/








































































Автор: Ластовский Р.П. Поспелов А.М.
Теги: органическая химия химия химическая промышленность химические реакции
Год: 1965




Текст
М И Н И С Т Е Р с т в о
ХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ
МЕТОДЫ ПОЛУЧЕНИЯ
ХИМИЧЕСКИХ РЕАКТИВОВ
И ПРЕПАРАТОВ
Выпуск 13
ВСЕСОЮЗНЫЙ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ ВЕШЕСТВ
МОСКВА —-1065
Редакционная коллегия
Проф. Р. П. Ластовский (гл. редактор),
инженер-технолог А. М. Поспелов (зам. гл. редактора),
канд. хим. наук Е. А. Божевольнов,
проф. А. В. Бромберг, канд. техн, наук В. Г. Брудзь,
доктор хим. наук В. М. Дзиомко.
канд. хим. наук Г. А. Певцов
Редактор Б. Г. Козлов
Техн, редактор А. И. Пирожкова
Корректор Л. Я- Кузнецова
Сдано в набор 5.5.65 г. Подписано к печати 17. 11.65 г.
Формат-б5Тмаги 6Ох9О]/10 Печ. л. 9 • -'Уч нэл. 6.6
Л120941 Заказ 684 Тираж 1000 экз. Цена 46 кон.
Типография ВАХЗ
СОДЕРЖ А НИЕ
Алкильные эфиры салициловой кислоты. Р. М. Гельштейн,
Н. Ф. Кривошеева.........•........................ . 5
8-(4-Амииофенил) этиловый спирт. Л. П. Финн, Э. Б. Грекова,
М. Я- Романкевич........................................ 7
N-Ацетилдифеииламин. Р. М. Гелыитейн, Г. А. Креймер ... 10
М-Ацетил-Е-фенилалаиил-Е-тирозил-Э-глюкозамип. Г. Н. Коше-
лева, Г. Н. Налецкая, Л. М. Гинодман............. . . 12
₽(5-Ацетилфурил)-2-акриловая кислота. Л. В. Дуленко, Г. Н.
Дорофе.енко.......................................... 17
1-Беизоил-2-(я-диметиламинофенил)-1,2-дигидрохниолин. А. И.
Прилепская, А. К. Шейнкман, Н. Ф. Казаринова........... 19
7-Бутил-6,8-тридекандион. В, М. Дзиомко, О. В. Иванов ... 21
Бутирилхолинйодид. И. Д. Сапожкова, Н. II. Смирнова ... 23
4,4'-Диацетоксидибутиловый эфир. Б. А. Розенберг... 27
4-(я-Димстиламиностирил)пиридин. А. К. Шейнкман, Л. Г. Пи-
отрковская ............................................ 29
2,6-Диметил-З-гексилииридин. В. И. Дуленко, Г. И. Дорофеенко 32
5,7-Диметил-8-оксихипальдиновый альдегид. И. А. Красавин.
В. М. Дзиомко, Т. И. Егорова........................... 34
3,4-Диметоксиацетофенои. В. И. Дуленко, Г. Н. Дорофеенко 38
Динитрил малоновой кислоты. С. С. Райле-Десятник, Н. И.
Розенфельд ......... ................. 40
2,8-Диоксихинолнн. В. М. Дзиомко. И. А. Красавин, Ю. П.
Радин ........................... 44
Ди-н-пропиловый эфир 2-пиридилметилфосфииовой кислоты.
В. И. Карбан, Г. Ф. Дрегваль ............... 47
4,4'-Дихлордифенилоксид. 3. В. Воронкова, Г. М. Бессуднова,
И. В. Хвостов .......................... 49
Изобутиловый эфир уксусной кислоты Р. М. Гелыитейн,
Е. И. Маловер......................................... .51
2-Ме’ил-З, 4. 5, 6-(бис-триметилен)пиридин. В. И. Дуленко,
Н. В. Коваленко, Г. Н. Дорофеенко...................... 53
2-Метил-5-и-бутилпиридин. Ю. И. Чум /ков, В. П. Шерстюк . 55
З-Метил-6-стирилпиридин. А. К. Шейнкман, А. И. Розенберг 58
4-(₽-Мстилстирил) пиридины. А. К. Шейнкман, А. Н. Розенберг 61
я-Метоксиацетофенон. В. И. Дуленко, Г. И. Дорофеенко ... 66
Метосульфат 8-окси-1-метоксихинолиния. И. А. Красавин,
В. М. Дзиомко, Ю. 11. Радин............................ 68
Моноэтиланилин. И. Е. Задорожная.................. 70
^-Нафтиловый эфир № -бензоил-D, L-фепилалаиина. Э. А. Баш-
кир, Я. И. Лапук .............. ....................... 73
я-Нитроанилид № -бензоил-D, L-аргинина. Э. А. Башкир, Я. И.
Лапук, В. М. Степанов ..................... 76
3
5-Нитрофуран-2-карбоноаая кислота. Б. В. Курган, А. А.Гру-
зе, С. А. Гиллер ...........................................
Р-(5-Нитрофурил)-2-аллиловый спирт. М. Я Берклава, С. А.
Гиллер .....................................................
5-Нитрофурфуриловый спирт. М. Я- Берклава, С. А. Гиллер .
Нитхромазо В. И Кузнецов, Н. Н. Басаргин.................
2-Оксипропиламии и ди-(2-оксипропил) амин. А. М. Самуилов,
Г. Ф. Дрегваль .............................................
8-Оксихинальдонитрил и 8-оксихинальдамид. И. А. Красавин,
В. М. Дзиомко, Ю. Л. Радин..................................
Пикрамин Р. Ю. М, Дедков, С. Б. Саввин ..........
2-Стирилпиридины. А. К. Шейнкман, А. И. Розенберг, Л, С.
Емельянова..................................................
Сульфохлорфенол С. С. Б. Саввин, Ю. Л1. Дедков...........
Тетраизопропиллифенил. Е. П. Бабин, А. А. Кузьменков . . .
2, 5, 7-Трнметил-8-ацетокснхинолин. В. М. Дзиомко, И. А.
Красавин, Т. Н. Егорова.....................................
2, 2, 4-Триметил-1, 3-диоксолаи. Г. Ф. Дрегваль, Н. А. Рыбак
2, 5. 7-Триметил-8-оксихинолин. И. А. Красавин, В. М. Дзиом-
ко, Т. Н. Егорова...........................................
2, 5, 7-Триметил-8-оксихинолин-1-оксид. В. М. Дзиомко, И. А.
Красавин, Т. Н. Егорова............................. ......
5-Фенил-акридиновый оранжевый хлоргидрат. <). Г. Лушина,
А. А. Прянишников, М. И. Герман .............
Фузариновая (5-н-бутнл-2-пиридинкарбоновая) кислота. Ю. И.
Чумаков, В. П. Шерсшюк......................................
2-Хлорметилпиридин. В. И. Карбан, Г. Ф. Дрегваль.........
Циклогексилсульфаминовая кислота. Р. Г. Вдовина, А. В. Кар-
пова, И. А. Редькин........................................
Я-Этилпиридин. Ю. И. Чумаков, В. П. Шерстюк..............
Этилфенилкетои. Е. П. Крысин. С. Е. Чеснокова..........
80
82
84
87
92
94
99
102
105
1С9
115
119
121
125
128
130
134
140
УДК 547.587.11
АЛКИЛЬНЫЕ ЭФИРЫ САЛИЦИЛОВОЙ КИСЛОТЫ
Общий метод их получения
Р. М. ГЕЛЬШТЕЙН, И. Ф. КРИВОШЕЕВА
HOC6H4COOR
Общеизвестный типовой метод получения алкильных эфи-
ров салициловой кислоты заключается во взаимодействии са-
лициловой кислоты с соответствующими спиртами в присут-
ствии серной кислоты в качестве водоотнимающего агента [1].
Однако для получения высших алкилсалицилатов этот спо-
соб малопригоден, поскольку высшие спирты под действием
серной кислоты легко окисляются и дегидратируются.
В связи с тем, что синтез по классическому методу прово-
дится без водоотделения, длительность этерификации состав-
ляет 10—12 часов.
Дыханов и Шилов осуществили синтез нормального и изо-
амилового эфира салициловой кислоты нагреванием ее с ами-
ловыми спиртами в присутствии каталитических количеств
арилсульфохлоридов [2].
Этот способ распространен нами и на получение гексило-
вого и гептилового эфиров салициловой кислоты, в процессе
которого оказалось возможным уменьшить количество загру-
жаемого спирта (в расчете на 1 Л4 салициловой кислоты) и
синтез проводить с водоотделителем.
СХЕМА СИНТЕЗА ЭФИРОВ САЛИЦИЛОВОЙ КИСЛОТЫ
HOC6H4COOH+ROH HOCeH4COOR4-H2O
Характеристика основного сырья
Салициловая кислота, ч., ГОСТ 5844—51.
n-Толуолсульфохлорид, ч., МРТУ 6-09-447—63.
5
Гексиловый спирт, ч., СТУ 12-10-154—61.
Гептиловый спирт, ч., СТУ 12-10-157—61.
Амиловый спирт, ч., СТУ 12-10-158—61.
Условия получения
В круглодонную колбу емкостью 500 мл, снабженную ме-
шалкой и обратным холодильником с водоотделителем, поме-
щают 138 г (1 М) салициловой кислоты, 1,48 М соответству-
ющего спирта и 13,3 г (0,07 М) п-толуолсульфохлорида.
При энергичном перемешивании нагревают колбу на гли-
цериновой бане до 160° и при этой температуре выдержива-
ют смесь, продолжая перемешивание, в течение 8 часов; прн
этом водоотделение заканчивается в первые 4 часа.
Затем реакционную массу охлаждают до 40° и обрабаты-
вают (при энергичном перемешивании) 100—200 мл 10%-ного
раствора соды. После добавления всего раствора соды пере-
мешивание продолжают еще в течение 30 минут, после чего
водный слой отделяют от эфира и последний промывают во-
дой до нейтральной реакции промывных вод на универсаль-
ную бумажку.
Очистку технического продукта проводят перегонкой под
вакуумом при остаточном давлении 8—10 мм.
Выход чистых эфиров составляет 55—60% от теоретичес-
кого. Весовые количества соответствующих спиртов, темпера-
туры кипения и выход полученных алкильных эфиров сали-
циловой кислоты приведены в таблице.
Наименование эфиров салициловой кислоты Количество со- ответствующего спирта, г Выход чистого продукта, % Температура ки- пения при оста- точном давлении 8—10 JAM Удельный вес Показатель пре- ломления
Амиловый 130,4 57,5 155-163° 1,054 __
Гексиловый 151,2 55,0 155-160° 1,035 —
Гептиловый 171,9 60,8 180-190° 1,018 I,5006
ЛИТЕРАТУРА
1. Препаративная органическая химия. Перевод с польского
В. В. Шпанова и В. С. Володиной, М„ Госхимиздат, 1959. стр. 366.
2. Н. Н. Дыха нов, В. Р. Шилов, Е. И. Зорина. Авт. свид.
162122; Бюлл. изобр., № 9 (1964).
Поступила в сентябре 1964 г.
ЦЗЛ
6
УДК 547.262.07
р- (4-АМИНОФЕНИЛ)ЭТИЛОВЫЙ СПИРТ
Л. П. ФИНН, Э. Б. ГРЕКОВА, М. Я. РОМАНКЕВИЧ
СН,СН2О11
//\
I II
ч/
I
NH,
C8HnNO
М. в. 137,18
р-(4-Аминофенил) этиловый спирт является промежуточ-
ным продуктом в синтезе 4-аминостирола.
По литературным данным, 0-(4-аминофенил) этиловый
спирт получают путем восстановления [3-(4-нитрофепил)эти-
лового спирта цинковой пылью в водном растворе хлористо-
го кальция [1], каталитическим гидрированием в присутствии
черной платины Адамса [2], металлическим или двухлори-
стым оловом в соляной кислоте [3].
В отличие от указанных методов нами предложен синтез
Р-(4-аминофенил)этилового спирта из нитроэфира р-(4-нит-
рофенил)этилового спирта с последующим восстановлением
его гидратом гидразина в присутствии никеля Ренея. Восста-
новление проводится аналогично описанным в литературе ме-
тодам получения ароматических аминов из нитросоединений
[4, 5]. Преимуществами этого метода являются сокращение
количества стадий синтеза (см. примечание 1), значительное
увеличение выхода (до 90%) и устранение дополнительной
очистки продукта.
СХЕМА СИНТЕЗА ^-(4-АМИНОФЕНИЛ)ЭТИЛОВОГО СПИРТА [6]
СН.2СН2ОН CH2CH2ONO2 CHaCH2OH
|^Ч|| HNO3 н- \,/ Ч/ Ni’' j A ч/
NO., nh2
Характеристика исходного сырья
Р-Феиилэтиловый спирт, ч., СТУ 79-564-Х—60.
Азотная кислота, ч., уд. в. 1,5, ГОСТ 701—58,
Спирт метиловый, ч., ГОСТ 6995—54.
Гидразин-гидрат, ч., ГОСТ 5832—51.
Никель Ренея (см. примечание 2).
Условия получения
Синтез нитроэфира р-(4-нитрофенил)этилового спирта. В
трехгорлый реактор, снабженный механической мешалкой,
капельной воронкой и термометром, помещают 330 мл охла-
жденной до минус 30° азотной кислоты и медленно при хоро-
шем перемешивании прикапывают 50 мл [3-фенилэтилового
спирта. Прибавление р-феиилэтилового спирта ведут так, что-
бы температура реакционной смеси не поднималась выше ми-
нус 5°, По окончании прикапывания содержимое реактора
еще перемешивают один час при температуре ниже 0°. Затем
выливают в десятикратное количество воды со льдом и обра-
зовавшиеся кристаллы нитроэфира |3-(4-нитрофеиил) этилово-
го спирта отфильтровывают и промывают четыреххлористым
углеродом. Масло (2- и 3-изомеры) отделяют от воды на де-
лительной воронке, водный слой обрабатывают эфиром, сое-
диненные эфирные вытяжки и масло сушат прокаленным
сульфатом натрия.
Выход нитроэфира р-(4-нитрофенил) этилового спирта ра-
вен 34,5 г, что составляет 39% от теоретического, а его изо-
меров— 53,2 г, что составляет 60% от теоретического.
Температура плавления нитроэфира |3-(4-нитрофенил) эти-
лового спирта после двукратной перекристаллизации из ме-
танола 56—57°.
Найдено, %: N- 13,58; 13,61.
CeHeN2O6. Вычислено, %: N-13,21.
Получение $-(4-аминофенил)этилового спирта. В трехгор-
лый реактор, снабженный шариковым обратным холодильни-
ком со счетчиком пузырьков отходящего газа, штуцером для
подвода азота и воронкой для прибавления катализатора, по-
8
мещают раствор 165 г нитроэфира [}-(4-нитрофенил)этилово-
го спирта в 1500 мл метанола (см. примечание 3) и 370 мл
гидрата гидразина. Систему герметизируют и в течение 10—
15 минут пропускают азот для полного удаления воздуха из
системы. Затем вносят катализатор—никель Ренея до начала
реакции, раствор вспенивается, и начинается бурное выделе-
ние газов (по счетчику пузырьков, см. примечание 4). По ме-
ре замедления реакции добавляют новые порции катализато-
ра. К концу реакции (прекращение вспенивания при добав-
лении повой порции катализатора) раствор нагревают на
водяной бане до кипения для полного удаления растворен-
ных в нем газов. Охлажденный раствор быстро отфильтровы-
вают от никеля. В токе азота из маточника отгоняют мета-
нол, а затем в вакууме водоструйного насоса — остатки ме-
танола и реакционную воду. Образовавшийся светло-кофей-
ного цвета кристаллический продукт сушат в вакууме водо-
струйного насоса, нагревая па водяной бане, при температу-
ре 80—90° в течение 3 часов, а затем в вакууме при остаточ-
ном давлении 4—5 мм в течение часа.
Выход р-(4-аминофенил)этилового спирта равен98—105г,
что составляет 92—98% от теоретического. Полученный про-
дукт после двукратной перекристаллизации из этилового
спирта имеет температуру плавления 107—108°. По литера-
турным данным, т. пл. продукта 108° [1}.
Примечания:
1. Во всех вышеописанных методах перед нитрованием необходимо
защищать спиртовую группу ацетильной и омылять ее после нитрования.
2. Катализатор никель Ренея готовят по методике, описанной в лите
ратурс [7], и хранят под слоем метанола.
3. Раствор иитроэфпра р- (4-нитрофенил) этилового спирта в метаноле
готовят нагреванием на водяной бане.
4. Так как реакция идет с выделением азота и водорода, подачу азо-
та извне следует прекратить до конца реакции, как только газы переста-
нут выделяться.
ЛИТЕРАТУРА
]. Б. Ferber. Вег., 62. 189 (1929).
2. М. Martvnolf. Bull. Soc chim. France, 22, 374 (1935).
3. H. M. Woodburn, C. F. Stutz J. Amer. Chem. Soc., 72, 1362
(1950).
4. D. Balcom A. F u r s t. J. Amer. Chem. Soc., 75, 4334 (1953).
5. Л. M. Литвиненко, А. П. Греков. Ж. общ. химии, 26, 2528
(1956).
6. П. М. Кочергин, Л. С. Блинова. Авт. свид. 128011, РЖхим,
1961, 14Л165.
7. А. А. Р a v 1 i с, Н. A d k i ti s. J. Amer. Chem. Soc., 68, 1471 (1946).
Поступила в июле 1964 г.
Институт высокомолекулярных
соединений АН УССР
УДК 547.551.2.07
N- АЦЕТИЛДИФЕНИЛАМИН
Дифенилацетамид
/>. Л-1. ГЕЛЬШТЕЙН, г. а. КРЕЙМЕР
сн3—с=о
CUH13NO М. в. 211,26
Дифениламин является одним из наиболее трудноацети-
лируемых ароматических аминов. Ацетилирование его уксус-
ным ангидридом требует длительного кипячения реакционной
массы [1]. Однако, как показал Берлин [1], в присутствии ка-
талитических количеств хлорной кислоты дифениламин аце-
тилируется в течение нескольких минут при 80—90°. Недос-
татком этой методики является большой расход уксусного
ангидрида и неудобный для производства метод очистки про-
дукта перекристаллизацией из воды (большие объемы рас-
творов). Мы значительно уменьшили загрузку уксусного ан-
гидрида и предложили метод очистки продукта перекристал-
лизацией из изопропилового спирта.
СХЕМА СИНТЕЗА АЦЕТИЛДИФЕНИЛАМИНА
^_^-NH-^_^+(CH3CO)2O —
— ^_^-N-^_^4-CH3COOH
сн3-с=о
10
Характеристика основного сырья
Дифениламин, техн., ГОСТ 194—41.
Уксусный ангидрид, техн., ГОСТ 787—55.
Хлорная кислота, 30%-ная, ТУ МХП ОРУ 87—57.
Изопропиловый спирт, ГОСТ 9805—61.
Условия получения
В трехгорлую круглодонную колбу емкостью 250 мл, снаб-
женную мешалкой, термометром, обратным холодильником и
установленную на водяной бане, загружают 75 г (0,44 М) ди-
фениламина, 58 г (0,57 М) уксусного ангидрида (концентра-
ция не ниже 92%) и 1,2 г 30 %-ной хлорной кислоты. Смесь
нагревают до 80—90° и после 10-минутной выдержки при этой
температуре выливают на лед. Выпавшие кристаллы ацетил-
дифениламина отжимают и промывают водой до нейтральной
реакции промывных вод. Технический продукт перекристал-
лизовывают из 120 мл изопропилового спирта, промывают
50 мл спирта и сушат при 50—60°.
Выход ацетилдифениламина с температурой плавления
101 —102° равен 68 г, что составляет 72,6% от теоретического.
ЛИТЕРАТУРА
1. А. А. Берлин. Ж. общ. химии, 14, 438 (1944).
Поступила в октябре 19С4 г.
цзл
УДК 547.587.42.07
। /I
I он
N-АЦЕТИЛ -L-ФЕНИЛАЛАНИЛ -L-ТИРОЗИЛ -
D-ГЛЮКОЗАМИН
Г. Н. КОШЕЛЕВА, Г. И. НАЛЕЦКАЯ, Л. М. ГИНОДМ.АН
СН2ОН
/ °-
NH-COCHg Но~\^
СН,--СН—CONH—CH—CO—NH
' 1
СН2^-\_ / -ОН
C26H33N3O, М. в. 531,57
N-Ацетил-Е-фенилаланил-Е-тирозил-В-глюкозамии может
использоваться в качестве водорастворимого субстрата при
изучении кинетики действия протеолитического фермента пеп-
сина.
N-Ацетил-Е-фенилаланил-Е-тирозил-В-глюкозамин (1)
был впервые синтезирован из N-ацетил-Е-фенилаланил-Е-ти-
розина (II) карбодиимидпым методом аналогично синтезу
пептидных производных В-глюкозамина, осуществленному
ранее [1].
Исходный пептид N-ацетил-Е-фенилаланил-Е-тирозин был
получен взаимодействием азлактона а-ацетаминокоричной ки-
слоты (III) и тирозина (IV) [2, 3] с последующим восстанов-
лением а-ацетаминоциннамоил-Е-тирозина (V) палладиевой
чернью и делением полученных изомеров [4].
В методику синтеза пептида нами были внесены измене-
ния: мы вводили в реакцию на ’А весовую часть азлактона
меньше, т. е. брали больший избыток тирозина, чем по лите-
ратурным данным [4]. Только в этом случае удалось получить
чистый а-ацетаминоциннамоил-Е-тирозин, который затем хо-
рошо гидрировался и делился на L- и В-изомеры. Кроме то-
го, нами были изменены условия очистки а-ацетаминоцинна-
моил-Е-тирозина.
12
СХЕМА СИНТЕЗА N- АЦЕТИЛ-L ФЕНИЛАЛАНИЛ -L- ТИРОЗИЛ-
D- ГЛЮКОЗАМИНА
У-СН=С-СО +НО-^ ^-CHj-CH-COOH
N О
СН;
^_)>-CH=C-CONH- сн-соон
NHCOCHs СН2-^“)-он
V
СН=С—CONH— СН-СООН+Н, Pd
I I z-x.
NHCOCH3 СН2-^ j>-OH
V —
СН2—CH —CONH —CH—СООН
II
^~^-ch2-ch-conh-ch-cooh
NHCOCHs CH2-(-)-OH
II “
CH2OH
HO—ink I C6H„N=C=N-CgHlt
ф YKoh
HCl-HsN
CH2OH
V- CHj—CH—CO—NH—CH—CONH
• / I I
NHCOCHs CHS-^ ^-OH
Характеристика основного сырья
Азлактон ct-ацетаминокоричной кислоты [2, 3], т. пл. 146-
148°.
Палладий хлористый, ч., ТУ МХП 2323—50.
L-Тирозин, ч., ВТУ РУ 723—52.
Глюкозамин хлоргидрат [5], [а]о20 =74° (С = 2, вода).
Дициклогексилкарбодиимид [6], т. кип. 148711 мм;
т. пл. 35°.
Условия получения
Получение а-ацетаминоциннамоил-Ь-тирозина. Растворя-
ют 10 г (0,054 Л4) L-тирозина в смеси 55 мл 1н. раствора ед-
кого натра, 100 мл воды и 200 мл ацетона; перемешивают,
дают постоять 10—15 минут, прибавляют 8,6 г (0,046 М) аз-
лактона ацетаминокоричной кислоты и взбалтывают 2 часа.
В течение этого времени азлактон растворяется, но в осадке
остается около 3 г тирозина. Раствор отфильтровывают и к
фильтрату прибавляют 56 мл 1н. раствора соляной кислоты.
Затем большую часть ацетона и часть воды отгоняют в
вакууме (температура бани 40°) и остаток, около 'Л
первоначального объема, ставят на ночь в холодильник. Вы-
павший а-ацетаминоциннамоил-Ь-тирозин отфильтровывают в
вакууме, сушат на воздухе и затем перекристаллизовывают,
растворяя его в 120 мл метанола (температура бани 40—45°)
и добавляя к раствору равный объем воды. Смесь выдержи-
вают в холодильнике в течение ночи. Выпавшие желтые кри-
сталлы отфильтровывают, сушат на воздухе, а затем в ваку-
ум-эксикаторе над фосфорным ангидридом.
Выход а-ацетаминоциннамоил-Ь-тирозина равен 8,4 г, что
составляет 50% от теоретического; т. пл. 217°; [а]2д =48°',
(С = 2,5; абс. пиридин).
По литературным данным, т. пл. продукта 217—218°;
[а]2о =471о [4].
Приготовление палладиевой черни. В колбе емкостью 0,5 л
растворяют 1,5 г хлористого палладия в 13 мл 1 н. раствора
соляной кислоты. К смеси приливают 0,3 мл муравьиной кис
лоты, 75 мл кипящей воды и 2н. раствор едкого натра до ще-
лочной реакции по фенолфталеину. Затем к горячей реакци-
онной массе по каплям добавляют 1—1,2 мл муравьиной кис-
лоты. Последние капли кислоты вызывают бурное выделение
газа.
Объемистый черный осадок палладия промывают водой
путем декантации и хранят под слоем воды.
Получение П-ацетил-Ь-фенилаланил-Ь-тирозина. В сосуд
для гидрирования помещают 20 г ацетаминоциннамоил-Ь-ти-
розина, 50 мл ледяной уксусной кислоты, 50 мл воды, 100 мл
спирта и 1 г палладиевой черни. Вытесняют из системы воз-
14
дух, пропуская 6-кратный объем водорода, и затем гидриру-
ют при непрерывном встряхивании до прекращения поглоще-
ния водорода. Гидрирование заканчивается приблизительно
за 13 часов. Отфильтровывают от палладиевой черни и D-изо-
мера (осадок 1). Фильтрат упаривают в вакууме при темпе-
ратуре бани 30—35° до объема 15—20 мл и охлаждают. От-
фильтровывают через пористый фильтр № 2 основную часть
L-изомера (осадок 2) и сушат его па воздухе.
Маточник помещают на ночь в холодильник. Из него полу-
чают дополнительное количество L-изомера.
Общий выход М-ацетил-Е-фепилаланил-Е-тирозина со-
ставляет 9,7 г; [а]д =15° (С = 2; абс. пиридин).
По литературным данным, ]а|у =14,5° (С = 2; абс. пири-
дин) [7].
Для выделения D-изомера осадок 1 обрабатывают таким
количеством метанола, чтобы весь продукт растворился. Рас-
твор D-изомера отфильтровывают от палладиевой черни.
Фильтрат упаривают в вакууме досуха при температуре бани
40°.
Выход М-ацетил-О-фепилаланил-Ь-тирозина равен 9,5 г;
[а]д=25° (С = 2,5; абс. пиридин).
По литературным данным, [а]®6 =24,9° (С = 2,5; абс. пи-
ридин) [7].
Получение П-ацетил-Ь-фенилаланил-Ь-тирозил-П-глюкоз-
амина. Растворяют 2,16 г (0,01 М) хлоргидрата глюкозами-
на в 5 мл воды и при перемешивании и охлаждении льдом до 0°
прибавляют 5 мл 2и. раствора едкого кали. Продолжают пе-
ремешивание смеси в ледяной бане еще 15—20 минут (тем-
пература в колбе 0—5°). Затем прибавляют 3,7 г (0,01 М)
М-ацетил-Ь-фенилалапил-Ь-тирозина и 5 мл перегнанного пи-
ридина. Добиваются растворения пептида и к полученному
раствору прибавляют раствор 3,1 г (0,015 М) дициклогексил-
карбодиимнда в 20 мл пиридина.
Реакционную смесь перемешивают 1—2 часа и оставляют
на сутки при комнатной температуре. Затем прибавляют
100—120 мл волы и отфильтровывают дициклогексилмочеви-
ну. Фильтрат экстрагируют эфиром (дважды по 50 мл) и вод-
ный слой обрабатывают активированным углем, после чего
его упаривают на роторном испарителе при температуре бани
40°. Продукт выпадает сначала в виде геля, а затем превра-
щается в порошок. Его сушат на воздухе. Сухой продукт пе-
рекристаллизовывают, растворяя в теплом метаноле и доба-
вляя этилацетат (метанол:этилацетат= 1:3). Осадок хорошо
отжимают на фильтре от растворителя и промывают этилаце-
татом. Сушат в вакуум-эксикаторе над фосфорным ангидри-
дом.
15
Выход М-ацетил-Е-фенилаланил-Е-тирозил-О-глюкоз амина
равен 5,3 г, что составляет 99,6% от теоретического; т. пл. пре-
парата 161 —162° (разл.); [а]д=49,8° (С=1; метанол).
Найдено, %: С—57,85; 58,03; Н—6,66; 6,50; N-7,38; 7,35.
C2(iH33N3O9' Н2О. Вычислено, %: С—57,76 % ; Н—6,33%; N—7.77%.
При нанесении на хроматограмму 100 мкг вещества в си-
стеме н.бутанол—ледяная уксусная кислота—вода (4:5:1) и
проявлении нингидрином примеси не обнаружены.
При действии карбоксипептидазы на синтезированный пре-
парат примесь исходного пептида N-ацетил-Е-фенилаланил-Е-
тирозипа не обнаружена.
ЛИТЕРАТУРА
1. Н. К. Кочетков, В. А. Деревицкая, Н. В. Молодцов.
Ж- общ. химии. 32, 2500 (1962).
2. Синтезы органических препаратов, Сб. 2, М., ИЛ, 1949, стр. 69.
3. -Синтезы органических препаратов, Сб. 2, М., ИЛ, 1949, стр. 72.
4. М. Bergmann, F. Stern, Ch. Witt. Liebigs Ann. Chem., 449
277 (1926).
5. Г. В. Л а з у p ь e в с к и й, И. В. Терентьева, А. А. Шамшу-
рин. Практические работы по химии природных соединений, ДА., Высшая
школа, 1961, стр. 61.
6. О. A mi a rd, R. Hey mis. Bull. Soc. chlm. France, 1360 (1956).
7. L. Backer. J. Biol. Chem., 193, 813 (1951).
Поступила в сентябре 1964 г.
Институт природных
соединений АН СССР
УДК 547.391.1.07
8-(5-АЦЕТИЛФУРИЛ)-2-АКРИЛОВАЯ КИСЛОТА
Л. В. ДУЛЕИКО, Г. И. ДОРОФЕЕНКО
СН3СОО / х О / ''-СН—СН-СООН
С9Н8О4 М. в. 180,16
5-Аи.етнлфурил-2-акриловая кислота представляет интерес
как потенциальное физиологически активное вещество.
СХЕМА СИНТЕЗА 5-АЦЕТИЛФУРИЛ-2-АК.РИЛОВОЙ КИСЛОТЫ [1]
\ Л (СН-.СО)„О
0х 'СН —СН-СООН-НтЬоГ—
is ;
-> СН3СООХХО / 4 сн сн- соон
Характеристика основного сырья
Фурилакриловая кислота, ч., ТУ TCP 461—61.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Хлорная кислота, х. ч., 57%-ная, ТУ ОРУ 87—57.
Условия получения
К суспензии 6,9 г (0,05 Л4) фурилакриловой кислоты в
50 мл уксусного ангидрида медленно, по каплям, добавляют
смесь 0,28 г (0,002 М) 70 %-ной хлорной кислоты (см. приме-
чание) и 1 мл уксусного ангидрида, при этом все переходит в
раствор, который окрашивается в фиолетовый цвет. После
часовой выдержки раствор выливают в 100 мл воды п от-
2 Зак. 684 17
фильтровывают выделившуюся смолу. Фильтрат упаривают
досуха и получают светло-желтый твердый продукт. Отфиль-
трованную смолу обрабатывают кипящей водой, упаривают
водный раствор и выделяют еше немного желтого продукта.
Общий выход 5-ацетилфурил-2-акриловой кислоты равен
4,5 г, что составляет 50% от теоретического; т. пл. 188—189°.
Примечание. 70%-ная хлорная кислота получена отгонкой воды
от 57%-ной хлорной кислоты в вакууме.
ЛИТЕРАТУРА
1. Г. Н. Дорофеенко, В. И. К арбан, Л. В. Дуленко,
В. Н. Новиков. Изв. ВУЗ СССР, Химия и хим. технология, 7, 432
(1964).
Поступила в октябре 1964 г. Донецкий филиал ИРЕА,
Ростовский-на-Дону
госуниьерситет
УДК 547.831.3.07
1-БЕНЗОИЛ-2-(п-ДИМЕТИЛАМИНОФЕНИЛ)-1,2-
ДИГИДРОХИНОЛИН
А. Н. ПРИЛЕПСКАЯ, А. К- ШЕИНКМАН, Н. Ф. КАЗАРИНОВА
l/'U— -<_>N(CH3)2
СО
СвН6
C21H22N2O М. в. 354,45
В литературе имеется одно сообщение о синтезе 1 -бензо-
ил-2- (л-диметил аминофенил )-1,2-дигидрохинолина. Синтез
продукта проводился авторами в течение 3 дней при комнат-
ной температуре [1].
Предлагаемая нами методика получения 1-бензоил-2-(п-
диметиламинофенил)-1,2-дигидрохинолина является более
удобной. Из него можно получить 2-(п-диметиламинофенил) -
хинолин, который используется как промежуточный продукт
для синтеза различных физиологически активных препара-
тов и красителей.
СХЕМА СИНТЕЗА 1-БЕНЗОИЛ-2-(п-ДИМЕТИЛАМИНОФЕНИЛ)-1,2-
ДИГИДРОХИНОЛИНА
С6Н5СОС1
4/W
I || Cl
I
- occ5H5-
C6H5N(CH3h r
T9
/\/Ч Н -
I II I/__// _м (гн \
4/\NZ----\=Z n(ghb)«
co
С,н6
Характеристика основного сырья
Хинолин, ч., ТУ МХП 93—47.
Хлористый бензоил, ч., ТУ МХП 92—51.
М,Хт-Диметиланилин, ч„ ГОСТ 5855—51.
Условия получения
В трехгорлую круглодонную колбу емкостью 500 мл, снаб-
женную механической мешалкой с затвором и обратным хо-
лодильником с хлоркальциевой трубкой, помещают 180,6 г
(1,4 А1) хинолина, 98 г (0,7 М) хлористого бензоила, 84,6 г
(0,7 М) диметиланилина. Все реагенты загружают при тща-
тельном перемешивании. Реакционную смесь выдерживают в
течение 8 часов на кипящей водяной бане при постоянном
размешивании. После нагрева содержимое колбы превраща-
ется в темно-коричневую вязкую массу, которую затем под-
вергают перегонке с водяным паром для освобождения от не-
прореагировавшего хинолина, диметиланилина и образую-
щихся бензойного альдегида и бензойной кислоты.
Красно-коричневый осадок, оставшийся в колбе, отделяют
от темно-коричневого раствора, очищают его перекристалли-
зацией из пиридина, затем из этилового спирта.
Выход 1 -бензоил-2- (и-диметиламинофенил) -1,2-дигидрохи-
нолина равен 87,5 г, что составляет 35% от теоретического;
по внешнему виду это бесцветные иглы, т. пл. 180—181°.
ЛИТЕРАТУРА
1. W. Е. Me Ewen, R. Н. Terss, 1. W. Elliott. J. Amer.
Chem. Soc., 74, 3605(195'2).
Поступила в октябре 1964 г. Донецкий филиал ИРЕА
УДК 547.295.2:66.095.11
7-БУТИЛ-6.8-ТРИДЕКАНДИОН
В. М. ДЗИОМКО, О. В. ИВАНОВ
СН2СН,СН2СН3
СНз iCH2)4COCHCO(CH2)4CH8
C„H„O2 М. в. 268,44
В литературе описан синтез 7-бутил-6,8-тридекандиона
путем самоконденсации ангидрида капроновой кислоты в при-
сутствии борного ангидрида как катализатора [1, 2].
Нами разработан новый способ получения 7-бутил-6,8-
тридекандиона ацилированием капроновой кислоты хлоран-
гидридом в присутствии безводного хлористого алюминия как
катализатора.
СХЕМА СИНТЕЗА 7-БУТИЛ-6.8-ТРИДЕКАНДИОНА
СН3 (CHS)4 СООН -2СНз(£Н2)«СОС1,А1С13
СН2СНаСН2СН3
СН3 (СН2)4 СОСНСО (СН3)4СН3
Характеристика основного сырья
Алюминий хлористый, безводный, МРТУ 6-09-125—63.
Капроновая кислота, ч., ТУ ГКХ 1530—61.
Серная кислота, ч., ГОСТ 4204—48.
Условия получения
В трсхгорлую колбу, снабженную мешалкой, обратным
холодильником, защищенным хлоркальциевой трубкой, и ка-
21
пельной воронкой, помещают 32 г (0,24 А4) безводного хлори-
стого алюминия и к нему при перемешивании прикапывают
за 15 минут 23,2 г (0,2 М) сухой перегнанной капроновой ки-
слоты. Смесь выдерживают 15 минут на бане с температурой
60—70° и к реакционной массе прикапывают 53,8 г (0,4 М)
хлористого капроиила (см. примечание) в течение 40 минут.
Затем температуру бани поднимают за 1 час до 140—150° и 5
часов выдерживают реакционную смесь при этой температу-
ре.
Далее содержимое колбы охлаждают, прибавляют 150 мл
хлороформа и перемешивают до образования гомогенного
раствора.
К этому раствору прибавляют по каплям 400 мл 15 %-ной
серной кислоты, а затем 200 мл воды. Органический слой от-
деляют и высушивают прокаленным сульфатом натрия, хло-
роформ отгоняют, а остаток фракционируют в вакууме.
Выход сырого продукта, кипящего в пределах 146—
15373 мм равен 34,5 г, что составляет 64% от теоретического.
После вторичной перегонки при 139—14272 мм выход со-
ставляет 31,5 г (58,6%); По—1,4460.
По литературным данным, т. кип. продукта 175—
178710,5 мм; 130—13271 мм; Пр—1,4480 [1, 2].
Примечание. Хлористый капронил с выходом 85—87% получен
действием хлористого тионнла на капроновую кислоту в присутствии дп-
метилформамида в качестве катализатора; т, кип. 150—153’
ЛИТЕРАТУРА
I. Англ. пат. 893327; РЖхим, 63, I3H50.
2. Герм. пат. 1118840; С. А., 56, 14085.
Поступила в марте 1965 г.
УДК 547.539.4.07
БУТИРИЛХОЛИНЙОДИД
Н. Д. САПОЖКОВА, Н. П. СМИРНОВА
I(CH?)3NCH8CH8OCOCH8CH8CH8]J-
C9H30NO2J М. в. 301,16
Бутирилхолиийодид применяется в биохимии в качестве
субстрата при определении псевдохолииэстеразы [1].
Разработанный нами трехстадийиый метод синтеза бути-
рилхолинйодида дает более высокий выход продукта (80%
вместо 56%) и обеспечивает полную безопасность процесса,
тогда как в двухстадийном методе [2] могут образоваться
взрывоопасные соедииеиия фосфора с водородом.
СХЕМА СИНТЕЗА БУТИРИЛХОЛИНЙОДИДА
NaJ СН,СНХН2СОС1
СН2СН3С1ОН—> JCH2CH2OH ——:~
ch.ch2ch2cooch2ch2j — •
- [(CH3)3NCH2CH2OCOCH3CH2CH3]J-
Характеристика основного сырья
Этиленхлоргидрин, ч., МРТУ 609-451—63.
Натрий йодистый, ч., ГОСТ 8422—57.
Хлорангидрид масляной кислоты, ТУ 79П 621—61.
Бензол, ч.д.а., ГОСТ 5955—51.
Натрий двууглекислый, ч., ГОСТ 4201—48.
Натрий сериоватистокислый, ч., ГОСТ 4215—48.
Натрий сернокислый, ч., ГОСТ 4166—48.
Спирт метиловый, ГОСТ 6995—54.
Эфир диэтиловый, медицинский, ГОСТ 6265—52.
23
Триметиламин ТУ МХП 2624—51, 20%-ный спиртовой рас-
твор.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Условия получения
Синтез этиленйодгидрина. В колбу емкостью 1 л, снабжен-
ную обратным холодильником и капельной воронкой, вносят
342 г (2,28 уИ) йодистого натрия и 356 г (11,12 М) метилово-
го спирта. Смесь нагревают на водяной бане до закипания
спирта, при этом основная масса йодистого натрия растворя-
ется. После этого добавляют 147 г (1,73 Л4) этиленхлоргидри-
на порциями по 10—15 мл в течение 1 часа 30 минут. По до-
бавлении всего количества этиленхлоргидрина нагревание на
водяной бане продолжают еще в течение 4,5 часа. По окон-
чании нагревания метиловый спирт отгоняют. Остаток
экстрагируют абсолютным серным эфиром в количе-
стве 500 мл порциями по 100 мл. Эфир отгоняют на водяной
бане и остающееся темно-окрашенное масло разгоняют в ва-
кууме. Отбирают фракцию, кипящую при 65—67°/12 мм.
Выход этиленйодгидрина равен 200 г, что составляет 64%,
считая на этиленхлоргидрип; nD —1,о7.
Получение р-йодэтилового эфира масляной кислоты [3]. В
грехгорлую колбу емкостью 0,5 л, снабженную термометром,
обратным холодильником и капельной воронкой, вносят 90г
(0,56 М) хлорангидрида масляной кислоты и 90 мл бензола.
Смесь нагревают до 60—65° и по каплям добавляют 120 г
(0,46 /И) этиленйодгидрина в течение 2 часов. Затем темпера-
туру в колбе поднимают до 90° и выдерживают смесь при
этой температуре в течение 1 часа. После этого реакционной
массе дают охладиться до комнатной температуры, переносят
ее в делительную воронку и встряхивают с 50 мл 10%-ного
водного раствора гипосульфита натрия до обесцвечивания
бензольного слоя. Отделяют раствор гипосульфита и экстракт
промывают несколько раз 100 мл насыщенного раствора дву-
углекислой соды, а затем дистиллированной водой. Бензоль-
ный раствор р-йодэтилового эфира масляной кислоты остав-
ляют на ночь для высушивания над 10 г безводного сернокис-
лого натрия. Отгоняют растворитель и остаток' перегоняют в
вакууме, собирая фракцию с температурой кипения 80—
82°/6 мм.
Выход (3-йодэтилового эфира равен 124 г, что составляет
73%, считая на этиленйодгидрин.
Получение бутирилхолинйодида. К 212 мл 20%-ного спир-
тового раствора триметиламина (0,57 М, 100%-ного), нахо-
дящегося в однолитровой конической колбе из стекла «пи-
рекс», приливают 124 г (0,51 М) р-йодэтилового эфира мас-
ляной кислоты, перемешивают и добавляют 212 мл (2 М) су-
24
хого серного эфира, не содержащего перекисных соединений
(см. примечания 1 и 2). Колбу герметично закрывают и ос-
тавляют при комнатной температуре в темном месте на пять
суток. По истечении указанного срока выпавший в осадок бу-
тирилхолипйодид отфильтровывают на воронке Бюхнера и
промывают (на фильтре) смесью абсолютного этилового
спирта (1 часть) с безводным серным эфиром (3 части) три
раза порциями по 50 мл.
Выход бутирилхолинйодида равен 211 г, что составляет
80%, считая на р-йодэтиловый эфир масляной кислоты; т. пл.
продукта 89—90°.
Перекристаллизация бутирилхолинйодида. В конической
колбе емкостью 0,75—1,0 л, снабженной капельной воронкой
и обратным холодильником, защищенным прокаленным хло-
ристым кальцием, растворяют при легком нагревании (25—
30°) 100 г бутирилхолинйодида в 250 мл абсолютного этило-
вого спирта. Затем колбу помешают в заранее нагретую до
45° водяную баню. Через 10—15 минут из капельной воронки
добавляют эфир при легком встряхивании порциями, пример-
но 50 мл, следя за полным растворением выпавшего осадка.
Всего добавляют 275 мл эфира, предварительно проверенно-
го на отсутствие перекисных соединений. Колбу вынимают
из водяной бани, закрывают пробкой и оставляют раствор
кристаллизоваться при комнатной температуре (15—25°) на
2—3 часа, после чего колбу помещают в холодильник на ночь
при температуре от 0 до 5°. На следующий день осадок бути-
рилхолинйодида отфильтровывают па воронке Бюхнера, от-
жимают стеклянной пробкой и промывают четыре раза по
50 мл смесью эфира (3 части) и спирта (1 часть). Отжатый
осадок бутирилхолинйодида переносят на фильтровальную
бумагу и сушат на воздухе до исчезновения запаха эфира,
после чего переносят в фарфоровую чашку и сушат в эксика-
торе над хлористым кальцием. Высушенный препарат не дол-
жен содержать желтых включений. До использования его сле-
дует хранить в темных склянках с притертой пробкой.
Выход бутирилхолинйодида после перекристаллизации ра-
вен 85—90 г, что составляет 85—90%; т. пл. 89—91°.
Примечания:
1. Для удаления перекисных соединений равные объемы этилового
эфира и насыщенного водного раствора тиомочевины встряхивают в тече-
ние 5 минут в делительной воронке, отделяют эфир и промывают его три
раза равным объемом дистиллированной воды. Отмытый эфир оставляют
на ночь над прокаленным хлористым кальцием, перегоняют и проверяют
на полноту удаления перекисных соединений.
2. Методика контроля очистки этилового эфира от перекисных соеди-
нений заключается в следующем: 10 мл эфира встряхивают в конической
колбе с притертой пробкой с 1 мл 10%-ного свежеприготовленного рас-
твора йодистого калия и с 0,5 мл свежеприготовленного 1%-ного водного
раствора крахмала в течение 3 минут и наблюдают за окраской водного
слоя. Параллельно к 1 мл йодистого калия прибавляют 0,5 мл крахмала
25
и сравнивают полученные окраски визуально. Если в испытуемом раство-
ре окраска более интенсивна, этиловый эфир подвергают обработке насы-
щенным раствором тиомочевины.
ЛИТЕРАТУРА
1. К. В. A u g u s t i n s s о n. Acta physiol, scand.. 15, Suppl. 52, 1948.
2. H. E. Кожевникова и др. Методы получения химических реак-
тивов и препаратов, вып. 2, М..ИРЕА, 1961, стр. 15.
3. F. г a u г п е a u, J. Page. Bull. Soc. chim., 15, 547 (1914).
Поступила в январе 1965 г. ИРЕА
УДК 547.272’264.07
4,4’-ДИАЦЕТОКСИДИБУТИЛОВЫЙ ЭФИР
Б. А. РОЗЕНБЕРГ
О О
II II
СН3СО-(СН2)4-О-(СН3)4-ОССН3
С12Н23О5 м. в. 246,30
Получение 4,4’-диацетоксидибутилового эфира из тетра-
гидрофурана через 4,4’-дихлордибутиловый эфир [1] является
трудоемким двухстадийным процессом. Выход 4,4’-диацеток-
сидибутилового эфира при этом не превышает 50% (в расче-
те на тетрагидрофуран). Описанный в литературе прямой спо-
соб получения 4,4’-диацетоксидибутилового эфира путем вза-
имодействия тетрагидрофурана с уксусным ангидридом в при-
сутствии каталитических количеств хлорной кислоты [2] при-
водит к низким выходам этого продукта вследствие значи-
тельного расхода тетрагидрофурана в параллельно протекаю-
щей реакции полимеризации последнего.
Нами показано [3], что эта реакция практически может
быть подавлена и выход 4,4’-диацетоксидибутилового эфира
при этом составляет 65—70%. Указанный способ может быть
рекомендован как простой препаративный метод получения
4,4’-диацилоксидибутиловых эфиров, которые могут быть ис-
пользованы в качестве пластификаторов и исходных веществ
для синтеза различных бифункциональных производных.
СХЕМА СИНТЕЗА 4,4’-ДИАЦЕТОКСИДИБУТИЛОВОГО ЭФИРА
2 J + (СН3СО)аО --------------,
о о
II II
СН3СО-(СН2)4-О-(СН3)4-ОССН3 27
Характеристика основного сырья
Тетрагидрофуран, техн., т. кип. 64—66°.
Уксусный ангидрид, ч.д.а., т. кип. 140°, ГОСТ 5815—52.
Хлорная кислота, х. ч„ 57%-ный раствор, ТУ ОРУ 87—57.
Условия получения
В круглодонную колбу, снабженную обратным холодиль-
ником, загружают 81,3 мл (1 М) предварительно обезвожен-
ного и перегнанного над металлическим натрием тетрагидро-
фурана, 99,5 мл (1 Л4) уксусного ангидрида и 1 г 57 %-ной
хлорной кислоты. После двухчасового нагревания реакцион-
чой смеси отгоняют непрореагировавшие тетрагидрофуран и
уксусный ангидрид. Остаток охлаждают, выливают в воду и
экстрагируют 150 мл этилового эфира. Экстракт промывают
разбавленным раствором бикарбоната натрия, водой и высу-
шивают над прокаленным хлористым кальцием. После отгон-
ки растворителя остаток перегоняют в вакууме.
Выход 4,4’-диацетоксидибутилового эфира равен 49 г, что
составляет 68% от теоретического; т. кип. 168—170°/9 мм".
1,0139; 1,4361.
Найдено, %: С-58,20; 58,12; Н-9,04; 9,15.
С12Н22О6. Вычислено, %; С 58,54; Н—8,94.
ЛИТЕРАТУРА
1. К. Alexander, L. Schniepp. J. Amer. Chem. Soc., 70, 1839
(1948).
2. T. S h о m о, T. T s u j 1 n o, G. Hachihama. J. Chem. Soc. Japan,
61, 1347 (1958).
3. Б. А. Розенберг, 3. А. Фишилевич, E. П. Бабин. Авт.
свид. 165177 (1963); Билл, изобр., № 18 (1964).
Поступила в октябре 1964 г.
Донецкий филиал ИРЕА
УДК 647.821.07
4-(л-ДИМЕТИЛАМИНОСТИРИЛ)ПИРИДИН
А. К. ШЕИНКМАН, Л. Г. ПИОТРКОВСКАЯ
/ СН=СН-^_____________N(CH3)a
C16HleNa М. в. 224,30
4-(п-Диметиламиностирил) пиридин может быть использо-
ван для получения изоникотиновой кислоты как комплексооб-
разователь и для синтеза различных физиологически активных
препаратов и красителей.
В литературе описано три общих метода получения раз-
личных стирилпиридинрв, замещенных в бензольном ядре [1]:
а) конденсация метилйодидов пиколинов с ароматически-
ми альдегидами в спирте с последующим пиролизом образу-
ющихся метилйодидов стирилпиридинов в соответствующие
свободные основания;
б) конденсация пиколинов с альдегидами в кипящем ук-
сусном ангидриде;
в) нагревание пиколинов и альдегидов в запаянных ам-
пулах при высокой температуре в присутствии хлористого
цинка.
Для получения 4-(н-диметиламиностирил) пиридина пер-
вые два метода оказались непригодными [1], по третьему спо-
собу удается получить продукт с небольшим выходом (20—
25%).
Недавно был описан синтез этого соединения конденсаци-
ей N-окси у-пиколина и н-диметиламинобензальдегида [2], од-
нако для его проведения необходимо получать N'-окись у-пи-
колина.
Предлагаемый нами оригинальный метод [3] заключается
в том, что у-пиколин вначале переводят в N-ацильное произ-
водное обработкой хлористым бензоилом (пригодны и любые
Другие хлорангидриды карбоновых кислот) в инертном рас-
29
творителе и полученное производное без выделения конден-
сирует с диметиламинобензальдегидом. Это существенно уп-
рощает процесс, повышает выход продукта и сокращает время
проведения реакции.
СХЕМА СИНТЕЗА 4-(п- ДИМЕТИЛАМИНОСТИРИЛ)ПИРИДИНА
СН3
I
Пс1’
Ч-n z
ос-с8н6
ОНС—C8Ht-N(CH3)2
пиридин
СН = СН-^ ^-N(CH3)2
\\ С1“
сн=сн
N(CH3)2
Характеристика основного сырья
Пиридин, ГОСТ 1625—61 (см. примечание 1).
у-Пиколин, техн., тщательно высушенный, свежеперегнан-
ный, т. кип. 147° (см. примечание 1).
Хлористый бензоил, ч., свежеперегнанный, ТУ МХП92—51
(см. примечание 2).
п-Диметиламинобензальдегид, ч., ТУ МХП 2679—51.
Условия получения
К 5 мл (0,053 Л4) у-пиколииа при интенсивном, перемеши-
вании и охлаждении ледяной баней прикапывают 5,75 мл
(0,04 Л1) хлористого бензоила. К выпавшему осадку светло-
желтого цвета приливают раствор 9 г п-диметиламииобенз-
альдегида в 10 мл абсолютного свежеперегнанного пириди-
на, при этом появляется красное окрашивание и температура
реакционной смеси самопроизвольно поднимается до 45—50°.
После прекращения разогревания реакционную смесь выдер-
живают 5 часов на масляной бане при температуре 155°±2°,
затем разлагают концентрированной соляной кислотой, отго-
30
няют с водяным паром образующуюся прн гидролизе бен-
зойную кислоту и бензальдегид, подщелачивают и вновь отго-
няют с водяным паром. Осадок в перегонной колбе отделяют,
многократно отмывают горячей водой от неорганических со-
лей, кипятят с активированным углем в растворе соляной кис-
лоты, затем осаждают раствором аммиака, отделяют и тща-
тельно высушивают вначале на пористой пластинке, а затем в
вакууме при 100° до постоянного веса.
Выход сырого продукта равен 7,65 г, что составляет 68%
от теоретического; т. пл. 228—230°. После перекристаллизации
из спирта т. пл. 4-(n-диметиламиностирил) пиридина 238—
239°; Хмакс 370 ммк, lg Е—4,34 (в спирте), что соответствует
литературным данным [2]. При проведении реакции в диме-
тилформамиде значительно облегчается выделение 4-(н-диме-
тиламиностирил)пиридина, так как он сразу выпадает в кри-
сталлическом состоянии при охлаждении реакционной смесн
(см. примечание 3).
Примечания:
1. Реакция очень чувствительна к присутствию влаги. Поэтому пири-
дин и 4-метилпиридин следует тщательно высушивать перед употребле-
нием (кипячение с окисью кальция и хранение над едким кали, затем
фракционированная перегонка на эффективной колонке).
2. Вместо хлористого бензоила можно использовать хлорангидриды
любых карбоновых кислот.
3. Аналогично получают 4-(п-диметнламииостирнл) пиридин с исполь-
зованием других хлористых ацилов. Выходы продукта реакции при этом
составляют: с применением хлористого ацетила—40%; хлористого пропио-
нила 52,5%; хлористого бутирила—53,5%; хлористого нзовалернла—
66,8%; хлористого капроила — 67%; /i-нитробензоилхлорида—68%.
ЛИТЕРАТУРА
1. J. Williams, R. Adel, J. Carlson, Ci. Reynolds,
D. Borden, J. Ford. J. Organ. Chem., 28, 3»7 (1963).
2. L. P e n 11 in a 111. Tetrahedron, 14, № 3-4, 151 (1961).
3. А. К. Ш e й н к м а и. Авт. свнд. 158576; Бюлл. изобр., К» 22 (1963).
Поступила в сентябре 1964г.
Донецкий филиал ИРЕА
УДК 547.821.412.6.07
2,6-ДИМЕТИЛ-З-ГЕКСИЛ ПИРИДИН
В. И. ДУЛЕНКО, Г. Н. ДОРОФЕЕНКО
/ ЧрСН2СН2СН2СНаСН3СН3
CH3/'4n/4'CH3
C13H2IN
М. в. 191,31
2,6-Диметил-З-гексилпиридин представляет интерес как
потенциальное физиологически активное вещество.
СХЕМА СИНТЕЗА 2,6-ДИМЕТИЛ-З-ГЕКСИЛПИРИДИНА
СвН13СН3СН=СН3
(СН;,СО)2О,НС1О4
Характеристика основного сырья
Нопеп-1, ч., свежеперегнанный.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Хлорная кислота, 57%-ная, ТУ ОРУ 87—57.
Условия получения
Смешивают 12,6 г (0,1 М) нонена-1 с заранее приготов-
ленным раствором 14 г (0,1 М) 70%-ной хлорной кислоты в
55 мл уксусного ангидрида (см. примечание). Реакционную
смесь нагревают на кипящей водяной бане в течение часа, по-
32
еле чего выливают в 50 мл холодной воды. При охлаждении
постепенно приливают избыток концентрированного водного
раствора аммиака и через 10—15 минут реакционную смесь
экстрагируют бензолом, а из бензольной вытяжки основание
извлекают 10%-ным раствором соляной кислоты. При ней-
трализации солянокислого раствора избытком едкого натра
выделяется 2,6-ди.метил-З-гексилпиридин, который экстраги-
руют эфиром, высушивают и перегоняют.
Выход продукта равен 8 г, что составляет 42% от теоре-
тического; т. кип. 247—253°; cL?°—1,5010; nf—0,9177.
По аналогичной методике получают при ацетилировании
[И:
а) гексена-1—2,6-диметил-З-пропилпиридин с т. кип. 203—
205°; выход равен 28%;
б) гептена-1—2,6-диметил-З-бутилпиридин с т. кип. 221 —
223°; выход равен 30%;
в) децена-1—2,6-диметил-З-гептилпиридин с т. кип. 269—
271°; выход равен 23%.
Примечание. 70%-ная хлорная кислота получена отгонкой воды
от 57%-ной хлорной кислоты в вакууме.
ЛИТЕРАТУРА
I. В. И. Дуленко, ['. Н. Дорофеенко. Докл. АН УССР, 1963,
78.
Поступила в октябре 1964 г. Донецкий филиал ИРЕА.
Ростовский-на-Дону
госуниверсигет
УДК 547.831.8.07
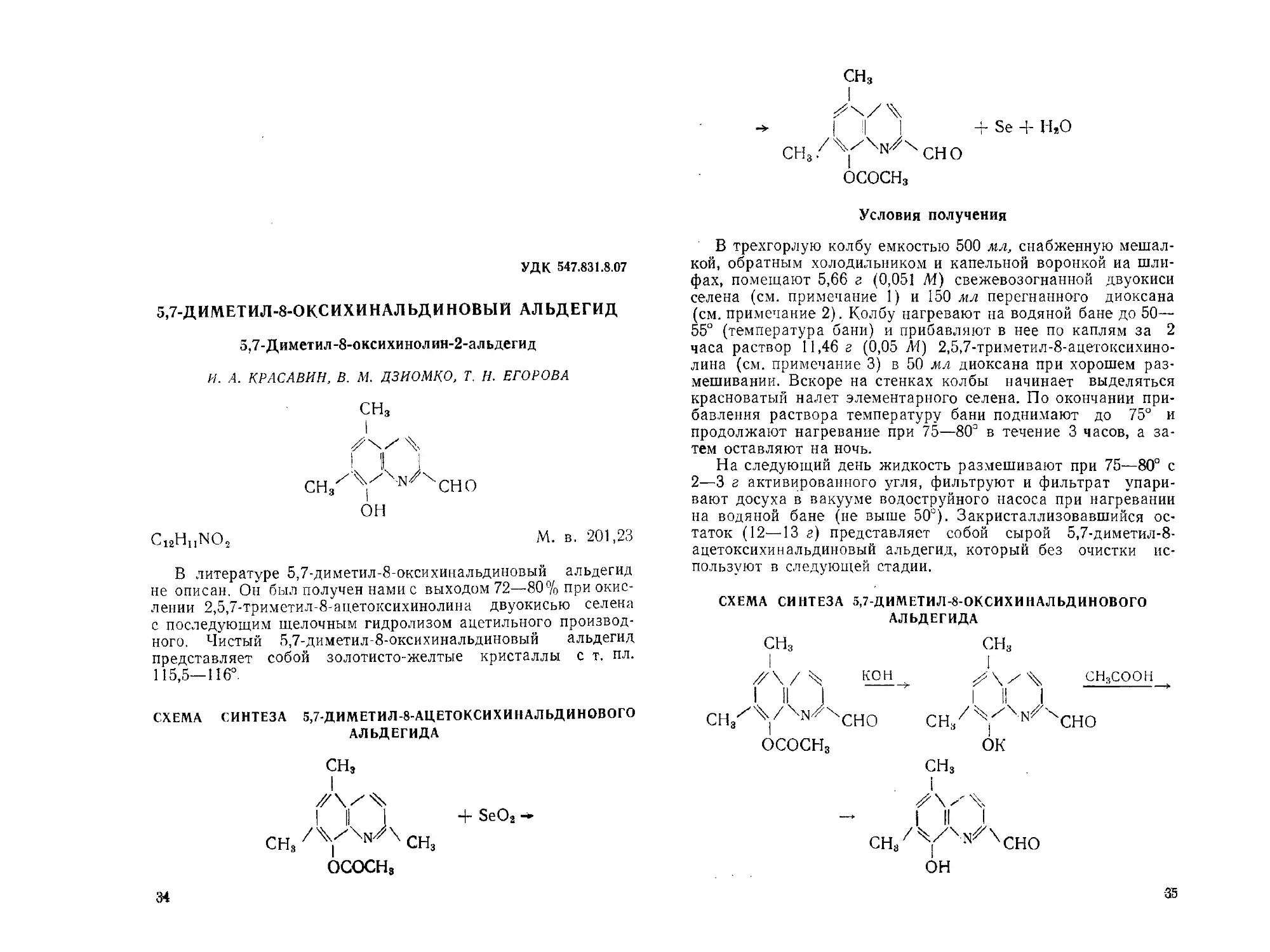
5,7-ДИМЕТИЛ-8-ОКСИХИНАЛЬДИНОВЫЙ АЛЬДЕГИД
5,7-Диметил-8-оксихинолин-2-альдегид
И. А. КРАСАВИН, В. М. ДЗИОМКО, Т. Н. ЕГОРОВА
СНз
он
Ci2HhNO2 М. в. 201,23
В литературе 5,7-диметил-8-оксихинальдиновый альдегид
не описан. Он был получен нами с выходом 72—80% при окис-
лении 2,5,7-триметил-8-ацетоксихинолина двуокисью селена
с последующим щелочным гидролизом ацетильного производ-
ного. Чистый 5,7-диметил-8-оксихинальдиновый альдегид
представляет собой золотисто-желтые кристаллы с т. пл.
115,5—116°.
СХЕМА СИНТЕЗА 5,7-ДИМЕТИЛ-8-АЦЕТОКСИХИНАЛ ЬДИНОВОГО
АЛЬДЕГИДА
СНЭ
I
I || + SeO2—
СН8 СНз
ОСОСНз
34
СНз
1 !! J
CHg/'Y'^^CHO
ОСОСНз
+ Se + Н,0
Условия получения
В трехгорлую колбу емкостью 500 мл, снабженную мешал-
кой, обратным холодильником и капельной воронкой иа шли-
фах, помещают 5,66 г (0,051 М) свежевозогнанной двуокиси
селена (см. примечание 1) и 150 мл перегнанного диоксана
(см. примечание 2). Колбу нагревают на водяной бане до 50—
55° (температура бани) и прибавляют в нее по каплям за 2
часа раствор 11,46 г (0,05 Л1) 2,5,7-триметил-8-ацетоксихино-
лина (см. примечание 3) в 50 мл диоксана при хорошем раз-
мешивании. Вскоре на стенках колбы начинает выделяться
красноватый налет элементарного селена. По окончании при-
бавления раствора температуру бани поднимают до 75° и
продолжают нагревание при 75—80° в течение 3 часов, а за-
тем оставляют на ночь.
На следующий день жидкость размешивают при 75—80° с
2—3 г активированного угля, фильтруют и фильтрат упари-
вают досуха в вакууме водоструйного насоса при нагревании
на водяной бане (не выше 50°). Закристаллизовавшийся ос-
таток (12—13 г) представляет собой сырой 5,7-диметил-8-
ацетоксихинальдиновый альдегид, который без очистки ис-
пользуют в следующей стадии.
СХЕМА СИНТЕЗА 5,7-ДИМЕТИЛ-8-ОКСИХИНАЛЬДИНОВОГО
АЛЬДЕГИДА
СНз
//\/ \ кон
1 11 J
CH3/'YXn^XCHO
ОСОСНз
СНз
!
СНзСООН
1)11
CH3/4:|/Xn/XCHO
ок
СНз
он
35
Условия получения
Сырой 5,7-диметил-8-ацетоксихинальдиновый альдегид
растворяют при подогревании в 100 мл толуола, обрабатыва-
ют активированным углем и фильтруют. Фильтрат помеща-
ют в трехгорлую колбу емкостью 2 л с мешалкой, термомет-
ром и капельной воронкой и охлаждают до —3°. При интен-
сивном размешивании прибавляют за 40—50 минут 536 мл
5%-ного раствора едкого кали, предварительно охлажден-
ного до 0°. Колбу сильно охлаждают снаружи, чтобы темпе-
ратура смеси не поднималась выше 2°, и размешивают жид-
кость при 0° еще 10—20 минут. Затем щелочной слой темно-
красного цвета отделяют, быстро фильтруют с отсасывани-
ем (см. примечание 4) и фильтрат помещают в коническую
колбу, погруженную в ледяную воду. Толуольный слой экст-
рагируют 3—5 порциями холодного 5%-ного раствора ед-
кого кали (по 100 мл) до тех пор, пока экстракты будут
лишь слабо окрашиваться в красный цвет. После отделения
каждой порции ее быстро фильтруют и присоединяют к ос-
тальным фильтратам, охлаждаемым в конической колбе (см.
примечание 4). Объединенные щелочные растворы нейтра-
лизуют 20%-ной уксусной кислотой до pH 7, причем крас-
ная окраска исчезает и выделяется желтый осадок. Его от-
сасывают, промывают водой, отжимают и высушивают вва-
кумм-эксикаторе над серной кислотой.
Выход неочищенного альдегида составляет 7,3—8,1 г
(72,6—80,5%, считая на 2,5,7-триметил-8-ацетоксихиполип);
т. пл. 107—108°. Его перекристаллизовывают из 230—250 мл
циклогексана с применением активированного угля и получа-
ют 4,7—6,3 г (46,7—62,6%) альдегида ст. пл. 109—110°. Из
маточного раствора можно выделить еще 0,6—1,3 г менее чи-
стого вещества.
Общий выход 5,7-диметил-8-оксихинальдинового альдеги-
да равен 6,0—6,9 г (59,6—68,6% от теоретического).
Более чистое вещество можно получить перекристаллиза-
цией из ацетона (см. примечания 5 и 6).
Примечания:
1. Применялась двуокись селена, активированная [1] и возогнанная в
специальном стакане [2] непосредственно перед употреблением. Двуокись
селена ядовита, поэтому работа с ней требует осторожности.
2. Перед перегонкой диоксан должен быть проверен на отсутствие
перекисных соединений (проба с йодистым калием).
3. Получение 2,5,7-триметил-8-ацеюкснхинолина описано в настоящем
сборнике.
4. Экстракцию толуольного слоя и переработку щелочных растворов
следует проводить быстро, а температуру растворов поддерживать около
0°. Если это не удается, то лучше нейтрализовать каждую порцию в от-
дельности. Длительное пребывание альдегида в щелочной среде приводит
к снижению выхода и ухудшению качества продукта.
5. Лучшая очистка вещества достигается при перекристаллизации из
ацетона, однако растворимость альдегида в нем слишком велика. Хоро-
36
шие результаты можно получить, если растворить сырой продукт в теп-
лом ацетоне, обработать активированным углем, профильтровать и оста-
вить фильтрат в глубокой чашке. По мере испарения ацетона выделяют-
ся пушистые кристаллы, которые можно отделить вручную от более за-
грязненного вещества.
6. Чистый образец 5,7-диметпл-8-окс11хинальдпнового альдегида в виде
золотисто-жслтых кристаллов с т. пл. 115,5—116° был приготовлен пере-
кристаллизацией из ацетона, а затем—из циклогексана и проанализиро-
вав.
Найдено, %: С-71,47; 71,61; Н —5,15; 5,32; N—7,20; 7,20.
Ci,HllNO2. Вычислено, %: С—71,63; Н—5,51; N 6,96.
ЛИТЕРАТУРА
1. В. М. Родионов. Синтезы органических соединений, сб. 2, Изд.
А.Н СССР, М„ 1952, стр. 118.
2. И. А. Красавин, О. В. Иванов, В. М. Д з и о м к о. /Методы
получения химических реактивов и препаратов, вып. 9, М., ИРЕА, 1964.
стр. 11, примечание 3.
Поступила в марте 1965 г. ИРЕА
УДК 547.572.3.07
3,4-ДИМЕТОКСИАЦЕТОФЕНОН
В. И. ДУЛЕНКО, Г. Н. ДОРОФЕЕНКО
СН3О^
CHsO-/~^-COCH9
CioHjaOa
М. в. 180,20
3,4-Диметоксиацетофенон получают путем ацетилирования
вератрола уксусным ангидридом или хлористым ацетилом 8
присутствии катализаторов реакции Фриделя—Крафтса.
Нами в качестве катализатора использован безводный
перхлорат магния (ангидрон) [1].
СХЕМА СИНТЕЗА 3,4-ДИМЕТОКСИАЦЕТОФЕНОНА
сн3о.
сн3о - \ + (сн3со)2о
СН3Оч
СН3О-^^--СОСН3 + СНзСООН
Характеристика основного сырья
Вератрол, ч., ТУ РУ 452—51.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Магний хлорнокислый, безводный (ангидрон), ч., ВТУ
МХП 3139—57.
38
Условия получения
Смесь 63 мл (0,5 М) вератрола, 63 мл (0,63 М) уксусного
ангидрида и 3,34 г (0,015 А1) ангидрона нагревают до кипе-
ния в течение 30 минут. После охлаждения реакционной сме-
си добавляют 3 г тщательно измельченного ацетата калия или
поташа и перемешивают в течение 30 минут, затем добавляют
100 мл диэтилового эфира и выделившийся осадок отфильт-
ровывают. Эфир и уксусную кислоту (30—35 мл) Отгоняют,
а остаток перегоняют в вакууме. Собирают 8—10 мл обрат-
ного вератрола при 8579 мм и 63 а (70%) 3,4-диметоксиаце-
тофенона при 159°/9 мм. При охлаждении кетон затвердевает;
т. пл. 49° (из эфира).
По литературным данным, вещество имеет т. кип. 286°;
т. пл. 49—51° [2].
ЛИТЕРАТУРА
1. Г. Н. Дорофеенко, В. И. Д у л е н к о. Авт. свид. 140054; Бюлл.
изобр., № 15, 1961.
2. Словарь органически* соединений, 1, М„ ИЛ, 1949, стр. 18.
Поступила в октябре 1964 г.
Донецкий филиал ИРЕА,
Ростовский-на-Дону
госуниверситет
УДК 547.461.3'052.2
ДИНИТРИЛ малоновой кислоты
Малононитрил, пропандинитрил, метилендицианид
С. С. РАДЛЕ-ДЕСЯТНИК, И. И. РОЗЕНФЕЛЬД
C=N
I
сн2
I
C=N
C3H2N2 М.в. 66,06
Динитрил малоновой кислоты является исходным продук-
том для получения тетрацианэтилена, тетрациаппропана и
ТД'.й.в'-тетрацианхинодиметана.
В литературе описано получение динитрила малоновой
кислоты дегидратацией диамида малоновой кислоты пяти-
окисью фосфора |1] и цнанацетамида пятиокисыо фосфора
[1], пятихлористым фосфором [2] и хлорокисью фосфора
|3, 4]. Нами уточнены условия синтеза, время ведения реак-
ции, оптимальные соотношения компонентов для обоих спо-
собов получения динитрила малоновой кислоты, а также ме-
тодика нейтрализации кислого нитрила. Диамид малоновой
кислоты мы синтезировали из малонового эфира и аммиа-
ка [5|.
СХЕМА СИНТЕЗА ДИНИТРИЛА МАЛОНОВОЙ КИСЛОТЫ
1. 2NCCH2CONH2+POC13 — 2CH2(CN)2+HPO3 + ЗНС1
2. 3CH2(CONH2)2+2P2O5 -> 3CH2(CN)2 + 4H3PO4
Характеристика основного сырья
Диамид малоновой кислоты, ч., ВТУ РУ 1360—57.
Цианацетамид, ч., ТУ 79-8—61.
40
Фосфорный ангидрид, ч., МРТУ 6-09-22—62.
Фосфор хлорокись, ч., МРТУ 6-09-337—63.
Дихлорэтан, ч., ГОСТ 5840—51.
Магний окись, ч., ГОСТ 4526—48.
Натрий хлористый, ч., ГОСТ 4233—48.
Условия получения
1. Синтез динитрила малоновой кислоты из цианацетами-
да. В литровую трехгорлую колбу, снабженную интенсивна
работающей мешалкой, ртутным затвором, обратным холо-
дильником и термометром, помещают 63 г (0,75 М) циан-
ацетамида, 50 г хлористого натрия и 250 мл дихлорэтана. Со-
держимое колбы перемешивают в течение 15 минут, после че-
го вносят 71 г (42 мл, 123% от стехиометрии) технической
хлорокиси фосфора. Колбу с содержимым при перемешива-
нии подогревают на глицериновой бане до кипения (83—85°).
Кипятят при интенсивном перемешивании в течение 8 часов.
В процессе реакции дегидратации выделяется газообраз-
ный хлористый водород, который поглощается водой в ловуш-
ке, соединенной с обратным холодильником.
После восьмичасового кипячения содержимое колбы ох-
лаждают до комнатной температуры, отфильтровывают оса-
док и промывают его на фильтре 30 мл дихлорэтана. Фильт-
рат нейтрализуют порошкообразной окисью магния до pH
5—6 по универсальной индикаторной бумажке. Сначала от-
гоняют дихлорэтан в вакууме при 40—50725 мм, а затем при
100—105710—12 мм—конечный продукт.
Выход динитрила малоновой кислоты равен 33,2—34,6 г,
что составляет 68—70% от теоретического; температура кри-
сталлизации 30—32°; кислотность продукта 0,1—0,2% (в пе-
ресчете на НО).
2. Получение динитрила малоновой кислоты из диамида
малоновой кислоты. Получение малононитрила осуществля-
ется в приборе, схема которого изображена на рисунке*.
В круглодонную колбу 1 емкостью 3 л загружают 153 г
(1,5 М) сухого диамида малоновой кислоты и 426 г (ЗМ) фос-
форного ангидрида. Колбу закрывают резиновой пробкой,
тщательно встряхивают в течение 30 минут и закрепляют на
штативе. На резиновой пробке присоединяют к колбе воз-
душный холодильник 2, который вторым концом соединяется
через шлиф с приемником нитрила 3. Последний в свою оче-
редь соединяется со вторым приемником 4 и далее через при-
емник 5 сообщается с вакуум-насосом (см. примечание 1).
Затем создают вакуум в системе с остаточным давлением по-
рядка 2—3 мм. Колбу с реакционной смесью подогревают
* Размерность дана в миллиметрах.
41
пламенем двух горелок (одна из них с воздушным дутьем).
Сначала обогревают горло и верх колбы небольшим пламе-
нем до пожелтения поверхности реакционной массы, далее
пламя увеличивают и обогревают реакционную смесь—сна-
чала с боков колбы, а затем, по мере того, как идет реакция,
обогревают дно колбы. Реакционная масса при этом пенится,
чернеет, а образующийся динитрил малоновой кислоты возго-
няется и перегоняется в приемник 3.
Прибор для получения динитрила малоновой кислоты из диамида мало-
новой кислоты:
/—реакционная колба; 2—холодильник;' 3,4,5— приемники
Холодильник время от времени подогревают слабым пла-
менем горелки, чтобы расплавить застывающий в нем нитрил,
обогревая одновременно колбу и реакционную массу.
Концом реакции следует считать визуальное окончание
выделения паров малононитрила из реакционной массы (см.
примечание 2).
Выход динитрила малоновой кислоты равен 44—50 г, что
составляет 45—50% от теоретического; температура кристал-
лизации продукта 30—32°.
Примечания:
1. Основным приемником продукта является приемник 3. Приемники
4 и 5 предназначены для улавливания несконденсировавшпхся паров ди-
нитрила малоновой кислоты.
2. В некоторых случаях в конце реакции масса сильно пенится. Для
предотвращения переброса ее в приемник следует соединить систему с
атмосферой.
42
ЛИТЕРАТУРА
1. A. L. Henry. Conipt. rend.. 102, 1344 (1886).
2. Б. К и P с о и. Синтез органических препаратов, 2, 356 (1949).
3. A. R. Surrey. J. Amer. Chem. Soc.. 70, 2471 (1943).
4. Пат. США 2459128 (1949).
5. С. С. Р а д л е-Д е с я т н и к. Методы получения химических реак-
тивов и препаратов, вып. 6, М., ИРЕА, 1962, стр. 101.
Поступила в январе 1965 г. ИРЕА
УДК 547.831.7.07
2,8-ДИОКСИХИНОЛ ИН
8-Оксикарбостирил
В. М. ДЗИОМКО, И. А. КРАСАВИН, Ю. П. РАДИН
I II । I II I
ОН он н
C9H7NO2 М. в. 161,16
2,8-Диоксихинолин образуется с выходом 70% при нагре-
вании 8-оксихинолина с расплавленной щелочью до 380°;
т. пл. продукта выше 260°. с разложением 1.1].
Другой путь, которым был синтезирован 2,8-диоксихино-
лин (т. пл. 250е), заключается в обработке 8-оксихиполин-1-
оксида уксусным ангидридом с последующим кислотным гид-
ролизом образовавшегося 8-ацетоксикарбостирила [2].
Как было нами установлено, 2,8-диоксихинолин может
быть получен с выходом 63—65% при действии раствора ще-
лочи на метосульфат 8-окси-1-метоксихинолииия. 2,8-Диок-
сихинолин мало растворим в обычных органических раство-
рителях и очищается с трудом. После двукратной перекри-
сталлизации из диметилформамида вещество приобрела вид
зеленовато-серых кристаллов с т. пл. 288—289,5°. Строение
продукта реакции подтверждается тем, что его ИК-спектры
тождественны спектрам 2,8-диоксихинолина, полученного по
литературным данным [2], а моноацетильное производное
(т. пл. 249,5—250°) не дает депрессии в пробе смешения с
8-ацетоксикарбостирилом (т. пл. 249—250°) [2].
Наряду с диоксипроизводным в реакции образуется (с
выходом 20—23%) не описанный в литературе 8-окси-2-ме-
токсихинолин, т. пл. 50,5—51° (из гептана).
44
СХЕМА СИНТЕЗА 2,8-ДИОКСИХИНОЛИНА
1 II + I +2К0Н
x\/\bZ ch3so4-
он осн3
— | II I + KCH3SO4 + снаон + нао
ч/ w\0H
ок
| II I +СН3СООН-Ч II I -1 СНзСООК
4\/\n^\qh Y^^014
ок он
Условия получения
В трехгорлой колбе емкостью 250 мл с мешалкой, термо-
метром и капельной воронкой растворяют 8,4 г (0,15 М) х.ч.
едкого кали в 75 мл воды. Охлаждают раствор на ледяной
бане и прибавляют к нему по каплям за 2 часа раствор 14,4 г
(0,05 Л4) метосульфата 8-окси-1-метоксихинолиния (см. при-
мечание 1) в 25 мл воды при 3—5°. Продолжают перемеши-
вание при 3—5° еще 3 часа и оставляют на ночь при комнат-
ной температуре. На следующий день раствор обрабатывают
активированным углем без нагревания, фильтруют и фильт-
рат нейтрализуют при охлаждении 10%-ной уксусной кисло-
той до pH 7. Выпавший осадок отсасывают, промывают водой
и высушивают над серной кислотой.
Сухой продукт (7,3—7,4 г) кипятят в течение 30 минут с
100 мл н-гептана в колбе с обратным холодильником. Нерас-
творившийся остаток отфильтровывают (см. примечание 2) и
высушивают над парафиновыми стружками.
Выход сырого 2,8-диоксихинолина равен 5,1—5,2 г, что со-
ставляет 63,3—64,5% от теоретического; т. пл. 275—280° (с
разложением и предварительным потемнением при 260°) .
Продукт перекристаллизовывают из 35 мл диметилформ-
амида с применением активированного угля и получают
3,35—3,4 г 2,8-диоксихинолина (41,6—42,2%) в виде зелено-
вато-серых кристаллов с т. пл. 285—286° (с разл.) (см. при-
мечание 3).
Примечания:
1. ^Применялся сырой метосульфат 8-окси-1-метоксйхииолипия, полу-
ченный, как описано в настоящем сборнике.
2. Гептановый экстракт содержит около 2 г побочного продукта—
о-окси-2-метокспхпнолина. Он перегоняется с водяным паром, легко рас-
45
творим в органических растворителях и кристаллизуется из гептана в
бесцветных иглах с т. пл. 50,5—51°.
3. Повторная перекристаллизация из диметилформамида дала обра-
зец с г. пл. 288—289,5° (с разл.).
Найдено, %: N—8,61; 8,45.
C9HtNO2. Вычислено, %: N—8,69.
ЛИТЕРАТУРА
1. J. Di am ant. Monatsh., 16, 760 (1895).
2. J. Р. Phillips, Е. М. Barral, R. Breese. Trans. Kentucky
Acad. Scl., 17, 135 (1956); Chem. Abstis, 51, 11349a (1957).
Поступила в марте 1S65 г.
УДК 547.821.41.07
ДИ-н-ПРОПИЛОВЫЙ ЭФИР
2-ПИРИДИЛМЕТИЛФОСФИНОВОИ КИСЛОТЫ
В. И. КАРБАН, Г. Ф. ДРЕГВАЛЬ
I II /О
L J—сн2р^
Xn' \ос8н,)2
CisHjqNOjP
М. в. 257,27
По литературным данным, ди-н-пропиловый эфир 2-пири-
дилметилфосфиновой кислоты получают из ди-н-пропилфос-
фита и хлоргидрата 2-хлорметилпиридина [1]. При проверке
этой прописи синтезировать указанный эфир не удалось.
Мы изменили условия получения и уточнили методику
[1]. Ди-н-пропиловый эфир 2-пиридилметилфосфиновой кисло-
ты находит применение в аналитической химии для экстрак-
ции редких элементов.
СХЕМА СИНТЕЗА ДИ-Н-ПРОПИЛОВОГО ЭФИРА
2-ПИРИДИЛМЕТИЛФОСФИНОВОЙ КИСЛОТЫ
/О
(н-С,Н7О)2 Р( + Na -> (н-С3Н7О)2Р< + ]/2 Н2
г! 'Na
О \
(н-С3Н7О)2Р^ + С1СН8—I J
xNa
// \ о
-* I II_СН2Р ^аС1
W 2 ^(ОСзН^
47
Характеристика основного сырья
Ди-н-пропилфосфит, свежеперегнанный, т. кип. 8776 мм;
п*> —1,4172.
2-Хлорметилпиридин, свежеперегнанный, т. кип. 45—
4772 мм; п$ —1,5360.
Натрий металлический, ч., ТУ МХП 1664—50.
Условия получения
К раствору 16,6 г (0,1 М) ди-н-пропилфосфита в абсолют-
ном эфире при перемешивании и охлаждении прибавляют
2,3 г (0,1 М) металлического натрия.
После того, как весь натрий прореагировал, осторожно
прибавляют раствор 13 г (0,1 М) 2-хлорметилпиридина в
30 мл диэтилового эфира и нагревают на кипящей водяной
бане в течение 2 часов. Затем реакционную смесь промывают
водой, эфирный слой отделяют, из водного слоя экстрагиру-
ют эфиром дополнительное количество вещества. Органиче-
ский слой, соединенный с эфирными вытяжками, сушат суль-
фатом натрия, перегоняют и получают 7,6 г ди-н-пропилового
эфира 2-пиридилметилфосфиновой кислоты, что составляет
58.6% от теоретического; т. кип. 188—192710 мм; <Д24—1,0808;
п* —1,4836.
По литературным данным, т. кип. продукта 134—135° при
0,6 мм; <725—1Д818;^ —1,4860 [1].
ЛИТЕРАТУРА
1. Е. Matuszewska-Wieczorkowska, J. Michalski,
A. Skowronsica. Roczniki chemii, 30, 1197 (1956).
Поступила в октябре 1964 г.
УДК 547.562.4’562.1
4,4’-ДИХЛОРДИФЕНИЛОКСИД
4,4’-Дихлордифениловый эфир
3. В. ВОРОНКОВА, Г. М. БЕССУДНОВА, И. В. ХВОСТОВ
С12Н8ОС12 М. в. 239,21
Впервые 4,4’-дихлордифенилоксид был получен при хло-
рировании дифенилового эфира в присутствии йода [1]. При
хлорировании дифенилового эфира в растворе уксусной кис-
лоты образуется преимущественно 4-хлордифенилоксид и не-
которое количество 4,4'-дихлордифенилоксида и 3,4,4'-три-
хлордифенилоксида [2].
4,4'-Дихлордифенилоксид может быть получен и другим
путем, по реакции Зандмейра, из 4-амино-4'-хлордифенило-
вого эфира [3]. Однако этот метод ввиду многостадийности
синтеза менее удобен для препаративного получения продук-
та.
Нами разработан простой способ получения 4,4'-дихлор-
дифеиилоксида из дифенилового эфира действием на него
хлористого сульфурила.
СХЕМА СИНТЕЗА ДИХЛОРДИФЕНИЛОКСИДА
\ О - 5 + 2SO2C13 ->
-> С1-^ ^-Cl + 2SO2 •- 2НС1
Характеристика основного сырья
Дифенилоксид, ч., ВТУ 2787—51.
Хлористый сульфурил, ч., ВТУ МХП 3591—52.
49
Условия получения
В четырехгорлую колбу, снабженную мешалкой, термо-
метром, капельной воронкой, обратным холодильником, вно-
сят 172 г (1,01 М) дифепилоксида. Включают мешалку, на-
гревают массу на масляной бане до 80° и при этой темпера-
туре в колбу прибавляют 145 г ( ~ 1,07 М) хлористого суль-
фурила в течение 30 минут. Реакционную смесь нагревают
до 120° и при этой температуре размешивают ее 5 часов. За-
тем массу охлаждают до 80° и в течение 30 минут вносят до-
полнительно 145 г ( -^1,07 М) хлористого сульфурила, разме-
шивают при 120° еще 5 часов. По окончании выдержки реак-
ционную массу перегоняют под вакуумом. При температуре
150—155°/3—5 мм собирают дихлордифенилоксид. Выход
продукта 170 г, что составляет ~ 70% от теоретического,
считая на исходный дифенилоксид.
Непрореагировавшие дифенилоксид и монохлордифснил-
оксид, получающиеся при вакуумной разгонке реакционной
массы, могут быть использованы в последующих синтезах
для получения дихлордифенилоксида.
После повторной разгонки дихлордифенилоксид имеет
следующие константы: т. кип. 150—15573 мм; т. кип. 310—-
312°/760 мм; d420—1,297—1,300; ri% — 1,609—1,611.
Найдено, %: 61—28,5—29,5.
С1;2Н8ОС1о. Вычислено, %: CI—29,2.
По литературным данным, дихлордифенилоксид имеет
т. кип. 312—3147760 мм [1]; 168—17277 мм [2]; 1,3164
[1]; 4° -1,611.
ЛИТЕРАТУРА
1. М. М а 11 h е, М. Murat. Bull. Soc. chim. France, (IV), 11, 328
(1912).
2. R. Brewster, G. Stevenson. J. Auier. Chem. Soc., 6?,
3144 (1946).
3. R. La Fevre, S. Saunders, E. Turner. J. Chem. Soc.,
1172 (1927).
Поступила в декабре 1964 г.
УДК 547.292
ИЗОБУТИЛОВЫЙ ЭФИР уксусной кислоты
Изобутилацетат
Р. М. ГЕЛЪШТЕИН, Е. И. МАЛОВЕР
СН3
I
СНз-СО-СНг-СН
II I
О сн3
CeH)2O2 М. в. 116,16
Существующий метод получения эфиров уксусной кислоты
основан на действии уксусного ангидрида или уксусной кис-
лоты на соответствующие спирты в присутствии серной кис-
лоты. Возможность побочных процессов, трудность очистки от
не вступившего в реакцию спирта являются недостатками
метода гомогенного кислотного катализа.
В последнее время разработан метод получения некоторых
эфиров уксусной кислоты этерификацией уксусной кислоты
соответствующими спиртами в присутствии ионообменной
смолы КУ-2 [1—3].
Использование ионитов как катализаторов реакции этери-
фикации позволяет применить эквивалентные количества реа-
гирующих веществ, что значительно облегчает очистку гото-
вого продукта.
СХЕМА СИНТЕЗА ИЗОБУТИЛОВОГО ЭФИРА УКСУСНОЙ КИСЛОТЫ
CH3COOH+HOCH2CH(CHS)3 - СН3СООСН2СН(СН3)3+Н3О
Характеристика основного сырья
Уксусная кислота, техн., ГОСТ 7077—54.
Изобутиловый спирт, техн., ТУ МХП 1704—47.
51
Ионообменная смола КУ-2, ВТУ ГХПК М-661—55, исполь-
зуется без предварительной обработки.
Условия получения
В круглодонную колбу емкостью 1 л, снабженную мешал-
кой с затвором, обратным холодильником и водоотделителем,
загружают 360 г уксусной кислоты, 440 г изобутилового спир-
та и 44 г смолы КУ-2 с 20%-ным содержанием влаги. Со-
держимое колбы при энергичном перемешивании кипятят в
течение 5—6 часов, до прекращения отделения воды (темпе-
ратура бани 110—120°). Затем реакционную массу охлажда-
ют до комнатной температуры, фильтруют и фильтрат промы-
вают сначала 250 мл воды, затем 250 мл 10%-ного раствора
соды и вновь водой до нейтральной реакции промывных вод.
Промытый эфир сушат хлористым кальцием. Выход техниче-
ского продукта равен 550—570 г. Очистку изобутилацетата
проводят перегонкой, собирая фракцию с температурой кипе-
ния 115—118°.
Выход чистого продукта составляет 470 г или 67,5% от
теоретического; пг° — 1,390; dt2a — 0,8710.
По литературным данным, n!J>7S —1,39066; d4M—0,8712 [4].
ЛИТЕРАТУРА
1. Н. Г. Полянский. Успехи химии, 31, 9, 1046 (1962).
2. Н, С. Рабо век а я. Ж. физ. химии, 33, 2467 (1959).
3. Т. И. Андрианова, Б. П. Брунс. Кинетика и катализ, 1, 440
(1960).
4. СОС, т. II, стр. 412.
Поступила в октябре 1964 г.
УДК 547.821.41
2-МЕТИЛ-3,4,5,6-(БИС-ТРИМЕТИЛ ЕН)ПИРИДИН
В. И. ДУЛЕНКО. Н. В. КОВАЛЕНКО, Г. И. ДОРОФЕЕНКО
c12h16n
М.в. 173,25
2-Метил-3,4,5,6- (бис-триметилен) пиридин был получен
Чичибабиным путем конденсации циклопентанона с паральде-
гидом в присутствии ацетата аммония [1]. Нами предложен
способ получения этого реактива из циклопентилиденцикло-
пентанона через промежуточное образование пирилиевой со-
ли [2], отличающийся простотой и удобством синтеза 2-метил-
3,4,5,6- (бис-триметилен) пиридина.
СХЕМА СИНТЕЗА 2-МЕТИЛ-3,4,5,6-(БИС-ТРИМЕТИЛЕН)ПИРИДИНА
р:НзСО),о
нсю4
НзС сю4-
Характеристика основного сырья
Циклопентилиденциклопентанон, т. кип. 250—254° (см.
примечание 1).
53
Хлорная кислота, 57%-ная, ТУ ОРУ 87—57.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Условия получения
К 15 г (0,1 М) циклопентилиденциклопентанона постепен-
но при перемешивании прибавляют смесь 40 мл (0,4 М) ук-
сусного ангидрида и 4 мл (0,05 М) 70%-ной хлорной кислоты
(см. примечания 2 и 3). Разогревшуюся реакционную массу
выдерживают при комнатной температуре 1 час, после чего
добавляют 50—70 мл холодной воды, а затем медленно при-
ливают концентрированный раствор аммиака до сильно ще-
лочной реакции. Органический слой отделяют и экстрагируют
10%-ным раствором соляной кислоты. После добавления к
солянокислому раствору избытка едкого натра пиридин из-
влекают эфиром, высушивают, растворитель отгоняют, а про-
дукт перегоняют в вакууме, собирая фракцию при 135—
140°/5 мм.
Выход 2-метил-3,4,5,6-(бис-триметилен) пиридина равен
5,2 г, что составляет 30% от теоретического; т. кип. препара-
та 274—2767760 мм.
По литературным данным, вещество имеет т. кип. 275° [1].
По аналогичной методике при использовании ангидрида
масляной кислоты может быть получен 2-пропил-3,4,5,6-(бис-
триметилен)пиридин с выходом 24%; т. кип. 293—296°; т. пл
пикрата 111°.
Примечания:
1. Циклопентилиденциклопентанон был получен автокопденсацпей
цнклопентанона в присутствии едкого кали [3].
2. 70%-ная хлорная кислота получена упариванием в вакууме 57°/>
ной хлорной кислоты.
3. Смесь готовят постепенным добавлением хлорной кислоты к уксус-
ному ангидриду при перемешивании и охлаждении льдом.
ЛИТЕРАТУРА
1. А. Е. Ч и ч и б а б и н. Bull. Soc. chim. France, [5], 4, 1826 (1937).
2. Г. И. Дорофеенко, В. И. Ду.тенко. Ж- общ. химии, 32, 3445
(1962).
3. J. Р 1 е s е k. Cliem. listy, 50,252 (1956).
Поступила в октябре J£>64 г. Донецкий филиал ИРЕА,
Ростове кий-на-Дону
госуииверситет
УДК 547.821.4.07
2-МЕТИЛ-5-Н-БУТИЛ ПИРИДИН
Ю. И. ЧУМАКОВ, В. П. ШЕРСТЮК
(¥
/W
СН3
C10H15N М. в. 149,23
2-Метил-5-и-бутилпиридин является исходным продуктом
в синтезе антибиотика фузариновой кислоты. 2-Метил-5-н-бу-
тилпиридин может быть получен из 2-метил-5-этилпиридина
[1, 2], 2,5-диметилпиридина [3] или 2-метил-5-этинилпиридина
[4].
По разработанной нами схеме 2-метил-5-н-бутилпиридин
был синтезирован из метилвинилкетона и гексен- 1-илэтилово-
го эфира. Полученный из этих соединений конденсацией
Дильса—Альдера 2-этокси-3-н-бутил-6-метил-3,4-дигидро-1,2-
пиран омылялся в соответствующее 1,5-дикарбонильное про-
изводное, которое, без выделения из реакционной массы, вво-
дилось в конденсацию с гидроксиламином с образованием
2-метил-5-н-бутилпиридина [5].
СХЕМА СИНТЕЗА 2-МЕТИЛ-5-Н-БУТИЛ ПИРИДИНА
^ОС2Н5
СН8(СН2)4СП
хосан5
MgHPO4
и. С4Н9-СН=СН-ОС2Н5
н сн2
I
/ \/н-С*Нэ
II I
/\о/"-ос2н5
55
_ СНЭ J СНз
Характеристика исходного сырья
Бромистый амил, ч., т. кип. 126—127,5°; /г‘д —1,4425.
Метилвинилкетон, техн., т. кип. 81—83°.
Ортомуравьиный эфир, ч., СТУ 79-570-Х—60.
Гидроксиламин солянокислый, ч., ГОСТ 5456—51.
Магний фосфорнокислый двузамещенный, ч., ТУ МХП
336—41.
Условия получения
Получение гексен- 1-илэтилового эфира. Гексен-1-илэтило-
вый эфир синтезируют из ацеталя капронового альдегида (см.
примечание 1) на каталитической установке, используя в ка-
честве катализатора двузамещенный фосфат магния на жид-
ком стекле (см. примечание 2) в условиях, описанных в [6].
Выход составляет 75—78% от теоретического; т. кип. продук-
та 143—146°; «д' —1,4183; d420—0,7963 (см. примечание 3).
Получение 2-этокси-3-н-бутил-6-метил-3,4-дигидро-1,2-пи-
рана. Смесь 41,7 г (0,325 М) гексен-1-илэтилового эфира,
20,65 г (0,630 М) свежеперегнанного метилвинилкетона и
0,12 г гидрохинона (0,2% по весу от общего количества ис-
ходных реагентов) разливают в стеклянные ампулы из молиб-
денового стекла (б ампул, наружный диаметр 17 мм, толщи-
на стенки 2 мм), которые вставляют в специальный взрыво-
безопасный блок, и нагревают при температуре 185° в течение
8—10 часов. Реакционную массу перегоняют в вакууме вна-
чале из колбы Клайзена, а затем на ректификационной ко-
лонке высотой 50 см (см. примечание 4). В результате пере-
гонки получают 13,5 г 2-этокси-3-н-бутил-6-метил-3,4-дигидро-
1,2-пирана, что составляет 40% от теоретического, считая на
прореагировавший гексен-1-илэтиловый эфир; т. кип. продук-
та 101—102°/13 мм; п™ —1,4450; d?0—0,9078.
Получение 2-метил-5-н-бутилпиридина. 2-Метил-5-н-бутил-
пиридин получают по методике, описанной для получения
3-этилпиридина (см. примечание 5). Из 5 г (0,02 М) 2-этокси-
3-н-бутил-6-метил-3,4-дигидро-1,2-пирана получают 1,4 г 2-
метил-5-н-бутилпиридина, что составляет 37,5% от теоретиче-
ского; т. кип. 105—107°/19 мм; па —1,4911; df0—0,9068;
т. пл. пикрата, перекристаллизованного из спирта, 137—138°
(испр.).
56
По литературным данным, 2-метил-5-н-бутилпиридин име-
ет т. кип. 94—95711 мм [1]; 105—106720 мм [2].
Примечания:
1. Диэтилацеталь капронового альдегида получен реакцией Гриньяра
из бромистого амила и ортомуравьиного эфира по методике [7] с выходом
57—73%, а также из капронового альдегида по методике [8] с выходом
50%. Применявшийся диэтилацеталь капронового альдегида имел следу-
ющие константы: т. кип. 91—92731 .ч.и; Пр —1,4095.
2. Катализатор готовился следующим образом [9]. Двузамещениый
фосфат магния (100 г) смешивался с жидким стеклом (уд. вес 1,384;
250 мл). Через 15—20 минут смесь становится достаточно вязкой, чтобы
пропустить ее через сито или лучше через мясорубку. Полученная «лап-
ша» резалась на отрезки длиной около 5 мм и сушилась в сушильном
шкафу при 150°.
3. По литературным данным, гексен-1-илэтиловый эфир имеет т. кип.
144—147°; Пр5 —1,4160; dp—0,7915 [10].
4. Насадка—одиночные витки стеклянной спирали диаметром 4—5 мм.
Диаметр ректифицирующей части колонки 10 я.и. Разгонка проводилась
для отделения циклического димера метилвииилкетона, который имеет
следующие константы: т. кип. 71,2715 м.ч; Лд —1,4488; Ф*20—0,9840.
5. Подробно превращение алкилзамещенных 2-алкокси-3,4-дигидро-
1,2-пиранов в пиридины описано в статье «З-Этилпиридин» (см. настоящий
сборник).
ЛИТЕРАТУРА
1. A. Platter, W. Keller, A. Boiler. Helv. chlm. acta, 37,
1379 (1954).
2. T. M a ka sh 1 m a. J. Pharm. Soc. Japan, 75, 1012 (1955); РЖхим,
1956, 54454.
3. E. H a r d e g g e r, E. N i k 1 e s. Helv. chlm. acta, 40, 2428 (1957).
4. A. H. К о с т, П. Б. Терентьев, Л. В. Моше нцев а. Гетеро-
циклы в органическом синтезе (тезисы докладов), Киев, 1964, стр. 72.
5. Ю. И. Ч у м а к о в, В. П. Шерстю к, В. Ф. Новикова.
Е. П. Дзыгуи. Гетероциклы в органическом синтезе (тезисы докладов),
Киев, 1964, стр. 68.
6. Ю. И. Чумаков, В. П. Шерстю к. См. статью «З-Этилпиридин»
в настоящем сборнике.
7. Синтезы органических препаратов, т. 2, М., ИЛ, 1949, стр. 295.
8. Синтезы органических препаратов, т. 1, М., ИЛ, 1949, стр. 62.
9. И. Н. Назаров, С. М. Макин, Б. К. Крупцов, В. А. Миро-
нов. Ж- общ. химии, 39, 111 (1959).
10. Дж. Эриксон, М. Восков. Химия и хим. технология, Орга-
ническая химия, № 3, 1959, 17; J. Organ, Chem., 23, 670 (1958).
Поступила о январе 1965 г.
Киевский политехнический институт
УДК 547.821.2.07
3-МЕТИЛ-6-СТИРИЛ ПИРИДИН
.4. К. ШЕЙНКМАН, А. Н. РОЗЕНБЕРГ
CMHl3N
М. в. 195,26
З-Метил-6-стирилпиридин может быть использован как
комплексообразующий реагент и как промежуточное соеди-
нение в синтезе различных физиологически активных соеди-
нений и красителей.
Для синтеза стирилпиридинов обычно используют или ки-
пячение метилпиридинов с ароматическими альдегидами в
среде уксусного ангидрида [1, 2] или нагревание смеси метил-
пиридина и ароматического альдегида с хлористым цинком в
запаянной ампуле при высокой температуре.
Ранее нами был предложен способ активации метильных
групп пиридина путем перевода последнего в N-ацильную соль
действием галоидангидридов различных кислот [3]. Оказа-
лось, что этот метод применим и для активации а-метильной
группы 2,5-диметилпиридина. N-Ацильная соль 2,5-лутидина
гладко реагирует с бензальдегидом, образуя стирилпиридин
с высоким выходом.
СХЕМА СИНТЕЗА 3-МЕТИЛ-6-СТИРИЛ ПИРИДИНА
Н С—
8 I II С6Н5СОС1
\N/~CH3
ОС-СвН6
58
С6Н6СНО
СН=СН—У
С1 Н
I J—сн=сн—
Ж/ \=/
Характеристика основного сырья
2,5-Лутидин, безводный, свежеперегнанный, т. кип. 159—
160°; /1ц —1,5022 (см. примечание 1).
Хлористый бензоил, ч., свежеперегнанный, ТУ МХП 92—
51 (см. примечание 2).
Бензальдегид, ч„ ГОСТ 157—51.
Диметилформамид, ч., СТУ 79-606-Х—60.
Условия получения
В трехгорлую круглодонную колбу, снабженную капель-
ной воронкой, механической мешалкой и обратным холодиль-
ником с хлоркальциевой трубкой, помещают 6,9 мл (0,065 4-1)
2,5-лутидина и по каплям прибавляют при перемешивании и
охлаждении на ледяной бане 6,93 мл (0,05 М) хлористого
бензоила. Выдерживают реакционную смесь в течение 20—
30 минут, добавляют раствор 6,1 мл бензальдегида в 5 мл
диметилформамида и выдерживают в течение 5 часов на мас-
ляной бане при 150—170°.
Затем реакционную массу охлаждают, разлагают концент-
рированной соляной кислотой и отгоняют с водяным паром
бензойную кислоту.
Остаток подщелачивают едким натром и вновь перегоня-
ют с водяным паром для освобождения от непрореагировав-
шего лутидина и бензальдегида. Осадок в перегонной колбе
отделяют, отмывают горячей водой от неорганических солей,
кипятят в растворе соляной кислоты с активированным уг-
лем, затем осаждают аммиаком, отделяют и сушат до посто-
янного веса в вакууме при 50°.
Выход сырого З-метил-6-стирилпиридина равен 4 г, что
составляет 47% от теоретического. Очистка продукта дости-
гается перегонкой с перегретым паром. По внешнему виду это
серебристо-белые чешуйки с т. пл. 68—69°, Хмакс =308 ммк,
1g £'=4,23, что соответствует литературным данным [2].
59
Примечания:
1. 2,5-Лутидин тщательно сушат кипячением над окисью кальция,
хранят над плавленым едким калн и перед употреблением перегоняют на
колонке.
2. Хлористый бензоил можно заменить хлорангидридом любой карбо-
новой кислоты.
Л ИТЕРАТУРА
1. В. D. Shaw, Е. A. Wagstaff. J. Chem. Soc., 1933, 77.
2. М. Haring, В. Prifs, Н. Erl еп me у er. Helv. chim. acta, 37,
147 (1954).
3. A. H. К о ст, А. К. Шейнкман. Ж. общ. химии, 33, 2077 (1963).
Поступила в сентябре 1964 г.
Донецкий филиал ИРЕА
УДК 547.821.2.07
4-ф- МЕТИЛСТИРИЛ)ПИРИДИНЫ
А. К. ШЕИН КМ АН, А. Н. РОЗЕНБЕРГ
HsC-C=CH-^~^-N(CH3)2
4-(л-Диметиламино-р-метилстирил) пиридин
CijHigNj
М. в. 238,33
Н3С-С=СН-^ ^-N(C2H6)2
I
4-(и-Диэтиламино-р-метилстирнл) пиридин
М. в. 266,38
Н3С—С=СН-^ ^-ОСН3
4-(я-Метркси-р-метилстирил) пиридин
М. в. 225,29
61
CleH16NO
H3C—C=CH—^-N03
4-(я-Нитро-3-метилстирил) пиридин
c14h18n2o8
М. в. 240,26
HSC—С=СН —
//\
i II
4-ф-Метилстнрил) пиридин
CUH13N
М. в. 195,26
Замещенные в бензольном ядре |3-метилстирилпиридины
могут быть использованы для синтеза различных физиологи-
чески активных веществ и красителей.
Известен способ получения подобных соединений конден-
сацией 4-этилпиридина с ароматическими альдегидами в сре-
де уксусного ангидрида [1]. Ранее нами был предложен спо-
соб активации метильных групп, связанных с гетероциклом,
переводом последнего в N-ацильную соль действием различ-
ных галоидных ацилов [2]. Оказалось, что этот метод приме-
ним и для активации метиленовой группы 4-этилпиридина.
N-Ацильная соль 4-этилпиридина гладко реагирует с различ-
ными ароматическими альдегидами с образованием соответ-
ствующих стирилпиридинов, а ацильный остаток легко уда-
ляется после реакции.
СХЕМА СИНТЕЗА 4-(₽- МЕТИЛСТИРИЛ)ПИРИДИНОВ
Н5С- СН2
С,Н5СОС1
n-R—С6Н4—СНО
62
где R-H; N(CH3)2; N(C2H6)2; OCHS; N02
Характеристика основного сырья
4-Этилпиридин, тщательно высушенный, т. кип. 163—166°
(см. примечание 1).
Хлористый бензоил, техн., свежеперегнанный, т. кип. 194-
197° (см. примечание 2).
n-Диметиламинобензальдегид, ч., ТУ МХП 2679—51.
n-Диэтиламинобензальдегид, ч., т. пл. 40—41°, СТУ 79-517-
X—60.
n-Метоксибензальдегид, техн., свежеперегнанный, т. кип.
135722 мм.
л-Нитробепзальдегид, ч., ТУ МХП 2969—51.
Бензальдегид, ч., ГОСТ 157—51.
Пиридин, ч., ГОСТ 1625—61 (см. примечание 3).
Условия получения
Синтез 4-(п-диметиламино-$-метилстирил)пиридина. К
2,25 мл (0,02 М) 4-этилпиридина по каплям при охлаждении
добавляют 2,34 мл (0,02 М) хлористого бензоила. К выпав-
шему осадку приливают раствор 4 г л-диэтиламинобензаль-
дегида в 5 мл пиридина. Реакционную смесь выдерживают па
масляной бане при температуре 150—160° в течение 5 часов,
после чего выливают в 20%-ный раствор соды. Осадок от-
фильтровывают, сушат и перекристаллизовывают из пириди-
на, затем из этилового спирта. Т. пл. 162—163°.
Выход 4-(л-диметиламино-р-метилстирил) пиридина в виде
желтых игл равен 3,5 г, что составляет 69,5% от теоретичес-
кого; т. пл. 162—163°.
По литературным данным, т. пл. продукта 164—165° [1].
Получение 4- (п-диэтиламино-$-метилстирил)пиридина.
Продукт получают в условиях, описанных выше для 4-(л-ди-
метиламино-р-метилстирил) пиридина, исходя из 2,25 мл
4-этилпиридина, 2,34 мл хлористого бензоила и 4гп-диэтил-
аминобензальдегида в 7 мл пиридина.
Выход продукта реакции равен 4 г, что составляет 75%
от теоретического; после двух перекристаллизаций вначале из
пиридина, затем из спирта т. пл. 115—116°.
Получение 4-(п-метокси-$-метилстирил)пиридина. К.
2,25 мл 4-этилпиридина при интенсивном перемешивании и
охлаждении в ледяной бане прикапывают 2,34 мл хлористого
бензоила. К выпавшему осадку светло-желтого цвета прили-
вают раствор 3 г n-метоксибензальдегида в 5 мл абсолютного
свежеперегнанного пиридина, при этом появляется коричне-
вое окрашивание и температура реакционной смеси самопро-
извольно поднимается до 45—50°. После прекращения разо-
гревания реакционную смесь выдерживают 5 часов на масля-
ной бане при температуре 155—160°, затем разлагают кон-
центрированной соляной кислотой, отгоняют с водяным паром
образующуюся при гидролизе бензойную кислоту и бензаль-
дегид, подщелачивают и вновь отгоняют с водяным паром.
Осадок в перегонной колбе отделяют, многократно отмывают
горячей водой от неорганических солей, кипятят в растворе
соляной кислоты с активированным углем, затем осаждают
раствором аммиака, отделяют и тщательно высушивают до
постоянного веса.
Выход 4-(л-метокси-р-метилстирил)пиридина равен 2 а, что
составляет 44% от теоретического; т. пл. 80—81° (из спирта).
По литературным данным, т. пл. продукта 83—84° [1].
Синтез 4-(п-нитро-$-метилстирил)пиридина. По методике,
описанной выше, из 11,42 мл (0,1 М) 4-этилпиридина, 11,7 мл
(0,1 М) хлористого бензоила и 15,1 г (0,1 М) п-нитробепзаль-
дегида в 23 мл пиридина получают 22,65 а 4-(п-нитро-(5-ме-
тилстирил)пиридина, что составляет 94% от теоретического;
т. пл. 109—110° (из гептана).
По литературным данным, т. пл. продукта ПО—111° [1].
Синтез 4-($-метилстирил)пиридина. Его получают анало-
гичным путем из 5,7 мл (0,05 М) 4-этилпиридина, 5,85 мл
(0,05 М) хлористого бензоила и 5,06 мл (0,05 М) бензальде-
гида в 5 мл пиридина. С водяным паром из щелочного рас-
твора перегоняются белые игольчатые кристаллы с т. пл. 72—
73°.
Выход 4-(р-метилстирил)пиридина равен 3,5 а, что состав-
ляет 46% от теоретического.
По литературным данным, т. пл. продукта 72—73° [1].
Примечания:
1. 4-Этилпнриднн, полученный по ранее описанной методике [3], сушат
над едким кали, кипятят с окисью кальция и перегоняют на колонке, со-
бирая фракцию 163—166°.
2. Хлористый бензоил может быть заменен хлорангндрндом любой
другой карбоновой кислоты.
3. Пиридин следует тщательно абсолютировать, так как реакция очень
чувствительна к влаге.
84
ЛИТЕРАТУРА
1. А. Р. R hill ips. J. Amer. Chem. Soc., 76, 3986 (1954).
2. A. H. К ост, А. К. Шейнкман. Ж. общ. химии, 33, 2077 (1963).
3. Н. Ф. Казаринова, Е. П. Б а б и и, К. А. С о л о м к о,
М. И. Котеленец, А. А. Артамонов, А. К. Шейнкман. Ж-
прнкл. химии, 36, 649 (1963).
Поступила в сентябре 1964 г.
Донецкий филиал ИРЕА
УДК 547.577.07
n-МЕТОКСИАЦЕТОФЕНОН
В. И. ДУЛ ЕН КО, Г. Н. ДОРОФЕЕНКО
сн3о-^ )-сосн3
C9HI0O2 ~ М. в. 150,17
n-Метоксиацетофенон получают путем ацетилирования
анизола уксусным ангидридом, хлористым ацетилом или кете-
ном в присутствии катализаторов реакции Фриделя—Крафтса
[1].
Предлагаемый метод отличается от известных использо-
ванием в качестве катализатора безводного перхлората м;и-
ния (ангидрона).
СХЕМА СИНТЕЗА п- МЕТОКСИАЦЕТОФЕНОНА
СНзО-^ ^+(СН3СО)2О —
СН3О-^_^-СОСН3+СН3СООН
Характеристика основного сырья
Анизол, ч., ТУ МХП 2793—55.
Уксусный ангидрид, ч.д.а., ГОСТ 5815—52.
Магний хлорнокислый, безводный (ангидрон), ч., ВТУ
МХП 3139—57.
Условия получения
Смесь 54 г (0,5 М) анизола, 75 мл (0,75 М) уксусного ан-
гидрида и 2,23 г (0,01 М) ангидрона нагревают до кипения в
течение двух часов. Через 15—20 минут постепенно добавля-
66
ют 2,75 г тонкоизмельченного ацетата калия или поташа и
реакционную смесь перегоняют под вакуумом 300 жж (водо-
струйный насос). При температуре 90—110° собирают около
70 мл уксусной кислоты с примесью уксусного ангидрида и
анизола, после чего остаток в колбе, предварительно охлаж-
денный до 40°, перегоняют в вакууме (3—4 жж). Сначала при
40—45° отгоняется небольшое количество анизола, а затем
собирают кетон при температуре 108—109°. При охлаждении
продукта получают кристаллы с т. пл. 37°.
По литературным данным, п-метоксиацетофенон имеет
т. пл. 38—39°, т. кип. 258° [2].
Выход n-метоксиацетофенона равен 45 г, что составляет
60% °т теоретического.
ЛИТЕРАТУРА
1. Г. Н. Дорофеенко, Л. В. Полищук. Методы получения хи-
мических реактивов н препаратов, вып. 2. М., ИРЕА, 1961, стр. 41.
Поступила в октябре 1964 г.
Донецкий филиал ИРЕА,
Ростовский-на-Дону
госуииверситет
УДК 547.831.7.07
МЕТОСУЛЬФАТ 8-ОКСИ-1-МЕТОКСИХИНОЛИНИЯ
И. А. КРАСАВИН, В. М. ДЗИОМКО, Ю. П. РАДИН
I | ch8so4-
он ОСН3
CnHJ3NO6S М.в. 287,30
Метосульфат 8-окси-1-метоксихинолиния в литературе не
описан. Он был получен нами при нагревании 8-оксихинолин-
1-оксида с диметилсульфатом. Сырой продукт, образующийся
с выходом 90—92%, вполне пригоден для синтетических це-
лей. Он представляет собой желтое кристаллическое вещест-
во, плавящееся с разложением при 126—128°, очень легко
растворяющееся в воде, несколько труднее—в спиртах, не-
растворимое в неполярных органических растворителях. По-
сле однократной перекристаллизации из абсолютного эта-
нола с эфиром был получен образец с. т. пл. 143—144,5°, но
такая очистка нецелесообразна, так как сопровождается
слишком большими потерями.
Реакции метосульфата 8-окси-1-метокс.ихиполиния с раз-
личными нуклеофильными агентами позволяют синтезировать
целый ряд 2-замещенных производных 8-оксихинолина, мно-
гие из которых трудно получить другим путем.
СХЕМА СИНТЕЗА МЕТОСУЛЬФАТА 8-ОКСИ-1-МЕТОКСИХИНОЛ ИНИЯ
А\/А
| II | +(CH3)2SO4 — I II + I
%. / % / -
II I | CH8SO4
ОН о ОН осн,
68
Условия получения
Внимание'. Синтез необходимо проводить в хорошо рабо-
тающем вытяжном шкафу, чтобы избежать воздействия ядо-
витых паров диметил сульфата (см. примечание 1).
В круглодонную колбу емкостью 500 мл с пришлифован-
ным обратным холодильником, защищенным хлоркальциевой
трубкой, помещают 48,3 г (0,3 М) измельченного 8-оксихино-
лин-1-оксида (см. примечание 2) и 37,9 г (0,3 М) свежепере-
гнанного диметилсульфата (см. примечание 3). Смесь нагре-
вают на кипящей водяной бане в течение 2 часов, причем мас-
са вскоре разжижается и становится однородной. Затем ре-
акционную массу охлаждают и размешивают стеклянным
шпателем, пока она не закристаллизуется. Прибавляют 200ли
сухого диэтилового эфира, размешивают до полного затвер-
девания вещества и отсасывают. Продукт переносят в ступку,
растирают с 50—100 мл эфира, вновь отсасывают, промывают
на фильтре эфиром и высушивают в вакуум-эксикаторе.
Выход сырого метосульфата 8-окси-1-метоксихинолиния
равен 77,6—79,3 г, что составляет 90—92% от теоретического;
т. пл. около 126—128° (см. примечания 4 и 5).
Примечания:
1. Диметилсульфат очень ядовит, попадание его на кожу или вдыха-
ние паров может вызвать тяжелые последствия. Перегоику диметилсуль-
фата и все работы с ним, включая отвешивание, следует проводить в хо-
рошо действующем вытяжном шкафу. Специфическим нейтрализатором
является аммиак, концентрированный раствор которого необходимо посто-
янно иметь под рукой, чтобы обезвредить случайно пролитый диметил-
сульфат.
2. Применялся 8-оксихинолин-1-оксид с г. пл. 138,5—139°, получен-
ный по описанным в литературе методам [1, 2] и дважды перекристалли-
зованный из воды или бензола.
3. Применялся дпметилсульфат хорошего ка>1сства («чистый» или
«ч.д.а.»), переданный перед употреблением и имеющий нейтральную ре-
акцию.
4. Вещество гигроскопично и его следует предохранять от действия
влаги воздуха.
5. При перекристаллизации 20 г сырого продукта из 25 мл абсолют
кого этанола с добавлением 25 мл сухого эфира было получено всего
3,67 г более чистого вещества с т. пл. 143—144,5°; поэтому дальнейшая
очистка не производилась.
ЛИТЕРАТУРА
1. J. Р. Phillips, Е. М. Barral, R. Breese. Trans. Kentucky,
Acad. Sci., 17, 135 (1956); Chem. Abstrs, 51. 11349a (1957!.
2. I. M u r a s e, Y. Demura. Mem. Fac. Sci., Kyushu Univ., Ser. C 4
№ 3, 175 (1961); Chem. Abstrs, 58, 3390b (1963).
Поступила в марте 1965 г.
ИРЕА
УДК 547.551.3.07
МОНОЭТИЛАНИЛИН
N-Этиланилин
C8HuN
И. Е. ЗАДОРОЖНАЯ
^>-nh-c2h5
М. в. 121,18
При этилировании анилина бромистым этилом, диэтил-
сульфатом или этиловым спиртом под давлением образуется
смесь анилина, моно- и диэтиланилина [1—3].
Анилин и моноэтиланилин разделяются через соединение
с n-толуолсульфохлоридом или п-толуолсульфокислотой [1, 4,
5]. Диэтиланилин отгоняется с паром, а из оставшейся смеси
анилид n-толуолсульфокислоты отмывается раствором щело-
чи; моноэтиланилин выделяется омылением этиланилида
п-толуолсульфокислоты серной кислотой. Этот метод длите-
лен и неудобен для производства.
Нами разработана методика получения чистого моноэтил-
анилина путем этилирования анилида п-толуолсульфокислоты
бромистым этилом.
СХЕМА СИНТЕЗА МОНОЭТИЛАНИЛИНА
'^-NHs+ClSOg-^^-CH» Na2C°3 -»
-> х /—NH-SOa-<f /—СН3
\ X ' \ UorisDF
С2н5
O-XS0a-C>CH3
- / 4- HSO3-^_^-CH3
Характеристика основного сырья
Анилин, ч., ГОСТ 313—58.
Натр едкий, ч., ГОСТ 2263—59.
n-Толуолсульфохлорид, ч., МРТУ 6-09-447—63.
Соляная кислота, ч., ГОСТ 3118—46.
Этил бромистый, ч., ГОСТ 2658—56.
Серная кислота, ч., ГОСТ 4204—48.
Сода кальцинированная, ч., ГОСТ 5100—49.
Условия получения
Синтез п-толуолсульфанилида. В колбу емкостью 2 л, ус-
тановленную в водяной бане и снабженную мешалкой и тер-
мометром, загружают 140 г (в пересчете на 100%-ный) ани-
лина и 1 л воды. Размешивают и нагревают до 60°. За 3 часа
одновременно небольшими порциями прибавляют 318 г (в пе-
ресчете на 100%-ный) n-толуолсульфохлорида и 115 г соды,
следя за тем, чтобы реакция среды была нейтральной. Затем
смесь размешивают 2 часа, охлаждают до комнатной темпе-
ратуры и приливают соляную кислоту до кислой реакции по
конго (расходуется около 45 мл). После 30-минутного раз-
мешивания кристаллы отфильтровывают и промывают водой
до отсутствия кислой реакции по конго и сушат при 40—50°.
Выход n-толуолсульфанилида равен 346 г, что составляет
93% от теоретического, считая на анилин; т. пл. 99,5—101,5°.
Получение этиланилида п-толуолсульфокислоты. В колбу
емкостью 2 л с шариковым холодильником, помещенную в во-
дяную баню, загружают 346 г n-толуолсульфанилида, 418лл
20%-ного раствора едкого натра и 334 г бромистого этила.
Реакционную массу кипятят 10 часов (при температуре бани
40—50°). Выпавшие кристаллы отфильтровывают, промыва-
ют 3 раза по 260 мл 10%-ного раствора едкого натра и во-
дой до нейтральной реакции. Сушат при 40—50°.
Выход этиланилида n-толуолсульфокислоты равен 350 г,
что составляет 91% от теоретического; т. пл. 84,8-85,8°.
Получение моноэтиланилина. В колбу емкостью 0,5л с ша-
риковым холодильником, установленную в глицериновую ба-
ню, загружают 350 г этиланилида п-толуолсульфокислоты,
99 мл воды и 220 г концентрированной серной кислоты. Массу
нагревают до 160—170° и кипятят 8 часов, после чего отбира-
ют пробу для определения конца омыления. Когда взятая
проба будет полностью растворяться в воде, омыление ведут
еще 3—4 часа при кипении. Затем реакционную массу охлаж-
дают, выливают на 585 г льда и нейтрализуют 20%-ным рас-
твором щелочи до щелочной реакции на фенолфталеин (рас-
ходуется около 640 мл раствора).
Маслянистый слой отделяют, сушат едким натром 24 часа,
фильтруют и перегоняют без вакуума с елочной колонкой.
71
Выход моноэтилаиилина равен 61,5 г, что составляет 40%
от теоретического, считая на этиланилид п-толуолсульфокис-
лоты; т. кип. 205—205,2°.
Данные анализа:
содержание анилина—нет;
содержание моноэтилаиилина—95,5%;
содержание диэтиланилина—3,9%.
По литературным данным, т. кип. моноэтилаиилина
204,7° [6].
ЛИТЕРАТУРА
1. Г. Э. Фи рц-Д а в и д. Основные процессы синтеза красителей, М.,
ИЛ, 1957, стр. 122, 123.
2. Н. Н. Ворожцов. Основы синтеза промежуточных продуктов г.
красителей, М„ Госхимиздат, 1955, стр. 484.
3. Ю. К- Юрьев. Практические работы по органической химии,
вып. 2, М.. МГУ, 1957, стр. 203.
4. О. Hinsburg, J. Kessler. Вег., 38,906 (1905).
5. Л. Н. Николенко. Лабораторный практикум по промежуточ-
ным продуктам и красителям, М., Изд. «Высшая школа», стр. 120.
6. Справочник химика, т. П. М.—Л., Изд. «Химия», 1964, стр. 435,
Я> 486.
Поступила в феврале 1965 г. ЦЗЛ
УДК 547.586.2.07
р- НАФТИЛОВЫЙ ЭФИР
№ - БЕНЗОИЛ-D,L- ФЕНИЛАЛАНИНА
Э. А. БАШКИР, Я. И. ЛАПУ К
СвН5-СН3-СН-СОО-^\/:Ч|
I I II I
NH \/\//
I
с=о
С6н6
CjeH21NO3 М. в. 395,45
P-Нафтиловый эфир № -бензоил-D, L-фенилаланина при-
меняется в биохимии для колориметрического определения
активности протеолитического фермента химотрипсина [1].
Ниже приводится методика получения 0-нафтилового эфи-
ра № -бензоил-DJ.-фенилаланина, основанная на взаимодей-
ствии азлактона N" -бензоил-D,L-фенилаланина с р-нафтолом.
СХЕМА СИНТЕЗА ₽-НАФТИЛОВОГО ЭФИРА
№ -БЕНЗОИЛ -D.L- ФЕНИЛАЛАНИНА
СвН5-СН2-СН-СООН + СвН8СОС1
NH,
С6Н5-СНаСН-СООН
NH
С = О + NaCl + Н2О
CSH6
73
2. С6Н6-СНа-СН-СООН
(СН3СО)гО
NH
С-0
сл
СвН5СН2- СН-С=О + 2СН3СООН
N 6
с«н6
3. СвН6-СН.-СН-С=О + _
II I !! I
N О
ceHs
CJ^-CHs-CH-COO-^V^l
1 ill
NH Ч/W
L°
ceHs
Характеристика основного сырья
Д,Ь-Фенилаланин, ч., ТУМЗ 1382—51.
Бензоил хлористый, ч„ ТУ МХП 92—51.
р-Нафтол, ч.д.а., ГОСТ 5835—51.
Уксусный ангидрид, ч., ГОСТ 5815—52.
Условия получения
Получение № -бензоил-D,L-фенилаланина [2]. В трехгор-
лой колбе емкостью 0,5 л, снабженной мешалкой и двумя ка-
пельными воронками, растворяют 26 а (0,160 М) фенилалани-
на в 80 мл 2 н. раствора едкого натра. В течение 30 минут
при температуре 5—10° одновременно прибавляют 28 мл 6н.
раствора едкого натра и 19,5 мл (0,165 М) хлористого бензо-
ила. После часовой выдержки при температуре 5—10° реак-
ционную смесь подкисляют 28 мл концентрированной соляной
кислоты до pH 2 по универсальной индикаторной бумаге. За-
тем охлаждают до 4 , осадок отфильтровывают, промывают
74
200 мл эфира и сушат при комнатной температуре. Получен-
ный технический N* -бензоил-D,L-фенилаланин растворяют в
250 мл этилового спирта, отфильтровывают от примесей и ох-
лаждают до 5—0°. Выделившиеся при этом кристаллы ве-
щества отфильтровывают, сушат на воздухе и получают 30,2 г
продукта с т. пл. 182—183°.
Маточный раствор упаривают в вакууме до '/з объема, ох-
лаждают до 5—0° и получают еще 8,7 г вещества с т. пл. 179—
180°, которое перекристаллизовывают из 40 мл этилового спир-
та. Получают дополнительно 8 г бесцветных кристаллов с
т. пл. 182—183°.
Таким образом, общий выход продукта равен 38,2 г, что
составляет 90% от теоретического.
Получение 2-фенил-4-№ -бензил-5-оксазолона [2]. Смесь
38,2 г (0,137 Л4) № -бензоил-D,L-фенилаланина и 382 мл ук-
сусного ангидрида загружают в колбу емкостью 0,5 л, снаб-
женную мешалкой и обратным холодильником. Массу нагре-
вают до 100° и выдерживают при этой температуре в течение
2 часов. Полученный раствор фильтруют от нерастворимых
примесей и упаривают в вакууме. Образовавшееся масло рас-
творяют в 380 мл петролсйного эфира (т. кип. 60—110°) и ох-
лаждают до 5—0°. Получают кристаллы 2-фенил-4-№ -бен-
зил-5-оксазолона в виде длинных игл.
Выход продукта равен 23 г, что составляет 68% от теоре-
тического; т. пл. 69—70°.
Получение ^-нафтилового эфира Nx -бензоил-D,L-фенил-
аланина [5]. Смесь 23 г (0,91 М) 2-фенил-4-бензил-5-оксазо-
лона и 13,7 г (0,95 М) fj-нафтола нагревают в фарфоровой
чашке до плавления и выдерживают при НО—115° до затвер-
девания реакционной массы. Растирают с 230 мл метанола и
охлаждают до 5°. Осадок отфильтровывают, промывают хо-
лодным метанолом. После перекристаллизации из 790 мл эти-
лового спирта получают белые кристаллы fj-нафтилового эфи-
ра №-бензоил-D,L-фенилаланина с т. пл. 155—157°.
Выход продукта равен 26 г, что составляет 73% от теоре-
тического.
Найдено, %: С—79.11; Н -5,40; N—3,91.
C26H21NO3. Вычислено, %; С,—78,95; Н-5,32: N 3,54.
По литературным данным, т. пл. р-нафтилового эфира
N* -бензоил-D,L-фенилаланина 157° [1].
ЛИТЕРАТУРА
1- Н. A. Ravin, Ph. Bernstein, А М. S е 1 i g m a n. J. Biol
Chern., 208, 1 (1954),
2. P. Адамс. Органические реакции, сб. 3, стр. 194.
3. Р. А. Матвеева, Я. И. Л any к, В. М. Степанов. Изв. АН
СССР, Химич, серия, № 3, 501 (1964).
Институт химии природных
соединений АН СССР
75
УДК 547.495.9.07
и-НИТРОАНИЛ ИД
N- - БЕНЗОИЛ -D, L- АРГИНИНА
Э. А. БАШКИР, Я. И. ЛАПУК, В. М. СТЕПАНОВ
NH=C-NH-(CH2)3-CH-CO-NH-^ ^-no2
NHg-HCl NHCOCeH6
Ci9HMNeO4-HCl M. в. 434,84
За последнее время большое распространение получил ме-
тод определения активности протеолитических ферментов с
помощью хромогенных субстратов. Для трипсина, например,
употребляют н-нитроанилид №-бензоил-D,Ь-аргинина[1]. Ме-
тоды получения этого субстрата, описанные в литературе, ис-
пользуют труднодоступные реактивы, такие, как диэтилфос-
фин, три-н-бутиламин [2] и дициклогексилкарбодиимид [1].
В предложенном нами методе применяются этиловый эфир
полифосфорной кислоты и диметилформамид, исключены
труднодоступные реактивы и достигнуты хорошие выходы.
СХЕМА СИНТЕЗА п-НИТРОАНИЛИДА N* -БЕНЗОИЛ-
D.L-АРГИНИНА
1. 2NH=C-NH-(CH2)3-CH-COOH
| I +2CeH6COCl+3NaaCO9—»
NH2 NH2-HC1
—+2 NH=C—NH-(CH2)9—CH—COONa
| | -f*4 NaCl+3 COs4-3 H,0
NHa NHCOCeHB
2. NH=C—NH—(CH2)8—CH—COONa
l । 4*hci—»
NHS NHCOC6H6
76
- NH=C-NH-(CH,),-CH-COOH+NaCl
I I
NHg-HCl NHCOCeHs
3. NH=C-NH-(CH,)8-CH-COOH
I I +h2n-< >-no2-
NH2-HC1 NHCOCeHB x=/
Этиловый эфир
полифосфориой КИСЛОТЫ NH = C—NH — (CH2),—
NH2-HC1
- CH-CO-HN-^-NO2+H3O
I —
NH—COC6HS
Характеристика основного сырья
D.L-Аргинин солянокислый, ч., ВТУ Роспромсовета
1199—59.
Бензоил хлористый, ч., ТУ МХП 92—51.
n-Нитроанилин, ч., ТУ М.ХП 1461—46.
N.N-Диметилформамид, ч., СТУ 79-606-Х—60.
Пятиокись фосфора, ч., МРТУ 6-09-22—62.
Условия получения
Получение N* -бензоил-D,L-аргинина [3]. В трехгорлую
колбу на 1 л, снабженную мешалкой и двумя капельными во-
ронками, загружают раствор 31,8 г (0,146 Л4) О,Ь-аргининав
180 мл воды. Затем, при перемешивании и охлаждении на ле-
дяной бане, добавляют одновременно в течение 2,5 часов
19,2 мл (0,167 Л1) хлористого бензоила и раствор 25,8 г угле-
кислого натра в 250 мл воды. Перемешивают еще 1,5 часа.
Подкисляют 30 мл концентрированной соляной кислоты по
конго красному. Небольшой осадок днбензоиларгинина от-
фильтровывают. Фильтрат дважды экстрагируют серным
эфиром по 400 мл для удаления бензойной кислоты. Далее
раствор нейтрализуют 30 мл концентрированного аммиака до
pH 8 по универсальной бумаге или потенциометру ЛП-58.
Раствор упаривают в вакууме до */з объема. Осадок отфильт-
ровывают, промывают 300 мл холодной дистиллированной во-
ды и сушат при 105°.
Выход N"-бензоил-D,L-аргинина равен 21—23,5 г, что со-
ставляет 50—55% от теоретического; т. пл. продукта 265°.
Получение этилового эфира полифосфориой кислоты [4]. В
Двухлитровую колбу, снабженную холодильником с хлор-
кальциевой трубкой, загружают 300 г пятиокиси фосфора,
77
300 мл хлороформа и 600 мл серного эфира. Смесь нагревают
на водяной бане 48 часов до образования прозрачного рас-
твора. Растворитель отгоняют. Получают 300 г этилового эфи-
ра полифосфорной кислоты в виде густого темного масла.
Получение абсолютного N,N-диметилформамида. Смесь
900 мл диметилформамида и 450 мл высушенного бензола
перегоняют, собирают фракцию при 153—155°, сушат окисью
бария в течение 1—2 дней, отфильтровывают и снова перего-
няют, собирая фракцию при 153—155°.
Получение п-нитро анилид а №-бензоил-О,Ь-аргинина. В
колбу емкостью 0,5 л, снабженную мешалкой, холодильником
с хлоркальциевой трубкой и термометром, загружают 15,5 <
(0,0555 Л1) N“ -бензоил-О,Ь-аргииина, 11,6 г (0,084 А4) п-нит-
роанилина, 100 мл абсолютного диметилформамида и 16 мл
эфира полифосфорной кислоты. Смесь нагревают до темпе-
ратуры 145—150° и выдерживают при этой температуре в те-
чение 1 часа. Образовавшийся раствор упаривают в вакууме
на водяной бане, тщательно освобождаясь от следов раство-
рителя. К полученному маслу добавляют 200 мл 0,5н. соляной
кислоты, нагревают на водяной бане (70—80°), перемешивая
содержимое колбы до тех пор, пока не наступит полная кри-
сталлизация масла, и оставляют на ночь при 4°. На следую-
щий день осадок отфильтровывают, промывают небольшим ко-
личеством воды, тщательно отжимают на фильтре и снова
промывают последовательно сначала 200 мл этилацетата, а
затем 100 мл эфира. После перекристаллизации из этилового
спирта с углем получают желтоватые кристаллы с т. пл. 249—
250°.
Выход я-нитроанилида № -бензоил-О,Ь-аргинина равен
13,5 г, что составляет 55% от теоретического.
Найдено, %: N—19,52; 19,27.
C19H22N60i-HCl. Вычислено, %: N-19,30.
Контроль чистоты, полученного продукта методом электро-
фореза на бумаге. Электрофорез проводят в течение 2 часов
в пиридин-ацетатном буфере с pH 5,6 (4 мл пиридина, 1 мл
уксусной кислоты довести водой до 1 л) при напряжении
1000 в и длине полосы 45 см. Сила тока составляет 0,3 ма[см
ширины полосы. Высушенную на воздухе электрофореграмму
просматривают в УФ на хемископе, после чего проявляют
соединения, содержащие свободную гуанидогруппу реакти-
вом Сакагуши. Электрофореграмму погружают в 0,1%-ный
раствор 8-оксихинолина в ацетоне, высушивают на воздухе и
опрыскивают раствором гипобромита (1 мл брома в 500 мл
0,5 и. едкого натра). Бензоиларгинин и его я-нитроанилид
проявляются красноватыми пятнами.
78
Наименование веществ 1 Проявление
в УФ в реак- тиве Сака- ту ши
Бензоиларгинин 0,5 см — —
л-Нитроанилидбензоил- аргинина 5 см + +
л-Нитроанилин 0,5 см* + —
* Желтая окраска.
ЛИТЕРАТУРА
1. Н. Tuppy, U. Wlesbauer, Е. W I n t е г s b е г g е г. Z. Physiol.
Chem., 329, 278 (19b2).
2. В. Erlanger, N. Kokowsky, W. Cohen. Arch. Biochem. and
Biophys., 15, 271 (I* 1 2 3 4 61).
3. N. M. Bergman., J. S. F rut on, H. P о 11 о k. J. Biol. Chem.,
127, 643 (1939).
4. O. Schramm, H. Grotsch, W. I’ollinann. Angew. Che-
mie, 74, 53(1962).
Поступила в июне 1964 г.
Институт химии природных
соединений АН СССР
УДК 547.725.07
5-НИТРОФУРАН-2-КАРБОНОВАЯ КИСЛОТА
5-Нитропирослизевая кислота
Б. В. КУРГАН, А. А. ГРУЗЕ, С. А. ГИЛЛЕР
'/-СООН
'О 7
C6HsNO5 м. в. 157,08
Имеется несколько возможных способов получения 5-нит-
рофуран-2-карбоновой кислоты. Клинкхардт [1] получил ее
при нитровании дегидрослизевой кислоты дымящей азотной
кислотой. Невысокие выходы 5-нитрофуран-2-карбоновой
кислоты получены при нитровании фуран-2-карбоновой кис-
лоты [2, 3]. Нитрование 5-сульфофуран-2-карбоновой кисло-
ты дает значительное количество побочных продуктов [3—5].
Гильман и Райт [6] получили 5-нитрофуран-2-карбоновую
кислоту с выходом 57% при окислении 5-нитрофурфурола с
помощью двухромовокислого натрия.
Нами было найдено, что 5-нитрофуран-2-карбоновая кис-
лота может быть получена с 68%-ным выходом из 5-нитро-
фурфуролдиацетата, который является промышленным про-
дуктом.
Вместо двухромовокислого натрия как окислителя можно
употреблять хромовый ангидрид, двухромовокислый калий
[7], а также перекись водорода [8].
СХЕМА СИНТЕЗА 5-НИТРОФУРАН-2-КАРБОНОВОЙ КИСЛОТЫ
O2N-^ >-CH(OCOCH3)s
-> ^-сб° + 2СНаСООН
80 О ХН
3O2N-^ 5-C^° + Na2Cr2O7 + 4H2SO4
0 11
30,N-^ ^-COOH + 2NaCr (SO4). % 4H2O
0 '
Характеристика основного сырья
5-Нитрофурфуролдиацетат.
Натрий двухромовокислый, ч., ГОСТ 4237—48.
Серная кислота, х. ч., ГОСТ 4204—48.
Условия получения
В трехгорлую колбу емкостью 3 л, снабженную механи-
ческой мешалкой и термометром, вливают 1200 мл воды и
396 мл концентрированной серной кислоты. Поддерживая
температуру 80°, добавляют 364 г (1,5 Л4) 5-нитрофурфу-
ролдиацетата и перемешивают при 70—80° до исчезновения
хлопьев 5-нитрофурфуролдиацетата. Потом в течение 1,5—2
часов небольшими порциями добавляют 447 г (1,5 М) тщатель-
но измельченного двухромовокислого натрия так, чтобы тем-
пература реакционной смеси не превышала 80°. После при-
бавления всего двухромовокислого натрия смесь при той же
температуре перемешивают еще 30 минут. После полного ох-
лаждения (см. примечание) густую кристаллическую темно-
зеленую массу фильтруют с отсасыванием. Осадок промыва-
ют ледяной водой несколько раз для удаления хромовых со-
лей и высушивают на воздухе.
Выход 5-нитрофуран-2-карбоновой кислоты равен 150—
160 г, что составляет 64—68% от теоретического; т. пл. 183—
184°.
По литературным данным, т. пл. продукта 183° [1]; 185—
185,5 [9].
Примечание.
Для полного охлаждения реакционную массу желательно оставить
на ночь.
ЛИТЕРАТУРА
1. J. К 1 i п k h а г d t. J. Pract. Chem., 25, 41 (1882).
2. H. В. Hill, R. White. J. Amer. Chem. Soc., 27. 193 (1902).
3. T. Sasaki. Bull. Chem. Soc. Japan, 27, 395 (1954).
4. H. В. H i 11, A. W. P a I m e r. J. Amer. Chem. Soc., 10, 409 (1888).
5. Датский пат. 83633: С. A., 52, 4695 (1953).
6. H. Gilman, Q. Wright. J. Amer. Chem. Soc., 52, 2550 (1930).
7. С. А. Г и л л e p, Б. В. К У р г а н. Н. О. С а л д а б о л. Изв. АН Латв.
ССР, 1959, As 3, 49.
8. Чехосл. пат. 99611; С. А., 56, 12857 d (1962).
9. В. Т. F г е u г е, J.R. Johnson. J. Amer. Chem. Soc., 53, 1142
(1931).
Поступила в июле 1964 г. Институт органического синтеза
АН Латв. ССР
6 Зак. Й4 81
УДК 547.361.07
3-£5-НИТРОФУРИЛ)-2-АЛЛИЛОВЫЙ СПИРТ
М. Я. БЕРКЛАВА, С. А. ГИЛЛЕР
O2NЧ>-СН=СН-СНгОН
(V
C7H5NO4 М. в. 167,12
Известен способ получения р-(5-нитрофурил)-2-аллилово-
го спирта путем восстановления [3-(5-нитрофурил)-2-акроле-
ина нзопропилатом алюминия по Мейервейну—ГТонндорфу
[1]. Выход составляет 66%. Воспроизведенные нами опыты
подтвердили эти данные, однако получение р- (5-нитрофурил) -
2-аллилового спирта по этому способу требует длительного
времени и аппаратуры для отделения ацетона.
Эти недостатки устраняются при использовании боргидри-
да натрия в качестве избирательно действующего восстанав-
ливающего агента (см. примечания 1 и 2) [2]. В качестве рас-
творителя применялся диоксап (см. примечание 3). Чистота
получаемого р-(5-нитрофурил)-2-аллилового спирта вполне
достаточна для того, чтобы он мог быть использован для
дальнейших синтезов.
СХЕМА СИНТЕЗА fi- (5-НИТРОФУРИЛ)-2-АЛЛ ИЛОВОГО СПИРТА
4 O3N-f У-СН = СН-СНО + NaBH4 —
НС1
NaB(O2N-У 7~СН=СН-СН.О)4
4 О '
4O2N-€ ^-СН=СН-СН,ОН
О '
Характеристика основного сырья
р-(5-Нитрофурил)-2-акролеин (см. примечание 4).
Боргидрид натрия, техн.
Диоксап, ч., ТУ ГКХ 1558—61.
Условия получения
В круглодонную трехгорлую колбу емкостью 0,5 л, снаб-
женную мешалкой, пропущенной через шариковый холодиль-
ник, термометром и капельной воронкой, помещают 2,3 г
(0,06 М) 70,4%-ного боргидрида натрия в виде тонкорастер-
того порошка и прибавляют 40 мл диоксана. В течение 40—60
минут из капельной воронки при постоянном перемешивании
прибавляют раствор 10 г (0,06 А4) р-(5-нитрофурил)-2-акро-
леина (т. пл. 115—117°) в 125 мл диоксана, температура
при этом была на уровне 21—30°. В этих условиях продол-
жают перемешивать реакционную смесь в течение 1 часа. За-
тем образовавшийся комплекс разрушают добавлением по
каплям 10 мл 1 н. раствора соляной кислоты. Выпавший оса-
док отфильтровывают и диоксан отгоняют при вакууме водо-
струйного насоса. Остаток в количестве 9,1 г представляет
собой сырой р-(5-нитрофурил)-2-аллиловый спирт в виде
красно-коричневой вязкой жидкости, которая' в холодильнике
легко превращается в кристаллическую массу оранжево-жел-
того цвета. Продукт перекристаллизовывают из 27,3 мл
(9,1X3) 75%-ного этилового спирта с добавлением активиро-
ванного угля.
Выход чистого р-(5-нитрофурил)-2-аллилового спирта ра-
вен 5,5 г, что составляет 54,2% от теоретического; т. пл. 57—
58°.
При м е ч а н и я:
1. Применялся технический боргидрид натрия, содержащий 70—80%
чистого продукта.
2. Разложение избытка боргидрида натрия проводилось одновремен-
но с разрушением образовавшегося комплекса добавлением 1 н. соляной
кислоты до окончания выделения пузырьков водорода.
[ 3. Восстановление можно проводить также в среде тетрагидропираиа
4. ?- (5-Нитрофурил)-2-акролеин приготовлялся по способу Гил.тера и
Вентера [3].
ЛИТЕРАТУРА
1. Т. Sasaki. С. А., 48, 14727 (1954); Pharm. Bull., 1, 346 (1953).
2. М. Я. Берклава, С. А. Гил л ер. Изв. АН Латв. ССР, сер. хим.,
№ 3, 349 (1963).
3. С. А. Гил л ер, К. К- В е и т е р. Изв. АН Латв. ССР, № 12 (137),
115 (1958).
Поступила в июне 1964 г. Институт органического
синтеза АН Латв. Сг-1'
83
УДК 547.722.5.07
5-НИТРОФУРФУРИЛОВЫЙ СПИРТ
М. Я. БЕРКЛАВА, С. А. ГИЛЛЕР
o2n-(z %-ch2oj]
C5H6NO4 М. в. 143,09
Известны два способа получения 5-нитрофурфурилового
спирта. Первый заключается в гидролизе 5-нитрофурфурил-
ацетата 5%-пой серной кислотой с выходом продукта 49—55%
[1]. Недостатком этого метода является его многостадий-
ность.
По другому способу [2] 5-нитрофурфуриловый спирт полу-
чают восстановлением 5-нитрофурфурола пзопропилатом
алюминия по Мейервейну—Понндорфу. Проведенная нами
проверка полностью подтвердила эти данные (максимальный
выход равен 77%), однако получение 5-нитрофурфурилового
спирта по этому методу требует длительного времени и при-
менения специальной аппаратуры для отделения образующе-
гося в ходе реакции ацетона.
Мы нашли, что этих недостатков можно избежать, при-
меняя в качестве восстанавливающего агента боргидрид нат-
рия [3].
СХЕМА СИНТЕЗА 5-НИТРОФУРФУРИЛОВОГО СПИРТА
4O2N-^o^-CHO + NaBH4
— NaB|OsN-^ ^-CHSO| — 4O2N-f ^-СН,ОН
\ 'О Л ЧО/
84
Характеристика основного сырья
5-Нитрофурфурол (см. примечание 1).
Боргидрид натрия, техн.
Диэтиловый эфир.
Условия получения
В двухлитровую круглодонную колбу, снабженную мешал-
кой, пропущенной через шариковый холодильник, термомет-
ром и капельной воронкой, помещают 13,4 г (0,35 М) 70%-ко-
го боргидрида натрия (см. примечание 2) в виде тонкорас-
тертого порошка и прибавляют 170 мл диэтилового эфира
(см. примечание 3). В течение 1 часа из капельной воронки
при постоянном перемешивании добавляют раствор 50 г
(0,35 М) свежеперегнанного 5-нитрофурфурола в 500 мл ди-
этилового эфира (температура реакционной смеси при этом
была на уровне 18—21°), затем нагревают на водя-
ной бане при температуре кипения эфира в течение 2 часов.
После охлаждения реакционной смеси добавляют к пей по
каплям 56 мл 1 н. соляной кислоты до pH 6—7 (см. примеча-
ния 4 и 5) с такой скоростью, чтобы температура в колбе не
превышала 30’. Образовавшийся осадок отфильтровывают от
фильтрата, отделяют эфирный слой и экстрагируют водный
слой 50 мл эфира. Объединенные эфирные вытяжки промы-
вают водой (2 раза но 50лъг), сушат над безводным сульфа-
том натрия и удаляют эфир отгонкой. Остаток в количестве
42,3г (85,1% от теоретического) представляет собой сырой
5-нитрофурфуриловый спирт в виде красно-коричневой вяз-
кой жидкости. После перегонки в вакууме при 140—14]°/4 мм
получают чистый 5-нитрофурфуриловый спирт в виде светло-
желтой жидкости.
Выход продукта равен 25,4 г, что составляет 50,1% от тео-
ретического; ио—1,577 (4); d—1,4041.
Примечания:
1. 5-Нитрофурфурол получают из 5-нитрофурфуролдиацетата путем
гидролиза разбавленной серной кислотой.
2. Пригоден технический боргидрид натрия, содержащий 70—80% чи-
стого вещества.
3. Восстановление можно проводить также в среде тетрагидроппрана
[3].
4. При действии соляной кислоты одновременно с разрушением ком-
плекса происходит разложение избытка боргидрида натрия, сопровождаю-
щееся выделением водорода.
5. В случае образования комков следует механическую мешалку на
время выключить, пока комки не расплывутся.
85
литература
1. 11. Gilman, G. F. VI right. J. Amer. Chem. Soc., 53, 19'23
(1931).
2. T. Sasaki. C. A., 48, 14727 (1954); Pharm. Bull., 1, 343 (1953).
3. M. Я- Берклава, С. А. Г нллер. Изв. АН Латв. ССР, сер. хим.,
№ 3, 349 (1963).
Посту! ила в июле 1961 г.
Институт органического
синтеза АН Латв. ССР
УДК 547.655.1.07
НИТХРОМАЗО
Бис-[2,7-(4-нитро-2-сульфо-1-азобензол)]-
1,8-диоксинафтал ин-3,6-дисульфокислота
В. И. КУЗНЕЦОВ, Н. Н. БАСАРГИН
^SOjI I он ОН HO3S х
OtN-^ /-N==N~-p' 4'/^-n = N-^ ^--NO2
HO3SZ ' So:sH
C2SHI4N6OI8S4 M. b. 778,64
Нитхромазо был предложен в качестве металлоиндика-
тора на барий при объемном определении сульфатов в при-
сутствии арсенатов, фосфатов [1, 2]. Определению мало меша-
ют небольшие количества селенитов и хроматов. Индикатор
может быть использован при определении сульфатов в супер-
фосфате. С применением нитхромазо разработан метод опре-
деления серы в фосфор- и мышьяксодержащих органических
соединениях [3], а также метод определения небольших коли-
честв серной кислоты в экстракционной фосфорной кислоте,
используемой в производстве минеральных удобрений [4].
Следует отметить, что отличительной особенностью нового
индикатора является его способность давать чувствительную,
контрастную цветную реакцию с барием в кислой среде да-
же при pH 1,7. Эта особенность обусловила возможность пря-
мого объемного определения сульфат-ионов на фоне больших
количеств фосфат-ионов и двукратных количеств арсенат-
ионов по отношению к сульфат-ионам. Описанные до сих пор
в литературе металлоиндикаторы взаимодействуют с барием
в менее кислых средах и поэтому не позволяют проводить та-
кое определение без предварительных операций по разделе-
87
нию или отделению этих ионов. Фосфаты и сульфаты в слабо-
кислых и нейтральных средах титруются одновременно рас-
твором солей бария.
При титровании сульфат-ионов в присутствии нитхромазо
последний в одинаковой степени может использоваться как в
виде натриевой, так и кальциевой соли. В условиях титрова-
ния, т. е. при pH 2 кальций ие образует комплекса с нитхро-
мазо.
Синтез нитхромазо был впервые осуществлен авторами .с
использованием способа повышения реакционной способности
комплексообразующих веществ в присутствии металлов [5].
Реагент получают азосочетанием кальциевой соли хромотро-
повой кислоты с диазонием n-нитроаиилии-о-сульфоната нат-
рия в присутствии гидрата окиси кальция.
СХЕМА СИНТЕЗА НИТХРОМАЗО
zSOsNH4 /SO3Na
OaN_^’_'^-NH1-|-NaOH--O2N—'^-NH2+NH3‘фН2О
SO3Na
02j\j_—NH2 2HC1 NaNO3 —>
SO3Na
o2n-
N.,C1 -f- NaCl + 2H2O
Ca
OH OH
О О
NaO3S
SO3Na
/ SQ3Na
2O2N-^ ^--N,C1 -I-
SO3Na
NaO3S'
88
CaCU OjN~
/Ca\ OHOH/Ca
Характеристика основного сырья
/г-Нитроанилин-о-сульфокислоты аммонийная соль, ч.,
СТУ 79-543—60.
Хромотроповой кислоты динатриевая соль, ч., ТУ МХП
4045—53.
Окись кальция, ч., ГОСТ 8677—58.
Хлористый кальций, ч., ГОСТ 4141—48 (СаСЬ • 6Н2О).
Нитрит натрия, ч., ГОСТ 4197—48.
Условия получения
Диазотирование. Смешивают 23,5 г (0,1 ЛГ) аммонийной
соли п-нитроанилии-о-сульфокислоты с 50 мл воды, добавля-
ют 4,1 г едкого натра. Смесь нагревают до кипения и кипятят
до удаления основного количества аммиака, затем к смеси
добавляют 50 мл воды и нагревают до полного прекращения
выделения аммиака (контроль по влажной универсальной
индикаторной бумаге). К раствору вновь добавляют 50 мл
воды и горячим фильтруют от механических примесей. К
фильтрату, охлажденному до 20—25° и имеющему pH— 7, до-
бавляют 7,3 г (0,106 А4) нитрита натрия.
Полученный раствор вливают в 30 мл концентрированной
соляной кислоты, предварительно охлажденной в ледяной во-
де, при интенсивном перемешивании. В момент смешения об-
разуется осадок, который быстро растворяется, и далее вы-
падает кристаллический темно-желтый осадок соли диазо-
ния. В процессе диазотирования реакционную смесь охлаж-
89
дают в ледяной бане, а затем перемешивают в течение — 2 ча-
сов при температуре около 10°. Осадок соли диазония от-
фильтровывают.
Азосочетание. В 200 мл воды растворяют 20 г (0,05 Л1)
очищенной динатриевой соли хромотроповой кислоты. До-
бавляя несколько капель 30%-ного раствора едкого натра,
добиваются полного растворения хромотроповой кислоты и
устанавливают pH раствора — 5,0 (контроль по универсаль-
ной индикаторной бумаге). К раствору добавляют 25 г
(0,12 Л4) хлористого кальция, растворенного в 20 мл воды,
и доводят pH до~ 5,0. Суспензию соли диазония в 30 мл
воды быстро, в один прием, добавляют при интенсивном пе-
ремешивании к полученному раствору кальциевой соли хро-
мотроповой кислоты.
Смесь окрашивается в красный цвет вследствие образова-
ния моноазосоединения. Далее при перемешивании и темпе-
ратуре~15° небольшими порциями добавляют в течение 1,5
часа суспензию гидрата окиси кальция (см. примечание 1).
Смесь приобретает синюю окраску и загустевает, когда вве-
дено ~-70% всего количества гидрата окиси кальция. При
введении всей суспензии смесь вновь разжижается. Переме-
шивают около 3 часов и оставляют стоять на ночь при ком-
натной температуре. На следующий день реакционную смесь
перемешивают 2,5—3 часа и проводят качественную пробу
для оценки состава реакционной смеси (см. примечание 2).
Выделение продукта проводят подкислением реакционной
смеси 30 мл концентрированной соляной кислоты. Осадок
отфильтровывают и отжимают. Полученный продукт в воз-
душно-сухом состоянии весит 19,0—19,5 г. Он представляет
собой кальциевую соль нитхромазо по сульфогруппам с при-
месью моноазосоединения.
Для очистки от моноазосоединения весь продукт растворя-
ют при нагревании в 350 мл воды с добавлением 10 г без-
водного углекислого натрия, фильтруют раствор горячим че-
рез стеклянный фильтр № 2. Фильтрат при постепенном ох-
лаждении загустевает. Выделившийся реагент в виде кальци-
евой соли отфильтровывают через стеклянный фильтр № 2,
отжимают, промывают иа фильтре 20 мл холодной воды.
10.мл этилового спирта и затем 40 мл I М соляной кислоты.
Осадок промывают далее 7—10 мл воды и сушат при 60—70°.
Выход очищенного препарата составляет 14—14,5 г.
Нитхромазо в виде кальциевой соли содержит в молекуле
два атома кальция и представляет собой кристаллический по-
рошок с характерным темно-зеленым цветом и металлическим
отблеском. Препарат заметно растворяется в холодной воде,
однако менее растворим, чем натриевая соль или свободная
кислота. Водные концентрированные растворы препарата
имеют темно-синий цвет.
90
Для перевода кальциевой соли нитхромазо в свободную
кислоту все количество препарата растворяют при нагрева-
нии в 400—500 мл воды с добавлением нескольких капель
30%-ного раствора едкого натра. Раствор горячим выливают
при перемешивании в 0,5 л концентрированной соляной кис-
лоты. Выпавший осадок свободной кислоты нитхромазо от-
фильтровывают через стеклянный фильтр № 2, отжимают,
промывают вначале 20 мл 1 М соляной кислоты, затем 10 мл
0,1 М соляной кислоты и 7—10 мл спирта. Выход воздушно-
сухого препарата равен 12,5—13,0 г.
Нитхромазо в виде свободной кислоты представляет собой
кристаллический порошок темно-фиолетового цвета с брон-
зовым отблеском. Растворимость в воде намного больше, чем
кальциевой соли.
В виде тетранатриевой соли нитхромазо получают выса-
ливанием с помощью хлористого натрия из концентрирован-
ных растворов кислоты, имеющих pH 5—7,0.
Примечая и я:
1. Суспензию гидроокиси кальция ютовят смешением 7 г (0,12 Л1)
окиси кальция и 40 мл воды. Ее следует прибавлять равномерно и малы-
ми порциями. Введение сразу всего количества гидрата окиси кальция или
больших его порций приводит к разложению если диазония, так как ре-
акция азосочетания идет во времени.
2. Капилляром отбирают реакционную смесь и каплю ее помещают и
центр круга диаметром 8—10 с.н из фильтровальной или хроматографиче
ской бумаги. Затем промывают 0,1 Л1 соляной кислотой, вводя ее непре-
рывно из капилляра в центр образовавшегося пятна, пока не получат хро-
матографический круг диаметром 7- 9 см. При нормальном течении син-
теза в хроматограмме имеется внешнее кольцо, окрашенное в относи-
тельно слабый розово-красный цвет, соответствующий мопоазопродукту.
Центральная зона окрашена в сине-фиолетовый цвет, соответствующий
нитхромазо.
Если хроматограмма имеет в центре буро-красную, а на периферия
буро-желтую окраску, это указывает на наличие только моноазопродукта
и разложившегося диазосоединения.
ЛИТЕРАТУРА
1. В. И. Кузнецов, Н. Н. Басаргин. Авт. евпд. № 165566; Билл,
нэобр., К» 19 (1964).
2. В. И. Кузнецов, Н, II Басаргин. Заводск. лаборатория,
31, 538 (1965).
3. Н. Н. Басаргин, К. Ф. Новикова. Ж. аналит химии, 20
(1965).
4. Н. Н. Басаргин, Н. Л. Никитина. Заводск. лаборатория,
31 (1965).
5. В. И. Кузнецов, А. А. Н е м о д р у к. Ж. общ. химии, 26, 3285
(1956).
Поступила в феврали 1Й05 г. Институт геохимии и аналит. химии
им. В. И. Вернадского АН СССР
УДК 547.313.3.07
2-ОКСИПРОПИЛАМИН
и ДИ-(2-ОКСИПРОПИЛ)АМИН
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
-CH3-CH-CH2NH2
1
ОН
CsH9NO М. в. 75,11
/СНз-СН-СНзХ^К'П
I
I он /
\ /2
C6H1sNO2 М. в. 133,19
Реакция окиси пропилена с аммиаком описана Красуским
[1]. Нами проверены литературные данные [1, 2] и внесены
небольшие изменения и уточнения в методику получения ок-
сипропиламинов.
Оксипропиламины находят применение в качестве фото-
реагентов для руд цветных металлов, поверхностно-активных
веществ и для борьбы с вредителями сельского хозяйства.
СХЕМА СИНТЕЗА 2-ОКСИПРОПИЛАМИНА И ДИ-(2-ОКСИПРОПИЛ)
АМИНА
СН8-СН -CH, NH . - > СНз-СН-СН,- NH,
\о/ I
и ОН
CHbCH-CHs4CH3-CH-CH,-NH3-> /СН3-СН- CH,\=NH
\п/ I 1
° он он /
Характеристика основного сырья
Аммиак водный, ч.д.а., ГОСТ 3760—47, насыщенный рас
твор.
Окись пропндена, свежеперегнанная, т. кип. 35°,
92
Условия получения
В трехгорлую колбу, снабженную механической мешал-
кой, термометром и капельной воронкой, загружают 830 мл
насыщенного раствора аммиака (12 Л1) и медленно прибав-
ляют 180 мл (2,6 Л1) окиси пропилена. Во время прибавления
окиси пропилена температуру реакционной смеси поддержи-
вают на уровне 0—3°. После прибавления всего количества
окиси пропилена температуру реакционной смеси медленно, в
течение трех часов, повышают до 25° и выдерживают при этой
температуре 15—17 часов. Затем медленно отгоняют непро-
реагировавший аммиак и воду (см. примечание).
Получают 180,4 г «сырых» аминов, которые подвергают
разгонке:
2-оксипропиламип при 158 160' с выходом 65 г, что сос-
тавляет 34%; di20—0,9615;
ди-(2-оксипропил) амин при 150—151°/23 мм с выходом
56 г, что составляет 32%; т. пл. 44,5—45,5°;
три-(2-оксипропил) амии при 190—196°/23 мм с выходом
7 г, что составляет 4%; т. пл. 50—52°.
В остатке находятся полимерные оксипропиламины.
Примечание. Воду отгоняют до температуры 105° в парах. В рас-
творе в это время температура повышается до 125°.
ЛИТЕРАТУРА
1. К. А. К р асу ск и й. Ж. общ. химии, 6, 460 (1936).
2. М. С. Малиновский, Окиси олефинов п их производные, М.,
Госхимиздат, 1961.
Поступила в октябре 1964 г,
Донецкий филиал 11РВА
УДК 547.831.7.07
8-ОКСИХИ НАЛЬДОНИТРИЛ
и 8-ОКСИХИНАЛЬДАМИД
Нитрил и амид 8-оксихинальдиновой
(8-оксихинолин-2-карбоновой) кислоты
И. А. КРАСАВИН, В. Af. ДЗИОМКО. Ю. П. РАДИН
'Ч./ xNzZXcN
ОН
CI0HgN2O М. В. 170,17
I k 1
'у7 '''CONH.rH.O
ОН
C]nH,4N2O=-H2O М. в. 206,20
В литературе 8-оксихинальдоиитрил и 8-оксихинальдамид
не описаны. Они были получены нами одновременно с сум-
марным выходом 70,6—73% при взаимодействии метосульфа-
та 8-окси-1-метоксихинолиния с цианидами щелочных метал-
лов в водном растворе. Реакция этого типа, открытая в
1958—1959 гг. [1—3], к производным 8-оксихинолина еще не
применялась.
Чистый 8-оксихинальдонитрил представляет собой блед-
но-желтоватые иглы или пластинки с т. пл. 134,5—135,5°, хо-
рошо растворимые в большинстве органических растворите-
лей.
Очевидно, что 8-оксихинальдамид, присутствующий в про-
дукте реакции цианирования, возникает в результате споп-
94
тайного гидролиза нитрила в щелочной среде. При кратко-
временном кипячении 8-оксихинальдонитрила с разбавлен-
ной щелочью амид образуется с выходом 79—84%.
Чистый 8-оксихинальдамид представляет собой бледно-
желтые кристаллы с т. пл. 215,5—216°; в отличие от нитри-
ла он нерастворим в кипящем гептане.
Для доказательства того, что при цианировании солей
8-окси-1-метоксихинолиния циангруппа вступает именно во
2-е положение хинолинового ядра, продукт реакции был
превращен в его О-ацетилыгое производное (т. пл. 130,5—•
131°). Последнее оказалось идентичным с 8-ацетоксихиналь-
донитрилом (т. пл. 131 —131,5°), который был получен нами
независимым путем--при кипячении 8-оксихинальдоксима с
уксусным ангидридом. Расположение же заместителей в 8-ок-
сихинальдоксиме (т. пл. 167,5—168°) не вызывает сомнений,
так как он был синтезирован из 8-оксихинальдина [4].
Кроме того, строение 8-оксихинальдонитрила и 8-оксихи-
нальдамида подтверждено превращением их в известную 8-ок-
сихинальдиновую кислоту, т. пл. 218° с разл. (По литера-
турным данным, т. пл. 211° с разл. [5]; 218—219° |6]).
СХЕМА СИНТЕЗА 8-ОКСИХИНАЛЬДОНИТРИЛА
% CHjSO,
он осн3
ONa
.z \ / °-
| |1 | ЧСНэСООН I I | + CHgCOONa
Vх W \ CN Vх 4 n/- \CN
ONa ОН
Условия получения
Внимание! Во время синтеза возможно выделение ядови-
того цианистого водорода, поэтому работу необходимо прово-
дить в хорошо действующем вытяжном шкафу.
Трехгорлую колбу емкостью 1 л снабжают мешалкой с
ртутным затвором, капельной воронкой п отводной трубкой,
95
присоединенной к поглотительной склянке со щелочью. В кол-
бе растворяют 14,7 г (0,3 Af) чистого цианистого натрия (см.
примечание 1) в 60 .ил воды и охлаждают в бане с ледяной
водой. Прибавляют по каплям за 1 час раствор 28,7 г (0,1 Л4)
метосудьфата 8-окси-1-метоксихинолииия (см. примечание 2)
в 60 .ил воды при размешивании и выдерживают еще 1 —1,5
часа при 0—2°. Затем медленно добавляют 300—400 мл
30%-ной уксусной кислоты до pH 4—5, осадок отсасываю’
(см. примечание 3), промывают холодной водой и подсуши-
вают в течение ночи в вакуум-эксикаторе. Подсохшее веще-
ство (15,8—16,2 г) растворяют без нагревания в 600 мл хло-
роформа, раствор взбалтывают с 1 г активированного угля,
фильтруют и фильтрат экстрагируют 600 .ил холодного 2%’-
ного раствора едкого кали (2 раза по 200 мл и 2 раза по
100 мл). Щелочной экстракт взбалтывают с активированным
углем, фильтруют, нейтрализуют при охлаждении 10%-ной
уксусной кислотой и доводят pH до 5- 6. Выпавший осадок
отсасывают, промывают холодной водой н высушивают в ва-
кууме над серной кислотой.
Выход сухого продукта, представляющего собой смесь
нитрила и амида, равен 13,4—14,1 г. Его помещают в кол-
бу с обратным холодильником, кипятят с 1600—1800 мл
н-гептана и фильтруют в горячем виде от нерастворившего
ся 8-оксихннальдамида (2,6—3,2 г, 12,6—15,5% от теорети-
ческого; см. примечание 4). Кристаллы, выделившиеся из
фильтрата при охлаждении и стоянии, отсасывают, промы-
вают гептаном и высушивают в вакууме над парафиновыми
стружками.
Выход 8-оксихинальдонитрила с т. пл. 132,5—133,5° равен
8,9 9,7 г, что составляет 52,3—57% от теоретического. Упа-
ривание маточного раствора дает еще 0,5—0,9 г вещества с
т. пл. 129,5—130,5°. Общий выход нитрила равен 9,5—10,2
(55,8—60%).
После повторной перекристаллизации основной части про-
дукта из 900—1100 мл циклогексана получают 7,7—8,2 г ни-
трила с т. пл. 133,5—134° (см. примечание 5).
СХЕМА СИНТЕЗА 8-ОКСИХИНАЛЬДАМИДА
ОН ок
I
^/Xn^XCONH3-H2O
9G ОН
Условия получения
Колбу, содержащую 1,70 г (0,01 Л1) чистого 8-оксихиналь-
допитрила (см. примечание 6) и 30 мл 1и. раствора едкого
кали, снабжают термометром и помещают в баню, заранее
нагретую до 85—90°. Содержимое колбы нагревают с разме-
шиванием в течение 5 минут при 80° (см. примечание 7), ох-
лаждают до 5—8° и нейтрализуют 10°/о-ной уксусной кисло-
той до pH 7. Выпавший светло-серый осадок отфильтровыва-
ют, промывают водой и высушивают в вакуум-эксикаторе.
Выход сырого продукта равен 1,64—1,73 г, что составляет
79,6—84% от теоретического; т. пл. 212—212,5°.
Для очистки вещество растворяют в 60 мл кипящего аце-
тона, обрабатывают активированным углем и фильтруют. К
фильтрату прибавляют 180 мл воды и оставляют для кри-
сталлизации.
Получают 1,17—1,23 г (56,8—59,7%) 8-оксихинальдамида
с т. пл. 215—215,5° (см. примечание 8).
Примечания: .
1. Вместо цианистою натрия можно применить эквивалентное коли-
чество (19,5 г) цианистого калия.
2. Применялся сырой метосульфат 8-окси-1-метоксихинолипия, полу-
ченный, как описано в настоящем сборнике.
3. Жидкость содержит синильную кислоту, поэтому фильтрование
следует проводить с соответствующими предосторожностями, а фильтрат
обезвредить.
4. 8-Оксихпнальдамид можно очистить перекристаллизацией из 25%-
кого водного ацетона, как описано в следующем разделе.
5, Чистый образец 8-окспхинальдонитрила с т. пл. 134,5—135,5° был
приготовлен перекристаллизацией пз гептана п проанализирован.
Найдено, %: С-70,79; 70,85; Н-3,59;3,89; N-16,48; 16,76.
Ci0H6N2O. Вычислено, %: С—70,58; Н-3,55; N—16,46.
6. Выгоднее использовать неперскристаллизованный 8-оксихинальдо-
нитрнл. Например, сырой нитрил, полученный из 11,5 г (0,04 Л1) мето-
сульфата 8-оксн-1-метоксихинолнния и очищенный только экстракцией из
хлороформа, дал 5,1 г (61,8% от теоретического, считая на метосульфат)
амида с т. пл. 211,5—212,5°. После перекристаллизации пз 150 мл ацетона
и 600 мл воды было получено 3,8 г (46%) вещества с т. пл. 214,5—215,5°
и 0,6 г (из маточника) с т. пл. 213—213,5°. Общий выход равен 4,4 г
(53,3%, считая на метосульфат 8-окси-1-мстоксихинолиння).
7. Длительное нагревание со щелочью приводит к образованию 8-ок-
сихинальдиновой кислоты.
8. Чистый образец 8-оксихинальдамида с постоянной т. пл. 215,5—
216° был приготовлен перекристаллизацией из водного ацетона. Судя по
данным анализа, он кристаллизуется с одной молекулой воды.
Найдено, %: С- 58,72; 58,55; Н 5,07; 4,88; N —13,77;
13,57.
CioHjNoOo-Н2О, Вычислено, %: С - 58,25; Н—4,89; N—13,59.
ЛИТЕРАТУРА
1. W. Е Feelv, Е. М, Beavers. J. Amer. Chern. Soc., 81, 4004
(1959).
7 Зак. 684 97
2. Пат. США 2991285 (1958); Chem. Abstrs, 56, 7282 1 (1962).
3. Т. О к a m о t о, Н. Т a n i. Chem. Pharm. Bull. (Tokyo), 7, 130 (1959);
Chem. Abstrs, 54, 22644 d (1960).
4. В. M. Дзиомко, И. А. Красавин. Химические реактивы и
препараты, вып. 26, М., ПРЕД, 1964, стр. 29.
5. Н. М. Irving, A. R. Pinning ton. J. Chem. Soc., 1954, 3782.
6. A. B. Durkee. J. Agric. a, Food Chem., 6, № 3, 194 (1958);
РЖхим, 1959 , 2366.
Поступила в марте 1965 г. ИРЕА
УДК 547.541.21.07
ПИКРАМИН Р
2,4-Динитрофен ол-(6-азо-1 )-2-
нафтол-3,6-дисульфокислота
Ю. М. ДЕДКОВ, С. Б. САВВИН
CleHI0N4O12S2
М. в. 514,40
Пикрамин Р синтезирован в Институте геохимии и анали-
тической химии им. В. И. Вернадского и предложен в качест-
ве реактива для фотометрического и экстракционно-фотомет-
рического определения циркония и ряда других элементов.
Пикрамин Р получают азосочетанием диазотированной
пикраминовой кислоты с Р-солью в щелочной среде.
СХЕМА СИНТЕЗА ПИКРАМИНА Р
o2n он
NH8+2HC14-NaNO5—
OaNZ
O2N ОН
-> ^-N2Cl+NaCl+2HaO
99
o2n oh ho so3h
N2C1+ / +Na>c°3
О2/ х_ У
SOsH
o2n он но so8h
-> N=N-^_J+NaCl + NaHCO3
О2/ "СЗ
SO3H
Характеристика основного сырья
Пикраминовая кислота, ч., ТУ ГКХ 1576—61.
Р-соль (натриевая соль), ч., ВТУ ХСНХ 361—60.
Натрий углекислый, безводный, ч., ГОСТ 83—41.
Натр едкий, ч., ГОСТ 4328—48.
Нитрит натрия, ч., ГОСТ 4197—48.
Условия получения
Диазотирование. 100 г (0,44 М) пикраминовой кислот
суспендируют в 400 мл 50%-ного этанола, приливают 100м
соляной кислоты (уд. в. 1,19) и тщательно перемешивают
При постоянном перемешивании и при охлаждении до 15° к
суспензии в течение 1 часа приливают НО мл 5н. раствора
нитрита натрия, следя за тем, чтобы реакция среды все вре-
мя оставалась отчетливо кислой. Через 20 минут пробой с
йодкрахмальной бумажкой' убеждаются в наличии избытка
азотистой кислоты. Через 40 минут избыток азотистой кисло-
ты снимают, добавляя некоторое количество пикраминовой
кислоты.
Сочетание. Растворяют 200 г Р-соли (0,61 М в пересчете
на 100%-ную) в 2 л воды с добавлением углекислого натрия
до нейтральной реакции, раствор фильтруют и переносят в
емкость па 5 л. Туда же вносят еще 150 г углекислого натрия
и 10 г едкого натра, перемешивают раствор 10 минут и затем
в течение 2 часов при постоянном перемешивании вносят пор-
циями по 20—35 мл суспензию диазосоли из пикраминовой
кислоты. Перед внесением первой порции суспензии диазосо-
ли к раствору приливают 20 мл теплого 40%-ного едкого нат-
ра. Вторые 20 мл суспензии вносят только после начала ре-
100
акции, о чем судят по изменению окраски раствора в корич-
невую или малиново-коричневую. После внесения каждых
100 мл суспензии диазосоли в раствор вводят 10 мл 40%-ного
едкого натра. Реакция среды должна быть все время щелоч-
ной. Затем реакционную массу перемешивают еще 3 часа и
оставляют на ночь. После ночной выдержки смесь при посто-
янном перемешивании подкисляют соляной кислотой (уд. в.
1,09) до кислой реакции по конго, охлаждают и отсасывают
выделившийся реактив. Его дважды переосаждают, растворяя
в 3—4 л воды с добавлением 50 мл 25%-ного аммиака и вы-
деляя соляной кислотой и хлористым аммонием (в качестве
высаливателя). Сушат реактив на воздухе при 70°. Чистоту
реактива проверяют хроматографически на бумаге, а кон-
центрацию основного вещества—титрованием солями меди в
1н. соляной кислоте. Исходя из стехиометрического соотно-
шения реакции комплексообразования (реактив: Си=1:1)
рассчитывают процентное содержание основного вещества
(на кислоту или аммониевую соль).
Поступила в декабре 1964 г. ГЕОХИ АН СССР
УДК 547.821.2.07
2-СТИРИЛПИРИДИНЫ
>4. к. ШЕЙНКМАН, А. Н. РОЗЕНБЕРГ, Л. С. ЕМЕЛЬЯНОВА
1 11-СН=СН-^ ^—N(CH3)2
2-(га-Диметиламиностирил) пиридин
Cj6H1sN2 М.в, 224,30
i 11-сн=сн-^~хх_снз
2-(п-Метилстирил) пиридин
CUH13N М.в. 195,26
// X
1 II—СН=СН—')-СН(СНз),
xNZ х=/
2-(п-Изопропилостирил) пиридин
CleHrN М.в 233,31
f II-СН==СН—no2
XN/ \ = /
2-(п-Нитростирил) пиридин
C13Hi0N2O3 М.в 226,23
2-Стирилпиридины, замещенные в бензольном ядре, могут
быть использованы как комплексообразователи, для синтеза
пиридин-2-альдегида, пиколиновой кислоты и различных фи-
зиологически активных веществ,
102
Обычно 2-стнрилпиридины получают конденсацией а-пи-
колина с ароматическими альдегидами в среде уксусного ан-
гидрида либо нагреванием смеси пиколина и альдегида
хлористым цинком в запаянных ампулах [1].
Предлагаемый нами метод увеличения активности метиль-
ных групп, связанных с гетероциклом [2], оказался примени-
мым и в случае а-пиколина. Для облегчения конденсации
а-пиколина с ароматическими альдегидами его следует пере-
вести в N-ацильную соль, которая гладко реагирует с ними,
образуя соответствующие стирилпиридины. После проведе-
ния реакции ацильный остаток легко отщепляется кислым
или щелочным гидролизом. Метод весьма прост и удобен,
время реакции в ряде случаев значительно сокращено по
сравнению с другими методами.
СХЕМА СИНТЕЗА 2-СТИРИЛПИРИДИНОВ
С,,ЩСОС1
//
i II гн
I
осс6н6
n-R—СЙН4СНО
-сн=сн—
С1
JI-CH=CH-^ ^-R
где R —СН3; NO2; СН(СН3),; N(CH3)2.
Характеристика основного сырья
а-Пиколин, ч., ВТУ РУ 771—52 (см. примечание 1).
Бензоил хлористый, техн., свежеперегнанный, т. кип. 194—
97° (см. примечание 2).
и-Диметил аминобензальдегид, ч., ТУ МХП 2679—51.
n-Нитробензальдегид, ч., ТУ МХП 2969—51.
Кумиловый альдегид, свежеперегнанный, т. кип. 236—237°.
Толуиловый альдегид, свежеперегнанный, т. кип. 204—205°.
Условия получения
Синтез 2-(п-диметиламиностирил) пиридина. К смесн2лы
а-пиколина и 2,34 мл хлористого бензоила добавляют раствор
103
4 г n-диметиламинобензальдегида в 5 мл абсолютного а-пико-
лина. При этом появляется красное окрашивание и темпера-
тура реакционной смеси самопроизвольно поднимается до
45—50°. После прекращения разогревания реакционную смесь
выдерживают 5 часов на масляной бане при температуре 155”
(±2°), затем разлагают концентрированной соляной кисло-
той, отгоняют с водяным паром образующуюся при гидроли-
зе бензойную кислоту и бензальдегид, подщелачивают и
вновь отгоняют с водяным паром. Осадок в перегонной колбе
отделяют, многократно отмывают горячей водой от неоргани-
ческих солей, кипятят с активированным углем в растворе со-
ляной кислоты, затем осаждают раствором аммиака, отделя-
ют и тщательно высушивают вначале на пористой пластинке,
а затем в вакууме до постоянного веса. Выход 2-(и-диметил-
аминостирил) пиридина после перекристаллизации из пириди-
на равен 2,8 г, что составляет 62,5% от теоретического; т. пл.
137—139°. После вторичной кристаллизации продукта из
спирта получают светло-желтые пластинки с т. пл. 139° [1];
360 мм; lgЕ 4,27 (в спирте). При проведении реакции
в среде диметилформамида выходы не превышают 10—15%,
как это указано ранее [2].
Получение 2-(п-метилстирил)пиридина. По аналогичной
методике из 2 мл а-пиколина, 2,34 мл хлористого бензоила и
2,36 мл толуилового альдегида получают 1,43 г (36%) 2-(п-
метилстирил)пиридина с т. пл. 88° (из петролейного эфира).
Получение 2-(п-нитростирил)пиридина. Продукт получают
так же, как и 2-(n-диметиламиностирил) пиридин. Из 2 мл.
а-пиколина, 2,34 мл хлористого бензоила и 4 г гг-нитробенз-
альдегида выход 2- (n-нитростирил)пиридина равен-2,3 г, что
составляет 50% от теоретического; т. пл. 123° (из бензола).
Получение 2-(п-изопропилостирил)пиридина. Как описано
выше, из 4,85 мл а-пиколина, 5,85 мл хлористого бензоила и
7,41 г кумилового альдегида в среде а-пиколина (5 мл) полу-
чают 4 г 2-(п-изопропилостирил) пиридина, который очищают
перегонкой в вакууме.
Выход чистого 2-п-(изопропилостирил) пиридина равен
3,6 г, что составляет 32% от теоретического; т. пл. 46—47°, что
совпадает с литературными данными [3].
Примечания:
1. а- Пиколин следует тщательно сушить над едким кали н кипятить с
окисью кальция и перед использованием перегонять на колонке.
2. Вместо хлористого бензоила можно использовать любой имеющий-
ся хлористый ацил.
ЛИТЕРАТУРА
1. J. L. W i 11 i a m s, R. A. Adel, J. М. С a rl s о n, G. A. R е у п о 1 d s,
D. G. В о г d е n, J. A. F о г d. J. Organ. Chem., 28, 3S7 (1963).
2. A. H. К ост, А. К- Шейнкман. Ж- общ. химии, 33, 2077 (1963).
3. В а с к е. Вег., 34, 1894 (1901).
Поступила в сентябре 19G4 г,
104
Донецкий филиал ИРЕА
УДК 547.655.1.07
СУЛЬФОХЛОРФЕНОЛ с
2,7-Бис( азо-2-окси-3-сульфо-5-хлорбензол) -
1,8-диоксинафталин-3,6-дисульфокислота
С. Б. САВВИН, Ю. М. ДЕДКОВ
HO3Sx ^ОН НО\ хОН НО\ ^ЗОзН
HO3S SO3H
CaaHl4N4OieS4Cl2
М. в. 789,53
Сульфохлорфенол С [1—3] применяется для фотометриче-
ского и экстракционно-фотометрического определения цирко-
ния [2,4—6], ниобия [1,7] и скандия [3,8]. Особенностью реа-
гента при определении указанных элементов является его вы-
сокая чувствительность, возможность определения в сильно-
кислых средах, что обеспечивает хорошую воспроизводимость
и надежность результатов.
Синтез реагента осуществляется действием избытка соот-
ветствующего диазосоединения на хромотроповую кислоту в
среде карбоната натрия, едкого натрия или гидроокиси каль-
ция [9]. При синтезе сульфохлорфенола С азосочетание идет
хорошо в присутствии гидроокиси кальция даже при эквимо-
лярных соотношениях компонентов. В содовой среде или в
растворе едкого натра сочетание идет значительно труднее.
СХЕМА СИНТЕЗА СУЛЬФОХЛОРФЕНОЛА С
HO3S он
2 ^_^-NH2+2NaNO8+4HCl->-
&
105
HO8S он
-> 2 N2C1 + 4H,0 + 2NaCl
cf
HO OH HO3S OH
1 к х_/
+ 2^_^-NCl+ Ca(OH)2->
/4/w\ /“
HO3S SO3H Cl
HO3S OH HO OH HO SO8H
\_/ II \_/
-> ^-N =N-^X'|i/^-N=N-^ ^+CaClH-H2O
/= /VV\ =\
Cl HO3S SOsH Cl
Характеристика основного сырья
2-Амино-4-хлор-6-сульфофепол, ВТУ ТСНХ 67—59 (см.
примечание).
Нитрит натрия, ч., ГОСТ 4197—48.
Кальций окись, ч., ГОСТ 8677—58.
Хромотроповой кислоты динатриевая соль, ч„ ТУ МХП
4045—53.
Кислота соляная, ч., ГОСТ 3118—46.
Условия получения
Диазотирование. 14 г 2-амино-4-хлор-6-сульфофенола
(0,0564 М, считая на 90%-ный препарат, что соответствует
5%-ному избытку по отношению к хромотроповой кислоте)
тщательно растирают до получения однородной пасты с 20 мл
концентрированной соляной кислоты и 100—150 мл воды. По-
лученную пасту охлаждают в ледяной бане и медленно, при
хорошем размешивании, добавляют к ней через стеклянную
трубочку, опущенную до дна сосуда, раствор 3,9 г (0,0566 AI)
нитрита натрия в 10—20 мл воды.
К концу приливания раствора нитрита натрия процесс ди-
азотирования замедляется. Стакан вынимают из ледяной ба-
ни и после добавления очередной порции раствора нитрита
натрия и перемешивания дают постоять 5—10 минут. Коней
диазотирования устанавливают пробой с йодкрахмальной бу-
мажкой при наличии избытка азотистой кислоты. По мере
диазотирования вещество почти нацело переходит в раствор.
106
По окончании диазотирования раствор отфильтровывают, не-
большой осадок отбрасывают, а раствор используют для по-
следующего азосочетания. Хранить раствор диазосоставляю-
щей можно не более 2—3 часов, добавляя для охлаждения
небольшие кусочки льда.
Сочетание. Готовят пушонку. Для этого 20 г окиси каль-
ция постепенно вводят в стакан со 150 мл нагретой почти до
кипения воды при одновременном хорошем перемешивании.
Необходимо соблюдать осторожность, поскольку процесс га-
шения извести сопровождается нагреванием. Суспензию гид-
роокиси кальция в воде охлаждают до комнатной температу-
ры или до более низкой, используя ледяную баню.
Затем 10 г динатриевой соли хромотроповой кислоты
(0,0275 М, считая на 100%-ную) растворяют при слабом на-
гревании в 30—50 мл воды и прибавляют приблизительно
'/4—'/з часть приготовленной пушонки. Стакан помещают в
ледяную баню и при хорошем перемешивании добавляют од-
новременно раствор диазония и пушонку, следя за тем, чтобы
реакция среды была постоянно щелочной. Для предотвраще-
ния нагревания реакционной массы в стакан добавляют ку-
сочки дробленого льда. Раствор приобретает вначале фиоле-
тово-красный, затем синий цвет. Оставляют стоять при ком-
натной температуре па 1—2 часа.
Далее реакционную массу подкисляют, добавляя */б—'/«
часть по объему концентрированной соляной кислоты и тща-
тельно размешивают для растворения кальциевых солей.
Раствор должен отстояться, для чего его следует оставить на
ночь. На следующий день выпавший в осадок продукт от-
фильтровывают, 2—3 раза перекристаллизовывают, растворяя
его в воде с добавлением соды, фильтруя и затем осаждая
при медленном добавлении из капельной воронки соляной
кислоты.
Чистота реактива проверяется хроматографически на бу-
маге, а концентрация основного вещества—титрованием соля-
ми циркония в 1 н. соляной кислоте. Исходя из стехиометри-
ческого соотношения реакции комплексообразования (реак-
тив: Zr= 1:1), рассчитывают процентное содержание основ-
ного вещества (на кислоту или динатриевую соль).
Выход сульфохлорфенола равен 29 г 75%-ного продукта.
Примечание. 2-Ампно-4-хлор-6-сульфофенол должен быть пред-
варительно переосажден из воды с добавлением соды и выделен кислотой
после отфильтровывания.
107
ЛИТЕРАТУРА
1. С. Б. Саввин, Ю. М. Дедков. Сборник «Применение фотомет-
рических методов анализа при контроле качества материалов», Моск, дом
техники, М„ 1963, т. 1, стр. 78.
2. Ю. М. Дедков, С. Б. Саввин. Там же, стр. 87.
3. С. Б. Саввин, Ю. М. Дедков. Ж. аналнт. химии, 19, 21
(1964).
4. Ю. М. Дедков, Д. С. К а д а н е р. Н. Д. Писаренко,
А. С. Рябова, С. Б. С а в в п н. Заводск. лаборатория, 30, 654 (1964).
5. С. Б. Саввин, Ю. М. Дедков. Заводск. лаборатория, 30, 654
(1964).
6. Ю. М. Дедков, Д. И. Рябчиков, С. Б. Саввин. Ж. аналнт.
химии, 20, 574 (1965).
7. И. П. А л и м а р и н, С. Б. Саввин, Ю. М. Дедков. Ж. аналит.
химии, 19, 328 (1964).
8. Д. И. Рябчиков, С. Б. Саввин, Ю. М. Дедков. Заводск.
лаборатория, 31, 154 (1965).
9. В. И. Кузнецов, А. А. Н е м о д р у к. Ж. общ. химии, 26, 3285
(1956).
Поступила в июле 1964 г. ГЕОХИ АН СССР
УДК 547.621.07
ТЕТРАИЗОПРОПИЛДИФЕНИЛ
С24Н34
Е. П. БАБИН, А. А. КУЗЬМЕНКОВ
сн3 сн3 сн3 сн:
СН "сн
/=к
сн сн
сн3 СН3 СНз СН;
М. в. 322,53
В литературе известно получение моно-, ди- и триизопро-
пилдифенилов алкилированием дифенила пропиленом [1],
спиртами [2, 3] и изопропилхлоридом [4] с различными ката-
лизаторами.
Тетраизопропилдифенил синтезирован нами впервые. Для
его получения применили метод алкилирования дифенила в
присутствии хлористого алюминия. В качестве алкилирующе-
го агента использован пропилен.
СХЕМА СИНТЕЗА ТЕТРАИЗОПРОПИЛДИФЕНИЛА
Свн6-Свн5+ 4С3Н„ (ЛС3Н7)2СвН3-СеН3(/-С8Н,)2
Характеристика основного сырья
Дифенил, ч., т. пл. 68—7Г, ТУ 2666—51, остаток после
прокаливания 0,1 %•
Пропилен, 96—98%-ный, получен дегидратацией изопро-
пилового спирта на алюмосиликатном катализаторе.
Алюминий хлористый, безводный, технический.
109
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную механиче-
ской мешалкой (1500—2000 об/мин), обратным холодильни-
ком и барботером, помещают 154,2 г (1 М) дифенила и 13,4 е
(0,1 М) хлористого алюминия. Колбу нагревают на водяной
бане с температурой 100° и при интенсивном перемешивании
подают через барботер сухой пропилен в количестве 105—
НО г (2,5—2,6 М).
После охлаждения колбы алкилат переливают в делитель-
ную воронку и промывают несколько раз горячей водой до
нейтральной реакции промывных вод. Высушенный хлористым
кальцием алкилат перегоняют на вакуумной ректификацион-
ной колонке и отделяют фракцию тетраизопропилдифенила в
температурном интервале 187—20675 мм в количестве 150 г,
что составляет 58% от взятого алкилата. Полученный тетра-
изопропилднфенил представляет собой маслянистую жидкость
желтого цвета с константами: т, кип. 187—20675 мм; т. заст.
—21°; d420—0,9351; я20 —1,5410.
Найдено, %: С —89,64; Н—9,58.
C2iH31. Вычислено %: С—89,44; Н—10,56.
ЛИТЕРАТУРА
1. Ш. Ахмедов, Г. Гаджиев, Р. Ахмедова. Уч. зап. Азерб.
ун-та, сер. физ.-мат. и хим. наук, № 4, Баку, 1961.
2. И. Рома дан, С. Берга. Ж. общ. химии, 28, 413 (1958).
3. И. Рома дан, Г. Рендель. Ж- общ. химии, 26, 202 (1956).
4. A. Levine, О. Cass. Chem. Zbl„ I, 19, 39j1 (1939).
Поступила в апреле 1965 г.
Донецкий филиал ИРЕА
УДК 547.831.7.07
2,5,7-ТРИМЕТИЛ-8-АЦЕТОКСИХИНОЛ ИН
В. М. ДЗИОМКО, И. А. КРАСАВИН, Т. Н. ЕГОРОВА
сн3
ОСОСНз
C14Hl6NOa М. в. 229,28
В литературе 2,5,7-триметил-8-ацетоксихинолин не описан.
Он был получен нами с выходом 86—92% при нагревании
2,5,7-триметил-8-оксихинолипа с уксусным ангидридом. Чис-
тое вещество представляет собой желтые кристаллы с т. пл.
64—64,5°.
СХЕМА СИНТЕЗА 2Д7-ТРИМЕТИЛ-8-АЦЕТОКСИХИНОЛ И НА
+ (СН8СО)2О ->
ОСОСНз
+ СНзСООН
111
Условия получения
В круглодонную колбу емкостью 300 мл с пришлифован-
ным обратным холодильником, защищенным хлоркальциевой
трубкой, помещают 28,1 г (0,15 М) 2,5,7-триметил-8-оксихи-
нолина (см. примечание 1), 120 мл (1,27 М) свежеперегнан-
ного уксусного ангидрида и 24 мл сухого перегнанного пири-
дина. Смесь кипятят в течение 8—10 часов, затем отгоняют
пиридин, уксусную кислоту и избыток ангидрида на кипящей
водяной бане в вакууме водоструйного насоса. Остаток пере-
гоняют в вакууме (см. примечание 2) при 157—16073 мм.
Желтый густой дистиллат вскоре затвердевает. Выход сырого
продукта 29,5—31,8 г (85,8—92,4%). Его перекристаллизовы-
вают из 110—120 мл петролейного эфира (т. кип. 60—80°; см.
примечание 3) и высушивают в вакуум-эксикаторе над пара-
финовыми стружками.
Выход 2,5,7-триметил-8-ацетоксихинолина равен 25,8—
29,8 г, что составляет 75—86,6% от теоретического; т. пл. 62—
63° (см. примечание 4).
Примечания:
1. Применялся 2,5,7-триметил-8-оксихинолин с т. пл. 84—84,5°, полу-
ченный, как описано в настоящем сборнике.
2. При перегонке удобно пользоваться специальной колбой для за-
твердевающих веществ [1].
3. При охлаждении раствора начинает появляться эмульсия; в этот
момент следует вызвать кристаллизацию, внеся затравку или потирая
по стенкам колбы стеклянной палочкой.
4. Чистый образец 2,5,7-триметнл-8-ацетоксихннолииа с т. пл. 64—
64,5° был приготовлен перекристаллизацией из низкокипящего петролей-
ного эфира (т. кип. 35—50°) и проанализирован.
Найдено, %: С—73,77; 74,06; Н—6,62; 6,66; N—6,18; 6,43.
ChHjjNO;. Вычислено, %: С—73,34; Н—6,59; N—6,11.
ЛИТЕРАТУРА
I B M. Дзиомко, И. А. Красавин, О. В. Иванов. Методы
получения химически ч реактивов и препаратов, вып. 9, М., ИРЕА, 1964,
стр. 9, примечание 5.
Поступило в марте 1S65 г.
ИРЕА
УДК 547,313.07
2,2,4-ТРИМЕТИЛ-1,3-ДИОКСОЛАН
свн12о~
Г. Ф. ДРЕГВАЛЬ, Н. А. РЫБАК
CH3v ZO-CH3
ж 1
СП/ хо-сн-сн3
М. в. 116,16
Окиси олефинов при взаимодействии с кетонами в присут-
ствии катализаторов образуют циклические кетали. В качест-
ве катализаторов применяют хлорное олово, фтористый бор и
другие вещества—агенты [1, 2]. Обзор методов получения ци-
клических кеталей приведен в монографии [3]. Подробные ме-
тодики получения 2,2,4-триметил- и 2,4-диметил-2-этил-1,3-
диоксоланов в литературе отсутствуют. В качестве катализа-
тора нами использовано хлорное олово.
СХЕМА СИНТЕЗА 2,2,4-ТРИМЕТИЛ-1,3-ДИОКСОЛАНА
сн3.
)С=О + СН3-СН-СН2 ->
СНз7 \ /
О
СНз
сн3
O-CH.J
/С 1
\о-сн-сн3
Характеристика основного сырья
Ацетон, ч.д.а., ГОСТ 2603—63,
Бензол, ч.д.а., ГОСТ 5955.
Пропилена окись, свежсперегнанная, т. кип. 35°; Л20—
0,8300.
113
Олово хлорное, ч., ТУ МХП 3381—52.
Натр едкий, ч.д.а., ГОСТ 4328—48.
Натрий сернокислый, безводный, ч., ГОСТ 4166—48.
Условия получения
В трехгорлую колбу, снабженную капельной воронкой,
термометром и мешалкой, помещают 11,6 г (0,2 М) ацетона и
при перемешивании и охлаждении ледяной водой медленно
прибавляют 2,6 г (0,01 М) хлорного олова. К полученной сме-
си прибавляют 14,5 г (0,25 Л1) окиси пропилена с такой ско-
ростью, чтобы температура внутри колбы не превышала
40—50°. Затем реакционную смесь разбавляют равным
объемом бензола и промывают 10 мл 10%-ного едкого натра
для удаления хлорного олова и еще раз промывают 20 мл во-
ды. Бензольный раствор сушат сульфатом натрия. Бензол от-
гоняют на водяной бане и продукт перегоняют при атмос-
ферном давлении.
Выход 2,2,4-триметил-1,3-диоксолана равен 19,1 г, что со-
ставляет 29% от теоретического; т. кип. 182—183°; tin —
1,4369; i/425—1,027.
Найдено, %: С -61,97;
Н-10,50; - 30,60.
С(;Н15О2. Вычислено, %: С -62,07; И —10,50; MRD—30,75.
По аналогичной методике получают 2,4-диметил-2-этнл-1,3-
диоксолан с т. кип. 130 - 132J; rf?5—0,9415: п'г>—1,4108.
Выход продукта равен 15,8г (31%).
Найдено, %: С.—64,87; Н —10,78; МКр - 35.70.
C-Ht,O;. Вычислено, %; С-64,61; Н —10,76; MR0 -35.38.
Л И Т Е Р А Т У Р А
1. М. В og ett, R. Roblin. J. Amer. Chem. Soc., 55, 3741 (193.3).
2. А. А. Петров. Ж- общ. химии, 10, 9816 (1940); Ф. Я. Пер всей,
II. И. Кудряшова. Ж. общ. химии, 22, 1583 (1952).
3. М. С. М а л и п о в с к и й. Окиси олефинов и их производные, М ,
Госхимиздат, 1961, стр. 174.
Поступила н октябре 1964 г. Донецкий филиал ИРЕ,\
УДК 547.831.7.07
2,5,7-ТР ИМЕТИЛ-8-ОКСИХИ НОЛ И Н
И. А. КРАСАВИН, В. М ДЗИОМКО, Т. И. ЕГОРОВА
СНз
//\/
ch3'/'^1/XnZxch3
он
CiaHI3NO
М. в. 187,24
В литературе 2,5,7-трнметнл-8-оксихинолин не описан. Он
был синтезирован нами из 4,6-диметил-2-аминофенола по при-
веденной ниже методике, которая может служить примером
применения реакции Дёбнера—Миллера. Выход сырого про-
дукта (в виде гидрохлорида) достигает 1,32—1,39 моля на
1 моль исходного аминофенола; это объясняется тем, что ис-
пользуемый в качестве окислителя 4,6-диметил-2-ннтрофенол
в ходе реакции восстанавливается до амииа, который тоже
принимает участие в циклизации. В пересчете на суммарное
количество амино- и нитросоединений выход составляет 88,5—
92,6%.
Чистый 2,5,7-триметил-8-окснхинолин представляет собой
бесцветные игольчатые кристаллы с т. пл. 84,5—85,5°, т. кип.
136—140°/4 мм. По своим аналитическим реакциям он напо-
минает 8-оксихинальдин и 8-оксихинолин, но имеет перед ни-
ми ряд преимуществ.
115
СХЕМА СИНТЕЗА ГИДРОХЛОРИДА 2,5,7-ТРИМЕТИЛ-8-
ОКСИХИНОЛИНА
СНз СН3
/ I
I II Г СНз-СН-СН-СНО ; f II i НС1 -
СНз"^7 N’H2 Cl X NOS
OH OH
СНз
I
-> f II I -HC1
С.н/ W \ CI la
OH
Условия получения
В трехгорлую колбу емкостью 2—3 л с мешалкой, обрат-
ным холодильником и капельной воронкой помещают 41,2 г
(0,3 А1) измельченного 4,6-диметил-2-аминофенола (см. при-
мечание I) и 150 мл 35%-ной соляной кислоты. Нагревая
колбу на кипящей водяной бане, размешивают массу до об-
разования однородной суспензии, прибавляют 25 г (0,15 А1)
измельченного 4,6-дпметил-2-нитрофенола (см. примеча-
ние2) и перемешивают массу еще 10—15 минут. Добавляют
по каплям за 45 -50 минут 42 г (0,6 М) ректифицированного
кротонового альдегида (см. примечание 3), продолжая нагре-
вание на кипящей водяной бане и размешивание во время
прибавления альдегида, а затем—в течение еще 5 часов. Из-
быток нитросоединения отгоняют с водяным паром, причем
колбу подогревают с помощью горелки, чтобы объем жидко-
сти в кубе не превышал 800—900 мл. По окончании отгонки
(см. примечание 4) кубовый остаток частично нейтрализуют
осторожным добавлением 20%-ного раствора едкого натра до
pH 2, доводят объем водой до 1200 мл, кипятят в течение
20—30 минут с 5 г активированного угля и фильтруют в горя-
чем виде. Фильтрат оставляют для кристаллизации на 15—
20 часов в холодильном шкафу. Выделившиеся при стоянии
оранжевые игольчатые кристаллы гидрохлорида отсасывают,
промывают на фильтре небольшим количеством 10%-ной со-
ляной кислоты, отжимают и высушивают над щелочью.
Выход составляет 77—85,7 г (76,5—85,2%, считая на сум-
му амино- и ннтрофенолов; см. примечание 5).
Продукт перекристаллизовывают из 350—370 мл кипящей
10%-пой соляной кислоты с применением активированного уг-
11(5
ля и получают 72,5—73 г (72—72,5%) очищенного гидрохло-
рида, имеющего вид ярко-желтых игл.
СХЕМА СИНТЕЗА 2,5,7-ТРИМЕТИЛ-8-ОКСИХИНОЛИНА
СН3 СНз
I I
\ / % %" \ / '%
| II | -НС1 + NaOH > I II | % NaCl ICO
CIl/ %' N CH, Сн/уХг/ЧСН,
OH OH
Условия получения
Растворяют очищенный гидрохлорид (72,5—73 г) в 200 лд
горячей воды, слегка охлаждают и подщелачивают до
pH 8—9 добавлением 150 170 мл 10%-ного раствора едкого
натра (х. ч.). Охладив образовавшуюся суспензию до 25—30'.
приливают к ней 200 мл бензола и размешивают до раство-
рения большей части твердого вещества. Затем прибавляют
20%-ный раствор уксусной кислоты до pH 7 и перемешивают
слои до полного растворения осадка. Бензольный слой отде-
ляют, а водный (pH 7) экстрагируют двумя порциями бензо-
ла по 50 мл. Бензольные экстракты соединяют, фильтруют и
бензол отгоняют досуха. Остаток (60,5—61 г) перекристалли-
зовывают из 100—105 мл перегнанного этанола без примене-
ния активированного угля. При охлаждении и стоянии выде-
ляются бесцветные кристаллы, которые собирают и высуши-
вают в вакуум-эксикаторе.
Выход 2,5,7-триметил-8-оксихинолнна равен 56,1—57,7 г,
что составляет 66,6—68,5%, считая на сумму амино- и нитро-
фенолов; т. пл. 84—84,5° (см. примечание 6).
Примечай п я:
1. Применялся 4,6-диметпл-2-аминофенол (5-амипо-4-окс11-1,3-диметпл-
бензол) с т. пл. 134—134,5°, полученный по описанному ранее методу [1].
2. Применялся 4,6-диметпл-2-нитрофенол (5-нитро-4-окси-1,3-днметнл-
бензол) с т. пл. 70—70,5°, полученный по описанному методу [2].
3. Применялся кротоновый альдегид с т, кип. 101—102° (не испр.).
ректифицированный на лабораторной колонке с высотой насадки 400 ,и.н.
4. Перегонку с паром продолжают до тех пор, пока в погоне уже не
будет содержаться желтого нитросоединения. Объем дистиллята
6-8 л.
5. Если фильтрат нейтрализовать и выделившийся продукт перевести
в гидрохлорид, то общий выход сырой соли достигает 89—93,2 г, что со-
ответствует 1,32—1,39 моля па 1 моль исходного аминофенола или
88,5—92,6% от теоретически возможного, считая па суммарное количество
амино- и нитрофено.юв.
6. Вещество может быть очищено также перегонкой с водяным паром,
перегонкой в вакууме (г. кип. 136—14074 лпн) или возгонкой.
117
Чистый образец 2,5,7-тримеп1л-8-оксихиио.1Ина в виде бесцветных иг.:
с т. пл. 84,5—85,5° был приготовлен перегонкой в вакууме и перекристал-
лизацией из этанола и проанализирован.
Найдено, %: С-77,29; 77,08; Н—7,06;6,92; N 7,16;7,26.
CJ2H„NO. Вычислено, %: С-76,98; Н 7,00; N -7,48.
.'1 И Т Е Р А Т У Р А
I. В. М. Дзяомко, И. С. Маркович. Методы получения хими-
ческих реактивов и препаратов, вып. 2, М., ИРЕА, 1961, стр. 24.
2. В. М. Д з и о м к о, И. С. Маркович. Методы получения химиче-
ских реактивов и препаратов, вып. 2, М., ИРЕА, 1961, стр. 23.
Поступила и марте 1965 г.
ПРЕД
УДК 547.831.7.05
2Д7-ТРИМЕТИЛ-8-ОКСИХИНОЛ ИН-1-ОКСИД
N- Окись 2,5,7-триметил-8-оксихинолина
В. М. ДЗИОМКО, И. А. КРАСАВИН, Т. Н. ЕГОРОВА
сна
C12HnNO2
М.в. 203,24
В литературе 2,5,7-тримс.тил-8-оксихинолин-1-оксид не опи-
сан. Он был получен нами при окислении 2,5,7-триметил-8-
оксихинолина надуксусной кислотой. Чистое вещество пред-
ставляет собой ярко-желтые мелкие иголочки с т. пл. 126—
126,5°.
Приготовление раствора надуксусной кислоты
(СН3СО)2О + Н2Ог -» СН3СО3Н +СН3СООН
Охлаждают ледяной водой 10,2г (0,1 А4) перегнанного ук-
сусного ангидрида и прибавляют к нему понемногу 13,6 г
(0,12 ЛТ) 30%-ной перекиси водорода при перемешивании.
Смесь оставляют при комнатной температуре на 1—2
суток.
Получение 2,5,7-триметил-8-Оксихинолин-1-оксида
СН3
сн3
| j| ! -1-СНзСОзН -
сн3 | СПз
он
О 119
Растворяют 5,6 г (0,03 Л4) 2,5,7-триметил-8-оксихинолина
(см. примечание 1) в 30 мл чистого хлороформа и прибавля-
ют раствор надуксусной кислоты. Смесь перемешивают в те-
чение 3—4 часов и оставляют на ночь. На следующий день
органический слой отделяют, а водный разбавляют вдвое во-
дой и экстрагируют четырьмя порциями хлороформа по 15 мл.
Все хлороформные экстракты соединяют, встряхивают с на-
сыщенным раствором углекислого натрия (15 мл), затем
дважды промывают водой (15 мл) и сушат безводным суль-,
фатом натрия. Растворитель отгоняют досуха на кипящей во-
дяной бане, а остаток (4,5—4,7-г) перекристаллизовывают ил
20—25 мл этанола.
Выход 2,5,7-триметил-8-оксихинолин-1-оксида в виде яр-
ко-желтых мелких иголочек с т. пл. 125—126” составляв!
2,5—2,6 г (41—43%); из маточного раствора можно выделить
еще 1 —1,2 г менее чистого вещества.
Общий выход продукта равен 3,7 г, что составляет 61% 01
теоретического (см. примечание 2).
Примечания:
1. Применялся 2,5,7 тр11мет1ы-8-океихпнол1п1 с т. пл. 84—84,5’, полу-
ченный, как описано в настоящем сборнике.
2. Чистый образец 2,5,7-трпметил-8-оксихинолпн-1-оксида с постоянной
т. пл. 126 -126,5е был приготовлен перекристаллизацией из этанола и про-
анализирован.
Найдено, %: С 70,57; 70,87; 11-6,47; 6,61: N--6.9O; 6,92.
C|2H13NC4. Вычислено, %: С—70,92; Н—6,45; N—6,89.
Поступила в марте 1965 г.
ИРЕА
УДК 547.835.1.07
5 ФЕНИЛ-АКРИДИНОВЫЙ ОРАНЖЕВЫЙ
ХЛОРГИДРАТ
2,8-Бис-(диметил амино)-5-фенилакридин-гидрохлорид
О. Т. ЛУШИНА, А. А. ПРЯНИШНИКОВ, М. И. ГЕРМАН
N(CH3)2
c2,h24n3ci
М. в. 377,91
5-Фенил-акридииовый оранжевый хлоргидрат предложен
для применения в качестве красителя в флуоресцентной мик-
роскопии при определении липоидов. Его получают циклиза-
цией 2,2’-диамино-4,4’-бис- (диметиламиио) -трифенилметана
с последующим окислением образовавшегося лейкооснова-
ния красителя [1—3]. Исходное соединение получают конден-
сацией бензальдегида (1 М) с ж-аминодиметиланилином
(2 44) в спиртовой среде в присутствии соляной кислоты [1—3].
Нами были проверены и уточнены условия синтеза обоих
соединений.
121
СХЕМА СИН1 ЕЗА - 2,2 -ДИАМИНО-4,4 -БИС-(ДИМЕТИЛАМИНО)-
ТРИФЕНИЛМЕТАНА
(CH3)2Nx^/\/NH2 H2N\ _N(CH3).,
к/\ -на ^-1 -
\ /'
ЧС11 z
(CH3)2Nx nh2 H2N’x z ч z N(CH3)2
Jz )[ J + NaCI -L H,o
4\ /
4 CH 7
I
/4
» I
\//
Характеристика основного сырья
.w-Аминодиметиланилип, техн., РТУ 20—63.
Бензальдегид, ч., ГОСТ 157—51.
Соляная кислота, ч., ГОСТ 3118—46, уд. в. 1,185.
Этиловый спирт, ректифицированный, ГОСТ 5962—51.
Условия получения
В трехгорлую круглодонную колбу емкостью 1 л, снабжен-
ную мешалкой, термометром, обратным холодильником и ка-
пельной воронкой и помещенную в водяную баню, загружают
135 г (0,99 М) л-аминодиметиланилина, 105 мл (0,99 Л4)
30%-ной соляной кислоты и смесь нагревают при перемеши-
вании в течение 15 минут при температуре 50—60°,
122
За iем к реакционной смеси добавляют 250 мл этилового
спирта и, не прекращая нагревания и перемешивания, добав-
ляют 53 г (0,5 Л4) бензальдегида и продолжают нагревание
в течение 3 часов при температуре 70—80°.
После охлаждения реакционную смесь разбавляют 4 л во-
ды и выделяют 2,2/-диамино-4,4/-бис-(диметиламино)-трифе-
нилметап добавлением 10%-пого раствора едкого натра до
слабощелочной реакции.
Выпадает обильный хлопьевидный осадок серовато-розо-
вого цвета. Осадок отфильтровывают и промывают водой
(около 5 л) до светлых промывных вод, затем промывают
800 мл водного спирта (1:1), отжимают и сушат при темпе-
ратуре 50—60°.
Выход 2,2/-диамино-4,4/-бис- (диметиламино) -трифепилме-
тана равен 160 г, что составляет 90%, считая на м-аминоди-
метиланилин. Температура начала плавления не ниже 92—
93°.
СХЕМА СИНТЕЗА 5-ФЕНИЛ-АКРИД И НОВОГО ОРАНЖЕВОГО
ХЛОРГИДРАТА
(CH,),N NH, H,N N(CH3i2
H,SO„ Одо 130°
-ML
CH
(CH3)2N4/x yNH X/N(CH3)a
\ V \ Ч -Н,ЯО, EeCl3, NaCI
\ )
12
Характеристика основного сырья
2,2/-Диамино-4,4'-бис- (диметиламино) -трифеиилметан.
Серная кислота, ч., ГОСТ 4204—48, уд. в. 1,84.
Железо хлорное, ч., ГОСТ 4147—48.
Натрий хлористый, ч., ГОСТ 4233—48.
Условия получения
В фарфоровый стакан емкостью 4 л, снабженный мешал-
кой и термометром, загружают 60 г (0,17 М) 2,2'-диамино-
4,4/-бис/- (диметиламино) -трифенилметана, 2,4 л воды и 600 г
серной кислоты. Смесь упаривают на масляной бане при сла-
бом перемешивании в течение 14 часов до температуры 130°
в реакционной смеси. Жидкость при этом окрашивается в
красный цвет. Затем, после охлаждения до 80—90°, реакци-
онную массу разбавляют 1,5 л горячей воды (70—80°), мед-
ленно добавляют 240 мл 30%-иого раствора хлорного железа
и для выделения красителя добавляют 1,2 л насыщенного
раствора хлористого натрия.
После 20-часового стояния выпавший краситель темного
цвета отфильтровывают, промывают 100 мл 5%-ной соляной
кислоты, отжимают и сушат при температуре 60—70°.
Выход хлоргидрата 5-фенил-акридинового оранжевого ра-
вен 42-—45 г, что составляет 67—72%, считая на 2,2/-диами-
по-4,4'-бис-(диметиламино)-трифеиилметан ; Хчакг 490±5ммк.
Л И 'Г Е Р А Т У Р А
1. DRP 6890b; Frdl. 2. 104 (1890).
2. DRP 43714; 59179; 71362: Frdl. 3, 293.
3. Пат. США 503305
Поступил-! н январе 1965 г
УДК 547.826.2.07
ФУЗАРИНОВАЯ
(5-Н-БУТИЛ-2-ПИРИДИНКАРБОНОВАЯ) КИСЛОТА
Ю. И. ЧУМАКОВ, В. П. ШЕРСТЮК
u.CJIo
нс юс
C1(,H]3O2N М. и. 179,22
Фузариновая кислота является антибиотиком ряда пири-
дина. Она была выделена из культуральной жидкости Gibe-
rella fujikuroi в 1934 г. [1]. Фузариновая кислота была синте-
зирована исходя из 2-метил-5-этилпиридина [2]. Для ее син-
теза в качестве исходных продуктов использовались также
2,5-диметилпиридии [3], З-н-бутилпиридин [4], а также 2-ме-
тнл-5-этинилпиридии [5]. Представляет интерес опубликован-
ный недавно синтез фузариновой кислоты, включающий пол-
ное построение пиридинового цикла на основе реакции,пред-
ложенной Несмеяновым и Кочетковым [4]. Во всех перечис-
ленных синтезах, за исключением последнего, фузариновая
кислота получалась в конечном итоге окислением 2-метил-5-
и-бутилпиридииа.
Для получения фузариновой кислоты в описываемой нами
методике использовался 2-метил-5-и-бутилпиридин, получен-
ный из метилвинилкетона и гексен-1-илэтилового эфира [6].
Окисление проводилось двуокисью селена, которая избира-
тельно окисляет только метильную группу в положении 2, не
затрагивая и-бутильной группы. В основу был положен ме-
тод окисления двуокисью селена 2-метил-5-этилпиридииа в
5-этил-2-пиридинкарбоновую кислоту, описанный в литерату-
ре [7].
125
СХЕМА СИНТЕЗА 5-Н-БУТИЛ-2-ПИРИДИНКАРБОНОВОИ КИСЛОТЫ
н.С4Н„ н.С4Н9
' ^\/
2[. -f-3SeO2-* 2 | jl ~г 3Se + 2Н3О
сн3 ноос
Характеристика основного сырья
2-Метил-5-н-бутилпиридин (синтезирован согласно [8]).
Селен двуокись, ч., ТУ ГКХ 1502—61,
Пиридин, ч., ГОСТ 2747—44,
Условия получения
В трехгорлую колбу на шлифах емкостью 25 мл, снаб-
женную мешалкой с затвором КПГ, термометром и обратным
холодильником, защищенным трубкой с безводным едким ка-
ли, помещают 0,5 г (0,0033 ЛГ) 2-метил-5-н-бутилпиридина,
0,56 г (0,005 Л4) двуокиси селена и 5 мл пиридина (см. при-
мечание 1). Смесь нагревают на масляной бане до темпера-
туры кипения пиридина и перемешивают в течение 4 часов
(см. примечание 2). Затем содержимое колбы охлаждают,
отфильтровывают на пористом стеклянном фильтре № 4 оса-
док селена, который тщательно промывают горячей водой до
объема около 15—20 мл. Фильтрат и промывные воды объе-
диняют и перегоняют с водяным паром до тех пор. пока не
отгонится весь пиридин и непрореагировавший 2-метил-5-н-
бутилпиридии (70 мл дистиллата). Остаток в перегонной
колбе (см. примечание 3) переносят в прибор для непрерыв-
ной экстракции из жидкости и экстрагируют эфиром в тече-
ние 24 часов (см. примечание 4). Эфирный экстракт упарива-
ют, добавляют 50 мл бензола и кипятят с насадкой для водо-
отделения (насадка Дина--Старка) в течение 1—2 часов.
Бензол упаривают под небольшим вакуумом на водяной бане
досуха и остаток перекристаллизовывают из низкокипящего
петролейного эфира или н. гептана. Получают 0,26 г (43%)
5-н-бутил-2-пиридинкарбоновой кислоты с т. пл. 96—98°. По-
сле перекристаллизации из петролейного эфира и сублимации
в вакууме диффузионного насоса (0,1—0,01 мм) при темпе-
ратуре 80—90° получают фузариновую кислоту с т. пл. 100—
101° (испр.).
По литературным данным, т. пл. продукта 100—102° [2].
И р и м е ч а н и я:
1. Двуокись селена была предварительно высушена нагреванием при
200—250° в токе кислорода.
Пиридин высушен кипячением с плавленым едким кали в течение 2
часов и перегнан, т. кип. 114—115°.
126
2. В процессе окисления реакционная масса начинает краснеть, а за-
тем становится бурой. На стенках колбы, выделяется черный осадок селе-
на.
3. В результате перегонки с паром на стенках колбы для перегонки
оседает красный селен. Если содержимое колбы становится мутным, то
его дополнительно фильтруют.
4. Использовалась насадка Тилепапе для экстракции, описанная в
[9]. Основная масса фузариновой кислоты извлекается в первые 2—3 часа
экстракции, при этом на стенках колбы выделяются желтоватые кристал-
лы. При сокращении времени экстракции выходы снижаются.
Л II Т Е Р А Т У Р А
1. Т. Ya but а, К. К a ni Ь е, Т. Havashi. Nippon Nogei - Kagaku
Kaishi, 10, 1059 (1934); C. A., 29, 1132 (1935).
2. A. Platiner, W. Keller, A. Boiler. Helv. eliim. acta, 37,
1379 (1954).
3. E. Hardegger. E. N ikies. Helv. chim. acta, 40, 2428 (1057).
4. E. Hardegger, E. N i k 1 e s. Helv. chim. acta, 43, 1016 (1957).
5. A. H. Кос т, Г1. Б. Тереитье в, Л. В. М о ш е н ц е в а. Гетеро-
циклы в органическом синтезе (тезисы докладов), Киев, 1964, стр. 72.
6. Ю. И. Чумаков, В. П. Шерстю к, Е П. Д з ы г у н, В. Ф. Н о-
впкова. Гетероциклы в органическом синтезе (тезисы докладов), Киев,
1964, стр. 68.
7. D. Jerchel, J. Heidel. Liebigs Ann. Chem., 613, 153 (1958).
8. Ю. И. Чумаков, В. П, Шерсгюк. См. статью «2-Метил-5-и.
бутилпиридии» в настоящем сборнике,
9. “Organikum", 3 Auflage, DVW. Berlin, 1964, s. 61.
Поступила в январе 19(55 г. Киевский политехнический институт
УДК 547.821.41.07
2-ХЛОРМЕТИЛПИРИДИН
В. Я. КАРМИ, Г. Ф. ДРЕГВАЛЬ
сн2с!
CeHBNCl М. в. 127,0
По литературным данным, 2-хлорметилпиридин получают
из 2-пиридилметанола [1], N-окиси 2-метилпиридина [2] или
хлорированием 2-метилпиридина [3].
Мы выбрали последний путь и внесли лишь небольшие из-
менения в литературные данные.
2-Хлорметилпиридип представляет интерес в качестве ис-
ходного соединения для синтетических работ.
СХЕМА СИНТЕЗА 2-ХЛОРМЕТИЛПИРИДИНА
\ / \
Cl2+Na3CO3 N/-CH,C1 4- NaCI 4- СО.
Условия получения
В трехгорлую колбу помещают 93 г (1 М) 2-метилпири-
дина, 200 г соды и 500 мл четыреххлористого углерода (см.
примечание 1). При постоянном перемешивании через реак-
ционную смесь пропускают ток хлора в течение 12 часов при
температуре 60—70°. К охлажденной до комнатной темпера-
туры смеси при сильном перемешивании прибавляют 700—
800 мл воды и прибавлением 2 в. раствора едкого натра до-
водят pH раствора до 8—9. Затем органический слой отде-
ляют, а водный слой промывают четыреххлористым углеро-
дом дважды по 100 мл.
128
Продукт реакции извлекают из объединенного органиче-
ского раствора, промывая его последовательно по 200 мл 5н.
и 2п. раствором соляной кислоты (см. примечание 2).
Полученный солянокислый раствор нейтрализуют до
pH 6—7 40%-ным раствором соды. Образовавшийся масля-
нистый слой отделяют.
Из водного раствора, доведенного до pH 8, экстрагируют
бензолом дополнительное количество продукта реакции.
Из отделенного масла и бензольных вытяжек после пере-
гонки получают 2-хлорметилпиридин с выходом 51,5 г, что
составляет 40,5% от теоретического.
2-Хлорметилпиридин обладает раздражающим и слезото-
чивым действием. По внешнему виду вещество представляет
собой бесцветную быстротемнеющую жидкость с т. кип.
48—5072 лмг; ng—1,5361.
По литературным данным, т. кип. продукта 45—4771,5 мм;
ng—1,5365.
П р 11 м е ч а н 11 и:
1. Вместо чстыреххлористого углерода в качестве .растворителя мож-
но применять хлороформ.
2. 2-Хлорметилппрпдин можно получить п непосредственно из орга-
нического слоя, без обработки соляной кислотой и последующей нейтра-
лизации, но выход продукта при этом несколько снижается.
Л И Т Е Р А Т У Р А
1. Е. Matuszewska-Wieczorkowska, J. Michalski. Rocz-
niki cheiiilt, 31, 543 (1957).
2. John F. Vozza. J. Organ. Chem., 27, 3858 (1962).
3. W. Mathes, H. S chilly. Angcw. Chemie, 75, 235 (1963).
Поступила в октябре 1964 г.
Донецкий филиал ИРЕА
УДК 547.592.14.07
ЦИКЛОГЕКСИЛСУЛЬФАМИНОВАЯ КИСЛОТА
Р. Г. ВДОВИНА, Л. В. КАРПОВА, И. А. РЕДЬКИН
^-NHSO3H
C6H)aNO3S ~' М. в. 179,23
Циклогексилсульфаминовая кислота обладает бактери-
цидным действием, применяется в виде солей в качестве под-
слащивающего средства при диабете [1, 2].
Циклогексилсульфаминовую кислоту получают пропуска-
нием сернистого газа в суспензию циклогексилгидроксилами-
на в индифферентном растворителе [3] или действием хлор-
сульфоновой кислоты на циклогексиламин с последующим
разложением образующейся соли [4, 5].
Ниже описан разработанный нами упрощенный способ по-
лучения циклогексилсульфаминовой кислоты путем разложе-
ния триэтиламиновой или натриевой соли циклогексилсульф-
аминовой кислоты в водном растворе с помощью ионообмен-
ной смолы КУ-2.
СХЕМА СИНТЕЗА ЦИКЛОГЕКСИЛСУЛЬФАМИНОВОЙ КИСЛОТЫ
; -nh2 + ciso3h 4-2 (c3h5)8n-*
/ -NHSO3H .N(C2H6)34- (CaH6)3 N. HC1
(C2h6)3n+h2o+
-NHSOsNa
130
-NHSOsH
Характеристика основного сырья
Циклогексиламин, т. кип. 133—134°.
Хлорсульфоновая кислота, ч., ТУ НКХП 385—41.
Триэтиламин, ч., ТУ РУ 796—53, высушенный над едким
натром и перегнанный, т. кип. 88'.
Хлороформ, х. ч., ГОСТ 3160—51, высушенный над пота-
шом и перегнанный, т. кип. 61°.
Натр едкий, х. ч., ГОСТ 4328—48.
Метиловый спирт, ч., ГОСТ 6995—54.
Соляная кислота, х. ч., ГОСТ 3188—46.
Ионообменная смола КУ-2, ВТУ 661—55.
Условия получения
Получение триэтиламиновой соли циклогексилсулырами-
новой кислоты. В четырехгорлую круглодонную колбу емко-
стью 0,5 л, снабженную мешалкой, капельной воронкой, тер-
мометром и небольшим воздушным холодильником с хлор-
кальциевой трубкой, помещают 39,6 г (0,4 Af) циклогексил-
амина, 84 г (0,8 М) триэтиламина и 200 мл хлороформа. По-
лученный раствор охлаждают до 5—10° и к нему при разме-
шивании прибавляют по каплям раствор 46,8 г (0,4 А4)
хлорсульфоновой кислоты в 50 мл хлороформа (см. приме-
чание 1), следя за тем, чтобы температура не превышала
15°. Хлорсульфоновую кислоту прибавляют в течение 4—5 ча-
сов, при этом образуется белый осадок. Реакционную смесь
размешивают в течение 1 часа при 10—15°, оставляют на 20
часов при комнатной температуре и отфильтровывают (см.
примечание 2). Хлороформный раствор упаривают в вакууме
досуха. Остаток в виде коричневого цвета масла с кристал-
лами объединяют с осадком и смесь солей (триэтиламиновая
соль циклогексилсульфаминовой кислоты и хлоргидрат три-
этиламина) используют в дальнейшем синтезе.
Получение натриевой соли циклогексилсульфаминовой кис-
лоты. В круглодонную широкогорлую колбу емкостью 0,5 л,
снабженную мешалкой, помещают упомянутую выше смесь
солей и 350 мл 20%-ного едкого натра, перемешивают и на-
гревают на паровой бапе в течение 50 минут. Образовавший-
ся при этом триэтиламин улетучивается, и в колбе после ох-
лаждения в ледяной бане выпадает коричневый осадок нат-
риевой соли циклогексилсульфаминовой кислоты. Осадок от-
фильтровывают на воронке Бюхнера, промывают 2 раза по
15 мл холодным метиловым спиртом (см. примечание 3) и
сушат на воздухе.
Выход натриевой соли циклогексилсульфаминовой кислоты
равен 49,6 г, что составляет 62% от теоретического.
131
Подготовка ионообменной смолы КУ-2. В стакан емкостью
2 л помещают 200 г смолы КУ-2 и заливают на ночь 20%-ной
соляной кислотой. (Вся смола должна быть покрыта жидко-
стью.) Смолу отфильтровывают на воронке Бюхнера и про-
мывают дистиллированной водой до отсутствия кислой реак-
ции по конго.
Заполнение колонки смолой. Стеклянная колонка с внут-
ренним диаметром 35 мм и длиной 800 мм снабжена оттяну-
тым внизу концом диаметром 5 мм, на который надета ре-
зиновая трубочка с винтовым зажимом. В суживающуюся
часть колонки помещают фарфоровую или стеклянную пла-
стинку с отверстиями и кусочек гигроскопической ваты. Сма-
чивают вату дистиллированной водой и в колонку заливают
суспензию КУ-2 в воде. Воде дают стекать, не допуская раз-
рыва столба жидкости. Когда КУ-2 полностью перенесена в
колонку, доливают воду до верха колонки и вставляют в нее
резиновую пробку с делительной воронкой, заполненной ди-
стиллированной водой. Зажимом регулируют скорость выте-
кающей воды (1,5 мл в одну минуту), проверяя pH. В слу-
чае кислой реакции смолу в колонке промывают водой до
отрицательной реакции по конго.
Получение циклогексилсульфаминовой кислоты. Через ко-
лонку пропускают с той же скоростью раствор 49,6 г натри-
евой соли в 250 мл воды (см. примечание 4), собирая фрак-
цию, обладающую кислой реакцией по конго (см. примеча-
ние 5). Колонку затем промывают дистиллированной водой
до отсутствия кислой реакции (~1 л). Объединенные кислые
растворы упаривают в фарфоровой чашке на водяной бане до
образования густой бесцветной массы, отфильтровывают на
воронке Бюхнера, тщательно отжимают, промывают 10 мл
холодной воды (см. примечание 6) и сушат на воздухе до по-
стоянного веса.
Выход циклогексилсульфаминовой кислоты 29,9 г, что со-
ставляет 41,9% от теоретического (см. примечание 7); т. пл.
продукта 167—169°, что соответствует литературным данным
[4].
Примечай п я:
1. Работу с хлорсульфоиовой кислотой следует вести в вытяжном
шкафу, пользуясь резиновыми перчатками и защитной маской или очками.
2. Осадок, представляющий собой смесь триэтилампновой соли цик-
логексилсульфаминовой кислоты и хлоргидрата триэтиламина, отфильтро-
вывают, чтобы облегчить упаривание хлороформного раствора. В литера-
туре нс указана температура плавления триэтилампновой соли циклогек-
силсульфаминовой кислоты. Аналитический образец получен перекристал-
лизацией из воды (1:20), т. пл. 190—192°. По внешнему виду это бесцвет-
ные пластинки, растворимые в воде и спирте.
Найдено, %: С - 51,53; Н—9,91; N-9,88; S— 11,40.
C1sHs8NsO3S. Вычислено, %-. С—51,38; Н- 10,06; N-9,99; S-11,39.
3. Работа с метиловым спиртом проводится под тягой, не допуска-
ется попадания метилового спирта на кожу.
132
J. Ec.'ii! раствор получаем я окрашенный, его кипятят с углем п филь-
труют.
5. Если вытекающий раствор мутный, его осветляют встряхиванием
углем п фильтрованием через двойной складчатый фильтр.
6. Фильтрат и промывную воду можно упарить еще раз для получе-
ния дополнительного количества цпклогексплсульфаминовой кислоты.
7. Цик.тогекси.тсульфамиповую кислоту' можно получить непосредст-
венно пз смеси солей без предварительного получения натриевой соли.
В этом случае водный раствор триэтилампновой соли циклогексилсульф-
аминовой кислоты п хлоргидрата триэтиламина пропускают через ко-
лонку с КУ-2. Однако при этом требуется увеличенное количество КУ-2
и большее время пропускания раствора.
Выход цпклогексплсульфаминовой кислоты, считая на циклогексил-
ампн, составляет 33% вместо 42%, полученных- при работе с натриевой
солью.
ЛИТЕРАТУРА
1. С. L. Ни у с k. J. L. Maxwell. J. Amer. Pharmac. Assoc. Pract.
Pharmacy Edit.. 19, 142 (1958).
2 К. M. Beck. Food Technol., 11, 156(ls.57).
3. G. Soder, 11. Schnell. DBP„ 950369, kl. 120. Gr. 25 (1956).
4 H. Hopff, E. G a s se n tn e i e r. DBP., 874309, kl. 120, Gr. 2.3ОЗ
(1951).
5. Abbott Laboratories, North Chicago,. (1) E. P„ 669200 (1949).
Поступила в пютс I9ti l г. Институт биологической и
медицинской химии A 'ЧН CCC!J
УДК 547.821.42.07
3-ЭТИЛПИРИДИН
10 7/. ЧУМАКОВ, В. П. ШЕРСТЮК
с,н5
! 4 ;Г
Лх;/
C,H,N м. в. 107,10
З-Этилпиридин представляет интерес как исходный про-
дукт для синтезов в ряду пиридина. Обычно его получают
восстановлением по Кижнеру—Вольфу—Хуанг—Минлощ
3-ацетилпиридина, который синтезируют из этилникотината
конденсацией Клайзена [1]. Браун и Морфи получили 3-этил-
пиридин из 3-метилпиридина алкилированием последнего хло-
ристым метилом в присутствии амида натрия [2]. Недостат-
ками упомянутых методов являются или относительная слож-
ность синтеза, или трудность выделения индивидуального про-
дукта.
Нами разработан синтез 3-этилпиридина исходя из 2-этил-
акролеина и виннл-н.бутилового эфира (1-й способ) или из
акролеина и бутен-1-илэтилового эфира (2-й способ) через
соответствующие этилзамещенные 2-алкокси-3,4-дигидро-1,2-
пираны, получаемые в результате диенового синтеза. Послед-
ние при омылении легко дают 2-этилглутаровый альдегид, ко-
торый, не выделяя из реакционной массы, конденсируют с
гидроксиламином, получая 3-этилпиридин, свободный от при-
месей изомеров и гомологов (см. примечание 1).
134
СХЕМА СИНТЕЗА З-ЭТИЛ ПИРИДИНА
1-й способ
СП3-СН2-СНа-СНО 4- СН2О + (CHa)3NH-HCI —
c2Hs
1
-> CHSCHSCHCHO -> СНа—С—
1
CH,N (СНа>
+ (CHs)8NH-HC)
НС1
С2Н5 сн2
с,н5
с сн2 \ / \
I -4- II I. 1 ->
С СИ \0/\
/% | Он.СД
НО О н.С4Н„
2-й способ
q у ОС,Н5
СН3СН.СН2С^ + 2С2Н,ОН -п-n-* снвсн„сн2сн
м 4 ос2н5
-> СНаСН2СН =CH-OC2H54-C2HsOH
135
Характеристика основного сырья
Формальдегид, 37-40%-ный водный раствор.
Масляный альдегид, ч., ТУ 79П-621—61.
Акролеин, ч., ТУ МХП 2802—54.
Винил-н.бутиловый эфир, челн., свежеперегнанный, т. кии.
92-94.
Диметиламин солянокислый, ч., ВТУ МХП 2885—51.
Гидроксиламин солянокислый, ч., ГОСТ 5456—51.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Условия получения
1-й способ
Получение 2-этилакролеина. В трехгорлую колбу емкостью
250 мл, снабженную мешалкой, пропущенной через резиновый
обтюратор, обратным холодильником, термометром и капель-
ной воронкой, помещают смесь 28,8 г (0,4 /И) масляного аль-
дегида, 32,6 г (0,4 Л1) солянокислого диметиламина и 32,4 г
(0,4 М) водного 37%-кого раствора формальдегида (см. при-
мечание 2). Из капельной воронки по каплям осторожно при
энергичном перемешивании прибавляют 4н. водный раствор
едкого натра до тех пор, пока не начнется разогревание
(3—4 мл раствора едкого натра). При этом температура ре-
акционной массы поднимается до 70°. Как только началась
экзотермическая реакция, колбу охлаждают водой и поддер-
живают температуру в пределах 55—65° в течение 30 минут.
Далее реакционную массу охлаждают до комнатной темпе-
ратуры, переносят в колбу емкостью 1 л и перегоняют с водя-
ным паром до объема дистиллята 100—150 мл. Органический
слой отделяют, высушивают безводным сульфатом натрия и
перегоняют с елочным дефлегматором. После отгонки непро-
реагировавшего масляного альдегида (1,5 г) получают 24 г
2-этилакролеипа, что составляет 75% от теоретического, счи-
тая па прореагировавший масляный альдегид; т. кип. про-
дукта 91—92°; Лд —1,4207 (см. примечание 3).
Получение 2-н. бутокси-5-этил-3,4-дигидро-1,2-пирана. Во
вращающийся автоклав из нержавеющей стали емкостью
250 мл (см. примечание 4) помещают смесь 32,8 г (0,39 Л4)
2-этилакролеина, 43 г (0,43 М) винил-н.бутилового эфира и
0,07 г гидрохинона (0,1% по весу от общего количества ис-
ходных продуктов). Автоклав герметизируют и нагревают до
температуры 210° в течение двух часов под давлением, соз-
даваемым упругостью паров реагентов. Затем автоклав охла-
ждают, разбалчивают и содержимое отсасывают в колбу для
перегонки. При перегонке реакционной массы в вакууме с
елочным дефлегматором (20 см) было получено 54 г 2-п.бу-
136
токси-5-этил-3,4-дигидро-1,2-пирана с т. кип. 89-—92°/6 лмг,
Др—1,4390; Д,20—0,9582.
Выход равен 71% от теоретического.
Получение З-этилпиридина, В колбу на шлифе емкостью
250 мл, снабженную мешалкой, пропущенной через обрат-
ный холодильник, помешают 50 г (0,27 Л4) 2-н.бутокси-5-этил-
3,4-дигидро-1,2-пирапа, 9 мл воды и 20 мл уксусной кислоты.
Смесь нагревают на масляной бане до кипения при переме-
шивании в течение 40 минут до полной гомогенизации.
В трехгорлую колбу на шлифах емкостью 500 мл, снаб-
женную мешалкой, капельной воронкой и обратным холо-
дильником, помещают 18,5 г (0,27 /И) солянокислого гидрок-
силамина и 100 мл ледяной уксусной кислоты. Смесь нагре-
вают до кипения при перемешивании и к ней постепенно из
капельной воронки в течение двух часов (см. примечание 5)
прибавляют уксуснокислый раствор 2-этилглутарового альде-
гида, полученного омылением 2-н. бутокси-5-этил-3,4-дигид-
ро-1,2-пирана, как описано выше. По окончании прибавления
реакционную массу нагревают еще в течение 15 минут, а за-
тем охлаждают и перегоняют с водяным паром до объема ди-
стиллята около 200 мл (см. примечание 6). Остаток в колбе
охлаждают и при охлаждении сильно подщелачивают едким
натром. Щелочную реакционную массу перегоняют до объема
дистиллята около 30 мл. Дистиллят обрабатывают твердым
едким натром (около 60 г), отделяют органический слой, вы-
сушивают вначале получасовым стоянием с едким натром, а
затем кипячением о плавленым едким кали (1:5). В резуль-
тате перегонки получают 12 г (выход 42% от теоретического)
З-этилпиридина с т. кип. 91—92°/65 мм или 164—166°/750 лш;
4° —1,5002; Дн»-—0,9403; т. пл. пикрата 127,8—128,3°
(испр.).
По литературным данным, 3-этилпиридип имеет т. кип.
1657740 мм; т. пл. пикрата 128,1—128,5° [2].
2-й способ
Получение бутен-1-илэтилового эфира. Бутен- 1-илэтило-
'вый эфир получают из ацеталя масляного альдегида (см.
примечание 7) на каталитической установке, используя в ка-
честве катализатора двухзамещенный фосфат магния на жид-
ком стекле [3], при температуре 325—340°, высота слоя ката-
лизатора 30 см, объем 50 мл, высота зоны испарения 5 см\
объемная скорость 0,3 г!час.мл.
При пропускании через каталитическую установку НО е
диэтилацеталя масляного альдегида получают 24,8 г бу-
тен-1-илэтилового эфира (конверсия 24,8%); т. кип. 93—96°;
«д—1,4033.
137
По литературным данным, бутен-1-илэтиловый эфир име-
ет т. кип. 94°; Яд —1,4050 [3].
Получение 2-этокси-3-этил-3,4-дигидро-1,2-пирана. Смесь
8,8 г (0,157 Af) акролеина, 17,5 г (0,167 А1) бутен- 1-илэтило-
вого эфира и 0,025 г гидрохинона (0,1% по весу от общей мае
сы) нагревают в стеклянных ампулах, помещенных во
взрывобезопасный блок при температуре 180° в течение 4 ча-
сов. В результате перегонки реакционной массы после не-
большого предгона акролеина и бутен-1-илэтилового эфир<а
получают 16,5 г 2-этокси-3-этил-3,4-дигидро-1,2-пирана, кото-
рый имеет т. кип. 68—72°/20 мм; «о—1,4341.
Выход составляет 65,8% от теоретического, считая на за-
груженный акролеин.
Получение 3-этилпиридина. Из 13,3 г (0,08 М) 2-этокси-З-
этил-3,4-дигидро-1,2-пирана, 3 г уксусной кислоты и 1,5 мл
воды (на омыление), 50 мл уксусной кислоты (при цикли-
зации) и 5,8 г (0,08 А4) солянокислого гидроксиламина по ме-
тодике, описанной выше в 1-м способе, получают 3-этилпири-
дин с выходом 60% от теоретического; т. кип. 83—84°/50 лш;
—1,5000; %г0 — 0,9405; т. пл. пикрата 127,5—128,5°
(испр.).
Примечай и я:
1. Синтезы других замещенных 2-алкоксн 3,4-дпгидро-1,2-ппранов бы-
ли описаны рядом американских химиков [4, 5]. Возможность превраще-
ния их в пиридиновые основания была показана в работах [6, 7].
2. Концентрация формальдегида в техническом формалине предвари-
тельно определялась йодометрическим титрованием.
3. Ио методике [8], использующей для получения 2-этилакролеппа
реакцию Маннпха без добавления щелочи, приводится выход, равный
73%. Памп при повторении этого синтеза 2-этплакролеин был получен с
выходом лишь 12—15%. 2-Этил акр олеин недавно был получен по этой же
схеме с выходом 97,8% в автоклаве при строго контролируемом значении
pH [9].
4. Реакцию можно проводить также в стеклянных ампулах, помещен
ных в специальный взрывобезопасный блок, изготовленный пз стали или
алюминия.
5. При быстром прибавлении уксуснокислого раствора 2-этилглутаро-
вого альдегида выходы снижаются.
6. Перегонка с паром кислой реакционной массы имеет назначение
отделить продукты кислого и нейтрального характера. Перед перегонкой
рекомендуется добавить в реакционную массу 1 мл соляной кислоты.
7. Диэтилацеталь масляного альдегида получают из масляного альде-
гида и этилового спирта по методике [10].
ЛИТЕРАТУРА
1. F, Е. F a n d. С. F. Lu tom sky. J. Anter. Chem. Soc., 71, 2931.
(1949).
2 H. C. Brown, W. A. M ц г p h e y. J. Amer. Client. Soc., 73, 3308
(1951).
3. И. H. Назаров, С. M. Макин, Б. К- Крупцов, В. А. Мп
р о и о в. Ж. общ. химии, 39, 111 (1959).
138
4. R. I Longley. Jr., W. S. Emerson. J. Amer, Chcin. Ser., 72,
3079 (1950).
5. C. W. S m i t h. D. O. N о r t о n, S. A. В a 11 a r d. J. Amer. Cheni.
Soc., 73, 5267 (1951).
(>. B. D. Shaw. J. Cheni. Soc., 1937, 300.
7. Am. пат. 2748130 (1956); C. A., 50, 11369 (1956).
S. C. S. Marvel. R. L. Myers, J. H. Saunders. J. Amer.
Client. Soc., 70, 1694 (1948).
9. M. И. Фарберов, Г. С. Миронов, М. Л. Коршунов. Ж.
прикл. химии, 35, 2483 (1962).
10. Синтезы органических препаратов, т. 1, М., ИЛ, 1949, стр. 62.
Поступила в январе 1965 г.
Киевский политехнический институт
УДК 547.572.6,07
ЭТИЛФЕНИЛКЕТОН
Пропиофенон
£'. П. КРЫСИН, С. Е. ЧЕСНОКОВА
< л i..< 11 = <. . >
,3 ~ , \, /
Этилфенилкетон применяется для лабораторных работ.
Его получают окислением этилфенилкарбинола [1] или дейст-
вием смеси СгО3 и H2SO4 на вторичный а-этиленхлорид с
выходом до 75% [2]; присоединением воды к углеводородам
ацетиленового ряда (реакция Кучерова) с выходом до 30%
[3, 4]; взаимодействием ангидридов и хлорангидридов кисло г
с бензолом в присутствии безводного А1С13 с выходом 80%
|5—7, 21]; обработкой а-бромпропиофенона едким кали [8];
нагреванием (3-бензоилкротоновой кислоты с баритовой водой
[9]; действием натрия на эфирный раствор этилйодида и бен-
зоилхлорида [10]; взаимодействием бензоилхлорида с цинк-
этилатом в эфирном растворе [11 —13] (выход до 80%);
аналогично идет реакция с кадмийорганическими соединения-
ми [14, 15]; взаимодействием жирных нитрилов с магнийорга-
ническим реактивом Гриньяра (выход кетона достигает
83—90%) [16, 17, 22, 23].
Нами проверен и уточнен метод [5, 21], по которому этил-
фенилкетон получают из пропионилхлорида и бензола в при-
сутствии безводного треххлористого алюминия.
Пропионилхлорид получают из пропионовой КИСЛОТЫ и
треххлористого фосфора, и из реакционной смеси он не вы-
деляется.
1.40
СХЕМА СИНТЕЗА ЭТИЛФЕНИЛКЕТОНА
ЗСН3СН,СООН + РС13 -> ЗСН3СН3СОС1
А.1С1
CH3CHSCOC1 + C0HG СН3СН3-СО -СвН4Ч-НС1
Характеристика основного сырья
Пропионовая кислота, ч., ТУ МХП 2492—51,
Фосфор треххлористый, ч., ГОСТ 91—41.
Бензол безводный, ч„ ГОСТ 5955—51.
Соляная кислота, ч., ГОСТ 3118—46.
Алюминий хлористый, безводный, ч., ГОСТ 3759—47.
Условия получения
В круглодонную колбу емкостью 750 лы, соединенную с
обратным холодильником, загружают 74 г (I Л1) пропионо-
вой кислоты и 70 г (0,5 М) треххлористого фосфора.
Смесь нагревают в течение одного часа на кипящей водя-
ной бане и, не давая реакционной массе остыть, осторожно
прибавляют к ней 300 г бензола.
Реакционную массу с бензолом сливают от остающейся на
дне колбы фосфористой кислоты в другую колбу (емкостью
1.5 л), соединенную с обратным холодильником, который
снабжен изогнутой трубкой для поглощения выделяющегося
хлористого водорода (трубка находится над поверхностью
воды).
В бензольный раствор пропионилхлорида добавляют 150 г
треххлористого алюминия безводного. Если реакция идет
бурно, колбу охлаждают. После полного растворения хлори-
стого алюминия смесь кипятят на водяной бане в течение од-
ного часа (колба соединена с обратным холодильником). За-
тем содержимое колбы выливают на смесь мелкодробленого
льда (1 кг) и соляной кислоты (40 г).
Бензольный слой реакционной массы отделяют, а из вод-
ного слоя экстрагируют этилфенилкетон смесью бензола и
этилового эфира (100 мл), составленной в отношении 1:1.
Эфирно-бензольный слой промывают водой, сушат хлори-
стым кальцием и подвергают разгонке. Сначала отгоняется
на водяной бане эфир и бензол, а затем на масляной бане пе-
регоняют полученный этилфенилкетон, собирая фракцию, ки-
пящую при температуре 216—220°.
Выход этилфенилкетона равен 80 г, что составляет 60% or
теоретического, считая на пропионовую кислоту.
Этилфенилкетон представляет собой бесцветную или жел-
товатую жидкость, нерастворимую в воде, но растворимую в
спирте и эфире [19, 20].
141
По литературным данным, т. кип. продукта 2187760 лиг,
du20—1,013; «р —1,526 [18, 22]. Технические требования к
этилфенилкетонм по ТУ 79-647—61: т. кип. 216—220э; di20—
1010—1,014,
.’1 11 Т Е Р А Т У Р А
1. Р. Вагнер. ЖРФХО, 16 325 (1884); Вег.. 17, Ref.. 317. •
2 J. Pier’rov. Compt. rend., 188. 1501-4 (1929); С. A., 23, 4197(5).
3. M. Kutscherov. Liebigs Ann. Chem., 310, 333; Ber.. 17,3 (1884).
1. A. Korner. Ber.. 21, 277 (1888).
о. O. Pa tn pel, G. S c h i d t. Ber., 19, 2896 (1886).
6. A. P. Скол ди нов, К. А. Кочешков. Ж. общ. химии, 12, 398
(19421.
7. М. С. Малиновский, А. А. Ляпина. Ж. общ. химии, 11, 168
(1941).
8. J. R. Kohler. J Amer. Chem. Soc., 41, 427 (1919).
9. V. Pecliman. Ber.. 15, 891 (1882).
10. V. Becln. Ber., 12, 463 (1879).
II. Органические реакции, т. 8. стр. 64.
12. A. Freund. Liebigs Ann. Chem., 118, 20 (1861).
13. W. Kalle. Liebigs Ann. Chem., 119, 166 (1861).
14. A. Michael. J. Amer. Chem. Soc., 45, 423 (1901).
15. J. Cason. J. Amer. Chem. Soc., 68, 2078 (1946).
16. C. Be is. C. r„ 137, 576 (1903).
17. Charles R. Hauser. J. Atuer Chem. Soc., 70, 426 (1948).
18. Beilst., 7, 300.
19. Словарь органических соединений, т. 3, стр 520.
20. К. С Гн vers. Вег.. 45, 2771 (1912).
21 М. Tuot. М. Guard. Bull. Soc. cltlm. France, 1947, 1086 —96;
Rec. trav. chim., 48, 718 (1929).
22. T. A. T urner, P. Schrtner. J. Amer. Chem. Soc., 52, 1267
(19 0).
23. Charles R. Hauser, W i b b c r t J. H u m p h I e 11. J. Organ.
Chem., 15, 359(1950).
Поступила н феврале lf-65 r.
АЛФАВИТНЫЙ ПЕРЕЧЕНЬ СОЕДИНЕНИИ, ОПИСАННЫХ В
НАСТОЯЩЕМ ВЫПУСКЕ
Алкильные эфиры салициловой кислоты .............................. 5
.Амиловый эфир салициловой кислоты................................ 6
3-(4-Аминофенил)этиловый спирт •.................................. 7
т-Ацетаминоциннамонл-Е-тирозин ................................... 14
N-Ацетилднфенилзмнп............................................... 10
N-Ацети л-Е-фенилаланил-Е-тнрози.i-D-i л юкола мин................ 12
Х-Ацетил-Е-феннлаланнл-Е-тирозин.................................. 15
^-(5-Ацетилфурпл)-2-акриловая кислота ........................... 17
N’-Бензоил-D, L-артпии........................................... 77
1-Беизоил-2-(п-диметнлами1|офенпл)-1>2-ли1'илрохииолни........... 19
N“ -Бензонл-О,Е-фенилаланип...................................... 74
Бутен-1-илэтиловып эфир........................................ 137
7-Бутил-6,8-тридекандион......................................... 23
Бутирнлхолппйодид ............................................... 23
2-н-Бутокси-5-этил-3,4-дигидро-1,2-ниран . . 130
Гексен-1-нлэтн.товый эфир ...................................... 56
Гексиловый эфир салициловый кислоты .............................. 6
Гептиловый эфир салициловый кислоты............................... 6
2,2-Диамнно-4,4'-(бис-диметиламипо)трнфени.1Мета1> ............. 122
4,4'-Диацетоксидибутиловый эфир.................................. 27
4-(я-Диметиламино-р-метилстирил)ш1рпдин.......................... 61
2-(гс-Диметиламиностирнл)пиридин ............................... 102
4-(и-Диметиламиностирил)пириди1) ................................ 20
2,о-Диметил-3-бутилпиридин....................................... 33
2.6- Диметил-З-гексилпиридпн .................................... 32
2,6- Диметил-З-гептилпирндин . .................................. 33
5,7-Диметил-8-оксихинальдиновый альдегид........................ 34
2,6-Диметил-З-пропилпиридин...................................... 33
2,4-Диметил-2-этил-1,3-диоксолан............................... 114
•3,4-Диметоксиацетофепои....................................... . 38
Ди-(2-оксипропил)амин............................................ 92
2,8-Диоксихинолин................................................ 44
Ди-н-пропиловый эфир 2-пиридилметилфосфнновой кислоты ... 47
4,4'-Дихлордифенилоксид.......................................... 49
Изобутиловый эфир уксусной кислоты............................... 51
2-(и-Изопропилстирил)пиридин.................................... 102
^-Йодэтпловый эфир масляной кислоты.............................. 24
143
2-(п-Метилстирил)пиридин...................................... I'Jy
З-Метил-6-стирилпирндип......................................... 58
4-(Р-Метилстирил)пирндин........................................ 61,
2-Метпл-3,4Д6-(бис-трнметн |ен)ниридин............................ ’
л-Метоксиацетофеноп...................................... • 66
1-(п-Метокси-Ч-метилстирил)1111рид1Ш............................. 61
Метосульфат 8-оксп-1 -метоксихинолиния....................... 68
Моноэтиланилин................................................. <0
Натриевая соль циклогексилсульфамииовон кислоты ..............
ft-Нафтиловый эфир N1 -бензоил-1), L-фенилаланнна.............
ii-Ннтроанилид N’ -бснзоил-D, L-аргпинна . ...................
4-(л-Ни1ро-?-мети.тстирил)пиридпн.............................
5-Нитроиирослизевая кислота ...............................
2-(л-11итростирнл)пириди[.....................................
5-Ннтрофуран-карбоновая кислота ..............................
3-(Нитрофурил)-2-аллиловый спирт..............................
й-Нитрофурфуриловый спирт..................•..................
Нитроэфир 3-(4-питрофеиил)эти.1ово1 о спирта .................
Нитхромазо....................................................
131
73
<0
62
St)
102
SO
8'2
S4
S
57
2-Оксииропиламии ...........................................
8-Оксихинальдамид..........................................
8-Экснхинальдонитрпт .......................................
Пикрамин Р .......................................................
2-Прог|ПЛ-3,4,5,6-(бпс-тр|1метилен)ш1ридпн .......................
2-Стприлииридины .......................•................... ЮЗ
Сульфохлорфенол С.......................................... 165
Тетра изопропилдифен и. .......................................
?1-Толуо.тсульфанилид . .......................................
2,2,1-Триметил-1,3-диоксолан..................................... ".
2,5,7-Трнмепы-8-оксихпноли11...................................... 1*5
2,5,7-Тр11метил-8-окснхииоли11- 1-оксид ............................ НО
Три-(2-оксипропил)амин . -.................................... 63
Трнэтиламшювая соль циклогексилсульфаминовой кислоты .... 134
5-Фенил-акридиновып оранжевый хлоргидрат........................... 1'21
2-Феннл-4-№ -беизпл-5-оксазолон.................................... 75
Фузариновая (5-н-бутил-2-пиридннкарбоновая) кислота................ 125
2-Х.юрметнлпирндпп ......................................... 128
Циклогексилсульфаминовая кислота ............................ 130
2-Этнлакролеин ................................................
Этиланилид п-толуолсульфокислоты........................ . . .
Этиленйодгидрин......................................... •
Этилфенилкетон ................................................
Этиловый эфир полифосфорной кислоты ...........................
З-Этилпиридин .................................................
2-Этоксн-3-н-бутил-б-метил-3,4-дигидро-1,2-ниран.............• .
2-Этокси-3-этнл-3,4-дигидро-1.2-пирап .........................
136
71
24
110
77
134
56
138
144
Замеченные опечатки
Страница Строка Напечатано Следует читать
56 3 снизу п° Я20 nD
71 17 снизу 10%-кого раствора 10%-ным раствором
142 2 сверху «о nD „20 nD
142 4 снизу (19 0) (1930)