/

















































































































































































































































































































Автор: Пилипенко А.Т. Бабко А.К.
Теги: спектральные методы анализа оптические методы анализа химия химическая промышленность
Год: 1974




Текст
А-КБИБКО,А-Т-ПИЛИПЕНКО фото м ётрич ески й АНАЛИЗ
Я9яммямммм1авшв>ммв9ммм11ви9алмкам1
МЕТОДЫ ОПРЕДЕЛЕНИЯ НЕМЕТАЛЛОВ
| А. К, БАБКО I, A. T. ПИЛИПЕНКО
МЕТОДЫ ОПРЕДЕЛЕНИЯ НЕМЕТАЛЛОВ
МОСКВА ИЗДАТЕЛЬСТВО «ХИМИЯ» 1974
543
УДК 543.43 : 546.I/.2 Б 12
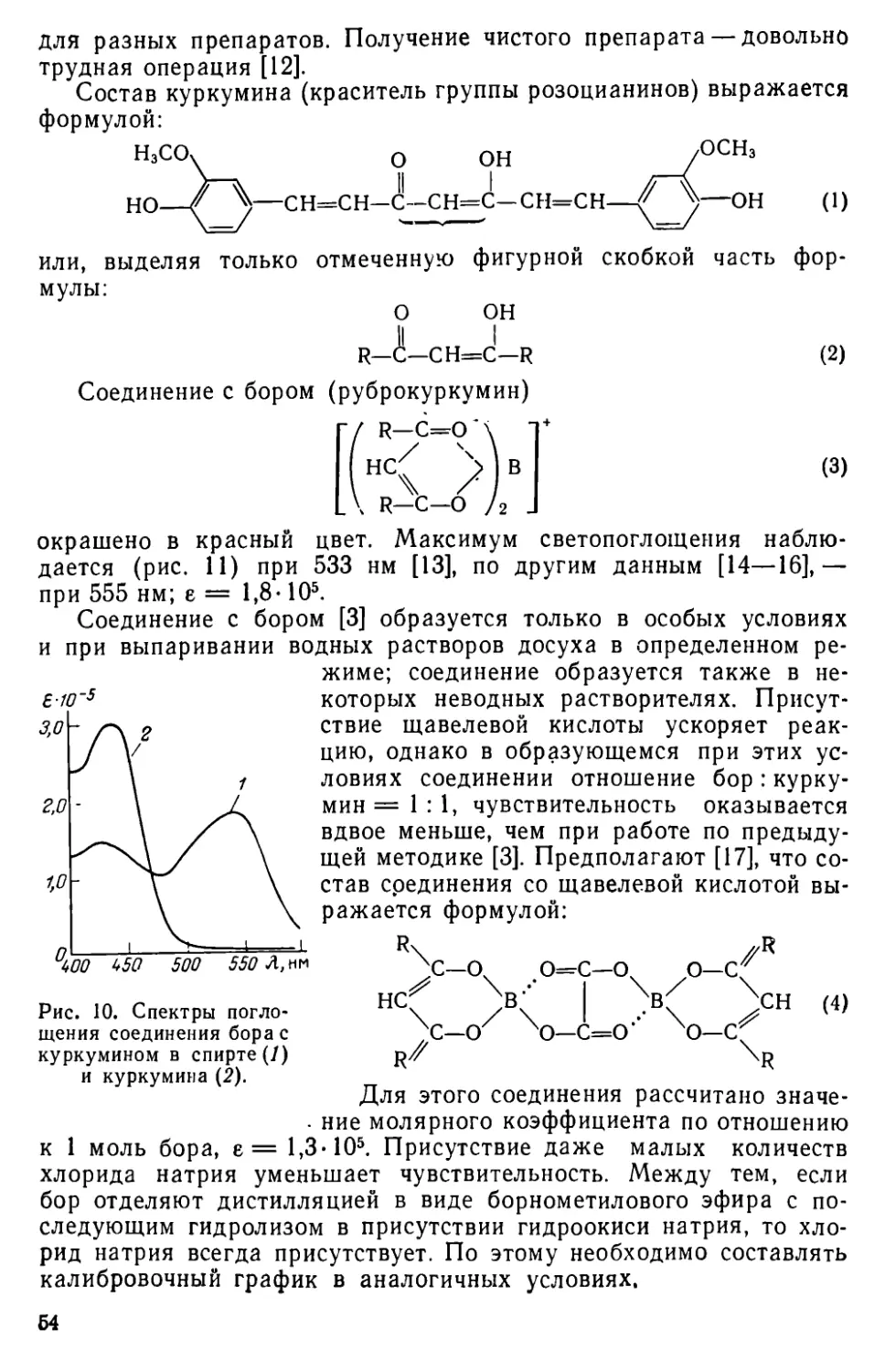
| А. К. Бабко |, А. Т. Пилипенко
Б 12 Фотометрический анализ. Методы определения неметаллов. М., «Химия», 1974. 360 с.; 12 табл.; 78 рис.; список литературы 1500 ссылок.
В монографии подробно изложены методы фотометрического определения азота, бора, кремния, фосфора, мышьяка, кислорода, серы, селена, теллура, фтора, хлора, брома и иода. Приведены спектры поглощения соединений, в виде которых проводят определение. Указаны чувствительность методов, мешающие определению ионы и способы их устранения. Описаны методы определения неметаллов в различных материалах.
Книга предназначена для работников химико-аналитических лабораторий различных отраслей промышленности и научных учреждений, а также для аспирантов, преподавателей и етудентов соответствующих вузов.
543
20506-027 ,050(01)-74
27-74
Редактор Л. Н. Овсянникрва
Технический редактор А. С. Кочетова
Художник Е. В. Бекетов
Корректоры Л. В. Лазуткина, М. С. Хрипунова
Т-20030 Сдано в наб. 17/VIII 1973 г. Подп. в печ. 24/XII 1973 г. Формат бумаги 60X90l/ie- Бумага тнп. № 2. Усл. печ. л. 22,5. Уч.-над. л. 25,04. тираж II 000 экз. Зак. 701. Изд. № 454. Цена 1 р. 75 к.
Издательство „Химия", 107076, Москва, Стромынка, 23
Ордена Трудового Красного Знамени Ленинградская типография № 2 имени Евгении Соколовой Союзполиграфпрома при Государственном комитете Совета Министров СССР по делам издательств, полиграфии и книжной торговли. 198052, Ленинград, Л-52, Измайловский проспект, 29.
© Издательство «Химия»
СОДЕРЖАНИЕ
Предисловие ..................................................... 9
Глава I. Азот....................................................11
I. 1. Общая характеристика методов определения аминного и общего азота .... .......... . 11
1.2. Разложение азотсодержащих веществ................. 12
1.3. Методы отделения . 16
1.4. Определение аммиака по Несслеру................... 18
1.5. Определение аммиака индофеноловым методом. 21
I. б. Другие методы определения аминного и общего азота .... 26
1.7. Определение нитрата........................ .... 27
1.8. Определение нитрита............................... 33
1.9. Другие методы определения нитрата и нитрита 35
Литература . ..................... 40
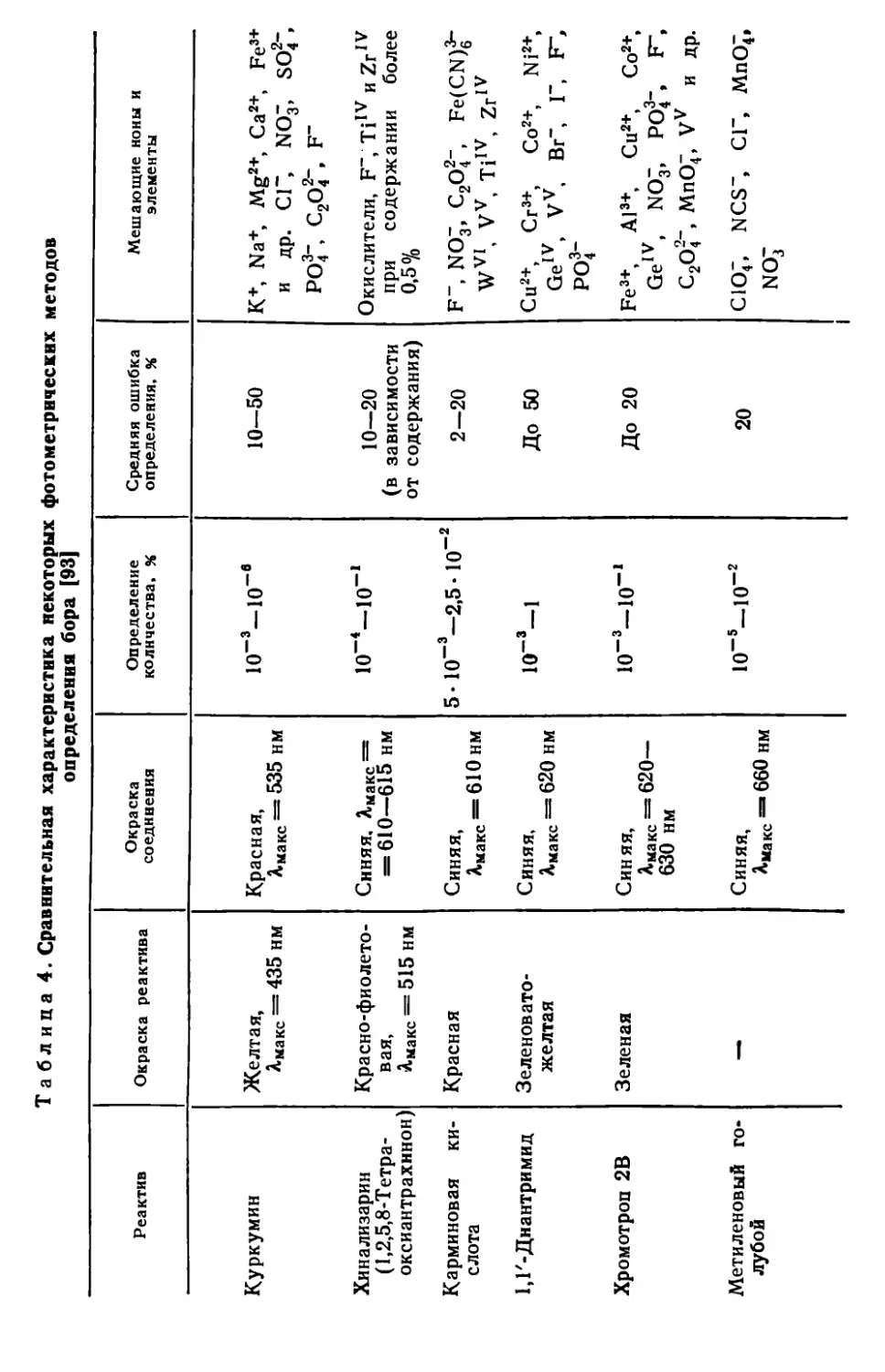
Глава II. Бор....................................................45
II. 1. Общая характеристика методов определения ... 45
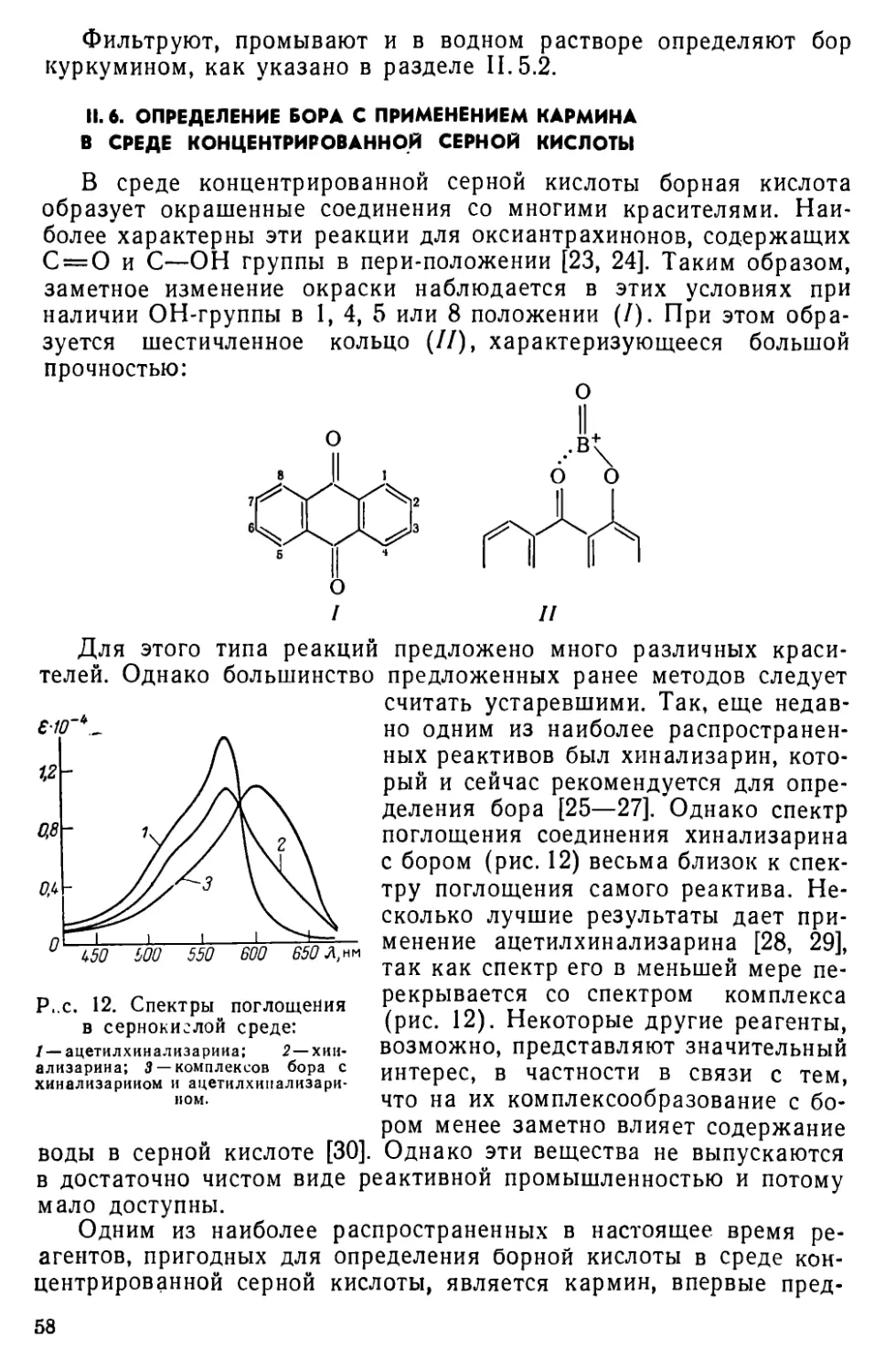
II. 2. Наиболее важные реакции и методы определения бора.....46
II. 3. Разложение борсодержащих материалов .............49
II . 4. Отделение бора ..................................... 50
II. 5. Определение бора с применением куркумина .............53
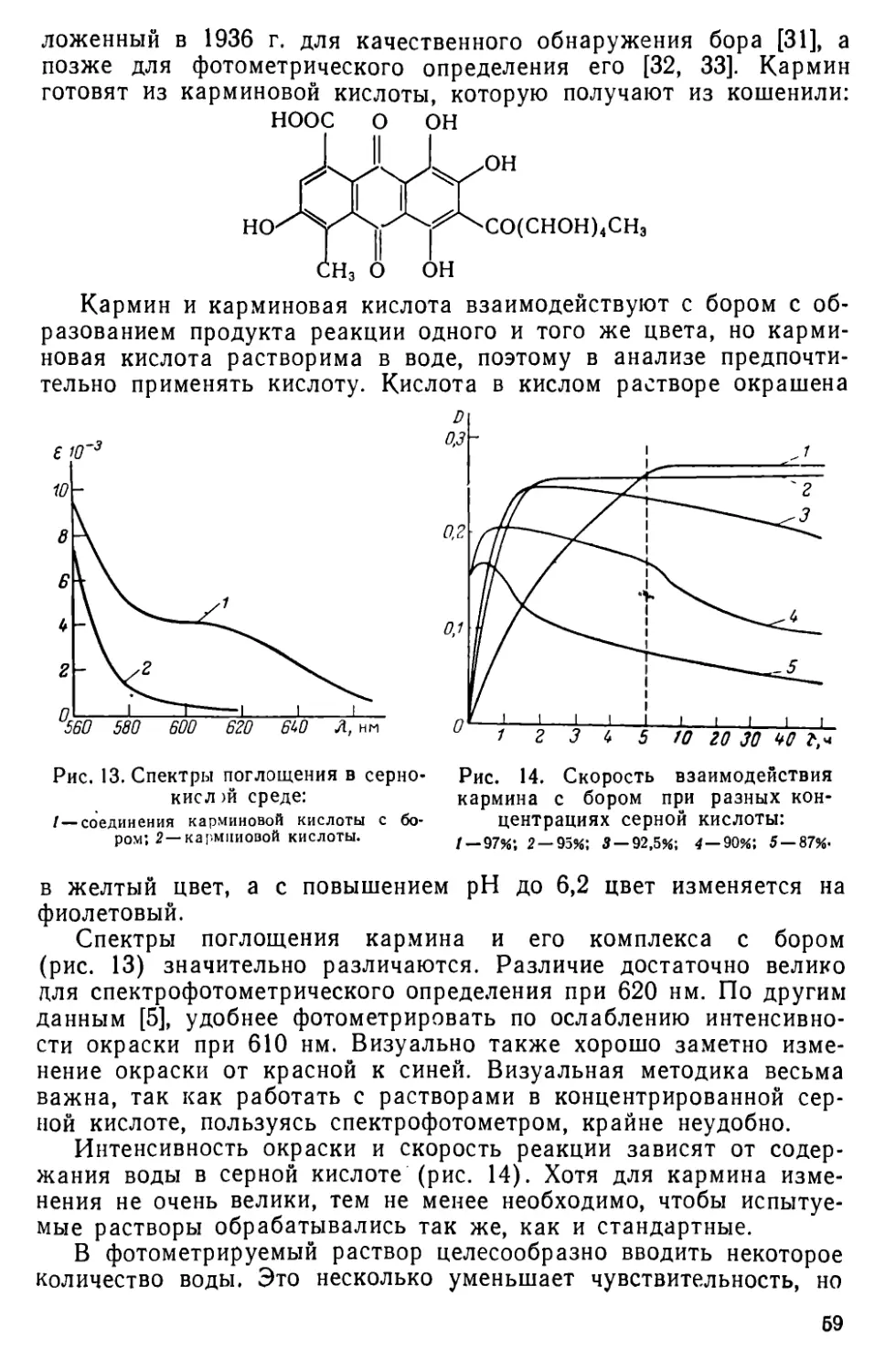
II. 6. Определение бора с применением кармина в среде концентрированной серной кислоты ......................................... 58
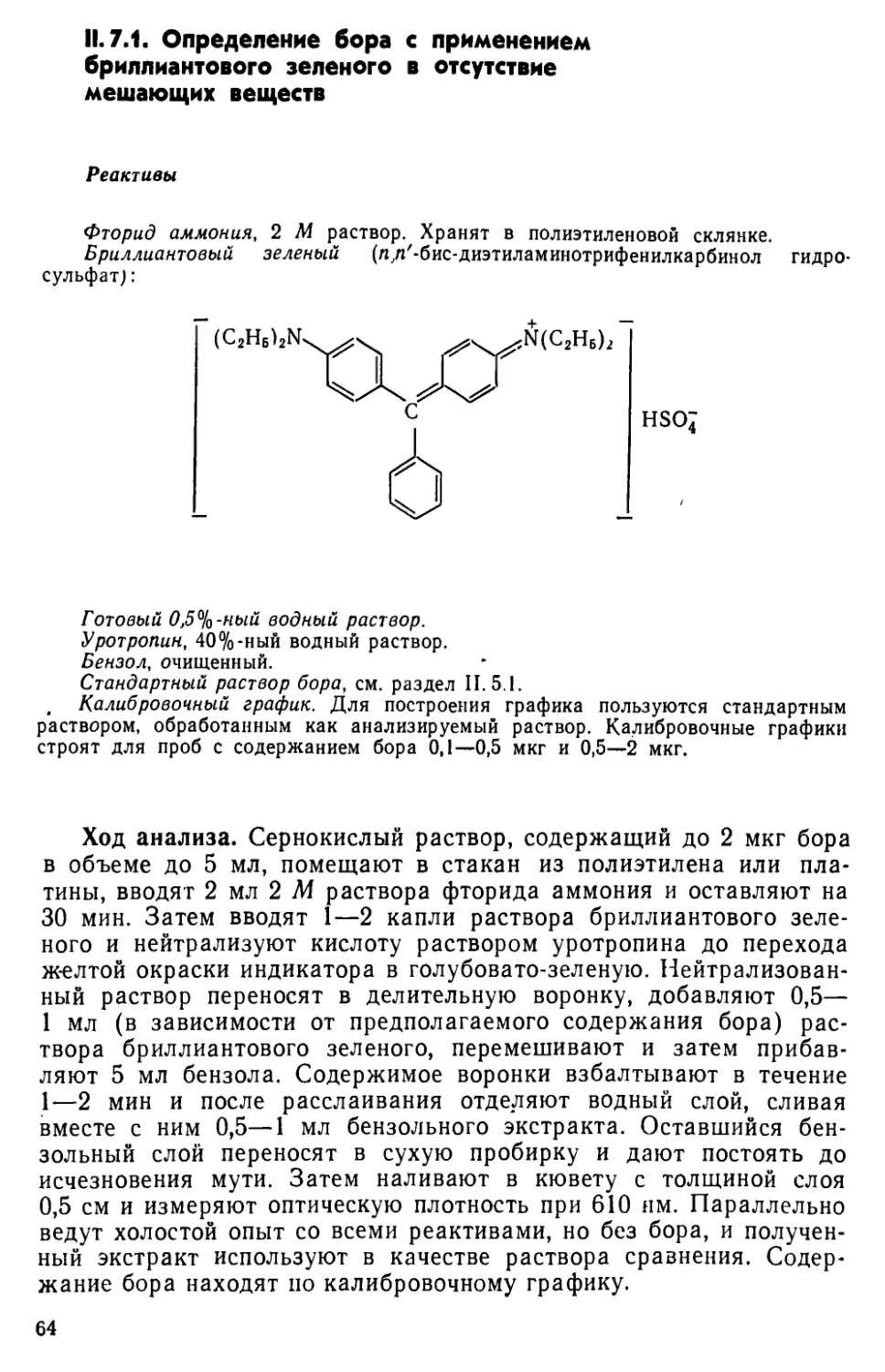
11.7. Определение бора в виде комплекса тетрафторида бора с бриллиантовым зеленым..........................................62
II. 8. Определение бора с применением салициловой кислоты и кристаллического фиолетового .... ............. . . .65
II. 9. Другие методы определения бора .......................68
Литература . .............................................70
Глава III. Фосфор и кремний......................................73
III. 1. Общая характеристика методов определения фосфора и кремния 73
III. 2. Подготовка материала к анализу и методы устранения мешающих веществ ... ... . 77
III. 3. Определение фосфора в виде желтой фосфорномолибденовой гетерополикислоты..................................... .... 80
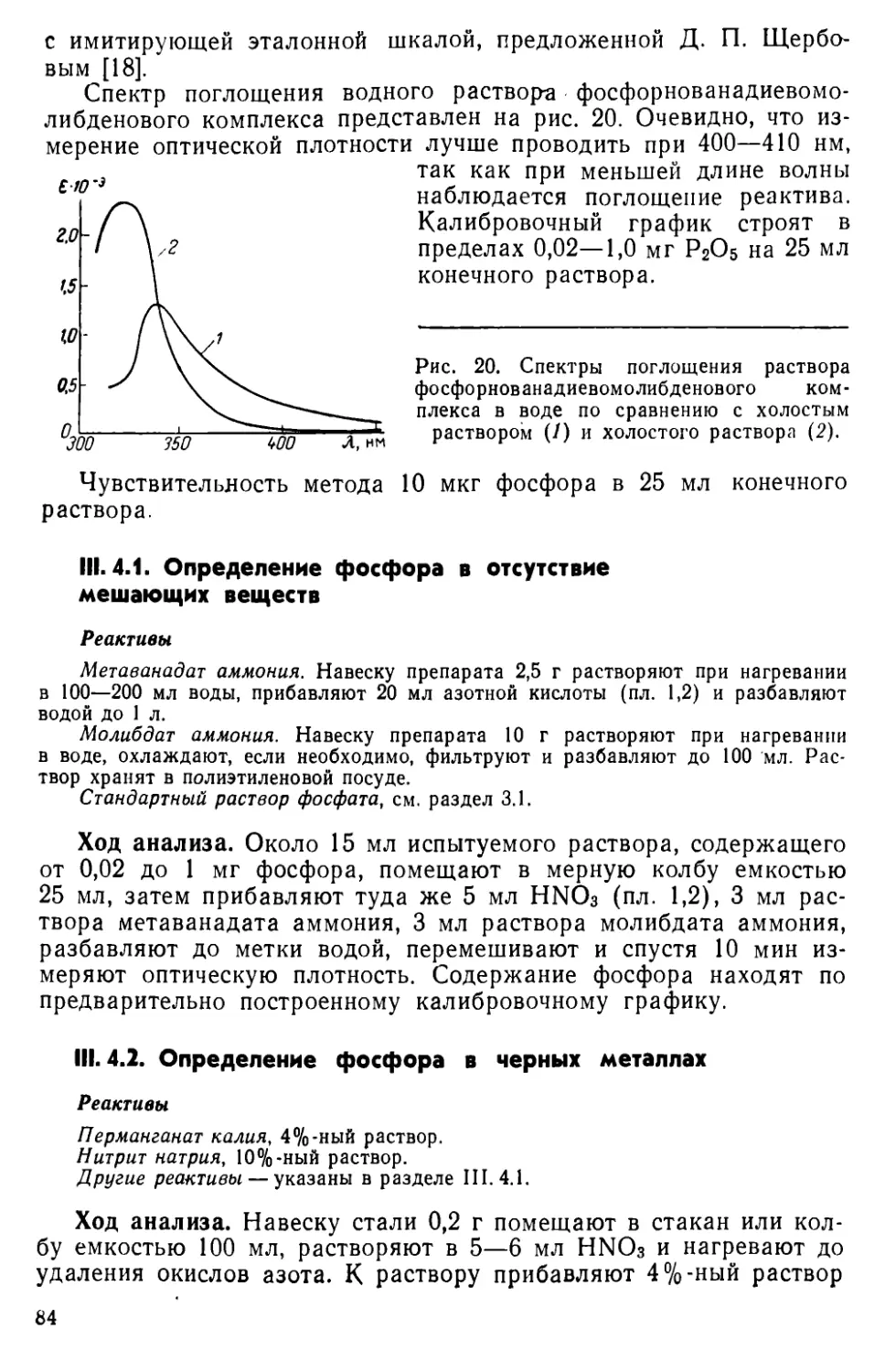
III. 4. Определение фосфора в виде желтого фосфорнованадиевомолиб-
денового комплекса ..... .... 83
III. 5. Экстракционно-фотометрический метод определения фосфора в виде фосфорнованадиевомолибденового комплекса ...... 87
III. 6. Определение фосфора в виде фосфорномолибденового комплекса с последующим восстановлением железом (II) и сульфитом . • 88
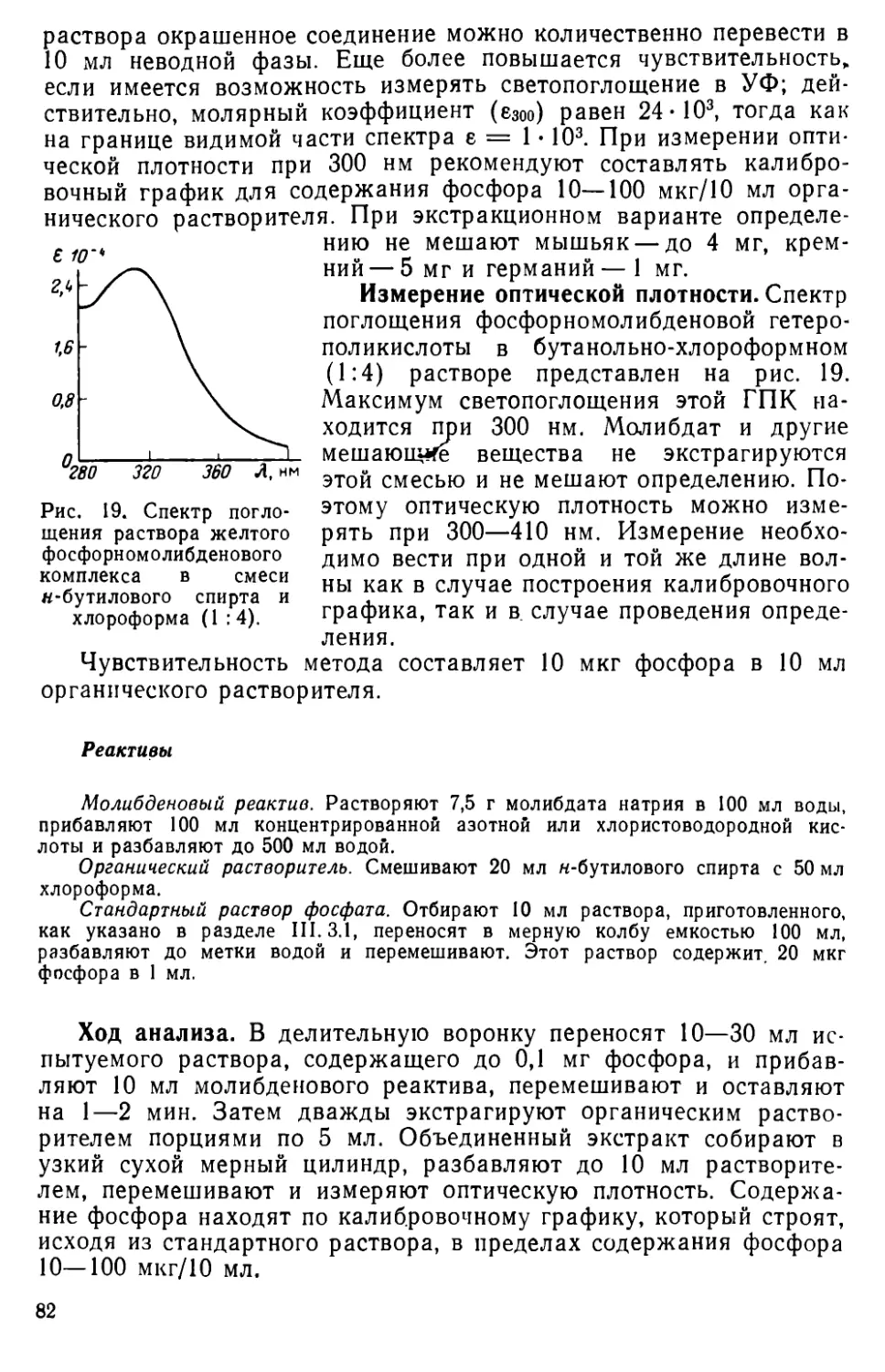
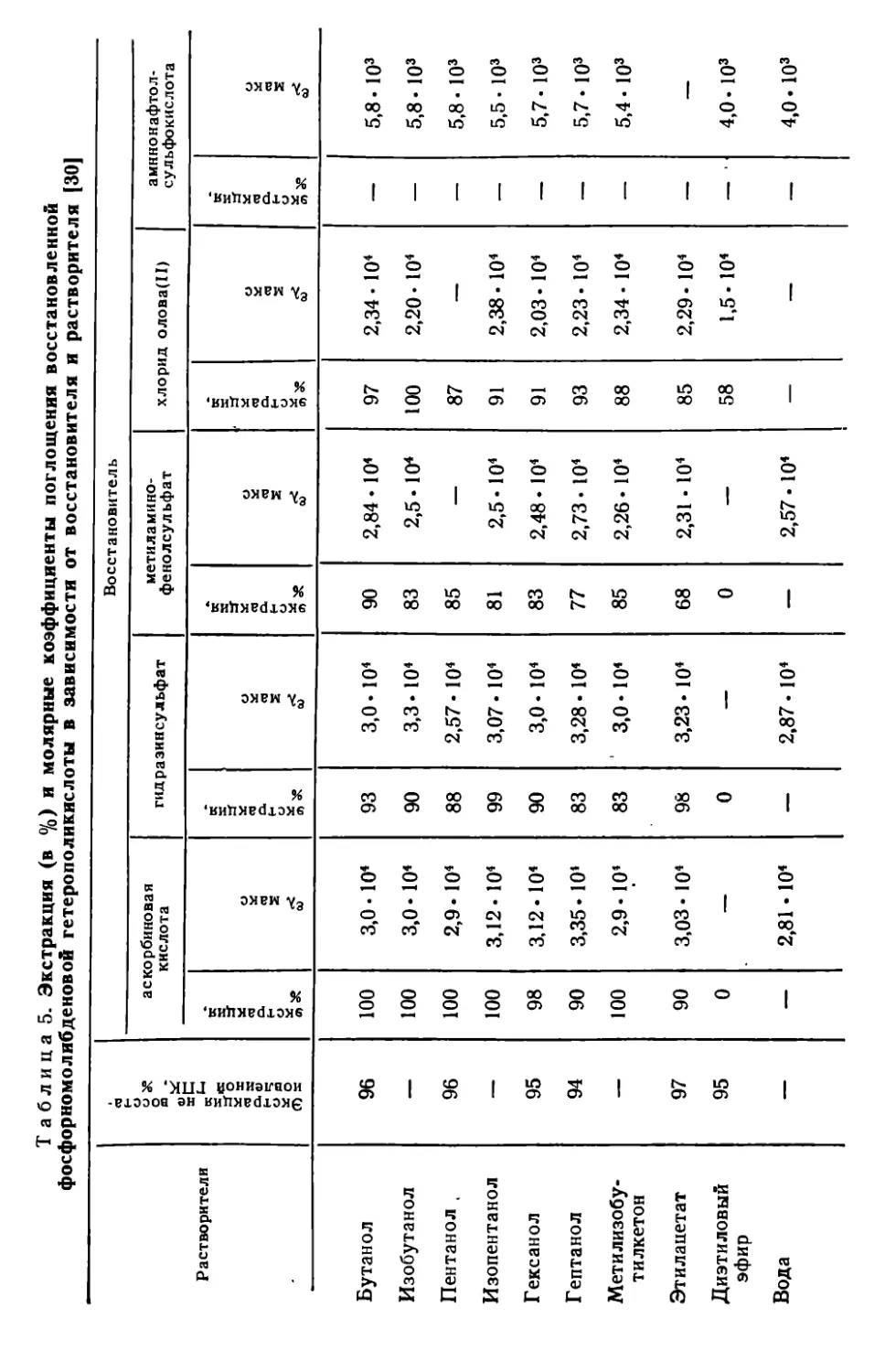
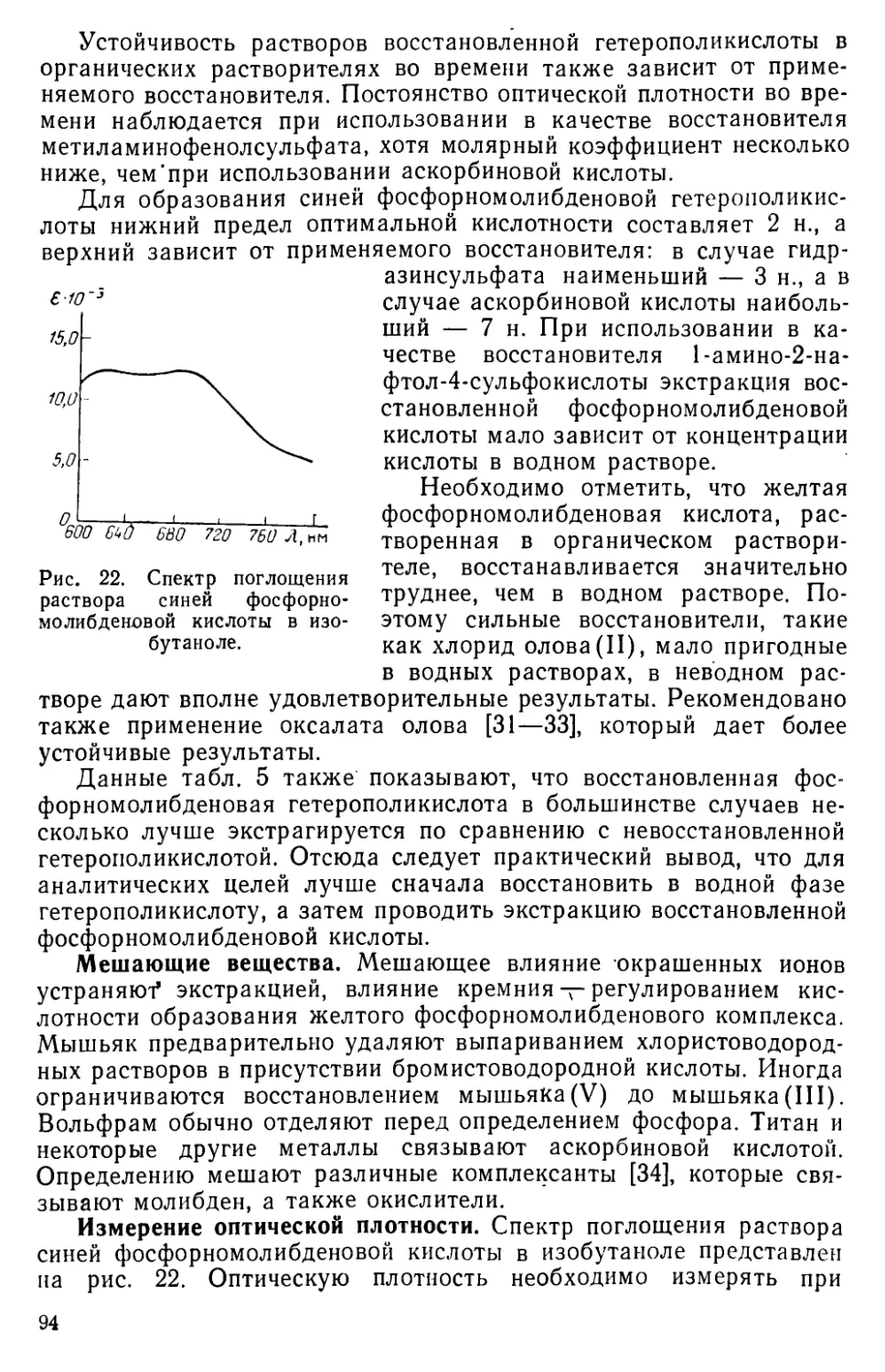
III. 7. Экстракционно-фотометрическое определение фосфора в виде синей фосфорвомолибденовой гетерополикислоты........................92
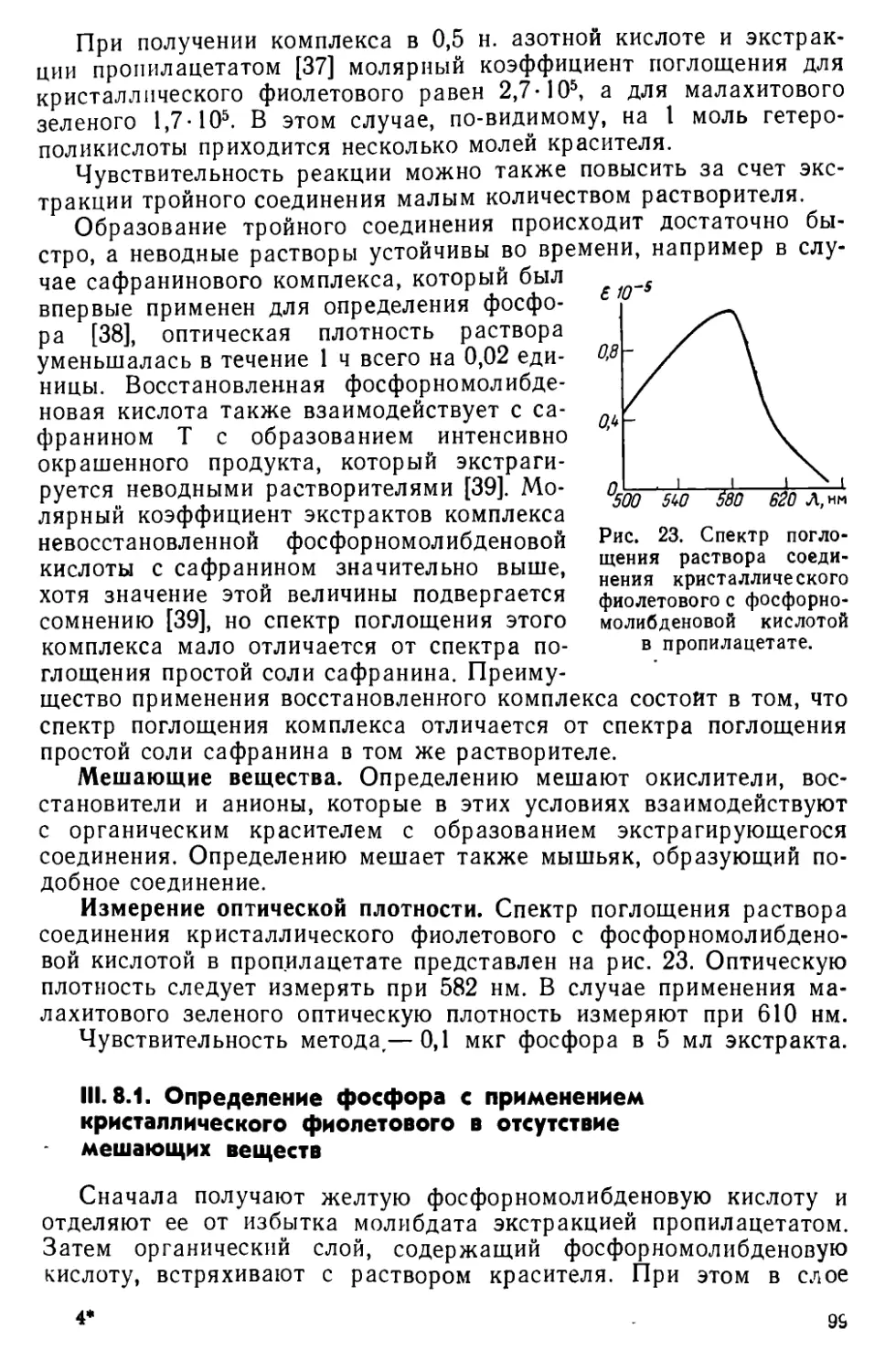
III. 8. Определение фосфора в виде соли фосфорномолибденовой кислоты с основными красителями..................................... 98
III. 9. Экстракционное отделение и определение фосфора в присутствии кремния и мышьяка ..............................................ЮЗ
III . 10. Другие методы определения фосфора.........................105
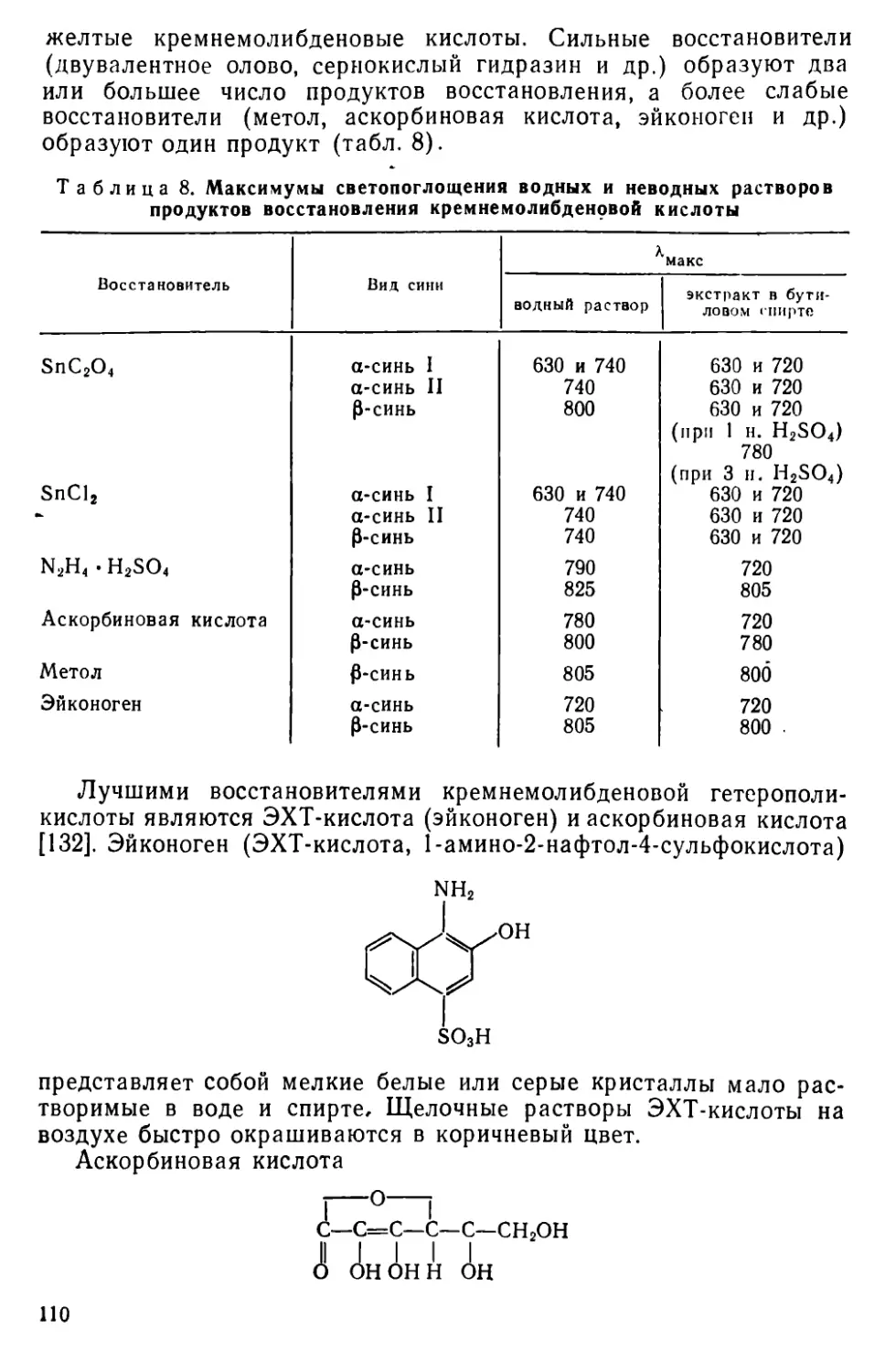
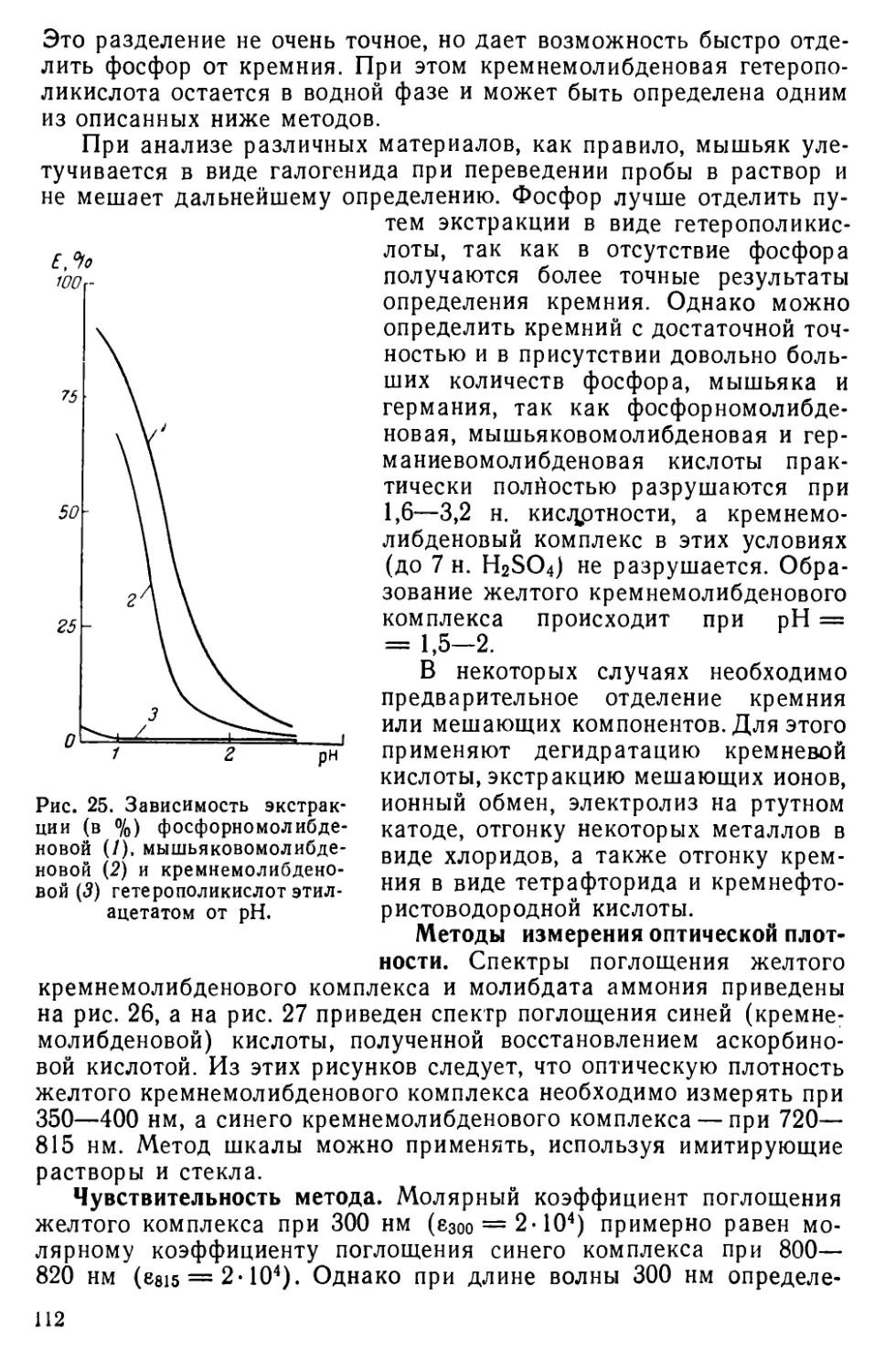
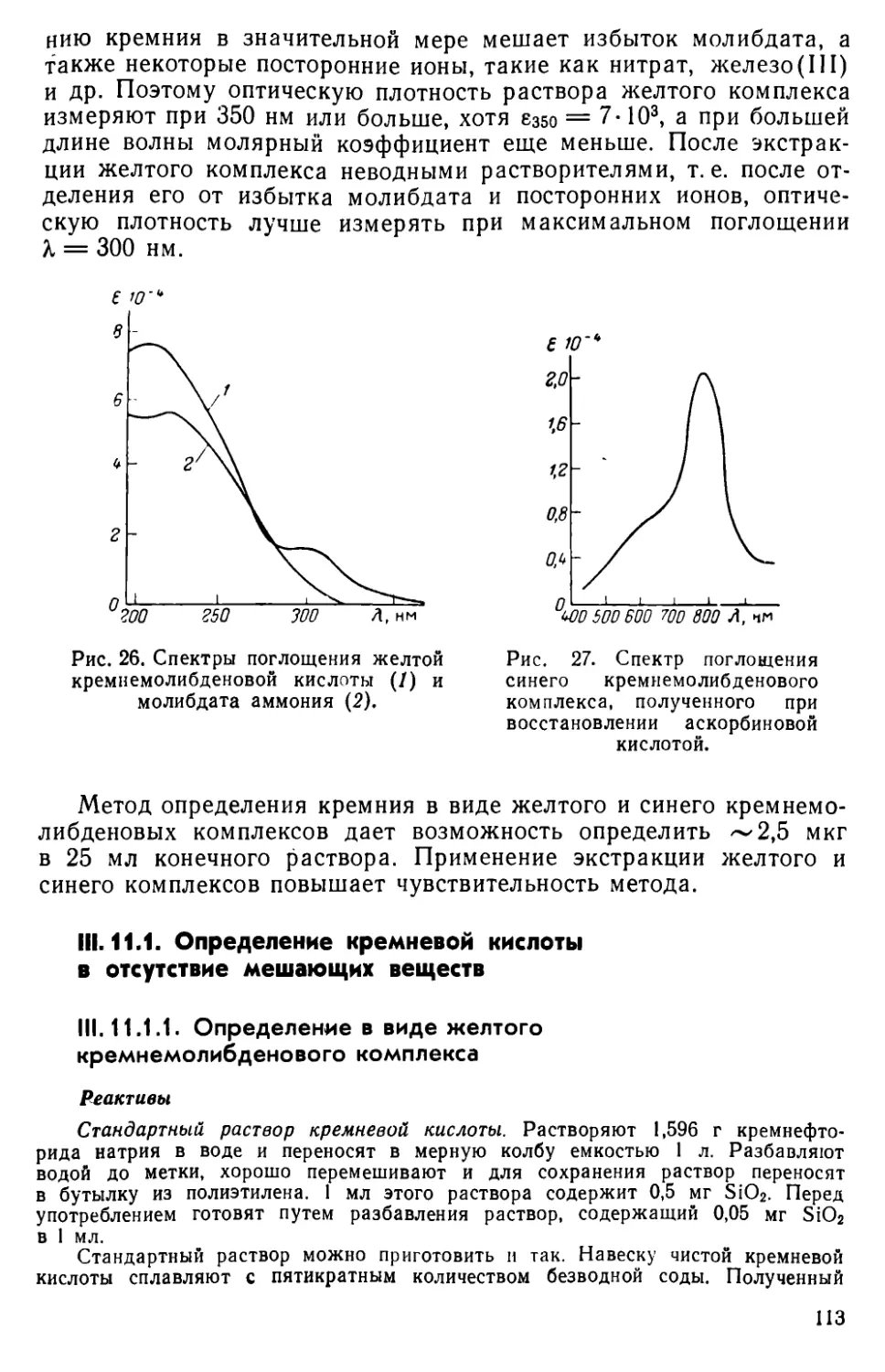
II I. 11. Методы определения кремния............................... 109
III . 12. Другие методы определения кремния....................... 116
Литература . 129
Глава IV. Мышьяк........................................................134
IV. 1. Методы отделения и концентрирования ....................... 134
IV. 2. Общая характеристика методов определения мышьяка.............141
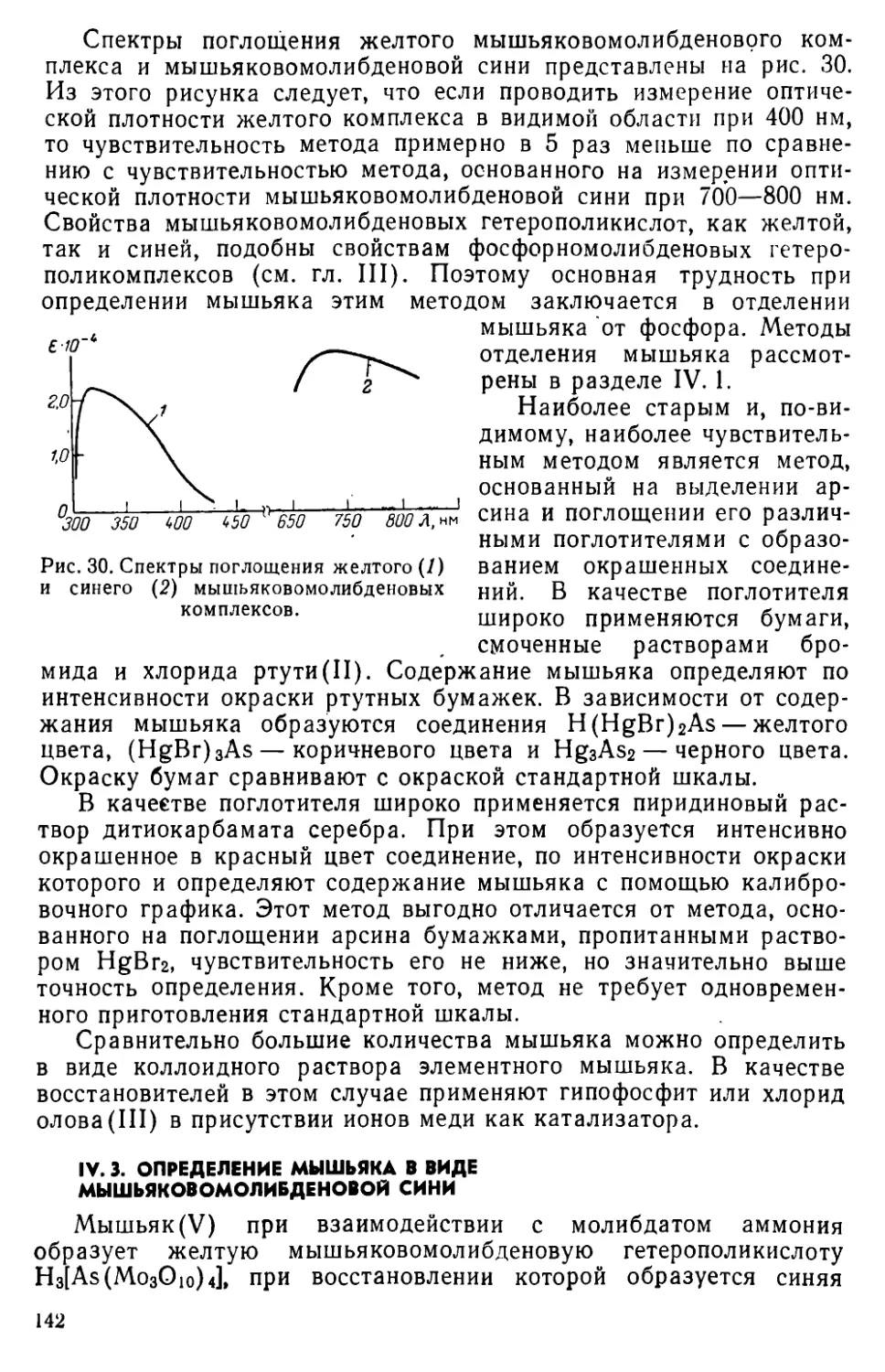
IV. 3. Определение мышьяка в виде мышьяковомолибденовой сяни • 142
IV . 4. Определение мышьяка с применением индикаторных бумаг . .158
IV. 5. Определение мышьяка в виде арсенидов серебра и ртути с последующим проявлением.............................................. 161
IV. 6. Определение мышьяка с применением диэтилдитиокарбамата серебра ........................................................... 163
IV. 7. Определение мышьяка с применением гипофосфита................165
IV. 8. Определение микроколичеств мышьяка с применением бутилрод-амина ............................................................168
IV. 9. Другие методы определения мышьяка.......................... 170
Литература . 171
Глава V. Кислород.....................................................175
V. 1. Общая характеристика методов определения кислорода ... .175
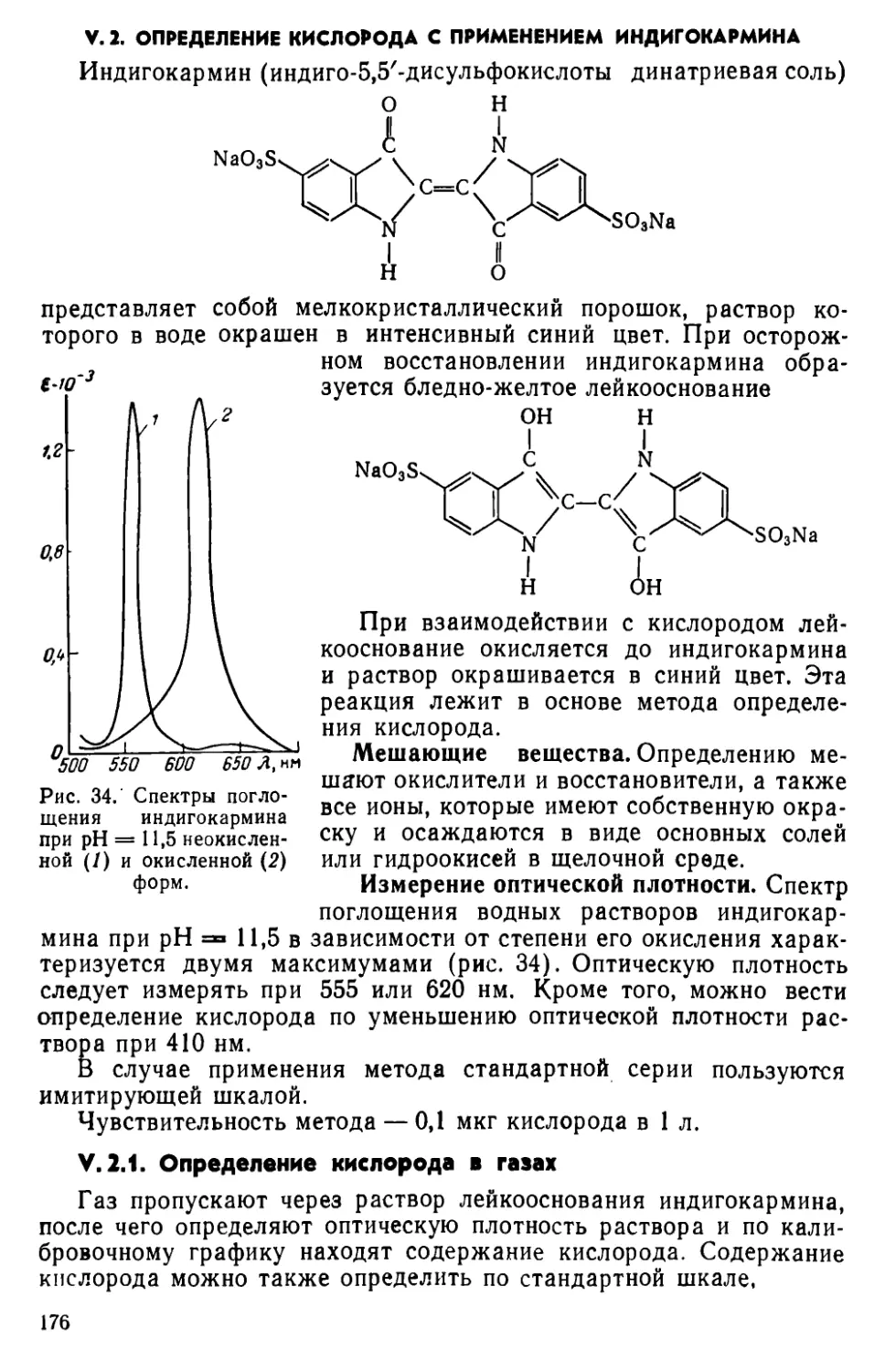
V. 2. Определение кислорода с применением индигокармина ..... 176
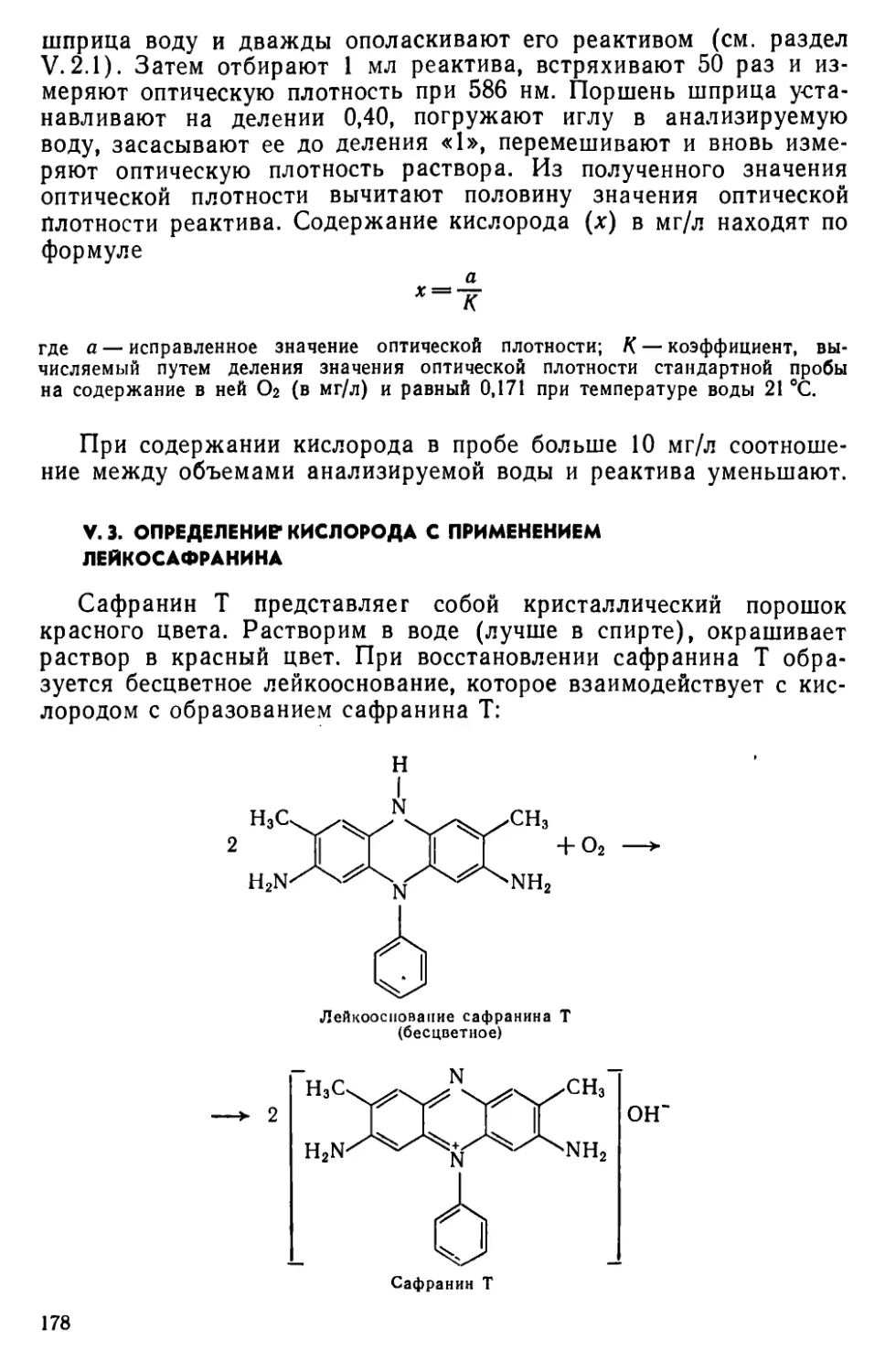
V. 3. Определение кислорода с применением лейкосафранина . . . .178
V. 4. Определение кислорода видоизмененным методом Винклера . . .179
V. 5. Определение кислорода по количеству окисленной меди .... 180
V. 6. Другие методы определения кислорода........................182
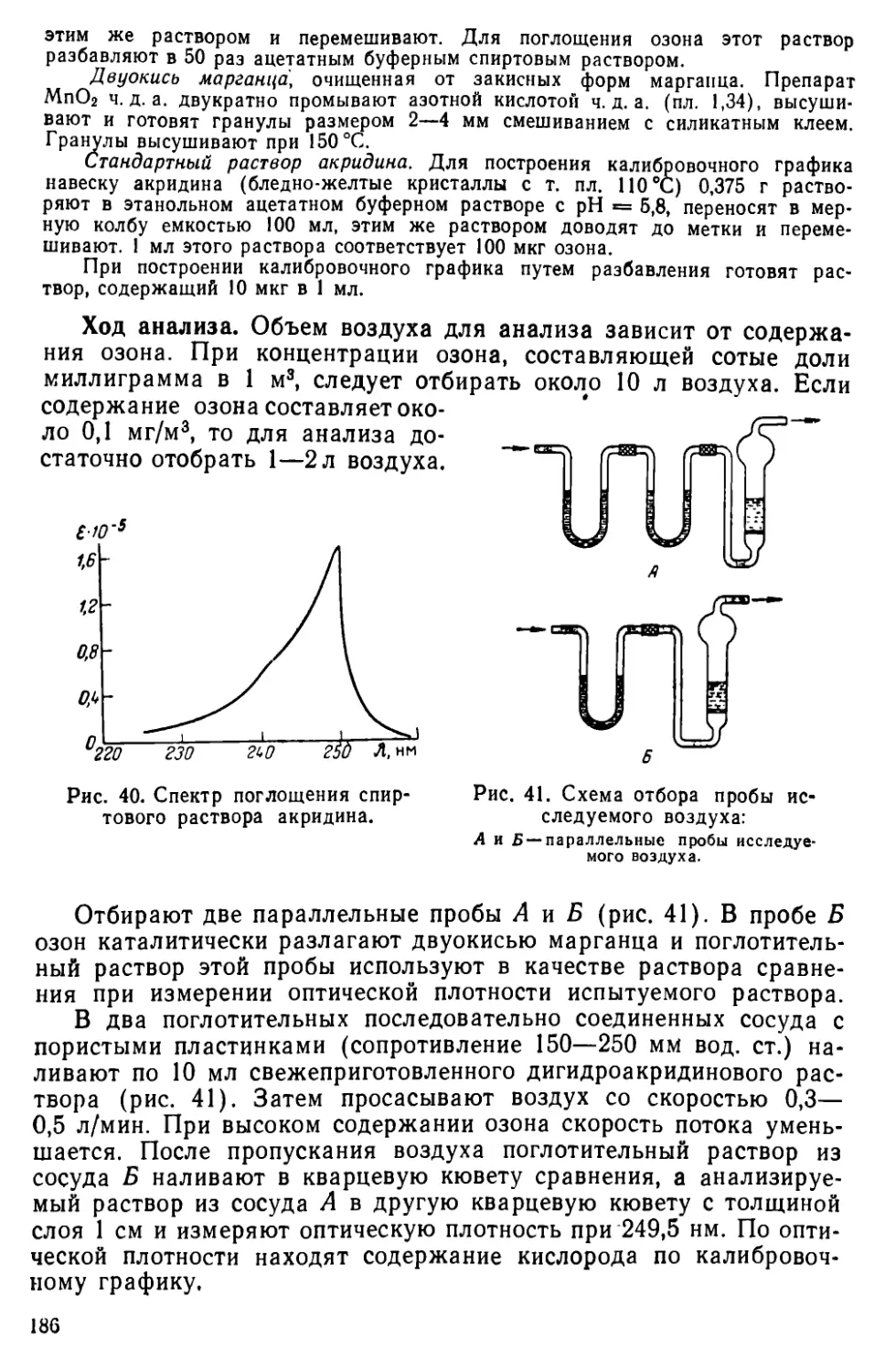
V. 7. Методы определения озона.....................................183
Литература..............................................................187
Глава VI. Сера..........................................................189
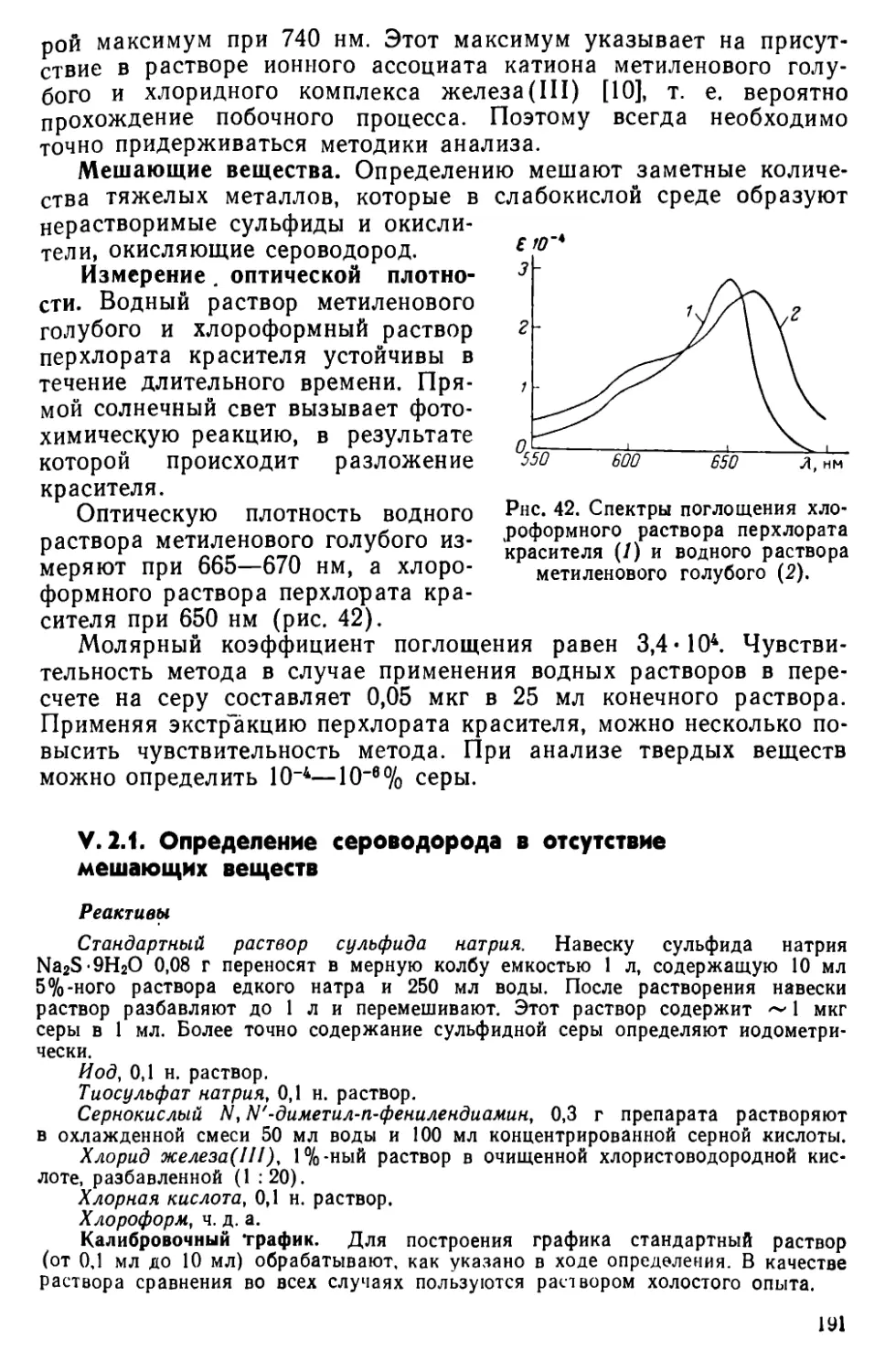
VI. 1. Методы отделения . . ....................189
VI. 2. Определение сероводорода в виде метиленового голубого . . . 190
VI. 3. Определение свободной серы ................... .... 196
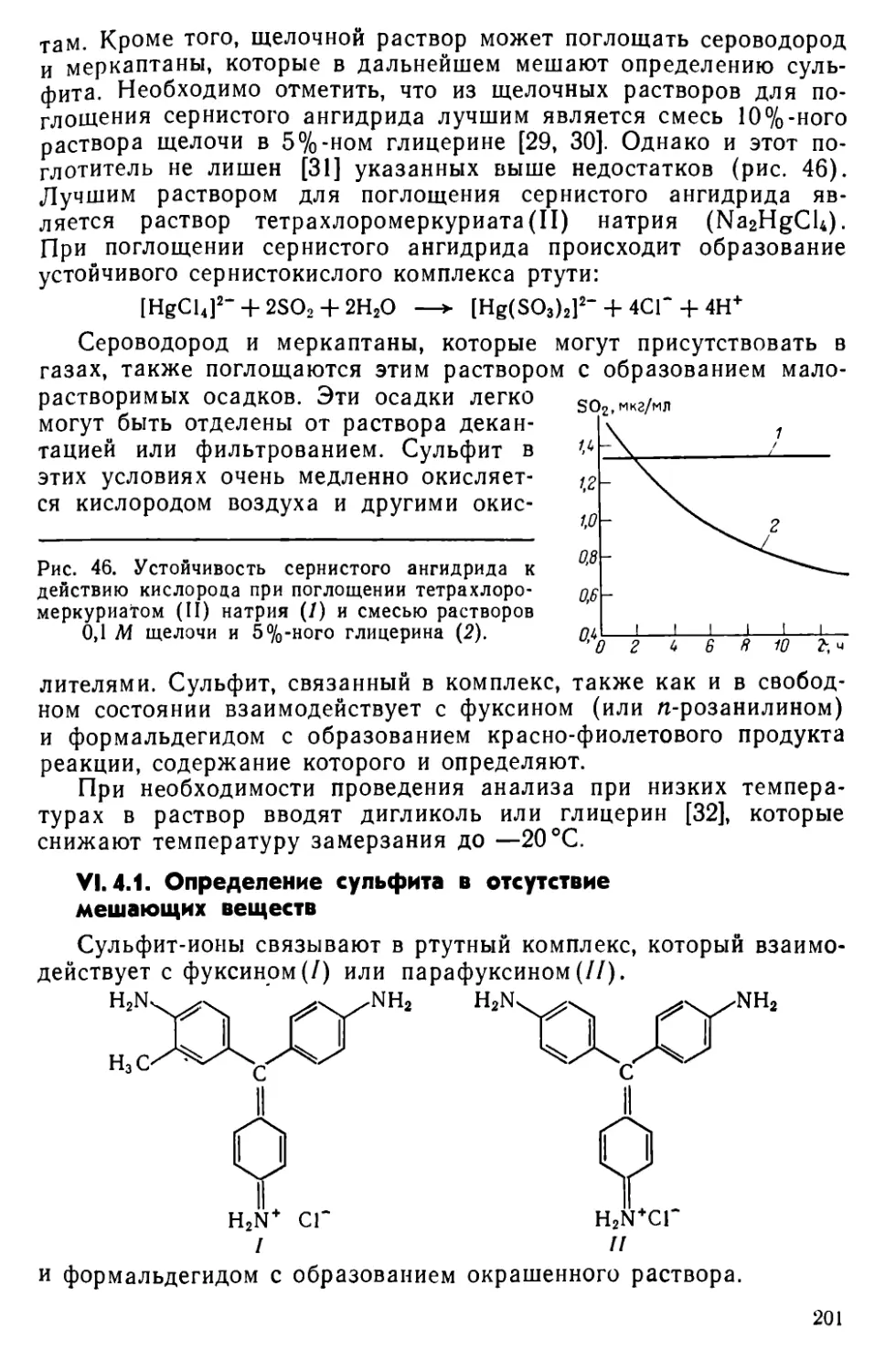
VI. 4. Определение сернистой кислоты..........................200
VI. 5. Определение роданида...................................203
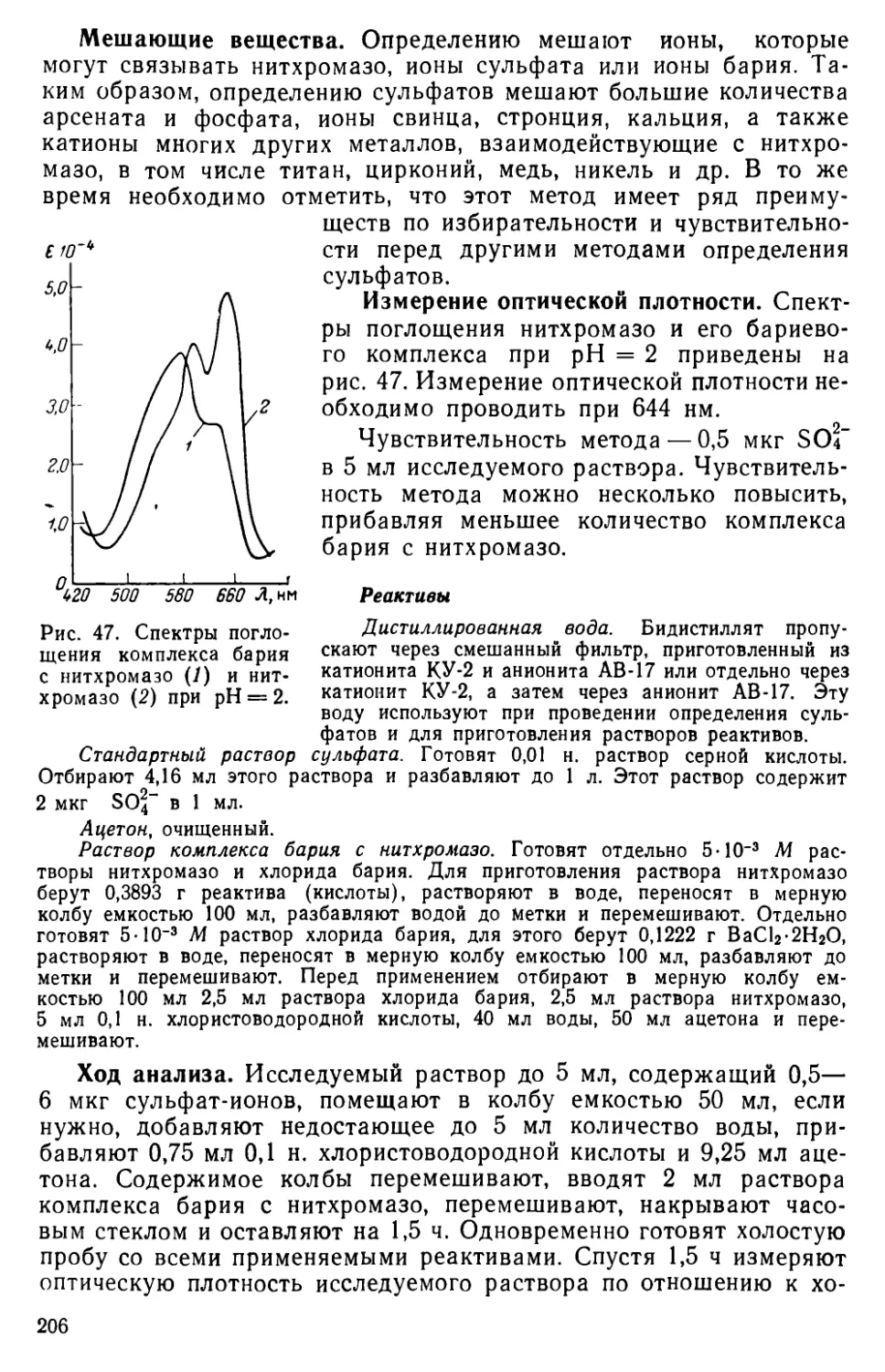
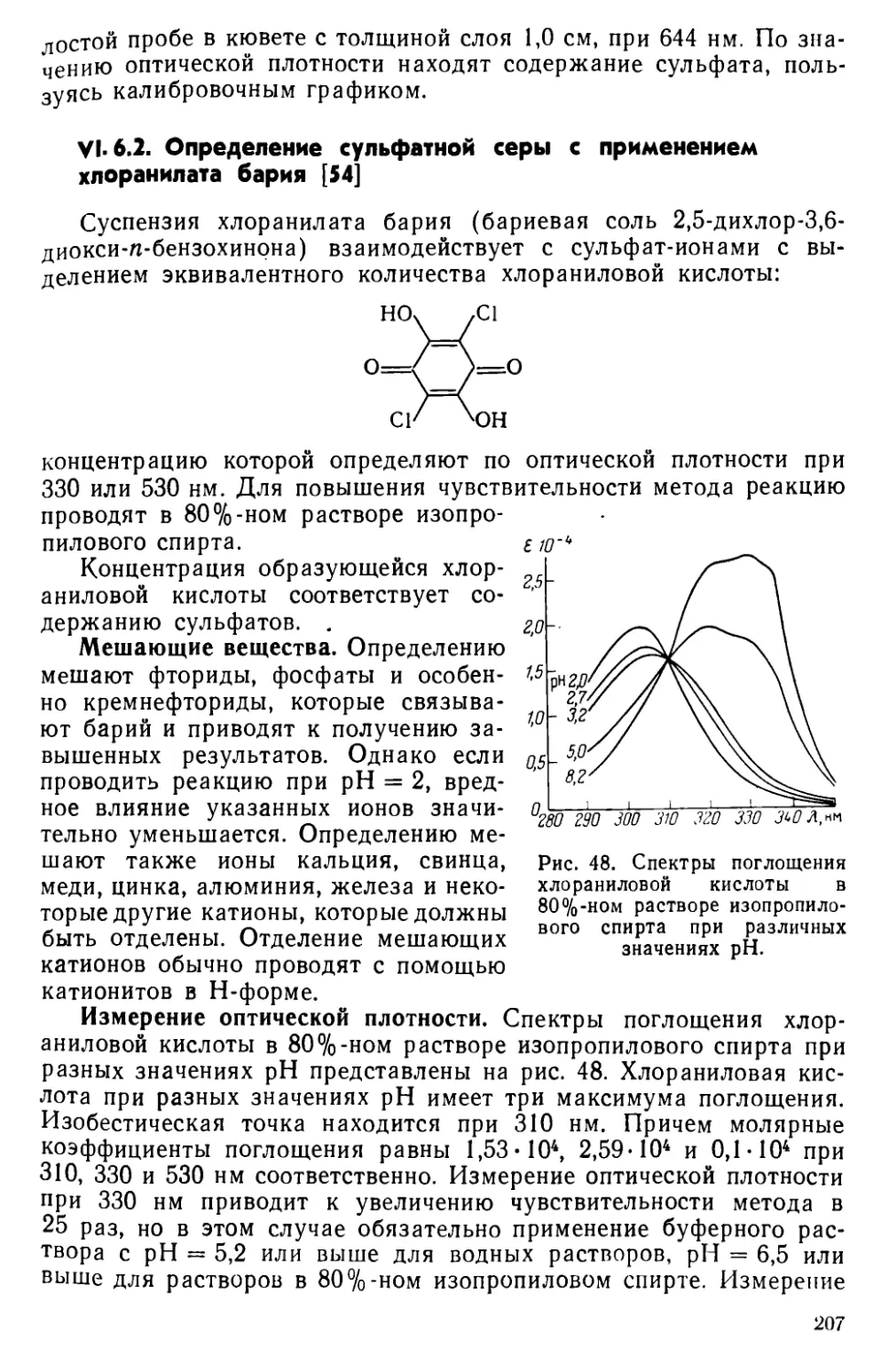
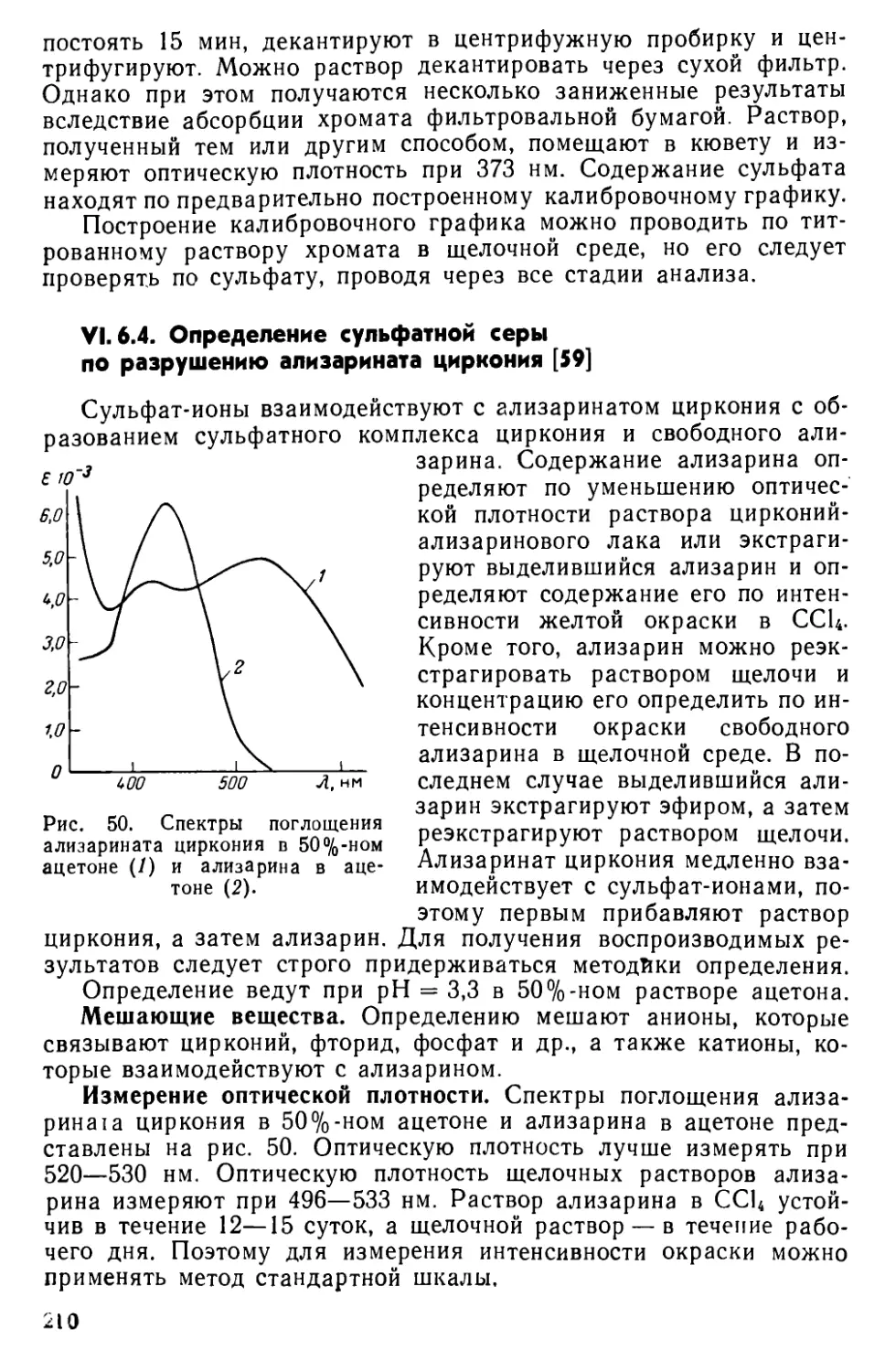
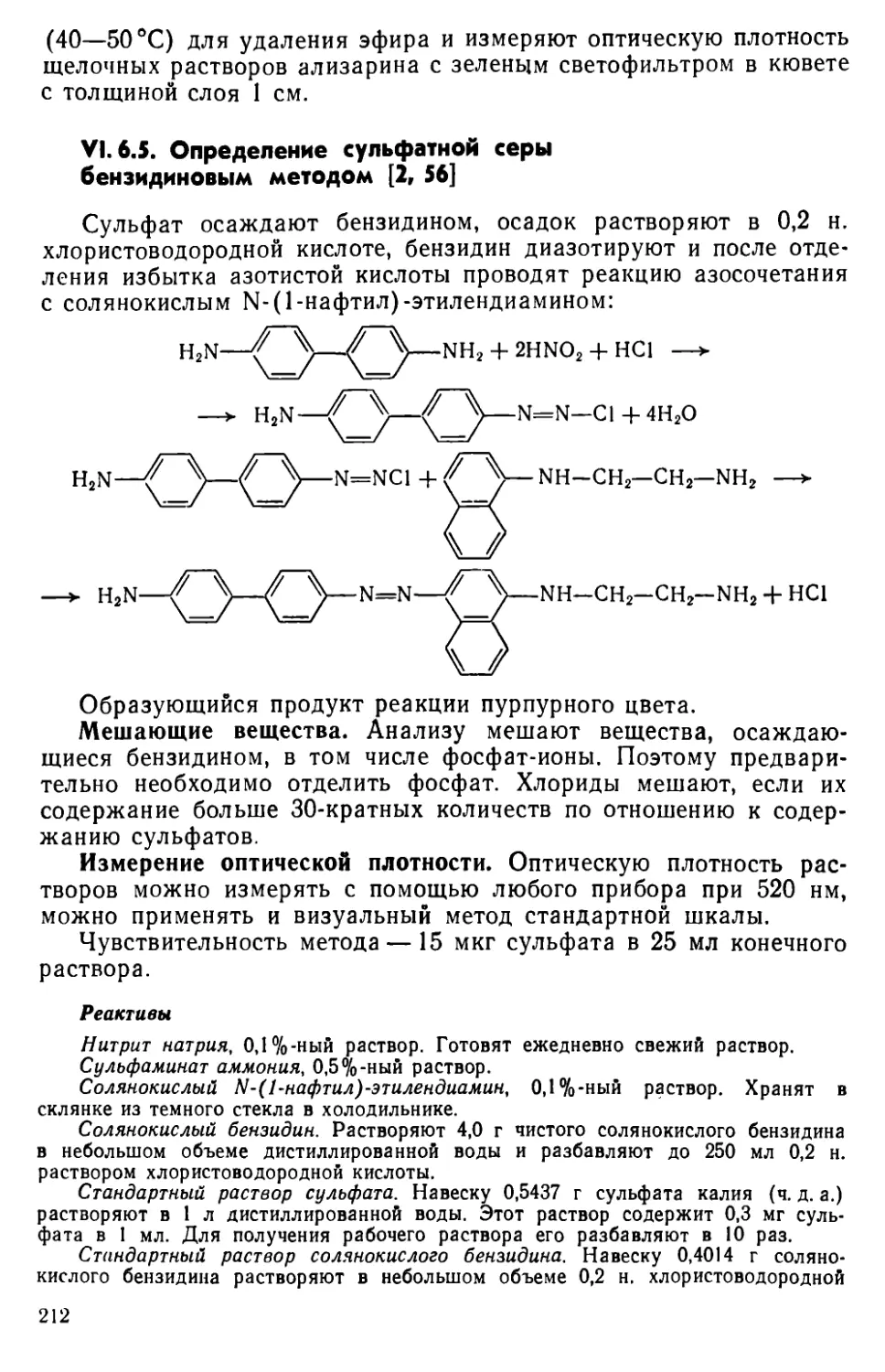
VI. 6. Определение сульфатной серы ......................204
VI. 7. Другие методы определения серы ... . . 215
Литература .'............................................................218
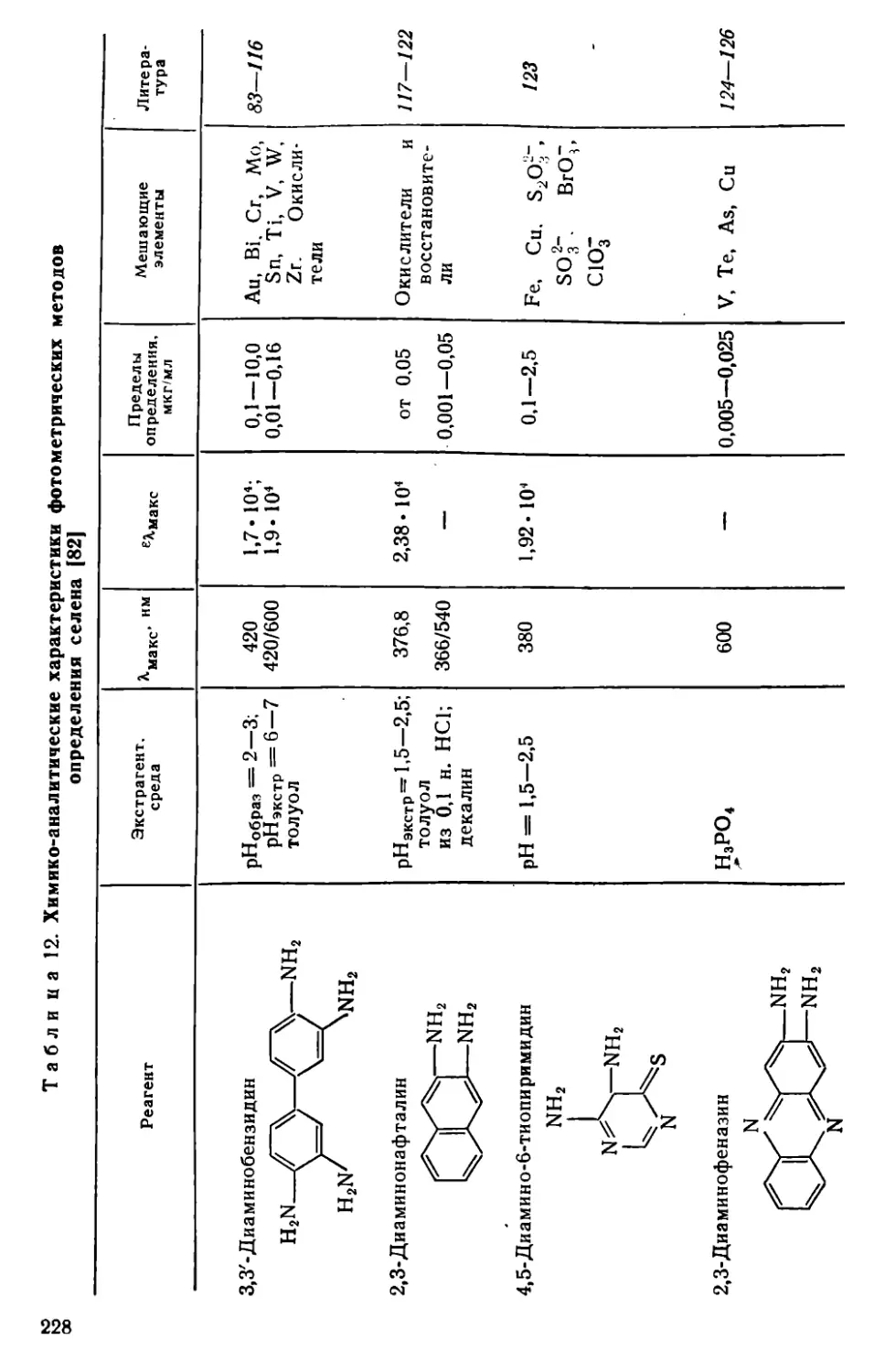
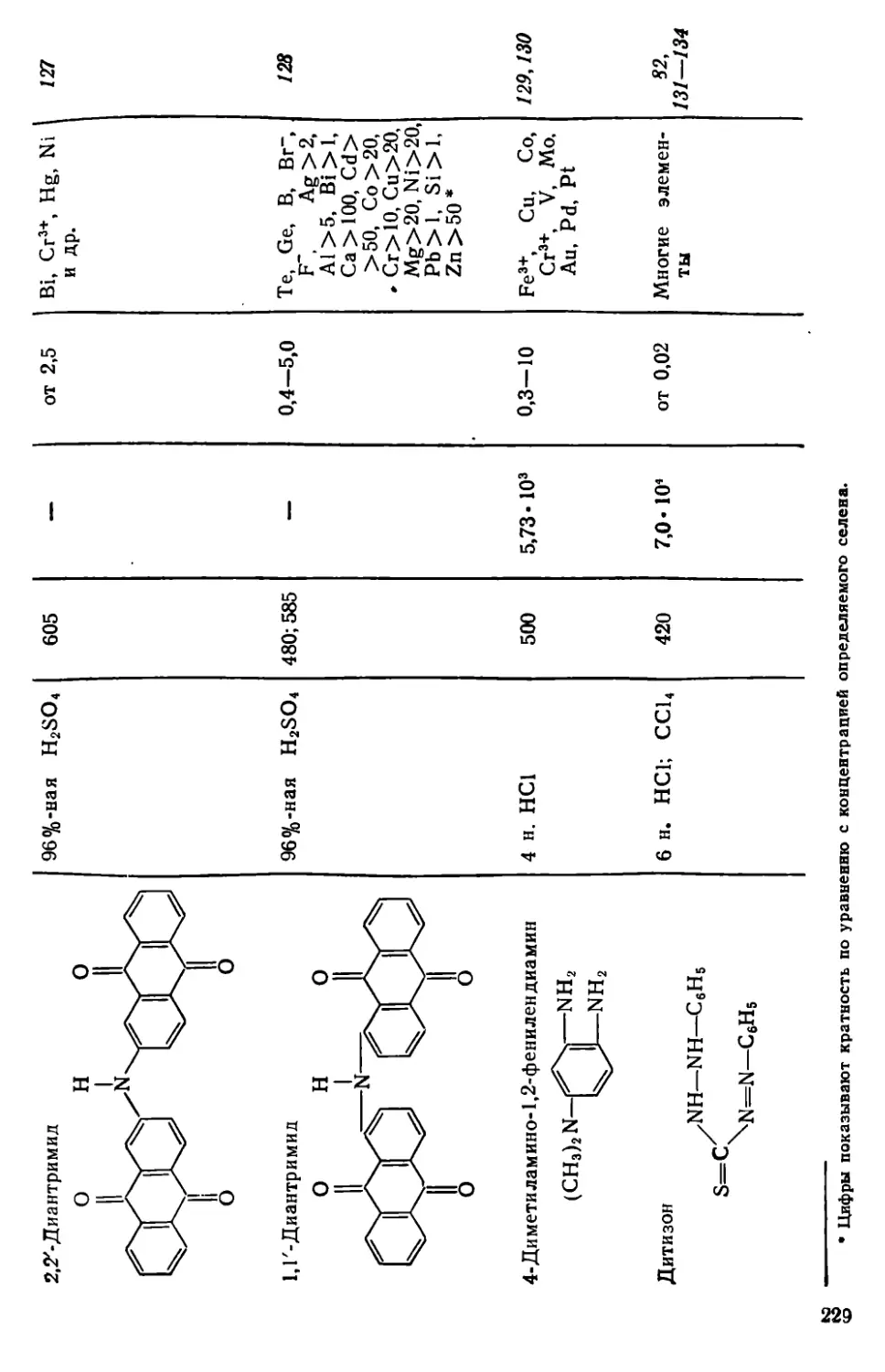
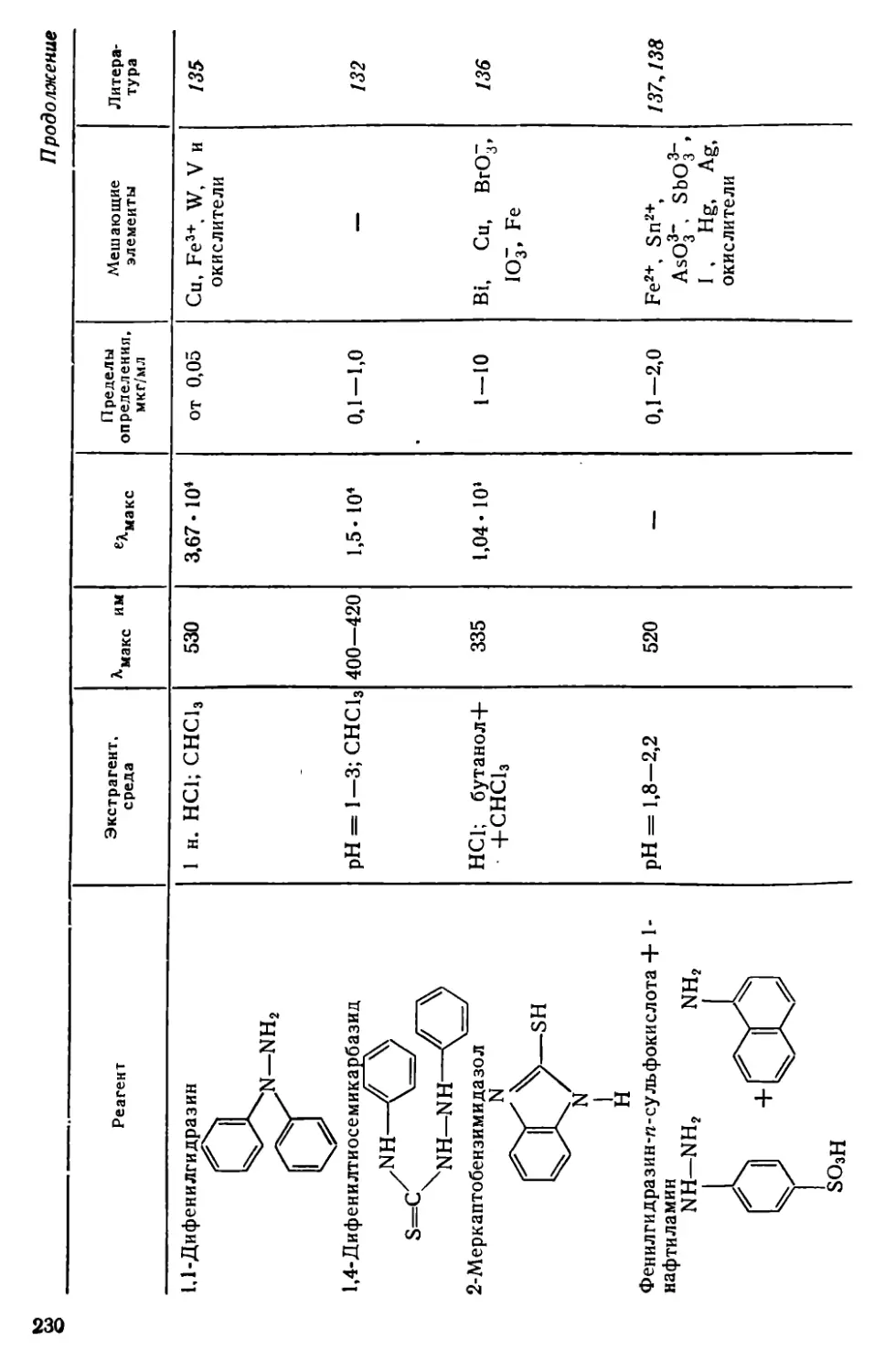
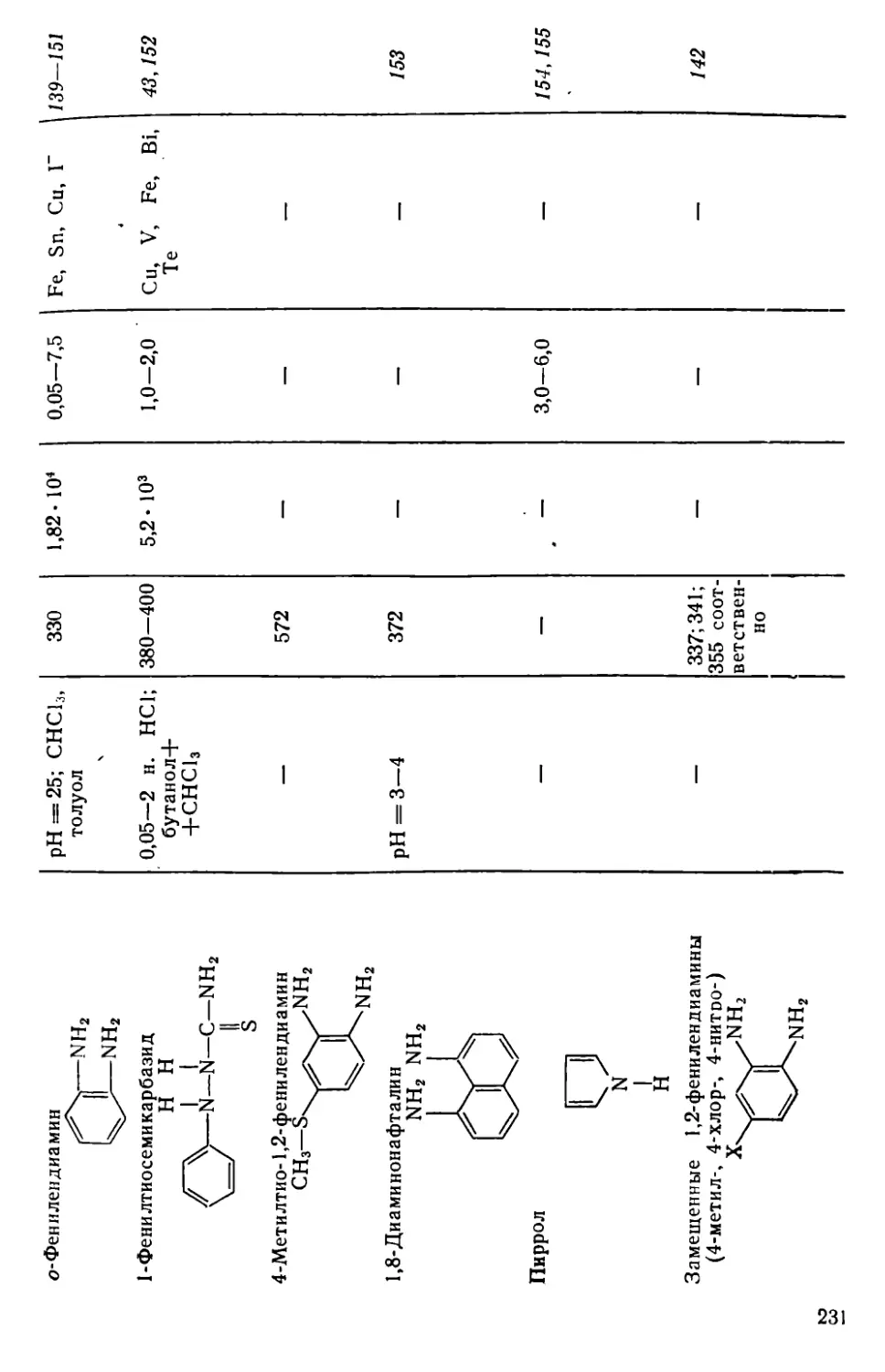
Глава VII. Селен и теллур . . . .........................................222
VII. 1. Методы отделения и разделения.......................... . 222
VII. 2. Общая характеристика методов определения селена и теллура • 226
VII. 3. Определение селена с применением З.З'-диаминобензидина . 230
VII. 4. Определение селена в виде дитизоната...................241
VII. 5. Определение селена с применением 1,4-дифенилтиосемикарбазида 243
VII. 6. Определение селена и теллура в форме золей.............246
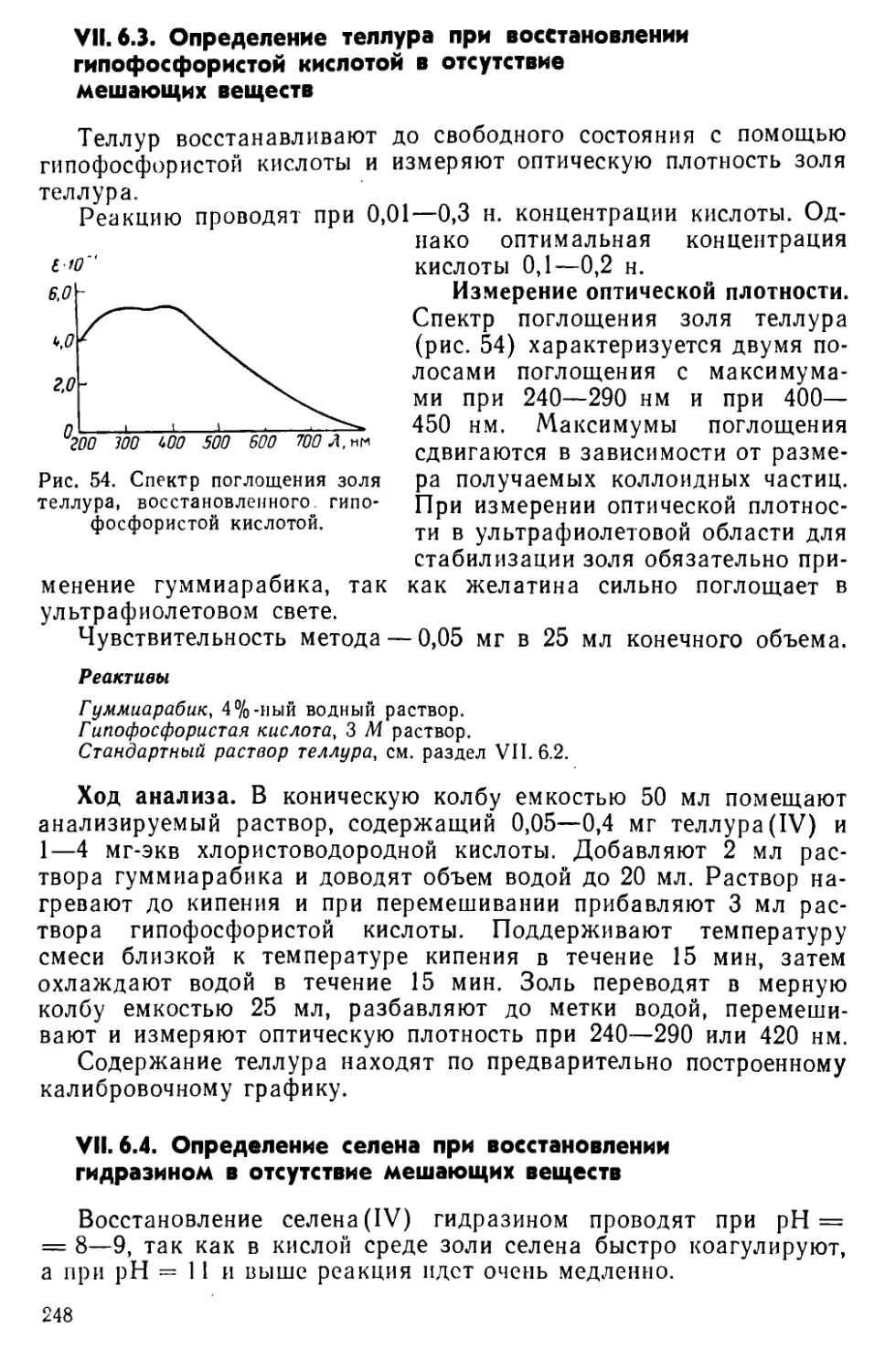
VII. 7. Определение селена по реакции окисления органических соединений ........................................................249
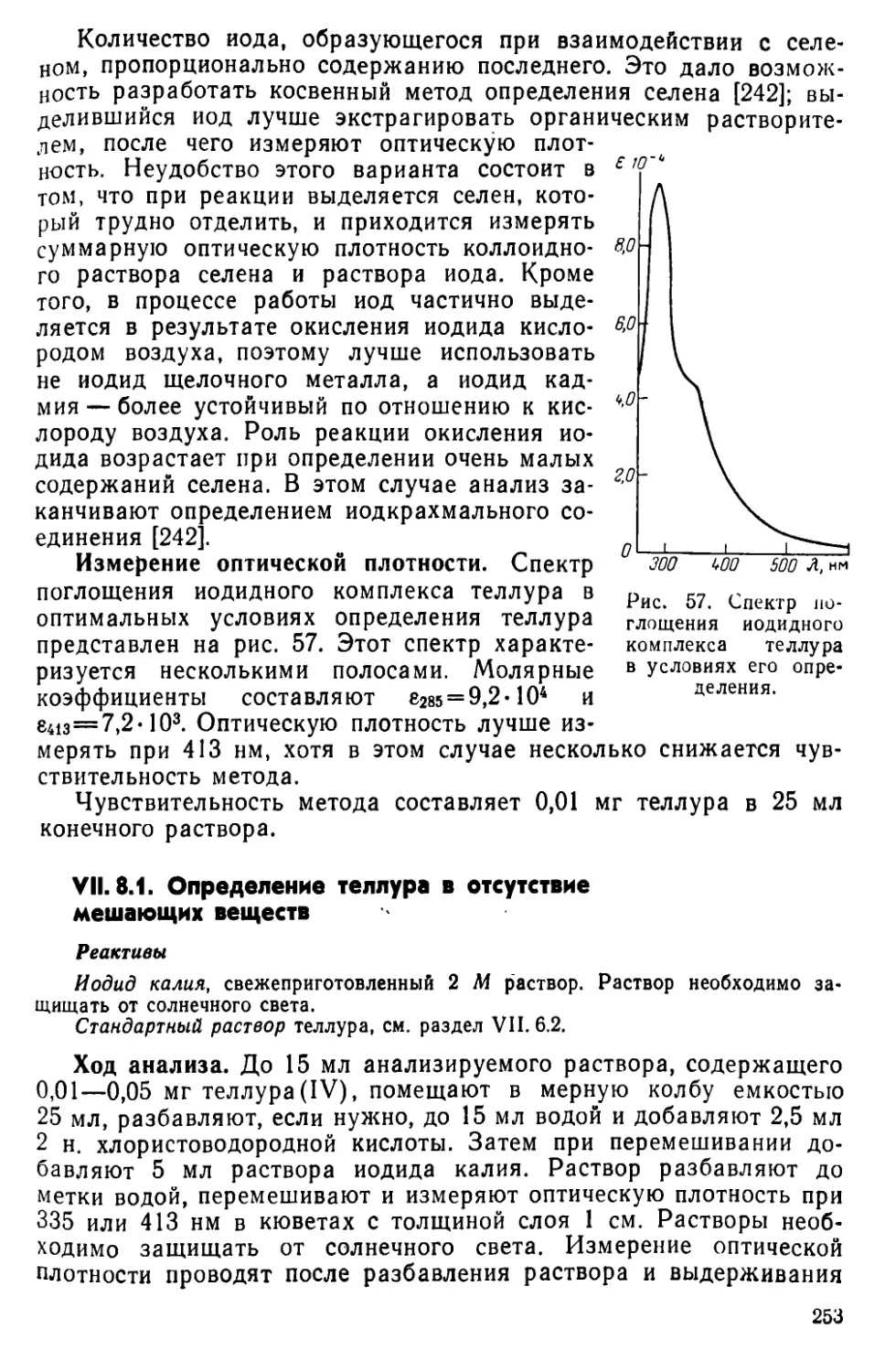
VII. 8. Определение теллура в виде иодидного комплекса...............252
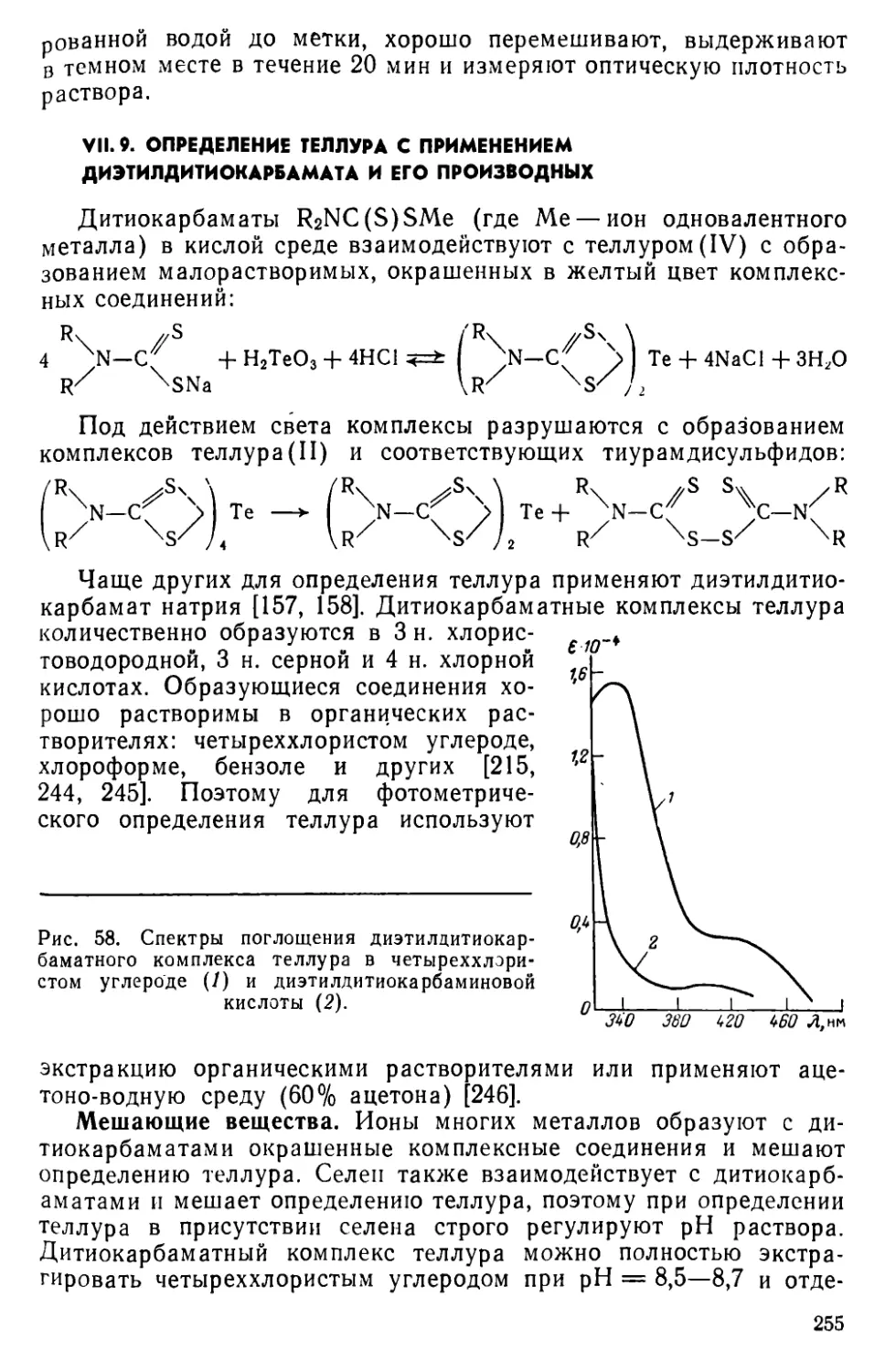
VII. 9. Определение теллура с применением диэтилдитиокарбамата и его производных...............................................255
VII. 10. Определение теллура с применением 3,5-дифенилпиразолин-1-дитиокарбамата натрия...........................................258
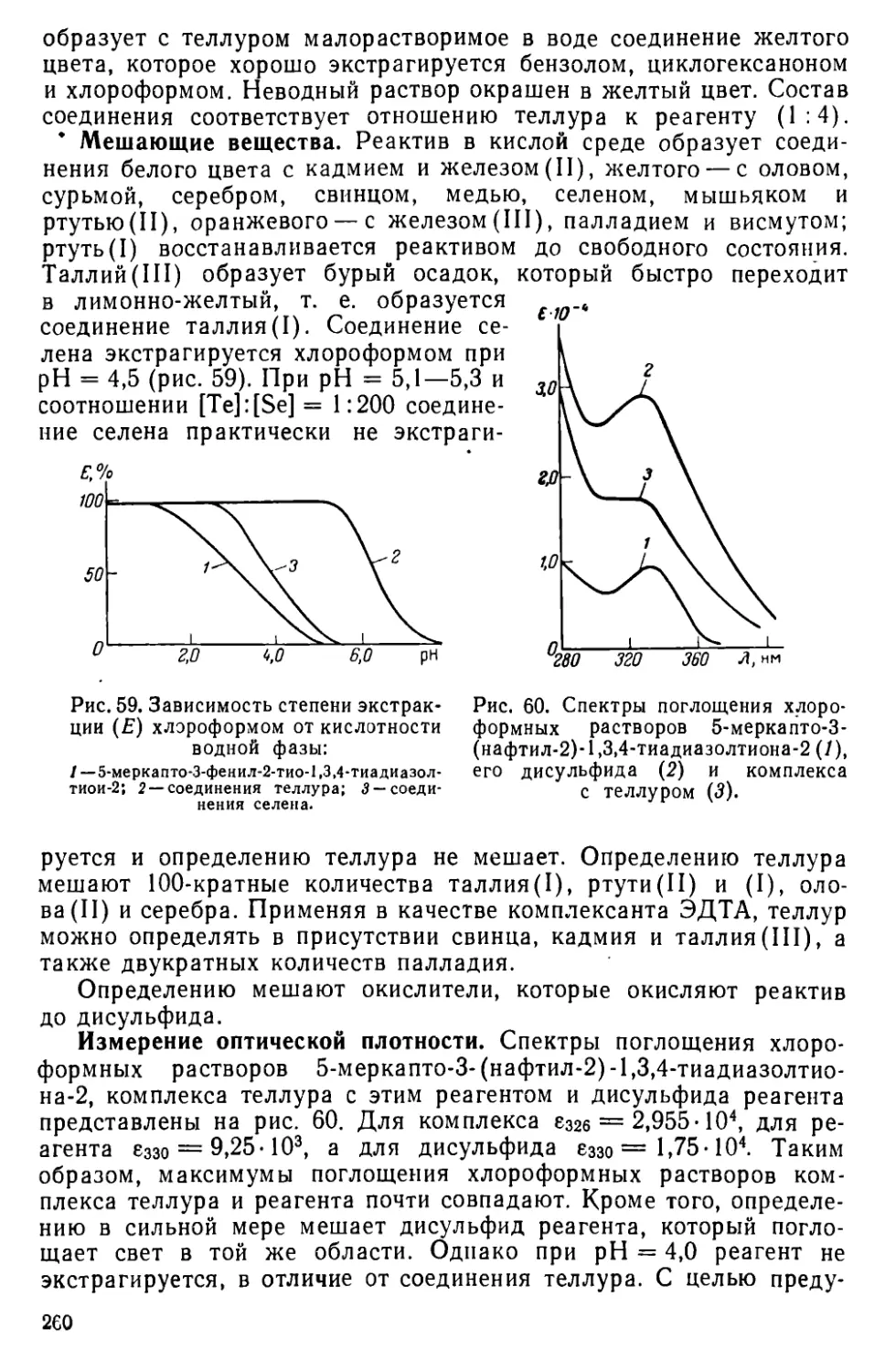
VII . 11. Определение теллура в виде комплекса с 5-меркапто-З-(нафтил-2)-1,3,4-тиадиазолтиона-2 ................................259
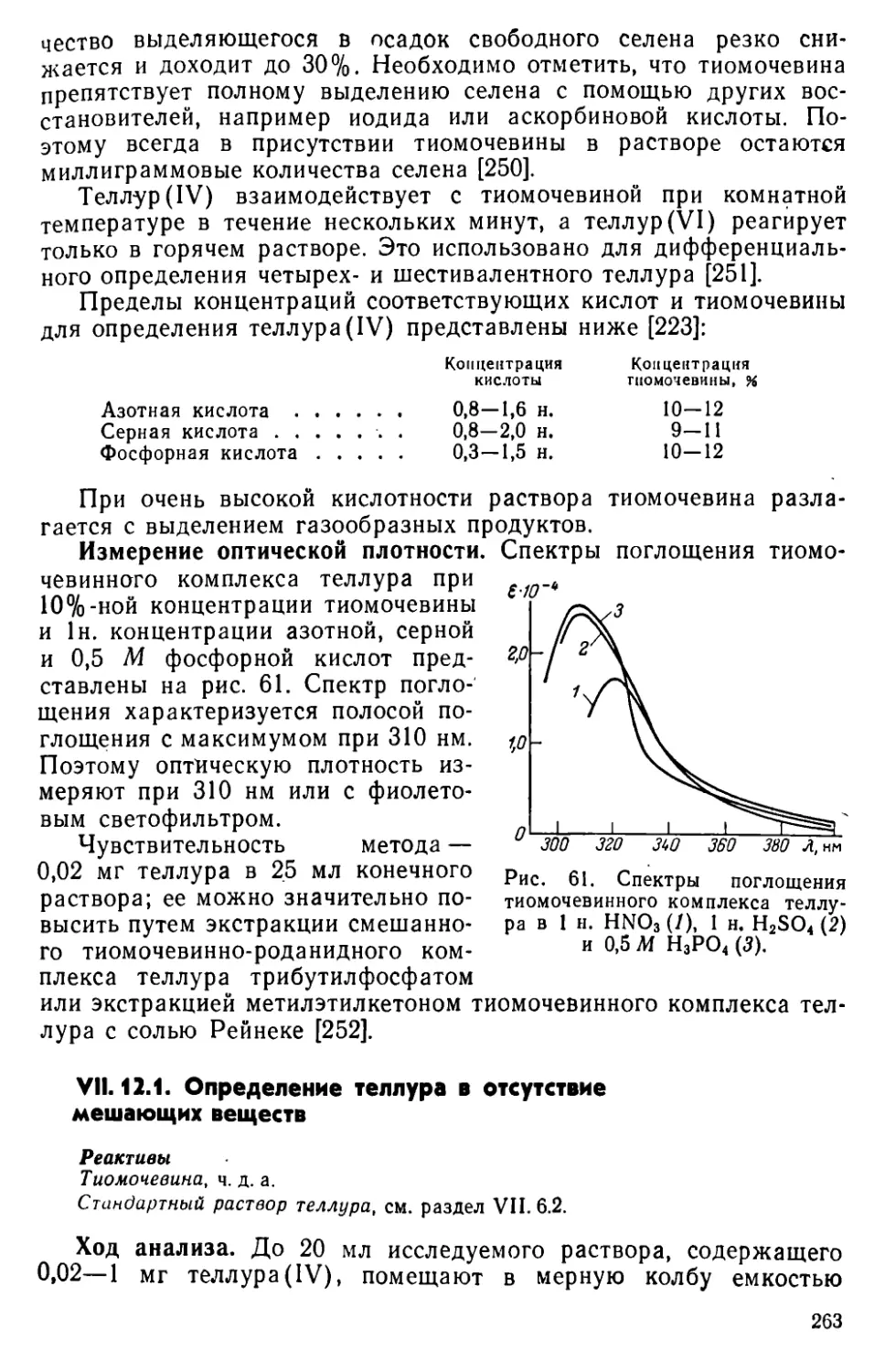
VII. 12. Определение теллура в виде тиомочевинного комплекса . . . 262
VII. 13. Определение теллура с применением производных пиразолона • 264
VII. 14. Определение теллура с применением родаминовых красителей . 269
VII. 15. Другие методы определения селена и теллура............273
•Литература ......................... 275
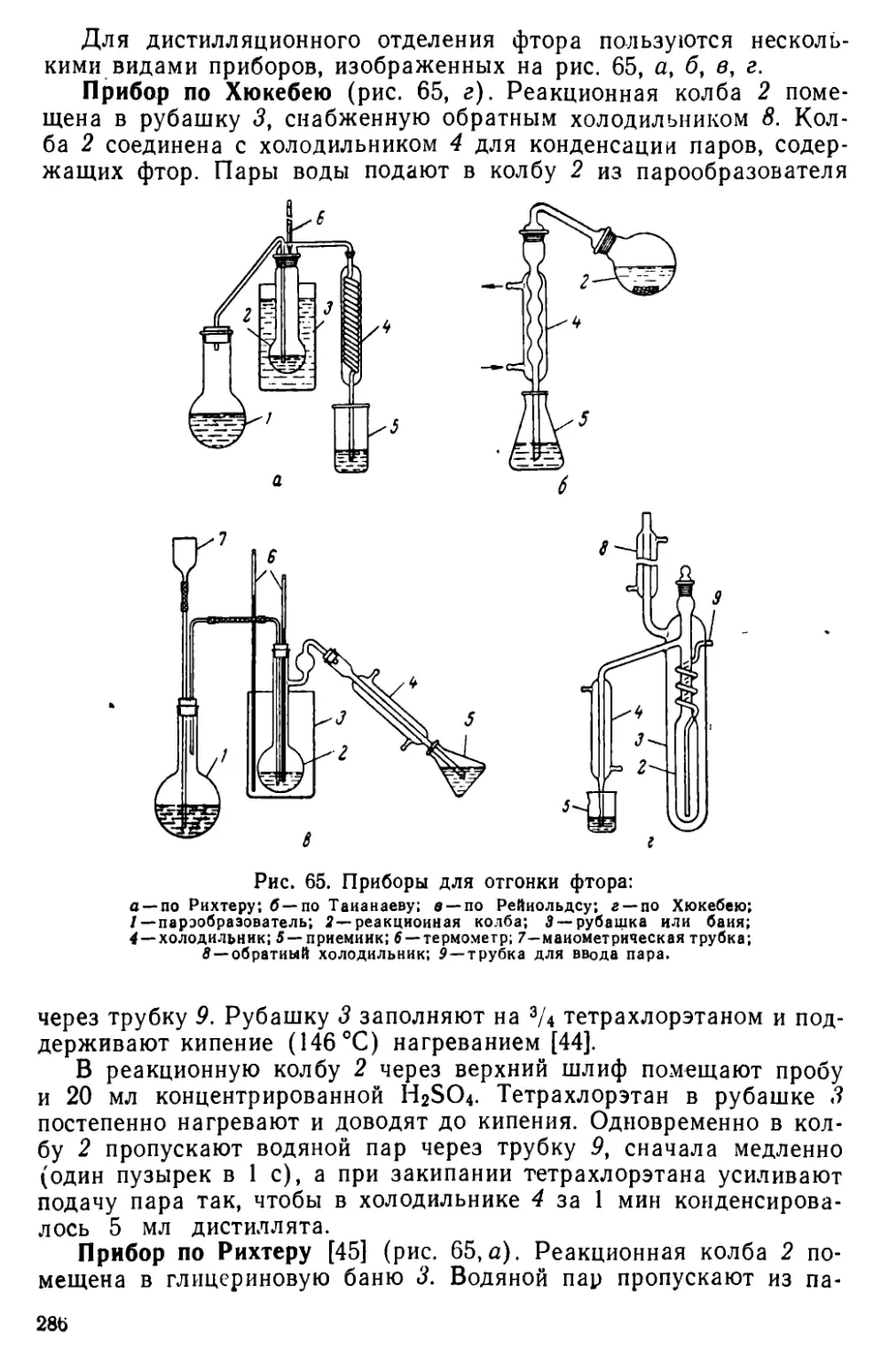
Глава VIII. Фтор.................................................282
VIII . 1. Общая характеристика методов отделения фторида..282
VIII. 2. Методы отделения фтора..............................285
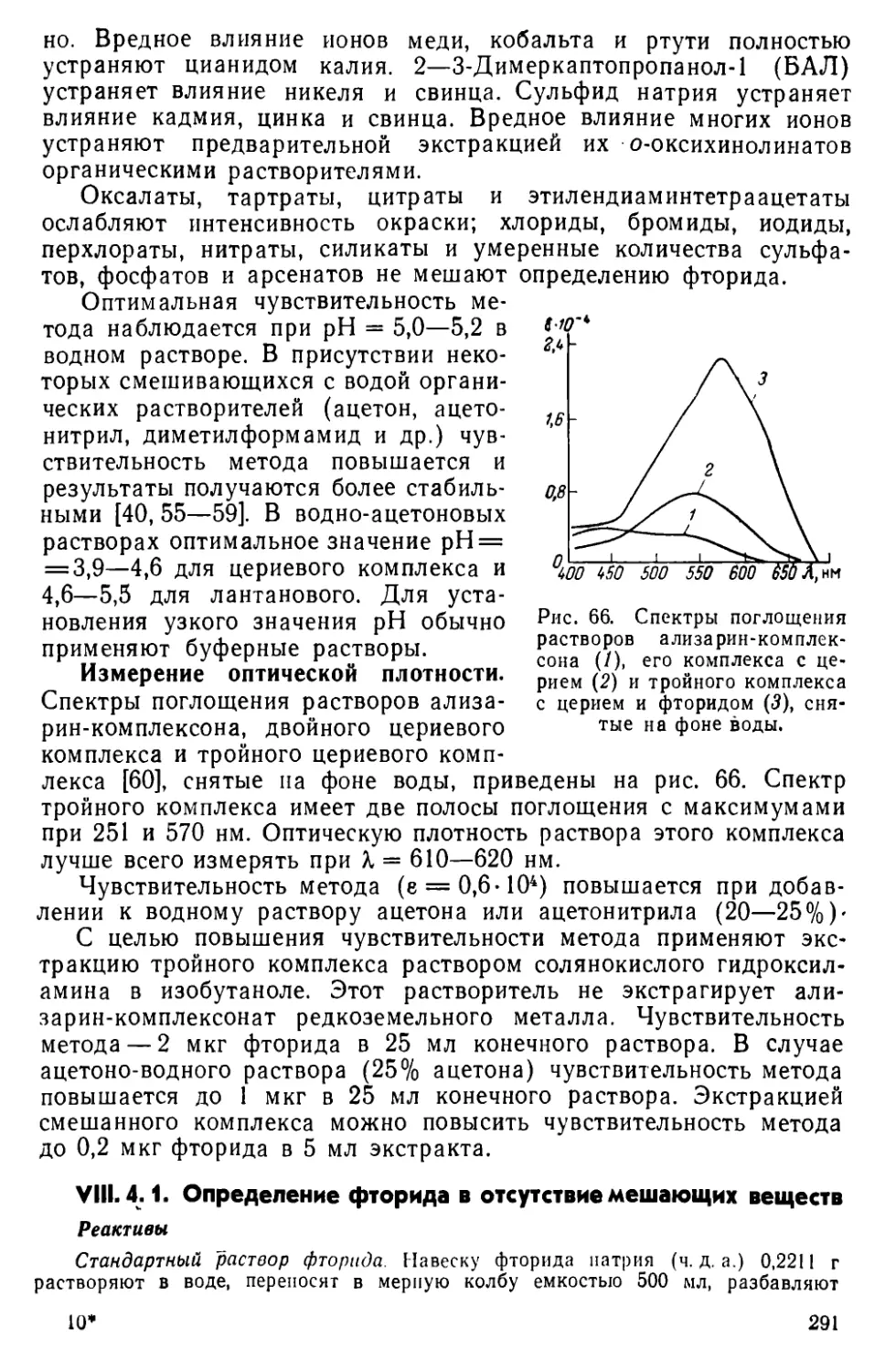
VIII. 3. Общая характеристика методов определения фторида .... 288
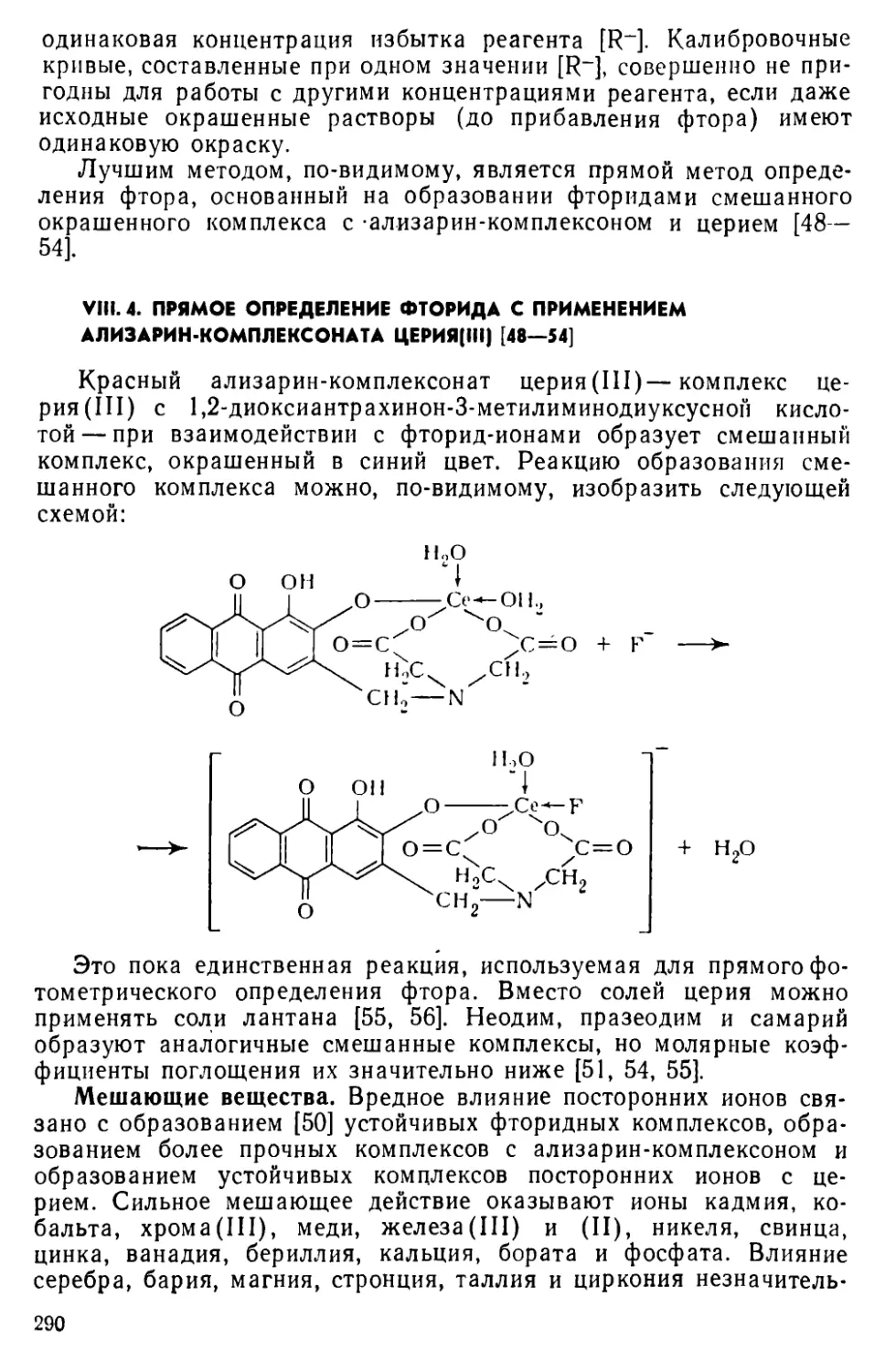
VIII. 4. Прямое определение фторида с применением ализарин-ком-
плексоната церия (III) .......................................290
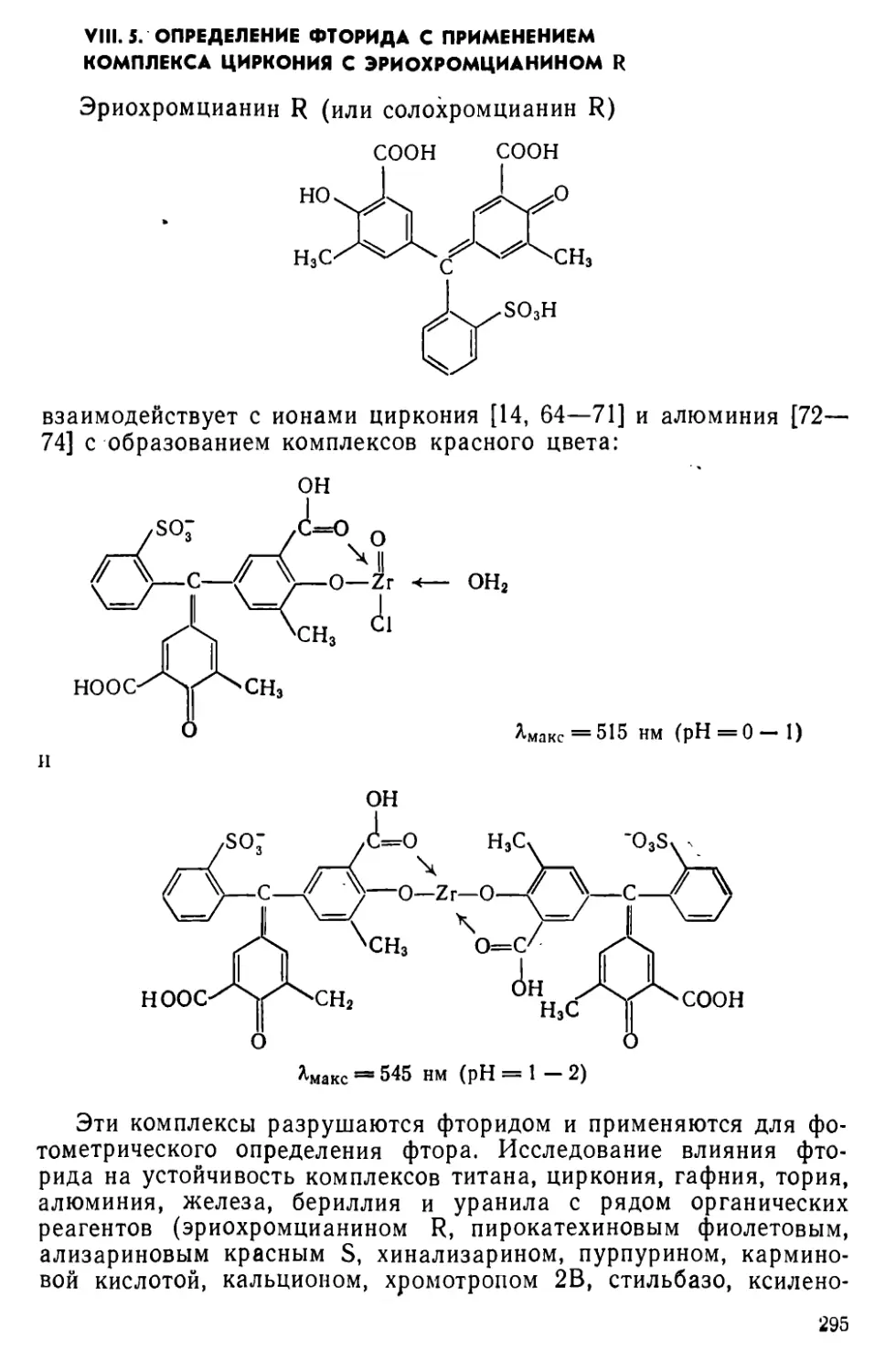
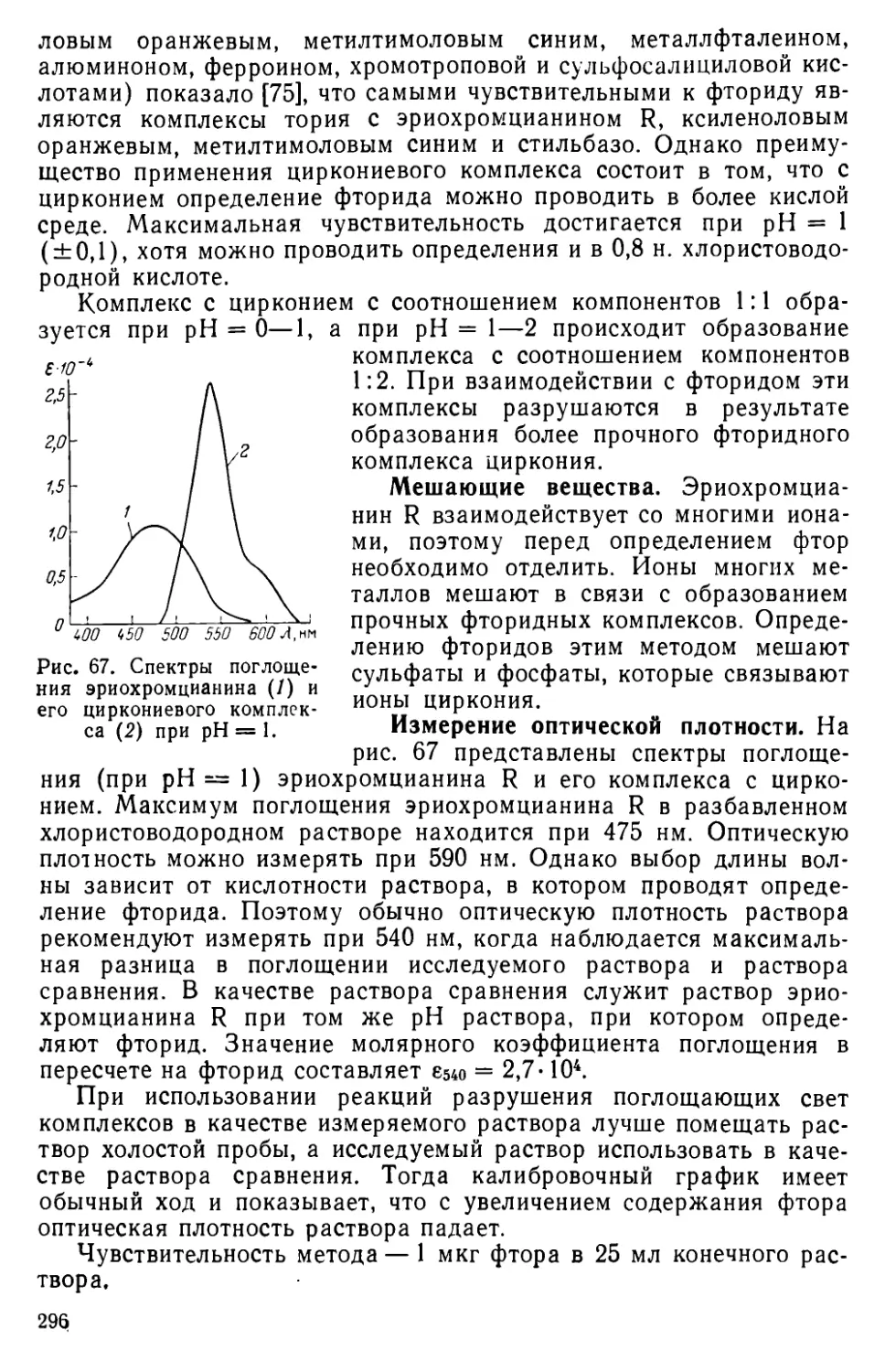
VIII. 5. Определение фторида с применением комплекса циркония с эрио-хромцианином R................................................295
VI II. 6. Определение фторида с применением цирконализаринового комплекса .......................................................298
VII I. 7. Определение фторида по реакций разрушения перекисноводородного комплекса титана.........................................300
VIII . 8. Другие методы определения фторида..........................302
Литература...............................................................303
Глава IX. Хлор...........................................................307
IX. 1. Общая характеристика методов определения и методы отделения 307
IX. 2. Определение хлорида по реакции разрушения комплекса ртути(П) с дифенилкарбазоном ...........................................307
IX. 8. Определение хлорида с применением хромата серебра.............310
IX. 4. Турбидиметрическое определение хлорида в виде хлорида серебра 311
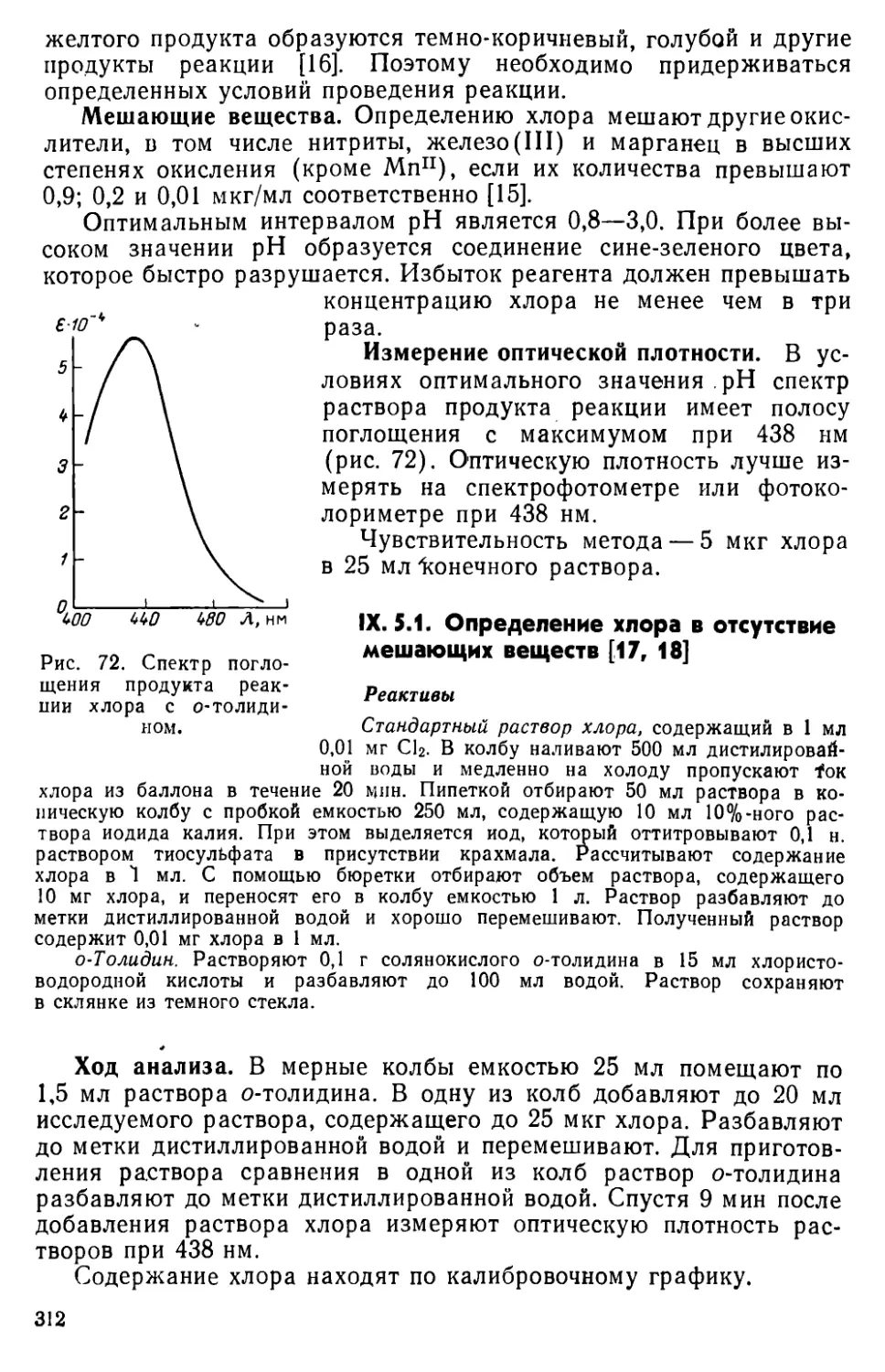
IX. 5. Определение свободного хлора с применением о-толидина • . .311
IX. 6. Определение хлора по реакции разрушения метилового оранжевого 313
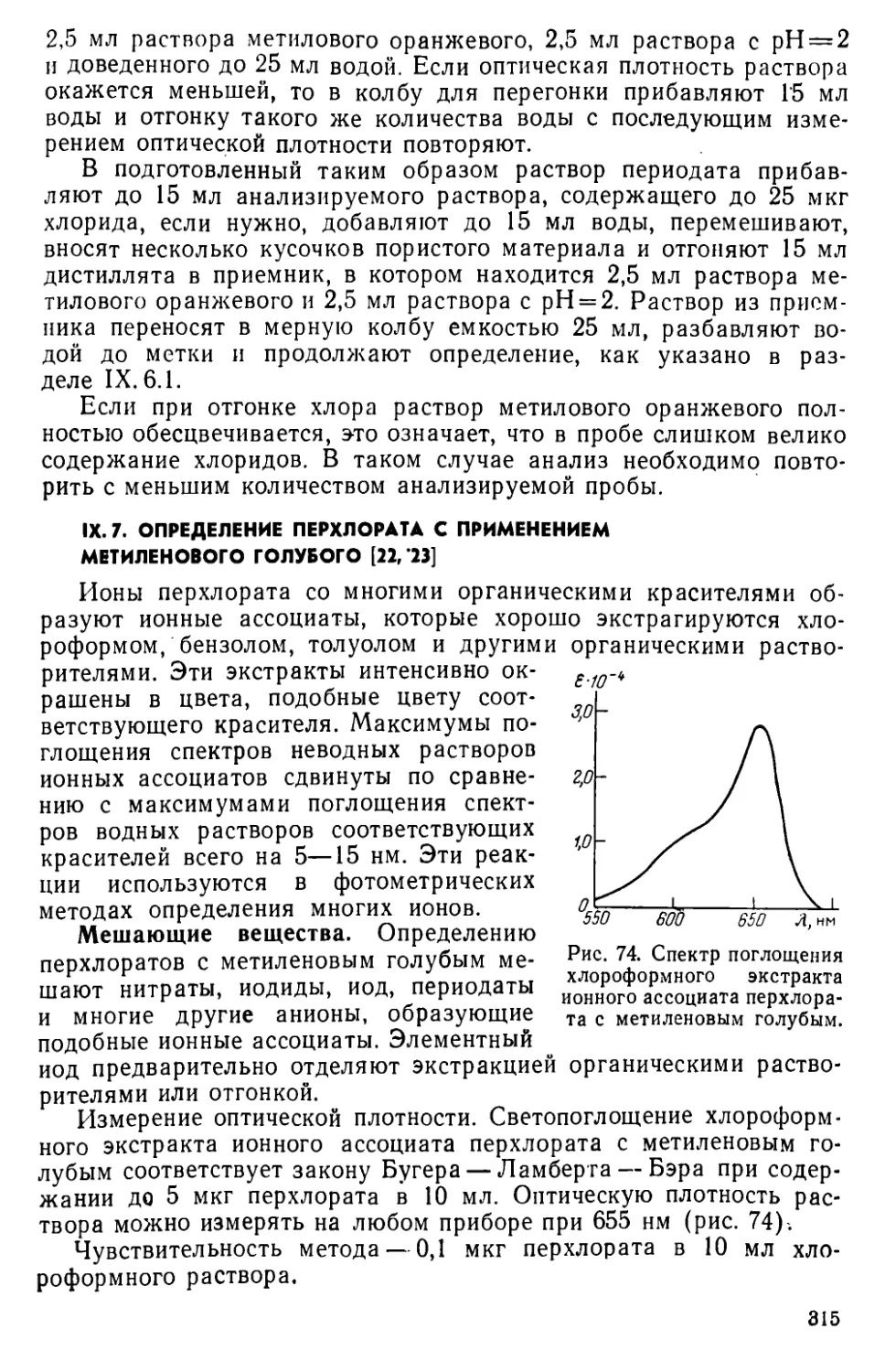
IX. 7. Определение перхлората с применением метиленового голубого . . 315
IX. 8. Другие методы определения различных форм хлора................316
Литература . ............. , ......... 318
Глава X. Бром...........................................................320
X. 1. Общая характеристика метода определения и методы отделения . 320
X. 2. Определение бромида по реакции разрушения метилового оранжевого ..............................................321
X. 3. Определение бромида с применением фенолового красного . . . 324
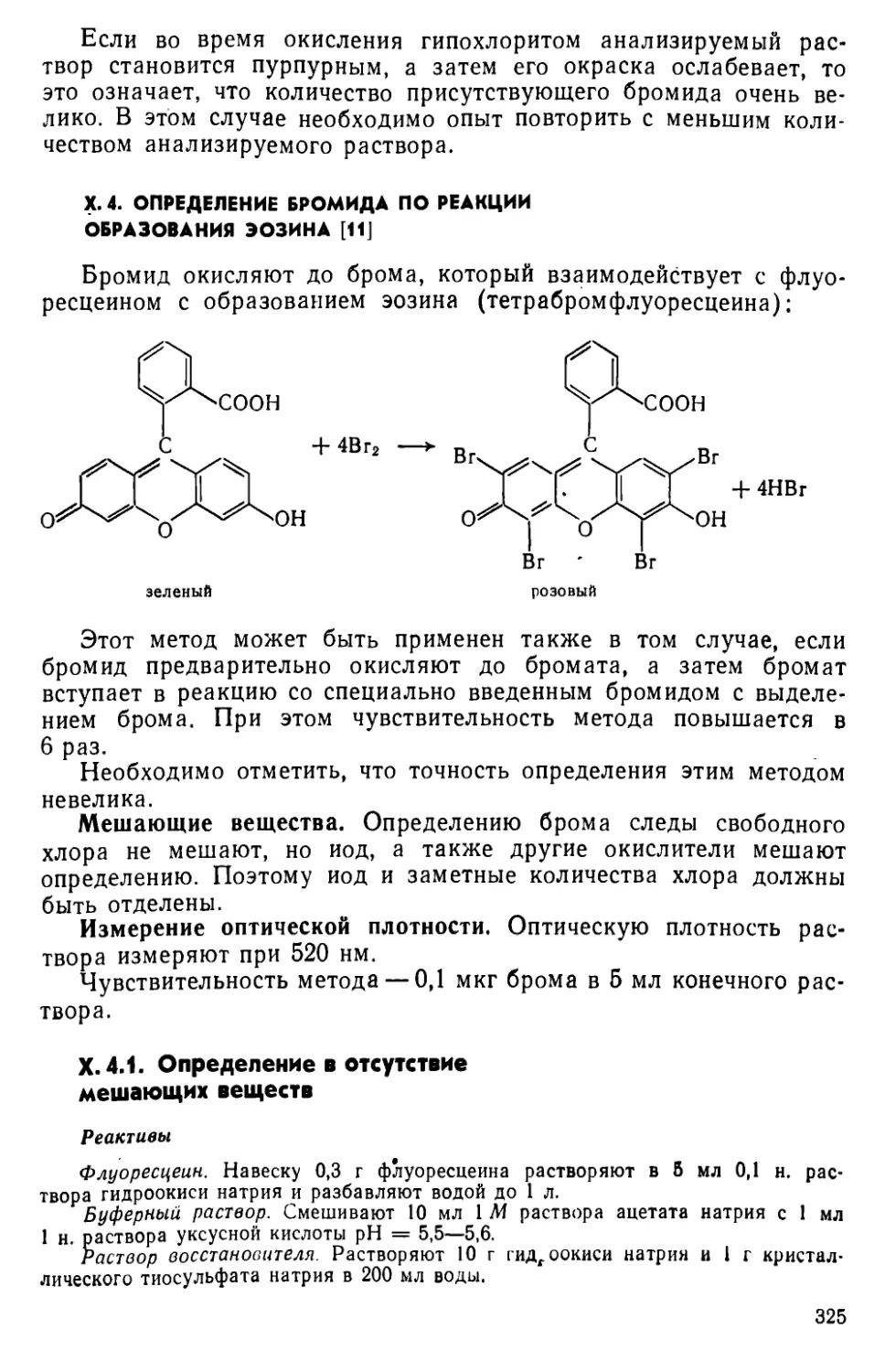
X. 4. Определение бромида по реакции образования эозина..............325
X. 5. Определение бромида с применением розанилина...................326
Х.6. Другие методы определения брома.................................327
Литература . ................................. . ... . 329
Глава XI. Иод...........................................................331
XI. 1. Общая характеристика, методы определения и методы отделения 331
XI. 2. Определение иода с применением крахмала..................332
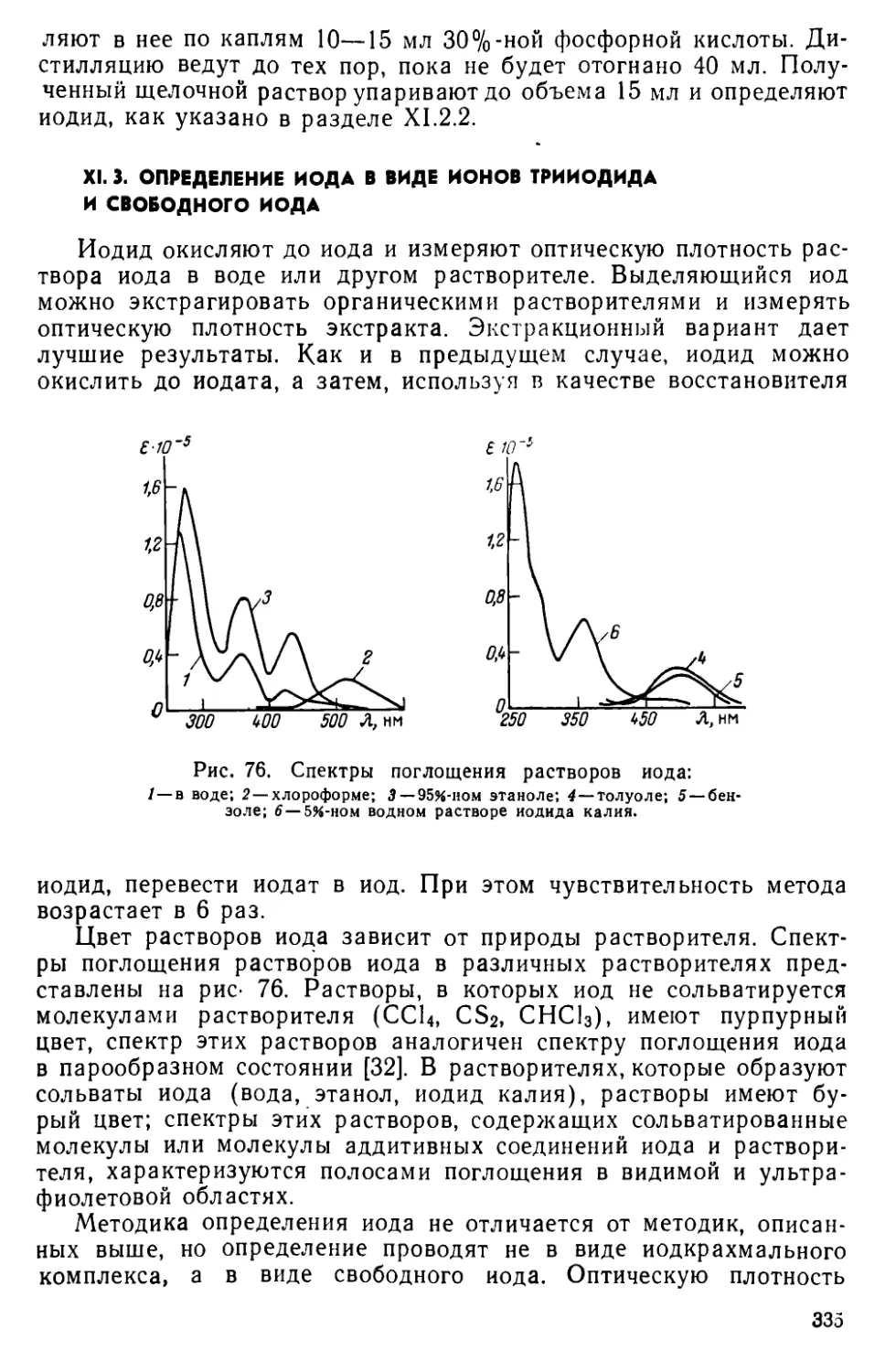
XI. 3. Определение иода в виде ионов трииодида и свободного иода • . 335
XI. 4. Определение иодида в виде ионного ассоциата трииодида с органическими красителями..............................338
XI. 5. Определение иодида с применением синего основного К • • . • 340
XI. 6. Определение иодида в виде ассоциата с ферроином..........341
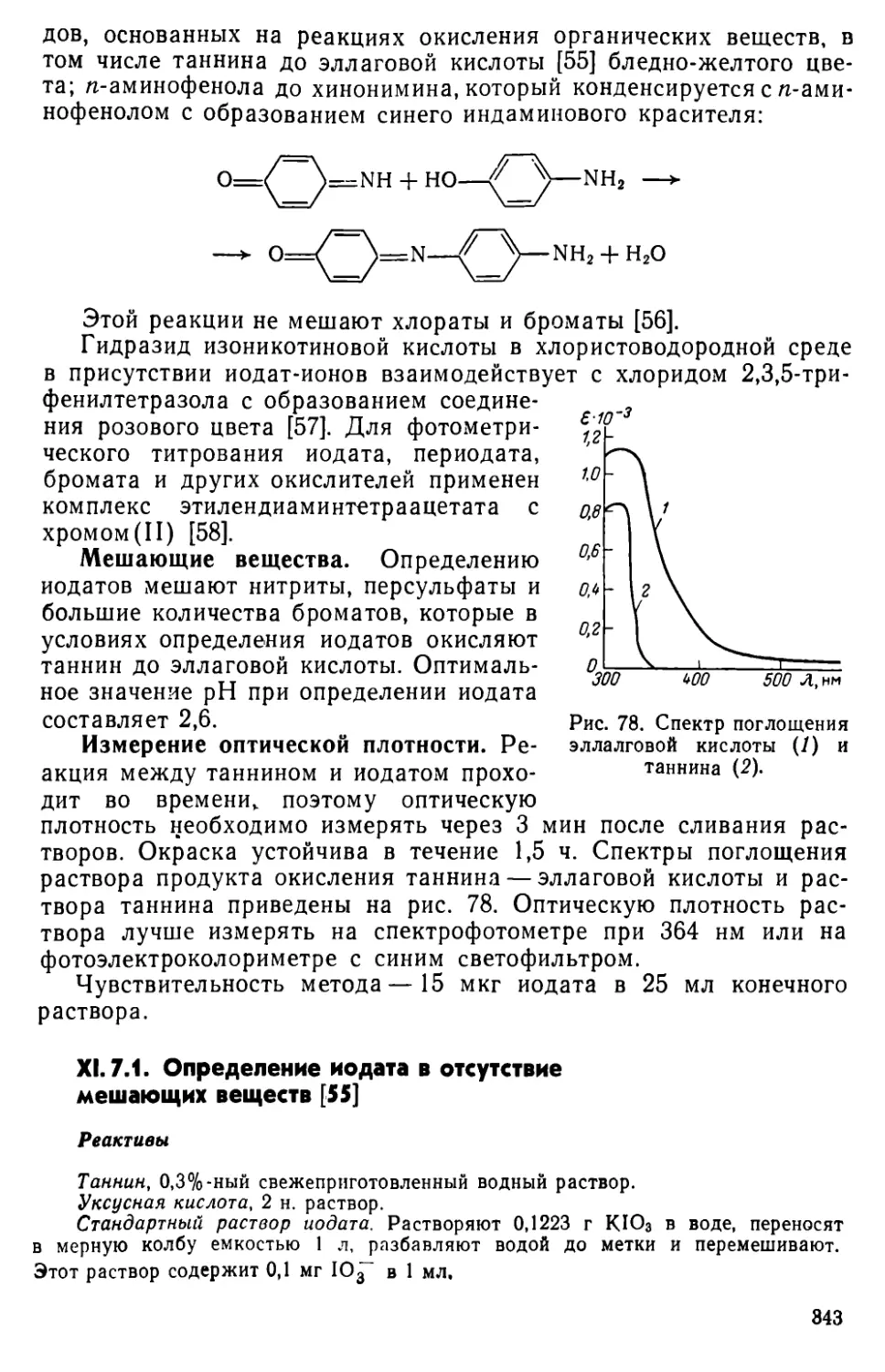
XI. 7. Определение йодата 342
XI. 8. Другие методы определения иода...........................344
Литература ......................... 345
ПРЕДИСЛОВИЕ
Фотометрические методы определения неметаллов являются менее разработанной областью фотометрического анализа по сравнению с методами определения металлов. Многие неметаллы не образуют окрашенных соединений, и фотометрические методы их определения основаны на реакциях разрушения окрашенных соединений. Большой интерес представляют реакции образования разнолигандных (тройных, смешанных) комплексов, которые в последние 10—15 лет широко применяются в анализе. Это направление оказалось ценным и для разработки новых фотометрических методов определения неметаллов. Так, единственным прямым методом определения ионов фтора является метод, основанный на реакции образования разнолигандного комплекса церия с ализа-рин-комплексоном и фторидом. Ряд методов экстракционно-фотометрического определения других неметаллов также основан на образовании соединений этой группы комплексов.
Данная книга представляет собой одновременно монографию и руководство для лабораторных работников, интересующихся определением неметаллов. В ней изложены фотометрические методы определения азота, бора, кремния, фосфора, мышьяка, кислорода, серы, селена, теллура, фтора, хлора, брома и иода.
Книга является продолжением монографии «Фотометрический анализ», первые две части которой уже вышли (в 1968 г. «Общие сведения и аппаратура», написанная А. К. Бабко и А. Т. Пилипенко, и в 1970 г. «Методы определения органических соединений», написанная И. М. Коренманом). Издательство предполагает издать еще одну часть монографии «Фотометрический анализ. Определение металлов».
Составление этой книги было начато вместе с моим учителем светлой памяти академиком АН УССР А. К. Бабко . Им были
частично написаны главы 1—3. С ним также согласован общий план книги.
Приведенные в книге методики были проверены сотрудниками кафедры химии и анализа редких элементов Киевского государственного университета и отдела аналитической химии ИОНХ АН УССР. Особенно большую работу провела ст. н. сотрудник кафедры Е. Г. Скороход; аспирант ИОНХ АН УССР тов. Гакал Р. К. проверила методики определения галогенид-ионов. При оформлении рукописи большую помощь оказали ст. лаборант Л. А. Криво-хижина и инженер В. А. Сацюк. Пользуясь случаем, всем им выражаю искреннюю благодарность.
Выражаю также искреннюю благодарность проф. А. И. Бусеву за ценные советы и замечания, сделанные им при просмотре рукописи.
Все критические замечания и пожелания читателей будут приняты с благодарностью.
А. Т. ПИЛИПЕНКО
Глава I
АЗОТ
1.1. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ ОПРЕДЕЛЕНИЯ АМИННОГО И ОБЩЕГО АЗОТА
Фотометрический анализ применяется обычно для определения аминного и общего азота, а также нитритов и нитратов. Аминный и общий азот определяется, главным образом, при анализе: а) природных и сточных вод или промышленных растворов на содержание ионов аммония или азотсодержащих органических веществ; б) металлов и сплавов (на содержание нитридов). Определение общего азота основано на переведении азота различных азотсодержащих соединений в аммоний, который затем образует окрашенные соединения с тем или другим реактивом.
Первый реактив на аммоний предложил в 1856 г. Несслер [1]; реактив представляет собой щелочной раствор иодидного комплекса ртути. При реакции образуется желтое нерастворимое в воде соединение. Определение выполняется либо по оптической плотности раствора, либо по интенсивности рассеивания света — нефелометрически. Нефелометрический метод более чувствителен, так как при боковом освещении слабый рассеянный свет наблюдается в темноте, т. е. сигнал измеряется почти при полном отсутствии фона. Однако если нет необходимости в большой чувствительности, т. е. если определяют около 1 мг азота в 1 л, применяют обычные фотометрические методы.
Определению аммиака по Несслеру мешают различные амины, ацетон, альдегиды. Кроме того, оптическая плотность или интенсивность рассеивания света зависит не только от общего количества продукта реакции, .но и от размера частиц коллоидной взвеси; размер частиц, в свою очередь, зависит от порядка сливания исследуемого раствора и реактива, от концентрации электролитов, от времени стояния и других причин. Несмотря на все это, метод Несслера наиболее распространен и хорошо проверен.
Довольно широко применяется метод, основанный на образовании индофенолового красителя при взаимодействии аммиака с фенолами и гипохлоритом (или гипобромитом). Этот метод предложен очень давно (Вертело, 1859 г.), но первые варианты его по чувствительности не превышали метод Несслера. Кроме того, образование красителя, как и многие реакции органического синтеза, сопровождается побочными процессами, и выход продукта реакции сильно зависит от температуры и других условий. Поэтому индофеноловый метод долгое время мало развивался. Позже вы
яснилось важное преимущество этого метода — возможность экстрагирования образующегося окрашенного соединения. При этом устраняется необходимость перегонки аммиака для отделения от ряда веществ, мешающих определению его собственной окраски или образующих осадок со щелочью.
Чувствительность обоих методов приблизительно одинакова, правда чувствительность второго метода можно повысить, применяя экстракцию окрашенного соединения небольшим объемом органической жидкости.
Время, затрачиваемое на определение, несколько меньше при анализе индофеноловым методом, хотя при массовых анализах с хорошим набором аппаратуры для дистилляции эта разница невелика.
Как сказано выше, методы вначале применялись для определения аммонийного азота. Позже стали объединять эти методы с определением азота по Кьельдалю. Азот, содержащийся во многих органических соединениях, переводится в NH^-hoh нагреванием с концентрированной серной кислотой в присутствии катализаторов. Затем прибавляется щелочь и отгоняется аммиак, что позволяет отделить его от окрашенных продуктов, образующихся при нагревании органических соединений с серной кислотой. Некоторые дополнительные операции позволяют перевести в аммонийную форму даже азот нитросоединений; для этой цели их предварительно восстанавливают металлическим цинком или иодистово-дородной кислотой.
Примеси азота (0,01% и менее) оказывают большое влияние на свойства металлов и сплавов. Особенно большое влияние азот оказывает на свойства титановых сплавов и ряда жаростойких сплавов. Небольшая часть растворенного азота выделяется при растворении металлов в кислотах и его определяют газоволюмометриче-ским методом. Наиболее важно установить содержание нитридного азота; для этого его так или иначе превращают в аммоний, после чего определяют, как описано выше.
Таким образом, оба описанных метода фотометрического определения, как и другие методы, например пиразолоновый, хингид-ринный и т. п., могут применяться для анализа различных материалов. Однако предварительная подготовка вещества к анализу, разумеется, различна. Поэтому вначале целесообразно рассмотреть методы обработки различных веществ для переведения азота в аммоний, а также методы отделения аммиака от мешающих компонентов.
I. 2. РАЗЛОЖЕНИЕ АЗОТСОДЕРЖАЩИХ ВЕЩЕСТВ
1.2.1. Органические вещества
Предложено много методик для определения азота по Кьельдалю. Сравнение этих методов [2] показывает, что все они дают близкие результаты. Наиболее часто применяют давно разрабо-
тайный и хорошо проверенный вариант, заключающийся в разложении навески серной кислотой в присутствии безводного сульфата калия. В ряде случаев отмечается, что определения без применения сульфата калия дают ошибки, обусловленные испарением значительной части серной кислоты. Не рекомендуется брать слишком много сульфата калия, так как в этом случае разложение идет при слишком высокой температуре, что приводит к потерям азота вследствие термической диссоциации сульфата аммония.
Катализатора, окиси ртути, вводят довольно много — больше, чем содержится определяемого азота. Чаще всего рекомендуют брать 50 мг окиси ртути; во многих методиках рекомендуется смесь окиси ртути (50 мг) с элементным селеном (25 мг). Предложено много и других катализаторов; однако проверка [3] показывает, что наиболее надежной является смесь окиси ртути (И) и элементного селена, но в большинстве случаев достаточно одной окиси ртути (II).
Разложение ведут при 350°C в течение 30—45 мин. Процесс можно ускорить до 7—15 мин, если нагревать в запаянной трубке при 470 °C [4]. Окисленные формы азота — в оксимах, карбазонах, нитросоединениях и других — предварительно восстанавливают. Восстановление цинком описано ниже. Из других методов восстановления можно назвать нагревание нитросоединений с салициловой кислотой в концентрированной серной кислоте. При этом образуется нитросалициловая кислота, которую восстанавливают до аминосоединения добавлением тиосульфата натрия. Еще лучше вести разложение нитросоединений нагреванием с тиосалициловой кислотой в среде серной кислоты [5].
В большинстве методик после разложения по Кьельдалю к пробе добавляют воду, избыток едкой щелочи и отгоняют аммиак.
Во многих статьях обращают внимание на необходимость высокой специальной очистки воды. Воду очищают вторичной перегонкой дистиллированной воды с добавлением в перегонную колбу перманганата и щелочи; первую часть бидистиллята отбрасывают.
В лаборатории, где ведут определение азота, не должно быть склянок с аммиаком. Содержание аммиака в воздухе может изменяться в зависимости от случайных причин, таких, как различная интенсивность работы вытяжной системы в данном или в соседнем помещении. Поэтому всегда необходимо ставить холостые опыты в день определения. Следует также иметь в виду, что во время переливания серной кислоты из запасных баллонов в склянки кислота может поглотить заметные количества аммиака.
Если в анализируемых материалах содержится много восстанавливающих веществ, то во время разложения серной кислотой с сульфатом калия добавляют немного перекиси водорода [6].
Разложение азотсодержащего органического вещества. Для переведения [7] азота органического соединения в аммиак в качестве катализатора служит смесь 150 г безводного сульфата калия, 10 г
окиси ртути и 5 г элементного селена. Смесь хорошо растирают и хранят в небольших склянках.
Навеску вещества, содержащую 0,1 — 1 мг азота, заворачивают в оловянную фольгу и помещают в перегонную колбу. Если предполагается присутствие окисленных форм азота, то вна^ле проводят его восстановление. Для этого прибавляют 1 мл безводной уксусной кислоты; если необходимо, нагревают для растворения навески. Охлаждают и прибавляют 200 мг цинка в виде порошка, 1,5 мл метанола и 4 капли концентрированной хлористоводородной кислоты. Нагревают 2—3 мин, после чего добавляют еще 4 капли той же кислоты и продолжают нагревать. Операцию повторяют еще 2 раза. Кипятят для удаления летучих растворителей, однако избегают выпаривания досуха. Если определяют аминный азот, эти операции пропускают, начиная сразу с обработки навески серной кислотой, как описано ниже.
Охлаждают, прибавляют 3 мл концентрированной серной кислоты и нагревают до удаления воды, пока смесь не начнет темнеть. Дают остыть и прибавляют 1,5 г катализатора (см. выше) и еще 1 мл концентрированной серной кислоты.
Кипятят до тех пор, пока раствор не обесцветится, и продолжают нагревать еще 30—45 мин. Прибавляют 25 мл воды и затем 12—15 мл 50%-ного раствора гидроокиси натрия и 2 г тиосульфата натрия. Присоединяют холодильник и приемник, содержащий 10 мл 0,01 н. серной кислоты. Пропускают пар, пока не соберется 35 мл дистиллята.
Далее определяют аммиак по Несслеру или другим методом. Этот метод разложения пробы применен для определения азота в растительных материалах и почве [8—13], в биологических материалах [14] и в сыре [15] с завершением анализа по методу Несслера, а также при анализе почвы [16, 17] по индофеноловому методу.
1.2.2. Неорганические вещества
При анализе углеродистых сталей скорость растворения зависит от содержания углерода. Образцы, содержащие меньше 0,1% углерода, быстрее растворяются в хлористоводородной кислоте, а образцы с содержанием углерода больше 0,1% быстрее растворяются в серной кислоте [18]. Однако в том и другом случае получаются сходные результаты. При растворении высоколегированных сталей остается нерастворимый осадок, состоящий из карбонитридов и нитридов. Для разложения таких проб применяют смесь серной и фосфорной кислот, иногда с прибавлением сульфата калия, металлической ртути или окиси ртути и других катализаторов. Иногда для окисления неразложившихся соединений применяют перекись водорода и другие окислители. При этом надо иметь в виду, что применение сильных окислителей, например хлорной кислоты, нежелательно, так как это может привести
к заниженным результатам вследствие окисления азота до элементного состояния. Некоторые авторы [19] рекомендуют отфильтровывать нерастворившийся в кислотах остаток и после озоления фильтра осадок сплавлять с гидросульфатом калия и несколькими каплями серной кислоты («мокрое сплавление»). Сплавление необходимо вести при как можно более низкой температуре, чтобы избежать потери азота. Надо также иметь в виду, что при озолении фильтра (450—500 °C) происходит частичное разложение нитридов железа и марганца, а при температуре выше 500 °C их разложение может достигать 99% [20]. Для растворения труд-норазлагающихся нитридов, особенно при анализе сталей, легированных кремнием, рекомендуется применять фтористоводородную кислоту. Достаточно быстрым методом разложения проб, содержащих труднорастворимые карбонитриды и нитриды, является метод сплавления навески со смесью едкого натра и карбоната натрия в никелевой лодочке, помещенной в никелевую трубку [21]. Выделяющийся аммиак поглощают раствором серной кислоты.
В ряде случаев отмечается, что гидроокись натрия часто содержит аммиак, поэтому рекомендуют предварительно нагревать ее со сплавом Деварда.
Иногда предлагают [22] выделить нитриды анодным растворением стали, причем химически несвязанный азот выделяется в виде газа. Остаток, содержащий нитриды, анализируют одним из описанных ниже методов.
В различных методиках много внимания уделяется очистке воды, для чего, в частности, кроме повторной перегонки предлагают катионитный метод или длительное (15 ч) нагревание с небольшим количеством щелочи и сплава Деварда.
Для определения азота по Кьельдалю вместо дистилляции аммиака предлагают осаждать аммоний в виде тетрафенилбората ЫН4[В(СбН5)4]. Правда, отделению мешает ртуть, которую обычно вводят в качестве катализатора при обработке материала серной кислотой. Поэтому необходимо связать ртуть иодидом калия и отделить на анионите. Методика проверена гравиметрически путем образования осадка аммония тетрафенилбората [23] лишь для определения сравнительно больших количеств азота.
Разложение пробы при определении связанного азота в железе и стали [24]. Определению 0,002—0,015% азота в чугунах и сталях посвящено много работ [19, 25—32]. Пробу обычно растворяют в хлористоводородной или серной кислотах. При этом образуются аммонийные соли:
2MN + 4H2SO4 —> (NH4)2SO4+ M2(SO4)3
Навеску стали 5 г помещают в колбу Кьельдаля емкостью 500 мл и обрабатывают 60 мл хлористоводородной кислоты (1 : 1). Растворяют в течение 10 мин при комнатной температуре, далее погружают в горячую воду до полного разложения. Дают остыть,
прибавляют 40 мл 50%-ного раствора винной или лимонной кислоты и пемзу или капилляры для облегчения кипения.
Прибавляют 100—200 мл воды и 60 мл 50%-ного раствора гидроокиси натрия. Перегоняют 150 мл в приемник, в котором содержится 7 мл 0,1 н. серной кислоты. Перед определением с реактивом Несслера раствор необходимо нейтрализовать щелочью.
Сплавы, содержащие кремний, ниобий и другие элементы, растворяют в хлористоводородной кислоте с добавлением 2—3 мл фтористоводородной кислоты. После растворения прибавляют 2 г борной кислоты и фильтруют. В фильтрате определяют аммиак, как указано ниже. В нерастворимом остатке может содержаться азот. Поэтому остаток вместе с фильтром нагревают с 200 мг порошка меди и 25 мл серной кислоты. После охлаждения прибавляют несколько кристаллов перманганата калия и отгоняют аммиак, как указано выше.
I. 3. МЕТОДЫ ОТДЕЛЕНИЯ
Фотометрические методы определения аммония основаны на реакциях, которые проходят в щелочной среде. Очень многие вещества мешают этим определениям, поэтому при проведении опре-
Рис. 1. Прибор для отгонки аммиака:
/ — промывные склянки (первые две —заполнены серной кислотой пл.-1,84 г/см8 для поглощения аммиака нз воздуха, третья —водой); 2— электроплитка; 3— реакционная (дистилляционная) колба;
4 — холодильник; 5—сосуд для улавливания аммиака.
деления в большинстве объектов аммиак предварительно отгоняют. Без отделения аммиака можно получить удовлетворительные результаты только в присутствии небольших количеств посторонних ионов, которые могут выпадать в виде гидроокисей в щелочной среде. Для связывания этих ионов также прибавляют винную кислоту или другие комплексанты. Такой вариант рекомендуется обычно при анализе вод.
Наиболее часто аммиак отгоняют из щелочного раствора. Лучшие результаты получаются при отгонке с водяным паром. Однако вполне удовлетворительные результаты получаются при обычйой отгонке в приборе, показанном на рис. 1. Для получения точных
Рис. 2. Прибор для отделения аммиака путем продувки воз-
результатов каждый дистилляционный аппарат предварительно проверяют с целью установления объема дистиллята при полной отгонке аммиака.
Необходимо всегда помнить, что для равномерного кипения раствора, особенно при наличии осадка гидроокисей, в дистилляционную колбу прибавляют кипятильники (шарики из неглазуро-ванного фарфора, стеклянные капилляры или кусочки стекла).
В качестве поглотителя можно применять воду при отгонке менее 0,2 мг аммиака, а при более высоких содержаниях отгоняемый аммиак поглощают избыточным количеством кислоты. При этом исходят из расчета, что 1 мл 0,01 н. кислоты стехиометрически может поглощать 0,14 мг аммиачного азота. Поэтому количество кислоты в приемнике должно всегда превышать рассчитанное.
Иногда для отделения аммиака применяют метод продувки воздуха (аэрация), для этого << раствору, содержащему аммоний, прибавляют насыщенный раствор карбоната калия или гидроокиси натрия и пропускают воздух для переведения газообразного аммиака в раствор кислоты. Этот процесс можно производить в очень простом приборе (рис. 2), который состоит из трех пробирок, соединен-ЛМх между собой стеклянными и резиновы-мр трубками и установленных в деревянный штатив. Концы входных стеклянных трубок заканчиваются барботерами и немного не достают до дна пробирок. Концы выводных трубок — на уровне нижнего основания пробки.
Сначала воздух при помощи насоса пропускают через пробирку 7, заполненную 1 н. раствором серной кислоты для улавливания следов аммиака из воздуха, а затем через пробирку 2 с исследуемым щелочным раствором. Перед продувкой воздуха к анализируемому раствору прибавляют 1 мл раствора парафина в толуоле* для уменьшения пенообразования. В пробирку 5, которая служит приемником, наливают 10 мл 0,01 н. серной кислоты. Продувку воздуха сначала ведут медленно, а затем поток воздуха увеличивают до умеренной скорости и пропускают воздух в течение 30 мин. Уменьшив поток воздуха, отсоединяют пробирки от насоса. Надо иметь в виду, что перед отсоединением пробирок необходимо давление в них уравновесить с атмосферным, иначе жидкость может перелиться из одной пробирки в другую.
Для приготовления этого раствора 5—10 г парафина с т. пл. 52—54 °C Растворяют в 100 мл толуола.
Отделение аммиака этим методом можно проводить при комнатной температуре, иногда температуру повышают до 50—70 °C [33].
Метод аэрации применяют, главным образом, при анализе биологических материалов.
Рекомендовано применять также микродиффузионный метод в различного типа приборах, состоящих из внутреннего и внешнего сосудов, помещенных в общую камеру. Обычно во внешний сосуд вводят пробу, наливают раствор карбоната калия и закрывают камеру. Во внутренний сосуд предварительно наливают 0,01 н. раствор серной кислоты. При комнатной температуре в течение 1,5 ч аммиак полностью переходит в раствор кислоты. Если повысить температуру до 38 °C, то время диффузии можно уменьшить до 1 ч.
Иногда при определении азота в металлах применяют осаждение катионов металлов щелочью в виде гидроокисей, отделяют осадок центрифугированием, отбирают аликвотную часть центри-фугата и определяют аммиак [30]. Однако такой метод дает весьма приближенные результаты и может быть применен лишь в том случае, когда не требуется большой точности анализа.
I. 4. ОПРЕДЕЛЕНИЕ АММИАКА ПО НЕССЛЕРУ
Аммиак реагирует с иодидом ртути и комплексными иодидами ртути, образуя различные соединения. Стехиометрические соотношения более точно установлены [34] для реакции в условиях фотометрического определения:
2K2HgI4 + 2NH3 —>NH2Hg2I3 + 4Kl +NH4I (1)
Содержание азота, ртути и иодида в осадке выражается отношением 1:2:3. Однако в состав образующегося продукта могут входить и другие соединения (NH2Hg2l2, NH2HgOI и т. д.) [35— 37]. Строение образующегося вещества не установлено. При большом избытке иодида осадок заметно растворяется, поэтому всегда при приготовлении реактива Несслера обращают внимание на то, чтобы не было значительного избытка иодида калия. При очень большом избытке едкой щелочи осадок разлагается с образованием окиси ртути, она также окрашена, но менее интенсивно, чем соединение NH2Hg2I3. Поэтому концентрация щелочи должна быть одинаковой при работе как со стандартным, так и с исследуемым растворами. Соединение, образующееся по указанной выше реакции, склонно к образованию коллоидных частиц с отрицательным зарядом. Для получения равномерной и устойчивой взвеси рекомендуется вводить защитный коллоид — желатин, оптическую плотность измерять через 10 мин после добавления реактива.
1.4.1. Определение аммония в отсутствие мешающих веществ
Для работы, равно как и для приготовления реактивов, применяют дважды перегнанную воду. При вторичной перегонке воду подкисляют серной кислотой; употребляют средний отгон.
реактивы
Щелочной раствор иодида меркуриата калия (реактив Несслера). Реактив можно приготовить двумя способами.
а. Растворяют 32 г иодида ртути(П) и 20 г иодида калия в 50 мл воды п разбавляют раствор до 200 мл; при этом выпадает небольшое количество избытка иодида ртути. Отбирают 20 мл прозрачного раствора и смешивают с 30 мл концентрированного раствора гидроокиси калия. Дают жидкости отстояться, сливают прозрачный раствор сифоном в темную склянку с притертой пробкой и хранят в темном месте.
б. Навеску 6 г хлорида ртути(II) растворяют в 50 мл горячей (80°C) воды в фарфоровой чашке, добавляют раствор 7,4 г иодида калия в 50 мл воды, охлаждают и дают отстояться. Жидкость сливают, а осадок иодида
ртути для удаления образовавшегося хлорида калия трижды промывают холодной водой порциями по 20 мл; промывную жидкость декантируют. К промытому осадку прибавляют 5 г иодида калия и немного воды, затем концентрированный раствор 20 г гидроокиси калия и по охлаждении разбавляют до 100 мл. Дают отстояться, сливают прозрачную жидкость в темную склянку с притертой пробкой и сохраняют в темном месте.
Раствор реактива Несслера устойчив длительное время, если его при хранении защищать от действия двуокиси углерода воздуха при помощи хлор-
кальциевой трубки.
Стандартный раствор аммонийной соли. Навеску х. ч. сульфата аммония 1,945 г. (или 1,481 г хлорида аммония) растворяют в 1 л воды и хорошо
перемешивают. Отбирают 1 мл этого раствора в мерную колбу емкостью 50 мл, разбавляют водой до метки и хорошо перемешивают. В 1 мл раствора содержится 10 мкг NH3.
При определении азота в металлах пересчет ведут на азот. Поэтому для приготовления стандартного раствора берут навеску 2,3571 г сульфата аммония (или 1,9125 г хлорида аммония). В 1 мл приготовленного раствора содержится 10 мкг азота.
Методы измерения оптической
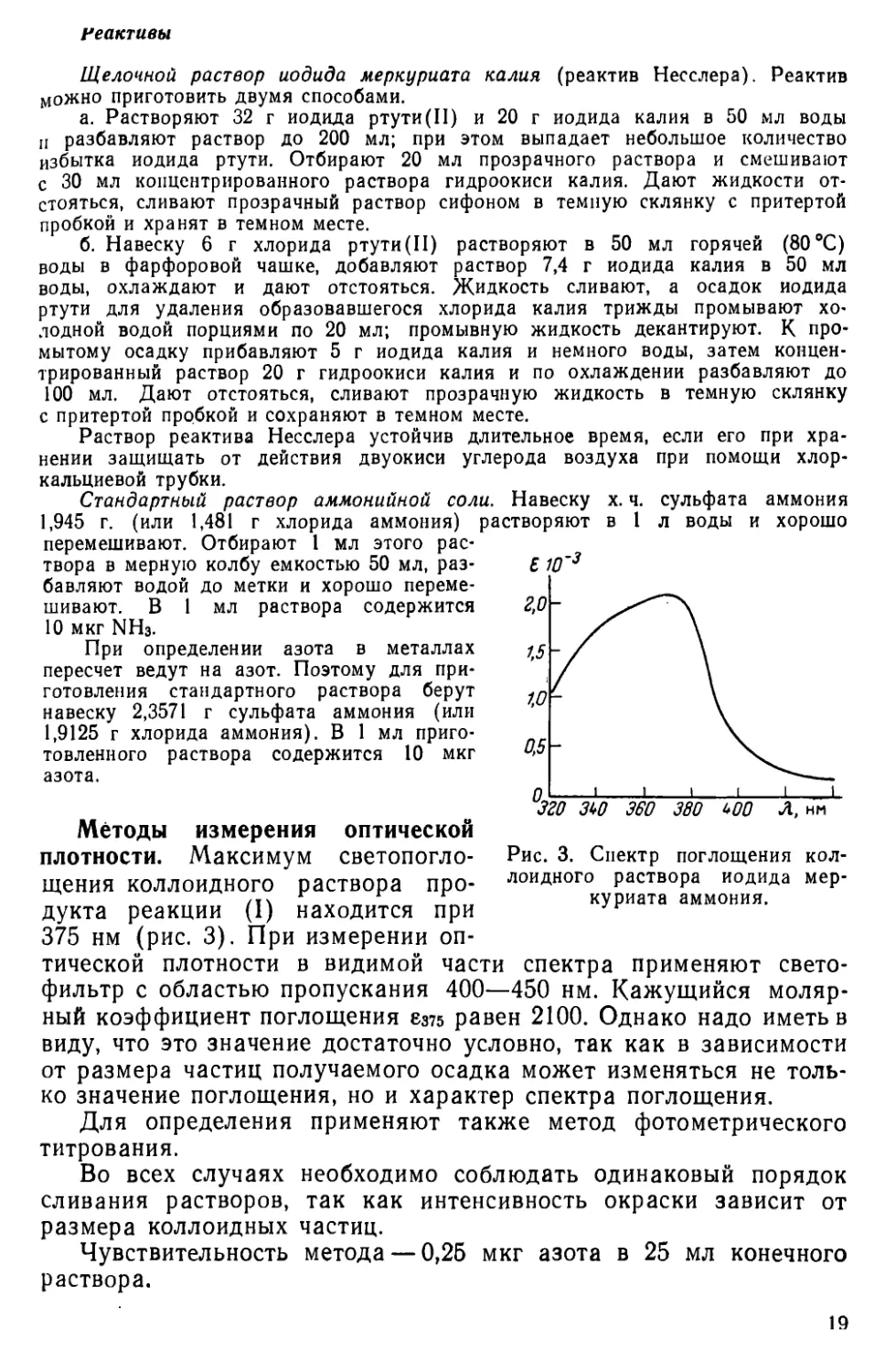
Рис. 3. Спектр поглощения коллоидного раствора иодида меркуриата аммония.
плотности. Максимум светопогло-щения коллоидного раствора продукта реакции (I) находится при 375 нм (рис. 3). При измерении оп-
тической плотности в видимой части спектра применяют светофильтр с областью пропускания 400—450 нм. Кажущийся молярный коэффициент поглощения 8375 равен 2100. Однако надо иметь в
виду, что это значение достаточно условно, так как в зависимости от размера частиц получаемого осадка может изменяться не только значение поглощения, но и характер спектра поглощения.
Для определения применяют также метод фотометрического титрования.
Во всех случаях необходимо соблюдать одинаковый порядок сливания растворов, так как интенсивность окраски зависит от размера коллоидных частиц.
Чувствительность метода — 0,25 мкг азота в 25 мл конечного
раствора.
Ход анализа. Испытуемый раствор, содержащий 2,5—25 мкг аммиака в 20 мл, помещают в мерную колбу емкостью 25 мл, прибавляют 1 мл реактива Несслера, разбавляют водой до метки, перемешивают и спустя 10 мин измеряют оптическую плотность коллоидного раствора. Содержание аммиака находят по предварительно построенному калибровочному графику.
1.4.2. Определение аммония в природной воде
Реактивы
Раствор тартрата калия-натрия. Растворяют 50 г препарата в 100 мл горячей воды. Раствор фильтруют, прибавляют 5 мл реактива Несслера (для предупреждения появления плесени), затем оставляют на 2—3 дня для отстаивания.
Ход анализа. Определение ведут так, как указано в разделе 1.4.1. В пробу воды перед прибавлением реактива Несслера вводят 0,1 мл 50%-ного раствора тартрата калия-натрия для маскировки посторонних катионов.
При анализе окрашенных вод берут определенный объем испытуемой воды, подщелачивают раствором ЫагСОз и, отогнав аммиак в приемник с дистиллированной водой, определяют его, как указано в разделе 1.4.1. Если в качестве поглотителя применяют серную кислоту, то перед прибавлением реактива Несслера ее предварительно нейтрализуют щелочью.
1.4.3. Определение азота в черных металлах
Ход анализа. Помещают 1 г стали в колбу для разложения навески, приливают 20 мл хлористоводородной кислоты (1:1) и немедленно закрывают пришлифованной пробкой с гидравлическим затвором, в резервуар которого предварительно наливают 1—2 мл хлористоводородной кислоты (1 : 1). Навеску растворяют сначала без нагревания, затем при умеренном нагревании.
После растворения колбу с раствором охлаждают, раствор из резервуара гидравлического затвора присоединяют к основному раствору и обмывают затвор 2—3 раза водой порциями по 2— 3 мл. К раствору прибавляют 10 мл H2SO4 (1:1) и нагревают до появления паров серной кислоты.
Если на дне колбы после разложения навески остались темные частицы нерастворившегося остатка, прибавляют в несколько приемов 2—3 г K2S2O8 и продолжают нагревание до исчезновения темных частиц. Содержимое колбы охлаждают. Соли растворяют в 30—50 мл горячей воды. Если раствор мутный, то прибавляют несколько капель хлористоводородной кислоты.
Если нерастворимый остаток этим способом разложить не удается, то поступают следующим образом: раствор выпаривают до появления паров серной кислоты, охлаждают, добавляют 5 г
K2SO4, 1 г CuSO4-5H2O, приливают 20 мл H2SO4 (1:1) и кипятят до полного растворения нерастворимого остатка. Раствор выпаривают до появления густых паров серной кислоты.
Стали, легированные кремнием, вначале растворяют без нагревания в 30 мл хлористоводородной кислоты (1:1), а затем при умеренном нагревании. После растворения (прекращение выделения пузырьков Н2) раствор из резервуара гидравлического затвора присоединяют к основному раствору и обмывают затвор 2—3 раза водой. Затем прибавляют при помешивании маленькими порциями 1 —1,5 г фторида натрия, после чего колбу вновь закрывают пришлифованной пробкой с гидравлическим затвором и продолжают нагревание. Фторид натрия прибавляют до полного растворения кремневой кислоты.
После растворения пробы раствор охлаждают, переносят в колбу для перегонки, нейтрализуют кислоту щелочью, прибавляют избыток ее и отгоняют аммиак в раствор серной кйслоты. Затем определяют его, как указано в разделе 1.4.1; перед прибавлением реактива Несслера раствор нейтрализуют щелочью.
В литературе описаны отдельные детали анализа различных материалов для определения аммиака, например сталей [30], белков [38], вод [39—43], порошка металлического вольфрама [44], титановой губки [45], металлического бериллия [46], атмосферных осадков [47], воздуха [43], бензгидроксамовой кислоты и ее производных [48], а также для определения гидроксиламина реактивом Несслера [49].
1.5. ОПРЕДЕЛЕНИЕ АММИАКА ИНДОФЕНОЛОВЫМ МЕТОДОМ
В качестве реактивов в индофеноловом методе применяют различные фенолы, чаще всего фенол [50—65] и тимол [66—68]; кроме гипохлорита применяют гипобромит [66, 67] и хлорамин [69, 70].
Обычно гипохлорит реагирует с аммиаком, окисляя его до азота:
3NaC10 + 2NH3 —> 3NaCl + ЗН2О + N3
Однако в присутствии избытка фенола происходит реакция образования красителя. Предполагается [71] следующая последовательность реакций. На первой ступени образуется п-аминофенол:
ОН ОН
|^j| + NaCIO + NH, —► |^J| + NaCI + H,0
NH>
л-Аминофенол, реагируя далее с гипохлоритом, образует хи нонхлоримин:
Хинонхлоримин реагирует со второй молекулой фенола, образуя краситель индофенол:
О ОН
NC1
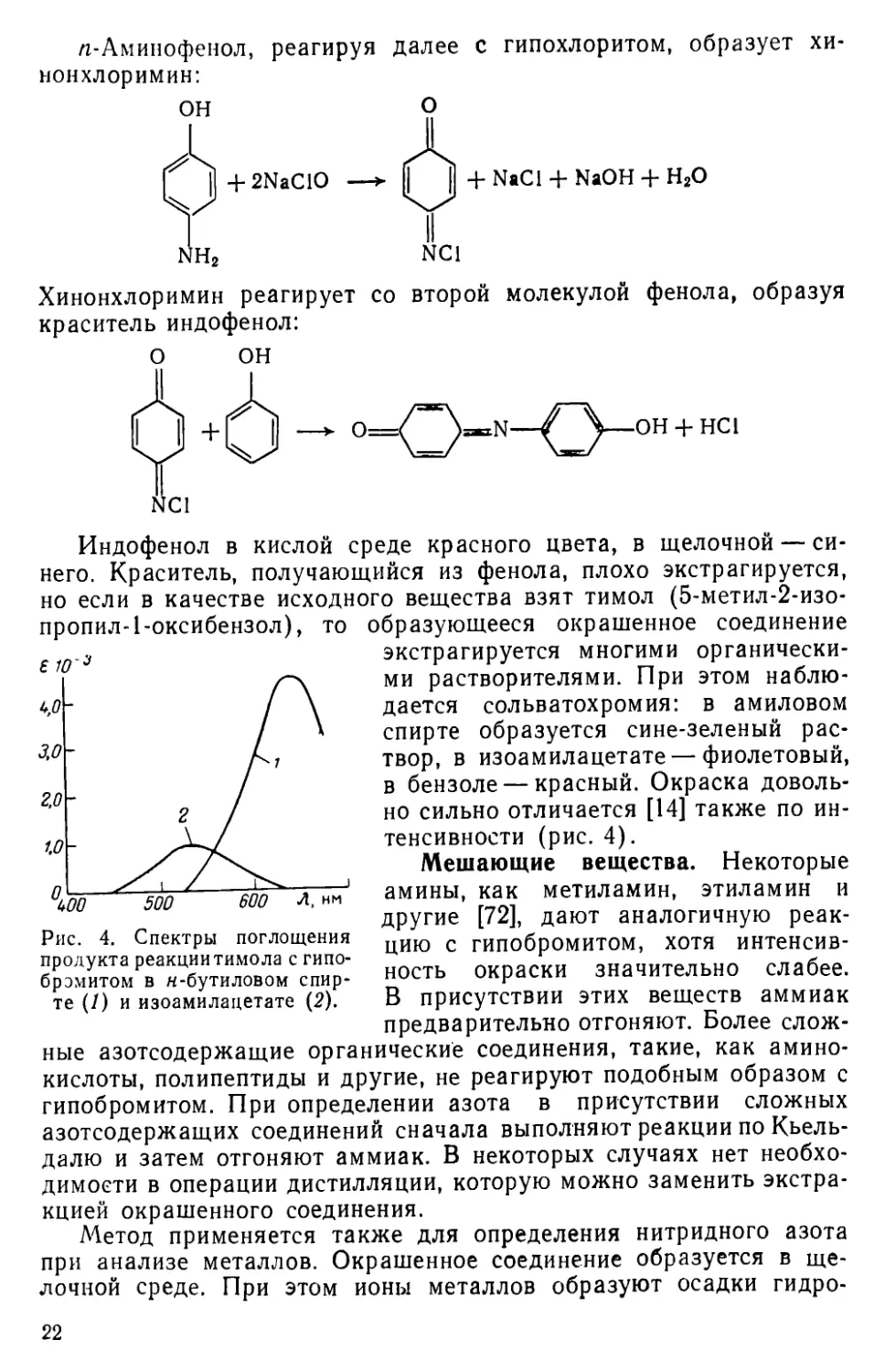
Индофенол в кислой среде красного цвета, в щелочной — синего. Краситель, получающийся из фенола, плохо экстрагируется, но если в качестве исходного вещества взят тимол (5-метил-2-изо-пропил-1-оксибензол), то образующееся окрашенное соединение
Рис. 4. Спектры поглощения продукта реакции тимола с гипо-брэмитом в н-бутиловом спирте (/) и изоамилацетате (2).
экстрагируется многими органическими растворителями. При этом наблюдается сольватохромия: в амиловом спирте образуется сине-зеленый раствор, в изоамилацетате — фиолетовый, в бензоле — красный. Окраска довольно сильно отличается [14] также по интенсивности (рис. 4).
Мешающие вещества. Некоторые амины, как метиламин, этиламин и другие [72], дают аналогичную реакцию с гипобромитом, хотя интенсивность окраски значительно слабее. В присутствии этих веществ аммиак предварительно отгоняют. Более слож
ные азотсодержащие органические соединения, такие, как аминокислоты, полипептиды и другие, не реагируют подобным образом с гипобромитом. При определении азота в присутствии сложных азотсодержащих соединений сначала выполняют реакции по Кьель-далю и затем отгоняют аммиак. В некоторых случаях нет необхо-
димости в операции дистилляции, которую можно заменить экстракцией окрашенного соединения.
Метод применяется также для определения нитридного азота при анализе металлов. Окрашенное соединение образуется в щелочной среде. При этом ионы металлов образуют осадки гидро
окисей, что затрудняет экстракционное разделение. Для связывания ионов многих металлов рекомендуют различные комплексообразующие вещества, такие как винная кислота, ЭДТА (этилен-диаминтетрауксусной кислоты динатриевая соль) и др. По некоторым данным [73], одним из наиболее подходящих маскирующих веществ является фторид калия, который связывает многие катионы и в то же время не оказывает влияния на реакцию образования индофенолового красителя. Для связывания 0,01 г железа или титана необходимо около 0,2—0,5 г фторида калия.
Методы измерения оптической плотности. Оптимальная длина волны для измерения оптической плотности зависит от взятого, экстрагента. Так, в н-бутиловом спирте Хмакс = 680 нм, евво = = 5,3-103, а в изопропиловом эфире Хмакс = 530 нм, е5зо = 5-1О2. Раствор в изоамиловом спирте устойчив в течение пяти месяцев [74].
Чувствительность метода — 2,5 мкг аммиака в 25 мл экстракта.
1.5.1. Определение аммония с применением тимола и гипобромита в отсутствие мешающих веществ
Реактивы
Стандартный раствор соли аммония. Раствор готовят, как указано в разделе 1.4.1.
Тимол. Навеску 2 г хорошо измельченного кристаллического тимола х. ч. растворяют в 10 мл 8%-ного раствора гидроокиси натрия и разбавляют водой до 100 мл.
Гипобромит натрия. К 100 мл насыщенной бромной воды прибавляют 35 мл 8%-ного раствора гидроокиси натрия и перемешивают. Раствор сохраняют в холодильнике при 5 °C. Концентрацию раствора устанавливают титрованием тиосульфатом. Наиболее приемлемая концентрация гипобромита приблизительно 0,3 н.
н-Бутиловый спирт.
Ход анализа. Испытуемый раствор, содержащий до 100 мкг аммиака, помещают в делительную воронку, если нужно, нейтрализуют кислотой или щелочью по фенолфталеину, прибавляют 5 мл тимола и далее по каплям 5 мл раствора гипобромита, а затем еще 5 мл тимола. Спустя 10 мин экстрагируют образовавшееся синее соединение н-бутиловым спиртом 2—3 раза порциями по 5—7 мл. Экстракты фильтруют через сухую вату в колбу емкостью 25 мл и разбавляют до метки н-бутиловым спиртом. Раствор перемешивают и измеряют оптическую плотность в кювете с толщиной слоя 1 см.
В случае измерения интенсивности окраски экстракта методом стандартных серий поступают следующим образом. Испытуемый раствор, содержащий до 100 мкг аммиака, помещают в одну из пробирок с притертыми пробками для колориметрирования, в остальные пробирки помещают различные количества стандартного раствора аммонийной соли. В пробирки прибавляют по 5 мл раствора тимола, затем по каплям 5 мл раствора гипобромита.
а затем еще 5 мл тимола и раствор перемешивают. Спустя 10 мин прибавляют 5 мл изопропилового эфира и экстрагируют окрашенное соединение, взбалтывая раствор в течение 3—5 мин. При этом эфирный слой окрашивается в розовый или красный цвет различных оттенков. Окраску эфирного слоя в исцытуемом растворе сравнивают с окраской шкалы.
1.5.2. Определение аммония с применением фенола и гипохлорита в отсутствие мешающих веществ
Установлено [75], что раствор фенолята надо готовить в день проведения анализа, так как при хранении он темнеет.
Калибровочные графики более постоянны, если запасной раствор фенола готовится на метиловом спирте. Установлено, что добавка ацетона значительно ускоряет реакцию образования окрашенного соединения и позволяет обойтись без применения катализатора (соль марганца) или нагревания, которое рекомендуется в некоторых методиках. В этих условиях калибровочный график линеен в интервале от 0 до 80 мкг азота в 25 мл.
Реактивы
Стандартный раствор соли аммония готовят, как указано в разделе 1.4.1.
Фенол. Навеску 62,5 г фенола растворяют в метиловом спирте, прибавляют 18,5 мл ацетона и разбавляют метиловым спиртом до 100 мл. Раствор хранят в холодильнике.
Фенолят натрия. Навеску 27 г гидроокиси натрия растворяют в 100 мл воды. Раствор хранят в холодильнике.
Ежедневно перед работой отбирают по 10 мл растворов фенола и фенолята натрия, разбавляют до 100 мл водой и перемешивают.
Гипохлорит натрия. Готовят 200 мл 10%-ного раствора гидроокиси натрия, в который пропускают на холоду хлор до нейтральной реакции, затем подщелачивают 30 мл 3%-ного раствора щелочи и перемешивают.
Ход анализа. Берут 10 мл испытуемого раствора, содержащего до 100 мкг азота, нейтрализуют по фенолфталеину, прибавляют 4 мл свежеприготовленного раствора фенолята натрия и перемешивают. Далее понемногу прибавляют 3 мл раствора гипохлорита натрия, разбавляют водой до 25 мл и оставляют на 20 мин при комнатной температуре.
Оптическую плотность измеряют в кювете с толщиной слоя 2 см при 610 нм.
Если применяют экстракцию, то реакцию ведут в делительной воронке, после прибавления гипохлорита выдерживают 20 мин и экстрагируют окрашенное соединение 5 мл изобутилового спирта в течение 1 мин, отделяют неводную фазу через сухой фильтр и измеряют оптическую плотность неводного слоя в кювете с толщиной слоя 1 см при 610 нм.
1.5.3. Определение аммиака в воздухе [58]
Пробу воздуха 100—1000 л, содержащую 1 — 100 мкг/мэ аммиака пропускают через 23 мл 0,01 н. раствора H2SO4 со скоростью 3-J-30 л/мин. Если в воздухе присутствуют большие количества сероводорода, то кислый раствор нагревают для его удаления. При содержании сероводорода меньше 60 мкг в пробе он не мешает определению аммония. Раствор переносят в мерную колбу емкостью 25 мл, доводят до метки водой и перемешивают. Отбирают аликвотную часть и определяют аммоний, как указано в разделе 1.5.2.
1.5.4. Определение аммония в природных водах [54]
Пробу воды, содержащую до 100 мкг аммония, помещают в делительную воронку и ведут определение, как указано в разделах 1.5.1 или 1.5.2.
1.5.5. Определение азота в черных металлах
Реактивы
Фторид калия, 20%-ный раствор.
Другие реактивы указаны в разделе 1.5.1.
Ход анализа. Тонкую стружку металла промывают чистым этиловым спиртом, затем диэтиловым эфиром и сушат при 50 °C не менее 30 мин. Навеску 1 г подготовленного образца, содержащую от 10 до 300 мкг азота, растворяют при нагревании в 25 мл хлористоводородной кислоты (пл. 1,12). Если проба металла не растворяется, то колбу охлаждают, прибавляют 5 мл H2SO4 (пл. 1,84), кипятят, если остаются нерастворимые частицы, вводят 1 г сульфата калия и нагревание продолжают при более высокой температуре. Затем раствор охлаждают, разбавляют водой и, если нужно, фильтруют в мерную колбу емкостью 100 мл, разбавляют водой до метки и перемешивают. Отбирают аликвотную часть, прибавляют 0,5 мл раствора фторида калия, нейтрализуют до розовой окраски по фенолфталеину и определяют аммоний, как указано в разделе 1.5.1.
Более точные результаты получаются после отгонки аммиака.
1.5.6. Определение азота в карбиде титана [73]
Навеску 0,1 г растворяют в 2,5 мл концентрированной серной кислоты с добавлением 2 г сульфата калия. Разбавляют до 100 мл и для анализа берут аликвотную часть, например 10 мл. Прибавляют 0,5 мл 20%-кого раствора фторида калия. Раствор нейтрализуют до розовой окраски по фенолфталеину и далее определяют, как указано в разделе 1.5.1.
1.5.7. Определение азота в растительных материалах
Навеску 0,1 г высушенного и измельченного материала помещают в коническую колбу емкостью 50 мл и прибавляют 2 мл концентрированной серной кислоты. Спустя 10 мин колбу постепенно нагревают до появления густых белых паров серной кислоты. Затем раствор охлаждают и прибавляют 0,5 мл 30%-ной перекиси водорода. Раствор нагревают до начала выделения пузырьков и после прекращения выделения пузырьков снова нагревают. Повторяют эту операцию с добавлением свежих порций перекиси водорода до полного или почти полного просветления жидкости. Раствор кипятят до полного разложения перекиси водорода и определяют аммоний, как указано в разделах 1.4.1, 1.5.1. или 1.5.2.
1.5.8. Определение азота в почве
Навеску пробы разлагают, как указано в разделе 1.2.1, отгоняют аммиак и определяют, как указано в разделах 1.4.1, 1.5.1 или 1.5.2. Для приближенного определения азота иногда делают кислотную или водную вытяжку, затем определяют азот после отгонки-или без отгонки, как указано в разделах 1.4.2 или 1.5.4.
I. 6. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ АМИННОГО И ОБЩЕГО АЗОТА
В литературе описан ряд других методов определения аммиака или азота в различных азотсодержащих соединениях обычно после переведения азота в аммонийную форму. Среди других методов определения азота необходимо назвать следующие.
Пиразолоновый метод [76—79], который основан на реакции между аммиаком, хлорамином Т (N-хлор-п-толуолсульфамид натрия) и пиразолоном с образованием рубазойной кислоты [1-фенил-З-метил-4-(Г-фенил-3,-метил-5'-пиразолонилиден-4,-амин) - пиразо-лон-5] синего или пурпурного цвета. Соединение экстрагируют четыреххлористым углеродом и измеряют оптическую плотность при 450 нм. Метод характеризуется более высокой чувствительностью по сравнению с описанными выше; однако точность метода меньше, а калибровочный график всегда нелинеен.
Хингидринный метод, основанный на образовании окрашенного соединения при взаимодействии хингидрина с аммиаком [80, 81] и измерении оптической плотности с желтым светофильтром.
В методе с применением гипобромита и бордо красного [бордо В, 1-(1-нафталиназо)-2-нафтол-3,6-дисульфокислота] к исследуемому раствору прибавляют титрованный раствор гипобромита, который взаимодействует с аммиаком. Избыток гипобромита определяют по разрушению бордо красного, оптическую плотность полученного раствора измеряют при 525 нм [82, 83].
В методе с применением гипохлорита [84] или гипобромита [85] переводят аммиак в NCI3 и ХВгз, полученные соединения взаимо
действуют с иодидом. Выделившийся иод определяют фотометри-ЧеСДминопирин-феноловый метод основан на образовании окрашенного в красно-оранжевый цвет соединения 4-(1',4'-бензохинон-4z-aMHHo)-антипирина при взаимодействии аминопирина с фенолом и аммиаком [86]. Соединение экстрагируют хлороформом и измеряют оптическую плотность при 458 нм относительно раствора аминопирина.
Определение аммиака в виде аммиаката меди [87] основано на образовании аммиаката меди и измерении оптической плотности при 700 нм. Определению мешают многие компоненты и особенно те ионы, которые образуют аммиакаты и гидроокиси.
Метод, основанный на измерении pH раствора, которым поглощают отогнанный аммиак [88], рекомендован для определения азота в сталях. Измерение pH проводится при помощи шкалы имитирующих стекол и универсального индикатора.
Метод, основанный на измерении спектров в ближней и инфракрасной области [89], рекомендован для определения аммиака в газовой смеси при повышенных давлении и температуре.
Для определения цианамидного азота рекомендовано цианамид при нагревании в кислой среде переводить в мочевину, которую определяют спектрофотометрически в виде комплекса с п-диметил-аминобенЭальдегидом [90].
1.7. ОПРЕДЕЛЕНИЕ НИТРАТА
Все нитраты хорошо растворимы в воде, поэтому при переведении анализируемой пробы в раствор не требуется особых приемов. Для фотометрического определения нитратов предложено много ‘.методов, из которых наиболее важное значение имеют следующие ^ри группы: 1) нитрование фенолов, а также хромотроповой кислоты, и фотометрическое определение образующихся нитрофенолов желтого цвета; 2) окисление нитратами некоторых органических соединений, например бруцина, дифениламина и т. п., с последующим фотометрическим определением окрашенных продуктов реакции; 3) восстановление до нитритов или до аммиака с последующим фотометрическим определением этих соединений.
1-7.1. Метод, основанный на нитровании фенолдисульфокислот
Принцип метода был предложен и усовершенствован еще в 1883—1885 гг. [91, 92]. Вначале предполагали, что при реакции нитрата с фенолом в среде концентрированной серной кислоты образуется пикриновая кислота. Однако подробное * исследование
* С использованием 48 литературных источников.
ею~3
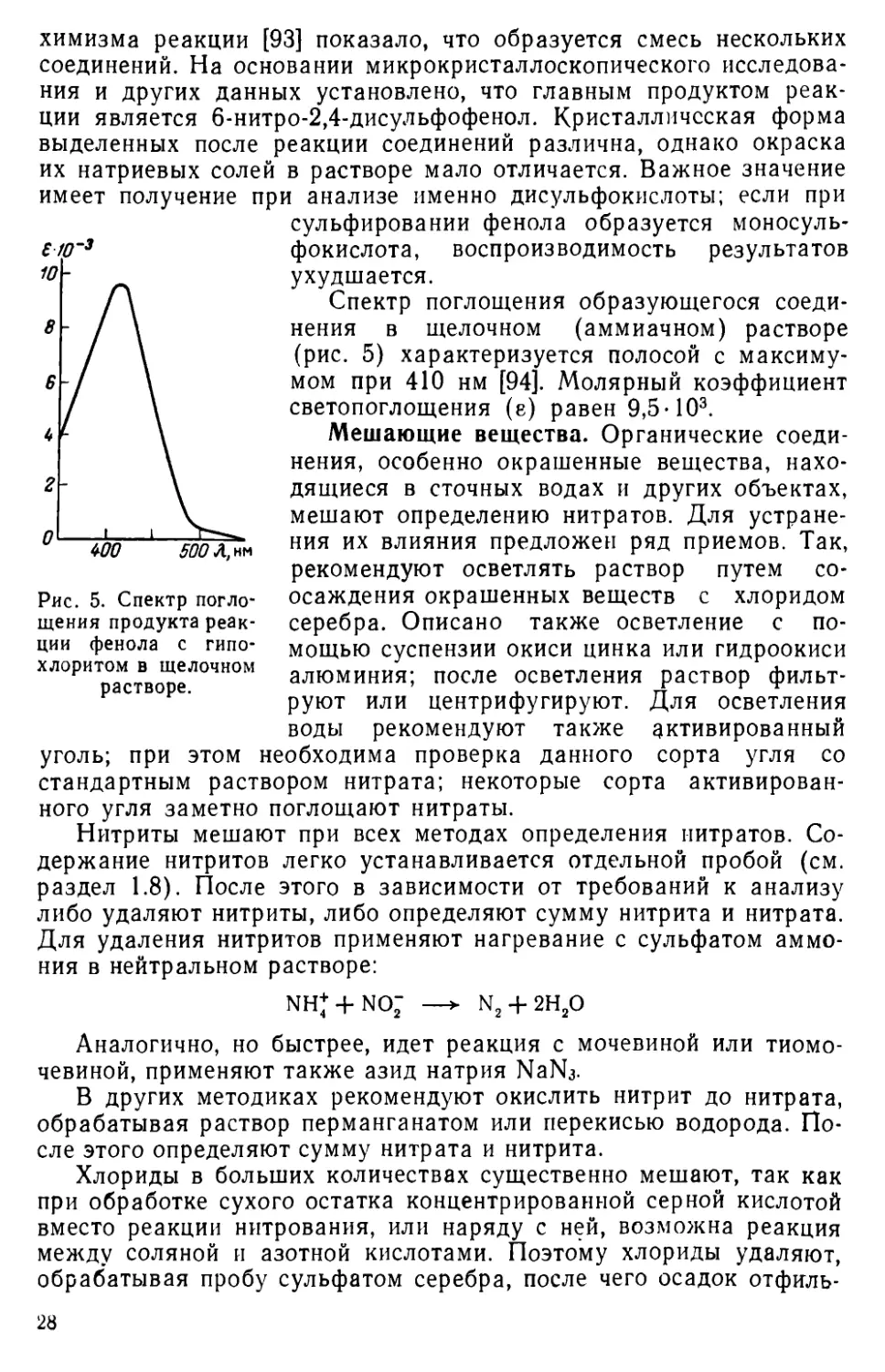
Рис. 5. Спектр поглощения продукта реакции фенола с гипохлоритом в щелочном растворе.
химизма реакции [93] показало, что образуется смесь нескольких соединений. На основании микрокристаллоскопического исследования и других данных установлено, что главным продуктом реакции является 6-нитро-2,4-дисульфофенол. Кристаллическая форма выделенных после реакции соединений различна, однако окраска их натриевых солей в растворе мало отличается. Важное значение имеет получение при анализе именно дисульфокислоты; если при сульфировании фенола образуется моносульфокислота, воспроизводимость результатов ухудшается.
Спектр поглощения образующегося соединения в щелочном (аммиачном) растворе (рис. 5) характеризуется полосой с максимумом при 410 нм [94]. Молярный коэффициент светопоглощения (е) равен 9,5-103.
Мешающие вещества. Органические соединения, особенно окрашенные вещества, находящиеся в сточных водах и других объектах, мешают определению нитратов. Для устранения их влияния предложен ряд приемов. Так, рекомендуют осветлять раствор путем со-осаждения окрашенных веществ с хлоридом серебра. Описано также осветление с помощью суспензии окиси цинка или гидроокиси алюминия; после осветления раствор фильтруют или центрифугируют. Для осветления воды рекомендуют также активированный
уголь; при этом необходима проверка данного сорта угля со стандартным раствором нитрата; некоторые сорта активированного угля заметно поглощают нитраты.
Нитриты мешают при всех методах определения нитратов. Содержание нитритов легко устанавливается отдельной пробой (см. раздел 1.8). После этого в зависимости от требований к анализу либо удаляют нитриты, либо определяют сумму нитрита и нитрата. Для удаления нитритов применяют нагревание с сульфатом аммония в нейтральном растворе:
NH^ + NO; —> N2 + 2Н2О
Аналогично, но быстрее, идет реакция с мочевиной или тиомочевиной, применяют также азид натрия NaNj.
В других методиках рекомендуют окислить нитрит до нитрата, обрабатывая раствор перманганатом или перекисью водорода. После этого определяют сумму нитрата и нитрита.
Хлориды в больших количествах существенно мешают, так как при обработке сухого остатка концентрированной серной кислотой вместо реакции нитрования, или наряду с ней, возможна реакция между соляной и азотной кислотами. Поэтому хлориды удаляют, обрабатывая пробу сульфатом серебра, после чего осадок отфиль-28
вывают. При большом числе анализов вследствие высокой стои-ТРо°сти солей серебра обычно отказываются от применения методов, основанных на нитровании фенолов. Из других методов в данном случае применяют восстановление в щелочной среде до аммиака и определение последнего с реактивом Несслера или с фенолом и гипохлоритом (см. выше).
Влияние различных примесей на определение нитратов по методу, основанному на нитровании фенолов, подробнее рассмотрено в литературе [95, 96].
Измерение оптической плотности. Оптическую плотность растворов измеряют при 410 нм. Можно применять также метод стандартных. серий. Приготовленной шкалой можно пользоваться в течение двух недель.
Реактивы
Фенолдисульфокислота. Навеску 25 г бесцветного фенола растворяют в концентрированной серной кислоте и добавляют 75 мл дымящейся серной кислоты, содержащей 13—15% серного ангидрида. Нагревают до 100 °C в течение 2 ч. Хранят в склянке с притертой пробкой. В некоторых методиках рекомендуют сразу готовить реагент в склянке с притертой пробкой и там же хранить.
Стандартный раствор нитрата. Растворяют 0,722 г сухого нитрата калия и разбавляют до 100 мл. Для приготовления рабочего раствора разбавляют 10 мл исходного раствора до 1 л; 1 мл разбавленного раствора содержит 0,01 мг нитратного азота или 0,044 мг нитрата.
Ход анализа. Нейтральный испытуемый раствор, содержащий 0,02—0,15 мг нитратного азота, выпаривают досуха на водяной бане. К сухому остатку прибавляют 2 мл фенолдисульфокислоты и перемешивают до растворения. Если растворение идет медленно, рекомендуют нагреть раствор на водяной бане; однако при этом возможно потемнение его в результате обугливания органических ^еществ.
? После охлаждения разбавляют до 20 мл водой, затем постепенно прибавляют около 7 мл концентрированного раствора аммиака. Если остаются взвешенные частицы, их удаляют фильтрованием. Разбавляют до 100 мл и измеряют оптическую плотность.
1.7.2. Определение нитрата с применением хромотроповой кислоты
Динатриевая соль хромотроповой кислоты—1,8-диоксинафта-л ин-3,6-дисульфокислоты
ОН ОН
в сернокислой среде взаимодействует с нитратом и нитритом с образованием окрашенного соединения с максимумом поглощения
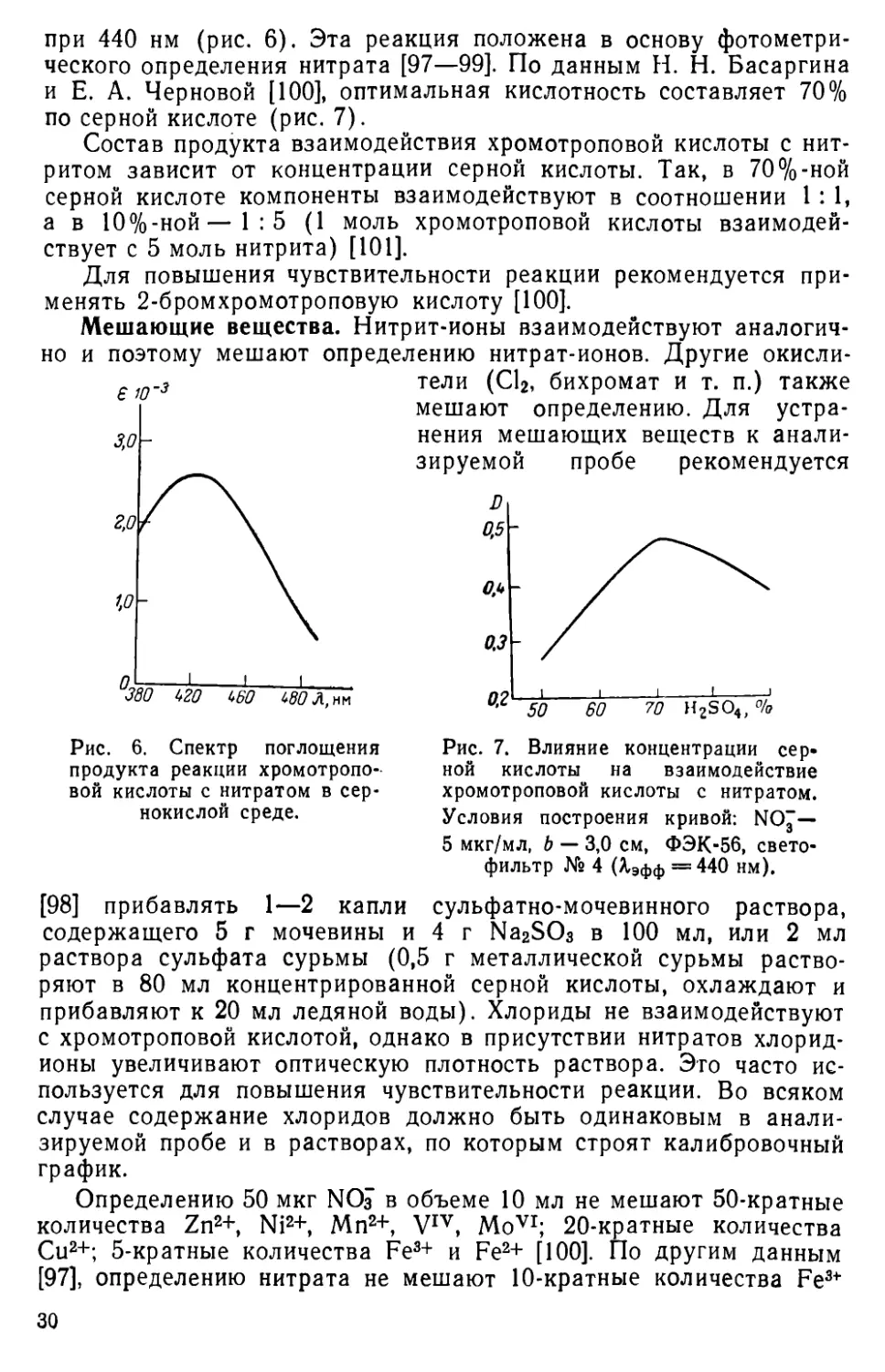
при 440 нм (рис. 6). Эта реакция положена в основу фотометрического определения нитрата [97—99]. По данным Н. Н. Басаргина и Е. А. Черновой [100], оптимальная кислотность составляет 70% по серной кислоте (рис. 7).
Состав продукта взаимодействия хромотроповой кислоты с нитритом зависит от концентрации серной кислоты. Так, в 70%-ной серной кислоте компоненты взаимодействуют в соотношении 1 : 1, а в 10%-ной—1:5 (1 моль хромотроповой кислоты взаимодействует с 5 моль нитрита) [101].
Для повышения чувствительности реакции рекомендуется применять 2-бромхромотроповую кислоту [100].
Мешающие вещества. Нитрит-ионы взаимодействуют аналогично и поэтому мешают определению нитрат-ионов. Другие окислители (С12, бихромат и т. п.) также мешают определению. Для устранения мешающих веществ к анализируемой пробе рекомендуется
Рис. 6. Спектр поглощения продукта реакции хромотроповой кислоты с нитратом в сернокислой среде.
Рис. 7. Влияние концентрации серной кислоты на взаимодействие хромотроповой кислоты с нитратом. Условия построения кривой: NOj — 5 мкг/мл, b — 3,0 см, ФЭК-56, светофильтр № 4 (Хэфф = 440 нм).
[98] прибавлять 1—2 капли сульфатно-мочевинного раствора, содержащего 5 г мочевины и 4 г Na2SO3 в 100 мл, или 2 мл раствора сульфата сурьмы (0,5 г металлической сурьмы растворяют в 80 мл концентрированной серной кислоты, охлаждают и прибавляют к 20 мл ледяной воды). Хлориды не взаимодействуют с хромотроповой кислотой, однако в присутствии нитратов хлорид-ионы увеличивают оптическую плотность раствора. Это часто используется для повышения чувствительности реакции. Во всяком случае содержание хлоридов должно быть одинаковым в анализируемой пробе и в растворах, по которым строят калибровочный график.
Определению 50 мкг NO3 в объеме 10 мл не мешают 50-кратные количества Zn2+, Ni2+, Mn2+, VIV, MoVI; 20-кратные количества Cu2+; 5-кратные количества Fe3+ и Fe2+ [100]. По другим данным [97], определению нитрата не мешают 10-кратные количества Fe3+
50-кратные количества Fe2+. При большом содержании железа И о отделяют с помощью катионита КУ-2.
еГ Определению сильно мешают кобальт и бор, поэтому их количества не должны превышать содержание нитрата.
Измерение оптической плотности. Измерение оптической плот* ности проводят при 440 нм.
1.7.2.1. Определение нитрата в отсутствие мешающих веществ
Реактивы
Хромотроповая кислота. Навеску препарата 100 мг (динатриевой соли) растворяют в 50—70 мл концентрированной H2SO4. Раствор должен быть бесцветным. Если он окрашивается в желтый цвет, то это означает, что в серной кислоте присутствует нитрат и такая кислота не пригодна для работы. С целью ускорения растворения смесь можно слегка нагреть в течение 3—4 мин на водяной бане до 70 °C. Длительное нагревание и более высокая температура могут привести к побурению раствора. После растворения навески раствор переносят в мерную колбу емкостью 100 мл и доводят до метки концентрированной H2SO4. Для приготовления рабочего раствора отбирают 10 мл этого раствора, переносят в мерную колбу емкостью 100 мл, добавляют 7 мл концентрированной хлористоводородной кислоты, доводят раствор до метки концентрированной H2SO4 и перемешивают. Растворы надо хранить в темной посуде с пришлифованными пробками, избегая действия прямых солнечных лучей. Раствор пригоден для работы в течение четырех дней.
Стандартный раствор нитрата готовят, как указано в разделе I. 7.1.1.
Построение калибровочного графика. В ряд стаканов емкостью 50 мл вводят стандартные растворы с содержанием 10—100 мкг нитрата, далее обрабатывают, как анализируемый раствор.
Ход анализа. Отбирают раствор пробы, содержащий до 100 мкг NO3 в 3 мл, и помещают в стакан емкостью 50 мл. Если надо, разбавляют водой до 3 мл и перемешивают. Затем добавляют 7 мл Рабочего раствора хромотроповой кислоты, перемешивают круговыми движениями стакана, оставляют на 30 мин для охлаждения до комнатной температуры, после чего измеряют оптическую плотность раствора при 440 нм. Раствором сравнения служит смесь 3 мл дистиллированной воды и 7 мл рабочего раствора хромотроповой кислоты. Содержание нитрата находят по калибровочному графику.
I. 7.2.2. Определение нитрата в почве [100]
Реактивы
Сульфат калия, 0,05%-ный раствор.
Другие реактивы — указаны в разделе I. 7.2.1.
Ход анализа. Образец анализируемой почвы растирают в фарфоровой ступке и просеивают через сито с отверстиями диаметром мм. Навеску почвы 10 г помещают в колбу емкостью 100 мл, До авляют 50 мл раствора H2SO4 и взбалтывают в течение 3 мин*
Затем смесь фильтруют через двойной складчатый фильтр (синяя лента). Если фильтрат мутный, то фильтрование повторяют. В приготовленном растворе определяют нитрат, как указано в разделе 1.7.2.1.
1.7.3. Определение нитрата с применением бруцина
Алкалоид бруцин (сильный яд!) в среде концентрированной серной кислоты образует при реакции с нитратом соединение красного цвета, который быстро переходит в желтый. Состав образующегося соединения не установлен. Бруциновый метод рекомендуют для быстрого определения нитратов в количестве 1 — 10 мг в пробе 10 мл, причем, в отличие от других методов, нет необходимости выпаривать раствор досуха.
/Мешающие вещества. Определению нитратов мешают нитриты. Поэтому их надо удалять или компенсировать их влияние, вводя нитриты в стандартные растворы, используемые при построении калибровочного графика [102].
Влияние различных примесей на определение нитратов по методу, основанному на реакции с бруцином, рассмотрено в литературе [95, 96].
Измерение оптической плотности. Спектр поглощения раствора характеризуется полосой с максимумом при 410 нм; молярный коэффициент поглощения в оптимальных условиях равен 1,5-103. Оптическую плотность измеряют при 410 нм. Окраска наиболее интенсивна, если к 1 объему испытуемого раствора прибавлено 2 объема концентрированной серной кислоты. Окраска недостаточно воспроизводима, поэтому обычно рекомендуют вести два параллельных определения и брать средний результат. Калибровочный график не прямолинеен.
Реактивы
Раствор бруцина. Навеску 5 г бруцина растворяют в 100 мл хлороформа.
Стандартный раствор нитрата приготовляют, как указано в разделе 1.7.1.
i
Ход анализа. Берут 10 мл испытуемого раствора, прибавляют! 0,20 мл раствора бруцина. Затем осторожно прибавляют 20 мл’ концентрированной серной кислоты. Через-5—10 мин разбавляют водой до 50 мл. Быстро охлаждают, доливают воду до метки’ (50 мл) и измеряют оптическую плотность.
1.7.4. Восстановление нитрата до нитрита
Для определения нитритов известно значительно больше про-! стых, надежных и чувствительных методов определения, чем для? определения нитратов. В связи с этим предложен ряд способов^
восстановления нитрата до нитрита, после чего определяют нитрит по одному из описанных ниже методов (см. раздел 1.8).
Вначале [ЮЗ] было предложено восстановление нитрата гидразином в присутствии катализатора — соли меди. Однако восстановление гидразином идет медленно, полное восстановление дости-1-ается только через 24 ч; при температуре ниже 26° С реакция еще более замедляется. Позже было предложено [104] восстановление цинковой пылью в аммиачной среде. Чтобы уменьшить побочные реакции, рекомендуется восстанавливать в ледяной бане, тем не менее выход составляет 85—90%.
Предложено также восстановление нитрата до нитрита цинковой пылью [105], металлическим цинком [106] и смесью цинковой пыли и порошка железа [107] в кислой среде, а также металлическим кадмием [108]. Наилучшие результаты получены при восстановлении амальгамированным кадмием [109—111].
Восстановление нитрата амальгамированным кадмием
Температура на процесс восстановления влияет мало. Рекомендуемый pH = 6,8—8,2. Восстановление идет на 91 ±2%, поэтому, несмотря на хорошую воспроизводимость, калибровочный график необходимо строить не по нитриту, а по нитрату, после его восстановления на колонке.
Восстановительная колонка
Восстановление ведут в колонке диаметром 0,8 см, длиной 22 см. Берут 200 г зерненного кадмия и встряхивают 3 мин с 200 мл 1°/о-ного раствора хлорида ртути(П). Слой кадмия в колонке должен быть не менее 7 см. Кадмий должен всегда находиться под слоем воды. Скорость прохождения анализируемого раствора через колонку 0,6 мл в 1 мин.
Ход определения нитратов в морской воде [108]. Берут 25 мл ^анализируемой воды и вносят ее в колонку с амальгамированным 1ЁЙдмием. Регулируют кран таким образом, чтобы весь объем воды прошел через колонку за 40—45 мин. Затем колонку споласкивают 4 раза дистиллированной водой порциями по 5 мл и в полученном растворе определяют нитриты, как указан) в разделе 1.8.1.
I. 8. ОПРЕДЕЛЕНИЕ НИТРИТА
Большинство методов определения нитритов основано на синтезе азокрасителей с участием нитрита. Нигриты реагируют с первичными ароматическими аминами, образуя соли диазония. Затем проводится сочетание соли диазония с ароматическим соединением, содержащим аминные или фенольные группы. Образующийся азокраситель определяют фотометрически.
2 Зак. 761
33
В качестве примера можно привести реакции с сульфаниловой кислотой и азосочетание с 1-нафтиламином:
Диазотирование необходимо вести в присутствии достаточного количества хлористоводородной или уксусной кислоты при комнатной температуре или при охлаждении. Кроме того, азосочетание необходимо проводить только после завершения диазотирования и при возможно более слабой кислотности раствора.
Калибровочный график прямолинеен даже при значительных количествах нитрата — до 150—300 мкг/л; чувствительность методов составляет несколько микрограммов нитритного азота в 1 л.
1.8.1. Определение нитритного азота с применением сульфаниловой кислоты и 1-нафтиламина
Мешающие вещества. Определению мешают окислители, восстановители и вещества, имеющие собственную окраску.
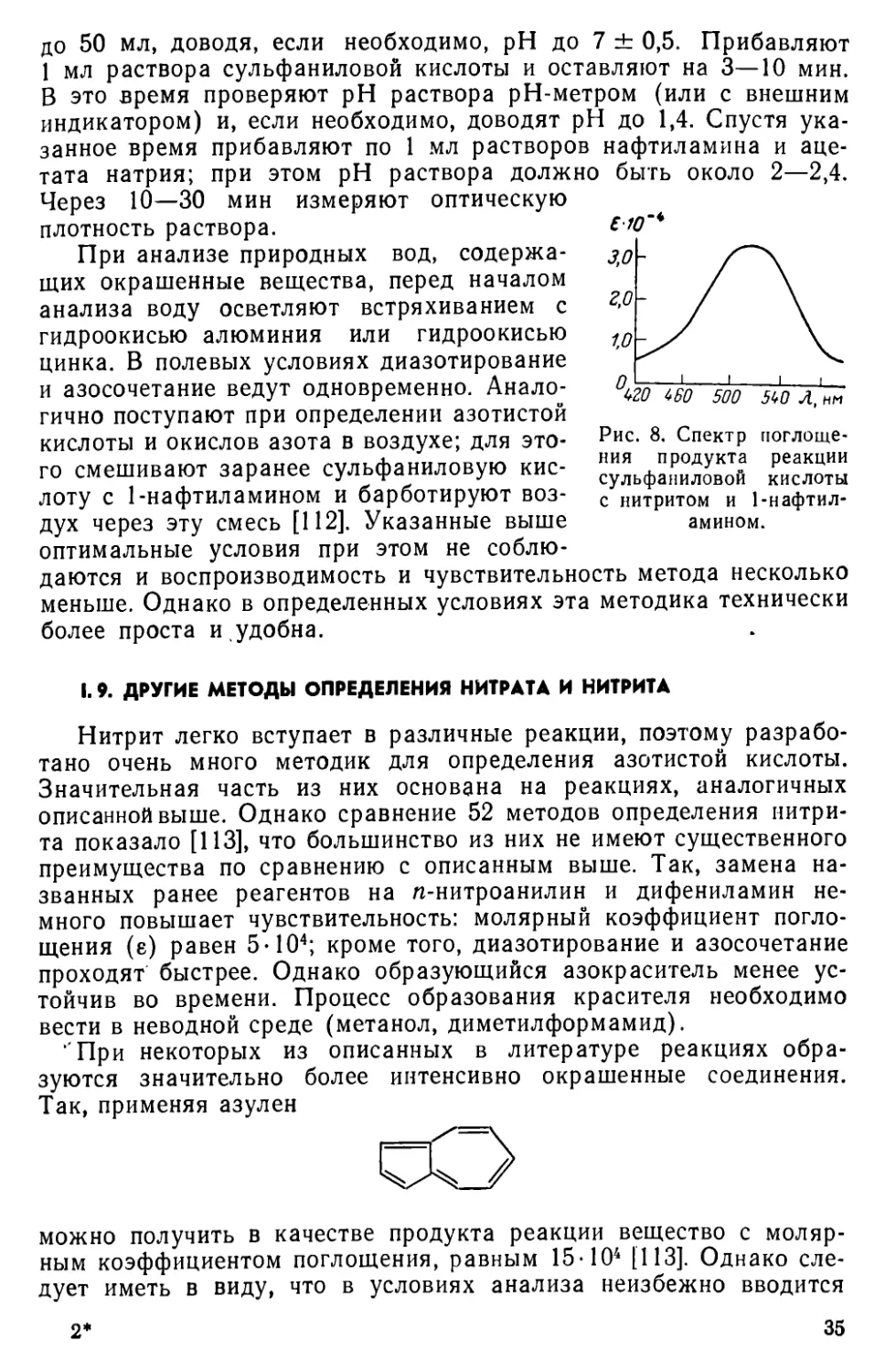
Измерение оптической плотности. Полоса поглощения образующегося азокрасителя характеризуется максимумом при 520 нм (рис. 8). Молярный коэффициент поглощения е составляет 3,3-104. Оптическую плотность раствора измеряют при 520 нм.
Реактивы
Дистиллированная вода. Вторично воду перегоняют в присутствии перманганата и щелочи.
Сульфаниловая кислота. Растворяют 0,6 г препарата в 70 мл воды, прибавляют 20 мл концентрированной хлористоводородной (или уксусной) кислоты и разбавляют до 100 мл.
1-Нафтиламин солянокислый. Растворяют 0,60 г солянокислого 1-нафтиламина в воде, добавив 1 мл концентрированной хлористоводородной кислоты, разбавляют до 100 мл и хранят в холодильнике; срок хранения 1 месяц.
Лцетат натрия, навеску в расчете на 16,4 г безводной соли растворяют в 100 мл.
Стандартный раствор нитрита. Растворяют 0,246 г нитрита натрия (соответствует 50 мг азота) в 1 л воды. Точное содержание нитрита устанавливают иодометрически. Рабочий стандартный раствор готовят разбавлением 10 мл запасного раствора до 1 л; раствор консервируют добавкой хлороформа.
Ход анализа. Для анализа берут 0,1—10 мл испытуемого раствора, содержащего не более 7 мкг нитритного азота. Разбавляют
до 50 мл, доводя, если необходимо, pH до 7 ± 0,5. Прибавляют 1 мл раствора сульфаниловой кислоты и оставляют на 3—10 мин.
В это время проверяют pH раствора pH-метром (или с внешним индикатором) и, если необходимо, доводят pH до 1,4. Спустя указанное время прибавляют по 1 мл растворов нафтиламина и аце-
тата натрия; при этом pH раствора должно быть около 2—2,4.
Через 10—30 мин измеряют оптическую плотность раствора.
При анализе природных вод, содержащих окрашенные вещества, перед началом анализа воду осветляют встряхиванием с гидроокисью алюминия или гидроокисью цинка. В полевых условиях диазотирование и азосочетание ведут одновременно. Аналогично поступают при определении азотистой кислоты и окислов азота в воздухе; для этого смешивают заранее сульфаниловую кислоту с 1-нафтиламином и барботируют воздух через эту смесь [112]. Указанные выше оптимальные условия при этом не соблю-
0 L----1-----1----1___I—_
Ь60 500 5М Л, нм
Рис. 8. Спектр поглощения продукта реакции сульфаниловой кислоты с нитритом и 1-нафтиламином.
даются и воспроизводимость и чувствительность метода несколько меньше. Однако в определенных условиях эта методика технически более проста и,удобна.
I. 9. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ НИТРАТА И НИТРИТА
Нитрит легко вступает в различные реакции, поэтому разработано очень много методик для определения азотистой кислоты. Значительная часть из них основана на реакциях, аналогичных описанной выше. Однако сравнение 52 методов определения нитрита показало [113], что большинство из них не имеют существенного преимущества по сравнению с описанным выше. Так, замена названных ранее реагентов на n-нитроанилин и дифениламин немного повышает чувствительность: молярный коэффициент поглощения (е) равен 5-Ю4; кроме того, диазотирование и азосочетание проходят быстрее. Однако образующийся азокраситель менее устойчив во времени. Процесс образования красителя необходимо вести в неводной среде (метанол, диметилформамид).
‘ При некоторых из описанных в литературе реакциях образуются значительно более интенсивно окрашенные соединения. Так, применяя азулен
можно получить в качестве продукта реакции вещество с молярным коэффициентом поглощения, равным 15* 104 [113]. Однако следует иметь в виду, что в условиях анализа неизбежно вводится
избыток реагента, между тем сам азулен довольно интенсивно окрашен.
В той же работе [113] описана оригинальная высокочувствительная автокаталитическая реакция на нитриты. Реактивом является 4,4'-бис-(диметиламино)-тиобензофенон (ДТБ). Последний реагирует с азотистой кислотой (в среде 0,15—0,25%-ной хлористоводородной кислоты) с образованием ряда промежуточных соединений. На определенной ступени этой реакции выделяется окись азота NO. Окись азота реагирует с растворенным в воде кислородом, причем частично снова образуется азотистая кислота, которая вступает в реакцию с новой порцией реактива. Таким образом, как обычно в каталитических реакциях, количество продукта реакции существенно превышает обычное стехиометрическое, что приводит к значительному увеличению чувствительности реакции. В данном случае кажущийся коэффициент светопоглощения, рассчитанный по отношению к 1 моль нитрита, равен 62-104. Это обусловлено участием нитрита в нескольких циклах образования окрашенного соединения.
Реакции, по-видимому, проходят по следующей схеме:
Спустя 40—50 мин реакция практически прекращается вследствие расхода нитрита на побочные процессы. После этого измеряют оптическую плотность.
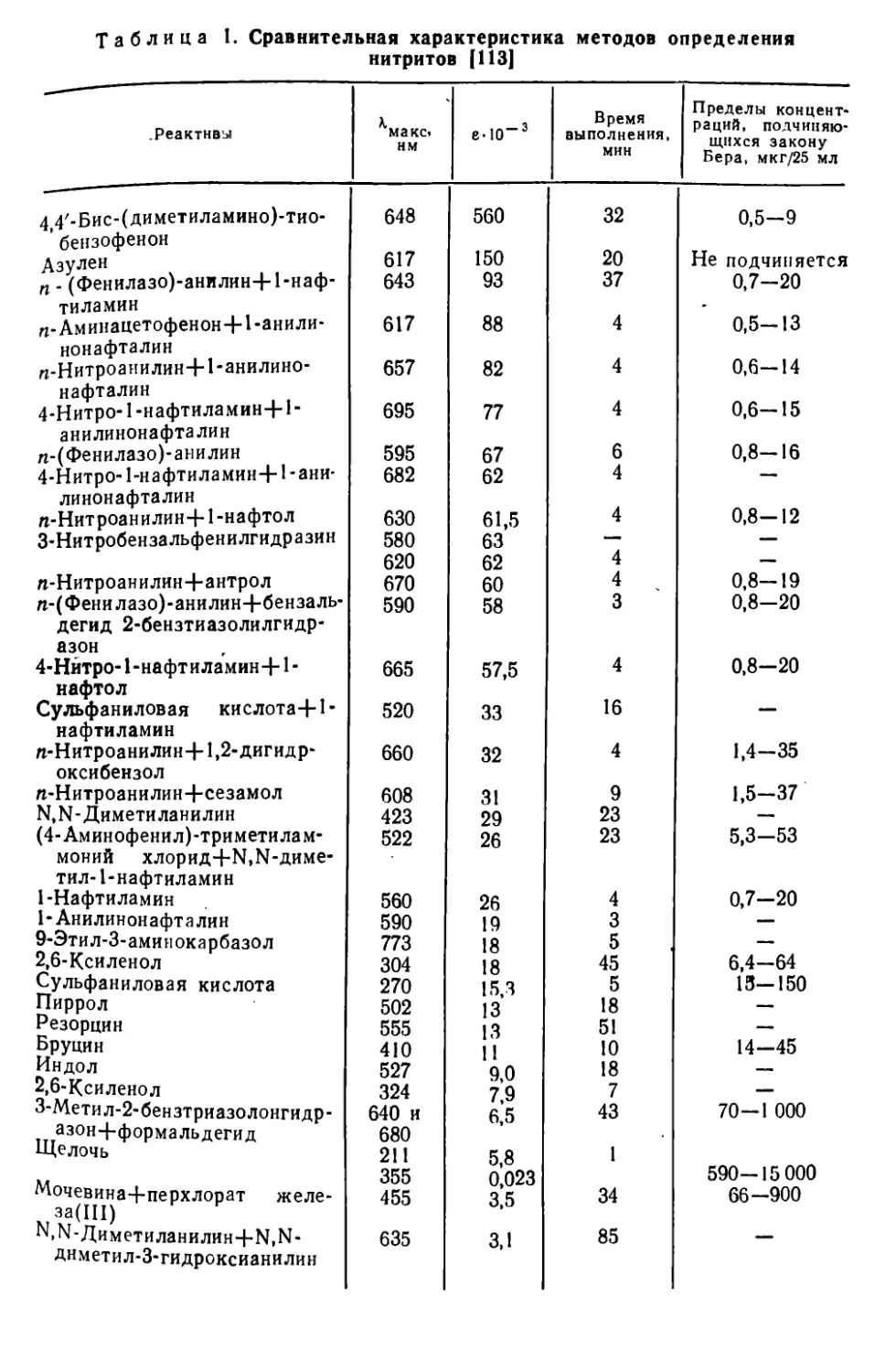
В табл. 1 приведена характеристика некоторых методов определения нитритов.
Описаны методики фотометрического определения нитритов и нитратов без применения реагентов, т. е. по собственному поглощению в ультрафиолетовом спектре [114—129] и в видимой области спектра [124, 125]. Однако эти методики весьма мало чувствительны, так, молярный коэффициент составляет всего 0,58-104 [113].
Таблица 1. Сравнительная характеристика методов определения нитритов [ИЗ]
.Реактивы К макс* нм е-10“3 Время выполнения, мин Пределы концентраций, подчиняющихся закону Бера, мкг/25 мл
4 4/-Бис-(диметиламино)-тио- 648 560 32 0,5-9
бензофенон
Азулен 617 150 20 Не подчиняется
п - (фенилазо)-анилин+1-наф- 643 93 37 0,7-20
тиламин п-Аминацетофенон+1-анили- 617 88 4 0,5-13
нонафталин
п-Нитроанилин+1-анилино- 657 82 4 0,6-14
нафталин
4-Нитро-1-нафтиламин+1- 695 77 4 0,6-15
анилинонафталин
п-(Фенилазо)-анилин 595 67 6 0,8-16
4-Нитро-1-нафтиламин4-!-ани- 682 62 4 —
линонафталин
п-Нитроанилин+1 -нафтол 630 61,5 4 0,8-12
З-Нитробензальфенилгидразин 580 63 — —
620 62 4 —
п-Нитроанилин+антрол 670 60 4 0,8-19
п-(Фенилазо)-анилин+бензаль- 590 58 3 0,8-20
дегид 2-бензтиазолилгидр-
азон
4-Нйтро- 1-нафтиламин+1 665 57,5 4 0,8-20
нафтол
Сульфаниловая кислота+1- 520 33 16 —
нафтиламин
п-Нитроанилин-|-1,2-дигидр- 660 32 4 1,4-35
оксибензол
п-Нитроанилин+сезамол 608 31 9 1,5-37
N.N-Диметиланилин 423 29 23 —
(4-Аминофенил)-триметилам- 522 26 23 5,3-53
моний хлорид+М.М-диме-
тил- 1-нафтиламин
1-Нафтиламин 560 26 4 0,7-20
1-Анилинонафталин 590 19 3 —
9-Этил-З-аминокарбазол 773 18 5 —
2,6-Ксиленол 304 18 45 6,4-64
Сульфаниловая кислота 270 15,3 5 15—150
Пиррол 502 13 18 —
Резорцин 555 13 51 —
Бруцин 410 11 10 14—45
Индол 527 9 0 18 —
2,6-Ксиленол 324 79 7 —
З-Метил-2-бензтриазолонгидр- 640 и 6,5 43 70-1 000
азон+формальдегид 680
Щелочь 211 58 1
355 0,023 590—15 000
Мочевина+перхлорат желе- 455 3,5 34 66—900
Мм(П1)
^.М-Диметиланилин+Ы.М- 635 3,1 85 —
Днметил-3-гидроксианилин
Продолжение
Реактивы ^макс» нм е-10” 3 Время выполнения, мин Пределы концентраций, подчиняющихся закону Бера, мкг/25 мл
п-Аминоацетофенон+антрол 619 57 4 0,8—20
4-Нитро-1-нафтиламин+ант- 720 56 4 0,82-20
рол 515
п-Нитроанилин+азулен 51 4 0,4-9
Антраниловая кислота+1- 571 50 16 0,4—9
анилинонафталин 655
п-Нитроанилин+дифениламин 50 4 1-24
п-Нитроанилин+бензальде- 595 49 3 —
гид 2-Бензтиазолилгидразон 685
4-Нитро-1-иафтиламин+ан- 47 4 1-24
трон
п-Аминоацетофенон+азулеп 521 47 4 0,4—10
п-Аминофенилсульфон+азу- 510 47 4 0,4-10
лен
Сульфаниловая кислота+N- 550 46 16 2,5-60
(1-нафтил)-этилендиамин
п-Нитроанилин+антрол 653 44 4 1-24
п- Фени лан и лин -j-азулен 549 44 4 0,4-10
п-Аминоацетофенон+антрол 597 41 4 1,1-24
4-Нитро-1-нафтиламин+дифениламин 705 40 4 1,2-30
Сульфаниламид и М-(1-наф- 550 40 34 5,8-35
тил)-этилендиамин
п- Аминоацетофенон+1 -нафтол 565 38 4 1,2-30
Хлор-п-фени лен диамин 345 37,7 11 6-60
4-Нитро-1-нафтиламин+бенз- 650 36,5 4 1,3-30
альдегид 2-бензтиазолил-гидразон
Бис-(4-аминофенил)-суль- 538 35 4 0,5-12
фид+азулен
п-Аминоацетофеноп+бензаль- 567 33,4 4 1,4-35
дегид 2-бензтиазолилгидр-аэон
Определение по собственному поглощению применяется для определения' окислов азота [126—128].
Для определения нитрита описана реакция с риванолом [129— 132], причем образуется окрашенное соединение с полосой поглощения при 515 нм, однако чувствительность определения мала.
Для определения нитратов по образованию нитросоединений предложен ряд методик, в которых сульфофенолы заменены другими, более доступными веществами. Так, описано применение сульфосалициловой кислоты [133, 134], n-аминосалициловой кислоты [135], 2,5-фенолдисульфокислоты [136] и др.
Предложен ряд веществ, образующих окрашенные продукты при окислении их азотной кислотой. Так, изучено применение дифениламина [137—141], однако в этом случае закон Бера соблю-38
дается значительно хуже. Много работ посвящено применению ксиленолов для определения нитрата [142—149].
Ряд работ посвящен созданию методов определения нитрата, основанных на восстановлении нитрата до нитрита цинковой пылью [150—154] в присутствии гидроокиси аммония или сернокислым гидразином [155, 156] и последующем определении нитрита.
В ряде случаев используется восстановление нитратов и нитритов до аммиака и определение последнего по Несслеру. В качестве восстановителя нитратов и нитритов до аммиака предложен титан(III) [157] и хлорид хрома(II) [158, 159]. По-видимому, проще и надежнее восстановление сплавом Деварда в щелочной среде [160].
Для определения нитритов предложен метод, основанный на нитровании антипирина с последующим измерением оптической плотности образующегося 4-нитрозоантипирина при 343 нм [161]. При определении двуокиси азота в газах применен тиокетон Мих-лера [162]. На реакции диазотирования бензидина и лг-фениленди-амина [163], производных бензидина [164], сульфаниламида [165], сульфаниловой кислоты [166], эйконогена [167], /г-розанилинметан-сульфокислоты [168], соединений ацилсульфоксония [169] с последующим сочетанием с разными реагентами предложены методы определения нитрита и окислов азота в разных объектах.
Реакция окислов азота с железом(II) применена для определения окислов азота в дыме табака [170] и в других газах [171, 172].
С целью повышения селективности и чувствительности определения примеси нитратов в серной кислоте предложен в качестве реагента хинализарин [173].
Более перспективным является развитие экстракционно-фотометрических методов определения нитрата и нитрита. Установлено [174], что нитрат количественно экстрагируется хлорбензолом в присутствии избытка кристаллического фиолетового при pH = = 5,0—7,0. Максимум поглощения находится при 595 нм. Предложен также метод экстракции нитрата из водного раствора раствором неокупроина (NK) в метилизобутилкетоне [175] в присутствии небольших количеств меди(1). Оптическую плотность экстракта измеряют при 456 нм. Авторы предполагают, что при этом экстрагируется соединение [Си (NKhNOg]. Для определения нитритов предложен экстракционный метод, основанный на реакции диазотирования 8-аминохинолина с образованием хинолиндиазония и последующем сочетании его с 8-аминохинолином, что приводит к образованию интенсивно окрашенного азосоединения [176], которое экстрагируют н-гептанолом и измеряют оптическую плотность при 465 нм. Лучшим экстракционно-фотометрическим методом, по-видимому, является метод определения нитрита в виде его соединения с красителем синим основным К [177]. Бензольный экстракт этого соединения окрашен в зеленый цвет, максимум поглощения около 650 нм.
Представляют большой интерес экстракционно-фотометрические методы определения нитрата с нильской голубой А [178], тетрафениларсонием [179], нитрата и нитрита в виде нитротолуола, получаемого предварительным нитрованием толуола [180], экстракционное определение нитрита в питьевой воде при помощи сульфо-ниламида и хлорида N-1-нафтилэтилендиамина в присутствии додецилбензилсульфоната [181].
На основе каталического действия нитрита на реакцию окисления комплекса марганца с ЭДТА перекисью водорода разработан фотометрический каталитический метод определения нитрита [182]. Экстракция диэтиловым эфиром соединения нитрита с родамином С применена для флуоресцентного определения нитрита [183].
ЛИТЕРАТУРА
1. Nessler J„ Chem. Centr., 27, Neue Folge, 1, 529 (1856).
2. Kirk P„ Anal. Chem, 22, 354, 611 (1950).
3. H i 11 e г A, Plazin J, Van Slyke D. D, J. Biol. Chem, 176, 1401 (1948). -
4. W h i t e L, L о n g M, Anal. Chem, 23, 368 (1951).
5. L a k e G. R. 1. e. a. Anal. Chem, 24, 1806 (1952).
6. M i 11 e r G, M i 11 e г E, Anal. Chem, 20, 481 (1948).
7. Fish V, Anal. Chem, 24, 760 (1952).
8. Курна ев В. T, Вести, с.-х. науки, № 10, 146 (1958).
9. Yren S. Н, Pollard A. G, J. Sci, Food and Agric, 5, 364 (1954).
10. С вяткин М. Т, Сб. науч. тр. Воен.-мед. фак. при Саратовском мед. ин-те, вып. I, 152 (1957).
11. Га росс И, Изв. АН ЛатвССР, № 12, 60 (1953).
12. В а 1 k s R, R е с к е г s J, Landwirtsch. Forsch, 8, 7 (1955).
13. Dost al A, Mastaiir L, Sb. Ceskosl. anad. Lemed. ved. Rostl. Vyroba, 5, 1331 (1959).
14. W i 1 c z о к T„ Chem. analit. (Polska), 4, 981 (1959).
15. Bernhard E, Mitt. Gebiete Lebensmitteluntersuch. und Hyg, 45, 115 (1954).
16. Schillak R, Roczn. gleborhancze, 7, Dod, 185 (1958).
17. Лапин Л. H, Заманов P. X, Макарова В. П, Почвоведение, № 4, 95 (1957).
18. И кэ га ми, Нагаока, Ямасаки. J. Iron and Steel Inst. Japan, 43, 951 (1953); цит. по РЖХим, 1958, 28498.
19. Федоров А. А, Кричевская А. М, Зав. лаб, 34, 1425 (1968).
20. А ф о н с к и й И. Ф, Вер О. И, Смирнов А. В. Теория и практика азотирования стали, М, ГНТИ по машиностроен, металлообработке и черной металлургии. 1933, стр. 27.
21. W й п s с h Н, Stahl и. Eisen, 83, 1705 (1963).'
22. Клячко Ю. А, Ларина О. Д, Зав. лаб, 26, 1047 (1960).
23. Crone F, Smith J. and Е, Anal. Chim. acta, 31, 258 (1964).
24. В ее gh 1 у H, Fu re g J, Anal. Chem, 24, 199 (1952).
25. Мальцев В. Ф, Герцман Ф. М, Те мир емко Т. П, Зав. лаб 15, 288 (1949).
26. Gyorbiro К., Romwalterne-Major Е, Kohasz. lapok, 11, 112 (1956)).
27. Winkler Н. J, Bach В. В, Brit. Gast. Iron Rec. Assoc. J. Res. and Develop, 4, 553 (1953).
28. Dubox E. J, Mayoral A. M, An. Asoc. quim. argent, 43, 227 (1955).
99 Namiki Michiko, Kakita Yachiyo, Goto Hidehiro, Taianta, ’ 11, 813 (1964).
30 К а м mo p и О и ко, Хияма Ясу о, Хотта Ватару, РЖХим, 1965, ‘ 22Г57.
31 Boalin R., Conlombean J., Jan don E., Rev. metallurgie (France), ’ 57, 623 (1960).
32. Л e в и Л. И., Китаев Я. А., Зав. лаб., 35, 1438 (1969).
33. Тарас М. Дж. В сб.: «Колориметрические (фотометрические) методы определения неметаллов». Пер. с анг. Под ред. А. И. Бусева. М., Издатин-лит, 1963. См. с. 78.
34. Nichols М„ W i 11 е t С., J. Amer. Chem. Soc., 56, 769 (1934).
35. S а г к а г Р. В., Gros k N. N., Anal. Chim. acta, 13, 195 (1955).
36. С io go lea Gh., Apreotesei C., Farmacia (RPR), 7, 399 (1959).
37. Moller G., Z. anal. Chem., 245, 155 (1969).
38. W i 11 i a m s P„ Analyst, 89, 276 (1964).
39 К i 11 n e r Z., К r a 1 A., Knizn. obzor. a ved. spis Vysok. uceni thechn, Brne, 5, 263 (1966).
40. Коннов В. A., Tp. ин-та океанол. АН СССР, 54, 123 (1962).
41. Батманова О. Я., Тр. Ленингр. сан.-гигиен, мед. ин-та, 11, 44 (1953).
42. Thielemann Н., Mikrochim. acta, № 6, 875 (1971).
43. Apostolache Seb., Rev. Chim. (RPR), 13, 615 (1962).
44. A was th i S. P., S. S a h a s z a n a m a n, M. Sun da resan, Analyst, 92, 650 (1967).
45. Cato in E., Badoin R., Dr a gut V., Di comun. ICECHIM, 1957—1962, Vol. 2, S. 1, Inst, cercetari chimice, s. a., 297.
46. W a 1 k d e n J., Atomic Energy Res. Establ., 1959, № AM43, 4p.
47. BurakowskiT., Asta geophys. polon, 8, 194 (1960).
48. C h w a 1 i n s k i S., Bizostak M„ Chem. analyst. (Polska), 12, 723 (1967).
49. F i s h b e i n W. N., Anal. Chim. acta, 37, 484'(1967).
50. S c h e u г e г P. G., Smith F., Anal. Chem., 27, 1616 (1955).
51. Chai u pa J., Vokounova-Gorovova E„ Ceskosl. Hyg., epidemiol., mikrobiol., imunol., 4, 273 (1955).
52. Bolleter W. T., Bushman C. J., Tidwell P. W., Anal. Chem., 33, 592 (1961).
53. Каплин В. T., Гидрохим. материалы, 32, 148 (1961).
54. Д а ц к о В. Г., Каплин В. Т. В сб.: «Совр. методы анализа природных вод». М„ АН СССР, 118 (1962).
55. Poskam R. Th., Lan gen D. de, Anal. Chim. acta, 30, 56 (1964).
56. S ch a ch H. G„ Limnologica, 4, 431 (1966).
57. SagiTakeshi, Oceanogr. Mag., 18, 43 (1966).
58. Lei the W., Pets chi G., Z. anal. Chem., 230, 344 (1967).
59. И в а н о в a H. Л., Улицкий P. И., Шматова А. К. В сб.: «Методы определения и исслед. состояния газов в металлах». М., «Наука». 1968. См. с. 190.
60. Н а г w о о d J. Е„ К й h n A. L., Water Res., 4, 805 (1970).
61. Gehrke Ch. W. e. a., J. Assoc. Offic. Anal. Chem., 54, 651 (1971).
62. S e 1 m e г - О 1 s e n A. R., Analyst, 96, 565 (1971).
63. Kammori Ohiko, Hiyama Yasuo, Hotta Wataru, Trans. Jap Inst. Metals, 8, 58 (1967).
64. S о 1 б г z a n о L., Limnol. and Oceanogr., 14, 799 (1969).
65. Kallmann S i 1 v e, 1. ol., Anal. Chem., 40, 332 (1968).
66. Куленок M. И., Изв. вузов. Хим. и хим. техн., 4, 558 (1961).
67. Гълъбов Ас. 3. Сб. тр. Высш. мед. ин-т, Пловдив, 12, 33 (1958, 1959).
68. Ковтун М. С., Гаухман М. С. В сб.: «Комплексообразов., межмол. взаимодействие и соосажд. в некотор. системах». Днепропетровск, 1970.
69. Goto Hidehiro, Kakita Jachiyo, Atsuya Ikuo, Sci. Rents Res. Insts Tohonu Unit., A19, 50 (1967).
70. Newell B. S., J. Marine Biol. Assoc., 47, 271 (1967).
71. Лапин Л. И. Z. anal. Chem., 98, 236 (1938); Тр. комиссии по аналит. хим. АН СССР, 5(8), 77 (1954).
72. Н a g a s h i М., Unemoto Т„ Miyaki К., Chem. and Pharmac. Bull., 7, 65 (1959).
73. К л и б у с А. X., Н а з а р ч у к Т. Н„ ЖАХ, 16, 79 (1961).
74. Вейнберг Г. Я., Зав. лаб., 9, 1073 (1940).
75. G г о w t h е г A., L а г g е R., Analyst, 81, 64 (1956).
76. К г u s е J., М е 11 о n М., Anal. Chem., 25, 1188 (1953).
77. Р г о с h a z к о v a L., Anal. Chem., 36, 865 (1964).
78. Ковамура Вар о, Ватанабэ Сиро, Ацубо Такаси, J. Iron and Steel Inst. Japan, 53, 555 (1967); цит. по РЖХим, 1968, ЗГПЗ.
79. Strickland J. D. H„ Austin К. H., J. Conseil perman. internal, explo-rat, 24, 446 (1959).
80. К а и ч у к А. А., Биохимия, 26, 393 (1961).
81. Moore S., Stein W. H„ J. Biol. Chem., 176, 367 (1948).
82. Кого left F., Internal. Council Explorat. Sea (Contribs)’, C.M. № 330, 2 (1960).
83. G i 11 b r i c h t M., Helgolander Wiss. Meeresuntersuch., 8, 58 (1961).
84. Z i t о m e r F., L a m b e r t J. J„ Anal. Chem., 34, 1768 (1962).
85. G r a s s h о f f K., Z. anal. Chem., 234, 13 (1968).
86. Kawamura K., Chem. and Pharmac. Bull., 16, 626 (1968).
87. В 1 i n u R. C., G u n t h e r F. A., Anal. Chem., 29, 1882 (1957).
88. П о н о м a p e в А. И., А с т а н и н а А. А., ЖАХ, 14, 234 (1959).
89. К о r e n J. G., A n d r e a t c h A. J., Anal. Chem., 37, 256 (1965).
90. Б о й ц о в E. H., Ширяева T. M., Мишина Э. А., Зав. лаб., 35, 35 (1969).
91. Sprengols H., Pogg. Ann., 121, 188 (1863); Ann. der Physik und Chemie, 121, 188 (1864).
92. Grandwol A„ H. Lajoux, J. Chem. Soc., 48, 1093 (1885).
93. C h a m u t E„ P r a 11 D., J. Amer. Chem. Soc., 31, 922 (1909).
94. Тарас M. Дж. В сб.: «Колориметрические (фотометрические) методы определения неметаллов». Пер. с анг. Под ред. А. И. Бусева, М., Издатин-лит, 1963. См. с. 140.
95. Nall С., Anal. Edit., 17, 426 (1945).
96. F a d г u s Н., М а 1 у J., Z. anal. Chem., 246, 239 (1969).
97. West Р. W., Lyles W. L„ Anal. Chim. acta, 23, 227 (1960).
98. West P. W., Ramachandran T. D., Anal. Chim. acta, 35, 317 (1966).
99. Clarke A. L., Lennings A. C., J. Agric. and Food Chem., 13, 174 (1965).
100. Басаргин H. H., Чернова E. А., ЖАХ, 23, 102 (1968).
101. R о b i n s о n J. W., H s u C. J., Anal. Chim. acta, 44, 51 (1969).
102. Wolf B., Anal. Edit., 16, 446 (1944).
103. Mu 11 i n J., P i 1 eу J., Anal. Chim. acta, 22, 464 (1955).
104. С г о w T., J о h n s t о n e M., Anal. Chim. acta, 27, 441 (1961).
105. Matsunaga K., Nishimura M., Anal. chim. acta, 45, 350 (1969).
106. Камацу Садакити, Хагино Кэн, РЖХим, 1968, 16Г93.
107. Кудеяров В. Н., Авт. св. СССР, кл. 421, 3/51 (gOln), № 217685, заявл.
12.04 67, опубл. 19.07.68. РЖХим, 1969, 65165П
108. Lambert R. S., Du В о is R. J., Anal. Chem., 43, 955 (1971).
109. М о г г i s A., R i 1 е у J., Anal. Chim. acta, 29, 272 (1963).
НО. Исаев А. Б., Богоявленский А. Н., Океанология, 8, 539 (1968).
111. Романов А. С., Новоселов А. А. В сб.: «Гидрофиз. исслед. Тихого и Атлантического океанов в кругосветн. плавании НИС «Михаил Ломоносов» (20 рейс). Севастополь, 1967. См. с. 82.
112. S а 1 z m а п В., Anal. Chem., 26, 1949 (1954).
113. Sawicki Е., Stonley Т., Pfoff J., D’Amico A., Taianta, 10, 641 (1963).
114. Armstrong A. F„ Anal. Chem., 35, 1292 (1963).
115. Ф и л и п п о в M. П. и др., Зав. лаб., 30, 1444 (1964).
116. С a w s е Р. A., Analust, 92, 311 (1967).
117. Navone R., J. Amer. Water Assoc., 59, 1193 (1967).
118. Mertens J., Massart D. L., Bull. Soc. chim. Belg., 80, 151 (1971).
119 Valente Soares M. I., Santos Pereira P. G., Antunes Ad. M., Rev. port, quim., 13, 151 (1971).
120. Рожковская Л. H., Цеханская Ю. В., Tp. н.-и. и проектн. ин-та азотн. пром-сти и продуктов орг. синтеза, 1970, вып. 5, ч. II, См. с. 22.
121 Р о 1 у d о г о р о u 1 о s С. N., Valiatis S. D., Anal. Chim. acta, 40, 170 ’ (1968).
122. McDonald J. C., Haddad L., Environ. Sci. and Technol., 4, 676 (1971).
123. W e 11 e г s J. H„ U g 1 u m K. L. Anal. Chim., 42, 335 (1970).
124 Phillijs J. F., McNutt R * C., Croomes E. F., Analyst, 94, 1047 ' (1969).
125. Wright Ch. M., Orr A. A., Balling W. C., Anal. Chem., 40, 29 (1968).
126 Norris M. S„ Fleck S. A., Lichtenfels, Anal. Chem., 27, 1565 ' (1955).
127. Comer S. W., J e n s e n A. V., Anal. Chem., 36, 799 (1964).
128. Семячкова А. Ф., Корсакова M. P., Авт. св. СССР, кл. Д о/п 421, 13/03, № 158448, Заявл. 20.10.61, опубл. 19.10.63, РЖХим, 1965, 1Г100П.
129. Svach М., Zyka J., Z. anal. Chem., 148, 2 (1955).
130. Jirele V., Vodni hospod., № 4, 167 (1959).
131. Багдасаров К. H., Коваленко П. Н., Изв. вузов. Хим. и хим. технол., 7, 736 (1964).
132. Бабкин М. П„ Гидрохим. материалы, 47, 157 (1968).
133. Васильева Л. А., Тр. Казанского хим.-технол. ин-та, 8, 48 (1940).
134. Kadic К., Chem. prumysl, 13, 363 (1963).
135. Суранова 3. П., Тр. Одесского ун-та, 146. Сер. хим. н., № 5, 69 (1956).
136. Богданова Е. А., Почвоведение, 8, 98 (1961).
137. PFeilsticker К., Z. anal. Chemie, 89, 1 (1942).
138. A t k i n s W. R. G., J. Conscil perman internat. explorat. mer., 20, 153 (1954).
139. Исаева А. Б., Тр. ин-та океанол. АН СССР, 19, 304 (1956).
140. Скородумов Н. Н., Гигиена и санитария, № 9, 81 (1958).
141. S z е k е 1 у Е„ Taianta, 14, 941 (1967).
142. Swain J. S„ Chemistry and Industry, № 16, 479 (1957).
143. Szekely E, Taianta, 15, 795 (1968).
144. Lewis D. G„ Anal. Chem., 33, 1127 (1961).
145. M о n t g о m e г у H. A., Dy mock J. F., Analyst, 87, 374 (1962).
146. Hartley A. M„ Asa i R. J., Anal. Chem., 35, 1214 (1963).
147. Hartley A. M., A s a i R. J., Anal. Chem., 35, 1207 (1963).
148. A n d г e w s D. W. W„ Analyst, 89, 730 (1964).
149. Nagasawa M., Bull. Univ. Osaka Prefect, B-19, 12 (1967).
150. Шаферштейн И. Я. и др., Тр. Тадж. с.-х. ин-та, 1, 3 (1958).
151. В а г к L. S., Acta chim. Acad, scient. Hung., 27, 391 (1961).
152. Chow T. J., Johnstons M. S., Anal. Chim. acta, 27, 441 (1962).
153. Шаферштейн И. Я., Савва И. Е., Лапкинд И. М., Почвоведение, № 9, 96 (1962).
154. Моримото Масахиро, Хиракоба Акира, Исибаси Рюго, РЖХим, 1969, 4Г103.
155. Р г о с h a z к о v a L., Z.. anal, chem., 167, 254 (1959).
156. Sawicki С. R., Scaringelli F. P., Microchem. J., 16, 657 (1971).
157. Wolf B„ Anal. Edit., 19, 334 (1947).
158. H а г г i s о n G. A. F., Taianta, 9, 533 (1962).
159. Grocneveld E. R., den Boef G., Z. anal. Chem., 238, 19 (1968).
160. Alberton F„ Analyst, 72, 349 (1947).
161. Weiss K. G., Boltz D.‘, Anal. Chim. acta, 55, 77 (1971).
162. Лямин И. А., Полетаев А. И., Пушко А. Г., Авт. св. СССР, кл.
Д 01 21/20, № 298874, заявл. 24.01.70, опубл. 21.04.71. РЖХим, 1972, 1Г162П.
163. Дубинский Р. А., Филиппов В. А., Изв. АН СССР, сер. хим., № 12, , 2835 (1968).
164. L i п Е у i h, С h е n g К- L„ Mikrochim. acta, № 4, 652 (1970).
165. М а с с h i G. R., C e s с о n В. S., Anal. Chem., 42, 1809 (1970).
166. Gonzalez G.-G. A., Inform, quim. analit, 23, 101 (1969).
167. Kieruczenkowa A., Chem. analit. (Polska), 12, 1031 (1967).
168. M a c h о v a 11 о n a • D о к 1 a d a 1 о v a J., Chem. Techn., 19, 767 (1967).
169. S о c z e w i n s к i E., Urban T., Wo Is к i T., Chem. analit (Polska), 12, 1231 (1967).
170. S h e г b а к M. P., S m i t h Th. A., Analyst, 95, 964 (1970).
171. Гоголь H. А., Смирнова Г. Д., Изв. АН КазССР, № 1, 60 (1972).
172. Halstead С. J., Nation G. Н„ Turner L., Analyst, 97, 55 (1972).
173. Шафран И. Г., Кураева А. В., Авт. св. СССР, кл. 421, 3/08 (g01), № 230500, заявл. 6.04.67, опубл. 17.03.69.
174. Jamamoto Juroku, Uchikawa Sumi о, Akabori Кого, Bull. Chem. Soc. Japan, 37, 1718 (1964)'.
175. Ямамото Юрок у, Окамото Нобукэ, Tao Эйдзи, РЖХим, 1968, 24Г117.
176. Foros A., Swelt Th. R„ Anal. Chem., 37, 701 (1965).
177. Подберезовска я H. К. В сб.: «Исслед. и разработка фотометрических методов для определения микроколичеств элементов минеральн. сырья». Алма-Ата, 1967. См. с. 105.
178. Pokorny G., Likussar W., Anal. Chim. acta, 42, 253 (1968).
179. Burns D. Th., Fogg A. G., Willcox A., Mikrochim. acta, № 1, 205 (1971).
180. Bhatty M. K., Townshend A., Anal. Chim. acta, 56, 55 (1971).
181. Matsunaga K., Oyama T., Nishimura M., Anal. Chim. acta, 58,' 228 (1972).
182. Клочковский С. П., Чистота В. Д., Сб. научн. тр. Магнитогорского горно-металлург. ин-та, вып. 87, 70 (1971).
183. Подберезская Н. К., Шиленко Е. А., Сушкова В. А. В сб.: «Исслед. в обл. хим. и физ. методов анализа минеральн. сырья». Алма-Ата, 1971. См. с. 93
Глава II
БОР
II. 1. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ ОПРЕДЕЛЕНИЯ
Для анализа после соответствующей подготовки получают обычно раствор борной кислоты или бората натрия. В разбавленных водных растворах борная кислота находится главным образом в виде НВО2 (рЛ = 9,2); в более концентрированных растворах образуются различные полиионы, например диборат, тетраборат и др. Связь между бором и кислородом довольно подвижна, и кислород легко замещается, на различные лиганды с образованием соединений ВХ3, В(ОН)Х3 и ВХч (где X — электроотрицательный лиганд, Например фторид-ион или салицилат-ион). Известны и промежуточные соединения, содержащие катион ВО+ (борил); так, при взаимодействии борной кислоты с метафосфорной в среде безводной уксусной кислоты образуется метафосфат борила (ВО)3РО3. Вероятно, этот же катион образует ряд комплексных соединений с глицерином и др.
При подготовке к анализу, особенно при разложении кислотами, выпаривании и т. п., необходимо иметь в виду летучесть некоторых соединений бора. Литературные данные о летучести борной кислоты довольно противоречивы, но эти противоречия обусловлены главным образом различными условиями работы. При кипячении водных растворов или выпаривании нейтральных растворов, содержащих большие количества бора (10—100 мг), относительные потери незначительны. Однако они становятся весьма значительными при работе с микрограммовыми количествами. Особенно большие потери наблюдались при выпаривании солянокислых и даже нейтральных растворов досуха [1]. Для устранения потерь рекомендуют выпаривать растворы с суспензией гидроокиси кальция.
Летучесть соединений бора при выпаривании можно также устранить добавлением маннита, так как борная кислота образует с ним прочные комплексы. Однако для дальнейшего определения приходится сжигать образовавшееся органическое вещество, что усложняет определение.
При сплавлении в кислых средах наблюдаются потери борной кислоты, поэтому такое разложение материалов обычно рекомендуют вести при низких температурах. Если необходимо разлагать материал при высокой температуре, рекомендуют сплавлять или
спекать его с карбонатами или другими материалами основного характера.
Летучесть борной кислоты приводит иногда к обратным результатам. Доказано получение в ряде случаев завышенных в 2—4 раза результатов; причиной здесь является улетучивание борной кислоты из материала муфеля и поглощение паров борной кислоты щелочным плавнем. Чтобы устранить это явление, необходимо производить сплавление или спекание в высоких тиглях, тщательно закрывая их крышками.
Расхождения в параллельных опытах и завышенные результаты иногда обусловлены тем, что стекло, в том числе обычное кварцевое, содержит бор, который выщелачивается. По этой же причине бор находится в качестве примеси в реактивах, особенно в кислотах, а также в дистиллированной воде. Поэтому при определении малых количеств бора всегда необходимо ставить холостой опыт, а нередко и специально очищать реактивы, перегонять и хранить воду в сосудах из пластмасс. В ряде работ рекомендуют не пользоваться платиновой посудой, так как она тоже содержит микроколичества бора.
Из других летучих соединений бора большое значение имеет борнометиловый эфир. При анализе сложных материалов часто приходится отделять бор; наиболее важным и общим методом отделения является перегонка его в виде борнометилового эфира. Эфир образуется довольно легко в присутствии серной, фосфорной и других кислот. Если необходимо просто удалить бор, например для очистки реактивов или для получения очищенных от бора образцов, рекомендуют выпаривание с метиловым спиртом и хлористоводородной кислотой. Для очистки метилового спирта от примесей бора предлагают перегонку в присутствии чистого металлического кальция.
Наконец, в некоторых методах анализа используется летучесть фтористого бора. Бор можно количественно удалить из раствора выпариванием с фтористоводородной кислотой.
II. 2. НАИБОЛЕЕ ВАЖНЫЕ РЕАКЦИИ
И МЕТОДЫ ОПРЕДЕЛЕНИЯ БОРА
Химия окрашенных соединений бора и многих его комплексов изучена мало; нередко не известны даже точные уравнения реакций.
Наиболее важными являются следующие реакции и методы.
1. Взаимодействие с различными одноатомными спиртами и фенолами (ROH). В этих реакциях борная кислота выступает как гидрат окиси борила (ВО)ОН, образуя соединения типа эфиров:
ROH + (ВО)ОН —> RO(BO) + H2O (1)
3ROH + (ВО)ОН —> (RO)3B + 2Н2О (2)
Обе реакции, особенно вторая, идут в присутствии водоотнимающих средств обычно в среде концентрированной серной кис
лоты или в присутствии хлорида кальция и т. п. На этих реакциях, в частности, основано отделение бора перегонкой.
Реакции с многоатомными спиртами и фенолами идут, по-видимому, несколько иначе:
R—ОН
I + (ВО)ОН —>
R—ОН
'R-Ov
I /В0 _R-OZ
+ Н+ + Н2О
(3)
На реакциях этого типа основаны многие методы качественного и фотометрического определения бора, в частности метод с применением цветных pH-индикаторов, которые изменяют окраску в связи с изменением кислотности среды. Эти же реакции являются основой известных методов алкалиметрического титрования борной кислоты в присутствии глицерина, глюкозы, маннита или других комплексообразователей этого типа.
2. Указанные выше реакции, особенно реакция (3), являются основой многочисленных методов, в которых вместо фенола используется соответствующий органический краситель, например полиоксиантрахинон, кармин или другие [2]. Эти реакции ведут обычно в среде концентрированной серной кислоты. Значение кислотной среды очень велико. Прежде всего она играет роль водоотнимающего средства; в среде концентрированной серной кислоты увеличивается относительная концентрация иона борила. Наконец, в этой среде не образуются окрашенные соединения многих металлов с применяемыми красителями. Конечно, повышенная кислотность несколько препятствует сдвигу равновесия (3) вправо, однако концентрированная серная кислота мало диссоциирует.
При введении даже небольших количеств воды (10—20%) образовавшиеся окрашенные соединения бора разлагаются.
Известны некоторые методы с применением неводных растворителей или концентрированной хлористоводородной кислоты вместо концентрированной серной. Во всех случаях интенсивность окраски сильно ослабляется при увеличении содержания воды в растворе.
3. Ряд красителей, имеющих хелатообразующие ОН-группы в орто- или пери-положении, реагируют с борной кислотой не только в среде концентрированной серной кислоты, но и в водных растворах, когда при повышении pH увеличивается относительная концентрация аниона красителя. Применение каждого реактива требует соблюдения особых условий и химизм этих реакций еще крайне мало изучен.
Ализарин S при pH > 5 в водном растворе образует краснофиолетовый анион, однако в присутствии борной кислоты в тех же условиях образуется желтый борно-ализариновый комплекс.
Желтый краситель куркумин образует красное соединение сбором; это соединение обычно получают путем выпаривания растворов в присутствии щавелевой кислоты.
Таблица 2. Характеристика групп методов определения бора
Тип реакции и условия Преимущества Главные ограничения
Реакция с многоатомными спиртами при рН-5, наблюдение изменения окраски рН-индикатора или иодо-метрическое определение кислотности Простота выполнения, хорошая чувствительность (до 0,02 мкг в 1 мл) Мешают буферирующие вещества, например растворимые карбонаты, ацетаты и т. п. Аналогичную реакцию с многоатомными спиртами дают некоторые металлы. Методики мало разработаны
Образование окрашенных соединений с кармином, ализарином S и т. п. в среде концентрированной серной кислоты Высокая чувствительность. Простота выполнения Технические трудности работы с концентрированной серной кислотой; сильное влияние небольших колебаний содержания воды в H2SO4. Мешают сурьма (V), цирконий, барий, свинеи, органические соединения, темнеющие в среде концентрированной серной кислоты; большие количества щелочных и других металлов, а также нитраты
Образование комплексов с куркумином и т. п. Высокая чувствительность, устойчивость окраски;работа в водной среде. Рекомендуется только после отделения- бора, например, дистилляцией Сложность выполнения и влияние многих недостаточно изученных факторов, как температура, неводные растворители, pH, продолжительность реакции. Мешают железо, молибден, титан, ниобий, германий, тантал и др.
Образование тройных комплексов с красителями, экстракция Высокая чувствительность и избирательность. Не мешает присутствие фторидов Недостаточная изученность условий образования экстрагируемого комплекса. Мешают тантал, рений, серная кислота в больших концентрациях
Образование люминес-цирующего соединения с бензоилом Очень высокая чувствительность Недостаточная надежность. Действие кислорода воздуха и фоторазложение бензоина
4. Большое значение имеет группа методов, основанных на образовании тройных комплексов. При этом бор сначала переводят в комплекс заменой кислорода борной кислоты на другие лиганды, как фторид или салицилат. Образующиеся комплексные кислоты, например HBF4, дают с рядом основных красителей новые соединения (тройные комплексы), мало растворимые в воде, но растворимые в некоторых органических растворителях. Подбирая кислотность и подходящий органический растворитель, можно найти условия, при которых свободный краситель не экстрагируется; тогда окраска неводного слоя пропорциональна содержанию бора в пробе.
В табл. 2 в краткой форме дана характеристика отдельных групп методов фотометрического определения бора.
II. Э. РАЗЛОЖЕНИЕ БОРСОДЕРЖАЩИХ МАТЕРИАЛОВ
В наиболее простых случаях для разложения борсодержащих материалов достаточно обработать их водой или разбавленными кислотами. При необходимости длительного кипячения с кислотами применяют обратный холодильник, чтобы избежать потерь борной кислоты. Выбор кислоты для растворения зависит от намеченного метода отделения мешающих компонентов. Для отгонки бора в виде борнометилового эфира следует применять при разложении только серную или фосфорную кислоту. В' большинстве случаев недопустимо присутствие азотной кислоты, часто, мешает и хлористоводородная кислота; поэтому при растворении металлов для определения бора применяют обычно серную кислоту, а в качестве окислителя вводят перекись водорода или перманганат калия и т. п. Только в том случае, если в дальнейшем намечается определять бор с применением куркумина или других реагентов в нейтральном или слабощелочном водном растворе, рекомендуют использовать для разложения материала хлористоводородную кислоту.
Некоторые материалы можно перевести в растворимое состояние обработкой реактивами, содержащими фтористоводородную кислоту. По ряду данных, в присутствии избытка HF летучесть бора невелика. Вместе с тем присутствие HF недопустимо при определении бора с применением кармина или в других случаях, когда окрашенное соединение образуется в среде концентрированной серной кислоты. Присутствие фторидов недопустимо также при определении бора с применением куркумина. Поэтому, если материал удобно переводить в раствор с помощью HF, особое преимущество имеют методы, основанные на экстракции соединения HBF4 с основными красителями. В случае необходимости проведения определения бора другими методами в присутствии фторида последний можно связать ионами алюминия и отогнать бор в виде борнометилового эфира.
Неразложимые кислотами рудные и силикатные материалы сплавляют с карбонатом натрия обычными методами. При малом
количестве кремневой кислоты в анализируемом материале рекомендуют перед сплавлением добавлять кварц. В этих условиях бор лучше переходит в раствор при выщелачивании плава водой, так как окислы Металлов, которые могли бы образовать с В2О3 малорастворимые бораты, образуют еще менее растворимые силикаты. Для некоторых материалов показано, что бор практически полностью переходит в раствор при выщелачивании содового плава водой. При этом анализ сильно облегчается, так как нерастворимый остаток гидроокисей, силикатов и карбонатов легко отделяется. Однако при анализе не изученных в этом отношении материалов содовый плав обрабатывают хлористоводородной или серной кислотами.
Разложение кремния. При анализе полупроводникового кремния необходимо определение до 10-в% бора и менее. Применение щелочного разложения вызывает затруднения, так как щелочные материалы обычно содержат значительно больше бора, чем кремний высокой чистоты. Рекомендуют разлагать кремний водой в присутствии лишь небольших количеств щелочи; такое разложение ведут в автоклаве при 354 бар и 350 °C. Вероятно, более удобным методом является разложение бромом или раствором брома в метаноле. Одна из таких методик описана ниже.
Разложение материалов, содержащих фтор. Во многих случаях эти материалы можно разлагать щелочным плавлением или кислотным разложением, в последнем случае, разумеется, избегая нагревания. Для дальнейшего анализа обычно используют реакцию HBF4 с основными красителями. Однако в некоторых случаях, например в присутствии тантала или большого количества сульфатов, фосфатов и других, этот метод неудобен, и материал разлагают кислотами, добавляя значительное количество хлорида алюминия. Хлорид алюминия связывает фтор, и это облегчает переведение в раствор основного материала; возможные потери бора в виде BF3 устраняются. Однако присутствие фтора и больших количеств алюминия затрудняет применение обычных методов отделения и определения бора. Поэтому в рассматриваемом случае рекомендуют извлекать борную кислоту из сильнокислого раствора диэтиловым эфиром или кетонами. Извлечение неполное; необходимо введение поправочного коэффициента.
II. 4. ОТДЕЛЕНИЕ БОРА
Только в небольшом числе материалов можно непосредственно определять бор. Посторонние элементы наименее влияют при использовании тех методов, которые основаны на экстракции соединения HBF4 или других ацидокомплексов бора с основными красителями.
Наиболее простым методом отделения мешающих ионов металлов является осаждение их карбонатом кальция или бария. Однако этот метод не всегда применим. Наиболее общим и хорошо 60
изученным методом отделения бора является отгонка его в Виде борнометилового эфира.
Дистилляция борнометилового эфира. В присутствии водоотнимающих средств борная кислота образует с метиловым спиртом легколетучий эфир:
НВО2 + ЗСН3ОН —> В(СН3О)з + 2Н2О
Температура кипения борнометилового эфира (69°C) лишь.ненамного превышает температуру кипения метилового спирта (65 °C). Этиловый спирт мало подходит для этого, так как он кипит при 78,4 °C, тогда как борноэтиловый эфир — при 120 °C.
В качестве водоотнимающего средства обычно применяют концентрированную серную кислоту; описаны удовлетворительные результаты, полученные при отгонке в присутствии концентрированной фосфорной кислоты. Можно также отгонять борнометиловый эфир в слабокислой среде, применяя хлорид цинка в качестве водоотнимающего средства.
Во всех случаях дистиллят собирают в приемник, содержащий водный раствор гидроокиси натрия или взвесь гидроокиси кальция. При нагревании борнометиловый эфир гидролизуется, а метиловый спирт испаряется; в остатке определяют бор тем или другим методом.
При определении микрограммовых количеств бора в процессе выпаривания раствора со щелочью наблюдаются потери вследствие гидролитического разложения бората с образованием Н3ВО3.
По данным ряда авторов, потери уменьшаются, если вместо гидроокиси натрия брать гидроокись кальция. Другие исследователи считают это недостаточным и прибавляют кроме щелочи также глицерин или маннит, образующие комплексы с борной кислотой; введение глицерина или маннита вызывает, в свою очередь, необходимость прокаливания сухого остатка. Бор улетучивается, по-видимому, только с водяными парами в виде борной кислоты; прокаливание остатка в присутствии избытка щелочей, как и сплавление со щелочными плавнями, не вызывает потерь бора.
Мешающие вещества. В материале перед отгонкой борнометилового эфира не должно быть легколетучих кислот, например НС1. При перегонке эти кислоты также частично переходят в приемник и нейтрализуют щелочь, используемую для гидролиза эфира и для удержания борной кислоты, в результате чего могут быть потери бора. Этот недостаток можно устранить, прибавляя больше щелочи (по индикатору) перед выпариванием. Тем не менее необходимо иметь в виду, что присутствие значительных количеств Щелочных или щелочноземельных металлов мешает фотометрическому определению бора с применением кармина или других реагентов, реакции с которыми проводятся в среде концентрированной серной кислоты. В присутствии летучих кислот рекомендуют вести перегонку в слабокислой среде, вводя избыток хлорида Цинка [3].
Должны отсутствовать окислители, так как метиловый спирт может окисляться до альдегида и далее, что искажает результаты фотометрического определения.
Наблюдаются потери в тех случаях, если в колбе для пере-j гонки образуется много геля кремневой кислоты. >
Многие органические вещества разлагаются при действии кон-’ центрированной серной кислбты, образуя летучие окрашенные про-' дукты, что сильно затрудняет фотометрическое определение. Поэтому подобные вещества следует предварительно сжигать, смешав анализируемый материал с окисью кальция.
Иногда метиловый спирт содержит летучие окрашенные примеси.
Рис. 9. Приборы для отгонки борнометилового эфира:
/ — колба для метилового спирта; 2—дистилляционная Колба; 3 — холодильники; 4 — приемник; 5—баня; 6—воронка со спиртом.
Серная кислота, хранившаяся в стеклянной посуде, всегда содержит примесь бора. Стеклянная посуда, применяемая при перегонке, также часто отдает значительные количества бора. Имеются сведения, что даже многие сорта платины содержат бор [4]. Поэтому всегда необходимо проводить холостые опыты; иногда следует специально очищать реактивы.
Для отделения бора перегонкой в виде борнометилового эфира можно пользоваться самыми различными приборами (рис. 9), простыми (а) и сложными (б).
Другие методы отделения бора. При анализе простых силикатных материалов, содержащих 0,02—1 % бора, наиболее целесообразно применять широко опробованный метод отделения бора от многих элементов, в частности от алюминия и железа.' Силикат сплавляют с карбонатом натрия, выщелачивают водой и отфильтровывают нерастворимый остаток. Фильтрат подкисляют хлористоводородной кислотой, затем прибавляют карбонат кальция и кипятят с обратным холодильником. При этом осаждаются гидроокиси алюминия и других металлов, а также переходят в осадок фосфаты и фториды, если они присутствовали в анализируемом
материале. После фильтрования в растворе фотометрически определяют бор. Разумеется, при этом необходимо иметь в виду, что в растворе присутствуют хлориды щелочных металлов и немного хлорида кальция. Обусловленные этим ограничения указаны выше.
Вероятно, значительно более перспективны методы, основанные на применении ионитов. Некоторые из таких методов уже разработаны [2].
В определенных условиях можно поглощать посторонние ионы смесью катионита и анионита, причем через такую колонку проходят борная, угольная и кремневая кислоты. При анализе наблюдаются завышенные результаты для бора, что объясняется выщелачиванием бора из стекла [5].
Весьма перспективны, хотя еще мало изучены, методы отделения бора поглощением борной кислоты на анибните (с последующим вымыванием). Более изучены методы, основанные на поглощении мешающих металлов на катионитах. Критическая проверка ряда методов [3] дала удовлетворительные результаты при поглощении борной кислоты из щелочных растворов на анионите. Однако этот метод неудобен в присутствии большого количества других солей; кроме того, при малых количествах борной кислоты становятся заметными потери вследствие неполного вымывания ее из анионита. В ряде лабораторий получены хорошие результаты при отделении бора электродиализом с использованием анионитной мембраны. Этот способ был предложен для определения бора в кремнии. В определенных условиях через анионитную мембрану проходит только борная кислота [6, 7].
Несомненный интерес представляет экстракция для отделения бора от многих мешающих элементов. Описаны методы экстракции солей фтороборной кислоты изобутилкетоном [8] и раствором 2-этил-1,3-гександиола в метилизобутилкетоне [9].
Для. отделения многих элементов от основной массы анализируемого материала широко применяются различные методы получения аналитических концентратов. Однако для бора такие методы пока разработаны мало, кроме рассмотренных выше методов дистилляции бора в виде борнометилового эфира. Между тем вполне возможно применение методов, основанных на соосаждении и осаждении соединений основных красителей с HBF4 и другими ацидо-комплексами бора. Из описанных в литературе методов можно отметить получение аналитических концентратов путем осаждения бариевой соли комплексов бора с винной кислотой совместно с. тартратом бария [10].
II. 5. ОПРЕДЕЛЕНИЕ БОРА С ПРИМЕНЕНИЕМ КУРКУМИНА
Реакция бора с куркумином описана в 1903 г. [11]. Куркумин — природный желтый краситель, получаемый из корня куркумы. Большая часть препаратов куркумина содержит примеси, в результате чего калибровочные кривые и стандартные растворы различны
для разных препаратов. Получение чистого препарата — довольно трудная операция [12].
Состав куркумина (краситель группы розоцианинов) выражается формулой:
Н,СО\ О он ,осн,
но ЛЛ—сн=сн—с!—сн=с— сн=сн— ОН (1)
или, выделяя только отмеченную фигурной скобкой часть формулы:
О ОН
R—С—СН=С—R (2)
Соединение с бором (руброкуркумин)
/ R—С=О‘\
(3)
окрашено в красный цвет. Максимум светопоглощения наблюдается (рис. 11) при 533 нм [13], по другим данным [14—16],— при 555 нм; е = 1,8« 105.
Соединение с бором [3] образуется только в особых условиях
и при выпаривании водных растворов досуха в определенном режиме; соединение образуется также в не-
Рис. 10. Спектры поглощения соединения бора с куркумином в спирте(/) и куркумина (2).
которых неводных растворителях. Присутствие щавелевой кислоты ускоряет реакцию, однако в образующемся при этих условиях соединении отношение бор : курку-мин =1:1, чувствительность оказывается вдвое меньше, чем при работе по предыдущей методике [3]. Предполагают [17], что состав соединения со щавелевой кислотой выражается формулой:
R\
НС\
/С—О' R^
,о=с—о
в\ I • \)—С=О’’
/О—с ^о—с
Для этого соединения рассчитано значе-
• ние молярного коэффициента по отношению
к 1 моль бора, е= 1,3-105. Присутствие даже малых количеств хлорида натрия уменьшает чувствительность. Между тем, если бор отделяют дистилляцией в виде борнометилового эфира с по-
следующим гидролизом в присутствии гидроокиси натрия, то хлорид натрия всегда присутствует. По этому необходимо составлять калибровочный график в аналогичных условиях,
Процесс образования окрашенного соединения (3) или (4) борной кислоты с куркумином весьма сложен и недостаточно изучен. Из многочисленных методик видно, что это соединение образуется только при выпаривании смеси компонентов, причем условия выпаривания сильно влияют на точность анализа. Температура выпаривания должна быть не выше 70 °C; иногда [18] указывают более строгие условия: 55 ± 3 °C. Рекомендуют не перемешивать раствор во время выпаривания, защищать от яркого света, от пыли и выпаривать не на водяной бане, а на плитке, чтобы устранить неравномерность выпаривания, обусловленную попаданием паров воды.
Чтобы устранить влияние многих факторов, рекомендуют готовить стандартные растворы тем же методом и из того же материала, что и анализируемый. Для этого анализируемый материал предварительно выпаривают несколько раз с хлористоводородной кислотой и метанолом для удаления примесей борной кислоты; затем добавляют известные количества бора для составления калибровочного графика.
Если бор предварительно отделяют перегонкой в виде борнометилового эфира, то иногда в дистилляте находятся темноокра-шенные органические примеси или образуются при выпаривании дистиллята досуха. В этих случаях определение повторяют, прибавляя в приемник глицерин (помимо щелочи). Далее дистиллят выпаривают досуха и прокаливают в платиновой чашке до темнокрасного каления. Остаток растворяют и анализируют.
Окрашенное соединение (4) борной кислоты с куркумином, образовавшееся при выпаривании дистиллята досуха, растворяют далее в спирте (не ниже 95°) и фотометрируют. Присутствующий избыток щавелевой кислоты предохраняет от изменений окраски, связанных с pH-индикаторными свойствами реактива.
Несмотря на сложность, куркуминовый метод широко применяется, характеризуется высокой чувствительностью и удовлетворительной воспроизводимостью результатов [19, 20]. Тем не менее в ряде случаев он вытесняется методами, основанными на образовании соединений основных красителей с HBF4 и другими ацидо-комплексами. Химизм образования этих соединений более изучен, поэтому легче установить оптимальные условия и предвидеть влияние отдельных факторов.
11.5.1. Определение бора в отсутствие мешающих веществ
11.5.1.1. Определение с применением спиртового раствора куркумина
Реактивы
Раствор куркумина. Берут 700 мл 96%-ного спирта, 15 г щавелевой кислоты, 1 г чистого куркумина (т. пл. 182 °C) и разбавляют водой до 1 л.
Стандартный раствор бора. Растворяют 0,5716 г борной кислоты в воде и разбавляют до 1 л. Этот раствор содержит 0,1 мг бора в 1 мл. Для приготовления
рабочего раствора отбирают 1,0 мл указанного раствора и разбавляют до 100 мл. Разбавленный раствор содержит 1 мкг бора в 1 мл.
Ход анализа. К нейтральному испытуемому раствору, содержащему 1—25 мкг бора, прибавляют 1 мл 0,1 н. едкого натра и выпаривают в платиновой чашке досуха. К сухому остатку прибавляют 10 мл реактива и выпаривают досуха при 55 °C. Затем прибавляют ГО мл спирта, фильтруют окрашенный раствор и промывают спиртом. После разбавления спиртом до 25 мл измеряют оптическую плотность при 535—550 нм.
11.5.1.2. Определение с применением спиртового раствора специального препарата куркумина
Предложен метод приготовления раствора куркумина с применением в качестве растворителя метилоксалата или этилоксалата [21]. По данным автора, такие растворы устойчивы в течение 6 месяцев, обладают большей чувствительностью к бору и дают хорошую воспроизводимость и точность результатов (ошибка ±3% при определении 0,05—2,5 мкг В).
Раствор куркумина. Растворяют 0,4 г куркумина в 0,4 М растворе этилоксалата; из полученного раствора готовят 0,04 %-ный раствор куркумина в этаноле или 0,4 г куркумина растворяют при нагревании в 0,4 М растворе метилоксалата. При охлаждении краситель выпадает в осадок, из которого готовят 0,04%ный раствор в метаноле или этаноле.
Ход анализа. К 2—4 мл анализируемого раствора, содержащего 0,05—2,5 мкг бора, прибавляют 4 мл 0,04%-ного раствора реактива и выпаривают досуха. После 30—40 мин охлаждения остаток растворяют в 20 мл этанола и измеряют оптическую плотность при 550 нм в кювете с толщиной слоя 1 см.
Для определения очень малых количеств бора (0,01—0,2 мкг) пользуются ацетоновым раствором куркумина без добавления щавелевой кислоты. Для растворения окрашенного соединения в этом случае рекомендуется также ацетон или смесь хлороформа со спиртом.
II. 5.2. Определение до 10~7% бора в полупроводниковом кремнии
Трудности определения бора в кремнии связаны не только с проблемами фотометрии малых количеств бора, но и с разложением материала. В процессе измельчения кремния, а также разложения щелочью происходит загрязнение бором. Поэтому был изучен ряд методов, которые позволяют переводить в раствор кристаллы кремния массой 1—20 г без предварительного измельчения. Наиболее подходящим оказалось разложение бромом при 600— 750 °C. Затем остаток брома и большую часть бромида кремния отгоняют. Остаток подвергают гидролизу, отфильтровывают гель кремневой кислоты и в водной фазе определяют бор куркумино-вым методом [22].
Аппаратура и реактивы
Прибор для бромирования. Собирают прибор (рис. 11) из кварца высокой чистоты. К колбе 1 емкостью 200 мл с помощью шлифа (№ 29), смазанного концентрированной серной кислотой, присоединяют вертикальную трубку 2 с перфорированным вкладышем 7 для навески анализируемого кремния. Трубку помещают в вертикальную трубчатую печь (на рисунке не показана). Пары брома и продуктов бромирования из трубки 2 поступают в обратный (вертикальный) холодильник 3 и после конденсации стекают в колбу 1. Нижний отвод 4 холодильника опущен почти до дна колбы 1 и погружен в жидкий бром; такое устройство обеспечивает циркуляцию брома. Для выравнивания давления обратный холодильник соединен с воздухом. Очень важно, чтобы в прибор не попадала влага, поэтому соединение с воздухом производится через хлоркальциевую трубку 5 и хлоркальциевую колонку 6.
Аппарат тщательно высушивают перед работой.
Бром. Бром предварительно сушат над концентрированной серной кислотой.
Все летучие реактивы перегоняют в кварцевой аппаратуре и сохраняют в полиэтиленовой или кварцевой посуде. Кремний перед анализом промывают смесью фтористоводородной и азотной кислот, затем бидистиллятом, метанолом и высушивают при 105 °C. При взятии навески пользуются полиэтиленовым пинцетом.
Ход анализа. После подготовки аппаратуры вносят в колбу 1 прибора бром из расчета 4,25 мл на 1г кремния. Затем присоединяют трубку 2 с навеской кремния, помещенную на перфорированный вкладыш 6, надвигают трубчатую печь и на-нагревают ее до 600—750 °C. Нагревают колбу с бромом и ведут бромирование 60—90 мин. После
окончания бромирования прибор охлаждают, трижды споласкивают верхнюю часть прибора порциями по 10 мл четыреххлористым углеродом, после чего снимают верхнюю часть прибора со шлифа. Вставляют в тот же шлиф колбы / прямой холодильник и отгоняют четырехбромистый кремний пока в колбе не останется около 4 мл жидкости. К остатку прибавляют 40 мл СС14, охлаждают льдом, включают магнитную мешалку и прибавляют 20 мл бидистиллята. Перемешивают до выпадения геля кремневой кислоты и декантируют органический растворитель. Затем прибавляют еще бидистиллят, несколько капель гидрата гидразина для восстановления остатков брома и доводят pH аммиаком до 1,5. Оставляют на 1 ч для более полного гидролиза и коагуляции геля кремневой кислоты.
Рис. 11. Прибор для бромирования пробы кремния:
/ — дистилляционная колба; 2— вертикальная трубка с керамической пластинкой; 3 —обратный (вертикальный) холодильник; 4 — нижний отвод холодильника; 5 —хлоркальциевая трубка (CaClj помещается между слоями кварцевого волокна); 6—хлоркальциевая колонка.
Фильтруют, промывают и в водном растворе определяют бор куркумином, как указано в разделе II. 5.2.
11.6. ОПРЕДЕЛЕНИЕ БОРА С ПРИМЕНЕНИЕМ КАРМИНА В СРЕДЕ КОНЦЕНТРИРОВАННОЙ СЕРНОЙ КИСЛОТЫ
В среде концентрированной серной кислоты борная кислота образует окрашенные соединения со многими красителями. Наиболее характерны эти реакции для оксиантрахинонов, содержащих С = О и С—ОН группы в пери-положении [23, 24]. Таким образом, заметное изменение окраски наблюдается в этих условиях при наличии ОН-группы в 1, 4, 5 или 8 положении (/). При этом образуется шестичленное кольцо (//), характеризующееся большой
прочностью:
II
Для этого типа реакций предложено много различных красителей. Однако большинство предложенных ранее методов следует
считать устаревшими. Так, еще недавно одним из наиболее распространенных реактивов был хинализарин, который и сейчас рекомендуется для определения бора [25—27]. Однако спектр поглощения соединения хинализарина с бором (рис. 12) весьма близок к спектру поглощения самого реактива. Несколько лучшие результаты дает применение ацетилхинализарина [28, 29], так как спектр его в меньшей мере ле-
p..с. 12. Спектры поглощения в сернокислой среде:
1 — ацетилхинализарина; 2 — хин-
ализарина; 3 —комплексов бора с хинализарином и ацетилхипализари-ном.
рекрывается со спектром комплекса (рис. 12). Некоторые другие реагенты, возможно, представляют значительный интерес, в частности в связи с тем, что на их комплексообразование с бо
ром менее заметно влияет содержание воды в серной кислоте [30]. Однако эти вещества не выпускаются в достаточно чистом виде реактивной промышленностью и потому
мало доступны.
Одним из наиболее распространенных в настоящее время реагентов, пригодных для определения борной кислоты в среде концентрированной серной кислоты, является кармин, впервые пред
ложенный в 1936 г. для качественного обнаружения бора [31], а позже для фотометрического определения его [32, 33]. Кармин готовят из карминовой кислоты, которую получают из кошенили:
ноос о ОН
Кармин и карминовая кислота взаимодействуют с бором с образованием продукта реакции одного и того же цвета, но карминовая кислота растворима в воде, поэтому в анализе предпочтительно применять кислоту. Кислота в кислом растворе окрашена
Рис. 13. Спектры поглощения в сернокисл )й среде:
I—соединения карминовой кислоты с бором; 2— карминовой кислоты.
Рис. 14. Скорость взаимодействия кармина с бором при разных концентрациях серной кислоты:
/ — 97%; 2 — 95%; 3 — 92,5%; 4 — 90%; 5 — 87%.
в желтый цвет, а с повышением pH до 6,2 цвет изменяется на фиолетовый.
Спектры поглощения кармина и его комплекса с бором (рис. 13) значительно различаются. Различие достаточно велико для спектрофотометрического определения при 620 нм. По другим данным [5], удобнее фотометрировать по ослаблению интенсивности окраски при 610 нм. Визуально также хорошо заметно изменение окраски от красной к синей. Визуальная методика весьма важна, так как работать с растворами в концентрированной серной кислоте, пользуясь спектрофотометром, крайне неудобно.
Интенсивность окраски и скорость реакции зависят от содержания воды в серной кислоте (рис. 14). Хотя для кармина изменения не очень велики, тем не менее необходимо, чтобы испытуемые растворы обрабатывались так же, как и стандартные.
В фотометрируемый раствор целесообразно вводить некоторое количество воды. Это несколько уменьшает чувствительность, но
ускоряет реакцию. Так, при 1 мл воды на 50 мл серной кислоты интенсивность окраски продолжает увеличиваться даже через 3 ч; между тем при добавлении 4 мл воды на 50 мл серной кислоты интенсивность окраски несколько меньше, но постоянная окраска устанавливается уже через 45 мин.
Изменение температуры в пределах 25—35 °C практически не влияет. При определении 1—40 мкг бора среднее отклонение составляет ±0,3 мкг [5].
Многие образцы серной кислоты содержат примесь бора, поэтому необходимо пользоваться одним препаратом серной кислоты.
Присутствие небольшого количества хлоридов не мешает. По данным некоторых авторов [34], азотная кислота мешает, но’ ее влияние устраняется добавкой нескольких капель хлористоводородной кислоты. Однако по более поздним данным, азотная кислота заметно мешает определению.
Измерение оптической плотности. Измерение оптической плотности проводят при 585 нм. Необходимо следить, чтобы концентрированная серная кислота не попала на наружные стенки кюветы.
Для измерения интенсивности окраски можно применять метод стандартной серии, которая устойчива в течение одного и более дней.
11.6.1. Определение бора в отсутствие мешающих веществ
Реактивы
Карминовая кислота. Растворяют 0,25 г карминовой кислоты в 1 л 96%-ной серной кислоты.
Стандартный раствор бора, см. раздел II. 5.1.
Ход анализа. Берут 2 мл испытуемого раствора, содержащего 1 —10 мкг бора, прибавляют 2—3 капли концентрированной хлористоводородной кислоты и 10 мл 96%-ной серной кислоты. Затем прибавляют 10 мл реактива и оставляют на 45 мин, после чего измеряют светопоглощение раствора.
11.6.2. Определение бора в природных водах [35]
Пробу воды от 5 до 50 мл подщелачивают карбонатом натрия до pH = 8, выпаривают в кварцевой чашке или в чашке из без-борного стекла Досуха и прокаливают в муфеле при 450 °C в течение 1,5 ч. Чашке дают остыть, прибавляют 5 мл концентрированной серной кислоты и нагревают на песочной бане при 100 °C до полного растворения. Затем содержимое охлаждают, прибавляют по 0,05 г хлорида натрия и солянокислого гидразина (для восстановления Fe3+) и нагревают до полного прекращения выделения
газов. Далее к охлажденному раствору прибавляют 5 мл раствора кремневой кислоты, переносят в пробирку из винипласта емкостью 20 мл и центрифугируют в течение 15 мин со скоростью 5000 об/мин. Прозрачный раствор переносят в кварцевую пробирку с притертой пробкой и оставляют на 3—5 ч или .на ночь. После чего измеряют оптическую плотность раствора и по предварительно построенному калибровочному графику находят содержание бора.
11.6.3* Определение бора в биологических материалах [36]
Реактивы
Карбонат калия, 1,2%-ный раствор.
Гидроокись кальция, насыщенный при 35 °C раствор.
Другие реактивы — указаны в разделе II. 6.1.
Ход анализа. Смешивают 0,5 г высушенной при 100 °C пробы с 10 мл раствора гидроокиси кальция, смесь выпаривают досуха и прокаливают при 450—500 °C в течение 2 ч. Остаток растворяют в 1 мл 3,5 н. хлористоводородной кислоты, раствор переносят в центрифужную пробирку с помощью 1 мл воды, прибавляют 0,5 мл раствора К2СО3, осадок центрифугируют и промывают 2 мл воды. Цен-трифугат и промывные воды выпаривают при 50 °C, остаток растворяют в 0,9 мл воды и 1—2 каплях концентрированной хлористоводородной кислоты и поступают, как указано в разделе II. 6.1.
далее
квар-с хо-
11.6.4. Определение бора в стали [37]
Реактивы
Перекись водорода, 30%-ный раствор.
Перманганат калия, 0,1 н. раствор.
Сульфат железа(П), 0,1 н. раствор.
Ход анализа. Навеску стали 1 г помещают в цевую колбу 1 емкостью 150 мл, соединенную
лодильником 2 (рис. 15), и растворяют в 10 мл H2SO4, разбавленной (1:4), затем через воронку 3 вводят по каплям 4 мл Н2О2 и продолжают кипячение до просветления раствора и разрушения карбидов. Через ту же воронку прибавляют 2,5 мл концентрированной Н3РО4 и продолжают нагревание 20—30 мин разрушения избытка перекиси водорода.
Рис. 15. Прибор для растворения стали:
/ — колба для разложения;
2 — хоюдиль-ник; 3 —ворон ка; 4— стеклянная пробка.
ДЛЯ Не снимая холодильника, колбу охлаждают и промывают ник 5 мл холодной дистиллированной воды. Отсоединяют дильник и прибавляют по каплям раствор КМпО4 до лающей окраски перманганата, хорошо перемешивают и избыток перманганата восстанавливают несколькими каплями раствора
холодиль-холо-неисче-
FeSO4. Р.аствор переводят в мерную колбу емкостью 25 мл, доводят дистиллированной водой до метки, перемешивают и фильтруют через сухой бумажный фильтр. Первые порции отбрасывают, отбирают аликвотную часть и определяют бор, как указано в разделе II. 6.1.
11.6.5- Определение бора в горных породах и минералах [35, 38]
Смешивают 0,1 г породы или минерала, тонкоизмельченных и высушенных при 70—80 °C, с пятикратным количеством безводной соды. Смесь помещают в платиновый тигель и сплавляют в течение 2 мин. Горячий тигель несколько раз погружают в холодную воду до растрескивания плава. Прибавляют по 5 мл H2SO4 (пл. 1,83) и подогревают до растворения плава. В тигель добавляют по 0,05 г хлорида натрия и солянокислого гидразина; раствор перемешивают кварцевым пестиком до полного выделения газов. После охлаждения добавляют 5 мл раствора карминовой кислоты, перемешивают и переносят в пробирку из винипласта для центрифугирования. Затем определяют бор, как указано в разделе II. 6.1.
II. 7. ОПРЕДЕЛЕНИЕ БОРА В ВИДЕ КОМПЛЕКСА
ТЕТРАФТОРИДА БОРА С БРИЛЛИАНТОВЫМ ЗЕЛЕНЫМ
Бор переводят в комплекс BF4 действием фтористоводородной кислоты или фторида аммония в кислой среде. При действии на полученное соединение основным красителем получают окрашенное соединение типа R[BF4] (где R — катион органического красителя). Такие соединения хорошо экстрагируются инертными органическими растворителями, в то время как сам органический краситель не экстрагируется. Таким образом, метод является достаточно специфичным, хотя присутствие некоторых ионов мешает определению [39]. Эта реакция была предложена Н. С. Полуэктовым с сотрудниками [40] и применена для анализа сталей А. К. Бабко и П. В. Марченко [41].
Необходимо иметь в виду, что образование ионов тетрафторида бора происходит медленно и наиболее полно эти ионы образуются в кислой среде. В слабокислой среде происходит гидролиз:
BF7 + HOH +=> BF3OH“ + HF
Несмотря на схожесть свойств BFF и BF3OH~ последний не образует с основными красителями экстрагирующихся комплексов [40], что приводит к уменьшению оптической плотности экстрактов, 62
Рис. 16. Спектры поглощения соединений тетрафторборидного комплекса с бриллиантовым зеленым в бензоле (/), метиловым фиолетовым в бензоле (2), метиленовым голубым в дихлорэтане (3).
т. е. к заниженным результатам. Поэтому необходимо строго придерживаться условий проведения реакции как при построении калибровочных графиков, так и при проведении определения.
Экстракцию окрашенного соединения проводят главным образом бензолом или толуолом.
Мешающие вещества. Определению мешает тантал, который с фторидом образует HTaFe, а последний взаимодействует с красителями с образованием экстрагирующегося соединения. Мешающее влияние других металлов в сильной мере зависит от присутствия анионов. Так, в присутствии галогенидов и роданидов определению бора мешают галий, индий, таллий, золото и др. Из анионов определению бора мешают роданид, иодид, нитрат и некоторые другие, образующие с красителями соли, экстрагирующиеся неводными растворителями.
Измерение оптической плотности.
Для измерения оптической плотности применяют спектрофотометры и фотоколориметры. Интенсивность окраски можно измерять на колориметрах или с помощью стандартной шкалы. Устойчивость шкалы зависит от применяемого красителя.
Чувствительность реакций зависит от выбранного красителя.
В табл. 3 приведены молярные коэффициенты поглощения комплексов тетрафтороборида с некоторыми красителями, которые характеризуют чувствительность реакций [41].
Т аблица 3. Кажущиеся молярные коэффициенты поглощения некоторых тройных соединений бора
Краситель Растворитель Кажущийся молярный коэффициент поглощения
Бриллиантовый зеленый Бензол 20 000
Метиловый голубой Дихлорэтан 14 000
Виктория голубой К Бензол: ацетон (10:1) 11 000
Метиловый фиолетовый Бензол 2 600
Спектры поглощения некоторых соединений представлены на рис. 16.
11.7.1. Определение бора с применением бриллиантового зеленого в отсутствие мешающих веществ
Реактивы
Фторид аммония, 2 М раствор. Хранят в полиэтиленовой склянке.
Бриллиантовый зеленый (п,л'-бис-диэтиламинотрифенилкарбинол гидросульфат) :
Готовый О,5°1о-ный водный раствор.
Уротропин, 40%-ный водный раствор.
Бензол, очищенный.
Стандартный раствор бора, см. раздел II. 5.1.
. Калибровочный график. Для построения графика пользуются стандартным раствором, обработанным как анализируемый раствор. Калибровочные графики строят для проб с содержанием бора 0,1—0,5 мкг и 0,5—2 мкг.
Ход анализа. Сернокислый раствор, содержащий до 2 мкг бора в объеме до 5 мл, помещают в стакан из полиэтилена или платины, вводят 2 мл 2 М раствора фторида аммония и оставляют на 30 мин. Затем вводят 1—2 капли раствора бриллиантового зеленого и нейтрализуют кислоту раствором уротропина до перехода желтой окраски индикатора в голубовато-зеленую. Нейтрализованный раствор переносят в делительную воронку, добавляют 0,5— 1 мл (в зависимости от предполагаемого содержания бора) раствора бриллиантового зеленого, перемешивают и затем прибавляют 5 мл бензола. Содержимое воронки взбалтывают в течение 1—2 мин и после расслаивания отделяют водный слой, сливая вместе с ним 0,5—1 мл бензольного экстракта. Оставшийся бензольный слой переносят в сухую пробирку и дают постоять до исчезновения мути. Затем наливают в кювету с толщиной слоя 0,5 см и измеряют оптическую плотность при 610 нм. Параллельно ведут холостой опыт со всеми реактивами, но без бора, и полученный экстракт используют в качестве раствора сравнения. Содержание бора находят по калибровочному графику.
11.7.2. Определение бора в воде
Бор в воде определяют аналогично тому, как указано в разделе II. 7.1. Однако если вода содержит кальций и другие катионы, которые связываются фторидом, то необходимо отделить осадок фторидов, что лучше провести путем центрифугирования раствора в центрифужных пробирках из пластмассы.
11.7.3. Определение бора в стали [41]
Навеску стали 0,5 г помещают в кварцевую колбу с обратным водяным или воздушным холодильником и растворяют при слабом нагревании в 12 мл серной кислоты, разбавленной (1:4). После полного разложения навески и охлаждения жидкости добавляют 0,5—2 мл пергидроля '(в зависимости от содержания углерода) для разрушения карбидов. В том же приборе жидкость кипятят 30 мин. После охлаждения холодильник осторожно промывают 10—12 мл бидистиллята. Раствор переносят в мерную колбу емкостью 25 мл, доводят до метки водой и перемешивают. Отбирают 5 мл раствора и поступают, как указано в разделе II. 7.1.
11.7.4. Определение бора в почвах и горных породах
Навеску пробы 0,1 г помещают в полиэтиленовый стакан, прибавляют 2,5 мл Юн. H2SO4 и 0,5 мл 40%-ной фтористоводородной кислоты. Содержимое стакана перемешивают, стакан накрывают полиэтиленовой пластинкой и выдерживают при 20 °C в течение 2 ч. Затем приливают 2 мл воды и еще выдерживают в течение 15—20 мин. Отбирают аликвотную часть раствора и определяют бор, как указано в разделе II. 7.1.
II. 8. ОПРЕДЕЛЕНИЕ БОРА С ПРИМЕНЕНИЕМ салициловой кислоты и кристаллического фиолетового
Бор переводят в борсалицилатный комплекс, который взаимодействует с органическими красителями с образованием окрашенных соединений, которые экстрагируют органическими растворителями. Оптическая плотность экстрактов пропорциональна содержанию бора. Эта реакция впервые была предложена В. А. Назаренко и С. Я. Винковецкой [42], а затем применена для определения бора в природных материалах [43, 44].
Мешающие вещества. Определению мешают многие ионы, которые образуют соединения с салициловой кислотой и красителями. Поэтому рекомендуется предварительное отделение бора отгонкой в виде борнометилового эфира или лучше с помощью ионного обмена [45, 46]. Избыток салициловой кислоты, мешающий
з Зак. 761
65
определению, отгоняют при нагревании на водяной бане или связывают ионами железа (III).
Измерение оптической плотности. Измерение оптической плотности следует проводить при 585 нм. Спектр поглощения раствора тройного комплекса бора с салициловой кислотой и кристаллическим фиолетовым в бензоле представлен на рис. 17.
Е-Ю'1*
Рис. 17. Спектр поглощения тройного комплекса бора с салициловой кислотой и кристаллическим фиолетовым.
Чувствительность метода — 0,1 мкг бора в анализируемой пробе.
11.8.1. Определение бора в отсутствие мешающих веществ
Реактивы
Салициловая кислота, насыщенный раствор.
Кристаллический фиолетовый, 0,2%-ный водный раствор.
Бензол, очищенный.
Стандартный раствор бора, см. раздел II. 5.1.
Калибровочные графики. Строят два калибровочных графика для содержания бора в анализируемой пробе от 0,1 до 1,0 мкг и от 1,0 до 10,0 мкг бора. Параллельно ведут холостой опыт со всеми реактивами; полученный экстракт используют в качестве раствора сравнения.
Ход анализа. Водный раствор с pH = 6,0, содержащий до 10,0 мкг бора, помещают в кварцевую чашку, прибавляют 2,0 мл салициловой кислоты, выпаривают на водяной бане досуха и сухой остаток выдерживают на водяной бане еще около 20 мин. К охлажденному остатку прибавляют 2,0 мл раствора кристаллического фиолетового и 10 мл 0,01 н. раствора соляной кислоты. Раствор переносят в делительную воронку, прибавляют 4 мл бензола и встряхивают. Водную фазу отделяют, а неводную переносят в сухую пробирку, оставляют до просветления и измеряют оптическую плотность. Содержание бора находят по значению оптической плотности, пользуясь предварительно построенным калибровочным графиком.
11.8.2. Определение бора в водах [43]
Реактивы
Катионит КУ-2. Колонку диаметром 1,5—1,8 см заполняют 10 г катионита в Н-форме.
Железоаммонийные квасцы, 0,5%-ный раствор.
Другие реактивы — указаны в разделе II. 8.1.
Ход анализа. Пробу воды от 1,0 до 10,0 мл нейтрализуют кислотой или щелочью до pH = 6, затем пропускают через колонку, наполненную катионитом КУ-2 в Н-форме, и промывают бидистиллятом. Общий объем жидкости не должен превышать 100 мл.
Аликвотную часть полученного раствора нейтрализуют щелочью до pH = 5—6 и ведут определение, как указано в разделе II. 8.1, с той разницей, что перед экстракцией вводят 0,5 мл раствора железоаммонийных квасцов с pH = 2.
11.8.3. Определение бора в горных породах, почвах, углях и растениях [43]
Навеску угля или растения смешивают с окисью кальция и озоляют при 500 °C. Затем золу (или навеску горной породы, почвы) сплавляют в платиновом тигле с пятикратным количеством \а2СОз. Плав охлаждают, выщелачивают водой и водную вытяжку переносят в мерную колбу емкостью 100 мл. Аликвотную часть раствора от 1 до 10 мл нейтрализуют соляной кислотой до pH = 6 и пропускают через ионообменную колонку. Далее ведут определение, как указано в разделе II. 8.2.
11.8.4. Определение бора в солях щелочных металлов [46]
Бор переводят в борноманнитовую кислоту и поглощают на анионите АВ-17 в С1- или СН3СОО-форме. Затем его элюируют хлористоводородной кислотой, выпаривают, прибавляют гидроокись кальция и прокаливанием разрушают маннит. Определяют бор в виде тройного комплекса бора с салициловой кислотой и кристаллическим фиолетовым.
Реактивы
Маннит, сухой и 0,5 М раствор.
Анионит АВ-17, колонку из кварца диаметром 1,5—1,8 см заполняют 8—10 г анионита в С1- или СН3СОО-форме.
Хлористоводородная кислота или уксусная кислота, 2 н. раствор.
Гидроокись кальция, насыщенный раствор.
Ход анализа. Пропускают 200 мл нейтрального раствора (приблизительно 1%-ный по анализируемому веществу и 0,5 М по манниту) через ионообменную колонку со скоростью 2 мл/мин. Затем колонку промывают 75 мл раствора маннита и элюируют бор 50 мл хлористоводородной или соответственно уксусной кислоты и 30 мл воды. Элюат собирают в платиновую чашку, где находится 0,5 мл раствора манндта, и упаривают досуха. Сухой остаток смачивают 0,2 мл раствора гидроокиси кальция, вновь упаривают досуха и прокаливают при 700—800 °C для разрушения органических веществ. Остаток увлажняют водой и ведут определение, как указано в разделе II. 8.1.
II. 9. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ БОРА
Красители антипиринового ряда рекомендованы для определения бора в сталях [47]; применяются также экстракционные варианты этих методов [48]. Бор переводят в борофторидный комплекс, который в дальнейшем взаимодействует с антипириновыми красителями с образованием экстрагирующегося бензолом интенсивно окрашенного тройного комплекса.
Рекомендовано определение бора в виде тройных (смешанных) комплексов борофторида с метиленовым голубым [49], который применяется для определения бора в сталях [50, 51], горных породах и почвах [52, 53], в металлическом цирконии и циркалое [54], кремнии [55], никелевых сплавах ЖС6К, ЖС6У, ВЖЛ [56], жаропрочных сплавах [57] и в окиси урана [58], с нильским синим А в водах [59, 60], с красно-фиолетовым [61, 62], кристаллическим фиолетовым [63], N-метилтионином (Азур С) в стеклах [64] и чистых соединениях урана [65], с ферроином [66].
Для определения очень малых количеств бора образование тройных комплексов имеет большое значение особенно при люминесцентном определении бора [67]. Так, в виде тройного комплекса бора с салициловой кислотой и родаминами люминесцентным методом можно определить 0,001 мкг бора [68], а боросалицилат с кристаллическим фиолетовым применен для фотометрического определения бора в хлоридах щелочных металлов [69].
Куркуминовый метод применен для определения бора в медных сплавах и никеле [70], хлорсиланах высокой чистоты [71], в алюминиевых сплавах [72], в присутствии металлов группы железа [73], в едком натре [74] и в водных растворах [75].
1,1-Диантримид в среде концентрированной серной кислоты образует окрашенное соединение с бором в соотношении 1 : 1 [76, 77]; этот реагент рекомендован для определения бора в алюминии [78], в сплавах на никелевой основе [79] и в речной воде [80].
Изучены производные антрахинона в качестве реагентов на борную кислоту [81]. Рекомендовано применение п-сульфофенил-азохромотроповой кислоты для определения бора в биологических материалах [82], азо- и аминопроизводных Аш- и К-кислот [83], Аш-резорцина [84] и других реагентов.
Разработано определение бора методом дифференциальной спектрофотометрии с применением диаминохризазина [85] и метод микроопределения бора в борорганических соединениях с применением азометина [86].
Для определения бора применены диаминоантрафурин [87], 1,1-бис(6-хлорантрахинолил)-амин [88], хромотроп 2В [89] и магнезон I [90].
Карминовый метод применен для определения малых количеств бора в водах, породах и минералах [91] и воднорастворимого бора в удобрениях [92].
Проведено сравнительное изучение методов определения бора [93] и сравнительная оценка чувствительности некоторых фотомет-68
Таблица 4. Сравнительная характеристика некоторых фотометрических методов определения бора [93]
Реактив Окраска реактива Окраска соединения Определение количества, % Средняя ошибка определения, % Мешающие ноны и элементы
Куркумин Желтая, Л*макс == 435 нм Красная, ^•макс == 535 нм 10“3—ю~в 10-50 К+, Na+, Mg2+, Са2+, Fe3+ и др. С1~, NO;, SO;j~, РО^, С2О?-, F’
Хинализарин (1,2,5,8-Тетра-оксиантрахннон) Красно-фиолетовая, А-макс == 515 НМ Синяя. А,макс == = 610—615 нм ю-4—ю-1 10—20 (в зависимости от содержания) Окислители, F~, TiIV и ZrIV при содержании более 0,5%
Карминовая кислота Красная Синяя, ^макс 610 нм 5 • 10“3—2,5 • 10~2 2—20 F”, NO’, С2О2-, Fe(CN)3“ WVI, Vv, TiIV, ZrIV
1,Г-Днантримид Зеленовато-желтая Синяя, А>макс == 620 нм 10~3—1 До 50 Cu2+, Cr3+, Co2+, Ni2+, GeIV, Vv, Вг~, Г, F", po|-
Хромотроп 2В Зеленая Синяя, А» макс = 620 630 нм 10“3—ю-1 До 20 Fe3+, Al3+, Cu2+, Co2+, GeIV, NO~ PO|~, F“, C2OJ-, MnO^, Vv и др.
Метиленовый голубой — Синяя, А-макс = 660 нм Ю-5—Ю“2 20 CIO;, NCS~, СГ, MnOp no;
рических методов определения бора [94]. Результаты сравнительного изучения некоторых фотометрических методов приведены в табл. 4.
Сравнительное изучение методов определения бора в среде концентрированной кислоты с ализарином, ализарином S хинализарином, 1,1'-диантримидом и карминовой кислотой показало, что лучшими реагентами являются 1,1-диантримид и карминовая кислота [95].
Люминесцентные методы менее разработаны, хотя в ряде случаев получены результаты, характеризующие высокую чувстви-дельность этих методов. Так, реакция с ализарином S может быть использована как люминесцентная, причем чувствительность повышается на несколько порядков.
Для флуориметрического определения бора применяются многие реагенты, в том числе окси- и аминоантрахиноны [96], бензоин [97, 98], фенилфлуорон [99, 100], морин [101], ацетилсалициловая кислота [102] и другие, описанные в специальных монографиях [103, 104].
Для аналитических целей предложены и некоторые другие комплексы бора, например борновольфрамовая кислота; однако эти соединения мало изучены; не ясны перспективы их применения.
ЛИТЕРАТУРА
1. F е 1 d m а n С., Anal. Chem., 33, 1916 (1961).
2. Немодрук А. А., Каралова 3. К. Аналитическая химия бора. М., «Наука», 1964. См. с. 55.
3. Schulek Е., Szakocs О. М., Z. anal. Chem., 151, 1 (1956).
4. W о 1 s z о n I., H a у e s J., Anal. Chem., 29, 829 (1957).
5. CollicoatD., Wolsszon, Anal. Chem., 31, 1435 (1959).
6. Morrison G., Rupp R„ Anal. Chem., 29, 892 (1957).
7. Newton A., Bonders J„ Tyrrell A., Analyst, 85, 870 (1960).
8. Maeck W„ Anal. Chem., 33, 1775 (1961).
9. Hofer A., Bros ch e E„ Heid in ger R., Z. anal. Chem., 253, 117 (1971).
10. В о v a 1 i n i E., P i a z z i M., Ann. Chimica, 48, 305 (1958).
11. C a s s a 1 C., G e r m a n H., Chem. News, 87, 27 (1903).
12. Г о л ь т м а н А. А., Укр. хим. ж., 24, 244 (1958).
13. В a ker A. S., J. Agric. and Food Chem., 12, 367 (1964).
14. Th i e r i g D., Umland F., Z. anal. Chem., 211, 161 (1965).
15. C r a w 1 e у R. H. A., Analyst, 89, 749 (1964).
16. G r i n s t e a d R. R., S n i d e r S., Analyst, 92, 532 (1967).
17. Speicer G., Strickland I., Anal. Chim. acta, 18, 231 (1958).
18. S i 1 v e r m a n L., T r e g о K., Anal. Chem., 25, 1264 (1953).
19. A n d r e w T. R., N i c h о 1 s P. N. R.f Analyst, 91, 664 (1966).
20. U m 1 a n d F„ Thierig D., Miiller G., Z. anal. Chem., 215, 401 (1966).
21. Миямото Масатоси. Яп. пат., кл. 113 А 22, № 9074, заявл. 21.02.63, опубл. 13.05.66, РЖХим, 1968, 7Г66П.
22. Pohl F. A., Kokes К., Bonsais W., Z. anal. Chem., 174, 6 (1960).
23. Ф а й г л ь Ф. Капельный анализ. М., ОНТИ, 1937. См. с. 398.
24. К о р е н м а н И. М„ ЖАХ, 2, 325 (1947).
25. W о j t о w i с z М., Kubica М., Chem. anal. (Polska), 13, 65 (1968),
26. Ушакова В. Ф., Гидрохим. материалы, 54, НО (1970).
27. G u p t a H. K. L., Boltz D. F., Mikrochim. acta, № 4, 577 (1971).
28. Буданова Л. M., Гуревич A. H., Зав. лаб., 32, 1208 (1966).
29. Д и в а к А. В., Зав. лаб., 35, 1443 (1969).
30. Шемякин Ф. М„ Барская С. И., Зав. лаб., 17, 515 (1951).
31. Зорин Ф. П., ЖПХ, 9, 1505 (1936).
32. Е v a n s С., Hargue М., J. Assoc. Offic. Agr. Chemistry, 30, 308 (1947).
33. К аза p ин о ва Ox н и н а В. А., Зав. лаб., 14, 263 (1948).
34. Н a t с h е г J., W i 1 с о х L„ Anal. Chem., 22, 567 (1950).
35. Ма л юг а Д. П„ Зав. лаб., 35, 279 (1969).
36. Dean-Duelbenzu М., Fontan-Candela J. L., An. Real acad. tarmac., 32, 301 (1966).
37. Марты нченко И. У., Бондаренко А. М., ЖАХ, 12, 495 (1957).
38. Fleet М. Е„ Anal. Chem., 39, 253 (1967).
39. Бабко А. К., Пилипенко А Т. Фотометрический анализ. М., «Химия». 1968. См. с. 353.
40. Полуэктов Н. С., Кононенко Л. И., Лауэр Р. С., ЖАХ, 13, 396 (1958).
41. Б а б к о А. К., М а р ч е н к о П. В., Зав. лаб., 26, 1202 (1960).
42. Назаренко В. А., Винковецька С. Я., Доп. АН УРСР, № 2, 196 (1960).
43. В а с и л е в с к а я А. Е., Ленская Л. К-, ЖАХ, 20, 747 (1965).
44. Василевская А. Е., Науч. тр. Всес. ин-та минер, ресурсов, № 5, 22 (1971).
45. Методы химического анализа минерального сырья, вып. 5, Госгеолтехиз-дат, М., 1959.
46. В и н к о в е ц к а я С. Я., Назаренко В. А., Зав. лаб., 32, 1202 (1966).
47. Б усе в А. И., Козина Г. В., Яковлев П. Я. Сб. тр. Центр, н.-и. ин-та черной металлургии, вып. 37, 57 (1964).
48. Бусев А. И., Яковлев П. Я., Козина Г. В., ЖАХ, 22, 1227 (1967).
49. Ицуми Сатори, Исодзаки А к и н о р и, РЖХим, 1967, 21Г170.
50. М г о z i n s k i J., Chem. anal. (Polska), 12, 93 (1967).
51. В 1 a z e j а к - D i t ge s D., Z. anal. Chem., 247, 20 (1969).
52. Stanton R. E., McDonald M. J„ Analyst, 91, 775 (1966).
53. Weir C. C„ J. Sci Food and Agr., 21, 545 (1970).
54. СудоЭмико, ИкидаСатико, РЖХим, 1969, 7Г144.
55. So Jung Yon, Jang Yong H wa n, Song Chae Ok, Пунсок хвахах, 9, 104 (1971); РЖХим, 1972, 9Г131.
56. Клитина В. И., Бакурова В. Ф., Технол. легких сплавов. Науч.-техн. бюл. ВИЛСа, № 2, 105 (1971).
57. Клитина В. И. В сб.:~ «Методы хим. анализа». М., 1969. См. с. 23.
58. ОнисиХироси, НагаиХитоси, РЖХим, 1969, 15Г135.
59. Gagliardi Е., Waif Е., Mikrochim. acta, № 1, 140 (1968).
60. G a g 1 i а г d i E., Hollinger W., Mikrochim. acta, № 1, 136 (1972).
61. Корен ман И. M., Сидоренко Л. В., Тр. по хим. и хим. технол. Горький, вып. 1(12), 105 (1965).
62. К о р е н м а н И. М., Сидоренко Л. В., Тр. по хим. и хим. технол., Горький, вып. 1, 173 (1970).
63. Д е й к о в а 3. Е., Докл. Моск. с.-х. акад. им. К- А. Тимирязева, вып. 103,
465 (1965).
64. G о у d i s h В. L., Microchem. J., 15, 572 (1970).
65. Federgriin L., A b г a о A., Publ. IEA, № 165, 1 (1968).
66. Archer Vernon S., Doolittle F. G., Young La Verne M., Ta-lanta, 15, 864 (1968).
67. Б а б к о А. К., Ч а л а я 3. И., Укр. хим. ж., 30, 268 (1964).
68. Бабко А. К., Василевская А. Е., Укр. хим. ж., 33, 314 (1967).
69. И в а н о в а В. С., П р е с е р Р. Ф. В сб.: «Методы анализа минерального сырья». Апатиты, 1971. См. с. 45.
70. PakalnsP., Analyst, 94, ИЗО (1969).
71. Marczenko Z., Mojski M., Kasiura К.» Chem. anal. (Polska), 14, 1331 (1969).
72. U m 1 a n d F, J a n s s e n A, Anal. Chem., 249, 186 (1970).
73. Pakalns P„ Met. and Metal Foem., 39, 308 (1972).
74. Ciecierska-Tworek Z. 1. ol, Chem. anal. (Polska), 13, 773 (1968).
75. Griffith G. R, Laird R. M, Anal. Chim. acta, 39, 302 (1972).
76. L a n g m у h r F. J., Arriesen R. T., Anal. Chim. acta, 29, 419 (1963).
77. G u p t a H. K. L., В о 11 z D. F, Anal. Lett, 4, 161 (1971).
78. Kerin D, Mikrochim. acta, № 5, 670 (1964).
79. В u г к e К. E, A 1 b г о g h t Ch„ Taianta, 13, 49 (1966).
80. L e v i n s о n A. A, Water Res, 5, 41 (1971).
81. Корепман И. M, Курина Н. В, Тр. по хим. и хим. технол, Горький, вып. 3, 573 (1958).
82. Truhaut R, Boudene С, Nguyen Р. L, Bull. soc. chim. France, № 8, 2551 (1966).
83. C a p e 11 e R, Anal. chim. acta, 24, 555 (1961).
84. Горбенко Ф. П, Надежина А. А, Целинский Ю. К. В сб.: «Методы анализа хим. реактивов и препаратов», вып. 15, 143 (1968).
85. Е b е г 1 е A. R, L е г n е г М. W, Anal. Chem, 37, 1568 (1965).
86. Шанина Т. М, Гельман Н. Э, Михайловская В. С, ЖАХ, 22, 782 (1967).
87. Wiinsch G, Z. anal. Chem, 258, 30 (1972).
88. Grob R. L. 1. ol. Anal. Chim. acta, 39, 115 (1967).
89 Андреева 3. Ф, Лейков a 3. E. Докл. Моск. с.-х. акад. им. К. А. Тимирязева, вып. 149, 293 (1969).
90. Н и к и т и н а Е. Н. В сб.: «Хим. свойства и методы анализа тугопл. соединений». Киев, «Наукова думка», 1969. См. с. 117.
91. М а л ю г а Д. П, Зав. лаб, 35, 279 (1969).
92. Peterson Н. Р, Zo гот ski D. W, Anal. Chem, 44, 1291 (1972).
93. П и л и п е н к о А. Т, Пинаева С. Г. В сб.: «Высокотемпературные неорганические соединения». Киев, «Наукова думка», 1965. См. с. 379.
94. Д е й к о в а 3. Е, Докл. Моск. с.-х. акад. им. К. А. Тимирязева, вып. 103,
459 (1965).
95. Ка siu га К., Chem. anal. (PRL), 16, 219 (1971).
96. Константинова-Шлезингер М. А. Люминесцентный анализ, М, Гос. изд-во физ.-мат. лит-ры, 1961. См. с. 170.
97. Щербаков Д. П, Кор ж ев а Р. Н, Пономаренко А. И. Методы люминесцентного анализа. Минск, изд-во АН БССР, 1960. См. с. 37.
98. Лельчук Ю. Л, Ивашина В. А, Изв. Томск, политех, ин-та, 148, 152 (1967).
99. Подчайнова В. Н, Скорнякова Л. В, Двиняников Б. Л, Изв. вузов. Хим. и хим. технол, 11, 241 (1968).
100. Назаренко В. А, Винковецкая С. Я, ЖАХ, 26, 802 (1971).
101. Dabrowski J, Pszonicki L, Chem. anal. (PRL), 16, 51 (1971).
102. Щербов Д. П. Флуориметрия в химическом анализе минерального сырья. М, «Недра», 1965. См. с. 215.
103. Божевольнов Е. А. Люминесцентный анализ неорганических веществ. М, «Химия», 1966. См. с. 273.
104. Столяров К. П., Григорьев Н. Н. Введение в люминесцентный анализ неорганических веществ. Л, «Химия», 1967. См. с. 245.
Глава III
ФОСФОР И КРЕМНИЙ
III. 1. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ ОПРЕДЕЛЕНИЯ ФОСФОРА И КРЕМНИЯ
Определение малых количеств фосфора и кремния имеет очень большое значение для многих производств, а также для различных геохимических, биохимических, агрохимических и других исследований. В связи с этим описано огромное количество методов определения, эти методы основаны главным образом на образовании гетерополикомплексов (ГПК), где центральным атомом является кремний или фосфор, а координированными группами — полианионы молибдата. Общая характеристика химических свойств и спектров поглощения ГПК рассмотрена в первом томе [1] этой монографии. Здесь приводятся лишь данные, имеющие непосредственное отношение к методикам анализа.
Гетерополикомплексы образуют не только фосфор и кремний, но также мышьяк, германий и многие другие элементы, однако для них имеется немало других методов определения. Кроме того, мышьяк и германий легко удаляются, например, в виде летучих хлоридов AsCh и GeCL или экстракцией их хлоридов. Поэтому методы, основанные на образовании ГПК, описываются для фосфора и кремния; для определения микроколичеств фосфора и кремния эти методы являются практически почти единственными.
Среди очень большого числа методов можно выделить две группы: а) методы, основанные на образовании желтых ГПК при взаимодействии фосфата или силиката с молибдатом в кислой среде; б) методы, основанные на образовании'синих соединений (церулеокомплексов) при взаимодействии желтых ГПК фосфата или силиката с восстановителями.
В общем, при выполнении анализа в наиболее простых условиях первая группа методов менее чувствительна, хотя результаты более надежны. По сравнению с чувствительностью второй группы меньшая чувствительность первой группы методов связана с тем, что желтая окраска обусловлена лишь частью полосы поглощения ГПК- Максимум этой полосы находится при 310—350 нм (по данным различных авторов, изучавших комплексы в разных условиях). Однако измерять поглощение в ультрафиолетовом спектре в условиях анализа неудобно, так как здесь сильно поглощает избыток молибдата. Вследствие симбатного хода кривых абсолютная разность оптических плотностей (£>Гпк— D^o) почти не изменяется в области коротких волн. Более того, заметное поглощение молибдена в УФ приводит к увеличению значения фона по отношению к сигналу, так как отношение Dmo : £>гпк растет.
Не связанный в комплекс молибдат практически не поглощает в видимой части спектра, а полоса поглощения ГПК, как уже отмечалось, немного сдвинута в видимую часть спектра, поэтому в видимой части спектра определение более надежно. Большая надежность определения в виде желтых ГПК по сравнению с определением в виде синих обусловлена тем, что желтые ГПК являются соединениями определенного состава, хорошо растворимыми в воде.
Вторая группа методов в общем более чувствительна, особенно при измерении светопоглощения в инфракрасной области спектра. Определение в виде синих ГПК часто более удобно в присутствии больших количеств железа или окрашенных в желтый цвет органических соединений, которые иногда находятся в природной или технической воде, в почвенных вытяжках и т. д. При введении восстановителя, необходимого для образования синих ГПК, же-лезо(Ш), имеющее желтую окраску и поглощающее в ближнем ультрафиолетовом спектре, восстанавливается. Далее, присутствие желтых органических соединений в некоторых материалах не мешает, так как эти соединения поглощают в коротковолновой части спектра, а концентрация синего ГПК измеряется в красной или инфракрасной области спектра.
Однако методы этой группы часто дают менее точные результаты, так как образующиеся синие ГПК имеют непостоянный состав. В спектрах поглощения синих ГПК обычно наблюдается не менее двух максимумов, что указывает на наличие по крайней мере двух хромофорных центров. Интенсивная синяя окраска обусловлена присутствием в этих соединениях двух валентных форм молибдена Mov и MoVI. Соотношение форм зависит от выбора восстановителя, от кислотности раствора и многих других условий. Кроме того, синие ГПК, по-видимому, малорастворимы в воде и находятся в коллоидном состоянии. Это усиливает влияние электролитов и температуры на окраску раствора и требует очень точного соблюдения всех условий определения. Несмотря на ряд недостатков, метод довольно широко применяется, особенно для определения фосфора в стали.
Для каждой из названных выше двух групп методов известны экстракционные варианты. Как обычно, экстракция позволяет увеличить чувствительность и устранить влияние некоторых окрашенных компонентов. Кроме того, экстракция создает ряд преимуществ, особенно при использовании первой группы методов. При надлежащем подборе органического растворителя можно экстрагировать ГПК так, чтобы свободный молибдат (избыток реагента) остался в водной фазе. Это позволяет измерять светопоглощение при 310 нм, где находится максимум полосы поглощения ГПК. Молярный коэффициент поглощения (по отношению к фосфору) в этих условиях равен 2- 104, т. е. почти не отличается от коэффициента для синего ГПК в красном и инфракрасном участке спектра.
Далее, применение экстракционных методов дает возможность увеличить чувствительность определения желтых ГПК и без измерений в УФ. В определенных условиях можно получить и экстрагировать соединение аниона ГПК с катионом основного красителя. В соответствии с основностью ГПК на один атом фосфора в таком соединении приходится 3 молекулы красителя. Кроме того, органические красители сильнее поглощают свет, чем молибдат. Предложены и другие варианты подобных сочетаний ГПК с органическими компонентами и реагентами.
Некоторое повышение чувствительности определения фосфора (а также мышьяка) без экстракции достигается введением в реактив ванадия, причем образуется желтый фосфорнованадиевомо-либденовый комплекс, который поглощает при 420 нм несколько сильнее, чем обычный фосфорномолибденовый комплекс. Кромё того, фосфорнованадиевомолибденовый комплекс, по ряду литературных данных, более устойчив к небольшим колебаниям кислотности и других условий. Кремний, в отличие от фосфора и мышьяка, по-видимому, не образует подобного комплекса с участием ванадия. В литературе иногда встречаются указания, что определению фосфора в виде фосфорнованадиевомолибденового комплекса не мешает мышьяк. Однако это относится лишь к мышьяку(III), который не образует ГПК. Мышьяк(У) участвует в образовании ГПК; ниже описана методика его определения в виде мышьяковованадиевомолибденового комплекса.
Относительная прочность ГПК и общие принципы определения фосфора, кремния и мышьяка при их взаимном присутствии рассмотрены в I томе [1] данной монографии. Наиболее точные результаты получаются при определении фосфора и мышьяка. Кремний образует по крайней мере две модификации ГПК, заметно отличающиеся по оптическим свойствам. Скорость перехода одной модификации в другую (как для желтых, так и для синих кремнемолибденовых комплексов) зависит от ряда факторов, например от концентрации электролитов. Кроме того, реакционная способность кремневой кислоты по отношению к молибденовому реагенту зависит от размера частиц золя кремневой кислоты и от предварительных условий образования. В частности, при анализе металлов кремний, который входит в состав силицидов, определяется надежно, тогда как кремний шлаковых включений обычно не реагирует с молибдатом. В то же время образовавшийся кремнемолибденовый комплекс не разлагается при действии довольно концентрированных кислот, ряда посторонних комплексантов, в том числе фосфорной кислоты, которая разрушает фосфорномолибденовый комплекс с образованием бесцветных 9-молибденовых комплексов.
Для образования синих ГПК предложено очень много различных восстановителей, например хлорид олова(II), гидразин, тиомочевина, метол и т. п. Основное требование к восстановителю заключается в том, чтобы он реагировал только с тем молибде-
ном, который связан в комплекс. Выполнение этого условия зависит не только от выбора восстановителя, но и от условий определения, прежде всего — от кислотности раствора; В общем, сильные восстановители и слабая кислотность мало пригодны, так как при этом может образоваться молибденовая синь за счет несвязанного в комплекс молибдата (избыток реагента). Вместе С тем сильные восстановители в сильнокислой среде могут привести к возникновению более слабой окраски, так как наиболее интенсивно поглощающие сини возникают при условии, что лишь часть молибдена, входящего в ГПК, переходит в пятивалентное состояние.
Широкое применение методов определения фосфора и кремния привело к многочисленным исследованиям и предложению множества методик. Результаты применения различных методов, в общем, мало отличаются друг от друга, в большинстве случаев выбор восстановителя не имеет значения. Только при анализе черных металлов необходимо принимать во внимание присутствие больших количеств железа(III). В связи с этим применение некоторых восстановителей менее удобно. Так, при работе с хлоридом олова(II) необходимо вводить его в избытке для того, чтобы полностью восстановить железо(Ш); в то же время избыток олова (II) может привести к указанным выше осложнениям. Некоторые органические восстановители образуют с железом(III) окрашенные соединения или окисляются до окрашенных продуктов. Поэтому для определения в черных металлах фосфора и кремния в виде синих ГПК чаще в качестве восстановителя применяют сульфит. Применение сульфита в присутствии железа(II) в строго определенных условиях кислотности дает хорошие результаты.
Описано для тех же случаев применение тиомочевины; тиомочевина удобна также для определения фосфора в сплавах цветных металлов при большом содержании меди, так как медь восстанавливается до бесцветного тиомочевинного комплекса меди(1).
Для определения фосфора в виде синего ГПК в разнообразных биологических и других материалах часто применяют восстановители, используемые как проявители в фотографии: метол, гидрохинон, эйконоген и др. Все они мало отличаются по конечному эффекту: следует лишь выбирать препараты, очищенные от темных продуктов окисления кислородом воздуха. Очищают препараты перекристаллизацией, если необходимо, с предварительной обработкой растворов активированным углем и др.
Мешающие вещества. Имеется много различных данных о влиянии посторонних элементов на определение фосфора, кремния и мышьяка в виде ГПК. Методы устранения влияния железа и меди указаны выше. Мешают определению ниобий и титан, которые образуют более сложные соединения, содержащие наряду с фосфором, кремнием или мышьяком также ниобий или титан. Эти комплексы сильнее поглощают свет в видимой части спектра, чем простые ГПК- Влияние титана и ниобия не удается устранить 76
экстракцией. Сложные ГПК не экстрагируются неводными растворителями. Наиболее надежным методом устранения влияния титана и ниобия является введение небольших количеств фторидов, которые связывают эти, а также некоторые другие элементы в устойчивые бесцветные комплексы.
Отделение ниобия, тантала, титана, олова, вольфрама, молибдена, ванадия, висмута, сурьмы, железа и меди от фосфора и кремния можно сравнительно легко осуществить экстракцией их купферонатов хлороформом. Для этого в холодный 3,5—4,0 н. по серной кислоте раствор прибавляют при перемешивании 5%-ный водный раствор купферона до прекращения выделения осадка и 2—3 мл избытка реактива. Раствор перемешивают и оставляют на 5 мин. Затем купферонаты металлов и избыток купферона экстрагируют отдельными порциями хлороформа до тех пор, пока прибавленная порция хлороформа не будет оставаться бесцветной. Перед определением фосфора или кремния растворенный в водной фазе хлороформ удаляют нагреванием раствора.
Некоторые ионы мешают лишь собственной окраской (никель, кобальт, хром); это влияние проще всего устраняется экстракцией ГПК. О влиянии многих других ионов имеются обширные, хотя и не строго обоснованные данные. Не мешают щелочные и щелочноземельные элементы, а также алюминий, цинк, марганец, свинец, ртуть и кадмий (не слишком большие количества). Во многих работах отмечается мешающее влияние висмута, циркония, тория, олова, сурьмы, вольфрама (более 25 мг), фтора (большие количества), а также бора. Оксалат и тартрат разрушают фос-форномолибденовый комплекс, но не разрушают кремнемолибденовый, а также фосфорнованадиевомолибденовый и мышьякова-надиевомолибденовый комплексы.
III. 2. ПОДГОТОВКА МАТЕРИАЛА К АНАЛИЗУ
И МЕТОДЫ УСТРАНЕНИЯ МЕШАЮЩИХ ВЕЩЕСТВ
Наиболее просто определение фосфата или силиката в природных иди технических водах, пробы которых часто можно непосредственно обрабатывать кислым раствором молибдата для образования ГПК- Однако даже в этих случаях определение кремния иногда более сложно: состояние .кремневой кислоты влияет на ее реакционную способность, в частности, по отношению к молибдату [2]. При pH = 1—8 в разбавленных растворах, содержащих до 100 мг/л кремневой кислоты, она находится в активной форме [3], а в более концентрированных растворах находятся полимерные формы этой кислоты, которые реагируют с молибдатом медленно. Состояние кремневой кислоты и скорость реакции с молибдатом зависят от многих условий. Для определения общего содержания кремневой кислоты в водах и т. п. рекомендуют предварительно нагреть пробу с раствором щелочи для переведения в силикат или обработать ее небольшим количеством фтористоводородной кислоты^
Оптимальными условиями образования кремнемолибденовой гетерополикислоты является слабая кислотность (pH = 1,5). В этих условиях, а также при более высокой кислотности (оптимальная кислотность ~0,25 н. кислота) происходит образование и фос-форномолибденовой кислоты. Таким образом, если вести определение фосфора в кислой среде, то его определению кремний не мешает. Образовавшаяся же при низкой кислотности кремнемолибденовая кислота устойчива к действию минеральных кислот. Поэтому если после образования кремнемолибденовой и фосфор-номолибденовой кислот подействовать на их смесь минеральной кислотой, то фосфорномолибденовая разрушается, на кремнемолибденовую кислоту повышение кислотности до 4 н. не действует. На этих свойствах ГПК основано определение фосфора в присутствии кремния и кремния в присутствии фосфора.
Влияние различных посторонних веществ на определение кремневой кислоты описано во многих исследованиях (см., например, [4]). При анализе нерастворимых в воде веществ подготовка раствора для анализа весьма сложна. Силикатные породы сплавляют с карбонатом натрия и выщелачивают водой; однако при этом часть кремневой кислоты остается в осадке. При определении кремния в металлах и шлаках следует пользоваться только строго проверенными методиками переведения вещества в раствор. Возможности применения фотометрического метода для определения кремния в каждом новом виде материалов требуют тщательной проверки и сравнения с обычными гравиметрическими методами. Для хранения всех растворов и реактивов следует пользоваться посудой из полимерных материалов.
Определение фосфатов обычно не требует строгого соблюдения условий и различные методики дают достаточно близкие результаты, так как реакционная способность фосфата мало зависит от условий переведения материала в раствор. Тем не менее определение фосфата даже в таких простых материалах, как карбонатные породы, часто показывает хорошую воспроизводимость в одной лаборатории, но результаты различных лабораторий нередко сильно отличаются. Таким образом, и в этом случае необходимо внимательное отношение к различным деталям методики.
В металлах фосфор содержится в виде фосфидов. Поэтому растворяют металлы обычно в присутствии окислителей. Имеются данные, что в присутствии азотной кислоты часть фосфора остается в низших валентных формах. Поэтому в большинстве методик для полного окисления фосфора до Н3РО4 обычно рекомендуют добавлять перманганат калия и нагревать некоторое время; избыток перманганата или образующийся осадок двуокиси марганца восстанавливают нитритом или перекисью водорода.
В некоторых случаях необходимо отделение кремния или фосфора от других элементов. Для отделения фосфата от меди, никеля, хрома и т. п. рекомендуют осаждать фосфаты с гидратами окисей алюминия или железа при pH = 5—6; при повышении pH
фосфат переходит в раствор. На основании исследований по методу радиоактивных изотопов установлено [5], что степень осаждения фосфата с гидратом окиси железа при pH = 5—7 составляет 92—97%. По тем же данным, осаждение в виде аммонийной соли фосфорномолибденовой кислоты составляет лишь 90%. Интересно, что экстракция фосфата в виде ГПК смесью бутанола и хлороформа приводит к извлечению лишь 87% фосфата; однако если при этом повысить содержание азотной кислоты до 8% (по объему), то наблюдается почти полное извлечение фосфора.
Для получения аналитических концентратов кремневой кислоты в литературе рекомендуется соосаждение с двуокисью марганца, которая образуется при pH = 2—3 в результате реакции между перманганатом и солью марганца(II). Для анализа сплавов описан метод перегонки кремния в виде фторида, причем в определенных условиях [6] достигается также отделение от бора.
Для отделения от ряда мешающих ионов применяют часто экстракцию фосфорномолибденовой кислоты. Наиболее полное извлечение достигается обычно при экстракции спиртами, хотя, по некоторым данным [7], более высокая избирательность наблюдается при использовании эфиров.
Мешающие анализу тяжелые цветные металлы, как медь, олово, висмут и др., часто целесообразно удалять из кислого раствора электролизом на ртутном катоде. Мышьяк можно удалить (и определить) экстракцией мышьяковомолибденовой кислоты (см. ниже). Однако в большинстве случаев проще удалить мышьяк, а также германий в виде легколетучих хлоридов AsCh и GeCk.
Методики определения кремния и фосфора при их совместном присутствии довольно хорошо разработаны. Однако большие количества кремния надежнее удалять в виде летучего фторида или обычным путем в виде кремневой кислоты.
Описано чрезвычайно много методик определения фосфора и кремния в виде комплексов с молибдатом. В некоторых методиках имеются противоречивые указания. Это обусловлено сложным характером влияния различных факторов на протекание реакции. Следует иметь в виду, что молибденовый ангидрид является амфолитом, для которого изоэлектрическая точка, наименьшая растворимость и наибольшая степень полимеризации соответствуют pH = 1. Приблизительно этим же условиям отвечает и оптимум образования ГПК; однако по ряду причин при анализе различных материалов рекомендуют другие значения pH, например, чтобы уменьшить гидролиз солей железа и др. Наконец, сложность реакций обусловлена также тем, что кремнемолибденовый [8] и фос-форномолибденовый комплексы [9] существуют по крайней мере в двух модификациях, отличающихся по интенсивности светопо-глощения [10], по равновесию диссоциации и другим свойствам.
Большинство исследователей обращает внимание лишь на отдельную группу факторов, а о других иногда не приводятся даже необходимые сведения или не указываются допустимые колебания
концентрации реактивов. Скорость и степень реакции образования ГПК зависят от концентрации молибдата и от кислотности раствора, а также от присутствия железа или других элементов, связывающих фосфат, а в некоторых случаях и молибдат.
В различных методиках указывается разный состав наиболее важного реактива — раствора молибдата. В некоторых случаях рекомендуют готовить его из молибдата аммония, который является солью изополикислоты; приблизительный состав обычного препарата отвечает формуле (NH4)eMo7O24«4H2O или 5(NH4)2O• 12МоО3-7Н2О. При определении больших количеств фосфата возможны осложнения в связи с осаждением аммонийной соли фосфорномолибденовой кислоты. Некоторые препараты молибдата аммония, особенно после длительного хранения, содержат нерастворимый в воде остаток. Для анализа рекомендуют растворить молибдат аммония в воде с добавкой небольшого количества NH4OH, отфильтровать нерастворимый остаток и осадить препарат прибавлением равного объема спирта.
Ввиду ряда неудобств работы с молибдатом аммония во многих методиках рекомендуют готовить раствор из молибдата натрия Na2MoO4 • 2Н2О. Наконец, иногда готовят раствор молибденового ангидрида, который можно получить при слабом прокаливании молибдата аммония; молибденовый ангидрид легко растворяется в едких щелочах. В литературе нет каких-либо указаний на то, что способ приготовления молибденового реактива заметно влияет на результат.
III. 3. ОПРЕДЕЛЕНИЕ ФОСФОРА В ВИДЕ ЖЕЛТОЙ ФОСФОРНОМОЛИБДЕНОВОЙ ГЕТЕРОПОЛИКИСЛОТЫ
В методе использована реакция образования желтой фосфорномолибденовой гетерополикислоты Н3[Р (МозОю) 4]. Оптимальная концентрация азотной или хлорной кислоты для образования фосфорномолибденовой кислоты 0,25 н., а концентрация молибдата около 0,04 М.
Мешающие вещества. Определению мешают многие ионы, которые образуют в этих условиях гетерополимолибденовые кислоты (мышьяк, германий и др.), восстановители, различные комплек-санты, связывающие молибден, а также ионы, имеющие собственную окраску.
Измерение оптической плотности. Спектр поглощения водного раствора фосфорномолибденовой гетерополикислоты, полученный при использовании в качестве раствора сравнения раствора молибдата, представлен на рис. 18. Для комплекса характерна полоса поглощения с максимумом при 315 нм. Однако в этой области заметно поглощает молибдат, который применяется в качестве реагента. Поэтому если определение фосфора ведут без применения 80
экстракции фосфорномолибденового комплекса, то оптическую плотность измеряют при, 380—410 нм.
Чувствительность метода — 0,5 мг фосфора в 25 мл конечного объема.
III. 3.1. Определение фосфора в отсутствие мешающих веществ
К раствору, содержащему фосфат, прибавляют молибдат аммония, создают определенную кислотность и измеряют оптическую плотность желтого раствора.
Реактивы
Молибдат аммония, 5%-ный водный раствор. Навеску 50 г перекристалли-
зованного (с прибавлением спирта) молибдата нагревании, фильтруют и разбавляют до 1 л водой.
Стандартный раствор фосфата. Навеску однозамещенного фосфата калия (КН2РО4) 0,4393 г переносят в мерную колбу емкостью 0,5 л, растворяют в воде и разбавляют до метки водой. Этот раствор содержит 0,2 мг фосфора в 1 мл.
Ход анализа. Около 12 мл испытуемого нейтрального раствора, содержащего 0,5—5 мг фосфора, помещают в мерную колбу емкостью 25 мл, прибавляют 6 мл 1,25 н. раствора азотной кислоты, 5 мл раствора молибдата аммония, разбавляют до метки водой, перемешивают и измеряют оптическую
аммония растворяют в воде при
Рис. 18. Спектр поглощения желтого фосфорномолибденового комплекса в водном растворе (по сравнению с раствором молибдата).
плотность.
Содержание фосфора находят по калибровочному графику, который строят по стандартному раствору в пределах содержания фосфора 0,5—5 мг/25 мл.
III. 3.2. Определение фосфора в водах, карбонатных породах и солях
Навеску карбоната переводят в раствор с помощью кислот, соли растворяют в воде или отбирают пробу природной воды и определяют фосфор, как описано в разделе III. 3.1.
III. 3.3. Определение фосфора в виде желтой фосфорномолибденовой гетерополикислоты с экстракцией
Методику определения фосфата с экстракцией рекомендуют в случае содержания в испытуемом растворе примесей, мешающих собственной окраской. Экстракция желтого фосфоромолиб-дата не смешивающимся с водой растворителем позволяет простым приемом увеличить чувствительность, так как из 50—100 мл
раствора окрашенное соединение можно количественно перевести в 10 мл неводной фазы. Еще более повышается чувствительность, если имеется возможность измерять светопоглощение в УФ; дей-
ствительно, молярный коэффициент (езоо) равен 24-Ю3, тогда как на границе видимой части спектра е = 1 • 103. При измерении оптической плотности при 300 нм рекомендуют составлять калибровочный график для содержания фосфора 10—100 мкг/10 мл органического растворителя. При экстракционном варианте определе-£ нию не мешают мышьяк — до 4 мг, крем-
I ний — 5 мг и германий — 1 мг.
2>k у \ Измерение оптической плотности. Спектр
\ поглощения фосфорномолибденовой гетеро-
45- \ поликислоты в бутанольно-хлороформном
\ (1:4) растворе представлен на рис. 19.
о,8- \ Максимум светопоглощения этой ГПК на-
ходится при 300 нм. Молибдат и другие я!_____।___мешающее вещества не экстрагируются
гео 320 360 л, нм это^ смесью и не мешают определению. По-
Рис. 19. Спектр поглощения раствора желтого фосфорномолибденового комплекса в смеси w-бутилового спирта и хлороформа (1 :4).
Чувствительность
этому оптическую плотность можно измерять при 300—410 нм. Измерение необходимо вести при одной и той же длине волны как в случае построения калибровочного графика, так и в. случае проведения определения.
метода составляет 10 мкг фосфора в 10 мл
органического растворителя.
Реактивы
Молибденовый реактив. Растворяют 7,5 г молибдата натрия в 100 мл воды, прибавляют 100 мл концентрированной азотной или хлористоводородной кислоты и разбавляют до 500 мл водой.
Органический растворитель. Смешивают 20 мл н-бутилового спирта с 50 мл хлороформа.
Стандартный раствор фосфата. Отбирают 10 мл раствора, приготовленного, как указано в разделе III. 3.1, переносят в мерную колбу емкостью 100 мл, разбавляют до метки водой и перемешивают. Этот раствор содержит. 20 мкг фосфора в 1 мл.
Ход анализа. В делительную воронку переносят 10—30 мл испытуемого раствора, содержащего до 0,1 мг фосфора, и прибавляют 10 мл молибденового реактива, перемешивают и оставляют на 1—2 мин. Затем дважды экстрагируют органическим растворителем порциями по 5 мл. Объединенный экстракт собирают в узкий сухой мерный цилиндр, разбавляют до 10 мл растворителем, перемешивают и измеряют оптическую плотность. Содержание фосфора находят по калибровочному графику, который строят, исходя из стандартного раствора, в пределах содержания фосфора 10—100 мкг/10 мл.
III. 3.4. Определение фосфора в водах, карбонатных породах и солях [11]
Навеску карбонатной породы растворяют в азотной или хлористоводородной кислоте, а навески солей — в воде, пробу воды берут без всякой подготовки и определяют фосфор, как указано в разделе 3.3.
III. 4. ОПРЕДЕЛЕНИЕ ФОСФОРА В ВИДЕ ЖЕЛТОГО ФОСФОРНОВАНАДИЕВОМОЛИБДЕНОВОГО КОМПЛЕКСА
Как было отмечено выше, некоторое повышение чувствительности определения фосфата (без экстракции) может быть достигнуто применением смешанного ванадато-молибдатного реактива. Действительно, молярный коэффициент светопоглощения (еио) фосфорпованадиевомолибденового комплекса на границе видимой части спектра [12] равен 2,1 • 103, т. е. приблизительно вдвое выше, чем подобная характеристика для простого фосфорномолибдено-вого комплекса (см. раздел III. 3). Для тройного соединения е430 = 1,3 - 103. Измерение при длине волны ниже 400 нм нецелесообразно, так как здесь заметно начинает накладываться свето-поглощепие реактива.
Метод предложен давно [13—16], затем были описаны различные варианты. Общий состав образующегося комплекса (Р2О5-УгО5-22МоОз-пН2О) установлен в 1947 г. [17], но строение тройного соединения остается неясным; известно, что фосфорно-ванадиевый комплекс в концентрированных растворах окрашен в коричневый цвет, а фосфорномолибденовый комплекс в желтый, однако по интенсивности светопоглощения тройное соединение значительно превышает каждое из этих двойных.
Мешающие вещества. Определению мешают хром, никель, кобальт и другие ионы, имеющие собственную окраску. В анализируемой пробе допустимо содержание кобальта и никеля до 5 мг, вольфрама, титана, урана до 10 мг; мышьяк в равных или вдвое больших количествах не мешает. Цирконий уменьшает оптическую плотность раствора. Сильно мешают восстановители.
Оптимальная кислотность при Ьпределении соответствует 0,5 н. раствору HNO3, при более высокой кислотности образование комплекса замедляется.
Оптимально определяемое количество фосфорной кислоты при введении 5 мг ванадия — от 0,02 до 1,0 мг Р2О5 на 25 мл конечного объема пробы. Верхний предел может быть несколько выше, но тогда необходимо вводить большее количество реактива.
Измерение оптической плотности. Оптическую плотность измеряют, сравнивая со стандартным раствором, содержащим приблизительно равное количество фосфора, так как раствор комплекса не вполне подчиняется закону Бэра. Для колориметрического определения применяют также метод стандартных серий
с имитирующей эталонной шкалой, предложенной Д. П. Щербо-
вым [18].
Спектр поглощения водного раствора фосфорнованадиевомо-либденового комплекса представлен на рис. 20. Очевидно, что из
мерение оптической плотности лучше проводить при 400—410 нм,
так как при меньшей длине волны наблюдается поглощение реактива. Калибровочный график строят в пределах 0,02—1,0 мг Р2О5 на 25 мл конечного раствора.
Рис. 20. Спектры поглощения раствора фосфорнованадиевомолибденового комплекса в воде по сравнению с холостым раствором (/) и холостого раствора (2).
Чувствительность метода 10 мкг фосфора в 25 мл конечного раствора.
III. 4.1. Определение фосфора в отсутствие мешающих веществ
Реактивы.
Метаванадат аммония. Навеску препарата 2,5 г растворяют при нагревании в 100—200 мл воды, прибавляют 20 мл азотной кислоты (пл. 1,2) и разбавляют водой до 1 л.
Молибдат аммония. Навеску препарата 10 г растворяют при нагревании в воде, охлаждают, если необходимо, фильтруют и разбавляют до 100 мл. Раствор хранят в полиэтиленовой посуде.
Стандартный раствор фосфата, см. раздел 3.1.
Ход анализа. Около 15 мл испытуемого раствора, содержащего от 0,02 до 1 мг фосфора, помещают в мерную колбу емкостью 25 мл, затем прибавляют туда же 5 мл HNO3 (пл. 1,2), 3 мл раствора метаванадата аммония, 3 мл раствора молибдата аммония, разбавляют до метки водой, перемешивают и спустя 10 мин измеряют оптическую плотность. Содержание фосфора находят по предварительно построенному калибровочному графику.
III. 4.2. Определение фосфора в черных металлах
Реактивы
Перманганат калия, 4%-ный раствор.
Нитрит натрия, 10%-ный раствор.
Другие реактивы — указаны в разделе III. 4.1.
Ход анализа. Навеску стали 0,2 г помещают в стакан или колбу емкостью 100 мл, растворяют в 5—6 мл HNO3 и нагревают до удаления окислов азота. К раствору прибавляют 4 %-ный раствор
КМпО4 до выпадения двуокиси марганца, которую переводят в раствор прибавлением по каплям раствора NaNO2 и нагревают до разложения избытка нитрита. Раствор охлаждают и переносят в мерную колбу емкостью 25 мл. Затем прибавляют туда же 3 мл раствора метаванадата аммония, 3 мл раствора молибдата аммония и доводят водой до метки. Раствор перемешивают, оставляют на 10 мин и измеряют оптическую плотность.
При анализе чугуна навеску 0,05—0,1 г растворяют в 6 мл HNO3, отфильтровывают кремний и графит, после чего поступают так же, как при анализе сталей.
III. 4.3. Определение фосфора в карбонатных породах [19]
Навеску образца от 0,5 до 1 г растворяют в 10—15 мл HNO3, выпаривают досуха и анализируют, как указано в разделе III. 4.1.
III. 4.4. Определение фосфора в силикатных породах
Навеску образца 0,25 г сплавляют в платиновом тигле с шестикратным количеством карбоната натрия. Сплав выщелачивают горячей водой и переносят в фарфоровую чашку. Раствор нейтрализуют азотной кислотой, прибавляют избыток кислоты и выпаривают досуха. Сухой остаток растворяют при нагревании в 5—6 мл азотной кислоты и 5 мл воды. Кремневую кислоту отфильтровывают и раствор анализируют, как указано в разделе III.4.1.
При анализе силикатных пород подготовку пробы можно проводить и кислотным разложением. Для этого навеску 0,5 г растертого анализируемого образца помещают в платиновую чашку, приливают 4 мл HNO3 (пл. 1,4) и 5 мл концентрированной фтористоводородной кислоты, затем медленно упаривают, а остаток высушивают при 200—250 °C. После охлаждения приливают 4 мл HNO3 (пл. 1,4) и 2 мл фтористоводородной кислоты, упаривают досуха, еще приливают 4 мл HNO3 (пл. 1,4) и опять выпаривают. Для удаления HF остаток высушивают в течение 30 мин, смачивают 4 мл HNO3 (пл. 1,4), разбавляют до ~25 мл и нагревают на паровой бане до растворения остатка. В присутствии больших количеств титана раствор кипятят несколько минут и отфильтровывают осадок TiO2. Фильтрат переносят в мерную колбу емкостью 50 мл, разбавляют до метки водой и перемешивают. Отбирают аликвотную часть и определяют фосфор, как указано в разделе III. 4.1.
III. 4.5. Определение фосфора в марганцевых и железных рудах
Для разложения навеску руды 0,25 г помещают в стакан емкостью 100 мл, прибавляют 10 мл хлористоводородной кислоты (пл. 1,19), нагревают на песочной бане и выпаривают в случае
марганцовых руд досуха, а в случае железных руд до сухой консистенции. Остаток смачивают 2—3 мл азотной кислоты (пл. 1,4), добавляют 20 мл горячей воды, нагревают до растворения солей и отфильтровывают кремневую кислоту. К фильтрату прибавляют 10 мл HNO3 (пл. 1,2) и выпаривают до объема 2—3 мл. Это повторяют еще 2—3 раза для удаления хлористоводородной кислоты. Затем добавляют 3—4 мл HNO3 (пл. 1,2) и определяют фосфор, как указано в разделе III. 4.1.
III. 4.6. Определение фосфора в ванадиевых рудах
Разложение навески (в случае сажистой руды предварительно прокаленной) проводят, как указано в разделе III. 4.5, а определение ведут, как указано в разделе III. 4.1, с той лишь разницей, что ванадия вводят меньше, так чтобы общее количество его соответствовало количеству ванадия, вводимого по методике III. 4.1.
III. 4.7. Определение фосфора в почве [20]
Навеску почвы 5 г помещают в колбу емкостью 100—150 мл, добавляют несколько' стеклянных шариков, вводят 25 мл HNO3 (пл. 1,4) и нагревают до прекращения вспенивания. Затем вводят 5—7 мл 70%-ной НС1О4 и нагревают до обесцвечивания смеси. Если необходимо, добавляют еще 5 мл хлорной кислоты и нагревание повторяют. Содержимое колбы охлаждают, добавляют 20 мл воды, доводят до кипения, охлаждают, фильтруют в мерную колбу емкостью 50 мл и промывными водами доводят до метки. Раствор перемешивают, отбирают аликвотную порцию и определяют фосфор, как указано в разделе III.4.1.
III. 4.8. Определение фосфора в удобрениях и сырье [21]
Навеску пробы 0,25—1,0 г помещают в колбу емкостью 100— 150 мл, прибавляют 20 мл HNO3 (пл. 1,4), при большом содержании органических веществ добавляют 30 мл HNO3 и упаривают при кипячении до 5—10 мл. Слегка охлаждают, добавляют 10 мл 70—72 %-ной НСЮ4, кипятят до обесцвечивания раствора и выпаривают до появления белых паров. Содержимое колбы охлаждают, добавляют 50 мл воды и кипятят 10 мин для удаления окислов азота. Охлаждают, разбавляют до 200 мл и фильтруют через сухой фильтр. Отбирают аликвотную порцию и определяют фосфор, как указано в разделе III. 4.1.
При определении только водорастворимого [22] фосфата навеску удобрения 10 г прибавляют к 400 мл воды, взбалтывают в течение 30 мин, разбавляют до 500 мл, взбалтывают, фильтруют и в аликвотной части определяют фосфор, как указано в разделе III.4.1.
111.5. ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ФОСФОРА В ВИДЕ ФОСФОРНОВАНАДИЕВОМОЛИБДЕНОВОГО КОМПЛЕКСА
Фосфорнованадиевомолибденовый комплекс экстрагируется изоамиловым или бутиловым спиртом. Это дает возможность повысить чувствительность и устранить влияние некоторых окрашенных компонентов.
Измерение оптической плотности лучше проводить при 380 нм. Чувствительность метода 5 мкг фосфора в 10 мл экстракта.
III. 5.1. Определение фосфора в отсутствие мешающих веществ
Реактивы
Ванадатно-молибдатный раствор. Растворяют 0,5 г ванадата аммония в 150 мл воды и 70 мл концентрированной азотной кислоты. К раствору прибавляют 20 г молибдата аммония, растворенного в 200 мл воды. Смесь разбавляют водой до 500 мл и перемешивают.
Изоамиловый спирт, чистый.
Стандартный раствор фосфата, см. раздел III. 3.1.
Перманганат калия, 4%-нын раствор.
Нитрит натрия, 10%-ный раствор.
Ход анализа. К нейтральному или слабокислому раствору, содержащему от 5 до 500 мкг фосфора, прибавляют 10 мл азотной кислоты (1:1) и раствор перманганата калия до розовой окраски. Раствор для окисления фосфора нагревают в течение нескольких минут. Затем по каплям прибавляют раствор нитрита натрия до исчезновения осадка двуокиси марганца. К горячему раствору приливают 15 мл ванадатно-молибдатного раствора и оставляют на 10 мин. Раствор разбавляют до 30—40 мл водой, переносят в делительную воронку и дважды экстрагируют изоамиловым спиртом порциями по 5 мл. Экстракты объединяют в сухой пробирке, доводят .до 10 мл изоамиловым спиртом, фильтруют через сухой фильтр и фотометрируют. Содержание фосфора находят по калибровочному графику, построенному в пределах содержания фосфора 5—500 мкг в 10 мл изоамилового спирта.
III. 5.2. Определение фосфора в чугунах [23]
Реактивы
Смесь кислот. К 50 мл воды прибавляют 25 мл хлористоводородной кислоты (пл. 1,19) и 15 мл азотной кислоты (пл. 1,4) и перемешивают.
Другие реактивы — указаны в разделе III. 5.1.
Ход анализа. Навеску чугуна 0,1 г растворяют в 10 мл смеси кислот и выпаривают до объема 3 мл. Затем прибавляют 10 мл азотной кислоты (1:1) и продолжают определение, как указано в разделе III. 5.1.
III. 5.3. Определение фосфора в сталях [23]
Навеску стали 0,25 г при содержании фосфора 0,025—0,045% растворяют в смеси 5 мл хлористоводородной кислоты (пл. 1,19) и 5 мл HNO3 (пл. 1,4). Затем добавляют 5 мл хлорной кислоты (пл. 1,54), упаривают до появления паров и нагревают еще 10— 15 мин. Стакан накрывают стеклом, охлаждают и добавляют 25 мл воды. Если присутствует вольфрам, то разбавляют 10 мл теплой воды, фильтруют и промывают 30 мл теплой воды. В присутствии хрома вводят 10 мл насыщенного раствора H2SO3, кипятят 10 мин. К кипящему раствору прибавляют 5 мл 20%-ной азотной кислоты и кипятят 2—3 мин. Затем определяют фосфор, как указано в разделе III. 5.1, минуя окисление перманганатом и восстановление двуокиси марганца.
III. 5.4. Определение фосфора в медных сплавах
Навеску сплава 0,25 г растворяют в смеси 5 мл хлористоводородной кислоты (1:1) и 5 мл HNO3 (1 : 1), разбавляют до 25 мл водой и определяют фосфор, как указано в разделе III. 5.3.
III. 6. ОПРЕДЕЛЕНИЕ ФОСФОРА В ВИДЕ
ФОСФОРНОМОЛИБДЕНОВОГО КОМПЛЕКСА С ПОСЛЕДУЮЩИМ ВОССТАНОВЛЕНИЕМ ЖЕЛЕЗОМ(Н) И СУЛЬФИТОМ
Описанные выше методы, основанные на образовании желтых ГПК, вызывают иногда трудности при массовых анализах черных металлов, так как невозможно избежать появления различных гидролизованных форм железа(III), которые также имеют желтую окраску. Поэтому для определения фосфора в черных металлах часто предпочитают применять методы, основанные на образовании восстановленных (синих) ГПК. Из огромного количества восстановителей наиболее часто применяют сульфит, который восстанавливает железо(Ш) до железа(II). Многие органические восстановители при реакции с железом(III) образуют окрашенные соединения; другие восстановители, как, например, хлорид олова(II), сильно изменяют свой потенциал при накоплении олова(IV) в результате реакции с железом (III), при этом воспроизводимость определения становится плохой. Применение сульфита в качестве восстановителя устраняет эти и некоторые другие затруднения, однако нельзя считать, что рассматриваемый метод свободен от недостатков.
Метод был предложен давно [24] для анализа чугунов и сталей. Позже он много раз применялся для анализа различных материалов, содержащих много железа, в том числе и для анализа железных руд [25].
В качестве восстановителей фосфорномолибденовой гетерополикислоты, кроме указанных выше, предложены также гидрохинон с сульфитом натрия, аминонафтолсульфоновая кислота и бисуль
фит, монометил-п-аминофенолсульфонат в присутствии сульфита и бисульфита, гидразин, тиомочевина и другие. Довольно часто в качестве восстановителя применяют хлорид олова(II), но этот вариант является одним из худших, так как SnCb может восстанавли-
вать не только молибден, связанный в гетерополикислоту, а также молибден, который всегда в условиях проведения анализа находится в избытке. В отсутствие избытка молибдата избыток хлорида олова(II) восстанавливает молибден синей фосфорномолибденовой кислоты до низшей валентности, что приводит к образованию двух
модификаций фосфорномолибденовой кислоты [26], характеризующихся различными спектрами поглощения [27]. Достаточно удобным восстановителем является железо (II) в присутствии сульфита.
Мешающие вещества. Определению мешают вольфрам, ванадий, кремний, окислители, а также сильные восстановители. Влияние кремневой кислоты устраняется регулированием кислотности раствора (см. § 3).
Измерение оптической плотности.
Спектр поглощения раствора синей фосфорномолибденовой кислоты, полученной восстановлением желтой фосфорномолибденовой гетерополикислоты сульфатом железа(II) в присут-
£7Z7'*
О -----1----1---------1-----1____।
580 620 660 700 7Ц) 780 Л, нн
Рис. 21. Спектр поглощения раствора синего фосфорно-молибденового комплекса,полученного восстановлением желтой фосфорномолибденовой гетерополикислоты сульфатом железа(П) в присутствии сульфита.
ствии сульфита, представлен на рис. 21. Измерение оптической плотности лучше проводить при 650—700 нм. По различным данным, молярный коэффициент поглощения для фосфорномолибденовой сини (еб50-700) равен 2-Ю4.
При анализе различных материалов калибровочные графики лучше строить, по стандартным образцам.
При колориметрическом определении одновременно с испытуемым раствором готовят стандартный раствор с определенным содержанием фосфора. Метод колориметрического титрования и метод стандартной шкалы неприменимы.
Чувствительность метода — 5 мкг фосфора в 25 мл конечного объема.
111.6.1. Определение фосфора в отсутствие мешающих веществ
К раствору, содержащему фосфор, прибавляют железоаммонийные квасцы, сульфит, кислоту и нагревают до восстановления железа *. При правильном проведении анализа почти тотчас после
* Для анализа материалов, не содержащих железа, надежнее применять другие методы, в частности восстановление до фосфорномолибденовой сини посредством тиомочевины, гидразина и т. п.
нагревания раствор обесцвечивается. Зеленоватый цвет раствора указывает на недостаточность восстановителя, а желтовато-золотистый цвет указывает на недостаточную кислотность раствора. В первом случае необходимо прибавить еще сульфита, а во втором — хлористоводородной кислоты. Если не удается получить совершенно бесцветный раствор, пробу необходимо отбросить и взять другую навеску. Затем к раствору пробы после восстановления железа прибавляют молибдат аммония, взбалтывают и измеряют оптическую плотность.
Реактивы
Сульфит натрия, 20%-ный раствор. Навеску Na2SO3-7H2O 100 г растворяют в 450 мл воды, спустя два дня фильтруют через два плотных фильтра и разбавляют до 500 мл водой. Раствор хранят в склянке с притертой пробкой. Он годен к употреблению в течение 2—3 недель. Сульфит в растворе довольно быстро окисляется кислородом воздуха, поэтому спустя две педели восстановительная способность падает на 20%.
Молибдат аммония, 5%-ный водный раствор. Растворяют 50 г перекристаллизованного (с прибавлением спирта) молибдата аммония в воде при нагревании, фильтруют и разбавляют до 1 л водой.
Железоаммонийные квасцы. Навеску 216 г железоаммонийных квасцов растворяют при нагревании в воде, к которой прибавлено 5 мл хлористоводородной кислоты (пл. 1,19), и разбавляют водой до 1 л.
Стандартный раствор фосфата, см. раздел III. 3.1.
Аммиак, разбавленный раствор.
Ход анализа. Приблизительно 15 мл испытуемого раствора, содержащего от 5 до 500 мкг фосфора в виде фосфата, помещают в коническую колбу емкостью 50 мл, прибавляют 2 мл раствора железоаммонийных квасцов и затем раствор аммиака по каплям до начала выпадения гидрата окиси железа. Образующийся осадок растворяют в нескольких каплях 4 н. хлористоводородной кислоты и прибавляют 1 мл избытка кислоты. Затем прибавляют 2 мл раствора сульфита натрия и кипятят 1 .мин для восстановления железа.
Раствор охлаждают, прибавляют 3 мл хлористоводородной кислоты, затем по каплям при взбалтывании 2 мл раствора молибдата аммония. Раствор переносят в мерную колбу емкостью 25 мл, разбавляют водой до метки и перемешивают, через 5—10 мин измеряют оптическую плотность.
III. 6.2. Определение фосфора в чугунах и простых углеродистых сталях
Навеску пробы растворяют в азотной кислоте, окисляют перманганатом и определяют фосфор, как указано в разделе III. 6.1.
Реактивы
Перманганат калия, 4%-ный раствор.
Другие реактивы — указаны в разделе III. 6.1.
Ход анализа. Навеску стали (чугуна) 0,1 г при содержании фосфора до 0,2% и 0,05 г при более высоком содержании его помещают в коническую колбу емкостью 50 мл и растворяют при нагревании в 2—3 мл HNO3, разбавленной (1:1). К раствору прибавляют 2 мл раствора КМпО4 и кипятят до исчезновения фиолетовой окраски перманганата. Для переведения в раствор осадка двуокиси марганца прибавляют 1 мл раствора сульфита натрия и кипятят. Раствор охлаждают, переносят в мер-ную колбу емкостью 25 мл и определяют фосфор, как описано в разделе III. 6.1, не прибавляя раствор железоаммонийных квасцов.
III. 6.3. Определение фосфора в нержавеющей высокохромистой стали *
Навеску стали растворяют в смеси азотной .и хлористоводородной кислот, хром окисляют персульфатом аммония в присутствии нитрата серебра и фосфор отделяют в виде фосфата железа. При этом большие количества хрома и никеля остаются в растворе. Осадок гидрата окиси и фосфата железа растворяют в хлористоводородной кислоте и определяют фосфор, как указано в разделе III. 6.2.
Реактивы
Нитрат серебра, 0,1 н. раствор.
Ход анализа. Навеску стали 0,1 г помещают в небольшой стакан и растворяют в 5 мл смеси (1:1) концентрированных азотной и хлористоводородной кислот. Раствор выпаривают почти досуха, прибавляют 5 мл HNO3 (пл. 1,4) и выпаривание повторяют. Остаток растворяют при нагревании в 1 мл HNO3 и 20 мл воды. К раствору прибавляют 2 мл 0,1 н. раствора AgNO3. В другой небольшой стакан помещают 20 мл H2SO4, разбавленной (1:1), нагревают до кипения и растворяют в ней 3 г персульфата аммония. Испытуемый раствор нагревают до кипения и прибавляют постепенно при помешивании к кипящему сернокислому раствору персульфата. Смесь кипятят 2—3 мин, затем прибавляют 2 мл 4 н. раствора НС1 и продолжают кипячение еще около 10 мин. Прибавляют аммиак до появления запаха.
Осадок гидроокиси и фосфата железа отфильтровывают, промывают горячей водой и небольшим количеством воды переносят в тот же стакан, в котором проводилось осаждение; фильтр промывают 1 мл горячей хлористоводородной кислоты (пл. 1,19), а затем водой, сливая промывную жидкость в тот же стакан. Содержимое стакана кипятят до полного растворения бурого осадка, отфильтровывают от AgCl и промывают небольшими порциями горячей 4 н. хлористоводородной кислоты. Раствор собирают в мерную колбу емкостью 25 мл, разбавляют водой до метки, перемешивают и отбирают 12,5 мл в другую мерную колбу емкостью 25 мл.
В дальнейшем в одной порции определяют фосфор, как указано в разделе III. 6.1., а в другую порцию прибавляют все реактивы, кроме молибдата аммония, и этот раствор используют в качестве «нулевого» раствора.
III. 6.4. Определение фосфора в железных и марганцевых рудах
Навеску руды 0,1 г при содержании фосфора до 0,2% и 0,05 г при более высоком его содержании помещают в стакан емкостью 20—30 мл и растворяют в 3 мл хлористоводородной кислоты (пл. 1,19) и нескольких каплях HNO3 (пл. 1,4). Содержимое стакана нагревают до удаления окислов азота, разбавляют небольшим количеством воды и отфильтровывают нерастворимый остаток. В фильтрате определяют фосфор, как указано в разделе III. 6.2. При анализе железных руд берут соответственно меньшее количество железоаммонийных квасцов.
III. 7. ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ФОСФОРА В ВИДЕ СИНЕЙ ФОСФОРНОМОЛИБДЕНОВОЙ ГЕТЕРОПОЛИКИСЛОТЫ
Экстракция синей фосфорномолибденовой гетерополикислоты имеет большое значение для определения фосфора, поскольку при этом устраняется вредное влияние ионов, имеющих собственную окраску, а также увеличивается чувствительность определения. Однако большинство экстракционных методов основано на предварительной экстракции желтой фосфорномолибденовой кислоты с последующим ее восстановлением и определением в органической фазе [7]. Применение экстракционно-фотометрического определения фосфора весьма важно для анализа многих цветных металлов и легированных сталей. Первые указания на возможность экстракции восстановленных гетерополикислот имеются в работе Дениже [28]. Экстракционно-фотометрический метод определения фосфора в виде сини впервые был разработан С. Б. Шнеерсоном [29] в 1936 г. По этой методике восстановленную гетерополикислоту экстрагируют диэтиловым эфиром и измеряют интенсивность окраски методом стандартной шкалы. Наиболее полно экстракция синей фосфорномолибденовой гетерополикислоты изучена И. П. Алима-риным с сотрудниками [30]. По их данным, эффективным экстрагентом для восстановленной фосфорномолибденовой гетерополикислоты, как и в случае невосстановленной гетерополикислоты, являются бутанол и изобутанол (табл. 5).
Из данных табл. 5 следует, что степень экстракции и молярный коэффициент поглощения зависят от применяемых восстановителя и растворителя. Лучшим восстановителем, применение которого приводит к устойчивым результатам, является аскорбиновая кислота.
Таблица 5. Экстракция (в %) и молярные коэффициенты поглощения восстановленной фосфорномолибденовой гетерополикислоты в зависимости от восстановителя и растворителя [30]
Растворители Экстракция не восстановленной ГПК, % Восстановитель
аскорбиновая кислота гидразинсульфат метиламинофенолсульфат хлорид олова(П) амннонафтол-сульфокислота
экстракция, % еХ макс экстракция, % еЛ макс экстракция, % еХ макс экстракция, % еЛ макс экстракция, % еХ макс
Бутанол 96 100 3,0 • 104 93 3,0 - 104 90 2,84- 104 97 2,34- 104 — 5,8- 103
Изобутанол — 100 3,0- 104 90 3,3 - 104 83 2,5- 104 100 2,20- 104 — 5,8- 103
Пентанол , 96 100 2,9- 104 88 2,57- 104 85 — 87 — — 5,8- 103
Изопентанол — 100 3,12- 104 99 3,07- 104 81 2,5- 104 91 2,38 • 104 — 5,5 • 103
Гексанол 95 98 3,12- 104 90 3,0- 104 83 2,48- 104 91 2,03- 104 — 5,7- 103
Гептанол 94 90 3,35- 10‘ 83 3,28 • 104 77 2,73 • 104 93 2,23- 104 — 5,7- 103
Метилизобу-тилкетон — 100 2,9 • 10’ 83 3,0-Ю4 85 2,26- 104 88 2,34- 104 — 5,4 • 103
Этилапетат 97 90 3,03- 104 98 3,23- 104 68 2,31 • 104 85 2,29- 104 — —
Диэтиловый эфир 95 0 — 0 — 0 — 58 1,5-104 — ' 4,0- 103
Вода — — 2,81 • 104 — 2,87- 104 — 2,57 • 104 — — — 4,0 • 103
15,0-
0 1---L_—-I I f
SOO Ш 680 720 760 Л,мм
Рис. 22. Спектр поглощения раствора синей фосфорномолибденовой кислоты в изобутаноле.
Устойчивость растворов восстановленной гетерополикислоты в органических растворителях во времени также зависит от применяемого восстановителя. Постоянство оптической плотности во времени наблюдается при использовании в качестве восстановителя метиламинофенолсульфата, хотя молярный коэффициент несколько ниже, чем'при использовании аскорбиновой кислоты.
Для образования синей фосфорномолибденовой гетерополикислоты нижний предел оптимальной кислотности составляет 2 н., а верхний зависит от применяемого восстановителя: в случае гидразинсульфата наименьший — 3 н., а в случае аскорбиновой кислоты наибольший — 7 н. При использовании в качестве восстановителя 1-амино-2-на-фтол-4-сульфокислоты экстракция восстановленной фосфорномолибденовой кислоты мало зависит от концентрации кислоты в водном растворе.
Необходимо отметить, что желтая фосфорномолибденовая кислота, растворенная в органическом растворителе, восстанавливается значительно труднее, чем в водном растворе. Поэтому сильные восстановители, такие как хлорид олова(II), мало пригодные в водных растворах, в неводном рас
творе дают вполне удовлетворительные результаты. Рекомендовано также применение оксалата олова [31—33], который дает более устойчивые результаты.
Данные табл. 5 также показывают, что восстановленная фосфорномолибденовая гетерополикислота в большинстве случаев несколько лучше экстрагируется по сравнению с невосстановленной гетерополикислотой. Отсюда следует практический вывод, что для аналитических целей лучше сначала восстановить в водной фазе гетерополикислоту, а затем проводить экстракцию восстановленной фосфорномолибденовой кислоты.
Мешающие вещества. Мешающее влияние окрашенных ионов устраняют’ экстракцией, влияние кремния — регулированием кислотности образования желтого фосфорномолибденового комплекса. Мышьяк предварительно удаляют выпариванием хлористоводородных растворов в присутствии бромистоводородной кислоты. Иногда ограничиваются восстановлением мышьяка(V) до мышьяка(III). Вольфрам обычно отделяют перед определением фосфора. Титан и некоторые другие металлы связывают аскорбиновой кислотой. Определению мешают различные комплексанты [34], которые связывают молибден, а также окислители.
Измерение оптической плотности. Спектр поглощения раствора синей фосфорномолибденовой кислоты в изобутаноле представлен на рис. 22. Оптическую плотность необходимо измерять при
625—7,25 нм. Применение колориметров требует параллельного приготовления стандартного раствора из образца с близким содержанием фосфора. Метод стандартной шкалы наименее удобен.
Чувствительность метода — 0,01 мкг фосфора в 10 мл экстракта.
III. 7.1. Определение фосфора в отсутствие мешающих веществ
Для проведения определения получают желтую фосфорномо-либденовую кислоту, восстанавливают ее до синего комплекса и экстрагируют этот комплекс органическими растворителями. Можно также предварительно экстрагировать фосфорномолибденовую кислоту, а затем проводить восстановление.
Реактивы
Молибдат аммония. Растворяют 50 г молибдата аммония (МН4)бМоО24-4Н2О в 400 мл воды, прибавляют 230 мл серной кислоты, разбавленной 1:1, разбавляют до 1 л водой и перемешивают.
Хлорид олова(Н). Растворяют 2,5 г SnCl2-2H2O в 170 мл концентрированной хлористоводородной кислоты и доводят дистиллированной водой до 1 л. В раствор вводят несколько гранул металлического олова.
Изобутанол.
Стандартный раствор фосфата, см. раздел 3.1.
Ход анализа. Вариант 1. Помещают в делительную воронку емкостью 100 мл до 15 мл нейтрального исследуемого раствора, содержащего до 10 мкг фосфора, прибавляют 5 мл 10 н. серной кислоты, если надо, разбавляют дистиллированной водой до 20 мл, вводят 5 мл раствора молибдата аммония, перемешивают и оставляют на 10 мин. Затем добавляют 10 мл раствора хлорида олова (II), перемешивают, прибавляют 10 мл изобутанола и взбалтывают в течение 0,5—1 мин. Отделяют и отбрасывают водный слой, а изобутанольный слой сливают в сухой тонкий мерный цилиндр емкостью 10 мл. Воронку споласкивают 0,5 мл изобутанола и доводят объем раствора в цилиндре до 10 мл. Раствор перемешивают, наполняют кювету и измеряют оптическую плотность.
Содержание фосфора находят по предварительно построенному калибровочному графику.
Вариант 2. До 15 мл нейтрального раствора, содержащего до 10,0 мкг фосфора, помещают в делительную воронку емкостью 100 мл, прибавляют 5 мл серной кислоты, если надо, дистиллированной водой разбавляют до 20 мл, приливают 5 мл раствора молибдата аммония, перемешивают и оставляют на 10 мин. Добавляют 10 мл изобутанола, встряхивают 1 мин, нижний водный слой сливают и отбрасывают. Экстракт два раза промывают дистиллированной водой порциями по 10 мл. Воду сливают и отбрасывают, а затем круговыми вращениями воронки собирают капли воды и отделяют.
К раствору желтой фосфорномолибденовой кислоты в изобутаноле добавляют 10 мл раствора хлорида олова(II) и взбалтывают 1 мин. Отделяют водный слой, а изобутанольный слой сливают в сухой тонкий мерный цилиндр емкостью 10 мл. Воронку споласкивают 0,5 мл изобутанола и этим же растворителем доводят раствор в цилиндре до 10 мл. Раствор перемешивают и измеряют оптическую плотность.
Содержание фосфора находят по предварительно построенному графику.
III. 7.2. Определение фосфора с применением в качестве восстановителя оксалата олова
Этот метод не отличается от описанного в разделе III. 7.1, только в качестве восстановителя применяют [32] оксалат олова (II), а в качестве экстрагента — раствор диоктиламина в смеси (1:1) хлороформа и изоамилового спирта. Оксалат олова(II) является более устойчивым по сравнению с хлоридом олова(II). Кроме того, применение оксалат олова(II) приводит к образованию синей фосфорномолибденовой кислоты, характеризующейся более высоким максимумом светопоглощения. Оптическую плотность лучше измерять при 740 нм.
Чувствительность метода — 0,01 мкг фосфора в 5 мл неводного раствора.
Реактивы
Молибдат натрия. Растворяют 18 г Na2MoO4-2H2O в 1800 мл дистиллированной воды, затем вводят 48 г HCOONa-2H2O, после растворения прибавляют 21 мл 99%-ной муравьиной кислоты и 92 мл серной кислоты, разбавленной (1 : 1).
Оксалат олова(П). Растворяют 33 г Н2С2О4 2Н2О в 1900 мл дистиллированной воды, затем прибавляют 48 г HCOONa-2H2O, после растворения добавляют 21 мл 99%-ной муравьиной кислоты и 17 г SnC2O4. Перемешивают до полного растворения оксалата олова (II).
Фторид натрия, 1%-ный раствор. Хранят в полиэтиленовой посуде.
Диоктиламин, 0,5%-ный раствор в смеси (1:1) хлороформа и изоамилового спирта.
Ход анализа. Две равные порции исследуемого раствора, содержащие по 0,01—5 мкг фосфора, помещают в две делительные воронки емкостью 100 мл, если надо, разбавляют дистиллированной водой до 12,5 мл, а затем в одну воронку, раствор в которой служит анализируемым раствором, прибавляют 5 мл формиатного раствора молибдата натрия, а в другую воронку, раствор в которой служит холостым раствором, прибавляют 5 мл формиатного раствора оксалата олова. Растворы перемешивают в течение 5 мин, в первую воронку вводят 5 мл формиатного раствора оксалата олова, а в другую — с холостым раствором—5 мл формиатного раствора молибдата натрия. Растворы перемешивают, вводят по 12,5 мл раствора фторида натрия, перемешивают и добавляют по
10 мл концентрированной серной кислоты и по 5 мл раствора диок-тиламина, взбалтывают в течение 30 сек. Органические фазы сливают в центрифужные пробирки, центрифугируют в течение 10 мин и измеряют оптическую плотность анализируемого экстракта относительно холостого при 740 нм с толщиной слоя 1 см. Содержание фосфора находят по предварительно построенному калибровочному графику.
III. 7.3. Определение фосфора в стали
Навеску стали 0,1 г помещают в стакан емкостью 50 мл и растворяют при нагревании в 10 мл азотной кислоты, разбавленной (1 :2). Раствор выпаривают до малого объема, охлаждают, нейтрализуют аммиаком до начала выпадения гидроокисей, которые растворяют в нескольких каплях хлористоводородной кислоты, переносят в делительную воронку и анализируют, как указано в разделе III. 7.1.
III. 7.4. Определение фосфора в высоколегированной стали [35]
Навеску стали переводят в раствор, удаляют хром в виде хлористого хромила при выпаривании с хлорной кислотой, вольфрам отделяют в виде вольфрамовой кислоты, мышьяк — выпариванием с бромистоводородной кислотой, титан связывают аскорбиновой кислотой и после получения желтой фосфорномолибденовой кислоты экстрагируют ее изобутанолом. Затем в неводном растворе восстанавливают желтый комплекс до синего с помощью хлорида олова(II) и измеряют оптическую плотность неводного слоя.
Реактивы
Аскорбиновая кислота, 20%-ный раствор.
Ход анализа. Помещают 0,2 г пробы в стакан емкостью 50 мл и растворяют в 6 мл смеси (1:2) концентрированных азотной и хлористоводородной кислот. К раствору прибавляют 5 мл концентрированной НС1О.1, нагревают до начала выделения белого дыма и еще 1 мин. Выпавшие соли растворяют в 5 мл воды, прибавляют 5 мл 48%-ной бромистоводородной кислоты, выпаривают до белого дыма, а затем до образования солей или сиропообразного состояния. Соли растворяют в 5 мл воды, прибавляют 5 мл раствора аскорбиновой кислоты. Полученный раствор анализируют, как указано в разделе III. 7.1, вариант 2.
Если присутствуют ниобий, тантал и вольфрам, то после выпаривания раствора до начала выделения белого дыма хлорной кислоты остаток разбавляют водой до 30 мл и выпавший осадок отфильтровывают.
Метод позволяет определить до 4 • 10—4% фосфора.
4 Зак. 761 У7
III. 8. ОПРЕДЕЛЕНИЕ ФОСФОРА В ВИДЕ СОЛИ ФОСФОРНОМОЛИБДЕНОВОЙ КИСЛОТЫ С ОСНОВНЫМИ КРАСИТЕЛЯМИ
Наиболее чувствительным методом определения фосфора является экстракционный метод, в котором применена реакция образования соли фосфорномолибденовой кислоты с основными красителями. Этот метод еще удобен и тем, что в условиях определения фосфора небольшие количества кремния не образуют подобного соединения и не мешают определению фосфора. Если соединение молибдата с органическим красителем или сам краситель экстрагируется неводным растворителем, их можно отделить от соединения фосфоромолибдата с тем же красителем промывкой экстракта разбавленной азотной кислотой или раствором перманганата калия [36]. Молярные коэффициенты поглощения основных красителей достаточно высокие, часто достигают 105, а 1 моль фосфорномолибденовой кислоты может присоединять до 3 молей основного красителя, что приводит к резкому повышению чувствительности метода.
Характеристика соединений фосфорномолибденовой кислоты с некоторыми органическими красителями приведена в табл. 6.
Таблица 6. Сравнительная характеристика соединений фосфорномолибденовой кислоты с некоторыми органическими красителями (экстракция проведена при pH =1,3 [36]; отношение красителя и фосфорномолибденовой кислоты 2:1)
Краситель Экстрагент ^макс* нм
Отношение D холостого раствора к D исследуемого ратвора. %
Кристаллический фиолетовый ( Бутанол — циклогек- 582 8,1 < 1 *
санол (1:1) . Метилпропилкетон 582 5,0 < 1 *
Метиловый фио- Бутанол — циклогек- 582 6,1 < 1 *
летовы й санон (1 : 1) Метилпропилкетон 610 6,0 < 1 *
Циклогексанон 610 5,2 < 1 *
Малахитовый зе- Метилпропилкетон 610 7,3 < 1 *
леный Иодный зеленый Бутанол — циклогек- 610 3,1 4,4
санон (1 :1)
Аурамин Метилбутилкетон 440 4,6 < 1 *
Родамин 6Ж Метилпропилкетон 490 1,5 26,3
Нейтральный Диизопропилкетон 540 3,6 6,7
красный
’ Метилпропилкетон 490 3,9 45,4
Метилбутилкетон 490 3,5 48,0
Сафранин Ацетофенон — дихлорэтан (3:1) 490 1,8 71,8
Ацетофенон — бензол 490 3,5 44,3
(3:1)
* В анализируемом
растворе избыток красителя
разрушают перманганатом
калия.
E f(T5
0,8
0,Ь
и__________J----1__2-L
500 5W 580620 А, нм
Рис. 23. Спектр поглощения раствора соединения кристаллического фиолетового с фосфорномолибденовой кислотой в пропилацетате.
При получении комплекса в 0,5 и. азотной кислоте и экстракции пропилацетатом [37] молярный коэффициент поглощения для кристаллического фиолетового равен 2,7-105, а для малахитового зеленого 1,7-105. В этом случае, по-видимому, на I моль гетерополикислоты приходится несколько молей красителя.
Чувствительность реакции можно также повысить за счет экстракции тройного соединения малым количеством растворителя.
Образование тройного соединения происходит достаточно быстро, а неводные растворы устойчивы во времени, например в случае сафранинового комплекса, который был впервые применен для определения фосфора [38], оптическая плотность раствора уменьшалась в течение 1 ч всего на 0,02 единицы. Восстановленная фосфорномолибде-новая кислота также взаимодействует с сафранином Т с образованием интенсивно окрашенного продукта, который экстрагируется неводными растворителями [39]. Молярный коэффициент экстрактов комплекса невосстановленной фосфорномолибденовой кислоты с сафранином значительно выше, хотя значение этой величины подвергается сомнению [39], но спектр поглощения этого комплекса мало отличается от спектра поглощения простой соли сафранина. Преиму
щество применения восстановленного комплекса состоит в том, что
CL
спектр поглощения комплекса отличается от спектра поглощения простой соли сафранина в том же растворителе.
Мешающие вещества. Определению мешают окислители, восстановители и анионы, которые в этих условиях взаимодействуют с органическим красителем с образованием экстрагирующегося соединения. Определению мешает также мышьяк, образующий подобное соединение.
Измерение оптической плотности. Спектр поглощения раствора соединения кристаллического фиолетового с фосфорномолибденовой кислотой в пропилацетате представлен на рис. 23. Оптическую плотность следует измерять при 582 нм. В случае применения малахитового зеленого оптическую плотность измеряют при 610 нм.
Чувствительность метода,— 0,1 мкг фосфора в 5 мл экстракта.
III. 8.1. Определение фосфора с применением кристаллического фиолетового в отсутствие мешающих веществ
Сначала получают желтую фосфорномолибденовую кислоту и отделяют ее от избытка молибдата экстракцией пропилацетатом. Затем органический слой, содержащий фосфорномолибденовую кислоту, встряхивают с раствором красителя. При этом в слое
органического растворителя образуется окрашенное соединение фосфорномолибденовой сини с красителем. В качестве органического красителя можно применять кристаллический фиолетовый или малахитовый зеленый [37], иодный зеленый [36], бриллиантовый зеленый [40] и другие.
Реактивы
Кристаллический фиолетовый. Растворяют 0,041 г реактива в воде, переносят в мерную колбу емкостью 500 мл, разбавляют водой приблизительно до 400 мл, прибавляют 63 мл 5 н. HNO3 и доводят водой до метки.
н-Пропилацетат или н-бутилацетат.
Ацетон.
Стандартный раствор фосфата, содержащий 0,5 мкг фосфора в 1 мл, готовят путем разбавления раствора, приготовленного, как указано в разделе III. 5.1.
Молибдат аммония. Растворяют 24 г молибдата аммония в воде, прибавляют 50 мл 5 н. раствора HNO3 и разбавляют водой до 1 л.
Ход анализа. В делительную воронку помещают до 20 мл нейтрального исследуемого раствора, содержащего до 1 мкг фосфора, прибавляют 2,5 мл молибдата аммония, 2 мл 5 н. азотной кислоты, разбавляют водой до 25 мл и экстрагируют 5 мл пропилацетата или бутилацетата. Водную фазу отделяют и отбрасывают. Экстракт промывают 5 мл 0,5 н. HNO3; водную фазу после отделения отбрасывают. К органической фазе прибавляют 10 мл раствора красителя и встряхивают. Водную фазу отделяют и отбрасывают, а органическую фазу промывают 5 мл 0,5 н. HNO3. Органическую фазу отделяют в сухую пробирку, доводят ацетоном до 10 мл, перемешивают и измеряют оптическую плотность при 582 нм.
Содержание фосфора находят по предварительно построенному калибровочному графику, для построения которого используют стандартный раствор.
III. 8.2. Определение фосфора в виде комплекса восстановленного фосфоромолибдата с сафранином [39]
Фосфорномолибденовую гетерополикислоту восстанавливают аскорбиновой кислотой и получают соединение сафранина с восстановленным фосфоромолибдатом, которое экстрагируют метил-изобутилкетоном; содержание фосфора находят по значению оптической плотности экстракта.
Преимущество этого метода состоит в том, что максимум поглощения комплекса сдвинут в длинноволновую область по сравнению с максимумом поглощения свободного красителя или его комплексом с молибдатом, которые экстрагируются неводными растворителями. Поэтому избыток красителя и молибдата не мешает определению. Однако молярный коэффициент поглощения комплекса в данном случае ниже по сравнению с комплексом невосстановленной фосфорномолибденовой кислоты с сафранином.
Спектры поглощения раствора комплекса восстановленного фосфоромолибдата с сафранином в метилизобутилкетоне (кривая /)
Рис. 24. Спектры поглощения раствора комплекса восстановленного сафранинфосфоро-молибдата в метилизобутилке-тоне (/) и смеси (1:3) монохлорбензола и ацетофено-
и смеси (1:3) монохлорбензола и ацетофенона (кривая 2) представлены на рис. 24. Измерение оптической плотности следует проводить при 780—785 нм.
Реактивы
Молибдат аммония. Растворяют 0,3126 г (NHJeMoyOj^HjO в 250 мл би-днстиллята.
Сафранин Т, раствор с концентрацией 0,2 г/л.
Аскорбиновая кислота, 1%-ный раствор.
Ацетофенон, перегнанный (т. кип. 78 ЬС). Монохлорбензол, перегнанный (т. кип. 208 °C).
Метилизобутилкетон, ч. д. а.
Стандартный раствор фосфата. Раствор, содержащий 5 мкг/мл фосфора, готовят разбавлением раствора, указанного в разделе III.5.1.
Ход анализа. В калиброванную пробирку емкостью 25 мл помещают до 10 мл нейтрального исследуемого раствора, содержащего 0,05—0,6 мкг фосфора, прибавляют 5 мл серной кислоты, 2 мл раствора молибдата аммония и 0,5 мл раствора аскорбиновой кислоты. Раствор перемешивают и нагревают на кипящей водяной бане в течение 10 мин. Затем охлаждают, прибавляют 0,5 мл раствора сафранина Т, выдерживают при комнатной температуре 10 мин и взбалтывают 5 мин с 10 мл метилизобутилкетона или смеси (1:3) монохлорбензола с ацетофеноном. После разделения слоев органическую фазу центрифугируют и измеряют оптическую плотность прозрачного раствора относительно холостого экстракта в кюветах с толщиной слоя 1 см. Содержание фосфора находят по предварительно построенному калибровочному графику.
III. 8.3. Определение фосфора с бриллиантовым зеленым в нитрате натрия [40]
Получают желтую фосфорномолибденовую гетерополикислоту, экстракцией отделяют ее от избытка молибдена и получают соединение ее с сафранином Т в неводном растворе. Затем измеряют оптическую плотность.
Определению фосфора не мешают 1000-кратное по отношению к фосфору количество кремния, а также титан, тантал, ниобий, мышьяк, сурьма, олово, свинец, бор, индий, таллий, галлий, алюминий, кальций, магний, никель, марганец, медь, железо, ртуть и серебро, если их количества не превышают 250-кратного по
отношению к содержанию фосфора. Цирконий мешает определению, поэтому допустимо присутствие его в равных или только лишь несколько больших по отношению к фосфору количествах. Влияние циркония устраняют путем связывания его в комплекс с ЭДТА перед получением фосфорномолибденовой кислоты.
Оптическую плотность измеряют при 560 нм. Колориметрическое определение можно проводить с помощью стандартной шкалы.
Чувствительность метода — 5- 10-7%.
Реактивы
Бриллиантовый зеленый. Растворяют 0,1 г реактива в бутаноле, разбавляют бутанолом до 500 мл и перемешивают.
Молибдат аммония, 10%-ный водный раствор.
Этилендиаминтетрацетат натрия (ЭДТА), 0,1 М раствор.
Мочевина, 5%'-ный раствор.
Четыреххлористый углерод и бутиловый спирт, смесь (2:1).
Нитрат нлтрия. Для очистки от фоефора 1 кг препарата растворяют в 1110 мл воды и нагревают до комнатной температуры. Полученный насыщенный раствор нагревают до 60 °C, прибавляют 10 мл 0,1 М раствора железоаммонийных квасцов и аммиака до слабого запаха и оставляют на ночь. Осадок гидрата окиси железа, с которым соосаждается примесь фосфата, отфильтровывают через плотный фильтр.
Стандартная серия. Серию готовят одновременно с проведением анализа. Для приготовления серии стандартных растворов в делительные воронки вносят по 65 мл очищенного от фосфора раствора нитрата натрия, 11 мл воды и 0,0—0,02—0,4—0,6—0,8 мкг фосфора. Все полученные растворы обрабатывают, как указано в разделе «ход анализа».
Ход анализа. В делительную воронку емкостью 150—200 мл вносят 65 мл анализируемого раствора, содержащего 42—43 г нитрата натрия и до 0,8 мкг фосфора, доводят объем до 75 мл водой, добавляют 0,5 мл раствора ЭДТА, 8,3 мл 5 н. азотной кислоты, 5 мл раствора молибдата аммония и 3 мл раствора мочевины. После прибавления каждого реактива раствор тщательно перемешивают. Затем прибавляют 4 мл смеси ч^тыреххлористого углерода и бутилового спирта и содержимое воронки перемешивают (не встряхивать!) 2 мин. После разделения фаз органический слой сливают в сухую пробирку из бесцветного стекла, добавляют 0,3 мл раствора бриллиантового зеленого и перемешивают. Спустя 15—20 мин окраску раствора сравнивают с окраской серии стандартных растворов.
III. 8.4. Определение микроколичеств фосфора в никеле и его сплавах
Для определения фосфора в никеле и его сплавах [41] применен метод, описанный в разделе III. 8.3. Установлено, что вольфрам и рений при их содержании ^10% не мешают определению фосфора.
Реактивы
Перманганат калия, 4%-ный раствор.
Другие реактивы — указаны в разделе III. 8.3.
Калибровочный график. Для построения калибровочного графика готовят серию стандартных растворов нитрата никеля, очищенного от фосфора. Для этого растворяют 50 г металлического никеля в 300 мл 5 н. азотной кислоты, добавляют 15 мл нитрата марганца, 80 мл 4%-ного раствора КМпО4, нагревают до кипения и выдерживают на горячей плитке 15 мин. Отфильтровывают раствор в мерную колбу емкостью 1 л, доводят до метки водой и перемешивают. График строят в интервале 0,0—0,8 мкг фосфора.
Ход анализа. При анализе чистого никеля навеску его 1 г растворяют в смеси 10 мл HNO3 (пл. 1,4) и 2 мл раствора КМпО4. При анализе сплавов никеля с рением и никеля с вольфрамом 1 г сплава растворяют в смеси концентрированных азотной и хлористоводородной кислот (1:3) и 2 мл раствора 1<МпО4. Раствор упаривают до консистенции густого сиропа, прибавляют 50 мл 1 н. HNO3, по каплям перекись водорода до обесцвечивания и нагревают при 100 °C в течение 1 ч. При анализе сплавов никеля раствор после растворения навески упаривают досуха, добавляют 1 мл HNO3 (пл. 1,4) и снова упаривают. Эту операцию повторяют трижды, после чего прибавляют 50 мл 1 н. HNO3 и нагревают в течение 1 ч. Затем раствор нейтрализуют едким натром до pH = — 6—7. Если раствор имеет бурый цвет или содержит осадок, прибавляют по каплям перекись водорода до полного обесцвечивания.
Нейтрализованный раствор разбавляют 20 мл воды, прибавляют 1 мл 0,1 М раствора ЭДТА, 10 мл HNO3, разбавленной (1:1), 5 мл раствора молибдата аммония, 3 мл раствора мочевины и содержимое перемешивают. Раствор доводят водой до 50 мл, переносят в делительную воронку, добавляют 2 мл бутилового спирта и энергично встряхивают для растворения бутанола. Затем добавляют 10 мл смеси четыреххлористого углерода и бутанола (2: 1), перемешивают в течение 2 мин, не встряхивая. Органический слой сливают в сухую пробирку, не допуская попадания в него воды.
Отбирают 8 мл экстракта и добавляют 0,6 мл бриллиантового зеленого. Спустя 10 мин измеряют оптическую плотность раствора при 560 нм.
При определении более 1 мкг фбсфора количество бутилового спирта уменьшают до 1 мл, а количество смеси для экстракции гетерополикислоты—до 1,5 мл. Для фотометрирования в этом случае отбирают 1 мл, к которому добавляют 0,1 мл бриллиантового зеленого. Параллельно проводят холостой опыт.
III. 9. ЭКСТРАКЦИОННОЕ ОТДЕЛЕНИЕ И ОПРЕДЕЛЕНИЕ ФОСФОРА
В ПРИСУТСТВИИ КРЕМНИЯ И МЫШЬЯКА
Определению фосфора в виде гетерополикислоты мешает мышьяк, а во многих случаях и кремний. Метод количественного разделения фосфорной, мышьяковой и кремневой кислот экстрак
цией в виде их гетерополимолибденовых комплексов разработал Р. И. Алексеев [42]. Сначала все три компонента обработкой раствором молибдата аммония в кислой среде переводят в гетерополикислоты. Затем экстрагируют смесью бутилового спирта и хлороформа; эта смесь извлекает только фосфорномолибденовую кислоту. К оставшемуся водному раствору прибавляют смесь бутилового спирта и этилацетата, при этом извлекается кремнемолибденовая и мышьяковомолибденовая гетерополикислоты. Если к этому раствору прибавить хлороформ, то кремнемолибденовая кислота переходит в водную фазу и может быть определена или в водной фазе, или в экстракте бутилового спирта после подкисления до получения 0,8 н. раствора кислоты.
Ортофосфат в присутствии пирофосфата можно определить аналогичным путем, экстрагируя фосфорномолибденовую кислоту смесью бутилового спирта и хлороформа [43].
Описанный метод широко применяется при анализе различных материалов [44—48]. Этим методом можно определить фосфор в присутствии 4 мг мышьяка, 5 мг кремния и 1 мг германия. Преимущество метода состоит также в том, что при экстракции фос-форномолибденовая гетерополикислота отделяется от избытка молибдата и измерение оптической плотности можно проводить в УФ при 310 нм, где наблюдается максимальное поглощение фосфорно-молибденового комплекса.
Мешающие вещества. Определению фосфора не мешают ионы аммония, натрия, калия, лития, магния, стронция, бария, бериллия, кадмия, кальция, хрома(Ш), кобальта, меди(П), марганца (II), никеля, ртути (II), а также анионы — ацетат, борат, бромид, хлорид, иодат, иодид, нитрат и селенит. Ионы золота(III), висмута, бихромата, свинца, нитрита, роданида, тиосульфата, тория, уранила и цирконила должны отсутствовать. Могут присутствовать в количестве до 1 мг ионы фторида, перйодата, перманганата, ванадата и цинка. Наличие алюминия, железа(III) и вольфрамата не должно превышать 10 мг в пробе.
Измерение оптической плотности. Наибольшая чувствительность наблюдается при измерении оптической плотности при 310 нм.
Для колориметрического определения можно применять метод стандартной шкалы.
III. 9.1. Определение фосфора экстракцией фосфорномолибденовой кислоты
Реактивы
Молибдат натрия. Навеску двуводного молибдата натрия Na2MoO<-2H2O 7,5 г растворяют в 200 мл воды, добавляют 100 мл концентрированной хлористоводородной кислоты и разбавляют до 500 мл. Раствор сохраняют в полиэтиленовой посуде.
Экстрагент. Смешивают 100 мл бутанола и 400 мл хлороформа.
Стандартный раствор фосфата. Раствор готовят, как указано в разделе III. 5.1, и путем разбавления получают рабочий раствор, содержащий в 1 мл 20 мкг фосфора. Хранят раствор в закрытом полиэтиленовом сосуде.
Ход анализа. До 10 мл нейтрального исследуемого раствора, содержащего до 20 мкг фосфора, помещают в делительную воронку емкостью 100 мл и прибавляют 10 мл раствора молибдата натрия. Раствор перемешивают и оставляют на 5 мин. Затем прибавляют 5 мл экстрагента, встряхивают в течение 1 мин, дают фазам разделиться и нижний слой сливают в сухой тонкий мерный цилиндр емкостью 10 мл. К водной фазе прибавляют еще 5 мл экстрагента и экстракцию повторяют. Вторую порцию экстракта переносят в мерный цилиндр, доводят до 10 мл экстрагентом, перемешивают и измеряют оптическую плотность раствора при 310 нм.
Содержание фосфора находят по предварительно построенному калибровочному графику.
III. 9.2. Определение фосфора в стали
Навеску стали 0,2 г растворяют при нагревании в 20 мл азотной кислоты, разбавленной (3:2), прибавляют 1—2 мл концентрированной азотной кислоты и нагревают до тех пор, пока раствор не станет прозрачным. Упаривают до удаления окислов азота. К охлажденному раствору прибавляют раствор аммиака до появления слабой мути, которую растворяют прибавлением нескольких капель азотной кислоты. Раствор переносят в делительную воронку и продолжают определение фосфора, как указано в разделе III. 9.1.
III. 10. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ФОСФОРА
Среди других методов определения фосфора необходимо упомянуть метод, основанный на экстракции пропилацетатом или смесью циклогексанона и бензола (1:2) фосфоромолибдата окси-хинолиния в присутствии избытка оксихинолина и молибдата [49]. Определение ведут или по светопоглощению суспензии образующегося соединения с молярным коэффициентом (е), равным 2,4-104, или по светопоглощению красителя, образующегося по реакции между оксином и диазотированной сульфаниловой кислотой, молярный коэффициент поглощения в пересчете на фосфор при этом равен 5,4-104 при 490 нм.
Разработан косвенный метод определения пирофосфата в присутствии фосфата [50]. Для этого пирофосфатный комплекс железа экстрагируют 0,1 М хлороформным раствором н-додециламина и определяют железо в виде сульфосалицилата. Метод позволяет определить ц-10-4 г-ион/л пирофосфата с точностью ±15 отн. %.
Показано, что фосфат в присутствии молибдата и железа(III) образует тройной окрашенный комплекс [51]. Эта реакция применена в новом фотометрическом методе определения фосфора в виде желтого тройного комплекса.
Наиболее чувствительным методом определения фосфора является косвенный метод, основанный на получении фосфорномо-либденового комплекса, экстракции его, реэкстракции буферным раствором с высоким значением pH и определении молибдена, связанного в гетерополикислоту. Молибден можно определять непосредственным спектрофотометрированием молибдата при 230 нм или любым другим фотометрическим методом определения молибдена, например с 2-амино-4-хлорбензолтиолом [52], хром фиолетовым К, хром черным специальным [53], с фенилфлуороном [54] или роданидным методом [55].
Молярные коэффициенты поглощения растворов некоторых соединений фосфора, применяемых для фотометрического определения его, приведены в табл. 7. По значению молярных коэффициентов поглощения можно характеризовать чувствительность методов.
Для определения микроколичеств фосфора в тонких магнитных пленках применен экстракционный метод, в котором экстрагируют соединения фосфорномолибдата с кристаллическим фиолетовым [56].
Таблица 7. Значения молярных коэффициентов поглощения растворов некоторых соединений фосфора, применяемых для фотометрического определения его [39]
Соединение Среда X, нм е
Фосфорнованадиевомолибдено- Водный раствор 460 560
вая кислота Желтая фосфорномолибденовая Водный раствор 345 700
кислота Бутанол — хлороформ 310 24 400
Синяя фосфорномолибденовая (1:4) Водный раствор 830 26 800
кислота Молибдат после разрушения фос- Водный раствор с рН = 9 230 57 400
форномолибденовой кислоты Молибдат после разрушения Водный pacTBOp+SnCl2 475 185 000
фосфорномолибденовой кисло-ты+NCS" Комплекс невосстановленного Дихлорбензол — ацето- 532 190 000
фосфоромолибдата с сафранином Комплекс восстановленного фос- фенон (1: 3) Дихлорбензол — ацето- 780 33 500
форомолибдата с сафранином фенон (1 :3) Метилизобутилкетон 785 33 000
Для определения фосфора в почве [57], удобрениях [58], медных сплавах, содержащих олово [59], высоколегированных сталях [60] и в медных сплавах [61] используется образование желтого фосфорнованадиевомолибденового комплекса. При анализе сплавов, содержащие олово, увеличивают концентрацию хлористоводородной кислоты для уменьшения гидролиза солей олова. При ана
лизе сталей, высоколегированных хромом, его предварительно удаляют отгонкой в виде хлористого хромила. Для отделения меди при определении фосфора в медных сплавах применяют катионит.
Описан метод определения фосфора в виде желтого фосфорно-молибденового комплекса в почве и растениях [62] и в водном аммиаке особой чистоты [63]. Для уменьшения диссоциации комплекса рекомендовано применять ацетоновые растворы [64]. При определении фосфора в феррониобии, ферротитане и в ниобиевой руде [65] титан и ниобий маскируют фторидом, а фосфорномолибденовую кислоту экстрагируют метилизобутилкетоном.
При определении фосфора в моторном бензине фосфорномолибденовую кислоту экстрагируют смесью бутанола и хлороформа [66]. Для определения больших количеств фосфора в двойном суперфосфате в виде желтого фосфорномолибденового комплекса применяют дифференциальную фотометрию [67].
Наиболее часто для определения фосфора фосфорномолибде-новый комплекс восстанавливают, применяя различные восстановители и разные методы отделения мешающих ионов; синий комплекс экстрагируют органическими растворителями, а нередко определение проводят без экстракции этого комплекса. Так, при определении фосфора в материалах с высоким содержанием вольфрама [68] и в металлическом хроме [69] рекомендовано предварительно отделить фосфор осаждением его в виде фосфата кальция в щелочной среде, а затем определять фосфор в виде синего комплекса.
Во многих случаях применяют восстановление с помощью хлорида олова(II). Так, рекомендован метод определения фосфора в сталях, высоколегированных хромом и никелем [70], в золе твердого топлива [71] и в почве [72]. Однако применение в качестве восстановителя хлорида олова(II) без предварительной экстракции желтого комплекса или без экстракции восстановленного комплекса приводит к большим ошибкам определения фосфора.
Хорошие результаты получены при восстановлении смесью хлорида олова(II) и сернокислого гидразина [73—80]. Для восстановления желтой фосфорномолибденовой кислоты применяют также сернокислый гидразин. При определении фосфора в присутствии нитрат-ионов [76] и в быстрорежущих и нержавеющих сталях [77] для восстановления фосфорномолибденовой кислоты применяют сернокислый гидразин [94], а также аскорбиновую кислоту [78, 79], хлорид титана(III) [80], ферроцен [81], соль Мора [82—85]. Для определения фосфора в виде синей фосфорномолибденовой кислоты рекомендовано применение смешанного реагента, содержащего молибден(V) и молибден(VI) в отношении 2:3 [86].
Лучшие результаты получаются с применением экстракции [87, 88]. Этот метод применен для определения фосфора в присутствии больших количеств ванадия [89], в водяном паре [90], в мышьяке и трехокиси мышьяка [91]. Мышьяк предварительно отделяют отгонкой или экстракцией галогенида мышьяка четыреххлористым
углеродом. Метод дает возможность определить 1 • 10-5% фосфора. При определении фосфора в пятиокисях ниобия и тантала рекомендовано применять в качестве восстановителя сернокислый гидразин и хлорид олова(II) [92]. При определении фосфора в органических соединениях проводят окисление перманганатом в сернокислой среде, получают желтую фосфорномолибденовую кислоту, которую восстанавливают метолом и бисульфитом [93].
Разработаны методы определения фосфора в углях и коксе [95, 96], в почве [97] и других материалах [98] с применением в качестве восстановителя сульфита или смеси сульфита и аминонафтолсульфокислоты [97]. С целью определения фосфора в биологических материалах в качестве восстановителя применяют метол [99], а в метилтрихлорсилане — гидразин [100]. В последнем случае кремний отгоняют в виде тетрафторида, а мышьяк в виде AsCU Мягким восстановителем является тиомочевина [101], которая рекомендована при определении фосфора в присутствии вольфрама, титана и ниобия [102]. Как отмечалось выше, лучшим восстановителем является аскорбиновая кислота [103, 104]. В качестве катализатора в этом случае рекомендовано применять антимонилтартрат калия [105]. Применение аскорбиновой кислоты рекомендовано при определении до 10“8% фосфора в четыреххлористом германии [106]. Германий предварительно отделяют экстракцией четыреххлористым углеродом.
Экстракция желтой фосфорномолибденовой кислоты с последующим ее восстановлением применена для определения фосфора в олове высокой чистоты [107], окиси свинца [108], алюминии, медных сплавах, белых металлах и сталях [109], сплавах алюминия и кремния [НО], плавиковом шпате [111], воздухе [112] и других материалах [113—115].
Изучено влияние мышьяка на определение фосфора [116]. При этом показано, что применение предварительного восстановления мышьяка(V) до мышьяка(III) дает удовлетворительные результаты.
Изучение реакции восстановления фосфоромолибдата сульфитом и солью Мора показало, что ионы меди(II) катализируют эту реакцию [117]. Проведено изучение влияния некоторых комплексо-образователей на восстановление фосфорномолибденовой кислоты [34]. При этом показано, что щавелевая, винная и лимонная кислоты, а также ЭДТА сильно влияют на полноту восстановления гетерополикислоты, хотя и не изменяют характера восстановления. Изучению экстракции фосфорно- и кремнемолибденовых кислот бутанолом посвящена работа [118], в которой показано, что в разбавленных растворах возможно разделение фосфора и кремния путем экстракции их гетерополикислот бутанолом.
Для определения фосфора широкое распространение получила реакция образования фосфорованадиевой гетерополикислоты. Исследование состава этого комплекса показало, что отношение ванадия к фосфору в комплексе равно 1 : 12 [119]. Этот метод применен 108
для определения фосфора в конденсированных фосфатах [120], сплавах алюминия, меди и никеля и белых металлах [121], ниобии, цирконии, титане и вольфраме [122], сталях и сплавах [123], а также в чугунах [124]. В последнем случае применен дифференциальный способ без взятия навески (бесстружковый вариант метода).
Экстракция фосфора в виде ионного ассоциата с кристаллическим фиолетовым применена при определении фосфора в силуминах [125] и в металлических производственных материалах [126]. Ионные ассоциаты фосфора с хинином [127] и пропиленкар-бонатом [128] экстрагируются хлороформом и применяются для определения фосфора. Для определения микроколичеств фосфора применен фотометрический вариант кинетического метода получения фосфорномолибденовой сини при восстановлении аскорбиновой кислотой [129].
111.11. МЕТОДЫ ОПРЕДЕЛЕНИЯ КРЕМНИЯ
В существующих методах определения кремния использовано образование желтого кремнемолибденового комплекса H4[Si (Mo3Oia)4]-xH2O. Кремний определяют или непосредственно в виде этого комплекса, или после восстановления его до синего комплекса. Состав желтого комплекса, по И. В. Тананаеву [130], отвечает отношению [Mo]: [Si] = 12 : 1. Как отмечалось в разделе III. 1, желтая кремнемолибденовая кислота существует в. а- и p-формах. Устойчивым является только a-комплекс. Образование этого комплекса происходит при pH = 2,3—3,9 при нагревании. р-Комплекс образуется в более кислой среде (pH = 1,5—1,7, по другим данным,— 1,5—2,0) и постепенно переходит в а-комплекс, который окрашен слабее. Поэтому, определяя кремний, необходимо строго придерживаться одинаковых условий проведения реакции (pH и времени) как при построении калибровочного графика, так и при проведении определения. Восстановление a-формы сопровождается образованием восстановленных продуктов двух типов в зависимости от количества восстановителя [131]. Так, в случае применения в качестве восстановителя хлорида олова (II) добавление менее 4 г-экв SnCb на 1 моль a-кремнемолибденовой кислоты приводит к образованию продукта голубого цвета, спектр поглощения которого имеет два максимума при 630 и 740 нм, — a-синь I. Если прибавить 4 г-экв и более восстановителя, то образуется синий продукт, спектр которого имеет размытый максимум при 730— 740 нм, — a-синь II. Этот продукт устойчив на воздухе в течение 7—10 ч, после чего спектр поглощения его изменяется с появлением максимума при 630 нм, т. е. спектр становится сходным со спектром a-сини I. Процесс перехода a-сини II в a-синь I ускоряется при подкислении раствора.
Восстановление желтых комплексов до синей практически не зависит от кислотности в интервале концентраций от 0,01 до 7 н. серной кислоты. Разные восстановители по-разному восстанавливают
желтые кремнемолибденовые кислоты. Сильные восстановители (двувалентное олово, сернокислый гидразин и др.) образуют два или большее число продуктов восстановления, а более слабые восстановители (метол, аскорбиновая кислота, эйконоген и др.) образуют один продукт (табл. 8).
Таблица 8. Максимумы светопоглощения водных и неводных растворов продуктов восстановления кремнемолибденовой кислоты
Восстановитель Вид сини ^"макс
водный раствор экстракт в бутиловом спирте
SnC2O4 a-синь I 630 и 740 630 и 720
а-синь II 740 630 и 720
0-синь 800 630 и 720
(при 1 н. H2SO4)
780
(при 3 н. H2SO4)
SnCl2 а-синь I 630 и 740 630 и 720
а-синь II 740 630 и 720
0-синь 740 630 и 720
n2h4 . H2SO4 а-синь 790 720
0-синь 825 805
Аскорбиновая кислота а-синь 780 720
0-синь 800 780
Метол 0-синь 805 800
Эйконоген а-синь 720 720
0-синь 805 800
Лучшими восстановителями кремнемолибденовой гетерополикислоты являются ЭХТ-кислота (эйконоген) и аскорбиновая кислота [132]. Эйконоген (ЭХТ-кислота, 1-амино-2-нафтол-4-сульфокислота)
NH2
SO3H
представляет собой мелкие белые или серые кристаллы мало растворимые в воде и спирте. Щелочные растворы ЭХТ-кислоты на воздухе быстро окрашиваются в коричневый цвет.
Аскорбиновая кислота
—°—
С—С=С—С—С—СН2ОН
(Ч (Ihohh он
белое кристаллическое вещество, хорошо растворимое в воде. В твердом состоянии устойчива, в водных растворах постепенно окисляется, поэтому применяют свежеприготовленные растворы.
Кроме того, как было отмечено выше, для восстановления кремнемолибденовой гетерополикислоты применяют ряд других восстановителей, в том числе сульфит натрия, сульфат железа(II), тиомочевину, оксалат олова(II) и хлорид олова(II). Необходимо всегда помнить, что в зависимости от применяемого восстановителя получаются синие кремнемолибденовые гетерополикомплексы, спектры поглощения которых могут довольно сильно отличаться.
При восстановлении p-кремнемолибденовой гетерополикислоты образуется p-синь, свойства которой отличаются от свойств а-сини, т. е. от продуктов восстановления а-кремнемолибденовой гетерополикислоты.
Оптимальные условия восстановления желтых кислот до синей разные для разных восстановителей. В связи с этим необходимо строго придерживаться условий проведения определения, указанных в методиках.
Восстановленные кремнемолибденовые гетерополикислоты устойчивы к действию минеральных кислот в концентрации до 7 н., что дает возможность определять кремний в присутствии фосфора, но при высокой кислотности частично может восстанавливаться молибден, не связанный в кремнемолибденовую кислоту.
Как было отмечено выше, желтая кремнемолибденовая гетерополикислота является малопрочным комплексом. Поэтому при проведении определения необходимо вводить большой избыток молибдата аммония для практически полного переведения кремневой кислоты в желтый кремнемолибдат.
Мешающие вещества. Определению мешают большие количества фторида, образующего тетрафторид кремния и кремнефтористоводородную кислоту, а также цирконий, титан и олово(IV), которые в условиях образования кремнемолибденовой гетерополикислоты гидролизуют и увлекают кремневую кислоту в осадок. Кроме того, определению кремния мешают анионы, образующие с молибдатом гетерополикислоты (фосфаты, арсенаты, германаты, ванадаты).
Вредное влияние фторидов устраняют прибавлением борной кислоты (20 частей на 1 часть F-) или солей алюминия. При этом фторид образует борофторидный или фторидный комплекс алюминия. Мешающее влияние высокозаряженных ионов устраняют прибавлением фторида. При этом образуются прочные растворимые комплексы титана, циркония и других металлов, а избыток фторида связывают борной кислотой.
Фосфор и мышьяк можно отделить экстракцией их гетерополикислот неводными растворителями. Так, при pH = 1,2 фосфор-номолибденовая и мышьяковомолибденовая гетерополикислоты экстрагируются этилацетатом. При большей кислотности экстрагируется также кремнемолибденовая гетерополикислота (рис. 25).
ние. 25. Зависимость экстракции (в %) фосфорномолибденовой (/), мышьяковомолибде-новой (2) и кремнемолибденовой (3) гетерополикислот этилацетатом от pH.
Это разделение не очень точное, но дает возможность быстро отделить фосфор от кремния. При этом кремнемолибденовая гетерополикислота остается в водной фазе и может быть определена одним из описанных ниже методов.
При анализе различных материалов, как правило, мышьяк улетучивается в виде галогенида при переведении пробы в раствор и не мешает дальнейшему определению. Фосфор лучше отделить путем экстракции в виде гетерополикислоты, так как в отсутствие фосфора получаются более точные результаты определения кремния. Однако можно определить кремний с достаточной точностью и в присутствии довольно больших количеств фосфора, мышьяка и германия, так как фосфорномолибде-новая, мышьяковомолибденовая и гер-маниевомолибденовая кислоты практически полйостью разрушаются при 1,6—3,2 н. кислотности, а кремнемолибденовый комплекс в этих условиях (до 7 н. H2SO4) не разрушается. Образование желтого кремнемолибденового комплекса происходит при pH = = 1,5—2.
В некоторых случаях необходимо предварительное отделение кремния или мешающих компонентов. Для этого применяют дегидратацию кремневой кислоты, экстракцию мешающих ионов, ионный обмен, электролиз на ртутном катоде, отгонку некоторых металлов в виде хлоридов, а также отгонку кремния в виде тетрафторида и кремнефтористоводородной кислоты.
Методы измерения оптической плотности. Спектры поглощения желтого кремнемолибденового комплекса и молибдата аммония приведены на рис. 26, а на рис. 27 приведен спектр поглощения синей (кремнемолибденовой) кислоты, полученной восстановлением аскорбиновой кислотой. Из этих рисунков следует, что оптическую плотность желтого кремнемолибденового комплекса необходимо измерять при 350—400 нм, а синего кремнемолибденового комплекса — при 720— 815 нм. Метод шкалы можно применять, используя имитирующие растворы и стекла.
Чувствительность метода. Молярный коэффициент поглощения желтого комплекса при 300 нм (езоо = 2-1О4) примерно равен молярному коэффициенту поглощения синего комплекса при 800— 820 нм (seis = 2-104). Однако при длине волны 300 нм определе-
нию кремния в значительной мере мешает избыток молибдата, а также некоторые посторонние ионы, такие как нитрат, железо(III) и др. Поэтому оптическую плотность раствора желтого комплекса измеряют при 350 нм или больше, хотя езбо = 7• 103, а при большей длине волны молярный коэффициент еще меньше. После экстракции желтого комплекса неводными растворителями, т. е. после отделения его от избытка молибдата и посторонних ионов, оптическую плотность лучше измерять при максимальном поглощении X = 300 нм.
Рис. 26. Спектры поглощения желтой кремнемолибденовой кислоты (/) и молибдата аммония (2).
Рис. 27. Спектр поглощения синего кремнемолибденового комплекса, полученного при восстановлении аскорбиновой кислотой.
Метод определения кремния в виде желтого и синего кремнемолибденовых комплексов дает возможность определить ~2,5 мкг в 25 мл конечного раствора. Применение экстракции желтого и синего комплексов повышает чувствительность метода.
III. 11.1. Определение кремневой кислоты в отсутствие мешающих веществ
III. 11.1.1. Определение в виде желтого кремнемолибденового комплекса
Реактивы
Стандартный раствор кремневой кислоты. Растворяют 1,596 г кремнефторида натрия в воде и переносят в мерную колбу емкостью 1 л. Разбавляют водой до метки, хорошо перемешивают и для сохранения раствор переносят в бутылку из полиэтилена. 1 мл этого раствора содержит 0,5 мг SiO2. Перед употреблением готовят путем разбавления раствор, содержащий 0,05 мг SiO2 в 1 мл.
Стандартный раствор можно приготовить и так. Навеску чистой кремневой кислоты сплавляют с пятикратным количеством безводной соды. Полученный
плав выщелачивают водой, прибавляют 3—5 г щелочи и разбавляют до 1 л. Путем разбавления этого раствора 1%-ным раств.ором карбоната натрия готовят раствор, содержащий 0,05 мг SiCh в 1 мл, а при анализе чугунов — 0,02 мг в 1 мл. Растворы хранят в полиэтиленовых бутылках.
Молибдат аммония, 5%-ный раствор. Препарат молибдата аммония растворяют в воде и осаждают спиртом, прибавляя его в количестве */з от общего объема. Переосаждение повторяют дважды, после чего готовят 5%-ный раствор, который хранят в полиэтиленовой посуде.
Ход анализа. Пробу 20 мл испытуемого нейтрального раствора, содержащего от 0,02 до 0,5 мг SiOs, помещают в мерную колбу емкостью 25 мл, прибавляют 1,5 мл 2 н. раствора серной кислоты, 1,5 мл раствора молибдата аммония, разбавляют водой до метки и хорошо перемешивают. По истечении 10 мин измеряют интенсивность окраски. По полученным данным находят содержание кремния по калибровочному графику, который строят, пользуясь стандартным раствором.
Можно также экстрагировать желтый кремнемолибденовый комплекс бутиловым спиртом и измерять оптическую плотность экстракта при 350 нм.
III. 11.1.2. Определение кремневой кислоты в виде синего кремнемолибденового комплекса в отсутствие мешающих веществ с применением в качестве восстановителя ЭХТ-кислоты
К слабокислому раствору кремневой кислоты прибавляют молибдат аммония, после образования желтого кремнемолибденового комплекса восстанавливают его до синего кремнемолибденового комплекса и измеряют интенсивность окраски.
Реактивы
Стандартный раствор кремневой кислоты — см. раздел III. 11.1.1.
1-Амино-2-нафтол-4-сульфокислота, 0,25%-ный раствор, содержащий 15 г бисульфита натрия и 0,5 г сульфита натрия в 100 мл.
Молибдат аммония. Растворяют 7,5 г крупнокристаллического молибдата аммония в воде, добавляют 9 мл концентрированной серной кислоты и разбавляют до 100 мл.
Винная или щавелевая кислота, 10%-ный раствор.
Ход анализа. В мерную колбу емкостью 25 мл помещают до 20 мл испытуемого нейтрального раствора, содержащего до 20 мкг кремния, разбавляют, если нужно, до 20 мл, добавляют 0,5 мл молибдата аммония, а спустя 5 мин добавляют 1 мл винной (щавелевой) кислоты и раствор перемешивают. Затем прибавляют 0,5 мл раствора восстановителя, разбавляют до метки водой и перемешивают. Спустя 20 мин измеряют оптическую плотность при 815 нм относительно раствора сравнения, который готовят одновременно и аналогично, но без прибавления раствора, содержащего кремневую кислоту.
Содержание кремния находят по предварительно построенному калибровочному графику.
111.11.1.3. Определение кремневой кислоты в виде синего комплекса при восстановлении аскорбиновой кислотой
Реактивы
Аскорбиновая кислота, 1%-ный раствор.
Другие реактивы — указаны в разделе III. 11.1.2.
Ход анализа. В мерную колбу емкостью 25 мл помещают 20 мл испытуемого нейтрального раствора, содержащего до 20 мкг кремния, и устанавливают pH раствора 1—2. Затем прибавляют 1 мл молибдата аммония, оставляют на 10 мин, к раствору приливают 2 мл 8 н. серной кислоты, 1 мл аскорбиновой кислоты, доводят объем до метки и перемешивают. Через 20 мин измеряют оптическую плотность по отношению к раствору сравнения при 800 нм и находят содержание кремния по предварительно построенному калибровочному графику.
III. 11.1.4. Определение кремневой кислоты
в виде синего комплекса при восстановлении солью Мора
Реактивы
Соль Мора. Растворяют 6 г соли в 100 мл воды, содержащей 1 мл H2SO4, разбавленной (1 :25).
Другие реактивы — указаны в разделе III. 11.1.2.
Ход анализа. Переносят в мерную колбу емкостью 25 мл до 15 мл нейтрального испытуемого раствора, содержащего до 40 мкг кремния, прибавляют 3 мл раствора молибдата аммония и оставляют на 10 мин, затем приливают 3 мл раствора щавелевой кислоты, перемешивают и сразу добавляют 3 мл раствора соли Мора, разбавляют до метки и перемешивают. Аналогично готовят раствор сравнения из раствора железа, не содержащего кремния.
Оптическую плотность раствора измеряют спустя 10 мин при 800 нм и по предварительно построенному калибровочному графику находят содержание кремния.
III. 11.1.5. Определение кремневой кислоты в виде синего комплекса при восстановлении хлоридом олова(П)
Реактивы
Хлорид олова(П), 0,5%-ный раствор.
Другие реактивы — указаны в разделе III. 11.1.2.
Ход анализа. До 15 мл испытуемого нейтрального раствора, содержащего до 50 мкг кремневой кислоты, помещают в мерную колбу емкостью 25 мл, прибавляют 0,5 мл 5 н. H2SO4 и 2 мл раствора молибдата аммония. Раствор перемешивают и оставляют на
10 мин для полного образования желтого кремнемолибденового комплекса. Затем прибавляют небольшими порциями 2 мл 8 н. H2SO4. Разбавляют до 22 мл и прибавляют по каплям 2 мл раствора хлорида олова(II). Раствор разбавляют водой до метки, перемешивают и через 5 мин измеряют оптическую плотность при 800 нм по отношению к раствору сравнения, который готовят аналогично, только без введения кремневой кислоты.
Содержание кремния находят по предварительно построенному калибровочному графику.
III. 11.1.6. Определение кремневой кислоты в виде синего комплекса с применением экстракции [32]
Реактивы
Стандартный раствор кремневой кислоты, см. раздел III. 11.1.1.
Молибдат аммония, 10%-ный водный раствор. Хранят в полиэтиленовой посуде.
Оксалат олова(П), формиатный раствор. В 190 мл дистиллированной воды последовательно растворяют: 3,3 г Н2С2О4-2Н2О; 4,8 г HCOONa-2H2O; 2,1 мл 99%-ной НСООН; 1,7 г SnC2O4 и перемешивают.
Экстрагент, 0,5%-ный раствор диоктиламина (ДОА) в смеси хлороформа с изоамиловым спиртом (1 : 1).
Ход анализа. Две равные порции исходного нейтрального раствора (1—6 мкг кремния) помещают в две делительные воронки емкостью 100 мл; разбавляют водой до 30 мл, прибавляют в одну воронку (анализируемый раствор) 5 мл раствора молибдата аммония, а в другую воронку (раствор сравнения) —5 мл H2SO4, перемешивают в течение 5 мин и в воронку с исследуемым раствором вводят 5 мл H2SO4, разбавленной (1:3), а в воронку с раствором сравнения — 5 мл раствора молибдата аммония, перемешивают, в обе воронки прибавляют по 5 мл формиатного раствора оксалата олова, снова перемешивают, вводят по 5 мл экстрагента и взбалтывают в течение 30 с. Затем сливают органические фазы в сухие пробирки, центрифугируют 10 мин и измеряют оптическую плотность анализируемого экстракта относительно раствора сравнения при 740 нм и толщине слоя в кювете 1 см.
Содержание кремния находят по предварительно построенному калибровочному графику.
III. 12. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ КРЕМНИЯ
III. 12.1. Определение кремния в стали и чугуне
Навеску стали растворяют в смеси разбавленной азотной кислоты с перекисью водорода, после чего получают кремнемолибденовый комплекс, который восстанавливают до молибденовой сини [133, 134]. При анализе высоколегированных сталей навеску металла сначала обрабатывают смесью (1:1) хлористоводородной и
азотной кислот, а затем содержимое колбы количественно переносят в фторопластовую чашку (можно в платиновую), в которую предварительно помещают 10%-ный раствор щелочи с таким расчетом, чтобы после нейтрализации кислоты оставался избыток щелочи. Щелочную жидкость нагревают в течение 3—4 мин, нейтрализуют избыток щелочи до слабощелочной среды азотной кислотой, переносят в мерную колбу, подкисляют до кислой среды и доводят до метки водой. В аликвотной части определяют кремний [135]. В некоторых случаях сталь растворяют в смеси серной кислоты и перекиси водорода. Для разрушения фосфорномолибдено-вого комплекса применяют щавелевую кислоту или оксалат аммония [136—138] или сильно повышают кислотность после образования гетерополикислот.
Реактивы
Солянокислый гидроксиламин, 1%-ный раствор.
Железоаммонийные квасцы, нулевой раствор. Навеску 216 г железоаммонийных квасцов растворяют при нагревании в воде, в которую прибавлено 5 мл серной кислоты (пл. 1,84), и разбавляют до 1 л водой. Этот раствор содержит 25 мг Fe в 1 мл.
Хлорид олова(П), 0,5%-ный раствор.
Пергидроль.
Молибдат аммония, 5%-ный раствор.
Калибровочный график. Для построения калибровочного графика применяют стандартный раствор, содержащий 0,2 мг кремния в 1 мл, который обрабатывают так же, как при анализе чугуна, только вместо навески чугуна берут 4 мл раствора железоаммонийных квасцов и добавляют 1 мл 4%-ного раствора двузамещенного фосфата натрия.
Ход анализа стали [139]. Навеску стали 0,1 г помещают в коническую колбу емкостью 100 мл, прибавляют 5 мл предварительно нагретой на водяной бане азотной кислоты, разбавленной 1 :3,5, 3—4 капли пергидроля и нагревают на водяной бане до полного растворения (3—5 мин). Раствор переносят в мерную колбу емкостью 100 мл, разбавляют до метки водой и хорошо перемешивают. Отбирают 2 мл раствора в мерную колбу емкостью 25 мл, прибавляют туда же 5 мл воды, 1 мл раствора солянокислого гидроксиламина, 2 мл 8 н. раствора H2SO4 и 3 мл раствора молибдата аммония. Смесь оставляют на 3 мин для полного образования желтого кремнемолибденового комплекса, затем прибавляют 7 мл 8 н. H2SO4 для разрушения фосфорномолибденового комплекса, перемешивают, прибавляют 2 мл раствора SnCl2, разбавляют до метки водой и измеряют интенсивность окраски.
Ход анализа чугуна [139]. Навеску чугуна 0,1 г помещают в коническую колбу емкостью 100 мл, смачивают 3 каплями пергидроля, прибавляют 10 мл горячей H2SO4 (1:8) и нагревают на кипящей водяной бане. По растворении основной массы навески прибавляют 10 мл горячей HNO3, разбавленной 1 :3,5, и нагревают до полного растворения пробы. Раствор переносят в мерную колбу емкостью 250 мл, разбавляют водой, перемешивают и анализируют, как описано при анализе стали.
III. 12.2. Определение кремния в черных металлах при восстановлении желтого комплекса сульфатом железа [138]
Навеску образца переводят в раствор, получают кремнемолибденовую гетерополикислоту, прибавляют щавелевую кислоту или оксалат аммония для разрушения фосфорномолибденовой кислоты и восстанавливают желтый кремнемолибденовый комплекс в синий, применяя в качестве восстановителя сульфат железа(II).
Реактивы
Молибдат аммония, 5%-ный раствор.
Оксалат аммония или щавелевая кислота, 3%-ный раствор.
Соль Мора, 6%-ный водный раствор, содержащий 1 мл H2SO4, разбавленной 1 : 25, на 100 мл раствора.
Железоаммонийные квасцы. Растворяют 54 г квасцов в 150—200 мл воды, подкисленной 1 мл H2SO4, разбавленной 1:1. После охлаждения фильтруют в мерную колбу емкостью 250 мл и разбавляют водой до метки.
Ход анализа стали. Навеску стали 0,1 г помещают в коническую колбу, приливают 10 мл H2SO4, разбавленной 1 :8, закрывают часовым стеклом и ставят на кипящую водяную баню. По окончании растворения снимают часовое стекло и приливают 5 мл азотной кислоты, разбавленной 1 :4. Нагревание на водяной бане продолжают в течение нескольких минут для удаления образующихся окислов азота, переносят раствор в мерную колбу емкостью 100 мл, разбавляют водой до метки и перемешивают. Затем отбирают две порции по 3 мл в мерные колбы емкостью 25 мл, прибавляют по 10 мл воды и в одну из них прибавляют 3,0 мл раствора молибдата аммония; спустя 3 мин, не обращая внимания на выпадение осадка, приливают 5 мл раствора оксалата аммония или щавелевой кислоты в зависимости от того, с каким из этих реактивов построен калибровочный график, и перемешивают до полного растворения осадка, образовавшегося после прибавления молибдата. Сразу же прибавляют 4 мл раствора^ соли Мора и перемешивают. В другую колбу, содержащую аликвотную часть раствора образца, одновременно вводят те же реактивы и в том же порядке, за исключением молибдата аммония.
Оптическую плотность раствора измеряют в кюветах с толщиной слоя 2,0 см по отношению раствора сравнения.
Содержание кремния находят по калибровочному графику, который строят по стандартным образцам стали.
Ход анализа чугуна. Навеску 0,05—0,1 г в зависимости от предполагаемого содержания кремния растворяют так же, как и при анализе стали. Если для анализа взято 0,05 г, то к навеске прибавляют 2 мл раствора железоаммонийных квасцов. Раствор после окисления и выдерживания на водяной бане переводят в мерную колбу емкостью 250 мл, разбавляют водой до метки и перемешивают. Отбирают по 3 мл раствора и продолжают определение кремния, как указано при анализе стали.
Ход анализа жароупорных сталей. Навеску 0,1 г растворяют в смеси 15 мл хлористоводородной кислоты, разбавленной (1:2), и 5 мл HNO3, разбавленной (1:2). После растворения навески выдерживают на водяной бане дополнительно в течение 5 мин и продолжают анализ, как указано выше.
III. 12.3. Определение кремния в ферросилиции [140]
Пробу сплавляют со щелочью в железном тигле, плав выщелачивают водой, нейтрализуют серной кислотой и в аликвотной части раствора определяют кремний в виде синего кремнемолибденового комплекса, применяя в качестве восстановителя соль Мора.
Реактивы — см. раздел III. 12.2.
Ход анализа ферросилиция с высоким содержанием кремния. Навеску 0,05 г тонко измельченного образца, пропущенного через сито в 200 меш (6400 отв-см2), помещают в железный тигель, прибавляют 5 г твердого едкого натра, покрывают тигель зачищенной железной пластинкой, ставят на электрическую плитку, покрытую асбестом, и нагревают 15—20 мин. Щелочь при этом плавится и проба разлагается. Тигель охлаждают, содержимое его выщелачивают горячей водой в стакане емкостью 500 мл. Сначала обмывают тигель и крышку теплой водой, а затем теплой серной кислотой (1 :8) над тем же стаканом и прибавляют 200 мл теплой серной кислоты (1 :8), включая то количество, которое израсходовано на обмывание тигля и крышки. Жидкость нагревают при помешивании стеклянной палочкой до полного посветления. После охлаждения раствор переносят в мерную колбу емкостью 500 мл, доводят до метки водой, тщательно перемешивают и отбирают пипеткой 10 мл в мерную колбу емкостью 100 мл. Приливают в колбу около 80 мл воды, перемешивают, прибавляют 4 мл серной кислоты (1:8), разбавляют водой до метки и хорошо перемешивают. Из этого раствора отбирают пипеткой 4 пробы по 4 мл в мерные колбы емкостью 25 мл, приливают в каждую по 10 мл воды и далее ведут определение, как указано в разделе III. 12.2. Одна проба служит раствором сравнения. Из полученных результатов берут среднее значение.
Калибровочный график строят по стандартным образцам ферросилиция.
При анализе ферросилиция со средним содержанием кремния увеличивают навеску пробы до 0,1 г.
Определение кремния можно проводить и в хлористоводородной среде, при этом ход работы аналогичный, но тигель и крышку обмывают 2—3 раза хлористоводородной кислотой (1:3) и приливают в стакан при помешивании палочкой 100 мл хлористоводородной кислоты (пл. 1,19), включая то количество, которое израсходовано на обмывание тигля.
III. 12.4. Определение кремния в меди и сплавах на медной основе
При определении кремния в меди и сплавах на медной основе лучшие результаты получаются с предварительным электролитическим отделением меди. Однако описан ряд методик, в которых предлагают медь связывать тиомочевиной или применять экстракцию восстановленной кремнемолибденовой гетерополикислоты.
III. 12.4.1. Определение кремния с предварительным отделением меди [141]
Реактивы
Смесь кислот. Смешивают 40 мл HNO3, 60 мл Н2О и 5 мл H2SO4.
Молибдат аммония, 10%-ный раствор.
Лимонная кислота, 25%-ный раствор.
Восстановитель, растворяют в воде 2 г Na2SO3, 0,4 г 1-амино-2-нафтол-4-сульфокислоты и 25 г пиросульфита натрия (Na2S20s). Раствор доводят водой до 250 мл и перемешивают.
Ход анализа. Навеску 2 г меди или бронзы растворяют при нагревании в 10 мл смеси кислот. Раствор разбавляют до 70—80 мл и электролитически выделяют медь, пользуясь платиновыми электродами, при силе тока 3—4 А, до получения бесцветного раствора. Электроды обмывают водой, раствор охлаждают, переносят в мерную колбу емкостью 100 мл, разбавляют водой до метки и перемешивают. Отбирают две аликвотные части по 10 мл в мерные колбы емкостью 25 мл. В одну колбу прибавляют 5 мл раствора молибдата аммония и спустя 5 мин — 5 мл лимонной кислоты. В другую колбу вводят те же реактивы, но в обратном порядке. Через 5 минут в обе колбы вводят по 3 мл раствора восстановителя. Растворы выдерживают в водяной бане при 50°C в течение 15 мин, охлаждают и разбавляют водой до метки, перемешивают, наполняют кюветы с толщиной слоя 4—5 см и измеряют оптическую плотность первого раствора по отношению второго при 820 нм.
Содержание кремния находят по калибровочному графику, для построения которого пользуются стандартными образцами меди или соответствующего сплава на медной основе.
III. 12.4.2. Определение кремния без предварительного отделения меди [142]
При содержании кремния 0,1 — 1% его определяют в виде желтой кремнемолибденовой кислоты без выделения меди. При анализе сплавов с содержанием кремния 0,002—0,02% его определяют в виде синего комплекса. Желтую кремнемолибденовую гетерополикислоту восстанавливают тиомочевиной, которая одновременно связывает медь в бесцветный комплекс и устраняет вредное влияние ионов меди.
Реактивы
Персульфат аммония, 25%-ный раствор.
Молибдат аммония, 10%-ный раствор.
Стандартный раствор кремния, см. раздел III. 11.1.
Тиомочевина, 15%-ный раствор.
Сульфат меди. Растворяют 200 г CuSO4-5H2O в воде, подкисленной 1 мл H2SO4, разбавляют до 1 л и перемешивают.
Ход анализа при содержании кремния 0,1—1%. Навеску сплава 0,1 г растворяют в смеси 15 мл раствора персульфата аммония и 3 мл HNO3 (пл. 1,4) вначале на холоду и далее при нагревании до 100°C в течение 5—10 мин. Раствор охлаждают, переносят в мерную колбу емкостью 25 мл, разбавляют водой до метки и перемешивают. Отбирают 10 мл раствора в колбу емкостью 25 мл и прибавляют 5 мл воды, 5 мл раствора молибдата аммония, 3 мл H2SO4 разбавленной (1:5), доливают до метки водой, перемешивают и измеряют оптическую плотность раствора желтого комплекса.
Содержание кремния находят по калибровочному графику, который строят по стандартному раствору кремния с добавлением в каждую пробу 2 мл раствора сульфата меди.
Ход анализа при содержании кремния 0,002—0,1%. Навеску сплава 0,5 г растворяют в 5 мл HNO3 (пл. 1,4) при комнатной температуре. Если сплав не растворился, его нагревают в течение 5 мин при температуре не выше 80°C. Затем раствор охлаждают, переводят в мерную колбу емкостью 100 мл, доводят водой до метки и перемешивают. Отбирают 5 мл аликвотного раствора в мерную колбу емкостью 25 мл, приливают 4 мл воды и 2 мл молибдата аммония. Спустя 10 минут приливают 5 мл серной кислоты, разбавленной (1:5), 5 мл тиомочевины и хорошо перемешивают. Через 5 мин разбавляют раствор до метки, перемешивают и измеряют оптическую плотность синего раствора.
Содержание кремния находят по калибровочному графику, который строят по стандартному раствору в тех же условиях.
При анализе оловофосфористых бронз марки БОФ пробу растворяют в смеси (3:1) азотной и хлористоводородной кислот с целью удержания в растворе олова. В дальнейшем проводят анализ, как описано выше. При добавлении молибдата может выпасть осадок, но это не мешает образованию кремнемолибденовой гетерополикислоты. Этот осадок полностью растворяется при подкислении серной кислотой. После прибавления тиомочевины сразу измеряют оптическую плотность раствора.
III. 12.5. Определение кремния в магниевых сплавах
Навеску металлического магния растворяют в смеси азотной кислоты и перекиси водорода и в аликвотной части раствора определяют кремний в виде кремнемолибденовой сини.
Ход определения. Навеску сплава 0,1 г помещают в коническую колбу емкостью 100 мл, прибавляют 1 мл 3%-ного раствора Н2О2, 10 мл 2 н. HNO3 и нагревают в течение 10 мин на кипящей водяной бане. Затем раствор переводят в мерную колбу емкостью 100 мл, доливают водой до метки и перемешивают. Из этого раствора отбирают аликвотную часть и определяют кремний одним из указанных выше методов — см. раздел. III. 11.
III. 12.6. Определение кремния в алюминиевых сплавах [143]
Навеску сплава растворяют в щелочи, нейтрализуют азотной кислотой и в аликвотной порции раствора определяют кремний в виде желтого или синего кремнемолибденового комплекса.
Реактивы
Гидроокись натрия, 30%-ный раствор хранят в-полиэтиленовом сосуде. Перманганат калия, 0,1 н. раствор.
Щавелевая кислота, 1%-ный раствор.
Ход анализа. В никелевый тигель емкостью 50 мл помещают навеску сплава 0,1 г, добавляют 10 мл раствора NaOH и закрывают крышкой. По прекращении реакции тигель осторожно нагревают до полного растворения пробы. Раствор охлаждают и содержимое тигля переносят в стакан емкостью 150 мл,.в который предварительно наливают 12,5 мл азотной кислоты. Стакан накрывают стеклом и осторожно нагревают до посветления раствора. Если сплав содержит много марганца, то раствор иногда приобретает коричневато-желтый оттенок от взвешенной двуокиси марганца. Для устранения этой окраски прибавляют несколько капель свежеприготовленного сульфита, избыток которого устраняют прибавлением 0,1 н. раствора перманганата калия до слабого розового окрашивания. Затем прибавляют по каплям 1%-ный раствор щавелевой кислоты до полного восстановления перманганата калия.
Раствор охлаждают, переносят в мерную колбу емкостью 100 мл, разбавляют до метки водой и перемешивают. Отбирают аликвотную часть раствора и определяют кремний одним из Методов, описанных выше (см. раздел III. 11). Одновременно ведут холостую пробу со всеми реактивами, отбирают такую же аликвотную часть и используют в качестве раствора сравнения.
Для построения калибровочного графика пользуются стандартным раствором и чистым алюминием или раствором нитрата алюминия.
III. 12.7. Определение кремния в цирконии
Навеску образца растворяют в фтористоводородной кислоте [143] или разлагают сплавлением с пиросульфатом [144]. При использовании фтористоводородной кислоты возможны потери крем-122
ния в виде тетрафторида кремния. Поэтому при проведении анализа необходимы особые предосторожности. Нагревать раствор можно не выше 60—65 °C. В этом случае фторид связывают борной кислотой и определяют кремний в виде синего кремнемолибденового комплекса.
В случае сплавления циркония с пиросульфатом при содержании кремния более 0,3% часть его остается в виде SiOs, что усложняет анализ.
Реактивы
Молибдат аммония, 10%-ный раствор. Растворяют 25 г крупнокристаллического молибдата аммония в 200 мл бидистиллята, добавляют 20 мл H2SO< (пл. 1,84) и после охлаждения разбавляют до 250 мл.
Винная кислота, 20%-ный раствор, к которому прибавлено несколько кристаллов тимола в качестве антикоагулянта.
Аммиак, раствор, не содержащий кремневой кислоты. Получают насыщением бидистиллята в пластмассовых сосудах газообразным аммиаком.
Борная кислота, насыщенный раствор.
Фтороцирконат. Растворяют 28 г хлорида цирконила октагидрата в бидистилляте, доводят объем до 384 мл, переносят в пластмассовый сосуд и добавляют 16 мл 48%-ной фтористоводородной кислоты.
Восстановитель. Растворяют 11 г бисульфита натрия, 0,8 г гидроокиси натрия и 0,2 г 1-амино-2-нафтол-4-сульфокислоты в бидистилляте и разбавляют до 100 мл.
Ход анализа с применением фтористоводородной кислоты. Навеску 0,25 г помещают в пластмассовый стакан, наливают 5 мл бидистиллята и прибавляют 3 капли серной кислоты, разбавленной (1:1). Стакан накрывают пластмассовой крышкой и растворяют пробу, постепенно прибавляя около 1 мл 48%-ной фтористоводородной кислоты. После растворения большей части пробы раствор нагревают до 60—65°C до полного растворения. Раствор охлаждают, добавляют 10 мл раствора борной кислоты и перемешивают. Добавляют 1 мл раствора молибдата аммония и аммиаком доводят pH до 1,2—1,3. Спустя 10 мин добавляют 1 мл винной кислоты и перемешивают. Затем добавляют 0,5 мл восстановителя, переносят в мерную колбу емкостью 25 мл, разбавляют до метки и перемешивают. Через 20 мин измеряют оптическую плотность при 810— 815 нм относительно раствора сравнения, который готовят параллельно из раствора фтороцирконата.
Содержание кремния находят по калибровочному графику, построенному по стандартному раствору и раствору фтороцирконата.
Аналогично определяют кремний в окиси циркония и других соединениях циркония.
Ход анализа с применением сплавления с пиросульфатом. Навеску 0,2 г помещают в платиновый тигель и сплавляют с 1,5 г пиросульфата калия. Плав растворяют в 40—50 мл горячей воды, содержащей 1,5 мл H2SO4 (пл. 1,84), переносят в мерную колбу емкостью 100 мл, разбавляют до метки водой и перемешивают.
При содержании .кремния более 0,3% часть его после выщелачивания пйросульфатного плава может остаться в виде осадка
кремневой кислоты. Этот осадок отфильтровывают, переносят вместе с фильтром в платиновый тигель, подсушивают и сжигают. Остаток сплавляют с небольшим количеством безводного Na2CO3 или KNaCO3, охлаждают, сплав растворяют в воде, нейтрализуют серной кислотой, разбавленной (1:1), и присоединяют к первоначальному раствору, а затем доводят объем до метки водой и перемешивают. Отбирают аликвотную часть раствора и определяют кремний, как указано в разделе.III. 11.3. Раствором сравнения служит раствор холостой пробы, которую ведут параллельно, вводя все реактивы.
Для построения калибровочного графика пользуются стандартными образцами или раствором соли циркония и стандартным раствором кремневой кислоты.
III. 12.8. Определение кремния в бериллии и его соединениях
Бериллий растворяют в щелочи, нейтрализуют серной кислотой, прибавляют фтористоводородную кислоту, которую связывают борной кислотой, и определяют кремний в виде синего кремнемолибденового комплекса.
Ход анализа. Навеску 0,05 г бериллия помещают в пластмассовый стакан и растворяют в 5 мл водного раствора щелочи, содержащего 3 г гидроокиси натрия в 20 мл. Подкисляют раствор разбавленной серной кислотой, добавляют 0,2 мл 24%-ной фтористоводородной кислоты и нагревают в течение 30 мин при 60—65 °C. Охлаждают, добавляют 10 мл насыщенного раствора борной кислоты, переносят в мерную колбу емкостью 25 мл и продолжают определение, как описано в разделе III. 12.7.
Ход анализа окиси бериллия. Навеску 0,25 г помещают в пластмассовый стакан и растворяют в минимальном количестве серной или хлористоводородной кислоты. Прибавляют 5 мл воды, 0,2 мл 24%-ной фтористоводородной кислоты и продолжают анализ, как описано для металлического бериллия.
III. 12.9. Определение кремния в титане
Пробу переводят в раствор с помощью фтористоводородной кислоты, избыток которой связывают борной кислотой. Для удержания титана в растворе применяют винную кислоту или глицерин [145]. Кремний определяют в виде синего кремнемолибденового комплекса в водной фазе или после экстракции изоамиловым спиртом.
III. 12.9.1. Определение кремния без применения экстракции
Реактивы
Восстановитель. Растворяют 15 г бисульфита натрия, 0,5 г безводного сульфита натрия и 0,25 г 1-амино-2-нафтол-4-сульфокислоты в бидистилляте и разбавляют до 100 мл. Храпят в пластмассовой посуде.
Перманганат калия, 3%-ный раствор.
Молибдат аммония, 5%-ный раствор.
Винная кислота, 20%-ный раствор.
Ход анализа. Навеску, содержащую 0,01—0,14 мл кремния, помещают в пластмассовую колбу, добавляют 5 мл воды и 0,2 мл 24 %-ной фтористоводородной кислоты. Колбу накрывают и оставляют на ночь для растворения. Затем добавляют 10 мл насыщенного раствора борной кислоты и перемешивают. Титан(Ш) окисляют 3%-ным раствором перманганата калия, прибавляя его до розовой окраски, и еще 2 капли избытка. Обмывают стенки колбы малым количеством бидистиллята, погружают в кипящую воду и нагревают 90 мин, время от времени вращая колбу. Колбу вынимают из кипящей воды, охлаждают до комнатной температуры, фильтруют раствор в мерную колбу емкостью*25 мл, прибавляют 3 мл 5%-ного молибдата аммония и оставляют на 10 мин. По истечении этого времени добавляют 2 мл 20%-ного раствора винной кислоты, перемешивают, добавляют 0,5 мл восстановителя, разбавляют до метки водой и раствор перемешивают. Спустя 20 мин измеряют оптическую плотность при 700 нм относительно раствора сравнения, который готовят одновременно, используя все применяемые реактивы.
Содержание кремния находят по калибровочному графику, который строят по стандартным образцам титана или по стандартному раствору кремневой кислоты с прибавлением раствора титана в количестве, соответствующем содержанию титана в навеске.
III. 12.9.2. Определение кремния с экстракцией кремнемолибденовой сини
Реактивы
Хлорид олова(П), 1%-ный раствор в 0,25 н. хлористоводородной кислоте. Глицерин, х. ч.
Перманганат калия, 0,1 н. раствор.
Молибдат аммония, 10%-ный раствор с pH = 6,5.
Изоамиловый спирт, х. ч.
Ход анализа. Навеску титана 0,2 г помещают в полиэтиленовый сосуд емкостью 100 мл, смачивают 3 мл бидистиллята, постепенно приливают 2 мл концентрированной фтористоводородной кислоты «особой чистоты», закрывают полиэтиленовой крышкой и оставляют до полного растворения навески. Сосуд помещают в горячую воду, прибавляют 2—3 капли концентрированной азотной кислоты для окисления Tim и после обесцвечивания раствора приливают 40 мл раствора борной кислоты, 5 мл глицерина и нагревают на кипящей водяной бане в течение 3 мин. Приливают 5 мл 10%-ного раствора молибдата аммония с pH = 6,5 и продолжают нагревание в течение 10 мин. Раствор охлаждают до комнатной температуры, приливают 12,5 мл 12 н. серной кислоты, 0,3 мл 0,1 н. раствора перманганата калия и после перемешивания переносят
раствор в мерную колбу емкостью 100 мл. Приливают 2 мл раствора хлорида олова(II) и доводят до метки бидистиллятом. Раствор перемешивают, отбирают 50 мл, помещают в делительную воронку и экстрагируют синий кремнемолибденовый комплекс 10 мл изоамилового спирта в течение 30 с. Спиртовый раствор сливают в мерный цилиндр емкостью 10 мл, доводят объем изоамиловым спиртом до метки, перемешивают и измеряют оптическую плотность при 650—720 нм.
Одновременно проводят холостой опыт.
Содержание кремния находят по калибровочному графику. Для построения графика используют раствор, не содержащий титана.
111.12.10. Определение кремневой кислоты в шлаках медной и свинцовой плавок [146]
Шлак переводят в раствор сплавлением с гидроокисью натрия и перекисью натрия, выщелачивают водой, нейтрализуют хлористоводородной кислотой и в аликвотной части раствора определяют кремний в виде синего кремнемолибденового комплекса.
Реактивы
Молибдат аммония, 5%-ный раствор.
Ход анализа. В железный тигель помещают 3 г гидроокиси натрия и нагревают до расплавления. Тигель охлаждают и на застывшую щелочь насыпают 0,5 г анализируемого шлака, а сверху 2 г перекиси натрия. Тигель ставят на край открытого муфеля, нагретого до 600—700 °C; когда сплав перестанет пениться, его перемешивают, покачивая тигель, и передвигают в более горячую зону муфеля. Спустя 7—8 мин тигель вынимают и опускают на 1 мин в холодную воду так, чтобы вода не попала в тигель. При этом тигель очищается от окалины. Затем его погружают в стакан, в котором содержится 150—160 мл горячей воды, накрывают часовым стеклом и нагревают несколько минут на теплой водяной бане. После окончания выщелачивания тигель извлекают стеклянной палочкой, обмывают горячей водой и в стакан приливают в один прием 20 мл хлористоводородной кислоты (пл. 1,19). Жидкость кипятят 1—2 мин, охлаждают, переливают в мерную колбу емкостью 500 мл, доливают до метки водой и перемешивают. Отбирают 25 мл раствор’а в мерную колбу емкостью 500 мл, в которую предварительна наливают 300—400 мл воды и 5 мл хлористоводородной кислоты (пл. 1,19). Жидкость разбавляют до метки водой, перемешивают, отбирают 2,5 мл в мерную колбу емкостью 25 мл, прибавляют 2,5 мл воды, 2 мл 0,5 н. серной кислоты и 2 мл 5%-кого раствора молибдата аммония. После прибавления каждого реактива раствор перемешивают; спустя 10 мин добавляют около 10 мл воды и 3,5 мл 8 н. H2SO4. После перемешивания прибавляют 1 — 1,5 мл 0,5%-ного раствора SnCl2 в 20%-ной хлористоводородной
кислоте, доводят водой до метки и перемешивают. Через 10 мин измеряют оптическую плотность при 650—720 нм в кювете с толщиной слоя 1 см.
Содержание кремневой кислоты находят по калибровочному графику, который строят по стандартному образцу шлака или по стандартному раствору кремневой кислоты.
111.12.11. Определение кремневой кислоты во флюсах, применяемых при электросварке [147]
Флюсы, применяемые при электросварке, содержат фториды, поэтому для предотвращения потери кремния при разложении пробы ее сплавляют со смесью карбоната калия и тетрабората натрия. После разложения пробы и переведения ее в раствор в аликвотной части раствора определяют кремневую кислоту в виде синего кремнемолибденового комплекса. В качестве восстановителя применена тиомочевина.
Реактивы
Тиомочевина, 7%-ный раствор.
Молибдат аммония, 5%-ный раствор.
Сульфат меди. Готовят раствор, содержащий 5 мг меди в 1 мл.
Ход анализа. Навеску тонкоизмельченного флюса 0,1 г смешивают в платиновом тигле с 4 г К2СО3 и 2,5 г тетрабората натрия, накрывают крышкой и сплавляют в течение 20 мин при 950— 1000 °C. Тигель извлекают из муфеля и держат в наклонном положении до застывания плава. Затем тигель переносят в стеклянный стакан емкостью 800—1000 мл, куда предварительно наливают 350 мл дистиллированной воды, вводят 7 мл концентрированной серной кислоты и прибавляют 6 мл профильтрованного 7%-кого раствора тиомочевины. Содержимое стакана помешивают стеклянной палочкой до полного разложения плава. Тигель и крышку извлекают из стакана стеклянной палочкой и обмывают водой. Раствор переносят в мерную колбу емкостью 500 мл, доводят водой до метки и перемешивают. Отбирают 1 мл раствора в мерную колбу емкостью 25 мл, прибавляют 5 мл 0,15 н. серной кислоты, 2 мл 5%-ного раствора молибдата аммония и оставляют на 3 мин для образования желтого кремнемолибденового комплекса. Затем прибавляют 3,5 мл 8 н. серной кислоты для разрушения фосфорномо-либденового комплекса, вводят 2 мл раствора сульфата меди, содержащего 5 мг меди в 1 мл, и 6 мл раствора тиомочевины, взбалтывают, доводят водой до метки, перемешивают и спустя 3 мин измеряют оптическую плотность раствора в кювете с толщиной слоя 1 см.
Одновременно ставят холостой опыт с использованием всех реактивов; полученный раствор служит раствором сравнения.
Содержание кремния находят по калибровочному графику. Для построения /рафика готовят раствор флюса с известным содержанием кремневой кислоты.
III. 12.12. Определение кремния в других объектах
Содержание кремния в некоторых полупроводниковых материалах очень мало, поэтому при анализе сурьмы, галлия, индия и таллия [148] предварительно отделяют основные компоненты, а затем определяют кремний в виде синего кремнемолибденового комплекса после экстракции его изоамиловым спиртом. При этом сурьму отгоняют в виде трехбромистой, отделяют галлий в виде оксихино-лината, индий в виде трихлорида, а таллий в виде окиси. При определении кремния в силуминах в качестве восстановителя применяют эйконоген — ЭХТ-кислоту [149]. Рекомендовано при определении кремния в чистой меди [150] применять раствор молибдата аммония с определенным значением pH. Разработаны методы определения кремния в продуктах цинкового производства [151] и экстракционно-фотометрический метод определения кремния в ниобии, тантале [152] и металлическом никеле [153]. Экстракцию проводят н-бутанолом, хотя удобнее применять изоамиловый спирт. Экстракция применена также при определении кремния в чистой воде [154], в морской воде [155], в железе и стали [156], в хроме высокой чистоты [157], в плавиковом шпате [158] и других объектах.
Разработаны методы определения кремния в никеле и никелевых сплавах [159], в присутствии больших количеств хлорида натрия [160], в соединениях урана [161], в биологических материалах и минералах [162], в кремнийорганических соединениях [163—165], трихлорсилане [166], в моче [167] и других веществах.
Разработаны методы определения кремния в виде желтого кре-мнёмолибденового комплекса в полупроводниковых материалах [168], минеральном сырье [169], в присутствии германия [170], болгарском баршге [171], природных силикатах [172] и в промывочных растворах в присутствии фторидов [173].
Для определения кремния значительно чаще применяют синий кремнемолибденовый комплекс. В виде этого комплекса определяют кремний в чистом теллуре [174], в воде бойлеров и накипи [175], в пробах с высоким содержанием кремния [176], огнеупорных материалах [177], глиноземе [178,] воде [179, 180], растворах нитрата уранила [181], ферросиликохроме [182], плавиковом шпате и флюоритовом концентрате [183], стекле [184], неметаллических включениях [185], окиси бора [186], техническом перборате [187], железных рудах и других продуктах металлургического производства [188], химических реактивах [189], двуокиси урана [190], сталях, алюминии, цирконии, титановой губке, сплавах кремния и никеля, урана и кремния, бифториде калия [191], хроматах кальция и магния [192], минеральном сырье [193] и в других объектах [194—197].
Применяется дифференциальный метод определения кремния в глинах и в материалах металлургического производства [198]. Косвенные методы определения кремния основаны на определении молибдена, связанного в кремнемолибденовый комплекс в виде пероксимолибденовой кислоты [199], а также с использованием 2-амино-4-хлортиофенола [200]. Однако наиболее чувствительным является метод, основанный на применении ионных ассоциатов с родамином Б [201] и другими красителями.
При определении микрограммовых количеств кремния его переводят в SiF4 и током воздуха отгоняют в специальный поглотительный раствор, содержащий молибдат аммония [202]. Для отделения кремневой кислоты применяют также отгонку кремния в виде кремнефтористоводородной кислоты. При этом пользуются серебряной аппаратурой [203]. Описан интересный метод отделения кремния экстракцией кремнефтористоводородной кислоты триоктиламином [204]. Этот метод может быть использован для отделения кремния от многих элементов, в том числе и от ниобия и тантала.
ЛИТЕРАТУРА
1. Бабко А. К., Пилипенко А. Т. Фотометрический анализ. Общие сведения и аппаратура. М., «Химия», 1968. См. с. 258.
2. Егорова Е. И., Методы выделения и аналитического определения кремнезема. М., Изд-во АН СССР, 1959.
3. Go vet t G., Anal. chim. acta, 25, 69 (1961).
4. W e b b e r H., W i 1 s о n A., Analyst, 89, 632 (1964).
5. Арефьева Г. E. и др. В сб.: «Современные методы анализа в металлургии». М., Металлургиздат, 1955. См. с. 96.
6. W i с k b о 1 d R., Z. anal. Chem., 171, 81 (1959).
7. Ал и мар ин Й. П., Судаков Ф. П., Клитина В. И., Усп. хим., 34, 1368 (1965).
8. S t г i с k 1 a n d J., J. Amer. Chem. Soc., 74, 862, 872 (1952).
9. Chalmers R., Singlair A., Anal. chim. acta, 33, 348 (1965).
10. Бабко А. К., Ш кара вский Ю. Ф., ЖНХ, 8, 934 (1963).
11. Wa 1 den C„ Mel 1 a n M„ Anal. Chem., 25, 1668 (1953).
12. L i n d 1 e у G., Anal. chim. acta, 25, 334 (1961).
13. M i s s о n G., Chemiker-Ztg., 32, 633 (1908).
14. Милославский H. M., Вавилова E. Г., Зав. лаб., 4, 1450 (1938).
15. Murray W„ Ashley S., Anal. Edit., 10, 1 (1938).
16. К о н к и н В. Д., Зав. лаб., 8, 322 (1939).
17. Максимова M. В., Козловский М. Т„ ЖАХ, 2, 353 (1947).
18. Щ е р б о в Д. П„ ЖАХ, 4, 152 (1949).
19. Щ е р б о в Д. П„ Бюл. ВЙМС, 9, 69 (1948).
20. Wenzel W., Pflanzenernahr. Z„ Diing:, Bodenkunde, 75, 216 (1956).
21. Rickey G. F., Avens A. W„ J. Assoc. Offic. Agric. Chemists, 38, 898 (1955).
22. Kowalscki W., Szwanenfeld M., Przem. chem., 11, 698 (1955).
23. E 1 w e 111 W„ Wilson H., Analyst, 81, 139 (1956).
24. Б у p с у к А. Я., Зав. лаб., 8, 12, 1939; 9, 95 (1940).
25. Ляликов Ю. С. и др., Анализ железных, марганцевых руд и агломератов. М., «Металлургия», 1966. См. с. 140.
26. Namiki Н., Bull. Soc. Chem. Japan, 37, 484 (1964).
27. Судаков Ф. П., Клитина В. И., Селиванова С, П., Вести. Моск, ун-та. Сер. хим., 2, 83 (1966),
28. Deniges G., C. r„ 173, 802 (1920).
29. Ш н e e p с о н С. Б., Зав. лаб., 3, 21 (1934).
30. К л и т и п а В. И., Судаков Ф. II., Ал им арии И. П., ЖАХ, 20, 1145 (1965).
31. I n g а ш е 1 1 s С. О., Chemist-Analyst, 45, 10 (1956).
32. Судаков Ф. П., К литии а В. И., Маслова Н. Т., Вести. Моск, ун-та. Сер. хим. и., № 1, 98 (1966).
33. Судаков Ф. П., Клитина В. И., Маслова Н. Т., ЖАХ, 21, 1089 (1966).
34. С у д а к о в Ф. Щ Б у то р о в а Л. В., ЖАХ, 23, 721 (1968).
35. Theakston Н М., Bandi W. R., Anal. Chem., 38, 1764 (1966).
36. Бабко А. К., Ш к a p а в с к и й Ю. Ф., Кулик В. И., ЖАХ, 21, 196 (1966).
37. Б а б к о А. К., Шкаравский Ю. Ф., Ивашкович Е. М., Укр. хим. ж., 33, 951 (1967).
38. D и с г е t L., D г о i 11 a s М., Anal. Chim. acta, 21, 86 (1959).
39. Судаков Ф. П., Клитина В. И., Даньшова Т. Я., ЖАХ, 21, 1333 (1966).
40. Чалая 3. И., Я к у м о в а М. Н., Зав. лаб., 32, 792 (1966).
11. Флидлидер Г. В., Харченко Л. В., Зав. лаб., 34, 925 (1968).
42. Алексеев Р. И., Зав. лаб., 11, 122 (1945).
43. М е р в е л ь Р. В., Зав. лаб., 11, 135 (1945).
44. W a d е 1 i п О., М е 11 о n М. G., Anal. Chem., 25, 1668 (1953).
45. Ur а М., Japan Analyst, 7, 420 (1958); цит. по Anal. Abstr., 6. 1745 (1959).
46. Филиппова Н. А., Кузнецова Л. И., Зав. лаб., 16, 536 (1950).
47. Ж а р о в с к и й Ф. Г., Костышина А. П., Укр. хим. ж., 19, 201 (1953).
48. Hes lop R. В., Pearson Е. F., Anal. Chim. acta, 39, 209 (1967).
49. Бабко А. К., Шкаравский Ю. Ф., Ивашкович Е. М., Укр. хим. ж., 33, 397 (1967).
50. Шевчук И. А., Скрипник Н. А., Енальева А. Я., Зав. лаб., 33, 288 (1967).
51. Дорохова Е. Н., Шахова 3. Ф., Сазонова Л. А., ЖАХ, 21, 884 (1966).
52. Djurkin V., Ki г kb right G. F„ West T. S., Analyst, 91, 89 (1966).
53. Арсланова H. В., Мясоедов а А. С., Судаков Ф. П., ЖАХ, 26, 947 (1971).
54. H a 1 6 s z A., Polyak К., P u n g о r E., Magy. Kem. folydirat, 76, 545 (1970).
55. Лельчук Ю. Л., Соколович В. Б., Деткова Г. А., Изв. Томского политех, ин-та, 148, 144 (1967).
56. Бабенко А. С., В о л о д ч е н к о Т. Т„ ЖАХ, 23, 1237 (1968).
57. Е р м е к о в М. А., Доп. АН УРСР, 2, 41 (1959).
58. Bridger G. L., Boylan D. R., Markey J. W., Anal. Chem., 25, 336 (1953).
59. Lutwak H. K., Analyst, 78,661 (1953).
60. S i n о g 1 Z., Hutn. listy, 16, 435 (1961).
61. Bilinska U., Terpilowski J., Chem. anal. (Polska), 5, 17 (I960).
62. Мельцер P. А., Почвоведение, № 6, 103 (1960).
63. Шафран И. Г., Павлова М. В., Долженко Т. Г., В сб.: «Методы анализа хим. реактивов и препаратов», М., изд. ИРЕА, вып. 18, 127 (1971).
64. В е г п h а г t D. N., W г е a t h A. R„ Anal. Chem., 27, 440 (1955).
65. СимандкиТанаси, РЖХим, 1969, 1Г130.
66. A gг a w а 1 В. В., F i s h S. F., Z. anal. Chem., 255, 36 (1971).
67. Шарапова Г. H., Тр. Уральского н.-и. хим. ин-та, вып. 19, 177 (1970).
68. Федоров А. А., Сб. тр. Центр, н.-и. ин-та черной металлургии, в. 19, 22 (1960).
69. ФедоровА. А., Линкова Ф. В., Зав. лаб., 25, 535 (1960).
70. Куsil В., Vobora J., Hutn. listy, 14, 1081 (1959).
71. F 1 u m Z., Paliva, 39, 126 (1959).
72. В e p и д з e A. E., Почвоведение, № 11, 101 (1964).
73. Г о p ю ш и н а В. Г., Есенина H. В., С н e с a p e в К. А., ЖАХ, 24, 1699 (1969).
74. Ц а п М. Л. и др., Агрохимия, № 6, 127 (1971).
75 Цап М. Л. и др., Авт. св. СССР, кл. g01 п31/00, g 01 п 33/00, № 310175, заявл. 10.04.69, опубл. 29.09.71. РЖХим, 1972, 12Г110П.
76. D u f f Е. J., S t u а г t J. L., Analyst, 96, 802 (1971).
77. Hodnfk J., Rud-met. zb., № 4a, 11 (1966/67).
78. Ш а ф p а н И. Г. и др., В сб.: «Методы анализа хим. реактивов и препаратов». М„ изд. ИРЕА, вып. 19. 67 (1971).
79. Kirk В. Р., Wilkinson Н. С., Taianta, 19. 80 (1972).
80. С у д а к о в Ф. П., Таланкина Н. Ф., X а м р а к у л о в а М. И., ЖАХ, 25, 548 (1970).
81. Судаков Ф. П„ Твердо в а Н. Л., ЖАХ, 26, 573 (1971).
82. Яковлева Е. Н., Мазо Л. Я., Ротенберг И. Л., То. Всес. н.-и. ин-та хим. реактивов и особо чистых хим. веществ, вып. 31, 239 (1969).
83. Narasaraju Т. S. В., Singh R. Р„ Rao V. L. N., Z. anal. Chem., 251, 300 (1970).
84. П о л я н с к и й Н. Г., А к о п о в Г. А., И с а е в а Г. Я., ЖАХ, 26, 1854 (1971).
85. В о л к о в а Г. П., Федоров А. А., Черняховская Ф. В., Сб. тр. ЦНИИ черн. металлургии, вып. 79, 45 (1972).
86. Аликина Н. А., Барковский В. Ф., Шва рев В. С., ЖАХ, 24, 1848 (1969):'
87. Schaffer F. L„ Fong J., Kirk P. L., Anal. Chem., 25, 343 (1953)
88. Ruf E., Z. anal. Chem., 151, 169 (1956).
89. Горюшина В. Г., Бирюкова-Гейлис Е. Я., Зав. лаб., 24, 402 (1958).
90. П а р in у т и н а М. В., Наладочные и эксперим. работы ОРГРЭС, вып. П, 34 (1955).
91. Г о р ю ш и н а В. Г., Е с е н и н а Н. В.. ЖАХ, 21, 239 (1966).
92. Забоева М. И., Спицын П. К., Зав. лаб.. 33, 554 (1967).
93. М а н д е л ь б а у м Я. А., Т р а к о в А. Ф., Щ у к о в а А. Л., ЖАХ, 20, 873 (1965).
94. Н о d n i k J., Rud-metal zv„ № 4а, 11 (1966/67).
95. Эдельман И. И., Забора Л. С., Хижняк Н. Д., Зав. лаб., 25, 159 (1959).
96. В о й н а л о в и ч М. Р.’ С и н о в с к и й Г. В., Докл. АН ТаджССР, 2, 11 (1959).
97. Б а р а н г у л о в а М. Н., Галимов Г. Ф., Изв. Сиб. отд. АН СССР, № 2, 97 (1960).
98. G i п g N. S.. Anal. Chem., 28, 1330 (1956).
99. Р i 1 z W., Mikrochim. acta., № I, 34 .(1965).
100. Буянова M. M„ Изв. Сиб. отд. АН СССР, № 4 (1967). Сер. хим. н., вып. 2, 176.
101. Мартинсон Э.. В и л л а к о Л., Лаб. дело, № 2, 30 (1961).
102. Никитина Е. И., Зав. лаб., 22, 1027 (1956).
103. Jean М„ Anal. Chem. acta, 14, 172 (1956).
104. Шафран И. Г. и др.. Тр. Всес. н.-и. ин-та хим. реактивов и особо чистых хим. веществ, вып. 28, 56 (1965).
105. Шафран И. Г., Павлова М. В., Титова С. А., Тр. Всес. н.-и. ин-та хим. реактивов и особо чистых хим веществ, вып. 28, 66 (1965).
106 Шафран И. Г., Кураева А В., В сб.: «Методы анализа хим. реактивов и препаратов». вып. 15, М.. 134 (1968).
107. Соколов им В Б., Лельчук Ю. Л., Дрегуна О. А. Изв. Томского политех, ип-та. 148. 162 (1967).
108. Шафран И. Г., Павлова М. В., Сухарева 3. С., В сб.: «Методы анализа хим. реактивов и препапатов», вып. 19. 71 (1971).
109. Рак a Ins Р., McAllister В. R., Met. and Metal Form., 38, 220 (1971).
110. Mu к a i К., Taianta, 19, 489 (1972).
111. Heegn H., Oehme M.. Freiberg. Forschungsh., A, № 499, 77 (1971).
112. P г u s z у ri s к a J.. Chem. analyt. (Polska), 12, 1129 (1967).
113. Горюшина В. Г.. Есенина Н. В., С нес а рев К. А., ЖАХ, 25, 1610 (1970).
*14 Р а к а 1 n s Р., Anal. Chim. acta, 40, 1 (1968).
115. Kirsten W„ Microchem. J., 12, 307 (1967).
116. D u v a 1 L., Chim. anal., 49, 307 (1967).
117. P e з н и к Б. E., Г а н з б у p г Г. М., Цыганок Л. П., Укр. хим. ж., 30, 1099 (1964).
118. Ш к а р а вский Ю. Ф., Укр. хим. ж., 30, 670 (1964).
119. R i р a n R., С о г d i s V., Rev. Roum. chim., 16, 1711 (1971).
120. Калмыков С. И., Малахова К. И., Шевченко Н. П., Изв. АН КазССР. Сер. хим., № 3, 1 (1971).
121. Р a k а 1 n s Р., Anal. chim. acta, 51, 497 (1970).
122. Р а к а 1 n s Р., Anal. chim. acta., 50, 103 (1970).
123. Schwarz H., Mikrochim. acta, № 4, 677 (1969).
124. Нен ахов а К. В., Харькова И. В., Коробова И. А., Изв. высш, учебн. заведений. Хим. и хим. технол., 15, 306 (1972).
125 Шкаравский Ю. Ф., Лынчак К. А., Черно горенко В. Б., Зав. лаб., 36, 524 (1970).
126. TrautnerA., Meta 11, 26, 467 (1972).
127. Kirkbright G. F., Narayanaswamy R., West T. S., Analyst, 97, 174 (1972).
128. J a k u b i e c R. J., Boltz D. F., Mikrochim. acta, № 6, 1199 (1970).
129. Я ци мирский К. Б., Росоловски Щ. К., Крисс Е. Е., ЖАХ, 25, 324 (1970).
130. Т а н а н а е в И. В., Зав. лаб., 11, 246 (1945).
131. Шахова 3. Ф., Дорохова Е. Н., Чуян Н. К., ЖАХ, 21, 707 (1966).
132. Мустафин И. С., Молот Л. А., Чепурова Ю. А. Ассортимент реактивов на кремний. М., НИИТЭХИМ, 1969. См. с. 20.
133. Фогельсон Е. И., Козачкова Ф. С., Зав. лаб., 13, 565 (1947).
134. Мальцев В. Ф., Сыч В. Я., Зав. лаб., 14, 768 (1948).
135. Мальцев В. Ф., Зав. лаб., 22, 1044 (1956).
136. Алима рин И. П., Зверев В. С., Тр. ин-та прикладной минералогии, вып. 63, 12 (1934).
137. Scherrington, J. of Soc. Chem. Ind., 65, 90 (1946).
138. Фогельсон E. И., Зав. лаб., 22, 163 (1956).
139. Давыдов Л. А., Резник Ю. Э., Вайсберг 3. М., Зав. лаб., 8, 1033 (1939).
140. Фогельсон Е. И., Козачкова Ф. С., Зав. лаб., 23, 24 (1957).
141. Sturton J. М., Anal. chim. acta, 32, 394 (1965).
142. Никитина Е. И., Зав. лаб., 24, 398 (1958).
143. Петтер Г. В. Колориметрические (фотометрические) методы определения неметаллов. Пер. с англ. Под ред. А. И. Бусева. М., Издатинлит, 1963. См. с. 62.
144. Елин сон С. В., По бе дин а Л. И., Зав. лаб., 25, 909 (1959).
145. Барковский В. Ф., Радовская Т. Л., Зав. лаб., 35, 160 (1969).
146. Файнберг С. Ю., Бляхман А. А., Станкова С. М., Зав. лаб., 23, 647 (1957).
147. Мальцев В. Ф., Лукьяненко Л. П., Зав. лаб., 24, 537 (1958).
148. Назаренко В. А., Флянтикова Г. В., Зав. лаб., 24, 663 (1958).
149. Толмачев В. Н., За тучна я Л. А., Зав. лаб., 23, 152 (1957).
150. Р a k а 1 n s Р., Anal. chim. acta, 40, 328 (1968).
151. С о й ф е р м а н И. А., Зав. лаб., 25, 418 (1959).
152. Курбатова В. И., Калашникова Л. Я., Тр. Всес. н.-и. ин-та стандартных образцов и спектр, эталонов, 4, 174 (1968).
153. Н а г г i s о n F. Н„ Metallurgia, 66, 300 (1962).
154. Sturla A., Acgua industr. inguinam., 10, 14 (1968).
155. S ch ink D. R., Anal. Chem., 37, 764 (1965).
156. Kakita Yachiyo, Goto Hidehiro, Taianta, 14, 543 (1967).
157. M i n с c z e w s k i J., Chwaetowska J., Chem. analit., 6, 715 (1961).
158. Fresenius W„ Schneider W., Z. anal. Chem., 214, 341 (1965).
159. A n d г e w T. R., Analyst, 82, 423 (1957).
160. К г a t о c h v i 1 V., Chem. listy, 60, 1238 (1966).
161. Lozenzini L., Stoppa C., Barbieri W., Metallurgia ital., 54, 387(1962).
162. Toma J., Mikrochim. acta, № 3, 513 (1962).
163. Терентьев А. П., Бондаревская E. А., Градскова H. А. ЖАХ 24, 753 (1969).
164. HI а н и н a T. M., Гельман H. Э., Кипаренко Л. M., ЖАХ, 20, 118 (1965).
165. Фогел ьсон E. И., Зав. лаб., 23, 1427 (1957).
166. Криворучко Ф. Д., Зав. лаб., 27, 290 (1961).
167. Иванов В. И., Розенберг П. А., Гигиена труда и профзаболеваний, № 4, 39 (1960).
168. Ревенко В. Г. и др., ЖАХ, 26, 2235 (1971).
169. Д о л а б е р и д з е Л. Д., Камкамидзе Д. К., Та у гл их П. А., Тр. Кавказ, ин-та минеральн. сырья, вып. 8 (10), 151 (1970).
170. Sorrentino F. A., Witiak N. J., Paul J., Microchem. J., 15, 441 (1970).
171. Вахранеева И. В., Лиц Л. Б., Хим. пром-сть Укр., № 1 (49), 43 (1970).
172. Langer К., Z. anal. Chem., 245, 139 (1969).
173. Рычкова В. И., Энергетика, № 2, 17 (1968).
174. Dobrowolski J„ Kwiatkowska-Sienkiewicz К., Chem. anal. (PRZ), 15, 647 (1970).
175. Mohyuddin W., A g 1 a n S., Pakistan J. Sci., 22, 96 (1970).
176. Цолов Ц., Ган ев П., Тр. н.-и. ин-та черной металлургии, 2, 415 (1966/69).
177. S t о b а г t J. A., Analyst, 94, 1142 (1969).
178. Зинченко В. А., Васильева К. С., Зав. лаб., 35, 1317 (1969).
179. Morrison I. R., Wilson A. L., Analyst, 94, 54 (1969).
180. Kosfeld Е. G., Techn. Uberwachung, 8, 133 (1967).
181. Немо друк А. А., Палей П. H., Без рогов а Е. В., Радиохимия, 11, 716 (1969).
182. Барбаш Т. Л., Курбатова В. И., Силаева Е. В., Всес. н.-и. ин-т стандарта, образцов и спектр, эталонов, 3, 94 (1967).
183. Шарапова Г. Н., Тр. Уральского н.-и. хим. ин-та, вып. 17, 122 (1968).
184. W i n t е г Е., Glastechn. Вег., 41, 522 (1968).
185. Ершова К. Н., Орлова Г. М., Зав. лаб., 34, 276 (1968).
186. Лапицкая Е. В., Горбенко Ф. П., В сб.: «Методы анализа хим. реактивов и препаратов», вып. 18, 133 (1971).
187. Куторкина А. К., Сб. тр. Уральского н.-и. хим. ин-та, вып. 26, 104 (1971).
188. Bhargava О. Р., Pitt G. F., Hines W. G., Taianta, 18, 793 (1971).
189. Шафран И. Г. и др., В сб.: «Методы анализа хим. реактивов и препаратов», вып. 19, 74 (1971).
190. R a j к о v i с D., Z. anal. Chem., 255, 190 (1971).
191. Р а к а 1 n s Р., Anal. chim. acta, 54, 281 (1971).
192. А л e ш e ч к и н а А. Е., Булычева И. Б., Масалович В. М., Сб. тр. Уральского н.-и. хим. ин-та, вып. 26, 14 (1971).
193. Д о л а б е р и д з е Л. Д., Камкамидзе Д. К., Тр. Кавказского ин-та минеральн. сырья, вып. 8 (10), 170 (1970).
194. Н е м о д р у к А. А., Без рогов а Е. В., ЖАХ, 24, 1704 (1969).
195. Немодрук А. А., Безрогова Е. В., ЖАХ, 24, 1704 (1969).
196. Trudell L. A., Boltz D. F„ Anal. chim. acta, 52, 434 (1970).
197. Деткова Г. А., Лельчук Ю. Л., Изв. Томского политех, ин-та, 185, 72 (1970).
198. Еремин Ю. Г., Коробейникова 3. И., Троп кина М. Н., Изв. высш, учебн. заведений. Хим. и хим. технол., 12, 402 (1969).
199. Trudell L. A., Boltz D. F., Anal. Lett., 3, 465 (1970).
200. Т г u d е 11 L. А., В о 11 z D. F„ Taianta, 19, 37 (1972).
201. Golkowska A., Chem. anal. (Polska), 14, 803 (1969).
202. Ch. H о z d i c, Anal. Chem., 38, 1626 (1966).
203. Ehrlich P., Keil Th., Z. anal. Chem., 166, 254 (1959).
204. Пальшин E. С. и др., ЖАХ, 24, 797 (1969).
Глава IV
МЫШЬЯК
IV. 1. МЕТОДЫ ОТДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ
Некоторые соединения мышьяка легко улетучиваются, что приводит к потере его в ходе анализа. Потери мышьяка наблюдаются при нагревании соединений мышьяка(III) и даже мышьяка(V) в присутствии хлоридов и бромидов, а также при сплавлении пробы с содой. Значительные потери мышьяка происходят при действии сильных восстановителей, которые могут восстановить соединения мышьяка до арсина.
Летучесть некоторых соединений мышьяка способствует сравнительно легкому отделению его от других элементов. Поэтому определение малых количеств мышьяка не представляет больших затруднений.
Отделение проводят путем отгонки хлорида мышьяка(III), который кипит при 130 °C. Отгонку ведут из хлористоводородного раствора [1, 2]. Бромид мышьяка(Ш) также летучее соединение, поэтому отгонку мышьяка часто проводят в присутствии бромида. В отличие от AsCls бромид мышьяка (V)—летучее соединение [3], поэтому его можно отогнать из раствора, содержащего бромистоводородную кислоту, смесь бромистоводородной и хлорной кислот или бромистоводородной и серной кислот, а также бромистоводородной, фосфорной и хлорной кислот. Очень малые количества мышьяка отгоняют в виде арсина (мышьяковистого водорода).
Широкое распространение получили экстракционные методы отделения мышьяка. С этой целью чаще других применяют экстракцию хлорида мышьяка(III) четыреххлористым углеродом из хлористоводородных растворов; мышьяк(III) экстрагируют также в виде иодида. Кроме того, применяют экстракционное отделение мышьяка в виде дитиокарбамата.
Экстракционное разделение мышьяка, фосфора и кремния проводят в виде их гетерополикислот [4]. При этом используют разное отношение их к некоторым органическим растворителям. В ряде случаев для отделения мышьяка можно применять ионообменники.
Для концентрирования малых количеств мышьяка и отделения его от многих посторонних веществ применяют соосаждение арсената совместно с фосфатом магния и аммония или .соосаждение арсената железа совместно с гидроокисью железа.
IV. 1.1. Отгонка мышьяка в виде хлорида
Рис. 28. Прибор для отгонки хлорида мышьяка (III):
1 — капельная воронка; 2 — предохр анительная склянка; 3— реакционная колба; 4 — холодильник.
Мышьяк отгоняют из хлористоводородного раствора в присутствии восстановителя. Хлорид мышьяка (III) отгоняется при 130 °C, но и при более низкой температуре летучесть его довольно значительная.
Процесс отгонки мышьяка подвергался неоднократной проверке. По одним данным, потери мышьяка происходят в приемнике, так как при высокой концентрации хлористоводородной кислоты в приемнике часть хлорида мышьяка (III) улетучивается [5]. Поэтому было рекомендовано прибавлять в приемник перекись водорода для окисления мышьяка(III) до мышьяковой кислоты. Однако позже было показано, что в приемнике потерь мышьяка не наблюдается [6] и прибавление перекиси водорода в приемник является излишним. В ряде работ также показано, что при отгонке мышьяка нет необходимости в окислителях [1, 2, 7, 8].
Этим методом можно отделить мышьяк с ошибкой 3—10%, если содержание мышьяка не превышает 1—3 мкг, при большем содержании ошибка уменьшается.
Мешающие вещества. Вместе с мышьяком отгоняются хлорид германия (IV) (т. кип. 86°C) и частично хлорид олова(IV) (т. кип. 115 °C), хлорид сурьмы (т. кип. 200°C), а также хлорид селена(IV). Отделению мышьяка в сильной мере мешают окислители, в том числе нитрат-ионы. Поэтому окислители необходимо предварительно удалить или перевести в восстанов
ленную форму. Для отделения от германия мышьяк(Ш) окисляют до мышьяка (V), затем отгоняют хлорид германия (IV), прибавляют восстановитель и отгоняют хлорид мышьяка(III). Олово удерживают в растворе фосфорной кислотой; эту кислоту прибавляют также при отгонке мышьяка в присутствии вольфрама(VI), чтобы разрушить ионы мышьякововольфрамовой гетерополикислоты [As(W3Oio)4]3- Однако необходимо учитывать, что в дистиллят частично попадает фосфорная кислота, в результате требуется повторная отгонка.
Поведение сурьмы и олова в процессе отгонки хлорида мышьяка (III) изучалось с применением меченых атомов 7eAs, 122Sb и 121Sn [9], а также 74As и 124Sb [10]. При этом показано, что из раствора, содержащего 11 н. хлористоводородную кислоту, мышьяк полностью отгоняется при 109 °C [9], а сурьма и олово остаются в растворе. В случае дистилляции хлорида мышьяка(III) из сер-
нокислого раствора в присутствии хлорида калия рекомендовано нагревание до 140 °C [10]. При этом потери мышьяка не превышают 3%. Однако большинство авторов рекомендуют отгонять мышьяк в присутствии сурьмы при температуре, не превышающей 107 °C, а при содержании сурьмы в пробе больше 2 мг отгонку мышьяка рекомендуется повторить [11].
В качестве восстановителя для перевода мышьяка (V) в мышь-як(Ш) в большинстве случаев применяют сернокислый гидразин. С целью ускорения дистилляции через раствор пропускают двуокись углерода или азот. Отгонку проводят в приборе, состоящем полностью из стекла, собирая дистиллят в холодную воду.
Ход отделения. В дистилляционную трубку (рис. 28) помещают 10—15 мл раствора, прибавляют 2 мл бромистоводородной кислоты, 0,3 г сернокислого гидразина (если необходимо, берут больше) и 10 мл концентрированной хлористоводородной кислоты. В маленький стакан наливают 10 мл холодной воды и погружают в него конец холодильника. Раствор в колбе медленно нагревают до 107 °C, пропуская через раствор медленный ток (2—3 пузырька в 1 с) двуокиси углерода или азота. Собирают 10—15 мл отгона и определяют мышьяк.
IV. 1.2. Отделение мышьяка в виде арсина
Мышьяк(III) в щелочной или кислой среде восстанавливают металлическим цинком, алюминием, амальгамой натрия [12] или электролитически [13—15] до мышьяковистого водорода, который поглощают растворами окислителей, дитиокарбамата серебра или хлорида (бромида) ртути, и определяют мышьяк тем или иным методом.
Мышьяк(V) восстанавливается значительно труднее, а в щелочной среде практически не восстанавливается до арсина [12, 16]. Поэтому перед проведением реакции мышьяк(V) восстанавливают до мышьяка(III) хлоридом олова(II) в присутствии иодида, сульфатом хрома(II) или другими восстановителями.
Арсин поглощают раствором иода в присутствии бикарбоната натрия, а также растворами перманганата калия, хлорида ртути (II), гипобромита натрия [17]. Главными источниками ошибок в этом методе отделения мышьяка являются неполное образование арсина и неполное его поглощение.
Мешающие вещества. Сурьма в условиях определения образует сурьмянистый водород и улетучивается вместе с арсином. При определении мышьяка с помощью солей ртути и серебра сурьмянистый водород реагирует подобно арсину и мешает определению. В щелочной среде мешающее влияние сурьмы значительно меньше.
Под действием кислоты и металла-восстановителя, кроме сурьмянистого водорода, образуются фосфористый водород, германи-стый водород и сероводород, которые могут мешать определению 136
мышьяка. Сероводородпоглощают ацетатом свинца, для этого дистиллят пропускают через тампон из ваты, смоченный 10%-ным раствором ацетата свинца. Если дистиллят пропускать через раствор меди в хлористоводородной кислоте [50 г хлорида меди(1) в 100 мл хлористоводородной кислоты, разбавленной (1:1)], то можно удалить значительные количества сероводорода, фосфори-
стого и сурьмянистого водорода.
Присутствие ионов тяжелых металлов, которые выделяются на металлах, применяемых для восстановления мышьяка, в сильной мере задерживает выделение мышьяка. В анализируемом растворе может находиться до 50 мг ионов свинца и до 10 мг серебра. Присутствие 50 мг ионов меди при определении 5—10 мкг мышьяка приводит к ошибке, составляющей 20%. В присутствии ионов никеля и кобальта получаются заниженные результаты.
Ионы железа, хрома и молибдена могут находиться в растворе в довольно заметных количествах (до 50 мг), селен не мешает отгонке. Окислители мешают выделению арсина.
Реактивы
Иодид калия, 20%-ный раствор.
Хлорид олова(П), 0,5%-ный раствор в концентрированной хлористоводородной кислоте.
Рис. 29. Прибор для отгонки мышьяка в виде
Ход отделения. Отгонку ведут в приборе, показанном на рис. 29. До 20 мл анализируемого раствора, содержащего до 30 мкг мышьяка, помещают в колбу для восстановления /, прибавляют 20 мл 8 н. серной
арсина:
1 — колба для восстановления; 2—капельная воронка;
3 — приемник.
или 3 н. хлористово-
дородной кислоты и, если надо, разбавляют дистиллированной водой до 40 мл. К раствору прибавляют 4 мл 20%-ного раствора иодида калия, 2—5 капель 0,5%-ного раствора хлорида олова(II) и слабо нагревают в течение 10 мин для восстановления мышьяка (V) до мышьяка(III). Охлаждают раствор ледяной водой до 5 °C, насыпают 10—15 г цийка, не содержащего примеси мышьяка, быстро закрывают колбу и конец отводной трубки погружают в смесь растворов иода и бикарбоната или любого другого указанного выше поглотительного раствора. Раствор оставляют в ледяной воде на 10—15 мин, а затем выдерживают при комнатной температуре в течение 1 ч.
Одновременно проводят холостой опыт со всеми используемыми реактивами, но не вводят исследуемый раствор.
Ход дальнейшего определения зависит от применяемого поглотителя (см. ниже).
Выделение арсина можно проводить также из щелочного раствора. Отгонка арсина из щелочного раствора, а также электроли-
тическое выделение арсина описаны ниже.
IV. 1.3. Экстракция хлорида мышьяка (III)
Хлорид мышьяка(III) хорошо экстрагируется из хлористоводородных растворов четыреххлористым углеродом [18—20], бензолом и хлороформом. Лучшими экстрагентами являются бензол и хлороформ [20]. Степень экстракции зависит от концентрации хлористоводородной кислоты. Так, из 12—13 М раствора при равных объемах водной и неводной фаз экстрагируется 77% мышьяка. Снижение концентрации хлористоводородной кислоты до 9 М приводит к уменьшению экстракции мышьяка на 4% (73%). При проведении экстракции из раствора, состоящего из воды, концентрированных серной и хлористоводородной кислот в объемных отношениях 2:2:7, степень экстракции мышьяка достигает 88%.
Мешающие вещества. Экстракции мышьяка(III) мешают окислители, которые окисляют его до мышьяка (V). Вместе с мышьяком, но значительно более полно, экстрагируется германий. Селен (IV) экстрагируется примерно на 0,1%, а сурьма(III) при кислотности от 6 до 13 М — менее чем на 0,01%. Другие элементы практически не экстрагируются. Для отделения германия мышь-як(Ш) окисляют до мышьяка(У) и экстрагируют германий из 9—10 Л4 раствора по хлористоводородной кислоте. Затем мышьяк восстанавливают иодидом калия и экстрагируют мышьяк(Ш). Этот метод приводит к практически полному разделению мышьяка и германия даже при соотношении [Ge]: [As] = 106.
Ход отделения. К исследуемому раствору, содержащему 1 — 60 мкг мышьяка, прибавляют концентрированные серную и хлористоводородную кислоты с таким расчетом, чтобы объемные отношения воды, серной и хлористоводородной кислот составили 2:2:7, не допуская разогревания жидкости. Затем жидкость переводят в делительную воронку, прибавляют равный объем хлороформа и жидкости перемешивают осторожным переворачиванием делительной воронки в течение 2 мин. Воронку оставляют до разделения жидкости и хлороформный слой отделяют. Экстракцию повторяют дважды с половинными объемами хлороформа. Хлороформные растворы переносят в другую делительную воронку, прибавляют 10—15 мл дистиллированной воды, взбалтывают и оставляют до разделения жидкостей. Хлороформ отделяют и реэкстракцию мышьяка дважды повторяют, применяя по 5 мл воды. Водные фазы сливают и определяют мышьяк.
Если нет уверенности, что мышьяк находится в трехвалентном состоянии, к кислому раствору перед экстракцией прибавляют иодид калия.
IV. 1.4. Экстракция иодида мышьяка (III) [21]
Мышьяк(Ш) экстрагируют в виде иодида с помощью четыреххлористого углерода или хлороформа [22, 23]. При содержании 10—30 мкг мышьяк из пробы практически полностью отделяется 138
трехкратной экстракцией. Описан также метод экстракционного отделения мышьяка (V) четыреххлористым углеродом из 14—17 н. раствора по H2SO4 в присутствии бромида [24]. На 100 мкг мышьяка в 25 мл берут 200 мг КВг. При кислотности меньше 10 н. по H2SO4 экстракция не происходит, а при кислотности больше 17 н. выделяется бром и бромистоводородная кислота.
Мешающие вещества. Отделению мышьяка мешают окислители. Нс мешает присутствие до 5 мг молибдена, 5 мг вольфрама, 5 мг Р2О5, 100 мг ванадия, 100 мг кремния, 100 мг селена и 100 мг ртути.
Реактивы
Иодид калия, 0,1 М раствор в очищенной хлористоводородной кислоте (пл. 1,19).
Ход отделения. До 10 мл испытуемого раствора, содержащего 5—45 мкг мышьяка, помещают в делительную воронку емкостью 100 мл и прибавляют трехкратное количество относительно взятого раствора очищенной хлористоводородной кислоты (пл. 1,19), содержащей 0,1 моль/л KI. Затем прибавляют 20 мл СС14 и взбалтывают в течение 2 мин. Органическую фазу переносят в делительную воронку емкостью 50 мл и промывают 5—10 мл 9 н. очищенной хлористоводородной кислотой, встряхивая в течение 20 с. Промытый экстракт сливают в делительную воронку и взбалтывают в течение 2 мин с 15 мл воды. При этом мышьяк переходит в водный слой. Извлечение мышьяка повторяют, добавив 10 мл воды. Водные растворы объединяют и определяют мышьяк.
IV. 1.5. Экстракция мышьяка с применением реактивов, содержащих тиольную и тионную группы
Для отделения мышьяка широко используются экстракционные методы с применением реактивов, содержащих тиольную и тионную группы: диэтилдитиокарбамат [25—32], пирролидиндитиокарбамат [33], гексаметилендитиокарбамат гексаметиленаммония [34, 35], ксантогенат [36—38] и тионалид [39].
Экстракция с применением диэтилдитиокарбамата. Диэтилди-тиокарбаматный комплекс мышьяка(III) экстрагируется хлороформом в широких пределах кислотности (1 —10 н. H2SO4).
Мешающие вещества. При кислотности выше 1 н. по минеральной кислоте цинк, никель, железо(II) и кадмий не экстрагируются хлороформом и не мешают определению. Для отделения от свинца необходимо создавать 2 М кислотность хлористоводородной кислотой и выше. В присутствии щавелевой кислоты германий экстрагируется в очень малых количествах, поэтому при отделении мышьяка германий связывают щавелевой кислотой. Медь, висмут и ртуть предварительно отделяют от мышьяка экстракцией в виде дитиокарбаматов в присутствии перекиси водорода для удерживания мышьяка в пятивалентном состоянии.
Реактивы
Диэтилдитиокарбамат диэтиламмония. Растворяют 1 г чистого препарата в 100 мл перегнанного хлороформа, хранят в темной склянке не более 1 суток. Вместо этого раствора можно пользоваться свежеприготовленным хлороформным раствором диэтилдитиокарбаминовой кислоты.
Пергидроль, х. ч.
Хлороформ, очищенный.
Бромная вода, насыщенный раствор брома в воде.
Тиогликолевая кислота, 10%-ный водный раствор.
Смесь аскорбиновой кислоты и иодида калия. Растворяют 15 г иодида калия и 2,5 аскорбиновой кислоты в 100 мл. Раствор готовят ежедневно.
Нитрат магния, 2%-ный водный раствор.
Ход отделения. К 16 мл испытуемого раствора, содержащего до 500 мкг мышьяка (V), прибавляют 2—4 мл концентрированной серной кислоты и переносят в делительную воронку. К раствору добавляют’' 0,5 мл пергидроля, 5 мл раствора дитиокарбамата или раствора дитиокарбаминовой кислоты в хлороформе и встряхивают в течение 30 с. Органическую фазу отбрасывают, а водную промывают без перемешивания 1 мл хлороформа, который также отбрасывают. Затем экстрагируют 2—3 мл раствора диэтилдитиокарбамата и снова промывают 1—2 мл хлороформа. Таким образом можно отделить 1—2 мг меди и висмута. Если второй экстракт окрашен, то экстракцию повторяют еще раз.
Кислый водный раствор переносят в колбу, добавляют 0,5 мл пергидроля и осторожно кипятят в течение 15 мин. Затем приливают 0,5 мл бромной воды и продолжают кипятить до удаления брома. Если бром сразу восстанавливается, то это означает, что в растворе присутствует перекись водорода. В таком случае кипячение продолжают несколько минут, прибавляют 0,5 мл бромной воды и кипятят до удаления брома. Нагревание прекращают, добавляют 2 мл раствора тиогликолевой кислоты и 0,5 мл раствора смеси аскорбиновой кислоты и иодида калия. Спустя 15 мин раствор охлаждают и добавляют 1 мл раствора смеси аскорбиновой кислоты и иодида калия.
Добавляют 5 мл дитиокарбамата и экстрагируют мышьяк(Ш), встряхивая в течение 10 с. При этом одновременно экстрагируется сурьма и олово. Хлороформный раствор сливают в колбу емкостью 50 мл. Водную фазу промывают без перемешивания тремя порциями хлороформа по 1 мл, который прибавляют к хлороформному раствору. Экстракцию повторяют еще с 5 мл раствора дитиокарбамата и промывают водный раствор 2 мл хлороформа, сливая хлороформные растворы в ту же колбу. Если в анализируемой' пробе содержится фосфат, то объединенный хлороформный экстракт встряхивают в течение нескольких секунд с 5 мл 1 н. серной кислоты для удаления следов фосфата.
Хлороформный экстракт помещают в маленькую фарфоровую чашку, в которую добавляют 1 мл дистиллированной воды так, чтобы она распределилась кольцом по окружности чашки над хлороформным раствором, и выпаривают на водяной бане до удале
ния хлороформа. Приливают 2 мл концентрированной азотной кислоты и выпаривают на водяной бане досуха. Затем смачивают сухой остаток 2 мл раствора нитрата магния и упаривают до образования густой массы, которую смачивают 0,5 мл концентрированной азотной кислоты и осторожно нагревают до растворения солей. Раствор переносят в маленький платиновый тигель с помощью 2 мл воды, выпаривают досуха, подсушивают и прокаливают при 500 °C. Остаток растворяют в 0,2 мл 6 н. серной кислоты и нескольких каплях воды. Содержание мышьяка определяют одним из описанных ниже методов.
IV. 1.6. Другие методы отделения и концентрирования мышьяка
Мышьяк количественно экстрагируется в виде фторидного комплекса H[AsFe] из концентрированного раствора фтористоводородной кислоты [40], а также в виде смешанного комплекса ферроина с гексафторидом мышьяка [41]. Фосфор отделяют экстракцией фосфорномолибденовой гетерополикислоты амиловым спиртом. Мышьяковомолибденовая гетерополикислота при этом остается в водной фазе [42—45].
Отделение мышьяка от многих компонентов можно проводить осаждением его сероводородом из сильно кислых хлористоводородных растворов.
• Соосаждение мышьяка проводят в виде арсената железа вместе с гидратом окиси железа, а также в виде арсената магния и аммония вместе с фосфатом магния и аммония [46].
Мышьяк от ионов металлов часто отделяют с помощью ионо-обменников [47]. Для этого кислый раствор (0,3 н. по НС1), содержащий мышьяковую кислоту, пропускают через катионит (КУ-1, КУ-2 и др.) в Н+-форме. При этом катионы остаются на катионите, а мышьяковая кислота остается в элюате.
Для отделения мышьяка применяют также тонкослойную [48, 49] и бумажную хроматографию [50—52]. Отделение катионов металлов от мышьяка можно также проводить экстракцией в виде купферонатов, оксихинолинатов и других соединений с органическими реагентами.
IV. 2. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ ОПРЕДЕЛЕНИЯ МЫШЬЯКА
В наиболее удачном методе определения мышьяка использована реакция образования желтой мышьяковомолибденовой. гетерополикислоты, которую затем восстанавливают. до так называемой мышьяковомолибденовой сини. Однако можно проводить определение мышьяка и по оптической плотности желтого комплекса [53].
Спектры поглощения желтого мышьяковомолибденового комплекса и мышьяковомолибденовой сини представлены на рис. 30. Из этого рисунка следует, что если проводить измерение оптической плотности желтого комплекса в видимой области при 400 нм, то чувствительность метода примерно в 5 раз меньше по сравнению с чувствительностью метода, основанного на измерении оптической плотности мышьяковомолибденовой сини при 700—800 нм. Свойства мышьяковомолибденовых гетерополикислот, как желтой, так и синей, подобны свойствам фосфорномолибденовых гетерополикомплексов (см. гл. III). Поэтому основная трудность при определении мышьяка этим методом заключается в отделении _4 мышьяка от фосфора. Методы
£ 10 ----отделения мышьяка рассмот-
/ г рены в разделе IV. 1.
Наиболее старым и, по-ви-
। X димому, наиболее чувствитель-
\ ным методом является метод,
Х^ основанный на выделении ар-
°зоо 350 doo dov65O 750 ‘воо'л,™ сина и поглощении его различными поглотителями с образо-Рис. 30. Спектры поглощения желтого (/) ванием окрашенных соедине-и синего (2) мышьяковомолибденовых ний. В качестве поглотителя комплексов. широко применяются бумаги,
смоченные растворами бромида и хлорида ртути(II). Содержание мышьяка определяют по интенсивности окраски ртутных бумажек. В зависимости от содержания мышьяка образуются соединения H(HgBr)2As— желтого цвета, (HgBr)3As— коричневого цвета и Hg3As2 — черного цвета. Окраску бумаг сравнивают с окраской стандартной шкалы.
В качестве поглотителя широко применяется пиридиновый раствор дитиокарбамата серебра. При этом образуется интенсивно окрашенное в красный цвет соединение, по интенсивности окраски которого и определяют содержание мышьяка с помощью калибровочного графика. Этот метод выгодно отличается от метода, основанного на поглощении арсина бумажками, пропитанными раствором HgBr2, чувствительность его не ниже, но значительно выше точность определения. Кроме того, метод не требует одновременного приготовления стандартной шкалы.
Сравнительно большие количества мышьяка можно определить в виде коллоидного раствора элементного мышьяка. В качестве восстановителей в этом случае применяют гипофосфит или хлорид олова (III) в присутствии ионов меди как катализатора.
IV. 3. ОПРЕДЕЛЕНИЕ МЫШЬЯКА В ВИДЕ МЫШЬЯКОВОМОЛИБДЕНОВОЙ СИНИ
Мышьяк (V) при взаимодействии с молибдатом аммония образует желтую мышьяковомолибденовую гетерополикислоту H3[As(Mo3O 10)4], при восстановлении которой образуется синяя
мышьяковомолибденовая гетерополикислота, которую часто называют мышьяковомолибденовой синью. Интенсивность окраски сини пропорциональна содержанию мышьяка.
Детальное исследование реакции образования мышьяковомолибденовой сини показало, что при восстановлении желтой мышьяковомолибденовой гетерополикислоты проходят параллельно еще два процесса: образование простой молибденовой сини и частичное разрушение мышьяковомолибденовой сини [54]. Поэтому при определении мышьяка необходимо строго придерживаться методики анализа.
В качестве восстановителя применяют сернокислый гидразин [29, 55—68], хлорид олова(II) [69—75], гидрохинон [76, 77], аскорбиновую кислоту [78], диметилдиамид пиразолин-3,4-дикарбоновую кислоту (препарат ИЭМ-335) [79] и др.
Этот метод применен для определения мышьяка в металлическом германии; предварительно мышьяк(Ш) окисляют до мышьяка (V) и основную массу германия отгоняют в виде тетрахлорида. Затем восстанавливают мышьяк до трехвалентного с помощью иодида калия или сульфита натрия, экстрагируют диэтилдитио-карбаматный комплекс мышьяка четыреххлористым углеродом и определяют его в виде мышьяковомолибденовой сини [80].
Для определения мышьяка в уране предварительно отделяют его в виде арсина и затем определяют в водном растворе [81] или экстрагируя синь с помощью метилизобутилкетона [82]. При определении мышьяка в селене последний предварительно выделяют в элементном состоянии, обрабатывая навеску сернистым ангидридом, и в фильтрате определяют мышьяк [83] или предварительно экстрагируют из фильтрата мышьяк в виде диэтилдитиокарбамата, а затем проводят определение [84]. В случае анализа серы мышьяк предварительно извлекают раствором аммиака и определяют в виде мышьяковомолибденовой сини [85].
Прямое определение мышьяка проводят в серной кислоте [86]. В силикатных горных породах мышьяк определяют после предварительного выделения в виде арсина [70, 87], арсената железа [88] или экстракцией с помощью диэтила^моний-диэтилдитиокарбама-та [89]. Для разложения угля навеску пробы спекают с окисью магния и отгоняют мышьяк в виде бромида [70], затем определяют его в виде сини, применяя в качестве восстановителя хлорид олова (II). Хлорид олова(II) также применяется в качестве восстановителя при определении мышьяка в производственных и сточных водах металлургических заводов [90] и в атмосферном воздухе [91]. При анализе нефтяных продуктов и катализаторов риформинга [92] пробу разлагают перекисью водорода и серной кислотой, а затем отгоняют хлорид мышьяка(III) и определяют его в дистилляте.
Мешающие вещества. Определению мешают фосфорная, кремневая и германиевая кислоты, образующие с молибдатом соответствующие гетерополик!глоты. Поэтому мышьяк предварительно отделяют от кремния, фосфора и германия. Мышьяк можно
определить и в присутствии германия,, но восстановление в этом случае проводят при более высокой кислотности [79]. Оптимальная кислотность для образования синей мышьяковомолибденовой гетерополикислоты соответствует 0,25 н. по серной кислоте, хотя и при более высокой кислотности происходит практически полное восстановление желтой мышьяковомолибденовой гетерополикислоты до синей. Однако во всех случаях необходимо поддерживать постоянную кислотность и концентрацию молибдата аммония.
Измерение оптической плотности. Максимум светопоглощения восстановленной мышьяковомолибденовой сини зависит от применяемого восстановителя. Поэтому оптическую плотность раствора необходимо измерять в области 700—840 нм (см. рис. 30). Молярный коэффициент поглощения мышьяковомолибденовой сини (ез-ю) равен 2,5-104.
Методы стандартной шкалы и колориметрического титрования неприменимы, так как реакция идет медленно, а окрашенный продукт реакции неустойчив во времени.
Метод стандартной шкалы можно применять в случае экстракции мышьяковомолибденовой сини изоамиловым спиртом [28]. Нагревание раствора ускоряет процесс образования мышьяковомолибденовой сини, после чего раствор становится устойчивым достаточно длительное время.
IV. 3.1. Определение мышьяка в отсутствие мешающих веществ с применением в качестве восстановителя сернокислого гидразина
Реактивы
Стандартный раствор арсената. Навеску мышьяковистого ангидрида (х. ч.) 0,132 г растворяют в 2—3 мл 1 и. раствора щелочи, переносят в мерную колбу емкостью 100 мл, подкисляют хлористоводородной кислотой до кислой реакции, прибавляют 0,5 г хлората калия, разбавляют водой до метки и хорошо перемешивают. Такой раствор содержит 1 мг мышьяка в 1 мл. Из этого раствора разбавлением готовят стандартный раствор с содержанием мышьяка 10 мкг в 1 мл.
Стандартный раствор можно приготовить из арсената натрия, титр такого раствора устанавливают весовым методом, осаждая арсенат в виде магний-аммоний арсената и взвешивая в виде Mg2A32O7.
Молибдат алцнония, растворяют 1 г молибдата аммония в 100 мл 6 н. серной кислоты и сохраняют в полиэтиленовой посуде.
Сернокислый гидразин. Растворяют 0,15 г сернокислого гидразина в 100 мл воды.
Ход анализа. До 20 мл нейтрального испытуемого раствора, содержащего от 1 до 50 мкг мышьяка, помещают в колбу емкостью 30 мл, прибавляют 1 мл раствора молибдата аммония и 0,5 мл раствора сернокислого гидразина. Колбу устанавливают в кипящую водяную баню и выдерживают в течение 15 мин. Время отсчитывают с момента начала кипения воды после погружения колбы. Затем колбу вынимают, охлаждают под краном, переносят раствор в мерную колбу емкостью 25 мл, колбу споласкивают
2 раза небольшим количеством воды, которую добавляют к основному раствору, разбавляют до метки водой, перемешивают и измеряют оптическую плотность при 840 нм.
По предварительно построенному калибровочному графику находят содержание мышьяка.
IV. 3.2. Определение мышьяка в отсутствие мешающих веществ с применением в качестве восстановителя хлорида олова (II)
Реактивы
Стандартный раствор арсената, см. раздел IV. 3.1.
Хлорид олова(П), 25%-ный раствор в хлористоводородной кислоте. Из этого раствора перед употреблением путем разбавления готовят 0,5%-ный раствор.
Молибдат аммония, 5%-ный раствор в воде.
Ход анализа. До 20 мл нейтрального испытуемого раствора, содержащего от 1 до 50 мкг мышьяка, помещают в мерную колбу емкостью 25 мл, прибавляют 1 мл хлористоводородной кислоты, разбавленной (1 : 1), 2 мл раствора молибдата аммония, если необходимо, разбавляют до 20 мл и перемешивают. Затем прибавляют 2 мл раствора SnCh, помещают в кипящую водяную баню и нагревают в течение 15 мин, раствор охлаждают, если нужно, разбавляют до метки водой, перемешивают и измеряют оптическую плотность.
Содержание мышьяка находят по калибровочному графику.
IV. 3.3. Определение мышьяка в черных металлах
При переведении в раствор пробы черных металлов могут быть значительные потери мышьяка. Это зависит от способа переведения металла в раствор, что, в свою очередь, зависит от предполагаемого способа отделения мышьяка. Кроме того, при одном и том же способе растворения пробы потери мышьяка зависят от содержания компонентов металла. Так, при растворении металлов с высоким содержанием связанного углерода в царской водке мышьяк восстанавливается углеродом и наблюдаются значительные потери его в виде AsCl3. Поэтому рекомендовано растворять навеску металла в азотной кислоте, смеси азотной и хлористоводородной кислоты (2:1), в серной кислоте в присутствии перманганата калия [93] или в смеси азотной, хлорной кислот и воды [94] в соотношении 1:1:1.
Обычно при анализе черных металлов мышьяк предварительно отделяют, однако определение мышьяка в стали можно провести и без предварительного отделения. Для этого ведут анализ двух одинаковых объемов раствора: в один не добавляют сульфит, во второй добавляют. В первом случае образуется смесь фосфорномолибденовой и мышьяковомолибденовой сини, т. е. оптическая
плотность раствора будет соответствовать содержанию суммы мышьяка и фосфора. Во втором случае сульфит восстанавливает мышьяковую кислоту до мышьяковистой и образование мышьяковомолибденовой гетерополикислоты не происходит. Поэтому определяют только содержание фосфора. По разности значений оптических плотностей в первом и втором случаях находят оптическую плотность раствора, соответствующую содержанию мышьяковомолибденовой сини, а по этому значению находят содержание мышьяка по калибровочному графику [94]. Однако такой вариант применим только в том случае, если соотношение [P]:[As] не превышает 1 : 2. При большем содержании мышьяка его следует отделить.
Реактивы
Перманганат калия, насыщенный раствор.
Ход анализа. Первый вариант. В стакан емкостью 100— 150 мл помещают навеску 0,2 г металла (при содержании 0,1 — 0,15% мышьяка, при меньшем содержании увеличивают навеску) и растворяют в 7—10 мл азотной кислоты, разбавленной (1:1). После растворения жидкость выпаривают досуха при ГЗО—150 °C, охлаждают и с помощью 30 мл хлористоводородной кислоты, разбавленной (1:1), переводят содержимое стакана в дистилляционную колбу емкостью 100 мл. Затем прибавляют 0,5 г бромида калия, 1,0—1,5 г сернокислого гидразина и мышьяк отгоняют, как указано в разделе IV. 1.1.
Второй вариант. В круглодонную дистилляционную колбу емкостью 100 мл помещают 0,2—0,5 г металла (в зависимости от содержания мышьяка), добавляют 20 мл насыщенного раствора перманганата калия, 10 мл серной кислоты, разбавленной (1 : 1), и нагревают на водяной бане до полного растворения навески. При растворении следят, чтобы все время был избыток перманганата. После растворения навески прибавляют небольшими порциями сернокислый гидразин для удаления избытка перманганата, что хорошо видно по обесцвечиванию раствора. Затем добавляют 0,5 г бромида калия, 1,0—1,5 г сернокислого гидразина, 10 мл концентрированной хлористоводородной кислоты и отгоняют мышьяк, как указано в разделе IV. 1.1.
В обоих вариантах собранный дистиллят переносят в мерную колбу емкостью 100 мл, обмывают стакан 2—3 раза небольшим количеством воды, прибавляют две капли фенолфталеина и нейтрализуют концентрированным раствором аммиака, периодически охлаждая раствор, до появления устойчивой розовой окраски. Затем разбавляют дистиллированной водой до метки и тщательно перемешивают. Отбирают аликвотную часть раствора и определяют мышьяк, как указано в разделах IV. 3.1 или IV. 3.2.
Третий вариант. По этому варианту после переведения пробы в раствор соосаждают мышьяк вместе с гидратом окиси
железа и проводят экстракционное отделение мышьяка в виде иодида [95]. Навеску пробы растворяют в азотной кислоте, разбавленной (1 : 1), как указано в первом варианте. После выпаривания раствора сухой остаток растворяют в минимальном количестве концентрированной хлористоводородной кислоты, разбавляют водой до 200 мл, нейтрализуют аммиаком до pH = 2,8—3,0. При этом образуется осадок гидрата окиси железа, с которым практически полностью соосаждается мышьяк в виде арсената железа. Осадок растворяют в минимальном количестве концентрированной хлористоводородной кислоты, раствор переносят в делительную воронку и прибавляют равный объем 1 н. раствора иодида калия. Из полученного раствора мышьяк экстрагируют бензолом, реэкстрагируют водой, окисляют до арсената и определяют, как указано в разделе IV. 3.1 или IV. 3.2.
IY. 3.4. Определение мышьяка в железных, марганцевых рудах и агломератах
IV. 3.4.1. Первый вариант
Пробу спекают со смесью карбоната натрия и окиси цинка [96, 97]. Затем спекшуюся массу измельчают, переносят в дистилляционную колбу, прибавляют хлористоводородной кислоты и сернокислого гидразина и мышьяк отгоняют. Затем определяют мышьяк в виде мышьяковомолибденовой сини.
Реактивы
Смесь для спекания, перемешивают тонкоизмельченный карбонат натрия ч. д. а. и прокаленную окись цинка ч. д. а. в весовом отношении 1 :2. Хранят в закрытой банке.
Окись магния или окись цинка ч. д. а., прокаленные и измельченные.
Ход анализа. Навеску 0,5 г, содержащую до 100 мкг мышьяка, смешивают с 1 г смеси для спекания и заворачивают в папиросную бумагу. В фарфоровый тигель № 3 с неповрежденной глазурью насыпают, уплотняя, 1 г окиси цинка или 0,3 г окиси магния; помещают завернутую в бумагу смесь навески со смесью для спекания и нагревают при 850—900 °C в течение 15—20 мин. Тигель охлаждают, спекшуюся массу измельчают в тигле стеклянным пестиком и порошок пересыпают в колбу для перегонки. В гигель, где проводилось спекание, наливают несколько миллилитров горячей воды и оставляют на некоторое время, помешивая палочкой. Когда остатки сплавленной массы полностью отделятся от тигля, его содержимое выливают в колбу со спеком и доводят объем воды в колбе до 10 мл.
Если материал содержит много марганца, то к спеку в колбе сразу добавляют в зависимости от содержания марганца 0,2— 0,5 г щавелевой кислоты.
При анализе окисленных руд можно навеску сразу, не спекая, переносить в колбу для перегонки и дальше поступать так, как описано ниже.
В колбу со снеком добавляют 0,5 г бромида калия, 0,5 г сернокислого гидразина, быстро вливают 15—20 мл концентрированной хлористоводородной кислоты, немедленно соединяют колбу с холодильником аппарата для отгонки и проводят отгонку, как указано в разделе IV. 1.1. Далее определяют мышьяк, как указано в разделе IV. 3.3.
IV. 3.4.2. Второй вариант
Мышьяк из пробы извлекают азотной кислотой, избыток которой удаляют выпариванием на водяной бане. Сухой остаток растворяют в концентрированной хлористоводородной кислоте, восстанавливают мышьяк(V) до мышьяка (III) сернокислым гидразином и хлорид мышьяка (III) экстрагируют четыреххлористым углеродом. Затем реэкстрагируют водой, мышьяк(Ш) окисляют до мышьяковой кислоты бромной водой, избыток брома удаляют и определяют мышьяк в виде мышьяковомолибденовой сини [98].
Реактивы
Четыреххлористый углерод, ч. д. а.
Ход анализа. Навеску измельченной руды 0,5—1,0 г помещают в стакан емкостью 100 мл, прибавляют 10—15 мл азотной кислоты, разбавленной (1 : 1), накрывают стакан часовым стеклом и кипятят до исчезновения окислов азота. Стекло снимают, обмывают водой, сливая воду в стакан, добавляют в стакан еще примерно равный объем воды, фильтруют в фарфоровую чашку и фильтрат выпаривают на водяной бане досуха. Остаток растворяют в 20 мл концентрированной хлористоводородной кислоты, раствор переносят в делительную воронку, добавляют 0,5 г сернокислого гидразина, 20 мл четыреххлористого углерода и содержимое воронки встряхивают в течение 1 мин. Органический слой отделяют в другую чистую воронку, содержащую 40 мл воды, и встряхивают в течение 1 мин. После расслаивания жидкостей четыреххлористый углерод сливают в первую воронку, а водную фазу собирают в мерную колбу емкостью 100 мл. Экстракцию и реэкстракцию повторяют еще 4 раза, применяя для реэкстракции дистиллированную воду порциями по 10 мл. К водной вытяжке добавляют немного бромной воды и нагревают на водяной бане до удаления брома. Раствор охлаждают, разбавляют водой до метки, перемешивают и определяют мышьяк, как указано в разделах IV. 3.1 или IV. 3.2.
IV. 3.5. Определение мышьяка в продуктах цветной металлургии
Пробу разлагают азотной кислотой, которую удаляют выпариванием с серной кислотой. Затем иодид мышьяка(III) экстрагируют четыреххлористым углеродом, реэкстрагируют водой, окисляют мышьяк(III) до мышьяковой кислоты перманганатом и определяют мышьяк в виде сини [21, 86, 99].
Реактивы
Хлористоводородная кислота, очищенная от мышьяка. Для этого в 500 мл концентрированной хлористоводородной кислоты растворяют 10 г иодида калия, переносят раствор в делительную воронку, прибавляют 25 мл СС14 и встряхивают в течение 2 мин. После отстаивания слой четыреххлористого углерода отделяют и экстракцию мышьяка повторяют еще с 25 мл СС14.
Смесь кислот. Готовят смеси хлористоводородной и азотной кислот в соотношении (1 : 3).
Ход анализа. Навеску пробы 0,5—1,0 г, в зависимости от содержания мышьяка, помещают в коническую колбу емкостью 150—200 мл, приливают 10—15 мл азотной кислоты (пл. 1,40), нагревают до растворения пробы, добавляют 10 мл серной кислоты, разбавленной (1:1), и выпаривают до появления густых белых паров. Если материал содержит значительные количества германия, то навеску разлагают 15 мл смеси кислот. После охлаждения стенки колбы обмывают небольшим количеством воды и выпаривание с H2SO4 до появления паров повторяют. Добавив 0,5 г сернокислого гидразина, продолжают выпаривание еще в течение 5—10 мин. Охлажденные соли растворяют при нагревании в 30— 40 мл воды, отфильтровывают нерастворимый остаток и промывают на фильтре 2—3 раза водой. Если проба содержит меньше 0,005% мышьяка, то фильтрат упаривают до 5 мл. При большем содержании мышьяка раствор, не фильтруя, переносят в мерную колбу емкостью 100 мл, разбавляют до метки водой и хорошо перемешивают.
В делительную воронку емкостью 100 мл переводят весь испытуемый раствор или его аликвотную часть (5—10 мл) так, чтобы в ней содержалось 5—50 мкг мышьяка, и прибавляют трехкратное количество относительно взятого объема раствора очищенной концентрированной хлористоводородной кислоты, содержащей 0,1 моль/л иодида калия. Из полученного раствора экстрагируют иодид мышьяка(III) 20 мл четыреххлористого углерода, встряхивая в течение 2 мин. Органический слой переносят в другую делительную воронку емкостью 50 мл и промывают 5—10 мл 9 н. очищенной хлористоводородной кислоты, встряхивая в течение 20— 30 с. Промытый экстракт сливают в делительную воронку и ре-эстрагируют мышьяк 15 мл воды, встряхивая в течение 2 мин. Извлечение мышьяка водой повторяют еще раз.
В объединенных водных вытяжках мышьяк (III) окисляют перманганатом и определяют, как указано в разделах IV. 3.1 или IV. 3.2.
IV. 3.6. Определение мышьяка в металлической меди и ее сплавах
Можно рекомендовать два варианта подготовки пробы к анализу при определении мышьяка в меди и ее сплавах. Лучшие результаты дает метод с предварительным электролитическим выделением меди [100], последующей экстракцией иодида мышьяка(III) хлороформом п определением его в виде мышьяковомолибденовой сини. По другому, варианту, пробу растворяют в смеси хлористоводородной кислоты и пергидроля, избыток последнего удаляют кипячением и восстановлением гипофосфитом [101, 102], а затем экстрагируют хлорид мышьяка(III) и продолжают определение, как и в первом варианте. Этот метод дает удовлетворительные результаты.
Реактивы
Сульфат железа(Ш), раствор, содержащий 5 мг железа в 1 мл.
Хлорид титана(Ш), 35% -ный раствор в концентрированной хлористоводородной кислоте.
Иодид калия, 40%-ный раствор.
Фосфорная кислота, 50%-ный раствор.
Ход анализа. Навеску сплава 1 г при содержании мышьяка 0,1% или 0,1 г при его содержании больше 0,1% растворяют в 10 мл азотной кислоты (пл. 1,33). Если сплав содержит олово, то навеску пробы растворяют в смеси 10 мл азотной кислоты (пл. 1,33), 10 мл 4%-ной борной кислоты, 1,5 мл фтористоводородной кислоты, разбавленной (1:1) и 5 мл раствора сульфата железа (III). В том и другом случае раствор после полного растворения пробы разбавляют водой до ~200 мл и выделяют медь электролитически, с вращающимся анодом при силе тока 5 А. Затем электроды обмывают водой. Если в пробе присутствует свинец, то он выделяется на аноде. Этот осадок растворяют в растворе, из которого проводили выделение меди, и разбавляют раствор до 250 или 500 мл. К аликвотной части раствора, содержащей до 100 мкг мышьяка, прибавляют 2 мл серной кислоты, разбавленной (1 : 1), и выпаривают до появления ее паров. Остаток растворяют при нагревании в 10 мл хлористоводородной кислоты, разбавленной (1 : 1), охлаждают, вводят 2 мл 35%-ного раствора хлорида тита-на(Ш) и 2 мл раствора иодида калия,.раствор перемешивают и выдерживают в течение 5—10 мин. Затем его переводят в делительную воронку, обмывая стакан 35 мл концентрированной хлористоводородной кислоты, и дважды экстрагируют иодид мышьяка хлороформом. Первый раз берут 25 мл, а второй раз— 10 мл хлороформа. Объединенные экстракты помещают в делительную воронку и реэкстрагируют мышьяк 15 мл воды. Далее ведут определение, как указано в разделах IV. 3.1 или IV. 3.2.
Второй вариант. Навеску образца 1 г или меньше (при большом содержании мышьяка) помещают в коническую колбу
емкостью 100 мл, прибавляют 5 мл концентрированной хлористоводородной кислоты, охлаждают в холодной воде, вводят сначала 3 мл, а затем еще 7 мл пергидроля, кипятят в течение 10 мин, охлаждают, переносят раствор в делительную воронку с помощью 50 мл концентрированной хлористоводородной кислоты и прибавляют 4 мл 50%-ного раствора фосфорноватистой кислоты (Н3РО2). Спустя 5 мин прибавляют 10 мл концентрированной хлористоводородной кислоты и экстрагируют хлорид мышьяка(III) хлороформом. После реэкстракции хлорида мышьяка водой окисляют мышь-як(Ш) иодом и определяют, как указано в разделах IV. 3.1 и IV. 3.2.
IV. 3.7. Определение мышьяка в чистых металлах
При анализе чистых металлов предварительное выделение мышьяка проводят в виде арсина, а также экстракцией дйэтилди-тиокарбамата мышьяка хлороформом из сильнокислой среды. Малые количества мышьяка определяют в виде мышьяковомолибденовой сини в небольшом объеме изоамилового спирта. Этот метод дает возможность определить 0,2 мкг мышьяка в 10 мл экстракта [103], а при уменьшении объемов водной и неводной фаз — 0,1 мкг мышьяка в 0,4 мл экстракта. Измерение интенсивности окраски проводят методом стандартной шкалы.
Ниже приведены методы определения мышьяка в сурьме, ниобии, ванадии, кремнии, галлии, индии и таллии, разработанные В. А. Назаренко с сотрудниками [28], а также в цинке и кадмии [104]. При анализе сурьмы, ниобия, кремния и ванадия мышьяк выделяют в виде арсина, который поглощается смесью растворов хлорида ртути(II) и перманганата. В случае анализа сурьмы и ниобия мышьяк(\9 предварительно выделяют вместе с магний-аммоний фосфатом. Для определения мышьяка в галлии, индии и таллии мышьяк(V) предварительно восстанавливают аскорбиновой кислотой и экстрагируют хлороформом в виде диэтилдитио-карбаматного комплекса, после разложения которого определяют мышьяк в виде мышьяковомолибденовой сини.
IV. 3.7.1. Определение мышьяка в сурьме
Навеску металла разрушают азотной кислотой, сурьмяную кислоту связывают тартратом и мышьяк выделяют вместе с магний-аммоний фосфатом. После чего отделяют мышьяк в виде арсина и определяют в виде мышьяковомолибденовой сини с применением экстракции изоамиловым спиртом.
Недостатком данного метода является то, что при осаждении мышьяка вместе с магний-аммоний фосфатом содержание мышьяка в холостой пробе оказывается весьма значительным. Даже в случае применения специально очищенных реактивов результат холостого опыта составляет до 0,5 мкг. Таким образом, этим методом
можно определить не менее 0,3 мкг мышьяка, хотя чувствительность метода позволяет определять 0,1 мкг мышьяка.
Реактивы
Фосфат калия, однозамещенный, 2%-ный раствор. Растворяют 20 г однозамещенного фосфата калия в воде, разбавляют до 1 л и перемешивают.
Магнезиальная смесь, растворяют 55 г хлорида магния MgCl2-6H2O и 105 г хлорида аммония NH4C1 в воде, добавляют 350 мл концентрированного раствора аммиака и разбавляют водой до 1 л. Дают раствору отстояться в течение 3—4 дней и фильтруют.
Стандартный раствор арсената, см. раздел IV. 3.1.
Иодид калия, 15%-ный раствор.
Хлорид олова(П), 0,5%-ный раствор в хлористоводородной кислоте, разбавленной (1 :'1).
Перманганат калия, 0,1 н. раствор.
Молибдат-гидразиновый реактив. Растворяют 1 г молибдата аммония в 100 мл 5 н. серной кислоты и 0,15 г сернокислого гидразина в 100 мл воды. Разбавляют 10 мл раствора молибдата аммония до 90 мл водой, добавляют 1 мл раствора сернокислого гидразина и водой разбавляют до 100 мл. Эта смесь неустойчива, и поэтому ее готовят ежедневно или в ходе анализа отдельно прибавляют соответствующее количество растворов молибдата аммония и сернокислого гидразина.
Поглотительная смесь. Смесь состоит из 1 мл 1,5%-ного раствора HgCl2, 0,2 мл 6 н. серной кислоты и 0,2 мл раствора перманганата калия.
Стандартная шкала. Помещают в ряд пробирок с притертыми пробками стандартный раствор арсената, содержащий соответственно 0; 0,5; 1,0 ... 5 мкг мышьяка, доводят объем до 2 мл водой, вводят все те реактивы, которые введены в поглотительную смесь, но раствора перманганата калия вводят 0,1 мл, нагревают в кипящей воде 5 мин и оставляют охлаждаться. Далее стандартные растворы обрабатывают, как анализируемый раствор (см. ниже).
Ход анализа. Навеску 1 г растертого в агатовой ступке металла (или 0,2—0,5 г при содержании мышьяка выше 0,0005%) помещают в стакан емкостью 100 мл, добавляют 5 мл азотной кислоты (пл. 1,4), накрывают стеклом и нагревают на плитке, осторожно покачивая стакан, избегая бурного кипения и излишнего испарения кислоты. Нагревают до превращения металла в белую массу. Охлаждают, снимают стекло, дают улетучиться окислам азота, насыпают 6 г тартрата аммония (при навеске 0,5 г сурьмы достаточно 5 г тартрата), приливают воды до 80 мл и нагревают при помешивании до растворения сурьмяной кислоты. Раствор переливают в стакан емкостью 300 мл и разбавляют водой до 150 мл. Охлаждают, добавляют 10 мл раствора однозамещенного фосфата калия, 10 мл магнезиальной смеси и при помешивании — раствор аммиака до слабого запаха и начала образования осадка. Затем приливают еще 10 мл раствора аммиака, перемешивают и оставляют на 7—12 ч. Осадок отфильтровывают через бумажный фильтр (7 см) и 15—16 раз промывают малыми порциями 1%-ного раствора аммиака. Осадок на фильтре и стенках стакана растворяют в горячей серной кислоте, разбавленной (1 : 10), собирая раствор в колбу прибора для отгонки арсина. Фильтр и стакан обмывают той же кислотой, сливая раствор в колбу прибора для отгонки арсина. На растворение и промывание должно быть израсходовано 50 мл серной кислоты. К еще теплому раствору прибавляют 2 мл 152
раствора иодида калия и 0,5 мл раствора хлорида олова(II) и оставляют на 30 мин. К холодному раствору прибавляют 5 г гранулированного цинка и закрывают колбу пробкой с насадкой, соединенной с поглотительным прибором (рис. 31). Капилляр 2 мед
ленно погружают в поглотительную смесь, находящуюся в центри-
фужной пробирке. Образующийся в колбе газ пропускают через поглотительный раствор в течение 1,5 ч.
После окончания пропускания газа, оставляя капилляр в поглотительной жидкости, переливают жидкость из поглотителя в пробирку с притертой пробкой и споласкивают поглотитель и капилляр два раза по 2,5 мл молибдат-гидразиновым реактивом, а затем 2 мл воды, сливая промывные жидкости в ту же пробирку. Во все пробирки со стандартными растворами приливают по 5 мл молибдат-гидразинового реактива и все пробирки с испытуемым и стандартными растворами погружают на 15 мин в кипящую воду (время отсчитывают с момента закипания воды после погружения пробирок). Затем пробирки охлаждают в холодной воде, приливают по 1 мл изоамилового спирта, закрывают их пробками и сильно встряхивают 10 раз. После
I
Рис. 31. По-
глотитель-ный прибор для отгонки микроколичеств мышьяка в виде арсина:
/—трубка с ватой; 2 — капилляр; 3 — уплотнительная
трубка;4—центрифужная пробирка.
разделения слоев сравнивают синие окраски верхних слоев, наблюдая их сбоку.
Обязательно проводят холостой опыт; при этом в поглотительную жидкость прибавляют не 0,20 мл, а 0,15 мл раствора перманганата калия. Однако во всех случаях следят, чтобы в поглотительной смеси после пропускания газа содержался еще неразложившийся перманганат. Полное разложение перманганата указывает на высокое содержание мышьяка в пробе или на попадание больших количеств сурьмы в фосфатный осадок. В этом случае опыт следут повторить с мень-
шей навеской металла. Одновременно в поглотительном приборе следует заменить ватный тампон в трубке 1.
IV. 3.7.2. Определение мышьяка в ниобии
Лучшим растворителем для ниобия является смесь серной кислоты и сульфата калия, но при растворении теряется до 20% мышьяка в виде арсина, поэтому полученный результат необходимо умножить на коэффициент 1,25.
Ход анализа. Навеску 1 г тонко нарезанного ниобия помещают в колбу Кьельдаля емкостью 50 мл, добавляют 4 г сульфата калия и 10 мл серной кислоты (пл. 1,84), помещают в углубление проволочной сетки и нагревают на пламени газовой горелки, избегая слишком бурной реакции. Горло колбы оставляют открытым. Растворение металла заканчивается через 10—15 мин, после чего жидкости в колбе дают остыть.
Взвешивают 7 г однозамещенного тартрата аммония, переносят в стакан емкостью 300 мл, растворяют при нагревании в 100 мл воды и приливают к раствору 2 мл 30%-ной перекиси водорода. Отливают около 20 мл раствора для промывания колбы Кьельдаля. Раствор в стакане нагревают до 60—70 °C и наливают в него небольшими порциями при помешивании раствор из колбы Кьельдаля, помешивая палочкой до растворения образующегося вначале осадка ниобиевой кислоты. Следующую порцию жидкости из колбы Кьельдаля вносят после растворения осадка, образовавшегося после внесения предыдущей порции. Колбу Кьельдаля обмывают жидкостью из стакана, растворяя образующийся на стенках колбы осадок ниобиевой кислоты. После этого колбу промывают отделенной ранее порцией раствора тартрата аммония и, наконец, небольшим количеством воды. Полученный желтый опалесцирующий раствор охлаждают, добавляют 10 мл раствора дигидрофос-фата калия и далее ведут определение, как указано в разделе IV. 3.7.1. Время отгонки арсина сокращают до 1 ч.
IV. 3.7.3. Определение мышьяка в ванадии
Навеску ванадия растворяют в азотной кислоте, которую удаляют выпариванием с серной кислотой. Восстановление ванадия и разложение азотной кислоты проводят щавелевой кислотой.
Реактивы
Хлорид олова(П), 0,5%-ный раствор.
Иодид калия, 15%-ный раствор.
Ход анализа. Навеску ванадия 1 г помещают в колбу прибора для выделения арсина емкостью 200—250 мл, прибавляют 10 мл концентрированной азотной кислоты и растворяют вначале на холоду, а затем при нагревании. Когда весь металл растворится, в колбу наливают 10 мл серной кислоты (пл. 1,84) и нагревают до растворения ванадиевой кислоты и прекращения выделения окислов азота. Колбу охлаждают, прибавляют 10 г щавелевой кислоты, горло и стенки колбы обмывают небольшим количеством воды, накрывают колбу часовым стеклом и осторожно нагревают через асбестированную сетку, периодически помешивая. После прекращения выделения окислов азота снимают стекло, обмывают горло и стенки колбы водой и жидкость нагревают до выделения паров серной кислоты. Если при этом выпадает осадок сульфата ванадия, прибавляют небольшое количество воды и нагревают до его растворения. Разбавляют водой до 100 мл, охлаждают, добавляют 1 мл раствора хлорида олова(II) и 0,2 мл раствора иодида калия и оставляют на 30 мин. Затем помещают в колбу 7 г гранулированного цинка, закрывают колбу насадкой и далее ведут определение, как указано в разделе IV. 3.7.1,
IV. 3.7.4. Определение мышьяка в кремнии
Навеску кремния растворяют в щелочи, подкисляют хлористоводородной кислотой (но не серной!) и отгоняют мышьяк в виде арсина. Затем определяют мышьяк в виде,мышьяковомолибденовой сини.
Навеску растертого в порошок кремния 1 г помещают в платиновую чашку, приливают 10—13 мл концентрированного раствора гидроокиси натрия и нагревают на водяной бане до растворения кремния, периодически покачивая чашку. В ходе растворения несколько раз прибавляют по 2—3 мл воды для возмещения испарившейся. Полученный раствор переносят с помощью 30 мл воды в колбу прибора для выделения арсина, приливают 15 мл хлористоводородной кислоты, 2 мл 15%-ного раствора иодида калия и 0,5 мл раствора хлорида олова(II) и оставляют на 15 мин. Насыпают 5 г гранулированного цинка, закрывают колбу пробкой с насадкой и продолжают определение, как указано в разделе IV. 3.7.1.
IV. 3.7.5. Определение мышьяка в галлии и индии
Мышьяк экстрагируют в виде диэтилдитиокарбаматного комплекса из сильнокислого раствора хлороформом. После удаления хлороформа выпариванием диэтилдитиокарбамат мышьяка разрушают азотной кислотой и определяют мышьяк в виде мышьяковомолибденовой сини с применением экстракции изоамиловым спиртом.
Реактивы
Пиросульфит натрия. Насыщенный раствор.
Иодид калия, 20%-ный раствор.
Аскорбиновая кислота, 5%-ный раствор.
Нитрат магния, 7%-ный раствор.
Молибдат аммония, 1%-ный раствор.
Сернокислый еидразин, 0,15%-ный раствор.
Стандартная шкала. Для приготовления шкалы в ряд пробирок вводят различное количество стандартного раствора мышьяка от 0,0 до 1,0 мл (1 мкг мышьяка в 1 мл) и разбавляют водой до 3 мл. Далее стандартные растворы обрабатывают, как указано ниже.
Ход анализа. Навеску металла 0,4 г помещают в коническую колбу емкостью 100 мл, смачивают 2 мл воды и прибавляют 3 мл серной кислоты (пл. 1,84), 2 мл концентрированной азотной кислоты и осторожно нагревают до растворения металла и начала выделения солей. Приливают 5 мл воды, нагревают до растворения и снова уйаривают до начала выделения соли. Такое выпаривание повторяют еще 3 раза, каждый раз обмывая горло колбы водой. Затем прибавляют 5 мл воды и 0,25 мг пиросульфита натрия, нагревают до кипения и кипятят до полного исчезновения запаха сернистого ангидрида, периодически добавляя в колбу воду. После этого разбавляют водой до 40 мл и к раствору, имеющему температуру 40—45-°C, прибавляют 1,5 мл раствора иодида калия и 1 мл
свежеприготовленного раствора аскорбиновой кислоты. Оставляют на 15 мин, холодный раствор переносят в делительную воронку, приливают 5 мл диэтилдитиокарбаминовой кислоты в хлороформе и взбалтывают в течение 30 с. После разделения жидкостей хлороформный слой сливают в делительную воронку емкостью 25 мл, а водную фазу еще раз экстрагируют 5 мл диэтилдитиокарбаминовой кислоты. Второй экстракт присоединяют к первому. Соединенные экстракты два раза промывают 1 н. серной кислотой порциями по 2,5 мл и хлороформные экстракты переливают в маленькую фарфоровую чашку. Добавляют в чашку 1 мл воды с тем, чтобы она распределилась кольцом по окружности чашки, и выпаривают на водяной бане до удаления хлороформа. Приливают 1,5 мл концентрированной азотной кислоты и выпаривают на водяной бане досуха. К остатку приливают 2 мл раствора нитрата магния и упаривают до образования густой массы. Смачивают 0,5 мл азотной кислоты, нагревают до растворения и раствор переносят в маленький платиновый тигель с помощью 2 мл воды. Выпаривают досуха, подсушивают на плитке и прокаливают в муфельной печи при 500°C. Остаток после прокаливания растворяют при нагревании в 0,2 мл 6 н. серной кислоты и нескольких каплях воды. Раствор переносят с помощью 3 мл воды в колориметрическую пробирку с притертой пробкой.
Во все пробирки с анализируемыми и стандартными растворами приливают по 0,3 мл свежеприготовленного раствора молибдата аммония в 6 н. серной кислоте и по 0,1 мл раствора сернокислого гидразина. Перемешивают и опускают на 5 мин в кипящую воду (время отсчитывают с момента закипания воды после погружения пробирок). Охлаждают в холодной воде, приливают во все пробирки по 0,4 мл изоамилового спирта, закрывают пробками и встряхивают 10 раз. После разделения жидкостей сравнивают синие окраски верхних спиртовых слоев.
IV. 3.7.6. Определение мышьяка в таллии
Навеску металла переводят в раствор, после удаления азотной кислоты экстрагируют диэтилдитиокарбаматный комплекс мышьяка, разрушают его и определяют мышьяк в виде синего мышьяко-вомолибденового .комплекса с применением экстракции.
Навеску металла в 0,4 г помещают в коническую колбу емкостью 100 мл, приливают 1,5 мл азотной кислоты и растворяют металл при слабом нагревании. Приливают 1,2 мл серной кислоты (пл. 1,84), 5 мл воды и нагревают до начала выделения паров серной кислоты, дают остыть, приливают 5 мл воды, обмывая горло колбы, и снова выпаривают до появления паров серной кислоты. Такую обработку повторяют еще 4 раза. Затем добавляют 5 мл воды, 0,25 г пиросульфита натрия и кипятят до исчезновения запаха сернистого ангидрида, время от времени добавляя воду. После 156
этого разбавляют водой до 40 мл, охлаждают, переливают в делительную воронку, добавляют 5 мл раствора диэтилдитиокарбамино-вой кислоты и далее поступают, как указано в разделе IV. 3.7.5.
IV. 3.7.7. Определение мышьяка в цинке и кадмии
Навеску металла в 0,1—0,5 г растворяют в смеси азотной и серной кислот и определяют мышьяк, как указано в разделе IV. 3.7.6.
IV. 3.8. Определение мышьяка в металлическом золоте
Навеску золота растворяют в царской водке, золото выделяют с помощью сернокислого гидразина в щелочной среде, отгоняют мышьяк в виде арсина, поглощают сернокислым раствором HgCls и перманганата и определяют мышьяк в виде мышьяковомолибденовой сини [105].
Ход анализа. Навеску золота 2—25 г растворяют в 10—20 мл царской водки и выпаривают почти досуха. Остаток растворяют в воде, подкисленной несколькими каплями хлористоводородной кислоты. Затем прибавляют 20 мл 10%-ного раствора КОН и нагревают до кипения. К раствору прибавляют твердый сернокислый гидразин до полного осаждения золота. Осадок отфильтровывают, промывают 0,5%-ным раствором КОН. Фильтрат осторожно при охлаждении нейтрализуют хлористоводородной кислотой и переносят в прибор для отгонки арсина. Арсин поглощают сернокислым раствором HgCl2 и перманганата и определяют мышьяк, как указано в разделе IV. 3.1.
IV. 3.9. Определение мышьяка в органических веществах
Наиболее трудной задачей при анализе органических веществ на мышьяк является разложение образца. Простым и в то же время дающим удовлетворительные результаты является метод сожжения пробы азотной и серной кислотами при нагревании.
Реактивы
Оксалат аммония, насыщенный раствор.
Ход анализа. Навеску образца от 1 до 20 г помещают в круглодонную колбу с длинным горлом, которую ставят на песочную баню, приливают в колбу 25—30 мл азотной кислоты (пл. 1,4), а затем 15 мл серной кислоты (пл. 1,84). Закрывают колбу воронкой и нагревают до полного разрушения органического вещества. Если наблюдается вспенивание или побурение раствора, то нагревание ведут с перерывами. После разложения пробы, которое можно заметить по посветлению раствора, осторожно прибавляют 50 мл воды, 15 мл раствора оксалата аммония и выпаривают до появления паров серной кислоты. Раствор охлаждают, разбавляют водой
до определенного объема, в аликвотной части определяют мышьяк, как указано в разделе IV. 3.1, или отделяют, если нужно, мышьяк одним из описанных методов и затем определяют.
IV. 4. ОПРЕДЕЛЕНИЕ МЫШЬЯКА С ПРИМЕНЕНИЕМ ИНДИКАТОРНЫХ БУМАГ
Мышьяк в кислой или щелочной среде восстанавливают металлическим цинком, алюминием, амальгамой натрия или электролитически до арсина, который при взаимодействии с хлоридом ртути(II), бромидом ртути(II), нитратом серебра или диэтилдитиокарбаматом серебра образует окрашенные соединения (см. IV-6). Интенсивность окраски пропорциональна содержанию мышьяка.
Метод очень чувствительный, но применение его требует исключительного внимания к условиям проведения эксперимента; Концентрация кислоты, количество и размер зерен или стружки металла, температура, продолжительность опыта должны быть совершенно идентичны как для испытуемого раствора, так и для раствора сравнения.
Мешающие вещества. Металлы, выделяющиеся на цинке или алюминии, замедляют процесс их растворения и мешают определению. Поэтому допустимо лишь небольшое количество их в исследуемом растворе. Окислители препятствуют выделению арсина. Железо(Ш) и селен(1У) понижают чувствительность метода.
При анализе германия рекомендовано отделять мышьяк соосаж-дением его с двуокисью марганца [106]. О мешающем влиянии других компонентов см. раздел IV. 1.2.
Измерение интенсивности окраски. В случае использования бумаги, смоченной растворами солей ртути или серебра, для измерения интенсивности окраски применим метод стандартных серий. Шкалу эталонов лучше всего готовить одновременно. Однако можно пользоваться и предварительно приготовленной шкалой, но соблюдение идентичности условий приготовления обязательно. Сохранять шкалу лучше всего в конверте из черной бумаги в эксикаторе над хлоридом кальция. Прямой солнечный свет обесцвечивает как парафинированные, так и непарафинированные шкалы полностью в течение одних суток, рассеянный комнатный свет— в течение двух суток. Во влажном воздухе в отсутствие воздействия света интенсивность окрасок шкалы в течение суток уменьшается на 25—50% [107].
Измерение интенсивности окраски пятен можно проводить методом отражательной спектрофотометрии [108, 109], для чего пользуются специальными насадками к спектрофотометрам.
Рекомендовано также применять невыцветающую искусственную шкалу, приготовляемую нанесением легкой штриховки цветными карандашами на белой бумаге [ПО].
Чувствительность метода — 0,01 мкг мышьяка в 25 мл конечного раствора.
IV. 4.1. Определение мышьяка в отсутствие мешающих веществ
Реактивы
Серная кислота, разбавленная (1:4) и не содержащая мышьяка.
Вата и бумага, смоченные 5%-ным раствором ацетата свинца и высушенные.
Фильтровальная бумага, пропитанная 5%-ным раствором хлорида или бромида ртути(II) и высушенная («сулемовая» бумага).
Ход анализа. Определение ведут в приборе, изображенном на рис. 32.
Коническая колба емкостью 50—100 мл при помощи стеклянной
(хуже резиновой) пробки соединена со стеклянной трубкой длиной
150 мм, диаметром 10—15 мм. Трубку заполняют ватой и фильтровальной бумагой, пропитанными раствором ацетата свинца для поглощения сероводорода, сверху трубку закрывают газовым фильтром [111, 112]. В дне стакана имеется отверстие диаметром 5—7 мм, которое закрывают «сулемовой» бумагой. На эту бумагу помещают бумажную шайбу и все прижимают плотно ко дну фильтра ниппелем. .
Испытуемый раствор (или навеску образца), содержащий до 10 мкг мышьяка, помещают в коническую колбу, приливают 10—15 мл серной кислоты, осторожно наклонив колбу, вводят туда же 5 г металлического цинка (не содержащего мышьяка) и быстро закрывают колбу пробкой с приготовленной трубкой. Реакцию ведут в течение 1 —1,5 ч, после чего прибор разбирают и сравнивают интенсивность окраски «сулемовой» бумаги
Рис. 32. Прибор для определения мышьяка:
а —прибор; б —детали газового фильтра; / — коническая колба; 2 — трубка с ватой и бумагой; 3 — газо >ый фил >тр; 4—металлический или пластмассовый стакан; 5 —ниппель; 6 — шайба.
с окраской таких же
стандартных бумажек, приготовленных в точно таких же условиях,
при различном содержании стандартного раствора соли мышьяка.
IV. 4.2. Определение арсина в воздухе
Пробу воздуха пропускают через поглотительные растворы, содержащие перманганат калия и серную кислоту, а также бромную воду. Затем поглотительные растворы объединяют, восстанавливают мышьяк до арсина и определяют его по почернению ртутно-бромидной бумаги [113].
Реактивы
Перманганат калия, растворяют 0,32 г КМпО4 в 1 л воды.
Серная кислота, 0,3 мл концентрированной H2SO4 прибавляют к воде и разбавляют до 1 л.
Бромная вода, дистиллированную воду насыщают бромом.
Перекись водорода, 1%-ный раствор,
Ход анализа. Пробу исследуемого воздуха пропускают последовательно через два поглотителя Рихтера, в которые изливают по 16,5 мл поглотительного раствора, состоящего из 7,5 мл раствора перманганата калия, 7,5 мл раствора серной кислоты и 1,5 мл бромной воды, со скоростью .2 л/мин. Можно также пропускать пробу воздуха через два поглотителя Зайцева, в которые наливают смесь по 2,5 мл растворов перманганата и серной кислоты и 0,5 мл бромной воды. Растворы из поглотителей объединяют, прибавляют раствор Н2О2 до о.бесцвечивания перманганата и выпаривают почти досуха. Остаток с помощью серной кислоты переносят в сосуд для отгонки арсина и проводят определение, как указано в разделе IV. 4.1.
IV. 4.3. Определение мышьяка в серной кислоте
Определение мышьяка в серной кислоте чаще всего проводят, восстанавливая его металлами до арсина, как указано в разделе IV. 4.1.
IV. 4.4. Определение мышьяка в свинце повышенной чистоты [46]
Навеску пробы переводят в раствор, соосаждают арсенат железа с гидроокисью железа в присутствии комплекса Са-ЭДТА для связывания свинца. Затем осадок растворяют в хлористоводородной кислоте, восстанавливают мышьяк до арсина и определяют с помощью ртутно-бромидной бумаги.
Реактивы
Азотная кислота, разбавленная (1 : 3).
Нитрат железа(Ш). Растворяют в воде, подкисленной азотной кислотой 6 г Fe(NO3)3-6H2O и водой разбавляют до 100 мл.
Этилендиаминтетрауксусной кислоты динатриевая соль (ЭДТА), 7%-ный водный раствор.
Нитрат кальция 7,5%-ный водный раствор.
Ход анализа. Навеску свинца 1 г растворяют при нагревании в 110 мл азотной кислоты, удаляют кипячением окислы азота, прибавляют 1,5 мл раствора нитрата железа(III), 40 мл раствора ЭДТА и 45 мл раствора нитрата кальция. Нагревают раствор до 60—70°C и осаждают железо аммиаком, прибавляя небольшой избыток его. Раствор с осадком оставляют на горячей водяной бане в течение 1,5—2 ч, осадок отфильтровывают, промывают его горячей водой и переносят в сосуд, в котором проводилось осаждение. Фильтр промывают сначала хлористоводородной кислотой, разбавленной (1:1), потом несколько раз горячей водой, присоединяя все промывные воды к осадку гидроокиси железа. Нагревают до растворения этого осадка, прибавляя, если надо, еще хлористоводородной кислоты, и повторяют осаждение аммиаком без добавле
ния ЭДТА и нитрата кальция. Осадок отфильтровывают, промывают горячей водой, растворяют на фильтре в 14 мл горячей хлористоводородной кислоты, разбавленной (1:1), и промывают фильтр горячей водой в таком количестве, чтобы общий объем полученного раствора не превысил 50 мл. Раствор переносят в колбу прибора для отгонки арсина и определяют мышьяк, как указано в разделе IV. 4.1.
IV. 5. ОПРЕДЕЛЕНИЕ МЫШЬЯКА В ВИДЕ АРСЕНИДОВ СЕРЕБРА И РТУТИ С ПОСЛЕДУЮЩИМ ПРОЯВЛЕНИЕМ
Чувствительность реакций на мышьяк можно значительно повысить, если арсин пропустить через бумагу, смоченную раствором бромида ртути, а затем бумагу с продуктом реакции обработать раствором иодида калия [114] или если арсин пропустить через бумагу, смоченную раствором нитрата серебра, а затем полученный продукт реакции проявить [14, 15]. Метод фотографического проявления более чувствителен, однако требует соблюдения ряда предосторожностей, кроме того, в этом случае наблюдается высокое значение холостой пробы. В связи с тем, что-очистить цинк или алюминий от столь малых количеств мышьяка очень трудно, для получения арсина применяют электролитическое восстановление мышьяка из щелочного раствора.
Электролитически до арсина восстанавливается только мышь-як(Ш), поэтому предварительное восстановление мышьяка(V) до мышьяка(III) проводят сульфатом хрома(II).
IV. 5.1. Определение мышьяка в щелочи особой чистоты [14, 15]
Реактивы
Сульфат хрома(П), 2°/о-ный раствор. В толстостенную склянку емкостью Ю—20 мл, снабженную полиэтиленовой пробкой, помещают 1—2 г металлического цинка, не содержащего мышьяка, 5 мл металлической ртути, 1—2 мл хлористоводородной кислоты, разбавленной (1 : 1), очищенной холодной перегонкой, и взбалтывают для образования амальгамы. Затем прибавляют 2%-ный раствор Cr2(SO4)3 так, чтобы склянка была полной, и взбалтывают до перехода зеленой окраски раствора в светло-голубую.
Винная кислота, 10%-ный раствор.
Реактивная бумага, фильтровальная бумага, смоченная 5%-ным спиртовым раствором HgBr2.
Иодид калия, 5%-ный раствор.
Нитрат серебра, 10- и 1%-ный растворы.
Проявитель (раствор I). Растворяют в воде 20 г метола, 100 г лимонной кислоты, 15 г нитрата натрия, разбавляют до 1 л и перемешивают.
Гидроокись натрия, раствор, очищенный длительным электролизом.
Ход анализа. Пробу раствора щелочи до 25 мл, содержащую до 0,5 мкг мышьяка, помещают в колбу прибора (рис. 33), прибавляют 2 мл винной кислоты и 1 мл раствора CrSO4. Прибор устанавливают на подставке магнитной мешалки, в крышку прибора
помещают индикаторную бумагу и проводят электролиз в течение 30—60 мин.
При использовании в качестве индикатора раствора нитрата серебра в крышку прибора, предварительно промытую азотной кислотой, водой и высушенную фильтровальной бумагой, вкладывают бумагу, смоченную 0,02 мл 1%-ного раствора AgNOa. На трубку с крышкой для защиты от света надевают картонный колпачок, оклеенный внутри черной бумагой. После проведения электролиза индикаторную бумагу помещают в проявитель, приготовленный непосредственно перед проявлением. Для этого к 10 мл раствора I прибавляют 0,1 мл 10%-jioro раствора нитрата серебра.
Рис. 33. Прибор для отгонки микроколичеств мышьяка в виде арсина:
/ — реакционная колба; 2—катод;3 — анод; 4 — ватный тампон; 5—поглотительный сосуд; 6—крышка с индикаторной бумагой.
Проявление ведут 2 мин, затем проявитель сливают, промывают бумагу дистиллированной водой, высушивают и сравнивают с бумагами стандартной серии. Всю работу, начиная с закладывания бумаги и кончая ее промывкой, проводят при красном свете.
В случае применения бромида ртути(II) бумагу смачивают 5%-ным спиртовым раствором бромида ртути(II), закладывают в крышку и проводят электролиз при обычном освещении. Затем бумагу обрабатывают в течение 1 мин 5 мл раствора иодида калия, промывают водой, высушивают и сравнивают с бумагами стандартной серии.
При навеске щелочи 15—20 г метод позволяет определить 3—5*10“7% мышьяка. Продолжительность анализа 1,5 ч.
IV. 5.2. Определение мышьяка в
кремнии и двуокиси кремния особой чистоты [115]
Навеску образца растворяют в щелочи, в щелочном растворе восстанавливают мышьяк до арсина, который пропускают через бумагу, смоченную раствором бромида ртути(II) или нитрата серебра. Затем продукт реакции проявляют и содержание мышьяка находят по стандартной шкале.
Реактивы
Гидроокись натрия, 30%-ный раствор, очищенный электролитически.
Другие реактивы — указаны в разделе IV. 6.1.
Ход анализа. Навеску анализируемого вещества 0,05 г растирают во фторопластовой ступке, помещают в колбу прибора (см. рис. 33) и растворяют при нагревании на водяной бане в 20 мл раствора NaOH. После полного растворения добавляют 2 мл 2%-нрго раствора сульфата хрома(II), закрывают колбу, в крышку прибора помещают индикаторную бумагу и включают ток. Электролиз продолжают в течение 45—50 мин при напряжении на электродах 8 В. Затем ведут определение, как указано в разделе IV. 5.1.
IV. 6. ОПРЕДЕЛЕНИЕ МЫШЬЯКА С ПРИМЕНЕНИЕМ ДИЭТИЛДИТИОКАРБАМАТА СЕРЕБРА
Мышьяк восстанавливают до арсина, который отгоняют и поглощают пиридиновым [116] или хлороформным [117,118] растворами диэтилдитиокарбамата серебра. Применение хлороформного раствора удобнее, так как пиридин обладает неприятным запахом. В хлороформный раствор прибавляют органическое основание, лучше /-эфедрин. В зависимости от соотношений между серебром и мышьяком окраска растворов различна — темно-желтая при трехкратном избытке серебра, красно-коричневая при 5—10-кратном, коричневая при 22—50-кратном и красная при больше чем 60-крат-ном избытке серебра. При этом происходит реакция [119]:
AsH3 + 6(C2H6)2NCSSAg —> 6Ag + 3(C2HB)2NCSSH + [(C2HB)2NCSS]3As
Таким образом, определение основано на измерении окраски золя элементного серебра.
Этот метод применен для определения мышьяка в водах [120], сере, серной и фосфорной кислотах [121] и других материалах.
Описан микрометод определения мышьяка в растворе с использованием диэтилдитиокарбамата серебра [122]. При определении мышьяка в органических и неорганических веществах навеску от 5 до 50 мг нагревают со смесью магния и окиси магния в запаянной трубке в течение 5 мин. Затем продукт реакции разлагают серной кислотой и выделившийся арсин поглощают раствором диэтилдитиокарбамата серебра [123, 124].
При анализе меди и ее солей предварительно выделяют мышьяк в виде магний-аммонийарсената совместно с магний-аммонийфосфатом, осадок растворяют в хлористоводородной кислоте и выделяют арсин, который поглощают раствором диэтилдитиокарбамата серебра [125].
Для определения 10“5—1О"6о/о мышьяка в металлическом серебре и его солях особой чистоты серебро выделяют в виде амальгамы, восстанавливая его в азотнокислой среде муравьиной кислотой. Затем определяют мышьяк с помощью диэтилдитиокарбамата серебра [126].
При анализе серы на мышьяк его экстрагируют щелочью в присутствии перекиси водорода и раствор выпаривают досуха. К остатку прибавляют серную кислоту, переносят в прибор для
отгонки арсина и определяют мышьяк по реакции с диэтилдитиокарбаматом серебра [127].
Диэтилдитиокарбамат серебра применен также для определения мышьяка в фтористоводородной кислоте [128], в хлориде натрия [129], в угле и дегте [130]. В случае анализа угля и дегтя пробу сплавляют с карбонатом натрия и окисью магния, а затем ведут определение.
Мешающие вещества. Определению мешают вещества, указанные в разделе IV. 4.
Измерение оптической плотности. Измерение оптической плотности производят при 540—560 нм.
Чувствительность метода — 0,1 мкг мышьяка в 10 мл конечного раствора.
IV. 6.1. Определение мышьяка в отсутствие мешающих веществ
Реактивы
Диэтилдитиокарбамат серебра. Растворяют 1 г диэтилдитиокарбамата серебра в 200 мл химически чистого пиридина. Раствор фильтруют и сохраняют в темной склянке с притертой пробкой. Вместо пиридинового раствора можно применять раствор 2,5 г диэтилдитиокарбамата серебра и 1,65 г /-эфедрина в 200 мл хлороформа.
Диэтилдитиокарбамат серебра готовят следующим образом. К раствору 2,25 г диэтилдитиокарбамата натрия в 100 мл воды приливают 100 мл водного раствора нитрата серебра, содержащего 1,70 г нитрата серебра. Образовавшийся желтый осадок диэтилдитиокарбамата серебра промывают несколько раз дистиллированной водой путем декантации, удаляют по возможности полнее воду и высушивают в вакуум-эксикаторе.
Присутствие в реактиве нитрата серебра оказывает значительное влияние на определение, поэтому препарат не должен содержать избытка нитрата серебра [131].
Иодид калия, 15%-ный раствор.
Хлорид олова(П), 40%-ный раствор в хлористоводородной кислоте, разбавленной (1 : 1).
Ход анализа. Раствор, содержащий до 20 мкг мышьяка, помещают в колбу прибора для отгонки арсина (см. рис. 31), прибавляют все реактивы, как указано в разделе IV, 4, и ведут отгонку мышьяка в виде арсина, который поглощают 5—10 мл пиридинового раствора диэтилдитиокарбамата серебра или хлороформного раствора диэтилдитиокарбамата серебра и эфедрина. По окончании отгонки арсина, продолжающейся 1 —1,5 ч, измеряют оптическую плотность поглотительного раствора при 560 нм.
Содержание мышьяка находят по предварительно построенному калибровочному графику. Для построения калибровочного графика пользуются стандартным раствором (см. раздел IV. 3.1) и проводят определение, как указано выше.
При малых содержаниях мышьяка берут более разбавленный раствор диэтилдитиокарбамата.
IV. 6.2. Определение мышьяка в черных металлах
Навеску металла растворяют в азотной кислоте и для предотвращения потери мышьяка прибавляют перекись водорода. Затем прибавляют серную кислоту и выпариванием удаляют азотную кислоту. В полученном растворе мышьяк(V) восстанавливают до мышьяка(Ш), а затем до арсина. Арсин отгоняют и поглощают пиридиновым или хлороформным раствором диэтилдитиокарбамата серебра [132, 133].
Ход анализа. Навеску 0,1 г железа, чугуна или стали растворяют в 10—20 мл азотной кислоты, разбавленной (1 : 10), с добавлением 1 мл пергидроля. Для ускорения растворения нагревают и в несколько приемов прибавляют от 5 до 25 мл концентрированной азотной кислоты. После окончания растворения для удаления азотной кислоты добавляют 1 мл концентрированной серной кислоты и нагревают до выделения паров серной кислоты. Затем чашку охлаждают, стенки смывают 10 мл воды, прибавляют еще 1 мл концентрированной H2SO4 и снова упаривают до выделения паров серной кислоты. Чашку охлаждают, содержимое разбавляют 10 мл воды, раствор переносят в колбу прибора для отгонки арсина и продолжают определение, как указано в разделе IV. 6.1.
IV. 6.3. Определение мышьяка в двуокиси германия
Пробу двуокиси германия растворяют при нагревании в соляной кислоте в атмосфере хлора. При этом четыреххлористый германий отгоняется, а мышьяк определяют с помощью пиридинового раствора диэтилдитиокарбамата серебра [134].
Ход анализа. Навеску двуокиси германия 2 г помещают в кварцевую колбу Кьельдаля емкостью 150 мл с меткой, соответствующей 10 мл, приливают 50 мл хлористоводородной кислоты и пропускают через кварцевую трубку ток хлора со скоростью два пузырька в 1 с.
Спустя 5 мин содержимое колбы осторожно подогревают на слабом пламени, не допуская вспенивания во избежание потерь. Когда жидкость посветлеет, нагревание усиливают, не прекращая подачи хлора, пока объем раствора в колбе не составит 10 мл. Затем вводят 25 мл хлористоводородной кислоты, упаривают раствор точно до 10 мл и охлаждают холодной водой при непрерывном подводе хлора. Раствор переносят в колбу для отгонки арсина и продолжают анализ, как указано в разделе IV. 6.1.
IV. 7. ОПРЕДЕЛЕНИЕ МЫШЬЯКА С ПРИМЕНЕНИЕМ ГИПОФОСФИТА
Метод основан на получении золя элементного мышьяка. Для этого к хлористоводородному испытуемому раствору прибавляют гипофосфит, который восстанавливает мышьяк до элементного
состояния. По интенсивности коричневой окраски коллоидного раствора мышьяка определяют его содержание [135, 136].
Восстанавливать мышьяк можно и хлоридом олова (II), но при этом необходимо проводить реакцию при высокой концентрации хлористоводородной кислоты, что неудобно. Введение 0,05— 0,5 моль/л иодида ускоряет процесс восстановления мышьяка [137]. В этих условиях можно вести восстановление в 3 н. хлористоводородной кислоте при использовании гипофосфита и в 5 н. хлористоводородной кислоте при использовании хлорида олова(II).
Мешающие вещества. Из элементов, которые встречаются при определении мышьяка, мешают молибден и ртуть, восстанавливающиеся гипофосфитом, молибден — до молибденовой сини, а ртуть — до металла. Поэтому в их присутствии мышьяк предварительно отделяют. Для отделения от молибдена мышьяк осаждают в виде арсената железа в аммиачной среде, а для освобождения от ртути отгоняют в виде АэОз или сплавляют пробу со щелочью. При сплавлении ртуть улетучивается. Сплавление ведут в железном тигле с 6—10-кратным количеством щелочи.
Измерение оптической плотности. Окраска устойчива в течение 3 ч, после чего наступает коагуляция золя мышьяка. Измерение оптической плотности проводят при 612 нм.
Применяют также метод стандартных серий. Метод колориметрического титрования неприменим.
Чувствительность метода — 10 мкг мышьяка в 20 мл конечного раствора.
IV. 7.1. Определение мышьяка в отсутствие мешающих веществ
Реактивы
Гипофосфит натрия или кальция. Насыщенный на холоду раствор NaH2PO2 или Са(Н2РО2)2 в хлористоводородной кислоте, разбавленной (1 : 1).
Сульфат меди (CuSO4-5H2O), 1%-ный раствор.
Ход анализа. До 10 мл испытуемого хлористоводородного раствора, содержащего от 10 до 100 мкг мышьяка, помещают в плоскодонную пробирку емкостью 30 мл или узкий стакан емкостью 50 мл и прибавляют 3—4 капли раствора хлорида железа (III). Надобность в последнем исключается при наличии железа в самом растворе, что часто имеет место при анализе различных материалов. Затем прибавляют 2 мл раствора сульфата меди и 5 мл раствора гипофосфита. Пробирку или стакан накрывают часовым стеклом и нагревают на кипящей водяной бане в течение 30 мин. После охлаждения разбавляют водой до определенного объема, перемешивают и измеряют оптическую плотность.
IV. 7.2. Определение мышьяка в воде [138, 139]
Ход анализа. Пробу воды, содержащую до 100 мкг мышьяка, помещают в стакан, подкисляют хлористоводородной кислотой, вводят 3—4 капли раствора хлорида железа(III), нагревают раствор до 70—80 °C и осаждают гидрат окиси железа аммиаком, избегая избытка последнего. Стакан устанавливают на теплой водяной бане для коагуляции. Затем осадок отфильтровывают, растворяют в горячей хлористоводородной кислоте (пл. 1,1) и определяют мышьяк, как указано в разделе IV. 7.1.
IV. 7.3. Определение мышьяка в цинке [140, 141]
Ход анализа. Навеску металла, содержащую 10—100 мкг мышьяка, растворяют в азотной кислоте, прибавляют 0,5 г персульфата аммония и кипятят несколько минут для полного окисления мышьяка(III) до мышьяка(V). Затем осаждают мышьяк в виде арсената железа вместе с гидроокисью железа и продолжают анализ, как указано в разделе IV. 7.1.
IV. 7.4. Определение мышьяка в меди и ее сплавах
Навеску металла растворяют в смеси серной и азотной кислот и восстанавливают мышьяк хлоридом олова(II) [141] или гипофосфитом [142]. При работе с SnCb рекомендовано проводить предварительное выделение мышьяка в виде магний-аммоний арсената с использованием в качестве коллектора магний-аммоний фосфат.
Реактивы
Перманганат калия, 1%-ный раствор.
Персульфат аммония, насыщенный раствор.
Ход анализа. Навеску металла 0,05 г растворяют в 25 мл серной кислоты, разбавленной (1:4), и 3—4 мл концентрированной азотной кислоты. После растворения нагревают до удаления азотной кислоты, прибавляют 2 мл раствора перманганата калия и кипятят до полного выделения МпО2. Прибавляют 1—2 мл раствора персульфата аммония, упаривают до выделения паров серной кислоты, охлаждают и разбавляют водой до 100 мл. Отбирают 10 мл и определяют мышьяк, как указано в разделе IV. 7.1.
IV. 7.5. Определение мышьяка в железной руде и стали [143]
Руду растворяют в хлористоводородной кислоте с прибавлением азотной кислоты, после отделения кремневой кислоты прибавляют тартрат натрия или винную кислоту для удержания железа, фосфат
натрия и осаждают магний-аммоний арсенат вместе с магний-аммоний фосфатом. Осадок отделяют, растворяют в хлористоводородной кислоте, восстанавливают мышьяк до элементного состояния и измеряют оптическую плотность полученного раствора.
Разложение руды можно также вести путем сплавления с перекисью натрия [144]. f
Реактивы
Двузамещенный фосфат натрия, 1%-ный раствор.
Ход анализа. Навеску 0,2 г руды (при содержании мышьяка меньше 0,01% необходимо брать большую навеску) растворяют при нагревании в хлористоводородной кислоте с добавлением небольшого количества азотной кислоты до полного растворения пробы. После растворения руды прибавляют 30—50 мл горячей воды и отфильтровывают выделившуюся кремневую кислоту. Последнюю промывают на фильтре (3—4 раза) горячей 2%-ной хлористоводородной кислотой. К фильтрату прибавляют 0,6 г тартрата натрия (или винной кислоты), 5 мл раствора двухзамещенного фосфата натрия, 15 мл магнезиальной смеси и раствор аммиака до сильного запаха. Осадку дают отстояться в течение 30 мин, затем его отфильтровывают, промывают, растворяют в горячей хлористоводородной кислоте, разбавленной (1 : 1). Мышьяк определяют, как указано в разделе IV. 7.1.
IV. 7.6. Определение мышьяка в других материалах
При анализе продуктов медного и свинцово-цинкового производства рекомендуется предварительное восстановление мышьяка в сильнокислой среде хлоридом олова(II) с последующим растворением элементного мышьяка в растворе перекиси натрия. Затем мышьяк восстанавливают сернокислым гидразином и измеряют оптическую плотность золя мышьяка [145].
Гипофосфитный метод-предложен для определения мышьяка в свинце, применяемом для изготовления аккумуляторных батарей [146], в минеральном сырье [147], в вольфрамовых рудах и концентратах [148] и в металлической сурьме, содержащей селен и теллур [149]. Селен и теллур предварительно выделяют в более мягких условиях хлоридом олова (II), а затем гипофосфитом выделяют мышьяк в присутствии сульфата меди в качестве катализатора.
IV. 8. ОПРЕДЕЛЕНИЕ МИКРОКОЛИЧЕСТВ МЫШЬЯКА
С ПРИМЕНЕНИЕМ БУТИЛРОДАМИНА
Метод основан на образовании соединения мышьяковомолибде-нового комплекса с бутилродамином, флотации его диэтиловым эфиром с последующим растворением в смеси эфира с ацетоном и 168
измерении оптической плотности раствора [150]. Оптимальная концентрация серной кислоты равна 0,2—0,4 н., молибдата аммо-нИЯ — 4. Ю-4 М и бутил родамина — 3- 10~e М.
Мешающие вещества. Аналогичную реакцию дают кремний и фосфор, поэтому при содержании фосфора больше 0,05 мкг и кремния больше 8 мкг в анализируемой пробе они мешают определению мышьяка. Многие металлы при их 200-кратном содержании по отношению к мышьяку не мешают определению.
Измерение оптической плотности. Оптическую плотность измеряют при 560 нм.
Чувствительность метода — 0,04 мкг мышьяка в 35 мл анализируемого раствора.
IV. 8.1. Определение мышьяка в отсутствие мешающих веществ
Реактивы
Молибдат аммония, 0,01 М водный раствор.
Бутилродамин, 0,02%-ный водный раствор.
Все реактивы, кроме бутилродамина, хранят в полиэтиленовой посуде.
Ход анализа. До 35 мл анализируемого раствора, содержащего 0,04—1 мкг мышьяка, помещают в делительную воронку емкостью 100 мл, прибавляют 1,5 мл 8 н. серной кислоты, 1,3 мл раствора молибдата аммония, 0,3 мл водного раствора бутилродамина и оставляют на 10 мин. Затем к раствору приливают 7 мл диэтилового эфира и взбалтывают в течение 1 мин. После разделения жидкостей эфирный слой и сфлотированную пленку соединения мышьяка отделяют от водной фазы и дважды промывают от избытка красителя небольшим объемом воды. К эфирному слою добавляют 2 мл ацетона и‘перемешивают. Интенсивность окраски эфирно-ацетоновых растворов сравнивают визуально с окраской серии стандартных эталонов или измеряют оптическую плотность растворов.
IV. 8.2. Определение мышьяка в хлориде натрия
Выделяют предварительно мышьяк экстракцией в виде диэтил-дитиокарбаматного комплекса, а затем определяют его с помощью бутилродамина.
Реактивы
Иодид калия, 20%-ный раствор.
Аскорбиновая кислота, 5%-ный раствор.
Диэтилдитиокарбаминовая кислота, 1%-ный раствор.
Молибдат аммония, 0,01 М раствор.
Бутилродамин, 0,02%-ный водный раствор.
Ход анализа. В стеклянный стакан помещают 10 г хлорида натрия и растворяют в 35 мл бидистиллята (при анализе
производственных рассолов берут соответствующий объем рассола), нагревают до 40—45°C и к раствору приливают 5 мл 8 н. H2SO4. Мышьяк(У) восстанавливают до мышьяка(III), прибавляют 1,5 мл раствора иодида калия и 1 мл свежеприготовленного 5%-ного раствора аскорбиновой кислоты и оставляют на 15 мин. Затем холодный раствор переливают в делительную воронку, добавляют 5 мл раствора диэтилдитиокарбаминовой кислоты в хлороформе и взбалтывают 30 с. После разделения слоев хлороформ сливают в делительную воронку емкостью 25 мл, а водную фазу еще раз встряхивают с 5 мл хлороформного раствора диэтилдитиокарбаминовой кислоты. Экстракты соединяют и дважды промывают 1 н. серной кислотой порциями по 2,5 мл, затем выливают в маленькую фарфоровую чашку, добавляют 3—5 мл воды так, чтобы она распределилась кольцом по окружности чашки, и выпаривают на водяной бане до удаления хлороформа. К сухому остатку приливают 2 мл азотной кислоты (пл. 1,4) и выпаривают на водяной бане досуха. Прибавляют к остатку 1,5 мл 8 н. серной кислоты, 1,3 мл 0,01 М раствора молибдата аммония, 0,3 мл 0,02%-ного водного раствора бутилродамина и ведут определение, как указано в разделе IV. 8.1.
IV. 9. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ МЫШЬЯКА
Кверцетин взаимодействует с мышьяком (V) с образованием соединения желтого цвета с максимумом поглощения при 398 нм, е = 2,5-104. Эта реакция предложена для спектрофотометрического определения мышьяка [151, 152].
Куркумин в присутствии щавелевой кислоты взаимодействует с мышьяком (V) с образованием окрашенного соединения, которое растворяется в н-бутаноле и характеризуется максимумом поглощения при 545 нм. На основе этой реакции предложен метод фотометрического определения мышьяка [153}. При взаимодействии арсина с щелочным раствором л-сульфамоилбензоата серебра образуется золь серебра с максимумом поглощения при 420 нм, е = = (18—51) • 103 (в зависимости от условий проведения опыта). По оптической плотности золя серебра определяют содержание мышьяка [154, 155].
а-Дитионафтол тетраметиламмония [156] и 8-меркаптохинолин [157] взаимодействуют с мышьяком (III) с образованием малорастворимых соединений желтого цвета. Первое соединение экстрагируется хлороформом, а второе — четыреххлористым углеродом. Эти реакции положены в основу фотометрических методов определения мышьяка.
Предложен косвенный метод фотометрического определения малых количеств мышьяка, основанный на окислении мышьяка (III) трииодидом [158] или иодметиленовым голубым [159]:
C.eH18N,SI7 + As111 —► С HI8N3S + ЗГ + Asv ID 10 о □ • Id lo 3 • 1
и последующем фотометрировании окраски метиленового голубого, выделяющегося в количестве, эквивалентном содержанию мышьяка [158]. Метод пригоден для определения 0,1 —1,4 мкг мышьяка в 1 мл. *
Из других косвенных методов следует отметить определение мышьяка по количеству молибдена, связанного в мышьяковомо-либденовую кислоту [160], по количеству K4[Fe(CN)6], образующегося при окислении мышьяка(III) с помощью K.3[Fe(CN)e] [161], а также по количеству выделенного дитизона при взаимодействии диэтилдитиокарбамата мышьяка (III) с дитизонатом серебра [162]. При этом образуется разнолйгандный (смешанный) комплекс ди-тиокарбаминовой кислоты и двузамещенного дитизоната с выделением соответствующего количества свободного дитизона.
Элементный мышьяк восстанавливает желтую фосфорномолибденовую кислоту до синей фосфорномолибденовой кислоты. Эта реакция применена для фотометрического определения мышьяка [163].
Ферроин при pH = 2,2—10,6 взаимодействует с гексафторид-ным комплексом мышьяка (AsFe) с образованием окрашенного соединения, которое экстрагируется н-бутиронитрилом и применяется для фотометрического определения мышьяка [164].
При взаимодействии ванадата натрия с мышьяковомолибденовой гетерополикислотой образуется желтая мышьяковомолибдено-ванадиевая кислота, в виде которой определяют мышьяк [165].
ЛИТЕРАТУРА
1. R о h г е К.. Z. anal. Chem., 65, 109 (1924).
2. Schaaf Е., Z. anal. Chem., 126,298 (1943). -
3. Magnuson H. J., Watson E. B., Ind. Eng. Chem., Anal. Ed., 16, 339 (1944).
4. А л e к с e e в P. И., Зав. лаб., 11, 122 (1945).
5. Финкельштейн Д. Н., Крючкова Г. М., ЖАХ, 12, 196 (1957).
6. Бутенко Г. А., Корж В. П„ Родионова Е. М., ЖАХ, 16, 692 (1961).
7. Н u b b а г d D. М„ Ing. Eng. Chem., Anal. Ed., 13, 915 (1941). ,
8. Bart let J. C., Wood M„ Chapman R. A., Anal. Chem., 24, 1821 (1952).
9. de Bruyne P., Hoste J., Bull. Soc. Chim. Belg., 70, 221 (1961).
10. К а л и н и н А. И., ЖАХ, 17, 390 (1962).
11. Ploum H., Arch. Eisenhiittenwesen, 32, 71 (1961).
12. К о з л о в с к и й M. Т., Ваганова Р. 3., За валище в а Н. Н., Зав. лаб., 13, 549 (1947).
13. R о g е г s D„ Н е г о n А. Е., Analyst, 71, 414 (1946).
14. Б а б к о А. К, Пилипенко А. Т., Розенфельд Г. Л., Доп. АН УРСР, № 8, 969 (1962).
15. Бабко А. К., Пилипенко А. Т., Розенфельд А. Л., Зав. лаб., 30, 1060 (1964).
16. Leo yd W. V., Trans. Faraday Soc., 27, 89 (1931).
17. M i 11 о m R„ D u f f i e 1 d W. D„ Analyst, 67, 279 (1942).
18. G r e e n M., К a f a 1 a s J. A., J. Chem. Phys., 22, 760 (1954).
19. F1 s c h e r W. e. ol„ Angew. Chem., 66, 165 (1954).
20. В r i n k G. O. e. ol„ J. Am. Chem. Soc., 79, 1303 (1957).
21. Милаев С. M„ Ворошнина К. П., Зав. лаб., 29, 410 (1963).
22. N i v i е г е Р., Chim. anal., 47, 448 (1965).
23. N i g е о п E., N i g е о n J., Chim. anal., 43, 276 (1961).
24. S t u d 1 а г К., Taianta, 8, 272 (1961).
25. W у a 11 P. F„ Analyst, 78, 656 (1953).
26. W у a 11 P. F., Analyst, 80, 368 (1955).
27. Str af fort N., Wyatt P. F., Kershaw F. G„ Analyst, 70, 232 (1945).
28. Назаренко В. А., Флянтикова Г. В., Лебедева Н. В., Зав. лаб., 33,891 (1957).
29. Fawler Е. W., Analyst, 88, 380 (1963).
30. В и х м Н., Педак Э„ Уч. зап. Тартуского ун-та, вып. 193, 115 (1963).
31. PayneS. Т., Analyst, 77, 278 (1952).
32. Luke С. L., Campbell М. Е., Anal. Chem., 25, 1588 (1953).
33. Kovacs Е., Guger Н., Ziischer W., Z. anal. Chem., 208, 321 (1965).
34. Ко ст A. H., Терентьев П. Б., Бы-рько В. M., Вести. Моск, ун-та. Сер. хим., № 4, 195 (1959).
35. К о м и с с а р о в а О. Н., Зав. лаб., 34, 410 (1968).
36. Klein А. К., Vorhes F. A., J. Assoc. Official Agr. Chem., 22, 121 (1939).
37. Klein A. K-, Vorhes F. A., J. Assoc. Official Agr. Chem., 25, 274 (1942).
38. Alteresck G., Rev. chim. (RPR), 16, 221 (1965).
39. H а к а я С e д з о, РЖХим, 1964, 2Г142.
40. L о u n a m a a К., Z. anal. Chem., 146, 422 (1955).
41. А г c h e г V. S., D о о 1 i 111 e Er. G., Taianta, 14, 921 (1967).
42. L i sk D. J., J. Agric. and Eood Chem., 8, 121 (1960).
43. D a n i e 1 s M., Analyst, 82, 133 (1957).
44. J о k о s u k a S., Japan Analyst, 5, 395 (1956).
45. Sugawara Ken, Kanamori Satoru, Bull. Chem. Soc., Japan, 37, 1358 (1964).
46. Л у p ь e Ю. Ю., M и н e н к о A. H., Зав. лаб., 23, 785 (1957).
47. Odencrants J. T., Riemann W., Anal. Chem., 22, 1066 (1950).
48. Vukcevic-Kovacevic V., Seremat S., Scientia pharmac., 36, 26 (1968).
49. Ikeda Nagao, Sukawa Tadaaki, Aguma Koichi, Radioisotopes, 17, 68 (1968).
50. Miketukova V., Kohlicek J., Kacl K., J. Chromatogr., 34, 284 (1968).
51. El be in I. I., Elbakry A., Aly M. M., Chemist-Analyst, 56, 48 (1967).
52. Garcia CH. A., Rev. Fac. farmac. v. biochim., 29, 22 (1967).
53. Heinz P., Z. anal. Chem., 134, 177 (1951).
54. Duval L., Chim. anal., 51, 415 (1969).
55. Б у т e н к о А. Г., Зав. лаб., 18, 275 (1952).
56. В er h out H. W., Jon gen G. H., Chemist-Analyst, 43, 60 (1954).
57. Berhout H. W., Chemist-Analyst, 45, 24 (1956).
58. Круглова M. H., Тр. Всес. н.-и. ин-та стандартн. образцов и спектр, эталонов, 3, 59 (1967).
59. Goszczynska Н., Kowalczyk М., Chem. anal. (Polska), 12,1261 (1967).
60. В а 1 a b а п о f f К. L., Salgado М. Н., Rev. Real. acad. cienc. exact., fis. у natur. Madrid, 62, 591 (1968).
61. S p о u s t о v a J., Hutn. Listy, 25, 129 (1970).
62. Hasek Z., Hutn. Lfsty, 26, 669 (1971).
63. Johnson D. L., Pilson M. E., Anal. chim. acta, 58, 289 (1972).
64. Бескова Э. С., Журавлев Г. И., Дорофеева В. И., Зав. лаб., 36, 541 (1970).
65. N а 11 W. R., Analyst, 96, 398 (1971).
66. Fogg A. G., Marriott D. R., Burns D. Th., Analyst, 97, 657 (1972).
67. Курденкова T. Л., Технол. легких сплавов. Научи.-техн. бюл. ВИЛСа, № 3, 122 (1972).
68. Marczenko Z., Mojski М., Chem. anal. (Polska), 14, 495 (1969).
69. Б а б к о А. К., Пилипенко А. Т. Колориметрический анализ. М., Гос-химиздат. 1951. См. с. 274.
70. Ed combe L. J., Gol d H. K., Analyst, 80, 155 (1955).
71. On is hi H., San dell E. B., Mikrochemie ver Mikrochim. Acta, ,№ 1—2, 34 (1953),
72. Р а к a 1 n s P., Anal. chim. acta, 47, 225 (1969).
73 Николов T., йосифова M„ Ляков H., Хим. и индустрия, 41, 468 (1969).
74. Щербаков В. Г., Онучина Г. В. В сб.: «Хим. свойства и методы анализа тугоплавк. соедин.». Киев, «Наукова думка», 1969. См. с. 128.
75. Курбатова В. И. и др., Тр. Всес. н.-и. ин-та стандартн. образцов и спектр, эталонов, 4, 95 (1968).
76. Rosi D., Metallurgia ital., 45, 166 (1953).
77. Di Вассо D., Boll. chim. tarmac., 93, 88 (1954).
78. Ш а ф p а н И. Г. и др. Труды ИРЕА, вып. 30, 25 (1967).
79. Жуковский Ю. Г., ЖАХ, 19, 1361 (1964).
80. lomo Kakuma, Sci Repts Res. Insts Tohoku Univ., A8, № 3, 243 (1956).
81. Моримото Иосио, Асидзава Нуеси, РЖХим, 1962, 10Д119.
82. Исии Дайдо, Такэути Цуги о, РЖХим, 1962, 19Д76.
83. Reed J. F„ Anal. Chem., 30, 1122 (1958).
84. Ebner E., Z. anal. Chem., 206, 106 (1964).
85. A p а н о в и ч Б. С., Искусств, волокно, вып. 10, 85 (1955).
86. Накая Седзо, РЖХим, 1962, 7Д122.
87. О н и с и, РЖХим, 1957, 23477.
88. Sugawara К., Tanaka М., N aka тог i S., Bull. Chem. Soc. Japan, 29, 670 (1956).
89. L о u п a m a a К., Z. anal. Chem., 146, 422 (1955).
90. К у ш а к о в с к и й Л. Н., Позднякова Т. Г., Гигиена и санитария, № 4, 46 (1955).
91. Алексеева М. В., Информ.-методические материалы Гос. н.-и. санит. ин-та, № 5, 19 (1954).
92. L i е d е г m a n D., Bowen J. Е„ М i 1 п е г О. J., Anal. Chem., 30, 1543 (1958).
93. Бутенко Г. А., Родионова-Е. М., Корж В. П., Зав. лаб., 27, 808 (1961).
94. Bohnstedt U., Budenz R., Z. anal. Chem., 159, 95 (1957).
95. T а н а к а Кацу, Japan Analyst, 10, 612 (1961); цит. по РЖХим, 1962, 8Д125.
96. T к а ч e н к о H. С., Саку нов В. И., Тр. науч.-техн, общества черной металлургии, Укр. респ. правление, 4, 126 (1956).
97. Л я л и к о в Ю. С. и др. Анализ железных марганцевых руд и агломератов. М., «Металлургия», 1966. См. с. 226.
98. F е j f а г V., Р е с h а г F„ Chem. prumysl., 13, 80 (1963).
99. М и л а е в С. М., Ворошнина К. П., Металлургия и хим. пром-сть Казахстана, Науч.-техн. сб. № 5 (21), 33 (1962).
100. N i v i<6 г е Р., Chim. anal., 47, 448 (1965).
101. Scholes I. R., W a t e г w a n W. R., Analyst, 88, 374 (1963).
102. В a 1 a b a no ft K. J., Salgado M. H., Rev. Real acad. scienc. exact., fis. у Natur. Madrid, 62, 591 (1968).
103. Назаренко В. А., Бык Г. И., Укр. хим. ж., 22, 234 (1956).
104. Макс ай Л. И., Силаева А. А., Сб. тр. Всес. н.-и. горно-металлургич. ин-та цвет, мет., № 7, 345 (1962).
105. Р е г z у n a J. О., Bol. Soc. guim. Peru, 21, 47 (1955).
106. Туманов А. А., Сидоренко А. Н., Тараденкова Ф. С., Зав. лаб., 30, 652 (1964).
107. Быстров С. П., Егоров Н. В., Паршикэв Ю. И., Науч, работы студ. Моск, фармацевт, ин-та, вып. I, 113 (1967).
108. Кавасир о, Окада, Като, РЖХим, 1959, 60543.
109. К. Тоттори, РЖХим, 1961, 19Д110.
НО. Теодорович И. Л., Зав. лаб., 18, 374 (1952).
111. 3 у с с е р Е. Е., Зав. лаб., 12, 640 (1946).
112. Федосов М., Зав. лаб., 13, 1138 (1947).
113. А л е к с е е в а М. В., Информ.-метод, материалы Гос. н.-и. сан. ин-та, 5, 16 (1954).
114. Коренман И. М. и др., Тр. по хим. и хим. технол. Горьковского гос. ун-та и Горьковского отд. ВХО им. Д. И. Менделеева, 2, 314 (1960).
115. Риг ин В. И., Мельниченко Н. Н., Зав. лаб., 32, 394 (1966).
116. V а § a k V., S е d i v е с V., Chem. listy, 46, 341 (1952).
117. Bode H., Hachmann K-, Z. anal. Chem., 229, 261 (1967).
118. Hu la nick! A., Glab S., Chem. analit. (PRL), 15, 1089 (1970).
119. Bode H., Hachmann K., Z. anal. Chem., 241, 18 (1968).
120. Thiel R., Carpentier G., Bull. Cent. rech. Pau, 4, 243 (1970).
121. Bahr H., Bahr Henryk, Chem. anal. (PRL), 16, 427 (1971).
122. Martin F., Floter A., Bull. Soc. chim. France, № 2, 404 (1965).
123. J u гe ёе к M„ J e n i к J., Chem. listy, 49, 264 (1955).
124. Juredek M., Jenik J., Сб. чехословацк. хим. работ, 20, 550 (1955).
125. Meyer J., Z. anal. Chem., 210, 84 (1965).
126. Meyer J., Z. anal. Chem., 231, 241 (1967).
127. S te i nke, Z. anal. Chem., 240, 184 (1968).
128. M e у e г J., Z. anal. Chem., 229, 409 (1967).
129. Агата Нобору, Симидзу Кадзуо, РЖХим, 1966, 10Г177.
130. F 1 u m Z.. Paliva, 37, 33 (1957).
131. Dubois L., Teichman T., Baker C. J., Zdrojewski A., Monk-m a n J. L., Mikrochim. acta, № 1, 185 (1969).
132. Vecer-a Z„ В i e b e г B., Slevarenstvi, 4, 366 (1956).
133. V e с e г a Z„ В i e b e г B„ GieBereitechnik, 3, 61 (1957).
134. РжезачЗ., Дитз Ю., Зав. лаб., 29, 1176 (1963).
135. Е v a n s R. S„ Analyst, 54, 524 (1929).
136. Цывина Б. С., Добкина Б. М., Зав. лаб., 8, 1116 (1938).
137. Бабко А. К., Марченко П. В., Укр. хим. ж., 22, 665 (1956).
138. Адамович В. И., Рыбникова А. И., Зав. лаб., 13, 487 (1947).
139. Крапивина С. С. В сб.: «Вопросы изучения курортных ресурсов СССР». М„ Медгиз, 1955. См. с. 189.
140. ГОСТ 2085—48. Цинк. Методы химического анализа.
141. М а р ч е н к о П. В., Укр. хим. ж., 20, 77 (1954).
142. S t е е 1 е М. С., Е n g 1 a n d L. J., Analyst, 82, 595 (1957).
143. Жаровский Ф. Г., О л ом у цк а я М. Б., Укр. хим. ж., 19, 672 (1953).
144. Wrzesihska Е„ Ргасе Inst, hutn., 12, 289 (1961).
145. Бабенко Н. Л., Тр. Алтайского горнометаллург. н.-и. ин-та, 14, 129 (1963).
146. Cyrankowska М., Chem. anal. (Polska), 8, 679 (1963).
147. ЩербовД. П. В сб.: «Современные методы анализа в металлургии», М., Металлургиздат. 1955. См. с. 160.
148. Андреев А. С., Зав. лаб., 19, 769 (1953).
149. Кипарисова Л. С., Толмачева Л. П., Бюл. н.-т. информ, центр, н.-и. ин-тя олова, сурьмы, ртути, Na 4, 59 (1961).
150. Бабко А. К-, Чалая 3. И., Ми китчен ко В. Ф., Зав. лаб., 32, 270 (1966).
151. Танака Такаси, Хииро Кадзуо, РЖХим, 1963, 21Г100.
152. ТанакаТакаси, ХиироКадзуо, РЖХим, 1964, 10Г105.
153. Хииро Кадзуо, Танака Такаси, РЖХим, 1963, 22Г70.
154. Ciuhandu G., Roscovanu A., Cutui М„ Chimie anal., 50, 489 (1968).
155. Ciuhandu Ch., R о c s i n M., Z. anal. Chem., 172, 268 (1960).
156. Гертнер M. Д., Янсон Э. Ю. Авт. св. СССР, кл. 321, 3/08, gOln, № 243248, заявл. 14.09.67, опубл. 18-09.69. РЖХим, 1970, 13Г150П.
157. S t а г а V., S t а г у J., Taianta, 17, 341 (1970).
158. Като Т„ РЖХим, 1961, 23Д82.
159. De Oliveira М. J., Sartori Platnicki С. М., Rev. quim. ind., 39, 11 (1970).
160. Криста л ев П. В., Криста лев а Л. Б., Шор Н. А., Тр. комиссии по анал. хим. АН СССР, 16, 19 (1968).
161. Cosovic С., Karas-Gasparec V., Farmac. glasnik., 24, 413 (1968).
162. S ebest a F„ Stary J., Collect. Czechosl. Chem. Communs., 33, 3895 (1968),
163. Бабко A. K-, Марченко П. В., Зав. лаб., 19, 1015 (1953).
164. А г с h е г V. S., Doolittle Fr. G„ Taianta, 14, 921 (1967).
165. G u 11 s t г о m D. K-, Mellon M. G., Anal. Chem., 25{ 1809 (1953),
Глава V
КИСЛОРОД
V. 1. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ ОПРЕДЕЛЕНИЯ КИСЛОРОДА
Кислород приходится определять в воде, в различных газовых смесях, технологических растворах, металлах, сплавах и многих других объектах. Кроме того, часто возникает необходимость в определении озона, который является более сильным окислителем по сравнению с кислородом. Для определения кислорода и озона, часто пользуются фотометрическими методами.
Фотометрические методы определения кислорода основаны на реакциях окисления органических и неорганических веществ. Из органических веществ, которые взаимодействуют с кислородом и применяются для его фотометрического определения, следует прежде всего назвать окислительно-восстановительные индикаторы: индигокармин в лейкоформе [1—5], антрахинон-а-сульфокислота [6—8], сафранин Т [9], метиленовый голубой [10] и др. Основными недостатками применения окислительно-восстановительных индикаторов для фотометрического определения кислорода является небольшая чувствительность, неустойчивость аналитических форм и не всегда достаточная специфичность.
Пирогаллол, пирокатехин, 2,4-диаминофенол и другие ароматические окси- и диоксисоединения взаимодействуют с кислородом с образованием окрашенных продуктов реакции. Из этих реагентов для фотометрического определения кислорода наиболее часто применяется пирогаллол.
Большое число фотометрических методов определения кислорода основано на реакциях окисления неорганических соединений, которые затем взаимодействуют с органическими и неорганическими веществами с образованием окрашенных соединений. В качестве восстановителей кислорода в щелочной среде часто применяются соли марганца(П), железа(П), хрома(П) и (III), одновалентной или металлической меди. После поглощения кислорода определяют окисленные формы этих элементов и пересчитывают на содержание кислорода. Достаточно распространенным является метод, основанный на окислении Мп11 в щелочной среде с последующим определением Mniv. После взаимодействия марганца (IV) в кислой среде с иодидом измеряют оптическую плотность раствора выделившегося иода.
V. 2. ОПРЕДЕЛЕНИЕ КИСЛОРОДА С ПРИМЕНЕНИЕМ ИНДИГОКАРМИНА
Индигокармин (индиго-5,5'-дисульфокислоты динатриевая соль)
представляет собой мелкокристаллический порошок, раствор которого в воде окрашен в интенсивный синий цвет. При осторожном восстановлении индигокармина образуется бледно-желтое лейкооснование
ОН н
Рис. 34. Спектры поглощения индигокармина при pH = 11,5 неокислен-ной (/) и окисленной (2) форм.
А 4н
При взаимодействии с кислородом лейкооснование окисляется до индигокармина и раствор окрашивается в синий цвет. Эта реакция лежит в основе метода определения кислорода.
Мешающие вещества. Определению мешают окислители и восстановители, а также все ионы, которые имеют собственную окраску и осаждаются в виде основных солей или гидроокисей в щелочной среде.
Измерение оптической плотности. Спектр поглощения водных растворов индигокар
мина при pH = 11,5 в зависимости от степени его окисления характеризуется двумя максимумами (рис. 34). Оптическую плотность следует измерять при 555 или 620 нм. Кроме того, можно вести определение кислорода по уменьшению оптической плотности раствора при 410 нм.
В случае применения метода стандартной серии пользуются имитирующей шкалой.
Чувствительность метода — 0,1 мкг кислорода в 1 л.
V. 2.1. Определение кислорода в газах
Газ пропускают через раствор лейкооснования индигокармина, после чего определяют оптическую плотность раствора и по калибровочному графику находят содержание кислорода. Содержание кислорода можно также определить по стандартной шкале,
{{ндигокармин, 0,1 %-ный кислый раствор. Сухой препарат тщательно измельчают, берут навеску 1 г и помещают в маленький стакан, прибавляют 4 мл концентрированной серной кислоты и нагревают на водяной бане при частом перемешивании до полного растворения навески. В колбу емкостью 1 л наливают 500—600 мл дистиллированной воды, туда же осторожно переносят содержимое стакана, обмывают стакан водой в ту же колбу, разбавляют до метки водой, перемешивают и оставляют на 5—6 дней. Затем раствор фильтруют и устанавливают Тог титр по кислороду (мг/мл) титрованием 0,01 н. раствором перманганата до желтого цвета:
_ ^КМпО4 ^КМпО4 ‘ 8 Ч” V------------------
к инд
где УкМпО4—°бъем перманганата, израсходованного на титрование; ЛГКМп0< — нормальность перманганата; Уивд — объем индигокармина по кислороду.
Из кислого раствора готовят лейкооснование следующими способами.
1. Индигокармин восстанавливают, прибавляя по каплям 5%-ный раствор дитионита натрия (На282О4-2НгО) до перехода синей окраски в бледно-желтую.
2. К кислому раствору индигокармина прибавляют рассчитанное количество 5—6 н. раствора аммиака с тем, чтобы раствор стал 0,2 н. по аммиаку. Аммиачный раствор неустойчив и хранить его больше суток не следует. Далее проводят восстановление аммиачного раствора амальгамированным цинком. Для этого обычную бюретку заполняют до половины амальгамированным цинком, затем наливают аммиачный раствор индигокармина и оставляют до перехода синего цвета раствора в желтый.
3. Готовят 1%-ный раствор по индигокармину, глюкозе и карбонату калия. Этот раствор нагревают в атмосфере азота в течение 1 ч при 80—90 °C.
Восстановленный одним из этих способов индигокармин немедленно применяют для анализа.
Ход анализа. В атмосфере инертного газа отбирают измеренный объем восстановленного индигокармина, помещают в пробирку или кювету спектрофотометра и через раствор пропускают определенный объем исследуемого газа. Затем пробирку или.кювету устанавливают в спектрофотометр и измеряют оптическую плотность.
Содержание кислорода находят по предварительно построенному калибровочному графику, для построения которого определяют кислород в газовых смесях с известным содержанием О2. Все операции необходимо проводить в атмосфере инертного газа (азота).
V. 2.2. Определение кислорода в воде
Пробу анализируемой воды смешивают с раствором индигокармина в шприце, перемешивают и измеряют оптическую плотность раствора. По оптической плотности раствора находят содержание кислорода.
Ход анализа [5]. Стеклянный шприц емкостью 1 мл с ценой деления в 0,01 мл устанавливают в фиксированном положении внутри кюветы (толщина слоя 1 см) спектрофотометра. Шприц заполняют анализируемой водой, помещают в кювету и шкалу спектрофотометра устанавливают в положение «0». Выливают из
шприца воду и дважды ополаскивают его реактивом (см. раздел V. 2.1). Затем отбирают 1 мл реактива, встряхивают 50 раз и измеряют оптическую плотность при 586 нм. Поршень шприца устанавливают на делении 0,40, погружают иглу в анализируемую воду, засасывают ее до деления «1», перемешивают и вновь измеряют оптическую плотность раствора. Из полученного значения оптической плотности вычитают половину значения оптической Плотности реактива. Содержание кислорода (х) в мг/л находят по формуле
где а — исправленное значение оптической плотности; К — коэффициент, вычисляемый путем деления значения оптической плотности стандартной пробы на содержание в ней СЬ (в мг/л) и равный 0,171 при температуре воды 21 °C.
При содержании кислорода в пробе больше 10 мг/л соотношение между объемами анализируемой воды и реактива уменьшают.
V. 3. ОПРЕДЕЛЕНИЕ* КИСЛОРОДА С ПРИМЕНЕНИЕМ ЛЕЙКОСАФРАНИНА
Сафранин Т представляет собой кристаллический порошок красного цвета. Растворим в воде (лучше в спирте), окрашивает раствор в красный цвет. При восстановлении сафранина Т образуется бесцветное лейкооснование, которое взаимодействует с кислородом с образованием сафранина Т:
Н
Лейкооснование сафранина Т (бесцветное)
Эта реакция используется в колориметрическом определении кислорода [9, 10—13]. Разработан непрерывный, автоматический метод определения кислорода с применением сафранина [14].
Мешающие вещества, см. раздел V.2.
Измерение оптической плотности. Спектр поглощения раствора
сафранина Т представлен на рис. 35. Оптическую плотность растворов лучше измерять при 520 нм. ,s
Чувствительность метода — 0,2 мкг кисло-
рода в 1 л. Л
°>Б' / I
V. 3.1. Определение кислорода в воде /
Реактивы / I
Сафранин Т. Навеску сафранина Т 1 г растворяют / I
в спирте и переносят в колбу "емкостью 1 л. В колбу 0,2- / I вводят аммиак, спирт и воду с таким расчетом, чтобы / \
получить 50%-ный раствор спирта и 3%-ный раствор / \.
аммиака. Полученный раствор перед определением про- 0^—-------6ДО лГнм
пускают через редуктор с амальгамированным цинком
до получения бесцветного раствора, который используют для определения.
Рис. 35. Спектр поглощения сафранина Т.
Ход анализа. В мерную колбу емкостью 50 мл, заполненную азотом, наливают 1,5 мл восстановленного реактива и доводят анализируемой водой до метки. Колбу закрывают хорошо притертой пробкой и содержимое перемешивают в течение 3 мин. Наполняют раствором кювету, закрывают крышкой и быстро измеряют оптическую плотность.
По значению оптической плотности находят содержание кислорода по калибровочному графику.
V. 4. ОПРЕДЕЛЕНИЕ КИСЛОРОДА ВИДОИЗМЕНЕННЫМ МЕТОДОМ ВИНКЛЕРА
В щелочной среде кислород быстро реагирует с марганцем (П) с образованием окислов марганца(III) и (IV):
2Мп(ОН)2 + О2 —► 2МпО2 • 2Н2О
Затем при взаимодействии этих окислов с иодидом в кислой среде выделяется иод:
МпО2 + 2KI + 4НС1 —► МпС12 + 2КС1 + 12 + 2Н2О
Выделившийся иод по методу Винклера титруют тиосульфатом, а в случае фотометрического варианта определение заканчивают измерением оптической плотности раствора иода [15—19]. Для увеличения чувствительности метода выделившийся иод экстрагируют органическим растворителем и измеряют оптическую плотность экстракта.
Мешающие вещества — см. раздел V.2.
Измерение оптической иода представлен на рис.
ЕЮ'г
плотности. Спектр поглощения раствора 36. Оптическую плотность измеряют при 510 нм в неводном растворе и при 450 и 350 в водных растворах.
Чувствительность метода — 0,15 мкг кислорода в 1 л.
9,0
0,0
3,0
Я, нм
рас-
Рис. 36. Спектры поглощения твора иода:
/ — в воде; 2—в СС14 (в CHCI3).
°300 350 ыоо Ь5О 500 550
v. 4.1. Определение кислорода в воде
К определенному объему исследуемой воды прибавляют раствор хлорида марганца, щелочь и иодид калия. После перемешивания раствор подкисляют и измеряют оптическую плотность
раствора выделившегося иода или выделившийся иод экстрагируют CCI4 и измеряют оптическую плотность экстракта.
Реактивы
Раствор МпС12-4Н2О, 400 г в 1 л воды.
Ход анализа. В колбу с притертой пробкой емкостью 500 мл помещают 45 мл анализируемой воды, с помощью пипетки вводят на дно колбы 0,5 мл 10 н. раствора щелочи, раствор 0,5 г иодида калия в минимальном объеме воды и 0,5 мл раствора хлорида марганца. Колбу закрывают пробкой и энергично встряхивают. В делительную воронку помещают 3 мл концентрированной хлористоводородной кислоты, 10 мл четыреххлористого углерода и переносят раствор из колбы. Колбу обмывают 2 мл концентрированной хлористоводородной кислоты, затем 5 мл воды и присоединяют промывную жидкость к раствору в делительной воронке. Содержимое воронки взбалтывают до полной экстракции выделившегося иода. После отстаивания раствор иода в ССк сливают в кювету прибора и измеряют оптическую плотность при 510 нм.
Содержание кислорода находят по предварительно построенному калибровочному графику, для построения которого пользуются раствором иода, определенной концентрации в иодиде калия.
V. 5. ОПРЕДЕЛЕНИЕ КИСЛОРОДА ПО КОЛИЧЕСТВУ
ОКИСЛЕННОЙ меди
Кислород взаимодействует с металлической медью или с солями меди(1) в аммиачном растворе с образованием аммиаката меди(П):
2Cu + О2 + 8NH3 + 4Н+ —> 2[Cu(NH3)4]2++ 2Н2О
4СиС1 + О2 + I6NH3 + 4Н+ —* 4ICu(NH3)4]2++ 4С1Ч-2Н2О
Измеряя оптическую плотность раствора аммиаката меди, можно определить содержание кислорода в газах и жидкостях [20—28].
Мешающие вещества — см. раздел V. 2.
Измерение оптической плотности. Спектр поглощения раствора аммиаката меди представлен на рис. 37. Оптическую плотность
лучше измерять при 600 нм.
Чувствительность метода — 0,2 мл кислорода в пробе.
V. 5.1. Определение кислорода в газах [23]
Анализируемый газ пропускают через аммиачный раствор хлорида меди(1), которым наполняют специальную кювету — барботер (рис. 38), затем измеряют оптическую плотность раствора аммиаката Меди(II) и по оптической плотности на
Рис. 37. Спектр поглощения раствора аммиаката меди.
ходят содержание кислорода.
Кювета-барботер — цилиндр с плоскопараллельными окошками из оптического стекла, впаянными в корпус кюветы или укрепленными на ее торцах клеем. Размер кюветы соответствует размеру
Рис. 38. Кювета-барботер для определения кислорода в газах: 1— оптическая кювета; 2—стеклян' ный фильтр; 3, 5 — трехходовые краны; 4—трубка; б — барботажная трубка.
Рис. 39. Схема расположения аппаратуры при определении кислорода в газах:
1 — баллон с анализируемым газом; 2—кювета-барботер; 3— газовые часы; 4—реометр.
кюветодержателя фотоколориметра ФЭК-М. В барботажную трубку впаян стеклянный фильтр, пористость которого равна пористости фильтра <№ 4.
Этот метод дает возможность определить 10“5—10“3 объемн.% кислорода в газах. Однако при продувке раствора газом может удаляться аммиак, что приведет к ошибкам определения,
Поглотительный раствор. Помещают 12 г CuCl и 36 г NH4C1 в мерную колбу емкостью 1 л, в которой находится 1,15 мл 4%-ного раствора аммиака. Соли растворяют добавлением воды, доводят до метки дистиллированной водой и перемешивают. Раствор хранят в бутыли, заполненной спиральками из медной проволоки, и предохраняют от попадания кислорода при помощи склянки с пирогаллолом и резиновой камерой, заполненной аргоном.
Стандартный раствор. В мерную колбу емкостью 1 л помещают 36 г NH4C1, 0,892 г CuSO4-5H2O, 115 мл 4%-ного раствора аммиака, разбавляют до метки водой и хорошо перемешивают.
Калибровочный график-можно построить по смесям газов с различным содержанием кислорода. В этом случае пользуются поглотительным раствором и при построении калибровочного графика используют разность значений оптической плотности поглотительного раствора после поглощения кислорода и до пропускания газа.
Ход анализа. Из кюветы (см. рис. 40) удаляют атмосферный воздух анализируемым газом, заполняют ее поглотительным раствором и измеряют первоначальную оптическую плотность на ФЭК с применением красного светофильтра (ЛЭфф = 600 нм). Затем барботируют (рис. 39) через поглотительный раствор в кювете определенный объем анализируемого газа со скоростью 4—5 л/ч и измеряют оптическую плотность. По разности оптических плотностей находят содержание кислорода по предварительно построенному калибровочному графику.
V. 6. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ КИСЛОРОДА
Индигокарминовый метод применен для определения кислорода в воде [29, 30]. В качестве растворов сравнения рекомендована имитирующая шкала [31], состоящая из растворов хлорида кобальта, хлорида железа (III) и сульфата меди. Индигокарминовый метод применен также для определения кислорода в столовых напитках [32].
Кислород поглощают гидратом окиси железа (II), который получают действием щелочи на соль Мора в отсутствие кислорода. Затем раствор подкисляют и образовавшееся железо(III) определяют в виде сульфосалицилата [33, 34] или в виде роданидного комплекса [35].
Предложен метод определения кислорода, основанный на окислении в щелочной среде марганца(II) до марганца(III), определении Мпш с применением о-толидина [36, 37] или в виде комплекса с тра«с-1,2-диаминциклогексантетрауксусной кислотой [38]. Фор-мальдоксим при взаимодействии с солями марганца(II) и кислородом образует красно-оранжевый продукт, по интенсивности окраски которого определяют содержание кислорода [39]. Темно-фиолетовый раствор 4,4'-бис(дифенилметил)-дифенила обесцвечивается под действием кислорода в результате образования бесцветной вы-сокополимерной перекиси. Эта реакция использована при колориметрическом определении кислорода [40].
При взаимодействии кислорода с иодидом выделяется иод, который определяют по реакции с 3,3'-диметилнафтидином, по количеству иода вычисляют содержание кислорода [41, 42].
Реакция кислорода с хлоридом меди(1) в аммиачной среде применена для определения кислорода в водах, богатых органическими веществами [27].
Для определения несвязанного кислорода в металлическом цирконии [43] навеску сплава растворяют в фтористоводородной кислоте, а выделившийся при этом кислород определяют видоизмененным методом Винклера (см. V. 4).
Для определения связанного кислорода в металлах рекомендованы косвенные методы. Так, при определении кислорода в стали ее восстанавливают алюминием’ и окисленный алюминий определяют с помощью стильбазо [44]. При определении кислорода в гидриде титана и металлическом титане [45] отгоняют металлический титан в токе сухого хлористого водорода, а в остатке определяют окислы титана фотометрическим методом. Определение связанного кислорода в металлическом натрии основано на проведении реакции Вюрца между н-амилхлоридом и металлическим натрием, при этом примесь кислорода связывается в виде NagO. Окись натрия действием двуокиси углерода переводят в карбонат натрия, количество которого определяют спектрофотометрическим методом при 11,38 мк [46].
Для определения кислорода в газах применяют серный метод [11], основанный на взаимодействии кислорода при 360°C с парами серы с образованием сернистого ангидрида, который определяют фуксин-формальдегидным реактивом. Микровариант этого метода применен для определения кислорода в тонких напыленных слоях стибнита [47]. ♦
Предложен спектрофотометрический метод определения кислорода в смесях с азотом, основанный на взаимодействии с N.N-ди-метиланилином [48]. Оптическую плотность продукта реакции измеряют при 347 нм.
В газообразной смеси кислород определяют косвенным методом — измерением при 253,7 нм оптической плотности озона, образующегося при пропускании анализируемой газовой смеси через трубку с электрическим разрядом [49].
V. 7. МЕТОДЫ ОПРЕДЕЛЕНИЯ ОЗОНА
Озон является более сильным окислителем по сравнению с кислородом, поэтому все методы, применяемые для определения кислорода, могут быть использованы для определения озона. Озон поглощает свет в ультрафиолетовой области и может быть определен по измерению поглощения света при 248—250 нм. Измерение поглощения в ультрафиолетовой области положено в основу непрерывного метода определения озона в газовых смесях [50]. Кроме того, озон можно определить в присутствии кислорода по
реакции окисления меди(П) до меди(Ш) в присутствии перйодата [51], железа(П) до железа(Ш) [52] или марганца(П) до марганца (III) в пирофосфатных комплексах [53, 54]. Последний метод дает возможность определять озон в присутствии хлора и других хлорсодержащих окислителей.
Реакция окисления озоном красителей типа фталеинов [55], метилового красного [56] до бесцветных соединений использована для фотометрического определения его.
V. 7.1. Определение озона в водных растворах [51]
В присутствии перйодата озон окисляет медь(П) до меди(Ш): 2Си2+ + О3‘ —> 2Си3+ + О2 + 2ОН“
Спектр поглощения перйодата меди(III) характеризуется максимумом при 415 нм.
Мешающие вещества. Метод характеризуется большой специфичностью. Растворенный в воде кислород не мешает определению.
Измерение оптической плотности. Оптическую плотность измеряют при 415 нм.
Чувствительность метода — около 5 мкг озона в 10 мл.
Реактивы
Раствор перйодата и сульфата меди. Готовят 0,03-М раствор CuSO4, 0,15 М раствор HsIO6 и 1,5 М раствор КОН. Все три раствора смешивают в равных объемах и перемешивают.
Ход анализа. Пробу воды до 20 мл, содержащую до 20 мкг озона, помещают в мерную колбу емкостью 25 мл, прибавляют 5 мл раствора перйодата и сульфата меди, если нужно, разбавляют водой до метки, перемешивают и измеряют оптическую плотность раствора при 415 нм. Содержание озона находят по калибровочному графику.
V. 7.2- Определение озона в присутствии соединений хлора [53]
Озон в сернокислой среде в течение 3 мин количественно окисляет марганец(П) в пирофосфатном комплексе марганца(II) до пирофосфатного комплекса марганца(III):
2[Мп(Н2Р2О7)8]4' + Оз + 2Н+ —► 2[Мп(Н2Р2О7)3]3-+О2 + Н2О
Эта реакция применена для фотометрического определения озона.
Мешающие вещества. Пирофосфатный комплекс марганца(II) не реагирует с НСЮ, СЮз и CIOJ, поэтому определение озона можно проводить в присутствии хлорсодержащих окислителей. Исключением является СЮг, однако реакция окисления пирофосфатного комплекса марганца(II) до комплекса марганца(III) с
помощью С1О2 происходит очень медленно и практически не мешает определению озона.
Измерение оптической плотности. Измерение оптической плотности производят при 518—524 нм.
Чувствительность метода — около 1 мг/л озона.
Реактивы
Пирофосфатный комплекс марганца. В мерную колбу емкостью 100 мл вводят 15 мл 1 М раствора MnSO4, 60 мл 0,75 М раствора ЫагНгРгО?, 12,5 мл H2SO4, разбавленной (1 :4), добавляют дистиллированную воду до метки и перемешивают.
Бихромат калия, 0,005 н. раствор для построения калибровочного графика. Навеску в 0,2451 г К2СГ2О7 растворяют в воде, переносят в мерную колбу емкостью 1 л, разбавляют водой до метки и перемешивают. 1 мл этого раствора эквивалентен 0,120 мг озона.
Ход анализа. К 100 мл анализируемой воды прибавляют 5 мл раствора пирофосфатного комплекса марганца(II), растворы перемешивают и оставляют на 5 мин. Затем наливают раствор в кювету с толщиной слоя 20 см и измеряют оптическую плотность при 520 нм. Содержание кислорода находят по калибровочному графику.
V. 7.3. Определение озона в воздухе [57]
Воздух пропускают через спиртовой раствор дигидроакридина в присутствии ацетатного буферного раствора (pH = 6,0). При этом дигидроакридин окисляется до акридина:
Н
Н
Акридин окрашивает раствор в слабый желтый цвет, но сильно поглощает при 249,5 нм, е249,5 = 1,69-105 (рис. 40).
Мешающие вещества. Определению мешают окислители, наличие которых в воздухе учитывают проведением холостой пробы. Для этого после каталитического разложения озона пропускают воздух через гранулы с двуокисью марганца.
Измерение оптической плотности. Оптическую плотность измеряют на спектрофотометре при 249,5 нм.
Чувствительность метода — 4 • 10-3 мкг/мл.
Реактивы
Ацетатный буферный раствор. Смешивают 250 мл 0,2 М спиртового раствора ацетата натрия с 250 мл 0,02 М спиртового раствора уксусной кислоты (pH = 5,8).
Поглотительный раствор. Навеску дигидроакридина 0,100 г растворяют в буферном ацетатном этанольном растворе (pH = 5,8), доводят объем до 1 л
этим же раствором и перемешивают. Для поглощения озона этот раствор разбавляют в 50 раз ацетатным буферным спиртовым раствором.
Двуокись марганца, очищенная от закисных форм марганца. Препарат МпОг ч. д. а. двукратно промывают азотной кислотой ч. д. а. (пл. 1,34), высушивают и готовят гранулы размером 2—4 мм смешиванием с силикатным клеем. Гранулы высушивают при 150 °C.
стандартный раствор акридина. Для построения калибровочного графика навеску акридина (бледно-желтые кристаллы с т. пл. 110°С) 0,375 г растворяют в этанольном ацетатном буферном растворе с pH = 5,8, переносят в мерную колбу емкостью 100 мл, этим же раствором доводят до метки и перемешивают. 1 мл этого раствора соответствует 100 мкг озона.
При построении калибровочного графика путем разбавления готовят раствор, содержащий 10 мкг в 1 мл.
Ход анализа. Объем воздуха для анализа зависит от содержания озона. При концентрации озона, составляющей сотые доли миллиграмма в 1 м3, следует отбирать около 10 л воздуха. Если содержание озона составляет около 0,1 мг/м3, то для анализа достаточно отобрать 1—2 л воздуха.
Рис. 41. Схема отбора пробы исследуемого воздуха:
А и Б — параллельные пробы исследуемого воздуха.
Рис. 40. Спектр поглощения спир' тового раствора акридина.
Отбирают две параллельные пробы А и Б (рис. 41). В пробе Б озон каталитически разлагают двуокисью марганца и поглотительный раствор этой пробы используют в качестве раствора сравнения при измерении оптической плотности испытуемого раствора.
В два поглотительных последовательно соединенных сосуда с пористыми пластинками (сопротивление 150—250 мм вод. ст.) наливают по 10 мл свежеприготовленного дигидроакридинового раствора (рис. 41). Затем просасывают воздух со скоростью 0,3— 0,5 л/мин. При высоком содержании озона скорость потока уменьшается. После пропускания воздуха поглотительный раствор из сосуда Б наливают в кварцевую кювету сравнения, а анализируемый раствор из сосуда А в другую кварцевую кювету с толщиной слоя 1 см и измеряют оптическую плотность при 249,5 нм. По оптической плотности находят содержание кислорода по калибровочному графику,
Содержание кислорода в миллиграммах на 1 м3 (х) можно также рассчитать по формуле:
D • 0,284 • V х~ v0
где D — оптическая плотность поглотительного раствора при 249,5 нм; V — объем поглотительного раствора, мл; Vo — объем просасываемого исследуемого воздуха, приведенного к объему при нормальных условиях (0 °C, 760 мм рт. ст.), л.
V. 7.4. Другие методы определения озона
Для определения озона в воде предложены методы, основанные на реакции окисления иодида до иода в присутствии крахмала и измерении оптической плотности синего продукта реакции при 570 нм [58, 59].
Разработан метод определения озона в атмосфере, основанный на взаимодействии его с 4-аллил-2-метоксифенолом (эвгенолом). Выделяющийся при этом формальдегид определяют спектрофотометрическим методом с применением дихлорсульфитомеркуриата натрия [60].
В щелочной среде (pH = 13,5) хром(III) окисляется озоном до хромата, по образовавшемуся количеству которого определяют содержание озона [61]. Флуоресцентный метод [62], основанный на окислении озоном дигидроакридина до акридина, предложено использовать как фотометрический [57]. Измерение оптической плотности производят при 249,5 нм, 6249,5= 1,69*105.
ЛИТЕРАТУРА
1. Константинова-Шлезингер М. А. Тр. Эльбрусской экспедиции 1934—1935 г. М., Изд-во АН СССР, 1936. См. с. 49.
2. Macura Н., Werner G., Die Chemie, 56, 90 (1943).
3. Loomis W. F., Anal. Chem., 26, 402 (1954).
4. Бабкин P. Л., Электрич. станции, 1, 16 (1954).
5. Loomis W. F., Anal. Chem., 28, 1347 (1956).
6. Brady I. Y., Anal. Chem., 20, 1033 (1948).
7. StruszynckiM. e. ol., Przem. chem., 9, 449 (1953).
8. Stafford C. e. ol., Chem., 27, 2012 (1955).
9. Ковальцев В. А., Алесковский В. Б. Определение растворенного в воде кислорода. М., Госхимиздат, 1961. См. с. 34.
10. Д а в д а р и а н и И. В. и др., Теплоэнергетика, № 10, 76 (1970).
J1. Митрофанова Т. П„ Орлов Ю. Ф., Плоткина Е. И., Зав. лаб., 36, 161 (1970).
12. Алесковский В. Б., Ковальцов В. А. Авт. свид. СССР 144046, 27.01.62.
13. Мустафин И. С. и др. Ассортимент реактивов на кислород. М., Изд-во НИИТЭХИМ, 1968.
14. Алесковский В. Б. и др., Зав. лаб., 28, 1440 (1962).
15. Ovens ton Т. С. J., Watson J. Н. Е., Analyst, 79, 383 (1954).
16. Trotti L., Sacks D., Atti Acad, liqure sci. elettere, 18, 255 (1961/1962).
17. Trotti L., Sacks D., Rapp, et proc.-verb. reun. Commiss. internal, explorat. scient. Mer. Mediterr., 16, 673 (1961).
18. Смирнов Э. В. В сб.: «Мор. гидрофиз. исслед.» (Севастополь), № 2 (48).
144 (1970).
19. BroenkowW, W., Cline J. D., Limnol and Oceanogr., 14, 450 (1969).
20. Mugdam M., S i x t J., Angew. Chem., 46, 90 (1933).
21. Uh rig K., Roberts F. M., Levin H., Ind. Eng. Chem., Anal. Ed., 17, 31 (1945).
22. P о w e 11 J. S, J о у P. C„ Anal. Chem., 21, 296 (1949).
23. Deinum H. W., D a m J. W., Anal. Chim. acta, 3, 353 (1949).
24. Скрипка В. Г., Дых но Н. М., Зав. лаб., 28, 1439 (1962).
25. Pilarczyk Н., Miller Gr., Paterok N., Chem. anal. (Polska), 12, 899 (1967).
26. Малкова Э. M. и др., Измерит, техника, № 5, 54 (1963).
27. Р е з а е в а Л. Т„ ЖАХ, 17, 874 (1962).
28. Березина Ю. И., Ко росты ле в а М. М., Бройт В. В., ЖАХ, 20, 262 (1965).
29. М е у 1 i n g А. Н„ F г a n k G. Н., Analyst, 87, 60 (1962).
30. John Р. A. St., Wine for dner J. D., Silver W. S., Anal. chim. acta, 30, 49 (1964).
31. Buchoff L. S., Ingber N. M., Brady J. H., Anal. Chem., 27, 1401 (1955).
32. von Canwenberghe H., de Clerck J., Internat. tijdsch. brouw. en mout., 16, 66 (1956/57).
33. А л e к с а н д p о в A. H., Левина И. Л., Скоп С. Л. В сб. «Методы исследования продуктов нефтепереработки и нефтехим. синтеза». Л., Гостоп-техиздат. 1962. См. с. 145.
34. Аршанський I. М., Наук. зап. Чершвецьк. ун-ту, 11, 135 (1955).
35. S h a w J. A., Ind. Eng. Chem., Anal. Ed., 14, 891 (1942).
36. L e i t h e W., H о f e r A., Mikrochim. acta, 5, 1066 (1968).
37. W i c k e r t K-, I p a c h E., Z. anal. Chem., 140, 350 (1953).
38. S as t г у G. S., H a mm R. E., Anal. Chem., 41, 857 (1969).
39. TanakaMotoharu, Mikrochim. acta, № 5—6, 1048 (1955).
40. M й 11 e r E., M e t z g e r H., Chemiker-Ztg, 78, 317 (1954).
41. Banks J., Analyst, 79, 170 (1954).
42. Fadrus H., Maly J., Analyst, 96, 591 (1971).
43. Silverman L., Bradshaw W., Anal. chim. acta, 18, 253 (1958).
44. Д а и e н к о О. В. и др., Зав. лаб., 26, 1109 (1960).
45. Нечина В. Р., Козырева Э. А., Богатова 3. С., ЖАХ, 23, 563 (1968).
46. de В г u i n Н. J., Anal. Chem., 32, 360 (1960).
47. Надеждина Л. С. и др., Тр. комиссии по аналит. хим. АН СССР, 16, 227 (1968). •
48. Bh a tty М. К., Townshend A., Anal. Lett., 4, 357 (1971).
49. К а у е S., К о е п с у J. Е., Anal. Chem., 41, 1491 (1969).
50. Ф а с то в с к и й В. Г., Ров и некий А. Е., Зав. лаб., 21, 1030 (1955).
61. Wagnerova D. М., Eckschlager К., Veprek-Siska J., Collect. Czechosl. Chem. Communs, 32, 4032 (1967).
52. Cohen I. R., Rufalini J. J., Environ. Sci. and Thechnol., 1, 1014 (1967).
53. H о f m a n n P., S t e r n P., Anal. chim. acta, 45, 149 (1969).
54. H о f m a n n P„ S t e r n P., Anal. chim. acta, 47, 113 (1969).
55. Planta Ch. А., пат. США, кл. 23—232, 3397966, заявл. 17.11.64, опубл. 20.08.68.
56. Czerniec J., Gregorowicz Z., Fligier J., Czichon P., Chem. anal. (PRL), 16, 1125 (1971).
57. Манита M. Д., Румянцева M. В., Эглита M. Э., Гигиена и санитария, № 5, 56 (1967).
58. Wiezbicki T., Pieprzyk H., Гигиена и санитария, 15, 1041 (1970).
59. Штрек кер Г. П. и др., Тр. н.-и. и проектн. ин-та по обогащ. руд. цветн. мет. «Казмеханобр», сб. 6, 259 (1971).
60. S а с h d е v S. L., L о d g e J. P., W e s t Ph. W., Anal. chim. acta, 58, 141 (1972).
61. Кикут и Синъити, Судзуки Син, Коянаги Конти, РЖХим, 1968, 5Г120.
62. Константинова-Шлезингер М. А., Изв. АН СССР, № 2 (1937).
Глава VI
СЕРА
VI. 1. МЕТОДЫ ОТДЕЛЕНИЯ
В соединениях сера проявляет валентность от —2 до +6. На практике приходится определять серу в различных степенях окисления. Все фотометрические методы определения серы требуют предварительного ее отделения. Методы отделения серы зависят от характера соединения, в виде которого находится сера в анализируемом образце, а также от состава образца. Чаще других для отделения серы применяются методы дистилляции ее в виде сероводорода или сернистого ангидрида. Отгонку сероводорода проводят в токе инертного газа (аргона, азота или двуокиси углерода), чтобы предотвратить окисление сероводорода кислородом воздуха. Выделение сероводорода из растворов не представляет трудностей. Для этого обычно подкисляют раствор хлористоводо-оодной кислотой и пропускают газ-носитель. При анализе твердых веществ необходимо иметь в виду, что не все сульфиды растворяются в хлористоводородной кислоте. Так, стали, закаленные при температуре выше 1200 °C, содержат много Fe2S3, которое мало растворяется в этой кислоте, и результаты анализа получаются заниженными.
Для поглощения сероводорода можно применять раствор щелочи, однако лучшим поглотителем сероводорода является 2%-ный раствор ацетата цинка [1]. Восстановление серы до сероводорода обычно проводят в две стадии. В щелочной среде восстанавливают станнитом, а затем в хлористоводородной среде металлическим алюминием. При восстановлении сульфатов используют смесь йодистого водорода, муравьиной кислоты и красного фосфора [2]. Выделяющийся сероводород вместе с азотом, который используют в качестве газа-носителя, пропускают через раствор пирогаллола и фосфата натрия, а очищенный сероводород поглощают раствором ацетата цинка.
Сульфидную серу отделяют также осаждением с помощью ацетата цинка, что дает возможность отделить ее от многих мешающих определению веществ и концентрировать. Коллектором в этом случае оказывается гидроокись цинка, образующаяся при подщелачивании раствора ацетата цинка.
Для определения сульфатной серы применяют различные косвенные методы или восстанавливают сульфат до сероводорода и
определяют его. Для отделения и концентрирования сульфатной серы ее осаждают с помощью хромата бария при pH около 1. При этом хромат бария играет роль осадителя и коллектора. Сульфат можно восстановить до сероводорода нагреванием в атмосфере водорода и паров хлористого водорода.
Заметные количества сульфата отделяют с помощью солянокислого бензидина при pH = 3 и температуре около О °C. Для уменьшения растворимости сернокислого бензидина прибавляют органические растворители, а отфильтрованный осадок промывают 80%-ным этанолом.
Серу, а также некоторые ее соединения можно выделять экстракцией органическими растворителями. Выбор экстрагента зависит от объекта анализа и от состояния серы в данном объекте. Так, при выделении серы часто применяют сероуглерод, а при выделении серы из углеводородов лучшим растворителем является ацетон [3]. Хорошие результаты получены при экстракции серы пиридином [4].
Газообразные соединения часто концентрируют сорбцией на силикагеле, а затем после десорбции определяют тем или другим методом. Необходимо иметь в виду, что сероводород и сероуглерод не сорбируются на силикагеле.
Обзор фотометрических методов определения серы приведен в работах [5, 6].
VI. 2. ОПРЕДЕЛЕНИЕ СЕРОВОДОРОДА В ВИДЕ МЕТИЛЕНОВОГО ГОЛУБОГО
В основе метода лежит реакция между сероводородом и солянокислым М,М-диметил-п-фенилендиамином в присутствии хлорида железа (III). При этом образуется метиленовый голубой:
• НС! + 6FeCl3 + H2S —>
2(CH3)2N
СГ + 6FeCl2 + NH4C1 + 7НС1
Определение заканчивают измерением оптической плотности водного раствора метиленового голубого [7] или хлороформного экстракта перхлората красителя [8], а также нитробензольного экстракта метиленового голубого [9].
Исследование взаимодействия сероводорода с М,М'-диметил-п-фенилендиамином и ионами железа(III) в растворах хлористоводородной кислоты показало, что спектр поглощения продукта реакции, кроме максимума при 670 нм, типичного для образующегося в результате реакции метиленового голубого, имеет еще вто-190
слабокислой среде образуют
Рнс. 42. Спектры поглощения хлороформного раствора перхлората красителя (/) и водного раствора метиленового голубого (2).
рой максимум при 740 нм. Этот максимум указывает на присутствие в растворе ионного ассоциата катиона метиленового голубого и хлоридного комплекса железа(III) [10], т. е. вероятно прохождение побочного процесса. Поэтому всегда необходимо точно придерживаться методики анализа.
Мешающие вещества. Определению мешают заметные количества тяжелых металлов, которые в нерастворимые сульфиды и окислители, окисляющие сероводород.
Измерение. оптической плотности. Водный раствор метиленового голубого и хлороформный раствор перхлората красителя устойчивы в течение длительного времени. Прямой солнечный свет вызывает фотохимическую реакцию, в результате которой происходит разложение красителя.
Оптическую плотность водного раствора метиленового голубого измеряют при 665—670 нм, а хлороформного раствора перхлората красителя при 650 нм (рис. 42).
Молярный коэффициент поглощения равен 3,4 • 104. Чувствительность метода в случае применения водных растворов в пересчете на серу составляет 0,05 мкг в 25 мл конечного раствора. Применяя экстракцию перхлората красителя, можно несколько повысить чувствительность метода. При анализе твердых веществ можно определить 10-4—10-в% серы.
V. 2.1. Определение сероводорода в отсутствие мешающих веществ
Реактивы
Стандартный раствор сульфида натрия. Навеску сульфида натрия ЫагБ-ЭНгО 0,08 г переносят в мерную колбу емкостью 1 л, содержащую 10 мл 5%-ного раствора едкого натра и 250 мл воды. После растворения навески раствор разбавляют до 1 л и перемешивают. Этот раствор содержит ~ 1 мкг серы в 1 мл. Более точно содержание сульфидной серы определяют иодометрически.
Иод, 0,1 н. раствор.
Тиосульфат натрия, 0,1 н. раствор.
Сернокислый N, N'-диметил-п-фенилендиамин, 0,3 г препарата растворяют в охлажденной смеси 50 мл воды и 100 мл концентрированной серной кислоты.
Хлорид железа(Ш), 1%-ный раствор в очищенной хлористоводородной кислоте, разбавленной (1 :20).
Хлорная кислота, 0,1 н. раствор.
Хлороформ, ч. д. а.
Калибровочный трафик. Для построения графика стандартный раствор (от 0,1 мл до 10 мл) обрабатывают, как указано в ходе определения. В качестве раствора сравнения во всех случаях пользуются раствором холостого опыта.
Ход анализа. В мерную колбу емкостью 25 мл помещают 0,5 мл раствора NaOH и 0,5 мл раствора п-амино-М.М'-диметиланилина. Затем прибавляют испытуемый раствор, содержащий от 0,05 до 10 мкг серы в виде сульфида, разбавляют дистиллированной водой до метки и колбу помещают в водяную баню, нагретую до 80— 90°C. Выдерживают в течение 5 мин, охлаждают, наливают в кювету прибора и измеряют оптическую плотность при 670 нм. По значению оптической плотности находят содержание сульфидной серы по предварительно построенному калибровочному графику.
Если применяют экстракцию, то полученный раствор переносят в делительную воронку емкостью 100 мл, прибавляют 2 мл хлорной кислоты и трижды экстрагируют хлороформом порциями по 3 мл. Экстракты сливают в пикнометр или узкий мерный цилиндр емкостью 10 мл, разбавляют хлороформом до 10 мл, перемешивают, заполняют кювету прибора и измеряют оптическую плотность хлороформного экстракта при 650 нм.
VI. 2.2. Определение сероводорода в газах
Определенный объем анализируемого газа пропускают через раствор ацетата цинка для поглощения сероводорода, а затем определяют ,сульфидную серу в виде метиленового голубого. В качестве поглотителя сероводорода рекомендуется также раствор соли кадмия в присутствии цитрата и триэтаноламина [11].
Реактивы
Ацетат цинка, 2%-ный раствор, подкисленный несколькими каплями уксусной кислоты.
Другие реактивы — указаны в разделе VI. 2.1.
Ход анализа. В бутыль с широким горлом помещают 50 мл раствора ацетата цинка и барботируют через него измеренный объем анализируемого газа со скоростью не более 10 л в 1 ч до появления слабой мути. Раствор из бутыли переносят в мерную колбу емкостью 1 л, промывая бутыль раствором ацетата цинка. Затем разбавляют дистиллированной водой до метки, перемешивают, отбирают аликвотную часть и определяют, как указано в разделе VI. 2.1.
VI. 2.3. Определение сульфидов в воде [12г 13]
К анализируемой пробе прибавляют раствор ацетата цинка и соосаждают сульфид цинка с гидроокисью цинка. Затем в осадке определяют сульфидную серу в виде метиленового голубого.
Реактивы
Карбонат натрия, 1 М раствор.
Ацетат цинка, 2%-ный раствор.
Другие реактивы — указаны в разделе VI. 2.1.
Ход анализа. Определенный объем анализируемой воды помещают в стакан и на каждые 250 мл воды прибавляют 1 —1,5 мл раствора ацетата цинка и такое же количество раствора карбоната натрия. При низком содержании сульфидов прибавляют 0,5— 1 мл раствора гидроокиси натрия на каждые 250 мл анализируемой воды. Если анализируемая вода имеет щелочную среду, то количество карбоната натрия или щелочи может быть уменьшено. Раствор перемешивают до выпадения хлопьевидного осадка, дают осадку осесть, раствор декантируют и определяют серу, как указано в разделе VI. 2.1.
VI. 2.4. Определение серы в металлах [14—19]
Навеску металла растворяют в кислоте, образующийся сероводород отгоняют в раствор ацетата цинка, а затем определяют серу в виде метиленового голубого. Для растворения металла лучше применять ортофосфорную кислоту, так как при этом вся находящаяся в металле сульфидная сера переходит в сероводород. При использовании других кислот возможна побочная реакция с образованием (CH3)2S, что приводит к заниженным результатам. Многие металлы восстанавливают сульфатную серу до сульфидной, поэтому при анализе металлов обычно получают суммарное содержание серы. При определении очень малых количеств серы в металлах особенно важным является очистка газа-носителя от кислорода.
Метод дает возможность определить до 10~4 % серы.
Реактивы
Медь, мелкая стружка, снятая фрезой. Отработанную медь можно регенерировать восстановлением в токе водорода при 500—600 °C или выдерживанием в хлористоводородной кислоте (пл. 1,19). В последнем случае стружку промывают водой, затем спиртом и высушивают при 100—105 °C.
Пирогаллол. В закрытой колбе растворяют 50 г пирогаллола при сильном встряхивании в 100 мл воды. В других 100 мл воды растворяют 50 г КОН, раствор охлаждают и смешивают его с водным раствором пирогаллола. Реактив можно применять в течение 7 суток.
Цинк, гранулированный. Перед употреблением цинк обрабатывают хлористоводородной кислотой, разбавленной (1 :2), в течение 3—5 мин.
Хлорид хрома(П1), 50 г электролитического хрома растворяют в 600 мл кислоты, разбавленной (1 : 1). Раствор" отфильтровывают через слой ваты, разбавляют водой до 1 л и перемешивают.
Сульфат меди, CuSO4-5H2O, 20%-ный раствор.
Хлорид натрия, сухая соль.
Ортофосфорная кислота, ос. ч., пл. 1,60, содержащая не более 2-10—Б% SO^".
Ацетат цинка, 2%-ный раствор.
Конго, индикаторная бумага.
Хлористоводородная кислота, пл. 1,19, перегнанная в стеклянном перегонном аппарате, не имеющем резиновых соединений.
Другие реактивы — указаны в разделе IV. 2.1.
Ход определения. Разложение пробы проводят на установке, показанной на рис. 43. Навеску металла для растворения
Рис. 43. Схема установки для определения малых количеств серы в металлах:
I—баллон с азотом; 2—реометр; 3 — трубчатые печи; 4—реостат; 5—гальванометр; 6— кварцевые трубки; 7—термопара; 8 и 19 трехходовые краны; У—склянки Тищенко, заполненные щелочным раствором пирогаллола; 10—склянки Тищенко, заполненные водой; 11— редуктор; 12 и 14— стеклянные тройники; /3 — холодильник; 15—стеклянные краны; 16—промывные сосуды; /7 — барботеры; 18—склянки Тищенко, заполненные раствором сульфата меди; 20—воронка; 21 — кварцевая колба; 22—электроплитка; 23— кварцевая трубка, присоединенная к пришлифованной пробке; 24— пробирка для поглощения сероводорода; ,25—стеклянный барботер со шлифом; 26—термометр; 27 — стакан с охлаждающей смесью; 28—шлифы; 29—платиновые проволочки.
помещают в сухую кварцевую колбу 21 и закрывают пришлифованной пробкой. Эта колба соединена с сухим барботером 25, опущенным в пробирку 24, в которую наливают 10 мл раствора ацетата цинка и 1 мл 5%-ного раствора гидроокиси натрия. Пробирку 24 помещают в стакан 27 емкостью 1 л с охлаждающей смесью (1—5°C). Для приготовления смеси в стакан наливают 400 мл воды, прибавляют лед и хлорид натрия.
Собрав установку, краном 8 соединяют систему с атмосферой и пропускают ток азота со скоростью 1 л/мин в течение 5 мин. Затем поворотом крана соединяют кварцевую трубку с остальной частью установки. Включают печь 3, доводят температуру до 600 °C и поддерживают ее в интервале 600—650 °C. При пропуска-’ нии азота в кварцевых трубках 6 (длина 1 м и диаметр 25 мм), заполненных стружкой металлической меди, происходит поглощение кислорода. Концы трубок выступают из печи на 200 мм. Ток азота пропускают со скоростью 0,3 л/мин в течение 20 мин. Не прекращая тока азота, заполняют промывные сосуды 16 раствором хлорида хрома(II).
Для приготовления раствора хлорида хрома (II) непосредственно перед определением восстанавливают раствор хлорида хрома (III) в редукторе 11. Редуктор представляет собой толстостенный стеклянный сосуд (общая высота 310 мм, высота широкой части 240 мм, диаметр 75 мм), нижний патрубок которого присоединен к тройнику 12. Через верхний патрубок сосуд приблизительно на половину заполняют гранулированным цинком и наливают 150—200 мл раствора хлорида хрома(III). Раствор выдерживают в редукторе 5—10 мин до перехода зеленого цвета раствора в синий, что указывает на восстановление хрома(III). Краном 19 соединяют систему с атмосферой. Из воронки 20 наливают в кварцевую колбу 21 30 мл ортофосфорной кислоты. Кран воронки закрывают и краном 19 соединяют кварцевую колбу с системой очистки азота. Включают электроплитку 22 и замедляют ток азота до 1—2 пузырьков в 1 с.
Содержимое колбы нагревают до полного растворения навески и далее до появления слабых паров фосфорной кислоты (обычно нагревание колбы до этого момента занимает около 40 мин). После разложения пробы ток азота увеличивают до 3—5 пузырьков в 1 с и пропускают его еще в течение 5 мин. Барботер 25 отъединяют от установки и ополаскивают над пробиркой 24 водой, дают осадку осесть, раствор декантируют и определяют серу, как указано в разделе VI. 2.1.
V I. 2.5. Определение серы в сульфидных минералах [20, 21]
Навеску образца разлагают хлористоводородной кислотой в атмосфере азота, сероводород отгоняют в раствор ацетата цинка и затем определяют сульфидную серу в виде метиленового голубого.
Реактивы
Олово, в виде фольги, свободное от серы и свинца.
Другие реактивы — указаны в разделе VI. 2.4.
Ход анализа. Навеску образца (измельченного до 100 меш) 0,1 — 1 мг взвешивают на кусочке 1 см2 оловянной фольги, заворачивают в фольгу и вносят в реакционную колбу 21 (см. рис. 43), в которую предварительно наливают 15 мл воды. Далее ведут определение, как указано в разделе VI. 1.4, но разложение пробы проводят не ортофосфорной, а концентрированной хлористоводородной кислотой (15 мл), прибавляя ее по каплям.
V I. 3. ОПРЕДЕЛЕНИЕ СВОБОДНОЙ СЕРЫ
Раствор серы в ацетоне при встряхивании с раствором щелочи образует окрашенное соединение [22]. Окраска раствора зависит от концентрации серы: при содержании серы больше 0,01 % раствор окрашивается в зеленый цвет, а при меньших содержаниях — в синий. Окраска растворов ослабевает со временем. Чувствительность реакции составляет 0,0002% серы. Однако эта реакция используется для качественного или полуколичественного определения.
Прямое спектрофотометрическое определение свободной серы основано на измерении оптической плотности раствора серы в н-гексане при 276 нм [23] или в хлороформе при 290 нм [24]. Однако, по данным [25], лучшие результаты получены при экстракции свободной серы трихлорэтиленом и измерении оптической плотности раствора при 264, 310 и 330 нм. Свободную серу определяют также с помощью ацетата таллия(I) [26], металлической ртути [27] или по реакции образования роданида при взаимодействии с цианидом [3] и последующем определении роданида с помощью железа (III). Широкое распространение получили косвенные методы, которые основаны на окислении серы до сернокислого ангидрида или восстановлении до сероводорода с последующим фотометрическим определением.
V I. 3.1. Определение свободной серы в воздухе с применением ацетата таллия (I) [26]
Воздух просасывают через фильтровальную бумагу, предварительно смоченную 0,5%-ным раствором ацетата таллия(1) и высушенную. Содержание серы находят по интенсивности окраски желтого пятна на фильтровальной бумаге.
Мешающие вещества. Определению мешают темные масла и пыль. Масло удаляют промывкой пятен бензолом или спиртом, а на присутствие пыли вводят поправку, пропуская такое же количество воздуха через фильтр, не пропитанный раствором ацетата таллия (I), 196
Мешает определению селен.
Измерение интенсивности окраски. Стандартную шкалу готовят, исходя из стандартного раствора серы в пиридине.
Чувствительность метода — 5-10—6 % в 0,028 м3 воздуха.
Реактивы
Реактивная бумага. Кружки фильтровальной бумаги диаметром 7 см смачивают 0,5%-ным водным раствором ацетата таллия(I). Бумагу подвешивают на горизонтальных стеклянных палочках в сушильном шкафу и высушивают при 80—105 °C. Хранят в закрытых стеклянных банках.
Чувствительность бумаги при хранении понижается, но ею можно пользоваться в течение нескольких недель.
Стандартный раствор серы. Берут капельницу и определяют точное количество капель в 1 мл пиридина. Для работы пригодны капельницы, с помощью которых можно получить более 40 капель в 1 мл. Готовят растворы серы в пиридине с содержанием от 1,0 до 200 мг/мл так, чтобы содержание в 1 капле в последующем растворе увеличивалось в 1,5 раза.
Стандартная шкала. На фильтры наносят по одной капле растворов серы в пределах ожидаемых концентраций. Пятнам дают просохнуть, обрызгивают пиридином и высушивают почти досуха. Затем обрабатывают вместе с пробой анализируемого раствора.
К вакуум-насосу
Рис. 44. Прибор для определения серы в газах.
Ход анализа. В приборе (рис. 44) зажимают листок бумаги, пропитанной раствором ацетата таллия(I), с таким расчетом, чтобы обнаженной оставалась площадь, равная площади, смоченной одной каплей раствора из выбранной капельницы. Через фильтровальную бумагу просасывают вакуум-насосом воздух, измеряя объем его с помощью счетчика. Затем листок бумаги извлекают и обрызгивают пиридином из специального распылителя, в котором нет резиновых частей, соприкасающихся с пиридином. Бумаге дают просохнуть и вместе с эталонами стандартной шкалы помещают в банку с сероводородом приблизительно на 30 с, затем удаляют сульфид таллия(I) в 0,5 н. азотной кислоте. Отмывают кислоту, перенося бумагу, подвешенную на стеклянной палочке, в сосуд с дистиллированной водой. Вынимают бумагу из сосуда и помещают на белую фарфоровую поверхность для визуального сравнения пятен полисульфида.
V I. 3.2. Определение свободной серы в углеводородах [1,3]
Раствор элементной серы в ацетоне при взаимодействии с цианидом образует роданид:
S + KCN —► KNCS
Образовавшийся продукт реакции определяют в виде роданидного комплекса железа(III).
Мешающие вещества. Меркаптаны в щелочных растворах взаимодействуют с элементной серой и мешают определению. Сероводород, сульфиды и меркаптаны удаляют встряхиванием пробы с раствором нитрата серебра или хлорида ртути(II).
Измерение оптической плотности. Оптическую плотность измеряют при 450 нм. Колориметрический метод шкалы неприменим, так как роданид железа(III) быстро разрушается вследствие окислительно-восстановительного процесса между роданидом и железом (III), особенно под влиянием прямых солнечных лучей.
Чувствительность метода — 1 мкг серы в 1 мл.
Реактивы
Ацетон. К 50 мл воды добавляют до 1 л технического ацетона и перемешивают.
Цианид натрия. Растворяют 0,1 г NaCN в 100 мл водного ацетона. Дают постоять некоторое время для осветления.
Хлорид железа(Ш). Растворяют 0,4 г РеС13-6Н2О в 100 мл водного ацетона. Оставляют на 24 ч для выделения осадка, затем декантируют прозрачный раствор в сухую склянку. Раствор можно хранить в течение нескольких недель.
Хлорид ртути(П). Около 20 г хлорида ртути(П) и 20 г КС1 растворяют в 1 л дистиллированной воды.
Стандартный раствор серы. Растворяют 50 мг порошка черенковой серы точно в 1 л петролейного эфира (т. кип. 77—ПО °C, пл. 0,73 при 25 °C). Полученный раствор содержит 50 мкг/мл серы.
Ход анализа. Помещают 5 мл анализируемого бензина, содержащего 5—50 мкг/мл серы (если необходимо, разбавляют петро-лейным эфиром), в мерную колбу емкостью 25 мл. Добавляют 15 мл раствора цианида натрия, перемешивают и оставляют на 2 мин. Разбавляют до 25 мл водным ацетоном и перемешивают. К аликвотной части 5 мл добавляют точно 5 мл раствора хлорида железа(III). В течение 10 мин измеряют оптическую плотность в кювете с толщиной слоя 1 см по отношению к нулевому раствору, приготовленному смешением 5 мл водного ацетона с 5 мл раствора FeCU. Если анализируемый бензин имеет собственную окраску, то вносят поправку. Для получения этой поправки проводят контрольный опыт, обрабатывая аликвотную часть раствора пробы 5 мл теми же реагентами, но без цианида.
Содержание серы находят по калибровочному графику, который строят, пользуясь стандартным раствором серы.
Если анализируемые углеводороды содержат сульфиды, дисульфиды или меркаптаны, то предварительно их удаляют. Дл<г этого к 20 мл анализируемой пробы прибавляют 50 мл раствора хлорида ртути (II) и тщательно встряхивают. Если образуется небольшой и быстро оседающий осадок, то для анализа берут пробы прозрачного раствора по 5 мл. В случае образования большого осадка его отфильтровывают и берут для анализа пробы фильтрата.
VI. 3.3. Восстановление свободной серы до сероводорода
Для восстановления серы до сероводорода чаще применяется металлический цинк [23]. Однако более равномерное протекание процесса наблюдается при использовании в качестве восстановителя металлического хрома. Восстановление проводят в хлористоводородной среде в атмосфере двуокиси углерода [28].
При определении свободной серы навеску пробы обрабатывают ацетоном и экстрагируют серу, которую затем восстанавливают металлическим хромом до сероводорода, а последний определяют в виде метиленовой сини.
Реактивы
Ацетон, очищенный и высушенный.
Пиридин, очищенный.
Хром в виде чешуек или зерен.
Другие реактивы — указаны в разделе VI. 2.1.
Ход анализа. Навеску анализируемого образца помещают в бумажную гильзу и вводят в патрон экстракционного аппарата с обратным холодильником.
Если анализируемое вещество кроме свободной серы содержит сульфидную серу, то ее предварительно удаляют обработкой разбавленной хлористоводородной кислотой в токе двуокиси углерода.
Пробу в экстракторе заливают ацетоном, наливают его в колбу прибора и экстрагируют свободную серу на слабо кипящей водяной бане не менее 16 ч. В случае необходимости перерыва экстракции надо, чтобы бумажная гильза в патроне экстрактора находилась целиком в ацетоне. По окончании экстракции ацетоновый раствор переводят в мерную колбу емкостью 50 мл и разбавляют ацетоном до метки. Отбирают пипеткой аликвотную часть раствора, содержащую до 2 мг серы, в реакционный сосуд прибора (рис. 45), не взмучивая возможного небольшого осадка. В качестве реакционного сосуда применяют колбу емкостью 150 мл, диаметр горла 22 мм, высота 230 мм или пробирку диаметром 24 мм, высотой 235 мм.
Реакционный сосуд 1 устанавливают на водяную баню с температурой 55—58 °C и упаривают раствор досуха, отсасывая пары ацетона водоструйным насосом. К сухому остатку прибавляют 2— 3 мл пиридина и перемешивают до растворения. Затем прибавляют 5 мл этанола и содержимое перемешивают. Вводят 0,3 г металлического хрома и вновь присоединяют сосуд к прибору.
Для вытеснения воздуха пропускают через систему СОг в течение 3—5 мин. Затем, прекратив ток СОг, открывают кран капельной воронки и прибавляют в реакционный сосуд 15 мл хлористоводородной кислоты (пл. 1,19). Закрывают кран воронки и пропускают через систему СОг со скоростью 60—80 пузырьков в 1 мин в течение 10—15 мин. Затем нагревают содержимое реакционного
сосуда до кипения и слабо кипятят в течение 35—40 мин. После этого нагревают до кипения содержимое промывной склянки 5 и пропускают СОг еще 5 мин. Окончание восстановления контролируют с помощью свинцовой бумаги, отъединив поглотители. Для поглощения сероводорода в первый поглотительный сосуд наливают 25—30 мл раствора ацетата кадмия и разбавляют водой до 80—90 мл; во второй поглотительный сосуд вводят 15 мл того же раствора ацетата кадмия, разбавляют водой до 50—60 мл и добавляют 2 мл уксусной кислоты. В промывную склянку 5 помещают 70 мл дистиллированной воды, предварительно прокипяченной для удаления кислорода и охлажденной в токе СОг.
Рис. 45. Прибор для восстановления серы металлическим хромом:
/—реакционный сосуд (колба или пробирка); 2 — шариковый обратный холодильник; 3 — газопроводящая трубка; 4 — капельная воронка с краном; 5—.промывная склянка с дистиллированной водой для поглощения хлористого водорода; 6, 7—поглотительные сосуды, заполненные раствором ацетата кадмия.
По окончании восстановления поглотительные сосуды отъединяют, сливают оба поглотительные растворы в мерную колбу емкостью 200 мл, разбавляют водой до метки, перемешивают, отбирают аликвотную часть и определяют сульфидную серу, как указано в разделе VI. 2.1.
VI. 4. ОПРЕДЕЛЕНИЕ СЕРНИСТОЙ КИСЛОТЫ
Для фотометрического определения сернистой кислоты наибольшее распространение получил метод, основанный на реакции между сернистой кислотой, фуксином или n-розанилином и формальдегидом. Другие методы основаны на реакциях окисления сернистой кислоты до серной с последующим нефелометрическим определением либо на восстановлении сернистой кислоты до сероводорода и определением его в виде метиленового голубого.
Отделение сернистого ангидрида проводят отгонкой. Для его поглощения предлагались разные поглотители. Однако раствор щелочи, как и различные смеси раствора щелочи с глицерином или спиртом, мало пригодны, так как сульфид, который при этом образуется в щелочной среде, легко окисляется кислородом воздуха и другими окислителями, что приводит к заниженным результа-200
там. Кроме того, щелочной раствор может поглощать сероводород и меркаптаны, которые в дальнейшем мешают определению сульфита. Необходимо отметить, что из щелочных растворов для поглощения сернистого ангидрида лучшим является смесь 10%-ного раствора щелочи в 5%-ном глицерине [29, 30]. Однако и этот поглотитель не лишен [31] указанных выше недостатков (рис. 46). Лучшим раствором для поглощения сернистого ангидрида является раствор тетрахлоромеркуриата(П) натрия (Na2HgC14). При поглощении сернистого ангидрида происходит образование устойчивого сернистокислого комплекса ртути:
[HgCl4]2- + 2SO2 + 2Н2О —► [Hg(SO3)2]2“ + 4СГ + 4Н+
Сероводород и меркаптаны, которые могут присутствовать в
газах, также поглощаются этим раствором растворимых осадков. Эти осадки легко могут быть отделены от раствора декантацией или фильтрованием. Сульфит в этих условиях очень медленно окисляется кислородом воздуха и другими окис-
Рис. 46. Устойчивость сернистого ангидрида к действию кислорода при поглощении тетрахлоро-меркуриатом (II) натрия (/) и смесью растворов 0,1 М щелочи и 5%-ного глицерина (2).
с образованием мало-
лителями. Сульфит, связанный в комплекс, также как и в свободном состоянии взаимодействует с фуксином (или п-розанилином) и формальдегидом с образованием красно-фиолетового продукта реакции, содержание которого и определяют.
При необходимости проведения анализа при низких температурах в раствор вводят дигликоль или глицерин [32], которые снижают температуру замерзания до —20°C.
VI. 4.1. Определение сульфита в отсутствие мешающих веществ
Сульфит-ионы связывают в ртутный комплекс, который взаимодействует с фуксином (/) или парафуксином (//).
H2N+ СГ H2N+C1“
/ II
и формальдегидом с образованием окрашенного раствора.
Мешающие вещества. Определению мешает двуокись азота, а также сероводород, меркаптаны и другие сильные восстановители. Сероводород и меркаптаны при взаимодействии с тетрахлоромер-куриатом(П) образуют малорастворимые соединения, которые отделяют декантацией или фильтрованием.
Измерение оптической плотности. Реакция образования окрашенного продукта происходит в течение 20—30 мин. Измерение оптической плотности проводят при 560 нм.
Чувствительность метода 1 мкг SO2 в 12 мл конечного раствора.
Реактивы
Тетрахлоромеркуриат(П) натрия, 0,1 М раствор. Навеску 27,2 г HgCl2 и 11,7г хлорида натрия растворяют в воде, переносят в мерную колбу емкостью 1 л, разбавляют дистиллированной водой до метки и перемешивают.
Солянокислый п-розанилин. Навеску 42,5 мг солянокислого п-розанилина растворяют в 6 мл концентрированной хлористоводородной кислоты и разбавляют дистиллированной водой до 100 мл.
Формальдегид, 0,2%-ный раствор. Берут 5 мл 40%-ного формальдегида и дистиллированной воде, i разбавляют до 1 л.
Стандартный раствор бисульфита натрия. Готовят раствор бисульфита натрия, титр устанавливают иодометрически. Из полученного раствора готовят стандартный раствор в 0,1 М растворе тетрахлоромеркуриата(II) натрия с содержанием 10 мкг SO2 в 1 мл. Раствор устойчив в течение нескольких дней.
Ход анализа. К испытуемому раствору, содержащему от 1 до 25 мкг сернистого ангидрида, прибавляют хлористоводородную кислоту и отгоняют сернистый ангидрид в цилиндр, содержащий 10 мл раствора тетрахлоромеркуриата(II) натрия. Затем к полученному раствору прибавляют 1 мл n-розанилина и 1 мл формальдегида. Содержимое цилиндра перемешивают и оставляют. Спустя 30 мин измеряют оптическую плотность раствора при 560 нм по отношению к холостой пробе, которую готовят аналогично без сернистого ангидрида. По значению оптической плотности находят содержание сернистого ангидрида по калибровочному графику.
V I. 4.2. Определение двуокиси серы в воздухе [33]
Определенный объем анализируемого газа пропускают через 10 мл раствора тетрахлоромеркуриата(II) натрия и определяют двуокись серы, как указано в разделе VI. 4.1.
V I. 4.3. Определение серы в металлах [34]
Навеску металла сжигают в токе кислорода и отходящие газы пропускают через раствор тетрахлоромеркуриата(II) натрия. В поглотительном растворе определяют сернистый ангидрид, как указано в разделе VI. 4.1.
V I. 4.4. Определение двуокиси серы
в пиве и солоде [35]
Пробу пива или солода подкисляют хлористоводородной кислотой, отгоняют сернистый ангидрид, который поглощают раствором тетрахлоромеркуриата(II) натрия и затем определяют, как указано в разделе VI. 4.1.
Ход анализа. Пробу пива 300 г (или 50 г солода, растворенного в ~ 300 мл воды) продувают в течение 5 мин азотом (для удаления кислорода), затем вводят 20 мл концентрированной хлористоводородной кислоты, нагревают в течение 45 мин, пропуская ток азота со скоростью 75 мл/мин. Выделяющийся сернистый ангидрид поглощают раствором тетрахлоромеркуриата(II) натрия и затем ведут определение, как указано в разделе VI. 4.1.
V I. 4.5. Определение серы в стибните [36]
Для определения следов свободной серы в стибните ее отгоняют в вакууме, сжигают и образовавшийся сернистый ангидрид определяют, как указано в разделе VI. 4.1.
V I. 5. ОПРЕДЕЛЕНИЕ РОДАНИДА
Для определения роданида наиболее распространенной реакцией является взаимодействие с железом(III). Однако этот метод малочувствителен, более чувствительными являются методы с применением метиленового голубого [37], комплекса ртути с метилтимоловым синим [38] и фенантролината железа(II) [39].
Из косвенных методов определения роданида можно указать метод, основанный на образовании роданидного комплекса меди, экстракции его хлороформом, превращении в медь-роданид-пири-динатный комплекс с последующим измерением оптической плотности при 407 нм [40]. Кроме того, предложен метод, основанный на обесцвечивании дитиофлуоресцеина бромистым цианом [BrCN], который образуется при действии на роданид бромной воды [41].
V I. 5.1. Определение роданида с применением метиленового голубого
Метиленовый голубой с роданидом образует соединение с соотношением компонентов (1 : 1). Это соединение из кислого раствора экстрагируется дихлорэтаном и окрашивает раствор в интенсивный голубой цвет.
Мешающие вещества. Определению мешает присутствие более чем 100-кратных количеств фторида, йодата, цианида и фосфата, более 40-кратных количеств хлоридов, 4-кратных количеств бромидов, хлоратов, броматов, нитратов, перйодатов и нитритов, а также равные количества иодида и перхлората.
Измерение оптической плотности. Оптическую плотность экстракта измеряют при 675 нм.
Чувствительность метода — 0,1 мкг NCS- в 5 мл экстракта.
Реактивы
Стандартный раствор роданида. Готовят 10~3 М раствор роданида и устанавливают концентрацию объемным методом. Перед применением готовят путем разбавления раствор, содержащий 10 мкг роданида в 1 мл.
Хлороформ, перегнанный.
Сульфат натрия, безводная сухая соль Na2SO4.
Метиленовый голубой, 0,5%-ный раствор. Навеску 0,05 г метиленового голубого растворяют в воде и разбавляют до 100 мл водой.
Ход анализа. Исследуемый раствор до 10 мл, содержащий 0,1—5,0 мкг роданида, помещают в делительную воронку емкостью 50 мл, разбавляют, если нужно, до 10 мл дистиллированной водой, прибавляют 0,5 мл H2SO4, 3 мл раствора метиленового голубого, 5 мл хлороформа и перемешивают на механическом встряхива-теле 40 с. Слой хлороформа отделяют в сухую пробирку, в которую предварительно помещают 0,5 г безводного сульфата натрия. Содержимое пробирки перемешивают, наливают раствор в кювету с толщиной слоя 1 см и измеряют оптическую плотность при 675 нм.
По значению оптической плотности находят содержание роданида по калибровочному графику.
V I. 6. ОПРЕДЕЛЕНИЕ СУЛЬФАТНОЙ СЕРЫ
Сульфат-ион не образует окрашенных соединений с каким-либо реактивом. Поэтому все фотометрические методы определения сульфатов основаны на реакциях разрушения окрашенных соединений. В основе лучших методов определения сульфатов лежит реакция разрушения окрашенного соединения комплекса бария с нитхромазо [42] и ортаниловым Б [43, 44]. Нитхромазо был синтезирован и применен в качестве индикатора на ионы бария при объемном определении сульфатов в присутствии фосфатов и арсенатов [45] и фосфор- и мышьяксодержащих органических соединений [46]. Этот метод применен для определения серы в биологических материалах [47].
Широкое распространение получили фотометрические методы, основанные на реакциях разрушения суспензий хлоранилата и хромата бария при взаимодействии этих соединений с сульфат-ионами. При разрушении хлоранилата бария в раствор переходят анионы хлораниловой кислоты, интенсивность окраски которых пропорциональна содержанию сульфатов [48—50]. Этот метод применен для определения сульфатов в воде [51], почвенных вытяжках [52], нефти [53] и каменноугольной золе [54]. Реакция между хроматом бария и сульфат-ионами используется при определении сульфатной серы. Определение ведут по интенсивности окраски хромата, бихромата или применяют любой другой фото-204
метрический метод определения хромата, выделяющегося при реакции между хроматом бария и сульфат-ионами. Этот метод применен для анализа многих материалов, в том числе для определения сульфатов в почвах [55].
Реакция осаждения сульфатов с помощью бензидина также применена для фотометрического определения сульфат-ионов. При этом осаждают и отделяют сульфат бензидина, затем после отделения и растворения осадка в хлористоводородной кислоте бензидин диазотируют и проводят реакцию сочетания с солянокислым N-(l-нафтил) -этилендиамином. Продукт реакции окрашен в пурпурный цвет, интенсивность которого пропорциональна содержанию серы [56].
Описаны методы определения серы, основанные на разрушении лаков тория [57], циркония, тория и церия [58, 59], а также на разрушении сульфат-ионами роданида железа [60], родизоната бария [61], комплексов тория с ксиленоловым оранжевым [62]. Рекомендуются также нефелометрические или турбидиметрические методы определения сульфатов в различных вариантах [2]. Для определения малых количеств сульфатной серы ее восстанавливают до сероводорода с помощью хлорида олова (II) [63], смесью иодида и гипофосфита [64, 65] или другими восстановителями с последующим определением сульфидной серы в виде метиленового голубого.
V I. 6.1. Определение сульфатной серы с применением нитхромазо* [42]
Метод основан на разрушении сульфат-ионами комплекса бария с нитхромазо 2,7-бис[(4-нитро-2-сульфофенил)-азо]-1,8-диоксинафт ал ин-3,6-дисульфокислота
zso3H ОН ОН HOjS
O,N——N=N——N=N——NO,
в слабокислой среде (pH = 2). При разрушении комплекса цвет раствора изменяется от голубого до фиолетового. Реакция проходит во времени и для полного выпадения сульфата бария требуется 6 ч. Однако реакция проходит на 98% в течение первых 1,5 ч. Этого времени достаточно для практически полного прохождения процесса, так как в дальнейшем увеличение оптической плотности соизмеримо с ошибкой ее измерения. С целью уменьшения растворимости определение ведут в 50—70%-ном водно-ацетоновом или водно-спиртовом растворе. Соотношение ионов бария и нитхромазо должно быть 1:1.
* Реактив выпускают в виде кислоты и двукальциевой соли. Лучше применять кислоту.
Мешающие вещества. Определению мешают ионы, которые могут связывать нитхромазо, ионы сульфата или ионы бария. Таким образом, определению сульфатов мешают большие количества арсената и фосфата, ионы свинца, стронция, кальция, а также катионы многих других металлов, взаимодействующие с нитхромазо, "
время
в том числе необходимо
титан, цирконий, медь, никель и др. В то же отметить, что этот метод имеет ряд преимуществ по избирательности и чувствительности перед другими методами определения сульфатов.
Измерение оптической плотности. Спектры поглощения нитхромазо и его бариевого комплекса при pH = 2 приведены на рис. 47. Измерение оптической плотности необходимо проводить при 644 нм.
Чувствительность метода — 0,5 мкг SO4” в 5 мл исследуемого раствора. Чувствительность метода можно несколько повысить, прибавляя меньшее количество комплекса бария с нитхромазо.
Рис. 47. Спектры поглощения комплекса бария с нитхромазо (/) и нит-хромазо (2) при pH = 2.
Реактивы
Дистиллированная вода. Бидистиллят пропускают через смешанный фильтр, приготовленный из катионита КУ-2 и анионита АВ-17 или отдельно через катионит КУ-2, а затем через анионит АВ-17. Эту воду используют при проведении определения сульфатов и для приготовления растворов реактивов. сульфата. Готовят 0,01 н. раствор серной кислоты.
Стандартный раствор
Отбирают 4,16 мл этого раствора и разбавляют до 1 л. Этот раствор содержит 2 мкг 864“ в 1 мл.
Ацетон, очищенный.
Раствор комплекса бария с нитхромазо. Готовят отдельно 5-Ю-3 М растворы нитхромазо и хлорида бария. Для приготовления раствора нитхромазо берут 0,3893 г реактива (кислоты), растворяют в воде, переносят в мерную колбу емкостью 100 мл, разбавляют водой до Метки и перемешивают. Отдельно готовят 5-10-3 М раствор хлорида бария, для этого берут 0,1222 г ВаСЬ^НаО, растворяют в воде, переносят в мерную колбу емкостью 100 мл, разбавляют до метки и перемешивают. Перед применением отбирают в мерную колбу емкостью 100 мл 2,5 мл раствора хлорида бария, 2,5 мл раствора нитхромазо, 5 мл 0,1 н. хлористоводородной кислоты, 40 мл воды, 50 мл ацетона и перемешивают.
Ход анализа. Исследуемый раствор до 5 мл, содержащий 0,5— 6 мкг сульфат-ионов, помещают в колбу емкостью 50 мл, если нужно, добавляют недостающее до 5 мл количество воды, прибавляют 0,75 мл 0,1 н. хлористоводородной кислоты и 9,25 мл ацетона. Содержимое колбы перемешивают, вводят 2 мл раствора комплекса бария с нитхромазо, перемешивают, накрывают часовым стеклом и оставляют на 1,5 ч. Одновременно готовят холостую пробу со всеми применяемыми реактивами. Спустя 1,5 ч измеряют оптическую плотность исследуемого раствора по отношению к хо
лостой пробе в кювете с толщиной слоя 1,0 см, при 644 нм. По значению оптической плотности находят содержание сульфата, пользуясь калибровочным графиком.
VI. 6.2. Определение сульфатной серы с применением хлоранилата бария [54]
Суспензия хлоранилата бария (бариевая соль 2,5-дихлор-3,6-диокси-п-бензохинона) взаимодействует с сульфат-ионами с выделением эквивалентного количества хлораниловой кислоты:
Рис. 48. Спектры поглощения хлораниловой кислоты в 80%-ном растворе изопропилового спирта при различных значениях pH.
концентрацию которой определяют по оптической плотности при 330 или 530 нм. Для повышения чувствительности метода реакцию проводят в 80%-ном растворе изопропилового спирта.
Концентрация образующейся хлораниловой кислоты соответствует содержанию сульфатов. .
Мешающие вещества. Определению мешают фториды, фосфаты и особенно кремнефториды, которые связывают барий и приводят к получению завышенных результатов. Однако если проводить реакцию при pH = 2, вредное влияние указанных ионов значительно уменьшается. Определению мешают также ионы кальция, свинца, меди, цинка, алюминия, железа и некоторые другие катионы, которые должны быть отделены. Отделение мешающих катионов обычно проводят с помощью катионитов в Н-форме.
Измерение оптической плотности. Спектры поглощения хлораниловой кислоты в 80%-ном растворе изопропилового спирта при разных значениях pH представлены на рис. 48. Хлораниловая кислота при разных значениях pH имеет три максимума поглощения. Изобестическая точка находится при 310 нм. Причем молярные коэффициенты поглощения равны 1,53• 104, 2,59-104 и 0,1-104 при 310, 330 и 530 нм соответственно. Измерение оптической плотности при 330 нм приводит к увеличению чувствительности метода в 25 раз, но в этом случае обязательно применение буферного раствора с pH = 5,2 или выше для водных растворов, pH = 6,5 или выше для растворов в 80%-ном изопропиловом спирте. Измерение
при 310 нм можно проводить при любом значении pH, так как эта длина волны отвечает изобестической точке перехода молекулярной формы кислоты в ионную. Если вести измерение при 530 нм, то для водных растворов необходимо поддерживать pH = 2, а для растворов в 80%-ном изопропиловом спирте — pH = 3—4. Оптическую плотность измеряют на спектрофотометре при 330 или 310 нм, но можно и на любом другом приборе при 530 нм. В этом случае чувствительность метода уменьшается.
Чувствительность метода — 0,1 мкг SO4" в 10 мл 80%-ного изопропилового спирта в случае измерения оптической плотности при 330 нм.
Реактивы
Хлоранилат бария. Смешивают один объем 0,1%-ного водного раствора хлораниловой кислоты и один объем 5%-ного водного раствора хлорида бария и смесь оставляют при комнатной температуре. После выделения осадка декантируют раствор через фильтр и промывают водой до исчезновения хлорид-ионов. Воду удаляют промыванием этанолом и затем один раз диэтиловым эфиром и осадок высушивают при 60 °C в вакуумном шкафу. Встряхивают 25 г хлоранилата бария с 200 мл 80%-ного изопропилового спирта в течение 1 ч. Отфильтровывают с отсасыванием, промывают этанолом и высушивают под вакуумом.
Буферный раствор. Навеску 0,945 г хлоруксусной кислоты или 0,58 мл ледяной уксусной кислоты разбавляют до 50 мл дистиллированной водой и нейтрализуют щелочью до определенного pH раствора. Затем разбавляют до 100 мл водой и контролируют pH с помощью рН-метра.
Стандартный раствор сульфата. Навеску 0,7399 г высушенного при 400 °C сульфата натрия растворяют в воде, переносят в мерную колбу емкостью 500 мл, разбавляют дистиллированной водой до метки и перемешивают. Этот раствор содержит 1,0 мг SO^~ в 1 мл. Перед применением этот раствор разбавляют до необходимой концентрации.
Можно воспользоваться стандартным раствором серной кислоты (см. раздел VI. 6.1).
Таблица 9. Условия построения калибровочных графиков при определении сульфата хлоранилатным методом
Содержание сульфата в 10 мл 80%-ного изопропилового спирта, мкг Сульфат в виде H2SO4 Сульфат в виде Na2SO4
50-100 530 нм; Ь = 1 см 530 нм; b = 1 см Необходимо введение буферного раствора pH = 3—4
5-100 310 нм; b= 1 см 310 нм; b = 1 см 330 нм; b = 1 см
0-10 310 нм; b = 2 см 310 нм; Ь = 2 см 330 нм; b = 2 см
Ход анализа. Исследуемый раствор 2 мл с содержанием от 0,1 до 1000 мкг сульфата помещают в пробирку или маленькую колбу, в которую введено 25 мг хлоранилата бария и 8 мл изопро
лилового спирта. Смесь встряхивают в течение 30 мин, декантируют раствор и центрифугируют в течение 2 мин при 2000 об/мин. Оптическую плотность полученного раствора измеряют при 530 или 310 нм по отношению к воде. Одновременно ведут две холостые пробы, после измерения оптической плотности холостых проб берут средний результат и вводят соответствующую поправку.
Содержание сульфата находят по калибровочному графику, который строят, пользуясь стандартным раствором сульфата. В зависимости от содержания сульфатов строят калибровочные графики в пределах, указанных в табл. 9.
VI. 6.3. Определение сульфатной серы с помощью хромата бария
Исследуемый раствор пересыщают кислым раствором хромата бария. Затем раствор нейтрализуют и избыток хромата бария вместе с сульфатом бария отфильтровывают, а в фильтрате опреде-
ляют хромат, содержание которого эквивалентно содержанию сульфата.
Мешающие вещества. Определению мешают кремнефториды, фосфаты, фториды, карбонат, которые связывают барий, что приводит к завышенным результатам.
Измерение оптической плотности. Спектр поглощения хромата калия приведен на рис. 49. Оптическую плотность раствора лучше измерять с помощью спектрофотометра при 373 нм, но можно применять и любой другой прибор или визуальный метод стандартной шкалы.
Чувствительность метода — 2,5 мкг в 25 мл конечного раствора. Если измере-
Рис. 49. Спектр поглощения хромата калия.
ние оптической плотности проводить в видимой области, то чув-
ствительность метода уменьшается в пять раз.
Реактивы
Хромат бария, 1%-ный раствор в 0,2 и. хлористоводородной кислоте.
Гидроокись натрия, не содержащая карбоната, 5%-ный раствор.
Стандартный раствор сульфата. Навеску 1,8152 г сульфата калия растворяют в воде, разбавляют до 1 л и перемешивают. 1 мл этого раствора содержит 1 мг ионов сульфата
Ход анализа. Исследуемый раствор 15 мл, содержащий до 10 мг сульфата, помещают в мерную колбу емкостью 25 мл и прибавляют 5 мл раствора хромата бария. Раствор оставляют на 30 мин на теплой водяной бане, нейтрализуют раствором щелочи по внешнему индикатору и сверх этого прибавляют несколько капель щелочи. Раствор разбавляют до метки, перемешивают, дают
постоять 15 мин, декантируют в центрифужную пробирку и центрифугируют. Можно раствор декантировать через сухой фильтр. Однако при этом получаются несколько заниженные результаты вследствие абсорбции хромата фильтровальной бумагой. Раствор, полученный тем или другим способом, помещают в кювету и измеряют оптическую плотность при 373 нм. Содержание сульфата находят по предварительно построенному калибровочному графику.
Построение калибровочного графика можно проводить по титрованному раствору хромата в щелочной среде, но его следует проверять по сульфату, проводя через все стадии анализа.
VI. 6.4. Определение сульфатной серы по разрушению ализарината циркония [59]
разованием
Рис. 50. Спектры поглощения ализарината циркония в 50%-ном ацетоне (/) и ализарина в ацетоне (2).
Сульфат-ионы взаимодействуют с ализаринатом циркония с об-сульфатного комплекса циркония и свободного ализарина. Содержание ализарина определяют по уменьшению оптической плотности раствора цирконий-ализаринового лака или экстрагируют выделившийся ализарин и определяют содержание его по интенсивности желтой окраски в ССЦ. Кроме того, ализарин можно реэк-страгировать раствором щелочи и концентрацию его определить по интенсивности окраски свободного ализарина в щелочной среде. В последнем случае выделившийся ализарин экстрагируют эфиром, а затем реэкстрагируют раствором щелочи. Ализаринат циркония медленно взаимодействует с сульфат-ионами, поэтому первым прибавляют раствор
циркония, а затем ализарин. Для получения воспроизводимых ре-
зультатов следует строго придерживаться методики определения. Определение ведут при pH = 3,3 в 50%-ном растворе ацетона. Мешающие вещества. Определению мешают анионы, которые связывают цирконий, фторид, фосфат и др., а также катионы, которые взаимодействуют с ализарином.
Измерение оптической плотности. Спектры поглощения ализаринам циркония в 50%-ном ацетоне и ализарина в ацетоне представлены на рис. 50. Оптическую плотность лучше измерять при 520—530 нм. Оптическую плотность щелочных растворов ализарина измеряют при 496—533 нм. Раствор ализарина в СС14 устойчив в течение 12—15 суток, а щелочной раствор — в течение рабочего дня. Поэтому для измерения интенсивности окраски можно применять метод стандартной шкалы.
Чувствительность метода без экстракции ализарина составляет 0,5 мкг SO?" в пробе, по желтой окраске ализарина в ССЦ составляет 10 мкг БОГ в 5 мл раствора, а по синей окраске ализарина в щелочной среде — 5 мкг SO?- в 5 мл.
Реактивы
Нитрат циркония, 0,1 М раствор в 2 н. азотной кислоте.
Ализарин, 0,024%-ный (0,01 М) раствор ализарина в ацетоне. Этот раствор устойчив в течение 2—3 месяцев.
Буферный раствор. Смешивают 0,2 М растворы хлористоводородной кислоты и гликоля в соотношении (1 : 9), pH такого раствора 3,3.
Ацетон, очищенный.
Четыреххлористый углерод, очищенный.
Стандартный раствор сульфата, 0,001 М сульфата калия.
Диэтиловый эфир, очищенный.
Калибровочный график. Для построения калибровочного графика в ряд пробирок наливают различные объемы от 0,01 до 1 мл раствора сульфата калия, дистиллированной водой доводят объем до 2 мл и приливают по 2 мл раствора нитрата циркония, 2 мл буферного раствора и 4 мл ацетона. Перемешивают, приливают по 2 мл ацетонового раствора ализарина и снова перемешивают. Затем пробирки с растворами помещают на 15 мин в стакан с горячей водой (50—53 °C). После охлаждения измеряют оптическую плотность растворов.
Ход анализа. В пробирки наливают до 2 мл исследуемого раствора, содержащего до 100 мкг БОГ, если нужно доводят дистиллированной водой до 2 мл, приливают 2 мл раствора нитрата циркония, 2 мл буферного раствора и 4 мл ацетона. Жидкость перемешивают и, прилив 2 мл ацетонового раствора ализарина, снова перемешивают, после чего помещают пробирку на 15 мин в стакан с водой при 50—53 °C. После охлаждения жидкость наливают в кювету с толщиной слоя 2 см и измеряют оптическую плотность при 520—530 нм (зеленый светофильтр).
Ход анализа. (Экстракционный вариант.) Растворы, подготовленные, как указано выше, переносят в делительные воронки емкостью 20—50 мл, приливают по 2 мл четыреххлористого углерода и энергично встряхивают 3—5 мин. В неводный слой переходит выделившийся при реакции ализарин, окрашивая слой CCI4 в желтый цвет. После разделения фаз отделяют неводный слой. К растворам ализарина в четыреххлористом углероде приливают ацетон ду 5 мл и измеряют оптическую плотность на фотоэлектроколориметре с синим светофильтром (№ 2) в кювете с толщиной 1 см или измеряют интенсивность окраски визуальным методом стандартных серий.
Для повышения чувствительности ализарин извлекают не четыреххлористым углеродом, а 2—3 мл диэтилового эфира, затем к эфирным растворам ализарина приливают по 5 мл раствор.а щелочи и энергично встряхивают. При этом эфирный слой обесцвечивается, а водный окрашивается в синий цвет. Затем пробирки с растворами помещают на 5—7 мин в стакан с горячей водой
(40—50 °C) для удаления эфира и измеряют оптическую плотность щелочных растворов ализарина с зеленым светофильтром в кювете с толщиной слоя 1 см.
VI. 6.5. Определение сульфатной серы бензидиновым методом [2, 56]
Сульфат осаждают бензидином, осадок растворяют в 0,2 н. хлористоводородной кислоте, бензидин диазотируют и после отделения избытка азотистой кислоты проводят реакцию азосочетания с солянокислым М-(1-нафтил)-этилендиамином:
H2N
H2N
N=N
nh-ch2—ch2—nh2
NH2 + 2HNO2 + НС1 —>
N=N—Cl + 4H2O
N=NC1 +
H2N
NH—CH2—CH2—NH2 + HC1
Образующийся продукт реакции пурпурного цвета.
Мешающие вещества. Анализу мешают вещества, осаждающиеся бензидином, в том числе фосфат-ионы. Поэтому предварительно необходимо отделить фосфат. Хлориды мешают, если их содержание больше 30-кратных количеств по отношению к содержанию сульфатов.
Измерение оптической плотности. Оптическую плотность растворов можно измерять с помощью любого прибора при 520 нм, можно применять и визуальный метод стандартной шкалы.
Чувствительность метода — 15 мкг сульфата в 25 мл конечного раствора.
Реактивы
Нитрит натрия, 0,1%-ный раствор. Готовят ежедневно свежий раствор.
Сульфаминат аммония, 0,5%-ный раствор.
Солянокислый Н-(1-нафтил)-этилендиамин, 0,1%-ный раствор. Хранят в склянке из темного стекла в холодильнике.
Солянокислый бензидин. Растворяют 4,0 г чистого солянокислого бензидина в небольшом объеме дистиллированной воды и разбавляют до 250 мл 0,2 н. раствором хлористоводородной кислоты.
Стандартный раствор сульфата. Навеску 0,5437 г сульфата калия (ч. д. а.) растворяют в 1 л дистиллированной воды. Этот раствор содержит 0,3 мг сульфата в 1 мл. Для получения рабочего раствора его разбавляют в 10 раз.
Стандартный раствор солянокислого бензидина. Навеску 0,4014 г солянокислого бензидина растворяют в небольшом объеме 0,2 н. хлористоводородной 212
кислоты и разбавляют до 100 мл той же кислотой; 1 мл этого раствора эквивалентен 1,5 мг сульфата. Путем разбавления этого раствора в 100 раз 0,2 н. хлористоводородной кислотой готовят рабочий раствор, 1 мл которого соответствует 0,015 мг сульфата.
Смесь ацетона и этанола. Смешивают (1:1) ацетон (ч. д. а.) и 95%-ный этанол.
Ход анализа. В центрифужную пробирку емкостью 15 мл помещают анализируемый раствор, содержащий 0,015—0,15 мг сульфата, добавляют 1 мл ледяной уксусной кислоты и 1 мл раствора солянокислого бензидина. Перемешивают, добавляют 2 мл смеси ацетона и этанола и выдерживают при 0°С в течение 30 мин для полноты осаждения. Центрифугируют при 2500 об/мин 10 мин, полностью удаляют жидкость и осадок дважды промывают смесью ацетона и этанола. Осадок растворяют в 2 мл 0,2 н. хлористоводородной кислоты, раствор охлаждают до 0°С и добавляют 1 мл свежеприготовленного раствора нитрита натрия. Перемешивают и оставляют на 3 мин, затем добавляют 1 мл раствора сульфами-ната аммония. Спустя 2 мин добавляют 1 мл водного раствора солянокислого 1Ч-(1-нафтил)-этилендиамина, оставляют на 20 мин, после чего переносят в мерную колбу емкостью 25 мл, разбавляют до метки водой, перемешивают и измеряют оптическую плотность при 520 нм (зеленый светофильтр).
Содержание сульфата находят по калибровочному графику.
V I. 6.6. Турбидиметрическое определение сульфата
Сульфат-ионы в кислой среде осаждают хлоридом бария и измеряют оптическую плотность суспензии. Для получения устойчивой суспензии пользуются смесью дипропиленгликоль— этанол — вода [66].
Мешающие вещества. Растворы хлорида бария не должны содержать даже следов сульфата, так как в этом случае при хранении раствора хлорида бария в водно-этанольно-дипропиленглико-левой смеси образуются центры кристаллизации сульфата бария, при использовании такого раствора кристаллы сульфата бария образуются очень быстро и большего размера. Катионы первой аналитической группы (NHj, Na+, К+),кальция, железа(III), а также хлориды, нитраты и фосфаты практически не мешают определению.
Измерение оптической плотности. Измерение оптической плотности производят в видимой области спектра.
Чувствительность метода — 0,1 мкг SOlj в 10 мл конечного раствора.
Реактивы
Вода, не содержащая сульфат-ионов. Дистиллированную воду пропускают через слой анионита высотой 25 см или дважды перегоняют.
Смесь этанола и дипропиленгликоля, 40%-ная. Смешивают 450 мл абсолютного спирта и 550 мл дипропиленгликоля, затем 400 мл этанольно-дипропилен-гликолыюй смеси смешивают с 500 мл дистиллированной воды, не содержащей
сульфата, добавляют 150 ммоль азотной кислоты, охлаждают и разбавляют до 1 л дистиллированной водой.
Хлорид бария. Готовят 100 мл 1,34 Л4 раствора хлорида бария в дистиллированной воде. Спустя не менее 3 ч раствор фильтруют через стеклянный фильтр № 4. Берут 6 мл этого раствора и переносят в мерный цилиндр емкостью 250 мл, разбавляют до 120 мл водой, а затем добавляют 80 мл смеси этанола и дипропиленгликоля. Раствор оставляют на 3 ч. Приготовленный раствор пригоден для работы в течение не более 40 ч.
Стандартный раствор сульфата — см. раздел VI. 6.5.
Ход анализа. В пять пробирок помещают 1, 2, 3, 4 и 5 мл стандартного раствора сульфата и добавляют до 5 мл смеси этанола и дипропиленгликоля. Аналогичную серию готовят из анализируемого раствора и раствора смеси этанола и дипропиленгликоля. В каждую из 10 пробирок по возможности быстро добавляют по 5 мл раствора хлорида бария. Содержимое пробирок перемешивают вращением и спустя 15 мин измеряют оптическую плотность.
Полученные точки должны укладываться на две прямые линии. Содержание серы вычисляют, пользуясь пропорцией: содержание серы в анализируемом растворе равно содержанию серы в стандарте, умноженному на отношение оптических плотностей анализируемого и стандартного растворов.
V I. 6.7. Определение сульфатной серы в виде метиленового голубого
Сульфатную серу, а также другие соединения серы, восстанавливают до сульфидной серы, которую определяют в виде метиленового голубого, как описано в разделе VI. 2.
В качестве восстановителя применяют раствор хлорида олова (II) в фосфорной кислоте [67]. При определении серы в сульфатах бария, магния, цинка, натрия [63, 68], а также при анализе сульфидных руд, тиосульфата и других серусодержащих материалов [69] раствор хлорида олова (II) и фосфорной кислоты предварительно нагревают до удаления хлористого водорода. Восстановление этой смесью детально изучено, и усовершенствован способ приготовления реагента для восстановления [70]. Для восстановления серы рекомендовано также применять металлические титан, хром, молибден, ванадий или вольфрам в присутствии фосфорных кислот и их солей [71]. Чаще других металлов рекомендуется применение металлического хрома в присутствии фосфорной кислоты, этот восстановитель применен для определения серы в феррохроме, металлическом хроме [14] и хлориде титана(IV) [72]. Широко распространен метод восстановления серы смесями иодистоводород-ной и фосфорноватистой кислот [73], иодистоводородной кислоты и гипофосфита натрия в присутствии уксусной [64], муравьиной [74] и хлористоводородной [75—77] кислот. Кроме того, рекомендована смесь иодистоводородной и муравьиной кислот и красного фосфора [78], а также смесь сульфата титана(III) и фосфорной кислоты [79].
Метод с применением гипофосфита натрия дает возможность определить практически все виды серы и применен для ее определения в нефтяны'х водах [75], полупроводниковых материалах типа А3В5 [80], в индии, таллии и других металлах [76], и полупроводниковых материалах [77], хлориде титана (IV) [72], селене [81], физиологических веществах [74] и других материалах.
Восстановление серы проводят в токе азота, очищенного от сероводорода и кислорода.
Реактивы
Восстановительная смесь. В коническую колбу емкостью 2 л помещают 240 г гипофосфита натрия (Nab^PC^-lbO), приливают 400 мл хлористоводородной кислоты, разбавленной (1 : 1), и 800 мл иодистоводородной кислоты. Полученный раствор упаривают в токе азота до 750 мл, охлаждают и хранят в склянке из темного стекла.
Поглотительный раствор ацетата цинка, 20%-ный раствор в 2%-ном растворе ацетата натрия.
Другие реактивы — указаны в разделе VI. 2.1.
Ход анализа. Пробу до 5 мл, содержащую до 100 мкг серы, помещают в круглодонную колбу, присоединенную к поглотительному сосуду, в который налит раствор ацетата цинка. В колбу вводят 20 мл восстановительной смеси, закрывают и через систему пропускают ток азота, предварительно очищенного пропусканием через 2%-ный раствор перманганата калия, содержащий HgCU. Содержимое колбы быстро (~ 1 мин) доводят до кипения и кипятят в течение 10 мин. Образующийся сероводород отгоняют через холодильник, промывая перед поглощением слабым раствором хлористоводородной кислоты. Затем сульфидную серу определяют, как указано в разделе VI. 2.1.
V I. 7. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ СЕРЫ
Для фотометрического определения микроколичеств серы применена иод-азидная реакция [82, 83]:
2NaN3 + 12 —► 2NaI + 3N2
Эту реакцию ускоряют растворимые и нерастворимые сульфиды, сероуглерод, а также тиосульфат- и роданид-ионы. Оптическую плотность раствора иода измеряют через определенный промежуток времени при 420 нм, иодкрахмальных растворов — при 680 нм. Метод позволяет определить 10-2 мкг серы в 10 мл конечного раствора. Этот метод применен для определения серы в черных металлах, цинке и кадмии [82], в титане и цирконии [84]. Иод-азидная реакция применена также для определения сероуглерода в растворах [85].
Каталитическое действие сульфидной серы на реакцию восстановления ионов серебра железом(II) применено для определения
малых количеств серы [86]. Чувствительность этого метода — 5-10-4 мкг сульфидной серы в 5 мл.
Предложен фотометрический метод определения сульфид-ионов, основанный на реакции окисления сульфида раствором окислителя, избыток которого определяют по количеству окисленного иода. Последний определяют фотометрически по реакции с красителями трифенилметанового ряда [87, 88]. Реакция восстановления молибдена (VI) до молибдена(V) в присутствии роданида применена для фотометрического определения сульфидной серы [89—91]. С целью стабилизации растворов использован ацетон [92].
При анализе смесей сероводорода и двуокиси серы, а также сероуглерода и сероокиси углерода предложено применять различные поглотители с последующим определением отдельных компонентов [93].
Тиофлуоресцеин обладает интенсивной голубой окраской, но образует бесцветный комплекс с ионами серебра. Комплекс с серебром менее прочен, чем сульфид или цианид серебра, поэтому при действии сероводорода или цианида на тиофлуоресцеиновый комплекс серебра он разрушается и появляется интенсивная голубая окраска свободного тиофлуоресцеина. На основе этой реакции разработан косвенный метод определения сульфидов и цианидов [94—99]. Для определения сероуглерода в воздухе его поглощают этанольным раствором щелочи. Образовавшийся ксантогенат подвергают гидролизу до сероводорода, который определяют в виде метиленового голубого [100].
Определение тетратионата основано на реакции взаимодействия его с цианидом в щелочной среде:
S4Og“ + 3CN” + Н2О —> S2O32" + SO24“ + 2HCN + NCS-
Образующийся роданид определяют спектрофотометрически с ионами железа (III) и вычисляют содержание тетратионата [101].
Для определения тиосульфата его переводят в роданид, который определяют фотометрически с железом (III). Мешающие ионы маскируют HgCl2 [102]. Однако определению тиосульфат-ионов с помощью роданида серебра мешают политионаты и сернистая кислота. Прибавление формальдегида устраняет вредное влияние только небольших количеств сернистой кислоты. Лучшим реактивом на тиосульфат в этом случае оказался n-бензохинон, который взаимодействует с тиосульфатом с образованием гидрохинонмонотиосульфокислоты, восстанавливающей избыток n-бензохинона до бесцветного гидрохинона и окисляющейся при этом до п-хинонмо-нотиосульфокислоты [103].
Для определения серы в металлах [104] и в селене [105, 106] рекомендован метод с применением астразонового розового Fg. Анализируемую пробу сжигают в токе кислорода, если анализи-216
руют селен, двуокись селена конденсируют, а двуокись серы поглощают раствором астразонового розового Fg. Содержание серы находят по ослаблению окраски раствора красителя. Определение свободной серы в сульфидах цинка, кадмия и индия основано на экстракции серы органическими растворителями и последующем измерении оптической плотности в УФ [107].
Фотометрический метод определения двуокиси серы основан на восстановлении диметилглиоксиматного комплекса меди(II).
Максимум поглощения [CuHDm(NH3)2]+ при 310 нм, поскольку двуокись серы восстанавливает медь(II) до меди(1), происходит образование комплекса меди(1), который характеризуется максимумом поглощения при 270 нм. По уменьшению оптической плотности раствора при 340 нм определяют содержание двуокиси серы [108]. Для определения двуокиси серы применена реакция восстановления железа(III) двуокисью серы с последующим определением содержания железа(II) в виде комплекса с а,а'-дипиридилом [109] или 1,10-фенантролином [НО] по поглощению света при 510 нм. Для фотометрического определения двуокиси серы рекомендованы ц-нитроанилин [111], п-аминоазобензол [112], n-розанилин с формальдегидом [113—115], о-ксилол [116], пиридиновый раствор нитропруссида [117, 118], органические дисульфиды [119], а также определение по собственному поглощению в ультрафиолете [120, 121] и др.
По разрушению комплексов тория с производными триокси-флуорона предложен метод флуорометрического определения серы [122]. Метод рекомендован для определения сульфата в двуокиси германия. Чувствительность метода — 0,1 мкг сульфат-ионов в 1 г пробы.
Реакции разрушения сульфат-ионами окрашенного лака тория с амарантом [123], комплекса тория с морином [124], хлоранилата трис-(1,10-фенантролината) двухвалентного железа [125], роданидного комплекса железа(III) [60] положены в основу фотометрических методов определения сульфат-ионов.
Разработан автоматический фотометрический хлоранилатный метод определения сульфатов [126]. Хлоранилатный метод применен для определения сульфатов в каменноугольной золе и подобных материалах [127], в нефти [128] и в почвенных вытяжках [129]. Для определения 1—400 мг/л сульфата применены производные хлораниловой кислоты [130].
Предложен простой и быстрый метод определения сульфата в виде FeSOj. Оптическую плотность растворов измеряют при 225 нм. Этот метод применен для определения сульфата в природных водах [131].
Показано, что свежеприготовленные растворы оксихлорида Циркония быстро взаимодействуют с метилтимоловым синим, а по мере старения оксихлорида циркония скорость реакции падает. Реакция с постоявшим раствором оксихлорида циркония катализируется субстехиометрическими количествами сульфат-ионов. Это
свойство использовано для разработки фотометрического определения сульфат-ионов [132].
Нефелометрический метод предложен для определения сульфата в почвенных вытяжках [133]. Для стабилизации суспензии сульфата бария предложен монолауринат полиоксиэтилсорбита (Твин-20) [134] и камедь [135]. Для нефелометрического определения сульфата предложен также 4-амино-4'-хлордифенил в присутствии пептина и гуммигута, играющих роль стабилизаторов [136].
Реакция между сульфат-ионами и ионами бария проходит во времени. Если к испытуемому раствору прибавлять хлорид бария одной и той же концентрации и с одинаковой скоростью, то скорость образования сульфата бария зависит от концентрации ионов сульфата. Эта зависимость выражается уравнением:
1g / = а + b 1g С где t — время, с; а и b — постоянные; С — концентрация сульфат-ионов.
Измеряя время образования определенного количества сульфата бария, суспензия которого характеризуется определенным значением оптической плотности, можно определить содержание сульфата в анализируемой пробе. Этот метод назван авторами фотохронометрическим [137] и применен для определения сульфатов в бихромате натрия и хромовом ангидриде [138], фосфате хрома [139] и фосфорной кислоте [140].
Предложен метод определения сульфид-, тиосульфат-, сульфит-и сульфат-ионов по собственному поглощению при 230, 220, 240 и 210 нм соответственно [141].
ЛИТЕРАТУРА
1. Johnson С. М., N i s h i t a H., Anal. Chem., 24, 736 (1952).
2. Паттерсон Г. Д. В сб. «Колориметрические (фотометрические) методы определения неметаллов». Пер. с англ. Под ред. А. И. Бусева. М., Издат-инлит, 1963. См. с. 308.
3. В а г 11 е 11 J. К-, Skoog D. A., Anal. Chem., 26, 1008 (1954).
4. Sommer H., Ind. Eng. Chem., Anal. Ed., 12, 368 (1940).
5. С а в в и н С. Б., Акимова Т. Г., Дедкова В. П. Органические реагенты для определения ионов бария и сульфата. М., «Наука», 1971. См. с. 136.
6. Камаева Л. В., Подчайнова В. Н., Студенская Л. С., Тр. ВСес. н.-и. ин-та стандартн. образцов и спектр, эталонов, 4, 46 (1968).
7. Mecklenburg W., Rosenkranzer F., Z. anorg. Chem., 86, 143 (1914).
8. Marczenko Z., Choluj-Lenarczuk L., Chem. anal. (Polska), 10, 729 (1965).
9. Китагава, Сибат а, РЖХим, 1958, 81346.
10. Hofmann К., Hamm R., Z. anal. Chem., 232, 167 (1967).
11. Haun R., Z. ges. Hyg., 14, 93 (1968).
12. За вод нов С. С. В сб.: «Соврем, методы анализа природных вод». М., АН СССР, 1962. См. с. 63.
13. С 1 i n е J. D., Limnol. and Oceanogr., 14, 454 (1969).
14. Яковлев П. Я., Федоров А. А., Буянов Н. В. Анализ материалов металлургического производства. М., «Металлургиздат», 1961. См. с. 35.
15. Т у о и Р„ Н u m b 1 е t L„ Taianta, 3, 232 (1960).
16. Kriege Owen H., W о 1 f e A. L„ Taianta, 5, 673 (1962).
17. Ciuhandu G., Dewaid A., Studii si cercetari chim., 3, 293 (1955).
18. Федоров А. А., Кричевская A. M., Зав. лаб., 36, 1433 (1970).
19. Miznike Atsushi, Kondo Akio, Mikrochim. acta., № 6, 841 (1971).
20. В u d d M. S„ В e w i c k H. A., Anal. Chem., 24, 1536 (1952).
21. van Loon J. C„ Parissis С. M., Kingston P. W., Anal. chim. acta, 40, 334 (1968).
22. Urbanski!., Taianta, 9, 799 (1962).
23. M a u г i с e M. J., Anal. chim. acta, 16, 574 (1957).
24. К о г e n J. G., Appl. Spectrosc., 23, 275 (1969).
25. Л a a a p e в В. И., Ко стр и ко в В. И., ЖАХ, 25, 553 (1970).
26. М a g i 11 Р. L., Ro Is ton M. V., В rem пег R. W., Anal. Chem., 21,
1411 (1949).
27. U h г i g K-, L e v i n H., Anal. Chem., 23, 1334 (1951).
28. Волков И. И., Жабина Н. Н., ЖАХ, 26, 359 (1971).
29. Atkin S., Anal. Chem., 22, 947 (1950).
30. Алексеева М. В., Само род ин а Р. Я., Гигиена и санитария, № 10, 42 (1953).
31. W е s t Р. W., G а е k е G. С., Anal. Chem., 28, 1816 (1956).
32. Huhn J., Wagner J., Fahnert R., Chem. Techn., 19, 564 (1967).
33. H e 1 w i g H. L., G о г d о n Ch. L., Anal. Chem., 30, 1810 (1958).
34. В u r ke K. E„ Da v i s С. M„ Anal. Chem., 34, 1747 (1962).
35. В eetch E. B., Octzel L. I., J. Agric. and Food Chem., 5, 951 (1957).
36. H а д e ж и н а Л. С., Б e с п а л e н к о в а Е. К., Г р и н з а й д Е. Л., ЖАХ.,
23, 787 (1968).
37. Koh Tomozo, Iwasaki Iwaji, Bull. Chem. Soc. Japan, 40, 569 (1967).
38. Номура Тосики, РЖХим, 1968, 9Г113.
39. Yamamoto Yuroku, Tarumoto Tsunehiko, Hanamoto Yasu-ko, Bull. Chem. Soc. Japan, 42, 268 (1969).
40. Danch.ik R. S., Boltz D. F., Anal. Chem., 40, 2215 (1968).
41. W г о n s k i M., Chem. anal. (Polska), 14, 1183 (1969).
42. Б а с a p г и н H. Н. и др., ЖАХ, 23, 732 (1968).
43. А к и м о в а Т. Г., Д е д к о в а В. П., Саввин С. Б. В сб.: «Применение
органических реагентов в химико-аналитическом контроле качества материалов». М., изд. Моск, дома н.-т. пропаганды им. Ф. Э. Дзержинского, № 2, 9 (1967).
44. С а в в и н С. Б., Дедкова В. П., Акимова Т. Г., Тр. комиссии по аналит. хим. АН СССР, 17, 322 (1969).
45. Кузнецов В. И., Басаргин Н. Н., Зав. лаб., 24, 524 (1958).
46. Б а с а р г и н Н. Н., Новикова К. Ф-, ЖАХ, 21, 473 (1966).
47. Покровская Е. И., Терещенко А. П., ЖАХ, 23, 1245 (1968).
48. Bertolacini R. J., Barney J. Е., Anal. Chem., 29, 281 (1957).
49. Carlson R. M., Rosel 1 R. A., Vallejos W., Anal. Chem., 39, 688 (1967).
50. Gales M. E., Kaylor W. H„ L о n g b 6 11 о m J. E., Analyst, 93, 97 (1968).
51. P г о c h a z k о v a H., Z. anal. Chem., 182, 103 (1961).
52. Ma gar W. J., Pollard A. G., Chem. and Ind., № 16, 505 (1961).
53. К 1 i p p R. W„ В a r n e у J. E„ Anal. Chem., 31, 596 (1959).
54. S c h a f e r H. N. S„ Anal. Chem., 39, 1719 (1967).
55. Ц а п M. Л., Ю н i к HI. M., Бюл. наук, жформ. по землеробству, № 5, 22 (1959).
56. Klein В., Ind. Eng. Chem., Anal. Ed., 16, 536 (1944).
57. Кузнецов В. M., ДАН СССР, 77, 61 (1951).
58. Бабко А. К., Маркова Л. В., Тр. комиссии по аналит. хим. АН СССР, 11, 309 (1960).
59. Б а б к о А. К , М а р к о в а Л. В., Зав. лаб., 24, 524 (1958).
60. Бабко А. К., Маркова Л. В., Укр. хим. ж., 25, 505 (1959).
61. Б а б к о А. К., Л и т в и н е н к о В. А., ЖАХ, 18, 237 (1963).
62. Р а 1 a t у V., Taianta, 10, 307 (1963).
63. К i b а Т„ К i s h i J., Bull. Chem. Soc. Japan, 30, 44 (1957).
64. QustafssonL, Taianta, 4, 227 (I960).
65. Q u s t a f s s о n L., Taianta, 4, 236 (1960).
66. Toennies G., В а к а у В., Anal. Chem., 25, 160 (1953).
67. Rancke-MadsenE., Acta chem. scand., 3, 773 (1949).
68. Ki ba T„ Takagi T., Yoshimura Y., Kishi I., Bull. Chem. Soc. Japan, 28, 641 (1955).
69. К i b a T., Akaza Y., Sugishita N., Bull. Chem. Soc. Japan, 30, 972 (1957).
70. Волков И. И., Остроумов Э. А., ЖАХ, 6, 686 (1958).
71. Suzuki Susumu e. a., Bull. Chem. Soc. Japan, 30, 771 (1957).
72. 3 и н ч e н к о В. А., Г e p ц e в a H. M., ЖАХ, 22, 1080 (1967).
73. Luke C„ Anal. Chem., 21, 1369 (1949).
74. Roth H., Mikrochemie, 36—37, № 1, 379 (1951).
75. Nedorost M., Paralova J., Chem. prumysl., 14, 428 (1964).
76. Г о p ю ш и н а В. Г., Бирюкова E. Я. Науч. тр. Гиредмета, M., Метал-лургиздат, т. 3, 1961. См. с. 95.
77. Горюшина В. Г., Бирюкова Е. Я., Зав. лаб., 31, 1303 (1965).
78. Lora nt I. S., Z. physiol. Chem., 185, 245 (1929).
79. Q u a г t e г m a i n P. G., Hill A. G., Analyst, 85, 211 (1960).
80. Adler S. J., P a f f R. J., Compounds semiconduet. Vol. 1, New York, Reinhold Publ. Corp., London, Champan and Holl, Ltd. 1962, p. 123.
81. S j d b о г g B. L., Taianta, 14, 693 (1967).
82. Бабко A. K-, Маркова Л. В., Зав. лаб., 25, 1283 (1959).
83. Б а б к о А. К-, М а р к о в а Л. В., Укр. хим. ж., 27, 796 (1961).
84. М а р к о в а Л. В., Зав. лаб., 27, 379 (1961).
85. Wojciac W., So leek i R., Kurzawa Z., Chem. anal. (Polska), 12, 849 (1967).
86. Бабко А. К., Максименко T. С., ЖАХ, 22, 570 (1967).
87. Б у н и к e н e Л. В. и др., ЖАХ, 23, 1679 (1968).
88. Раманаускас Э. И., Григонене К. М., Науч. тр. высш, учебн. заведений ЛитССР. Хим. и хим. технол., 13, 21 (1971).
89. Koren Н., Gier linger W., Mikrochim. acta, 3, 220 (1953).
90. Pepkowitz L. P., Shirley E. L., Anal. Chem., 23, 1709 (1951).
91. Назаренко В. А., Полуэктов H. С. Краткое руководство по качественному и количественному анализу руд и минералов в полевых условиях. Томск, Изд-во Зап.-Сиб. геол, упр., 1943. См. с. 98.
92. Назаренко В. А., Ш у с т о в а М. Б., ЖАХ, 11, 489 (1956).
93. Ким Юн Ван, Ли Дон Дюн, Тен Дек Бон, Пусок хвахак, 5, 37 (1966); цит. по РЖХим., 1967, 14Г87.
94. W г о n s k i М„ Chem. anal. (Polska), 5, 457 (1960).
95. Oliveira M. J„ Antonio C. L., Rev. quim. ind., 39, 14 (1970).
96. R a h i m S. A., W e s t T. S., Taianta, 17, 851 (1970).
97. Г о p ю ш и н а В. Г., Бирюкова E. Я., Зав. лаб., 35, 1163 (1969).
98. Горюшина В. Г., Бирюкова Е. Я., Науч. тр. н.-и. и проектн. ин-та редкомет. пром-сти, 29, 141 (1970).
99. Humphrey R. Е., Hinze W., Jenkines W. М., Anal. Chem., 43, 140 (1971).
100. Dutkiewicz T., Jaszka B., Rocan. Panstw. zakl. hig., 12, 441 (1961).
101. Nietzel O. A., Desesa M. A., Anal. Chem., 27, 1839 (1955).
102. Straub G., Kiss A. S., Magyar kem. folyoirat, 61, 43 (1955).
103. SchoonN. H„ Acta chem. scand., 13, 525 (1959).
104. Fischer W., Bastius H., Mehl horn R., Neue Hiitte, 8, 35 (1963).
105. Fischer W., Bastius H., Mehlhorn R., Neue Hiitte, 9, 434 (1964).
106. Odzawa Takedir o, J. Chem. Soc. Japan, Pure Chem. Soc., 87, 578 (1966).
107. Туманов A. A„ Шахверди H. M., Тр. no хим. и хим. технол., Горький, вып. I, 134, (1962).
108. Fujita Yoshio, Technol. Repts Osaka Univ., 16, March, 225 (1966)
109. Stephens B. G., Lin ds ton F., Anal. Chem., 36, 1308 (1964),
110. Atta г i A., I gielsk i T. P„ Anal. Chem., 42, 1282 (1970),
111. BethgeP. O., Carlson M., Taianta, 16, 144 (1969),
112. К n i s e I е у Sh. J., Т h г о о р L., Anal. Chem., 38, 1270 (1966).
113 Янагисава Сабур о, Мицудзава Сюмпо, Мори Масаки, РЖХим., 1969, 20Г156.
1(4. Arikawa Y os hi ко, Ozawa Takejiro, Iwasaki Iwaji, Bull. Chem. Soc. Japan, 41, 1454 (1968).
115. Pugh H., Waterman W. R., Anal. chim. acta, 55, 97 (1971).
Ц6. Bh a tty M. K., Townshend A., Anal. chim. acta, 55, 401 (1971).
117. Cavdarova R., Докл. Болг. АН, 23, 1263 (1970).
118. Bourbon P., Malbosc R., Albert M„ Франц, пат., кл. gOln, № 1542703, заявл. 5.09.67, опубл. 9.09.68. РЖХим., 1969, 23Г107П.
119. Humphrey R. Е., Ward М. Н., Hinze W., Anal. Chem., 42, 698 (1970).
120. S с о g g i n s M. W., Anal. Chem., 42, 1091 (1970).
121. Bhatty M. K., Townshend A„ Anal. chim. acta, 55, 263 (1971).
122. Назаренко В. А., Шустова M. Б., Зав. лаб., 24, 1344 (1958).
123. Lambert J. L., Jasuda К., Grotheer М. Р., Anal. Chem., 27, 800 (1955).
124. Nasy Toshiko, Japan Analyst, 18, 1183 (1969); РЖХим., 1970, 10Г132.
125. Yamamotu Yurok u, eningo Kadruo, Sanaka sa kasha, Japan Analyst, 17, 206 (1968); РЖХим, 1968, 24Г118.
126. Gales M. E., Kaylor W. H., Longbottom J. E., Analyst, 93, 97 (1968).
127. Sch a f er H. N. S„ Anal. Chem., 39, 1719 (1967).
128. К1 i p p R. W., В а г n e у J. E., Anal. Chem., 31, 596 (1959).
129. Ma gar W. J., Pollard A. G., Chem. and Ind., № 16, 505 (1961).
130. Prochazkova L., Z. anal. Chem., 182, 103 (1961).
131. GoguelR., Anal. Chem., 41, 1034 (1969).
132. Hems R. V., Kir kb right G. F., West T. S., Taianta, 16, 789 (1969).
133. Koter M., Grzesiuk W., Roczn. gleboznawcze, 18, 25 (1967).
134. Blanc P., Bertrand P., Liandier L. M., Chim. anal., 37, 305 (1955).
135. Martin J. M., Stephen W. J., Anal. chim. acta., 39, 525 (1967).
136. Martin J. M., Stephen W. J., Anal. chim. acta., 39, 175 (1967).
137. Пономарева Л. К., Соколова А. А., Зав. лаб., 32, 16 (1966).
138. Мошкина А. А., Пономарева Л. К-, Козлова И. Г., Зав. лаб., 32, 1329 (1966).
139. Мошкина А. А., Пономарева Л. К-, Козлова И. Г., Тр. Уральского н.-и. хим. ин-та, вып. 19, 185 (1970).
140. Масалович В. М., Агасян П. К-, Николаева Е. Р., Тр. Уральского н.-и. хим. ин-та, вып. 19, 173 (1970).
141. Еремин Ю. Г., Киселева К. С., ЖАХ, 24, 1201 (1969).
Глава VII
СЕЛЕН И ТЕЛЛУР
VII. 1. МЕТОДЫ ОТДЕЛЕНИЯ И РАЗДЕЛЕНИЯ
Характерным для селена и теллура является образование соединений, в которых степень окисления равна +4 и +6. Двуокись селена легко возгоняется и хорошо растворима в воде с образованием аналога сернистой кислоты — селенистой кислоты H2SeO3. Двуокись теллура практически не летуча и плохо растворима в воде. При нагревании двуокиси теллура в присутствии воды до 600°C происходит образование летучей ТеО(ОН)2 [1]:
ТеО2 + Н2О —> ' ТеО(ОН)21
Теллуристая кислота очень слабая и существует преимущественно в виде гидратированной НвТеОв.
По комплексообразующей способности селен и теллур резко различаются. Теллур легко образует комплексы со многими лигандами, селен проявляет значительно более слабые комплексообразующие свойства [2]. Разница в их способности к комплексообразованию широко используется в аналитической химии для разработки методов обнаружения и определения селена и теллура [3].
Селен легко, а теллур несколько труднее восстанавливаются до свободного состояния. Селен обычно выделяется в виде аморфной красной формы, а при нагревании выделяется серая форма.
При действии на селен и теллур азотной кислоты они легко окисляются до соединений со степенью окисления +4. Окисление до четырехвалентного состояния происходит и Нри действии брома, но при этом образуются также соединения шестивалентных селена и теллура. Чтобы направить реакцию окисления только до образования четырехвалентных селена и теллура, необходимо в раствор ввести избыток бромистоводородной кислоты.
При работе с растворами селена и теллура в присутствии гало-генидных кислот необходимо иметь в виду, что селен и теллур легко образуют соединения типа МХ4 и МОХ2 или МО2-2НХ, которые сравнительно легко улетучиваются, что может привести к значительным потерям при проведении различных манипуляций в процессе переведения пробы в раствор и других подготовительных операциях. Селенистая кислота при взаимодействии с галогенводородными кислотами и их солями образует также нестойкие га-логенидные гидроксокомплексы вероятного состава [4]: Se(OH)2Cl2 и Se(OH)2Bra.
Методы выделения и разделения. Одним из наиболее распространенных методов выделения и разделения селена и теллура является их осаждение в элементном состоянии с помощью различных восстановителей. Однако необходимо иметь в виду, что ионы платиновых металлов, золота, серебра и меди мешают выделению селена и теллура, так как при этом возможно выделение благородных металлов в элементном состоянии или в виде их теллуридов и селенидов;
2CuC12 + ТеОС12 + 4SnCl2 + 2НС1 —► Cu2Te ф + 4SnCl4 + Н2О 2HAuC14 + ТеОС12 + 5SnCl2 —► Au2Te; + 5SnCl4 + H2O
Для осаждения селена и теллура применяют сернистый ангидрид, гидроксиламин, гидразин, гипофосфорную кислоту, хлорид олова(II) и другие восстановители.
Двуокись серы практически количественно осаждает селен и теллур из 3—5 н. растворов по хлористоводородной кислоте [5]. Снижение кислотности нежелательно, так как при этом увеличивается соосаждение металлов, особенно меди, кадмия, висмута, сурьмы, олова и молибдена [6]. Повышение кислотности приводит к потере теллура, который при кислотности выше 8 н. по хлористоводородной кислоте пактически полностью остается в растворе. Это свойство используется для разделения селена и теллура, так как при такой кислотности происходит осаждение очень чистого селена [7]. Как уже отмечалось выше, соединения селена легко улетучиваются, а в процессе восстановления возможно образование легколетучего монохлорида селена. Поэтому температура при восстановлении селена не должна превышать 30 °C [8]. С целью предупреждения образования монохлорида селена прибавляют большой избыток восстановителя.
При осаждении селена и теллура хлоридом олова(II) их осадки в большей степени загрязняются сопутствующими металлами. Однако осаждение селена и теллура SnCU имеет преимущество по сравнению с осаждением сернистым ангидридом, так как реакция восстановления происходит значительно быстрее.
При фотометрическом определении теллура рекомендовано выделение селена с помощью иодида [9].
Для количественного осаждения селена в присутствии теллура используют также гидроксиламин. При этом осаждение ведут из горячей 17%-ной хлористоводородной кислоты или в присутствии лимонной кислоты [10].
Для концентрирования микропримесей селена и теллура при их определении в различных материалах широко применяют флотацию из несмешивающихся с водой органических растворителей, например хлороформа [9], или применяют различные коллекторы. В качестве коллектора для выделения следов теллура применяют селен [11], а при осаждении селенистой и теллуристой кислот рекомендовано применять гидроокиси алюминия и железа, но в
последнем случае полное выделение селена и теллура не достигается [12]. Для соосаждения селена применяют сульфат свинца [13].
Сорбционные методы отделения и разделения. Выше отмечалось, что теллур легче образует комплексные соединения по сравнению с селеном. Так, теллур образует хлоридный комплекс уже в 2 н. хлористоводородной кислоте, а селен образует комплексы только в 6 н. хлористоводородной кислоте. Это свойство используется для их разделения с помощью различных сорбентов. В табл. 10 приведены некоторые условия отделения селена и теллура с помощью сорбентов [14].
Таблица 10. Условия отделения селена и теллура от других ионов сорбцией
Разделяемые элементы Ионит Среда Литература
Поглощение TeW и отделение его от TeVl, Bi, Си, Fe, Se IPA-120, КУ-2, СДВ-3 0,3 н. НС1 рН~0,4—0,5 15
Поглощение TeJV, отделение его от Al, Си, Fe, Sn, Sb ЭДЭ-10П 0,05-10 н. HCI 16. 17
Поглощение UVI, отделение его от TelV Дауэкс-2 1 н. Н3РО4 18. 19
Поглощение ВПН, отделение его от Teiv ЭДЭ-10П 1 н. НС1 20
Для поглощения селена ионитами пользуются растворами, содержащими карбонаты [14]. По поглощающей способности селена из карбонатных растворов иониты располагают в следующий ряд [14]: ВП-1А > ЭДЭ-10П = АЭ-1 = ВП-1 > АВ-17 = AM > Вофа-тит N = АМП.
Таким образом, лучшим сорбентом для селена из карбонатных растворов является сильноосновной ионит типа ВП-1 А, с помощью которого селен можно отделить от теллура, так как теллур в этих условиях не поглощается.
Разделение селена и теллура возможно также с помощью тонкослойной хроматографии на А120з. Для этого применяют смеси хлористоводородной кислоты и метанола, этанола, пропанола и изопропанола. При отношении объема спирта к объему концентрированной хлористоводородной кислоты, равном (1:3), Rf для TeIV достигает ~0,90, селен (IV) в этих условиях перемещается незначительно [21].
Рекомендованы методы разделения селена и теллура хроматографическим методом на бумаге [22] с последующим определением селена и теллура визуальной колориметрией непосредственно в зонах адсорбции. Метод применен для определения селена и теллура в промышленных объектах, содержащих платиновые металлы и золото [23]. В качестве растворителя в этом случае применен «-бутанол, насыщенный 10%-ным раствором нитрата натрия, а для 224
Таблица 11. Условия экстракционного отделения селена и теллура от других элементов
Разделяемые элементы Растворитель Кислотность раствора и комплексанты Литература
Экстракция TelV, отделение его от Se, Al, Bi, Cr, Co, Метилизобутил-кетон 4,5 н. НС1 25
Cu, Ni
Экстракция FelH, отделение Метилбутилкетон 6 н. НС1 и К2Сг2О7 26
его от TelV
Экстракция TelV, отделение от TeVl и SelV Трибутилфосфат (ТБФ), 30%-ный раствор в керосине 4-5 н. НС1 27
Экстракция SO?’, отделение ,То же 4 н. НС1 28
от TelV
Экстракция TelV, отделение 100% ТБФ 0,1 н. НС1 и KSCN 29
от TeVl 30 -
Экстракция TelV, отделение То же 4 н. НС1
от TeVl и иода 31
Экстракция TelV » 0,6-1,7 н. НС1 и тиомочевина
Экстракция SelV, отделение от SO?’ ТБФ в СС14 Концентрированная HCI 32
Экстракция TelV, отделение от Fe и Au ТБФ в диэтиловом эфире 2 н. НС1 33
Экстракция SelV Изобутанол 4,6 н. HNOa 34
Экстракция TelV, отделение от SelV и платиновых ме- Диэтилдитиокарбамат в СС14 0,05—10 н. НС1 35
таллов
Экстракция TelV, отделение SelV N-Додеценил-три-алкилметил-амин, трибензиламин в СНС13 4 н. НС1 36
Экстракция SelV, отделение от TelV 3,3'-Диаминобензидин в толуоле 0,05-10 н. HCI 37,38
Экстракция TelV Эфиры фосфорной кислоты 0,05-10 н. НС1 39
Экстракция Bi, отделение от TelV Смесь амилового спирта и этилацетата (3:1) 1-2 н. НС1, 0,1 М KI 9
Экстракция TelV Смесь диэтилового эфира и амилового спирта ( I . 1-2 н. НС1, 0,6А4 KI 40
Экстракция FelH, AsV, Sbv, AulH, Til", Bi, SniV, SelV, TelV ( 1 . Диизопропиловый эфир 8 н. НС1 41
Экстракция TelV Метилизобутил-кетон 4 н. НС1 41
Экстракция купферонатов Ag, То же pH =3-5 41
Al, Cd, Co, Cu, Ni, Pb, Zn, отделение от TelV 8—10 н. НС1
Экстракция Se'V Метилэтилкстоп 42
8 Зак. 761
Разделяем.ые элементы Растворитель Кислотность раствора и комнлексанты Литература
Экстракция SelV, отделение ст As, Pb, Zn, In, Те и других элементов Бутанол и СНС13 (1:4) 0,1 н. НС1, 1-фе-нилтиосеми-карбазид 43
Экстракция TelV, отделение ст Se!V и других ионов Дихлорэтан 5—7М НС1, диан-типирилпропил-метан 44—46
Экстракция TeW, отделение от многих элементов 1,1,2,2-Тетрахлор-этан НО, разбавленная (1:1). 4,4-метилендиантипирин 47
проявления зон пользуются хлоридом олова(II). Для отделения селена от ионов Fe, Си, Zn, Pb, Bi и Те применен метод хроматографии на бумаге, импрегнированной вольфраматом олова(IV) [24].
Экстракционные методы отделения и разделения. Ряд комплексов теллура и некоторые комплексы селена легко экстрагируются неводными растворителями. Это используется в экстракционных методах отделения и разделения селена и теллура. Краткая характеристика некоторых экстракционных методов отделения и разделения селена и теллура приведена в табл. 11.
Экстракция селена лучше происходит из сильно кислых растворов (ЗМ по H2SO4 или >6н. НО), а теллур экстрагируется и при более низкой кислотности.
Некоторые другие экстракционные методы и детальные методики будут рассмотрены ниже при описании отдельных методов отделения селена и теллура.
Дистилляционные методы. Для отделения селена и теллура рекомендованы также дистилляционные методы. Так, селениды и теллуриды отгоняются в токе хлора при нагревании. Хлориды селена (IV) и теллура (IV) отгоняют в токе хлористого водорода [48]. Необходимо иметь в виду, что летучесть галогенидных соединений теллура значительно понижается в присутствии серной или фосфорной кислот, а также при разбавлении водой. В настоящее время дистилляционные методы отделения селена и теллура применяются редко.
VII. 2. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ ОПРЕДЕЛЕНИЯ
СЕЛЕНА И ТЕЛЛУРА
Одними из наиболее старых и до недавнего времени наиболее распространенных методов определения селена и теллура являются методы, основанные на получении золей селена и теллура и измерении поглощения света их коллоидными растворами. Отличие различных методик состоит в применении различных восстанови-226
телей, стабилизаторов [49] или в способах подготовки пробы к анализу.
В качестве восстановителя для селена используют хлорид олова (II) [50—55], тиомочевину [56], аскорбиновую кислоту [57^—59], солянокислый гидразин [60—65], ацетон в хлористоводородной среде [66], а для теллура — хлорид олова (II), гипофосфит натрия и гипофосфористую кислоту [67—69] и др. Для стабилизации золей используют гуммиарабик [62, 67, 70—72] и желатин [35, 61, 71, 73—76]. Для увеличения чувствительности фотометрического определения селена и теллура вводят при восстановлении сенсибилизирующие добавки в виде ионов меди, сурьмы и висмута [35, 68, 74, 75, 77—80].
Описанные методы обладают недостаточной чувствительностью и имеют ряд недостатков, связанных со строгой регламентацией условий получения золей. Кроме того, эти методы могут быть применены после разделения селена и теллура.
Более чувствительные и более специфические методы определения селена и теллура основаны на реакциях образования комплексных соединений с органическими и неорганическими лигандами. Для определения селена применяются два типа органических реактивов: серусодержащие и о-диамины. Особенностью действия о-диаминов является избирательность их к селену, благодаря чему их широко применяют для определения селена в присутствии теллура. Серусодержащие реагенты в большинстве случаев взаимодействуют с теллуром и селеном, кроме того, они, как правило, характеризуются малой чувствительностью по отношению к селену.
Способность о-диаминов к образованию соединений с селеном (IV) была установлена давно [81]. В настоящее время для фотометрического определения селена предложен ряд различных производных диаминов. Некоторые характеристики этих реагентов представлены в табл. 12.
Лучшим из этой группы реактивов является 3,3'-диаминобензи-дин [156]. Из реактивов для флуоресцентного определения селена лучшим, по-видимому, является 2,3-диаминонафталин [118], его соединение с селеном образуется в кислой среде. Флуоресценцию измеряют при 306 нм.
Чувствительность определения составляет 0,002 мкг селена в 5 мл экстракта. Теллур не мешает определению селена.
Органические реагенты, содержащие тионную и тиольную группы, применяются для определения селена сравнительно недавно. Одним из первых реактивов этого типа был, по-видимому, диэтилдитиокарбамат натрия [157, 158]. Однако этот реагент малочувствителен и неспецифичен. Было предложено несколько серу-содержащих реагентов на селен [43, 137, 152, 159—164], в том числе дитиофосфорная кислота [159], 2-меркаптобензимидазол [137, 162], n-анизидин и п-толуидин [160], дитизон [164, 165] и фенилтиосемикарбазид [43]. Все эти реагенты, кроме фенилтиосемикарбазида, взаимодействуют также с теллуром и не могут быть применены для
228
Табли и а 12. Химико-аналитические характеристики фотометрических методов определения селена [82]
Реагент Экстрагент, среда Ьмакс’ нм е^макс Пределы определения, мкг/мл Мешающие элементы Литература
3,3'-Диаминобензидин H2N /"А \ nh2 H2N' 'NH2 рНобраз = 2 3; pH экстр = 6 7 толуол 420 420/600 1,7 • 104; 1,9- 104 0,1 — 10,0 0,01—0,16 Au, Bi. Сг, Мо, Sn, Ti, V, W, Zr. Окислители 83—116
2,3-Диаминонафталин NH2 —NH2 pH экстр ~ 1’5 2,5, толуол из 0,1 н. НС1; декалин 376,8 366/540 2,38- 104 от 0,05 0,001—0,05 Окислители и восстановители 117—122
4,5-Диамино-6-тиопиримидин nh2 NH2 pH = 1,5—2,5 380 1,92- 104 0,1—2,5 Fe, Си. S2O:“, SO|~. BrO" CIO3 123
2,3-Диаминофеназин N А/ NH? nh2 N н3ро4 600 — 0,005—0,025 V, Те, As, Си 124—126
96 %-на я H2SO4
605
от 2,5 Bi, Сг3+, Hg, Ni
127
Й^-Диантримид
1,Г-Диантримид
4- Диметиламино- 1,2-фенилен диамин
(CH3)2N——nh2
—NH2
Дитизон
NH—NH—C6H5
S=C\
\n=n—C6H5
и др.
96%-ная H2SO4
4 н. HC1
6 н. HC1; CC14
Цифры показывают кратность по уравнению с концентрацией
480;585 0,4—5,0
500 5,73-103 0,3—10
420 7,0-104 от 0,02
селена.
Те, Ge, В, Вг~, .
F". Ag>2,
Al >5, Bi > 1, Са>100, Cd> >50, Со >20, » Cr> 10, Cu>20,
Mg>20, Ni>20, Pb>l, Si > 1, Zn>50*
Fe3+, Cu, Co, Cr3+, V, Mo.
Au, Pd, Pt
Многие элементы
128
129,130
82, 131—134
to
w
о
Реагент
Экстрагент, среда
1 н. НС1; СНС13
Фенилгидразин-п-сульфокислота + 1-нафтиламин
SO3H
pH = 1—3; СНС13
НС1; бутанол+ 4-СНС1з
pH = 1,8—2,2
*макс им е> лмакс Пределы определения, мкг/мл Мешающие элементы Литература
530 3,67- 104 ОТ 0,05 Cu, Fe3+, W, V и окислители /35
400—420 1,5- 104 0,1-1,0 — 132
335 1,04- 10‘ 1-10 Bi, Си, ВгО^, IO3, Fe 136
520 — 0,1—2,0 Fe2+, Sn2+, AsO’~, SbO?~, 1 . Hg, Ag, окислители 137,138
о-Фенилендиамин
pH = 25; СНС13, толуол
1-Фенилтиосемикарбазид Н Н
4-Метилтио- 1,2-фенилендиамин
1,8-Диаминонафта лин
NH2 nh2
Пиррол
N I Н
0,05-2 н. НС1; бутанол+ +СНС13
pH =3-4
Замещенные 1,2-фенилендиамины (4-метил-, 4-хлор-, 4-нитоо-)
ьэ
с*э
330 1,82- 104 0,05—7,5 Fe, Sn, Cu, 1 139—151
380-400 5,2- 103 1,0-2,0 Cu, V, Fe, Bi, Те 43,152
572 — — —
372 — — — 153
— — 3,0-6,0 — 154,155
337; 341; 355 соответственно — — 142
определения селена в присутствии теллура. Метод определения селена с помощью дитизона характеризуется высокой чувствительностью (е42о = 7,0 -104), но мало специфичен, и, кроме того, реагент неустойчив на свету. Дифенилтиокарбазид также взаимодействует с селеном [132], но он еще менее устойчив на свету в присутствии кислорода и других окислителей. Значительно более устойчив 1,4-дифенилтиосемикарбазид, с помощью которого можно определять селен в присутствии более чем 100-кратных количеств теллура [166]. Правда, чувствительность метода в данном случае несколько ниже (ез25 = 3,2-104 и е4ю = 1,6-104), но метод позволяет определить селен в рудах [167, 168] и дает хорошо воспроизводимые результаты.
При взаимодействии селенистой кислоты с тиомочевиной образуется элементный селен [169], что дает возможность определять теллур в присутствии селена. Избыток тиомочевины растворяет элементный селен с образованием комплексного соединения Se11 [170]. В 8,5 н. растворе по хлористоводородной кислоте образуется растворимый тиомочевинный комплекс селена, который осаждается солью Рейнеке [171]. Из этой группы реагентов на селен рекомендована также тиогликолевая кислота [172], однако этот реагент не имеет преимуществ по сравнению с другими серусодержащими реагентами.
Селен является сравнительно сильным окислителем (^Seiv/Seo = = 0,74 В) и окисляет многие органические соединения с образованием яркоокрашенных продуктов, восстанавливаясь до элементного состояния. Эти реакции применяются для фотометрического определения селена. Так как £Teiv/Teo = 0,53В, то он не взаимодействует со многими соединениями и не мешает определению селена. Примером реакций такого типа является взаимодействие селена(IV) с 1,Г-дифенилгидразином [135], а также реакции с фенилантраниловой кислотой [173, 174], пирролом [154] и др.
В отличие от селена теллур образует более прочные комплексы, которые, широко применяются для его экстракционно-фотометрического определения. Из комплексов теллура с неорганическими лигандами, применяемыми в фотометрическом анализе, следует прежде всего назвать галогенидные комплексы [175] и гетерополикислоты [176—181], в том числе теллуристомолибденованадиевую кислоту [182]. В гексагалогенидтеллуратах Ме2ТеХ6 центральным атомом является теллур(IV) (Me — катион одновалентного металла; X—С1“, Вг", I- и SCN-).
В молибденовотеллуровой и вольфрамотеллуровой кислотах, которые являются производными теллуровой кислоты Н6ТеОб, роль центрального атома также играет теллур: МебТе(МоО4)б и MeeTe(WO4)6 (где Me — катионы одновалентных металлов, аммоний или гуанидиний).
При восстановлении желтой теллурито-12-вольфрамовой гетерополикислоты образуется теллуритовольфрамовая синь [183].
В водных растворах теллур (IV) вытесняет кремний из кремнемолибденовой гетерополикислоты и желтая окраска раствора ослабляется [184]. На этом основан косвенный фотометрический метод определения теллура.
При взаимодействии теллура с фосфорномолибденовой гетерополикислотой образуется желтая фосфорнотеллуристомолибдено-вая гетерополикислота с отношением Р : Те : Mo = 1 : 1 : 11. Эта реакция предложена для фотометрического определения теллура [185]. Селен при содержании не более 20-кратных количеств по сравнению с теллуром не мешает определению.
Значительно больший интерес представляют фотометрические методы определения теллура, основанные на реакциях образования комплексных соединений с органическими лигандами. При этом главным образом используются смешанные комплексы гексагалогенидов теллура(IV) с органическими основаниями и комплексы с серусодержащими лигандами.
Изучение смешанных комплексов проводилось давно. Так, было изучено взаимодействие галогенидов теллура (IV) с анизолом, фе-нитолом и другими подобными соединениями [186]. Позже изучено взаимодействие бромида теллура(IV) с органическими основаниями в неводных растворах [187]. Однако для экстракцирнно-фо-тометрического определения теллура смешанные комплексы были применены позже. Особый интерес представляют соединения галогенидов теллура с родаминами [80, 82, 188—197], метиловым фиолетовым [196] и виктория голубой [197]. Лучшие результаты определения теллура с помощью флуоресцентного метода дает применение бутилродамина [190]. Этот метод позволяет определить 1О'4о/о теллура в минеральном сырье из навески 0,5—1,0 г.
Бромидный комплекс селена также взаимодействует с основными красителями, однако описана лишь одна такая реакция — с виктория голубой Б [198]. Детальное изучение этой реакции [199] показало, что она не представляет практического интереса [193].
Для фотометрического определения селена использована реакция окисления селенистой кислотой иодида до иода, который разрушает трифенилметановые красители [200—202].
Большой интерес представляют реакции между галогенидными комплексами теллура и производными пиразолона, например, реакции образования галогенидных комплексов теллура с дианти-пирилметаном [203], диантипирилметилметаном, диантипирилпро-пилметаном, диантипирилфенилметаном [44—46, 204—206]. Хло-ридный и бромидный комплексы селена (IV) также взаимодействуют с производными пиразолона, однако в отличие от соединений теллура (IV) хлоридный комплекс селена (IV) не экстрагируется органическими растворителями. Это используется для разделения селена и теллура при определении последнего [207].
Многие методы отделения и фотометрического определения теллура основаны на реакциях с серусодержащими органическими лигандами [208—214]. С этой целью широко применяют
диэтилдитиокарбамат натрия (ДДТК) [157, 158, 215, 216], который образует с теллуром малорастворимые в воде соединения. При pH = 3 эти соединения практически полностью извлекаются этилацетатом, толуолом, четыреххлористым углеродом и хлороформом. При pH = 8,5—8,7 экстрагируется только соединение теллура, а селен остается в водной фазе. Предложен метод фотометрического определения теллура в водно-ацетоновой среде [217]. С этой целью применяют также ряд других дитиокарбаматов [218]. Однако ди-диокарбаматные комплексы теллура обладают малой устойчивостью по отношению к окислителям, в том числе к кислороду воздуха. Особенно быстро происходит разрушение комплексов под действием света. Более устойчивым является комплекс с 3,5-ди-фенилпиразолин-1-дитиокарбаматом натрия [207].
Калиевая соль 5-меркапто-3-фенил-2-тио-1,3,4-тиадиазолон-2(висмутол II) при pH = 3,3—4,3 образует с теллуром(IV) соединение желтого цвета [219], которое растворяется в спиртах, кетонах, хлороформе, четыреххлористом углероде и бензоле.
Тиомочевина в кислой среде взаимодействует с теллуром (IV) с образованием малорастворимого в воде соединения желтого цвета [220]. Этот комплекс хорошо растворяется в неполярных растворителях. Установлено [221], что соотношение в комплексе теллура к тиомочевине равно 1 :4. В процессе реакции происходит восстановление теллура(IV) до теллура(II), который с избытком реагента образует комплексное соединение [222—229].
5-Меркапто-З-(нафтил-2)-1,3,4-тиадиазолтион-2 в широком интервале кислотности от 18 н. по серной кислоте до pH = 6 образует с теллуром(IV) желто-оранжевый комплекс, практически нерастворим в воде, но хорошо растворим в хлороформе, бензоле и других органических соединениях. В интервале кислотности от 2 н. НС1 до pH = 2 реагент взаимодействует также с селеном(IV) с образованием подобного соединения. Эти реакции применены для определения теллура и селена в самородной сере и пылях свинцового производства [227].
VII. 3. ОПРЕДЕЛЕНИЕ СЕЛЕНА С ПРИМЕНЕНИЕМ
3,3'-ДИАМИНОБЕНЭИДИНА
Селен (IV) взаимодействует с 3,3'-диаминобензидином с образованием пиазселенола, окрашенного в интенсивный желтый цвет:
реакция идет довольно медленно, поэтому раствор после сливания реагентов выдерживают в течение 30—50 мин. Пиазселенол
плохо растворим в воде, в связи с этим в водных растворах можно определить не более 2,5 мкг/мл Se. В случае определения больших количеств необходимо применять экстракцию толуолом. Реакцию проводят при pH = 2—3, а экстракцию толуолом— при pH = == 6—7, хотя рекомендованы и другие условия (pH проведения реакции 0,8—1,1, а экстракцию проводить при pH = 8,0—8,5) [228].
Мешающие вещества. Этот метод является одним из наиболее
специфичных. Теллур(IV) не мешает определению. Однако окисли-
тели, в том числе ванадий (IV), желе-зо(Ш) и медь(Н), образуют окрашенные продукты при взаимодействии с 3,3'-диами-нобензидином и мешают определению селена. Определению также мешают вещества, которые связывают или восстанавливают селен (Sn2+, 1“, аскорбиновая кислота и др.). Для устранения вредного влияния железа его связывают фторидом, медь маскируют оксалатом. Лучше все мешающие ионы, кроме ванадия(V), маскировать ЭДТА.
Измерение оптической плотности. Продукт реакции — пиазселенол — устойчив во
Рис. 51. Спектры поглощения раствора пиазселенола в воде(/) и в толуоле (2).
выраженным макси-
времени, поэтому для измерения интенсивности окраски можно применять любой метод. Спектры поглощения раствора пиазсе-ленола в воде и в толуоле представлены на рис. 51. Водные растворы пиазселенола характеризуются полосой поглощения с явно
мумом при 340 нм (е34о = 3,6-103) и размытой полосой с максимумом при 400 нм. Для толуольных растворов характерно снижение полосы поглощения с максимумом при 340 нм (ез4о = 1,1 • 103) и повышение поглощения при 425 нм (6420 =0,7 • 103). Измерять оптиче-
скую плотность водных растворов лучше при 340 нм, а толуольных растворов при 425 нм, так как при 340 нм наблюдается значительное поглощение свободного реактива.
Метод колориметрического титрования не применим, а метод шкалы в этом случае мало удобен в работе.
Чувствительность метода — 1 мкг селена в 6 мл экстракта. Оптимальные концентрации определения 1—5 мкг селена в 6 мл.
VII. 3.1. Определение селена в отсутствие мешающих веществ
Реактивы
Стандартный раствор селена. Навеску 0,281 г двуокиси селена, очищенной возгонкой и высушенной над Р2О5, растворяют в воде и разбавляют до 1 п дистиллированной водой. Этот раствор содержит 0,2 мкг селена в I мл. Перед работой разбавлением готовят раствор меньшей концентрации Необходимо иметь в виду, что при длительном хранении раствора, особенно при попадании
прямого солнечного света, селен частично восстанавливается примесями органических веществ до элементного состояния и стенки сосуда покрываются красным налетом селена.
Для очистки двуокись селена растирают, помещают в фарфоровую чашку и смачивают азотной кислотой. В воронку несколько большего диаметра, чем диаметр чашки, вставляют пробку из стеклянной ваты. В эту воронку вставляют другую, несколько меньшего диаметра, и накрывают воронками чашку с двуокисью селена. Переносят собранный прибор под хорошую тягу (пары двуокиси селена ядовиты!) и нагревают на открытом пламени для возгонки двуокиси селена. Температура возгонки 317 °C. Очищенный препарат хранят в эксикаторе над Р2О5 или над серной кислотой.
Стандартный раствор селена можно готовить из металлического селена или селенита натрия Na2SeO3. В последнем случае титр водного раствора устанавливают титриметрическим методом [229].
Солянокислый 3,3'-диаминобензидин, 0,5%-ный водный раствор. Реактив очищают переосаждением из 3 н. хлористоводородной кислоты. Для этого растворяют 50—100 г солянокислого З.З'-диаминобензидина в 200—300 мл воды, переносят в делительную воронку емкостью 500 мл, прибавляют активированный уголь и взбалтывают в течение 3 мин. Затем раствор быстро фильтруют. Операцию очистки повторяют до получения почти бесцветного раствора. К фильтрату прибавляют хлористоводородную кислоту до 3 н. кислотности раствора и оставляют в темном месте на 1,5—2 ч. Осадок фильтруют под вакуумом, промывают на фильтре 3 н. хлористоводородной кислотой и затем высушивают под вакуумом в эксикаторе. Высушенные кристаллы хранят в холодильнике в темной склянке.
Муравьиная кислота, 2,5 М раствор.
Толуол, ч. д. а.
Ход анализа. До 20 мл анализируемого раствора, содержащего от 0,5 до 5 мкг селена(IV), помещают в стакан емкостью 50 мл. К раствору прибавляют 1 мл раствора муравьиной кислоты и доводят реакцию раствора аммиаком или кислотой до рН = 2—3. Добавляют 1 мл раствора 3,3'-диаминобензидина и оставляют на 50 мин. Затем раствор разбавляют до 25 мл, переносят в делительную воронку емкостью 50—100 мл, аммиаком доводят pH раствора до 6,0—7,0 и экстрагируют 10 мл бензола в течение 30 с.
Бензольный слой отделяют и измеряют оптическую плотность при 425 нм в кювете с толщиной слоя 1 или 5 см.
При малом содержании селена экстракцию проводят 5 мл толуола. Более воспроизводимые результаты получаются, если предварительно толуольный раствор высушить безводным хлоридом кальция. В качестве холостого раствора применяют толуольный экстракт, приготовленный экстракцией раствора сравнения, содержащего все реактивы кроме селена.
Содержание селена находят по калибровочному графику, построенному по значениям оптической плотности, полученным при определении селена в растворах с различным содержанием стандартного раствора.
V II. 3.2. Определение селена в сталях [43, 230]
Реактивы
Этилендиаминтетрауксусной кислоты динатриевая соль (ЭДТА), 0,1 М раствор.
Другие реактивы — указаны в разделе VII. 3.1.
Ход анализа. Навеску 0,5—1 г (при содержании 0,5—0,25% Se) помещают в колбу, вставляют в нее вертикальный холодильник, добавляют 10 мл концентрированной хлористоводородной кислоты и 3 мл концентрированной HNO3, присоединяют к холодильнику барботер, погружают в приемник с 20 мл 1%-ной HNO3 и нагревают. После окончания растворения смесь охлаждают, промывают барботер и холодильник водой, затем поглотительный раствор и промывные воды присоединяют к раствору в колбе. Холодильник снимают, и раствор осторожно кипятят для удаления окислов азота. Охлажденный раствор фильтруют, фильтр промывают 1%-ным раствором НС1, переносят в мерную колбу емкостью 200 мл и разбавляют водой до метки. После перемешивания отбирают аликвотную порцию 10 мл, добавляют 2 мл раствора ЭДТА и продолжают анализ, как указано в разделе VII. 3.1.
V II. 3.3. Определение селена в металлической меди
Навеску меди 1 г растворяют в 10 мл азотной кислоты, разбавленной (1:1). Если необходимо, добавляют еще небольшое количество кислоты. Раствор упаривают, переносят в мерную колбу емкостью 50 мл, разбавляют до метки водой и перемешивают. Отбирают 10 мл раствора, добавляют 2 мл раствора ЭДТА и продолжают анализ, как указано в разделе VII. 3.1.
V II. 3.4. Определение селена в полупроводниковых мышьяке, индии, висмуте, галлии и сурьме [231]
Реактивы
Этилендиаминтетрауксусной кислоты динатриевая соль (ЭДТА), 8%-ный раствор.
Другие реактивы — указаны в разделе VII. 3.1.
Ход анализа. Навеску исследуемого материала 0,5—1,0 г помещают в стакан емкостью 50 мл и растворяют при осторожном нагревании на песочной бане в 5 мл HNO3 (пл. 1,40) при анализе индия, висмута и сурьмы или в 5 мл смеси (1:1) концентрированных хлористоводородной и азотной кислот при анализе мышьяка, галлия и сплава галлий-мышьяк. Полученный раствор осторожно упаривают почти досуха.
При анализе мышьяка, индия и висмута остаток после упаривания растворяют на холоду в 6—8 мл хлористоводородной кислоты, разбавленной (1:3), и доливают водой до 30 мл. При анализе индия и висмута в раствор предварительно вводят раствор ЭДТА в количестве, необходимом для связывания всего металла. Затем к раствору прибавляют 2 мл муравьиной кислоты, разбавленной (1:9), 1 мл раствора ЭДТА и аммиак до pH = 1. Далее продолжают анализ, как указано в разделе VII. 3.1.
Для’анализа галлия и сурьмы остаток, полученный после упаривания, растворяют в 40 мл НС1 (1 : 1), прибавляют раствор мышьяка (5 мг), 0,1 г CuSO4, немного бумажной массы и нагревают до 80—90 °C. В горячий раствор добавляют небольшими порциями при перемешивании 3 г NaH2PO2, раствор нагревают до кипения, кипятят 15—20 мин и оставляют до следующего дня. Осадок отфильтровывают через плотный фильтр и промывают на фильтре 5—6 раз горячей хлористоводородной кислотой, разбавленной (1:7), и затем 3 раза горячей водой. Осадок растворяют на фильтре в два приема в 3 мл смеси азотной и хлорной кислот (10:1) и промывают фильтр горячей водой. Полученный раствор упаривают до появления паров хлорной кислоты, охлаждают, разбавляют до 35 мл водой, прибавляют 2 мл муравьиной кислоты, 1 мл ЭДТА, доводят аммиаком pH до 2—3 и продолжают анализ, как указано в разделе VII. 3.1.
V II. 3.5. Определение селена в рудах [232, 233]
Реактивы
Хлорид олова(П), 40%-ный раствор.
Этилендиаминтетрауксусной кислоты динатриевая соль (ЭДТА), 0,1 М раствор.
Ход анализа. Навеску руды (железные, медные сульфидные и и др. руды) 0,05—0,5 г помещают в фарфоровую чашку, прибавляют 5—10 мл концентрированной HNO3 и нагревают до разложения пробы. Затем прибавляют 10 мл H2SO4, разбавленной (1 : 1), и выпаривают до появления паров серной кислоты. Содержимое чашки охлаждают, стенки смывают водой и выпаривание до паров серной кислоты повторяют. Остаток обрабатывают 15—20 мл воды, нагревают до кипения и фильтруют. Осадок промывают 5—6 раз небольшими порциями 2%-ной серной кислоты. Если содержание селена в пробе составляет меньше 100 мкг, то к фильтрату прибавляют раствор теллура, содержащий 100 мкг Те. Раствор нагревают, прибавляют 40%-ный раствор SnCl2 в 20%-ной хлористоводородной кислоте до восстановления Fe3+ и еще 2 мл, вводят 1 г солянокислого гидразина, кипятят 2—3 мин и оставляют до следующего дня. Затем осадок селена и теллура отфильтровывают через бумажную массу и промывают 10—12 раз 10%-ной хлористоводородной кислотой, содержащей 0,5% NH2NH2-2HCL Осадок вместе с бумажной массой помещают в колбу, в которой проводилось осаждение селена, вводят 10 мл концентрированной хлористоводородной кислоты, 3—5 капель концентрированной HNO3 и нагревают на кипящей водяной бане 30 мин. После растворения селена добавляют 10—15 мл воды и раствор фильтруют. Аликвотную часть фильтрата, содержащую 5—30 мкг селена, нейтрализуют до pH = 2—3, добавляют 3 мл 0,1М раствора ЭДТА, 2 мл 2,5Л4 раствора муравьиной кислоты и ведут определение, как указано в разделе VII. 3.1.
V II. 3.6. Определение селена в теллуре [90]
Реактивы
Лимонная кислота, 40%-ный раствор.
Другие реактивы — указаны в разделе VII. 3.1.
Ход анализа. Навеску тонкоизмельченного теллура 0,5 г помещают в термостойкий стакан емкостью 100 мл и растворяют в 10 мл HNO3, разбавленной (1:1), если необходимо, добавляют еще небольшое количество кислоты. Раствор упаривают почти досуха, прибавляют 25 мл 2 н. раствора NaOH и 10 мл 40%-ного раствора лимонной кислоты (с целью предотвращения гидролиза теллура), добавляют хлористоводородной кислоты до pH = 2—3 и ведут определение, как указано в разделе VII. 3.1.
V II. 3.7. Определение четырех-и шестивалентного селена в теллуровой кислоте[91]
Соединения четырех- и шестивалентного селена последовательно выделяют, затем растворяют соединение селена (VI) и определяют его в виде пиазселенола. Солянокислый гидразин количественно осаждает селен (IV) в виде элементного селена в 0,2—2,0 н. растворе по хлористоводородной кислоте. Селен(VI) практически не восстанавливается из нейтрального раствора, но количественно выделяется из раствора, содержащего больше 2 моль/л хлористоводородной кислоты. Во всех случаях вместе с селеном выделяется небольшое количество теллура.
Реактивы
Солянокислый гидразин, насыщенный раствор.
Ход анализа. До 2 г анализируемой пробы растворяют в воде и объем раствора доводят до 50 мл. При малом содержании селена навеску увеличивают до 17 г и после растворения пробы разбавляют раствор до 250 мл.
В первом случае к раствору добавляют 2 мл насыщенного раствора NH2NH2-2HG1, а во втором — 4 мл. Смесь нагревают до кипения, выдерживают в течение 3 ч при 40—60 °C, затем еще 10— 12 ч, фильтруют, промывают водой, растворяют в 5 мл концентрированной HNO3 и определяют селен(VI), как указано в разделе VII.3.6 и VII.3.1.
Для определения селена (VI) фильтрат после отделения осадка элементного селена и промывную жидкость упаривают до 100 мл, вводят хлористоводородную кислоту с таким расчетом, чтобы создать концентрацию кислоты, равную 2,1 моль/л. Затем добавляют 2—6 мл насыщенного раствора солянокислого гидразина и осаждают селен, как описано выше.
Большие количества теллуровой кислоты затрудняют осаждение селена, поэтому если для анализа взята большая навеска
пробы (17 г), то водный раствор подкисляют уксусной кислотой и проводят предварительное разделение селена и теллура с помощью ионитов. ,
Предварительное выделение основной массы теллура [92] можно производить также путем гидролиза его до теллуристой кислоты (Н2ТеО3) при pH = 3—4.
V II. 3-8. Определение селена в почвах [234]
Пробу разлагают смесью азотной и хлорной кислот. Затем селен осаждают с помощью гипофосфита, применяя в качестве со-осадителя мышьяк(III). После отделения и растворения выделенного селена его определяют с помощью 3,3'-диаминобензидина.
Реактивы
Смесь кислот. Смешивают 800 мл концентрированной HNO3 с 200 мл 60%-ной хлорной кислоты.
Арсенит натрия. Навеску 250 мг AS2O3 и 2 г NaOH растворяют в воде и разбавляют до 200 мл.
Фосфористая кислота, 50%-ный водный раствор.
Другие реактивы — указаны в разделе VII. 3.1.
Ход анализа. Навеску пробы 1 г помещают в фарфоровую чашку и обрабатывают 10 мл смеси кислот. После окончания бурной реакции добавляют еще 10 мл смеси кислот, после чего выпаривают до 1—2 мл, но не досуха. Чашку с содержимым охлаждают, прибавляют 10 мл хлористоводородной кислоты, разбавленной (1 : 1), нагревают до кипения, фильтруют и промывают той же кислотой. К фильтрату прибавляют 2 мл раствора арсенита натрия, 5 мл раствора Н3РО2, осторожно кипятят до коагуляции осадка, фильтруют и промывают осадок хлористоводородной кислотой. Фильтр с осадком обрабатывают 20 мл смеси кислот и выпаривают до 1—2 мл. После охлаждения раствор обрабатывают 3—5 мл дистиллированной воды, прибавляют 1 мл 0,05 н. раствора ЭДТА и определяют селен, как указано в разделе VII. 3.1.
V II. 3.9. Определение селена в растительных материалах [228, 235]
Пробу разлагают азотной, а затем хлорной кислотами, удаляют азотную кислоту выпариванием и определяют селен с помощью 3,3'-диаминобензидина. Для маскирования мешающих ионов применяют раствор ЭДТА.
Реактивы
Этилендиаминтетрауксусной кислоты динатриевая соль (ЭДТА), 0,1 М раствор.
Ход анализа. Навеску пробы 2—10 г сухого растительного материала помещают в колбу Кьельдаля емкостью 200 мл и разлагают при нагревании с 10—30 мл концентрированной HNO3 в те-240
чение 2—3 ч. Затем прибавляют несколько миллилитров концентрированной НС1О4 и постепенно усиливают нагревание до обильного выделения паров хлорной кислоты. Остаток охлаждают, разбавляют 10—20 мл воды и операцию выпаривания до белого дыма повторяют до тех пор, пока полностью не будет удалена азотная кислота. К охлажденному раствору прибавляют 5 мл концентрированной хлористоводородной кислоты и нагревают несколько минут для восстановления селена(VI) до селена(IV). Раствор разбавляют до 25 мл или 50 мл, берут аликвотную часть, прибавляют 10 мл 0,1 М раствора ЭДТА и определяют селен, как указано в разделе VII. 3.1.
Окончание определения можно также проводить люминесцентным методом [236]. При этом чувствительность определения селена повышается примерно на порядок.
V II. 4. ОПРЕДЕЛЕНИЕ СЕЛЕНА В ВИДЕ ДИТИЗОНАТА [82]
В сернокислой (1—13 н.) и солянокислой (1—8 н.) средах селен и теллур взаимодействуют с дитизоном с образованием окрашенных внутрикомплексных соединений. Дитизонаты селена и теллура количественно экстрагируются четыреххлористым углеродом из 11 — 12 н. H2SO4 и 6—7 н. НС1.
Мешающие вещества. В кислой среде с дитизоном взаимодействуют ионы многих металлов (Те, Ag, Cu, Hg, Аи и др.), мешающие определению селена. Перманганат, хромат, ванадат, Fe3+ и другие окислители окисляют дитизон, т. е. мешают определению селена. Высокозарядные ионы, которые легко подвергаются гидролизу (W, Nb, Та, Bi, Sn) также мешают определению селена.
Лучшим вариантом для устранения мешающего действия посторонних ионов является экстракция дитизонатов при pH = 2 (отделение Ag, Bi, TeIV, Hg, Cu и др.). Селен при этом экстрагируется не более чем на 1,5%. Можно проводить предварительное осаждение селена и теллура совместно с мышьяком. При этом необходимо иметь в виду, что совместно с селеном и теллуром осаждаются также платиновые металлы, золото, серебро и частично медь. При содержании теллура до 10 мкг в пробе он не мешает определению селена. При большем его содержании учитывают поглощение дитизоната теллура. Ртуть можно удалить путем предварительного кратковременного нагревания пробы с порошком металлического железа, золото — восстановлением до металла, а серебро — осаждением в виде хлорида. С целью отделения меди, цинка и свинца экстракт дитизонатов встряхивают с раствором гексацианоферрата (II) калия.
Измерение оптической плотности. Спектры поглощения дитизоната селена и дитизоната теллура представлены на рис. 52. Полосы поглощения дитизонатов селена и теллура практически совпадают (X = 405—420 нм), но комплекс селена поглощает значительно сильнее (еса4 = 7,55 • 104).
Дитизонат селена разлагается под действием прямого солнечного света, поэтому необходимо работать в затемненном помещении или при электрическом освещении. В этом случае дитизонат селена практически устойчив вте-
Рис. 52. Спектры поглощения растворов дитизоната селена (/) и ди-тизоната теллура (2) в СС14.
шого внимания при выполнении многие вещества.
чение 3—4 ч.
Для определения можно применять также метод стандартной шкалы, однако ее необходимо готовить каждые 3—4 ч.
Значение молярного коэффициента поглощения показывает, что дитизонатный метод является одним из наиболее чувствительных. Метод позволяет определить 0,1 мкг селена в 5 мл экстракта. Однако этот метод требует боль-анализа, и определению мешают
V II. 4.1. Определение селена в отсутствие мешающих веществ
Реактивы
Дитизон, 0,01%-ный раствор в СС14 сохраняют в склянке из темного стекла под слоем сернистой кислоты. Перед применением из этого раствора путем разбавления готовят 0,002%-ный раствор.
Аммиак, водный раствор, разбавленный (1 : 100).
Гексацианоферрат(П) калия, 25%-ный водный раствор.
Калибровочный график. Для построения калибровочного графика берут растворы, содержащие 0; 0,5; 1,0; 2,0; 5,0; 10,0 мкг селена в 25 мл 11 н. серной кислоты, проводят через ход определения, начиная с экстракции дитизоном, одновременно с пробами. Экстракты проб и шкалы должны находиться в одинаковых условиях освещенности.
Ход анализа. В делительную воронку емкостью 100 мл помещают 5 мл исследуемого раствора, содержащего от 0,2 до 10 мкг селена, прибавляют, если нужно, воды до 5 мл и разбавляют 14 н. серной кислотой и водой до 25 мл с таким расчетом, чтобы раствор стал 11 н. по серной кислоте. Затем приливают 15 мл 0,002%-ного свежеприготовленного раствора дитизона и встряхивают в течение 30 с. После расслоения фаз сливают органический слой в другую делительную воронку, добавляют 20 мл раствора гексацианоферрата (II) калия и встряхивают в течение 2 мин. Еще раз сливают слой органического растворителя в чистую делительную воронку, добавляют раствор аммиака и встряхивают 10—20 с для удаления избытка дитизона. После разделения фаз органический слой переносят в кювету с толщиной слоя 0,3—1 см и измеряют оптическую плотность раствора при 420 нм.
Содержание селена находят по предварительно построенному калибровочному графику.
V II. 4.2. Определение селена в рудах [82, 190]
Селен из пробы руды извлекают азотной кислотой, выделяют его совместно с мышьяком, растворяют в смеси азотной и хлористоводородной кислот, выпаривают с серной кислотой и определяют селен в виде дитизоната.
Реактивы
Арсенат меди, раствор, содержащий в 1 мл 1 мг мышьяка и 100 мг сульфата меди.
Ход анализа. Навеску руды 2 г или меньше помещают в стакан емкостью 150 мл, прибавляют 35—40 мл азотной кислоты (пл. 1,4), при анализе сульфидных руд добавляют 0,05—0,1 г иодида калия или 0,1—0,2 г бертолетовой соли и оставляют на сутки. Затем упаривают на умеренно нагретой бане до 5—10 мл, приливают 10 мл серной кислоты, разбавленной (1:1), и упаривают до появления белых паров. Охлаждают, обмывают стенки стакана 5— 10 мл воды и повторяют упаривание. Приливают 25—30 мл воды, нагревают до кипения и нерастворимый остаток отфильтровывают, собирая фильтрат в стакан емкостью 150 мл. Фильтр с осадком промывают 2—3 раза небольшими порциями воды. Объем фильтрата не должен превышать 50 мл. Добавляют равный объем хлористоводородной кислоты (пл. 1,19), 1 мл раствора арсената. Затем в раствор вводят немного бумажной массы, нагревают до 80— 90 °C, в горячий раствор добавляют при перемешивании небольшими порциями гипофосфит калия или натрия до восстановления железа (III) и еще 1—2 г избытка. Нагревают до кипения, кипятят 10 мин и оставляют на 2—3 ч или до следующего дня.
Осадок отфильтровывают, промывают 4—5 раз горячей 3%-ной хлористоводородной кислотой и 3—4 раза горячей водой и на фильтре растворяют горячей смесью кислот (2—3 капли хлористоводородной и 5 мл азотной кислоты), приливая эту смесь небольшими порциями. На растворение обычно расходуется не более 5—8 мл смеси кислот.
Промывают фильтр небольшим количеством горячей воды, добавляют в фильтрат 2 мл серной кислоты, разбавленной (1 : 1), отмеривая пипеткой или бюреткой, и упаривают до появления паров серной кислоты. Ополаскивают стенки стакана водой и повторяют упаривание. После охлаждения жидкость переводят в пикнометр емкостью 10 мл или калиброванную пробирку емкостью 10 мл, доводят объем до метки водой, перемешивают, отбирают аликвотную часть и определяют селен, как указано в разделе VII. 4.1.
V II. 5. ОПРЕДЕЛЕНИЕ СЕЛЕНА С ПРИМЕНЕНИЕМ 1,4-ДИФЕНИЛТИОСЕМИКАРБАЗИДА [167, 237]
1,4-Дифенилтиосемикарбазид в слабокислой (хлористоводородной, сернокислой или азотнокислой) среде взаимодействует с селеном (IV) с образованием соединения, окрашенного в красно-бурый
цвет. Оптимальный pH для проведения реакции — 0—2. Образующееся соединение мало растворимо в воде, но хорошо экстрагируется органическими растворителями (дихлорэтан, хлороформ, четыреххлористый углерод, бензол, толуол и др.), образование его используется для фотометрического определения селена. В соединении соотношение селена и реагента равно 1 :2.
Мешающие вещества. 1,4-Дифенилтиосемикарбазид образует окрашенные комплексные соединения с Cu, Au, Pt, Pd, Rh, Cd, Ag,
Hg и другими металлами. Образующиеся соединения экстрагируются органическими растворителями и мешают определению селена. В определенных условиях теллур не мешает определению селена. Таким образом, при анализе технических объектов не-
S-7Z?-*
°2w 260 280 300 320 яо1&ол,нм обходимо предварительное отделение селена.
Рис. 53. Спектры поглощения Измерение оптической плотности.
хлороформных растворов 1,4- ~ .
дифенилтиосемикарбазида (/) Спектры поглощения хлороформных и его комплекса с селеном (2). растворов 1,4-ДИфенилтиосемикарбази-
да и его комплекса с селеном(IV)
представлены на рис. 53. Спектр поглощения хлороформного раствора комплекса характеризуется полосой поглощения с максимумами при 275 и 320 нм. При 275 нм сильно поглощает свет реагент, поэтому измерение оптической плотности растворов следует проводить при 320 или 410 нм (езао = 4,4 • 104; епо = 6,94-103).
Чувствительность метода составляет 4 мкг селена в 10 мл водного раствора или в 4 мл хлороформного экстракта.
VII. 5.1. Определение селена в отсутствие мешающих веществ
Реактивы
1,4-Дифенилтиосемикарбазид, насыщенный раствор (~10-2 М) в уксусной кислоте, разбавленной (3: 1).
Хлороформ, ч. д. а.
Стандартный раствор селена, см. раздел VII. 3.1.
Ход анализа. До 9 мл исследуемого раствора, содержащего 4—50 мкг селена (IV), помещают в делительную воронку емкостью 50 мл, прибавляют 0,5 мл 1 н. хлористоводородной кислоты, доливают водой до объема 9,5 мл. Добавляют 0,5 мл уксуснокислого раствора 1,4-дифенилтиосемикарбазида, перемешивают и оставляют на 10—15 мин, затем в воронку прибавляют 4 мл хлороформа и экстрагируют окрашенное соединение, встряхивая 1—2 мин. Хлороформный слой отделяют и измеряют оптическую плотность по отношению к хлороформному раствору сравнения, содержащему
все реагенты, кроме селена, в кювете с толщиной слоя 0,5 см при 320 нм или на ФЭК со светофильтром № 2.
Содержание селена находят по предварительно построенному калибровочному графику.
V II. 5.2. Определение селена в сульфидной руде [167]
Пробу руды растворяют, выделяют селен и теллур в элементном состоянии с помощью хлорида олова (II), осадок отделяют, переводят в раствор и определяют с помощью 1,4-дифенилтиосеми-карбазида.
Реактивы
Хлорид олова(П), 50%-ный раствор в хлористоводородной кислоте, разбавленной (1 : 1).
Мочевина, ч. д. а.
Ход анализа. Навеску руды 1—2 г помещают в термостойкую колбу емкостью 250 мл и маленькими порциями добавляют 15 мл концентрированной азотной кислоты. После прекращения бурной реакции приливают 30 мл концентрированной серной кислоты и нагревают до выделения ее паров. Охлаждают, осторожно добавляют 20 мл дистиллированной воды, после чего снова упаривают до появления паров серной кислоты.
К охлажденному остатку осторожно добавляют 10 мл дистиллированной воды и 10 мл хлористоводородной кислоты (пл. 1,19), нагревают для растворения сульфатов, отфильтровывают нерастворимый остаток и промывают его горячей хлористоводородной кислотой, разбавленной (1:20). Фильтрат нагревают до кипения, добавляют бумажную массу и осаждают селен и теллур раствором хлорида олова(II). Сначала приливают раствор SnCh до обесцвечивания раствора (восстановление железа), а затем избыток его 2—3 мл. Раствор кипятят в течение 3—5 мин для коагуляции осадка селена и теллура. Оставляют на холоду в течение 5 мин, после чего осадок селена и теллура отфильтровывают через неплотный бумажный фильтр и промывают горячей хлористоводородной кислотой, разбавленной (1 :20).
Фильтр с осадком переносят в колбу, в которой вели осаждение, и обрабатывают при осторожном нагревании 10 мл хлористоводородной кислоты (пл. 1,19) и 5—7 каплями HNO3 (пл. 1,40). После растворения осадка разбавляют раствор дистиллированной водой до 100 мл и отфильтровывают бумажную массу, промывая ее горячей хлористоводородной кислотой, разбавленной (1:20). К полученному раствору добавляют 1 г мочевины и немного отмытой бумажной массы, раствор нагревают и снова осаждают селен и теллур хлоридом олова(II).
Получённый осадок отфильтровывают, промывают горячей хлористоводородной кислотой, разбавленной (1:20), и растворяют в смеси концентрированных хлористоводородной (10 мл) и азотной
(5—7 капель) кислот. Раствор разбавляют дистиллированной водой и отфильтровывают бумажную массу, собирая фильтрат в мерную колбу емкостью 50, 100 или 200 мл в зависимости от содержания селена и теллура. Доводят дистиллированной водой до метки и хорошо перемешивают.
Из мерной колбы отбирают 5, 10, 20 или 25 мл раствора в зависимости от содержания селена в склянку с притертой пробкой емкостью 100 мл или в делительную воронку. Добавляют 0,1—0,2 г мочевины и хорошо взбалтывают. Прибавляют 1 каплю фенолфталеина и нейтрализуют концентрированным раствором аммиака до появления розовой окраски.
Избыток аммиака нейтрализуют 1 н. хлористоводородной кислотой до обесцвечивания, затем добавляют на каждые 10 мл раствора 1,0—1,5 мл 1 н. хлористоводородной кислоты и продолжают определение, как указано в разделе VII. 5.1.
V II. 6. ОПРЕДЕЛЕНИЕ СЕЛЕНА И ТЕЛЛУРА В ФОРМЕ ЗОЛЕЙ
Метод основан на том, что селен и теллур выделяют в элементном состоянии с образованием золей:
H2SeO3 + 2SnCl2 + 4НС1 —> Se + 2SnCl4 + ЗН2О
Н2ТеО3 + 2SnCl2 + 4НС1 —> Те + 2SnCl4 + ЗН2О
В качестве защитного коллоида применяют гуммиарабик или желатин. Кроме хлорида олова(II) [50] в качестве восстановителя применяют гипофосфит, а для восстановления селена и гидразин [50]. При восстановлении хлоридом олова(II) золи селена и теллура содержат значительные количества окислов олова [52].
Восстановление хлоридом олова(II) проводят в слабокислой среде с применением ацетатной буферной смеси или в солянокислой среде (5—6 н. НС1). В качестве катализатора чаще применяют сульфат меди, хотя рекомендовано применение также солей висмута, сурьмы и др.
Мешающие вещества. Определению селена и теллура мешают высокозарядные ионы, которые подвергаются гидролизу и выделяются в виде основных солей. Если вести восстановление в очень кислой среде, то вместе с селеном и теллуром может выделяться мышьяк. При содержании золота свыше 10 г/т его необходимо предварительно отделить. Чаще всего для этого применяют гидрохиноновый метод [50, 237].
Измерение оптической плотности. Оптическую плотность золей селена лучше измерять при 390 нм, а теллура — при 440 нм. Методы колориметрического титрования и разбавления не могут быть применены, а метод стандартной шкалы в этом случае мало пригоден. Цвет золя зависит от размера частиц, поэтому всегда необходимо строго придерживаться одних и тех же условий получения коллоидных растворов
Чувствительность метода — 0,03 мкг селена и 0,05 мкг теллура.
V II. 6.1. Определение селена в отсутствие мешающих веществ
Реактивы
Хлорид олова(П), 25%-ный раствор в 20%-ной хлористоводородной кислоте.
Желатина, 0,5%-ный раствор.
Сульфат сурьмы(Ш), 0,1 %-ный раствор в серной кислоте.
Стандартный раствор селена, см. раздел VII. 3.1.
Ход анализа. До 10 мл анализируемого раствора, содержащего 0,03—0,2 мг селена и 0,03—0,3 мг теллура, помещают в мерную колбу емкостью 25 мл, добавляют 10 мл хлористоводородной кислоты (пл. 1,19), 2 капли азотной кислоты (пл. 1,14), доводят водой до ~20 мл и перемешивают. Прибавляют 0,5 мл раствора сульфата сурьмы(III), 2 мл раствора желатины и 3—4 капли раствора хлорида олова(II), доводят до метки водой и перемешивают. Спустя 40 мин раствор наливают в кювету с толщиной слоя 2 см и измеряют оптическую плотность при 390 нм или пользуются синим светофильтром, если применяют ФЭК-М.
Содержание селена находят по предварительно построенному калибровочному графику, для построения которого пользуются стандартным раствором селена.
V II. 6.2. Определение селена и теллура в хлористоводородном растворе в отсутствие мешающих веществ
Реактивы
Стандартный раствор селена, см. раздел VII. 6.1.
Стандартный раствор теллура. Навеску 0,050 г металлического теллура растворяют в 10 мл концентрированной азотной кислоты и осторожно кипятят до удаления бурых паров. Раствор охлаждают, переносят в мерную колбу емкостью 500 мл, разбавляют до метки водой и хорошо перемешивают. Раствор содержит 100 мкг теллура в 1 мл.
Хлорид олова(П), 10%-ный раствор в 20%-ной (по объему) хлористоводородной кислоте.
Гуммиарабик, 4%-ный водный раствор.
Ход анализа. До 12 мл анализируемого раствора в 3 н. кислоте, содержащей 0,05—0,5 мг теллура(IV) или 0,1 —1,0 мг селена(IV), переносят в мерную колбу емкостью 25 мл, разбавляют, если нужно, до ~ 12 мл. 3 н. хлористоводородной кислотой и прибавляют при перемешивании 1 мл раствора хлорида олова(II). После выделения селена или теллура добавляют 2 мл раствора гуммиарабика, разбавляют водой до метки и перемешивают. Наполняют раствором кювету с толщиной слоя 1 см и измеряют оптическую плотность.
Содержание селена или теллура находят по предварительно построенным калибровочным графикам.
Аналогично готовят нулевой раствор со всеми реактивами, кроме селена или теллура.
V II. 6.3. Определение теллура при восстановлении гипофосфористой кислотой в отсутствие мешающих веществ
Теллур восстанавливают до свободного состояния с помощью гипофосфористой кислоты и измеряют оптическую плотность золя теллура.
Реакцию проводят при 0,01—0,3 н. концентрации кислоты. Од-
Рис. 54. Спектр поглощения золя теллура, восстановленного, гипофосфористой кислотой.
менение гуммиарабика, так ультрафиолетовом свете.
Чувствительность метода
нако оптимальная концентрация кислоты 0,1—0,2 н.
Измерение оптической плотности. Спектр поглощения золя теллура (рис. 54) характеризуется двумя полосами поглощения с максимумами при 240—290 нм и при 400— 450 нм. Максимумы поглощения сдвигаются в зависимости от размера получаемых коллоидных частиц. При измерении оптической плотности в ультрафиолетовой области для стабилизации золя обязательно при-как желатина сильно поглощает в
-0,05 мг в 25 мл конечного объема.
Реактивы
Гуммиарабик, 4%-ный водный раствор.
Гипофосфористая кислота, 3 М раствор.
Стандартный раствор теллура, см. раздел VII. 6.2.
Ход анализа. В коническую колбу емкостью 50 мл помещают анализируемый раствор, содержащий 0,05—0,4 мг теллура (IV) и 1—4 мг-экв хлористоводородной кислоты. Добавляют 2 мл раствора гуммиарабика и доводят объем водой до 20 мл. Раствор нагревают до кипения и при перемешивании прибавляют 3 мл раствора гипофосфористой кислоты. Поддерживают температуру смеси близкой к температуре кипения в течение 15 мин, затем охлаждают водой в течение 15 мин. Золь переводят в мерную колбу емкостью 25 мл, разбавляют до метки водой, перемешивают и измеряют оптическую плотность при 240—290 или 420 нм.
Содержание теллура находят по предварительно построенному калибровочному графику.
VII. 6.4. Определение селена при восстановлении гидразином в отсутствие мешающих веществ
Восстановление селена (IV) гидразином проводят при pH = = 8—9, так как в кислой среде золи селена быстро коагулируют, а при pH = 11 и выше реакция идет очень медленно.
Теллур не восстанавливается гидразином, но оптическая плот-
ность золя селена несколько повышается в присутствии теллура, возможно, вследствие образования соединения Se^Te [64].
Для стабилизации коллоидного раствора применяют гуммиарабик или желатину.
Спектр поглощения золя селена, восстановленного гидразином,
приведен на рис. 55. Спектр характеризуется с максимумом при 260—310 нм. Оптическую плотность растворов измеряют при 260 нм.
Чувствительность метода — 0,05 мг в 25 мл конечного раствора.
Реактивы
Гидразин гидрат, 85%-ный (17 Л1) раствор.
Гуммиарабик, 4%-ный водный раствор.
Стандартный раствор селена, см. раздел VII. 6.1.
Ход анализа. До 20 мл исследуемого раствора, содержащего 0,05—0,5 мг селена (IV), помещают в коническую колбу емкостью 50 мл, нагревают до кипения и быстро прибавляют при помешивании 1 мл раствора гидразин гидрата. Затем прибавляют 2 мл раствора гуммиарабика и про-
полосой поглощения
Рис. 55., Спектр поглощения золя селена, восстановленного гидразином.
должают нагревание раствора еще в течение
2—5 мин. Раствор охлаждают до комнатной температуры водой,
переносят в мерную колбу емкостью 25 мл, разбавляют водой до метки и перемешивают. Наполняют кювету с толщиной слоя 1 см и измеряют оптическую плотность при 260 нм. Измерение можно проводить в более длинноволновой области спектра, но при этом умень-
шается чувствительность определения.
Содержание селена находят по предварительно построенному калибровочному графику.
VII. 7. ОПРЕДЕЛЕНИЕ СЕЛЕНА ПО РЕАКЦИИ ОКИСЛЕНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Арилгидразины окисляются селенистой кислотой до диазоние-вых солей, которые при взаимодействии с ароматическими аминами образуют окрашенные азосоединения [138, 238, 239]. Из арил-гидразинов чаще других применяют фенилгидразин-м-сульфоно-вую кислоту, которая в кислой среде окисляется селенистой кислотой:
HO3S-
NH-NH2 • HCI + HSeO; + н
HO3S—С \—NE=NCr + Se + 3H2O
Эта реакция протекает в сильносолянокислом растворе (>4,5 н.) в течение 35—60 мин. Затем диазониевая соль сочетается с нафтиламином с образованием азокрасителя (4-амино-4'-сульфобензол-азо-нафталин):
продукта вой соли кислоты
сочетания диазоние-фенил-п-сульфоновой с аминонафталином.
Реакция образования красителя проходит при pH = 1,8—2,2.
Мешающие вещества. Определению селена мешают окислители (Сг2О|", Юз, СЮз, MnOJ, ВгО;, УОз, МпОГ, Fe3+ и др.), восстановители (Fe2+, Sn2+, S2 , I , S2O2 , SO2 , As111, SbIn, аскорбиновая кислота и др.), а также Сг3+, Со2+, Cu2+, Au111, Pt2+, Ag+, Hg2+. Теллур (VI) в 100-кратном избытке не мешает, а влияние теллура(IV) даже 50-кратных количеств необходимо учитывать. Селен(IV) реагирует аналогично селену(^), но более слабо. Железо и медь предварительно отделяют экстракцией в виде купферо-натов.
Измерение оптической плотности. Спектр поглощения продукта сочетания диазониевой соли с аминонафталином (рис. 56) характеризуется полосой поглощения с максимумом при 520 нм. Реагенты при этой длине волны свет. Полученный краситель устойчив
практически не поглощают в течение 6 ч.
Чувствительность определения — 0,25 мкг селена в 25 мл конечного раствора.
VII. 7.1. Определение селена в отсутствие мешающих веществ
Реактивы
Фенилгидразин-п-сульфоновая кислота, 0,5%-ный раствор.
1-Нафтиламин, 0,5%-ный раствор.
Ацетат натрия, 4 М раствор.
Стандартный раствор селена, см. раздел VII. 3.1.
Ход анализа. До 2 мл исследуемого раствора, содержащего 2—40 мкг селена, помещают в стакан емкостью 50 мл, добавляют 250
4 мл концентрированной хлористоводородной кислоты и 3 мл раствора фенилгидразин-/г-сульфоновой кислоты. Раствор оставляют на 40 мин. Затем добавляют 2 мл раствора 1-нафтиламина и 10 мл раствора ацетата натрия. Доводят pH до 1,8—2,2 и проверяют pH на потенциометре. Раствор переводят в мерную колбу емкостью 25 мл, доводят до метки водой, перемешивают и оставляют на 10 мин. Оптическую плотность измеряют при 520 нм по сравнению с раствором холостого опыта.
Содержание селена находят по предварительно построенному калибровочному графику.
V II. 7.2. Определение селена в присутствии
меди и железа
Медь и железо предварительно отделяют экстракцией в виде купферонатов и определяют селен, как указано в разделе VII. 1.
Реактивы
Купферон, 5%-ный свежеприготовленный раствор.
Хлороформ, ч. д. а.
Другие реактивы — указаны в разделе VII. 7.1.
Ход анализа. До 5 мл исследуемого раствора переносят в делительную воронку емкостыб 50 мл, добавляют хлористоводородной кислоты до 0,1 н., 1 мл свежеприготовленного раствора купферона и экстрагируют хлороформом двумя порциями по 10 мл. Затем к водной фазе добавляют хлористоводородной кислоты до 1 н., 1 мл раствора купферона и экстрагируют хлороформом 3 раза порциями по 10 мл.
Водную фазу переводят в стакан и нагревают до удаления хлороформа, охлаждают, доводят объем до 10 мл, перемешивают, берут аликвотную часть раствора и определяют селен, как указано в разделе VII. 7.1.
V II. 7.3. Определение селена в германии и двуокиси германия особой чистоты [238]
Пробу переводят в раствор и отгоняют германий в виде тетрахлорида в присутствии гидразина. Селен при этом находится в остатке. Затем селен переводят в раствор и отгоняют в виде тетрабромида, который поглощают водным раствором аммиака. Раствор упаривают и определяют селен, как указано в разделе VII. 7.1.
Реактивы
Гидразин гидрат, 25%-ный раствор.
Перекись водорода, 30%-ная.
Аммиак, 5%-ный водный раствор.
Бромид калия, 10%-ный раствор.
Другие реактивы — указаны в разделе VII. 7.1.
Ход анализа. Навеску анализируемого металла 2—5 г растирают в халцедоновой ступке и помещают в колбу кварцевого дистилляционного аппарата. Приливают 10 мл перекиси водорода и нагревают на водяной бане до полного окисления металла до двуокиси.
Выпаривают на водяной бане почти досуха, охлаждают и приливают по каплям при перемешивании 1 мл раствора гидразин гидрата. Оставляют до прекращения выделения пузырьков газа, приливают 5 мл раствора гидразин гидрата и выпаривают на водяной бане почти досуха. Наливают в колбу 20 мл 8 н. хлористоводородной кислоты и 2 мл раствора гидразин гидрата, присоединяют колбу к'Перегонному аппарату и отгоняют пока в колбе не останется 1—2 мл жидкости. Заменяют холодильник перегонного аппарата ловушкой с раствором аммиака и по каплям приливают 3 мл раствора бромида калия и 5 мл концентрированной серной кислоты. Отгоняют SeBr4 до тех пор, пока объем раствора в колбе не станет равным 5 мл.
Аммиачный раствор, содержащий селен, упаривают до объема 3—5 мл и определяют селен, как указано в разделе VII. 7.3.
V II. 8. ОПРЕДЕЛЕНИЕ ТЕЛЛУРА В ВИДЕ ИОДИДНОГО КОМПЛЕКСА
Теллур(IV) в солянокислом растворе взаимодействует с иодидом с образованием иодтеллуритных комплексов, растворы которых окрашены в желтый цвет [240]. Спектр поглощения и интенсивность окраски зависят от состава образующегося комплекса. Спектр поглощения комплексов с большим координационным числом сдвинут в длинноволновую область, а интенсивность окраски зависит от концентрации иодида. Наиболее интенсивно окрашен гексаиодидный комплекс теллура (IV). Общая константа нестойкости равна 4,5-10-8 [241].
Мешающие вещества. Определению мешают окислители, которые окисляют иодид, в том числе Cu2+, Fe3+, МпО4, СГ2О7, 5еОз~ и другие, выделяющие свободный иод, а также висмут, образующий окрашенный иодидный комплекс, сурьма(III) и другие ионы, образующие малорастворимые . окрашенные и неокрашенные иодиды (Pb2+, Hg?\ Hg2+, Ag+).
При определении теллура в присутствии селена его предварительно отделяют любым из методов, в том числе и иодидным. При концентрации иодида 0,02 н. и 0,5—1,0 н. хлористоводородной кислоте теллур не образует интенсивно окрашенного комплекса, но селен в этих условиях в течение 1 ч практически полностью выделяется. Необходимо иметь в виду, что теллур(IV) слабо растворим в воде, но при его концентрациях, которые обычно применяются в фотометрическом анализе, осадок теллура не образуется.
Количество иода, образующегося при взаимодействии с селеном, пропорционально содержанию последнего. Это дало возможность разработать косвенный метод определения селена [242]; выделившийся иод лучше экстрагировать органическим растворителем, после чего измеряют оптическую плотность. Неудобство этого варианта состоит в том, что при реакции выделяется селен, который трудно отделить, и приходится измерять суммарную оптическую плотность коллоидного раствора селена и раствора иода. Кроме того, в процессе работы иод частично выделяется в результате окисления иодида кислородом воздуха, поэтому лучше использовать не иодид щелочного металла, а иодид кадмия— более устойчивый по отношению к кислороду воздуха. Роль реакции окисления иодида возрастает при определении очень малых содержаний селена. В этом случае анализ заканчивают определением иодкрахмального соединения [242].
Измерение оптической плотности. Спектр поглощения иодидного комплекса теллура в оптимальных условиях определения теллура представлен на рис. 57. Этот спектр характеризуется несколькими полосами. Молярные коэффициенты составляют е285 = 9,2* 104 и 6413=7,2* 103. Оптическую плотность лучше из
мерять при 413 нм, хотя в этом случае несколько снижается чувствительность метода.
Чувствительность метода составляет 0,01 мг теллура в 25 мл конечного раствора.
V II. 8.1. Определение теллура в отсутствие мешающих веществ
Реактивы
Иодид калия, свежеприготовленный 2 М раствор. Раствор необходимо защищать от солнечного света.
Стандартный раствор теллура, см. раздел VII. 6.2.
Ход анализа. До 15 мл анализируемого раствора, содержащего 0,01—0,05 мг теллура (IV), помещают в мерную колбу емкостью 25 мл, разбавляют, если нужно, до 15 мл водой и добавляют 2,5 мл 2 н. хлористоводородной кислоты. Затем при перемешивании добавляют 5 мл раствора иодида калия. Раствор разбавляют до метки водой, перемешивают и измеряют оптическую плотность при 335 или 413 нм в кюветах с толщиной слоя 1 см. Растворы необходимо защищать от солнечного света. Измерение оптической плотности проводят после разбавления раствора и выдерживания
Рис. 57. Спектр поглощения иодидного комплекса теллура в условиях его определения.
в темном месте в течение 20 мин. В качестве раствора сравнения используют аналогично приготовленный холостой раствор, без теллура.
Содержание теллура находят по предварительно построенному калибровочному графику.
V II. 8.2. Определение теллура в стали [243]
Сталь растворяют в серной кислоте, при этом теллур остается в нерастворимом остатке. Остаток отфильтровывают и растворяют в смеси хлористоводородной и азотной кислот. Теллур восстанавливают хлоридом олова(II), отделяют, переводят в раствор и определяют в виде иодидного комплекса.
Реактивы
Хлорид олова(П). Растворяют при нагревании 100 г БпСЬ-ЗНгО в 250 мл концентрированной хлористоводородной кислоты и разбавляют водой до 1 л.
Стандартный раствор теллура, см. раздел VII. 3.1.
Ход анализа. Навеску стали 0,5—1,0 г растворяют при умеренном нагревании в 50—100 мл серной кислоты. После полного растворения стружки нерастворимый остаток отфильтровывают и промывают несколько раз горячей водой. Фильтр с осадком помещают в колбу, в которой проводили растворение навески, добавляют 10 мл концентрированной хлористоводородной кислоты и 5—6 капель концентрированной азотной кислоты и нагревают 2—3 мин, время от времени встряхивая колбу, до полного разложения нерастворимого остатка. Если нужно, повторно добавляют азотной кислоты и нагревание повторяют. Добавляют 25—30 мл горячей воды и отфильтровывают бумажную массу, которую промывают несколько раз горячей разбавленной хлористоводородной кислотой.
Фильтрат нагревают до кипения, добавляют 3 г мочевины и раствор хлорида олода(П) сначала до обесцвечивания раствора и затем еще 5 мл избытка, кипятят 10 мин для коагуляции элементного теллура. Добавляют небольшое количество бумажной массы. Доводят раствор до кипения и оставляют на 10—15 мин в теплом месте, после чего осадок отфильтровывают через фильтр «белая лента» и промывают горячей хлористоводородной кислотой, разбавленной (1 :20). Фильтр с осадком помещают в колбу, в которой вели осаждение, добавляют 10 мл концентрированной хлористоводородной кислоты и 5—6 капель концентрированной HNO3. Раствор нагревают, время от времени энергично встряхивая колбу. После полного растворения теллура раствор разбавляют дистиллированной водой до 20—30 мл, фильтруют в мерную колбу емкостью 50 мл и промывают фильтр горячей водой. Фильтрат в колбе охлаждают водопроводной водой под краном, добавляют 1,5 г мочевины, 5 мл раствора иодида калия, разбавляют дистилли
рованной водой до метки, хорошо перемешивают, выдерживают 0 темном месте в течение 20 мин и измеряют оптическую плотность раствора.
V II. 9. ОПРЕДЕЛЕНИЕ ТЕЛЛУРА С ПРИМЕНЕНИЕМ ДИЭТИЛДИТИОКАРБАМАТА И ЕГО ПРОИЗВОДНЫХ
Дитиокарбаматы R2NC(S)SMe (где Me — ион одновалентного металла) в кислой среде взаимодействуют с теллуром (IV) с образованием малорастворимых, окрашенных в желтый цвет комплексных соединений:
R\ //S /R^ /s\ \
4 )N-C\ + H2TeO3 + 4HC1 I Te + 4NaC1 + 3H^°
RZ ZSNa \RZ ^SZ/2
Под действием света комплексы разрушаются с образованием комплексов теллура(II) и соответствующих тиурамдисульфидов: /R\ ^Sx X /Rx ^Sx \ Rx ZS ZR
XN—Cx A Те —> XN—> Te+ N—С" /C—N\
\RZ ZSZ \RZ ZSZ J2 RZ ZS-SZ ZR
Чаще других для определения теллура применяют диэтилдитиокарбамат натрия [157, 158]. Дитиокарбаматные комплексы теллура количественно образуются в 3 н. хлористоводородной, 3 н. серной и 4 н. хлорной кислотах. Образующиеся соединения хорошо растворимы в органических растворителях: четыреххлористом углероде, хлороформе, бензоле и других [215, 244, 245]. Поэтому для фотометрического определения теллура используют
Рис. 58. Спектры поглощения диэтилдитиокар-баматного комплекса теллура в четыреххлэри-стом углероде (/) и диэтилдитиокарбаминовой кислоты (2).
экстракцию органическими растворителями или применяют ацетоно-водную среду (60% ацетона) [246].
Мешающие вещества. Ионы многих металлов образуют с дитиокарбаматами окрашенные комплексные соединения и мешают определению теллура. Селен также взаимодействует с дитиокарбаматами и мешает определению теллура, поэтому при определении теллура в присутствии селена строго регулируют pH раствора. Дитиокарбаматный комплекс теллура можно полностью экстрагировать четыреххлористым углеродом при pH = 8,5—8,7 и отде-
лить таким образом от селена, который взаимодействует с дитиокарбаматами только в кислой среде. При этом значении pH ЭДТА полностью маскирует Zn, Mn2+, Cd, Ni, Hg11, Со, Pb, WVI, MoVI. Смесь цианида и ЭДТА при pH = 8,5—8,7 практически полностью маскирует все ионы, в том числе и платиновые металлы [247], кроме меди, железа(III), олова, сурьмы и частично висмута.
Измерение оптической плотности. Спектр поглощения диэтил-дитиокарбаматного комплекса теллура в четыреххлористом углероде (рис. 58) характеризуется полосой поглощения с максимумом при 340 нм. Оптическую плотность измеряют при 340 нм, а также при 380—400 нм (ез4о = 1.55-104 и 8415 = 0,35-104).
Чувствительность метода—1 мкг теллура в 20 мл экстракта.
V II. 9.1. Определение теллура в отсутствие мешающих веществ
Реактивы
Стандартный раствор теллура, см. раздел VII. 6.2.
Диэтилдитиокарбамат натрия. Растворяют 1 г реактива в 100 мл воды, если нужно, раствор фильтруют, отделяя нерастворившийся остаток. Применяют свежеприготовленный раствор.
Хлороформ, ч. д. а.
Сульфат натрия, Na2SO4, безводный.
м-Крезоловый пурпурный. Растворяют 0,1 г ^-крезолового пурпурного и одну таблетку NaOH в 10 мл воды при нагревании. Раствор разбавляют до 100 мл.
Ход анализа. Исследуемый раствор, содержащий до 150 мкг теллура, помещают в коническую колбу емкостью 100 мл, прибавляют 0,25 мл НСЮ4 и 1,0 мл серной кислоты. Раствор упаривают до объема 1 мл, охлаждают, добавляют 50 мл воды и охлаждают до комнатной температуры. Затем прибавляют 2 капли раствора л4-крезолового пурпурного и нейтрализуют раствором аммиака до начала перехода красного цвета в желтый. Раствор переносят в делительную воронку емкостью 100 мл из темного стекла.
Добавляют 2 мл раствора диэтилдитиокарбамата натрия, раствор перемешивают, добавляют 20 мл хлороформа, закрывают пробкой, встряхивают, приоткрывают пробку для уменьшения давления и затем энергично взбалтывают в течение 30 с. Дают фазам разделиться и сливают большую часть хлор.оформного раствора в коническую колбу из темного стекла емкостью 50 мл, в которую помещено около 3 г сульфата натрия. Содержимое колбы перемешивают вращательным движением и измеряют оптическую плотность при 415 нм в кювете с толщиной слоя 5 см относительно хлороформного раствора сравнения, полученного аналогично, но без прибавления раствора теллура.
Содержание теллура находят по предварительно построенному калибровочному графику.
V II- 9.2. Определение теллура в свинце
Пробу переводят в раствор, теллур выделяют гипофосфористой кислотой, применяя в качестве коллектора соединение мышьяка (III). Затем теллур отделяют, переводят в раствор и определяют в виде диэтилдитиокарбамата.
Реактивы
Арсенит натрия. Растворяют 0,25 г AS2O3 в 10 мл концентрированного раствора гидроокиси натрия при нагревании. Раствор разбавляют до 200 мл.
Этилендиаминтетрауксусной кислоты динатриевая соль (ЭДТА). Растворяют 5 г в 250 мл воды при нагревании.
Смесь кислот. Хлорную и азотную кислоты смешивают в отношении (5:1) и перемешивают.
Другие реактивы — указаны в разделе VII. 9.1.
Ход анализа. Навеску анализируемой пробы 10 г помещают в коническую колбу емкостью 300 мл, добавляют 15 мл смеси кислот, накрывают и осторожно нагревают до растворения металла. Для удаления азотной кислоты раствор упаривают до появления обильных паров хлорной кислоты. Охлаждают, добавляют 50 мл воды, нагревают до слабого кипения и затем добавляют 50 мл хлористоводородной кислоты. Продолжают нагревание около 1 мин, время от времени перемешивая вращательным движением для предотвращения толчков, до коагуляции осадка. Охлаждают до 10—15 °C в бане со льдом, фильтруют через фильтр с белой лентой в коническую колбу емкостью 250 мл и промывают осадок 3—4 раза, добавляя хлористоводородную кислоту, разбавленную (1 : 1), тонкой струей. Осадок отбрасывают.
К фильтрату добавляют 2 мл раствора арсенита натрия, а затем 50%-ную гипофосфористую кислоту, перемешивают вращательным движением, накрывают часовым стеклом и осторожно кипятят в течение 5 мин. Охлаждают до 70—80°C, фильтруют через фильтр с синей лентой, промывают колбу и фильтр 3—4 раза хлористоводородной кислотой, разбавленной (1 : 1), при комнатной температуре. Фильтр с осадком, переносят обратно в колбу, в которой проводилось осаждение теллура. Собирают остатки теллура со стеклянной воронки кусочком фильтровальной бумаги, который помещают в ту же колбу. Добавляют 10 мл концентрированной HNO3 и 4 мл концентрированной НСЮ4, накрывают часовым стеклом и нагревают до разрушения бумаги. Когда окисление закончится, раствор упаривают до объема 2 мл, добавляют 2 мл H2SO4, упаривают до 1 мл и продолжают анализ, как описано в разделе VII.9.1..
V II. 9.3. Определение теллура в меди
Анализируемый образец переводят в раствор, выделяют теллур совместно с мышьяком с помощью гипофосфористой кислоты, затем теллур и мышьяк растворяют и определяют теллур в виде Диэтил дитиокарбамата.
Реактивы — указаны в разделе VII. 9.2.
Ход анализа. Навеску исследуемого образца меди 5 г помещают в коническую колбу емкостью 200 мл, медь растворяют в 25 мл смеси хлорной и азотной кислот и упаривают до появления обильных паров для удаления азотной кислоты. После охлаждения добавляют 50 мл воды и нагревают до растворения. Приливают 50 мл хлористоводородной кислоты, разбавленной (1 : 1), 2 мл раствора арсенита натрия, 15 мл гипофосфористой кислоты и затем продолжают, как указано в разделе VII. 9.2, но промывают осадок мышьяка и теллура хлористоводородной кислотой, разбавленной (1 : 1), до тех пор, пока в очередной порции промывного раствора при добавлении аммиака до сильно щелочной реакции не будет появляться голубая окраска. Это значит, что в осадке и в колбе, в которой велось осаждение мышьяка и теллура, остается не более 1 мг меди.
V II. 10. ОПРЕДЕЛЕНИЕ ТЕЛЛУРА С ПРИМЕНЕНИЕМ
3,5-ДИФЕНИЛ ПИРАЗОЛ ИН-1 -ДИТИОКАРБАМАТА НАТРИЯ [248]
Пиразолиндитиокарбамат натрия - (3,5-дифенилпиразолин-1-ди-тиокарбамат натрия) образует с теллуром более устойчивое соединение, чем диэтилдитиокарбамат натрия. Этот комплекс хорошо экстрагируется хлороформом в широких пределах кислотности от pH = 9 до 12 н. по хлористоводородной кислоте.
Мешающие вещества. Определению теллура мешают многие ионы, в том числе селен. Однако если вести определение при pH = = 8 в присутствии тартрата, то возможно определение теллура при стократном избытке селена. Применение маскирующей смеси, состоящей из тартрата, ЭДТА и цианида при pH = 8,0—9,0 позволяет определять теллур в присутствии Zn, Ni, Мп11, Fe111, Cu, Cd, Bi, As, Sn, Sb, Pd и Se. Таллий(I) и (III) необходимо предварительно отделить, что можно выполнить экстракцией с помощью этой же маскирующей смеси при pH = 14.
Измерение оптической плотности. Спектр поглощения раствора пиразолиндитиокарбамата теллура в хлороформе имеет полосу поглощения с максимумом при 413 нм (е = 2,04• 104). Оптическую плотность растворов измеряют при 413 нм.
Чувствительность метода — 0,5 мкг теллура в 25 мл экстракта.
VII. 10.1. Определение теллура в отсутствие мешающих веществ
Реактивы
3,5-Дифенилпиразолин-1 -дитиокарбамат натрия, 1%-ный водный раствор. Хлороформ, ч. д. а.
Стандартный раствор теллура, см. раздел VII. 6.2.
Ход анализа. До 50 мл исследуемого раствора, содержащего до 3 мкг теллура, помещают в делительную воронку емкостью 200 мл, устанавливают pH = 8,7—8,9, прибавляют 1—2 мл рас-256
твора 3,5-дифенилпиразолин-1-дитиокарбамата натрия и экстрагируют хлороформом порциями по 10, 10 и 5 мл. Экстракты фильтруют через сухой фильтр в мерную колбу емкостью 25 мл. Доводят до метки хлороформом, перемешивают и измеряют оптическую плотность раствора в кювете с толщиной слоя 5 см при 413 нм. При высоком содержании теллура берут кювету с толщиной слоя 1 см.
Содержание теллура находят по предварительно построенному калибровочному графику.
V II. 10.2. Определение теллура в селенистой кислоте
Мешающие ионы связывают маскирующей смесью (NaCN, тартрат и ЭДТА) и экстрагируют теллур в виде пиразолиндитиокарб-амата хлороформом.
Реактивы
Маскирующая смесь, 1 г ЭДТА, 5 г тартрата натрия и 10 г цианида натрия растворяют в воде и разбавляют до 100 мл.
Другие реактивы — указаны в разделе VII. 10.1.
Ход анализа. Навеску 0,5—1,0 г селенистой кислоты растворяют в минимальном количестве воды, добавляют 5 мл маскирующей смеси, доводят pH до 8,0—9,0 добавлением щелочи или кислоты, прибавляют 1—2 мл раствора реагента и продолжают определение, как указано в разделе VII. 10.1.
V II. 10.3. Определение теллура в селене
Навеску 0,5—1,0 г металлическдго селена в виде порошка растворяют в HNO3 (пл. 1,42) при очень слабом нагревании на водяной бане, добавляя несколько капель хлористоводородной кислоты. Кислый раствор переносят в делительную воронку, нейтрализуют раствором NaOH, затем доливают маскирующую смесь и доводят pH до ~8,5, добавляют 1—2 мл раствора 3,5-дифенилпиразолин-1-дитиокарбамата натрия и определяют, как указано в разделе VII. 10.1.
V II. 11. ОПРЕДЕЛЕНИЕ ТЕЛЛУРА В ВИДЕ КфМПЛЕКСА
С 5-МЕРКАПТО-3-(НАФТИЛ-2)-1,3,4-ТИАДИАЗОЛТИОНОМ-2 [227]
Аналог висмутола I и висмутола II — 5-меркапто-З-(нафтил-2'
1,3,4-тиадиазолтион-2
образует с теллуром малорастворимое в воде соединение желтого цвета, которое хорошо экстрагируется бензолом, циклогексаноном и хлороформом. Неводный раствор окрашен в желтый цвет. Состав соединения соответствует отношению теллура к реагенту (1:4).
* Мешающие вещества. Реактив в кислой среде образует соединения белого цвета с кадмием и железом (II), желтого — с оловом,
сурьмой, серебром, свинцом, медью, селеном, мышьяком и ртутью(П), оранжевого — с железом (III), палладием и висмутом; ртуть(I) восстанавливается реактивом до свободного состояния. Таллий(III) образует бурый осадок, который быстро переходит в лимонно-желтый, т. е. образуется £ „
соединение таллия(I). Соединение селена экстрагируется хлороформом при
pH = 4,5 (рис. 59). При pH = 5,1—5,3 и /
соотношении [Te]:[Se] = 1:200 соедине-
ние селена практически не экстраги- \ \
Рис. 59. Зависимость степени экстракции (Е) хлороформом от кислотности водной фазы:
1— 5-меркапто-3-фенил-2-тио-1,3,4-тиадиазол-тиои-2; 2 — соединения теллура; 3 — соединения селена.
Рис. 60. Спектры поглощения хлороформных растворов 5-меркапто-З-(нафтил-2)-1,3,4-тиадиазолтиона-2 (/), его дисульфида (2) и комплекса с теллуром (3).
руется и определению теллура не мешает. Определению теллура мешают 100-кратные количества таллия(1), ртути(Н) и (I), олова (II) и серебра. Применяя в качестве комплексанта ЭДТА, теллур можно определять в присутствии свинца, кадмия и таллия(III), а также двукратных количеств палладия.
Определению мешают окислители, которые окисляют реактив до дисульфида.
Измерение оптической плотности. Спектры поглощения хлороформных растворов 5-меркапто-З-(нафтил-2)-1,3,4-тиадиазолтио-на-2, комплекса теллура с этим реагентом и дисульфида реагента представлены на рис. 60. Для комплекса езгв = 2,955-104, для реагента еззо = 9,25-103, а для дисульфида еззо = 1,75-104. Таким образом, максимумы поглощения хлороформных растворов комплекса теллура и реагента почти совпадают. Кроме того, определению в сильной мере мешает дисульфид реагента, который поглощает свет в той же области. Однако при pH = 4,0 реагент не экстрагируется, в отличие от соединения теллура. С целью преду
преждения образования дисульфида применяют свежеприготовленный раствор реагента в небольшом избытке.
Оптическую плотность хлороформного раствора измеряют при 326 нм или со светофильтром № 2 (ФЭК-56) относительно раствора сравнения.
Чувствительность метода — 5 мкг теллура в 10 мл экстракта.
V II. 11.1. Определение теллура в отсутствие мешающих веществ
Реактивы
5-Меркапто-3-(нафтил-2)-1,3,4-тиадиазолтион, 0,1 %-ный свежеприготовленный водный раствор.
Буферный раствор, pH = 5,1—5,3. Смешивают 56,4 мл 0,2 н. СН3СООН и 45,6 мл 0,2 н. раствора аммиака.
Этилендиаминтетрауксусной кислоты динатриевая соль (ЭДТА), 10%-ный раствор.
Хлороформ, ч. д. а.
Стандартный раствор теллура, см. раздел VII. 6.2.
Ход анализа. До 40 мл нейтрализованного исследуемого раствора, содержащего 5—100 мкг теллура, помещают в делительную воронку емкостью 100—150 мл, прибавляют 10 мл буферного раствора, необходимое количество реактива и разбавляют, если необходимо. водой до 50 мл. Раствор перемешивают и трижды экстрагируют хлороформом порциями 5, 3 и 2 мл, экстракт сливают в узкий цилиндр емкостью 10 мл, разбавляют, если необходимо, до 10 мл хлороформом, перемешивают и измеряют оптическую плотность раствора при 326 нм относительно раствора сравнения.
Содержание теллура находят по предварительно построенному калибровочному графику.
V II. 11.2. Определение теллура в свинцовом концентрате
Навеску образца переводят в раствор кислотным разложением, прибавляют 100-кратные количества по отношению к теллуру селена и выделяют свободные селен и теллур при помощи гидразина и SnCl2. Осадок отделяют, растворяют в хлористоводородной кислоте в присутствии нескольких капель азотной кислоты, отбирают аликвотную часть раствора, нейтрализуют до pH = 5,1—5,3, прибавляют 5 мл 10%-ного раствора ЭДТА и определяют теллур, как указано в разделе VII. 11.1.
V II. 11.3. Определение теллура в сере
Навеску тонкорастертой черенковой серы 0,3—0,9 г смачивают водой, приливают 5—10 мл HNO3 (пл. 1,4) и осторожно прибавляют несколько капель брома. Содержимое нагревают на водяной
бане и продолжают прибавление брома до полного разложения навески. Добавляют 2—3 мл концентрированной H2SO4 и нагревают до появления белых паров. Остаток охлаждают, переносят в мерную колбу емкостью 25 мл и доводят водой до метки. Отбирают аликвотную часть раствора, нейтрализуют до pH = 5,3 по pH-метру, добавляют 5 мл раствора ЭДТА и ведут определение, как указано в разделе VII. 11.1.
V II. 12. ОПРЕДЕЛЕНИЕ ТЕЛЛУРА В ВИДЕ ТИОМОЧЕВИННОГО КОМПЛЕКСА
Теллур(IV) в кислой среде взаимодействует с тиомочевиной с образованием растворимого желтого комплекса теллура(II}:
zNH2 / znh2\2+
ТеО2- + 4S=C\ + 4Н+ —> Те S=C^ +
^NH2 \ \nh2/2
HN4 ^NH
+ V-s-s-c^ + 3H2O h2nz ^nh2
Константа нестойкости тиомочевинного комплекса теллура -2-Ю"3 [249].
Скорость реакции теллурита с тиомочевиной зависит от pH раствора, концентрации тиомочевины и температуры. Максимальный выход тиомочевинного комплекса теллура наблюдается в 0,8 н. НСЮ4, насыщенной тиомочевиной.
Мешающие вещества. Определению теллура с тиомочевиной мешают Se, [Hg2]2+, Pd, Os, Ph, Pt, Bi и большие количества Sb и Sn. Медь образует устойчивый тиомочевинный комплекс и не мешает определению. Селен(IV) при взаимодействии с тиомочевиной выделяется:
/NH2
H2SeO3 + 4S=C\ + 4НС1 —>
^nh2
2
' HN^ /NH
j;C—S—S—-2HC1
_H2n/ \NH2
+ Se 3H2O2
Течение л ой реакции зависит от кислотности и концентрации тиомочевины. Так, в 3 н. хлористоводородной кислоте и 10%-ном растворе тиомочевины выделение селена практически прекращается [249]. С повышением концентрации хлористоводородной кислоты количество тиомочевины, достаточное для полного удержания селена в растворе уменьшается. Выделение селена тиомочевиной в сильной мере зависит от температуры. При оптимальных концентрациях тиомочевины и хлористоводородной кислоты в интервале 20—60 °C выделяется примерно 80% от теоретически рассчитанного количества селена, С повышением температуры коли
чество выделяющегося в осадок свободного селена резко снижается и доходит до 30%. Необходимо отметить, что тиомочевина препятствует полному выделению селена с помощью других восстановителей, например иодида или аскорбиновой кислоты. Поэтому всегда в присутствии тиомочевины в растворе остаются миллиграммовые количества селена [250].
Теллур(IV) взаимодействует с тиомочевиной при комнатной температуре в течение нескольких минут, а теллур (VI) реагирует только в горячем растворе. Это использовано для дифференциального определения четырех- и шестивалентного теллура [251].
Пределы концентраций соответствующих кислот и тиомочевины для определения теллура(IV) представлены ниже [223]:
Концентрация кислоты
Концентрация гномочевины, %
Азотная кислота ..........
Серная кислота ........ . .
Фосфорная кислота .........
0,8-1,6 н. 10-12
0,8-2,0 н. 9-11
0,3-1,5 н. 10-12
При очень высокой кислотности раствора тиомочевина разлагается с выделением газообразных продуктов.
Измерение оптической плотности. Спектры поглощения тиомо-
чевинного комплекса теллура при 10%-ной концентрации тиомочевины и 1н. концентрации азотной, серной и 0,5 М фосфорной кислот представлены на рис. 61. Спектр поглощения характеризуется полосой поглощения с максимумом при 310 нм. Поэтому оптическую плотность измеряют при 310 нм или с фиолетовым светофильтром.
Чувствительность метода — 0,02 мг теллура в 25 мл конечного раствора; ее можно значительно повысить путем экстракции смешанного тиомочевинно-роданидного комплекса теллура трибутилфосфатом
Рис. 61. Спектры поглощения тиомочевинного комплекса теллура в 1 н. HNO3 (/), 1 н. H2SO4 (2) и 0,5 М Н3РО4 (3).
или экстракцией метилэтилкетоном тиомочевинного комплекса теллура с солью Рейнеке [252].
VII. 12.1. Определение теллура в отсутствие мешающих веществ
Реактивы
Тиомочевина, ч. д. а.
Стандартный раствор теллура, см. раздел VII. 6.2.
Ход анализа. До 20 мл исследуемого раствора, содержащего 0,02—1 мг теллура(IV), помещают в мерную колбу емкостью
25 мл. Добавляют достаточное количество концентрированной кислоты для доведения суммарной кислотности до указанных выше пределов, т. е. 1,25—2,5 мл концентрированной HNO3, 0,5—1,5 мл концентрированной H2SO4 или 0,5—2,5 мл 85%-ной H3PO4. Добавляют 2,5 г тиомочевины, разбавляют водой до метки, перемешивают и измеряют оптическую плотность раствора при 310 нм или с фиолетовым светофильтром в случае применения других приборов.
V II. 13. ОПРЕДЕЛЕНИЕ ТЕЛЛУРА С ПРИМЕНЕНИЕМ ПРОИЗВОДНЫХ ПИРАЗОЛОНА
Теллур (IV) в присутствии бромида взаимодействует с произ-
водными пиразолона (антипирин, диантипирилметан, диантипирил-
пропилметан и др.) с образованием малорастворимых смешанных комплексов. Состав комплексов с диантипирилметаном соответствует формуле (С2зН24О2М4Н)2[ТеВг6], с диантипирилметилмета-
V80 У,им
Рис. 62. Спектр поглощения раствора смешанного дианти-пирилметанбромидно-
ном—(С24Н3о0^4Н)2[ТеВгб], с диантипирил-пропилметаном — (С2бНзо02^4Н)2[ТеВг6] [44, 46, 203]. Эти соединения из солянокислых растворов (5—7 н. НС!) экстрагируются дихлорэтаном и окрашивают раствор в желтый цвет. Эта реакция использована в фотометрических методах определения теллура.
Мешающие вещества. Производные пиразолона взаимодействуют со многими комплексными галогенидными ионами с образованием малорастворимых смешанных комплексов. Поэтому спределению мешают кадмий, таллий(III), висмут, сурьма, золото и другие ионы металлов. Применение ЭДТА устраняет
го комплекса теллура вредное влияние многих посторонних ионов, в дихлорэтане. кроме золота(Ш). Золото(Ш) осаждается
производными пиразолона из кислых бромидных и хлоридных растворов, а теллур(IV) не осаждается из солянокислых растворов. Это позволяет предварительно отделить золото экстракцией из солянокислых растворов, а затем определять теллур из бромидных растворов. Хотя бромидный комплекс селена
не осаждается основаниями, но в условиях экстракции теллура частично экстрагируется, поэтому его предварительно осаждают аскорбиновой кислотой.
Бромидные комплексы теллура взаимодействуют с производными пиразолона при 1,5 — 6 М концентрации бромистоводородной
кислоты.
Измерение оптической плотности. Спектр поглощения дихлор-этапового раствора смешанного диантипирилметанбромидного комплекса теллура представлен на рис. 62. Спектр характеризуется
полосой поглощения с максимумом при 450 нм. Оптическую плотность растворов измеряют на спектрофотометре при 450 нм или с синим светофильтром в случае применения других приборов.
Чувствительность метода — 10 мкг теллура в 5 мл экстракта.
V II. 13.1. Определение теллура в отсутствие мещающих веществ [44]
Реактивы
Бромистоводородная кислота, 40%-ный раствор.
Бромид калия, 5 М раствор.
Диантипирилметан, 1%-ный раствор в 0,5 н. H2SO4. Вместо диантипирил-метана можно брать такой же концентрации раствор диантипирилметилметана, диантипирилпропилметана или диантипирилфенилметана в уксусной кислоте, разбавленной (1 : 1).
Дихлорэтан, ч. д. а.
Стандартный раствор теллура, см. раздел VII. 6.2.
Ход анализа. До 10 мл исследуемого раствора, содержащего 10—100 мкг теллура, помещают в делительную воронку емкостью 50 мл, добавляют 5 мл бромистоводородной кислоты, 4 мл раствора КВг (концентрация бромид-ионов в конечном растворе должна быть не меньше 5 М, кислотность не ниже 1,5 н.) и 1 мл раствора диантипирилметана.
Избыток реагента не влияет на полноту экстракции соединения теллура. Разбавляют, если нужно, водой до 20 мл, перемешивают, прибавляют 5 мл дихлорэтана и встряхивают в течение 2 мин. Слой дихлорэтана отделяют и измеряют оптическую плотность раствора при 450l нм в кювете с толщиной слоя 1 см относительно дихлорэтана.
Содержание теллура находят по предварительно построенному калибровочному графику.
V II. 13.2. Определение теллура в
свинце [44]
Пробу свинца переводят в раствор, теллур соосаждают с гидроокисью железа, золото отделяют осаждением смешанного диан-типирилметанхлоридного комплекса из солянокислого раствора, железо(Ш) восстанавливают аскорбиновой кислотой и экстрагируют теллур в виде смешанного диантипирилметанбромидного комплекса.
Реактивы
Хлорид железа(Ш), 3%-пый раствор в 10%-ной хлористоводородной кислоте.
Диантипирилметан, 1%-ный раствор в 1 н. хлористоводородной кислоте.
Окись цинка, свежеприготовленная.
Аскорбиновая кислота, ч. д. а.
Винная кислота, ч. д. а.
Другие реактивы — указаны в разделе VII. 13.1.
Ход анализа. Навеску металлического свинца 10 г растворяют в азотной кислоте, разбавленной (1:1), к раствору добавляют 1 мл раствора хлорида железа (III), осаждают гидроокиси сначала раствором аммиака, а затем окисью свинца, создавая pH = 5. Осадок отфильтровывают через бумажный фильтр, промывают горячей водой и растворяют гидроокиси в хлористоводородной кислоте, разбавленной (1:1), в колбе, где проводилось осаждение. Далее раствор разбавляют водой, чтобы концентрация хлористоводородной кислоты была около 3 М, добавляют 10 мл раствора диантипирилметана, выпавшему осадку дают созреть в течение 5—10 мин, затем отфильтровывают его через бумажный фильтр, промывают 5%-ной хлористоводородной кислотой с небольшой добавкой осадителя. Фильтрат собирают в мерную колбу емкостью 100 или 200 мл в зависимости от содержания теллура. В делительную воронку отбирают аликвотную часть 5 мл, добавляют 5 мл воды, сухую аскорбиновую кислоту в десятикратном количестве по отношению к железу, немного винной кислоты и затем продолжают определение, как указано в разделе VII. 13.1.
V II. 13-3. Определение теллура в селене [253]
Хлоридный комплекс теллура(IV) с диантипирилпропилмета-ном количественно экстрагируют дихлорэтаном из солянокислых растворов в широких пределах кислотности. Хлоридный комплекс селена (IV) при этом полностью остается в водной фазе. Таким образом можно отделить любые количества теллура от селена.
Для определения теллура хлоридный комплекс переводят в бромидный, экстрагируют смешанный диантипирилпропилметан-бромидный комплекс и измеряют оптическую плотность полученного раствора.
Реактивы
Диантипирилпропилметан, 5%-ный и 1%-ный растворы в уксусной кислоте, разбавленной (1 : 1).
Этилендиаминтетрауксусной кислоты динатриевая соль (ЭДТА), 0,02 М раствор.
Аскорбиновая кислота, свежеприготовленный раствор, содержащий 1 г в 20 мл воды.
Другие реактивы — указаны в разделе VII. 13.1.
Ход анализа. Навеску металлического селена 0,5 г растворяют в 15—20 мл концентрированной азотной кислоты, добавляют 5 мл H2SO4 и выпаривают до появления белых паров. После охлаждения осторожно прибавляют 5 мл воды и снова выпаривают до появления паров серной кислоты, охлаждают и остаток переводят в мерную колбу емкостью 50 мл хлористоводородной кислотой, разбавленной (1:1). Отбирают аликвотную часть 5 или 10 мл (в зависимости от содержания теллура) полученного раствора в делительную воронку емкостью 50 мл, прибавляют 1—2 мл 5%-ного раствора диантипирилпропилметана, хлористоводородную кислоту 266
до объема 20 мл, 5 мл дихлорэтана и встряхивают в течение 1 мин.
После разделения фаз экстракт сливают в чистую делительную воронку емкостью 50 мл. К оставшейся водной фазе добавляют еще 5 мл дихлорэтана и снова встряхивают в течение 1 мин, после чего экстракт отделяют. Дихлорэтановые экстракты объединяют, водный раствор отбрасывают, а экстракты промывают 2 раза хлористоводородной кислотой, разбавленной (1 : 1), порциями по 10 мл.
Теллур извлекают из объединенного экстракта встряхиванием с 10 мл раствора ЭДТА. После разделения фаз дихлорэтан сливают. К оставшемуся водному раствору добавляют 5 мл НВг и оставляют на 1 мин. Затем прибавляют 0,5 мл раствора аскорбиновой кислоты, 1 мл 1%-ного раствора диантипирилпропилметана и продолжают анализ, как указано в разделе VII. 13.1.
V II. 13.4. Определение теллура в свинце и висмуте [46]
Пробу металла растворяют в азотной кислоте и экстрагируют теллур в виде хлоридного комплекса с диантипирилпропилметаном в дихлорэтан, связывая металлы.ЭДТА. Затем реэкстрагируют теллур раствором ЭДТА в водную фазу и определяют в виде дианти-пирилпропилметанбромидного комплекса.
Реактивы
Этилендиаминтетрауксусной кислоты динатриевая соль, 0,1 М и 0,02 М растворы.
Диантипирилпропилметан, 5 и 1%-ный растворы в уксусной кислоте, разбавленной (1 : 1).
Дихлорэтан, ч. д. а.
Стандартный раствор теллура, см. раздел VII. 6.2.
Бромистоводородная кислота, 40%-ная перегнанная.
Ход анализа. Навеску металла 0,2—0,5 г растворяют в 20— 25 мл азотной кислоты. Раствор упаривают почти досуха, обмывают стенки колбы водой и выпаривают досуха. Сухой остаток растворяют в 15—40 мл 0,1 М раствора ЭДТА при нагревании. Добавляют концентрированную хлористоводородную кислоту в объеме, равном объему прибавленного раствора ЭДТА, с тем, чтобы среда по хлористоводородной кислоте была (1 : 1). Переносят в делительную воронку емкостью 100 или 200 мл, добавляют 2 мл 5%-ного раствора диантипирилпропилметана, перемешивают, добавляют 5 мл дихлорэтана и встряхивают в течение 1 мин.
После разделения фаз экстракт сливают в чистую делительную воронку емкостью 50 мл. К оставшейся водной фазе добавляют еще 5 мл дихлорэтана и встряхивают в течение 1 мин. Дихлорэтановые экстракты объединяют и дважды промывают 10 мл хлористоводородной кислоты, разбавленной (1:1); водный слой отбрасывают.
Теллур из органической фазы дважды экстрагируют 0,02 М раствором ЭДТА порциями по 10 мл. После расслаивания жидкостей дихлорэтан сливают, водные растворы объединяют, переводят в мерную колбу емкостью 50—100 мл и доводят до метки 0,02 М раствором ЭДТА. Отбирают из колбы аликвотную часть 5 мл, добавляют 5 мл 0,02 М раствора ЭДТА, 10 мл бромистоводородной кислоты, 1 мл 1%-ного раствора диантипирилпропилме-тана, 5 мл дихлорэтана и встряхивают в течение 1 мин. После разделения фаз экстракт фильтруют через сухой фильтр и измеряют оптическую плотность раствора относительно холостого раствора при 450 нм в кювете с толщиной слоя 1 см.
Содержание теллура находят по предварительно построенному калибровочному графику.
V II. 13.5. Определение теллура в меди [46]
Пробу растворяют в азотной кислоте и экстрагируют теллур в виде хлоридного комплекса с диантипирилпропилметаном в дихлорэтане. Затем теллур реэкстрагируют водой и определяют в виде диантипирилпропилбромидного комплекса.
Реактивы
Аскорбиновая кислота, свежеприготовленный раствор, содержащий 1 г в 20 мл воды.
Бромид калия, насыщенный водный раствор.
Другие реактивы, кроме растворов ЭДТА, указаны в разделе VII. 13.4.
Ход анализа. Навеску 10 г металлической меди растворяют в 50 мл азотной кислоты, разбавленной (1:3), раствор переносят в мерную колбу емкостью 200 мл, доводят до метки водой и перемешивают. Большая навеска пробы необходима в связи с тем, что теллур неравномерно распределяется в металлической меди. Отбирают 10,0 мл раствора, упаривают с 5 мл серной кислоты, разбавленной (1 : 1), до появления ее паров. Обмывают стенки водой и вновь упаривают до появления паров серной кислоты. Остаток растворяют в 20 мл хлористоводородной кислоты, разбавленной (1 : 1), раствор переносят в делительную воронку емкостью 100 мл, прибавляют 2 мл 5%-кого раствора диантипирилпропилметана и 5 мл дихлорэтана, встряхивают в течение 2 мин. После разделения фаз органический слой сливают в чистую делительную воронку. К оставшейся водной фазе добавляют еще 5 мл дихлорэтана и встряхивают в течение 1 мин.
Дихлорэтановые экстракты объединяют и дважды промывают 10 мл хлористоводородной кислоты, разбавленной (1:1), а водный раствор отбрасывают. Затем теллур реэкстрагируют 10 мл воды. Реэкстракцию повторяют 2—3 раза, водные растворы собирают в мерную колбу емкостью 50 мл, разбавляют до метки водой и перемешивают. При содержании теллура 0,01—0,02% отбирают 5 мл из мерной колбы, прибавляют 8 мл раствора бромида калия, 268
0,5 мл раствора аскорбиновой кислоты, перемешивают, прибавляют 3 мл бромистоводородной кислоты, 1 мл 1 %-кого раствора диан-типирилпропилметана и 5 мл дихлорэтана и встряхивают в течение 1 мин. Экстракт отделяют от водного раствора, фильтруют и заканчивают определение, как указано в разделе VII. 13.4.
У II. 13.6. Определение теллура в цинковом электролите [46]
Теллур предварительно отделяют с помощью хлорида олова (II), а затем определяют в виде диантипирилпропилбромидного комплекса.
Реактивы
Аскорбиновая кислота, 5%-ный раствор.
Бромид калия, насыщенный раствор.
Другие реактивы — указаны в разделе VII. 13.4.
Ход анализа. Отбирают 500 мл нейтрального электролита и прибавляют концентрированную хлористоводородную кислоту до 10%-кого ее содержания. Нагревают до кипения, прибавляют бумажную массу и осаждают теллур и селен 20 мл хлорида олова (II). Раствор осторожно кипятят до коагуляции осадка и оставляют на 8—10 ч. Осадок отфильтровывают через маленький (5—7 см) фильтр с бумажной массой, промывают 4—5 раз 5%-ной хлористоводородной кислотой, растворяют на фильтре в 20 мл горячей хлористоводородной кислоты с 8—10 каплями азотной кислоты. Раствор собирают в колбу емкостью 100 мл, прибавляют 2 мл серной кислоты, разбавленной (1:1), и выпаривают до паров серной кислоты. Охлаждают, стенки колбы осторожно обмывают водой и вновь выпаривают до паров серной кислоты.
Охлажденный раствор переносят в мерную колбу емкостью 25 мл при помощи серной кислоты, разбавленной (1 :8), этой же кислотой доводят до метки и перемешивают. Отбирают 5 мл раствора, помещают в делительную воронку емкостью 50 мл, добавляют 0,5 мл аскорбиновой кислоты, 5 мл 0,02 М раствора ЭДТА, 7 мл насыщенного раствора бромида калия, 10 мл бромистоводородной кислоты, 1 мл 1 %-кого раствора диантипирилпропилме-тана, 5 мл дихлорэтана и встряхивают в течение 1 мин. Органическую фазу фильтруют через сухой фильтр и заканчивают определение, как указано в разделе УП. 13.4.
V II. 14. ОПРЕДЕЛЕНИЕ ТЕЛЛУРА С ПРИМЕНЕНИЕМ
РОДАМИНОВЫХ КРАСИТЕЛЕЙ
Родаминовые красители взаимодействуют с хлортеллуритом или бромтеллуритом с образованием комплексных соединений. Цвет комплексов подобен цвету красителей. Эти комплексы хорошо экстрагируются низкополярными органическими растворителями и
применяются в фотометрическом анализе. Впервые было предложено экстрагировать хлортеллурит родамина С из 2,0—2,5 н. хлористоводородной кислоты смесью бензола и эфира (3:1) и определения заканчивать люминесцентным методом [189]. Лучшими из родаминовых красителей оказались бутилродамин С и этилрод-амин С [254]. Экстракцию бромтеллуритного комплекса с бутил-родамином С проводят смесью бензола и бутилацетата (5:1) из 10 н. по H2SO4 и 0,18 н. по КВг раствора [190]. В случае применения этилродамина С (этилового эфира родамина С) максимальное извлечение теллура происходит из раствора, содержащего в 1 л 9,0—9,5 г-экв H2SO4, 0,13—0,15 г-экв/л В г- и 0,01 %-кого раствора красителя. Заканчивают определение люминесцентным или спектрофотометрическим методом.
Мешающие вещества. Смесью бензола и бутилацетата в условиях экстракции комплекса бутилродамина с бромтеллуритом экстрагируются и мешают определению теллура индий, тал-лий(Ш), сурьма(У), золото(Ш), ртуть(1) и (II), железо(Ш), медь(1), олово(П) и (IV), серебро и висмут [191]. Селен(1У) в этих условиях не мешает определению теллура, поэтому при определении теллура предварительно отделяют теллур и селен гипофосфитом. При этом в осадок переходят золото, ртуть и частично со-осаждается железо.
Для удаления ртути предварительно прокаливают навеску пробы с восстановленным водородом металлическим железом или разлагают ее щелочным сплавлением. Если содержание ртути до 100 мкг в пробе, то она легко улетучивается в виде хлорида при обработке сернокислого раствора концентрированной хлористоводородной кислотой, одновременно удаляется и селен. Золото выделяется в виде металла при разложении пробы серной кислотой и упаривании до паров серной кислоты, его отфильтровывают вместе с нерастворимым остатком пробы. Железо(Ш) восстанавливают аскорбиновой кислотой до железа(II). Однако при этом происходит медленный процесс восстановления теллура. Таким образом, экстракцию следует проводить сразу после прибавления аскорбиновой кислоты.
Важными являются тщательное разрушение органических веществ, которые окрашивают экстракт, и отделение следов меди. Медь(II) в процессе определения восстанавливается до меди(1), которая мешает определению. Органические вещества обычно разрушают при разложении пробы.
В случае применения этилродамина С небольшие количества мешающих ионов устраняют промывкой экстрактов серной кислотой. При этом теллур переходит в водную фазу, а металлы, которые образуют прочные бромидные комплексы, остаются в органической фазе в виде комплексов с красителем. Затем к водной фазе, содержащей теллур, прибавляют бромид и краситель, экстрагируют комплекс теллура органическим растворителем и определяют теллур.
Рис. 63. Спектр поглощения бензольного раствора бромтеллурита с бутилродамином С.
Измерение оптической плотности. Спектр поглощения бензольного раствора бромтеллурита с бутилродамином С представлен на рис. 63, он характеризуется полосой поглощения с максимумом при 565 нм. Поэтому оптическую плотность экстрактов измеряют при 565 нм. Условные молярные коэффициенты бензольных растворов комплексов равны ебв5 = 7,7-104 для бутилрод-аминового С комплекса и Б5во = 6,5-1О4 для этилродаминового С комплекса. Однако необходимо иметь в виду, что теллур в этих условиях экстрагируется не полностью, поэтому приведенные молярные коэффициенты поглощения являются условными.
Чувствительность метода — 0,1 мкг в 5 мл экстракта. Такая высокая чувствительность метода не может быть практически реализована при анализе различных материалов, так как погрешности, связанные с неполным отделением мешающих элементов, иногда достигают значений, эквивалентных 0,2—0,3 мкг теллура. Поэтому ность метода — 0,5 мкг теллура в 5 мл экстракта.
реальная чувствитель-
V II. 14.1. Определение теллура в отсутствие мешающих веществ
Реактивы
Этилродамин С, 10%-ный раствор.
Аскорбиновая кислота, ч. д. а.
Бензол, ч. д. а.
Стандартный раствор теллура. Помещают 10 мл стандартного азотнокислого раствора теллура (см. раздел VII. 6.2) в стакан емкостью 100 мл, прибавляют 5 мл H2SO4, разбавленной (1 :8), и упаривают до паров серной кислоты. Охлаждают, осторожно обмывают стенки стакана небольшим количеством воды и выпаривают до паров серной кислоты, затем эту операцию повторяют. Остаток переносят в мерную колбу емкостью 100 мл с помощью той же кислоты, которой доводят раствор до метки. Этот раствор содержит 10 мкг теллура в 1 мл.
Бромид калия, 9%-ный раствор.
Ацетон, ч. д. а.
Ход ацализа. До 12 мл исследуемого 14,8 н. по H2SO4 раствора, содержащего от 0,5 до 12 мкг теллура, помещают в делительную воронку емкостью 50 мл, если нужно, разбавляют 14,8 н. серной кислотой до 12 мл, приливают 12 мл бензола, 4 мл раствора этил-родамина С и 4 мл раствора бромида калия; экстрагируют в течение 30 с. Переводят водный раствор в другую воронку, приливают 12 мл бензола и встряхивают 30 с. Тщательно отделяют водную фазу и объединяют экстракты в одной из воронок. Приливают 12 мл 14,8 н. серной кислоты, 4 мл раствора этилродамина С и, встряхивая 30 с, реэкстрагируют теллур. Отделяют реэкстракт и
два раза промывают его 10—12 мл бензола. Переводят раствор в пробирку с притертой пробкой, приливают 12 мл бензола, 4 мл раствора бромида калия и экстрагируют в течение 30 с. Спустя 10—15 с отбирают пипеткой* с грушей 10 мл экстракта в предварительно высушенную пробирку, содержащую 5 мл апетона. Перемешивают и измеряют оптическую плотность при 560 нм в кювете с толщиной слоя 3 см.
V II. 14.2. Определение теллура в рудах и других материалах
Предварительно выделяют теллур и селен из 6 н. по хлористоводородной кислоте раствора гипофосфитом совместно с мышьяком, используемым в качестве коллектора. Из мешающих элементов в осадок попадают ртуть, золото, платина. Кроме того, осадок всегда содержит часть адсорбированных примесей: железо, медь, олово (до 0,5% относительно содержания в исходном растворе) и сурьма (0,05% относительно содержания в исходном растворе). Выделенный теллур переводят в раствор и* определяют с помощью этилродамина С.
Реактивы
Бромистоводородная кислота, 7 н. раствор.
Арсенит натрия, раствор, содержащий 1 мг мышьяка в 1 мл.
Другие реактивы — указаны в разделе VII. 14.1.
Ход анализа. Навеску 2—3 г при анализе горных пород или 1 г при анализе руд и минералов помещают в стакан емкостью 100 мл, прибавляют 35—40 мл азотной кислоты (пл. 1,4) (при анализе сульфидных руд добавляют 0,05—0,1 г иодида калия или 0,1—0,2 г бертолетовой соли) и оставляют на сутки. При высоком содержании ртути навеску пробы предварительно смешивают с порошком восстановленного водородом железа и прокаливают или разлагают навеску пробы щелочным сплавлением. Раствор пробы упаривают на умеренно нагретой бане до 5—10 мл, приливают 10 мл серной кислоты, разбавленной (1:1), и упаривают до появления белых паров. Охлаждают, обмывают стенки стакана 5 — 10 мл воды и повторяют упаривание. Приливают 25—30 мл воды, нагревают до кипения и отфильтровывают нерастворимый остаток, собирая фильтрат в стакан емкостью 150 мл. Промывают фильтр с осадком 2—3 раза небольшими порциями горячей воды. Объем фильтрата не должен превышать 50 мл. Добавляют равный объем хлористоводородной кислоты (пл. 1,19), 1 мл раствора арсенита натрия и 0,1 г сульфата меди. Затем вводят немного бумажной массы, нагревают до 80—90 °C, в горячий раствор добавляют небольшими порциями при перемешивании гипофосфит натрия или калия до восстановления железа(III) и еще 1—2 г избытка. Нагревают до кипения, кипятят 10 мин и оставляют па 2—3 ч или до следующего дня. Остаток отфильтровывают, промы-272
вают 4—5 раз горячей 3%-ной хлористоводородной кислотой и 3—4 раза водой и растворяют на фильтре горячей смесью концентрированных кислот (2—3 капли хлористоводородной кислоты и 5 мл азотной кислоты), приливая эту смесь небольшими порциями. На растворение осадка обычно расходуется не более 5— 8 мл смеси кислот.
Фильтр промывают небольшим количеством горячей воды, добавляют в фильтрат 12 мл 15 п. H2SO4, упаривают до появления белых паров; добавляют 7—8 мл воды и 3—4 мл 7 н. НВг. Снова медленно упаривают до появления паров; упаривание повторяют после вторичного добавления воды и НВг, при этом отгоняются летучие бромиды сурьмы и ртути. Переводят раствор в делительную воронку, доводят объем водой до 12 ± 1 мл, приливают 12 мл бензола и продолжают анализ, как описано в разделе VII.14.1.
V II. 15. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ СЕЛЕНА И ТЕЛЛУРА
Металлические селен и теллур растворяются в концентрированной серной кислоте с образованием окрашенных продуктов присоединения ТеБОз и SeSO3 [255]. Спектр соединения селена характеризуется двумя полосами поглощения с максимумами при 420 и 680 нм, теллура — полосой поглощения с максимумом при 520 нм. Спектры поглощения соединений селена и теллура перекрываются, поэтому при определении необходимо предварительное их разделение. Метод характеризуется малой чувствительностью и требует строгого соблюдения условий проведения определения.
2-Меркаптобензтиазол в солянокислой среде (6,5—10 н. НС1) взаимодействует с селеном (IV) с образованием комплексного соединения желтого цвета [НО, 256]. В сернокислом растворе молярный коэффициент поглощения меньше; в азотнокислом растворе происходит выделение свободного селена. В нейтральной и щелочной средах селен не взаимодействует с 2-меркапгобензтиазолом.
Раствор комплекса поглощает в ультрафиолетовой области (е370 = 9,48-103). Комплекс экстрагируется СС14, но при этом чувствительность реакции понижается. Определению мешают многие ионы.
2-Меркаптобензимидазол с селеном (IV) образует соединение желтого цвета (еззо = 1,05-104), которое экстрагируется смесью (1:5) бутилового спирта и хлороформа [136, 152]. Определению мешают многие ионы.
Тиогликолевая кислота и ее производные [152, 160, 172] в. 1—5 М растворе хлористоводородной кислоты взаимодействуют с теллуром (IV) и селеном(IV) с образованием комплексных соединений, окрашенных в желтый цвет. Эти комплексы экстрагируются эфиром, н-бутанолом, амилацетатом и бутилметилкетоном. Спектры растворов этих комплексов характеризуются полосами поглощения с максимумом при 260 нм (егбо = 1,6-103 для селена и
6260 = 3,3-102 для теллура). Однако селен мешает фотометрическому определению теллура, а теллур — определению селена. Кроме того, их определению мешают многие другие ионы.
Висмутиол II и его производные в 2 н. хлористоводородной или ,серной кислотах взаимодействуют с селеном(IV) и теллуром (IV) с образованием соединений желтого цвета, которые экстрагируются четыреххлористым углеродом и хлороформом и используются для фотометрического определения этих элементов [217, 257]. Определению мешают многие ионы; селен мешает определению теллура и наоборот.
4,5-Диамино-6-тиопиримидин при pH = 1,5—2 взаимодействует с селеном(1\т) с образованием растворов, окрашенных в желтый цвет [258]. Селен при этом, по-видимому, восстанавливается до элементного состояния. Спектр характеризуется полосой поглощения с максимумом при 380 нм (езво = 1,92-104). Определению селена этим методом мешают многие ионы.
Циклогексанон в среде 7 н. хлористоводородной кислоты образует с селеном(IV) кетоно-хлорокомплекс, окрашенный в желтый цвет (ез45 = 5,6-103) [259]. Определению мешают Те, Со, Fein и Си11.
Селеномолибденовая гетерополикислота образуется в азотнокислой среде с соотношением Se : Mo = 1 : 6 [260]. Теллур (IV) в оптимальных условиях образования селеномолибденовой кислоты не образует с молибденом гетерополикислоты и не усиливает интенсивность желтой окраски раствора селеномолибденовой гетерополикислоты [261], но ускоряет образование осадка из исследуемых растворов* при стоянии. По-видимому, в данном случае имеет место образование тройного гетерополикомплекса селен (VI)-теллур (IV) -молибдат.
Спектр раствора селеномолибденовой кислоты характеризуется полосой поглощения с максимумом при 380 нм.
Тетраметилтиурамдисульфид (или бис-диметилтиокарбамидди-сульфид)
S S
II И
(CH3)2N—С—S—S—С—N(CH3)2 (I)
и тетраэтилтиурамдисульфид (или бис-диэтилтиокарбамиддисуль-фид)
S S
(C2H6)2N—(I— S—S—(!—N(C2H6)2 (II)
взаимодействуют с теллуром(IV) с образованием желтых нерастворимых в кислых водных растворах соединений. Соединение теллура с реагентом I хорошо экстрагируется хлороформом, бензолом и ксилолом [262], а с реагентом II — четыреххлористым углеродом [262], окрашивая растворы в желтый цвет. Раствор соединения теллура с реагентом I характеризуется спектром с двумя 274
полосами поглощения с максимумами при 380 и 390 нм (езво = = 6,35-103, езэо = 4,25 • 103). Раствор соединения теллура с реагентом II характеризуется спектром поглощения с максимумом при 278 нм, а максимум поглощения реактива — при 274 нм.
Определению теллура с реагентом I мешают Hg, Си, Bi, Fe, Cr, Sb, с реагентом II —Си, Se,v, Hg2+, NO3. В последнем случае допустимо присутствие 15-кратных количеств Bi, 50-кратных — Со, 100-кратных — Ni и 200-кратных — Fe111.
Теллуритовольфрамовая гетерополикислота H^TeO^FbWO^a]-• 13НгО восстанавливается до теллуритовольфрамовой сини. При этом желтый цвет раствора переходит в синий. В качестве восстановителей применяют углеродистую сталь и другие мягкие восстановители в среде 0,04—0,1 н. хлористоводородной кислоты [183]. Сильными восстановителями (гидрохинон, гидразин, SnCk) теллуритовольфрамовая гетерополикислота разрушается с выделением элементного теллура.
Определению мешают многие компоненты, образующие с вольфрамом прочные гетерополикислоты.
Фосфоротеллуристомолибденовая гетерополикислота окрашивает раствор в желтый цвет и применяется для фотометрического определения теллура [185]. Состав комплекса соответствует соотношению [Р]:[Те]:[Мо] = 1:1: 11.
Производные триоксифлуорона взаимодействуют с теллуром с образованием малорастворимых соединений с соотношением [Те]: реагент—1:2 [263, 264]. В качестве стабилизатора применяют желатину.
Определению теллура мешают многие ионы.
Для определения теллура используются также косвенные методы. Феррин — комплекс 1,10-фенантролина с Feni, восстанавливается теллуром(IV) до ферроина [265, 266]:
TeOj" + 2Fe(C12H8N2)®+ + Н2О —> ТеО2" + 2Fe(C12H8N2)2+ + 2Н+
Эта реакция используется в косвенном' фотометрическом определении теллура. Определению теллура мешают многие ионы.
*
ЛИТЕРАТУРА
1. GlemserO., HaeselerR., Naturwis., 47, 467 (1960).
2. Рябчиков Д. И., Назаренко И. И., Усп. хим., 33, 108 (1964).
3. Назаренко И. И., Ермаков А. Н. Аналитическая химия селена и теллура. М., «Наука». 1971. С. 252.
4. Б а б к о А. К , М и т ю р е в а Т. Т., ЖНХ, 6, 421 (1961).
5. Lenh е г V., Honiberger A. W., J. Am. Chem. Soc., 30, 387 (1908).
6. Bode Н., Z. anal. Chem., 153, 335 (1956).
7. Lenh er V., К а о C. H., J. Am. Chem. Soc., 47, 769 (1925).
8. Lenh er V., Kao С. H., J. Am. Chem. Soc., 47, 772 (1925).
9. Johnson R. A., Kwan F. R., Anal. Chem., 23, 651 (1951).
10. Lenh er V., Kao C. H., J. Am. Chem. Soc., 47, 2454 (1925).
11. Kotowska M„ Szczepk'owska-Mamczarczyk L, Rudy i metale niezel., 13, 410 (1968).
12. A a rem ae A., Assarssoh G. O., Anal. Chem., 27, 1155 (1955).
13. Jackwerth E., Z. anal. Chem., 235, 235 (1968).
14. Тимофеева В. К., Сб. науч. тр. Гос. н.-и. ин-та цвет, мет., 27, 22 (1967).
15. Vydra F., Pribil R., Acta Chim. Acad. Sci. Hung., 28, 297 (1961).
16. Furco A., Scatturin V., Giacometti G., Nature, 183, 601 (1959).
17. Cotton F. A., Bannister E., Barnes R., Holm R., Proc. Chem. Soc., May, 158 (1959).
18. Issleib V., Doll G., Z. anorg. allgem. Chem., 305, 1 (1960).
19. Issleib V., Wenschu G., Z. anorg. allgem. Chem., 305, 15 (1960).
20. Д a p б и н я н M. В., Капанцяп Э. E., Арм. хим. ж., 21, 103 (1968).
21. Гайбакян Д. С., А т у р я и М. М., Арм. хим. ж., 21, 1015 (1968).
22. Г е л ь м а н Е. М„ ЖАХ, 23, 736 (1968).
23. Павлова В. Н:, Мурашова В. И., Зав. лаб., 34, 392 (1968).
24. Q u г е s h i, М a t h и г К. N., Anal. chim. acta, 41, 560 (1968).
25. S a b и г о I., К е n d z i г о Н., Japan Analyst, 12, 257 (1963).
26. U r u m a s a, Bull. Chem. Soc. Japan, 36, 301 (1963).
27. I n a r i d a M., J. Chem. Soc. Japan, 80, 273 (1959).
28. I n а г i d a M., J. Chem. Soc. Japan, 80, 280 (1959) .
29. Ina rid a M., Hikime S., Yosduda H., Urunasa X., Japan Analyst, 8,531 (1959).
30. I n a r i d a M., J. Chem. Soc. Japan, 79, 721 (1958).
31. Hikime S., Bull. Chem. Soc. Japan, 33, 761 (1960).
32. Стрельникова H. П., Зав. лаб., 28, 1193 (1962).
33. T h о m p s о n С. E., Zakin H. W., Geol. Surv. Profess. Paper, № 475-B, 164 (1963).
34. A n a 1 у G., Acta Chim. Acad. Sci. Hung., 25, 391 (1960).
35. Павлова В. П., Васильева Н. Г„ Кашли некая С. Э., Зав. лаб., 27,965 (1961).
36. N a k a g a w a G., J. Chem. Soc. Japan, 81, 1255 (1960).
37. W е а 11 е С. Z„ Analyst, 85, 133 (1960).
38. Barcza Z., Madyar Kern, folyoirat, 69, 248 (1963).
39. Pohl F. A., H а к n e с к t T., P c e s e M., пат. 1077645, 17/111, 1960.
40. H a n s о n С. K., Anal. Chem., 29, 1204 (1957).
41. Jordanov N., Havesonl., Z. anal. Chem., 248, 296 (1969),
42. Jordanov N., F u t e к о v L., Taianta, 12, 371 (1965).
43. БусевА. И., ХоангМиньТяу, ЖАХ, 18, 1370 (1963).
44. Б у се в А. И, Бабенко H. Л„ ЖАХ, 18, 972 (1963).
45. Б у с е в А. И., Бабенко Н. Л., Хоанг Минь Т я у, ЖАХ, 18, 1094 (1963).
46. БусевА. И., Бабенко Н. Л„ Ч е п и к М. Н., ЖАХ, 19, 871 (1964),
47. Р о 1 i о с k Е. N., Anal. Chim. Acta, 40, 285 (1968).
48. Гиллебранд В. Ф. и др. Практическое руководство по неорганическому анализу. Пер. с англ. Под ред. Ю. Ю. Лурье, Госхимиздат, 1957. См. с. 354.
49. М у р а ш о в а В. И., С у ш к о в а С. Г., ЖАХ 24, 729 (1969).
50. ВолковС. Т„ Зав. лаб., 5, 1438 (1936).
51. 3 е м е л ь В. К-, Зав. лаб., 5, 1438 (1936).
52. Ш а хов а А. С., Зав. лаб., 11, 893 (1945).
53. Файн бе р г С. Ю. Анализ руд цветных металлов. М., Металлургиздат, 1953. См. с. 542.
54. Футеков Л. П., Научн. тр. Высш. пед. ин-т, Пловдив, 9, 107 (1971).
55. М a n е s с h i S., G а 11 a z z i C., Anal. chim. acta, 54, 461 (1971).
56. Jilek A., V rest al J., Ha viz J., Chem. Zvesti, 10, 110 (1956).
57. 3 а й к о в с к и й Ф.* В. Современные методы анализа в металлургии. М., Металлургиздат. 1955. См. с. 223.
58. С а х а р о в А. А., ЖАХ, 15, 614 (I960).
59. D i n g w а 11 D., Williams W. D., J. Pharmacy and Pharmac, 13, 12 (1961).
60. Корен май И. М., Глазунова 3. И., Тр. по хим. и хим. технол., Горьковского ун-та, вып. 2, 368 (1962).
61. Т а р а я н В. М„ А в а к я н Т. Т., Зав. лаб., 27, 967 (1961).
62. Echegaray R. М., Qu ins ре Р. Z., Bol. Soc. quim, Peru, 25, 179 (1959).
63. F e i g 1 F„ Anal. chim. acta, 24, 501 (1961).
64. L e о n t о w i t c h N., Chim. Anal., 41, 56 (1959).
65. Leonto witch N., Chim. Anal., 42, 329 (1960).
66. Futekov L., Atanasova B., Natura. Ecole norm, super. Plovdiv, 3, 65 (1970).
67. Zagorski Z. P., Cyrankowska M. M., Taianta, 2, 380 (1959).
68. T a p а я н В. M., А руста м я н Ж. М., Изв. АН АрмССР. Сер. хим. наук, 15, 415 (1962).
69. Johnson R. A., Kwan F. Р., Wes tl aka G. D., Anal. Chem., 25, 1017 (1953).
70. К i b a T., An az a J., Ha chi no H., Bull. Chem. Soc. Japan, 32, 454 (1959).
71. Wild F. E., Atomic Energy. Res. EstabL, 1959, N : C/R 2666; РЖХим, 1960, 51706.
72. J о h n s о n R. A., Andersen B. R., Anal. Chem., 27, 120 (1955).
73. Б e л о п о л ь с к а я Т. Л. В сб.: «Материалы по геологии и полезным ископаемым Северо-Востока СССР», вып. 14, Магадан, 1960. См. с. 165.
74. Павлова В. Н., Зав. лаб., 28, 166 (1962).
75. Kumpikaite V., Salkauskas М., Тр. Каунасского политехи, ин-та, 11, 49 (1959).
76. Г л а д ы ш е в а В. П., Зав. лаб., 24, 275 (1958).
77. Б л ю м И. А., Глазкова А. Ф. В сб.: «Химические, физико-химические и спектральные методы исследования руд редких и рассеянных элементов». М., Госгеолтехиздат, 1961. См. с. 8.
78. М а р к о в а Н. В. В сб.: «Химические, физико-химические и спектральные методы исследования руд редких и рассеянных элементов». М., Госгеолтехиздат, 1961. См. с. 75.
79. Б л ю м И. А., Глазкова А. Ф. В сб.: «Химические, физико-химические и спектральные методы исследования руд редких и рассеянных элементов». М., Госгеолтехиздат, 1961. См. с. 13.
80. Васильева П. И., Воронкова М. А., Бюл. науч.-техн, информ. Гос. геол, ком-та СССР, ВИМС, № 1 (56), 14 (1965).
81. Hinsberg О., Вег., 22, 2895 (1889).
82. Щербов Д. П., Иванкова А. И., Гладышева Г. П., Зав. лаб., 33, 683 (1967).
83. Ш к р о б о т Э. П., Шабаршина Н. И., Сб. науч. тр. Гос. н.-и. ин-та цветн. мет., № 28, 18 (1968).
84. Н о s t е J., G i 11 i s J., Anal. chim. acta, 12, 158 (1955).
85. Hoste J., Anal. Chim. Acta, 2, 402 (1948).
86. Cheng K. L„ Anal. Chem., 28, 1738 (1956).
87. S t a n s о n R. E., McDonald A. J., Analyst, 90, 497 (1965).
88. C h a n Y. K-, R i 1 e у J. P., Anal. chim. acta, 33, 36 (1965).
89. Бело польская T. Л., Тр. Всес. н.-и. политех, ин-та, 117, 85 (1964).
90. V е а 1 е С. R„ Analyst, 85, 130 (1960).
91. Vea 1е С. R., Analyst, 85, 133 (1960).
92. Kujawa R„ Z. Phys. Chem., 221, 269 (1962).
93. Tacko Danzuka, Keihci Ueno, Anal. Chem., 30, 1370 (1958).
94. В a r c z a L., Z s i n d e 1 у S., Z. anal. Chem., 199, 117 (1964).
95. G eb a u h г W., S p a n g A., Z. anal. Chem., 175, 175 (1960).
96. Cheng K. L., Chem. Anal., 45, 67 (1956).
97. Luke C. L„ Anal. Chem., 31, 572 (1959).
98. 3 а й к о в с к и й Ф. В., Бюл. н.-т. информации Мин-ва геол, и охраны недр СССР, № 6, 72 (1959).
99. Jamamoto М., Sakai Н., Anal. chim. acta, 32, 370 (1965).
100. Ягнятинская Г. Я., Шитарева Г. Г., В сб.: «Металлургия и химическая промышленность Казахстана», № 2, 131 (1962).
101. Васильев П. И., Воронкова М. А., В сб.: «Методы химического анализа минерального сырья», № 8, 171 (1965).
102. Иванова А. И., Блюм И. А., Зав. лаб., 27, 371 (1961).
103. Handley R., Johnson С. М., Anal. Chem., 31, 2105 (1959).
104. Dye W. В. и др., Anal. Chem., 35, 1687 (1963).
105. Kelleher W. J., Johnson M. J., Anal. Chem., 33, 1429 (1961).
106. W a t к i n s о n J. K., Anal. Chem., 32, 981 (1960).
107. P а гкег C. A., Harvey Z. G., Analyst, 86, 54 (1961).
108. West Ph. W„ Ci merman Ch., Anal. Chem., 36, 2013 (1964).
109. Barcza L., Z. anal. Chem., 199, 10 (1964).
110. Barcza L., Sommer L., Z. anal. Chem., 192, 304 (1963).
111. Bock R, Detlef J., Z. anal. Chem., 200, 81 (1964).
112. Джонсон P. А. В кн.: «Колориметрические (фотометрические) методы определения неметаллов». Пер с анг. Под ред. А. И. Бусева. М., Издатин-лит, 1963. См. с. 383.
113. Н а з а р е н к о И. И. В сб.: «Новые методы исслед. по анализу редкоме-тальн. минералов руд и горн, пород», М., «Наука», 1970. См. с. 41.
114. Russell В. G. е. ol„ Taianta, 14, 957 (1967).
115. D о m е n ес h R., Chim. anal., 51, 440 (1969).
116. Стенина Н. И. и др., Сб. тр. Уральского н.-и. хим. ин-та, вып. 26, 88 (1971).
117. Р а г к е г С. А., Н е г v е у L. G., Analyst, 87, 558 (1962).
118. Lott Р. F. е. о1., Anal. Chem., 35, 1159 (1963).
119. W a t к i n s о п J. Н., Anal. Chem., 38, 92 (1966).
120. Орлова E. С., Назаренко И. И., Баренбаум М. Е., Зав. лаб., 36, 926 (1970).
121. Н а 11 R. J„ G u р t а Р. L., Analyst, 94, 292 (1969).
122. Dickey D. W. е. ol., Mikrochim. acta, № 3, 605 (1969).
123. F г а п к - С h a u L., Taianta, 11, 1019 (1964).
124. Tanaka М„ Kawashima Т., Bull. Chem. Soc. Japan, 37, 1085 (1964).
125. Крейнгольд С. У., Сосенкова Л. И., В сб.: «Методы анализа хим. реактивов и препаратов». М., вып. 19, 37 (1971).
126. Назаренко И. И., Кислов А. М„ Кислова И. В., В сб.: «Методы анализа хим. реактивов и препаратов». М., вып. 19, 32 (1971).
127. Langmyhr F. J., Dahl J., Anal. Chim. Acta, 29, 377 (1963).
128. Langmyhr F. J., Omang S. H., Anal. Chim. Acta, 23, 565 (1960).
129. Demeye re D„ Hoste J., Anal. chim. Acta, 27, 288 (1962).
130. Sawicki E., Anal. Chem., 29, 1376 (1957).
131. Mabuchi H., Nakahara H., Bull. Chem. Soc. Japan, 36, 151 (1963).
132. Сушкова С. Г., Мурашова В. И., В сб.: «Исследование в области аналитической химии редких и благородных металлов». Свердловск, Уральский политех, ин-т, сб. 163, 35 (1967).
133. lordanov N., Daskalova К., Докл. Болг. АН, 23, 969 (1970).
134. Kasterka В., Doborowlski J., Chem. anal. (PRL), 15, 303 (1970).
135. Мурашова В. И., Сушкова С. Г., ЖАХ, 19, 1503 (1964).
136. Бусев А. И., ХоангМинь Тяу, ЖАХ, 17, 1091 (1962).
137. Kirktright F. J., Н. J. Uoe, Anal. Chem., 35, 808 (1963).
138. Ригин В. И., Мельниченко Н. Н., Яницкий В. К-, Зав. лаб., 33, 1370 (1967).
139. Ar iy oshi Hisae, Kiniwa Michi о, Toci Kyoji, Taianta, 5, 112, (1960).
140. Throo p L. J., Anal. Chem., 32, 1807 (1960).
141. Toci K-, Ito K., Taianta, 12, 773 (1965).
142. Tanaka M., Kawashima T., Taianta,. 12, 211 (1965).
143. Щ e p б о в Д. П., Иванова А. И„ Гладышева Г. П. В сб.: «Исслед. и разработка фотометрич. методов для определения микроколичеств элементов в минеральн. сырье». Алма-Ата, 1967. См. с. 10.
144. К u n t е N. S., R a n a d е S. N., Indian J. Chem., 8, 370 (1970).
145. Goszczynska Н., Kowalczyk М., Chem. anal. (PRL), 15, 561 (1970).
146. В a res el D., Corinth U., Stahr А., пат. ФРГ, кл. 421, 3/01, gOln), № 1270842, заявл. 28.07.64, опубл. 16.01.69; РЖХим, 969, 22Г170П.
147 Ш к р о б о т Э. П., Ш е б а р ш и н а Н. И., Зав. лаб., 35, 417 (1969).
148. Zs in del у S., Barcza L., Acta chim. Acad, scient. hung., 62, 125 (1969).
149. Nesvadba P., Acta geol. et georg. Univ. Comemianae. Geol., № 15, 165 (1968).
150. Лебедь H. Б., Панталер P. П., Семенова Л. H., Тр. комиссии no аналит. хим. АН СССР, 16, 223 (1968).
151. Smoczkieurczowa A., Augustyniak J., Meissner W., Chem. anal. (Polska), 12, 629 (1967).
152. В u s e v A. I., Taianta, 11, 485 (1964).
153. Murakami T., Ishii E., Koguo Kagaki Zasshi, 66, 1652 (1963); C. A., 1964, 15133h.
154. Suzuki M„ J. Chem. Soc. Japan, Ind. Chem. Sect., 56, 323 (1953); цит. no РЖХим, 1956, 1169.
155. MandravelC., Gutul M., Rev. chim., 19, 734 (1968).
156. M i 1 a z z о G., M e z i E., Ann. Chimica, 52, 858 (1952).
157. Bode H., Z. anal. Chem., 143, 182 (1954).
158. Bode H., Mosenthin H., Z. anal. Chem., 164, 232 (1958).
159. БусевА. И., ХоангМинь-Тяу, ЖНХ, 7, 88 (1962).
160. Б у с е в А. И., X о а н г М и н ь - Т я у, ЖАХ, 18, 361 (1963).
161. Вега В. С., С h a k г a b а г 11 у М. М., Analyst, 93, 50 (1968).
162. Туткувене В. Е., Раманаускас Е. И., Зав. лаб., 35, 1045 (1969).
163. Looyenga R. W., Boltz D. F., Anal. Letters, 2, 491 (1969).
164. Mabuchi H„ Nakahara H., Bull. Chem. Soc. Japan, 36, 151 (1963).
165. Marhenke K-, San dell E. B., Anal. Chim. Acta, 38, 421 (1967).
166. Сушкова С. Г., Мурашова В. И., ЖАХ, 21, 1475 (1966).
167. Мурашова В. И., Сушкова С. Г., Бакунина Л И., Зав. лаб., 33, 280 (1967).
168. Степин В. В. и др. Тр. Всес. н.-и. ин-та станд. образцов и спектр, эталонов. М., «Металлургия», 1967. См. с. 109.
169. Овсеп ян К- Н., Тараян В. М., Шапошникова Г. Н., Изв. АН СССР. Отд. хим. н., 18, 225 (1965).
170. О в с е п я н Е. Н., Шапошникова Г. Н., Г а л ф а я и Н. Г., ЖНХ, 12, 2411. (1967).
171. Багбанлы Н. Л. и др., Уч. зап. Азерб. ун-та. Сер. хим. наук, № 1, 18 (1966).
172. Kirkbrigt J. F., Anal. Chim. Acta, 35, 116 (1966).
173. Пацук В. В., ЖАХ, 12, 509 (1967).
174. Пацук В. В., Субботина Т. С., Сб. науч. тр. Свердловск. Филиал Моск, ин-та народного хозяйства, вып. 1, 42 (1967).
175. W h е 11 е г L„ Z. anorg. Chem., 3, 428 (1893).
176. Rosen geim A., Traube A., Z. anorg. Chem., 91, 75 (1915).
177. Klein D., Bull. Soc. Chim. France, 2, 169 (1884).
178. Г а н e л и н a E. III., ЖНХ, 7, 1570 (1962).
179. Procad S., Pothak K-, J. Indian Chem. Soc., 42, 373 (1965).
180. Ганелина E. ILL, Резникова T. H.. ЖНХ, 8, 1891 (1963).
181. Ганелина E. Ш„ Нервяткин a H. И., ЖАХ, 10, 894 (1965).
182. Полотебнова H. А., Деркач Л. В., Тр. комиссии по аналит. хим., АН СССР, 16, 30 (1968).
183. Га не л и н а Е. Ш„ ЖНХ, 18, 555 (1963).
184. Ку Тхань Лонг, Судаков Ф. П., Шахова 3. Ф., ЖАХ, 19, 734 (1964).
185. Ку Тхань Лонг, Судаков Ф. П., Шахова 3. Ф., ЖАХ, 19, 968 (1964).
186. Rust Е„ Вег., 30, 2828 (1897).
187. Lony A., Dundrook R., J. Amer. Chem. Soc., 44, 614 (1922).
188. Щербов Д. П., Иванкова А. И., Зав. лаб., 24, 1346 (1958).
189. Щербов Д. П., Иванкова А. И., Зав. лаб., 24, 667 (1958).
190. Иванкова А. И., Блюм И. А., Зав. лаб., 27, 371 (1961),.
191. Владимирова В. М. и др Зав. лаб., 29, 1419 (1963).
192. Шитарева Г. Г., Уч. зап. центр, н.-и. ин-та оловянной промышл., 2 45 (1963),
193. Блюм И. А. Экстракционно-фотометрические методы анализа с применением основных красителей. М, «Наука», 1970. См. с. 142.
194. Р а м а н а у с к а с Э. И, Ж и л е н а й т е М. В., Науч. тр. высш, учебн. заведений ЛитССР. Хим. и хим. технол., № 12, 27 (1970).
195. Мурашова В. И, Скрип чук В. Г., ЖАХ, 27, 340 (1972).
196. Раманаускас Э. И, Жиле найте М. В., Науч. тр. высш, учебн. заведений ЛитССР. Хим. и хим. технол., 11, 57 (1970).
197. Киш П. П., К р е м е п е в а С. Г., ЖАХ, 25, 2200 (1970).
198. Б л ю м И. А, Душина Т. К. Тезисы докладов V конференции работников лабораторий Казахстана и Средней Азии. Алма-Ата, 1958. См. с. 79.
199. Иванкова А. И. В сб. «Исследования и разработка фотометрических методов определения микроколичеств элементов в минеральном сырье». Алма-Ата, 1967. См. с. 98.
200. Шулюнене А. К-, Бу нике не Л. В., Раманаускас Э. И., Тр. по хим. и хим. технол., Горький, вып. 3 (24), 135 (1969).
201. Р а м а н а у с к а с Э. И., Шулюнене А. К., ЖАХ, 23, 1859 (1968).
202. Шулюнене А. К-, Раманаускас Э. И., Тарозайте Р. К., Науч тр. высш, учебн. заведений ЛитССР. Хим. и хим. технол., 11, 63 (1970).
203. Пилипенко А. Т., Капустин А. И., Укр. хим. ж., 30, 9 (1964).
204. Б у се в А. И., Акимов В. А., Гусев С. И., Усп, хим., 34, 565 (1965).
205. Б у се в А. И., Бабенко Н. Л., ЖАХ, 18, 926 (1963).
206. Б у се в А. И., Бабенко Н. Л, Чепик М. Н, ЖАХ, 19, 1057 (1964).
207. Б у се в А. И., Бабенко Н. Л. В сб.: «Методы анализа веществ высокой
чистоты». М., «Наука», 1965. См. с. 451.
208. Бусев А. И, Симонова Л. Н, 3 а ю к о в а Н. Д., Вести. Моск, ун-та хим. наук, № 1, 119 (1968).
209. Р а м а н а у с к а с Э. И., Гро д ник ас Ш. Г., Научи, тр. высш, учебн. заведений ЛитССР. Хим. и хим. технол., № 12, 33 (1970).
210. Poliock Е. N, Anal. chim. acta, 40, 285 (1968).
211. Туткувене В. Е. и др., Научн. тр. высш, учебн. заведений ЛитССР. Хим и хим. технол., № 9, 55 (1968).
212. Раманаускас Э. И., Гро д ник а с Ш. Г., Науч. тр. высш, учебн заведений ЛитССР. Хим. и хим. технол., № 11, 11 (1970).
213. Loouenga R. W., Boltz D. F., Mikrochim. acta, № 3, 507 (1971).
214. Бусев А. И, Симонова Л. H., Заюкова H. Д., Тр. комиссии no аналит. хим. АН СССР, 17, 218 (1969).
215. Bode Н„ Z. anal. Chem., 144, 90 (1955).
216. В о d e H, Z. anal. Chem, 144, 165 (1955).
£17. Hikime S, Voshida H, Yomamiti M, Japan Analyst, 10, 112 (1961)
218. Yoshida H, Japan Analyst, 11, 549 (1962).
219. lankovsky I, OtakarK., Taianta, 5, 238 (1960).
220. F a 1 c i о 1 a P, Ann. Chim. applic, 17, 359 (1927).
221. Тара ян В. M, Саркисян А. А., ЖНХ, 10, 2684 (1965).
222. N i 1 s с h W, G i е f е г Z„ Z. anal. Chem, 145, 347 (1955).
223. Nilsch W, Giefer Z. Z. anal. Chem, 155, 401 (1957).
224. N i Isch W, Z. anal. Chem, 144, 191 (1955).
225. Aynsley E, Campbell W, J. Chem. Soc, 3290 (1958).
226. VfeStal I, Collect. Zechosl. Chem. Common, 25, 443 (1960).
227. Бусев А. И, Симонова Л. H, ЖАХ, 22, 1850 (1967).
228. Ковалевский В. В, Ермаков В. В, ЖАХ, 21, 447 (1966).
229. Кольт го ф И. М, Стенгер В. А. Объемный анализ. М, Госхимиздат, 1952. См. с. 357.
230. N i v i е г е Р, Chim. Anal, 47, 125 (1965).
231. В л а д и м и р о в а В. М, Кучм иста я Г. И, Зав. лаб, 30, 528 (1964).
232. Гурьев С. Д, Бляхман А. А, Лутченко Н. Н, Сб. тр. Гос. н.-и. ин-та цвети, мет, № 19, 661 (1962).
233. Б о я д ж и е в а Р, Тр. геол. Бълг. Сер. геохим. и полезни ископаемо, кн. 3, 281 (1962).
234. Stanton R. Е, McDonald A. J, Analyst, 90, 497 (1965).
235. Sluzewska L, Roczn. Panstw. Zakl. hig, 15, 303 (1964).
936 Рябчиков Д. И., Назаренко И. И., Аникина Л. И., ЖАХ, 23, 1242 (1968).
237. Волков С. Т. Определение селена и теллура в рудах и концентратах, содержащих золото. М., Госгеолиздат. 1945. См. с. 5.
238. Риги п В. И., Мельниченко Н. Н„ Яиицкий В. К., Зав. лаб., 33, 1370 (1967).
239. Ра trow sky J. J., Lu go wk in В. P., Mardryk G. T„ Ber., 69, 1913 (1936).
240. Brown E. C„ Analyst, 79, 50 (1954).
241. Мурашова В. И., ЖАХ, 21, 345 (1966).
242. Lambert J., Arthur P., Moore T., Anal. Chem., 28, 1101 (1951).
243. Мурашова В. И., ЖАХ, 17, 80 (1962).
244. Черни хов Ю. А., Добкина Б. М„ Зав. лаб., 15, 1143 (1949).
245. В о d е Н., Z. anal. Chem., ИЗ, 3 (1954).
246. Hikime S., Yoshida H., Uzumasa Y., Japan Analyst, 8, 531 (1959).
247. Павлова В. H., Васильева Н. Г., Кашли некая С. Э., Зав. лаб., 27,965 (1961).
248. Б усе в А. И., Бырько В. М., Прохорова Л. Н. В сб.: «Методы определения и анализа редких элементов». Изд. АН СССР, 1961. См. с. 626.
249. Т а р а я н В. М., С а р к и с я н А. А., ЖНХ, 10, 2684 (1965).
250. Овсепян Е. Н., Тараян В. М„ Шапошникова Г. Н., Арм. хим. ж., 20, 184 (1967).
251. J i 1 е k А., V г е s t а 1 J., Chem. Zvesti, 7, 33 (1953).
252. Б у се в А. И., Гусейнов И. К-, Багбанлы И. Л., Азерб. хим. ж., 6, 92 (1966).
253. Б у се в А. И., Бабенко Н. Л. В сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965. См. с. 451.
254. Ч у в и л е в а А. И., Блюм И. А., Зав. лаб., 35, 1153 (1969).
255. Wiberley S. Е., Bassett L., Burrill А. М., Lyng Н., Anal. Chem., 25, 1586 (1953).
256. Вега В. С., Chakrabarthy М. М., Analyst, 93, 50 (1968).
257. Navratil О., Sort a J., Coll. Czech. Chem. Comm., 34, 975 (1969).
258. С h a n F. L„ Taianta, 11, 1019 (1964).
259. Gresser M. S., West T. S., Taianta, 16, 416 (1969).
260. P г a n d 11 W„ Z. anorg. Chem., 93, 45 (1915).
261. Тутку вене E. E., Раманаускас Э. И., ЖАХ, 21, 564 (1966).
262. Yamamoto M., Sakai H., Anal. chim. Acta, 32, 370 (1965).
263. Шитарева Г. Г., Зав. лаб., 27, 1196 (1961).
264. Шитарева Г. Г., Назаренко В. А., Укр. хим. ж., 26, 368 (1960).
265. Etten N., Musch a wee k J., Z. anal. Chem., 206, 17 (1964).
266. J е п к i n s J., J. Am. Chem. Soc., 74, 1353 (1952).
Глава VIII
ФТОР
VIII. 1. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ ОТДЕЛЕНИЯ ФТОРИДА
Дистилляция. Лучшими методами отделения фторида являются дистилляционные, основанные на отгонке фтороводорода, четырехфтористого кремния и кремнефтористоводородной кислоты из растворов хлорной, серной или фосфорной кислот вместе с парами воды [1], с воздухом [2—5], азотом [6, 7] или без принудительной отгонки [8—11]. О составе продуктов, в виде которых происходит дистилляция фторидов, существуют различные мнения. Долгое время считалось, что фторид отгоняется в виде кремнефтористоводородной кислоты и тетдафторида кремния. Летучесть тетрафторида кремния не вызывает сомнений, а чистая H2SiF6 полностью улетучивается уже при 40 °C. При этом соотношение между HF и SiF4 меняется в зависимости от концентрации компонентов. Имеются данные о том, что при концентрации 13,3% H2SiF6 состав паров при 720 мм рт. ст. соответствует 1 моль SiF4 и 2 моль HF[12]. По данным физико-химического анализа системы, состоящей из фторида, кремневой кислоты и минеральной кислоты [13], и исследования полученного дистиллята [14], кремнефтористоводородная кислота и тетрафторид кремния полностью гидролизуются, а образующаяся фтористоводородная кислота отгоняется. Однако эти данные вызывают возражения [15]. По-видимому, отгонка фторида происходит главным образом в виде фтористого водорода, но нельзя отрицать дистилляцию фторида в виде тетрафторида и кремнефтористоводородной кислоты, что подтверждается известной качественной пробой помутнения капли воды в ушке платиновой нити [16].
Преимущество применения хлорной кислоты при отгонке фторида состоит в том, что она не образует малорастворимых соединений с ионами металлов. Она также не образует устойчивых комплексов с ионами металлов и в этом отношении уступает серной и фосфорной кислотам, анионы .которых образуют комплексы со многими металлами и способствуют разрушению комплексных фторидов, например циркония, ниобия, тантала и многих других. В случае применения хлорной кислоты необходимо следить, чтобы в раствор не попали органические вещества, приводящие к взрыву. Применяя для отгонки фтора серную кислоту, лучше при определении фторидов не пользоваться соединениями циркония, гаф-282
ния и других ионов, которые образуют комплексы с сульфатом,
увлекаемым парами в дистиллят.
Сопоставление методов отгонки фторида в присутствии циркония показало [17], что отгонка в присутствии серной кислоты дает
худшие результаты, чем в присутствии фосфорной. Это объясняется неполной отгонкой фтора при низкой температуре, а также слабым связыванием циркония сульфат-ионами.
При анализе малорастворимых в кислотах кремнийсодержащих
материалов их предварительно сплавляют с содой или щелочью.
Большие количества геля кремневой кислоты затрудняют отгонку фторида. В этом случае отгонку проводят при более высокой температуре.
Пирогидролиз. Метод состоит в том, что на измельченную пробу действуют перегретым паром при 700— 1200 °C. Выделяющийся фтористый водород и тетрафторид кремния охлаждаются в холодильнике и поступают в приемник. Разложение проводят в термоустойчивой трубке, помещенной в трубчатую печь. Лучшие результаты получаются в случае применения платинового прибора (рис. 64).
Температуру, при которой проводят пирогидролиз, устанавливают в зависимости от состава проб [18]. Фториды
Рис. 64. Стандартный платиновый прибор для проведения-пирогидролиза:
/ — конусный шлнф с трубкой для ввода пара; 2 —термоустойчивая трубка; 3— платиновая холодильная трубка; 4 — стеклянная рубашка;
5—полиэтиленовый наконечник.
250-
щелочных и щелочноземельных метал-
лов не разлагаются даже при 1200 °C, поэтому для разложения их, а также для разложения комплексных фторидов, содержащих щелочные металлы [19], в пробу необходимо добавлять А120з, WO3 или UO3. При этом происходят следующие реакции:
2NaF + Н2О + А12О3 —►
2NaF + Н2О + UO3 —► 2NaF + Н2О + WO3 —►
CaF2 + Н2О + WO3 —►
2NaA102 + 2HF
Na2UO4 + 2HF
Na2WO4 + 2HF
CaWO4 + 2HF
Пру разложении фторида алюминия добавляют двуокись титана [20]. В случае проведения пирогидролиза в присутствии кремневой кислоты или при проведении процесса в кварцевой трубке образуется силикат натрия и тетрафторид кремния:
2NaF + SiO2 + Н2О —> Na2SiO3 + 2HF
4NaF + 3SiO2 —> SiF4f + 2NaxSiO3
Фториды делят [20] на легко гидролизующиеся (A1F3, BiF3, MgF2, ThF4, UF, UF3, UO2F2, VF3, ZnF2, ZrOF2, ZrF4, фториды редкоземельных элементов) и трудно гидролизующиеся (фториды щелочных и щелочноземельных элементов и BeF2).
Результаты исследования процесса пирогидролиза некоторых фторидов представлены в табл. 13.
Таблица 13. Результаты исследования пнрогидролиза некоторых фторидов [21]
Соединение Время, мин Гидроли-зовано, % Соединение Время, мин Гндроли-зовано, % Соединение Время, мин Гидролизовало, %
ZnF2 4 100 A1F3 15 100 CaF2 — 30
ThF4 — 100 CeF3 — 100 LiF — 18
UO2F2 — 100 MgF2 — 100 BeF2 — 13
uf4 6 100 BiF3 25 100 SrF2 — 8
UF3 12 100 BeF2 60 97 NaF — 6
Иногда для разложения пробы пирогидролизом применяют смесь водяного пара и кислорода [22]. При разложении фторидов элементов в низшей валентности одновременно с разложением фторида происходит окисление металла [23]:
2UF4 + 4Н2О + О2 —> 2UO3 + 8HF
При разложении пробы пирогидролизом вместе с фтористым водородом может отгоняться тетрафторид германия и бор в виде BF3 или борной кислоты. Для связывания галогенидов вводят перхлорат или сульфат серебра [8].
Экстракция. Одним из наиболее перспективных и удобных методов разделения является экстракция. Однако для отделения фторидов методы экстракции разработаны слабо.
Наибольший интерес представляет экстракция фторидов в силъ-нокислой среде с помощью арил- и алкилсиланолов [24, 25]. Например, триэтилсиланол взаимодействует с фторидом с образованием триэтилфторсилана:
R3SiOH + HF —> R3SiF + Н2О
Полученные кремнийорганические соединения экстрагируются неводными растворителями и, таким образом, фторид переходит в органическую фазу. Этот метод позволяет избирательно извлекать фтор из растворов, содержащих почти все катионы и анионы, мешающие его определению. Фтор реэкстрагируют раствором гидрокарбоната натрия и затем определяют. Однако следует отметить, что реэкстракция фторида проходит трудно и требует усовершенствования.
Фтор экстрагируется также в виде окрашенного комплекса с церийализаринкомплексоном азотсодержащими экстрагентами [26, 27]. Мешающее влияние посторонних ионов, к сожалению, при этом снижается недостаточно.
Экстракция фтористоводородной кислоты аминами, трибутилфосфатом и другими экстрагентами не перспективна, так как H2F2 экстрагируется мало и не избирательно, экстракции мешают многие ионы. Слабо экстрагируется фтор в виде фторидов ди-цикло-пентадненил-титана [28], триалкилсвинца и олова с дитизоном [29]. Экстракцию фторида тетрафенилстибонием [30, 31] используют для выделения фтора [28]. Коэффициент распределения фтора в присутствии высаливателя достигает 40, раствор щелочи легко реэкстрагирует фтор. Однако для аналитических целей этот метод не применим, так как уже небольшие количества алюминия и железа подавляют экстракцию. Фториды тетрафенилсульфония и тетрафенилфосфония экстрагируются незначительно [32, 33]. При точном соблюдении концентрационных условий удовлетворительные результаты получены при экстракции фторида с помощью раствора ди-(2-этилгексил)-фосфата циркония в гексане [34]. Процесс экстракции можно изобразить схемой:
[ZrXfHR^^+FJ^ [2гН(нкг)э]орг + х;од
(где X — анион минеральной кислоты). Реэкстракция щелочью изображается схемой:
[2гр(НШрГ + 0Н»л -* [Zr(OH)(HR2)3Jopr + р;«л
В водной фазе определяют фтор фотометрическим методом.
Исследована экстракция фторид-ионов из растворов фторида аммония [35] сульфатом триалкиламина [(RaNH^SOJ, представляющим смесь 65% третичных и 20% вторичных аминов фракции С7—Сэ при pH = 2—3. Этот метод, по-видимому, не может быть применен для отделения фторидов в присутствии ионов металлов, которые образуют прочные фторидные комплексы.
Хроматографические методы. Газо-жидкостная хроматография фторидов широко применяется в атомной технике. Газовая хроматография использована для анализа многих фторидов, с различными температурами кипения [18]. Описано отделение фторидов ионообменной хроматографией [36—43].
Осаждение. Осадительные методы отделения фтора мало пригодны для последующего фотометрического определения.
VIII. 2. МЕТОДЫ ОТДЕЛЕНИЯ ФТОРА
VIII. 2.1. Дистилляция фтора из кислых растворов водяным паром
Фториды разлагаются минеральными кислотами и гидролизуются при действии водяного пара:
MeFrt +/гН2О —► Me(OH)rt4-nHF
SiO2 + 6HF —► H2SiF6 + 2H2O
Me(OH)ft + nHC104 —> Me(C104)rt + nH2O
Для дистилляционного отделения фтора пользуются несколькими видами приборов, изображенных на рис. 65, а, б, в, г.
Прибор по Хюкебею (рис. 65, г). Реакционная колба 2 помещена в рубашку 3, снабженную обратны.м холодильником 8. Колба 2 соединена с холодильником 4 для конденсации паров, содержащих фтор. Пары воды подают в колбу 2 из парообразователя
Рис. 65. Приборы для отгонки фтора:
а —по Рихтеру, б—по Таианаеву; в —по Рейнольдсу; а —по Хюкебею; /—парообразователь; 2—реакционная колба; 3 — рубашка или баия; 4 — холодильник; 5 —приемник; 6 — термометр; 7—манометрическая трубка; 8—обратный холодильник; 9—трубка для ввода пара.
через трубку 9. Рубашку 3 заполняют на 3/4 тетрахлорэтаном и поддерживают кипение (146 °C) нагреванием [44].
В реакционную колбу 2 через верхний шлиф помещают пробу и 20 мл концентрированной H2SO4. Тетрахлорэтан в рубашке 3 постепенно нагревают и доводят до кипения. Одновременно в колбу 2 пропускают водяной пар через трубку 9, сначала медленно (один пузырек в 1 с), а при закипании тетрахлорэтана усиливают подачу пара так, чтобы в холодильнике 4 за 1 мин конденсировалось 5 мл дистиллята.
Прибор по Рихтеру [45] (рис. 65,а). Реакционная колба 2 помещена в глицериновую баню 3. Водяной пар пропускают из па
рообразователя 1 емкостью 1,5 л, который соединен с колбой 2 через пришлифованную пробку, снабженную термометром и имеющую отвод в холодильник 4.
В реакционную колбу 2 вносят пробу в смеси с порошком кварца. Вводят 25 мл 30%-ной НС1О4 и сосуд закрывают. Глицериновую баню 3 нагревают до 180 °C и одновременно пропускают пар, собирая конденсат в приемник 5.
Прибор Рейнольдса [46] (рис. 65, в) применяют для серийных анализов. В воздушную баню 3, выполненную в виде длинного ящика (7X15X75 см) и нагреваемую хромоникелевой спиралью с сопротивлением ~50 Ом, можно установить восемь дистилляционных аппаратов. Каждый из них состоит из парообразователя 1 емкостью 300 мл с манометрической трубкой, реакционной колбы 2 емкостью 50 мл, холодильника 4 и приемника 5. Температуру нагревания контролируют термометром, установленным в середине бани 3.
В реакционную колбу 2 помещают исследуемую пробу, измельченный кварц и 60%-ную НС1О4 (органические вещества должны отсутствовать, иначе произойдет взрыв). Температура в колбе поддерживается 135—140 °C. В течение 1 ч собирают в каждом приемнике 150 мл конденсата, содержащего фтор.
V III. 2.2. Дистилляция фтора без пропускания водяного пара
По И. В. Тананаеву, отгонку фтора проводят из сернокислых, растворов без принудительной отгонки [8—11]. Прибор (рис. 65,6) состоит из холодильника 4 и круглодонной колбы 2 емкостью 100—150 мл из йенского стекла. Длина соединительной трубки между колбой и холодильником не должна превышать 10—15 см. Нагревание наиболее удобно проводить на песочной бане. Полнота выделения фтора для 0,1 г криолита и фторида кальция составляет 96,5—99,4%.
Ход анализа. В реакционную колбу 2 помещают анализируемую пробу, смешанную с растертым кварцем и 20—40 мл H2SO4. Концентрация применяемой серной кислоты зависит от содержания фтора. При малых содержаниях фтора применяют H2SO4, разбавленную (1 : 1), при большем содержании фтора применяют H2SO4, более разбавленную (1:2). В этом случае уменьшается выделение в отводную трубку кремневой кислоты, которая сорбирует кремнефтористоводородную кислоту.
Этот вариант использован при анализе фосфатов разложением фосфорной или хлорной кислотой [47], но в дистиллят переходит часть фосфата, хотя, по данным авторов, это не отражалось па дальнейшем ходе анализа.
Навеску исследуемой пробы в 10 г вносят в колбу 2 (емкость ее должна быть 300—350 мл), в которой находятся стеклянные шарики и 20 мл воды. Затем добавляют в реакционную колбу 40 мл 60%-ной НС1О4. Фтор отгоняют при 135 °C.
V III. 2.3. Отделение фтора пирогидролизом
Прибор для отделения фтора пирогидролизом изображен па рис. 64. Часть прибора, связанная с подачей пара, представляет собой притертый к трубке 2 конусный шлиф 1 с отростком для ввода пара. Пар с продуктами гидролиза поступает в платиновую холодильную трубку 3, помещенную в стеклянную водяную рубашку 4. Конец платиновой холодильной трубки имеет полиэтиленовый наконечник 5, опущенный в приемник до поверхности находящейся в нем жидкости.
Ход анализа. Анализируемый образец помещают в платиновую лодочку, которую вставляют в нагреваемую часть трубки. Затем прибор полностью собирают, нагревают печь до 700—1200 °C (в зависимости от состава анализируемой пробы) и пропускают водяной пар. Продукты пирогидролиза собирают и определяют фтор одним из описанных ниже методов.
Если применяют платиновый прибор, то при анализе проб, не содержащих кремневой кислоты, в дистилляте находится чистая фтористоводородная кислота.
V III. 2.4. Экстракция фторид-ионов ди-(2-этилгексил)-фосфатом циркония [34]
Реактивы
Ди-(2-этилгексил)-фосфат циркония, 0,5 н. раствор в гексане, содержащем 0,450 г циркония в 100 мл.
Ход анализа. Около 10 мл исследуемого раствора 2—5 н. по серной, хлористоводородной или хлорной кислоте помещают в делительную воронку емкостью 50 мл и перемешивают в течение 1 мин с 10 мл раствора ди-(2-этилгексил)-фосфата циркония. Экстракт отделяют в другую такую же воронку и дважды промывают в течение 1 мин 5 мл 2 н. хлористоводородной кислоты. Фтор реэкстрагируют смесью 2 мл трибутилфосфата, 1 мл ацетона и 10 мл 0,35 н. NaOH, затем 10 мл 0,1 н. NaOH в течение 15 мин. Объединенный реэкстракт подкисляют 2 мл 5 н. хлористоводородной кислоты, разбавляют до 50 мл, перемешивают, фильтруют и отбирают аликвотную часть для фотометрического определения фтора.
При .построении калибровочного графика экстрагируют известные количества фтора из 2 н. хлористоводородной кислоты.
V III. 3. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ
ОПРЕДЕЛЕНИЯ ФТОРИДА
Фторид не образует окрашенных соединений, кроме некоторых смешанных комплексов, поэтому почти все фотометрические методы определения фторида основаны на разрушении окрашенных соединений различных лигандов с ионами металлов, которые об-288
разуют достаточно прочные фторидные комплексы. Общую схему фотометрического определения фторида можно представить следующим образом:
F" + MeR —> MeF + R"
(где MeR — окрашенный комплекс металла, a MeF — бесцветный фторидный комплекс этого металла). Фторид образует комплексы с рядом катионов (железо, титан, цирконий, торий, алюминий и др.). Эти катионы дают окрашенные комплексы со многими реагентами. Однако далеко не все окрашенные комплексы таких металлов пригодны для фотометрического определения фторида. Прочность фторидных комплексов не очень велика; их константы нестойкости порядка 10-5—10-9. Таким образом, для определения фторида можно применять только сравнительно мало прочные окрашенные комплексы металлов или необходимо создать такие условия реакции, при которых уменьшается их прочность. Основным условием применения окрашенного комплекса для определения ионов фтора является КмеН^> Кмег-
В связи с невысокой прочностью и ступенчатым комплексообразованием фторидных комплексов не наблюдается прямой пропорциональности между общей концентрацией и оптической плотностью раствора. Поэтому фотометрическое определение фторида требует особых условий измерения оптической плотности. Наиболее пригоден для этого метод фотометрического титрования, при котором отклонение от закона Бера не имеет значения. Метод шкалы мало удобен. В случае применения циркониевых, ториевых и других «лаков», когда окрашен не только комплекс, но и реагент, метод шкалы дает более удовлетворительные результаты. Шкалу необходимо готовить в день определения.
Применение методов измерения оптической плотности требует построения калибровочных графиков по многим точкам, так как зависимость оптической плотности от концентрации фторида не выражается прямой линией.
Наиболее благоприятная кислотность при определении фторида соответствует pH от 2 до 3. При более высокой кислотности значительная часть ионов фтора связывается в молекулу, константа диссоциации которой равна 10-3.
При повышении pH окрашенные комплексы (соли слабых кислот) часто становятся настолько прочными, что не реагируют с ионами фтора, другие окрашенные комплексы (как роданид железа) с повышением’ pH разрушаются вследствие гидролиза.
Для фотометрического определения обычно применяют не готовый комплекс MeR, а готовят его, смешивая растворы соли металла Меп+ с некоторым избытком соответствующего реагента (Rn-). Выше было отмечено, что при фотометрическом определении фторида комплекс MeR должен быть не очень прочным. Окраска заметно зависит от избытка реагента. Поэтому необходимо, чтобы в испытуемом и в стандартном растворах была
одинаковая концентрация избытка реагента [R-]. Калибровочные кривые, составленные при одном значении [R-], совершенно не пригодны для работы с другими концентрациями реагента, если даже исходные окрашенные растворы (до прибавления фтора) имеют одинаковую окраску.
Лучшим методом, по-видимому, является прямой метод определения фтора, основанный на образовании фторидами смешанного окрашенного комплекса с -ализарин-комплексоном и церием [48— 54].
V III. 4. ПРЯМОЕ ОПРЕДЕЛЕНИЕ ФТОРИДА С ПРИМЕНЕНИЕМ АЛИЗАРИН-КОМПЛЕКСОНАТА ЦЕРИЯ(Ш) [48—54]
Красный ализарин-комплексонат церия(Ш)—комплекс церия (III) с 1,2-диоксиантрахинон-З-метилиминодиуксусноп кислотой— при взаимодействии с фторид-ионами образует смешанный комплекс, окрашенный в синий цвет. Реакцию образования смешанного комплекса можно, по-видимому, изобразить следующей схемой:
Это пока единственная реакция, используемая для прямого фотометрического определения фтора. Вместо солей церия можно применять соли лантана [55, 56]. Неодим, празеодим и самарий образуют аналогичные смешанные комплексы, но молярные коэффициенты поглощения их значительно ниже [51, 54, 55].
Мешающие вещества. Вредное влияние посторонних ионов связано с образованием [50] устойчивых фторидных комплексов, образованием более прочных комплексов с ализарин-комплексоном и образованием устойчивых комплексов посторонних ионов с церием. Сильное мешающее действие оказывают ионы кадмия, кобальта, хрома(Ш), меди, железа(Ш) и (II), никеля, свинца, цинка, ванадия, бериллия, кальция, бората и фосфата. Влияние серебра, бария, магния, стронция, таллия и циркония незначитель
но. Вредное влияние ионов меди, кобальта и ртути полностью устраняют цианидом калия. 2—З-Димеркаптопропанол-1 (БАЛ) устраняет влияние никеля и свинца. Сульфид натрия устраняет влияние кадмия, цинка и свинца. Вредное влияние многих ионов устраняют предварительной экстракцией их о-оксихинолинатов
органическими растворителями.
Оксалаты, тартраты, цитраты и этилендиаминтетраацетаты ослабляют интенсивность окраски; хлориды, бромиды, иодиды,
перхлораты, нитраты, силикаты и умеренные количества сульфатов, фосфатов и арсенатов не мешают определению фторида.
Оптимальная чувствительность метода наблюдается при pH = 5,0—5,2 в водном растворе. В присутствии некоторых смешивающихся с водой органических растворителей (ацетон, ацетонитрил, диметилформамид и др.) чувствительность метода повышается и результаты получаются более стабильными [40, 55—59]. В водно-ацетоновых растворах оптимальное значение рН = = 3,9—4,6 для цериевого комплекса и 4,6—5,5 для лантанового. Для установления узкого значения pH обычно применяют буферные растворы.
Измерение оптической плотности. Спектры поглощения растворов ализа-рин-комплексона, двойного цериевого
Рис. 66. Спектры поглощения растворов ализарин-комплек-сона (/), его комплекса с церием (2) и тройного комплекса с церием и фторидом (3), снятые на фоне воды.
комплекса и тройного цериевого комп-
лекса [60], снятые иа фоне воды, приведены на рис. 66. Спектр
тройного комплекса имеет две полосы поглощения с максимумами при 251 и 570 нм. Оптическую плотность раствора этого комплекса лучше всего измерять при X = 610—620 нм.
Чувствительность метода (е = 0,6-104) повышается при добавлении к водному раствору ацетона или ацетонитрила (20—25%)' С целью повышения чувствительности метода применяют экстракцию тройного комплекса раствором солянокислого гидроксиламина в изобутаноле. Этот растворитель не экстрагирует али-зарин-комплексонат редкоземельного металла. Чувствительность метода — 2 мкг фторида в 25 мл конечного раствора. В случае ацетоно-водного раствора (25% ацетона) чувствительность метода повышается до 1 мкг в 25 мл конечного раствора. Экстракцией
смешанного комплекса можно повысить чувствительность метода до 0,2 мкг фторида в 5 мл экстракта.
V III. 4.1. Определение фторида в отсутствие мешающих веществ Реактивы
Стандартный раствор фторида. Навеску фторида натрия (ч. д. а.) 0,2211 г растворяют в воде, переносят в мерную колбу емкостью 500 мл, разбавляют
до метки водой, перемешивают и переносят в полиэтиленовый сосуд. Этот раствор содержит 0,2 мг фтора в 1 мл. Перед применением готовят разбавленный раствор, содержащий 10 мкг в 1 мл.
Составной реактив для определения фтора с pH = 5,0—5,2. Навеску али-зарин-комплексона 0,0772 г переносят в мерную колбу емкостью 200 мл и растворяют в 60 мл дистиллированной воды, содержащей 0,4 мл концентрированного раствора аммиака, затем приливают 80 мл ацетона, 30 мл буферного раствора (pH = 5,0—5,2) и перемешивают. К раствору прибавляют 10 мл 0,02 М раствора нитрата церия(III), разбавляют до метки дистиллированной водой и перемешивают. Этот составной реактив пригоден для применения в течение 5 дней.
Нитрат церия, 0,1 М водный раствор. Готовят по навеске нитрата церия(III). Титр устанавливают комплексонометрически в присутствии ксиленолового оранжевого. Путем разбавления дистиллированной водой готовят 0,02 М раствор.
Буферный раствор с pH = 5,0—5,2. Растворяют 100 г трехводного ацетата натрия в воде, добавляют 11,0 мл ледяной уксусной кислоты и разбавляют до 1 л.
Буферный раствор с pH = 4,1. Растворяют 85 г трехводного ацетата натрия в воде, добавляют 115 мл ледяной уксусной кислоты и разбавляют дистиллированной водой до 1 л.
Ацетон, ч. л а.
Ход анализа. Определение в водных растворах. В мерную колбу емкостью 25 мл помещают 5 мл составного реактива, до 18 мл нейтрального исследуемого раствора, содержащего до 20 мкг ионов фтора, разбавляют дистиллированной водой до метки и перемешивают. Раствор оставляют на 25 мин, затем заполняют кювету с толщиной слоя 1 см и измеряют оптическую плотность раствора при 620 или 281 нм. В последнем случае пользуются кварцевой кюветой. Раствором сравнения служит аналогично приготовленный раствор, не содержащий фторида.
Определение в ацетоно-водном растворе. В мерную колбу емкостью 25 мл помещают 5 мл раствора ализа-рин-комплексоната церия (III), 6 мл ацетона, 1 мл буферного раствора (рН = 4,1), до 12 мл нейтрального исследуемого раствора, содержащего до 15 мкг ионов фтора, разбавляют до метки дистиллированной водой и перемешивают. Далее ведут определение, как указано выше.
V III. 4.2. Определение фторида в флюсах [54, 61]
Флюсы обычно содержат значительные количества фтора; для его определения применяют дифференциальный фотометрический метод. Навеску флюса разлагают сплавлением с содой и последующим выщелачиванием водой. При этом в водную фазу частично попадает алюминий, который отделяют экстракцией хлороформом в виде о-оксихинолинатного комплекса. Если сплавление' пробы с содой проводить в присутствии кремневой кислоты, то алюминий практически не попадает в водную фазу при выщелачивании водой и экстракционное отделение алюминия можно ис* ключить.
Реактивы
Фенолфталеин, 1%-ный спиртовый раствор.
о-Оксихинолин, 2%-ный раствор в хлороформе.
Другие реактивы— указаны в разделе VIII. 4.1.
Калибровочный график. Для построения калибровочного графика в мерные колбы емкостью 25 мл вводят порции раствора, содержащего от 50 до 75 мкг ионов фтора. Добавляют 10 мл реактива, доводят до метки водой и спустя 25 мин фотометрируют, как описано выше. По полученным значениям оптических плотностей строят калибровочный график.
Ход анализа. Определение с отделением алюминия. Навеску флюса (0,1 г для флюса АН-26 и 0,3 г для ФЦЛ-2). сплавляют с десятикратным количеством соды, выщелачивают 250—300 мл горячей воды и, не фильтруя, переносят в мерную колбу емкостью 500 мл. Отбирают в делительную воронку 25 мл этого раствора, нейтрализуют по фенолфталеину уксусной кислотой и отделяют алюминий двукратной экстракцией раствором о-оксихинолина (10 и 15 мл). Водный слой количественно переносят в мерную колбу емкостью 50 мл, разбавляют водой до метки и перемешивают. Отбирают отфильтрованную аликвотную часть раствора в мерную колбу емкостью 25 мл, прибавляют 10 мл раствора ализарин-комплексоната церия (111), разбавляют водой до метки и перемешивают. Одновременно готовят раствор сравнения, содержащий 50 мкг фтор-иона и такое же количество реактива. Спустя 25 мин измеряют оптическую плотность раствора в кювете с толщиной слоя 1 см при 610—620 нм или со светофильтром № 7.
Определение без экстракционного отделения алюминия. Навеску флюса 0,1 г перемешивают в платиновом тигле с 0,25 г двуокиси кремния и 1 г карбоната калия-натрия и сплавляют при 900 °C в течение 30 мин. Плав выщелачивают 100— 150 мл горячей воды и, не фильтруя, переносят в мерную колбу емкостью 500 мл. Раствор охлаждают, доводят до метки водой и хорошо перемешивают. В мерную колбу емкостью 50 мл отбирают 25 мл этого раствора, нейтрализуют уксусной кислотой по фенолфталеину, доводят до метки водой, перемешивают и фильтруют через сухой фильтр в сухую колбу. Аликвотную часть профильтрованного раствора до 10 мл вводят в мерную колбу емкостью 25 мл, приливают 15 мл составного реактива и воду до метки. Одновременно готовят раствор сравнения, содержащий 50 мкг ионов фтора и то же количество составного реактива. Спустя 25 мин измеряют оптическую плотность при 610 нм в кювете с толщиной слоя 1 см.
При работе на фотоколориметре уменьшают количество составного реактива до 10 мл при толщине слоя 0,5 см, а нулевой раствор должен содержать 25 мкг ионов фтора.
V III. 4.3. Определение фтора в природных водах
Пробу воды нейтрализуют по фенолфталеину и отделяют мешающие ионы сорбцией на катионите КУ-2. В элюате определяют фтор в виде смешанного комплекса в ацетоно-водной среде. Для этого применяют комплекс церия [62] или лантана [63].
Реактивы
Ализарин-комплексонат церия(Ш). При постоянном перемешивании к 165 мл ацетона приливают последовательно 34 мл буферного раствора (см. ниже), 5мл ализарин-комплексона (0,643 г ализарин-комплексона суспендируют в 50 мл воды, добавляют 0,25 мл концентрированного раствора аммиака и 0,25 мл ледяной уксусной кислоты и разбавляют до 100 мл водой), 5 мл раствора нитрата церия(Ш) и разбавляют водой до 250 мл. Раствор пригоден для работы в течение 5 дней.
Нитрат церия(Ш), 0,0167 М раствор.
Буферный раствор. Растворяют 60 г CHaCOONa-ЗН2О и прибавляют 115 мл ледяной уксусной кислоты. Раствор разбавляют водой до 1 л и перемешивают.
Катионит, КУ-2.
Другие реактивы — указаны в разделе VIII. 4.1.
Ход анализа. Пробу воды до 10 мл с содержанием до 15 мкг ионов фтора нейтрализуют, если нужно, по фенолфталеину и пропускают через колонку с катионитом КУ-2 со скоростью 5 мл/мин. Колонку промывают небольшим количеством воды до получения общего объема элюата до 17 мл, который помещают в мерную колбу емкостью 25 мл. Прибавляют 7 мл составного реактива, если необходимо, доводят до метки дистиллированной водой, перемешивают и спустя 25 мин измеряют оптическую плотность раствора.
Содержание фтора находят по калибровочному графику.
V III. 4.4. Определение фторида в органических веществах
Пробу сжигают, в атмосфере кислорода, продукты сжигания поглощают водой и определяют фторид, как указано в разделе VIII.4.1.
Реактивы
Полиэтиленовая пленка, толщиной 0,01 мм, нарезанная на квадраты со стороной 1,5 см.
Хлопчатобумажная нить.
Другие реактивы — указаны в разделе VIII. 4.1.
Ход анализа. Навеску образца 30—80 мкг, взятую на микровесах, помещают на полиэтиленовый квадратик. Завертывают образец, обматывают пакет двойным витком хлопковой нити, служащей запалом. Закрепляют в платиновом держателе. В коническую или круглодонную колбу емкостью 25 мл из кварцевого стекла наливают 2—3 мл дистиллированной воды, наполняют кислородом и сжигают образец, как обычно. Тщательно встряхивают колбу в течение 5—10 мин. Переносят раствор в мерную колбу емкостью 50 мл, доводят дистиллированной водой до метки, перемешивают, отбирают аликвотную часть и определяют фтор, как указано в разделе VIII.4.1 (2),
V III. 5. ОПРЕДЕЛЕНИЕ ФТОРИДА С ПРИМЕНЕНИЕМ КОМПЛЕКСА ЦИРКОНИЯ С ЭРИОХРОМЦИАНИНОМ R
Эриохромцианин R (или солохромцианин R)
взаимодействует с ионами циркония [14, 64—71] и алюминия [72— 74] с образованием комплексов красного цвета:
и
ОН2
Л-макс=515 нм (pH =0—1)
^макс = 545 нм (pH = 1—2)
Эти комплексы разрушаются фторидом и применяются для фотометрического определения фтора. Исследование влияния фторида на устойчивость комплексов титана, циркония, гафния, тория, алюминия, железа, бериллия и уранила с рядом органических реагентов (эриохромцианином R, пирокатехиновым фиолетовым, ализариновым красным S, хинализарином, пурпурином, карминовой кислотой, кальционом, хромотропом 2В, стильбазо, ксилено-
зуется при рн = и—1, а
Рис. 67. Спектры поглощения эриохромцианина (/) и его циркониевого комплекса (2) при pH = 1.
ловим оранжевым, метилтимоловым синим, металлфталеином, алюминоном, ферроином, хромотроповой и сульфосалициловой кислотами) показало [75], что самыми чувствительными к фториду являются комплексы тория с эриохромцианином R, ксиленоловым оранжевым, метилтимоловым синим и стильбазо. Однако преимущество применения циркониевого комплекса состоит в том, что с цирконием определение фторида можно проводить в более кислой среде. Максимальная чувствительность достигается при pH = 1 (±0,1), хотя можно проводить определения и в 0,8 н. хлористоводородной кислоте.
Комплекс с цирконием с соотношением компонентов 1:1 обра-при pH = 1—2 происходит образование комплекса с соотношением компонентов 1:2. При взаимодействии с фторидом эти комплексы разрушаются в результате образования более прочного фторидного комплекса циркония.
Мешающие вещества. Эриохромциа-нин R взаимодействует со многими ионами, поэтому перед определением фтор необходимо отделить. Ионы многих металлов мешают в связи с образованием прочных фторидных комплексов. Определению фторидов этим методом мешают сульфаты и фосфаты, которые связывают ионы циркония.
Измерение оптической плотности. На рис. 67 представлены спектры поглоще
ния (при pH — 1) эриохромцианина R и его комплекса с цирконием. Максимум поглощения эриохромцианина R в разбавленном хлористоводородном растворе находится при 475 нм. Оптическую плотность можно измерять при 590 нм. Однако выбор длины волны зависит от кислотности раствора, в котором проводят определение фторида. Поэтому обычно оптическую плотность раствора рекомендуют измерять при 540 нм, когда наблюдается максимальная разница в поглощении исследуемого раствора и раствора сравнения. В качестве раствора сравнения служит раствор эриохромцианина R при том же pH раствора, при котором определяют фторид. Значение молярного коэффициента поглощения в пересчете на фторид составляет 6540 = 2,7-104.
При использовании реакций разрушения поглощающих свет комплексов в качестве измеряемого раствора лучше помещать раствор холостой пробы, а исследуемый раствор использовать в качестве раствора сравнения. Тогда калибровочный график имеет обычный ход и показывает, что с увеличением содержания фтора оптическая плотность раствора падает.
Чувствительность метода — 1 мкг фтора в 25 мл конечного раствора.
V III. 5.1. Определение фтора в отсутствие мешающих веществ [76]
Реактивы
Эриохромцианин R, 0,004 М раствор. Навеску реагента 0,536 г растворяют в дистиллированной воде с добавкой 2,5 мл 1 н. хлористоводородной кислоты. Раствор переносят в мерную колбу емкостью 250 мл, разбавляют до метки водой и перемешивают.
Хлорид циркония. Раствор 1, 0,005 М раствор в 4 н. хлористоводородной кислоте. Навеску хлорида или нитрата циркония, содержащую ~ 0,5 г циркония, растворяют в 25 мл хлористоводородной кислоты, разбавленной (1 : 1), выпаривают па водяной бане до появления кристаллов соли и разбавляют 4 н. хлористоводородной кислотой до 1 л. Содержание циркония в растворе определяют гравиметрическим или комплексонометрическим методом и после этого разбавляют раствор 4 н. хлористоводородной кислотой с таким расчетом, чтобы концентрация циркония-в растворе была точно 0,005 М (0,4561 г циркония в 1 л).
Раствор 2, 0,001 М раствор циркония в 2 н. хлористоводородной кислоте. Отбирают 50 мл раствора 1 в мерную колбу емкостью 250 мл и разбавляют до метки точно 1,5 н. хлористоводородной кислотой.
Цирконэриохромцианиновый реагент, свежеприготовленный раствор с соотношением [Zr]: [ER] =1:4. К 25 мл раствора эриохромцианина R добавляют при перемешивании 25 мл 0,001 М раствора циркония.
Ход анализа. В мерную колбу емкостью 25 мл наливают 2,5 мл цирконэриохромцианинового реагента и при взбалтывании прибавляют нейтрализованный по фенолфталеину исследуемый раствор. Затем раствор разбавляют до метки, перемешивают и измеряют оптическую плотность при 540 нм. Для приготовления раствора сравнения отбирают 2,5 мл цирконэриохромцианинового реагента, переносят в мерную колбу емкостью 25 мл, разбавляют дистиллированной водой до метки и перемешивают.
V III. 5.2. Определение фтора в воде
Для получения хороших результатов при определении фтора в воде его необходимо предварительно отделить методом отгонки, как описано в разделе VIII. 2. Однако в воде удовлетворительные результаты получаются и при определении фтора без предварительной отгонки [76, 77]. В этом случае измерение оптической плотности ведут не сразу, а спустя некоторое время, чтобы фторид, связанный в комплекс с алюминием или ионами других металлов, прореагировал с цирконэриохромцианиновым реагентом. Методика определения фтора подобна описанной в разделе VIII. 5.1, но оптическую плотность измеряют спустя 5 мин. Если проба воды содержит хлор, то к ней сначала прибавляют 2 капли 0,2 н. раствора тиосульфата на каждый 1 мг С1г-
V III. 5.3. Определение фтора в силикатных породах
Хорошие результаты определения фтора в любом объекте получаются после предварительной отгонки его [78]. Рекомендованы также методы отделения многих ионов в виде гидроокисей после
сплавления пробы с содой и окисью цинка. В этом случае оптическую плотность рекомендовано измерять спустя 1 —1,5 ч [79]. Ниже приведена методика определения фтора в силикатах без предварительной отгонки его.
Ход анализа. Навеску измельченной до частиц 90 меш и высушенной пробы 0,5 г сплавляют при 900 °C в платиновом тигле с 3,5 г карбоната натрия и 0,6 г окиси цинка в течение 20—25 мин. Плав охлаждают, прибавляют 10 мл воды и 3 капли 95%-ного этанола, кипятят в течение 1 мин и фильтруют. Остаток промывают 5 раз водой порциями по 2 мл и собирают фильтрат и промывные воды в колбу емкостью 50 мл. К раствору осторожно прибавляют 4,1 мл концентрированной азотной кислоты, размешивают до удаления СОг (среда должна быть слабокислой), разбавляют раствор до метки дистиллированной водой и перемешивают. Одновременно готовят холостую пробу с 3,5 г Ка2СОз и 0,6 г ZnO в таком же объеме воды, содержащем такое же количество азотной кислоты. В дальнейшем ведут определение, как указано в разделе VIII. 5.1.
V III. 6. ОПРЕДЕЛЕНИЕ ФТОРИДА С ПРИМЕНЕНИЕМ ЦИРКОНАЛИЗАРИНОВОГО КОМПЛЕКСА
Окрашенный цирконализариновый комплекс («лак») взаимодействует с фторидом с образованием более прочного фторидног^ комплекса циркония. Содержание фтора определяют по уменьшению оптической плотности раствора. В связи с тем, что реагент имеет собственную окраску, при разрушении «лака» фторидом происходит не обесцвечивание, а изменение цвета раствора. Наибольшая чувствительность определения фторида наблюдается при использовании циркон-ализаринового лака, не содержащего избытка ни реагента, ни циркония [80]. Оптимальная кислотность — в пределах 0,3—0,8 н. по хлористоводородной кислоте.
Вместо цирконализаринового комплекса применяют также торий [81—90] и лантанализариновый комплекс [91, 92].
Мешающие вещества. Определению мешают катионы, связывающие фторид (титан, торий, железо, алюминий), а также анионы, связывающие цирконий (оксалаты, тартраты, фосфаты и др.).
Методы измерения оптической плотности. Как было отмечено выше, при данном методе происходит лишь изменение окраски, а не обесцвечивание раствора. Спектры поглощения цирконализаринового комплекса и ализарина в условиях определения фтора представлены на рис. 68. Оптическую плотность раствора лучше измерять при 532 нм. Для этого применяют спектрофотометры или
Рис. 68. Спектры поглощения цирконализаринового комплекса (/) и ализарина (2) в условиях определения фтора.
фотоэлектроколориметры с использованием соответствующих светофильтров. Хорошие результаты дает также "метод стандартной шкалы.
Чувствительность метода — 0,5 мкг фтора в 25 мл конечного раствора.
V III. 6.1. Определение фтора в отсутствие мешающих веществ
При анализе различных материалов такие растворы получают обычно путем отгонки фторида в виде фтористоводородной кислоты или другими методами, описанными в разделе VIII. 2.
Реактивы
Цирконализариновый комплекс. Готовят из запасных растворов смешиванием равных объемов растворов циркопийоксихлорида и ализарина (или ализарина S) одинаковых концентраций (1,5—2,5)-Ю'4 М и нагревают в течение 15 мин при 60 °C. Такой раствор пригоден для применения в течение суток.
Стандартный раствор фторида, см. раздел VIII. 4.1.
Ход анализа. В колбу емкостью 25 мл к кислому (0,6—0,8 н. по HCI) исследуемому раствору (до 20 мл), содержащему от 0,5 до 100 мкг фтора, прибавляют 5 мл раствора цирконализаринового комплекса, разбавляют, если нужно, дистиллированной водой до метки, перемешивают и оставляют на 1 ч. В присутствии фторидов розово-фиолетовый цвет раствора переходит в слабо-розовый или желтый в зависимости от содержания фтора. Одновременно готовят раствор сравнения, содержащий такое же количество реактивов за исключением фторида. Спустя 1 ч измеряют оптическую плотность раствора сравнения по отношению к исследуемому раствору.
Содержание фтора находят по предварительно построенному калибровочному графику.
V III. 6.2. Определение фтора в силикатах
При анализе горных пород, силикатов и руд для отделения фтора от сопутствующих элементов применяют метод отгонки фтора в виде SiF4 или H2SiF6, а также метод разложения образца щелочным сплавлением. При этом кремневую кислоту, полуторные окислы и другие мешающие компоненты отделяют при помощи карбоната аммония.
Подготовка пробы методом сплавления. Навеску тонко измельченного образца 0,5—1 г сплавляют с 4-кратным количеством KNaCOa и сплав выщелачивают водой. К раствору, не обращая внимания на осадок, прибавляют 2—4 г (МН^гСОз. Смесь нагревают несколько минут, а затем выпаривают на водяной бане до малого объема и разложения карбоната аммония. При этом осаждаются кремневая кислота (частично) и полуторные окислы
Осадок отфильтровывают и промывают водой. В раствор сравнения вводят такое же количество KNaCOg. Оба раствора нейтрализуют, вводя по каплям хлористоводородную кислоту, и анализируют, как указано в разделе VIII. 6.1.
Если проба плохо разлагается при сплавлении с KNaCOa, то сплавление ведут с перекисью натрия в никелевых или железных тиглях. [93].
Подготовка пробы методом отгонки. Навеску 0,5—1 г тонко растертого испытуемого образца переносят в круглодонную колбу емкостью 100 мл. Через ту же широкогорлую воронку насыпают в колбу 1 г мелко истертого кварца. После перемешивания в колбу приливают 30 мл H2SO4, разбавленной (1:2). В приемник вводят несколько миллилитров воды. Затем содержимое колбы кипятят до полной перегонки всей воды. Конец перегонки определяют по остыванию трубки возле пробки, соединяющей трубку с холодильником. К этому времени в колбе остается 98,5%-ная серная кислота,, т. е. перегонка заканчивается при температуре выше 300 °C. Время перегонки 45 мин— 1 ч. Колбу снимают, внутреннюю трубку холодильника промывают 5—8 мл хлористоводородной кислоты, разбавленной (1:15), промывную жидкость собирают в тот же приемник и определяют фтор, как указано в разделе VIII. 6.1, но в качестве реагента применяют алюминий-ализариновый комплекс.
V III. 6.3. Определение фтора в воде [94—97]
Ход анализа. Испытуемую воду (1 л) подщелачивают и выпаривают досуха. Сухой остаток слегка прокаливают для разложения органических веществ, растворяют в небольшом количестве воды и переносят в колбу для отгонки. Если в воде присутствуют большие количества хлоридов, в дистилляционную колбу вводят Ag2SO4. В дальнейшем поступают, как указано при анализе силикатов и горных пород.
V III. 7. ОПРЕДЕЛЕНИЕ ФТОРИДА ПО РЕАКЦИИ РАЗРУШЕНИЯ ПЕРЕКИСНОВОДОРОДНОГО КОМПЛЕКСА ТИТАНА
Фторид разрушает желтый перекисноводородный комплекс титана; в разбавленных растворах реакция идет с образованием мо-нофторидного комплекса титана:
[TiO(H2O2)]2++ F’ ч=± [TiOF]+ + Н2О2
В присутствии значительных количеств перекиси водорода равновесие устанавливается при значительной концентрации не вступивших в реакцию фторид-ионов. Поэтому график зависимости обесцвечивания от содержания ионов F- криволинеен.
Мешающие вещества. Железо, цирконий, торий, алюминий и другие элементы, образующие устойчивые комплексы с фторидом, мешают определению. Ванадий, молибден и церий мешают вследствие образования окрашенных комплексов с перекисью водорода. 300
Фосфаты, оксалаты, тартраты и другие комплексообразователи, связывающие титан, также мешают определению. Большие количества солей щелочных металлов частично разрушают перекисное соединение титана.
Измерение оптической плотности. Определение лучше вести по методу колориметрического титрования. Можно применять и непосредственное измерение оптической плотности, пользуясь точной калибровочной кривой, построенной по большому количеству точек.
Раствор перекисноводородного комплекса титана и избыток перекиси водорода при определении должны быть такими же, как при построении калибровочного графика. Оптическую плотность раствора измеряют при 400 нм.
Большой избыток перекиси водорода уменьшает чувствительность реакции, однако применение эквивалентного количества Н2О2 (в отношении титана) недопустимо, так как перекись водорода разлагается при стоянии, что приводит к неправильным результатам. Лучше всего применять 3—5-кратный избыток Н2О2.
Метод стандартной шкалы мало удобен, так как фториды при стоянии разъедают стекло, а перекись водорода легко разрушается.
Чувствительность метода — 5 мкг фтора в 25 мл конечного раствора.
VIII. 7.1. Определение фтора в отсутствие мешающих веществ
Реактивы
Сульфат титана. Навеску двуокиси титана от 0,2 до 0,3 г сплавляют в пла гиновом (можно в фарфоровом) тигле с 2—3 г гидросульфата калия и после охлаждения плав выщелачивают 10%-ным раствором H2SO4. Несплавленный остаток двуокиси титана, не растворившийся в серной кислоте, отфильтровывают, фильтрат разбавляют до 0,5 л кислотой и водой с таким расчетом, чтобы после разбавления раствор содержал 5% серной кислоты. Отбирают пробы по 50 мл и гравиметрическим или объемным (восстановление в редукторе с последующим титрованием трехвалентным железом) методом устанавливают титр раствора. Если сплавление с KHSO4 проводили в фарфоровом тигле, то титр раствора соли титана устанавливают только объемным методом. Затем полученный раствор разбавляют 5%-ной серной кислотой с таким расчетом, чтобы 1 мл раствора содержал 0,1 мкг титана.
Раствор соли титана можно также приготовить растворением точной навески х. ч. двуокиси титана в смеси серной и фтористоводородной кислот с последующим нагреванием до полного растворения двуокиси титана и выпари ванием с большим избытком H2SO4 для удаления фтористого водорода. Для полного удаления фтористого водорода выпаривание необходимо продолжать до выделения паров серной кислоты. Затем раствор охлаждают, разбавляют водой и выпаривание повторяют. Полученный раствор разбавляют водой и серной кислотой с таким расчетом, чтобы раствор после разбавления содержал 5% H2SO4 и 0,1 мг титана в 1 мл.
Наконец, стандартный раствор можно приготовить растворением навески сульфата титана в серной и фтористоводородной кислотах при нагревании и двухкратным выпариванием до выделения паров серной кислоты. Полученный раствор разбавляют и устанавливают его титр, как указано выше.
Стандартный раствор фторида, см. раздел VIII. 4.1.
Перепись водорода, 3%-ный раствор.
Ход анализа. Метод колориметрического титрования. Готовят раствор комплекса титана, добавляя к 40—45 мл раствора сульфата титана 3 мл 3%-ной перекиси водорода и разбавляя до 50 мл. Раствор перемешивают и наливают в бюретку. Испытуемый минеральнокислый раствор 25—30 мл помещают в цилиндр для колориметрирования и титруют из бюретки раствором перекисного соединения титана до заметной желтой окраски. В другой цилиндр для колориметрирования наливают из бюретки точно израсходованное количество раствора перекисного соединения титана и титруют стандартным раствором фторида до уравнивания окрасок испытуемого и стандартного растворов.
Необходимо проверить кислотность раствора сравнения, так как стандартный раствор фторида имеет делочпую реакцию и в процессе титрования кислотность раствора сравнения уменьшается. Если нужно, раствор подкисляют серной кислотой, разбавленной (1:4). Кислотность раствора должна соответствовать значению pH не выше 2,5.
Чувствительность такого варианта определения — 10 мкг фтора в 50 мл конечного раствора.
Измерение оптической плотности при помощи спектрофотометров и фотоколориметров. Берут две мерные колбы емкостью 25 мл, в одну из них наливают испытуемый раствор, содержащий 5—100 мкг фтора, затем в обе колбы наливают по 5 мл приготовленного раствора перекисного комплекса титана. Оба раствора разбавляют водой до метки и перемешивают. Приготовленные растворы наливают в кюветы и измеряют оптическую плотность раствора сравнения по сравнению, с исследуемым раствором при 400 нм.
Для построения калибровочного графика поступают точно так же, как описано выше, только вместо исследуемого раствора берут различные количества стандартного раствора фторида.
VIII. 8. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ФТОРИДА
Цирконализариновый комплекс применяется для определения фторида в стекле [98], природных фосфатах [99], окислах и шлаках [100], силикатах и фосфатных породах, смолах и каменных метеоритах [101, 102], хлорированной воде [103], морской соли [104], напитках [105], лекарствах [106], водных растворах [107] и в других материалах [108, 109].
Фотометрическое титрование лантаном, церием и иттрием применено для определения фторидов. В качестве индикатора применяют 4-(2-пиридилазо)-резорцин [ПО].
Арсеназо I образует окрашенные комплексы с рядом металлов, в том числе с цирконием и алюминием. Реакции разрушения этих комплексов применяются для фотометрического определения фторида [111 —113]. Этот метод применен для определения фтора в горных породах [114], апатито-нефелиновых рудах и апатитах [115], 302
растворах, содержащих фосфаты [116], и в подземных водах [117]. Подобно арсеназо I взаимодействует торон, ториевый комплекс которого применяется для определения фтора [118].
Пирокатехиновый фиолетовый и ксиленоловый оранжевый образуют интенсивно окрашенные комплексы с рядом металлов. Фотометрическое определение фтора основано на реакциях разрушения этих комплексов фторид-ионами. Для этого применяют комплексы циркония [119—123], тория [124] и скандия [125, 126].
Реакция разрушения фторидом комплексов тория [127, 128] и скандия [129] с метилтимоловым синим также применена для фотометрического определения фторидов.
Разработано много других методов определения фтора, основанных на разрушении различных комплексов металлов. Кроме описанных выше для определения фтора применяют реакции разрушения комплексов циркония с хромфиолетовым [130], тория с шиффовыми основаниями [131], 4-(о-арсонофеиилазо)-1Ч-(1-на-фтил)-этилендиамином [132], хромазуролом S [133], 2-(1,8-диокси-3,6-дисульфо-2-нафтилазо)-феноксиуксусной кислотой [134], амарантом [135], хлоранилатом [136], СПАДНС [137], алюминия с гематоксилином [138—140], алюминоном [141], морином [142], хинализаринсульфонатом [143], лантана с хлораниловой кислотой [144], ализаринфлуориновым синим [145], титана с аскорбиновой кислотой [146], хромотроповой кислотой [147, 148], перекисью водорода [149—151], железа с ацетилацетоном [152], 2,4-диоксиацетофе-ноном [153], резацетофеноном, 5-фенилсалициловой кислотой и Р-резорцилальдоксимом [154], сульфосалициловой кислотой [155], 7-иод-8-оксихинолин-5-сульфокислотой [156] и с роданидом [157].
Разработан косвенный метод определения очень малых количеств фторида, основанный на отгонке тетрафторида кремния, проведении гидролиза и определении кремневой кислоты в виде кремнемолибденовой сини [158, 159].
Для фотометрического определения фтора предложена каталитическая реакция между цирконием и метилтимоловым синим [160]. Скорость реакции определяют по увеличению оптической плотности растворов комплекса метилтимолового синего с цирконием при 586 нм.
ЛИТЕРАТУРА
1. Willard Н. Н., Winter О. В., Ind. Eng. Chem., Anal. Ed., 5, 7 (1933).
2. P e n f i e 1 d S. L„ Z. anal. Chem., 21, 120 (1882).
3. Magerhofer A., Wasitzky A., Korn N., Mikrochemie, 20, 29 (1936).
4. Hartmann H., Ch у trek E., Ammon R., Z. phys. Chem., 265, 52 (1940).
5. P о з а н о в С. H., Зав. лаб., 3, 791 (1934).
6. W a g n e r C. R., Ross W. H., Ind. Eng. Chem., 9, 1116 (1917).
7. Armstrong W. D., Ind. Eng. Chem., Anal. Ed., 5, 300 (1933).
8 T а н а н a e в И. В., ЖПХ, 5, 884 (1932).
9 Та п а па ев И. В., ЖОХ, 8, 1120 (1938).
10. Т а в а п а е в И. В., А б и л о в С. Т„ ЖПХ, 15, 61 (1942).
11. Та на наев И. В., Левина М. И., Зав. лаб., 9, 805 (1945).
12. Deussen Н., Anorg. Chem., 10, 18 (1905).
13. Fox Е. G., J а с к s о n W. A., Anal. Chem., 31, 1657 (1959).
14. Marczenko Z., C h о 1 u j - L e n a r c z у k, Chem. anal., 13, 405 (1968).
15. S h e 11 H. R„ Anal. Chem., 32, 1529 (1960).
16- Тредвелл Ф. П„ Голл В. T. Качественный анализ. М. Госхимиздат, 1946. См. с. 477.
17. Годнева М. М., Мотов Д. А., Зав. лаб., 36, 151 (1970).
18. Николаев Н. С. и др. Аналитическая химия фтора. М., «Наука», 1970. См. с. 62.
19. S i 1 v е г m a n Н. Р., Anal. Chem., 31, 1960 (1959).
20. A n s о 11 Е. Y., Е 11 i s W. Р„ Anal. Chem., 28, 393 (1956). •
21. Wart J„ Cline W. D., Те veb a u gh R. D., Anal. Chem., 26, 342 (1954).
22. G a m b 1 e L. W., Anal. Chem., 32, 189 (1960).
23. Johnson C. A., Leonard M. A., Analyst, 86, 101 (1961).
24. Bock R., Semmler H., Ben rends K-, Naturwis., 53, 305 (1966).
25. В о c k R., S e m m 1 e r H., Z. anal. Chem., 230, 161 (1967).
26. Хи рано Садзо, Каски Тосидзи, цит. по РЖХим, 1967, 13Г101.
27. Сох F. Н„ Pharmac. Weekbl., 99, 801 (1964).
28. W i n k h a u s е J., U h г i g H., Z. anal. Chem., 200, 14 (1964).
29. I r w i n g H., С о x J., J. Chem. Soc., 1470 (1961).
30. Moffett K., Simmler J., H. Rotretz H., Anal. Chem., 28, 1356 (1956).
31. Bowen U. H., Rood R. T., J. Inorg. and Nucl. Chem., 39, 1776 (1967).
32. В о c k R., J a i n z J., Z. anal. Chem., 198, 315 (1963).
33. В о c k R., В e i 1 s t e i n J., Z. anal. Chem., 198, 44 (1963).
34. К лете ник Ю. Б., Быховская И. А., ЖАХ, 25, 351 (1970).
35. Тиганов Г. П. и др., Сб. науч. тр. Всес. н.-и. горно-металлургического ин-та цветн. мет., № 19, 93 (1970).
36. Фунасака Кананэ, Кадзима Мацуда, Бунсэки Кагаку, 4, 514 (1955); цит. по РЖХим, 1956, № 13, 246.
37. F г а п k W. В., F о s t е г L. М., J. Phys. Chem., 64, 310 (1960).
38. R о b s о n J. W., A s к e w W. B., J. Chromatogr., 7, 409 (1962).
39. Shehyn H., Anal. Chem., 29, 1466 (1957).
40. Y a m a m u r a S. S., Wade M. A., Sikes J. H., Anal. Chem., 34, 1308 (1962).
41. Yamamura S. S., Anal. Chem., 34, 1312 (1962).
42. Y a m a m u г a S. S., К u s s у M. E., R e a n J. E., Anal. Chem., 33, 1635 (1961).
43. Martini M., Tonani F., Atti Accad. naz. Lincei. Rend. Cl. Sci. Fis., mat. e natur, 42, 683 (4967).
44. H u с к a b a у W. B., Welch E. T., Metier A. V., Anal. Chem., 19, 154 (1947).
45. R i c h t e r F., Z. anal. Chem., 124, 161 (1942).
46. Reynolds D. S., Kerschaw J. K. D., J. Assoc. Off. Agr. Chem., 19, 156 (1936).
47. Harris S. E., Christiansen W. G., J. Amer, pharm. Assoc., 25, 306 (1936).
48. Belcher R., Leonard M. A., West T. S„ J. Chem. Soc., 3577 (1959).
49. Belcher R., Leonard M. A., West T. S., J. Chem. Soc., 4477 (1960).
50. Belch er R, West T. S., Taianta, 8, 853 (1961).
51. Belcher R., West T. S., Taianta, 8, 863 (1961).
52. Мустафин и др. Ассортимент реактивов на фтор, М., НИИТЭХИМ, 1970. См. с. 29.
53. М и н и н А. А., Филиппова Л. П. Ализариновый комплексон. Реактив для определения фтора, М., НИИТЭХИМ, 1971. См. с. 18.
54. N о w i с k а - J а п о w з к а Т., Chem. anal. (PRL), 16, 961 (1971).
55. Greenh а 1 gh R., Riley J. P., Anal. Chim. Acta, 25, 179 (1961).
56. H a n о c q M., M о 11 e L., Anal. chim. Acta, 40, 13 (1968).
57. H a n о c q M., M о 11 e L., Anal. chim. Acta, 42, 349 (1968).
58 M о 1 1 e L., H a n о c q M., Pharmac. Weekbl., 104, 743 (1969),
59. Brunzie G. F., Pflaum R. T., Proc. Jowa Acad. Sci., 69, 186 (1963).
60. Kubota H„ Mikrochem. J., 12, 525 (1967).
61. Минин А. А., Бармина Г. А., Филиппова А. П., Уч. зап. Пермского гос. ун-та, № 141, 247 (1966).
62. Kletsch R. A., Richard Fr. A., Anal. Chem., 42, 1435 (1970).
63. Киселева Е. К. Анализ фторсодержащих соединений. Л., «Химия», 1966. См. с. 219.
64. К е m р t Th., J. anal. Chem., 244, 113 (1969).
65. V a 1 a c h R., Taianta, 9, 341 (1962).
66. Megregian S., Anal. Chem., 26, 1161 (1954).
67. T h a t c h e r L. L„ Anal. Chem., 29, 1709 (1957).
68. Oelschlager W., J. anal. Chem., 191, 408 (1962).
69. S a r m a P. L., Anal. Chem., 36, 1684 (1964).
70. Dixon E. J., Analyst, 95, 272 (1970).
71. P i n x t e r e n J. A. C., Pharmac. Weekbl., 94, 649 (1959).
72. T h r u n e W. E., Anal. Chem., 22, 918 (1950).
73. Visintin B., Monteriolo S., Rend. Inst, super, sanita, 21, 338 (1958).
74. Macnulty B. J., Hunter G. J., Barrett D. G„ Anal. Chim. Acta, 14, 368 (1956).
75. Kiciak S„ Rozpr. Politechn. poznanska, № 21, 70 (1967).
76. Гицова С., Хигиена и здравеоназване, 13, 198 (1970).
77. Bender H., Arch. Hyg. und Bakteroil, 145, 586 (1961).
78. Ewans W. H., Sergeant G. A., Analyst, 92, 690 (1967).
79. Hu a n g W. H, Joh ns W. D., Anal. chim. Acta, 37, 508 (1967).
80. Коренман И. M„ Корнакова А. А., Тр. по химии и хим. технол., Горький, вып. 3, 584 (1959).
81. G u n t з А. А., А г е n е М., Anal. chim. Acta, 19, 580 (1958).
82. 3 е н и н Л. А., Гидрохим. материалы гидрохим. ип-та АН СССР, 22, 129 (1954).
83. Lothe J. J., Anal. Chem., 28, 949 (1956).
84. Ш а п и р о М. Я., Колесникова В. Г., ЖАХ, 18, 507 (1963).
85. Y a s u d a S. К-, Lambert J. L., Anal. Chem., 30, 1485 (1958).
86. Гурьев С. Д., Иоффе В. П., Сб. науч. тр. Гинцветмета, № 14, 61 (1958).
87. Р i с k h а г d t W. Р., Anal. Chem., 34, 863 (1962).
88. Guntz A. A., Arene M., Chem. anal. (Polska), 40, 39 (1958).
89. I с к e n J. M„ В 1 a n к В. M„ Anal. Chem., 25, 1741 (1953).
90. Заманов P. X., Рафикова P. Ш., Сб. науч. тр. Самаркандского мед. ип-та, 37, 298 (1967).
91. Evans L., Hoyle R. D., Ma саз kill J. B., N. Z. J. Sci., 13, 143 (1970).
92. Quentin К. E., Ro so pul о A., Z. anal. Chem., 241, 241 (1968).
93. Isharan R., Adati T., Denki Seiko, Electr. Furrrance Steel, 38, 66 (1967).
94. Виноградов А. П., Данилов В. В., Селиванов А. С., ДАН СССР, 14,361 (1937).
95. М е у 1 i n g А. Н., Analyst, 88, 84 (1963).
96. Shaw W. М, Anal. Chem., 26, 1212 (1954).
97. Danielsen M. E., Arbok Univ. Bergen Naturvidensk, № 9, 1 (1953/1954).
98. Allison R. S., J. Soc. Glass Technol., 37, № 177, 213T (1953).
99. V i g i e r R., Bull. Soc. Chim. France, № 2, 160 (1957).
100. Bohnstedt Uirich, DEW-Techn. Ber., 2, 34 (1962).
101. SenGuptaJ. G., Anal. chim. Acta, 42, 119 (1968).
102. Hall A., Walsh J. N., Anal. Chim. Acta, 45, 341 (1969).
103. W i e r z b i c k 1 T., Pawlita W., Chem. anal. (Polska), 14, 1193 (1969).
104. Venkatarayachari K., Rao R. J., Naidu M. G. Ch., Salt. Res. and Ind., 6, 79 (1969).
105. C h i о f f i V., Boll. Lab. Chim. provinc., 5, 9 (1954).
106. Vodelenzang E. H., Pharmac. Weekbl., 91, 905 (1956).
107. Л у б я н с к а я Е. Н., Зав. лаб., 22, 921 (1956).
108. Гурин П. А., Науч. тр. Иркутского н.-и. ин-та редких мет., № 10, 87 (196).
109. Масалович В. М„ Тр. Уральского н.-и. хим. ин-та, вып. 174 141 (1968).
ПО. Harzdorf Cl., Z. anal. Chem., 233, 348 (1968).
111. Кутейников А. Ф., ЖАХ, 16, 327 (1961).
112. К у т е й н и ко в А. Ф., Бродская В. М., Ланской Н. А., ЖАХ, 17, 87 (1962).
113. Шемякина М. А., Вопр. прикл. геохим., вып. 1, М., «Недра», 1966. См. с. 128.
114. Курильчикова Г. Е., ЖАХ, 25, 563 (1970).
115. И ж а к И. Г., Зав. лаб., 28, 907 (1962).
116. Жендарева О. Г., Прохорова В. Г., Конышева Л. И. В сб.: «Физ.-хим. методы анализа мет. и сплавов». М., 1969. См. с. 59.
117. Мальков Е. М., Косарева В. Г. В сб. «Новые методы анализа хим. состава подземных вод». М., 1967. См. с. 59.
118. Ingrem В. L., Geol. Surv. Profess. Paper, № 450-Е, 130 (1963).
119. Krahulec L., Ceskosl. hyg. epidemiol. Mikrobiol., imunol., 4, 367 (1955).
120. Ruziska J. A., Collect. Czechosl. Chem. Communs, 30, 2717 (1965).
121. Шпак И. Г., Власова Л. Г. В сб. «Методы анализа руд Кольского полуострова», «Апатиты», 1970. См. с. 194.
122. Кукишева Т. Н., Синицына Е. С. Авт. св. СССР, кл. 42в, 3/08, (gOln), № 234732, заявл. 2.11.62, опубл. 24.07.70.
123. С a b е 11 о - Т о m a s М. L., W е s t Т. S., Taianta, 16, 781 (1969).
124. R е z а с Zd„ D i t z J., Z. anal. Chem., 186, 424 (1962). 4
125. M a r c z e n k о Z., Lenarczyk L., .Chem. anal. (PRL), 15, 607 (1970).
126. Смирнов С. M. В сб. «Микроэлементы в биосфере и применение их в. с. х. и мед. Сиб. и Дальн. Вост.», Улан-Удэ, 1971. См. с. 504.
127. Н 1 и с h а п Е., М а у е г J., Chem. Zvesti, 17, 569 (1963).
128. Uh Hr H., Chem. Zvesti, 18, 756 (1964).
129. M a r c z e n k о Z., Lenarczyk L., Chem. anal. (PRL), 15, 753 (1970).
130. Жендарева О. Г., Лисенкова Л. П. Авт. свид. СССР, кл. 421, 3/08, (gOln), № 194405, заявл. 22.03.66, опубл. 27.05.67.'
131. Р о d d а г S. N., D е у К-, J. Indian. Chem. Soc., 41, 731 (1964).
132. L i d d е 11 L., Analyst, 78, 494 (1953).
133. R e v i n s о n D., Harley J. H., Anal. Chem., 25, 794 (1953).
134. T о c i К у о j i e. oL, Bull. Chem. Soc. Japan, 39, 638 (1966).
135. L a m d er t, Anal. Chem., 26, 558 (1954).
136. H e n s 1 e у A. L., В a r n e у J. E„ Anal. Chem., 32, 828 (1960).
137. В a n e r j e e G., Anal. chim. Acta, 13, 409 (1955).
138. Product. Group. U. K. Atomic Energu Author. Rept, 1961, № 191 (CA), p. 8.
139. McNulty B. J., Hunter G. J., Anal. chim. Acta, 9, 425 (1953).
140. Beveridge J. S., Hunter G. H., McNulty B. J., Anal. chim. Acta, 9, 330 (1953).
141. H й b ne r M„ Chem. Erde, 22, 264 (1962).
142. Beck M., Acad. Sci. Hung., 4, 223 (1954).
143. N e w m a n A. C. D„ Anal. chim. Acta, 19, 580 (1958).
144. Hayashi K-, Danzuka T., Ueno K., Taianta, 4, 126 (1960).
145. Leonard M. A., Nagi F. L, Chem. Communs, 20, 1224 (1968).
146. Schall E. D., Williamson H. G., J. Assoc. Offic. Afric. Chemists, 38, 454 (1955).
147. Новак В. П., Б о г о в и п а В. И., Мальцев В. Ф., Зав. лаб., 31, 278 (1965).
148. С к а н а в и М. Д., Зав. лаб., 24, 683 (1958).
149. К о г i t п i g S., Z. anal. Chem., 193, 1 (1950).
150. Клейнер К. E., Зав. лаб., 17, 398 (1951).
151. L a s i e w icz К., Przem. Chem., 10, 36 (1954).
152. McKaveneyJ. P., Anal. Chem., 40, 1276 (1968).
153. Liang Sh. Ch., Hsieh K., Scientia Sinica, 6, 1039 (1957).
154. N i c h о 1 s M. L., С о n d о A. C., Anal. Chem., 26, 703 (1954).
155. Sen B., Z. anal. Chem., 153, 168 (1956).
156. Liang Sh. C h e n g - у ii, Scientia Sinica, 6, 259 (1957).
157. Фельдман M. M., Зав. лаб., 10, 208 (1941).
158. C u г г у R. P., M e 11 о n M. G., Anal. Chem., 28, 1567 (1956).
159. П e p e г у д E. А., Б о н к и н а Б. С., ЖАХ, 17, 611 (1962).
160. Hems R. V., Ki г kb right G. F., West T. S„ Taianta, 17, 433 (1970),
Глава IX
ХЛОР
IX. 1. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ ОПРЕДЕЛЕНИЯ
И МЕТОДЫ ОТДЕЛЕНИЯ
Хлор в различных соединениях находится в степени окисления — 1, +1, +3, +5 и -j-7. Прямых фотометрических методов определения ионов хлора нет, поэтому для их определения обычно применяют либо турбидиметрический (или нефелометрический) метод или пользуются косвенными фотометрическими методами, основанными на разрушении соединений, поглощающих свет. Во всех случаях ионы хлора необходимо предварительно отделять, особенно необходимо отделение от бромидов, иодидов и роданидов, которые всегда мешают определению ионов хлора.
Для отделения бромидов и иодидов применяют селективное их окисление до свободного состояния и последующее отделение путем отгонки или экстракции. Селективное окисление иодидов и бромидов проводят, перманганатом в смеси с сульфатом марганца, предотвращающим окисление хлоридов, а также йодатом калия в 0,2—0,4 М растворе азотной кислоты или перйодатом калия в сернокислой среде.
После удаления бромидов и иодидов хлориды можно осадить в виде хлорида серебра. Однако такой метод мало удобен, так как для дальнейшего определения хлоридов необходимо переведение в раствор малорастворимой соли; определение хлоридов можно проводить и по количеству перешедшего в осадок серебра. Хлорид можно отделить также методом отгонки в виде хлористого водорода из сернокислой среды при 150 °C.
Для окисления хлорида до свободного хлора применяют перманганат и хромат в сернокислой среде. Образующийся хлор отгоняют в токе азота. Этот метод удобен тем, что дальнейшее фотометрическое определение хлора является более легкой задачей, чем определение хлоридов.
IX. 2. ОПРЕДЕЛЕНИЕ ХЛОРИДА ПО РЕАКЦИИ РАЗРУШЕНИЯ КОМПЛЕКСА РТУТИ [II] С ДИФЕНИЛКАРБАЗОНОМ [1—11]
В методе использована реакция между комплексом ртути(П) с дифенилкарбазоном и хлорид-ионами:
Комплекс ртути(II) с дифенилкарбазоном при взаимодействии с хлорид-ионами разрушается и интенсивность красной окраски уменьшается.
Мешающие вещества. Определению хлоридов мешают ацетаты, бромиды, иодиды, роданиды, оксалаты и сульфиды, которые, так же как хлорид-ионы, разрушают комплекс ртути(II) с дифенилкарбазоном, а также ионы меди, железа, кобальта, цинка, кадмия и свинца, которые взаимодействуют с дифенилкарбазоном с образованием окрашенных соединений. Поэтому указанные ионы должны быть предварительно удалены. Небольшие количества меди можно замаскировать триэтаноламином. Реакцию проводят при pH = 3, при этом мешающее действие ионов металлов уменьшается.
Измерение оптической плотности. Спектр поглощения хлороформного раствора комплекса ртути(II) с дифенилкарбазоном представлен на рис. 69. Оптическую плотность
измеряют при 520
Рис. 69. Спектр поглощения хлороформного раствора комплекса ртути(П) с дифенилкарбазоном.
нм. Используется также метод стандартной
шкалы.
Чувствительность метода — 0,8 мкг хлорида в 5 мл экстракта.
IX. 2.1. Определение хлорида в отсутствие мешающих веществ
Реактивы
Стандартный раствор хлорида, содержащий в 1 мл 1 мг С1". Навеску хлорида натрия 1,6486 г, прокаленного при 400—500 °C, растворяют в воде, переносят в мерную колбу емкостью 1 л, разбавляют до метки водой и перемешивают. Рабочие растворы готовят путем разбавления этого раствора.
Дифенилкарбазон, 1%-ный раствор очищенного препарата в этаноле. Торговый препарат часто содержит примесь дифенилкарбазида. Для его очистки навеску препарата 5 г растворяют в 75 мл теплого этанола и прибавляют 25 г карбоната натрия, растворенного в 400 мл воды. После охлаждения экстрагируют 6 раз диэтиловым эфиром: порциями 100 мл, два раза по 50 мл и 3 раза по 35 мл. Эфирный экстракт, содержащий дифенилкарбазид, отбрасывают. Щелочной раствор подкисляют 3 н. азотной кислотой или ледяной уксусной кислотой, которые осаждают дифенилкарбазон. Выпавший продукт фильтруют через воронку Бюхнера. Очищенные кристаллы дифенилкарбазона с т. пл. 118— 120 °C устойчивы при комнатной температуре в течение года.
Нитрат ртути(Н), 0,001 М раствор. Сначала готовят 0,05 М раствор. Содержание ртути устанавливают титрованием хлоридом в присутствии дифенилкарбазона. Затем разбавлением готовят 0,001 М раствор.
Хлороформ, ч. д. а.
Буферная смесь, pH = 3,4. Навеску 5,2 г формиата натрия растворяют в 700 мл воды, 1 н. раствором HNO3 доводят pH до 3,4, разбавляют водой до 1 л и перемешивают.
Ход анализа. До 30 мл исследуемого раствора, содержащего до 50 мкг ионов хлора, нейтрализуют азотной кислотой или раствором щелочи до pH = 3, прибавляют 5 мл буферной смеси и раствор переносят в делительную воронку емкостью 100 мл. Затем прибавляют 1 мл раствора нитрата ртути(II), раствор перемешивают, прибавляют 1 мл раствора дифенилкарбазона, перемешивают и спустя 5 мин прибавляют 3 мл хлороформа. Содержимое воронки встряхивают, оставляют до разделения фаз и слой хлороформа сливают в мерную пробирку емкостью 5 мл. Раствор разбавляют хлороформом до 5 мл, перемешивают и измеряют оптическую плотность при 520 нм.
Содержание хлорида находят по предварительно построенному калибровочному графику (рис. 70).
IX. 2.2. Определение хлорида в воде [11, 12]
Тяжелые металлы предварительно экстрагируют хлороформом в виде комплексов с дифенилкарбазоном. Затем определение хлоридов проводят, как указано в разделе IX. 2.1.
Рис. 70. Калибровочный график для определения хлоридов по реакции разрушения комплекса ртути(П) с дифенилкарбазо-пом.
в
IX. 2.3. Определение хлорида в двуокиси титана особой чистоты [13]
Навеску двуокиси титана 1 г помещают пробирку с притертой пробкой, прибавляют 5мл
0,01 н. азотной кислоты и встряхивают в течение 2—3 ч. Затем раствор центрифугируют и в центрифугате определяют хлорид, как указано в разделе IX. 2.1.
Метод дает возможность определить до 10-4% хлорида.
IX. 2.4. Определение хлорида в чистой сере [14]
Навеску серы 4 г помещают в склянку с притертой пробкой, смачивают этанолом, прибавляют 10 мл бидистиллята и встряхивают 2 ч. Раствор центрифугируют и в центрифуге определяют хлорид, как указано в разделе IX. 2.1.
IX. 2.5. Определение хлорида в сыворотке крови [6]
В центрифужную пробирку емкостью 12 мл вводят 0,1 мл сыворотки, 7,2 мл воды, 1,0 мл 0,3 н. раствора гидрата окиси бария и 1,0 мл 5%-ного раствора сульфата цинка. Центрифугируют 3— 5 мин, отбирают 1 мл прозрачного раствора, разбавляют водой до 15 мл, нейтрализуют 0,05 н. азотной кислотой и продолжают определение, как указано в разделе IX. 2. 1.
IX. 3. ОПРЕДЕЛЕНИЕ ХЛОРИДА С ПРИМЕНЕНИЕМ ХРОМАТА СЕРЕБРА
Хлорид в нейтральной среде взаимодействует с твердым хроматом серебра с образованием малорастворимого осадка AgCl:
Ag2CrO4 + 2Cl —> 2AgCl + CrO4-
Перешедший в раствор хромат определяют по поглощению его при 373 нм или по реакции с дифенилкарбазидом.
Мешающие вещества. Определению хлоридов мешают все анионы, соли которых с серебром малорастворимы в воде, в том числе
бромид, иодид, роданид, сульфид, фосфат, тиосульфат, оксалат и др. Кроме того, определению мешают ионы, которые образуют малоррстворимые в воде хроматы (бария, стронция), а также ионы, имеющие собственную окраску.
Измерение оптической плотности. Для измерения оптической плотности лучше применять спектрофотометр (ХМакс = 373нм), других приборах. С увеличением оптической плотности растворов
Рис. 71. Спектр поглощения хромата в аммиачной среде.
но можно вести измерение и на длины волны при измерении
хроматов снижается чувствительность определения хлоридов (рис. 71).
Чувствительность метода — 0,03 мг хлорида в 25 мл раствора.
IX. 3.1. Определение хлорида в отсутствие мешающих веществ [15]
Реактивы
Хромат серебра. К 100 мл кипящего 10%-ного раствора нитрата серебра медленно при перемешивании прибавляют 200 мл 5,5%-ного раствора хромата калия.
Выпавший темно-бурый осадок отфильтровывают с отсасыванием, промывают 2 л воды и высушивают над серной кислотой в вакууме. Сухой порошок пропускают через сито 100 меш и хранят в банке из желтого стекла.
Стандартный раствор хлорида, см. раздел IX. 2.1.
Ход анализа. В мерный цилиндр с притертой пробкой емкостью 25 мл помещают навеску 0,05 г порошка хромата серебра и наливают 25 мл нейтрального исследуемого раствора. Раствор энергично перемешивают в течение 5 мин, дают отстояться и отфильтровывают аликвотную часть раствора.
Фильтрат наливают в кювету спектрофотометра и измеряют оптическую плотность раствора при 373 нм по отношению к воде.
Содержание хлорида находят по предварительно построенному калибровочному графику.
IX. 4. ТУРБИДИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ХЛОРИДА
В ВИДЕ ХЛОРИДА СЕРЕБРА [15]
В методе использована реакция образования взвеси хлорида серебра:
СГ + Ag+ —► AgCl
Помутнение раствора измеряют визуально или с помощью фотоколориметров. Определению мешают анионы, образующие малорастворимые в разбавленной азотной кислоте соли серебра.
Чувствительность метода — 5 мкг хлорида в 25 мл конечного раствора.
Реактивы
Стандартный раствор хлорида, см. раздел IX. 2.1.
Эталонный раствор. В мерную колбу емкостью 25 мл прибавляют стандартный раствор хлорида, содержащего 0,4 мг хлорида натрия, и разбавляют водой до 20 мл; затем прибавляют 2,5 мл азотной кислоты, разбавленной (1 : 160), и 2,5 мл 0,1%-ного раствора нитрата серебра.
Азотная кислота, 1 мл концентрированной азотной кислоты приливают к 160 мл воды и перемешивают.
Нитрат серебра. Растворяют 0,1 г AgNCh в 100 мл воды.
Ход анализа. В пять мерных колб емкостью 25 мл наливают по 2,5 мл азотной кислоты. В четыре колбы добавляют разные количества стандартного раствора хлорида, а в пятую—до 20 мл исследуемого раствора, содержащего от 5 до 15 мкг хлорида. Все растворы разбавляют водой до ~22 мл, добавляют в каждую колбу по 2,5 мл раствора нитрата серебра, доводят водой до метки и перемешивают. Содержимое колб нагревают в течение 30 мин при 40°C на водяной бане, затем быстро охлаждают до комнатной температуры в смеси льда с водой.
Помутнение растворов измеряют на фотоэлектрическом нефелометре и по стандартным растворам строят калибровочный график, который используют для нахождения содержания хлоридов в исследуемом растворе.
IX. 5. ОПРЕДЕЛЕНИЕ СВОБОДНОГО ХЛОРА
С ПРИМЕНЕНИЕМ о-ТОЛИДИНА
В кислом растворе хлор взаимодействует с о-толидином с образованием продукта реакции, окрашенного в желтый цвет:
H2N
Химизм этой реакции окончательно не выяснен. В зависимости от кислотности раствора и соотношения хлора и о-толидина кроме
желтого продукта образуются темно-коричневый, голубой и другие продукты реакции [16]. Поэтому необходимо придерживаться определенных условий проведения реакции.
Мешающие вещества. Определению хлора мешают другие окислители, в том числе нитриты, железо(III) и марганец в высших степенях окисления (кроме Мп11), если их количества превышают 0,9; 0,2 и 0,01 мкг/мл соответственно [15].
Оптимальным интервалом pH является 0,8—3,0. При более высоком значении pH образуется соединение сине-зеленого цвета, которое быстро разрушается. Избыток реагента должен превышать
концентрацию хлора не менее чем в три раза.
Измерение оптической плотности. В условиях оптимального значения pH спектр раствора продукта реакции имеет полосу поглощения с максимумом при 438 нм (рис. 72). Оптическую плотность лучше измерять на спектрофотометре или фотоколориметре при 438 нм.
Чувствительность метода — 5 мкг хлора в 25 мл ^конечного раствора.
Рис. 72. Спектр погло-
IX. 5.1. Определение хлора в отсутствие мешающих веществ [17, 18]
щения продукта реак- Реактивы пии хлора с о-толиди-
ном. Стандартный раствор хлора, содержащий в 1 мл
0,01 мг С12. В колбу наливают 500 мл дистилировай-ной воды и медленно на холоду пропускают Ток хлора из баллона в течение 20 мин. Пипеткой отбирают 50 мл раствора в коническую колбу с пробкой емкостью 250 мл, содержащую 10 мл 10%-ного раствора иодида калия. При этом выделяется иод, который оттитровывают 0,1 н. раствором тиосульфата в присутствии крахмала. Рассчитывают содержание хлора в 1 мл. С помощью бюретки отбирают объем раствора, содержащего 10 мг хлора, и переносят его в колбу емкостью 1 л. Раствор разбавляют до
метки дистиллированной водой и хорошо перемешивают. Полученный раствор содержит 0,01 мг хлора в 1 мл.
о-Толидин. Растворяют 0,1 г солянокислого о-толидина в 15 мл хлористоводородной кислоты и разбавляют до 100 мл водой. Раствор сохраняют
в склянке из темного стекла.
Ход анализа. В мерные колбы емкостью 25 мл помещают по 1,5 мл раствора о-толидина. В одну из колб добавляют до 20 мл исследуемого раствора, содержащего до 25 мкг хлора. Разбавляют до метки дистиллированной водой и перемешивают. Для приготовления раствора сравнения в одной из колб раствор о-толидина разбавляют до метки дистиллированной водой. Спустя 9 мин после добавления раствора хлора измеряют оптическую плотность растворов при 438 нм.
Содержание хлора находят по калибровочному графику.
IX. 6. ОПРЕДЕЛЕНИЕ ХЛОРА ПО РЕАКЦИИ РАЗРУШЕНИЯ МЕТИЛОВОГО ОРАНЖЕВОГО [19, 20]
Метиловый оранжевый разрушается под действием хлора:
HO3S——N=N-----------N(CH3)2 + C12 + H2O —
—> (CH3)2N——Cl + НО——SO3H + N2 + НС1
аналогично протекает реакция с метиловым красным [21]
В результате окисления индикаторов хлором интенсивность окраски их растворов уменьшается. Это используется для косвен
ного фотометрического определения хлора. Интенсивность окраски
индикаторов зависит от pH раствора. В кислой среде интенсивность окраски выше. Уменьшение интенсивности окраски зависит также от порядка смешивания растворов. При добавлении хлора и перемешивании раствора наблюдается более значительное ослабление окраски по сравнению с ослаблением окраски в том случае, если раствор хлора прибавляют без пере
мешивания.
Мешающие вещества. Определению хлора мешают другие окислители, которые взаимодействуют аналогично, например бром, поэтому окислители должны отсутствовать. Преимуществом с о-толидиновым методом является то,
Рис. 73 Спектр поглощения раствора метилового оранжевого при pH = 2 (/) и метилового красного в 2 н. H2SO4 (2).
этого метода по сравнению что железо(III) и некото-
рые вещества, содержащие активный хлор, например хлорамин,
не мешают определению хлора.
Измерение оптической плотности. В случае применения метилового оранжевого оптическую плотность измеряют при 505 нм, а в случае метилового красного — при 515 нм (рис. 73). В том и другом случае лучше применять спектрофотометры или фотоэлектроколориметры.
Чувствительность метода — 6 мкг хлора в 25 мл конечного
раствора.
IX. 6.1. Определение хлора в отсутствие мешающих веществ
Реактивы
Стандартный раствор хлора, см. раздел IX. 5.1.
Метиловый оранжевый. Навеску препарата 0,06 г растворяют в воде и разбавляют до 1 л.
Раствор с pH = 2. В мерную колбу емкостью 1 л наливают 37 мл 0,2 н. хлористоводородной кислоты и 250 мл 0,2 н. хлорида калия, разбавляют водой до метки и перемешивают.
Ход анализа. В мерную колбу емкостью 25 мл помещают по 2,5 мл раствора метилового оранжевого и примерно 10 мл раствора с рН = 2, а затем при энергичном перемешивании до 10 мл исследуемого раствора. Доводят до метки раствором с pH = 2, перемешивают и измеряют оптическую плотность при 505 нм относительно воды.
Содержание хлора находят по предварительно построенному калибровочному графику (стандартный раствор добавляют до 2,5 мл; обработка эталонных растворов аналогична обработке исследуемого раствора).
IX. 6.2. Определение хлорида
Хлорид окисляют до хлора, который отгоняют, поглощают раствором метилового оранжевого и концентрацию неразложившегося метилового оранжевого определяют фотометрически. Хлорид окисляют раствором перйодата калия в смеси с серной кислотой.
Реактивы
Стандартный раствор хлорида, см. раздел IX. 2.1.
Метиловый оранжевый, см. раздел IX. 6.1.
Раствор перйодата. К 300 мл воды осторожно прибавляют 200 мл концентрированной серной кислоты. Раствор охлаждают, переносят в колбу для перегонки емкостью 750 мл и добавляют 10 г КЮ4 (ч. д. а.). После растворения соли в колбу вносят несколько кусочков пористого материала, подсоединяют колбу к холодильнику и отгоняют ~200 мл воды, которую отбрасывают. Раствор в колбе охлаждают, прибавляют еще 200 мл воды й после присоединения колбы к холодильнику вновь отгоняют воду (~200 мл). Такая отгонка гарантирует полную очистку от хлоридов. Охлажденный раствор переносят в склянку для хранения и закрывают пробкой.
Раствор с pH = 2, см. раздел IX. 6.1.
Ход анализа. В колбу для перегонки емкостью 150—200 мл вносят 50 мл раствора перйодата, добавляют 15 мл воды, перемешивают, опускают несколько кусочков пористого материала и отгоняют 15 мл воды в приемник, в котором находится 2,5 мл раствора метилового оранжевого и 2,5 мл раствора с рН = 2. Раствор из приемника переносят в мерную колбу емкостью 25 мл, доводят водой до метки, перемешивают и измеряют оптическую плотность относительно воды. Оптическая плотность этого раствора должна быть равной оптической плотности раствора, приготовленного из
2,5 мл раствора метилового оранжевого, 2,5 мл раствора с рН = 2 и доведенного до 25 мл водой. Если оптическая плотность раствора окажется меньшей, то в колбу для перегонки прибавляют Г5 мл воды и отгонку такого же количества воды с последующим измерением оптической плотности повторяют.
В подготовленный таким образом раствор перйодата прибавляют до 15 мл анализируемого раствора, содержащего до 25 мкг хлорида, если нужно, добавляют до 15 мл воды, перемешивают, вносят несколько кусочков пористого материала и отгоняют 15 мл дистиллята в приемник, в котором находится 2,5 мл раствора метилового оранжевого и 2,5 мл раствора с рН = 2. Раствор из приемника переносят в мерную колбу емкостью 25 мл, разбавляют водой до метки и продолжают определение, как указано в разделе IX. 6.1.
Если при отгонке хлора раствор метилового оранжевого полностью обесцвечивается, это означает, что в пробе слишком велико содержание хлоридов. В таком случае анализ необходимо повторить с меньшим количеством анализируемой пробы.
IX. 7. ОПРЕДЕЛЕНИЕ ПЕРХЛОРАТА С ПРИМЕНЕНИЕМ МЕТИЛЕНОВОГО ГОЛУБОГО [22, ’23]
Рис. 74. Спектр поглощения хлороформного экстракта ионного ассоциата перхлората с метиленовым голубым.
Ионы перхлората со многими органическими красителями образуют ионные ассоциаты, которые хорошо экстрагируются хлороформом, бензолом, толуолом и другими органическими растворителями. Эти экстракты интенсивно окрашены в цвета, подобные цвету соответствующего красителя. Максимумы поглощения спектров неводных растворов ионных ассоциатов сдвинуты по сравнению с максимумами поглощения спектров водных растворов соответствующих красителей всего на 5—15 нм. Эти реакции используются в фотометрических методах определения многих ионов.
Мешающие вещества. Определению перхлоратов с метиленовым голубым мешают нитраты, иодиды, иод, перйодаты и многие другие анионы, образующие подобные ионные ассоциаты. Элементный
иод предварительно отделяют экстракцией органическими растворителями или отгонкой.
Измерение оптической плотности. Светопоглощение хлороформного экстракта ионного ассоциата перхлората с метиленовым голубым соответствует закону Бугера — Ламберта — Бэра при содержании до 5 мкг перхлората в 10 мл. Оптическую плотность раствора можно измерять на любом приборе при 655 нм (рис. 74).
Чувствительность метода — 0,1 мкг перхлората в 10 мл хлороформного раствора.
IX. 7.1. Определение перхлората в отсутствие мешающих веществ
Реактивы
Стандартный раствор перхлората. Навеску перхлората натрия (NaClOJ 0,1232 г помещают в мерную колбу емкостью 100 мл, растворяют в воде, разбавляют водой до метки и перемешивают. Этот раствор содержит 10 мг ионов перхлората в 1 мл. Перед применением путем разбавления готовят раствор с меньшей концентрацией. В случае применения моногидрата или днгидрата перхлората берут навески 0,1412 г или 0,1594 г соответственно.
Метиленовый голубой, 1,6%-ный водный раствор.
Хлороформ, ч. д. а.
Ход анализа. В делительную воронку емкостью 100 мл помещают до 50 мл исследуемого раствора, содержащего до 5 мкг перхлората, и доводят pH раствора щелочью или серной кислотой до 6—7. Если нужно, разбавляют водой до 50 мл. Прибавляют 1 мл раствора метиленового голубого, взбалтывают и образующийся ассоциат экстрагируют три раза хлороформом порциями по 3 мл. Экстракты сливают в мерный цилиндр емкостью 10 мл, разбавляют хлороформом до 10 мл, перемешивают и измеряют оптическую плотность раствора при 655 нм.
Содержание перхлората находят по предварительно построенному калибровочному графику.
IX. 8. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ
РАЗЛИЧНЫХ ФОРМ ХЛОРА
Хлорид-ионы определяют по реакции вытеснения роданид-ионов из роданидного комплекса ртути:
Hg(NCS)2 + 2Cr —HgCl2-f-2NCS~
Выделившийся роданид определяют в виде роданидного комплекса железа [24—33]. Этот метод применен для определения хлоридов в металлическом титане [26], металлических бериллии, тории, уране и их окислах [25].
Выделившийся роданид определяют также путем экстракции нитробензолом разнолигандного комплекса фенантролината железа с роданидом и измерении оптической плотности при 516 нм [34].
Предложены также методы определения хлоридов, основанные на реакциях разрушения хлоранилата ртути и комплекса ртути с метилтимоловым синим. Выделившиеся хлораниловую кислоту [35] и метилтимоловый синий [36] определяют фотометрически.
Косвенные методы определения хлоридов основаны на взаимодействии хлороформного раствора HgCl2Py* с дитизоном [37], на разрушении комплексов ртути с азопроизводными [38], на определении серебра дитизоном после осаждения хлорида серебра [39], на реакции разрушения AgIO3 [или твердого Hg(IO3)2] хлоридом
* Ру — пиридин.
с последующим определением выделившегося йодата [40] или на измерении оптической плотности раствора хлорида ртути в ультрафиолете [41], на определении содержания хрома после отгонки хлорида в виде хлористого хромила [42].
Для прямого определения хлоридов предложено измерять светопоглощен и е дибромфлуоресцеина в присутствии хлорида серебра [43] при 550 нм, а также в виде хлоридного комплекса железа [44].
При определении в ядерном горючем хлориды окисляют перманганатом в сернокислой среде до хлора, который определяют в виде полиметинового красителя (ЛОпт = 580 нм), образующегося при взаимодействии хлора со смесью пиридина и барбитуровой кислоты [45].
Для определения активного хлора в воздухе использована реакция окисления иодида с последующим фотометрическим определением элементного иода [46, 47]. о-Толуидин и диметил-п-фе-нилендиамин применены для определения хлора в стерилизованной [48] и сточной [49] водах, в селене, теллуре, галлии и висмуте [50].
Для определения хлора в воде [51] использована реакция образования хлорциана и окрашенного продукта с барбитуровой кислотой [52]. Реакция с фенолом и анилином [53] применена для определения хлора в хлорной извести [54]. Реакция обесцвечивания фуксина применена для определения хлора в воздухе [55]. Дана оценка о-толидинового метода определения хлора в воздухе [56] и предложен лучший вариант проведения определения. Для определения хлора в воде разработан метод с применением 3,3'-диметилнафтидина [57], а также тетра-(п-диметиламинофе-нил)-этилена [58] и п-аминодиэтиланилина [59].
При определении хлора в присутствии других окислителей его отделяют экстракцией в ССЦ и измеряют оптическую плотность раствора при 327 нм [60].
Для определения хлора в воздухе при его содержании больше 1 мкг/л предложен метод с применением индикаторных бумаг. При этом через индикаторные бумажные кружки, пропитанные л-ди-метиламинобензальдегидом и дифенилам'ином, пропускают анализируемый воздух и сравнивают их окраску со стандартами, полученными при пропускании воздуха с определенным содержанием хлора [61] через такие же бумажные кружки.
Большинство методов определения перхлорат-ионов основано на экстракции органическими растворителями ионных ассоциатов с бриллиантовым зеленым [62, 63], малахитовым зеленым [64], кристаллическим фиолетовым [65, 66], 2,9-диметил-1,10-фенантроли-ном [67], тетрафениларсонием [68] и измерении оптической плотности экстрактов при соответствующей длине волны. Другие методы, основанные на взаимодействии перхлората с тетрапиридиновым комплексом меди [69] или восстановлении перхлората сульфатом титана(III) и определении его избытка [70], в настоящее время не применяется.
Разработан ряд методов определения хлорат-ионов, основанных па реакции с фенилантраниловой кислотой [71], по ослаблению окраски комплекса рения с а-фурилдиоксимом [72], образовании ионных ассоциатов с трифенилметановыми красителями [73] и др. [74—76].
Для определения двуокиси хлора применяют прямце методы при анализе воды [77] и сложных смесей [78], а также метод с применением кислотного хромфиолетового К [79] и другие методы [80, 81]. На реакции окисления в кислых растворах двуокисью хлора 1,10-фенантролината железа(II) основан фотометрический метод определения двуокиси хлора по уменьшению оптической плотности 1,10-фенантролината железа(II) [82]. Предложен также фотометрический метод определения двуокиси хлора с помощью тирозина [83, 84].
ЛИТЕРАТУРА
I. F е i g 1 F., N e u b e г F., Z. anal. Chem., 62, 370 (1923).
2. Clarke F. E., Anal. Chem., 22, 553 (1950).
3. К о 11 h о f f I. M„ Chem. Weekblad, 21, 20 (1924).
4. Литвиненко П. M., Розовский В. С., Гигиена и санитария, № 1, 52 (1954).
5. Scott A., J. Am. Chem. Soc., 51, 3351 (1929).
6. G е г 1 a ch J. Z„ F г a z 1 е г R. G., Anal. Chem., 30, 1142 (1958).
7. Novak J., Hauptman Z., Z. anal. Chem., 217, 340 (1966).
8. Utsumi S., Okutani R., J. Chem. Soc. Japan, Pure Chem. Sect., 85, 543 (1964).
9. Tamonari A., J. Chem. Soc. Japan, Pure Chem. Sect., 82, 864 (1961).
10. Kemula W., Hulanicki A., Janowski A., Chem. anal., 3, 581 (1958).
11. Kemula W., Hulanicki A., Janowski A„ Taianta, 7, 65 (1960).
12. Быстрицкий А. Л., Алесковский В. Б., Дегтяренко А. П., Изв. вузов. Хим. и хим. технол., 8, 555 (1965).
13. Туманов А. А., Сидоренко А. Н., Тр. по хим. и хим. технол., Горький, 2, 378 (1962).
14. Коренман И. М. и др., Тр. по хим. и хим. технол., № 2, 336 (1961).
15. Болц Д. Ф., Холланд Дж. В кн.: «Колориметрические (фотометрические) методы определения неметаллов». Пер. с англ. Под ред. А. И. Бу-сева. М., Издатинлит., 1963. См. с. 180.
16. J о h п s о п J. D., О v е г b у R., Anal. Chem., 41, 1744 (1969).
17. Grant J., Chemist-Analyst, 26, 59 (1937).
18. Marks H. C„ Joiner R. R., Anal. Chem., 20, 1197 (1948).
19. S ch m i d t M. P., J. prakt. Chem., 85, 235 (1912).
20. Taras M., Anal. Chem., 19, 342 (1947).
21. Marczenko Z., C h о 1 u j - L e n a r c z у k L., Chem. anal., 11, 1221 (1966).
22. H ah n F. L., Z. angew. Chem., 39, 451 (1926).
23. К г u g e г D., T s c h i г c h E., Z: anal. Chem., 85, 171 (1931).
24. Utsumi S., J. Chem. Soc. Japan, Pure Chem. Sec., 74, 611 (1953).
25. W a 1 k d e n J., Atomic Energy Res. Establ., 1959, AM42, 6 pp.
26. Iwasaki J., Ta cur о T., Ozawa T., Japan Analyst, 5, 275 (1956).
27. I w a s a k i J. e. ol., Bull. Chem. Soc. Japan, 29, 860 (1956).
28. I w a s a k i J. e. oL, Bull. Chem. Soc. Japan, 78, 468 (1957).
29. I w a s a k i J. e. ol., Bull. Chem. Soc. Japan, 78, 474 (1957).
30. I w a s a k i J. e. ol., Japan Analyst, 14, 12 (1965).
31. Mestres R., m-me Chave Chr., Claparede G., Barthes F., Trav. Soc. pharmac. Montpellier, 24, 304 (1964).
32. Utsumi S., J. Chem. Soc. Japan. Pure Chem. Sect., 74, 608 (1953),
33. Rodabaugh R. D., Upperman G. T., Anal. chim. acta, 60, 434 (1972).
34. Y a m a m о t о Y. e. ol., Anal. chim. Acta, 50, 433 (1970).
35. Barney J. E., В er to 1 acini R. J., Anal. Chem., 29, 1187 (1957).
36. Nomura Joshiaki, Komatsu Sumio, J. Chem. Soc. Japan, Pure Chem. Sec., 90, 168 (1969).
37. Einaha Kishachiko, Isij Chodchima, Japan Analyst, 18, 1211 (1969).
38. Мустафин И. С., Си в а нов а О. В., Изв. вузов, Хим. и хим. технол., 5, 875 (1962).
39. S u t е г Н., Н a d о г n Н., Z. anal. Chem., 160, 335 (1958).
40. L a m b е г t J. L., Y a s u d a S. К- Anal. Chem., 27, 444 (1955).
41. H u m p h г e у R. E. e. ol., Anal. Chem., 44, 1299 (1972).
42. M a q u i n a у A., J. pharmac. Belg., 14, 280 (1959).
43. Nash T„ Analyst, 85, 515 (1960).
44. West Ph. W., С о 11 H., Anal. Chem., 28, 1834 (1956).
45. E h n E., M e i e г K-, Radiochim. acta (BRD), 8, 219 (1967).
46. Полежаев H. Г., Гигиена и санитария, № 11, 46 (1955).
47. Парзоменко Г. В., Теплоцков Г. А., Тр. метрол. ин-та СССР, вып. 96, 158 (1968).
48. L е р р s G., G а е г t n е г Р., Z. anal. Chem., 139, 188 (1953).
49. Serfass Е. J., Muraca R. F., Gardner D. G., Plating, 42, 401 (1955).
50. S c h e u b e c k E„ E г n s t O., Z. anal. Chem., 254, 185 (1971).
51. Webber H. M„ Wheeler E. A., Analyst, 90, 372 (1965).
52. A s m u s E., G a г s c h a g e u H., Z. anal. Chem., 138, 404 (1953).
53. Кульберг Л. M„ Борзова Л. Д„ ЖАХ, 11, 470 (1956).
54. Кульберг Л. М., Борзова Л. Д„ Гигиена и санитария, № 9, 50 (1955).
55. Литвинова Н. С., Злобин Н. Я., Гигиена и санитария, № 4, 45 (1953).
56. Wojciech К., Kesy-Debrowska К., Chem. anal. (Polska), 9, 1063 (1964).
57. Belcher R., Nutten A. J., Stephen W. J., Anal. Chem., 26, 772 (1954).
58. Кульберг Л. M. и др., Уч. зап. Саратовского ун-та, 71, 251 (1959).
59. В io г k lu nd J. G., Rand М. С., J. Amer. Water Works Assoc., 60, 608 (1968).
60. Sherman M. I., Strickland J. D. H„ Anal. Chem., 27, 1778 (1955).
61. К u b a 1 s к i J. e. ol., Farmac. polska, 25, 991 (1969).
62. Г о л о с и ц к а я В. А., П e т p а ш e н ь В. А., Тр. Новочеркасского политех, ин-та, 141, 65 (1964).
63. R е u s m а п п G., Z. anal. Chem., 226, 346 (1967).
64. Голосицкая В. А., Петрашень В. И., Тр. Новочеркасского политех, ин-та, 141, 73 (1964).
65. UvhikawaSumio, Bull. Chem. Soc. Japan, 40, 798 (1967).
66. UvhikawaSumio, J. Sci. Hiroshima Univ., 33, 169 (1969).
67. С о 111 i n s о n W. J., В о 11 z D. F., Anal. Chem., 40, 1896 (1968).
68. Going J. E., Pflaum R. T., Proc. Jowa Acad. Sci., 72, 117 (1967).
69. Bodenheimer W., Weiler H., Anal. Chem., 27, 1293 (1955).
70. Forster C. F., Analyst, 79, 90 (1954).
71. Билык H. А., Тр. Одесского ун-та, Сб. хим. фак-та, 3, 127 (1953).
72. Trant we in N. Z., Gugon J. C., Anal. chim. Acta, 41, 275 (1968).
73. P а м а н а у с к а с Э. И. и др., ЖАХ, 24, 244 (1969).
74. U г о n a R., В о n d e E., Anal. Chem., 32, 1666 (1960).
75. В a n d e г i s A., J. Sci. Food and Agric., 16, 558 (1965).
76. C h e n T u n g - h o, Anal. Chem., 39, 804 (1967).
77. Hoggden H. W„ In go Is R. S., Anal. Chem., 26, 1224 (1954).
78. Hong C. C„ Rapson W. H., Canad. J. Chem., 46, 2061 (1968).
79. Masschelein W., Anal. Chem., 38, 1839 (1966).
80. Щ и г о л ь Ш. С., Зав. лаб., 24, 1043 (1958).
81. Щ и го ль Ш. С., Тр. по хим. и хим. технол. Горький, № 1, 148 (1958).
82. Н о w е 11 J. A., L i п i п g t о п G. Е., Boltz D. F., Microchem. J., 15, 598 (1970).
83. Туманов Т. А., Пахомова Л. Н., Майорова Л. П., Зав. лаб., 36, 1036 (1970).
84. Ascik К., Techn. wlok., 21, 92 (1972).
Глава X
БРОМ
X. 1. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДОВ ОПРЕДЕЛЕНИЯ И МЕТОДЫ ОТДЕЛЕНИЯ
Бром образует соединения, в которых проявляет степень окисления — 1, + 1, +5 и +7. Бромид-, как и хлорид-ионы, не образует окрашенных соединений, кроме некоторых броморганических соединений. Поэтому в фотометрических методах определения бромида используются реакции разрушения окрашенных комплексных соединений, а также реакции окисления бромида до брома, который затем определяют, применяя реакции бромирования или окисления окрашенных органических соединений.
Лучшим методом отделения от мешающих ионов является окисление бромида до брома с последующей отгонкой в токе воздуха, азота или двуокиси углерода. Для разделения хлоридов, бромидов и иодидов часто применяют селективное их окисление [1, 2]. Легче других окисляется иодид, в среде фосфорной кислоты (pH « 1) его можно окислить перекисью водорода до иода и отделить отгонкой. Бромиды окисляются до брома азотной кислотой при ее концентрации (1 :3) — (1 :6). В этих условиях ионы хлора не окисляются. Удобным методом отделения брома и иода является экстракция их четыреххлористым углеродом, хлороформом и другими органическими растворителями [3]. В присутствии хромовой кислоты, цианидов и разбавленной серной кислоты бромид образует летучий бромциан CNBr. Эта реакция применена для отделения бромида [4]. Кроме того, описаны ионообменные методы разделения галогенид-ионов [5], а также методы осаждения бромидов ионами серебра.
Бромид можно окислить до бромата с помощью гипохлорита при pH = 5,5—7,0. Для поддержания соответствующего pH применяют буферные растворы. Для последующего разрушения гипохлорита используют формиат натрия или перекись водорода. При окислении бромида до бромата гипохлоритом иод окисляется до йодата, который мешает дальнейшему определению брома. Поэтому в случае применения такого варианта определения бромидов необходимо предварительно отделить иодиды. Определение бромида после его окисления до бромата заканчивают путем выделения свободного брома и действия его на метиловый оранжевый или розанилин. Для выделения брома к подкисленному раствору бромата прибавляют бромид. Необходимо, чтобы кислотность рас-320
твора при этом не превышала 3 н., иначе хлорат, образующийся при кипячении гипохлорита, будет окислять бромид до брома.
Преимуществом броматного метода является то, что чувствительность реакции повышается в 6 раз, так как каждому определяемому микрограмму бромида в конце определения соответствует 6 мкг брома:
вг' + зсю’ —> зсг + вго;
ВгО; + 5Вг~ + 6Н+ —> ЗВг2 + ЗН2О
Органические вещества могут окисляться бромом, поэтому в случае применения этого метода они должны отсутствовать.
X. 2. ОПРЕДЕЛЕНИЕ БРОМИДА ПО РЕАКЦИИ РАЗРУШЕНИЯ МЕТИЛОВОГО ОРАНЖЕВОГО [6]
Бромиды окисляют до брома, который взаимодействует с метиловым оранжевым с образованием бесцветных продуктов реакции:
(CH3)2N——N=N——SO3H + 2Вг2 —>
—> (CH3)2N——NBr2 + Br2N——S°3H
Содержание брома находят по уменьшению интенсивности розовой окраски раствора.
Для увеличения чувствительности реакции бромид предварительно окисляют до бромата, который восстанавливают специально введенным бромидом с выделением брома. В качестве катализатора реакции между бромидом и броматом применяют молибдат. Чувствительность реакции при этом увеличивается в 6 раз.
Мешающие вещества. Определению брома мешает хлор, который взаимодействует аналогично, поэтому процесс окисления бромида необходимо проводить так, чтобы хлорид при этом не окислялся. Иодид не мешает определению, но если бром выделяют в виде бромата, то в процессе окисления бромида до бромата иодид также окисляется до йодата, который медленно реагирует с бромидом и количество выделившегося брома увеличивается, что приводит к ошибочным результатам. Завышение результатов по бромиду, в этом случае, обычно достигает 7%. Особенно мешает иодид в случае применения в качестве катализатора молибдата, который в сильной мере катализирует реакцию между бромидом и йодатом.
Большие количества хлоридов мешают при отгонке брома.
Измерение оптической плотности. Определение брома по обесцвечиванию раствора метилового оранжевого ведут при pH = 2. Оптическую плотность измеряют при 505 нм (см.' рис. 73).
Чувствительность метода — 1 мкг бромида в 25 мл конечного раствора.
X. 2.1. Определение бромида в отсутствие мешающих веществ
Реактивы
Окислитель. К 20 г хромового ангидрида прибавляют 40 мл концентрированной серной кислоты и эту смесь осторожно переносят в стакан, содержащий 120 мл воды.
Гипохлорит натрия, 1 н. раствор. В двухгорлую колбу помещают 40 г МпОг и 130 мл концентрированной хлористоводородной кислоты (х. ч.). К одному горлу колбы присоединяют выходную трубку, а к другому — трубку, доходящую до дна второй двухгорлой колбы, в которой находится около 100 мл воды. Ко второй колбе присоединяют трубку, доходящую до дна третьей двухгорлой колбы, в которой находится 500 мл 1,1 н. раствора NaOH. Колбу со щелочью погружают в ледяную баню. Хлор, выделяющийся в первой колбе, очищается, проходя через воду во второй колбе, и поглощается в третьей колбе.
» Для проверки степени образования гипохлорита натрия периодически берут 1 мл щелочного раствора, добавляют 5 мл 3%-ной перекиси водорода для разложения гипохлорита натрия и титруют 0,1 н. кислотой. Когда на титрование пробы расходуется I мл 0,1 н. кислоты, приготовление раствора гипохлорита натрия считается оконченным. Раствор переносят в склянку из коричневого стекла; при хранении в холодильнике раствор устойчив неопределенно долгое время.
Формиат натрия. Навеску 85 г соли растворяют в 250 мл воды.
Метиловый оранжевый, 0,05%-ный раствор. Перед анализом этот раствор разбавляют в 25 раз и таким образом готовят 0,002%-ный раствор.
Бромид калия, 20%-ный раствор с pH = 1,1.
Стандартный раствор бромида калия. Навеску бромида калия 0,149 г растворяют в воде, переносят в мерную колбу емкостью 1 л и доводят водой до метки. Этот раствор содержит 0,1 мг ионов бромида в 1 мл. Для анализа раствор разбавляют в 100 раз и получают раствор с содержанием 1 мкг ионов бромида в 1 мл.
Молибдат натрия, 1%-ный раствор.
Ход анализа. Отгонка брома при окислении бромида. Исследуемый образец, содержащий от 3 до 900 мкг брома, растворяют в 7 мл воды в конической колбе емкостью 100 мл, снабженной пробкой с двумя отверстиями. Наклоняют колбу под углом 60° для обеспечения максимальной аэрации. В пробку вставляют две трубки; одна из них слегка оттянута и достигает дна наклоненной колбы, другая согнута под углом 60° и присоединена к сосуду, в котором находится поглотитель брома. По стенке очень медленно добавляют в колбу 2,5 мл концентрированной серной кислоты в течение не менее 10 мин при постоянном охлаждении водопроводной водой. Добавляют 4 мл окислителя. Вставляют пробку и пропускают воздух 1 ч со скоростью 150 мл/мин. Меняют поглотитель и продолжают аэрацию еще 2 ч. Выделение галогена в течение второго периода аэрации указывает на присутствие слишком больших количеств хлорида для того, чтобы можно было достичь полного разделения этим методом.
Если хлорид присутствует в количестве 5 мг или менее, то метод позволяет количественно отгонять до 900 мкг брома. Отогнанный бром определяют одним из описанных ниже методов.
Окисление бромида до бромата. В стакан емкостью 50 мл помещают анализируемый раствор, содержащий до 8 мкг 322
бромида, и доводят pH до 3 (хлористоводородной кислотой или щелочью). Добавляют 1 мл 1 н. раствора гипохлорита натрия. При этом pH раствора не должен быть ниже 4 во избежание разложения гипохлорита. Добавляют, если нужно, хлористоводородную кислоту до pH менее 6. Затем добавляют около 0,1 г карбоната кальция и несколько кусочков неглазурованного фарфора, накрывают стакан и кипятят раствор в течение 8 мин. Промывают стенки стакана и часовое стекло малым объемом бидистиллята. Добавляют 2,5 мл раствора формиата и кипятят 8 мин. Охлаждают раствор до комнатной температуры. Доводят pH до 1,1 хлористоводородной кислотой и добавляют 5 мл раствора метилового оранжевого, 0,5 мл молибдата натрия и 2 мл раствора бромида калия в указанном порядке. Раствор перемешивают и спустя 5 мин после добавления бромида калия доводят pH до 2,0. Переносят раствор в мерную колбу емкостью 25 мл и разбавляют до метки водой при рН = 2,0. Измеряют оптическую плотность при 505 нм (зеленый светофильтр).
Содержание брома находят по калибровочному графику.
X. 2.2. Окисление бромида до бромата в отсутствие молибдата [7—9]
Реактивы
Формиат натрия, 20%-ный раствор.
Метиловый оранжевый — см. раздел Х.2.1.
Бромид калия, раствор, содержащий в 1 мл 0,01 г КВг.
Ход анализа. К'анализируемому раствору бромида добавляют 4 мл насыщенного раствора борной кислоты и 1 мл раствора гипохлорита и кипятят на водяной бане в течение 5 мин. К раствору прибавляют 2 мл раствора формиата натрия и продолжают кипятить еще 5 мин для разрушения избытка гипохлорита. Переносят раствор в мерную колбу емкостью 25 мл, добавляют 5 мл 1,5 М раствора серной кислоты, раствор метилового оранжевого (от 0,2 до 0,5 мл в зависимости от содержания бромата), 1 мл раствора бромида калия, бидистиллятом доводят до метки и перемешивает. Спустя 1 ч раствор наливают в кювету с толщиной слоя 5 см и измеряют оптическую плотность при 490 нм. Чувствительность определения — 0,02 мкг в 25 мл конечного раствора.
йодат в малых концентрациях медленно взаимодействует с бромидом, поэтому пятикратные количества иодида практически не мешают определению бромида. Большие количества иодида мешают. Молибдат примерно в два раза ускоряет реакцию между бромидом и броматом. Однако молибдат в сильной мере катализирует также реакцию между бромидом и йодатом. Поэтому в присутствии молибдата даже равные количества иодида мешают определению-
X. 2.3. Определение бромида в природных водах
Пробу воды выпаривают до небольшого объема и отфильтровывают раствор от выпавшего осадка. Затем ведут определение, как указано в разделе X. 2.2.
X. 3. ОПРЕДЕЛЕНИЕ БРОМИДА С ПРИМЕНЕНИЕМ
ФЕНОЛОВОГО КРАСНОГО [10]
Бромид окисляют до брома гипохлоритом кальция, избыток которого разрушают арсенитом натрия, а образующийся бромат взаимодействует с феноловым красным с образованием тетрабромсульфофталеина.
Мешающие вещества. Определению мешают сильные окислители и восстановители. Предварительно лучше отделить бром, как указано в разделе Х.2. Отгоняемый бром поглощают раствором сульфита натрия. После отделения бромид вторично окисляют смесью СгО3 и H2SO4 и выделившийся бром экстрагируют четыреххлористым углеродом. Затем бром извлекают из органического слоя 2 н. раствором аммиака и определяют, как описано ниже.
Измерение оптической плотности. Оптическую плотность измеряют при 580 нм. При визуальном определении по интенсивности окраски используют метод стандартной шкалы. Молярный коэффициент поглощения esso = 1,14-104.
Чувствительность метода — 1 мкг бромида в 25 мл конечного раствора.-
Реактивы
Гипохлорит кальция. Встряхивают препарат хлорной извести с водой и доводят фильтрат до концентрации приблизительно 0,1 н.
Феноловый красный. Навеску 10 мг препарата растворяют в 1 мл 0,1 н. раствора NaOH и разбавляют до 100 мл.
Боратный буферный раствор. Готовят насыщенный водный раствор тетрабората натрия при 25 °C и фильтруют. Доводят pH раствора до 8,8 серной кислотой.
Ацетатный буферный раствор. Смешивают 68 г тригидрата ацетата натрия и 30 мл химически чистой ледяной уксусной кислоты, с водой так, чтобы общий объем составлял 1 л; pH раствора должен быть 4,6—4,7.
Арсенит натрия, приблизительно 0,1 н. раствор.
Стандартный раствор бромида калия, см. раздел Х.2.1.
Ход анализа. Анализируемый раствор, содержащий до 15 мкг бромида, помещают в мерную колбу емкостью 25 мл. При необходимости доводят водой до 10 мл. Добавляют 0,2 мл фенолового красного и 2 мл боратного буферного раствора. Прибавляют 0,2 мл раствора гипохлорита кальция и оставляют точно на 4 мин, периодически встряхивая содержимое колбы. Затем прибавляют 0,5 мл раствора арсенита и 1,5 мл ацетатного буферного раствора. Доводят до метки водой, pH раствора должен быть равен 5,0—5,4. Измеряют оптическую плотность при 580 нм и находят содержание бромида по калибровочному графику,
Если во время окисления гипохлоритом анализируемый раствор становится пурпурным, а затем его окраска ослабевает, то это означает, что количество присутствующего бромида очень велико. В этом случае необходимо опыт повторить с меньшим количеством анализируемого раствора.
X. 4. ОПРЕДЕЛЕНИЕ БРОМИДА ПО РЕАКЦИИ
ОБРАЗОВАНИЯ ЭОЗИНА [11]
Бромид окисляют до брома, который взаимодействует с флуоресцеином с образованием эозина (тетрабромфлуоресцеина);
Этот метод может быть применен также в том случае, если бромид предварительно окисляют до бромата, а затем бромат вступает в реакцию со специально введенным бромидом с выделением брома. При этом чувствительность метода повышается в 6 раз.
Необходимо отметить, что точность определения этим методом невелика.
Мешающие вещества. Определению брома следы свободного хлора не мешают, но иод, а также другие окислители мешают определению. Поэтому иод и заметные количества хлора должны быть отделены.
Измерение оптической плотности. Оптическую плотность раствора измеряют при 520 нм.
Чувствительность метода — 0,1 мкг брома в 5 мл конечного раствора.
X. 4.1. Определение в отсутствие мешающих веществ
Реактивы
Флуоресцеин. Навеску 0,3 г флуоресцеина растворяют в 5 мл 0,1 н. раствора гидроокиси натрия и разбавляют водой до 1 л.
Буферный раствор. Смешивают 10 мл 1 М раствора ацетата натрия с 1 мл 1 н. раствора уксусной кислоты pH = 5,5—5,6.
Раствор восстановителя. Растворяют 10 г гидроокиси натрия и 1 г кристаллического тиосульфата натрия в 200 мл воды.
Ход анализа. К 3 мл нейтрализованного анализируемого раствора, содержащего до 1 мкг свободного брома, прибавляют 3 капли раствора флуоресцеина и 0,4 мл буферного раствора. Раствор перемешивают и оставляют на 1 —1,5 мин. Затем реакцию останавливают прибавлением 3 капель раствора тиосульфата натрия, разбавляют раствор до 5 мл в пробирке с делениями, перемешивают и измеряют оптическую плотность раствора при 520 нм.
X. 5. ОПРЕДЕЛЕНИЕ БРОМИДА С ПРИМЕНЕНИЕМ
РОЗАНИЛИНА [10]
Бромид окисляют до брома, который при взаимодействии с розанилином образует красного цвета тетрабромрозанилин или пента-бромрозанилин, их спектры поглощения имеют полосу поглощения с максимумом при 570 нм. Продукты бромирования розанилина нерастворимы в воде, поэтому для фотометрического определения вводят смешивающиеся с водой органические растворители или экстрагируют окрашенное соединение несмешивающимися органическими растворителями, например бензиловым спиртом.
Мешающие вещества. Определению брома мешают окислители, в том числе хлор. Кроме того, розанилин слабо окрашен и мешает собственной окраской. Поэтому'лучше применять экстракционно-фотометрический вариант, позволяющий отделить броми-рованный продукт от реагента. Если применяют водно-органические растворы, то определение проводят в среде 3—7 н. серной кислоты. При такой кислотности уменьшается интенсивность окраски реактива.
Измерение оптической плотности. Оптическую плотность измеряют при 585 нм (е585 = 6,8-104). Окраска устойчива в течение нескольких часов.
Чувствительность метода—0,2 мкг брома в 5 мл конечного раствора.
X. 5.1. Определение бромида в отсутствие мешающих ионов
Реактивы
Гипохлорит натрия, см. раздел X. 2.1,
Формиат натрия, раствор 50 г в 100 мл воды.
Буферный раствор с pH = 6,35. Растворяют 18,2 г дигйдрата однозамещенного фосфата натрия и 3,6 г гидроокиси калия в воде и доводят объем до 100 мл.
Розанилин. Навеску 6 мг розанилина растворяют в 100 мл 2 н. серной кислоты. При хранении раствора в склянке из темного стекла он пригоден к работе в течение нескольких недель.
Смесь бромида и молибдата. Растворяют 0,15 г бромида калия и 3 г молибдата аммония в воде и разбавляют до 100 мл.
Серная кислота, 14 и.
трет-Бутанол, содержащий 5% абсолютного бутанола для предотвращения замерзания.
Бензиловый спирт, ч. д. а.
Стандартный раствор бромида калия, см. раздел X. 2.1,
Калибровочный график. Готовят серию стандартных растворов, содержащих до 25 мкг бромида, и проводят через весь ход анализа параллельно с анализируемым раствором.
Следует иметь в виду, что во все растворы необходимо добавлять одинаковые количества смеси бромида и молибдата и серной кислоты.
Ход анализа. В пробирку вводят 1 мл буферного раствора, затем анализируемый бромидный раствор и прибавляют воду до общего объема 4,5 мл. Затем добавляют 0,25 мл раствора гипохлорита, перемешивают и погружают пробирку в кипящую водяную баню на 10 мин. Добавляют 0,25 мл раствора формиата натрия для удаления избытка гипохлорита, перемешивают и снова помещают пробирку в кипящую воду на 5 мин. Пробирку охлаждают, добавляют некоторое количество воды, чтобы возместить потери при испарении, и перемешивают.
В другую пробирку помещают 0,1 мл раствора смеси бромида и молибдата, по 0,1 мл раствора розанилина на каждый микрограмм брома в виде бромата, не более 0,4 мл воды, 0,4 мл серной кислоты и 1 мл приготовленного раствора бромата. Общий объем раствора должен быть 2 мл. Оставляют смесь на 3 мин при 20— 30 °C и добавляют 2 мл трет-бутанола и 1 мл серной кислоты. Перемешивают и измеряют оптическую плотность при 570 нм.
Окрашенное соединение можно предварительно экстрагировать 5 мл бензилового спирта. В этом случае оптическую плотность измеряют при 585 нм.
X. 5.2. Определение бромида в рассоле
В две пробирки помещают реагенты, как указано в разделе X. 5.1, считая, что объем анализируемого рассола 0,1 мл, а также, что одна пробирка должна содержать 0,05 мл, а другая — 0,5 мл розанилина. После этого добавляют по 0,1 мл анализируемого раствора в каждую пробирку и далее поступают, как указано в разделе X. 5.1. Таким путем находят приблизительное количество присутствующего бромида, если только количество определяемого бромида не превышает 250 мкг. После этого берут различные количества анализируемого раствора и соответствующие количества розанилина до тех пор, пока не будет установлена пропорциональность между поглощением и взятыми порциями анализируемого раствора.
Если свободный бром присутствует в большом избытке по отношению к розанилину, то результаты будут заниженными вследствие того, что образующийся бромрозанилин обесцвечивается бромом.
X. 6. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ БРОМА
Бромциан взаимодействует с пиридином и ароматическими аминами с образованием интенсивно окрашенных соединений [4, 12, 13]:
CBH6N + CNBr —> C6H6NCNBr
CeH6NCNBr + 2RNH2 —► RNH2BrCH=CH=CH=CHCH=NR + CNNH2
Продукт реакции имеет бледно-красный цвет и используется для фотометрического определения бромида (еыакс = 530 нм). Метод довольно чувствительный, но его применение ограничено в связи с большой токсичностью цианидов. Этот метод применен для определения бромида в муке [10].
Метиловый оранжевый использован для определения бромида каталитическим фотометрическим методом [14]. Бромид окисляют броматом, выделяющийся бром разрушает метиловый оранжевый. Этот реагент применен, для определения бромида в крови [10].
о-Толидин применяется для определения малых количеств бромида и хлорида после их окисления перманганатом калия до свободного состояния [15]. Оптическую плотность образующегося желтого продукта измеряют при 450 нм. Определение бромида и хлорида основано на создании условий последовательного селективного их окисления. Толидиновый метод применен для определения брома в присутствии бромистого водорода [16].
Розанилин применен для определения бромида в водах [17]. Установлены соотношения для многих ионов, в которых они не мешают определению бромида.
Реакция образования тетрабромсульфонафталина применена для автоматического определения бромида с помощью автоматического анализатора [18]. Предварительное окисление бромида до брома проводят с помощью хлорамина Т.
Хромотроп 2В окисляется бромом, образуя окрашенное соединение с максимумом поглощения при 511 нм. Эта реакция использована для определения бромида [19]. Для окисления бромида применяют перекись водорода.
Этилентриаминтетрацетат ртути при pH = 5,0—8,3 взаимодействует с бромидом и иодидом с образованием комплексов состава [HgYBr]3- и [HgYI]3-, сильно поглощающих ультрафиолетовый свет (е250 = 2650 для бромида и егво = 4200 для иодида). Этот реагент рекомендован для фотометрического определения бромида и иодида [20].
Фуксинсернистая кислота использована для определения бромида [21]. Предварительно бромид окисляют до брома хлорной водой. Этот метод применен для анализа солей в калийной промышленности и определения бромида в водах нефтяных месторождений [22].
Трифенилметановые красители применены для фотометрического определения бромида после окисления до брома перманганатом. Полученный бром экстрагируют толуолом, затем к толуольному раствору брома прибавляют трифенилметановые красители, которые образуют интенсивно окрашенные соединения [23, 24].
Трифенилбензилфосфоний в присутствии двуокиси серы образует с бромидом и иодидом ионные ассоциаты, которые экстрагируются 1,2-дихлорэтаном и применяются для фотометрического определения бромида и иодида [25].
Подавление реакции хлорирования аммония бромидом используется для фотометрического определения бромида [26].
Реакция бромирования фенолового красного применена для определения бромида в окислах и фторидах урана [10, 27].
Реакции разрушения дитизоната серебра [28] и дифенилкарба-зоната ртути [29] бромидом используют для фотометрического его определения.
Каталитическая реакция окисления иода до йодата с помощью перманганата в сернокислой среде применена для фотометрического определения бромида [30].
Для анализа газовой смеси нитрозилбромида (NOBr) и брома в присутствии окиси азота ведут измерения оптической плотности газовой смеси при 338 и 416 нм, а затем рассчитывают содержание каждого компонента [31].
n-Диметиламинобензилиденроданин взаимодействует с броматом с образованием окрашенного продукта реакции с максимумом поглощения при 400—410 нм. Эта реакция используется для определения бромата [32].
Кристаллический фиолетовый при взаимодействии с перброматом образует ионный ассоциат, который экстрагируется хлорбензолом и применяется для экстракционно-фотометрического определения пербромата [33]. Присутствие меньше чем 10-3 г-ион/л бромата не мешает определению.
Бромиды определяют также по поглощению HgBr2 в ультрафиолете [34].
ЛИТЕРАТУРА
1. К a h а n е Е., К a h а n е М., Bull. Soc. Chim. France, 1954, 396.
2. Yates E. D., Biochem. J„ 27, 1763 (1933).
3. Collins A. G., Watkins J. W., Anal. Chem., 31, 1182 (1959).
4. V a n P i n k f e r r e n J. A. C., Anal., 77, 367 (1952).
5. Winefordner J. D., Mau ng Tin, Anal. Chem., 35, 382 (1963).
6. Taras M„ Anal. Chem., 19, 342 (1947).
7. L a i t i n e n H. А., В о у e r K- W-, Anal. Chem., 44, 920 (1972).
8. ТамарченкоЛ. M„ Гигиена и санитария, № 10, 53 (1968).
9. Тамарченко Л. М., Торопова В. Ф., ЖАХ, 23, 1028 (1968).
10. Р а й т Е. Р., С м и т Р. А., Б л э к С. В сб. «Колориметрические (фотометрические) методы определения неметаллов». Пер. с англ. Под ред. А. И. Бу-сева. М., Издатинлит, 1963. См. с. 198.
11. Р о h I F. A., Z. anal. Chem., 149, 69 (1956).
12. A s m u s E., G a r s h a g e n H., Z. anal. Chem., 136, 269 (1952).
13. M i 11 о n R. F., Nature, 164, 338 (1949).
14. Бабкин M. П. В сб. «Комплексообразование, межмолекулярное взаимодействие и соосаждение в некоторых системах». Днепропетровск, 1970. См. с. 209.
15. S с h е u b е с k Е., Е г n s t О., Z. anal, chem., 249, 370 (1970).
16. С г е i f z Е. С., Anal. Chem., 37, 1690 (1965).
17. М о 1 d а 1 п В., Z у к a J., Mikrochem. J., 13, 357 (1968).
18. А г ch i m b a u d M., Bertrand M. R., Chim. anal., 52, 521 (1970).
19. Elbein I. I. M., El-Sirafy A. A., Chemist-Analyst., 54, 8 (1965).
20. Komotsu Sumi о, Nomura Toshiaki, J. Chem. Soc. Japan, Pure Chem. Sec., 88, 63 (1967).
21. Sell R., Kali und Steinsalt, 4, 240 (1966).
22. P а к о в и ч X. А., Новости нефт. техн. Геология, 6, 37 (1957).
23. Савичев Е. И., Матренкин В. Ф., Авт. св. СССР, кл. 421, 3/08; 12i, 7/10 (gOln), Colb, № 199487, заявл. 1.12.65, опубл. 28.08.67. РЖХим, 1968, 17Г102П.
24. Буникене Л., Раманаускас Э., Янкаускайте 3., Науч. тр. высш, учебн. заведений ЛитССР. Хим. и хим. технол., 10, 37 (1969).
25. Behrends К., Klein Н., Z. anal. Chem., 249, 165 (1970).
26. Z i t о m е г T.j L a m b е г t J. J., Anal. Chem., 35, 1731 (1963).
27. L a rs en R. P., I n gber N. M., Anal. Chem., 31, 1084 (1959).
28. К i г s t e n W. J., Mikrochem. Acta, 1955, 1986.
29. To mon a ri A„ J. Chem. Soc. Japan, Pure Chem. Sec., 83, 459 (1962).
30. Jonehara Nori no bu, Ntsumi Satori, Iwasaki Iwaji, Bull. Chem. Soc. Japan, 38, 1887 (1965).
31. Eden Ch., Reilchenfald H., Manof Sh., Anal. Chem., 41, 1150 (1969).
32. Bosch S. F., Bol. Soc. guim. Oeru, 35, 14 (1969).
33. В г о w n L. С., В о у d G. F., Anal. Chem., 42, 291 (1970).
34. Кремер В. А. и др. В сб. «Контроль и автоматизация в процессах обога« щения полезных ископаемых». М., «Наука», 1969. См. с. 57.
Глава XI
ИОД
XI. 1. ОБЩАЯ ХАРАКТЕРИСТИКА, МЕТОДЫ ОПРЕДЕЛЕНИЯ И МЕТОДЫ ОТДЕЛЕНИЯ
Степень окисления иода в соединениях равна —1, +1, +3, +5 и 4-7. В зависимости от степени окисления иод может быть восстановителем или окислителем. Окислительные свойства иода слабее, чем у хлора и брома. Поэтому при действии свободного хлора на иодид происходит выделение свободного иода.
Фотометрические методы определения иодида можно разделить на две группы: методы, основанные на разрушении иодидом окрашенных комплексных соединений, и методы, основанные на реакциях окисления — восстановления с образованием иода, йодата или перйодата с последующим их определением. Для их определения в виде иода или ионных ассоциатов с красителями предложены экстракционно-фотометрические методы, а также методы, основанные на окислении таннина. Свободный иод, особенно малые его количества, определяют по реакции с крахмалом.
В большинстве случаев определению иодида мешают многие ионы, поэтому его лучше предварительно отделить от сопутствующих веществ. Наиболее надежным методом отделения соединений иода является экстракция органическими растворителями после предварительного образования иода. Это один из старых методов отделения иода [1, 2]. Необходимо отметить, что иодид калия удерживает иод в водной фазе и из 5%-ного водного раствора иодида калия элементный иод экстрагируется слабо [3]. Иод экстрагируют четыреххлористым углеродом [4, 5], хлороформом [6—10], толуолом [3], бензолом [3], сероуглеродом [И] и другими растворителями.
Широко используется также дистилляция иода [12—15]. Для этого ионы иода предварительно окисляют или восстанавливают (в зависимости от валентного состояния иода в соединениях) до иода. В качестве окислителей иодида до иода применяют хлор, нитрит, хромовый ангидрид, перманганат в сернокислой среде и др.
• Для разложения анализируемых образцов применяют сплавление с карбонатом натрия [16], смесью карбоната и гидроокиси натрия [17], гидроокисью натрия и нитрита калия [18] или с гидроокисью калия [19] при 600—650 °C. При этом происходит полное разложение органических веществ, но потерь иода не наблюдается, что установлено при помощи меченого иода [20].
Иодид определяют в подкисленном растворе спека.
Минерализацию органических соединений можно проводить также мокрым путем, применяя смесь хлорноватой и хромовой кислот [21, 22] или хлорноватую кислоту [23]. При этом образуется иодат.
Наиболее простым и наименее чувствительным методом определения иода является метод, основанный на измерении интенсивности окраски водного раствора трииодида или раствора иода в органическом растворителе после экстракции. Значительно более чувствительным является метод определения иода с помощью крахмала. Широкое распространение получили каталитические методы определения иода, основанные на реакции между церием (IV) и мышьяковистой кислотой.
XI. 2. ОПРЕДЕЛЕНИЕ ИОДА С ПРИМЕНЕНИЕМ КРАХМАЛА
Иодид окисляют до иода, который взаимодействует с крахмалом с образованием соединения, окрашенного в интенсивный синий цвет. Окраска иодного крахмала обусловлена тем, что цепочка полимера-крахмала обвивается вокруг молекулы иода, сильно
сжимая ее и вытягивая в одном направлении. Поэтому окраска в сильной мере зависит от температуры и при 40—50 °C практически исчезает [24].
С целью увеличения чувствительности этого метода иодид окисляют до йодата, при последующем добавлении к раствору иодида происходит выделение иода:
Рис. 76. Спектр поглощения Ю~ + 51“ + 6Н+ —> 312 + ЗН2О
водного раствора иодкрах-
мального комплекса. Таким образом, чувствительность метода повышается в шесть раз.
В качестве окислителей чаще других применяют бромную воду [25—27]. Избыток брома удаляют кипячением или связывают фенолом в трибромфенол. При окислении иодида перманганатом в щелочной среде образуется иодат [28]. Избыток перманганата восстанавливают нитритом, а избыток последнего восстанавливают мочевиной. Для окисления иодида до иода применяют нитриты или соли железа(III).
Мешающие вещества. Определению мешают многие окислители и восстановители, а также ионы, имеющие собственную окраску. Хлорид- и бромид-ионы не мешают определению. Однако если определение проводят с отгонкой иода, то большие количества хлоридов мешают. ,
Измерение оптической плотности. Спектр поглощения раствора иодкрахмального комплекса представлен на рис. 75. Оптическую плотность таких растворов измеряют при 575 нм. Необходимо от
метить, что спектр поглощения иодкрахмального комплекса зависит также от качества крахмала. Крахмал содержит амилозу (нерастворимый крахмал 10—20%) и амилопектин (растворимый крахмал 80—90%). При действии иода амилоза (или линейный крахмал) дает соединение синего цвета, а амилопектин (нелинейный крахмал) —от фиолетового до красно-фиолетового.
Молярный коэффициент поглощения иодкрахмального комплекса (6575) равен 1,8-104. Если проводить реакцию через иодат, тогда е575 = 1,08-105.
Чувствительность метода — 0,5 мкг иода в 25 мл конечного раствора.
XI. 2.1. Определение иодида в отсутствие мешающих веществ
Ниже приведены два варианта определения иодида: после окисления до иода и до йодата.
Реактивы
Стандартный раствор иодида. Навеску 0,6541 г иодида калия (не содержащего йодата) растворяют в воде, переносят в мерную колбу емкостью 500 мл, разбавляют водой до метки и перемешивают. Этот раствор содержит 1 мг иода в 1 мл. Раствор хранят в темном месте. Растворы с меньшим содержанием иодида готовят путем разбавления исходного раствора водой.
Крахмал, 1%-ный раствор. Смешивают 1 г крахмала с 5 мл воды, полученную смесь при перемешивании сливают в 50 мл кипящей воды, добавляют 50 мл глицерина, нагревают и выдерживают при слабом кипячении. Такой раствор можно хранить в течение нескольких недель. Для консервации крахмала применяют также салициловую кислоту.
Если иодид определяют, предварительно окисляя его до йодата, то лучше применять раствор крахмала в смеси с иодидом кадмия, который готовят следующим образом. Навеску 11 г иодида кадмия растворяют в 300—400 мл дистиллированной воды и умеренно кипятят в течение 15 мин. Разбавляют кипящей дистиллированной водой до 800 мл и к слабо кипящему раствору при перемешивании медленно добавляют 2,5 г крахмала. Раствор фильтруют и разбавляют до 1 л водой.
Фенол, 20%-ный раствор в ледяной уксусной кислоте.
Иодид калия, 0,5%-ный свежеприготовленный раствор.
Бромная вода, насыщенный раствор.
Нитрит натрия, 1 М раствор.
Мочевина, 5 М раствор.
Ход анализа. Определение иодида после окисления до йодата. Нейтральный анализируемый раствор до 15 мл, содержащий до 25 мкг иода в виде иодида, помещают в мерную колбу емкостью 25 мл и прибавляют 1 мл 1 н. H2SO4, 2 капли бромной воды, перемешивают и оставляют на 1 мин. Затем прибавляют 2 капли фенола и спустя 1 мин приливают 1 мл раствора иодида калия, 1 мл раствора крахмала, разбавляют водой до метки, перемешивают и измеряют оптическую плотность при 575 нм по отношению к раствору холостого опыта.
Содержание иода находят по калибровочному графику.
Определение иодида после окисления до иода. Начало определения аналогично описанному выше. Для окисления
иода вместо бромной воды прибавляют 2 мл раствора нитрита натрия и оставляют на 2 мин. Затем прибавляют 5 капель раствора мочевины для разрушения избытка нитрита, 1 мл раствора крахмала (без иодида), разбавляют водой до метки, перемешивают и измеряют оптическую плотность раствора при 575 нм.
Содержание иода находят по калибровочному графику.
XI. 2.2. Определение иодида после окисления до йодата перманганатом калия [29]
Реактивы
Перманганат калия, 0,2 н. раствор.
Фосфорная кислота, 28%-ный раствор.
Иодид калия, 0,25%-ный раствор.
Стандартный раствор иодида, см. раздел XI. 2.1.
Крахмал, 0,25%-ный раствор.
Ход анализа. Анализируемый раствор, содержащий до 5 мкг иодида, помещают в стакан емкостью 50 мл и добавляют 1 мл 0,2 н. раствора гидроокиси натрия. Добавляют 1—2 капли раствора перманганата калия и упаривают до 2—3 мл. Содержимое стакана переносят в калиброванную пробирйу емкостью 5 мл- Помещают на 2 мин в ледяную баню и затем добавляют 0,05 мл раствора иодида калия, 1 мл фосфорной кислоты и 0,5 мл раствора крахмала. Раствор перемешивают, разбавляют до метки и спустя 2 мин измеряют оптическую плотность при 575 нм.
Содержание иода находят по калибровочному графику.
XI. 2.3. Определение иода в породах, почвах, растительных и животных тканях [30, 31]
Пробу разрушают хромовым ангидридом в серной кислоте, иод отгоняют, затем окисляют перманганатом до йодата и восстанавливают до иода с помощью • иодида, заканчивая определение иода в виде иодкрахмального комплекса.
Ход анализа. В колбу прибора для дистилляции иода, содержащую 1—5 г образца, добавляют от 10 до 30 мл насыщенного раствора хромового ангидрида, а затем 5 мл концентрированной серной кислоты на каждый миллилитр раствора хромового ангидрида. Серную кислоту вначале вводят по каплям, а после прекращения бурной реакции добавляют кислоту большими порциями. Нагревают раствор до 220 °C и поддерживают эту температуру в течение 5 мин для разложения избытка хромового ангидрида. Затем раствор охлаждают до 100°C, добавляют 50 мл дистиллированной воды, содержимое колбы хорошо перемешивают и колбу присоединяют к перегонному аппарату. Приемником служит сосуд емкостью 50 мл, содержащий 1 мл 1 н. раствора гидроокиси натрия; конец холодильника погружают в поглотительный раствор. Содержимое колбы нагревают до кипения и через капельную воронку добав
ляют в нее по каплям 10—15 мл 30%-ной фосфорной кислоты. Дистилляцию ведут до тех пор, пока не будет отогнано 40 мл. Полученный щелочной раствор упаривают до объема 15 мл и определяют иодид, как указано в разделе XI.2.2.
XI. 3. ОПРЕДЕЛЕНИЕ ИОДА В ВИДЕ ИОНОВ ТРИИОДИДА И СВОБОДНОГО ИОДА
Иодид окисляют до иода и измеряют оптическую плотность раствора иода в воде или другом растворителе. Выделяющийся иод можно экстрагировать органическими растворителями и измерять оптическую плотность экстракта. Экстракционный вариант дает лучшие результаты. Как и в предыдущем случае, иодид можно окислить до йодата, а затем, используя в качестве восстановителя
Рис. 76. Спектры поглощения растворов иода:
1 — в воде; 2 —хлороформе; 3 — 95%-ном этаноле; 4 — толуоле; 5 —бензоле; 6— 5%-ном водном растворе иодида калия.
иодид, перевести иодат в иод. При этом чувствительность метода возрастает в 6 раз.
Цвет растворов иода зависит от природы растворителя. Спектры поглощения растворов иода в различных растворителях представлены на рис- 76. Растворы, в которых иод не сольватируется молекулами растворителя (СС14, CS2, СНС13), имеют пурпурный цвет, спектр этих растворов аналогичен спектру поглощения иода в парообразном состоянии [32]. В растворителях, которые образуют сольваты иода (вода, этанол, иодид калия), растворы имеют бурый цвет; спектры этих растворов, содержащих сольватированные молекулы или молекулы аддитивных соединений иода и растворителя, характеризуются полосами поглощения в видимой и ультрафиолетовой областях.
Методика определения иода не отличается от методик, описанных выше, но определение проводят не в виде иодкрахмального комплекса, а в виде свободного иода. Оптическую плотность
водных растворов иода измеряют при 420—430 нм, а экстрактов иода в хлороформе при 510 нм.
Ниже приведен экстракционный вариант метода определения иодида в виде иода как более чувствительный.
XI. 3.1. Определение иодида в отсутствие мешающих веществ [33]
Реактивы
Хлороформ, ч. д. а.
Другие реактивы — указаны в разделе XI. 2.1.
Ход анализа. Нейтральный анализируемый раствор, содержащий до 1 мг иода в виде иодида, подкисляют 2 мл 1 н. серной кислоты, добавляют 10 капель бромной воды, перемешивают и оставляют на 1 мин. Избыток брома удаляют нагреванием раствора. Раствор охлаждают, прибавляют 1 каплю раствора фенола для связывания остатков брома, спустя 1 мин приливают 2 мл раствора иодида калия и выделившийся иод экстрагируют дважды хлороформом порциями по 10 мл, встряхивая в течение 1 мин. Экстракты сливают в колбу емкостью 25 мл, доливают до метки хлороформом, перемешивают и измеряют оптическую плотность раствора при 510 нм по отношению к хлороформу.
Содержание иода находят по калибровочному графику-
XI. 3.2. Определение иодида и бромида в нефтяных водах [17, 34]
йодиды окисляют до иода нитритом, иод экстрагируют четыреххлористым углеродом и измеряют оптическую плотность при 517 нм. После отделения иодидов бромиды окисляют до брома гипохлоритом (см. гл. X), экстрагируют четыреххлористым углеродом и измеряют оптическую плотность раствора при 417 нм.
Чувствительность и точность метода составляют: для иодидов — 0,2 и 0,5, а для бромидов — 5 и 4,6 мг/л. В случае анализа более разбавленных растворов их концентрируют путем упаривания подщелоченных проб.
XI. 3.3. Определение иода в горных породах [35]
Иод окисляют до йодата в щелочной среде, затем вводят иодид, который окисляется йодатом до иода. Иод экстрагируют четыреххлористым углеродом и измеряют оптическую плотность при 517 нм.
Реактивы
Флюс. Смешивают 400 г К2СО3, 400 г Na2CO3 и 100 г MgO.
Сульфат олова(П), 0,3 н. раствор SnSO4 в H2SO4, разбавленной (1 :4).
Нитрит натрия, 0,4 М раствор.
Четыреххлористый углерод, ч. д. а.
Перманганат калия, 0,2 н. раствор.
Мочевина, 4%-ный раствор.
Ход анализа. Навеску пробы 1 г смешивают с 4,5 г флюса и нагревают в открытом тигле при 750 °C. К спеку прибавляют 30— 40 мл воды и оставляют на ночь на водяной бане. Затем нерастворимый осадок отфильтровывают и промывают горячей водой. Фильтрат упаривают приблизительно до 20 мл, вводят 3 капли раствора перманганата калия и нагревают до кипения. Раствор охлаждают на водяной бане, нейтрализуют по лакмусу серной кислотой, разбавленной (1 : 1), прибавляют 3,5 мл избытка этой кислоты й перемешивают до удаления СОг. Затем для восстановления йодата до иодида прибавляют по каплям раствор сульфата олова (II) до исчезновения розовой окраски и 1 мл избытка этого раствора. Спустя 1 мин прибавляют 1 мл раствора нитрита натрия для окисления иодида до иода, перемешивают и оставляют на 1 мин. Затем вводят 1 мл раствора мочевины, переводят раствор в делительную воронку и выделившийся иод экстрагируют 3 мл четыреххлористого углерода в течение 1 мин. Экстракт переводят в кювету и измеряют оптическую плотность раствора при 517 нм относительно СС14.
Содержание иода находят по калибровочному графику-Чувствительность метода—1 • 10“4% иода в навеске 1 г.
XI. 3.4. Определение иода в кремнии [36]
Пробу растворяют в щелочи, нейтрализуют серной кислотой, окисляют иодид до иода нитрозой, экстрагируют выделившийся иод бензолом, реэкстрагируют раствором щелочи, окисляют бромной водой до йодата, который восстанавливают до иода, экстрагируют бензолом и измеряют оптическую плотность при 517 нм.
Реактивы
Нитроза. Растворяют при нагревании 2 г нитрата натрия в 100 мл концентрированной серной кислоты. Раствор годен в течение 4 ч.
Бензол, ч. д. а.
Бромная вода, свежеприготовленная.
Сульфосалицилат натрия, 1%-ный раствор.
Иодид калия, 0,5%-ный раствор.
Ход анализа. Навеску 0,5—1 г мелко растертого в агатовой ступке кремния помещают в стакан и растворяют в 20 мл 3 н. раствора гидроокиси натрия при нагревании. После растворения прибавляют 5 мл серной кислоты, разбавленной (1:1), и доливают водой до 150 мл. Переносят в делительную воронку, в которую приливают 1 мл нитрозы, перемешивают и дважды экстрагируют бензолом порциями по 10 мл. Экстракты три раза промывают водой, дважды реэкстрагируют 0,1 н. раствором NaOH порциями по 1 мл, сливая нижние слои в фарфоровую чашку. Щелочной реэкстракт подкисляют 1,5 мл 3 н. серной кислоты, добавляют 0,5 мл бромной воды и кипятят до исчезновения желтой окраски жидкости. Остывший раствор переносят в маленькую делительную воронку,
разбавляют до 3 мл водой, прибавляют 0,5 мл раствора сульфосалицилата натрия, перемешивают и прибавляют 0,5 мл раствора иодида калия. Содержимое воронки перемешивают и дважды экстрагируют иод бензолом порциями по 1 мл. После объединения экстрактов измеряют оптическую плотность при 517 нм.
Содержание иода находят по калибровочному графику.
XI. 3.5- Определение иода в водах и рассолах [17]
К 50 мл воды, 1 —10 мл минеральной воды или 100 мл рассола прибавляют 6 г карбоната натрия и упаривают до получения вязкой массы. Охлаждают раствор и добавляют 100 мл этанола. Смесь нагревают, перемешивают и фильтруют. Обработку твердого остатка повторяют таким же образом и фильтраты объединяют. Добавляют несколько капель концентрированного раствора гидроокиси натрия и упаривают до получения вязкой массы. Обрабатывают остаток этанолом, нагревают, перемешивают и фильтруют. К фильтрату добавляют несколько капель раствора гидроокиси натрия и выпаривают досуха. Остаток растворяют в воде, переносят в делительную воронку, прибавляют 10 мл хлороформа и 20 мл перекиси водорода. После встряхивания слой хлороформа отделяют и экстракцию иода повторяют. Далее заканчивают определение, как указано в разделе XI. 3.1.
XI. 4. ОПРЕДЕЛЕНИЕ ИОДИДА В ВИДЕ ИОННОГО АССОЦИАТА ТРИИОДИДА С ОРГАНИЧЕСКИМИ КРАСИТЕЛЯМИ
Трииодид взаимодействует с органическими красителями с образованием ионных ассоциатов, которые экстрагируются органическими растворителями. Эти растворы интенсивно окрашены и широко применяются для экстракционно-фотометрического определения иодида. Так, ионные ассоциаты трииодида с метиловым фиолетовым экстрагируются бензолом и толуолом [37], с ферроином— нитробензолом [38, 39], бриллиантовым зеленым — толуолом [40—44], родаминовыми красителями [45] и викторией голубой Б [46] — бензолом. Иодхлорид (hCl-) образует ионный ассоциат с кристаллическим фиолетовым [41], метиловым фиолетовым [48], иодбромид (12Вг_)—с синим основным К [49—51], с кристаллическим фиолетовым [52, 53] и др. Иодид и бромид образуют с нильским голубым [54] ионные ассоциаты, которые экстрагируются хлороформом и применяются для фотометрического определения бромидов и иодидов.
Мешающие вещества. Определению мешают многие ионы, которые также образуют ионные ассоциаты с красителями, экстрагирующиеся органическими растворителями совместно с триио-дидом.
Измерение оптической плотности. Рабочая длина волны для измерения оптической плотности зависит от выбранного красителя.
Так, в случае применения бриллиантового зеленого оптическую плотность толуольного раствора измеряют при 610 нм.
Чувствительность метода — 0,5 мкг в 5 мл толуольного экстракта.
XI. 4.1. Определение иодида в отсутствие мешающих веществ с применением бриллиантового зеленого
Реактивы
Нитрит натрия, 0,5%-ный раствор.
Бриллиантовый зеленый, 0,5%-ный раствор.
Толуол, ч. д. а.
Стандартный раствор иодида, см. раздел XI. 2.1.
Ход анализа. Нейтральный анализируемый раствор около 20 мл, содержащий до 100 мкг иодида, помещают в делительную воронку, добавляют 1 мл хлористоводородной кислоты, разбавленной (1:1), и 0,5 мл растЬора нитрита натрия. Содержимое воронки взбалтывают, спустя 5 мин добавляют 0,5 мл раствора бриллиантового зеленого и встряхивают. Затем к оранжево-желтой жидкости приливают 10 мл толуола и энергично взбалтывают в течение 30 с. При этом слой толуола окрашивается в зеленый цвет. Спустя 30 с слой толуола отделяют и измеряют интенсивность окраски при 610 нм по отношению к толуолу.
Содержание иодида находят по калибровочному графику.
XI. 4.2. Определение иода в питьевой воде [31, 42]
Реактивы
Хлорид натрия, 20%-ный раствор.
Нитрат натрия, 0,5%-ный раствор.
Бриллиантовый зеленый, 0,5%-ный раствор.
В высокий термостойкий стакан емкостью 1 л помещают 500 мл анализируемой воды и подщелачивают 10%-ным раствором карбоната калия. Воду осторожно выпаривают до 10—15 мл и количественно переносят ее в термостойкую колбу Кьельдаля емкостью 100 мл, содержащую небольшую бусинку. Стакан два раза прополаскивают 10 мл дистиллированной воды, стараясь стеклянной палочкой возможно полнее снять осадок, образовавшийся на дне и стенках сосуда. Промывные воды переносят в ту же колбу. Содержимое колбы выпаривают досуха, прокаливают при 450—480°C для сожжения органических веществ, находящихся в осадке. После охлаждения в колбу с осадком приливают 15 мл раствора хлорида натрия, 0,5 мл раствора нитрита натрия и 0,5 мл раствора бриллиантового зеленого. Взбалтывают, приливают 5 мл толуола, а затем из бюретки прибавляют 2 мл сиропообразной фосфорной кислоты. В связи с тем, что кислотность среды оказывает большое влияние на образование ассоциата, количество прибавляемой
ортофосфорной кислоты следует предварительно точно вычислить. Оно слагается из 2 мл кислоты, необходимых для создания нужной для реакции кислой среды, и количества кислоты, необходимой для нейтрализации прибавленного К2СО3 и имеющихся в воде карбонатов.
После прибавления ортофосфорной кислоты колбу» энергично встряхивают в течение 1—2 мин, переливают ее содержимое в коническую центрифужную пробирку и центрифугируют около 1 мин при 2000 об/мин. Образовавшийся в верхней части пробирки слой толуола, окрашенный в зеленый цвет, переливают в кювету и измеряют оптическую плотность при 610 нм по отношению к толуолу.
Содержание иода находят по калибровочному графику.
XI. 4.3. Определение иода в хлориде натрия [31, 41]
Навеску 50 г соли растворяют в 200 мл воды и добавляют 0,51 окиси кальция (ч.д. а.) для осаждения присутствующего железа. Содержимое стакана количественно переносят в мерную колбу емкостью 250 мл и доводят до метки водой. Полученный раствор фильтруют через складчатый фильтр в сухую колбу. Отбирают 20 мл фильтрата и определяют иодид, как указано в разделе XI.4.1.
XI. 5. ОПРЕДЕЛЕНИЕ ИОДИДА С ПРИМЕНЕНИЕМ СИНЕГО ОСНОВНОГО К [50, 51]
В методе использована реакция образования соединения иодида с красителем синим основным К, которое экстрагируется бензолом. Реакция образования экстрагирующегося соединения происходит в присутствии бромида. Концентрация бромида должна составлять 27 вес.°/о, а концентрация красителя — 0,015 вес.%. Оптимальная кислотность проведения реакции—1 н. серная кислота.
Мешающие вещества. Краситель образует окрашенные соединения при содержании в анализируемой пробе 1 мкг золота (III) и таллия(1), 10 мкг хрома(У1), индия(Ш), сурьмы(У), олова(1У) и ванадия(V), 500 мкг железа(III). Поэтому мешающие ионы при большом их содержании необходимо предварительно отделять. Определению иода также мешают нитрит-ионы при их содержании больше 5 мкг в анализируемой пробе. Определению не мешают до 5 мг таллий, рений, теллур, ртуть; до 10 мг медь, цинк, свинец, олово(П), сурьма(Ш), марганец(П), мышьяк, титан, цирконий, иттрий, церий; до 100 мг литий, рубидий, алюминий, кобальт, никель, кадмий, хром (III), железо (II), молибден, висмут; до 200 мг щелочные и щелочноземельные металлы-
Измерение оптической плотности. В бензольном растворе ассоциат устойчив в течение 6 ч; оптическую плотность лучше измерять при 650 нм. л
Чувствительность метода — 0,5 мкг иодида в 6 мл бензольного экстракта.
XI. 5.1. Определение иодида в отсутствие мешающих веществ
Реактивы
Бромид калия, навеску соли 50 г растворяют в 100 мл воды.
Синий основной К, 0,3%-ный раствор.
Бензол, ч. д. а.
Стандартный раствор иодида, см. раздел XI. 2.1.
Ход анализа. В пробирку емкостью 25 мл с меткой 10 мл вводят до 2 мл анализируемого раствора, содержащего до 10 мкг иода, тгриливают 7 мл раствора бромида калия, 0,5 мл 20 н. серной кислоты, 0,5 мл раствора красителя, доводят до метки (10 мл) дистиллированной водой, перемешивают, прибавляют из бюретки 6 мл бензола, экстрагируют в течение 1,5—2,0 мин. Спустя 15 мин синий бензольный слой сливают с помощью делительной воронки и измеряют оптическую плотность при 650 нм.
Содержание иодида находят по калибровочному графику.
XI. 5.2. Определение иодида в водах и рапе
Вариант А (для аликвотных частей до 2 мл). Определение проводят, как указано в разделе XI.5.1.
Вариант Б (для аликвотных частей более 2 мл). Взятый объем анализируемой воды помещают в узкогорлую коническую колбу из жаростойкого стекла емкостью 100 мл и подщелачивают 10%-ным раствором карбоната калия. Необходимое количество К2СО3 предварительно устанавливают титрованием отдельной аликвотной части анализируемого раствора в присутствии фенолфталеина до получения четкой розовой окраски. При анализе очень кислых вод, требующих большого количества карбоната калия, его очищают от иодида.
Раствор осторожно выпаривают досуха, не допуская вскипанил во избежание разбрызгивания. В охлажденную колбу с сухим остатком приливают 7 мл раствора бромида калия, 2 мл дистиллированной воды, 0,5 мл серной кислоты, 0,5 мл раствора красителя и продолжают анализ, как указано в разделе XI.5.1.
XI. 6. ОПРЕДЕЛЕНИЕ ИОДИДА В ВИДЕ АССОЦИАТА
С ФЕРРОИНОМ [38]
В. методе использована реакция между иодидом и ферроином (1,10-фенантролинатным комплексом железа) с образованием ассоциата, который хорошо экстрагируется органическими растворителями с образованием интенсивно окрашенного раствора. Оптимальное значение pH образования ассоциата — 4,5. Лучшим экстрагентом является нитробензол.
Мешающие вещества. Определениюиодида мешают 100-крат-
ные количества ионов роданида и перхлората.
Измерение оптической плотности. Спектр раствора ассоциата в нитробензоле характеризуется полосой поглощения с максиму-
мом при 516 нм (рис. 77). Оптическую плотность нитробензольного экстракта измеряют при 516 нм. Спектр водного
Рис. 77. Спектр поглощения нитробензольного раствора ассоциата иодида с ферроином.
раствора ассоциата характеризуется полосой поглощения с максимумом при 510 нм.
Чувствительность метода—1 мкг иода в 10 мл экстракта.
XI. 6.1. Определение иода в отсутствие мешающих веществ
Реактивы
1,10-Фенантролин, 0,01 М раствор.
Железоаммонийные квасцы, 0,005 М раствор.
Буферный ацетатный раствор, 3 М раствор (pH = 4,5).
Нитробензол, очищенный.
Сульфат натрия, безводный.
Стандартный раствор иодида, см. раздел XI. 2.1.
Ход анализа. В мерную колбу емкостью 25 мл прибавляют 2 мл раствора 1,10-фенантролина, 1,0 мл раствора железоаммонийных квасцов, 2 мл буферного раствора, исследуемый раствор, содержащий до 5 мг иода, и водой разбавляют до метки. Содержимое колбы перемешивают, переносят в делительную воронку и экстрагируют 10 мл нитробензола, встряхивая в течение 20 мин. После разделения слоев нитробензольный раствор переносят в колбу, в которую предварительно помещают 1 г сульфата натрия. Содержимое колбы встряхивают для поглощения воды, отфильтровывают часть раствора через сухой складчатый фильтр и измеряют оптическую плотность раствора при 516 нм по отношению к раствору нитробензола.
Содержание иода находят по калибровочному графику.
XI. 7. ОПРЕДЕЛЕНИЕ ИОДАТА
Иодат можно определить одним из описанных выше методов после проведения реакции с иодидом, в результате чего выделяется иод. Кроме того, для определения йодата разработан ряд мето-
дов, основанных на реакциях окисления органических веществ, в том числе таннина до эллаговой кислоты [55] бледно-желтого цвета; n-аминофенола до хинонимина, который конденсируется с п-ами-нофенолом с образованием синего индаминового красителя:
Этой реакции не мешают хлораты и броматы [56].
Гидразид изоникотиновой кислоты в хлористоводородной среде
в присутствии иодат-ионов взаимодействует с хлоридом 2,3,5-три-
фенилтетразола с образованием соединения розового цвета [57]. Для фотометрического титрования йодата, перйодата, бромата и других окислителей применен комплекс этилендиаминтетраацетата с хромом(II) [58].
Мешающие вещества. Определению йодатов мешают нитриты, персульфаты и большие количества броматов, которые в условиях определения йодатов окисляют таннин до эллаговой кислоты. Оптимальное значение pH при определении йодата
Рис. 78. Спектр поглощения эллалговой кислоты (/) и таннина (2).
составляет 2,6.
Измерение оптической плотности. Реакция между таннином и йодатом прохо-
дит во времени,, поэтому оптическую
плотность необходимо измерять через 3 мин после сливания растворов. Окраска устойчива в течение 1,5 ч. Спектры поглощения раствора продукта окисления таннина — эллаговой кислоты и раствора таннина приведены на рис. 78. Оптическую плотность раствора лучше измерять на спектрофотометре при 364 нм или на фотоэлектроколориметре с синим светофильтром.
Чувствительность метода—15 мкг йодата в 25 мл конечного
раствора.
XI. 7.1. Определение йодата в отсутствие мешающих веществ [55]
Реактивы
Таннин, 0,3%-ный свежеприготовленный водный раствор.
Уксусная кислота, 2 н. раствор.
Стандартный раствор йодата. Растворяют 0,1223 г КЮ3 в воде, переносят в мерную колбу емкостью 1 л, разбавляют водой до метки и перемешивают. Этот раствор содержит 0,1 мг в 1 мл.
Ход анализа. В мерную колбу емкостью 25 мл помещают до 15 мл анализируемого раствора, содержащего 15—750 мкг йодата, прибавляют 7,5 мл раствора таннина, 2 мл уксусной кислоты, разбавляют до метки водой и перемешивают. Спустя 3 мин измеряют оптическую плотность раствора при 364 нм по отношению к раствору сравнения, который готовят аналогично, но без прибавления йодата.
Содержание йодата находят по калибровочному графику.
XI. 8. ДРУГИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ИОДА
Для определения иодида в воде применен метод окисления его до йодата с помощью бихромата, восстановления йодата до иоди-стоводородной кислоты и ее отгонки, затем иодид окисляют до йодата и после взаимодействия с иодидом измеряют оптическую плотность раствора иода [59] или определяют по иодкрахмальной реакции [60]. С целью определения иодида в продуктах переработки пылей свинцового производства применена экстракция свободного иода четыреххлористым углеродом [61].
Разработан метод определения иодида, основанный на образовании окрашенного ассоциата тетраиодидного комплекса ртути с метиленовым голубым [62] и иода с бис(бензтиазолил)-2,2'-дисульфидом и ацетонитрилом [63], а также на реакции образования тройного комплекса ртути с гександиаминтетраацетатом и иодидом [HgYI]3-, спектр поглощения которого имеет максимум при 255 нм [64].
Ряд методов основан на реакциях разрушения иодидом окрашенных комплексов, например дифенилкарбазоната ртути [65], дитизоната ртути [66], диэтилдитиокарбамата серебра [67] и др.
При взаимодействии перхлората 4-циано-1-метилпиридиния с иодидом образуется иодид 4-циано-1-метилпиридиния, который под действием света переходит в интенсивно окрашенный комплекс с переносом заряда с максимумом поглощения при 340—350 нм (е = 2,6«104). Этот метод применен для определения иодида в воде [68].
Иодкрахмальная реакция использована для разработки термоколориметрического метода определения иода [69].
Для определения перйодата использована реакция окисления о-дианизидина (3,3'-диметоксибензидина) в присутствии «-пропанола или н-бутанола. В результате этой реакции образуется бис-(3,3'-диметокси-4-амино)-азофенил, спектр которого характеризуется полосой поглощения с максимумом при 445 нм [70].
Для определения йодата в иодидах натрия, цезия, кальция и в монокристаллах иодидов натрия и цезия, активированных таллием, применен метод, основанный на измерении оптической плотности раствора иода, выделяющегося в кислой среде [71]. Перйодат в слабокислой среде образует с бензоилгидразином окрашенное соединение с максимумом поглощения при 405 нм, в виде которого 344
определяют перйодат [72]. Перйодат при pH = 4,0—4,2 окисляет окрашенный комплекс железа(II) с 2,4,6-три-2-пиридил-сшюи-три-азеном до бесцветного комплекса железа(III) с этим реагентом. Эта реакция применена для косвенного определения перйодата [73].
Широко применяется реакция между церием (IV) и мышьяком (III), которая катализируется иодидом [74—79]:
2CeIV + As111 оранжевый бесцветный
2Ceni + Asv бесцветный бесцветный
В присутствии иодида скорость реакции пропорциональна его концентрации, что дает возможность определять иодид с чувствительностью 0,005 мкг, но специфичность реакции невелика [80]. Эта реакция применена для определения иода в кремнии и германии [81], воде [82, 83], в почвах [84], в хлориде натрия [85] и в других объектах. Для каталитического определения иодида применены реакции обесцвечивания пирокатехинового фиолетового перекисью водорода [86], реакция между 3,3'-диметилнафтидином и перекисью водорода [87], роданидным комплексом железа и нитритом [88, 89], хлорамином Т и М,М-тетраметилдиаминодифенилметаном [90], ка-дионом и персульфатом калия [91] и ряд других каталитических методов [92—98].
ЛИТЕРАТУРА
1. RobourdinS. М., Ann. Chem., Liebigs, 76, 375 (1850).
2. С h a t i n A., Compt. rend., 32, 669 (1851).
3. C u s t e r J. J., N a t e 1 s о n S., Anal. Chem., 21, 1005 (1949).
4. Almquist H. J., Givens J. W., Ind. Eng. Chem., Anal. Ed., 5, 254 (1933). 5. Fraps G. S., Fudge J. E., J. Assoc. Offic. Agr. Chemists, 23, 164 (1940). 6. von FellenburgT., Biochem. Z., 139, 371 (1923).
7. von FellenburgT., Biochem. Z„ 152, 116 (1924).
8. M a 1 i j a г о v K. L., M a t s k i e v i c h W. B., Mikrochem., 13, 85 (1933).
9. Robin M. S., Anal. Chem., 22, 480 (1950).
10. W i n t e r s t e i n e r E„ Z. physiol. Chem., 104, 54 (1918).
11. McHargue J. S., Young D. W., Cal fee R. K., Ind. Eng. Chem., Anal. Ed., 6, 318 (1934).
12. Gross W. G., Wood L. K., McHargue J. S., Anal. Chem., 20, 900 (1948).
13. Shahrokh В. K., Chesbro R. M., Anal. Chem., 21, 1003 (1949).
14. Houston F. G., Anal. Chem., 22, 493 (1950).
15. C h e n g K. L„ G о у d i s h B. L., Anal. Chem., 35, 1965 (1963).
16. Waleszek-Piotrowski, Koch F. C., J. Biol. Chem., 194, 427 (1952).
17. 3 а к Б. В Сб. «Колориметрические (фотометрические) методы определения неметаллов». Пер. с англ. Под ред. А. И. Бусева, М., Издатинлит, 1963, См. с. 219.
18. S a 1 ten W. Т., МсКа у E. A., Endocrinology, 35, 380 (1944).
19. M с С u 11 a g h D. R., J. Biol. Chem., 107, 35 (1934).
20. D e c k e r J. W., Hayden H. S., Anal. Chem., 23, 798 (1951).
21. L e i p e r t T., Biochem. Z., 261, 436 (1933).
22. Z a k B., Willard H. H., Myers G. B., Boyle A. J., Anal. Chem., 24, 1345 (1952).
23. Shahrokh В. K., J. Biol. Chem., 147, 109 (1943).
24. L a m b e r t J. L., Anal. Chem., 23, 1251 (1951).
25. Crouch W. H„ Anal. Chem., 34, 1698 (1962).
26. S u g a w a r a K., Koya ma T., Terada K-, Bull. Chem. Soc. Japan, 28, 494 (1955).
27. T s u n о g a i S., Anal. chim. acta, 55, 444 (1971).
28. C u s t e r J. J., N a t e 1 s о n S., Anal. Chem., 21, 1005 (1949).
29. H о u s t о n F. G„ Anal. Chem., 22, 493 (1950).
30. Thrum W. E., Proc. Indiana Acad. Sci., 59, L55 (1950).
31. Мустафин И. С. и др. Ассортимент реактивов на иод. М., НИИТЭХИМ, 1968. См. с. 25.
32. BrodeW. R., J. Am. Chem. Soc., 48, 1877 (1926).
33. Марченко 3. Фотометрическое определение элементов. Пер. с польского. Под ред. Ю. А. Золотова. М., «Мир», 1971. См. с. 189.
34. С о 11 i n s A. G„ W a t k i n s J. W., Anal. Chem., 31, 1182 (1959).
35. Grimaldi F. S., Schnepfe M. M., Anal. chim. acta, 53, 181 (1971).
36. Назаренко В. A., HI у с т о в а М. Б., Зав. лаб., 27, 15 (1961).
37. Савичев Е. И., Васильева И. Г., Головин Е. И., Зав. лаб., 29, 1433 (1963).
38. Yamamoto Y., К i n u w a k i S., Bull. Chem. Soc. Jap., 37, 434 (1964).
39. Yamamoto Y., Ta rum о to T., Tsuboughi M., Bull. Chem. Soc. Jap., 44, 2124 (1971).
40. Проскурякова Г. Ф., HI e й к и н а Р. В., Ч е р н а в и н а М. С., Известия вузов, Хим. и хим. технолог., 6, 729 (1963).
41. Л а п и н Л. Н., Изв. АН УзбССР. Сер. мед., 6, 60 (1959).
42. Л а п и н Л. Н., Рейс Н. В., Лаб. дело, 8, 21 (1961); Вопр. питания, 26, № 2, 26 (1967).
43. Панталер Р. П., Шершнева Л. А., Шерщуков В. М. В сб. «Методы анализа галогенидов щелочн. и щелочноземельн. мет. высокой чистоты», ч. 2. Харьков, 1971. См. с. 154.
44. Раманаускас Э. И., Буникене Л. В., Науч. тр. высш, учебн. заведений ЛитССР. Хим. и хим. технол., № 9, 17 (1968).
45. П о д б е р е з с к а я Н. К., Сушкова В. А., Шиленков Е. А. В сб. «Исслед. в области хим. и физ. методов анализа минеральн. сырья». Алма-Ата, 1971. См. с. 87.
46. П о д б е р е з с к а я Н. К., И в а н к о в а А. И. В сб. «Исслед. и разработка
фотометрии, методов для определения микроколичеств элементов в минеральном сырье». ОНТИ КазИИС, Алма-Ата, 1967. См. с. 101.
47. М о г s с h е s В., Т б 1 g G., Z. anal. Chem., 200, 20 (1964).
48. Ш в е й к и н а Р. В., Изв. вузов. Хим. и хим. технолог., 10, 1288 (1967),
49. Her г is D. G., Richards F, М., Anal. Chem., 36, 1156 (1964).
50. Под берез ск а я Н. К. Авт. св. СССР, кл. 421, 3/08; (gOln), № 167670, заявл. 23.01.64, опубл. 27.01.65. РЖХим, 1966, 14Г94.
51. П о д б е р е з с к а я Н. К., Ш и л е н к о Е. А., Зав. лаб., 32, 918 (1966).
52. Раманаускас Э. И., Бунине не Л. В., Жиленайте М., Науч. тр. вузов ЛитССР. Химия и хим. технол., № 9, 29 (1968).
53. Б у н и к е н е Л. В., Раманаускас Э. И., ЖАХ, 23, 1364 (1968).
54. Likussar W., Pokorny G., Zechmann H., Anal. chim. acta, 49, 97 (1970).
55. Вильборг С. С., Дроздов В. А., Изв. вузов. Хим. и хим. технолог., 3, 75 (1960).
56. Fuchs J., J u n g г е i s Е., В е n - D о г L., Anal. chim. acta, 31, 187 (1964).
57. Н a s h m i M. H., Ahmad H„ Rashid A., Azam F., Anal. Chem., 36, 2471 (1964).
58. G г о e n e v e 1 d E. R., den Boef G., Z. anal. Chem., 237, 85 (1968).
59. Matthews A. D., Riley J. P., Anal. chim. acta, 51, 295 (1970).
60. Д и м и т p о в Г., Науч. тр. Высш. пед. ин-та, Пловдив, 4, 141 (1966).
61. X р и с т о ф о р о в Б. С., Чепик М. Н., Металлург, и хим. пром. Казахстана. Науч.-тех. сб., № 2 (12), 74 (1961).
62. Gantschewa A., Mikrochim. acta, № 4, 601 (1967).
63. Kujawa R., Mikrochim. acta, № 1, 193 (1969).
64. Nomura Toshiaki, J. Chem. Soc. Japan, Pure Chem. Sec., 88, 199 (1967).
65. О k u t a m T a d a o, J. Chem. Soc. Japan, Pure Chem. Sec., 88, 737 (1967).
66. A gt e r d e n b о s J., Jiitte В. A. H. G., Elberse P. A., Taianta, 17, 1085 (1970).
67. Komatsu Sumio, Nomura Toshiaki, Ushui loshiko, J. Chem. Soc. Japan, Pure Chem. Sfec., 88, 1164 (1967).
68. P о z i о m e k E. J., Reger D. W., Anal. chim. acta, 58, 459 (1972).
69. Зиновьев А. И., Зав. лаб., 19, 793 (1953).
70. G u e r n e t M., Bull. Soc. Chim. France, № 3, 478 (1964).
71. Манжелей Л. С., Булгакова A. M., Никитина Л. M. В сб. «Методы анализа галогенидов щелочи, и щелочноземельн. мет. высокой чистоты», ч. 2, Харьков, 1971. См. с. 146.
72. Escarrila А. М., Maloney Р. F., Maloney Р. М., Anal. chim. acta, 45, 199 (1969).
73. Avigad G., Carbohydrate Res., 11, 119 (1969).
74. Sandell E. B., Kolthoff I. M., J. Am. Chem. Soc., 56, 1426 (1934).
75. San dell E. B., Kolthoff I. M., Mikrochim. Acta, 1, 9 (1951).
76. Pen A., Zb. Biotehn. fak. Univ. Ljubljani, 18, № 9—11, 23 (1971).
77. E r n s t O., S z о 1 n о k i G., Z. anal. Chem., 258, 263 (1-972).
78. Kurbart H., Quiroga S., Caro R. A., Radicell a R., Informes. Comis. nac. energ. atom., 308, 11 (1972); РЖХим, 1972, 18Г98.
79. P a 1 e 11 a B„ Mikrochim. acta, № 5, 956 (1969).
80. Stoic V., Z. anal. Chem., 183, 262 (1961).
81. Туманов А. А., Сидоренко A. H., Коренман Я. И., Зав. лаб., 30, 1058 (1964).
82. Станчева М., Химия и индустрия (Болг.), 39, 112 (1967).
83. Revel J., Cahiers Oceanogr., 21, 273 (1969).
84. Ceausescu D., Acta chim. Acad, scient. Hung., 28, 165 (1961).
85. D a 1 i b о r W., Chem. listy, 62, 684 (1968).
86. Oiwa Koimura e. a., Japan Analyst, 17, 805 (1968),
87. В о h n a r J., N a g у L., Mikrochim. acta, № 1, 108 (1969).
88. Ottawa у J. M., Fuller C. W., Rows ton W. B., Anal. chim. acta, 45, 541 (1969).
89. П p о с к у p я к о в а Г. Ф. Тр. Свердловского с.-х. ин-та, 15, 322 (1969).
90. Jungreis Е., G е d а 1 i a I., Mikrochim. acta, № 1, 145 (1960).
91. ТамарченкоЛ. М., ЖАХ, 25, 567 (1970).
92. Yonehara Norinob u, Bull. Chem. Soc. Japan, 37, 1101 (1964).
93. В a 11 c z о H., Z. anal. Chem., 245, 20 (1969).
94. F r i t s c h e U., Mikrochim. acta, № 6, 1322 (1969).
95. Belcher R., Ha my a J. W., Townshend A., Chim. anal. (RSR), 1,
23 (1971).
96. В о g n a r J., Mikrochim. acta, № 3, 455 (1968).
97. Я Ц и м и р с к и й К. Б. Кинетические методы анализа. М., «Химия», 1967. См. с. 199.
98. Funahashi S., Tabata М., Tanaka М., Anal. chim. acta, 57, 311 (1971).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Агломераты, определение мышьяка 147
Азот 11 сл. .
определение аминного И, 26
— аминопиридин-феноловым методом 27
— аммонийного 12
— общего 11, 26
— окислов 39
— пиразолоновым методом 12, 26
— по Кьельдалю 12, 15
— по собственному поглощению 38
— с бордо В 26
--- гипохлоритом и гипобромитом 26
— характеристика методов L1, 26
— хингидронным методом 12, 26
— цианамидного 27
отгонка 18
отделение 16 сл.
Азотная кислота, определение 38
Азотсодержащие вещества, определение азота 12 сл.
Алюминий
определение кремния 128
— фосфора 108
Аммиак
определение 18 сл.
— в виде аммиаката меди 27
— индофеноловым методом И, 21 сл.
— по Несслеру И, 14, 18 сл.
— фосфора 107
Аммиак
отгонка 16 сл.
отделение 12, 17 сл.’
Аммоний
определение в отсутствие мешающих веществ 18 сл.
— с тимолом и гипобромитом 23 сл.
---- фенолом и гипохлоритом 24 сл.
— характеристика методов 1I сл. Апатиты, определение фтора 302
Барит, определение кремния 128 Белки, определение аммиака 21 Бензгидроксамовая кислота и ее производные, определение аммиака 21
Бензин, определение фосфора 107 Бериллий
определение аммиака 21
— кремния 124
— хлорида 316
Биологические материалы, см. также
Растительные материалы определение бора 61, 68 — иода 334 — кремния 128 — серы 204 — фосфора 108
Бор 45 сл. определение в отсутствие мешающих веществ 55, 60, 66
— в присутствии фторида 49 — люминесцентное 68, 70 — методы и реакции 46 сл.
Бор
определение с азометином 68
— — азуролом С 68
— — ализарином 70
— — аминоантрахиноном 70
— — антипириновыми красителями 68
— — ацетилсалициловой кислотой 70
----Аш-резорцином 68
— — бензоином 70
— — 1,1,-бис(6-хлорантрахино-лил) -амином 68
— — бриллиантовым зеленым 62 сл., 64
— — диаминоантрафурином 68
----диаминохризазином 68
----1,1,-диантримидом 68, 70
— — карминовой кислотой 69, 70 кармином 51, 58 сл., 70
— — К-кислоты производными 68
— — красно-фиолетовым 68
— — кристаллическим фиолетовым 68
— — куркумином 53 сл., 69
----магнезоном I 68
----метиленовым голубым 68, 69
---- морином 70
----нильским сипим А 68
— — оксиантрахиноном 70
----производными Аш-кислоты 68
— — рН-индикаторами 47
— — салициловой кислотой и кристаллическим фиолетовым 65 сл.
----п-сульфофенилазохромо-троповой кислотой 68
— — фенилфлуоропом 70
— — ферроином 68
— — флуориметрическое 70
— — хинализарином 70
хромотропом 2В 68
— — хромотропом ЗВ 69
— характеристика методов 45 сл., 68 сл.
отделение 46, 50 сл.
Бора о^ись, определение кремния 128 Борил-ионы определение с кармином 47 ' — — полиоксиантрахиноном 47
Борная кислота
определение с ализарином S 47
— — комплексообразующими веществами 47
— — куркумином 47
— — образованием тройных комплексов 49
— — производными антрахинона 68
— со спиртами 46
— с фенолами 46
Борнометиловый эфир, дистилляция 51 Борсодержащие материалы, определение бора 49 сл. Бром 320 сл. в смеси с нитрозилбромидом, анализ 329
определение с хромотропом 2В 328
— характеристика методов 320 отделение 320
Бромат, определение 329
Бромид, см. также Бром окисление до бромата 323 определение 320 сл. — в отсутствие мешающих веществ 322, 325, 326 — на автоматическом анализаторе 328 — по подавлению реакции хлорирования аммония 329 --------реакции разрушения дити-зоната серебра 329 —-------дифенилкарбазоната
ртути 329
----светопоглощению HgBrj 329
— с метиловым оранжевым 321 сл., 328
---- образованием эозина 325
---- пиридином и цианидом 327
----розанилином 326—328
----тетрабромсульфонафталином 328
Бромид
определение с о-толидином 328
— — трифенилбензилфосфонием 328
— — трифенилметановыми красителями 328
---феноловым красным 324, 329
--- фуксинсернистой кислотой 328
— — этилентриаминтетрацетатом ртути 328
отделение 307, 320
Ванадий, определение мышьяка 154
Висмут
определение селена 237
— теллура 267
— хлора 317
Воды
определение аммиака 21
— аммония 20, 25
— бора 60, 65, 68
— бромида 324, 328, 336
— двуокиси хлора 318
— иода 338, 339, 345
— иодида 336, 341, 344
— кислорода 175, 177—180, 182, 183
— кремния 128
— мышьяка 163, 167
— озона 184, 187
— сероводорода 192
— серы 215
— сульфата 204, 217
— фосфора 81, 83
— фтора 293, 297, 300, 303
— фторида 302
— хлора 317
— хлорида 309
очистка от соединений азота 13, 15
Воздух
определение аммиака 21, 25
— арсина 159
— двуокиси серы 202
— озона 185
— сероуглерода 216
Воздух
определение серы 196
— фосфора 108
— хлора 317
Вольфрам, определение аммиака 21
Газовые смеси
определение аммиака 27
— кислорода 175, 176, 181, 183
— озона 183
— сероводорода 192
Г аллий
определение мышьяка 155
— селена 237
— хлора 317
Германий
определение иода 345
— мышьяка 143, 165
— селена 251
— сульфата 217
Гидроксиламин, определение 21
Глинозем, определение кремния 128, 129
Деготь, определение мышьяка 164
Зола каменноугольная, определение сульфата 204, 217
Золото, определение мышьяка 157
Известь хлорная, определение хлора 317
Индий
определение мышьяка 155
— селена 237
— серы 215, 217
Иод 331 сл.
определение 332 сл.
— в виде трииодида 335
---отсутствие мешающих веществ 342
— с ацетонитрилом 344
---бис(бензтиазолил)-2,2'-ди-
сульфидом 344
— — крахмалом 332 сл.
— термоколориметрическое 344
— характеристика методов 331
сл., 344
Иодат
определение 342 сл.
— в отсутствие мешающих веществ 343
Иодид определение в виде ассоциата с органическими красителями 338 сл.
---------- с ферроином 341 — в отсутствие мешающих веществ 333, 336, 339, 341, 343
— иодкрахмальной реакцией 344 — каталитической реакцией 345 — по оптической плотности раствора иода 344
— после окисления 334
— с бриллиантовым зеленым 339
— — гександиаминтетрацетатом ртути 344
----З.З'-диметилнафтидином 345
-----дитизон атом ртути 344
— — дифенилкарбазоном ртути 344
----диэтйлдитиокарбаматом серебра 344
---- кадионом и персульфатом калия 345
•---комплексом метиленового
голубого 344
----перхлоратом 4-циано-1-ме-тилпиридиния 344
---- пирокатехиновым фиолетовым 345
----роданидным комплексом железа и нитритом 345
-----синим основным К 340 сл.
----трифенилбензилфосфонием 328
----хлорамином Т и N.N-тетра-метилдиаминодифенилкетоном 345
----этилентриаминтетрацетата 328
отделение 307, 331
Кадмий определение мышьяка 157
Кадмий
определение серы 215, 217 Карбазоны, определение азота 13 Катализаторы риформинга, определе-
ние мышьяка 143
Кислород 175 сл.
определение методом Винклера 179 сл.
— по количеству окисленного железа 182
---------- марганца 182
-------окисленной меди 180 сл.
— с 4,4'бис(дифенилметил)-ди-
фенила 182
----индигокармином 176 сл., 182
---- иодидом 183
----лейкосафранином 178 сл.
— характеристика методов 175 сл.
Кокс, определение фосфора 108 Кремневая кислота
определение 77 сл., 109 сл.
— в виде желтого комплекса 113
-------синего комплекса 114сл.
— в отсутствие мешающих веществ 113
получение аналитических концентратов 79
Кремний 73 сл.
определение 109 сл.
— бора 50, 53, 56, 68
— в присутствии германия 128
------- меди 120
-------фосфора 78, 79
-------фосфора и мышьяка 75
-------фторида 128
— иода 337, 345
— мышьяка 155, 162
— с 2-амино-4-хлортиофенолом 129
---- родамином В 129
— фосфора 108
— характеристика методов 73 сл.
отделение 79, 129
Кремнийорганические соединения, оп-
ределение кремния 128
Кремния бифторид, определение кремния 128
Кровь
определение бромида 328
— хлорида 309
Лекарства, определение фторида 302
Медь
определение кремния 120, 128
— мышьяка 150, 163, 167, 168
— селена 237
— теллура 257, 268
Металлы
определение азота 12, 20, 25
— бора 49
— кремния 76, 78, 118, 175, 183 •
— мышьяка 145, 149, 151, 165
— нитридов 11, 12, 22
— серы 193, 202, 215, 216
— фосфора 76, 84, 109
Метеориты, определение фторида 302 Метилтрихлорсилан, определение фосфора 108 Минералы
определение бора 62
— кремния 128
— мышьяка 168
— серы 195 Молибден
определение с 2-амино-4-хлорбен-золотиолом 106
--- роданидом 106
---фенилфлуороном 106
---хром фиолетовым К 106
-------хром черным специальным 106 Мышьяк 134 сл.
концентрирование 134 сл.
определение 142 сл., 170 — в виде арсенидов 161 сл. -------мышьяковованадиево-
молибденового комплекса 75
—-------мышьяковомолибдено-
вой сини 142
— в отсутствие мешающих веществ 144, 145, 159, 164, 166, 169
Мышьяк
определение в присутствии фосфора и кремния 75
— с бутилродамином 168
----- ванадатом натрия 171
-----гексацианоферратом (III) калия 171
-----гипофосфитом 165 сл.
----- дитизонатом серебра 171
-----а-дитионафтол тетраметил-аммония 170
----- диэтилдитиокарбаматом серебра 163 сл.
— селена 237
— с индикаторными бумагами 158
-----иодметиленовым голубым 170
----- кверцетином 170
----- куркумином 170
----- 8-меркаптохинолином 170 ----- молибденом 171
-----сернокислым гидразином 144
-----n-сульфамоилбензоатом серебра 170
----- трииодидом 170
----- ферроином 171
----- хлоридом олова 145
— характеристика методов 141 отгонка в виде арсина 136 ------- хлорида 135 отделение 135 сл.
— хроматографическое 141
— экстракционное 138, 139,
141
Накипи, определение кремния 128 Напитки, определение фторида 302 Натрий и его соединения определение бора 68 — иода 340, 345 — йодата 344 — кислорода 183 — мышьяка 164, 169 — сульфата 218
Неметаллические включения, определение кремния 128
Неорганические вещества определение азота 14 сл. — мышьяка 163
Нефть, определение сульфата 204, 217
Нефтяные продукты, определение мышьяка 143
Никель
определение кремния 128
— фосфора 102
Ниобий определение кремния 128 — мышьяка 152 — фосфора 108
Нитрат определение 27 сл. — в отсутствие мешающих веществ 31
— по образованию нитросоединений 27 сл., 38
— — собственному поглощению 36
— с бруцином 32
— — восстановлением до аммиака 39
--------— нитрита 32, 39
---- ксиленолами 39
---- неокупроином 39
----нильским голубым А 40
--- образованием азосоединения 39
— — тетрафен ил арсонием 40
— — толуолом 40
— — хинализарином 39
---хромотроповой кислотой 29
— характеристика методов 11 Нитриды, определение 11, 12, 14 сл. Нитрит
определение 33 сл.
— каталитическое 40
— по собственному поглощению 36
— с азуленом 35
---- антипирином 39
---- бензидином и л-фенилен-диамином 39
— — восстановлением до аммиака 39
---- н-гептанолом 39
Нитрит
определение с ДТБ 36
—---n-нитроанилином и дифе-
ниламином 35
----производными бензидина 39
---- риванолом 38
----га-розанилинметансульфо-кислотой 39
— — синим основным К 39
----соединениями ацилсульфоксон ия 39
----сульфаниламидом 39, 40
---- сульфаниловой кислотой 34, 39
толуолом 40
эйконогеном 39
— флуоресцентное 40
— характеристика методов 11,37 Нитрозилбромид в смеси с бромом, анализ 329
Нитросоединения, определение азота 43
Огнеупорные материалы, определение кремния 128
Озон
определение 175, 183 сл.
— в присутствии кислорода 184
— — — соединений хлора 184
— по реакции окисления иодида 187
— с дигидроакридином 187
эвгенолом 187
Окислы, определение фторидов 302
Оксимы, определение азота 13
Олово, определение фосфора 108 Органические вещества
определение азота 12, 13
— бора 68
— мышьяка 157, 163
— фосфора 108
— фторида 294
Осадки, определение аммиака 21
Пар, определение фосфора 107
Перборат, определение кремния 128
Пербромат, определение с кристаллическим фиолетовым 329
Перйодат
определение с бензоилгидразином 344
---(3,3'-диметокси-4-амино) -азофенилом 344
---2,4,6-три-2-пиридил-силл<-
триазеном 344
Перхлорат
определение в отсутствие мешающих веществ 316
— с бриллиантовым зеленым 317
----2,9-диметил-1,10-фенан-
тролином 317
--- кристаллическим фиолетовым 317
----малахитовым зеленым 317
----метиленовым голубым 315
---- тетрафениларсонием 317
Пиво, определение двуокиси серы 203
Пирофосфат, определение 105
Пленки магнитные, определение фосфора 106
Полупроводники
определение кремния 128
— селена 237
— серы 215
Породы
определение бора 62, 65, 67, 68
— иода 334, 336
— мышьяка 143
— фосфора 78, 81, 83, 85
— фтора 297, 302
Почвы
определение азота 26
— бора 65, 68
— иода 334, 345
— нитрата 31
— селена 240
— сульфата 204, 217, 218
— фосфора 86, 106—108
Пыли свинцового производства определение иодида 344 — теллура и селена 230
Рапа, определение иодида 341
Рассол
определение бромида 327
— иода 338
Растительные ткани определение азота 26 — иода 334 — селена 240 — фосфора 107
Реактивы, определение кремния 128
Роданид, определение 203
Руды
определение бора 49
— кремния 128
— мышьяка 147, 167, 168
— селена 232, 238, 243, 245
— теллура 272
— фосфора 85, 86, 92, 107
— фтора 302
Свинец
определение мышьяка 160, 168
— теллура 257, 261, 265, 267
— фосфора 108
Селен 222 сл. »
восстановители 237 определение 230 сл., 273 сл. — в виде дитизоната 241 сл. ----отсутствие мешающих веществ 235
-----присутствии меди и железа 251
форме золя 246 сл.
— мышьяка 143
— по реакции окисления органических соединений 249 сл.
— реагенты 227
— с висмутолом II 274
гидразином 248
----З.З'-диаминобензидином
227, 228, 230 сл.
----диаминонафталином 228,
231
-----4,5-диамино-6-тиопирими-дином 228, 274
----2,3-диаминофеназином 228
----диантримидом 229
----диметиламино-1,2-фели-
лендиаминами 229, 231
---- дитизоном 229
----1,1'-дифенилгидразином
230, 232
Селен
определение с 1,4-дифенилтио-семикарбазидом 230, 243 сл.
— серы 215, 216
— с 2-меркаптобензимидазолом 230, 273
— — 2-меркаптобензтиазолом 273
---4-метил-2-фенилендиамй-ном 231
---молибденом 274
— — пирролом 231, 232
— — реагентами, содержащими тионную и тиольную группы 227
--- теллуром 247
--- тиогликолевой кислотой и ее производными 273
--- фенилантраниловой кислотой 232
--- фенилгидразином 230
— — о-фенилендиамином 231
--- 1-фенилтиосемикарбазидом 231
— — циклогексаноном 274
— теллура 259, 266
— характеристика методов 226 сл.
— хлора 317
осаждение 223
отделение 222 сл.
Селенистая кислота, определение теллура 259
Сера 189 сл., 215 сл.
восстановление до сероводорода 199
определение 190 сл., 196
— иод-азидной реакцией 215
— мышьяка 143, 163
— с ацетатом таллия 196
— селена 230
— теллура 230, 261
— хлорида 309
отделение 189 сл.
Сера сульфатная
определение 204 сл.
— в виде метиленового голубого 214
— малых количеств 205
Сера
определение по разрушению али-зарината циркония 210
— с бензидином 205, 212
--- комплексом тория 205
--- лаками 205
---нитхромазо 204, 205
--- ортаниловым Б 204
---роданидом железа 205
--- родизонатом тория 205
---хлоранилатом бария204,207
---хроматом бария 204, 209
Серебро, определение мышьяка 163
Серная кислота, определение мышьяка 143, 160, 163
Сернистая кислота, определение 200
Сероводород
определение в отсутствие мешающих веществ 191
— — смеси с двуокисью серы и сероокисью углерода 216
— с Н,Ы'-диметил-/г-фениленди-амином 190
Сероуглерод, определение 215
Серы двуокись, определение 216, 217
Силикаты
определение бора 49, 52
— кремния 77, 78, 128
— мышьяка 143
— фосфора 85
— фтора 297, 299
— фторида 302
Силумины
определение кремния 128
— фосфора 109
Смолы, определение фторида 302
Соли
определение бора 67, 68
— бромида 328
— йодата 344
— фосфора 81, 83
— фторида 302
Солод, определение двуокиси серы 203
^плавление мокрое 15
Сплавы
определение азота 11, 16
— бора 68
Сплавы
определение кислорода 175
— кремния 120—122
— мышьяка 150, 167
— фосфора 88, 102, 106, 109
Стали
определейие азота 14, 15, 27
— бора 61, 65, 68
— кислорода 183
— кремния 116, 118, 128
• — мышьяка 167
— селена 236
— теллура 254 сл.
— фосфора 88, 90, 91, 96, 105, 107
Стекла
определение бора 68
— кремния 128
— фторида 302
Стибнит, определение серы 203 Сульфат
определение с хлоранилатом 217
— турбидиметрическое 213
Сульфид, определение 216
Сульфит, определение 201
Суперфосфат двойной, определение фосфора 107
Сурьма
определение мышьяка 151, 168
— селена 237
Сырье, определение фосфора 86
Таллий
определение мышьяка 166
— серы 215
Тантал
определение кремния 128
— фосфора 108
Теллур 222 сл.
восстановители 227
определение 252 сл., 273 сл.
— в виде иодидного комплекса 252 сл.
------— фосфоротеллуристомо-либденовой кислоты 273 сл.
— — отсутствие мешающих веществ 253, 256, 258, 261, 263, 265, 271
Теллур
определение в форме золя 246 сл.
— кремния 128
— реагенты 227
— с виктория голубой 233
— — висмутолом II 230, 274
— — вольфрамом 275
— — гипофосфористой кислотой 248
---3,5-дифенилпиразолин-
1-дитиокарбамат натрия 258 сл.
— — диэтилдитиокарбаматом и его производными 255 сл.
— селена 239
— — 5-меркапто-З-(нафтил-2) -
1,3,4-тиадиазолтионом 259 сл.
---метиловым фиолетовым 233
— совместно с селеном 247
— с производными пиразолона 233, 264 сл.
-------триоксифлуорона 275
---родаминами 233, 269 сл.
---серусодержащими лигандами 233
--- тетраэтил(метил)тиурам-дисульфидом 274
--- тиогликолевой кислотой и ее производными 273
— — тиомочевиной 234, 262 сл.
— — феррином 275
— — фосфорномолибденовой кислотой 233
— характеристика методов 226 сл.
— хлора 317
отделение 222 сл.
Теллуровая кислота, определение селена 239
Тетратионаты, определение 216
Тиосульфат, определение 216
Титан
определение азота 25
— аммиака 21
— кислорода 183
— кремния 124 сл., 128
— серы 215
— хлорида 309, 316
Топливо твердое, определение фосфора 107
Торий, определение хлорида 316
Трихлорсилан, определение кремния 128
Углеводороды, определение серы 197 сл.
Углерода сероокись, определение с сероводородом 216
Уголь
определение мышьяка 164
— фосфора 108
Удобрения
определение бора 68
— фосфора 86, 106
Уран и его соединения
определение бора 68
— бромида 329
— кремния 128
— мышьяка 143
— хлорида 316
Феррониобий, определение фосфора 107
Ферросиликохром, определение кремния 128
Ферросилиций, определение кремния 119
Ферротитан, определение фосфора 107
Флюоритовый концентрат, определение кремния 128
Флюсы
определение кремния 127
— фторида 292
Фосфаты
определение фосфора 109
— фтора 303
— фторида 302
отделение 78
Фосфиды, определение 78
Фосфор 73 сл., 105
коэффициент поглощения соединений 93, 106
определение в виде ГПК 73 сл.
-------- тройного комплекса 105
—------фосфорнованадиевомо-
либденового комплекса 83 сл., 87
Фосфор
определение в виде фосфорномолибденовой кислоты 80 сл., 88,92
— влияние мышьяка 108
— в отсутствие мешающих веществ 81, 84, 87, 89, 95
----- присутствии ванадия 107
--------вольфрама 107
-------вольфрама, ниобия и титана 108
-------- кремния 78
--------кремния и мышьяка 75,
103
— микроколичеств 109
— с бриллиантовым зеленым 101
---- кристаллическим фиолетовым 99, 109
----- оксалатом олова 96
---- оксихинолином и молибдатом 105
----основными красителями 98
----- пропиленкарбонатом 109
----- сафранином 100
----- хинином 109
— характеристика методов 73
экстракция 93, 104
Фосфорная кислота
определение мышьяка 163
— сульфатов 218
Фтор, см. также Фторид
определение 282 сл., 302
— в отсутствие мешающих веществ 297, 299, 301
— с комплексом ализаринфлуо-ринового синего 303
--------алюминона 303
-------- амаранта 303
------- аскорбиновой кислоты 303
--------ацетилацетон а 303
-------- гематоксилина 303
--------2,4-диоксиацетофенона
303
--------2-(1,8-диокси-3,6-ди-
сульфо-2-нафтилазо) -фенокси-уксусной кислоты 303
--------7-иод-8-оксихинолина-
5-сульфокислоты 303
Фтор определение с комплексом ксиленоловым оранжевым 303 -----------морина 303
— — — перекиси водорода 303
------- пирокатехинового фиолетового 303
— — — резацетофенона 303
-------0-резорцилальдоксима 303
—-------роданида 303
-------СПАДНС 303 -------- сульфосалициловой кислоты 303
--------г торона 303
—-------5-фенилсалициловой
кислоты 303
— ------ хинализаринсульфона-
та 303
-------- хлоранилатат 303
— ------ хромотроповой кислоты
303
--------хромфиолетового 303
-------шиффового основания 303
отделение 285 сл.
Фторид, см. также Фтор определение 282, 302 сл. — в виде кремнемолибденовой кислоты 303
— в отсутствие мешающих веществ 291
— с ализарин-комплексонатом церия 290 сл.
— — арсеназо I 302
-----комплексом 4-(о-арсонофе-нилазо) -N- (1 -нафтил) -этилендиамина 303
-------- метилтимолового синего 303
--------перекиси водорода и титана 300 сл.
-------- хромазурола S 303
--------цирконализарина 298,302
— ------ эриохромцианина R
295 сл.
-----4-(2-пиридилазо)-резорцином 302
Фторид
определение, характеристика методов 288 сл., отделение 282 сл.
Фтористоводородная кислота, определение мышьяка 164
Фторсодержащие вещества, определение бора 50
Хлор, см. также Хлорид определение 307 сл. — в отсутствие мешающих веществ 312, 314
— по реакции разрушения метилового оранжевого 313
— различных форм 316 сл.
— с га-аминодиэтиланилином 317 ----- барбитуровой кислотой 317 — '— З.З'-диметилнафтидином 317
-----диметил-га-фенилендиами-ном 317
-----индикаторными бумагами 317
— — тетра-(га-диметиламино-фенил)-этиленом 317
-----о-толидином 311, 317
----- о-толуидином 317
-----фенолом и анилином 317
----- хлорцианом 317 — характеристика методов 307 сл.
Хлора двуокись определение с кислотным хром-фиолетовым К 318
----- тирозином 318
-----1,10-фенантролинатом же-леза(П) 318
Хлорат
определение с комплексом а-фу-рилдиоксима 318
-----трифенилметановыми красителями 318
----- фенилантраниловой кислотой 318
Хлорид, см. также Хлор окисление до хлора 307 определение в виде полиметино-вого красителя 317
Хлорид
определение в отсутствие мешающих веществ 308, 310
— по реакции вытеснения рода-нид-ионов 316
— с дибромфлуоресцеином 317
— — дитизоном 316
— — комплексами азопроизводных 316
------- йодата 316
— ----- метилтимолового синего
316
-------ртути (II) и дифенил-карбазона 307 сл.
---метиловым оранжевым 314
---о-толидипом 328
--- хлоранилатом ртути 316
— — хроматом серебра 310
— турбидиметрическое 311
ртути, определение 317
Хлорсиланы, определение бора 68
Хром и его соединения
определение кремния 128
— сульфата 218
— фосфора 107
Цезий, определение 344
Цианид, определение 216
Цинк и его соединения определение кремния 128 — мышьяка 157, 167 — серы 215, 217 — теллура 269
Циркалой, определение бора 68
Цирконий
определение бора 68
— кислорода 183
— кремния 122, 128
— серы 215
Чугун
определение азота 15
— кремния 116, 118 — фосфора 87, 88, 90, 109
Шлаки
определение кремния 78, 126
— фторида 302
Шпат плавиковый определение кремния 128 — фосфора 108
Щелочь, определение мышьяка 161
Ядерное горючее, определение хлорида 317
Анатолий Кириллович Бабко, Анатолий Терентьевич Пилипенко
фотометрический анализ
методы определения неметаллов