/






































































































































Автор: Большаков Г.Ф. Гулин Е.И. Торичнев Н.Н.
Теги: технология топлив физика химия топливо
Год: 1965




Текст
(
87 коп.
.u
- J
К
И 3 Д А Т Е Л
« ХИМ
! 9 G 5
Г. Ф. БОЛЬШАКОВ, Е. И. ГУЛИН, Н. Н. ТОРИЧНЕВ
ж,/у/
ФИЗИКО-ХИМИЧЕСКИЕ
ОСНОВЫ ПРИМЕНЕНИЯ
МОТОР Н Ы X,
РЕАКТИВНЫХ
И РАКЕТНЫХ
ТОПЛИВ
Под редакцией
засл. деят. науки проф. К. К. ПАПОК
и канд. техн, наук П. И. ДАВЫДОВА
V.JT н г
•> • *
ИЗДАТЕЛЬСТВО «ХИМИЯ»
МОСКВА • 1965 • ЛЕНИНГРАД
. . 7
. . 9
. . 9
. . 12
. . 14
. . 19
. . 22
. . 23
. . 24
. . 26
. . 26
. . 27
. • 41
. . 44
. . 45
. . ”47
. . 47
. . 49
. . 52
. . 57
. • 66
. . 69
72
. . 72
. . 79
. . 81
. . 88
. . 93
. . 94
. . 94
. . 96
. . 98
. . 104
. . 105
3
В книге излажены физико-химические основы
применения моторных, реактивных и ракетных
топлив. Рассмотрены основные эксплуатацион-
ные свойства топлив — прокачиваемость, испа-
ряемость, воспламенение и горение, стабиль-
ность и коррозионность. Отдельная глава посвя-
щена токсичности топлив.
Книга рассчитана на инженеров и научных
сотрудников, работающих в области применения,
хранения и транспортирования топлив и эксплу-
атации двигателей и может быть полезной для
студентов высших учебных заведений.
ОГЛАВЛЕНИЕ
Предисловие ....................................................... 7
Глава I. Условия применения топлив...............•................. 9
Автомобильные и авиационные бензины............................ 9
Дизельные топлива ............................................ 12
Реактивные топлива................,........................... 14
Ракетные топлива ............................................. 19
Литература ....................................................... 22
Глава II. Прокачиваемость топлив.................................. 23
Основные факторы, влияющие на прокачиваемость топлив.......... 24
Прокачиваемость топлив при низких температурах................ 26
Охлаждение топлив при хранении и применении................. 26
Свойства топлив, определяющие их прокачиваемость............ 27
Фильтруемость топлив ....................................... 41
Гидравлические потери в трубопроводах при подаче топлив ... 44
Улучшение прокачиваемости топлив............................ 45
Прокачиваемость топлив при высоких температурах............... 47
Нагревание топлив при хранении и применении................. 47
Фильтруемость топлив ....................................... 49
Улучшение прокачиваемости топлив............................ 52
Прокачиваемость топлив при пониженном давлении.............. 57
Методы оценки прокачиваемости топлив.......................... 66
Литература .................-................................... 69
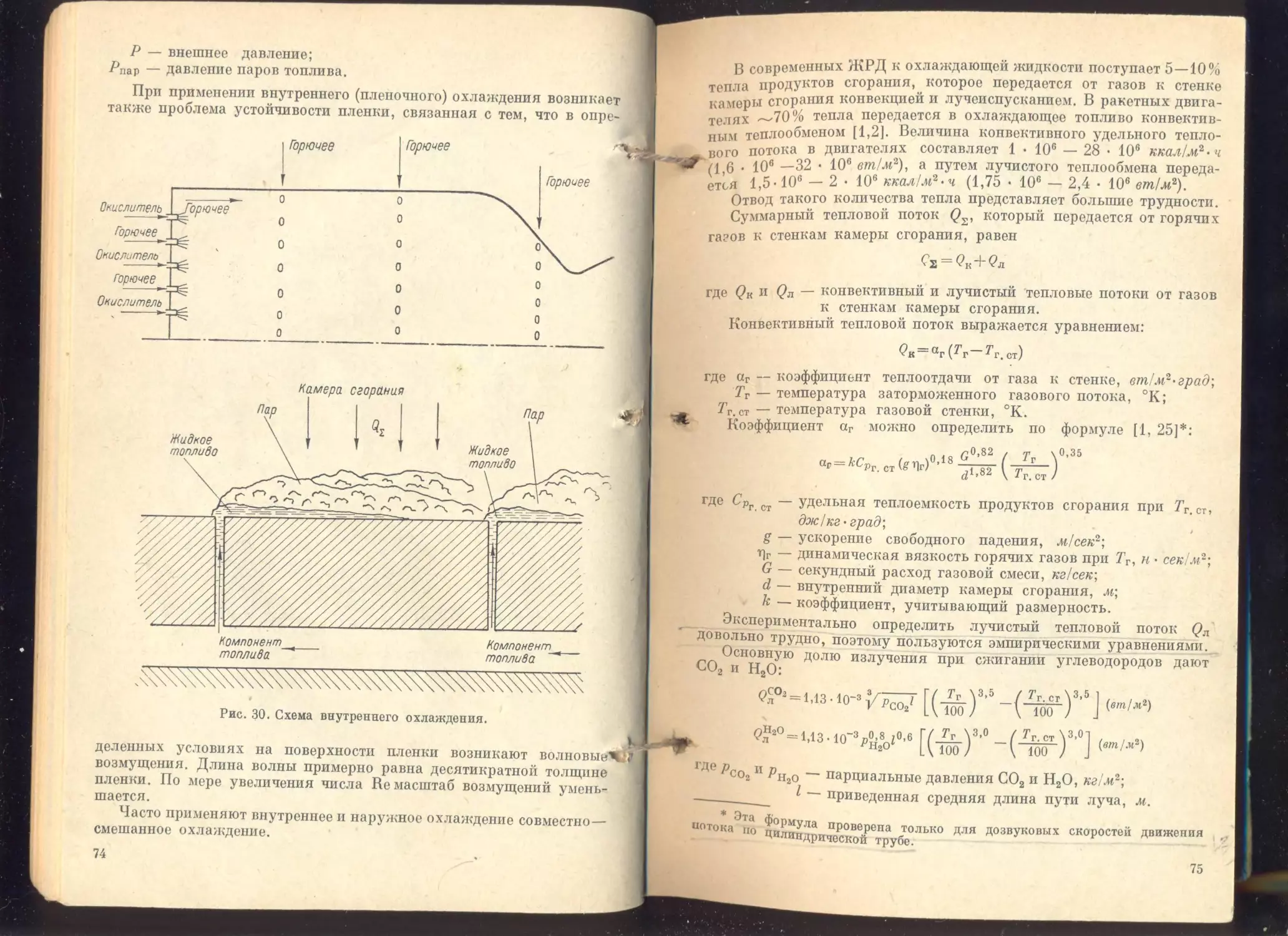
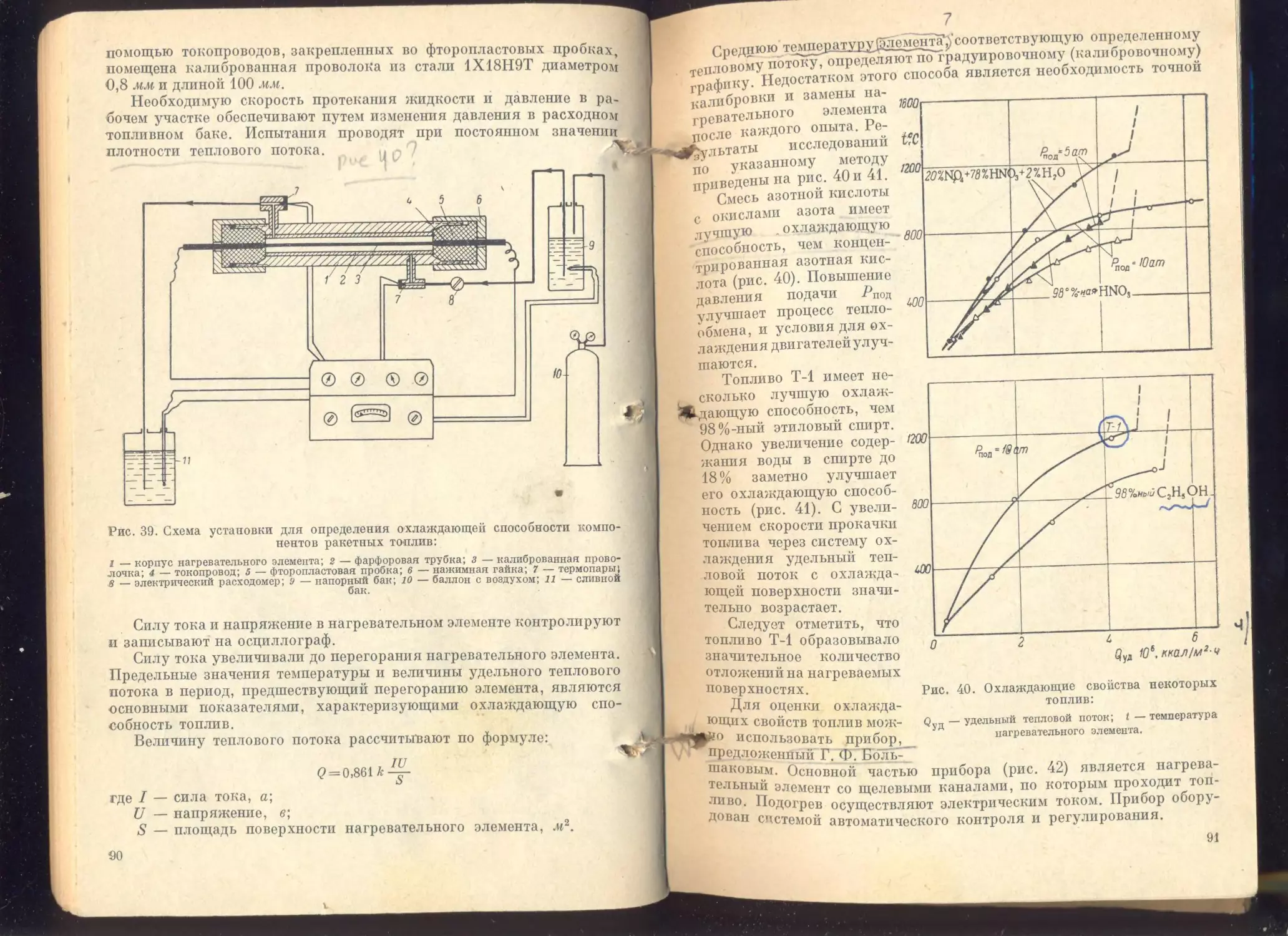
Глава III, Охлаждающие свойства топлив.................... 72
Некоторые представления о процессе теплообмена..............
Факторы, влияющие на процесс охлаждения.....................
Свойства топлив, определяющие их охлаждающую способность . .
Методы оценки охлаждающей способности топлив................
Литература .....................................................
Глава IV. Испаряемость топлив ...................................
Скорость испарения ...........................................
Статическое испарение ..................................... ;
Зависимость скорости испарения от свойств топлива...........
Зависимость скорости испарения от внешних условий...........
Динамическое испарение........................................
72
79
81
88
93
94
94
96
98
104
105
3
Зависимость скорости испарения от качества распиливания
топлива ...................................................
Зависимость скорости испарения от внешних условий..........
Методы расчета скорости испарения капель в потоке воздуха . . .
Методы оценки испаряемости топлив............................
Влияние испаряемости топлиц на рабочий процесс двигателя . . .
Литература ......................................................
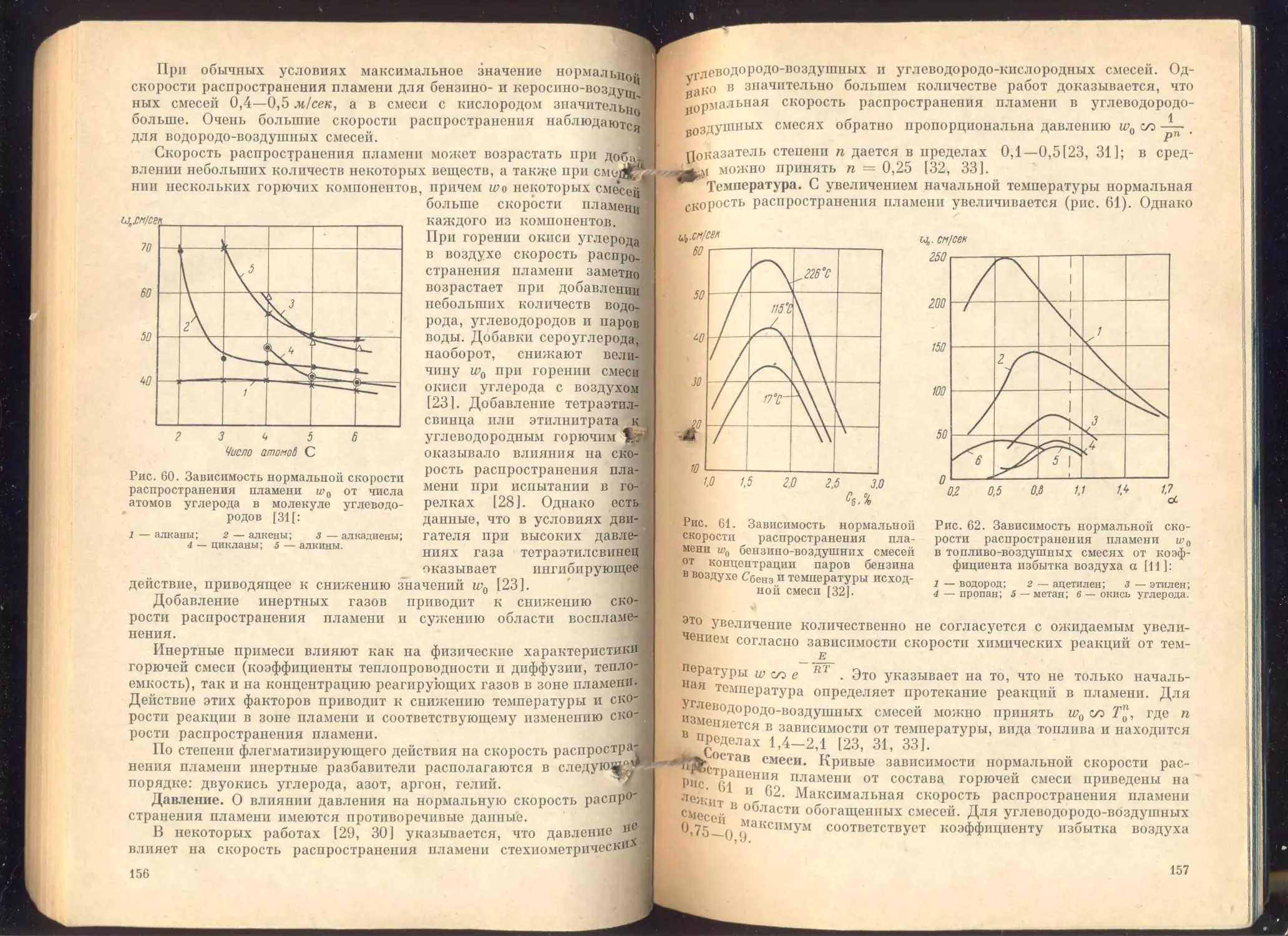
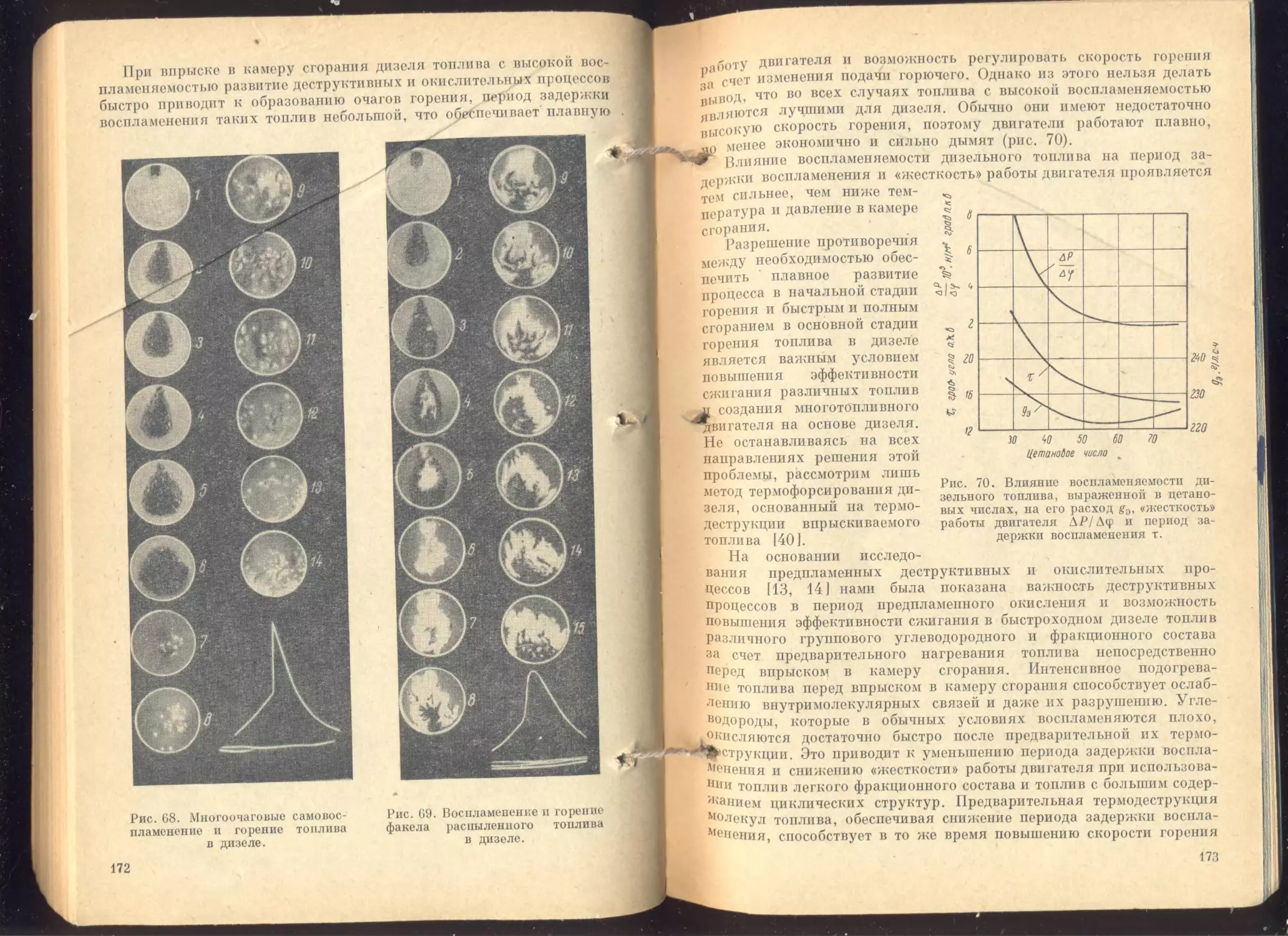
Глава V. Воспламенение и горение топлив..........................
Термохимия горения ..........................................
Основные положения цепной теории окисления...................
Условия воспламенения смесей горючего и окислителя...........
Механизм воспламенения ......................................
Влияние свойств топлива и условий воспламенения на параметры про -
цесса .......................................................
Методы оценки воспламеняемости топлив................... . . .
Параметры, характеризующие процесс горения . *...............
Нормальное распространение пламени.........................
Распространение пламени в турбулентном потоке..............
Детонационное распространение пламени......................
Виды горения топлив в двигателях..........»..................
Турбулентное горение однофазных горючих смесей.............
Турбулентное горение двухфазных горючих смесей.............
Методы определения скорости горения в двигателях . . . . . .
Зависимость рабочего процесса двигателей от химических свойств
топлива .....................................................
Литература ................................•.....................
Глава VI. Стабильность топлив ...................................
Химическая стабильность топлив...............................
Некоторые представления об окислении нефтяных топлив в жидкой
фазе ......................................................
Влияние углеводородного состава на стабильность топлив ....
Влияние гетероорганических соединений на стабильность топлив
О механизме образования смол и нерастворимых осадков в нефтя-
ных топливах ..............................................
Химическая стабильность компонентов ракетных топлив . . . .
Методы улучшения стабильности топлив.........................
Подбор оптимального химического состава топлив.............
Применение присадок .......................................
Методы определения стабильности топлив.......................
Лабораторные методы .......................................
Стендовые и эксплуатационные испытания.....................
Литература ......................................................
Глава VII. Коррозионность топлив ................................
Виды коррозии и механизм коррозионных процессов..............
Химическая коррозия .......................................
Электрохимическая коррозия ................................
Факторы, влияющие на коррозионность..........................
Коррозионность ракетных топлив...............................
Коррозионность нефтяных топлив...............................
О механизме образования отложений на металлах в углеводородных
топливах ....................................................
Противоизносные свойства топлив..............................
106
111
112
114
117
122
123
124
129
134
137
143
149
152
153
158
161
162
162
164
166
167
176
178
179
179
185
187
191
204
206
206
207
211
212
216
217
218
218
219
221
226
231
234
241
248
4
Способы уменьшения коррозионности топлйв..................... 250
Методы определения коррозионности топлив.................... 253
Лабораторные методы ....................................... 255
Эксплуатационные и стендовые испытания..................... 257
Литература
257
Глава VIII- Токсичность топлив....................................
Виды поражения организма токсическими веществами..............
Оценка токсичности топлив.....................................
Определение токсических веществ в воздухе.....................
Защита человека от поражения токсическими веществами..........
Оказание помощи при отравлениях.................................
259
259
260
263
265
268
Литература
269
Соотношение единиц международной системы (СИ)
с единицами других систем, использованными в книге
Единица измерения
Величина
Переводные коэффи-
циенты в единицы СИ
СИ
другие системы
Вязкости коэффи-
циент динамиче-
ский
Вязкости коэффи-
циент кинематиче-
ский
Давление, напряже-
ние
Мощность, тепловой
поток
н • сек/м2
м2/сек
н/м2
вт
Плотность
Работа, энергия, ко-
личество теплоты
Сила (в том числе
сила тяжести)
Температуропровод-
ности коэффициент
Теплоемкость удель-
ная
Теплопроводности
коэффициент
кг/м3
дж
н
м2/сек
дж/кг • град
вт/ м. • град
пз
спз
кгс • сек/м2
ст
сст
кгс/см2, ат
мм рт. ст.
мм вод. ст.
кгс • м/сек
л. с.
ккал/ч
кал/сек
г /см3
кет • ч
л. с. ч.
кал
ккал
дин
кгс
м2/ч
кал/г • град
ккал/м • ч • град
0,1 н • сек/м2
0,001 н-сек/м2
9,80665 н-сек/м2
10“4 м2/сек
10~6 м2/сек
9,80665 • 104 н/м2
133,322 н/м2
9,80665 н/м2
9,80665 вт
735,499 вт
1,163 вт
4,1868 вт
Ю3 кг/м3
3,6 • 106 дж
2,64780 -106 дж
4,1868 дж г/
4,1868 • 103 дж
10“5 н
9,80665 н
0,278 • 10~3 м2/сек
4,1868 дж/кг • град
1,163 ет/м • град
ПРЕДИСЛОВИЕ
С развитием моторной, реактивной и ракетной техники во много
раз возросла потребность в топливах; качество их улучшается,
появляются новые сорта.
В связи с повышением требований к качеству моторных, реактив-
ных и ракетных топлив усложнилось их хранение, транспортиро-
вание и применение, возросло значение систематического контроля
качества.
Возникла и непрерывно развивается новая отрасль знания —
применение топлив, в которой тесно переплетаются вопросы, отно-
сящиеся к химии и технологии топлив и эксплуатации двигателей.
Значение этой области науки исключительно велико, так как пра-
вильное применение топлив в двигателях обеспечивает их надежную
п длительную работу.
В настоящее время теоретическая и практическая разработка
новых топлив является исключительно сложной задачей, так как
к каждому пз них предъявляется комплекс специфических требо-
ваний, вытекающих из особенностей рабочего процесса двигателей,
условий хранения и транспортирования. Топливо должно обеспе-
чивать долговечную и надежную работу двигателя при одновремен-
ном уменьшении эксплуатационных расходов и упрощении хране-
ния, транспортирования и перекачки. Производство топлива должно
базироваться на недефицитном сырье, быть наиболее экономичным,
облегчать унификацию существующего ассортимента продуктов
и не вызывать усложнения конструкции и удорожания производства
двигателей.
Чтобы решать все эти сложные проблемы в комплексе, необхо-
димо систематически и глубоко изучать поведение топлив непосред-
ственно в двигателях, в эксплуатационных условиях, непрерывно
вести исследования в области химико- и физико-моторных и эксплу-
атационных свойств топлив.
Все это выполнимо только в том случае, если указанные проблемы
будут разрабатываться целенаправленно, высококвалифицирован-
ными специалистами и если будет обеспечена хорошая координация
работ по качеству и применению топлив, проводимых в нефтяной
7
и в машиностроительной промышленности и в организациях, эксплу-
атирующих технику. *
Поэтому совершенно правильно и своевременно подняты вопросы
о значении науки химмотологии, которая изучает эксплуата-
ционные свойства топлив и рассматривает наиболее целесообразные
пути их применения с целью обеспечения долговечной, надежной
и экономичной работы двигателей.
В данной книге рассматриваются физико-химические основы
применения моторных, реактивных и ракетных топлив их основ-
ные эксплуатационные свойства (прокачиваемость, испаряемость,
воспламенение и горение, стабильность, коррозионность и токсич-
ность). В начале книги рассмотрены условия применения автомо-
бильных и авиационных бензинов, дизельных, реактивных и ракет-
ных топлив. . .
Авторы приносят глубокую благодарность профессору, доктору
технических наук Я. Б. Черткову за помощь при подготовке этой
книги.
*К. К. Папок, Химия и технология топлив и масел, № 6 (1964).
Глава I
УСЛОВИЯ ПРИМЕНЕНИЯ ТОПЛИВ
В настоящее время выпускается значительное количество разно-
образных сортов и марок топлив для двигателей. Разнообразие топлив
объясняется различными условиями их применения в двигателях.
Свойства их во многом зависят от качества и технологии переработки
сырья, применяемых присадок и различных добавок.
Все существующие жидкие топлива по условиям применения
можно разделить на 6 групп:
1) автомобильные и авиационные бензины — для поршневых
двигателей с искровым зажиганием;
2) дизельные топлива — для дизелей;
3) реактивные топлива — для воздушно-реактивных двигателей;
4) ракетные топлива — для ракетных двигателей;
5) топлива для газовых турбин;
6) мазуты — для котельных установок.
Рассмотрим особенности устройства и рабочего процесса порш-
невых, реактивных и ракетных двигателей, условия применения
и требования к качеству топлив. Топлива для газовых турбин и
мазуты в книге не рассматриваются.
АВТОМОБИЛЬНЫЕ И АВИАЦИОННЫЕ БЕНЗИНЫ
В поршневых двигателях с искровым зажиганием бензин из бака
подается по топливопроводам с помощью насоса через фильтр-отстой-
ник в карбюратор. В нем происходит образование топливо-воздуш-
ной смеси, которая поступает затем во впускной трубопровод, где
заканчивается испарение топлива и образование горючей смеси
с воздухом. Горючая смесь поступает в цилиндр двигателя в такте
всасывания.
Весь процесс смесеобразования, который начинается в карбюра-
торе, продолжается во впускном трубопроводе и заканчивается
в такте сжатия, происходит достаточно быстро (0,05—0,1 сек в дви-
гателях с числом оборотов 2500 в минуту). Основная часть топлива
9
уносится во впускной трубопровод, где крупные капли оседают 1
на стенки и образуют жидкую пленку, которая обычно полностью 1
испаряется к концу впускного трубопровода. Однако иногда окон-
чательное испарение бензина происходит в цилиндре двигателя >
в момент всасывания и сжатия. ; -1
К моменту воспламенения необходимо полное испарение топлива.Д*
Наличие жидкой фазы ухудшает процесс горения. Возрастает удель-
ный расход топлива, увеличивается нагарообразование. Капли |
неиспарившегося бензина стекают по стенкам цилиндров в картер |
двигателя, смывая смазочное масло и вызывая этим повышенный
износ цилиндров и поршневых колец. <
Наличие жидкой пленки ухудшает распределение бензина по
цилиндрам двигателя; в одни цилиндры жидкая пленка поступает
в большем количестве и тогда рабочая смесь в них оказывается обо-
гащенной, в другие — в меньшем и рабочая смесь в них оказывается
обедненной. То и другое приводит к плохому сгоранию смеси, умень-
шению мощности и снижению экономичности двигателя.
Поступившая в цилиндры смесь сжимается до 6—12 ат (6 • 10°—
1,2 • 106 н/м2) и за 15—25° до верхней мертвой точки поджигается
от искры [1, 2]. В результате сгорания смеси резко повышается
давление, под воздействием которого поршень двигателя производит
механическую работу.
В двигателях с непосредственным впрыском бензина время,
отводимое на процесс Испарения, значительно меньше. Оно опреде-
ляется моментом от начала впрыска до воспламенения и составляет
0,02—0,03 сек. В такте впуска факел распыленного бензина омы-
вается потоком поступающего воздуха. Значительная скорость вих-
ревого движения воздуха, повышенная температура остаточных
газов и низкое давление в камере сгорания являются благоприят-
ными факторами, обеспечивающими высокую скорость испарения
бензина и перемешивания его паров с воздухом. Экспериментально
установлено, что в такте впуска испаряется — 80% бензина.
Во время сжатия топливо-воздушной смеси снижается
рость вихревого движения воздуха и повышается давление
ухудшает испаряемость бензина. Кроме того, температура достигает
400—450° С. Повышение температуры оказывает на испарение большее
влияние, чем замедляющий эффект повышенного давления, и поэтому
доиспарение бензина в такте сжатия происходит практически пол-
ностью.
Энергия, выделяющаяся при сгорании бензинов, может быть
использована достаточно эффективно только при нормальном сго-
рании.
При нормальном сгорании (рис. 1, а) после зажигания топливо-*
воздушной смеси от свечи начинается процесс горения, сопрово-
ждающийся плавным повышением давления. Скорость распростра-
нения фронта пламени составляет 20—30 м/сек; температура газов
в цилиндре двигателя 2500—2700° С. Давление в камерах сгорания
достигает максимума: — 40 am (4- 106 н/м2) для автомобильных
ско-
нто
10
«- и _go ат для авиационных двигателей (8 • 10е н/м2).
сгорания 0,002-0,010 сек.
’ _____ А ЕГ Z Г\ /_ хч ГЪ. лгтт Г ТТ П Т\ П ЛЛППТТТТ ГТ
- в современных форсированных двигателях составляет
двигателей
—
Время средней скорости сгорания 15—Уд м/сек скорость нарастания
При f ________Л>ГЛТтттт_тл’ Лппеппппяштиу пппгатрлях составляет
TTlfCI 13 CVJ-D |На‘Н {^АААА LJA-iv xj, wjy р V/ —*- ГЛ-
давлен* пт 7 . ю5—2,4 • 105 н/м2) на Г поворота коленчатого
Г | ‘ ) j
ВаЛВ определенных условиях нормальное
в детонационное, которое сопровождается
сгорание переходит
появлением
ударных
Н ВМ.Т р| ВМТ
Рис. 1. Развернутые диаграммы рабочего процесса четырехтактного карбюра-
торного двигателя при нормальном (а) и детонационном (б) сгорании.
I — первая фаза — период предпламенных реакций; II — вторая фаза —период видимого
сгорания. -
волн (рис. 1, б); скорость сгорания возрастает и составляет
1500—2500 м/сек. Тепловой режим двигателя нарушается, мощность
его уменьшается. Возникновение детонационного сгорания во многом
зависит от химического состава бензинов. Октановое число бензинов,
характеризующее их антидетоиационные свойства, должно быть
оптимальным и не вызывать нарушения нормального сгорания на
всех режимах работы двигателя.
Таким образом, для обеспечения надежной и экономичной работы
поршневых двигателей с искровым зажиганием в любых условиях
эксплуатации бензины должны удовлетворять следующим важней-
шим требованиям:
1) иметь оптимальную испаряемость (бензин должен полностью
испариться до воспламенения, но испаряемость не должна быть
чрезмерно высокой из-за возможности образования паро-воздушных
пробок в системе питания);
2) сгорать без детонации;
И
3) не образовывать отложений и нагаров при применении в дви-
гателях;
4) не вызывать коррозии металлических деталей двигателя;
5) иметь высокую химическую и физическую стабильность;
6) не содержать воды и механических примесей.
ДИЗЕЛЬНЫЕ ТОПЛИВА
Отличительной особенностью рабочего процесса дизеля является
воспламенение топлива без внешнего источника зажигания в среде
воздуха, сильно нагретого за счет сжатия в цилиндре двигателя.
Давление в цилиндре в конце такта сжатия составляет 30—40 ат
(3 • 106 — 4 • 106 н/ж2), а в двигателях с наддувом 70 ат
(7 • 106 н!м2) и более. Температура воздуха в конце сжатия дости-
гает, как правило, 500° С [2].
Топливо подается через фильтры (центрифуги) во всасывающую
полость насоса высокого давления или в насос-форсунку, а затем
через распыливающие устройства в камеру сгорания. Для обеспе-
чения хорошего смесеобразования впрыск осуществляется под давле-
нием 100—1500 ат (9,8 • 106 — 1,4 • 108 н/м2).
Процесс смесеобразования включает: распылпвание определен-
ного количества топлива, вводимого в камеру сгорания; распреде-
ление его элементарных объемов в камере сгорания; испарение и диф-
фузию паров в окружающую среду (воздух). Время смесеобразования
и сгорания в быстроходных дизелях 0,003—0,006 сек,
С началом воспламенения топливо-воздушной смеси процессы
смесеобразования не успевают еще полностью завершиться во всем
объеме камеры сгорания и протекают одновременно с процессом
горения.
Физико-химические превращения, которым подвергается рабочая
смесь в двигателях от начала поступления топлива в камеру сгора-
ния до выхлопа, представляют собой единый непрерывный процесс.
При исследовании этих превращений процессы воспламенения и горе-
ния в дизеле удобно разделить на отдельные фазы.
К первой фазе относят процессы, протекающие от начала впрыска
топлива до образования очагов пламени и резкого повышения давле-
ния газов в камере сгорания. Это время называется периодом за-
держки воспламенения.
Топливо, поступающее в этот период в камеру сгорания, распы-
ливается, испаряется и смешивается с воздухом. Под воздействием
температуры и давления происходят реакции окисления и разрушения
химических соединений, входящих в состав топлива. К началу
воспламенения испариться и смешаться с воздухом успевает только
часть поступившего топлива, в которой и происходит затем воспла-
менение.
Длительность периода воспламенения определяется скоростью
физических процессов подготовки топлива к воспламенению (нагрев,
распыливание, испарение и диффузия) и скоростью предпламенного
окисления, зависящей в основном от химического состава то-
плива.
Длительность периода задержки воспламенения оказывает боль-
шое влияние на характер развития
Сущность второй фазы — процесса
: по
последующей фазы.
горения — составляет распро-
топливо-воздушной смеси от
При ' очень большой
повышения давления
странение турбулентного горения
одного или нескольких первона-
чальных очагов пламени.
Большая склонность топлива
к низкотемпературному окислению
в фазе турбулентного горения ока-
зывается излишней и нежелатель-
ной. Топлива с очень хорошей вос-
пламеняемостью имеют сравни-
тельно невысокую скорость
горения, и использование их уве-
личивает продолжительность вто-
рой фазы.
При нормальной работе дви-
гателя скорость горения и, соот-
ветственно, скорость нарастания
давления после воспламенения не
превышают определенной вели-
чины для двигателя данной кон-
струкции,
скорости
работа двигателя становится «же-
сткой»: силовые нагрузки на ос-
новные детали поршневой и
шатунно-кривошипной групп уве-
личиваются. Это приводит к повышению деформации и местному
перегреву колец и поршня, к нарушению нормальных условий
работы коренных и шатунных подшипников. «Жесткая» работа
нередко приводит к поломкам поршневых колец и иногда порш-
невых пальцев.
Важнейшим фактором, оказывающим влияние на «жесткость»
работы двигателя, является период задержки воспламенения. С уве-
личением его в камере сгорания к моменту начала воспламенения
накапливается большое количество топлива. Переход от предпла-
менных стадий реакции к воспламенению в основной массе топливо-
воздушного заряда сопровождается резким нарастанием давления,
определенное влияние на скорость нарастания давления оказывает
закон подачи топлива (рис. 2).
Увеличением до определенного предела «жесткости» работы
двигателя происходит увеличение его мощности и уменьшение удель-
н°го расхода топлива. Только «жесткая» работа при очень большом
Р задержки воспламенения сопровождается ухудшением этих
зателей, так как при этом сильно возрастают затраты энергии
Рис. 2. Влияние закона подачи то-
плива на скорость нарастания да-
dP
вления в камере сгорания:
1,2 — кривые, характеризующие подачу
топлива G по углу поворота коленчатого
вала ф; 5, 4 — индикаторные диаграммы,
соответствующие кривым 1 и 2.
13
на трение, а общая продолжительность процесса горения увеличи-
вается настолько, что он занимает значительную часть хода расши-
рения.
Для большинства быстроходных дизелей при скорости нараста-
ния давления в начале второй фазы более 4—7 ат (3,8 • 105 —
6,8- 10б н/м2) на 1° поворота коленчатого вала работа двигателя ста-
новится «жесткой». Для отдельных типов форсированных двигателей
вполне допустима скорость нарастания давления более 10 ат на Iе
поворота коленчатого вала. Эти двигатели, как правило, являются
более быстроходными. Таким образом, в более быстроходных дизе-
лях можно и нужно допустить большую скорость нарастания
давления.
Фазы воспламенения и горения не всегда можно четко разделить
по времени. После возникновения первоначальных очагов воспламе-
нения и начавшегося распространения турбулентного пламени
возможно образование дополнительных очагов, от которых, в свою
очередь, начинается распространение пламени.
Таким образом, для обеспечения надежной и экономичной работы
дизелей, а также нормальных условий применения, хранения и транс-
портирования топливо должно отвечать следующим основным тре-
бованиям:
1) хорошо прокачиваться в различных условиях эксплуатации;
2) хорошо распиливаться, легко испаряться и сгорать с большой
скоростью;
3) воспламеняться с минимальной задержкой воспламенения;
4) не образовывать смолистых отложений и осадков в системе
питания;
5) не образовывать лаков и нагаров в цилиндро-поршневой
группе двигателя;
6) не вызывать коррозии деталей двигателя, средств заправки,
перекачки и
хранения,
РЕАКТИВНЫЕ ТОПЛИВА
распространение воздушно-реактивных двигателей
объясняется следующими преимуществами их перед
. Широкое
(в авиации)
поршневыми:
1) повышением коэффициента полезного действия, силы тяги
и тяговой мощности двигателя с увеличением скорости полета, что
позволяет осуществить близкие к звуковым и сверхзвуковые ско-
рости полета летательных аппаратов;
2) простотой конструкции — отсутствием сложного и тяжелого
кривошипно-шатунного механизма с неуравновешенными силами
третьего и четвертого порядков;
3) сравнительно небольшим удельным весом двигателя по отно-
шению к общему полетному весу летательного аппарата;
4) возможностью применения более дешевых сортов топлива,
с меньшей пожароопасностью по сравнению с высокооктановыми
авиационными бензинами.
14
Применение воздушно-реактивных двигателей стало необходи-
мым условием развития авиации, так как поршневые двигатели не
могли обеспечить требуемых скоростей и потолка летательных
аппаратов. Приближение к звуковому порогу явилось пределом
применения поршневых двигателей в авиации (рис. 3).
Рис. 3. Области применения летательных аппаратов с порш-
невыми (I), турбовинтовыми (2), турбореактивными (5), тур-
бореактивными с дожиганием (4), прямоточными (5) и ра-
кетными (6) двигателями. М — число Маха *.
осу-
Полет летательных аппаратов с реактивными двигателями
Ществляется в среде, имеющей различные температуры и давления
13, 4], поэтому температура топлива может изменяться от —50° С
* М — число Маха. М =
аппарата, км/ч; 1194,5 — скорость распространения звука при 0° С и нормаль-
ном атмосферном давлении, км/ч.
—где V — скорость полета летательного
1194,5
км/ч.
15
Принцип работы современного турбореактивного двигателя
состоит в следующем [3].
Воздух через входной диффузор (рис. 4) направляется в компрес-
сор, где сжимается до 3,5—4,5 ат (3,3 • 105 — 4,3 • 105 н/м2). Затем
ЭШЕ
часть его попадает в камеру сгорания, куда через форсунки под
давлением 50—60 ат (5 • 106 — 6 • 106 н/м2) подается топливо. Осо-
бенность работы реактивных двигателей заключается в том, что
Рис. 4. Принципиальная схема турбореактивного двигателя с дожиганием:
I — топливный бак; 2 — насос низкого давления; 3 — фильтр грубой очистки; 4 — баростат;
5 _ насос высокого давления; 6 — фильтр тонкой очистки; 7 — насос системы дожигания;
g —регулируемое сопло; 9 —форсунки системы дожигания; 10 —турбина; 11 камеры
сгорания; 12 — компрессор; 13 — система выдвижного конуса; 14 — диффузор; 15 — система
для перепуска воздуха.
топливо перед поступлением в камеру сгорания проходит сложную
систему приборов командного агрегата с малыми зазорами трущихся
прецизионных пар. В камере сгорания происходит образование топ-
ливо-воздушной смеси и ее горение, причем температура газов
повышается до 1400—1700° С.
Рабочий процесс в воздушно-реактивных двигателях происходит
непрерывно в потоке воздуха и газа. При установившемся режиме
процессы испарения топлива, смесеобразования и горения топливо-
воздушной смеси происходят одновременно, испарение и смесеобразо-
вание не заканчиваются к моменту поджигания смеси факелом пла-
мени и практически продолжаются в зоне горения. Фронт пламени
в камере сгорания должен быть устойчивым на всех режимах работы
двигателя. Затухание и срыв пламени могут произойти при чрез-
мерном обеднении или обогащении рабочей смеси, или же когда
скорость газового потока превышает скорость распространения
фронта пламени. — ’ * } •
Для обеспечения устойчивого фронта пламени, имеющего особое
значение в условиях полета, необходимо надежное регулирование
подачи топлива и воздуха, хорошее воспламенение в широких пре-
делах состава рабочей смеси. Температуру газов, выходящих
из камеры сгорания, понижают путем смешения их с холодным возду-
16
хом из второго контура. Эта газовая смесь вращает турбину, вслед-
ствие чего температура и давление смеси несколько понижаются. За-
тем газовый поток направляется в диффузор, при выходе из которого
расположена форсажная камера, где сжигается еще некоторое коли-
чество топлива, благодаря чему создается дополнительная тяга.
На выходе из сопла образуется мощный газовый поток большой
скорости, который и создает реактивную тягу.
Существуют разные типы воздушно-реактивных двигателей. Кроме
турбореактивных известны турбовинтовые и прямоточные двигатели.
Рис. 5. Принципиальная схема турбовинтового двигателя:
1 — винт; 2 — компрессор; з — камера сгорания; 4 — газовая турбина; 5 — сопло;
6 — редуктор.
В турбовинтовом двигателе (рис. 5) перед входным диффузором
имеется винт, который приводится во вращение через понижающий
редуктор от вала турбины. При этом около 80% мощности расхо-
дуется на привод винта и лишь 15—20% на создание реактивной
тяги. Турбовинтовые двигатели весьма экономичны и выгодны при
околозвуковых (Л/ = 0,8—0,9) скоростях полета.
Для больших сверхзвуковых скоростей полета применяют прямо-
точные воздушно-реактивные двигатели и их комбинации с компрес-
сорными воздушно-реактивными двигателями. В прямоточных дви-
гателях воздух при торможении сжимается и затем под давлением
поступает в камеры сгорания (рис. 6). В камеры сгорания через
форсунки подается топливо, которое распыливается, испаряется,
смешивается с воздухом, воспламеняется и сгорает. Продукты сго-
рания вытекают из сопла, создавая реактивную тягу. Перед посту-
плением в камеры сгорания воздух необходимо сжать в 2—3 раза,
то осуществимо только ПрИ значительной сверхзвуковой скорости
олета (3—4 Л/). Таким образом, прямоточные двигатели предназ-
ены только для больших сверхзвуковых скоростей.
it v ‘°А* Из ос°бенностей эксплуатации летательных аппаратов
ВИи рабочего процесса воздушно-реактивных двигателей, для
2 Закаа 782. ‘ 17
обеспечения надежной и безотказной работы двигателей на всех
режимах, реактивные топлива должны:
1) надежно прокачиваться по топливоподающей системе при
высоких и низких температурах, а также при пониженном внешнем
давлении;
2) полностью испаряться в камерах сгорания;
*
Рис. 6. Устройство прямоточного воздушно-реактивного
двигателя (а) и принципиальная схема изменения харак-
теристик газового потока в нем (б):
1 — диффузор; 2 — камера сгорания; 3 — сопло; 4 — стабилизатор
пламени; 5 — форсунки; 6 — топливный насос.
3) воспламеняться в широких пределах состава топливной смеси
с небольшим периодом задержки воспламенения;
4) устойчиво, полно, с высокой скоростью сгорать при большом
избытке воздуха, без образования нагаров;
5) иметь высокую теплоту сгорания, как весовую, так и объ-
емную; о J
6) не корродировать деталей топливной системы; й
7) не изменять своих свойств в условиях хранения и транспорти-
рования;
8) иметь достаточные противоизносные свойства (особенно для
топлив облегченного фракционного состава).
18
Реактивные топлива, применяемые в сверхзвуковых летательных
аппаратах, наряду с указанными требованиями должны иметь высо-
кую термоокислительную стабильность и температуру начала кипе-
ния выше температуры возможного нагревания топлива в полете.
РАКЕТНЫЕ ТОПЛИВА
Потребность в мощных силовых установках, которые могут
работать в отсутствие атмосферного кислорода (в условиях космоса)
и позволяют достигать огромных высот и скоростей, привела к со-
зданию ракетных двигателей. Русские ученые и инженеры сыграли
выдающуюся роль в развитии и совершенствовании ракетной тех-
ники. В нашей стране впервые в мире была высказана мысль о воз-
можности применения ракетных двигателей для летательных аппа-
ратов, созданы первые двигатели. Русские и советские ученые раз-
работали п по существу первыми применили на практике теорию
реактивного движения и ракетных двигателей.
Ракетные двигатели по сравнению с другими типами двигателей
имеют особые свойства и преимущества, которые вкратце сводятся
к следующему:
1) исключительно большая мощность — десятки миллионов
лошадиных сил и более, вследствие чего ракеты могут достигнуть
Увесьма больших скоростей (50 000 км/ч — для жидкостных ракет-
ных двигателей; 100 000 км/ч — для двигателей с термоядерными
силовыми установками; более 100 000 км/ч— для фотонных двига-
телей);
2) простота конструкции и небольшой удельный вес, приходя-
щийся на единицу тяги;
3) ракетные двигатели могут работать в безвоздушном простран-
стве и под водой;
4) тяга не зависит от скорости полета.
Существующие ракетные двигатели характеризуются кратко-
временностью работы и большой теплонапряженностью камер сго-
рания. Рассмотрим особенности рабочего процесса и условия при-
менения топлив только в жидкостных ракетных двигателях (ЖРД).
Жидкие ракетные топлива делятся на однокомпонентные и много-
компонентные (как правило, применяют двухкомпонентные топлива),
которые, в свою очередь, могут быть самовоспламеняющимися и неса-
мовоспламеняющимися. Жидкие однокомпонентные топлива практи-
чески не нашли широкого применения в ракетных двигателях [5].
Рассмотрим основные физико-химические превращения двухком-
плектных топлив в ЖРД. Химические реакции в камерах сгорания
Должны осуществляться с большой скоростью и возможно более
полно с выделением максимального количества тепла, а процесс
истечения газов должен происходить с наименьшими потерями
энергии.
На коэффициент полезного действия двигателя весьма большое
влияние оказывают теплопроизводителыюсть топлива, степень
2*
19
газообразования, молекулярный состав образующихся продуктов
сгорания, давление в камере сгорания.
Основными процессами, протекающими в ЖРД, являются: подача
компонентов в камеры сгорания в строго определенных количествах;
охлаждение топливом (как правило) камер сгорания; распиливание
и испарение топлива; смешение паров топливных компонентов;
химическое взаимодействие горючего и окислителя; воспламенение
и горение топливной смеси; истечение продуктов сгорания из сопла.
Рис. 7. Принципиальная схема рабочего процесса и ха-
рактеристика газового потока жидкостного ракетного
двигателя:
Зоныз I — смесеобразования; II —горения; III —догорания;
IV — упорядочения движения продуктов сгорания; V — истече-
ния и расширения газов.
В соответствии с этими процессами объем камеры сгорания и реак-
тивного сопла ЖРД можно условно разделить на зоны (рис. 7).
Физико-химические превращения топлива происходят в основном
в первых трех зонах.
В зоне смесеобразования происходят подогрев, распыливание,
испарение и смешение топлива. К ней прилегает зона горения, а на
границе зон происходит воспламенение топлива.
Подготовленное и воспламенившееся топливо поступает в зону
горения, где развиваются реакции взаимодействия между горючим
и окислителем, начавшиеся в зоне смесеобразования. В зоне горения
происходит выделение большого количества энергии, давление и тем-
пература смеси резко возрастают. Образующиеся раскаленные
продукты сгорания направляются в зону упорядочения движения
и далее в сопло. Зона истечения и расширения продуктов сгорания
включает пространство от критического сечения до наружного среза
сопла.
20
Исходя из особенностей эксплуатации и рабочего процесса ракет-
ных двигателей, для обеспечения надежной и безотказной работы
двигателей в период запуска и на основных режимах, ракетные
топлива должны отвечать следующим основным требованиям:
1) надежно прокачиваться по топливоподающей системе при
низких и высоких температурах, при пониженном внешнем давлении
и в условиях космического пространства;
2) иметь хорошую охлаждающую способность;
3) возможно лучше воспламеняться (необходимо, чтобы при
воспламенении скорость нарастания давления в камере не превышала
определенных пределов, зависящих от прочности двигателя и ста-
бильности сгорания);
4) иметь максимальное парообразование;
5) сгорать при оптимальной температуре с большой скоростью
и полнотой;
6) обладать максимально высокой объемной и весовой теплопроиз-
водительностью; р
7) иметь высокую степень газообразования;
8) отношение теплоемкостей продуктов сгорания должно быть
как можно больше, так как с увеличением его при прочих равных
условиях удельная тяга увеличивается;
9) не изменять своих физико-химических свойств в условиях
хранения и транспортирования;
10) иметь возможно малую токсичность и взрывоопасность;
11) быть доступными для получения, иметь достаточно широкую
сырьевую и промышленную базу.
В дальнейшем с развитием ракетной техники требования к ракет-
ным топливам безусловно будут уточняться и изменяться.
Как следует из приведенного материала, хотя в различных типах
двигателей рабочий процесс и имеет свои характерные особенности,
все же можно выделить общие для всех двигателей внутреннего
сгорания физико-химические превращения топлив.
Во всех двигателях топливо подается из баков через фильтры,,
систему регулирования подачи и т. п. в камеры сгорания, причем
они часто используется для охлаждения различных агрегатов дви-
гателя. В камерах сгорания происходит превращение химической
энергии топлива в тепловую, _
На первой стадии топливо распыливается, испаряется, затем
происходит диффузия образовавшихся паров в окружающую среду
г и образование смеси горючего и окислителя.
В смеси протекают сложные химические процессы окисления
топлива, сопровождающиеся выделением тепла и появлением пла-
мени — процессы воспламенения и сгорания.
При применении топлив в двигателях происходит изменение
их состава и свойств; некоторые топлива способны оказывать
21
коррозионное действие на конструкционные материалы двигателей,
средств хранения, транспортирования и перекачки.
Таким образом, основными эксплуатационными свойствами топлив
являются прокачиваемость, охлаждающее действие, испаряемость,
воспламенение и горение, стабильность и коррозионность. Суще-
ственное значение имеют также токсические свойства моторных, реак-
тивных и особенно ракетных топлив.
ЛИТЕРАТУРА
1. Моторные, реактивные и ракетные топлива, сб. под ред. проф. К. К. Па-
пок и Е. Г. Семенидо, Гостоптехиздат, 1962.
2. Н. Н. Бобров, П. И. В о ро пай, Применение топлива и смазоч-
ных материалов, Гостоптехиздат, 1962.
3. Я. М. П а у ш к и н, Химия реактивных топлив, Изд. АН СССР, 1962.
4. Я. Б. Чертков, Г. Ф. Большаков, Е. И.‘Гулин,
Топлива для реактивных двигателей, Изд. «Недра», 1964.
5. А. В. Серегин, Жидкие ракетные топлива, Воениздат, 1962.
Глава II
ПРОКАЧИВАЕМОСТЬ ТОПЛИВ
и ракетных топ-
свойств, опреде-
Под прокачиваемостью моторных, реактивных
лив понимают комплекс пх физико-химических
ляющих процесс перекачки по трубопроводам и топливной системе
двигателей.
Условия перекачки топлив по трубопроводам в разных климати-
ческих условиях различны. В жарких странах, вследствие испарения
легких компонентов топлив, возможно образование паро-топливной
смеси; в условиях холодного климата — кристаллизация и потеря
текучести.
Прокачиваемость моторных, реактивных и ракетных топлив
имеет большое практическое значение. Необходимым условием нор-
мальной работы всех двигателей является бесперебойное поступление
топлива в камеры сгорания в соответствии с заданным законом
подачи. Нарушение подачи топлива приводит к обеднению или чрез-
мерному обогащению топливной смеси и снижает мощность двига-
телей, а нарушение нормальной подачи топлива в камеры сгорания
ракетных двигателей может привести к взрыву ракетной системы.
Современные двигатели имеют сложное устройство. Зазоры
между трущимися деталями в узлах топливной системы некоторых
двигателей (реактивных, дизельных, ракетных) весьма невелики
и составляют величину порядка 7—10 жя. Поэтому попадание более
крупных частиц примесей в топливную систему в период эксплуата-
ции может привести к нарушению нормальной работы двигателей
и повышенному износу деталей.
Нормальная работа двигателей нарушается не только при наличии
твердых частиц в топливах. В результате повышения вязкости топлив
возрастают гидравлические потери в трубопроводах и уменьшается
производительность перекачивающих механизмов. Затруднения при
перекачке могут возникнуть п вследствие образования отложений
углеводородов с высокой температурой кристаллизации, кристал-
лов льда, продуктов коррозии, нерастворимых осадков и смол на
фильтрах и стенках трубопроводов. На больших высотах и при
2а
высоких температурах эксплуатации наземной техники могут воз-
никнуть явления кавитации из-за образования паро-воздушной
смеси в топливной системе.
ОСНОВНЫЕ ФАКТОРЫ, ВЛИЯЮЩИЕ НА ПРОКАЧИВАЕМОСТЬ ТОПЛИВ
Необходимое давление подачи топлива РПОд при перекачке
в общем случае равно:
где
леи
где
up । чр । труи । per г фл
ср — давление в среде, куда перекачивается
топливо, w/ж2;
А^Ф, АРтруб, АРрег, АТ^фл — потери давления в форсунках, трубо-
проводах и системе охлаждения, регу-
лирующей системе и на фильтрах, н/м2.
При перекачке по трубопроводам на большие расстояния давле-
ние подачи (напор) в основном определяется гидравлическими поте-
рями в труоопроводах АРТруб, поскольку остальные составляющие
уравнения относительно малы.
При подаче топлива из баков в камеры сгорания двигателей при-
ходится принимать во внимание потери давления на фильтрах,
в системе охлаждения, регулирующей системе и др. Рс^ определяется
давлением в камере сгорания.
Потери давления в форсунках АТ^ф зависят от типа распылите-
форсунок и определяются следующим выражением:
дрл, —,
ф 2i|)252Qg
Сф расход топлива через форсунку, кг/сек*,
ф — коэффициент расхода, учитывающий сужение струи и
уменьшение действительной скорости течения по сравнению
с теоретической;
5 площадь поперечного сечения отверстия в форсунке, м2\
к — коэффициент пропорциональности;
q — плотность топлива, кг/м3-,
g ускорение земного притяжения, м/сек2.
Потери давления (напора) на трение в трубопроводах или охла-
ждающей системе АТ^-руб определяют по следующей формуле *:
ДРтруб = Х-£е^_
где L длина трубопровода (охлаждающего тракта), м\
d3 эквивалентный диаметр трубопровода или охлаждающего
тракта, который определяют как отношение учетверенной
площади приведенного сечения к периметру сечения, м\
В случае подачи по трубопроводам и охлаждающей системе со сложным
переменным периметром расчет производят по отдельным приведенным уча-
сткам.
q — плотность топлива, кг/м3-,
w — скорость перекачки, м/сек-,
g — ускорение земного притяжения, м/сек2-,
X — коэффициент потерь на трение.
Коэффициент X зависит от характера потока и формы канала
(коэффициент ф). Характер потока определяется числом Рейнольд-
са Re:
при
ЗШЕ
ламинарном потоке, когда Re < 2320
i-Ji
Re
при турбулентном потоке, когда 2320 < Re < 10б
' 0,3164
Л = ---—‘ Ф
це0,/5 т
при Re > 105
X = (0,0032 + 0,221 Re- 0,237)
Ниже показана зависимость ф от соотношения сторон прямо-
угольных каналов:
Соотношение Соотношение
сторон сторон Ф
0 Л 1,32 0,5 0,97
0,2 1,25 0,7 0,91
0,3 1,10 1,0 0,90
0,4 1,03
Для круглых каналов ф = 1, а для кольцевых ф = 1,5.
Число Re подсчитывают по формуле:
V
где v — кинематическая вязкость, м2/сек.
Потери давления в регулирующей системе подачи топлива ДРрег
определяют по формуле:
per = cfq ——
где а — коэффициент местного сопротивления;
w — скорость топлива после фильтра, м/сек.
Потери давления на фильтрах, когда они не засорены, опреде-
ляются по такой же формуле. По мере засорения фильтров их про-
пускная способность уменьшается. Коэффициент сопротивления а
не поддается пока теоретическому расчету и может быть определен
только экспериментально.
Таким образом, на подачу топлив по трубопроводам и топливным
системам оказывают влияние: конструкция систем перекачки, усло-
вия эксплуатации, физико-химические свойства топлив.
Конструкция систем перекачки оказывает весьма большое влияние
на прокачиваемость ракетных топлив и нефтепродуктов. Например,
форма, длина, диаметр трубопроводов влияют на гидравлические
потери напора; конструкция насосов, особенно всасывающих поло-
стей, оказывает влияние на возникновение кавитации в топливной
системе. Оборудование фильтров подогревательными устройствами
улучшает фильтруемость топлив при низких температурах, обору-
дование топливной системы специальными изолирующими экранами,
уменьшающими нагрев топлива при сверхзвуковых скоростях полета
(в условиях повышенных температур), улучшает прокачиваемость
топлив, поскольку уменьшается осадкообразование в топливах.
Например, при создании двигателя для одного из транспортных
гражданских самолетов обнаружилась ненормальная работа дви-
гателя на стандартном топливе ТС-1 из-за повышенного осадко-
и смолообразования в области входов в форсунки. После того как
установили специальную тепловую защиту, уменьшающую нагрев
топлива, двигатель стал работать нормально.
Из условий эксплуатации на прокачиваемость топлив оказывают
влияние: температура, давление, влажность, загрязненность воздуха,
относительная площадь соприкосновения с воздухом, концентрация
кислорода над поверхностью топлива, скорость перекачки топлива.
К физико-химическим свойствам моторных, реактивных и ракет-
ных топлив, определяющим их прокачиваемость, относятся: вязкость,
температуры помутнения, кристаллизации и застывания, испаря-
емость, загрязненность топлива, стабильность (в первую очередь
при повышенных температурах) и плотность.
ПРОКАЧИВАЕМОСТЬ ТОПЛИВ ПРИ НИЗКИХ ТЕМПЕРАТУРАХ
Охлаждение топлив при хранении и применении
При хранении, транспортировании, перекачке, а также при
заправке и эксплуатации техники [1—3] температура топлива может
значительно снизиться.
Охлаждение топлива при хранении зависит от климатического
пояса, а также от времени года. Скорость охлаждения зависит от
объема хранящегося топлива. В мелких резервуарах охлаждение его
происходит довольно быстро.
Так, при хранении реактивного топлива ТС-1 в резервуарах
до Ю.и3 температура топлива уже через сутки становится равной
температуре окружающего воздуха. При хранении топлив в крупных
резервуарах их охлаждение, вследствие тепловой инерции, проис-
ходит медленнее. Таким образом, максимальные колебания темпе-
ратуры топлив, и в том числе наиболее глубокое охлаждение, про-
исходят при хранении в мелких наземных резервуарах и в баках
машин. При хранении в крупных подземных резервуарах колебания
температуры топлива незначительны и не превышают 10е С [4 J.
В эксплуатационных условиях охлаждение топлив может происхо-
26
дить и зимой, и летом, как в северных, так и в южных (в том числе
тропических) районах. Например, на высоте 12—15 км температура
окружающего воздуха достигает —60° С [1—3], а температура
в космосе близка к абсолютному нулю.
В качестве примера можно привести данные [5] по охлаждению
топлива в самолете типа ТУ-114 во время полета на высоте 10—12 км
со скоростью около 800 км/ч (табл. 1).
Таблица 1
Температура топлива в топливной системе реактивного пассажирского
самолета типа ТУ-114 во время полета
Температура, °C
Время полета,
мин
топлива
наружного
воздуха
стенок
топливных
баков
в подвесном
баке
во внутрен-
нем баке
в трубопро-
; воде
0
10
20
40 )
120
160
220
300
340
—25
-40
-54
-67
-63
-55
-56
—5
-5
-20
—40
-34
—31
—28
-34
-10
+15
+10
+10
-5
—30
-30
—23
-18
-27
+15
+10
+ 10
-5
-30
-30
-27
-28
-30
+15
+10
+6
—5
-10
-10
—20
—30
-27
Из данных, приведенных в табл. 1, видно, что охлаждение топлива
в самолете происходит достаточно быстро. Уже через 2 ч полета
температура топлива в баках самолета снизилась на 45° С и достигла
минимальной величины —30° С. При дальнейшем полете темпера-
тура топлива не снижалась. Аналогичные результаты были полу-
чены при полетах нескольких самолетов [1—3, 5]. Очевидно, можно
считать, что при дозвуковой скорости полета на высоте 12—15 км
топливо может охладиться до —35° С. При отрицательных темпе-
ратурах может наступить нарушение нормальной подачи топлива
вследствие повышения вязкости, выпадения кристаллов льда и угле-
водородов. Повышение вязкости топлива влияет, в основном, на
увеличение гидравлических потерь в трубопроводах, а выпадение
кристаллов льда и углеводородов — на пропускную способность
фильтров [6, 7].
Свойства топлив, определяющие их прокачиваемость
Прокачиваемость топлив при низких температурах определяется
в основном их вязкостью, помутнением, кристаллизацией и засты-
ванием [5, 8—10].
Вязкостно-температурные свойства. Вязкость топлив при дан-
ной температуре определяется их химическим составом. У углеводо-
родов с одинаковым молекулярным весом при одинаковой темпера-
27
туре вязкость повышается в следующем ряду: нормальные алканы,
изоалканы, цикланы, ароматические углеводороды. С увеличением
молекулярного веса вязкость возрастает.
Вязкость топлив с повышением температуры уменьшается, при-
чем у индивидуальных веществ меньше, чем у смесей. Среди угле-
водородов алканы имеют наиболее пологую вязкостно-температур-
ную кривую, а ароматические — наиболее крутую. Зависимость
вязкости углеводородов от температуры описывается в основном
эмпирическими формулами [5, 8, 10—17].
Известна формула Вальтера [18]:
lg 1g (100v + 0,8) = А — В 1g Т
где v — кинематическая вязкость, м21сек\
Т — температура, °К;
И, В — постоянные, зависящие от свойств жидкости.
Для расчетов пользуются также формулой Рамайя [19]:
/ D \2
1пц=^С + —J
где т) — динамическая вязкость, н • сек/м2\
Т — температура, °К;
С, D — постоянные.
Применяют также формулу Бачинского [20]:
(Т4-а)п
где ц — динамическая вязкость, н-сек/м2*,
Т — температура, °К;
Л, а, п — постоянные.
Для характеристики вязкостно-температурных свойств часто
пользуются различными коэффициентами [8, 11, 12, 21]. Для этой
цели, например, используют вязкостно-температурный коэффициент
Пинкевича (ВТК):
BTK = [lglg (v2 + 0,8) — Iglg (Vi + 0,8)] • 100
Чем больше ВТК, тем интенсивнее повышается вязкость при
понижении температуры.
Применяется также температурный коэффициент вязкости (ТКВ):
ткв0_100 =
(Ур —Уюо) • 100 vp у юр
У50 (ЮО — 0) У50
Т^В20-100 ~
(V20 — У100) ' 1QQ _ У20 V100
у so (100 — 20) у go
1,25
В табл. 2 приводятся данные по изменению вязкости топлив
с температурой.
28
Таблица 2
Зависимость вязкости топлив (в сст *) от температуры [11, 21—25]
ТС-1
Б-70
Топливо
Температура, °C
мепгльи-Диметил-
гидразин . . .
Смесь 78% HNO3,
20% N2O4, 2%
Смесь 70% HNO3,
г 28% N2O4, 2%
Тонка-250 • (50%
триэтиламина и
50% ксилиди-
нов) ...........
-60 -50 -40 -30 -20 -10 0 + 10 + 20
43-71 16-24 8-10 5,0— 5,5 3,3- 3,8 2,3- 2,7 1,7— 2,0 1,5- 1,6 1,2- 1,3
6,4 (при —57°) 4,2 2,7 1,6 1,4 1,1 0,9 0,8 0,7
9 7 5 4 3 2 1,7 1,5 1,3
29 16 10,2 7,0 5,0 4,0 3,0 2,5 2,0
300 61 30 15 9,5 7,0 5,0 4,0 3,0
8,8 6,8 4,9 3,7 2,8 1,8 1,6 1,4 1,2
6,3 4,5 1,7- 2,5 1,5— 2,0 1,3- 1,8 1,5 1,2- 0,8 1,0- 0,8 0,8- 0,7
35 10,7 5,80 3,5 2,60 1,8 1,45 0,90 0,75
45 12,3 7,7 4,5 3,3 2,2 1,8 1,4 1,2
* 1 сст = 1О“6 м^/сек.
Вязкость углеводородных топлив зависит
и описывается следующим уравнением [5]:
также от давления
(1+« Р)
где т)0 — динамическая вязкость при нормальном давлении,
н • сек/м2;
Р — избыточное давление, н/ж2;
а — коэффициент, учитывающий изменение вязкости на 1 н!м2.
Таблица 3
Зависимость кинематической вязкости N2O4 на линии насыщения
от температуры [11, 21, 25 — 27]
С °с • V, сст * С °с V, сст * С °C V, сст * t, °C v, сст *
-ю 0,41 30 0,28 70 0,21 110 0,17
0 0,37 40 0,26- 80 0,20 120 0,16
10 0,33 50 0,24 90 0,19 130 0,15
20 0,30 60 0,22 100 • 0,18 140 0,14
* 1 сстп = 10-вл12/сек.
29
Имеются сообщения о применении в качестве окислителей ракет-
ных топлив окислов азота [22, 23]. В табл. 3 приводятся данные
по изменению вязкости окислов азота с температурой.
Динамическая вязкость т] N2O4 может быть рассчитана по сле-
дующей эмпирической формуле:
Т]~------2,81о (н^сек/м2) \ ’
(140,89 + 0 ’7 5
где t температура, °C. |
Динамическая вязкость газообразного кислорода г|° при нор-
мальных условиях равна 1,92 • 10“4 г/см • сек (1,92• 10’1 н-сек/м2)
и может быть рассчитана при любой температуре по формуле [28]:
t о
Л =Л
где Т — температура, ° К.
Зависимость вязкости жидкого и газообразного кислорода от
температуры и давления показана на рис. 8, 9.
Динамическая вязкость жидкого фтора [97, 98] в интервале
температур от —204 до —192° С выражается уравнением: |
5 +
1]—2,43*10 (и • сек/м2)
В табл. 4 приведена зависимость вязкости жидкого и газообраз-
ного фтора от температуры. Я
Таблица 4
Зависимость динамической вязкости
фтора от температуры [21]
1, °C Т), спз * t, °C Л* 10~з# спз *
0,414
0,076
0,143
0469
-183
—104
-73
-16
—200,0
-197,9
-195,0
—192,3
-190,0 .
0,275
0,257
+153
+198
0,309
* 1 спз — 1 0 “8 н • сек / Л12.
Вязкость газообразного водорода при повышении температуры
от —260° до +30° С увеличивается почти в 20 раз. Вязкость жидкого
водорода при повышении температуры от —258° до —252° С умень-
шается в ~2 раза с 2,29 • 106 до 1,36 • 106 Н'сек/м2 [29—33].
30
Структурная вязкость. Для углеводородных топлив при опре-
деленных условиях характерно появление резко выраженной ано-
малии вязкости, когда часть топлива переходит в коллоидальное
состояние [9, 34].
Рис. 8. Зависимость вязкости жидкого кислорода от темпе-
ратуры.
Аномалия вязкости объясняется кристаллизацией парафина
и объединением кристаллов в структурную сетку. Температура,
|при которой образуется структура, зависит от концентрации пара-
финовых углеводородов в топливе и от их температуры плавления.
Образование структуры наблюдается за 1—2° С до выпадения кри-
сталлической фазы. В отличие от истинной, ньютоновской вязкости,
являющейся при данной температуре величиной постоянной и не
. зависящей от градиента скорости, аномальная вязкость определяется
31
, спз (10'3н-сек/м2)
Рис. 9. Зависимость вязкости кислорода от температуры и да-
вления.
32
величиной градиента скорости, т. е. вязкость нефтепродукта при
данной температуре становится величиной переменной, зависящей
от условий ее определения. Структурной вязкостью объясняются
специфические механические свойства системы, проявляющиеся
в возникновении предельного напряжения сдвига, а также в тиксо-
тропии [9].
Структура, образующаяся в топливах, разрушается довольно
легко вследствие малой их вязкости, поэтому предельная темпера-
тура прокачиваемости лежит ниже температуры их застывания.
Температуры помутнения, кристаллизации и застывания. Помут-
нение углеводородных топлив при охлаждении обычно связано
с выделением микрокапель воды или микрокристаллов льда. Это
объясняется тем, что при понижении температуры растворимость
воды в углеводородах резко уменьшается и она выпадает в виде
второй фазы. Содержание воды в топливе зависит от внешних усло-
вий (температуры, влажности воздуха и др.).
Как видно из табл. 5, температура помутнения ароматических
углеводородов значительно выше температуры помутнения исходных
топлив. Это объясняется большой гигроскопичностью ароматических
углеводородов. Температура помутнения алкано-циклановых угле-
водородов остается практически незамеченной — сразу же происхо-
дит кристаллизация фракций.
► Таблица 5
Температуры кристаллизации и помутнения реактивных топлив
и выделенных углеводородных групп [5]
Групповой химический состав, вес. % Температура, °C
Топливо ароматиче- ские углево- дороды алкановые циклановые алкеновые начала кри- сталлизации кристалли- зации (за- мерзания) помутнения •
Исходное топливо .......
Алкано-циклановая фрак-
ция ....................
Ароматическая фракция. .
20,6
0
98,0
21,7
48,6
0
57,0
51,4
1,0
0,7
0
1,0
-61
-57
-64
-63
-55
—95
(излом)
-41
Не заме-
чено
-12
исходное топливо ........
Алкано-циклановая фрак-
ция . . . ...............
«Ж
Ароматическая фракция
-64
-57
-67
-55
Не заме-
чено
-25,
3 Заказ 792.
зз;
Под температурой начала кристаллизации понимают максималь-
ную температуру, при которой в топливе невооруженным глазом
обнаруживаются кристаллы.
Для индивидуальных веществ температура начала кристалли-
зации совпадает с их температурой замерзания. Поскольку угле-
водородные топлива являются сложной смесью соединений с раз-
личными температурами замерзания, то кристаллообразование в нйй
происходит в некотором интервале температур. Раньше всего при
охлаждении выделяются кристаллы соединений с высокой темпе-
ратурой кристаллизации — некоторые углеводороды, гетерооргани-
ческие соединения, вода и др.
С увеличением молекулярного веса температура кристаллизации
углеводородных топлив повышается.
Среди углеводородов, присутствующих в топливах, максималь-
ную температуру кристаллизации имеют нормальные алканы; с уве-
личением разветвленности алканов температура кристаллизации
понижается. Наиболее низкими температурами кристаллизации
обладают изоалканы Т- и П-образной структуры.
По сравнению с алканами циклановые углеводороды, особенно
с короткими несимметрично расположенными разветвленными цепями,
замерзают при более низкой температуре.
Среди ароматических углеводородов наименьшую температуру
кристаллизации имеют моноциклические углеводороды (арень.А>
с короткими разветвленными боковыми цепями.
Температура начала кристаллизации углеводородных топлив
на несколько градусов выше температуры кристаллизации. Эта
разница увеличивается с повышением содержания ароматических
углеводородов (аренов) и достигает максимума у чистых ароматиче-
ских углеводородов. Температура начала кристаллизации и тем-
пература полной кристаллизации алкано-циклановых углеводородов
совпадают (табл. 5). По абсолютному значению более низкие темпе-
ратуры кристаллизации имеют ароматические углеводороды, наи-
более высокие — алкано-циклановые углеводороды. Их смеси зани-
мают промежуточное положение.
Вполне очевидно, что температура начала кристаллизации сухих
нефтепродуктов определяется присутствием алканов и бицикличе-
ских углеводородов. Высказываются предположения, что различие
в температурах начала кристаллизации и замерзания зависит не
только от углеводородного состава топлив, но и от присутствия
сернистых соединений [9].
Форма выделяющихся кристаллов сильно зависит от химического
состава углеводородной среды. Скорость их роста зависит от несколь-
ких факторов и приближенно выражается следующей формулой
[8]:
и> = ±(С-В)
где w — скорость роста кристаллов, кг/сек',
34
к___константа, зависящая от скорости охлаждения и характера
перемешивания, кг2/сек • м\
«п _ вязкость среды, н • сек/м2;
С __концентрация алканов нормального строения, вес. %;
р - растворимость алканов нормального строения при данной
температуре, вес. %.
г Из формулы следует, что скорость роста- кристаллов прямо про-
порциональна концентрации алканов нормального строения и обратно
пропорциональна вязкости среды.
О механизме кристаллизации топлив в настоящее время нет
единого мнения [9, 35, 36, 12]. Однако большинство исследователей
считают, что замерзание светлых нефтепродуктов обусловливается
выпадением из них прежде всего алканов нормального строения,
образующих группы ориентированных молекул, которые в даль-
нейшем являются центрами образования зародышей кристаллов.
Механизм замерзания топлив в настоящее время объясняется
двумя теориями: кристаллизационной и коллоидной [9]. Очевидно,
наиболее правильно считать, что замерзание (кристаллизация)
нефтепродуктов в зависимости от внешних условий и химического
состава происходит в результате образования либо кристаллической
сетки, либо сольватных оболочек (коллоидной структуры), или
сверхмицеллярной структуры, либо при действии этих трех (или
двух) факторов одновременно.
Температура замерзания ракетных топлив не нефтяного проис-
хождения, в которых вода растворяется неограниченно, зависит
от количества растворенной воды (табл. 6, 7).
Таблица б
Температуры замерзания (в °C) азотных окислителей в зависимости
от содержания в них воды и окислов азота [21, 100]
Содержание
воды, %
Содержание окислов азота, %
17 20 22,5 24 26 28
—58 —62 —62 -61 -59 -56
-61 -64 -63 —61 -51 -55
-66 -65 -63 —— - *
‘ z Таблица 7
Температуры начала кристаллизации (в °C) гидразинов в зависимости
от содержания в них воды [99]
Содержание воды, %
Вещество
0,2 1,0
2,0 3,0 4,0 7,0 10,0
несилин-Диметилгидразин
Метилгидразин.........
3*
35
Как видно из данных, приведенных в табл. 7, температура начала
кристаллизации т/есижж-диметилгидразина с увеличением содержа-
ния воды до 4 % понижается, а затем резко возрастает. При увеличе-
нии содержания воды в метилгидразине от 0,2 до 10% температура
начала кристаллизации этих смесей понижается.
Рис. 10. Зависимость растворимости воды С в топливе ТС-1 от
относительной влажности воздуха ip.
Температура замерзания 100%-ной перекиси водорода составляет
—0,86°, 98%-ной —2,91°, а 90%-ной -11,30° С.
Гигроскопичность топлив. Моторные, реактивные и многл^
компоненты ракетных топлив ограниченно растворяют воду. Напри-
мер, в смеси 50% триэтиламина и 50% ксилидина при 100% влаж-
ности воздуха и 20° С может раствориться до 9,0% воды. В этих же
условиях в изопропилнитрате растворяется до 0,06% воды.
36
Растворимость воды в топливах прямо пропорциональна влаж-
ности окружающей среды (рис. 10). Влажность атмосферного воздуха
постоянно меняется, поэтому изменяется и содержание воды в топлп-
-30 -20 -10 0 10 20 30 UQ 50
t,°C
Рис. 11. Зависимость растворимости воды в топливах от темпе-
ратуры:
1 тонка-250; 2 — изопропилбензол; 3 — бензин Б-100/130; 4 — изо-
пропилнитрат; 5 — топливо ТС-1; 6 — топливо Т-1; 7 — топливо Т-5.
вах. Таким образом, вода, растворенная в топливах, находится
в постоянном равновесии с влагой, находящейся в атмосферном
воздухе.
На растворимость воды в топливах существенное влияние оказы-
вает температура. Прп прочих равных условиях с повышением тем-
пературы растворимость воды возрастает, при понижении — умень-
шается (рис. И). Содержание воды в углеводородных топливах
37
зависит не только от абсолютного значения температуры, но и от раз-
ности температур топлива и воздуха (табл. 8).
Таблица 8
Зависимость содержания воды в реактивных топливах ТС-1 и Т-1
от разности температур топлива и воздуха [5, 7, 9]
Топливо
Температура, °C Относительная влажность воздуха, %
топлива воздуха
Содержание
воды, %
ТС-1
40 21,5 60 0,0031
—7 —2 54 * 0,0028
—7 —2 85 0,0041
13 23 66 0,0073
18 24 53 0,0061
2 26,5 10 0,0024
4,5 26 . ю 0,0073
40 25,5 88 0,0068
С увеличением разности температур топлива и воздуха конден-
сация водяных паров из воздуха в топливо происходит при меньшей
относительной влажности.
При уменьшении внешнего давления содержание растворенной
в топливе воды уменьшается, причем это происходит более интенсивно
в воздухе с повышенной относительйой влажностью.
Скорость насыщения водой углеводородных топлив довольно
велика и зависит от относительной влажности воздуха, площади
контакта топлива с воздухом, толщины слоя и некоторых других
факторов (рис. 12). Особенно интенсивно происходит насыщение
топлива водой в первые минуты контакта с водой или влажным возду-
хом. Следует отметить, что скорость насыщения при контакте с водой
намного меньше, чем из влажного воздуха.
Таблица 9
Гигроскопичность реактивных топлив [5]
• Топливо Углеводородный состав, вес. % Содержание воды, вес. %
аромати- ческие алкены цикланы алканы
ТС-1 18,6 2,2 19,8 * 59,3 0,010
Т-5 20,6 0,7 57,0 21,7 0,005
Алкано-циклаповая фрак- ция ТС-1 0 0 22,0 78,0 0,002
Т-5 0 0 51,4 48,6 0,001
Ароматическая фракция ТС-1 97,8 0,1 1,2 0,9 0,031
Т-5 98,0 1,0 • 1,0 0,027
38
Растворимость воды в углеводородных топливах зависит от их
химического состава и молекулярного веса. С повышением молеку-
лярного веса гигроскопичность топлив уменьшается. Таким образом,
ю 20 30
Т . мин
Рис. 12. Скорость насыщения керосина водой из воздуха и из водя-
ной подушки при 17—18° С:
з насыщение из воздуха при относительной влажности соответственно
100, 95 и 30%; 4 — насыщение из водяной подушки.
максимальную гигроскопичность имеют бензины и минимальную —
тяжелые топлива. По гигроскопичности углеводороды различных
ipynn располагаются в следующий возрастающий ряд: алканы,
Т Гн 1? ТТ О ТТТ Т гч w V
ipynn располагаются г
» Цикланы, арены.
39
В табл. 9 приведены средние данные по содержанию в топливах
растворенной воды.
С увеличением содержания ароматических углеводородов гигро-
скопичность топлив увеличивается.
Растворимость воды С в углеводородных топливах можно выра-
зить уравнением [9]:
где Ci — содержание воды в исходном топливе, %;
b — коэффициент, зависящий от химической природы вещества;
Т — температура, °К.
Условия выделения из углеводородных топлив растворенной
воды и образования кристаллов льда. В тех случаях, когда содержа-
ние воды в углеводородных топливах превышает растворимость при
данных условиях, вода выделяется в виде капель. Это происходит
в основном при понижении температуры.
При потеплении также наблюдается помутнение топлива за счет
перехода воды из атмосферы в жидкость. Микрокапли воды могут
выделяться из топлива при понижении относительной влажности.
Это следует из закона Генри:
С=кР
где С — концентрация воды в углеводородах, %;
Р — парциальное давление паров воды, н/ж2;
к — коэффициент пропорциональности.
Показано, что
= Гмйкс “п—
* нас
Следовательно
б’макс^ .1,
G — й----— ь макс V •
t нас
где при данной температуре:
Смаке — максимально возможная концентрация воды в углеводо-
родных топливах, %;
Р — парциальное давление паров воды, н/м2\
Рыас — давление насыщенных паров воды, н/м2}
ф — относительная влажность воздуха, %. -
Если микрокапли воды выделяются при температуре ниже 0° С,
то они образуют кристаллы льда. Однако это происходит не всегда.
Часто наблюдается переохлаждение растворов воды в топливе. При
быстром охлаждении топлив выделяется вода, которая в определен-
ных условиях может превратиться в кристаллы льда. Поэтому очен^
благоприятные условия для выделения кристаллов льда имеются при
перекачке топлив из подземных емкостей в топливозаправщики
зимой.
В жидких ракетных топливах, в которых вода растворяется неог-
раниченно, выделение последней в виде кристаллов не происходит.
40
Такие сильно обводненные топлива замерзают, как правило, при
определенной температуре. Это относится прежде всего к азотным
окислителям (см. табл. 6), перекиси водорода, азотоводородным
горючим (см. табл. 7).
В процессе перекачки топливо проходит по трубопроводам, рас-
пределительным и регулирующим устройствам, фильтрам и т. п.,
причем физико-химические процессы, происходящие в топливе
на фильтрах и влияющие на прокачиваемость топлив, существенно
отличаются от процессов, протекающих в трубопроводах и распре-
делительных устройствах при низких температурах.
Поэтому удобно рассмотреть отдельно фильтруемость топлив
и процессы, связанные с гидравлическими потерями при перекачке
топлив по трубопроводам.
Фильтруемость топлив
При прохождении через фильтр топливо разделяется на множе-
ство мелких потоков. При понижении температуры скорость про-
хождения топлива через фильтрующие элементы может существенно
уменьшиться вследствие выделения твердой фазы — кристаллов
самого топлива и инородных примесей с высокой температурой
замерзания, например воды [21, 22, 25], и за счет увеличения проч-
ности межмолекулярных связей топлива. Поэтому разделение топлива
на более мелкие струйки (процесс фильтрации) будет затруднено.
Таким образом, фильтруемость топлив при низких температурах
зависит от температур помутнения и начала кристаллизации и в мень-
шей степени от вязкости.
С увеличением температур помутнения, начала кристаллизации
и вязкости фильтруемость * топлив ухудшается (рис. 13). Среди
углеводородных топлив худшую фильтруемость при одинаковых
условиях имеют дизельные топлива; наилучшую — бензины. Однако
с повышением температуры различие в фильтруемости топлив умень-
шается [7 ]. На установке, моделирующей топливную систему лета-
тельных аппаратов, была исследована фильтруемость различных
топлив.
Нарушение нормальной пропускной способности фильтра (35 мк)
при перекачке топлива Т-5 обнаруживается уже около —20° С; при
58° С подача топлива Т-5 через 12 мин уменьшается в 2 раза.
Нарушение нормальной фильтруемости реактивных топлив Т-1
и ГС-1 происходит в интервале от —30 до —40° С. Топливо Т-2 хорошо
фильтруется при охлаждении до —45° С. Лишь при —50° С после
1 ч работы наблюдается уменьшение пропускной способности фильтра
на 30/о. Хорошую фильтруемость имеют бензины.
фракции алкано-циклановых и ароматических углеводородов,
а также их смеси хорошо фильтруются до —30° С. С пониже-
ФильтРУемость
засорения фильтра.
определяли по уменьшению расхода топлива вследствие
% ‘шлэчцо * пдтшиош vgoxQvd эпнэтчнднь
42
нием температуры фильтруемость углеводородных фракций и их
смесей резко ухудшается. Худшей фильтруемостью обладают арома-
тические углеводороды, лучшей — алкаио-циклановые. При добав-
ттенпи к топливам предварительно выделенных ароматических угле-
«мттлпопов их фильтруемость ухудшается пропорционально коли-
Плохую
Рис. 14. Фильтруемость углеводородных фракций и их
смесей с топливом Т-1 при —50° С [7]:
1 — ароматическая фракция; 2, 3, 4, и 5 — смесь топлива Т-1 с 60, 30,
20 и 10 % ароматических углеводородов соответственно; 6 — алкано-
циклановая фракция.
фильтруемость ароматических углеводородов следует объяснить
их большой гигроскопичностью.
Из всех производных гидразина нес^иж-диметилгидразин имеет
лучшую фильтруемость при низких температурах (он хорошо филь-
труется до —55° С).
Аминные горючие по фильтруемости приближаются к углеводо-
родному топливу Т-1. При перекачке влажной смеси 50 % триэтила-
мина и 50% ксилидина (тонка-250) при —50° С расход уменьшается
наполовину за 35 мин работы установки (рис. 13, б), это же горючее
в сухом виде фильтруется значительно лучше.
Вообще следует отметить, что сухие топлива (в том числе и аро-
матические) хорошо фильтруются даже при глубоком охлаждении.
Это связано с их способностью к переохлаждению, которое может
достигать 20°. Однако в присутствии даже небольшого количества
воды наблюдается резкое ухудшение фильтруемости топлив, связанное
43
очевидно, с тем, что кристаллы образующегося льда инициируют
образование кристаллов углеводородов [5, 7, 9]. Предельное содержа-
ние воды, определяющее нормальную фильтруемость топлива, должно
уменьшаться с повышением содержания высокоплавких углеводо-
родов в топливе.
Наличие в топливе механических примесей, частицы которых
меньше отверстий фильтрующих элементов, неблагоприятно сказы-
вается на его фильтруемости. Вероятно, механические примеси
могут быть центрами кристаллизации не только углеводородов
и гетероорганических соединений, но и воды.
Гидравлические потери в трубопроводах при подаче топлив
Как уже отмечалось, прокачиваемость топлив по трубопроводам
определяется прежде всего их вязкостно-температурными свой-
ствами; при данной температуре вязкость определяет подвижность
и легкость прохождения топлива по топливной системе, вплоть до
камер сгорания. Перекачка высоковязкого топлива приводит к гид-
равлическим потерям в системе, что влечет за собой некоторое умень-
шение производительности топливных насосов и, как следствие
этого, понижение давления впрыска и ухудшение качества распили-
вания топлив. При подаче чрезмерно вязкого продукта, оказыва-
ющего большое сопротивление при протекании по подающей системе,
может произойти разрыв струи жидкости и нарушение нормальной
подачи топлива или полное прекращение подачи.
Для каждой топливной системы существует оптимальное значение
вязкости, которое обеспечивает бесперебойную работу двигателей.
Максимальное значение вязкости, при которой топливо перестает
поступать в количестве, обеспечивающем номинальную мощность
двигателя, можно условно назвать предельной вязкостью. В связи
с тем, что топливные системы различных машин конструктивно
отличаются друг от друга, и значения предельной вязкости для
каждой системы различны. Усложнение топливной системы, как
правило, вызывает снижение предельной вязкости топлив. При подаче
топлива с повышенной вязкостью главным образом увеличиваются
гидравлические потери в трубопроводах.
Что касается снижения производительности насосов за счет повы-
шения вязкости, то для горючих: тонка-250 (50% ксилидинов и 50%
триэти ламина), несилм^-диметилгидразина, мети л гидразина, азотных
окислителей (за исключением окислов азота), жидкого кислорода,
жидкого водорода, реактивных топлив Т-1, ТС-1 и Т-2 и бензинов
это снижение несущественно и практически на подачу топлив не
влияет. Например, установлено (рис. 15), что при подаче топлив по
топливопроводу длиной 25 м наиболее существенные гидравлические
потери наблюдаются только для дизельных и тяжелых реактивных
топлив. Для горючих Т-1, ТС-1, Т-2, несшш-диметилгидразина
и тоика-250 гидравлические потери в интервале температур от —50
до —60° С не превышают 300 мм рт. ст (4 • 104 н/м1).
Прокачиваемость таких продуктов определяется в основном их
фильтруемостью. Однако следует учитывать влияние вязкости на
другие эксплуатационные свойства топлив, в частности на степень
распыливания топлива форсунками. Известно, что для хорошего
распыливания вязкость топлива не должна превышать 15 сст
Рис. 15. Зависимость гидравлических потерь в трубопро-
воде ДРтруб (длиной 25 м и диаметром 0,005 лг) при пе-
рекачке различных топлив от температуры [5J.
(0,015 и» сек/м2). Таким образом, верхний предел вязкости топлив
ограничивается именно этим требованием, а не условиями прокачи-
ваемости.
*
Улучшение прокачиваемости топлив
Многие компоненты ракетных топлив имеют достаточно хорошую
прокачиваемость при низких температурах, например несимм-дяме-
тклгидразин, аминные горючие, окислители на основе азотной кис-
лоты и др. Температура начала кристаллизации их составляет —57СС
45
и ниже. Вязкостно-температурные свойства перечисленных ком-
понентов удовлетворительные, поэтому улучшать прокачиваемость
этих продуктов не требуется.
Однако некоторые компоненты ракетных топлив (окислы азота,
перекись водорода и др.) имеют высокую температуру начала кри-
сталлизации. Способы улучшения прокачиваемости этих компо-
нентов в настоящее время не разработаны.
Высокую температуру начала кристаллизации имеют углеводо-
родные топлива, в которых присутствуют нормальные алканы,
а также топлива со значительным содержанием би- и трициклических
ароматических углеводородов определенного строения.
Прокачиваемость топлив при низких температурах может быть
улучшена одним из следующих способов [5]:
1) удалением из топлив соединений с высокой температурой
кристаллизации;
2) применением присадок, улучшающих вязкостно-температур-
ные свойства;
3) применением присадок, понижающих температуру начала
кристаллизации топлив;
4) применением присадок, предотвращающих выпадение из
топлив воды в виде льда.
Удаление из топлив соединений с высокой температурой кристал-
лизации осуществимо только в заводских условиях и по экономи-
ческим соображениям не всегда выгодно, поскольку ухудшает в не-
которых случаях эксплуатационные свойства топлив и уменьшает
их выход на сырье.
Присадки, улучшающие вязкостно-температурные свойства
топлив, в настоящее время практически не применяются.
Используя такие присадки, как парафлоу, сантопур, алкилфе-
нолы и депрессатор АзНИИ, понижающие температуру замерзания
топлив, можно улучшить фильтруемость топлив [37, 38]. Все эти
вещества состоят из молекул несимметричного строения с высоким
молекулярным весом [39, 40]. Наиболее подробно исследовано
влияние парафлоу — продукта, получающегося в результате кон-
денсации хлорированного парафина и нафталина. Парафлоу в коли-
чествах до 1 % эффективно понижает температуру кристаллизации
топлив [9].
В настоящее время депрессаторы не применяют для понижения
температуры кристаллизации углеводородных топлив, однако их
применение позволит улучшить фильтруемость реактивных и дизель-
ных топлив.
Наиболее эффективным средством улучшения прокачиваемости
углеводородных топлив (в первую очередь реактивных и ракетных)'
при низких температурах является применение специальных при-
садок, которые предотвращают выделение льда из топлива при охла-
ждении. Наилучшим оказался применяемый в СССР и США моноэти-
ловый эфир этиленгликоля [9, 11, 41], который хорошо растворяет
кристаллы льда, выделившиеся пз топлива, и тем самым резко улуч-
46
тает фильтруемость топлива при низких температурах (рис. 16).
Добавление присадки обеспечивает нормальную фильтруемость
топлив в интервале температур от —50 до —60° С. Лишь при даль-*
нейшем понижении температуры наблюдается частичная забивка
фильтра.
Присадка прежде всего улучшает фильтруемость ароматических
"углеводородов, а следовательно, топлив, в которых содержится
значительное количество воды. Однако она не изменяет фильтру-
емости, если последняя ухудшилась за счет выпадения кристаллов
Рпс. 16. Влияние моноэтилового эфира этиленгликоля
на фильтруемость реактивных топлив [5, 7]:
1 — TC-1; 2 — то же с 0,1 % присадки; з — Т-2; 4 — то же
с 0,1 % присадки..
в сухой углеводородной среде. При введении присадки в топливо
(обычно до 0,3%) содержание воды в нем несколько увеличивается,
поскольку сама присадка содержит 1—2% воды.
ПРОКАЧИВАЕМОСТЬ ТОПЛИВ ПРИ ВЫСОКИХ ТЕМПЕРАТУРАХ
Нагревание топлива при хранении и применении
При хранении максимальное разогревание топлив наблюдается
в летний период в южных районах, где воздух днем может нагреваться
до +70° С. Учитывая тепловую инерцию, температура топлива
может достигнуть 50—60° С.
Длительное хранение нефтепродуктов при таких температурах
связано с накапливанием нерастворимых осадков и смол, которые
отрицательно влияют на эксплуатационные свойства. В некоторых
►компонентах ракетных топлив (производные гидразина, перекись
водорода, азотные окислители и др.) под воздействием повышенных
температур происходит разложение, в результате чего резко ухуд-
шаются их эксплуатационные свойства.
г хранении нефтяных топлив прямой перегонки даже при
а 60 С окисление происходит медленно; нефтепродукты, получен-
47
।
ные путем термического крекинга, окисляются быстрее, в них ин-
тенсивно накапливаются смолы и осадки.
Современные летательные аппараты с ВРД имеют скорость полета
в 2,5 раза и более превышающую скорость звука [42—57, 58, 59].
В летательных аппаратах со сверхзвуковой скоростью наблюдается
образование в топливе осадков, так как в полете происходит значи-
тельный аэродинамической нагрев вследствие адиабатического сжа-
тия воздуха перед летательным аппаратом. В этом «заторможенном
потоке» за счет сжатия температура резко возрастает. Так, например,
при скорости полета 3 М температура заторможенного потока
воздуха на высоте И км будет —330° С, а при скорости 4 М ~630° С.
Вес топлива в реактивных летательных аппаратах составляет
45—55% от всего полетного веса. При небольших сверхзвуковых
скоростях полета (до 2000 км/ч) топливо используется для охлажде-
ния масла, радарной установки, гидравлической системы, установки
для кондиционирования воздуха и др. Поэтому топливо дополни-
тельно нагревается. Установлено, что при скорости полета 2,8 М
топливо в баке (30 in) за 20 мин полета нагревается на 70°; за счет
нагревания в подкачивающем насосе и в распределительных и регу-
лирующих устройствах — еще на 56—70°. С учетом нагрева в тепло-
обменнике и других агрегатах температура топлива перед форсун-
ками может составить 200—250° С [5].
Нагрев топлива в баках самолетов может быть рассчитан ио
следующей формуле:
^к = Тт+(7н-Тт).
ф к т S
GC~P
где Гк, ?н — конечная и начальная температура топлива, °К;
— температура торможения, °К;
G — масса топлива, кг;
Ср — удельная теплоемкость топлива, дж!кг • град;
к — коэффициент теплопередачи, вт!м2 *град;
т — время полета, сек;
S — площадь поверхности баков, м2;
ф — коэффициент, учитывающий размерность.
Коэффициент теплопередачи к рассчитывают по формуле:
где ах — коэффициент теплоотдачи от воздуха к стенкам топливного
бака, вш!м2-град;
I — толщина стенок бака, м;
X — коэффициент теплопроводности материала бака, вт!м*град;
а2 — коэффициент теплоотдачи от стенок бака к топливу,
вт/м2 • град.
48
Температуру торможения Тт можно рассчитать по следующей
формуле:
где Т — температура окружающего воздуха, °К;
к — коэффициент теплопередачи;
г — коэффициент восстановления температуры;
М — скорость полета в числах Маха.
За коэффициент восстановления г принимают отношение дейст-
вительного повышения температуры в пограничном слое у стенки
к адиабатическому:
Для ламинарного течения в пограничном слое г = 0,85, для
турбулентного г = 0,88—0,89.
Аэродинамический нагрев летательных аппаратов при определен-
ной скорости полета ^увеличивается до тех пор, пока не устанавли-
вается равновесие между притоком тепла от трения корпуса аппарата
о воздух и его отводом в окружающую среду [60]. Аэродинамический
нагрев зависит как от скорости, так и от высоты полета.
Так, например, при скорости полета 471/ на высоте 6100 м равно-
весная температура устанавливается через 90 сек и составляет 680° С.
При такой же скорости на высоте 36 600 м равновесная температура
устанавливается через 30 мин и равна всего 310° С. В американском
самолете А-Т1 и других аналогичных самолетах температура топлива
перед форсунками двигателей составляет 250—280° С [5, 14, 28,
61, 62-66].
При температурах выше 100° С скорость окисления углеводород-
ных топлив Т-1, ТС-1, Т-2, ^1-5 возрастает настолько, что в них об-
разуются нерастворимые осадки и смолы, которые забивают филь-
трующие элементы двигателей, откладываются в топливной системе,
нарушают нормальную работу командных агрегатов, зазоры между
трущимися деталями которых составляют 5—9 мк [66—71, 72,
73-76].
Прокачиваемость топлив при высоких температурах определяется
в основном их химической стабильностью и температурой кипения.
Фильтруемость топлив
пРименении топлив в условиях повышенных температур
I
ТВеРД°й фазой, образующейся в результате окисления топлив и
г°рючих и некоторых окислителях нерастворимых продуктов
। в течение
количество нерастворимых продуктов, которое
наибольшая^ опасность заключается в возможности засорения филь-
ров твердой фазой, образующейся в результате окисления топлив и
р?Р?г23УОНЫОГО износа деталей топливной системы. Скорость образо-
достаточно велика. Например, при нагревании Т-1 (150° С)
ооразуется такое --------------
Уменьшить пропускную способность современных фильтров на
4 Заказ 792. zn
5—7%. На рис. 17 представлены фильтрующие элементы сверх-
звуковых самолетов после работы на топливе ТС-1.
На рис. 18 приведены данные по прокачиваемости топлив (ДА,
ДС, Т-1 и ТС-1) в различных условиях. Ниже показаны температуры,
при которых топлива имеют наихудшую фильтруемость:
ДА.......................... 160-190° С
Т-1 ........................ 140-150° С
ТС-1.......................... 160 °C
С изменением температуры меняется фильтруемость топлив.- Это
объясняется тем, что для каждого топлива существует определен-
I
Рис. 17. Микрофотографии фильтрующих элементов из топливной системы
сверхзвуковых самолетов после работы на топливе ТС-1 (хЗОО):
а — размер отверстий 25 jhk; б — размер отверстий 15 мк.
ная температурная зона, при которой наблюдается максимальное
осадкообразование.
Так, например, топлива ДС, ДА, ТС-1 и Т-1 хорошо фильтру-
ются до 110—120° С. При дальнейшем повышении температуры филь-
труемость их ухудшается, а после того, как топливо нагреется
выше «опасной зоны» (максимального осадкообразования) филь-
труемость резко улучшается. Таким образом, если бы удалось
быстро нагреть топливо выше температуры максимального осадко-
образования, прокачиваемость его могла бы быть улучшена. Однако
па практике это трудно осуществить, поскольку нагрев в летатель-
ных аппаратах происходит постепенно, во всяком случае, в течение
нескольких десятков минут.
Фильтруемость углеводородных топлив при высоких температу-
рах зависит от их химического состава [5, 77, 78, 79—84]. Присут-
50
ствие в топливе ароматических бициклических, ароматических
с ненасыщенными боковыми цепями, непредельных углеводородов,
а также различных гетероорганических соединении значительно
ухудшает его фильтруемость.
Среди гетероорганических соединений наибольшее влияние на
прокачиваемость оказывают сернистые соединения и смолистые
Рис. 18. Зависимость прокачиваемости
реактивных и дизельных топлив от тем-
пературы.
Т. мин
Рис. 19. Зависимость прокачивае-
мости топлива ТС-1 при 150° С от
содержания в нем меркаптановой
серы.
вещества. Действие других гетероорганических соединений еще не
изучено. Влияние сернистых соединений обусловливается строением
их углеводородного скелета и характером функциональных групп.
Например, с повышением содержания меркаптанов фильтруемость
резко ухудшается, что объясняется интенсивным осадкообразованием
при нагревании топлив, содержащих меркаптаны (рис. 19).
При увеличении содержания смолистых соединений в топливах
и Углеводородных фракциях наблюдается значительное ухудшение
loo] их фильтруемости (рис. 20). Однако полное удаление из топлив
смолистых веществ нецелесообразно, поскольку в оптимальных коли-
чествах (0,04—0,069 вес. %) естественные смолистые вещества спо-
собны задерживать окисление топлив [86, 87].
Поскольку фильтруемость топлив при высоких температурах
|ooiC1IT от интенсивности процессов окисления кислородом воздуха
’ то вполне естественно, что в среде инертных газов она
4*
51
значительно улучшится (рис. 21). В отсутствие кислорода фильтру-
емость топлив становится удовлетворительной при нагревании в тече-
ние 10 ч [88].
Рис. 20. Зависимость прокачиваемости Рис. 21. Фильтруемость топлива Т-1
топлива Т-1 от содержания в нем смо- через фильтр 30 мк при 160 G в
листых веществ: среде различных газов:
1 — обессмоленное' топливо; 2, 3 — топливо 1 — азот; 2 — воздух; 3 — кислород,
с 0,04 и 0,1 % смол.
Улучшение прокачиваемости топлив
Существуют следующие пути улучшения прокачиваемости угле- I
водородных топлив при высоких температурах: J|
1) глубокая очистка от непредельных и некоторых ароматических
углеводородов и большинства гетерооргаинческих соединений; 1
2) тщательная и возможно более тонкая фильтрация перед за-
правкой летательных аппаратов, механические примеси должны
быть удалены возможно полнее;
3) применение присадок, предотвращающих осадко- и смолооб-
разование при нагреве; f
4) удаление воздуха из топливной системы и замена его инертным "й ♦
газом. 1
Были предложены следующие способы очистки: сернокислотная,
селективная, очистка адсорбентами, гидроочистка и др. При об-
работке топлива активированной окисью алюминия и концентриро-
ванной серной кислотой фильтруемость его улучшалась в несколько
52
раз. Очистка сернистым ангидридом хотя и способствует улучшению
фильтруемости топлив, однако этот способ не совсем приемлем.
Наиболее перспективным является применение каталитической
гидроочистки. Для получения высококачественных топлив для сверх-
звуковой авиации предложен метод каталитического крекинга в при-
сутствии водорода — гидрокрекинг. Реактивное топливо, получен-
ное с помощью гидрокрекинга,
при высоких температурах имеет
значительно лучшую фильтру-
емость, чем аналогичные топ-
лива, полученные методом пря-
мой перегонки (рис. 22).
С увеличением степени очи-
стки от серы, а также степени
гидрирования ароматических
углеводородов фильтруемость
топлив улучшается (табл. 10);
кроме того, они имеют высокую
плотность и большую теплоту
сгорания [5].
На фильтруемость топлив
при повышенных температурах
оказывает большое влияние
предварительная очистка топли-
ва от механических примесей.
Речь идет уже не только о тех
механических примесях, отсут-
ствие которых контролируется
согласно ГОСТу, но и о микро-
примесях, не видимых нево-
оруженным глазом. При нагре-
вании топлив протекают слож-
ные процессы, связанные в
первую очередь с окислением нестабильных компонентов топлив и
приводящие к образованию твердой фазы. На образование твердой
фазы (осадков) оказывают влияние микрозагрязнения, присут-
ствующие в топливе.
Перед применением топлив механические примеси могут быть
удалены из них отстаиванием или фильтрацией.
Скорость осаждения механических примесей V выражается фор-
мулой:
Рис. 22. Прокачиваемость топлива,
полученного методом прямой пере-
гонки (7) и гидрокрекинга (2).
„ Ы2 / 1
h • г = Ч8^(етв-е?Л
ГДС л коэффициент, учитывающий размерность;
средний диаметр механических примесей, м;
Л — динамическая вязкость жидкости, н-сек/м2;
Qtb — плотность твердых частиц механических примесей, кг/м3;
Qm плотность жидкости, кг/м3.
53
Таблица 10
Зависимость фильтруемости ароматических концентратов
от степени гидрирования и содержания серы
в гидрогенизате [89] *
Степень гидрирова- ния, % Содержание серы в гидро- генизате, вес. % Фильтруемость топлив
время испытания, лшн перепад давления на фильтре, мм pm. ст.
99 0,001 300 5
88 0,033 300 405
83 0,020 300 500
79 0,056 125 625
испытаний в подогревателе поддерживалась температура
* Во время испытаний в подогревателе поддерживалась температура
232° С, а на фильтре 260° С.
Однако процесс отделения механических примесей отстаиванием
длителен по времени. Он может быть ускорен фильтрацией, скорость
которой определяется уравнением:
Nk &PS
где ip — скорость фильтрации, кг/сек-м1-, v ।
N — проницаемость фильтра, зависящая от природы фильтру- 1
ющпх элементов; 1
к — коэффициент пропорциональности; 1
ДР — уменьшение давления на фильтре, н!м\ !
S — поверхность фильтрующего слоя, ж2; j
L — толщина фильтрующего слоя, м; ]
ц — динамическая вязкость жидкости, н-сек/м2. J
Повышение термоокислительной стабильности топлив с помощью ]
присадок является одним из путей эффективного улучшения их экс- 1
плуатационных свойств [90—95]. В настоящее время проводятся I
весьма интенсивные работы по синтезу новых соединений, работоспо- 1
собных по крайней мере до 200—250° С. Большое количество приса- I
док разработано и применяется для повышения стабильности нефте- I
продуктов в условиях длительного хранения при умеренных темпе- I
ратурах.
Присадки разделяют на следующие группы: I
1) ароматические амины; |
2) аминофенолы; 1
3) фенолы.
Однако все известные присадки такого строения оказались^?* I
малоэффективными при температурах выше 100° С. Большинство I
из них при нагреве подвергаются термоокислительным превращениям I
и образуют нерастворимые осадки и смолы. I
Таким образом, для повышения термоокислительной стабильности ]
реактивных топлив нужно было синтезировать новые вещества. 1
Совершенно очевидно, что эти присадки должны быть:
1) антикоррозионными, чтобы изолировать влияние металла и
нагретого топлива друг на друга;
2) антиокислительными, чтобы предотвратить окисление неста-
бильной части топлива;
3) диспергирующими, чтобы предотвратить укрупнение образу-
ющихся частиц осадка;
4) противоизносными.
Снижение положительного катализирующего влияния металлов
на окисление топлив достигается тем, что присадки образуют на
поверхности металлов защитные пленки. Такую функцию могут
выполнить вещества, имеющие в своем составе полярные группы
(например —ОН, —SH, —NH2 и др.). Часть присадки, не пошедшая
на образование защитных пленок, вследствие специфического стро-
ения должна выполнять антиокислительные функции. Если анти-
коррозионных и антиокислительных свойств недостаточно для эффек-
тивного предотвращения осадкообразования, то присадка должна
иметь еще диспергирующие свойства.
Действие диспергирующих присадок можно объяснить образо-
ванием вокруг первоначально появляющихся мельчайших частиц
осадков оболочек из молекул поверхностно-активной присадки,
ориентированной своей полярной группой к частице и радикалом
к топливу. Эта оболочка препятствует дальнейшей агрегации мель-
чайших частиц в более крупные, размер которых остается меньше
отверстий фильтрующих элементов. Присадки должны иметь высокую
термоокислительную стабильность и хорошо растворяться в топливе.
Установлено, что алифатические амины [91] существенно по-
вышают термоокислительную стабильность топлив (табл. 11).
Таблица 11
Зависимость термоокислительной стабильности реактивных топлив
ТС-1 и Т-2 от содержания в них алифатических аминов (150° С, 2 ч)
Присадка Формула присадки Концен- трация присад- ни, % Корро- зия, г/At2 Отложе- ния, г/лг2 Осадки, мг/100 мл
Топливо ТС-1
C18H37NH2 0,05
0,005
. (C18H37)2NH 0,05
0,01
Топливо Т-2
8,1 1,1 5,4
7,3 0,5 3,0
7,0 0,4 2,7
7,4 0,5 5,2
6,0 0,5 3,3
7,5 1,2 4,2
8,7 0,7 2,8
8,3 0,7 1,6
7,2 0,5 9,8
7,5 0,7 6,2
Исходное топливо
Окта деци л амин
Диоктадецил амин
Исходное топливо
Октадециламин
Диоктадецил амин
C18H37NH2
(C18H37)2NH
0,05
0,005
0,05
0,01
55
Наиболее эффективными из алифатических аминов оказались
амины следующего строения:
2?i+ 1
NH
« = 154-12
з
Были синтезированы [21 ] аминоалкилфенолы, которые уменьшили
образование нерастворимых осадков в топливе Т-1 в 2—3 раза
(табл. 12). .
Таблица 12
Зависимость термоокислительной стабильности топлива Т-1
(150° С, 2 ч) от содержания в нем алкилфенолов
и аминоалкилфенолов
Присадки
Концентра-
ция присад-
ки, %
Осадок,
мг/100 мл
Кислот-
ность, мг
КОН/100 мл
Исходное топливо ..........
2-Амино-4-пропилфенол . .
2-Амино-4-«грг«г-бутилфенол
2-Амино-4-«грг«г-амилфенол .
Диметилфенил-З-амино-4-ок-
сифенилметап ..............
2,6-Диамино-4-тргт-бутил-фе-
нол .......................
0,05
0,012
0,012
0,012
0,05
2,8
1,19
1,43
1,78
0,83
0
Как видно из табл. 12, достаточно эффективными присадками
алкилфенолы и аминоалкилфенолы признать нельзя.
Хорошими диспергирующими присадками оказались незольные
присадки типа полярных полимеров. Сополимеры термически ста-
бильны и эффективны в широком температурном интервале. Если
в составе сополимеров нет несгорающих элементов, то они беззольны,
что является большим преимуществом. В настоящее время для син-
теза присадок сополимерного типа имеется значительное количество
различных соединений. Наибольшие трудности заключаются в раз-
работке технологического процесса хорошо управляемой сополиме-
ризации, при котором получался бы стабильный сополимер заданного
молекулярного веса и структуры с нужными активными функциональ-
ными группами.
Известна целая группа сополимерных присадок. Например,
в США используется присадка FOA-2, которая представляет собой
сополимер метакриловых эфиров лаурилового спирта и диэтилэта.^
коламина. Физико-химические свойства присадки FOA-2 следующие;
Молекулярный вес....................... 50 000
Плотность 5............................. 0,902
Температура вспышки в открытом тигле, °C 68
56
Вязкость, ест, при
—17,8° С.............................. 6000
0° С............................. 1800
98,9° С.............................. 65
Температура застывания, °C................. —28,9
Присадка FOA-2 хорошо растворяется в углеводородах и органи-
ческих растворителях. Растворимость в воде 0,01%. При содержании
0,1—0,05% этой присадки в топливах последние выдерживают
испытания в течение 30 мин при 204° С; при 260 С соединение де-
полимеризуется, а при 426° С — разлагается. Ориентировочно фор-
мула FOA-2 может быть представлена в виде:
СН3 СН3 л
I I
—СН2-С—СН2-С—сн2-
I I
(С2Н6)2 NCH2CH2OOC СООС12Н.,6 J
X
Добавлением к присадке FOA-2 деактиватора металла в различ-
ных соотношениях получают трехфункциональную присадку, кото-
рая выполняет дополнительные функции. Созданы и известны под
шифром вещества FOA-208 и FOA-212, в которых кроме присадки
FOA-2 содержатся соответственно 8 и 12% деактпватора металла.
В ближайшем будущем ожидается применение соединений с более
разнообразным действием: они будут одновременно улучшать сго-
рание топлив и повышать их термоокислительную стабильность.
В последнее время Ф. Ю. Рачинским, Г. Ф. Большаковым,
Ю. А. Брук, Н. М. Славачевской [90, 95] синтезированы и испытаны
новые присадки, которые оказались весьма эффективными (табл. 13).
Как видно из табл. 13, наиболее эффективным соединением ока-
зался 2-фенил-2-меркаптобутиламин. Остальные присадки тоже до-
вольно эффективны. При содержании 2-фенил-2-меркаптобутил-
амина в количестве 0,05% топливо после нагревания при 150° С
в течение 6 ч остается прозрачным, бесцветным, практически не содер-
жащим нерастворимых осадков и смол.
Несомненно, что вопросу создания новых эффективных присадок
в дальнейшем должно быть уделено еще больше внимания.
ПРОКАЧИВАЕМОСТЬ ТОПЛИВ ПРИ ПОНИЖЕННОМ ДАВЛЕНИИ
При увеличении высоты полета летательных аппаратов, а также
при действиях наземной техники в горах происходит уменьшение
внешнего давления на топливо, заправленное в машины. Так, напри-
мер, на высоте 18 км давление составляет 60 мм рт. ст. При умень-
шении внешнего давления возрастает склонность топлива к испаре-
нию, улучшаются условия выделения из него растворенного воздуха
Других газов. Выделяющиеся газы и пары образуют пробки в раз-
личных местах топливной системы (в подкачивающем насосе, изгибах
вопровода и др.), что приводит к уменьшению напора и расхода
57
4 Таблица 13
Влияние некоторых присадок (0,05%) на термоокислительную
стабильность реактивного топлива ТС-1 при 150° С [90, 95]
Формула присадки Осадок, мг/100 мл Коррозия, г/м9 Отложения на бронзе, г/л12 Оптическая плотность D Кислотность, мг КОН /100 мл
Исходное топливо (без присадки) СбН5ч /CH2NH2 >С< C2H5Z xsh f V сн2 . - ч 1 к /\ /сн2 4NH ч он 1 (СН3)зС-^Х-С (СНз)з и 1 ch2nh2 он 1 (СНз)зС-С (СН3)з 1 CH2N(CH3)2 он 1 (СН3)зС-^Х-С.(СН3)3 .. V 1 /—\ ch2-nh-<; н > он он 1 1 (СНз)зС-Z4-С (СН3)3 (СН3)3С-^\-С (СН3)3 V 1 1 СН2 = s сн2 он он 1 1 (СН3)3С-/^-С (СН3)3 (СН3)3С-^Х -С (СН3)3 V v 1 1 СН2 S S сн 2 5,8 0,3 0,8 0,7 0,5 0,6 0,5 • 0,6 0,70 0,1 0,4 0,3 0,2 0,1 0,2 • 0,3 1,05 0,1 0,2 0,2 0,2 0,2 0,2 0,3 0,105 0 0 0 0 0 0 0 1,6 0 0 0 0 0 0 0
58
топлива, к нарушению нормальной работы топливного насоса. Обра-
зование паровых пробок (кавитация)* наиболее легко происходит
во всасывающем трубопроводе подкачивающего насоса.
Возможность кавитации вытекает из уравнения Бернулли:
При полном напоре топлива Ро и при отсутствии потерь стати-
ческое давление Р в потоке будет равно:
где g — ускорение свободного падения, м/сек2;
q — плотность топлива, кг/м3;
w — скорость потока, м/сек.
После увеличения скорости потока до величины, при которой
статическое давление Р становится меньше нуля.
При таких условиях в топливе появляются пузырьки вскипающего
топлива. В пузырьках устанавливается давление, равное давлению
насыщенных паров топлива при данной температуре Рнас.
Как видно из приведенной выше формулы, кавитации при прочих
равных условиях способствует увеличение относительной скорости,
потока w. При этом возможно образование пустот за счет отрыва
топлива от стенок. Разность между давлением на входе в насос
Рвх и давлением насыщенных паров Рнас (Рвх — Рнас) обычно
называют кавитационным запасом. С уменьшением (Рвх — РНас)
возможность кавитации возрастает, вместе с топливом к насосу
будут поступать его пары и газы, нормальное течение потока нару-
шается, количество подаваемого топлива уменьшается. Теоретически
подача топлива прекращается при Рвх = РНас- Если учесть гидра-
влические сопротивления всасывающего трубопровода, прекращение
подачи топлива к насосу произойдет раньше, чем давление насыщен-
ных паров достигнет значения Рвх. Следует учитывать, что с подъ-
емом на высоту Рвх будет уменьшаться, если топливные баки имеют
дренаж. При кавитации по трубопроводу перемещается не сплошной
поток жидкости, а смесь топлива с паро-газовой смесью. Из-за
сжимаемости такой смеси возникает пульсация давления в топливной
системе и снижается давление подачи.
Определим основные свойства жидкости, которые влияют на
прокачиваемость топлив при пониженном внешнем давлении.
► , Наиболее опасным участком в топливных системах летательных
аппаратов является 'линия всасывания (при отсутствии соответ-
ствующего подпора). Максимальное разрежение ДРмакс, которое
или называется возникновение в потоке жидкости разрывов
У » которые заполняются парами перекачиваемой жидкости.
59
возникает при обтекании потоком жидкости лопаток колеса, должно
быть меньше кавитационного запаса (РВх — РНас)-
&Рмакс Рвх — Рнас
Известно, что наибольшее разрежение
г макс — —Г Л Q"~ — ^77
где -----скоростной напор при абсолютной скорости потока,
н!м2\
q — плотность перекачиваемой жидкости, кг/л3;
е — абсолютная меридиональная скорость потока, м!сек\
X -----доля скоростного напора при относительной скорости
потока w при входе в колесо насоса,
— коэффициент, равный 0,2—0,4.
Таким образом, при определенном коэффициенте запаса кавитации
ср, равном обычно 1,2—1,4, можно написать:
(с2 + X IV2)
Рвх — ф (f2 ~Ь X iv2) + Рнас
или
Если иметь в виду, что квадрат относительной скорости движения
топлива w равен сумме квадратов меридиональной скорости с и
окружной скорости и, то
ДЛиакс = [с2(1 + *т«2]^-
Выразим окружную скорость и через основные параметры насоса:
и =
л DYn
60 ’
где Di — диаметр расположения входных кромок лопаток, ж;
п — число оборотов колеса, об!мин.
При этом надо иметь в виду, что
где Q — производительность насоса, м']!сек\
kQ — коэффициент, равный 4,5—5,0.
Таким образом:
л А'о з/ -г
“ = -60"^
60
Меридиональная скорость поступления топлива на лопатки колеса
7Г У <?"2
Zc
о
фл
где ф — коэффициент «стеснения» топлива на входе в колесо, ф << 1.
Следовательно
АР макс
16______
гг 7.2 \ 2
t к2
__о
60
«?»•)«
где
е=(1+М
16
-7.2X2
к2 V
;_о_
60 /
Как видно, е зависит только от конструкции насоса и нами рас-
сматриваться не будет.
Учитывая приведенные выше формулы, можно написать:
Рвх — Рнас = ф е у (Qn2)2
или
22L _ = <р е у
Рнас
—' с - = #нас и выразив //вх и HaiC в мет-
вх
Обозначив ——
рах столба перекачиваемого топлива, получим:
Явх-ЯНас = ф (Qn2)2
Максимально допустимое число оборотов насоса на заданном
топливе
3
__/ (Рвх — Рнас) 2g\ 4
"макс —
ф8 Q2 /
Напор 7/вх.необх и давление Азх. необх, которые необходимо соз-
дать при заданном числе оборотов при перекачке заданного топлива,
будут выражены следующим образом;
Рвх. необх+Рнас + -тт— У(Qn2)2
Рвх. необх — Q Рвх. необх — Рнас 4“ Фе (Qn2)2
в спогЛ~ЧеПНЬГе закономеРности справедливы для перекачки топлив
тон >чОИНОМ состоянии; в момент старта и полета летательных аппара-
ту111 характеристики несколько изменяются.
Давление и напор топлива на входе в насос Рвх и Явх будут:
Вх —
АРП
61
Рис. 23. Изменение высоты столба топ-
лива Н и осевой перегрузки ракеты
во время полета т.
где Pq — абсолютное давление в баке над свободной поверхностью
топлива, н/м2\
Рст — давление столба топлива, н/м2;
ДРП — гидравлические потери при перекачке топлива по трубопро-
водам, н/м2\
q — плотность топлива, кг/м3*
Н — высота столба топлива, м.
В ракетах топливные баки всегда расположены выше турбонасос-
ного агрегата и двигателя, и Рст —
летах баки могут располагаться
ниже насоса и двигателя, в этом
случае Рет будет величиной от-
рицательной.
При движении летательного
аппарата с ускорением i и при
угле наклона его оси к гори-
зонту а получим:
РСТ=Я Q (y + sina)
где ---осевая перегрузка лета-
тельного аппарата, н.
Приблизительное изменение
высоты II, давления Рст столба
» i
топлива, осевой перегрузки —
и давления на входе в насос Рвх
показано на рис. 23. Минималь-
ное давление столба топлива
будет сразу после запуска дви-
гателя. *
Если определена Рвх. необх
(рис. 23), то необходимое давление в баке
Рб = -Рвх. необх — Рст. мин 4~ А Рп
Давление в баках летательных аппаратов обычно создают путем
наддува отработанными газами.
Таким образом, с увеличением давления насыщенных паров
топлива и уменьшением его плотности максимально допустимое
число оборотов насоса уменьшается. Чем выше давление насыщенных
паров, тем больше нужно создавать давление на входе в насос.
Для улучшения прокачиваемости топлив при пониженном внеш-
нем давлении необходимо применять топлива с меньшим давлением
насыщенных паров. Улучшить прокачиваемость топлив можно кон-
структивными мероприятиями, а именно:
1) увеличивают давление наддува в топливных баках;
2) выносят лопатки колеса насоса во входную часть, что умень-
шает относительную скорость входа топлива в насос;
62
3) включают дополнительно осевое колесо на входе в центробежный
насос, что позволяет повысить давление топлива на входе в насос.
На рис. 24—26 приводятся данные авторов, характеризующие
прокачиваемость топлив с различным давлением насыщенных паров
при пониженном внешнем давлении. С увеличением давления насы-
щенных паров допустимая вы-
сота полета уменьшается.
Допустимая высота полета
зависит не только от давления
насыщенных паров, но и темпе-
ратуры топлив. Например, если
в летательный аппарат заправ-
лено топливо с температурой
20° С и давлением насыщенных
паров 60 мм рт. ст., то в этом
топливе бурное выделение паро-
газовой смеси начнется на
высоте 19 км, однако это же
топливо, имеющее температуру
10° С, «закипит» на высоте 21 км.
Давление насыщенных паров
интенсивно увеличивается при
нагреве топлив в летательных
аппаратах со сверхзвуковой ско-
ростью полета.
Склонность топлив к обра-
зованию паро-газовых пробок
можно примерно определить по
Рис. 24. Влияние высоты полета на ка-
витацию во всасывающей системе при
перекачке смеси азотной кислоты (78%),
окислов азота (20%) и воды (2%) при
давлении во всасывающей системе, рав-
ном внешнему давлению.
Длина всасывающего трубопровода 3,4 м,
перепад давления 1 кгс/см2 (9,8-104 н/м2).
отношению объемов жидкой
и паровой Vn фаз. Установ-
лено, что образование паро-газо-
вых пробок нарушает нормаль-
ную^ работу двигателя, если
17п При температуре
топлива 38° С нормальная ра-
бота насосов на топливе Т-1
нарушается на высоте более 17 км, а в случае применения Т-2 —на
высоте 10-12 км.
Образование паро-газовых пробок можно уменьшить созданием
давления в топливной системе, охлаждением топлива перед полетом
И другими способами.
еоиходимое избыточное давление, которое нужно создать для
р р Дотвращения образования паро-газовых пробок, определяется
тр\ 10сть1° между давлением насыщенных паров топлива при данной
д > атУР° 11 величиной атмосферного давления. Однако повышение ,
допол В топливн°й системе вызывает необходимость установления
плат’’ 1тельного оборудования па летательном аппарате и увеличи-
ает его полетный вес.
63
ГД(
'//v ' //
св
64
Таблица 14
Устранение «кипения» и потерь топлива с помощью
охлаждения или создания избыточного давления в баках
самолета при высоте 18 км [3, 5J
Давление
паров топли-
ва,
Pm- сгп'
Температу-
ра топлива,
°C
Потери топлива, вес. %
в баках,
сообщаю-
щихся с ат-
мосферой
в баках под
избыточным
давлением
100 ммрт.ст.
Необходимое
избыточное
давление,
мм рт. ст.
Необходимая
температура,
°C, не выше
15,5
37,8
15,5
37,8
9,6
15,0
0
1,0
0
5,8
0
0
90
235
0
10
-23
+30
Предварительное охлаждение топлива более доступно и, кроме
уменьшения давления насыщенных паров топлива, позволяет увели-
чивать количество топлива, заправляемого в топливные баки.
На склонность топлива к образованию воздушных пробок боль-
шое влияние оказывает растворенный в нем воздух. Растворимость
воздуха в топливе зависит от внешнего давления. При подъеме
самолета воздух, растворенный в топливе, начинает выделяться и
может привести к нарушению прокачиваемости реактивных топлив
(табл. 15).
Как видно из табл. 15, при подъеме на высоту 22 км почти весь
воздух, содержавшийся в топливах, выделится. Выделившийся
воздух занимает значительный объем. Например, при емкости баков
Выделение воздуха, растворенного в топливах,
при подъеме летательного аппарата [3, 5]
Таблица 15
Высо- та по- лета, КЛ1 Атмосферное давление, мм рт. ст. Количество воздуха, выделившегося из топлива, объемы. % Содержание воздуха в топливе, объемы. %
Т-1 Т-2 Т-1 4 Т-2
по высоте всего по высоте всего •
0
5,4
13,5
13,5
13,5
20,8
5,4
13,5
10,3
0
5,4
18,9
32,4
45,9
66,7
72,1
85,6
95,9
11,5
10,8
9,0
7,5
6,0
4,5
3,0
1,8
0,2
18,5
17,5
15,0
12,5
10,0
6,0
5,0
2,5
0,6
0 . 760 0 0
0,7 700 6,1 6,1
1,8 600 15,6 21,7
3,4 Г* п 500 13,0 34,7
в* 5,3 400 13,0 47,7
7,2 10,0 14,2 22,0 300 200 100 30 13,0 ' 13,0 10,4 13,9 60,7 73,7 84,1 98,0
6 Заказ
792.
65
30 м3 объем выделившегося воздуха будет ~5 м3. Этот воздух
выходит через дренажную систему, унося с собой пары топ-
лива, а в топливной системе может вызвать нарушение нормальной
подачи топлива.
МЕТОДЫ ОЦЕНКИ ПРОКАЧИВАЕМОСТИ ТОПЛИВ
Прокачиваемость моторных, реактивных и ракетных топлив
оценивают по физико-химическим показателям: вязкости, темпера-
турам кристаллизации, помутнения и кипения, фракционному соста-
ву, термоокислительной стабильности и содержанию механических
примесей. Однако анализ физико-химических свойств топлив поз-
воляет получить лишь относительное представление о прокачивае-
мости продуктов в условиях их реального применения. Более полное
представление о прокачиваемости топлив получают при испытаниях
на специальных установках, моделирующих топливную систему
летательных аппаратов и боевых машин, или в реальных усло-
виях.
В США фильтруемость топлив определяют на установке CFR.
Топливо нагревают до 150—230° С в алюминиевой трубке с электро-
подогревом и пропускают через фильтр с отверстиями 20 мк, изгото-
вленный из сплавленного порошка нержавеющей стали. Держатель
фильтра может нагреваться до 425° С. Топливо подается с постоянной
скоростью 2,7 кг/ч при давлении 10,5 ат (1,03 • 106 н/м2).
В США применяют и другие методы исследования прокачива-
емости топлив при повышенных температурах. Известен прибор,
работающий при низком давлении, который дает результаты, хорошо
согласующиеся с испытаниями на полноразмерной установке. Основ-
ным элементом прибора является трубчатый змеевик из стекла
«Пирекс» (диаметр трубки 4 мм, длина 2 м), по которому топливо
движется под давлением 0,2 кгс/см2 (1,96 • 104 н!м2), Змеевик погру-
жен в масляную баню при 204° С; о прокачиваемости топлив судят
по количеству отложений в баллах, образующихся в змеевике за
определенное время. Испытание длится 3—4 ч, и па него расхо-
дуется около 4 л топлива.
В СССР также применяют установки, предназначенные для
исследования прокачиваемости топлив.
На установке, разработанной А. А. Гуреевым и другими, иссле-
дуемое топливо непрерывно прокачивается (без циркуляции) с посто-
янной скоростью через нагреватель и фильтр до тех пор, пока перепад
давления на фильтре достигнет 0,45 ат (4,4 • 104 н!м2). В качестве
фильтра применяется металлическая сетка квадратного плетения
с отверстиями 30—32 мк. Температура топлива на выходе из нагре-
вателя поддерживается с точностью до ± 2° С. Основным крите«яй
рием для оценки стабильности топлива при данной температуре
является продолжительность испытания (в мин) до забивки филь-
тра, т. е. до достижения предельной величины перепада давления на
фильтре.
66 1
Была создана установка [5, 7], моделирующая работу топливной
системы летательных аппаратов, на которой можно исследовать
прокачиваемость топлив при температурах от —60 до 4-250° С.
Установка (рис. 27) состоит из следующих основных узлов:
топливного бака,' насоса, фильтра грубой очистки, охладителя,
клапан; 5
8
Рис. 27. Принципиальная схема установки для исследования про-
качиваемости топлив:
*----г-1 3 — топливный бак; 4 — редукционный
ктгпй 6 —дифференциальный манометр; 7 — охладитель;
— мотор; 15 — насос.
тапяи^Л?ы с_г?з°м; 2 — ресивер;
~ ш-ихЖрГ?^ - да^ик
фильтр грубой очистки; 13 — сливной бак; 14 - Р’
лировагшяЯ’ 0ПЫТНЫХ Фильтров, сливного бака и автомата для регу-
такжо п„ РаСХ0Да -топлива- Подачу топлива можно осуществлять
ОхлаПИеВМаТИЧеСКИ’
теле, в \4тогПе“Т°ПЛИВа производится в специальном бачке-охлади-
честве охпа; И погРУжается змеевик с опытным фильтром. В ка-
кдающей смеси применяют смесь этилового спирта и
67
твердой углекислоты. Нагревание топлива осуществляют в баке,
а также в змеевике.
Для замера расхода топлива установка оборудована штихпро-
бером и электрическим расходомером. Перепад давления на фильтре
и гидравлические потери в змеевике охладителя измеряют дифферен-
f3 Ik Я /4 15
Рис. 28. Схема установки для исследования прокачиваемости топлив
циркуляционной прокачкой:
2 — клапан перепуска;
6 — холодильник; {
; 3 — ротаметры; 4 — фильтр;
__________*___ ____ 7—подкачивающий топливный
насосредуктор; 9,’ 10 — электромоторы; и — редуктор; 12 — топливные
иярппм пыр.пкпгп давления; 13 — тахометр; 14 — манометр; 15 термопара.
1 — топливный бак;
5—дроссельный кран;
насосы высокого давления; 13 — тахометр; 14
циальными манометрами, температуру в основных узлах термо-
метрами и хромель-копелевыми термопарами, а абсолютное давление
в системе — образцовыми манометрами.
На установке, разработанной во ВНИИ НИ, прокачиваемость^
топлив при нагреве оценивают циркуляционной прокачкой через
насосы реактивных двигателей (рис. 28). Испытание заключается
в циркуляционной прокачке топлива (60 л) 8-часовыми этапами при
температуре на входе в насос 50° С.
68
Гопливо через каждые 8 ч заменяют свежим, и за все время испы-
В промежутках между
, заполненные испытуемым топливом,
- ‘О п
^11осле 50, 100 и 200 ч испытания насосы подвергают частичной
разборке и осмотру. Особое внимание обращают на наличие на дета-
осадков и отложений смолистых веществ. Общая максимальная
^Длительность испытания одного образца топлива (6 месяцев) сопо-
ставима с реальным периодом эксплуатации двигателя.
Окончательно о прокачиваемости ракетных топлив и нефтепро-
дуктов судят после проведения эксплуатационных испытаний на
натурных объектах.
тання (25 этапов) его расходуется 1500 л.
этапами (5-6 суток) насосы
выдерживают в термостате при 40 С.
ЛИТЕРАТУРА
1. Oil a. Gas J., 54, № 7, 69 (1955).
2. J. Е. W а 1 к р г у, J. Roy. Aer. Soc., 56, № 501 (1952).
3. Н. А. Рагозин, Реактивные топлива, Гостоптехиздат, 1963.
4. И. П. Б у д а р о в, Потери от испарения моторных топлив при хра-
нении, ВНИИСТ, 1961.
*5. Я. Б. Чертков, Г. Ф. Большаков, Е. И. Гулин,
Топлива для реактивных двигателей, Изд. «Недра», 1964.
6. Д. Л. Гольштейн, 3. В. Векслер, Г. Е. Журавлев,
сб. «Низкотемпературные свойства нефтепродуктов», Гостоптехиздат, 1949.
• 7. М. В ы т р ж е н с, Исследование прокачиваемости топлив для воздушно-
реактивных двигателей, Автореф. дисс., Л., 1959.
8. Моторные топлива, масла и жидкости, т. I, Гостоптехиздат, 1957.
• 9. Б. А. Энглин, Применение моторных топлив при низких темпера-
турах, Гостоптехиздат, 1961.
10. Т. П. Жузе, сб. «Низкотемпературные свойства нефтепродуктов»,
Гостоптехиздат, 1949.
11. Моторные, реактивные и ракетные топлива, под ред. К.’К. Папок
и Е. Г. Семенидо^ Гостоптехиздат, 1962.
12. Б. В. Л о с и к о в, Н. Г. Пучков,
применения нефтепродуктов, Гостоптехиздат, 1955.
13. Я. Б. Чертков, В. Н. 3 р е л о в, Нефт. хоз., № 11 (1953).
14. Wehr u. Wirtschaft, ноябрь 1963.
15. Д. Кеи, Т. Л э б и, Справочник физика-экспериментатора, 1959.
” ” ) т, В. П. Борисоглебский, Инж.-физ. ж.,
n g t о n, Proc. ASTM, 37, 380-384 (1937).
------, Petrol. Z., 26, 755—757 (1930).
С. Р а м а й я, сб. «Вязкость жидкостей и коллоидных растворов»,
178, Изд. АН СССР, 1944.
Ф- Дубовкин, Справочник по углеводородным топливам и их
сгорания, Госэнергоиздат, 1962.
Б. П а у ш к и н, Химия реактивных топлив, Изд. АН СССР, 1962.
49, № 9 (1957) rgenti’ G‘ Sachse 1, Ind. Eng. Chem., 52, № 2, 101 (1960);
Д* Whittaker, R. Sprague, S. S k о 1 n i k, J. Smith,
Soc., 74, 4794 (1952).
TiJ, Chemistry and Physics, 37-th Ed., 1955-1956.
1 n k l'ockets' and Propellants, New York — London, 1960.
^n nston, G r i 11 y, J. Chem. Phys., 14, 233 (1946).
-* n, H. H a i n e m a n, Ind. Eng. Chem., 49, № 4,
апрель 1963.
А. Энглин, Основы
№ I, 12 (1961).
17. С. “
18. С.
- 19. К.
т. II, вып.
яродуктам
23.
. Am.
24.
Walter
26.
97 П 7. v vx r 1
664 (1957). H‘ S 1 e v e n « о
28. All Hands
69
29. W li i t e, Hu, J о nston, J. Am. Chem. Soc., 76, 2584 (1954).
30. Franck, Naturwiss., 41, 37 (1954).
31. Cole, Farber, Elverum, J. Chem. Phys., 20, 586 (1952).
32. R u s s e 1, Scott, Cryog. Eng., 289—307 (1959).
33. Itterbeek, Paemal, Phisika, 7, 208 (1940); 8, 133 (1941).
34. А. И. До ла дугин, M. С. Солодовник, Б. А. Энглин,
Нефт. хоз., № 4 (1935). I
35. В о n d у, Ref., IX, 97 (1943). >
36. С о 1 v о г t h, J. Petr. Inst., XII (1937).
37. H. J. S t r a w s о n, J. Inst. Petr., 45, № 425 (1959).
38. C. J. Davies, Roy. Aer. Soc., 57, № 515 (1953); H. Strawson,
Shell Aviat. News, № 210, 8 (1955).
39. Д. О. Го льдберг, Аз. нефт. хоз., № 7 (1939).
40. Г. И. Фукс, сб. «Присадки к смазочным маслам», Гостоптехиздат,
1946.
41. Б. А. Энглин, Вести, воздушн. флота, № 1 (1956).
42. Aeroplane, № 2549 (26/VIII 1960).
43. Aviation Week, № 4 (25/VII 1960).
44. Aviation Week, № 5 (1960).
45. Missiles a. Rockets, № 6 (1960). *
46. Flugkorper, № 8 (1960).
47. Interavia, № 4552 (19/VIII 1960).
48. Aviation Week, № 1 (4/VII I960).
49. Flight, № 2677 (1/VII 1960).
50. Interavia, № 4521 (7/VII 1960).
51. Flight, № 2675 (17/VI 1960).
52. Flight, № 2683 (12/VIII 1960).
53. Interavia, № 4548 (13/VI 1960).
54. Interavia, № 4537 (29/VI 1960).
55. Interavia, № 4540 (3/VIII 1960).
56. Interavia, № 4541 (4/VIII 1960).
57. Сб. «Эксплуатационные свойства реактивных топлив при повышенных
температурах», под ред. Я.-Б. Черткова, ГОСИНТИ, 1959.
58. Сверхзвуковые самолеты, ИЛ, 1958.
59. American Aviation, 15, № 18, 43 (1954).
60. Л. Г. Райков, Нагрев летательных аппаратов в полете, Воен-
издат, 1962.
61. Naval Revien, 1962—1963.
62. Naval Revien, 1964.
63. Navy Times, 22/V 1963.
64. Naval Aviation News, май 1963.
65. J. All the World’s Aircraft, 1963—1964. ; ’
66. С. K. Johnson, D. E. Fink, A. C. Nixon, Ind. a. Eng.
Chem., 46, № 10, 2166 (1954).
67. SAE J., 12, 39 (1955).
68. Chem. a. Eng. News, 10, 4502 (1955).
69. К. M. Barringer, M. I. С о r z i 1 i о s, I. D. Rogers, Pet-
rol. Processing, 10, № 12, 1909 (1955).
70. Keif, American Aviation, 20. № 11 (1956).
71. А. А. Гуреев, 3. А. Саблина, Новости нефтяной техники,
сер. Нефтепереработка, № 2 (1959).
72. World Petroleum, 26, № 9, 69 (1955).
73. Я. Б. Чертков, В. Н. 3 р е л о в, К. В. Рыбаков,
В. М. Щагин, Б. А. Фомишиенко, Химия и технология топли^
и масел, № 10, 56 (1962).
74. Я. Б. Чертков, В. М. Щагин, Химия и технология топлив
и масел, № 11 (1959).
75. М. О. Scott, J. Inst. Petrol., 37, 333 (1951).
76. L. D. Derri, E. B. Evans, B. A. F о u 1 k n e r, J. Inst. Pet-
rol., 38, 343 (1952).
70
69, № 2, 557 (1954).
77. Г. Ф» Большаков, Е. А. Глебовская, Гетероорганиче-
ские соединения реактивных топлив, Гостоптехиздат, 1962.
78 Е. С. Davidson, Schweizer, Arch, angew. Wiss. u. Technik,
69 №2, 557 (1954).
79. E. P. Терещенко, M. E. Тарарышкин, сб. «Химия cepa-
и язотопганических соединений, содержащихся в нефтях и нефтепродуктах»,
т. III, Изд. АН СССР, 1960.
80. Б. Ф. Коробов, Б. И. Комаров, Химия и технология топлив
масел, № 4, 51 (1958).
81. Е. Р. Терещенко, М. Е. Тарарышкин, А. И. Туров,
В И. 3 р о л о в, Б. Н. Баранов, сб. «Химия сераорганических соеди-
нений, содержащихся в нефтях и нефтепродуктах», т. IV, Гостоптехиздат, 1961.
82. 3. А. Саблина, А. А. Гуреев, Химия и технология топлив
и масел, № 2 (1959).
83. Е. Р. Терещенко, М.
логия топлив и масел, № 2 (1959).
84. А. С. Nixon, Н. В. М i
(1956).
85. 3. А. Саблина,
и масел, № 7, 33 (1960).
86. П. И. Давыдов,
топлив и масел, № 10 (1960).
А. Тарарышкин, Химия и техно-
п о г, Ind. a. Eng. Chem. 48, № 10, 1909
Гуреев, Химия и технология топлив
Р. Большаков, Химия и технология
Химия и технология топлив и масел, № 1
Мурзабулатов, Химия и техно-
техиздат, 1963.
91. В.
(1964).
88. И. H. Данилов,
логия топлив и масел, № 2, 44 (1960).
89. М. Е. Conn и др. SAE Annual Meet Preprints, 1959.
90. Г. Ф. Большаков, П. И. Давыдов, Т. Г. Потапенко,
Ф. 10. Рачинский, Н. М. Славачевская, сб. «Химия сераорга-
нических соединений, содержащихся в нефтях и нефтепродуктах», т. V, Гостоп-
Н. 3 р е л о в, В. М. Мелехин, сб. «Химия сераорганических
4п^Инени”’ содержащихся в нефтях и нефтепродуктах», т. IV, Гостоптехиздат,
J. Fro mi ng, С. Robbins, Ind.
Гуреев, Присадки к
11, И. В. Minor,
1961.
92. С. В i s w е 11, Catlin,
a. Eng. Chem., 47, № 8, 1998 (1955).
93. 3. А. Саблина, А. А.
вам, Гостоптехиздат, 1959.
94. A. A. N i х о n, С. А. С с
Data, 4, № 2, 187 (1959).
95. Г. ф. Большаков, Ю. А. Брук, Ф. Ю. Рачи
Авт. свид. СССР № 159591. р у , ч iv. гачи
96. Справочник по разделению газовых смесей, 1953.
Stober, Z. Naturforsch., № 7, 822 (1952).
W i с k е, Z. Elektrochem., 55, № 7, 643 (1951).
моторным трпли-
J. Chem. a. Eng.
97.
98. ______, _
im’ ьЛ (пД ри,Т’ Химия гидразина, ИЛ, 1954-
WO. Jet Propulsion, 26, № 9, 741—744 (1956).
ОХЛАЖДАЮЩИЕ СВОЙСТВА ТОПЛИВ
НЕКОТОРЫЕ ПРЕДСТАВЛЕНИЯ О ПРОЦЕССЕ ТЕПЛООБМЕНА
В некоторых машинах топливо используют для охлаждения
различных вспомогательных агрегатов, особенно широко это при-
меняют в летательных аппаратах [1, 2]. Так, в реактивных самолетах
со скоростью полета до 2М топливом охлаждают масляный радиатор
и различные вспомогательные системы приводов. Большое значение^?
имеют охлаждающие свойства топлив в ракетной технике, поэтому
основное внимание будет уделено охлаждающим свойствам ра-
кетных топлив. В связи с большой теплонапряженностью ракетных
двигателей их камеры сгорания необходимо интенсивно охлаждать.
Использование топлива для охлаждения выгодно тем, что: 1) для
охлаждения камер сгорания не требуется специальных жидкостей
и 2) топливо поступает в камеру сгорания нагретым и более
подготовленным к воспламенению и сгоранию.
Охлаждение в ЖРД осуществляется чаще одним из компонентов
топлива (рис. 29, а), реже — двумя компонентами (рис. 29, б). Охла-
ждающие тракты обычно выполняют в виде винтовых щелевых кана-
лов [1]. I
В ракетных двигателях большой мощности требуется увеличивать
давление в камере сгорания и удельные расходы компонентов, при-
менять топлива с повышенной теплопроизводительностью [1—3].
В связи с этим сильно возрастают удельные тепловые потоки, кото-
рые снять наружным охлаждением (рис. 29) не представляется воз-
можным. Для улучшения отвода тепла применяют внутреннее охла-
ждение (рис. 30), заключающееся в том, что около стенки камеры ,
сгорания создают слой газа, имеющего более низкую температуру ;
за счет испарения топлива, которое подается на стенку через спе-
циальные отверстия. Л
Следует учесть, что для достаточной эффективности наружного
охлаждения необходимо подавать на внутреннюю стенку до 10%
компонента (от общего расхода). При этом происходит уменьшение
72
удельной тяги ракетного двигателя. Условия, при которых пленка
не уносится потоком, а остается на внутренней поверхности камеры
Охлаждающая жидкость
Горшее
Окислитель
Рис. 29. Схемы наружного охлаждения одним (а) и дву-
мя (б) компонентами ракетных топлив:
3 — стенка камеры
5 — головка камеры сгорания
двигателя.
•L „ входн°й коллектор; 2 — выходной коллектор;
сгорания; 4 — охлаждающий тракт;
сгорания, выражается уравнением [1, 2]:
Re®’8 Re*,3C*’3
бж^Ж
Де ог, иГ) Rer плотность, скорость и число Рейнольдса газового
У потока;
Q'K, .к, Неж те параметры для топлива в момент подачи
через стенку; ,
коэффициент кавитации, равный
Рж ^ж .
73
Р — внешнее давление;
Aiap — давление паров топлива.
При применении внутреннего (пленочного) охлаждения возникает
также проблема устойчивости пленки, связанная с тем, что в опре-
Горючее
Окислитель
Горючее
Окислитель
горючее
Окислитель
орючее
Горючее
о
О
о
О
о
о
Камера сгорания
Рис. 30. Схема внутреннего охлаждения.
деленных условиях на поверхности пленки возникают волновые
возмущения. Длина волны примерно равна десятикратной толщине
пленки. По мере увеличения числа Re масштаб возмущений умень-
шается.
Часто применяют внутреннее и наружное охлаждение совместно—
смешанное охлаждение.
74
В современных ЖРД к охлаждающей жидкости поступает 5 —10 %
тепла продуктов сгорания, которое передается от газов к стенке
камеры сгорания конвекцией и лучеиспусканием. В ракетных двига-
телях —70% тепла передается в охлаждающее топливо конвектив-
ным теплообменом [1,2]. Величина конвективного удельного тепло-
вого потока в двигателях составляет 1 • 106 — 28 • 106 ккал/м2 -ч
/1 6 • 10б —32 • 10° вт/м2), а путем лучистого теплообмена переда-
ется 1,5 • 106 — 2 • 106 ккал/м2-ч (1,75 • 106 — 2,4 • 106 вт/м2).
Отвод такого количества тепла представляет большие трудности.
Суммарный тепловой поток Q^, который передается от горячих
газов к стенкам камеры сгорания, равен
Сх — Ск + (?л
где Qk и Qn — конвективный и лучистый тепловые потоки от газов
к стенкам камеры сгорания.
Конвективный тепловой поток выражается уравнением:
(?к аг (7% Т’г >ст)
где аг — коэффициент теплоотдачи от газа к стенке, вт/м2*град;
Тт — температура заторможенного газового потока, °К;
7\ ст — температура газовой стенки, °К.
Коэффициент аг можно определить по формуле [1, 25]*:
0,82
аг —ст
0,35
Т’г. ст
гДе Срг ст — удельная теплоемкость продуктов сгорания при 7\ст,
дж/кг •град]
g — ускорение свободного падения, м/сек2;
т]г — динамическая вязкость горячих газов при ТГ, н • сек/м2;
G — секундный расход газовой смеси, кг/сек;
d — внутренний диаметр камеры сгорания, м;
к — коэффициент, учитывающий размерность.
Экспериментально определить лучистый тепловой поток Qn
довольно трудно, поэтому пользуются эмпирическими уравнениями.
Основную долю излучения при сжигании углеводородов дают
= 1,13.10-3 ^рС02
3,5
(вт/м2)
^2О=1,1з.10-3^8ог0-б ’
У г \ 3,0 / Т г< ст
ioir
н2о парциальные давления СО2 и Н2О, кг/м2;
приведенная средняя длина пути луча, м.
потока по Цилиндрической^та gT0JIbK0 для Д°ЗВУКОВЬ1Х скоростей движения
ГДе -Рсо2
100
3,01
(вт /.и2)
75
Таким образом, суммарный тепловой поток к стенке камеры его
рання
<h = «г (Тт - Гг. ст) + Q+ eS°2 =
— А-С (an \0.18 С°’82 ( Гг \0,35 (Т т ) I
-*Срг.ст (?Пг) —(Гг-уг.ст) +
Характер распределения теплового потока неодинаков по длине
камеры сгорания (рис. 31).
Начальный период работы двигателя характеризуется нестацио-
нарным режимом охлаждения. В период запуска стенки камеры и
сопла еще холодные и охлаждающему компоненту будет передаваться
Рис. 31. Распределение тепловых пото-
ков по длине камеры сгорания ракет-
ного двигателя [1].
*
сгорания и через них передается
чество тепла, которое передается
топливу, выражается уравнением:
<?2=а F (Т
~ Ж Ж \ J
только часть тепла, а осталь-
ная часть теплового потока пой-
дет на разогревание стенок ка-
меры сгорания двигателя.
С течением времени уста-
навливается равновесие, при
котором охлаждающая жидкость
снимает весь тепловой поток,
поступающий от горячих про-
дуктов сгорания в стенку ка-
меры сгорания. С этого момен-
та устанавливаются и остаются
постоянными тепловой поток
(на данном режиме работы дви-
гателя) и температуры газовой
и жидкостной стенок камеры
двигателя. Такой режим назы-
вается стационарным режимом
охлаждения.
Физическая картина про-
цесса теплообмена в ЖРД с
жидкостным охлаждением по-
казана на рис. 32.
Суммарный тепловой поток
направляется к стенкам камеры
< охлаждающему топливу. Коли-
от стенки камеры сгорания к
— Г }
. ст. ж. ср/
где аж — коэффициент теплоотдачи, вт/м2 • град',
F}K — поверхность охлаждения, м2;
_ температура стенки камеры сгорания со стороны топ-
лива, °К;
— средняя температура охлаждающего топлива, СК,
7 Ж. СТ
7\н.ср
камера
сгорания
Гж
Фронт пламени
охл
Рис. 32. Принципиальная схема процесса теплообмена в ракетном двигателе
при наружном охлаждении:
L’ 2 наружная и внутренняя стенки камеры сгорания; 3 — охлаждающая жидкость.
2 г температура газов в камере сгорания; Тг> ст —температура внутренней поверхности
камеры сгорания; Тж Ст — температура поверхности камеры сгорания со стороны охла-
ждения; средняя температура охлаждающего компонента; Твх, Твых — температура
топлива на входе и выходе из системы охлаждения.
Коэффициент теплопередачи аж не может быть определен теорети-
чески. Известно эмпирическое соотношение:
0,8 / £ПжСрж Y>>43 у/ jT
\ А» ж / 1'
ж э
Аж
/рж
о ж
эквивалентный диаметр поперечного сечения канала
коэффициент теплопередачи
вт/м2 • град;
теплопроводность топлива, вт/м* град;
-Динамическая вязкость топлива, н-сек/м2;
Удельная теплоемкость топлива, дж/кг • град;
__плотность охлаждающей жидкости, кг/м3;
__Ускорение свободного падения, м/сек2;
_ коэффициент, учитывающий размерность;
от стенки
м;
топливу,
X ж
77
1Ш1
Р — коэффициент, учитывающий неизотермичность теплового
потока -------------------—-------------
S Лж
^ж. ст \0,25
8 Лж. ст^Рж.Ст /
где Щк. ст
X ж. ст
— динамическая вязкость топлива при температуре
н*сек/м2;
— удельная теплоемкость топлива при температуре
дж/кг* град}
— теплопроводность топлива при температуре
ет/м*град.
Величину подсчитывают из выражения
d - 4F’«
“Э ~ “77
ж. ст»
ж. ст,
ж. ст,
Рж. ст
где Fm—площадь поперечного сечения охлаждающего тракта,
ж2;
П — полный (смоченный) периметр сечения независимо от того,
какая часть этого периметра участвует в теплообмене, м.
Таким образом:
а =0,021 X®'67 СРж<ежЦ00,81
(^ж)°’3Ч0’2
По уравнению неразрывности:
Лк £ж w — £охл
ИЛИ
СЖ^=-7Г-
Г Ж
где G
рубашку охлажде-
охл — секундный расход охладителя через
ния, кг/сек}
Fm — площадь поперечного сечения охлаждающего тракта
ж2;
w — скорость движения топлива, м/сек.
Обозначим
сРж '
(S'1!*)0,8
тогда
«ж = Z р • 0,021
аэ
^охл
F ж
0,8
Передаваемый от стенки суммарный тепловой поток CL
= / °’5? 7
Ж (?Пж)0'4
Р 0,021 (%^)0’8/’;к(Г!к.ст-Лк.ср)
dz* \ ^ж / F
о
78
направляется к охлаждающему, топливу. При нормальной работе
двигателя охлаждающее топливо должно воспринять это количество
тепла:
I = (^вых-^вх)
где Сг* к — теплоемкость охлаждающего топлива при температуре
7’ж.ср = г’вых^-7’вх , дж 1кг-град-,
7'вх, Т’вых — температуры топлива па входе и выходе из системы
охлаждения, °К.
Температура топлива на выходе из системы охлаждения
1 7 ВЫХ —"7, „-Ч-Т’вх
ьАнОохл
Таким образом, температура топлива на выходе 7ВЫХ должна
быть ниже его температуры кипения 7кИП при данном давлении
в системе охлаждения, т. е. 7ВЫХ < TKIin. Кроме того, 7ВЫХ ограничи-
вается химической стабильностью топлива. Выше определенных
температур некоторые компоненты начинают интенсивно разлагаться
(гидразин и его производные, окпслы азота, тетранитрометан, угле-
водороды и др.). В присутствии кислорода воздуха (который всегда
находится в растворенном состоянии в жидком топливе) протекают
термоокислительные превращения топлив. В углеводородных горю-
чих компонентах (например, в присутствии гетероорганических
и непредельных соединений) при температуре выше 100° С начинают
интенсивно образовываться нерастворимые осадки и смолы, которые
откладываются на поверхности охлаждения и ухудшают процессы
охлаждения камеры сгорания двигателей. При высоких температурах
интенсивно осмоляются амины, особенно ароматические.
Как следует из приведенных выше уравнений, охлаждающее
топливо должно иметь максимальную теплоемкость, большую тепло-
проводность. Чем выше теплоемкость и теплопроводность топлива,
тем лучше его охлаждающие свойства.
ФАКТОРЫ, ВЛИЯЮЩИЕ НА ПРОЦЕСС ОХЛАЖДЕНИЯ
На процесс охлаждения ЖРД оказывают влияние: 1) к он стр у к-
ия системы охлаждения; 2) условия охлаждения; 3) свойства охла-
ждающей жидкости (топлива).
• пев конструктивных факторов важнейшими являются: скорость
констаЧКИ топлива w по системе охлаждения, теплопроводность
жденияГ/^Н0ПП0Г0 матеРиала камеры сгорания, поверхность охла-
сечения ’ толп*ина стеиок камеры сгорания дСт, форма поперечного
тУРбулеи1^0ЛеВ0Г0 канала охлаждения. С увеличением скорости и
мого 'гЛтг?ПОсти Движения охлаждающего топлива количество отводи-
тспла возрастаете____________---- ------- -------------------
79
1
Процесс теплоотдачи в конвективной области теплообмена (до
'В ст = 633°К) описывается уравнением Михеева:
рг0,25 ли
Nu = 0,021 Re°>8 Pr0’43 ———
_ pr°>2®
Г Л* 7 $ . ггст 1
где Nu, Re, Pr — числа Нуссельта, Рейнольдса и Прандтля для
топлива; Я
Ргст — число Прандтля, вычисленное для условий движе-
ния топлива у стенки.
Известна также следующая зависимость:
Nu = к Re0,8 Pr0’33 V’
хЛст /
1Н
где 1] — динамическая вязкость топлива
Лет — Д
Предыдущее уравнение можно написать также в виде:
iyu —Q \0,8 / Л \Q>зз X / л \°,н
\ Л / \ X / \ Пет /
н- сек/м2;
[намическая вязкость топлива у стенки, н*сек/м2.
где q — плотность топлива, кг/м3;
w — скорость движения топлива, м/сек;
Ср — удельная теплоемкость, дж/кг • град;
X —коэффициент теплопроводности, вт/м'град;
к — коэффициент пропорциональности.
В этом выражении величины вязкости, теплоемкости и теплопро-
водности вычисляются обычно при средней температуре пограничного
слоя топлива:
Если температура топлива повысится настолько, что начнется
термический распад, то наблюдается нарушение нормального режима
теплообмена. В этом случае справедлива эмпирическая формула
[1.2 8]:
где — перепад температур на границе стенка — топливо, °К;
Т — температура топлива, °К;
q — удельный тепловой поток, вт/м2;
w — скорость движения жидкости, м/сек;
В — эмпирический коэффициент, °К.
Большое значение для процесса охлаждения имеют также такие
конструктивные элементы, как форма поперечного сечения щелевого
канала охлаждения, поверхность охлаждения, толщина стенок ка-
меры сгорания. Форма канала охлаждения должна быть такой,
чтобы обеспечить максимальное соприкосновение охлаждающего
плива с поверхностью охлаждения. Стенки камеры сгорания нужно
пелать как можно тоньше. Однако нужно учесть, что с уменьшением
Толщины стенок прочность камеры сгорания уменьшится, а суммар-
ный* тепловой поток передаваемый к охлаждающему топливу,
возрастет.
На условия охлаждения решающее влияние оказывает режим
Г аботы двигателя (давление в камере сгорания, скорость движения /7
газов и их турбулентность, температура). С увеличением давления и
температуры в камере сгорания возрастает.
СВОЙСТВА ТОПЛИВ, ОПРЕДЕЛЯЮЩИЕ
ИХ ОХЛАЖДАЮЩУЮ СПОСОБНОСТЬ
Охлаждающая способность топлив определяется теплоемкостью,
теплопроводностью, температурой кипения (для индивидуальных
веществ) и фракционным составом (для сложных, многокомпонентных
топлив), стабильностью, плотностью и вязкостью топлива..
Теплоемкость. Теплоемкость углеводородных топлив [4] при
20° С и 760 мм рт. ст. составляет 0,4—0,5 ккал!кг* град (1,6—
2,0 кдж 1кг* град) и с повышением температуры значительно возра-
стает (рис. 33). ~ ~
’ С увеличением отношения С/Н, а также с увеличением разветвлен-
^ности углеводородов, входящих в состав топлив, теплоемкость
уменьшается. Таким образом, наибольшую теплоемкость имеют
алкановые углеводороды нормального строения.
Теплоемкость углеводородных топлив Со с пределами кипения
100—300° С можно рассчитать по формуле:
/ л / Г 0)403
(кдж/кг • град) или с0 =
(ккал/кг • град)
/е
гдед — плотность топлива при 15° С, кг/м\
Для расчета средней теплоемкости жидких углеводородных
топлив СРср можно применить уравнение:
^Рср = ^0 (1 + а 0
где а — коэффициент, при t = 0—200° С равный 0,001;
• 60 — теплоемкость топлива при 0° С и 760 мм рт. ст.
Азотная кислота имеет значительно меньшую теплоемкость,
Чем Углев°дородные топлива (рис. 33), особенно при высоких
ф ^1еРатУрах. Теплоемкость N2O4 при 20° С составляет [5—8]
ккал 'кг'град (2 кдж/кг * град) и может быть рассчитана по сле-
дующему уравнению [9]:
^-«868 (4Д+ф1 ™+ф1»+ф>№1
+ 0,7 • 10’4) (кдж/кг • град)
Ункции Эйнштейна (pi, (р2, <рз вычисляют по таблицам [10].
Заказ 792.
81
gvdz гн/ждн <dj
QV02 2»!U Ш 1 d0
Рис. 33. Зависимость теплоемкости топлив от Рис. 34. Зависимость теплоемкости топлив от тем-
температуры [3, 4, 20, 24]. пературы [3, 4, 20J- •- .
82
Зависимость теплоемкости //ссгш.и-диметилгидразина от темпера-
туры (°C) выражается следующим уравнением [11 ]:
Ср = 2,565 + 0,00376 t (кд^/кг-град)
Теплоемкость смеси 50/6 трпэтиламина и 50% ксилидина при
|Гмпературах от —50 до +80° С рассчитывают по уравнению:
Ср = 2,209+ 0,00271 t (кдж/кг • град)
—0,6 ккал!кг*град и с повышением температуры
Высокую теплоемкость имеют спирты, причем с увеличением
обводненности последних их теплоемкость возрастает. Весьма вы-
сокая теплоемкость и у жидкого водорода (рис. 34). Следует отметить,
что теплоемкость жидкого водорода значительно увеличивается (при-
мерно в 1,5 раза) при повышении температуры от —260 до —253° С.
Теплоемкость большинства других жидких ракетных топлив лежит
в пределах 0,2—0,6 ккал/кг- град и с повышением температуры
изменяется незначительно (рис. 34).
В системе охлаждения двигателей поддерживается повышенное
давление, которое оказывает определенное влияние на теплоемкость
углеводородных и других топлив, причем с повышением давления
теплоемкость немного уменьшается (рис. 35). При увеличении давле-
ния до 100 атм теплоемкость уменьшается в среднем на 5—6%.
чЬ повышением температуры и давления теплоемкость уменьшается
более резко.
Теплопроводность. Как было показано выше, коэффициент те-
плоотдачи от горячей поверхности к топливу ож зависит от коэффи-
циента теплопроводности £ и оказывает значительное влияние на
количество тепла, передаваемое в охлаждающее топливо. Тепло-
проводность имеет определенное значение для улучшения процесса
теплоотдачи при турбулентной скорости движения охлаждающего
топлива. Чем выше теплопроводность топлива, тем лучше его охлаж-
дающие свойства при прочих равных условиях.
Теплопроводность углеводородных топлив зависит от химического
состава и температуры. С повышением температуры коэффициент
теплопроводности уменьшается, причем это уменьшение происходит
заметнее для топлив, выкипающих при более низкой температуре
Давление незначительно влияет на коэффициент теплопровод-
2°1f)G Увеличении давления от 1 до 20 ат (от 0,1 • 10° до
и н/м ) коэффициент теплопроводности увеличивается при нагре-
< нии до 573° К в среднем на 0,5%, а при дальнейшем повышении
Лрпоратуры до 673° К - на 1 %. •
2730 Р^СЧета коэффициента теплопроводности Хо при 760 мм рт. ст.
К используют следующую формулу [25]:
1 117’2 / /
Ао = —-— (вт/м • град)
ГДС 6 ~ плотность топлива при 288° К, кг/м\
I
83
4,05
84
Кроме того, коэффициент теплопроводности %0 при 273' К
может быть вычислен по формуле:
%0 = 0,0132 —0,0000260 «ср. кип (вт/м • град)
— средняя температура кипения фракций, С.
также уравнение, по которому можно рассчитать тепло-
углеводородных топлив при различных температурах
где • tcp. кип
Известно
проводность
U=kACp^l3M 3
где А
__ __ универсальная величина для всех жидкостей при
о — плотность, кг/м3;
М — молекулярный вес;
Ср — теплоемкость при Т = — 7кр, дж/кг>град;
Ткр — критическая температура, °К.
Температура кипения, фракционный состав и давление насыщен-
ных паров. Чем выше температура кипения, тем лучше охлажда-
ющая способность топлив при прочих равных условиях. Температура
кипения (для простых топлив) и температура начала перегонки
(для сложных топлив) не должны быть ниже температуры топлива
на выходе из системы охлаждения с учетом давления в системе.
Относительно высокие температуры начала кипения имеют углеводо-
родные горючие (—200° С), некоторые азотные окислители — тетра-
нитрометан (126° С), азотная кислота, причем добавление окислов
азота к последней понижает ее температуру кипения. Низкие темпе-
ратуры кипения имеют водород, кислород, фтор, поэтому их применен
Пйе в качестве охлаждающих жидкостей ГлтрУпнено—
С уменьшением давления насыщенных паров топлив их охлажда-
ющие свойства улучшаются. Углеводородные, аминные и некоторые
другие горючие компоненты ракетных топлив имеют довольно низкое
давление насыщенных паров, поэтому их успешно применяют для
охлаждения. Напротив, большая часть окислителей имеет значитель-
ное давление насыщенных паров и их применение в качестве охлаж-
дающих жидкостей затруднено.
Давление паров (в мм рт. ст.) некоторых окислителей в зависи-
мости от температуры (°К) может быть вычислено, по следующим
Формулам [15—18[*
Г 1g PFa = 7,08718-^-^ 1013
lg ^Н2О2 = 8,853-^
lg pfcio3=4>46862--^7^ (р в ато)
85
Для кислорода давление насыщенных паров равно
22 800ммрт, ст. (при 144° К) и 11,4 мм рт. ст. (при 53° К).
Стабильность. Стабильность является одним из основных свойств,
определяющих охлаждающие свойства топлив. При применении
в качестве охлаждающих жидкостей малостабильных компонентов
процессы теплообмена могут существенно ухудшиться.
При термоокислительных превращениях в топливе образуются,
как правило, твердые и газообразные продукты. Твердые продукты
откладываются на горячих поверхностях, в щелевых трактах
охлаждения и из-за малых теплопроводности и теплоемкости резко
нарушают процессы теплопередачи от охлаждаемой поверхности
к топливу. Образующаяся газовая фаза нарушает режим охлаждения
и может привести к изменению закона подачи топлива в двигатель.
Например, азотная кислота при повышенной температуре раз-
лагается по следующей схеме:
2HNO3<zt H2O + N2O5
N2O5 2NO2+-i-O2
£
В этом случае твердая фаза не образуется (если исключить воз-
можность взаимодействия с материалами системы охлаждения),
однако образуется газовая фаза, состоящая из кислорода и окислов
азота.
Термодинамически неустойчивыми являются гидразин и его
производные [12]. Термическое разложение гидразина может идти
по следующим направлениям:
N2H4->NH3 + N2
n2h4->n2+h2
n2h4-_>nh3+n2+h2
Полное разложение гидразина сопровождается выделением
~41,86 кдж/кг. Распад явсилм«-диметилгидразина описывается сле-
дующим образом [13]:
(СН3)2 NNH2 -> ch3nh + nh2 + ch2
NH2 + (CH3)2 NNH2 =-> nh3 + ch3nnh2
I i
•CH2
(CH3)2 N + (CH3)2 NNH2-> (CH3)2 NH + CH3CH2NNH
ch3nnh2 -> CH3N=CH2+nh2
•CH. qjj
CH3NNH2 + NH2^CH3N = GH2 + N2H4 J
I
•CH2
(CH3)2 NNH =^> CHs+CH4-FN2
86
(CH3)2 NNH2 + (CH3)2N (СН3)2 NH + (CH3)2 nnh
СН3 + (СНз)2 nnh2 ch4+ch3nnh2
I
•CH2
При распаде несылм«-диметилгидразина получаются также смо-
дустые вещества [9, 13, И].
V, см 1
Рис. 37. Инфракрасный (а) и ультрафиолетовый (б) спектры поглоще-
ния отложений, образовавшихся в системе охлаждения ракетного
двигателя, работающего на углеводородном горючем п жидком кис-
лороде.
При взаимодействии с кислородом алкилпроизводных гидразина
образуются диалкиламины, вода, азот, тетраалкилтетразены, диал-
килнитрозоамин [19].
С углекислым газом гидразин и его производные образуют соли
алкилгидразинкарбоновой кислоты, например R2NNH2 • СО2,
а также продукты состава R2NNH2 • СО2 и R2NNH2 • 2СО2, легко
растворимые в воде. Эти соли представляют собой твердые вещества,
которые могут нарушить режим охлаждения двигателей [8, 19].
В присутствии меди разложение гидразина и его алкплпроиз-
водных значительно ускоряется.
Образование отложений в углеводородных горючих при нагре-
вании до 300° С связано с присутствием в их составе гетерооргани-
ческих и непредельных соединений, которые имеют малую стабиль-
сть и при нагревании, особенно в присутствии кислорода, образуют
ваю Тельное количество нерастворимых осадков и смол, отклады-
на охлаждаемых поверхностях.
метода Большаковым и Е. А. Глебовской [14] спектральными
ного ля ОЫли изУчоны отложения из системы охлаждения ракет-
горючем*1 Г7""’ РаботаюЩог° на жидком кислороде и углеводородном
Дс (рис. 37).
Е. А. Глебовской [14] спектральными
87
Интенсивное поглощенц^
хорошо
Отложения характеризуются очень слабым поглощением связен
СН (1385, 1460, 2870, 2930, 2960 слг1) и интенсивным поглощением
кислородсодержащих структурных групп в областях 3200—3470 смг1
(группы ОН) п 1080 — 1200 слг1. Имеется также интенсивное
поглощение при 630, среднее поглощение при 1610—1720 и неболь-
шое поглощение при 880 и 770 см'1.
в области 1080—1120 см'1 в сочетании с полосой 630 см'1
соответствует характеристическим частотам колебаний ионизирован-
ных сульфатов (1080—1130 и 680—610 елг1) и ионизированных
сульфонатов (1080—1010 и 700—600 слс1).
Кроме гетероорганических соединений, в образовании нераство-
римых осадков принимают участие ароматические углеводороды,
что подтверждают ультрафиолетовые спектры отложений (рис. 37).
Плотность. Плотность оказывает прямое влияние на охлажда-
ющие свойства топлив. С увеличением плотности, как правило, воз-
растает коэффициент теплопередачи особенно в области 40—50° С;
как следует из приведенного вышё^уравнения (стр. 77), коэффициент
теплопередачи зависит от плотности теплоемкости и теплопроводности.
Причем с увеличением плотности коэффициент теплопередачи аж
возрастает в большей степени, чем при увеличении теплоемкости и
теплопроводности. Следовательно, с увеличением плотности охла-
ждающие свойства топлив улучшаются. Зависимость плотности
от температуры для некоторых топлив показана на рис. 38.
Вязкость. С увеличением вязкости коэффициент теплоотдачи
аж уменьшается. При применении топлив с меньшей вязкостью
облегчается возникновение турбулентного режима, наиболее благо-
приятного для процессов охлаждения.
Оценка охлаждающей способности топлив по физико-химическим
показателям не дает окончательного ответа о пригодности топлив
для охлаждения двигателей. Полное представление об охлаждающих
свойствах топлив можно получить только после испытаний на специ-
альных установках и в реальных двигателях.
МЕТОДЫ ОЦЕНКИ ОХЛАЖДАЮЩЕЙ СПОСОБНОСТИ ТОПЛИВ
Е. И. Гулин и Д. Лука разработали установку, которая позво-
ляет оценивать охлаждающую способность топлив по критической
температуре поверхности охлаждения и по критическому удельному
тепловому потоку. Схема установки представлена на рис. 39.
Установка состоит из нагревательного элемента, системы труб<х
проводов, напорного и сливного баков, баллона со сжатым воздухом
и измерительной аппаратуры, которая позволяет определять темпе-
ратуру, давление и скорость охлаждающего топлива.
В корпусе нагревательного элемента, выполненного из стали
1Х18Н9Т, установлена фарфоровая трубка, внутри которой с
88
(ri"/2*-e0l)
Рис. 38. Зависимость плотности топлив от температуры (4, 7, 8, 11—13»
19—23].
89
помощью токопроводов, закрепленных во фторопластовых пробках,
помещена калиброванная проволока пз стали 1Х18Н9Т диаметром
0,8 мм и длиной 100 мм, Л
Необходимую скорость протекания жидкости и давление в ра-
бочем участке обеспечивают путем изменения давления в расходном
топливном баке. Испытания проводят при
плотности теплового потока.
постоянном значении
Рис. 39. Схема установки для определения охлаждающей способности компо-
нентов ракетных топлив:
1 — корпус нагревательного элемента; 2 — фарфоровая трубка; 3 — калиброванная прово-
лочка; 4 — токопровод; 5 — фторопластовая пробка; 6 — нажимная гайка; 7 — термопары;
в — электрический расходомер; 9 — напорный бак; 10 — баллон с воздухом; 11 — сливной
бак.
Силу тока и напряжение в нагревательном элементе контролируют
и записывают на осциллограф.
Силу тока увеличивали до перегорания нагревательного элемента.
Предельные значения температуры и величины удельного теплового
потока в период, предшествующий перегоранию элемента, являются
основными показателями, характеризующими охлаждающую
собность топлив.
Величину теплового потока рассчитывают по формуле:
IU
(2 = 0,861 к —
спо-
где I — сила тока, а;
U — напряжение, в;
S — площадь поверхности нагревательного элемента, м2.
50
Среднюю темтературу Элемента1,/соответствующую определенному
тепловому потоку? определи ют по градуировочному (калибровочному)
рафику. Недостатком этого способа является необходимость точной
---------nnirniTTJ ТТЛ —
калибровки и замены на-
гревательиого элемента 10ии
после каждого опыта. Ре-
тэты исследований
п0 указанному методу
приведены на рис. 40 и 41.
Смесь азотной кислоты
с окислами азота имеет
лучшую . охлаждающую
способность, чем концен-
трированная азотная кис-
лота (рис. 40). Повышение
давления подачи Рпод
улучшает процесс тепло-
обмена, и условия для ох-
лаждения двигателей улуч-
шаются.
Топливо Т-1 имеет не-
сколько лучшую охлаж-
Ж^дающую способность, чем
98%-ный этпловый спирт.
Однако увеличение содер-
жания воды в спирте до
18% заметно улучшает
его охлаждающую способ-
ность (рис. 41). С увели-
чением скорости прокачки
топлива через систему ох-
лаждения удельный теп-
ловой поток с охлажда-
ющей поверхности значи-
тельно возрастает.
Следует отметить
топливо Т-1 образовывало
значительное количество
отложений на нагреваемых
поверхностях.
Для оценки охлажда-
^1ощих свойств топлив мож-
использовать прибор,
предложенный Г. Ф. Боль-
шаковым. Основной частью
? • 5 ат
ПОД
800
non
1200
800
M
О
Цуд 10*, ккал/м2-ч
ЧТО
Рис. 40. Охлаждающие свойства некоторых
топлив:
<2уд — удельный тепловой поток; /—температура
нагревательного элемента.
прибора (рис. 42) является нагрева-
тельный элемент со щелевыми каналами, по которым проходит топ-
‘ во- ^°Д°гРев осуществляют электрическим током. Прибор обору-
н системой автоматического контроля и регулирования.
ш
91
78 %HNO3+ 2MN,<V2%Hn
Принцип работы прибора заключается в следующем. Топливо
строго определенной начальной температурой 24° С подается
с давлением газа из топливного бака через входной патрубок
Л нагревательный элемент и проходит по щелевому каналу.
В Включают нагревательный элемент, производят нагрев до 1300 ±
ч- 2° С и выдерживают прибор в течение 15 мин при этой температуре.
Л1сле этого под давлением газа осуществляют подачу топлива
вщелевой канал нагревательного элемента. Начальная температура
топлива и давление подачи являются постоянными. Одновременно
с подачей топлива нагрев автоматически выключается.
Топливо проходит по щелевому каналу и охлаждает нагреватель-
ный элемент. Чем лучше охлаждающие свойства топлив, тем больше
охладится нагревательный элемент. Уменьшение температуры
(Тнач — Укон — ДУ), отмечаемое с помощью термопар, и является
критерием оценки охлаждающих свойств топлив.
ЛИТЕРАТУРА
1. Г. Б. С и н я р е в, М. В. Добровольский, Жидкостные
ракетные двигатели, Оборонгиз, 1957.
h 2. А. В. Болгарский, В. К. Щукин, Рабочие процессы в жидко-
стно-реактивных двигателях, Оборонгиз, 1953.
3. Н. Г. Чернышев, Химия ракетных топлив, Госэнергоиздат, 1948.
> 4. Справочник физических, химических и технических величин, 1927.
* 5. Э. В. Б р и ц к е и др., Термические константы неорганических веществ,
Изд. АН СССР, 1949.
6. Производство жидкой N2O4 в США. Miss. Roc., 5, № 12, 46 (1959).
7. Исследования жидкой N2O4. Redsone Arsenale. Miss. Roc., 4, № 3, 27
(1958). •
•**8. M. Б a p p e p, А. Ж о м о т т и др., Ракетные двигатели, Оборонгиз,
1962.
X/ 9. Ю. А. Л у к о м с к и й, И. Н. Г о дне в, Труды Ивановского хими-
ко-техпологич. ин-та, вып. 5, 1956.
10. В. М. Грязнов, А. В. Фрост, Статические методы расчета
термодинамических величин, Москва,
•' 11. A s t о n, J. Am. Chem. Soc.,
12. Н a n г a t t у, J. Eng. Chem _________
13. U. F. Cordes, J. Phys. Chem., 9, 65 (1961).
1949.
75, № 23, 6202 (1953).
, 11113 (1951V_
13. U. F. Cordes
V 14. г. Ф. Большаков, Е. А. Глебовская, Тезисы докладов
на XVII совещании по спектроскопии, Изд. АН СССР, 1965.
15. Hu, White, Jo ns ton, «. ™
16. Cady Hildebrandt, J. Am. Chem. Sos
17. Г’
18. J а г г у
— 19. Л. Од
20. Я. M.
Jo ns ton, J. Am. Chem. Soc., 75,5642 (1953).
----.........u. 52, 3839 (1930).
Clausen, J. Am. Chem. Soc., 56, 614 (1934).
, J. Phys. Chem., 61, 489 (1957).
рит, Химия гидразина, ИЛ, 1954.
П аушкип, Химия реактивных топлив, Изд. АН СССР, 1962.
» Miller, J. Am. Chem. Soc., 78, 1552 (1956).
з, Rudge, J. Chem. Soc.,’Jan., 191 (1950).
J. Phys.. Chem., 61, 503—504 (1957).
чття a\ Ф. Большаков,
25 А ТХгДВИГаТелей’ Нзд. Недра», 1964.
ппи Tnii/,»* 1 Ухман, H. В. Илюхин, Основы учения о теплообмене
‘ тетении газа с большой скоростью, Машгиз, 1951.
1
, Топлива
Глава IV
ИСПАРЯЕМОСТЬ ТОПЛИВ
Испаряемость топлив имеет большое значение при применении
их в двигателях, в процессе хранения, транспортирования и за-
правки машин.
Потери легкоиспаряющихся топлив могут достигать очень боль-
ших величин. Например, если принять, что с момента получения бен-
зина на заводе до применения в двигателях он около 10 раз пере-
качивается из одной емкости в другую и хранится 3 месяца в резер-
вуарах, то потери бензина могут составить до 5% от всего вырабо-
танного количества. С учетом потерь в баках и карбюраторах рлв
время работы машин общие потери от испарения бензина достига/оТ
10% и более [1]. Наряду с этим ухудшается качество топлив вслед-
ствие испарения наиболее легких его компонентов. Испарение то-
плив создает также условия, при которых возможно возникновение
пожара, взрывов и отравление обслуживающего персонала. 1
Исключительно велика роль испаряемости топлив для нормаль-
ного протекания процесса горения в двигателях. Во всех двигателях
внутреннего сгорания воспламенение и горение топлив происходит
в паровой фазе при определенном соотношении между горючим
и окислителем. Процесс испарения не только предшествует процес-
сам воспламенения и горения, но в значительной мере определяет
характер их протекания. Например, высокая скорость испарения
в начальной стадии процесса горения в дизеле повышает жесткость
работы двигателя, хотя и способствует более полному горению.
В возду
«I!
до-реактивных двигателях улучшение испаряемости топлив
повышает устойчивость и полноту горения.
С испаряемостью связаны образование паро-воздушных пробок
в трубопроводах и явление кавитации в насосах, оказывающие
влияние на процесс перекачки топлив.
СКОРОСТЬ ИСПАРЕНИЯ
Распространение паров может осуществляться в виде молеку-
лярного переноса (молекулярная диффузия) и в виде переноса частиц
или объемов, имеющих значительно большие размеры, чем моле-
кулы — молярные объемы (конвективная диффузия).
94
В общем случае
количество испаряющегося топлива определяется
выражением.
где
0с и
JG = Pc S (Сп — Со) dr — fipS (Рп Ро) dr
поверхность испарения, л«2;
концентрации паров топлива на поверхности испарения
и в окружающей среде, кг/м3;
давления паров топлива на поверхности испарения
и в окружающей среде, н/м2;
время испарения, сек;
коэффициенты испарения, отнесенные к разности кон-
центраций или разности парциальных давлений пара
на поверхности испарения и в окружающей среде,
м/сек и кг/н- сек.
Коэффициенты испарения 0С и 0Р учитывают условия испарения
и физические свойства жидкости. Они численно равны количеству
вещества, испаряющегося с единицы поверхности в единицу времени
при разности концентраций или давлений, равной единице.
Следовательно, скоростьиспарения — количество вещества, испа-
ряющегося с единицы поверхности в единицу времени, — опреде-
ляется как:
^исп = Рс (^п со) = рр(^п Ро)
Впервые этот закон испарения был экспериментально открыт
Дальтоном в 1803 г.
В случае свободного испарения, т. е. когда испарение происходит
в неограниченный объем, концентрацию и давление паров в окружа-
ющей среде можно принять равными нулю, и тогда скорость испаре-
ния
LrT- ^исп = ₽с = Рр Рп
При испарении в замкнутом сосуде в начальный момент Ро = 0,
и скорость испарения будет максимальной. В дальнейшем скорость
уменьшится и будет пропорциональна разности (Рп — Ро). При
Рп = Ро наступает равновесие между жидкой и паровой фазами,
и скорость испарения становится равной нулю.
Коэффициенты испарения рс и рр легко можно вычислить, в боль-
шинстве случаев используют экспериментальные данные, получен-
ные на основе теории подобия. Результаты экспериментальных
исследований обычно даются в виде таких зависимостей, как
Nu = /(Re, Pr); М= <р (Re, Рг)
Зпа* Для заданных условий испарения величину критерия Нус-
’( 1,11 или Маргулиса М, можно определить коэффициенты испа-
W НИЯ из выражений:
Nu Dc Nu Dp
„ » Pp~ ~ ’
= М iv
определяющий размер, например при испарении ка-
пель — диаметр, м\
где
95
Re — критерий Рейнольдса;
Pr — критерий Прандтля; I
w — линейная скорость потока, м/сек\ I
Dc и Dp — коэффициенты диффузии, отнесенные к градиентам
концентраций и давления, м2!сек и кг • м!н* сек, 3
Наиболее сложно процесс испарения протекает при распылила-
пии горючего и окислителя в двигателе в условиях турбулентно/^
потока. На скорость процесса оказывают влияние химические реак-
ции взаимодействия горючего и окислителя при воспламенении, а за-
тем процесс горения предшествующих порций впрыснутого топлива.
При этом процессы тепло- и массообмена протекают одновременно.
Тепло, подводящееся к каплям из окружающей среды, затрачивается
на нагревание и испарение жидкости и перегрев пара, диффунди-
рующего в окружающую среду. 1
В условиях адиабатического процесса при установившемся ре-
жиме количество тепла, подводимого к каплям, и количество испа-
рившегося вещества связаны соотношением:
* dQ — dG Дг Я
или
а 5 (/q £п) = Рр5 (Рп — Рq) Si
где Д/ — увеличение теплосодержания испаряющегося вещества
при нагревании жидкости, изменении агрегатного coi
стояния и перегреве паров, дж!кг\ 1
а — коэффициент теплопередачи, вт/м2• град\ 1
t0 и tn — температуры окружающей среды и поверхности испаря-
ющейся жидкости, °C.
Приведенное уравнение характеризует зависимость между тем-
пературой среды и температурой поверхности испаряющегося то-
плива в условиях изотермического испарения. Определив из этого
выражения, например методом подстановки, температуру изотерми-
ческого испарения, можно рассчитать количество испаряющегося
топлива.- 1
В зависимости от условий, испарение может быть статическим
и динамическим. * ' j
СТАТИЧЕСКОЕ ИСПАРЕНИЕ
Испарение жидкости с неподвижной поверхности в среду покоя-
щегося газа называется статическим. j
Экспериментально установлено, что в условиях статического
испарения коэффициенты испарения рс и рр связаны с соответству-
ющими коэффициентами диффузии Dc и Dp и общим давлением Р0ош
газов и паров, находящихся над жидкостью, выражениями:
Р общ
2 Робщ
где Ki и К2 — коэффициенты пропорциональности.
96
На практике в чистом виде статическое испарение не встречается.
конвективные потоки. Причиной образования таких конвек-
потоков могут быть: разность молекулярных весов пара
окружающей среды, наличие температурного градиента в погранич-
Вещества с молекулярным весом меньше молекулярного веса
П испарении жидкостей даже в неподвижной среде обычно обра-
зуются
тивпых
м слое, непосредственно примыкающем к поверхности испарения.
Вещества с молекулярным весом меньше молекулярного веса
Опушающей среды движутся вверх. Под влиянием разности темпе-
ратур в пограничном слое происходит перенос вещества по потоку
тепла (термодиффузия). Если паровая фаза состоит из веществ
с различными молекулярными весами, то вещество с большим моле-
кулярным весом передвигается в направлении потока тепла, а с мень-
шим — против этого потока (относительная термодиффузия).
Эффект термической диффузии оценивается величиной разделе-
ния:
ML АА. = С2-С1
где Ci и С2 — объемные концентрации легкого компонента в обла-
, °К.
и концентрационного
стях с температурами Т± и Т2
Величина разделения ДХ определяется выражением:
а;.=а'г1п^1
Г _ 1 2
L DT
где Кт = -р----отношение термического
коэффициентов диффузии.
По экспериментальным данным коэффициент испарения в усло-
виях свободной конвекции с учетом конвективных токов, образу-
ющихся в результате различия в молекулярных весах диффундиру-
емого вещества и среды, определяется выражением:
г
R _ AZ>pPo6nxMn(Gr, Рг)0-25
где К
Мп
Gr
коэффициент пропорциональности, н/кг;
молекулярный вес паров;
— критерий Грасгофа, характеризующий движение среды
из-за разностей молекулярных весов и температуры
пограничного слоя;
— критерий Прандтля, характеризующий физические свой-
ства системы;
Давление паров топлива на поверхности испарения, н/м2;
Длина поверхности жидкости, м.
испапения11^ Факторами, определяющими скорость статического
f 11 являются:
паров, коэфАии 1К11аРЯ101Це*’ся ЖИДКОСТИ — давление насыщенных
теплонровопппг ИеНТ ДИФФУЗИИ, теплота испарения, коэффициенты
«ности и теплоемкости;
7 о 7
* Заказ 792.
97
2) условия испарения — температура топлива и окружающей
среды, давление среды, форма, размеры и материал резервуара. |
Учесть совместное влияние всех указанных факторов на скорость
статического испарения не всегда представляется возможным. Рас-
смотрим влияние каждого фактора отдельно. 1
Зависимость скорости испарения от свойств топлива w
Давление насыщенных паров. Основным параметром, определя-
ющим скорость испарения, является давление насыщенных паров.
Чем выше эта величина, тем интенсивнее (при прочих равных усло-
виях) происходит испарение топлива. Давление насыщенных паров
зависит прежде всего от химического состава топлива и температуры.
Для индивидуальных веществ величина давления насыщенных
паров при неизменной температуре является постоянной и не зави-
сит от количества взятой жидкости, от количества пара и от наличия
и концентрации воздуха или другого газа, инертного по отношению
к данному пару.
Для компонентов ракетных топлив и нефтепродуктов, предста-
вляющих собой, как правило, сложные смеси различных веществ,
давление насыщенных паров зависит от отношения паровой и жидкой
фаз 7П/Уж. «
Давление, развиваемое парами жидких топлив, равно сумме па^
циальных давлений компонентов, входящих в состав топливной смесЖ’
Робщ = Р1Х1~}~ Р2Х2 + • • • “bPn^n I
где pi, ръ, ..., рп — давление паров отдельных компонентов топли-
ва в чистом виде при заданной температуре;
— молярные концентрации компонентов в жидкой?
фазе. 11
При испарении сложных смесей вначале испаряются преимуще-
ственно низкокипящие компоненты, обладающие высоки
паров, что приводит к обогащению жидкой фазы тяжелый
тами. С увеличением отношения Уп/^ж повышается соде]
•ровой фазе высококипящих компонентов с неболы
паров. Таким образом, давление насыщенных паров сложных смесей,
будет тем меньше, чем больше отношение объемов паровой и жид-
кой фаз. ‘ еИ
Зависимость давления насыщенных паров различных топлив от
температуры показана на рис. 43. Для нефтяных топлив давление j
насыщенных паров РНас определялось при Уп/Уж = 4. I
Как видно из рис. 43, общий вид зависимостей Ляас =
приближается к кривым показательной функции: &
*Г1, ^2»
давлением
компонен-
ание в па-
м* давлением
пп
или
98
где
Т — температура, °К;
5 ^4 н ___постоянные коэффициенты.
’Для смесей углеводородов при VJV
25 прямые 1g Рнас =
j имеют примерно одинаковый наклон, пе зависящий от
вменения отношения Vn/^ж. При значениях 7п/Пж £> 25 можно
700
. 600
80 voHNO3+ 20%N&
300
200
100
% COO
CL1
Рис. 43. Зависимость давления насыщенных паров Рнас не-
которых горючих и окислителей от температуры.
Для бензина Б-70 и топлива Т-1 даны две кривые, в зависимости от со-
става топлив.
Ю 20 30 СО 50 60 70 80
800
pax-^HNOj
приблизительно применять линейную зависимость 1g jPHac от
у п/ У ж.
Зависимость Рнас от Т, выраженная уравнением
нас —
T
для сравни практического использования, но является справедливой
значения 30Л1’П0 Узких пределов температур. В табл. 16 приведены
7™»™* коэффициентов Л В,
«’о. раке;1„™ив
по которым можно
насыщенных паров (мм рт. ст.) для компо-
топлив и нефтепродуктов.
• 99
’ Таблица 16
Значения коэффициентов А и В в уравнении
пас = — -уп+А для компонентов ракетных топлив
и нефтепродуктов [1—61
Вещество
Топливо Т-1 ........JL • •
Метиловый спирт...........
Гидразин..................
Четырехокись азота........
Концентрированная пере-
кись водорода ............
Кислород (жидкий).........
Фтор (жидкий).............
в Температурные пределы опре- деления, ° К
6,961 1730 273-573
8,802 1989 263-353
8,589 2207 273—373
8,814 1746 265-316
8,853 2585 283-363
4,129 374 90—154
9,720 462 50-90
более точно выражают температур
1
• Из многих формул, которые
ную зависимость .Рнас и применимы в широком интервале темпера
тур, простейшей является:
lgPHac = -4+>Ug7’ + C
где А, В и С — постоянные коэффициенты.
При расчетах процесса испарения в условиях высоких давлений,
например при испарении топлива в камере сгорания двитзтрля,
вместо давления насыщенных паров топлива пользуются фугитив-
ностью.
Для нахождения фугитивности составлены графики (рис. 44),
на которых отношение фугитивности к давлению изображено в виде
функции приведенных давлении и температур.^ у
Приведенное давление представляет собой отношение давлений
топлив к их критическому давлению: I
кр
Аналогично определяются приведенные температуры:
т
^пр— т»
1 кр
Зависимость фугитивности от температуры
ворительно выражается уравнением:
1g Рф ~ — "F + А
достаточно удовлетт
Коэффициент диффузии. Количество паров,
с поверхности испарения в окружающую среду
которое переносится
за единицу времени,
100
ЛИГ
возрастает
давления насыщенных паров.
V г _____гттж/ЬЯКхГОТ/ПТ
зпастает с увеличением коэффициента диффузии. Однако влияние
В°ого параметра па скорость испарения не так велико, как влияние
давления насыщенных паров.
1 Коэффициент диффузии зависит от свойств испаряющейся жидко-
_ газа в котором происходит диффузия, температуры и давления.
СТ * • *
Рис. 44. График для определения фугитивности.
На кривых показаны значения приведенной температуры.
центрации
Зависимость коэффициента диффузии, отнесенного к градиенту кон-
, от температуры и давления выражается формулой:
где
с искомый коэффициент диффузии, отнесенный к градиен-
ту концентраций;
° коэффициент диффузии при Го = 273°
__________________ ~ „ К и давлении
Л) = 760 мм рт. ст.;
давление и температура, при которых определяется
коэффициент диффузии;
постоянные коэффициенты,
топлив можно принять т = 2
, отнесенный к градиенту концентраций,
узии, отнесенным к градиенту пар-
яг
кЛЯд хуглевоД°Родных
ЦПязапФФИЦИ1НТ ДИФФУЗИИ
ЦИаллпЛ КоэФФи Ди оптом ди
Х Давлений, уравнением:
лиг
= DpRT
гДе R __ га
темп аЯ постоянная> Н'м!кг»град;
оратура поверхности испаряющейся жидкости, °
101
lull
Коэффициенты диффузии ракетных топлив и нефтепродуктов изу.
-“в ХТпртввТеив коэффициенты диффузии для паров веко-
торых углеводородов и топлив в воздухе. Таблица п
Коэффициенты диффузии веществ в мздухе при 273° К
и 760 мм рт. ст. [1—7]
Вещество
см2 / сек
Температура
определения,
Гептан ........................
Бензол ........................
Толуол ........................
Анилин .I......................
Этиловый спирт ................
Метиловый спирт ...............
Бензин Б-70 ..................
Топливо Т-5 . . . ............
Дизельное топливо ДА...........
Концентрированная перекись водо-
рода • • •;....................
Кислород (газ).................
Водород (газ)..................
0,0594
0,0783
0,0709
0,0609
0,1020
0,1325
0,0845
0,0287
0,0730
0,1260
0,1860
0,6110
1,60
1,89
1,90
2,0
2,0
2,0
2,0
1,96
2,0
2,0
1,92
2,0
373—573
273-617
273—332
523—673
77—353
m
с поглощением
—ура
ерда ” испаряющейся жидкости понижается, причем
Таблица 18
Теплота испарения некоторых горючих веществ
и окислителен 11—7]
Вещество
Теплота
испарения,
кд ж I к г
Температура
определения,
OR
Бензин Б-70..........
\1 Топливо Т-1.........
Бензол ..............
Анилин ...............
Гидразин..............
Этиловый спирт ....
Азотная кислота ....
Концентрированная пере
кись водорода ....
Фтор..................
Кислород .............
397,3
1274,9
906,4
481,5
1381,6
172,3
213
453,15
456
351,3
90,00
102
рость
больше, чем выше теплота испарения жидкости. Следовательно,
пли прочих равных условиях с повышением теплоты испарения ско-
ость испарения уменьшается. Теплота испарения данной жидкости
зависит от температуры. В области низких давлений пара умень-
павнительно медленно. С повышением давления теплота испарения
гменьшается .очень сильно и при критической температуре стано-
вится равной нулю.
В табл. 18 приведены значения теплот испарения ряда компонен-
тов ракетных топлив и нефтепродуктов.
шение теплоты испарения с повышением температуры происходит
сравнительно медленно. С повышением давления теплота испарения
уменьшается .очень сильно и при критической температуре стано-
вится равной нулю.
В табл. 18 приведены значения теплот испарения ряда компонен-
Теплопроводность и теплоемкость. Коэффициент теплопровод-
ности и теплоемкость косвенно оказывают влияние на скорость
испарения, поскольку они определяют время прогрева или охлажде-
ния, а следовательно, и температуру испаряющейся жидкости при
изменении внешних условий.
Зависимость теплоемкости и теплопроводности от температуры
может быть выражена следующими уравнениями:
Ср—a -f-bt
— А,2о {1 + Р — 20)]
НИК
где а, b и р — постоянные коэффициенты;
t — температура, °(
Значение коэффициента р может быть как положительным, на-
пример для воды, глицерина, так и отрицательным — для керосина,
толуола, этилового спирта.
Для определения коэффициента теплопроводности можно вос-
пользоваться эмпирическими формулами. Для нормальных и ассо-
циированных жидкостей % определяется выражением:
к ± --
Х= — ВСр^М 3
а
ив а коэффициент, учитывающий степень ассоциации жидко-
_ стеи (для ассоциированных жидкостей а = 1,0);
универсальная для всех жидкостей величина при Т =
= ~ Т •
0 плотность топлива, кг/м3-
молекулярный вес топлива;
— теплоемкость топлива при Т — у^кр,
коэффициент пропорциональности,
ности величин
Зна *
торых компонент ^ИЦИеНТСВ теплопРовоДн°сти и теплоемкость неко-
в табл. 19. °в Ракетных топлив и нефтепродуктов приведены
р
дж/кг» град-,
учитывающий размер-
Таблица 19
Коэффициенты теплопроводности и теплоемкость некоторых
горючих веществ и окислителей [1—6]
Вещество
вт/м • град
кджJk2>град
Темпера-
тура опре-
деления,
°К
Этиловый спирт .
Гидразин . . . .
Бензин Б-70 . . .
Топливо Т-1
Топливо Т-5 •
Дизельное топливо . .
Кислород (жидкий) . .
Фтор (жидкий) . . . .
0,1686
0,5233
0,1204
0,1104
0,1192
0,1116
0,1186
0,1126
0,1169
0,1766
2,47
3,08
2,00
2,20
2,08
2,14
1,87
2,08
1,70
273
323
90,4
85
внешних
условии
Зависимость скорости испарения от
При повышении температуры окружающей среды и испаряющейся
жидкости увеличиваются давление насыщенных паров и коэффициент
диффузии, уменьшается теплота испарения, в результате чего испа-
рение происходит с большей скоростью.
При повышении давления среды, в которой происходит испаре-
ние, увеличивается число молекул, возвращающихся обратно в жид-
Таблица 20
Среднегодовые потери автомобильных и авиационных бензинов за 1 месяц
хранения в наземных полевых горизонтальных резервуарах без дыхательных
клапанов [1] I
хР
I
Объем, л<з
I
I
I
Климатиче-
ская зона
।
и
£ со
S о
И
Северная
Средняя
Южная
А-66
Б-70
Б-70
0,95
1,11
1,41
1,20
Б-70
Б-70
А-66
Б-70
Б-70
0,60
1,40
0,50
1,00
370
330
500
159
388
137
17
16
50
17
50
50
50
54
30
16
1,10
2,72
1,63
1,03
1,41
94
43
95
95
94
86
95
94
95
11 700
10900
34'700
И 800
31 000
34 300
31400
36 300
20 800
11000
18000
6,77
14,6'
10,00
13,40
9,72
104
снижается коэффициент диффузии, в результате скорость
испарения уменьшается.
1 На скорость испарения влияют форма, размеры, материал и окра-
пезервуаров, в которых хранятся или транспортируются топлива,
сК от этих факторов зависит температура парового простран-
таК и верхнего слоя жидкости. Например, с уменьшением объема
£ТВеРвуаров скорость прогрева топлива возрастает и испарение про-
исходит более интенсивно.
Влияние свойств топлива и условий хранения на потери от испа-
рения иллюстрируют опытные и расчетные данные, приведенные
в табл. 20 и 21.
Таблица 21
Потери топлива за 1 месяц хранения, кг на 1 3t3 парового
пространства в средней климатической зоне [1]
Условия хранения
Топливо
Наземные горизонтальные резерву-
ары:
зимний период ...............
весенне-осенний.......... .
летний.......................
Наземные вертикальные резервуары:
зимний период ...................
весенне-осенний...............
летний ..............
Полуподземные резервуары:
зимпий период ...................
весенне-осенпий..............
летний ......................
Подземные резервуары:
зимний период....................
весенне-осенний..............
летний
1,43
8,78
42,00
0,951
4,910
16,00
0,614
2,160
5,420
0,192
0,534
1,010
0,344
1,970
6,570
0,22
1,14
3,58
0,131
0,424
0,971
0,056
0,174
0,352
0,023
0,124
0,369
0,011
0,059
0,179
0,007
0,023
0,055
0,003
0,010
0,019
ДИНАМИЧЕСКОЕ ИСПАРЕНИЕ
тополИНаМ11ЧеСКИМ пспаРепием называется такое испарение, при ко-
В >ки^кость и газовая среда движутся относительно друг друга,
верхнее 1хущемся пот°ке воздуха скорость испарения с плоской по-
1 увеличивается пропорционально скорости воздуха:
гДе К —
^исп = ^tpn (Р-Р0)
/ i льпый коэффициент пропорциональности,
KQl Н • М]
Пока°СТЬ воздуха> м1сек-,
затель, зависящий от скорости воздуха.
105
По многим экспериментальным данным значение коэффициента п
лежит в пределах 0,33—1,0. В работе Ирисова А. С. [7 J указывается,
что в области ламинарного потока п — —, а при испарении в турбу-
лентный поток п = 1,0. 1
Большое практическое значение в технике имеет испарение ка-
пель жидкости в движущемся потоке, поскольку этот вид испарения
наблюдается во всех типах двигателей внутреннего сгорания.
Выражать скорость динамического испарения капель топлива
количеством вещества, испаряющегося в единицу времени с единицы
поверхности, не представляется возможным, поскольку поверхность
испарения трудно определима и изменяется в процессе испарения.
Поэтому скорость динамического испарения распыленного топлива
оценивается количеством вещества, испаряющегося в единицу вре-
мени. В отдельных случаях скорость динамического испарения харак-
теризуется количеством вещества, испаряющегося в единицу времени
в единице объема резервуара или камеры, где происходит испарение.
Скорость испарения распыленного топлива в газовом потоке за-
висит прежде всего от разности парциальных давлений паров на
поверхности капель и в окружающей среде и условий обтекания ка-
пли газовым потоком. Парциальное давление паров определяется
температурой топлива и зависит от теплообмена с окружающей
средой и от свойств топлива — давления насыщенных паров, коэф-
фициента диффузии, теплоты испарения, теплоемкости и т. д. При
обтекании капель газовым потоком происходит срыв паровой обо-
лочки на одной стороне поверхности и образование области застоя
на другой. Вследствие этого с различных частей поверхности капли
испарение происходит неодинаково. Внутри вихревой зоны застоя
образуется насыщенное парами пространство, дальнейшее испарение
в котором идет медленно и мало зависит от скорости воздушного
потока. j
Вот почему, кроме рассмотренных выше факторов, влияющих на
скорость статического испарения, при динамическом испарении
необходимо учитывать влияние качества распыливания, определя-
ющего^ размер капель и суммарную поверхность испарения, скорость
и интенсивность турбулентности газового потока, состав смеси го-
рючего и окислителя. При динамическом испарении большее влия-
ние, чем при статическом испарении, оказывают давление и темпе-
ратура окружающей среды и такие свойства топлива, как вязкость,
поверхностное натяжение, теплопроводность и теплоемкость.
Зависимость скорости испарения от качества распыливания топлива
Общая характеристика процесса распыливания. Одним из важ-
нейших элементарных процессов смесеобразования является распи-
ливание топлива, качество которого определяется тонкостью и одно-
родностью распыливания, формой факела и его дальнобой-
ностью.
106
ь пли степень распыливания характеризуется диаметром
Тонкое меньше диаметр капель, тем тоньше распиливание,
капель: расПыливания определяется степенью отклонения раз-
Однород о? цХ среднеГо диаметра: чем уже пределы изменения
мерой на ,^педь> тем однороднее распиливание. О тонкости и одно-
диаметра пыд^вания можно судить по экспериментальным харак-
родносп КОТОрые могут быть представлены в виде числовых и
^ерИСТ ’ пягппеттеления
массовых кривых распределения
каПЧисловые кривые (рпс. 45)
выражают процентное отношение
количества капель определенного
диаметра к общему количеству всех
капель в факеле топлива.
100=над
Рис. 45. Числовая N и массовая G
кривые распределения капель при
струйном распиливании дизельного
топлива.
Диаметр распылителя 0,3 мм, давление
затяжки пружины форсунки 250 кгс/см*
(245- 105 н/м*).
где ni — число капель определен-
ного диаметра;
2V — общее число капель;
dK — диаметр капель.
Такие кривые удобно исполь-
зовать при исследовании влияния
качества распыливания на про-
цессы воспламенения в двигателе.
С изменением качества распыливания топлива изменяются и
условия воспламенения. При неоднородном распиливании создаются
самые различные местные концентрации паров горючего и окисли-
теля, в том числе и наиболее благоприятные для быстрого развития
предпламепных процессов окисления. При однородном распылпвапии
(как мелком, так и грубом) создаются менее благоприятные условия
для воспламенения. ••
При оценке скорости, продолжительности и полноты сгорания
необходимо учитывать, какая часть впрыснутого топлива превра-
щается в мелкие, средние и крупные капли. Для этого надо построить
массовые кривые распределения капель (рис. 45), выражающие про-
центное отношение массы капель различного диаметра к общей массе
всех капель в факеле:
— .100 = i|?(dK)
иК
»да«родвом
иг с более
Si масса капель определенного диаметра;
общая масса всех капель;
Диаметр капель.
мелком распыливании процесс горения про-
высокой скоростью и полнотой, чем при грубом.
107
Соответствующим подбором характеристик распыливаиия можно
регулировать протекание рабочего процесса в двигателях. Например,
одним из авторов [8] при исследовании влияния степени и однород-
пости распиливания топлива на процессы воспламенения и горения
в дизеле было показано, что за счет изменения степени и однород-
ности распиливания трудно воспламеняющегося топлива можно
добиться снижения скорости нарастания давления и, следовательно,
«жесткости» работы двигателя. 1
В современных жидкостных ракетных двигателях при распыли-
вании компонентов топлива большая его часть первоначально рас-
падается на капли диаметром 25—250 мк, в двигателях с воспламе-
нением от сжатия 5—100 мк. j
Для обеспечения более равномерного распределения топлива
в пространстве камеры сгорания форма и дальнобойность факела
распыленного топлива должны подбираться в соответствии с кон-
фигурацией и размерами камеры.
Качество распыливания зависит от конструкции распыливающих
устройств, давления впрыска, плотности и вязкости среды, в кото-
рую попадает впрыскиваемое топливо, а также от вязкости, поверх-
ностного натяжения п плотности топлива. i
Влияние конструкции распылителя на качество распыливания.
Распылитель создает струю определенной формы и вызывает перво-
начальный распад ее. Сопло, из которого топливо выходит компакт-
ной п плотной струей с малым конусом расширения, обеспечивает
большую дальнобойность по сравнению с соплом, по выходе из
которого топливная струя сильно расширяется. j
Сопло с винтовыми канавками обеспечивает больший угол конуса
струп по сравнению с нормальным цилиндрическим соплом. Увели-
чение конуса распыливания и уменьшение дальнобойности при при-
менении сопл с винтовыми канавками тем больше, чем интенсивнее
происходит закручивание струи. I
Форсунки с цилиндрическим соплом обеспечивают более тонкое
распиливание, чем форсунки с винтовыми канавками или камерами
закручивания. Большие гидравлические сопротивления в последних
приводят к уменьшению скорости струи. !
Центробежные форсунки оказываются удобными, когда требуется
получить достаточно хорошее распыливание при относительно малом
давлении и больших расходах топлива. 1
Значительное влияние на развитие топливного факела — степень
распыливания, дальнобойность и однородность распыливания — ока-
зывает диаметр соплового отверстия: чем он меньше, тем больше угол
конуса струи, выше степень и однородность распыливания. Глу-
бина проникновения вершины факела возрастает с увеличением диа-
метра отверстия.
В некоторых жидкостных ракетных двигателях распыливание
топлива осуществляется путем столкновения струй. Тонкость рас-
пыливания при этом получается меньше, но зато достигается хоро-
ший контакт компонентов в жидкой фазе, что имеет большое значе-
•звития процесса самовоспламенения топлив. В факеле
пне ДлЯ образовавшемся в результате столкновения струй,
распылива » ляется по объему достаточно равномерно, но даль-
ТО"''йность при этом снижается.
I,(H.R ше давления впрыска па качество распыливания. С увели-
*"ли явления впрыска при постоянном противодавлении степень
чеппем сть раСпыливания возрастают, при этом повышается ско-
^0Дь°истечения. Это содействует распаду струи из-за больших вихре-
РоСТ1> >И;кенпй внутри нее и на периферии, а также из-за увеличения
ВЫХ ействия па поверхность аэродинамических сил среды, в которой
происходит распыливание.
С повышением давления впрыска при одном и том же диаметре
сопла дальнобойность струи увеличивается до известного предела,
затем начинает уменьшаться. Вначале на дальнобойность реша-
ющее влияние оказывает скорость истечения топлива, а затем топ-
кость распыливания. С уменьшением размеров капель дальность их
полета уменьшается.
Влияние плотности газовой среды на качество распыливания.
С увеличением плотности среды, в которую производится впрыск,
возрастают силы аэродинамического действия среды на струю,
и если скорость струи не меняется или меняется мало, степень рас-
пиливания увеличивается. При чрезмерном повышении противода-
вления, когда вследствие этого сильно уменьшается скорость струи,
распыливание становится более грубым, так как значительно умень-
шается воздействие начальных возмущений, возникающих в струе
при истечении. 4
Увеличение давления среды, в которую производится впрыск,
приводит к повышению ее плотности. Возрастает сопротивление
движению струи и, следовательно, уменьшается дальнобойность
струи.•
Влияние свойств топлива на качество распыливания. Распыли-
вание жидких топлив связано с необходимостью преодоления сил,
противодействующих образованию новых поверхностей раздела фаз,
т- е. сил сцепления молекул топлива и поверхностного натяжения
Жидкости на границе с газовой средой. Таким образом, распыливание
стРуи топлива при прочих равных условиях зависит от вязкости
поверхностного натяжения топлива.
сти впРыске чеРез цилиндрическое сопло с увеличением вязко-
бой 10Плпва Уменьшается угол конуса струи, увеличивается дальне- i
В cj СТЬ’ Раслыливание становится более грубым и неоднородным. I
галитеа<3 ц,ептР°^ежных форсунок и форсунок со штифтовыми рас- I
Т(Л1 С Увеличением вязкости степень распыливания
мается меньшается, а угол конуса распыливания увеличи-
Дит срПИЬ1ливании топлива центробежными форсунками происхо-
кела, а мп’^ИЯ капель: крупные капли попадают на периферию фа-
Т()илива \1ЯПе в его Центральную часть. С увеличением вязкости
* еличиваются размеры капель, обладающих большой
109
инерцией и сохраняющих при движении значительное время то на-
правление, которое они получили при выходе из форсунки.
Повышение плотности топлива увеличивает пробивную способ-
ность капель и, следовательно, дальнобрйность факела.
Влияние качества распыливания на скорость испарения. При
впрыске одного и того же количества топлива суммарная поверх-
ность испарения может изменяться в зависимости от степени распы^,
ливания в десятки и сотни раз (табл. 22).
Таблица 22
Увеличение общей и удельной поверхности испарения жидкого
топлива при изменении диаметра капель
Диаметр капель,
с.и
Число капель
Общая поверх-
ность испаре-
ния, CJW2
Удельная поверх-
ность испарения (от-
ношение поверхности
к объему), ой-*
1
0,1
0,01
0,001
103
106
10е
3,14
31,4
314
3140
6
6-10
6 -102
6 • 103
Чем больше общая поверхность испарения,
тем выше скорость
динамического испарения. Повышение скорости испарения при более^
тонком распиливании происходит вследствие увеличения поверхно-
сти испарения и давления насыщенных паров над кривой поверхно-
стью капли, а также сокращения времени прогрева отдельных капель.
Количество пара <?0, диффундирующего за единицу времени через
сферическую поверхность, концентрическую с каплей, а следова- I
тельно, и количество жидкости, испаряющейся с поверхности капли
в единицу времени в неограниченную атмосферу газа, описывается
уравнением: I
dC
dr
где
г — радиус капли, м;
Dc — коэффициент диффузии, отнесенный к
траций, м2!сек\
С — концентрация паров топлива, кг!м3.
пт
градиенту концен-
1Ш1
Принимая концентрацию паров в окружающей среде на беско-
нечно большом расстоянии от поверхности испарения = 0, после
интегрирования получаем:
Со — 4л гT)cC^o.g — 4л г^рРнас
где Снас
нас — концентрация и давление насыщенных паров
у поверхности капли;
Dp — коэффициент диффузии, отнесенный к градиенту
давлений.
Ын
110
СлеДовательн0’ СК0Р0СТЬ испарения отдельных капель
_____________________ &о ___ ^cGiao DpPнас
^исп “ '4л7г “ 7_7
Динамическое испарение распыленных топлив определяется
ктке однородностью Распыливания. При одинаковой величине сред-
диаметра капель в случае неоднородного распыливания ско-
рость испарения в первый период (пока испаряются мелкие капли)
выше, а затем, по мере испарения крупных капель, снижается. Время
полного испарения при неоднородном распиливании больше, чем при
однородном.
Зависимость скорости испарения от внешних условий
На скорость динамического испарения большое влияние оказы-
вают скорость газового потока, его турбулентность, температура
и давление среды, а также состав смеси горючего и окислителя.
Аэродинамическое воздействие газового потока на струю рас-
пыленного топлива заключается в дроблении капель на более мелкие,
благодаря чему увеличивается поверхность испарения. Срыв паро-
вой оболочки с поверхности испаряющихся капель и перемешивание
^ускоряют процессы диффузии и тем самым скорость испарения.
Скорость диффузии и испарения в турбулентном потоке опреде-
ляются такими характеристиками, как интенсивность и масштаб
турбулентности.
Если средняя скорость в данном месте турбулентного потока
^ср> то действительная скорость w в произвольный момент времени
отличается от этой скорости. Величина w' = w — wcp называется
пульсационной скоростью.
Отношение среднеквадратичного значения пульсационной ско-
рости к средней скорости потока называется интенсивностью или
степенью турбулентности.
Масштаб турбулентности представляет собой некоторый эффек-
тивный размер перемещающихся объемов газа или жидкости, в ко-
ором в данный отрезок времени все частицы обладают одинако-
ДЛИИ <?КОростями Движения. Масштаб турбулентности называют еще
нее^)°И ПУТИ смешения, поскольку его можно представить как сред-
ппсг Расстояние, на которое перемещаются молярные объемы, смеши-
ваясь с окружающей средой.
ПрбуХне?сТПие/Пульсационпой скорости
< Ч<ЬАгЯЬаяется аналогом коэффициента диф
и тысяч КТПВНВ1й турбулентный коэффициент
<«Ффу31П,Раа бМЬШв
Пот°ка, Ка1
Jllll
турбул еАеппе пУльсационпой скорости потока w' на масштаб
- птности I носит название коэффициента турбулентного об-
коэффициента диффузии DT = w' I.
1 диффузии DT в сотни
соответствующего коэффициента молекулярной
в турбулентном потоке перенос вещества опре-
молекулярной диффузией, а такими характеристиками
пульсационная скорость и масштаб турбулентности,
111
в результате чего влияние свойств топлива на процессы переноса 1
испарившегося вещества проявляется в меньшей мере, чем в ламп-. 1
парном потоке. j
При динамическом испарении влияние давления и температуры
среды на скорость испарения проявляется не только за счет измене-
ния физических свойств топлива, по и вследствие воздействия на ха^
рактеристики турбулентного потока.
С повышением давления увеличивается пульсационная скорость
и уменьшается масштаб турбулентности. По экспериментальным
данным интенсивность турбулентности е зависит от давления следу- ]
ющим образом е ~ Рп, следовательно, при постоянной скорости J
потока w' ~ Рп.
О влиянии температуры на турбулентные характеристики потока .
в ряде работ указывается, что иУ ~ Т~т (коэффициенты п и т 1
меньше единицы). 11
Скорость динамического испарения смеси горючего и окислителя
в камере сгорания ракетного двигателя будет неодинакова для раз- I
личных ее составов. Чем меньше парциальное давление паров какого-
либо компонента топлива в горючей смеси, тем выше скорость его
испарения.
Методы расчета скорости испарения капель в потоке воздуха
Методы расчета скорости динамического испарения распылен-
ного топлива сложны, так как требуют учета множества различных
факторов. Введение ряда условных предпосылок и допущений, с уче-
том экспериментальных исследований, позволяет разработать мето-
дику расчета для определенных частных случаев. Одна из таких ме-
тодик разработана Д. Н. Вырубовым [9, 10]. Взаимное влияние про-
цессов тепло- и массообмена при больших разностях температур
и парциальных давлений в процессе испарения учитывается теорией
Аккермана [11]. По этой теории коэффициент испарения при сов-
местном протекании процессов тепло- и массообмена и больших
разностях парциальных давлений связан с коэффициентом испаре-
ния Рр при раздельном процессе и малых разностях парциальных
давлений соотношением: I
гр гр 1
где к — поправочный коэффициент. 1
Как следует из расчетов, величина к для углеводородных топлив ।
при температурах окружающей среды до 200° С мало отличаете
от единицы. При 700° С и давлении 4 ат (39,2 • 104 hIw?) к = 0,97?
т. е. тоже близок к единице. Таким образом, при расчете процессов
испарения, с достаточной степенью точности можно использовать 1
экспериментальные данные, полученные при малых разностях пар* 1
цпальных давлений и атмосферном давлении. 1
112
результаты этих
зависимостей:
3 пил теплообмена
исследовании даются в виде критериальных
*
Nu = -^- = /(Re, Рг)
Л
для
массообмена
Nu = -4£ = /(Re, Рг)
а__коэффициент теплопередачи, emlM2,-град;
гДе в _______коэффициент испарения, отнесенный к разности пар-
циальных давлений, кг/w-сек;
Л. — коэффициент теплопроводности, вт!м'град',
х — определяющий размер (диаметр) капли, м\
Nu Re, Рг — критерии Нуссельта, Рейнольдса и Прандтля.
При расчете испарения отдельных капель топлива величина коэф-
фициента испарения составляет:
₽p = -^Nu
После подстановки значения в уравнение скорости испарения
dG — Рр S (Рп — Р о) dx
<< последнее примет вид:
dG - Nu_ S (Рп-Р0) dx
X
При интегрировании этого уравнения получаем выражение, по-
казывающее характер изменения размеров капель со временем:
о-Бт
где z0 — диаметр капли в начальный момент, м;
х — диаметр капли через промежуток времени т, м;
В — коэффициент пропорциональности, м2/сек.
Например, для частного случая испарения при быстром тормо-
жении капель и небольших величинах массового потока можно
принять значение критерия Nu = 2. Тогда:
z? {Рп—Ро)
гДе q плотность топлива, кг/м3.
Вопрос о температурном режиме испарения в настоящее время
* '"Ц) не вполне выяснен. Д. Н. Вырубов [9, 10] предлагает считать,
ттТ° В первом приближении испарение происходит в условиях ста-
ционарного температурного режима и следует пренебрегать наличием
температурного поля в капле.
из емпеРатУРа испарения, как было указано выше, рассчитывается
А1гЛСЛ03Ия теплового баланса dQ = dG с учетом зависимостей
-^ькермана. '
& Заказ 792.
ИЗ
1Ш1
При расчете значения теплоты парообразования, теплоемкости,
давления насыщенных паров, плотности топлива относят к темпера-
туре испарения на поверхности капли; коэффициентов диффузии,
вязкости и теплоемкости паров — к среднелогарифмической темпе-
ратуре пограничного слоя; коэффициента теплопроводности — к тем-
пературе среды.
Для расчета испарения в факеле распыленного топлива должна
быть известна суммарная кривая распределения капель по размерам.
Такая кривая может быть задана графически или в виде приближен-
ного уравнения. -М
Для упрощения расчета вся совокупность капель разбивается на
отдельные узкие группы, для которых может быть принят общий сред-
ний диаметр. По этому среднему диаметру и производится
степени испарения данной группы капель.
Степень испарения всего факела йИсп определяется из
жения:
расчет
выра-
групп
Qncn—z । Qi Vi
где Q{, Vi — степень испарения и объемные доли отдельных
капель.
Зная объемную долю отдельных групп капель во всем факеле
и скорость испарения капель такого размера, определяют степень
испарения в факеле распыленного топлива.
нас* В зависимости от состава
определяют различными методами. Для
МЕТОДЫ ОЦЕНКИ ИСПАРЯЕМОСТИ ТОПЛИВ
Если топлива имеют близкие значения теплот испарения и коэф-
фициентов диффузии, то об их испаряемости можно судить по вели-
чине давления насыщенных паров Ряас*
н свойств топлива Рнас 1
многих компонентов ракетных топлив, представляющих собой инди-
видуальные вещества или смеси с узкими пределами выкипания,
удобной является методика, основанная на определении температуры
кипения при различных давлениях. Давление насыщенных паров
при этом равно внешнему давлению. Экспериментальные данные
могут быть выражены уравнением:
Для нефтепродуктов широкое применение нашел метод опреде-
ления давления насыщенных паров в статических условиях при соот-
ношении паровой и жидкой фаз 4 : 1 и температуре 38° С. Получен-
ные этим методом значения РНас характеризуют склонность топлива
к образованию паровых пробок в топливной системе двигателей $
пусковые качества и возможные потери при транспортировании
и хранении топлив.
Аппаратурное оформление метода может быть различным.
В СССР давление насыщенных паров в статических условиях опре-
деляют по ГОСТ 1756—52 и ГОСТ 6668-53.
114
Пия оценки склонности топлив к потерям от испарения И. П. Бу-
вым [11 предложен метод, сущность которого заключается в опрс-
Д\11 (ч1пи убыли веса продукта после продувки его строго определенным
эмом воздуха. В качестве стандартного принят метод продувки
10 /л топлива 10-кратиым объемом воздуха при 293° К.
Qg испаряемости топлив можно судить по температурам кипения
Жпакциопному составу. Фракционный состав топлива определяется
ем перегонки определенного количества топлива на стандартном
** иборе. На рис. 46 приведены кривые перегонки для некоторых
углеводородных горючих.
t,°C
Рис. 46. Кривые перегонки некоторых топлив.
размеров аппарата
наблюдений
Характер кривых зависит от конструкции и
скорости перегонки и других факторов. Этим объясняется строгая
стандартизация прибора, способа перегонки
(ГОСТ 2177—59).
По температуре выкипания определенного количества топлива
можно судить о поведении топлива при хранении, транспортирова-
нии и применении в двигателях. В качестве таких характерных то-
чек выбраны температуры начала, конца, 10, 50, 90% перегонки и
Другие, в зависимости от вида топлива.
Температура начала кипения указывает на присутствие легких
Фракций, характеризует огнеопасность и склонность к образованию
паровых пробок, но не дает представления о количестве этих фрак-
ции. О количестве легких фракций можно судить по температуре
©регонки 10% топлива. Температура выкипания 50% топлива ха-
топТеРИзУет сРеДпюю испаряемость. Температура перегонки 90%
лых1*11^ И конеп» кипения указывают па присутствие в топливе тяже-
Фрак Трудпоиспаряющихся фракций. Чем меньше содержится этих
ции в топливе, тем полнее оно испаряется в двигателе.
8*
115
Рядом исследований установлена связь между температурой вы-
кипания определенного количества топлива и легкостью пуска,
возможностью образования паровых пробок, «жесткостью» работы
двигателя и т. д. [7, 12, 13].
Оценка испаряемости топлива может быть осуществлена на стен-
довых лабораторных установках, воспроизводящих в той или иной
Рис. 47. Схема установки для определения испа-
ряемости бензинов в динамических условиях:
1 — топливный бак; 2 — расходомер; 3 — кран; 4 — всасы-
вающая труба; 5 — дроссельная шайба; 6 — дифференциаль-
ный манометр; 7— поплавковая камера; 8—диффузор; 9 —
подогреватель; 10 — термометр; 11 — манометр; 12 — улавли-
ватель неиспарившегося топлива; 13 — мерная склянка для
сбора нбиспарившегося топлива. 1
степени процесс испарения в двигателях. Скорость испарения вы-
ражается количеством горючего, испарившегося за определенные
промежутки времени, или в процентах от количества поданного
топлива. 1
Для примера на рис. 47 приведена схема установки для исследо-
вания испаряемости бензинов, воспроизводящая условия смесеобра-
зования в карбюраторных автомобильных двигателях. Установки
такого типа позволяют выявить зависимость скорости испарения от
свойств топлива, скорости газового потока, состава смеси, темперам
туры, давления среды, конструктивных особенностей системы пита-
ния и других факторов. Я
Стендовые лабораторные установки для оценки испаряемости
не могут быть универсальными, поскольку условия испарения то-
1Ва в различных типах двигателей не одинаковы. К недостаткам
аких установок следует отнести сложность аппаратуры и самого
Т „..orca испытаний.
ВЛИЯНИЕ ИСПАРЯЕМОСТИ ТОПЛИВА
НА РАБОЧИЙ ПРОЦЕСС ДВИГАТЕЛЯ
Испаряемость топлив приобретает большое значение при запуске
поршневых двигателей с искровым зажиганием и воздушно-реактив-
ных двигателей, особенно при их работе в условиях низких темпера-
тур окружающего воздуха. В этих условиях или при применении
ппохоиспаряющихся топлив состав образующейся горючей смеси
не соответствует пределам воспламенения.
Для обеспечения легкого запуска поршневых двигателей с искро-
вым зажиганием необходимо, чтобы топливо содержало возможно
больше низкокппящих углеводородов, о наличии которых судят
по температуре перегонки 10% топлива и давлению насыщенных
паров. Влияние испаряемости топлива на запуск карбюраторного
автомобильного двигателя показано на рис. 48 и в табл. 23.
Таблица 23
Влияние испаряемости бензина на время запуска
► двигателя автомобиля «Москвич» [14]
Температура
перегонки
10% бензина,
°C
Температура
воздуха, °C
Продолжи-
тельность
запуска, сек
Расход
бензина, мл
0
79
10,5
10
-16
0
-6
—16
515
29
678
30
В работах Н. А. Рагозина [16—17] и А. С. Ирисова [7] указы-
вается, что пусковые качества бензина определяются не только тем-
пературой перегонки 10% топлива, но имеют значение и более высо-
'окипящие фракции, определяющие температуру перегонки 50%
топлива.
п 1ем оыстрее прогревается двигатель, тем меньше расход топлива
топ8НОС АеТалеЙ‘ С понижением температуры перегонки 50 и 90 %
лви ИВа вРемя прогрева сокращается и улучшается приемистость
if * V J1 /f е
Пусковые
3°ваться
легче г
Mon Т. AVM Ml
кпт..ДЛя обеспечения
свойства реактивных топлив также могут характери-
температурой перегонки 10% топлива. Чем ниже она, тем
^шУск и тем меньше может быть давление подачи, необходи-
котором 'vvvuc’ieunn такого качества распыливания топлива, при
г происходит воспламенение.
117
Рис. 48. Зависимость между минималь-
ной температурой окружающего воздуха
fB, обеспечивающей легкий запуск кар-
бюраторного двигателя, коэффициентом
избытка воздуха а в пусковой период
и температурой перегонки 10% топлива
*<noz [15].
В работах [18, 19] приведены следующие данные: авиационный
бензин с температурой перегопки 10%, равной 71° С, обеспечивает
легкий запуск двигателя при температурах окружающего воздуха
до —65° С. При использовании топлива с температурой перегопки
10%, равной 190° С, запуск двигателя при —40° С затруднен. Пц
топливе с пределами кипения 240—365° С запуск двигателя был
невозможен, хотя он работал nS
этом топливе, будучи предва-
рительно запущен на керо-
сине. На более тяжелом топ-
ливе, выкипающем в пределах
256—396° С, двигатель не ра-
ботал и на режиме.
В табл. 24 приведены дан-
ные, характеризующие пуско-
вые свойства ряда топлив, по-
лученные при испытании на
малоразмерном двигателе. Оцен-
ка пусковых свойств топлив
проводилась при расходе воз-
духа через камеру 0,1 mlcei
и температуре на входе в ка-
меру 60° С. В качестве пара^
метра, характеризующего пу-
сковые свойства, принят опти-
мальный состав топливо-воз-
душной смеси, при котором про-
исходит
камере сгорания [20].
легче
больше
тем беднее состав топлпво-воз-
смеси, при котором происходит воспламенение в камере
ее воспламенение в
Чем
фракционный состав,
давление насыщенных
меньше вязкость топлива,
паров и
душной
сгорания, следовательно, лучше пусковые качества топлива.
Таблица 24
Пусковые свойства реактивных и дизельных топлив [20]
ТС-1
Топливо
Пределы
выкипания,
V при 20° С,
сст
•Рцас ПРИ
38° С,
мм рт. ст.
коэффициент
избытка воз-
духа, при к о*
тором проис-
ходит
воспламенение
Дизельное
78-237
140-232
148-238
185-355
1,09
4,47
100
50
118
Для облегчения запуска некоторых серийных воздушно-реактив-
лых двигателей применяют пусковые топлива — неэтилированные
бензины — в чистом виде или в смеси с реактивным топливом или
маслом. Однако значительное облегчение фракционного состава то-
плива без учета конструктивных особенностей двигателя может
дривести и к ухудшению запуска.
** На рис. 49 приведены пусковые характеристики топлив Т-2 и
Т-1, снятые на полноразмерной камере сгорания при температуре
воздуха на входе —35° С [20]. Как видно из рис. 49, пусковые свой-
ства топлива Т-2 лучше, чем Т-1. Запуск двигателя на топливе Т-2
Рис. 49. Пусковые характери-
стики топлив Т-1 и Т-2, сня-
тые на полноразмерной камере
сгорания.
Рис. 50. Влияние фракционного состава
дизельного топлива на продолжитель-
ность пуска х двигателя ЯАЗ-204 при раз-
личных температурах воздуха.
По опытным данным И. А. Трактовенко [12].
возможен при большей скорости воздуха на входе в камеру сгорания
вх на данной высоте Н или на больших высотах, при заданной ско-
рости воздуха.
Чем легче фракционный состав дизельного топлива (в известных
Ределах), тем меньше время запуска двигателя (рис. 50). Однако
мпература перегонки 10% не характеризует пусковых качеств
топлива* Наличие легких бензиновых фракций резко
в каЧа^'Т 010 воспламеыяемость» кроме того приводит к образованию
возчу1С*^° оСГ0Рания до начала воспламенения однородной топливо-
плал! П°й смеси» что неблагоприятно сказывается на развитии пред-
j-j Ныых процессов окисления.
Ьи тг(^°ЛЬ1е качества дизельного топлива улучшаются при пониже-
но топлива, при условии, что тем-
остается выше 200° С, а воспламеняемость
вх па данной высоте Н или на больших высотах, при заданной ско-
рости воздуха.
Чем легче фракционный состав дизельного топлива (в известных
Ределах), тем меньше время запуска двигателя (рис. 50). Однако
мпература перегонки 10% не характеризует пусковых качеств
топлива* Наличие легких бензиновых фракций резко
в 010 воспламеняемость, кроме того приводит к образованию
воздушной смеси
I
Гг XJ_
А1Усковые г- ______ ________
ПепатлМПеРатУРы перегонки 50 и 90%----------- —
Toii;IUppa ПеРегонки 10% । ' '
Заметно не снижается.
ДеигаТс°1К^ИВПОСТЬ 0СУЩествления процесса горения во всех типах
т°в йен- °И В значительной степени определяется скоростью и полно-
аРепия топлива в камере сгорания.
119
ОС
Рис. 51. Характеристики полноты сгорания
различных топлив, снятые на малоразмерной
камере сгорания [20].
В карбюраторных двигателях при недостаточной полноте испа-
рения топлива горючая смесь распределяется по цилиндрам нерав-
номерно. Наибольшая неравномерность имеет место при наличии
топливной пленки на стенках впускного трубопровода и неиспа-
рившихся капелек топлива в рабочей смеси. Неравномерное рас-
пределение топлива по цилиндрам и неполное его испарение, приводи
дящее к увеличению расхода топлива, особенно сильно проявляются^
для автомобильных бензинов с концом кипения выше 200° С. ЦЩ
Чем легче фракцион-
ный состав, больше да- I
вление насыщенных паров
и меньше вязкость топли-
ва, тем выше полнота его
сгорания в воздушно-ре-
активных двигателях. При-
чем с изменением условий
воспламенения и горения
влияние свойств топлива
проявляется не в одинако- <
вой мере. Например, чем
тяжелее фракционный со-
став топлива, тем сильнее^ -
снижается полнота сгора-*4
нпя при ухудшении усло-
вий воспламенения и сго-
рания, особенно на боль- I
ших высотах. На рис. 51
показано, как изменяется
полнота горения топлив
различного фракционного состава при изменении коэффициента из-
бытка воздуха в условиях испытаний на установке с малоразмер-
ной камерой. 1 ]
При применении тяжелых топлив в быстроходных дизелях про-
цессы распыливания, испарения и смешивания с воздухом ухудша-
ются, что приводит к снижению скорости и полноты сгорания. 1
С полнотой сгорания топлив тесно связана возможность образова-
ния нагара и появления смолистых, углистых и коксообразных от-
ложений в двигателях. Особенно опасно образование отложений
в воздушно-реактивных двигателях. При образовании нагара на стен-
ках жаровых труб камер сгорания увеличиваются гидравлические :
сопротивления, ухудшается качество смесеобразования и снижается *1
эффективность использования топлива. Нагар на стенках жаровых |
труб вызывает местный перегрев, что может привести к короблепшЗ^
и растрескиванию труб. Частицы нагара, отрываясь от стенок жаро- ]
вых труб, уносятся газовым потоком и оказывают разрушающее I
действие на лопатки турбины.
Нагарообразующая способность топлива зависит от его испаряе- I
мости. Из данных табл. 25 видно, что по склонности к нагарообра- |
120 1
ию различные топлива располагаются в соответствии с фрак-
3°ИаП ым составом: чем легче топливо, тем меньше оно дает
Сложений В камере сгорания.
° Влияние испаряемости топлива на процесс сгорания наиболее
но проявляется в дизелях и воздушно-реактивных двигателях,
лива облегченного фракционного состава способствуют усилению
А°есткостп» работы дизелей, так как они имеют худшую воспламе-
пяемость и, вследствие этого, боль-
шой период задержки воспламене-
Ц11Н • —
шения п испарения
топлив
указанных
В то же время процессы сме-
---------------------- дЛЯ легких
протекают быстрее. Оба
- обстоятельства способ-
ствуют повышению скорости пара-
Таблица 25
Нагарообразующая способность различных
топлив при испытании на установке
с малоразмерным воздушно-реактивным
двигателем [20]
Количество нагара
в камере
12
Рис. 52. Изменение пределов ус-
тойчивого горения топлив, выра-
женных предельными значениями
коэффициента избытка воздуха а,
в зависимости от давления воз-
духа на входе в камеру сгора-
ния Рвх (по данным авторов).
1.20 1.25 1.30 1.35 1k0 М
> кгс/см 2
Б-70
ТС-1
Топливо
пливу Т-1
1,69
Дизельное . .
2,90
3,80
58
76
100
199
в г
стапия давления в
к «жесткой» работе двигателя
начальной
стадии
процесса горения и приводят
X" v ААJL A JL VA. Л. VVA J Л •
Нормальное сгорание топлива в воздушно-реактивных двигателях
характеризуется устойчивым факелом пламени, при этом сгорание
роисходит без пульсаций, срывов и затухания пламени. Срыв пла-
п ни может быть при очень высокой скорости движения газового
в 3°Ка в камере сгорания, а также при уменьшении тепловыделения
обе°НС» 10Репия пз’за снижения температуры и давления или резкого
щсния пли обогащения топливо-воздушной смеси, поступающей
зону горения.
ста1иСИаРЯеМ0СТЬ РеактпвнЬ1Х топлив влияет на равномерность со-
| 1КН П '?оне горения, а также на степень обеднения или обога-
теля * ('<НЮМ(?п смеси ПРИ резком изменении режима работы двига-
УстойЧп УлУчшением испаряемости реактивных топлив область
6олрр ° Г0Рения расширяется, причем главным образом в сторону
ее бедных смесей (рис. 52).
121
Следует отметить, что лучшая испаряемость топлив как для порщ.
невых, так и для воздушно-реактивных двигателей может быть
полезной только при учете условий применения и других эксплуата-
ционных свойств топлив. Например, надо учитывать, что топлива
облегченного фракционного состава склонны к образованию паро-
воздушных пробок, испаряются при хранении, транспортировке
и применении и имеют более низкую объемную теплоту сгорание^
ЛИТЕРАТУРА
1. И. П. Б у д а р о в, Потери от испарения моторных топлив при хра-
нении, ВНИИСТ, 1961. 4
2. Справочник по теплофизическим свойствам газов и жидкостей, Физмат-
гиз, 1963. Я
3. Теплофизические свойства некоторых авиационных топлив в жидком
и газообразном состоянии, Оборонгиз, 1961. Я
4. Д. К э й, Т. Л э б и, Справочник физика-экспериментатора, ИЛ,
5. Я. Б. Чертков, Г. Ф. Большаков, Е. И. Гулин,
Топлива для реактивных двигателей, Изд. «Недра», 1964. 1
6. Техническая энциклопедия. Справочник физических, химических и тех-
нологических величин, т. 7, 1931. '1
7. А. С. Ирисов, Испаряемость топлив для поршневых двигателей
и методы ее исследования, Гостоптехиздат, 1955. ]
8. Е. И. Гулин, Влияние степени и однородности распыливания топлива
на процессы воспламенения и горения в дизеле, в кн.: «Сгорание и смесеобразо^л.
вание в дизелях», Изд. АН СССР, 1960.
9. Д. Н. Вырубов, О методике расчета испарения топлива, в кн.:
«Двигатели внутреннего сгорания», Машгиз, 1954 (Труды ‘МВТУ, вып. 25).
10. Д. Н. Вырубов, Смесеобразование в двигателях дизеля, в кн.:
«Рабочие процессы двигателей внутреннего сгорания и их агрегатов», Машгиз,
1946. W
И. G. Ackermann, VDI Forsch., № 382 (1937). И
12. Б. В. Л о с и к о в, Н. Г. Пучков, Б. А. Энглин,' Основы
применения нефтепродуктов, Гостоптехиздат, 1959. I
13. Г. Б р а у н, Испаряемость моторных топлив, ОНТИ, 1935. Я
14. Н. И. А л ь д о х и н, Нефт. хоз., № 8, 42 (1948). V
15. Tornton, Aero Res. Lab. Techn., T-43, 15/9 (1945). i
16. Моторные, реактивные и ракетные топлива, под ред. К. К. Папок и
Е. Г. Семенидо, Гостоптехиздат, 1962. .1
17. Моторные топлива, масла и жидкости, т. I, под ред. К. К. Папок
и Е. Г. Семенидо, Гостоптехиздат, 1953. Я
18. Я. М. Паушкин, Химический состав и свойства реактивных
топлив, Изд. АН СССР, 1958. 1
19. J. Sharp, Journal of the Royal Aeronautical Soc., 58, 813, 1954.
20. E. P. Терещенко, Б. Д. Залога, С. M. Максимов,
Химия и технология топлив и масел, № 11 (1960). 1
ВОСПЛАМЕНЕНИЕ И ГОРЕНИЕ ТОПЛИВ
Горение представляет собой сложный физико-химический про-
цесс, в основе которого лежит быстро протекающая химическая
реакция, сопровождающаяся интенсивным выделением тепла и из-
лучением света. Горение развивается в условиях прогрессирующего
самоускорения химической реакции, связанного с накоплением
в реагирующей системе тепла и активных промежуточных продуктов.
Для горения необходимо наличие окислителя и окисляемого
^вещества — горючего. В качестве окислителей могут быть исполь-
зованы фтор, кислород, хлор и некоторые их соединения друг с дру-
гом или с другими элементами, в качестве горючих — соединения,
содержащие водород, углерод, азот и другие элементы; обычно это
углеводороды, азотоводородные соединения и их производные.
Горение является энергетическим процессом, при котором про-
исходит разрушение электронных оболочек исходных компонентов
топлива и образование молекул продуктов сгорания. Кроме основ-
ных химических превращений реагирующих веществ, в процессе
горения протекают побочные процессы, связанные с преобразованием
химической энергии топлива в другие виды энергии, главным об-
разом в тепловую и световую.
В зависимости от физического состояния компонентов топлива
горение может протекать в гомогенных и гетерогенных системах.
Примерами горения в гомогенной системе является сжигание горю-
чей смеси, состоящей из газообразного горючего и окислителя, в ге-
терогенной — сжигание распыленных жидких топлив в среде воздуха.
Во всех случаях собственно горению предшествует процесс вос-
пламенения (или самовоспламенения), представляющий комплекс
<|.ш зико-химических процессов, развитие которых обеспечивает в
^ДОльнейшем быстрое протекание реакции окисления. Таким образом,
оспламенение есть начальная стадия процесса горения.
Значение процессов воспламенения и горения определяемся преж-
ПовВсего тем» что работа двигателей внутреннего сгорания всех ти-
в основана на использовании тепловой энергии, выделяющейся
Результате горения. В современных двигателях внутреннего
123
сгорания полезно используется до 30—40% тепла, выделяющегося
при горении топлива. Я
Среди сложных физико-химических превращений, которые пре-
терпевает топливо в двигателях, процессу горения принадлежит
определяющая роль. Дальнейшее совершенствование двигателей
внутреннего сгорания в значительной мере зависит от возможности
управления процессами воспламенения и горения топлив. От скр/
рости и полноты сгорания топлива в основном зависят устойчивость
и надежность работы двигателя, а также его мощностные и экономи-
ческие показатели.
Скорость и полноту горения в двигателях в первую очередь опре-
деляют физико-химические свойства топлива. Большое значение
имеют свойства, определяющие нормальное развитие процесса го-
рения, при котором наиболее полно используется тепловая энергия,
выделяющаяся при горении топлива. При одном и том же запасе
химической энергии эффективность использования топлива в двига-
теле зависит от того, как будут развиваться и протекать процессы
воспламенения и горения. Для каждого типа двигателя существуют
оптимальные условия, определяющие время и скорость развития
этих процессов. Уменьшение и значительное увеличение скорости
горения могут вызвать серьезные нарушения в рабочем процессе
двигателя; особенно опасно возникновение взрывного неуправляД
емого воспламенения и горения. *
Характеристики процессов воспламенения и горения топлив —
количество выделившегося тепла и скорость тепловыделения — за-
висят не только от свойств топлива, но и от условий, в которых
происходят воспламенение и горение. 1
В различных типах двигателей процессы воспламенения и горе-
ния имеют свои характерные особенности, но тем не менее можно вы-
делить основные общие для всех двигателей закономерности. Из-
учение основных закономерностей, определяющих воспламенение
и горение топлив, необходимо для понимания особенностей протека-
ния рабочего процесса в том или ином типе двигателей и обоснова-
ния требований к качеству топлив для них, а также для правиль-
ного решения ряда практических вопросов применения топлив,
их хранения и транспортирования. 1
Исключительно большое практическое значение имеет создание
таких условий хранения, транспортирования и перекачки топлив,
особенно ракетных, при которых исключалась бы возможность воз-
никновения пожаров и взрывов. Эти вопросы также можно успешно
решить; опираясь на знание основных закономерностей развития
процессов воспламенения и горения. • Я
ТЕРМОХИМИЯ ГОРЕНИЯ
При оценке энергетических свойств смесей горючего и окисли-
теля необходимо знать тепловой эффект реакции горения. В про-
цессе горения протекание химической реакции между горючим
124
кислителем сопровождается уменьшением внутренней химиче-
И й энергии реагирующих тел, в результате чего выделяется тепло
СК<лпопсходит изменение объема реагирующих веществ. Величина
П нового эффекта зависит от условий протекания реакции — тем-
пеоатуры, давления и объема.
Ес in реакция происходит при постоянном давлении, то умень-
r io внутренней химической энергии определяет не только вели*
ШСцу теплового эффекта Qp, ио и внешнюю работу L. Реакция при
постоянном объеме сопровождается увеличением теплового эффекта
О так как внешняя работа будет равна нулю, а уменьшение внутрен-
ней энергии будет определять только величину теплового эффекта
реакции. Тепловые эффекты химических реакций, протекающих
ппп постоянном объеме V = const и постоянном давлении Р = const,
связаны между собой соотношением:
• •Il I
Таким образом, тепловой эффект химического процесса при
у = const больше, чем при Р = const, на величину работы, затра-
чиваемой на расширение газа.
Тепловой эффект реакции горения, отнесенный к 1 моль, 1 кг
массы или 1 м3 горючего, называется теплотой сгорания.
Тепловой эффект экзотермической реакции окисления между го-
рючим и окислителем, идущей до образования наиболее устойчивых
окислов, отнесенной ко всему количеству реагирующих веществ,
называется теплопроизводительностью.
Размерность теплоты сгорания и теплопропзводительности:
кдж/молъ, кдж/кг, кдж/м3.
Зависимость теплового эффекта реакции от температуры опре-
деляется разностью теплоемкостей исходных и конечных продуктов
реакции:
dQp__р „
dT ~GMi gM2
где Mi и — суммарные мольные теплоемкости исходных и ко-
нечных продуктов реакции.
В зависимости от условий протекания реакции температурный
коэффициент теплоты реакции может быть определен для реакций
при V = const или Р ~ const, при этом вместо См^ и подста-
зляются суммарные теплоемкости при постоянном давлении Ср
I при постоянном объеме Cv. Необходимо учитывать, к какой тем-
РатУРе отнесен тепловой эффект реакции: окружающей среды,
Г ' ^Ч^Г0 тела или принятой в стандартах. . •
к ста °ольшинстве справочников теплоты реакций Qv и Qp отнесены
(1 niQor Р™Ь1М Условиям, соответствующим давлению 760 мм рт. ст.
’ Тепло* Ю5 7//ле2) и температуре 293° К.
Горюч(‘гоТа СГ0Рания горючих и теплопроизводительность смесей
Динамо t И. окислителя могут быть определены на основании термо-
(ских расчетов или экспериментально, причем необходимо
учитывать условия, к которым относится тепловой эффект реакции.
Продукты сгорания могут находиться в различном агрегатном состо-
янии, и теплота их образования будет неодинакова.
В соответствии с этим различают высшую и низшую теплопроиз-
водительность и высшую и низшую теплоту сгорания. Высшие те-
плопроизводительность и теплота сгорания включают количество*
тепла, которое выделяют продукты горения, конденсирующиеся
в условиях определения теплового эффекта реакции. При определе-
нии низших теплопроизводительности и теплоты сгорания тепло
конденсации продуктов сгорания не учитывается.
Рассмотрим методику определения теплопроизводительности для
частного случая реакции горения, протекающей по уравнению:
CnHmOpNft + Хо H/NuOJ)Cg = (п + Хо 9) СО2+0,5 (т + Хо О Н2О + 0,5 (к + Хо «) N2
где CnHmOpNft и H(NuOt>Cg — химические формулы горючего и
окислителя;
— теоретическое количество окислите-
ля, необходимое для полного сгора-
ния горючего, моль на 1 моль горю-
чего.
Из уравнения реакции горения
Хо =
Тепловой эффект данной реакции
Величина QM представляет собой теплоту сгорания 1 моля го-
рючего или теплопроизводительность, отнесенную к общему числу
молей (1 +хо) исходных продуктов, взятых в стехиометрическом
соотношении.
Следовательно, теплопроизводительность 1 тез и 1м
рючего и окислителя равна:
3 смеси го-
(кдж/кг);
- -77———77- (кдж/м3)
VT“ Мр + Хо^о 4 " МГ + ХОМОУ
Теплота сгорания горючего, отнесенная к 1 кг и 1 ж3 горючего
= (кдж/кг); q'*= (кдж/м3)
где Мг и Mq — молекулярные массы горючего и окислителя;
рт — плотность смеси горючего и окислителя, кг/м3\
Qr — плотность горючего, кг/м\.
126
соа
теплота сгорания 1 кг
Ппи определении теплопроизводительности смеси горючего и оки-
1 еля и теплоты сгорания горючего можно воспользоваться и дру-
сЛ11Т расчетными формулами. При полном сгорании горючего
c’ilmOpNfc образуются С02, Н20 и N2.
Теплота образования двуокиси углерода при 298,15 К Q
393522 кдж/кг • моль [1]. Таким образом, 1
углерода будет равна
” <2с=. п аГГ = 32763 кдж/кг
12,011
э>.
Теплота образования воды в газообразном состоянии при 298,15° К
п == 241827 кдж/кг • моль [1].
VH2O ___________ ППТТПППТТЛ
~ ~ 241827 кдж 1кг*моль [1].
Следовательно, низшая теплота сгорания водорода
Сн2=-^пг =119954 кдж/кг
В результате низшая теплота сгорания 1 кг горючего равна:
<2г= 328Сг+120бНг±<2обр (кдж/кг)
!НГ — содержание углерода и водорода в горючем, %;
<?обр — теплота образования горючего, кдж/кг.
Если при образовании горючего из элементов тепло выделяется,
то в приведенной выше формуле (?обр имеет отрицательное значение;
если образование горючего происходит с поглощением тепла — поло-
жительное.
Для окислителя HiNuOvCg можно найти:
<2o=328Co + 1200Ho±<2g6p (кдж/кг)
Величина Qo обозначает количество тепла, которое затрачивается
или выделяется при использовании в реакции горения 1 кг окисли-
теля. При этом за начало реакции принимается температура 298,15° К.
Теплопроизводительность смеси горючего и окислителя при сте-
хиометрическом соотношении компонентов определяется по следу-
ющим формулам:
где Сг и
QT — ^r±vo<2o ^к^Ж/Кг)
• YT l+v0
<?М = (?т(Мг + ХоМо) (кдж/кг-моль)
гДе v0 — теоретически необходимое количество окислителя для
полного сгорания 1 кг горючего, кг.
Если известен элементарный состав горючего и окислителя, то
4-Сг+8Нг-Ог
•»
v0=----------------------------
3
Но — содержание соответствующих элемен-
тов в горючем и окислителе, %.
'о —8Н0
О? ^0,
127
Когда в качестве горючего или окислителя используются сжижен-
ные газы, при определении теплопроизводительности следует учесть я
тепло, расходуемое на испарение и нагревание продуктов до началь- 1
ной температуры определения. Я
При использовании веществ, содержащих в своем составе воду, t
надо учесть тепло, пошедшее на растворение вещества в воде и на
испарение воды. При определении теплопроизводительности смесм^Д
горючего и окислителя нестехиометрического состава необходимо
учесть избыток или недостаток окислителя соответствующими коэф- I
фициентами. 'Я
Теплопроизводительность — одна из основных характеристик pa- w
кетных топлив, поскольку она определяет запас химической энер- 1
гии. Обязательным условием эффективного использования химиче- Д
ской энергии топлива является наличие массы, воспринимающей эту 1
энергию. Самое теплопроизводительное топливо не обеспечит тяги 1
при сжигании в двигателе, если не будут образовываться газообраз- I
ные продукты сгорания. Работоспособность массы газов, вытека- I
ющих из сопла реактивного двигателя, определяется температурой I
п газообразованием, зависящим, в свою очередь, от состава продуктов |
сгорания.
Степень газообразования характеризуется количеством газооб
разных продуктов, получающихся при сжигании 1 кг или 1м
при 273° К и 760 мм рт. cm.^k
горения горючего CnHmOpNft и окислителя
H/NuOuCg, приведенной на стр. 126, степень газообразования опре-
деляется выражением: М
Т7 22,4 [(п + Хо 4) +0>5 (пг + Хо Q+ 0.5 (А’ + Хо ы)] Я
3
окислителя
смеси горючего и
(1,01325-105 н/м2).
Для реакции
?
Мг +Xo Mq
V'0 = V0Q (mW)
где 22,4 — объем, занимаемый 1 кг-моль вещества при нормальных
условиях, ж3; 1
q — плотность топлива, кг/м3. |
В табл. 26 приведены основные термохимические характеристики
ряда смесей горючих и окислителей. ।
Термохимические характеристики не дают еще окончательного
представления о целесообразности применения того или иного то-
плива. Для наилучшего использования энергетических свойств то-
плива, осуществления рабочего процесса с высоким коэффициентом
полезного действия и обеспечения надежности работы двигателя
необходимо учитывать свойства топлива, определяющие его подачу;/
в двигатель, охлаждающую способность, пусковые качества, пол
ноту, скорость и устойчивость сгорания. Большое значение имеют 1
также свойства, влияющие на условия транспортиро1анпя и храпе- |
ния топлива. Я
128 I
Таблица 26
трпмохимические характеристики некоторых смесей горючих
Основные тер и 0Ю1СЛИтелей
Окислитель Горючее • Теоретически необходимое ко- личество окисли- теля vn, кг на 1 кг горючего Низшая теплопроиз- водитель- ность QT, кдж/кг Степень газо- образования Уо, мз/кг
Толуол 4,92 6150 0,747
Азотная кислота Гидразин 1,57 5983 1,030
Четырехокись Толуол 4,50 7113 0,686
азота Гидразин 1,44 6653 1,000
Толуол 3,14 9540 0,650
Кислород ч Гидразин 1,00 8117 , 1,050
Толуол 7,43 11213 0,433
г Фтор Гидразин 2,37 10096 1,040
ОСНОВНЫЕ ПОЛОЖЕНИЯ ЦЕПНОЙ ТЕОРИИ ОКИСЛЕНИЯ
лярного кислорода, „
еория окислительных
' I — — — Ji' X XX X X V. X С. А I |
ДРУг <-1ЯХ И °ыла сФормулироваиа ничти одг
вания т Д1)уга академиком А. Н. Бахом [2]
пая процессов
Рганизмах, ;
Р зом Неорганических
Зэказ 792.
Процессы воспламенения и горения топлив подчиняются основ-
ным закономерностям химической кинетики и цепной теории окисле-
ния. Кроме процессов воспламенения и горения в соответствии
с цепной теорией окисления протекают и процессы, развивающиеся
в топливах при контакте их с кислородом воздуха при храпении,
транспортировании, заправке машин, перекачке и подаче по топлив-
ной системе двигателя.
Одним из интересных и важных разделов учения о цепных про-
цессах является автокаталитическое окисление углеводородов. Из
юючисленных теорий, предложенных для объяснения механизма
ляпСЛИТеЛЬНЫХ пР°Цессов> совершающихся под влиянием молеку-
Трпп °Г0 КислоР°Да, наибольшее признание получила перекисная
0Кислительных процессов.
рекисная теория возникла на основе учения об окислительных
почти одновременно и независимо
J, проводившим исследо-
медлепного окисления в животных и растительных
и К. Энглером [3], изучавшим окисление главным об-
— веществ. Перекисная теория автокаталити-
129
ческого окисления углеводородов получила дальнейшее развит^
па основе положений цепной теории химических реакций, развитой
в работах Н. Н. Семенова [4, 5].
Основное положение перекисной теории окисления заключается
в том, что под влиянием источников энергии, например тепла или
свободной энергии окисляемых веществ, происходит разрыв одной
из связей в молекуле кислорода
О=О->—О-О-
Активный кислород присоединяется к окисляемому веществу Д,
образуя продукты присоединения АО2, называемые перекисями.
Перекиси содержат реакционноспособную группу —О—О— и
легко реагируют с другими веществами: • К
АО2 + А->2АО
ао2+в—>ао + во
По химической структуре органические перекиси можно рас-
сматривать как производные перекиси водорода Н—0—0—Н, в
которой один или оба атома водорода замещены органическими
радикалами. К
При замещении одного атома водорода радикалом получаются
гидроперекиси. Гидроперекиси могут содержать не одну, а несколько
перекисных групп. При замещении двух атомов водорода образуются
диалкилперекиси. J^^H
• Многие перекисные соединения очень неустойчивы и в чистом
виде или в концентрированных растворах взрывают при ударе
или сравнительно небольшом нагревании. Освобождающаяся при
разложении перекисей энергия ускоряет дальнейший процесс
окисления. 'К
Для понимания сложного комплекса превращений, претерпевае-
мых углеводородными топливами в процессе их окисления, необхо-
димо прежде всего выяснить направление первоначального действия
молекулярного кислорода на углеводороды различного строения.
Экспериментальные работы, проведенные К. И. Ивановым [6], по-
зволили установить направление первичных реакций автоокисле-
ния углеводородов в жидкой фазе. 1
При автоокислении углеводородов различного строения и их
кислородных производных в жидкой фазе молекулярный кислород
первоначально присоединяется только по алифатическим и ацикли-
ческим связям С—Н с образованием гидроперекисей. КН
Эта закономерность наблюдается в случае окисления молекУ']
лярным кислородом в жидкой фазе при умеренной температж^
алкановых, циклановых, циклано-ароматических, ароматическим
углеводородов с алифатическими боковыми цепями, алкеновых
циклоалкеновых углеводородов, а также простых алифатический]
эфиров.
130
Вк ночевие кислорода по связям С—С с образованием перекисей
F д______0-^0—R в начальной стадии окислительного процесса
Т,^П мио не наблюдается. Также не обнаружены (в качестве первичных
°оЬ1т vKTOb окисления) соединения, в которых кислород присоеди-
нится по двойным связям с разрывом их и образованием цикличе-
ских перекисей типа
R—CH—CH—R
.0
0
Таким образом, общим и характерным направлением для началь-
ной стадии автоокисления всех видов углеводородов является вклю-
чение молекулярного кислорода по связям С—Н с образованием
гидроперекисей.
Следующей после образования перекисей ступенью окисления
углеводородов являются превращения образовавшихся перекисных
соединений. Одним из главных превращений перекисей при авто-
окислении является их распад.
Распад перекисей и образование устойчивых продуктов проис-
ходит при не очень глубоком окислении; в этих условиях возможна
полимеризация перекисей. Процессы эти играют важную роль при
смолообразовании в топливах во время их хранения. Высокотемпе-
'Ж)атУРное окисление, протекающее в паровой фазе, происходит преи-
мущественно с образованием свободных радикалов, способствующих
дальнейшему развитию процесса.
При автоокислешш углеводородов возможны такие превращения
первичных продуктов окисления, как повторная пероксидация
гидроперекисей, присоединение карбонильных соединений с образо-
ванием оксиалкильных перекисей, внутримолекулярные перегруп-
пировки с образованием сложных перекисных соединений.
Направление промежуточных реакций окислительного процесса
углеводородов и его механизм в паровой фазе начальной (предпла-
менпой) стадии взаимодействия с кислородом требует уточнения.
Решение этого вопроса затруднено отсутствием достаточного коли-
чества экспериментальных данных, особенно для условий развития
процесса в двигателях, очень сильно влияющих на направление
Механизм первичных и промежуточных химических реак-
В большинстве реакций окисления в качестве главных активных
ромежуточпых продуктов образуются гидроперекиси. Цепь с уча-
I А рМуПеРекисного и углеводородного радикалов была разработана
. ' Уо(>ел°Де [7], который считает, что протекающая в объеме глав-
'-ХНМПЧеская реакция окисления насыщенного углеводорода начи-
Деп СЯ С °бРазования гидроперекиси, распадающейся затем на аль-
’прег^т.Н ВОДУ- Общую реакцию углеводорода с кислородом можно
RCH3 + O2—>RCH2—О—О—Н—>RCHO +Н20
9*
131
А. Р. Уббелоде считает, что цепной механизм окисления алканов
связан с цепным механизмом окисления альдегидов, протекающие
через радикал R—С=0, который образуется в реакциях:
PqZ активация
О
Радикал R—С=О реагирует с кислородом
Радикалы
взаимодействуют с углеводородами,образуя алкилрадикалы,которые,
в свою очередь, реагируют с кислородом, давая радикал перекиси,
снова взаимодействующий с углеводородом:
r R—СН2 F О2—>ИСН2—О—О— ^j|
R-CH2-0-0 F RCH3—>R—CH2-O-O-H + rch2- 1
Разветвление цепи может идти по реакции
/Н
R—СН2—О—О—+ RCH3-^R—С—О—н н—OH4-RCH2— 1
а также в результате распада перекиси:
RCH2—О—О—Н—>R—C<fН н—ОН
| хон
rch2—о—о—h->rch2—о— н—он
Схема окисления А. Р. Уббелоде в. дальнейшем подвергалась
уточнениям, изменениям и дополнениям.
А. С. Соколик [8, 9] приводит схему окисления углеводородов
с двумя путями вырожденного разветвления: 1) через образование
алкильных гидроперекисей и 2) через образование высших альдеги-
дов
RCH3 + 02->Н02- + RCH2— «
RCH2—+ 02->RCH2—О—О— I
RCH2—0-0— —~ R—СН2— O-O-Н —- R—CH2— О— + — ОН
(перекисная схема)
132
о
I RCH2-0-0-----> R^H^O-H-GHgCHO—-СН3С~О-О-Н
сн3соонн—о—
г Льюис и Г. Эльбе [10] разработали сложный цепной механизм
успения углеводородов, имеющий для высших алканов следующий
ВП^Возиикновение цепи:
стенка^ RC<^0_0_H
* R--1—ОН (альдегидная схема)
rcho + o2
стенка
О
Цепь окисления углеводорода:
-ОН + RCH3-»RCH2—Н Н2О
RCH2— + О2—>RCH2— О—О—
RCH2-0-0--->RiCHO +сн3о- —- со + Н2О + -ОН
. Цепь окисления альдегида:
-ОН + RCHO -> Н2О + RC=O -- НСНО + СО Н—ОН
В- I
£ -ОН + НСНО->НС=О + Н2О
нс=о+о2->н—0-0—+со
Н—0-0—1-НСН0-»Н20+С0Н—он
Обрыв цепи:
„тт стенка _тт
—ОН------* гибель ОН-радикалов
Разветвление цепи может идти через перекисные радикалы:
RCH2-O-O- + RCHO->RCH2~O-O-C-R — . —ОН
рк ‘' I
ОН
В этой схеме учитываются результаты исследования природы
перекисей, образующихся при окислении углеводородов, и характер
их термических превращений. Б. Льюис и Г. Эльбе за первичную
принимают реакцию образования альдегидов, являющуюся, по-ви-
Димому, каталитической. В. Иост [И] считает это возможным при
ольших задержках воспламенения, когда стенка играет заметную
Роль и малые добавки альдегидов сильно ускоряют реакцию и сокра-
~уают период задержки воспламенения. При высоких начальных
_ Пературах в условиях двигателя следует принять другой меха-
с • м возникновения цепей. Термический распад углеводородов
Усл ^а3°Ванием РаДикалов, видимо, в большей мере соответствует
ням воспламенения в двигателях. Исследования предпламен-
133
ных превращений углеводородов в условиях двигателя [12—14]
показали, что процессы деструкции углеводородов являются первич-
ным актом. Окисление осколков молекул в виде радикалов, непре-
дельных и активированных молекул — вторичное явление, идущее
по цепному механизму. Процессы деструкции находятся в непосред-
ственной связи с прочностью молекулярных структур углеводородов.
УСЛОВИЯ ВОСПЛАМЕНЕНИЯ СМЕСЕЙ ГОРЮЧЕГО И ОКИСЛИТЕЛЯ
Для осуществления воспламенения необходимы следующие усло-
вия:
1) горючая смесь должна быть в паро- или газообразном состоя-
нии;
2) вполне определенное соотношение между горючим и окисли-
телем;
3) в какой-то части или во всем объеме горючей смеси должна быть
достигнута такая температура или накопиться такое количество
активных промежуточных продуктов реакции, при которых возмож-
но быстрое прогрессивное самоускорение реакции окисления до
воспламенения.
Хотя воспламенение горючей смеси происходит в паровой фазе,
при определенных условиях большое значение для развития процесса
воспламенения приобретают жидкофазные химические процессы,
роль которых особенно важна для самовоспламеняющихся ракетных
топлив. При жидкофазном контакте некоторых горючих и окисли-
телей выделяется довольно значительное количество тепла и проис-
ходит образование активных промежуточных продуктов, переходя-
щих затем в паровую фазу. Таким образом, состав паровой фазы,
в которой происходит воспламенение, может отличаться от состава
горючего и окислителя в жидкой фазе.
Смесь горючего и окислителя может воспламеняться только при
определенных соотношениях между горючим и окислителем.
Высшим концентрационным пределом воспламенения называется
концентрация паров горючего в смеси с окислителем, при превышении
которой воспламенения смеси не происходит.
Низшим концентрационным пределом воспламенения называется
концентрация паров горючего в смеси с окислителем, при уменьше-
нии которой воспламенения также не происходит.
Состав смеси и пределы воспламенения могут быть выражены при
помощи трех различных величин:
1) концентрации паров горючего в окислителе или воздухе,
в % от общего объема;
2) содержания горючего в окислителе или воздухе, в г!м3\
3) коэффициента, представляющего собой отношение количества
окислителя или воздуха в горючей смеси к количеству, соответству-
ющему стехиометрическому соотношению между горючим и окисли-
телем.
134 "Я
И .воспламеняемость очень богатых и очень бедных смесей объяс-
* . । тем, что в этих смесях скорость протекания реакции окисле-
Г,НР,< W3ко падает вследствие того, что концентрация реагирующих
1,11)1 !тв и активных промежуточных продуктов реакции невелика,
ВС1()ТВ<)Д тепла в окружающую среду превышает тепловыделение
ппГреакЦИИ окисления.
1 На пределы воспламенения оказывают влияние химический со-
I в горючего и окислителя, температура, давление и турбулент-
сТ‘\’ь среды, концентрация и род инертного разбавителя, а также
Яощпость источника зажигания при принудительном воспламе-
нении .
Влияние химического состава топлива на пределы воспламенения
доказано в табл. 27.
Таблица 27
ппрпрлы воспламенения в воздухе для некоторых горючих веществ при 20° С
иреД [И, 15]
Вещество
Водород
Ацетилен .
Этилен . .
Пропилен .
Метан . . .
Пропан . .
Гексан . .
Циклогексан
Бензол . .
Пределы воспламе- нения, объемы. % Вещество Пределы воспламе- нения, объемы. %
нижний верхний нижний верхний
4,0 74,2 Толуол 0,92 5,0
2,5 80,0 Анилин 1,31 4,2 \
2,75 28,6 Аммиак 15,5 27,0
2,0 11,0 Диэтиламин . . . 2,2 14,9
5,0 15,0 Триэтиламин . . 1,5 6,1
2,37 9,5 Ксилолы (смесь
1,25 6,9 изомеров) . . . 1,15 5,53
1,33 8,35 Спирт метиловый 6,0 34,7 \
1,1 6,8 » этиловый 3,3 18,4 1
Наиболее широкие пределы воспламенения из приведенных соеди-
нений имеют ацетилен и водород.
Смеси алкановых, циклановых и ароматических углеводородов,
входящих в состав бензиновых и керосиновых фракций, имеют близ-
кие пределы воспламенения. Более широкие пределы имеют ненасы-
щенные соединения.
Повышение температуры горючей смеси увеличивает скорость
химических реакций, предшествующих воспламенению, и расши-
Ряет пределы воспламенения (рис. 53).
типичные кривые зависимости пределов воспламенения от давле-
ния представлены на рис. 54. Как видно из рисунка, существует опре-
деленное минимальное давление, ниже которого при любом составе
горючей смеси воспламенение невозможно. В этом случае нижний
орхний пределы воспламенения совпадают друг с другом. С повы-
шен температуры смеси это давление понижается.
ства аВ11СП“10СТЬ пРеДелов воспламенения от давления для большин-
аых СМесе® Углеводородов с воздухом имеет несколько четко выражен-
Л1111шмумов. Наличие нескольких значений предельного давле-
135
ния в области богатых смесей связано с образованием и гибелью ак-
тивных промежуточных продуктов предпламеиного окисления. При
повышении давления пределы воспламенения приближаются к не-
которым постоянным значениям. Я
С увеличением турбулентности горючей смеси пределы воспламе-
нения расширяются, если при этом в зоне воспламенения интенсифи-
цируются процессы передачи
оборот, они сужаются, если
тепла и активных продуктов
из зоны реакции окисления.
Пр ед ел ы во сп л а мен ени я
могут значительно изме-
няться при добавлении не-
которых веществ, которые
будут либо ускорять развитие
цепных реакций, приводя-
щих к воспламенению, либо
тормозить. При добавлении
тепла и активных продуктов, и, на-
турбулизация смеси вызывает отвод
Рис. 54. Зависимость пределов воспла-
менения углеводородов, выраженных коэф-
фициентами избытка воздуха а, от да-
вления:
Рис. 53. Зависимость пределов
воспламенения водорода и ме-
тана в воздухе от темпера-
туры.
1 — метан; 2 — бутан; 3 — гексан.
инертных разбавителей, способствующих увеличению теплоем-
кости и теплопроводности горючих смесей, пределы воспламенения
сужаются. 1
Увеличение энергии источника зажигания расширяет пределы
воспламенения. В литературе имеется мало сведений о пределах вос-
пламенения двухфазных смесей.
Пределы воспламенения туманообразиых горючих смесей при-
мерно такие же, как и смесей полностью испарившегося топлива
с воздухом.
Пределы воспламенения в гетерогенных смесях шире, чем в го-
могенных. Факел распыливания дает самые разнообразные местные
соотношения паров горючего и окислителя, в том числе и самые бла-
гоприятные для воспламенения. - ’
136
МЕХАНИЗМ ВОСПЛАМЕНЕНИЯ
г ’ тествуют два способа воспламенения горючих смесей. Первый
Г заключается в том, что горючая смесь зажигается в одной
спосо(> пр0СТранстВа посредством какого-либо высокотемператур-
Т°ЧЬ<источника и дальнейшее воспламенение всего объема газообраз-
горючей смеси происходит без внешнего вмешательства, само-
Н°И тзвольно — принудительное воспламенение.
ДР°Во втором способе всю горючую смесь доводят до такой темпе-
ы при которой самопроизвольное бурное развитие реакций
₽киспения приводит к воспламенению без внешнего воздействия —
самовоспламенение. „
Следует отметить, что воспламенение может произойти и без
предварительного температурного воздействия на горючую смесь,
а в результате накопления в системе активных промежуточных про-
дуктов реакции. Этот тип воспламенения возможен только для
вполне определенных горючих смесей.
Несмотря на различие в параметрах, характеризующих процессы
принудительного воспламенения и самовоспламенения, физико-хи-
мическая природа их одинакова. Воспламенение горючей смеси свя-
зано с очень быстрым переходом от медленной реакции к резкой,
почти мгновенной. Это происходит тогда, когда в реагирующей
смеси создаются условия, при которых возможно прогрессивное
ускорение химических реакций.
В зависимости от того, какие причины вызывают прогрессиру-
ющее самоускорение реакции, различают следующие виды воспла-
менения: тепловое, цепное и цепочно-тепловое.
Сущность теплового воспламенения можно рассмотреть на следу-
ющем примере. Пусть некоторый объем горючей смеси V заключен
в сосуд, стенки которого имеют постоянную температуру TQ. В ре-
зультате химической реакции между горючим и окислителем темпе-
ратура смеси возрастает и делается равной Т. Возрастание темпера-
туры реагирующей смеси вызывает увеличение скорости реакции
по закону Аррениуса пропорционально е . Следовательно, коли-
чество тепла, выделяющегося при реакции в единице объема в еди-
ницу времени, равно:
qL = Ae RT
где А коэффициент пропорциональности.
?1асть тепла, выделяющегося при реакции, идет на нагревание
Другая отводится через стенки сосуда.
оличество тепла, отводимого из реакционного сосуда в единицу
смени с единицы объема, будет:
<7г =(Г - Го)
137
где « — коэффициент теплоотдачи от газа к стенке, вт/м2-град; I
S — поверхность сосуда, ж2; ’.Я
V — объем реакционного сосуда, м3. 1
Тепловой баланс реакции может оказаться как благоприятным, j
так и неблагоприятным для ее развития и стабилизации. В этом легко i
уоедиться, сравнивая характер температурных зависимостей тепло’
выделения и теплоотвода. Зависимость
Т — экспоненциальная.
q2 от T линейная
а
71 от
Рис. 55. Изменение скоростей выделе-
ния и отвода тепла в процессе химиче-
ских превращений различных горючих
смесей при постоянной температуре
стенок сосуда.
ратуры Тъ -------
отвод. В точке 1 ± нагревание прекратится и система придет в равно-
Наклон прямой теплоотвода зависит от численного значения
множителя —р- , а местонахождение линии теплоотвода в системе
координат q — Т от температуры стенки 710.
На рис. 55 представлена система кривых qY = f (71) для трех
различных горючих смесей при постоянных условиях отвода тепла
Рис. 56. Изменение скоростей выде-
ления и отвода тепла в процессе хи-
мических превращений горючей сме-
си при различных температурах
стенок реакционного сосуда.
Для случая, когда выделение тепла определяется кривой 7/ =
— / (7), начиная с температуры То смесь разогревается до темпе-
ратуры 71т так как до этого момента выделение тепла превышает
отвод. В точке 7 х нагревание прекратится и система придет в равно-
весие. Реакция, дальше будет идти сначала с постоянной скоростью,
а затем при снижении концентрации исходных веществ скорость
реакции начнет уменьшаться.
Если выделение тепла при реакции характеризуется кривой
71 — /(7), то теплоотвод меньше тепловыделения и температура
реагирующей смеси будет непрерывно возрастать; в результате ско-
рость реакции быстро достигнет очень высоких значений и вызовет
самовоспламенение смеси.
138
Таким образом, существует определенное условие перехода от
случая ограниченного повышения температуры реагирующей смеси
к случаю, когда ее температура быстро растет и достигает весьма
высокого значения, близкого к теоретической температуре горения.
Это условие соблюдается для средней кривой q{ = j (71). В точке
касания кривых qY и q2 имеют место равенства:
ff
Эти два условия однозначно определяют температуру воспламе-
нения или самовоспламенения горючей смесп 7ЛВ.
В рассмотренном примере неизменной поддерживалась темпера-
тура стенки сосуда То. При заданной температуре стенок сосуда
только при определенных значениях интенсивности реакции, со-
става горючей смеси и давления может наступить самопроизвольное
возрастание температуры и быстрое ускорение реакции до воспламе-
нения.
На рис. 56 приводятся зависимости и q2 от температуры для
случая, когда при заданном составе горючей смеси и давлении ме-
няются условия отвода тепла за счет изменения температуры стенок
сосуда. При температурах стенки выше То происходит воспламене-
ние, при температурах ниже То возможен лишь стационарный разо-
грев.
В практике за температуру воспламенения часто принимают ту
наинизшую температуру стенок сосуда, при которой в данных усло-
виях происходит воспламенение заданной горючей смесп. В рас-
смотренных примерах такой температурой является температура
70. Очевидно, что 7В и 70 отличаются друг от друга, причем всегда
Т^в > 70.
Температура воспламенения не является константой, характе-
ризующей горючую смесь, так как опа зависит от условий определе-
ния. Точка касания кривых qr и q2 может смещаться в зависимости
от величин S и а, характеризующих параметры экспериментальной
установки, начальных давлений, температуры и других факторов.
Гаким образом, сущность теплового воспламенения заключается
в следующем. Постепенное повышение температуры горючей смеси
при постоянном давлении или повышение давления при постоянной
температуре могут привести к такому возрастанию скорости хими-
ческой реакции окисления, что скорость выделения тепла начнет
превышать скорость теплоотдачи в стенки. В результате этого нач-
^^||( |(я прогрессирующий саморазогрев горючей смеси, приводящий
ч к еще большему самопроизвольному возрастанию скорости реакции
практически к мгновенному выделению теплоты реакции в неболь-
шом объеме газа — воспламенению [16].
Ьольшинство химических реакций, в том числе и горение, яв-
Ются сложными и идут через посредство активных промежуточных
к пРоДуктов.
139
В гомогенных реакциях первой стадией является обычно образова-
ние из исходных веществ тех или иных активных промежуточных
продуктов, природа которых может быть различной. Во многих слу-
чаях роль активных промежуточных продуктов играют свободные
радикалы или молекулы повышенной реакционной способности
например органические перекиси. J
Цепные процессы могут протекать по двум различным режимам:
стационарному и нестационарному. Изменение концентрации
активного промежуточного продукта X со временем подчиняется
кинетическому уравнению:
dX
dx
= n0 + fX-gX
где n0, f,g — константы скорости зарождения, разветвления и об-
рыва цепей;
т — время реакции.
При g >> / имеет место стационарный режим протекания реак-
ции. Концентрация активного продукта X с течением времени стре-
мится к стационарному значению:
После достижения этого значения концентрация активного про-
дукта будет оставаться постоянной, а реакция будет протекать
с неизменной скоростью:
iv = KX
где К — константа скорости продолжения цепи.
Величины /г0, / и g зависят от концентрации исходных веществ
и поэтому изменяются по мере протекания реакции, а вместе с ними
изменяется величина X. Поэтому последнюю правильнее называть
не стационарной, а квазистационарной концентрацией активного
продукта. При / > g имеет место нестационарное протекание
реакции.
Решение уравнения
имеет вид:
dX
dx
[е(/-г)т_и
Начальный период реакции, когда концентрация активного про-
дукта неизмеримо мала, называется периодом задержки. Продолжи-
тельность этого периода по времени порядка —Д— . Далее при т
*
концентрация активного продукта и скорость реакции будут
возрастать со временем по экспоненциальному закону — пропор-
ционально величине , ,1
140
р считать концентрации исходных веществ постоянными, то
‘ возрастание будет неограниченным. Фактически предел ему
эТ° л положен израсходованием запаса исходных веществ.
» ^Если с изменением условий протекания реакции начинает уве-
чиваться константа /, то при / = g будет происходить резкое из-
лИ ние скорости реакции — переход от стационарного к нестацио-
^^ппому режиму — цепному воспламенению.
Яа Чисто цепной механизм воспламенения наблюдается при сравни-
льно низких давлениях и только для некоторых горючих смесей.
Воспламенение при атмосферном и более высоком давлениях является
тепловым или, чаще всего, цепочно-тепловым.
Сущность цепочно-теплового воспламенения заключается в сле-
дующем. Если цепная реакция развивается очень медленно, то ис-
ходные продукты успевают израсходоваться настолько, что скорость
процесса начнет снижаться, не достигнув критического взрывного
значения. Такой процесс Н. Н. Семенов назвал «вырожденным взры-
вом». Это имеет место, когда разветвление цепи происходит не через
активные центры, образующиеся непосредственно в реакционных
звеньях цепи, а в результате распада относительно устойчивых про-
межуточных продуктов. Причем образуются свободные радикалы,
которые начинают новые цепи, но развитие цепей пойдет тем мед-
леннее, чем устойчивее промежуточные продукты. Однако в резуль-
тате медленно нарастающей скорости цепного процесса может быть
достигнута скорость тепловыделения, превышающая скорость тепло-
отдачи в окружающую среду. При таких условиях становится воз-
можным прогрессивное саморазогревание смеси и дальнейшее уско-
рение реакции не от разветвления цепи, а от повышения температуры.
Такой вид воспламенения, при котором медленно развивающаяся
цепная реакция самоускоряется сначала за счет образования актив-
ных продуктов, а затем за счет выделяющегося тепла, называется
цепочно-тепловым. При цепочно-тепловом воспламенении цепное
ускорение реакции играет роль предварительного процесса, созда-
ющего условия для теплового воспламенения.
В зависимости от условий воспламенения и химической природы
топлива цепочно-тепловое воспламенение имеет различную физико-
химическую основу и кинетический механизм.
При низких давлениях в начальной стадии процесса большую
рольиграют диффузия и гибель активных центров реакции на стен-
ках, а тепловая сторона явлений не имеет большого значения из-за
значительного превышения теплоотдачи над тепловыделением.
Воспламенение, происходящее при атмосферном и более высоких
давлениях,
Т(Ламя в°зникает после непрерывного самоускоренпя реакции
сад >М паиболее длительную часть x^xx.^v,
Част?( |\°Рение» а тепловое занимает сравнительно незначительную
ным * 1 ВЬ1сокотемпературном воспламенении становится возмож-
своб РазРыв внутримолекулярных связей молекул и образование
Дных радикалов, которые являются начальными центрами —
имеет свои особенности. При высоких температурах
:, в ко-
по времени составляет цепное
141
инициаторами развития цепной реакции, ускоряющейся во времени I
Чем выше начальная температура реагирующей смеси, тем больнш
образуется начальных активных центров реакции. При высоко!
температурном воспламенении происходит непрерывное самоуско-
рение реакции, обусловленное вначале главным образом развитием
цепного процесса, а затем разогреванием смеси. *4
В этом процессе нельзя провести границу, отделяющую цепное
самоускорение от теплового. Поэтому его называют одностадийным
процессом воспламенения. Высокотемпературное воспламенение _
всегда точечное, а не объемное. Предпламенные реакции высокотем-
пературного воспламенения имеют высокую энергию активации.
Вследствие этого, если в смеси имеются даже относительно неболь-
шие колебания температуры, то скорость реакции будет настолько
снижаться с понижением температуры, что воспламенение всегда
возникнет в точке максимальной температуры раньше, чем в осталь-
ном объеме. *
В области относительно низких температур процесс воспламене-
ния развивается за несколько стадий. При низких температурах
невозможен распад молекул и образование таким путем значитель-
ного числа активных радикалов. Вместо этого возникают активные
• промежуточные соединения. |
При самовоспламенении углеводородных горючих в среде кисло^
рода при условии низких начальных температур «горячее» пламя
возникает после предварительного образования так называемого
«холодного» пламени. Развитие предпламенных реакций в этом слу-
чае можно представить в виде следующей примерной схемы. 1
В результате редких взаимодействий углеводородных молекул
с кислородом создается некоторое количество свободных радикалов-
обломков углеводородных молекул, обладающих высокой химиче-
ской активностью: / ЛЯ
R-CH3 + О2-»НО2— 4 R-CH2— 1
Радикалы, реагируя с кислородом, дают перекисные радикалы:
R—СН2---------|-О2—>R—СН2—О—О— (зарождение цепи) I
9
Перекисные радикалы при взаимодействии с углеводородной
молекулой образуют гидроперекиси: J
R—СН2—О—О—J-RjH—>R—СН2—О—О—H-f-Ri— (продолжение цепи)
Гидроперекиси — относительно устойчивые активные промежу-
точные продукты; при их распаде образуется несколько свободных
радикалов, дающих начало новым самостоятельным реакционным
цепям, например: $
ц_СН2—О—О—Н—>—OH + R—J-HCHO i
R-CH2-О-О-НН—OH->R—+ Н—О—О—4-СНз—О— Ч
сн3—о—+о2—>со + Н20 4-—ОН я
СН3—О—+ о2—►HCIIO-j-H—О—О— я
F Г >иные радикалы, образующиеся при распаде перекисей,
С®01" ' расПад новых молекул перекисей при столкновении с ними.
выз1’11’2 3"1 ‘кисец дополнительно усиливается под влиянием разогре-
распад 1 Когда в ходе реакции распад перекисей приобретает
/бразный характер, образуется «холодное» пламя. При этом
ла»я”° т*я относительно небольшая доля энергии сгорания топлива
уделяй _ даль11ейшее развитие окислительного процесса и на-
тепла приводят к воспламенению и появлению обычного
«горячего» пламени.
ВЛИЯНИЕ СВОЙСТВ ТОПЛИВА И УСЛОВИЙ ВОСПЛАМЕНЕНИЯ
НА ПАРАМЕТРЫ ПРОЦЕССА
К При любом механизме процесса воспламенения появлению пла-
ни предшествует период относительно медленного нарастания
^орости реакции, соответствующий разогреванию горючей смеси
в условиях медленного повышения температуры при тепловом воспла-
менении и изотермическому саморазгону реакции вследствие раз-
ветвления цепей при цепном воспламенении. Этот период называется
периодом задержки воспламенения.
Температура воспламенения и период задержки воспламенения
являются важнейшими параметрами, характеризующими воспламе-
няемость топлива. Однако обе эти величины не являются физическими
константами, так как зависят не только от свойств топлива, но и от
состава смеси, начальных значений давления и температуры, условий
теплообмена, размеров и материала аппаратов, в которых происхо-
дит воспламенение, а также мощности источника зажигания (при
принудительном воспламенении).
Склонность-горючих смесей к воспламенению зависит от свойств
как горючего, так и окислителя. Смеси горючего и окислителя, об-
ладающие большой реакционной способностью, обеспечивающие
быстрое образование активных промежуточных продуктов реакции,
воспламеняются с небольшой задержкой.
Самовоспламенению горючих при контакте с окислителями пред-
втествует ряд промежуточных реакций, которые для самовоспламе-
няющихся горючих смесей начинаются при обычных условиях и про-
текают с большой скоростью. В результате первоначального энергич-
ного разогревания реакционной смеси инициируются последующие
нредпламенные реакции окисления, приводящие к самовоспламене-
нию.
Например, методом спектрального анализа [171 установлено,
при жидкофазном контакте между азотной кислотой и аминами
^повременно протекают три типа реакций.
ими ^Чеиь быстрая реакция нейтрализации с образованием нитрат-
2) Реакции ионного характера, которые приводят к образованию
У тР?наиных и питрозированных алифатических нитратов, мало-
То,,чпвых при повышенной температуре. Эти соединения можно
143
рассматривать как инициаторы воспламенения, причем они активцЛ
как в жидкой, так и в паровой фазах. Необратимые реакции ионного
характера протекают с меньшей скоростью, чем реакции нейтрали,
зации, и имеют в качестве инициаторов катион NO2, содержащийся
в HNO3, и катион NO+, который образуется при восстановлении
азотной кислоты с помощью аминов и нитрозированных производи}.^
3) Реакция восстановления окислителя, взаимодействующего
с ионом NO+ при окислении амина и нитрозированных производных.
Взаимодействие в жидкой фазе между азотной кислотой и три-
этиламином может быть представлено следующей упрощенной схе-
мой [17]:
С2Н5
5 ^2^5
+ HNO3?+ \
Триэтиламин может вступать в реакцию с NO2 и NO , образуя
инициаторы воспламенения — нитрозоамин и нптратэтил. Скорость
этих реакций очень мала. Ж
При образовании соли нитрата триэтиламина выделяется тепла
достаточно для того, чтобы началось разложение образовавшихся
инициаторов воспламенения и определенное количество HNO3 ii
(C2H5)3N превратилось в пар. По достижении определенной темпе-
(C2H5)3N превратилось в пар. По достижении определенной
ратуры реагирующие компоненты воспламеняются. 1
Взаимодействие в жидкой фазе между азотной кислотой и кси-
лидином может быть представлено следующей схемой [17]: I
Ксилидины и нитраты ксилидинов при взаимодействии с NO2
и NO+ образуют достаточно большое количество инициаторов воспла-
менения в жидкой фазе в результате реакций нитрования и нитро-
зирования. Тепла, выделяющегося при образовании соли нитрата
ксилидина, недостаточно, чтобы произошло воспламенение смеси,
а образование инициаторов воспламенения дает необходимое раз-
витие реакции до воспламенения.
Зависимость между периодом задержки самовоспламенения и хи-
мическими свойствами топлива не всегда может быть точно опреде-
лена вследствие того, что трудно учесть влияние гидродинамическиflj
факторов на период задержки самовоспламенения в различных усло-
виях испытания. Наиболее сложно эта зависимость проявляется
при самовоспламенении распыленных жидких компонентов ракет-
ных топлив. Тем не менее на основании имеющихся эксперименталь-
ных данных можно вывести некоторые общие закономерности.
144
состав горючих, образующих самовоспламеняющиеся топлива
>тной кислотой, могут входить алифатические и ароматические
° а гы гидразин и его алкилзамещенные производные, многоатом-
аМИ фенолы, некоторые гетероциклические соединения, ненасыщен-
ные углеводороды с несколькими непредельными связями и другие
вещества [18].
' Первичные ароматические амины с азотной кислотой самовоспла-
еняются лучше, чем вторичные и третичные. У алифатических ами-
ов наоборот, лучше самовоспламеняются с HNO3 третичные амины,
чем вторичные и первичные. Углеводороды с одной двойной или
тпойной связью не дают хорошего самовоспламенения с азотной
О _ я-Я л л ГЧ гг Ъ'У я-r wwr -W -» к-r-r ТТ гч ТТ ТЧ XX ГТ ZX. ТТ Т ТТ Т ТТ гтт XX Гл ГТ о ГТ ж я тж
В последнее время уделяется боль-
кислотой, а соединения с сопряженными непредельными связями
самовоспламеняются хорошо. В последнее время уделяется боль-
шое внимание самовоспламеняющимся топливам на основе азотной
кислоты, окислов азота и горючих, имеющих в своем составе гидра-
зин и метилгидразины [19—21 ], так как они обладают очень хорошей
воспламеняемостью. Из алкилзамещенных гидразинов большой ин-
терес представляют метилгидразин, симм- и нвсггл<.и-диметилгидра-
зин. Хорошее самовоспламенение с азотной кислотой дают ненасы-
щенные амины и меркаптаны.
Следует отметить, что многокомпонентные горючие иногда имеют
иную способность к реакции, чем их отдельные составляющие.
Например, смесь анилина с циклогексиламином воспламеняется
с азотной кислотой лучше, чем каждый из компонентов в отдельности.
Добавка небольшого количества анилина к винилбутиловому эфиру
увеличивает скорость реакции с азотной кислотой, а добавка оле-
финов или виниловых эфиров к аминам тормозит реакцию [18].
Таблица 28
Период задержки воспламенения для топлив на основе азотной
кислоты при испытании на малоразмерном ракетном двигателе [22]
Горючее
Окислитель
Начальная
температура
топлива, °C
Период
задержки вос-
пламенения,
т• 10_3 сек
Гидразин
Гидразин
Фурфуриловый
спирт
Аммиак+ 14,1 %
гидразина
Аммиак+5,о%
гидразина
96%-ная HNO3 +
+ NO2 (76 : 24)
96%-ная HNO3
96%-иая HNO3
96%-ная HNO3 +
+ NO2 (76:24)
96%-ная HNO3 +
+ NO2 (76 : 24)
21
21
21
-36
от —37 до
-47
3,1 ±1,4
5,0 ±1,7
16,6 ±2,4
6-10
37
Влияние
Нос состава самовоспламеняющихся топлив
в за^еР>кк*и воспламенения иллюстрируют данные
ь табл. 28.
на длитель-
при веденные
Ю Заказ 792
145
Способность к самовоспламенению можно улучшить за счет при-
садок к топливу. В качестве присадок, катализирующих процесс
окисления, могут быть использованы некоторые неорганические
и органические соли металлов с переменной валентностью. Неорга-
нические соли растворяются в окислителе, а органические — в го-
рючем. В том и другом случаях каталитическое действие оказывают
катионы металлов. 4
В табл. 29 приведены некоторые катализаторы, которые вводятся
в окислитель для улучшения воспламенения ракетных топлив,
а в табл. 30 показана эффективность действия двух катализаторов.
Таблица 29
Катализаторы, улучшающие воспламенение ракетных топлив [18]
Катализатор
Формула
Топливо
Хлорное железо
Нитрат железа
Соли органических
кислот
Me дно синеродистый
калий
Окислы п соли меди
FeCl3-6H2O
Fe(NO3)3
Ме(СОО)2_3
K3[Cu(CN)4]
Азотная кислота —
амины, непредельные
углеводороды
То же
»
CuO, Cu2Cl2
Перекись водорода —
гидразингидрат, ме-
тиловый спирт
Азотная кислота — пе-
рекись водорода, пе-
' предельные углеводо-
роды
Таблица 30
Влияние катализаторов на период задержки воспла-
менения топлива (90%-ная Н2О2 и N2H4-H2O) при
начальной температуре топлива 20° С [23]
Катализатор
Концентрация ионов
Gu2+ или Fe3+, г/л
Период задержки
самовоспламенения,
т • 10-3 сек
K3[Cu(CN)4]
Na2[Fe(CN)5NO]
0,01
0,10
1,00
0,19
0,47
0,93
1,87
Нет воспламене-
ния
90
21
87
67
36
22
Влияние химического строения углеводородов на температуру
и период задержки воспламенения в среде воздуха изучено доста-
точно подробно. Наиболее низкие температуры воспламенения /в
имеют алкановые углеводороды, паиболее высокие — ароматиче-
ские. При увеличении молекулярного веса углеводородов 7В умепь-
146
>тСя Температура воспламенения алцанов быстро понижается
щченпем числа атомов углерода в молекуле от 3 до 8; дальней-
с ^увеличение молекулярного веса от С8 до С16 практически не со-
111 лвождается изменением Тв. С увеличением разветвления цепей 7В
ПдКапов и алкенов повышается; для углеводородов с прямой цепью
а пичие двойной связи повышает температуру воспламенения. Тем-
----------------------- ------ -------------- ------- -----чем
Для алкилбензолов Тв пара-изомеров
чем для соответствующих орто-производных.
снижению температуры воспламенения и сокращению
р* )атура воспламенения сильно разветвленных алкенов ниже
соответствующих алканов. Д™ т
выше,
L Зависимость периода задержки самовоспламенения от числа ато-
• мов углерода в молекуле алкановых, алкеновых и циклановых угле-
водородов представлена на рис. 57.
Основными факторами, определяющими условия воспламенения,
являются: начальные температура и давление, состав смеси, гидро-
динамические факторы, влияющие на подачу и смешение жидких
топлив, а также подвод и отвод тепла в ходе химических реакций.
Период задержки самовоспламенения жидких ракетных топлив
включает в себя время развития физических процессов Тф начальной
стадии процесса воспламенения и время протекания химических
реакций тх процесса воспламенения.
С изменением начальной температуры горючей смеси изменяется
^скорость химических реакций. Повышение температуры увеличи-
вает скорость предпламенных реакций окисления и скорость смеши-
вания при воспламенении распыленных жидких топлив, что при-
водит к
длительности задержки воспламенения. Влияние начальной темпера-
туры на период задержки воспламенения особенно сильно проявляется
при низких температурах: оно тем сильнее, чем хуже воспламеняе-
мость топлива. При высоких температурах влияние химической при-
роды топлива проявляется в меньшей мере, чем при низких. В случае “
воспламенения распыленных жидких топлив при низких температу-
рах большую роль играет Тф, т. е. время, необходимое на физические
процессы подготовки топлива к воспламенению. Эта величина за-
висит от физических свойств топлива. При низких температурах
сильно увеличиваются вязкость, поверхностное натяжение, умень-
шается давление насыщенных паров и в результате этого уменьшается
I эффективность смешения.
Существенное влияние на параметры воспламенения оказывает
Давление. Выполнено большое количество экспериментальных ис-
следований для изучения влияния низких начальных давлений ок-
ружающей среды па период задержки самовоспламенения. Устано-
лЛеиО’ Что У самовоспламеняющихся топлив наблюдается значитель-
Увеличение задержки воспламенения с уменьшением давления,
I при очень малых давлениях топлива не воспламеняются.
акая же картина наблюдается при самовоспламенении капель
|на^ЮЧеГ° В В03ДУХе (рис. 58). Изменение давления сильнее влияет
Те Тф 11 Меныпе на тх. Понижение давления ухудшает условия про-
t НИя химических реакций как в жидкой, так и в паровой фазах.
I
13 работе [24] приведена зависимость между периодом задержки
воспламенения т и давлением Р для некоторых ракетных топлив.
пример, для топлива триэтиламин — азотная кислота тР1,2 = const
Снижение концентрации реагирующих веществ и менее тесный
контакт между молекулами горючего и окислителя, обусловленные
понижением давления, уменьшают
скорость предпламенных процол)
Рис. 57. Зависимость периода за-
держки самовоспламенения т угле-
водородов от числа атомов углерода
в молекуле: испытание в бомбе при
+582° С и давлении 20,7 • 105 н/л2
1 — н-алканы; 2 — циклогексаны;
3 — н-алкены.
2
Р, атм
Рис. 58. Зависимость периода за-
держки самовоспламенения т капель
керосина с начальным диаметром
70 мк от давления при различных
температурах воздуха [18].
сов окисления, вызывая повышение температуры воспламенения
газообразных горючих смесей.
С изменением состава горючей смеси значительно изменяются
условия воспламенения, и прежде всего химическая активность и фи-
зические свойства горючей смеси. Наиболее оптимальные условия
воспламенения создаются в несколько обогащенных смесях. Обога-
щение и обеднение смеси относительно оптимального состава увели-
чивает период задержки воспламенения. Особенно сильно изменяется
период задержки воспламенения вблизи пределов воспламенения.
Имеющихся экспериментальных данных по влиянию гидродипам.й|/
ческих факторов на период задержки воспламенения недостаточно
для того, чтобы дать количественную оценку. Уменьшение длитель-
ности задержки самовоспламенения с увеличением давления впрыска
наблюдалось в определенном интервале давлений при испытании
в закрытых камерах для различных горючих с азотной кислотой
148
„рстве окислителя, а также для системы гидразин — перекись
Р ГОГ I Qnn/^гт, ГаЛлЛагчТТТп ЛПГТППТТ Т> ТТТТ ГГТТКАВ1 ГТ<ГГТЛАТТТТТГП»<Т1Т7/хЛТ,т1х,
подороДа
фактор»1’
ванне
[25 ]. Этот эффект связан с влиянием гидродинамических
С повышением давления впрыска улучшаются распылп-
и смешение компонентов, происходит нагревание струй при
Однако очень мелкое распыливание компонентов самовоспла-
^яюгцихся топлив ухудшает условия развития жидкофазных реак-
Ш.. и приводит к увеличению задержки самовоспламенения. '
ТуРбУлизация ПРИ смешении горючего и окислителя как в паро-
так и жидкой фазах может либо ускорять развитие предпламен-
химических процессов, либо их тормозить (в зависимости от
ого, как осуществляются подвод и отвод тепла и активных промежу-
точных продуктов в зоне воспламенения), поэтому турбулизация
горючей смеси в одних условиях приводит к увеличению периода
задержки воспламенения, а в других — к уменьшению.
вс и,
цых
2)
МЕТОДЫ ОЦЕНКИ ВОСПЛАМЕНЯЕМОСТИ ТОПЛИВ
Для оценки воспламеняемости топлив необходимо определить:
1) пределы воспламенения;
температуру воспламенения;
период задержки воспламенения;
минимальную критическую энергию зажигания в случае при-
нудительного воспламенения.
Трудность непосредственной оценки воспламеняемости топлив
по этим параметрам заключается в том, что они, как было указано
выше, сильно зависят от условий определения. Для более полной
оценки необходимо определить эти параметры при разных значениях
температуры, давления, состава смеси и т. д.
В настоящее время предложено большое количество эксперимен-
тальных методов для оценки воспламеняемости топлив.
Пределы воспламенения определяют в сосудах или бомбах,
в которых горючую смесь определенного состава нагревают до само-
воспламенения или поджигают принудительно при заданных темпе-
ратуре и давлении. О воспламенении судят по изменению темпера-
туры и давления или появлению пламени.
Для определения температуры самовоспламенения газовых горю-
чих смесей используются следующие три основных метода:
J) метод впуска заранее подготовленной горючей смеси в нагре-
тый сосуд;
2) метод нагревания компонентов смеси раздельно с последующим
Решением внутри нагретого сосуда;
°) метод адиабатического сжатия заранее подготовленной
WBch.
Ряд других методов являются по существу разновидностями
УИазанпых выше.
°рвый метод наиболее прост и заключается в том, что смесь
J состава с определенной начальной температурой и давле-
яоступает в нагретый сосуд, из которого выкачан воздух.
Энного
иием
Нагревая сосуд, находят такую температуру, при которой процСуД
дит воспламенение. \
По второму методу компоненты горючей смесп пропускают череД
отдельные трубки, в которых они нагреваются до одинаковой темп^Я
ратуры. На выходе из трубок оба компонента смешиваются в сосуд0ш
и, если температура достаточна, происходит воспламенение. Тем^Д
ратура, соответствующая моменту появления пламени, принимает^}*
за температуру самовоспламенения.
При определении температуры самовоспламенения методом адиа-
батического сжатия горючую смесь определенного состава вводя?
в металлический цилиндрический сосуд с поршнем. При быстром
движении поршня смесь нагревается за счет работы сжатия. Повы-
шая степень сжатия, можно вызвать самовоспламенение смеси. За
температуру самовоспламенения принимают низшую температуру
конца сжатия горючей смеси, при которой смесь воспламеняется.
При оценке полученных данных надо учитывать возможность
искажения результатов испытаний за счет воздействия посторонних
факторов и изменения условий воспламенения. Я
Большие трудности вызывает определение периода задержки
воспламенения. Особенно трудно определить момент начала уско-
рения реакций взаимодействия между горючим и окислителем,
поэтому в большинстве случаев определяется условный перр^ч I
задержки воспламенения. В зависимости от условий и способа воспла-
менения за начало периода берется момент поступления смеси в со-
суд, момент начала контактирования горючего и окислителя, момент
включения источника зажигания и т. д. Начало воспламенения
фиксируется фотографированием или с помощью фотоэлементов.
Для этой цели можно использовать также ионизацию смеси при
воспламенении. Наиболее часто в практике применяют метод изме-
рения температуры или давления реагирующей смеси. Начало вос-
пламенения определяют по линии перехода от плавного к резкому!
изменению этих величин. Я
Наиболее объективные данные о воспламеняемости топлив, харак-11
теризующие его поведение в двигателе, дают испытания на СП0Д
цпальных установках с малоразмерными двигателями. На такие
установках, кроме периода задержки воспламенения, можно опр1Я
делять целый ряд других параметров, характеризующих воспламе-Я
пение топлива в двигателе, например скорость нарастания давления,
максимальное давление при воспламенении и др. Я
Для относительной оценки воспламеняемости топлив широко^
применение находят условные показатели, определяемые в стандарте
пых условиях воспламенения. Например, вместо концентрационная
пределов воспламенения в ряде случаев удобнее и проще определЯу i
температурные пределы взрываемости. *Я
Нижним температурным пределом взрываемости называется цЯ
низшая температура горючего, при которой его насыщенные парЧ
образуют с воздухом в замкнутом сосуде смесь, способную воснЛЯЯ
меняться от источника зажигания. Концентрация паров при нижНеЯ
150
натурном пределе взрываемости является нижним концентра-
ционным пределом.
Д Непхним температурным пределом взрываемости называется та
пая температура горючего, при которой насыщенные пары его
чзуют с воздухом в замкнутом сосуде смесь, способную воспла-
X» яться от источника зажигания. Концентрация паров при верх-
ам температурном пределе взрываемости является верхним кон-
центрационным пределом.
Д Зная давление насыщенных паров при температуре, соответству-
ющей температурному пределу взрываемости, можно определить
концентрационные пределы:
„ Pt •100
Где (J ___ концентрационный предел взрываемости, объемн. %;
pt — давление паров горючего при температуре, соответству-
ющей температурному пределу взрываемости;
— атмосферное давление при определении температурных
пределов.
Нижний температурный предел взрываемости по существу совпа-
дет с понятием температуры вспышки. Температура вспышки опре-
деляется в открытых или закрытых приборах. Для горючих с темпе-
ратурой вспышки более 70° С определение производится в открытых
приборах.
' Температура вспышки лежит в основе классификации жидкостей
по степени их пожароопасности. Жидкости с температурой вспышки
ниже 45° С относятся к легковоспламеняющимся, а выше 45° С —
к горючим.
I В качестве характеристики огнеопасности определяют темпера-
туру воспламенения, как самую низкую температуру горючего,
при которой смесь его паров с воздухом в стандартных условиях
испытания после поджигания горит ровным пламененем не менее
5 сек.
Для легковоспламеняющихся жидкостей
_ j° выше температуры
температура воспла-
вспышки, для оста л ь-
20°.
взрывоопасности для
Значения основных показателей огне-
менения обычно на 1—5° выше температуры
ых этот интервал может колебаться от 5 до
I п ^наченпя основных показателен огне- и
Ряда горючих приведены в табл. 31.
т ^гне- и взрывоопасность компонентов ракетных топлив зависят
fa 5Ке от их склонности к детонационным взрывам под воздействием
Цж°ТоРЬ1Х внешних импульсов. Взрывные свойства этих компонен-
к Мпли^0ВеРяют по восприимчивости к внешним воздействиям —
к трению, прострелу пулей и дето-
Пя1,.'Х'11111ЧесК1,м УДарам на копре,
«’Дионны
м импульсам.
о6РетатьТОРЬ1е горючие
ЯП||
склонные к электризации, способны при-
иотенциал электрического заряда при трении, распылива-
РУн и ударе о твердую поверхность, а также при прохождении
151
7 блица ^2
Показатели огне- и взрывоопасности для некоторых горючих [15]
Горючее
7 Анилин.........
Бензол..........
Ксилидины (смесь
- меров) ....
Метиловый спирт
Этиловый спирт .
Триэтиламин . .
Диэтиламин . . .
Автобензин А-74
» А-66
Авиабензин Б-70
Топливо Т-1 . .
Дизельное топливо
» »
Мазут флотский
Авиационное масло
МК-22 .........
ИЗО-
ДЗ
Температура
самовоспла-
Темнература
Температурные пределы
взрываемости, °с
менения па- вспышки, по
ров в воздухе ГОСТ 6356—52,
(метод
«капли»), °C
620
500
510
300
300
240
310
390
380
°C
79
-14
98
13
-12
-36
-39
78
71
158
нижний
270
-14
80
11
-12
-39
69
106
верхний
90
12
103
39
40
14
8
57
119
100
133
через пористые и сетчатые преграды, например фильтры. Этой
способностью обладают жидкости-диэлектрики, например углеводо-
родные горючие. В
ПАРАМЕТРЫ, ХАРАКТЕРИЗУЮЩИЕ ПРОЦЕСС ГОРЕНИЯ
Вопрос о распространении пламени в горючей смеси является
одним пз наиболее сложных в теории горения. Горение может про-
исходить в неподвижных средах, при ламинарном течении горючей
смеси и в турбулентном потоке, при предварительном перемешивании
паров горючего и окислителя или при раздельной их подаче. Про-
цесс горения по существу представляет химический процесс, однако
скорость его часто определяется физическими процессами — испаре-
нием, передачей вещества и тепла.
Наиболее сложным видом горения является горение жидких
распыленных ракетных топлив. Сложный вид горения изучается
на основе закономерностей более простых видов горения, в част-
ности горения однородных, заранее перемешанных горючих смесей.
Основными параметрами, характеризующими процесс горешЯ?
являются: фронт пламени, ширина фронта пламени, скорость рас-
пространения фронта пламени. Фронтом пламени называют зону»
где происходит превращение химической энергии топлива в тепло-
вую. Фронт пламени отделяет область еще не прореагировавшей
исходной смеси от области продуктов сгорания. В практике даШе
152 Я
г гОрении однородных горючих смесей редко приходится иметь
И со сплошным фронтом пламени. Обычно наблюдаемое пламя
^fToiiT из многих очагов и распространение зоны горения от них
С° исходит с различными скоростями.
nP°j3 зависимости от химической природы горючей смеси и условий
пения, механизм распространения пламени может быть различ-
1 L Существует нормальное (тихое), турбулентное и детонационное
йзрьтвяое) распространение пламени.
Нормальное распространение пламени
Механизм нормального распространения пламени связан с пере-
дачей тепла посредством молекулярной теплопроводности или с диф-
фузией активных продуктов реакции из зоны горения в свежую
смесь.
Важной характеристикой такого вида горения является нормаль-
ная или фундаментальная скорость распространения пламени.
Нормальной скоростью распро-
странения пламени называется ско-
рость распространения зоны реакции
по отношению к неподвижной горю-
чей смеси в направлении, перпенди-
кулярном поверхности фронта пла-
вни. Эта величина при горении
в ламинарном потоке или в непо-
движной горючей смеси является ве-
личиной постоянной.
Количество горючей смеси, сго-
рающее в единицу времени на единицу
поверхности фронта пламени, назы-
вается массовой скоростью горения
^гор, которая связана с нормальной
скоростью распространения пламени
выражением:
£гор — Q
где q — плотность исходной горю-
чей смеси, кг/ж3;
wo — нормальная скорость рас-
пространения пламени,
Направление движения
горючей смеси
Рис. 59. Изменение температуры
при нормальном горении горю-
чей смеси:
I — зона нагревания горючей смеси;
II — зона горения; III — зона про-
дуктов сгорания.
м/сек.
Согласно приближенной теории
рамени принимается, что быстрая
нормального распространения
______________________________А_„._ химическая реакция горения
7^ * В110м иДет ПРИ температурах, весьма близких к максимальной
Ющеле/)аТ^^е горения. Сущность этой теории заключается в следу-
гор^°П^СТИМ’ что в неподвижную зону горения поступает свежая
Рас о 4 Смесь с постоянной скоростью, равной нормальной скорости
Р°с гранения пламени (рис. 59). Горючая смесь нагревается за
счет передачи тепла из зоны горения от начальной температуры у
до температуры воспламенения Тв. Дальнейшее разогревание горю0
чей смеси идет не только за счет передачи тепла из зоны горений
но и за счет тепла, выделяющегося при воспламенении поступившей!
горючей смеси.
Если принять, что все количество тепла, требуемое для нагревав
ния горючей смеси от начальной температуры То до температуре
воспламенения Тв, передается из зоны пламени, то количество тепла I
расходуемое на единице поверхности фронта пламени за единицу |
времени qv составит:
7i— — T.) w
скорость распространения пламени, м1сек*
горючей смеси, жд!кг • град', j
где wo — нормальная
Ср — теплоемкость
q — плотность горючей смеси, кг/м3. 'fl
Количество тепла д2, передаваемое через зону пламени без учета
теплового излучения и при условии, что тепловой поток из пламени
расходуется только на нагревание смеси, выражается следующим
образом: I
<12
\ о л /макс
* •
где X — теплопроводность горючей смеси, вт/м*град', ЧИ
/ дТ \ Я
( —- — максимальное значение градиента температур в зоне
\ /макс пламени, град!м,
( дТ \ ТГ-ТВ ~ Гв-Т0 Я
\ дХ /макс A2 Xi
где ТГ — максимальная температура горения, °К;
А\ — ширина зоны горения, в которой происходит изменение
температуры от начальной до температуры воспламене-
ния; м .
Х2 — ширина зоны горения, в которой протекает быстрая хими-
ческая реакция и температура смеси повышается от тем-
пературы воспламенения до максимальной, м. Я
Так как = q2, то I
CPQIVO(TB-TO) = X- fl
I
где т — время химической реакции при максимальной темпера^/*?
пламени, сек,
Следовательно, нормальная скорость распространения пламеВ^Ч
в
^0
154
fj[<i ° CpQ
— физическая константа горючей смеси, которая носит
название коэффициента температуропроводности,
м2!сек.
Отсюда следует, что нормальная скорость распространения пла-
и зависит не только от химического состава смеси
«^ояроводности, теплоемкости и плотности
тепловой зоны горения определяется
но также от
горючей смеси. Вели-
уравнением:
а
Если химические реакции в зоне горения являются автокатали-
тическими, то распространение пламени связано не только с переда-
чей тепла из зоны пламени к несгоревшей смеси, но и с диффузией
из этой зоны активных продуктов реакции. В определенных усло-
виях диффузия активных продуктов реакции может быть причиной
распространения пламени независимо от передачи тепла.
При нормальном распространении пламени скорость процесса
горения можно представить выражениями:
grop =" р— ° (кг/м3 • сек) и grop S Q (кг/сек)
где S — общая поверхность фронта пламени, м2;
ip0 — нормальная скорость распространения пламени, м!сек\
I р — плотность исходной горючей смеси, кг/м3;
г V — объем камеры сгорания, м3.
Рассмотрим влияние различных факторов на нормальную ско-
рость распространения пламени.
Химическая природа топлива. В работах [26, 27] приведены ре-
зультаты исследований влияния структуры углеводородов на мак-
симальную нормальную скорость распространения пламени при
горении в воздухе. На рис. 60 показана зависимость скорости рас-
ространения пламени в смеси с воздухом от числа атомов углерода
и насыщенности углеводородов.
Нормальная скорость распространения пламени w0 при горении
углеводородов увеличивается в таком порядке: алканы, алкены,
лкадиены, алкины. z
увеличением длины цепи алифатических углеводородов w0
» увеличение ненасыщенности соединений повышает
горения. Пяти- и шестичленные циклоалканы имеют
распространения пламени, близкую соответствующим
снижается
скорость
скорость
Н°Д1Г|ВС 1ИЧеИИе длины Цепи кислородсодержащих соединений при-
Ки1л к уменьшению нормальной
По данным работы [27]
скОро’ Как 11 в случае углеводородов
Для р( Ти РаспР°отранения пламени, по данным раооты iz/j
аль p'j (1,оР°Асодержащ11х соединений уменьшается в ряду: кетоны,
V ИДЫ» спирты, простые эфиры.
155
При обычных условиях максимальное значение нормально-
скорости распространения пламени для бензино- и кероспно-воздущ!
ных смесей 0,4—0,5 м/сек, а в смеси с кислородом значительна
больше. Очень большие скорости распространения наблюдаются
для водородо-воздушных смесей. d
Скорость распространения пламени может возрастать при добц||
влении небольших количеств некоторых веществ, а также при смей£ ।
нии нескольких горючих компонентов, причем шо некоторых смесей
больше скорости пламени
каждого из компонентов. |
При горении окиси углерода
в воздухе скорость распро-
страпеипя пламени заметно
возрастает при добавлении
небольших количеств водо-
рода, углеводородов и паров
воды. Добавки сероуглерода,
наоборот, снижают вели-
чину w0 при горении смесп
окиси углерода с воздухом
[23]. Добавление тетраэтил-
свинца пли этил нитрата к
углеводородным горючим
оказывало влияния на ско-
Число атомов С
Рис. 60. Зависимость нормальной скорости
распространения пламени от числа
атомов углерода в молекуле углеводо-
родов [31 [:
1 — алканы; 2 — алкены;
4 — цикланы; 5 — алкины.
з — алкадиены;
действие, приводящее к снижению
Добавление инертных
рости распространения
газов
пламени
рость распространения пла-
мени при испытании в го-
релках
данные, что
гателя при
нпях газа
оказывает
значений [23]. Я
приводит к снижению ско-
и сужению области воспламе-
[28]. Однако есть
в условиях дви-
высоких давле-
тетраэтилсвинец
ингибирующее
нения. Д
Инертные примеси влияют как на физические характеристики
горючей смеси (коэффициенты теплопроводности и диффузии, тепло-
емкость), так и на концентрацию реагирующих газов в зоне пламени.
Действие этих факторов приводит к снижению температуры и ско-
рости реакции в зоне пламени и соответствующему изменению ско-
рости распространения пламени. 1
По степени флегматизирующего действия на скорость распрострач
нения пламени инертные разбавители располагаются в следуюа^
порядке: двуокись углерода, азот, аргон, гелий.
Давление. О влиянии давления на нормальную скорость распр0"
странения пламени имеются противоречивые данные. 1
В некоторых работах [29, 30] указывается, что давление Я I
влияет на скорость распространения пламени стехиометрически 1
гЛеводородо-воздушных п углеводородо-кислородных смесей. Од-
13 знаЧ11Тельно большем количестве работ доказывается, что
нормальная скорость распространения пламени в углеводородо-
воздушных смесях обратно пропорциональна давлению ш0 оо .
Показатель степени п дается в пределах 0,1—0,5[23, 31]; в сред-
Хм можно принять п = 0,25 [32, 33].
Температура. С увеличением начальной температуры нормальная
скорость распространения пламени увеличивается (рис. 61). Однако
Рис. 61. Зависимость нормальной
скорости распространения пла-
мени бензино-воздушнпх смесей
от концентрации паров бензина
в воздухе Сбенз п температуры исход-
ной смеси [32].
Рис. 62. Зависимость нормальной ско-
рости распространения пламени
в топливо-воздушных смесях от коэф-
фициента избытка воздуха а [11]:
1 — водород; 2 — ацетилен; 3 — этилен;
4 — пропан; 5 — метан; 6 — окись углерода.
из '"н^ди-виздушных смесей можно принять со 10, где п
в пп^ЯеТСЯ в. зависим°сти от температуры, вида топлива и находится
|нЛл)став ।
г^^странепия
Рис. 61 ;
||rp 1Л1<л/А VU'JjWVllJ pr LA КУ Л А р, V/кУ JL 1А V» JL1 /1 Л1 </1 СА 1Н II £1
СА1ессй В °^ласти обогащенных смесей. Для углеводородо-воздушных
ЭТо увеличение количественно не согласуется с ожидаемым увели-
чением согласно зависимости скорости химических реакций от тем-
братуры w co е RL . Это указывает на то, что не только пачаль-
я температура определяет протекание реакций в пламени. Для
и.э^В(}Д()РОД°"ВОЗДУШНЫХ смесей можно принять со Z™, где п
| _ ----------— — — w —. у л-r ал.^-^4^4 Л.ЛЧЛ. Л.Л. 4^4. ЖЖ JLX. 4.4. ЖВ. VZ^-^ЛЛ Л. \~/ Л Л-
У пРеДелах 1,4—2,1 [23, 31, 33].
--1 смеси. Кривые зависимости нормальной скорости рас-
- пламени от состава горючей смеси приведены на
\ 62. Максимальная скорость распространения пламени
• - ........ :
максимум соответствует коэффициенту избытка воздуха
ее толщине увеличивает площадь
Распространение пламени в турбулентном потоке 3
На практике процесс сгорания всегда осуществляется при той
пли иной степени турбулентности горючей смеси, причем турбулент-
ность увеличивает скорость горения. 1
Количество горючей смеси, сгорающее в единицу времени Во
фронте пламени, пропорционально площади фронта пламени. 4
Всякое искривление поверхности зоны горения при постоянной*
фронта пламени и, следовательно,
общее количество сгоревшей смеси
(рис. 63). В
Отсюда вытекает закон, извест-
ный под названием закона площа-
дей, который гласит, что при
всяком увеличении поверхности
фронта пламени турбулентная
скорость распространения пла-
мени относительно свежей
горючей смеси возрастает в том
же отношении, как увеличивается
поверхность фронта пламени S
относительно плоской поверх-
ности 50, перпендикулярной к на^
правлению распространения, т. е.
5 1
^т = ^ю -тг-
.Ж
Впервые этот закон горения
в движущемся газе был установлен
русским физиком В. А. Михель-
соном, который доказал, что если
нормаль к поверхности фронта
образует угол ср с направлением
распространения фронта пламени, то скорость распространения пла-
мени увеличивается обратно пропорционально косинусу угла ф
(рис. 63, а): В
Рис. 63. Нормальная (а) и наблюда-
емая (б) скорость распространения
пламени.
COS (р
Движение горючей смеси при наличии стенок происходит с раз-
ными скоростями в разных точках, что искривляет фронт пла-
мени. Поэтому наблюдаемая скорость распространения пламени
всегда больше нормальной. В турбулентных средах скорость рас-
пространения пламени должна определяться процессами турбулшЖз
ной теплопроводности, турбулентной диффузии и химического
превращения в зоне пламени. Турбулентность может увеличить
скорость распространения пламени в десятки и сотни раз. Но
механизм распространения пламени при турбулентном горении
158 1
цЩ
тол
в
полностью не выяснен. Из фотографий турбулентного пламени
по, что фронт пламени имеет размытые контуры и значительную
in|,пну (вместо тонкого и четко очерченного фронта пламени при
ламинарном течении газа). Фронт пламени может быть разорван,
’ ним могут быть очаги еще не воспламенившейся смеси, поэтому
определение величины поверхности турбулентного пламени за-
труднено.
г В настоящее время существует несколько моделей турбулентного
горения, на основе которых и строятся теоретические положения.
Первая и наиболее распространенная модель усложненного ламинар-
ного фронта пламени представляет турбулентное горение горючих
смесей с поверхности. Вторая модель — турбулентное горение
объеме горючей смеси в процессе перемешивания. Смешивание
исходной смеси с горячими продуктами реакции вызывает протека-
ние гомогенных химических реакций.
Е. С. Щетинков [34] предлагает следующую модель турбулент-
ного горения. Отдельные объемы газа (турбулентные моли), состоя-
щие из сгоревшей пли полусгоревшей горючей смеси, выбрасыва
за счет пульсаций газа из глубины зоны горения и воспламеняют
моли свежей смеси, которые могут начать гореть с поверхности.
Турбулентное смешение в одном месте приводит к затуханию реак-
ции в объеме, в другом же, наоборот, ускоряет реакцию и вызывает
^воспламенение, если температура в этом объеме высокая и время
существования его достаточно, чтобы произошло воспламенение.
Различие между двумя указанными выше схемами турбулентного
горения заключается в том, что в первой схеме превалирует процесс
распространения пламени, а во второй — процесс самовоспламене-
ния. Ни та, ни другая модель в полной мере не объясняют всех
особенностей турбулентного горения. При построении теории турбу-
лентного горения необходимо совместное рассмотрение как распро-
странения пламени, так п объемных реакций, протекающих в тех
зонах, где турбулентное смешение опережает распространение пла-
мени. А. Н. Воинов [9] указывает, что возможность объемного
горения сильно возрастает с повышением давления и что роль объем-
ных реакций, завершающихся самовоспламенением, должна сильно
проявляться в форсированных камерах сгорания при высоких давле-
ниях. Возникновение очагов самовоспламенения в процессе горения
является одной из вероятных причин появления элементарных удар-
ных волн, вызывающих нарушение нормального развития процесса
прения в двигателях внутреннего сгорания.
гэг!3 со°тветс5гвии с теорией поверхностного горения Дамкелера
°'J I влияние турбулентности па скорость горения можно предста-
• ь следующим образом. Если масштаб турбулентности, характе-
ризующийся длиной пути смешения Z, больше ширины зоны горения,
фронт пламени искривляется (см. рис. 63, б). Таким образом,
Ри крупномасштабной турбулентности увеличивается поверхность
на еДиниДУ площади поперечного сечения турбулент-
0 потока, поэтому увеличивается и наблюдаемая скорость
ни другая модель в полной мере не объясняют всех
159
распространения пламени. При этом изменения структуры пламени
и нормальной скорости распространения мгновенного фронта пла-
мени не происходит. Но это наблюдается при относительно неболь-
шой интенсивности турбулентности, когда деформированный фронт
пламени является непрерывным. Большая интенсивность турбулент-
ности способствует разрушению поверхности фронта пламени, от
которого начинают отрываться газовые объемы и появляются отделкам
пые беспорядочно движущиеся объемы невоспламененной и горящей *
смеси [36]. В раздробленной зоне горения каждый элементарный
объем горючей смеси сгорает с поверхности со скоростью wQ. Возраста-
ние скорости турбулентного горения определяется только увеличе-
нием поверхности горения. Л
Когда масштаб турбулентности мал по сравнению с шириной
зоны горения, поверхность фронта пламени не может искривляться
и увеличиваться, при этом эффект турбулентности проявляется
в увеличении интенсивности процессов перемешивания внутри зоны
горения. Благодаря интенсификации процессов переноса тепла
и активных продуктов посредством микротурбулизацпи в самой
зоне горения, нормальная скорость распространения пламени резко
увеличивается. Для ламинарного потока нормальная скорость рас-
пространения пламени пропорциональна корню квадратному из
величины а = —
туропроводность во много раз больше, чем в ламинарном, она равна
пульсационной скорости w', умноженной на длину пути смешения I:
(см. стр. 154). В турбулентном потоке темпера-
а
Следовательно, турбулентная скорость распространения пламени
Увеличение температуропроводности при всех прочих равных
условиях приводит к увеличению ширины зоны пламени. Тепловая
ширина зоны пламени
(Iгр
Т “
При увеличении ширины зоны пламени возрастает количество
сгорающей смеси. К. И. Щелкин [36] при анализе влияния мелко-
масштабной турбулентности на скорость распространения пламени
учел процессы не только турбулентного, но и молекулярного пере-
носа. Для относительного увеличения скорости пламени в турбу-
лентном потоке им предложена формула: м
Для относительного увеличения скорости пламени в турбу-
lw'
а
W'o
В отсутствие турбулентности, когда w' = 0, получаем wT = ^о*
1G0
Детонационное распространение пламени
При определенных условиях в некоторых горючих смесях
зможно распространение пламени с громадной скоростью
1000—3500 м/сек. Зону горения, распространяющуюся с такой
6 тыпой скоростью, называют детонационной волной.
► Скорость детонационного распространения пламени есть физи-
ческая константа газовой горючей смеси, зависящая преимуще-
ственно от химического состава смеси. При детонационном распро-
странении пламени воспламенение горючей смеси происходит вслед-
ствие сжатия ее в ударной волне.
Ударная волна, проходя по горючей смеси, вызывает ее нагрева-
ние, интенсивность которого зависит от характеристик ударной
волны (табл. 32).
Таблица 32
Характеристики ударных волн [31]
Отношение давления
за ударной волной
к начальному давле-
нию
Скорость удар-
ной волны, м/сек
Температура за
ударной волной, °C
2
5
10
50
100
452
698
978
2149
3020
63
209
432
1988
3588
Когда по горючей смеси проходит ударная волна со степенью сжа-
тия, достаточной для ее воспламенения, то возникает детонационная
волна, представляющая собой распространение механической удар-
ной волны совместно с фронтом пламени. Это означает, что при
давлении и температуре ударной волны горючая смесь воспламе-
няется с задержкой, не превышающей времени пребывания смеси
в зоне сжатия ударной волной:
где X — ширина зоны сжатия (фронта ударной волны), м\
и — скорость распространения ударной волны, м/сек*,
т — задержка воспламенения горючей смеси, сек.
Ширина фронта ударной волны весьма мала, и химическая реак-
не успевает пройти по всему фронту. За фронтом ударной волны
Располагается растянутая зона химической реакции, ширина кото-
ром ограничена. При очень большой ширине зоны реакции затраты
омергцц на Трение и теплоотдачу в окружающую среду будут на-
столько велики, что заметно снизят температуру во фронте волны
1 Распространение детонационной волны сделается невозможным.
П Заказ 792. 161
ВИДЫ ГОРЕНИЯ ТОПЛИВ В ДВИГАТЕЛЯХ
Процессы горения в двигателях внутреннего сгорания протекают
сложно, и для каждого типа двигателей имеется много характерных I
особенностей, поэтому определение количественных характеристик I
процесса возможно при его экспериментальном исследовании в усло-1
виях двигателя. Однако полезно выделить отдельные виды горени)|й(
которые схематично дают представление об особенностях протека- ।
ния горения в различных типах двигателей.
В зависимости от свойств топлива, начальной температуры и да-
вления, конструкции камеры сгорания, способа подачи в нее горю-
чего и окислителя преобладающее влияние на скорость сгорания
могут оказывать как гидродинамические факторы и процессы пере-
мешивания горючего и окислителя, так и факторы, связанные
с химическими превращениями при окислении топлива. 1
Можно выделить два основных вида горения топлив в двигате-
лях: турбулентное горение однофазных (однородных) горючих сме-
сей и турбулентное горение двухфазных горючих смесей. I
Турбулентное горение однофазных горючих смесей
К этому виду можно отнести процесс горения топливо-воздушных
смесей в карбюраторных двигателях. На отдельных режимах работ#
воздушно-реактивных двигателей процесс горения можно также рас-
сматривать с точки зрения горения однофазной горючей смеси.
При горении однородной горючей смеси возможно: 1) турбулент-
ное фронтальное сгорание и 2) последовательное воспламенение
отдельных объемов горючей смеси по мере поступления в зону пла-
мени. Основными характеристиками турбулентного горения одно-
родной горючей смеси являются турбулентная скорость распростра-
нения пламени wT, ширина зоны горения Хт и время горения в тур-
булентном пламени тт- I
Многочисленные экспериментальные исследования показывают,
что указанные параметры зависят от пульсационной скорости потока
и , масштаба турбулентности I и нормальной скорости распростра-
нения пламени шо. Однако по виду функциональные зависимости,
полученные разными авторами, существенно отличаются друг от
друга. Почти все исследователи признают, что турбулентная ско-
рость распространения пламени возрастает с увеличением пульса-
ционной скорости потока и нормальной скорости распространения
пламени. Обобщая результаты многих экспериментальных иссле-
дований, можно написать следующую эмпирическую зависимость:
wT (ю')П (ш0)т
Значение показателей п и т зависит от степени турбулентности.
Когда w' < wo турбулентная скорость распространения пламени
wT wo. я
162 . Я
При большой интенсивности турбулентности, когда w' шо,
г булентная скорость распространения пламени зависит главным
Тглазом от и в меныпей степени от wo. При этом разные исследова-
те-in принимают « = 0,6—1,0, а т = 0,2—0,4.
Т К И. Щелкни считает, что при w' ш0 турбулентность увели чи-
аеТ поверхность горения не только вследствие искривления фронта
замени, но и вследствие раздробления объемов несгоревшей смеси,
павшей в зону горения. Фронт горения приобретает значительную
— заполняется множеством горящих с поверхности и
ьшающихся по размерам объемов несгоревшей смеси.
горючей
в более
ширину, он
умен:—
Время горения раздробленных объемов
К И. Щелкин [37] принимает равным Тт ~ —
работе [36] — равным времени смешения гт Ширина зоны
смеси
ранней
турбулентного горения ХТ штгт. При сильной турбулентности
ш', следовательно ХТ wTrT -— w'
Большой практический интерес представляет выяснение зависи-
мости скорости распространения турбулентного пламени и ширины
зоны горения от давления и температуры исходной смеси.
Экспериментальные исследования показывают, что с понижением
давления скорость распространения турбулентного пламени умень-
шается, а ширина зоны горения увеличивается. Многие исследова-
тели считают, что влияние давления па турбулентную скорость
распространения пламени и ширину зоны турбулентного горения свя-
зано главным образом с влиянием давления на характеристики тур-
булентности — пульсационную скорость и масштаб турбулентности.
Имеющиеся в литературе данные по этому вопросу относятся
к давлениям ниже атмосферного и далеки от условий сгорания
в двигателях. Например, В. Е. Дорошенко и А. И. Никитский [32]
установили, что пульсационная скорость за турбулентными сетками
при давлениях ниже атмосферного w' /А25.
Как указывалось выше, для углеводородо-воздушных смесей
нормальная скорость распространения пламени wo ~р-0’25. Подста-
вив зависимости wo и wf от давления в выражение
(Шо)т
что турбулентная скорость распространения
h
гДе п J72, видим, что турбулентная скорость распространения
пламени с повышением давления должна возрастать в функциональ-
ной зависимости где к — величина порядка 0,05—0,25.
опытным данным [32] к 0,5.
Масштаб турбулентности с понижением давления становится
дольше, что приводит к увеличению тт и Хт.
п емпература смеси оказывает слабое влияние на скорость рас-
пространения турбулентного пламени и довольно значительное —
воПп1Р11нУ зоны горения, причем как температура, так и давление
•Действуют на турбулентность потока w' и нормальную скорость
И*
163
распространения пламени ip0. с повышением температуры газовой
горючей смеси пульсационная скорость уменьшается в функциональ-
ной зависимости w' у0-42 [32]. Однако wr с увеличением темпе-
ратуры не падает, так как wT хотя и в небольшой степени, по все же
зависит от wо, a wо зависит от температуры смеси в степени п =
= 1,3—2,0. - ,'W
С повышением температуры смеси ширина зоны горения уменД^.
шается, вследствие значительного увеличения нормальной скорости
распространения пламени. ЗЯ
Процесс стационарного горения в потоке заранее подготовленной
газообразной смеси возможен тогда, когда скорость потока не пре-
вышает скорости распространения пламени. Если скорость газооб-
разной горючей смеси больше скорости распространения пламени,
то процесс стационарного горения возможен только при непрерыв-
ном поджигании смеси каким-либо источником зажигания. I
Турбулентное горение двухфазных горючих смесей
При горении распыленных топлив в жидкостных и воздушно- I
реактивных двигателях, а также в двигателях с воспламенением 1
от сжатия образуется двухфазная смесь, в которой наряду с испарен- ]
ным топливом присутствуют капли жидкого топлива. Для характе- 1
ри стики этой смеси большое значение имеют качество распыливания*^.
и скорость испарения топлива. ЯВ
Фотографирование процесса горения двухфазной смеси показы- j
вает, что поверхность горения более размыта, чем фронт пламени I
однородной смеси. В зоне горения двухфазной смеси отчетливо 1
видны следы горящих капель топлива. 'В
При низких температурах смеси и малом содержании в ней испа- |
репного топлива непрерывный фронт пламени отсутствует и горение I
происходит в зонах, окружающих отдельные капли и их скопления. 1
При высоких температурах наблюдается сплошной фронт пламени и, 1
кроме того, очаги горения отдельных капель топлива за фронтом, j
Горение двухфазных смесей может происходить при суммарном
коэффициенте избытка окислителя или воздуха, значительно пре- 1
вышающем пределы воспламенения для однородных горючих смесей.
В двухфазных смесях происходит воспламенение отдельных групп •
капель как за счет распространения зоны горения от соседних капель, I
так и счет самовоспламенения в пространстве между каплями. 1
Расчеты распределения температур вокруг отдельных капель керо-
сина в неподвижном воздухе, выполненные на основе диффузион- 1
ной теории горения без учета диссоциации продуктов сгорания, J
показывают, что на расстоянии около 30 диаметров капли от ее по-
верхности температура среды ~2200° С. При такой высокой темп$^ |
ратуре возможно надежное воспламенение смеси паров топлива |
с воздухом, образующейся при испарении соседних капель. В двух-
фазных смесях скорость распространения пламени мало зависит
от общего суммарного коэффициента избытка воздуха. И
164
П п раздельной подаче горючего и окислителя в камеру сгора-
ли горение может протекать в диффузионной или кинетической
области.
При высоких температурах и концентрациях и при достаточной
гтп вностп компонентов горючей смеси химическая реакция окисле-
' тОплива начинает протекать со столь значительной скоростью,
физические факторы, определяющие подготовку горючей смеси,
начинают тормозить процесс горения. К ним относятся, например:
качество распыливания топлива, характер течения газо-воздушного
потока, распределение концентраций и температур в потоке, форма
и размеры камеры сгорания, распределение тепла внутри потока,
а также между потоком и внешней средой. Именно эти процессы при
высокотемпературном горении начинают отставать по скорости про-
текания, регулируя ход всего процесса в целом.
Такой вид горения, когда время смешения намного больше вре-
мени химических превращений и когда общая скорость процесса
определяется в основном скоростью смешения топлива и воздуха,
называется диффузионным горением. При диффузионном горении
скорость процесса определяется главным образом факторами гидро-
динамического характера и меныпую роль играют свойства топлива.
Распространенным видом диффузионного горения, для которого
разработана теория, является микроди ффузионное турбулентное
Горение.
При микродиффузпонном турбулентном горении жидкое топливо
посредством форсунок или столкновения струй распыливается на
отдельные малые объемы, беспорядочно распределенные в потоке
воздуха. Сгорание происходит одновременно с процессом турбулент-
ного микросмешения отдельных малых объемов топлива с окружа-
ющим воздухом.
В теории микродиффузионпого горения, разработанной
Д. А. Франк-Каменецким и Е. М. Минским [38], принимается, что
скорость горения определяется скоростью микросмешения отдельных
малых объемов испарившегося топлива с .окружающим воздухом.
Если масштаб дробления б, представляющий средний линейный
размер тех объемов, на которые раздроблено топливо в потоке воз-
духа, равен Масштабу турбулентности I или больше него, то переме-
шивание происходит посредством турбулентной диффузии. Время
смешения в этом случае * • :
1 урбулентиая скорость распространения пламени при микро-
Днффузионном' процессе может быть найдена из выражения для нор-
р-тьной скорости распространения пламени, в котором коэффи-
циент температуропроводности следует заменить коэффициентом
УРоулентного обмена, а время химической реакции — временем
Решения. Тогда:
Соответственно, для ширины зоны пламени получаем:
Таким образом, при микродиффузионном горении в турбулентном
потоке скорость распространения пламени прямо пропорциональна
пульсационной скорости потока и — при постоянной степени тур-
булентности — скорости потока. Это обеспечивает устойчивость горе-
ния и возможность увеличения скорости горения за счет скорости
потока. |
Устойчивость диффузионного горения объясняется тем, что при
любой концентрации в смеси одновременно имеются самые различные
местные концентрации, в том числе и наиболее благоприятные для
горения. Ускорение микродиффузионного горения под действием
турбулентности не может быть беспредельным. При очень большой
скорости потока и степени турбулентности микродиффузионное
горение переходит в кинетическое.
Кинетическим горением называется такой вид горения, когда
скорость смешения велика и процесс в целом лимитируется только
скоростью химических превращений. Форсирование процесса кине-
тического горения за счет увеличения скорости потока может при-
вести к потере устойчивости горения и срыву пламени. Для микро-
смешения увеличение скорости потока только благоприятно, но
даже при самом совершенном микросмешении горение будет устой-1
чивым лишь при условии, если будет обеспечена передача тепла
п активных продуктов реакции из зоны горения к свежей смеси.
При очень больших скоростях потока передача тепла и активных
продуктов из зоны пламени в свежую смесь будет отставать от про-
цессов микросмешения и произойдет срыв пламени.
Методы определения скорости горения в двигателях
Теоретический расчет скорости горения в двигателях с учетом
всех факторов, определяющих развитие процесса во времени, очень
сложен. При исследовании процесса сгорания в двигателях широко
применяются экспериментальные методы, дающие относительную
оценку скорости горения по скорости нарастания давления и распро-
странения пламени, времени развития отдельных фаз процесса
и т. д.
Наиболее распространенными являются следующие эксперимен-
тальные методы. !
1) Скоростное фотографирование, позволяющее исследовать раз-
витие процесса сгорания и определять одну из важнейших характе-
ристик процесса — скорость распространения пламени. aj
2) Измерение давления в камере сгорания практически безынер^
ционными электрическими индикаторами давления, дающее воз-
можность определить общие количественные и качественные харак-
теристики физико-химических превращений топлива в двига-
телях.
166 Я
3) Измерение температуры газов в камере сгорания с последу-
ющим исследованием процесса воспламенения и сгорания по линии
температур, позволяющее произвести ряд кинетических расчетов
п дать количественную и качественную характеристику процесса
и целом.
4) Скоростные спектроскопические и химико-аналитические ме-
^годы исследования превращений горючей смеси в камере сгорания
двигателей, позволяющие определить наличие и концентрацию про-
межуточных продуктов реакции и оценивать характер кинетических
превращений в процессе воспламенения и сгорания топлива.
Для общей характеристики процесса исследуются продукты
сгорания с помощью газоанализаторов.
Для исследования процессов смесеобразования, воспламенения
и сгорания в двигателях используются также методы с применением
радиоактивных изотопов.
ЗАВИСИМОСТЬ РАБОЧЕГО ПРОЦЕССА ДВИГАТЕЛЕЙ ОТ ХИМИЧЕСКИХ
СВОЙСТВ ТОПЛИВА
Влияние химической природы топлива на запуск наиболее сильно
проявляется в двигателях, не имеющих устройств принудительного
зажигания. Осооенно большое значение имеют пусковые характери-
4в*тпкп для самовоспламеняющихся ракетных, а также дизельных
топлив.
Рис г/ тт- времр
веа/т/1’ Измеясыпе давления в камере сгорания малоразмерного жидкостно-
I М явного двигателя в период пуска и выхода на режим для различных топлив:
Tii4eei"“eaMm>on«o"/V ,?8 %;иая а'<0™ая кислота; 2 - смесь алифатических (50 %), арома-
пия; р „я^,, %? 98 %"ная азотная кислота; т — период задержки самовоспламене-
макс — максимальное давление при пуске; Рср — давление на установившемся режи-
ме работы двигателя.
за , еМ лучше воспламеняется топливо, тем легче осуществляется
/".'•к жидкостно-реактивного двигателя, более плавно и быстро
* ‘"навливается заданный режим его работы. При плохой воспла-
Дав,ЯеМ0СТИ топлива скорость нарастания давления и максимальное
Be I '1111е В каме₽е сгорания при запуске могут достигнуть таких
•"‘чин, при которых возможно разрушение двигателя.
' pi ' 1'ачествб примера на рис. 64 приведены индикаторные диа-
1 "мы, Показывающие изменение давления в камере малоразмер-
1G7
ного двигателя в период запуска и выхода на режим на топливах раз-
личной воспламеняемости.
Пусковые характеристики самовоспламеняющихся ракетных топ-
лив могут быть представлены в виде графиков, показывающих измене-
ние периода задержки самовоспламенения т, максимальной скорости
/ dP \
нарастания давления в период запуска I и отношений
\ «Т /макс
максимального давления в камере сгорания при запуске к давлению
на установившемся режиме в зависимости от условий пуска.
* ср "Я
На рис. 65 показано, как изменяются основные параметры,
характеризующие пусковые качества самовоспламеняющихся ра-
Рис. 65. Пусковые характеристики топ-
лива ксилидин и98%-ная азотная кисло-
та, снятые на малоразмерном двигателе.
кетных топлив, с изменением
коэффициента избытка окисли-
теля. 1
С улучшением воспламеня-
емости дизельных топлив сокра-
щается период задержки само-
воспламенения и облегчается
запуск двигателя. На рис. 66
приведены пусковые характери-
стики дизельных топлив различ-
ного группового углеводород-’
но^о состава. Топлива с большим
содержанием ароматических
углеводородов вызывают за-
труднения при запуске двига-
теля, особенно при низких тем-
пературах окружающего воз-
духа. Наилучшие пусковые
качества имеют топлива алка-
нового основания, однако вы-
сокая температура застывания горючих, содержащих большое коли-
чество алканов, ограничивает возможность использования таких
топлив в зимних условиях.
Во всех типах двигателей внутреннего сгорания химические
свойства топлива оказывают влияние на полноту и устойчивость
процесса сгорания.
Основные нарушения нормального развития рабочего процесса
в поршневых двигателях с искровым зажиганием связаны с возник-
новением детонационного сгорания, в дизелях — с появлением
неуправляемого быстрого горения в начальной стадии процесса,
в воздушно-реактивных и жидкостных ракетных двигателях — с явлс^
нием срыва пламени и вибрационным горением. Указанные наруше-
ния в рабочем процессе всех типов двигателей приводят к снижению
эффективности использования энергии, выделяющейся при горении
топлива, а в отдельных случаях могут вызвать и механические повре-
ждения двигателя.
168
При нормальном сгорании в поршневых двигателях с искровым
и ганием после образования первоначального очага пламени,
счет принудительного воспламенения, происходит турбулентное
^• спространение пламени в объеме камеры сгорания со средней
Скоростью 15—40 м/сек.
При детонации развитие процесса сгорания вначале ничем не
л и чается от нормального. Изменения в характере сгорания,
сопровождающиеся резкими местными повышениями температуры,
давления и скорости распростра-
нения пламени, наблюдаются в той
части топливо-воздушной смеси,
которая сгорает в последнюю оче-
редь. В этой части смеси в резуль-
тате развития предпламенных
окислительных процессов возни-
кают очаги самовоспламенения.
Взрывной процесс самовоспламе-
нения распространяется на доста-
точно большой объем частично
прореагировавшей горючей смеси.
При этом на границах воспламене-
ния объемов горючей смеси возни-
кает резкий перепад давления —
ударная волна.
При определенных условиях
ударные волны, проходя по горю-
чей смеси, могут вызвать появле-
ние детонационных волн. Возник-
новение ударных волн — наиболее •
специфический признак детона-
Шел
50г
-20 -10 0 + 10 +20
tX
Рис. 66. Влияние группового угле-
водородного состава дизельного топ-
лива и температуры окружающего
воздуха t на продолжительность
пуска двигателя т:
ционного сгорания в двигателе.
Ударные волны, в основном, вызы-
вают и, наиболее важные внешние
признаки детонационного сгора-
1 — топливо алканового основания;
2 — топливо циклано-алканового основа-
ния; 3 — топливо ароматического осно-
вания.
пия — перегрев двигателя, снижение мощности и экономичности.
Ударные волны, периодически отражаясь от стенок, вызывают коле-
бания газов и усиленную теплоотдачу от газов к стенкам. Это при-
водит к снижению температуры выпускных газов и перегреву двига-
теля. Перемешивание полностью не прореагировавшей смеси с про-
дуктами сгорания и диссоциация, вызываемая резким повышением
температуры, во фронте ударных волн также не позволяют наиболее
полно и эффективно использовать химическую энергию топлива.
Поскольку возникновение детонационного сгорания связано
<‘о скоростью развития предпламенных окислительных процессов
в горючей смеси, то повышение устойчивости к окислению в таких
Условиях, т. е. улучшение антидетонационных качеств (детонационной
стойкости) топлив, позволяет обеспечивать нормальное развитие
процесса горения при оптимальных и достаточно высоких темпера-
туре и давлении в камере сгорания. Это, в свою очередь, дает
возможность увеличить мощность и повысить экономичность двига-
теля, например, за счет повышения степени сжатия (рис. 67).
Чем лучше детонационная стойкость топлива, тем при более
высокой степени сжатия в двигателе можно обеспечить нормальное
развитие процесса горения и, следовательно, повысить мощность
Рис. 67. Изменение интенсивности дето-
нации /ц эффективной мощности УУЭ и
удельного расхода топлива g3 в зависи-
мости от степени сжатия в двигателе и
детонационной стойкости топлива:
1 — топливо с большим содержанием н-алканов,
октановое число 70; 2 — топливо с большим содер-
жанием изоалканов, октановое число 100; з —
смесь сильно разветвленных изоалкановых угле-
водородов.
п экономичность двигателя^
Следует отметить, что повы-
шать детонационную стой-
кость топлива целесообразно
до определенных пределов.
Топливо не должно вызывать
нарушения в нормальном раз-
витии горения в двигателе в
условиях, соответствующих
наиболее высокому эффек-
тивному коэффициенту по-
лезного действия двигателя.
Например, топливо 3 (рис. 67)
сгорает нормально при боль-
ших степенях сжатия, но
применение его уже не дает
заметного повышения эффек-1
тивной мощности и сниже-
ния экономичности двига-
теля.
Нарушение в нормальной!
развитии процесса горения
в дизелях связано с возник-
новением неуправляемого
быстрого горения в началь-
ной стадии процесса горе-
ния. В результате резкого
перехода от предпламенных
процессов окисления к мас-
совому горению резко возра-
стает скорость нарастания давления по углу поворота коленчатого
вала и возникает «жесткая» работа двигателя. При этом повыша-
ются ударные нагрузки на шатунно-кривошипный механизм, воз-
никает опасность механических повреждений — разрушения подшип-
ников, деформации поршневых колец и пальцев.
Повышение «жесткости» работы двигателя до определенного
предела является полезным, так как повышается мощность и улуч-
шается экономичность двигателя. Это объясняется тем, что процесс!
сгорания осуществляется в более выгодных термодинамических
условиях, приближаясь к процессу горения при V = const.
Физико-химическая сущность явлений при неуправляемом взрыв-
ном начальном горении и нормальном развитии процесса горения
.170
дизеле изучалась многими исследователями, причем процессы
воспламенения и горения объясняются с различных точек зре-
1 Большой интерес представляют работы, выполненные в Инсти-
туте нефтехимического синтеза АН СССР Ф. А. Лавровым с сотруд-
никами, в которых применялось скоростное фотографирование рабо-
^рго процесса в дизеле, имевшем прозрачные окна. Один из авторов
этих работ Б. Н. Баранов [39] показал, что в зависимости от свойств
топлива и условий воспламенения в дизеле можно выделить два
вида горения:
1) многоочаговое самовоспламенение топлиР.ч (рис. 68);
2) самовоспламенение на периферии факела и прение в струе
распыленного топлива (рис. 69).
При неблагоприятных условиях воспламенения и пло^й вос_
пламеняемости топлива предпламенные химические превраЩстТИЯ
развиваются медленно; за это время происходит развитие фа-
кела топлива (рис. 68, 7—5) и выравнивание состава горючей
смеси (6).
В таких условиях предпламенные процессы окисления разви-
ваются в больших объемах топливо-воздушной смеси и, когда ско-
рости химических реакций достигнут величин, соответствующих
цепочно-тепловому взрыву, возникают почти одновременно много-
^чп елейные очаги самовоспламенения (рис. 68, 7—9). Возможно, что
эти очаги образуются вблизи отдельных крупных капель топлива
и их скоплений. Одновременно с развитием турбулентного горения
от этих очагов пламени во всем объеме камеры сгорания воз-
никают все новые и новые мелкие очаги горения (10—14). На
кадре 12 видно, что вблизи стенки камеры сгорания, на кото-
рую была направлена струя распыленного топлива, формируются
несколько довольно крупных очагов пламени. Эти очаги быстро
развиваются и образуют мощный турбулентный факел пламени,
охватывающий непрореагировавшую и частично прореагировавшую
горючую смесь во всем объеме камеры сгорания (кадры 73, 14).
При таком горении начальная скорость нарастания давления может
быть очень большой, что видно на индикаторной диаграмме, приве-
денной на рис. 68.
При хорошей воспламеняемости топлива, высоких температуре
и давлении в камере сгорания предпламенные процессы подготовки
топливо-воздушной смеси развиваются достаточно быстро в наиболее
благоприятных условиях на периферии факела. На кадрах 3 и 4
рис. 69 видно возникновение очагов пламени на периферии факела,
когда развитие его еще продолжается. В дальнейшем наблюдается
Немедленное обгорание» факела (кадры 5—14). Развитие процесса
торения в объеме камеры сгорания происходит за счет турбулентного
1 ирония на периферии факела распыленного топлива, а также вслед-
ствие образования новых очагов самовоспламенения. Такой вид
прения соответствует нормальному — плавному горению топлива
н Двигателе.
171
%
При впрыске в камеру сгорания дизеля топлива с высокой вое-
пламеняемостью развитие деструктивных и окислительных процессов i
быстро приводит к образованию очагов горения, период задержки
воспламенения таких топлив небольшой, что обеспечивает плавную .
й»:
fe « i
f}
Рис. 69. Воспламенение и горение
факела распыленного топлива
в дизеле.
Рис. 68. Многоочаговые самовос-
пламенение и горение топлива
в дизеле.
172
боту двигателя и возможность регулировать скорость горения
чеТ изменения подачи горючего. Однако из этого нельзя делать
1В()Д, что во всех случаях топлива с высокой воспламеняемостью
н’в 1ЯЮТСЯ лучшими для дизеля. Обычно они имеют недостаточно
высокую скорость горения, поэтому двигатели работают плавно,
’ менее экономично и сильно дымят (рис. 70).
F Влияние воспламеняемости дизельного топлива на период за-
воспламенения и «жесткость» работы двигателя проявляется
держки
тем сильнее, чем ниже тем-
пература и давление в камере
сгорания.
Разрешение противоречия
между необходимостью обес-
печить плавное развитие
процесса в начальной стадии
горения и быстрым и полным
сгоранием в основной стадии
горения топлива в дизеле
является важным условием
повышения эффективности
сжигания различных топлив
и создания многотопливного
двигателя на основе дизеля.
Не останавливаясь на всех
Цетанобое число
Рис. 70. Влияние воспламеняемости ди-
зельного топлива, выраженной в цетано-
вых числах, на его расход £э, «жесткость»
работы двигателя ДР/ Дер и период за-
держки воспламенения т.
д3. ул с.ч
направлениях решения этой
проблемы, рассмотрим лишь
метод термофорсирования ди-
зеля, основанный на термо-
деструкции впрыскиваемого
топлива [40].
На основании исследо-
вания предпламенных деструктивных и окислительных про-
цессов [13, 14] нами была показана важность деструктивных
процессов в период предпламеиного окисления и возможность
повышения эффективности сжигания в быстроходном дизеле топлив
различного группового углеводородного и фракционного состава
за счет предварительного нагревания топлива непосредственно
перед впрыском в камеру сгорания. Интенсивное подогрева-
ние топлива перед впрыском в камеру сгорания способствует ослаб-
лению внутримолекулярных связей и даже их разрушению. Угле-
водороды, которые в обычных условиях воспламеняются плохо,
окисляются достаточно быстро после предварительной их термо-
|кструкции. Это приводит к уменьшению периода задержки воспла-
менения и снижению «жесткости» работы двигателя при использова-
нии топлив легкого фракционного состава и топлив с большим содер-
жанием циклических структур. Предварительная термодеструкция
Молекул топлива, обеспечивая снижение периода задержки воспла-
менения, способствует в то же время повышению скорости горения
173
в основной стадии процесса, что приводит к повышению экономич-
ности. Нагревание топлива должно осуществляться быстро и непо-
средственно перед впрыском топлива в камеру сгорания, так как
нагревание топлива выше 150° С вызывает образование осадков
в системе питания и нарушения в работе топливной аппаратуры.
Нами осуществлялся подогрев топлива непосредственно в форсунку
до 200-230° С. , 5а
Влияние термофорсирования на экономичность работы двигателя
при применении топлив различного группового углеводородного
показано на
Термофорси-
оказалось
Рис. 71. Зависимость удельного расхода топли-
ва g3 от эффективной мощности двигателя Nd
для различных топлив:
--- при обычных условиях работы;----при термофор-
сировании; 1 — 5!) % алканов и цикланов, 41 % аромати-
ческих углеводородов; 2 — 35 % алканов и цикланов,
65 % ароматических углеводородов.
состава
рис. 71.
рование
особенно эффективным
для топлива с большим
содержанием аромати-
ческих углеводородов.
Применение специ-
альных форсунок по-
зволяет увеличить пред-
варительный подогрев
топлива и эффект тер-
модеструкции. <
Одно ИЗ ОСНОВНЫХ
требований к качеству
реактивных топлив —
способность обеспечи-
вать в различных ус-
ловиях работы воз-
ii устойчивое сгора-
душно-реактивных двигателей полное
ние.
Исследованиями влияния химического состава топлива на устой-
чивость горения в условиях, имитирующих горение в прямоточных
воздушно-реактивных двигателях, установлено, что наиболее широ-
кие пределы устойчивого горения имеют алкановые углеводороды,
наименьшие — ароматические. С повышением температуры топлива
пределы устойчивого горения ароматических углеводородов увели-
чиваются более значительно, чем для циклановых и алкановых.
В наших исследованиях па малоразмерных двигателях в условиях,
имитирующих высотиые, было обнаружено, что наиболее широкими
пределами устойчивого горения обладают топлива циклапо-алкапо-
вого основания с небольшим содержанием ароматических углеводо-
родов. Узкую область устойчивого горения имеют топлива с содер- t
жанием 60% и более ароматических углеводородов.
На расчетных и близких к расчетным режимах работы двигателя'
изменение группового углеводородного состава топлива не ока-
зывает большого влияния на полноту горения. Однако в менее
благоприятных условиях, например при понижении давления и
температуры воздуха на входе в камеру сгорания и при значительном
174
чении коэффициента избытка воздуха, влияние химического
топлива проявляется довольно сильно.
Пои больших коэффициентах избытка воздуха температура газов
ь _ --------что уМеНыпает полноту сгорания, осо-
у вел11
состава
зоне горения снижается
Г ио при использовании топлив ароматического основания.
условиях низких температур и давления, а также с уменьше-
скорости движения топлива и воздуха, что имеет место на боль-
высотах, наибольшей полнотой сгорания обладают топлива
JRiCM
ши*
Рис. 72. Зависимость полноты сгорания топлив различного группового угле-
Ь водородного состава от высоты полета самолета Н [41]:
1—эталонное топливо — 41,4% алканов, 39,2% цикланов, 19,4% ароматических; 2 —
алкановое топливо — 92,2 % алканов, 5,2% цикланов, 2,6% ароматических; з —циклано-
алкановое топливо — 46,7 % алканов, 51,9 % цикланов, 1,4 % ароматических; 4 — аромати-
ческие основания — 13,4% алканов, 11,4% цикланов, 75,2% ароматических.
алканового и цикланового оснований (рис. 72). Полнота сгорания
топлив различного группового углеводородного состава, предста-
вленных на рис. 72, сравнивалась с полнотой сгорания эталонного
топлива на высоте 6096 м [41]. Испытания этих топлив при различ-
ных составах смеси показали, что топливо алканового основания
обеспечивает лучшую полноту сгорания при меньшем отношении
воздуха к топливу, чем все остальные.
В жидкостных ракетных двигателях одним из видов нарушений
нормального развития процесса сгорания является вибрационное
горение, при котором наблюдаются колебания давления газов
в камере сгорания, снижение полноты сгорания, а в отдельных слу-
чаях и срыв пламени. Колебания давления газов в камере при вибра-
ционном горении могут иметь различные амплитуды и частоту.
Вибрационное горение зависит от свойств топлива, от условий
воспламенения и горения топлива в двигателе. Низкочастотные коле-
2^ния с частотой менее 300 гц возникают при понижении давления
температуры газов в камере сгорания или давления подачи компо-
нентов топлива и связаны с колебаниями в системе питания двига-
теля и периодом задержки воспламенения топлива [42—44].
Чем лучше воспламеняемость топлива, тем при меньшем давлении
в камере сгорания и давлении подачи компонентов сохраняется
175
нормальное устойчивое сгорание без возникновения низкочастотных
колебаний газов. 1
Высокочастотная неустойчивость горения характеризуется регу-
лярными колебаниями с частотой 1000—12 000 гц. Амплитуда коле-
баний может изменяться в пределах от 1 до 100% величины нормаль-
ного давления в камере сгорания. Установлено, что интенсивность
высокочастотных колебаний возрастает с увеличением давлений,
в камере сгорания [43, 44], причем возникновение высокочастотных
колебаний связано с особенностями протекания рабочего процесса
жидкостных ракетных двигателей. В условиях высоких давлении
и температур в камере сгорания при больших скоростях смешения
горючего и окислителя возникают очаги самовоспламенения турбу-
лентных объемов горючей смеси, в результате чего возникают удар-
ные волны. I
В соответствии с таким объяснением механизма высокочастотной
неустойчивости, более склонны к этому виду нарушения процесса
горения самовоспламеняющиеся топлива. Практика подтверждает
это положение. Например, для того, чтобы вызвать высокочастотную
неустойчивость смеси керосин — жидкий кислород (несамовоспламе-
няющейся) необходимо более высокое давление в камере сгорания,
чем для смеси алифатических и ароматических аминов и концентри-
рованной азотной кислоты (самовоспламеняющейся). |
Таким образом, химический состав и структура молекул топливу
определяют возможность обеспечения нормального развития рабо-
чего процесса того или иного двигателя в условиях, когда наиболее
полно используется химическая энергия топлива.
ЛИТЕРАТУРА
1. Термодинамические свойства индивидуальных веществ, под ред.
В. П. Глушко, т. 1 и 2, Изд. АН СССР, 1962.
2. А. Н. Б а х, ЖРФХО, 29, 373 (1897); 44, прилож. 1—79 (1912).
3. С. Engler, J. W е i s s b e r g, Kritische Studien liber die Vorgange
der Autoxydation, Braunschweig, 1904.
4. H. H. Семенов, Цепные реакции, Госхимиздат, 1934. 1
5. Н. Н. Семенов, О некоторых проблемах химической кинетики
и реакционной способности, Изд. АН СССР, 1958. j
6. К. И. Иванов, Промежуточные продукты и промежуточные реак-
ции автоокисления углеводородов, Гостоптехиздат, 1949.
7. A. R. U b b е 1 о h d е, The Science of Petroleum, т. IV, Oxford, 1938.
8. Сб. «.Поршневые двигатели внутреннего сгорания», Изд. АН СССР, 1956.
9. Сб. «Сгорание и смесеобразование в дизелях», Изд. АН СССР, 1960.
10. Б. Льюис, Г. Эльбе, Горение, пламя и взрывы в газах, ИЛ,
1948. !
V 11. В. И ост, Взрывы и горение в газах, ИЛ, 1952.
12. Б. Г. Гаврилов, Е. И. Гулин, А. П. ЛссникогЬу
А. К. Тарасов, ЖПХ, XXXIII (1960).
13. Б. Г. Гаврилов, Е. И. Гулин, А. П. Лесников,
Н. В. Шамрова, сб. «Низкотемпературные каталитические превращения
• углеводородов», Изд. ЛГУ, 1962.
14. М. К. Бычкова, Б. Г. Гаврилов, Е. И. Гулин,
А. П. Лесников, ЖПХ, XXXV (1962). 3
176
г Легковоспламеняющиеся и горючие жидкости. Справочник, 1956.
16 Н И. Семенов, Успехи хим. наук, XXIII, вып. 3; XXIV, вып. 4
<194ОЪ р. Schneebeli, Н. Moutet, Recherche aeronautique, № 87,
qq_46 (1962). v
1Я Я. M. П а у ш к и н, Химический состав и свойства ракетных
„ ’ Нал АН СССР, 1958; Химия ракетных топлив, Изд. АН СССР, 1963.
Т0П Ifl’ Пат.’ США 2712496, 5. VII. 1955.
20. L. Aston, Е. Rock, S. Isserow, J. Am. Chem. Soc., 74, 2484
^^°21. H a r s h m a n, Jet Propulsion, 27, № 4, 398 (1957).
22^ Жидкие и твердые ракетные топлива, Сб. переводов, ИЛ, 1959.
23. Процессы горения, под ред. Б. Льюис, Р. Н. Пиз, X. С. Тэйлор, Физ-
МаТГ24.’ Kogyo Kagaku Zasshi, 63, 1879-1883 (1960).
25. S. V. Gunn, J. Am. Rocket Soc., 22, 33 (1952).
96 M. Gerstein, 0. Levine, E. L. Wong, NACA RM, ESO
G 24~(1950); J. Am. Chem. Soc., 73, № 1, 418—422 (1951); Ind. Eng. Chem., 43,
Ko 12, 2770—2772 (1951).
27. P. Wagner, G. L. Dugger, J. Am. Chem. Soc., 77, № 1, 227—
231 (1955).
28. H. S a c h s s e,
E. В a r t h о 1 m e, Z. Elektrochem., 53, № 4, 183—
190 (1949). „ •
29. H. S. Pickering, J. W. Linnet,
Trans. Faraday Soc., 47,
1101—1106 (1951).
30. H. G. W о 1 f h a r d, Z. Tech. Phys., № 9, 206-211 (1943).
31. Основы горения углеводородных топлив, ИЛ, 1960.
32. Горение при пониженных давлениях и некоторые вопросы стабилизации
яламени в однофазных и двухфазных системах, Изд. АН СССР, 1960.
33. Б. В. Р а у ш е и б а х, С. А. Белый, И. В. Беспалов,
В. Я. Б о р о д а ч е в, М. С. Волынский, А. Г. Прудников,
Физические основы рабочего процесса в камерах сгорания воздушно-реактивных
двигателей, Изд. «Машиностроение», 1964.
34. Сб. «Горение в турбулентном потоке», Изд. АН СССР, 1959.
35. G. D a m к б h 1 е г, Z. Electrochem., 46, № И, 601—626 (1940).
36. К. И. Щелкин, ЖЭТФ, 13, № 9—10 (1943).
37. К. И. Щелкин, Я. К. Трошин, Газодинамика горения, Изд.
АН СССР, 1963.
38. Е. М. М и н с к и й, Д. А. Ф р а н к - К а м е н е ц к и й, ДАН СССР,
50, 353 (1945).
39. Б. Н. Баранов, Автореф. канд. дисс., Калинин, 1955.
40. Б. Г. Гаврилов, Е. И. Гулин, А. П. Лесников,
Т. А. Новиков а, ЖПХ, XXXVI (1963).
41. М. О. Scott, R. Stansfield, Т. Tait, J. Inst. Petrol., 37,
№ 333, 487—509 (1951).
42. Л. К p о к к о, И. Чжень Синь, Теория неустойчивости горе-
ния в жидкостных ракетных двигалелях, ИЛ, 1958.
43. Вопросы ракетной техники, № 4, 49 (1959).
44. Ю. X. Ш г у л о в, М. О. Лернер, Горение в жидкостных ракет-
ных двигателях, Оборонгиз, 1961. *• ‘
12
Заказ 792.
СТАБИЛЬНОСТЬ ТОПЛИВ
Стабильностью топлив называется их способность сохранять без
изменения свои эксплуатационные свойства в условиях хранения,
транспортирования и применения.
Стабильность определяется физико-химическими свойствами то-
плив и внешними .условиями, из которых наибольшее влияние ока-
зывают температураг площадь контакта с кислородом воздуха,
свет, каталитическое действие металлов.
Долговечность и экономичность работы любого двигателя обеспе-
чивается применением топлива с определенными эксплуатационными
свойствами [1]. Поэтому для каждого топлива устанавливаются
соответствующие требования к качеству, которые излагаются
в ГОСТах или технических условиях на топливо. При недостаточ-
ной стабильности топлив к моменту применения их качество не
будет соответствовать требованиям ГОСТа, и при работе двигателя
возникнут серьезные неполадки. Так, применение нефтяных топлив,
в которых увеличилась кислотность, значительно ускоряет износ
двигателя; потеря легких фракций в бензинах ведет к усложнению
запуска двигателя; повышение содержания смол в топливах вызы-
вает нарушение нормальной подачи топлива в двигатель, повышает
нагарообразование; изменение качества ракетных топлив часто
вызывает нарушение режима работы двигателя, вплоть до взрыва.
В результате физических или химических процессов происходит
изменение эксплуатационных свойств топлив. Исходя из этого,
введены понятия о физической и химической стабильности топлив.
Физически стабильными называются топлива, в которых под
воздействием изменяющихся внешних условий, не протекают физи-
ческие процессы, способные изменить физико-химические свойства
топлив. Химически стабильными называются топлива, в который
при хранении и эксплуатации не происходит химических реакций,
ведущих к изменению физико-химических свойств.
Физическими процессами, вызывающими изменение эксплуата-
ционных свойств топлив, будут: изменение агрегатного состояния
178
I
.ход из жидкого состояния в твердое или в пар), поглощение
(шп изменение вязкости; химическими — окисление, полимери-
₽ЛП1Я разложение, взаимодействие с конструкционными материа-
33 hi и посторонними примесями в топливе.
11 Необходимо учитывать, что и при физических, и при химических
оцессах изменяются как физические, так и химические свойства
пив. Так, при чисто физическом процессе испарения в остающейся
ч ичкой фазе сложного топлива происходит изменение химического
состава, поскольку испарение отдельных составных частей топлива
различно. В процессе окисления топлива появляются новые вещества,
которые изменяют не только химические свойства топлива, но и такие
чисто физические свойства, как плотность, вязкость, температура
кипения и т. д.
Сущность физических процессов и их влияние на изменение
свойств топлив рассмотрены в предыдущих главах.
ХИМИЧЕСКАЯ СТАБИЛЬНОСТЬ ТОПЛИВ
Некоторые представления об окислении нефтяных топлив
в жидкой фазе
Нефтяные топлива представляют собой сложную смесь угле-
водородов различного строения и некоторого количества сернистых,
азотистых, кислородных и металлорганическнх соединений.
В условиях хранения и транспортирования наиболее химически
активная часть этих соединений может вступать в реакции окисле-
ния, полимеризации и конденсации. Основной реакцией, вызыва-
ющей изменение эксплуатационных свойств углеводородных топлив,
является реакция окисления.
Жидкофазное окисление углеводородов протекает по цепному
механизму с «вырожденным разветвлением» (см. гл. V). Промежу-
точными продуктами, способными давать начало новым реакцион-
ным цепям окисления, являются перекиси. Образующиеся при
окислении перекиси либо подвергаются дальнейшей оксидации
(пероксидации), либо вступают в реакцию с другими промежуточ-
ными продуктами окисления, либо распадаются на два активных
радикала, дающих начало «вырожденному разветвлению» окисли-
тельных цепей.
Скорость и направление превращений перекисей определяется
их химической природой и условиями окисления, причем особенно
большое влияние на направление дальнейших превращений пере-
। кисей оказывает температура. При высоких температурах преобла-
НЙ^ет реакция пероксидации с последующим термическим распадом
образующихся многоатомных перекисей и, по мере роста темпера-
туры реакции окисления, возрастает количество кислот и окси-
,уислот, образующихся при распаде многоатомных перекисей. При
Достаточно высоких температурах многоатомные \ перекиси распа-
даются до мелких осколков; процесс этот сопровождается выделе-
12*
179
пнем такого количества энергии, что происходит воспламенение
продуктов распада. При пониженных температурах скорость пер-
оксидации мала, и основными продуктами распада моноперекпсей
являются спирты и карбонильные соединения (альдегиды и
кетоны). $
Распад перекисей идет по двум направлениям: 1) с образованной
новых, как правило, более стойких продуктов и 2) с образованием
радикалов. Оба вида распада протекают одновременно, по при более
низких температурах в жидкой фазе преобладает первый вид,
при более высоких — второй. «
Примером распада перекисей па радикалы может служить рас-
пад перекиси третичного бутила: ;
СН3 СН.) сн3 сн,
II I I I
СНз-С-О-О-С-СН3 -> 2СН3-С-О-----» 2СН3-С=О+2С1Г Я
сн3 сн3 сн3
При распаде перекисей с образованием новых продуктов характер
последних определяется степенью пероксидации и структурой пере-
кисей. Моногидроперекпси распадаются с образованием спиртов
и карбонильных соединений. При этом первичные моногидропере-
киси образуют альдегиды и воду: j
R-CH2— О-О-Н
Вторичные моногидроперекпси образуют кетон и воду:
R—СНООН—СН3
Н-С-СН3+Н2О
о
Третичные моногидроперекпси образуют кетон и спирт, либо
спирт и атомарный кислород: ,
Rx / ^C=O + R2OH
кДсоон< R1
r2z \ R\
^Ri-^COHH-O
rZ
Дигидроперекиси распадаются с образованием кислот, альдеги-
дов, непредельных соединений, воды и перекиси водорода:
СН3 ,0 /ОН ♦
I *CH3-C<f +R-С< +Н2О
Н-С-ООН' хн -о
। ' Я
Н—С—ООН. /ОН
I ^CH2=CH2 + H2O2 + R-C/
R О
180
Г Альдегиды, образующиеся при распаде перекисей, легко взаимо-
•• вуют с кислородом, образуя нестойкие перкислоты, распада-
Д<1циеся на кислоту и атомарный кислород:
Ю /О /О /О
w Кетоны окисляются значительно труднее альдегидов и длящих,
в основном, характерны реакции конденсации:
R-CH2-C-R1 + R-CH2-C-R1->H2O + R-CH2-C=C-C~ Rr
О О Ri R О
Кислоты способны к дальнейшей пероксидации с образованием
оксикислот:
/О. /О ZO
I R—CH2-C<f + О2->R—CH-C<f ->R—CH-C<f +0
XOH “ | XOH | XOH
OOH OH
Образующиеся при распаде перекисей вещества могут вступать
и в другие реакции, образуя конечные продукты жидкофазного
окисления. Так, альдегиды и кетоны, реагируя с перекисями, обра-
зуют оксиалкилперекиси, распадающиеся с разрывом С—С связи
спирты и кислоты:
z0
/ОН
—>Rr —О—О—CH—R—>RXOH + R—С<
| х0
он
Кислоты, реагируя со спиртами, образуют сложные эфиры:
>/0
R-C<f +r1oh->h2o + r-c-o-r1
Х0Н II
с оксикислотами эфирокислоты:
/О /О /О
R-C<f + RX—CH—C<f —>R—С—О—CH-C<f +Щ0
ХОН | ХОН || | ХОН
ОН О Ri
Оксикислоты в результате распада и конденсации образуют
непредельные кислоты, лактоны, лактиды и эстолиды.
Перекиси, креме распада, могут претерпевать следующие превра-
щения;
Дальнейшая пероксидация
R-CH2-CH2OOH + O2->R-CHOOH-CH2OOH
полимеризация с карбонильными соединениями:
ZR1
ROOH + Ri—С—R2->R—0-0—С<
II I XR2
О он
181
окисление исходных веществ:
^\-ООН
О
-ОН
внутримолекулярная перегруппировка:
/О-О-
СН3-СН(
о X
п /СНз-СН-ООН
п
он
полимеризация с непредельными углеводородами с разрушением
и без разрушения перекисной группы: ' J
ОН ,1
з
О
R—СН=СН2+СН3—СН—Rx—
СНз-СН-R
оон
R-CH-CH
о
СН3—CH-R,
Первичное присоединение кислорода к атому углерода опреде-
ляется реакционной способностью водородных атомов, их количе-
ством при данном атоме углерода и в некоторых случаях структурно-
объемными особенностями строения молекулы. В общем случае
возможность первичного присоединения кислорода возрастает в ряду:
первичный, вторичный и третичный атом углерода.
Работами К. И. Иванова [2, 3] установлены следующие законо-
мерности направления первоначального присоединения кислорода
к углеводородам различного строения.
1. У насыщенных углеводородов первоначальное присоединение
кислорода идет к третичному атому углерода, более редко — к вто-
ричному и еще реже к первичному. Наличие четвертичного атома
углерода рядом с третичным снижает активность атомов водорода
и значительно замедляет процесс окисления:
сн3
СН3
СН3—СН + о
2--> СНз-С-О-О-Н
сн2
сн
сн3
2. У алкановых углеводородов нормального строения присоеди-
нило кислорода происходит преимущественно к 0-углероду:
СН3-СН3-(СН2)Х-СН3 + О2---СН3 —СН —(СН2)Х—СН3
I
ООН
3. У циклано-ароматическпх углеводородов без боковых цепей
*)jfсоедипение происходит к углероду цикланового кольца в со-
положении: z , -
/О-О-Н
СН СН2 СН СН
4. У ароматических углеводородов присоединения кислорода
к атомам углерода бензольного кольца не происходит, однако аро-
матическое ядро активизирует процесс окисления и ароматические
углеводороды с боковыми цепями окисляются быстрее других угле-
водородов, за исключением ненасыщенных. Разница в окислении
первичного, вторичного и третичного углерода при этом проявляется
Ш‘нее четко. При наличии двух боковых цепей первоначальное
окисление происходит в более короткой боковой цепи:
/СНа-СНз /СН—СН3
С С ООН
НС СН НС СН
г I II О2---* | ||
НС сн НС сн
сн ^сн
сн3 Н2СООН
С ' с
HCZ сн нсГ ^сн
I II + о2 ► I и
нс сн НС сн
С CZ
I СН3—сн—сн3 сн3—сн—сн3
У ненасыщенных углеводородов окисление идет очень легко*
пРи низких температурах. Первоначальное присоединение
’“'орода происходит без разрыва двойной связи к углероду, сосед-
й°му с двойной связью:
СН2=СН-СН2-СИ2-СН3 + О2->СН2=СН-СН-СН2-СН3
ООН
I
183
Стабильность углеводородов к окислению, под которой подразу-
мевается минимальная глубина и скорость окислительных процессов
при контакте с кислородом, определяется не только легкостью обра-
зования перекиси в начальной стадии окисления, но также характе-
ром продуктов превращения перекисей, их способностью к развет-
влению окислительных цепей.
Скорость окисления и характер конечных продуктов различу
при окислении углеводородов в жидкой и паровой фазах. При окис-
лении в жидкой фазе молекулы кислорода контактируют в основном
с молекулами углеводородов, расположенных у поверхности раздела
жидкость — воздух, и только незначительная часть молекул
углеводородов внутри объема жидкости может контактировать с
кислородом, растворенным в нефтепродукте. Вследствие высокой
концентрации исходных веществ в жидкой фазе длина окислитель-
ных цепей должна быть большой, и возникновение первоначальных
перекисей будет вызывать развитие длительных окислительных це-
пей по всей массе продукта. д
Низкие температуры окисления в жидкой фазе способствуют
развитию реакций полимеризации и конденсации. Реакции перокси-
дации и распада протекают в незначительной степени. Поэтому
углеводороды, окисляющиеся при нормальных температурах, в ос-
новном образуют кислородные соединения и смолистые вещества
€ большим молекулярным весом, чем у исходных углеводородов
При окислении в паровой фазе за счет более высокой темпера-
туры преобладают реакции пероксидации и распада окисляющихся
молекул на более легкие. При достаточном количестве кислорода
и высокой температуре окисление в паровой фазе заканчивается
образованием СО2 и Н2О. При недостатке кислорода или недоста-
точно высокой температуре окисление углеводородов не протекает
до конца и образуются вещества с высоким содержанием углерода.
Стабильность к окислению в жидкой фазе у углеводородов пони-
жается в следующем ряду: ароматические углеводороды без боковых
цепей; циклановые и алкановые углеводороды нормального строе-
ния; углеводороды, содержащие в своей молекуле циклановые
и ароматические кольца; ароматические углеводороды с боковыми
цепями; ненасыщенные углеводороды с одной двойной связью;
ненасыщенные углеводороды с двумя двойными связями. Указанный
ряд справедлив для углеводородов приблизительно одинакового
молекулярного веса. При большом различии в молекулярном весе,
в силу того что с повышением его стабильность к окислению у всех
групп углеводородов снижается, указанный выше порядок может
быть нарушен. J
В результате окисления в жидкой фазе алкановых и циклановых^
углеводородов, в основном, образуются спирты, кислоты, оксикКЗй
лоты и карбонильные соединения. Продукты полимеризации и кон-
денсации получаются в небольшом количестве при длительном окис-
лении циклановых углеводородов. При окислении ароматических
углеводородов с короткими боковыми цепями основными конечными
184
/
унтами окисления являются продукты уплотнения (смолы и
Пр()Дие высокомолекулярные соединения). С увеличением длины
цепей наряду со смолами образуются и кислые продукты.
И Насыщенные соединения при окислении образуют продукты поли-
ризации, составляющие основную часть смолистых веществ.
При совместном присутствии углеводородов различных групп
, например, в нефтяных топливах) малостабильные углеводо-
оды, легко вступая в окислительные реакции, инициируют окисление
более стабильных углеводородов. Вследствие цепного механизма
окисления даже небольшое количество нестабильных углеводородов
способствует значительному окислению всего топлива. Так, смесь
бензина прямой перегонки с 68% алкеновых углеводородов в отсутст-
вие ненасыщенных углеводородов с двумя связями имела индукцион-
ный период 7 ч. Этот же бензин с 31 % алкеновых углеводородов
(гептена-2), но с добавкой 2% 2,3-диметилбутадиена-1,3 практически
не имел индукционного периода при окислении.
Влияние углеводородного состава на стабильность топлив
Влияние углеводородного состава на стабильность топлив было
детально изучено на реактивных топливах Т-1, TG-1 и Т-5 [4, 5 К
Предварительно обессмоленные при помощи силикагеля исходные
топлива хроматографическим путем разделялись на алкано-цикла-
новую фракцию (АЦ), ароматическую (Ai), состоящую из моноци-
клических ароматических углеводородов с боковыми цепями, и аро-
матическую (А2), содержащую би- и трициклические ароматические
углеводороды с боковыми цепями. Сернистые соединения из арома-
тических фракций удалялись перекисью водорода и при помощи
никеля Ренея. При удалении сернистых соединений на никеле Ренея
непредельные углеводороды не разрушались.
Исходное топливо и полученные фракции нагревали при 150° С
в течение 6 ч, при этом за счет значительной интенсификации про-
цесса окисления образовывались продукты окисления в виде кислых
соединений, смол и нерастворимых осадков.
Алкано-циклановая фракция реактивных топлив в этих условиях
нерастворимых осадков и отложений не образует, однако появление
после нагревания кислотности свидетельствует о протекании про-
цесса окисления. В алкано-циклановой фракции топлива Т-5, содер-
жащей углеводороды с более тяжелым молекулярным весом, наблю-
дается появление и фактических смол в количестве до 1 мг на 100 мл
Фракции. Как показали исследования, проведенные Н. ,П. Галичем
18L при более длительном нагревании алкано-циклановой фракции
^кет появляться некоторое количество осадков (особенно при
окислении углеводородов с большим молекулярным весом) в резуль-
тате вторичных реакций продуктов окисления.
При нагревании ароматической фракции образуется значитель-
ное количество нерастворимых осадков и смол, а также карбоновых
Кислот. Увеличение цикличности ароматических углеводородов
185»
I
приводит к возрастанию осадко- и смолообразования. Наличие в аро-
матических углеводородах боковых цепей с ненасыщенными связями
также увеличивает их способность образовывать нерастворимые
в топливах осадки. Увеличение молекулярного веса ароматических
углеводородов ведет к повышению количества образующихся при
нагреве смол и осадков. Так, во фракции Ai из топлива ТС-1 пос^<
нагревания образовалось 2,8 мг осадков и 6,3 мг фактических смол
на 100 мл, а фракция Ai топлива Т-1 имела 3,1 мг осадков и 9,1 мг
смол. Поэтому более тяжелые топлива, даже при одинаковом соот-
ношении углеводородных групп, должны иметь худшую стабильность
по сравнению с топливами более легкого фракционного состава.
Таблица 33
Стабильность углеводородных фракций топлива ТС-1
Фракция
Осадок,
Л!г/ 10 0 мл
Смолистые
отложения
на бронзе,
г / м 2
Фактические
смолы,
мг 1100 мл
Кислотность,
мг КОН /100 At л
Исходное обессмоленное
топливо ТС-1 . . . .
АЦ....................
Aj после удаления сер-
нистых соединений на
никеле Ренея . . . .
Aj после удаления сер-
нистых соединений пе-
рекисью водорода. .
А2 после удаления сер-
нистых соединений на
никеле Ренея . . . .
А2 после удаления сер-
нистых соединений пе-
рекисью водорода . .
4,0
О
2,8
2,0
0,2
0
0,2
0,2
4,9 * 0,5
4,0
0,4
8,3
0
6,3
6,0
10,1
9,4
1,2
2,7
2,3
2,1
2,4
2,0
Таблица 34
Влияние ароматических углеводородов на стабильность
топлив
Ароматические фракции, добавляемые
к алканоциклановой фракции
Содержание арома-
тических фракций
в смеси, вес. %.
О‘С а д о к, лгг/100
Aj топлива ТС-1 после удаления
сернистых соединений на никеле
Ренея ..........................
А2 топлива Т-1 после удаления
сернистых соединений на ни-
келе Ренея .....................
мл
0,8
5
ю
0
186
алдпмому, именно этим следует объяснить меньшее осадкообразо-
qinie при нагревании в топливах типа ТС-1 по сравнению Т-1 и Т-5,
в< сМотря на несколько большее содержание в ТС-1 сернистых соеди-
н *»
яеНд* табЛе 33 приведены данные анализа фракций топлива ТС-1
после нагревания.
велпченпе содержания ароматических углеводородов в топли-
закономерно приводит к росту количества образующихся при
продуктов окисления
вах
наГревании растворимых и нерастворимых
(табл. 34).
Влияние гетероорганических соединений на
стабильность топлив
Гетероорганические соединения оказывают значительное влияние
на стабильность нефтяных топлив. Наиболее детально изучено влия-
ние сернистых и азотистых соединений, смолистых веществ [4—14].
В нефтяных топливах присутствуют следующие сернистые соеди-
нения: меркаптаны, сульфиды, дисульфиды, тиофаны и тиофены.
Кроме того, в небольших количествах могут находиться сероводород
п элементарная сера. Строение остальных сернистых соединений,
входящих в состав нефтяных топлив, точно не установлено.
Элементарная сера в реактивных и дизельных топливах содер-
жится в пределах десятитысячных долей процента и практически
отсутствует в бензинах; в таких малых концентрациях ее влияние
на стабильность топлив почти не сказывается. Однако необходимо
учитывать, что повышение содержания серы ускоряет смолообразо-
вание в нефтяных топливах (табл. 35).
Таблица 35
Влияние элементарной серы на смолообразование в топливах при 120° С
в течение 1 ч
Топлива, к которым добавлялась сера
Содержание серы, вес. %
0 0,002 0,004 0,006 0,008 0,01
Смолистые отложения на бронзе,
айт-спирит .•.....................
Дизельное топливо из бакинских неф-
тей ...............................
Реактивное топливо из волжских неф-
тей .
0
0,2
0,2
1,0
1,1
1,2
10,0
3,6
8,0
г /м2
13,0
14,5
18,3
16,0 24,0
17,8 29,0
19,8 24Д
Меркаптаны присутствуют во всех нефтяных топливах и содержа-
ще их может достигать сотых долей процента. Вследствие высокой
Омической активности меркаптаны снижают химическую стабиль-
ность топлив и при нагревании способствуют увеличению осадко-
11 смолообразования в топливах [4, 5]. На рис. 73 показано содержа-
ние смол в бензине при добавлении различных меркаптанов в коли-
честве 0,1 вес. % (в пересчете на серу).
187
Однако следует учитывать, что в малых концентрациях сераорга*
нические соединения способны оказывать ингибирующее действие
па процессы окисления топлив. *1
На рис. 74, 75 приводятся данные Г. Ф. Большакова, который
изучал влияние сераоргаипческих соединений на термоокислитель-
ную стабильность углеводородных топлив.
Из приведенных данных видно, что в присутствии меркаптайЛ
уменьшается поглощение кислорода и
ВО
5 10 15 20
время хранения Вензина при 70 'С, ч
Рис. 73. Зависимость смолообразования
в бензине от добавок различных меркап-
танов:
1 — исходный бензин без добавки; 2 — с амил-
меркаптаном; з — с тиофенолом; 4 — с доде-
цилмеркаптаном; 5 — с тетрадецилмеркаптаном;
6 — с гексадецилмеркаптаном.
при определенных концентра-
циях — образование нерас-
творимых осадков.
Интересно отметить, что
структура углеводородного
радикала существенно влияет
па образование нераствори-
мых осадков. В алифатиче-
ских углеводородах меркап-
таны с алифатическим ради-
калом образуют осадков
меньше, чем меркаптаны с
ароматическим радикалом.
Наоборот, в ароматических
углеводородах в присутствии
ароматических меркаптан Л*
образуется меньше осадков,
чем в присутствии алифати-
ческих меркаптанов. Ана-
логичные явления происхо-
дили и при окислении угле-
водородов с другими сера-
органпческими соединениями
[13, 14].
Однако при повышенных концентрациях сернистых соединений
стабильность топлив резко ухудшается.
Все сернистые соединения по их влиянию на уменьшение термо-
окислительной стабильности при концентрациях 0,2% S и более
располагаются в следующий ряд: меркаптаны, дисульфиды, суль-
фиды, тиофаны, тиофены.
В табл. 36 приведены обобщенные данные по предельному содер-
жанию сернистых соединений в топливах при 100 и 150° С. После
превышения указанных концентраций сернистых соединений наблю-
дается интенсивное уменьшение стабильности топлив [4].
В настоящее время весьма актуальным является вопрос о пере-
смотре допустимого содержания общего количества сернистых соеди-
нений в топливах, а также ограничения содержания сернистых соеди-
нений по группам.
Смолистые вещества также оказывают отрицательное влияние
на стабильность топлив, причем с повышением содержания их ста-
бильность ухудшается (табл. 37).
188
Таблица 36
Предельное содержание сернистых соединении
Сернистые соединения
I
Температура,
°C
Предельно допустимая
концентрация (в пере-
счете на серу), вес. %
Меркаптаны •
Тиофены .................
Сульфиды ароматические .
Сульфиды алифатические .
Дисульфиды ..............
100
150 и более
100
150
150
100
150
100
150
0,005
0,002
0,20
0,10
0,05
0,10
0,08
0,05
0,02
Содержание фенил меркаптана > %
74. Зависимость термоокислитель-
ной стабильности некоторых углеводо-
родов от содержания в них н-нонил-
меркаптана:
1 — цетан; 2 — изопропилбензол; 3 — ги-
дрированное топливо.
Рис. 75. Зависимость термоокисли-
тельной стабильности некоторых
углеводородов от содержания в них
фенилмеркаптана:
1 — цетан; 2 — изопропилбензол;
3 — гидрированное топливо.
189
При хранении в топливах интенсивно накапливаются смолы,
строение которых существенно отличается от первоначальных смол,
переходящих в топлива из нефти (табл. 38).
Таблица 37
Зависимость стабильности топлива Т-1 от содержания
в нем смолистых веществ
Концентрация
смолистых
веществ до
нагревания,
мг/100 мл
Нерастворимые продукты окисления после
6 ч нагревания при 150° С
осадок, мг/100 мл
смолистые отложе-
ния иа’бронзе, г/м2
0
50
100
150
10,4
0,3
0,6
Образующиеся при хранении смолы характеризуются большими 1
иодными и кислотными числами. Содержание серы и азота в смолах, 1
образующихся при хранении, по сравнению со смолами, выделен-
ными из исходного топлива, уменьшается, а содержание кислорода
возрастает. Молекулярный вес, плотность и показатель преломлен
изменяются незначительно.
Таблица 38
Характеристика смолистых веществ, выделенных из топлива ТС-1 [9]
Смолы, выделенные из топлива
после 12 месяцев хранения
Показатели
VI ,20
Плотность а.
20
Показатели преломления
Молекулярный вес . .
Иодное число, г J2/100 з
Кислотное число, мг КОН/1 г
Поверхностное натяжение,
дин/см ....................
Элементарный состав, %
углерод ...................
водород ...............
сера...................
азот...................
кислород ..............
Выход, вес. % ...........
свеже-
получеи-
ного
0,9573
1,4957
185,4
2,61
27,9
78,04
9,18
0,045
С
предвари-
тельно
обессмолен-
ного
0,9671
1,5137
192,3
90,8
76,31
9,02
0,08
10,21
0,183
дважды
обессмо-
ленного
0,9689
1,5184
198,4
110,4
40,4
30,1
77,57
8,91
1,21
0,04
12,30
трижды обес-
смоленного
0,9678
1,5117
197,5
115,3
63,8
77,59
8.51 * '
0,80 '
не обнаружено
13,16
0,598
190
I ц своему влиянию на стабильность топлива смолистые вещества
весьма
янно увели'
Jm, хранении, в то время
осадка в присутствии смол,
выделенных из свежеполу-
Г.пного топлива, имеет
минимум при концентра-
циях 0,04-0,0/ вес. о
(рис. 76).
L nnvr от друга. Осадкообразование в топливе посто-
ОТЛИЧИ**! /Аг? ' г
швается с повышением концентрации смол, образующихся
как кривая образования нерастворимого
О механизме образования
смол и нерастворимых
осадков в нефтяных
топливах
Образование смол
нерастворимых осадков
нефтяных топливах приво-
дит к значительному ухуд-
шению их эксплуатацион-
ных свойств. Нераствори-
мые осадки вызывают
засорение фильтров и ухуд-
шают прокачиваемость топ-
лив, смолы увеличивают
нагарообразование топлив,
а при отложении на стейках
шение нормальной работы
в карбюраторных двигателях па штоках и тарелках впускных кла-
панов препятствует нормальной посадке клапанов в гнездах
II
Содержание смол, Вес. %
выделенные
выделенное
Рис. 76. Влияние смолистых веществ раз-
личного состава на образование осадков
при нагревании топлива ТС-1 [9]:
7 — смолы, выделенные из свежего топлива;
2 — смолы, выделенное после года хранения
обессмоленного топлива; 3 — смолы, выделенные
после года
топлива;
хранения дважды обессмоленного
лива; 4 — смолы, выделенные после года
хранения трижды обессмоленного топлива.
топливных систем вызывают нару-
двигателя. Так, отложение смол
и вызывает их зависание, что ведет к уменьшению давления
в камерах сгорания и к нарушению фаз газораспределения.
Выделение продуктов окислительной полимеризации в дизельных
топливах вызывает зависание игл распылителей форсунок, вследст-
вие чего топливо либо вовсе перестает поступать в камеру сгорания,
либо не распыляется и подается сплошной струей. На поршне
двигателя, на внутренних стенках цилиндров и головок двига-
телей, на впускных клапанах образуются отложения, ускоряющие
износ двигателя и вызывающие пригорание поршневых колец.
В табл. 39 приведены данные по влиянию содержания смол
в бензинах на работоспособность автомобильных двигателей [15].
Увеличение содержания смол в нефтяных топливах и появление
твердых осадков при хранении и применении топлив происходит
в первую очередь за счет окисления нестабильных компонентов
топлив 15, 16]. Исходные свежеполученные топлива можно рассмат-
ривать как истинный раствор сернистых, кислородных, азотистых,
Металлорганических соединений и смолистых веществ в углеводород-
191
Таблица 39
Влияние содержания смол в бензинах на состояние и работоспособность
автомобильных двигателей
Содержание
фактических
смол,
мг 1100 мл
Состояние двигателей после 50 ч работы
Возможный пробег
автомобилей до по-
явления неисправ-
ностей в двигателе,
тыс. км - •
до 10
11-15
16-20
26-30
Во впускной системе и в цилиндрах отложе-
ний нет
Слабые следы отложений на клапанах и стен-
ках впускной трубы
Небольшие отложения на стенках впускной
трубы, камере сгорания и на клапанах
Заметные отложения в узлах системы пита-
ния и камере сгорания
Значительные отложения на стенках впуск-
ной трубы, камеры сгорания и клапанах.
Уменьшение сечения впускной трубы на
20-25%
Неограниченный
16
До 5
В определенных условиях при повышении
температуры
нои среде.
и при контакте с кислородом нестабильные углеводороды и гетеро-
органические соединения окисляются, что приводит к увеличению их
молекулярного веса.
। .
— I —
— I —
Рис. 77. Схема экспериментальной установки
для исследования интенсивности светорассея-
ния в реактивных и ракетных топливах при
высоких температурах: ' I
1 — электронный потенциометр; 2 — блок питания
и усиления;
4 — фотоумножитель;
ризатор;
3 — предварительный усилитель;
5 — спектрограф; 6 — поля-
7 — ртутная лампа; 8 — кювета. л
Первичные продукты окисления полностью растворимы в топливах,
но в дальнейшем, по мере увеличения глубины окисления и услож-
нения состава, становятся нерастворимыми. Вследствие этого в топ-
ливах гомогенная система превращается в гетерогенную или коллот^
ную, в которой постепенно увеличиваются размеры частиц второй
фазы. Увеличение размеров коллоидных частиц происходит в резуль-
тате агрегации мелких частиц в более крупные. Продукты окисле-
ния всегда полярны и имеют значительный дипольный момент. Эти
частицы несут, как правило, определенный электрический заряд.
192
> ше электрического заряда частиц объясняется сле-
Возникиовш на поверхности коллоидной частицы, диссоцпи-
мо раствор ионы одного знака, а ионы протпвополож-
nvH» посылают в
|О1.о знака остаются на по-
верхности частицы. Дпссоцп-
аппи, очевидно, подвергаются
^.рерву10 очередь молекулы с
конными связями (меркаптиды,
сульфиды металлов и др.). Из-
менение фазового состояния топ-
лив при нагревании изучалось
на специальной установке, по-
казанной на рис. 77 117]. По
изменению интенсивности пер-
пендикулярной составляющей
рассеянного света при наблю-
дении под углом 90° к пада-
ющему свету были обнаружены
частицы с размерами значи-
тельно меньшими, чем длина
волны. Частицы, соизмеримые
с длиной волны падающего
Й^ета, изменяют также интен-
сивность и параллельной соста-
вляющей рассеяного света. По
характеру перпендикулярной и
параллельной
65
15
10
го
60 60 100 120 1^0 160 L°C
Рис. 78. Зависимость интенсивности
светорассеяния I в исходном (7) и
очищенном (2) топливах TG-1 от тем-
пературы.
-----------составляющих
можно судить о величине 117]
образующихся нерастворимых
частиц (рис. 78).
Значительное влияние па
коагуляцию мелких частиц в
более крупные оказывает внеш-
нее электрическое поле. В ре-
альных топливных системах,
особенно при повышенных тем-
пературах, иногда наблюдается
возникновение различных элек-
трических полей. Особенно ча-
сто возникают так называемые
термотоки, что обусловливается
нагреванием двух контактпру-
• ^цих металлов. Эти термотоки
!^ц,их металлов. Эти термотоки c,,ot °'полярность, большой
Очевидно, что молекулы, имеющие >ц Удь 1 группу, будут
Дипольный момент и активную фунщ ТПГПРЯ нерастворимого
также способствовать укрупнению ооразую е укрупнение
осадка. Именно этим следует об^яс“’”Ь “”1ИЧества меркаптанов,
осадка в присутствии определенно! 4 * <
193
13 Заказ 792.
которые имеют чрезвычайно активную группу SH, обусловливающую,
кроме того, высокую полярность молекулы. Меркаптаны активно
взаимодействуют с металлами, особенно с медью, с образованием
\),СП
Рис. 79. Инфракрасные спектры поглощения нерастворимых осадков, образо-
вавшихся при нагревании топлив в течение 4 ч при 150° С:
1.2 — топлива Т-1 и ТС-1 в присутствии меди; з — СиЗО^бНгО; 4, 5 — топлива Т-1
и ТС-1 в стеклянном сосуде. Осадок высушен при следующих температурах; -----40° С;
-------125° С; —.—.—300° С; * — полосы, относящиеся к возелиновому маслу. •
соответствующих меркаптидов. Меркаптиды в дальнейшем могут
диссоциировать на ионы, что является очень важным для обра-
зования двойного электрического слоя коллоидных частиц.
194
льфпдах атом серы экранирован углеводородными радика-
’ ” СУ. ~miv в их присутствии частицы нерастворимого осадка коагу-
лпруют’в меньшей степени.
' р писульфидах два атома серы также экранированы углеводород-
и радикалами, 110 связи междУ атомами серы являются непроч-
\),СН
Рис. 80. Инфракрасные спектры поглощения осадков, образовавшихся
в топливной системе реактивного двигателя:
2 на фильтрах; 2 — перед форсункой; 3 — на кольцах форсунки.
ными. При температуре 150° С и выше дисульфиды начинают распа-
даться с образованием меркаптанов и сероводорода, которые вызы-
вают интенсивное осадкообразование.
Были исследованы состав и структура осадков, а также изучена
кинетика накопления их в топливах и углеводородных фракциях
^лабораторных и эксплуатационных условиях методами инфракрас-
ном, ультрафиолетовой,
V1AVAAA1U1A VIA VAX A j* V 11 U
структурного анализа [7, 13, 14, 18, 21]. Показано
осадков содержатся сульфон
оповых кислот, сульфаты
ПАплг..- ~ *
эмиссионной спектроскопии и рентгено-
________________________________________________•, что в составе
ы, соли сульфокислот, тиокислот, кар-
оси, 7 сложные эфиры тио- и оксикислот, кар-
е кислоты, окси-, тио- и сульфокислоты (рис. 79, 80).
13* •
195
Рентгенографическими исследованиями [13] строения осадков
установлено, что в отсутствие металлов в топливах образуются осадкц
в основном аморфного строения, содержащие только 2—3% кргь
сталлических веществ, в присутствии же металлов они содержат
40—50% кристаллических веществ, состоящих в основном из солей,
в первую очередь серной кислоты.
На основании изучения состава и структуры осадков, а таюцв
промежуточных продуктов окисления [13, 14, 18] предложена сле-
дующая схема образования осадков при нагревании углеводородных
топлив, содержащих сернистые соединения (стр. 197).
Справедливость указанной схемы была проверена Г. Ф. Больша-
ковым и Е. А. Глебовской на многих модельных системах и реаль-
ных топливах. В качестве примера можно привести результаты окис-
ления п-гексадекана, а-гексплтиофана и их смеси.
Окисление чистого п-гексадекана характеризуется образованием
и накоплением кислородных соединений (рис. 81): групп С = 0 (1720
и 1420 см"1), групп ОН (3400 см"1), связей С—О (1280, 1240 и
1170 слг1). Соотношение интенсивностей полос поглощения указывает,
что окисление идет в направлении, показанном на схеме (стр. 202).
При окислении в течение 4 ч количество кислот увеличивается
(1720, 1420, 940-960 см'1).
Содержание гидроксильных групп (3440 ем-1) оказывается наи-
большим после 2 ч окисления. Это соответствует интенсивному обра^.
зованию гидроперекисей, которые затем подвергаются дальнейшему
окислению (полоса 3440 см
ее интенсивность),
лекулы п-гексадекана с отщеплением крайних углеродных ато-
мов, о чем свидетельствует уменьшение поглощения групп СН2
(720 см-1). |
Инфракрасные спектры поглощения продуктов окисления чистого
а-гексилтиофана представлены на рис. 82. Отчетливо видно окисле-
ние бокового радикала с образованием групп С = О (1720, 1420,
940 см-1), групп ОН (3500—3400 си"1). Инфракрасные спектры сви-
детельствуют о разрыве пятичленпого кольца по сопряженной с ра-
дикалом связи С—S (обращение максимума 1260 см-1, исчезновение
полосы поглощения связи С—С 630 см"1). Одновременно происходит
окисление атома серы (после 70 мин), в заметном количестве возни-
кают группы \s=O(1050 II 1025
становится более размытой п изменяется
При окислении происходит деструкция мо-
см-1), а после 4 ч — группы
(1290 и 1160 см-1)
(1290 п 1160 см-1) и структуры О—SO2, О—SO2—О— (1200,
1370 п 1420 см'1). По-видимому, первоначально образовав-
шиеся сульфоокиси окисляются далее в сульфоны и сульфокис-
лоты.
Разрыв кольца по связи С—S обусловлен низкой энергией связи
С—S (56,7 ккал, связь СН 98 ккал) и подтверждается образованием
непредельных связей (1640—1630, 540 см"1). Полосы 700 и 980 см"1,
интенсивность которых растет со временем окисления, могут быть
196
3
н
аз
197
Рис. 81. Инфракрасные спектры поглощения продуктов окисления
и, ом'
н-гексадекана в кислороде.
Снято с компенсацией в кювете переменного слоя относительно исходного н-гексадекана.
--------Исходный н-гексадекан.
198
* ис* 82. Инфракрасные спектры поглощения продуктов окисления а-гексил-
тиофана в кислороде при 150° С.
Снято с компенсацией в кювете переменного слоя относительно а-гексилтиофана.
----Исходный а-гексилтиофан.
199
отнесены к непредельным связям типа CHRi=CHR2 (цис- и тринс-
соответственно), подтверждая распад кольца по схеме:
СН ---СН
.-(СН2)6СН3—’\с СН=СН-R —► \в СН=СН-R--
s/ \ (Ao J
' —HOSO2-(CH2)3CH=CH-R
Появление полосы 820 см"1 и увеличение ее интенсивности со
временем окисления указывает на образование непредельных соеди-
нений типа CRjRg = CHR3 и отсутствие структур CHRX = СН2
и CRxR2 = СН2. Это свидетельствует о том, что отделения углеводо-
родного радикала от тиофанового кольца в процессе окисления не
происходит. '
Интенсивное образование групп С = О (1720 слС1) происходит,
очевидно, по второму углеродному атому от конца цепи. Число групп
СН2 в длинных цепях уменьшается очень медленно (обращение мак-
симума 720 еж-1), что вполне понятно, так как одновременно идут
два противоположных процесса — уменьшение общего числа групп
СН2 в цепи за счет окисления и увеличение их числа за счет раскры-
тия кольца. Образование групп ОН также выражено слабо. По-види-
мому, основная масса карбонильных групп входит в структуры алг.С
дегидов (2720 сж“1) и кетонов. Таким образом, присутствие атома
серы в самой молекуле уже задерживает окисление, не допуская
образования кислот, которые в большом количестве образуются при
окислении нормального гексадекана.
Результаты окисления смеси а-гексилтиофана с н-гексадеканом
показывают, что присутствие а-гексилтиофана существенно изменяет
течение процессов окисления обоих компонентов смеси. Из приве-
денных на рис. 83 инфракрасных спектров видно, что, как и в случае
окисления смеси с изопропилбензолом [18], совершенно прекра-
щается образование групп ОН (нет поглощения 3500—3400 слг1).
Очевидно, цепь окисления обрывается на гидроперекиси. Если бы
это обусловливалось преимущественным окислением атома серы, то
продукты, содержащие связи S—О, выявились бы значительно
ярче. В действительности в спектрах смеси можно обнаружить погло-
щение, по-видимому, относящееся к группам ^)S=O (1050 и 1030 см"1},
но не наблюдается поглощения, указывающего на образование
групп ^>S = O. Эти продукты, очевидно, не образуются. Образование
групп С = О также резко заторможено.
Разрыв пятичленного кольца выявляется отчетливо (обращение
максимума 1260 о4), но гораздо менее интенсивно, чем при окисле-
нии чистого а-гексилтиофаиа. Таким образом, можно говорить о
торможении процессов окисления в смеси.
200
Т ij основании экспериментальных данных можно представить
ВТ ^юший механизм окисления я-гексадекана, а-гексилтиофана и
сЛСД\К( П справедливость которого была подтверждена и детальными
1,Х пческими исследованиями (см. схему на стр. 202).
хиМ|1рИ окислении чистого я-гексадекана образования твердой фазы
> происходит, однако при окислении смеси а-гексилтиофана с н-гек-
Рис. 83. Инфракрасные спектры
н-гексадекана с
поглощения продуктов окисления 150° С
а-гексилтиофаном в кислороде.
Снято с компенсацией в кювете переменного слоя относительно исходной смеси.
садеканом возникает твердый осадок за счет взаимоействпя продук-
тов окисления а-гексилтиофана и я-гексадекана. Количество осадка
возрастает с увеличением времени окисления:
Количество осадка,
мг /100 мл
Время окисления,
ч-
0,5
4 12,3
6 16,0
Образующиеся промежуточные продукты взаимно тормозят окис-
нпе исходных компонентов.
рЬ1^ е110сРодственное участие в осадкообразовании металлов, с кото-
тон Коптактирует топливо, доказывается тем, что при нагревании
ив в контакте с бронзой в осадках обнаружено 3—10% меди,
201
контакте
ИР гте со сталью 1—3% железа и с дюралюминием 1—3%
алюминии 17 b пдастмасс в качестве внутренних покрытий резервуа-
ПРиме^1пения ТОплив уменьшает скорость образования смоли-
ЯхЛЛвЯеЩеств (табл. 40). Таблица 40
№ чяписимость смолообразования в бензине А-72 при хранении
I Зависимое внуТреннего покрытия баков [1]
Характер внутреннего покрытия бака
Освинцованный . . .
Бакелитированный . .
Бакелитированный . .
Концентрация кислорода,
Емкость
бака,
л
150
150
60
обмен
Содержание фактических смол
в топливе при хранении, мг/100 мл
исходное
газовой
через 9 ме-
сяцев
через 14,5
месяцев
8-10
8-10
среды,
15-17
8-11
соотношение
жидкой и паровой фаз [1], как уже отмечалось, в значительной сте-
пени влияют на скорость образования смол и осадков (табл. 41, 42).
Таблица 41
Влияние герметизации бака автомобиля ЗИЛ-150 на процесс
смолообразования
Содержание фактических смол в бензине при
хранении, лгг/100 мл
Бак
исходный
через
1. месяц
через
2 месяца
через
3 месяца
через
4,5 месяца
обычной пробкой . . .
герметической пробкой
16
Таблица 42
Зависимость скорости смолообразования от степени
заполнения тары
Заполнение
емкости, %
100
исходный
Содержание фактических смол в бензине
при хранении, мг! 100 мл
через 3 ме- сяца через 6 ме- сяцев через 10 ме- сяцев
6 8 18
12 36 78
Присутствие воды в топливах ускоряет процесс смолообразования
(табл. 43), поэтому при хранении топлив необходимо периодически
удалять влагу, накапливающуюся в емкостях вследствие обратимой
гигроскопичности нефтяных топлив [1].
Таблица 43
Влияние воды на смолообразование
Топливо Условия хранения — Содержание смол в топливе при хранении, лгг/ЮО мл . 4
исходное через 1 месяц через 2 месяца через 3 месяца через 4 месяца через 6 месяцев
Керосин Без воды 8 — 84 116 156
С водой 8 — - 154 - — 206 291
Бензин Без воды 4 4 — - О’ . 8
С водой 4 < 6 ‘ 10 — - 22
В образовании нерастворимых в топливах осадков, кроме серни-
стых и других гетероорганических соединений, весьма активную
роль играют непредельные углеводороды и металлорганические со-
единения, присутствующие в топливе [5, 7]. Подтверждением этого,
является обнаружение в составе золы нерастворимых осадков боль-
шей части зольных элементов исходных топлив. Мельчайшие частицы^
продуктов коррозии металлов и окружающей пыли, [проникающих'
в топлива, являются как бы центрами, вокруг которых агрегируются
частицы высокомолекулярных гетероорганических соединений. Уда-
ление зольных элементов из состава топлив привело бы к значитель-
ному уменьшению осадкообразования.
Химическая стабильность компонентов ракетных топлив
Химическая стабильность компонентов ракетных топлив опреде-
ляется способностью веществ, входящих в их состав, вступать в хими-
ческое взаимодействие друг с другом и с посторонними веществами,
например с кислородом воздуха, металлами и т. д. Для некото-
рых компонентов изменение их свойств может наступать за счет
химического разложения.
При взаимодействии компонентов ракетных топлив с металлами
образуются соединения, как правило, плохо растворимые в основном
продукте, выпадающие в осадок и вызывающие загрязнение продукта.
При хранении компонентов ракетных топлив в емкостях из метал-
лов, слабо подверженных коррозии, сроки хранения определяются
в основном временем, в течение которого физико-химические свойства
топлив не изменяются сверх допустимых пределов. Для увеличение
времени хранения вибираются металлы, наиболее стойкие к воздей-
ствию данного компонента, и добавляются вещества, тормозящие
взаимодействие металла с компонентом. Удачный выбор таких
веществ (ингибиторов) позволяет длительное время хранить в метал-
лических резервуарах весьма коррозионные компоненты ракетных
204
топлив-
ем
окислители на основе азотной кислоты с добавкой
Так, ки0 годами хранить в алюминиевых емкостях и
11нгибиторо1^^циальных сортов легированных сталей. При отсутст-
вХ” ов резк0 сокращаются сроки хранения. Например,
вии ингиои 1 в ОцИНКованных и алюминиевых емкостях не раз-
хранение вПосУ{ОЛЬКу спирты легко реагируют с цинком и алюминием,
ле1и,',еТ1,,’,1К0Г0ЛЯТы металлов, выпадающие в осадок, а эффективных
РбРпбиторов против этой реакции не найдено.
ШПОчень часто металлы, не реагирующие с ракетными топливами,
°iBawT каталитическое действие на реакцию разложения или
0Ка3ления этих топлив. Так, при хранении горючих на основе аминов
°Кльзя допускать их контакта с медью или ее сплавами, поскольку
Ноны меди являются сильнейшими катализаторами процесса окисле-
ния аминов кислородом воздуха. Ионы многих тяжелых металлов
катализируют процесс разложения перекиси водорода.
Некоторые компоненты ракетных топлив легко окисляются кисло-
родом воздуха, и длительное хранение их возможно только в атмо-
сфере азота или в герметично закрытых резервуарах. Например,
горючие компоненты на основе аминов, гидразина и его производных
окисляются значительно более энергично, чем углеводороды. Реакция
окисления безводного гидразина идет настолько бурно, что он при
^оптанте с воздухом воспламеняется и горит фиолетовым пламе-
нем. Диметилгидразин при контакте с воздухом очень быстро окис-
ляется с образованием диметиламина и воды. Кроме того, диметил-
гидразин реагирует с двуокисью углерода, имеющейся в воздухе,
с образованием солей состава [(CHs^N — NH2]n«CO2, нераство-
римых в диметилгидр азине и выпадающих в виде твердого
осадка.
Реакции разложения характерны для компонентов ракетных топ-
лив, представляющих собою эндотермические вещества. Как правило,
они могут длительное время храниться без разложения при нормаль-
ных температурах, но при повышении температуры или при воздей-
ствии катализатора начинается разложение, самоускоряющееся под
воздействием выделяющегося при этом тепла. Так, гидразин, нагре-
тый до 350° С, полностью разлагается на азот й аммиак, причем
гораздо интенсивнее в присутствии окпслов железа, хрома, меди и
Других катализаторов. Характерным веществом, способным к разло-
жению с выделением тепла, является перекись водорода. В чистом
виде она довольно устойчива и только при нагревании свыше 140° С
начинает разлагаться на воду и кислород с выделением тепла. Абсо-
лютно чистая Н2О2 может быть нагрета до кипения (151,4° С) и пере-
гоняться без разложения, однако даже малейшие царапины на стен-
ок сосуда, в котором нагревается перекись водорода, могут явиться
^Ричиной ее разложения. Скорость разложения перекиси зависит
ее концентрации, величины pH, температуры, природы и количе-
а катализирующих разложение примесей или стабилизаторов,
w ическои и химической природы поверхности сосудов, в которых
находится Н2О2.
205
При разбавлении перекиси водорода водой ее чувствительность
к катализаторам разложения понижается, уменьшается скорость
распада и опасность взрыва, так как при разложении водных раство-
ров выделяется меньше
ния водных растворов Н2О
2
С, вес.
100
90
80
70
30
тепла. Ниже приведены теплоты разложе-
в зависимости от ее концентрации:
о/
/о
Q, дж/ кг
2893
2600
2307
2022
* 867
Наиболее стабильны слегка кислые растворы перекиси водорода
(pH = 4—6). При изменении pH раствора Н2О2 скорость распада
увеличивается.
МЕТОДЫ УЛУЧШЕНИЯ СТАБИЛЬНОСТИ ТОПЛИВ
Подбор оптимального химического состава топлив
лимическии состав сложных топлив, в частности нефтяных,
определяется выбором соответствующего сырья для их производства
и технологическими процессами его переработки. При производстве
нефтяных топлив технологическими процессами, улучшающими сц
став, являются каталитический риформинг, депарафинизация и раз-^*
личные методы очистки — щелочные, сернокислотные, гидроочистка.
В процессе карбамидной депарафинизации, который в настоящее
время применяется для получения дизельных топлив с низкими
температурами застывания, происходит удаление высокомолекуляр-
ных алкановых углеводородов нормального строения (парафинов)
с высокими температурами застывания, вследствие чего улучшается
физическая стабильность дизельных топлив при низких температу-
рах. J
При очистке топливных дистиллятов серной кислотой их стабиль-
ность улучшается за счет извлечения ненасыщенных соединений.
В процессе гидроочистки все непредельные соединения гидри-
руются до предельных, а сернистые соединения — до сероводорода,
который затем легко удаляется из топлив фенолятной или этанол-
аминной очисткой. В настоящее время процесс гидроочистки исполь-
Таблица 44
Влияние гидроочистки на термическую стабильность топлива ТС-1
Оптическая
плотность
Топливо
Осадок,
мг/100 мл
Смолистые отложе-
ния на пластинке,
г/м2,
Негидрированное . . 4,5
Гидрированное ... 0,6
0,7
0,1
0,102
0,010
206
тсЯ для улучшения качества дизельных и реактивных топлив»
комическая стабильность реактивных топлив после гидроочистки
Рачительно повышается (табл. 44).
31 Умелое использование технологических процессов и применение
пециальных методов очистки получаемых продуктов дает возмож-
ность значительно улучшать стабильность топлива.
Применение присадок
Химическая стабильность топлив, изменяющаяся в основном за
счет процессов окисления, может быть значительно улучшена до-
бавкой в топлива противоокпслптельных присадок (антиоксидантов)
и деактиваторов металлов, снижающих каталитическое действие
металлов на процесс окисления топлив. Применяются также при-
садки, улучшающие стабильность против разложения.
Наличие в топливах веществ, способных реагировать с промежу-
точными продуктами окисления, разрушая их и образуя при этом
неактивные вещества, ведет к обрыву окислительных цепей и замед-
ляет процесс окисления. Вещества, способные замедлять процесс
окисления, называются антиоксидантами, пли ингибиторами окисле-
ния. Добавка таких веществ к топливам значительно повышает их
химическую стабильность.
Действие антиоксидантов может быть представлено следующей
схемой:
I rooh+ah=roh+aoh
и
R’ + AH = RH + A'
где ROOH — перекись;
АН — молекула антиоксиданта;
А* — малоактивный радикал, быстро исчезающий путем ре-
комбинации, раньше чем произойдет его реакция с моле-
кулой окисляемого вещества.
Согласно исследованиям К. И. Иванова и Е. Д. Вилянской [3],
антиоксиданты по своему действию могут быть отнесены к одному из
следующих типов:
1) реагирующие с радикалами, заменяя их на неактивные ради-
калы; они тормозят окисление только на стадии инициирования
окислительных цепей, т. е. в начале окисления;
2) реагирующие как с радикалами, так и с перекисями, разрушая
их без образования новых радикалов; они тормозят процесс окисле-
ния как на стадии инициирования, так и на стадии автокаталитиче-
ского развития процесса;
3) реагирующие с радикалами и умеренно способствующие раз-
ложению перекисей; они способны тормозить процесс окисления
в начальной стадии, а в случае повышенных концентраций — и на ста-
дии автокаталитического развития.
Опыт классификации антиоксидантов по характеру их действия
кинетику процесса окисления в различных стадиях его развития
207
Классификация антиоксидантов [3]
Таблица 45
Тип
анти-
окси-
данта
Название
Дифениламин
Фенил-р-иафтиламин
п-0 ксидифе ниламин
п-Оксифенил-р-нафтиламин
а-Нафтил амин
а-Нафтол
п-Фенилендиамин
I
п-Аминофенол
Гидрохинон
Бензидин
о-По лидин
Р-Нафтиламин
р-Нафтол
л^-Фенилендиамин
о-Аминофенол
Формула
он
-nh2
- nh2
—nh2
СН3
-он
Испытанная
концентра-
ция антиок-
сиданта,
0,017
0,023
0,018
0,036
0,10
0,01
0,01
0,11
0,036
0,042
0,10
0,10
0,01
0,016
/
208
Продолжение табл. 45
Название
Формула
оксп-
Испытанная
концентра-
ция анти-
оксиданта,
о-Диэтиламинофенол
он
/С2Н5
0,07
он
Резорцин
0,11
?н сн3
2,6-Ди-третп-бутил-4-метил-
фенол
сн3-с
0,05
сн3
приведен в табл. 45. Как видно из таблицы, антиоксидантами явля-
ются органические вещества, содержащие группы — ОН и NH2.
Аминопроизводные ароматических углеводородов менее активны,
чем фенолы, но одновременное присутствие окси- и аминогрупп
Рачительно повышает эффективность антиоксиданта. Противоокис-
лптельная активность фенолов повышается при их алкилировании.
Так, индукционный период бензинов с добавкой 5-бутил-2,6-диизобу-
тилфенола был в 8 раз больше, чем после добавки такого же количе-
ства фенола. Одним из наиболее активных антиоксидантов для бен-
зинов является м-оксидифениламин.
Сама по себе химическая природа функциональной группы,
входящей в состав антиоксиданта, не определяет его принадлеж-
ности к тому или иному типу по действию на процесс окисления,
более важное значение имеет положение функциональной группы.
Так, отличительной особенностью антиоксидантов 2-го типа является
то, что аминная группа в них имеет первичный характер и находится
в а-положении. При совместном присутствии двух функциональных
гРУпп они всегда находятся в пара-положении. Если же функцио-
нальная группа находится в р-положении, а две группы в орто-или
Мета-положении, то такое вещество действует как антиоксидант 3-го
типа.
Эффективность действия антиоксидантов помимо их природы
11 строения зависит также от природы топлива, наличия в нем продук-
тов окисления, от температуры и т. д. Наиболее эффективно анти-
•Лриданты любого типа действуют при добавке к неокисленным топли-
вам, поэтому антиокислители вводят в топлива непосредственно на
вводах. Выбор наиболее эффективного антиоксиданта производится
Для каждого вида топлива, определенных условий хранения и при-
менения. Так, присадки, стабилизирующие бензины, не совсем пр’и-
ГоДНы для стабилизации керосинов или дизельных топлив; присадки,
14 Заказ 792.
209
замедляющие окисление топлив при нормальных температурах,
неэффективны при повышенных температурах.
В процессе хранения топлива антиоксиданты непрерывно рас-
ходуются. Уменьшение содержания присадок зависит от химического
состава топлива, условий хранения и активности присадок. В про-
цессе хранения можно добавлять присадки, это позволит увеличить
сроки хранения топлив. Однако к глубоко окисленным топлива
содержащим значительное количество смол, добавлять присадки
нецелесообразно.
Антиоксиданты должны удовлетворять следующим требованиям:
1) быть эффективными в малых количествах;
2) хорошо растворяться в топливах;
3) не вымываться водой;
4) не вступать в реакции с компонентами топлива и с материалом
тары; . '
5) быть стабильными при повышенных температурах.
Подобрать антиоксиданты, полностью удовлетворяющие всем
перечисленным требованиям, не всегда удается.
В настоящее время наиболее широко используются в качестве
антиоксидантов вещества следующих групп.
Алкилфенолы — универсальные антиоксиданты, пригодные для
топлив до -(-120° С. Очень хорошо растворимы в топливах, устойчивы
в углеводородной среде при разных температурах, практически
нерастворимы в воде.
Аминофенолы — весьма эффективные антиоксиданты даже в ма-
лых концентрациях. Могут применяться для стабилизации этилиро-
ванных бензинов, реактивных топлив. Плохо растворяются в высоко-
молекулярных углеводородах и хорошо — в воде, особенно при
повышении температуры.
Амины. Рекомендуются для продуктов с большим содержанием
ненасыщенных углеводородов (продуктов крекинга) и реактивных
топлив. Ограниченно растворимы в топливах, особенно в тяжелых
фракциях (керосинах, дизельных топливах).
Полифенолы. Обладают относительно невысокой эффективностью
и поэтому их следует добавлять в значительных количествах (до
0,05—0,06%). Необходимость добавки полифенолов в больших коли-
чествах вызывается также тем, что в промышленных.антиоксидантах
содержится менее 50% активного вещества (полифенолов). Одним из
первых противоокислителей для стабилизации автомобильных бен-
зинов, приготовляемых в промышленном масштабе, был древесно-
смольный антиоксидант Б, являющийся дистиллятом древесной
смолы. Получают его извлечением фенольной фракции из древесных
смол сухой перегонки главным образом березы и бука. Этот антиокси-
дант хорошо растворим в бензинах и воде. При наличии воды на дй ,
резервуаров антиоксидант переходит из топлива в воду, и вследствие
этого стабильность топлива значительно снижается.
Для снижения каталитического действия ионов металлов на про-
цесс окисления, вызывающего ускоренный расход антиоксидантов,
яименяется добавка специальных деактиваторов, образующих
11 ионами металлов комплексы.
С Наиболее эффективными деактиваторами металлов являются
аЛпцплидены — продукты конденсации салицилового альдегида
с 0-аминофенолами пли диаминами, например салицилиден-о-амино-
*110Л он •
-он
Совместное применение антиоксидантов с деактиваторами метал-
лов является эффективным способом стабилизации топлив, особенно
тех, которые содержат ненасыщенные углеводороды и легко подвер-
гаются .окислению. Деактиваторы добавляются в малых количест-
вах — от 0,005 до 0,01%.
Каталитическое действие ионов металлов на процесс окисления
обусловливается реакциями между ионами металлов и продуктами
промежуточного окисления. Так медь, реагируя с перекисями, обра-
зует поны двухвалентной меди и свободные радикалы, являющиеся
началом новых окислительных цепей:
ROOH + Cu^CuO4-R* + OH’
В присутствии антиоксиданта двухвалентная медь восстанавли-
вается и снова может вступать в реакцию с перекисями. Поэтому
наличие в топливах ионов металлов приводит к быстрому расходу
антиоксиданта.
В табл. 46 показана эффективность совместного применения
антиоксидантов и деактиваторов металлов.
Таблица 46
Эффективность совместного применения
антиоксидантов с деактиваторами [20]
Индукционный период
окисления бензина, мин
Металл
с антиокси-
с анти- дантом
оксидантом и деакти-
ватором
Без металла...........
Медь ..... . . .' . . .
Латунь................
Сталь ................
340
85
120
240
350
280
325
350
МЕТОДЫ ОПРЕДЕЛЕНИЯ СТАБИЛЬНОСТИ ТОПЛИВ
Определение стабильности топлив производится лабораторными
Методами, испытанием на специальных стендах и в эксплуатационных
Условиях.
211
14*
Лабораторные методы
Эти методы позволяют в относительно короткое время при исполь-
зовании небольших количеств испытуемых топлив исследовать их
стабильность.
В лабораторных условиях стабильность топлив оценивают по тем
изменениям, которые происходят в них при воздействии факторов
ускоряющих окисление и другие химические реакции (высокая
температура, контакт с кислородом при повышенном давлении,
каталитическое действие металлов и т. д.). Кроме того, о стабиль-
ности можно судить по содержанию в топливах малостабильных
соединений.
Химическая стабильность топлив оценивается по химическому
составу и содержанию фактических смол, по индукционному периоду,
максимальной величине некоптящего пламени, периоду стабиль-
ности до выпадения осадков, устойчивости к образованию осадков
при нагревании и по скорости поглощения кислорода.
Ненасыщенные соединения, количество которых определяют
по иодному или бромному числу, легко окисляются, вступают в ре-
акции полимеризации, вызывая быстрое изменение физико-химиче-
ских свойств топлив. Для определения их содержания используется
реакция присоединения к ним иода или брома, которая легко про-
текает при нормальных температурах:
R—СН=СН2 +J* = R—CHJ’—СН J’
z 1 2 2
Для определения иодного числа навеску топлива вносят в спир-
товый раствор иода. Часть иода при этом связывается с ненасы-
щенными соединениями, а избыток оттптровывается тиосульфатом
натрия. Начальное количество иода в спиртовом растворе опреде-
ляют титрованием холостой пробы, без топлива. Бромное число
определяют, титруя спиртовый раствор навески топлива бромид-
броматным раствором известной концентрации. Конец титрования
фиксируется по изменению потенциала раствора топлива.
Процентное содержание ненасыщенных соединений может быть
вычислено по иодному пли бромному числу, если предположить,
что в каждой молекуле ненасыщенных соединений имеется только
одна двойная связь:
И. ч. М Б.ч. М
“ 254 ’ “ 160
где X — содержание ненасыщенных соединений, %;
И. ч. и Б. ч. — иодное и бромное числа;
М — молекулярный вес топлива;
254 и 160 — молекулярный вес иода и брома.
Качественное определение содержания ненасыщенных соедшге-
ний производится по изменению цвета раствора перманганата калия
или по обесцвечиванию бромной воды при смешении их с топливом:
R-CH=CH2 + КМпО4 + Н2О —>МпО2 + КОН + R-CHOH-CH2OH
R—СН=СН2 +Br2—>R—СНВг—СН2Вг
212
I
Определение содержания ненасыщенных соединении дает возмож-
сть делать только самые общие выводы о химической стабильности
11 иного топлива, поскольку различные ненасыщенные соединения
^падают неодинаковой химической активностью, а указанными
Методами определяется только их суммарное содержание, без учета
\сличил в химической активности.
Содержание фактических смол (в мг на 100 мл топлива) опре-
деляется взвешиванием стакана до и после испарения опреде-
ленного объема топлива. Испарение топлива ведут при помощи
стрУи воздуха, нагретого до 150° С, или водяным паром (по Буда-
рову).
Количество фактических смол позволяет судить о происшедшем
изменении состава топлива, но не дает возможности определить
склонность топлив к смолообразованию, т. е. как долго можно
хранить данное топливо до появления смол.
Склонность топлив к окислению, а значит, и его стабильность
при хранении в контакте с кислородом воздуха оценивают по
величине индукционного периода.
При контакте нефтяных топлив с кислородом реакция вначале
идет медленно вследствие наличия в топливах природных веществ,
тормозящих окисление, и малой концентрации перекисей. По мере
-расходования противоокислительных веществ и накопления пере-
писей реакция окисления ускоряется. Таким образом, процесс окис-
ления делится на два периода: период подготовки топлива к быст-
рому окислению (индукционный) и период интенсивного окисления.
В первый период топливо сохраняет свои эксплуатационные каче-
ства, во втором — быстро увеличивается его кислотность и возра-
стает содержание смол. В дальнейшем, когда наиболее активные
углеводороды прореагируют, процесс окисления снова замедляется,
но к этому времени топливо уже становится непригодным к употреб-
лению из-за большого содержания в нем смол. Индукционный
период топлив может длиться месяцы, а для более стабильных даже
годы. Увеличивая концентрацию кислорода и температуру, можно
резко сократить индукционный период.
Для определения индукционного периода в лабораторных усло-
виях стаканчик с навеской испытуемого топлива помещают в бомбу,
заполняют ее кислородом под давлением в 7 атм (6,9 • 105 н!м2)
и опускают в кипящую воду. Следя за показанием манометра на
бомбе, определяют время, прошедшее от момента погружения бомбы
п горячую воду до начала падения давления в бомбе, указывающего
па начало быстрого окисления. Время (в мин), необходимое для
подготовки топлива к быстрому окислению в данных условиях,
’•Называется индукционным периодом. Чем больше индукционный
период, тем более стабильно топливо.
В этилированных бензинах, в которых возможно разложение
тетраэтилсвинца с выделением осадков, стабильность определяется
временем (в ч) до начала выпадения осадка при нагревании топлива
до Цо° С. Этот показатель называется стабильностью против выпаде-
213
I • * i
214
1Т11я осадка. Для его определения испытуемый бензин наливают
р герметически закрываемый сосуд и помещают в термостат, нагре-
тый до 110° С (прибор ЛСА). Через специальное окно в приборе
наблюдают за появлением в топливе осадка. Установлено, что бен-
зины, имеющие стабильность в условиях испытания 20—24 ч, можно
<панить в южной зоне до 3 лет, не опасаясь образования осадков,
угри стабильности в 4 ч срок хранения сокращается до 1 года, а бен-
зины со стабильностью менее 4 ч необходимо использовать немед-
ленно, так как в них осадок от разложения тетраэтилсвинца может
появиться в течение ближайших месяцев.
В приборах ЛСАРТ, не имеющих окон для наблюдения, бензин
заливают в стаканы, которые устанавливают в герметически закры-
ваемые металлические бомбы. Бомбы со стаканами помещают в кар-
маны нагретого прибора и выдерживают определенное вре-
мя, после чего вынимают, охлаждают и проверяют наличие
осадка.
Для определения термоокислительной стабильности реактивных
топлив применяют прибор ЛСАРТ, представляющий собой металли-
ческую алюминиевую баню с электрическим нагревателем и внеш-
ней теплоизоляцией. Топливо нагревают в металлической «бомбе»,
которая представляет собой цилиндр из нержавеющей стали, герме-
тч1чески закрывающийся специальным устройством. Внутри «бомбы»
устанавливают стеклянный стакан, в который заливают испытуемое
топливо.
После нагревания в течение 1 ч при 150° С выключают электро-
подогрев, вынимают «бомбу» из прибора и охлаждают до комнатной
температуры. Охлажденное топливо фильтруют через доведенные до
постоянного веса фильтры. Осадок на фильтре промывают изопента-
ном или петролейным эфиром до удаления следов топлива. После
окончания промывки каждый фильтр высушивают в сушильном
шкафу ДО постоянного веса.
Термоокислительную стабильность испытуемого топлива выра-
жают в мг осадка на 100 мл топлива.
По такому же принципу стабильность топлив определяют и на
приборе ЛТС, схема которого показана на рис. 84. Однако этот
прибор [8] выгодно отличается от прибора ЛСАРТ тем, что можно
одновременно испытывать с параллельными пробами 8 образцов
топлив и, самое главное, в процессе испытаний осуществляется пере-
мешивание топлива внутри сосудов.
Для определения стабильности топлив по скорости поглощения
кислорода используются приборы, в которых при помощи специаль-
ной системы измеряется количество кислорода, поглощаемого топли-
[19].
На основании полученных данных строят, кинетическую зависи-
мость количества поглощенного кислорода от времени. Сравнивая
кинетические кривые, определяют окисляемость топлив разного
химического состава.
215
Стендовые и эксплуатационные испытания
Наиболее достоверно стабильность топлив может быть оценена
в условиях хранения, транспортирования и применения. Это требует
больших расходов испытуемых топлив, а также продолжительного
времени. Однако для выявления действительной стабильности то-
плив и сопоставления ее с величиной, полученной лабораторный
методами, такие испытания проводятся для каждого вида топлив.
Испытания, проводимые на специально оборудованных стендах,
предшествуют эксплуатационным испытаниям и только в отдельных
случаях используются для контроля качества уже принятых на
снабжение топлив.
Стендовые испытания проводятся на полноразмерных двигателях
или агрегатах и на специальных установках, моделирующих условия
работы испытуемого топлива в реальных двигателях. Такие испыта-
ния подразделяются на краткосрочные функциональные и длитель-
ные. При краткосрочных функциональных испытаниях оцениваются
отдельные свойства испытуемых топлив, а при длительных произ-
водится комплексная оценка всех эксплуатационных свойств топлива.
Стабильность топлив при стендовых испытаниях оценивается по
количеству отложений в топливоподающих системах двигателей,
а для ракетных топлив — ив системах охлаждения, кроме того по
изменению прокачиваемости при изменении внешних условий рабо^
топлив.
Оценка стабильности в условиях эксплуатационных испытаний
производится по количеству отложений в топливных системах двига-
теля, количеству осадков в самих топливах, по неизменности физико-
химических свойств испытуемых топлив.
Эксплуатационные испытания являются заключительными и
позволяют окончательно оценить качество топлив. Эти испытания
проводят путем систематического наблюдения за работой автомобилей,
тракторов, самолетов и другой техники до выработки определенного
моторесурса в разных условиях. Испытуемое топливо при этом под-
вергается систематическому лабораторному анализу.
Перед проведением эксплуатационных испытаний новых сортов
топлив или старых сортов, но предназначенных для применения
на новой технике, проводят пробеговые или летные испытания.
От эксплуатационных испытаний пробеговые отличаются тем, что
последние проводятся на специально оборудованных машинах с при-
менением необходимых контрольно-измерительных приборов, уста-
навливаемых на машинах дополнительно (термопар, термометров,
манометров и т. д.). Водители выбираются из числа наиболее квали-
фицированных и им дается подробный инструктаж по правилам
эксплуатации машин при данных испытаниях. Перед началом испы-
таний и после их окончания двигатели разбираются, детали осматри-
ваются, производится измерение или взвешивание наиболее ответ-
ственных деталей. Проверяют состояние систем питания и смазки.
Все особенности поведения испытуемого топлива и случаи нарушения
216
реальной работы двигателя или машины в целом в процессе прои-
^ого пробега (полета) записывают в журнал наблюдения.
Я Стабильность топлив в условиях хранения определяют путем
рытного хранения топлив в разных климатических условиях в
емкостях различного объема, изготовленных из различных матери а-
Физико-химические свойства испытуемых топлив системати-
проверяют лабораторными методами. Сроки хранения каждого
сорта топлив устанавливают только на основании данных опытного
хранения.
ЛИТЕРАТУРА
1. Моторные, реактивные и ракетные топлива, нод ред. К. К. Папок,
Е Г. Семенидо, Гостоптехиздат, 1962.
2. К. И. Иванов, В. К. Савинова, В. П. Ж а х о в с к а я,
ДАН СССР, 72, 903 (1950).
3. К'. И. Иванов, Промежуточные продукты и промежуточные реак-
ции автоокисленпя углеводородов, Гостоптехиздат, 1949.
4. Я. Б. Чертков, Г. Ф. Большаков, Е. И. Гулин,
Топлива для реактивных двигателей, Изд. «Недра», 1964.
5. Г. Ф. Большаков, Е. А. Глебовская, Гетероорганическне
соединения реактивных топлив, Гостоптехиздат, 1962.
6. Сб. «Эксплуатационные свойства реактивных топлив при повышенных
температурах», под ред. Я. Б. Черткова, Гостоптехиздат, 1959.
7. Г. Ф. Большаков, Химия и технология топлив и масел, № 5 (1963).
^8. П. И. Давыдов, Г. Ф. Большаков, Химия и технология
Жлив и масел, № 10 (I960).
9. Г. Ф. Большаков, Химия и технология топлив и масел, № 1 (19Ьч).
А. А. Гуреев, Химия и технология топлив
В. М. Шагин, Химия и технология топлив
И. В. Рожков, А. А. Гуреев, Химия
10. 3. А. Саблина,
и масел, № 2 (1959).
И. Я. Б. Чертков,
и масел, № 11 (1959).
12. 3. А. Саблина,
и технология топлив и масел, № 6 (1957).
13. Г. Ф. Большаков, Е. А. Глебовская, 3. Г. Каплан,
Восьмая научная сессия по химии сераорганических соединений нефти и нефте-
продуктов, Уфа, 1964.
14. Г. Ф. Большаков, Е. А. Глебовская, 1 еохимическии
сборник, № 9, Труды ВНИГРИ, вып. 227, 1964.
15. А. С. И р и с о в, Авт. транспорт, № 4 (1957).
16. Я. Б. Чертков, Химия и технология топлив и масел, № 9 (19а9).
17. Г. Ф. Большаков, Л. И. Л и с н я н с к и й, Тезисы докладов
VIII научной сессии по химии сераорганических соединений нефтей и нефте-
продуктов, Изд. АН СССР, 1964.
18. Г. Ф. Большаков, Е. А. Глебовская, 1езисы докладов
на XVII совещании по спектроскопии, Изд. АН СССР, 1965.
19. Г. Ф. Большаков, Нефть и газ, № 10 (1964).
20. 3. А. Саблина, А. А. Гуреев, Присадки к моторным топли-
вам, Гостоптехиздат, 1959.
21. Г. Ф. Большаков, Е. А. Глебовская, 3. Г. Каплан,
Нефть и газ, № 8 (1965).
Глава VII
КОРРОЗИОННОСТЬ топлив
ВИДЫ КОРРОЗИИ И МЕХАНИЗМ КОРРОЗИОННЫХ ПРОЦЕССОВ
Коррозией называется разрушение материалов при химическом
или электрохимическом взаимодействии их с окружающей средой.
Скорость и величина коррозии обусловливаются как свойствами
корродируемых материалов, так и свойствами окружающей среды.
Топлива — при хранении, перекачке и транспортировании, а также
в процессе применения — и продукты сгорания топлив, взаимодей-
ствуя с конструкционными материалами, могут вызывать их разру-
шение. Способность топлив разрушать конструкционные материалы
при контакте с ними называется коррозионностью топлив [1—3].
Коррозия конструкционных материалов приводит к износу и
снижению прочности изделий, что' сокращает сроки эксплуатации
двигателей, средств хранения, перекачки и транспортирования то-
плив. Продукты коррозии загрязняют топлива и ухудшают их экс-
плуатационные качества [4—8].
По характеру разрушений чаще всего встречаются следующие
виды коррозии [1—3].
1. Сплошная, или равномерная, коррозия (рис. 85, а). Разруше-
ние конструкционного материала происходит равномерно по всей
поверхности соприкосновения его с агрессивной средой. После уда-
ления продуктов коррозии поверхность материала становится шеро-
ховатой, с большими или меньшими углублениями.
2. Местная, или локальная, коррозия (рис. 85, б). Разрушаются
только отдельные участки поверхности материала. Глубина местных
поражений может быть равномерной и неравномерной.
3. Точечная коррозия (рис. 85, в). Разрушения появляются на
отдельных маленьких площадках, но имеют большую глубину про-
никновения. Характерна для металлических деталей, работающих
при высоких температурах или высоких давлениях.
4. Межкристаллитная коррозия (рис. 85, а). Разрушения про-
исходят по границам между кристаллами металла. При такой кор-
розии внешний вид изделия часто остается без видимых изменений,
218
jio его прочность резко снижается и изделие может быть разрушено
при небольшом внешнем механическом воздействии.
5. Подповерхностная коррозия (рис. 80, д). Разрушенные места
скрыты под поверхностью материала и соединяются с ней узкими
каналами. Этот вид коррозии часто наблюдается в сплавах алю-
миния.
^^Очень часто различные виды коррозии сочетаются между собою
и наблюдаются на данной поверхности материала одновременно.
Рис. 85. Виды коррозии:
У//////\ Металл
I-Продукты коррозии
а — сплошная; б — местная; в — точечная; г — межкристаллитная; д — подповерхностная.
Так, сплошная коррозия может сочетаться с межкристаллитной,
местная — с подповерхностной и т. д.
Наиболее опасными видами коррозии являются межкристаллит-
ная, точечная и подповерхностная. Эти виды коррозии характерны
тем, что при сравнительно небольшом общем количестве разрушен-
ного материала и незначительном внешнем изменении поверхности
его сильно снижается механическая прочность изделий.
Химическая коррозия
При химической коррозии разрушение материалов происходит
в результате химического взаимодействия их с окружающей средой.
Продукты коррозии образуются непосредственно на тех участках
поверхности конструкционного материала, которые реагируют с агрес-
е^ными веществами окружающей среды.
К химической коррозии относятся такие типы коррозии:
1. Газовая коррозия — разрушение металлов газами или парами
при температурах выше температуры конденсации влаги на поверх-
ности металла. Этот тип коррозии наблюдается при соприкосновении
219
деталей двигателей с нагретыми продуктами сгорания то-
пли в.
2. Жидкостная коррозия металлов в неэлектролитах. Происхо-
дит при контакте металлов с химически активными веществами, не
проводящими электрический ток. Несмотря на то, что углеводороды,
составляющие основную часть нефтяных топлив, являются пеэлектро-i
литами, коррозия металлов нефтепродуктами не может рассма<у^?А
ваться как пример чисто химической коррозии. В нефтепродуктах
всегда содержится большее пли меньшее количество растворенной
воды и неорганических соединений, п поэтому практически корро-
зия металлов нефтепродуктами вызывается не только химическими,
но и электрохимическими процессами.
3. Коррозия неметаллических материалов. При контакте неме-
таллических материалов с агрессивными веществами величина и ско-
рость разрушения определяются преимущественно характером хими-
ческого взаимодействия, независимо от того, проводит окружающая
среда электрический ток пли не проводит.
Существенное влияние на скорость и величину всех видов
коррозии оказывают свойства пленки, образующейся на поверхности
корродируемого материала пз продуктов коррозии. Чем прочнее
образующаяся пленка и чем меньше ее проницаемость, тем меньше
скорость и глубина коррозии данного материала.
Защитные свойства пленок проявляются в том случае, если
дукты коррозии образуют на поверхности корродируемого материала
прочно удерживающуюся пленку, малопроницаемую как для ионов,
атомов или молекул материала, так и для молекул внешней среды.
Эффективно защищающие от коррозии пленки должны иметь
близкий с материалом коэффициент теплового расширения и быть
достаточно пластичными, с тем чтобы при изменении температуры
не происходило растрескивания пленки.
Пористые пленки, не защищающие материал от дальнейшей кор-
розии, образуются в том случае, если объем продуктов коррозии
меньше объема исходного, вступившего в реакцию взаимодействия
с окружающей средой материала. Через пористые пленки окружа-
ющая среда легко проникает к корродируемому материалу. Коли-
чество разрушенного материала, а значит, и толщина пленки в этом
случае пропорциональны продолжительности контакта материала
с агрессивной средой:
h — kx
где h — толщина пленки;
к — постоянная величина для данных условий коррозии;
т — время коррозии.
Если образующаяся из продуктов коррозии пленка затруд^^т
доступ внешней среды к корродируемому материалу, то по мере роста
толщины пленки скорость коррозии снижается
dh , 1
--= к • —
dx h
220
Решение этого уравнения относительно h дает параболическую
зависимость между толщиной пленки и временем коррозии:
h2~2kx-\-A или h2 = к' т+ А
где А — константа интегрирования.
Лучшими защитными свойствами обладают пленки, у которых
рост их толщины происходит по логарифмической зависимости от
времени:
д = 1п (к т)
Электрохимическая коррозия
Разрушение металлов в электролитах происходит в результате
действия на поверхности металла большого числа микроскопических
короткозамкнутых гальванических элементов, возникающих вслед-
ствие неоднородности металла или среды, окружающей металл.
При электрохимической коррозии разрушение металлов сопрово-
ждается переносом электрических зарядов от одной части металла
к другой, т. е. протеканием электрического тока. Реакция взаимо-
действия металла с окружающей средой разделяется на два процесса:
анодный — переход металла в раствор в виде гидратированных ионов
составлением эквивалентного количества электронов в корродиру-
гшом металле — и катодный — ассимиляция избыточных электронов
в металле молекулами или ионами раствора, восстанавливающимися
на катоде.
При погружении металла в электролит, вследствие притяжения
ионов металла дипольными молекулами воды, всегда будет происхо-
дить реакция:
Ме£±Ме+ + е~
Положительные ионы Ме+ переходят в окружающую среду,
а поверхность металла приобретает отрицательный заряд. В резуль-
тате электростатического притяжения большинство гидратированных
ионов металла располагается непосредственно у поверхности металла,
Далее их концентрация будет снижаться и на каком-то удалении
сравняется с концентрацией их в растворе, окружающем металл.
На границе металла с электролитом образуется двойной слой отрица-
тельно заряженных электронов и положительно заряженных гидра-
тированных ионов Ме+ (рис. 86).
При погружении металла в раствор электролита, содержащего
ионы Ме+, может происходить процесс осаждения их и приобретение
Металлом вследствие этого положительного заряда. Величина заряда,
£^еделяющая равновесный потенциал металла, зависит от свойств
wkob Ме+ и от их концентрации в растворе электролита. Эта зависи-
мость выражается уравнением Нернста:
„ „ . 2,303 ЯГ , ... +1
Е ~ Ео --------1g [ Ме+ ]
221
где Е—равновесный электродный потенциал, в\
Ео — нормальный или стандартный электродный потенциал (по-
тенциал металла, погруженного в раствор, содержащий
ионы данного металла в концентрации 1 моль/л), в\
R — газовая постоянная (8,314 дж/град • моль)\
п — число электронов, освобождающихся при переходе одного
атома металла в ион; .
F — число Фарадея (96500); ^7
Т — температура, °К.
Сольбатиробанные ионы Me
Металл
Электролит
Рис. 86. Схема двойного слоя на поверхности ме-
талла в электролите.
Значения стандартных электродных потенциалов для некоторых
металлов приведены в табл. 47.
Переход ионов Ме+ в раствор электролита является процессом
разрушения, или коррозии металла. Для того чтобы ионы Ме+ могли
отрываться от поверхности металла, необходима ассимиляция нака-
пливающихся в металле свободных избыточных электронов, которая
может производиться каким-либо веществом из окружающей среды,
способным к восстановлению. Так, при погружении в раствор серной
кислоты соединенных между собою пластинок железа и цинка будет
происходить быстрое разрушение цинковой пластинки. Поскоф’%' ’
цинк обладает более отрицательным потенциалом, чем железо, па
цинковой пластине будет протекать анодный процесс перехода попов
металла в раствор. Накапливающиеся избыточные электроны будут
перетекать к железной пластине и на ее поверхности ассимилироваться
Таблица 47
Стандартные электродные потенциалы некоторых металлов
Электродный
процесс
Стандартный
электродный
потенциал,
Ъ
Электродный
процесс
Стандартный
электродный
потенциал,
в
Mg—>Mg2+ + 2е~
Al->А13+ + Зе~
Mn—>Мп2+ + 2<?~
Zn->Zn2+ + 2e"
Сг->Сг3+ + Зе-
Fe—>Fe2+ +2<?“
Cd—>Cd2+4-2e-
-2,36
-1,66
-1,18
-0,76
-0,74
—0,44
-0,40
Ni —>Ni2++2e-
Sn—>Sn2++2e“
Pb->Pb2++ 2e~
Cu->Cu2+ + 2*r
Cu—>Cu+ + r
Ag-»Ag+ + <r
-0,25
-0,136
-0,126
-0,153
+0,52
+0,80
ионами водорода из окружающей среды. Ионы водорода, связывая
электроны, восстанавливаются до водорода, который и будет выде-
ляться с железной пластины в виде газа. Если взять пару пластин
никель — железо, то растворяться будет железо, имеющее больший
отрицательный потенциал, чем никель. Катодный процесс связывания
электронов и выделение газообразного водорода в этом случае будут
v£)onсходить на никеле.
Площадки с разными потенциалами при контакте с электролитом
могут появляться и у изделий из одного металла или сплава. Эта
разность потенциалов возникает в результате наличия посторонних
включений, неоднородности структуры металла или в результате
неоднородности внешней среды. Так, Эвансом бцло установлено,
что при неравномерном доступе воздуха к разным участкам поверх-
ности металла возникает разность потенциалов. Если к одной из
железных пластин, опущенных в электролит, подвести больше кис-
лорода, чем к другой, то возникает электрический ток, причем элек-
трод, соприкасающийся с более аэрируемым раствором, является
катодом. Это явление названо неравномерной пли дифференциаль-
ной аэрацией.
Неравномерной аэрацией Эванс объяснил целый ряд случаев
разрушения металлов, причина которых ранее была неясна. Так,
например, известна низкая коррозионная устойчивость пористого
литья и плохо проваренных, неплотных сварных швов. По Эвансу это
объясняется тем, что металл в порах и трещинах имеет более отрица-
тельный потенциал вследствие меньшей аэрации, чем прилегающие
Участки поверхности изделия.
При отсутствии ассимиляции избыточных электронов на катоде,
"^исходит постепенное смещение потенциала катода в сторону
отрицательных значений и приближение его значения к потенциалу
ан°да. При равных значениях потенциалов катода и анода гальвани-
ческий элемент перестает работать. Изменение потенциала анода
в сторону положительных значений и приближение его к потенциалу
,уатода происходит при увеличении концентрации ионов металла
223
вблизи электрода. Изменение потенциала при прохождении тока
в процессе работы гальванического элемента называется поляриза-
цией. Процесс ассимиляции электронов на катоде, поддерживающий
постоянство разницы потенциалов катода и анода, называется де-
поляризацией. Окислители, связывающие электроны на катоде,
называются деполяризаторами. Катодными реакциями связывания
электронов могут быть, например, следующие:
2Н+ + 2*-->2Н—>Н2
O2 + 2H2O+4e"-->4OH-
Fe3++e-->Fe2+
С12+2е-->2С1“
Наиболее часто встречаются реакции восстановления иона водорода
до газообразного водорода и реакции ионизации кислорода с образо-
ванием ионов гидроксила.
Характер и скорость катодного процесса оказывают значительное,
часто решающее, влияние на большинство электрохимических кор-
розионных процессов.
При электрохимической коррозии имеет место целый ряд сопря-
женных между собою процессов: анодный процесс, который сопро-
вождается переходом металла в электролит, перенос электронов
в металле от анодного к катодному участку, выделение газообраЛ
ного водорода или пополнение убыли кислорода, участвующих в ка-
тодном процессе. Скорость наиболее медленного из этих процессов
и определяет общую скорость коррозии. Из указанных выше про-
цессов медленнее всего идут процессы диффузии кислорода к катод-
ным участкам или водорода — от катодных участков.
Количество растворяющегося на аноде металла по закону Фара-
дея пропорционально силе тока и времени:
где Q — количество металла, растворяющегося на аноде;
Э — грамм-эквивалентный вес металла;
F — число Фарадея;
I — сила тока;
т — время.
По закону Ома: 1
Е
г _ г\ с!
= Ё
где Е — э. д. с. гальванического элемента;
и 2?" — потенциалы катода и анода; -
R — сопротивление. 1
Величины Е™ и необходимо брать с учетом поляризации,
происходящей при работе элемента. Скорость растворения металла
на аноде определяется величиной установившейся разности потен-
224
цпалов анода и катода, которая значительно отличается от началь-
ной разности потенциалов вследствие поляризации электродов при
протекании тока.
Поскольку поляризация электродов зависит от тока, можно
записать: г '
E* = EK-kJK El = E* + kfa
п К V K ci а 1 2
где £*а и Ек — начальные равновесные потенциалы анода и катода;
/к и /а — плотности тока на аноде и катоде;
кг и к2 — коэффициенты.
Тогда:
где 5К и 5а — площади катода и анода.
к к
Величины —А- и определяют изменение потенциала катода
*->К ДОа
или анода при поляризации током равным единице и называются
поляризуемостью катода или анода. Для сокращения записи обозна-
чаем их через /?к и ра. Тогда:
Е" = Е-1рК ^а^а + ^а
Подставляя полученные выражения в уравнение силы тока галь-
ванического элемента, ролучаем:
а
к
и окончательно
Это уравнение является основным уравнением скорости электро-
химической коррозии.
Из уравнения видно, что скорость коррозии возрастает при
уменьшении ри и ра, которое происходит с увеличением доступа
Деполяризатора (при перемешивании окружающей металл среды,
введении в нее окислителя и т. д.) и с увеличением поверхности
катодных или анодных участков. Скорость коррозии в широких
пРеделах не зависит от сопротивления, так как значение R мало по
^Равнению с рЕ +/?а. Скорость коррозии тем выше, чем больше разность
г>^ЙЙрльных равновесных потенциалов катода и анода. Вследствие
Исключительно большого влияния на поляризацию свойств пленок
113 продуктов коррозии, отлагающихся на поверхностях участков
катода или анода, скорость электрохимической коррозии, так же как
11 Химической, зависит от физико-химических свойств образующихся
Ри коррозии пленок.
Заказ 792. 225
ФАКТОРЫ, ВЛИЯЮЩИЕ НА КОРРОЗИОННОСТЬ
Процессы коррозии происходят па границе между корродируемым
веществом и внешней средой в результате сложного химического
или электрохимического взаимодействия между ними. Поэтому
оценка коррозионности топлив может производиться только по
ношению к конкретному веществу в определенных условиях.
Основными факторами, определяющими величину и скорость
коррозии, являются: .
1) концентрация коррозионно-агрессивных веществ в топливах;
2) химические и физические свойства корродируемого вещества;
3) концентрация ионов водорода пли окислителя в растворе элек-
тролита; •
4) концентрация присадок, замедляющих или ускоряющих
процессы коррозии;
5) степень/пассивации металлов;
6) температура и продолжительность контакта веществ, подвер-
гающихся коррозии, с топливом.
Свойства корродируемых веществ. Скорость и величина коррозии
в значительной степени определяются физико-химическими свойства-
ми корродируемого вещества. Коррозионная стойкость металла не
определяется его положением в периодической системе Д. И. Менде-
леева. Большинство наименее устойчивых металлов расположены
в I группе периодической системы (Li, Na, К, Rb, Cs), а наиболее
устойчивые находятся в VIII группе (Pd, Os, Ir, Pt), однако и в I
группе имеются стойкие ко многим агрессивным веществам металлы
(Au, Ag, Си), а в VIII группе есть металлы легко поддающиеся
коррозии (Fe).
Коррозионная стойкость металлов не зависит от их положения
в ряду напряжений. Так, алюминий (Ео = —1,67 в) и свинец (EQ =
= 0,12 в) устойчивы в разбавленной серной кислоте, а железо
(Ео = 0,44 в) неустойчиво. В растворах едкого натра алюминий
неустойчив, а магний и железо — устойчивы и т. д.
Стойкость к коррозии для металлов в основном определяют за-
щитные свойства пленок продуктов коррозии, образующихся при
взаимодействии металла с окружающей средой. Так, устойчивость
свинца в серной кислоте объясняется образованием нерастворимой
в кислоте пленки сульфата свинца, устойчивость железа в щелочах —
образованием пленки из гидрата окиси железа и т. п.
Структура металла также оказывает влияние на скорость корро-
зионных процессов. Укрупнение кристаллов металла вызывает значи-
тельное усиление межкристаллитной коррозии. Коррозия, проис-
ходящая с выделением водорода, при прочих условиях тем интен&*
нее, чем неоднороднее структура металла. Коррозионные процессы,
связанные с поглощением кислорода, не зависят от неоднород-
ности структуры, если эта неоднородность не вызывает изменения
в равномерности отложений на поверхности металла продуктов
коррозии и не ослабляет защитных свойств образующейся пленки.
226
Если сплав металлов состоит из двух фаз и основной является
катодная фаза, а количество и площадь анодной невелики, то с воз-
никновением коррозии анодные включения быстро растворяются и
кЪррозия резко замедляется. Когда же количество анодной составля-
ющей относительно велико и распространяется на весь сплав, раз-
рушение металлического сплава будет очень интенсивным. Весьма
^’благоприятен случай расположения анодных составляющих вдоль
границ зерен, так как при этом происходит межкристаллитное
разрушение сплава. Важную роль играют электрохимические
свойства включений: если включения легко поляризуются, коррозия
может быть слабой даже при большой неоднородности сплава.
Напряжения и деформации увеличивают скорость коррозионных
процессов и вызывают межкристаллитную коррозию.
Состояние поверхности металла также влияет на скорость корро-
зии. Металлы с гладкой, полированной поверхностью корроди-
руют медленнее, чем с грубо шлифованной, шероховатой. На гладкой
поверхности при коррозии образуется более совершенная защитная
пленка, чем на поверхности шероховатой. Разница в скорости раз-
рушения изделий с различно обработанной поверхностью особенно
заметна при медленной коррозии, например атмосферной. При
коррозии сильно агрессивными веществами с большой скоростью
уже через короткое время различие в состоянии поверхности исчезает.
Концентрация ионов водорода. Изменение концентрации ионов
водорода в окружающем металл растворе — один пз существенных
факторов, влияющих на скорость электрохимической коррозии.
Изменение скорости коррозии различных металлов в зависимости
от pH раствора показано па рис. 87. Только для таких стойких к кор-
розии металлов, как золото и платина
чески не зависит от pH среды.
Амфотерные металлы, такие как цинщ алюминий, свинец, под-
вергаются сильной коррозии как в кислых, так и в щелочных средах
вследствие растворимости их окислов.,и в кислотах и в щелочах.
Скорость коррозии железа и магния очень мала в щелочных раство-
рах, в которых образующиеся гидроокиси этих металлов нераство-
римы и дают прочную пленку на поверхности металла. Никель и
кадмий устойчивы в нейтральных и щелочных растворах и быстро
корродируют в кислых.
Влияние pH среды на скорость коррозии может маскироваться
влиянием других факторов, таких как разрушение пленки, деполя-
ризация катода и т. д. Так, алюминий при одинаковых значениях
pH быстрее корродируется соляной кислотой и значительно медлен-
нее азотной. Это объясняется тем, что соляная кислота с алюминием
"^разует хлористый алюминий, растворимый в кислоте, а азотная
создает на поверхности алюминия прочную пленку из окиси алюми-
Ни я.
Содержание воды. Скорость коррозии металлов значительно уве-
личивается при наличии в топливах воды. Особенно сильно увели-
чивается коррозия в том случае, если содержание воды в топливах
15* 227
скорость коррозии практи-
превышает ее растворимость и вода выделяется в виде отдельной
фазы. Например, первые признаки коррозии стали Ст. 20 керосином,
содержащим растворенную воду, обнаруживаются через 7—10 суток,
а при наличии слоя воды — уже через сутки.
Ускорение процессов коррозии в присутствии воды объясняется
Как возможностью накапливания в ней агрессивных веществ, вымк-
Pt; Au
Рис. 87. Зависимость скорости коррозии различных
металлов от pH среды.
Ваемых водой из нефтепродуктов, так и тем, что в присутствии воды
процессы из чисто химических переходят в электрохимические,
протекающие со значительно большей скоростью. Кроме того, вода
способствует окислению металлов, а реакции взаимодействия слабых
органических кислот с окислами металлов протекают значительно
быстрее, чем с металлами. Вода часто способствует растворению вф:'
дуктов коррозии и тем самым препятствует образованию защитной
пленки на поверхности металла. В топливах, имеющих в своем
составе электролиты, увеличение содержания воды повышает их
диссоциацию, что способствует ускорению электрохимических про-
цессов коррозии, и только значительное разбавление водой, ведущее
228
к снижению концентрации агрессивных веществ, приводит к замед-
лению коррозии.
Содержание ингибиторов и активаторов коррозии. Скорость кор-
розии изменяется в присутствии различных добавок к коррозионной
среде [6, 9—15]. Вещества, тормозящие процессы разрушения мате-
риалов в агрессивных средах, называются ингибиторами или пас-
,^Лваторами, а ускоряющие коррозионные процессы — актива-
Порами коррозии. Ингибиторы и активаторы по характеру своего
действия делятся на вещества, способствующие созданию или раз-
рушению защитной пленки на корродируемом материале (1 тип);
уменьшающие или увеличивающие агрессивность среды (2 тип);
одновременно воздействующие на прочность защитной пленки и на
агрессивность среды (3 тип).
К 1-му типу относятся большинство ингибиторов, применяемых
для снижения коррозии металлов сильными неорганическими кис-
лотами. В основном это сильные окислители, способствующие со-
зданию защитных пленок из окислов металлов, — бихромат калия,
хромат натрия, йодноватая кислота — или вещества, сами образу-
ющие с металлом прочную защитную пленку — ортофосфорная
кислота и многие органические соединения, содержащие серу или
фосфор. В водных средах в качестве ингибитора 1-го типа часто упо-
требляют нитрит натрия.
Ингибиторами 2-го типа в применении к нефтепродуктам являются
вещества, снижающие скорость окисления нефтепродуктов и,
тем самым, скорость образования агрессивных органических
кислот.
В качестве примера ингибиторов 3-го типа можно назвать карбо-
наты щелочных металлов (обычно Na2CO3), добавляемые в водные
растворы для уменьшения коррозии сталей. Защитное действие
карбонатов заключается в снижении концентрации ионов водорода
в растворе (изменение агрессивности среды) и в создании на стали
тонкой пленки нерастворимых карбонатов железа (создание защит-
ной пленки).
Пассивация металлов. Перевод химически нестойких в данных
условиях металлов в состояние высокой стойкости против коррозии
называется пассивацией металлов. Наиболее характерные случаи
пассивации относятся к металлам группы железа (железо, кобальт,
никель), а также к хрому, танталу, ниобию, молибдену, алюминию,
Например, железо быстро разрушается при воздействии азотной
кислоты. С повышением концентрации кислоты скорость коррозии
Увеличивается. Однако при достижении определенной концентрации
скорость коррозии резко снижается вследствие перехода железа
•^пассивное состояние.
Пассивный металл при изменении внешних условий может
снова переходить в активное состояние (депассивация, активация).
Необходимо различать условия наступления пассивного состояния
Металла и сохранения пассивности. Так; железо пассивируется при
Действии 60—80%-ной азотной кислоты, но после этого концентра-
229
ция азотной кислоты может быть снижена до 30%, без нарушения
пассивного состояния железа в течение длительного времени.
Пассивное состояние металлов, как правило, вызывают сильные
окислители. Так, железо пассивируется достаточно концентрирован-
ными растворами HNO3, ШО3, КС1О3, К2Сг2О7. Концентрация
окислителя, необходимая для пассивации, должна быть тем больше,
чем выше температура среды. Таким образом, температура являете^
активизирующим коррозию фактором, действие которого должно
компенсироваться повышением концентрации окислителя.
Грубообработанные поверхности пассивируются труднее, чем
тонкообработанные. Устойчивость пассивного состояния зависит от
характера пассиватора, условий пассивации (температуры, давления),
структуры и состояния поверхности металла и времени действия
пассиватора.
Пассивный металл быстро активируется во влажном воздухе,
особенно если воздух содержит углекислый газ. Сильными актива-
торами являются также ионы F~, С1~, Br~, J". Механические повре-
ждения металла (царапины) в сухом воздухе приводят к потере пас-
сивности только на поврежденном участке, а в растворах — к актива-
ции всей поверхности металла.
Наиболее обоснованным представлением о пассивном состоянии
является предположение о том, что па поверхности металлов обра-
зуется защитная пленка. Согласно этим представлениям, на поверх-
ности металла при взаимодействии его с окислителями возникает
прочная окисная пленка толщиной 20—30 А. Поры образовавшейся
пленки играют роль анодных участков. Поскольку площадь пор
мала, они находятся под действием анодного тока большой плот-
ности. Явление пассивности следует рассматривать как результат
появления на поверхности металла защитной пленки высокой плот-
ности, с малой площадью пор. Пока металл сохраняет свою пассив-
ность, должно сохраняться равновесие между силами, создающими
пленку (действие окислителя), и силами, нарушающими ее целост-
ность. При достаточно большой концентрации окислителя в защит-
ной пленке имеется малое количество пор и скорость коррозии мала.
Со снижением концентрации окислителя число пор и их общая по-
верхность увеличиваются, и при достижении определенной величины
наступает переход металла из пассивного состояния в активное,
происходит быстрое разрушение защитной пленки.
Гипотеза об образовании защитной пленки еще далека от того,
чтобы объяснить всю сложную совокупность явлений, происходящих
при переходе металла из активного состояния в пассивное и обратно.
Температура. Повышение температуры вызывает ускорение про-
цессов коррозии. Однако, вследствие изменения под влиянием тем-
пературы действия других факторов коррозии, встречаются случаЪ*л
более сложной зависимости скорости коррозии от температуры.
Так, если под влиянием температуры изменяются свойства образу-
ющихся на поверхности металла пленок, то изменение скорости кор-
розии может определяться изменением защитного действия пленки.
230
Например, коррозия цинка в дистиллированной воде при температуре
нпе 50° С начинает сильно возрастать, достигает максимума и после
qQ° С резко падает почти до нуля. Это объясняется тем, что при тем-
пературе ниже 50° и выше 90° С продукты коррозии образуют пленку
с высокими защитными свойствами, а в интервале 50—90° С обра-
зуется пленка пористая с плохими защитными свойствами.
Под влиянием температуры может изменяться и характер корро-
зионных процессов. Например, при сгорании сернистых топлив
в двигателях имеет место газовая коррозия деталей серным и сер-
нистым ангидридами при высоких температурах, а при понижении
температуры происходит жидкофазная коррозия серной и сернистой
кислотами. Поэтому при понижении температуры скорость коррозии
деталей двигателя вначале уменьшается, достигает минимума, а
затем начинает резко увеличиваться. Происходит это потому, что
вначале с понижением температуры замедляется скорость газовой
коррозии, однако дальнейшее понижение температуры приводит
к конденсации системы SO2—SO3—Н2О и образованию серной и
сернистой кислот, которые ускоряют процессы коррозии в жидкой
фазе.
Продолжительность коррозии. Увеличение продолжительности
контакта с коррозионной средой ведет к увеличению количества
разрушенного вещества. Зависимость скорости коррозии от времени
Ж>жет быть различной. Если факторы, ускоряющие коррозию,
и факторы, замедляющие ее, уравновешиваются друг другом,
скорость коррозии остается постоянной. Если в ходе коррозионного
процесса появляется защитная пленка или происходит увеличение
и изменение структуры ранее существовавших пленок, то скорость
коррозии со временем уменьшается и может достигнуть весьма малых
величин. При разрушении защитных пленок, увеличении числа мик-
роэлементов или увеличении температуры в ходе коррозионного
процесса скорость коррозии будет непрерывно увеличиваться.
Практически такая зависимость имеет место при коррозии большин-
ства металлов кислотами и алюминия — щелочами.
Если со временем происходит изменение в соотношении факторов,
ускоряющих или замедляющих процесс коррозии, то кривая зависи-
мости скорости коррозии от времени может иметь минимум или мак-
симум.
КОРРОЗИОННОСТЬ РАКЕТНЫХ топлив
Коррозионность ракетных топлив определяется содержанием
в них веществ, способных к химическому взаимодействию с конструк-
ционными материалами. Некоторые компоненты ракетных топлив
^^ладают высокой агрессивностью как к металлическим, так и неме-
таллическим материалам. Такие компоненты имеют высокую корро-
зионность, и для их хранения и транспортирования необходимо
подбирать материалы с высокой стойкостью к химическому воздей-
ствию агрессивных веществ и добавлять специальные присадки.
231
Особенно высокой коррозионностью обладают окислители ра-
кетных топлив на основе азотной кислоты и окислов азота, фтора
и его соединений.
Азотная кислота при 20—25° С разрушает почти все металлы,
за исключением золота, платины, родия, иридия, тантала и титана.
При взаимодействии HNO3 с металлами образуются нитриты или
окислы металлов и выделяется NO с небольшими примесями N2O и
3Me + 4n HNO3 -> rcNO+3Me(NO3)n + 2rc Н2О
4Me + 6rcHNO3 -> nNO2 + nNO + 4Me(NO3)n + 3« Н2О
6Me + 2nHNO3 -> 2nNO+6MeO+«H2O
10Me + 2«HNO? —> тг N2-|-10MeO+« H2O
где п — количество электронов, отдаваемых каждым атомом металла
при окислении.
При действии разбавленной кислоты на легкие металлы, например
цинк, восстановление азотной кислоты может происходить даже до
аммиака:
4Zn + 9HNO3 NH3 + 4Zn(NO3)2 + 3H2O
Степень восстановления азотной кислоты при реакциях с метал-
лами зависит от целого ряда факторов — концентрации кислоты,
природы металла, температуры и т. д.
Образование нитратов следует рассматривать как двухстадийну^
реакцию. При взаимодействии кислоты с металлами на первой ста-
дии образуются окислы, которые затем реагируют с азотной кислотой
и дают нитраты. Так, например, реакция меди с азотной кислотой
проходит через следующие стадии:
3Cu + 2HNO3 -> 3CuO + 2NO + H2O
3CuO + 6HNO3 -> 3Cu(NO3)2-f-3H2O
3Cu + 8HNO3 -> 3Cu (NO3)2 + 2NO + 4H2O
Реакция останавливается на первой стддии в тех случаях, когда
образующиеся окислы металлов не растворяются в азотной кислоте.
Так, при действии крепкой азотной кислоты на алюминий на поверх-
ности металла образуется тонкая плотная пленка окислов, нераство-
римая в концентрированной HNO3 и поэтому защищающая алюми-
ний от дальнейшей коррозии.
Скорость коррозии металлов с увеличением концентрации HN03
в большинстве случаев возрастает только до определенного значения
концентрации, а затем начинает уменьшаться. Наибольшие скорости
коррозии металлов наблюдаются при действии 20—40 %-ной кислоты.
Снижение скорости коррозии с дальнейшим ростом концентрации
кислоты объясняется пассивацией металлов, наступающей вследствие
образования на поверхности металлов пленок окислов, плохо
творимых в концентрированной кислоте.
Легкость пассивации и устойчивость пассивного состояния
зависят от состава металла. Наиболее легко пассивируются железо,
хром, молибден, алюминий, никель. Пассивное состояние металла
и / Л
Г ________ I
сохраняется и после изменения окружающей металл среды. Так,
щелезо, запасснвированное крепкой HNO3 сохраняет устойчивость
против коррозии не только в разбавленной HNO3, но даже в течение
некоторого времени в воде, водяных парах и других средах. Процесс
пассивации облегчается при понижении температуры. Так медь,
яе пассивирующаяся азотной кислотой при комнатной температуре,
-^»жет быть запассивирована при температуре ниже —1Г С, после
чУго остается пассивной длительное время и при комнатной темпера-
туре.
Большое влияние на скорость коррозии металлов азотной кисло-
той оказывает присутствие некоторых солей. Так, нитрит натрия
заметно ускоряет скорость коррозии металлов HNC)3. Это объяс-
няется тем, что окисление HNO3 является автокаталитической реак-
цией, в которой роль катализатора играет образующаяся при вос-
становлении кислоты двуокись азота. При добавке нитрита натрия
к HNO3 выделяется свободная азотистая кислота, разлагающаяся
с выделением окиси азота и двуокиси азота.
Обработка, которой подвергаются металлы, также влияет на
скорость их коррозии азотной кислотой. Так, в резервуарах корро-
зии прежде всего подвергаются места сварки.
Особенно большое влияние на скорость коррозии азотной кисло-
той оказывает температура. Ниже приведены данные, показывающие
сияние температуры на скорость коррозии легированной стали
'концентрированной HNO3 с 6,5% двуокиси азота: [3, 9]:
tt° с Глубина коррозии, мм/год
27 0,040
54 0,078
80 5,080
97 * 7,62
НО* ' 12,7
120 * ' _• 30,5
* Определения проводились в закрытом сосуде под да-
влением.
Для изготовления емкостей, средств перекачки и деталей ракет-
ных двигателей, контактирующих с окислителями на основе азотной
кислоты, используются алюминий п его сплавы, высококремнистое
железо и легированные стали, содержащие хром, никель, титан.
Сроки хранения концентрированной HNO3 в резервуарах из этих
металлов определяются не величиной коррозии металлов и потерей
из-за этого прочности стенок емкостей, а количеством осадков ми-
неральных солей, накапливающихся в кислоте в результате медлен-
ной коррозии металла.
При хранении азотной кислоты в металлических резервуарах
иначале выпадают мягкие и студенистые осадки, затем они уплот-
^Вотся и превращаются в твердые.
Азотная кислота энергично воздействует и на большинство
неметаллических материалов. Органические вещества, как правило,
Реагируют с HNO3 настолько бурно, что происходит их воспламе-
нение.
23а
Четырехокись азота по отношению к металлам значительно менее
активна, чем азотная кислота. Для хранения и перевозки приме-
няются, так же как и для HNO3, емкости из алюминия, сплавов
алюминия и из легированной стали.
Жидкий фтор является одним из наиболее реакционноспособных
химических элементов. Медленно реагируют с фтором или совсем
не реагируют инертные газы, фториды металлов, фторопласты^-^
металлы: висмут, золото, платина, олово и цинк. Медь, хром, мар-1
ганец, никель, легированная сталь и алюминий в отсутствие воды
практически стойки при контакте с фтором в результате образования
на их поверхности защитной пленки фторидов. При повышенных
температурах удовлетворительной стойкостью обладают никель,
его сплавы и легированные стали. Жидкий фтор хранят в резерву-
арах из алюминия или легированных сталей. Еще более энергично,
чем азотная кислота, фтор разрушает большинство неметаллических
материалов. Пластмассы в контакте с фтором воспламеняются. Жид-
кий и газообразный фтор не оказывает коррозионного воздействия
на некоторые керамические материалы.
Механизм коррозии металлов перекисью водорода мало изучен.
При контакте перекиси водорода с металлами на первое место ста-
вится требование минимального каталитического воздействия метал-
лов на разложение перекиси. Такими металлами являются пассиви-
рованный алюминий и нержавеющие стали, которые в то же врем;^
весьма стойки в среде перекиси водорода. Остальные металлы вы-
зывают ускоренное разложение перекиси и поэтому даже в случае
их высокой коррозионной стойкости не могут применяться в ка-
честве конструкционных материалов для изделий, контактирующих
с перекисью водорода.
Горючие компоненты ракетных топлив обладают значительно
меньшей коррозионностью по сравнению с окислителями и не тре-
буют подбора специальных коррозионностойких конструкционных
материалов. Их корродирующее действие возрастает с увеличением
содержания воды.
КОРРОЗИОННОСТЬ НЕФТЯНЫХ топлив
Главной составной частью нефтяных топлив являются угле-
водороды, не обладающие способностью к химическому взаимодей-
ствию с конструкционными материалами и поэтому не являющиеся
коррозионно-агрессивными веществами.
Коррозионность нефтяных топлив обусловливается наличием
в них некоторых кислородных и сернистых соединений, а такж$
водорастворимых кислот или щелочей [8, 16, 17].
Водорастворимые кислоты энергично корродируют все металлы
и поэтому присутствие в топливах даже их следов вызывает быстрое
разрушение емкостей, трубопроводов, насосов, деталей топливной
системы двигателей. По техническим условиям на все виды нефтяных
234 ! I
топлив требуется полное отсутствие водорастворимых кислот и
щелочей.
• Кислородными соединениями в нефтяных топливах, способными
^рродировать металлы, являются перекиси и органические кислоты.
Перекиси образуются в нефтяных топливах как первичные проме-
жуточные продукты окисления углеводородов топлива кислородом,
^ррозиониое действие перекисей на металлы еще мало изучено.
Органические кислоты, преимущественно нафтеновые, переходят
из нефти, а также образуются в топливах при окислении. В свеже-
приготовленных топливах наименьшее количество кислот содержится
н бензинах. С повышением пределов выкипания топлива содержание
в нем органических кислот увеличивается, однако в масляных фрак-
циях содержание органических кислот снова уменьшается.
Органические кислоты, переходящие в топлива из нефти, пред-
ставляют собою нафтеновые кислоты — труднолетучпе, обычно жид-
кие, малорастворимые в воде вещества со структурной формулой:
/°
Н2С—CH-(CH2)X-C<f где х>1
I I Ч)Н
Н2С СН2
Хсн2
Нафтеновые кислоты сильно корродируют цветные металлы
(медь, свинец, цинк, олово и т. д.) и значительно слабее алюминий
п черные металлы.
Кислоты, образующиеся при окислении топлив, более агрессивны,
чем нафтеновые, так как в их составе имеются алифатические низко-
молекулярные органические кислоты и сульфокислоты. Содержание
агрессивных органических кислот в окисленных топливах может
быть довольно значительным. Часть из этих кислот растворима
в воде. При наличии в топливах воды водорастворимые кислоты
концентрируются в ней и вызывают сильную коррозию металлов.
Коррозия металлов кислотами в присутствии воды является электро-
химической, поэтому наличие в топливных системах различных ио
природе сплавов или металлов способствует усилению коррозии.
В табл. 48 и 49 приведены данные, характеризующие скорость
коррозии металлов в присутствии и в отсутствие воды.
Приведенные в табл. 48 и 49 данные убедительно показывают,
что вода является основным фактором, ускоряющим коррозию
металлов в нефтяных топливах.
Продукты коррозии металлов органическими кислотами под
^воздействием двуокиси углерода из воздуха частично превращаются
г’^^карбонаты с выделением свободных органических кислот, что
нэдёт к дальнейшей коррозии металлов.
С понижением молекулярного веса кислот растворимость их
солей в топливах ухудшается. Нерастворимые соли отлагаются на
стенках тары или остаются во взвешенном состоянии. При подаче
в Двигатель топлива, содержащего продукты коррозии во взвешенном
состоянии, происходит быстрая забивка фильтров, жиклеров
в карбюраторах или форсунок.
При отложении продуктов коррозии на металле в виде прочной
пленки каталитическое действие металлов на процесс окисления
углеводородов топлив снижается и скорость дальнейшей коррозии
замедляется.
*
Таблица
Время появления первых признаков коррозии (в сутках)
в зависимости от обводненности топлива Т-1 [5, 7]
поверхности металла прочную пленку, предохраняет металл от кор-
розии и замедляет процесс окисления топлив.
Время появления первых признаков коррозии металлов (в сутках)
в керосине и в системе керосин — вода [5, 7]
/
Сталь Ст. 20
Электрон МА-5
Среда
керосин
граница
раздела
водный
слой
керосин
граница
р аздела
водный
слой
Керосин . . .
Вода — керосин
7-10
1 ч
ускорение окисления прямогонных топлив ме-
Каталитическое
талламп приводит к образованию продуктов, которые, в свою очередь,
взаимодействуют с металлами. Так, электрон МА-5 корродируется
органическими кислотами значительно сильнее, чем сталь Ст. 20.
Однако при испытании коррозионного действия керосинов прямой
перегопки на эти металлы оказалось, что пластинки из стали Ст. 20
корродировали значительно сильнее пластинок из электрона МА-5.
Это объясняется тем, что в керосине, хранившемся без металлических
пластинок, и в керосине с пластинками из электрона МА-5 кислдС
ность за время хранения не изменилась, а в образцах керосина,
хранившегося со стальными пластинками, вследствие каталити-
ческого действия стали на процесс окисления, кислотность за 6 ме-
сяцев возросла с 0,56 до 14,5 мг КОН/ЮО мл топлива.
»
г
Вследствие того, что продукты термического крекинга, богатые
ненасыщенными углеводородами, легче окисляются кислородом
«оздуха они являются более коррозионными по сравнению с продук-
тами прямой перегонки. Поэтому крекинг-бензины довольно зна-
чительно корродируют медь, ее сплавы и углеродистые стали. Стой-
vVmh против коррозии в крекинг-бензинах являются алюминий,
сплавы и хром-никелевые стали.
Разнообразно влияние на коррозионность нефтяных топлив
сернистых соединений [17—19]. Только некоторые серусодержа-
щие соединения в топливах вызывают коррозию металлов при кон-
такте с жидкими топливами, но абсолютно все сернистые соединения
после сгорания топлив в двигателях, превращаясь в SO3 и SO2,
вызывают резкое усиление коррозионности продуктов сгорания
топлив.
В условиях хранения и транспортирования металлы корроди-
руются сероводородом, меркаптанами и элементарной серой. Эти
соединения условно называют активными сернистыми соединениями.
Кроме них коррозию могут вызывать серная и сернистая кислоты,
сульфокислоты, средние и кислые эфиры серной кислоты.
Активные сернистые соединения с металлами образуют сульфаты,
сульфиды и меркаптиды. Последние выделяются из топлив в виде
студенистых осадков, быстро забивающих фильтры и форсунки,
анализ отложений на фильтрах, напоминающих пластические смазки,
показал, что в основном они состоят из топлива, продуктов
его окисления и меркаптидов меди или кадмия, играющих роль
загустителей. Типичный состав отложений, образующихся при
контакте сернистого топлива с медью или ее сплавами, следующий
(вес. %)*.
Топливо............................................ 95
Меркаптид меди................... 1,8
Продукты окисления углеводородов 2,2
Вода................................ 1
Экспериментально доказано, что увеличение коррозии конструк-
ционных металлов при низких температурах вызывается в основном
присутствием меркаптанов, сероводородов и элементарной серы.
Так, при увеличении содержания в топливах меркаптановой серы
с 0,01 до 0,13% коррозия стали возрастала в 15 раз, а меди — в 36
Раз. Влияние меркаптанов [17] на коррозионную активность реак-
тивного топлива ТС-1 показано на рис. 88.
Коррозионная активность меркаптанов определяется их стро-
ением. Алифатические меркаптаны значительно более агрессивны,
ароматические, что объясняется способностью последних обра-
^|ывать на поверхности металлов смолистые отложения. В присут-
ствии алифатических меркаптанов отложений образуется значи-
тельно меньше [17].
При сгорании в двигателях все серусодержащие соединения
°празуют серный и сернистый ангидриды. Поскольку SO3 и образу-
ющаяся из него H2SO4 более коррозиоины, чем SO2 и H2SO3, на
237
величину коррозии металлов продуктами сгорания сернистых топлив
влияет соотношение между SO3 и SO2 в этих продуктах. Оно опре-
деляется температурой и давлением в камере сгорания и составом
рабочей смеси. Чем выше температура и давление и чем беднее ра-
бочая смесь, тем больше содержание SO3 и меньше SO2. В выхлопных
Рис. 88. Зависимость коррозионности
гидрированного топлива TG-1 от содер-
жания меркаптанов (150° С за 6 ч):
2 — втор-октилмеркаптан; 2 — фенилмеркап-
тан.
В выхлопных
газах дизелей, работающих при
высоких температурах и давТ?#
ниях и на бедных смесях, со-
держится больше SO3, чем в
выхлопных газах карбюратор-
ных двигателей (60—90% от
общего количества окислов
серы).
При высоких температурах
окислы серы вызывают газовую
коррозию металлов. При низ-
ких температурах окислы серы,
реагируя с водой, образуют
кислоты и вызывают кислотную
3
5
Тенператцро дбигатем
Рис. 89. Зависимость износа двига-
теля от температуры при работе на
сернистом топливе.
1 — кислотная; 2 — минимальная;
3 — газовая.
температурах, когда взаимодеи-
конденсации паров
коррозию. При промежуточных
ствие металлов с окислами замедляется,
еще не происходит, коррозия, обусловленная продуктами сгорания
сернистых соединений, минимальна (рис. 89).
Преобладающее влияние на износ цилиндров двигателей при
работе на сернистых топливах оказывает кислотная коррозия,
величина которой зависит от соотношения между SO3 и SO2. В при-
сутствии SO3 повышается точка росы, т. е. та минимальная темпера-
тура, при которой происходит конденсация водяных паров, а По-
этому чем выше содержание SO3, тем при более высоких температу-
рах начинается кислотная коррозия.
Для смеси Н2О + H2SO4 температура кипения определяется
соотношением компонентов, причем точка росы, в соответствии с за-
коном фазового равновесия, будет выше, чем температура кипения.
Температура конденсации зависит от парциального давления пара
j3 газовой смеси и является функцией состояния газа. Изменение
состояния системы Н2О + H2SO4 при изменении температуры при-
ведено на рис. 90. Пар с небольшим содержанием H2SO4 (точка
jvохлаждается, не изменяя своего
’сЯстава, до точки росы (точка 2).
Затем пар становится насыщен-
ным, образует капли, которые
обогащаются серной кислотой до
тех пор, пока содержание ее в
каплях не достигнет предела, со-
ответствующего температуре ки-
пения дайной смеси (точка 3).
Таким образом, несмотря на не-
большое содержание окислов серы
в продуктах сгорания, продукты
конденсации могут содержать
капли серной кислоты высокой
концентрации, обладающие силь-
ным коррозионным действием.
Проникая через зазоры порш-
невых колец в картер, окислы
серы образуют в смазочных мас-
лах сульфокислоты и смолистые
вещества. Ускоряя полимеризацию
продуктов окисления смазочных
Рпс. 90. Линия насыщения и точка
росы смеси НоО + H2SO4.
масел 'и коагулируя вейщства, диспергированные в маслах, окислы
серы значительно увеличивают лако- и нагарообразование в двигате-
лях, что ведет к ускоренному износу деталей.
Ускорение образования лака под воздействием окислов серы
происходит либо вследствие каталитического действия серной кис-
лоты на конденсацию ароматических углеводородов с альдегидами
и перекисями, либо вследствие образования высокомолекулярных
соединений типа диарилсульфонов.
Образование диарилсульфонов происходит через сульфокислоты:
SO2 + АгН—> ArSO3H
ArSO3H + АгН —>ArSO2 Аг + Н2О
Сульфокислоты образуются при жидкофазном окислении топлив,
и их количество определяется содержанием серы в топливах. Схема
Образования сульфокислот следующая:
* ROOH + RSR-»ROH + R— S—R
О
R-S-R-J-ROOH —R-S-R + ROH
Il
О 0 0
239
-S-R + O2
R—S—CH—Ri +1100-
R-S-CH-R1 + O2
о 0 ’1
—> R-S—CH—Rx *RCH0 +SO3 + R1-CH2-
• Л I /
0 0 0-0 /
^R-S—C-Rx —> R-S-0H-f-C0 4-Ri
//K I A
о 0 0-0—н о 0
В процессе сгорания топлив сульфокислоты образуют серный
ангидрид:
RSO3H—>RH + SO3
RSO3H + O2
✓О
Ri-CC +SO3 + H2O
41
Износ двигателей при работе на сернистых топливах тем выше,
чем больше содержание общей серы в топливах. Опыты на судовом
двигателе показали, что за 500 ч работы на топливе с 0,9% cepjl
износ был в 2—4 раза выше, чем при работе на топливе с содержанием
серы 0,2%. При работе двигателя Я АЗ-204 на топливе с 1,3% серы
отмечено быстрое изнашивание гильз цилиндров, поршневых колец,
ухудшение экономичности и значительное ускорение старения сма-
зочного масла [7]. - -
Отложения на деталях двигателя при работе на сернистых топли-
вах более прочны и труднее удаляются. В табл. 50 приведены данные,
характеризующие зависимость износа деталей от содержания серы
в топливах [20].
Таблица 50
Износ цилиндров двигателя (в мк) в зависимости от общего содержания серы
Тип двигателя
Длительность
испытаний
Содержание серы в топливах, %
Стендовые испытания
ЯАЗ-204
Д-35 .
Д-54 .
КДМ-46
550 ч
1000 »
1000 »
1000 »
100
39
30
48
115
140
70
Эксплуатационные испытания
ЯАЗ-204
Д-35 .
1000 км
1000 »
5,6
130
4,5
280
8,6
440
240
Поскольку величина коррозии деталей двигателя при работе
па сернистом топливе определяется не только количеством серы,
яо и тепловым режимом работы двигателя, составом рабочей смеси
п т. д., то для различных двигателей и различных режимов работы
^опускается разное содержание общей серы в топливах. Так, работа
Lflree на малосернистом топливе при низкотемпературном режиме,
4^(стых остановках и запусках двигателя на ходу сопровождается
усиленным износом цилиндров двигателя за счет их коррозии окис-
ламн серы. И, наоборот, при использовании высокосернистого топлива
на двигателях с хорошей компрессией при высокотемпературном
режиме работы вредное действие окислов серы незначительно.
Установлено, что топлива с содержанием общей серы до 1%
при отсутствии активных сернистых соединений не оказывают суще-
ственного коррозионного действия на топливную аппаратуру двига-
телей и на стенки емкостей при хранении. Эксплуатация двигателей
7о серы, приводит к
повышенному
на сернистых топливах, содержащих более 1 %
ускоренному износу основных деталей двигателя
отложению нагара и лака, пригоранию поршневых колец. Примене-
ние присадок к маслам или топливам позволяет в значительной мере
нейтрализовать вредное действие продуктов сгорания сернистых
соединений. При использовании масла с эффективными присадками
^парообразование и износ деталей некоторых типов дизелей при
работе на топливах с содержанием серы до 1 % становится таким же,
как и на малосернистых топливах.
•При изготовлении топлив стремятся к возможно полному удале-
нию из них сернистых соединений, однако, учитывая, что любые
способы очистки ведут к удорожанию топлив, в технических усло-
виях допускается следующее содержание серы (в %):
Авиабензины................................ 0,05
Автобензины................................ 0,15
Реактивные топлива
из несернистых нефтей .................. 0,1
из сернистых нефтей.................... 0,25
Дизельные топлива
из’ несернистых нефтей.................. 0>2
из сернистых нефтей.................... до 1
При использовании сернистых дизельных топлив обязательно
применение масел с многофункциональными присадками.
О МЕХАНИЗМЕ ОБРАЗОВАНИЯ ОТЛОЖЕНИЙ НА МЕТАЛЛАХ
В УГЛЕВОДОРОДНЫХ ТОПЛИВАХ
При контакте углеводородных топлив с металлами, особенно при
повышенной температуре, на поверхности последних образуются
отложения., . '
Способность органических соединений топлив образовывать
11 ленки и отложения на металлах во многом определяется их поляр-
16 Заказ 792.
241
ностью и прочностью связей в молекулах нестабильных соединений
топлива, а также дефектами кристаллических решеток металлов.
В несовершенных кристаллах в отличие от идеальных * имеются
нарушения порядка расположения атомов пли ионов в решетке [21].
Для стехиометрических кристаллов известны два типа дефектов:
1) незанятый (вакантный) узел решетки и 2) атомы из одного угды
решетки смещены в другой. Оба эти дефекта являются причин1^
смещения соседних атомов или ионов решетки, которое определяется
зарядом, размером атома или иона. Более важную группу дефектных
кристаллов представляют кристаллы нестехиометрического состава.
Нестехпометрические кристаллы характеризуются излишком пли
недостатком одного из компонентов по отношению к стехиометри-
ческой формуле состава, хотя это и не приводит к изменению фазового
состояния. Действительно, большинство окислов металлов имеет
непостоянный состав, особенно металлов с переменной валентностью
(Fe, Си, А1 и др.).
Из изложенного выше следует, что в дефектных кристаллах
металлов имеются узлы с неравномерно распределенным электри-
ческим полем. Например, вакантные анионные узлы действуют
в кристалле, как центры с эффективным положительным зарядом
п создают кулоновское потенциальное поле, способное связывать
электроны. В других металлах образуются вакантные места в катиоц-
ной решетке, которые будут действовать в кристаллах как центра
с эффективным отрицательным зарядом и давать кулоновское поле,
способное связывать положительные заряды.
Резко выраженные дефекты кристаллов имеют медь, свинец,
железо. Следовательно, у них должны быть наиболее неравномерное
распределение центров с разными энергетическими уровнями, мини-
мальная стойкость к коррозионному воздействию полярных соедине-
ний (сернистых, азотных, металлорганпческих и др.) с низкими
уровнями энергии активации. Сильнее всего подвергается коррозии
медь, на ней легче всего образуются пленки и отложения.
Добавление к металлам различных компонентов, которые умень-
шают неравномерность распределения вакантных мест и узлов с по-
вышенной электрической плотностью, должно приводить к повыше-
нию устойчивости металлов против коррозионного воздействия
гетероорганических соединений. Действительно, сплавы меди с цин-
ком (бронзы, латуни) имеют лучшую коррозионную стойкость, чем
чистая медь; добавление к железу небольших количеств хрома,
ванадия, никеля, углерода и других приводит к резкому повышению
коррозионной стойкости этих сплавов. 1
В присутствии кислорода в углеводородной среде, содержащей
сернистые соединения, образуются разнообразные промежуточное
полярные продукты окисления, которые могут адсорбироваться ДМ
поверхности металлов с дефектными кристаллическими решетками.
* Идеальные кристаллы характеризуются тем, что каждый их атом или ион
находится в положении, которое имеет минимум потенциальной энергии.
242 1
Состав и структура этих отложений были изучены спектроскопи-
ческими методами [22].
На рис. 91 и 92 приведены инфракрасные спектры поглощения
отложений, образовавшихся на меди при нагревании н-гексадекана
и изопропилбензола с добавками ароматических сернистых соедине-
ний. Этими данными подтверждается присутствие в составе отложе-
Шй следующих структур;
RSCH2Rx; RSO2C—Rj Rx-O-S-O-Ra;
О ОН О
Me (SOR)n;
Me (SOoR)n; Me(OSO2R)n; RC<^
4OH
O'
Me; Me (SO4)n; MenO
О n
и некоторых других.
На основании исследования структуры и состава отложений
методами инфракрасной, ультрафиолетовой и рентгеновской спектро-
скопии [22] можно в первом приближении представить схему обра-
зования первичных слоев на поверхности металла (рис. 92). Образо-
вание ориентированных молекул на поверхности металла приводит
к перераспределению энергии связи в молекулах, вследствие чего
может наступить разрушение молекул (сольватационный отрыв)
и свободные радикалы будут переходить в раствор, инициируя
развитие окислительной цепи. Осколки молекул типа SO’, S”n другие
ориентируются около вакантных узлов кристаллической решетки,
проникают в межкристаллическое пространство, образуя прочную
пленку на поверхности металла. Одновременно с этим катионы
металла будут переходить в топливо. Адсорбция полярных (в боль-
шинстве случаев окисленных) молекул (радикалов) не ограничива-
ется первичньш актом, а продолжается дальше и приводит к
образованию следующих слоев отложений; вполне естественно,
что структура этих отложений будет иной по сравнению с первич-
ным слоем. Возможная структура отложений на металле показана
на рис. 93. »
Исследованиями поперечных срезов пластинок из меди (рис. 94)
с отложениями на них установлено, что на поверхности меди имеется
относительно толстый слой отложений (до 200—300 мк), который
состоит из веществ аморфного и кристаллического строения. Рентге-
Д^рструктурным анализом установлено, что доля кристаллической
^зы в отложециях по мере приближения к металлу возрастает.
Верхние слои в большинстве случаев легко удаляются механи-
ческим путем.
Отложения, образовавшиеся на меди при нагревании топлив
с Добавками ароматических и алифатических сернистых соединений,
1G*
243
по внешнему виду резко отличны друг от друга [17]. В первом слу-
чае — это рыхлые, легко снимающиеся отложения желтого и корич-
невого цвета, во втором — плотные блестящие, иногда матовые,
10
20
; 70
U. СП
Рис. 91. Инфракрасные спектры поглощения отложений, образовавшихся на
меди при нагревании (150° С) изопропилбензола с добавкой фенилмеркаптана.
отложения, которые, как правило, трудно удаляются механически.
Но и среди ароматических сернистых соединении склонность к обра-
зованию отложений неодинакова. Максимальное количество отло-
жений образуется в присутствии фенилмеркаптана [17].
Под верхним рыхлым слоем отложений располагаются пленки,
которые невозможно удалить механическим путем; они не раство-
ряются п в органических растворителях. Можно предположить, что
в данном случае происходит внедрение осколков полярных молекул
244
Рис. 92. Инфракрасные спектры поглощения отложений, образу
ющихся на медной пластинке при нагревании (150° С) цетана с добав
кой 0,2% (в пересчете на серу) дифенилдисульфида.
245
Таблица 51
Состав отложений, образующихся на металлах при их контакте
с раствором тиофенола в среде н-гексадекана, вес. % [231
Контактирующие металлы
Элементы
медь электро-
литическая
бронза ВБ-24 латунь Л-62
дюралюми-
ний ДТТ
Углерод
Водород
Сера . .
Азот . .
Медь . . .
Алюминий
Магний
ДинкЗ>.
Кремний
Марганец
Титан
29,9
6,81
6,35
Следы
4,5
0,001
0,2
0,01
0,3
0,001
0,001
30,41
5,38
5,40
Следы
4,0
0,003
0,3
1,0
0,3
0,002
0,0001
32,82
7,42
5,11
Следы
3,5
0,001
0,2
0,8
0,3
0,01
0,0001
34,48
10,11
6,81
Следы
0,01
5,6
0,4
0,01
0,2
0,003
0,2
В- OSO?H
В—С—О—Мг —
в - с<
хон
В - SO
R-S-CH.-R
R-SO.CH-R,
0-0
в
MeSO
-С—С—СН, —R,
Me=S Me О
H О
O = S = O
!(Z\o
МехгО
С-0
О =S = O
о
h J /У' |^/>
Зона внедрения промежуточных продуктов
превращения гетероорганических соединений б металл
Рис. 93. Возмож-
ное строен ие^отло-
жений на метал-
лах.
246
(серооргапическпх соединений) в дефектные
основания утверждать, что ионы сульфокислоты адсорбируются на
/сСроорганических соединении) в дефектные кристаллические
решетки металлов (?) и межкристаллпческое пространство. Имеются
анодных участках и изолируют активную поверхность металла [22].
Исследование элементарного состава отложений позволило уста-
^х>вить присутствие в них различных элементов, являющихся компо-
нентами контактирующего металла (табл. 51).
Рис. 94. Микрофотография поперечного среза бронзо-
вой пластинки с образовавшимися на ней отложе-
' ниями (X 300).
Специальным анализом было установлено, что сера проникает
в металл на значительную глубину (до 30--50 мк) [24].
Из сказанного следует, что при воздействии сернистых нефте-
продуктов на металлы одновременно с коррозионным разрушением
последних протекают процессы образования защитных пленок [24].
Если бы удалось образовать на поверхности металлов прочные
защитные пленки, то коррозия была бы предотвращена или в значи-
тельной степени уменьшена. Эту задачу можно решить введением
♦углеводородную среду присадок.
Логично предположить, что в составе присадок должны оыть
Функциональные группы с небольшими величинами энергии разрыва
связей между атомами, остальные связи должны быть достаточно
прочными. Молекулы присадок должны иметь ярко выраженную
Полярность и большой дипольпыи момент. Такими присадками
247
могут быть, например, некоторые азотистые, серу-, кислород-
и азотсодержащие соединения и, возможно, металлорганпче-
ские. 1
Действительно, в группах N—Н, С—S, S-Н связи между атомами
значительно менее прочны, чем в исходных углеводородах. Вероятно *
по этим связям идет разрушение молекул и последующее их внедрс|«^
нпе в дефектную кристаллическую решетку металлов. Кроме того,
эти соединения должны обладать антпокислительными свойствами
[13—15, 25]. ]
ПРОТИВОИЗНОСНЫЕ СВОЙСТВА топлив
Срок службы механизмов в основном зависит от интенсивности
износа трущихся деталей. Износ трущихся деталей происходит
в результате: 1) механического истирания; 2) коррозионного пораже-
ния поверхности; 3) проникновения между трущимися поверхно-
стями деталей микрочастиц, играющих роль абразива.
Механическое истирание уменьшается, если сопряженные поверх-
ности деталей тщательно обработаны и трение между ними осуще-
ствляется через слой смазки. В узлах трения и сопряженных дета-
лях топливной системы ракетных, реактивных и некоторых поршне-
вых двигателей смазка осуществляется топливом. Смазывающая
способность топлив зависит от уровня вязкости, характера изменений >
вязкости с температурой и от наличия в топливах поверхностно-
активных веществ. Однако смазывающие свойства топлив значительно
хуже, чем специально приготовленных масел и смазок, так как
углеводородные топлива по сравнению с маслами имеют меньшую
вязкость п содержат меньше полярных соединений, способных об-
разовать на поверхности металла прочную пленку. Смазывающие
свойства углеводородных топлив ухудшаются по мере понижения их
температуры кипения (пределов выкипания). Также плохие протпво-
износные свойства имеют азотнокпслотные окислители, перекись
водорода, гидразин и некоторые другие компоненты ракетных
топлив. I
Коррозионное поражение трущихся деталей, так же как и при
контакте неподвижных металлических деталей с топливами, опреде-
ляется коррозионным действием топлива на данный металл. Однако
для трущихся деталей механизмов коррозионное действие топлив
усугубляется тем, что продукты коррозии могут переходить в среду
топлива в виде твердой фазы и вызывать дополнительно абразивный
износ трущихся металлических поверхностей деталей, работающих
в маловязкой среде топлива.
В условиях повышенных температур, аэрации при перемещенитт ч
топлива и контакта с каталитически активными металлами ускоряв Ч
ются процессы окисления нестабильных соединений топлив, обра-
зование мелкодисперсной твердой фазы за счет смолистых продуктов
окисления, влаги и минеральных микропримесей с «зольными»
элементами. В 1 мл топлива обычно насчитываются тысячи частиц
размером до 5 мк, которые кроме того, что являются центрами по-
>ледующей коагуляции, сами играют роль абразивов, способству-
ющих ускорению износа трущихся деталей.
При нагревании системы на поверхностях металлических деталей
откладываются смолистые вещества, что ведет к сильному ухудше-
нию условий трения, уменьшает зазоры прецезионных пар, вызывает
задиры и даже заклинивание трущихся деталей. Скорость накопле-
ния смолистых отложений зависит от стабильности топлива. Поэтому
почти все антиокпслительные присадки и присадки, улучшающие
Рис. 95. Принципиальная схема установки ПСТ-1:
I — горячий контур; II — холодный контур. 1 — топливная
аппаратура ТРД; 2 — дроссельный кран; з — топливная ко-
лонка; 4—электроподогреватель; 5—регулировочный кран;
6 — холодильник; 7 — топливный бак; 8 — подкачивающий
насос; 9 — кран-клапан; 10 — ресивер.
термическую стабильность топлив, являются одновременно и про-
ти в о и зносными.
Интенсивность износа трущихся деталей механизмов при кон-
такте с топливом определяется:
1) противоизьЬсными свойствами топлива;
2) содержанием в топливе микрозагрязнений (почвенная пыль,
продукты износа и коррозии металлов);
3) составом металлов или сплавов, из которых изготовлены тру-
щиеся детали, тщательностью обработки их поверхностей, величиной
и характером нагрузок (постоянных или переменных);
4) температурным режимом работы деталей и температурой
топлива.
Противоизносные свойства топлив зависят от их коррозионных
свойств, стабильности и смазывающей способности и определяются
величиной износа трущихся деталей в среде топлива. Так, на стенде
ПСТ-1 (рис. 95) противоизносные свойства топлив оценивают по
износу сферической поверхности вставки-свидетеля, находящейся
249
в насосе вместо подпятника [26]. Исследования, проведенные с реак-
тивными топливами на этом стенде, показали, что износ трущихся
деталей зависит от физико-химических свойств топлива. Легкое то-
пливо Т-2 вызывало наибольший износ (рис. 96). С повышением тем-
пературы, когда снижается вязкость топлив и ухудшается их ста-
бильность, противоизносные свойства топлив резко ухудшаются.
К
Гдердость СсгШки
Рис. 96. Влияние некоторых реактивных топлив на
износ вставки в установке ПСТ-1.
Несколько иной критерий для оценки противоизносных свойств
топлив использован в приборе КВ-1, представляющем собой машину
трения, в которой одним из элементов трущейся пары является спи-
ральный виток проволоки, закрепленный на образующей поверхности
диска, а вторым — неподвижный цилиндрический ролик. Трущуюся
пару помещают в ванну с испытуемым топливом. Противоизносные
свойства топлива определяются величиной нагрузки (в кг), при
которой в условиях испытания происходит разрушение граничного
слоя и начинается катастрофический износ трущейся пары [27].
и
Уменьшение коррозионности топлив может быть достигнуто уда-
лением из топлив коррозпонно-агресспвных веществ и применением
присадоц.
9
Очистка топлив. Органические кислоты, сероводород и меркап-
таны извлекают из нефтяных топлив щелочной очисткой. Эти веще-
ства, реагируют со щелочью, образуют соли, растворимые в воде
п легко удаляющиеся с водой:
RCOOH + NaOH<z>RCOONa + H2O
H2S + NaOH <=> NaHS + H2O
RSH + NaOH<=>RSNa + H2O
Однако при щелочной очистке вследствие гидролиза невозможно
достигнуть полного удаления меркаптанов и органических кислот.
Чем больше молекулярный вес органических кислот или меркапта-
нов, тем труднее они извлекаются из топлива. Так, при щелочной
очистке из нефтяного топлива можно извлечь 97,1% этилмеркаптана
и только 33% изоамилмеркаптана.
При сернокислотной очистке удаляются частично сернистые со-
единения, органические кислоты и асфальто-смолистые вещества.
Сернистые соединения или непосредственно растворяются в серной
кислоте, или образуют с ней соединения, растворимые в H2SO4.
Сероводород окисляется серной кислотой до серы с образованием
сернистого ангидрида и. воды:
H2S + HaSOi->S+SOa + 2H2O
Меркаптаны с H2SO4 образуют дисульфиды, сернистый ангидрид
и воду:
2RSH + H2SO4—>RSSR + SO2 + 2Н2О
Тиофен и его гомологи образуют хорошо растворимую в серной
кислоте тиофенсульфокислоту:
СН—СН
СН СН
НС---С—SOsH
+ H2SO4_-> II II +ы2о
НС СН
Сульфиды, дисульфиды и тпофаны не реагируют с H2SO4, но
растворяются в ней и поэтому частично извлекаются из нефтяных
топлив при сернокислотной очистке.
Сернистые соединения можно удалять пз топлив при помощи
селективных растворителей и адсорбцией твердыми адсорбентами.
При очистке от сернистых соединений необходимо учитывать, что
во время удаления неразрушенных сернистых соединений различ-
ными реагентами (серной кислотой, селективными растворителями,
адсорбентами и т. п.) происходят большие потери углеводородной
части топлива.
Наиболее эффективным методом очистки топлив от сернистых со-
единений является каталитическое гидрирование. При гидроочистке
сернистые соединения разрушаются водородом в присутствии катали-
затора с образованием углеводородов и сероводорода. Большая часть
251
сероводорода удаляется из топлива при перегонке, а остатки его —
после щелочной (этаноламинной или фенолятной) очистки. В этих
процессах очистка основана на обратимости реакции сероводорода
с этаноламином или фенолятом натрия при разных температурах:
HOCH2CH2NH2 + H2S hoch2ch2nh4s .J
110° С чг
л
on —qno г*
CeH5ONa + H2S С6Н5ОН + NaHS
Применение присадок. В качестве присадок, уменьшающих кор-
розионное разрушение металлов топливами, применяются вещества,
способные образовывать на поверхности металла прочную защитную
пленку, или вещества, реагирующие с коррозионно-агрессивными
веществами топлив и нейтрализующие их. Например, в качестве
антикоррозионных присадок (ингибиторов коррозии) к окислителям
на основе азотной кислоты используются ортофосфорная и фтористо-
водородная кислоты, которые, реагируя с металлами, образуют проч-
ные пленки фосфатов или фторидов, предохраняющие металлы от
коррозии азотной кислотой. В качестве антикоррозионной присадки
к азотным окислителям применяют также иод. Ингибирующий
эффект иода проявляется лишь в том случае, когда он находится
в виде кислородных соединений HJO3, J2O5 и др. Протпвокоррозпон>^
ный эффект соединений пода прекращается, когда потенциал системы **
меньше пли равен 1,405 в; последнее объясняется тем, что эти кисло-
родные соединения восстанавливаются до чистого пода. Коррозион-
ность углеводородных топлив снижается при добавке противоокпс-
лительных присадок, замедляющих окисление углеводородов топлив
и тормозящих процессы образования коррозионно-агрессивных
веществ в топливах.
При перевозке водным транспортом, для уменьшения коррозии
танкеров в топлива добавляются ингибиторы ржавления, например
соли некоторых нафтеновых кислот, эфиры пентаэритритового
спирта, п-диокспбензофенон и т. д. [9]. Для снижения коррозии
при попадании в топлива морской воды рекомендуется добавка смеси
алкилмеркаптоуксусной кислоты с кислыми эфирами монолаурил-
фосфорной или диоктилфосфорной кислоты. Очень часто в качестве
ингибиторов ржавления используются аммонийсульфонаты и орга-
нические фосфорные соединения [9].
Коррозионное действие продуктов сгорания сернистых соедине-
ний также снижается при добавке в топлива присадок; одним из
первых был предложен нафтенат цинка [И]. Его действие заклю-
чается в том, что при сгорании присадки в двигателе образующаяся
окись цинка реагирует с окислами серы и дает сульфат цинка, уже"4 .
не являющийся коррозионным продуктом. Однако следует иметь
в виду, что ZnSO4, отлагаясь на днище поршня и на клапанах, изме-
няет тепловой режим двигателя и нарушает работу клапанов.
Б. В. Лосиковым и С. Э. Крейном предложена аминная присадка
в топлива, создающая щелочную среду и способствующая восста-
252
повлению серного ангидрида в сернистый [8]. Эффективным оказа-
лось также действие газообразного аммиака, подаваемого с воз-
духом в цилиндры двигателя.
Поскольку в выхлопных газах не обнаружено соединений аммо-
ния, нельзя объяснять действие аммияйса*'простой нейтрализацией
а^нслов серы. Проведенными исследованиями установлено, что
При добавке аммиака в цилиндры двигателя в продуктах сгорания
снижается содержание серного ангидрида. По-видимому, аммиак
реагирует с перекисями сернистых соединений и разрушает их с об-
разованием менее агрессивного сернистого ангидрида:
• уО
R-SOr-CH-Rj+NHa
I
О-О-
R—S02—СН—Rx + NH * —* R +SO2 + CO + H2O + R’+N
0-0-
МЕТОДЫ ОПРЕДЕЛЕНИЯ КОРРОЗИОННОСТИ ТОПЛИВ
Коррозионность ракетных топлив и нефтепродуктов не является
/абсолютной величиной и изменяется в зависимости от свойств ве-
ществ, с которыми контактируют топлива, и от внешних условий,
в которых происходит это контактирование. Оценку коррозионности
топлив проводят, как правило, только по отношению к материалам,
с которыми топливо должно контактировать в процессе хранения,
транспортирования и применения. Чтобы оценить коррозионное
действие топлива на данный материал, необходимо выбрать соот-
ветствующие условия испытания и метод определения величины кор-
розии. Коррозия чаще всего определяется потерей веса образцов ма-
териала, контактирующего с топливом (в г/м1 • ч). Кроме этого,
она может определяться глубиной разъедания металла (в мм1год),
изменением механических свойств металла, изменением электриче-
ского сопротивления образцов металла и целым рядом других пока-
зателей.
Точность метода определения величины коррозии по потере веса
зависит от полноты удаления продуктов коррозии с испытуемого
образца материала с минимальными потерями самого материала.
В случае межкристаллитной или подповерхностной коррозии, когда
большая часть'продуктов коррозии расположена под поверхностью
металла, откуда их никакими способами очистки удалить не удается,
метод оценки величины коррозии по потере веса неприемлем.
' Удаление продуктов коррозии производится механически. Можно
также применять любой растворитель, не реагирующий с металлом:
воду, бензин, спирт, ацетон и др. Если продукты коррозии не уда-
ляются полностью механически, то прибегают к химическим методам
очистки металлов, при чем обязательно проводится холостое опреде-
ление для проверки воздействия химических реагентов на свежие
253
образцы металла. Химический реагент для удаления продуктов кор-
розии считается приемлемым в том случае, если потери веса образцов
металла в холостом определении меньше 10% от средних потерь
в весе, вызываемых коррозией испытуемым топливом. Величину
потери веса в холостом опыте учитывают при определении корро-
зионных потерь веса металла; После травления образцов химиче-
скими реагентами окончательное удаление продуктов коррозии при-
водят промывкой водой или спиртом. Взвешивают образцы после
промывки не ранее чем через 5—10 ч с выдержкой очищенных образ-
цов в эксикаторе.
Наиболее надежными реагентамп для удаления продуктов корро-
зии являются: с железных и стальных образцов — 10%-ный лимон-
нокислый аммоний в растворе аммиака, 10%-ная серная кислота
с добавкой 1% формалина или тиомочевины (при 25° С), 5% раствор
едкого натра с цинковой стружкой (при 80—90° С); с оцинкованных
и цинковых образцов — 1 % раствор персульфата аммония, насы-
щенный раствор уксуснокислого аммония; со свинцовых образцов —
насыщенный раствор уксуснокислого аммония; с оловянных и мед-
ных, — 5% раствор соляной кислоты; с алюминиевых и магниевых —
5% раствор азотной кислоты, 20% раствор окиси хрома с 1% азот-
нокислого серебра (очищать в кипящем растворе 1 мин).
Таблица 52
Десятибалльная шкала коррозионной стойкости
металлов (при равномерной коррозии)
Глубина
коррозии,
мм / год
Балл
Группа стойкости
< 0,001
0,001—0,005
0,005—0,01
0,01—0,05
0,05-0,1
0,1-0,5
0,5-1,0
1,0-5,0
5,0-10,0
10,0
1 — совершенно стойкие
2 — весьма стойкие
3 — стойкие
4 — относительно стойкие
5 — малостойкие
6 — нестойкие
Для оценки коррозионной стойкости металлов предложены раз-
личные шкалы. В Советском Союзе стандартизирована десятибалль-
ная шкала, разработанная Институтом физической химии АН СССР
(табл. 52). Коррозионная стойкость металлов по этой шкале опре-
деляется глубиной коррозии (в мм/год). Для пересчета потери веса
с каждого квадратного метра в 1 ч на глубину коррозии (в мм/год),
пользуются следующей формулой: ч
8,76-А- *
е .
где П — проницаемость коррозии, мм/год;
К — потеря веса металла, г/м2 • ч;
q — плотность металла, г/см3.
Оценка коррозионной стойкости металлов как по потере веса,
Так и по проницаемости применима только для равномерной корро-
зии. При неравномерной и местной коррозии эти показатели харак-
теризуют только усредненную скорость коррозии, в то время как на
отдельных участках скорость отличается от этого значения. Осо-
бенно трудно оценить коррозионную стойкость металлов при меж-
кристаллитной коррозии. В этих случаях прибегают к определению
механической прочности образцов до и после коррозии.
Исходя из условий испытаний, оценка коррозионной агрессив-
ности топлив может производиться при помощи лабораторных или
натурных испытаний. При лабораторных испытаниях оценивают
коррозию различных образцов металлов в условиях контакта их
с топливом при различных режимах. Натурными испытаниями опре-
деляют коррозию емкостей, насосов, двигателей данным топливом
в различных условиях эксплуатации. При этом объектами испытаний
могут быть как промышленные образцы, так и изготовленные спе-
циально. Величина коррозии испытуемых изделий оценивается ви-
зуально и путем замера линейных размеров отдельных деталей,
а также по потере веса отдельными деталями при испытании. В дви-
гателях коррозионность топлив может определяться увеличением
количества металлических примесей в смазочном масле и в отложе-
ниях на фильтрах.
Лабораторные методы
Перед началом лабораторных испытаний необходимо иметь пол-
ную характеристику как испытуемого топлива, так и образцов метал-
лов, на которых проводится определение. Для топлива должен быть
указан его химический состав, количественное содержание корро-
зионно-агрессивных веществ и пр. Для образцов металлов указы-
вается химический состав, структура, технологическая характери-
стика, характер и степень деформации, величина и состояние поверх-
ности и ее обработка, форма.
Наиболее простым и широко используемым лабораторным методом
оценки коррозионности топлив является метод испытаний в открытом
сосуде, который имеет много разновидностей в зависимости от спо-
соба погружения образцов металла, способов регулировки темпера-
туры, формы и размеров сосудов и т. д.
При испытаниях в открытых сосудах необходимо учитывать, что
образцы металла нельзя подвешивать на металлических проволоках,
так как при этом возникает гальванический ток, усиливающий кор-
розию. Образцы металла должны укрепляться па стеклянных крюч-
ках или подставках. В одном сосуде можно испытывать два-три об-
разца, но только из одного и того же материала. Количество корро-
дирующего продукта должно быть таким, чтобы за время испытания
его состав не претерпел существенных изменений. Обычно это соста-
вляет от 20 до 200 мл топлива на 1 см2 поверхности погружаемых
образцов. Перемешивание испытуемого продукта в сосуде способствует
»
отводу от поверхности металла продуктов коррозии и подводу 'све-
жего топлива, что ускоряет коррозию и дает возможность сократить
Рис. 97. Прибор для иссле-
дования коррозионности
топлив в жидкой и паровой
фазах:
1 — топливо; 2 — пластинка из
исследуемого металла; 3 — ре-
акционный сосуд; 4 — обратный
холодильник.
В электрохимических
сроки испытания. Еще большее сокраще- 1
ние времени испытаний достигается при ]
переменном погружении образцов метал-
ла в испытуемое топливо и извлечении
их па воздух. Ускорение коррозии в эту/Хл
случае происходит за счет энергичного г
окисления топлива в тонком слое и об-
разования в нем агрессивных веществ, а
также частично за счет разрушения плен- |
ки на поверхности металла при перемене
условий коррозии. 1
На рис. 97 показан прибор, предло- I
женный Г. Ф. Большаковым и Е. И. Гули-
ным, на котором можно определять кор-
розионные свойства разных топлив в жид-
кой и паровой фазах. Прибор помещают
в термостат, который поддерживает задан-
ную температуру испытаний с точностью
± 0,1°. Пластинки из исследуемого мате-
риала подвешивают на держателе так,
чтобы две пз них находились в жидко^
фазе и две — в паровой. Коррозию опре-'
деляют по изменению
пок до и после
в г/м2.
Определение
нентов ракетных
в закрытых ампулах. Для этого образцы
металлов помещают в ампулы, заливают
исследуемым продуктом и ампулы запаи-
вают.
При определении газовой коррозии
образцы металла помещают в электриче-
ские печи, через которые продуваются газы
соответствующего состава. Для упрощения
конструкции прибора газовую среду мож-
но получать сжиганием топлива в лампах.
Предложен прибор, дающий возможность
получать сравнительные данные по кор-
розионности продуктов сгорания в зави-
симости от содержания в топливах серы,
тетраэтилсвинца и т. д.
методах определения коррозионности из-
меряется величина потенциала па границе металл — топливо пли
электрическое сопротивление металла, изменяющееся за счет роста
пленки продуктов коррозии на поверхности металла. Простой одно-
веса пласти-
испытаний п выражают
коррозионности компо-
топлив часто проводят
зпачпон связи между потенциалом и скоростью коррозии не суще-
ствует, поэтому метод измерения потенциалов может применяться
только для определения характера коррозионного воздействия
п изучения хода процесса. При этом получают кривую в координатах
потенциал — время. Обычно через некоторое время после начала
процесса коррозии кривая потенциала приходит к постоянному зна-
ы**нию. Конечные, установившиеся величины потенциала характе-
ризуют коррозионный процесс. Для измерения потенциалов исполь-
зуются потенциометрические установки различных типов, электрон-
ные вольтметры и электростатические измерительные приборы.
Эксплуатационные и стендовые испытания
При определении коррозионности лабораторными методами не-
возможно создать полного соответствия реальным условиям, в кото-
рых происходит контакт материалов с топливами при хранении,
транспортировании и применении. Так, в двигателях большая часть
деталей подвергается трению, что коренным образом изменяет усло-
вия создания пленок на поверхности металла. Возможность меж-
кристаллитной коррозии алюминия, его сплавов и нержавеющих
сталей, а также влияние на величину коррозии методов обработки
и напряжений в металлах не позволяют точно определить коррозию
деталей по результатам лабораторных испытаний отдельных образ-
/ диН металла. Все это вызывает необходимость проводить испытания
коррозионности топлив непосредственно в эксплуатационных усло-
виях на натурных объектах, представляющих собою либо полные
конструкции двигателей и резервуаров, либо стенды, имеющие
отдельные натурные детали или узлы двигателей и их топливных
систем.
При эксплуатационных и стендовых испытаниях определяют сум-
марное воздействие коррозионности топлив на механический износ
деталей или замером и взвешиванием изнашиваемых деталей, или
по количеству металла, переходящего за определенное время в еди-
ницу объема топлива.
Величину износа и коррозии можно также определять при помощи
радиоактивных изотопов. Детали или отдельные узлы перед испыта-
нием облучают до образования в них радиоактивных изотопов. При-
меняется вместо облучения вставка изотопов в исследуемые детали.
Во время испытания поверхности деталей разрушаются и изотопы
переходят в топливо пли масло. По величине радиоактивности то-
плива или масла определяют их коррозионность в условиях
испытания по отношению к меченым деталям.
ЛИТЕРАТУРА
7
1. Н. Д. Томашев, Теория коррозии и защиты металлов, Изд. АН
СССР, 1960.
2. Коррозия металлов и борьба с ней, под ред. С. Г. Веденкпна, И. А. Ле-
вина, Оборонгиз, 1955.
17 Заказ 792.
257
I
3. Коррозия металлов, под ред. В. В. Скорчеллетти, Госхимиздат, 1952.
4. Физико-химические и эксплуатационные свойства сернистых котельных
и дизельных топлив, ГОСИНТИ, 1958.
5. Моторные, реактивные и ракетные топлива, под ред. К. К. Папок, Е. Г. Се-
менпдо, Гостоптехиздат, 1962.
6. И. В. Рожков и др. Защита от коррозии и зачистка резервуаров
и тары, Воениздат, 1963.
7. М. С. Смирнов, Физико-химические и эксплуатационные свопер
сернистых котельных и дизельных топлив, ГОСИНТИ, 1958. 1
8. Б. В. Л о с и к о в, Н. Г. Пучков, Б. А. Энглин, Основы
применения нефтепродуктов, Гостоптехиздат, 1955.
9. И. Н. Путилова, С. А. Балезин, В. П. Баранник,
Ингибиторы коррозии металлов, Госхимиздат, 1958.
10. 3. А. Саблина, А. А. Гуреев, Присадки к моторным топли-
вам, Гостоптехиздат, 1959.
11— 12. Б. В. Л о с и к о в, М. С. С м и р н о в и др., Борьба с коррозией
двигателей внутреннего сгорания и газотурбинных установок, Машгиз, 1962.
13. Ф. 10. Рачинский, Ю. А. Брук, Г. Ф. Большаков,
Авт. свид., № 159591.
14. Г. Ф. Большаков, П. И. Давыдов, Ф. Ю. Р а ч и н-
с к и й и др., Химия сераорганических соединений, содержащихся в нефтях
и нефтепродуктах, Гостоптехиздат, 1963.
15. Ф. Ю. Рачинский, Г. Ф. Большаков, Ю. А. Брук,
Доклад на VIII научной сессии по химии сераорганическихсоединений, содер-
жащихся в нефтях и нефтепродуктах, Уфа, 1964.
16. П. И. Давыдов, Г. Ф. Большаков, Химия и технология
топлив и масел, № 5 (1961).
17. Г. Ф. Большаков, Е. А. Глебовская, Гетерооргани^-
ские соединения реактивных топлив, Гостоптехиздат, 1962.
18. Сб. «Химия сераорганических соединений, содержащихся в нефти и неф-
тепродуктах», Изд. АН СССР, 1958. *
19. Сб. «Химия сераорганических соединений, содержащихся в нефти и неф-
тепродуктах», Изд. АН СССР, 1959.
20. Н. В. Б р у с я н ц е в, Автомобиль, № 3 (1946).
21. А. Риз, Химия кристаллов с дефектами, ИЛ, I960.
22. Г. Ф. Большаков, Е. А. Глебовская, 3. Г. Каплан,
Тезисы докладов на VIII научной сессии по химии сераорганических соединений,
содержащихся в нефтях и нефтепродуктах, Изд. АН СССР, 1964.
23. Г. Ф. Большаков, Химия и технология топлив и масел, № О
(1963).
24. Г. Ф. Большаков, Е. А. Глебовская, Борьба с корро-
зией. Материалы Всесоюзной конференции, декабрь, 1963, Изд. «Недра», 1965.
25. П. И. Давыдов, Г. Ф. Большаков, Химия и технология
топлив и масел, № 10 (1960).
26. А. П. Грязнов, И. В. Рожков, Химия и технология топлив
и масел, № 4 (1964).
27. К. И. Климов, А. В. Виленкин, Авт. свид. № 121967.
Глава VIII
ТОКСИЧНОСТЬ топлив
ВИДЫ ПОРАЖЕНИЯ ОРГАНИЗМА ТОКСИЧЕСКИМИ ВЕЩЕСТВАМИ
Токсичностью или ядовитостью называется способность веществ
причинять вред живым организмам, вызывая местное раздражение
или общее заболевание организма. Вещества, обладающие токспч-
уостью, называются токсическими веществами или ядами [1, 2].
Поражение живых организмов токсическими веществами может
быть местным, ведущим к ожогам, язвам, экземам и другим пора-
жениям кожи, и общим, в результате которого происходит наруше-
ние нормальной деятельности нервной системы, органов дыхания,
кровообращения и других внутренних органов, сопровождающееся
болезненными явлениями различной тяжести, от слабого недомога-
ния до смертельного (летального) исхода.
Поражения живых организмов токсическими веществами делятся
на хронические и острые. Хроническое поражение возникает при
систематическом проникновении в организм малых количеств токси-
ческих веществ в течение длительного времени. Острое поражение
вызывается однократным попаданием в организм большой дозы токси-
ческого вещества [3, 4].
Некоторые соединения способны накапливаться в организме. Явле-
ние постепенного накапливания токсических веществ внутри орга-
низма называется кумуляцией токсических веществ. Вещества, спо-
собные к кумуляции, в малых концентрациях не оказывают вред-
ного действия в течение одного или нескольких рабочих дней. IIрп
Длительной работе в атмосфере даже с малой концентрацией кумули-
рующихся вредных веществ наступает сильное отравление организма.
правило, в этом случае в организме обнаруживается большое
количество токсического вещества, кумулированного в одном из
внутренних органов.
Кроме непосредственного накопления вещества в организме,
может происходить постепенное усиление изменений в состоянии
организма, вызываемое действием отдельных малых порций ракетных
17* - - ’259
*
топлив и нефтепродуктов. Такое явление называется функцио- J
налъной кумуляцией. При функциональной кумуляции чувствитель-
ность организма к токсическому веществу возрастает настолько,
что малейшие дозы вещества, ранее не вызывавшие никакой реакции
со стороны организма, приводят к тяжелому расстройству деятель-
ности организма. KzX
Оценка токсичности топлив
Токсичность топлив оценивается характером их действия на
живые организмы и концентрациями, при которых вызываются
болезненные изменения в организме при действии на него в течение
определенного времени.
По характеру воздействия на организм токсические вещества
можно разделить на пять групп.
1. Вещества, действующие на поверхность кожи или другие
наружные ткани организма и вызывающие химические ожоги, дер-
матиты, экземы и другие виды поражения. Такое действие оказывают
кислоты, едкие щелочи, нефтепродукты и т. д.
2. Вещества, вызывающие поражение дыхательных путей и лег-
ких, в результате чего появляется воспаление или отек легких.
К ним относятся окислы азота, пары ^азотной кислоты и перекис.; /
водорода и т. д.
3. Вещества, поражающие нервную систему и вызывающие
психическое возбуждение, нарушение сердечной деятельности, оста-
новку дыхания: синильная кислота и ее соли, бензол, бензин, керо-
син, гликоли, спирты, соли свинца и т. д.
4. Вещества, вызывающие изменение состава крови, так назы-
ваемые яды крови: амины, окись азота, иод и т. д.
5. Вещества, нарушающие обменные и окислительные процессы
в организме: окись углерода, окислы азота, бензол и т. д.
Большинство токсических веществ вызывают комбинированное
поражение организма. Так, бензины и керосины при попадании на
кожу вызывают химические ожоги, а проникая внутрь организма
через кожу или дыхательные пути, действуют на нервную систему.
Действие токсических веществ часто усиливается в присутствии
других веществ. Например, этиловый спирт значительно усиливает
отравляющее действие бензина, бензола и ряда других веществ.
Четыреххлористый углерод принадлежит к ядам, кратковременная
работа с которыми мало опасна, однако если после вдыхания паров
четыреххлористого углерода выпить хотя бы немного этилового
спирта, появляется сильное отравление, которое может окончиться u
даже смертью. Действие смеси окиси углерода и окислов азо^:
вызывает значительно более сильное отравление, чем действие
каждого вещества в отдельности.
При хранении некоторых веществ в результате Процессов окисле-
ния или разложения могут образовываться вещества значительно бо-
лее токсичные, чем основной продукт. Так, при хранении хлороформа
2G0
(
образуется продукт его окисления — фосген, обладающий- очень вы-
сокой токсичностью.
Иногда действие некоторых ядов усиливается в результате реак-
ций, происходящих с ними уже внутри организма. Так, при попада-
нии внутрь организма этиленгликоля происходит быстрое окисление
..чяг'о щавелевой кислоты. Образовавшаяся щавелевая кислота в по-
чсадых канальцах реагирует с солями кальция и отлагает кристаллы
оксалата кальция. В сосудах мозга также отлагаются кристаллы,
напоминающие кристаллы оксалата кальция и вызывающие пораже-
ние нервной системы. При попадании в организм человека около
100 мл этиленгликоля эти процессы вызывают смерть. Окисление
пли разложение некоторых токсических веществ, наоборот, приводят
к их обезвреживанию. Так, синильная кислота, окисляясь в орга-
низме, образует безвредные вещества, поэтому попадание синиль-
ной кислоты в организм в несмертельной дозе, после острых явлений
отравления, проходит без дальнейших осложнений. Однако уже
0,1 г синильной кислоты достаточно для полного паралича дыхатель-
ного центра, ведущего к смерти раньше, чем синильная кислота
успеет разложиться в организме.
Разделение веществ на токсические и безвредные не является без-
условным. Некоторые вещества в небольших дозах необходимы орга-
но му и способствуют его жизнедеятельности, а в повышенных дозах
вызывают отравление. Так, соли фтора в малых дозах необходимы
для нормальной жизнедеятельности организма, в больших же —
вызывают быстрое разрушение зубов. Многие вещества, относящиеся
к сильно токсическим, в ничтожных дозах стимулируют деятельность
организма и часто применяются с лечебной целью (препараты мышь-
яка, стрихнин, соли ртути п т. д.). И, наоборот, вещества, счита-
ющиеся безвредными, попадая в организм в больших дозах, могут
вызвать смертельное отравление, Так, хлористый алюминий, практи-
чески считающийся безвредным, при попадании внутрь в количе-
стве 15—17 г вызывает смерть. Сульфат меди вызывает смертельное
отравление при попадании в организм в количестве 1—2 г.
Очень часто повышенную токсичность веществ вызывают примеси,
попадающие в пего при получении вещества из загрязненных продук-
тов. Так, водород, получаемый действием кислоты на цинк, может
оказаться высокотокспчным при наличии в цинке соединений фос-
фора или мышьяка. В этом случае получаемый водород будет содер-
жать примесь сильно токсических фосфористого или мышьяковистого
водорода.
Возможность значительного повышения токсичности веществ
за счет наличия в них примесей вызывает необходимость очень осто-
^Чл-Пого обращения со всеми химическими веществами и строгого
соадюдения правил безопасности при работах в химических лабора-
ториях.
Специальные лабораторные и клинические исследования позво-
ляют установить для токсических веществ определенные концентра-
ции, вызывающие изменения в организме при различном времени
261
пх воздействия. Обычно определяют предельно допустимую концен-
трацию, концентрацию, допустимую для кратковременного воз-
действия, опасную и летальную (смертельную) концентрацию. 1
Предельно допустимой концентрацией токсического вещества
в воздухе считается концентрация, при которой не возникает ника- !
кпх изменений в организме в условиях ежедневного контакта с токси-
ческим веществом в течение неограниченного времени. Установление g
этой концентрации требует продолжительного наблюдения за мно- 1
гпми лицами, работающими с данным веществом. я
В табл. 53 приводятся предельно допустимые концентрации ]
некоторых топлив и их компонентов [5—8].
Таблица 53
Предельно допустимые концентрации вредных веществ в воздухе
производственных помещений (СН 245—63)
Вещества
Амиловый спирт . .
Аммиак ............
Ацетон.............
Бензин ...........
Бензол...........-.
Бутиловый спирт . .
Гексиловый спирт . .
Гидразин и его произ
водные.............
Двуокись хлора . . .
Диметилбензиламин .
Динитробензол . . .
Динитро-о-крезол . .
Дпнитротолуол . . .
Дихлоргидрин . . .
Диэтилампн.........
Изопропилнитрат . .
Иод................
Керосин (топлива Т-1,
Т-2, Т-5 в пересчете
на С)...............
К силидин............
Предельно
допустимая
концентра-
ция, Л12/Л13
100
20
200
100
20
200
100
0,1
0,05
30
300
Вещества
Ксилол ..............
Лигроин (в пересчете
на С)............
Метиловый спирт
Метилфторфенилднх
силан ..........
Монобутиламин .
Нитроксилол . .
Озон.............
Окислы азота (в пере
счете на N2O5) . .
Скипидар ..........
Тетранитрометан . .
Тетрахлоргептан . .
Тетраэтилсвинец . .
Тиофен ............
Трихлорбензол . . .
Триэти ламин ....
Фосген.............
Фтористый водород .
Хлористый метилен .
Этиловый спирт . . .
лор
Предельно
допустимая
концентра-
ция, Л12/.МЗ
50
300 V
50 >
10
0,1
300 •
0,3
0,005
10
10
50
1000
I
для кратковременного
пребывания,
Концентрацией, допустимой
считается концентрация, позволяющая проведение аварийных работ
без применения защитных средств в течение короткого времени и не
вызывающая при этом никаких вредных изменений в органп&че.
Обычно такая концентрация в 10—20 раз выше предельно дот.у-
стимой.
Опасной концентрацией считается концентрация, способная вы-
звать болезненные явления в организме даже при условии кратко-
временного воздействия. При наличии веществ в воздухе в таких коп-
262
центрациях категорически запрещается проведение любых работ
без применения средств защиты.
Летальной концентрацией является концентрация, при которой
наступает смертельное отравление при самом кратковременном воз-
действии. Эта концентрация устанавливается с учетом минимальной
д^чротивляемости организма.
/yZ иД’ Кроме концентраций токсических веществ в воздухе устанавли-
ваются также те минимальные количества вещества в граммах,
которые при попадании внутрь организма вызывают болезненные изме-
нения или смертельное отравление. Эти величины называют токси-
ческой или смертельной дозой данного вещества.
Определение токсических веществ в воздухе
Чтобы избежать несчастных случаев и отравлений при работе
с токсическими веществами, необходимо систематически контроли-
ровать их содержание в воздухе производственных помещений
[8,9]. . < '
Анализ воздуха на содержание токсических веществ проводится
либо при помощи газоанализаторов, либо после отбора пробы воз-
духа методами химического анализа.
Для определения содержания в воздухе токсических веществ
^ помощью газоанализаторов используются различные физические
и физико-химические явления, обусловленные наличием искомых
веществ в воздухе. Наиболее широко в газоанализаторах исполь-
зуются следующие явления, связанные с содержанием в воздухе
токсических веществ:
1) изменение электросопротивлении нагретой платиновой прово-
локи вследствие повышения ее температуры при каталитическом
сжигании анализируемой пробы на поверхности проволоки (газоана-
лизатор ПГФ-2-ВЗГ);
2) изменение электросопротивления нагретой платиновой про-
волоки при отводе тепла проходящими газами (газоанализатор
ГЭУК-21);
3) изменение температуры катализатора, на поверхности кото-
рого происходит сжигание анализируемой пробы воздуха (газоанали-
затор ЛИОТ);
4) изменение окраски фильтров-индикаторов при прохождении
через них анализируемого воздуха.
Концентрацию токсических веществ при хранении и применении
топлив определяют.по изменению окраски индикаторов. При помощи
сильфона или ручного насоса просасывают определенное количество
^ здуха через стеклянные трубки, наполненные индикаторным по-
рошком. Индикаторный порошок готовится нанесением на инертное
твердое вещество (фарфор, силикагель и т. п.) реактива, образу-
ющего с искомым веществом цветные продукты реакции. Сопоста-
вляя длину пли интенсивность окрашенной части индикаторной
трубки с заранее приготовленной шкалой, определяют концентрацию
263
4
данного вещества в воздухе. Подбором индикаторных трубок с раз-
личными реагентами можно определять концентрацию в воздухе раз-
личных веществ. Имеющиеся сейчас газоанализаторы УГ-1 и УГ-2
. снабжены набором трубок, дающих возможность определить кон-
центрацию в воздухе паров бензина, окиси углерода, аммиака, серо-
углерода и окислов азота.
Газоанализаторы, построенные на принципе изменения элецк.^"’ .
«сопротивления или температуры катализатора, дают возможность
осуществлять непрерывный контроль воздуха с включением свето-
вой или звуковой сигнализации при повышении концентрации токси-
ческого вещества сверх предельно допустимой. Однако такие газо- :
анализаторы не могут быть универсальными, они обязательно раз-
рабатываются и применяются для определения концентрации только
одного токсического вещества с учетом его физико-химических
свойств, и это значительно сужает область их применения. I
Значительно более универсальным, позволяющим определять
в воздухе содержание любого токсического вещества, является
весовой, объемный или колориметрический анализ проб воздуха.
Способы, место и время отбора проб могут быть различными в за-
висимости от цели анализа. Перед отбором пробы рекомендуется про-
вести качественное определение наличия в воздухе искомого веще-
ства. 11
Присутствие многих токсических веществ может быть обна* ч- .»
жено по запаху или по раздражающему действию на слизистые
оболочки, вызывающему слезотечение или кашель. Однако в боль-
шинстве случаев токсические вещества обнаруживаются органами
чувств человека в концентрациях значительно больших, чем пре-
дельно допустимые. Для обнаружения токсических веществ, не дей-
ствующих на органы чувств человека или действующих в концен-
трациях выше предельно допустимых, необходимо применение спе-
цифичных реактивов, например образующих с топливом цветные
продукты реакции. Очень чувствительным
является реактив Грисса — Илосвая, образующий с ним азокра-
ситель розового цвета, к аминам — хлорат алюминия (синяя окра-
ска), к аммиаку — солянокислый раствор нптроанплина с несколь-
кими крупинками азотистокислого натрия (красный цвет)
водороду — раствор ацетата свинца (черная окраска), к углероду —
5% раствор хлористого палладия (черная окраска).
Если для данного вещества подобраны высокочувствительные
реактивы, то достаточно развесить в обследуемом помещении полоски
бумаги, смоченные реактивом, чтобы определить распространение
паров токсического вещества в воздухе помещения. Если выбранные
реактивы не обладают необходимой чувствительностью, тогда их
заливают в пробирку или помещают туда бумажку, смоченную р<Чй’-
тпвом, и грушей для пульверизатора прокачивают воздух через
пробирку в течение 1—2 мин.
Для отбора проб чаще всего используется метод аспирации
(просасывацие измеряемого количества воздуха через жидкие пли
окислению азота
к серо-
нердые поглотители) и метод отбора проб в бутылки или газовые
пипетки.
Метод аспирации применяется в тех случаях, когда для прове-
ряя анализа требуется взять большое количество воздуха или
<оГда в воздухе возможно присутствие нескольких токсических ве-
^й^в со сложными химическими свойствами и требуется их раздель-
7 определение. Смесь таких веществ вначале адсорбируют на твер-
дом поглотителе, протягивая анализируемый воздух через трубки,
заполненные поглотителем. После накопления на поглотителе доста-
точного количества искомых веществ проводят ступенчатую десорб-
цию, продувая через поглотители чистый воздух при различных тем-
пературах. Разделенные таким образом искомые вещества порознь
улавливаются в поглотителях с жидким раствором, в котором затем
и проводится их определение химическими методами.
При отборе проб методом аспирации необходимо иметь соответ-
ствующие поглотители для каждого анализируемого вещества.
Воздух через поглотители протягивается аспираторами или вакуум-
насосдм. Аспираторы одновременно служат для протягивания воз-
духа и для замера количества протянутого воздуха, а также для
установления нужной скорости просасывания.
После отбора пробы в бутыли или газовые пипетки, определяемое
Vх ‘ество для дальнейшего анализа переводят из отобранной пробы
воздуха в раствор жидкого поглотителя. Это осуществляется одним
из следующих способов:
1) к короткой трубке в пробке бутыли присоединяют 2—3 погло-
тителя с жидким раствором, через длинную трубку в бутыль зали-
вают нейтральную жидкость до полного вытеснения пробы воздуха
из бутыли; • ,
2) пробу воздуха из бутыли вытесняют чистым воздухом, для
чего к длинной трубке в бутыли присоединяют поглотительные
колонки, а к короткой трубке — поглотители с раствором, и при
помощи аспиратора протягивают десятикратный объем воздуха;
3) поглотительный раствор вносят непосредственно в бутыль
с отобранной пробой — это самый простой и в большинстве случаев
наиболее надежный способ поглощения искомого вещества.
Дальнейший ход химического анализа устанавливается в зависи-
мости от физико-химических свойств искомого вещества.
ЗАЩИТА ЧЕЛОВЕКА ОТ ПОРАЖЕНИЯ ТОКСИЧЕСКИМИ ВЕЩЕСТВАМИ
Безопасность человека при работе с токсическими веществами
обеспечивается соблюдением мер предосторожности, не допуска-
к<..^их попадания токсических веществ в окружающую атмосферу,
и применением индивидуальных средств защиты. Тара, в которой
хранятся токсические вещества, трубопроводы и средства перекачки
Должны быть тщательно герметизированы. Пары, выделяющиеся
через дыхательные устройства резервуаров, необходимо отводить
за пределы рабочей зоны в устройства для нейтрализации. Перед
265
эксплуатацией или ремонтом технических средств треоуется тщатель-
ная нейтрализация деталей, загрязненных токсическими веще-
ствами. 1
При работе с токсическими веществами па открытом воздухе об-
служивающий персонал должен размещаться с наветренной стороны,
с тем чтобы в аварийных случаях уменьшить опасность поражеш?^.
В закрытых помещениях при работах с токсическими вещества^
обязательно включение приточно-вытяжной вентиляции, обеспечи-
вающей создание концентраций токсических веществ в воздухе ниже
предельно допустимых. Для веществ с относительно малыми токсич-
ностью или летучестью (нефтепродукты, спирты, перекись водорода
и т. п.) достаточно двухкратного обмена воздуха в час. При работе
с более токсическими веществами пли с веществами с высокой упру-
гостью паров, требуется десятикратный обмен воздуха в час.
Лабораторные помещения, помимо общей вентиляции, оборуду-
ются вытяжными шкафами с верхним и нижним отсосом. Скорость
протягивания воздуха в открытых створках шкафа должна быть
в пределах 1—1,2 м/сек. Все работы с токсическими веществами в ла-
бораториях проводятся только в вытяжных шкафах.
При случайном проливе токсических веществ необходимо при-
нимать срочные меры для их нейтрализации. На складах и пунктах
погрузки должны быть заранее приготовленные стоки для отвода
пролитых веществ в нейтрализационные устройства. А
Индивидуальными средствами защиты являются специальная
защитная одежда (брюки, куртки, комбинезоны, фартуки), сапоги,
перчатки, защитные очки и противогазы. Защитная одежда изго-
тавливается из материалов, не разрушаемых токсическими веще-
ствами и не впитывающих этих веществ. После работы защитную
одежду всегда нужно проветривать, а если на нее попадали жидкие
токсические вещества, то вымыть в горячей воде с мылом.
При всех работах с токсическими веществами необходимо иметь
противогаз, который надевают как только концентрация паров токси-
ческого вещества в воздухе превысит предельно допустимую. Про-
тивогазы по своей конструкции бывают изолирующие, шланговые
п фильтрующие.
Изолирующие противогазы состоят из маски и баллонов с запасом
кислорода и регенератора выдыхаемого воздуха.
При вдохе воздух из дыхательного мешка проходит под маску
п далее в легкие. В легких кровью поглощается 5% вдыхаемого
кислорода, а оставшаяся часть возвращается с выдыхаемым возду-
хом. Выдыхаемый воздух, содержащий увеличенное количество
двуокиси углерода и паров воды, поступает в клапанно-распредели-
тельную коробку и через выдыхательный клапан направляемая
в регенеративный патрон. В регенеративном патроне из воздуха погло-
щается двуокись углерода и пары воды. Через нижнюю соединитель-
ную трубку регенерированный воздух проходит в дыхательный ме-
шок и снова насыщается кислородом, поступающим туда из баллона
через редуктор. При каждом вдохе и выдохе цикл действия протнво-
за повторяется до израсходования запаса кислорода в баллоне.
Изолирующий противогаз весит свыше 7 кг и имеет срок действия от
Z1q до 120 мин.
[Иланговые противогазы имеют маску с клапанами и длинный
шланг (15—18 м), с помощью которого под маску подается чистый
.-опух. Применяются шланговые противогазы чаще всего при
®8рстке резервуаров и железнодорожных цистерн.
При пользовании изолирующим и шланговым противогазами
человек совершенно не зависит от состава окружающей среды,
и поэтому такие противогазы применимы при любых концентрациях
токсических веществ в воздухе.
фильтрующие противогазы, состоящие из маски, соединительного
шланга и металлической коробки с поглотителями вредных веществ,
применяются только в случаях, когда окружающий воздух имеет
достаточное для дыхания количество кислорода (не менее 16 вес. %)
п если концентрация токсического вещества в воздухе не превышает
2%. Время действия фильтрующих противогазов ограничено адсорб-
ционной способностью поглотителя.
Основным поглотителем в фильтрующих противогазах является
активированный древесный уголь с суммарной поверхностью пор
800—1000 м2 на 1 г веса. Кроме активированного угля в металличе-
ские коробки противогазов помещают также химические поглоти-
т4А< и противодымный фильтр из пористой целлюлозы. Химиче-
ские поглотители предназначены только для определенных веществ,
поэтому противогазы выпускаются нескольких марок с соответству-
ющим обозначением; например, для кислых веществ применяют по-
глотители щелочного состава: натронную известь, соду и т. п.;
для щелочных веществ — пемзу, пропитанную серной кислотой.
В универсальных противогазах, предназначенных для работы в ат-
мосфере как кислых, так и щелочных веществ, поглотитель состоит
из активированного угля с нанесенными на него содой и окисламп
металлов. Номенклатура промышленных противогазов приведена
в табл. 54.
После окончания работ с отравляющими веществами необходимо
снять защитную одежду, принять душ или помыть с мылом лицо
и руки, прополоскать рот водой и надеть чистую одежду.
Противогазы после пользования необходимо дезинфицировать.
Для этого маску противогаза выворачивают внутренней стороной
наружу и протирают ватой, смоченной спиртом или 2% раствором
формалина. После дезинфекции маску тщательно просушивают.
В силу того, что фильтрующие противогазы имеют ограниченный
срок защиты от ядовитых веществ, на каждый противогаз нужно
^естч учетную карточку с записями времени пользования противо-
Противогазы, бывшие в употреблении, хранить отдельно
от новых.
Защитную одежду по окончании работ проветривают в течение
2—3 ч и только после этого убирают в специально отведенное поме-
щение. При попадании на средства защиты жидких токсических
26
Номенклатура промышленных противогазов [10]
Таблица 5^
Марка
противо-
газа
Цвет коробки
Поглощаемые вещества
Среднее время
защитного
действия,
мин
Коричневый
Желтый
Желтый с чер-
ной полосой
Черный
Пары органических веществ (нефте-
продукты, ацетон, бензол, спир-
ты, эфиры, амины, нитро соеди-
нения и т. п.)
Кислые пары и газы (окислы азота,
галогеиоводороды, сернпстые га-
зы, сероводород, пары различных
кислот и т. п.)
Пары ртути
120
60
100
КД
со
м
БКФ
Зеленый
Серый
Белый
Красный
3 ащитный
Мышьяковистые и фосфористые со-
единения
Аммиак
Аммиак и сероводород в смеси
Окись углерода
У ниверсальный
Кислые газы, мышьяковистый водо-
род, дымы, туманы и синильная
кислота
пх необходимо
360
200
150
90
70
веществ
в горячей воде с 2,5%
30 мин, с трехкратной
ополаскиванием в чистой воде. При сильном загрязнении одежду
перед стиркой пропаривают при 120—130° С в течение 2 ч.
нейтрализовать, а затем вымыть
раствором соды. Мыть необходимо в течение
сменой моющего раствора и с последующим
спецодежду
ОКАЗАНИЕ ПОМОЩИ ПРИ ОТРАВЛЕНИЯХ
J
Для оказания первой помощи до прибытия врача или доставки
пострадавшего в лечебное учреждение необходимо удалить постра-
давшего из зараженной зоны, принять меры к удалению яда из орга-
низма или для нейтрализации яда внутри организма и поддерживать
сердечную деятельность и дыхание пострадавшего. '
Если отравление произошло через желудочно-кишечный тракт,
для удаления яда необходимо вызвать рвоту и промыть же-
лудок. |
При отравлении целым рядом веществ (азотной кислотой, этилен-
гликолем, этиловой жидкостью и т. д.) необходимо сразу же про-
мыть желудок водой. Промывание желудка полезно проводить
с одновременным осаждением яда или его нейтрализацией. Болй
ство органических ядов хорошо поглощается животным углем,
который добавляют к промывной воде по 2 столовых ложки на 1 л
воды. Легко окисляющиеся яды нейтрализуются промыванием же-
лудка 0,04% раствором перманганата калия. При отравлении кис-
268
потами на 1 л промывной воды следует добавить 20—30 г окиси
магния.
Обезвреживание ядов внутри организма может ныть достигнуто
приемом внутрь противоядия. При отравлении органическими веще-
сТвами полезно принять 20—30 г животного угля на стакан воды,
рвотный уголь быстро поглощает пз желудка попавший туда яд,
< штем выводится через кишечник. При отравлении кислотами
^’.пппмают окись магния в виде 15% взвеси по столовой ложке
через 5—10 мин или известковую воду в тех же дозах, щелочами — ,
2% раствор уксусной, винной или лимонной кислоты; при отравле-
нии ядами, разрушаемыми при окислении, —0,4% раствор перман-
ганата калия по чайной ложке через 10—15 мин.
Все эти виды помощи оказывают пострадавшему после выноса
его на свежий воздух. При отравлении веществами, способными сор-
бироваться тканями одежды, необходимо, после того как постра-
давший вынесен из зараженной зоны, снять с него одежду и надеть
чистую. Одежда, в которой проводилась работа в зараженной зоне,
может выделять адсорбированный ею яд и явиться причиной повтор-
ного отравления уже вне зараженной зоны.
В некоторых случаях, когда яд вызывает нарушение теплорегу-
лирования и приводит к усиленной теплоотдаче (при отравлении
этиловым спиртом, окисламп азота и т. д.), необходимо обогревать
^страдавшего при помощи теплых ванн, грелок, укутывания
в теплую одежду.
При остановке дыхания следует немедленно делать искусствен-
ное дыхание. При расстройстве и затруднении дыхания нужно
применять средства, возбуждающие деятельность дыхательного
центра, например лобелии, вдыхание смеси 5% двуокиси углерода
с кислородом и т. п.
При расстройстве сердечной деятельности (частом и неправиль-
ном пульсе) рекомендуется вводить подкожно 2—4 мл стерильного
камфорного масла или 1—2 мл кофеина. Хорошее действие при
незначительных нарушениях сердечной деятельности оказывает
смесь из настоек валерианы, адониса и белладонны.
В местах работ с токсическими веществами должна быть аптечка
первой помощи, состав которой согласовывается с врачом. Состав
аптечек определяется характером работ в данном помещении и свой-
ствами токсического вещества, с которыми ведутся работы. Кроме
аптечек на местах работ обязательно должны быть нейтрализующие
растворы для данного токсического вещества с тем, чтобы при слу-
чайном проливании или при попадании на кожу можно было
немедленно нейтрализовать ядовитое вещество.
% ЛИТЕРАТУРА
А
1. Вредные вещества в промышленности, ч. I и II, Справочник под
ред. Н. В. Лазарева, Госхимиздат, 1965.
2. Л. К. X о цен о в, Р. Г. Л е й т и с, Б. И. Марципков-
с к и й, Гигиена труда, Машгиз, 1958.
269
3. К. К. Папок, И. Г. Барон, Ядовитость топлив, масел и техни-
ческих жидкостей, Воениздат, 1960. ]
4. И. Я. Сосновик, Клиническая симптоматика и первая помощь
при профессиональных отравлениях, Медгиз, 1955. ]
5. Е. А. Перегуд, Е. В. Г е р н е т, Химический анализ воздуха
промышленных предприятий, Изд. «Химия», 1965. П
6. Д. Росс, Вопросы ракетной техники, № 1 (1951). I
7. С. Крон, Вопросы ракетной техники, № 6 (1955). г?Ф<
8. Санитарные нормы проектирования промышленных предприятий Н
54, 1954; Дополнения к Н 101—54, 1959.
9. И. С. Р о й з е н, Техника безопасности и противопожарная техника
в химической промышленности, Госхимиздат, 1951. |
10. Номенклатура промышленных противогазов и респираторов Госхпм-
издат, 1951. И
Геннадий Федорович Большаков, Евгений Ильич Гулин,
Николай Николаевич Торичнев
ФИЗИКО-ХИМИЧЕСКИЕ ОСНОВЫ
ПРИМЕНЕНИЯ МОТОРНЫХ, РЕАКТИВНЫХ
II РАКЕТНЫХ ТОПЛИВ
с. 272. . УДК 662.75
Издательство «Химия», Ленинградское отделение
Невский пр., 28
Редактор издательства В. Д. Пиастра
Технический редактор 5. Е. Маркова
Корректор В. Б. Генгут
Обложка художника А. П. Рыбакова
___________ _______________________-_
Сдано в набор 5/VI 1965 г. Подписано к печати 1/XII 1965 г.
Бумага 60 х ЭО1/^. Уч.-изд. л. 17,32. Печ. л. 17,0. М-27172
Тираж 35 00 экз. Цена 87 коп. Заказ 792
Ленинградская типография №14 «Красный Печатник»
Главполиграфпрома Государственного комитета
Совета Министров СССР по печати. Московский пр., 91
ИЗДАТЕЛЬСТВО „ХИМИЯ"
ГОТОВЯТСЯ К ПЕЧАТИ
Лосиков Б. В., Виппер А. Б., Виленкин А. В. Зарубеж-
ные методы испытания моторных масел па двигателях.
20 л., 4000 экз., цена 1 р. 30 к. в пер. (IV кв.).
В книге систематизированы данные о зарубежных методах
испытания моторных и трансмиссионных масел. Описаны
методы испытания масел на модельных установках, на одно-
цилиндровых и полноразмерных двигателях, на специальных
стендах п в эксплуатационных условиях и применяемая
для этого аппаратура. Сопоставлены й проанализированы
результаты испытания масел различными методами. Рас-
смотрены тенденции зарубежной техники в области определе-
ния эксплуатационных свойств моторных и трансмиссионных
масел.
Книга предназначена для инженерно-технических и науч-
ных работников, связанных с применением и испытанием
моторных и трансмиссионных масел, и для инженеров нефте-
перерабатывающей промышленности.
Новейшие достижения нефтехимии и нефтеперера-
ботки, т. 7—8. Под ред. Дж. Мак-Кета (Нью-Йорк, 1963,
1964). Перевод с английского под ред. Абрамсона И. И.
45 л., 4000 экз., цена 3 р. 48 к. в пер. (IV кв.).
В книге освещены вопросы экономики и перспективы раз-
вития нефтехимической и нефтеперерабатывающей промыш-
ленности, рассмотрены новые процессы, аппараты и оборудо-
вание нефтеперерабатывающих и нефтехимических заводов,
использование достижений науки и промышленности.
В данной книге уделено большое внимание присадкам
к смазочным маслам, применению пенного и эмульсионного
фракционирования, гидрированию сырья для каталитического
крекинга, использованию ионизирующего излучения в нефте-
химических производствах, влиянию радиоактивных излу-
чений на смазочные материалы, высокотемпературным тех-
нологическим процессам и другим актуальным вопросам.
Книга предназначена для широкого круга инженеров
предприятий нефтеперерабатывающей, нефтехимической и
химической промышленности, работников научно-исследо-
вательских и проектных учреждений, занимающихся разра-
боткой и внедрением новой техники.