, чтобы подготовить продукт к экстракции аро-
матических углеводородов. (См.зв0)
Разработана и проверена в полупромышленном мас-
штабе схема получения малосернистого котельного
топлива из высокосернистых( нефтей с высоким со-
381
382
383
384
385
40 390-430 — А1-]-Мо + Со
— — — —
1,5-1,8 70-90 450—920 Палладиевый
100 180-200 — Ni + Cr
50 и 30 380-400 ,2 и 5—6 А1 + Со-|-Мо
— — — То жэ
28-84 204-371 — На основе благородных металлов
Ниже обычно- го 177-316 6,0 —
держанием металлов. Схема включает деасфальтиза-
цию мазута (в которой удаляется 90—95% Ni, V, пор-
фиринов и асфальтенов) и гидрирование деасфальти-
зата (ср.380) с последующим смешением с гидроочищен-
ным газойлем. Выходы (на нефть): 24,7% бензинов,
5,4% легкого газойля, 25,0% газойля, 39,6% котель-
ного топлива с 1% серы. Расход водорода 0,45%.
Катализаторы новые, разработанные на основе стан-
дартных Al + Ni + Мо и А1 + Со + Мо
На примере гидроочистки сырой хайфинской нефти
(см.364) сравнены катализаторы с различной кажущейся
плотностью. Лучшим катализатором оказался катали-
затор с меньшей кажущейся плотностью
При гидродеалкилировании алкилароматических сое-
динений в качестве донора водорода использован 9,10-
дигидрофенантрен ,
Достигается полное гидрирование диенов (бутади-
ена-1,3) при гидрировании фракции С4 газов крекинга,
н-бутилен и изобутилен затрагиваются незначительно
Осуществлено селективное гидрирование циклопен-
тадиена и его димера в циклопентен. Гидрирование
побочных продуктов дает бензин с октановым числом 76
Глубоким гидрированием бензинов вторичных про-
цессов получено высококачественное сырье для ката-
литического риформинга с содержанием серы менее
0,003%, азота 0,0002%
Доказано, что осернение катализатора сероводородом
снижает коксообразование на 25—30% и увеличивает
межрегенерационные пробеги в процессах гидроочистки
и гидрокрекинга
Рекламируется новый процесс Unisar — гидрирова-
ние ароматических углеводородов в реактивном то-
пливе. Катализатор не требует предварительной гид-
роочистки сырья
Рекламируется новый процесс глубокого гидрирова-
ния для получения высококачественных керосинов
и реактивных топлив. Катализатор регенерируемый,
с длительным сроком службы
386
387
388
389
390
391
392
393
dfc
а
Давле- ние, кгс/см2 Темпера- тура, еС Объемная скорость (проточи, уст.), Ч-1 Катализаторы
Ниже 56 275—315 6,0 Благородный металл на специальном носителе
30-40 30-50 130—250 20 350 — А1+Со+Мо Pd Со+ Мо
350 445 — Fe (I); FeO + NiS с до- бавками W, Сг, Zn (II); Al+Co+Mo
40 410—430 0,3—1,0 Al -J- Co Mo
25—100 150 375—450 0,4 Al+Ni + Mo; Al + Mo + + Co Нечувствительный к сере и азоту (I); расщепляющий (II)
20-175 330-360 0,5-2,5 А1 +Со+Мо (осерненный)
Продолжение табл. 4
Основные результаты
Указывается, что при получении керосинов и реак-
тивных топлив (см. выше) почти не образуется газооб-
разных продуктов; можно получать реактивное топливо
с высотой некоптящего пламени до 44 мм и большой
ассортимент растворителей с содержанием ароматиче-
ских углеводородов менее 1%
Описаны различные варианты гидростабилизации
бензинов пиролиза (см.360). Достигается полное удале-
ние диенов; индукционный период бензинов возрастает
до >• 360 мин
Приведены более подробные данные о комбиниро-
ванном способе переработке нефтяных остатков
(см.381),. Содержание серы в бензине и реактивном то-
пливе 0,27%, в дизельном топливе 0,67%, в остатке
1,04%
Продолжение исследований по прямой гидроочистке
хайфинской нефти (см.386). Делается вывод о необхо-
димости предварительного удаления асфальтенов и при-
менении широкопористых катализаторов
Изучалось обессеривание нефтяных остатков из нефти
АРЕ. Достигнуто снижение содержания серы до 1%
Описаны два варианта гидрокрекинга тяжелых ди-
стиллятов коксования и каталитического крекинга
по обычной двухступенчатой и «совмещенной» схеме,
в которой гидрогенизат первой ступени подается на
вторую без разделения продуктов. Выход дизельных
топлив от 74,5 до 83,5%. Содержание серы снижается
с 2,39 до 0,01%
Изучено гидрирование ароматических углеводородов
Лите-
рату-
ра
394
395
396
397
398
399
400
40 — 1,0 Pt на цеолитах разного состава
Повы- 205-288 Pt на А12О3 (обработан-
шенное ный НС1, SO2C1, и снова
НС1)
200 387
Со + Мо на А12О3
35—200 343—455
Умерен- 343—400
ное
0,3—3,0
Ni + V
и их смесей в присутствие 0,1 —20,0% водяного пара.
Присутствие последнего тормозит гидрирование за счет
адсорбционного вытеснения; в наибольшей степени
тормозится гидрирование моноциклических углеводо-
родов, за счет чего повышается селективность гидриро-
вания бициклических углеводородов
Изучен гидрокрекинг легких парафиновых углево-
дородов с целью получения изобутана и изопентана,
используемых в синтезе мономеров для синтетического
каучука. Наиболее подходят катализаторы со средней
гидрирующей активностью
В мягких условиях из пентана, гексана и нафты
получают сжиженный бытовой газ (соответственно 76,
80 и 102 объемн. % за проход)
Сопоставлены результаты гидрокрекинга различного
сырья на стационарном и движущемся катализаторах.
Первые более эффективны по удалению серы, азота
и кислорода при низких объемных скоростях, вторые —
при более высоких. По выходу нафты катализаторы
различаются только при низких объемных скоростях
Описано модифицирование носителя кобальтмолибде-
нового катализатора для гидроочистки мазутов (см.38Ь)
добавками 1,5—3,1 %• металлов второй группы. Окиси
Be, Mg, Са, Sr, Zn и Cd увеличивают объем микропор
и активность катализатора, а окись Ва — уменьшает
Изучалось прямое обессеривание тяжелых масел
и сырых нефтей на катализаторе повышенной активно-
сти в системе с движущимся слоем катализатора. Ак-
тивность катализатора повышается с увеличением со-
держания СоО и МоО3. Из остатка с 4,26% серы полу-
чен продукт, содержащий 0,9% серы
Рекламируется процесс гидроочистки мазутов Gulf-
HDS. В промышленных условиях содержание серы
снижено с 4 до 0,87%
Рекламируется применение процесса «изомакс» фир-
мы Chevron Research для гидроочистки нефтяных ос-
татков. Содержание серы снижено с 3 до 1% и ниже
Описывается новый процесс гидроочистки нефтяных
остатков. Содержание серы снижено с 5,2 до 1%
401
402
403
404
405
406
407
408
Давле- ние, кгс/см’ Темпера- тура, °C Объемная скорость (протоны, уст.), ч~> Катализаторы
— — —
70 371-427 0,25—2,0 NiCU на алюмосиликате с 6% А12О3 + НС1
Продолжение табл. 4
Основные результаты
Лите-
рату-
ра
Для улучшения качества реактивного топлива ре-
кламируется процесс (фирма Shell Oil) глубокого (на
90%) гидрирования ароматических углеводородов до
нафтенов. Используется регенерируемый катализатор,
содержащий благородный металл; срок работы 3—
5 лет. Допустимое содержание серы в сырье —0,0005% .
Гидрогенизат используется для компаундирования (1:1)
с прямогонным керосином
Катализатор, содержащий хлор, лучше удаляет
азот, чем обычные никелевые, молибденовые и вольфра-
мовые катализаторы. Из сырья с 0,315% азота и 0,888%
серы удалено до 95% азота. Азот выделяется в виде
NH4Cl. Для поддержания активности катализатора
добавляется газообразный хлористый водород в отно-
шении Cl : N = 9,4
Обобщены данные о масштабах использования гид-
рокрекинга: имеется 50 установок мощностью 122 тыс.
м3/сут и строится еще 17 мощностью 58,5 тыс. м3/сут.
Отмечается продолжающаяся тенденция (см.1в) вовлече-
ния в переработку все более тяжелого сырья. Приве-
дены типичные выходы продуктов гидрокрекинга: при
получении сжиженного газа (97,9 объемн. % углево-
дородов С3 С4, 32,3 объемн. % углеводородов С5 +
+ Се), при получении бензина (122,3 объемн. % фрак-
ции С4 — 204,4 °C), реактивного топлива (суммарный
выход 123,2 объемн. %, в том числе 58,9 объемн. %
реактивного топлива). Указывается на некоторые успехи
усовершенствования катализаторов и доведение меж-
регенерацпонных пробегов до двух п более лет. Кратко
характеризуются тенденции сочетания гидрокрекинга
с другими процессами: с каталитическим риформингом,
409
410
411
105-280
каталитическим крекингом и с исчерпывающим гидри-
рованием турбинного топлива, а также использование
его вместо каталитического риформинга
Отмечается, что при гидрокрекинге остаточного
сырья, в отличие от дистиллятного, имеют место
затруднения, связанные с присутствием в остато-
чном сырье повышенных количеств серы и азота,
а также асфальтенов, солей и металлоорганических сое-
динений. Сообщается об испытании в процессе H-Oil
(см-333) нового микросферического катализатора: из
остатка с 2,08% серы (75% кипит выше 524 °C) при рас-
ходе водорода 116 м3/м3 получено 103,1 объемн. %
продукта с 0,44% серы, в том числе 8% бензина, 33,9%
дизельного топлива, 32,4% газойля и 25,7% котельного
топлива (>524 °C) с 0,93% серы
Описывается опыт работы первой промышленной
установки процесса RCD-Isomax (см. збв) и дальнейшие
усовершенствования катализатора этого процесса. Ус-
тановка построена фирмой Idemitsu (Япония), мощность
6360 м3/сутки. В испытательном пробеге при расходе
водорода 148 м3/м3 получено 1,1% бензина, 6,1 %
среднего дистиллята (0,03% серы) и 94,1% котельного
топлива (1,02% серы) из отбензиненной нефти, содер-
жавшей 3,90% серы. Все данные относятся к выработке
котельного топлива с 1% серы. Если вырабатывать
топливо с 1,4% серы, срок работы катализатора про-
длится до 11 месяцев. Пущены еще две установки,
которые работают удовлетворительно. На одной из них
из нефти с 3,99% серы получено 98% котельного то-
плива с 1% серы
Обобщаются данные по применению гидрокрекинга
для производства смазочных масел (см.331,3’1-376).
К середине 1970 г. работали три промышленные уста-
новки. Гидрокрекинг несколько выгоднее экстракции
растворителями, причем это преимущество тем больше,
чем выше индекс вязкости масла. Высокоиндексные
масла можно получать только гидрокрекингом. Лучшее
сырье — парафинистое
412
413
414
g
Давле- ние, кгс/см2 Темпера- тура, ®G Объемная скорость (проточи, уст.), кЧ'1 Катализаторы
— — — —
40 320-400 0,25-1,0 Pt на цеолите
50 625—700 0,75-4,1 Без катализатора; А1 -|- + С04-М0; Alp-Ni + Mo; Со-|-Мо на цеолите
100 400—425 1,4 АЦ-Со + Мо
Продолжение табл. 4
Основные результаты
Лите-
рату-
ра
Приводится обзор патентов по катализаторам гидро-
крекинга дистиллятов и мазутов.
Сопоставлены выходы легких пзоалканоп — сырья
для получения мономеров для СК — из парафинов,
нафтенов и ароматических углеводородов Св — Сп.
Лучшее сырье — парафины С9—Св> но приемлемы и
смеси углеводородов С, — С, t (бензин с т. кип. 85 —
200 °C). При переработке широкой фракции катализа-
тор должен обладать высокой гидрирующей актив-
ностью (ср.401)
Концентрат ароматических углеводородов, получен-
ный экстракцией керосино-газойлевой фракции высо-
косернистой нефти, перерабатывается в высокоаромати-
зированный бензин, пригодный для извлечения аро-
матических углеводородов Св—С10 или как высокоокта-
новый (октановое число 100,3) компонент. Лучший из
катализаторов — Мо на цеолите, ускоряющий гидро-
крекинг и гидродеалкилирование при более низкой
температуре
Осуществлен гидрокрекинг остаточного сырья в пи-
лотной установке в трехфазном псевдоожиженном
слое (см. также 377). Содержание серы снижено с 3,8%
в сырье до 1,0% в маловязком котельном топливе.
Образование кокса не более 0,2% на сырье. При 400 °C
выход малосернистого котельного топлива из гудронов
ромашкинской и арланской нефти соответственно 90,4
и 86,3%; при 425 °C он уменьшается почти вдвое, за
счет чего увеличивается выход летнего дизельного
топлива
415
416,
417
418,
419
420,
421
150 380—450 1,0 Al + Mo -|- N i
100 410 1,0 To же
Без указания условий и катализатора рекламируется
процесс переработки нефтяных остатков, разработан-
ный Французским Институтом нефти и включающий
стадию «предварительной обработки», в которой адсор-
бируются поликонденсированныо соединения. Содержа-
ние серы снижается во фракции 376—550 °C с 3,45 до
0,30%, в остатке >550 °C — с 5,8 до 2,35%. Без пред-
варительной обработки снижение меньше — 0,37 и
2,85% соответственно
Па лабораторной установке сравнивались различные
катализаторы обессеривания остаточных фракций хай-
финской и кувейтской нефтей. Изучались методы внесе-
ния активных компонентов и температуры прокалива-
ния. Активны катализаторы, приготовленные из у-А12О3
с малой кристалличностью и преимущественным радиу-
сом пор >100А. В длительных пробегах (1000—3000 ч)
производительность на опытных образцах катализато-
ров составила 6,0—9,2 м3 сырья на 1 кг против 5,6 м3/кг
у промышленных образцов
Наиболее активные образцы катализаторов получены
двухстадийной пропиткой (сначала наносится МоО3).
Оптимальная температура прокаливания 600 °C, про-
должительность прокаливания подбирается так, чтобы
не вызывать физических изменений структуры катали-
затора. Введение Pd и Ст понижает активность, введе-
ние MgO — повышает. Количество Мо03 — 12—15%,
NiO —4—5%. См. также предыдущую аннотацию
Сообщается о работе установки гидроочистки
нефтяных остатков в Японии мощностью 1,57 млн. т/год.
Из сырья с 4,0% серы в трех пробегах полу-
чено котельное топливо с 1,0 и 1,5% серы. В уста-
новке используется процесс Gulf-HDS. Катализатор
служил от 6 до 12 месяцев, за это время температура
повышалась на 100—120 °C для компенсации его дез-
активации (см, также *м)
422
423,
424
425
426
Ь5
Давле- ние, кгс/см? Темпера- тура, °C Объемная скорость (проточи, уст.), 4-i Катализаторы
Mo-j-Al; Мо 4“ Со 4" Al;
Go + Ni + Al; Pt на . A12O3
П родолжение табл. 4
Основные результаты
Лите-
рату-
ра
Приводятся данные об использовании двух процессов
производства малосернистых котельных топлив, раз-
работанных фирмами Esso Res. и Union Oil. (см. так-
же 21). Процесс GO-fining очищает вакуумный дистил-
лят от серы на 87%, после смешения очищенного проду-
кта с остатком котельное топливо содержит 1,72% серы
против 3% в сырье. Процесс Residfining очищает оста-
ток непосредственно и дает котельное топливо с 1%
серы. Первый процесс реализован на 4 заводах, пуска-
ются еще 5; их суммарная мощность 21,2 млн. т/год.
Второй процесс проходит пилотные испытания; создан
новый катализатор, удаляющий меньше металлов,
с большим сроком службы
Продукт риформинга, содержащий 68% ароматиче-
ских углеводородов и олефины (бромное число 1048 мг
Вг2/100 мл), селективно очищают от последних. Лучшие
результаты получены на катализаторе А1 + Со + Мо
при 350 9С, 25 кгс/см2 и 3 ч'1: бромное число 34 мг
Вг2/100 мл при том же содержании ароматических
углеводородов
Сопоставляются варианты включения в схему нефте-
перерабатывающего завода процесса гидрокрекинга
атмосферного остатка (RDS-Isomax) или сырой нефти
(CDS-Isomax). Второй вариант несколько экономичнее.
Особенностью защиты против отложения металлов
является применение форкатализатора и тщательное
обессоливание сырья. Катализаторы испытывались в те-
чение 2000 ч
427
428
429
и уступают по активности сульфидным катализаторам *, но зато
легко и полностью восстанавливают активность при регенера-
ции 35 9’ 430. Во многих процессах применяют и сульфидные ката-
лизаторы * **.
Процессы гидроочистки дистиллятного сырья проводятся как
в паровой, так и в жидкой фазе.
Парофазные процессы используют для получения бессернистых
бензинов и облагораживания сырья каталитического рифор-
минга 359- з90- 428, 4301 431, получения чистой нафты — сырья для
пиролиза и газификации, а также чистых технических растворите-
лей ***, очистки ароматических углеводородов от сернистых
соединений, в первую, очередь для очистки бензола от тио-
фена 199> 2051419,43°, очистки и стабилизации бензина пиро-
лиза **** и бензинов вторичного происхождения на нефтеперерабаты-
вающих заводах 5*. В двух последних группах процессов трудной и
еще не решенной до конца задачей является селективная очистка
бензинов от сернистых соединений с сохранением ценных олефино-
вых углеводородов.
Жидкофазные процессы разработаны для очистки кероси-
нов 2211 243> 35 91 430, 431, дизельных топлив6* и смазочных масел7*
от сернистых соединений. Гидроочистка смазочных масел сопрово-
ждается существенным изменением химического строения сырья
(в том числе деструкцией), что приближает ее к процессам гидрокре-
кинга и гидроизомеризации при получении высокоиндексных ма-
сел 361.
Данные табл. 4 (см. также обобщающие статьи и моногра-
фии 359> 4301 431) позволяют сделать вывод, что проблемы селективной
гидроочистки любых дистиллятных продуктов от сернистых, азоти-
стых и смолистых веществ в основном решены. Разработаны теоре-
тические основы управления этими процессами путем варьирования
технологических параметров 431; в случае «трудного» сырья, т. е.
сырья, содержащего много смолистых и ароматизированных компо-
нентов, помимо более жестких условий используется противоток
жидкого сырья, улучшающий его контакт с водородом 204> 23°, а также
добавка доноров водорода 274> 298’ 358. В целях уменьшения расхода
водорода процессы проводят в условиях, при которых наряду с гид-
рогенолизом сернистых соединений происходит дегидрирование наф-
генов, дающее дополнительный источник водорода. Таким образом
иожет быть обеспечена автогидроочистка бензинов, керосинов и
♦ См. ссылки 3, 6, 151, 415, 431, 432.
** См. ссылки239, 241, 250, 257, 270, 288, 291, 323, 338, 347, 360,
173 375.
•••’ См. ссылки 236, 258, 265, 271, 295, 296, 359, 430.
**** См. ссылки 281, 301, 360, 364, 384, 395, 430.
ь* См. ссылки 190, 193, 194,208, 211, 213, 230, 232, 337, 359, 428,430,
131.
•* См. ссылки 198, 214, 227, 243, 261, 359, 430, 431.
1* См. ссылки 241, 245, 246, 282, 294, 323—325, 347, 361, 371, 374, 375,
44, 431.
93
дизельных топлив или сочетание гидроочистки и автогидроочистки
дизельных топлив, т. е. процессы, в которых водород совсем не под-
водится извне или его расход резко уменьшается *.
Хорошо проработаны различные сочетания процессов гидроочи-
стки с другими процессами нефтепереработки, в результате чего от-
крываются возможности резкой интенсификации последних. Это
относится в первую очередь к процессам риформинга (см. стр. 11)
и каталитического крекинга **. При каталитическом крекинге
облагороженного сырья увеличивается выход бензина, возрастает
его октановое число и производительность установок. Показана также
целесообразность сочетания гидроочистки с висбрекингом 2981 299,
в том числе с добавками доноров водорода 358.
Гидроочистка применяется для облагораживания не только чисто
топливных, но и коксохимических продуктов 200’ 211, парафинов и
восков 270’ 2801 334> 373, синтетического кумола 292 и др. Следует отме-
тить, что советские технологи были в числе пионеров разработки
этих столь важных процессов гидроочистки. Первые работы в этом
направлении относятся еще к 1935 г.433, а принципиальные резуль-
таты, легшие в основу промышленных процессов, как указывается
в работах 2271 245, были получены в 1949—1952 гг.
Второе место (после гидроочистки) занимают процессы, в которых
осуществляется более или менее глубокая деструкция сырья, —
процессы гидрокрекинга. Они также представлены большим
числом фирменных модификаций, но практически могут быть раз-
делены на две группы: одно- и двухступенчатые процессы. В пер-
вых используется относительно легкорасщепляемое сырье, а целе-
выми продуктами являются более тяжелые дистилляты, например
дизельное топливо, во вторых — стадии насыщения сырья (гидриро-
вание) и расщепления разделены. Подготовка сырья в первой сту-
пени позволяет применять во второй ступени более активные рас-
щепляющие катализаторы, вследствие чего двухступенчатые схемы
более гибки, позволяют перерабатывать неблагоприятное сырье
и получать бензин и другие низкокипящие товарные продукты.
Данные табл. 4 иллюстрируют важные тенденции в эволюции про-
цессов гидрокрекинга: гидрокрекинг, появившийся сначала как
вспомогательный процесс бензинообразования и призванный
дополнять каталитический крекинг, становится универсальнее
и приспосабливается к переработке все более тяжелого
сырья 303, 316, 383, 431
Таким образом, в настоящее время процесс гидрокрекинга стал
одним из методов переработки нефтяных остатков, в том числе пере-
работки их в низкосернистое котельное топливо.
Помимо возможности применения неблагоприятного сырья, уни-
версальность гидрокрекинга позволяет перерабатывать почти любое
сырье в любые товарные продукты. Благодаря этому нефтепере-
* См. ссылки 207, 218, 219, 243, 431.
** См. ссылки 228, 248, 273, 284, 431.
94
рабатывающий завод, имеющий установку гидрокрекинга, может
всегда приспособиться к сезонным или конъюнктурным колеба-
ниям рынка. Кроме того, на таком заводе установка гидрокре-
кинга может освободить другие установки от переработки не-
благоприятного сырья. Так, например, высокосернистые и тяжелые
газойли могут направляться на гидрокрекинг, более легкие —
на каталитический крекинг, а остатки гидрокрекинга тяжелых ва-
куумных газойлей — на каталитический крекинг 431. Считают, что
установка универсального гидрокрекинга типа «изомакс» может
быть даже головной на нефтеперерабатывающем заводе 434.
Повышение универсальности процессов гидрокрекинга и вовле-
чение в их сырьевую базу тяжелых дистиллятов, остатков и сырой
нефти определили необходимость подбора усовершенствованных ста-
ционарных катализаторов гидрокрекинга * с целью получения мало-
сернистого котельного топлива, а также разработки специальных
технологических схем, позволяющих непрерывно регенерировать
катализатор. Это так называемые системы с трехфазным псевдоожи-
женным слоем, разрабатываемые в США и СССР 328, 329> звз> 3’7,
и деструктивная гидрогенизация в циркулирующем потоке катали-
затора **, создаваемая в СССР. В этих процессах тяжелое сырье об-
разует жидкую фазу со взвешенным катализатором, в которую
подается сжатый водород. Катализатор либо непрерывно отбирается
для регенерации, а в систему добавляется регенерированный и све-
жий через специальное устройство (процессы H-.Oil, Ну-С, Hy-G
и др.), либо непрерывно циркулирует между реактором и регенера-
тором (процесс ИНХС АН СССР). Эти процессы, как видно из табл. 4,
также прошли большой путь, видоизменяясь и приспосабливаясь
к все менее благоприятному сырью ***. Как и в процессах со стацио-
нарным слоем, решающим направлением было усовершенствование
катализаторов. Так, например, разработка специального микросфе-
рического катализатора для процесса H-Oil303, 412 позволила значи-
тельно упростить процесс, увеличить глубину превращения сырья,
снизить капитальные затраты.
Несколько особняком стоит способ гидрокрекинга с добавкой
доноров водорода (способ Варга). В этом процессе используются3 04’ 3 7 9
неактивные катализаторы старого жидкофазного процесса деструк-
тивной гидрогенизации и, видимо, поэтому он не находит про-
мышленного использования, хотя идея добавки доноров водорода
универсальна и применяется в других процессах.
Благодаря успехам развития процессов гидроочистки и гидро-
крекинга можно считать почти решенной проблему прямого обес-
серивания нефти и нефтяных остатков с получением малосер-
нистых котельных топлив ****. Выше (см. стр. 13) показаны огромная
* См. ссылки 21, 339, 353, 355, 404, 406, 407, 413, 423—425, 427, 429.
** См. ссылки 222, 223, 252—254, 263, 264, 290, 307, 378.
См. ссылки 318, 330, 331, 341, 378.
**** См. ссылки 385, 405—407, 412, 413.
95
социальная значимость этой проблемы и значительные перспективы
распространения таких процессов.
Процессы гидрокрекинга находят и некоторые специфические
области применения: производство сжиженных газов за1> 102, про-
изводство легких изопарафиновых углеводородов — сырья для по-
лучения мономеров для синтетического каучука4 °1’416’417, сочетание гид-
рокрекинга с дегидрированием для производства бензинов 367 и др.
Специфическими процессами гидроочистки и гидрокрекинга не
исчерпывается интерес к гидрогенизационным процессам. Суще-
ственные успехи достигнуты в разработке процессов «чистого» гидри-
рования, т. е. таких процессов, где нужно избежать побочных реак-
ций изомеризации и гидрогенолиза.
Наиболее распространены процессы гидрирования бензола в цик-
логексан высокой чистоты *. В разработке этих процессов конкури-
руют две идеи: создание стойких к ядам и селективных (т. е. не-
изомеризующих и нерасщепляющих) катализаторов и осуществле-
ние процесса в одну ступень или, наоборот, разделение процесса
на несколько ступеней, с тем чтобы повысить эффективность каждой
операции, т. е. подготовки сырья и его исчерпывающего гидрирова-
ния. Эти же принципы используются при гидрировании нафталина
с целью получения ценных растворителей — тетралина и дека-
лина 25 °’ 338. Глубокое гидрирование ароматических углеводородов
используется для улучшения качества (повышения теплотворной
способности на единицу массы) реактивных топлив 392> 393> 409.
«Чистое» (селективное) гидрирование начинает все шире исполь-
зоваться и для получения других индивидуальных соединений.
Можно упомянуть селективное гидрирование циклопентадиена в ци-
клопентен 389 и гидрирование других диенов в моноолефины 435, вос-
становление кислородсодержащих соединений на сульфидных ката-
лизаторах 436 и гидрирование альдегидов в спирты 437’ 438.
Созданы и широко применяются процессы, в которых осу-
ществляется максимальное торможение реакций гидрирования при
интенсивном ускорении реакций гидрогенолиза. К ним относятся
в первую очередь промышленные процессы гидродеалкилирования
гомологов бензола ** и нафталина ***.
Таким образом, из всего вышесказанного видно, что процессы
гидрогенизации играют важнейшую роль в современной топливной
и нефтехимической промышленности и, что наиболее существенно,
эта роль еще больше возрастет в будущем.
Сопоставление основных тенденций развития гидрогенизационных
процессов убеждает в их растущей специализации. Увеличивается
число процессов, в которых необходима высокая селективность,
т. е. определенное соотношение между скоростями различ-
ных реакций: собственно гидрирования (гидрирование конден-
* См. ссылки 220, 285, 291, 338, 348, 349.
** См. ссылки 302, 310, 326, 333, 350, 431.
*** См. ссылки 276, 278, 293, 311, 312, 431.
96
сированных ароматических углеводородов, моноциклических аро-
матических углеводородов, диенов, олефинов и др.), реакций вос-
становления (восстановление карбоксильных, эфирных, гидроксиль-
ных и других кислородсодержащих групп, восстановление амино-
групп и т. д.), реакций изомеризации и гидрогенолиза (гидро-
генолиз связей С—О, С—N, С—S в сравнении с гидрогенолизом
связи С—С).
Разработка способов управления скоростями этих реакций долж-
на служить теоретическим фундаментом для создания новых и усовер-
шенствования известных процессов переработки твердых горючих
ископаемых, смол, нефтей и нефтепродуктов. Очевидно, что управле-
ние этими реакциями и поиски важнейшего инструмента такого
управления — катализаторов — немыслимы, без точных и. деталь-
ных знаний химии и элементарных стадий превращения компонентов
сырья в условиях различных гидрогенизационных процессов.
ЛИТЕРАТУРА
1. Pja и о и о р т И. Б. Искусственное жидкое топливо. М., Гостонтехиздат,
1955. 546 с.
2. VIII Мировой нефтяной конгресс. Москва, 1971. Препринты симпозиумов
№ 14. См. с. 11; № 12. См. с. 33; № 10. См. с. 58 и 88.
3. Лозовой А. В., Дьякова М. К. Гидрогенизация топлива в СССР.
М. — Л., Изд. АН СССР, 1940. 269 с.
4. П е т р о в А. Д. Очерки по химии моторных топлив и смазочных масел.
М., Изд. АН СССР, 1941. 308 с.
5. Немцов М. С., Усн. хим., 7, 1635 (1938).
6. К г б и I g W. Die katalitische Druckhydrierung von Kohlen, Teeren und
Mineralolen. Berlin, Springer Verlag, 1950. 266 S.
7. П e т p о в А. Д. Химия моторных топлив. М., Изд. АН СССР, 1953. 512 с.
8. S h i b a t а К., Chem. Eng., 60, 214 (1953); С. А., 47, 6634 (1953).
9. F i е 1 d n е г А. С., Fuel, 28, 19 (1949).
10. Brennstoff-Chemie, 41, 14, 53 (I960).
И. Sherwood Р. W., Petrol. Ref., 31, № 5, 161 (1952).
12. G а 1 1 D., Rev. of Petrol. Techn., Inst, of Petrol. (London), 14, 179 (1955).
13. К а л e ч и ц И. В. Современные тенденции разработки процессов получе-
ния малосернистых котельных топлив. М., ЦНИИТЭнефтехим, 1969. См.
с. 4—5.
14. О b о 1 е n t s е v R. D. VII World Petroleum Congress. Mexico. Proceedings.
V. 2. London, Elsevier Publ. Corp., 1967. See p. 109.
15. Б p e д л и В. E., Хендрикс Дж. Б., Хуфман Г. К. 'IV Между-
народный нефтяной конгресс. Т. 4. М., Гостоптехиздат, 1956. См. с. 233.
16. S с о 11 J. М., Paterson N. J. VII Wold Petroleum Congress. Mexico.
Proceedings. V. 4. London, Elsevier Publ. Corp., 1967. See p. 97.
17. Б a p а л В. И., Гуфман Г. С. VIII Мировой нефтяной конгресс.
Москва, 1971. Препринт симпозиума № 12. См. с. 3.
18. С i а р е 11 a F. G. VII World Petroleum Congress. Mexico. Proceedings.
V. IB. London, Elsevier Publ. Corp., 1967. See p. 139.
19. S 1 a d j 1 s k u E. M., Maples R. E., Oil a. Gas J., 66, № 18, 55 (1968).
20. C h о p e у N. P., Chem. Eng., 74, № 9, 92 (1967).
21. Oil a. Gas J., 65, № 38, 55 (1967).
22. Oil a. Gas Internat., 7, № 8, 67 (1967).
23. Brennstoff-Chemie, 38, 66W (1957).
24. W e i s s e r O., Landa S. Sulphide Catalysts, Their Properties and
Applications. Prague, Academia, 1972. See p. 17.
25. Schnabel B., Erdol u. Kohle, 11, 647 (1958).
7 Заказ 271
97
26. Ч ж ан Д а - юй, Чжу Бао-лин, Изв. СО АН СССР, № 2, 70 (1959).
27. Санчес Р., Ж у р а д о Дж. Р. IV Международный нефтяной кон-
гресс. Т. 4. М., Гостоптехиздат, 1956. См. с. 469.
28. Б р у н и К. Э., В а с к о н с е л о с Д., П а д у л а В. Т. VIII Мировой
нефтяной конгресс. Москва. 1971. Препринт симпозиума № 10. См. с. 20.
29. К а р ж е в В. И., Сильченко Е. И. В сб. «Труды ВНИГИ». Вып. 1.
М., Гостоптехиздат, 1948. См. с. 5.
30. Дьякова ,М. К., Давтян Н. А., ЖПХ, 21, 113 (1948).
31. С у д з и л о в с к а я М. С. В сб. «Труды ВНИГИ». Вып. 1., М., Гостоп-
техиздат, 1948. См. с. 47.
32. О г с h i n М., Storch Н. Н., Ind. End. Chem., 40, 1385 (1948).
33. P e 1 i p e t z M. G., Kuhn E. M. et al., Ind. Eng. Chem., 40, 1259
(1948).
34. W e 11 e r S., P e 1 i p e t z M. G. et al., Ind. Eng. Chem., 41, 972 (1949).
35. Markovits J. A., Meeh. Eng., 71, 553 (1949).
36. К a s t e n s M. L., Hirst L. L., Chaffee С. C., Ind. Eng. Chem.,
41, 870 (1949).
37. О г c h i n M., Storch H. H., J. Soc. Chem. Ind. (London), 69, 121
(1950).
38. W e 1 1 e r S., P e 1 i p e t z M. G. et al., Ind. Eng. Chem., 42, 330 (1950).
39. W e 11 e r S., Clark E. L., P e 1 i p e t z M. G., Ind. Eng. Chem.,
42, 334 (1950).
40. С 1 a r k E. L., Pelipetz M. G. et al., Ind. Eng. Chem., 42, 861 (1950).
41. Hirst L. L., Chaffee С. C., Ind. Eng. Chem., 42, 1607 (1950).
42. С у д з и л о в с к а я М. С., П р о к о п е ц Е. И. В сб. «Труды ВНИГИ».
Вып. 3. М., Гостоптехиздат, 1951. См. с. 26.
43. Weller S., Pelipetz М. G., Ind..Eng. Chem., 43, 1243 (1951).
44. Weller S., Pelipetz M. G., Friedman S., Ind. Eng. Chem.,
43, 1572 (1951).
45. К a n d i n e r H. J., Hit eshue R. W., Clark E. L., Chem.
Eng. Progr., 47, 455 (1951).
46. Clark E. L., Hiteshue R. W., Kandiner H. J., Chem. Eng.
Progr., 48, 15 (1952).
47. P e 1 i p e t z M. G., Salman J. R. et al., Ind. Eng. Chem., 45, 806
(1953).
48. Chaffee С. C., Hirst L. L., Ind. Eng. Chem., 45, 822 (1953).
49. P e 1 i p e t z M. G., Frank L. V. et al., Chem. Eng. Prog., 50, 626
(1954).
50. К а з а н с к и й Б. А., Гоникберг M. Г. и др. В сб. «Труды ИГИ
АН СССР». Вып. 6. М., Изд. АН СССР, 1955. См. с. 3.
51. С у д з и л о в с к а я М. С., Р о б о ж е в а Е. В. В сб. «Труды ВНИГИ».
Вып. 6. М., Гостоптехиздат, 1954. См. с. 30.
52. Р е 1 i р е t z М. G., Salman J. R. et al., Ind. Eng. Chem., 47, 2101
(1955).
53. Szucs M., Magyar tudomanyos akad. Kem. tudomanyok osztalyanan kozlemi-
nyei, 6, 375 (1955); C. A., 50, 6019 (1956).
54. 43 hannabasappa К. C., Linden H. R., Ind. Eng. Chem., 48,
900 (1956).
55. Д ь я к о в а М. К. В сб. «Химическая переработка топлив». М., Изд. АН
СССР, 1957. См. с. 261.
56. Д ь я к о в а М. К., Воль-Эпштейн А. Б., Советова Л. С.
В сб. «Химическая переработка топлив». М., Изд. АН СССР, 1957. См. с. 276.
57. N о d a Sh., Matsuda Т. et al., Нэнрё Кёкайси, 36, 696 (1957); С. А.,
52, 3307 (1958).
58. Hiteshue R. W., Anderson R. В., Schlesinger М. D.,
Ind. Eng. Chem., 49, 2008 (1957).
59. Chen Kuo-Chua n, Chang Fung-Liah, Acta focalia Sinica,
3, 23 (1958).
60. Channabasappa К. C., Linden H. R., Ind. Eng. Chem., 50,
637 (1958).
98
61. Блонская А. И., Лозовой А. В. и др. В сб. «Труды ИГИ АН
СССР». Вып. 9. М., Изд. АН СССР, 1959. См. с. 50.
62. К р и ч к о А. А., К о и я ш и н а Р. А. В сб. «Труды ИГИ АН СССР».
Вып. 9. М., Изд. АН СССР, 1959. См. с. 62.
63. М а р к о в Л. К., Оречкин Д. Б. В сб. «Труды ВСФ СО АН СССР».
Вып. 18. М., Изд. АН СССР, 1959. См. с. 64, 70.
64. В о s е S. К., В a s a k N. G., Lahiri A., J. Sci. Ind. Rea., 18В,
255 (1959); С. А., 54, 3919 (1960).
65. Р у г с о с h Е. J., Linden Н. *R., Ind. Eng. Chem., 52, 590
(1960).
66. H i t e s h u e R. W., Anderson R. B., Friedman S., Ind.
Eng. Chem., 52, 577 (1960).
67. G i n s b e г g H. H., Friedman S. et al., U. S. Bur. Mines, Report
Invest., № 5673 (1960); C. A., 55, 4924 (1961).
68. G i n s b e r g H. H., Lewis P. S. et al., U. S. Bur. Mines, Report
Invest. № 5676 (1960); C. A., 55, 4923 (1961).
69. L e w i s P. S., H i t e s h u e R. W., Ind. Eng. Chem., 52, 919 (1960).
70. S a к a b e T., О g о Y. et al., Когё кагаку дзасси, 63, 545 (1960); С. А.,
57, 14089 (1962).
71. К a s h i m а К., О s a d а Т., Когё кагаку дзасси, 64, 916 (1961); С. А.,
57, 3669 (1962).
72. L е w i s Р. S., Ginsberg Н. Н., Hiteshue R. W., U, S. Bur.
Mines, Report Invest., № 5908 (1961); C: A., 56, 6276 (1962).
73. H а г a H., Kudo H., Mizushima К., Нэнрё кёкайси, 40, № 411,
545 (1961); С. A., 61, 1669 (1964).
74. К а з а к о в Е. И., Мелентьев И. Н. В сб. «Труды ИГИ АН СССР».
Вып. 17. М., Изд. АН СССР, 1962. См. с. 43.
75. Н i t е s h u е R. W., Friedman S., Madden R., U.S. Bur.
Mines, Report. Invest., № 6027 (1962); C. A., 57, 12804 (1962).
76. Hiteshue R. W., Friedman S,, Madden R., U.S. Bur.
Mines, Report. Invest., № 6112 (1962); C. A., 58, 5418 (1963).
77. L e w i s P. S., Kawa W., Hiteshue R. W., U.S. Bur. Mines,
Report. Invest., № 6022 (1962); C. A., 57, 12804 (1962).
78. S a k a b e T., О g о a Y. et al., Когё кагаку дзасси, 65, 297 (1962); С. Ar,
57, 15434 (1962).
79. А г i с h G., Cocco A., Univ. Studi Trieste, Fac. Ind. First Chim. Appl.,
№ 12 (1963); C. A., 60, 7838 (1964).
80. F e 1 d k i r c h n e r H. L., . Linden H. R., Ind. Eng. Chem. Proc.
Design, a. Develop., 2, 153 (1963).
81. L e f о r t M., Bayer A. F., Payer P., Bull. Soc. Chim. France,
1963, 1589.
82. S a k a h e T., О g о Y., Когё кагаку дзасси, 66, 1875 (1963); С. А., 61,
1670 (1964).
83. В 1 a n k s R. F., Prausnitz J. М., Ind. Eng. Chem., Fundamental,
3, 1 (1964).
84. F г i e d m a n S., Hiteshue R. W., Schlesinger M. D., U. S.
Bur. Mines, Report. Invest., № 6470 (1964); C. A., 61, 6823 (1964).
85. F г i d у R. W., Kawa W., Hiteshue R. W., U. S. Bur. Mines,
Report. Invest., № 6563 (1964); C. A., 62, 3851 (1965).
86. H i t e s h u e R. W., Friedman S.,. Madden R., U.S. Bur.
Mines, Report. Invest., № 6376 (3) (1964); C. A., 60, 14295 (1964).
87. Shih Mei-Jen, Yang Hsueh-Jen, Peng Shao - I’K’o
Hsueh T’ung Pao, № 9, 808 (1964); C. A., 62, 8897 (1965).
88. Hawk С. O., Schlesinger M. D. et al., U. S. Bur. Mines,Tleport.
Invest., № 6548 (1965); C. A., 62, 8896 (1965).
89. H e 11 w i g К. C., Johnson E. S. et al., Hydrocarh. Proc. a. Petrol.
Ref., 45, № 5, 165 (1966).
'90. Brit. Chem. Eng., № 3, 309 (1968).
91. Hellwig К. C., Chtfiwenak M. C. et al., Chem. Eng. Prog., 85,
. 64, 98 (1968).
7* 99
92. С т р у с Б. В. Материалы сессии ЮНИДО в Баку. 1969. Препринт ID/WG
43/53.
93. A n g е 1 о v i с h J. М., Pastor G. R., Silver Н. F., Ind. Eng.
Chem., Proc. Design a. DeveL, 9, 106, 609 (1970).
94. Г о p и н Э., Лебо в иц Г. и др. VIII Мировой нефтяной конгресс.
Москва, 1971. Препринт симпозиума № 10. См. с. 83.
95. Джонсон К., Гельвиг К. и др. VIII Мировой нефтяной конгресс.
Москва, 1971. Препринт симпозиума. № 10. См. с. 110.
96. Лозовой А. В. В сб. «Труды ИГИ АН СССР». Вып. 5. М., Изд. АН
СССР, 1955. См. с. 115.
97. Donath Е. Е., Ind. Eng. Chem., 46, 2032 (1954).
98. W i 1 1 i a m s P. E., Erdol u. Kohle, 11, 394 (1958).
99. Chem. Prod., 22, 271 (1959); РЖХим., 23828 (1960).
100. H a m m a r C. G. B. Ill World Petroleum Congress. Hague. Proceedings.
Sec. IV. London, Elsevier Publ. Corp., 1951. See p. 295.
101. Perna F., Pelcik J., Paliva, 31, 151 (1951); C. A., 47, 10200 (1953).
102. Clark E. L., Hiteschue R. W. et al., Ind. Eng. Chem., 43, 2173
(1951).
103. Marceaunt P., Oil Shale a. Cannel Coal Conf., Inst, of Petrol. (Lon-
don), 2, 673 (1951); C. A., 47, 1364 (1953).
104. Pelipetz M. G., Wolfson M. L. et al., Chem. Eng. Progr., 48,
353 (1952).
105. Smith W. M., Landrum T. C., Phillips G. E., Ind. Eng.
Chem., 44, 586 (1952).
106. Perna F., Pelcik J., Paliva, 33, 126 (1953); РЖХим., 1391 (1953).
107. Чжен Лу-бинь, Дунбей Кэсюэ Цзюсо Хуайбао, 4, 260 (1953).
108. В i г t h 1 е г R., Freiberger Forschungshefte, А17, 29 (1953).
109. Gunther G., Freiberger Forschungshefte, A17, 38 (1953).
110. В i 1 1 e R., Iva, 24, 124 (1953); C. A., 47, 10203 (1953).
111. X э Сюэ-лунь, Чень Го-цюань, Цзян Бин-нан ь.
В сб. «Труды совещания по исследованию жидкого топлива». Дальний,
Изд. Института нефти АНК, 1954. См. с. 78.
112. X э Сюэ-лунь, Чень Го-цюань, Цзян Бин-нань.'
Там же. См. с. 92.
113. В г а а е В., Acta Polytech. Chem. Met., Ser. 3, № 9 (1954); C. A., 49, 601
(1955).
114. В i r t h 1 e r R., Szkibik C., Chem. Techn. (Berlin), 6, 373 (1954).
115. Jensen H. B., Barnet W. J., Murphy W. J. R. U.S. Bur.
Mines, Report. Invest., № 533 (1954); C. A., 48, 11765 (1954).
116. Калечиц И. В., Салимгареева Ф. Г. Веб. «Труды ВСФ
АН СССР». Вып. 3. М., Изд. АН СССР, 1955. См. с. 79.
117. Глушенкова Е. В., Семенов С. С. В сб. «Труды ВНИИПС».
Вып. 4. Л., Гостоптехиздат. 1955. См. с. 167.
118. Birth ler R., Szkibik C. Freiberger Forschungshefte, A36, 42
(1955).
119. Schmidt R., G ii n t h e r G., Chem. Techn. (Berlin), 7, 316 (1955).
120. Дьякова M. К., В о л ь - Э п ш т е й н А. Б; и др. Химия и тех-
нология топлива, № 9, 44 (1956).
121. Ли Цзя-пэн, Рефераты докладов на конференции Дальнинского
отделения КХО. 1956. См. с. 40.
122. С а г р е n t е г Н. С., Hopkins С. В. et al., Ind. Eng. Chem., 48,
1139 (1956).
123. Cottingham P. L., Antweiler J. C. et al., Ind. Eng. Chem.,
48, 1146 (1956).
124. С г e с e 1 i u s R. L., Knidschy E. O. et al., Petrol. Ref., 35, № 6,
171 (1956).
125. Л e в ч e н к о Д. H., Агафонов А. В. и др. Химия и технология
топлива, № 8, 65 (1957).
126. Д ь я к о в а М. К., Во ль-Эпштейн А. Б. и др., Химия и тех-
нология топлива, № 4, 28 (1957).
100
127. Д ь я к о в а М. К., Воль-Эпштейн А. Б. и др., ЖПХ, 30, 1056
(1957).
128. Л о з о в о й А. В., Крич к о А. А., Михеева Р. А., Хийия
и технология топлива, № 5, 32 (1957).
129. Кричко А. А., Ковяшина Р. А., Лозовой А. В. В сб.
«Труды ИГИ АН СССР». Вып. 9. М., Изд. АН СССР, 1959. См. с. 68.
130. Clough Н., Ind. Eng. Chem., 49, 673 (1957).
131. С о 11 i n g h a m P. L., White E. R., Frost С. M., Ind. Eng.
Chem., 49, 679 (1957).
132. Landa S., Mostecky J., Chem. prumysl, 7, 393 (1957).
133. Г о и ч a p о в a H. В., Кривозубова H. В. и др., Химия и тех-
нология топлива, № 12, 15 (1958).
134. Ф е о ф и л о в Е. Е., Гарновская Г. Н., В сб. «Труды ВНИИПС».
Вып. 6. Л.; Гостоптехиздат, 1958. См. с. 183.
135. Цзян Бин-нан ь, Лин Ли-у и др. Шиюляньчжи, № 2, 6 (.1958).
136. SihTsu-wei, Kalechita I. V. Acta focalia Sinica, 3, 300 (1958).
137. Глушенкова E. В., Семенов С. С. Веб. «Труды ВНИИПС».
Вып. 6. Л., Гостоптехиздат, 1958. См. с. 206.
138. Birthler R., Dentloff Е., Szkibik С., Chem. Techn. (Ber-
lin), 10, 75 (1958).
139. S v a j g 1 O., Erdol u. Kohle, 11, 647 (1958).
140. Блонская А. И., Лозовой А. В.'и др. В сб. «Труды ИГИ АН
СССР». Вып. 9. М., Изд. АН СССР, 1959. См. с. 5.
141. Кричко А. А., Лозовой А. В., Пчел ин а Д. П. В сб.
«Труды ИГИ АН СССР». Вып. 9. М., Изд. АН СССР, 1959. См. с. 37.
142. Салимгареева Ф. Г., Давидович Б. В., КалечицИ. В.
В сб. «Труды ВСФ СО АН СССР». Вып. 18. М., Изд. АН СССР, 1959. См. с. 87.
143. Калечиц И. В. н др. В сб. «Труды ВСФ СО АН СССР». Вып. 18. М.,
Изд. АН СССР, 1959. См. с. 107.
144. Цзян Бин-нан ь, Вей Ши-пин и др. Изв. СО АН СССР, № 2,
81 (1959).
145. Ц з я н Бин-нан ь, Калечиц И. В., Вей Ши-пин, Химия
и технология топлива, № 10, 16 (1959).
146. Барышников Л. И., Казаков Е. И. В сб. «Труды ИГИ АН
СССР». Вып. 9. М., Изд. АН СССР, 1959. См. с. 86.
147. Carpenter Н. С., Cottingham Р. L., U. S. Bur. Mines, Re-
port Invest., № 5533 (1960); С. A., 54, 5057 (1960).
148. Schnabel В., Chem. Prumysl, 9, 10 (1959).
149. Schultz E. B., Linden H. R., Ind. Eng. Chem., 51, 573 (1959).
150. С а л и м г a p e e в а Ф. Г., Давидович Б. В., Иванова M. Ф.
В сб. «Труды ВСФ СО АН СССР». Вып. 18. М., Изд. АН СССР, 1959. См. с. 95.
151. Цзян Бин-нан ь, Вей Ши-пин, Чжоу Фын-лен. Веб.
«Труды ВСФ СО АН СССР». Вып. 26. М., Изд. АН СССР, 1959. См. с. 128.
152. Huang Chih - Yu an, Wei Yu-Hain, Schapiro M. D.,
Acta focalia Sinica, 5, 21 (1960).
153. L ii d e r H., D г e s h e r K., Chem. Techn. (Berlin), 12, № 1, 16 (1960).
154. Frost С. M., Carpenter H. C. et al., U. S. Bur. Mines, Report
Invest, № 5574 (1960); C. A., 54, 11447 (1960).
155. Левин С. 3., Д и н e p И. С. В сб. «Труды ВНИИТ». Вып. 9. Л.,
Гостоптехиздат, 1960. См. с. 65.
156. Семенов С. С., Глушенкова Е. В. В сб. «Труды ВНИИТ».
Вып. 9. Л., Гостоптехиздат. 1960. См. с. 99.
157. Radomyski В., Т о m a s i k Z., Chem. Stosowana, 4, 501 (1960);
C. A., 56, 7582 (1962).
158. Б о p ц А. Г., Кричко А. А. и др. В сб. «Труды ИГИ АН СССР».
Вып. 17. М., Изд. АН СССР, 1962. См. с. 250; Кокс и химия, № 10, 53 (1961).
159. Е р у И. И., Ланге А. А. и др., Кокс и химия, № И, 41 (1961).
160. Л о з о в о й А. В., Муселевич Д. Л. и др., ЖПХ, 34, 1200, 2295
(1961).
101
161. Кацо башвили Я. Р., Гарбер Ю. Н. и др., Кокс и химия,
№ 10, 48 (1961).
162. 3 а й д м а н Н. М., Гладовская М. Ф., Химия и технология
топлив и масел, № 6, 27 (1961).
163. Цзян Бин-нан ь, Вей Ши-пин и др., Химия и технология
топлив и масел, № 2, 21 (1961).
164. Cherodame R., Letort М., Centre Etudes Rech. Charb. Fr., Papp
1960; C. A., 61, 14433 (1964).
165. G i 1 b e r t G., Weil R. C., Hunter R. H., Ind. Eng. Chem., 53,
993 (1961).
166. Schlesinger M. D., Hiteshue R. W., U. S. Bur. Mines, Re-
port Invest., № 5902 (1962); C. A., 56, 10471 (1962).
167. V a i d у e s w a r a n R., Krishna M. G., Zaheer S. H., J. Sci.
Ind. Research (India), 20D, 369 (1961); C. A., 56, 11927 (1962).
168. В о л ь - Э п ш т е й н А. Б., Григорьев С. М. и др. В сб. «Труды
ИГИ АН СССР». Вып. 17. М., Изд. АН СССР, 1962. См. с. 269.
169. Воль-Эпште'йн А. Б., К р и ч к о А. А. В сб. «Производство
бензола». Л., Гостоптехиздат, 1962. См. с. 161.
170. Лозовой А. В., Мусалевич Д. Л. и др. В сб. «Труды ИГИ
АН СССР». Вып. 17. М., Изд. АН СССР, 1962. См. с. 174.
171. Graves R. D., Kawa W., Hiteshue R. W., U. S. Bur. Mines,
Report Invest., № 6124 (1963); C. A., 58, 5420 (1963).
172. Jelinek J. F., Sb. Praci Vyzkumu Chem. Vynziti Uhli, Dehtu, Ropy,
2, 70 (1962); C. A. 62, 5116 (1965).
173. Jelinek J. F., Chem. prumysl, 12, № 1, 4 (1962).
174. Letort M., Hydrocarb. Proc. a. Petrol. Ref., 41, № 7, 83 (1962).
175. Matic D., Mijatovic I., Kolojnbo M., Tehnika (Beograd),
17, 1761 (1962); C. A., 61, 4116 (1964).
176. S v a j g 1 O., Sb. Praci Vyzkumu Chem. Vynziti Uhli, Dehtu, Ropy, 2,
21 (1962); C. A., 62, 5120 (1965).
177. Carpenter H. C., Cottingham P. L. et al. U. S. Bur. Mines,
Report Invest., № 6237 (1963); C. A., 59, 2555 (1963).
178. К а ц о б а ш в и л и Я. Р., Эльберт Е. И., Смирнов В. К.,
Химия и технология топлив и масел, 9, № 2, 5 (1964).
179. Aitken A. R., Merrill W. Н., Р 1 е е t М. Р., Can. J. Chem.
Eng., 42, № 5, 234 (1964); С. А., 61, 15905 (1964).
180. Feldkirchner Н. L., Linden Н. R., Ind. Eng. Chem., Proc.
Design a. Develop., 3, 218 (1964).
181. Kostova H., К u b i c k a R., К v a p i 1 Z., Sb. Praci Vyzkumu
Chem. Vynziti Uhli, Dehtu, Ropy, 3, 97 (1964); C. A., 62, 6314 (1965).
182. Logwinuk A. K., Fridman L., Weiss A. H., Erdol u. Kohle,
17, 532 (1964).
183. Saxena E. R., Narashimha Rao T. L. et al. Low-Temp. Carb.
Non — Coking Coals, Lignites, Briquetts, Coal Fines, Symp. Hyderabad,
№ 2, 242 (1964); C. A., 61, 13092 (1964).
184. К u b i c k a R., Kvapil Z., Brennstoff-Chemie, 47, № 5, 21 (1966).
185. Chem. Eng. News, 44, № 40, 79 (1966).
186. Mirza A., Masood M. A., Mallikarjunan M. M. et al.,
Brennstoff-Chemie, 48, № 10, 310 (1967).
187. Mirza A., Masood M. A., et al., Chem. Age India, 19, № 7 (1968);
Экспресс-информация. Сер. «Химия и переработка нефти и газа». Вып. 47.
М., ВИНИТИ, 1968. См. с. 512.
188. Q a d е г S. A., W i s е г W. Н., Н i 1 1 G. R., Ind. Eng. Chem., Proc.
Design a. Develop., 7, № 3, 390 (1968).
189. К а м e p о н P. Д. VIII Мировой нефтяной конгресс. Москва, 1971.
Препринт симпозиума № 10. См. с. 42.
190. С о 1 е R. М., Davidson D. D., Ind. Eng. Chem., 41, 2711 (1949).
191. Hale J. H., Simmons M. C., Wi senhunt E. P., Ind. Eng.
Chem., 41, 2702 (1949).
102
192. V о о r h i e s A., Smith W. М., Ind. Eng. Chem., 41, 2708 (1949).
193. Eagle S., Rudy Ch. E., Ind. Eng. Chem., 42, 1294 (1950).
194. Nonnenmacher H., Brennstoff-Chemie, 31, 138 {1950).
195. H о о g H., Reman G. H., S m i t h a у s e r W. С. B. Ill World
Petroleum Congress. Hague. Proceedings. Sect. IV. London, Elsevier Publ.
Corp., 1951. See p. 282.
196. Becker R., Ill World Petroleum Congress. Hague. Proceedings. Sect. IV.
London, Elsevier Publ. Corp.; 1951. See p. 68.
197. Pier M., F u e n e r W. V. et at. Ill World Petroleum Congress. Hague.
Proceedings. Sect. IV. London, Elsevier Publ. Corp., 1951. See p. 81.
198. Oettinger W., Erdol u. Kohle, 6, 693 (1953).
199. Дунбей Кэсюэ Тун сюинь, 1, вып. 11, 530 (1951).
200. Urban W., Erdol u. Kohle, 4, 279 (1951).
201. Каржев BJ И. В сб. «Труды ВНИГИ». Вып. 4. М., Гостоптехиздат,
1952. См. с. 87.
202. О р о ч к о Д. И., Сильченко Е. И., Шаволина Н. В.
В сб. «Труды ВНИГИ». Вып. 4. М., Гостоптехиздат, 1952. См. с. 97.
203. Pier М., Z. Elektrochem., 57, 456 (1953).
204. Н о о g Н., Klinkert Н. G., Schaafsma A., Petrol. Ref.,
32, № 5, 137 (1953).
205. Novak Н., L i е b i с h H. G., Brennstoff-Chemie, 35, 308 (1954).
206. V arg a J. Magyar tud. akad. Kem. tud. oszt. Koze, 5, 167 (1954);
РЖХим., 20229 (1956).
207. Гайд Д. В., Портер Ф. В. Б. IV Международный нефтяной кон-
гресс. Т. 4. М., Гостоптехиздат, 1956. См. с. 366.
208. К о m а г е w s к у V. I., К n a g g s Е. А., В г a g g С. J., Ind. Eng.
Chem., 46, 1689 (1954).
209. К а ц о б а ш в и л и Я. Р., Куркова Н. С. В сб. «Труды Инсти-
тута нефти АН СССР». Вып. 6. М., Изд. АН СССР, 1955. См. с. 100.
210. A b b о 11 М. D., Liedholm G. Е., Sarno D. H., Petrol. Ref.,
34, № 6, 118 (1955).
211. N о n n e n m a c h e r H., Reitz O., Schmidt P., Erdol u. Kohle,
8, 407 (1955).
212. McAfee J., Montgomery C. W. et al., Petrol. Ref., 34, № 5,
156 (1955).
213. C a s a g r a n d e R. M., Meerbott W. K. et al., Ind. Eng. Chem.,
47, 744 (1955).
214. О d a s z F. B., Scheffield J. V., Oil a. Gas J., 53, K° 46, 203
(1955); Petrol. Ref., 34, № 9, 158 (1955).
215. Varga J., R a b 6 G., Steingaszner P., Acta China. Acad. Sci.
Hung., 10, 245 (1956).
216. V a r g a J., R a b о G., Zalai A., Brennstoff-Chemie, 37, 244
(1956).
217. Law R. D., Petrol. Ref., 34, № 11, 219 (1955).
218. Л о б e e в M. В., Сулимов А. Д., Агафонов А. В., Нефт.
хоз., № 1, 59 (1955).
219. Сулимов А. Д., Л о б e e в М. В. и др. Химия и технология топлива,
№ 9, 1 (1956).
220. С у л и м о в А. Д., Каржев В. И. и др. Химия и технология то-
плива, № 1, 33 (1956).
221. Г о л ь д ш т е й н Д. Л., Гусенкова Е. А., Новости нефтяной
техники. Нефтепереработка, № 1, 22 (1956).
222. К а ц о б а ш в и л и Я. Р., Куркова Н. С., Химия и технология
топлива, № 3, 31 (1956).
223. Кацобашвили Я. Р. Веб. «Химическая переработка топлив».
М., Изд. АН СССР, 1957. См. с. 159; в сб. «Переработка нефтяных остатков».
М., ГОСИНТИ, 1958. См. с. 190.
224. Лавровский К. П., Макаров Д. В., Назарова Л. М.
В сб. «Труды Института нефти АН СССР». Вып. 8. М., Иад. АН СССР,
1956. См. с. 145.
103
225. Dent F. J., Edge R. F. et al., Gas Council, Res. Comm. № GC37
(1956); C. A., 51, 3123 (1957).
226. Ппнчук Л. В., Овсяников Л. Ф. и др. В сб. «Труды ВСФ
АН СССР». Вып. 4. М., Изд. АН СССР, 1956. См. с. 137.
227. Г о л ь д ш т е й н Д. Л., Агафонов А. В. и др., В сб. «Химическая
переработка топлив». М., Изд. АН СССР, 1957. См. с. 245.
228. Е Ъ е г 1 i n е С. R., Wilson R. Т., Larson L. G., Ind. Eng.
Chem., 49, 661 (1957).
229. Storey G. R., Inst. Pert. Rev., 11, № 122, 35 (1957).
230. H о f f m a n E. J., Lewis E. W., Wadley E. F., Ind. Eng.
Chem., 49, 656 (1957).
231. Shiba T., Nagata T., Коацу Газу Кёкайси, 21, 364 (1957); С. А.,
52, 5798 (1958).
232. Heinemann Н., Kirsch F. W., В u r t i s T. A., Erdol u.
Kohle, 10, 225 (1957).
233. Pichler H., Chervenak M. etal., Petrol. Ref., 36, № 9, 201 (1957).
234. G w i n G. T., Heinrich. R. L. et al., Ind. Eng. Chem., 49, 668 (1957).
235. Varga J., Karolyi J. et al., Petrol. Ref., 36, № 9, 198 (1957).
236. Shultz E. B., Linden H. R., Ind. Eng. Chem., 49, 2011 (1957);
Petrol. Ref., 36, № 9, 205 (1957).
237. Stevenson D. H., Heinemann H., Ind. Eng. Chem., 49, 664 (1957).
238. К a p ж e в В. IL, Касаткин Я. Ф., Op очко Д. И., Химия
и технология топлива, № 12, 3 (1958).
239. Макаров И. А. В сб. «Химическая переработка топлив». М., Изд. АН
СССР, 1957. См. с. 146.
240. Макаров И. А., Н и з я е в В. М., Химия и технология топлива,
№ 1, 9 (1958); № 5, 1 (1958).
241. Калашникова Н. И., Оречкин Д. Б., Химия и технология
топлива, № 4, 47 (1958).
242. М у ш е н к о Д. В. В сб. «Переработка нефтяных остатков». М.,
ГОСИНТИ, 1958. См. с. 181.
243. Сулпмов А. Д., Лобеев М. В. и др. В сб. «Химия сераоргани-
ческих соединений, содержащихся в нефтях и нефтепродуктах». Т. 1. Уфа,
Изд. Башк. филиала АН СССР, 1958. См. с. 135.
244. Рогов С. П., Рысаков М. Ф., Фер шт И. Я., Химия и тех-
нология топлива, № 10, 29 (1958).
245. Дружинина А. В., Рысаков М. В. и др. В сб. «Труды
ВНИИНП». Вып. 7- М., Гостоптехиздат, 1958. См. с. 166.
246. Гольдштейн Д. Л., Рысаков М. В. и др. В сб. «Труды
ВНИИНП». Вып. 7. М., Гостоптехиздат, 1958. См. с. 245.
247. Carlson С. S., Langer A. W. et al., Ind. Eng. Chem., 50, 1067
(1958).
248. Агафонов А. В., Абаева Б. Т. и др. Химия и технология
топлива, № 4, 18 (1959).
249. Николаева Д. X. В сб. «Труды ВСФ СО АН СССР». Вып. 18. М.,
Изд. АН СССР, 1959. См. с. 78.
250. О в с я н и к о в Л. Ф., Оречкин Д. Б. В сб. «Труды ВСФ
СО АН СССР». Вып. 26. М., Изд. АН СССР, 1959. См. с. 63.
251. Оречкин Д. Б., Овсяников Л. Ф., Богданова Т. А.
В сб. «Труды ВСФ АН СССР». Вып. 26. М., Изд. АН СССР, 1959. См. с. 71.
252. Голосов С. А., Кацобашвили Я. Р., Изв. АН СССР, ОТН,
№ 5, 168 (1959).
253. Кацобашвили Я. Р., Куркова Н. С., Кухтичева В. Ф.
В сб. «Труды Института нефти АН СССР». Вып. 13. М., Изд. АН СССР,
1959. См. с. 196.
254. Кацобашвили Я. Р., Волынский Н. П., ЖПХ, 32, 2290
(1959); в сб. «Труды института нефти АН СССР». Вып. 13. М., Изд. АН
СССР, 1959. См. с. 213.
255. Макаров И. А. В сб. «Труды ВСФ СО АН СССР». Вып. 26. М., Изд.
АН СССР, 1959. См. с. 86.
104
256. Макаров И. А. Труды ВСФ СО АН СССР. Вып. 26. М., Изд. АН
СССР, 1959. См. с. 92.
257. Веселов В. В., Оречкин Д. Б. и др. В сб. «Труды ВСФ СО
АН СССР». Вып. 26. М., Изд. АН СССР, 1959. См. с. 135.
258. Зырянов Б. Ф., Калашникова Н. И., Оречкин Д. Б.
В сб. «Труды ВСФ СО АН СССР». Вып. 26. М., Изд. АН СССР, 1959. См.
с. 141.
259. Yang J ah-shu, Liang Chuan, Acta focalia Sinica, 4, 408 (1959).
260. В i г t hl er R., Denthoft E. u. a., Erdol u. Kohle, 12, 71 (1959).
261. W e 1 1 e г H., Erdol u. Kohle, 12, 551 (1959).
262. Stormont D. H., Oil. a. Gas J., 57, № 44, 48 (1959).
263. Кацобашвили Я. P., Голосов С. А., Химия и технология
топлива, № 1,8 (1960).
264. Кацобашвили Я. Р., Кузьмина Т. Н. и др., Изв. АН СССР,
ОТН, сер. «Металлургия и топливо», № 2, 159 (1960).
265. Schultz Е. В., Mechales N., Linden Н. R., Ind. Eng. Chem,
52, 580 (1960).
266. Oil a. Gas J., 58, № 16, 104; № 21, 102 (1960).
267. Варга И., Б пртл ер Р. и др., Химия и технология топлива, № 10,
И (1960).
268. Вишневский Н. Е., Майоров Д. М., Мушенко Д. В.
В сб. «Труды ВНИИнефтехим». Вып. 3. Л., Гостоптехиздат, 1960. См. с. 183.
269. Oraby М. A., J. Chem. UAR, 3, 175 (1960); С. А., 55, 10858 (1961).
270. Веселов В. В., Оречкин Д. Б. и др., Химия и технология
топлива, № 8, 11 (1960).
271. Султанов А. С., Васильева Н. В., Сафаев А. С., ДАН
УзССР, № 5, 37 (1960).
272. Поюжаев В. Д., Заглодпн Л. С. и др., Химия и технология
топлива, № 7, 1 (1960).
273. Мушенко Д. В. В сб. «Труды ВНИИнефтехим». Вып. 3. Л., Гостоп-
техиздат, 1960. См. с. 157.
274. Левина М. И., Мушенко -Д. В., Рысаков М. В. Веб.
«Труды ВНИИнефтехим». Вып. 3. М., Гостоптехиздат, 1960. См. с. 178.
275. Thonon С., Alexandre М. et al. V World Petroleum Congress
New York. Proceedings. V. 3. London, Elsevier Publ. Corp., 1960. See p. 71.
276. Friedman B. S., Kovach S. M. V World Petroleum Congress.
New York. Proceedings. V. 4. London, Elsevier Publ. Corp., 1960. See p. 17.
277. Chervenak M. C., Johnson E. S. et al., Oil a. Gas J., 58, № 35,
80 (1960).
278. Hi gu chi K., Kanai H., Кору тару, 12, 371 (1960); C. A., 61, 2879
(1964).
279. Оболенцев P. Д., Машкина А. В., Михеев Г. M. В сб.
«Химия сераорганических соединений, содержащихся в нефти и нефте-
продуктах». Т. 4. Уфа, Изд. Башк. филиала АН СССР, 1961. См. с. 184.
280. Фершт И. Я., Рогов С. П. и др., Нефтепереработка и нефтехимия,
№ 3, 19 (1961).
281. Воль-Эпштейн А. Б., Кричко А. А., Химия и технология
топлив и масел, № 3, 14 (1961).
282. Герасименко Н. М., Ястребов Г. И. и др., Химия и тех-
нология топлив и масел, № 4, 27 (1961).
283. Осипов Л. Н., Фершт И. Я. и др., Химия и технология топлив
и масел, № 5, 15 (1961).
284. Масагутов Р. М., Берг Г. А., Волкова Л. И., Химия
и технология топлив и масел, № 8, 8 (1961).
285. Лавровский К. П., Макаров Д. В., Ф и ш Ю. Л., Нефтехи-
мия, 1, 509 (1961).
286. В eu t her Н., Flinn R. A., Rice Т., Proc. Am. Petrol. Inst.,
41, 228 (1961); C. A., 58, 1285 (1963).
287. H e n d r.i k s W. J., V 1 u g t e r J. C., Waterman H. I., Brenn-
stoff-Chemie, 42, 1 (1961).
105
288. Langer A. W. Stewart J. et al., Ind. Eng. Chem., 53, 27 (1961).
289. Visser G. H., Bull, assoc. Franc, techn. petrole, №147, 371 (1961);
C. A., 55, 21559 (1965).
290. Кацобашвпли Я. P., Брун-Цеховой A. P., Черны-
шова M. M., Химия и технология топлив и масел, № 8, 17 (1962).
291. Левицкий И. И., Удальцова Е. А., Г он и к бе р г М. Г.,
ЖПХ, 35, 204 (1962).
292. Антоновский А. Д., Л я т и е в Г. Г., ЖПХ, 35, 2074 (1962).
293. Крич ко А. А., Лозовой А. В., Советов а Л. С., Хим.
пром., № 10, 387 (1962).
294. Агафонов А. В., Хавкин В. А. и др., Нефтепереработка и
нефтехимия, № 9, 3 (1962).
295. Оречкин Д. Б., Мальцев И. Г. и др., Нефтепереработка и
нефтехимия, № 9, 15 (1962).
296. Масагутов Р. М., Султанов А. С. и др., ДАН УзССР, 19,
№ 9, 49 (1962).
297. Kozlowski R. Н., Mason Н. F., Scott J. М., Ind. Eng.
Chem., Proc. Design a. Develop., 1, 276 (1962).
298. Hydrocarb. Proc. a. Petrol. Ref, 41, № 2, 45 (1962).
299. Langer A. W., Stewart J. etal., Ind. Eng. Chem., Proc. Design
a. Develop., 1, 309 (1962).
300. Peralta B., Reeg С. P. et al., Chem. Eng. Progr., 58, №4, 41
(1962).
301. Peterson H. G., Jamieson G. P., Am. Chem. Soc., Div. Petrol.
Chem., Preprints, 7, № 3, 23 (1962); C. A., 61, 501 (1964).
302. Weiss A. H., Maerker J. В., V e w i r t h R., Oil a. Gas J.,
60, № 4, 64 (1962).
303. Petrol. Ref., 41, № 9, 153 (1962).
304. Zalai A., Birthler R., Acta Chim. Acad. Sci. Hung., 31, 301 (1962).
305. Оболенцев P. Д., Михеев Г. M., Кузыев А. Р. В сб.
«Химия сераорганических соединений, содержащихся в нефти и нефте-
продуктах». Т. 5. Уфа, Изд. Башк. филиала АН СССР, 1963. См. с. 42.
306. Оболенцев Р. Д., Михеев Г. М., Кузыев А. П. В сб.
«Химия сераорганических соединений, содержащихся в нефти и нефте-
продуктах». Т. 5. Уфа, Изд. Башк. филиала АН СССР, 1963. См. с. 47.
307. Кацобашвили Я. Р., Брун-Цеховой А. Р. и др. В сб.
«Химия сераорганических соединений, содержащихся в нефти и нефте-
продуктах». Т. 5. Уфа, Изд. Башк. филиала АН СССР, 1963. См. с. 54.
308. Масагутов Р. М., Берг Г. А. и др. В сб. «Труды Баш. НИИ НИ».
Вып. 6. Уфа. Баш. обл. изд., 1963. См. с. 5.
309. Масагутов Р. М., Берг Г. А. и др., Химия и технология то-
плив и масел, № 12, 7 (1963).
310. Hydrocarb. Proc. a. Petrol. Ref., 42, № 3, 121 (1963).
311. World Petrol., 34, № 4, 30 (1963).
312. Берти В., Падовани К. П., Тодеска Ф. Труды VI Между-
народного нефтяного конгресса. М., ЦНИИТЭнефтегаз, 1965. См.
с. 21.
313. Бейт ер X., Шмидт Б. Труды VI Международного нефтяного кон-
гресса. М., ЦНИИТЭнефтегаз, 1965. См. с. 106.
314. Birthler R., Kahl Е., Erdol u. Kohle, 16, 281 (1963).
315. Karolyi J., Zalai A. et al., Mag. Kem. Lapja, 18, № 5, 212 (1963);
C. A., 60, 3928 (1964).
316. X e н з e л В., Политцер Э. Л., Уоткинс Ц. Г. Труды
VI Международного нефтяного конгресса. М., ЦНИИТЭнефтегаз, 1965. см.
с. 94.
317. В о о р х п с А., Смит У. М., Макларен Д. Д. Труды
VI Международного нефтяного конгресса. М., ЦНИИТЭнефтегаз, 1965.
См. с. 76.
318. Chervenak М. С., Feigelman S. et al., Oil a. Gas J., 61, № 41,
227 (1963); Chem. Eng. Progr., 59, № 2, 53 (1963).
106
319. Horing M., Oettinger W., Reitz O., Erdol u. Kohle, 16,
361 (1963).
320. Mr az V., Krafft О., К u b i с к a R., Acta Chim. Acad. Hiing.,
36, 269 (1963); C. A., 59, 4939 (1963).
321. Beuther H., Flinn R. A., Ind. Eng. Chem., Prod. Res. Develop.,
2, 53 (1963).
322. Кулаков В. H., Нефтепереработка и нефтехимия, № 4, 30 (1964).
323. Расулов А. М., Черножуков Н. И. и др., Химия и техноло-
гия топлив и масел, № 12, 11 (1964).
324. Beuther Н., Donaldson R. Е., Henke А. М., Ind. Eng.
Chem., Prod. Res. Develop., 3, № 3, 174 (1964).
325. Keil G., Roth H., Wenzel B., Chem. Techn., 16, 545 (1964).
326. Natori B., De Madle M., Hydrocarb. Proc. a. Petrol. Ref., 43,
№ 12, 91 (1964).
327. Gardner L. E., Hutchinson W. M., Ind Eng. Chem., Prod.
Res. Develop., 3, № 1, 28 (1964).
328. Van Driesen R. P., Stewart N. C., Oil a. Gas J., 62, № 20,
100 (1964).
329. Johnson A. R., Rapp L. M., Hydrocarb. Proc. a. Petrol. Ref.,
43, № 5, 165 (1964).
330. Beuthlr H., Larson O. A., Ind. Eng. Chem., Proc. Design a.
Develop., 4, 174 (1965).
331. Hydrocarb. Proc. a. Petrol. Ref., 44, № 6, 161 (1965).
332. Delhaye J., Du tri an R., Rev. Inst. Franc. Petrol. Ann. Com-
bust. Liquides, № 1, 160 (1965).
333. Craig R. G., D о e 1 p L. C., L i g w i n u k A. K., Erdol u. Kohle,
18, 527 (1965).
334. Schneider W., Teufel J., Schmiedel R., Chem. Techn.,
17, 577 (1965).
335. 'Oettinger W., Reitz O., Erdol u. Kohle, 18, 267 (1965).
336. Агафонов А. В., Осипов Л. H. и др. В сб. «Проблемы пере-
работки высокосернистых нефтей». М., ЦНИИТЭнефтехим, 1966. См. с. 177.
337. Агафонов А. В., Осипов Л. Н. и др. Сб. «Проблемы перера-
ботки высокосернистых нефтей». М., ЦНИИТЭнефтехим, 1966. См., с. 192.
338. Каржев В. И., Шаволина Н. В. и др. В сб. «Исследование
и применение гидрогенизационных процессов в нефтеперерабатывающей
и нефтехимической промышленности». М., ЦНИИТЭнефтехим, 1966. См.
с. 8.
339. Watkins С. Н., F о г t m a n J. Т., Oil a. Gas J., 64, № 11, 102
(1966).
340. Billon A., D е г г i е n et al., Hydrocarb. Proc. a. Petrol. Ref., 45,
№ 3, 129 (1966).
341. Craig R. G., White E, A., Hydrocarb. Proc. a. Petrol. Ref., 45,
№ 5, 159 (1966).
342. Ruedisuly M. M. J., Chem. Age India, 17, 264 (1966).
343. Oil a. Gas J., 64, № 4, 112 (1966).
344. Rutkowski A., Rutkowski M., T о m a s i k Z., Nafta, 22,
№ 2, 44 (1966); Экспресс-информация. Сер. «Химия и переработка нефти
и газа», № 17, 167 (1966).
345. Пережиг ина И. Я., Рогов С. П. и др., Химия и технология
топлив и масел, № 3, 1 (1967).
346. Пестриков С. В., Козик Б. Л. и др., Нефтецереработка и неф-
техимия, № 12, 10 (1967).
347. Novak V., К uh i с k a R., Brennstoff-Chemie, 48, 258 (1967).
348. Peet W. A., W i n s 6 г J., Oil a. Gas J., 65, № 10, 107 (1967).
349. Field S., Da Is on M. H., Can. Petrol., 8, № 12, 52 (1967).
350. Masa m‘u n e Sh., Fukuda J., Katada Sh., Hydrocarb. Proc. a.
Petrol. Ref., 46, № 2, 155 (1967).
351. Henke A. M., Schmid В. K., Strom J. R., Chem. Eng. Progr.,
63, № 5, 51 (1967).
107
352. Kremer H., Gas-Warme Internet., 16, 513 (1967).
353. World Petrol., 38, № 13, 14 (1967).
354. Ohtsuka T., Hasegawa Y. etal., Bull. Japan Petrol. Inst., 9,
1 (1967); РЖХим., 8П216 (1968).
355. Gould G. D., Paterson N. J. et al., Oil a. Gas J., 65, № 14, 164
(1967); Chem. Eng. Progr., 63, № 5, 6 (1967).
356. Haensel V., Stine L. O., Hydrocarb. Proc. a. Petrol. Ref., 46,
№ 6, 155 (1967).
357. Nelson W. L., Oil a. Gas J., 65, № 34, 101 (1967).
358. MusialE., Rutkowski M., Tomasik Z., Nafta, 23, № 1,
15 (1967); РЖХим, 8П152 (1968).
359. Docksey P., Gilbert R. J. H., VII World Petroleum Congress.
Mexico. Proceedings. V. 4. London, Elsevier Publ. Corp., 1967. See p. 153.
360. Watkins Ch. H. VII World Petroleum Congress. Mexico. Proceedings.
V. 4. London, Elsevier Publ. Corp., 1967. See p. 207.
361. Gilbert J. B., Kartzmark R. VII World Petroleum Congress.
Mexico. Proceedings. V. 4. London, Elsevier Publ. Corp., 1967. See p. 193.
362. А г e у W., Blackwell N. E., Reichle A. D. VII World Pet-
roleum Congress. Mexico. Proceedings. V. 4. London, Elsevier Publ. Corp.,
1967. See p. 167.
363. Van Driesen R. P., Rapp L. M. VII World Petroleum Congress.
Mexico. Proceedings. V. 4. London, Elsevier Publ. Corp., 1967. See p. 261.
364. Беренц А. Д., Воль-Эпштейн А. Б. и др. В сб. «Труды
ИГИ МУП СССР». Т. 24. М., «Недра», 1968. См. с. 181.
365. Курганов В. М., Мынова 3. А. и др., Нефтепереработка
и нефтехимия, № 7, 13 (1968).
366. Масагутов Р. М., Берг Г. А. и др. В сб. «Труды БашНИИНП».
Вып. 8. Уфа, Башк. обл. изд., 1968. См. с. 126.
367. Велькер И., Израель Г. и др. В сб. «Исследование и примене-
ние гидрогенизационных процессов в нефтеперерабатывающей и нефтехими-
ческой промышленности». М., ЦНИИТЭнефтехим, 1968. См. с. 124.
368. Осипов Л. Н., Хавкин В. А. и др. В сб. «Исследование и при-
менение гидрогенизационных процессов в нефтеперерабатывающей и нефте-
химической промышленности». М., ЦНИИТЭнефтехим, 1968. См. с. 68.
369. Бугай Е. А., Агафонов А. В. и др., Нефтепереработка и нефте-
химия, № 5, 6 (1968).
370. Пестриков С. В., Козик Б. Л. и др. В сб. «Исследование и при-
менение гидрогенизационных процессов в нефтеперерабатывающей и нефте-
химической промышленности». М., ЦНИИТЭнефтехим, 1968. См. с. 160.
371. Гольдштейн Д. Л., Гусенкова Е. А. и др. В сб. «Исследо-
вание и применение гидрогенизационных процессов в нефтеперерабаты-
вающей и нефтехимической промышленности». М., ЦНИИТЭнефтехим,
1968. См. с. 238.
372. Шаволина Н. В.. Кар ж е в В. И. и др., Химия и технология
топлив и масел, № 5, 6 (1968).
373. К а р ж е-в В. И., Жердева А. Г. и др. В сб. «Исследование и при-
менение гидрогенизационных процессов в нефтеперерабатывающей и неф-
техимической промышленности». М., ЦНИИТЭнефтехим, 1968. См. с. 176.
374. Кулиев Р._ Ш., Садыкова Б. А. и др. В сб. «Исследование
и применение гидрогенизационных процессов в нефтеперерабатывающей
и нефтехимической промышленности». М., ЦНИИТЭнефтехим, 1968. См.
с. 257.
375. Гусенкова Е. А., Гольдштейн Д. Л. и др. В с б. «Исследова-
ние и применение гидрогенизационных процессов в нефтеперерабатыва-
ющей и нефтехимической промышленности». М., ЦНИИТЭнефтехим, 1968.
См. с. 228.
376. Пережигина И. Я., Агафонов А. В. и др. В'сб. «Исследова-
ние п применение гидрогенизационных процессов в нефтеперерабатыва-
ющей п нефтехимической промышленности». М., ЦНИИТЭнефтехим, 1968.
См. с. 59.
108
377. Маншилин В. В., Манаков Н. X. и др. В сб. «Исследование
и применение гидрогенизационных процессов в нефтеперерабатывающей
и нефтехимической промышленности». М., ЦНИИТЭнефтехим, 1968. См.
с. 86.
378. Кацобашвили Я. Р., Брун-Цеховой А. - Р., Голо-
сов С. А. В сб. «Исследование и применение гидрогенизационных про-
цессов в нефтеперерабатывающей и нефтехимической промышленности». М.,
ЦНИИТЭнефтехим, 1968. См. с. 10.
379. Томасик 3., Рутковский А. Веб. «Исследование и примене-
ние гидрогенизационных процессов в нефтеперерабатывающей и нефтехими-
ческой промышленности». М., ЦНИИТЭнефтехим, 1968. См. с. 108.
380. Новак В., Кубичка Р. Веб. «Исследование и применение гидро-
генизационных процессов в нефтеперерабатывающей и нефтехимической
промышленности». М., ЦНИИТЭнефтехим, 1968. См. с. 114.
381. Brit. Chem. Eng., 13, № 5, 609 (1968).
382. D о u w е s С. T. t’ Hart M., Erdol u. Kohle, 21, 202 (1968).
383. Oil a. Gas J., 66, № 37, 136 (1968).
384. Griffits D. J., Luntz D. M., J a m e s J. K., Oil a. Gas J.,
66, № 8, 107 (1968).
385. К u b i c k a R., C i r J. et al., Brennstoff-Chemie, 49, 308 (1968).
386. Ohtsuka T., Hasegawa Y., Takanari N., Bull. Japan"
Petrol. Inst., 10, № 5, 14 (1968).
387. Hill J. C., Engelbrecht R. M. etal., Am. Chem. Soc., Div.
Petrol. Chem., Preprint 13, № 3, 183 (1968); C. A., 72, 1236 (1970).
388. ШумовскийВ. Г., ДорогояинскийА. 3., Химия и техноло-
гия топлив и масел, № 8, 24 (1969).
389. Марданов М. А., Велиев К. Г. и др., Азерб. хим. журн:, № 4,
41 (1969).
390. Эйгенсон А. С., Берг Г. А. и др., Химия и технология топлив
и масел, № 4, 1 (1969).
391. Курганов В. М., Мынова 3. А. и др., Нефтепереработка
и нефтехимия, № 1,4 (1969).
392. Hansford R. С., G a u d i о D. A. et al., Oil a. Gas J., 67, № 18,
134 (1969).
393. Oil a. Gas J., 67, № 50, 50 (1969).
394. Debus H. R., C a h e n R. M., Aga R. L., Hydrocarb. Proc. a.
Petrol. Ref., 48, № 9, 137 (1969).
395. Kronig W., Hal cour K., Brennstoff-Chemie, 50, 258 (1969).
396. Q и i n s e у D. H., M e r r i 1 W. H. et al., Can. J. Chem. Eng., 47,
418 (1969).
397. О h t s и k a T., Hasegawa Y., Ono T., Bull. Japan Petrol. Inst.,
11, 3 (1969).
398. Elkady T. Y., Hassan H. A., Abdon J. K., J. Inst. Petrol., '
55, 338 (1960).
399. Агафонов А. В., Хавкин В. А. и др., Химия и технология
топлив и масел, № 11, 6 (1970).
400. Зю ба Б. И., Крич к о А. А., Нефтехимия, 10,813 (1970).
401. Окружной А. М., Вольфсон И. С., Нефтепереработка и нефте-
химия, № 9, 21 (1970).
402. Gianetti J. Р., Mcllvried Н. G., Sebulsky R. Т., Ind.
Eng. Chem., Proc. Design a. Develop., 9, 473 (1970).
403. Qader S. A., Wiser W. H., Hill G. R., Erdol u. Kohle, 23,
801 (1970).
404. J i r i с e k B., Ropa a. Uhlie, 12, № 1, 30 (1970); C. A., 73, 17008 (1970).
405. Kotera Y., Нэнрё Кёкайси, 49, № 516, 197 (1970); С. A., 73, 27262
(1970).
406. Hanke A. M., Oil Week, 21, № 16, 44 (1970); Oil a. Gas J., 68, № 46,
115 (1970).
407. Scott J. W., Bridge A. G. et al., Oil a. Gas J. 68, № 22, 72
(1970).
109
•408. Ragford H. D. Rigg R. G., Hydrocarb. Proc. a. Petrol. Re f-
49, № 11, 187 (1970).
409. Van der Giessen J. A., Hydrocarb. Proc. a. Petrol. Ref., 49, № 8,
1131 (1970).
410. McCondless F. P., Berg L., Ind. Eng. Chem., Proc. Design a. De-
velop., 9, № 1, 110 (1970).
411. Б a p а л В. И., Гуфман Г. С. VIII Мировой нефтяной конгресс.
Москва. 1971. Препринты симпозиума № 12. См. с. 3.
412. Гальбрит Р. Б., Ван-Дризен Р. П. VIII Мировой нефтя-
ной конгресс. Москва. 1971. Препринты симпозиума № 12. См. с. 31.
413. Ку бот а К., Карнер В. М. VIII Мировой нефтяной конгресс.
Москва. 1971. Препринты симпозиума № 12. См. с. 61.
414. Джпльберт Д. Б., Уокер Д. VIII Мировой нефтяной конгресс.
Москва. 1971. Препринты симпозиума № 12. См. с. 81.
415. Флюгтер И. К., Ван-Спийкер П. VIII Мировой нефтяной
конгресс. Москва. 1971. Препринты симпозиума № 12. См. с. 119.
416. Вольфсон И. С., Окружной А. М., Нефтепереработка и неф-
техимия, № 3, 32 (1971).
417. Вольфсон И. С., Окружной А. М., Нефтепереработка и неф-
техимия, № 8, 22 (1971).
418. К р и ч к о А. А., Садыкова С. Р. и др. В сб. «Труды ИГИ МУП
СССР». Вып. 26. № 1. М., «Недра», 1971. См. с. 100.
419. Крич к о А. А., Каганер Г. С. и др. Химия и технология топлив
и масел, К» 8, 4 (1971).
420. Маншилин В. В., Вайль Ю. К. и др., Химия и технология
топлив и масел, № 7, 1 (1971). ।
421. Маншилин В. В., Вайль Ю. К. и др., Химия и технология
топлив и масел, № 11, 7 (1971).
422. Audibert F., Lavergne J. С., Chem. Eng. Progr., 67, №8,
71 (1971).
423. I n о g u c h i M., Nishiyama R. et al., Bull. Japan. Petrol Inst.;
13, № 1, 3 (1971).
424. I n о g u c h i M., I n a b a K. et al., Bull. Japan. Petrol. Inst., 13, № 1,
11 (1971).
425. I n о g u c h i M.f M i z u t о r i T. et al., Bull. Japan Pertol. Inst., 13,
№ 1, 19 (1971).
426. McKinney J. D., Stipanovich J., Int. Hydrocarb. Proces-
sing, 50, № 5, 97 (1971).
427. Moritz К. H., S a r a g e H. R. et al., Chem. Eng. Progr., 67, № 8,
63 (1971).
428. Sv ajgl O., Ropa a. Uhlie, 13, № 7, 359 (1971); РЖХим, 24П261 (1971).
429. Paradis S. G., Could G. D. et al., Chem. Eng. Prog., 67, № 8,
57 (1971).
430. Schuman S. C., Shalit H., Catalysis Rev., 4 (2), 245 (1970).
431. Op очко Д. И., Сулимов А. Д., Осипов Л. Н. Гидрогени-
зацпонные процессы в нефтепереработке. М., «Химия», 1971. 352 с.
432. Лозовой А. В., Сенявин С. А., Воль-Эпштейн А. Б.,
ЖПХ, 28, 175 (1955).
433. Пучков П. В., Нефт. хоз., № 3, 51 (1935).
434. Nelson W. L., Oil a. Gas J., 66, № 3, 99 (1968).
435. Moore В. J., Trimble R. A., Greensfelder B. S., J.
Am. Chem. Soc., 74, 373 (1952).
436. Landa S., Weisser O., Mostecky J., Chem. listy, 52, 60
(1958).
437. ЛевинС. 3., Динер И. С., Карпов А. 3., Химия и техноло-
гия топлива, № 8, 8 (1956).
438. Кецлах М. М., Рудковский Д. М., Химия и технология то-
плива, № 3, 1 (1957).
ГЛАВА 2
НЕКОТОРЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
ПРОТЕКАНИЯ ИОННЫХ И РАДИКАЛЬНЫХ РЕАКЦИЙ
Процессы переработки топлив под давлением водорода весьма
многообразны по своим целям и, соответственно, по выбору ката-
лизаторов и технологических параметров, поэтому в них могут иметь
место различные реакции гидрирования, изомеризации и расще-
пления.
группе реакции гидрирования следует различать реакции
присоединения водорода к диенам, алкенам, ароматическим угле-
водородам. Эти реакции, протекая обычно по сходным механизмам,
могут значительно различаться между собой по скорости, которая
определяется, с одной стороны, строением гидрируемого вещества,
а с другой — свойствами катализатора, в первую очередь его хими-
ческим составом *. Хотя реакции гидрирования являются своего
рода стандартными реакциями, на которых изучался и развивался
катализ 2,% детали элементарных актов этих реакций, особенно
взаимодействие с катализатором, продолжают изучаться 4. Наи-
более общей схемой механизма реакции гидрирования является
представление .о хемосорбции гидрируемого вещества и водорода
на поверхности катализатора и их последующем взаимодействии.
Образование промежуточных соединений, адсорбированных на
поверхности катализатора, доказано в основном изучением реакций
дейтерообмена. Вещество может быть моно- и диадсорбированным,
причем в последнем случае различают а,а-, а,0--и а,у-диадсорбцию 5:
I
моноадсорбция
С или С
У ' \
а.а-диадсорбция
а,Р-диадсорбция
а.у-диадсорбция
Ненасыщенная связь (в простейшем случае, олефиновая) легче
всего образует л-комплекц, который может переходить в а,0-диад-
сорбированный о-комплекс или, значительно труднее, — в а,у-диад-
сорбированный о-комплекс:
111
Ароматические соединения также образуют л-комплексы в, пере-
ходящие в о-комплексы с двух- или шеститочечнои ориентацией.
В рамках терминологии мультиплетной теории А. А. Балан-
дина 2 реакции гидрирования будет соответствовать следующее
превращение индексной группы:
Н
С-Н
I I
н С-Н
Влияние заместителей при индексной группе проявляется через
изменение электронной плотности двойной связи, а также через
изменение'энергий взаимодействия промежуточных соединений с ка-
тализатором.
Эти положения можно пояснить на примере реакции
н н
Х\
сн н
II + I
,сн н
X'/
хх I I /X'
>с-с<
Kz хк
Х\
ХСН2
“к /СН,
X'/
к
н
+ 2 I
к
где X и X' — замещающие (внеиндексные) группы (в общем случае
они могут входить в состав одного цикла), К — активные участки
поверхности катализатора.
В соответствии с принципом энергетического соответствия муль-
типлетной теории 2 энергия образования промежуточного комплекса
равна
— (?с=с — Он—н + Ок-снх—+ Ок—СНХ'—+2(?к-н
где (?к-снх-, (?к-снх'-> (?к-н — энергии связей осколков молекул
с катализатором; Qc=c, (?н-н — энергии связей в реагирующих
молекулах.
Энергия распада промежуточного комплекса на продукты реак-
ции составит соответственно
Е’= (?н-снх- + Оц-снх'——(<?к-снх- + <2к-снх'-+2<?к-н)
В зависимости от величин Е' и Е" одна из стадий будет лимити-
ровать реакцию в целом, а наибольшая из величин Е' и Е" войдет
в уравнение энергии активации
е = А — уЕ
где е — энергия активации, А — постоянная величина, обраща-
ющаяся в нуль для простых реакций 2, у — коэффициент, равный
по А. А. Баландину 0,75, а по Н. Н. Семенову 7 — 0,75 для эндо-
термических и 0,25 для экзотермических реакций. Влияние заме-
стителей X и X', следовательно, может сказываться на скорости
реакции либо через величину <2с=с, либо через величины (?к-снх-
и (?к-снх'-- Кроме того, на скорость реакции оказывают
112
влияние яисто стерические факторы, а также соотношение между
степенями покрытия поверхности гидрируемым веществом и водо-
родом 3.
Подробно влияние структуры гидрируемого вещества на скорость
его превращения рассматривается в гл. 3.
В группе реакций изомеризации и расщепления возможно ана-
логичное влияние строения молекулы и характера замещающих
групп на скорость процесса. Однако особенности таких реакций
определяются в первую очередь образованием в качестве промежу-
точных продуктов заряженных или незаряженных частиц, т. е. ионов
или радикалов.
Деление химических реакций на гомолитические и гетеролити-
ческие в настоящее время общепринято и распространяется также
на гетерогенные каталитические реакции Ч
Наиболее распространенным типом связи является двухэлек-
тронная связь (п-связь). В зависимости от электроотрицательности
атомов, образующих эту связь, электронные орбитали могут быть
симметричными (при равной электроотрицательности) или смещен-
ными так, что электронная плотность будет выше у более электро-
отрицательного атома. Смещение электронной плотности может
иметь место и в случае связи, образованной одинаковыми атомами,
которые соединены с атомами или группами атомов, имеющими
разную электроотрицательность (индуктивный эффект). Так, на-
пример, двойная связь в бутене-2 не поляризована, а в пропилене
и хлористом аллиле — поляризована:
Г» О
сн3— сн=сн—сн3 сн3—СН=СН2 СН2=СН—СН2С1
Разрыв связи может произойти либо с разрывом электронной
пары, т. е. с образованием двух неспаренных электронов (гомолиз),
либо с ее переходом к одному из атомов, который приобретает отри-
цательный заряд (гетеролиз). Естественно, что второй партнер при-
обретает положительный заряд. Направление разрыва зависит от
строения вещества, т. е. от степени поляризации разрываемой связи,
а также, что существенно для рассматриваемого вопроса, от природы
катализатора.
При гомолитическом разрыве необходимая для этого энергия
должна быть компенсирована образованием новых электронных
пар с участием неспаренных электронов катализатора. Такие неспа-
ренные электроны легче всего поставляют переходные элементы
с незаполненными d- и /-оболочками Ч
Катализаторы, ускоряющие гетеролитический разрыв, должны
обладать способностью к образованию координационной связи за счет
отдачи или присоединения электронной пары. Они могут быть,
в частности, протонными или апротонными кислотами или основа-
ниями, причем наличие элементов с незаполненными d- и /-оболоч-
ками в них не обязательно Ч
8 Заказ 271 ИЗ
По Г. К. Борескову \ реакции гидрирования ненасыщенных
соединений (олефинов, бензола, фенола, анилина) и гидрогенолиз
связей углерод—гетероатом (обычно С—S) относят к группе гомо-
литических каталитических реакций, в то время как реакции изо-
меризации и расщепления — к группе гетеролитических. Это не
строгая классификация и есть группа процессов, в том числе и про-
мышленно важных, в которых наблюдаются и гомолитический,
и гетеролитический катализ г. К ним, в частности, относятся про-
цессы каталитического риформинга и гидрокрекинга, осуществля-
емые на полифункциональных катализаторах.
Б. С. Гринсфельдером 8 подсчитаны изменения энтальпии двух
гипотетических реакций — прямого гомо- и гетеролитического рас-
щепления углерод-углеродной связи в гексане:
—► СН3-СН2-СН2 + СН2-СН2-СН3 1
СН3—СН2—СН2: СН2-СН2-СН3—
ккал/моль
--► СН3-СН2-СН2 + СН2-СН2-СН3 2
АЯ=—260 ккал/моль
Изменения энтальпии показывают, что реакции типа (1) могут
происходить в результате прямого нагревания, что й наблюдается,
например, в термических процессах нефтепереработки. Осуществле-
ние реакций типа (2) только при нагревании невозможно — гораздо
раньше произойдет гомолитический разрыв, поэтому для образования
ионов необходимо снижение энергии активации.
Для рассмотрения химических превращений компонентов топлив
в условиях гидрогенизационных процессов важно отметить, что
независимо от путей возникновения ионы несут несравненно боль-
шую энергию, чем свободные радикалы. Следовательно, они гораздо
более реакционноспособны, что не может не отразиться на течении
реакций ионного и радикального расщепления.
Теория цепных радикальных реакций достаточно хорошо раз-
работана 7. Положения этой теории можно привлечь и для объясне-
ния закономерностей радикального расщепления в условиях гидро-
генизационных процессов. Радикальная реакция складывается из
стадий инициирования, роста и обрыва цепей. Стадия инициирования
в процессе гидрогенизации может быть осуществлена или путем
взаимодействия с катализаторо.м, или в результате прямого гомо-
литического расщепления п-связи при достаточно высоких темпе-
ратурах.
В стадии роста цепей происходит взаимодействие радикалов с мо-
лекулами реагирующего вещества. Радикал обладает большой энер-
гией, поэтому он реагирует очень быстро, отрывая от молекулы
атомы, находящиеся во внешнем слое: в углеводородах это атомы
водорода. Таким образом, хотя связи С—Н прочнее связей С—С,
при взаимодействии радикалов с углеводородами, тем - не менее,
отрываются атомы водорода. Но, как ни велика энергия радикала,
114
энергии связей С—Н соизмеримы с ней или даже выше; поэтому
не каждое взаимодействие углеводорода с радикалом приводит к от-
рыву атомов водорода.
Наиболее прочные связи С—Н должны быть атакованы наиболее^
богатыми энергией радикалами, содержание которых в системе
невелико. Прочность связи С—Н уменьшается от первичного атома
углерода к вторичному и далее — к третичному. Если принять веро-
ятность отрыва атома водорода от первичного атома углерода за
единицу, то вероятность отрыва от вторичного и третичного атомов
может быть вычислена по формуле
AQ
W = eRT 3
где А(? — разность энергий связи, принимаемая 9 за 1,2 ккал/моль
для вторичного углеродного атома и 4,0 ккал/моль для третичного.
Помимо прочности связи значительную роль в направленности
роста цепей играет большая или меньшая компенсация энергии
неспаренного электрона во вновь образованном радикале за счет
соседних связей. Соответственно, третичные радикалы будут ста-
бильнее вторичных, а вторичные — первичных 10:
'С—CH—С;
Еще более стабилизируется радикал, если в 0-положении к не-
спаренному электрону находится двойная связь; в этом случае
с неспаренным электроном сопрягаются более подвижные л-элек-
троны:
-^С- дн^СН^СНг
Для учета влияния двойной связи на вероятность отрыва атома
водорода предлагают11 в формуле 3 увеличивать AQ на 1,2 ккал/молй,
если двойная связь находится в 0-положении, и уменьшать на эту
величину, если она находится в a-положении. Так, например, для
парафина и олефина одной структуры величины AQ составят:
О 4,0 1,2 1,2 0
СН3-СН-СН2-СНа-СН3
I
сн3
о
1,2 О 2,4 0
сн3-с=сн-сн2-сн3
I
сн3
1,2
формулу 3, получим следующие
Подставляя эти величины в
вероятности отрыва одного атома водорода от углеродных атомов
115
в 2-метилпентане и 2-метилпентене-2, например для температуры
427 °C:
1 17,4 2,4 2,4 1 2,4 1 6,5 1
сн3-сн-сн2-сн2-сн3 сн3-с=сн-сн2-сн3
I I
сн3 сн3
1 2,4
Соответственно, образование из 2-метилпентана радикала
(СН3)2С—С3Н7 при данной температуре будет в 5,8 раза более веро-
ятно, чем образование радикала (СН3)2СНСН2СН2СН2. В слу-
чае 2-метилпентена-2 вероятность образования радикала
(СН3)2С=СН—СН—СН3 в 3,7 раза выше, чем вероятность образова-
ния радикала (СН3)2С—СН—СН2—СН2.
Интересные данные, качественно подтверждающие такие вероят-
ности отрыва атомов водорода, были получены 12 при определении
реакционной способности различных атомов водорода в углеводородах
по отношению констант скоростей реакций:
CeHs’+RH —► CgHg-f-R1
CeH5-+CCl4 —► СвН5С1 + СС13.
Радикалы СвН5 • генерировались разложением фенилазотрифенил-
метана СвН6—N=N—С(СвН5)3 в смеси четыреххлористого углерода
и исследуемого углеводорода. Отношение констант скоростей опре-
делялось отношением количеств образующихся бензола и хлор-
бензола. В зависимости от строения исходного углеводорода эти
отношения составили:
С-СН2—Н......... 0,0098
С—СН—Н..........0,091
I
С
с
I
С-С-Н...........0,43
I
С
с
I
С-С-СН2-Н........0,0117
I
С
С=С—СН2—Н . . . 0,145
С=С—СН—Н. . . . 0,30
I
С
с
С=С—С—Н......1,20
I
С
<7>-н......1181
f^\-H......0,87
Приведенные данные не относятся к процессам гидрогенизации,
но представляют интерес для их понимания, так как позволяют
предвидеть наиболее вероятное положение неспаренного электрона
в реагирующей молекуле. Это имеет большое значение, так как
радикалы не только участвуют в реакциях передачи цепи, но и под-
вергаются реакциям расщепления, образуя олефин и радикал мень-
116
шего молекулярного веса. А так как в радикале неспаренный электрон
сопряжен с электронами связей, находящихся в 0-положении, именно
эти связи будут легче всего расщепляться.
Это было подтверждено 13 расчетами энергий разрыва связей
в алифатических радикалах (в ккал/моль):
• С—C-J-C .........15
• С—C-j-C—С ...... 8,5
• С—C4-R............8,0
• С—C4-R..........10,0
I '
С
• С—СуС...........18,0
Видно, что легче всего протекает расщепление с образованием
осколков С2, причем тем легче, чем больше молекулярный вес
радикала.
Применимость общих положений теории радикальных реакций
к процессам гидрогенизации можно иллюстрировать примером де-
струкции тетралина и бутилбензола под высоким давлением водо-
рода. Во многих работах было показано 14, что тетралин расще-
пляется по связи, примыкающей к бензольному кольцу. Это объяс-
няли 15 присоединением атомарного водорода, генерируемого
в условиях высоких давлений реакцией молекулярного водорода
с радикалами:
R- + H2 —> RH + H-
S\/\ ^\ЩС\
| || 1+Н. —> | | | —> | | 4
'W' \/-х/ \/\/
Как видно из схемы, связь, примыкающая к бензольному кольцу,
во-первых, превращается из более прочной связи между алифати-
ческим и ароматическим атомами углерода в обычную связь двух
алифатических атомов углерода и, во-вторых, она^находится в 0-по-
ложении к неспаренному электрону. Казалось бы, на основании этих
данных можно считать доказанным, что превращение тетралина
под давлением водорода идет с промежуточным образованием бутил-
бензола, т. е. так, как считали раньше (см., например, 14):
(СН2)3СН3
<СН2)2СН3
Однако экспериментально показано, что в продуктах деструкции
тетралина и декалина преобладает этилбензол, тогда как в продуктах
деструкции бутилбензола — толуол 1в, т. е. эта схема неточна. Для
объяснения такого различия нужно обратиться к рассмотрению
строения промежуточно образующихся радикалов. Образование
этилбензола из тетралина понятно из схемы (4), так как в радикале
бутилбензола связь, находящаяся в у-положении к бензольному
кольцу, занимает [J-положение по отношению к неспаренному элек-
трону. Образование толуола из бутилбензола заставляет предполо-
жить, что наиболее вероятным положением неспаренного электрона
при деструкции бутилбензола является ^-положение:
C-j-C-C-C
Еще более стабилен радикал с неспаренным электроном в a-поло-
жении
С-С-рС-С
поскольку в этом случае неспаренный электрон сопряжен с л:-элек-
тронами кольца. Однако энергия радикала при этом уменьшается
и, соответственно, уменьшается вероятность разрыва связи, находя-
щейся в у-полцжении к бензольному кольцу.
Вероятность образования радикала
С-С-С-С
почти равна вероятности образования у-радикала, но разрыв а-
и 6-связей затруднен, так как первая прочнее других, а при
разрыве 6-связи должен оторваться метильный радикал (см. расчеты
на стр. 117).
Хотя радикалы обладают значительной энергией, ее недостаточно
для скелетной изомеризации. Единственно возможным видом изо-
меризации является внутримолекулярное превращение первичного
радикала во вторичный:
СН2
СН2
Н2С СН2 —► Н2С СН2
Н2С • СН2—сн3
Н3С -сн—сн3
Следовательно, деструкция органических молекул через про-
межуточное образование радикалов, не будет сопровождаться
изомеризацией.
118
Сравнение изменения энтальпии реакций гомо- и гетеролити-
ческого расщепления С—С-связи в гексане (см. стр. 114) показывает,
что ионы несут в несколько раз больше энергии, чем радикалы.
Гипотеза о промежуточном образовании карбониевых ионов 17t
плодотворно примененная для объяснения механизма многих
реакций в органической химии10, успешно использована и для
объяснения механизма ионных реакций, протекающих в процессах
переработки нефти. Основные обобщения сделаны применительно
к каталитическому крекингу 81 *• 18, но могут быть, с опреде-
ленной ревизией, использованы и для процессов гидрогенизации.
Эти обобщения, получившие название карбониево-ионной теории,
в первую очередь должны были объяснить различия протекания
каталитического и термического крекинга. ’
Отличительными особенностями каталитического крекинга по
сравнению с термическим являются:
а) преобладание в газообразных продуктах углеводородов Са
и изо-С4, а не С2 с примесью Сх и С3;
б) протекание реакций изомеризации;
в) расщепление алкилбензолов по а-связи;
г) большая легкость расщепления олефинов по сравнению с пара-
финами, а также углеводородов изостроения по сравнению с угле-
водородами нормального строения.
Так как каталитический крекинг протекает на кислых катализа-
торах, способных поставлять протоны, для объяснения его особен-
ностей и была принята гипотеза о промежуточном образовании
карбониевых ионов.
Карбониево-ионная теория сначала развивалась путем вывода
эмпирических правил на основе сопоставления экспериментальных
данных, лишь в последнее время в нее внесены расчеты8. Так
были сформулированы основные постулаты теории, предусматрива-
ющие следующие типы реакций карбониевого иона:
а) перемещение пары электронов от соседней л-связи (аллильный
перенос):
R—С=С—С— R—С—С=С—
III III
б) перемещение пары электронов вместе с водородом (гидридный
перенос):
R\ И + /Н R4 + Н н
>С-С< —> )С-С<
Н3С/ ^СН3 H3CZ ХСН3
в) перемещение пары электронов вместе с алкильной группой
(алкильный перенос):
R\CH3+ СНз r • СНзСН3
>С—С< —> >С-С<
H3CZ \н ЩСХ ' \Н
г) стабилизация карбониевого иона путем отщепления протона:
RCH2-SHR' ---> RCH=CHR' +Н+
11»
д) стабилизация карбониевого иона путем отщепления иона
меньшей величины:
RCH2-CH-CH3 —> r+ + ch2=gh-gh3
е) стабилизация карбониевого иона путем отщепления гидрид-
ного иона или иного аниона от другой молекулы:
RCH2-CH—СН3 RCH2-CH2—GH3 + R3G+
Эти реакции протекают с соблюдением так называемого 0-пра-
вила, т. е. преимущественно разрывается связь, стоящая в 0-поло-
жении к заряду, поскольку наличие в цепи положительного заряда
вызывает сопряжение с ним электронных зарядов всех связей, сто-
ящих в 0-положении. Такое сопряжение, естественно, сильнее, чем
сопряжение с неспаренным электроном. В отношении скоростей
и превалирования того или иного типа превращения были сформули-
рованы следующие правила:
1. Аллильный перенос осуществляется быстрее других вследствие
высокой подвижности л-электронов; во многих случаях предпола-
гается существование равновесия с равномерным распределением
электронной плотности:
+ в<- б h +
R—С—С=С— R—С—С—G— R G=G-C—
III III III
2. Гидридный и алкильный переносы более предпочтительны
в тех случаях, когда первичный ион превращается во вторичный,
а вторичный в третичный, а не наоборот.
Для обоснования этого правила были вычислены 8 теплоты гипо-
тетических реакций гетеролитического отщепления гидрид-иона:
СН3СН2СН2СН3-
СН3
I
СН3СНСН2СН3
СН3СН2СН2СН2 + Н- ДЯ=-268 ккал/моль
СН3СН2СНСН3 + Н" ДЯ = —241,5 ккал/моль
сн2
I
-->- СН3СНСН2СН3 + Н- ДЯ = —263 ккал/моль
—>- СН3СНСНСН3-|-Н_ ДЯ = —237,5 ккал/моль
СН3
—>- СН3ССН2СН3-|-Н_ ДЯ=—227,5 ккал/моль
I
СН3
. 120
Из сопоставления изменения энтальпии приведенных выше реак-
ций видно, что превращение первичного иона во вторичный дает
выигрыш энергии 26,5 ккал/моль, превращение первичного в тре-
тичный — 35,5 ккал/моль, а вторичного в третичный — 10 ккал/моль.
Величины такого же порядка наблюдаются при сравнении энтальпии
образования ионов другой структуры, т. е. различие в энергиях
первичных, вторичных и третичных ионов является общим правилом.
3. При отщеплении олефина и образовании карбониевого иона
с меньшим молекулярным весом на скорость реакции влияет
структура образующегося иона.
Для реакции
RCH2-CH-CH3 —> R+ + C3He
вычислены 8 величины энергии активации:
Еакт,
R+ ккал/моль
Третичный ... 14,5
Нторичный . . 30,5
Первичный . . 45,0
4. При цепном распространении ионной реакции легче всего
отщепляется водород от третичных атомов углерода, далее от вто-
ричных и затем от первичных. Это согласуется с данными о том, что
каталитический крекинг интенсивно протекает только при наличии
разветвленных углеводородов.
Сформулированные таким образом основные постулаты карбо-
ниево-ионной теории позволили объяснить отмеченные выше основ-
ные особенности каталитического крекинга.
Поскольку прямой гетеролитический распад из-за большого
энергетического барьера невозможен (см. стр. 114), зарождение
цепей объясняют появлением следов олефинов за счет термического
крекинга.
Донором протонов является кислый катализатор. Присоединение
протона к олефину энергетически очень выгодная реакция, причем
тепловой эффект увеличивается в ряду: первичный <; вторичный <
< третичный ионы, например:
।—► СН3—СН2—СН2 ДЯ = 4-165 ккал/моль
СН3-СН=СН2 + Н+ —
'—> СН3—СН—СН3 ДЯ= 4-181 ккал/моль
Образовавшийся первоначально карбониевый ион вступает в об-
менные реакции с исходным углеводородом. Этот обмен облегчается,
если в исходном углеводороде есть третичные углеродные атомы.
Так как образование первичных ионов энергетически невыгодно,
их концентрация мала; кроме того, они легко переходят во вторичные
или третичные ионы в результате гидридного переноса:
111+ I I ♦ I
R—С—С—С—С-------► R-C-C-C-C —
1111 1111
121
Поэтому расщепление по 0-правилу дает главным образом угле-
водороды С3. За счет алкильного переноса образуется также большое
количество углеводородов С4 изостроения:
I I + I I + I I
R-C-C-C-C- R-C-C-C : С----------►
Illi 1111'
R-C-C-C+ -->
Следовательно, относительная легкость алкильных переносов
объясняет легкость изомеризации.
Крекинг ароматических углеводородов по a-связи объяснялся
присоединением протона к двойной связи:
Этот механизм, долгое время фигурировавший во многих статьях
и монографиях, обоснован недостаточно. В настоящее время пола-
гают 10, что образуется л-комплекс, который стабилизуется, от-
щепляя не протон (наибольшая электрофильность), а карбониевый
ион. Цепная реакция продолжается за счет стабилизации отщеплен-
ного положительно заряженного иона, например:
СН3-СН-СН3 —> СН3-СН=СН2+Н+
Таким образом, хотя и несколько спекулятивно, положения
карбониево-ионной теории объяснили особенности каталитического
крекинга. Существенным успехом этой теории было вычисление
состава продуктов крекинга цетана 8, который удовлетворительно
совпал с экспериментальными данными. Следует, однако, отметить,
что эти вычисления сделаны после опыта, а не до него; карбониево-
ионная теория хорошо объясняет, но плохо предсказывает течение
ионных реакций.
Слабой стороной этой теории является допущение строго лока-
лизованных зарядов или так называемых классических карбониевых
ионов (сг-комплексов), поскольку такая локализация требует слиш-
ком большой энергии. В настоящее время считают 101 19, что, по-види-
мому, более устойчивы «неклассические» ионы или ионы с мостиковой
структурой, например для случая миграции метильной группы
122
Слабость этой части теории можно проиллюстрировать на при-
мере изомеризации циклогексана и метилциклопентана. С позиций
описанных выше постулатов очень логично превращение цикло-
гексана в метилциклопентан, включающее гидридный перенос 9:
Но в условиях, например, платформинга идет Обратная изомери-
зация. Для объяснения механизма этой реакцйи с помощью клас-
сических карбониевых ионов нужно предусмотреть гидридный пере-
нос от третичного атома углерода к первичному:
R+
-НН"”
Между тем, для подобного переноса необходима энергия порядка
30 ккал/моль (ср. реакции на стр. 120). Поэтому такого рода превра-
щения объясняют, впрочем без доказательств, образованием мости-
ковых ионов:
Несмотря на перечисленные выше слабые стороны и умозритель-
ный характер отдельных положений •* представления карбониево-
ионной теории играют большую положительную роль в понимании
существа ионных реакций изомеризации и расщепления.
Применение основных положений этой теории к процессам, проис-
ходящим при переработке топлив под давлением водорода, позволяет
по характеру получаемых продуктов судить об ионном или радикаль-
ном течении реакций изомеризации и расщепления и с большой
* Приложение представлений о классических и мостиковых ионах и крити-
ческое их рассмотрение применительно к реакциям изомеризации нафтенов
читатель найдет в монографии А. А. Петрова20.
123.
степенью обоснованности предполагать наиболее энергетически вы-
годные пункты локализации заряда или неспаренного электрона
в реагирующих молекулах.
Первые представления о возможности протекания в условиях
гидрогенизации радикально-цепных реакций развиты Н. Н. Семе-
новым, В. В. Воеводским и Ф. Ф. Волькенштейном 21. Согласно этим
взглядам, активные участки поверхности катализатора можно рас-
сматривать как фиксированные радикалы, дающие начало цепным
превращениям, например:
+ СН2=СН2
СН2—СН2
---+ СгНб
Известным подтверждением такого или аналогичного цепного
механизма могут служить случаи, когда концентрация катализатора
входит в кинетическое уравнение в степени большей, чем единица
(например, для случая гидрирования толуола на никеле Ренея
показатель степени для концентрации катализатора составил 22
1,52—1,67;.
Важным критерием для оценки степени участия радикалов или
ионов в реакции расщепления является состав газообразных про-
дуктов: образование углеводородов С4, С2 характерно для расщепле-
ния радикалов, а С3, С4 — для расщепления ионов.
Отношение С3 С4 к С4 -|- С2 используется для характеристики
катализаторов гидрогенизационных процессов, что важно при их
подборе. Примером может служить оценка трех различных катали-
заторов гидрокрекинга по составу гидрогенизатов индивидуальных
углеводородов (см. табл. 5).
Из данных, приведенных в табл. 5, видно, что при расщеплении
большинства углеводородов в присутствии алюмокобальтмолибдено-
вого катализатора углеводородов С4 и С2 образуется больше, чем
углеводородов С3 и С4 (отношение С3 С4 к С4 + С2 меньше еди-
ницы). В случае никелевого и платинового катализаторов на кислых
носителях это отношение больше единицы, т. е. среди газообразных
продуктов преобладают углеводороды С3 и С4. Исключение соста-
вляет лишь бутилбензол, расщепление боковой цепи которого про-
текает как по ионному, так и по радикальному механизмам (см.
выше реакции на стр. 117 сл. и 122). Следовательно, отношение
(С3 -l-CJ^C! + С2) может быть надежным индикатором протекания
реакций по ионному или радикальному механизму, а его большая
величина будет указывать на кислотную функцию катализатора
или носителя.
Изучению кислотных свойств алюмосиликатных и цеолитовых
носителей посвящена обширная литература (см. обзоры 24, 25), рас-
424
смотрение которой выходит за рамки настоящей монографии. Доста-
точно указать, что алюмосиликаты и цеолиты обладают как протон-
ными, так и апротонными кислотными центрами, структура и стро-
ение которых все еще вызывают споры, вследствие чего
их исследования продолжаются. В одной из работ 28 на основании
изучения инфракрасных спектров хемосорбированного пиридина
(на цеолите) дается следующая схема образования кислотных цен-
тров по мере активации (прокаливание) цеолита:
О NH4 О
0 0 0 0
Протонные
центры
(центры Бренстеда)
Апротонные
кислотные центры
(центры Льюиса)
По изменению интенсивности полос поглощения хемосорбирован-
ного пиридина сделан вывод, что центров Бренстеда больше, но
с повышением температуры прокаливания их число уменьшается,
доходя до нуля при 800 °C. Число центров Льюиса монотонно воз-
растает с повышением температуры прокаливания.
Итак, поскольку алюмосиликаты и цеолиты обладают кислотными
участками структуры, их участие в ускорении ионных реакций
понять легко. Однако явление взаимосвязи кислотности катализатора
с его способностью ускорять ионные реакции в ходе процессов гидро-
генизации много сложнее. Нужно принять во внимание, во-первых,
что некоторые катализаторы, достаточно хорошо ускоряющие ионные
реакции изомеризации и расщепления, не содержат в своем составе
алюмосиликатов или цеолитов (например, WS2, MoS2 и др.). Во-
вторых, как отмечалось уже на ранних ступенях разработки ката-
лизаторов гидрокрекинга 27, активные катализаторы должны обла-
дать не только кислотной, но и гидрирующей активностями, т. е.
обе активности должны быть выше определенного критического
уровня. Весьма активные алюмосиликаты, использованные в каче-
стве носителей, давали недостаточно активные катализаторы гидро-
крекинга (Pt на А12О3 -f- SiO2) при малых содержаниях платины;
с увеличением содержания платины их активность росла, но только
до определенного предела.
125
Таблица 5. Состав газов гидрокрекинга индивидуальных углеводородов
на различных катализаторах
(моль на 100 моль исходного углеводорода 23)
Исходный углеводород С, с, с, с4 0,4-0,
Al + Co + Mo
н-Гексадекан 35,9 43,1 21,5 13,1 0,44
Метилциклопентан 24,7 4,8 — 1,9 0,06
Циклогексан 9,7 38,4 3,9 2,9 0,14
Метилциклогексан 16,0 3,0 2,9 1,6 0,24
Изодурол 121,8 — — Следы —
Бутилбензол 8,8 11,8 40,5 42,2 4,01
NiS на Al2O3+SiO2
н-Гептан Следы Следы 82,0 69,0 —
н-Гексадекан 5,8 6,6 59,4 55,4 9,26
Метилциклопентан 7,4 37,8 60,6 36,7 2,16
Циклогексан 4,7 34,0 52,2 24,3 1,97
Метилциклогексан 1,2 4,0 35,7 39,8 14,52
Изодурол 29,8 51,4 39,4 66,2 1,30
Бутилбензол 2.7 16,8 29,6 41,1 3,62
Pt на цеолите
н-Гептан . 40,4 11,0 49.0 44,6 1,82
н-Гексадекан 9,4 14,8 92,5 86,1 7.35
Метилциклопентан 11,7 24,0 30,5 30,0 1.69
Циклогексан 11,4 17,9 29,9 19,8' 1,70
Изодурол 76,3 20,4 26,5 29,7 0,57
Бутилбензол 8,5 25,4 38,2 42,1 2,35
При изучении группы катализаторов Pt на алюмосиликате и Ni
на А12О3 были определены 28 общая поверхность катализатора,
поверхность, занятая металлами, и средний размер кристаллитов.
На основании этих данных вычислено расстояние между актив-
ными центрами платины, которое оказалось равным примерно
1500 А. Между тем расстояние между кислотными центрами соста-
вляет только 10 А. Следовательно, металлические активные центры
окружены кислотными. В опытах наблюдалась прямолинейная кор-
реляция между константой скорости гидрокрекинга и величиной
поверхности, занятой платиной. Был сделан вывод 28, что роль
платины — предотвращение (за счет гидрирования) закоксовывания
кислотных центров. Активные центры платины могут защитить
только близлежащие кислотные центры, поэтому скорость гидро-
крекинга коррелирует с величиной поверхности платины, а не с сум-
марной поверхностью катализатора. В случае никелевых катализа-
торов картина осложняется взаимодействием никеля с окисью алю-
миния и с серой сырья. Но защита кислотных центров — не главная
функция гидрирующих центров, основной их ролью является облег-
чение образования карбониевых ионов (см. стр. 121), т. е. образо-
вание олефинов. На гидрирующих центрах, по мнению некоторых
126
авторов 29, протекают и реакции дегидрирования, дающие олефины,
которые необходимы для образования карбониевых ионов. По нашему
мнению, более логично предположить образование «затравочного»
количества олефинов в результате неполного гидрирования арома-
тических углеводородов. (например, промежуточное образование
циклогексена из бензола). Вопросы взаимосвязи гидрирования и
ионных реакций еще до конца не выяснены, их изучение продол-
жается (более подробное обсуждение уже известных данных приве-
дено в гл. 5). Сейчас важно отметить как доказанный факт, что
кислотная функция катализатора может проявиться в условийх-
гидрогенизационных процессов только в определенном сочетании
с гидрирующей функцией.
Все сказанное выше относится к кислотным катализаторам. Труд-
нее представить механизм образования карбониевых ионов на ката-
лизаторах без кислых носителей. Здесь можно обратиться к элек-
тронным представлениям в катализе 30, согласно которым вслёдствие
дефектов решетки полупроводника и наличия примесей могут обра-
зовываться узлы решетки с избытком электронов или с их недостат-
ком («дырки»). При наличии таких «дырок» нетрудно представить
себе, что хемосорбированный радикал с одноэлектронной связью
с поверхностью катализатора или хемосорбированный водород при
взаимодействии с «дыркой» передадут ей электрон и превратятся
соответственно в карбониевый ион или протон:
R* Н«
R* Н+
R+ Н«
Известно, что отрицательные (т]~), нейтральные (т]°) и положи-
тельные центры (т]+) — «дырки» находятся в равновесии и доля
каждой формы определяется уровнем Ферми 30. Уровень Ферми
повышается при наличии донорных примесей и понижается в при-
сутствии акцепторных. Имеются данные, что сульфидные катализа-
торы, в частности WS2, представляют собой «-полупроводники,
в которых есть избыточная нестехиометрическая сера — акцепторная
примесь 311 32. Вполне возможно, что сера может играть аналогич-
ную роль и в других сульфидных катализаторах.
Отмечалось 33, что суммарная активность никелевых катализа-
торов гидрокрекинга увеличивается в ряду
алюмосиликат < Ni на алюмосиликате < NiS на алюмосил икате
127
а доля изопарафинов (показатель активности ускорения ионных
реакций) — в ряду
Ni на алюмосиликате < алюмосиликат < NiS на алюмосиликате
Таким образом, никель увеличивает суммарное расщепление,
но снижает скорость изомеризации (и, следовательно, ионное рас-
щепление). Это объясняют тем, что никель блокирует кислотные
центры, а сероводород, который образуется при гидрировании сер-
нистых соединений, добавляемых к сырью, превращая Ni в NiS,
восстанавливает эти центры 33.
На рис. 2 показана зависимость отношения количеств изопентана
и пентана в продукте гидрокрекинга, как меры интенсивности про-
текания ионных реакций, от количества добавленной серы.
Рис. 2. Гидрокрекинг
м-декана с постепенным
введением в систему серы
(в % отстехиометрически
требуемого для образо-
вания сульфида никеля
Ni2S3)33.
Заслуживает внимания прямая пропорциональность этой зависи-
мости, а также то, что интенсивное протекание реакций изомериза-
ции (отношение кзо-С8 : н-С5 равно 8—10) имело место при добавке
серы в количествах больших, чем нужно для образования NiS.
Следовательно, в этой области часть серы была избыточной (несте-
хиометрической), что согласуется с ранее сделанными предположе-
ниями.
ЛИТЕРАТУРА
1. Боресков Г. К. Катализ. Ч. 1,2. Новосибирск, «Наука», 1971. 267 с.
2. Б а л а н д и н А. А. Современнное состояние мультиплетной теории ге-
терогенного катализа. М., «Наука», 1968. 202 с.
3. С о к о л ь с к и й Д. В., Д р у з ь В. А. Теория гетерогенного ката-
лиза. Алма-Ата, «Наука», 1968. 390 с.
4. Боресков Г. К. В кн. «Основы предвидения каталитического дей-
ствия». Т. 2. М., «Наука», 1970. См. с. 437.
5. Кембол Ч. В кн. «Основы предвидения каталитического действия».
Т. 2. М., «Наука». 1970. См. с. 5.
6. Rooney J. J., Webb G., J. Catalysis, 3, 488 (1966).
7. С e м e н о в H. H. О некоторых вопросах химической кинетики. №.,
Изд. АН СССР, 1955. 686 с.
8. Гринсфельдер Б. С. В кн. «Химия углеводородов нефти». Т. 2.,
М., Гостоптехиздат, 1958. См. с. 114.
9. Хансфорд Р. Ц. В кн.: «Физическая химия углеводородов». Л.,
Гостоптехиздат, 1957. См. с. 172.
128
10. Реутов О. А. Теоретические основы органической химии. М., Иад.
МГУ, 1964. 700 с.
11. Bass С. J.,Vlugter I. С., Brennstoff-Chemie, 45, 321 (1964).'
12. Bridger В. F., Russel G. A., J. Am. Chem. Soc., 85, 3754 (1963).
13. Воеводский В. В., ДАН СССР, 79, 455 (1951).
14. Рапопорт И. Б. Искусственное жидкое топливо. М., Гостоптехиздат^
1955. 546 с.
15. Г о н и к б е р г М. Г., Никитенков В. Е., Изв. АН СССР,
ОХН, 1956, 56.
16. Калечиц И. В., Павлова К. А. и др. В сб. «Труды ВСФ СО АН
СССР». Вып. 38. М., Изд. АН СССР, 1961. См. с. 31.
17. Whitmore F. С., J. Am. Chem. Soc., 54, 3274 (1932).
18. V о ge Н. Н. «Catalysis». Ed. by Р. Н. Emmett. V. 6. New York, Rein-
hold Publ. Corp., 1958. See p. 407.
19. Б p e с л о у P. Механизмы органических реакций. М., «Мир», 1968.
См. с. 121 сл. г
20. Петров Ал. А. Химия нафтенов. М., «Наука», 1971. См. с. 153 сл.
21. В о е в о д с к и й В. В., В о л ь к е н ш т е й н Ф. Ф., Семенов Н. Н.
В сб. «Вопросы химической кинетики, катализа и реакционной способ-
ности». М., Изд. АН СССР, 1955. См. с. 423.
22. L i 11 m a n H., Bless H., Ind. Eng. Chem., 51, 659 (1959).
23. К а з а н ц e в а В. M., Неудачина R. И., Калечиц И. В..,
Нефтепереработка и нефтехимия, № 8, 25 (1968).
24. Л и т в и н е н к о А. Г., Гельме И. Э. и др. В кн. «Производство
и применение катализаторов в нефтеперерабатывающей промышленности
за рубежом». М., ЦНИИТЭнефтехим, 1970. 92 с.
25. М и н а ч е в X. М., Исаков Я. И. Приготовление, активация и ре-
генерация цеолитных катализаторов. М., ЦНИИТЭнефтехим, 1971. 85 с.
26. W а г d J. W., J. Catalysis, 9, 225 (1967).
27. С о о п г a d t Н. L., С i а р е 11 a F. G. et al., Am. Chem. Soc., Div.
Petr. Chem., Preprints, 5, № 4, B35 (1960); C. A., 55, 2040(1(1961).
28. Beuther H., Larson O. A., Ind. Eng. Chem., Proc. Design a.
Develop., 4, № 2, 177 (1965).
29. Thomas C. L., M c N e 1 i s E. VII World Petroleum Congress. Mexico.
Proceedings. V. IB. London, Elsevier Publ. Corp., 1967. See p. 161.
30. Волькенштейн Ф. Ф. Электронная теория катализа на полу-
проводниках. М., Физматгиз, 1960. 187 с.
31. Rohlander W. Dissertation. Halle-Leuna, 1963.
32. К а 1 е с i с I. V., Derjagina Е. N. et al. Symposium uber hydro-
katalytische Prozesse in der Erdolverarbeitung und Petrolchemie, Leuna.
1966. B. 2. S. 76.
33. Langlois G. E.,SullivanR. F., EganC. I., J. Phys. Chem., 70,
3666 (1966).
9 Заказ 271
ГЛАВА 3
ПРОЦЕССЫ НИЗКОТЕМПЕРАТУРНОЙ
ГИДРОГЕНИЗАЦИИ
Превращения многих компонентов сырья в промышленных гидро-
генизационных процессах начинаются с реакций насыщения водо-
родом непредельных и ароматических связей. Во многих препаратив-
ных, а также в промышленных процессах (например, получение
чистого циклогексана из бензола, см. гл. 1, ссылки 22°- 338> 348- 349)
эти реакции являются целевыми, и их стремятся осуществить строго
селективно. Поэтому реакции собственно гидрирования не могут
не представить интерес для понимания механизма и взаимосвязи
всего комплекса реакций, протекающих в условиях гидрогениза-
ционных процессов. Кроме того, реакции собственно гидрирования
всегда привлекали внимание исследователей возможностями изуче-
ния на них закономерностей катализа вообще. Вследствие этого
рассмотреть все аспекты данной проблемы в рамках настоящей моно-
графии не представляется возможным *.
Каталитическое гидрирование в паровой фазе при атмосферном
давлении над восстановленным никелем было открыто Сабатье5.
Вскоре В. Н. Ипатьев 6 впервые применил гидрирование в жидкой
фазе под давлением водорода. За почти семидесятилетний период
развития и изучения реакций гидрирования было открыто много
весьма активных катализаторов 4, позволявших работать при очень
мягких условиях: никелевые катализаторы на носителях, хромит-
медные катализаторы, окись платины, платиновая чернь и др. Боль-
шое значение, в том числе и промышленное, получили так называ-
емые скелетные никелевые катализаторы («никель Ренея»)7. К насто-
ящему времени ряд катализаторов значительно пополнен,
а известные катализаторы усовершенствованы. Так, например, очень
активными катализаторами являются сплавы4 никеля и родия,
платины и рутения, модифицированные катионами палладиевые
катализаторы и др. Скелетные катализаторы значительно улучшены
промотированием 3, а приготовление катализаторов усовершенство-
вано так, что платиновая чернь, например, может быть получена
с поверхностью до 200 м2/г, в то время как в прошлом лучшие
образцы имели поверхность 4 не более 50—60 м2/г.
На скорость гидрирования влияют химическая природа (хими-
ческий состав) катализатора, строение ненасыщенного соединения,
условия процесса (давление, температура, растворитель в случае
* Обстоятельные обзоры и монографии в этой области см. 1 4.
130
жидкофазного процесса, присутствие других соединений, скорость
поступления водорода). Для целей настоящей монографии наиболь-
шее значение представляет второй фактор — строение соединения.
Соответственно, ниже последовательно рассматриваются современ-
ные представления о механизме гидрирования бензола, затем влияние
усложнения ароматической структуры при введении заместителей,
переходе к конденсированным ароматическим системам, введении
гетероатомов. Влияние других факторов будет рассматриваться
только в связи с этими аспектами.
ГИДРИРОВАНИЕ БЕНЗОЛА
На поверхности катализатора бензол может адсорбироваться
либо всей плоскостью, либо одним из ребер. По А. А. Баландину 8
это будут соответственно секстетная и дублетная модели. В случае
плоскостной хемосорбции (секстетная модель) размеры молекулы
бензола и расстояния между атомами металла должны соответство-
вать друг другу. Мультиплетная теория А. А. Баландина 8 по пара-
метрам решеток металлов постулирует, что катализаторами гидриро-
вания и дегидрирования могут быть только металлы: никель, ко-
бальт, медь, рутений, иридий, палладий, платина, родий, осмий,
.рений. Это подтверждено экспериментально, за исключением меди,
на которой гидрирование бензола часто не наблюдалось. Однако
считают 8, что это исключение кажущееся и незначительная актив-
ность меди объясняется энергетическими факторами.
При реберной ориентации (дублетная модель) должны проис-
ходить три последовательных акта присоединения трех молекул
водорода:
Изменения свободной энергии для этих стадий и для прямого
(например, по секстетной модели) гидрирования бензола приведены
в табл. б.
Таблица 6. Изменение свободной энергии при
гидрировании бензола 9
Температура, К Изменение свободной энергии, ккал/моль
стадия 1 стадия 2 стадия 3 прямое гидриро- вание (стадия 4)
296,16 13,2 -17,9 -18,7 -23,4
400 15,7 -14,8 —15.2 —14,3
550 19,6 -10.2 -9.4 0,0
700 23,5 —5,5 -4,4 13,6
1000 31,3 4,2 6,5 42,0
9
131
Данные табл. 6 иллюстрируют давно известную закономерность,
что для гидрирования благоприятны низкие температуры. Во всем
интервале изученных температур селективное гидрирование до цикло-
гексадиена (стадия 1) термодинамически невозможно; эта ступень
должна немедленно компенсироваться следующими, энергетически
выгодными ступенями. В соответствии с этим диены не были найдены
в составе продуктов гидрирования бензола, тогда как небольшие
количества циклогексена в отдельных случаях были обнаружены 4.
Так, например, в работе 10, специально посвященной этому вопросу,
циклоалкены были обнаружены при гидрировании ароматических
углеводородов на рутениевых и родиевых катализаторах, в меньшем
количестве — на никелевых. Особенно четко сохранялся при гидриро-
вании о-ксилола пространственно-затрудненный 1,2-диметилцикло-
гексен-1:
/\/СНз
J II
Х/ХСН3
Для выяснения вероятности промежуточного образования цикло-
алкенов и, соответственно, соотношения между дублетной и секстет-
ной моделями был использован 111 12 метод меченых атомов. Для
случая дегидрогенизации взаимопревращения циклогексана, цикло-
гексена и бензола можно выразить схемой:
Тогда, последовательно вводя метку в циклогексан и цикло-
гексен, можно вычислить раздельно величины wlf w[, w2 и wa, ха-
рактеризующие скорость превращения по дублетному (и?! и w3)
или секстетному (lp2) механизмам, а также возможное участие в этих
превращениях реакции диспропорционирования циклогексена
(по
На рениевом катализаторе 11 при 336 °C 77% радиоактивного
углерода, введенного в небольшом количестве циклогексена, пере-
ходило в циклогексан и только 17% — в бензол. Следовательно,
к?! w3, и реакция диспропорционирования не может играть суще-
ственной роли. Расчеты показали, что w2 wa, т. е. реакция про-
текает в основном по секстетному механизму.
На окиси хрома при 450 °C в бензол переходило 12 до 90% радио-
активного углерода, т. е. w3 "%> ш2, и реакция протекала в основном
по дублетному механизму.
Позднее аналогичные исследования были проведены 13 на плати-
новом катализаторе при парциальном давлении водорода
20 кгс/см2 и парциальном давлении углеводородов 5 кгс/см2 й вы-
132
числены относительные скорости превращений в отсутствие диффу-
зионных осложнений (схема а) и при их наличии (схема б):
0,0070
0,0134^
0,0019
1,379
а
Т 1,0 I
0,0027
При наличии диффузионных осложнений возрастают скорости
прямых взаимопревращений бензола и циклогексана:
б
Из приведенных выше данных видно, что на платине прямое
гидрирование бензола в циклогексан по крайней мере в 5 раз более
вероятно, чем через промежуточное образование циклогексена при
протекании процесса в кинетической области и в 58 раз — в диф-
фузионной области. Следовательно, на платине преобладает сек-
стетный механизм. Отметим также, что скорость гидрирования цикло-
гексена по сравнению со скоростью гидрирования бензола в кине-
тической области выше на два порядка, в диффузионной — на один
порядок. Этим объясняется невозможность образования больших коли-
честв циклогексена при гидрировании бензола.
Влияние природы катализатора может сказываться (помимо
соответствия геометрических размеров) и энергетически, облегчая
или затрудняя образование хемосорбированного промежуточного
комплекса и его распад. Для случая гидрирования этилена по ду-
блетному механизму соответствующие уравнения мультиплетной
теории запишутся следующим образом.
а) Стадия образования промежуточного комплекса:
\с=с/ н—н н \с—cZ н
/ X —> I /| Iх I
Me Me Me Me Me Me Me Me
E' — *?C=C — *?H— н + 2*?Ме-с + 2*?Ме-Н 1
б) Стадия распада комплекса:
Н
I
Me
^с-с/
Х1 1Х
Me Me
Н
I '
Me
\с—с/
/| Iх
н н
Me Me Me Me
— +2<?C-H — 2<?Me—C“ 2@Me-H %
133
Величину суммы энергий реагирующих связей при образовании
промежуточного комплекса (в данном случае 2^ме-с + 2(?ме-н)
А. А. Баландин 8 обозначает q и называет адсорбционным потенци-
алом катализатора. В координатах [Е, q] уравнения 1 и 2 дадут
прямые линии
£' = q —s/2 + “/2
£" = -5 + s/2 + u/2
где и — тепловой эффект реакции, s — сумма энергий реагирующих
связей:
—и = <2С==С + <?н„н — 2<?С-н
s= *?с=с + <2н-н + 2<?с-н
Эти функции, получившие по А. А. Баландину 8 название «вул-
канообразных кривых», показаны на рис. 3. Оптимальный катализа-
Рис. 3. Вулканообраз-
ные кривые:
I — эндотермические реак-
ции; II — экзотермические
реакции.
и
тор будет отвечать точке пересечения прямых — точке z с коорди-
натами [s/2, и/2]. Другими словами, в случае оптимального ката-
лизатора Е' — Е", а адсорбционный потенциал катализатора равен
среднему из энергий разрываемых и вновь возникающих связей.
Для оценки различных катализаторов в рамках этих предста-
влений необходимы данные об энергиях связи металл—углерод
и металл—водород для данных условий. В настоящее время многие
из этих величин определены 8, но не всегда можно применять вели-
чины, определенные для одной реакции, при изучении другой.
Можно пользоваться и таким показателем, как теплота сублима-
ции металла (1к), поскольку, с одной стороны, она связана с такими
характеристиками металла, как незаполненность d-электронных
уровней и параметры кристаллической решетки, а с другой сто-
роны 8, пропорциональна величине q.
На рис. 4 и 5 показаны вулканообразные кривые для реакций
гидрирования этилена и бензола. Аналогичные кривые получаются,
если по оси абсцисс откладывать величину энергии связи металл —
водород (£ме-н) (рис. 6 и 7). Из приведенных данных видно, что
лучшим катализатором гидрирования этилена является родий, а бен-
зола — платина.
Аналогичные результаты были получены17 при определении
удельных активностей платины, никеля и кобальта в реакции гидри-
134
рования бензола. Показано, что до НО—115 °C платина активнее
никеля и кобальта и только при более высоких температурах усту-
пает никелю.
2*, ккал/г-атом
Рис. 5. Вулканообразная
кривая для гидрирова-
ния бензола1*.
Рис. 4. Вулканообразная кривая для
гидрирования этилена14, 16.
По оси ординат отложена температура, при
которой достигается равное фиксированное
превращение (1g г = —0,8).
Рис. 6. Вулканообразная кривая для Рис. 7. Вулканообразная кривая
гидрирования этилена3'16-1’. для гидрирования бензола3'16.
Кинетика гидрирования бензола изучалась многими авторами *.
Как правило, реакция в газовой и в жидкой фазах имеет нулевой
порядок по бензолу и первый порядок по водороду.
Для случая жидкофазной гидрогенизации Д. В. Сокольский 4
приводит уравнение Чао и Эмметта 18, выведенное на основе изо-
термы Ленгмюра
dC __ abC
dt 14- ЬС
где
AGp
« = -|*1Рн2,е RT
AGp
b = RT
*2
135
AGi« — свободная энергия активации для перехода водорода из
газовой фазы в раствор, AGp — то же, из раствора в газовую фазу,
W — количество катализатора.
Константы а и Ъ зависят от давления и температуры, а пропор-
циональна скорости перехода водорода из газовой фазы в раствор,
Ь представляет собой отношение скорости поглощения водорода
к скорости выхода его в газовую фазу. Константа Ь может быть
очень мала (мало катализатора, гидрируемое вещество хорошо
растворимо) и тогда величиной ЬС можно пренебречь. Уравнение 3
принимает вид
AGsq|
__4£ = аЬс=-^к.е“^г"wc
dt к2 Нг
где Д6бО1 = ДСр — ДК;о — стандартная свободная энергия рас-
твора водорода, т. е. реакция будет иметь первый порядок и по веще-
ству, и по водороду.
Если, наоборот, Ь велика, то ЬС )§> 1 и уравнение 3 обращается в
AGf о
d С 1 р j,
---Г“ = а = -7Г^1?Н *е К1
dt 3 Нг
т. е. реакция имеет нулевой порядок по веществу и первый — по
водороду. По-видимому, этот случай реализуется чаще всего, но
действительная кинетика сложнее.
Для парофазного гидрирования (проточный реактор, 120—192 °C,
варьирование парциальных давлений бензола и водорода) обнару-
жено 19, что реакция имеет порядок по бензолу от 0,20 до 0,48, а по
водороду от 0,65 до 2,5.
Исходя из предположения о том, что лимитирующей стадией
является присоединение первого атома водорода, поскольку при
этом должна быть компенсирована энергия сопряжения бензольного
кольца, выведено 29 уравнение для скорости гидрирования бен-
зола с использованием изотермы Фрейндлиха:
V = kCmP^t
Экспериментально определены показатели степеней (т и п) при
использовании различных катализаторов:
Палладий (10%) на угле . . .
Платина (10%) на угле . . .
Рутений..................
Никель Ренея ............
7П
0,08 ± 0,04
0,10 ± 0,05
0,10 ±0,10
0,30 ±0,08
0,76 ± 0,02
0,71 ±0,03
0,90 ±0,10
0,64 ±0,02
Аналогично были определены 21 значения тип для серии кобаль-
товых катализаторов (дифференциальный проточный реактор, по-
стоянные парциальные давления бензола и водорода, степень пре-
вращения 0,3—1,1%). Значения т составили 0,1—0,5, а п — от 1
до 2. Близость значения п к двум авторы объясняют образованием
л-комплекса бензола с металлом, медленно гидрируемого до цикло-
гексена. Предполагают, что гидрирование циклогексена до цикло-
гексана идет быстро и не влияет на скорость всей реакции.
136
Таким образом, современные представления о механизме и кине-
тике гидрирования бензола, хотя и имеют определенные противо-
речия и неясности, приводят к ряду общепринятых основных выво-
дов. К ним относятся обязательность геометрического соответствие
структуры металла и ароматического соединения при плоскостной
адсорбции, невозможность или трудность образования промежу-
точных продуктов, определение скорости процесса большею частью
парциальным давлением водорода и, наконец, представление об
образовании л-комплексов ароматического соединения с переход-
ными металлами 22, причем присоединение первого атома водорода
является лимитирующей стадией процесса в целом. Все эти выводы
сделаны в отношении гетерогенных катализаторов.
В последние годы разработано много комплексных гомогенных
катализаторов на основе переходных металлов, изучение которых
пролило дополнительный свет на механизм гидрирования бензола.
Очевидно, что изучение реакции гидрирования в присутствии
комплексных соединений переходных металлов ценно тем, что про-
цесс не осложнен явлениями диффузии внутри пор, характером
и параметрами поверхности и др., что имеет место в случае гетеро-
генного катализа.
В самом деле, если гидрирование протекает через образование
л-комплексов, то катализаторами этой реакции должны быть все
переходные металлы. Было показано 23, что в присутствии комплексов
никеля, кобальта, железа, марганца, хрома и ванадия, активиро-
ванных алюмоорганическими соединениями, олефины гидрируются
с высокими скоростями, т. е. зти комплексы достаточно хорошо
активируют водород. Основываясь на этом, удалось показать м, что
бензол гидрируется в присутствии комплексных металлоорганиче-
ских катализаторов на основе всех переходных металлов четвертого
периода.
Если принять каталитическую активность ацетилацетоната ни-
келя за единицу, то относительные активности других катализаторов
составят:
Дициклопентадиенилдихлорйд
титана ................0,07
Диметилглиоксимат никеля . 0,08
Ацетилацетонат ванадия . . . 0,11
Ацетилацетонат хрома . . . , 0,13
Ацетилацетонат марганца . . 0,16
Дициклопентадиенилжелезо . 0,19
Ацетилацетонат железа . ^ . 0,22
Ацетилацетонат меди .... 0,26
Ацетилацетонат кобальта . . 0,90
По каталитической активности переходные металлы четвертого
периода в комплексах с одинаковыми лигандами (ацетилацетонаты)
образуют ряд:
Ni Со > Си Fe > Мп > Cr > V
Порядок металлов в этом ряду совпадает с порядком для реакции
гидрирования олефинов 23 (за исключением меди), а также с порядком
активностей гетерогенных катализаторов 15:
Ni3sCo»Fe
137
Эта закономерность совпадает с изменением числа d-электронов
переходного металла (см. рис. 4, 5) и прочности связей Me—Н и
Me—непредельное соединение (см. рис. 6, 7).
Все эти данные, полученные для гомогенных катализаторов, т. е.
для условий, не осложненных явлениями диффузии, адсорбции
и десорбции, характеризуют явление в более чистом виде, подтвер-
ждая, в частности, промежуточное образование л-комплексов. Харак-
терно, что гидрирование бензола на меди, хроме, марганце, ванадии,
титане отмечается 24 впервые, оно не наблюдалось на гетерогенных
катализаторах.
Показана 25 возможность гидрирования бензола и других не-
насыщенных соединений на комплексных катализаторах, получаемых
восстановлением солей трехвалентного родия в присутствии арома-
тических аминокислот и пептидов.
Кроме гидрирования ароматических углеводородов на гомогенных
катализаторах удалось осуществить самые разнообразные реакции
гидрирования 26. Результаты этих работ были использованы для
объяснения закономерностей гетерогенного катализа 26. Наиболее
важными выводами являются следующие.
1. Процесс гидрирования включает стадию активации молеку-
лярного водорода, реагирующего с активным соединением с образо-
ванием гидридного комплекса, который обычно и играет роль ката-
лизатора.
2. Наиболее активны металлы в состояниях низших валент-
ностей, которые стабилизируются л-донорными лигандами, т. е.
образование л-комплексов энергетически выгодно для протекания
гидрирования.
ГИДРИРОВАНИЕ ГОМОЛОГОВ БЕНЗОЛА
Поскольку, как показано в предыдущем разделе, важнейшими
этапами реакции гидрирования бензольного кольца являются стадии
образования л-комплекса, а также образования и разрушения связи
атома углерода с металлом, присутствие в бензольном кольце заме-
стителей, оказывающих влияние на распределение электронной
плотности или, другими словами, на потенциал ионизации, обяза-
тельно должно сказываться на скорости гидрирования.
Изучение этого вопроса началось задолго до сформирования со-
временных представлений о механизме и элементарных актах гидри-,
рования бензольного кольца. Пионерами этих исследований
были А. В. Лозовой и М. К. Дьякова 27’ 28, которые изучали вли-
яние структуры и размеров алкильных заместителей, а также числа
метильных заместителей в ароматических углеводородах на скорость
их гидрирования (катализатор — восстановленный Ni на А12О3,
30 кгс/см2, 140 °C). Было показано, что увеличение длины али-
фатической цепи (от толуола до октилбензола) оказывает лишь очень
небольшое влияние на скорость гидрирования. С примерно одинако-
выми скоростями протекает также гидрирование н-амил- и изоамил-
138
бензолов, 1-метил-4-пропил- и 1-метил-4-изопропилбензодов. Важ-
нейшим выводом явилось доказательство того, что скорость гидри-
рования определяется не структурой заместителя, а числом
заместителей. Например, гидрирование о-ксилола, тетралина и 1-ме-
тил-2-пропилбензола протекает с примерно одинаковыми скоро-
стями. Полученные данные, а также данные других исследо-
вателей 4> 29> 30 приведены в табл. 7.
Таблица 7. Относительные скорости гидрирования гомологов бензола
на различных катализаторах
Углеводород I Ni на А12Оа (30 кгс/см2, 140 °C) 27,28 Pt (4 кгс/см2, 30 °C) 29,30 Rh-t-Pt Вычислено по формуле 4
и о О СП (40 °C, без растворите- ля) 4
Бензол 100 100 100 100 100
Толуол 50 62 60 38 50
Этилбензол . . . 43 45 72 32 50
н-Пропилбензол 45 41 — — - 50
Изопропилбензол — 33 — — 50
н-Бутилбензол 44 38 — — 50
втор-Бутилбензол — 29 — — 50
mpem-Бутилбензол — 26 — — 50
Изобутилбензол — 23 — — 50
н-Амилбензол 41 41 — — 50
Изоамилбензол 40 42 — — 50
н-Гексилбензол — 39 — — 50
н-Октилбензол 40 50
н-Нонилбензол — 39 — — 50
о-Ксилол 24 32 31 7 25
л-Ксилол 23 49 47 23 25
п-Ксилол 31 65 50 30 25
1-Метил-2-пропилбензол 22 — — — 25
1-Метил-4-пропилбензол 34 — — — 25
1-Метил-4-изопропилбензол .... 33 43 — — 25
Тетралин 23 — — — 25
1,2,3-Триметилбензол 14 14 —. — 12,5
1,2,4-Триметилбензол — 29 — — 12,5
1,3,5-Триметилбензол 10 58 — — 12,5
1,2,3,4-Тетраметилбензол — 10 — — 6,3
1,2,3,5-Тетраметилбензол — 11 - — 6,3
1,2,4,5-Тетраметилбензол ...... 3,8 18 — — 6,3
Пентаметилбензол 0,5 3,5 — — 3,2
Гексаметилбензол Очень мала 0,2' — — 1,6
Из данных табл. 7 четко видно, что скорость гидрирования умень-
шается по мере увеличения числа заместителей. Особенно наглядно
это показано в серии опытов с полиметилбензолами аз.
Эту закономерность объясняли 28 тем, что заместители затруд-
няют доступ водорода к гидрируемому бензольному кольцу. Была
139
предложена эмпирическая формула расчета относительной скорости
гидрирования гомологов бензола:
Р„ = 2-пГбенз 4
где V„ — относительная (к бензолу) скорость гидрирования гомо-
лога бензола; п — число заместителей; Кбенз — скорость гидриро-
вания бензола.
Позднее было показано \ что стерические препятствия про-
являются сильнее, если боковая цепь разветвлена (скорость гидри-
рования изопропилбензола ниже скорости гидрирования пропил-
бензола, а в случае изобутил- и mpem-бутилбензолов ниже, чем
для н-бутилбензола), а также при несимметричном расположении
заместителей (скорости гидрирования о-ксилола, 1,2,3-триметил- и
1,2,3,4-тетраметилбензолов ниже, чем скорости гидрирования соот-
ветствующих симметричных изомеров). Следовательно, формула 4,
хорошо передавая качественную закономерность, как видно из
табл. 7, часто дает слишком большие количественные отклонения.
Уменьшение скорости гидрирования при наличии алкильных заме-
стителей качественно подтверждено п в ряде других исследований
по гидрированию полиалкилбензолов на низкотемпературных
катализаторах х.
Интересным подтверждением этого правила явились резуль-
таты 31, полученные при гидрировании 2-алкилнафталинов на хро-
митмедном катализаторе при 220 °C и начальном давлении 117 кгс/см2
в автоклаве. Авторы 31 изучали соотношение между тетрагидро-
производными типов I и II:
I и
В данном случае «слияние» в одной молекуле бензольного и алкил-
бензольного колец исключало какие-либо ошибки опыта, возможные
при раздельном гидрировании, и адсорбционную конкуренцию при
гидрировании парных смесей. Соотношение типов I : II было близко
к 1 : 2, т. е. полностью соответствовало 27 формуле 4.
Исследование 32’ 33 закономерностей влияния заместителей на
скорость гидрирования бензольного кольца в присутствии высоко-
температурных сульфидных катализаторов дало результаты, резко
отличные от результатов работ с низкотемпературными катализа-
торами.
Как видно из данных, приведенных в табл. 8, в случае катализа-
тора WS2 введение заместителей увеличивает скорость гидрирования.
Однако скорость гидрирования гексаметилбензола меньше скорости
гидрирования пентаметилбензола. Таким образом, в случае WS2
имеет место явление, противоположное тому, что наблюдается на
самых различных низкотемпературных катализаторах. Катализатор
MoS2 занимает промежуточное положение.
140
Таблица 8. Относительные скорости гидрирования гомологов бензола
в присутствии высокотемпературных сульфидных катализаторов
(420 ° С, 200 кгс/см2)
Углеводород SW, MoSi Углеводород WS, MoS,
Бензол 10 100 1,3,5-Триметилбензол 430 111
Толуол 230 99 Пентаметилбензол . . . 630 92
^Этилбензол 140 78 Гексаметилбензол . . . 150 —
•и-Ксилол - 330 108 Тетралин . 250 287
Позднее при изучении составов продуктов гидрирования аромати-
ческих углеводородов в присутствии различных промышленных
вольфрамовых катализаторов 34-36 вывод работы 33 был подтвержден
для катализаторов WS2 и WS2 -f- NiS на А12О3. При гидрированйи
на катализаторе WS2 на терране наличие заместителей снижает
скорость гидрирования (см. табл 9).
Таблица 9. Количества непрореагировавших ароматических углеводородов
(в %) при гидрировании над промышленными катализаторами
(420 °C, 300 кгс/см2, автоклав, 3 ч)
Катализатор Бензол Толуол Этил- бензол о-Ксилол м-Квилол
ws24 NiS на А12О3 . . 28,3 5;6 19,2 — 3
WS2 30,8 — 6,0 5,3
WS2 на терране .... 64,1 76,1 75,5 —
Необычное поведение высокотемпературных катализаторов объяс-
няли 37 различной продолжительностью пребывания углеводородов
в адсорбированном состоянии, что зависит от их строения. Увеличе-
ние времени пребывания вещества на поверхности катализатора
ускоряет гидрирование, если катализатор недостаточно активен
г отношении гидрирования данной связи (гидрирование бензола и его
гомологов над WS2 37), и замедляет гидрирование, если катализатор
высокоактивен (гидрирование над WS2 конденсированных аромати-
ческих углеводородов 37). Эта точка зрения не объясняет, почему
на WS2 на терране — гораздо менее активном катализаторе, чем
WS2, — введение заместителей не ускоряет, а замедляет процесс
гидрирования.
Это различие никелевых, вольфрамовых и молибденовых катали-
заторов было объяснено тем, что на них имеют место разные лимити-
рующие стадии, т. е. они находятся на разных половинах вулкано-
образной кривой (уравнения на стр. 134). К этим представлениям
близко подходят представления 22 о большей или меньшей легкости
образования л-комплексов, примененные к рассматриваемому слу-
чаю гидрирования алкилбензолов 39> 4°. Кинетические параметры
Таблица 10. Кинетика гидрирования ароматических углеводородов
на катализаторе Ni + MgO (144 °C, атмосферное давление)
и относительная стабильность л-комплексов за. 40
k—константа скорости гидрирования; А-предэкспоненциальный множитель,
Е—энергия активации
НС1
Относительная стабиль-
ность л-комплекса при
действии;
Углеводород
пикрино-
вой
кислоты
Бензол......................
Толуол .....................
п-Ксплол ...................
Этилбензол .................
1,3,5-Трпметилбензол . . . .
18 2,4 5,8 14,2 9,24 0,66 0,83
16 2,1 5,4 13,5 8,22 1,00 1,00
10 1,3 4,0 11,1 8,44 1,09 1,19
10 1,3 3,7 10,4 8,77 1,15 0,88
5 0,7 1,8 8,0 8,39 1,73 1.33
0,92
1,00
1,92
5,10
этого процесса, потенциалы ионизации и относительная стабиль-
ность л-комплексов приведены в табл. 10.
Из данных по стабильности комплексов, несмотря на некоторый
разброс данных, следует, что устойчивость л-комплексов должна воз-
растать от бензола к 1,3,5-триметилбензолу. Это легко понять, так
как алкильные группы повышают электронную плотность в кольце.
Соответственно, в ряду бензол — 1,3,5-триметилбензол уменьшается
энергия активации (Е), что говорит о легкости образования л-ком-
плексов, но скорость гидрирования падает, т. е. лимитирующей ста-
дией является распад л-комплекса.
Уменьшение скорости гидрирования при введении алкильных
заместителей объясняли41 стерическими препятствиями на стадии
адсорбции. Были вычислены кинетические константы в опытах гид-
рирования пар углеводородов и относительные коэффициенты адсорб-
ции углеводородов (см. табл. 11).
На основании экспериментальных данных было показано 41, что
не существует прямой зависимости между величиной предэкспонен-
циального множителя и строением полиметилбензолов, но можно
связать в одно уравнение предэкспоненциальный множитель, энер-
гию активации и число замещающих групп (п):
1g А = 0,68£ + 2,178 - о,33п ± 0,129
Таким образом, подводя итоги изложенному выше, можно сделать
вывод, что следует учитывать по крайней мере два аспекта
влияния алкильных заместителей: энергетический, проявляющийся
в изменении электронной плотности в кольце, вследствие чего облег-
чается или затрудняется образование л-комплекса с катализатором,
и стерический, проявляющийся в затруднении доступа водорода
к реагирующей молекуле.
142
Таблица 11. Кинетические и адсорбционные константы
гидрирования метнлбензолов 41-
Катализатор Rh на А1,Оа, 25 °C, 80 кгс/смг
Углеводород Энергия актива- ции (£), ккал/моль Логарифм предэкспоненци- ального множи- теля (1g А) Относительная константа ско- рости гидриро- вания Относительный коэффициент адсорбции '
Бензол 7,8 ±0,4 7,5 ±0,2 1,00* 1,00
Толуол 8,1 ± 0,5 7,3 ± 0,3 0,43 0,65
о-Ксилол 6,2 ± 0,6 5,7 ± 0,3 0,25 0,34
ж-Ксилол 8,7 ± 0,4 7,3 ± 0,3 0,16 0,23
«-Ксилол 9.2 ± 0,5 7,9 ±0,3 0,22 0,12
1,2,3-Триметилбензол . . . 5,7 ± 0,9 4,9 ± 0,6 0,08 0,18
1,2,4-Трпметилбензол 7,9 ± 0,8 6,4 ± 0,5 0,07 0,092
1,3,5-Триметилбевзол 8,4 ±0,6 7,1 ±0,4 0,15 0,023
1,2,3,4-Тетраметилбензол 6,5 ± 0,4 4,9 ±0,2 0,027 0,096
1,2,3,5-Тетраметилбензол 9,5 ±0,6 7,4 ±0,3 0,048 0,023
1,2,4,5-Тетраметилбензол 7,5 ± 2,4 6,0 ±1,6 0,057 0,015
Пентаметилбензол 9,3 ± 2,0 6,6 ± 1,3 0,011 0,036
Гексаметилбензол .... 4,7 ± 0,03 3,2 ±0,1 0,010 0,0072
* К21 для бензола, условно принятая за единицу, равна 58,4 мл/(мин-г).
Необходимо также учитывать роль адсорбции, так как адсорб-
ционная активность зависит от электронной плотности ароматиче-
ского кольца. При этом необходимо иметь в виду, что с введением
алкильных заместителей в молекуле появляется неполяризуемая
часть, что может сказаться на адсорбционных и десорбционных
свойствах.
По-видимому, было бы неправильно сводить влияние алкильных
заместителей к какому-либо одному эффекту. Так, например, отме-
ченные выше различия между высокотемпературными катализато-
рами нельзя объяснить лишь стерическими факторами, поскольку
они действуют только в условиях низких температур. Если же все
свести к энергетическим факторам, то следовало бы ожидать, что
заместители, имеющие положительный или отрицательный индуктив-
ные эффекты, оказывают противоположное влияние. Между тем
из кинетических данных 1 следует, что аналогичное алкильным заме-
стителям влияние оказывают не только гидроксильные и метоксиль-
ные, но и фенильные и карбоксильные группы (см. табл. 12).
Скорость гидрирования уменьшается по мере накопления этих
групп, причем сильнее в случае 1,2-изомеров, чем в случае 1,3-
и 1,1низомеров. Если бы скорость гидрирования снижалась при повы-
шении электронной плотности в кольце, то введение фенильных
и карбоксильных групп привело бы к увеличению скорости гидриро-
вания. Если предположить, что электроотрицательные заместители
затрудняют стадию распада активированного комплекса, то это также
143
Таблица 12. Константы скорости гидрирования
некоторых ароматических углеводородов
Платиновый катализатор, 30 °C
Углеводород Константы скорости гидрирования
х = н х=сна х = соон Х = С«Н„
Х-/~ \ 0,288 ' 0,188 0,112 0,054
Н3С\
— 0,092 0,031 —
/СН3-
Х~\_ — 0,142 0,081 —
Х-<^_ ^-СН3 — 0,186 0,094 —
н3сх“ /СН3
x-d — 0,041 0,019 —
н3сх~
х-с ^-СН3 — 0,084 0,029 —
/СН3
х-<2 — 0,166 0,064 —
\сн3
невероятно, так как из данных табл. 12 видно, что по мере перехода
от моно- к ди- и тризамещенным карбоксильные и метильные группы
оказывают совершенно аналогичное влияние на скорость гидриро-
вания. По-видимому, в рассматриваемом случае превалируют все же
стерические факторы.
Таким образом, вопрос влияния алкильных заместителей на
скорость гидрирований бензольного кольца достаточно сложен.
В этой связи значительный интерес представляет использование гомо-
генных катализаторов, гидрирование в присутствии которых сво-
бодно от адсорбционных осложнений.
Действительно, изучение кинетики гидрирования бензола и его
гомологов на ряде таких катализаторов показало, что механизм
гидрирования зависит от природы катализатора 42.
В табл. 13 представлены кинетические константы гидрирования
некоторых ароматических углеводородов. Из приведенных данных
видно, что экспериментально определенная кажущаяся энергия акти-
вации реакции гидрирования бензола уменьшается в ряду
Си > Ni > Fe > Сг > Мо
144
Таблица 13. Кинетические характеристики гидрирования
ароматических углеводородов в присутствии
комплексных металлоорганических катализаторов 42
Углеводород IgA Е, ккал/моль Углеводород IgA Е, ккал/моль
(С2Н5)3А1 + Сг(С5Н,О2)3(А1:( > = 8: 1) (С2Н3)зА1+Ni(C5H7O2) 2(Al:Ni=8:1)
Бензол 1,927 7,8 ±0,4 Бензол . . 3,351 9,3 ± 0,5
Толуол 1,985 8,2 ±0,4 Толуол 2,814 8,9 ±0,8
о-Ксилол 1,806 8,2 ± 0,4 о-Ксилол 1,963 7,7 ±0,6
Мезитилен 2,060 9,1 ± 0,5
V (C2H5)3A1+Cu(C5H7O2)2 (А1: Си = 6 :1)
(С2Н5)3А1 + Fe(C5H,O2)3 (Al: те = 8: 1) Бензол 3,788 11,0 ±0,5
Мезитилен 0,898 6,0 ±0,4
Бензол 2,792 9,0 ± 0'8
Толуол 2,234 84±о;5 (С2Н5)3А1+МоО2(С5Н7О2)2(А1 : Мо=6 :1)
о-Ксилол 1,602 7,9 ±0,4 Бензол . 1,080 6,2 ±0,4
Мезитилен 0,654 6,7 ±0,4 Мезитилен 2,753 11,0 ±0,9
При гидрировании на катализаторах (С2Н5)3А1 -|- Ni(C5H7O2)2
и (С2Н5)3А1 -|- Fe(C5H7O2)3 энергия активации падает с увеличением
числа заместителей в бензольном кольце. Однако на катализаторах
(С2Н5)3А1 + Сг(С5Н7О2)3и (С2Н5)3А1 + МоО2(С5Н7О2)2 энергия акти-
вации растет по мере перехода от бензола к 1,3,5-триметилбензолу.
В табл. 14 приведены скорости гидрирования ароматических
углеводородов по сравнению с бензолом на различных катализато-
рах. Как видно из этих данных, скорость гидрирования уменьшается
с увеличением числа заместителей в ароматическом кольце, однако
соотношения между скоростями на различных катализаторах имеют
резко различный характер.
Предполагают, что при гидрировании ароматических углеводоро-
дов на комплексных металлоорганических катализаторах происходит
промежуточное образование л-комплекса между молекулой аромати-
ческого углеводорода и атомом переходного металла и последователь-
ное присоединение атомов водорода а2.
Возможно, сам факт гидрирования ароматических углеводородов
на таких металлах, как Си, Сг, V, Ti, косвенно подтверждает эту
гипотезу.
В этом случае процесс гидрирования может быть изображен
следующей схемой:
10 Заказ 271
145
н н
+ 2Н2
*4
быстро
О + •••ме—н
где . . .Me—Н — комплексный гидрид металла, образующийся при
гидрогенолизе алкилированного триэтилалюминием переходного ме-
талла.
Таблица 14. Скорости гидрирования ароматических углеводородов
иа комплексных металлоорганических катализаторах при 150 °C
(в моль/мин на 1 моль переходного металла)
Углеводород W Относите; рость гид экспери- менталь- ная ьная ско- рирования расчет- ная
(С2Н5)3А1 + Сг(С3Н7О2)3 (Al: Cr = 8 : 1)
(гидрирование в 1 стадию)
Бензол 0,400 100 100
Толуол 0,297 74 74
о-Ксилол 0,204 51 51
Мезитилен 0,121 30 30
(С2Н5)зА1 + Ре(С5Н7О2)3 (Al: Fe = 8 : 1)
(гидрирование в 2 стадии)
Бензол 0,7^7 100 100
Толуол 0,435 57 59
о- Ксилол 0,169 22 25
Мезитилен 0,082 11 11
(С2Н5)3А1 + Ni(C5H7O2)2 (Al: Ni = 8 : 1)
(гидрирование в 3 стадии)
Бензол 2,078 100 100
Толуол 0,972 47 45
о-Ксилол 0,260 13 13
Мезитилен 0,071 4 4
Если допустить, что концентрация промежуточного комплекса А
мала по сравнению с концентрацией других промежуточных частиц,
то скорость процесса можно выразить уравнением:
АР1А:2ЫС6Н6][Н2][С]
W~(K^ [С6Н6] + 1)(А3[Н2]4-А_2) 5
146
При fc_2 к3 и KVI (С6НВ1 1 уравнение принимает вид:
»=Кр,МН,]|С1
где Хр, — константа равновесия первой стадии; KPi — константа
равновесия второй стадии; [С] — концентрация катализатора.
Данные, полученные при гидрировании бензола на катализаторе
(С2Н6)3А1 + Ni(C5H,O2)2 при 150° С, удовлетворяли этому уравне-
нию.
Из уравнения 5 следует, что порядок по водороду и бензолу может
меняться от нулевого до первого в зависимости от условий опыта.
Так, например, с понижением температуры экспериментально наблю-
далось понижение порядка по водороду.
Рис. 8. Корреляция
между энергией актива-
ции гидрирования и по-
тенциалом ионизации
ароматических углево-
дородов 42;
1 — бензол; 2 — толуол; з—
о-ксилол; 4 — мезитилен;
I — (C.eH6), А1Ni(C.6H7O2)2;
II —(С,Н,),А1+Сг(С,Н,О,)1.
Z7, к кал/моль
Если теперь вернуться к рассмотрению механизма влияния ал-
кильных групп в бензоле, то следует в первую очередь отметить, что
их накопление приводит к уменьшению потенциала ионизации и уве-
личению электронодонорности кольца, а это облегчает образование
л-комплексов. Следовательно, стабильность л-комплексов возрастает
от бензола к мезитилену. Между тем считают 39> 4°, что гидрирование
протекает тем легче, чем устойчивее комплекс катализатора с гидри-
руемым веществом. Данные, полученные при гидрировании на ката-
литических системах триэтилалюминий — ацетилацетонаты железа
и никеля, подтверждают это предположение. Однако в случае ката-
литических систем триэтилалюминий — ацетилацетонаты хрома и мо-
либдена увеличение числа алкильных групп л бензольном кольце
приводит к увеличению кажущейся энергии активации, хотя устой-
чивость л-комплексов при этом должна расти в том же ряду (рис. 8).
Иными словами, данные рис. 8 можно интерпретировать в свете
мультиплетной теории так, что прямые 1 и 2 (для никелевого и хромо-
вого катализаторов) представляют собой разные «склоны» двух
различных «вулканообразных» кривых. Экстремальный характер
зависимости кажущейся энергии активации от потенциала ионизации
10*
147
можно объяснить тем, что увеличение прочности л-комплексов поло-
жительно влияет только до определенного предела, после которого
энергия активации определяется стадией, включающей разрушение
этих комплексов.
Однако интерпретированная таким образом разница в энергетике
образования и разрушения активного комплекса в случае хромового
и молибденового катализаторов, с одной стороны, и никелевого и желез-
ного, с другой — еще не объясняет ни качественно одинакового явления
уменьшения скорости гидрирования в ряду бензол — толуол — кси-
лолы — триметилбензол, ни того, что это уменьшение более резко
проявляется в случае никелевого катализатора и наименее резко —
в случае хромового (см. табл. 14). Интересно отметить, что на хромо-
вом катализаторе 1,3,5-триметилбензол гидрируется медленнее бен-
зола в 3,3 раза, на железном в 9,3 раза, а на никелевом в 28 раз.
Поскольку при гидрировании присоединяются три молекулы водо-
рода, то лимитировать процесс может либо присоединение второго
атома водорода 22, либо группа медленных стадий присоединения
водорода 43.
Как видно из табл. 14, экспериментальные данные удовлетвори-
тельно совпадают с расчетными при предположении разного числа
лимитирующих стадий на различных катализаторах.
Таким образом, эксперименты с гомогенными катализаторами
показывают, что энергетические факторы играют большую роль,
но из-за различия механизмов могут проявляться по-разному. Одним
из показателей этого является прямо противоположное влияние уве-
личения числа алкильных групп на величину энергии активации
(см. рис. 8).
ГИДРИРОВАНИЕ ПОЛИЦИКЛИЧЕСКИХ
АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
Конденсация двух или нескольких бензольных колец нарушает
их симметрию и равенство прочности и электронной плотности связей
колец44. Так, например, уже в простейшем конденсированном угле-
водороде — нафталине
8 9 1
5 10 4
связи между углеродными атомами 1 и 2, 3 и 4, 5 и 6, 7 и 8 более
реакционноспособны и приближаются по своим свойствам к этилено-
вой связи. Так, нафталин присоединяет типичные реактивы на двой-
ную связь — озон и четырехокись осмия 44, 45.
Современные методы квантово-химических расчетов, хотя и вклю-
чают в себя известные упрощения, позволяют рассчитывать длины
и порядки связей. Ниже приведены результаты некоторых из таких
148
расчетов для полициклических углеводородов 44’ 48, а также для
бензола и бутадиена-1,3:
1,40а; 1,667
1,36 А*,
1,395 А;
1»6уЭ
СН2=СН—СН=СН2
1,ЗЗА 1.47А 1,ЗЗА
1,89 1,45 1,89
1,623
Зависимость между длиной связи и ее кратностью показана на
рис. 9.
Как видно из приведенных выше молекулярных диаграмм, значе-
ния длин и порядка укороченных связей некоторых конденсирован-
ных ароматических углеводородов занимают промежуточное положе-
ние между бензолом и этиленом. Связи в сопряженных диенах уко-
рочены по сравнению с изолированной двойной связью.
Рис. 9. Зависимость
между кратностью и дли-
ной связи.
Легче всего гидрируются самые укороченные тройные связи,
затем одна из двух сопряженных двойных связей4,4в, затем изо-
лированные двойные и, наконец, ароматические связи 3’ 8’ 37.
Конденсированные ароматические углеводороды, следовательно,
должны гидрироваться быстрее моноциклических ароматических
углеводородов, но медленнее олефинов.
При изучении кинетики гидрирования полициклических углеводо-
родов нужно учитывать их пространственное строение, изменения
149
энергии сопряжения и строения в ходе гидрирования и другие фак-
торы. Вследствие этого пока трудно говорить о четкой корреляции
между скоростью гидрирования полициклических ароматических
углеводородов и их строением.
В случае нафталина оба кольца одинаковы и первоочередность
гидрирования одного иэ них наблюдается только при наличии заме-
стителей (быстрее насыщается незамещенное кольцо 11 31). Если заме-
стителями являются функциональные группы (гидроксильная, окси-
метильная), более сильно влияющие на адсорбируемость, то при гид-
рировании на никелевом катализаторе направление гидрирования
определяется характером среды: в щелочной среде гидрируется заме-
щенное кольцо с образованием 1,2,3,4-тетрагидропроизводных, в кис-
лой или нейтральной — незамещенное кольцо с образованием
5,6,7,8-тетрагидропроизводных Нафтойные кислоты гидрируются
с образованием в первую очередь 5,6,7,8-тетрагидропроизводных.
Конденсированные ароматические углеводороды с тремя и более
кольцами гидрируются последовательно так, что для осуществления
каждой следующей стадии нужны все более и более жесткие условия.
Так, например, при гидрировании антрацена на окисномедном ката-
лизаторе эта последовательность может быть представлена схемой
I II I I
115 кгс/см2;
120- 150 °C;
Зч
146 кгс/см2;
200-220 °C;
4,5 ч
152 кгс/см2;
255 °C;
7,5 ч
182 кгс/см2;
24 ч
Аналогичная закономерность отмечалась в случае фенантрена,
пирена, хризена, бензпирена. Труднее всего гидрируется последнее
кольцо, а в перилене и декациклене оно совсем не гидрируется \
например:
Последовательность гидрирования колец (показана цифрами)
в структурах флуорантена, рубицена и декациклена иллюстрируется
следующей схемой (Н — прогидрировать не удалось):
150
Систематическое изучение кинетики гидрирования полицикличе-
ских систем проводилось как на низкотемпературных, так и на высо-
котемпературных катализаторах (табл. 15).
Таблица 15. Относительные скорости гидрирования
некоторых ароматических углеводородов
на низко- и высокотемпературных катализаторах 37
Реакция Относительная скорость гидрирования
Ni на А12Оа 120-200 °C; 30—50 кгс/см2 MoS, 420 °C; 200 кгс/см2 WS, 400 °C; 150 кгс/см* *
Бензол -> Циклогексан 100 100 100
Нафталин -+ Тетралин 314 1409 2300
Тетралин -> Декалин 24 287 250
Антрацен->9,10-Дигидроантрацен . . 9,10-Дигидроантрацен -> Тетрагидро- 326 — 6210
антрацен Тетрагидроантрацен -> Октагидроантра- 308 — 1380
цен Октагидроантрацен -> Пергпдроантра- 147 — 460
цен 4,4 — 299
Хризен -> Тетрагидрохризен — — 80
Тетрагидрохризен -> Октагидрохризен — — 75
Октагидрохризеп -> Додекагидрохрпзен — — 95
Нетрудно заметить, что влияние насыщенных колец на скорость
гидрирования аналогично влиянию метильных групп, причем зто
Таблица 16. Относительные скорости гидрирования
некоторых ароматических углеводородов
Углеводород
Бензол.....................................
1,2-Диметнлбензол .........................
Тетралин ..................................
1,2,4,5-Тетраметилбензол...................
1,2,3,4,5,6,7,8-Октагидроантрацен ......
Относительная ско-
рость гидрирования
Ni WS,
100 100
24 (330) *
24 250
3,8 (530) **
4,4 299
* Для м-ксилола (см. табл. 8).
** Среднее между значениями для 1,3,5-триметил- и пентаметилбензолов.
влияние проявляется различным образом в случае низко- или высоко-
температурных катализаторов 37 (табл. 16).
151
Однако при гидрировании нафталина, тетрагидроантрацена и окта-
гидрохризена на высокотемпературном катализаторе (WS2) скорость
гидрирования убывает по мере усложнения молекулы и не зависит
от увеличения числа насыщенных колец (см. табл. 15).
Сравнительные кинетические данные по гидрированию полицикли-
ческих углеводородов были получены и при использовании дру-
гих катализаторов: MoS247, никеля Ренея 48, железного жидкофаз-
ного катализатора 48 и платины Адамса 49, 5°. Эти данные приведены
в табл. 17.
Таблица 17. Относительные скорости гидрирования полициклических
ароматических углеводородов на различных катализаторах
Бензол ......................
Дифенил .....................
Нафталин ....................
Антрацен.....................
Фенантрен....................
Тетрацен ....................
1,2-Бензантрацен.............
Хризен ......................
Пирен........................
Коронен .....................
670
164
100
124
24
135
77
29
38
14
23
100
338
59
19
88
12
32
. 33
100
104
152
— 7
100 100
300 -
50 —
4 -
- 48
100 100
270 923
89
Не гид-
рируется
Полученные данные очень противоречивы и не дают возможности
сделать общие выводы о зависимости скорости гидрирования поли-
циклических ароматических углеводородов от длины и кратности их
связей.
Подтверждением бблыцей реакционной способности конден-
сированных углеводородов служит тот факт, что на всех катализато-
рах кроме платинового скорости гидрирования нафталина и антра-
цена были выше скоростей гидрирования бензола и дифенила.
Кинетические характеристики реакций гидрирования некоторых
ароматических углеводородов на платиновом катализаторе приве-
дены в табл. 18.
Следует отметить, что при гидрировании полициклических угле-
водородов на платиновом катализаторе сохраняются многие законо-
мерности, отмеченные выше. В самом деле, по мере насыщения моле-
кулы водородом скорости гидрирования падают, а энергии актива-
ции возрастают. Образование полиметиленовых колец сильно тормо-
зит гидрирование, что можно объяснить пространственными затруд-
152
Таблица 18- Константы скоростей гидрирования, энергии активации
и порядки связей некоторых углеводородов во
40° С, 2,5 кгс/см2
Реакция Средняя константа ско- рости KJa-iO*, мин-1 Е, ккал/моль Наибольший порядок связи в исходном углеводороде Число связей в исходном углеводороде
с наибольшей крат* ностью в т. я. способных гидрироваться на Pt
Бензол Циклогексан 315,0 7,4 1,667 6 3
Дифенил фенилциклогексан 76,8 6,6 1,667 12 4
Нафталин Тетралин 47,0 7,0 1,725 4 2
Тетралин -> Декалин ...... 5 25,9 11,4 — — —
2,6-Диметилнафталин 2,6-Дпметилтетралин 35,1 6,7 — — —
2,6-Диметилтетралин 2,6-Диметил дек алии 21,9 9,6 — — —
2,7-Диметилнафталин 2,7-Диметилтетралин 37,0 7,1 — — —
2,7-Диметилтетралин 2,7-Диметилдекалин 22,1 9,8 — — —
Антрацен Дигидроантрацен 58,5 8,9 1,738 4 2
Дпгидроантрацен -+• Тетрагидроантрацен . . 13,2 10,2 — —• —
Тетрагидроантрацен -+• Октагидроантрацен * 3,1 13,5 — — —
Тетрацен -+• Гидропроизводное 63,2 — 1,746 4 2
Фенантрен -> Дигидрофенантрен 11,1 7,1 1,775 1 0,5
Дпгидрофенантрен -► Тетрагидрофенантрен 4,8 9,9 — — —
1,2-Бензантрацен -> Дигидро-1,2-бензантра- цен 36,4 5,3 1,783 1 0,5
Дигидро-1,2-бензантрацен -> Тетрагидро-1,2- бензантрацен 18,9 11,2 — <— —
Хризен -► Дигидрохризен 14,5 <— 1,750 2 1
Пирен -> Дигидропирен 17,5 6,9 1,682 6 1
Дигпдропирен -> Гексагидропирен * 8,9 8,3 — — —
Коронен Гидропроизводное 6,6 — 1,385 ♦» 6 1
* Константа скорости гидрирования этого соединения очень мала.
** Длина укороченной связи, А.
нениями. Так, например, константа скорости гидрирования быстро
падйет в рядах:
153
Аналогично, но менее резко проявляется влияние метильных
групп: нафталин гидрируется быстрее, чем 2,6- и 2,7-диметилнафта-
лины:
47,0
37,0
35,1]
Если рассматривать только начальные стадии гидрирования, то
изученные углеводороды по скорости гидрирования на платиновом
катализаторе образуют ряд:
Бензол > Дифенил > Тетрацен > Антрацен >> Нафталин >
> 1,2-Бензантрацен > Пирен > Хризен > Фенантрен > Коронен
Из приведенных данных видно, что при аналогии структур ско-
рость гидрирования снижается с уменьшением порядка наиболее
короткой связи, например в рядах (цифры под формулами — порядки
наиболее коротких связей):
Z\Z\Z\Z\
I II I I I i
1,746
Z\Z\Z\
I II I I > I ' ll ।
\Z\Z\Z \Z\Z
1,738 1,725
I II I II
/Л Z\Z\Z Z\Z\Z
I II I > I II I
\Z\Z\Z '4Z\Z
1,783 1,775
Однако это справедливо не для всех углеводородов. Особенно
непонятна высокая скорость гидрирования бензола и дифенила, не
имеющих укороченных связей, а также большая скорость гидрирова-
ния антрацена по сравнению с фенантреном, а тетрацена — по срав-
нению с 1,2-бензантраценом, хотя у ангулярных углеводородов есть
связи с большей кратностью.
Можно попытаться объяснять эти аномалии, если рассмотреть
геометрическое расположение углеводородов на кристаллической
решетке платины в соответствии с принципами мультиплетной теории
А. А. Баландина 8. При этом нужно учитывать взаимное положение
активных центров, занятых углеводородами, и свободных, на кото-
рых может активироваться водород. У конденсированных углеводо-
родов в первую очередь следует учитывать наличие свободных для
водорода центров против укороченных связей с повышенной электрон-
ной плотностью. В этом случае локализация л-электронов потребует
наименьшей затраты энергии.
Возможные случаи адсорбции углеводородов на платине изобра-
жены на рис. 10. Размеры кристаллической решетки и длины связей
углеводородов даны в сравнимых масштабах.
154
Если сравнить адсорбцию бензола и дифенила, легко заметить,
что в то время как против всех трех активируемых связей бензола
имеются свободные центры катализатора, на которых может активи-
роваться водород, против связей дифенила таких центров только
четыре. Этим, вероятно, частично объясняется разница в скоростях
гидрирования этих углеводородов на платине.
Рис. 10. Модели плоскостной адсорбции углеводородов на решетке платины.
При адсорбции линеарных углеводородов (аценов) (рис. 10 а, б, в)
на площадке, занимаемой углеводородом, находятся два активных
центра катализатора, против которых расположены активируемые
связи с повышенной электронной плотностью. Таким образом, эти
углеводороды находятся в сравнимых условиях, и скорость гидриро-
вания определяется кратностью связи.
При адсорбции фенантрена (рис. 10 г) и 1,2-бензантрацена
(рис. 10 д) возможны два варианта, причем только в одном из них
против наиболее активной связи в среднем кольце находится актив-
155
ный центр катализатора. Именно поэтому эти углеводороды, имея
связи еще более непредельные, чем у тетрацена, антрацена и нафта-
лина, гидрируются медленнее последних.
Пирен и хризен занимают промежуточное положение (рис. 10 е, ж).
Более симметричные молекулы пирена и особенно коронена
(рис. 10 з) характеризуются меньшими отклонениями в длинах свя-
зей и в их кратности. Это объясняет относительно низкие скорости
гидрирования как пирена, так и коронена.
В соответствии с рассмотренными особенностями геометрии ад-
сорбции на платине в табл. 18 показано число связей, способных
гидрироваться. С учетом этих связей становится понятно, почему
ангулярные и симметричные (пери-конденсированные) углеводороды
гидрируются медленнее линеарных.
Однако геометрия адсорбции не является, по-видимому, един-
ственным фактором, влияющим наряду с наличием укороченных свя-
зей на скорость гидрирования, в частности на платиновом катализа-
торе. Различия в геометрии, очевидно, недостаточны для объяснения
значительной разницы в скоростях гидрирования бензола, дифенила
и нафталина. Необходимо учитывать влияние продуктов гидрирова-
ния и соотношения между гидрируемым веществом и водородом на
поверхности катализатора. В случае гидрирования на платиновом
катализаторе в проточной установке 50 бйли вычислены коэффициенты
торможения (Р) по уравнению Фроста 51
vo ln l_-y==^voy + a
где v0 — скорость подачи сырья, у — степень превращения, а —
константа скорости.
Рассчитанные по этому уравнению коэффициенты торможения
гидрирования нафталина и дифенила соответственно равны 0,91
и 0,71, т. е. декалин тормозит гидрирование нафталина сильнее, чем
дициклогексил гидрирование дифенила. Кроме того, опытами гидри-
рования нафталина и дифенила с добавлением конечных продуктов
гидрирования 50 показано, что добавление 2% декалина при гидри-
ровании нафталина снижает скорость реакции более чем в 1,5 раза.
Добавление такого же количества дициклогексила снижает скорость
гидрирования дифенила лишь незначительно. Нафталин не гидри-
руется в чистом декалине, в то время как дифенил в дициклогексиле
гидрируется (/С40 0,0139). »
Отмеченные закономерности наблюдаются при давлении водо-
рода порядка 2—3 кгс/см2, когда на поверхности катализатора нехва-
тает водорода. В этом случае реакция имеет первый порядок по водо-
роду и нулевой — по веществу. При высоком давлении водорода
(50—300 кгс/см2) наблюдается обычно первый порядок по веществу
и нулевой — по водороду. Большую скорость гидрирования поли-
циклических углеводородов (например, нафталина и антрацена)
по сравнению с бензолом при высоком давлении водорода37, 48
можно объяснить тем, что с ростом давления доля поверхности ката-
156
лизатора, занятая водородом, увеличивается, и водород становится
доступным для всех укороченных связей.
Это подтверждается, в частности, результатами, полученными при'
гидрировании нафталина и дифенила на никелевом катализаторе 60
(рис. 11).
Как видно из рис. 11, на никелевом катализаторе при повышенных
давлениях нафталин гидрируется много быстрее дифенила, но в об-
ласти давлений порядка 2-^-3 кгс/см2, характерных для платинового
катализатора, имеет место обратная зависимость.
Рис. 11. Зависимость
скоростей гидрирования
нафталина (7) и дифе-
нила (2) от давления
водорода.
Константа скорости гидриро6ания(#-1О3)
Таким образом, из всего рассмотренного материала Можно сделать
несколько достаточно определенных выводов: на скорость гидрирова-
ния полициклических углеводородов оказывает влияние наличие
в них связей с повышенной электронной плотностью, вследствие чего
ди- и трициклические углеводороды, а также большая часть более
конденсированных углеводородов гидрируются, как правило, быстрее
моноциклических; линеарные конденсированные углеводороды гидри-
руются быстрее ангулярных и симметричных: гидрирование, как
драбило, протекает ступенчато, причем скорость каждой п ос лед у-
Кнцей ступени меньше предыдущей; заместители и гидрированные
кольца тормозят гидрирование.
Этц закономерности, как показано выше, могут нарушаться, на-
пример, из-за торможения продуктами реакции, недостатка водо-
рода, особенностей адсорбции вещества. Поэтому особенно интересно
применение для гидрирования полициклических ароматических угле-
водородов гомогенных комплексных катализаторов, при использова-
нии которых не имеют место осложняющие явления, связанные с ад-
сорбцией и десорбцией на катализаторе. Эти катализаторы появились
недавно, а применение их для гидрирования полициклических угле-
водородов описано пока только в одной работе 28. Катализатор был
приготовлен на основе родия и N-фенилантраниловой кислоты. На
примере антрацена опытами с дейтерированием и определением места
дейтерия в прореагировавшей молекуле было показано, что в данном
случае не происходит промежуточного образования 9,10-дигидро-
157
антрацена. Механизм превращения антрацена в присутствии фенил-
антранилата родия может быть представлен следующей схемой:
Лимитирующей здесь является первая стадия, так как олефины
в присутствии такого катализатора гидрируются во много раз быс-
трее. Кроме того, реакция имеет первый, а не второй порядок по водо-
роду. Из схемы видно, что какие-либо стерические препятствия при
образовании л-комплексов любыми полициклическими углеводоро-
дами маловероятны.
Примененные для гидрирования на этом катализаторе углеводо-
роды по относительным скоростям гидрирования образовали ряд
(за 100 принята скорость гидрирования нафталина):
158
Важно отметить, что в соответствии с сформулированными выше
выводами в этом ряду все полициклические углеводороды гидрирова-
лись быстрее бензола, трициклические — быстрее нафталина, а пирен
и хризен — медленнее нафталина.
ГИДРИРОВАНИЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИИ
АРОМАТИЧЕСКОГО ХАРАКТЕРА
Пяти- и шестичленные гетероциклы, содержащие кйслород, азот
или серу, энергетически менее стабильны, чем бензол, и легче гидри-
руются. Так, например, фуран и его производные, содержащие
метильную, карбоксильную, оксиметильную, карбоксиметильную
группы и др., гидрируются на платиновом катализаторе Адамса
в среднем в 2,7 раза быстрее бензола и его аналогичных производ-
ных х. Введение заместителей, как и в случае бензола, уменьшает
скорость гидрирования фуранового кольца. Аналогично, скорость
гидрирования метил- и полиметилпиридинов меньше скорости гидри-
рования пиридина х. Однако введение метильных заместителей в пир-
рольное кольцо ускоряет гидрирование х. Возможно, что в этом слу-
чае, как полагает автор х, могли быть получены неверные результаты
из-за чрезвычайно легкой окисляемости пирролов на'воздухе. При
гидрировании соединений, содержащих два кольца — бензольное
и гетероциклическое, — бензфурана, индола, бензтиофена (тионаф-
тена) и хинолина на высокотемпературных сульфидных катализато-
рах, как правило, в первую очередь гидрируется гетероциклическое
кольцо 52> 53. Однако в этих условиях процесс гидрирования ослож-
няется процессами разложения, в ходе которых гидрированное гете-
роциклическое кольцо подвергается деструкции с элиминированием
гетероатома в виде воды, аммиака или сероводорода (механизм таких
превращений рассмотрен в гл. 4). Поэтому вполне возможно, что
равновесие реакции гидрирования могло быть сдвинуто. Вследствие
этого для сопоставления скоростей гидрирования гетеро- и карбо-
циклических соединений ароматического характера лучше использо-
вать низкотемпературные катализаторы. Так как сернистые соедине-
ния необратимо отравляют последние, опыты проводились с бенз-
фураном, хинолинами и индолами.
Гидрирование метилхинолинов54 осуществляли в присутствии
никелевого катализатора при 200 °C и 20 кгс/см2, метилиндолов —
при 220—250 °C 55, бензфурана и изохинолина 1 — на платиновом
катализаторе при 20—50 °C (см. табл. 19).
Сопоставляя данные табл. 19, можно достаточно уверенно рас-
пространить на гетероциклические соединения основную закономер-
ность — уменьшение скорости гидрирования бензольного кольца
при наличии метильных заместителей. При отсутствии заместителей
или равном их количестве в бензольном и гетероциклическом коль-
цах гидрируется, только или в основном, гетероциклическое кольцо.
При наличии заместителей только в гетероциклическом кольце начи-
нает гидрироваться бензольное кольцо, причем этот процесс легче
159
Таблица 19. Гидрирование гетероциклических соединений 1-5*, 55
Соединение Степень восста- новления, % Соединение Степень восста- новления, %
бензоль- ное кольцо гетеро- цикличе- ское . кольцо бензоль- ное кольцо гетеро- цикли- ческое кольцо
Бензфуран ...... 0 100 8-Метилхинолпн . . 0 100
Индол 0 100 2-Пропилхинолин . . 35 65
2-Метилиндол .... 50 50 2,3-Димети лхинолин 44 56
З-Метилпндол .... 90 10 . 2,4-Диметил хинолин 80 20
1,2-Диметилиндол . . 100 0 2,6- Диметил хинолин 1,5 98,5
Изохпнолин 0 100 2-Метил-4-фенилхи-
Хинолин . 0 100 ноллн 84 16
2-Метилхинолпн . . 4 96 2,4,5,8-Тетраметилхи-
4-Метилхинолин . . . 33 66 нолин 4 96
6-Метилхннолин . . 0 100 2,4-Диметил-7,8-тет-
7-Метилхинолпн . . 0 100 раметпленхинолин 0 100
проходит в индолах. Действительно, для селективного гидрирования
только бензольного кольца в индолах достаточно двух заместителей
в пиррольном кольце, а в хинолинах — необходимо введение трех
заместителей.
Тормозящее влияние метильных заместителей наблюдается и при
гидрировании метилхинолинов; только 8-метилхинолин гидриро-
вался быстрее хинолина т.
Скорости гидрирования некоторых азотсодержащих гетероцикли-
ческих соединений и соответствующих карбоциклических соединений
над платиновым катализатором Адамса (40 °C, 2,5 кгс/см2) 58 приве-
дены в табл. 20.
Таблица 20. Кинетические константы гидрирования азотсодержащих
гетероциклических соединений и их карбоциклических аналогов
Соединение Константа скорости Mg- 10s, МИН"1 Энергия активации, ккал/моль 1 Соединение Константа скорости Мо-Ю', МИН”1 Энергия активации, ккал / моль
Бензол 315,0 7,4 2-Метилхинолпн 79,5 ' 5.3
Пиридин 191,2 4,9 1 Дифенил 76,8 6,7
Нафталин .... 47,0 7,0 2,2'-Дипиридил 14,6 5,4
Хинолин 103,4 6,2
Данные табл. 20 удовлетворительно совпадают с данными, приве-
денными выше: хинолин гидрируется быстрее нафталина, введение
метильного заместителя уменьшает скорость гидрирования. Однако
пиридин гидрируется медленнее бензола, а дипиридил — дифенила.
Было установлено, что эти отклонения объясняются специфическими
160
условиями гидрирования на платиновом катализаторе Адамса в рас-
творе уксусной кислоты. При этом, если в процессе гидрирования
образуются сильные основания, то они нейтрализуют кислоту и тор-
мозят гидрирование. Действительно, инфракрасные спектры гидро-
генизатов показывали изменения интенсивности и положения полосы
поглощения карбоксильной группы. Кроме того, изменение скоростей
гидрирования дифенила и дипиридила в нейтральном растворителе —
декалине дало обратный порядок констант: к^0 • 103 для дипиридила
составила 6,2, а для дифенила — 3,3 мин-1.
Таким образом, общей закономерностью является более легкое
гидрирование гетероциклических колец.
ЛИТЕРАТУРА
1. Смит X. А. В кн. «Катализ в нефтеперерабатывающей и нефтехимиче-
ской промышленности». М., Гостоптехиздат, 1959. См. с. 184.
2. В о n d G. С. Catalysis by Metals. New York, Academie Press, 1962. 519 p.
3. Сокольский Д. В., Сокольская A. M. Металлы — катали-
заторы гидрогенизации. Алма-Ата, «Наука», 1970. 436 с.
4. Сокольский Д. В. Оптимальные катализаторы гидрирования в рас-
творах. Алма-Ата, «Наука», 1970. 112 с.
5. Сабатье П. Катализ в органической химии. Л., Госхимтехиздат, 1932.
418 с.
6. Ипатьев В. Н., ЖРФХ.О, 38, 75 (1906).
7. Raney М., Ind. Eng. Chem., 32, 1199 (1940).
8. Баландин А. А. Современное состояние мультиплетной теории гетеро-
генного катализа. М., «Наука». 1968. 202 с.
9. JanzG. J., J. Chem. Phys., 22, 751 (1954).
10. Н а г t о g F., Zweitering P., J. Catalysis, 2, 79 (1963).
11. ДербенцевЮ. И., Баландин А. А., Исагулянц Г. В.,
Кинетика и катализ, 2, 741 (1961).
12. И с а г у л я н ц Г. В., Б а л а н д и н А. А. и др., Кинетика и катализ,
5, 171 (1964).
13. S m i t h R. L., P г a t e r C. D., Chem. Eng. Progr., 63, 105 (1967).
14. S c h u i t G. C., R e у e n L. L., S a c h t 1 e r W. M., Actes II Congress
International Catalyse. V. 1. Paris, Technip, 1961. P. 893.
15. С к e й т Г., В а н - P e й н Л. В сб. «Катализ. Исследование поверх-
ности катализаторов». М., Издатинлит, 1960. См. с. 153.
16. РешетниковС. М., Сокольская А. М., Кинетика п катализ,
8, 224 (1967).
17. Т а у 1 о г W. F., J. Catalysis, 9, 99 (1967).
18. J а о Н i - С h, Е m m е t t Р. Н., J. Am. Chem. Soc., 81, 4125 (1959);
83, 796 (1961); 84, 1086 (1962).
19. Germain J. E., M a u r e 1 R. et al., J. Chim. Phys., 60, 1219 (1963).
20. HartogF., TabbenJ. H., W e t e r i n g s С. A. M. Ill Interna-
tional Congress on Catalysis. Proceedings. V. 2. Amsterdam, North Holland
Publ. Corp., 1965. See p. 1210.
21. T а у 1 о r W. F., S t a f f i n К. H., J. Phys. Chem., 71, 3314 (1967).
22. Roon e у J. ,T., Webb J., J. Catalysis, 3, 488 (1964).
23. К а л e ч и ц И. В., Шмидт Ф. К., Кинетика и катализ, 7, 614 (19661.
24. Л и п о в и ч В. Г., Ш м и д т Ф. К., Калечиц И. В., Кинетика/и
катализ, 8, 939 (1967).
25. Ефимов О. Н., Еременко О. Н. и др., Изв. АН СССР, Сер. хим.,
1969, 855.
26. В о л ь п и н М. Е., К о л о м н и к о в И. С., Усп. хим., 38, 561 (1969).
27. Л о з о в о й А. В., Д ь я к о в а М. К., ЖОХ, 7, 2964 (1937); 9, 26, 895
(1939); 10, 1 (1940).
И Заказ 271
161
28. Л о з о в о й А. В., Д ь я к о в а М. К. Гидрогенизация топлива в СССР.
М. —Л., Изд. АН СССР, 1940. См. с. 49.
29. Smith Н. A., Pennekamp Е. F. Н., J. Am. Chem. Soc., 67,
276 (1945).
30. Smith Н. A., Pennekamp Е. F. Н., J. Am. Chem. Soc., 67,
279 (1945).
31. В a i 1 е у A. S., S т i t h J. C., S t a v e 1 e у С. M., J. Chem. Soc.,
1956, 2731.
32. Л о а о в о й А. В., С е н я в и н С. А., ЖОХ, Сб. 1, 254 (1953).
33. Л о а о в о й А. В., СенявинС. А. ЖОХ, Сб. 2, 1035 (1953).
34. Калечиц И. В., П а в л о в а К. А. В сб. «Труды ВСФ СО АН СССР».
Вып. 38. М., Изд. АН СССР, 1961. См. с. 15.
35. Павлова К. А., Калечиц И. В. Там же. См. с. 19.
36. Калибердо Л. М., Калечиц И. В. Там же. См. с. 25.
37. Лозовой А. В., СенявинС. А. В сб. «Химическая переработка
топлива». М., Изд. АН СССР, 1957. См. с. 180.
38. Баландин А. А., Хидекель М. Л., Патрикеев В. В. ДАН
СССР, 123, 83 (1958).
39. V о 11 е г J., J. С a t а 1 у s i s, 3, № 3, 297 (1964).
40. V б 11 е г J., L a n g е В., К u h n W., Z. anorg. Chem., 340, 253 (1965).
41. S m i t h Н. A., C a m p b e 11 W. E. Ill International Congress on
Catalysis, Proceedings. V. 2. Amsterdam. North Holland Publ. Corp., 1965.
See p. 1373; RaderC. P., S m i t h H. A., J. Am. Chem. Soc., 84,
1443 (1962).
42. Л и n о в и ч В. Г., Ш м и д т Ф. К., К а л е ч и ц И. В., Кинетика и
катализ, 8, 1300 (1967).
43. Снаго вский Ю. С., Любарский Г. Д., Островский Т. М.,
Кинетика и катализ, 7, 258 (1966).
44. Реутов О. А. Теоретические основы органической химии. М., Изд.
МГУ, 1964. См. с. 454—457.
45. Эфрос Л. С., Усп. хим., 29, 162 (1960).
46, Ф р е й д л и н Л. X., Полковников Б. Д., Изв. АН СССР, ОХН,
1956. 1171; 1959, 1106.
47. Немцов М. С., Усп. хим., 7, 1635 (1938).
48. Калечиц И. В., Окладникова 3. А,, Николаева Д. X.
В сб. «Труды ВСФ СО АН СССР». Вып. 38.М., Изд. АН СССР, 1961.
См. с. 112.
49. К а л е ч и ц И. В., Н а х м а н о в и ч А. С., ДАН СССР, 148, 835 (1963).
50. К а л е ч и ц И. В., НахмановичА. С., Казанцева В. М.,
Кинетика и катализ, 4, 395 (1963).
51. ФростА. В. Вести. МГУ, № 3—4, 111 (1946).
52. Л о з о в о й А. В., Д ь я к о в а М. К. Гидрогенизация топлив в СССР.
М., Изд. АН СССР, 1940. 269 с.
53. Рапопорт И. Б. Искусственное жидкое топливо. М., Гостоптехиздат,
1955. 546. с.
54. V о n BraunJ., G m е 1 i n W., PetzololA., Вег., 55B, 3779
(1922); 56B, 1338 (1923); 57B, 382 (1924).
55. Von BraunJ., Bayer O., Blessing G., Вег., 57B, 392 (1924).
56. H a x M а н о в и ч А. С., К а л е ч и ц И. В. В сб. «Каталитические
реакции в жидкой фазе». Алма-Ата, Изд. АН КазССР, 1963. См. с. 166.
ГЛАВА 4
ЖИДКОФАЗНАЯ ВЫСОКОТЕМПЕРАТУРНАЯ
ГИДРОГЕНИЗАЦИЯ
Процесс жидкофазной гидрогенизации, который хотя и не нашел
широкого практического использования, продолжает изучаться при-
менительно к переработке ненефтяного сырья, из которого могут быть
получены ценные химические продукты.
Кроме того, этот процесс — самый надежный и дешевый, хотя
и малоэффективный, — позволяет перерабатывать сырье с высоким
содержанием минеральных компонентов или трудногидрируемых
смолистых и высокомолекулярных веществ. Именно поэтому он был
применен для переработки высокосмолистых нефтей (гидрокрекинг
по методу Варга 11 2) и может рассматриваться как возможный метод
утилизации различных смол, образующихся в качестве побочных
продуктов при процессах газификации3, коксования, пиролиза и т. д.
В отдельных случаях при переработке более благоприятного
сырья могут применяться и стационарные окисные регенерируемые
катализаторы4. Однако протекающие при этом процессы очень
близки к процессу жидкофазной гидрогенизации, так как идут почти
исключительно по радикальному механизму.
Накопленные к настоящему времени данные по химии процесса
жидкофазной гидрогенизации и взаимосвязи реакций, имеющих
место в этом процессе, представляют значительный интерес. Целесо-
образно рассмотреть сначала общую динамику изменения группового
состава типичного сырья в процессе жидкофазной гидрогенизации
с тем, чтобы выяснить закономерности превращений одних групп
компонентов в другие, а затем перейти к уточнению и детализации
химии превращений каждого класса соединений — нейтральных соеди-
нений, кислотных компонентов (фенолов) и азотсодержащих соедине-
ний. Превращения соединений, содержащих серу, вследствие специфич-
ности процессов гидрообессеривания топлив рассматриваются в гл. 6.
СОСТАВ СЫРЬЯ И ПРОДУКТОВ
ЖИДКОФАЗНОЙ ГИДРОГЕНИЗАЦИИ.
ОБЩАЯ СХЕМА ПРОЦЕССА
%
Вопросам изучения химии жидкофазной гидрогенизации было
посвящено немало работ еще в первые периоды развития процесса
деструктивной гидрогенизации. Результаты этих работ обобщены
в ряде монографий и обзорных статей 5‘8; их сопоставление позволяет
сделать следующие выводы ’.
11* 16»
Й:
1. Парафиновые углеводороды в условиях жидкофазного процесса
подвергаются крекингу с образованием парафиновых углеводородов
более низкого молекулярного веса.
2. Олефиновые углеводороды превращаются в соответствующие
парафиновые углеводороды, которые подвергаются дальнейшим
превращениям.
3. Полициклические ароматические углеводороды гидрируются
с образованием структур, включающих ароматические и гидрирован-
ные кольца.
4. Нафтены расщепляются с образованием нафтенов и парафинов
более низкого молекулярного веса и частично изомеризуются (гекса-
метиленовые превращаются в пентаметиленовые).
5. Кислородсодержащие соединения восстанавливаются до соот-
ветствующих углеводородов и частично расщепляются с образованием
более низкомолекулярных кислородсодержащих соединений.
6. Соединения, содержащие азот или серу, восстанавливаются до
соответствующих углеводородов.
Эти обобщения хотя и дают общую характеристику процессов,
протекающих при жидкофазной гидрогенизации, не раскрывают взаи-
мосвязи или взаимной конкуренции отдельных реакций, а также
важные детали химизма превращений компонентов.
Очевидно, что систематически изучать зти вопросы можно только
сопоставлением данных по составу сырья й полученных из него гидро-
генизатов. Такого рода исследования начались относительно недавно,
когда были разработаны хроматографические методы анализа полу-
коксовых смол и сходных с ними по составу продуктов 9-15. Широкое
применение хроматографических методов в сочетании с экстракцией,
ректификацией, спектральными и другими физико-химическими мето-
дами позволило идентифицировать в составе угольных, полукоксовых
и сланцевых смол большое число индивидуальных соединений 16-18.
Так, например, только в углеводородных фракциях эстонской слан-
цевой смолы идентифицировано 288 индивидуальных углеводородов
и 8 сопутствующих им соединений, содержащих серу 17.
Эти же методы исследования были использованы для анализа гид-
рогенизатов смол. Так, при помощи хроматографического метода
определен групповой состав жидкофазного гидрогенизата низко-
температурной смолы из черемховского угля 19’ 20, состав асфальте-
нов 21, выделенных из угольного гидрогенизата. Из жидкофазного
гидрогенизата бурого угля удалось выделить 22, 23 8 парафиновых
углеводородов, 6 полициклических углеводородов, 20 азотсодержа-
щих соединений, 9 фенолов. Подробно исследован 24, 25 состав
низкотемпературного гидрогенизата (процесс ТТН) буроугольной
смолы.
Сравнительное изучение составов сырья и продуктов жидкофаз-
ной гидрогенизации было предпринято 26-30 на примере смолы из
черемховского угля, а затем и самого черемховского угля. Исход-
ным сырьем была низкотемпературная смола, полученная полукоксо-
ванием угля при 650 °C, среднетемпературная смола (полукоксова-
164
ние при 850 °C) и гидрогенизаты этой смолы. Для сравнения изу-
чен также жидкофазный гидрогенизат угля.
Состав низкотемпературной смолы и ее гидрогенизата2в> 27.
Результаты, полученные при сравнительном изучении составов низко-
температурной смолы и ее гидрогенизата, приведены в табл. 21, 22.
Гидрогенизат был получен на четырехлитровой опытной установке при
650 кгс/см2 и 485 °C в присутствии промышленного железного катализатора
(1,5% от массы свежего сырья).
Сопоставление аналитических данных и данных материального
баланса (табл. 21) показывает, что больше всего увеличивается коли-
чество моноциклических ароматических углеводородов, возрастает
также количество парафинов и нафтенов. Содержание бициклических
ароматических углеводородов несколько уменьшается. Если учесть,
что значительное количество бициклических ароматических углево-
дородов должно было бы образоваться при гидрировании пековых
фракций и нейтральных кислородсодержащих соединений, то даже
небольшое уменьшение общего количества бициклических углеводо-
родов говорит о значительно большей скорости гидрирования этих
углеводородов по сравнению со скоростью гидрирования моноцикличе-
ских ароматических углеводородов.
Таблица 21. Изменение группового состава рабочего сырья
в условиях жидкофазной гидрогенизации
Компоненты Содержание, % Измене- ние содержа- ния. % '
рабочее сырье гидро- генизат
Углеводороды • • • 48,3 56,6 +17,8
парафины н нафтены 11,6 15,0 +23,9
моноциклические ароматические 9,4 17,5 +88,0
бициклические ароматические 27,3 24,1 —11,3
Нейтральные кислородсодержащие соединения 19,8 8,3 —51,1
Карбоновые кислоты 0,2 0,0 —100,0
Пековые фракции 26,2 21,1 -20,8
в т. ч. асфальтены 13,6 0,2 -98,5
Неучтенные соединения 5,5 6,1 —
Газы 0,0 5,5 —
Вода и потери 0,0 2,4 —
Итого 100,0 100,0
Эти закономерности отмечались как при жидкофазном гидрирова-
нии индивидуальных углеводородов 8, так и при гидрогенизации
нефтяного сырья 31, т. е. они носят общий характер.
В ходе гидрогенизации, естественно, уменьшается содержание
соединений с двойными связями, содержание серы, азота, функцио-
нальных групп, но с различной интенсивностью (табл. 22). Из данных
165
табл. 22 видно, что наиболее трудно удаляется азот, легче
всего — сера. В случае кислородсодержащих соединений наиболее
резко снижается число эфирных групп, особенно в высших фракциях;
количество гидроксильных групп больше всего снижается в легких
фракциях; полностью восстанавливаются карбоксильные группы.
В низкокипящих фракциях появляется небольшое количество карбо-
нильных соединений.
Таблица 22. Сравнительная характеристика фракций
полукоксовой смолы и гидрогенизата (в %)
Показатели Фракции смолы (°C) Фракции гидрогенизата (°C)
200- 250 250- 3 00 300- 350 200- 250 250 — 300 300- 350
Двойные связи в смеси парафинов и нафтенов 44 40 40 5 6 4
в моноцпклических ароматических углеводородах 37 41 42 2 5 5
в бициклических ароматических углеводородах 39 36 29 2 4 5
в неуглеводородных соединениях 40 55 88 31 31 37
Сера в смеси парафинов и нафтенов 0,04 0,04 0,07 0,00 0,02 0,00
в моноцпклических ароматических углеводородах 0,07 0,53 0,34 0,00 0,03 0,02
в бициклических ароматических углеводородах 1,30 0,96 — 0,00 0,05 0,02
в неуглеводородных соединениях 0,00 0,02 0,00 0,00 — 0,00
Функциональные группы в нейтраль- ных неуглеводородных соединениях эфирные группы 19 25 54 12 2 0
гидроксильные группы ацетили- руемые) 69 66 10& 0 0 59
гидроксильные группы (по Те- рентьеву) 80 65 90 — — 64
карбонильные группы 15 0 0 0 11 7 0
Азот (в неуглеводородных соедине- ниях) 0,53 0,87 1,20 0,50 0,84 0,65
Состав среднетемпературной смолы и ее гидрогенизатов 28-3°.
Гидрогенизата были получены при температурах 450 и 473 °C, что
позволило охарактеризовать влияние температуры на интенсивность
отдельных реакций в условиях промышленного процесса.
Материальные балансы этих процессов приведены в табл. 23 и 24.
В этих балансах учитывается циркуляция тяжелых фракций, кипя-
щих выше 320 °C (шлам), но не учитываются расход водорода и вы-
ходы газов и воды. (Выходы жидких продуктов — средние за дли-
тельный срок работы).
Как видно из приведенных данных, основным направлением изме-
нения состава сырья в процессе деструктивной гидрогенизации яв-
166
Таблица. 23^ Упрощенный материальный баланс процесса
жидкофазной гидрогенизации смолы при 473 °C и v =1,0 ч*1
Компоненты Введено, кг Выведено, кг Разница
тяже- лое масло шлам ИТОГО широ- кая фрак- ция шлам ИТОГО кг % ОТ ИСХОД- НОГО количе- ства
н-Парафины .... Изопарафины и наф- 52,5 20,5 73,0 35,6 20,4 56,0 -19,0 -26,0
тены 24,0 13,0 37,0 48,0 12,5 60,5 +23,5 +63,5
Олефины Ароматические моно- 51,0 0,0 51,0 0,9 0,0 0,9 —50,1 -98,2
циклические угле- водороды .... 43,5 70,5 114,0 100,2 68,5 168,7 +54,7 +47,0
Ароматические би- и
полициклические углеводороды . . 63,0 121,0 184,0 127,1 118,2 245,3 +61,3 +33,3
Итого . . . 234,0 225,0 459,0 311,8 219,6 531,4 +72,4 +15,8
Фенолы 59,5 23,0 82,5 44,5 22,2 66,7 —15,8 —19,2
Азотистые основания 6,5 8,0 14,5 15,1 8,0 23,1 +8,6 +59,1
Водорастворимые ве- 1,0
щества — 1,0 0,0 0,0 0,0 -1,0 -100,0
Асфальтены .... Вещества, кипящие 41,0 — 41,0 0,0 0,0 0,0 —41,0 -100,0
выше 520 °C ... — 27,5 27,5 4,4 26,7 31,1 +3,6 —
Твердые вещества Нейтральные неугле- — 116,0 116,0 0,0 113,0 113,0 -3,0 -2,6
водородные соеди- нения 126,0 71,0 197,0 24,0 67,7 91,7 -105,3 -53,5
ацетилируемые — 28,5 — 13,3 27,6 40,9 — —
карбонильные — 13,5 — 2,7 12,4 15,1 — —-
Итого . . . 468,0 470,5 938,5 399,8 457,2 857,0 -81,1 —
Потери при анализе 32,0 29,5 61,5 4,4 28,6 33,0 — —
Всего . . . . . 500 500 1000 404,2 485,8 890,0 -110,0 -11,0
ляется восстановление неуглеводородных соединений, вследствие
чего возрастает количество углеводородов и меняется их состав.
Рассмотрим превращения углеводородов. Парафиновые углеводо-
роды подвергаются деструкции и тем сильнее, чем выше температура
(содержание их уменьшаетсяна 26,0% при 473 °C и на 6,5 % при 450 °C).
Большая склонность парафиновых углеводородов к деструкции отме-
чалась 81 82 при исследовании скорости разложения различных инди-
видуальных углеводородов.
167
Таблица 24. Упрощенный материальный баланс процесса
жидкофазной гидрогенизации смолы при 450 °C и v = 0,5 ч-1
Компоненты Введено, кг Выведено, кг Разница
тяже- лое масло шлам ИТОГО широ- кая фрак- ция шлам ИТОГО кг О/ /0 от ИСХОД- НОГО количе- ства
н-Парафпны .... 30,0 46,4 76,4 28,2 43,2 71,4 -5,0 6,5
Изопарафины п наф- тены 13,7 31,4 45,1 43,2 28,2 71,4 +26,3 +58,4
Олефины 29,2 0,0 29,2 1,0 0,0 1,0 -28,2 -96,6
Ароматические мопо- цпклпческпе угле- водороды 24,9 102,9 127,8 71,1 , 96,2 167,3 +39,5 +31,1
Ароматические поли- циклические угле- водороды 36,0 215,8 251,8 47,2 200,8 248,0 -3,8 -1,5
Итого . . . 133,8 396,5 530,3 190,7 368,4 559,1 +28,8 +5,4
Фенолы 34.0 75,0 109,0 41,4 70,0 111.4 +2,4 +2,2
Азотистые основания 3,7 13,6 17,3 9,6 12,5 22.1 +4,8 +27.8
Водорастворимые ве- щества 0,6 0,6 0,0 0,0 0,0 -0,6 —100,0
Асфальтены .... 23,4 — 23,4 0,0 0,0 0,0 -23,4 —100,0
Вещества, кипящие выше 520 °C ... — 60,0 60,0 9,6 55,8 65,4 +5,4
Твердые вещества — 72,8 72,8 0,0 68,3 68,3 -4,5 -6,2
Нейтральные неугле- в о до родные сое дп- неппя 72,1 60,0 132,1 32,6 56,7 89,3 —42,8 —32,4
ацетилируемые — 39,2 — 30,2 36,5 56,7 — —
карбонильные —. 11,4 — 3,8 11,5 15,3 — —
Итого . . . 267,6 677,9 945,5 283,9 631,7 915,6 — —
Потери при анализе 18,4 36,1 54,5 13,1 31,3 44,4 — —
Всего 286,0 714,0 1000 297,0 663,0 960,0 -40,0 -4,0
Олефиновые углеводороды восстанавливаются практически на-
цело, причем уже при 450 °C скорость гидрирования двойной связи
очень велика. Так как непредельные углеводороды полукоксовой
смолы не содержат значительных количеств циклоалкенов, гидриро-
вание олефинов должно давать в основном парафины. Следовательно",
фактически расщепление парафинов (с учетом вновь образовавшихся)
проходит при 450 °C на 31,8%, а при 473 СС на 56,1%.
Значительнее всего увеличивается количество нафтенов и изо-
парафинов, второе место занимают моноциклические ароматические
168
углеводороды. Количество бициклических ароматических углеводо-
родов при 450 °C уменьшается, а при 473 °C возрастает. Это объяс-
няется тем, что при 450 °C интенсивность восстановления неуглеводо-
родных соединений значительно меньше, чем при 473 °C. Если при
450 °C восстанавливается 21,1% неуглеводородных соединений (фе-
нолы, азотистые основания, водорастворимые вещества, асфальтены
и нейтральные неуглеводородные соединения), то при 473 °C —
45,9% этих веществ.
Так как в рассматриваемом случае все типы неуглеводородных
соединений представлены высококипящими веществами, при их вое
становлении должны образоваться не алифатические и моноцикличе-
ские, а би- и полициклические углеводороды. Следовательно, исходное
количество последних должно быть увеличено настолько, насколько
восстановились неуглеводородные соединения. Полное превращение
би- и полициклических ароматических углеводородов при 473 °C
составит 27,5%, а при 450 °C — 20,4%, т. е. при 473 °C би- и поли-
циклические углеводороды превращаются существенно быстрее.
Соответственно этому возрастает количество моноциклических арома-
тических углеводородов и нафтенов, причем несколько больше при
473° С. Следовательно, повышение температуры процесса ускоряет
гидрирование ароматических углеводородов, хотя, и в заметно мень-
шей степени, чем процессы восстановления, которые более подвер-
жены влиянию температуры.
Чтобы оценить соотношение между долями ароматических и наф-
теновых углеводородов в ходе жидкофазной гидрогенизации смолы,
был применен кольцевой анализ 281 29. По аналогичной методике
проанализирован гидрогенизат черемховского угля. Полученные
данные приведены на рис. 12, 13 и в табл. 25,
Диаграммы, приведенные на рис. 12,13, отражают ход анализа: экстракция
кислотой (основания), экстракция щелочью (фенолы), ректификация нейтрального
остатка на фракции, хроматографическое разделение на группы углеводородов
и нейтральные кислородные соединения (ИКС) с последующим кольцевым ана-
лизом углеводородов (А — ароматическое кольцо, N — нафтеновое кольцо,
цифра указывает число колец данного типа).
Из данных табл. 25 видно, что угольный гидрогенизат отличается
от смоляных повышенным содержанием кислородсодержащих соеди-
нений, высокомолекулярных компонентов, полициклических угле-
водородов.
Среди ароматических углеводородов, как видно из рис. 12 и 13,
обращает на себя внимание явное преобладание структур с одним
гидрированным кольцом. Объяснение этой закономерности нужно
искать в отношении скоростей гидрирования и расщепления колец.
Было показано 8, что скорость гидрирования ароматических угле-
водородов увеличивается с ростом числа колец (трициклические
гидрируются быстрее бициклических, а бициклические — быстрее
моноциклических) и уменьшается с глубиной гидрирования. Нао-
борот, скорость расщепления возрастает с увеличением степени
169
ИнтерВалы каления фракций, “С
80 150 210 320 потери^
Состав Фракций, % . Q) ХЛ Сь <<, С; ©э ©э ©э ©> ^5 ©з S J 1 1 1 ) 1 1 1 L 1 тяжелые основания | тяжелые фенолы олефины по- тери Кен ЗОЛЫ 1А nomew пот ери потери
ИКС ИКС НКС
ГМН тетра- лины 1A + 1N 2А 2A*1N нкс
1A+1N инданы
2A*1N ЗА *1N фенилтетра- лины* флуорены* бензврлуорены 2A + W ЗА *1N пирены и ИКС
44 4A*1N 3A-1N за*2н 2 >ъ Б & й %
нафталины 1A + 2N 2А 2A+1N
h h 1A+1N тетралины
1А бен- золы пирены и другие углево- дороды
I s. олефин* У M+1N 2А+ IN 2A+2N 1А + 2N
нафте- ныб-чл инданы
нафте ныб-чл. 1А бензолы
1-лпилфенол о изопара- фины
ксале- НОЛЫ изо- ин-пар
нафтены
нафтены
п'крезол
н-парафины .
м-крезол
н-парасри ны
о-крезал н-пара- фины
н-пара финн
фенол
и -ГГТ---1-------------1------]-----1----—~1 — 1---------1-----1—-----1
Лцтшны\ 10 20 30 40 50 60 70 80 90 100
лутидины Состав широкой фракции, °/о
а
Интервалы кипения фракций, аС
39 170 230_______280 ________320 400 520
Состав фракций, % I I 1 L I 1 | | I I | основания I тяжелые фенолы потер разде- ления потери разделен потери разделения потери потери | соединения* кипящие выше 520аС J | ипнзи-ареой пфэшои
нкс НКС
НКС
цуи НИС
2А
QDOM9 • тика 1А +2N
1A*W 2 A 4N
оле финь/
2А 2А 1A*-1N 2A* IN 1A+2N
нафтены 5-и. 6-членные
1АЧЫ 2А-2И ЗА*2Н 3MN 4A*1N
JA бензопь! 1А 4N 1А +2N
изо- и н- парафины нафтены 2A+1N ЗА* IN
1А бензолы
нафтены
п-зтилфенол
нафтены нафтены
ксиленолы
наф- тены
п- крезол н-парафины !ЧНПф ! 1хФи-н\\
м-крезол изо Я н-парф финн н-парафины
о-крезол н-парафины
фенол
10 20 30 40 50 60 70 80 9Q 100 Состав широкой фракции, %
Рис. 12. Состав жидкофазного гидрогенизата среднетемпературной смолы:
а — 473 °C, 500 кгс/см2, 1,0 ч~>; 6 — 450 °C, 500 кгс/см2, 0,5 ч~2.
100
до-
80-
70-
13
5 4-/7-
<5 зо-
20-
10-
0--
о
Интервалы кипений фракций,"С
57 150 210 320
бензолы
олефины.
фенолы
10
boo ! 500
НКС
1А
потери
нкс
<ь
м
потери
разделения
— - нафталины
инданы
и
тетра
НКС
потери
потери
/А
- §
II
нкс
нке
, 1А + 1Ы
аепэслы
2А
2AHN
олефины
1А
1A+1N
2А
1A+2N
бензолы 1А
2AHN
3AHN
4Д
2A+1N
ЗАЧЫ
заны ;
ОА
6А*1Ы
изопор а-
фины —
кпара-
финь/
нафтены и изопарафины
нафтены и изопарафине.
н-парафины
н-парафины
(I
* 3
I
Из
сэ
н-пара
фаны
30 60 50 60 70
Состав широкой фракции , °/а
Рис. 13. Состав жидкостного гидрогенизата угля (476 °C, 500 кгс/сма, 1,0 ч'1).
20
80
90 100
I
CJ
о
h
и
в
I
4)
I
I
й
£
Таблица 25. Состав промышленных жидкофазных гидрогенизатов
смолы и угля (в %)
I—473 °C, 500 кгс/см’, 1,0 ч-1; II—450 °C, 500 кгс/см’, 0,5 ч-1;
III—467 °C, 500 кгс/см’, 1,0 ч-‘
Компоненты Гидрогенизат смолы Гидро- генизат угля
I II III
Парафины и нафтены .............................
н-парафины..................................
изопарафины ................................
нафтены.....................................
Ароматические углеводороды .....................
моноциклическне ............................
бициклические...............................
полициклические ............................
Неуглеводородные соединения................ . .
фенолы ......................................
в т. ч. с т. кип. до 220 °C . .............
азотистые основания .........................
нейтральные кислородсодержащие соединения
ацетилируемые ................................
с карбонильной группой .....................
другие ...................... ..............
Твердые вещества................................
Смолы ..........................................
Соединения, кипящие выше 520 °C ..................
Всего охарактеризовано .........................
13,1 15,0 10.9
6,3 7,5 5,0
0,9 5,9 )« 5,9
46,6 43,2 41,8
19,0’ 17.4 10,7
17,5 12,4 12,1
10,1 13,4 19,0
17,7 22,5 24,0
7,5 11,6 13,0
1,2 0,9 3,5
2,6 2,3 3,0
4,6 5,5 3,4
1,7 1,6 1.4
1,3 1,8 3,2
12,7 7,1 4.6
2,7 — —
3,5 6,3 10,1
96,3 94,1 91,4
171
насыщения водородом ароматического углеводорода. Этими соотно-
шениями объясняется обычно наблюдаемая в процессах гидриро-
вания последовательность превращений полициклических аромати-
ческих углеводородов 31 (см. также гл. 1):
Полициклические --> Бициклические ---> Моноциклические и т. д.
Если по данным кольцевого анализа определить количество аро-
матических и гидроароматических углеводородов каждого типа, то
несмотря на всю его условность можно оценить главные и второстепен-
ные пути их превращений. Так, для процесса гидрогенизации смолы,
протекающего при 473 °C, эти превращения выразятся схемой
2A2N
0,8 4
1A2N
2N + IN,
5,2
4А—> ЗА 1N
0,5 5,2 '
2A1N
у 14,6 >
1A1N
14,04'
2А
0,9
1А
2x0
где жирными стрелками показаны главные направления; А — арома-
тическое кольцо, N — гидрированное кольцо; цифры означают содер-
жание углеводородов данного типа в гидрогенизате (в %).
В условиях проведения процесса при 450 °C схема превращений
несколько изменяется:
2A2N
1 A2N
2N + 1N
4А—у 3A1N
0,9 8,3
2A1N
^Х 9,9 ''
ЗА
0,3
1 A1N
15,7
2А 1 а
°,2 0,7
Процесс гидрогенизации угля можно изобразить схемой:
2A2N 1A2N 2N + 1N
^Х о-1 Ху 0,1 6,о
(4А)—>3A1N 2A1N 1A1N
3,5 11,7\ 7’°\
ЗА^ 2А^ 1А
5,6 3,5 2,7
Во всех случаях процесс превращения полициклических углеводо-
родов складывается из чередующихся стадий гидрирования и рас-
щепления колец. Процессы, протекающие при 473 и 450 °C, разли-
чаются в основном направлением превращения углеводородов
типа 3A1N. В первом случае главным направлением является рас-
щепление этой системы колец, во втором — гидрирование. Кроме
того, при 450 °C на каждом этапе процесса доли продуктов расщепле-
ния ниже, а доли продуктов гидрирования выше, чем в первой схеме.
Следовательно, повышение температуры, изменяя скорости процессов
расщепления больше, чем скорости процессов гидрирования, в извест-
172
ной степени меняет и механизм суммарного процесса превращения
полициклических ароматических углеводородов. Это положение ста-
нет более наглядным, если изобразить аналогичной схемой процесс
гидрирования над стационарным катализатором 2i.
Применение гораздо более активного катализатора позволяет
проводить процесс гидрирования смол при еще более низкой темпера-
туре (400 °C). Протекающие при этом превращения отличаются
от описанных выше и могут быть представлены следующей схемой:
*1A3N< 1 /3N-^.
(4А) —► (3A1N) —► 2A2N < 2 *1A2N< 6 ’ 2N —> IN
oo4 ^2A1Nz ’ *1A1NZ 37
7 10
В этом гидрогенизате (процесс ТТН) уже совсем отсутствуют чисто
ароматические соединения и появляются соединения с тремя и че-
тырьмя гидрированными кольцами.
Таким образом, из сопоставления рассмотренных выше схем пре-
вращения полициклических углеводородов видно, что, изменяя тем-
пературу процесса, можно направлять его в сторону преимуществен-
ного образования тех или иных углеводородов.
Неуглеводородные соединения помимо основного процесса —
восстановления — также подвергаются расщеплению (см. табл. 26).
Таблица 26. Превращения неуглеводородных соединений
(в % к содержанию в рабочем сырье)
Соединения Прй 450 °C При 473 “С
степень превращения продукты восстановле- ния (по разности) продукты деструкции степень превращения продукты восстановле- ния (по разности) продукты деструкции
Фенолы Азотистые основа- 35,8 — 38,0 73,1 19,2 53,9
НИЯ Нейтральные соедп- 27,2 — 55,3 44,8 — 104,0
пения 57,0 32'4 24,6 65,6 53,4 12,2
Как видно из данных табл. 26, повышение температуры резко
увеличивает степень превращения всех классов неуглеводородных
соединений. Правда, долю продуктов восстановления азотистых
оснований (как при 450, так и при 473 °C) и фенолов (при 450 °C)
подсчитать невозможно, так как увеличивается их общее количество.
Следовательно, часть нейтральных неуглеводородных соединений
в процессе гидрирования превращается соответственно в фенолы или
основания.
Таким образом, в условиях деструктивной гидрогенизации основ-
ные компоненты подвергаются главным образом деструкции и, в мень-
173
шей степени, восстановлению. Для кислотных компонентов также более
характерны процессы деструкции, но доля процессов восстановления
больше, чем в случае основных компонентов. Нейтральные компо-
ненты интенсивно восстанавливаются, в значительно меньшей степени
подвергаются деструкции, а также приобретают основные или кислот-
ные свойства, превращаясь соответственно в основания и фенолы. Этот
последний процесс более характерен для нейтральных азотсодержа-
щих соединений, чем для кислородсодержащих. Из кислородсодержа-
щих соединений легче всего превращаются карбоновые кислоты,
затем эфиры, а наиболее устойчивы фенолы.
ПРЕВРАЩЕНИЯ НЕЙТРАЛЬНЫХ СОЕДИНЕНИЙ
Химическая природа нейтральных соединений, входящих в со-
став типичного сырья жидкофазной гидрогенизации — полукоксо-
вых смол, изучена недостаточно. Лучше и полнее изучены углеводо-
роды и легкие фракции, значительно меньше — нейтральные кисло-
родсодержащие соединения, составляющие значительную часть высо-
кокипящих фракций.
Не подлежит сомнению, что в этих фракциях преобладают арома-
тические структуры, причем в тяжелых — структуры с несколькими
конденсированными кольцами 33. Так, например, было показано 34,
что в асфальтенах и смолистых веществах полукоксовой смолы содер-
жатся три- и тетрациклические соединения.
Из тяжелых фракций смол кроме небольшого количества парафи-
нов нормального строения 35 были выделены нафталин и его гомологи,
дифенил, аценафтен, антрацен и его' гомологи, труксен 12, пирен,
флуорен, хризен, 3,4-бензфлуорен, 4,5-иминофлуорен, гомологи
пирена, бенз[6]нафт[2,1-</]тиофен, 2,13-бензфлуорантен, тетрацен зв.
Из продуктов гидрогенизации угля 37 были выделены пирен, 1- и 4-
метилпирены, 4,9-диметилпирен, хризен, фенантрен и антрацен 38.
Сильно ароматизированы и тяжелые фракции сланцевых смол,
в которых 50—60% составляют три- и тетрациклические соедине-
ния 391 40. Высокомолекулярные соединения нефти также очень
сложны по составу и ароматизированы 41, но для них более харак-
терны длинные цепи с ароматическими, в том числе конденсирован-
ными, системами у одного или обоих концов цепи 42.
Кроме углеводородов в состав нейтральных соединений входят
различные кислородсодержащие соединения 14 (эфиры, гидроксил-
содержащие и карбонильные). Из индивидуальных соединений были
выделены только альдёгиды й кетоны С2—С9, этилпропионат,. все
в ничтожных количествах 12. Наличие эфирных групп было дока-
зано 43 путем омыления нейтральных кислородсодержащих соедине-
ний, в результате которого были получены фенолы (9,5%) и карбоно-
вые кислоты (7,8%). О наличии значительных количеств эфиров сви-
детельствуют также данные по определению эфирных чисел. Кроме
того, в ИК-спектрах этих соединений обнаружены интенсивные
полосы поглощения, характерные для эфирных групп 44. Поскольку
174
в процессе гидрогенизации нейтральные кислородсодержащие соедине-
ния частично могут приобретать кислотный характер, вполне вероятно,
что в их состав входят фенолы, содержащие большое число замести-
телей и не способные вследствие этого экстрагироваться водными рас-
творами щелочей, но способные ацетилироваться и титроваться орга-
ническими основаниями.
В состав неуглеводородных соединений входят также азотсодер^
жащие соединения нейтрального характера 18, 33, соединения, содер-
жащие серу, и, вероятно, гетероциклические кислородсодержащие
соединения. Весьма возможно также наличие простых эфиров, при-
сутствие которых в сланцевых продуктах доказано спектральными
методами 45.
Химия превращений нейтральных соединений рассматривается ниже в раз-
делах, посвященных полициклическим ароматическим углеводородам, слож-
ным и простым эфирам и карбонильным соединениям. Превращения сернистых
соединений рассматриваются в гл. 6.
Изучение превращений нейтральных соединений проводилось
с помощью индивидуальных соединений, так или иначе моделиру-
ющих соответствующие группы компонентов сырья. Эти соединения
имеют более простое строение и меньший молекулярный вес, чем ком-
поненты реального сырья. Такой подход был, естественно, вынужден-
ным из-за невозможности выделения индивидуальных соединений
из высококипящих погонов смол. Превращения нейтральной части
реального сырья изучать существенно сложнее.
Общая динамика изменения группового состава нейтральных
веществ уже охарактеризована выше (стр. 166 сл.). Гидрогенизация же
высокомолекулярной, наименее изученной части этого сырья, освобо-
жденной тщательной экстракцией от кислотных и основных компонен-
тов, позволяет уточнить зту динамику 4в.
Результаты, полученные при изучении состава гидрогенизатов
нейтральных компонентов пека (автоклав, 300 кгс/см2, 485 °C, ката-
лизатор — Fe на полукоксе), приведены в табл. 27. Как сырье, так
и гидрогенизаты разделялись хроматографически; все легкие фрак-
ции, выкипающие до 325 °C, отбрасывались, т. е. характеризовались
только изменения состава высокомолекулярной части сырья.
Общие закономерности изменения группового состава нейтраль-
ных компонентов в основном аналогичны закономерностям, наблюда-
емым в процессе гидрогенизации всего сырья (см. стр. 173).
Весьма характерно изменение химической природы компонентов
сырья в ходе гидрогенизации (табл. 28).
Прежде всего можно отметить значительно меньшую устойчивость
эфирных групп по сравнению с гидроксильными (быстрое уменьше-
ние эфирного числа и медленное — ацетильного). Из приведенных
дайных видно, что в начале процесса наблюдается рост среднего
числа колец, затем уменьшение: быстрое — в случае неуглеводород-
ных соединений и медленное — для углеводородов, наиболее бедная
водородом фракция которых характеризуется наличием 4—5 колец.
175
Таблица 27. Изменение состава нейтральных компонентов пека
в процессе деструктивной гидрогенизации
в зависимости от продолжительности процесса
(в % от исходного количества)
Компоненты Содержание в пеке, % Содержание в гидрогенизате (в %) при времени контакта:
ним QH i нии о VI \ 200 мин 260 мин
Углеводороды 38,0 38,1 43,3 34,1 30,7
парафины и нафтены 16,2 6,1 5,6 3,1 1,1
моноциклические ароматические . . . 2,4 7,0 1 21,8 18,2 18,9
бициклические ароматические .... 10,2 13,3 /
Смолы 62,0 24,0 11,6 11,1 8,9
десорбируемые * бензолом 39,6 12,4
десорбируемые * ацетоном 22,4 11,6
* При хроматографическом анализе.
Таблица 28. Изменение химической природы нейтральных
компонентов пека в ходе жидкофазной гидрогенизации
Характеристика фракций В сырье ' - В гидрогенизате при времени контакта:
ПО мин 14 0 мин 200 мин 260 мин
Полициклические ароматические уг-
леводороды [CnH2n_x]
МОЛ. в 260 273 266 233 258
X Неуглеводородные соединении, де- 19,98 23,04 23,13 20,06 22,00
сорбируемые бензолом [CnH2n_x O(N, S)^]
МОЛ. в 240 258 257 224 196
X 16,27 18,47 19,42 15,52 13,13
эфирное число 84,7 72,9 14,9 12,9 4,5
ацетильное число Неуглеводородные соединении, де- 261 250 216 187 180
сорбируемые ацетоном
[CnHin_xO(N, S),]
МОЛ. в 260 260 264 — 210
X 17,18 18,74 18,28 — 11,88
эфирное число 50,2 46,3 29,7 — 7,5
ацетильное число 252 227 199 — 124
Данные элементарного анализа целиком подтверждаются спектраль-
ными данным44: в УФ-спектрах сначала увеличиваются, а затем
уменьшаются оптические плотности соответствующих фракций, в ИК-
спектрах сначала увеличивается, затем уменьшается поглощение
в области 9,5—9,7 мк.
176
Эти наблюдения следует объяснить тем, что в условиях жидкофаз-
ной гидрогенизации соединения с четырьмя-пятью циклами превра-
щаются медленнее, чем соединения с двумя и, особенно, с тремя
циклами, что приводит к обогащению высокомолекулярных фракций
соединениями с четырьмя-пятью циклами. В соответствии с этим
в УФ-спектрах углеводородных фракций гидрогенизатов отсутствуют
максимумы поглощения, характерные для антрацена, но появляются
и с ростом времени контакта становятся все более четкими максимумы
поглощения, характерные для пирена.
В УФ-спектре гидрогенизата, полученного при времени контакта
260 мин, обнаружены полосы поглощения, характерные для хризена
и других тетра- и пентациклических ароматических систем, выделен-
ных из циркуляционного газойля каталитического крекинга 47.
Обогащение высокомолекулярных фракций гидрогенизата такими
полициклическими углеводородами доказывает их высокую устой-
чивость, что требует специального рассмотрения.
ПРЕВРАЩЕНИЯ ПОЛИЦИКЛИЧЕСКИХ
АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
Результаты изучения реакций собственно гидрирования аромати-
ческих углеводородов с применением высокоактивных катализаторов
в большинстве случаев при низких температурах (см. гл. 3) не могут
быть целиком перенесены на условия жидкофазной гидрогенизации,
которая из-за использования малоактивного железного катализатора
проводится при высоких температурах и давлениях. Наиболее важ-
ными вопросами, которые следует осветить в настоящем разделе,
являются следующие:
1) влияние термодинамических ограничений в условиях высокой
температуры;
2) зависимость скорости гидрирования от наличия в структуре
углеводорода укороченной связи, более непредельной, чем «стандарт-
ная» ароматическая связь в бензоле, а также закономерность более
быстрого гидрирования линеарных структур по сравнению с ангуляр-
ными и центрированными;
3) механизм расщепления гидрированных колец.
О термодинамике гидрирования полициклических ароматических
углеводородов можно судить только качественно, так как не опреде-
лены термодинамические функции многочисленных промежуточных
продуктов гидрирования этих углеводородов, да и для самих исход-
ных веществ, начиная с углеводородов с тремя-четырьмя ароматиче-
скими кольцами, точные термодинамические константы отсутствуют.
Поэтому опубликованы константы равновесия лишь некоторых
реакций гидрирования полициклических углеводородов (табл. 29).
Из табл. 29 следует, что при температуре жидкофазного процесса
порядка 485 °C все реакции гидрирования могут протекать только
при повышенном давлении. Нетрудно подсчитать также, что чем
глубже степень гидрирования, тем меньше константа равновесия
12 Заказ 271
177
Таблица 29. Термодинамика гидрирования полициклических углеводородов
Реакция lgKp = f (T) Примечание Литера- тура
л / \
1 II 1 - -> 1 il 1 -13,65 + Термодинамический 48
\/\/ + 7350/7 расчет
-13,13 + Определено 49
+ 7000/7 экспериментально
-12,86 + То же 50
1 II 1 - + 6814/7
->111 -34,70 + Расчет по спектро- 51
\/ \z' + 18 537/7 скопическпм данным
(ЦЦС- + транс-)
-21,71 + Определено 52
+ 11 750/7 экспериментально
- / \ / \ —44,08 + То же 52
\/ ^x—X + 23 500/7
-6,11 + 52
\ / + 2600/7
-13,25 + 52
+ 6565/7
—26,38 + » 52
-> \—+ / + 13 030/7
/ ^x / ''x. -46,49 + 52
+ 23 190/7
(для фенантрена, например, логарифмы констант равновесия гидри-
рования до ди-, тетра-, окта- и пергидропроизводных при 485 °C
составляют соответственно —2,68, —4,59, —9,19 и —15,90).
Для более высокомолекулярных углеводородов приходится поль-
зоваться приближенными методами. А. Бонди53 предложил сле-
дующий набор формул для вычисления констант равновесия:
(Л//„)о-А//„ + 2032га — 745m
1g Ар = 0,2186 [(А^С)°4- razj (1,95-0,017) + га(16,5 + 0,5337I/j)]
lg KP(Ai) = 1g —0,2186m )
lg/C₽(A2) = lg/C'’-°’4372ra('D
lgtf(p) = lg Ap + ralgp
178
Здесь (ДЯ’)0 и ЛЯ£ — соответственно теплоты гидрогенизации
неизвестного и известного ароматического углеводорода; Д№ —
теплота сгорания; п — число молекул водорода, присоединяющихся
согласно стехиометрическому уравнению; mr — число колец, гидри-
руемых четырьмя атомами водорода; пг2 — число колец, гидрируемых
шестью атомами водорода; т — общее число гидрируемых колец
(гп = mi 4-тп2); Ах и А2 — гомологи; содержащие соответственно
1 и 2 заместителя.
Зависимость констант равновесия от температуры 83 показана на
рис. 14.
Можно отметить значительные численные расхождения между кон-
стантами, приведенными на рис. 14, и константами, вычисленными
Рис. 14. Зависимость констант
равновесия некоторых реакций
от температуры:
з — нафталин тетралин; 2 — тетра-
лин^декалин; з —фенантрен^2 окта-
гидрофенантрен; 4 — октагидрофенан-
трен ^пергидрофенантрен; 5 — хри-
эен^± додекагидрохризен.
по формулам табл. 29, но качественные выводы одни и те же. Важней-
шим выводом является уменьшение величины констант с ростом числа
ароматических колец. Так, например, при 497 °C (1/Г = 1,3) и
300 кгс/см2 равновесная доля гидрированного углеводорода с одним
ароматическим кольцом убывает с 86,5% Для нафталина (нафталин —►
-> тетралин) до 13,7% для фенантрена (фенантрен -> октагидрофенан-
трен). Для углеводородов с четырьмя и пятью кольцами (и более)
следует считаться с возможными термодинамическими ограничениями:
равновесная доля гидрированного производного очень невелика,
а дальнейшее превращение исходного углеводорода будет протекать
только по мере расщепления этого производного. Это подтверждается
экспериментальными данными. Так, например, коронен
12*
179
был прогидрирован 54 на 97% при 287 °C и 250 кгс/см2, а при 408 СС
и том же давлении происходит дегидрирование пергидрокоронена 55.
Относительные скорости превращения индивидуальных углеводо-
родов в условиях жидкофазной гидрогенизации (480 °C, 300 кгс/см2)46
составили:
Нафталин ............100 Фенантрен ... 89
Дифенил............. 48 Коронен . Не гидрируется
Антрацен ...........923
Результаты, полученные при совместном гидрировании нафталина
и его гомологов, антрацена и пирена над железным катализатором
(табл. 30), полностью соответствуют результатам гидрирования этих
углеводородов над никелевым катализатором (см. гл. 3): антрацен
превращается полностью уже при малом времени контакта; количе-
ство пирена уменьшается наиболее медленно, медленнее нафталина
и его гомологов. В то время как за 260 мин прореагировало только
39% пирена, степень превращения нафталина и его гомологов соста-
вила 53%, а всех бициклических ароматических углеводородов —
59%.
Таблица 30. Изменение состава смеси углеводородов
в ходе деструктивной гидрогенизации
480 °C, начальное давление 140 кгс/смг, катализатор Fe на полукоксе (5%)
Углеводород
Пирен .......................................
Гидропирены..................................
Антрацен.............................: ... .
Тетрагидроантрацен ..........................
Нафталин и его гомологи .....................
Продукты расщепления октагидроантрацена . . .
Моноциклнческие ароматические углеводороды . .
4А 9,0 7,2 5,5
4A1N — 1,8 2,3
ЗА 20,5 0,0 0,0
2A1N •— 13,5 7,2
2А 20,5 14,3 9,7
1A1N — 2,7 7,0
1А — 3,7 9,4
Кроме того, данные табл. 30 хорошо подтверждают тот факт, что
в условиях жидкофазного процесса практически не образуются гидро-
производные с двумя и более гидрированными кольцами.
Соотношение скоростей гидрирования индивидуальных углеводо-
родов в присутствии железного катализатора жидкофазного процесса
качественно аналогично соотношениям, рассмотренным в гл. 3: не
гидрируется совсем коронен, медленнее всех гидрируется пирен (сим-
метричные углеводороды); фенантрен (ангулярная структура) гидри-
руется медленнее антрацена (линеарная структура). По скорости
гидрирования в условиях жидкофазного процесса углеводороды можно
расположить в следующем ряду:
Олефины > Би- и трициклические ароматические углеводороды )>
> Тетрациклические ароматические углеводороды
> Моноциклнческие ароматические углеводороды
180
Необычное положение в этом ряду тетрациклических углеводоро-
дов может определяться двумя причинами: 1) тем, что для угольных
смол нехарактерно присутствие линеарных углеводородов и 2) воз-
можными термодинамическими ограничениями.
Очень малая скорость гидрирования моноциклических ароматиче-
ских углеводородов (см. табл. 30) наблюдалась 66 также при прямом
гидрировании бензола в присутствии железного катализатора при
420—480 °C и начальном давлении 127—154 кгс/см2. В гидрогенизате
обнаруживались только очень небольшие количества циклогексана,
метилциклопентана и гексанов.
При гидрировании полициклических углеводородов на промыш-
ленных катализаторах жидкофазного процесса расщепление проте-
кает настолько интенсивно, что в молекуле углеводорода, как пра-
вило, образуется не более одного гидрированного кольца. Типичной
реакцией расщепления является разрыв гидрированного кольца
в системе частично гидрированного ароматического углеводорода
типа xAlN, где х = 1, 2, 3 и т. д. Простейшим углеводородом такого
типа является тетралин, на примере которого и уточнялся механизм
реакции расщепления в условиях жидкофазного процесса.
При гидрировании тетралина на малоактивных катализаторах 51 7
и в отсутствие катализатора 67 было установлено, что расщепление
гидрированного кольца протекает по связи, примыкающей к арома-
тическому кольцу. Эта точка зрения справедлива и для случая жидко-
фазной гидрогенизации.
При гидрировании нафталина при 470 °C и 450 кгс/см2 в присут-
ствии катализатора Fe на полукоксе в течение 3 ч был получен жидкий
продукт (79,2%), состоящий нацело из ароматических углеводоро-
дов 58. Последние содержали 1% нафталина, 6,5% тетралина, 6,5%
производных индана и 86,0% моноциклических ароматических угле-
водородов (Св — 18,8, С7 — 21,2, С8 — 31,2, С1о — 14,8%), среди
которых преобладали монозамещенные производные. Из последних
были выделены и идентифицированы толуол, этил- и н-бутилбензолы.
Доминирование монозамещенных производных бензола указывает
на преобладание разрыва по схеме:
В гл. 2 (стр. 117) дано объяснение механизма этого процесса,
включающего присоединение атомарного водородапо двойной связи 67:
Ниже показано, что в условиях
ный механизм весьма вероятен для
соединений и позволяет объяснить
их структуры.
жидкофазного процесса радикаль-
превращения и неуглеводородных
некоторые особенности изменения
181
ПРЕВРАЩЕНИЯ СЛОЖНЫХ ЭФИРОВ
Изучение превращений сложных эфиров в условиях жидкофазного
процесса было предпринято, чтобы проверить возможность образова-
ния из них фенолов59, поскольку, как было показано (см. выше),
фенолы образуются из нейтральных веществ смол, в которых были
обнаружены эфирные группы.
Основным вопросом химии превращений сложных эфиров в про-
цессе жидкофазной гидрогенизации является направление расщепле-
ния сложноэфирной группы:
Аг—С—О—Ar'(R)
Разрыв связи 3 наименее вероятен, а направление разрыва по свя-
зям 1 или 2 определяется тем, этерифицировалась ли кислота спиртом
или фенолом: в первом случае главным образом рвется связь 1,
во втором — 2.
Продукты, образующиеся при гидрогенизации алкиловых (на при-
мере метилбензоата) и ариловых (на примере фенилбензоата) слож-
ных эфиров, приведены ниже (цифры над стрелкой показывают разры-
ваемую связь):
182
Результаты, полученные при деструктивной гидрогенизации неко-
торых сложных эфиров, и вычисленные соотношения между вероят-
ностями разрыва связей приведены в табл. 31, 32 (за единицу принята
вероятность разрыва молекулы по связи 2) 69.
Таблица 31. Продукты деструктивной гидрогенизации сложных эфиров
485 °C, начальное давление 160 кгс/см*, продолжительность 3 ч,
количество катализатора 5%
Исходный сложный эфцр Продукты гидрогенизации, % от исходного вещества
бензол толуол бензой- ная кислота Z фенол
Дпметилфталат 31,5 5,6 0,8 —
Дпэтилфталат 32,8 3,0 0,7 —
Дпбутилфталат 21,8 5,5 0,7 —
Метплбензоат 43,7 8,6 1,3 —
Этилбеизоат 37,4 6,0 1,2 —
Фенилбензоат 19,8 16,6 4,4 24,4
Фенилсалицплат 12,8 — 0,9* 48,9**
Фенилацетат 13,0 5,2 — 39,4
4 Салициловая кислота.
** Фенол + о-крезол.
Приведенные в табл. 32 соотношения вероятностей разрывов по
связям 1, 2, 3 следует рассматривать лишь как приближенные, по-
скольку нельзя учесть протекание возможных вторичных реакций.
Вероятнее всего, что значения интенсивности разрыва по связи 1
несколько завышены, а по связям 2 и 3 несколько занижены из-за
протекания этих реакций. Вторичными реакциями, видимо, объяс-
няется также большая разница в мольных выходах толуола и фенола
при гидрогенизации фенилбензоата, хотя при разрыве по связи 2
они должны были бы образоваться в эквимолцных количествах.
Однако выход бензола при деструкции алкиловых эфиров во всех
опытах в несколько раз больше выхода толуола, что не оставляет
сомнения в доминировании разрыва по связи 1, а при деструкции
ариловых эфиров выход бензола гораздо меньше выходов толуола
и фенола, т. е. преобладает расщепление по связи 2.
Таким образом, из приведенных выше данных видно, что деструк-
тивная гидрогенизация ариловых сложных эфиров может быть одним
из путей образования фенолов из нейтральных кислородсодержащих
соединений.
183
Таблица 32. Соотношения между интенсивностями разрыва по связям 1, 2 и 3
Исходный сложный эфир Разрыв по связи 1 Разрыв по связи 2 Разрыв по связи 3 Соотношение между интенсивностями разрывов по связям 1 ; 2 : з
вещество, по кото- рому определена интенсивность разрыва его выход, мол. % вещество, по которому определена интенсивность разрыв з его выход, мол. % вещество, по которому определена интенсивность разрыва его выход, мол. %
Димстнлфталат Бензол 70,3 Толуол 10,6 Бензойная кислота 1,1 6,6 : 1 : 0,1
Дмэтллфталат » 93,3 7,2 » 1,3 13,0 : 1 : 0,2
Дибутилфталат » 77,5 » 16,6 » 1,6 ' 4.1 : 1 : 0,1
Метилбензоат » 77,6 » 12,7 » 1,5 6,1 : 1 : 0,1
Этилбепзоат » 71,9 » 8,5 ' » 1,5 8,5 ; 1 : 0,2
Фенилбензоат » 50,3 » 35,7 » 7,2 0,6 ; 1 : 0,2 *
» — — Фенол 51,3 — — 0,4 :1 ; 0,1 *
Фенилсалицп- лат Бензол 35,1 Фенол + о-крезол 110,2 Салициловая и бензойная кислоты 1,5 0,8 :1 : 0,04**
Фенил ацетат » 22,6 Фенол 57,0 Бензол — 0,4 : 1 ***•
* Для вычисления интенсивности разрыва по связи 1 из мольного выхода бензола вычтен мольный выход бензойной кислоты, так как
ее образование сопровождается образованием бензола, а остаток уменьшен вдвое (при разрыве по связи 1 образуются две молекулы
бензола).
** Для вычисления интенсивности разрыва по связи 1 из мольнбго выхода бензола вычтен мольный выход кислот; для вычисления
интенсивности разрыва по связи 2 из мольного выхода фенола вычтен мольный выход бензола, так как в этом случае образуются бензол
и фенол, а остаток уменьшен вдвое, таг: как наряду с фенолом образуется о-крезол.
*** Поскольку бензол образуется при разрыве ио связям 1 и 3, в соотношении дана сумма интенсивностей.
ПРЕВРАЩЕНИЯ ПРОСТЫХ ЭФИРОВ
Источником образования фенолов при деструктивной гидрогени-
зации могут быть и простые эфиры фенолов.
Гидрирование простых эфиров фенолов изучалось главным обра-
зом с применением низкотемпературных катализаторов. При низких
температурах, как правило, протекает не деструкция связей С—О,
а гидрирование бензольного кольца 60, хотя при гидрировании кума-
рина — сложного эфира — над хромитом меди при 250 °C был полу-
чен 61 3-(о-оксифенил)-пропанол-1.
При использовании активных высокотемпературных катализато-
ров простые эфиры дают фенолы. Еще в ранних работах по деструк-
тивной гидрогенизации отмечалось образование незначительных
количеств фенолов из дифенилового эфира (9%) 62 и циклического
эфира — пентаметилтетрагидродиоксиксантена 63. Небольшое коли-
чество фенола было получено 63 при гидрогенизации дифенилоксида
при высоком давлении и высокой температуре в црисутствии
(NH4)2MoS464.
Фенолы образуются из простых эфиров фенолов не только в усло-
виях гидрогенизации, но обязательно при высоких температурах:
при нагревании в запаянной трубке 65, при нагревании в присутствии
алюмосиликатного катализатора 66 и окиси алюминия 67, 68. В послед-
нем случае отщепляющийся олефин алкилирует образовавшийся
фенол. Было показано 69, что в присутствии катализатора Ni на
А12О3 деструкция анизола может идти не только по связи О—СН3,
но и по связи О—С6Н5.
Первой работой, в которой превращения простого эфира изуча-
лись в условиях, моделирующих промышленный процесс, было сооб-
щение 7°. В этой работе изучалось влияние анизола на гидрогени-
зацию ароматических углеводородов и показано, что анизол менее
стабилен, чем фенол, образование которого в гидрогенизатах дохо-
дило до 63%.
Таким образом, как и в случае сложных эфиров, возможность
образования фенолов из простых эфиров определяется направлен-
ностью деструкции связей С—О в молекуле простого эфира:
Следует ожидать, что более прочной является связь I за счет
л,р-сопряжения.
В соответствии с этим при гидрогенизации простых эфиров фено-
дов мольные выходы фенола всегда выше, чем мольные выходы бен-
зола (см. табл. 33), кроме опытов с анизолом и дифениловым эфи-
ром 71. В дифениловом эфире связи 1 и 2 равноценны и их деструкция
должна давать фенол и бензол в эквимольных количествах; поэтому
больший выход бензола можно объяснить только вторичной реакцией
восстановления части образовавшегося фенола. Возможно, что вос-
становлением фенола объясняется и заниженный выход фенола (по
185
сравнению с бензолом) при гидрогенизации анизола и фенетола, т. е.
можно предполагать, что в этих случаях также преобладает деструк-
ция по связи 2.
В ряду анизол — третп-бутилфениловый эфир отношение мольных
долей фенола и бензола возрастает с увеличением числа углеродных
атомов в алкильном остатке, причем особенно быстро, если этот оста-
ток разветвлен. В опытах с изопропил- и шреш-бутилфениловым
эфирами выход бензола настолько незначителен, что можно полагать
полное отсутствие разрыва по связи 1, так как такое небольшое коли-
чество бензола вполне могло образоваться за счет восстановления
фенола.
Таблица 33. Продукты деструктивной гидрогенизации
простых эфиров фенолов
485 °C, начальное давление 160 кгс/см2, 3 ч, количество катализатора 5%
Исходный эфир Степень превра- щения. % Выход основных продуктов, мол. % Отноше- ние количеств фенола и бензола
бензол (разрыв по 1) фенол (разрыв по 2)
Анизол 97,8 35,6 23,0 0,65
Фенетол 87,9 22,4 24,3 1,08
н-Пропилфениловый эфир 99,7 16,4 36,2 2,20
Изопропилфениловый эфир 99,3 5,2 55,0 11,58
mpem-Бутилфениловый эфир 100,0 4,0 62,4 15,60
Дифениловый эфир 49,0 58,5 31,7 0,54
Следовательно, механизмы расщепления простых эфиров, содер-
жащих неразветвленные и разветвленные алкильные остатки, раз-
личны. Это различие показано также при изучении поведения в усло-
виях жидкофазного процесса не только модельных простых эфиров,
но и возможных промежуточных продуктов — углеводородов и спир-
тов 72 (табл. 34).
Как видно из данных, приведенных в табл. 34, в ряду анизол —
шреш-бутилфениловый эфир уменьшаются отношения количеств
бензола и фенола, а также отношения суммы ароматических углеводо-
родов к сумме фенолов. Следовательно, по мере усложнения алкиль-
ного остатка все больше доминирует разрыв по связи 2. Это подтвер-
ждается также величинами энергий расщепления связей 1 и 2 в соот-
ветствующих простых эфирах фенолов (табл. 35).
Из приведенных в табл. 35 данных можно сделать вывод, что рас-
щепление фенилалкиловых эфиров протекает в основном по углерод-
кислородным связям, давая в качестве промежуточных продуктов
оксифенильный, фенильный, алкильные и оксиалкильные радикалы.
Промежуточное образование фенильного и алкильных радикалов
доказывается выделением дифенила, толуола и о-крезола из гидроге-
низата анизола, этилбензола из гидрогенизата фецетола.
Следует отметить незначительную роль катализатора, что видно
из сопоставления результатов, полученных при гидрировании ани-
186
Таблица 34. Продукты деструктивной гидрогенизации (моль на 100 моль исходного вещества)
485 °C, 160 кгс/см', 3 ч, катализатор Fe на полукоксе
Продукты гидрогенизации Анизол Анизол * Анизол * * Фенетол н-Пропилфенило- вый эфир н-Пропилфенило- вый эфир *** тпрет-Бутилфени- ловый эфир Пропиловый спирт Толуол Этилбензол н-Йропилбензол 1 м-ПропилбеН’ зол ***
Метан . 72,7 96,5 76,3 12,0 6,8 3,4 11,2 14,0 40,7 5,9 31,6 51,0
Этан . 10,6 11,2 1,8 89,7 36,2 45,8 7,0 53,0 4,7 78,5 67,5 56,1
Пропан 11,9 12,9 1,2 3,2 82,1 60,0 16,0 48,5 1,4 1,3 51,7 20,5
Бутаны Парафины 1,8 — — 0,6 1,6 2,6 101,1 2,4 — 0,4 0.4 —
Сб 0,4 — —- — — —— -*Т —— —— —— — —
св 3,4 1,9 1,3 9,7 10,9 2,5 11,2 — 0,05 0,7 0,53 0,45
с, 0,5 0,4 0,1 — 0,6 — — — 0,2 0,04 — —
Св — — — — — — — — — ' 0,3 —
Бензол 54,2 53,1 23,8 38,9 30,4 25,6 22,2 — 43,2 58,2 83,5 51,1
Толуол 7,0 3,5 14,1 10,1 11,1 13,7 0,5 —- 55,0 0,4 2.2 10,3
Этилбензол — — — 1,6 —. — — —. — 33,2 5,25 29,5
н-Пропилбензол — — — — — — — — — 0,85 3,2
Изопропилбензол — — — — — — — — — — 0,86 3,3
Дифенил 0,8 1,2 — — — — — — — 0,6 — —
Фенол 23,6 30,7 48,0 34,7 35,4 53,6 51,3 — — — — —
о-Крезол 0,8 0,6 ' — — — — — — — — — —
Окись углерода 4,9 18,0 8.9 8,7 15,5 6,8 8,6 12.8 — — — —
Двуокись углерода 2,9 4,3 1,2 4,3 8,0 7,4 1,7 9,7 — — — —
Непревращенный эфир Отношения количеств — 0,02 9,7 — — — — — — — —
бензол : фенол . . '. . 2,3 1,7 0,5 1,1 0,8 0,5 0,4 — — — — —
бензол + толуол : фенол + крезол . . 2,5 1,8 0,8 1,4 1,26 0,73 0,4 — — — — —
* В отсутствве катализатора.
** Без выдержки при рабочей температуре.
*** Начальное давление водорода 50 кгс/см*.
Таблица 35. Энергии расщепления связей
в молекулах простых эфиров при 25 °C (в ккал/моль) 73,74
Эфир Е' (связь 1) Е" (связь 2) Е' — Е*
Анпзол 85,9* 86,0 —0.1
Фенетол 85,3* 83,0 +2,3
н-Пропилфениловып эфир 86,9 81,0 +5,9
т pm-Бутилфепиловый эфир 84,6 77,3 + 7,3
П римечание. Величины, отмеченные знаком *, взяты из работы ”.
зола в присутствии катализатора и без него. Действительно, при срав-
нении составов продуктов гомогенного гидрирования алкилбензолов
при разных давлениях водорода было показано 75, что при повыше-
нии давления разрывается главным образом связь в а-положении
к бензольному кольцу. Это было объяснено 75 присоединением атомар-
ного водорода к бензольному кольцу:
На примере изопропилбензола 75 показано, что изменение давле-
ния резко меняет соотношение как между ароматическими угле-
водородами, так и между газообразными продуктами. В случае же
пропилфенилового эфира пропан остается главным компонентом газо-
образных продуктов и при начальном давлении 50 кгс/см2. Однако
повышение давления существенно увеличивает долю пропана и умень-
шает выход фенола, что может указывать на известную роль про-
цесса, аналогичного предыдущей схеме:
Дальнейшие превращения продуктов деструкции простых эфиров
изучены в опытах с анизолами, меченными 14С в кольце (положение 1)
или метильной группе 76 (см. табл. 36).
Из данных, приведенных в табл. 36, видно, что жидкие парафино-
вые и нафтеновые углеводороды образуются практически из фениль-
ного остатка и, следовательно, метильная или оксиметильная группы
в их образовании не участвуют. Исключение составляют метилцикло-
гексан и толуол, из величин активностей которых следует, что они
образуются в результате реакции метилирования. Следует отметить,
что если удельные радиоактивности гексанов, а особенно циклогек-
сана и бензола, близки к расчетным, то активность пентанов ниже
расчетной, что подтверждает частичное протекание реакции глубокого
распада оксифенильного радикала с выделением 14СО. Возможность
такого распада была обнаружена ранее в опытах с фенолом 75.
188
Таблица 36. Выходы и удельные активности продуктов
деструкции анизола, меченного 14С
осн
Соединение Выход, моль на 100 моль превра- щенного анизола Удельная радиоак- тивность, имп/мг ВаСО« в мин Доля суммарной радиоактивности, %
ДЛЯ продукта из I для продукта из II Для продукта из I Для продукта из II
Анизол (исходный) ’ — 134 123 /
Метан 72,7 10 577 0,9 46,0
Этан 10,6,
Пропан 11,9 10 595 0,8 41,8
Бутаны 1,8
Пентаны 0,4 79 2 0,2 0,0
Гексаны 123 6
Циклогексан [ 3,4 145 1 3,2 0,0
Метплциклопентан ....
Метилциклогексан 0,5 100 85 0,5 0,3
Бензол 54,2 149 1 58,1 0,3
Толуол 7,0 107 125 6,3 6,7
Фенол 23,6 152 — 25,8 —
Крезолы 0,81
Дифенил . . 0,8' 115 28 2,1 0,5
Окись углерода 4,9 229 457 1,3 2,5
Двуокись углерода 2,9 232 587 0,8 1,9
Близкие значения удельных радиоактивностей СО и СО2 свидетель-
ствуют о том, что они образуются из одного источника и, скорее
всего, СО2 является продуктом, дальнейшего превращения СО по
реакциям:
2СО —> СО2 + С СО + Н2О —>- со2 + н2
Газообразные углеводороды С4—С4 также имеют близкие актив-
ности, что указывает на один и тот же путь их образования в процессе
деструктивной гидрогенизации анизола.
Из приведенных в табл. 36 данных видно, что 67% метана обра-
зуется из метильного (или оксиметильного) радикала и 33% — из
углеродных атомов бензольного кольца, в том числе 1% из углерод-
ного атома, связанного с кислородом; 69% углеводородов С2—С4
образуется из метильного (или оксиметильного) радикала и 31% —
путем распада кольца.
189
Интересно отметить, что мольное соотношение углеводородов
С4 : С2 : С3 : С4 в продуктах деструкции анизола составило.
1 : 0,146 : 0,164 : 0,025, а в продуктах деструкции толуола (в тех же
условиях) 1 : 0,116 : 0,034 : 0,00, т. е. в этом случае количество
углеводородов С2—С4 (особенно С3 и С4), получающихся через про-
межуточное образование метильного радикала, значительно ниже.
Поэтому следует предположить, что эти углеводороды образуются
в результате дальнейших превращений оксиметильного радикала.
Сравнение удельных радиоактивностей продуктов деструктивной
гидрогенизации анизола позволяет представить себе общую схему
дальнейших превращений радикалов оксифенила, фенила, оксиме-
тила и метила, которые образуются в данных условиях примерно
в эквимольных количествах (при этом необходимо учесть, что фенол
на 30% восстанавливается в бензол и соответственно его выход дол-
жен быть увеличен, а выход бензола — уменьшен).
Из фенильного радикала образуется главным образом бензол,
а также толуол (~10%) и дифенил (~1%).
Из оксифенильного радикала получается в основном фенол
С6Н5О. + Н2 —> с6н5он + н-
а часть его ('—30%) гидрируется
CeHjO^-j-SHa --> GgHjiO-
причем половина этой части расщепляется до СО и углеводородов
С4—С5, а вторая половина восстанавливается с образованием воды
и насыщенных углеводородов Св (часть их метилируется).
Тот факт, что источником образования побочных продуктов яв-
ляется оксифенильный радикал, а не фенол, подтверждается тем, что
при гидрогенизации фенола в идентичных условиях 70% его остается
неизменным, а количество насыщенных углеводородов составляет
доли процента. Источником насыщенных углеводородов не может
быть также и фенильный радикал, так как при гидрировании аромати-
ческих углеводородов доля насыщенных углеводородов тоже весьма
незначительна (0,05—0,7%). В отсутствие катализатора (см. табл. 34)
доля насыщенных углеводородов уменьшается до 2,3%, следова-
тельно, малоактивный железный катализатор может ускорять реак-
ции гидрирования только очень реакционноспособных соединений,
каким и является оксифенильный радикал. Предположение о более
высокой реакционной способности радикала оксифенила по сравне-
нию с фенилом подтверждается также и термодинамическими данными:
энтальпия образования радикала оксифенила 32,1 ккал/моль, а для
радикала метила — 32,6 ккал/моль ”.
Метильный радикал превращается в метан, небольшая его часть
расходуется на реакции рекомбинации, приводящие к этану, толу-
олу, метилциклогексану ио-крезолу.
Оксиметильный радикал восстанавливается в метан, примерно
десятая его часть расщепляется с образованием СО, а значительное
количество участвует в образовании углеводородов С3—С4.
190
Упрощенная схема превращений простых эфиров на примере
н-пропилфенилового эфира представлена ниже
w*
X
СО-]- с2 + с3
СО -f- Cj
X X X X
СО С1 с, с3
0,225 0.14 0,53 0.48
где х — расщепление по связи 2; у — расщепление по связи 1\
z — стабилизация -ОС3Н7; z' — расщепление -ОСзН?; и — стабили-
зация -С3Н7; и’ — расщепление -С3Н7; v — гидрирование СвН5О-;
v' — стабилизация CeH5O'; w — образование циклоалканов; w' —
расщепление промежуточного продукта гидрирования оксифениль-
ного радикала. Вычисленные таким образом доли 1 различных
реакций, протекающих при дегидрогенизации некоторых простых
эфиров фенолов, приведены в табл. 37.
Таблица 37. Доли реакций при деструктивной гидрогенизации
алкилфениловых эфиров (при 160 кгс/смз)
Реак- ции Ани- зол Фене- тол н-Пропил- фениловый эфир трет-Бутилфени- ловый эфир Реак- ции Ани- зол Фене- тол н-Пропил- феииловый эфир mpem-Бутилфени- ловый эфир
160 кгс/см’ 50 кгс/см’ 160 кгс/см* 50 кгс/см’
X 0,483 0,634 0,720 0,826 0,948 и' 0,000 0,060 0,080 0,358 0,055
У 0,508 0,357 0,263 0,163 0,007 V 0,249 0,214 0,296 0,071 0,227
Z 1,000 0,742 0,620 0,437 0,239 v' 0,751 0,786 0,704 0,929 0,773
z' 0,000 0,258 0,380 0,563 0,761 w 0,350 0,715 0,540 0,427 0,520
и 1,000 0,940 0,920 0,642 0,945 0,650 0,285 0,460 0,573 0,480
191
Из приведенных данных видно, что доля реакций расщепления по’
связи 2 растет, а по связи 1 падает в ряду анизол — щреиг-бутилфе-
ниловый эфир. Это еще раз иллюстрирует вывод о преобладании раз-
рыва по связи 2 с увеличением молекулярного веса эфира. Доля реак-
ций стабилизации радикала оксиалкила уменьшается при переходе
от оксиэтила (z = 0,742) к окси-щреш-бутилу (z = 0,239), что
объясняется уменьшением стабильности оксирадикалов (табл. 38).
Таблица 38. Теплоты образования простейших алкильных
п окепалкильных радикалов при 25 °C
Радикал АН0, ккал/моль Литера- тура Радикал ДН°, ккал/моль Литера- тура
СН3. 32,6 77 СН3О- -0.5 73, 77
С2Н5. 25,7 77 С2Н5О- -8,5 73
н-С3Н7- 19,3 77 H-CgHjO* .... —13,0 73
изо-С3Н7- .... 16,4 77 uao-C3H7O. . . . —15,0 73
трет-СдН^' . . . 5,1 77 mpem-CaHeO- . . —25,0 74
Во всех случаях, в хорошем соответствии с данными термодина-
мики, алкильные радикалы стабилизируются значительно интенсив-
нее, чем оксиалкильные.
Соотношение реакций распада гидрированного кольца и образова-
ния циклоалканов изменяется незакономерно и с большим разбросом
данных, так как это третья ступень превращения и слишком велико
влияние трудно учитываемых факторов.
ПРЕВРАЩЕНИЯ СПИРТОВ
И К АРБОНИ ЛСО ДЕРЖАЩИХ СОЕДИНЕНИИ
Спирты и карбонильные соединения не характерны для реаль-
ного сырья, но могут быть промежуточными продуктами его пре-
вращений. Они весьма реакционноспособны и поэтому данные об
их устойчивости относятся обычно к мягким условиям.
Алициклические и алифатические кетоны в присутствии WS2
при 80—120 кгс/см2 и 320—325 °C в течение 30 мин восстанавли-
ваются 78 на 89—93%. Из пинаколина в присутствии MoS2 при 340—
350 °C и 80 кгс/см2 получен 79 2,3-диметилбутан с выходом 87%.
В присутствии WS2 простые эфиры восстанавливаются при несколько
меньшей температуре — 300—320 °C; при 340—350 °C они превра-
щаются уже в течение 10 —15 мин, спирты в течение 30 мин, окиси
занимают промежуточное положение 78. По реакционной способности
кислородсодержащие соединения могут быть расположены в сле-
дующий ряд 78179:
Сложные эфиры > Кетоны > Простые эфиры > Спирты > Фенолы
На основании результатов гидрирования в более мягких условиях
в присутствии MoS3 (температура < 350 °C, начальное давление
100 кгс/см2) 80 был выведен несколько иной ряд:
Альдегиды > Кетоны > Карбоновые кислоты > Фенолы > Эфиры > Спирты
192
Были определены 81
торых спиртов при 250—350 °C
31 кгс/см2 в присутствии MoS2:
относительные скорости восстановления неко-
и начальном давлении водорода
Циклопентанол.......... 100
Циклогексанол.......... 100
н-Гексиловый спирт . . . 7—11
н-Октиловый спирт . . ,Ц
«mop-Октиловый спирт . 19—23
Фенол ................ 3
Легче всего, следовательно, восстанавливаются вторичные цик-
лические, затем вторичные алифатические и,4 несколько труднее,
первичные алифатические спирты, медленнее всего идет восстано-
вление фенола.
В мягких условиях (270—300 °C, 30 кгс/см2, WS2) происходит
не только восстановление спиртов, но и образование простых эфиров,
являющихся промежуточными продуктами 82.
Результаты, полученные при гидрогенизации некоторых карбо-
нилсодержащих соединений 83, приведены в табл. 39.
Таблица 39. Гидрогенизация карбонилсодержащих соединений
485 °C, начальное давление 160 кгс/см1, 3 ч, количество катализатора 5%
Исходное вещество Степень превра- щения, % Выход гидроге- низата, % Состав гидрогенизата
Бензофенон . . . 100,0 97,5 Дифенилметан
Бензальдегид . . 100,0 88,6 Кислые вещества (2,8%, гл. обр. бен- зойная кислота), бензол (4,3%), то- луол (45,1%), продукты конденсации, выкипающие при 114—254 °C (36,4%)
Циклогексанон 100,0 54,2 Циклопентан (8,4%), метилциклопентан (17,6%), циклогексан (17,1%), остаток с т. кип. 80 °C (11,1%)
Из данных табл. 39 видно, что карбонилсодержащие соединения
в условиях жидкофазного процесса превращаются полностью с глу-
боким восстановлением карбонильной группы. В случае аромати-
ческого кетона конечным продуктом восстановления является угле-
водород, при восстановлении алициклического кетона протекают
более сложные превращения, идущие, вероятно, по радикальному
механизму:
Большая вероятность такого или аналогичного механизма обра-
зования метилциклопентана и циклопентана при восстановлении
13 Заказ 271
193
циклогексанона по сравнению с путем, включающим промежуточное
образование циклогексана
О
II
доказывается тем, что в параллельном опыте с циклогексаном выход
жидкого продукта был выше (86,7%), причем, хотя содержание в нем
метилциклопентана составляло 30%, циклопентана оказались только
следы.
Главным продуктом превращения бензальдегида является толуол,
частично деметилирующийся в бензол. Однако одновременно про-
текают реакции конденсации и реакция Тищенко — Канниццаро,
приводящая к бензойной кислоте и бензиловому спирту. Оба эти
вещества, особенно бензиловый спирт, очевидно, быстро восста-
навливаются.
ПРЕВРАЩЕНИЯ ФЕНОЛОВ И ДРУГИХ КИСЛЫХ СОЕДИНЕНИИ
) Фенолы были в числе первых соединений, в опытах с которыми
было начато изучение гидрогенизации. Объясняется это тем, что
фенолы уже очень давно выделяют из каменноугольных смол и исполь-
зуют в промышленности, а в полукоксовых смолах фенолы являются
одной из основных составных частей. Кроме того, интерес к гидро-
генизации фенолов вызывался, ка*М?кгЯ5^плкавано н перспек-
тивностью топливно-химических вариантов переработки полу-
коксовых смол. При этом фенолы, будучи склонны к реакциям кон-
денсации и уплотнения, вызывают технологические осложнения 84,
поэтому скорости их превращения всегда были предметом детального
изучения.
В силу этих причин литература по гидрогенизации- фенолов
весьма обширна 5> 7- 83, 86. Ниже приведены лишь наиболее общие
выводы всех предыдущих работ и данные, появившиеся в печати
за последние 10 —12 лет.
Следует упомянуть, что вслед за работой Сабатье и Сандерана 87,
прогидрировавших фенол в паровой фазе, в России было впервые
осуществлено гидрирование фенола в жидкой фазе под давлением 88.
Гидрирование фенолов
ных катализаторов протекает по различным механизмам. При низких
температурах в присутствии Pt, Ni, Си Сг главным продуктом
являются соответствующие циклогексанолы; процесс идет через
промежуточное образование циклогексанона 88, что доказано резуль-
татами кинетических исследований 6°' 85:
ОН
194
Этот процесс широко применяется в промышленности как на-
чальная ступень производства капролактама и адипиновой кислоты.
В последнее время удалось разработать катализаторы, селективно
ускоряющие гидрирование только на первой ступени. На специальном
катализаторе при 140—180 °C и атмосферном давлении достигнут
90—95%-ный выход циклогексанона 89._1
Аналогично протекает низкотемпературное гидрирование би-
циклических фенолов 6 °.
При гидрировании фенолов в присутствии высокотемпературных
катализаторов или в термическом процессе без катализатора спирты
не образуются совсем; основными продуктами являются аромати-
ческие и алициклические углеводороды, а также небольшое коли-
чество циклоалкенов 86, 90. Образование циклоалканов и циклоалке-
нов нельзя объяснить гидрированием промежуточно образующегося
бензола, так как бензол гидрируется медленнее фенола, а в специ-
ально подобранных условиях не гидрируется совсем. Если в этих
условиях гидрировать фенол, то все равно образуется 10—30%
циклогексана 80. Было предложено 90* 91 объяснение, основанное
на протекании двух параллельных реакций:
В более мягких условиях (236 °C, 300 кгс/см2) на катализаторе
WS2 NiS на А12О3 из смеси технических крезолов были получены
все возможные продукты восстановления (32% метилциклогексана,
5,9% метилциклогексенов, 5,8% толуола) и гидрирования (56,3%
метилциклогексанолов) 92.
При более высоких температурах возможна еще одна реакция :—
деалкилирование производных фенола. Именно это превращение
больше всего привлекало исследователей, поскольку получение
ценных низших фенолов из малоценных высших, содержащихся
в угольных и сланцевых смолах, являлось важным составным звеном
получения химических продуктов при гидрогенизационной пере-
работке этих типов сырья. г.
Исследования в этом направлении были впервые начаты в СССР;
При гидрогенизации фенолов, кипящих в пределах 230 — 250 °С,;
при 470—520 °C и давлении 75 кгс/см2 в присутствии паров воды
были получены 93 низшие фенолы с выходом 27,4%. Из о-крезола
в присутствии железного катализатора было получено до 25% фе-
нола, а из гидрогенизата челябинской смолы (катализатор МоО)3,
150 кгс/см2, 430—450 °C) — 14—40% низших фенолов ’• 94 .
13*
195
Интерес к процессам превращения высших фенолов в низшие
методами деалкилирования или диспропорционирования не ослабел
до настоящего времени *.
Для превращения высших фенолов в низшие были применены
также термический крекинг как при атмосферном 95, так и при повы-
шенном давлении 9в, и каталитический крекинг в присутствии алюмо-
силикатных катализаторов 97.
Таким образом, к настоящему времени сформировались следу-
ющие направления исследований по превращению высших фенолов
в низшие.
1. Негидрогенизационное, в присутствии различных главным
образом кислотных катализаторов типа катализаторов каталитиче-
ского крекинга; его преимущества — простота, отсутствие дорогого
оборудования высокого давления, недостаток — обильное образова-
ние кокса 98.
2. Термическое деалкилирование под давлением водорода; его
преимущество — отсутствие дезактивации катализаторов, недоста-
ток— дорогая аппаратура.
3. Жидкофазная гидрогенизация с плавающим железным ката-
лизатором, недостаточно активным, чтобы восстанавливать фе-
нолы, но достаточно активным для предотвращения реакций уплот-
нения. Преимущества этого направления — совмещение получения
низших фенолов с переработкой смол в целом и возможность исполь-
зования имеющегося оборудования старых заводов деструктивной
гидрогенизации; недостаток — относительно низкие скорости пре-
вращения, недостаточная избирательность катализатора.
4. Жидкофазная гидрогенизация с применением других, более
активных, обычно стационарных и регенерируемых катализаторов.
Следует отметить, что превращения высших фенолов в присут-
ствии кислотных катализаторов протекают сложно, очевидно по ионным
механизмам. При этом идет не только деалкилирование, но и реакции
изомеризации и диспропорционирования. Реакции диспропорциони-
рования являются основными, например, при проведении процесса
при 70—215 °C в присутствии серной кислоты ", при более высоких
температурах (до 400 °C) на кислотном катализаторе BF3 на А12О3 реак-
ции деалкилирования усиливались, но все еще не были главными —
преобладала изомеризация 10°. Поэтому для деалкилирования фено-
лов стремились работать при еще более высоких температурах, не
придавая большого значения кислотным свойствам катализаторов.
Были испытаны 101 очень многие катализаторы (А12О3, алюмо-
силикаты с разным содержанием А12О3, окислы W, Mo, Сг, Ni, V,
Fe, Pt на А12О3), но выходы низших фенолов из высших были не-
велики. Таким образом, на пути негидрогенизационного превраще-
ния высших фенолов в низшие пока нет существенных успехов.
* См. в гл. 1 ссылки 26, 35, 36, 37, 71, 92, 97, 99, 101, 106, 116, 117,
126, 128, 129, 134, 136, 137, 140, 141, 142, 148, 150, 155, 170, 172, 173,
176, 184.
196
Гидрогенизация высших фенолов в отсутствие катализатора из-
учена много полнее. Термодинамика этого процесса была исследо-
вана 102, 103 на примере крезолов. Показано, что наиболее вероятным
продуктом является бензол, далее толуол, наименее вероятным —
фенол; наиболее реакционноспособным является орто-изомер, наи-
менее реакционноспособным — мета-изомер; теоретический выход
фенола из о-крезола 46% , из п-крезола 39% , из .м-крезола 33% (600 °C,
70 кгс/см2). Делались неоднократные попытки проводить высоко-
температурный процесс деалкилирования (70 кгс/см2, 600 °C) в при-
сутствии катализаторов. Лучшие результаты были получены на при-
родном каолините, активированном HF; выход фенола из техниче-
ских крезолов составил 104 13,1—14,0%. Позднее было показано,
что лучшим катализатором является смесь Fe2O3 и 7,3% окиси
хрома 105.
Простота условий гидрогенизации без катализатора позволила
наиболее подробно изучить механизм деструкции фенолов под высо-
ким давлением водорода 75. Было доказано, что' деструкция идет
по радикально-цепному механизму 1бв.
а) Зарождение цепи:
С6Н5-ОН — ► С6н5о-+н- 1
б) Рост цепи:
► С6н5.+Н2о 2
Н- + СвНБон - > С6Н6+НО. 3
—> R- + CO 4
свн5-+н3 — -> CgHe+H- 5
R • + Н3 —> R-H + H- 6
в) Обрыв цепи:
SCgHj- > С6Н5-СвН5 7
СвН5-+СвН5О. —> (СвН5)2О —> (СвН4)3О+Н3 8
Протекание реакций 4, 7, 8 было подтверждено идентификацией
в продуктах процесса окиси углерода, дифенила, дифенилового
эфира и окиси дифенилена 10в. Предполагают 781 10в, что протекание
реакций 2, 3 и 4 объясняется присоединением атомарного водорода
к двойной связи бензольного кольца с образованием неустойчивых
радикалов
Н ОН
С6Н5. + Н3О
СвНб + НО.,
ОН
он
/\/н
—> ( 1\н —> CO+R.
где R — углеводородные радикалы.
197
Подтверждением вероятности такого рода реакции является
аналогия ее кинетики с кинетикой гидрирования алкилбензо-
лов 107 — возрастание степени превращения с повышением давления
водорода (см. также стр. 117 и 181), а также образование 14СО из
СвН5ОН с 14С в положении 1 10в.
Промежуточное образование радикалов было показано также
в работах по гомогенной гидрогенизации гомологов фенола108-110.
По составу продуктов гидрогенизации крезолов и пропилфенолов
(были выделены не только фенол и бензол, но и толуол, этилбензол,
крезолы, зтилфенол) было предложено объяснять зарождение цепей
гомолитическим расщеплением наименее прочной связи.
В крезолах это связь С—Н в метильной группе
НО-СвЩ— СН3 —> НО— СвЩ—СН2 + Н-
а в пропилфенолах — углерод-углеродная связь:
НО—СвЩ—С3Н7 —> НО-СвЩ—СН2+СН3—СН2
Опыты с бутилфенолами110 также показали, что скорость де-
струкции определяется энергией наименее прочной связи (в бутил-
фенолах р- или у-связь). В гидрогенизатах о- и n-бутилфенолов были
обнаружены орто- и пара-изомеры крезола, а также этил- и пропил-
фенолов. Показано, что с ростом длины алкильного остатка растет
тенденция к его полному отрыву:
Дополнительным доказательством в пользу радикально-цепного
механизма процесса явилось торможение реакции при добавке окиси
азота 75. Представления о радикальном механизме имеют, по-види-
мому, общий характер. С их помощью, например, легко понять
результат одной из ранних работ Холла 1П, который из гидрогенизата
нафтолов выделил ди-Р-нафтиловый эфир, оба динафтиленоксида
и Р-динафтил (см. реакции 7, 8 на стр. 197).
Расщепление более сложных фенолов [например, п-(а, а-диметил-
бензил)-фенола 112] также хорошо объясняется (по 75) промежуточным
расщеплением С—С-связи.
Деструктивная гидрогенизация фенолов в присутствии промыш-
ленного плавающего железного катализатора была впервые изучена
в работах 1131 114. Было показано, что при гидрогенизации
(450 °C, 350 кгс/см2, 3 ч, 5% железного катализатора) количество
низших фенолов возрастает в 1,5 раза, а наиболее вероятным источ-
ником их образования являются высшие фенолы и кислые асфаль-
тены, хотя небольшое количество низших фенолов образуется и из
нейтральных веществ (см. стр. 173 сл.).
198
Аналогичные выводы можно сделать на основании данных мате-
риальных балансов гидрогенизации тяжелого остатка полукоксовой
смолы из гуачендзыского угля в крупной опытной установке (емкость
реактора 3,6 л) 115. Результаты трех пробегов, в том числе одного
с циркуляцией, приведены в табл. 40.
Таблица 40. Материальные балансы деструктивной гидрогенизации
тяжелого полукоксового сырья
Железный катализатор, 200 кгс/см2, 475—480 °C, объемная скорость
по свежему сырью 1,0 количество водорода 1200 — 13 00 й/кг свежего сырья
Компоненты Введено, вес. ед. Выведено, вес. ед.
I пробег II пробег III пробег (с циркуля- цией) I пробег П пробег III пробег (с циркуля- цией)
Жидкие продукты (всего) . . 100,0 100,0 133,9 79,9 80,3 101,4
в том числе:
остаток смолы 325 °C остаток гидрогенизата, выкипающий < 325 °C 100,0 100,0 65,0 — — —
— — 35,0 38,8 38,7 42,7
гидрогенизат, выкипа-
югций <325 °C .... — — 33,9 41,1 41,6 58,7
Водород 11,7 13,6 11,2 — — —
Вода — — — 3,1 2,6 2,7
Газы потери — — — 28,7 30,7 41,0
Итого ... 111,7 113,6 145,1 111,7 113,6 145,1
В составе жидких
продуктов:
Кислотные компоненты, вы- кипающие ^>325 °C ... . Кислотные компоненты, вы- 9,4 9,4 9,7' 4,0 5,1 5,0
кппающие <325 °C ... — — 4,6 5,4 5,4 8,5
в том числе:
низшие фенолы .... — — 2,4 2,9 2,8 4,6
фенол — — 0,40 0.48 0,45 0,75
о-крезол • •••«••• — — 0,24 0,30 0,27 0,42
л-крезол — — 0,51 0.61 0,58 1,01
п-крезол — — 0,26 0,31 0,30 0,43
ксиленолы — — 0,99 1,20 1,20 1,99
Из данных, приведенных в табл. 40, видно, что и в проточной
системе образуется значительное количество низших фенолов. Из
9,4% кислотных компонентов сырья образуется 5,4% фенолов,
содержащихся в широкой фракции. В то же время в тяжелой части
гидрогенизата остается 4,0—5,1% кислотных компонентов, т. е.
их общее количество не только не уменьшается^, а даже возрастает.
199
В гидрогенизатс(х, полученных как на проточной установке, так
и в автоклавах, главным компонентом крезольной фракции является
лт-крезол. Интересно сравнить соотношения изомерных крезолов
в сырье (полукоксовая смола из гуачендзыских углей), прямопро-
ходном гидрогенизате и гидрогенизате опыта с циркуляцией 115:
В сырье В гидрогенизате
I пробега III пробега
(с цирку-
ляцией)
о-Крезол : n-Крезол . . 0,73 0,95 0,98
.и-Крезол : n-Крезол . . 1,04 1,98 2,33
.и-Крезол : о-Крезол . . 1,42 2,08 2,38
Отношения количеств мета- и пара-, а также мета- и орто-изоме-
ров увеличиваются наиболее заметно, т. е. наиболее устойчив из них
лт-крезол, значительно менее устойчивы о- и п-крезолы.
Для выяснения основных химических закономерностей жидко-
фазного процесса были исследованы составы продуктов гидрогениза-
ции индивидуальных фенолов 118 (табл. 41).
Таблица 41. Продукты деструктивной гидрогенизации
индивидуальных фенолов
485 °C, 300 кгс/см1, 5% железного катализатора
Исходное вещество Выход продуктов, % от исходного вещества Отношение доли продуктов восстановле- ния к доле продуктов расщепления
неизме- нившееся вещество ней- тральные продукты кислые продукты расщеп- ления
Фенол 63,5 30,6
.ч-Крезол 44;7 33.7 11,5 2,9
о-Крезол 34,2 39,5 15,0 2,6
n-К резол 22,8 43,3 17,9 2,4
п-Пропилфенол * 13,8 19,0 28,5 0,77
3,5-Ксиленол 30,4 30,5 24,8 1,2
Тимол 0,0 26,3 44,6 0,6
я-Бензилфенол 0,0 56,4 23,7 2,4
п-Фенилфенол 52,5 20.3 0,9 22,5
а-Нафтол 1,7 87,6 6,0 14,6
0-Нафтол 7,2 83,8 4,7 17,8
Инданол-5 * 32,4 26,5 20,9 1,3
Тетрагидро-Р-нафтол * 18,3 39,0 15,9 2,0
Пирокатехин 1,6 45,2 21,8 —
Резорцин — 39,1 4,8 —
Гидрохинон 1,3 59,9 12,0 —
* Гидрирование этих фенолов проводилось при 475 °C и 250 кгс/см!, вследствие чего
скорости их восстановления и деструкции по сравнению с другими фенолами занижены.
Из полученных данных видно, что введение в фенол алкильных
заместителей и второй гидроксильной группы ускоряет и восстано-
вление, и деструкцию. Это ускорение тем интенсивнее, чем больше
200
число заместителей и чем выше их индуктивный эффект. Так, интен-
сивность восстановления увеличивается в рядах:
Фенол < л-Крезол < 3,5-Ксиленол
Фенол < n-Крезол ге-Пропилфенол
Фенол Крезолы Диоксибензолы
Интенсивность деструкции увеличивается в рядах:
л-Крезол < 3,5-Ксиленол < Тимол
ге-Крезол < ге-Пропилфенол
Влияние длины заместителя по аналогии с опытами гидрирования
без катализатора 108, 110 следует объяснить тем, что связи С—G
в алкильных заместителях расщепляются тем легче, чем больше
молекулярный вес алкильного остатка, давая начало цепям ра-
дикальных превращений. Однако восстановление ускоряется
и заместителями, не содержащими С—С-связей, — гидроксильными
группами, что показывает важную роль индуктивного эффекта. Это
следует объяснить тем, что заместители, обладающие положительным
индуктивным эффектом, повышая плотность электронов в кольце
фенола, облегчают присоединение атомарного водорода (см. реакции
на стр. 197).
Наоборот, при введении в молекулу фенола фенильного замести-
теля, не обладающего ни легко расщепляемыми связями, ни поло-
жительным индуктивным эффектом (случай фенияфенола), интен-
сивность восстановления несколько снижается.
С учетом индуктивного эффекта можно попытаться объяснить
и различие в реакционной способности крезолов. Вопрос этот из-
учался давно и, хотя данные отдельных авторов иногда противо-
речивы, в большинстве случаев отмечалась наименьшая реакционная
способность л-крезола (см. табл. 42).
Однако эта закономерность пока не была объяснена. Очевидно,
что на скорости деструкции и восстановления фенолов должна влиять
электронная Плотность связей углерода кольца и гидроксильной
и метильной групп. В случае л-крезола электронная плотность этих
связей должна быть выше, чем у его изомеров. В самом деле, в кре-
золах имеют место два сопряжения л-электронов кольца: с р-электро-
нами гидроксильной группы и с ст-электронами С—Н-связей ме-
тильной группы. Эти два сопряжения должны ослаблять друг друга,
так как в них участвуют одни и те же электроны кольца:
201
Таблица 42. Сопоставление интенсивностей различных реакций изомерных крезолов
Условия процесса Реакции Суммарное превращение Литература
катализатор температура, °C давление, кгс/см2 восстановле- ние деметилиро- вание гидрирование кольца
— 700 1 — п > о > Jt 117
— 800 1 м п — О о> п — 118
Ni 65—150 Повышенное — — >Н О У> п — 119
Мо8» 350 31 п м У> о — — •— 120
Ni 60 130-150 — — П^>М^>0 — 121
— 816 1 о > .и > п о^>гг^>м — О У> п м 95
— 600—650 70 — — — о^> п 96
Ni 60—80 10-110 — — п У> о > м 122
Жидкофазная гидрогенизация 475-485 300 п 3> о Л1 — п^>о^> м 116
— 490 100-330 0 > п м — о^> п^>м 108
— 650 30 — О П У> м — — 103
Термодинамический расчет — о^>п^>м __ — 102
Косвенным подтверждением того, что в jw-крезоле р—л- и ст—л-
сопряжения в наименьшей степени «мешают» друг другу, является
большая кислотность его по сравнению с орто- и пара-изомерамй,
а также разная скорость обмена водорода в феноле на дейтерий
в орто- и пара- и мета-положениях 1аз.
В табл. 43 приведены индивидуальные вещества, выделенные
из жидкофазных гидрогенизатов индивидуальных фенолов 11в. Дан-
ные о количествах выделенных низших фенолов не только иллю-
стрируют большую устойчивость мета-изомеров, но и позволяют
развить представления о деструкции как о термической реакции,
протекающей по радикальному механизму.
Таблица 43. Продукты гидрогенизации некоторых фенолов
(в % от исходного вещества)
Продукты гидрогенизации п-Пропил- фенол Тимол п-Бензилфе- нол а-Нафтол р-Нафтол Тетрагидро- р-нафтол Инданол-5
Фенол 15,9 11,7 16,6 0,5 1,5 0,9
о-К резол — — — 0,3 — — —
•м-К резол — 24,7 — 0,6 0,4 2,0 6,6
п-Крезол 4,6 — — — — 1,0 3,8
.и-Этилфенол — — — 0,8 1,3
п-Этилфенол 8,0 — — — 1,0 | 0,8
и-Пропилфенолы (м- и п-) . . — — — — — — 7,5
га-Изоиропилфенол 2,7 — — — — — —
Бутвлфенолы — — — — — 9,4 —
Р-Нафтол — — — —— — 1,8 —
Инданол-4 — — — . — — — 2,3
Метилинданолы — — — — — 5,9 —
Прежде всего следует отметить, что в продуктах гидрогенизации
исследованных алкилфенолов преобладают продукты расщепления
по связи, примыкающей к бензольному кольцу (a-связь): л-крезол
из тимола, фенол из пропилфенола и бензилфенола. Из бицикличе-
ских фенолов почти не образуется фенолов с двумя заместителями,
т. е. разрыв гидрированного кольца происходит по связи, при-
мыкающей к бензольному, причем легче с образованием мета-
изомеров.
Однако соотношение между алкилфенолами в гидрогенизатах
моноциклических и бициклических фенолов различно: в гидрогени-
затах первых преобладает фенол, в то время как в гидрогенизатах
нафтолов и тетрагидронафтола — зтилфенолы, а в гидрогенизате
инданола — крезолы. Если принять сумму образовавшихся
203
моноалкилфенолов за 100, то мольные доли каждого из них в гидро-
генизатах соответствующих фенолов составят:
НО
Фенол n-Крезол л-Этилфенол
^/^СЩ-СЩ-СНз
\/\/
а 0
НО
\/\/\
I II IV
\/\/
а р
В случае n-пропилфенола преобладает а-расщепление, в случае
инданола-5 — [3- и для тетрагидронафтола — у-расщепление. Объяс-
няется это тем, что в первом случае расщепляется сам п-пропил-
фенол (равно, как и тимол, и другие алкилфенолы), а в случае би-
циклических фенолов — радикал:
Н0\/\/\ Н2 \/\,/\ Н. Н0\/\-/\
III— I й I — I I
^/\/ \/|\/
II
Эти схемы аналогичны схеме расщепления тетралина (см. стр. 117).
В заключение следует отметить, что, судя по составу продуктов,
в данных условиях идут и реакции изомеризации (хотя и с незначи-
тельной интенсивностью): изомеризация гидрированного кольца
(наличие метилинданолов в гидрогенизате тетрагидро-Р-нафтола),
изомеризация боковой цепи (м-изопропилфенол в гидрогенизате
м-пропилфенола) и изменение положения гидроксильной группы
(инданол-4 в гидрогенизате инданола-5). Возможно, что перемещение
гидроксильной группы объясняется чередованием стадий разрыва
и замыкания кольца:
204
Другое вероятное объяснение этого типа изомеризации может
заключаться в прямой миграции гидроксильной группы. Возмож-
ность внутримолекулярной миграции заместителей доказана в случае
а-метилнафталина, изомеризующегося в 0-метилпафталин 124.
Недостаточная активность железного плавающего катализатора
побудила испытать для процессов деалкилирования высших фенолов
более активные стационарные катализаторы. Это направление в на-
стоящее время привлекает наибольшее внимание. При использова-
нии таких катализаторов давление и температуру понижали до таких
пределов, чтобы не восстанавливался целевой продукт — фенол.
Так, например, при гидрировании смолы, рбразующейся при синтезе
фенола кумольным методом, в составе которой содержится 2-(п-окси-
фенил)-2-фенилпропан, на катализаторе WS2 + NiS на А12О3 для
сохранения образующегося фенола (выход 23,5%) 125 температуру
понижали до 275 °C, а давление до 50 кгс/см2.
Однако сульфидные катализаторы оказались недостаточно селек-
тивными и значительно большее внимание привлекли окисные ката-
лизаторы. При деструкции фенолов на катализаторе Ni (8,4%) на
А12О3 при 350 °C было показано 12в, что легче всего отщепляются
алкильные остатки С4—С4 в орто-положении к гидроксильной
группе, а интенсивность деалкилирования возрастает с увеличением
числа углеродных атомов в алкильном остатке. Однако наряду
с деалкилированием весьма интенсивно протекало диспропорциони-
рование, а частично — восстановление.
Было показано 4, что на алюмокобальтмолибденовом катализа-
торе фенол при 325 °C и 10—60 кгс/см2 восстанавливается с малой
скоростью. Метилфенолы при 350—425 °C и 10—60 кгс/см2 легче
всего подвергаются диспропорционированию. Наиболее подвижны
метильные заместители в орто-положении, наименее подвижны —
в мета-положении, т. е. наблюдается та же закономерность, что
и в высокотемпературном процессе с железным катализатором или
без катализатора. Соотношение между различными возможными
направлениями превращений крезолов приведено 99 в табл. 44.
Кинетические константы реакций метилфенолов приведены
в табл. 45.
Таблица 44. Превращения крезолов в присутствии
алюмокобальтмолибдеиового катализатора при 350—400 °C и 10 кгс/см^
Крезолы Степень превра- щения, % Доли реакций (моль на 100 моль превра- щенного вещества)
изомери- зация диспропор- ционирова- ние деметили- рование восстанов- ление
о-Крезол 5-50 8 56 20 20
п-Крезол 10—50 17 46 10 31
л«-Крезол ......... 13-23 19 20 — 61
205
Таблица 45. Кинетические константы различных реакций крезолов в условиях гидрогенизации 89
Реакции Предэкспоненциа- льный множитель, С-1 Константа скорости ( х 10*)» с~* Энергия активации, ккал/моль
350 °C 375 °C 400 °C
Изомеризация 29,7
о-крезол-*-гс-крезол 6,3 Ю6 0,2 0,6 1,2
-и-крезо л ->о-крезо л — — 0,3 1,3 —
л«-крезол-*-п-крезол — — — 0,8 —
л-крезол-ки-крезол — — 0,25 2,8 —
Восстановление
о-крезол-*-толуол 4,0 • 107 0,6 2,1 3,8 30,5
.и-крезол-*-толуол .... 6,3 109 0,7 2,9 6,5 36,6
п-крезол-*-толуол 3,2-108 1,0 3,0 7,3 32,8
Деметилирование о-крезол-»-фенол 1,6 -108 0,8 2,7 5,2 32,0
n-крезо л -«-фено л —. — 0,03 2,8 —
Диспропорционирование 33,5
о-крезол-»-2,6-ксиленол 8 • 109 10 39 76
о-крезол-»-2,4-ксиленол 4 Ю9 5 18 38 33,5
о-крезол-*2,5-ксиленол 2 1О10 л • моль"1 2,4 л/моль-1 10 л/моль-1 22 л/моль-1 36,6
и-крезол-»-2,5-ксиленол 1 • 101* — 3,8 17 49
п-крезол-*2,4-ксиленол , 6-lOib 4,4 22 130 56
Восстановление ксиленолы-^-ксилолы (сумма) 3 Ю7 2 5 10 30,3
фенол-«-бензол i 5 • 106 1 2,1 4,1 26,5
С повышением давления скорости всех реакций, кроме реакции
восстановления, уменьшаются ". При добавлении к ксиленолам
фенола основной реакцией становится диспропорционирование с об-
разованием крезолов. Более высокую реакционную способность о-
. и n-крезолов в сравнении с ле-крезолом автор " объясняет большей
легкостью образования л-комплексов с катализатором. Интересно
отметить, что правило наибольшей устойчивости мета-изомеров
полностью сохраняется в ряду ксиленолов (табл. 46).
Таблица 46. Продукты гидрогенизации индивидуальных ксиленолов
алюмокобальтмолибденовый катализатор—30 объема. %, 425 °C, начальное давление
водорода в автоклаве 7 кгс/см4, продолжительность опыта 60 мин
Исходный ксиленол Степень превраще- . НИЯ, % Углеводо- роды, % Крезолы, % Изомерные ксиленолы, % Триметил- фенолы, %
2,3- 90,5 9,7 20,0 32,7 26,6
2,4- ' 83,8 10,6 19,9 23,7 27,7
2,5- 81,1 7,7 21,2 23,6 27,6
2,6- 82,0 8,6 20,4 27,5 24,3
3,4- 73,0 9,2 19,2 18,3 24,9
3,5- 42,4 11,2 10,4 9,0 10,0
В самом деле, наиболее устойчив 3,5-ксиленол, в котором все
заместители находятся по отношению друг к-другу в мета-поло-
жениях.
Эта же закономерность сохраняется в ряду этилфенолов
(n-этилфенол превращается в 5,8, а о-этилфенол в 8,2 раза быстрее
мета-изомера), а также в рядах высших алкилфенолов. Скорость
превращения возрастает при увеличении длины алкильного замести-
теля (от С3 до С4), а также при переходе от н- к втор- и трет-алктр-
фенолам.
Относительные скорости превращений пара- и орто-изомеров
пропил- и бутилфенолов приведены в табл. 47.
В заключение интересно сопоставить превращения фенолов с пре-
вращениями других кислых соединений — возможных компонентов
различных смол. Их простейшими моделями являются тиофенол
и бензойная кислота. Было показано, что бензойная кислота менее
устойчива, чем фенол. В качестве катализаторов использовали
МоО3 на угле 127, а также WS2 и железный катализатор при низкой
температуре 7°. В присутствии MoS2 тиофенол восстанавливается
в 28,5 раз быстрее фенола 120. Эти закономерности сохраняются при
использовании промышленного железного катализатора (см. стр. 165,
изменение группового состава сырья в ходе жидкофазной гидрогени-
зации). Опыты с индивидуальными соединениями дают тот же резуль-
тат: если фенол превращается при 300 кгс/ck2 И 485 °C на 36,5%
за 3 ч, то бензойная кислота превращается нацело, давая 62,3%
бензола, 26,4% толуола и 0,4% нафтеновых углеводородов
207
Таблица 47. Относительные скорости превращений изомеров пропил-
и бутилфенолов
Относительные скорости
Алкилфенол
суммарного
превращения
деалкили-
рования
Пара-изомеры
и-Пропилфенол ............................
Изопропилфенол ...........................
йтор-Бутилфенол...........................
mpem-Бутилфенол ..........................
Орто-изомеры
и-Пропплфенол ...........................
Изопропилфепол ..........................
йтор-Бутилфеиол ..........................
тпрет-Бутилфенол .........................
1
1
1.3
16
0,7
1
2,1
12
0,6
1
1,3
35
0,5
1
4
38
Тиофенол превращается настолько быстро, что уже при 300 °C реа-
гирует полностью за 30 мин, тогда как из фенола даже при 400 °C
в течение 1 ч образуется только 2,4% продуктов восстановления.
Изложенные выше данные о химии превращений фенолов в усло-
виях гидрогенизационных процессов послужили теоретической осно-
вой для многочисленных технологических разработок (см. гл. 1,
табл. 3), некоторые из которых уже реализованы. К ним относятся
процессы переработки высших фенолов буроугольной смолы 9в,
а также процессы переработки смол, получаемых при производстве
фенола кумольным методом ".
ПРЕВРАЩЕНИЯ АЗОТСОДЕРЖАЩИХ СОЕДИНЕНИЙ
Азотсодержащие соединения в гидрогенизационных процессах
играют весьма важную роль, так как даже небольшое количество их
снижает активность стационарных, особенно расщепляющих, ката-
лизаторов (см. гл. 1). Поэтому в первых ступенях процесса стреми-
лись возможно полнее разрушить азотсодержащие соединения, что
контролировалось по общему содержанию азота. Превращения этих
соединений изучались также в первую очередь с целью выяснения
условий их полной деструкции или моделирования превращений
аналогичных соединений, содержащихся в угле и смолах.
При рассмотрении процесса гидрирования азотсодержащих соеди-
нений в присутствии низкотемпературных катализаторов (см. гл. 3)
было показано, что гетероциклические соединения менее устойчивы,
чем соответствующие карбоциклические. Так, пиридин и пиррол
гидрируются, как правило, быстрее бензола, а если в соединении
есть и бензольное, и гетероциклическое кольцо, то в первую очередь
208
гидрируется последнее 8 °. Эти закономерности сохраняются и при
гидрировании на низкотемпературных катализаторах при высоком
давлении водорода. Так, например, в присутствии никеля Ренея 128
в хинолине и изохинолине гидрируется пиридиновое кольцо. Заме-
стители в пиридиновом кольце тормозят гидрирование и при низком
(1,1—4,5 кгс/см2, Pt) 60 и при высоком (начальное давление
95 кгс/см2, Ni Ренея) 129 давлении.
В высокотемпературных процессах, в том числе и в процессах
жидкофазной гидрогенизации, основной технологической целью
является подготовка сырья к последующим стадиям глубокой пере-
работки на стационарных катализаторах. А так как они весьма
чувствительны к азотсодержащим соединениям, то в жидкофазных
процессах стремились обеспечить максимальную деструкцию этих
соединений.
Поскольку прямая деструкция последних невозможна из-за
их высокой стабильности, удаление азота в гидрогенизационных
процессах обязательно должно включать стадию насыщения кольца.
Так, для пиридина предложена следующая схема деструкции 88:
//\ /\
I II —► I I —► CH3(CH2)4NH2 —► C5H12 + NH3
N NH
Эта схема подтверждена результатами кинетических исследова-
ний 13°. Концентрация пиперидина проходит через максимум, т. е.
гидрирование пиридина протекает значительно быстрее деструкции
пиперидина, а расщепление образовавшегося н-пентиламина идет
настолько быстро, что не может влиять на суммарную скорость.
Процесс проводили при 315 °C и 50—100 кгс/см2 в присутствии
осерненного катализатора — 2,25% Ni -(-1,25% Со 4-11% Мо
на А12О3.
Кинетика реакции отщепления азота описывается уравнением
первого порядка
0,89npHpN • 10'4
(!+«) (1 + 1,57Др)
где pjv — парциальное давление пиперидина, кгс/см2; л — общее
давление, кгс/см2; рн — парциальное давление водорода, кгс/см2;'
рР — парциальное давление пиридина, кгс/см2; So — объемная
скорость, ч-1; R — скорость подачи водорода, моль/моль жидкости.
При изучении кинетики деструктивной гидрогенизации некоторых
азотсодержащих гетероциклов и их гидропроизводных (372 °C,
17 кгс/см2) показано131, что превращения шестичленных гетеро-
циклов описываются уравнениями первого, а пятичленных — вто-
рого порядка.
Поскольку все превращения азотсодержащих соединений начи-
наются с гидрирования, целесообразно начать рассмотрение химии
деструктивной гидрогенизации этих соединений с динамики измене-
ния их состава в промышленном процессе. Уже при сопоставлении
14 Заказ 271 20»
составов полукоксовой смолы и ее гидрогенизата было показано,
что в ходе гидрогенизации меняется основность азотсодержащих
соединений. Более детальное изучение превращений одних классов
азотсодержащих соединений в другие и сопоставление их реакцион-
ной способности проведено в работе 132. Гидрогенизацию полукоксо-
вой смолы осуществляли на полупромышленной установке с реакто-
ром емкостью 3,6 л. Исследуемые пробы экстракцией разделялись
на фенолы, вещества, экстрагируемые 10%-ной H2SO4, вещества,
экстрагируемые 60%-ной H.2SO4, и нейтральное масло. Результаты
анализов и материальные балансы пробегов установки приведены
в табл. 48.
Таблица 48. Материальные балансы превращений азотсодержащих
соединений в процессе деструктивной гидрогенизации полукоксовой смолы
(кг на 100 кг фракций >325 °C)
200 кгс/см2, 480 СС, объемная скорость 1 ч-1, 5% железного катализатора
Показатели Введено Выведено
пробег 1 пробег 2 пробег 3 (с цир- куляцией) пробег 1 пробег 2 1 пробег з (с цир- куляцией)
Содержание азота 1,100 1,100 1,443 0,836 0,853 1,059
в том числе:
во фракциях > 325 °C 1,100 1,100 1,181 0,516 0.531 0.601
во фракциях < 325 °C — -—. 0,262 0,320 0,322 0,458
Соединения, экстрагируемые 10%-ной H2SO4 1,500 1,500 3,755 3,250 3,430 4,168
в том числе:
из фракций > 325 °C 1,500 1,500 2,165 1,320 1,520 1,238
из фракций 325 °C — — 1,590 1,930 1,910 2,930
Соединения, экстрагируемые 60%-ной H2SO4 0,500 0,500 0,668 0,401 0,526 0,597
в том числе:
из фракций >325 °C 0,500 0,500 0,465 0.155 0,194 0,128
из фракций <325 °C — — 0,203 0,246 0,332 0,469
Из данных табл. 48 видно, что хотя общее количество азота
уменьшается (в прямопроходных опытах на 23,4—23,9%, в опыте
с циркуляцией — на 26,6%) значительно более, чем в два раза,
количество соединений, экстрагируемых 10%-ной H2SO4, т. е.
азотистых оснований, увеличивается. Количество соединений, ха-
рактеризующихся слабоосновными свойствами (экстрагируются
60%-ной H2SO4), изменяется незначительно.
Увеличение количества азотистых оснований отмечалось
и ранее 133, 134. Для того чтобы установить, из каких классов азот-
содержащих соединений они образуются, был использован 132 метод
210
титрования перхлорной кислотой 136, фиксирующий только основной
азот. Сочетание этого метода с экстракцией может дать следующую
классификацию азотсодержащих соединений по их основности:
А —все соединения основного характера, т. е. титруемые
НС104;
Б — сильноосцовные соединения, экстрагируемые 10 %-ной H2SO4
(обычно пиридины, пиперидины, амины);
В — менее сильные основания, экстрагируемые 60 %-ной H2SO4
(обычно пирролы);
Г — соединения нейтрального характера, не титруемые НС1О4;
содержание азота в них определяется по разности между
общим и титруемым азотом.
Балансы распределения этих типов соединений (по содержанию
азота) в полукоксовой смоле и ее гидрогенизате приведены
в табл. 49.
Таблица 49. Балансы распределения азота между продуктами
деструктивной гидрогенизации полукоксовой смолы
Тип соединения Единица расчета 1 J\ft пробега Введено Выведено
во фракции <325 °C во фракции >325 “С восстановилось (по разности)
Соединения, титруе- мг азота на 1 кг сырья 1 6300 2060 2910 1330
мне НС1О4 (Л) 2 6300 1955 3057 1288
3 6283 2365 2329 1589
% к содержанию дан- 1 100 32,7 46,2 21,1
ного типа азота в 2 100 31,0 48,5 20,5
сырье 3 100 37,6 37,1 25,3
Соединения, экстра- мг азота на 1 кг сырья 1 746 1731 1005 —1990
гируемые 10%-ной 2 746 1625 1165 -2044
H2SO4 (Б) 3 2105 1855 958 -708
% к содержанию дан- 1 100 232 135 —267
ного типа азота в 2 100 218 156 —274
сырье 3 100 88 46 -34
Соединения, экстра- мг азота на 1 кг сырья 1 225 168 96 -39
гируемые 60%-ной 2 225 232 98 -105
H2SO4 (В) 3 274 240 51 —17
% к содержанию дан- 1 100 75 42 -17
ного типа азота в 2 100 103 44 -47
сырье 3 100 87 19 -6
Нейтральные' соеди- мг азота на 1 кг сырья' 1 4700 1154 2250 1296
нения (Г) 2 4700 1165 2245 1290
3 4504 1051 2169 1284
% к содержанию дан- 1 100 24i6 47,9 27,5
ного типа азота в 2 100 24,8 47,8 27,4
сырье 3 100 23,3 48,2 28,5
14*
211
Из приведенных данных видно, что содержание азота типа А
гораздо выше, чем типов Б и Б. В процессе гидрогенизации коли-
чество соединений типов А та Г уменьшается, а Б и В растет, т. е.
соединения слабоосновного и нейтрального характера приобретают
сильноосновные свойства. Было показано 132 также, что этот процесс
сопровождается уменьшением молекулярного веса слабоосновных
соединений и заканчивается довольно быстро.
К увеличению основности могут вести многие реакции, например
процессы изомеризации
процессы гидрирования пиридиновых колец в пиперидиновые и пир-
рольных в пирролидиновые, образование первичных или вторичных
алифатических аминов в результате расщепления пиперидиновых
и пирролидиновых колец, например:
И, наконец, можно предположить, что в процессе деструктивной
гидрогенизации происходит упрощение структуры азотсодержащих
соединений (устранение пространственных затруднений), что также
приводит к повышению основности, например:
Реакции изомеризации включают стадии дегидрирования и по-
этому маловероятны в условиях высокого давления водорода. Про-
текание реакций образования первичных и вторичных аминов дока-
зывается тем, что примерно половина оснований, экстрагируемых
10%-ной H2SO4, ацетилируется, т. е. представляет собой смесь
первичных и вторичных аминов 132. Однако эти реакции не могут
играть большой роли в явлении повышения основности азотсодержа-
щих соединений, так как производные пиридина и хинолина и до
гидрирования являются сильными основаниями, а содержание произ-
водных пиррола невелико (см. табл. 49).
Следовательно, рассматривая химию превращений индивидуаль-
ных азотсодержащих соединений, в числе других вопросов необхо-
димо осветить пути превращения слабоосновных азотсодержащих
соединений в сильноосновные.
212
Индивидуальные вещества, выделенные из продуктов деструк-
тивной гидрогенизации азотсодержащих соединений, приведены
в табл. 50.
Данные, приведенные в табл. 50, подтверждают для пиридина
предложенную выше (стр. 211) схему деструкции и в то же время
показывают, что основные реакции сопровождаются реакциями алки-
лирования, конденсации и, в небольшой степени и не всегда, изо-
меризации. Для пиридинов, в отличие от фенолов, нехарактерны
реакции деметилирования. Так, в продуктах гидрирования а-пико-
лина найдено лишь очень небольшое количество пиридина, а в гидро-
генизате 2-метилхинолина не удалось обнаружить хинолин141;
из 2-метилиндола получено только 3,1 % индола 148.
Образование N-циклопентилпиперидина и дипиперидилпентана
из пиперидина, циклогексиламйна и циклогексана из а-пиколина
указывает на радикальный характер этих процессов:
21Х
Таблица 50- Состав продуктов деструктивной гидрогенизации некоторых индивидуальных
азотсодержащих соединений
Исходное вещество Условия опыта Компоненты гидрогенизатов, присутствие которых Литература
катализатор температу- ра, °C давление, кгс/см2 доказано неполностью доказано предполагается
Пиридин Ni 200 200 Пиперидин, бутиламин, а-пиколин — Пиррол, (3- и у-пиколии 136
Ni на А12Оз 150 1 Пиперидин, н-амиламин, а-пиколин, 2-аминопири- дин, пиррол у-Пиколин Р-Пиколии, 2,2'-Дипирп- дил 137
Ni 200 1 Пиперидин, а-пиколин, 3-метилпиррол, 2-про- пилпиперидин, N-цикло- пентилпиперидин 138
Пиридин MoSa 280 100 (начальное) н-Пентан, циклопентен, циклопентан, N-метил-, N-этил-, N-пропил-, N-бутил-, N-амил- и N-циклопентилпипериди- ны, 1,5-дипиперидилпен- тан 139
Пиперидин Ni 200 200 (начальное) N-н-Бутил-, N-н-амил- и N-циклопентилпипериди- ны 2-Метплпиридин N-Метилпиридин, пир- рол, 1,5-дипиперидил- пентан 136
а-Пиколин Ni 200, 250 200 2-Метил- и N-амилпипери- дины н-Пентан, н-гек- сан, цикло пентан, метилциклопен- тан 140
» Ni 200 1 2-Метилпиперидин, 3-ме- тилпиррол — — 138
а-Пиколин Fe 460 2.30-260 Пирпдпп, 2-метилпипери-
дли, н-гексан, циклогек- сан
Хинолин MoS2 450 80 (начальное) —
» MoS2 210— 100—110 1,2,3,4-Тетрагидрохинолин
220
» Fe 460 230-260 1,2,3,4- и '5,6,7,8-тетрагид- рохинолины, о-толуидин, анилин, метил- этил- и пропилциклогексаны, то- луол, этил- и пропил- бензолы
2-Метилхи- нолин Fe 460 230-260 2-Метил-1,2,3,4-тетрагидро- хинолин, 2-метил-5,6,7,8- тетрагидрохинолин
1,2,3,4- Тетрагид- рохинолин MoS2 420— 450 80 (начальное) Бензол, толуол
» MoS2 500 100 (начальное) Этилбензол
215.
Цпклогексттл- амип — 141
Анилин, о-толу- идин, о-этил- анилин, о-пропил- анилин, цикло- гексан, метил-, этил- и пропил- циклогексаны Метил-и этил-о-толуиди- ны, метилиндол 143
— — 144
Индол, цикло- гексан, бензол — 145
— — 141
Этил-и пропил- бензолы Тетрагидроанилин, этилдигидроанилин, этил-и пропиланили- ны, циклоалканы (Се—С9), олефины 144
Пропиланилин, 1,4- дигидрокарб- азол Метил- и этилциклопен- таны, циклогексан, метил-, этил- и i про- пи лциклогексаны, 1 пропилбензол, метил- и этил анилины, метил- и этилциклогексилами- ны, циклогексиламин, дигидроиндол, метил- индол, метилдигидро- индол 146
hродолжение табл. SO
Исходное вещество Условия опыта Компоненты гидрогенизатов, присутствие которых Литература
катализатор температура, °C давление, кгс/см* доказано неполностью доказано предполагается
1,2,3,4- Тетрагид- рохинолин Fe 460 230—260 5,6,7,8-Т етр агидрохинолин (8,8%) — — 145
Изохино- лин Ni 450 80 (начальное) Пиридин, толуол, ксилол Николины, лу- тидины, индан 5,6,7,8-Тетрагпдроизохи- нолип, 2,3-лутидин, Р-пиколин, пиридин, о-ксилол, толуол, бен- зол 143
» Cu+Cr 180 120 1,2,3,4-Тетрагидроизохпно- лин — — 147
Индол ? 315 20 Дшидроиидол, фенилэтил- амин, циклогексилэтил- амин, 2-этилциклогексил- амин, октиламин, 2- этиланилип, 1,2,3,4-тет- рагидрохинолин, хино- лин, диметилхинолины Анилин, о-толуидпп, 2-ме- тилгидроиндол, индол, 3-метилиидол 1,3-Диметнл-2- этилиндол, 1-этил- пндол, 3-нро- ПИЛИИДОЛ, 3-ИЗО- пронилиндол, 2-т. /iem-бути ли н- ДОЛ 142
2-Метил- индол Fe 460 230—260 — 148
Карбазол MoS2 430— 440 100 (начальное) — Диметилдицик- лопентил Дициклогексил 149
(NH4)2- • MoS.j 450— 470 100 (начальное) Индол о-Толуидип, о-этилани- лин, диметилдицикло- пентил, гомологи ин- дола 149
Превращения хинолина много сложнее и в прошлом вызывали?
споры. Так, например, в продуктах гидрогенизации хинолина были:
найдены циклогексан и его гомологи 143, 144 14в. Считали, что они
образуются либо путем деструкции декагидрохинолина 14в, либо
через промежуточное образование октагидрохинолина. Между тем
в гидрогенизате не было доказано присутствие ни окта-, ни дека-
гидрохинолинов .
И. Б. Рапопортом в обзорной монографии 7 была предложена
следующая схема:
Гидропроизводные
t
В этой схеме предполагается либо гидрирование бензола и его
производных, что мало вероятно, либо образование декагидро-
хинолина.
Исследование продуктов гидрогенизации хинолина показало,,
что как в присутствии катализатора, так и в его отсутствие частично-
гидрируется бензольное кольцо хинолина с образованием 5,6,7,8-те-
трагидрохинолина. Рацее при использовании в качестве катализа-
торов соединений никеля, молибдена и вольфрама образование этого-
продукта не наблюдалось. Количество 5,6,7,8-тетрагидрохинолина
растет с увеличением продолжительности опыта, а в мягких усло-
виях (430 °C, без выдержки) хинолин превращается только
в 1,2,3,4-тетрагидрохинолин. Эти наблюдения дают возможность,
предположить, что 5,6,7,8-тетрагидрохинолин образуется за счет
изомеризации 1,2,3,4-тетрагидрохинолина. Прямой опыт гидрогени-
зации последнего подтвердил возможность такой изомеризации 144.
Характерно, что в этом гидрогенизате не был обнаружен
217
хинолин. Доказательство изомеризации 1,2,3,4-тетрагидрохино-
лина в 5,6,7,8-тетрагидрохинолин уточняет предложенную выше
схему превращении хинолина и объясняет присутствие небольшого
количества низших гомологов пиридина:
Образование в предлагаемой схеме -5,6,7,8-тетрагидрохинолина
не противоречит энергетической неравноценности пиридинового и хи-
нолинового колец. 5,6,7,8-Тетрагидрохинолин, видимо, быстро ги-
дрируется до декагидрохинолина, который затем расщепляется.
Поэтому количество гомологов циклогексана равно или даже не-
сколько больше количеств соответствующих гомологов бензола.
Другое направление превращения 5,6,7,8-тетрагидрохинолина —
образование гомологов пиридина — имеет подчиненное значение
и проходит в очень небольшой степени. Аналогично должна^ по дан-
ным работы 142, выглядеть схема превращений индола:
При использовании в условиях жидкофазного процесса желез-
ного катализатора маловероятны стадии гидрирования фенилэтил-
218
амина и этиланилина, но возможно, что гидрирование происходит
в момент образования радикалов, как это было рассмотрено выше
для случая нейтральных кислородсодержащих соединений и фенолов.
Кроме того, показано 148, что для гомологов индола возможна изо-
меризация — превращение 2-метилиндола в 3-метилиндол.
Все приведенные выше данные и схемы иллюстрируют, что основ-
ность азотсодержащих соединений повышается в ходе гидрогениза-
ции за счет накопления первичных и вторичных аминов. Однако эти
схемы не раскрывают механизма превращения в основания нейтраль-
ных соединений. Для попытки объяснения этого вопроса в табл. 51
сведены данные работ 132, 141> 145’ 148, характеризующие соотношение
между основными и нейтральными компонентами гидрогенизатов
азотсодержащих соединений различных типов.
Таблица 51. Состав продуктов деструктивной гидрогенизации
индивидуальных азотистых соединений
(вес. % от исходного вещества)
5% катализатора, 460 °C, 230-260 кгс/см’, выдержка 1 ч
Вещества, экстрагируемые
10%-ной HjSO,
Исходное вещество
а
а-Пиколин.......... . . . .
а-Пиколин а...............
Хинолин ..................
Хинолина .................
1,2,3,4-Тетрагпдрохииолин . .
2-Метилхинолин ...........
2-Стирилхинолин ......
Бензонитрил...............
Карбазол .................
2-Метилиндол .............
89,5
89,5
98
97,5
86
96
98
96
100
91
44,9
77,9
7,1
31,3
9,7
5,1
90
22,6
1,9
32,7
36,2
29,1
7,1'
5,4
1,2
8,8
21,7
58,0'
5.0*
0,8'
2,50
0,4
0,5
0,2
3,0
0,9
0,5
5,1
0,1
в — в отсутствие катализатора; б — включая продукты гидрирования; в — суммарное
содержание веществ, экстрагируемых 10%-яой H,S04; г — фракция 2-метилдигидроиндола.
Типичное нейтральное вещество — бензонитрил дает очень мало
соединений основного характера. Для нитрилов наиболее характерен
процесс восстановления с образованием углеводородов и аммиака.
То же имеет место и при низкотемпературном гидрировании 1В0,
и при высокотемпературном в присутствии сульфидных катализато-
ров 151. Из слабых оснований, не экстрагируемых 10%-ной H2SO4, —
карбазола, 2-метилиндола и 2-стирилхинолина — большое количе-
219
ство сильноосновных соединений дает только последний, значи-
тельно меньше их образуется за счет гидрирования пиррольного
кольца 2-метилиндола, а карбазол весьма устойчив в условиях
жидкофазного процесса.
Эти опыты подтверждают вывод о том, что основным путем об-
разования сильноосновных компонентов является деструкция
сложных соединений, .в которых высокомолекулярные заместители
понижают основность гетероциклического ядра или препятствуют
экстрагированию.
* *
*
В заключение можно отметить, что жидкофазное гидрирование
представляет сложный процесс, в котором протекают чередующиеся
и часто взаимосвязанные реакции гидрирования и расщепления,
алкилирования и деалкилирования, изомеризации положения за-
местителей, функциональных групп, водорода. Закономерности
насыщения ароматических карбо- и гетероциклических систем часто
объясняются большей или меньшей электронной плотностью арома-
тических связей за счет конденсации или введения гетероатома,
а закономерности изомеризации и расщепления — радикальным
механизмом этих реакций.
[ЛИТЕРАТУРА
1. Varga I., RaboG., Zalai A., Brennstoff-Chemie, 37, 244 (1956).
2. Варга И., БпртлерР. и др., Химия и технология топлив и масел,
№ 10, 11 (1960).
3. Г л у ш е и к о в а Е. В. В сб. «Труды ВНИИПС». Вып. 6. Л., Гостоп-
техиздат, 1958. См. с. 163.
4. Воль-Эпштейн А. Б., Жарова М. Н., Суровцева В. В.,
Хим. пром., № 2, 88 (1962).
5. Лозовой А. В., ДьдковаМ. К. Гидрогенизация топлива в СССР.
М. — Л., Изд. АН СССР, 1940. 269 с.
6. К г б n i g W. Die katalytische Druckhydrierung von Kohlen, Teeren und
Mineralolen. Berlin, Spinger Verlag, 1950, 266 S.
7. Рапопорт И. Б. Искусственное жидкое топливо. М., Гостоптех-
издат, 1955. 546 с.
8. Л о з о в о й А. В., С е н я в и н С. А. В сб. «Химическая переработка
топлив». М., Изд. АН СССР, 1957. См. с. 180.
9. V a n Meter R. А., В a i 1 е у С. Q. et al., Anal. Chem., 24, 1758 (1952).
10. Dinneen G. U., Ball J. S. et al., Ind. Eng. Chem., 44, 2632,
2647 (1952).
11. M c N e i 1 D., V a u g h e n G. A., Gas World, 138, 1385 (1953); 139,
137 (1954).
12. К а л e ч и ц И. В. Изв. ФХНИИ при Ирк. Гос. Унив., Вып. 1. Иркутск,
Обл. книжн. изд., 1953. См. с. 197.
13. Jager A., Kattwinkel G-, Brennstoff-Chemie, 37, 371, 372, 375
(1956).
14. 3 о и о в а 3. Т., Л а н и н В. А. Изв. АН СССР, ОТН, № 4, 170 (1957).
15. Григорьеве. М., 3 о и о в а 3. Т., Матвеева М. Д. Изв. АН
СССР, ОТН, сер. «Металлургия и топливо», № 1, 164 (1960).
16. К а г г С., Е s t е г Р. A. et al., U. S. Bur. Mines. Bull. № 637 (1967);
РЖХпм. 4П78 (1968).
220
17. Э йз ей О. Г., Р а н г С. А. Симпозиум ООН по разработке и исполь-
зованию горючих сланцев. Таллин. 1968. Препринт.
18. Lang К. F., Eigen I., Fortschr. Chem. Forsch., 7, № 1, 172 (1966);
8, № 1, 91 (1967).
19. К н я з e в a M. С., Ланин В. А. и др.. Изв. АН СССР, ОТН, № 4, 142
(1955).
20. Ланин В. А., П р о н и н а М. В., К н я з е в а М. С. В сб. «Хими-
ческая переработка топлив». М., Изд. АН СССР, 1957. См. с. 231.
21. Э д е м с к а я Н. Д. В сб. «Труды ИГИ АН СССР». Вып. 6. М., Изд.
АН СССР, 1955. См. с. 35.
22. Runge F., Meckelburg A., Freiberger Forschungshefte, А17, 54
(1953).
23. В u n g е F., Freitag J., Kolbe J., Chem. Ber., 87, 873 (1954).
24. KuhnhaussG., Rosner H. u. a., Erdol u. Kohle, 10, 372 (1957).
25; Gunter H., Kiihnhauss G., Huttig E., Chem. Techn. (Ber-
lin), 7, 656(1955).
26. Калечиц И. В., Попова Н. И., Салимгареева Ф. Г.
В сб. «Химическая переработка топлив». М., Изд. АН СССР, 1957. См.
с. 216.
27. Калечиц И. В., Салимгареева Ф. Г. и др. В сб. «Труды
ВСФ АН СССР». Вып. 3. М., Изд. АН СССР, 1955. См. с. 19, 25.
28. Сидоров Р. И., Троценко 3. П., X в о с т и к о в а А. А. В сб.
«Труды СО АН СССР». Вып. 18. М., Изд. АН СССР, 1959. См. с. 3, И.
29. К а л е ч и ц И. В., С и д о р о в Р. И. В сб. «Труды ВСФ СО АН СССР».
Вып. 18. М., Изд. АН СССР. 1959. См. с. 41; Металлургическая и хими-
ческая промышленности Казахстана, № 2, 76 (1959).
30. Князева М. С., Ланин В. А., Пронина М. В. Изв. АН СССР,
ОТН, № 4, 169 (1957).
31. О р о ч к о Д. И., Сильченко Е. И., Ш а в о л и н а Н. В. В сб.
«Труды ВНИГИ». Вып. 4. М., Гостоптехиздат, 1952. См. с. 97.
32. Л о з о в о й А. В., С е н я в и н С. А., ЖПХ, 18, 35, 43 (1945).
33. Калечиц И. В., Попова Н. И. Труды комиссии по аналитиче-
ской химии, VI/IX, 97 (1955). ‘
34. Эдельштейн Н. Г., Ланин В. А. Веб. «Труды ИГИ АН СССР».
Вып. 5. М., Изд. АН СССР, 1955. См. с. 144.
35. Попова Н. И., Курбан-Галеева Д. X. Веб. «Труды ВСФ
АН СССР». Вып. 3. М., Изд. АН СССР, 1955. См. с. 63.
36. К г u b е г О., G г i g о 1 е i t G., Chem. Вег., 87, 1895 (1954).
37. Сидоров P. И., Троценко 3. П., Н и к о л а е в а Д. X. В сб.
«Труды ВСФ СО АН СССР». Вып. 18. М., Изд. АН СССР, 1959. См. с. 32.
38. Сидоров Р. И., Хвостикова А. А. и др. В сб. «Труды ВСФ
СО АН СССР». Вып. 38. М., Изд. АН СССР, 1961. См. с. 95.
39. К у к Г. Л., Д ж е н с е н Г. Б., Д и и н и н Г. У.' Симпозиум ООН по
разработке и использованию горючих сланцев. Таллин. 1968. Препринт.
40. Зеленин Н. И., ФайнбергВ. С., Чернышева К. Б. Хи-
мия и технология сланцевой смолы. Л., «Химия», 1968. 308 с.
41. С е р г и е н к о С. Р., Итоги науки, 2, 199 (1958).
42. Hood A., Clark R. J., O’N е i 1 М. I., J. Inst. Petrol. (London),
45, 168 (1957).
43. П о п о в a H. И., Ш о р о х о в а М. В. В сб. «Труды ВСФ АН СССР».
Вып. 3. М., Изд. АН СССР, 1955. См. с. 40.
44. Окладникова 3. А., Нахман ович А. С., Шер г и н а Н. И.
В сб. «Труды ВСФ СО АН СССР». Вып. 26. М., Изд. АН СССР, 1959. См.,
с. 39.
45. Казаков Е. И., Л и и в Э. X. ЖПХ, 31, 1125 (1958).
46. Калечиц И. В., Окладникова 3. А. Веб. «Труды ВСФ СО
'АН СССР». Вып. 18. М., Изд. АН СССР, 1959. См. с. 49.
47. Charlet Е. М., Lanneou К. Р., Johnson F. В., Anal. Chem.,
26, 861 (1951).
48. RaboG., Szekely A., Acta Chim. Hung., 5, 453 (1955).
221
49. W i 1 s о n T. P., C a f 1 i s h E. О., H u г 1 e у G. F., J. Phys. Chem.,
62, 1059 (1958).
50. В о й т e x о в А. А., О p о ч к о Д. И. и др., ЖОХ, 38, 1963 (1968).
51. Miyazawa Т., Pitzer К., J. Am. Chem. Soc., 80, 60 (1958).
52. Fry е С. G., J. Chem. a. Eng. Data, 7, 592 (1962).
53. Bondi A., Petrol. Bef., 38, № 2, 161 (1959).
54. В о e n t e L. Brennstoff-Chemie, 36, 210 (1955).
55. Donath E. E., Adv. in Catalysis, 8, 239 (1956).
56. Самойлов С. M., Петрова В. H. Изв. СО АН СССР, № 9,
119 (1961).
57. Гаврилова А. Е., ГоникбергМ. Г. и др. Изв. АН СССР,
ОХН, 1958, 981.
58. Вей Шин-пин, Цзян Бин-нан ь. Веб. «Труды ВСФ СО АН
СССР». Вып. 38. М., Изд. АН СССР, 1961. См. с. 58.
59. К а л е ч и ц И. В., СалимгарееваФ. Г. и др. Изв. СО АН
СССР, № 1, 44 (1961).
60. Smith Н. A. In «Catalysis». Ed. by P. H. Emmett. V. 5. New York.
Beinhold Publ. Corp., 1957. P. 175.
61. C h a t e 1 u s G., C. r., 224, 201 (1946).
62. И п а т ь e в В. H., О p л о в H. A., Ber., 60, 1963 (1927).
63. Ипатьев В. H., Орлов Н. А., Лихачев Н. Д., ЖРФХО, 61,
1339 (1929).
64. Н а 1 1 О. С., С a w 1 е у С. М., J. Soc. Chem. Ind., 58, 7 (1939).
65. Bamberger E., Ber., 19, 1818 (1886).
66. Тиц-Скворцова И. П., Вести. МГУ. Сер. физ.-мат. и ест. наук,
№ 6, 81 (1954).
67. Ш у й к и н Н. И., Эриванская Л. А., Кузнецова Р. Е.,
Вести. МГУ, Сер. физ.-мат. и ест. наук, №. 3, 135 (1957).
68. Шуйкин Н. И., Эриванская Д. А., Ан А. В., Вести. МГУ,
Сер. физ.-мат. и ест. наук, № 5, 125 (1957).
69. Шуйкин Н. И., Эриванская Л. А., К о н ц о в а В. В., ЖОХ,
26, 2562 (1956).
70. К л и м о в Б. К., Б о г д а и о в И. Ф. В сб. «Труды ИГИ АН СССР».
Вып. 3. М., Изд. АН СССР, 1954. См. с. 151.
71. Калечиц И. В., СалимгарееваФ. Г. и др. Изв. СО АН СССР,
№ 4, 54 (1961).
72. Т р ж ц и и с к а я Б. В., Калечиц И. В., Кинетика и катализ, 6,
346 (1965).
73. G г а у Р., Williams A., Trans. Farad. Soc., 55, 775 (1959).
74. Веденеев В. И., ГурвичЛ. В. и др. Энергия разрыва химиче-
ских связей, потенциалы ионизации и сродство к электрону. М., Изд. АН
СССР, 1962. См. с. 136.
75. ГоникбергМ. Г. Химическое равновесие и скорость реакций при
высоких давлениях. М., «Химия», 1969. См. с. 370—378.
76. К а л е ч и ц И. В., Т р ж ц и н с к а я Б. В. и др. Изв. вузов. Химия
и хим. технол., 10, 530 (1967).
77. Кондратьев В. Н., Усп. хим., 24, 861 (1957).
78. Landa S., W е i s s е г О., Chem. listy, 50, 569 (1956); Collect. Cze-
chosl. Chem. Communs., 22, 93 (1957).
79. Молдавский Б. Д., Низовкина T. В., ЖОХ, 10, 653 (1940).
80. S h о п о Sb., I tabash i et al., Когё кагаку дзасси, 64, 1357 (1961); С.
А., 58, 4390 (1963).
81. Л о з о в о й А. В. В сб. «Труды ИГИ АН СССР». Вып. 9, М., Изд. АН
СССР, 1959. См. с. 148.
82. W е i s s е г О., Landa S., Р е с h к а К., Chem. Techn., 16, 463 (1964).
83. СалимгарееваФ. Г., Иванова М. Ф. и др., Изв. СО АН СССР,
№ 5, 115 (1961).
84. Patera Е., Freiberger Forschungshefte, А36, 21 (1955).
85. Шуйкин Н. И., Эриванская Л. А., Усп. хим., 29, 648 (1960).
86. CawleyC. М., Research (London), 1, 553 (1948).
222
87. Сабатье П. Катализ в органической химии. Л., Госхимтехиздат,
1932. См. с. 109.
,38. Ипатьев В. И., ЖРХО, 38, 89 (1906).
89. Schaefer Н. Symposium uber hydrokatalytische Prozesse in der Erdol-
verarbeitung und Petrolchemie. Leuna. B. 3. 1966. S. 106.
90. Молдавский Б. Л., Лифшиц С. E., Ж0Х, 3, 603 (1933)-.
91. Алексеева К. А., Молдавский Б. Л., Химия и технология
топлива, № 1, 43 (1959).
92. Dahlke В., Guenther G., Chem. Techn. (Berlin), 14, lf>0 (1962).
•93. Кузнецов М., Белов К., Химия твердого топлива, 4, 592 (193Й).
94. Рапопорт И. В., М а с и н а М. П. и др., Химия твердого топлива,
7, 545, 694 (1936).
95. JonesB. М., Neuworth Н. В., Ind. Eng. Chem., 44, 2872 (1952).
"96. Kubicka R., Freiberger Forschungshefte, A36, 95 (1955).
97. Ворожцов H. H., Лисицын В. H. и др., Кокс и химия, № 11,
42 (1958).
98. Дьякова М. К., Воль-Эпштейн А. Б. и др., ЖПХ, 32, 2120,
(1959).
99. Воль-Эпштейн А. Б., Юлин М. К. и др., Нефтехимия, 7,
609 (1967).
100. Kraus М., В a z a n t V., Coll. Czech. Chem. Comm., 28, 1877 (1963).
101. BerberJ. S., Little L. R., U. S. Bur. Mineg, Report. Investig.,
№ 6585 (1965); C. A., 62, 8896 (1965).
102. J e 1 i n e k J., Chem. prumysl., 7, 4 (1957).
103. Wells G. L., Long R., Ind. Eng. Chem., Proc. Design a. Develop.,
1, 79 (1962).
104. J elinek J., Chem. prumysl., 12, 4 (1961).
105. J elinek J., Coll. Czech. Chem. Comm., 28, 504 (1963).
106. Г о н и к б e p г M. Г., Ли Гуан-нянь, ДАН СССР, 120, 1259
(1958).
107. ГоникбергМ. Г., НикитенковВ. Е., Изв. АН СССР. ОХН,
1954. 936.
108. ГоникбергМ. Г., Ли Гуан-нянь, ДАН СССР, 130, 763
(1960); Изв. АН СССР, ОХН, 498 (1960).
109. ГоникбергМ. Г., Ли Гуан-нянь, Изв. АН СССР, ОХН,
1961, 491.
ПО. Г а в р и л о в а А. Е., Г о н и к б е р г М. Г., Изв. АН СССР, ОХН,
1961, 1961.
111. HaUC. С., Fuel, 12, 419 (1933).
112. П л а т е А. Ф., К и к о т ь Г. С., ЖОХ, 32, 1828 (1962).
113. Калечиц И. В., СалимгарееваФ. Г. В сб. «Труды ВСФ
АН СССР». Вып. 3. М., Изд. АН СССР, 1955. См. с. 79.
114. Калечиц И. В., СалимгарееваФ. Г. В сб. «Труды ВСФ
АН СССР». Вып. 4. М., Изд. АН СССР, 1956. См. с. 5.
115. Sih Tsu-wei, Kalechits I. V., Acta focalia Sinica, 2, 333 (1957).
116. Калечиц И. В., Си Цзу-вей, СалимгарееваФ. Г. В со.
«Труды ВСФ СО АН СССР». Вып. 26. М., Изд. ДН СССР, 1959. См. с. 45.
117. Nakai R., Bull. Chem. Soc. Japan, 5, 136 (1930).
118. К os ak a Y., J. Soc. Chem. Ind. Japan, 34, 10 (1931).
119. Yamamoto K., J. Soc. Chem. Ind. Japan, 60, 451 (1959).
120. Бобышев В. E., Дьякова M. К., Лозовой А. В., ЖПХ,
13, 942 (1940).
121. U n g n a de Н. Е., Nightingall D. V., J. Ащ. Chem. Soc.,
66, 1218 (1944).
122. Tsutsumi Sh., Akatsuka H., Sakamoto Y., Techn. Repts.
Osaka Univ., 6, J63 (1956); C. A., 51, 8678 (1957).
123. Шатенштейн А. И., Веденеев А. В., Алиханов П. П.,
ЖОХ, 28, 2638 (1958).
124. Ворожцов H. Н., Коптюг В. А., Хим. наука и пром., 4, 409 (1959).
223
125. В о л ь - Э п ш т е й н А. Б., Жарова М. Н., Суровцева В. В.
В сб. «Труды ИГИ АН СССР». Вып. 17. М., Изд. АН СССР, 1962. См. с. 262.
126. Beranek L., Kraus М., BazantV., Coll. Czech. Chem. Comm.,
29, 239 (1964).
127. CawleyC. M., Fuel, 12, 366 (1933).
128. H i b i n о О., К i n о s h i t a K. at al., Когё кагаку дзассп, 68,
1703 (1965); С. А., 64, 3472 (1966).
129. М а г и о k а М., Ниппон кагаку дзасси., 82, 1257 (1961); С. А., 58, 11324
(1963).
130. Mcllvried Н. G., Ind. Eng. Chem. Proc. Design Develop., 10, 125
(1971).
131. Сох К. E., Berg L., Chem. Eng. Progr., 58, № 12, 54 (1962).
132. Yeh Tsu-hang, Kalechits I. V., Acta focalia Sinica, 2, 341 (1957).
133. Дариев А. Д. Кандидатская диссертация. ЛенНИИ. 1955.
134. Birthler R., Szkibik Ch., Freiberger Forschungshefte, A36, 42
(1955).
135. Moore R. T., McCutebau P.,. Young D. A., Anal. Chem.,
23, 1639 (1951).
136. J ones J. S., .J. Chem. Soc., 1950, 1392.
137. Шуйкин H. И., Тулупов В. А., Вести. МГУ. Сер. физ.-мат.
и ест. наук, 2, 73 (1956).
138. Шуйкин Н. И., Брусникина В. М., Изв. АН СССР, ОХН,
1959, 1288.
139. L a n d a S., К a f k a Z. et al., Coll. Czech. Chem. Comm., 34, 3588 (1969).
140. J ones J. J., Lindsey A. S., J. Chem. Soc., 1952, 3261.
141. Yeh Tsu-hang, Kalechits I. V., Acta focalia Sinica, 4, 59 (1959).
142. H a r t 1 u n g G. K., J e w e 1 1 D. M. et al., J. Chem. Eng. Data, 6,
477 (1961).
143. J amaguchi Sh., Bull. Chem. Ind. Japan, 9, 303 (1934).
144. Рапопорт И. Б., ЖПХ, 8, 1457 (1936).
145. Yeh Tsu-hang, К al e ch i ts I. V., Acta focalia Sinica, 3, 305 (1958).
146. E p у И. И., С a x н о в с к а я E. М., ПычкоВ. А., ЖОХ, 8,
1563 (1938).
147. -Cromwell N. Н., Cram D. I., J. Am. Chem. Soc., 65, 301 (1943j.
148. Yeh Tsu-hang, Kalechits I. V., Acta focalia Sinica, 4, 134 (1959).
149. Орлов H. А., И p о к о п e ц E. И., Еру И. И., ЖПХ, 4, 831 (1931).
150. Metayer К., Ann. chim< [12], 4, 196 (1949); С. А., 44, 3922 (1950).
151. I tab ash i К., Юки госей кагаку кёкайси, 17, 330 (1959); РХЖим,
51972 (1960).
ГЛАВА 5
ПАРОФАЗНАЯ ГИДРОГЕНИЗАЦИЯ
Стационарные катализаторы на основе сульфидов или окисей
переходных металлов гораздо более активны, чем плавающие желез-
ные катализаторы. Поэтому важнейшей тенденцией технологических
исследований последних десятилетий была замена плавающих ката-
лизаторов более активными стационарными катализаторами. Есте-
ственно, что параллельно развивалось исследование самих катали-
заторов и их усовершенствование. В этих направлениях можно
отметить две основные линии — стремление максимально повысить
стойкость стационарных катализаторов к ядам, чтобы обеспечить
длительный срок их работы, и разработка регенерируемых катали-
заторов^ И в том и в другом направлении достигнуты большие
успехи.
При переработке нефтяных фракций протекают не только реак-
ции собственно гидрирования, но и различные реакции изомеризации
и расщепления. Большею частью они связаны между собой общ-
ностью механизма или общими промежуточными продуктами. Между
тем в технологических целях важно обеспечить высокую селектив-
ность катализаторов: в одних случаях они должны интенсивно гидри-
ровать полициклические ароматические углеводороды и сохранять
моноциклические (производство бензинов), в других — обеспечивать
глубокую изомеризацию (производство низкозастывающих дизель-
ных и реактивных топлив), в третьих — сохранять углеродный
скелет сырья без изомеризации и т. д. Очевидно, что для обеспечения
селективности процесса нужно создавать катализаторы с различ-
ными сочетаниями гидрирующей, изомеризующей и расщепляющей
активностей. Первым шагом на пути разработки таких катализаторов
должно было явиться изучение химии превращений различных
классов углеводородов в присутствии промышленных катализаторов.
ГИДРОГЕНИЗАЦИЯ БЕНЗОЛА
Уже в первых работах с высокотемпературными сульфидными
катализаторами было показано, что при деструктивной гидрогени-
зации ароматических углеводородов образуются не только цикло-
гексаны, но и циклопентаны и парафины х. Протекание реакций
изомеризации и расщепления было установлено в опытах гидрогени-
зации бензола 21 3 и циклогексана 4~ *. Однако точный состав жидких
продуктов не был установлен: доказано присутствие только метил-
15 Заказ 271 215
циклопентана 2-41 6, циклопентана и изопентана 3. Позднее анало-
гичные исследования были проведены другими авторами 7-1 °. Наи-
более полого были исследованы 9 продукты деструктивной гидро-
генизации бензола в присутствии МоО3 при 550 °C и 250 кгс/см2.
При этом были выделены циклогексан, метилциклопентан, пентан
и 2-метилбутан; кроме того, вероятно присутствие гексана, 2-метил-
пентана и циклопентана.
Представления о химизме процесса, предложенные на основании
этих работ, были противоречивы. Предполагали314, что изогексаны
образуются из метилциклопентана, а н-гексан из циклогексана,
тогда как в работах111 12 предполагалась дециклизация только
метилциклопентана. Однако позднее вновь предположили10 воз-
можность образования гексана в процессе деструктивной гидро-
генизации путем расщепления как метилциклопентана, так и ци-
клогексана. Эта точка зрения была, видимо, принята и в последнее
время приводилась в обзорных работах 13-15. Все эти данные отно-
сятся к молибденовым (сульфидным и окисным) катализаторам.
Для более часто применяемого катализатора WS2 сообщалось 16
только, что главным продуктом деструктивной гидрогенизации бен-
зола является метилциклопентан.
На ранних стадиях оставалось также невыясненным место раз-
рыва кольца метилциклопентана 81 9, так как точное соотношение
между изомерными гексанами установить не удавалось 81 9. Между
тем точные данные о составе и количественном соотношении продук-
тов изомеризации и расщепления в ходе гидрогенизации бензола
позволили бы раскрыть особенность этих реакций и сопоставить их
течение с механизмом мягкого гидрогенолиза.
Гидрогенолиз циклопентанового кольца был изучен Б. А. Ка-
занским и сотр. 17, которые подробно исследовали превращения
циклопентана и его гомологов в условиях дегидрогенизационного
катализа. В этих работах установлены основные закономерности
зависимости места раскрытия кольца от строения исходных цикло-
пентанов, определены вероятности (в %) разрыва различных связей 17:
бД^б \/ 5 Н 14 | 14
33| |33 37| |37 42| |5 45| |14
22 26 42 ~14
Из приведенных данных видно, что преобладает разрыв в 0-пол о-
жении по отношению к третичному атому углерода.
Эта закономерность соблюдается также при гидрогенизации
углеводородов Св на различных высокотемпературных катализато-
рах 18~22. Полученные результаты приведены в табл. 52.
Как видно из данных табл. 52, составы гидрогенизатов отли-
чаются друг от друга лишь количественно, качественно они практи-
чески состоят из одних и тех же углеводородов. Это свидетельствует
об общности механизмов гидрогенизации бензольного" кольца в при-
сутствии различных катализаторов. Только в гидрогенизатах, полу-
ченных в присутствии катализатора WS2 на терране, найдены
226
Таблица 52. Составы гидрогенизатов некоторых углеводородов Св начальное давление 140 кгс/см*, продолжительность реакции 3 ч
сл Я Состав гидрогенизата, %
Исходный углеводород Катализатор Температура, °C Выход гидрогени; та, % бензол циклогексан метил циклопен- тан циклопентан гексаны пентаны другие идентифицированные углеводороды
Бензол Ni на А1а03 WSa » MoSa » » WSa+NiS на А1аО3 То же » WSa на терране 420 420 450 500 420 450 500 420 450 500 420 79,9 95,0 93,0 80,0 99,0 88,0 74,0 99,0 93,0 79,0 85,7 76.8 32,5 23,1 49,3 54,6 48,6 72,6 28,6 23,3 27,2 74,8* 16,8 25,5 21,5 10,8 29,1 25,6 10,3 60,3 65,8 32,3 1,2 3,9 37,5 47,7 32,0 13,2 22,6 10,3 9,8 10,2 33,0 13,0 1.4 0,5 5,0 0,4 2,3 6,1 0,1 5,5 0,5 0,7 3.8 6,1 2,0 2,7 0,6 0,5 0,8 0,7 1,4 2,7 0,4 0,7 1,1 0,9 0,3 0,2 0,4 0,6 3,2 2-Метилбутан, пентан 2-Метилпентан 2-Метилпентан 2-Метилпентан Толуол, дифенил, циклоалканы С? и выше (4,6%)
Цикло- гексан WSa на терране То же 420 450 84,0 73,4 — 34,8 12*3 51,2 44,6 0,7 1,5 8,9 14,4 4,4 16,9 2-Метилбутан, 2- и 3-метилпентаны 2-Метилбутан, пентан, 2- и 3-мети л пентаны, циклоалканы Су и выше (10,3%)
Мотил цик- лопентан WSs на терране То же 420 450 77,0 52,0 — 12,1 1,5 75,1 34,5 0,5 1,3 8,8 18,8 3,5 32,5 2-Метилбуган, пентан, 2- и 3-метилпентаны 2-Метилбутан, пентан, 2- и 3-метилпентаны, циклоалканы С7 и выше (12,1%)
Гексан WSa на терране 420 84,0 — — — — 94,0 6,0 2-Метилпентан (5,8%), 3-метилпентан (4,6%), гексан (83,6%), изопентан (5,1%), пентан (0,9%)
* В^том'числе 6,9% толуола и ксилолов.
продукты большего молекулярного веса (толуол, дифенил, цикло-
алканы С7 и выше).. Это отличие объясняется, очевидно,
значительно большей расщепляющей активностью данного катали-
затора при очень небольшой гидрирующей активности, вследствие
чего образующиеся осколки молекул успевают рекомбинироваться.
Аналогичные результаты были получены 23 при гидрогенизации
циклогексана, метилциклопентана и алканов на катализаторе Pt
на А12О3 в жестких условиях (460 °C, 15—20 кгс/см2). Повышение
температуры, естественно, сказывается на увеличении доли продук-
тов изомеризации и расщепления. Эти продукты образуются в замет-
ных количествах даже в присутствии никельглиноземного катализа-
тора Зелинского, обычно применяемого в качестве неизомеризующего
низкотемпературного катализатора.
Важно отметить, что из двух возможных путей расщепления
метилциклопентана — деметилирования с образованием циклопен-
тана и раскрытия кольца с образованием гексанов — при низких
температурах преобладает второй, а при высоких (500 °C) — пер-
вый 231 24. Деметилирование более характерно для термического
процесса или для процессов, протекающих в присутствии катализа-
торов, но при высоких температурах и малых давлениях 231 24.
Раскрытие кольца более характерно для мягких условий. Зако-
номерность направленности раскрытия циклопентанового кольца17
сохраняется полностью. В самом деле, во всех гидрогенизатах
во фракции гексанов преобладает 2-метилпентан. Если принять
его количество за единицу, то количества 3-метилпентана варьируют
от 0,26 до 0,70, а количества н-гексана — от 0,30 до 0,61. Аналогич-
ное преобладание 2-метилпентана в продуктах расщепления бензола
и циклогексана отмечено и для других высокотемпературных катали-
заторов 25. Так, например, при гидрогенизации бензола в присут-
ствии катализатора W + Сг + Zn S F на алюмосиликате было
выделено 4,1% циклогексана, 31,2% метилциклопентана, 7,9%
2-метилпентана, 3,8% н-гексана, 1,5% 3-метилпентана, а также
продукты расщепления (Сх — С5) и, частично, алкилирования
(С7 - С8).
Если бы в соответствии с выводами работ 31 11 образование изо-
гексанов происходило за счет изомеризации н-гексана, получа-
ющегося из циклогексана
| || -> I I ---> w-C(jHu -> иао-С8Н14
V
то в гидрогенизате w-гексана было бы обнаружено значительно боль-
шее количество изогексанов, чем приведено в табл. 52.
При гидрогенизации бензола на WS2 и циклогексана на катали-
заторе WS2 на терране не отмечалось 261 27 образования заметных
количеств н-гексана, а когда его доля становилась заметной (повы-
шение объемных скоростей от 0,5 до 6,0 ч-1), содержание 2-метил-
228
пентана было все же значительно выше. Следовательно, более пра-
вильной была бы следующая схема:
СбН14
Было найдено 26, что изменения концентраций бензола, цикло-
гексана и метил циклопентана в процессе гидрогенизации прибли-
женно описываются уравнениями первого порядка. В работе 28
при использовании катализатора WS2 порядок по бензолу составлял
0,45, а по водороду — около 1,5, а в случае MoS2 порядок по бензолу
был близок к нулевому, а по водороду — близок к первому. При
изучении кинетики гидрирования бензола в присутствии MbS2
в проточной установке найдено 29, что наиболее совпадающие резуль-
таты дает уравнение первого порядка по бензолу и нулевого по
водороду, если мольное отношение водорода к бензолу не меньше
трех.
Однако точные кинетические Исследования заставляют пред-
положить, что гидрогенизация как на сильно изомеризующих суль-
фидных катализаторах, так и на низкотемпературных катализаторах
протекает более сложно. В табл. 53 приведены соотношения про-
дуктов гидрогенолиза и энергии активации при гидрогенизации
метил- и этилциклопентанов на платинированном угле 30.
Таблица 53. Соотношение продуктов гидрогенолиза и энергия активации
при гидрогенизации метил- и этилциклопентанов
Состав гидрогенизата Соотно- шение продуктов Еакт’ ккал/моль Состав гидрогенизата Соотно- шение продуктов Е .акт’ ккал/моль
Для метилциклопеитана Для эти л циклопентана х,
м-Гексан .... 0,1 39,4 м-Гептан 0,3 41,6
2-Метцлпентан . . 1,0 37,0 З-Метилгексан . . 1,0 38,9
З-Метилпентан . . 0,8 36,1 З-Этилпентан . . 0,9 39,4
На основании этих данных был сделан вывод30, что лимитиру-
ющей стадией является образование' промежуточного комплекса
на поверхности катализатора. Одновременно было показано 31, что
образование трех изомерных гексанов из метилциклопентана про-
текает много быстрее, чем изомеризация каждого из них в два других
гексана, что также дало возможность предположить образование
общего промежуточного комплекса при реакции гидрогенолиза.
Кроме весьма вероятного образования общих промежуточных
продуктов схема механизма деструктивной гидрогенизации должна
учитывать отмеченную ранее 18, 32 особенность этого процесса —
образование изомеризованных углеводородов в количествах, пре-
вышающих термодинамически равновесные. Зависимость содержания
229
изобутана в бутановой фракции гидрогенизата среднего масла от
температуры (в сравнении с термодинамически равновесным) пока-
зана на рис. 15.
Повышение температуры должно ускорять реакции изомеризации,
поэтому увеличение содержания н-бутана с ростом температуры
может быть интерпретировано как ускорение реакции изомеризации
изобутан -> н-бутан, т. е. изобутан первоначально образуется в сверх-
равновесных количествах, а затем часть его (и тем большая, чем выше
температура) изомеризуется в н-бутан. Сверхравновесное образова-
ние изомеризованных (точнее, содержащих третичные углеродные
атомы) углеводородов является общей закономерностью 1в' 22, 25, 32
и, следовательно, должно учитываться схемой механизма.
Поскольку реакции изомеризации углеродного скелета про-
текают по ионным механизмам и концентрация изоалканов часто
Рис. 15. Содержание
изобутана в продуктах
деструктивной гидроге-
низации среднего масла
на катализаторе WS2 на
терране:
1 — бутановая фракция гид-
-рогенизата; 2 — термодина-
мически равновесная смесь
н-бутана и изобутана.
превышает равновесную, образование изоалканов нужно связывать
с промежуточным образованием наиболее устойчивых третичных
карбониевых ионов. Цепи ионных превращений инициируются
присоединением протонов к олефинам. Следовательно, реакции
изомеризации должны быть реакциями дегидрогидрирования, что
подтверждается 33 снижением скорости реакций изомеризации с ро-
стом давления, особенно если давление создается водородом, тормо-
зящим стадию дегидрирования.
Таким образом, схема превращений бензола в условиях гидро-
генизации должна учитывать и кажущуюся последовательность
превращений компонентов, и сверхравновесное образование метил-
щиклопентана, и возможность образования общих промежуточных
продуктов гидрирования, изомеризации и расщепления. Предпола-
гают34, что общими промежуточными продуктами являются цикло-
гексен и метилциклопентен.
Изучение механизма превращений бензола, циклогексана и ме-
тилциклопентана с помощью меченых атомов (гидрогенизации под-
вергались смеси этих веществ, в которых один из компонентов ме-
тился радиоактивным углеродом 38-37) показало (см. табл. 54), что
в начальный период действительно происходит накопление цикло-
гексана, но при длительном контакте его количество уменьшается.
Однако если меченым компонентом является бензол, удельная
радиоактивность метилциклопентана выше активности циклогексана
230
Таблица 54. Деструктивная гидрогенизация смесей углеводородов
катализатор WS., 42 0 °C, 350 кгс/см1
Номер опыта Исходный меченый компонент Время контакта, ч Состав исходной смеси, % Совтав гидрогенизата, % Удельная радиоактивность, % от радиоактивности исходного меченого компонента Доля суммарной радиоактивности, %
с.н. с.н,, С.Н.СН. С.Н. С.н,. С.Н.СН. парафины С, —С, С.Н,, С.Н.СН, парафины С,—С. С.Н,. Я о Я 1? парафины Ci—С»
1 CgHg 3 52,3 47,7 2,3 34,2 45,6 15,9 f 43 50 66 24,7 39,5 18,3
2* » — 49,8. 50,2 — 32,7 56,0 10,6 0,7 20 65 85 19,1 11,9 0,9
3* » ——. 50,2 49,8 — 33,1 57,3 8,5 1,1 9 49 62 9,9 7,4 1,2
4* » г— 33,9 66,1 — 22,3 71,0 6,4 0,3 6 46 47 11,4 7,9 0,4
5 1,5 — 50,1 49,9 36,67 55,3 7,1 93 25 47 68,3 26,8 6,3
6* » — 49,9 50,1 ——. 25.5 65,15 8,2 1,2 69 48 54 77,7 6,8 1,1
7* » — 50,2 49,8 — 32,5 58,35 8,1 1,0 79 34 36 90,7 5,5 0,7
8 с5н9сн3 0,5 48,75 48,80 2,45 24,3 58,1 14,8 2,8 0,5 16 14 11,8 89,1 14,7
9 » 0,5 36,6 55,0 8,4 17,4 63,45 16,3 2,8 0,6 45 28 4,6 88,0 9,2
10 » 1,5 — 57,5 42,5 — 36,30 56,9 6,8 7 62 52 5,7 77,6 7,5
11 * свн10 ** 44,3 43,7 — 29,6. 59,9 8,8 1,7 17 16 13 82,2 Ю,7 1,8
12* » — 43,9 44,2 39,2 57,1 0 0 15 0 0 71,2 0 0
* В этих опытах автоклав охлаждался сразу после достижения рабочей температуры.
** В опытах 11 и 12 кроме бензола и циклогексана вводилось 12,0—11,9% меченого циклогексена. В опыте И циклогексен превра-
тился полностью, в опыте 12 (без катализатора) в гидрогенизате осталось 3.7% циклогексена.
и. составляет примерно от половины до двух третей активности
бензола.
Следовательно, схема 1 (стр. 229) не отражает действительного
хода процесса, и большая часть метилциклопентана образуется
непосредственно из бензола, минуя стадию десорбции с катализатора.
В опытах, где меченым компонентом был циклогексан, только 5,5—
6,8% радиоактивности перешло в метилциклопентан, а удельная
радиоактивность метилциклопентана составила меньше половины
активности исходного циклогексана. Это хорошо согласуется с ре-
зультатами опытов, в которых меченым компонентом был бензол,
и подтверждает схему 2, согласно которой метилциклопентан может
образоваться как из бензола (реакции 1—4), так и из циклогексана
(реакции 5—4):
I JJ ^11 2
\/\сН? Ч'/\СН8
При длительном времени контакта удельные радиоактивности
циклогексана и метилциклопентана почти выравниваются, но доля
суммарной радиоактивности, перешедшей в метилциклопентан,
больше. Эти результаты можно интерпретировать следующим обра-
зом: при большой глубине превращения доля бензола в реагирующей
смеси уменьшается и соответственно в большей степени начинает
превращаться циклогексан. Вследствие этого основным источником
образования метилциклопентана становится немеченый цикло-
гексан; новые порции метилциклопентана разбавляют образова-
вшиеся ранее, которые имели более высокую по сравнению с цикло-
гексаном радиоактивность. Таким образом, эти данные обосновывают
кажущуюся последовательность процесса.
Результаты, полученные при введении в реакционную смесь
меченого циклогексана, в известной степени подтверждают роль
циклогексена как общего промежуточного продукта реакций гидри-
рования и изомеризации. Правда, из-за высокой реакционной способ-
ности циклогексен успевает гидрироваться уже в начальной стадии
опыта (более 80% радиоактивности попадает в циклогексан). Однако
в отсутствие катализатора почти треть циклогексена остается не-
измененной, на основании чего можно сделать вывод, что в присут-
ствии катализатора по крайней мере треть циклогексена превраща-
лась каталитическим путем. В этом случае 10,7% радиоактивности
перешло в метилциклопентан.
На основании материальных балансов и балансов распределения
радиоактивности можно вычислить мольные доли углеводородов,
превращающихся по отдельным реакциям.
232
Если упростить схему 2
то можно вычислить мольные доли х, у, z и и, составив необходимое
количество уравнений, например: 1) х -|- У = а, где а — экспери-
ментально определяемое количество превратившегося бензола, моль;
2) = Ь, где b — экспериментально определяемое отношение долей
радиоактивности, перешедшей в циклогексан и в метилциклопентан
в опытах с меченым бензолом; 3) z = с, где с — экспериментально
определяемая доля радиоактивности, перешедшая в метилцикло-
пентан и продукты расщепления Cj — Св в опытах с меченым цикло-
гексаном; 4) х — z = d, где d — экспериментально определяемое
количество вновь образовавшегося циклогексана, моль; 5) у -|-
z — и = е, где е — экспериментально определяемое количество
образовавшегося метилциклопентана, моль.
Подобные же уравнения можно составить и решить в случае
смесей бензола, циклогексана и метилциклопентана, определив ско-
рости их изомеризации друг в друга и скорости расщепления. Резуль-
таты расчетов приведены в табл. 55.
Таблица 55. Мольные доли веществ, превращающихся
в реакциях процесса деструктивной гидрогенизации бензола
Номер опыта * Р адиоактивный компонент X У 2 W ** U
2 С6нв 0,239 0,149 0,106 — 0,015
3 » 0,218 0,176 0,047 — 0,023
4 » 0,129 0,090 0,030 — 0,005
5 СбН12 — — 0,327 0,053 0,070
6 » 0,454 0,134 0,080 —. 0,028
7 » 0,270 0,134 0,067 — 0,021
9 С6НцСН3 — — 0,241 0,052 0,086
* См. табл. 54.
** Реакция изомеризации исходного метилциклопептана в циклогексан.
Из соотношения удельных радиоактивностей циклогексана, ме-
тилциклопентана и продуктов расщепления можно сделать вывод,
что реакции изомеризации и расщепления связаны между собой
общими промежуточными продуктами так же, как и реакции гидри-
рования и изомеризации. Связи между удельными радиоактивно-
стями циклогексана ' и продуктов расщепления не наблюдается,
233
а между удельными радиоактивностями метилциклопентана и про-
дуктов расщепления прямая зависимость существует только в тех
случаях, когда метилциклопентан образовывался вновь, т. е. его
не было в исходной смеси. Подобная-зависимость может означать,
что продукты расщепления образуются преимущественно из метил-
циклопентана или же, что более вероятно, метилциклопентан и про-
дукты расщепления образуются через один и тот же промежуточный
продукт. В опытах, в которых метилциклопентан был компонентом
исходной смеси (опыты 5, 8, 9, 10), не наблюдалась пропорциональ-
ность радиоактивности метилциклопентана и продуктов расщепле-
ния, поэтому первое предположение должно быть исключено.
Невероятность промежуточного образования метилциклопентана
и его десорбции в объем следует также из сопоставления распре-
деления полной и удельных радиоактивностей продуктов гидрогени-
зации смесей циклогексана и метилциклопентана (опыты 5, 10).
Если исходным радиоактивным компонентом был циклогексан, сум-
марная активность метилциклопентана больше, чем для продуктов
расщепления, но удельная активность последних выше; если исход-
ным радиоактивным компонентом был метилциклопентан, суммарная
активность продуктов расщепления больше, чем активность цикло-
гексана, но удельная радиоактивность их была меньше
активности метилциклопентана. Если -бы процесс превращения
циклогексана складывался из ступеней: адсорбция — превращение
в метилциклопентан — десорбция — адсорбция метилциклопен-
тана — превращение в продукты расщепления — десорбция, то де-
сорбируемый метилциклопентан ' смешивался бы с находящимся
в объеме непрореагировавшим метилциклопентаном, что привело бы
к уменьшению активности смеси. Следовательно, продукты расщепле-
ния образуются не только из метилциклопентана, но и из цикло-
гексана (без выхода в объем промежуточных продуктов).
Согласно современным представлениям о реакциях изомеризации
и расщепления как об ионных реакциях, общим промежуточным
продуктом при образовании из циклогексана как метилциклопен-
тана, так и продуктов расщепления должен быть карбониевый ион.
Наиболее вероятно, что он является метилциклопентилкарбониевым
ионом. Этот ион энергетически более выгоден, чем циклогексил-
карбониевый ион, поэтому равновесие
должно быть сдвинуто в сторону циклопентановой структуры сильнее,
чем термодинамическое равновесие метилциклопентан # циклогексан
при высоких температурах. Если стабилизация ионов протекает
быстро, а это должно происходить в условиях высоких давлений
водорода за счет реакций типа
В+ + Н2 —> RH + H+
234
то легко объяснить, почему количество метилциклопентана в гидро-
генизатах часто превышает равновесное. Аналогично объясняется
также преобладание парафиновых углеводородов С4 — Се с третич-
ными углеродными атомами (а не с четвертичными, количества кото-
рых ничтожны), концентрация которых превышает термодинами-
чески равновесную.
Таким образом, применение меченых атомов позволяет разгра-
ничить отдельные реакции, составляющие сложный процесс деструк-
тивной гидрогенизации, сравнить их скорости, получить представле- '
ние о характере взаимосвязи этих реакций.
. Результаты опытов показывают, что быстрее всего идет гидри-
рование бензола, затем гидроизомеризация, изомеризация и наи-
более медленно — расщепление.
Процесс деструктивной гидрогенизации бензола может быть
представлен следующей схемой
где в скобках указаны превращения, протекающие на поверхности
катализатора. Схема хорошо согласуется с перечисленными выше
особенностями процесса в целом.
Представления о взаимосвязи реакций гидрирования, изомери-
зации и расщепления имеют, по-видимому, и общее значение. При
изучении кинетики гидрогенолиза метилциклопентана и бензола
в условиях платформинга было показано 38' зя, что изобутан, изо-
пентан и 2-метилпентан образуются в количествах, превышающих
равновесные. Давление водорода увеличивало скорость расщепления,
что авторы связывали с увеличением концентрации протонов 38.
Поэтому был предложен механизм, включающий равновесное обра-
зование циклических гидрированных промежуточных продуктов,
235
которые затем подвергаются реакции раскрытия кольца — стадии,
определяющей суммарную скорость процесса 39.
Позднее выводы о взаимосвязи реакций гидрирования и изомери-
зации, а также о циклогексене как общем промежуточном продукте
этих реакций были сделаны также на основании чисто кинетических
измерений40: одинаковый выход метилциклопентана достигался
при пропускании над бифункциональным катализатором Pt на
SiO2 + А12О3 циклогексена со скоростью 2,4 моль/ч, бензола со
скоростью 1,91 моль/ч и циклогексана со скоростью 0,56 моль/ч.
Для реакций, протекающих в процессе платформинга, была
предложена 41 следующая схема:
Эта схема предполагает наличие в катализаторе платформинга
двух типов активных центров [дегидрирования -Ди изомериза-
ции — И (кислотный)] и миграцию реагирующего метилциклопен-
тана от центра Д к центру И и снова к центру Д. Иногда эту схему
распространяют на все катализаторы, обладающие свойствами уско-
рять как реакции гидрирования и дегидрирования, так и реакции
изомеризации (см., например, обзор 42). Однако наличие двух родов
активной поверхности в одном катализаторе вряд ли является рас-
пространенным явлением43 и такие представления подвергались
справедливой критике. Тем более невероятно наличие двух центров
в катализаторе без носителя (WS2). Схема на стр. 235 предпола-
гает, что все превращения, отмеченные в скобках, идут на одной и
той же активной поверхности катализатора. Это доказывается экспе-
риментально получением метилциклопентана из бензола, минуя про-
межуточное образование циклогексана и десорбцию с этой активной
поверхности.
ГИДРОГЕНИЗАЦИЯ НЕАРОМАТИЧЕСКПХ УГЛЕВОДОРОДОВ
Многие закономерности превращений циклоалканов, циклоалке-
нов и алифатических углеводородов выяснены при изучении пре-
вращений ароматических углеводородов, рассмотренных выше.
Однако, поскольку разработаны промышленные процессы гидро-
генизационной переработки неароматического (в том числе алифати-
ческого) сырья, представляется целесообразным обобщить резуль-
таты работ, посвященных изучению химии и механизма таких про-
цессов.
Цели этих процессов различны: очистка олефинов от примесей
диенов, увеличение содержания изопарафинов (для повышения
236
октановых чисел) в крекинг-бензинах и прямогонных фракциях
С5—Св, понижение молекулярного веса парафинистого сырья иногда
, с максимальной (для производства высокоиндексных масел), а иногда
с минимальной (для целей нефтехимической переработки) изомери-
зацией углеродного скелета.
Как и в низкотемпературных процессах (см. гл. 3, стр. 149),
разница в скоростях гидрирования олефинов и диенов очень велика.
Так, например, отношение констант скоростей гидрирования сопря-
женных диенов и олефинов 44 на катализаторе WS2 + NiS в интер-
вале температур 205—290 °C при давлении 3—14 кгс/см2 составляет
от 103 при 205 °C до 65,7 при 290 °C. Поэтому технологическое осу-
ществление процессов гидроочистки олефинов от диенов достигается
относительно легко.
Представление об изомеризации как о процессе дегидрогидриро-
вания и ионной реакции является в настоящее время общепринятым;
соответствующие равновесия можно представить48 следующим
образом:
н-П 4224' н-0 ;—иао-0 4722' изо-П
или
н-П ;—н-0 47224 н-П+ 47774 иао-П+ 47224 изо-0 ; изо-П
где П — парафин, О — олефин, П+ — карбониевый ион.
Допускается и прямое образование карбониевого иона из пара-
фина (типа реакции Фриделя — Крафтса) или более сложная схема
(с двумя маршрутами) 46:
н-П 42224 н-П+ 4724 иао-П+ 42224 изо-П
н-0 иао-0
н-П изо-П
Во всех случаях изомеризация олефинов протекает много легче,
чем изомеризация парафинов, и процессы гидрорзомеризации про-
дуктов крекинга, в которых олефины прямо превращаются в изо-
парафины, разработаны во многих вариантах 46, 47. Подбор темпе-
ратур и давлений в основном определяется свойствами катализато-
ров 48~52. На сильно изомеризующих (кислотных) катализаторах
происходит не только изомеризация парафина, но и его расщепление
с образованием изоолефинов, что приводит к снижению выхода
и, видимо, к снижению активности катализатора из-за повышения
концентрации олефинов.
Скорости изомеризации растут с повышением молекулярного веса:
если скорость изомеризации пентана принять за единицу, то скорости
изомеризации гексана, гептана и октана при 340 °C составят 81 2,1,
3,1 и 4,2, а скорости изомеризации гексана и гептана при 360 °C —
U 237
1,9 и 2,9. Скорости расщепления также растут с увеличением молеку-
лярного веса. Относительные скорости разрыва различных связей
в н-гексане и н-гептане и на типично ионном катализаторе (Pt на
алюмосиликате) и типично радикальном катализаторе (Pt на окиси
алюминия) приведены 53 в табл. 56.
Таблица 56. Относительные скорости разрыва связей
в н-гексане и н-гептане
Углеводород Связи Относительная скорость разрыва на катали- заторе
Pt на Al2Os+aiO, Pt на А12О1
н-Гексан Ci—с2 1.2 ±0,2 1,7
С2—с3 1 1
С3 С4 2,9 ± 0,4 2,1
и-Гептан Ci—с2 1 1
с2—с3 1,9 ±0,6 1,2 ±0,1
С3 С4 - 9,6 ± 2,6 1,7 ±0,1
Из приведенных данных видно, что в ионном процессе скорость
разрыва резко возрастает при переходе от гексана к гептану. По-
этому, если нужно избежать нежелательного расщепления, необ-
ходимо регулировать активность (кислотность) катализатора и темпе-
ратуру процесса.
Так, при получении высокоиндексных масел из высокомолеку-
лярных парафинов 54 хорошие результаты (умеренное расщепление
при глубокой изомеризации) были получены на катализаторе Pt
на А12О3. Для получения из высокомолекулярных парафинов более
ценных низших парафинов, выкипающих при 180—230 и 230—320 °C
(сырье для окисления), оказался выгодным 52 выбор высокой темпе-
ратуры и активного дегидрогидрирующего катализатора — Pt на
цеолите 13Х. При низких температурах и на менее активных (по рас-
щеплению) катализаторах успевала, по мнению авторов, пройти
скелетная изомеризация. Следовательно, скелетная изомеризация
должна быть (по Б2) если не лимитирующей, то довольно медленной
стадией. При изучении изомеризации пентана на катализаторе Pt
на А12О3 при 375—425 °C показано 4Б, что суммарная скорость про-
цесса не зависит от общего давления, т. е. лимитирующей стадией
не является стадия дегидрирования, но зависит от добавок хлора,
что прямо указывает на роль кислотности катализатора. Авторы 45
считают поэтому лимитирующей стадию изомеризации, но выделить,
какой из актов — адсорбция, собственно изомеризация или десорб-
ция — является самым медленным, невозможно: стадия ионного
равновесия н-П + изо-П+ должна быть быстрой. Очевидно, что
в стадии изомеризации лимитирующими должны быть, скорее всего,
другие элементарные акты. Во многих случаях лимитирующей ста-
дией является образование олефинов, что доказывается тормозящим
влиянием водорода 33, в том числе и в опытах5Б, моделирующих
238
промышленный процесс изомеризации гексана на WS2 при 330—
400 °C и 30—108 кгс/см2.
Изомеризация неразветвленных цепей протекает, как правило,
неглубоко, образование углеводородов с четвертичными углерод-
ными атомами или с несколькими разветвлениями отстает от термо-
динамического равновесия и в заметной степени наблюдается только
при глубоком превращении. Это легко объясняется положениями
карбониево-ионной теории. Так, например, при изомеризации гек-
сана образование 2- и 3-метилпентанов энергетически выгодно,
так как в конечном счете вторичный карбониевый ион заменяется
третичным:
С-С—С-С-С
I
С
п
с-с-с-с-с-с с-с-с-с-с-с С-С—С-С-С
с
It
+l + +
с-с-с-с с-с-с—с-с
I I
с-с с
Образование 2,3-диметилбутана и, особенно, 2,2-диметилбутана
не дает выигрыша энергии, так как при этом третичный ион превра-
щается в третичный же или первичный:
С
+ I
С-С-С-С-С С-С-С-С
I I
С с+
It
о
с-с—с-с-с С-С-С-С с-с-с-с
I 'll II
с с с с с
В соответствии с этим 2- и 3-метилпентаны образуются быстро
и так же быстро устанавливается равновесие между ними, в то время
как 2,2- и 2,3-димети лбу таны образуются из них медленно58. Даже
в оптимальных условиях (см., например4в) концентрация 2,2-
и 2,3-диметилбутанов ниже равновесной. Для повышения выхода
этих, наиболее ценных, компонентов фракций Св предлагается51
отбирать из продуктов изомеризации низкокипящуЮ часть, возвра-
щая погоны, обогащенные метилпентанами, в цикл. Таким способом
из гексана получено 28,8% 2,2- и 16,7% 2,3-диметилбутанов и только
27,5% метилпентанов 61.
239
ГИДРОГЕНИЗАЦИЯ ГОМОЛОГОВ БЕНЗОЛА
И ЦИКЛОГЕКСАНА
В ранних работах было установлено, что при деструктивной гидро-
генизации толуола, метилциклогексанах, этилбензола57 и гекса-
гидромезитилена 58 в присутствии MoS2 протекают процессы изо-
меризации, деалкилирования и раскрытия колец, но индивидуальных
углеводородов было выделено мало (пентан, бензол, метилцикло-
гексан, концентраты 1,2- и 1,3-диметилциклопентанов). При гидро-
генизации на никелевых, платиновых и палладиевых катализаторах
при высоких температурах (460 °C) и небольшом давлении идут
сложные радикальные реакции, приводящие к образованию мети-
леновых радикалов, а также к метилированию, деметилированию
и изомеризации 231 24, 691 60.
При изучении химии парофазной гидрогенизации на промыш-
ленных сульфидных катализаторах наибольший интерес предста-
вляет выяснение влияния заместителей на скорость и направление
отдельных реакций процесса — реакций гидрирования, изомериза-
ции и расщепления. Влияние алкильных заместителей на скорость
гидрирования бензольного кольца рассмотрено выше (см. гл. 3,
стр. 140сл.). О влиянии заместителей на интенсивность реакций изо-
меризации и расщепления можно судить-по данным табл. 57.
Изомеризация — ионная реакция, но расщепление при темпера-
туре 420 °C и выше может протекать не только как ионная, но и как
радикальная реакция. Для оценки интенсивности протекания этих
реакций целесообразно выделить продукты деметилирования — ти-
пично радикальной реакции — и продукты раскрытия кольца —
реакции, чаще протекающей по ионному механизму. На скорость
деметилирования больше всего влияет температура, что видно по
выходам нафтенов С6 и Св, достигающих значительных величин
при 500 °C.
Выходы продуктов изомеризации и расщепления при деструктив-
ной гидрогенизации некоторых ароматических углеводородов и наф-
тенов приведены в табл. 58, 59.
Из данных, приведенных в табл. 58 и 59, видно, что введение
заместителей ускоряет и изомеризацию, и расщепление. Это, оче-
видно, нужно связывать с появлением одного или двух третичных
карбониевых ионов, что ускоряет передачу и зарождение цепей:
—> ГУ+вн
Можно также отметить, что процессы расщепления протекают
сложно: доли продуктов «чистого» раскрытия кольца и «чистого»
деметилирования невелики по сравнению с общим выходом продук-
тов расщепления. Количество продуктов деметилирования в
2'<0
16 Заказ 271
Таблица 57. Составы гидрогеиизатов гомологов бензола и циклогексана 20, ei-e«
начальное давление 140 кгс/см1, время реакции 3 ч
Исходный углеводород Катализатор Температура, °C Выход жидкого гидрогенизата, % Состав гидрогенизата, %
ароматические углеводороды циклогексаны циклопентаны парафины
С. с, С» а> п и S с, с, с, С. с. с, « ® и s С» с. с, С.
Толуол ws2 420 94 20,4 0,1 20,7 . 0,1 0,6 46,1 4,4 1,1 6,5
» 450 94 — 28,3 — — 0,4 23,6 — 0,2 4.5 35,5 — 4,9 1,1 1,5 —
500 72 23,8 56,3 — — 1,2 1,4 — 1,9 4,5 3,5 — 6,3 0,4 0,7 —
WS24-NiS на А12О3 420 99 — 5,6 — — 1,3 79,7 — — 0,7 12,3 — — 0,1 0,3 —
» То же 450 97 — 12,8 — — 1,1 73,5 — 0,3 0,6 10,9 — 0,4 0,3 0,1 —
» 500 66 4,9 32,3 — — 7,4 12,3 — 8,0 21,9 5,7 —- 5,6 0,4 1,5
» ws2 на терране 420 90,6 11,9 66,1 3,9 2,1 — — — 0,2 2,0“ 12,6е — 1,0 0,2 — —
о-Ксилол ws2 420 93 — — 6,4 — 0,3 1,2 15,7 0,1 1,9 3,8 55,0 3,8 0,2 1,9 9,7
.м-Ксилол Этилбензол » ws2 на терране 420 420 92 86,6 11,0 2,4 5,8 58,8е 15,0 0,7 12,8® 1,8 14,0 0,2 4,2 7,5 45,1 4,9 1,9 1,2 12,7
Метилцикло- гексан То же 420 81,0 — 2,6 — — 1,8 254 — 0,1 5,0 48,6 — 5,7 2,8 8,3 —
То же » 450 60,8 — 1,0 2,2 — 0,7 8,7 2,4 0,9 9,0 39,8 — 15,9 10,1 9,3 —
Этилциклогек- » 420 62,6 — — — — 0,5 1,3 23,3е 1,1 3,8 8,0 30,4 9,4 6,9 3,2 12,1
сан
1,2-Диметил- » 420 68,7 — — — — 0,7 1,2 15,8 0,6 1,6 5,0 24,9 30,1 8,3 2,5 9,3
циклогексан
1,3-Диметил- циклогексан » 420 69,7 — — — — — 1,2 18,5 1,2 3,4 4,6 32,8 14,4 8,1 3,1 12,7
а—включая циклогексаны; б—включая циклогексаны и парафины; в—в том числе 1,9% ксилолов; г—суммарное содержание цикло-
гексанов, циклопентанов и парафинов; д—в том числе 13,3% неизмененного этилциканалогекс.
Таблица 58. Сравнение интенсивности реакций изомеризации
и расщепления при деструктивной гидрогенизации
ароматических углеводородов при 420 °C
Исходный углеводород Катализатор Отношение количества продуктов расщепления и изомеризации к количеству продуктов гидрирования Выход продуктов изомеризации, % Выход продуктов расщепления, %
суммарный продукты 1 раскрытия гидрированного кольца продукты деметилирования
Бензол ws2 1,64 37,5 9,2 3,6
» WS2 + N1S на А12О3 0,18 9,8 2,3 0,8 0,1
Толуол ws2 2,62 46,1 18,0 6,1 0,8
WS2 + NiS на А12О3 0,19 12,3 3,4 0,3 2,0
л-Ксилол WS2 4,67 45,1 40,3 11,7 13,2
о-Ксплол » 4,43 55,0 28,2 9,1 6,7
Таблица 59. Сравненце интенсивностей реакций изомеризации
и расщепления при деструктивной гидрогенизации нафтенов Ce—С3
420 °C, катализатор WS, на терране
Исходный углеводород Количество ! неизмененного вещества, % Отношение количества продуктов расщепления и изомери- зации к количеству шестичлен- ных нафтенов Выход продуктов изомеризации, % Выход продуктов расщепления, %
суммарный продукты раскрытия кольца продукты деметилиро- вания
Циклогексан 29,2 1,87 43,1 27,7 7,5 0,6
Метилциклогексан 20,4 2,72 39,4 40,2 6,7 5,6
Этилциклогексан 14,6 3,06 19,4 66,0 7,6 9,3
1,3-Диметилцик логексан 12,9 4,08 22,8 64,3 8,9 7,3
1,2-Диметплциклогексан 10,7 4,58 17,1 72,2 6,4 6,3
большинстве случаев меньше, чем количество продуктов раскрытия
кольца. Это обстоятельство, а также симбатность изменения интен-
сивностей изомеризации и расщепления различных углеводородов
указывают на то, что преобладающим типом расщепления является
раскрытие пентаметиленового кольца, образовавшегося за счет
изомеризации гексаметиленового кольца. Этим и следует объяснить
высокий выход низкомолекулярных продуктов расщепления, потому
что после раскрытия кольца легко образуются характерные для ион-
ного расщепления осколки С3 цзо-С4.
Для катализаторов, содержащих WS2 (табл. 58, 59), характерно
ионное течение реакций расщепления. Разница между ионным и ра-
242
дикальным расщеплением будет отчетливой, если сравнить суль-
фидный и окисный катализаторы на примере превращения
этилдиклогексана в присутствии катализаторов WS2 на терране 88
и МоО3 на А12О3 66 (см. табл. 60).
Выходы продуктов превращения при этом составили (в %):
Суммарный выход . .
Продукты изомеризации
Продукты ароматизации
Продукты расщепления
WS, МООа
на терране на А1,О<
88,0 49,8
38,3 22,1
Следы 2,6
49,7 25,1
Таблица 60. Превращение этилциклогексана в присутствии
сульфидного и окисного катализаторов
WSj на терране, 400 °C, 300 кгс/см2; МоО» на А11О«, 300 °C, 300 кгс/см2
WS, на терране МоО> на А1:О«
выход продуктов, выход продуктов,
Компоненты гидрогенизата моль на 100 моль моль на 100 моль
этилциклогексана этилциклогексана
введенно- расщеп- введенно- расщеп-
го ленного го ленного
Метан 5,6 22,1
Этан .— —. 25,8 100,3
Пропан 6,9 13,9 7,1 28,3
Бутан 7,7 15,5 1,5 6,0
Изобутан 12,7 25,6 1,7 6,8
П ентан 2,5 5,0 — —.
Изопентан 14,9 30,0 1.2 4,8
Циклопентан 3,0 6.0 2,4 9,6
Гексаны 6.6 13,3 0,4 1,6
Метилциклопентан 5,1 10,3 5,1 20,3
Циклогексан 2,0 4,0 2,8 11,1
Гептаны 2,9 5,8 — —
Диметилциклопентаны 3,3 6,7 1,8 7,2
Метилциклогексан 8,5 17,1 4,0 15,9
- Триметилциклопентан 9,5 — 1,5 —
Диметилциклогексаны 28,8 20,6 —
Доказательством явного преобладания ионного расщепления на
катализаторе WS2 на терране и радикального на катализаторе МоО3
на А12О3 может служить преобладание в первом случае осколков
С3 пзо-С4, во втором — Сх +С2. Можно отметить, что в первом
случае сохраняются заметные количества парафинов, иногда даже
большие, чем количества соответствующих циклопентановых угле-
водородов, а во втором — парафинов С5 — С7 очень мало. Очевидно,
что в ионном процессе раскрытие кольца и его последующее рас-
щепление протекают со сравнимыми скоростями, а в радикальном —
кольцо раскрывается медленно, но образовавшиеся при этом али-
фатические радикалы очень быстро отщепляют осколки С2 и Сх.
16*
243
Ускоряющее влияние заместителей на скорость реакции изомери-
зации в присутствии сульфидных катализаторов позволяет пред-
положить, что в стадии ионного равновесия заряд в основном должен
находиться у третичного углеродного атома. Это обстоятельство
делает метилциклогексан, имеющий только один третичный атом
углерода, очень удобным объектом для изучения механизма изо-
меризации. В самом деле, если процесс сжатия кольца идет только
через сг-комплексы, то наиболее вероятной должна быть реакция
т. е. в первую очередь должен образоваться 1,3-диметилциклопентан.
Если, наоборот, изомеризация идет через л-комплекс
то в первую очередь образуется этилциклопентан.
В присутствии WS2 при 420 °C и 300 кгс/см2 реакции изомери-
зации идут с измеримой скоростью, поэтому можно проследить
соотношение между изомерами метилциклогексана (табл. 61).
Таблица 61. Соотношение между продуктами изомеризации
метилциклогексана
[в % на превращенный метилциклогексан (без продуктов расщепления)]
Время выдержки, мин Степень превра- щения, % Выход продуктов изомеризации, %
этил- цикло- пентан транс- 1,2-диме- тилцикло- пентан транс- 1,3-диме- тилцикло- пентан -цис- 1,3-диме- тилцикло- пентан 1,1-диме- тилцикло- пентан
0 3,50 45,7 18,8 16,6 13,3 5,6
30 13,60 44,2 19,3 17,1 13,6 5,8
60 21,80 43,0 19,0 17,4 14,6 6,0
70 23,81 42,0 20,4 17,5 13,8 6,2
90 29.20 41,0 20,8 17,3 14,2 6,9
120 34,40 41,2 19,6 16,4 15,5 7,5
150 38,20 39,3 19,0 18,4 16,7 6,6
180 41,10 39,0 20.4 17,0 16,7 6,9
360 62,20 31,5 22,8 18,6 ' 17,5 9,6
540 70,60 28,3 25,9 20,8 16,2 8,8
720 72,70 24,0 26,6 22,6 17,3 9,5
Термодинамически равновесная смесь 73,10 21,8 26,4 22,9 15,5 13,4
244
Из приведенных в табл. 61 данных видно, что по скорости обра-
зования изомеры метилциклогексана образуют ряд
Следовательно, при гидрогенизации метилциклогексана главную
роль играет превращение через л-комплекс (см. стр. 244). При второ-
степенных положениях заряда
1,1-диметилциклопентан образуется с вероятностью вдвое меньшей,
чем 1,2- и 1,3-диметилциклопентаны, что хорошо корреспондирует
с данными табл. 61.
Однако при изомеризации метилциклогексана определенную роль
играют и ст-комплексы. Была предпринята попытка сопоставить
роль ст- и л-комплексов с помощью измерения скорости обмена атома
углерода метильной группы метилциклогексана с атомами углерода
кольца,стоящими в положениях 1, 2, 3 и 4, причем в параллельных
опытах радиоактивным углеродом метились эти положения 68-7 °. Если
ограничить равновесия сжатия — расширения цикла только двумя
стадиями, то при ст-механизме наиболее интенсивно должен обмени-
ваться атом углерода в положении 3:
245
Можно подсчитать, что вероятность обмена с углеродом метиль-
ной группы в случае о-механизма (при двух стадиях) для Ci равна О,
для С2 и С4 равна 1, а для С3 — 3.
При расчете по схемам с л-комплексами для атома Ст вероятность
обмена равна 0, для всех остальных — вероятности равны. Следова-
тельно, о числе стадий можно судить по выходу в метильную группу
атома Сх — он не должен выходить совсем или в очень малой степени.
В экспериментах, где степень превращения составляла около 60%,
было получено следующее распределение радиоактивности 7°:
Положение 14С
3 2 4 1
Суммарная радиоактивность ме-
тильной группы, % .... 6,0 4,5 2,3 1,0
При незначительном протекании вторичных и третичных реакций
(только 1% суммарной активности атома Cj попадает в метильную
группу) участие атомов 2, 3 и 4 в обмене с метильной группой не-
одинаково, причем первое место занимает атом С3. Следовательно,
хотя л-комплексы и играют, по-видимому, главную роль в ионных
изомеризационных превращениях, небольшая часть последних про-
текает и через ст-комплексы. Этому выводу соответствует и несколько
более высокая скорость образования 1,3-диметилциклопентана по
данным табл. 61.
ГИДРОГЕНИЗАЦИЯ БИЦИКЛИЧЕСКИХ УГЛЕВОДОРОДОВ
Полициклические ароматические углеводороды являются-важной
составной частью большинства типов сырья для деструктивной
гидрогенизации, поэтому изучение их превращений начато очень
давно.
В первой схеме, предложенной для объяснения превращений
нафталина, предполагали 71, что гомологи бензола образуются как
непосредственно из тетралина, так и из продукта его изомеризации —
метилиндана. В последующих работах, видимо вследствие исполь-
зования малоактивных (по изомеризующей способности) катализа-
торов, изомеризация рассматривалась как побочный процесс 72, 73.
Для расщепления би- и трициклических углеводородов были пред-
ложены 741 75 схемы с непосредственным расщеплением по связи,
примыкающей к ароматическому кольцу:
з
246
В статье Донаса ,в, обобщающей многолетние работы немецких
исследователей, схема процесса (для декалина) уже включает из®-
меризацию:
I . I —> | | -|-СН3
Замещенные
циклопентаны
Однако предположение о промежуточной изомеризации шести-
членного кольца в пятичленное не имело экспериментальных доказа-
тельств и было основано только на том, что в газообразных продуктах
деструкции отношение пзо-С4 : н-С4 выше термодинамически равно-
весного (см. стр. 230).
Была предложена14 также следующая схема:
I —► | | I —> | I —> Парафины
СН3
Н3С СН3
Эта схема мало вероятна 7в, так как в гидрогенизате присутствуют
гомологи как циклогексана, так и циклопентана, а образование
первых этой схемой не объясняется.
В табл. 62 приведены (в хронологическом порядке) результаты
исследования состава гидрогенизатов различных бициклических
углеводородов, на основании которых можно ответить на вопросы:
предшествует ли изомеризация расщеплению гидрированного кольца
и по каким связям разрывается гидрированное кольцо.
О месте разрыва связей в гидрированном кольце можно судить
по наличию в продуктах расщепления моно- или диалкильных произ-
водных бензола. Можно отметить, что моноалкилпроизводные обна-
руживались главным образом в присутствии малоактивных (особенно
в отношении изомеризации) катализаторов — металлических или
окисных — при высоком давлении водорода. При гомогенном -де-
структивном гидрировании тетралина в отсутствие катализатора 86
также преобладали монозамещенные производные, а содержание
дизамещенных было ничтожно мало. Только одни диалкилпрбизвод-
ные получались при атмосферном давлении в присутствии метал-
лических катализаторов. При тщательном анализе. гидрогенизата,
полученного в присутствии катализатора WS2, обладающего доста-
точно высокой изомеризующей активностью, был сделан вывод
о возможности всех путей расщепления:
а) расщепление без изомеризации, с разрывом связи, примыка-
ющей к бензольному кольцу (бутилбензол, бутилциклогексан, 1-ме-
тил-2-бутилциклопентан, а также, косвенно, пропилбензол, бутил-
циклопентан, пропилциклогексан, этилциклогексан, этилбензол,
пропилциклопентан);
247
Таблица 62- Составы гидрогенизатов полициклических углеводородов
Исходный углеводород Темпера- тура, °C Давле- ние, кгс/см2 Катализатор ' Выделенные вещества или данные об их структуре Лите- рату- ра
Тетралин 450—480 100 Ni на А12О3 о-Ксилол и другие моно- и дизамещенные бен- золы 77
Фенантрен 440—450 75 Ni на глине (З-Этилнафталин. Окисление фракции моноцик- лических ароматических углеводородов дало бензойную и о-фталевую кислоты 78
Нафталин, тетралин, а- и (3-метилнафта- лины, 2,6-диметил- нафталин 450-500 100-120 (NH4)2MoS4 на активированном угле* Этилбензол, бутилбепзол, 1-метил-З-этилбензол, метилтетралины. Окисление дало только бен- зойную кислоту, а в случае метилнафтали- нов — изофталевую кислоту 74, 79
Хризен 440—450 85 FeCl3 Окисление фракции моноциклических аромати- ческих углеводородов дало бензойную и фта- левую кислоты 80
Нафталин 500 100 MoS3 + S WO3 + S Окисление фракций, выкипающих до 120 и 120 —140 °C, дало только бензойную кислоту 73
» * 510 120 MoS2 на A12O3 То же 73
Гидриндан 300 1 Pt 1 -Метил-2-этилбензол 81
Нафталин 500 Повышен- ное Отсутствие Бензол, толуол, этилбензол 82
Октагидроантрацен 485-490 100 MoS2 Окислением фракций 130—150 и 150—180 ?С по- лучены бензойная, изофталеная и терефтале- вая кислоты 75
Нафталин 500 100 M0S2 на активиро- ванном угле Этилбензол 83
а-Метилнафталин 400 140 Ni 1,2-Диметилциклогексан, 1-метил-2-этилцикло- гексан 84
Гидриндан 325 100 Си -j- Al на кизельгуре То же 85
Тетралин 440, 462 230-1200 Отсутствие Бензол, толуол, этилбензол, о-ксилол, «-про- пил- и изопропилбензолы, 1-метил-2-этилбен- зол, н- и emop-бутилбензолы, индан, а- и 0- метилинданы, траис-декалин; доминируют и-бутилбензол, этилбензол, втор-бутилбензол, н-пропилбензол, изопропилбензол; дизамещен- ных производных бензола менее 1% 86
Тетралин 400 300 WS2 па терране
» 206—250 — Гумбрин
Нафталин 470 450 Fe на полукоксе
420' 300 WS2
» 420 300 WS2 + NiS на А12О3
420 300 WS2 на терране
Тетралин 420 300 WS2
» 420 300 WS2-|-NiS на А12О3
420 300 WS2 на терране
Декалин 420 300 ' ws2
» 420 300 WS2 + NiS на А12О3
» 420 300 WS2 на терране
а- и р-метилнафтали- ны 420 300 WS2
Метилциклогексан, транс-1,3-ДИметилциклогек-
сан, толуол, этил-, н-пропип- и н-бутилбен-
золы
Бензол, циклогексан, метилциклопентан. Оки-
слением ароматической фракции С1о получены
бензойная и о-фталевая кислоты
Бензол, толуол, этил- и н-бутилбензолы, 1-ме-
тилиндан, тетралин
о-Ксилол
1,3-Диметилциклопентан, этилциклогексан, ди-
метилциклогексаны, бутилциклогексан, тет-
ралин, декалин, бензол, толуол, этилбензол,
о-цимол, гидр индан, метилгидриндан, индан,
бутилбензол
1,2- и 1,3-диметилциклопентаны, декалин, бу-
тилциклогексан, бензол, толуол, этилбензол,
и-ксилол, бутил-, изобутил- и mpem-бутил-
бензолы, метилиндан, тетралин, 1- и 2-метил-
тетралины
2-Метилпентан, метилциклопентан, 1,2-ДИметил-
циклопентан, циклогексан, метилциклогексан,
метилиндан, бутилбензол
Циклопентан, метилциклопентан, 1,2-диметил-
циклопентан, циклогексан, бутилциклогексан,
бутилбензол, декалин
2-Метилпентан, метилциклопентан, бензол, то-
луол, о-ксилол, индан, 1-метилиндан
Изопентан, пентан, 2- и 3-метилпентаны, метил-
циклопентан, 1,2- и 1,3-диметилциклопентаны,
циклогексан
1,2-Диметилцик лопентан, бутилциклогексан,
метилгидриндан
Изопентан, пентан, 2- и 3-метилпентаны, ме-
тилциклопентан, 1,2-, 1,3- и 1,4-Диметилцик-
лопентаны, бутилциклогексан, толуол, о-кси-
лол ч
В гидрогенизате преобладают 5,6,7,8-тетрагид-
ропроизводные (насыщается незамещенное
кольцо)
65
87
88
88
88
88
88
88
88
88
88
88
89
Исходный углеводород Темпера- тура, °C Давле- ние, кгс/см2 Катализатор
Нафталин 460 250 Fe на активирован- ном угле
Нафталин, фенан- трен, пирен 205-370 68 Pd на А12О3; Pt(+F + Cl) на А12О3; WS2 + NiS на А12О3; Co-j-Mo на А12О3
Дифенилметаны и дп- фенилэтаны 300-450 100 МоО3 на А12О3; МоО3 на SiO2; МоО3 на SiO24-Al2O3; Ni на А]2О3
Нафталин 4- тетр алии 420 300 WS2
Инден, 1-метилинден 250-450 1 Ni на А12О3
Продолжение табл. 62
Выделенные вещества или данные об их
структуре
Лите-
рату-
ра
Бензол, толуол, эт-ил- и бутилбензолы 90
При температурах выше 320—330 °C при данном 91
давлении глубина пренращения зависит от
структуры углеводорода (из-за термодинами-
ческих ограничений)
Парафины Ci—С8, циклоалканы С5—С7, арома- 92
тические углеводороды Св—С10
Приведен состав гидрогенизата, выкипающего
выше 100 °C:
н-пропил-, изопропил-, н-бутилциклопентапы,
1-метил-1-этил-, 1-метил-2-этил- и 1-метил-
3-этилциклопентаны, 1-метил-2-пропил- и
, 1-метил-2-бутилциклопентаны, 1,1,2-, 1,1,3-,
1,2,3-, 1,2,4- и 1,3,5-триметилциклопентаны;
этил-, пропил-, изопропил- и бутилциклогек-
саны, 1,2-, 1,3- и 1,4-диметилциклогексаны,
1-метил-2-пропил-, 1-метил-4-пропил-, 1-метил-
2-изопропилциклогексаны, диметилэтилцикло-
гексаны; 2,2-, 2,4- и 2,5-Диметилгексаны,-
3-этилгексан, 2-, 3- и 4-метилгептаны, н-ок-
тан, 2,4-Диметилоктан, 2-, 3-, 4- и 5-метилно-
наны; бензол, толуол, этил-, пропил- и изо-
пропилбензолы, ксилолы, 1-метил-2-пропил-
бензол, 1,2,4- и 1,3,5-триметилбензолы, 1,4-
диметил-2-этилбензол, 1,2,3,5-тетраметилбен-
эол, декалин, диметил пента ланы (7 изомеров),
1-, 2-, 4- и 5-метилинданы
эасщепляются (1- и у-связи пятичленного кольца
93—
95
96
б) расщепление с предшествующей изомеризацией и с разрывом
связи, примыкающей к бензольному кольцу (изопропилбензол, изо-
пропилциклогексан, изопропилциклопентан);
в) расщепление без изомеризации по связи, не примыкающей
к соседнему кольцу (1-метил-2-пропилбензол, 1-метил-2-пропилцик-
логексан, а также, косвенно, 1,2-диметилциклогексан, 1-метил-
2-этилцик лопентан);
г) расщепление с предшествующей изомеризацией и с разрывом
связи, не примыкающей к соседнему кольцу (1-метил-2-изопропил-
циклогексан).
Следовательно, процесс вообще очень сложен (было выделено
около 100 индивидуальных углеводородов, из них 62 были иденти-
фицированы 94-95 ) и в зависимости от условий может протекать
по тому или иному механизму.
В отсутствие изомеризующих катализаторов и npii высоком давле-
нии водорода преобладает раскрытие гидрированного кольца (без
предшествующей изомеризации) по связи, примыкающей к арома-
тическому кольцу. Это расщепление характерно для жидкофазного
процесса и протекает по радикальному механизму (см. стр. 117
и 181).
В присутствии катализаторов, обладающих высокой расщепля-
ющей активностью и не обладающих изомеризующей актив-
ностью, при невысоком давлении водорода или без избыточного
давления разрыв связи, примыкающей к бензольному кольцу, проис-
ходит редко, гидрогенолизу подвергаются менее прочные связи
в гидрированном кольце:
В присутствии сульфидных катализаторов, т. е. в условиях про-
мышленного парофазного процесса, расщепление должно быть свя-
зано с изомеризацией, как это было показано ранее для моноцикли-
ческих углеводородов. Для иллюстрации этого в табл. 63 приведен
групповой состав гидрогенизатов бициклических углеводоро-
дов 88.
Из приведенных данных видно, что при гидрогенизации нафта-
лина и тетралина в присутствии активных гидрирующих катализа-
торов WS2 и WS2 NiS на А12О3 производных бензола образуется
мало, интенсивно гидрируется бензольное кольцо, быстро идет изо-
меризация и количество углеводородов рядов бицикло [3,3,0]ок-
тана (пенталана) и бицикло (4,3,01нонана (гидриндана) в несколько
251
Таблица 63. Групповой углеводородный состав гпдрогенизатов бициклических углеводородов
420 °C, начальное давление 140 кгс/см2, время реакции 3 ч
Исходный углеводород Катализатор Выход гидроге- низата. % Состав гидрогенизата, %
пара- фины нафтены ряда ароматические углеводороды ряда
цикло- пентана цикло- гексана пснтала- на гидринда- на дека- лина бензо- ла индана тетрали- на нафтали- на
Нафталин WS2 97,4 1,6 7,8 8,2 15,9 5,1 28,9 10,5 4,7 16,7 0,6
Тетралин 89,0 14,2 6,3 7,3 19,0 8,3 29,6 5,7 4,5 5,1 —
Декалин » 65,0 43,2 22,0 5,6 9,9 5,5 13,8 — — — —
Нафталин WS2 + NiS па А12О3 99,0 0,2 0,8 7,8 2,4 6,7 61,1 3,7 1,3 14,2 1,8
Тетралин То же 100,0 — 1,2 5,8 4,1 11,3 73,6 0,7 — 3,3 —
Декалин » 98,0 — 0,8 4,2 4,9 3,5 86,6 — — — —
Нафталин WS2 на терране 94,2 1,2 5,2 3,2 3,5 0,7 9,4 11,8 2,7 60,5 1,8
Тетралин То же 93,3 10,0 14,3 3,8 6,0 3,9 2,1 17,5 4,7 36,7 —
Декалин 60,6 27,2 31,7 7,7 10,8 3,4 7,5 И, 7 (суммар ное содер жанпе)
раз больше, чем производных бензола. Катализатор WS2 на тер-
ране обладает слабой гидрирующей активностью и процесс гидро-
генизации идет в основном с образованием тетралина и расщеплением
его гидрированного кольца. Если гидрогенизации подвергается насы-
щенный углеводород — декалин, также образуется большое количе-
ство продуктов изомеризации.
Таким образом, из сопоставления данных по групповому составу
углеводородов видно, что в условиях парофазного процесса реакции
изомеризации проходят значительно интенсивнее, чем в процессах
без катализатора и в жидкофазном процессе, и, следовательно, рас-
щеплению будут подвергаться в основном изомеризованные угле-
водороды.
Важной особенностью превращений углеводородов в присутствии
высокотемпературных сернистых катализаторов является сущест-
венное преобладание в газообразных продуктах пропана и изобутана,
что подтверждает протекание этого процесса по ионному механизму.
Так, например, если мольное отношение углеводородов С4 : С3 : С2
в продуктах деструктивной гидрогенизации тетралина в присутствии
железного катализатора 90 равно 1 : 2,8 : 11,3, то в продуктах дест-
руктивной гидрогенизации тетралина в присутствии катализатора
WS2 на терране оно составляет 85 1,00:0,31:0,03, а для дека-
лина — 1,00 : 0,30 : 0,02. Аналогично этому из мольных отношений
углеводородов Св : С7 : С8 (см. табл. 64) видно 85,88, что в продуктах
расщепления больше всего образуется углеводородов Св и С,.
Таблица 64. Мольные отношения углеводородов С8 : С- : С8
(в гидрогенизатах бициклических углеводородов С1о)
Исходный- углеводород Катализатор Мольное отношение С« : С? : Св
для нафтеновых углево- дородов для ароматических углеводородов
Тетралин WS2 1,00 : 0,81 : 0,76
Декалин 1,00 : 0,72 : 0,17 —
Нафталин WS2 на терране 1,00 : 1,06 : 0,92 1,00 : 0,62 : 0,55
Тетралин То же 1,00 : 2,24 : 0,97 1,00 : 0,11 : 0,17
Декалин » 1,00 : 0,36 : 0,03 —
Тетралин * » 1,00 : 0,60 : 0,19 1,00 : 0,38 : 0,33
Декалин * 1,00 : 0,86 —
* По данным работы
Таким образом, расщепление циклических углеводородов в при-
сутствии сульфидных катализаторов, с одной стороны, должно
включать промежуточную изомеризацию, а с другой — протекать
с отщеплением осколков С3 4- С4. Последнее обстоятельство прямо
указывает на разрыв одной или двух связей, примыкающих к бен-
зольному кольцу. Было бы неправильно на основании только этих
253
данных, не учитывая возможности глубокой изомеризации, пола-
гать 6В-’6, что расщепление, например тетралина, идет через проме-
жуточное образование бутилбензола:
—>• + С4Н10
^\/СНз
—► I 11 + С3Н8
Хотя углеводороды, указанные в приведенной выше схеме, и
содержатся в гидрогенизатах, их количество невелико. Так, напри-
мер, в гидрогенизате тетралина, полученном в присутствии катали-
затора WS2 на терране с наиболее сильной расщепляющей актив-
ностью, мольное отношение моноциклических нафтенов (Св: С7 : С8 :
: С9 : С1о) и гидринданов равно 1,00 : 2,24 : 0,97 : 0,23 : 0,62 : 2,86,
т. е. содержание гидринданов почти в пять раз больше, чем содер-
жание бутилциклогексанов, причем в последних велика доля изобу-
тилциклогексанов 88. Кажущееся противоречие между представле-
ниями о разрыве наименее прочной связи с фактом доминирования
разрыва по связям, примыкающим к бензольному кольцу, можно
устранить, если привлечь для объяснения положенйя карбониево-
ионной теории.
Реакции, протекающие при гидрогенизации тетралина, можно
представить схемой, приведенной на стр. 255 (жирными стрелками
показаны превращения, протекающие более легко или преимуще-
ственно).
В случае декалина можно полагать, что превращения идут
по схеме, приведенной на стр. 256 (некоторые стадии объеди-
нены).
Из приведенных схем видно, что в ионных превращениях преобла-
дают разрывы менее прочных связей в гидрированных кольцах,
но в конечном счете смещения зарядов приводят к отрыву фрагмен-
тов С3 и uso-Ci после изомеризации. Следует подчеркнуть, что почти
все структуры, показанные в этих схемах, подтверждены выделением
из гидрогенизатов соответствующих индивидуальных углеводо-
родов.
В упрощенном виде взаимные превращения отдельных классов
углеводородов можно представить схемой (стр. 257), которая
254
ГГ’+с.н. . ^/^СНз ^\/сНз ^\/СНз ^\/СМз и — 1 II —► I II —► 1 || + СзН8 | ||+ ИЗ0-С4Н10 Х^/\(СН2)2СН2 ^/^СНаСНСНз t . 1 ^\^сн2 ^/СНз ’ f ^\/СН2С(СН3)2 ^/^(СН^гСНг ^/^СНаСНСНз ^^/^СНз
сн2сн=сн2
сн3
СН2 /Х/СН2С(СН3)2
* II I------------------► I II ---► I 1+ИЗО-С4Н10
СН2СНСН3 \/\/\сН3 \/
X
Н2С । i 1 i i ________1 ij CH2C(CH3)2
СНзСН-НгС/^^СНз НзС'/''ч/\/\сН3
I I+USO-C4H10
подтверждается наличием в гидрогенизатах как углеводородов ряда
циклогексана, так и углеводородов ряда пенталана:
В этой схеме обычно R = R' = H и R = H, R' = СН3.
Таким образом, предположение о протекании процесса по ион-
ному механизму объясняет все экспериментальные данные; при этом
закономерности превращения бициклических углеводородов пол-
ностью совпадают с закономерностями превращения моноцикличе-
ских углеводородов: непосредственное расщепление гексаметилено-
вого кольца невероятно, ему предшествует изомеризация; расщепле-
ние кольца протекает главным образом по связям между вторичными
углеродными атомами, приводя к образованию разветвленных угле-
водородов. Лишь наличие двух третичных углеродных атомов в кон-
денсированной системе приводит к последующему разрыву связи
между вторичным и третичным углеродными атомами с отрывом
трех или четырех атомов углерода.
Все вышеизложенное относится к главным путям превращений
бициклических углеводородов. Детальное изучение гидрогенизата
смеси нафталина и тетралина 93-95 позволяет отметить некоторые
специфические особенности. К ним, в частности, относится наличие
4- и 5-метилинданов в количествах, превышающих даже количества
продуктов первичной изомеризации тетралина — 1- и 7-метилинда-
нов. Образование 4- и 5-метилинданов, а также три- и тетраметил-
бензолов в результате вторичной реакции метилирования мало
вероятно, так как осколки Ct в ионном процессе не образуются и,
кроме того, количество индана — исходного вещества для метили-
рования — мало (1,6%). Более вероятна прямая изомеризация 1-
и 7-метилинданов:
СНз СН3
17 Заказ 271
257
Механизм такой изомеризации неясен. Можно только предполо-
жить, что при этом либо происходит перегруппировка, аналогичная
гидриндановой перегруппировке 97
либо миграция метильного заместителя, как это было показано 98
для случая а- и Р-метилнафталинов.
Образование заметных количеств три- и тетраметилбензолов так-
же следует объяснять не метилированием, а внутримолекулярной
перегруппировкой, например:
СН3
I II —> I II —> I II —> I II
НзС/'^/\с2Н5 НзС/^/^СНз
Для уточнения механизма расщепления представляет также
интерес групповой состав продуктов гидрирования нафталина и тет-
ралина (табл. 65) и распределение в гидрогенизате углеводородов
состава Сх — С10.
Таблица 65. Групповой состав продуктов гидрирования
нафталина и тетралина (в %) вз-65
Углеводороды ряда Суммарное содержание Состав фракции
С. С. С, с. с. С1о
парафинов 15,42 1,02 3,30 1,41 1,00 0,61 8,08
циклопеитана 23,99 0,45 4,52 2,11 2,04 4,30 10,55
циклогексана 11,85 — 0,45 1,07 1,55 0,91 7,89
пенталана 24,27 — — — 0 0 24,27
гидриндана 7,69 —- — — — 0 7,69
декалина 16,78 — — — — — 16,78
Всего 100,00 .,47 8,27 4,59 4,59 5,82 75,26
Обращает на себя внимание неравномерное распределение пара-
финов: их очень мало во фракциях С8 и Св и много во фракциях Св
и С10. Совершенно нет гидринданов и метилпенталанов, а также
самих пенталанов. Очевидно, что быстрее всего идут реакции изо-
меризации — сжатия колец, которые затем раскрываются. Отщепле-
ние осколков С3 — С4 дает углеводороды состава Св — С?, у которых
продолжается процесс раскрытия циклов. Отсутствие во фракциях
С8 и Св углеводородов рядов пенталана и гидриндана указывает, что
258
скорости раскрытия колец подчиняются тем же закономерностям, что
и для циклопентацовых углеводородов 17, т. е. по мере накопления
заместителей скорость раскрытия кольца уменьшается:
Бициклические нафтены С8 и Сд, скорость образования которых
невелика (реакция деметилирования не характерна для ионного про-
цесса), не успевают накапливаться в реакционной среде, так как
у них раскрытие цикла происходит с большой скоростью.
Распределение в гидрогенизате углеводородов С2 — С10 приве-
дено ниже: Содержание в гидрогенизате Содержание в гидрогенизате вес. % мол. %
вес. % МОЛ. %
Ci . • 0,2 1,8 Сю 62,6 45,7
С2 • • 1,3 4,4 в том числе:
С3 . 3,4 7,6 декалин .... 14,0 10,3
с* . 9,5 16,5 гидринданы +
с5 . . 3,7 5,2 -f- пенталаны . 26,6 19,3
Св . . 6,9 8,2 циклогексаны +
С, . • 3,8 3,9 +циклопентаны -]-
С8 - 3,8 3,4 . пар афины • 22,0 16,1
с9 . . . 4,9 3,8
ла основании этих данных можно составить следующий
превращений:
Выход продукта
баланс
превращения
а) Сщ ----► иао-Сю................... • • • 19,3
б) Сю ----► Продукты раскрытия одного или
двух колец.....................16,1
* С7+С3....................... 3,9
---► С6 иао-С<................... 8,2
'10 ——> 2Сз + С4 .................. 4,0
— 2С5 2,6
I * Cg-pCi...................... 3,8
‘10 —
I---► Cg-f-Cg..................... 3,4
в)
г)
вес. %
26,6
22,0
26,4
11,0
Схемы а — в представляют собой ионные превращения, схема ,
г — реакции расщепления с отрывом одного или двух атомов угле-
рода — типично радикальные превращения. Рассматривая получен-
ные результаты, можно сделать выводы о том, что происходит очень
глубокое превращение, так как лишь 14% структуры декалина
остается неизменной. При этом легче всего протекает изомеризация
(75 вес. %) и преобладают ионные процессы. Радикальные процессы
17* 259
деметилирования и отщепления С2 составляют лишь около 11%.
Деметилирование происходит незначительно, так как содержание
углеводородов С1о примерно в 13 раз больше, чем углеводородов С9
(62,6 и 4,9%).
Из гидрогенизата декалина было выделено 7 индивидуальных
диметилпенталанов 94. Два из них оказались стереоизомерами ",
остальные были изомерами строения. На основании их физических
констант, а также строения идентифицированных малоразветвленных
парафинов С1о им были приписаны структуры 1,5-, 1,6-, 1,3-, 1,4-
и 2,5-диметилпенталанов. Образование этих соединений в ходе изо-
меризации можно представить схемой:
Влияние метильных заместителей в ряду бициклических аромати-
ческих углеводородов аналогично их влиянию в ряду моноцикличе-
ских ароматических углеводородов. Так, из приведенных в табл. 66
данных видно, что содержание ароматических углеводородов в гидро-
генизатах метилнафталинов меньше, чем в гидрогенизате нафталина.
Введение заместителей увеличивает скорость гидрирования, но в
первую очередь насыщается незамещенное кольцо. Введение замести-
теля в P-положение сказывается больше, чем введение в «-положе-
ние, особенно на интенсивности реакции изомеризации и расщепле-
ния. Так, отношение суммы количеств продуктов изомеризации (угле-
водороды рядов пенталана, гидриндана и индана) к сумме неизо-
меризованных углеводородов (декалин и тетралин) 89 составляет
в гидрогенизатах нафталина 1,47, в гйдрогенизатах а-метилнафта-
лина — 1,55 и в гидрогенизатах [3-метилнафталина — 5,40.
260
Таблица 66. Групповой состав гидрогенизатов нафталина
и метилиафталинов (в %) 89
420 °C, начальное давление 140 кгс/см2, время реакции 3 ч, катализатор
WS,(30%)
Состав гидрогенизата Исходный углеводород ,
'нафталин а-метил- нафталин р-метил- нафталин
Парафины 3,1 5,8 13,0
Нафтены ряда 5,6
циклопентана 1,4 1,8
циклогексана 0,7 1,5 1,2
пенталана 15,9 14,4 16,6
гидриндана 15.7 ' 14,6 20,7
декалина 9,4 10,1 4,2
Ароматические углеводороды ряда
бензола 2,1 3,2 4,5
индана . 18,0 16,6 21,1
тетралина 33,7 32,0 13,1
КАТАЛИЗАТОРЫ ПАРОФАЗНОЙ ГИДРОГЕНИЗАЦИИ
Из изложенного выше материала видно, как сложно и взаимо-
связанно протекают реакции гидрирования, изомеризации, расщепле-
ния и, иногда, алкилирования в процессах парофазной гидрогениза-
ции. Для технологических целей нужно усиливать одни и подавлять
другие реакции. Это достигается подбором катализаторов, варьиро-
ванием и модифицированием их состава, изменением пористости,
активной поверхности и других свойств, введением добавок в реак-
ционную смесь, варьированием условий процесса. Естественно по-
этому, что крайне важны данные о гидрирующей, изомеризующей и
р асщепляющей активности наиболее употребительных катализато-
ров и о влиянии состава и свойств этих катализаторов на интенсив-
ность отдельных реакций.
Результаты, полученные при изучении химии превращений раз-
личных классов соединений в присутствии катализаторов парофаз-
ной гидрогенизации, дают возможность сделать вывод о двух соста-
вляющих их активности: способности ускорять гомолитические реак-
ции (собственно гидрирование и радикальное расщепление) и способ-
ности ускорять гетеролитические реакции (изомеризация и ионное
расщепление). Связь между составом и свойствами катализаторов
и их способностью ускорять эти два основных класса реакций,
следовательно, могла бы служить важным ориентиром для техноло-
гических исследований.
Разработанные в основном эмпирическими методами современ-
ные промышленные сульфидные и окисные катализаторы более под-
робно стали изучаться относительно недавно. Так, была описана 100
кристаллическая структура катализаторов WS2 и MoS2 и показано
261
послойное распределение в них атомов серы и металла. Почти одно-
временно была опубликована 18 статья о генезисе сульфидных ката-
^лизаторов, в которой впервые делалась попытка связать их физиче-
ские и каталитические свойства. В этой работе приводились первые
данные об адсорбционных свойствах WS2 и рентгенограммы актив-
ных и неактивных образцов катализаторов. По' мнению Пира 1в,
активными являются микрокристаллы WS2, сохранившие при син-
(NH4)2WS< —> WS2 + (NH4)2S2
кристаллическую структуру тиомолибдата аммония. Под действием
высоких температур кристаллики WS2 упорядочиваются, образуя
стабильную кристаллическую структуру, и теряют при этом катали-
тическую активность. В этой же статье 18 были обобщены некоторые
закономерности модифицирования каталитических свойств WS2
путем нанесения на носители: нанесение WS2 на основные (А12О3)
носители понижает изомеризующую и повышает гидрирующую актив-
ность, а нанесение на кислотные (террана) носители понижает гидри-
рующую и повышает расщепляющую активность. Аналогичные вы-
воды были сделаны ранее для молибденовых катализаторов 101.
Позднее для объяснения большей или меньшей гидрирующей актив-
ности промышленных катализаторов впервые были использованы 102
результаты электрономикроскопических исследований. В этой ра-
боте более высокая гидрирующая активность катализатора WS2 -}-
+ NiS на А12О3 по сравнению с чистым WS2 была объяснена тем,
что WS2 в нем тонко распределен на большой поверхности А12О3
и доступность его кристаллов значительно больше. Кроме того,
кристаллы WS, в катализаторе WS2 + NiS на А12О3 имеют значи-
тельно меньшие размеры, они как бы деформированы, что и было
подтверждено 102 злектрономикроскопическими исследованиями при
увеличении в 14 000 раз. Подобные выводы и наблюдения делались
до последнего времени вне связи с химией ускоряемых данными ката-
лизаторами процессов. Поэтому даже в наиболее тщательных и пол-
ных обзорах (например, обзор 7в) не дается разграничения ионных и
радикальных реакций, различных типов реакций изомеризации и
расщепления.
В настоящее время катализаторы на основе окислов и сульфидов
вольфрама, молибдена и никеля применяются не только в процес-
сах гидрогенизации, но и в процессах гидроочистки, гидрокрекинга,
изомеризации фракций СБ — Св, поэтому их значение еще больше
возрастает.
у/ Очевидно, что прежде всего нужно оценить уже известные катали-
заторы с тем, чтобы показать влияние главных факторов — химиче-
ского состава и природы носителей — на активность катализатора.
Было предложено 103 оценивать активность катализаторов парофаз-
ной гидрогенизации по отношению продуктов превращения простей-
шего ароматического углеводорода бензола к количеству неизменен-
ного бензола. Было показано 104, что качественная оценка актив-
262
ности катализаторов таким методом практически всегда совпадает
и в случае замены бензола на другие моно- и бициклические аромати-
ческие углеводороды или реальное промышленное сырье. Получен-
ные результаты приведены в табл. 67—69.
Гидрирующая активность катализатора оценивалась по отношению суммы
количеств продуктов гидрирования и расщепления к количеству непревращен-
ного бензола, изомеризующая активность — по отношению суммы количеств
метилциклопентана и продуктов расщепления к количеству циклогексана,
расщепляющая активность — по отношению количества продуктов расщепле-
ния к количеству метилциклопентана.
Таблица 67. Гидрирующая активность высокотемпературных катализаторов
' (гидрирование бензола)
в автоклаве: давление 300 кгс/см*, время контакта 3 ч; в проточной установке:
давление 200 кгс/см2, объемная скорость 3 ч-1
Катализатор Гидрирование в автоклаве при Гидриро- вание в проточной установке
42 0 °C 450 °C 500 °C
WS2 + NiS на А12О3 2,50 3,40 2,68 499,0
Pt на А12О3 (фторированный) — — — 249,0
WS, 2,08 3,32 1,03 6,20
MoS? 0,83 1,07 0,38 5,33
MoS, на активированном угле .... — — — 1,97
Со-|-Мо на А12О3 — — — 0,87
WS2 на терране 0.36 — — 0,04
Ni на А12О3 0,30 — — —
* в опытах в автоклаве, использовали невосстановленный катализатор (МоЗз), в про-
точной установке—восстановленный катализатор (MoS2).
Таблица 68. Изомеризующая активность высокотемпературных катализаторов
давление в автоклаве 300 кгс/см2, в проточной установке —200 кгс/см*
Катализатор Гидрирование бензола в авто- клаве при Гидрирование в про- точной установке при 420° С
420 °C 450 °C 500 °C бензола циклогек- сана
WS2 на терране 10,82 12,0 2,50
WS2 1,47 2,24 3,41 3,31 2,92
MoS2 0,45 0,88 1,00 1,53 1,10
Pt на А190я (фторированный) — — — 1,36 0,09
Ni на А12О3 0,23 — — —
WS2 + NiS на А12О3 0,16 0,16 1,02 0,24 0,07
(Jo —J—Мо на AI2O2 — — — 0,01 0,01
MoS2 на активированном угле — — — 0,001 0,01
263
Таблица 69. Расщепляющая активность высокотемпературных катализаторов
давление в автоклаве 300 кгс/см2, в проточной установке—200 кгс/см2
Катализатор Гидрирование бензола в авто- клаве при Гидрирование в про- точной установке при 420° С
420° С 450° С 500’ С бензола циклогек- сана
Ni на А12О3 0,53 —. .
-WS2 на терране 0,25 —' — 0,45 0,54
Pt на А12О3 (фторированный) — — — 0,25 0.04
WS2 0,10 0,14 0,22 0,10 0,16
MoSj 0,23 0,13 0,65 0,07 0,03
WS2 + NiS на А12О3 0,09 0,08 0.20 0,13 0,17
Co-f-Mo на А12О3 — — — 0,01 0,01
MoS2 на активированном угле — — 0,01 . 0,01
* См. примечание к табл. 66.
Из сопоставления данных, приведенных в табл. 67—69, видно,
что порядок активностей катализаторов совпадает как для опытов
в автоклавах, так и в проточной установке. Испытанные катализа-
торы можно расположить в следующие ряды (для температуры
420 °C).
По гидрирующей активности:
WS«-|-NiS на А12О3 > Pt на А12О3 (фторированный) >•
WS2 > MoS2 > MoS2 на активированном угле ~>
> Со-|-Мо на А12О3 > WS2 на терране > Ni на А12О3
По изомеризующей активности:
WS2 на терране > WS2 > MoS2 > Pt на А12О3 (фторированный) >
Ni на А12О3 > WS2 + NiS на А12О3 >>
>Со-[-Мо на А12О3 >> MoS2 на активированном угле
По расщепляющей активности:
Ni на А12О3 > WS2 на терране > Pt на А12О3 (фторированный) >
> WS2 > MoS2 > WS2-i-NiS на А12О3 >
> Co-f-Mo на А12О3 > MoS2 на активированном угле
По результатам опытов в автоклаве при разных температурах
можно отметить, что положение катализаторов WS2, MoSa и WS2 4-
-4- NiS на А12О3 в рядах мало меняется с температурой (особенно
по изомеризующей активности).
Тот же порядок активностей катализаторов наблюдался при
оценке некоторых из приведенных катализаторов на стандартном
промышленном сырье 161 102, а также в опытах гидрогенизации дру-
264
гих углеводородов — этилбензола и цетана 105. Аналогичный по-
рядок активностей катализаторов парофазной гидрогенизации соблю-
дается и при оценке их в качестве катализаторов изомеризации
и гидрокрекинга. Так, вольфрамовые и молибденовые катализаторы
в процессе изомеризации гексана при 400—460 °C и 20—50 кгс/см2
по изомеризующей активности могут быть расположены в следующий
ряд108:
WS2 > WS2 на А12О3 >> MoS2 на А12О3 > Ni + Mo на А12О3 £>
> Со-(-Мо на А1аО3 > МоО3 на А12О3 > WO3 на А12О3
Клайн и Коллонтиш 107 в обзорной статье о вольфрамовых ката-
лизаторах гидроочистки, гидрокрекинга и изомеризации подчерки-
вают высокую изомеризующую активность WS2, в то же время
вольфрамовые катализаторы на носителях уступают платиновым и
сульфидированным никелевым катализаторам. Для усиления изо-
меризующей активности W и Мо наносятся на кислотные носители —
алюмосиликаты и цеолиты. Эти выводы 107 являются свидетельством
известной универсальности и достоверности оценки активности
катализаторов таким методом.
Рассматривая приведенные выше ряды активностей катализато-
ров, можно сделать очень важный вывод, что гидрирующая, изоме-
ризующая и расщепляющая активности окисных катализаторов
ниже активностей сульфидных катализаторов, а расщепляющая
активность металлических катализаторов может быть даже выше,
чем активность сульфидных катализаторов. Чтобы оценить роль
носителя, целесообразно определить удельную активность различ-
ных катализаторов — отношение степени превращения веществ в
реакциях гидрирования, изомеризации и расщепления к одинако-
вому числу атомов Мо или W на поверхности катализатора, условно
принимая во всех случаях мономолекулярный слой. Полученные дан-
ные приведены в табл. 70.
Эти данные в некоторых отношениях неточны (превращение бен-
зола в случае WS2 на терране, выход продуктов расщепления для
некоторых катализаторов), так как в отдельных случаях при малых
превращениях большую ошибку вносит неточность определения
выхода гидрогенизата. Тем не менее можно отметить важные законо-
мерности.
Нанесение WS2 и MoS2 на носители повышает удельную гидри-
рующую активность, что видно из сопоставления активностей катали-
заторов WS2 и WS2 -f- NiS на А12О3 1в* 78 и катализаторов MoS2
и MoS2 на активированном угле. Это явление следует объяснить,
с учетом данных18,1оа, тем, что тонкое диспергирование частичек
сульфида металла на поверхности носителя облегчает их доступ-
ность для водорода и гидрируемых веществ.
При нанесении WS2 и MoS2 на носители с большой поверхностью
уменьшаются удельные изомеризующая и расщепляющая актив-
ности, однако в случае катализатора WS2 на терране, наоборот,
265
Таблица 70. Удельные активности некоторых катализаторов
' (средние данные)
Показатели Катализатор
WS2 WS2 + NiS на А12О2 WSt на терране MoS2 MoS2 на активиро- ванном угле
Содержание активного агента в
10 мл катализатора, г Удельная поверхность катализато- 20,4 2,27 0,75 11,0 0,60
ра, м-/г Число атомов металла на поверх- 52 139 87 46 190
ностп катализатора (Х1021) • • Степень превращения 12,06 5,4-7 1,88 5,84 2,27
бензола 88,2 99,7 13,6 85,2 68,2
циклогексана 74,5 10,6 75,0 53,2 1,5
Суммарный выход продуктов рас-
щепленпя
из бензола 12,1 3,7 9,2 9,2 4,6
из циклогексана 10,0 5,3 30,7 3,5 0
Удельная активность катализатора
гидрирующая 7,3 18,2 7,4 14,6 30,0
изомеризующая 6,2 1,9 41,0 9,1 0,7
расщепляющая -
по бензолу 1,0 0,7 5,0 1,6 2,2
ио циклогексану 0,8 1,0 16,9 0,6 0
изомеризующая и расщепляющая активности увеличиваются. Оче-
видно, это следует объяснить тем, что в данном случае носитель —
террана обладает сильнокислотными свойствами, так как алюмо-
силикат обработан плавиковой кислотой 7в. Нанесение WS2 на при-
родный алюмосиликат, обладающий низкой кислотностью, давало
умеренно активный как по гидрирующей, так и по изомеризующей и
расщепляющей активностям катализатор 104.
Многочисленные экспериментальные данные, изложенные в пре-
дыдущих разделах, приводят к несомненному доказательству ионного
механизма изомеризации и расщепления на сульфидных каталйза-
торах. Поэтому причиной отмеченного выше явления понижения
изомеризующей и расщепляющей активностей сульфидов вольфрама
и молибдена при нанесении их на носители, не обладающие кислот-
ными свойствами (далее для краткости называемое «эффектом раз-
ведения»), должно быть то, что на поверхности таких катализаторов
адсорбированное гидрируемое вещество гораздо труднее приобре-
тает положительный заряд. Очевидно, что большую - и меньшую
легкость образования заряженного комплекса при адсорбции гидри-
руемого вещества следует рассматривать только в связи с электрон-
ными свойствами катализатора.
В связи с этим нужно вновь вернуться к сопоставлению сульфи-
дов и окислов молибдена и вольфрама, а также к вопросу о роли
серы в WS, и MoS2. В обычном промышленном катализаторе WS2
266
содержание серы всегда превышает стехиометрическое. Известно,
что уменьшение количества последней понижает активность катали-
заторов 13- 7в. Первые наблюдения об активирующей роли нестехио-
метрической серы описаны 108 еще в 1935 г. При рентгеноструктур-
ном анализе образцов катализаторов WS2 с разным содержанием
серы было показано 109, что они независимо от соотношения W : S
состоят из одной фазы сернистого вольфрама с одинаковыми поряд-
ками решетки. Эти данные указывают на то, что нёстехиометриче-
ская сера, по крайней мере большая ее часть, не входит в кристал-
лическую решетку, и, следовательно, эта часть серы может быть
легко удалена. Было проведено 110 систематическое изучение влия-
ния содержания серы в молибденовом и вольфрамовом катализато-
рах на их гидрирующую и изомеризующую активности (см. табл. 71
и 72).
Таблица 71. Характеристика каталитической активности
катализаторов MoS2 и MoS2 на активированном угле
Катализатор Степень превраще- Удельная активность, отнесенная к 1% Мо
НИЯ • %
условия во ни восстано- витель сстановле- я продол- житель- ность, ч содержа- ние MoS12 % S : Мо (мольное) бензола цикло- гексана гидриру- ющая изомери- зующая
__ 100 — _ __ 11.6 9,1
н2 6 10 2,17 74,6 6,2 12,4 1,0
Н2 6 5 2,10 69,8 3,4 23,3 1,1
н2 6 1 2,00 18,1 1,3 30,2 —
Н2 6 0,1 9,0 0,9 150,0 —
Н2 6 0,01 — 8,6 0,1 —- —
H2S 8 10 —— 80,1 5,7 13,3 1,0
Н2 4 10 2,03 72,2 2,4 12,0 0,4
Н2 1 ' 10 — 76,0 2,4 12,7 0,4
Н2 0,5 10 — 78,2 2,7 13,0 0,5
H2S 8 10 2,28 70,0 10,0 11,7 1,7
H2S 4 10 —- 72,9 6,3 12,1 1,0
H2S 1 io — 76,6 4,5 12,8 0,8
Из приведенных в табл. 71 данных видно, что вынесение сульфида
молибдена на активированный уголь увеличивает его удельную
гидрирующую активность и понижает удельную изомеризующую
активность в сравнении с чистым MoS2. При этом нужно учитывать,
что величины превращения циклогексана, вероятно, завышены из-за
возможности чисто термической изомеризации, которая в холостых
опытах без катализатора составляла 0,1—0,3%. «Разведение» облег-
чает, судя по мольному отношению S : Мо, удаление нестехиометри-
ческой серы (свежеприготовленный катализатор имеет формулу
MoS22 — MoS23).
267
Таблица 72. Влияние нестехиометрической серы
на каталитическую активность сульфида вольфрама
Номер опыта Содержание серы Превра- щение бензола, о/ /о Состав парафино-нафтеновой части гидро- генизата, %
в катали- заторе, % в газах гидриро- вания, мг парафины с6-св цикло- пентан метил- циклипен- тан циклогек- сан
серия опытов
1 27,7 240 88,1 4,6 0,4 86,5 8,5
2 — 9,6 — 1,4 ’ 0,1 76,2 22,3
3 — 6,0 85.1 1,1 — 64,3 34,6
4 — 0,22 86,7 1.3 ' — 57,2 41,5
5 24,0 0,1 86,9 1,1 — 56,5 43,3
6* 27,8 1093 89,4 3,2 — 72.4 24,4
7 — 3 — 1.7 — 68,3 30,0
8 24,1 Следы 88,2 1,2 — 63,4 35,4
9* 27,8 761 92,1 7,4 — 79,3 13,3
10 24,5 19 88,1 3,7 — 62,2 34,1
11 23,5 6 — 2,8 — 60,0 37,2
II серия опытов**
1 24,35 65,6 76,5 15,0 63.8 21,2
2 — 0,32 73,5 13,5 — 62,9 23,6
3 — 0,19 67,8 12,0 — 57,4 30,6
4 22,3 0,27 73,7 7,3 — 51,5 41,2
5* — 470 72,4 7,7 — 59,7 32,6
6 — 104 72.3 5,4 — 54,1 40,5
7 — 9,6 72,3 4,2 — 50,3 45,5
8 — 1,8 69,0 4.8 — 44,5 40,7
9 — 0,3 70,1 6,6 — 44,0 49,4
10 21,3 0,1 — 4,9 — 43,2 51,9
I II серия О П Ы Т О в
1 25,64 322,4 78,0 8,5 0,8 67,0 23,7
2 — 14,4 74,2 5,5 0,5 62,8 31,3
3 — 3,6 74,7 4,4 — 58,6 37,0
4 23,8 2,5 74,2 4,3 — 53,1 42,6
5* 26,3 1250.2 76,6 6,8 0,5 65,2 27,5
6 23,9 31,3 74,8 6,4 0,3 57,9 35,4
7 23,6 4,0 74,9 6,4 0,3 52,4 40,9
* С добавкой CS2, содержащего e5S-
** Процент серы в катализаторах после гидрирования занижен из-за механических
примесей.
Сравнение катализаторов, при получении которых в качестве
восстановителя использовали водород или сероводород, показывает,
что при обработке водородом изомеризующая активность ниже, чем
при обработке сероводородом. Влияние восстановителя и продолжи-
тельности восстановления на гидрирующую активность относительно
невелико, а активности одних и тех же образцов, восстановленных
сероводородом или водородом в течение одинакового времени, сов-
падают. Восстановленный в среде сероводорода катализатор сохра-
няет большее количество нестехиометрической серы, чем восстано-
вленный водородом.
Таким образом, показано, что имеется определенная связь между
изомеризующей активностью сульфида молибдена и количеством
нестехиометрической серы. Так как даже после длительного вос-
становления водородом катализаторы все же содержат небольшое
количество нестехиометрической серы, напрашивается предположе-
ние, что эта неудаляемая восстановлением избыточная сера не
механически увлечена в процессе приготовления катализатора и не
физически адсорбирована на поверхности катализатора, как боль-
шая, легко удаляемая часть нестехиометрической серы, а удержи-
вается более прочными силами.
Для проверки этого предположения были проведены опыты с до-
бавкой сернистых соединений, меченных радиоактивной серой
(табл. 72). В качестве модельной реакции выбрана та же реакция
гидрогенизации бензола, а в качестве катализатора — аналог сер-
нистого молибдена — промышленный катализатор WS2. Гидриро-
вание бензола проводилось в каждой серии опытов без смены
катализатора, в необходимый момент в очередную порцию бензола на
гидрогенизацию добавлялся меченый сероуглерод. По выходам ци-
клогексана и метилциклопентана изучалась динамика изменения
гидрирующей и изомеризующей активности WS2 как следствия изме-
нения содержания в нем нестехиометрической серы.
Как видно из таблицы, в каждой серии опытов с разными образ-
цами промышленного катализатора WS2 качественная картина одна
и та же: при использовании свежего катализатора количество серо-
водорода в газах гидрирования велико (первый опыт), затем оно
резко падает и, уменьшаясь от опыта к опыту, достигает ничтожной
величины. Параллельно уменьшается содержание серы в катализа-
торе, т. е. определенная часть серы выделяется из катализатора легко,
но затем выделение серы идет с трудом и почти прекращается.
После введения в гидрируемое сырье (бензол) примеси сероуглерода
содержание серы в катализаторе сначала увеличивается, но потом
снова снижается.
Сопоставлением радиоактивности серы в газе и в катализаторе
после введения меченого сероуглерода показано, что до 10,6% серы
в катализаторе после удаления избыточной серы было радиоактив-
ным, т. е. произошел обмен между серой сырья и серой катализатора.
Эти данные однозначно доказывают, что подвижность избыточ-
ной серы в катализаторе неодинакова. В опытах со свежими образцами
269
катализатора (первые опыты каждой серии) и в опытах с добав-
кой CS2 глубина изомеризации гораздо выше, чем в опытах, в кото-
рых избыточная сера удалена восстановлением (ср., например,
выходы метилциклопентана в опытах 1, 2, 6 и 9 первой серии с выхо-
дами в опытах 5, 8, 11 той же серии). В то же время превращение
бензола во всех опытах каждой серии изменяется незначительно.
Следовательно, наличие или удаление избыточной серы больше
всего сказывается на изомеризующей активности, т. е. на способ-
ности ускорять ионные реакции.
Гидрирование смесей бензола и циклогексана, в которых один
из компонентов метился радиоактивным углеродом иС, позволило
вычислить (как это описано на стр. 233) мольные доли всех реакций,
протекающих в процессе гидрогенизации бензола (см. табл. 73):
I I
Гидрирование осуществлялось на катализаторе WS2 с атомар-
ным отношением S : W, равным 2,3, 2,0 и 1,9, т. е. с избытком, сте-
хиометрическим количеством и недостатком серы ш.
Из данных, приведенных в табл. 73, видно, что при переходе
от WS23 к WSlj9 суммарная гидрирующая активность практически
не меняется, а изомеризующая активность уменьшается, причем
больше всего за счет собственно изомеризации (z в табл. 73). Оче-
видно, что для понимания этой специфической роли нестехиометри-
Ческой серы нужно изучить и пути ее образования. Для этого были
приготовлены образцы катализаторов WS2, в которых радиоактив-
ной серой 35S метилась сера сульфовольфрамата (образец а) или серо-
водорода (образец б), в присутствии которого раздагался сульфо-
вольфрамат 112:
a (NH4)2WS4-|-H2S
б (NH4)2WS4+H2S
Установлено, что основная часть подвижной серы образуется за
счет сероводорода. Опыты по кинетике обмена серы сырья с серой
катализатора показали, что равновесие достигается при обмене
12,5% серы.
В соответствии с этим интересно отметить, что на поверхности
катализатора находится 9,1% (расчет по размерам элементарной
ячейки) или 11,5% (расчет по молекулярному объему WS2) молекул
WS2, что указывает на возможность обмена с C?5S2 всех атомов серы
поверхности. Предпринятое параллельно изучение полупроводнико-
270
Таблица 73. Мольные доли превращений в ходе процесса гидрогенизации
на катализаторах с различным содержанием ееры
* *
I—смесь С«Н« и C«Hi2; II—смесь С«Н< и С»Нц
Превращение I и Средние данные
WS,., WS, WS,,, ws,,, WS, WS,,, ws„, ws, WS,,,
Гидрирование бен- зола в цикло- гексан (х) . . . ' 0,030 0,032 0,058 0,020 0,031 0,0345 0,025 0,0315 0,046
Гпдроизомериза- ция бензола в метилциклоиен- тан (у) 0,015 0,010 0,03 0,022 0,015 0,0045 0,0185 0,0125 0,004
Изомеризация циклогексана в метилциклопен- тан (z) 0,022 0,008 0,001 0,006 0,004 0,0005 0,014 0,006 0.001
Суммарная гидри- рующая актив- ность (х 4-1/) . . 0,045 0,042 0,061 0,042 0,046 0,039 0,0435 0,044 0,050
Суммарная изоме- ризующая ак- тивность (г/-)-2) 0,037 0,018 0,002 0,028 0,019 0,005 0,0325 0,0185 0,004
вых свойств катализаторов WS2 с недостатком, избытком и стехио-
метрическим количеством серы показало, что электропроводность,
работа выхода электрона, энергия активации электропроводности
зависят от количества серы. Оказалось, что WSa является п-полу-
проводником, а нестехиометрическая сера в нем — акцептор-
ной примесью пз. Аналогичный вывод был сделан в рцботе 114.
Эти данные позволили связать наличие серы в катализаторе WSa
с механизмом изомеризации в ходе деструктивной гидрогенизации.
При избытке нестехиометрической серы увеличивается доля
положительно заряженных участков поверхности, взаимодействие
которых с хемосорбированным атомарным водородом может давать,
согласно положениям теории катализа на полупроводниках, протоны.
Исходя из этих положений (см. также стр. 114,127), можно понять
не только роль нестехиометрической серы — акцепторной примеси,
понижающей уровень Ферми, но и описанный выше «эффект разве-
дения». Очевидно, что тонкое диспергирование сульфида металла на
носителе, создавая широкоразветвленную поверхность, облегчает
удаление избыточной серы.
Все приведенные выше данные относятся к вольфрамовым и,
частично, к другим сульфидным катализаторам. Однако модифици-
рование изомеризующей активности акцепторными примесями имеет,
по-видимому, общее значение. Изменение свойств никелевых катали-
заторов добавкой серы так, что они начинают вести себя как
сильнокислотные, было описано 115 еще в 1957 г. В настоящее время
271
сульфидированные никелевые катализаторы широко применяются в
промышленных процессах гидрокрекинга 116.
Модификация свойств сульфидных катализаторов отмечалась
и в случае других добавок. Часто добавки просто конкурируют
с водородом и гидрируемым веществом при адсорбции на поверх-
ности катализатора. Так, активность WS2 снижается в присутствии
основании 131 76. При изучении влияния добавок фенола, пиридина
и хинолина на гидрирование бензола и его смеси с нафталином пока-
зано 117, что фенол не оказывал существенного влияния, основания
же тормозили гидрирование как бензола, так и фенола. При высоких
температурах, когда адсорбирующиеся основания гидрировались
так же быстро, это влияние было незначительным. Введение неболь-
шого количества воды тормозит гидрирование ароматических угле-
водородов 118. Это явление можно использовать, например, в тех
случаях, когда сырье содержит и бициклические, и моноциклические
ароматические углеводороды и желательно сохранение последних
при селективном превращении первых.
В некоторых случаях примесь действует более глубоко. Так,
например, показано 119, что тщательная очистка сырья и водорода
от следов воды и генерирующего ее кислорода резко изменяет актив-
ность катализатора: возрастает его гидрирующая и понижается изо-
меризующая активность. При этом на -катализаторе WS2 + NiS
на А1аО3 можно было получить из бензола почти чистый циклогек-
сан без заметных количеств метилциклопентана. Поскольку коли-
чества воды ничтожны, ее действие можно объяснить только пониже-
нием уровня Ферми, т. е. изменением полупроводниковых свойств
катализатора.
В модификации активности катализаторов могут играть роль
и физические факторы. Среди них первостепенную роль играет
величина поверхности. Так, при сравнении в реакции гидрирования
фенола различных образцов WS2, освобожденных от физических
загрязнений (в том числе от механически увлеченной избыточной
серы) прокаливанием в вакууме, показано 12°, что активность ката-
лизатора была прямо пропорциональна его удельной поверхности.
Следовательно, развитая поверхность — обязательное условие полу-
чения активного катализатора. В ходе эксплуатации поверхность
катализатора уменьшается за счет упорядочения кристаллической
структуры и образования углистых отложений. Считают 109, что
упорядочение кристаллической структуры протекает не вследствие
перехода из моноклинной в гексагональную систему, как полагали
ранее 16, так как все образцы катализаторов независимо от отноше-
ний S : W состояли из одной фазы с одинаковыми порядками решетки.
Свежий катализатор представляет собой небольшие тонкие пакеты,
образованные беспорядочно смещенными по отношению друг к другу
слоями WS2. Упорядочение при кратковременном нагревании про-
исходит только при температуре выше 700 °C. При этом быстро
уменьшается удельная поверхность 121, в основном за счет пор радиу-
сом 20—80 к. По этой же причине уменьшается и поверхность ката-
272
Таблица 74. Константы скоростей отдельных реакций гидрогенизации
над образцами WS2 различной активности
Катализатор WSi Углево- дород Константа скорости реакции Уменьшение конс- танты скорости по сравнению с ис- ходным катализато- ром в реакции Уменьшение удельной поверхности катализа- , тора по сравнению с исходным образцом
гидриро- I вания изомери- зации расщеп- ления 1_ гидриро- вания изомери- зации । расщеп- ления
Исходный Бензол 2,48 2,52 0,90
Дезактивированный длительной работой Прокаленный » 0,90 2,13 0,58 2,76 1,18 1,55 2,1
до 600 °C )) 1,96 2,69 0,60 1,26 0,94 1,50 1,22
до 750 °C » 0,78 2,13 0,51 3,18 1,18 1,76 2,87
до 1000 °C » 0,17 1,55 0,51 14,59 1,62 1,76 22,0
Исходный Прокаленный Цикло- гексан — 2,08 1,14 — — — —
до 750 °C » — 0,71 0,44 — 2,93 2,59 2,87
до 1000 °C » — 0,11 0,40 — 18,91 2,86 22,0
лизатора, бывшего в длительной работе 122. Оценка каталитической
активности дезактивированных образцов WS2 приведена в табл. 74.
Снижение гидрирующей активности при гидрировании бензола
примерно пропорционально уменьшению величины удельной поверх-
ности, изомеризующая и расщепляющая активности понижаются
в меньшей степени. При изомеризации циклогексана, наоборот,
снижение изомеризующей активности пропорционально уменьшению
удельной поверхности. Эти результаты легко интерпретировать
с позиций представлений о взаимосвязи реакций гидрирования,
изомеризации и расщепления (см. стр. 232 сл.). В самом деле, при гид-
рировании бензола промежуточно образуемый циклогексен, присое-
диняя протон, дает начало ионным превращениям. При гидрогениза-
ции циклогексана первой стадией, затрудненной в той же степени,
что и реакция гидрирования, будет дегидрирование (см. схему на
стр. 235), поэтому уменьшение константы скорости изомеризации
в этом случае пропорционально уменьшению удельной поверхности.
* *
*
В заключение можно отметить, что сложные и взаимосвязанные
превращения углеводородов в ходе парофазной гидрогенизации
определяются и объясняются различным соотношением ионных
и радикальных реакций. Направленность и интенсивность этих
превращений в первую очередь определяются активностью катали-
затора в отношении ускорения гомолитических (радикальных) и
18 Заказ 271 273
гетеролитических (ионных) реакций. Во многих случаях прослежи-
вается связь между химическим составом и свойствами катализато-
ров и химизмом ускоряемых ими превращений углеводородов. Соот-
ветственно намечаются пути модификации катализаторов для реше-
ния технологических задач.
Так, например, для разработки технологических процессов
гидрирования сырья без его изомеризации и расщепления удобнее
применять окислы, а не сульфиды переходных металлов, наносить
гидрирующий агент на носители, лишенные кислотных свойств,
устранять примеси, могущие быть акцепторными добавками (сера,
вода, кислород). В этих условиях реакции изомеризации и рас-
щепления, протекающие по ионному механизму, будут подавлены.
Для максимальной изомеризации и расщепления сырья будут вы-
годны противоположные меры — использование сульфидов вместо-
окислов, применение кислотных носителей, добавка электроноакцеп-
торных веществ. Многие из этих приемов, как это видно из таблиц
первой главы, уже применяются на практике.
ЛИТЕРАТУРА
1. Лозовой А. В., Дьякова М. К. Гидрогенизация топлива в СССР.
М.-Л., Изд. АН СССР, 1940. 269 с.
2. Пучков П. В., ЖОХ, 8, 1677 (1938).
3. Прокопец Е., Филаретов А., Богуславский С., ЖПХ,
И, 1475 (1938).
4. Пучков П. В., Николаева А. Ф., ЖОХ, 8, 1153 (1938).
5. Прокопец Е., Филаретов А., Пычко В., ЖПХ, 11, 1626
(1938).
6. Прокопец Е., Филаретов А., ЖПХ, 11, 1631 (1938).
7. Ando S., J. Soc. Chem. Ind. Japan, Suppl. Bind., 42, 322 (1939).
8. Mac л янский Г. H., ЖОХ, 11, 1221 (1941).
9. Масл янский Г. Н., ЖОХ, 14, 148 (1944).
10. С a w 1 е у С. М., Н а 1 1 С. С. J. Soc. Chem. Jnd., 63, 33 (1944).
11. Н и к о л а е в а А. Ф., ЖОХ, 12, 240 (1942).
12. Николаева А. Ф., Фрост А. Е., ЖОХ, 12, 6464 (1942).
13. Рапопорт И. Б. Искусственное жидкое топливо. М., Гостоптехиздат,
1955. 546 с.
14. Cawley С. W. Research (London), 1, 553 (1948).
15. Хорн У. А., Макафи Дж. В кн. «Новейшие достижения нефтехимии
и нефтепереработки. Т. 3. М., Гостоптехиздат, 1962. См. с. 116.
16. Pier М., Z. Elektrochem., 53, 291 (1949).
17. К а з а н с к и й Б. А., Проблемы кинетики и катализа, 6, 223 (1949);
Казанский Б. А., Либерман А. Л. VIII Мировой нефтяной
конгресс. Москва. 1971. Обзорный доклад. См. с. 8.
18. К а л е ч п ц И. В., С т р а х о в а К. А., С к в о р ц о в Ю. М. В сб.
«Труды ВСФ АН СССР». Вып. 3. М., Изд. АН СССР, 1955. См. с. 88.
19. К а л е ч и ц И. В., К а т к о в а Л. М., Б л и н о в В. Н. В сб. «Труды
ВСФ АН СССР». Вып. 3. М., Изд. АН СССР, 1955. См. с. 84.
20. Калечиц И. В., Каткова Л. М. В сб. «Труды ВСФ АН СССР».
Вып. 3. М., Изд. АН СССР, 1955. См. с. 99.
21. К а л е ч и ц И. В., П а в л о в а К. А. В сб. «Труды ВСФ АН СССР».
Вып. 4. М., Изд. АН СССР, 1956. См. с. 81.
22. К а л е ч п ц И. В., С т р а х о в а К. А., К а т к о в а Л. М. В сб.
«Химическая переработка топлива». М-, Изд. АН СССР, 1957. См. с. 206.
274
23. Ш у йкин Н. И. IV Мировой нефтяной конгресс. Т. 4. М., Гостоптех-
издат, 1956. См. с. 165.
24. Шуйкин Н. И., Черкашин М. И., Изв. АН СССР, ОХН. 1957, 878.
25. Лозовой А. В., СенявинС. А., Советовал. С. В сб.
«Труды ИГИ АН СССР». Вып. 9. М., Изд. АН СССР, 1959. См. с. 122.
26. К а л е ч и ц И. В., П а в л о в а К. А. В сб. «Труды ВСФ АН СССР».
Вып. 4. М., Изд. АН СССР, 1956. См. с. 87.
27. К а л и б е р д о Л. М., К а л е ч и ц И. В. В сб. «Труды ВСФ СО АН
СССР». Вып. 38. М., Изд. АН СССР, 1961. См. с. 152.
28. Л о з о в о й А. В., С е н я в и н С. А. В сб. «Химическая переработка
топлива». М., Изд. АН СССР. 1957. См. с. 180.
29. С h g е n Lu-bin, Ly an Czuan, Acta focalia Sinica, 2, 136 (1957).
30. Л и б e p м а н А. Л., Брагин О. В. и др., Изв. АН СССР, Сер. хим.,
1963. 1737.
31. Barron Y., С о г n е t D. et al., J. Catalysis, 2, № 2, 152 (1963).
32. Pier M., Brennstoff-Chemie, 32, 129 (1951).
33. Г о н и к б е р г М. Г. Вопросы химической кинетики, катализа и реак-
ционной способности. М., Изд. АН СССР, 1955. См. с. 374.
34. К а л е ч и ц И. В. и др., Изв. СО АН СССР, № 10, 3 (1958).
35. К а л е ч и ц И. В., Липович В. Г., В ыхованец В. В. ДАН
СССР, 138, 381 (1961).
36. Калечиц И. В., Л и п о в и ч В. Г., В ы х о в а н е ц В. В-. В сб.
«Труды ВСФ СО АН СССР». Вып. 38. М., Изд. АН СССР, 1961. См. с. 5.
37. К а л е ч и ц И. В., Л и п о в и ч В. Г. и др. Кинетика й катализ, 2,
748 (1961).
38. S inf elt J. Н., RohrnerJ. С., J. Phys. Chem., 65, 2272 (1961).
39. R о h г е г J. С., SinfeltJ. Н., J. Phys. Chem., 66, 1190 (1962).
40. Clement C., Derrien M., Montarnal R., C. r., 258, 919 (1964).
41. Mills G. A., Heinemann H. et al., Ind. Eng. Chem., 45,. 134
(1953).
42. C i a p e t t a E. G., D о b r e s R. M., В aker R. W. In «Catalysis».
Ed. by P. H. Emmett. V. 6. New York, Reinhold publ. Corp., 1958. See
p. 495.
43. Боресков Г. К. В кн. «Гетерогенный катализ в химической промыш-
ленности». М., Госхимиздат, ' 1955, См. с. 5.
44. Н о f f m a n n E. L, L e w i s E. W., Wadley E. F., Ind. Eng.
Chem., 49, 656 (1957).
45. Hosten L. H., Fro men t G. F., Ind. Eng. Chem., Proc. Design
a. Develop., 10, 280 (1971). ,
46. Kouwenhoven H. W., Van Zijll Laugh ou t W. C., Chem.
Eng. Progr., 67, № 4, 65 (1971).
47. Pla tteuw J.C., de R u i t e r H. et al., Ind. Eng. Chem., Prod.
Res. Develop., 6', 76 (1967).
48. В a r b i d g e B. W., Rolfe J. R. E., Hydrocarbon Proc., 45, 169 (1966).
49. Erickson R. A., A s s e 1 i n G. F., Chem. Eng. Progr., 61, № 3,
53 (1965).
50. R awlings A. A., L a w r e n с e P. A. VII World Petroleum Congress.
Mexico. Proceedings. V. 4. London, Elsevier Publ. Corp., 1967. See p. 135.
51. МаслянскийГ. H., БурсианН. P. и др., Химия и технология
топлив и масел, № 1, 11 (1961).
52. Welker J., Chem. Techn., ¥1, № 4, 213 (1965).
53. М е у е г s С. G. , Munns G.W., Ind. Eng. Chem., 50, 1727 (1958).
54. К a p ж e в В. И., Сильченко E. И. и др. В сб. «Исследование и
применение гидрогенизационных процессов в нефтеперерабатывающей
и нефтехимической промышленности». М., Изд. ЦНИИТЭнефтехим, 1968.
См. с. 18.
55. М а с л я н с к и й Г. Н., БурсианН. Р., ЖПХ, 36, 1549 (1963).
56. К о н д о н Ф. В кн. «Катализ в нефтехимической и нефтеперерабатыва-
ющей промышленности». Т. 2. М., Гостоптехиздат, 1961. См. с. 48.
57. Пучков П. В., Нико л а е в а А. Ф., ЖОХ, 9, 280 (1939).
18* 275
58. Пучков П. В., Николаева А. Ф., ЖОХ, 8, 1'939 (1958).
59. Шуйкин Н. И., Бердникова Н.Г., Изв. АН СССР, ОХН,
1957. 455.
60. Шуйкин Н. И., Миначев X. М. и др., Изв. АН СССР, ОХН,.
1957, 598.
61. Калечиц И. В., Кал ибердо Л. М. В сб. «Труды ВСФ СО АН
СССР» Вып. 4. М., Изд. АН СССР, 1956. См. с. 94.
62. К а л ечиц И. В.( П а в л о в а К. А. В сб. «Труды ВСФ СО АН СССР».
Вып. 38. М._, Изд. АН СССР, 1961. См. с. 15.
63. Павлов а К. А., Калечиц И. В. Там же. См. с. 19.
64. К а л и б е р д о Л. М., Калечиц И. В. Там же. См. с. 25.
65. 3 а х а р е н к о В. А. В сб. «Труды ИГИ АН СССР». Вып. 9. М-, Изд.
АН СССР, См. с. 107.
66. Ц и р л п н а Р. Н. Там же. См. с. 139.
67. В ы х о в а и е ц В. В., М о р ж е й В. В. и др. Нефтепереработка и неф-
техимия, № 8, 49 (1965).
68. В ы х о в а н е ц В. В., Л и п о в и ч В., Г. и др., Журн. ВХО им.
Д. И. Менделеева, 10, 465 (1965).
69. В ы х о в а н е ц В. В., Ч е н е ц В. В. и др., Изв. вузов. Химия и хим.
технол., № 3, 432 (1965).
70. В ы х о в а н е ц В. В., К а л е ч и ц И. В. В сб. «Применение меченых
атомов для изучения нефтехимических процессов». М., ВНИИОЭНГ, 1965.
См. с. 59.
71. О г 1 о f f N. A., L i ch a t s ch ef f N. D., Ber., 63, 2179 (1930).
72. Прокопец E. И., Хаджинов В. H., Химия твердого топлива,
6, 347 (1935).
73. Еру И. И., ЖПХ, 7, 145 (1934).
74. На 11 С. С., Fuel, 3, 76 (1933).
75. П р о к о п е ц Е. И., ЖПХ, 10, 126 (1937).
76. Donath Е. Е., Adv. in Catalysis, 8, 239 (1956).
77. И п а т ь е в В. Н., К л ю к в и н Н. А., ЖРФХО, 56, 245 (1924).
78. О г 1 о f f N. A., Ber., 60, 1950 (1927).
79. Hall С. C., J. Soc. Chem. Ind., 54, 208T (1935).
80. О r 1 о f f N. A., Lichatscheff N. D. Ber. 62, 719 (1929).
81. T у p о в a - П о л я к M. Б., ЖОХ, 6, 947 (1936).
82. НемцовМ. С., Усп. хим., 7, 1635 (1938).
83. К Osaka Y., Dan Т., J. Soc. Chem. Ind. Japan, Suppl. Bind., 44,
21 (1941).
84. ShanfieldH., Smith E. A., Can. J. Res., 26B, 763 (1948).
85. I p a t i e f f V. N., Pines H., Meisinger E. E., J. Am. Chem.
Soc., 71, 2685 (1949).
86. Гаврилова A. E., ГоникбергМ. Г. и др., Изв. АН СССР,
ОХН, 1958, 981.
87. Тищенко В. В., Перин Ю. И., ЖПХ, 32, 2204 (1959).
88. Калечиц И. В., Павлова К. А. и др. В сб. «Труды ВСФ СО АН
СССР». Вып. 38. М., Изд. АН СССР, 1961. См. с. 31.
89. Павлова Н. А., Калечиц И. В. Там же. См. с. 61.
90. В е й Ш и - п и н, Цзян-Бин-нань. Там же. См. с. 58.
91. Н о g a n R. I., М i 1 1 s К. L., Lanning W. С., J. Chem. Eng. Data,
7, 148 (1962).
92. TsugeO., YanagiK., Tashiro M., Кору тару, 14, 507 (1962).
93. Богданова T. А., Калечиц И. В., Нефтепереработка и нефте-
химия, № 12, 27 (1966).
94. Калечиц И. В., Богданова Т. А., Нефтепереработка и нефте-
химия, № 1, 22 (1967).
95. Богданова Т. А., Л и п о в и ч Т. В., К а л е ч и ц И. В., Нефте-
химия, 6, 27 (1966).
96. PencevV., Davidova N., Erdol u. Kohle-Brennstoffchemie, 23,
727 (1970).
97. Петров А. А. Химия нафтенов. M., «Наука», 1971- См. с. 209—213.
276
38. Ворожцов Н. Н., Контюг В. А., ЖОХ, 30, 999 (1960).
39. Богданова Т. А., Моржей В. В., Калечиц И. В., ДАН
СССР 149 .461 HQ641
100. Griff i th R. Н., Adv. in Catalysis, 1, 106 (1948).
101. Масл янский Г. H. Шендерович Ф. С., ЖФХ, 14, 1301 (1940)-
102. Лозовой А. В., Сенявин С. А., Воль-Эпштейн А. Б.,
ЖПХ, 28, 175 (1955).
ЮЗ. Калечиц И. В. Проблемы кинетики и катализа, 10, 121 (1960).
104. Калечиц И. В. Докторская диссертация, МИТХТ им. М. В. Ломоно-
сова, 1962.
105. Захаренко В. А., Л о з о в о й А. В. В сб. «Труды ИГИ АН СССР».
Вып. 9. М., Изд. АН СССР, 1959. См. с. 96.
106. Маслянский Г. Н., Б у р с и а н Н. Р., ЖПХ, 35, 816 (1962).
Ю7. К 1 i n е С. Н., Kollonitsch V., Ind. Eng. Chem., 57, № 7, 53 (1959).
108. Прокопец E. И., E p у И. И., Химия твердого топлива, 6, 67 (1935).
109. Самойлов С. М., Рубинштейн А. М., Изв. АН СССР, ОХН,
1959, 1905.
110. Павлова К. А., Пантелеева Б. Д. и др., Кинетика и катализ,
6, 493 (1965).
111. Калечиц И. В., Дерягина Э. Н., Л и п о в и ч В. Г. В сб.
«Применение меченых атомов для изучения нефтехимических процессов».
М., ВНИИОЭНГ, 1965. См. с. 53.
112. КалечпцИ. В., Дерягина Э. Н., Кинетика и катализ, 8, 604
(1967).
ИЗ. Kalechits I. V., Derjaginaetal. Symposium iiher hydrokatalyti-
sche Prozesse in der Erdolverarbeitung und Petrolchemie. Leuna. 1966.
B. 2. S. 76.
114. R о h 1 a n d e г W. Dissertation. Halle-Leuna. 1963.
115. P i n e s H., P о s t W. S., J. Am. Chem. Soc., 79, 1769 (1957).
116. Флюгтер И. К., Ван-Спийкер П. VIII Мировой нефтяной
конгресс. Москва. 1971. Препринт № 12. См. с. 119.
117. К а р ж е в В. И., Орочко Д. И. и др., Химия и технология топлива,
№ 12, 29 (1956).
118. 3 го б а Б. И., К р и ч к о А. А., Нефтехимия, 10, 813 (1970).
119. Левицкий И. И., Г о н и к б е р г М. Г., ДАН СССР, 137, 609 (1961).
120. Самойлове. М. Рубинштейн А. М., Изв. АН СССР, ОХН,
1960, 427.
121. Калечиц И. В., Павлова К. А., С а м о й л о в С. М. В сб.
«Труды ВСФ СО АН СССР». Вып. 18. М., Изд. АН СССР, 1959. См. с. 81.
122. К а л е ч и ц И. В., П а в л о в а К. А., С а м о й л о в С. М. В сб.
«Труды ВСФ АН СССР». Вып. 4. М., Изд. АН СССР, 1956. См. с. 123.
ГЛАВА 6
ПРОЦЕССЫ ГИДРООЧИСТКИ
Процессы гидроочистки углеводородного сырья, нефтяных фрак-
ций и нефти являются в настоящее время, как показано в гл. 1,
самыми распространенными гидрогенизационными процессами. Их
быстрое развитие было предопределено в основном двумя факто-
рами: 1) вредным действием сернистых соединений, содержащихся
в моторных топливах, в ходе эксплуатации двигателей и загрязне-
нием атмосферы сернистым газом после сгорания этих соединений и
2) значительным удельным весом сернистых нефтей в общем балансе
нефтедобычи. Вследствие этого в разработке и освоении процессов
гидроочистки уже достигнуты существенные успехи и еще более
благоприятные перспективы их развития можно ожидать в будущем
(см. стр. 10, 12 сл.). Поскольку гидроочистке подвергаются разные
виды сырья с различным не только количественным, но й качественным
содержанием сернистых соединений, процессы гидроочистки много-
образны (см. гл. 1-) и столь же многообразны чисто химические во-
просы, которые нужно решить для понимания механизма известных
и создания новых процессов гидроочистки. Основными из этих воп-
росов являются: природа и реакционная способность сернистых
соединений нефтей, а также особенности механизма и энергетики
гидрогенолиза С —S-связей, поскольку необходима селективность
их разрыва без затрагивания в одних случаях ординарных связей,
в других случаях — ароматических или олефиновых связей и т. д.
Очевидно, что вопросы химии превращений сернистых соединений
было бы полезно связать со свойствами и составом применяемых ката-
лизаторов. Эти вопросы и будут рассмотрены ниже. Что касается
технологии процессов гидроочистки, они весьма полно рассмотрены
в обзорных работах, например 1-3.
СЕРНИСТЫЕ СОЕДИНЕНИЯ, ВХОДЯЩИЕ
В СОСТАВ НЕФТЕЙ,
И ИХ РЕАКЦИОННАЯ СПОСОБНОСТЬ
Сернистые соединения нефтей, как правило, являются сложными
смесями, состоящими из меркаптанов (тиолов), сульфидов (с откры-
той цепью и циклических), а также дисульфидов и гетероцикличе-
ских соединений. Групповой состав сернистых соединений нефти
весьма различен. Помимо элементарной серы и сероводорода в сырых
нефтях идентифицировано4 111 сернистых соединений.
278
Сравнение групповых составов сернистых соединений 17 образцов
нефтей США и Ирана показало 4, что сероводород содержится только
в одном образце (1,2%), элементарная сера — в семи, причем в
пяти из них от 0,1 до 1,2%, а в двух — 34,6 и 42,5%. Количество
тиолов варьирует от 0 до 45,9%, дисульфидов от 0 до 22,5%, али-
фатических и алициклических Сульфидов от 0 до 20,9%, аромати-
ческих сульфидов и тиофенов от 3,0 до 41,5%. Однако еще очень
большая часть сернистых соединений нефтей не изучена.
В нефтях СССР (Казево, Арлан, Западный Сургут, Новосерафи-
мовка, Яблоневка, Шугурово, Восточная Черновка, Веденовка,
Ишимбай, Туймазы-Бавлы) идентифицировано 18 тиолов, 22 алифа-
тических сульфида, 20 циклических сульфидовв. Подавляющее
большинство всех выделенных соединений относится к легким фрак-
циям.
Фракции, выкипающие до 160 °C, содержат тиолы, алифатические
и алициклические сульфиды, а в болёе высококипящих фракциях
присутствуют замещенные тиофены и бициклические сульфидыа.
Исследование состава высококипящих сернистых соединений
представляет собой трудную задачу, так как большей частью нет
модельных индивидуальных соединений для идентификации. Было
установлено®, что в прямогонном остатке 50% серы входит в состав
тиофеновых колец. Описано 7 подробное исследование фракции
вакуумного газойля 425—455 °C с 2,85% серы.
Сернистые соединения были переведены окислением в сульфоны-или сульфо-
ксиды, последние разделены элюэнтной хроматографией и исследованы спектро-
скопическими и масс-спектроскопическими методами.
Было найдено, что 35,36% всех сернистых соединений содержали
два ароматических кольца (группа бензтиофенов), 51,55% — три
кольца (группа дибензтиофенов) и 13,09% — четыре и более. На
основании полученных результатов сернистым соединениям, входя-
щим в состав вакуумного газойля, были приписаны.7 следующие
структуры:
279
Связь сера—углерод менее прочна, чем связь углерод—углерод
(по усредненным данным 8 соответственно 54,3 и 79,3 ккал/моль).
Однако не это определяет высокую реакционную способность связи
С—S. Прочность связи нужно оценивать с учетом компенсации энер-
гии, идущей на ее разрыв, и энергии образования новой связи с ка-
тализатором в переходном комплексе (см. стр. 112). Так, например,
на никеле 9 энергии разрыва связей С—С, С—N и С—S составляют
(в ккал/моль):
С-С .... 48,8
С—N .... 26,0
С—S .... 5,0
При гидроочистке дистиллятных продуктов удается обеспечить
почти количественную деструкцию связей С—S, практически не
затрагивая связей С—С, т. е. без заметной деструкции сырья 21 3.
Удаление азота протекает много труднее. В работе с модельными
соединениями — дибензтиофеном и 3-метплхинолином, добавляемыми
к лигроину, — показано, что в обычных условиях гидроочистки
(Со -)-Мо на А12О3, 380 °C, 114 кгс/см2) энергия активации реакций
обессеривания составляла только 3,8 ккал/моль, а энергия актива-
ции реакции удаления азота 20,0 ккал/моль. При удалении 90%
серы, удалялось только 40% азота, при удалении 99,5% серы — 75%
азота 10. В другой работе показано, что азот удалялся не только
труднее серы, но и труднее кислорода, диенов и олефинов п.
Термодинамика реакций деструкции сернистых соединений очень
благоприятна х: все реакции тиолов, алифатических и циклических
сульфидов имеют положительные значения логарифма константы
равновесия до 900 К. Только константа равновесия реакции
II || —> C4H10 + H2S
s’/
быстро уменьшается с ростом температуры, ее логарифм достигает
нуля при 540 °C, но в температурных пределах гидроочистки и она
имеет положительное значение логарифма константы равновесия
(рис. 16).
В условиях гидроочистки сернистые соединения алифатического
ряда настолько нестабильны, что термодинамически вероятен их
распад на сероводород и олефин, а также образование сульфидов:
2RSH —> R-S-R + H2S
280
Поскольку логарифм константы равновесия реакции циклизации
бутантиола
C4H9SH || й '
S
при 427 °C равен примерно +0,6, такого рода реакции могут проте-
кать при термическом воздействии на нефть. Этим объясняется тот
факт, что нефтепродукты вторичного происхождения обычно более
богаты тиофенами, чем продукты первичного происхождения 2.
Рис. 16. Зависимость кон-
станты равновесия реакции
восстановления сернистых со-
единений водородом с образо-
ванием насыщенных углеводо-
родов и сероводорода от тем-
пературы:
1 — этантиол; 2 — тиациклогексан;
3 — 2-тиабутан; 4 — тиофен; 5 —
3,4-Дитиагексан.
Исследование кинетики гидрогенолиза сернистых соединений
различного строения показало, что по мере усложнения молекулы
особенно при сопряжении р-электронов атома серы с л-злектронами
бензольных колец, реакционная способность уменьшается. Если
принять за единицу скорость гидрогенолиза дибензтиофена, то отно-
сительные скорости гидрогенолиза сернистых соединений других
классов составят 12:
Производные тиофена и диарилсульфиды . . 1,0—2,0
Алкилсульфиды
первичные ............................ 3,2
вторичные ..........................4,3—4,4
Тиациклопентан и его производные .... 3,8—4,1
Дибензилсульфид........................... 7,0
Меркаптаны (тиолы) ....................... 7,0
В границах одного класса соединений разница в скоростях пре-
вращений невелика. Так, например, константы скорости гидрогено-
лиза семи различных по строению тиациклопентанов и тиациклогек-
санов составляют 13 от 0,26 до 0,78 (при 400 °C), тогда как для
2-метилтиофена при 450 °C — 0,02, и только для цисг2,5-дипро-
пилтиациклопентана — 1,27. Гидрогенолиз диалкилсульфидов над
MoS2 при 100 кгс/см2 и 100—300 °C также показал и, что скорость
281
гидрогенолиза не зависит от величины алкильного остатка и воз-
растает незначительно в ряду:
Изобутил- ч-Бутил- < втор-Бутил- <; тргт-Бутпл-
Превращения моно- и дисульфидов: 3-тиапентана, 5-тианонана,
8-тиапентадекана, 5,6-дитиадекана, тиациклопентана, 2,5-диметил-
тиацикл опентана, 2-гексилтиациклопентана, 2-(3'-фенилпропил)-тиа-
циклопентана и декантиола также протекают с примерно одинако-
выми скоростями 15.
В среднем можно считать, что связь С—S ароматического харак-
тера в 3—4 раза прочнее связи С—S алифатического характера.
Рпс. 17. Хроматограммы вакуумного газойля
п продуктов его гпдроочисткп.
Накопление ароматических колец в молекуле сернистого соединения
уменьшает его реакционную способность. Так, например, скорость
гидрогенолиза тетрафенилтиофена в три раза меньше, чем в случае
дибензтиофена 16. Очевидно, с еще меньшей скоростью будут пре-
вращаться соединения типов, представленных на стр. 279, 280,
поскольку среди них преобладают полициклические системы. При
совместном превращении соединений, содержащих различное число
ароматических колец, это различие проявляется еще четче.
Так, например, отмечали, что в условиях промышленного про-
цесса гидроочистки тиофены и бензтиофены удаляются более трудно,
чем меркаптаны и сульфиды 17, а наиболее трудно удаляется послед-
няя часть серы 18: после80%-нойдесульфуризации наблюдается пере-
лом кривой потребления водорода как функции удаления серы,
т. е. для удаления последней части серы нужны очень глубокие пре-
образования структуры вещества. Трудность удаления высокомоле-
кулярных и ароматизированных сернистых соединений можно также
проиллюстрировать рис. 17, на котором показаны хроматограммы
вакуумного газойля и полученного из него гидрогенизата 7. Хотя
282
общее содержание серы уменьшилось с 2,85% до 0,40%, содержание
дибензтиофенов изменилось гораздо меньше, чем содержание бензтио-
фенов и тиофенов.
Между тем, как показывают многочисленные анализы, сернистые
соединения нефти почти всегда концентрируются в тяжелых фрак-
циях и, следовательно, представлены в значительной степени гетеро-
циклическими соединениями ароматического характера (табл. 75).
Таблица 75. Распределение серы во фракциях дистиллятов
и нефтяных остатков нефтей СССР 5 (в %)
Группы нефтей Группы нефтей
Фракции, °C Сибирь Урал—Волга Фракции, °C Сибирь Урал—Волга
Я 1рбон И о 0 0 S а nogdi Ч Я о ю а Я о я € р. я о
S X а н X X 8 я к
Нефти с —1% серы
До 200 0,8 1,1 1,1 — 8,4
200-300 7,2 7,9 10,5 — 8,6
>300 92,0 91,0 88,4 — 83,0
>400 68,5 72,6 69,0 — —
Нефти с ~2% серы
До 200 0,5 1,2 1,6 8,1 2,7
200-300 4,0 9,1 10,3 10,0 8,2
Нефти с ~2%' серы
>300 95,5 89,7 88,1 81,9 89,1
>400 80,5 75,5 - -
Нефти с —3% серы
До 200 0,4 1,6 1,9 7,6 2,7
200—300 4,5 9,6 9,8 8,1 9,7
>300 95,1 88,9 88,3 84,3 87,6
>400 — 76,7 — — —
Как видно из данных, приведенных в табл. 75, примерно 82—
96% всей серы остается в погонах, выкипающих выше 300 °C, и
69—81% в остатках, кипящих выше 400 °C. Содержание азотистых
соединений в высококипящих фракциях нефтей также выше, чем'
в низкокипящих. Между тем в присутствии азотсодержащих соеди-
нений на всех обычных катализаторах гидроочистки скорости гидро-
генолиза сернистых соединений и гидрирования ненасыщенных
связей уменьшается (см. стр. 295 сл.).
Таким образом, деструкция высокомолекулярных сернистых
соединений применительно к технологическим задачам гидроочистки
сырых нефтей и нефтяных остатков является довольно трудным про-
цессом и его механизм и особенности еще предстоит изучить.
МЕХАНИЗМ ПРЕВРАЩЕНИЙ СЕРНИСТЫХ СОЕДИНЕНИЙ
В УСЛОВИЯХ ГИДРООЧИСТКИ
Вследствие непрочности связи С—S и относительной легкости ее
деструкции в низкомолекулярных сернистых соединениях превра-
щения этих соединений под давлением водорода протекают относи-
тельно просто: гомолитически разрывается связь С—S, свободные
283
связи насыщаются водородом. Продуктами реакции являются, соот-
ветственно, сероводород и насыщенный углеводород.
Так, тиолы: 1-гептантиол, 1-октантиол и циклогексантиол при
126—217 кгс/см2 и 260 °C в присутствии MoS3 количественно превра-
щаются соответственно в гептан, октан и циклогексан 19. Точно
также нацело превращается над MoS3 при 20 кгс/см2 и 200 °C тио-
фенол в растворе циклогексана 2 °. Из 1-октадекантиола на катали-
заторе Со 4- Мо был получен только октадекан 21, тогда как на
V2O3 при 400 °C и атмосферном давлении из 1-бутантиола образова-
лась смесь 80,1% бутана и 19,9% бутена 22. Из нонантиола кроме
нонана был получен динонилсульфид (14%) 23.
Следовательно, тиолы обычно превращаются нацело:
RSH —5. RH + H2S
В мягких условиях или при недостатке водорода возможна реак-
ция
RSH -5- RH+R'CH=CHR' + H2S
а также реакция образования сульфидов:
2RSH —> R—S—R+H2S
Дисульфиды также превращаются нацело: из диоктилдисульфида
получен октан 21. В мягких условиях 23 из динонилдисульфида
в качестве промежуточных продуктов выделены нонантиол и динонил-
сульфид, из дифенилдисульфида — тиофенол и дифенилсульфид.
Следовательно, первичная деструкция дисульфидов идет по связи
S—S, а образовавшиеся тиолы превращаются далее:
R-S-S-R — + 2RSH —- 2RH + 2H2S
R—S— r_i-h2S
Ациклические сульфиды, в том числе жирноароматические суль-
фиды (бензил-к-октил- и фенил-н-децилсульфиды), в растворе при
50 кгс/см2 и 375 °C превращаются полностью, давая соответствующие
углеводороды 21. Такие же результаты получены с дигептил-19
и диоктилсульфидами 21.
В мягких условиях 21 сульфиды (динонилсульфид и дифенил-
сульфид) частично дают соответствующие тиолы:
R—S— R —С R-S—H+RH -iU- 2RH+H2S
Алициклические сульфиды ведут себя подобно ациклическим:
тиациклопентан дает бутан 24, его гомологи и тиациклогексаны —
соответствующие углеводороды и не более 2,5% тиолов 13. Из про-
дуктов гидрогенизации тиациклогексана выделен циклопентан 2Б
284
{его количество невелико, главный продукт — пентан), что доказы-
вает радикальный механизм гидрогенолиза:
н,
-H2S^
СН2(СНг)3СН3 —> | |
Тиофены и бензтиофены также, в конечном счете, давали соответ-
ствующие насыщенные или ароматические углеводороды: тиофен —
бутан 20, 261 27, дибензтиофен — смесь циклогексилбензола и дици-
клогексила 21 или дифенил 28, бензтиофен — этилбензол 27 и т. д.
Первые представления о механизме деструкции тиофена включа-
ли промежуточное образование тиациклопентана 201 2в:
н2)
—+ h-C4H9SH —t м-С4Ню+Н23
Следует отметить, что в качестве катализатора использовали MoS3,
причем присутствие тиациклопентана было доказано 20. Однако при
использовании промышленных алюмокобальтмолибденовых катали-
заторов тиациклопентан в продуктах гидрогенизации не был обна-
ружен 29. В связи с этим было высказано предположение 6 необяза-
тельности промежуточного образования тиациклопентана 29. Это
предположение обосновывалось большей скоростью гидрообессери-
вания тиофена по сравнению со скоростью гидрирования олефинов,
тогда как теоретически следовало бы ожидать меньшей скорости
гидрирования ароматической системы по сравнению со скоростью
гидрирования изолированной двойной связи. Кроме того, на
возможность разрыва С—S-связей в тиофене без насыщения двой-
ных связей указывали известные работы Ю. К. Юрьева 30 по
взаимному превращению тиофена, пиррола и фурана в отсут-
ствие водорода.
Гипотеза, обоснованная в работе 29, была затем подтверждена
экспериментально. В продуктах превращения тиофена на хромовом
и алюмокобальтмолибденовом катализаторах в импульсном микро-
реакторе не были обнаружены ни тиациклопентан, ни меркаптаны 31.
Единственными продуктами были бутан и бутены, а на хромовом
катализаторе — также и бутадиен. Было показано 32, что тиацикло-
пентан не только превращается в бутены и бутан, но дегидрируется
в тиофен; распад тиациклопентана идет через бутантиол, т. е. по
иному пути, чем распад тиофена. Кинетические исследования33
также подтвердили прямое образование бутадиена, а затем бутенов и
бутана из тиофена. Бутадиен был обнаружен 34, 33 и в продуктах
деструкции тиофена на окислах и сульфидах кобальта и молибдена.
285
В соответствии с вышеизложенным можно представить, что пре-
вращение тиофена идет по следующей схеме 31:
... || II —> СН2=СН-СН=СН2
|
У X
СН2=СН-СН2-СН3 СН3-СН=СН-СН3
I трам-
сн3—сн=сн-сн3
цис-
I
> СН3-СН2-СН2-СН3
Скорости отдельных реакций этой схемы (в ммоль/сна 1 г катали-
затора) приведены ниже:
Для хро- Для алюмо-
мового кобальтмо-
натализа- либденового
тора катализатора
Тиофен —>- Бутадиен ... 18
Бутадиен ----> Смесь бутенов 110
Бутен-1 транс-Еутен-2 80
транс-Бутен-2 i/uc-By-
тен-2 .................. 100
Бутены ----> Бутан.......... 2,5
80
> 160
Быстро
»
20
В отдельных случаях изучались вопросы тонкого механизма и
кинетики реакций на поверхности катализаторов. При изучении
механизма гидрогенолиза простейших тиолов (метан- и этантиолы)
на катализаторах MoS2 и WS2 (измерялась скорость образования
различных продуктов и скорость дейтеробмена) показано 36, 37, что
водород в группе SH обменивался на обоих катализаторах, а на
MoS2 — частично и в группе СН3. Легкость активации связей изме-
няется зв> 37 в ряду:
Н—Н
H-S
С—S
С-Н
Превращения этантиола авторы зв* 37
схемой (а — адсорбированное состояние,
представляют следующей
г — газовая фаза):
C2H5SH (Г) . с2н6 (Г) С2Щ (г)
•IX 1'
3 в
C2H5S- с2н5- С2Н4
4 7
(а) (а) (а)
В соответствии с указанным выше стадии 1 и 2 протекают быстрее
любых других, а стадии 8 и 9 лишь немного превосходят по скорости
стадии 6 и 7. Стадии 3 и 4 идут несколько быстрее, чем стадии 6 и 7,
а стадия 5 является самой медленной.
286
Вопросы соотношения реакций гидрирования и гидрогеноййза
вызвали дискуссию: считают, что эти реакции могут идти на разных
или на одних и тех же активных центрах.
Было показано 31, что предварительная адсорбция сероводорода
на катализаторе тормозит превращение тиофена и особенно сильно —
гидрирование бутенов. Однако обработка сероводородом не влияет
на цис-транс-изомеризацию, миграцию двойной связи и гидриро-
вание бутадиена. При изучении зависимости активности катализа-
торов от времени их работы было найдено 35, что активность непре-
рывно уменьшается вследствие отравления сероводородом. Алюмо—
кобальтмолибденовый катализатор отравляется также тиофеном,
метилтиофеном, пиридином и аммиаком 32. Реакция гидрирования
тормозилась этими добавками, а гидрогенолиз тиофена — аммиаком.
Все эти данные привели 31, 32, 35 к постулированию наличия у
десульфирующих катализаторов активных центров двух типов —
сильнокислотных и слабокислотных, имеющих низкое сродство к
электронам. Первые сильно отравляются донорами электронов (серо-
водородом, тиофеном и пиридином), вторые — слабее. На первых
гидрируются олефины и эта реакция сильно подавляется при отра-
влении сероводородом, тиофеном и пиридином. Десульфуризация же
протекает и на слабокислотных центрах, причем идет двумя путями:
присоединением водорода к связи С—S, что приводит к удалению
серы в одну стадию, или путем серии реакций на поверхности.
Известным подтверждением такой двухЦентровой теории явились
кинетические исследования 33, на основании которых выведены сле-
дующие уравнения, коррелирующие с уравнением Ленгмюра — Хин-
шельвуда — уравнение 1 для скорости исчезновения тиофена и урав-
нение 2 для скорости образования бутана:
г =______^РтРн_____
т (i+kTpT + ksps)i
г fe,pBPH 2
с 1 + къРв + кзРз
где /си к' — константы скорости реакции, fop, кд, к% — коэффи-
циенты адсорбции тиофена, сероводорода и бутенов, а р?, рн, рд,
Рв — парциальные давления тиофена, водорода, сероводорода и
бутенов.
Уравнение 2 дает возможность предположить, что бутен десорби-
руется с места своего образования, а затем адсорбируется и гидри-
руется на другом центре. Если данные, полученные в работе 33,
обрабатывать по уравнению, учитывающему образование бутана как
за счет десульфуризации, так и за счет гидрирования бутена на од-
аом центре, то оно не выполняется. Полагают 33, что участки поверх-
ности, занятые тиофеном, относительно неэффективны для гидриро-
вания бутенов.
287
Нужно отметить, в согласии с авторами обзора2, что выводы рас-
смотренных выше работ о двухцентровом механизме не могут прямо
переноситься на реальные условия гидроочистки. Эти опыты прово-
дились при атмосферном давлении и все наблюдаемые закономер-
ности вполне могли объясняться недостатком водорода на поверх-
ности катализатора. Слабой стороной двухцентровой теории является
также отсутствие связи между фактом «наличия» двух видов центров
и составом катализатора. Кроме того, дифференциация центров по
кислотности должна бы приводить к некой гетеролитической модели
разрыва связей, что представляется невероятным, особенно в свете
результатов работ 38, 37: при наличии заряда наблюдался бы быст-
рый дейтерообмен. Вопрос о том, идет ли гидрогенолиз тиофена на
тех же центрах, что реакции гидрирования, или на других центрах,
имеет принципиальное, не только теоретическое, но и технологиче-
ское значение. Если бы была правильной двухцентровая гипотеза,
то открывалась бы возможность обеспечить строгую селективность ги-
дрогенолиза в присутствии олефинов, отравив гидрирующие центры.
На основании данных, полученных при изучении кинетики гидро-
генолиза сланцевого бензина в условиях гидроочистки был сделан
вывод 38, что реакции гидрирования олефинов и гидрогенолиза сер-
нистых соединений сланцевого бензина не влияют друг на друга
и проходят на разных активных центрах 38. Предложены уравнения
скоростей этих реакций, выведенные на основе уравнения адсорбции
Ленгмюра:
^1аОаН
ГО= (1 + аоко + аркр)^
^'aaSaH
т (1+aH2s*H2s)n
где «о, ан. а-p, ад, aH!S соответственно означают активности олефи-
нов, водорода, парафинов, сернистых соединений и сероводорода,
a Zcq , кр и Zch2s — соответствующие коэффициенты адсорбции.
Однако в этой работе варьировалась только объемная скорость, а это
часто может дать несколько кривых и несколько уравнений, особен-
но если условия изменялись незначительно 39. В самом деле, если
по данным работы 38 построить график зависимости lgao от объемной
скорости, получается прямая линия, что можно было бы интерпрети-
ровать как подчинение скорости реакции классическому уравнению
для мономолекулярных нетормозящихся реакций. Кроме того, не
была учтена роль диффузии, хотя гидрирование проводилось на табле-
тированном катализаторе, не варьировалось отношение количеств
олефинов и сернистых соединений. Поэтому вывод о двух родах
активных центров и независимости реакций гидрирования и гидро-
генолиза 38 не может считаться доказанным.
Взаимная связь реакций гидрогенолиза и гидрирования в усло-
виях гидроочистки была изучена на примерах гидрирования гептена
и гидрогенолиза тиофена в присутствии продуктов гидрирования на
288
промышленном алюмокобальтмолибденовом катализаторе 4°’ 41. При
этом в целях достижения максимальной достоверности все пара-
метры процесса варьировались в широких пределах: давление от 8
до 48 кгс/см2, температура от 250 до 475 °C, парциальные давления
тиофена от 0 до 0,1 кгс/см2, олефинов от 0,32 до 1,80 кгс/см2, парафи-
нов от 2,52 до 16,3. кгс/см2, водорода от 4,2 до 45 кгс/см2. В отсут-
ствие тиофена скорость гидрирования олефинов С7 подчиняется сле-
дующему уравнению:
(р \п
3
причем п = 0,35 и 0,55 при 250 и 375 °C соответственно.
При совместном превращении олефинов и тиофена были полу-
чены следующие уравнения.
Для 250 °C:
^&>вР°й9
г _ — - 5
г РоРп
Для 375 °C:
^зРоРн »
Г°“ Р°т'2РЙв
^4Р^вР^3
Гт~_Р°п4
В приведенных выше уравнениях г0 и гт означают соответственно
скорости превращения олефинов С7 и тиофена, к0 — ki — кон-
станты скорости реакций, р0, рп, рт и рп — парциальные давле-
ния олефинов, водорода, тиофена и парафинов. В уравнении 7 в зна-
менателе опущено парциальное давление олефинов, так как его вели-
чина близка к единице (показатель степени равен 0,04).
Из сопоставления этих уравнений видно, что присутствие тиофена
оказывает влияние на скорость гидрирования олефинов, а присут-
ствие олефинов (но не при 375 °C) — на скорость гидрогенолиза тио-
фена. Очевидно, что при их совместном превращении процесс идет
достаточно сложно.
Влияние температуры на скорости гидрирования олефинов и
гидрогенолиза тиофена иллюстрируется рис. 18. Прямолинейный
характер зависимости, а также значения модуля диффузии и коэф-
фициента использования поверхности доказывают отсутствие диффу-
зионных ограничений для обеих реакций.
Когда реакции гидрирования олефинов и гидрогенолиза тиофена
протекают в кинетической области (рис. 18), углы наклона кри-
вых близки, т. е. с повышением температуры скорости обеих реак-
ций изменяются в одинаковой степени. Следовательно, изменением
19 Заказ 271
289
температуры невозможно добиться избирательности той или иной реак-
ции. Участие парциальных давлений тиофена и олефинов в уравне-
ниях 4—6 свидетельствует о конкуренции этих двух реакций, что
следует интерпретировать протеканием их на одних и тех же актив-
ных участках катализатора.
Скорость гидрирования олефинов в отсутствие тиофена при 250 °C
меньше, чем в присутствии тиофена, а при 375 °C — больше. Это
указывает на то, что даже при низкой температуре в присутствии
тиофена происходив определенное изменение характера активных
центров. С повышением же температуры, очевидно, увеличиваются
скорости обеих реакций, и тиофен начинает тормозить превращение
олефинов.
Рис. 18. Зависимость ско-
рости гидрогенолиза тио-
фена (7) и гидрирования
олефинов (2) от темпера-
туры.
Уравнение 3, описывающее гидрирование олефинов в отсутствие
тиофена, хорошо корреспондирует с уравнением, выведенным42
на основе изотермы Фрейндлиха, если предположить, что лимитиру-
ющей стадией является хемосорбция водорода.
При гидрировании олефинов в присутствии тиофена, во-первых,
скорость гидрирования олефинов при 250 °C возрастает, а при 375 °C
уменьшается по сравнению с аналогичными опытами без тиофена;
во-вторых, значительно уменьшается кажущаяся энергия актива-
ции процесса: 14,5 ккал/моль против 22,2 ккал/моль и, в-третьих,
показатели степеней в уравнениях 4 и 6 отличны от таковых в урав-
нении 3. Вследствие разницы в показателях степеней ро и рп в урав-
нениях 4 и 6 последние уже не корреспондируют с выводимым по
Квану 42 уравнением, аналогичным уравнению 3, и, следовательно,
в присутствии тиофена наиболее медленной ступенью не является
хемосорбция водорода. Все эти явления нельзя объяснить влиянием
тиофена, как такового; наоборот, он должен бы во всех случаях тор-
мозить гидрирование олефинов, занимая часть активной поверхности
и потребляя водород, нужный для обеих реакций.
Наиболее вероятно (и это соответствует представлениям о непо-
средственном разложении тиофена до гидрирования), что присутст-
290
ше тиофена изменяет активные центры катализатора; окислы пре-
щащаются (вероятно частично, на наиболее активных участках
юверхности) в сульфиды, гидрирующая активность которых выше
см. стр. 264). В результате этого легко хемосорбирующиеся на по-
1ерхности олефины приобретают способность взаимодействовать
с молекулярным водородом. Скорость гидрирования возрастает и по-
[ижается кажущаяся энергия активации. Однако при повышении
емпературы одновременно усиливается и гидрогенолиз. Тиофен
гожет конкурировать с олефинами путем вытеснения их с поверх-
1ости и потребления адсорбированного на поверхности водорода.
1оэтому, хотя скорость гидрирования олефинов в присутствии тио-
фена при 375 °C много выше, чем при 250 °C, она несколько меньше,
сем в отсутствие тиофена, несмотря на то что в последнем случае
зеакция протекает на неактивированном серой катализаторе. Сопо-
ставление уравнений 4 и 6 с различными вариантами механизма
'идрирования 42 приводит к единственной аналогии — уравнению,
выведенному из предположения о поверхностной реакции олефина
с водородом, как наиболее медленной стадии процесса.
Таким образом, механизм гидрогенолиза сернистых соединений
звляется сложным превращением, в котором возможна прямая, без
1редварительного насыщения водородом деструкция С—S-связей;
сера влияет на активность катализатора, а весь процесс склады-
зается из многих взаимно влияющих и конкурирующих реакций.
КИНЕТИКА ПРОМЫШЛЕННЫХ ПРОЦЕССОВ
ГИДРООЧИСТКИ. СЕЛЕКТИВНОСТЬ
ГИДРОГЕНОЛИЗА СЕРНИСТЫХ СОЕДИНЕНИЙ
Сложность процесса гидроочистки и взаимосвязанность (или
взаимная конкуренция) протекающих в нем реакций етавит вопрос
э влиянии условий и катализаторов процесса на селективность
'идрогенолиза сернистых соединений. Как правило, целью процесса
гидроочистки (кроме процессов гидрокрекинга мазутов для получе-
гия малосернистых котельных топлив) является устранение серни-
стых соединений без гидрирования ароматических колец и олефинов,
г также гидрогенолиза связей С—С. Избежать гидрогенолиза свя-
зей С—С относительно легко; поскольку в условиях преимуществен-
гого протекания радикальных реакций их скорости определяются
температурой.
Что касается гидрирования ароматических и олефиновых связей,
избежать его, очевидно, можно только создав условия, при которых
скорости этих реакций различны. Олефины гидрируются много
быстрее, чем ароматические соединения, а диены с сопряженными
связями — много быстрее олефинов (см. стр. 149, 168, 237). Между
гем, гидрогенолиз самой устойчивой системы — тиофена — протекает,
как было показано в предыдущем разделе, через прямую деструкцию
2—S-связей с последующим гидрированием диеновой системы. Если
же сернистое соединение (тиолы, сульфиды, дисульфиды и др.)
19*
291
имеет насыщенный характер, то гидрогенолиз связей С—S протекает
с наименьшей затратой энергии, что объясняется компенсацией ее
при образовании переходного комплекса (см. стр. 134, 280). Сле-
довательно, обеспечить селективный гидрогенолиз сернистых соеди-
нений теоретически можно, причем особенно легко по отношению
Рис. 19. Зависимость селективности катализаторов
от условий процесса:
1 — СоМоО. на А13О3; 2 — МоО3 на А13О3; 3 — WS3 + NiS'
4 — WS3 + NiS на A13O3; 5 — Pt + F на A13O3; 6 — Ni на
A13O3 + SiO3; 7 — CoMoO, на A1,O3 (гидрирование олефинов).
к ароматическим соединениям и труднее по отношению к олефинам.
Последняя задача осложняется тем, что, как показывают кинети-
ческие исследования 331 41, сернистые соединения и олефины тормозят
превращения друг друга.
На рис. 19 показана 29 зависимость селективности различных
катализаторов при гидрировании бензола и тиофена от условий
процесса. Видно, что между скоростями гидрирования бензола и
гидрогенолиза тиофена разница настолько велика, что подбором
условий на каждом катализаторе можно обеспечить почти количест-
венное удаление тиофена без затрагивания или с минимальным затра-
292
гиванием бензола. Катализаторы СоМоО4 на А12О3, Ni на SiO2 4-
4- А12О3 и МоО3 на А12О3 имеют удовлетворительную селективность
в очень широком интервале объемных скоростей при давлении 7—
17 кгс/см2. Условия удовлетворительной селективности других
катализаторов более узки. Несколько менее селективны катализа-
торы WS2 4- NiS и WS2 4- NiS на А12О3, обладающие наиболее
высокой гидрирующей активностью. Легкость подбора условий селек-
тивной очистки ароматических продуктов определила создание
Рис. 20. Диаграмма селективности катализаторов по отношению к гидроочистке
олефинов:
I — Pt + F на А1аО3; II — NiS на AltO, Si О,; III — Ni на А1,О, ЙО,; IV — МоО,
на А1,О3; V — СоМо04 на А1,О3;
1 — 17 кгс/см’, 375 °C, 3 ч"»; 2 — 7 кгс/см’, 375 °C, 10 ч" 3 — 2,5 кгс/см', 315 °C; 8 ч-1;
4 — 100 кгс/см’, 360 °C, 1,5 ч-’; 5 — 4 кгс/см’, 300 °C, 6 ч~>; 6 — 7 кгс/см', 490 °C, 1,2 ч-’;
7 — 17 кгс/см’; 250 °C, 3 ч-’; 4 — 7 кгс/см’, 375 °C, 10 ч">; 9 — 2,5 кгс/см’, 315 °C, 8 ч*>;
10 — 7 кгс/см’, 375 °C, 10 ч--1; 11 — 2,5 кгс/см’, 315 °C, 8 ч-1; 12 — 17 кгс/см’, 250 °C, 3 ч* ;
1з — 2,5 кгс/см’, 315 °C, 8 ч'1; 14 — 7 кгс/см’, 375 °C, 10 ч-1. Точки без номера относятся
к условиям, когда превращение обоих компонентов больше 90%.
ряда процессов гидроочистки коксохимического бензола и концентра-
тов ароматических углеводородов /см. гл. 1).
На рис. 19 показано для сравнения изменение селективности
при гидрировании олефинов одного из лучших катализаторов
гидроочистки СоМоО4 на А12О3. Эта кривая лежит выше всех
других кривых, иллюстрируя тем самым трудность обеспечения
селективного гидрогенолиза тиофена с сохранением олефинов,
что хорошо согласуется с указанными выше особенностями
механизма его превращения. Эта трудность иллюстрируется
также диаграммой 29, приведенной на рис. 20; болыпин-
293
ство точек, соответствующих различным катализаторам и разным
условиям, располагается в правом верхнем углу диаграммы, что
соответствует почти полному превращению как олефинов, так и тио-
фена. Однако в более мягких условиях возможно разделение испытан-
ных катализаторов по селективности. Как видно из рис. 20, селек-
тивность платинового катализатора неудовлетворительна: его ак-
тивность к гидрогенолизу тиофена снижается быстрее, чем активность
к гидрированию олефинов. Катализаторы WS.2 + NiS на А12О3
и WS2 NiS даже в мягких условиях дают почти количественное
превращение обоих компонентов. Линии, характеризующие изме-
нение селективности других катализаторов, имеют наклон более
45°, т. е. при смягчении условий скорость гидрирования олефинов
уменьшается быстрее, чем скорость гидрогенолиза. Катализатор
СоМоО4 на А12О3 несколько лучше других, но даже самый лучший
результат, полученный при 7 кгс/см2, 375 °C и 10,0 ч-1 (точка 14),
дает 81,6% превращения тиофена при сохранении только 54,3%
олефинов.
На основании данных по кинетике одновременного гидрирования
олефинов и гидрогенолиза тиофена (уравнения 4—7) можно вывести
уравнения зависимости селективности катализатора от условий про-
цесса.
Введем обозначения: л — общее давление (в кгс/см2); п — моль-
ное отношение водорода к жидкому продукту; ср, ст — доли моле-
кул олефинов и тиофена в общем числе молекул (без водорода).
По уравнению материального баланса:
dcO _ ГО
dcrjt т
Подставляя значения го и гт 113 уравнений 4—7 и вводя принятые
обозначения, получим следующие выражения.
Для 250 °C:
dcO __ к0 Ср58(1—Ср)2.37”0,55 / Л \0,70
dcT , кт 4’» \ га + 1 ) 9
Для 375 °C:
dcO _ кЬ е^по,б5 , л . 0 36
dcT к'т ' 4-05 (1 - С0)«,26 \«+1 / °
При 250 °C превращение тиофена слишком мало, и анализ урав-
нения 9 ничего не даст. Анализ уравнения 10 показывает, что селек-
тивность прямо пропорциональна величине
Это значит,
что при прочих равных условиях, если мы хотим увеличить селек-
тивность в 4 раза (например, с 1,0 до 0,25), то давление должно быть
уменьшено примерно в 50 раз. Очевидно, что это нереально, так как
294
в вакууме процесс вообще не будет протекать. Аналогичные рассужде-
ния можно сделать и в отношении варьирования величины п. Выше
было показано, что и варьирование температуры не может дать су-
щественного эффекта. Следовательно, варьированием условий нельзя
или очень трудно обеспечить достаточно полное превращение тио-
фена без существенного гидрирования олефинов.
Действительно, еще в 1949 г. при гидродесульфуризации бен-
зинов крекинга отмечали 43 одновременное весьма существенное сни-
жение бромных чисел. С тех пор опубликовано много работ, причем
направление исследований можно условно разбить на две группы:
1) проведение процесса при высоких температурах, термодинами-
чески более благоприятных для сохранения олефинов и 2) подбор
таких мягких условий процесса, чтобы скорость гидрирования оле-
финов снижалась больше, чем скорость гидрогенолиза сернистых
соединений. Из публикаций первого направления можно упомянуть
работу 44, в которой указывается, что при гидроочистке сланцевого
бензина на катализаторе СоМоО4 на А1203 выгоднее применение
высоких температур, сообщение 45 о методе гидроочистки буроуголь-
ного легкого масла НТМ (катализатор WS2 4- NiS на А12О3, да-
. вление 25—50 кгс/см2, температура 440—530 °C); сообщение48 о гид-
роочистке колорадской сланцевой смолы. Что касается второго на-
правления, то была показана 47 целесообразность применения сред-
них давлений (порядка 30 кгс/см2) для селективного удаления серы,
найдено48, что на катализаторе WS2 -j-NiS снижение давления до
5 кгс/см2 уменьшает скорость гидрирования олефинов больше, чем
скорость гидрогенолиза сернистых соединений. Аналогичное на-
блюдение было сделано 38 ранее для катализатора СоМоО4 на А12О3.
Однако данз 90%-ной селективности до сих пор достигнуть не уда-
лось. Лучший из опубликованных результатов принадлежит А. В. Ага-
фонову с сотр. Им удалось добиться снижения содержания серы
в бензине вторичного происхождения с 0,41 до 0,12% (обессерива-
ние 69%) при уменьшении иодного числа с 90 до 70 мг 12 на 100 мл
(гидрирование олефинов 22%)49.
Для скоростей гидрирования олефинов и гидродесульфуризации
были выведены 50 эмпирические уравнения первого порядка. Уста-
новлено, что гидрирование идет легче в первом слое катализатора,
а десульфуризация — медленнее и протекает во всем каталитиче-
ском объеме.
Для селективной очистки олефинового сырья (см. также2)приме-
нялись специальные приемы. Так, поскольку сернистые соединения
концентрируются в основном в высококипящей части бензина, схема
гидроочистки включала 51 отгонку малосернистой легкой части, гид-
роочистку тяжелой (с применением ингибитора реакции гидриро-
вания) и затем их смешение. Однако эта схема непригодна для бен-
зинов с равномерным распределением сернистых соединений, к ко-
торым, в частности, принадлежат сланцевые бензины.
Позднее было изучено52 влияние азотсодержащих соединений
(добавок пиридина) на селективность гидроочистки олефинов. Однако
295
даже лучшие результаты были все же недостаточно хороши: на
никелевых катализаторах при 371 °C и 20 кгс/см2 при глубине
обессеривания 63,6% гидрировалось все же 23,9% олефинов.
При полном предотвращении гидрирования олефинов обессеривание
составляло только 45%. Подбор различных азотсодержащих соедине-
ний (природных, выделенных из сланцевых смол и индивидуальных)
не дал существенных результатов вз. Все они тормозили обе реак-
ции примерно пропорционально своей основности, но селективность
достигалась только при незначительных степенях превращения.
Малоэффективным оказалось также внесение в алюмокобальтмолиб-
деновой катализатор основного компонента — окиси бария В3.В луч-
ших случаях удавалось сохранить 75—85% олефинов, но при этом
удалялось только 56—47% серы. Тормозящее влияние азотсодер-
жащих соединений на реакции гидрирования и гидрогенолиза сле-
дует связывать, видимо, с конкурирующей адсорбцией, поскольку
они оказывают влияние на обе реакции. Так, например, изучалось 84
влияние азотистых соединений различных классов (аминов, пир-
рола, хинолина) на скорость гидрирования средних дистиллятов
и на скорость удаления азота. Влияние основности соединений было
незначительным, но труднее удалялись азотсодержащие соединения
более высокого молекулярного веса, т. е. более прочно адсорбиру-
емые.
Таким образом, селективность гидроочистки ароматического
сырья обеспечивается легко, а олефинового — очень трудно.
Кинетика гидроочистки реальных промышленных видов сырья
весьма сложна. Сложность определяется различием в скоростях
превращения различных классов сернистых соединений (иногда на
порядок и больше), а также изменением активности катализатора в х о-
де процесса (см. стр. 282, 291). Кроме того, всегда, особенно в случае
тяжелых продуктов, приходится считаться с большой вероятностью
диффузионных ограничений. Наконец, явления торможения реак-
ции сероводородом, отмеченные при гидрогенолизе индивидуальных
соединений 31-35, наблюдаются и в условиях промышленного про-
цесса 2’ вв. Несмотря на все перечисленные трудности было выведено
много кинетических уравнений для расчета скоростей гидроочистки.
В одной из первых работ было предложено 86 уравнение первого
порядка:
1 J-1-k
*ps PHtPO
11
где ps и *Ps — парциальное давление сернистых соединений в гид-
рогенизате и в сырье, ро — константа скорости реакции при
данных температуре, парциальных давлениях водорода и паров
сырья, т — условное время контактирования. Было показано 6в,
что до глубины обессеривания 95% и в случае узких фракций это
уравнение удовлетворительно описывает скорость процесса. Однако
для широких фракций оно неприменимо, так как в этом случае
скорость десульфуризации является суммой различных скоростей
296
в уравнениях первого порядка для узких фракций. Константы ско-
ростей десульфуризации, экстраполированные к нулевому парци-
альному давлению жидких продуктов (бесконечное разбавление водо-
родом), мало зависели от давления водорода, а соответствующие кон-
станты при парциальном давлении жидких продуктов 2,5 кгс/см2—
весьма существенно. Это интерпретировалось как явление более
предпочтительной адсорбции жидких продуктов, вследствие чего
при высоких парциальных давлениях последних поверхность ката-
лизатора становится труднодоступной для водорода и его давление
начинает определять скорость реакции.
Хотя по мнению других авторов 2, эти опыты были недостаточно
строги методически, так как часть сырья оставалась в жидкой фазе
и не достигалась нужная глубина обессеривания, качественные выводы
этой работы нашли подтверждение в последующих исследованиях:
первый кинетический порядок по веществу, логически вытекающий
из механизма реакции гидрогенолиза, соблюдается только в более
простых случаях, а для сложного по составу сырья уравнение пре-
вращается в сумму разных уравнений. —
Позднее уравнение 11 было упрощено (не учитывалось влияние
циркулирующего водорода) 10:
где х0 и х — концентрация серы в сырье и продукте, a Sa — объем-
ная скорость подачи сырья.
Было предложено 67 также уравнение, аналогичное уравнению 11,
но включающее вместо т плотность сырья, его средний молекуляр-
ный вес, объемную скорость, отношение водород : жидкие продукты,
общее давление. При больших отношениях водород : жидкие про-
дукты порядок по водороду был нулевым 68.
Наконец была показана Б9 применимость уравнения первого
порядка как по сырью, так и по водороду, выведенного на основе
изотермы Ленгмюра. Однако приложение его к скоростям гидроге-
нолиза индивидуальных соединений (триметилбензтиофенов и ди-
бензтиофена) показало столь значительную разницу, что уравнение
пришлось сильно усложнить. Поэтому для промышленного сырья,
особенно для сырья широкого состава или высококипящего, под-
бирали любые эмпирические уравнения, лишь бы они давали луч-
шую сходимость, чем уравнения первого порядка.
Так, на основании результатов опытов обессеривания вакуум-
ного остатка кувейтской нефти с 5,45% серы при 35 и 70 кгс/см2
было выведено следующее уравнение в0:
V*
— к • —
1 — й V
где с = 1 — (отношение содержания серы в продукте к содержа-
нию ее в сырье), к — константа скорости реакции; г? — объемная
скорость. При этом авторы 60 не считают, что второй кинетический
297
порядок—истинный, просто он является лучшим приближением
суммы многих уравнений первого порядка для отдельных классов
и групп сернистых соединений. Вывод о кажущемся втором порядке
подтвержден и в других работах 81-83.
Обработка многочисленных данных по гидроочистке вакуум-
ного газойля 61 со степенями обессеривания от очень малых до 90—
95% по трем уравнениям первого порядка (Фроста 81, Вильсона 10
и Хуга 56), не дала линейной зависимости в полулогарифмических
координатах. Напротив, уравнение Бойтера и Шмида 8°, давало
линейную зависимость в координатах [1^7- v]- °днако кон-
станта скорости не давала прямой в координатах ^lg &, Энер-
гии активации для различных температурных интервалов составили
(в ккал/моль):
350-380 °C .... 33,0
380—410 °C ... 26,0
410-430 °C ... 5,3
По мнению авторов в1, при высоких температурах, когда ско-
рость обессеривания резко возрастает, суммарное превращение
определяется диффузией. Это было подтверждено уменьшением диа-
метра частиц катализатора с 0,3 до 0,05 см, после чего резко воз-
росла скорость реакции, удовлетворяя уравнению второго порядка.
Диффузионные ограничения отмечались и во многих других рабо-
тах 85~88. При этом в ряде работ было показано, что на скорость
реакции не влияют условия течения масла 88- 89, т. е. диффузионные
ограничения не относятся к области внешней диффузии. Эффектив-
ность использования поверхности катализатора существенно не из-
менялась со степенью десульфуризации, а также с изменением гра-
ниц кипения сырья, что, возможно, указывает (по 2) на то, что с ро-
стом молекулярного веса уменьшение реакционной способности
отдаляет момент перехода в диффузионную область. Эти данные,
а также данные работ 88< 88 указывают на внутренние диффузионные
ограничения.
Подобные диффузионные ограничения были тщательно изучены
в работе 85, в которой использовались образцы катализатора с раз-
личными суммарным объемом пор (размер таблеток 3x3 мм) и
их структурой. Экспериментальные данные в области обессеривания
75,0—97,5% давали семейство кривых в полулогарифмических ко-
ординатах 4")’ г^е > ^пр — соответственно содержание
серы в сырье и продукте, v — объемная скорость. Если, согласно
теории диффузии в порах ’°, объемную скорость заменяли скоррек-
тированной объемной скоростью, равной
V
spo.s
(где v — объем-
ная скорость, s — удельная поверхность, р — средний радиус
иор), то кривые трансформировались в прямые. Даже в случае наи-
более слабо спрессованных таблеток (плотность 0,60 г/см3) лимити-
298
рующей стадией была диффузия, что подтверждено опытами с из-
мельченными таблетками.
На основании полученных результатов авторы 69 сделали выводы
не только о применимости теории диффузии Тиле — Уиллера к про-
цессу гидроочистки, но и о необходимости использования бидисперс-
ных катализаторов с «серией узких пор, чтобы создать достаточную
поверхность, и с серией широких пор, чтобы сделать эту поверх-
ность доступной». Большие поры были ими произвольно определены,
как поры с радиусом более 400 А. По-видимому, эти выводы имеют
большое практическое значение, так как начат выпуск подобных
бидисперсных катализаторов 2.
КАТАЛИЗАТОРЫ ГИДРООЧИСТКИ
Наиболее употребительным катализатором гидроочистки яв-
ляется алюмокобальтмолибденовый. Лишь в специальных случаях
применяются сульфидные катализаторы, а в последнее время'стали
использоваться алюмоникельмолибденовые 2. Иногда алюмокобальт-
молибденовый и алюмоникельмолибденовый окисные катализаторы
называют молибдатами кобальта или никеля. Фактически они в про-
цессе гидроочистки образуют сложные системы, содержащие Co(Ni),
Мо, кислород и серу. Данные о генезисе и природе активных компо-
нентов этих катализаторов весьма ограниченны.
На основании изучения магнитных свойств несульфидированных
катализаторов был сделан вывод 71, что их компонентами являются
А12О3, СоА12О4, СоО, МоО3, СоМоО4 и некий «комплекс окисей ко-
бальта и молибдена». Активными составляющими являются те,
которые образуют окисные ионы октаэдрической формы, т. е. СоО,
СоМоО4 и «комплекс». СоО и СоМоО4 только умеренно активны,
а главным носителем активности является упомянутый комплекс,
которому приписывается 71 «дефектная» структура. Найдено 72, что
двухвалентный кобальт распределен равномерно между тетраэдри-
ческой и октаэдрической формами. По величине доли кобальта,
способного восстанавливаться водородом, и величине магнитного
момента катализатора, можно (по 71) вычислить долю «активного
кобальтового комплекса».
На рис. 21 сопоставляются величины магнитных моментов, доли
восстанавливаемого кобальта и «активного комплекса» как функции
температуры прокаливания катализатора. Ход кривых рис. 21
интерпретируется 71 так, что при более низких температурах ко-
бальт образует СоО и СоА12О4. С ростом температуры концентрация
тетраэдрического Со растет за счет СоО, вследствие чего магнитный
момент падает. При температуре выше 650 °C начинает образовы-
ваться СоМоО4, конкурируя с СоА12б4, и магнитный момент возра-
стает. Экстремальное изменение магнитных свойств отмечено и
в работе 72. Концентрация «активного комплекса» зависит от на-
чального атомарного отношения кобальта и молибдена, давая мак-
симум при отношении Со : Мо, равном 0,3—0,4 при температуре
прокаливания 538 °C 71.
299
Однако между концентрацией «активного комплекса» и активно-
стью катализатора зависимость не прямолинейная, а экстремальная
(рис. 22): максимум активности наблюдается при содержании актив-
ного комплекса 0,18%. Вычислено71, что такая доля активного
комплекса получается при начальном атомарном отношении Со :
: Мо, равном 0,2.
Прохождение кривой через максимум возможно связано с изме-
нением состава катализатора в ходе процесса. Полагают 71, что
в условиях процесса катализатор состоит из смеси А12О3, СоА1204
(оба неактивны), Co9S8 (слабо активен), MoS2 (умеренно активен
Температура прокаливания, °C
Рис. 21. Зависимость магнитного
момента и долей кобальта, входя-
щего в активные и неактивные фор-
мы. от температуры прокаливания 71.
Рис. 22. Зависимость константы ско-
рости гидроочистки западно-техасского
газойля с 3,0% серы при 400 °C от со-
держания «активного комплекса» в раз-
личных образцах алюмокобальтмолиб-
денового катализатора.
но промотируется кобальтом) и, возможно, МоО2 (слабо активен).
Заключительный вывод автора 71 все-таки неопределенен: «Настоящий
катализатор — MoS2, промотированный кобальтом. Последний дол-
жен рассматриваться как «активный кобальт» и является предпо-
ложительно невосстановленной или сульфидированной формой ко-
бальта. Его промотирующий эффект может быть некоей формой
«допинга» MoS2 или следствием образования смешанного сульфида
Со и Мо, точное строение которого не установлено».
При помощи современных физико-химических методов (рентгено-
структурный анализ, спектроскопия, парамагнитный резонанс и
др.) единственным соединением, которое удалось обнаружить в чи-
стом (не на носителе) алюмокобальтмолибденовом катализаторе 73,
был молибдат кобальта — СоМо04, существующий в двух модифи-
кациях, переходящих друг в друга приблизительно при 35 и 420 °C.
Молибден в обеих модификациях находится в октаэдрических си-
стемах. Одна из модификаций, менее симметричная, имеет «незави-
300
симые Мо—О-связи», которым авторы 73 приписывают важную роль
в каталитической активности. Однако в промышленном (на носителе)
катализаторе не был обнаружен 73 и СоМоО4. Полагают, что в этом
случае молибден присутствует в виде МоО3, образует мономолеку-
лярный слой и занимает примерно 20% поверхности катализатора.
Таким образом, проведенные до сих пор исследования вскрывают
крайнюю сложность структуры и состава алюмокобальтмолибдено-
вых катализаторов, указывают на несомненное взаимное влияние
структуры и состава и роль серы в их модифицировании. Однако
основной вопрос о природе Главного активного компонента и при-
чине его большей активности остается еще без ответа.
НЕКОТОРЫЕ ПРОБЛЕМЫ ДАЛЬНЕЙШЕГО РАЗВИТИЯ
ПРОЦЕССОВ ГИДРООЧИСТКИ
В гл. 1 (см. стр. 10—13) показано, что значимость процессов
гидроочистки будет, видимо, бурно возрастать по мере вовлечения
в переработку огромных масс сернистых нефтяных остатков. Хотя
такие процессы уже применяются в промышленных масштабах,
многие задачи, в первую очередь предотвращение дезактивации
катализаторов, еще ждут своего решения.
Особое затруднение представляет высокое содержание металлов
в нефтяных остатках 74, что приводит к высокой концентрации ме-
таллов в катализаторе — около 15—20% (до 50%) от массы свежего
катализатора. Это понижает активность катализатора и очень за-
трудняет его регенерацию. Замена дезактивированного катализа-
тора свежим увеличивает стоимость процесса, которая растет с по-
вышением содержания металлов в сырье.
Другой проблемой является наличие асфальтенов и высокомоле-
кулярных соединений ароматического характера, которые дезак-
тивируют катализатор, образуя на его поверхности кокс. Исключи-
тельная важность борьбы с коксообразованием побудила начать
систематическое изучение химической природы асфальтенов и вы-
сокомолекулярных соединений нефти, а также механизма образова-
ния кокса. К настоящему времени сделаны лишь первые шаги, но
следует ожидать быстрого развития такого рода исследований.
Важно отметить, что содержание в нефтяных остатках асфальтенов,
серы и металлов изменяется обычно симбатно. Это увеличивает* тех-
нологические трудности, ас чисто химической точки зрения позволяет
предположить, что металлы и сера входят в состав асфальтенов.
Асфальтены дают четкие сигналы ЭПР 2, указывающие на наличие
в них металлов с неспаренными электронами. Найдено 75, что пор-
фирины, входящие в состав асфальтенов, могут иметь один или болбе
атомов серы в основной структуре.
Что касается склонности к образованию кокса, то наибольшую
опасность должны представлять сложные, непредельные, но труд-
ногидрируемые структуры или структуры, легко дающие непре-
дельные, полимеризующиеся осколки. Показано 7в, что образование
301
кокса ускоряют сернистые соединения, содержащие связь сера — али-
фатический углерод, но не ускоряют дифенилсульфид и дибензтио-
фен. Эти явления нужно связывать с легкостью деструкции связи
С—S и вступлением ненасыщенных остатков в реакции конденсации.
В работе 77 это доказано прямыми опытами измерения радиоактив-
ности кокса на катализаторе гидроочистки, что дало возможность
подсчитать вклад тех или иных соединений (в смеси с бензолом)
в коксообразование. Оказалось, что стирол образует кокс в 1,3—
33 раза больше, чем гептан, а тиофен — в 39—270 раз больше геп-
тана. Эти данные легко понять, если учесть, что при деструкции
тиофена образуется такой легко полимеризующийся остаток, как
бутадиен (см. стр. 285).
Отложение кокса может нейтрализоваться соответствующим по-
вышением давления от 100 до 200 кгс/см2 и повышением температуры
от 400 до 470 °C. Однако при повышении давления свыше 68 кгс/см2
его влияние проявляется незначительно в0, а стоимость гидро-
очистки остатков растет с ростом давления больше, чем экономия
на сроке службы катализатора 78. Отложение металлов может до
некоторой степени понижаться подбором катализатора и условий
процесса.
Несмотря на эти затруднения патентные и рекламные публика-
ции о возможности прямой гидроочистки остатков появляются
во все возрастающих количествах, что характеризует интерес к
этой проблеме во всем мире.
К настоящему времени наметились три принципиальных под-
хода к проблеме переработки нефтяных остатков в малосернистое
котельное топливо: 1) возможно большая часть остатка перего-
няется, дистиллят гидроочищается обычными методами и смеши-
вается с остатком перегонки (этот вариант может быть дополнен де-
асфальтизацией остатка перегонки с добавкой деасфальтизата к
гидроочшцаемому дистилляту); 2) то же плюс коксование остатка
и гидроочистка коксового дистиллята и 3) прямое гидрообессери-
вание сырой нефти или нефтяных остатков.
Выбор того или иного метода будет определяться качеством нефти
и экономикой. Первый метод наиболее дешев, но наименее эффекти-
вен. По расчетам 7Э, он не может дать котельное топливо с содержанием
серы менее 0,87%, если исходный остаток содержал 2,6% серы.
Следовательно, этот метод применим только к относительно малосер-
нистым нефтяным остаткам. Третий метод наиболее радикален, но
и наиболее дорог. Кроме того, при современном развитии процессов
гидроочистки, он еще не может применяться к нефтяным остаткам
с высоким содержанием металлов и асфальтенов. Поэтому второй
метод может оказаться не только промежуточным между первым
и третьим, но и единственным методом переработки неблагоприят-
ного сырья. Его существенный недостаток — образование не нахо-
дящего сбыта высокосернистого кокса, являющегося отходом про-
изводства.
302
В заключение следует отметить, что если химия процессов гидро-
очистки дистиллятного сырья в основном сложилась, химия про-
цессов гидроочистки нефтяных остатков только начинает склады-
ваться. Все еще крайне недостаточны сведения о природе и структуре
высокомолекулярных сернистых соединений нефти, хотя они пред-
ставляют одну из основных опасностей закоксовывания катализа-
торов.
Кроме сернистых соединений вредной составной частью мазутов
являются металлы, особенно ванадий. Для борьбы с дезактивиру-
ющим отложением металлов на катализаторах нужно искать но-
вые, более стабильные катализаторы. Это, скорее всего, должны
быть широкопористые контакты, содержащие промоторы, подавля-
ющие блокировку активных центров высокомолекулярными ком-
понентами, особенно азотистыми основаниями. Для предотвращения
коксообразования из-за «водородного» голодания катализаторы не
должны иметь высокой кислотности и ярко выраженного ионного
характера. Они должны отличаться очень высокой гидрирующей
активностью.
Таким образом, ключом к появлению новых, более эффективных
и универсальных в отношении сырья процессов гидроочистки должно
быть создание новых, стабильных к отравлению высокомолекуляр-
ными компонентами и металлами, механически прочных катализа-
торов. Для их разработки предстоит глубоко изучить генезис уже
применяемых катализаторов и его связь с их каталитической ак-
тивностью.
ЛИТЕРАТУРА
1. Мак Кинли Д. Б. В кн. «Катализ в нефтехимической и нефтепере-
рабатывающей промышленности». Т. 1. М., Гостоптехиздат, 1959. См. с. 339.
2. SchumanS. С., Shalit Н., Catalysis Rev., 4, 245 (1970).
3. О р о ч к о Д. И., Сулимов А. Д., Осипов Л. Н. Гидрогениза-
ционные процессы в нефтепереработке. М., «Химия», 1971. 352 с.
4. X а р т у Г. Д. В кн. «Новейшие достижения нефтехимии и нефтепере-
работки». Т. 3. М., Гостоптехиздат, 1962. См. с. 257.
5. Obolentsev R. D. VII World Petroleum Congress. Mexico. Proceedings.
V. 2. London, Elsevier Publ. Corp., 1967. See p. 109.
6. McCoy R. N., Weiss F. T., Anal. Chem., 26, 1928 (1954).
7. Drushel H. V., Sommers H. L., Anal. Chem., 39, 1819 (1967).
8. Реутов О. А. Теоретические основы органической химии. М., Изд.
МГУ, 1964. См. с. 664.
9. Б а л а н д и н А. А. Современное состояние мультиплетной теории гетеро-
генного катализа. М., «Наука», 1968. См. с. 120—136.
10. Wilson W. А., V о г е с k W. Е., М а 1 о R. V., Ind. Eng. Chem., 49,
657 (1957).
11. R u е d i s u I j М. М. J., Chem. Age India, 17, 264 (1966).
12. О б о л e н ц e в P. Д., Машкина А. В. и др. В сб. «Химия сераорга-
нических соединений, содержащихся в нефтях и нефтепродуктах». Т. 4.
Уфа, Изд. Башк. филиала АН СССР, 1961. Си. с. 166.
13. Оболенцев Р. Д., Дронов В. И., ДАН СССР, 130, 98 (1960).
> 303
14. Landa S., Mrnkova A., Gruskova A., Sb. «Vysoke skoly chem.
techn. Praze, D13, 151 (1967); РЖХим., 18П152 (1968).
15. ОболенцевР. Д., Кузыев A. P., Михеев Г. M. В сб. «Химия
сераорганических соединений, содержащихся в нефтях и нефтепродуктах».
Т. 6. Уфа, Изд. Башк. филиала АН СССР, 1964. См. с. 331.
16. Сергиенко С. Р., ПерченкоВ. Н., Михневская А. А. В
сб. «Химия сера- и азоторганических соединений, содержащихся в нефтях
и нефтепродуктах. Т. 3. Уфа, Изд. Башк., филиала АН СССР, 1960. См.
с. 353.
17. Welker Y., Freiberforschungshefte, А264, 63 (1963).
18. В е u t h е г Н., Flinn R. A., R i с е Т., Am. Petrol. Inst. Proc.,
Sect. Ill, 41, 228 (1961); С. А., 58, 1285 (1963).
19. Соре А. С., Farkas Е., J. Org. Chem., 19, 385 (1954).
20. С a w 1 е у С. М., Н а 11 С. С., J. Soc. Chem. Ind. (London), 62, 116 (1943).
21. H о о g Н., Rec. trav. chim., 69, 1289. (1950).
22. К о m a r e w s k у V. I., К n a g g s E. A., Ind. Eng. Chem., 43, 1414
(1951).
23. Рыбникова А. А., Тиц-Скворцова И. H., Нордов Е.,
Нефтехимия, 1, 100 (1961).
24. G г i f f i t h R. H., Marsh J. D. F., N e w 1 in g W. B. S., Proc. Roy.
Soc. (London), A197, 194 (1949).
25. В i r c h S. F., D e a n R. A., Ann., 585, 234 (1954).
26. Молдавский Б. Л., Прокопчук H., ЖПХ, 5, 619 (1932).
27. Молдавский Б. Л., Кумари 3. И., ЖОХ, 4, 298 (1934).
28. Сергиенко С. P., ПерченкоВ. Н., Итоги науки, 2, 113 (1958).
29. Калечиц И. В., Инь Юань-ген ь. Веб. «Труды ВСФ СО АН
СССР». Вып. 26. М., Изд. АН СССР, 1959. См. с. 108.
30. Юрьев Ю. К. Уч. зап. МГУ. Вып. 79. М., Изд. МГУ, 1945. См. с. 1.
31. Owens P.J., Amberg С. Н., Adv. Chem. Ser., 33, 182 (1961).
32. DesikanP., Amberg С. H., Can. J. Chem., 42, 843 (1964).
33. Satterfield Ch. H., R о b e r t s G. W., AIChE Journ., 14, № 1,
159 (1968).
34. Mann R. S., Can. Oil a. Gas Ind., 15, № 14, 7 (1962).
35. KolboeS., Amberg С. H., Can. J. Chem., 44, 2623 (1966).
36. W i 1 s о n R. L., Kemball C., J. Catalysis, 3, 426 (1964).
37. Kieran P., Kemball C., J. Catalysis, 4, 380 (1965).
38. H a m m a r C. G. B. Ill World Petroleum Congress. Hague. Proceedings.
V. 4. London, Elsevier Publ. Corp., 1951. See p. 295.
39. P r a t e r C. D., Lago R. M., Adv. in Catalys., 8, 284 (1956).
40. К а л e ч и ц И. В., Инь Ю а н ь - г е н ь, ЖФХ, 34, 2687 (1960).
41. Калечиц И. В., Инь Юань-ген ь, ЖФХ, 35, 501 (1961).
42. Kwan Т., J. Phys. Chem., 60, 1033 (1950).
43. С о 1 е R. М., D a v i d s о n D. D., Ind. Eng. Chem., 41, 2711 (1949).
44. Гу Чунь, Ли Цзя-пен. Рефераты докладов на конференции Даль-
пинского отделения КХО. 1956. См. с. 40.
45. Schmidt R., Guenther G., Chem. Techn., 7, 316 (1955).
46. С о t t i n g h a m P. L., A n t w e i 1 e r J. C. et al., Ind. Eng. Chem.,
48, 1146 (1956).
47. A b b о t M. D., L u d h о 1 m G. E., S a r n о D. H., Petrol. Ref., 34,
№ 6, 118 (1955).
48. Casagrande R. M., Meerbott W. K. et al., Ind. Eng. Chem., 47,
744 (1955).
49. А г а ф о н о в А. В., О с и п о в Д. H. и др. В сб. «Проблемы перера-
ботки высокосернистых нефтей». М., ЦНИИТЭнефтехим, 1966. См. с. 192.
50. К a b u г a k i Т., I п о n е S. et al., Кору тару, 15, 466 (1963); С. А., 61,
61820 (1964).
51. Heinemann Н., Kirsch F. W., В u г t i s Т. A., Erdol и. Kohle,
10, 225 (1957).
52. К i г s с h F. W., SchalitH., Heinemann H., Ind. Eng. Chem.,
51, 1379 (1959).
304
53. К а л е ч и п И. В., Инь Ю а н ь - г е н ь. В сб. «Труды ВСФ СО АН
СССР». Вып. 26. М., Изд. АН СССР, 1961. См. с. 121.
54. Flinn R. A., Larson О. A., Beuther Н., Hydrocarb. Proc. а.
Petrol. Ref., 42, № 9, 129 (1963).
55. Metcalfe T. В., Chim. et ind., 102, 1300 (1969).
56. H о og H., J Inst. Petrol., 36, 738 (1950).
57. S h i b a T., N a g a t a T., Коацу Газу Кёкайси, 21, 364 (1957); С. А.,
52, 5798 (1958).
58. OhtsukaT., Shimazu Sh. et al., Bull. Japan Petrol. Inst., 2, 13
(1960); C. A., 55, 959 (1961).
59. Frey C. G., M о s b у J. F., Chem. Eng. Progr., 63, № 9, 66 (1967).
60. Бейтер X., Шмидт Б. Труды VI Международного нефтяного коц.-
гресса. Вып. 2—7. М., ЦНИИТЭнефтегаз, 1965. См. с. 106.
61. MassagutovR.M., В е г g G. A. et al. VII World Petroleum Cong-
ress. mexico. Proceedings. V. 4. London, Elsevier Publ. Corp., 1967. See.
p. 177.
62. OhtsukaT., Hasegawa Y. et al., Bull. Japan Petrol. Inst., 9,
1 (1967); РЖХим., 8П216 (1968). '
63. Shimizu Y.< Inoue К. et al., Bull. Japan Petrol. Inst., 12, 10 (1970);
РЖХим., 2П183 (1971).
64. ФростА. В. Вест. МГУ, № 3—4, 111 (1946).
65. Van Zoonen D., D о ikw e s С. T., J. Inst. Petrol., 49, 383 (1963).
66. V a n De emet er J. J. Chemical Reaction Engineering. New York,
Pergamon Press, 1965. See. p. 215; цитируется по ссылке 2. z
67. A d 1 i n g t о n D. G., Chem. Eng. Practice, 8, sect. 4—5 (1965); цитируется
по ссылке 2.
68. А г e у W. F., В 1 a c k w e 1 1 N. E., R e i c h 1 e A. D. VII World Pet-
roleum Congress. Mexico. Proceedings. V. 4. London, Elsevier Publ. Corp.,
1967. See. p. 167.
69. Adlington D., Thompson F., Third European Symposium on
Chem. Reaction Engineering. Amsterdam. 1964; цит. по ссылке 2.
70. W h e e 1 e r A., Adv. in Catalysis, 3, 250 (1951).
71. R i c h a r d s о n J. T., Ind. Eng. Chem., Fund.,’3, 154 (1964).
72. A s h 1 e у J. H., M i t c h e 11 P. С. H., J. Chem. Soc., (A), 1968, 2821.
73. L i p s c h J. M. J. G., Schuit G. C. A., J. Catalysis, 15, 163, 174,
179 (1969).
74. С о r t e 1 у о n C. G., Mallatt R. C., Meredi th H. H., Chem.
Eng. Progr., 64, № 11, 53 (1968).
75. Larson O. A., Beuther H., Am. Chem. Soc. Preprints, 11, B95
(1966); цитируется по ссылке 2.
76. T а у 1 о r W. F., W a 1 1 a с e T. J., Ind. Eng. Chem., Prod. Res. a. Deve-
lop., 7, 198 (1968).
77. Титушкин В. А. Кандидатская диссертация. Уральский научный
центр АН СССР. Свердловск. 1972.
78. Blume J. Н., М i 11 е г D. R., N i с о 1 a i L. A., Hydrocarb. Proc.,
48, № 9, 131 (1969).
79. S 1 a d j е s k i Е. М., M a p 1 e s R. E., Oil a. Gas J., 66, № 8, 55 (1968)
20 Заказ 271
ГЛАВА 7
ПРОЦЕССЫ ГИДРОКРЕКИНГА
Процессы гидрокрекинга за исключением специальных способов
переработки тяжелого остаточного сырья в малосернистые котель-
ные топлива 1 (см. гл. 6) являются частным, но значительно мо-
дернизированным случаем парофазной гидрогенизации.
Создание новых, более активных и селективных, но менее под-
верженных отравлению катализаторов позволило сократить число
стадий процесса, часто путем объединения в одну стадию стадий
гидрирования и расщепления сырья. Двухступенчатые схемы приме-
няются обычно только для переработки низкокачественного сырья,
например высококипящего или с высоким содержанием азотистых
соединений. Современные процессы гидрокрекинга с более эффек-
тивными катализаторами проводятся при значительно меньшем
расходе водорода, а следовательно, и при более низком давлении,
что значительно упрощает аппаратурное оформление и снижает
себестоимость продуктов.
Химия процессов гидрокрекинга во многом аналогична химии
процессов парофазного гидрирования (см. гл. 5). Однако обнару-
жены специфические реакции и закономерности гидрокрекинга,
что обосновывает выделение описания этих процессов в специаль-
ней). главу. В обзорных работах по процессам гидрокрекинга2 '5
эти специфические закономерности, обычно освещены очень коротко,
основное внимание уделено вопросам технологии.
В химии гидрокрекинга много общего с каталитическим кре-
кингом, но от последнего его резко отличает присутствие водорода,
тормозящего все реакции, протекающие через промежуточное об-
разование олефинов 4.
Как и при изучении парофазной гидрогенизации, наиболее де-
тальные представления о процессе дает исследование превращений
индивидуальных углеводородов. Полученные результаты приведены
(в хронологическом порядке) в табл. 76.
Данные, приведенные в табл. 76, а также и ряд других данных,
не включенных в эту таблицу, позволяют перечислить особенности
гидрокрекинга, присущие как каталитическому крекингу и паро-
фазной гидрогенизации, так и специфичные только для этого про-
цесса.
Как и в процессах парофазной гидрогенизации, неуглеводород-
ные примеси легко образуют продукты расщепления в результате
деструкции по связям углерод-гетероатом (см. выше, стр. 280),
306
поскольку эти связи менее прочны 4. Деструкция азотсодержащих
соединений затруднена только в том случае, если в сырье присут-
ствуют ароматические соединения, вытесняющие их с поверхности
катализатора13. Это обстоятельство существенно для процессов
переработки тяжелого сырья.
Из данных табл. 76 следует, что характер реакций гидрокрекинга
весьма сложен: наряду с расщеплением и гидрированием протекают
реакции изомеризации, разрыва и сжатия — расширения колец,
алкилирования, гидродеалкилирования и т. д.
При гидрокрекинге парафиновых углеводородов в присутствии
катализаторов кислотного типа образуется большое количество
низкомолекулярных углеводородов изостроения; количество раз-
ветвленных углеводородов обычно выше равновесного. Очевидно,
что образующиеся в ходе процесса олефины разветвленного строения
тотчас насыщаются и уже не могут изомеризоваться. Эта законо-
мерность характерна и для парофазной гидрогенизации в присут-
ствии катализаторов, ускоряющих ионные реакции.
Интересно отметить, что менер активные катализаторы (вольфра-
мовые, молибденовые) дают более высокое отношение низкомоле-
кулярных изопарафинов к н-парафинам по сравнению с более ак-
тивным платиновым катализатором, который успевает вызвать
изомеризацию изопарафинов в н-парафины. В гидрогенизатах прак-
тически отсутствуют углеводороды с четвертичными атомами угле-
рода. Это также указывает на ионный характер изомеризации, так
как образование четвертичного углеродного атома требует энерге-
тически невыгодного перехода третичного иона во вторичный, а
затем в первичный. В присутствии алюмокобальтмолибденового
катализатора в газовой части содержатся главным образом метан
и этан. В основном получаются углеводороды нормального строения
меньшего молекулярного веса. Очевидно, этот катализатор обладает
ярко выраженным свойством ускорять радикальные реакции.
Интересно сравнить данные о выходах продуктов расщепления
цетана, полученные в разных процессах (см. табл. 77).
Из данных табл. 77 видно, что при гидрокрекинге протекает
сложный комплекс реакций, но на катализаторах кислотного типа
(NiS на А12О3 -|- SiO2, Pt на цеолите) процесс идет в основном по
карбониево-ионному механизму. Напротив, алюмокобальтмолибде-
новый катализатор дает продукт, более близкий к продукту терми-
ческого крекинга.
Для цетана 29 показано, что первоначальный разрыв в наиболь-
шей степени протекает по центральным связям. Аналогичный вывод
сделан и для других алифатических углеводородов 4- * 13*1б,
т. е. он имеет довольно общий характер для условий как ионного,
так и радикального гидрокрекинга, а также термического 27 и ката-
литического 28 крекинга.
При гидрокрекинге парафинов обычно не наблюдается изомери-
зации исходного сырья (катализаторы WS2, WS2 на алюмосиликате,
NiS на алюмосиликате,’ А1 Со + Мо) ’ 10. Однако в случае
20*
30?
80S
Таблица 76. Гидрокрекинг индивидуальных углеводородов
Исходные вещества Темпера- тура, °C Катализатор Основные результаты исследования Литера- тура
н-Пент ан, н-гек ;>ан, w-гептан 371 Pt на алюмосиликате Разрыв происходит главным образом по централь- ной связи 6
н-Парафпны, олефины Ci—С1В 375 WS2 на активирован- ной глине В основном образуются углеводороды Cg—Cji для углеводородов С1о—Cjg отношение количеств по- лучающихся углеводородов С3 : С< : С6 : Св равно 2 : 5 : 3 : 2. Олефины дают сходные продукты, но их превращения протекают быстрее. Отношение изопарафины : н-парафины выше равновесного 7
н-Парафины (цетан, ок- тан), н-бутилбензол, тетралин 387 Окись или сульфид Ni на алюмосили- кате В продуктах расщепления нормальных парафинов олефины отсутствуют. Отношение изопарафины : : н-парафины выше равновесного для uao-Ci : н-Сд в 6 раз, для uso-Сб : н-СБ в 3 раза. Отсутствуют углеводороды с четвертичным углеродным ато- мом. Непревращенное сырье не изомеризовано. Бутилбензол в основном деалкилируется с обра- зованием бутанов и бензола, а также толуола и пропана. В тетралине происходит раскрытие кольца и дегидрогенизация с образованием зна- чительного количества нафталина 8
Гексаметилбензол 315—375 NiS на алюмосили- кате Главный ациклический продукт — изобутаи; арома- тические углеводороды в основном Сщ— Сц, наф- тены Су—Сд. Отношение изопарафины: к-иара- фины значительно выше термодинамически рав- новесного. Хотя, судя по высоким выходам аро- матических углеводородов Сю—Сц, должно иметь место деметилирование, отсутствие замет- ных количеств метана (всего 3,6—10,3 моль на 100 моль превращенного продукта), этана, бен- зола, толуола указывает на необычную реакцию «спаривания» метильных заместителей в осколки Сд И U3O-C4 9
Алкилциклогексаны Се —С12 230-290 NiS на алюмосили- кате Главный ациклический продукт—изобутан, основ- ные циклические продукты содержат на 4 атома 10
Бензол 371 Pt на алюмосилика-
Пентен-1, пентен-2, 250—350 те; NiS+WSa на алюмосиликате; Co-|-Mo + S на алюмосиликате Ni на алюмосили-
2-метилбутан кате
Гептан Условия и катализатор про- цесса «Изомакс»
Этилбензол «-Гептан, «-гексадекан, «-гексадецен, «-доко- зан 400-450 371 Ni и NiS на алюмо- силикатах Pt на алюмосили- кате
/ Гексай, парафины Сц—Си «-Бутилбензол, н-децил- бензол, гексаэтилбен- зол, тетралин, фе- нантрен, антрацен, пирен 350—475 290 Pd на AljOa-j-BgOg NiS на алюмосили- кате
углерода меньше, чем исходный продукт. Реак-
ция разрыва кольца происходит в незначитель-
ной степени. В продуктах реакции преобладают
разветвленные алканы. Отношение метилцикло-
пентан : циклогексан выше равновесного
Скорость гидрирования бензола изменялась в ряду
Pt > Ni+ W>Co + Mo; гидрогенизаты содержали
8—14% парафинов, остальное—нафтены
Гидроизомеризация олефинов, т. е. прямое прев-
ращение их в изопарафины протекает только на
сульфидированном катализаторе. В отсутствие
серы идет только миграция двойной связи и ди-
спропорционирование. Если в качестве носителя
использовать SiO2 гидроизомеризация не идет
Главные продукты —углеводороды Cg + Ca. Изу-
чено влияние азотсодержащих соединений на
скорость гидрокрекинга
Присутствие серы понижает кажущуюся энергию
активации с 44 до 36 ккал/моль
Гексадекан быстрее всего расщепляется до Cg.
Только после 100%-ного превращения в заметной
степени протекают вторичные реакции, приводя-
щие к углеводородам (%—С12 (преобладают С7 —
Са). Циклизация незначительна (12—16 моль на
100 моль превращенного сырья). н-Гептаи дает
в основном продукты С3 — С4. У докозана более
заметны вторичные реакции. Гексадецен превра-
щается аналогично гексадекану. Непревращенное
сырье изомеризовано
Расщепление происходит в основном по централь-
ным связям
Для н-бутилбензола преобладают реакции деалки-
лирования; н-децилбензол На 27% превращается
в низкомолекулярные углеводороды; на 35% де-
алкилируется с образованием в основном алканов
11
12
13
14
15
16
17
310
Исходные вещества Темпера- тура, °C Катализатор
Циклододекан, цпкло- пентадекан 290 NiS на алюмосили- кате
н-Декан Ni на алюмосили- кате и тот же ка- тализатор, обрабо- танный H2S
Изопентан, н-лелтан, н-гексан 400-480 Катализаторы кис- лотного типа
н-Гептан, гексадекан, циклоалкапы C# — С7, н-бутилбепзол, изо- дурол 425 Ni на алюмосили- кате
Продолжтие табл. 76
Основные результаты исследования
Литера-
тура
С10 и бензола и на 39% циклизуется с обра-
зованием тетралина. Гексаэтилбензол деалкили-
руется, образуя при малом времени контакта
этан и полиэтилбензолы, а при длительном
контакте углеводороды С10 — Cj2 рядов тетралина
и индана. Тетралин дает в числе других углево-
дородов трициклические пергидроароматические
углеводороды, фенантрен образует тетралин и
метилциклогексан, антрацен превращается в тет-
ралин, а пирен—в тетралин и метилциклопен-
тан
Получаются в основном изобутан, изопентан
и циклические продукты состава С?—С8 (алкил-
цпклопентаны и алкилциклогексаны)
На катализаторе, не обработанном H2S, образу-
ются продукты, состоящие в основном из н-па-
рафинов с большим содержанием СЬЦ. При до-
бавлении же 20%'серы достигается максималь-
ное увеличение активности катализатора; обра-
зуются диметилоктаны
Пентаны довольно устойчивы. Для гексана харак-
терно образование двух молекул пропана. Бу-
таны и пентаны образуются в большем коли-
честве, чем можно ожидать в результате отрыва
осколков Сх и С2
Парафины расщепляются, образуя углеводороды
Для гексадекана велика роль вторичных
реакций расщепления и изомеризации образую-
щихся осколков. Отношения изо-Сд: H-C4,
изо-С5 : н-С5 и изо-Сь : к*Св превышают термоди-
намически равновесные в 4,7, 2,5 и 2,4 раза со-
ответственно. Циклоалканы расщепляются, обра-
зуя осколки С2— С4; шестичленные углеводороды
18
19
20
21.
22
н-Гептан, гексадекан,
циклоалканы С8 —С7,
бензол, н-бутилбен-
зол, изодурол
Дурол, метильные груп-
пы которого содержат
14С
Смеси дурола с бензо-
лом, толуолом, ксило-
лом и триметилбензо-
лом, меченными 14С
(в кольце)
Изопропилбензол
425
425
425
300—500
Pt на цеолите; А1 +
+ Со + Мо
NiS на алюмосили-
кате
Al + Со + Мо
Мо на А1О3; Мо + Со
на А12О3
w
в значительной степени изомеризуются в пяти-
тленные; происходит раскрытие цикла без рас-
щепления (особенно у метилциклонентана).
При гидрокрекинге нафтенов образуется значи-
тельно больше продуктов изостроения, чем из
нормальных парафинов. Для изодурола харак-
терно образование значительных количеств изо-
бутана. н-Бутилбензол деалкилируется, образуя
в основном бутаны и бензол, в меньшей степени—
этилбензол и толуол
На катализаторе Pt на цеолите превращения по-
добны превращениям углеводородов на указан-
ном выше никелевом катализаторе, но расщеп-
ление происходит в большей степени. На ката-
лизаторе А1 + Со-}-Мо в газовой части преобла-
дают метан и этан. Углеводороды, образуемые в
результате расщепления, в основном неизомери-
зованы. При гидрокрекинге изодурола основными
продуктами являются лгетан и триметилбензолы.
н-Бутилбензол расщепляется, образуя главным
образом пропан, бутан, бензол и толуол
Все образующиеся продукты расщепления имеют
приблизительно одинаковую радиоактивность
Доказано, что при гидрокрекинге протекают ре-
акции не только деметилирования, но и метили-
рования образующихся углеводородов
21,
23
24
25
На катализаторах, не обработанных H2S, при по-
ниженных давлениях и повышенных температу-
рах происходит частичное гидрирование арома-
тического кольца и образование, помимо пропа-
на, метана и этана. В обычных условиях и при
добавке серы гидрирование кольца незначитель-
но, а расщепление идет только по связи, при-
мыкающей к кольцу 1
26
Таблица 77. Выход продуктов расщепления цетана
в различных процессах
(в моль на 100 моль превращенного сырья)
Продукты расщеп- лении Терми- ческий крекинг (500 °C)27 Каталитиче- ский крекинг (500 °C)28 Гидрокре- кинг (NiS на Al.OH-SiO,, 425 °C)21 Гидрокре- кинг (Pt на цеолите, - 425 °С)2|> Гидрокре- кинг (Al+Co + Mo, 425 °C)22 Расчет по ионному механиз- - му 28
С1 53 5 6 9 36 0
с2 130 12 7 15 43 0
С3 60 97 59 92 21 95
с4 23 102 55 86 13 97
С5 9 64 86 68 30 72
с 8 24 50 76 64 52 41
С, 16 8 27 18 29 7
С8 13 8 11 4 35 6
применения платинового катализатора^ продукты, не подвергнув-
шиеся расщеплению, оказываются высокоизомеризованными. Одно
из возможных объяснений этого факта предлагается в работе 15
на основе соотношения гидрирующей и кислотной активностей ката-
лизатора. Если гидрирующая активность катализатора ниже кис-
лотной, преобладают реакции расщепления и конечный продукт
мало или вообще не изомеризован. Так, показано, что гидрирующая
активность катализатора и степень изомеризации исходного сырья
изменяются симбатно в ряду:
Pt > Со + мо > Мо
Такое объяснение является, по-видимому, общепринятым. Од-
нако во многих работах, в том числе и в рассматриваемой работе 15,
обязательно предполагается раздельное существование гидриру-
ющих и кислотных центров с миграцией реагирующего вещества
с одного центра на другой. Между тем, главной причиной взаимо-
связи реакций гидрирования и изомеризации является наличие
общих промежуточных продуктов (см. стр. 232 сл.).
Предположение о раздельном существовании гидрирующих и
кислотных центров побудило даже экспериментально определять
необходимый минимум гидрирующей активности катализаторов
(между 20%-ным превращением бензола на катализаторе Со +'
+ Мо + S на А12О3 + SiO2 и 43%-ным на катализаторе Ni + W +
4- S на А12О3 + SiO2) u, хотя несомненно, что действительная кар-
тина влияния химического состава катализатора на их активность
много сложнее. Так, например,’ один из лучших катализаторов —
Ni + W + S на алюмосиликате — имел наименьшую поверхность,
умеренную гидрирующую и нулевую дегидрирующую активности.
Из важных специфических особенностей гидрокрекинга парафи-
нов следует отметить, что в процессе гидрокрекинга по сравнению
с каталитическим крекингом образуются осколки большего моле-
кулярного веса.
312
При расчете выхода продуктов каталитического крекинга при- ,
нимается 7, что расщепление карбонйевого иона идет цо 0-правилу
так, что отщепляются осколки не менее С3 и до тех пор, пока в ионе
не останется шесть углеродных атомов. В случае Тидрокрекинга
принимается 7, что отщепляются осколки не менее С4 и до тех пор, '
пока в ионе не останется семь атомов углерода. Была определена 20
зависимость интенсивности гидрокрекинга узких фракций от их
среднего молекулярного веса и показано, что с максимальной интен-
сивностью расщепляются фракции С8. Углеводороды с числом угле-
родных атомов менее 8 устойчивее и тем более, чем ниже их моле-
кулярный вес. Уменьшение конверсии в случае углеводородов С»
и С10 связывается с образованием осколков С8, устойчивых к даль-
нейшему распаду. Кроме того отмечается 20, что отношения мольных
выходов продуктов С4 и С4 в случае гидрокрекинга углеводородов С6
и продуктов С2 и С4 в случае углеводородов Св отличны от 1 :
т. е. процесс сложнее, чем обычный распад. Авторы 20 предпола-
гают, что этот процесс идет через присоединение ионов Сз к проме-
жуточно образуемым олефинам (С”), например:
cj+c4 —> CJ" —> Cj’+Cj’
CJ + Cg —> —>• CJ+CT
Однако было показано 30, что образование больших количеств
олефинов из парафинов того же молекулярного веса в условиях
гидрокрекинга термодинамически невозможно.
Был термодинамически обоснован вероятный механизм образова-
ния больших количеств изопарафинов 30. Он включает стадии обра-
зования олефинов нормального или изостроения, изомеризацию
углеводородного, скелета первых и превращение изоолефинов в изо-
парафины. Олефины образуются при крекйнге парафиновых угле-
водородов большего молекулярного веса. При этом возможна .про-
межуточная изомеризация исходного «-парафина:
, н-Сп --► мао-Сл --► изо-Сп-т~Ь~Ст
ИЛИ
я-С Л > н-Сп-т Г m
Эти реакции могут протекать практически нацело.
'J При гидрокрекинге ароматических углеводородов сохраняются
общие закономерности легкого отрыва боковых цепей С4 и больше
и возрастающей с уменьшением числа углеродных атомов стабиль-
ности цепей С3 и меньше 4> 8> 17> 81. Правда, в продуктах гидрокре-
кинга децилбензола 17 было обнаружено до 39% продуктов циклиза-
ции, однако в реальном сырье ароматические-углеводороды с таким
большим числом углеродных атомов в боковой цепи практически
не содержатся ®х.
Точно также в условиях гидрокрекинга сохраняется зако-
номерность чередования реакций гидрирования и расщепления
313
гидрированных колец в случае полициклических ароматических
углеводородов 171 31, например:
ЗА --->- 2A1N --> 2А ---> 1A1N ---> 1А
Возможно 31, что процесс идет и несколько иначе через насы-
щение центральных ароматических колец, последующее расщепле-
ние которых дает сразу два ароматических осколка, например:
I II I I
нг
сн3
\/\/\^
Прямых доказательств такой реакции нет; при гидрокрекинге
антрацена 17 получены тетралин и метилциклогексан.
Неконденсированные полициклические ароматические углево-
дороды типа полифениленов легко расщепляются с образованием
моноциклических ароматических углеводородов и дифенилов 31.
При гидрокрекинге 1,2-диарилэтана
(400—450 °C) получены псевдокумол
и дурол 32.
Специфической особенностью гидро-
крекинга является селективность гид-
рирования ароматических углеводоро-
дов: в то время как би- и полицикли-
ческие системы превращаются с высокой
скоростью, моноциклические — гидри-
руются незначительно А 31. Вследствие
этого, а также в результате сохране-
ния осколков изо-С5 — изо-Сч, бензины
гидрокрекинга обладают высокими ок-
тановыми числами.
Для гидрокрекинга характерна
относительно незначительная глубина
вторичных процессов 15> 31. На рис. 23
показано изменение фракционного со-
става (сырье — фракция 204—371 °C),
как функции степени превращения 33.
Из приведенных данных видно, что
при степени превращения 2,4 моль/моль сырье полностью перерабаты-
вается, но и после этого газообразование незначительно. Это по-
зволило разработать методы предсказания и расчета выхода про-
дуктов гидрокрекинга, а также потребления водорода 31> 33.
Совершенно неожиданные результаты обнаружены 9 при гидро-
крекинге гексаметилбензола в присутствии катализаторов кислот-
ного типа: образуются в основном изобутан и ароматические угле-
водороды С10. Авторы 9 предлагают механизм, включающий частич-
ное гидрирование ароматического кольца, его изомеризацию в пяти-
членное кольцо, сопровождающуюся ростом боковой цепи, вновь
образование шестичленного кольца и так много раз с обрывом цепи,
Степень превращения,
моль на моль сырья
Рис, 23. Зависимость фракци-
онного состава продукта гидро-
крекинга от степени превра-
щения.
314
«собранной из метильных групп». Таким образом для объяснения-
необычной реакции «спаривания» потребовалось предположение
о многостадийности этого процесса:
СН3
Нз<\Д^СН3 _п+
НзС/^/^СНз
СН3
Н СН3
н3с-/'У'\сн3
СН3
СН-СНз
Н3С-|—jj—СНз
Нзс/у\сн3
СН3
С Н3—СН-СНз
•|—си,
н3с/'Ухсн3
СПз
(СН3)2СН (СН3)2СН
НзС/'У'СНз НзС/^^СНз
СН3 СНз
СН(СН3)2
НзС\А
I I-
H3CZY^
СНз
(СН3)2СН-СН
НзС-j—| _
НоС-/444^
СН3
СН(СН3)2
НзС\/\
- II I
Н3с/\^
СН3
(СН3)2С-СН2
н.с-|-|
HaC/'Y
СНз
СН(СНз12
СН3
НзС || |+ + СН2=С(СН3)2
н3с/Х^
СН3
I .. .
СН3-СН(СН3)2
Н3с/чу
СНз
Для гексаэтилбензола первичными реакциями в тех же условиях
были исключительно реакции деалкилирования с образованием
больших количеств этана 17.
Предложенная схема носит несколько спорный характер. Была
предпринята попытка выяснить механизм подобных реакций при
помощи меченых атомов 24. Данные радиометрического анализа
продуктов гидрокрекинга дурола, содержащего 14СН3, показали
почти статистическое распределение радиоактивности по всем
углеродным атомам (в том числе, что особенно необычно, и в арома-
тических углеводородах С8 — С9), что делает представление об
образовании изобутана посредством «сбора» метильных групп в
длинную боковую цепь маловероятным. Это подтверждается и
крайне малым количеством бензола в продуктах гидрокрекинга.
Очевидно этот процесс протекает сложнее, чем его можно предста7
вить приведенной выше схемой. Авторы 24 предполагают, что при
315
активированной адсорбции ароматических углеводородов связи в
них настолько активируются, что становятся возможными реакции
обмена атомов углерода. Эти активированные углеводороды, гид-
рируясь, расщепляются в основном по ионным схемам, а чем свиде-
тельствует большое количество образовавшихся продуктов С3 и С4
по сравнению с С1; и С2.
Существует еще одно объяснение механизма реакции «спарива-
ния» (без доказательств) 33: при адсорбции полиметилбензола в по-
рах метильные заместители сближаются, давая сначала этильные
заместители, а при последующей диффузии в еще более узкую пору —
изобутильный заместитель. Если бы этот механизм был справедлив,
то в условиях работы 24 вся радиоактивность была бы сосредоточена
в газообразных продуктах С3 -4- пзо-С4, а бензол был бы неактив-
ным. Однако этого не наблюдалось, следовательно предложенный
.механизм 33 мало вероятен.
Реакции «спаривания» наблюдаются и для полиметилциклогек-
санов (1,2,4,5-тетраметил, пентаметил- и гексаметилциклогекса-
нов) 10. Циклододекан и циклопент а дек ан в аналогичных условиях
также образуют в основном изобутан, изопентан, алкилциклопен-
таны и алкилциклогексаны, имеющие семь или восемь углеродных
атомов 18. Очевидно, на поверхности катализатора происходит бы-
строе сжатие колец циклододекана и цикпопентадекана с образова-
нием алкилциклопентанов и алкилциклогексанов. Полученные изо-
меры селективно расщепляются, давая изобутаны и изопентан.
По-видимому, гидрокрекинг алкилциклогексанов также протекает
очень сложно и включает скелетную изомеризацию. Это подтвер-
ждается и работой по изучению гидроизомеризации меченных 14С
этилциклогексанов на никелевых катализаторах 34. При этом на-
блюдалось образование изомерных диметилциклогексанов с почти
статистическим распределением радиоактивности между кольцом
и боковыми цепями.
Интересно отметить, что при гидрокрекинге дурола и изодурола
на катализаторе (Al J- Со -|-Мо), ускоряющем радикальные реак-
ции, образуются в основном метан и триметилбензолы, т. е. в данном
случае протекает ярко выраженная реакция деметилирования.
Испольруя метод меченых атомов, удалось показать, что при гидро-
крекинге дурола протекают как реакции деметилирования, так
и реакции метилирования полученных углеводородов 25. При этом
оказалось, что в ряду С6 — С10 относительные скорости реакций
метилирования уменьшаются, а относительные скорости деметилиро-
вания возрастают.
Доказана возможность не только последовательного пути пре-
вращения, но и непосредственного, без десорбции в объем промежу-
точных продуктов, хотя последний путь имеет меньшее значение 25.
Таким образом, показано, что превращения индивидуальных угле-
водородов- при гидрокрекинге протекают очень сложно.
Благодаря применению новых, более совершенных методов ана-
лиза можно получать определенную информацию и при изучении
316
продуктов гидрокрекинга смесей углеводородов. Так,
нии гидрокрекинга н-декана, декалина и их смеем на мордешДОЙ&
катализаторах 35 было отмечено, что декалин в смеси превращавши*
быстрее, чем взятый отдельно. Исследовайие масс-спектрометриче>-
ским методом бензинов, полученных в результате гидрокрекинг^
вакуумного дистиллята, привело к выводу об отсутствии в них дйе-
новых, циклоалкеновых и алкенилароматических углеводородов
и о незначительном содержаний (<10%) олефинов; высоким было
содержание бициклоалканов, тетралиновых и индановых углеводо-
родов зв. На основании структурно-группового анализа концент-
ратов изопарафинов 37, выделенных из продукта гидрокрекинга
высококипящих парафиновых углеводородов установлено, что во
всех случаях отсутствуют углеводороды е четвертичными углерод-
ными атомами.
При исследовании масс-спектрометрическим методом продуктов
превращений вакуумного дистиллята показано,-что би- и полицикли-
ческие углеводороды подвергаются при гидрокрекинге глубокому
распаду с образованием моноциклических систем 38. Из анализа
изменения группового состава сырья в ходе гидрокрекинга видно,
что количество моноциклических насыщенных углеводородов и
парафинов уменьшается, а количество полициклических насыщен-
ных углеводородов и полициклических ароматических углеводо-
родов проходит через максимум 38. Эти данные коррелируют с пред-
ставлениями о последовательности процессов гидрирования колец
(см. стр. 314).
Механизм превращений соединений, содержащих серу или азот,
в процессе гидрокрекинга, по-видимому, аналогичен их превраще-
ниям в условиях гидроочистки. Полагают40, что разрыв связей
С—S и С—N идет в основном по радикальному механизму:
RCH2—SR' —► RCH2 + R'S- 1
R’CHz—NHR'" —► R'CH2 + R"'NH 2
R's- + h2 —► r'- + h2s i
R"'NH + H2 —► R"'--|-NH3 4
RCH2(R'-, R"CH2, R'"-) + H2 —► RCH3(R'H, R"CH3, R"H)+H- S
Роль радикала H • в развитии цепей радикальных реакций неясна,
но можно полагать, что реакции передачи цепи начинаются с атаки
гетероатома, например:
R\ R\H-
>S:4-H- —► >S —► RH + R'S- 6
R'Z R'Z
Такое предположение представляется вероятным, поскольку
повышение давления водорода, т. е. повышение его концентрации,
облегчая реакции 3—6, ускоряет гидрогенолиз, правда, по данным 4в,
лишь незначительно. .
317
Соединения, содержащие серу, явно участвуют в коксообразо-.
вании. При спектральном изучении состава коксовых отложений,
экстрагированных растворителями41 после гидрокрекинга (давле-
ние 30 кгс/см2) нефтей и тяжелых фракций, установлено, что в них
содержатся парафиновые и циклопарафиновые углеводороды, про-
изводные бензола, гомологи дифенила, би- и трициклические аро-
матические углеводороды и ароматические соединения, содержащие
серу42. В экстрактах обнаружены также соединения молибдена
и кобальта, образовавшиеся, очевидно, из активных компонентов
катализатора, но не найдены продукты уплотнения. Они, вероятно,
образуются на последних стадиях процесса, так как с переходом
к «сухому коксу» увеличивается число ароматических колец, резко
возрастает отношение С : Н.
Исследовалось влияние условий процесса на отдельные направле-
ния превращений. Так, при крекинге высококипящих парафинов
повышение давления до определенной величины (50 кгс/см2) увели-
чивает скорости реакций расщепления и изомеризации 43. Дальней-
шее повышение давления тормозит эти реакции, причем более ин-
тенсивно реакции изомеризации. Эту зависимость можно объяснить,
очевидно, тем, что, как показал еще М. Г. Гоникберг 44, изомери-
зация насыщенных углеводородов является реакцией дегидрогидри-
рования, т. е. включает стадию дегидрирования, которая, согласно
закону действующих масс, должна тормозиться при повышении
давления водорода. Позднее это было подтверждено работами
А. А. Петрова 45 и других исследователей. При исследовании кине-
тики гидрокрекинга этана и пропана на никелевом катализаторе
также было найдено4в, что реакция ингибируется водородом.
Повышение температуры увеличивает скорость как расщепления,
так и обессеривания.
Что касается кинетики гидрокрекинга, то уже из описанной выше
сложности механизма этого процесса следует, что в условиях про-
мышленного процесса она может описываться только приближенными
уравнениями.
Так, например, были определены порядки различных превраще-
ний в ходе гидрокрекинга туймазинской нефти47: образование
кокса описывалось уравнением нулевого порядка, образование
продукта, выкипающего в пределах вакуумного дистиллята, —
уравнением третьего порядка, а образование моторных топлив —
уравнением первого порядка, в то время как общая конверсия —
уравнением второго порядка. Второй порядок (кажущийся) был
предложен 48 и для процесса H-Oil, причем это уравнение хорошо
подошло и для описания скоростей гидроочистки (см. стр. 297 сл).
Для другого процесса гидрокрекинга — процесса Ну-С найдено,
что превращения продуктов, кипящих выше 350 ®С, описываются
уравнениями нулевого порядка (по сырью), порядок по водороду
составил 1,4. В то же время расход водорода на реакцию раскрытия
колец был постоянным в равные промежутки времени, т. е. реакция
раскрытия колец имела нулевой порядок по водороду 40.
318
Еще сложнее кинетика гидрокрекинга ароматических углеводо-
родов 351 50. Удовлетворительные результаты получаются при
расчете превращений только по отдельным реакциям (разрыв
С—С-связей35) или для отдельных классов углеводородов50. При
сравнении скоростей различных реакций с помощью дейтерообмена
было сделано заключение51, что скорость реакций в процессе
гидрокрекинга в одних случаях определяется десорбцией, в дру-
гих — наиболее медленной ступенью является разрыв углерод-
углеродных связей. Энергии активации реакций гидрогенолиза
относительно невелики, составляя, например, в процессе
H-Oil 14 ккал/моль 48. Температурные коэффициенты реакций изо-
меризации (1,54—1,69) и расщепления (1,2—1,3) парафиновых угле-
водородов при гидрокрекинге их на алюмоплатиновом катализаторе
и энергии активации этих реакций (соответственно 45>7 и
17,0 ккал/моль) значительно выше 37.
Изучение кинетики гидрокрекинга газойля 39 показало, что
гидрокрекинг, десульфуризация и удаление азота проходят как
реакции первого порядка, а константы скорости (в ч-1) могут быть
представлены уравнениями:
А-г= 1 107 . e-21 000/НТ
As = 0,6814-105 е~16800/лт
/cN = 0,8253 Ю5 е-17 400/ЙГ
где кг, к$ и kN — константы скорости гидрокрекинга, десульфури-
зации и удаления азота соответственно. Рассчитанные значения
энтальпии для этих реакций равны соответственно 19,0, 16,5 и
16,0 ккал/моль, а значения энтропии составили —4418, —4435,
-4240 э. е.
По мнению авторов 39, высокие отрицательные значения А5
указывают на то, что диссоциация при адсорбций происходит с об-
разованием неподвижного активированного комплекса с малым
числом степеней свободы.
Сведения о катализаторах гидрокрекинга весьма ограничены.
По патентным данным 52, наиболее распространены катализаторы
гидрокрекинга, содержащие в качестве гидрирующих компонентов
металлы VI и VII групп периодической системы элементов, их суль-
фиды или окислы, осажденные на различных носителях (в зависи-
мости от направленности процесса). Катализаторы содержат также
активирующие добавки — другие металлы, серу, галогены. Роль
каждого из компонентов катализатора не может считаться до конца
ясной, тем более, что несомненно взаимодействие активного агента
с добавками и носителем, а также изменение всего катализатора
в целом под влиянием среды,' компонентов сырья и высокой темпе-
ратуры.
Роль металлов в катализаторе, как правило, связывают с обеспе-
чением его гидрирующей активности. Так, например, при сравнении
319
гидрирующей и расщепляющей активностей катализаторов
МоО3 на А12О3 и NiO на А12О3, а также одного носителя было по-
казано 63, что расщепление сырья было во всех случаях примерно
одинаковым, но обессеривание наблюдалось только на катализато-
рах, причем молибденовые были активнее никелевых.
Были сравнены 11 12 катализаторов гидрокрекинга, содержащих
Pt, Ni —-W, СоО -|-МоО3, МоО3 на носителях с 75—90% SiO2
с добавками А12О3 или ZrO2, причем носители обрабатывались
хлором. Критерием служил выход бензина из стандартного газойля
при наименьшем газообразовании и минимальной температуре.
По начальной активности лучшими катализаторами были Pt и Ni
-t- W, но по стабильности, оцениваемой по скорости повышения
температуры для поддержания равного выхода бензина, лучшими
оказались окисные катализаторы, в частности СоО 4-МоО3, худ-
шими — платиновые катализаторы.
При сравнении таких катализаторов гидрокрекинга, как иридий,
осмий, платина, рутений и родий на кислотных носителях было
показано, что при содержании металлов в катализаторе в количе-
стве 0,5% высшей активностью обладал родиевый катализатор,
однако наибольший выход углеводородов С5 получен на плати-
новом катализаторе.
Обычно при малых концентрациях - гидрирующего агента его
количество влияет на гидрирующую активность катализатора:
чем больше количество гидрирующего агента, тем выше активность.
Однако после определенного предела дальнейшее повышение со-
держания гидрирующего агента не оказывает влияния. Эта законо-
мерность отмечалась для платины 54 и окиси молибдена 55, а для
окиси никеля 66 после достижения определенного оптимума наблю-
далось даже падение активности.
•' Роль гидрирующего агента не ограничивается одним ускорением
реакций гидрирования. Он влияет также на интенсивность таких
типично ионных реакций, как изомеризация и расщепление. Ха-
рактерно, что не обнаружено корреляции между расщепляющей
активностью и числом кислотных центров 57, однако найдена зави-
симость скорости расщепления от площади, занимаемой металлом
(см. рис. 24) 58. Авторы 58 пришли к выводу, что активны лишь
кислотные центры катализатора, располагающиеся вблизи металли-
ческих кристаллитов, так как только эти центры не закоксовыва-
ются в процессе работы. Таким образом, основная роль металлов
в катализаторе гидрокрекинга состоит в том, чтобы сохранять ки-
слотные центры активными путем гидрогенизации соединений —
предшественников кокса.
Применяемые катализаторы могут предварительно осерняться 59
и модифицироваться различными добавками. Показано 59, что в слу-
чае алюмокобальтмолибденового катализатора наибольший эффект
наблюдался при осернении его сероводородом. При этом осернение
практически не изменяет расщепляющие свойства катализатора,
но заметно улучшает гидрирующие, вследствие чего уменьшается
320
образование кокса. Катализаторы, содержащие только никель,
снижают активность при осернении68. Сопоставление интеневЕв»
ности расщепления и изомеризации' на алюмосиликате, нйкеле^а
алюмосиликате и сульфиде никеля на алюмосиликате вскрыло
довольно сложную картину влияния осернения 1в, рассмотренную
выше в гл. 2 (см. стр. 127 сл).
Повышение скорости расщепления при добавке в катализатор
никеля объясняют 19 блокированием кислотных активных центров.
Эта интерпретация, вообще говоря, носит дискуссионный характер.
Можно предложить другое, более простое объяснение: чистый ме-
талл ускоряет только радикальные реакции (действительно, как
видно из данных работ 19, при гидрокрекинге цетана на катализа-
торе Ni на А12О3 -f- SiO2 велика доля метана и этана и очень мало
Рис. 24. Влияние продол-
жительности работы плати-
нового катализатора на его
активность.
Удельная поверхность платины
(м«/г): 1 — 0,05; 2 — о,10;
3 — 0,60.
Продолжительность работы ката-
лизатора , ч
изобутана), в то время как добавка серы превращает Металл в р-полу-
проводник, способный ускорять и ионные реакции.
Одним из распространенных способов активации катализатора
является его галогенирование ®2. Введение галогена улучшает
гидрирующую активность катализатора и активность к расщепле-
нию колец. Кроме того, галогенированные катализаторы более
устойчивы к дезактивации и лучше регенерируются.
Химический состав и структура носителя катализатора также
играют важную роль в формировании его активности и особенно
в его стойкости к отложению кокса и металлов. Так, на специаль-
ным образом подготовленной окиси алюминия (содержание гидрат-
ной воды 1,2—1,6 моль/моль) значительно уменьшается отложение
металла и углерода по сравнению с обычной у-окисыо алюминия ’°.
Отмечалось, что в алюмосиликатном катализаторе увеличение
доли силикатной части приводит к росту расщепляющей активности,
а увеличение доли глинозема — к уменьшению чувствительности
катализатора к отравлению азотсодержащими соединениями и удли-
нению срока его службы в1. Природные алюмосиликаты более чув-
ствительны к отравлению азотсодержащими соединениями, чем
синтетические ®а.
21 Заназ 271 ' '321
Изучение влияния содержания окиси кремния на свойства про-
мышленных алюмокобальтмолибденовых и алюмоникельмолибдено-
вых катализаторов показало, что введение SiO2 увеличивает объем
и средний радиус пор, повышает в 1,5 раза механическую прочность
катализатора. При этом возрастают расщепляющая и изомеризующая
активности катализаторов в3,
Большое значение в настоящее время уделяется катализаторам
на цеолитной основе. Эти катализаторы обладают высокой актив-
ностью и хорошей избирательностью, а кроме того позволяют часто
проводить процесс без предварительной очистки сырья от азотсо-
держащих соединений. Содержание в сырье до 0,2% азота практи-
чески не влияет на их активность 65. Применение цеолитных катали-
заторов часто позволяет проводить процесс при более низкой тем-
пературе 66. Повышенная активность катализаторов на основе
цеолитов объясняется более высокой концентрацией активных кислот-
ных центров в кристаллической структуре по сравнению с аморф-
ными алюмосиликатными катализаторами 67.
Весьма важны наблюдения о влиянии различных компонентов
сырья на активность катализаторов. В случае переработки тяжелого
сырья наибольшую опасность представляют асфальтены (см. стр. 320).
Кроме асфальтенов на активность катализатора влияют и другие
трудногидрируемые компоненты. Так, например, активность пла-
тиновых катализаторов снижалась при использовании сырья с вы-
соким содержанием серы, газойлей термического и каталитического
крекинга, тяжелых газойлей Б4. Весьма влияет на активность ката-
лизатора наличие в сырье азота. Показано 68, что гидрокрекинг аро-
матизированного сырья, содержащего 40 млн’1 азота и выкипающего
в пределах 205—321 °C, мог быть осуществлен без падения актив-
ности катализатора в течение 4 месяцев при давлении 100 кгс/см2.
При понижении содержания азота до 2 млн-1 можно было достичь
тех же результатов уже при 54 кгс/см2.
На механизм дезактивации катализаторов азотсодержащими
соединениями точки зрения исследователей практически едины.
Считают *’ в0, что высокомолекулярные азотсодержащие соедине-
ния прочно адсорбируются на кислотных центрах катализатора,
блокируя их и понижая расщепляющую активность. Более высокое
давление водорода повышает его концентрацию на поверхности
катализатора, вследствие чего ускоряются йроцессы гидрирования
и гидрогенолиза адсорбированных азотсодержащих соединений.
Роль кислородсодержащих соединений изучена относительно
мало. Однако показано п, что уменьшение удельной поверхности
катализатора гидрокрекинга Pt на алюмосиликате, модифицирован-
ного цирконием, не коррелирует ни с интенсивностью отложения
кокса (выжигаемого при регенерации), ни со структурой применя-
емого сырья и содержанием в нем азота (в виде пиридина) или серы
(в виде тиофена). Уменьшение удельной поверхности коррелирует
только с содержанием в сырье кислородсодержащих соединений.
На основании этого был сделан вывод, что причиной уменьшения
322
активности катализатора является образование воды в ходе гидро-
крекинга и. Это заключение было подтверждено и опытами прямой
добавки паров воды к водороду. Механизм влияния паров воды
неясен, это, вероятно, связано с прочной адсорбцией воды и ее хими-
ческим взаимодействием с носителем.
Данные работы11 о независимости уменьшения поверхности
и активности катализатора от присутствия соединений, содержащих
азот или серу, по-видимому, не противоречат выводам работ *• в0> в8,
приведенным выше. В работе 11 применялся высокоактивный гидри-
рующий катализатор, а добавки серы и азота вводились в виде низко-
молекулярных соединений. Поэтому они не могли затруднять ад-
сорбцию реагирующих углеводородов, а сами относительно быстро
превращались. Соединения, содержащие азот и серу, видимо пред-
ставляют наибольшую опасность при переработке прочно адсорби-
руемого сырья в условиях недостатка водорода на поверхности ка-
тализатора.
Вследствие опасности отравления катализаторов внимание ис-
следователей привлекает в первую очередь обеспечение их стабиль-
ности. Известно много публикаций, в которых утверждается, что
созданы стабильные к отравлению катализаторы. Сообщалось 68
о создании катализаторов, способных работать при 100 кгс/см2
на сырье с содержанием 1000—2000 млн-1 азота. В одном из реклам-
ных сообщений 89 указывается, что частичное восстановление ме-
талла в катализаторе Ni -|- W на А12О3 -|- SiO2 (6% восстановлен-
ного Ni и 19% W в виде сульфида) повысило его азотоустойчивость.
В присутствии этого катализатора удалялось 92,5% азота (содержа-
ние азота в перерабатываемом газойле 0,319%; давление 70 кгс/см2)
против 80% на алюмокобальтмолибденовом катализаторе; через
90 суток он еще удалял 75% азота.
О создании более стабильных катализаторов было сообщено
в докладах на VII и VIII Мировых нефтяных конгрессах 52’ в0’ 70,
а также в публикациях 71> 72. Утверждается 72, что создан катали-
затор, настолько устойчивый к отложению металлов и кокса, что
даже после 2 месяцев работы он еще сохранял 80% начальной ак-
тивности, тогда как активность обычного катализатора в этих усло-
виях терялась практически полностью.
* , *
В заключение можно отметить, что процессы гидрокрекинга
отличаются большой специфичностью в отношении характера
химических превращений сырья. Многие особенности этих превра-
щений объясняются соотношением ионных и радикальных реакций
в зависимости от природы катализатора и условий процесса. Неко-
торые необычные реакции, например «спаривание» метильных за-
местителей, скелетная изомеризация в ароматических углеводоро-
дах (обмен атомов углерода заместителей и кольца) характерны
только для условий гидрокрекинга и для новых катализаторов
21*
323
этого процесса. Эти реакции еще ждут детального изучения и интер-
претации.
Развитие и успехи гидрокрекинга тесно связаны с созданием
новых, более эффективных катализаторов 52> 60 и работы в этом
направлении несомненно будут продолжаться. Создание еще более
устойчивых к отравлению и стабильных катализаторов не только
понизит капитальные вложения, но и позволит расширить диапазон
гидрокрекируемого сырья до сырых нефтей и остатков.
ЛИТЕРА ТУРА
1. КалечицИ. В. Современные тенденции разработки процессов получе-
ния малосернистых котельных топлив. М., ЦНИИТЭнефтехим, 1969. 79 с.
2. Sherwood W., Petroleum, 25, № 4, 122 (1962).
3. Современные процессы гидрогенизации. Сер. «Нефтепереработка». Вып. 10.
М., ВНИИОЭНГ, 1965.
4. ВоорхисА., СмитУ. С. В кн. «Новейшие достижения нефтехимии
и нефтепереработки». Т. 7—8. М., «Химия», 1968. См. с. 250.
5. О р о ч к о Д. И., СулимовА. Д., Осипов Л. Н. Гидрогениза-
ционные процессы в нефтепереработке. М., «Химия», 1971. 350 с.
6. Myers С. G., Munns G. W., Ind. Eng. Chem., 50, 1727 (1958).
7. Archibald В. C., Greensf elder B.S. et al., Ind. Eng. Chem.,
52, 745 (1960).
8. Flinn R. A., Larson O. A., Beut-her H., Ind. Eng. Chem., 52,
153 (1960).
9. S u 11 i v a n R. F., E g a n C. J. et al., J. Am. Chem. Soc., 83, 4156 (1961).
10. E g a n C. J., L a n g 1 о i s G. E., White R. J., J. Am. Chem. Soc.,
84, 1204 (1962).
11. Myers C. G., G a г w о о d W. E. et al., J. Chem. Eng. Data, 7, 257 (1962)
12. Frye C. G., В a г g e г B. D. et al., Ind. Eng. Chem., Prod. Res. Deve-
lop., 2, 40 (1963).
13. ХензелВ., Политцер Э. Л., Уоткинс Ц. Г. Труды VI Меж-
дународного нефтяного конгресса. М., ЦНИИТЭнефтегаз, 1965. См. с. 94.
14. Clement С., Montarnal R., Bull. Soc. chim. France, 1964, 334.
15. Coonradt H. L., Carwood W. E., Ind. Eng. Chem., Proc. Design a.
Develop., 3, 38 (1964).
16. HartwigM., Brennstoff-Chemie, 45, 234 (1964).
17. S u 11 i v a n R. F., E g a n C. J., Langlois G. E., J. Catalysis, 3,
183 (1964).
18. White R. J., E g a n C. J., L a n g 1 о i s G. E., J. Phys. Chem., 68,
3085 (1964).
19. Langlois G. E., Sullivan R.F., E g a n C. J., J. Phys. Chem.,
70, 3666 (1966).
20. H e n k e A. M., S c h m i d В. K., Strom J. R., Chem. Eng. Progr.,
63, № 5, 51 (1967).
21. Казанцева В. M., H e у д а ч и н а В. И., К а л е ч п ц И. В.
Нефтепереработка и нефтехимия, № 8, 23 (1968).
22. Казанцева В. М., К а л е ч и ц И. В., Нефтепереработка и нефте-
химия, А° 11—12, 24 (1968).
23. Н е у д а ч и н а В. И., Калечиц И. В., Нефтепереработка и нефте-
химия, № 1, 21 (1969).
24. К а л е ч и ц И. В., К а з а н ц е в а В. М., Л и п о в и ч В. Г., Нефте-
переработка и нефтехимия, № 6, 31 (1969).
25. К а л е ч и ц И. В., Н е у д а ч и н а В. И., Л и п о в и ч В. Г., Нефте-
химия, № 1, 47 (1969).
26. М i k i Y., К a b е Т. et al., Сёкубай, 12, № 4, 63 (1970); РЖХим. 7П159
(1971).
324
27. C iapett a E. C., D о b г e s R. M., Baker R. W. In «Catalysis».
Ed. by P. H. Emmett. V. 6. New York., Reinhold Publ. Corp., 1958. See
p. 495.
28. Гринсфельдер С. Б. В кн. «Химия углеводородов нефти». Л.,
Гостоптехиздат, 1958. Т. 2. См. с. 114.
29. Н е у д а ч и н а В. И. Кандидатская диссертация. МИНХ и ГП
им. И. М. Губкина, 1968.
30. П а н ч е н к о в Г. М., Васильева И. И., Жо ров Ю. М. В сб.
«Кинетика каталитических процессов». Вып. 86. М., «Химия», 1969. См.
с. 196.
31. N е 1 s о n W. L., Oil a. Gas J., 66, № 12, 120 (1968).
32. Фарберов М. И., Ветрова В. В., М и р о н о в а Г. С., Ж. орг.
хпм., 4, 163 (1963).
33. N е 1 s о n W. L., Oil a. Gas J., 65, № 36, 194 (1-967).
34. Pines Н., Shaw A. W., J. Am. Chem. Soc., 79, 1479 (1957).
35. Beecher R., V о о r h i e s A., Eberly P., Ind. Eng. Chem., Prod.
Res. a. Develop., 7, 203 (1968).
36. X мельницкий P. А. и др., Химия и технология топлив и масел,
№ 2, 56 (1967).
37. Каржев В. И., Сильченко Е. И. и др. В кн. «Исследование и
применение гидрогенизационных процессов в нефтеперерабатывающей и
нефтехимической промышленности». М., ЦНИИТЭнефтехим, 1968. См.
с. 18.
38. Хавкин В. А., Хмельницкий Р. А. и др., Химия и технология
топлив и масел, № 1, 7 (1969).
39. X а в к и н В. А., О с и п о в Л. Н. и др. В кн. «Исследование и приме-'
нение гидрогенизационных процессов в нефтеперерабатывающей и нефте-
химической промышленности». М., ЦНИИТЭнефтехим, 1968. См. с. 33.
40. Q a d е г S. A., Hill G. Ц., Ind. Eng. Chem., Proc. Design a. Develop.,
8, 98 (1969).
41. КацобашвилиЯ. P., Нечаев В. И., Нефтепереработка и нефте-
химия, № 8, 6 (1969).
42. КацобашвилиЯ. P., Нечаев В. И., Нефтепереработка и нефте-
химия, № 9, И (1969).
43. Каржев В. И., Сильченко Е. И. и др., Химия и технология топ-
лив и масел, № 11, 4 (1965).
44. Г о и и к б е р г М. Г. В сб. «Вопросы химической кинетики, катализа и
реакционной способности». М., Изд. АН СССР, См. с. 374.
45. Петров Ал. А. Химия нафтеновых углеводородов. М., Изд. АН СССР,
1971. 338 с.
46. S infelt J. Н., Ind. Eng. Chem., 58, № 12, 18 (1962).
47. LaszlaA., Magyar M., S teingaszher P., Acta Chim. Acad.
Hung, 31, 137 (1962); C. A., 57, 3691 (1962).
48. Б e й т e p X., Ш м и д т Б. Труды VI Международного нефтяного кон-
гресса. Вып. 2—4. М., ЦНИИТЭнефтегаз, 1965. См. с. 106.
49. Chervenak М. С., F е i 1 i g m a n S. et al., Chem. Eng. Prog., 59,
К» 2, 53 (1963).
50. Ф и л и м о н о в В. A., П b п о в А. А. и др., Нефтепереработка и нефте-
химия, № 10, 4 (1970).
51. Anderson J. R., Baker В. C., Proc. Roy. Soc., Ser. A., 271402
(1962); C. A., 58, 4355 (1963).
52. ФлюгтерИ. К., Вант СпийкерП. VIII. Мировой нефтяной
конгресс. Москва 1971. Препринты симпозиума № 12. См. с. 119.
53. Кацобашви ли Я. Р., Химия и технология топлива, № 1, 8 (1960).
54. С о о n г a d t Н. L., С i а р е 11 a F. G. et al., Ind. Eng. Chem., 53,
727 (1961).
55. К а ц о б а ш в и л и Я. Р., ПоповА. А., ЖПХ, 33, 1613 (1960).
56. К а ц о б а ш в и л и Я. Р., ПоповА. А., ЖПХ, 33, 1706 (1960).
57. L а г s о п О. A. et al., Ind. Eng. Chem., Proc. Design a. Develop., 1, 300
(1962).
325
58. Beuther H., Larson 0., Ind. Eng. Chem., Proc. Design a. Develop.,
4, 177 (1965).
59. Курганов В. M. и др., Нефтепереработка п нефтехимия, № 1,4 (1969).
60. А г е у W. F., Blackwell N. Е., R е i с h 1 е A. D. VII World Pet-
roleum Congress. Mexico. Proceedings. V. 4. London, Elsevier Puhi. Corp.,
1967. See p. 168.
61. Nonnenmacher H. u. a., Erdol u. Kohle, 22, № 9, 15 (1969).
62. H о e r i n g M., Oettinger W., Reitz 0., Erdol u. Kohle, 16,
361 (1963).
63. Oettinger W., Reitz 0., Erdol u. Kohle, 18, 267 (1965).
64. П e p e ж и г и н а И. Я. и др., Химия и технология топлив и масел,
№ 3, 8 (1970).
65. Can. Petrol. Eng., 40, № 6, (1965).
66. Hatch L. F., Hydrocarb. Proc., 11, № 2, 77 (1969).
67. Пат. CHIA 3267022 (1967); Экспресс-информация Сер. «Химия переработки
нефти и газа», № 36 (1967).
68. V о о г h i е s А., S m i t h W. M., Am. Chem. Soc. Div. Petrol. Chem.,
Preprints, 7 (1), 187 (1962); C. A., 59, 12567 (1963).
69. Oil a. Gas J., 64, № 4, 112 (1966).
70. Кубота К., Карн ер В. М. VIII Мировой нефтяной конгресс.
Москва. 1971. Препринты № 12. См. с. 61.
71. Н о в а к В., К у б и ч к а Р. В сб. «Исследование и применение гидро-
генизациоппых процессов в нефтеперерабатывающей и нефтехимической
промышленности». М., ЦНИИТЭнефтехим, 1968. См. с. 114.
72. Oil a. Gas I., 65, № 8, 55 (1967).
ГЛАВА 8
ПРОЦЕССЫ ДЕМЕТИЛИРОВАНИЯ
Процессы деметилирования являются частным случаем процессов
парофазной гидрогенизации и гидрокрекинга, но их химические
цели — отщепление метильных заместителей без затрагивания аро-
матических ядер — заставляют проводить такие превращения в жест-
ких условиях, что накладывает на них некоторые специфические
особенности. В самом деле, ионное отщепление метильных замести-
телей энергетически почти невозможно из-за высокой энергии об-
разования иона Н3С+ (см. гл. 2), следовательно в процессах деме-
тилирования необходимо обеспечить исключительное протекание
радикальных реакций. Последние усиливаются больше всего с ро-
стом температуры так, что при 450 —500 °C начинают преобладать
даже процессы деметилирования циклоалканов (см. стр. 228). С дру-
гой стороны, рост температуры сдвигает равновесие
ароматический углеводород-!-водород 7~~*~ циклоалкановый углеводород
влево, т. е. тоже выгоден для целей процессов деметилирования.
Таким образом, процессы деметилирования представляют собой
высокотемпературные процессы гидрокрекинга, в которых созда-
ются максимально благоприятные условия для радикальных реак-
ций расщепления и всеми мерами предотвращается гидрирование
ароматических углеводородов. ,Разработано много модификаций
как каталитических, так и некаталитических процессов деметили-
рования (см. гл. 1, а также обзоры 11 2), различающихся сырьем
и технологическими параметрами. Применение катализаторов по-
зволяет снижать температуру процесса на 100—150 °C (500—550
против 650—700 °C), что в свою очередь снижает капитальные вло-
жения вследствие применения более дешевых металлов для изго-
товления оборудования, но повышает стоимость эксплуатации из-за
расходов на производство и регенерацию катализатора. В зависи-
мости от конкретных экономических условий применяются и катали-
тические, и некаталитические процессы; в настоящее время в ряде
стран до 20—25% бензола и более 50% нафталина получают при
помощи процессов гидродеалкилирования х. Все процессы проте-
кают под давлением водорода.
Было показано 3, что наиболее вероятным механизмом отщепле-
ния, например отщепления метильной группы в толуоле, является
превращение связи С6Н6—СН3, упрочненной а — л-сопряжением,
327
в обычную связь за счет присоединения атомарного водорода
(см. также стр. 117, 181):
^\/СПз ^\/Н •
| II +н. —> I |\сн —► I || + сн3
\/-снз
1
Повышение давления водорода увеличивает его относительную
концентрацию и облегчает генерирование атомарного водорода:
R.(CH3) + I-I2 —> НН(СН4)+Н- 2
Однако из-за опасности гидрирования и нецелесообразности
удорожания оборудования применяемые давления не превышают
10—50 кгс/см2. При таких давлениях уже в области температур
500—550 °C реакции гидрирования практически невероятны из-за
термодинамических ограничений.
При изучении термодинамики реакций деметилирования толу-
ола и ксилола вычислены 4 константы равновесия этих реакций:'
С6Н5-СН3Ч-Н2 5=± СвНв + СЕЦ 3
С6Н4(СН3)2 + 2Н2 С6Нв + 2СН4 4
Темпера- 1g К Темпера- 1g Ар
тура, для ДЛЯ тура, для ДЛЯ
К реакции «3 реакции 4 К реакции 3 реакции 4
300 7,564 16,409 700 3,088 7,618
400 5,699 12,699 800 2,605 6,738
.500 4,525 10,362 900 2,209 6,038
600 3,703 8,778 1000 1,879 5,471
Показано, что, хотя с ростом температуры константы равновесия
уменьшаются, равновесия реакций 3 и 4 вплоть до температур
800 °C сдвинуты вправо.
Превращение толуола по радикально-цепному механизму вклю-
чает следующие реакции.
Зарождение цепей:
СвН5-СН2-Н —> СвН8-СН2 + Н- 5
Продолжение цепей:
свнй-си3+н. —> С6Н5- + СН4 6
С6Н5-СН3+Н- ----> С6Н6+-СН3 7
С6Н5- + Н2 -> С6Нв-1-Н- 8
Обрыв цепей:
С6Н5-+Н- —> С6Нв 9
2С6Н5-СН2 —> С6Н5-СН2-СН2-С6Н5 10
2С«Н5. —> С6Н5-С6Н5 11
Предполагали 5, что лимитирующей стадией является диссоциа-
ция молекулы водорода на атомарный водород с последующими
328
стадиями 6. и 1. Однако зарождение цепей по реакцим-
роятно, так как связь СвН6СН2—Н в толуоле наименее
и гораздо менее прочна, чем связь Н—Н (83 и 103 ккал/мол ь'сйк
ответственно). “ ' ‘
В случае ксйлолов наименее прочной связью оказывается свяаь
СН3—СвН4—СН3, и процесс инициируется реакцией в> ’
СвН4(СН3)2 —► СН3С«Н4- + -СН3
с последующими реакциями типа 2.
Радикально-цепной механизм реакций деметилирования хорошо
корреспондирует с данными об ускорении этого процесса в присут-
ствии других, более легко расщепляемых углеводородов, что уве-
личивает скорость стадии зарождения цепей. Так, деметилирование
толуола ускоряется парафинами 8, а ксилолов — добавкой этил-
бензола ®, имеющего легко разрываемую связь CgHjCHj—СН8.
Предложена 9 также схема,. включающая (в каталитическом .про-
цессе деметилирования толуола) диссоциацию толуола при хемо-
сорбции на бензильный радикал и водород и взаимодействие их
с образованием бензола и метиленового радикала, который аатем
гидрируется водородом. ,
Важнейшим вопросом химии процессов Деметилирования явля-
ется влияние строения исходных углеводородов на скорость их
превращения. В табл. 78 сопоставляются 1 скорости превращений
моноциклических ароматических углеводородов в каталитическом
и термическом процессах.
Таблица 78. Относительные скорости реакций
гидродеалкилирования 6>#’14
исходный углеводород Каталитический процесс Термический процесс
относитель- ная скорость гидродеал- килирования литература относитель- ная скорость гидродеал- килирования литература
Толуол 1,0 9—12 1,0 5, 12—14
.и-Ксплол 2,6 9, 10 2,3 13
» 1,2 12 3,5 14
— — 1,5 12
п-Ксилол 2,4 9, 10 1,5 5
» 2,1 12 2,9 14
о-Ксилол 4,6 9, 10 2,5 5
» 3,5 12 6,4 14
» — — 0,8 12
Смесь С3 2,7 11 -— —
1,3,5-Триметилбензол 1,9 12 4,2 13
— — 3,4 121
1,2,4-Триметилбензол 2,9 12 '— —
1,2,3-Триметилбензол 5,4 12 — —
Смесь Се 4.4 11 — ' —
Как видно из табл. 78, общей закономерностью, отмечаемой и,
для каталитического, и для термического процесса, является уско-
рение реакции деметилирования по мере накопления метильных
заместителей.
Можно также отметить, что соотношения констант скорости ре-
акции деметилирования ксилолов и толуола составляют при 590 °C
для орто-, мета- и пара-изомеров 3,0, 2,0 и 4,4, а при 680 °C — 3,0,
2,1 и 2,2 соответственно 15. л-Ксилол деметилируется в 1,5—2,0
раза быстрее толуола в. Объяснение этой общей закономерности
следует искать не только в том, что с ростом числа заместителей
увеличивается число столкновений молекулы с атомарным водо-
родом, ведущих к отрыву метильной группы, но и в механизме ре-
акции 7: заместители, имеющие положительный индуктивный эф-
фект, повышают электронную плотность в бензольном кольце, за
счет чего облегчается присоединение атомарного водорода.
Тот факт, что электронная структура молекулы играет важную
роль в этом процессе, вытекает также из того, что изомеры с разным
расположением заместителей существенно различаются по реак-
ционной способности. По скорости деметилирования углеводороды
С8'п С9 могут быть расположены в следующие ряды (б1; б2, б3—допол-
нительные заряды, вызываемые метильными группами I, II и III):
Из приведенных данных видно, что в случае 1,3-диметилбен-
зола и 1,3,5-триметилбензола, скорости деметилирования которых
ниже, чем у других углеводородов соответствующего ряда, метиль-
ные заместители не дают дополнительных зарядов у углеродных
атомов, несущих другие метильные заместители. Характерно, что
1,2,4-триметилбензол давал при неглубоком превращении 1,5%
бензола, 6,9% толуола, 3,8% о-ксилола, 0% n-ксилола и 16,8%
л-ксилола 16, т. е. быстрее всего удалялась метильная группа I
(алюмомолибденовый катализатор, 520°С, 100 кгс/см2). Аналогично,
при гидрокрекинге изодурола (алюмокобальтмолибденовый ката-
330
лизатор, 425 °C, 50 кгс/см2) относительные скорости образования
различных продуктов деметилирования и изомеризации (миграция
метильной группы с образованием дурола) составили 17:
1,2,3-Триметилбензол . . 3,7 1,3,5-Триметилбензол . . 38,1
1,2,4-Триметплбензол . . 30,1 Дурол................ 7,1
Так как к образованию 1,2,4-триметилбензола ведет удаление
любого из двух метильных заместителей изодурола, стоящих в по-
ложениях I и III, скорость удаления метильной группы в положении
II, следовательно, более чем вдвое превосходит скорость удаления
соседних. Это можно связать с тем, что именно у второго атома
будет образовываться наибольший дополнительный заряд:
С(1)
ЛЛ хс (id
8|8з^Г|^8з84
М к
“V|C w?0*'"1
Кроме этих закономерностей довольно четко определяется еще
одна: с наибольшей скоростью идет деметилирование изомеров
с расположением заместителей у соседних атомов углерода. Объясне-
ние этого не может заключаться ни в индуктивном эффекте, ни в про-
странственных затруднениях. Характерно, что в термическом про-
цессе без катализатора эта закономерность выполняется не всегда.
Возможно, что объяснение заключается в максимальной асимметрии
изомеров такого строения и, следовательно, в наибольшем диполь-
ном моменте, облегчающем адсорбцию в случае каталитического
процесса.
Помимо деметилирования в условиях гидродеалкилирования
идет и метилирование, так как происходит образование изомерных
ароматических углеводородов того же молекулярного веса. Так
в продуктах превращения и-ксилола был найден о-ксилол, в слу-
чае 1,2,4-триметилбензолй — 1,3,5-триметилбензол и для 1,3,5-три-
метилбензола — 1,2,4-триметилбензол 12.
Соотношение между скоростями деметилирования и метилирова-
ния было определено 18 при гидрокрекинге в сравнимых условиях
(катализатор А1 + Со Мо, 425 °C, 50 кгс/см2) смесей дурола
с меченными 14С (в кольце) бензолом, толуолом, ксилолами и три-
метилбензолами. Радиометрический анализ позволил определить
долю радиоактивности, перешедшей как в продукты меньшего (де-
метилирование), так и большего молекулярного веса (табл. 79).
Данные, приведенные в табл. 79, хорошо совпадают с описан-
ной выше основной закономерностью: скорость деметилирования
быстро возрастает с накоплением метильных заместителей. Скорость
метилирования изменяется в данных условиях медленно и в протц,
воположном направлении.
Из других реакций в процессах гидродеалкилирования предста-
вляют интерес реакции гидрирования ароматических углеводоро-
331
Табл. 79. Распределение радиоактивности в продукте гидрокрекинга дурола
Углево- дороды Радиоактивная добавка Суммарная радиоактивность
С, Се С, С, продуктов метилиро- вания продуктов деметилиро- вания
Сю 0.151 0,044 0 0 0,661 *
с9 — 0,094 0,046 0,020 0,151 0,236
с8 0,161 — 0,083 0,035 0,138 0,175
С7 0,063 0,150 — 0,061 0,129 0,052
С6 0,012 0,025 0,052 — 0,116 —
* Превращение дурола.
также содержавшихся
дов и расщепления продуктов гидрирования, а
в сырье алициклических и алифатических примесей. Все эти реак-
ции нежелательны, так как увеличивается расход водорода, хотя
в отдельных случаях 112 для очистки бензола от примесей идут
на заведомый перерасход водорода. В обычных условиях гидроде-
алкилирования реакции гидрирования подавлены, но если при
520 °C повысить давление с 50 до 100 кгс/см* * 2 (катализатор МоО3
на А12О3), то скорости гидрирования возрастают 12 следующим
образом:
1,2,4-Триметилбензол .
о-Ксплол.............
п-Ксилсл.............
Относитель-
ная скорость
гидрирования
1
2
2,3
Относитель-
ная скорость
гидрирования
2,5
3,8
9,1
.м-Ксилол
Толуол
Бензол
Таким образом, влияние заместителей на скорость гидрирования
аналогично низкотемпературному процессу (гл. 3).
Поведение углеводородов различных классов изучалось 19 при
температуре 700 °C. Парафины расщеплялись, давая осколки
и С2, олефины гидрировались до парафинов и превращались по-
добно им, циклоалканы также давали осколки Сх и С2, тетралин
давал не только бензол, но и нафталин. Декалин превращался
как парафины, аценафтены давали бензол и алкилбензолы. Анало-
гичные выводы сделаны и в ряде других работ х. Все классы угле-
водородов превращались тем быстрее, чем выше был их молекуляр-
ный вес. Таким образом, почти все неароматические углеводороды под-
вергаются в условиях гидродеалкилирования глубокой деструкции.
Закономерности гидродеалкилирования нафталинов аналогичны
закономерности гидродеалкилирования метилбензолов. При 520 °C,
40 кгс/см2, в присутствии катализатора МоО3 на А12О3 относитель-
ные скорости деметилирования составили 2°:
2-Метилнафталин............1,0
1-Метилнафталин............1,5
2,3- и 2,6-Диметилнафталины 1,6
1,6- и 1,7-Диметилнафталины 1,6
1,5-Дпметилнафталин .... 1,9
1,2-Диметилнафталин .... 2,2
332
Приведенные цифры показывают, что ддметилнафталМЯ^ЩШННН
руются быстрее монометилнафталинов, при рядовом^ pgfenslSHHHI
заместителей скорость превращения несколько увеличйЁЙ^вй}^
Влияние условий процесса в основном хорошо согласуется-с
ложенными выше его химическими особенностями. Повышение
давления водорода, облегчая стабилизацию радикалов (реакция 2)
должно тормозить реакции конденсации типа 10, 11. Поэтому при-
меняются повышенные давления, но так, чтобы не уменьшить селек-
тивность *. Повышение температуры увеличивает выход продуктов
деметилирования как в каталитических, так и в термических про-
цессах. Однако одновременно растет выход продуктов конденсации
и усиливаются отложения кокса на катализаторе. Поэтому для
каждого катализатора подбирается оптимальная температура, со-
ставляющая 1 для хромового и молибденового катализаторов на ак-
тивированном угле 535—550 °C, для окисного алюмокобальтмолиб-
денового катализатора — 580—600 °C, для хромового катализатора
без носителя — 600—650 °C. Во многих процессах в сырье вводят
водяной пар, что уменьшает образование продуктов конденсации
и кокса. Такое действие пара объясняют 21 ассоциацией молекул
воды с радикалами, что снижает реакционную способность ради-
калов, но не в такой мере, чтобы препятствовать реакции 2.
Кинетика процессов деметилирования соответствует представле-
ниям о цепном радикальном процессе. Для толуола эксперимен-
тально было получено уравнение 22 r = fcp!p8 р^9 где рт ирН1 —
парциальные давления толуола и водорода соответственно, которое
в принципе совпадает с уравнениями полуторного порядка (первый
по толуолу и 0,5 по водороду), предложенными для термиче-
ского 13- 23 и каталитического 9 (35—65 кгс/см3, 600—650 °C, оки-
сный хромовый катализатор) процессов.
Аналогичное уравнение г=кртр^& удовлетворяет условиям
термического деалкилирования пироконденсата производства эти-
лена, содержащего до 30% неароматических углеводородов 24. Энер-
гии активации термического24125 процесса 50—55 ккал/моль, а ката-
литического 25 — 35 ккал/моль.
Роль катализатора в процессах деметилирования ограничена.
Он должен облегчать зарождение радикалов' и быть устойчивым
к отложению кокса. Поэтому важное значение придается удельной
поверхности и размерам пор катализатора. Так, считают 2в, что ката-
лизатор должен иметь минимальное количество микропор, средний
радиус которых увеличивается путем прокаливания. Для пониже-»
ния коксообразования уменьшают кислотность носителей и вводят
в них щелочные металлы з7. Уменьшению коксообразования способ-
ствует и вода, конкурируя в. адсорбции с предшественниками
кокса 21.
В последние годы к процессам гидродеалкилирования под давле-
нием водорода добавились разработки процесса деметилирования
водой 28. Этот процесс еще не реализован в промышленности, но
его принципиальная возможность показана. 'Он осуществляется
333
в присутствии никелевого или никельхромового катализатора при
атмосферном давлении и температуре около 375 °C:
С6Н5-СН3 + 2ЩО —> с6н6+со2 + зн2
Реализация этого процесса, не потребляющего, а генерирующего
водород, может составить серьезную альтернативу процессам гидро-
деалкилирования.
ЛИТЕРА ТУРА
1. К р и ч к о А. А., Макарьев С. В. и др. Производство бензола гидро-
деалкилированием ароматических углеводородов. М., ЦНИИТЭнефтехим,
1970. 72 с.
2. О р о ч к о Д. II., С у л и м о в А. Д., О с и п о в Л. Н. Гидрогениза-
ционные процессы в нефтепереработке. М., «Химия», 1971. 350 с.
3. ГоникбергМ. Г. Химическое равновесие и скорость реакций при
высоких давлениях. М., «Химия», 1969. 427 с.
4. М а й о р о в Д. М., ЖОХ, 30, 230 (1960).
5. S i 1 s b у R. I., Sawyer E.W., J. Appl. Chem., 6, 347 (1956).
6. ГоникбергМ. Г., Гаврилова А. Е., Н икитенков В. Е.,
Изв. АН СССР, ОХН, 1961.1711.
7. Weiss А. Н., Hydrocarbon Proc. a. Petrol. Ref., 41, № 6, 185 (1962).
8. Zimmermann С. С., York R., Ind. Eng. Chem., Proc. Design
Develop., 3, 254 (1964).
9. W e i s s A. H., D о e 1 p L. C., Ind. Eng. Chem., Proc. Design Develop.,
3, 73 (1964).
10. Pitts P. M., С о n n о r G. E., Ind. Eng. Chem., 47, 770 (1955).
11. D о e I p L. C., W e i s s A. H., Ind. Eng. Chem., Proc. Design Develop.,
4, 92 (1965).
12. К p и ч к о А. А., Советовал. С. В сб. «Исследование и применение
гидрогенизационных процессов в нефтеперерабатывающей и нефтехимиче-
ской промышленности. М., ЦНИИТЭнефтехим, 1968. См. с. 4.
13. Shull S. Е., Nixon A. N., Ind. Eng. Chem., Proc. Design Develop.,
5, 146 (1966).
14. В e t t s N. D., Popper F.,Silsby R. L., J. Appl. Chem., 7, 497 (1957).
15. T s u c h i у a A., Hashimoto A. et al., Bull. Japan Petrol. Inst.,
2, 85 (1960); C. A., 55, 964 (1961).
16. К p и ч к о А. А., С о в e т о в Л. С., ЖПХ, 37, 141 (1964).
/ 17. НеудачинаВ. И., Калечиц И. В., Нефтепереработка и нефте-
* химия, № 1, 21 (1969).
18. К а л е ч и ц И. В., Н е у д а ч и н а В. И., Л и и о в и ч В. Г. Нефте-
химия, Л“ 1, 47 (1969).
19. Fowle М. J., PittsP. М., Chem. Eng. Progr., 58, № 4, 37 (1962).
20. К р и ч к о А. А., Советова Л. С., Нефтехимия, 4, И (1964).
21. К р и ч к о А. А. В сб. «Труды ИГИ и МУП СССР». Вып. 24. М., «Недра».
1967. См. с. 116.
22. ГоникбергМ. Г., Ли Гуан-нянь, ДАН СССР, 130, 763 (1960).
23. М о г i i Н., Н a s h i m о t о A., T о m i n a g a H. VI World Petro-
leum Congress. Proceedings. Sect. IV. Humburg, 1963. See p. 131.
24. Masamune Sh., Fukuda Y., К ata da S h., Hydrocarb. Proc.,
46, № 2, 155 (1967).
25. C r a i g R. G., D о e 1 p L. C., L igwinuk A. K., Erdol u. Kohle,
18, 527 (1965).
26. Gardner L. K., Ind. Eng. Chem., Proc. Design Develop., 3, 28 (1964).
27. С у л и м о в А. Д., Кожина И. Н., Т р а х т е н б е р г Д. М. Хи-
мия и технология топлив и масел. № 1, 17 (1965).
28. В а 1 a n d i n А. А., М a s 1 у a n s к у G. N. et al. VII World Petroleum
Congress. Mexico. Proceedings. V. 5. London, Elsevier Publ. Corp., 1967.
See. p. 121.
ЗАКЛЮЧЕНИЕ
Фундаментальные теоретические исследования и технологические
разработки в области гидрогенизационных процессов развиваются
параллельно, взаимно влияя друг на друга и определяя задали
близкого и далекого будущего. Некоторые из этих задач вытекают
из обобщения материалов, сопоставленных в данной монографии.
Можно считать, что решены основные проблемы гидроочистки
любых дистиллятных продуктов, хорошо проработаны вопросы
сочетания гидроочистки и гидрокрекинга со многими другими про-
цессами нефтепереработки — каталитическим крекингом, риформин-
гом, висбрекингом и другими. В значительной степени решены
проблемы селективного гидрирования непредельных и ароматиче-
ских связей без изомеризации и расщепления, а также проблемы
селективного расщепления без насыщения водородом ароматических
колец. Близки к разрешению проблемы прямого обессеривания
нефти и нефтяных остатков. Продолжают разрабатываться и станут,
вероятно, в определенных экономических условиях конкуренто-
способными с нефтепереработкой процессы гидрогенизационной
переработки различных смол и даже твердых топлив. Но в то же
время во многих важнейших направлениях прогресса гидрогени-
зации остается не мало, а иногда и очень много нерешенных и не-
ясных вопросов, а также возможностей совершенствования.
Сопоставление основных тенденций развития гидрогенизацион-
ных процессов убеждает прежде всего в том, что растет их специали-
зация, т. е. возникают все более и более селективные процессы,
в которых интенсивно протекает какое-либо одно превращение
или одна реакция, в то время как другие возможные сопутствующие
реакции сводятся к минимуму. Такая селективность достигается
определенным соотношением между различными реакциями соб-
ственно гидрирования (гидрирование диенов, олефинов, конденси-
рованных ароматических углеводородов, моноциклических углево-
дородов и другие), реакциями восстановления различных типов
(восстановление кислотных, эфирных, гидроксильных и других
кислородсодержащих групп, восстановление аминогрупп и другие),
реакциями изомеризации и гидроизомеризации, реакциями гидро-
генолиза различных типов (гидрогенолиз связей С—О, С—N, С—S,
раскрытие алициклических колец, отщепление алифатических за-
местителей и другие).
Управление скоростями этих реакций является сложной проб-
лемой, в которую входит комплекс таких вопросов, как влияние
строения исходных веществ на скорости самих превращений, а также
на скорости адсорбции и десорбции, вопросов взаимосвязи строения
реагирующих веществ и катализатора процесса.
В отношении реакций собственно гидрирования важнейшей про-
блемой является выяснение влияния на скорость гидрирования
электронной плотности гидрируемой связи. В этом направлении
сделано многое, но еще предстоит связать в единую стройную
335
систему влияние электронной конфигурации полициклических систем
на термодинамику, адсорбционно-десорбционные процессы и кине-
тику собственно гидрирования. Не решена, но принципиально раз-
решима задача неполного гидрирования ароматических углеводо7
родов с получением ценных для нефтехимии циклоалкенов и даже
циклоалкадиенов. Значительные перспективы открывает появление
гомогенных металлокомплексных катализаторов гидрирования.
Другой до конца не решенной проблемой собственно гидрирова-
ния является выяснение влияния заместителей, "иногда ускорящих,
а иногда тормозящих реакцию. Это противоположное в различных
условиях влияние заместителей объясняется либо их воздействием
на электронную плотность гидрируемого ароматического кольца,
либо стерическими препятствиями, но для разграничения этих
двух влияний пока нет достоверных методов.
Сложные превращения веществ в реакциях изомеризации и рас-
щепления во многих случаях удовлетворительно объясняются соот-
ношением радикальных и ионных .реакций. Изменением состава
и свойств катализаторов уже возможно усиливать их способность
ускорять либо ионные, либо радикальные превращения. Однако
стройной и законченной системы взаимосвязи состава и свойств
гидрирующих катализаторов с их активностью и селективностью
нет. Во многих случаях эмпирически найденные весьма активные
катализаторы не изучены даже настолько, чтобы судить об их хими-
ческом и фазовом составах. Это направление исследований — изу-
чение взаимосвязи свойств катализаторов с механизмом и кинети-
кой протекающих в их присутствии реакций — является ключевым
для создания новых гидрогенизационных процессов, в том числе
процессов гидроочистки нефтей и нефтяных остатков и более селек-
тивных процессов гидрокрекинга.
Таким образом, несмотря на крупные успехи в развитии гидро-
генизационных процессов, выдвинувшихся благодаря этим успе-
хам в первые ряды важнейших промышленных каталитических
процессов, и ученым-теоретикам, и ученым-технологам предстоит
сделать еще очень многое.
В соответствии с этим данная монография не может считаться
законченным трудом, она лишь констатирует современные теорети-
ческие представления и достижения в области гидрогенизации.
Хотелось бы надеяться, что это обобщение поможет информации
всех специалистов, работающих в данной интересной и быстро про-
грессирующей области, и тем самым ускорит ее развитие.
ИГОРЬ ВАДИМОВИЧ КАЛЕЧИЦ
Химия гидрогенизационных процессов в переработке топлив