/













































Теги: химия химическая промышленность химические реакции
Год: 1963




Текст
ГОСУДАРСТВЕННЫЙ КОМИТЕТ ХИМИЧЕСКОЙ И НЕФТЯНОЙ
ПРОМЫШЛЕННОСТИ ПРИ ГОСПЛАНЕ СССР
МЕТОДЫ ПОЛУЧЕНИЯ
ХИМИЧЕСКИХ РЕАКТИВОВ
И ПРЕПАРАТОВ
Выпуск 7
ВСЕСОЮЗНЫЙ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ ВЕЩЕСТВ
МОСКВА- 1963
СОДЕРЖАНИЕ
8-Гидразинохинолин и его солянокислая соль. И. А. Красавин,
Б. В. Парусников, В. М. Дзиомко............................ 5
8-Ацетоксихинальдин. В. М. Дзиомко, И. А. Красавин, К). П. Радин 8
4,4'-Тетраметилднаминотиобензофенон. Е. П. Тупикова, В. Н. Воро-
нина ..................................................... Ю
2,2-Бис-(4'-оксифенил)-бутан. И. М. Билик, Л. М. Серебряный,
Р. Л. Глобус, В. Г. Брудзь................................ 12
3 3-Бис-(4‘-оксифенил)-пентан. И. М. Билик, А. Л-1. Серебряный,
Р. Л. Глобус, В. Г. Брудзь ............................... 14
5,5- Бис-(4‘-оксифенил)-ионан. И. М. Билик, А. Л!. Серебряный,
Р. Л. Глобус, В. Г. Брудзь ........... ...................... 15
Л’-Салицилоилфенилгидроксиламии. Р. П, Ластовский, В. Я. Тем-
кина, Г. Ф. Ярошенко .................... 17
Диоксиэтиламиноуксусная кислота. Р. П. Ластовский, В. Я. Тем-
кина, И. П. Фадеева ..................... 19
Триоксифлуороиы. В. А. Назаренко, Н. В. Лебедева, Л-1. Б. Шустова,
Е. А. Бирюк .............................................. 21
Сульфонафтолазорезорцин. С. Я. Винковецкая, Н. С. Полуэктов 25
Особенности работы с производными ряда пиридина. Ю. И. Чумаков 27
2-Метилпиридин. Ю. И. Чумаков ................. 30
2-Этилпиридин. Ю. И. Чумаков ................. 33
2-трет. Бутилпиридин. Ю. И. Чумаков, 3. М. Корсакова . ... . 35
Л
- и 3-(3‘-Пентеиил)-пиридины. 10. И. Чумаков, В. М. Ледовских 38
Смесь изомерных фенилпиридинов. Ю. И. Чумаков, Э. В. Лугов-
ской......................................................... 41
2- и 4-(2‘-Фенилэтил)-пиридины. Ю. И. Чумаков. Ю.П. Шаповало-
ва, В. М. Ледовских ..................... 44
2-, 3- и 4-(3‘-Фенилпропил)пиридины. Ю.П. Чумаков, В. М. Ледов-
ских .................................................... 46
Изохинолин. Ю. И. Чумаков, 3. П. Васильева .......... 49
1,3-Ди-(2-пиридил)-пропан. Ю. И. Чумаков, В. М. Ледовских,
Р. Е. Лохов, В. А. Ралко................................. 56
.V-Окиси алкилпиридинов. К). И. Чумаков ............ 58
“-Ацетоксиалкилпиридины. Ю. И. Чумаков, 3. Е. Столяров,
Ю. П. Шаповалова ............'......... 61
2-Оксиметилпиридины. Ю.И. Чумаков, З.Е. Столяров.......... 65
Диацетоксиметилпиридины. Ю. И. Чумаков, 3. Е. Столяров ... 69
2-Пиридинальдегид. Ю. И. Чумаков, 3. Е. Столяров.......... 72
Пиридинкарбоновые кислоты. Ю. И. Чумаков .......... 74
Никотиновая кислота. Ю. И. Чумаков, Л. А. Русакова, А. И. Меч-
ников, Р. И. Вирник.................................. 79
Изоникотиновая кислота. Ю. И. Чумаков, Е. Г. Чвырева, П. А. Ган-
грский ............................................... 82
3-Пиридинсульфокислота. Ю. И. Чумаков............. . . . 86
л-Метилстирбл. Э. Б. Грекова .................. 88
2-Йодфлуорен. Э. Б. Грекова ........... ................ 90
2-Фенил-5-фениламино-1, 3, 4-оксадиазол. А. П. Греков..... 92
Алфавитный перечень соединений, описанных в настоящем
выпуске............................................... 94
8 ГИДРАЗИНОХИНОЛИН И ЕГО солянокислая соль
И. А. КРАСАВИН, Б. В. ПАРУСНИКОВ, В. М. ДЗИОМКО
//\/\
i N I N
NH NH
I I
NHa NHa-2HCl-H2O
C9H9N3 M. в. 159,19 C9H13C13N3O M. b. 250,14
Впервые 8-гидразинохинолин был получен [1] диазотиро-
ванием 8-аминохинолина в солянокислой среде и восстанов-
лением диазосоединения с помощью хлористого олова. После
осаждения соединений олова сероводородом и упаривания
раствора образовались кристаллы дигидрохлорида
C9H9N3-2HCI. Свободное основание было выделено из этой
соли действием ацетата натрия при смешивании их концент-
рированных водных растворов. Выход веществ в этой [1] и
последующих [2, 3, 4] работах не указан и, по-видимому, мал.
Предметом недавно выданного патента [5] явился способ,
по которому восстанавливают (цинковой пылью) не само ди-
азосоединение, а продукт взаимодействия его с диметилфос-
фитом. Выход 8-гидразинохинолина (в виде солянокислой
соли) составляет 35%. Методика этого синтеза трудоемка,
так как требует выполнения большого числа последователь-
ных операций.
Перечисленные методы довольно неудобны, но главным их
недостатком является низкий выход 8-гидразинохинолина, что
особенно существенно из-за относительно малой доступности
исходного вещества — 8-аминохинолина.
Нами был разработан в 1959—1960 гг. способ получения
8-гидразинохиполина с хорошим выходом из вполне доступ-
ного 8-оксихинолина, состоящий в нагревании последнего с
5
избытком гидразин-гидрата в течение 40—50 часов. Реакция
проводится при температуре кипения гидразин-гидрата
(118°) в приборе, воздух из которого' вытеснен пропусканием
слабого тока азота. Одновременно была разработана простая
и удобная методика выделения продукта, который может быть
получен, по желанию, или в виде основания (выход 64%) или
в виде солянокислой соли (выход 69%). Ярко-желтая соляно-
кислая соль (гидрат дигидрохлорида 8-гидразинохинолиния
состава СдНдИз^НСЬНгО) легко очищается посредством кри-
сталлизации и, в отличие от основания, вполне устойчива при
хранении. Из нее действием раствора щелочи может быть по-
лучено, по мере надобности, основание 8-гидразинохинолина.
Метод применим и для получения других гидразинопро-
изводных. Например, из 8-оксихинальдина нами был синте-
зирован не описанный в литературе 8-гидразинохинальдин в
виде игл с т. пл. 67,8—68,3° и его солянокислая соль состава
CioHhN3-2HC1-H20.
Позднее нам стала известна работа [6], в которой описано
получение 8-гидразинохинолина с выходом 67 % (считая на
неочищенный дигидрохлорид) посредством нагревания 8-ок-
сихинолина в течение 120 часов с гидразин-гидратом, в кото-
ром был растворен сернистый ангидрид.
Следует отметить, что мы вначале также применяли до-
бавление солей сернистой кислоты (пиросульфита гидрази-
ния). Однако вскоре было обнаружено, что и в отсутствие
этого катализатора реакция проходит почти без снижения
выхода (75 и 69% соответственно). Это позволяет утверж-
дать, что она протекает в основном не по механизму реакции
Бухерера. Наконец, в наших опытах было установлено, что
увеличение длительности нагревания свыше 40—45 часов не
приводит к возрастанию выхода продукта.
Получение 8-гидразинохинолина основания.
//\/\ ^\/Ч
| || | + NHjNHj-HsO —► | || | + 2НаО
V/W 4/W
|N IN
ОН NH
NH2
В четырехгорлую колбу емкостью 0,75—1 л со шлифами
(см. примечание 1), снабженную термометром, короткой труб-
кой для введения азота, обратным холодильником (см. при-
мечание 2) и мешалкой (см. примечание 3), помещают 72,6 г
(0,5 А1) 8-оксихинолина и 250 мл (5,15 М) 100%-ного гидра-
зин-гидрата (см. примечание 4). Воздух из прибора вытесня-
ют азотом, подводимым из баллона через склянку Тищенко с
серной кислотой. Включив мешалку и продолжая пропускать
G
над поверхностью реакционной массы медленный ток азота,
нагревают колбу на масляной бане (около 135° в бане) при
слабом кипении гидразин-гидрата (118°) в течение 45 часов
(см. примечание 5). По истечении указанного срока реакци-
онную массу охлаждают до 50—60°, прибавляют _250_уил бен-
зола и кипятят при размешивании в течение 15—20 минут.
Затем еще теплую жидкость переливают в делительную во-
ронку емкостью 700—1000 мл, отделяют нижний слой, а верх-
ний слой промывают подогретым до 40—50° 5%-ным раство-
ром едкого натра (2 раза по 250 мл), потом — теплой водой
(2 раза по 200 мл). Кипятят экстракт с 2 г активированного
угля, фильтруют/й бензол отгоняют. Остаток перегоняют в
вакууме изгспециальной колбы Клайзена с длинным широким
отводом, который служит холодильником. Собирают вещество,
кипящее при 142—146°/2 мм или при 187—190°/13 мм рт. ст.
Дистиллат застывает в холодильнике и приемнике в светло-
желтую массу. Выход 47,8—51 г (60—64%), т. пл. 63,5—65°
(см. примечание 6).
Получение моногидрата дигидрохлорида 8-гидразинохино-
линия.
Х\/Х //^/Х
I I’ I + 2НС1 + Н2О - | || |
X/W WW
IN |N
NH NH
NH2 NH3-2HC1-H2O
К полученному, как описано выше, бензольному раствору
(после промывки щелочью и водой) прибавляют при 40—50°
100 мл концентрированной соляной кислоты (уд. веса 1,19).
Охлаждают при размешивании до 5—8°, через 30—40 минут
желтый осадок отфильтровывают, промывают на фильтре
концентрированной соляной кислотой (50 мл), отжимают и
высушивают в вакуум-эксикаторе над твердым едким натром.
Выход 83—83,5 г (66,4—66,8%).
Из солянокислого слоя фильтрата посредством нейтрали-
зации, экстракции бензолом и осаждения небольшим количе-
ством соляной кислоты можно выделить еще 3—3,5 г более
загрязненного вещества. Общий выход 86—86,5 г (68,8- -
69,2%).
Для очистки соль растворяют при нагревании в 100 мл
воды, обрабатывают активированным углем, фильтруют в го-
рячем виде и к еще теплому фильтрату добавляют 100 мл со-
ляной кислоты (уд. веса 1,19). После охлаждения и стояния
ярко-желтый осадок отфильтровывают, промывают соляной
кислотой (40 мл) и высушивают в вакууме над едким натром.
7
Выход очищенного вещества 74,4—75,7 г, что составляет
59,5—60,5% от теоретического (ом. примечание 7).
Примечания.
1. Нельзя применять резиновые или корковые пробки, так как
пары гидразин-гидрата сильно разъедают их. Для укрепления тер-
мометра, а также в качестве затвора для мешалки можно исполь-
зовать отрезки резиновых трубок.
2. В процессе реакции происходит возгонка вещества, которое
забивает обычный обратный холодильник. Чтобы избежать этого,
целесообразно применить специальный холодильник, изготовленный
из прямого форштосса, верхняя часть которого снабжена водяной
рубашкой, а нижняя (длиной 20—25 см) служит воздушным холо-
дильником.
3. Применение мешалки ие является необходимым. Можно ис-
пользовать прибор, в котором перемешивание реакционной массы
происходит за счет пробулькивапия азота через барботер, погружен-
ный до дна колбы.
4. Применялся 8-окснхинолин «чистый» и продажный 100%-ный
гидразин-гидрат. Прн увеличении количества гидразин-гидрата до
500 мл П0,3 А4) выход возрастает на 5—6%.
5. Реакцию можно вести с перерывами. Если сократить дли-
тельность нагревания до 30 часов, выход снизится на 5—6%.
6. Продукт неустойчив при хранении и начинает разлагаться
уже через несколько дней. По литературным данным, 8-гидразнно-
хинолин имеет т. пл. 64° [1,5], 05° [3], т. кип. 188—189713 мм [3].
7. Эта соль вполне устойчива и не изменилась при хранении в
закрытой банке в течение 4 лет.
ЛИТЕРАТУРА
1. S. F. D ufton, J. Chem. Soc., 59,756 (1891).
2. M. J. S. Dewar, J. Chem. Soc.. 1944, 615.
3. Ng. Ph. Bu u-H oi, F. Perin, P. Jacquignon, J. Chem. Soc.,
1960, 4500; 1962, 146.
4. H. Wieland, L. Horner, Liebigs Ann. Chem.,536, 89 (1938).
5. F. Suckfull, H. Haubrich, пат. ФРГ 1015 442 (1958); Chem.
Abstrs, 54, 413c (1960); РЖхим., 1959, 24392 П.
6. S. H u n i g, H. Werner, Liebigs Ann. Chem., 628, 46 (1959).
Поступила в марте 1963 г. ИРЕА
8-АЦЕТОКСИХИ Н АЛ ЬД И Н
В. М. ДЗИОМКО, И. А. КРАСАВИН, Ю. П. РАДИН
| N
CHSCOO
Cl2HnNO2 М. в. 201,23
8
Синтез 8-ацетоксихинальдина описан только в одной ра-
боте [1]. При кипячении 8-оксихинальдина в уксусном ангид-
риде в присутствии пиридина О-ацетильное производное было
получено с 77%-ным выходом. Нами была произведена про-
верка этой методики в увеличенном (в 10—25 раз) масштабе.
Получение 8-ацетоксихинальдина.
/Х/Ч /\/Ч
I || | + (СН3СО)2О - | || I
। N ^Пз I N ^Пз
ОН СН3СОО
+ СНдСООН
В круглодонную колбу емкостью 1 л, снабженную обрат-
ным холодильником на шлифе, помещают 100 г (0,628 М)
8-оксихинальдина (см. примечание 1), 500 мл (5,3 М) свеже-
перегнанного уксусного ангидрида, 80 мл сухого, перегнан-
ного пиридина и нагревают при кипении в течение 7—8 часов.
Затем отгоняют уксусную кислоту, пиридин и избыток ангид-
рида на кипящей водяной бане в вакууме водоструйного на-
соса. Остаток перегоняют из колбы Клайзепа в вакууме, со-
бирая вещество, кипящее при 115—132°/2 мм или 155—
173°/13 мм рт. ст. Желтый густой дистиллат закристаллизо-
вывается в приемнике. Выход сырого продукта 114—116 г
(90,2—92,5%). Его перекристаллизовывают из 1200—1400 мл
петролейного эфира (т. кип. 80—100°) с применением активи-
рованного угля. Выпавшие при стоянии кристаллы отфильтро-
вывают и промывают на фильтре небольшим количеством
петролейного эфира. Выход чистого продукта 88,5—94,8 г
(70—75%), т. пл. 64,5—65,5° (см. примечание 2). После кон-
центрирования маточника получают еще 9,5—12,5 г вещества.
Общий выход 8-ацетоксихинальдина 101—105 г, что состав-
ляет 80—83% от теоретического (см. примечания 3 и 4).
Примечания.
1. Применялся 8-оксихинальдин с т. пл. 71,0—71,5°, полученный
по методике [2]. Если при этом использовать ректифицированный
(на лабораторной колонке высотой 400 .ч.м) кротоновый альдегид с
т. кип. 101—102° (не испр.), то получается более чистый продукт с
выходом (в расчете на о-амииофенол) 84—88,5% вместо 62,5—65%
[2].
2. По литературным данным [1], 8-ацетоксихииальднн имеет
т. пл. 63—64°, т. кип. 169—171713 мм рт. ст.
3. Вещество неустойчиво к действию влаги п омыляется уже
при кипячении в течение нескольких минут с разбавленным спиртом
[1]. Его следует хранить в плотно закрывающихся банках.
4. При проведении синтеза с большими в 2,5 раза количествами
реагентов выход продукта (в %) остается примерно таким же.
9
ЛИТЕРАТУРА
1. J. В u с h 1, А, А е b i, A. D е f 1 о г i п, Н Н u г n 1, Helv. cliini. acta,
39, 1676 (1956).
2. Г. И. Михайлов, Труды ИРЕА, вып. 20, Госхимиздат, 1951,
стр. 266. '
Поступила в марте 1963 г. ИРЕА
4,4-ТЕТРАМЕТИЛДИАМИНОТИОБЕНЗОФЕНОН
(Тиокетон Михлера)
Е. П. ТУПИКОВА, В. Н. ВОРОНИНА
(CH3)2N-/ ’\-N(CHa)a
S
CijHaoNaS М. в. 284,42
В литературе имеются сведения о получении 4,4'-тетраме-
тилдиаминотиобеизофенона путем взаимодействия тиофосге-
на с диметиланилином и сероуглеродом [И. аурамина основа-
ния с сероуглеродом [2, 3], 4,4'-тетраметилдиаминодифенилме-
тана с серой [4], аурамина основания с сероводородом [5].
Авторы дают различную температуру плавления тиокето-
на Михлера: 164—166° [2], 196° [3], 194—196° (4], 202° [6] и не
всегда указывают количественный выход продукта.
Нами для получения тиокетона Михлера был проверен ме-
тод взаимодействия аурамина основания с сероводородом как
наиболее простой по технологическому процессу и дающий
хороший выход [5].
СИНТЕЗ ТИОКЕТОНА МИХЛЕРА
NH
(CH3)2N-/==^-C-/“^-N(CH3)s + NH3.
s
10
Характеристика основного сырья
Аурамин солянокислый, ч., ВТУ МХП 3327—52.
Натр едкий, х. ч„ ГОСТ 4328—48.
Железо сернистое, ТУ МХП 87—52.
Кислота соляная, х. ч„ ГОСТ 3118—46.
Условия получения
В трехгорлую колбу емкостью ~ 300 лгл, снабженную ме-
шалкой, термометром и газоподводящей трубкой, загружают
26,7 г (0,1 41) аурамина основания (см. примечание) и 75 мл
этилового спирта.
Смесь перемешивают, нагревают до 60° и по достижении
полного растворения аурамина через раствор начинают по-
степенно пропускать сероводород, при этом желтый цвет аур-
амина изменяется па пурпурно-красный и начинают выделять-
ся кристаллы тиокетона Михлера. Подача сероводорода про-
должается около 2 часов.
Затем реакционную массу перемешивают в течение 15—
30 минут, постепенно охлаждают до комнатной температуры
и фильтруют.
Осадок, представляющий собой кристаллы тиокетона Мих-
лера, промывают 25 мл спирта и сушат при температуре 60°.
Выход продукта около 24—25 г, что составляет 80—85% от
теоретического, т. пл. 181 —183°.
Очистка тиокетона производится путем перекристаллиза-
ции его из смеси бензола со спиртом. Для этого в колбу с об-
ратным холодильником помещают 25 г тиокетона и 300 мл
бензола, нагревают на водяной бане до полного растворения
продукта, быстро фильтруют, охлаждают фильтрат до комнат-
ной температуры и приливают к нему 50 мл этилового спирта.
Выпавшие кристаллы тиокетона отфильтровывают и су-
шат при 60°.
Температура плавления полученного тиокетона Михлера
192—193°.
Найдено, %: S—10,74; N —10.27.
Ci,H20N2S. Вычислено, %: S —11,26; N—9,84.
Примечание.
Аурамин основания получают взаимодействием солянокислого
аурамина с едким натром, т. пл. 136°.
ЛИТЕРАТУРА
1. A. Kern, DRP, 37730, 1887.
2. W. Fehrmann, Вег., 20, 2857 (1887).
3. С. О г а е b а, Вег., 20, 3266 (1887).
4. О. Wallach, Liebigs Ann. Chem., 259, 303 (1890).
5. W. F e h r m a n n, Ber, 20, 2859 (1887).
6. O. Baihter, Ber., 20, 3290 (1887).
Поступила в марте 1963 г. ИРЕА
11
2,2 БИС (4' ОКСИФЕНИЛ)-БУТАН
(4,4Г- диокси дифенилметилэтилметан)
И. М. БИЛИК, А. м. серебряный, р. л. глобус, в. г. брудзь
СНз
с2нб
С1вН18О2 М. в. 242,32
2,2-Бис-(4/-оксифенил)-бутан (ДФБ) получен впервые
конденсацией в течение одной недели метилэтилкетона с фе-
нолом в присутствии сухого хлористого водорода [1]. В даль-
нейшем этот метод был усовершенствован и выход продукта
доведен до 83% при продолжительности конденсации 50 часов
[2]. Более высокий выход ДФБ, равный 90%, получен в
условиях, когда длительность конденсации составила 20 не-
дель [3]. Другие авторы получали ДФБ с низкими выходами
[4, 5, 6]. В некоторых работах данные о выходах вообще от-
сутствуют [7, 8].
Нами показано, что применение в качестве конденсирую-
щего средства фтористого бора позволяет существенно сокра-
тить продолжительность конденсации фенола с метилэтил-
кетоном. В результате реакции получается ДФБ с количест-
венным выходом. Уже после однократной перекристаллизации
продукт имеет температуру плавления, указанную в литера-
туре.
СИНТЕЗ 2,2-БИС-(4'-ОКСИФЕНИЛ)-БУТАНА
ОН
A PF
2 || | % СН3СОС2Н5 —
сн3
- но-С>%--(7>-он+н,о
С,Н5
Характеристика основного сырья
Метилэтилкетон, ч„ ВТУ МХП 3024—55.
Фенол, ч, ГОСТ 6417—52.
Бор фтористый, технический.
12
Условия получения
К раствору 36 г фтористого бора в 288 г фенола при тем-
пературе не выше 25° быстро добавляют при перемешивании
28,1 а метилэтилкетона. Реакционную массу выдерживают
24 часа при той же температуре, после чего к пей добавляют
300 мл уксусной кислоты и перемешивают до полного раство-
рения осадка. Полученную смесь выливают в колбу, содер-
жащую 3 л воды. Образовавшуюся эмульсию перемешивают
до начала кристаллизации. Через 2 часа осадок отсасывают
на воронке Бюхнера и промывают водой до нейтральной ре-
акции и отрицательной пробы промывных вод на фенол (см.
примечание). Затем продукт сушат в вакуум-эксикаторе над
хлористым кальцием до постоянного веса. Выход ДФБ ко-
личественный. После перекристаллизации из шестикратного
количества хлорбензола выход ДФБ составляет около 60%,
т. пл. 125—125,5°. По литературным данным, т. пл. продукта
124 125° [6], 125° [5, 8].
2,2- Бис-(^-оксифенил)-бутан представляет интерес как
препарат для проведения исследовательских работ.
Примечание.
Промывные воды иа феиол проверяют следующим образом.
К 20 мл промывных вод добавляют 1,5 мл 0,1 н. раствора гипохло-
рита натрия и 1 мл 25%-иого раствора аммиака. При кипячении этой
смеси ие должно в отсутствие фенола возникать зеленое окрашива-
ние.
ЛИТЕРАТУРА
1. Т. Н. Z i п с k е, J. G old е man, Liebigs Ann. Chem., 362, 205
(1908).
2. J. V. В r a u n, Liebigs Ann. Chem., 472, 71 (1921).
3. E. E. Reid, E. Wilson, J. Amer. Chem. Soc., 66, 967 (1944).
4. A. Muller, Chem. Ztg., 45, 632 (1921).
5. M. E. Me G r e a 1, V. N ie d er 1, J. B. N ied er 1, J. Amer. Chem.
Soc., 61, 345 (1939).
6. A. P. T. E a s s о n, J. H ar r I s о n, B. A. Me Swiney, F. L. Ру-
та n, Quart. J. Pharmac. Pharmacol., 7, 509 (1934); цитировано no Zbl.,
1935, I, 3133.
7. Герм. пат. 494778; Zbl., 1930, II, 1291.
8. F. Leibnitz, K. Nauman, Chem. Tcchn., 3, 5 (1951).
Поступила в январе 1963 г. ИРЕА
3,3-БИС-(4'-ОКСИФЕНИЛ )-ПЕНТАН
(4,4'-дноксиднфенилдиэтилметан)
И. М. БИЛИК, А. м. серебряный, р. л. глобус, в. г. брудзь
C2HS
С2Н5
СПН2()О2 М. в. 256, 35
3,3-Бис- (4'-окснфенил)-пентан (ДФП) получается при
конденсации диэтилкетона с фенолом в присутствии соляной
кислоты [1, 2, 3, 4]. Нами было показано [5], что применение
серной кислоты вместо соляной улучшает выход целевого про-
дукта. В дальнейшей работе мы установили, что использова-
ние в качестве конденсирующего средства фтористого бора
повышает выход ДФП до 86% и улучшает его- качество.
СИНТЕЗ 3,3-БИС-(4'-ОКСИФЕНИЛ)-ПЕНТАНА
ОН
I
гП + С’Н>0
\ / СгН5
СгН6
- но~о Н~>“он+н,°
С,нв
Характеристика основного сырья
Диэтилкетон, я.
Фенол, ч., ГОСТ 6417—52.
Бор фтористый, технический.
Условия получении
К раствору 4 г фтористого бора в 32 г фенола, при тем-
пературе не выше 25°, быстро добавляют при перемешивании
3,7 г диэтилкетона. Реакционную массу выдерживают 24 ча-
са при той же температуре, после чего ее выливают в стакан
с 500 мл воды. Образовавшуюся суспензию подщелачивают
раствором едкого натра до pH 8, осадок отфильтровывают,
промывают водой до нейтральной реакции и отрицательной
14
пробы промывных вод на фенол (см. прим, к статье «2,2-Бис-
(Г-оксифенил)-бутан»), после чего высушивают при 80° до
постоянного веса.
Выход ДФП 9,4 г. что составляет 86%, считая иа диэтил-
кетон, т. пл. 192—196°, После перекристаллизации из водного
спирта (1:1) с добавкой бисульфита натрия продукт имеет
т. пл. 200,5—201,5°. По литературным данным, т; пл- препара-
та 198—200° [1}, 200° [2], 204“ [3].
3,3- Бис- (4/-окснфенил)-пентан представляет интерес как
препарат для проведения исследовательских работ.
ЛИТЕРАТУРА
1. А. П. Дианин, ЖРФХО, 23, 499 (1891).
2. А. Р. Т. Е a s s о n, J. Н а г г 1 s о п, В. А. М с S w i п е у, Р. L. Р у-
m a n, Quart. J. Pharmac, Pharmacol., 7, 509 (1934); Zhl., 1935. 1, 3133.
3. E. E. Reid, E. Wilson, J. Amer. Chem. Soc., 66, 967 (1944).
4. N. R. C a m p be 11, Proc. Roy. Soc., Lend., В 129. 528 (1940); но
С. A. 35, 43678 (1941).
5. И. M. Билик, А. М. Серебряный, Н. М. Бон дарец,
Р. Л. Г л о б у с, В. Г. Б р у д з ь, «Методы получения химических реакти-
вов и препаратов», вып. 2, стр. 113, ИРЕА, 1961.
Поступила в январе 1963 г.
ИРЕА
5,5 БИС-(4' ОКСИФЕНИЛ)-НОНАН
(4,4'-дноксидифеннлднбутилметан)
И. М. БИЛИК, А. М. СЕРЕБРЯНЫЙ, Р. Л. ГЛОБУС, В. Г. БРУДЗЬ
С4Н9
Н°-С)-ГО-ОН
С4Н9
CgjHggC^ М. в. 312, 46
В литературе имеются лишь два сообщения о синтезе
5,5-бис-(4'-оксифенил)-нонана (ДФН). В одном из них при-
веден синтез ДФН путем конденсации дибутилкетона с фено-
лом в присутствии хлористого водорода. Выход продукта 8%
[1]. В другом сообщении ДФН получался также конденсацией
дибутилкетона с фенолом, но в течение 20 недель и с выходом
90% [2].
15
Нами получен ДФН с выходом около 35% при конденса-
ции в течение 24 часов дибутилкетона с фенолам в присутст-
вии фтористого бора. Метод не требует большой затраты тру-
да и времени и может быть использован для синтеза ДФН.
СИНТЕЗ 5,5-БИС-(4'-ОКСИФЕНИЛ)-НОНАНА
ОН
_ С4Н9
но-0-с-0он+Н!°
С4Н9
Характеристика основного сырья
Дибутилкетон, ч.
Фенол, ч., ГОСТ 6417—52.
Бор фтористый, технический.
Условия получения
К раствору 8 г фтористого бора в 64 г фенола при темпе-
ратуре не выше 25° быстро добавляют при перемешивании
12 г дибутилкетона. Реакционную массу выдерживают 24 ча-
са при той же температуре и выливают в стакан с 1 л воды.
При этом выделяется масло, в котором вскоре появляются
кристаллы. Массу подщелачивают раствором едкого натра до
pH 8, кристаллы отфильтровывают, тщательно отжимают от
масла и промывают сначала петролейным эфиром, затем во-
дой до нейтральной реакции и отрицательной пробы промыв-
ных вод на фенол (см. примечание к статье «2,2-Бис- (^-окси-
фенил) -бутан»). Продукт сушат до постоянного веса в ва-
куум-эксикаторе над хлористым кальцием. После перекри-
сталлизации из водного спирта выход ДФН составляет 35%,
считая на дибутилкетон, т. пл. 167,5—168°. По литературным
данным, т. пл. препарата 165° [1], 170,5° [2].
5,5-бис-(4'-оксифенил)-нонан представляет интерес как
препарат для проведения исследовательских работ.
ЛИТЕРАТУРА
1. N. R. Campbell, Proc. Roy. Soc., bond., В 129, 528 (1940); no
C. A., 35, 4367s (1941).
2. E. E. Reid, E. Wilson, J. Amer. Chem. Soc., 66, 967 (1944).
Поступила в январе 1963 г. ИРЕА
16
А-САЛИЦИЛОИЛ ФЕНИЛ ГИДРОКСИЛАМИН
Р. П. ЛАСТОВСКИИ, В. я. ТЕМКИНА, Г. Ф. ЯРОШЕНКО
Ч 's у_>
оно он
C19HnO3N М.в. 229,23
Салицилоилфснилгидроксиламин является не описанным в
литературе оксипроизводным известного аналитического реа-
гента бензоилфенилгидроксиламина (БФГА).
Нами предложен метод получения салицилоилфенилгид-
роксиламина путем взаимодействия фенилгидроксиламина с
салицилоилхлоридом.
По литературным данным, исходный салицилоилхлорид
получают взаимодействием тионилхлорида с салициловой кис-
лотой [1] или салицилатом натрия [2]. Реакцию рекомендуется
проводить в присутствии третичных аминов в качестве ката-
лизатора [3, 4].
Проверка описанного метода получения салицилоилхло-
рида взаимодействием салициловой кислоты или салицилата
натрия с тионилхлоридом [1, 2] не дала положительных ре-
зультатов. Проведение же этой реакции в присутствии диэта-
ноламина [4] позволило получить салицилоилхлорид, однако
наилучшис результаты были достигнуты при использовании в
качестве катализатора диметилформамида.
СИНТЕЗ САЛИЦИЛОИЛФЕНИЛГИДРОКСИЛАМИНА
^- NO,4-2Zn-4-3H2O ^-NHOH + 2Zn(OH)2
С_^“СО()Н -ф SOC12 ^COCl -ф SO2 4- НС!
И 1
он он
—nhOh+^ ^COClv, —N—СО—^+НС1
~ ~ он Г
он он он
+ ОС0С1
он он он
2 Зак. 578
17
- \\„//“N-CO-VJ + HCl
I OH
j OH
c°-O
(побочный продукт)
0*-со-0+NHi0H
OH (in
* 4—^~N~CO—+H2O
ONH4 QJ_|
C_)-^“co~\Z)+HCI
onh4
- C>N-co-C)+NH<cl
0H OH
Характеристика основного сырья
Аммиак водный, ч. д. а., ГОСТ 3760—47.
Нитробензол, ч., ГОСТ 5846—51.
Салициловая кислота, ч., ГОСТ 5844—51.
Тионил хлористый, ВТУ МХП 3591—52.
Цинковая пыль, 85%-ная.
, Условия получения
Получение салицилоилхлорида. В круглодопную колбу,
снабженную мешалкой, обратным холодильником, капельной
воронкой и термометром, загружают 84 г (0,61 М) салициловой
кислоты и 3 мл диметилформамида. К смеси при размешива-
нии прибавляют в течение 15 минут 50 мл (0,69 А4) хлористого
тиснила. Реакционную массу нагревают до 50°, размешивают
при этой температуре 40 минут, после чего обратный холо-
дильник заменяют нисходящим и отгоняют избыток хлористо-
го тионил а. ;
Выход продукта 78,9 г, что составляет 83% от теоретиче-
ского, т. пл. 14,5—15,5°. По литературным данным, т. пл. са-
лицилоилхлорида 14—17° [3], 14,5° [4], 19—19,5° [7].
18
Получение салицилоилфенилгидроксиламина. В кругло-
донную колбу, снабженную механической мешалкой, обрат-
ным холодильником и термометром, загружают 13 г (0,12 М)
свежеприготовленного фенилгидроксиламина [5,6], 200 мл
сухого серного эфира и 4 г пиридина. К реакционной смеси
при размешивании прибавляют 15,7 г (0,1 Л1) салицилоилхло-
рида в течение 10—15 минут. После добавления всего коли-
чества салицилоилхлорида реакционную массу выдерживают
при размешивании в течение 1,5 часа, после чего отделяют на
делительной воронке солянокислый пиридин. Эфирный слой
обрабатывают 10 %-ной соляной кислотой для удаления из-
бытка пиридина, несколько раз промывают дистиллирован-
ной водой до отсутствия кислой реакции в промывных водах,
переносят в круглодонную колбу с нисходящим холодильни-
ком и отгоняют эфир. Полученную в результате реакции смесь
моносалицилоил- и дисалицилоилфенилгидроксиламипа раз-
деляют обработкой 100 мл водного раствора аммиака, в ко-
тором растворяется только моносалицилоилфенилгидроксил-
амин. Массу отфильтровывают и подкисляют при охлаждении
10%-ным раствором соляной кислоты до значения pH 2.
Полученные белые кристаллы салицилоилфенилгидроксил-
амина отфильтровывают и промывают холодной дистилли-
рованной водой до отсутствия кислой реакции в промывных
водах по универсальной индикаторной бумаге.
Выход полученного салицилоилфенилгидроксиламина 2 г.
Найдено, %: N -6,12; 6,23.
C1sHiiO8N. Вычислено, %: N—6,11.
ЛИТЕРАТУРА
1. DRP, 284161; Frdl 12, 667.
2. DRP, 262883; Frdl И, 211..
3. Пат. США 289945S; РЖхим, 85795 П (1960).
4. В. М. Дзномко, И. А. М а р к о в и ч, «Химические реактивы и
препараты», № 2, 27 (1961).
5. «Синтезы органических препаратов», сб. I. М., ИЛ, 1949, стр. 432.
6. А. М. Лукин, Е. Я. Яро в е н к о, «Методы получения химических
реактивов и препаратов», № 3, 12 (1961).
7. Словарь органических соединений, III, М., ИЛ, 1949, стр. 613.
Поступила в марте 1963 г. ПРЕЛ
ДИОКСИЭТИЛАМИНОУКСУСНАЯ КИСЛОТА
Р. П. ЛАСТОВСКИИ, В. я. ТЕМЦИНА, И. П. ФАДЕЕВА
CeH13O,N
2*
НО-СН2-СН2х
НО-СН2-СН2/
N-CH2-COOH
М. в.
163,17
19
По литературным данным, диоксиэтиламиноуксусная кис-
лота может быть получена взаимодействием диэтаноламина
с монохлоруксусной кислотой [1], диэтаноламина с формаль-
дегидом и цианистым калием [2], а также окиси этилена с
глицином [3].
Нами проверен первый метод получения диоксиэтилами-
ноуксусной кислоты и уточнены условия выделения препара-
та.
СИНТЕЗ СОЕДИНЕНИЯ
,сн2- СН2ОН
HNf + С1СН2—COOH-RNaOH ->
ЧСН2-СН3ОН
НОСН2-СН2
НОСН2-СН2
/N-СН2СООН + NaCl+HjO.
Характеристика основного сырья
Диэтаноламин, ВТУ РУ 614—52.
Кислота моиохлоруксусная, ч., ГОСТ 5836—51.
Уголь активированный, ГОСТ 8703—58—АР—3.
Спирт метиловый, ГОСТ 6995—54.
Условия получения
В трехгорлую колбу, снабженную обратным холодильни-
ком, термометром и мешалкой, помещают 14,4 г (0,137 /И)
диэтаноламина и добавляют при перемешивании 12,6 г
(0,133 М) монохлоруксусиой кислоты, предварительно нейтра-
лизованной 20%,-ным раствором едкого натра. Реакционную
массу выдерживают при перемешивании и температуре
90—95 в течение 4 часов, при этом pH раствора снижается
от 9 до 5,5—6 по универсальной индикаторной бумаге. После
окончания выдержки к горячему раствору добавляют 2—3 г
активированного угля, перемешивают в течение 15 минут и
затем фильтруют. Фильтрат упаривают до густой кашицеоб-
разной массы и затем прибавляют к ней 20 мл этилового спир-
та. Осадок отфильтровывают и промывают 1,0 мл спирта.
Для очистки продукта от хлористого натрия его дважды
перекристаллизовывают из 80%-иого метилового спирта.
Выход диэтаноламиноуксусной кислоты 7,17 г, что состав-
ляет 32% от теоретического.
Содержание основного вещества в полученном продукте
99% (определено методом неводного титрования).
Найдено, %: N—8,55; 8,45.
C6HlaO4N. Вычислено, %: N—8,59.
ЛИТЕРАТУРА
1. Н. В. Хромов-Борисов, А. Л. Ремизов, Ж. общ. химии, 23
598 (1953).
2. В. Ф Любомудров, Укр. хим ж., II, 119 (19S6).
3. Г. Киприанов, Укр. хим. ж., 2, 236 (1927).
Поступила в марте 1963 г.
ИРЕА
триоксифлуороны
(9-£-2,3,7-триокси-6-флу9роны)
В. А. НАЗАРЕНКО, Н. В. ЛЕБЕДЕВА, М. Б. ШУСТОВА, Е. А. БИРЮК
О О
h°--UUU~oh
С
I
R
Триоксифлуороны с различными алифатическими или аро-
матическими радикалами R применяются как реактивы для
фотометрического определения многовалентных металлов III,
IV, V и VI групп периодической системы элементов [1]: фе-
иилфлуорон (R = CeHs—) Для германия, циркония, олова и
др., л-нитрофенилфлуорон (R = n-O2NCeH4—) для олова,
о-иитрофеиилфлуорон (R=o-O2NC6H4—) для молибдена и
ниобия, л-диметиламииофенилфлуорон или диметилфлуорои
(R = /z- (CH3)2NCeH4—) для тантала, пропилфлуорон
(R=CH3CH2CH2—) для скандия, дисульфофенилфлуорои
(R = o-,/z-(HO3S)2C6H3—) для титана и т. д.
Триоксифлуороны впервые получены из оксигидрохинона и
соответствующего альдегида конденсацией при нагревании в
спиртово-водной среде в присутствии серной кислоты с после-
дующим гидролизом сульфата [2]. Вместо оксигидрохинона
предложено пользоваться более доступным и более устойчи-
вым к окислению триацетилоксигидрохиноном с конденсацией
при нагревании [3, 4] или на холоду [5—9]. Мы уточнили по-
следний вариант и распространили метод иа получение три-
оксифлуоронов из альдегидов, нерастворимых в разбавленном
спирте.
21
20
СИНТЕЗ ТРИОКСИФЛУОРОНОВ
СНзСОО- f \ -ОСОСНз н,5О.,Н,0
СН8СОО-1 ,
но—^\—он
HO-LJ
+ ЗСНзСООН
\ .ОН TJ
2H0JJ +rX0 + h-S0‘-
\х/
Характеристика основного сырья
Триацетилоксигидрохинон (пирогаллол А), ч. д. а., ГОСТ
6408—52. .
Кислота серная, х. ч., ГОСТ 4204—48.
Спирт этиловый, ГОСТ 5962—51.
Альдегид—см. таблицу.
Условия получения
В зависимости от того, растворяется или не растворяется
используемый альдегид в 50%-ном спирте, синтез проводит-
ся в водно-спиртовой или спиртовой среде.
В конической колбе на 500 мл растворяют при нагревании
25 г (0,2 М) триацетилоксигидрохинона в смеси из 150 мл
этилового спирта и 150 мл воды или в 300 мл спирта (см.
табл.) и осторожно добавляют 20 мл концентрированной сер-
ной кислоты. К горячему раствору добавляют 0,2 моля нуж-
ного альдегида (см. примечания 1,2 и таблицу), после охлаж-
22
дения закрывают колбу пробкой и оставляют стоять в темном
месте при температуре 25—30° до выпадения кристаллическо-
го осадка сульфата триоксифлуорония. Выпадение осадка в
зависимости от радикала триоксифлуорона практически за-
канчивается в разные сроки: фепилфлуорон — через 8—9 дней,
салицилфлуорон через 10—12 дней, остальные — до 3—4 не-
дель. Осадок отсасывают на воронке Бюхнера (см. примеча-
ние 3) и промывают на фильтре в несколько приемов 50 мл
смеси из равных объемов воды и спирта или 50 мл спирта,
если синтез проводится в спиртовой среде. Затем осадок пе-
реносят в стакан, наливают 150—200 мл воды и , нагревают
при помешивании до 70—80° в течение часа (ом. примеча-
ние 4). Отсасывают на воронке Бюхнера и промывают не-
сколько раз горячей (80°) водой, после чего сушат на воз-
духе.
Выход, в зависимости от триоксифлуорона, 20—30% от
теоретического. Полученные препараты применяются как ре-
активы без дальнейшей очистки.
Таблица
№ п/п Триоксифлуорон Альдегид Характе- ристика альдегида Среда ДЛЯ синтеза
1 Фенилфлуорои Бензойный ч„ГОСТ 157—51 Водно- спирт.
2 Сзлицил флуорон Салициловый ч.д.а., СТ гохп 27-1865
3 о-Нитрофенилфлуорон о-нитробензой- ный ч. спирт.
4 л-Нитрофенилфлуорон л-нитробензой- ный ч, ТУ МХП 99-51
5 п-Нитрофенилфлуорон п-нитробензойный ч„ ТУ МХП 2969-51 •
6 Динитрофенилфлуорон 2,4-ДИнитробен- зойный ч. »
7 Диметилфлуорон я-диметиламино- бензойный ч„ ТУ МХП 2671- 51 ВОДНО" спирт.
8 9 Дисульфофенилфлуорон Пропилфлуорон 2,4-дисульфо- бензойный н-масляный ч. ч„ ТУ МХП 1523-51 я
10 Трихлорметилфлуорон хлоральгидрат мед. »
23
Примечания.
1. Как видно из уравнения реакции конденсации, на 2 моля ок-
сигидрохипоиа требуется 1 моль альдегида. Однако при стехиомет-
рическом соотношении компонентов выход продукта снижается. Оп-
тимальным количеством альдегида является 1 моль на 1 моль окси-
гидрохинона. Дальнейшее увеличение количества альдегида не по-
вышает выход, а лишь загрязняет триоксифлуорон продуктами ос-
моления избыточного альдегида.
2. При синтезе дисульфофеиилфлуорона вместо чистого 2,4-ди-
сульфобензальдегида можно употреблять технический продукт—его
двузамещенную натриевую соль в смеси с сульфатом натрия, при-
сутствие которого не мешает.
3. Сульфат n-нитрофенилфлуорона выпадает в виде тонких зо-
лотистых кристаллов, которые плотно забивают фильтр при отсасы-
вании на воронке Бюхнера, и фильтрование проходит с трудом. Луч-
ше фильтровать через бумажный фильтр на обычной воронке.
4. Свободный дисульфофенилфлуорои, в отличие от других три-
окенфлуоронов, легко растворим в воде, так что гидролиз не дает
чистого продукта. Поэтому дисульфофенилфлуорои применяют в
виде сульфата CijHijOuSi - HzSO4.
5. При синтезе пропилфлуорона лучший выход получается при
уменьшении количества реактивов и объемов растворов в 5 раз про-
тив указанных выше.
ЛИТЕРАТУРА
1. В. А. Назаренко, Н. В. Л е б е д е в а, Е. А. Бирюк, Мате-
риалы VIII совещания работников, лабораторий геологических организа-
ций, вып. 3, М., 1961, стр. 3.
2. С. L i е b е г m a n n, S. L i п d е и b a u т, Вег., 37, 1171,2728 (1904).
3. R. Duckert, Helv. chim. acta, 20, 362 (1937).
4. К. Kimura, H. S а п о, M. A s a d a, Bull. Chem. Soc. Japan, 29, 640
(1956).
5. J. G i 11 i s, J. H о s t e, A. С 1 a e у s, Anal, chim acta. 1, 302 (1947).
6. J. G il 1 i s, A. С I a e у s, J. Hos t e, Anal, chim acta, 1, 421 (1917).
7. J. Gillis, Anal. Chim. acta, 8, 97 (1953).
8. В. A. II а з a p e н к о, M. Б. Шустова, Заводск. лаборатория, 24,
1344 (1958).
9. H. В. Л e б e д e в а, В. А. Назаренко, Труды комиссии по анали-
тцч. химии, XI, 287 (I960).
Поступила в феврале 1963 г.
ИОНХ АН УССР, Одесса
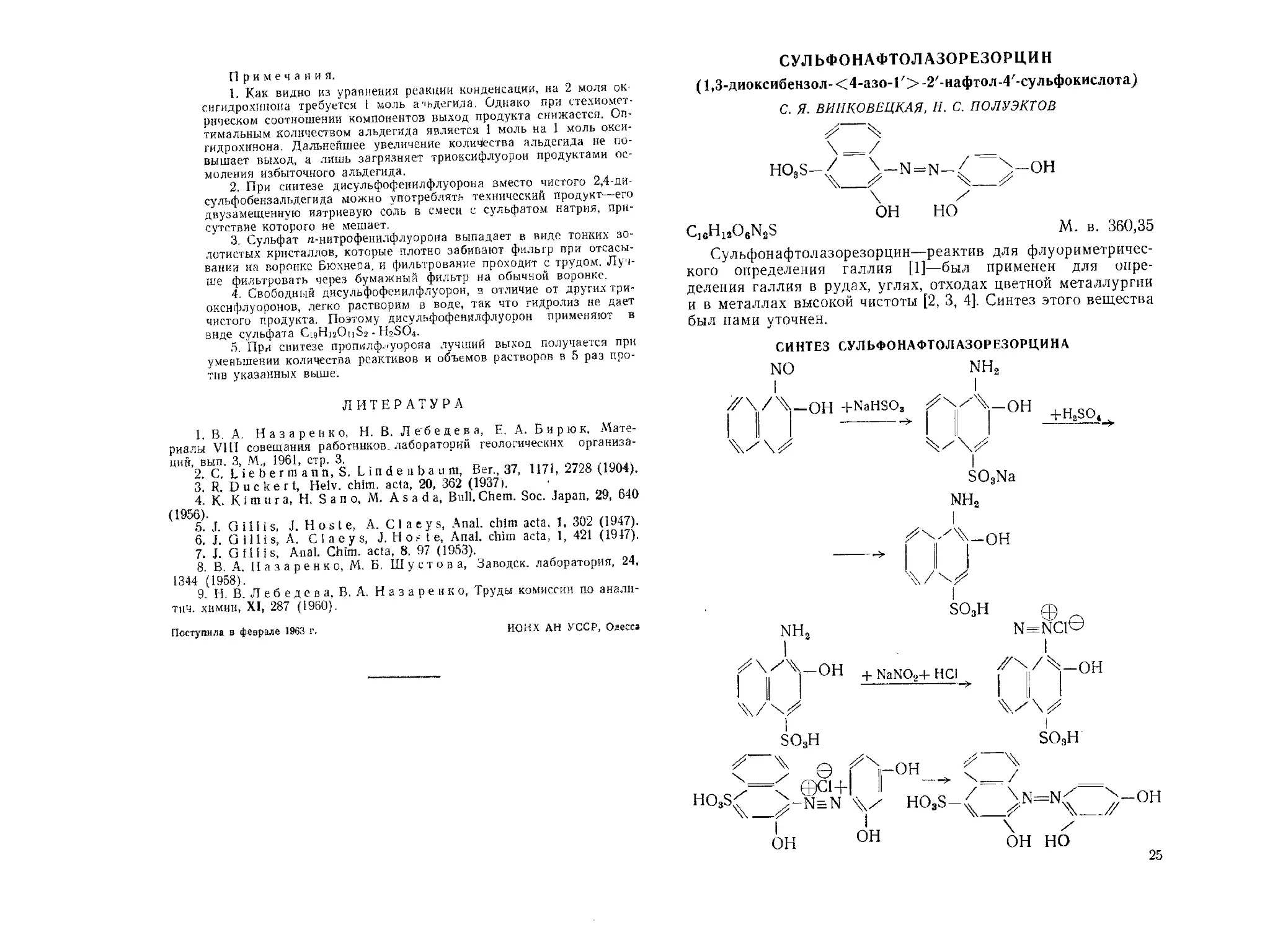
СУЛЬФОНАФТОЛ АЗОРЕЗОРЦИН
(1,3-диоксибензол-<4-азо-Г>-2/-нафтол-4/-сульфокислота)
С. Я. ВИИКОВЕЦКАЯ, Н. С. ПОЛУЭКТОВ
HO3S-.<
он но
с, 6H12OeN2S
М. в. 360,35
Сульфонафтолазорезорцин—реактив для флуориметричес-
кого определения галлия [1]—был применен для опре-
деления галлия в рудах, углях, отходах цветной металлургии
и в металлах высокой чистоты [2, 3, 4]. Синтез этого вещества
был нами уточнен.
СИНТЕЗ СУЛЬФОНАФТОЛАЗОРЕЗОРЦИНА
NO
I
^Х/^-ОН +NaHSO3
vv
nh2
I
мГ^г°н
SO3Na
+ H2SO4
25
Характеристика основного сырья
Натр едкий, ГОСТ 4328—48.
Бисульфит натрия, ТУ МХТ 1944—49.
Кислота соляная, ГОСТ 3118—46.
ГНитрозо-2-нафтол, порошок коричневато-бурого цвета,
свежеприготовленный [5].
Кислота серная, ГОСТ 4204—48.
Нитрит натрия, ГОСТ 4197—48.
Резорцин, ч. д. а.
Эфир этиловый, ГОСТ 5963—51.
Условия получения
Для синтеза сульфонафтолазорезорцина необходима све-
жеприготовленная 1-амино-2-нафтол-4-суль'фокислота.
Получение 1-амино-2-нафтол-4-сульф6кислоты. Сырую пас-
ту Гнитрозо-2-нафтола, полученную из 14,4 г (0,1 Л1) бета-
нафтола, размешивают в стакане с небольшим количеством
воды (около 20 мл) и охлаждают льдом до 5°. К однородной
массе приливают в один прием 65 г 37%-пого раствора мета-
бисульфита натрия. Нитрозонафтол растворяется. Если этого
не происходит, осторожно добавляют раствор едкого натра (5—
7%) для растворения нитрозонафтола. Раствор отделяют
фильтрованием от небольшого количества смолистых приме-
сей и подкисляют при 25° 35 мл 45%-кого раствора серной
кислоты. При этом реакция должна быть кислой по конго.
Через час раствор осторожно нагревают до 50° и оставля-
ют на ночь. Выпавшие в виде густой массы кристаллы 1-ами-
но-2-нафтол-4-сульфокислоты отсасывают и промывают во-
дой. Если полученные кристаллы темного цвета (загрязнены
смолистыми веществами), то их следует перекристаллизовать.
Для этой цели кислоту переносят в стакан, добавляют дис-
тиллированной воды до образования кашицеобразной массы
и растворяют главную массу кристаллов в 5%-ном растворе
едкого натра. Затем добавляют небольшое количество раство-
ра бисульфита натрия, активированного угля, кипятят до про-
светления раствора и фильтруют.
К полученному прозрачному фильтрату, прибавляют по
каплям концентрированную соляную кислоту при помешива-
нии. При этом выпадают кристаллы 1-амино-2-нафтол-4-суль-
фокислоты розоватого цвета в виде густой массы. Отсасыва-
ют, хорошо промывают дистиллированной водой и сушат на
воздухе до постоянного веса.
Получение 1,3-диоксибензол <4-азо~1'> 2'-нафтол-4'-
сульфокислоты. 3 г (0,012 М) свежеприготовленной чистой
1-амино-2-нафтол-4-сульфокислоты размешивают с 20 мл во-
ды, охлаждают льдом до 5° и прибавляют к полученной массе
26
J,25 г сульфата меди, растворенного в 5 мл воды, затем
раствор 0,9 г (0,013 М) нитрита натрия в 10 мл воды. Поме-
шивают до растворения. Затем раствор отфильтровывают и к
фильтрату прибавляют по каплям 2—3 мл концентрированной
соляной кислоты. При этом выпадает желтого цвета диазо-
окись. Выпавший осадок отфильтровывают на воронке Бюх-
нера и промывают 3—4 раза раствором соляной кислоты
(1:10).
Навеску 1,6 г (0,0145 М) резорцина растворяют в 20 мл
воды и в раствор вносят диазоокись, которая полностью рас-
творяется. Затем прибавляют при помешивании по каплям
30%-ный раствор едкого кали до окрашивания раствора в
темно-вишневый цвет и оставляют на ночь.
На следующий день краситель осаждают концентрирован-
ной соляной кислотой, добавляя ее по каплям до образования
густой массы. Последнюю отсасывают, промывают 3—4 раза
небольшим количеством разбавленной соляной кислоты (1:10),
затем эфиром и высушивают на воздухе до постоянного веса.
Выход 2,6 г, что составляет 60% от теоретического.
ЛИТЕРАТУРА
1. Н. С Полуэктов, Н. К. Киселева, Ж. аналит. химии, 13.
555 (1958).
2. В. А. Назаренко, С. Я. Винковецкая, Ж. аналит. химии,
13, 327 (1958).
3, Методы определения и анализа редких элементов, Изд. АН СССР,
М„ 1961, стр. 442.
4. В. А. Назаренко, С. Я. Винковецкая, Р. В. Р а в и ц к а я,
Укр. хим. ж., 28, 726 (1962).
,5 . В. М. Родионов и др., «Лабораторное руководство по химии
промежуточных продуктов и красителей», Госхимпздат, 1948, стр. 103.
Поступила в феврале 1963 г. ИОНХ АН УССР, Одесса
ОСОБЕННОСТИ РАБОТЫ С ПРОИЗВОДНЫМИ РЯДА
ПИРИДИНА
Ю. И. ЧУМАКОВ
Высушивание. Полное удаление влаги из пиридиновых ос-
нований, вследствие их значительной гигроскопичности, пред-
ставляет нелегкую задачу. Лучшими высушивающими веще-
ствами являются едкий натр и едкое кали. Можно применять
также окись бария и прокаленный поташ, но они менее эф-
фективны.
27
Если пиридиновое основание содержит много воды (20—
40%), то его обрабатывают щелочью (обычно более дешевым
едким натром) и отделяют верхний слой, представляющий
пиридиновое основание, от нижнего—водно-щелочного слоя.
Затем полученное основание высушивают над свежей порци-
ей плавленой щелочи.
Следует иметь в виду, что кратковременное высушивание
малоэффективно, необходимо сушить 5—10 дней. Ускорить
высушивание можно путем кипячения пиридинового основа-
ния и щелочи с обратным холодильником в течение 4—8 ча-
сов. Но даже при таком высушивании основание содержит
заметные количества влаги, в чем легко убедиться, подвергая
его перегонке на эффективной ректификационной колонке.
Во всех случаях в предгон вначале обязательно переходит за-
метное количество более низкокипящего азеотропа с водой
(часть этой воды находилась на стенках прибора).
Весьма эффективными высушивающими средствами яв-
ляются гидрид кальция и алюмогидрид лития. Но эти реак-
тивы сравнительно редко употребляются в обычной лабора-
торной практике.
Описанные приемы высушивания пригодны только для ал-
кил- и арилпиридинов или других пиридинов с индифферент-
ными по отношению к едким щелочам группами. Они непри-
менимы для высушивания эфиров пиридинкарбоновых кислот
или сложных эфиров оксиметилпиридинов. В этих случаях ис-
пользуют сульфат натрия или азеотропную перегонку с бен-
золом.
Для высушивания многих замещенных производных ряда
пиридина успешно может быть применен прокаленный поташ.
Хранение. Все пиридиновые основания при стоянии, осо-
бенно на свету, темнеют. Этот процесс, вероятно, связан с фо-
тохимическим расщеплением до глутаконового диальдегида
и его производных и осмолением последних. Поэтому хранить
пиридиновые основания следует в склянках из темного
стекла и в темном месте. Сильно потемневшие старые образ-
цы могут быть очищены простой перегонкой, причем потери
продукта за счет осмоления незначительны. Высококипящие
гомологи пиридина, начиная с изомерных этилпиридинов и
диметилпиридинов, при обычном давлении перегоняются ок-
рашенными в светло-желтый цвет и иногда при хранении быст-
ро темнеют. Для получения бесцветных продуктов эти осно-
вания лучше перегонять в небольшом вакууме.
Общий способ очистки. Многие пиридиновые основания
легколетучи с водяным паром *. Для. высококипящих произ-
водных пиридина (фенил-, нафтилпиридины) предпочтительна
* Замещенные производные пиридина, содержащие, полярные и гидро-
фильные группы, нелетучи с паром (оксиметилпиридины, пиридинкарбоно-
вые кислоты и т. п.),
28
перегонка с перегретым до 300—400° водяным паром. Это —
общий и весьма эффективный способ выделения пиридиновых
оснований из реакционной массы и отделения от загрязняю-
щих примесей, если последние нелетучи с водяным паром.
Если эти примеси летучи с паром, но не являются основания-
ми, то от них можно освободиться, переведя пиридиновое ос-
нование с избытком минеральной кислоты в соль, нелетучую
с водяным паром, и отогнать с водяным паром присутствую-
щие примеси неосновного характера, после чего выделить дей-
ствием едкого натра свободное основание и перегнать его так-
же с водяным паром.
Перегонка с паром особенно полезна тогда, когда при вы-
саливании пиридиновых оснований щелочью образуется стой-
кая эмульсия. В этом случае иногда помогает фильтрация, но
обычно лучше пиридиновое основание перегнать с водяным
паром и выделить его из дистиллата обработкой щелочью.
Значение эффективной ректификации. При приготовлении
высокоочищенных образцов пиридиновых оснований решаю-
щее значение имеет тщательная ректификация как исходных
смесей пиридиновых оснований, так и получаемых чистых
продуктов. Это вызывается тем, что пиридиновые основания,
даже со значительной разницей в температурах кипения, име-
ют тенденцию перегоняться друг с другом. В частности, имен-
но по этой причине при перегонке смеси таких оснований, как
пиридин и 2-метилпиридин, отличающихся друг от друга по
температурё кипения на 14°, ректификация па несовершенной
колонке (не говоря уже о дефлегматоре) не дает возможно-
сти получить чистые продукты, что, по-видимому, объясняет-
ся явлением полиазеотропии [1].
В этих случаях вполне достаточно использовать наиболее
доступные в обычных лабораторных условиях, простые в из-
готовлении и эксплуатации насадочные ректификационные
колонки эффективностью 20—30 теоретических тарелок с на-
садкой Левина.
Оценка содержания. Контроль чистоты азотсодержащих
оснований по пикратам, длительное время считавшийся наи-
более достоверным, едва ли следует считать надежным, тем
более, что получаемый пикрат, как правило, перед определе-
нием температуры плавления дополнительно перекристалли-
зовывался. В то же время известно, что перекристаллизация
пикратов сама по себе является методом очистки пиридино-
вых оснований. В статье «2—Метилпиридип» приведена спе-
циальная методика получения пикратов пиридиновых основа-
ний (с избытком пикриновой кислоты), которая позволяет по-
лучать незавышенные результаты.
Последним достижением в области анализа пиридиновых
оснований являются термометрический и спектроскопический
методы анализа. Спектроскопические методы требуют нали-
29
чия чистых образцов, довольно сложны и не всегда доступны
в рядовой синтетической лаборатории. Термометрический
контроль более доступен, но в том виде, в котором он описан
для пиридиновых оснований, является довольно сложным, так
как анализ производится по кривым замерзания [2, 3]. Слож-
ность усугубляется еще и тем, что пиридиновые основания
весьма гигроскопичны, а присутствие влаги существенно ис-
кажает температуры плавления. Ниже описан упрощенный
контроль их чистоты по температуре плавления (см. «Изохи-
нолин») .
Большим подспорьем при анализе пиридиновых оснований
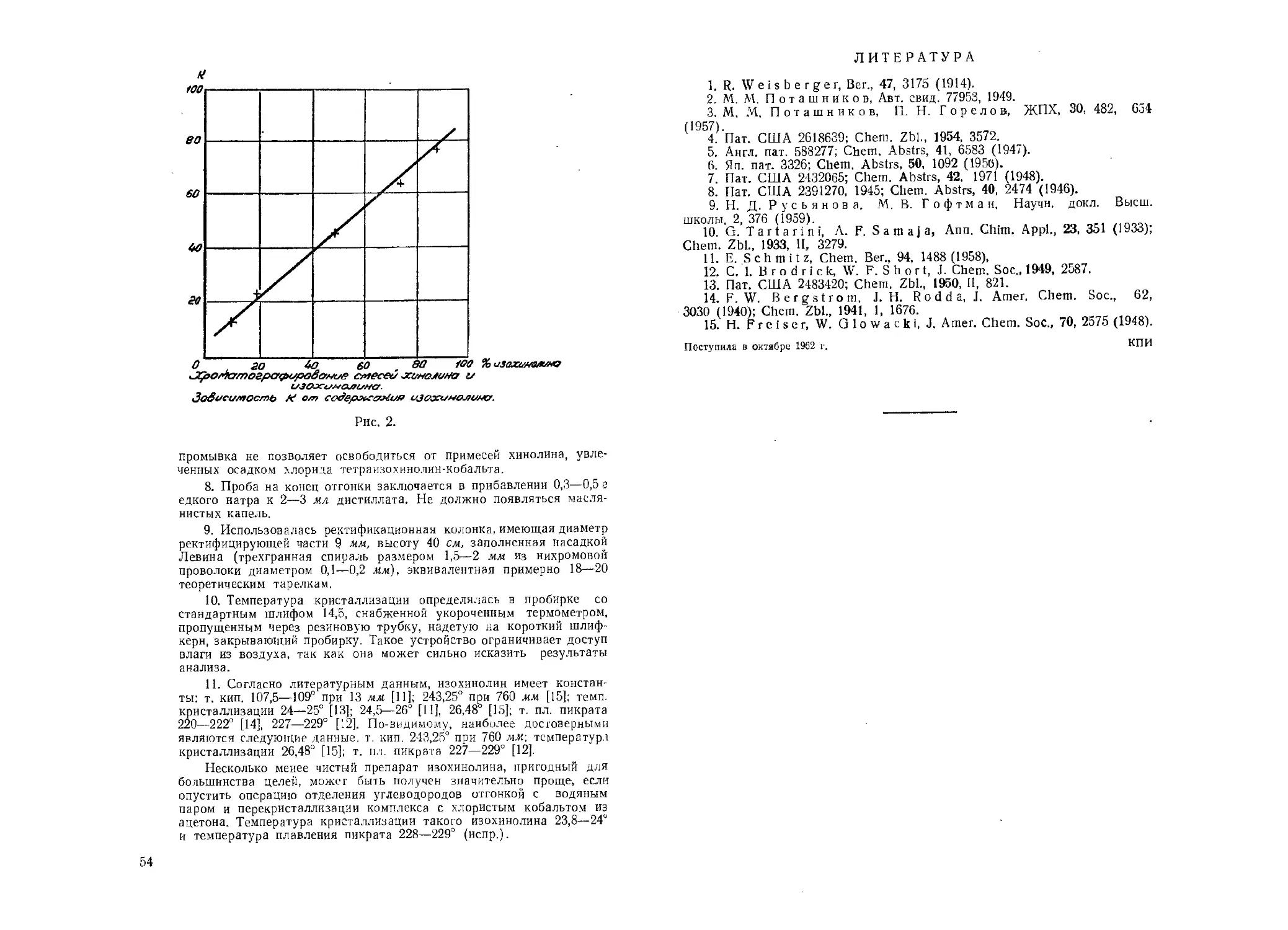
может явиться хроматографический метод. Его точность рав-
на 2—5%, что значительно ниже, чем у термометрического
метода. Тем не менее, по быстроте выполнения и надежности
результатов в тех случаях, когда он применим, его вполне до-
статочно. Особый интерес представляет применение его для
приблизительного количественного анализа смесей алкил-,
арил-, аралкил- и бензопиридинов (подробнее см. ст. «Изохи-
нолин»).
ЛИТЕРАТУРА
1. В. В. Свентославский, Физическая химия каменноугольной
смолы, ИЛ, Москва, 1958.
2. Е. A. Coulson, J. J. Jones, J. Soc. Chem. Ind., 65, 169 (1946).
3. D. 1. Biddiscombe, E. A. Coulson, R. Handley, E. F. G.
Herington, J. Chem. Soc., 1957, 1954.
Поступила в октябре 1962 г. Киевский Политехнический институт (КПИ)
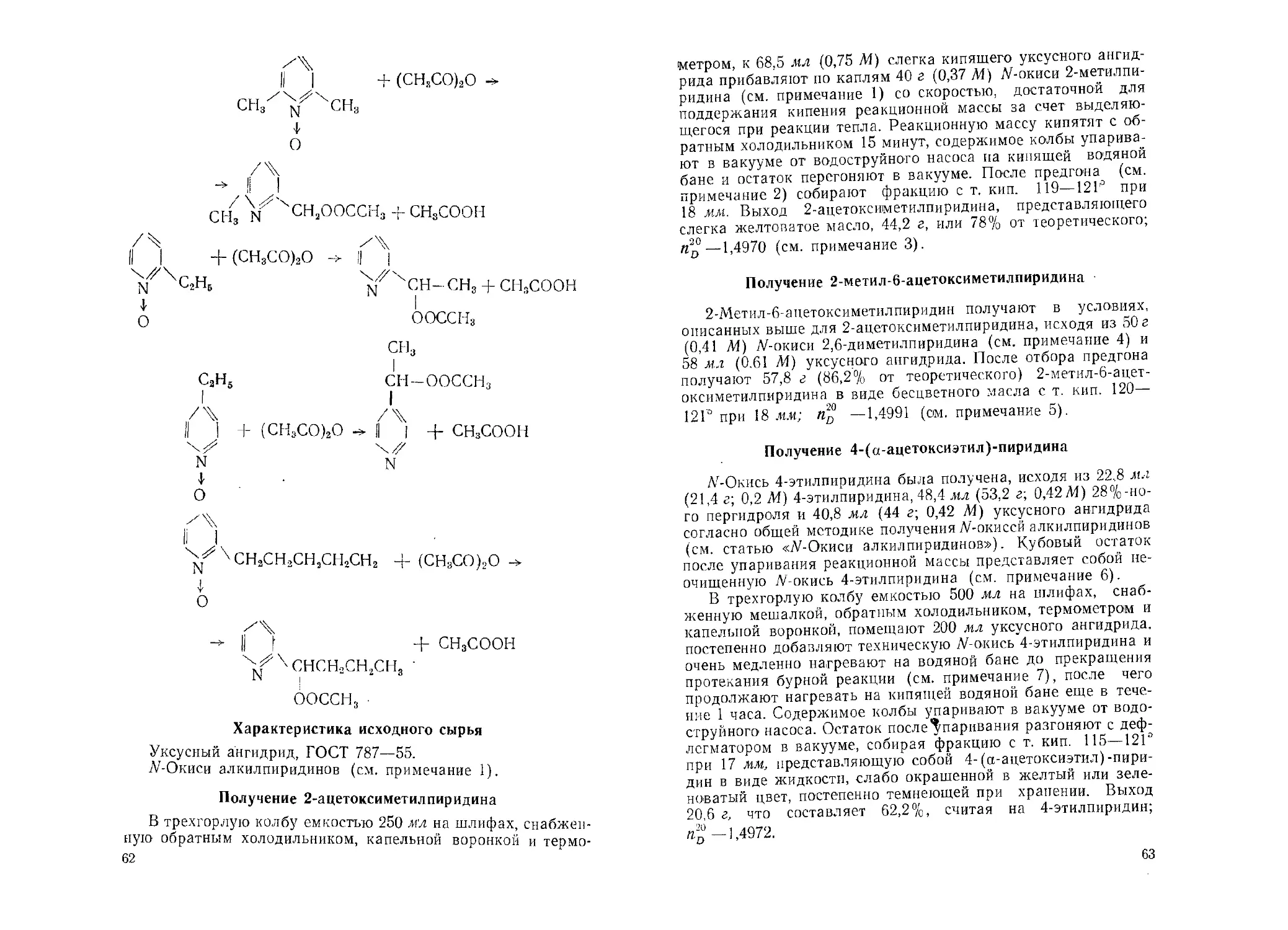
2-МЕТИЛ ПИРИДИН
(а-Пиколин, 2-пиколин)
Ю. И. ЧУМАКОВ
/Ч
V''Vh
C6H7N М. в. 93, 13
2-Метилпиридин является исходным продуктом в синтезах
пиколиновой кислоты, 2-(а-оксиэтил)-пиридина, 2-винилпири-
дина, 2-ацетоксиметилпиридина, 2-пиридинкарбинола, 2-пири-
30
динальдегида, 2-хлорметилпиридина и ряда других замещен-
ных в положении 2 производных пиридина.
Основным промышленным источником 2-метилпиридина
являются фракции пиридиновых оснований коксохимическо-
го происхождения [1]. Полный синтез свободного от изомеров
2-метилпиридина является многостадийным, сложным и прак-
тического значения не имеет [2J.
Для очистки 2-метилпиридина коксохимического проис-
хождения было предложено несколько способов, в том числе
осаждение в виде комплексных соединений с хлористым цин-
ком [3], персульфатом серебра [4], сулемой, платинохлористо-
водородной кислотой [5], осаждение в виде фталата [6, 7],
фракционированная кристаллизация [8] и ряд других спосо-
бов.
Приведенная ниже методика предусматривает предвари-
тельное освобождение 2-метилпиридина коксохимического
происхождения от примесей углеводородного характера и по-
следующую ректификацию на достаточно эффективной колон-
ке, что позволяет отделить примеси пиридина и вышекипящих
гомологов [8].
Характеристика основного сырья
2-Метнлпиридин, ЧМТУ 5044—55.
Условия получения
В колбу емкостью 1 л для перегонки с водяным паром по-
мещают 150 мл (139,5 г; 1,5 М) 2-метилпиридина и при пере-
мешивании и охлаждении постепенно прибавляют охлажден-
ный раствор 55 мл (100 г, 1 Л!) концентрированной серной
кислоты (d 1,83—1,84) в 100 мл воды. Водный раствор суль-
фата 2-метилпиридиния перегоняют с водяным паром до объ-
ема дистиллата 0,5—1,0 литра (примечание 1). Содержимое
колбы охлаждают, затем при охлаждении в ледяной бане и
перемешивании постепенно прибавляют раствор 320 г (8 М)
едкого натра в 400 мл воды. Выделившийся верхний слой 2-
метилпиридина отделяют и дополнительно высушивают его в
течение ночи 40—50 граммами плавленого едкого натра или
едкого кали. Для окончательного высушивания к 2-метилпи-
ридину, отделенному от щелочи, прибавляют дополнительно
25 г плавленого едкого натра или кали и кипятят с обратным
холодильником на масляной бане в течение 4—5 часов (см.
примечание 2).
Высушенный 2-метилпиридин ректифицируют на колонке
эффективностью не менее 18—20 теоретических тарелок, с
флегмовым числом 10—15 (см. примечание 3), отбирая фрак-
ции:
31
Т. кип. Давление Вес, г
1 фр. до 128,4° 752 мм 12,5
2 фр. 128,4-128,8° 752 мм 78,3
Первая фракция состоит из 2-метилпиридина и примесей
пиридина и воды. Вторая фракция представляет 2-метилпи-
ридин. Т. пл. пикрата 165,5—166° (примечание 4). В кубовом
остатке содержится 2-метилпиридин и примеси вышекнпящих
алкилпиридинов (3-, 4-метил- и 2,6-диметилпиридин).
Примечания.
1. Серная кислота берется с избытком против расчетного. Не-
смотря на это, некоторое количество 2-метилпиридина перегоняет-
ся с паром, вследствие частичного гидролиза сульфата 2-метилпири-
диния. Чтобы избежать чрезмерного накопления воды в перегонной
колбе, необходимо ее подогревать.
2. Холодильник должен быть защищен хлоркальциевой труб-
кой, заполненной гранулированным плавленым едким кали.
3. Ректификация производилась па обычной ректификационной
колонке «Лаборприбор» с высотой ректифицирующей части 40 см,
внутренним диаметром 8—10 мм, заполненной трехгранными спи-
ралями из нихромовой проволоки (диаметром 0,2 лш) размером
1,5—2 мм (насадка Левина), снабженной головкой полной конден-
сации обычной конструкции.
4. Согласно литературным данным, чистый 2-метилпиридин
имеет т. кип. 129,408° при 760 мм; т. крист. 66,7Г; Яд—1,50101;
— 0,94432 [8]; т. пл. пикрата 167—168° [9] или 169° [10].
Для получения пикрата использовалась нижеописанная
методика.
Точно взвешивают в бюксе 2 капли 2-метилпиридина и сра-
зу прибавляют 1 ял сухого спирта, чтобы не испарялось пи-
ридиновое основание. Для анализа на точных технических ве-
сах отвешивают больше на 10—15% против расчетного ко-
личества пикриновой кислоты и растворяют в спирте, кото-
рый берут по 0,5—1,0 мл на 0,1 г пикриновой кислоты. Для
полного растворения пикриновой кислоты бюкс слегка подогре-
вают на выключенной плитке. К спиртовому раствору пириди-
нового основания приливают спиртовой раствор пикриновой
кислоты, размешивают стеклянной палочкой, после чего при-
бавляют еще 2 мл диэтилового эфира. Дают постоять 20—30
минут, выпавший пикрат отфильтровывают, промывают па
фильтре 3 раза по 0,5 мл сухого эфира, высушивают при SO-
950 в течение получаса и определяют температуру плавления.
Обычно пикраты получают, используя эквивалентное количе-
ство пикриновой кислоты и перекристаллизацию перед опре-
делением температуры плавления. Такой способ не позволяет
судить о чистоте исходного пиридинового основания.
Для получения высокоочшценного 2-метилпиридина с со-
держанием 99,87% (молярных) применялась фракциониро-
ванная кристаллизация при температурах, близких к темпе-
32
ратуре плавления этого пиридинового основания. Однако эта
операция в обычных лабораторных условиях трудно выпол-
нима, поскольку необходимо работать при температурах око-
ло минус 70° и при полном отсутствии влаги. Присутствие по-
следней существенно искажает результаты вымораживания.
В то же время 2-метилпиридин так же, как сам пиридин и
его гомологи, является весьма гигроскопичным.
ЛИТЕРАТУРА
1. М. С. Литвиненко, И. М. Н о с а л е в и ч. Химические продук-
ты коксования для производства полимерных материалов, Металлургиз-
даг, Харьков, 1962.
2. Пат. США 2748130, 1953.
3. J. G. Heap, W. J. J о п е s, J. В. Speakman, J. Amer. Chem.
Soc., 43, 1936 (1926).
4. G. Tartarini, T. Satnaja, Ann. Chini. Applic., 23, 356 (1933);
Chem. Zbl., 1933, 11, 3279.
5. A. Ladenburg, Liebigs Ann. Chem., 247, 1 (1888).
6. Пат. США 2408975; Chem. Abstrs, 41. 938 (1947).
7. Пат. США 2458743; Chem. Abstrs, 43, 2240 (1949).
8. D. P. В i dd is com be, E. A. Coulson, R. H a n d 1 eу, E. F.
• Harington, J. Chem. Soc., 1957, 1954.
9. E. А. С оu 1 s о n, J. I. Jones, J. Soc. Chem. Ind., 65, 169 (1946).
10. В. С. Джонсон, И. Д. Шенн ан, Р.. А. Рид, Органические ре-
активы для органического анализа, ИЛ, Москва, 1948.
Поступила в октябре 1962 г. КПИ
2-ЭТИЛПИРИДИН
Ю. И. ЧУМАКОВ
C7H0N м. в. 107,15
2-Этилпиридин является исходным продуктом в синтезе
некоторых реактивов ряда пиридина, в частности, 2-этил-4-
пиридинкарбоновой кислоты.
Для получения 2-этилпиридина использовались: гидриро-
вание 2-винилпиридина, взаимодействие 2-пикодиллития с
иодистым метилом [1], а также восстановление по Кижнеру
2-ацетилпнридина [2].
з Зак. 576 33
Другие пути получения 2-этилпиридина, например по Ла-
денбургу [3, 4], малоудобны и приводят к продукту, с трудом
поддающемуся очистке.
Ниже описано получение 2-этилпиридина гидрированием
на никеле Ренея 2-винилпиридина [1] (в экспериментальной
части принимал участие В. М. Ледовских).
СИНТЕЗ 2-ЭТИЛПИРИДИНА
Ni /\
|| | +Н2--->|| |
^'СН=СН2 х/хс2н6
Характеристика основного сырья
2-Винилпиридин (см. примечание 1).
Водород, ГОСТ 3022—45.
Условия получения
Во вращающийся автоклав емкостью 700 мл . помещают
раствор 105 мл свежеперегнанного 2-винилпиридина (т. кип.
60—62° при 20 мм, см. примечание 1) в 100 мл метилового
спирта и 2 чайные ложки никеля Ренея. Надавливают в ав-
токлав азот до 2—3 атмосфер давления и спускают его для
удаления воздуха из автоклава. Затем надавливают водород
до 20—40 атмосфер и приводят автоклав во вращение при
комнатной температуре до поглощения расчетного количест-
ва водорода. Если требуется, то водород надавливают по-
вторно (см. примечание 2). Гидрирование занимает 40—60
минут, после чего через шланг давление спускают под тягу,
разбалчивают автоклав и содержимое отсасывают в колбу
Бунзена через стеклянную трубку и шланг. Стенки автоклава
обмывают 50 мл метанола и также отсасывают, присоединяя
их к реакционной массе после гидрирования. Никель Ренея
отфильтровывают и на фильтре промывают 15—20 мл мета-
нола, которым предварительно смывают склянку Бунзена (см.
примечание 3). Отгоняют метанол и остаток фракционируют
в вакууме с елочным дефлегматором высотой 20—30 см, со-
бирая фракцию с т. кип. 142—144°; nf—1,4950; т. пл. пик-
рата 106—108°. Выход 89 г, что составляет 88% от теоретиче-
ского.
Примечания.
1. Использовался технический 2-винилпиридин, полученный из
2-метилпиридина через 2-(|3 -оксиэтил)-пиридин. 2-Винилпирндип яв-
ляется лакриматором, поэтому работу с ним следует производить в
вытяжном шкафу.
34
2. Необходимое для гидрирования количество водорода рассчи-
тывается по формуле:
л М273 + 0
(1-0-273 ’
где Др — необходимое давление в атмосферах;
о —объем водорода, необходимый для гидрирования^ приве-
денный к нормальным условиям;
t — температура автоклава;
L — объем автоклава, л\
I — объем загрузки, л.
Если гидрирование пе начинается при комнатной температуре,
то содержимое автоклава нагревают до 50—70° и реакцию ведут
при этой температуре.
3. Никель Реиея может сохранить свою пирофорность и после
гидрирования, поэтому следует соблюдать необходимые меры пре-
досторожности.
4. Согласно литературным данным, 2-этилпиридин нмеет т. кип.
77— 79° при 80 мм: Лр —1,4978; т. пл. пикрата 108,5—110° [1].
ЛИТЕРАТУРА
1. Е. С. Gregg, D. Graig, J. Amer. Chem. Soc., 70, 3138 (1948).“
2. A. Furst, J. Amer. Chem. Soc., 71, 3550 (1949).
3. A. Laden burg, Liebigs Ann, Chem., 32, 44 (1889).
4. A. E. Чичибабин, О продуктах действия галоидных соединений
на пиридин и хинолин, 1962 г.
Поступила в октябре 1962 г. КПИ
2-трет. БУТИЛПИРИДИН
Ю. И. ЧУМАКОВ, 3. М. КОРСАКОВА
C9HiaN
у\с(сн3)3
М. в. 135, 21
2-трет. Бутилпиридин является алкилпиридином,.обладаю-
щим большей основностью, чем пиридин, и в то же время сла-
быми комплексообразующими свойствами в связи с наличием
объемистого заместителя в положении, ближайшем к атому
азота. 2-трет. Бутилпиридин используется для синтеза 2,6-ди-
трет. бутилпиридина [1].
3* 35
2-трет. Бутилпиридин может быть получен двумя способа-
ми: действием хлористого метила на 2-метилпиридин в при-
сутствии амида натрия [2] или взаимодействием пиридина и
трет, бутиллития [1]. Первый способ дает невысокие выходы и
очистка от примесей 2-изопропилпиридина затруднительна.
Ниже приведен синтез 2-трет. бутилпиридипа по реакции Циг-
лера-Цайзера кипячением смеси хлористого трет, бутиллития
и пиридина [1]. Третичный бутиллитий был получен в диэти-
ловом эфире при минусовых температурах согласно [3, 4, 5].
Этот путь получения дает более устойчивые результаты, чем
синтез третичного бутиллития в петролейном эфире [6].
СИНТЕЗ 2-mpem. БУТИЛПИРИДИНА
С(СН8)аС1 + 2Li (CH3)3CLi + L1C1
/Ч
II | + LiC(CH8)3 - I! I
Ухс(сн3)з + ин
Характеристика основного сырья
1. Пиридин, ГОСТ 1625—61.
2. Третичный хлористый бутил (см. примечание 5).
3. Литий металлический, ГОСТ 8774—58.
Условия получения
Прибор представляет четырехгорлую колбу емкостью 1 л,
снабженную саблевидной мешалкой с масляным затвором, об-
ратным холодильником с хлоркальциевой трубкой, трубкой
для ввода азота, термометром и капельной воронкой (см.
примечание I). Прибор высушивают, слегка нагревая колбу
газовой горелкой на сетке при работающей мешалке и одно-
временном пропускании тока сухого азота; температура на
термометре, не касающемся стенки колбы, равна 120—150°.
Убирают нагрев и дают прибору охладиться в токе сухого
азота (см. примечание 2).
В колбу помещают 400 мл сухого эфира (см. примечание
3) и через большую коническую воронку, вставленную в гор-
ло для капельной воронки, опускают в эфир нарезанный по-
лосками I X 10 мм металлический литий в количестве 16,7 г
(2,4 г-атома) (см. примечание 4), не прекращая при этом
пропускания тока азота. Заменяют коническую воронку на
капельную и содержимое колбы охлаждают до минус 35—
минус 40° в бане из сухого льда и ацетона. При этой темпе-
ратуре в течение 3 часов прибавляют 125 мл (103 г, 1,1 М)
третичного хлористого бутила (см. примечание 5), растворен-
36
ого в примерно равном по объему количестве сухого эфира.
1о прибавлении реакционную массу дополнительно выдержи-
ают. размешивая при той же температуре в течение получаса
см. примечание 6). Реакционную массу охлаждают до минус
О—минус 75° и при этой температуре в течение часа прибав-
1яют 56 мл (55 г; 0,7 (И) сухого пиридина (см. примечание
'), растворенного в равном по объему количестве сухого эфи-
ja. Перемешивают в течение 30 минут и оставляют на ночь
три той же температуре в сосуде Дьюара. Заменяют обрат-
чый холодильник на нисходящий и при работающей мешалке
отгоняют эфир (примерно 450 мл) до температуры в колбе
70—75°. Мешалку останавливают и реакционную массу вы-
держивают при этой же температуре в течение двух часов без
размешивания, затем еще 1 час при работающей мешалке.
Охлаждают до комнатной температуры и в токе азота, при
энергичном перемешивании и охлаждении водой со льдом,
постепенно прибавляют 300—350 лмводы. Для уничтожения
образовавшейся эмульсии содержимое профильтровывают на
воронке Бюхнера в слабом вакууме. Верхний эфирный слой
отделяют от нижнего щелочного, который дважды экстраги-
руют эфиром порциями по 50 мл и эфирные вытяжки присое-
диняют к основной массе эфирного раствора 2-трет. бутилпи-
ридина. Высушивают над хлористым кальцием и отгоняют
растворитель. Остаток перегоняют на колонке в вакууме, со-
бирая предгои с температурой кипения до 61,5° при 30 мм и
основную фракцию с температурой кипения 61,5—62° при
30 лш в количестве 39 г (39% от теоретического); Пр —
1,4890 (ом. примечание 8).
Примечания.
1. Использовался масляный затвор обычной конструкции, но
несколько больший по высоте. Между ним и колбой был помещен
короткий водяной холодильник длиной 10—12 см с целью предо-
хранить попадание эфира в масло. Раствор заполняют на 2/з вазе-
линовым маслом, предварительно прогретым с металлическим нат-
рием. Азот высушивался в трубках, заполненных хлористым каль-
цием и пятиокисью фосфора, нанесенной на стеклянную вату (спе-
циального удаления примеси кислорода не производилось).
Обратный холодильник устроен так, Что может быть превра-
щен в нисходящий без разбора прибора. В качестве счетчика пу-
зырьков азота служит склянка Тищенко, заполненная сухим вазе-
линовым маслом.
2. Для ускорения охлаждения целесообразно направить на при-
бор струю воздуха от вентилятора.
3. Использовался эфир, освобожденный от перекисей и высу-
шенный последовательно хлористым кальцием и металлическим нат-
рием. Перед использованием его отфильтровывают от взвеси гидро-
окиси натрия.
4. Металлический литий предварительно расплющивают молот-
ком _на наковальне до толщины 0,5—1,0 мм.
5. Хлористый третичный бутил должен быть тщательно высу-
шен над хлористым кальцием н свежеперегнан иа колонке или с
дефлегматором. В одном опыте при использовании третичного хло-
37
ристого бутила, долго хранившегося иад хлористым кальцием, син.
тез не удался.
6. Температура во время реакции не должна
во избежание разложения литий-алкила за счет
превышать —35°
взаимодействия с
ЭфИ 7° Пиридин был высушен кипячением с </> по весу едкого =
обратным .холодильником и перегнан (первые 15—20 мл отбрас
лись).
8. Согласно литературным данным, 2-трет. бутилпиридин име-
ет следующие константы: т. кип. 169° при 743 мм; —1,4891;
т. пл. минус 33,0 — минус 33,5°, т. пл. пикрата 104,6—105,2° [2].
ных н-амилпиридинов. Они также являются комплексообра-
зующими реагентами.
Единственным способом получения 2-(З'-пентенил)-пири-
дина является взаимодействие 2-метилпиридина с бутадие-
ном-1,3 в присутствии каталитических количеств металличе-
ского натрия [1, 2].
Нами было найдено, что этот способ применим для синте-
за З-(З'-пентенил)-пиридина.
СИНТЕЗ ИЗОМЕРНЫХ (3-ПЕНТЕНИЛ)-ПИРИДИНОВ
литература
I. Н. С. Braun,
2. Н. С. В г а и п,
В Каплет, J. Amer. Chem.
W. A. Murphey, J. Amer.
Soc., 75, 3865 (1953).
Chem. Soc., 73, 3308
(1951).
3. P. D. В a r 11 e t, S. Fri edm a n, M. Stiles, J. Amer. Chem. Soc.,
75, 1771 (1953).
4. P. D. В art le t, E. B. L e f f er t s, J. Amer. Chem. Soc., 77, 2804
|| +СН34-СН, = СН CH-=CHS
\//
N 2-или 3-изомер
(1955).
5. А. Д. Петров, E. Б. Соколова, Гао Чаи-лан, ЖОХ, 30, 1109
(I960).
6. Н. G i 1 m а п, F. W. М о г е, О. В a i n с, J. Amer. Chem. Soc., 63,
2479 (1941).
Поступила в октябре 1962 г. КПИ
/Ч
II +сна—сн2-сн=сн-сн3
N 2-или 3-изомер
/\ /сн2-сн=сн-сн3
II + сн
'ч/ хсн2-сн=сн-сн3
2- или 3-нзомер
2- и З-^-ПЕНТЕНИЛ) ПИРИДИНЫ
Ю. И. ЧУМАКОВ, В. М. ЛЕДОВСКИХ
х1/хсн2-сн1!-сн=сн -СН,
и /^CHj-CHg-CH^CH-CHa
и
N
C10HI3N М. в. 147,22
Изомерные 2- и З-(З'-пентенил)-пиридины используются
как исходные продукты в синтезах соответствующих изомер-
Характеристика основного сырья
Изомерные метилпиридины (см. примечание 1).
Бутадиен-1,3, 99%-ный.
Условия получения
1. Получение 2-(3'-пентенил)-пиридина и 2-(5'-нонадиен-
2’, 7'-ил)-пиридина. Во вращающийся автоклав из нержавею-
щей стали емкостью 300 мл помещают 49 мл (46,5 г; 0,5-Л4)
2-метилпиридина, 0,7 г металлического натрия, герметизируют
его и нагревают при перемешивании до 150° в течение 20—30
минут. Автоклав охлаждают последовательно водой, льдом и
твердой углекислотой с ацетоном, разбалчивают его и к полу-
ченному металлорганическому соединению (см. примечание 2)
приливают, в один прием, 41,5 мл (27 г; 0,5 М) сжиженного в
охладительной смеси —ацетон с твердой углекислотой—бу-
тадиена-1,3 (см. примечание 3). Автоклав быстро герметизи-
39
руют и нагревают в течение 20—30 минут до температуры
150—160°. Нагревание прекращают и дают автоклаву медлен-
но охлаждаться в течение 1 часа до температуры 100—110°.
Дальнейшее охлаждение производят вначале водой, затем
льдом. Автоклав разбалчивают, реакционную массу извлека-
ют отсасыванием в колбу Бунзена и осторожно обрабатывают
10 мл воды для разложения металлорганического соединения
(см. примечание 4). После отстаивания верхний слой отделя-
ют от нижнего, высушивают кипячением с едким кали в тече-
ние 2—3 часов с обратным холодильником (см. примечание 5).
Высушенный продукт декантируют или отфильтровывают и
промывают на фильтре 20 мл бензола. Бензол и 2-метилпири-
дин, не вступивший в реакцию, отгоняют при атмосферном
давлении, а остаток перегоняют в вакууме, получая 27,8 г
2-(З'-пентенил)-пиридина с температурой кипения 68—75°’при
4 мм, —1,5080 и 25,5 г 2-(5/-нопадиен-2/, 7'-ил)-пиридина
с температурой кипения 113—119° при 5—6 мм; —1,5118
(см. примечание 6). Общий выход 2-(З'-пентенил)-пиридина и
2-(5/-нонадиен-2/, 7'-ил)-пиридина составляет 63,3% от теоре-
тического без учета регенерации не вступившего в реакцию
2-метилпиридина (см. примечание 7).
2. Получение 3-(3'-пентенил)-пиридина и 3-(5'-нонадиен-
2', 7'-ил)-пиридина. Металлорганическое соединение получа-
ют из 47,4 мл (46,5 г, 0,5 Л4) 3-метилпиридина (см. примеча-
ние 1) и 0,7 г (0,03 г-атома) металлического натрия в усло-
виях, описанных выше. К полученному металлорганическому
соединению после охлаждения приливают 41,5 мл (27 а; 0,5 М)
сжиженного бутадиена-1,3. Автоклав герметизируют и нагре-
вают до 180° в течение 30 минут. Нагревание прекращают и
дают автоклаву медленно охлаждаться в течение 1 часа до
температуры НО—130°. Автоклав охлаждают вначале про-
точной водой, затем на ледяной бане. Извлеченные продукты
реакции обрабатывают согласно описанию, приведенному вы-
ше. Получают 13,2 г 3- (З'-пентенил) -пиридина с температурой
кипения 123—145° при 12 мм, п2$ —1,5133 и 8,2 г 3-(5'-но-
надиен-2', 7'-ил)-пиридина с температурой кипения 161 —170°
при 25 мм; п2® —1,5196. После повторной перегонки на рек-
тификационной колонке эффективностью 15—20 теоретичес-
ких тарелок З-(З'-пентенил)-пиридин кипит в пределах 97,6—
97,8° при 10 мм, т. пл. пикрата 59—60° (испр.), а 3-(5'-нона-
диен-2', 7'-ил)-пиридин — в пределах 132—133,2° при 9 мм.
Общий выход составляет 51,8% от теоретического с учетом
регенерации 3-метилпиридина (см. примечание 9).
Примечания.
1. Использовался 2-метилпиридин квалификации «чистый», вы-
сушенный кипячением в течение 4 часов с твердым едким кали и пе-
регнанный (первые порции погона отбрасывались). Использовавшие-
40
ся 3- и 4-метилпиридины были приготовлены согласно [3], тщатель-
но высушены и имели содержание 98—99%.
2. Образование металлоргапического соединения характеризу-
ется появлением интенсивного красно-бурого окрашивания.
3. Синтез можно проводить, загружая все исходные продукты
одновременно.
4. Воду необходимо прибавлять медленно, чтобы избежать бур-
ной реакции.
5. Для улучшения расслоения образующейся иногда эмульсии
к ней необходимо добавить небольшое количество твердого едкого
натра или едкого кали.
6. Перегонку можно проводить и при более высоком давлении.
По литературным данным, продукты конденсации 2-метилииридина и
бутадиена-1,3 имеют следующие константы: 2-(З'-пснтенил)-пири-
дин— т. кип. 93—94° при 12 мм или 70—80° при 5 мм; 2-(5'-нонади-
ен-2', 7'-ил)-пиридин — т. кип. 132—134° при 12 мм или ПО—114°
при 5 мм.
7. Выход может быть повышен за счет регенерации не вступив-
шего в реакцию 2-метилпириднна.
8. При разложении металлорганического соединения водой цвет
меняется от коричневого до зеленого.
9. Строение не описанного в литературе 3-(З'-пентеиил)-пири-
дина было доказано гидрированием его в 3 п. амнлпирндин н срав-
нением последнего с известным образцом. Аналогичная реакция кон-
денсации бутадиена в присутствии натрия имеет место также с 4-
метилпиридином.
ЛИТЕРАТУРА
1. R. W е g 1 е г, Q. Pieper, Chem., Вег., 83, 6 (1950).
2. Пат. ФРГ 831099; Chem. ZbL, 1952, 4815.
3. Ю. И. Чумаков, Сборник «Методы получения химических реак-
тивов и препаратов», ИРЕА, 4—5, 44, 50 (1963).
Поступила и октябре 1962 г. КПИ
СМЕСЬ ИЗОМЕРНЫХ ФЕНИЛПИРИДИНОВ
Ю. И. ЧУМАКОВ, Э. В. ЛУГОВСКОЙ
с6н5
Смесь фенилпиридинов используется как исходное сырье
для выделения индивидуальных изомеров фенилпиридина.
Последние представляют интерес для синтезов в ряду пириди-
на и как комплексообразующие реагенты.
Наиболее удобным путем приготовления смесей изомер-
ных фенилпиридинов является действие солей диазония на
пиридин — способ впервые предложенный Мёлау и Бергером
[1]. Позже этот способ был значительно усовершенствован в
работах А. Е. Чичибабина [2] и других авторов [3], благодаря
чему удалось существенно повысить выход и обеспечить бе-
зопасное проведение реакции. Мёлау н Бергер получали фе-
нилпиридины действием сухого хлористого фенилдиазония на
пиридин [1].
Другие способы получения смесей изомерных фенилпири-
динов используют разложение перекиси бензоила в пиридине
[4] или разложение 1-фенил-3,3-диметилтриазола в смеси пи-
ридина и бензола [5]. А. Е. Чичибабин и Панютин получили
одновременно 2- и 4-фенилпиридин при пропускании смеси
ацетальдегида и бензальдегида над окисью алюминия [6].
Нами было проверено получение смеси изомерных фенил-
пиридинов действием водного раствора хлористого фенилдиа-
зония на избыток пиридина, описанное группой английских
авторов [3].
СИНТЕЗ ИЗОМЕРНЫХ ФЕНИЛПИРИДИНОВ
. Ф
nh2 n---n
if ^1+2HCl+NaNO2 — |f C1 + NaC1 +H*°
• HC1
|| -j-QHs+NaOH >-|| +C6HB+NaCl+H2O
\//
N N
•HCi
Характеристика основного сырья
Пиридин, ГОСТ 2747—44.
Анилин, ГОСТ 313—41.
Нитрит натрия, ГОСТ 4197—48.
42
Условия получения
1. Получение хлористого фенилдиазония. В фарфоровом
стакане емкостью 2 л смешивают 580 мл воды и 220 г (185 мл)
соляной кислоты, уд. в. 1,19. В этой смеси растворяют 90 г
(0,97 44) свежеперегнанного анилина. Охлаждают стакан
смесью льда и соли до 0°, затем при интенсивном перемеши-
вании из капельной воронки медленно прибавляют раствор
66,8 г (0,97 Л4) нитрита натрия в 175 мл воды, следя, чтобы
температура не поднималась выше 10°. Полученный хлори-
стый фенилдиазоний необходимо держать во льду и исполь-
зовать немедленно во избежание разложения.
2. Получение смеси изомерных фенилпиридинов. В одно-
горлую круглодонную колбу емкостью 3 л помещают 900 мл
(884 г; 11,2 44) пиридина и при энергичном перемешивании
медленно, в течение 2 часов, прибавляют полученный ранее
раствор хлористого фенилдиазония (0,98 44) из воронки, ох-
лаждаемой смесью льда и соли. К концу прибавления реак-
ционная масса приобретает темную окраску. Далее смесь вы-
держивают на кипящей водяной бане 1 час с обратным холо-
дильником, охлаждают, после чего осторожно при охлажде-
нии и перемешивании обрабатывают 400 граммами твердого
едкого натра (см. примечание 1). Большую часть верхнего
слоя, содержащего смесь изомерных фенилпиридинов и не-
прореагировавший пиридин, декантируют, а остальную часть
отделяют на делительной воронке. Непрореагировавший пи-
ридин отгоняют на водяной бане в вакууме от водоструйного
насоса (т. кип. 35—40° при 28 мм) (см. примечание 2). Оста-
ток перегоняют в вакууме от масляного или водоструйного
насоса на коптящем пламени горелки с воздушным холодиль-
ником, собирая фракцию с т- кип. 120—130° при 5 мм или
145—185° при 17 мм. Выход смеси фенилпиридинов составляет
72,5 г, или 43,9%, считая на аналин (см. примечание 3).
Примечания.
1. Высаливание должно производиться тщательно, но количе-
ство едкого натра не должно превышать 400 г; в противном случае
наблюдается образование эмульсии, что делает невозможным раз-
деление.
2. Отгоняемый пиридин иногда окрашен в желтый цвет благо-
даря присутствию легко окисляющегося анилина. Отгоняемый пири-
дин, после определения содержания (титрованием) и добавления
недостающего количества пиридина, используется в следующих опы-
тах по получению фенилпиридинов. При этом не происходит сниже-
ния выхода. Попытка проведения синтеза с меньшим избытком пи-
ридина привела к резкому снижению выходов.
3. Твердый, спекшийся остаток на стенках перегонной колбы,
остающийся после перегонки, хорошо растворим в хлороформе.
Смесь фенилпиридинов представлеят собой светло-желтую, темнею-
щую при хранении жидкость. При стоянии из нее постепенно выкри-
сталлизовывается 4-фенилпиридин.
43
Согласно литературным данным, смесь фепилпиридинов, полу-
чаемая описанным путем, имеет температуру кипения 170—190° при
10—20 jhai и состоит из 55% 2-изомера, 22,5% 3-изомера и 22,5%
4-изомера [3].
ЛИТЕРАТУРА
1. R. Mohlau, R. Berger, Вег.. 26, 2003 (1893).
2. А. Е. Ч и ч и б а б и н, Вег., 37, 1373 (1904).
3. J. W. Н a w о г 1 h, J. М. Н е 11 b г о п, D Н. Hay, J. Chem. Soc.,
1940, 350.
4. R. Н. Pausacker, Austral. J. Chem., 11, 200 (1958); РЖхим, 1959,
6, реф. 19471.
5. J. Elks, D. H. Hey, J. Chem. Soc., 1943, 441.
6. A. E. Чичибабин, Панютни, ЖРФХО, 47, 711 (1904).
Поступила в октябре 1962 г. КПИ
М. в. 183, 27
2- и 4 (2х ФЕНИЛЭТИЛ) ПИРИДИНЫ
Ю. И. ЧУМАКОВ, Ю. П. ШАПОВАЛОВА, В. М. ЛЕДОВСКИХ
и
у\сн,сн,-/_)
C]3H13N
2- и 4- (2х-фенилэтил) -пиридины представляют интерес как
комплексообразующие реагенты и как исходные продукты для
синтезов в ряду пиридина.
Для получения 2- и 4- (2'-фенилэтил) -пиридинов обычно
использовалось взаимодействие 2- или 4-метилпиридина с
2-фенилэтилгалогенидами в присутствии амидов щелочных ме-
таллов в жидком аммиаке [1]. Аналогично протекает реакция
между литийорганическим производным в случае 2-метилпи-
ридина, которая приводит к получению 2-(2х-фенилэтил)-пи-
ридина [2].
Недавно был предложен новый синтез этих соединений,
заключающийся в действии смеси бензилового спирта и едко-
го кали на 2- и 4-метилпиридины [3]. В основу приведенной
ниже методики положен этот способ.
44
СИНТЕЗ 2- и 4 (2' ФЕНИЛЭТИЛ)-ПИРИДИНОВ
СН2ОН
I
/Ч кон
11^ сн3 +IM------------> 11^ +снаснэ + на0
N N
2-или 4-изомер 2-или 4-изомер
Характеристика исходного сырья
2- и 4-Метилпиридины, см. примечание 1 к ст. «2- и 3-(3Х-
Пентенил) -пиридины».
Бензиловый спирт, ГОСТ 8751—58.
Условия получения
1. Получение 2-(2'-фенилэтил)-пиридина. В круглодонную
колбу емкостью 250 мл, снабженную ловушкой Дина и Стар-
ка с обратным холодильником, загружают НО мл (114,4 г;
1,06 М ) бензилового спирта и 11,2 г (0,2 М) гранулирован-
ного едкого кали. Содержимое колбы кипятят до прекраще-
ния отделения воды в ловушке (см. примечание 1). Охлажда-
ют, прибавляют 9,5 мл (9,3 г; 0,1 М) 2-метилпиридина и ки-
пятят с обратным холодильником в течение 30 часов. Реак-
ционную смесь охлаждают и добавляют 60 мл воды и 320 мл
эфира. Эфирный раствор отделяют и промывают два раза во-
дой по 50 мл. Водные растворы объединяют и экстрагируют
130 мл эфира. Полученные эфирные экстракты высушивают
над безводным сульфатом натрия и фильтруют на складчатом
фильтре. Эфир отгоняют, а остаток подвергают вакуум-пере-
гонке. После отгонки не вступивших в реакцию 2-метилпири-
дина и бензилового спирта получают 9,5 г 2-(2,-фенилэтил)-
пиридина с т. кип. 163—164° при 25—27 мм; Ид —1,5566,
т. пл. пикрата 130—131° (см. примечание 2); выход 52% от
теоретического без учета регенерации 2-метилпиридина (см,
примечание 3).
2. Получение 4-(2'-фенилэтил)-пиридина. Обезвоженный
раствор бензилата калия в бензиловом спирте приготовляют
из НО мл (114,4 г; 1,06 М) бензилового спирта и 11,2 г (0,2 Л4)
едкого кали, как описано выше. Прибавляют 9,5 мл (9,3 г;
0,1 М) 4-метилпиридина и кипятят с обратным холодильни-
ком в течение 4 часов. Полученную смесь перегоняют, соби-
рая фракцию с т. кип. 95—145°, представляющую собой не
вступивший в реакцию 4-метилпиридин. Остаток обрабатыва-
ют водой и эфиром, как описано выше. После отгонки эфира
реакционную массу подвергают перегонке, получая 7,2 г
4-(2'-фенилэтил)-пиридина с т. кип. 158—163° при 13 мм (см.
45
примечание 4). 4-(2'-Фенилэтил)-пиридин представляет собой
белую кристаллическую массу с т. пл. 63—64,5° (см. примеча-
ние 5). Выход продукта составляет 39,4% от теоретического
без учета регенерации 4-метилпиридина, не вступившего в ре-
акцию (см. примечание 3).
Примечания.
1. После удаления всей воды отгоняют дополнительно 10 мл
бензилового спирта, чтобы обеспечить полное обезвоживание.
2. По литературным данным, 2- (2'-фенилэтил)-пиридин имеет
константы: т. кип. 289,5° при 766 мм; d,’’"— 1,0465; т. кип. 120—125°
при 2 мм; т. пл. пикрата 124—125,4° [2]; т. кип. 164—165° при 25 мм,
145—146° при 10 мм; т. пл. пикрата 128,5—130° [3].
3. Выход может быть повышен за счет регенерации не вступив-
шего в реакцию исходного пиридинового основания.
4. При перегонке, для предотвращения кристаллизации 4-(2'-
феннлэтил)-пиридина, используют холодильник с обогревом.
5. По литературным данным, 4-(2'-фенилэтил)-пиридин имеет
константы: т. пл. 65° (из спирта) [5], 69—71° [6], 70—70,8° [3]; т. пл.
пикрата 162—163° [5, 6].
ЛИТЕРАТУРА
1. F. W. Bergstro m, G. R. Norton, R. A. Seibert, J. Org.
Chem., 5, 1607 (1936).
2. С. О s и с h, R. Levine, J. Amer. Chem. Soc., 78, 1723 (1956).
3. M. Avramov, I. Sprinzak, J. Amer. Chem. Soc., 78, 4090
(1956).
4. H. В a u r a t h, Ber., 21, 821 (1888).
5. B. Fells, Ber., 37, 2147 (1904).
6. к. Friedlander, Ber., 38, 2837 (1905).
Поступила в октябре 1962 г. КПИ
2-, 3- и 4-(3' ФЕНИЛПРОПИЛ)-ПИРИДИНЫ
Ю. И. ЧУМАКОВ, В. М. ЛЕДОВСКИХ.
-СН2-СН2-СН2-"Х//1
||/Ч[ j|/4|-сн2—сн2-сн2-^|
\// \s
сн2-сн2-сн2-
c14h15n
М. в. 197, 28
Изомерные 2-, 3- и 4- (З'-фенилпропил)-пиридины являют-
ся комплексообразующими реагентами, а также служат ис-
ходными продуктами для синтеза производных ряда пири-
дина.
2-(З'-фенилпропил)-пиридин был впервые синтезирован
А. Е. Чичибабиным действием Р-фенилэтилбромида на 2-ме-
тилпиридин в присутствии амида натрия [1]. Бергстром ана-
логичным способом получил 4-(З'-фенилпропил)-пиридин [2].
В 1950 г. Веглер и Пипер предложили новый способ получе-
ния 2-(З'-фенилпропил)-пиридина конденсацией 2-метилпири-
дина н стирола в присутствии металлического натрия. Способ
является более удобным в препаративном отношении и дает
хорошие выходы продукта [3, 4].
Нами было проверено получение этим способом 2-(3'-фе-
пилпропил)-пиридина. Было показано, что таким путем могут
быть получены также 3- и 4-(З'-фенилпропил)-пиридины, при-
чем вместо металлического натрия могут быть использованы
металлический калий или амид натрия.
СИНТЕЗ 2-,3- И 4-(3'-ФЕНИЛПРОПИЛ)-ПИРИДИНОВ
/"''Г /=s. (Na)
II +сНз + снг=сн-4
. х// \\_/
N 2-, 3- или 4-изомер
-> II +СН2-СН9-СН2-^
\// ~
N
2-, 3- или 4-изомер
/\\ /СН2-СН2^_^
| +СН __
у сн„сн,-(_у
2-, 3-или 4-изомер
Характеристика основного сырья
Изомерные метилпиридины (см. примечание 1).
Стирол, ВТУ ЛУ 47- 53.
Условия получения
1. Получение 2-(3'-фенилпропил)-пиридина. В трехгорлую
колбу на шлифах емкостью 100 мл, снабженную обратным
холодильником, мешалкой, пропущенной через резиновый об-
47
тюратор, капельной воронкой, термометром и трубкой для
ввода азота, помещают 19,5 мл (18,6 г; 0,2 44) 2-метилпири-
дина и 0,2—0,3 г металлического натрия (см. примечание 2).
Содержимое колбы нагревают при перемешивании в токе
азота при 130° в течение 0,5—1 часа до появления интенсив-
ного красно-бурого окрашивания (натрийорганическое произ-
водное 2-метилпиридина), после чего прибавляют 23 мл (20,8 г;
0,2 44) стирола. Реакционную массу нагревают 2 часа при
130°. Охлаждают и осторожно, при перемешивании, прибав-
ляют 4 мл воды для разложения металлорганического соеди-
нения (см. примечание 3). Дают отстояться, после чего верх-
ний слой отделяют, а нижний, водный слой, экстрагируют
5—8 мл бензола (см. примечание 4). Бензольную вытяжку
присоединяют к основной массе продуктов реакции и высуши-
вают кипячением с гранулированным едким кали в течение
2 часов. Охлаждают, фильтруют на шоттовском фильтре и
промывают 5 мл бензола. Бензол отгоняют, а остаток пере-
гоняют в вакууме, получая 15,3 г 2-(З'-фенилпропил)-пириди-
на с температурой кипения 169,6—177° при 15—16 мм;
п2£ —1,5585 и 11,0 г 1,5-дифенил-3-(2-пиридил)-пентана с
температурой кипения 217—225° при 4—5 мм; —1,5773.
Общий выход составляет 56,8% от теоретического без учета
регенерации не вступившего в реакцию 2-метилпиридина.
2. Получение 3-(З'-фенилпропил)-пиридина. В прибор, опи-
санный выше, помещают 9,5 мл (9,3 г; 0,1 М) 3-метилпириди-
на (см. примечание 1) и 0,3 г металлического натрия. Содер-
жимое колбы нагревают при перемешивании в токе азота до
температуры 145°, после чего прибавляют в течение 5—10 ми-
нут 11,5 мл (10,4 г; 0,1 44) свежеперегнанного стирола. Реак-
ционную массу по прибавлении нагревают еще 2 часа при
температуре кипения 3-метилпиридина. Дальнейшую обра-
ботку реакционной массы производят аналогично описанному
выше. Получают 3,3 г 3-(З'-фенилпропил)-пиридина с темпе-
ратурой кипения 185—190° при 14—15 мм и 3,75 г фракции с
температурой кипения 250—265° при 11 —12 мм (см. приме-
чание 5). При повторной перегонке 3-(З'-фенилпропил)-пири-
дин кипит в пределах 123,7—127° при 2 мм; п™ — 1,5634;
d^0—1,0302. Общий выход, с учетом регенерации 3-метилпи-
ридина, составляет 40,9% от теоретического.
3. Получение 4-(3'-фенилпропил)-пиридина. В прибор, опи-
санный выше, помещают 9,5 мл (9,3 г; 0,1 44) 4-метилпириди-
на (см. примечание 1) и 0,2—0,3 г металлического натрия.
Содержимое колбы нагревают при перемешивании в токе азо-
та до 125°. К полученному металлорганическому соединению
прибавляют в течение 5—10 минут 11,5 мл (10,4 г; 0,1 44) сти-
рола. Нагревают еще 40 минут при 125°. Дальнейшую обра-
ботку ведут, как описано выше. Получают 5,8 г 4-(3'-фенил-
48
пропил)-пиридина с температурой кипения 185—187° при
18 мм, т. пл. пикрата 142—143° (испр.); п2® —1,5613;
du20— 1,0224 и 7,4 г 1,5-дифенил-З-(4-пиридил)-пентана с тем-
пературой кипения 260—270° при 18 мм, п™ —1,5816. После
очистки через хлоргидрат 1,5-дифенил-З-(4-пиридил)-пропан
кипит при 215—218° при 4 мм; п2£ —1,5810, d42n— 1,0450.
Примечания.
1. О 2-, 3- и 4-метилпиридинах, вводившихся в реакцию, см. при-
мечание 1 к статье «2- и 3-(3/-Пентенил)-пиридины».
2. Колбу предварительно тщательно высушивают нагреванием
до 120—150° в токе азота, пропускание которого ие прекращается и
при ее охлаждении.
3. Во избежание излишнего разогревания воду следует при-
бавлять постепенно.
4. Для улучшения расслоения реакционную массу подщелачи-
вают небольшим количеством едкого кали.
5. По всей вероятности, эта фракция представляет собой за-
грязненный 1,5-дифенил-3-(З-пиридил)-пеитан.
ЛИТЕРАТУРА
1. А. Е. Чичи бабин, Bull. Soc. chim. Fr., 5, 1607 (1936). •
2. F. W. В c r g s t г о ш, G. R. Norton, R. A. Seibert, J. Org.
Chem., 10, 452 (1945).
3. R. W e g 1 e r, G. Pieper, Ber., 83, 6 (1950).
4. R. W e g 1 e r, G. Pieper, Пат. ФРГ 831099, 1952; Chem. Zbl., 123,
4815 (1952).
Поступила в октябре 196'2 г. КПП
ИЗОХИНОЛИН
(3,4-бензопиридин)
IO. И. ЧУМАКОВ, 3. П. ВАСИЛЬЕВА
c9h7n
М. в. 129,16
Изохинолин является исходным веществом в синтезах фо-
тосенсибилизаторов, цинхомеровой кислоты и некоторых дру-
гих препаратов.
Источником изохинолина служит хинолин-изохинолиновая
фракция азотсодержащих оснований, выделяемая из камен-
4 Зак. 576 4 9
ноугольной смолы и выкипающая в пределах 230—245°. Син-
тетические методы получения изохинолина сравнительно
сложны и могут иметь лишь ограниченное применение.
Основную трудность выделения изохинолина из хинолин-
изохинолиновой фракции представляет близость свойств обо-
их компонентов. Разница в температурах кипения между ними
составляет 3—4°, вследствие чего разделение их прямой рек-
тификацией неосуществимо. Для выделения индивидуального
изохинолина из хинолин-изохинолиновой фракции было пред-
ложено использовать осаждение и последующую дробную
кристаллизацию сульфатов из спирта [1—3], осаждение суль-
фата в ледяной уксусной кислоте [4], осаждение хлоргидра-
та в безводной среде [5], разделение TV-окисей хинолина и
изохинолина [6], продуктов присоединения с фенолом и р-наф-
толом [7], осаждение продуктов присоединения с хлористым
кальцием [8], азеотропную ректификацию с этиленгликолем
[9], комплексообразование с роданидом никеля [10] и ряд дру-
гих методов.
Ниже приведен способ выделения чистого изохинолина из
хинолин-изохинолиновой фракции с применением реакции
комплексообразования с хлористым кобальтом. Нами было
найдено, что изохинолин, в противоположность хинолину, об-
разует более устойчивые комплексные соединения с целым ря-
дом солей тяжелых металлов, в том числе с хлористым ко-
бальтом, никелем или железом. Эта особенность позволяет
производить как грубое разделение хинолина и изохинолина,
так и эффективную очистку последнего.
ВЫДЕЛЕНИЕ ИЗОХИНОЛИНА
+ 2NaOH 4
| || + Со(ОН)а +• 2NaCl
\\/
50
Характеристика основного сырья
Хинолин-изохинолиновая фракция, т. кип, 230—238°.
Хлористый кобальт, ГОСТ 4528—48.
Условия получения
В колбу для перегонки с паром помещают раствор 25 мл
(46 г; 0,47 Л1) серной кислоты уд. в. 1,83—1,84 в 50 мл воды
и при охлаждении и перемешивании постепенно прибавляют
75 г хинолин-изохинолиновой фракции (0,58 М изомеров, см.
примечание 1). Содержимое колбы перегоняют с водяным па-
ром до объема дистиллата 800—1000 мл (см. примечание 2).
Остаток в колбе для перегонки охлаждают и при перемеши-
вании и охлаждении прибавляют раствор 80—100 г едкого
натра в 100—120 мл воды. Выделившуюся хинолин-изохино-
линовую фракцию, освобожденную от примесей углеводоро-
дов, дополнительно высушивают в течение 12—16 часов над
10- 15 г плавленого едкого натра или едкого кали (см. при-
мечание 3). Получают около 60 г хинолин-изохинолиновой
фракции, свободной от углеводородных примесей и содержа-
щей небольшое количество воды (см. примечание 4).
В трехгорлую колбу емкостью 250 мл, снабженную эффек-
тивной мешалкой, пропущенной через холодильник, термомет-
ром и капельной воронкой, помещают 4 мл 36%-ной соляной
кислоты (1,7 г 100%-ной НС1; 0,047 (И) (см. примечание 5) и
13,4 г (0,056 Л1) хлористого кобальта (см. примечание 6). При
перемешивании из капельной воронки постепенно прибавляют
60 г хинолин-изохинолиновой фракции, свободной от примесей
углеводородного характера и содержащей примерно 60% изо-
хинолина (36 а; 0,28 М), остальное—хинолин и хинальдин. За
счет тепла реакций нейтрализации и комплексообразования
температура реакционной массы повышается до 40—60°. Пос-
ле прибавления смеси оснований содержимое колбы при раз-
мешивании быстро доводят до интенсивного кипения, после
чего убирают нагрев и реакционной массе дают постепенно
охладиться при непрерывном перемешивании до 10—15°
(1—2 часа; к концу охлаждение ведут холодной водой). При
этой температуре реакционную массу размешивают дополни-
тельно 15 минут. Выпавший осадок комплекса изохинолина с
хлористым кобальтом отфильтровывают на воронке Бюхнера,
тщательно отжимают и на фильтре дважды промывают по
15—20 мл метанола (см. примечание 7). Получают 40 г хло-
рида тстраизохинолинкобальта в виде слегка смоченного
метанолом порошка розового цвета. Его перекристаллизовы-
вают из 140 мл ацетона. Получают 28,8 г очищенного комп-
лекса. Из ацетонового маточника после упаривания до ’/з
первоначального объема получают дополнительно 7,2 г про-
4*
51
дукта, который присоединяют к основному количеству ком-
плекса. Перекристаллизованный комплекс вносят в колбу с
40 мл 20%-ного водного раствора едкого натра и перегоняют
с перегретым паром до отсутствия изохинолина в погоне (см.
примечания 2 и 8). Дистиллат при охлаждении и перемешива-
нии обрабатывают твердым едким натром из расчета 20 г ед-
кого натра на каждые 100 мл. Выделившийся изохинолин от-
деляют от нижнего слоя и высушивают кипячением с плав-
леным едким кали или едким натром, из расчета 1 части ще-
лочи на 5 частей основания, в течение 3—4 часов с обратным
холодильником. Получают 20' г изохинолина, который оконча-
тельно очищают ректификацией на колонке в вакууме (см.
примечание 9), получая 18 г изохинолина с температурой ки-
пения 121 —121,Г при 22 мм. Температура кристаллизации
25,0—25,3° (см. примечание 10), температура плавления пи-
крата 228,5—229° (испр.). При хроматографировании па бу-
маге, импрегнированной хлорной медью в токе гептана или
петролейного эфира с т. кип. 75—100°, полученный изохино-
лин дает зону чистого голубого цвета (см. примечание 11).
Выход изохинолина составляет 60%, считая на изохинолин,
содержащийся в хинолин-изохинолиновой фракции, свобод-
ной от углеводородов.
Примечания.
1. Использовалась хинолин-изохинолнновая фракция коксохи-
мического происхождения с т. кип. 230— 238° и содержанием изохи-
нолнна 60%. Для быстрого определения процентного содержания изо-
хинолина в хинолин-изохинолиновой фракции использовалась хро-
матография медных комплексов на бумаге. Анализ заключается в
следующем.
Приготовление бумаги, импрегнированной хлорной медью. Бы-
стро фильтрующую хроматографическую бумагу замачивают в кю-
вете с раствором следующего состава: дистиллированной воды —
1000 мл, глицерина—150 мл, хлорной меди (СнСЬ . 2Н,О)—50 г,
после чего тщательно отжимают между листами фильтровальной
бумаги и высушивают на воздухе в течение 3—4 часов. Приготов-
ленная таким образом бумага по сравнению с неимпрегнированной
имеет голубоватый оттенок.
Методика хроматографирования. Хроматографирование произ-
водят на по.юсках импрегнированной бумаги размером 1,5—2 на
10—15 см в восходящем или нисходящем токе проявляющего рас-
творителя. Прибор — цилиндр со стеклянным крючком-держателем,
пропущенным через корковую пробку. Каплю исследуемой смеси ос-
нований наносят на расстоянии 1,5—2 см от нижнего края полоски
капилляром с оттянутым концом, диаметр которого около 0,5—1 мм,
после чего полоску погружают нижним концом на 0,5—1 см в эфир,
налитый слоем 3—5 см в цилиндр. Время проявления подбирают для
каждого индивидуального вещества в отдельности. Обычно для ка-
чественного анализа достаточно 3—5 минут, для количественного
анализа время удлиняют с целью более четкого разделения зон.
В большинстве случаев достаточно 15—30 минут. В качестве прояв-
ляющего растворителя используют петролейцый эфир с т. кип. 70—
100°; возможно также использование диэтилового эфира, н-октана,
н-гептана и подобных им углеводородных растворителей. Хромато-
52
граммы обычно имеют вид удлиненных, переходящих один в другой
и окрашенных в яркие цвета, языков. Зоны переходят одна в дру-
гую и наблюдаются визуально непосредственно после проявления
(см. рис. 1).
Количественный анализ искусственных смесей хинолина и изо-
хинолина производят с помощью графика зависимости отношения
длины зоны изохинолина к длине всей хроматограммы от его про-
центного содержании в смеси, используя отношение
К— -10°,
Ч 1 ‘2
длин
изо-
Рис. 1. Хроматограмма
смеси хинолина и изохи-
нолина (1:1):
1 — зона вымывания;
2 зона изохинолина (го-
лубая); зона хиноли-
на (коричневато-зеленая)
где /| — длина зоны изохинслика;
/з — длина зоны хинолина.
График зависимости отношения
зон К от процентного содержания
хинолина в смеси, построенный с по-
мощью искусственных смесей, представ-
лен на рис. 2.
Изохинолин и другие алкил-, арил-,
аралкил- или бензониридины, не имею-
щие заместителей в положениях 2 или
6, дают зону голубой окраски в проти-
воположность их 2-нзомсрам, которые
дают зону коричневато-зеленой (.хаки*)
или лиловой окраски, передвигающуюся
быстрее голубой зоны, соответствующей
3- или 4-изомерам.
2. Использовался пар, перегретый
до температуры 330—380° (измерено
термопарой). Колба дополнительно по-
догревалась газовой горелкой на сетке.
3. Описанный прием имеет целью
освободить исходную фракцию от при-
месей неосновного характера, прежде
всего нафталина.
4. Имеют место существенные поте-
ри хинолин-изохинолиновой фракции за
счет того, что при перегонке с паром,
несмотря на избыток серной кислоты,
происходит частичный гидролиз суль-
фатов хинолиния и изохинолиния. Отог-
нанная с водяным паром смесь хиноли-
на и изохинолина может быть выделена
насыщенным едким натром. Эта смесь содержит несколько большие
количества углеводородов, от которых она может быть освобож-
дена таким же приемом.
5. Соляная кислота берется в количестве, достаточном для свя-
зывания примерно 25% хинолина, присутствующего в исходной хи-
нолин-изохинолиновой фракции,
6. Хлористый кобальт (СоСМНаО) берется в количестве, не-
обходимом для вступления в реакцию комплексообразования
80%-ного изохинолкна, присутствующего в исходной хинолин-изохи-
нолиновой фракции.
7. Промывку производят следующим образом: отсоединяют ва-
куум, смачивают растворителем осадок, осторожно разминая его
шпателем, после чего дают постоять 5—10 минут до появления из
воронки Бюхнера первых капель фильтрата. Соединяют с вакуумом
и фильтруют, тщательно отжимая осадок на фильтре. Небрежная
53
dflOflKrmoepctffic/poooAAfe етесес/ тиомна г/
иЗОХсмалиИСГ.
Завиеитость X от cotfe/xwmdtM изохинолшю.
Рис. 2.
промывка не позволяет освободиться от примесей хинолина, увле-
ченных осадком хлорида тетраизохинолин-кобальта.
8. Проба на конец отгонки заключается в прибавлении 0,3—0,5 г
едкого натра к 2—3 мл дистиллата. Не должно появляться масля-
нистых капель.
9. Использовалась ректификационная колонка, имеющая диаметр
ректифицирующей части 9 мм, высоту 40 см, заполненная насадкой
Левина (трехгранная спираль размером 1,5—2 мм из нихромовой
проволоки диаметром 0,1—0,2 мм), эквивалентная примерно 18—20
теоретическим тарелкам.
10. Температура кристаллизации определялась в пробирке со
стандартным шлифом 14,5, снабженной укороченным термометром,
пропущенным через резиновую трубку, надетую на короткий шлиф-
керн, закрывающий пробирку. Такое устройство ограничивает доступ
влаги из воздуха, так как она может сильно исказить результаты
анализа.
11. Согласно литературным данным, изохинолин имеет констан-
ты: т. кип. 107,5—109° при 13 мм [11]; 243,25° при 760 мм [15]; темп,
кристаллизации 24—25° [13]; 24,5—26° [11], 26,48° [15]; т. пл. пикрата
220—222° [14], 227—229° [12]. По-видимому, наиболее достоверными
являются следующие данные, т. кип. 243,25° при 760 мм; температура
кристаллизации 26,48° [15]; т. п.т. пикрата 227—229° [12].
Несколько менее чистый препарат изохинолина, пригодный для
большинства целей, может быть получен значительно проще, если
опустить операцию отделения углеводородов отгонкой с водяным
паром и перекристаллизации комплекса с хлористым кобальтом из
ацетона. Температура кристаллизации такого изохинолина 23,8—24"
и температура плавления пикрата 228—229° (испр.).
54
ЛИТЕРАТУРА
1. R. Weisberger, Ber., 47, 3175 (1914).
2. M. M. Поташников, Авт. свид. 77953, 1949.
3. M. М. Поташников, П. Н. Горелов, ЖПХ, 30, 482, 634
(1957).
4. Пат. США 2618639; Chem. Zbl., 1954, 3572.
5. Англ. пат. 588277; Chem. Abstrs, 41, 6583 (1947).
6. Яп. пат. 3326; Chem. Abstrs, 50, 1092 (1950).
7. Пат. США 2432065; Chem. Abstrs, 42. 1971 (1948).
8. Пат. США 2391270, 1945; Chem. Abstrs, 40, 2474 (1946).
9. Н. Д. Р у с ь я н о в а. М. В. Г о ф т м а и. Научи, докл. Высш,
школы, 2, 376 (1959).
10. G. Т а г t а г i n 1, A. F. Sam aj a, Ann. Chim. Appl., 23, 351 (1933);
Chem. Zbl., 1933, II, 3279.
11. E. Schmitz, Chem. Ber., 94, 1488 (1958),
12. С. 1. В г о d r i c k, W. F. S h о r t, J. Chem. Soc., 1949, 2587.
13. Пат. США 2483420; Chem. Zbl., 1950, II, 821.
14. F. W. Bergstrom, J. H. R о d d a, J. Amer. Chem. Soc., 62,
3030 (1940); Chem. Zbl., 1941, 1, 1676.
15. H. Preiser, W. Q 1 о w a c k i, J. Amer. Chem. Soc., 70, 2575 (1948).
Поступила в октябре 1962 г. КПИ
1,3-ДИ-(2-ПИРИДИЛ)-ПРОПАН
(ди-а-пиколилметан)
Ю. И. ЧУМАКОВ, В. М. ЛЕДОВСКИХ, Р. Е. ЛОХОВ, В. А. РАЛКО
/Ч
^-сн2-сн2-сн2
N
C18HuN2 М. в. 198,27
1,3-Ди-(2-пиридил)-пропан является комплексообразую-
щим реагентом и служит исходным продуктом в ситезах неко-
торых производных ряда пиридина.
Единственный описанный синтез 1,3-ди-(2-пиридил)-про-
пана заключается во взаимодействии 2-винилпиридина с 2-ме-
тилпиридином в присутствии металлического натрия. Нами
был проверен этот способ [1].
СИНТЕЗ 1,3-ДИ-(2-ПИРИДИЛ)-ПРОПАНА
+
сн2=сн
СН2—СН2-СН
Характеристика основного сырья
2-Винилпиридин, см. примечание 1.
2-Метилпиридин, ЧМТУ 5044—55.
Натрий, ГОСТ 3273—55.
Условия получения
В трехгорлую колбу емкостью 250 мл, снабженную мешал-
кой, капельной воронкой, термометром, обратным холодиль-
ником, защищенным хлоркальциевой трубкой и трубкой для
ввода инертного газа, помещают 24,5 мл (23,2 г, 0,25 М)
2-метилпиридина (см. примечание 2) и в течение 20—30 минут
пропускают высушенный инертный газ (ом. примечание 3).
К 2-метилпиридину прибавляют 0,4 г мелконарезанного
металлического натрия и кипятят с обратным холодильником
56
в течение 1 —1,5 часа (см. примечание 4). К реакционной мас-
се, не охлаждая ее, постепенно, в течение 30—45 минут при-
бавляют 26,5 мл (26,5 г; 0,25 Л4) 2-винилпиридина (см. при-
мечание 1) и кипятят с обратным холодильником в течение
2 часов. Содержимое колбы охлаждают и при энергичном
перемешивании разлагают натрийоргаиическое производное
прибавлением 2—3 мл воды. Отделяют нижний водно-щелоч-
ной слой, а верхний высушивают плавленым едким кали ки-
пячением с обратным холодильником. Высушенный продукт
реакции перегоняют вначале при атмосферном давлении, со-
бирая фракцию с температурой кипения до 130°. Остаток пе-
регоняют в вакууме, получая 9 г 1,3-ди-(2-пиридил) -пропана
с температурой кипения 178—18Г при 16 мм (см. примеча-
ние 5); йр —1,5605; т. плавл. дипикрата 206—207° (испр.).
Выход 18,3% от теоретического (см. примечание 6).
Примечания.
1. Использовался высушенный, свежеперегнанный 2-винилпири-
дин, полученный дегидратацией 2-({1-оксиэтил)-пиридина и имевший
температуру кипения 79—82° при 29 мм.
2. В реакцию вводился свежеперегнанный 2-метилпиридин, пред-
варительно высушенный плавленым едким кали.
3. Реакцию проводят в токе высушенного метана из газовой се-
ти или азота. В качестве затвора использовался резиновый обтюра-
тор.
4. Реакционная масса приобретает темио-коричневую окраску
(2-пиколилнатрий).
5. В колбе Кляйзена остается значительное количество вышеки-
пящих соединений, по-видимому, представляющих собой продукт
конденсации двух молекул 2 випилпиридина с 2-метилпиридином.
6. Дипикрат был получен согласно примечанию 4 к статье.
«2-Метилпнрчдин» и перед определением температуры плавления не
перекристаллизовывался.
Согласно литературным данным, 1,3-ди-(2-пиридил) -пропан име-
ет константы: т. кип. 117° при 0,5 мм; —1,5607. Дипикрат име-
ет т. пл. 208—209° (после перекристаллизации из спирта) [1].
ЛИТЕРАТУРА
1. N. J. Leonard, J. Н. Bayer, J. Amer. Chem. Soc., 72, 4818 (1950).
Поступила в октябре 1962 г. КПП
Л'-ОКИСИ АЛКИЛПИРИДИНОВ
(У-окиси 2-, 3-, 4-метил- и 2,6-диметилпиридина)
10. И. ЧУМАКОВ
C6H,NO
М. в. 119,12
СНз/Х/ХСН3
О
C7H8NO М. в. 123,15
АДОкиси алкилпиридинов являются ключевыми исходными
соединениями в синтезах соответствующих им окси-, нитро- и
галоидзамещенных производных ряда пиридина,, а также пи-
ридина льдегидов.
Обычно используемым методом получения А-окисей ал-
килпиридинов является предложенное Охиаи длительное на-
гревание соответствующих алкилпиридинов с пергидролем в
присутствии большого избытка уксусной кислоты [1].
Ниже описано приготовление /V-окисей алкилпиридинов
действием уксусного ангидрида на смесь алкилпиридина и
пергидроля, ранее предложенное нами для получения JV-окиси
пиридина [2]. Этот путь-имеет своим преимуществом более ко-
роткое время реакции (1 —1,5 часа) *.
СИНТЕЗ Л'-ОКИСЕЙ 2-, 3-, 4-МЕТИЛ- И 2,В-ДИМЕТИЛ ПИРИДИНА
/X
II +СН3+Н2О2+(СН3СО)2О ->
II +СН3+СН3СООН
2-, 3- или 4-изомер
о
2-, 3-или 4-изомер
/X
|| 4-сн3 ->
гГ
•СН3СООН
2-, 3- или 4-изомер
II +СН3+СН3СООН
I
о
2-, 3- или 4-изомер
* В экспериментальной работе принимали участие: 3. А1. Корсакова,
3. Е. Столяров и В. В. Новикова.
58
II I +Н3О2+(СН3СО)2О - II | 4-снэсоон
СН3/Х^\СН3 СН3/Х''Г>^' 'СН3
ф
О-СН3СООН
/'V,
II | -> II I 4-сн3соон
ch3/W\Ch3 сн3/х^\сн3
I г
О-СН3СООН о
Характеристика основного сырья
Алкилпиридины, см. примечание 1.
Перекись водорода, 27—30%-ная, ГОСТ 177—55.
Уксусный ангидрид, ГОСТ 787—55.
Условия получения
Алкилпиридин (3 М) (см. примечание 1) помещают в
трехгорлую колбу емкостью 1 л, снабженную мешалкой, про-
пущенной через шариковый холодильник, термометром и ка-
пельной воронкой, и при перемешивании прибавляют 363 мл
(399 г) 27,5%-ного пергидроля (3,24 г-моля, причем темпера-
тура поднимается до 30—40°. Затем постепенно, при энергич-
ном перемешивании, начинают прибавлять 306 мл (330 г,
3,24 М) уксусного ангидрида. Температура за счет тепла ре-
акции быстро поднимается до 70°. Прибавление ведут с такой
скоростью, чтобы температура держалась в интервале 70—
75°. После прибавления уксусного ангидрида реакционную
массу выдерживают в течение 1 часа на кипящей водяной ба-
не, затем по каплям прибавляют 10 мл 40%-ного водного фор-
малина (см. примечание 2), продолжают нагревание на ки-
пящей водяной бане дополнительно в течение 1 часа. Полу-
ченный уксуснокислый раствор А-окиси алкилпиридина упа-
ривают на кипящей водяной бане в вакууме 20—60 мм (см.
примечание 3). Остаток перегоняют при 1—3 мм в вакууме
от масляного насоса (см. примечание 4). Вначале постепенно
отгоняется уксусная кислота, которую отдает при термической
диссоциации ацетат //-окиси алкилпиридина, затем перегоня-
ется А-окнсь алкилпиридина (см. примечание 5).
Средние выходы А-окисей алкилпиридинов составляют
70—80%. Из объединенных предгонов и кубовых остатков
может быть получено дополнительно до 15—20% выхода.
Получаемые А-окиси алкилпиридинов имеют константы:
59
/V-окись 2-метилпиридина — т. кип. 120—125° при 14 мм;
nf —1,5910;
JV-окись 3-метилпиридина— т. кип. 150—153° при 20 мм;
jV-окись 4-метилпиридина—т. пл. 183,5—184°;
.iV-окись 2,6-диметилпиридина—т. кип. 120—123° при21лш.
Примечания.
1. Использовался 2-метилпиридин квалификации «чистый». 3-.
4-Метил- и 2,6-диметилпиридины были приготовлены согласно [4] и
имели содержание 98—99% [2].
2. Формальдегид прибавляют для разрушения избытка переки-
си водорода, не вступившей в реакцию.
3. Внимание! Прежде чем начать перегонку в вакууме, необхо-
димо убедиться в отсутствии пепрореагировавшей перекиси водоро-
да, для чего производят анализ с йодистым калием и крахмалом. В
отсутствие перекисей раствор не изменит окраску.
4. В случае если используется масляный вакуум-насос, между
прибором для перегонки и вакуумным насосом необходимо помес-
тить две ловушки, охлаждаемые смесью метанол—твердая углекис-
лота, емкостью каждая не менее 100—150 мл. Ловушки должны
иметь широкие вводные трубки, иначе возможна закупорка их твер-
дой уксусной кислотой. В ловушках собирается уксусная кислота,
образующаяся при термической диссоциации А-окисей алкилпири-
динов.
5. А'-Окись 4-метилпирндина является кристаллическим веще-
ством и в этом случае вместо перегонки в вакууме используют пе-
рекристаллизацию из этилацетата или бензола остатка, полученного
после отгонки от реакционной массы в вакууме большей части из-
бытка уксусной кислоты.
6. Согласно литературным данным, Л'’-окиси алкклпиридинов
имеют следующие константы [3]:
Л'-Окись 2-метилпиридина — т. кип. 123—124° при 15 мм;
Пд— 1,5918; т. пл. пикрата 125—126,5°.
Л'-Окись 3-метилпиридина — т. кип. 146—149° при 15 мм; т. пл.
пикрата 138—139°.
Л'-Окись 4-метилпиридина—т. пл. 185—188°; т. пл. пикрата
158,7—159,7°.
Л’-Окись 2,6-диметилпиридина — т. кип. 115—119° при 18 мм;
т. пл. пикрата 127,5-129,0°.
ЛИТЕРАТУРА
1. Е. Ochiai, J. Org. Clietn., 18,531 (1953).
2. Ю. И. Чумаков, Методы получения химических реактивов и пре-
паратов, вып. 4—5, М;, ИРЕА, 1962.
3. V. В о с k е 1 h е j d е, М. .1. L i n u, J. Amer. Chem. Soc., 76, 1286
(1954).
Поступила в октябре 1962 г.
КПП
а АЦЕТОКСИАЛКИЛПИРИДИНЫ
[2-ацетоксиметилпиридин, 2-метил-6-ацетоксиметилпиридин,
2-(а-ацетоксиэтил)-пиридин, 4-(а-ацетоксиэтил)-пиридин и
2-(а-ацетокси-н-бутил)-пиридин]
Ю. И. ЧУМАКОВ, 3. Е. СТОЛЯРОВ, Ю. П. ШАПОВАЛОВА
/Ч
\^\сн2ооссн3
CsHsNO2 М. в. 151,17
II I
сн^ х/хсн2ооссн3
CgHjiNOa М. в. 165,19
\/4CH~CHs
N । s
ООССНз
C9HnNO2
М. в. 165,19
СН3
СНООССНз
I /Ч
/Ч II I
У'хсн-сн2сн2сн3
N ।
ООССНз
C9HuNO9 CuH16NO2
M. B. 165,19 M. в. 193,25
a-Ацетоксиалкилпиридины являются исходными продукта-
ми при получении соответствующих а-оксиалкилпиридинов
(см. статью «2-Оксиметилпиридины»).
Обычно применяемым методом получения а-ацетоксиал-
килпиридинов является перегруппировка У-окисей соответст-
вующих алкилпиридинов в уксусном ангидриде [1—6].
В настоящей работе проверены методики получения 2-ацет-
оксиметилпиридина, 2- (a-ацетоксиэтил) -пиридина, 2-(а-ацет-
окси-н-бутил) -пиридина, 2-мстил-6-ацеюксиметилпиридина и
4- (a-ацетоксиметил) -пиридина.
СИНТЕЗЫ 2-АЦЕТОКСИМЕТИЛПИРИДИНА, 2-МЕТИЛ-6-АЦЕТОКСИ-
МЕТИЛПИРИДИНА, 2 и 4-(« -АЦЕТОКСИЭТИЛ)-ПИРИДИНА И
2- (а -АЦЕТОКСИ-Н-БУТИЛ)-ПИРИДИНА
II I + (СН3СО)2О II j
^ХСН3 ^'/ХСН2ООССНз+СН3СООН
4-
о
61
сн/у^сНз
I
о
+ (СН3СО)3О
СН^ ¥ХСНаООССН3 4- СНзСООН
/Ч
II I +(СН8СО)2О -> II I
¥Хс2н6 ¥Хсн-снз+сн->соон
О ООССМз
сн3
I
С3Н5 СН-ООССНз
I I
/ч /\\
II i + (СН3СО)2О -й | + СНзСООН
\//
N N
I
о
^\СН2СН2СН,СН2СН2 + (СН3СО)2О ->
6
/ч
-> II | + СНзСООН
^СНСН3СН2СН3 •
N । 2 3
ООССН3
Характеристика исходного сырья
Уксусный ангидрид, ГОСТ 787—55.
Л?-Окиси алкилпиридинов (см. примечание 1).
Получение 2-ацетоксиметилпиридина
В трехгорлую колбу емкостью 250 jo на шлифах, снабжен-
ную обратным холодильником, капельной воронкой и термо-
62
метром, к 68,5 мл (0,75 Л4) слегка кипящего уксусного ангид-
рида прибавляют по каплям 40 г (0,37 М) JV-окиси 2-метилпи-
ридина (см. примечание 1) со скоростью, достаточной для
поддержания кипения реакционной массы за счет выделяю-
щегося при реакции тепла. Реакционную массу кипятят с об-
ратным холодильником 15 минут, содержимое колбы упарива-
ют в вакууме от водоструйного насоса па кипящей водяной
бане и остаток перегоняют в вакууме. После предгона (см.
примечание 2) собирают фракцию с т. кип. 119—12Г° при
18 мм. Выход 2-ацетоксиметилпиридина, представляющего
слегка желтоватое масло, 44,2 г, или 78% от теоретического;
«о°—1,4970 (см. примечание 3).
Получение 2-метил-6-ацетоксиметилпиридина
2-Метил-6-ацетоксиметилпиридин получают в условиях,
описанных выше для 2-ацетоксиметилпиридина, исходя из 50 г
(0,41 Л1) JV-окиси 2,6-диметилпиридина (см. примечание 4) и
58 мл (0.61 Л1) уксусного ангидрида. После отбора предгона
получают 57,8 г (86,2% от теоретического) 2-метил-6-ацет-
оксиметилпиридина в виде бесцветного масла с т. кип. 120—
121° при 18 мм; Яд —1,4991 (ом. примечание 5).
Получение 4-(а-ацетоксиэтил)-пиридина
Лг-Окись 4-этилпиридина была получена, исходя из 22,8 мл
(21,4 г; 0,2 М) 4-этилпиридина, 48,4 .ил (53,2 г; 0,42 М) 28%-ко-
го пергидроля и 40,8 мл (44 г; 0,42 Л1) уксусного ангидрида
согласно общей методике получения Л'-окиссй алкилпиридинов
(см. статью «JV-Окиси алкилпиридинов»). Кубовый остаток
после упаривания реакционной массы представляет собой не-
очищенную jV-окись 4-этилпиридина (см. примечание 6).
В трехгорлую колбу емкостью 500 мл на шлифах, снаб-
женную мешалкой, обратным холодильником, термометром и
капельной воронкой, помещают 200 мл уксусного ангидрида,
постепенно добавляют техническую JV-окись 4-этилпиридина и
очень медленно нагревают на водяной бане до прекращения
протекания бурной реакции (см. примечание 7), после чего
продолжают нагревать на кипящей водяной бане еще в тече-
ние 1 часа. Содержимое колбы упаривают в вакууме от водо-
струйного1 насоса. Остаток после^паривания разгоняют с деф-
легматором в вакууме, собирая фракцию с т. кип. 115—121°
при 17 мм, представляющую собой 4-(«-ацетоксиэтил)-пири-
дин в виде жидкости, слабо окрашенной в желтый или зеле-
новатый цвет, постепенно темнеющей при храпении. Выход
20,6 г, что составляет 62,2%, считая на 4-этилпиридин;
—1,4972.
63
Получение 2-(а-ацетоксиэтил)-пиридина
2- (а-Ацетоксиэти л)-пиридин получают в условиях, опи-
санных выше для 4-(а-ацетоксиэтил)-пиридина, исходя из
22,8 мл (21,4 г; 0,2 2W) 2-этилпиридина, 48,4 мл (53,2 г; 0,42 jW)
28%-пой перекиси водорода и 40,8 мл (44 г; 0,42 М) уксусно-
го ангидрида. На перегруппировку А-окиси 2-этилпиридина
берут 200 мл (216 г; 2,12 М) уксусного ангидрида. Выход
22,2 г, или 67,2%, считая па 2-этилпиридин. Т. кип. 1111 —116°
при 14 мм; я™ —1,4925 (см. примечание 8).
Получение 2-(а-ацетокси-н-бутил) пиридина.
2-(а-Ацетокси-н-бутил)-пиридин получают, исходя из 5 а
(0,037 Л1) 2-н.бутилпиридина, 9,9 г (8,9 мл\ 0,08 Л1) 28%-ной
перекиси водорода, 7,5 мл (8,2 г; 0,08 7И) уксусного ангидри-
да, в условиях, описанных выше для 4-(а-ацетоксиэтил)-пири-
дина. На перегруппировку А-окиси 2-н.бутилпиридина берут
18,8 мл (20,3 г; 0,2 М) уксусного ангидрида. Получают 4,7 г
2-(а-ацетокси-н-бутил)-пиридина в виде светло-желтой жид-
кости с т. кип. 115—125° при 7 мм; n2Q —1,4896. Выход 67%.
считая на 2-н.бутилпиридин (см. примечание 9).
Примечания.
1. Исходные А-окиси алкилпиридинов были получены действи-
ем пергидроля и уксусного ангидрида на соответствующие алкил-
пиридины. Они были только что перегнаны и имели константы, при-
веденные в статье «А-окиси алкилпиридинов».
2. Предгон образуется вследствие термической диссоциации аце-
тата 2-ацетоксиметилпиридина на уксусную кислоту и 2-ацетокси-
метилпиридин.
3. Константы 2-ацетоксиметилпиридина, по литературным дан-
ным: т. кип. 115—118° при 22 мм; п2^—1,4969, т. пл. пикрата
168—168,5" (после перекристаллизации из метанола) [1].
4. При проведении перегруппировки в условиях, указанных в
статье [1], возможен выброс реакционной массы.
5. Константы 2-метил-6-ацетоксиме1ИЛпиридина, по литературным
данным: т. кип. 110—114° при 15 мм [1], 100—105° при 4 .«.« [4];
л* —1,5043 [I].
6. Для перегруппировки . используется неочищенная Лг-окись
4-эти.ширидина, что упрощает работу и несколько увеличивает вы-
ход.
7. Если проводить перегруппировку с большими количествами
исходных- веществ, то при неосторожном нагреве возможен выброс
реакционной массы вследствие протекания реакции с саморазогре-
вом. Поэтому при больших исходных количествах целесообразно по-
степенно прибавлять А-окись 4-этилпиридипа к кипящему уксусному
ангидриду (см. выше).
8 По литературным данным, 2-(а-ацетоксиэтил)-пиридин имеет
следующие константы: т. кип. 109—111° при 16 мм [1], 89—93° при
3 'мм [6]; Лд —1.4937 [1].
9. По литературным данным, 2-(а-ацетокси-и-бутил)-пиридин име-
ет константы: т. кип. 135—137° при 1-5 мм; Пр—1,4895 [1J.
64
ЛИТЕРАТУРА
1. V. В о с к е 1 h е i d е, W. J. Linn, J. Amer. Chem. Soc., 76, 1286
(1954).
2. S. Oku da, Pharm. Bull. (Japan), 3, 316 (1955); Chem. Abstrs, 50,
13056 (1956).
3. G. К о b a у a s h i, S. Furukawa, Pharm. Bull. (Japan). I, 317
(1953); Chem. Abstrs, 49, 10948 (1955).
4. G. Kobayashi, S. Furukawa, G. Kawada, J. Pharm. Soc.
Japan, 74, 790 (1954); Chem. Abstrs, 49, 11646 (1955).
5. T. R. Govindachari, K. Nagarajan, S. Rajappa, J. Chem.
Soc., 1957, 551.
6. О. В u 11 i t, J. T. M a у n a r d, J. Amer. Chem. Soc., 76, 1370 (1954).
Поступила в октябре 1962 г. КПИ
2 ОКСИМЕТИЛПИРИДИНЫ
(2-Оксиметилпиридин, 2-метил-6-оксиметилпиридин и
2,6-диоксиметилпиридин)
Ю. И. ЧУМАКОВ, 3. Е. СТОЛЯРОВ
C6H,NO
\^\СН3ОН
М. в. 109, 13
СН2ОН
C7H9NO М. в. 123, 15
C7H9NO2
М. в. 139, 15
2-Оксиметилпиридины являются внутрикомплексообразую-
щими реагентами. Они также находят применение в качестве
исходных продуктов в синтезах соответствующих пиридиналь-
дегидов. 2,6-Диоксиметилпиридин, являясь бисфункциональ-
ным соединением, может быть использован в синтезах поли-
меров.
2-Оксиметилпиридин был получен восстановлением этило-
вого эфира 2-пиридинкарбоновой кислоты алюмогидридом
лития [1—4], восстановлением 2-пиридинкарбоновой кислоты
цинком в уксусной кислоте [5], омылением 2-ацетоксиметил-
5 Зак. 576 65
пиридина соляной кислотой [6]. 2-Метил-6-оксиметилпиридин
был получен омылением 2-метил-6-ацетоксиметилпиридина в
солянокислой среде [4] и восстановлением метилового эфира
6-метилпиколиновой кислоты алюмогидридом лития [8].
2,6-Диоксиметилпиридин получается с очень низким выхо-
дом восстановлением диэтилового эфира 2,6-пиридинкарбоно-
вой кислоты алюмогидридом лития {8, 9], омылением 2,6-ди-
ацетоксиметилпиридина концентрированной соляной кислотой
[6] или гидрированием на палладии труднодоступного 2,6-пи-
ридинальдегида [10].
Другие пути получения названных оксиметилпиридинов
малоудобны.
В приведенной ниже методике при получении оксиметиль-
ных производных нами было использовано щелочное омыление
легкодоступных в последние годы 2-(а-ацетоксиметил)-пири-
динов. Описанное ранее омыление этих же ацетоксиметилпи-
ридинов разбавленными растворами кислот [3, 4] дает менее
устойчивые выходы.
СИНТЕЗ 2-ОКСИМЕТИЛПИРИДИНОВ
|| | 4- NaOH -* || | + CHsCOONa
Х/ХСН2ООССН, ХХ\СН2ОН
N 2 3 N
|| I + NaOH —
CH3^W\CH3OOCCH3
[[ I + CHsCOONa
CHS/\^\CH2OH
|j I -4- 2NaOH
CH8COOCH2 /x^ \ CH2OOCCH3
|f f + 2CH3COONa
-> HOCH2/\/xCH2OH
N
Получение 2-оксиметилпиридина
В колбу емкостью 500 мл на шлифе, снабженную обрат-
ным холодильником, помещают 30 г (0,20 М) 2-ацетоксиме-
66
тилпиридина (см. примечание 1) и 225 мл 10%-ного раствора
едкого натра (0,56 Л1) и содержимое колбы кипятят в течение
6 часов на масляной бане (см. примечание 2). После охлажде-
ния каждые 100 мл реакционной смеси экстрагируют в экстрак-
торе непрерывного действия 50 мл хлористого метиле-
на в течение 10 часов (см. примечание 3). Экстракт сушат
поташом в течение суток, фильтруют через бумажный фильтр
от взвешенных частичек поташа, промывая поташ двумя пор-
циями по 10 мл сухого .хлористого метилена. Хлористый ме-
тилен отгоняют на водяной бане с елочным дефлегматором
высотой 25 см, а темпо-красный остаток подвергают перегон-
ке в вакууме, получая 5,7—5,8 г 2-оксиметилпиридина (вы-
ход 67—69%) в виде бесцветной жидкости с т. кип. 108—109°
при 15 мм; п^ —1,5430; т. пл. пикрата 157,5—158,0° (см.
примечание 4).
Получение 2-метил-6-оксиметилпиридииа
2-Метил-6-оксиметилпиридин получают омылением 2-ме-
тил-6-ацетоксиметилпиридина (см. примечание 5) в условиях,
описанных для 2-оксиметилпиридина, исходя из 9 г (0,055 М)
2-метил-6-ацетоксиметилпиридина и 62,2 мл (0,16М) 10%-ного
раствора едкого натра. Экстракцию проводят в экстракторе
непрерывного действия 40 мл хлороформа в течение 12 часов.
После обработки экстракта, как указано выше, растворитель
отгоняют с дефлегматором длиной 25 см в вакууме 40—60 мм,
а остаток перегоняют в вакууме, получая 40 г (60% от теоре-
тического) 2-метил-6-оксиметилпиридина в виде вязкого жел-
товатого масла с т. кип. 80—8Г при 5 мм. При охлаждении
в тающем льду продукт превращается в желтовато-белую кри-
сталлическую массу, расплывающуюся на воздухе (см. при-
мечание 6,7); Пр —1,5390 (переохлажденного).
Получение 2,6-диоксиметилпиридина
2,6-Диоксиметилпиридин получают омылением, исходя из
8 г (0,036 М) 2,6-ди-(ацетоксиметил)-пиридина (см. примеча-
ние 8) и 81 мл (0,20 Л4) 10%-пого раствора едкого натра в
условиях, описанных выше, экстракцию проводят в экстракто-
ре непрерывного действия 40—50 мл хлороформа в тече-
ние 15 часов. Экстракт высушивают над поташом в течение
суток, при этом частично выпадает 2,6-диоксиметилпиридин,
который растворяют добавлением дополнительного количест-
ва хлороформа и легким подогревом. Экстракт отделяют от
поташа, упаривают с дефлегматором длиной 25 см в вакууме
40—60 мм. После отгонки растворителя на стенках колбы ос-
таются желтоватые кристаллы 2,6-диоксиметилпиридина. Пос-
5* 67
ле двукратной перекристаллизации из бензола получают 30 г
(60% от теоретического) 2,6-диоксиметилпиридина в виде бе-
лых с легким желтоватым оттенком кристаллов с т. пл. 114—
114,5° (см. примечание 9).
Примечания.
1. Использовался свежеперегнанный 2-ацетоксиметилпиридин,
полученный перегруппировкой Л'-окиси 2-метилпиридина в уксус-
ном ангидриде (см. статью « «-Ацетоксиалкилпиридины»), Он име-
ет следующие константы: т. кип. 119—121° при 18 мм; nD —
1,4970.
2. Если после омыления в реакционной массе находятся взве-
шенные хлопья, то перед экстракцией их отфильтровывают на бу-
мажном фильтре.
3. Используется экстрактор непрерывного действия обы^ой кон-
струкции для экстракции тяжелыми жидкостями.
4. По литературным данным, 2-оксиметилпиридин имеет кон-
станты: т. кип. 103—105° при И мм [1], 105° при 12 мм [2],
111 —112° при 25 мм [6], 122—125° при 23 мм [3], 135—140° при 30 мм
[41; т. пл. пикрата 158° [1, 2], 157-158° [4], 153-156° [3], 159-161° [6].
5. Использовался свежеперегнанный -2-метил-б-ацетоксимстилпи-
ридин, полученный перегруппировкой М-окнсн 2,6-диметилпиридина с
уксусным ангидридом (см. статью «« -Ацетоксиалкилпиридины»). Он
имеет константы: т. кип. 124—125° при 25 мм; п 1,5004.
6. После перегонки 2-метил-6-оксиметилпиридина на стенках
колбы остается в виде кристаллического осадка 2,6-диметил-З-окси-
пиридин, который получается при омылении 2,6-диметил-З-ацетокси-
пиридипа, сопутствующего в качестве побочного продукта 2-метил-
6-ацетоксиметилпиридина (см. статью «а-Ацетоксиалкилпиридины»),
После перекристаллизации из бензола его т. пл. 212,5—213°.
7. По литературным данным, 2-метил-б-оксиметилпиридин име-
ет константы: т. кип. 104—110° при 14 мм; т. пл. пикрата 130—131°
(из этанола) [8].
8. Используется свежеперегнанный 2,6-ди-(ацетоксиметил)-пи-
ридин, полученный из 2-метил-6-ацетоксиметилпиридииа (см. статью
„а-Ацетоксиалкилпиридины “), который имеет константы: т. кип.
120—121° при 18 мм; п2° —1,4991.
9. 2,6-Диоксиметилпиридин, согласно литературным данным, име-
ет т. пл. 114—115° (из бензола) [6, 10].
ЛИТЕРАТУРА
1. V. М. Mi с о vic, М. L. Mich а По vic, Rec. trav. chim., 71
970 (1952).
2. R. Graf, G. P e r a t k о h e r, M. T a t z с 1, J. pract. Chem., 146,
88 (1936); Chem. Abstrs, 31, 382 (1937).
3. H. S. Mosher, J. E. Tcssieri, J. Amer. Chem. Soc., 73, 4925
(1951).
4. M. Protiva, Chem. listy, 45, 8998 (1951); Chem. Abstrs, 45, 8998
(1955).
5. F. S о r m, L. S e d i v y, Collection Czeclroslov. Chem. Communs.,
13, 289 (1948); Chem. Abstrs, 43, 2996 (1949).
6. V. Bockelheide, W. J. L i n n, J. Amer. Chem. Soc., 76, 1246
(1954).
7. G. Kobayashi, S. Furukawa, G. Kawada, J. Pharm.
Soc. Japan, 74, 790 (1954); Chem. Abstrs, 49, 11646 (1955).
68
8. S. N. Baker, K. N. Buggle, J. F. N. Omic, D. A. N.
Watkins, J. Chem. Soc., 1958, 3594.
9. R. А. В a r n e s, H. F a 11 e s, J. Amer. Chem. Soc., 75, 3830 (1913).
10. W. Mathes, W. S a n e r m 11 c h, F Klein, Ber., 86, 584 (1953).
Поступила в октябре 1962 г. КПП
ДИАЦЕТОКСИМЕТИЛ ПИРИДИНЫ
(2,6-диацетоксиметилпиридин и 2-диацетоксиметилпиридин)
Ю. И. ЧУМАКОВ, 3. Е. СТОЛЯРОВ
/X /Ч
II I ! I хооссна
СН3СООН2С/ч,</ХСН2ООССН3 х^хс^ооссн3
CnHi3O4N М. в. 223,23 С10НцО4У М. в. 209,20
2,6-Диацетоксиметилпиридин используют для синтеза
2,6-диацетоксиметилпиридина (см. статью «2-Оксиметилпири-
дины»), а также 2-ацетокси-6-ди-(ацетоксиметил)-пиридина,
который может быть превращен в 2-оксиметил-6-пиридиналь-
дегид [1]. Из 2-диацетоксиметилпиридина может быть получен
2-пиридинальдегид и ряд его производных [2].
Обычным методом получения 2,6-диацетоксиметилпириди-
на и 2-диацетоксиметилпиридина является перегруппировка
N-окисей соответствующих моно-ацетоксиметилпиридинов [2].
2,6-Диацетоксиметилпиридин может быть получен кипячением
2,6-диоксиметилпиридина с уксусным ангидридом [1].
В данной работе были проверены синтезы 2-диацетоксиме-
тилпиридина и 2,6-диацетоксиметилпиридина, исходя из
JV-окисей соответственно 2-метил-6-ацетоксиметилпиридина н
2-ацетоксиметилпиридина, Нами было найдено, что использо-
вание двукратного против теоретического избытка перекиси
водорода и уксусного ангидрида при получении У-окиси 2-ме-
тил-6-ацетоксиметилпиридина и полуторного избытка уксус-
ного ангидрида при перегруппировке полученной У-окиси
2-метил-6-ацетоксиметилпиридина позволяет получить 2,6-
диацетоксиметилпиридин с выходом 55% против 31%, полу-
ченных согласно [2].
69
СИНТЕЗ 2,6-ДИАЦЕТОКСИМЕТИЛПИРИДИНА И
2-ДИАЦЕТОКСИМЕТИЛ ПИРИДИНА
|| j + н2о2 + (СН3СО)2О
СН3 N СН2ООССНс
/\
-> || | +СН3СООН
Гз N СН,ООССН3
4
О
IM +(СН3СО)2О
ХСН,ООССН3
-4
о
II | + CHgCOOH
/\^\
СН3СООСН2 N СН2ООССН3
| -f- Н2О2 -j- (СН3СО)2О
'сн2ооссн3
/Ч|
w\
N СН2ООССН3
4
о
/ч
|| | + (СН3СО)2О ->
^'сн2ооссн3
4
о
/ч
II I + СНзСООН
\//\
N СН(ООССН3)2
70
Характеристика основного сырья
Ацетоксиметилпиридины (см, статью «а-Ацетоксиалкилпи-
ридины»).
Перекись водорода, ГОСТ 177—55.
Уксусный ангидрид, ГОСТ 787—55.
Получение 2,6-диацетоксиметилпиридина
В трехгорлую колбу емкостью 250 мл на шлифах, снабжен-
ную обратным холодильником, мешалкой с обтюратором из
эластичной каучуковой трубки, термометром и капельной во-
ронкой, помещают 20 г (0,12 34) свежеперегпапного 2-метил-
6-ацетоксиметилпиридина, добавляют 33 мл (0,26 34) 27,5 %-
ной перекиси водорода и перемешивают. Затем добавляют
при интенсивном перемешивании 25 мл (27 г; 0,26 34) уксус-
ного ангидрида в течение получаса. При добавлении первых
порций уксусного ангидрида температура реакционной массы
поднимается до 80° и в дальнейшем реакцию проводят при
этой температуре. После добавления уксусного ангидрида ре-
акционную массу выдерживают один час на кипящей водяной
бане, затем добавляют 5 мл 40%-ного формалина, вновь вы-
держивают в течение 1 часа при 100°, после чего производят
йодкрахмальную пробу на присутствие перекисных соедине-
ний в реакционной массе (см. примечание 1). Убедившись в
отсутствии перекисей, содержимое колбы тщательно упари-
вают в вакууме от водоструйного насоса на кипящей водяной
бане, а остаток в колбе подвергают перегруппировке с уксус-
ным ангидридом.
В трехгорлую колбу емкостью 150 мл на шлифах, снабжен-
ную обратным холодильником, термометром и капельной во-
ронкой, к 18,5 мл (20 г; 0,195 М) нагретого до 100° уксусного
ангидрида добавляют по каплям неперегнанную А-окись
2-метил-6-ацетоксиметилпиридина, поддерживая температуру
в колбе в пределах 105—115°. Реакционную массу выдержи-
вают 10 часов при 100° и упаривают в вакууме на кипящей
водяной бане. Остаток перегоняют в вакууме, получая 15 г
(55%, считая на 2-метил-6-ацетоксиметилпиридин) 2,6-ди-
ацетоксиметилпиридина в виде бледно-желтого масла ст. кип.
155—156° при 7 мм или 162—165° при 10 мм, которое быстро
желтеет; —1,4904, А?0— 1,1590 (см. примечание 2).
Получение 2-диацетоксиметилпиридина
В колбу на 250 мл помещают 20 г (0,13 34) 2-ацетоксиме-
тилпиридина, добавляют 80 мл (115 г; 1,42 34) ледяной уксус-
ной кислоты, 13,5 мл (0,13 34) 30%-ной перекиси водорода и
смесь нагревают с обратным холодильником 3 часа при 70е.
71
Добавляют 9,3 мл (0,09 Л4) 30%-ной перекиси водорода и вы-
держивают реакционную массу еще 9 часов при 70° (см. при-
мечание 3). Содержимое колбы тщательно упаривают в ваку-
уме от водоструйного насоса на кипящей водяной бане. В трех-
горлую колбу емкостью 150 мл на шлифах, снабженную об-
ратным холодильником, капельной воронкой и термометром,
к 25 мл (27 г; 0,26 А1) кипящего уксусного ангидрида добав-
ляют. по каплям остаток после упаривания (N-окись 2-ацет-
оксиметилпиридина). Реакционную массу кипятят 15 минут.
Содержимое колбы упаривают в вакууме на кипящей водяной
бане, а остаток перегоняют в вакууме, получая 13,8 г (50,3%,
считая на 2-ацетоксиметилпиридин) 2-диацетоксиметилпири-
дина в виде бесцветного масла с т. кип. 131 —133° при 3 мм;
—1-4890 (см. примечание 4).
Примечания.
1. Если присутствуют перекисные Соединения, необходимо до-
бавить еще 5 мл 40%-ного формалина, реакционную массу выдер-
жать в течение 1 часа на кипящей водяной бане и вновь произве-
сти пробу па присутствие перекисных соединений.
2. По литературным данным, 2,6-диацетоксиметилпиридин име-
ет следующие Константы: т. кнп. 135—136° при 0,3 мм; т. пл. пикра-
та ПО—110,5° (из этанола) [2].
3. А-окись 2-ацетоксиметилпиридина может быть получена в
тех же условиях, что и W-окись 2-метил-6-ацетоксиметилпиридииа.
4. Дважды перегнанный 2-диацетоксиметилпнридин закристал-
лизовывается в желто-белую воскообразную массу, со временем при-
обретающую желто-красный цвет.
По литературным данным, 2-днацетоксиметилпиридин имеет
следующие константы: т. кип. 160—162“ при 15 мм; «20—1,4872 [2].
ЛИТЕРАТУРА
1. W. В а к е г, К. М. В u g g 1 е, .!. F. W. М с Omic, D. А. М. Wa t-
kins, J. Chem. Soc., 3594 (1958).
2. U. В о с к e lh e i d e, W. J. Linn, J. Amer. Chem. Soc., 76, 1286
(1954).
Поступила в октябре 1962 г. КПП
2-ПИРИДИНАЛЬДЕГИД
/О. И. ЧУМАКОВ, 3. Е. СТОЛЯРОВ
ceH5NO
72
М. в. 107,11
2-Пиридинальдегид является исходным продуктом в син-
тезах ряда реактивов, в том числе йодметилата а-пиридин-
альдоксима [1].
Впервые 2-пиридинальдегид был получен озонолизом
2-стирилпиридина (а-стильбазола) [2]. Препаративно ценны-
ми способами получения 2-пиридинальдегида являются окис-
ление 2-оксиметилпиридина окисью селена [3,4], тетраацетатом
свинца [5], омыление его диацсталя [6], каталитическое окис-
ление 2-метилпиридина на ванадиево-молибденовом катализа-
торе [7„ 8].
В настоящей работе проверен способ получения 2-пиридин-
альдегида окислением 2-оксиметилпиридина селенистой кис-
лотой в диоксане {3].
СИНТЕЗ 2-ПИРИДИНАЛЬДЕГИДА
/\ч /Ч
2|| | +H2SeO3-2|| | о +Se+3H2O
У^сн,он УУсСн
Характеристика основного сырья
Диоксан, ТУ ГКХ 1558—61.
Получение 2-пиридинальдегида
В трехгорлую колбу емкостью 150 мл на шлифах, снаб-
женную термометром, обратным холодильником и мешалкой
(см. примечание 1), помещают 4,4 г (0,04 М) 2-оксиметилпи-
ридина и 40 мл (0,55 М) диоксана (см. примечание 2). К по-
лученному раствору добавляют 2,6 г (0,02 М) мелкоизмель-
ченной селенистой кислоты (см. примечание 3) и при быстро-
работающей мешалке начинают нагрев реакционной смеси.
При 60—65° начинается растворение селенистой кислоты и
содержимое в колбе приобретает красный цвет. Реакционную
массу нагревают до 80° и при непрерывном перемешивании
поддерживают эту температуру в течение 2 часов. После ох-
лаждения содержимое колбы фильтруют на шоттовском
фильтре, черный осадок селена в колбе промывают тремя пор-
циями диоксана по 5 мл, диоксановые вытяжки также филь-
труют и присоединяют к основной массе фильтрата. Раство-
ритель отгоняют с елочным дефлегматором длиной 25 см, а
остаток перегоняют в вакууме, собирая фракцию с т. кип.
54—55° при 3,5—4 мм. Выход 2-пиридинальдегида, представ-
ляющего бесцветное масло с резким характерным запахом,
составляет 2,6 г, или 60,5% от теоретического; ng —1,5386
(см. примечания 4 и 5).
73
Примечания.
1. В качестве затвора служит обтюратор из эластичной каучу-
ковой трубки. Все работы необходимо проводить в вытяжном шкафу.
Использовался 2-оксиметилпиридин, полученный щелочным омы-
лением 2-ацетоксиметилпиридина (см. статью «2-Окснметилпиридн-
ны»),
2. Необходимо предварительно убедиться в отсутствии в днок-
сане перекисных соединений (йод-крахмальная проба); в случае
присутствия перекисей их надо удалить кипячением с порошкооб-
разным едким кали (65—70 г на литр диоксана) в течение 6—8 ча-
сов.
3. Использовалась импортная селенистая кислота. Селенистая
кислота является сильным ядом и при соприкосновении с кожей
вызывает трудноизлечимые язвы, поэтому все работы с селени-
стой кислотой следует проводить в резиновых перчатках и соблю-
дать меры предосторожности.
4. Пиридинальдегиды являются лакриматорами.
5. Согласно литературным данным, 2-пиридинальдегид имеет
следующие константы: т. кип. 62—63° при 13—14 мм [1], 70—73° при
16 мм [4]; «д’5 —1,53886; d'f —1,1255 [1].
ЛИТЕРАТУРА
1. К. В е t h е, W. Г>. Е г m а п, L. Lendl е, G. Shinfdt, Arch-
Exptl. Phathol, Pharmacol., 231, 3 (1957); Chem. Abstrs, 51, 16929 (1957).
2. G. Harries, G. H. Lenart, Liebigs Ann. Chem., 95, 410 (1915).
3. S. Furukawa, V. Kuroiva, Pharm. Bull. Japan, 3, 232 (1955);
Chem. Abstrs, 50, 1092 (1956).
4. D. Jerchel, H. E. Hech, Liebigs Ann. Chem., 613, 1834 (1958).
5. V. M. M i с о v 1 с, M. L. M I h a i 1 о v i c, Rec. trav. chim., 71, 970
(1952).
6. J. P. Wibaut, P. Hu lies, Rec. trav. chim., 71, 1021 (1952).
7. W. M a t h e s, W. S a u e r m 11 c h, T. К 1 e i n, Chem. Ber., 84, 452
(1951).
8. Швейц, пат. 304383, 1955.
Поступила в октябре 1962 г. КПИ
пиридинкарбоновые КИСЛОТЫ
(пиколиновая, никотиновая, изоникотиновая и
дипиколиновая кислоты)
Ю. И. ЧУМАКОВ *
II +СООН
N
C6H5NO2 М. в. 123,11
НООС/Х^ХСООН
C,H6NO4 М. в. 167, 12
* В экспериментальной работе принимали участие Т. К- Жигач,
Н. Г. Нехаееа, Е. Г. Чвырева, Н. Г. Псковских. ‘
74
Пиридинкарбоновые кислоты используются как ключевые
исходные продукты в синтезах изомерных замещенных произ-
водных ряда пиридина (эфиров, галоидангидридов пиридин-
карбоновых кислот, амино-пиридинов, оксиметилпиридинов, га-
лоидпиридинов, ацетилпиридинов и др.).
Обычно используемым методом получения пиридинкарбо-
новых кислот является окисление алкилпиридинов различны-
ми окислителями. В основу описанной ниже методики поло-
жено окисление метилпиридинов перманганатом калия, под-
робно описанное в работе [1]. Обзор литературы по получению
незамещенных пиридинкарбоновых кислот приведен там же.
СИНТЕЗ ПИРИДИНКАРБОНОВЫХ КИСЛОТ
СН3
КМпО4
2-, 3- или 4-
изомер
> |] +СООК
N
2-, 3- или 4-
изомер
НС1
— || 4-соон
W
N
2-, 3- или 4-
изомер
|| | КМпО4
CHZ'^CHa
НС1
КООС/Х^ХСООК
Характеристика основного сырья
Метилпиридины, см. примечание 1.
Калий марганцовокислый, ГОСТ 5777—51.
Условия получения
Пиколиновая кислота. В трехгорлую круглодонную колбу
емкостью 2 л, снабженную мешалкой, термометром и обрат-
ным холодильником, помещают 99 мл (93 г; 1 М) 2-метилпи-
ридина (см. примечание 1) и 1000 мл воды. Нагревают на во-
дяной бане до 70—75°, через горло для термометра прибав-
ляют первую порцию марганцовокислого калия в количестве
41,8 г и перемешивают при этой температуре до полного рас-
кисления, которое занимает 30—60 минут (см. примечание 2),
после чего прибавляют очередную порцию марганцовокисло-
75
го калия. Всего прибавляют 10 порций марганцовокислого ка-
лия по 41,8 г каждая (всего 418 г; 2,64 Л1). Первые пять пор-
ций вводят в реакцию при 70—75°, следующие пять— при тем-
пературе 90—95“ (на кипящей водяной бане).. По раскисле-
нии последней порции марганцовокислого калия горячую ре-
акционную массу отфильтровывают на воронке Бюхнера. От-
фильтрованную двуокись марганца возвращают в колбу, при-
бавляют 500—600 мл горячей воды и перемешивают в течение
30—60 минут при нагревании до 90—100°. Вновь отфильтро-
вывают, тщательно отжимая на фильтре осадок двуокиси мар-
ганца. Объединенные фильтрат и промывные воды упарива-
ют на сетке до объема 500—600 мл (см. примечание 3). Оста-
ток в колбе охлаждают и при перемешивании постепенно ней-
трализуют до pH 3,2 (с помощью pH-метра или, что менее
точно, по универсальному индикатору) концентрированной
соляной кислотой. Полученный водный раствор пиколиновой
кислоты и хлористого калия упаривают в вакууме от водост-
руйного насоса на водяной бане досуха. Содержимое колбы
извлекают, измельчают в ступке или на шаровой мельнице,
переносят обратно в колбу Кляйзена и прогревают в вакууме
от водоструйного насоса на кипящей водяной бане в течение
1—2 часов (см. примечание 4). Полученную смесь пиколино-
вой кислоты и хлористого калия дополнительно измельчают
в шаровой мельнице и экстрагируют в аппарате Сокслета бен-
золом в течение 15—25 часов. К концу экстракции на стенках
колбы выпадают кристаллы пиколиновой кислоты. Бензол от-
гоняют досуха, остаток извлекают и растирают в ступке. По-
лучают 55—64 г слегка окрашенной пиколиновой кислоты с
т. пл. 135—137° (испр.). Выход 45—52%. В случае необходи-
мости ее перекристаллизовывают из горячего бензола, полу-
чая продукт с т. пл. 136—137,5° (испр.). в виде белого мелко-
кристаллического порошка (см. примечание 5). Из остатка
после экстракции, представляющего собой хлористый калий,
содержащий значительные количества пиколиновой кислоты,
может быть получено дополнительное количество последней.
Для этого остаток высушивают, вновь измельчают на шаро-
вой мельнице и повторно экстрагируют в аппарате Сокслета.
Никотиновая кислота. Никотиновую кислоту получают, ис-
ходя из 97,5 мл (93 г; 1 Л4) 3-метилпиридина (см. примечания
1 и 6) и 418 г (2,64 М) марганцовокислого калия, в условиях,
описанных выше для пиколиновой кислоты, за исключением
выделения никотиновой кислоты, которое производится сле-
дующим образом. В стакан емкостью 1 л помещают водный
раствор калиевой соли никотиновой кислоты, упаренный до
объема 800—900 мл, и постепенно при перемешивании при-
бавляют концентрированную соляную кислоту до pH 3,4 (см.
примечание 7). Содержимое стакана нагревают при переме-
шивании до растворения всего выпавшего осадка никотиновой
76
кислоты (см. примечание 8). Оставляют медленно охлаждать-
ся в течение ночи (см. примечание 9). Выпавшую никотиновую
кислоту отфильтровывают на воронке Бюхнера и на фильтре
дважды тщательно промывают 50—60 мл охлажденной до
5—7° воды (см. примечание 10). Высушивают в сушильном
шкафу при 95—105°. Получают 76—87,5 г (62—71% от теоре-
тического) никотиновой кислоты с т. пл. 236—237° (испр.).
Полученная никотиновая кислота содержит лишь следы хло-
ристого калия, от которого можно освободиться однократной
перекристаллизацией ее из 12- или 13-кратного количества
горячей воды (см. примечание 11).
Изоникотиновая кислота. Изоникотиновую кислоту полу-
чают, исходя из 97,5 мл (93 а; 1 ЛИ 4-метилпиридина (см.
примечания 1 и 13) и 418 г (2,64 М) марганцовокислого калия,
в условиях, описанных выше для никотиновой и пиколиновой
кислот. Выделение производят действием концентрированной
соляной кислоты до pH 3,6 на щелочной раствор изоникотина-
та калия, упаренный до объема 2000 мл (см. примечание 14).
Получают 64—71,5 г изоникотиновой кислоты (52—58% от
теоретического выхода) в виде белого кристаллического по-
рошка, имеющего т. пл. 320—323° с разложением (испр.) (см.
примечание 15).
Дипиколиновая кислота. Дипиколиновую кислоту получа-
ют в условиях, описанных выше для пиколиновой кислоты,
исходя из 58 мл (53,5 г; 0,5 Л1) 2,6-диметилпиридина в 1000.и л
воды (см. примечание 1) и 418 а (2,64 /И) марганцовокислого
калия, прибавляемого 10 порциями по 41,8 а. Водный рас-,
твор дипиколината калия упаривают в стакане, под тягой, до
объема 500 мл, охлаждают и осторожно, при перемешивании
прибавляют 300 мл концентрированной соляной кислоты (см.
примечание 16), нагревают раствор до кипения и оставляют
охлаждаться в течение ночи. Выпавшую дипиколиновую кис-
лоту отфильтровывают и на фильтре промывают двумя пор-
циями по 15 мл холодной (0—5°) воды. После высушивания
при 95—105° в сушильном шкафу получают 46,1—51,7 г
(55—62% от теоретического выхода) дипиколиновой кислоты
в виде белого кристаллического порошка, имеющего т. пл.
230—232° с разложением (испр.) (см. примечание 17).
Примечания.
1. Использовался 2-метилпиридин квалификации «чистый»; 3-,
4-метил- и 2,6-дпметилпнрндины были приготовлены согласно [2] и
имели содержание 98—99% [2].
2. Марганцовокислый калий берется в избытке против теоре<
тического (более 30%), тем не менее значительная часть исходных
алкилпиридинов не вступает в реакцию окисления.
Считают, что очередная порция перманганата калия раскисли-
ла», когда вокруг капли реакционной массы, нанесенной на филь-
тровальную бумагу термометром, вынутым из реакционной массы,
77
не будет фиолетового или розового окрашивания. На время взятия
пробы горло реакционной колбы закрывается запасной пробкой во
избежание потерь 2-метилпиридина в атмосферу.
3. Полученный дистилЛат содержит 8—15% от исходного 2-ме
тилпнридина, не вступившего в реакцию. Он может быть использо-
ван вместо воды в последующих опытах окисления.
4. Капилляр не должен доходить на 1 см до поверхности насы-
панного в колбу порошка. Если обезвоживание производить без ка-
пилляра, то на стенках колбы оседает влага, которую не удается
удалить и прн длительном нагревании.
5. По литературным данным, т. пл. пиколиновой кнслогы
136,5—138° [1].
6. На окисление может быть взят также 3-метилпиридин 93—
95%-ного содержания.
7. Нейтрализация сопровождается выделением углекислоты п
вспениванием,
8. Если необходимо, то прибавляют дистиллированную воду до
полного растворения осадка при кипячении.
9. Медленное охлаждение необходимо с целью уменьшить за-
грязнение никотиновой кислоты хлористым калием за счет соосаж-
дения.
10. Промывку производят следующим образом: отсоединяют ва-
куум, пропитывают осадок водой и осторожно стеклянной пробкой
разминают никотиновую кислоту на фильтре. При появлении первых
капель фильтрата присоединяют вакуум и отфильтровывают, тща-
тельно отжимая осадок никотиновой кислоты на фильтре. Количе-
ство воды на промывку ие должно превышать указанного, иначе
возможны потери за счет растворимости никотиновой кислоты.
11. Никотиновую кислоту растворяют в 15—20-кратном коли-
честве горячей воды, кипятят с углем, отфильтровывают и полу-
ченный раствор упаривают до объема, соответствующего 12—13-
кратному, по отношению к никотиновой кислоте, взятой на пере-
кристаллизацию.
По литературным данным, чистая никотиновая кислота имеет
т. пл. 235,5—236,5° (испр.) [3].
12. Если иа окисление берут низкопроцентный (87—93%-ный)
3-метилпиридин, то выходы никотиновой кислоты существенно ниже
и из маточников после выделении получают лишь небольшое количе-
ство никотиновой кислоты, которая может быть очищена перекри-
сталлизацией из воды только с большими потерями.
13. На окисление может быть взят также 4-метнлпиридин с со-
держанием 80—93% [2], поскольку изоникотиновая кислота легко
поддается очистке от изомерных никотиновой и пиколиновой кис-
лот, обладая наименьшей растворимостью в воде.
14. При кипячении водной суспензии, полученной при выделе-
нии изоникотиповой кислоты, последняя растворяется не полностью.
Тем ие менее этот прием так же, как и в случае никотиновой кис-
лоты, позволяет предупредить загрязнение кзонккотиновой кислоты
хлористым калием.
15. По литературным данным, изоннкотниовая кислота имеет
т. пл. 323—325° с разложением [1].
16. Дипиколиновая кислота соосаждает большие количества ка-
лия в виде дипиколината. Дли предупреждения соосаждения необ-
ходимо использовать значительный избыток соляной кислоты.
17. По литературным данным, дипиколиновая кислота имеет
т. пл. 232—233° с разложением [1]. Если полученный продукт не пла-
вится при этой температуре, то полученная дипиколиновая кислота
сильно загрязнена солями калия и ее необходимо перекристаллизо-
вать из горячей концеитрнроваиной соляной кислоты.
78
ЛИТЕРАТУРА
1. О. Black, Е. Depp, В. В. Corson, J. Org. Chem., 14, 14 (1949)
2. 10. И. Ч у м а к о в, Методы получения химических реактивов и пре-
паратов, ИРЕА, 4—5, 44 (1962).
3. R. Q or ding, L. A. F lexer, J. Amer. Pharm. Ass., 29, 230 (1940).
Поступила в октябре 1962 г. КПИ
НИКОТИНОВАЯ КИСЛОТА
(3-пиридинкарбоновая кислота)
Ю. И. ЧУМАКОВ, Л. А. РУСАКОВА, А. И. МЕДНИКОВ, Р. И. ВИРНИК
соон
СвНБМО, М. в. 123,11
Никотиновая кислота является исходным продуктом в
синтезах 3-замещенных производных пиридина, в том числе
3-аминопиридина, никотинамида, диэтиламида никотиновой
кислоты и других препаратов.
Обычно используемыми путями синтеза никотиновой кис-
лоты являются окисление 3-метилпиридина (р-пиколина), ни-
котина или окисление с последующим декарбоксилировани-
ем хинолина или 2-метил-5-этилпиридина. Известны другие
способы получения никотиновой кислоты (например, окисле-
нием анабазина через никотиннитрил, получаемый из 3-бром-
пиридина или 3-пиридинсульфокислоты), однако они препара-
тивно мепее удобны или исходят из труднодоступного сырья.
В основу методики, описанной ниже, положен известный
способ окисления никотина азотной кислотой [1—7]*.
Нами найдено, что окисление никотина протекает спокой-
но и с минимальным расходом окислителя в случае, если его
производить кипящей азотной кислотой при непрерывном ее
прибавлении к реакционной массе и с одновременной отгон-
* О получении никотиновой кислоты из 3-метилпиридина см. в статье
«Пиридинкарбоновые кислоты».
79
кой отработанной азотной кислоты [8]. Тем самым удается в
7—10 раз сократить объем реакционной массы. Если по из-
вестному методу [4] для окисления 210 а никотина необходи-
ма колба емкостью 5 л, то для окисления того же количества
никотина по способу, описанному ниже, требуется объем
0,5—0,7 л.
Существенной особенностью предлагаемого способа яв-
ляется то, что в противоположность описанным ранее спосо-
бам [3—5] окисление никотина в никотиновую кислоту проте-
кает спокойно без вспенивания или скрытого индукционного
периода, за которым следует бурная реакция.
Литературные данные о константах никотиновой кислоты
значительно расходятся. Так, для нее сообщались следующие
температуры плавления: 228—229° [1], 235<5—236,5° [9] и дру-
гие. В большинстве справочников для никотиновой кислоты
приводится слишком низкая температура плавления 232°.
Окисление никотина азотной кислотой — многократно прове-
ренный способ, впервые предложенный Вайделем [1] и тща-
тельно разработанный Макэлвином и Адамсом [3, 4], приво-
дит к недостаточно чистой никотиновой кислоте с температу-
рой плавления 230—232°.
Нами найдены условия выделения никотиновой кислоты из
реакционной массы после окисления азотной кислотой нико-
тина, позволяющие получить продукт с температурой плавле-
ния 235—236°, что хорошо совпадает с данными, полученными
в специальной работе по определению истинной температуры
плавления никотиновой кислоты [9], и удовлетворяет требова-
ниям Государственной Фармакопеи [10].
СИНТЕЗ НИКОТИНОВОЙ кислоты
/Ч/соон
+ HNO3 -> !| I Л!»£2»
•HNO3
Характеристика основного сырья
Азотная кислота, ТУ АУ 112—56.
Никотин-основание, ГОСТ 8839—58.
80
Условия получения
К 325 мл 56%-пой азотной кислоты при перемешивании и
охлаждении прибавляют 30 г никотина (см. примечание 1).
В прибор, состоящий из колбы, снабженной нисходящим хо-
лодильником, термометром, погруженным в реакционную мас-
су, и капельной воронкой (все соединения на шлифах), поме-
щают 75 мл 56%-ной азотной кислоты и нагревают на масля-
ной бане. При появлении первых капель погона (ПО—115°)
постепенно прибавляют приготовленный раствор никотина в
азотной кислоте с той скоростью, с которой происходит от-
гонка разбавленной азотной кислоты. По окончании прибав-
ления продолжают нагревание, упаривая реакционную массу
до объема 35—40 мл. Весь процесс окисления занимает 8—12
часов. Содержимое колбы медленно охлаждают при переме-
шивании. Выпавший осадок отфильтровывают, тщательно от-
жимая на фильтре. Получают около 35 г влажного светло-
желтого нитрата никотиновой кислоты. Его растворяют в
40 мл горячей воды и при температуре 70 —90° производят вы-
деление никотиновой кислоты, прибавляя соду до значения
pH 3,1—3,7. Реакционную массу охлаждают при размеши-
вании до 10—15°, дают постоять 1—2 часа при этой темпера-
туре, после чего выпавшую никотиновую кислоту отфильтро-
вывают и промывают двумя порциями по 10 мл холодной
воды с температурой 5—10°. Получают около 30 г технической
никотиновой кислоты. Ее растворяют в 15-кратном количест-
ве (по весу от содержания никотиновой кислоты в пасте) воды
и кипятят в течение 1 часа с 1 г активированного угля. Филь-
труют в горячем состоянии и остаток на фильтре промывают
15 мл горячей воды. Фильтрат упаривают до объема 140—
150 мл из расчета, чтобы полученный раствор содержал 10
частей воды па 1 часть сухой технической никотиновой кисло-
ты, взятой на перекристаллизацию. Упаренном5г раствору дают
постепенно охладиться до комнатной температуры. Выпавший
осадок отфильтровывают, на фильтре промывают двумя пор-
циями по 10 мл охлажденной до 5—10° воды и высушивают
при 90—100°. Получают в среднем 14,5 а никотиновой кисло-
ты с температурой плавления 235—236° (испр.). Маточные
растворы после перекристаллизации никотиновой кислоты от
пяти опытов объединяют и упаривают до 1/з первоначального
объема. При охлаждении выпадает никотиновая кислота, ко-
торую перекристаллизовывают из воды с активированным уг-
лем согласно приведенному выше описанию. Получают до-
полнительное количество никотиновой кислоты с температурой
плавления 235—236° (испр.). .
Средний выход никотиновой кислоты с т. пл. 235—236°,
удовлетворяющей всем требованиям Фармакопеи, составляет
68,4% (см. примечание 3). Из маточного раствора после от-
6 Зак. 576
деления нитрата никотиновой кислоты в последнем опыте и
из маточных растворов после выделения и перекристаллиза-
ции никотиновой кислоты может быть получено дополнитель-
ное количество никотиновой кислоты с несколько более низ-
кой температурой плавления (230—232°).
Примечания.
1. Использовался свежеперегнанный в вакууме технический ни
котин. Полученный раствор никотина в азотной кислоте необходимо
держать в ледяной воде, поскольку при хранении его иногда проис-
ходит саморазогревание с последующей бурной реакцией.
2. Второй и последующие опыты окисления никотина произво-
дили, используя отогнанную азотную кислоту от предыдущего опы-
та, укрепленную до 50—60% отгонкой воды с дефлегматором. Кро-
ме того, в начале опыта к 75 мл исходной азотной кислоты прибав-
ляли маточный раствор после отделения нитрата никотиновой кис-
лоты, полученный в предыдущем опыте окисления.
3. Согласно наиболее достоверным литературным данным, тем-
пература плавления чистой никотиновой кислоты составляет 235,5—
236,5° (нспр.) [9].
ЛИТЕРАТУРА
1. Н. Weldel, Ann., 165, 330 (1878).
2. A. Pictet, A. Rotschy, Ber., 34, 702 (1901).
3. S. Me El vain, R. Adams, J. Amer. Chem. Soc., 45,2738(1923).
4. Синтезы органических препаратов, 1, ИЛ, М., 288 (1949).
5. А. Ш м у к, ЖПХ, 20, 245 (1947).
6. И. А. Васюнина, А. А. Беэр, Н. А. Преображенский
ЖПХ, 16, 206 (1943).
7. Пат. США 2409345, 1946.
8. Ю. И. Ч у м а к о в, П. А. Г а н г р с к и й, Н. Н. Д ы х а и о в,
Л. А. Р у с а к о в а, А. И. М е д н и к о в, Р. И. Вирник, 3. В. Ворон-
кова, Т. С. Семеновых, Авт. свид. 109387, 1957.
9. R. Gordon, L. A. Flex er, J. Amer. Chem. Soc., 29, 230 (1940).
10. Государственная Фармакопея СССР, 8 изд., 26 (1952).
Поступила s октябре 1962 г. КП И, Никотиновый завод.
ИЗОНИКОТИНОВАЯ КИСЛОТА
(4-пиридинкарбоновая кислота)
Ю. И. ЧУМАКОВ, Е. Г. ЧВЫРЕВА, П. А. ГАНГРСКИЙ
СООН
I
/Ч
N
CtH5NO, М. в. 123,11
82
Изоникотиновая кислота является исходным продуктом в
синтезах 4-аминопиридина, эфиров изоникотиновой кислоты,
4-оксиметилпиридина и ряда других замещенных в 4-е поло-
жение производных пиридина.
Изоникотиновая кислота может быть получена окислени-
ем перманганатом калия 4-метилпиридина [ 1J, 4-этилпиридина
[2, 3] или окислением азотной кислотой оксиметильных про-
изводных 4-метилпиридина [4—8]. Другие методы получения
изоникотиновои кислоты препаративно малоудобны.
Ниже описано получение изоникотиновой кислоты окисле-
нием смеси оксиметильных производных 4-метилпиридина ки-
пящей азотной кислотой [7, 8]. Этот синтез имеет преимуще-
ством использование в качестве сырья легко доступной «0-пи-
колиновой фракции» и простоту выполнения отдельных опе-
раций. В способ по сравнению с ранее описанными [4—6]
внесен ряд изменений, в результате чего удалось улучшить
качество и повысить выход конечного продукта, а также су-
щественно уменьшить объем реакционной массы на стадии
окисления.
СИНТЕЗ НИКОТИНОВОЙ кислоты
СН2СН2ОН СН(СН2ОН)2 С(СН2ОН)з
огулен,
СН2СН2ОН
СН(СН»ОН)2
/\
II I
С(СН2ОН)3
\\ +HNO3
соон
соон
Характеристика основного сырья
«р-Пиколиновая фракция», техн,, ЧМТУ 3454—53.
Формальдегид, 40%-ный, ГОСТ 1625—61.
Азотная кислота, ТУ АУ 112—56.
Условия получения
1. Оксиметильные производные 4-метилпиридина. В колбу,
снабженную обратным холодильником и термометром, поме-
щают 110 г 35—40%-ного водного формальдегида, 81 г техни-
ческой «р-пиколиновой фракции» (см. примечание 1) и нагре-
вают до кипения (98—100° в реакционной массе). При этой
температуре реакционную массу выдерживают в течение
18—20 часов, после чего отгоняют с водяным паром не всту-
пившие в реакцию пиридиновые основания и избыток форм-
альдегида. Остаток, представляющий собой водный раствор
оксиметильных производных 4-метилпиридина, упаривают до
объема 75 мл (см. примечание 2),
2. Изоникотиновая кислота. В трехгорлую колбу емкостью
100 мл, снабженную двумя капельными воронками для окси-
метильных производных и азотной кислоты, нисходящим хо-
лодильником и термометром, погруженным в реакционную
массу, помещают 50 мл 55—60%-ной азотной кислоты и на-
гревают до кипения (см. примечание 3). При температуре
НО—115° к реакционной массе начинают одновременно при-
бавлять 375 мл 55—60 %-ной азотной кислоты и 75 мл окси-
метильных производных, полученных из 81 г «0-пиколиновой
фракции». Азотную кислоту прибавляют примерно в 5 раз
быстрее, чем оксиметильные производные, и с той скоростью,
с которой происходит отгонка отработанной азотной кислоты
(см. примечание 4). По окончании прибавления продолжают
нагревание, упаривая реакционную массу до объема 40—50 мл,
причем температура постепенно поднимается до 120—130° в
реакционной массе. К полученному раствору нитрата изонико-
тиновой кислоты в азотной кислоте прибавляют 100 мл воды,
переносят в стакан и нагревают до 50—70°. При этой темпе-
ратуре производят выделение изоникотиповой кислоты,
прибавляя небольшими порциями безводную соду до pH
3,3—3,9. Реакционную массу охлаждают при перемешивании
84
до 10—15° и дают постоять 1—2 часа. Изоникотиновую кисло-
ту отфильтровывают, промывают водой до отсутствия в про-
мывных водах нитрат-иона, используя для этого пробу с ди-
фениламином, после чего высушивают при 100—105°. Полу-
чают в среднем 21,4 а (в пересчете на 100%) изоникотиновой
кислоты й виде мелкокристаллического слегка желтоватого
порошка. Содержание изоникотиновой кислоты колеблется в
пределах 98—100%. Температура плавления продукта в за-
паянном капилляре 320—323° с разложением (см. примеча-
ние 5).
Примечания.
1. Использовались обычные технические образцы „{3-пиколино-
вой фракции» с температурой кипения 138—146°С, имеющие следу-
ющий примерный состав: 3-метилпиридина 25—30%, 4-метилпириди-
на 25—30%, 2,6-диметилпиридина 15—20%, пиридина, 2-метилпири-
дииа и 2,4-диметилпиридина в сумме 15—25%.
2. В реакцию с формальдегидом вступает исключительно 4-ме-
тилпиридип и практически не вступают 2-метилпиридии и 2,6-диме-
тилпиридин. Последние 2 основания, а также пиридин и 3-метилпи-
ридип отгоняют с паром, в то время как нелетучие с паром оксимс-
тильные производные 4-метилпиридина остаются в колбе.
3. Прибор собирается на нормальных шлифах.
4. Предварительное приготовление смеси оксиметильных произ-
водных и азотной кислоты оказалось невозможным, так как эта
смесь, приготовленная на холоду, быстро саморазогревается, вспе-
нивается и происходит экзотермическая реакция окисления с вы-
бросом реакционной массы.
5. Согласно наиболее достоверным литературным данным, изо-
никотиповая кислота имеет температуру плавления в запаянном
капилляре 322—325° с разложением [1]. Для получения совершенно
белого продукта полученную изоникотиновую кислоту 1—2 раза пе-
рекристаллизовывают с углем из кипящей воды (15—20 г кислоты на
1000 мл воды).
ЛИТЕРАТУРА
1. G. Black, Е. Depp, В, В. Corson, J. Org, Chem,, 14, 14
(1949).
2. J. Р. Wibaut, F. В. Ahrens, Rec. trav, cliim., 60, 119 (1941).
3, W. H. Linuel, A. F. V у a s, Quart. J. Pharm. Pharmacol., 20, 119
(1947); Chem. Abstrs, 41, 6885 (1947).
4. R. Graf, J, Prakt. Chem., 146, 88 (1936).
5. M. Л и д а к, С. Тиллер, П. Науменко, Изв. АН Латв. СССР,
83, 1 (1953).
6. М. В. Рубцов, Е. Е. М и х л и н а, В. Я. Ф у р ш т а т о в а, Авт.
свид. 97408, 1954.
7. Ю. И. Ч у м а к о в, II. А. Гангрский, Е. В. Швецова,
Е. Т. Ч вы рев а, Авт. свид. 111193, 1957,
8. П. А. Гангрский, Е. Г. Ч выреза, Ю. И. Чумаков, Мед.
пром. СССР, 3, 13 (1959).
Поступила в октябре 1962 г. КПП, Московский химико-фармацевтический
завод ,Акрихин-
3-ПИРИДИНСУЛЬФОКИСЛОТА
Ю. И. ЧУМАКОВ
SO3H
N
C5H5NOsS М. в. 159,18
3-Пиридинсульфокислота служит исходным продуктом в
синтезах 3-замещенных производных пиридина (3-оксипири-
дина, простых и сложных эфиров 3-оксипиридина, 3-цианпири-
дина и др.).
Обычно используемым методом получения пиридин-3-
сульфокислоты является сульфирование пиридина олеумом
различных концентраций в присутствии сульфата ртути как
катализатора [1, 2]. Описано сульфирование пиридина серным
ангидридом путем нагревания пиридинсульфотриоксида с сер-
ной кислотой также в присутствии ртути [3].
Ниже приведена методика, в основу которой положено
сульфирование 20—22°/о-ным олеумом [1].
СИНТЕЗ 3-ПИРИДИНСУЛЬФОКИСЛОТЫ
SO3H S03H
(Hg) /4/ /Ч/
!| I +H,SO4 + SO3 > !! I - II ] .
Х/'
N N N
•V2h2so4
Характеристика основного сырья
Пиридин, ГОСТ 2747—44.
Условия получения
В трехгорлую колбу емкостью 1 л на шлифах, снабженную
мешалкой, капельной воронкой и термометром, помещают
418 мл (800 а) 20—22%-ного олеума уд. в. 1,911—1,919 (см.
примечание 1) и при перемешивании и охлаждении (темпера-
тура в колбе невыше25—30°) прибавляют 158г (161мл; 2 /И)
сухого пиридина. Затем прибавляют 2,5 г сульфата ртути или
1,7 г металлической ртути. Мешалку удаляют и в центральное
горло колбы помещают воздушный холодильник, соединенный
через стеклянную трубку с пробиркой, в которую налито 10 мл
86
серной кислоты для улавливания серного ангидрида. Реакци-
онную массу нагревают на масляной бане в течение 24 часов
при температуре 220—230° в бане (см. примечание 2). Обрат-
ный холодильник заменяют на широкий нисходящий холо-
дильник на шлифах («мост»), в свободное горло колбы встав-
ляют капилляр. В качестве приемника используют другую
двухгорлую колбу емкостью 1 л, которую соединяют с ваку-
умной системой масляного насоса. Из реакционной массы от-
гоняют избыток серной кислоты при температуре около 180°
и вакууме 2 мм (см. примечание 3). Отгонку ведут до прекра-
щения погона. Темпо-коричневый остаток в колбе охлаждают
и при перемешивании прибавляют 400 мл метанола (см. при-
мечание 4). Охлаждают при перемешивании до 0—5° и дают
постоять при этой температуре 0,5—1 час. Полученную техни-
ческую пиридин-3-сульфокислоту отфильтровывают на ворон-
ке Бюхнера, тщательно отжимая на фильтре. Промывают
40—50 мл охлажденного метанола. Высушивание пиридин'-З-
сульфокислоты производят в вакууме в колбе, соединенной с
водоструйным насосом и подогреваемой на водяной бане. По
внешнему виду пиридин-3-сульфокислота представляет собой
светлый, с серым оттенком, кристаллический порошок. Полу-
чают 350—370 г технической пиридин-3-сульфокислоты. Даль-
нейшую очистку производят следующим образом. 100 а техни-
ческой пиридин-3-сульфокислоты растворяют при нагревании
в 100 мл воды, после чего прибавляют 500 мл метанола. Осаж-
дение начинается почти сразу. Охлаждают холодной водой,
выпавший продукт отфильтровывают на воронке Бюхнера и
промывают 3 раза по 70—100 мл метанола. После высушива-
ния получают пиридип-3-сульфокислоту в виде белого или
слегка сероватого мелкокристаллического порошка с т. пл.
342—346° (разд., см. примечание 5); выход около 190 г или
60% °т теоретического (см. примечание 6).
Примечания.
1. Если отсутствует олеум данной концентрации, то его готовят
разбавлением 60%-ного олеума серной кислотой 96%-ной концент-
рации.
2. Можно делать перерывы во время нагревания.
3. Между насосом и прибором обязательно должны быть поме-
щены две охлаждаемые раствором твердой углекислоты в метаноле
или ацетоне ловушки. Между насосом и этими ловушками помеща-
ют дополпитслыюл овушку с плавленым едким кали.
4. Остаток после отгонки в вакууме представляет собой свет-
ло-коричневую массу с легкой опалесценцией. При прибавлении ме-
танола происходит разогрев, в связи с чем необходимо использовать
перемешивание и охлаждение.
5. Температуру плавления определяют в запаянном капилляре
на укороченном термометре, в медном блоке с электрообогревом.
6. Полученная 3-пиридннсульфокислота содержит заметные ко-
личества ртути, которая не удаляется при переосаждении метанолом.
Для полного ее удаления водный раствор технической 3-пиридин-
сульфокислоты обрабатывают прн кипячении сероводородом в тече-
87
ние 3—4 часов, после чего отфильтровывают сульфид ртути, при-
бавляют метанол и дальнейшую обработку ведут, как описано выше.
По литературным данным, 3-пиридинсульфокислота имеет т. пл.
352—356° [1].
ЛИТЕРАТУРА
1. S. М, Me Elvain, М. A. Goese, J. Amer. Chem. Soc., 65, 2233
(1943).
2. Препаративная органическая химия, М., Госхимиздат, 275, 1959.
3. Англ. пат. 602882; Chem. Abstrs, 43, 696 (1949).
Поступила в октябре 1962 г. КПИ
га-МЕТИЛСТИРОЛ
Э. Б. ГРЕКОВА
сн=сн2
С9Н10
М. в. 118,17
В литературе описан ряд способов получения «-метилсти-
рола, который может быть синтезирован путем дегидратации
n-метилфенилкарбипола в жидкой [1J или паровой фазе [2],
путем декарбоксилирования и дегидробромирования ₽-бром-
₽-(«-метилфенил)-пропионовой кислоты [3]. «-Метилстирол по-
лучают также в результате двукратной перегонки л-метилфе-
нилметилкарбинола над пиросульфатом калия [4] или путем
крекинга несимметричного ди-(«-метилфенил)-этана [5, 6].
Мы синтезировали «-метилстирол по следующей схеме:
О
Br MgBr СН,С^
/Ч/ Mg /\\/
II I —* 1'1--------------->
сн3 сн3
СН2(ОН)СН3
сн8
88
Характеристика основного сырья
Эфир диэтиловый, ГОСТ 6265—52.
n-Бромтолуол, ч., т. кип. 184°, ВТУ У 280а—51.
Магний металлический, ГОСТ 804—56.
Уксусный альдегид, ч., т. кип. 20°, ТУ МХП 2633—51.
Соляная кислота, ч., ГОСТ 3118—46.
Бисульфит натрия, ч., ТУ МХП 1944—49.
Гидрохинон, х. ч., ГОСТ 2549—44.
Условия получения
Получение Бромистого п-метилфенилмагния. В трехгорлую
колбу емкостью 500 мл, снабженную механической мешалкой,
капельной воронкой и холодильником с хлоркальциевой труб-
кой, помещают 4,37 г магния в порошке, затем приливают
50 мл абсолютного эфира и прибавляют крупный кристалл
йода. Из капельной воронки приливают 5 мл раствора 30,78 г
n-бромтолуола в 50 мл абсолютного эфира. Колбу нагревают
на водяной бане до тех пор, пока не начнется реакция, при
этом эфир начинает мутнеть. Затем прибавляют из капель-
ной воронки раствор n-бромтолуола с такой скоростью, чтобы
эфир равномерно кипел; после введения всего п-бромтолуола
реакционную смесь нагревают 1,5—2 часа до полного раство-
рения магния. Полученный раствор бромистого п-метилфе-
нилмагния охлаждают и используют для последующего син-
теза.
Получение п-метилфенилметилкарбинола. 9 г уксусного
альдегида растворяют в 30 мл абсолютного эфира и получен-
ный раствор по каплям при механическом перемешивании и
охлаждении прибавляют к раствору бромистого п-метилфе-
нилмагния. После прибавления всего раствора уксусного аль-
дегида удаляют баню со льдом и реакционную смесь выдер-
живают, пока ее температура не сравняется с комнатной, по-
сле чего колбу нагревают в течение 15 минут на водяной ба-
не для окончания реакции. Затем реакционную массу снова
охлаждают льдом и по каплям прибавляют концентрирован-
ную соляную кислоту (уд. в. 1,19) в количестве 12 мл в \2мл
воды. При этом раствор становится прозрачным. Эфирный
слой отделяют, а водный слой дважды промывают эфиром по
12—20 мл. Соединенные эфирные вытяжки энергично встря-
хивают в делительной воронке с 10 мл 10%-ного раствора би-
сульфита натрия для удаления непрореагировавшего уксус-
ного альдегида, затем промывают небольшим количеством
раствора соды до щелочной реакции и водой, после чего вы-
сушивают хлористым кальцием. Эфир отгоняют, а оставшийся
в колбе n-метилфенилметилкарбинол перегоняют в вакууме
при остаточном давлении 10 мм рт. ст. и температуре 110°
Выход 20 г, что составляет 80% от теоретического.
89
Получение п-метилстирола. 20 г «-метилфенилметилкарби-
нола помещают в колбу Кляйзена. Туда же прибавляют 2 а
(10 весовых процентов от количества исходного карбинола)
прокаленного кислого сернокислого натрия (см. примечание 1)
и 0,2 г (1 весовой процент от количества «-метилфенилме-
тилкарбинола) гидрохинона (см. примечание 2). Затем соз-
дают в системе вакуум 100 мм рт. ст. и постепенно нагревают
колбу на масляной бане. В интервале температур 50—100°от-
гоняют воду, образовавшуюся при дегидратации, и получен-
ный «-метилстирол. Если температура поднимается выше 100°
(см. примечание 3), то вносят небольшое количество свеже-
прокаленного кислого сернокислого калия и продолжают пе-
регонку. Полученный «-метилстирол отделяют на делитель-
ной воронке от воды, сушат небольшим количеством хлори-
стого кальция и перегоняют при 10 мм рт. ст. и температуре
68—70°. Выход 10 г, что составляет 59% от теоретического.
Примечания.
1. Прокаленный кислый сернокислый калий получают нагрева
нием его в тигле на пламени до появления белого дымка, после чеп,
сразу же плав выливают в ступку и растирают в мелкий порошок.
2. Прибавление гидрохинона обязательно, в противном же слу-
чае образующийся n-метилстирол полимеризуется.
3. Повышение температуры выше 100° указывает на то, что де-
гидратация n-метилфенилметилкарбинола произошла не полностью.
ЛИТЕРАТУРА
1. М. Sulzbacher, Е. В е rg m а п, J. Org. Chem., 13, 303 (1948).
2. D. Mo wry, М. Ren о II, W. Н u b е г, J. Amer. Chem. Soc., 68,
1105 (1946).
3. K. Auwers, Ber., 45, 2764 (1912).
4. F. E1 s e r 1 о h r, L. Schulz, Ber., 57, 1808 (1924).
5. В. Л. В а й с e p, В. Д. Рябов, С. Ш. Соколина, Докл. All
СССР, 106 , 271 (1956).
6. J. D 1 x о п, К. Saunders, Ind. Eng. Chem., 46, 652 (1954).
Поступила в январе 1963 г. Институт химии полимеров и мономеров
АН УССР
2-ИОДФЛУОРЕН
Э. Б. ГРЕКОВА
сн2
М. в. 292,12
90
2-йодфлуорен обычно получают по реакции Зандмайера
путем нагревания флуорсндиазоний йодида-2 с йодистой медью
в йодистоводородной кислоте [1].
Нами 2-йодфлуорен был получен прямым йодированием
флуорена по схеме:
А
ХСН2Х
X J+HJ
хсн2/
Характеристика основного сырья
Уксусная кислота, х. ч., ГОСТ 61—51,
Флуорен, ч., т. пл. 114°.
Йод металлический, ч., ГОСТ 4159—48.
Серная кислота, ч., ГОСТ 4204—48.
Азотная кислота, ч., ГОСТ 4461—48.
Условия получения
В трехгорлую колбу емкостью 0,5 л, снабженную механи-
ческой мешалкой, капельной воронкой и термометром, загру-
жают 20 г флуорена, 150 мл ледяной уксусной кислоты, 15,9 г
йода и 13,5 мл серной кислоты (уд. в. 1,84). Реакпионную
массу нагревают на водяной бане до температуры 34—36°. При
этой температуре и постоянном механическом перемешивании
прикапывают 2,4 мл азотной кислоты (уд. в. 1,4) в течение
35 минут. После прибавления всего количества азотной кисло-
ты перемешивание продолжают еще 5 минут. Затем реакцион-
ную смесь приливают к 500 мл воды, выпавший осадок от-
фильтровывают и промывают до нейтральной реакции. Цвет
осадка слегка коричневатый от не вошедшего в реакцию йода
(см. примечание).
Выход 24 г, что составляет 70,6% от теоретического, т. пл.
121°.
С целью очистки 2-йодфлуорен перекристаллизовывают из
этилового или изопропилового спирта.
Примечание.
Для удаления йода осадок промывают па фильтре 300 мл рас-
твора бисульфита натрия.
ЛИТЕРАТУРА
1. A. Korzczynski, Bull. Soc. Chitn. France [4], 43, 346 (1929).
Поступила в январе 1963 г.
Институт химии полимеров и мовомеров АН УССР
2-ФЕНИЛ-5-ФЕНИЛАМИН0-1, 3, 4 ОКСАДИАЗОЛ
А. П. ГРЕКОВ
N-N
< \ с C-NH-/
\/ Х_Х
О
CUHUON,
М. в. 237,26
2-Фенил-5-фениламино-1, 3, 4-оксадиазол получают из
1-бензил-4-фенилтиосемикарбазида путем отщепления серо-
водорода при помощи окиси свинца [1] или под влиянием во-
доотнимающих средств [2].
Нами разработан препаративный метод получения 2-фе-
нил-5-фениламино-1, 3, 4-оксадиазола из 4-фенилсемикарба-
зида и бензоилхлорида по схеме:
/ ^-coci + h2nnhconh-4< \_<wl
./ '--CONHNHCONH-- + НС1
Т X X X
С~^-CONHNHCONH-/^^ —-
Х—Х Х_//
N-N
О
+ Н2О
Характеристика основного сырья
Пиридин, ч., ГОСТ 2747—44.
4-ФенилсемикарбазиД, ч., т. пл. 120—123°.
Бензоил хлористый, ТУ МХП 92—51.
Хлорокись фосфора, ч., ВТУ У 166—51.
Условия получения
Получение 1-бензоил-4-фенилсемикарбазида. В колбу ем-
костью 100 мл, снабженную механической мешалкой и капель-
ной воронкой, помещают 6 г 4-фенилсемикарбазида и 35 мл
сухого пиридина. В полученный раствор при перемешивании
добавляют по каплям в течение 20 минут 5,6 г хлористого
бензоила. При этом наблюдается незначительное повышение
92
температуры. По окончании прибавления всего бензоилхло-
рида продолжают перемешивание еще один час, после чего ре-
акционную массу приливают к 150 мл холодной воды. Выпав-
ший бесцветный кристаллический продукт отфильтровывают,
промывают на фильтре водой и сушат при температуре 100°.
После перекристаллизации из спирта выход продукта равен
4,8 а, что составляет 47,8% от теоретического, т. пл. 192—193’.
Получение 2-фенил-5-фениламино-1,3,4-оксадиазола. В
колбу емкостью 100 мл, снабженную обратным холодильни-
ком, помещают 4,6 г 1-бензоил-4-фенилсемикарбазида и 42 мл
хлорокиси фосфора. Полученную смесь кипятят до образова-
ния гомогенного раствора. Затем реакционную массу охлаж-
дают и осторожно, при энергичном перемешивании, прилива-
ют к 300 мл холодной воды. Выпавший бесцветный кристал-
лический осадок отфильтровывают, промывают на фильтре
водой до нейтральной реакции и сушат при температуре 100°.
После двукратной перекристаллизации из этанола и ледяной
уксусной кислоты выход продукта равен 1,8 г, что составляет
41 % от теоретического, т. пл. 212—213°.
ЛИТЕРАТУРА
1. R. St oil е, К. Fehrenbach, J. Pract. Chem., 122, 298 (1229).
2. Е. Hoggarth, J. Chem. Soc., 1949, 1918.
Поступила в январе 1963 г. Институт химии полимеров и мономеров АН УССР
АЛФАВИТНЫЙ ПЕРЕЧЕНЬ СОЕДИНЕНИЙ, ОПИСАННЫХ
В НАСТОЯЩЕМ ВЫПУСКЕ
1-Амиио-2-иафтол-4-сульфокислота................................. 26
а-Ацетоксиалкилпиридины . ........................................ 61
2-(а-Ацетокси-н-бутил)-пиридин.................................... 61
2-Ацетоксиметилпиридин............................................ 61
8-Ацетоксихинальдии............................................... 8
2-и 4-(а-Ацетоксиэтил)-пирнДин.................................... 61
2-(а-Ацетоксиэтил)-пиридин........................................ 61
4-(а-Ацетоксиэтил)-пиридин........................................ 61
1-Бензоил-4-фенилсемикарбазид.................................. 92
2,2-Бис-(4‘-оксифенил)-бутан...................................... 12
5,5-Бис-(4’-оксифенил)-ионан...................................... 15
3,3-Бис-(4’-оксифенил)-пентан.................................... 14
Бромистый /г-метилфепнлмагний.................................... 89
2-трет. Бутилпиридии............................................. 35
8-Гидразинохинолин и его солянокислая соль ....................... 5
1,3-Диоксибеизол<4-азо-Г'>2‘-нафтол-4,-сульфокислота............. 26
Диацетоксиметилпиридины ......................................... 69
2,6-Диацетоксиметилпиридин.................................... 71
2-Диацетоксиметилпиридии........................................ 71
2,6-Диоксиметилпиридин........................................... 67
Диоксиэтиламиноуксусная кислота................................. 19
Дипиколиновая кислота ........................................... 77
1,3-Ди-(2-пиридил)-пропан........................................ 56
Изоникотииовая кислота........................................... 82
Изохинолин....................................................... 49
2-Йодфлуореи..................................................... 90
2-Метил-6-ацетоксиметилпиридин................................... 63
2-Метил-б-оксиметилпиридин....................................... 67
2-Метилпиридин................................................... 30
п-Метилстирол.........................................• . . . . 88
Моногидрат дигидрохлорида 8-гидразинохиполиния................... 89
/г-Метилфеиилметилкарбинол ...................................... 89
Никотиновая кислота.............................................. 79
2-(5‘-ионадиен-2', 7'-ил)-нирндин................................ 39
3-(5'-ионадиен-2', 7'-ил)-пиридин................................ 40
2-Оксиметилпириднны.............................................. 65
Оксиметильиые производные 4-метилпиридииа........................ 84
V-Окиси алкилпиридинов.......................................... 58
34
2- и 3-(3’-Пентенил)-пириднны.................................. 38
2-Пиридинальдегид ............................................. 72
Пиколиновая кислота........................................... 7.5
Пиридиикарбоновые кислоты ..................................... 74
3-Пиридинсульфокислота......................................... 86
Смесь изомерных фенилпиридинов................................. 41
Салицилоилхлорид.............................................. 18
lV-Салицилоилфенилгидроксиламии ............................... 17
Сульфонафтолазорезорцин........................................ 25
4,4’-Тетраметилдиаминотиобензофенон . . -...................... 10
Триоксифлуороны................................................ 21
2-, 3- и 4-(3’-Фенилпропил)-пиридины........................... 46
2- и 4-(2’-Фенилэтил)-пирндины................................. 44
2-Фенил-5-фениламиио-1,3,4-оксадиазол.......................... 92
Хлористый феиилдиазоний........................................ 43
2-Этилпиридин................................................ 33