/















































































































Текст
УДК [543-4+542.22]: 001.8.546/5 47.0$ ?
М-54
В сборнике приведены проверенные авторами,
известные из литературных источников или оригиналь-
вые методы синтеза комплексонов, ароматических ами-
нов, фосфорорганических, гетероциклических и других
соединений.
В статьях описан синтез 279 органических соеди-
вений.
Книга представляет интерес для химиков, занятых
синтезом органических соединений, работников науч-
но-исследовательских и производственных лаборато-
рий, а также для преподавателей, аспирантов и сту-
дентов химических факультетов высших учебных
заведений.
РЕДАКЦИОННАЯ КОЛЛЕГИЯ
Р. П. Ластовский (гл. редактор). Е. А. Бо-
жевольно в, А. В. Бромберг, В. Г.
Брудзь, В. М. Дзиомк о, И. А, К Р а-
савин, Г. П. Михайло в, |Г, А. П е в ц о в|
2-5-5
97-69
СОДЕРЖАНИЕ
Алкилалкокситрихлорметилфосфинаты. С. 3. Ивин,
Л. Е. Дмитриева, К. В. Караванов................... 9
Алкилдибромфосфины. В. Г. Груздев, К. В. Караванов,
С. 3. Ивин........................................ 10
Алкилдииодфосфины. В. Г. Г руздев, К. В. Караванов,
С. 3. Ивин........................................ 12
Алкилированные ксиленолы. Л. В. Глушкова, В. И. Дере-
вякина, С. П. Старков, М. И. Волкотруб............ 14
Алкил(арил)тетрафторфосфины и диалкилтрифторфосфины.
К- В. Караванов, С. 3. Ивин....................... 18
Алкилцианацетаты. П. И. Родионов, Г. Я- Соломина . . 22
1-Амино(Ь1,Ь1-дикарбоксиметил)-8 - нафтол - 4,6-дисульфокис-
лота. В. Я. Темкина, Г. Ф. Ярошенко, И. Е. Кавченко,
Р. П. Ластовский .................. 24
1-Амино(М,М-дикарбоксиметил) - 2 - нафтол-4-сульфокнслота.
Г. Ф. Ярошенко, В. Я- Темкина, И. Е. Кавченко,
Р. И. Ластовский .................. 26
1-Аминоэтиламино-2-нафтол-4-сульфокислота и 1-аминоэтил-
амино-8-нафтол-4,6-дисульфокислота. В. Я- Темкина,
Г. Ф. Ярошенко, И. Е. Кавченко, Р. П. Ластовский 28
1-Ацетокси-1,1-дицианэтаи. Г. И. Михайлов, М. Ф. Кондра-
шова, И. А. Горкер................................ 30
Бензиловые эфиры алкилендиамино-бис-изопропилфосфоио-
вых кислот. Т. Я. Медведь, М. В. Рудо ми но .... 32
2-(2'-Бензимидазолил)-хинолин и 2-(2'-бензимидазолил)-4-фе-
нилхинолин. А. Л. Гершунс, А. И. Бризицкая ... 36
2-(2'-Бензоксазолил)-хинолин, 2-(2’ -бензоксазолил)-4-фенил-
хинолин и 2-(2'-бензоксазолил)-хиноксалин. А. Л. Гер-
шунс-, А. Н. -Бризицкая ............... 38
3
2-(2'-Бензтиазолил)-хинолин, 2-(2'-бензтиазолил)-4-фенилхи-
нолин и 2-(2'-бензтиазолил)-хиноксалин. А. Л. Гершунс,
А. Н. Бризицкая....................................... 41
6,6'-Бис-хинальдин. А. Л. Гершунс, И. -4. Растрепана 43
5-Бром-0,М-диацетилиндоксил В. М. Островская, И. А. Гор-
кер .................................................. 44
5-Броминдоксилацетат. В. М. Островская, И. А. Горкер 46
4-Бромфенилглицин-о-карбоновая кислота. В, М. Остров-
ская, И. А. Горкер.................................... 49
Винилиденцианид. Л. И. Богомолова, И. А. Горкер, С. А. Ко-
четкова, Г. И. Михайлов .............. 50
Винилсульфокислота. Е. Б, Тростянская, С. Б. Макарова,
И. Н. Мурашко......................................... 53
Гидрофильные гели на основе декстрана. С. Б. Макарова,
И. И. Мурашко, Л. Г. Голикова, Е. В. Егоров .... 56
Гидрохинон-2-метилениминодиуксусная кислота. В. fl. Тем-
кина, И. В. Цирульникова, И. П. Фадеева, Р. П. Ластов-
ский ......................... 58
4-Диазо-2,2,5,5-тетраалкилфуранидоны-3. И. К. Коробицина,
Л. Л. Родина.......................................... 59
К,М-Диалкиламиноизопропилдиалкилфосфаты. А. М. Самуи-
лов, Г. Ф. Дрегваль ................. 63
Диаллиловый эфир фенилфосфоновой кислоты. Е. Л. Гефтер 65
Диариловые эфиры винилфэсфэновой кислоты. Е. Л. Гефтер,
И. И. Бондарь......................................... 67
4,4'-Дибромднфениламин. О. М. Статкевич, Г. Т. Пилюгин 70
Дибутилацеталь ацетальдегида. Е. Л. Гефтер................ 71
Дибутиловый эфир метилфосфоновой кислоты. Е. Л. Гефтер 74
Диизоамиловый эфир 2,2'-бицинхониновой кислоты. А. Л. Гер-
шунс, А. А. Верезубова................................ 75
1-(М,Г4-Дикарбоксиметил)-аминометил-2-нафтол. В. Д. Тем-
кина, Г. Ф. Ярошенко, Л. М. Тимакова, Р. П. Ластов-
ский, Н. В. Цирульникова .............. 77
2,5-Диметилгексин-3-диол-2,5. Е. Л. Гефтер................ 78
Г,4-Диметил-2,5-диизопропилбензол. В. П. Марштупа,
Е. П. Бабин, Н. С. Морозова, О. Ф. Возиянова ... 82
М-(2',4'-Диметилфенил)-3-нитробензолсульфамид и Ы-(2'-ди-
фенил)-3-нитробензолсульфамид. И. С. Маркович,
И. В. Круглова, В. М. Дзиомко......................... 84
Динатриевая соль 3,3'-бис-[(Ь1,М-дикарбокснметил)-аминоме-
тил]-4,4'-диоксистильбена. В. Я. Темкина, Г. Ф. Яро-
шенко, Л. М. Тимакова, Р. П. Ластовский............... 86
2,2'-Диоксидифенилметан. А. В. Страшненко, А. М. Давы-
дова ................................................. 87
4
4 4'-Диоксистильбен. В. Я- Темкина, Г. Ф. Ярошенко,
’ Л. М. Тимакова, Р. П. Ластовский...................... 89
3 9-Ди-(4,4'’ОКСифенилэтил)-спиробиметадиоксан. Н. И. Ку-
цепко, А. И. Семенова............................... 91
п,п'-Дитолиламин. Г. Т. Пилюгин, Л. Е. Живоглазова ... 93
4,4,_Дифениламиндикарбоновая кислота. И. Л. Зотова,
А. А. Артамонов, Е. П. Бабин......................... 94
4,4/-Дифенил°ксиДДИкарбоновая кислота. М. С. Бегинина,
А. А. Артамонов, В. П. Марштупа, Е. П, Бабин ... 96
Дихлорангидрид винилфосфоновой кислоты. Е. Л. Гефтер,
И. А. Рогачева....................................... 98
Дихлорангидрид фенилфосфоновой кислоты. Е. Л. Гефтер 99
Дихлорангидрид p-хлорэтилфосфоновой кислоты. Е. Л. Геф-
тер, М. И. Кабачник, Л. С. Журавлева................ 102
Ди хлорид 2,3-дифеиил-5-1 Г-(п-бромфенил)-6'-бромхинолил-2']-
тетразолия. Г. Т. Пилюгин, В. В. Статкевич, С. В. Шин-
коренко, О. М. Статкевич............................ 104
Ди-р,р'-хлорэтиловый эфир винилфосфоновой кислоты.
Е. Л. Гефтер, П. А. Мошкин, Л. С. Журавлева . . . 108
Ди-р,р'-хлорэтиловый эфир фенилфосфонистой кислоты.
Е. Л. Гефтер, И. А. Рогачева....................... 111
Ди-р,р'-хлорэтиловый эфир p-хлорэтилфосфоновой кислоты.
Е. Л. Гефтер, Л. С. Журавлева....................... 113
Ди-р,р'-хлорэтилфосфористая кислота. Е. Л. Гефтер,
М. И. Кабачник...................................... 115
Диэтиламиноацеталь пропиленгликоля. Б. Г. Ясницкий,
С. А. СаркисянЦ, Е. Г. Иванюк....................... 116
1,3-Диэтилбензол. А. Б. Страшненко, Е. С. Эндельман 118
Комплексы о-оксифенилиминодиуксусной кислоты с медью
и свинцом. М. П. Русина, В. Я-Темкина, И. М. Дятлова 120
п-Крезол-2-метилениминодиуксусная кислота. В. Я- Тем-
кина, И. В. Цирульникова, Р. П. Ластовский ..... 122
Метилизоцианат. Ю. А. Наумов, Л. Г. Бажанова, А. П. Кня-
зева ............................................... 123
N-Метилкарбамнноилхлорид. Ю. А. Наумов, А. П. Князева,
Л. Г. Бажанова...................................... 125
N-Морфолил- и N-пиперидилизопропилдиалкилфосфаты.
А. М. Самуилов, Г. Ф. Дрегваль .......... 128
1-Нафтилфосфат. В. М. Островская, Т. А. Марьяшкина . . 130
2-Нафтилфосфат. В. М. Островская, Т. А. Марьяшкина . . 132
1-Нафтилфосфата динатриевая соль. В. М. Островская,
Т. А. Марьяшкина.................................... 134
5
2-Нафтилфосфата динатриевая соль. В. М. Островская,
Т. А. Марьяшкина .................. 135
Нитрил 2-хлорпентен-2-овой кислоты-5. Е. Л. Гефтер . . . 136
Оксиметилфосфоновая кислота. Е. Л. Гефтер, II. II. Бондарь 138
2-Окси-6-метилцинхониновая кислота. А. Л. Гершунс,
А. А. Верезубова................................... 141
Оксифенилди-а-тиенилметан и днарил-а-тиенилметаны.
И. И. Лапкин, М. И. Белонович....................... 142
8-Оксихинальдазин. В. М. Дзиомко, И. А. Красавин,
Ю. П. Радин......................................... 147
З-Пиридилсульфамид. А. М. Самуилов, Г. Ф. Дрегваль 149
Пикрамин С. Л. А. Оханова, С. Б. Саввин................. 151
Пиромеллитовая кислота. М. Т. Разумовская, А. И. Беля-
кова, Г. И. Карельская.............................. 154
N-Пропиламиноизопропилдиалкилфосфаты. А. М. Самуилов,
Г. Ф. Дрегваль...................................... 156
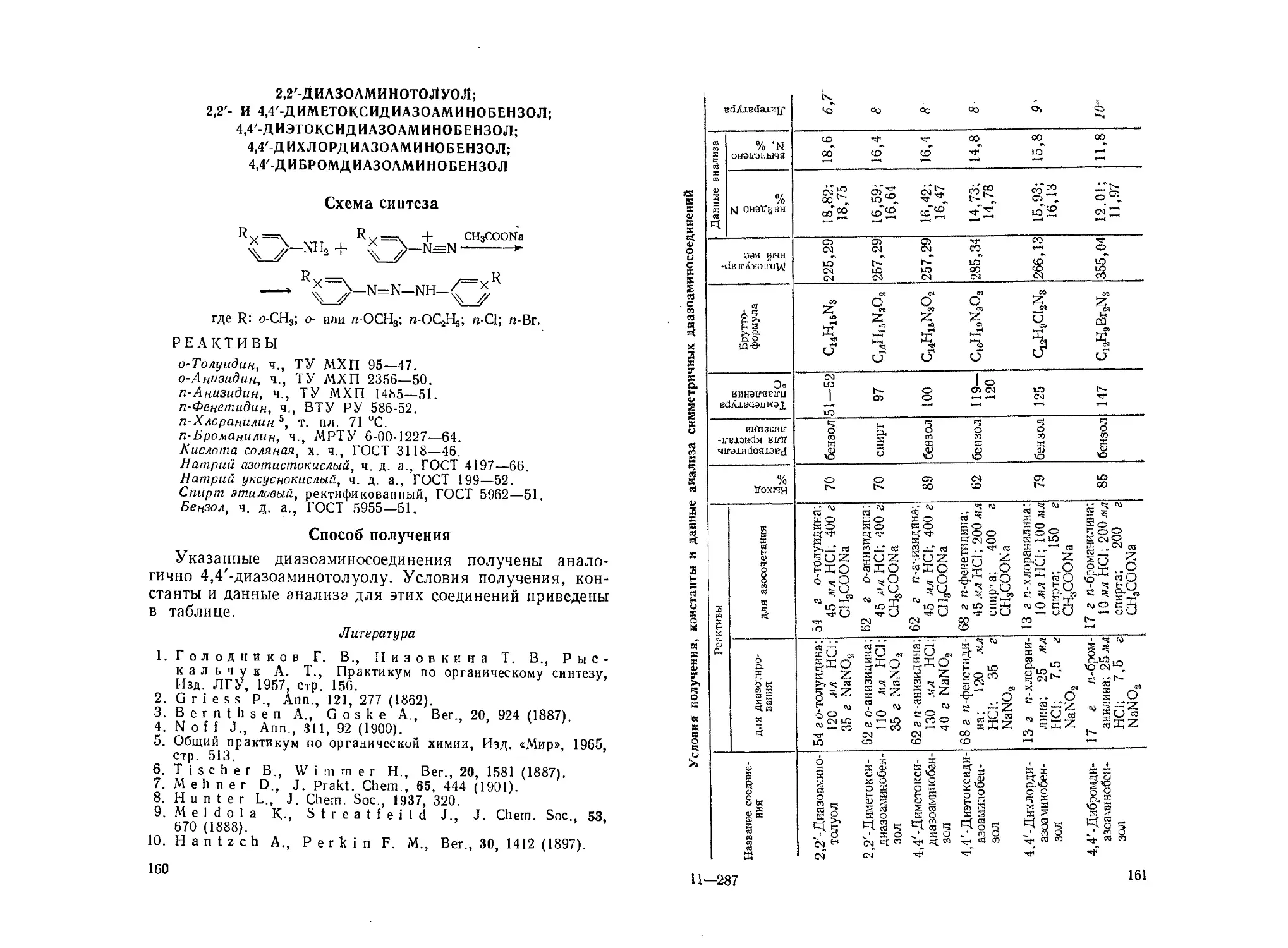
Симметричные диазоаминосоединения (диарилтриазены).
О. М. Статкевич, Г. Т. Пилюгин, В. В. Статкевич 158
Сложные эфиры а-тиенилглиоксалевой и а-тиенилгли колевой
кислот. И. И. Лапкин, Н. А. Караванов, Б. П. Дорми-
донтов.............................................. 162
Сульфиды фуранового и тиофенового рядов. И. И. Лапкин,
И. В. Богословский.................................. 166
2,2,4,4-Тетраалкилоксетан-З-карбоновые кислоты. И. К. Ко-
робицина, Л. Л. Родина.............................. 174
Тетрамид 3,3'4,4'-дифенилоксидтетракарбоновой кислоты.
М. Т. Разумовская, А. И. Белякова, Л. А. Егорова . . . 176
1,1,4,4-Тетрахлорбутан. Ю. М. Зиновьев, В. И. Кулакова,
Л. 3. Соборовский................................... 178
1,1,3,3-Тетрацианпропан. 17. М. Рыбкина, А. А. Былинкина,
Г. И. Михайлов ................... 180
2-(2'-Тозиламино-5'-бромфенил)-4Н-нафто-[2,3-<1] - [1,3]-окса-
зои-4. М. В. Лосева, В. Г. Брудзь, Б. М. Бо-
лотин............................................... 182
2-(3'-Тозиламино-2'-нафтил)-4Н-3,1 -бензоксазон-4. В. Г.
Брудзь, Б. М. Болотин, М. В. Лосева ........ 186
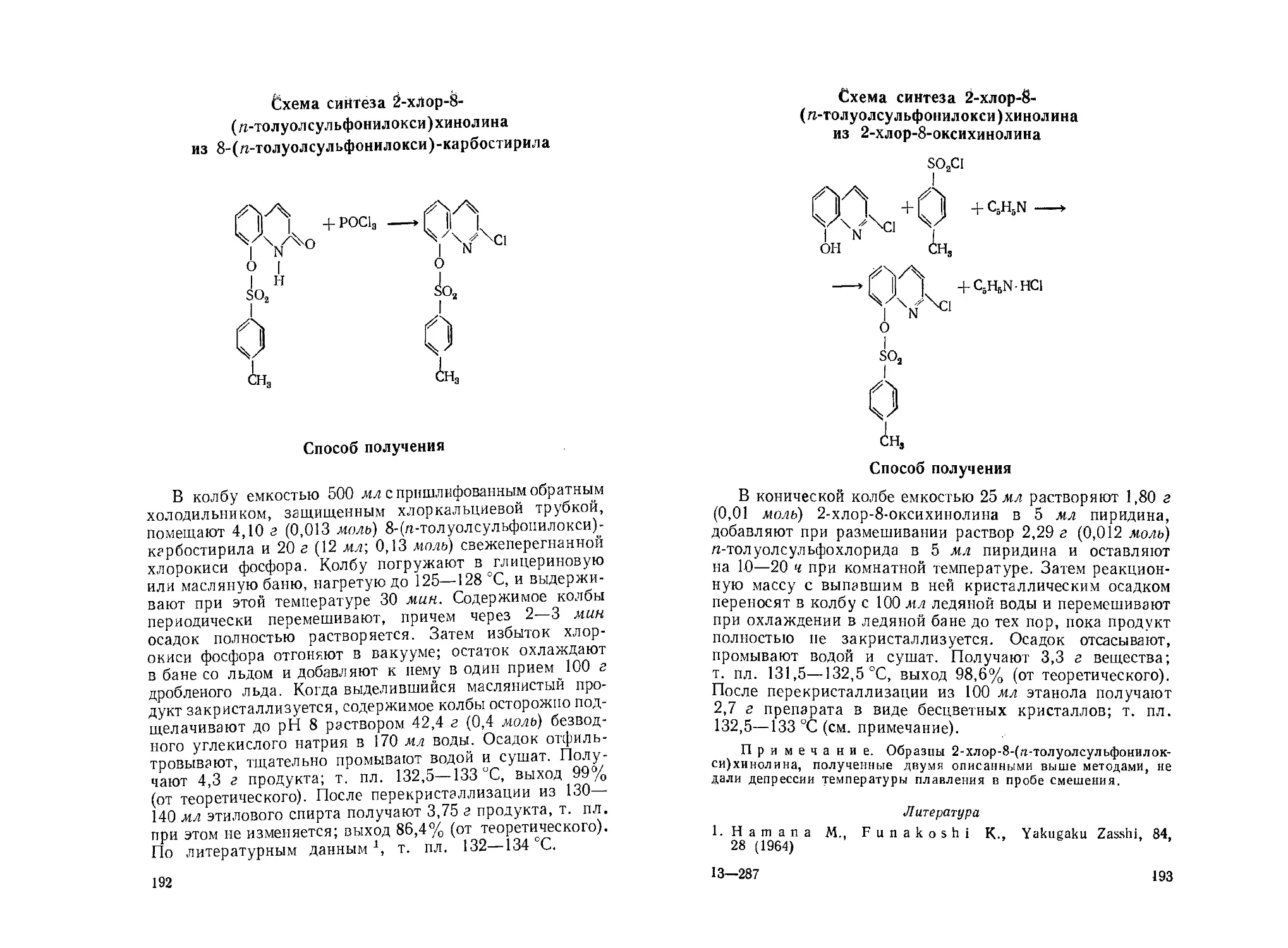
8-(гг-Толуолсульфонилокси)-карбостирил. В. М. Дзиомко,
И. А. Красавин, И. В. Рубцов........................ 189
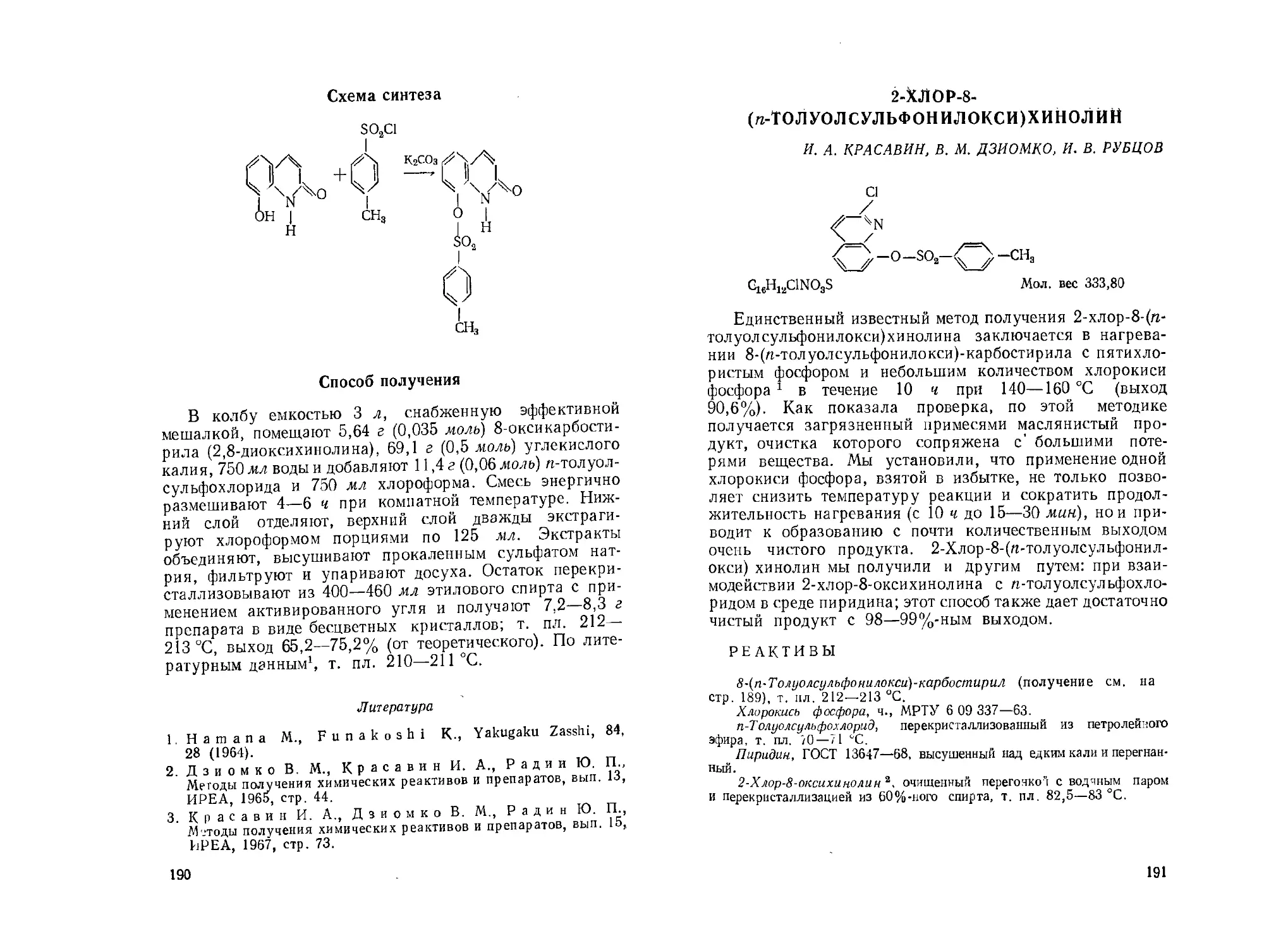
2-Хлор-8-(п-Толуолсульфонилокси)хинолин. И. А. Краса-
вин, В. М. Дзиомко, И. В. Рубцов.................... 191
6
УрамиЛдиуксусная кислота. В. ft. Темкина, Н. 4. Егоруш-
кина, Р. П. Ластовский ............... 194
2-Фенил-1,2,3-триазол. Ю. А. Наумов.................. 196
4-Фенилхинальдиновый альдегид. А. Л. Гершунс, И. А. Рас-
трепана ......................................... 198
2-Фторэтиловый эфир этиленгликоля. Ю. М. Зиновьев,
В. Н. Кулакова, Л. 3. Соборовский................ 200
2-Хиполилгндразои 8-оксихннальдинового альдегида.
В. М. Дзиомко, И. А. Красавин, Н. И. Дударева . . . 201
8-Хинолилгидразон 8-оксихинальдинового альдегида.
И. А. Красавин, В. М. Дзиомко, Б. В. Парусников,
Ю. П. Радин...................................... 203
5-Хлор-2-аминотолуол. Н. В. Донинг, В. В. Панова .... 205
Хлорангндрид дифенилфосфорной кислоты. А. Г. Фадеичева,
Л. Г. Руденко, Т. Н. Скуратовская................ 207
Хлорангидрид фенил-(3-хлорэтилфосфиновой кислоты.
Е. Л. Гефтер, И. А. Рогачева ............ 209
Хлорангидрид p-хлорэтилового эфира винилфосфиновой1 кис-
лоты. Е. Л. Гефтер, X. М. Зиявуддинов............ 210
Хлорангидриды метил- и этилтрихлорметилфосфоновых кис-
лот. С. 3. Ивин, Л. Е. Дмитриева, К. В. Караванов 212
Хлорацеталь этиленгликоля. Б. Г. Дсницкий, С. Л. Сарки-
сян, Е. Г. Иванюк .................. 213
2-Хлорпропилдихлортиофосфат. А. М. Самуилов, Г. Ф. Дрег-
валь ......................... 216
2-Хлорпропилдихлорфосфат. А. М. Самуилов, Г. Ф. Дрег-
валь ............................................ 218
2-Хлорпропилдихлорфосфит. А. М. Самуилов, Г. Ф. Дрег-
валь ......................... 219
З-Этил- и 3-н-бутиланилины. А. В. Страшненко, А. И. Алек-
сеенко, Р. К- Страшненко......................... 220
Этилендиамин-М,М'-диуксусная кислота. Н. В. Цирульни-
кова, В. ft. Темкина, Г. А. Громова, Р. П. Ластовский 223
Этиленднамино-бис-изопропилфосфоновая кислота, меченая
по углероду. Т. ft. Медведь, М. В. Рудонино...... 224
Этилендиамин-Ь1-(4 - сульфо-2-нафтол) - N,N',N' - триуксусная
кислота и этиленди 3mhh-N-(4,6-дисульфо-8-нафтол)-
М,М',М'-триуксусная кислота. Г. Ф. Ярошенко, В. ft. Тем-
кина, Н. Е. Хавчецко, Р. Л. Ластовский ....... 227
7
N-Этилениминоизопропилдиалкилфосфаты. A. М. Самуилов,
Г. Ф. Дрегваль..................................... 23&
N-Этнлениминопропанол-З. А. М. Самуилов, Г. Ф. Дрегваль 232
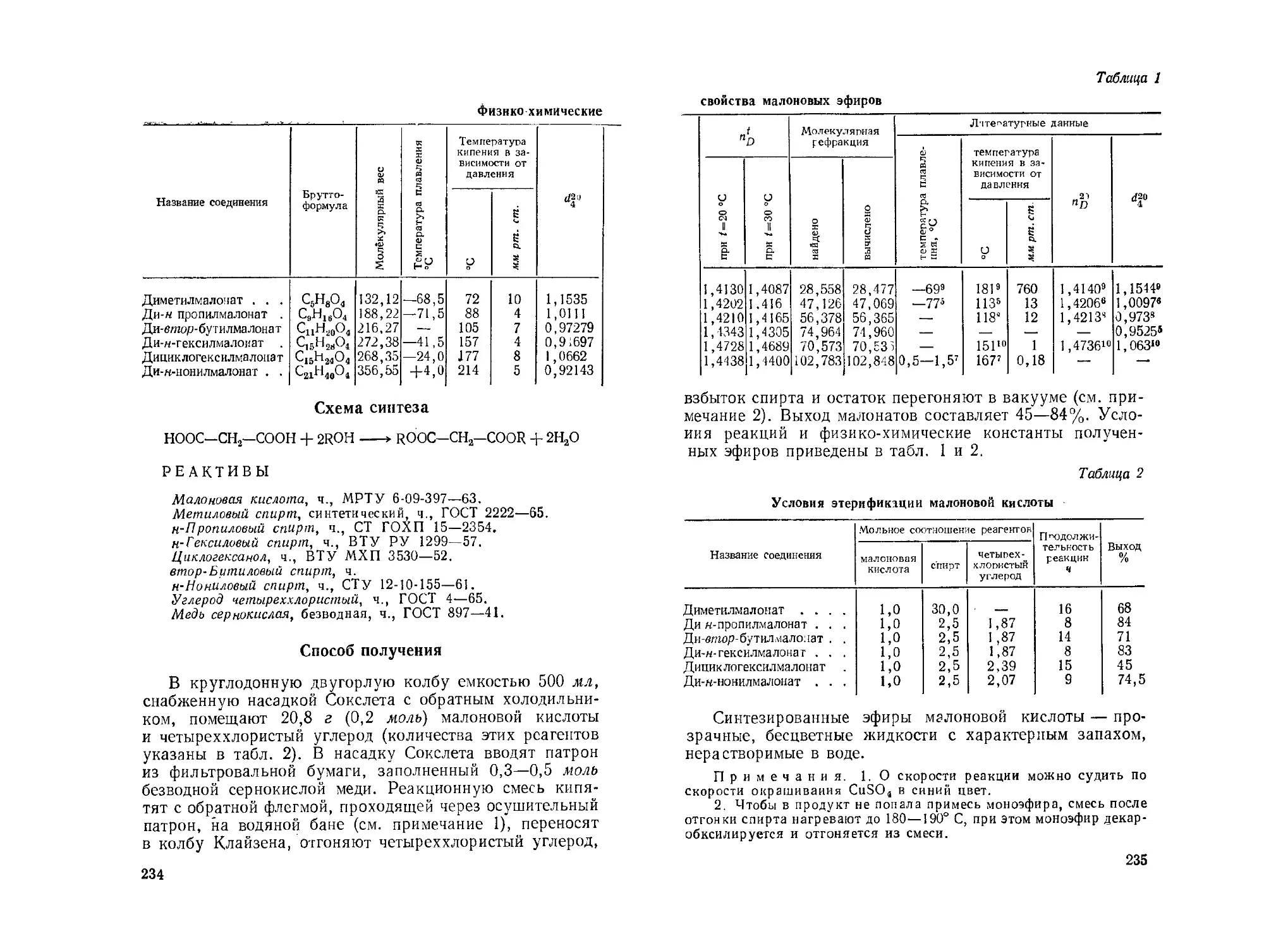
Эфиры малоновой кислоты. П. П. Родионов............... 233
Эфиры салициловой кислоты и нормальных алифатических
спиртов. П. П. Родионов, В. А. Масленников......... 236
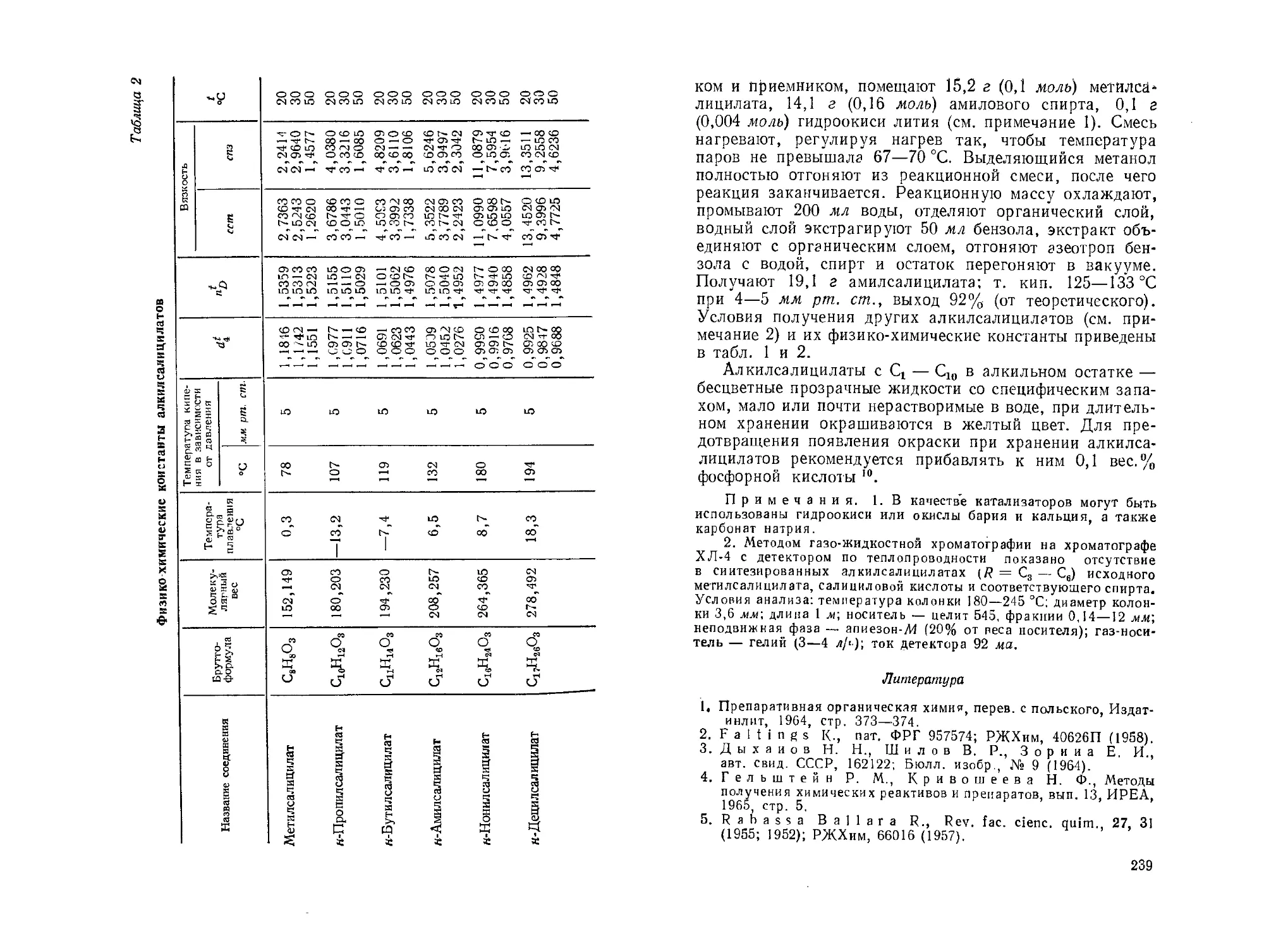
АЛКИЛАЛКОКСИТРИХЛОРМЕТИЛФОСФИНАТЫ
С. 3. ИВИН, Л. Е. ДМИТРИЕВА, К. В. КАРАВАНОВ
R4 /OR'
>р<
СС1/ О .
В литературе описаны 1-2 методы получения эфиров
фенил- и л-толилтрихлорметилфосфиновой кислоты, осно-
ванные на взаимодействии эфиров фенил- и п-толилфос-
финистой кислот с четыреххлористым углеродом. Мы раз-
работали новый способ получения О-этил- и О-изопро-
пилметилтрихлорметилфосфинатов, основанный на взаи-
модействии комплексного соединения метилтрихлорметил-
трихлорфосфина и хлористого алюминия со смесью спирта
и воды. Реакцию проводят в хлористом метилене.
О-ЭТИЛМЕТИЛТРИХЛОРМЕТИЛФОСФИНАТ
С4Н8С13О2Р Мол. вес 225,43
Схема синтеза
снзч
>РС13 А1С13 + С2Н5ОН + Н2О-»
СС13
СН3. уОС2Н5
---> >р< + А1С13-6Н,О
СС1/ Ч)
РЕАКТИВЫ
Комплексное соединение метилтрихлорметилтрихлорфосфина
с хлористым алюминием3.
Спирт этиловый ректификованный, ГОСТ 5962—51.
Метилен хлористый, ГОСТ 9968—62.
Способ получения
В фарфоровый стакан помещают 40,5 г (0,1 моль)
комплексного соединения метилтрихлорметилтрихлорфос-
?
фина с хлористым алюминием в 100 мл хлористого мети-
лена и при —20 °C постепенно добавляют из капельной
воронки смесь 4,6 г этилового спирта и 13,62 г (0,7 моль)
воды. Температуру реакционной смеси доводят до комнат-
ной, отфильтровывают осадок А1С13-6Н2О и отгоняют
хлористый метилен. Остаток перекристаллизовывают
из хлористого метилена и получают 10,9 г продукта;
т. пл. 40 °C, выход 50% (от теоретического).
Аналогично получают О-изопропилметилтрихлорме-
тилфосфинат; т. пл. 45 °C, выход 50% (от теоретического).
Литература
1. К а м а й Г. Д., АН СССР, 55, 3, 223 (1947),
2. X и с а м о в а 3. Л., ЖОХ, 20, 1162 (1950).
3. И в и и С. 3., К а р а в а н о в С. В., сб. «Методы получения
химических реактивов и препаратов», вып. 12, ИРЕА, 1965,
стр. 76.
АЛКИЛДИБРОМФОСФИНЫ
В. Г. ГРУЗДЕВ, К. В. КАРАВАНОВ, С. 3. ИВИН
/Вт
R—Р<
Вт
Алкилдибромфосфины являются полупродуктами в син-
тезе различных фосфорорганических соединений. Моно-
бромметилдибромфосфин с выходом до 50% образуется
при реакции диазометана с трехбромистым фосфором *.
Этилдибромфосфин был получен обменной реакцией тет-
раэтилсвинца или тетраэтилолова с трехбромистым фос-
фором 2. В 1958 году Майер 3 получил алкилдибромфос-
фины взаимодействием бромистых алкилов с красным
фосфором.
Перечисленные способы, как правило, малоудобны
в лабораторной практике. Главный недостаток большин-
ства из них — низкий выход алкилдибромфосфинов.
Мы разработали новый способ получения алкилдибром-
фосфинов, заключающийся в восстановлении металлами
комплексных соединений алкилтетрабромфосфинов с бро-
мистым алюминием в присутствии свежепрокаленного
10
бромистого калия. Выход алкилдибромфосфинов при
этом достигает 70%.
ЭТИЛДИБРОМФОСФИН
С2Н6Вг2Р Мол. вес 219,86
Схема синтеза
С2Н6РВг4-А1Вг3 + А1 4- КВг-> С2Н6РВг2 4- КВг-А1Вг3
РЕАКТИВЫ
Комплексное соединение этилтетрабромфосфина с бромистым
алюминием4.
Алюминий (пудра).
Калий бромистый, х. ч., безводный, свежепрокаленный,
ГОСТ 4160—65.
Способ получения
В колбу Клайзена^мкостью 0,5 л с прямым холодиль-
ником и присоединенной к горловине колбой помещают
тщательно перемешанную смесь 323,1 г (0,5 моль) комп-
лексного соединения этилтетрабромфосфина с бромистым
алюминием (см. примечание 1) и 59,5 г (0,5 моль) броми-
стого калия. В слегка расплавленную реакционную массу
из присоединенной колбы вводят небольшими порциями
(см. примечание 2) смесь 9 г (0,334 моль) алюминиевой
пудры и 39,8 г (0,334 моль) бромистого калия, одновре-
менно осторожно нагревая (см. примечание 3) и постоян-
но встряхивая содержимое колбы.
Реакция протекает со значительным выделением тепла.
После внесения всего количества смеси образовавшийся
продукт отгоняют (см. примечание 4). Получают 80,3 а
вещества; т. кип. 157—159 °C при 748 мм pm. ст., выход
73% (от теоретического), = 2,1145, п'в = 1,6073.
Примечания. 1. Комплексное соединение этилтетрабром-
фосфина с бромистым алюминием гигроскопично.
2. В случае добавления больших количеств смеси реакция
протекает бурно с выделением тепла, что может привести к обра-
зованию побочных Продуктов. Очередную порцию смеси вносят
после того, как прореагирует предыдущая.
3. Нагревание проводят так, чтобы через склянку Тищенко
с раствором перманганата калия, присоединенную в конце при-
бора, проскакивало как можно меньше пузырьков. Комплексное
соединение, нагретое выше температуры его плавления, разла-
гается .
11
4. Этилдибромфосфин легко окисляется на воздухе, поэтому
его получение н перегонку желательно проводить в токе азота
или любого другого инертного газа.
Литература
1. Якубович А. Я..Гинзбург В. А., М. а к а р о в С. П
ДАН СССР, 71, 303 (1950); Якубович А. Я., Гинз-
бург В. А., ЖОХ, 22, 1534 (1952).
2. Sacco A., Atti Accad. Line., Rend. Classe Sci. fis., mat. e nat.
11, 101 (1951); C. A. 49, 158 (1955).
3. M a i e r L., Angew. Chem., 71, 574 (1959); Helv. Chim. Acta,
46, 2026 (1963); швейц, пат. 375007 (1958).
4. П л о т н и к о в В. А., ЖРФХО, 48, 189 (1916).
АЛКИЛДИИОДФОСФИНЫ
В. Г. ГРУЗДЕВ, К. В. КАРАВАНОВ, С. 3. ИВИН
R-p<,'
Алкил- и арилдииодфосфины являются полупродук-
тами в синтезе различных фосфорорганических соедине-
ний. В литературе описаны различные способы получения
алкил- и арилдииодфосфинов: взаимодействие иодистых
алкилов с элементарным фосфором как в присутствии
катализатора, так и без него 2’ 3; реакция обмена
фенилдихлорфосфина с пропилтрииодсиланом4; восстанов-
ление метилтетраиодфосфина желтым фосфором в растворе
сероуглерода 6.
Перечисленные методы, как правило, малоудобны
в лабораторной практике.
Мы разработали 6 новый способ получения алкилдииод-
фосфинов, заключающийся во взаимодействии алкилди-
хлорфосфинов с иодидами щелочных металлов; при этом
выходы достигают 60—70%.
МЕТИЛДИИОДФОСФИН
СН312Р . Мол. вес 299,81
Схема синтеза
СН3РС12 + KI---. СН3Р12
12
РЕАКТИВЫ
Метилдихлорфосфин1 .
Калий иодистый, ч., безводный, свежепрокаленный, ГОСТ
4232—65.
Метилен хлористый, ч., ГОСТ 9968—62.
Способ получения
В круглодонную колбу с обратным ХОЛОДИЛЬНИКОМ
помещают 116,9 г (1 моль) метилдихлорфосфина* и 332,0 г
(2 моль) йодистого калия. Реакционную смесь нагре-
вают 3 ч (температура бани 160 °C). После охлаждения
до комнатной температуры в колбу приливают 250 мл
хлористого метилена и содержимое колбы перемешивают.
Осадок отфильтровывают через фильтр Шотта № 2 и про-
мывают хлористым метиленом 4 раза порциями по 25 мл.
От фильтрата отгоняют хлористый метилен, а остаток
перегоняют в вакууме. Получают 183 г продукта с т. кип.
74—76 °C при 5 мм рт. ст.; выход 61 % (от теоретическо-
го); т. пл. 30,5 °C (в запаянном капилляре), d'f = 2,7943.
По аналогичной методике можно получать этилдииод-
фосфин с выходом 56% ит. кип. 90—92 °C при 4 ммрт.ст.
или 103—104 °C при 9 мм pm. cm.; dl° 2,5245.
Литература
1. Bennett F. W., Е m е 1 е u s Н., Haszeldine R. N.,
J. Chem. Soc., 1953, 1565.
Burg A.В.,Mahler N.,Bilbo A., Haber C. P.,Hez-
z i n g D. L., J. Am. Chem. Soc., 79, 247 (1957).
2. E m e 1 e и s H., Smith, J. Chem. Soc., 1959, 375
3. Maier L., Helv. Chim. Acta, 4G, 2026 (1963); швейц, пат.
375007 (1958).
4. Anderson H. H., J. Am. Chem. Soc., 75, 1576 (1953).
5. Гинсбург В.А., Привезенцева Н.Ф., ЖОХ, 28, 736
(1958).
6. Г p у з д e в В. Г., К а р а в а н о в К. В., И в и н С. 3., авт.
свид. СССР 186467; Бюлл. изобр., № 19 (1966).
7. Ком ков И. П., Караванов К. В., Ивин С. 3.,
Методы получения химических реактивов и препаратов, вып. 12.
ИРЕА, 1965, стр. 5.
* Метилдихлорфосфин на воздухе легко окисляется.
13
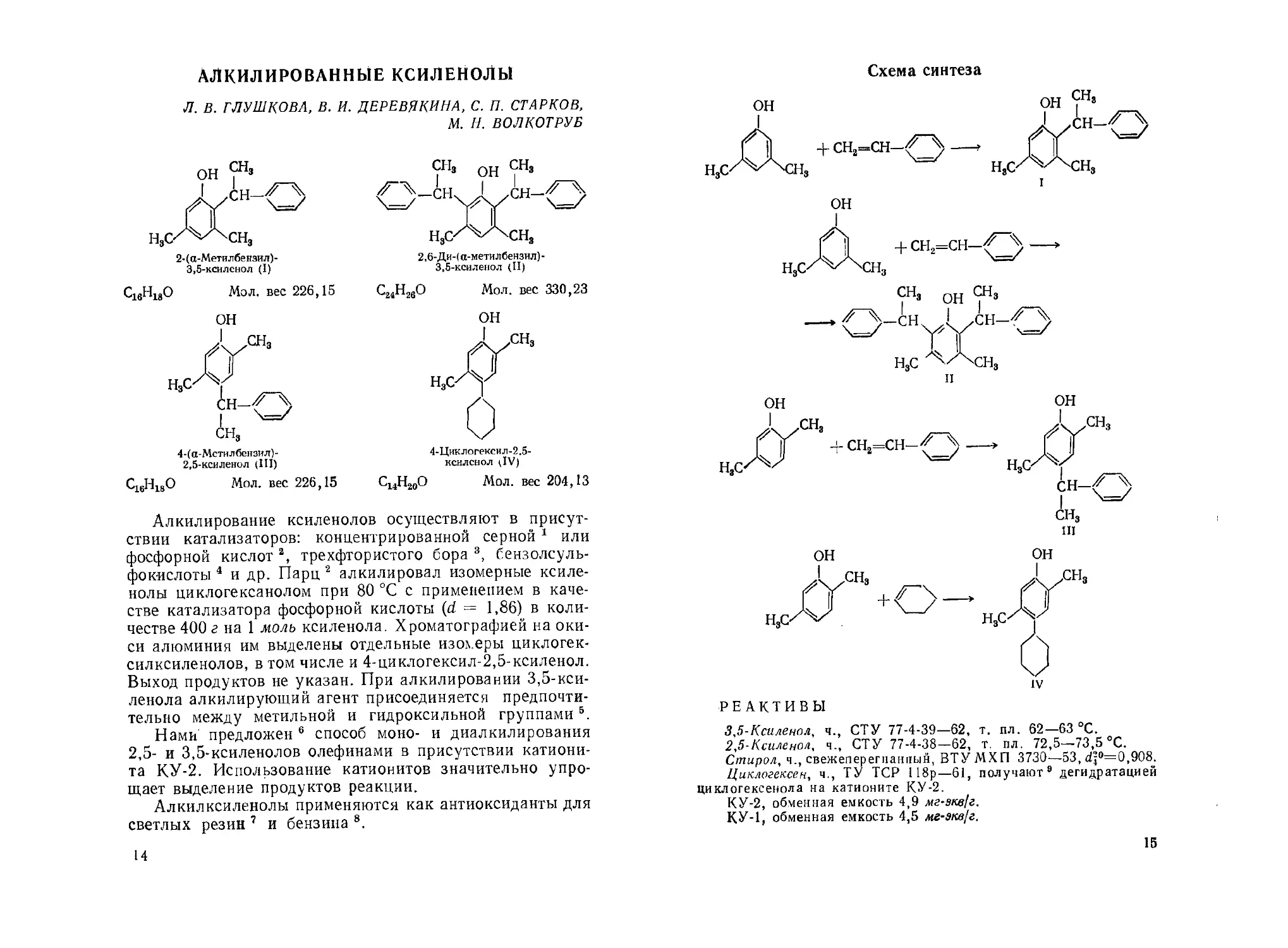
АЛКИЛИРОВАННЫЕ КСИЛЕНОЛЫ
Л. В. ГЛУШКОВА, В. И. ДЕРЕВЯКПНА, С. П. СТАРКОВ,
М. Н. ВОЛКОТРУБ
2-(«-Метилбензил)-
3,5-ксилснол (I)
С16Н18О Мол. вес 226,15
2,6-Ди-(а-метилбензил)-
3,5-ксиленол (П)
С24Н26О Мол. вес 330,23
ОН
СН3
4-(а-Мстилбензил)-
2,5-ксиленол (Ш)
4-Циклогексил-2.5-
ксилснол UV)
С16Н18О Мол. вес 226,15
С14Н20О Мол. вес 204,13
Алкилирование ксиленолов осуществляют в присут-
ствии катализаторов: концентрированной серной 1 или
фосфорной кислот 2, трехфтористого бора 3, бензолсуль-
фокислоты 4 и др. Парц 2 алкилировал изомерные ксиле-
нолы циклогексанолом при 80 °C с применением в каче-
стве катализатора фосфорной кислоты (d = 1,86) в коли-
честве 400 г на 1 моль ксиленола. Хроматографией на оки-
си алюминия им выделены отдельные изомеры циклогек-
силксиленолов, в том числе и 4-циклогексил-2,5-ксиленол.
Выход продуктов не указан. При алкилировании 3,5-кси-
ленола алкилирующий агент присоединяется предпочти-
тельно между метильной и гидроксильной группами5.
Нами предложен 6 способ моно- и диалкилирования
2,5- и 3,5-ксиленолов олефинами в присутствии катиони-
та КУ-2. Использование катионитов значительно упро-
щает выделение продуктов реакции.
Алкилксиленолы применяются как антиоксиданты для
светлых резин 7 и бензина 8.
14
Схема синтеза
РЕАКТИВЫ
3,5- Ксиленол, ч., СТУ 77-4-39—62, т. пл. 62—63 °C.
2,5- К.силенол, ч., СТУ 77-4-38—62, т. пл. 72,5—73,5 °C.
Стирол, ч., свежеперегпанпый, ВТУ МХП 3730—53, dj°=0,908.
Циклогексен, ч., ТУ TCP 118р—61, получают9 дегидратацией
циклогексенола на катионите КУ-2.
КУ-2, обменная емкость 4,9 мг-экв!г.
КУ-1, обменная емкость 4,5 ле-а/св/г.
15
Способ получения
Синтез 2- {а-м',тилбензил)-3,5, ксиленола (/). К смеси
29,32 г (0,24 коль) 3,5-ксиленола и 3 г КУ-1 при SC— 110 °C
в течение 30 кин прибавляют 20,83 г (0,18 коль) стирола.
После двухчасовой выдержки при этой температуре реак-
ционную смесь декантируют и перегоняют в вакууме.
Получают 25,3 г продукта в виде желтоватой масляни-
стой жидкости; т. кип. 160—170 °C при 3 кк рт. ст.,
выход 56% (от теоретического); d2° = 1,057, = 1,5830,
MRd = 71,44 (вычислено 70,41).
При длительном стоянии вещество кристаллизуется.
После перекристаллизации из н-гексена т. пл. препа-
рата 73—73,5 °C.
В ИК-спектре имеется сильная полоса поглощения
в области 848 см"1, соответствующая 1, 2, 3, 5-замещен-
ному бензолу, а также полосы поглощения в областях
710 и 750—775 сж-1, соответствующие монозамещенному
бензольному кольцу 10.
Состав синтезированного продукта (С1ВН18О):
с н
Найдено, %.......... 84,67 8.27
Вычислено, %........ 84,90 7,96
Синтез 2,6-ди-{а-кетилбензил)-3,5-ксиленола {II). Про-
дукт получают аналогично предыдущему, исходя из 24,43 г
(0,20 коль) 3,5-ксиленола, 52,07 г (0,45 коль) стирола
и 0,25 г концентрированной серной кислоты. Реакцион-
ную массу нейтрализуют 50%-ным раствором едкого нат-
ра, экстрагируют бензолом и экстракт сушат сульфатом
натрия. После отгонки бензола остаток перегоняют в ва-
кууме. Получают 55,5 г продукта в виде вязкой желтой
жидкости; т. кип. 200—210 °C при 3 кк рт.ст., выход
84% (от теоретического); d20 = 1,079, лЬ = 1,5970, MRD =
= 104,51 (вычислено 103,75).
В ИК-спектре имеется полоса поглощения средней
интенсивности в области 855 слг-', соответствующая пен-
тазамещенному бензолу и, а также полосы поглощения,
характерные для монозамещенных бензольных колец.
Состав синтезированного продукта (С24Н26О):
с н
Найдено, %.......... 87.03 7.99
Вычислено, %........ 87,10 8,15
16
Синтез 4-(а-метилбензил)-2,Г>-ксиленола (ИГ). Продукт
получают аналогично предыдущему из 12,21 г (0,1 моль)-,
2,5-ксиленола, 6 г КУ-2 и 10,41 г (0,1 моль) стирола.
Вещество представляет собой бесцветную маслянистую
жидкость; т. кип. 170—174 °C при 7 мм рт. ст.; выход
14,7 г — 65% (от теоретического). При стоянии кристал-
лизуется, т. пл. 90,5—91 °C (после перекристаллизации
из петролейного эфира).
В ИК-спектре обнаруживаются полосы поглощения
средней интенсивности в областях 860—870 см'1 и 900 см'1,
характерные для 1, 2, 4, 5-замещенного бензола и, а так-
же полосы поглощения, соответствующие монозамещен-
ному бензольному кольцу.
Синтез 4-циклогексил-2,5-ксиленола (/К). К смеси 36,64 г
(0,3 моль) 2,5-ксиленола и 18 г КУ-2 при 125 °C в течение
90 мин прибавляют 16,42 г (0,2 моль) циклогексена.
Реакционную смесь выдерживают 20 мин при этой тем-
пературе, декантируют и перегоняют в вакууме. Полу-
чают 29,4 г продукта с т. кип. 165—172 °C при
9 мм рт. ст.; выход 72% (от теоретического); т. пл. 73—
74 °C (после перекристаллизации из петролейного эфира).
По литературным данным 2, т. пл. продукта, очищенного
хроматографией па окиси алюминия, 84 °C. Продукт
полностью растворяется в 10%-ном водном растворе едко-
го натра, что указывает на отсутствие орто-изомера 12.
Состав синтезированного продукта (С14Н20О):
Найдено, %........................
Вычислено, %......................
с н он
82,02 10.14 6,58
82,30 9,67 6,10
Литература
1. Пат. США 2900362 (1959); РЖХим, 14П380 (1961).
2. Parc С., Rev. Inst, franc, petrole, 15, Xs 4, 680 (1960).
3. Фр. пат. 1128968 (1957).
4. Пат. США 2537636 (1946).
5. Р h. В и п - Н о i, Sy М., N i е 1 М., Coinpt. rend., 254,
i 4476 (1962).
6. Старков С. П., Глушкова Л. В., ЖПХ, 40, № 1
(1967).
7. Англ. пат. 723838 (1955); РЖХим, 37559 (1965).
8. Пат. США 2248827 (1946); Zbl., 1942, 2092.
9. Ш у й к и н Н. И., Поздняк Н. А., Д о б р ы н и-
н а Г. П., Изв. АН СССР, Сер. хим. вып. 9, 1705 (1964).
10. Н а к а н и с и К., Инфракрасные спектры и строение орга-
нических соединений. Изд. «Мир», 1965, стр. 32.
2-287
17
11. Рудольфи Т. А., ж. прикл. спектроскопии, 7, 366 (1965).
12. Беллами Л., Инфракрасные спектры сложных молекул,
Издатинлит, 1963.
АЛКИЛ (АРИЛ) ТЕТРАФТОРФОСФИНЫ
И ДИАЛКИЛТРИФТОРФОСФИНЫ
А. В. КАРАВАНОВ, С. 3. ИВИН
RPF4 и r2pf3
В литературе описаны два способа получения алкил-
(арил)тетрафторфосфинов и диалкилтрифторфосфипов.
Первый из них основан на взаимодействии алкил(арил)-
дихлорфосфинов и диалкилхлорфосфинов с фторидами
тяжелых металлов х. Существенным недостатком этого
способа является малая доступность исходных реаген-
тов. Второй способ основан на фторировании комплекс-
ных соединений алкил(арил)тетрахлорфосфинов и диал-
килтрихлорфосфинов и хлористого алюминия фтористым
водородом или фторидами тяжелых металлов *. Однако
в этом случае получаются низкие выходы продуктов,
а при использовании фтористого водорода реакция ста-
новится трудноуправляемой и требует особого оборудо-
вания, исключающего стеклянные детали.
Нами разработан2 новый, более доступный, способ
получения алкил(арил)тетрафторфосфинов и диалкилтри-
фторфосфинов, основанный на взаимодействии алкил-
(арил)дихлорфосфинсульфидов и диалкилхлорфосфинсуль-
фидов с фторидами тяжелых металлов, в частности с трех-
фтористой сурьмой. Реакция протекает в две стадии.
Вначале образуются алкил(арил)дифторфосфинсульфиды
и диалкилфторфосфинсульфиды:
R. .С1
>Р< +SbF3
ХС1
R4 .Cl
>Р< ' + SbF3
RZ Ч
которые при дальнейшем нагревании с трехфтористой
сурьмой превращаются в алкил(арил)тетрафторфосфины
18
и диалкилтрифторфосфины:
Выход продуктов составляет 80%.
МЕТИЛ ТЕТРАФТОРФОСФИН
ch3f4p
Мол. вес 122,00
Схема синтеза
+ SbF3--->CH3PF4
РЕАКТИВЫ
Метилдихлорфосфинсульфид *, перегнанный.
Трехфтористая сурьма, ч., безводная, свежепрокаленная,
МРТУ 6-09-524—63.
Способ получения
В колбу с капельной воронкой и обратным холодиль-
ником (длиной 350 лои), соединенным с ловушкой (охлаж-
денной до минус 30—40 °C), помещают 53,6 г (0,30 моль)
трехфтористой сурьмы и из капельной воронки медленно
прибавляют 33 г (0,22 моль) метилдихлорфосфинсульфида.
Затем содержимое колбы в течение 1 —1,5 ч нагревают
(температура бани 60—70 °C) так, чтобы образующийся
вначале метилдифторфосфинсульфид (т. кип. 61 °C при
760 мм рт. ст.) не отгонялся из реакционной массы,
а реагировал с трехфтористой сурьмой до образования
метилтетрафторфосфина. Последний по мере образования
отгоняется и конденсируется в ловушке. Собранный
в ловушке метилтетрафторфосфин перегоняют в токе
азота *. Получают 20,6 г продукта; выход 80% (от теоре-
тического), т. кип. 11—12 °C при 753 мм pm. cm.,dl =
= 1,4538.
* Метилтетрафторфосфин на воздухе гидролизуется.
2*
19
ЭТИЛТЕТРАФТОРФОСФИН
C2H5F4P Мол. вес 136,02
Схема синтеза
CjHj. , Cl
>Р< + SbF3---------CaHsPF4
<1
РЕАКТИВЫ
Этилдихлорфосфинсульфид \ перегнанный.
Трехфтиристая сурьма, ч., безводная, свежепрокаленная,
МРТУ 6-09-524—63.
Способ получения
В колбу с капельной воронкой и обратным холодиль-
ником (длиной 300 мм), на верхнем конце которого нахо-
дится насадка Вюрца с термометром, прямым холодиль-
ником с пауком и приемниками, помещают 32,5 г
(0,18 моль) трехфтористой сурьмы и из капельной ворон-
ки медленно прибавляют 20 г (0,12 моль) этилдихлорфос-
финсульфида. Затем содержимое колбы нагревают (тем-
пература бани 60—70 °C) так, чтобы образующийся этил-
тетрафторфосфин отгонялся. При повторной перегонке
получают 12,5 г продукта с т. кип. 33—34,5 °C; выход
75% (от теоретического), dj° = 1,3074.
ФЕНИЛТЕТРАФТОРФОСФИН
C6H5F4P
Мол. вес 184,07
Схема синтеза
С6Н5. С1
>Р< + SbF3----------» C6H5PF4
Cl
РЕАКТИВЫ
Фенилдихлорфосфинсульфид \ перегнанный.
Трехфтористая сурьма, ч., безводная, свежепрокаленная,
МРТУ 6-09-524—63.
Способ получения
В колбу с капельной воронкой и насадкой Вюрца
с термометром помещают 40 г (0,22 моль) трехфтористой
сурьмы и из капельной воронки медленно прибавляют
20
32 г (0,15 моль) фенилдихлорфосфинсульфида. После при-
бавления всего количества фенилдихлорфосфинсульфида
содержимое колбы нагревают и одновременно отгоняют
образующийся фенилтетрафторфосфин. При повторной
перегонке получают 21,3 г продукта с т. кип. 134—135 °C
при 750 мм рт. ст.-, выход 76% (от теоретического),
= 1,3888, п2в = 1,4245.
ДИМЕТИЛТРИФТОРФОСФИН
c2H6F3p
Мол. вес 118,03
Схема синтеза
+ SbF3
РЕАКТИ ВЫ
Диметилхлорфосфинсульфид \ перегнанный.
Трехфтористая сурьма, ч., безводная, свежепрокаленная,
МРТУ 6-09-524—63.
Способ получения
В колбу Клайзена с елочным дефлегматором (длиной
230 мм), прямым холодильником, термометром и капель-
ной воронкой помещают 26 г (0,15 моль) трехфтористой
сурьмы и из капельной воронки медленно прибавляют
16,7 г (0,13 моль) диметилхлорфосфинсульфида. Реакция
протекает с выделением тепла. Затем содержимое колбы
нагревают до температуры отгонки диметилтрифторфос-
фина (60—65 °C). При повторной перегонке получают
12 г продукта с т. кип. 60—61 °C при 750 мм рт. ст.,
выход 78% (от теоретического), а^0 = 1,2155,/?Ь"= 1,3230.
По описанным методикам, исходя из различных алкил-
(арил)дихлорфосфинсульфидов и диалкилхлорфосфинсуль-
фидов, можно получать соответствующие алкил(арил)-
тетрафторфосфины и диалкилтрифторфосфины.
Литература
1. Конков И. П , Ивин С. 3., Караванов К. В.,
Смирнов Л. Е., Методы получения химических реактивов
и препаратов, вып. 12, ИРЕА, 1965, стр. 14, 20, 23, 57, 59.
2. Караванов К. В., Ивин С. 3., Груздев В. Г.,
авт. свид. СССР 18485156; Бюлл. пзобр., № 1б" (1966).
21
АЛКИЛЦИАНАЦЕТАТЫ
л. п. Родионов, г. я. соломина
N=C—СН3—С—О—R
где R = С4Н9; С5Н13; С6Н13; С7Н1Б.
Алкилцианацетаты с Ct— С8 в алкильном остатке,
а также циклогексил-, циклопентил-, фенил- и алкоксиал-
килцианацетаты получают цианированием соответствую-
щих эфиров монохлоруксусной кислоты В 2. Метил-
и бутилцианацетаты были получены переэтерификацией
этилцианацетата в присутствии фторида калия 3. Нами
разработан способ получения алкилцианацетатов с С4— С,
в алкильном остатке переэтерификацией этилцианаце-
тата спиртами в присутствии кислых или щелочных ката-
лизаторов. В качестве щелочных катализаторов мы при-
менили алкоголяты натрия и соответствующих спиртов
(1—2 мол. %), в качестве кислого катализатора—п-толуол-
сульфохлорид.
Схема синтеза
N=C—СН2—С—О—С2Н5 + R-OH
/°
-------> N= С—СН2—С—О—R
РЕАКТИВЫ
Циануксусный эфир (этилциаиацетат), ч., РТУ 539—60.
Натр едкий, х. ч., ГОСТ 4328—66.
n-Толуолсульфохлорид, ч., МРТУ 6-09-447—63.
н-Бутиловый спирт, ч., ГОСТ 6006—51.
н-Амиловый спирт, ч., СТУ 10-158—61.
н-Гексиловый спирт, ч., СТУ 12-10-154—61.
н-Гептиловый спирт, ч., СТУ 12-10-157—61.
Способ получения
В трехгорлую колбу из термостойкого стекла емкостью
200 мл, снабженную термометром, елочным дефлегмато-
ром с термометром и нисходящим холодильником, поме-
щают 32,5 г (0,44 моль) н-бутилового спирта, 0,3 а
22
Условия получения алкилцианацетатов Таблица 1
23
(0,0075 моль) порошкообразного едкого натра или 0,476 г
(0,0025 моль) п-толуолсульфохлорида, нагревают до пол-
ного растворения катализатора, после чего добавляют
28,2 г (0,25 моль) этилцианацетата. Смесь кипятят
до выделения расчетного количества этанола 3—9 ч,
регулируя обогрев куба так, чтобы температура паров
не превышала 78—80 °C. Реакционную массу охлаждают,
добавляют 100 мл воды и встряхивают. Катализатор
нейтрализуют по универсальному индикатору либо 2—
5%-ным раствором серной кислоты (при щелочном ката-
лизе), либо 5%-ным раствором карбоната натрия. Затем
добавляют 20 мл бензола, отделяют органический слой,
водный слой экстрагируют 10 мл бензола и экстракт
присоединяют к органическому слою. Бензол, воду, избы-
ток спирта отгоняют в вакууме водоструйного насоса
и остаток перегоняют при остаточном давлении 10—
20 мм рт. ст. Аналогичным способом получены другие
алкилцианацетаты, условия синтеза и физико-химические
константы которых приводятся в Табл. 1 и 2. Полу-
ченные алкилцианацетаты прозрачные, бесцветные жид-
кости, нерастворимые в воде.
Литература
1. Пат. США 2985682 (1961); РЖХим, 14Л96 (1961).
2. Синтезы органических препаратов, сб. 1, Издатинлит, 1949,
стр. 560.
3. Minoru J., Hiroshi М., Shinichi A., J. Sci. Res. Inst., 52, 105
(1958); РЖХим, 38538 (1959).
1-AMHHO-(N, N-ДИКАРБОКСИМЕТИЛ)-
8-НАФТОЛ-4,6-ДИСУЛЬФОКИСЛОТА
В. Я. ТЕМКИНА, Г. Ф. ЯРОШЕНКО, Н. Е. ХАВЧЕНКО,
Р. П. ЛАСТОВСКИЙ
.СН2СООН
ОН N<
| I ХСН2СООН
;о3н
Мол. вес 435,38
24
1-Амино-(Ы,Ы-дикарбоксйметил) - 8 - нафтол-4,6-дисуль-
фокислота является новым флуоресцирующим комплек-
соном. Это соединение также может быть использовано
в качестве азосоставляющей для получения колориметри-
ческих реагентов. Комплексон получен взаимодействием
1-амино-8-нафтол-4,6-дисульфокислоты (К-кислоты) с мо-
нохлоруксусной кислотой при температуре 85,—90 °C
и pH 7—8 (для избежания образования нафтоксиуксусной
кислоты).
Схема синтеза
,СН2СООП
ОН NH, ОН N<
| | 1| ХСН2СООН
РЕАКТИВЫ
К-кислота, ч., МРТУ 6-09-1636—64.
Кислота монохлоруксусная, ч., ГОСТ 5836—51.
Натр едкий, ч., ГОСТ 4328—66, 20%-ный раствор.
Кислота соляная, ч., ГОСТ 3118—46.
Способ получения
В трехгорлую колбу, снабженную механической мешал-
кой, обратным холодильником и термометром, помещают
раствор 17 г (0,05 моль) К-кислоты в 400 мл воды и дово-
дят pH до 7 раствором едкого натра, после чего вводят
раствор 14,2 г (0,15 моль) монохлоруксусной кислоты
в 15 мл воды, предварительно нейтрализованной раство-
ром едкого натра. Реакционную массу перемешивают,
поддерживая температуру 90—95 °C и pH 7—8 (по уни-
версальной индикаторной бумаге). Реакцию ведут до пре-
кращения снижения pH и установления отрицательной
пробы на диазотирование. Горячий раствор фильтруют,
отгоняют в вакууме 200 мл растворителя, охлаждают
в бане со льдом и подкисляют до pH 1,5—2 концентри-
рованной соляной кислотой. Выпавший объемный осадок
перекристаллизовывают из 50 мл воды с добавлением
активированного угля. Полученный после перекристал-
лизации белый осадок отфильтровывают, промывают 80%-
25
ИЫм этиловым Спиртом до отрицательной пробы на СГ-
ионы и сушат в сушильном шкафу при 70—80 °C. Полу-
чают 8 г натриевой соли комплексона; выход 38% (от тео-
ретического).
Для получения комплексона в виде кислоты 2 г соли
растворяют в 200 мл воды и пропускают 1 через колонку
с катионитом КРС-5 (емкостью 50 мл, диаметром 14 мм),
отбирают фракцию со значением pH 1—2, отгоняют воду
в вакууме, в конце отгонки добавляют в несколько прие-
мов 100 мд абсолютного этанола и отгоняют досуха.
Полученный осадок сушат в сушильном шкафу при 60—
70 °C до постоянного веса. Выход комплексона 1,8 г.
Вещество растворяется в воде и щелочах, не раство-
ряется в органических растворителях.
Состав синтезированного продукта (C14Hi3NOnS2):
с н N
Найдено, %............... 39,08; 38,7 3,3; 3,59 3,45; 3,34
Вычислено, %............. 38,6 2,98 3,22
Литература
1. Авт. свид. СССР 178000; Бюлл. изобр., № 2 (1966).
1-AMHHO-(N, N-ДИКАРБОКСИМЕТИЛ)-
2-НАФТОЛ-4-СУЛБФОКИСЛОТА
Г. Ф. ЯРОШЕНКО, В. я. ТЕМКИНА, Н. Е. ХАВЧЕНКО,
Р. П. ЛАСТОВСКИЙ
c14h13no8s
Мол вес 355,29
1 - Амино - (N.N-дикарбоксиметил) - 2 -нафтол-4-сульфо-
кислота является новым флуоресцирующим комплексо-
ном, содержащим оксигруппу в орто-положении к хела-
тообразующей иминодиацетатной группе. Это соединение
26
образует прочные комплексы с металлами, что в ряде
случаев сопровождается гашением флуоресценции. Комп-
лексон получен взаимодействием 1-амино-2-нафтол-4-суль-
фокислоты (эйконоген-кислоты) с цианистым калием
и формальдегидом в щелочной среде С
Схема синтеза
,СН,СООН
N<
I ХСН2СООН
агн
SOsH
РЕАКТИВЫ
Эйконоген-кислота, ч., ВТУ 4178—54.
Цианистый калий, ч., ТУ А4ХП 3961—53.
Кислота соляная, ч., ГОСТ 3118—46.
Формальдегид, ч., ТУД1ХП 26.31—51.
Способ получения
Реакцию ведут в токе азота. В четырехгорлую колбу,
снабженную механической мешалкой, обратным холо-
дильником и термометром, вводят раствор 12 г (0,05 моль)
эйконоген-кислоты, 3 г едкого натра и 7,8 г (0,12 моль)
цианистого калия в 300 мл воды. Разбавляют 10,6 г
(0,12 моль) 37%-ного раствора формальдегида водой
до объема 100 мл и 20 мл полученного раствора вводят
в реакционную смесь при 75 °C в течение 1 ч. По оконча-
нии добавления формальдегида отгоняют в вакууме 20 мл
растворителя. Остаток формальдегида вводят в смесь
в четыре приема при 80, 85, 90 и 95 °C, каждый раз в тече-
ние часа, после каждой порции отгоняя в вакууме по 20 мл
растворителя. После добавления всего количества форм-
альдегида реакционную массу выдерживают 2 ч при
95 °C и перемешивании, охлаждают,'подкисляют соляной
кислотой до pH 2 и упаривают досуха в вакууме в токе
азота. Продукт отделяют от неорганического остатка
экстракцией абсолютным этиловым спиртом в аппарате
Сокслета. После отгонки спирта в вакууме получают 6 г
продукта в виде, темно-фиолетового порошка, хорошо
27-
растворимого в воде и спирте, нерастворимого в ацетоне,
бензоле, хлороформе. Выход 34% (от теоретического).
Состав синтезированного продукта (C14H13NO8S):
N
Найдено, %................3.72; 3,44
Вычислено, %.............3,94
Литература
1. A i k е n J. К., Chem. a. Ind., 17, 1334 (1956).
1-АМИНОЭТИЛАМИНО-2-НАФТОЛ-
4-СУЛЬФОКИСЛОТА И 1-АМИ НОЭТИЛАМИНО-
8-НАФТОЛ-4,6-ДИСУЛЬФОКИСЛОТА
В. Я- ТЕМКИНА, Г. Ф. ЯРОШЕНКО, Н. Е. ХАВЧЕНКО,
Р. П. Л ACT О ВС КИН
—NH—СН2—СН2—NH2
ОН
Ci2H14N2O4S
Мол. вес 282,31
HO.S.
О-°н
ho3s-<J>-nh-ch2-ch2-nh2
c12h14n207s2
Мол. вес 362,38
1-Аминоэтиламино-2-нафтол-4-сульфокислота и 1-ами-
ноэтиламино-8-нафтол-4,6-дисульфокислота являются ис-
ходными продуктами в синтезе флуоресцирующих комплек-
сонов. Эти два соединения получены по реакции Бухе-
рера1-3 взаимодействием 1-амино-2-нафтол-4-сульфокисло-
ты (эйконоген-кислоты) и 1-амино-8-нафтол-4,6-дисуль-
фокислоты (К-кислоты) с этилендиамином в присутствии
водного раствора бисульфита натрия. Место вступления
заместителя обусловлено положением сульфогруппы х.
28
Схема синтеза
R—NH2 + NH2—СН2—СН2—NH2 + NaHSO3-»
---> R—NH—CH2—CH2—NH2 + NH3 + SO3
OH
РЕАКТИВЫ
К-кислота, ч., МРТУ 6-09-1636—64.
Э йко ноге н-кис лота, ч., ВТУ 4178—54.
Этилендиамин, ч., ВТУ 327—58, 20%-ный раствор.
Бисульфит натрия, ч., ТУ МХП 1944—49, 40%-ный раствор
Способ получения
Смесь 6,8 г (0,02 моль) К-кислоты или 4,8 г (0,02 моль)
эйконоген-кислоты (препараты предварительно перекри-
сталлизованы), 40 мл раствора бисульфита натрия и 18 г
(0,06 моль) раствора этилендиамипа кипятят с обратным
холодильником (30—40 ч) д,о исчезновения исходных
аминов по диазопройе. После охлаждения реакционную
массу подкисляют концентрированной соляной кислотой
до кислой реакции по конго и кипятят до исчезновения
запаха сернистого ангидрида. Осадок отфильтровывают
на воронке Бюхнера. Для очистки продукты реакции
растворяют в 10%-ном растворе карбоната натрия и вто-
рично осаждают соляной кислотой. Получают 3,1 г
1-аминоэтиламино-2-нафтол-4-сульфокислоты (выход со-
ставляет 56% от теоретического) или 4,1 г 1-аминоэтилами-
но-8-нафтол-4,6-дисульфокислоты (выход составляет 57%
от теоретического).
Состав синтезированного продукта (C12H14NaO4S):
С Н N S
Найдено, % ........50,9; 50,8 4,7; 4,8 9,6; 9,6 11,0; 11,1
Вычислено, %............51,1 4,9 9,9 11,3
Состав синтезированного продукта (C12H14N2O7S2):
С Н N S
Найдено, %................39.5; 39,8 4,0; 4,2 7,2; 7,3 17.0; 16,9
Вычислено, %.............. 39,7 3,9 7,7 17,6
29
Литература
1. Органические реакции, сб. 1, Издатиилит, 1949, стр. 143.
2. Герм. пат. 467626; Frdl., 16, 508.
3. Пат. США 1543569; С. А. 19, 2345 (1925).
1-АЦЕТОКСИ-1,1-ДИЦИАНЭТАН
ДИАЦЕТИЛ ЦИАНИД
Г. И. МИХАЙЛОВ, М. Ф. КОНДРАШОВА, И. А. ГОРКЕР
C6H6N2O2
О CN
II I
сн3—с—о—с—сн3
I
CN
Мол. вес 138,12
1-Ацетокси-1,1-дицианэтан был получен впервые нз
уксусного ангидрида и цианистого натрия 1; в дальней-
шем были внесены некоторые изменения в методику выде-
ления и очистки вещества 21 3. Опубликован также пре-
паративный метод получения, основанный на взаимодей-
ствии уксусного ангидрида с цианистым калием в бен-
золе4 (продолжительность синтеза 5 ч; выход 60%).
Известен также метод получения 1-ацетокси-1,1-дициан-
этана из уксусного ангидрида и цианистого водорода8
(выход 41 %). После детального изучения методов синтеза
1-ацетокси-1,1-дицианэтана мы установили, что взаимо-
действие уксусного ангидрида с цианистым натрием в бен-
золе протекает быстро и для окончания реакции достаточ-
но 5—10 мин нагревать смесь (длительное нагревание
приводит лишь к осмолению продукта и снижению выхо-
да). Реакция сопровождается незначительным разогре-
ванием и не требует внешнего охлаждения.
Схема синтеза
О CN
сн3соч II I
>0 4- NaCN-> СН —с—О—С—СН3
СН3СО/ )
CN
30
РЕАКТИВЫ
Ангидрид уксусный, ч., ГОСТ 5815—52.
Цианистый натрий, ч., ТУ МХП 3362—53.
Бензол, ч., ГОСТ 5955—51.
Углерод четыреххлористый, ч., ГОСТ 5827—51.
Способ получения
Внимание! Всю работу по получению 1-ацетокси-1,1-ди-
цианэтана следует проводить в хорошо действующем вы-
тяжном шкафу.
В трехгорлую круглодонную колбу емкостью 1,5 л,
снабженную мешалкой и термометром, помещают 500 мл
бензола, 100 г (2 моль) цианистого натрия и при разме-
шивании добавляют в течение 5 мин 190 мл (2 моль)
уксусного ангидрида. При этом температура смеси повы-
шается до 65—70 °C. Для завершения реакции смесь
кипятят на водяной бане 5 мин и затем охлаждают до 10—
15 °C. Раствор фильтруют, осадок уксуснокислого натрия
промывают на фильтре бензолом два раза порциями
по 50 мл и тщательно отжимают. Бензол отгоняют в ваку-
уме, остаток выливают в 500 мл ледяной воды. Выпавшие
кристаллы отфильтровывают, тщательно отжимают, про-
мывают 50 мл воды, 50 мл четыреххлористого углерода
и сушат. Получают 87—90 г технического продукта с т. пл.
63—60 °C. Для очистки его перегоняют с водяным паром.
После перегонки получают 84 г продукта; т. пл. 69—70 °C,
выход 61 % (от теоретического). По литературным дан-
ным4,8 т. пл. 69 °C.
Литература
1. Kleeman S., Вег., 18, 256 (1885).
2. М а г ’’ е 1 3. S., Brace N. О., Miller Г. A., Jon-
son A. R., J. Am. Chem. Soc., 71, 34 (1949).
3. А г d i s А. Е., Averill S. J., Gilbert H., Mil-
ler F. F., Schmidt R. F., J. Am. Chem. Soc., 72, 1305
(1950).
4. Сёренсон У., Кемпбел T., Препаративные методы
химии полимеров, Издатиилит, 1963, стр. 259.
5. Т р е ш и л о в а А. Ф., Карпухин П. П., Труды Харь-
ковского политехнического института, 46, 22 (1963).
31
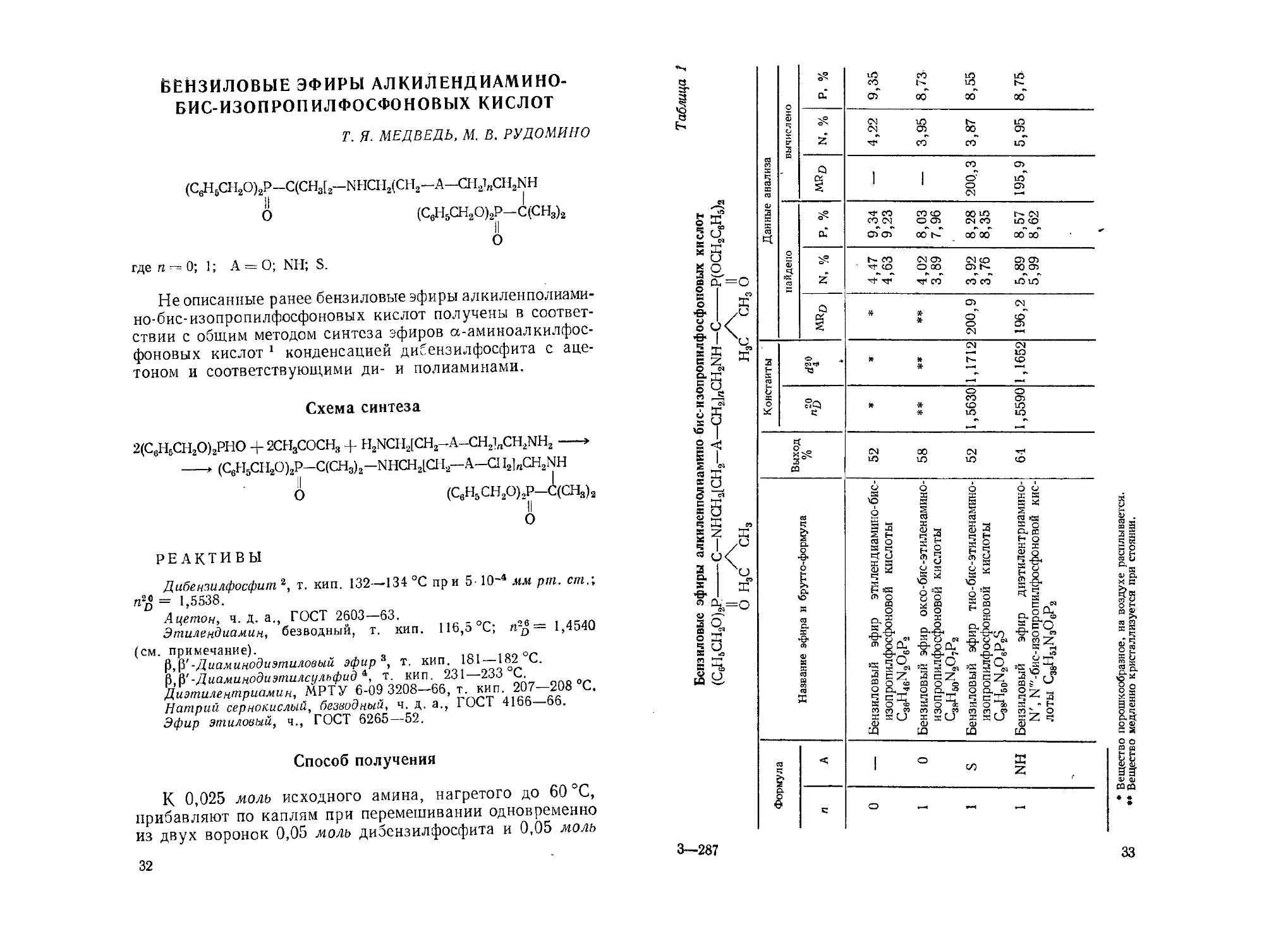
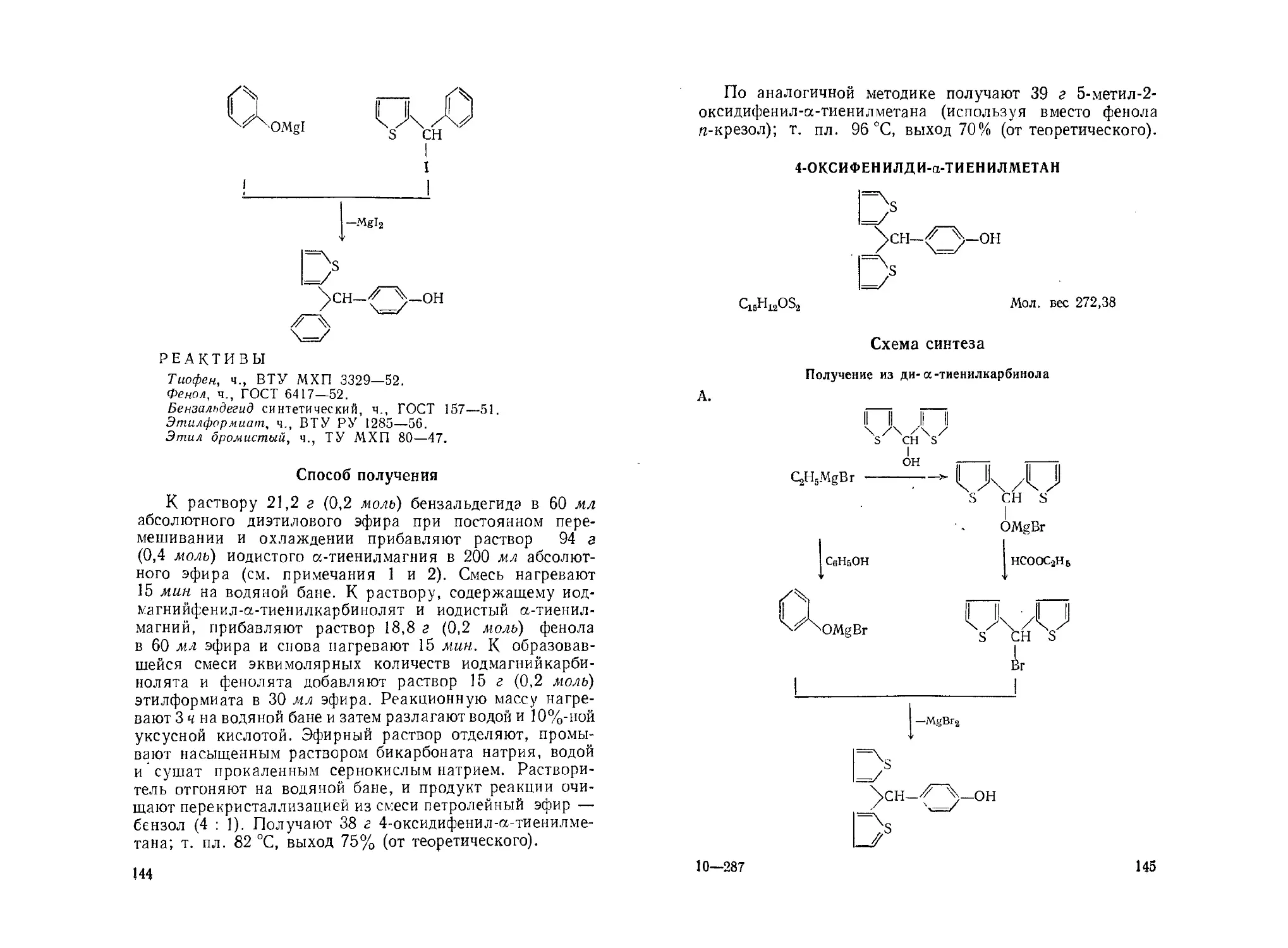
ЁЕЙЗИЛОВЫЕ ЭФИРЫ АЛКИЛЕНДИАМИНО-
БИС-ИЗОПРОПИЛФОСФОНОВЫХ КИСЛОТ
Т. Я. МЕДВЕДЬ, М. В. РУ ДОМИНО
(C,jH6CH2O)2P-C(CH3[2—NHCH2(CH2—a—ch2i„ch2nh
II I
О (С6Н5СН2О)2Р-С(СН3)2
II
о
где га-0; 1; А = О; NH; S.
Не описанные ранее бензиловые эфиры алкиленполиами-
но-бис-изопропилфосфоновых кислот получены в соответ-
ствии с общим методом синтеза эфиров а-аминоалкилфос-
фоновых кислот 1 конденсацией дибензилфосфита с аце-
тоном и соответствующими ди- и полиаминами.
Схема синтеза
2(С6Н6СН2О)2РНО + 2СН3СОСН3 + H2NCH2[CH2-A-CH21„CH2NH2-----»
---* (CeH5CH2O)2P-C(CH3)2-NHCH2lCH2-A-a I2]nCH2NH
II (
О (С6Н5СН2О)2Р—С(СН3)2
II
о
РЕАКТИВЫ
Дибензилфосфшп2, т. кип. 132—134 °C при 510~4 мм рт. ст.;
пг$ = 1,5538.
Ацетон, ч. д. а., ГОСТ 2603—63.
Этилендиамин, безводный, г. кип. 116,5 °C; п-- 1,4540
(см. примечание).
-Диаминодиэтиловый эфир3, т. кип. 181 —182 °C.
р,р'-Диаминодиэтилсульфид 4, т. кип. 231—233 °C.
Диэтилентриамин, МРТУ 6-09 3208—66, т. кип. 207—208 °C.
Натрий сернокислый, безводный, ч. д. а., ГОСТ 4166—66.
Эфир этиловый, ч., ГОСТ 6265—52.
Способ получения
К 0,025 моль исходного амина, нагретого до 60 °C,
прибавляют по каплям при перемешивании одновременно
из двух воронок 0,05 моль дибензилфосфита и 0,05 моль
32
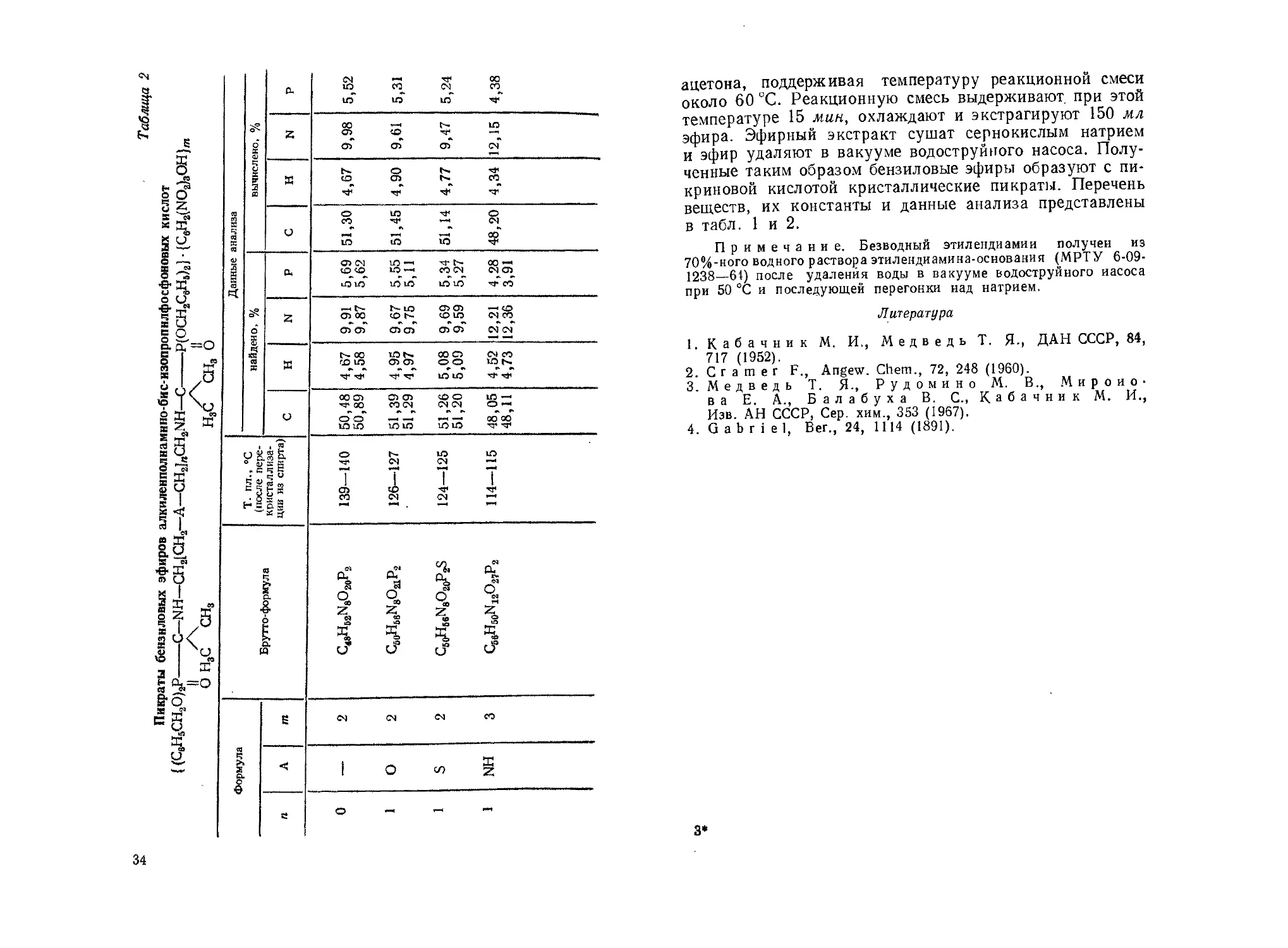
О S' 3 о ©° сц 9,35 8,73 1 8,55 8,75
в ф \О СМ 1С ш
СМ о 00 СП
X •ч •ч а.
F 3 Z тГ со со Ю
$ Й Q со О>
Ч а | 1
03 а: «3 S СП
<и
н Л La •ОТ со СО со 00 LO г- см
№ со О] О СП СМ СО Ш со
а *. *
<3 О ч CL. СП СП 00 ь- □0 00 □0 эо
о
о уо Ь- СО О1 СП СМ со СП СИ
S 2- Tt со о ао СП г- 00 СП
в (Х=О S3 Z Tf Tf тг СО СО СО lO ю
О г м
£ ж СП см
в- /О Q * •»
о <J<f | V,. 1 * S §
в см см
X К 5? ю
е Z к О 3 1 ог? » » * ь- <£>
о. гг 3: *>
и м «3
° ч 3= 05
О » * со ю
6 к & с а ю ю
х о
2 < § 1 О X CM СО см
2 '« 2 X 3 м ю 1С 1С СО
й - О сч 6 s о X о я О 6 X s
Е X ю S ё S «
Я 0 S S
ч X 03 X - X аз 2 ’S X о
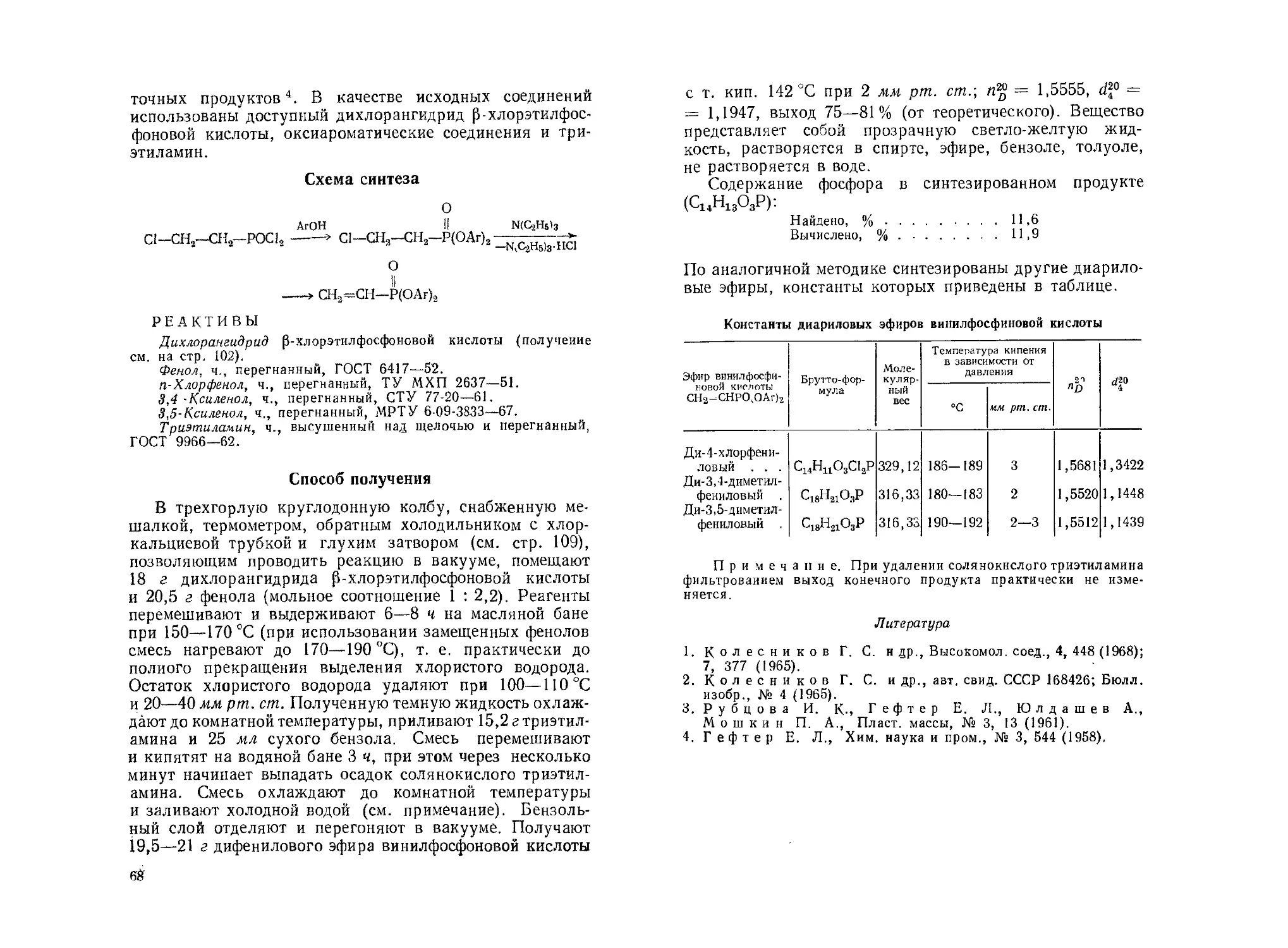
S Z X S 1 /с в о\ 11 ч S 1 т 1 б £ S н аз а S Ч Ч о X . 5 о ? 3 о s о л § о ь Ч СП о й * \о С1, £0 X S а> о й §-§
>& 1 Л Л CU =о « ч-/ о 2 О (Q Cl © X § ч Си О S ез Си X е- я> эфир эт фосфоновой >6Р2 8§ « Я ° ° 'm'S- о’§ £ О g s4 -В-сл w JZ.. о « §аГ о. 5 » ЙЕ О •У" о п т SZ X iH > л
5 X к 9S Ч ’Я « _ Ч О ч О 2 X »S я Хо
й Ч ез ей Й t=Z СО о О 3 CZ 2 Е7 са о о а о о ik U
св Е 2 ©-.Л Ч с X X О ® 2 & §о _J? о О, л ч §§ч gz з s - ь
со со со * О
х х О ?. Я J я s О х Z ч
а> а> из й
<s & <с | а СО NH
'
€ с
Вещество порошкообразное, на воздухе расплывается.
Вещество медленно кристаллизуется при стоянии.
3—287
33
Vo
£
s
as
g
s’
о
_,_РЭ
_js
о
X
?"ся
31
e
8
о О
Ч А
я U
g о
е-л— о
о I
1 ।
Ж
g и ,
к 3 >
s
д
2 З3
Е ГС
ж г?
iY
га I
15
15
о Z
>8
з
и
га
X
&o
Я -S1
а. 5,52 5,31 5,24 оо со ч-”
<? 0s- Z др о <5 ю
э £ сп о? о? сч
3 Е 3 а к 4,67 4,90 4,77 4,34 |
Я m S ч я к я и 51,30 51,45' I 51,14 48,20
Данные CL. О СЧ гЛЮ 5,55 5,11 Й сч Ю1Л 4,28 3,91
с g Z & СО СТ) о? 9,67 9,75 9,69 9,59 112,21 12,36
к « Я я к 4,67 | 4,58 - 4,95 4,97 СО О оо L0 Ю 4,52 4,73
00 о ч» 00 О СТ) СО СЧ <£> О сч сч 1Г S0
и о аг ю ю Ю io ю ю 48, 48,
Т- пл., °C (после пере- кристаллиза- ции из спирта) 139—140 । 126—127 124—125 114—115
Брутго-формула © О. X 64 5 Он сЗ О од Z «о Л % ''j. °ь ы О СО Z о“ а S о ГГ4 X 3 =- с/
S сч сч сч СО
Я 1 1 о СП NH
О — — —
ацетона, поддерживая температуру реакционной смеси
около 60 °C. Реакционную смесь выдерживают при этой
температуре 15 мин, охлаждают и экстрагируют 150 мл
эфира. Эфирный экстракт сушат сернокислым натрием
и эфир удаляют в вакууме водоструйного насоса. Полу-
ченные таким образом бензиловые эфиры образуют с пи-
криновой кислотой кристаллические пикраты. Перечень
веществ, их константы и данные анализа представлены
в табл. 1 и 2.
Примечание. Безводный этилендиамии получен из
70%-ного водного раствора этилендиамина-основания (МРТУ 6-09-
1238—61) после удаления воды в вакууме водоструйного иасоса
при 50 °C и последующей перегонки над натрием.
Литература
1. Кабачник М. И., Медведь Т. Я., ДАН СССР, 84,
717 (1952).
2. Cramer F., Angew. Chem., 72, 248 (1960).
3. Медведь Т. Я., Рудомино М. В., Мироно-
ва Е. А., Балабуха В. С., Кабачник М. И.,
Изв. АН СССР, Сер. хим., 353 (1967).
4. Gabriel, Вег., 24, 1114 (1891).
3*
34
2-(2'-БЕНЗИМИДАЗОЛИЛ)-ХИНОЛ ИН
И 2-(2'-БЕНЗИМИдАЗОЛИЛ)-4-ФЕНИЛХИНОЛИН
О 22^15^3
А. Л. ГЕРШУНС, 4. И. БРИЗИЦКАЯ
CigHnN3
Связанные азотсодержащие гетероциклические систе-
мы представляют интерес вследствие их способности к об-
разованию окрашенных комплексов с ионами металлов,
2-(2'-Бензимидазолил)-хинолин описан в литературех.
Его аналог, содержащий в четвертом положении фениль-
ную группу, является новым соединением. Описанный
в литературе, метод получения 2-(2'-бензимидазолил)-
хинолина заключается во взаимодействии гетероцикли-
ческого альдегида с о-фенилендиамином в присутствии
палладиевого катализатора х. Мы осуществили этот син-
тез более простым путем в присутствии ацетата меди
по аналогии с получением 2-(а-фурил)-бензимидазола 2,
причем в полтора раза увеличили выход 2-(2'-бензими-
дазолил)-хинолина.
Схема синтеза
где R = H;CeH6.
РЕАКТИВЫ
Хинальдиновый альдегид т. пл. 67—68 °C.
4-Фенилхинальдиновый альдегид, т. пл. 90 °C (получение см.
на стр. 199).
о-Фенилендиамин, ч., СТУ 79—507-Х-60.
Медь уксуснокислая, ч. д. а., ГОСТ 5852—51.
Спирт пропиловый, ч., СТ ГОХП 15—2354.
Спирт метиловый, ч., ГОСТ 6995—54.
36
Способ получения
В литровую круглодонную колбу, снабженную обрат-
ным холодильником, вносят раствор 10,8 г (0,1 моль}
о-фенилендиамина в 150 мл пропилового спирта, раствор
7,85 г (0,05 моль} хинальдинового альдегида в 100 мл
пропилового спирта и 30 г уксуснокислой меди, раство-
ренной в 300 мл воды. Смесь нагревают на водяной ба-
не 1 ч. Синяя окраска раствора постепенно исчезает
и выделяется коричневый осадок медной соли 2-(2'-бен-
зимидазолил)-хинолина. Раствор охлаждают, отфильтро-
ванный осадок размешивают с 300 мл 70%-ного горячего
метилового спирта и пропускают сероводород до полного
осаждения меди. Сульфид меди отфильтровывают, промы-
вают 75 мл горячего метилового спирта и спиртовую
вытяжку присоединяют к фильтрату. После добавления
200мл воды выпадает желтый осадок 2-(2'-бензимидазолил)-
хинолина. Продукт кристаллизуют из метанола. Полу-
чают 11,6 г вещества; т. пл. 219—220 °C, выход 95%
(от теоретического).
Аналогично синтезируют 2-(2'-бензимидазолил)-4-фе-
нилхинолин из 0,1 моль о-фенилендиамина и 0,05 моль
4-фенилхинальдинового альдегида. После перекристалли-
зации из метилового спирта получают 15,2 г продукта
с т. пл. 220—221 °C; выход 95% (от теоретического).
Оба вещества растворимы в спиртах, пиридине, хлорофор-
ме и плохо растворимы в воде.
Содержание азота в препарате (CleHuNg):
Найдено, %.........16,98; 17,01
Вычислено, % . . . . 17, 14
Содержание азота в препарате (C22Hi8N8):
Найдено, %.........13,38; 13,20
Вычислено, % . 13,08
Литература
1. Jerchel D., Krachf М., Krucker К., Ann., 590,
232 (1954).
2. Weidenhagen R., Вег., 69, 2263 (1936).
3. Kaplan H., J. Am. Chem. Soc., 63, 2654 (1941).
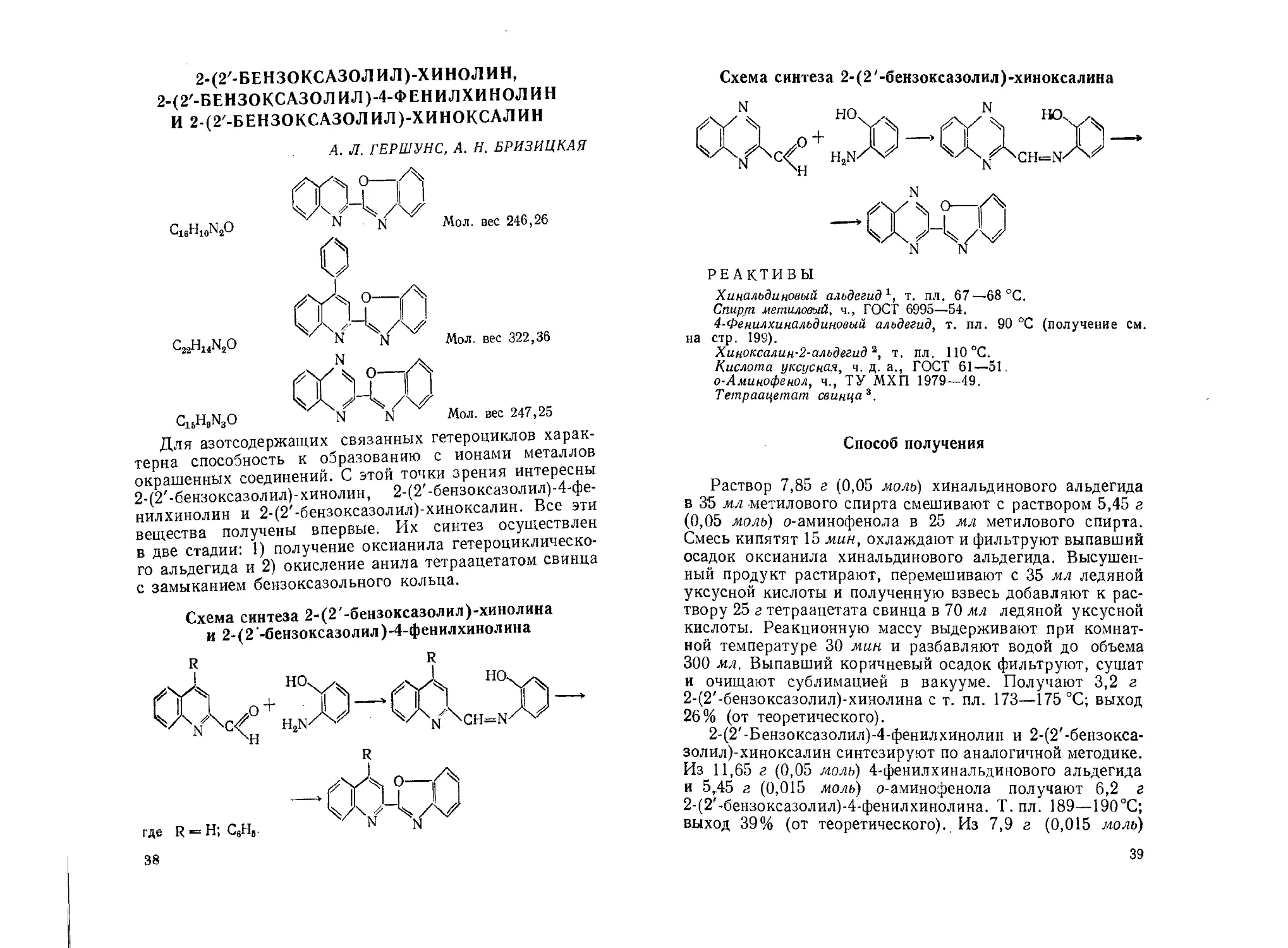
2-(2'-БЕНЗОКСАЗОЛ ИЛ)-ХИНОЛИН,
2-(2'-БЕНЗОКСАЗОЛИЛ)-4-ФЕНИЛХИНОЛИН
И 2-(2 -БЕНЗОКСАЗОЛИЛ)-ХИНОКСАЛИН
А. Л. ГЕРШУНС, А. И. БРИЗИЦКАЯ
CieH10N2O
C15H9W3O
Мол. вес 246,26
Мол. вес 322,36
Мол. вес 247,25
Для азотсодержащих связанных гетероциклов харак-
терна способность к образованию с ионами металлов
окрашенных соединений. С этой точки зрения интересны
2-(2'-бензоксазолил)-хинолин, 2-(2'-бензоксазолил)-4-фе-
нилхинолин и 2-(2'-бензоксазолил)-хиноксалин. Все эти
вещества получены впервые. Их синтез осуществлен
в две Стадии: 1) получение оксианила гетероциклическо-
го альдегида и 2) окисление анила тетраацетатом свинца
с замыканием бензоксазольного кольца.
Схема синтеза 2-(2'-бензоксазолил)-хинолина
и 2-(2 -бензоксазолил )-4-фенилхинолина
38
Схема синтеза 2-(2'-бензоксазолил)-хиноксалина
РЕАКТИВЫ
Хинальдиновый альдегид1, т. пл. 67—68 °C.
Спирт, метиловый, ч., ГОСТ 6995—54.
4-Фенилхинальдиновый альдегид, т. пл. 90 °C (получение см.
на стр. 199).
Хиноксалин-2-альдегид а, т. пл. ПО °C.
Кислота уксусная, ч. д. а., ГОСТ 61—51.
о-Аминофенол, ч., ТУ МХП 1979—49.
Тетраацетат свинца3.
Способ получения
Раствор 7,85 г (0,05 моль) хинальдинового альдегида
в 35 мл метилового спирта смешивают с раствором 5,45 г
(0,05 моль) о-аминофенола в 25 мл метилового спирта.
Смесь кипятят 15 мин, охлаждают и фильтруют выпавший
осадок оксианила хинальдинового альдегида. Высушен-
ный продукт растирают, перемешивают с 35 мл ледяной
уксусной кислоты и полученную взвесь добавляют к рас-
твору 25 г тетраацетата свинца в 70 мл ледяной уксусной
кислоты. Реакционную массу выдерживают при комнат-
ной температуре 30 мин и разбавляют водой до объема
300 мл. Выпавший коричневый осадок фильтруют, сушат
и очищают сублимацией в вакууме. Получают 3,2 г
2-(2'-бензоксазолил)-хинолина с т. пл. 173—175 °C; выход
26% (от теоретического).
2-(2'-Бензоксазолил)-4-фенилхинолин и 2-(2'-бензокса-
золил)-хиноксалин синтезируют по аналогичной методике.
Из 11,65 г (0,05 моль) 4-фенилхинальдинового альдегида
и 5,45 г (0,015 моль) о-аминофенола получают 6,2 г
2-(2'-бензоксазолил)-4-фенилхинолина. Т. пл. 189—190°С;
выход 39% (от теоретического). Из 7,9 г (0,015 моль)
39
хиноксалин-2-альдегида и 5,45 г (0,05 моль) о-аминофено-
ла получают 7 г 2-(2'-бензоксазолил)-хиноксалина. Т. пл.
183—184 °C, выход 57% (от теоретического).
Все полученные вещества представляют собой бесцвет-
ные кристаллические продукты, растворимые в спиртах,
эфире, дихлорэтане, ксилоле, пиридине; в воде раство-
римы плохо.
Содержание азота в препарате (CieHl0NaO):
Найдено, %...........11,01; 11,08
Вычислено, % ... . 11,38
Содержание азота в препарате (C22H14N2O):
Найдено, %...........8,55; 8,50
Вычислено, %..........8,69
Содержание азота в препарате (C15H9N3O):
Найдено, %...........17,41; 17,26
Вычислено, % ... . 17,00
Литература
1. Kaplan Н., J. Am. Chem. Soc., 63, 2654 (1941).
2. В о г s с h е С., D а с 1 1 е г Р., Ann., 39, 537 (1938).
3. Б р а у э р Г., Руководство по препаративной неорганической
химии, Издатинлит, 1965, стр. 367.
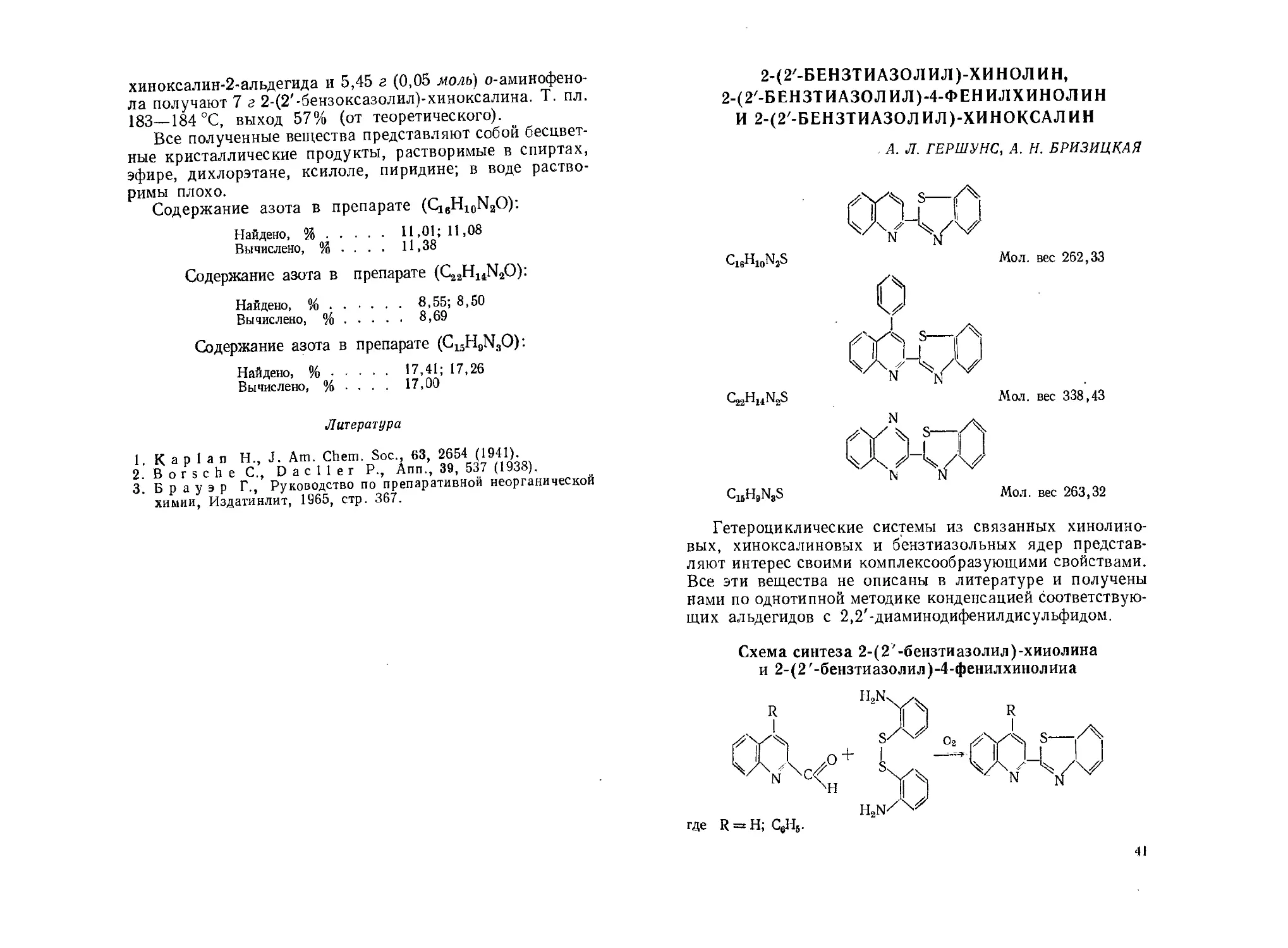
2-(2'-БЕНЗТИАЗОЛИЛ)-ХИНОЛ ИН,
2-(2/-БЕНЗТИАЗОЛИЛ)-4-ФЕНИЛХИНОЛИН
И 2-(2'-БЕНЗТИАЗОЛИЛ)-ХИНОКСАЛИН
. А. Л. ГЕРШУНС, А. Н. БРИЗИЦКАЯ
CieHioNjS
C22H14N2S
Мол. вес 262,33
Мол. вес 338,43
c15H9N3s
Мол. вес 263,32
Гетероциклические системы из связанных хинолино-
вых, хиноксалиновых и бензтиазольных ядер представ-
ляют интерес своими комплексообразующими свойствами.
Все эти вещества не описаны в литературе и получены
нами по однотипной методике конденсацией соответствую-
щих альдегидов с 2,2'-диаминодифенилдисульфидом.
Схема синтеза 2-(2'-бензтиазолил)-хииолина
и 2-(2'-бензтиазолил)-4-фенилхинолииа
где R = Н; СвН6.
41
Схема синтеза 2-(2'-бензтиазолил)-хиноксалина
РЕАКТИВЫ
Хинальдиновый альдегид1, т. пл. 67—68 °C.
2,2'-Диаминодифенилдисулъфид 2, т. пл. 93 °C.
Спирт метиловый, ч., ГОСТ 6995—54.
м-Ксилол, ч., ВТУ 279а—51.
4-Фенилхинальдиновый альдегид, т. пл. 90 °C (получение см. на
стр. 1S9).
Хиноксалин-2-альдегид3, т. пл. 110 °C.
Способ получения
В колбу с обратным холодильником помещают 7,85 г
(0,05 моль) хинальдинового альдегида, 12,4 г (0,05 моль)
2,2-диаминодифенилсульфида и 40 мл ксилола и кипятят
2 ч. После охлаждения фильтруют выпавший желтый оса-
док 2-(2'-бензтиазолил)-хинолина, промывают на фильтре
метиловым спиртом и кристаллизуют из ксилола. Полу-
чают 11 г продукта; т. пл. 199—200 °C, выход 84% (от
теоретического). Вещество представляет собой желтые
иглы; растворяется в ксилоле, эфире, дихлорэтане и пи-
ридине, в воде растворяется плохо.
2-(2'-Бензтиазолил)-4-фениДхинолин и 2-(2'-бензтиазо-
лил)-хиноксалин синтезируют по аналогичной методике.
Из 11,65 г (0,05 моль) 4-фенилхинальдинового альдегида
и 12,4 г (0,05 моль) 2,2'-диаминодифенилдисульфида полу-
чают 10,4 г 2-(2'-бензтиазолил)-4-фенилхинолина с т. пл.
240—241 °C; выход 62% (от теоретического). Из 7,9 г
(0,05 моль) хиноксалин-2-альдегида и 12,4 г (0,05 люль)
2,2'-диаминодифенилдисульфида получают 6,7 г 2-(2'-
бензтиазолил)-хиноксалина с т. пл. 227—228 "С, выход
51% (от теоретического).
Полученные вещества представляют собой светло-жел-
тые иглы; растворяются в ксилоле, эфире, дихлорэтане,
пиридине, в воде растворяются плохо.
42
Содержание азота и серы в препарате (CleH10NgS):
N S
Найдено, % ........... 10,75 12,00
Вычислено, % . . . . 10,67 12,21
Содержание азота и серы в препарате (C22HMN2S):
N з
Найдено, %............ 8,56 9,265
Вычислено, %........... 8,28 9,46
Содержание азота и серы в препарате (С 15HBN3S):
N S
Найдено, %..........16,15 11,88
Вычислено, % ... . 15,95 12,16
Литература
1. Kaplan Н., J. Am. Chem. Soc., 63, 2654 (1941).
2. В о г s с h е С., D а с 1 1 е г Р., Ann., 39, 537 (1938).
3. М 5 h 1 a u R., Beyschlag Н., К о h г е s Н., Вег.,
45, 133 (1922).
6,6'-БИС-ХИНАЛЬД ИН
А. Л. ГЕРШУНС, И. А. РАСТРЕПИНА
C2oHi6N2
Мол. вес 284,36
6,6'-Бис-хинальдин используется в синтезе макромо-
лекулярных комплексообразователей. В литературе при-
ведены два способа получения этого соединения с исполь-
зованием в качестве исходных веществ в первом случае
бензидина и паральдегида \ во втором — кротонового
альдегида 3. Однако неоднократные попытки получить
6,6'-бис-хинальдин по описанным в литературе методикам
не дали положительных результатов. Реакционная смесь
осмолялась и выделить необходимое вещество не удава-
лось. Поэтому мы разработали новый способ получения
6,G'-бис-хинальдина путем каталитического дегалоидиро-
вания 6-бромхинальдина.
43
Схема синтеза
РЕАКТИВЫ
б-Бромхинальдин*, т. пл. 96—97 °C.
Спирт метиловый, ч., ГОСТ 6995—54.
Гидразингидрат, ч., ГОСТ 5832—65.
Способ получения
В круглодонной колбе емкостью 0,5 л, снабженной
обратным холодильником, кипятят 4 ч смесь 11,1 г
(0,05 моль) 6-бромхинальдина, 15 г едкого кали, 150 мл
метилового спирта, 2,5 г гидразингидрата и 7,5 г палла-
диевого катализатора (0,5% Pd/CaCO3) 5. Горячий раствор
фильтруют; катализатор промывают на фильтре горячим
метиловым спиртом. Выпавший при охлаждении фильт-
рата белый осадок отделяют и добавлением воды осаж-
дают остальной продукт. Получают 5 г вещества с т. пл.
207 °C (что совпадает с литературными данными); выход
35% (от теоретического).
Литература
1. Hinz Е., Ann., 242, 326 (1887).
2. Герм. пат. 567273 (1931); С. А. 27, 1362 (1933).
3. Фр. пат. 739880 (1932); С. А. 27, 2164 (1933).
4. J о h п s о n J. R., Adams R., J. Am. Chem. Soc., 43,
2257 (1921).
5. Busch M., Stove H., Ber., 49, 1063 (1916).
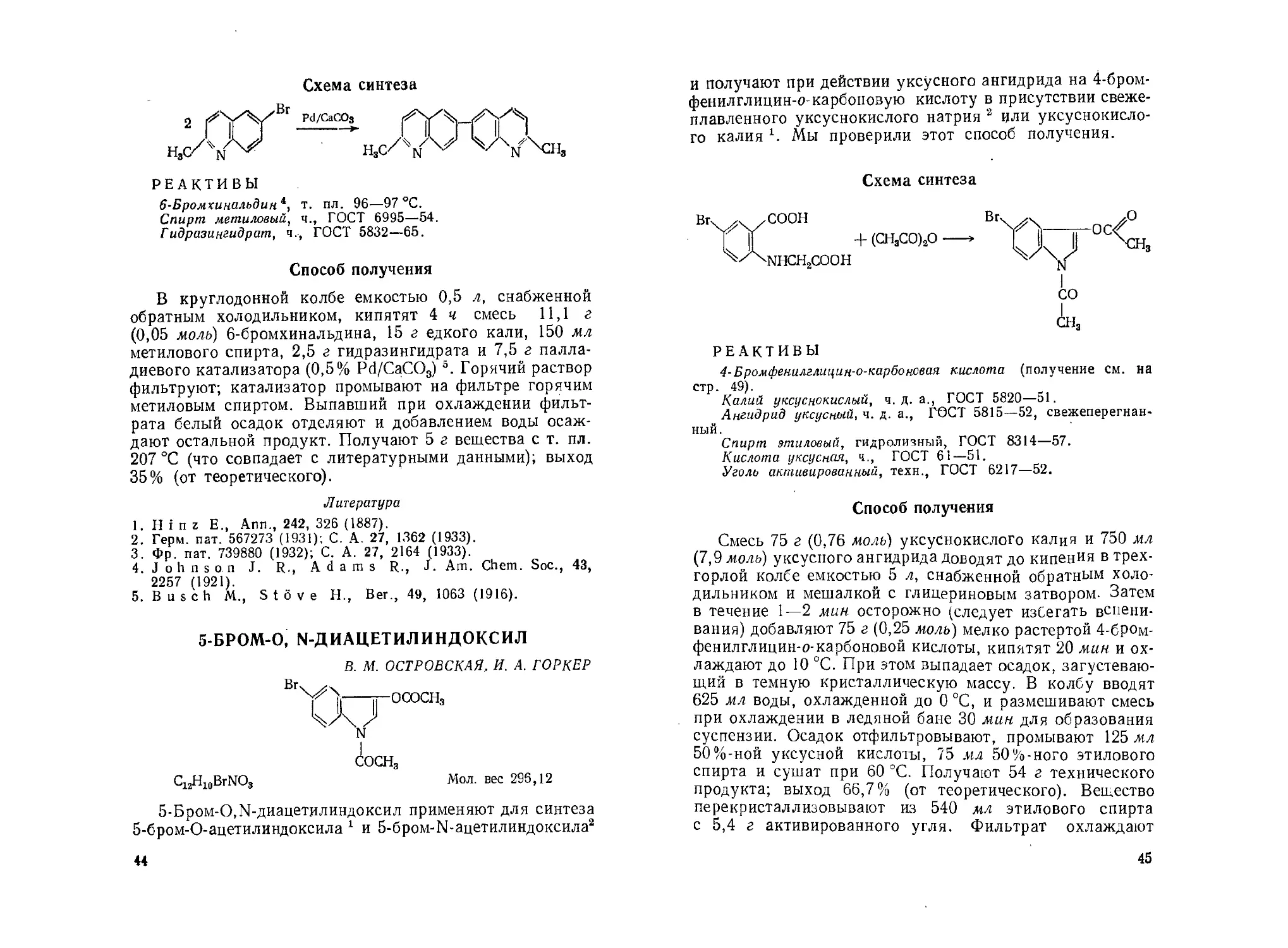
5-БРОМ-О, N-ДИАЦЕТИЛ ИНДОКСИЛ
В. M. ОСТРОВСКАЯ, И. А. ГОРКЕР
Br
OCOCHg
C12H10BrNO3
Мол. вес 295,12
5-Бром-О, N-диацетилиндоксил применяют для синтеза
5-бром-О-ацетилиндоксила 1 и 5-бром-Г\[-ацетилиндоксила2
44
и получают при действии уксусного ангидрида на 4-бром-
фенилглицин-о-карбоповую кислоту в присутствии свеже-
плавленного уксуснокислого натрия 2 или уксуснокисло-
го калия г. Мы проверили этот способ получения.
Схема синтеза
РЕАКТИВЫ
4-Бромфенилглицин-о-карбонсвая кислота (получение см. на
стр. 49).
Калий уксуснокислый, ч. д. а., ГОСТ 5820—51.
Ангидрид уксусный, ч. д. а., ГОСТ 5815—52, свежеперегнан-
ный.
Спирт этиловый, гидролизный, ГОСТ 8314—57.
Кислота уксусная, ч., ГОСТ 61—51.
Уголь активированный, техн., ГОСТ 6217—52.
Способ получения
Смесь 75 г (0,76 моль) уксуснокислого калия и 750 мл
(7,9 моль) уксусного ангидрида доводят до кипения в трех-
горлой колбе емкостью 5 л, снабженной обратным холо-
дильником и мешалкой с глицериновым затвором. Затем
в течение 1—2 мин осторожно (следует избегать вспени-
вания) добавляют 75 г (0,25 моль) мелко растертой 4-бром-
фенилглицин-о-карбоновой кислоты, кипятят 20 мин и ох-
лаждают до 10 °C. При этом выпадает осадок, загустеваю-
щий в темную кристаллическую массу. В колбу вводят
625 мл воды, охлажденной до 0 °C, и размешивают смесь
при охлаждении в ледяной бане 30 мин для образования
суспензии. Осадок отфильтровывают, промывают 125 мл
50%-ной уксусной кислоты, 75 мл 50%-ного этилового
спирта и сушат при 60 °C. Получают 54 г технического
продукта; выход 66,7% (от теоретического). Вещество
перекристаллизовывают из 540 мл этилового спирта
с 5,4 г активированного угля. Фильтрат охлаждают
45
в смеси льда и соли; выпавший осадок отфильтровывают
и сушат при 60 °C. Получают 45 г продукта с т. пл. 122,5—
124,0°С (интервал плавления 0,5—1,5 °C); выход 55,5%
(от теоретического). По литературным данным, т. пл.
вещества 124—124,5 °C 3; 123 °C4; 123—124 °C1; 123—
123,8 °C 2.
Литература
1. Н о 1 t S. J. in «General cytochemical methods», v. 1, Acad.
Press, ing. N. Y.a.L., 1958, p. 375.
2. S u H. C. F., T s о u K. S., J. Am. Chem. Soc., 82, 1187 (1900).
3. Holt S. J., Petrow V., J. Chem. Soc., 1947, 607.
4. H о 1 t S. J., S a d 1 er P. W., Proc. Roy Soc., 148 8, 465 (1958).
5-БРОМИНДОКСИЛАЦЕТАТ
В. M. ^ОСТРОВСКАЯ, и. А. ГОРКЕР
Br
рОСОСНз
I
C10H8BrNO2 H Мол. вес 254,09
5-Бром-О-ацетилиндоксил применяют в гистохимии для
исследования гидролитических ферментов 1-12. В лите-
ратуре описано два варианта одного метода получения
5-бром-О-ацетилиндоксила из 5-бром-О,М-диацетилиндок-
сила путем гидролиза и последующего ацетилирования
образующегося после гидролиза 5-броминдоксила а.
Мы проверили и уточнили один из этих вариантов 2,
причем очистка 5-бром-О-ацетилиндоксила перекристал-
лизацией из абсолютного этилового спирта предлагается
впервые.
Схема синтеза
46
РЕАКТИВЫ
5-Бром-О,Н-диацетилиндоксил, ч. (получение см. на стр. 44).
Натр едкий, ч. д. а., ГОСТ 4328—66.
Ангидрид уксусный, ч. д. а., ГОСТ 5815—52.
Спирт этиловый, абсолютный.
Уголь активированный, техн., ГОСТ 6217—52.
Способ получения
В четырехгорлую колбу емкостью 1 л, снабженную
обратным холодильником, мешалкой, капельной воронкой
и подающей азот трубкой (достигающей до дна), вводят
770 мл 2 н. раствора едкого натра. Затем в колбу подают
через щелочной раствор пирогаллола азот в течение 20—
25 мин для вытеснения воздуха, после чего быстро прибав-
ляют 42 г (0,141 моль) 5-бром-О,М-диацетилиндоксила
и снова пропускают азот. Реакционную массу кипятят
до растворения осадка, охлаждают до 0 °C и в течение
30 мин добавляют из капельной воронки при быстром
перемешивании 74,2 мл (0,784 моль) уксусного ангидрида,
после чего продолжают перемешивание реакционной сме-
си еще 20—30 мин. Выпавший осадок отфильтровывают,
промывают водой 3 раза порциями по 50 мл и сушат
в вакуум-эксикаторе над прокаленным хлористым каль-
цием. Получают 35,6 г технического продукта в виде
коричневого порошка; т. пл. 126—128,5 °C.
Для получения продукта квалификации ч. д. а. про-
водят трехкратную очистку переосаждением и двукратной
перекристаллизацией.
Переосаждение. Растворяют 35,6 г технического про-
дукта в 250 мл абсолютного этилового спирта, нагревают
до 60 °C, добавляют 11 г активированного угля (см. при-
мечание 1) и встряхивают 10 мин. Уголь отфильтровы-
вают. К фильтрату добавляют 250 мл дистиллированной
воды, нагретой до 70 °C, и полученный раствор охлажда-
ют до —10 °C. Выпавший осадок отфильтровывают и су-
шат в вакуум-эксикаторе над едким натром до постоян-
ного веса. Получают 27,7 г продукта с т. пл. 129—130 °C.
Первая перекристаллизация. 27,7 г продукта раство-
ряют в 118 мл абсолютного этилового спирта при 60 °C,
добавляют 0,5 г активированного угля и встряхивают.
Раствор фильтруют, охлаждают до —50 °C и выпавший
осадок отфильтровывают (см. примечание 2). Получают
23,7 г продукта с. т пл. 130—131 °C.
47
Вторая, перекристаллизация. 23,7 г продукта пере-
кристаллизовывают из 65 мл абсолютного этилового
спирта, как описано выше, но без угля и получают 21,5 г
5-броминдоксиланетата (квалификации ч. д. а.) в ви-
де белых кристаллов; т. пл. 131 —134 °C (интервал плав-
ления 1 °C). Оптическая плотность 2%-ного спиртового
раствора вещества не более 0,120 (определена на фото-
колориметре ФЭК-М).
По литературным данным, т. пл. 134 °C 13; 133—
133,5 °C1; 129—132 °C14; 130,5—132 °C 1S.
Примечания. 1. Перед употреблением 50 г активиро-
ванного угля кипятят в 500 мл дистиллированной воды, отфильтро-
вывают, промывают на фильтре 100 мл этилового спирта (гидролиз-
ного) и сушат при 150 °C.
2. Фильтраты после перекристаллизации 5-броминдоксилаце-
тата используют для переосаждения технического продукта
в последующих опытах. При этом выход продукта увеличивается
на 5%.
Литература
l. Su Н.С. F., TsouK.S., J. Am. Chem. Soc., 82Д187 (1960).
2. Holt S. J., in «General cytochemical methods», v. 1, Acad.
Press., ing. N.Y.a.L., 1958, p. 375.
3. Берстон M., Гистохимия ферментов, Изд. «Мир», 1965,
стр. 230.
4. П и р с Э., Гистохимия теоретическая и прикладная, Издат-
инлит, 1962, стр. 419.
5. В а г k а Т., Anderson Р. J., Histochemistry, theory,
practice, bibliography, N. Y., Evanston, London, 1963, p. 265.
6. H о 1 t S. J., Sadler P. W., Proc., Roy. Soc., 148B,
465 (1958).
7. С о t s о n S., H о 1 t S. J., Proc. Roy. Soc., 148B, 506 (1958).
8. Hess R., Pearse A.C.E., Brit. J. exp. Pathol., 39, 292
(1958).
9. T s о u K. S., S u H. C. F., J. Histochem. Cytochem., 11,
562 (1963).
10. S i u r a 1 a M., Niemi M., Sunb erg M., Ann. Med.
Internal Fenniae, 49, 39 (1960); C. A., 54, 19907d (1964).
11. T s о u K. S., S u H. C. F., P a w n s 1 e у H. M., J. Histo-
chem. Cytochem., 13, 10 (1965).
12. H о 1 t S. J., Withers R. F. J., Proc. Roy. Soc., 148B,
520 (1958).
13. Schuchardt Th., GMBH u co., Mtinchen, 1966, S. 161.
14. Light's organic chemicals biochemicals enzymes alkaloids-ste-
rotds photosensitizing dyes, London, 1963—64, p. 14.
15. Aldrich organic chemicals, 1966, p. 55.
4-БРОМФЕНИЛГЛИЦИН-о-КАРБОНОВАЯ
КИСЛОТА
4-БРОМ-2-КАРБОКСИФЕНИЛ ГЛИЦИН
В. М. ОСТРОВСКАЯ, И. А. ГОРКЕР
соон
nhch2cooh
C9H8BrNO4
Мол. вес 274,07
4-Бромфенилглин.ин-о-карбоновую кислоту используют
для синтеза 5-бром-о,Ы-диацетилиндоксила1 и 6-хлор-
4-бромфенилглиципо- карбоновой кислоты2 и получают
бромированием фенилглицин-о-карбоновой кислоты 3 или
ее монокалиевой соли 1 в ледяной уксусной кислоте.
Мы проверили и уточнили последний метод получения г.
Схема синтеза
СООН
н+
+ Вг2 —-
nhch2cook
соон
nhch2cooh
РЕАКТИВЫ
Фенилглицин-о-карбоновой кислоты монокалиевая соль 4, ч.
Бром, ч., ГОСТ 4109.—64.
Кислота уксусная, х. ч., ГОСТ 61—51.
Способ получения
В трехгорлую колбу емкостью 1 л, снабженную мешал-
кой, хл'оркальциевой трубкой и капельной воронкой,
помещают смесь 72 г (0,309 моль) тонко растертой моно-
калиевой соли фенилглицин-о-карбоновой кислоты
и 300 мл ледяной уксусной кислоты и добавляют в тече-
ние 15 мин при интенсивном перемешивании раствор
17 мл (0,325 моль) брома в 60 мл ледяной уксусной кисло-
4—287 49
ты. После размешивания (в течение 30 мин) вливают
1200 мл воды. Выпавший осадок отфильтровывают, про-
мывают 400 мл воды и сушат в сушильном шкафу при
60—70 °C до постоянного веса (2—3 суток). Получают
78,4 г продукта в виде желтовато-белого кристаллического
порошка; т. пл. 219—220 °C (с разложением); выход
92,5% (от теоретического). После перекристаллизации
из 50%-ного этилового спирта с добавлением активиро-
ванного угля температура плавления не изменяется.
По проверенным литературным данным1, т. пл. 220 °C
(с разложением); выход 87% (от теоретического). После
перекристаллизации из 50%-ного этилового спирта
т. пл. 238 °C (с разложением). Продукт пригоден для
получения 5-бром-О,Ь1-диацетилиндоксила.
Литература
1. Holt S. J., in General cytochemical methods, v. 1, Acad.
Press, ing. N.Y.a.L., 1958, p. 375.
2. Герм. пат. 226680 (1908); Frdl., 10, 351.
3. Герм. пат. 148615 (1901): Zbl., 1904, 1, 1045.
4. Островская В. М., Горкер И. А., Методы получе-
ния химических реактивов и препаратов, вып. 17, ИРЕА, 1968,
стр. 37.
ВИНИЛИДЕНЦИАНИД
1,1-ДИЦИАНЭТИЛ ЕН
Л. И. БОГОМОЛОВА, И. А. ГОРКЕР, С. А. КОЧЕТКОВА,
Г. И. МИХАИЛОВ
C4H2N2
(CN)2C=CH2
Мол. вес 78,08
В литературе описаны методы получения винилиден-
цианида на основе пиролиза 1,1,3,3-тетрацианпропа-
на а> 3, 1-ацетокси-1,1-дицианэтана 5 и 4,4-дициан-
циклогексана ®. Пиролиз 1,1,3,3-тетрацианпропана яв-
ляется наиболее удобным препаративным методом полу-
чения винилиденцианида. Этот метод мы проверили
и уточнили.
50
Схема синтеза
(CN)2CH-CH2—CH(CN)2 » (CN)2C=CHa + (CN)aCHa
РЕАКТИВЫ
1,1,3,3-Тетрацианпропан (получение см. иа стр. 180).
Ангидрид фосфорный, ч., МРТУ 6-09-22—62.
Способ получения
Внимание! Винилиденцианид ядовит, вызывает резкое
раздражение глаз и верхних дыхательных путей. Всю ра-
боту необходимо проводить в хорошо действующем вытяж-
ном шкафу.
Пиролиз 1,1,3,3-тетрацианпропана проводят в приборе,
собранном на шлифах. Прибор состоит из двугорлой колбы
емкостью 3 л (из стекла пирекс), в центральное горло
которой вставляют капилляр для подачи сухого азота
или воздуха, а боковое горло соединяют широким форш-
тоссом (шлиф № 29) с приемником. Прибор должен быть
пригоден для работы в вакууме при остаточном давле-
нии 2—4 мм рт. ст.; все его части необходимо хорошо
вымыть и высушить при 105—ПО °C. В колбу помещают
10—12 г порошкообразного фосфорного ангидрида (см.
примечание 1) и встряхиванием равномерно распреде-
ляют его по дну и стенкам колбы. В центральное горло
вставляют капилляр, доходящий до дна, а через короткое
боковое горло насыпают 400 г 1,1,3,3-тетрацианпропана
(см. примечание 2) и сверху осторожно насыпают еще
2—3 г фосфорного ангидрида. Боковое горло колбы
закрывают форштоссом, обмотанным асбестовым шну-
ром; другой конец форштосса соединяют с приемником.
В форштосс и приемник также помещают по 2 г фосфор-
ного ангидрида. Приемником служит колба Вюрца емко-
стью 0,5 л, погруженная в баню с охлаждающей смесью
из льда и соли. Колбу для пиролиза помещают в глубо-
кую баню с глицерином, доходящую до горла колбы.
В приборе устанавливают давление 2—3 мм рт. ст.,
затем глицериновую баню нагревают до 150 °C (см. при-
мечание 3). При этом начинается пиролиз 1,1,3,3-тетра-
цианпропана, сопровождающийся отгонкой смеси паров
винилиденцианида и динитрила малоновой кислоты. Реак-
ция продолжается 4—4,5 ч. После прекращения отгонки
4*
51
удаляют глицериновую баню, охлаждают реакционную
смесь до комнатной температуры, затем в систему впус-
кают сухой воздух, снимают приемник и быстро закры-
вают его стеклянной пробкой. Выход продуктов пиро-
лиза 240—260 г.
Для выделения и очистки винилиденцианида полу-
ченную после пиролиза смесь перегоняют в вакууме при
давлении 3—4 мм рт. ст., пропуская через капилляр
сухой воздух. Прибор перед перегонкой заполняют фос-
форным ангидридом. В зависимости от давления т. кип.
первой фракции, содержащей винилиденцианид, состав-
ляет 32—34 °C при 2 мм рт. ст., 34—36 °C при
3 мм рт. ст., 36—38 °C при 4 мм рт. ст. Т. кип. вто-
рой фракции, содержащей смесь винилиденцианида
и динитрила малоновой кислоты, 40—70 °C при
2—4 мм рт. ст. Т. кип. третьей фракции, содержа-
щей динитрйл малоновой кислоты, 70—95 °C при
2—4 мм рт. ст. Для предотвращения полимеризации
винилиденцианида систему охлаждают под вакуумом.
Получают 100—108 г продукта (первая фракция) в виде
бесцветной жидкости с т. затвердевания 8,8—9,5 °C; «Ь0
1,439—1,441; выход 46—50% (от теоретического). По лите-
ратурным данным, т. затвердевания 8 °C 2; 9 °C ®; 9,2 °C 6;
п'в = 1,441 5. Полученный винилиденцианид практиче-
ски пригоден для синтеза полимеров. Для получения
продукта с т. затвердевания не ниже 9,2 °C его следует
еще раз перегнать в вакууме. Готовый продукт стабили-
зируют фосфорным ангидридом или сухим бензолом.
После работы с винилиденцианидом вся лабораторная
посуда должна быть обработана шелочью и 10%-ным вод-
ным раствором сернокислого железа.
Примечания. 1. Винилиденцианид легко полимеризуется
в присутствии следов влаги, поэтому пиролиз 1,1,3,3-тетрацианпро-
пана, а также перегонку и хранение винилиденцианида ведут в при-
сутствии фосфорного ангидрида. При пиролизе через капилляр про-
пускают азот или воздух, предварительно высушенный в колонке
с фосфорным ангидридом.
2. Следует применять свежеполученный 1,1,3,3-тетрацианпро-
пан, высушенный до постоянного веса в вакуум-сушильном шкафу
(90—100 °C при 10 мм рт. ст.); т. пл. 132—137 °C.
3. Температура бани в зависимости от давления в приборе
поддерживается в следующих интервалах: 150—160 °C при 1 —
2 мм рт. ст.; 160—170 °C при 2—3 мм рт. ст.; 170—180 °C при
3—4 мм рт. ст.
52
Литература
1. Пат. США 2514387 (1950).
2. Якубович А., Разумовский В., Розен-
штейн С., ЖОХ 38, 2292 (1958).
3. Rosenberg A., Devlin J. Р., Spectrochim. Acta, 21,
1614 (1965).
4. Трещилова А. Ф., Карпухин П. П., Труды
Харьковского политехнического института, 46, 22 (1963).
5. Сёренсон У., Кемпбел Т., Препаративные методы
химии полимеров, Издатинлит, 1963, стр. 259.
6. Шервуд П., Химия и химическая технология, № 4 (99),
Издатинлит, 1958, стр. 64.
ВИНИЛСУЛЬФОКИСЛОТА
ЭТИЛЕНСУЛЬФОКИСЛОТА
Е. Б. ТРОСТ ЯНСКАЯ, С. Б. МАКАРОВА, И. И. МУРАШКО
сн2=сн
SO3H
<^H4O3S
Мол. вес 108,11
Винилсульфокислота и ее соли используются в синтезе
ионообменных смол заданной структуры, гомогенных
ионитовых мембран и растворимых полиэлектролитов.
В литературе предложено несколько методов синтеза
винилсульфокислоты: обработка абсолютного этанола1’ 2
или этилена 3 серным ангидридом, а затем щелочью
(выход 45 и 20% соответственно); взаимодействие 1,2-
дибромэтана и сульфита натрия с последующей обработ-
кой образующегося продукта щелочьюi (выход 41%)
или пятихлористым фосфором и водой в’ 6 (выход 65%);
взаимодействие этиленхлоргидрина с бисульфитом нат-
рия, а затем с пятихлористым фосфором 7 (выход 37%);
дегидратация натриевой соли оксиэтансульфокислоты
пирофосфорной кислотой 8 (выход 18%). Нами проверен
и уточнен наиболее простой способ получения винилсуль-
фокислоты, описанный в работах 6’ 6, который приводит
к высокому выходу продукта.
53
Схема синтеза
NajSOs PCij
ВгСН2—-CH2Br----- NaOjS—CH2—CH2—SO3Na--->
H2o
---> C1O2S—CH2—CH2—SO2C1--> CH2=CH—SOjH
РЕАКТИВЫ.
1,2-Дибромэтан, ч., ВТУ МХП 102—51.
Сульфит натрия, безводный, ч., ГОСТ 195—66.
Фосфор пятихлористый, ч., МРТУ 6-09-604—63.
Способ получения
Получение динатриевой соли 1,2-этандисульфокислоты.
В круглодонной колбе, снабженной обратным холодиль-
ником, мешалкой с ртутным затвором и капельной ворон-
кой, растворяют при нагревании 138 г (1,1 моль) безвод-
ного сульфита натрия в 400 мл дистиллированной воды.
Раствор энергично перемешивают, нагревают на масляной
бане до кипения (114 °C) и добавляют по каплям в течение
2 ч 94 г (0,5 моль) 1,2-дибромэтана. Реакционную смесь
кипятят до исчезновения органического слоя (3 ч), филь-
труют, не охлаждая, и оставляют на 10—12 ч. Выпавшие
крупные бесцветные кристаллы отфильтровывают и сушат
5 ч при 140 °C. Получают 50—58 г вещества. После упари-
вания маточного раствора выделяют еще 20—25 г продук-
та, загрязненного бромистым натрием (1—1,5%); для
дальнейшего использования его перекристаллизовывают
из воды. Общий выход около 70% (от теоретического).
Получение 1,2-этандисульфохлорида. Тщательно пере-
мешивают 100 г (0,54 моль) тонко измельченной натрие-
вой соли 1,2-этандисульфокислоты с 240 г (1,15 моль)
пятихлористого фосфора. Небольшое количество этой
смеси (5—10 г) помещают в литровую круглодонпую
колбу, снабженную обратным холодильником, мешалкой
с ртутным затвором и загрузочной воронкой, и осторожно
нагревают на открытом огне до начала реакции (около
5 мин). Реакция сопровождается разжижением реакцион-
ной массы и выделением хлорокиси фосфора. Затем колбу
быстро переносят в масляную баню, нагретую до 100 °C,
и вводят ранее приготовленную смесь с такой скоростью,
чтобы реакционная масса успевала разжижаться. По
окончании реакции основную часть хлорокиси фосфора
54
отгоняют, вязкий остаток охлаждают и небольшими пор-
циями растирают со льдом для разложения хлоридов
фосфора. Осадок отфильтровывают, хорошо отжимают
и сушат при комнатной температуре. Сухой 1,2-этанди-
сульфохлорид экстрагируют в течение 4 ч в аппарате
Сокслета 400 мл хлороформа или четыреххлористого
углерода. При медленном охлаждении экстракта выпа-
дают крупные чешуйчатые кристаллы. Кристаллы отфиль-
тровывают, получают 87 г продукта; т. пл. 91—92 °C;
выход 90% (от теоретического). По литературным дан-
ным, т. пл. 91.°C 6 и 98 °C’.
Содержание углерода, водорода и хлора в синтезиро-
ванном продукте (CaH4Cl2O4S2);
с н ci
Найдено, %. . . 10,72; 10,64 1,82; 1,82 31,20; 31,15
Вычислено, %I . . 10,57 1,77 30,84
Получение винилсульфокислоты. Растворяют 50 г 1,2-
этандисульфохлорида в 600 мл дистиллированной воды
при перемешивании и нагревании до 80 °C.
Полученный раствор перегоняют в вакууме при давле-
нии 10—15 мм рт. ст. до отрицательной реакции на Cl-
ион в отгоне. Для этого необходимо 3—4 раза доливать
в колбу по 300 мл воды. Перегонку желательно вести
в атмосфере азота. В процессе перегонки из реакционной
смеси удаляют сернистый ангидрид и хлористый водород.
Оставшуюся в колбе жидкость соломенного цвета, пред-
ставляющую собой концентрированный раствор винил-
сульфокислоты, выдерживают несколько дней в вакуум-
эксикаторе над фосфорным ангидридом. Выпавшие иголь-
чатые кристаллы 1,2-этандисульфокислоты отфильтровы-
вают. В фильтрате остается 19 г винилсульфокислоты.
Т. кип. 136 °C при 1,5 мм рт. ст.', = 1,4492, выход
88% (от теоретического). Содержание основного вещества
в продукте определяют бромид-броматным методом. По
литературным данным, т. кип. 124 °C при 1 мм рт. ст.,
114—115 °C при 0,5 мм рт. ст. 10; п'п = 1,4499 10, Иц =
= 1,4493 С Для более тщательной очистки от 1,2-этан-
дисульфокислоты фильтрат, содержащий 19 г винилсуль-
фокислоты, подщелачивают 40%-ным раствором едкого
натра до pH 10, добавляют 100 мл метанола и образую-
щуюся нерастворимую соль 1,2-этандисульфокислоты
отделяют фильтрованием. Фильтрат, представляющий
55
собой раствор натриевой соли винилсульфокислоты, при-
годен для использования в синтезе полимеров,
Винилсульфокислота — гигроскопичная прозрачная
жидкость, смешивается во всех соотношениях с водой,
спиртом, ацетоном и уксусной кислотой, трудно раство-
ряется в эфире и хлороформе. Винилсульфокислоту сле-
дует хранить в атмосфере азота в эксикаторе, в темном про-
хладном месте. Лучше всего сохраняются водные растворы
винилсульфокислоты или ее солей.
Литература
1. Br eslow D. S., Hulse G. Е., J. Am. Chem. Soc.,
76, 6369 (1954).
2. В r e s 1 о w D. S., J. Am. Chem. Soc., 76, 5361 (1954).
i 3. Br eslow D. S., J. Am. Chem. Soc., 79, 5000 (1957).
4. Марвел К., Синтезы органических препаратов, сб. 2,
Издатинлит, 1949, стр. 124.
5. К о h 1 е г Е. Р., J. Am, Chem. Soc., 19, 728 (1897).
6. Kohler Е. Р., J. Am. Chem. Soc., 20, 680 (1898).
7. Герм. пат. 569148 (1931).
8. Пат. США 2597696 (1953).
9. К u n 1 с h i k a S., К a t a g i г i Т., Bull. Inst. Chem. Res.,
39, 215 (1961).
10. Пат. США 2515714 (1950).
ГИДРОФИЛЬНЫЕ ГЕЛИ НА ОСНОВЕ
ДЕКСТРАНА
С. Б. МАКАРОВА, И. Н. МУРАШКО, Л. Г. ГОЛИКОВА,
Е. В. ЕГОРОВ
Декстрановые гели широко применяются для разде-
ления, очистки и идентификации различных органиче-
ских соединений методом гель-хроматографии 1-3. Наибо-
лее распространены Н 4> 5 гидрофильные декстрановые
гели, сшитые эпихлоргидрином (гели типа «Сефадекс»).
Мы проверили и уточнили условия получения подобных
гелей.
56
Схема синтеза
ROH
СН2-СН—СН2С1--* R-O-CH2—СН(ОН)—СН2С1--»
'Хо'/
ROH
---> R—О—СН2—ОН—СН2 , R—О—СН2-СН(ОН)—СН2—О—R
V
РЕАКТИВЫ
Декстран, мол. вес 40 000.
Эпихлоргидрин, ч., МРТУ 6-01-37—65.
Способ получения
Размешивают 15 г декстрана с 12 мл дистиллированной
воды, оставляют на 10 мин для набухания и добавляют
15 мл 5 н. раствора едкого натра. Смесь тщательно пере-
мешивают до образования прозрачного раствора. В реак-
ционный сосуд цилиндрической формы емкостью 500 мл
(см. примечание 1), снабженный многолопастной пропел-
лерной мешалкой, термометром и обратным холодиль-
ником, помещают 300 мл 3%-ного раствора полиметил-
метакрилата в эпихлоргидрине. Прибавляют раствор
декстрана и реакционную смесь при непрерывном пере-
мешивании выдерживают 8 ч при 40 °C, 3 ч при 70 °C
и затем охлаждают. Осадок в виде гранул (см. примеча-
ние 2) отфильтровывают, промывают дихлорэтаном, спир-
том, водой до нейтральной реакции и сушат в вакуум-
сушильном шкафу 5 ч при 70 °C. Выход продукта 95% (от
теоретического), считая на декстран. Коэффициент набу-
хания геля в воде 4 мл/г (см. примечание 3).
Примечания. 1. В качестве реакционного сосуда при-
меняется полимеризатор с соотношением диаметра к высоте 1 : 3.
2. Размер гранул (от 50 до 200 мк) регулируют изменением
скорости перемешивания и вязкости дисперсионной среды.
3. Используя декстраны различного молекулярного веса и изме-
няя их концентрацию, можно получить набор гелей различной
степени набухания.
57
Литература
1. F 1 о d i n P., Dextran gels and their applications in gel fil-
tration, Uppsala, Sweden, 1962.
2. Porath J., Biochim. et Biophys. Acta, 39, 193 (1960).
3. P о r a t h J., Clin. Chim. Acta, 4, 776 (1959).
4. Пат. США 3002823 (1961).
5. Аигл. пат. 974054 (1964).
ГИДРОХИНОН-2-МЕТИЛЕНИМИНОДИУКСУСНАЯ
КИСЛОТА
В. я. ТЕМКИНА, Н. В. ЦИРЮЛЬНИКОВА, И. П. ФАДЕЕВА,
Р. П. ЛАСТОВСКИЙ
СцН13ЬЮв
?н ,СП2СООН
^усН=Хсн.соон
Ан
Мол. вес 255,22
Гидрохинон-2-метилениминодиуксусная кислота (1,4-
диоксифенил-2-метилениминодиуксусная кислота) обра-
зует комплексы с рядом катионов. Синтез нового, не опи-
санного в литературе комплексона мы осуществили кон-
денсацией гидрохинона с формальдегидом и иминодиук-
сусной кислотой по реакции Манниха в среде ледяной
уксусной кислоты1.
Схема синтеза
ОН
СН2СООН
ОН
СН2СООН
/СН2СООН
СНа—N<
сн.соон
он
РЕАКТИВЫ
Гидрохинон, марка А, ГОСТ 2549—60.
Формальдегид, ч., ТУ МХП 2631—51.
Иминодиуксусная кислота, ч., ВТУ МГ УХП 190—58.
Кислота уксусная, ч., ГОСТ 61—51.
Натр едкий, ч., ГОСТ 4328—66.
58
Способ получения
В трехгорлую колбу, снабженную обратным холодиль-
ником, механической мешалкой и капельной воронкой,
помещают раствор 6,4 г (0,04 жоль) иминодиуксусной кисло-
ты в 5 мл дистиллированной воды. Кислоту нейтрали-
зуют при размешивании 30%-ным раствором едкого натра
до pH 7—8. Затем постепенно при размешивании и охлаж-
дении (температура не должна подниматься выше 30 °C)
вводят 50 мл уксусной кислоты и 4,4 г (0,04 моль) гидро-
хинона. Смесь охлаждают до 5—7 °C и при тщательном
перемешивании прибавляют по каплям 4 мл (0,04 моль)
35,7%-ного водного раствора формальдегида. Получен-
ную суспензию размешивают 1,5—2 ч при комнатной
температуре и оставляют на ночь. На следующий день
реакционную массу выдерживают при 55—60 °C 3—
3,5 ч и затем охлаждают. Осадок отфильтровывают, про-
мывают дистиллированной водой и сушат при 60—70 °C.
Получают 9 г продукта; выход 88,2% (от теоретического).
Вещество представляет собой порошок белого цвета,
плохо растворимый в воде, нерастворимый в спирте,
ацетоне, бензоле и хорошо растворимый в щелочах.
Состав синтезированного продукта (CUH13NO6):
с н N
Найдейо, % . . . 51,40; 51,45 5,23 ; 5,33 5,30; 5,37
Вычислено, % . .. 51,37 5,17 5,48
Литература
l.Schwarzenbach G., AndereggG., Sallmann
R., Helv. Chim. Acta, 35, 1785 (1952).
4-ДИАЗО-2,2,5,5 ТЕТРААЛКИЛФУРАНИДОНЫ-3
И. к. КОРОБИЦЫНА, Л. Л. РОДИНА
Нами разработан препаративный метод синтеза неиз-
вестного ранее класса соединений — 4-диазо-2,2,5,5-те-
59
траалкилфуранидонов-3, основанный на окислении моно-
гидразонов 2,2,5,5-тетраалкилфуранидиндионов-3,4 жел-
той окисью ртути С Интерес к этим соединениям обуслов-
лен их большой реакционной способностью, в частности,
возможностью получения из них путем фотохимической
перегруппировки труднодоступных кислот ряда оксе-
тана. Моногидразоны 2,2,5,5-тетраалкилфуранидиндио-
нов-3,4 получают взаимодействием эквимолярных коли-
честв 2,2,5,5-тетраалкилфуранидиндионов-3,4 с гидразин-
гидратом \
Схема синтеза
где R и R' = СН3 (соединение a) R+R' = —(СН2)4—(соединение в)
R=CH3; R' = С.2Н5 (соединение б) R+R' — — (СН2)5—(соединение г)
РЕАКТИВЫ
Тетраалкилфуранидиндионы-3,4 2> 3
Окись ртути, желтая, х. ч., ГОСТ 5230—50.
Кали едкое, х. ч., ГОСТ 4203—65.
Способ получения
Синтез моногидразона 2,2,5,5-тетраметилфуранидин-
диона-3,4 (II а). К раствору 15,61 г (0,1 моль) 2,2,5,5-
тетраметилфуранидиндиона-3,4 (1а) в 30 мл этилового
спирта при комнатной температуре прибавляют по кап-
лям 5 г (0,1 моль} гидразингидрата. Спирт отгоняют в ва-
кууме водоструйного насоса и к остатку добавляют абсо-
лютный эфир. Полученный эфирный раствор сушат про-
каленным сульфатом магния. После отгонки эфира из
6Р-
реакционной смеси при охлаждении выпадают желтые
кристаллы моногидразона. Остальные моногидразоны
(соединения II б, lie, II г) получают аналогично. Тем-
пература плавления, выходы продуктов и данные эле-
ментарного анализа синтезированных моногидразонов
2,2,5,5-тетраалкилфуранидиндионов-3,4 приведены в
табл. I.
Таблица 1
Данные анализа моногидразонов
2,2,5,5-тетраалкилфуранидиндионов 3,4
Моно гидразоны
2,2,5,5-тетраалкилфу-
ранидиндионов-3,4
Вы-
ход
%
Температура
плавления
°C
Найдено, % Вычислено, %
С Н N С Н N
2,2,5,5-Тетраметил
(11 а).............99
2,5-Диметил-2,5-ди-
этил (II б) . . . 94,2
2,5-Бнс-тетраметн-
лен(Не). . . . 90
2,5 Бнс-пентаметн-
лен (11 г) ... 90
103—103,5 56,57 8,43 16,37 56,45 8,29 16,46
56,52 8,49 16,27
34,5—35,5 60,499,25 14,16 60,37 9,15 14,13
60,70 9,24 14,09
71,5—72 64,668,39 12,71 64,83 8,16 12,60
64,758,40 12,68
73,5—74 67,308,96 11,11 67,168,87 11,20
67,52 9,00 11,21
Получение 4-диазо-2,2,5,5-тетраметйлфуранидона-З
(Ша). К раствору 17г(0,1 моль) моногидразона2,2,5,5-тет-
раметилфуранидиндиона-3,4 в 150—200 мл абсолют-
ного эфира добавляют 43,3 г (0,2 моль) желтой окиси
ртути и 1 мл насыщенного спиртового раствора едкого
кали. При этом реакционная смесь разогревается, желтый
цвет раствора переходит в красный и выпадает серый
осадок металлической ртути. Реакционную смесь встря-
хивают 2—6 ч на механической качалке (см. примеча-
ние 1) в толстостенной склянке, закрытой резиновой проб-
кой с краном. Кран периодически открывают и выпус-
кают пары эфира. Выделившуюся ртуть отфильтровы-
вают, эфирный раствор сушат над прокаленным сернокис-
лым магнием. После отгонки эфира остаток перегоняют
в вакууме. Остальные диазокетоны (соединения III б,
III в, III г) получают по аналогичной методике, Выходы
61
Константы и данные анализа дназокетонов
SS 2 16,65 14,27 12,71 11,28
о в ф I S О 8,21 7,32 ОО
3 СО О 57,12 61,20 64,43 67,71
2 16,61 16,53 14,20 14,17 12,75 12,82 сч ь- 0104
g га S Ю QO rf СО 8,53 8,40 О ОО хЬСО 1 8,20 8,28
м Q 57,39 57,39 61,29 61,50 65,30 65,60 67,84 67,79
а ф Е 5 га гмостн от давления мм рт. ст. СО
>. а g В 5 аз Я СО Й о 53—54 77—79 1 i 114—115
Температура плавления ; минус 5,5—4,5 1 24—25 40,5—41,5
Выход % ! 88,5 89,6 82 80
6-
*—<
4-Диазо-2,2,5.5-тетра а лк ранидоны-3 2,2,5,5-Тетраметнл (III § ю СЧ Ч 8 <U из см я § я <и & о 7 о S U) еГ я 1 СО н S с 6 S й
продуктов, константы и данные элементарного анализа
синтезированных 4-диазо-2,2,5,5-тетраалкилфуранидонов-3
приведены в табл. 2.
Примечание. Реакцию можно проводить в колбе при
энергичном перемешивании механической мешалкой.
Литература
1. Кор оби цы на И. К., Родина Л. Л., ЖОХ, 34
2851 (1964).
2. К о р о б и ц ы н а И. К., Юрьев Ю. К-, Нефедо-
ва О. И., ЖОХ, 24, 188 (1954).
3. Кор о би цы я а И. К., Юрьев Ю. К., Ч е б у р-
ков Ю. А., Лукина Е. М., ЖОХ, 25, 734 (1955).
N, N-ДИАЛКИЛАМИНОИЗОПРОПИЛ-
ДИАЛКИЛФОСФАТЫ
N, N-ДИЭТИЛАМИНОИЗОПРОПИЛДИЭТИЛ ФОСФАТ
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
>N—СН,—СИ—О—Р<
СзН/ Ч)С2Н6
qiH2eNO4P Мол. вес 267,30
Для биологии и токсикологии большой интерес пред-
ставляют фосфорорганические соединения, являющиеся
близкими структурными аналогами продуктов жизне-
деятельности организмов. К таким веществам относятся
фосфорные эфиры аналогов холина — N.N-диалкиламино-
изопропилдиалкилфосфаты, которые в литературе не опи-
саны и получены нами впервые *. Синтез этих соединений
заключается во взаимодействии алкоголятов натрия,
диэтиламинопропанола-2 и диизопропиламинопропанола-2
с диалкилхлорфосфатами. Подобные соединения приме-
няются в качестве инсектицидов2 и фармацевтических
препаратов ®.
63
62
Схема синтеза
о
СН3 СН, С1_р/ОС2Нб
| Na С2Н5 | ^ОС^На
>N-CH3-CH-OH-------> \N—СН,—CH—ONa------------►
С3Н/ С3Н/
СН3 О
С,н5ч I II -ОСА
---> )N-CH,-CH-O-P<
Сзн/ ХОД
РЕАКТИВЫ
Диэтиламинопропанол-2 *, т. кип. 157—159 °C; «д = 1,4250.
Натрий металлический, ч., ТУ МХП 1664—50.
Диэтилхлорфосфат, свежеперегнанный, т. кип. 81—83 °C при
10 мм рт. ст.
Бензол, ч. д. а., ГОСТ 5955—51.
Диизопропиламинопропанол-24, т. кип. 94—96 °C при
35 мм рт. cm., пр = 1,4340.
Способ получения
В трехгорлую колбу с обратным холодильником поме-
щают 13, 12 г (0,1 моль) диэтиламинопропанола-2, прибав-
ляют 50 мл сухого бензола и 2,3 г (0,1 моль) металличе-
ского натрия и при перемешивании нагревают реакцион-
ную массу до полного растворения натрия (60—70 °C).
Охладив полученный раствор алкоголята натрия до 5 °C,
прибавляют по каплям 17,25 г (0,1 моль) диэтилхлорфос-
фата с такой скоростью, чтобы температура реакционной
смеси не поднималась выше 10 °C. Затем смесь нагревают
30 мин на кипящей водяной бане, охлаждают до комнат-
ной температуры и выпавший осадок хлористого натрия
отделяют центрифугированием. Из фильтрата отгоняют
бензол и остаток перегоняют в вакууме. Получают 20 г
продукта с т. кип. 120—121 °C при 5 мм рт. ст.-, выход
75% (от теоретического), drf — 1,009, tiD = 1,4290.
Состав синтезированного продукта (C11H26NO4P):
с н р
Найдено, %................... 49,45 9.82 11.60
Вычислено, %................. 49,42 9,81 11,59
Аналогичным путем получают другие диалкиламино-
изопропилдиалкилфосфаты, константы которых приве-
дены ниже:
64
Название соединения
N, N-диэтиламиноизопропилдиизо-.
пропилфосфат..................
Ы.М-диизопропиламиноизопропил-
диэтилфосфат..................
Ь1,М-диизопропиламиноизопропил-
диизопропилфосфат.............
Выход кип- <°С> 20 ,2!)
о/ при давлении «£> а
° (мм ptn.ctn.) 4
69 119—120; 4 1,4267 0,9792
73 131—132; 4 1,4335 1,0130
73 151—152; 10 1,4309 0,9771
Литература
1. Дрегваль Г. Ф., Самуилов А. М., авт. свид.
СССР 182159; Бюлл. изобр., № И (1966).
2. Пат. ФРГ 1143512 (1957); РЖХим, 6Н426 (1965).
3. Фр. пат. 1072М (1962); РЖХим, 23Н128 (1963).
4. М а л и н о в с к и й М. С., Окиси олефинов и их производ-
ные, Госхимиздат, 1961, стр. 267.
ДИАЛЛИЛОВЫЙ ЭФИР ФЕНИЛФОСФОНОВОЙ
кислоты
Е. Л. ГЕФТЕР
C12H15O3P
,осн,сн=сн2
с£н5-р<
|| осн,сн=сн2
Мол. вес 238,22
Диаллилфенилфосфонат применяют для изготовления
огнестойких высокомолекулярных соединений, органи-
ческих стекол М 2 и получают взаимодействием дихлор-
ангидрида фенилфосфоновой кислоты с аллиловым спир-
том в пиридине 1 и в среде инертных растворителей 3,
а также взаимодействием дихлорангидрида с аллилатом
натрия \ Мы проверили и уточнили 4 методику, основан-
ную на взаимодействии дихлорангидрида с аллиловым
спиртом в пиридине.
Схема синтеза
С6Н6РОС12 + 2СН2=СНСН2ОН + 2C6H6N-»
---> С6Н5РО(ОСН2СН-СН.,)2 + 2C5H3N • на
5—287 65
РЕАКТИВЫ
Дихлорангидрид фенилфосфоновой кислоты, я., (см. стр. 99).
Аллиловый спирт, ч., ТУ МХП 1880—48.
Пиридин, ч., ГОСТ 2747—44. •
Способ получения
Прибор для синтеза состоит из круглодонной четы-
рехгорлой колбы емкостью 3 л, снабженной капельной
воронкой, обратным холодильником с хлор кальциевыми
трубками, мешалкой и термометром. В колбу помещают
315 г сухого аллилового спирта и 432 г сухого пиридина
(см. примечание 1) и вытесняют воздух из колбы сухим
N2 или СО2 (см. примечание 2). Колбу погружают в смесь
льда и соли и при перемешивании вводят по каплям в течение
6—8 ч 505 г свежеперегнанного дихлорангидрида фенил-
фосфоновой кислоты (мольное соотношение CeHsPOCl2:
:СН2 = СНСН2ОН : C5H5N = 1 : 2,1 : 2,1), поддерживая
температуру от 0 °C до 5 °C (см. примечание 3). При этом
выпадает осадок солянокислого пиридина. Реакционную
массу перемешивают 1 ч при комнатной температуре
и прибавляют холодную воду до растворения осадка. Орга-
нический слой отделяют, промывают 2%-ным раствором
карбоната натрия, водой до нейтральной реакции (инди-
катор — бумага конго), прибавляют 5 г порошкообраз-
ной соды (см. примечание 4) и перегоняют в вакууме.
Отгоняют легколетучую фракцию при 30—50 мм рт. ст.
н температуре бани 40—100 °C и немедленно (см. примеча-
ние 5) перегоняют продукт реакции. Перегонка ведется
на масляной или металлической бане (см. примечание 6)
нз колбы с низким дефлегматором при давлении
1—3 мм рт. ст. (см. примечание 7). Верхнюю часть
колбы н дефлегматор тщательно закрывают асбестом.
Получают 480—500 г продукта (см. примечание 8);
т. кип. 128—130 °C при 1 мм рт. ст. и 137—138 °C при
1,5—2 мм рт. ст.- пЪп = 1,5110—1,5119, d20 = 1,0750—
1,0830; выход 78—81 % (от теоретического). По литератур-
ным данным, т. кип. вещества 128 °C при 1 мм рт. ст.;
= 1,5128, df = 1,1097 >.
Диаллилфенилфосфонат представляет собой бесцвет-
ную прозрачную маслянистую жидкость, растворяется
в ряде органических растворителей, не растворяется
В воде.
66
Примечания. 1. При проведении реакции в среде инерт-
ных растворителей выход диаллилфенилфосфоната не повышается.
2. Влага, содержащаяся в воздухе, и особенно не вполне сухие
реагенты снижают выход продукта.
3. При более высокой температуре реакции (особенно выше
10 °C) выход продукта снижается.
4. Карбонат натрия является более надежным ингибитором
процессов полимеризации и разложения диаллилфенилфосфоната
при перегонке, чем другие вещества.
5. Если сырое вещество оставить даже на несколько часов, это
приведет к уменьшению выхода продукта (иногда на 8—10%) за счет
гидролиза.
6. Температуру в пределах 100—120 °C следует повышать
сравнительно медленно, в противном случае наблюдается разложе-
ние вещества.
7. Повышение давления влечет за собой уменьшение выхода
вследствие полимеризации диаллилфенилфосфоната при высокой
температуре.
8. Перегонку следует прекратить, как только начнется легкое
разложение продукта, т. е. в колбе появятся пары.
Литература
1. Toy A. D. F., J. Am. Chem. Soc., 70, 186 (1948).
2. Т о у A. D. F., Brown V., Ind. Eng. Chem., 40, 2276 (1948).
3. S h о k a 1 E. C., W h i t e h i 1 1 L. N., англ. пат. 645222;
С.A. 45, 4267 (1951).
4. Г e ф т e p E. Л., Пласт, массы, Ns 11, 38 (1961).
ДИАРИЛОВЫЕ ЭФИРЫ ВИНИЛФОСФОНОВОЙ
кислоты
Е. Л. ГЕФТЕР, Н. И. БОНДАРЬ
,ОАг
СН2=СН—Р<
|| хОАг
О
Дифениловый эфир винилфосфоновой кислоты поли-
меризуется и сополимеризуется с некоторыми мономе-
рами х. Он является исходным веществом при получении
огнестойких пенопластов 2 и отверждаемых фосфорсодер-
жащих полиэфиров 3. Для аналогичных целей могут при-
меняться другие эфиры винилфосфоновой кислоты. Мы
разработали простую методику синтеза диариловых эфи-
ров винилфосфоновой кислоты без выделения промежу-
5* 67
точных продуктов4. В качестве исходных соединений
использованы доступный дихлорангидрид (5-хлорэтилфос-
фоновой кислоты, оксиароматические соединения и три-
этиламин.
Схема синтеза
О
ArOH II ШЫЦЬ
Cl—СН2—СН2—РОС12 -----> С1—ОТ2—СН2—Р(ОАг)2
О
II
---» СН.2=СН—Р(ОАг)2
РЕАКТИВЫ
Дихлорангидрид [5-хлорэтилфосфоновой кислоты (получение
см. на стр. 102).
Фенол, ч., перегнанный, ГОСТ 6417—52.
п-Хлорфенол, ч., перегнанный, ТУ МХП 2637—51.
3,4 -Ксиленол, ч., перегнанный, СТУ 77-20—61.
3,5-Ксиленол, ч., перегнанный, МРТУ 6-09-3833—67.
Триэтиламин, ч., высушенный над щелочью и перегнанный,
ГОСТ 9966—62.
Способ получения
В трехгорлую круглодонную колбу, снабженную ме-
шалкой, термометром, обратным холодильником с хлор-
кальциевой трубкой и глухим затвором (см. стр. 109),
позволяющим проводить реакцию в вакууме, помещают
18 г дихлорангидрида Р-хлорэтилфосфоновой кислоты
и 20,5 г фенола (мольное соотношение 1 : 2,2). Реагенты
перемешивают и выдерживают 6—8 ч на масляной бане
при 150—170 °C (при использовании замещенных фенолов
смесь нагревают до 170—190 °C), т. е. практически до
полного прекращения выделения хлористого водорода.
Остаток хлористого водорода удаляют при 100—НО °C
и 20—40 мм рт. ст. Полученную темную жидкость охлаж-
дают до комнатной температуры, приливают 15,2 г триэтил-
амина и 25 мл сухого бензола. Смесь перемешивают
и кипятят на водяной бане 3 ч, при этом через несколько
минут начинает выпадать осадок солянокислого триэтил-
амина. Смесь охлаждают до комнатной температуры
и заливают холодной водой (см. примечание). Бензоль-
ный слой отделяют и перегоняют в вакууме. Получают
19,5—21 г дифенилового эфира винилфосфоновой кислоты
с т. кип. 142 °C при 2 мм рт. ст.-, п2° = 1,5555, d20 =
= 1,1947, выход 75—81 % (от теоретического). Вещество
представляет собой прозрачную светло-желтую жид-
кость, растворяется в спирте, эфире, бензоле, толуоле,
не растворяется в воде.
Содержание фосфора в синтезированном продукте
(С14Н13О3Р):
Найдено, %................11,6
Вычислено, %..............11,9
По аналогичной методике синтезированы другие диарило-
вые эфиры, константы которых приведены в таблице.
Константы диариловых эфиров винилфосфиновой кислоты
Эфир винилфосфи- повой КИСЛОТЫ СН3-СНР<ХОАг)2 Брутто-фор- мула Моле- куляр- ный вес Температура кипения в зависимости от давления nD
°C ММ рт. ст.
Ди-4-хлорфени- ловый . . . С14НиО3С12Р 329,12 186-189 3 1,5681 1,3422
Ди-3,4-диметил- фениловый . С18Н21О3Р 316,33 180—183 2 1,5520 1,1448
Ди-3,5-диметил- фениловый . ^‘18^21^3^ 316,33 190—192 2—3 1,5512 1,1439
Примечание. При удалении солянокислоготриэтиламина
фильтрованием выход конечного продукта практически не изме-
няется.
Литература
1. Колесников Г. С. н др., Высокомол. соед., 4, 448 (1968);
7, 377 (1965),
2. Колесников Г. С. и др., авт. свид. СССР 168426; Бюлл.
изобр., № 4 (1965).
3. Рубцова И. К., Г е ф т е р Е. Л., Юлдашев А.,
Мошкин П. А., Пласт, массы, № 3, 13 (1961).
4. Г е ф т е р Е. Л., Хим. наука и пром., № 3, 544 (1958).
4,4'-ДИБРОМДИФЕНИЛАМИН
О. М. СТАШКЕВИЧ, Г. Т. ПИЛЮГИН
Cl2HeBr2N
Мол. вес 327,03
Впервые 4,4'-дибромдифениламин был получен бро-
мированием бензоилдифениламина в среде уксусной кис-
лоты х. Позднее Фишер получил его с выходом 5% при
действии бромистоводородной кислоты на N-нитрозоди-
фениламин 2. В дальнейшем было показано 3, что дифе-
ниламин при бромировании в спиртовом растворе обра-
зует помимо дибромдифениламина тетрабромпроизвод-
ные, и было проведено 4 доказательство строения, т. е. точ-
ного положения атомов брома. Нами разработан метод
получения 4,4'-дибромдифениламина, основанный на бро-
мировании дифениламина элементарным бромом при по-
ниженной температуре; выход 30—35%. При этом 13—
20% дифениламина остается непрореагировавшим. Тетра-
бромпроизводные, образующиеся в значительном количе-
стве, легко отделяются от основного продукта.
Схема синтеза
Вг2 /==
(CeH6)2NH —>
РЕАКТИВЫ
Дифениламин, ч. д. а., ГОСТ 5825—51.
Эфир этиловый, ч., ГОСТ 6265—52.
Диоксан, ч., ВТУ МХП 3111-52.
Бром, ч. д. а., ГОСТ 4109—64.
Спирт этиловый, ректификованный, ГОСТ 5962—51.
Натр едкий, ч. д. а., ГОСТ 4328—66.
Способ получения
В трехгорлой колбе емкостью 1 л, снабженной меха-
нической мешалкой, капельной воронкой и поглотителем
для бромистого водорода, растворяют 34 г (0,2 моль)
дифениламина в смеси 250 мл эфира и 150 мл диоксана.
При энергично работающей мешалке к раствору, охлаж-
70
денному до минус 10—15 ЙС, по каплям в течение 4 ч при-
бавляют раствор 64 г (0,4 моль) брома в 250 мл эфира (бром
растворяют в зфире при температуре от 0 до —5 °C).
По мере прибавления раствора брома в колбе образуется
осадок белого цвета. Затем реакционную смесь переме-
шивают 2 ч при 0 °C, оставляют стоять 12 ч при комнатной
температуре, после чего фильтруют. Осадок суспенди-
руют в воде и суспензию обрабатывают 20%-ным рас-
твором едкого натра до сильнощелочной реакции. При
этом выпадает масло, которое при охлаждении застывает
в твердую массу. После двукратной кристаллизации из
этанола получают 20—23 г продукта в виде белых игл;
т. пл. 106—107 °C; выход 30—35% (от теоретического).
По литературным данным, т. пл. вещества 106,5—107 °C 4,
107 °C 3.
Содержание азота и брома в препарате (C12H8Br2N):
Найдено, % . .
Вычислено, % .
Br N
4S,C2; 49,22 4,32; 4,37
48,9 4,28
Литература
1. L е 1 1 m а п n Е., Вег. 15, 825 (1882).
2. Pischer Н., Вег. 45, 1103 (1912).
3. Galatis L., Megaloikonomos J., Zbl., 1934, 2974.
4. С г о u n s e N. N., Raiford C. L., J. Am. Chem. Soc., 67,
875 (1945).
ДИБУТИЛАЦЕТАЛЬ АЦЕТАЛЬДЕГИДА
E. Л. ГЕФТЕР
,ОС4Н9
X)C4H9
Мол. вес 174,28
Ацетальдегиддибутилацеталь является хорошим рас-
творителем для различных органических веществ. В част-
ности, он рекомендуется в качестве безопасного раство-
рителя, повышающего выходы при проведении реакции
Фаворского Ацетальдегиддибутилацеталь получают
взаимодействием ацетальдегида с избытком бутанола раз-
71
личными методами: в присутствий катализаторов (НО,
H2SO4, FeCl3, катионообменной смолы и др.), при высокой
температуре, в присутствии моногидрата хлористого каль-
ция 2-6; присоединением бутанола к винилбутиловому
эфиру 6; обработкой полуацилалей или ацетальдегида
бутанолом в присутствии катализаторов ,-9; взаимодей-
ствием а-галоидэтилбутиловых эфиров с диалкиланили-
нами или аммиаком в растворе бутанола или с бутилатом
натрия также в растворе бутанола 10>1Х. Мы использовали
видоизмененную методику 12 получения ацетальдегидди-
бутилацеталя. Увеличение количества хлористого кальция
в реакционной смеси позволило поднять выход продукта
с 64 до 80%.
Схема синтеза
СН3СНО + С4Н9ОН + СаС12-, СН3СН(ОС4НВ)2 + СаС1, 6Н2О
РЕАКТИВЫ
Ацетальдегид, ГОСТ 9585—61.
н-Бутанол, ч., ГОСТ 6006—51.
Кальций хлористый, прокаленный, х. ч., ГОСТ 4141—48.
Способ получения
Реакцию проводят в очень прочной бутыли емкостью
10—15 л, завернутой в частую металлическую сетку или
материю. В охлажденную до 5 °C смесь 3120 г (12 моль)
сухого н-бутанола и 575 а (1,45 моль) хлористого кальция
по стенке приливают 975 а (6,3 моль) охлажденного до
0—5 °C свежеперегнанного уксусного альдегида (см. при-
мечание). При этом наблюдается легкое вскипание.
Бутыль плотно закрывают, несколько раз встряхивают,
быстро открывают и вновь закрывают. При взбалтыва-
нии альдегидный слой почти мгновенно исчезает. Бутыль
встряхивают более энергично 2—3 мин, время от времени
открывая и закрывая ее. Через 8—15 мин прозрачная
желтоватая жидкость мутнеет, разогревается и через
20 мин на дне образуется жидкий слой, содержащий шести-
водный гидрат хлористого кальция. Раствор встряхивают
еще несколько раз и оставляют до следующего дня, после
чего верхний слой отделяют, трижды промывают водой,
сушат прокаленным поташем и перегоняют в вакууме,
.отбирая фракцию с т. кил. 60—64 °C при 6—7 мм рт. ст.
Получают 2880—2950 а продукта; выход 78—80%
72
(от теоретического), п2° = 1,4070—1,4072; df = 0,8345—
0,8349. Вещество представляет собой бесцветную про-
зрачную жидкость, растворяется в спирте, эфире, этилаце-
тате и не растворяется в воде. По литературным данным 7,
т. кип. 185—187 °C, п“ = 1,4090, d*> = 0,8315.
Состав синтезированного продукта (С10Н22О2):
с н
Найдено, %.................68,71 12,58
Вычислено, % . . . . 68,97 12,64
Примечание. Приведены оптимальные соотношения ком-
понентов. Если применять расчетное количество хлористого каль-
ция, то выход снижается на 12—16%.
Литература
1. Bergmann Е., Sulzbacher М., Herman D.F.,
J. Appl. Chem., 3, 39 (1953).
2 Weizmann C., Bergmann E., Sulzbacher M.,
J. Org. Chem., 15, 918 (1950).
3 Doi n i c k A. A., Potash M., пат. США 2566559;
C.A., 46, 2566 (1952).
4. Renault I., фр. пат. 868182; C.A., 43, 5034 (1949).
5 F r e n e 1 I.. К., H e d el u nd J. W., пат. США 2691685;
С.A., 49, 12530 (1955).
6. Шостаков ский М. Ф., Герштейн Н. А., Син-
тезы органических соединений, сб. 2. , ИОХ АН СССР, 1952,
стр. 154.
7. Шостаковский М. Ф., Герштейн Н. А., ЖОХ,
21, 1602 (1951),
8. Decker R., Holz Н., Пат. ФРГ 844441; С. А., 47, 11225
(1953).
9. С г о х'а 11 J., Glavis F. J., Neh ег Н. Т., J. Ат.
Chem. Soc., 70, 2805 (1948).
10. Шостаковский М. Ф., Богданова А. В., ЖОХ,
20, 1315 (1950).
И. Шостаковский М. Ф., Богданова А. В., ЖОХ,
17, 565 (1947).
12. Синтезы органических препаратов, сб. 1, Издатинлит, 1949,
стр. 62,
ДИБУТИЛОВЫЙ ЭФИР МЕТИЛФОСФОНОВОЙ
кислоты
Е. Л. ГЕФТЕР
/ОС4Н9
СН3-Р<
If ХОС411я
о
С9Н21О3Р Мол. вес 208,23
Дибутиловый эфир метилфосфоновой кислоты исполь-
зуется как растворитель и пластификатор с повышенной
огнестойкостью. Описан синтез этого эфира путем пере-
группировки трибутилфосфита в присутствии йодистого
метила 1 (перегруппировка Арбузова). Мы выбрали для
получения эфира реакцию этерификации дихлорангидри-
да метилфосфоновой кислоты бутанолом в присутствии
пиридина.
Схема синтеза
СН3РОС12 4- С4Н9ОН 4- C5H6N .—» СН3РО(ОС4Н9)2 + c5h6n.hci
РЕАКТИВЫ
Дихлорангидрид метилфосфоновой кислоты 2
н-Бутанол, ч., высушенный прокаленным сульфатом меди
и перегнанный, ГОСТ 6006—51.
Пиридин, ч., высушенный над щелочью и перегнанный, ГОСТ
2747—44.
Способ получения
Прибор для синтеза состоит из круглодонной четырех-
горлой колбы, снабженной капельной воронкой, обратным
холодильником с хлоркальциевыми трубками, мешалкой
и термометром. В колбу помещают 100 мл сухого бен-
зола, 171 г сухого, «-бутанола и 182 г сухого пиридина
(см. примечание 1). К этой смеси при интенсивном пере-
мешивании и охлаждении в бане со льдом и солью при-
бавляют по каплям в течение 3—5 ч раствор 153 г (1,15 моль)
дихлорангидрида метилфосфоновой кислоты в 80 мл сухого
бензола. При этом выпадает осадок солянокислого пири-
дина. Температура реакции поддерживается в пределах
2—5 °C (см. примечание 2). Смесь перемешивают 1 « при
комнатной температуре и приливают холодную воду до
растворения осадка. Органический слой отделяют,
74
отгоняют бензол и остаток перегоняют в вакууме (см, при-
мечание 3). Получают 190—205 г продукта с т. кип. 90—
92 °C при 2—3 мм рт. ст.-, выход 79—85% (от теоретиче-
ского), л® = 1,4252, df = 0,9921. По литературным дан-
ным1, т. кип. 135—136 °C при 10—12 мм рт. cm., пп —
= 1,4241, dj = 0,9962. Дибутиловый эфир метилфосфи-
новой кислоты растворяется в спирте, эфире, бензоле,
толуоле, не растворяется в воде.
Содержание фосфора в синтезированном продукте
(С9Н21О3Р):
Найдено, %................14,6
Вычислено, %..............14,8
Примечания. 1. При недостаточной сухости реактивов
выход продукта резко снижается.
2. Более высокая температура (особенно выше 10 °C) способ-
ствует снижению выхода.
3. Если перегонку производить не сразу, выход продукта
уменьшается вследствие гидролиза.
Литература
1. К а м а й Г., Труды КХТИ им. С. М. Кирова, № 10, 29 (1946).
2. К i n 1 а г А. М., Реггей Е. A., J. Chem. Soc., 1952,
3437.
ДИИЗОАМИЛОВЫЙ ЭФИР
2,2-БИЦИНХОНИНОВОЙ КИСЛОТЫ
А. Л. ГЕРШУНС, А. А. ВЕРЕЗУБОВА
uaoCjHuOOC СООС5Н1Гизо
Сз0Н32М2О4
Мол. вес 484,59
Диизоамиловый эфир 2,2'-бицинхониповой кислоты
образует интенсивно окрашенные комплексы с ионами
меди. Обладая такой же избирательной способностью,
как и 2,2'-дихинолил, этот реактив почти в два раза более
чувствителен. Аналогичные свойства имеют и другие
7.5
синтезированные нами диалкильные эфиры 2,2'-бицйн-
хониновой кислоты 1. Эти соединения можно получать
двумя методами: исходя из хлорангидрида 2,2'-бицинхо-
ниновой кислоты или этерификацией 2,2'-бииинхонино-
вой кислоты в присутствии серной кислоты. Последний
способ более удобен.
Схема синтеза
ноос соон
Ц- U30-C3HUOH
«зо-С6НиООС СООС5Нн-цзо
РЕАКТИВЫ
2,2’-Бицинхониновая кислота 2> 3, т. пл. 365 °C.
Спирт изоамиловый, ч. д. а., ГОСТ 5830—51.
Кислота серная, ч., ГОСТ 4204—66.
Способ получения
В колбу с обратным холодильником помещают 17,2 г
(0,05 моль) 2,2'-бицинхониновой кислоты, 400 мл изо-
амилового спирта, 15 мл серной кислоты и кипятят 5 ч
до полного растворения 2,2'-бицинхониновой кислоты.
Кристаллы, выпавшие после охлаждения реакционной
массы, отфильтровывают. Из фильтрата после разбавле-
ния метиловым спиртом выпадает дополнительное коли-
чество вещества. Продукт кристаллизуют из изоамило-
вого спирта; т. пл. 109—НО °C, выход 16,9 г—70% (от
теоретического). Кристаллы диизоамилового эфира 2,2'-
бицинхониновой кислоты представляют собой бесцвет-
ные длинные пластинки. Вещество хорошо растворяется
в пиридине, эфире, диоксане и хлороформе, в спиртах —
только при нагревании, в воде растворяется плохо.
Содержание азота в препарате (C3oH3aN204):
Найдено, %...........5,63; 5,69
Вычислено, %.........5,78
76
Литература
1. Гер шу нс А. Л., Верезубова А. А., Укр. хим. ж.,
30, 955 (1964).
2. Г е р ш у н с А. Л., Верезубова А. А., Тол-
стых Ж. А., Изв. высших учебных заведений, Химия и хим.
технол., 4, 25 (1961).
3. Гершунс А. Л., Верезубова А. А., Методы полу-
чения химических реактивов и препаратов, ИРЕА, вып. 9,
1964, стр. 78.
1-(N, N-ДИКАРБОКСИМЕТИЛ)-
АМИНОМЕТИЛ-2-НАФТОЛ
В. я. ТЕМКИНА, Г. Ф. ЯРОШЕНКО, Л. М. ТИМАКОВА,
Р. П. ЛАСТОВСКИЙ, Н. В. ЦИРУЛЬНИКОВА
zch2cooh
чсн,соон
c15h15N06
Мол. вес 289,28
1-(М,М-Дикарбоксиметил)-аминометил-2-нафтол, яв-
ляющийся новым флуоресцирующим комплексоном, обра-
зует прочные комплексы с металлами. В ряде случаев
комплексообразование сопровождается гашением флуо-
ресценции, что может быть использовано для определения
некоторых катионов комплексонометрическим и флуори-
метрическим методами. Комплексон получен конденса-
цией ^-нафтола с формальдегидом и иминодиуксусной
кислотой по реакции Манниха *.
Схема синтеза
ОН
ОН
СН2СООН
СН2СООН
РЕАКТИВЫ
^Нафтол, ч., ГОСТ 5835—51.
Кислота иминодиуксусная, ч., ВТУ МГУХП 190—58.
Формальдегид, ч., ТУ МХП 2631—51.
Кислота уксусная, ч., ГОСТ 61—51.
77
Способ получения
В трехгорлую колбу, снабженную механической ме-
шалкой, обратным холодильником и капельной ворон-
кой, помещают 7,3 г (0,055 моль) иминодиуксусной кис-
лоты, нейтрализованной 30%-ным раствором едкого натра
до pH 7—8, и добавляют раствор 7,2 г (0,05 моль) |3-
нафтола в 50 мл ледяной уксусной кислоты. К реакцион-
ной смеси, охлажденной до 5—7 °C, добавляют по каплям
при перемешивании 4,6 г (0,06 моль) 37%-ного раствора
формальдегида. После добавления всего количества форм-
альдегида температуру поднимают до 20—22 °C, и при-
мерно через 30 мин выпадает обильный белый осадок.
Реакционную массу выдерживают при 20—22 °C в тече-
ние 2 ч, нагревают до 55—60 ЭС и размешивают при этой
температуре еще 3 ч. По охлаждении осадок отфильтро-
вывают, промывают 0,5 л воды и сушат в сушильном
шкафу при 70 °C. Получают 7 г комплексона; выход 48%
(от теоретического).
Вещество белого цвета; растворяется в щелочах, не
растворяется в воде, ацетоне, хлороформе.
Состав синтезированного продукта (C16H15NO6):
с н N
Найдено, %...............62,2; 62,2 5.3; 5,7 4,9; 5,1
Вычислено, % . . ... 62,3 5,2 4,9
Литература
1. SchwarzenbachG., Ап d er egg G., Salman R.,
Helv. Chim. Acta, 35, 1785 (1952).
2,5-ДИМЕТИЛГЕКСИН-3-ДИОЛ-2,5
£. Л. ГЕФТЕР
сн, сн3
но—с—с=с—с—он
I I
CHS СН3
Мол. вес 142,19
78
2,5-Диметилгексин-3-диол-2,5 используется в качестве
полупродукта в различных синтезах и получается при
взаимодействии дима гиийдибр ома цетилена и ацетона 1;
карбида кальция, едкого кали и ацетона а; ацетилена,
едкого кали и ацетона3. Мы уточнили и отработали
последний метод. Синтез проводился в крупном мас-
штабе в металлической аппаратуре.
Схема синтеза
CHg СНд
кон | I н2о
(СН3)2СО + СН=СН-> ко—с—с=с—с—ок —»
СНд ^Нд
сн3 сн3
—»но-с—с=с-с—он
^Нд СНд
В качестве побочного продукта образуется диметил-
ацетиленилкарбинол по следующей схеме:
СН3 СН3
КОН I Н2О |
(СН3)2СО + СН=СН--* CHsC-C—ОК----> СН=С—С—ОН
ОД, ^Нд
РЕАКТИВЫ
Ацетилен, ГОСТ 5457—60.
Ацетон, марка А, ГОСТ 2768—60.
Кали едкое, ГОСТ 9285—59.
Бензол, ч., ГОСТ 9572—60.
Способ получения
Прибор для получения 2,5-диметилгексин-3-диола-2,5
представляет собой 10—11-литровый цилиндрический же-
лезный пятигорлый бачок (см. примечание 1), снабжен-
ный капельной воронкой, обратным холодильником с хлор-
кальциевыми трубками, мешалкой, термометром и труб-
кой для подачи ацетилена (см. примечание 2). В синтезе
используют: 1400—1500 г (3,22—3,44 моль) порош-
кообразного едкого кали, 300—320 л (350—370 г;
1,60—1,68 моль) ацетилена, 4,5 л (3900 г) бензола и 900 г
79
(2 моль) ацетона (см. примечание 3). Сначала в реакцион-
ный сосуд вводят едкое кали и бензол. Затем в систему
подают ацетилен, включают мешалку (см. примечание 4)
и по каплям прибавляют ацетон, причем наружным
охлаждением (смесь льда и соль) поддерживают темпера-
туру 8—12 °C (см. примечание 5). Скорость подачи аце-
тилена 1,4—2 л[мин (измеряется газовыми часами), ско-
рость подачи ацетона 21—28 г/мин. В этих условиях аце-
тилен поглощается практически полностью. Если ацети-
лен не поглощается полностью, следует ускорить подачу
ацетона. Через 3—4 ч, когда израсходуется около 300—
320 л ацетилена и около 500 г ацетона, подачу ацетилена
прекращают, и остальное количество ацетона добавляют
в течение 10—15 мин (см. примечание 6). Смесь переме-
шивают еще 1—2 ч и выдерживают в течение суток (см.
примечание 7). Затвердевшую массу охлаждают до 4—6 °C
и проводят гидролиз, вливая постепенно в течение 1—2 ч
4—4,5 л холодной воды (см. примечание 8). Бачок перио-
дически встряхивают для устранения местных перегре-
вов. Содержимое бачка выливают в делительную воронку,
бензольный слой отделяют, фильтруют, насыщают угле-
кислым газом до нейтральной реакции раствора по фенол-
фталеину. Бензол отгоняют на водяной бане (см. примеча-
ние 9). Затем в вакууме отгоняют остаток бензола и диме-
тилацетиленилкарбинол (30—60 г), постепенно снижая
давление от 100 до 10 мм и доводя температуру бани до
100 °C. Остаток, представляющий собой 2,5-диметилгек-
син-З-диол-2,5, при охлаждении превращается в желто-
коричневую массу. Выход продукта 600—650 г. Водный
слой дважды экстрагируют эфиром, из экстракта (после
перегонки) получают 100—115 г 2,5-диметилгексин-З-
диола-2,5. Общий выход технического продукта 700—
760 г (63—68%). После двукратной промывки петролей-
ным эфиром и последующего высушивания в вакууме
2,5-диметилгексин-3-диол-2,5 — это светло-желтое кри-
сталлическое вещество с запахом, несколько напоми-
нающим камфору, и т. пл. 90—92 °C. Перекристалли-
зованный из бензина или перегнанный при 120—125 °C
и 20 мм рт. ст. диол является белым кристаллическим
веществом с т. пл. 93,5—94 °C. По литературным данным \
т. кип. 207 °C, т. пл. 94—96 °C. Вещество растворяется
в воде, спирте, эфире, плохо растворяется в бензоле,
толуоле.
80
Примечания. 1. Применялся прибор, имеющий сле-
дующие размеры: высоту 250 мм, внутренний диаметр 240 мм,
длину лопастей мешалки 200 мм, ширину лопастей мешалки 30 мм,
скорость вращения мешалки 300—350 об/мин (менее мощная мешал-
ка не обеспечивает необходимой интенсивности перемешивания).
Мощность мотора составляет обычно 200—300 вт.
2. Трубку опускают ниже уровня жидкости, она должна быть
не слишком узкой во избежание закупоривания.
3. Едкое кали может быть чистым или техническим (любой
марки), не годится едкое кали полностью свободное от железа4.
Ацетон и бензол сушат и перегоняют. Ацетилен подают из баллона,
пропуская его через колонку с хлористым кальцием.
4. Если сначала включить мешалку, то трубка, подводящая
ацетилен, легко забивается щелочью.
5. Повышение температуры реакции до 20—22 °C несколько
снижает выход; при температуре выше 30 °C выход продукта резко
падает.
6. Этот прием, рекомендованный Захаровой и Ильиной s,
несколько повышает выход продукта.
7. Без выдерживания реакционной смеси выход 2,5-диметил-
гексии-З-диола-2,5 уменьшается.
8. Не следует прибавлять большее количество воды, так как
диол хорошо растворяется в воде.
9. Бензол после отгонки сушат прокаленным сульфатом меди
(не следует сушить хлористым кальцием) до тех пор, пока свежая
порция сульфата меди не перестанет окрашиваться в голубой цвет,
и используют в синтезе.
Литература
I. И о ц и ч Ж. И., ЖРХО, 34, 243 (1902).
2. К а з а р я н Л. 3., ЖОХ, 4, 1347 (1934).
3. Бабаян А. Т., Акопян Б., Гюли-Кевхян Р.,
ЖОХ, 9, 1631 (1939).
4. П е т р о в А. Д., Михайлова А. К., Митрофа-
нова Е. В., ДАН СССР, 91, 857 (1953).
5. Захарова А. И., Ильина Г. Д., ЖОХ, 24, 2144
(1954).
6-287
1,4-ДИМЕТИЛ-2,5-ДИИЗОПРОПИЛБЕНЗОЛ
В. П. МАР ШТУП А, Е. п. БАБИН, И. С. МОРОЗОВА,
О. Ф. В03ИЯН0ВА
Ci4H22
Мол. вес 190,32
1,4-Диметил-2,5-диизопропилбензол, аналогично дуро-
лу, может служить исходным продуктом для получения
пиромеллитовой кислоты или ее диангидрида. В литера-
туре описано получение 1,4-диметил-2,5-диизопропилбен-
зола с выходом до 60% при взаимодействии п-ксилола
и пропилена в присутствии хлористого алюминия 1 и дру-
гих катализаторов 2> 3.
Нами показано, что при алкилировании п-ксилола
пропиленом в присутствии хлористого алюминия при
температуре от 0 до -|-5 °C можно получить 1,4-диметил-
2,5-диизопропилбензол с высоким выходом (90—95%).
Схема синтеза
РЕАКТИВЫ
n-Ксилол, ч., ТУ МХП 3601—53.
Алюминий хлористый, безводный, ГОСТ 4452—66.
Пропилен, 96—98%-ный, получен дегидратацией изопропилово-
го спирта над природным алюмосиликатом.
Способ получения
В круглодониую трехгорлую колбу емкостью 1 л,
снабженную обратным холодильником, шнековой мешал-
кой, вращающейся со скоростью 1000—1500 об/мин и бар-
82
ботером для подачи пропилена, вводят 212,3 г (2 моль)
n-ксилола (высушенного над хлористым кальцием)
и 26,5 г (0,2 моль) хлористого алюминия. Колбу поме-
щают в водяную баню, нагретую до 5 °C (см. примеча-
ния 1 и 2), и пропускают через реакционную массу высу-
шенный над серной кислотой пропилен с такой скоростью,
чтобы он успевал поглощаться при минимальном проско-
ке. После подачи 90 л (168,3 г в пересчете на 100%-ный)
пропилена (см. примечание 3) реакционную смесь про-
мывают горячей (60—80 °C) водой до отрицательной
реакции промывных вод на СГ-ион и охлаждают до 2—
5 °C. Выпавший осадок 1,4-диметил-2,5-диизопропилбен-
зола отфильтровывают, фильтрат перегоняют, отбирая
фракцию, кипящую при 240—250 °C. Полученный в этой
фракции продукт снова охлаждают до 2—5 °C, и выпав-
ший осадок фильтруют. Выделенный 1,4-диметил-2,5-
диизопропилбензол кристаллизуют из горячего этилового
спирта. В фильтрате остается в основном 1,4-диметил-
2-изопропилбензол (около 32 г — 8,5% от теоретиче-
ского выхода). После кристаллизации получают 348 г
очищенного 1,4-диметил-2,5-диизопропилбензола; т. пл.
37,5 °C, выход 91,5% (от теоретического), т. кип. 245 °C
при 770 мм рт. ст.
По литературным данным 2, т. кип. 244 °C, т. пл. 37,4 °C.
Примечания. 1. При температуре выше 10 °C выход
1,4-диметил-2,5-диизопропилбензола уменьшается за счет увеличе-
ния скорости изомеризации п-ксилола в о- и .«-ксилол.
2. Во избежание кристаллизации п-ксилола перед тем, как
поместить реактор в водяную баню, через реакционную смесь про-
пускают слабый ток пропилена.
3. Если через взятое количество п-ксилола пропустить около
45 л пропилена, то, отбирая при перегонке фракцию с т. кип. 195—
198 °C, можно получить 1,4-диметил-2-изопропилбензол с выходом
До 60%.
Литература
1. Бабин Е. П., Хим. пром., № 6, 381 (1961).
2. Т о п ч и е в А. В., Волков Р. Н., Завгород-
ний С. В., ДАН СССР, 134, 844 (1960).
3. Авт. свид. СССР 195436; Бюлл. изобр., Ns 10 (1967).
6‘
М-(2', 4/-ДИМЕТИЛФЕНИЛ)-
3-НИТРОБЕНЗОЛСУЛЬФАМИД И
1Ч-(2'-ДИФЕНИЛ)-3-НИТР0БЕН30ЛСУЛЬФАМИД
И. С. МАРКОВИЧ, И. В. КРУГЛОВА, В. М. ДЗИ0МК0
СНз
Ci4H14N2O4S Мол. вес 306,34
N-(2' ,4'-Диметилфенил)-3-нитробензолсульфамид и
Ы-(2'-Дифенил)-3-нитробензолсульфамид в литературе не
описаны. Соединения получены взаимодействием соответ-
ствующих аминов с .и-нитробензолсульфохлоридом в при-
сутствии ацетата натрия.
Схема синтеза
РЕАКТИВЫ
м-Ксилидин, ч., ТУ МХП 197—51, свежеперегнанный.
2-Аминодифенил, ч., ТУ 6-09-1859—64.
м-Нитробензолсульфохлорид, ч., ТУ 79 777—62.
Натрий уксуснокислый, безводный, ГОСТ 199—52.
Кислота уксусная, ГОСТ 61—51.
84
Способ получения
Синтез N-(2' ,4'-диметилфенил)-3-нитробензолсульф-
амида. В трехгорлую колбу, снабженную мешалкой и обрат-
ным холодильником, помещают 13,3 г (0,11 моль), м-кси-
лидина, 22 г (0,10 моль) л-нитробензолсульфохлорида
и 16,4 г (0,20 моль) ацетата натрия. Смесь выдерживают
2 ч при перемешивании и температуре 100—ПО °C, после
чего оставляют на ночь при комнатной температуре. На
следующий день реакционную массу фильтруют, осадок
промывают горячей дистиллированной водой и перекри-
сталлизовывают из этилового спирта. Получают 13 г про-
дукта с т. пл. 125 °C, выход 40% (от теоретического).
Препарат представляет собой белое кристаллическое ве-
щество; растворяется в щелочах, ацетоне, бензоле, не рас-
творяется в воде.
Содержание углерода, водорода и серы в синтезиро-
ванном продукте (C14H14N2O4S):
Найдено, %.......................
Вычислено, %.....................
CHS
55,45 4,62 10,44
54,89 4,64 10,46
Синтез N-(2'-дифенил)-3-нитробензолсульфамида.
В трехгорлую колбу, снабженную обратным холодильни-
ком и мешалкой, помещают 19 г (0,11 моль) 2-аминодифе-
нила, 22 г (0,10 моль) л-нитробензолсульфохлорида,
16,4 г (0,20 моль) ацетата натрия и 50 мл ледяной уксус-
ной кислоты. В дальнейшем поступают так, как указано
в предыдущей методике. Получают 9,4 г продукта; т. пл.
127 °C, выход 25% (от теоретического). Препарат пред-
ставляет собой белое кристаллическое вещество; раство-
ряется в щелочах, ацетоне, четыреххлористом углероде,
бензоле, не растворяется в воде.
Содержание углерода, водорода и азота в синтезиро-
ванном продукте (C18H14N2O4S):
Найдено, %..........................
Вычислено, %........................
С Н N
60,93 3,05 7,64
61,00 3,98 7,90
ДИНАТРИЕВАЯ СОЛЬ 3,3' БИС-
((N, Ы-ДИКАРБОКСИМЕТИЛ)-АМИНОМЕТИЛ>
4,4'-ДИОКСИСТИЛЬБЕНА
В. Я. ТЕМКИНА, Г. Ф. ЯРОШЕНКО, Л. М. ТИМАКОВА,
Р. И. ЛАСТОВСКИЙ
HO-^-CH=CH-<VoH
НООСН2С. >=/ W ,СН2СООН
>N—Н,с/ \СН2—N<
NaOOCH2Cz X2H2COONa
Мол. вес 546,44
C24H24N2O2(,Na2
Динатриевая соль 3,3'-бис-[(М,М'-дикарбоксиметил)-
аминометил]-4,4'-диоксистильбена является новым флуо-
ресцирующим комплексоном. Синтез комплексона осно-
ван на взаимодействии 4,4'-диоксистильбена с формальде-
гидом и иминодиуксусной кислотой по реакции Манниха 1.
Схема синтеза
,сн,соон
НО-f ^-СН=СН-^ у—OH-f-HNQ +сн2о
\=/ \=/ '-CH2COONa
ПО—СИ =СН-^-ОН
НООСН2С >=/ \=< ZCH2COOH
► >N-H2CZ хсн2—n<
NaOOCH2Cz 4CH.2COONa
РЕАКТИВЫ
Кислота иминодиуксусная, ч., ВТУ МГУХП 190—58.
Формальдегид, ч., ТУ МХИ 2631—51.
Кислота уксусная, ч., ГОСТ 61—51.
Способ получения
В трехгорлую колбу, снабженную механической ме-
шалкой, обратным холодильником и капельной ворон-
кой, помещают 3,2 г (0,02 моль) иминодиуксусной кислоты,
нейтрализованной 30%-ным раствором едкого натра до
pH 7—8, и добавляют суспензию 2,1 г (0,01 моль) п-диок-
систильбена в 60 мл ледяной уксусной кислоты. К реак-
ционной смеси, охлажденной до 5—7 °C, добавляют по
каплям при перемешивании 4,6 г (0,06 моль) 37%-ного
86
раствора формальдегида и оставляют при комнатной
температуре на ночь. На другой день реакционную смесь
выдерживают 4 ч при 55—60 °C, одновременно размеши-
вая ее, и затем в течение часа поднимают температуру
до 80 °C. При этом вся масса постепенно растворяется.
Смесь выдерживают при 80 °C в течение 3—4 ч, по охлаж-
дении переносят в стакан и при перемешивании добав-
ляют 200 мл этилового спирта. Выпавший белый осадок
динатриевой соли комплексона фильтруют, промывают
50 мл спирта и сушат в сушильном шкафу при 50 °C.
Получают 3,2 г продукта; выход 59% (от теоретического).
Вещество хорошо растворяется в воде, не раство-
ряется в органических растворителях.
Содержание азота в препарате (C24H24N2O10Na2):
Найдено, %...........4,8; 4,7
Вычислено, %.........5,1
Литература
1. Schwarzenbach G., AndereggG., Sailman R.,
Helv. Chim. Acta, 35, 1785 (1952).
2,2'-ДИОКСИДИФЕНИЛМЕТАН
А. В. СТРАШНЕЙ ДО, A. M. ДАВЫДОВА
C13H12O2
4 \\zZ
Мол. вес 200,24
2,2'-Диоксидифенилметан представляет интерес как
исходный продукт в синтезе полимерных соединений; эпок-
сидных смол, поликарбонатов, полиэфиров и др. Известно
два способа получения 2,2'-диоксидифенилметана.
Первый способ 1 основан на дегалоидировании 2,2'-
диокси-3,5-дибром-5'-хлор-дифенилметана, образующегося
при конденсации 2-окси-3,5-дибромбензилового спирта
с n-хлорфенолом. Согласно второму способу 2, 2,2'-диок-
сидифенилметан получают взаимодействием формальде-
гида с фенолом. При этом наряду с 2,2'-изомером обра-
зуются 4,4'- и 2,4'-изомеры.
87
В настоящей работе 2,2'-диоксидифенилметан получен
восстановлением 2,2'-диоксибензофенона сплавом Ренея.
Исходный 2,2'-диоксибензофенон образуется при сплав-
лении ксантона с едким кали 3.
Схема синтеза
РЕАКТИВЫ
2,2'-Диоксибензофенон3, технический, т. пл. 59—60 °C.
Натр едкий, ГОСТ 4328—66.
Никель-алюминиевый сплав (сплав Ренея), технический.
Способ получения
В трехгорлую колбу емкостью 0,5 л, снабженную
мешалкой, холодильником и термометром, вводят 9 г
(0,042 моль) 2,2'-диоксибензофенона и 250 мл 10%-ного
раствора едкого натра и прибавляют при 90 °C в течение
2 ч 27 г растертого в порошок сплава Ренея. После окон-
чания прибавления реакционную смесь выдерживают
еще 2 ч, энергично перемешивая при температуре 90 °C,
охлаждают и фильтруют. Фильтрат подкисляют соляной
кислотой. Образовавшийся маслообразный осадок экстра-
гируют эфиром. Эфирный экстракт промывают водой
и сушат безводным сульфатом натрия. После отгонки
эфира остаток кристаллизуют из воды.
Получают 4,4 г продукта; т. пл. 118—119 °C, выход
(см. примечание) 52% (от теоретического). По литератур-
ным данным1, т. пл. продукта 118—119 °C.
Примечание. При восстановлении 2,2'-диоксибензофено-
па по Клемменсену амальгамой цинка и соляной кислотой выход
2,2'-диоксидифенилметана составляет всего 15%.
Литература
l.Zigeuner G., Sellinek К., Norman D., Е 1 -
b er K., Monatsch, 90, 477 (1959).
2. Фр. пат. 1111253 (1954); Zbl, 1958, 4327.
3. Qasue U., Katoo H., Bull. Nugoya Citv Univ. Pharm
School, 4, 26 (1956); C.A., 51, 16364 (1957).
88
4,4'ДИОКСИСТИЛЬЁЕН
В. я. ТЕМКИНА. Г. Ф. ЯРОШЕНКО. Л. М. ТИМАКОВА,
Р. П. ЛАСТОВСКИЙ
но—сн=сн-/~:Ч—ОН
CnHiaOj
Мол. вес 212,24
Из описанных в литературе способов получения 4,4'-
диоксистильбена практически используется лишь метод
восстановления диоксидифенилтрихлорэтана цинковой
пылью 1-1 и железным порошком Б. При восстановлении
железным порошком в среде уксусной кислоты, по дан-
ным авторов, и выход, и качество продукта повышаются.
Мы проверили этот метод получения 4,4'-диоксистильбена
и уточнили условия выделения и очистки продукта. Исход-
ный диоксидифенилтрихлорэтан получен по видоизменен-
ному методу Элбса В в.
Схема синтеза
НО. НО__/ \
но>сн-сс1з НО
НО—С //----------^>сн—СС13 »
н°~О-7
но—сн=сн-/~^—он
РЕАКТИВЫ
Фенол, ч., ГОСТ 6417—52.
Хлоральгидрат, ч., ВТУ РУ 428—51.
Кислота уксусная, ГОСТ 61—51.
Кислота серная, ч., ГОСТ 4204—66.
Бензол, х. ч., ГОСТ 5955—51.
Спирт этиловый, синтетический, ГОСТ 9674—61.
Натр едкий, ч., ГОСТ 4528—66.
Железо (порошкообразное), ч.
Способ получения
Синтездиоксидифенилтрихлорэтана. В фарфоровый ста-
кан с механической мешалкой помещают 99,2 г (0,6 моль)
хлоральгидрата и 112,8 г (1,2 моль) расплавленного фенола,
89
растворенного в 40 мл ледяной уксусной кислоты. К полу-
ченной смеси в течение 2 ч при непрерывном перемешива-
нии прибавляют по каплям 90 мл (165 г) концентрирован-
ной серной кислоты, следя за тем, чтобы температура
реакционной среды не превышала 20 °C. Перемешивание
продолжают еще 30 мин. Полученную темную тягучую
массу выливают в 1 л холодной воды, при этом масса
осаждается на дно, постепенно затвердевая. Образовав-
шиеся комки разбивают и оставляют на ночь. Получен-
ный после стояния хлопьевидный осадок розового цвета
отфильтровывают и дважды перекристаллизовывают: сна-
чала из смеси этиловый спирт — бензол (2 : 3), затем из
этилового спирта. Получают 111г продукта; т. пл. 202 °C,
выход 65% (от теоретического). По литературным дан-
ным 2> 6, т. пл. вещества 202 °C (с разложением).
Получение диоксистильбена. В трехгорлую колбу
с механической мешалкой и обратным холодильником
вводят раствор 28,5 г (0,1 моль) диоксидифенилтрихлорэтана
в 600 мл воды, 30 мл уксусной кислоты и 60 г железного
порошка. Смесь кипятят 48 ч, охлаждают и декантируют.
Из осадка экстрагируют 4,4'-диоксистильбен 6%-ным
раствором едкого натра. Полученный щелочной раствор
подкисляют соляной кислотой. Выпавший осадок повтор-
но растворяют в щелочи и выделяют при подкислении,
а затем перекристаллизовывают из спирта. Получают
6,4 г продукта; т. пл. 284 °C, выход 30% (от теоретиче-
ского). По литературным данным, т. пл. вещества 280—
281 °C С ь или 284 °C 3, 4.
Литература
1. Е 1 b s К., J. pract. Chem., [2] 47, 66 (1893).
2. Ter Meer E., Ber., 7, 1202 (1874).
3. Au wets G., Ber., 36, 1887 (1903).
4. Willstatter H., Benz F Ber., 39, 3490 (1906).
5. Z i n c k e F., Fries G., Ann., 325, 26 (1902).
6. T г о j n a M., H u b a c e k, Chem. Listy, 51, 752 (1957).
3,9-ДИ-(4,4'-ОКСИФЕН ИЛ ЭТИЛ )-
СПИРОБИМЕТАДИОКСАН
БИС-ФЕНОЛ
Н. И. КУЦЕНКО, А. И. СЕМЕНОВА
^23^28^6
Мол. вес 400,47
3,9-Ди-(4,4'-оксифенилэтил)-спиробиметадиоксан (бис-
фенол) является сырьем для получения новых видов
эпоксидных смол, поликарбонатов, полиарилатов и дру-
гих видов пластических масс. 3,9-Ди-(4,4'-оксифенил-
этил)-спиробиметадиоксан получен взаимодействием фе-
нола с диаллилиденпентаэритритом в присутствии кис-
лых катализаторов 1. Хорошие результаты получены при
проведении реакции алкилирования в токе азота, в среде
толуола и в присутствии п-толуолсульфокислоты.
Схема синтеза
РЕАКТИВЫ
Диаллилиденпентаэритрит 2, получают из акролеина и пента-
эритрита.
Фенол, ч., ГОСТ 236—54.
Толуол, ч., ГОСТ 1930—56.
п-Толуолсульфокислота, ч., ТУ МХП 3836—53.
Гидрохинон, ч., ГОСТ 2549—44.
Аммиачная вода, ГОСТ 9—57.
9}
Способ получения
В четырехгорлую колбу, снабженную капельной ворон-
кой, обратным холодильником, мешалкой и термометром,
помещают 564 г (6 моль) фенола, 920 г (10 моль) толуола
и 29,27 г (0,17 моль) n-толуолсульфокислоты (см. приме-
чание 1). При температуре 10—12 °C и работающей ме-
шалке в течение 5 ч равномерно добавляют 212 г (1 моль)
диаллилиденпентаэритрита, растворенного в 920 г (10 моль)
толуола с добавкой 1,1 г (0,01 моль) гидрохинона. Реак-
тив вводят со скоростью 266,2 г смеси в час (см. примеча-
ние 2), после чего перемешивание прекращают и реакцион-
ную смесь оставляют при комнатной температуре на
сутки. Затем смесь нейтрализуют аммиачной водой.
Образовавшийся осадок отфильтровывают. Избыток непро-
реагировавшего фенола отделяют перегонкой с паром.
После двойной перекристаллизации из хлорбензола полу-
чают 280 г продукта; т. пл. 214 °C; выход 68—70% (от
теоретического).
Состав синтезированного продукта (С23Н28О6):
с н он
Найдено, %.................. 69.26 6,9 8,4; 8,5
Вычислено, %.............. 69,0 6,9 8,5
Бис-фенол представляет собой кристаллическое вещество
белого цвета, без запаха; хорошо растворяется в ацетоне,
спирте, тетрагидрофуране, диметилформамиде; не раство-
ряется в ароматических и хлорированных углеводородах.
Примечания. 1. Увеличение количества п-толуолсульфо-
кислоты от 0,17 до 0,34 моль, считая на моль диаллилиденпента-
эритрита, вызывает интенсивное образование побочных продуктов
ал килирования.
2. Приведено оптимальное соотношение реагентов (диаллили-
денпентаэритрит : фенол =1:6). Увеличение этого соотношения
до 1 : 10 или уменьшение до 1 : 2 снижает выход бис-фенола.
Литература
1. Куценко Н. И., Семенова А. И., авт. свид. СССР
184824; Билл, изобр., № 16 (1966).
2. Куценко Н. И., Перцов Л. Д., Калинкин С. Ф.,
С к р и п к о С. Б., Пласт, массы, № 2, 47 (1967).
п, n-ДИТОЛИЛАМИН
Г. Т. ПИЛЮГИН, Л. Е. ЖИВОГЛАЗОВА
СцН1бН
Мол. вес 197,28
п,п'-Дитолиламин является исходным продуктом
в синтезе четвертичных гетероароматических солей хиналь-
диния и лепидиния. В литературе описаны методы полу-
чения п.п'-дитолиламинаП 2. Из них наиболее приемлем
способ \ заключающийся в конденсации солянокислого
n-толуидина с n-толуидином в запаянной ампуле при
230—240° С. Мы проверили условия получения /г,л'-дито-
лиламина по этому методу, отработали и уточнили время
нагревания смеси, что позволило увеличить выход про-
дукта.
Схема синтеза
МН
РЕАКТИВЫ
п-Толуидин, ч., ОРУ 47—56.
Солянокислый п-толуидин, ч.
Соляная кислота, ч., пл. 1,19, ГОСТ 3118—46.
Натр едкий, ч. д. а., ГОСТ 4328—66.
Спирт этиловый, ректификованный, ГОСТ 5962—51
Способ получения
В фарфоровой ступке тщательно растирают 10 г
(0,069 моль) солянокислого n-толуидина и 11,2г
(0,104 моль) /г-толуидина. Полученную смесь вносят в тол-
стостенную стеклянную ампулу и запаивают. Ампулу
выдерживают в трубчатой печи 35—37 ч при 230—240 °C.
Охлажденную ампулу осторожно вскрывают и извлекают
из нее темно-зеленую кристаллическую массу, которую
измельчают в ступке и обрабатывают 30-кратным количе-
9?
ством разбавленной (1 : I) соляной кислоты. При этом
n-толуидин, не вступивший в реакцию, переходит в рас-
твор. Образовавшийся во время реакции хлористый аммо-
ний также растворяется. Оставшийся в осадке солянокис-
лый дитолиламин отфильтровывают и промывают на филь-
тре водой. Для выделения основания осадок переносят
в стакан, обрабатывают 20 %-ным раствором едкого натра,
отфильтровывают и промывают на фильтре большим коли-
чеством воды. Полученный л,и'-дитолиламин перекристал-
лизовывают из смеси вода — спирт (1:2) и выделяют
6,9 г продукта в виде белых игольчатых кристаллов;
т. пл. продукта 78—79 °C, что совпадает с литературными
данными1; выход 53% (от теоретического).
Состав синтезированного продукта (C14H15N):
с н N
Найдено, %........ 85,03; 85,12 7,50; 7,75 6.97
Вычислено, % .... 85,27 7,61 7,12
Литература
1. L a i г е G., Girard С., Chapoteut Р., Ann. pharm.
fr., 140, 346 (1866).
2. Фр. пат. 1240704 (I960).
4,4-ДИФЕНИЛАМИНДИКАРБОНОВАЯ КИСЛОТА
Н. Л. ЗОТОВА, А. А. АРТАМОНОВ, Е. Л. БАБИН
НООС-<^>-NH— СООН
C14HuNO4 Мол. вес 257,24
4,4'-Дифениламиндикарбоновая кислота может исполь-
зоваться в качестве мономера при получении пленок,
волокон, а также в аналитической химии как реагент
для фотометрического определения окислителей Методи-
ка получения кислоты основана на взаимодействии калие-
вых солей амино- и галоидбензойных кислот в присутствии
порошкообразной меди 2.
94
Схема синтеза
НООС—^J/-NH2 + 1-^>-СООН + К2СО3
----» КООС—NH—-^2/-~с0°К---"
НООС—NH— -f^S-COOH
РЕАКТИВЫ
п-Аминобензойная кислота, ч., ТУ 2681—51, т. пл. 185—188 °C.
п-Иодбензойная кислота, получена диазотированием и-амино-
бензойной кислоты и последующей заменой диазогруппы на иод;
т. пл. 269—271 °C.
Медь (порошкообразная), ТУ 4451—54.
Калий углекислый, х.ч. ГОСТ 4221—65.
Способ получения
В двухгорлую колбу емкостью 250 мл, снабженную
механической мешалкой и обратным холодильником, поме-
щают 22,4 г (0,09 моль) n-иодбензойной кислоты, 27,2 г
(0,18 моль) n-аминобензойной кислоты, 48 г (0,33 моль)
карбоната калия, 0,1 г порошка меди и 35—40 мл воды.
Реакцию проводят 8 ч при энергичном перемешивании
и кипячении. После этого содержимое колбы разбавляют
горячей водой и фильтруют. Горячий фильтрат обрабаты-
вают 90 мл 10%-ной соляной кислоты. При этом осаж-
дается нерастворимая в воде 4,4'-дифениламиндикарбо-
новая кислота, а избыток n-аминобензойной кислоты
в виде хлоргидрата остается в растворе. Светло-коричне-
вый осадок отфильтровывают, промывают на фильтре
водой до нейтральной реакции промывных вод и сушат.
Получают 9,92 г продукта; т. пл. (после перекристалли-
зации из диоксана) 327—329 °C, выход 43% (от теорети-
ческого).
4,4'-Дифениламиндикарбоновая кислота представляет
собой светло-коричневый порошок; растворяется при
нагревании в диоксане, спирте, ацетоне; не растворяется
в воде, эфире, хлорбензоле.
Состав синтезированного продукта (Ci4HnNO4):
Найдено, %........................
Вычислено, %......................
сны
65.38 4,49 5.58
65,36 4,31 5,44
95
Кислотное число синтезированного продукта:
Найдено, %.................... 308,4
Вычислено, %..................311,2
Литература
1. Ф р у м и н а Н. С., Труды комиссии по аналитической химии.
АН СССР, т. XI, 1960, стр. 121.
2. Ullman F. и др., Ann., 355, 312 (1907).
4,4-ДИФЕН ИЛОКСИДДИ КАРБОНОВАЯ КИСЛОТА
М. С. БЕГИНИНА, А. А. АРТАМОНОВ, В. П. МАРШТУПА,
Е. П. БАБИН
с14н10о5
НООС-О——соон
Мол. вес 258,23
4,4'-Дифенилоксиддикарбоновая кислота является
сырьем для получения полимерных материалов.
В литературе описаны способы получения 4,4'-дифе-
нилоксиддикарбоновой кислоты из диметилолдифепилок-
сида с выходом 44 % 1 и из диметилдифенилоксида путем
окисления хромовой кислотой и марганцевокислым ка-
лием в среде пиридина 2> 3 (выход кислоты не указан).
Мы уточнили условия окисления диметилдифенилоксида
перманганатом калия и нашли оптимальные соотноше-
ния компонентов.
Схема синтеза
НдС-^у— О-^2у—сна -МП->
-» КООС—О— \~/~С00К ““*
--> HOOC-Q-O-Q-COOH
РЕАКТИВЫ
Калий марганцевокислый, х.ч., ГОСТ 4527—65.
Пиридин чистый, ГОСТ 2747—44.
Кислота соляная, х. ч., ГОСТ 3118—46.
96
Способ получения
В трехгорлую колбу емкостью 1,5 л, снабженную
мешалкой и обратным холодильником, вводят 30 г
(0,15 моль) диметилдифенилоксида, 240 мл пиридина,
700 мл воды и 80 г (третью часть общего количества)
измельченного марганцовокислого калия. Колбу погру-
жают в водяную баню, нагретую до 70 "С, включают
мешалку и постепенно нагревают реакционную массу
до 95—100 °C. После обесцвечивания раствора вводят
остальную часть перманганата калия (160 г) порциями
по 20—30 г. После введения всего количества перманга-
ната калия и обесцвечивания раствора реакционную массу
выдерживают при этой температуре еще 15—20 мин,
охлаждают и фильтруют на воронке Бюхнера. Осадок
двуокиси марганца тщательно промывают 500 мл горя-
чей воды. Фильтрат и промывные воды осторожно под-
кисляют разбавленной (1 : 1) соляной кислотой. Выпав-
шему осадку дают отстояться, отсасывают на воронке
Бюхнера, трижды промывают холодной водой и сушат
при 100—НО °C (см. примечание).
Получают 36,5—37,3 г продукта в виде белого аморф-
ного порошка; выход 93,5—95,5% (от теоретического);
т. пл. 331—332 °C. По литературным данным, т. пл. веще-
ства 32&—329 °C 1 и 329—330 °C4. Вещество раство-
ряется в диметилформамиде, плохо растворяется в мети-
ловом спирте, не растворяется в воде, бензоле, эфире
и ацетоне.
Состав синтезированного продукта (С14Н10О5):
с н
Найдено, %..............65.21; 65,40 3,94; 3,76
Вычислено, %............ 65,13 3,87
Примечание. Пиридин из фильтрата может быть регене-
рирован одним из обычных способов, например упариванием филь-
трата в вакууме, подщелачиванием остатка и отделением пиридина.
Литература
1. Р у д е н к о М. Г., Т у р я н ч и к И. Г., Нефтехимия, 5,
№ 2, 256 (1965).
2. Reilly J., Drumm Р. J., Barrett Н. S., J.Chem.
Soc., 1927, 69.
3. F u i i k a w a F., G u r u g i S., J. Pharm. Soc. Japan, 63,
341 (1943); C.A. 45, 512 (1951).
4. Ma ss ar ar i E., Nardi D., Mauri L., Gavalli-
r i O., Farmaco (Pavia), 18, 582 (1963).
7—287
97
Дихлорангидрид ВИНИЛФОСФОНОВОЙ
кислоты
Е. Л. ГЕФТЕР, И. А. РОГАЧЕВА
СН,
С2Н3С12ОР
Мол. вес 144,92
Дихлорангидрид винилфосфоновой кислоты представ-
ляет собой исходный продукт для получения различных
производных винилфосфоновой кислоты и отвержденных
фосфорсодержащих полиэфиров.
Описаны синтезы дихлорангидрида винилфосфоновой
кислоты дегидрохлорированием дихлорангидрида Р-хлор-
этилфосфоновой кислоты1’2, а также взаимодействием
пятихлористого фосфора с эфирами винилфосфоновой
кислоты 3. Предлагаемый нами метод получения заклю-
чается во взаимодействии пятихлористого фосфора с ди-
Р.Р'-хлорэтиловым эфиром винилфосфоновой кислоты.
Схема синтеза
CuCl или FeClg
СН2=СН—Р(ОСН2СН2С1)2 + РС16--------► СН2=СНРОС12
II
о
РЕАКТИВЫ
Ди-$,$'-хлорэтиловый эфир винилфосфоновой кислоты (получе-
ние см. на стр. 108).
Пятихлористый фосфор, ч., МРТУ 6-09-604—63.
Способ получения
Прибор для синтеза представляет собой четырехгорлую
круглодонную колбу, снабженную термометром, мешалкой
с глухим затвором (см. стр. 109), позволяющим проводить
реакцию в вакууме, и нисходящим холодильником, соеди-
ненным с приемником. В колбу помещают 1500 г (1 моль)
неперегнанного ди-Р,Р'-хлорэтилового эфира винилфос-
фоновой кислоты и 15 г однохлористой меди (см. приме-
чание 1). Смесь нагревают до 130—140 °C и при интен-
98
сивном перемешивании (см. примечание 2) прибавляют
довольно быстро порциями по 50—70 г 2800—2900 г (2,08—
2,16 моль) пятихлористого фосфора (см. примечание 3).
Во время быстро протекающей реакции отгоняются обра-
зующиеся дихлорэтан и хлорокись фосфора. Оставшийся
в колбе продукт реакции перегоняют в вакууме, собирая
фракцию с т. кип. 30—95 °C при 20 мм рт. ст. Получен-
ный в этой реакции продукт перегоняют и выделяют около
850 г дихлорангидрида винилфосфоновой кислоты; т. кип.
68—70 °C при 20 мм рт. ст., выход 70% (от теоретиче-
ского); п2£ = 1,4800, df = 1,4083. Дихлорангидрид ви-
нилфосфоновой кислоты — бесцветная прозрачная жид-
кость; растворяется в бензоле, толуоле, эфире, диоксане.
По литературным данным \ т. кип. вещества 67—69 °C
при 21 мм рт. ст.; п2° = 1,4808, d20 = 1,4092.
Содержание хлора в синтезированном продукте
(С2Н3С12ОР):
Найдено, %....................48,6
Вычислено, %..................48,9
Примечания. 1. В отсутствие катализатора выход про-
дукта резко падает.
2. Реакция вначале идет с выделением тепла; при температуре
реакционной смеси выше 150° С и плохом перемешивании выход
вещества значительно уменьшается.
3. Пятихлористый фосфор должен быть хорошего качества.
Увлажнившийся продукт применять нельзя.
Литература
1. Кабачник М. И., Медведь Т. Я., Изв. АН СССР,
ОХН, 1959, 2142.
2. Рубцова И. К., Гефтер Е. Л., Юлдашев А.,
Мошкин П. А., Пласт, массы, № 2, 22 (1961).
3. Пат. ФРГ 1020019, 1023034; РЖХим, 39595 (1959); 39631 (1960).
ДИХЛОРАНГИДРИД ФЕНИЛФОСФОНОВОЙ
кислоты
Е. Л. ГЕФТЕР
с6н5-р/С'
IIХС1
о
СаН6С12ОР Мол. вес 194,98
7*
99
Дихлорангидрид фенилфосфоновой кислоты является
исходным продуктом для получения фенилфосфина, произ-
водных фенилфосфоновой кислоты и третичных фосфинов,
содержащих фенильный радикал.
Дихлорангидрид фенилфосфоновой кислоты получают
обработкой фенилдихлорфосфина пятиокисью фосфора
и хлором \ хлористым сульфурилом 2, обработкой фенил-
фосфоновой кислоты пятихлористым фосфором 3, дейст-
вием сернистого газа на фенилтетрахлорфосфин или частич-
ным гидролизом фенилтетрахлорфосфина3 и, наконец,
прямым окислением фенилдихлорфосфина 3.
Мы получали дихлорангидрид фенилфосфоновой кис-
лоты взаимодействием фенилдихлорфосфина с хлором
с последующей обработкой образующегося при этом
фенилтетрахлорфосфина сернистым газом 4.
Схема синтеза
Cl2 so2
С6Н5РС12:--> С6Н6РС14-> СвН5РОС12
РЕАКТИВЫ
Фенилдихлорфосфин 4, ч.
Хлор (в баллоне), ГОСТ 6718—53.
Сернистый газ (в баллоне), ГОСТ 2918—45.
Углерод четыреххлористый, ГОСТ 5827—51.
Способ получения
Реакцию следует проводить в хоро-
шо действующем вытяжном шкафу.
Прибор для синтеза дихлорангидрида фенилфосфоновой
кислоты состоит из четырехгорлой круглодонной колбы
емкостью 4 л, снабженной термометром, мешалкой, нис-
ходящим холодильником и тройником. Через боковой
отвод тройника подается газ, а в верхнее отверстие встав-
ляется с помощью вакуумного каучука, смазанного гли-
церином, стеклянная палочка, предназначенная для
очистки вертикальной части тройника от образующихся
кристаллов фенилтетрахлорфосфина. Перед реакционной
колбой ставят предохранительную склянку (см. приме-
чание 1). В колбу помещают 1720 г фенилдихлорфосфина
и 1400 мл четыреххлористого углерода. Трубку для
подачи газа устанавливают над поверхностью жидкости
100
(см. примечание 2) и в колбу при хорошем перемешива-
нии смеси подают в течение 6—8 ч сухой хлор. Колбу
охлаждают в бане со льдом и солью, поддерживая темпе-
ратуру реакционной смеси в пределах 10—20 °C (более
высокая температура влечет за собой уменьшение выхода
продукта). Образующийся фенилтетрахлорфосфин выпа-
дает в виде желтовато-белых кристаллов. Вертикальную
часть тройника периодически прочищают палочкой. После
прекращения реакции, на что указывает резкое падение
температуры реакционной смеси, хлор пропускают еще
5—10 мин. Затем трубку для подачи газа погружают
в полужидкую массу и в систему при перемешивании
и охлаждении подают при 5—15° С сухой сернистый газ
до полного исчезновения кристаллов и превращения
реакционной смеси в гомогенную желтую жидкость. При
температуре бани 40—100 °C на водоструйном насосе
отгоняют легколетучую фракцию, состоящую из серни-
стого газа, хлористого тионила, хлористого сульфурила
и четыреххлористого углерода, а остаток перегоняют
в вакууме. Получают 1620—1650 г продукта; т. кип. 83—
84 °C при 1 мм рт. ст.; выход88—90% (оттеоретического),
= 1,5578, = 1,1977. По литературным данным,1
т. кип. продукта 137—138 °C при 15 мм рт. ст., п2£ =
= 1,5581, d25 = 1,197. Вещество представляет собой бес-
цветную прозрачную жидкость, дымящую на воздухе;
растворяется в некоторых органических растворителях,
энергично реагирует с водой, спиртами, аминами.
Примечания. 1. Следует ставить трехгорлую склянку,
через которую газ проходит в колбу. Одно горло склянки закрывает-
ся (не туго) притертой или резиновой пробкой. В случае забивания
тройника это горло является предохранительным клапаном.
2. Погружение тройника в жидкость вызывает немедленное
забивание трубки кристаллами.
Литература
l. Toy A. D. F., J. Am. Chem. Soc., 70, 186 (1948).
2. П е т р о в К. А. и др., авт. свид. СССР 143799; Билл, изобр.,
№ 1 (1962).
3. Kosolapoff G. М., Organophosphorus Compounds.,
New York (1950).
4. Г e ф т e p E. Л., ЖОХ, 28, 1338 (1958).
ДИХЛОРАНГИДРИД р-ХЛОРЭТИЛФОСФОНОВОЙ
кислоты
Е. Л. ГЕФТЕР, М. И. КАБАЧНИК, Л. С. ЖУРАВЛЕВА
С1СН2СН2-
о
С2Н4С13ОР
Мол. вес 181,38
Дихлорангидрид |3-хлорэтилфосфоновой кислоты яв-
ляется исходным веществом для получения различных
производных p-хлорэтилфосфоновой кислоты, а также
хлорангидрида винилфосфоновой кислоты. В литературе
описаны методы синтеза дихлорангидрида Р-хлорэтилфос-
фоновой кислоты, основанного на взаимодействии ди-
р,Р'-хлорэтилового эфира этой кислоты с пятихлори-
стым фосфором под давлением \ на окислительном фос-
финировании этилена 2’ а, а также на реакции треххло-
ристого фосфора с дихлорэтаном и хлористым алюми-
нием 4. Мы разработали два метода получения этого ди-
хлорангидрида, исходя из доступного ди-Р,р'-хлорэтило-
вого эфира p-хлорэтилфосфоновой кислоты5: 1) непо-
средственной обработкой этого вещества пятихлористым
фосфором без давления (А); 2) через промежуточное обра-
зование свободной кислоты (Б). Оба метода отличаются
простотой, безопасностью выполнения и высокими выхо-
дами.
Схема синтеза
CuCl или FeCIg
А. С1СН2СН2РО(ОСН2СН2С1)2 -I- РС15---------С1СН2СН2РОС12
HCI РС16
Б. С1СН2СН2РО(ОСН2СН2С1)2----. С1СН2СН2РО(ОН)2------
----> С1СН2СН2РОС13
РЕАКТИВЫ
Ди-$,Р'-хлорэтиловый эфир ft-хлорэтилфосфоцовой кислоты
(ДЭХК), ч. (получение см. на стр. ИЗ).
Фосфор пятихлористый, ч., МРТУ 6-09-604—63.
Способ получения
А. Прибор для синтеза — это четырехгорлая кругло-
донная колба, снабженная термометром, мешалкой с глу-
102
хим затвором *, позволяющим проводить реакцию в ва-
кууме, и нисходящим холодильником, соединенным с при-
емником. В колбу помещают 68 г ДЭХК и 0,9 г безвод-
ного хлорного железа или однохлористой меди (см. при-
мечание 1). Смесь нагревают до 140—150 °C и при энер-
гичном размешивании (см. примечание 2) прибавляют
ПО г пятихлористого фосфора порциями по 10—15 г.
При этом отгоняются образующиеся дихлорэтан и хлор-
окись фосфора. Оставшийся продукт реакции перего-
няют в вакууме, собирая фракцию с т. кип. 50—95 °C.
(3—5 мм рт. ст.). Эту фракцию перегоняют еще раз и выде-
ляют 35—36,5 г продукта; т. кип. 80—82°C при 4 мм рт. ст.,
= 1,4992; dl° = 1,5440, выход 70—80% от теорети-
ческого (см. примечание 3). Вещество представляет собой
бесцветную прозрачную жидкость; растворяется в бен-
золе, толуоле, эфире, диоксане. По литературным дан-
ным \ т. кип. продукта 68 °C при 2 мм рт. ст., =
= 1,4977, d>5 = 1,5430.
Б. Прибор такой же, как и в предыдущей методике.
В колбу помещают 135 г ДЭХК, нагревают до 150—160 °C
и при энергичном перемешивании пропускают избыток
сухого хлористого водорода (около 100 г) до прекращения
отгонки выделяющегося дихлорэтана. Оставшуюся в колбе
густую жидкость (^-хлорэтилфосфоновую кислоту) охлаж-
дают до комнатной температуры и к ней прибавляют не-
большими порциями 209 г пятихлористого фосфора (см.
примечания 4). При этом смесь разогревается до 50—
60 °C и наблюдается обильное выделение хлористого
водорода. Образовавшуюся хлорокись фосфора отгоняют,
нагревая баню до 120 °C. Остаток дважды перегоняют
в вакууме, как указано в предыдущей методике.
Получают 68—75 г продукта; выход 75—82% (от
теоретического). Константы вещества практически такие
же, как приведенные выше.
Примечания. 1. В отсутствие катализатора выход про-
дукта резко уменьшается.
2. Реакции вначале идет с выделением тепла. При температуре
реакционной смеси выше 160 °C и плохом перемешивании выход
продукта значительно уменьшается.
3. При использовании технического (т. е. неперегнанного)
ДЭХК выход дихлорангидрида (1-хлорэтилф)сфоиовой кислоты
составляет 65—70%. При увеличении количеств исходных веществ
в 10—15 раз выход вещества не уменьшается.
* Описание затвора приведено на стр. 109.
103
4. При отсутствии пятихлористого фосфора можно прибавить
в колбу 138 г треххлористого фосфора и медленно пропускать сухой
хлор. Добавление большого количества пятихлористого фосфора
влечет за собой сильное вспенивание и возможный выброс реакцион-
ной массы.
Литература
1. Кабачник М. И., Российская П. А., Изв, АН
СССР, ОХН, 1946, 515.
2. Соборовский Л. 3., Зиновьев Ю. М., Энг-
лин М. А., ДАН СССР, 67, 293 (1949).
3. V i 1 с s е k И., Rochlitz F., пат. ФРГ 1103922; С.А.,
56, 1482 (1962); Rochlitz F., Vilcsek И., Angew.
Chem., 74, 970 (1962).
4. К i n n е а г А. М., Р е г г е n Е. A., J. Chem. Soc., 1952,
3437
5, Г е ф т е р Е. Л., Кабачник М. И., Пласт, массы,
№ 1, 63 (1961).
ДИХЛОРИД г.З-ДИФЕНИЛ-б-Г^-Сн-БРОМФЕНИЛ)-
б'-БРОМХИНОЛ ИЛ-2']-ТЕТРАЗОЛИЯ
Г. Т. ПИЛЮГИН, В. В. СТАШКЕВИЧ, С. В. ШИНКОРЕНКО,
О. М. СТАШКЕВИЧ
CagHigBrgC 13N 5
Мол. вес 656,21
В данном сообщении приводится методика синтеза
неописанной в литературе тетразолиевой соли, которая
может найти применение в гистохимии как индикатор при
изучении окислительно-восстановительных процессов.
Предварительно были получены исходная четвертичная
соль и формазан. Исходная четвертичная соль — хлорид
1-(и-бромфенил)-6-бромхииальдиния — образуется в про-
цессе циклизации 4,4'-дибромдифениламина по реакции
Дебнера — Миллера, разработанной для случая вто-
10.4
ричных ароматических аминов *. Для синтеза формазана
использовано свойство четвертичной соли вступать по
реакционноспособной метильной группе в реакцию азо-
сочетания с солями диазония 2. При взаимодействии
1 моль четвертичной соли с 2 моль хлористого фенилдиа-
зония в щелочной среде образуется хлорид формазана
в виде темно-красных красивых кристаллов, плохо рас-
творимых в воде, но хорошо растворимых в большинстве
органических растворителей. Конечный продукт — соль
тетразолия — получается окислением формазана азотной
кислотой.
Схема синтеза
СН2=СН—О—С4Н9; HCI
РЕАКТИВЫ
4,4'-Дибромдифениламин, т. пл. 106—107 °C (получение см.
на стр. 70).
105
Эфир винилбутиловый3, т. кип. 93—93 7 °C.
Диоксан, ч., ВТУ МХП 3111—52.
Кислота соляная, х. ч., пл. 1,19, ГОСТ 3118—46.
Анилин, ч., ГОСТ 5819—51.
Натрий азотистокислый, ч. д. а., ГОСТ 4197—66.
Спирт этиловый, ректификованный, ГОСТ 5962—51.
Кислота азотная, ч. д. а., ГОСТ 4461—48.
Кали едкое, ч. д. а., ГОСТ 4203—65.
Способ получения
ррге'Г' : - -
Г' . '
Синтез хлорида 1-(п-бромфенил)-6-бромхинальдиния.
В ампулу вносят 15 г (0,06 моль) 4,4'-дибромдифениламина,
15 мл соляной кислоты, 15 мл винилбутилового эфира
и 30 мл диоксана. Ампулу запаивают и после тщатель-
ного взбалтывания нагревают на кипящей водяной бане
16—18 ч. Затем летучие компоненты отгоняют с водяным
паром. Массу фильтруют, фильтрат кипятят с неболь-
шим количеством активированного угля и снова филь-
труют. При охлаждении фильтрата до 0 °C выпадают
желтоватые кристаллы четвертичной соли. После пере-
кристаллизации из воды с добавкой активированного
угля получают 5,3—6,3 г продукта; т. пл. 262—263 °C,
выход 28—33% (от теоретического). ,
Содержание азота, брома и хлора в препарате
(C16HlaBr2ClN):
N Br+ С1
Найдено, %................3.32; 3,38 47,52; 47,65
Вычислено, %.............. 3,38 47,3
Синтез хлорида 1,5-дифенил-3-[Г-(п-бромфенил)-б'
бромхинолил-2'\-формазана. Растворяют 13,8 г (0,05 моль)
четвертичной соли в 150 мл этилового спирта и охлаж-
дают раствор до минус 5—10 °C. Отдельно готовят рас-
твор хлорида фенилдиазония. Для этого 5,6 г анилина
энергично перемешивают с 15 мл соляной кислоты и 30 мл
воды, охлаждают до 0 °C и диазотируют раствором 4,1 г
азотистокислого натрия в 20 мл воды. Растворы четвер-
тичной соли и соли диазония смешивают, охлаждают
до минус 5—10 °C и к этой смеси при энергичном пере-
мешивании прибавляют по каплям 10%-ный спиртовый
раствор едкого кали до начала реакции азосочетания,
что обнаруживается по появлению интенсивно красной
окраски. Смесь выдерживают 5 ч в ледяной бане и 12 ч
при комнатной температуре, после чего добавляют 200 мл
106
воды и выпавший осадок формазана фильтруют. После
перекристаллизации из этилового спирта получают 10 г
продукта в виде темно-красных кристаллов; т. пл. 195—
196 °C, выход 47,7% (от теоретического).
Содержание азота в синтезированном продукте
(Ca8HaoBraClN6):
Найдено, %...........11,18; 11,22
Вычислено, %........ 11,26
Получение дихлорида 2,3-дифенил-5-[Г-(п-бромфенил)-
6'-бромхинолил-2']-тетразолия. Смешивают 7,9 г
(0,012 моль) формазана с 300 мл азотной кислоты и нагре-
вают до кипения. По мере кипячения начинают выде-
ляться окислы азота, а раствор, вначале коричневый,
постепенно светлеет. Добавление 1—2 капель этилового
спирта значительно ускоряет реакцию окисления фор-
мазана. Полученный желтый прозрачный раствор охлаж-
дают, фильтруют и добавляют 100 мл воды и 20 мл соля-
ной кислоты. Через некоторое время выпадает соль тетра-
золия в виде желтых хлопьев. После перекристаллизации
из этилового спирта получают 5,47 г продукта в виде
бледно-желтого мелкокристаллического порошка; т. пл.
202—203 °C, выход 56,6% (от теоретического).
Содержание азота в синтезированном продукте
(C28H19Br2Cl2N5):
Найдено, %..............10,91; 10,85
Вычислено, % ... . 10,67
Литература
1. Пилюгин Г. Т„ Опанасенко Е. П., ЖОХ 27,
1015 (1957).
2. Пилюгин Г. Т., Шинкоренко С. В., ЖОХ, 28,
1313 (1958).
3. Шостаковский М. Ф., Простые виниловые эфиры, Изд.
АН СССР, 1952, стр. 33.
ДИ-3, З'-ХЛОРЭТИЛОВЫЙ ЭФИР
ВИНИЛФОСФОНОВОЙ кислоты
Е. Л. ГЕФТЕР, П. А. МОШКИН, Л. С. ЖУРАВЛЕВА
СН2.
•ОСН2СН2С1
|| чЭСН2СН2С1
о
Мол. вес 233,03
Ди-3,Р'-хлорэтиловый эфир винилфосфоновой кислоты
является перспективным мономером, способным к полиме-
ризации, сополимеризации, гомополиконденсации. Опи-
сано его применение в качестве компонента пластических
масс для придания им огнестойкости, в качестве добавки,
улучшающей свойства смазочных масел, в качестве стаби-
лизатора полимеров и пр.1-2. Описан синтез ди-р,р'-
хлорэтилового эфира винилфосфоновой кислоты дегидро-
хлорированием соответствующего эфира Р-хлорэтилфос-
фоновой кислоты спиртовым раствором щелочи 3 и три-
этиламииом 4. Ниже описан простой лабораторный метод
получения эфира, исходя из треххлористого фосфора
и окиси этилена без выделения промежуточных продук-
тов s.
Схема синтеза
термическая
изомеризация
РС13 + ЗСН2-СН2----. Р(ОСН2СН2С1)3----------»
ОСН2СН 2С1 CH3COOK(Na)
---> С1СН,СН2—р<---------------------->-
|| ХОСН2СН,С1
о
ZOCH2CH2C1
---> СН2=СН—Р<
II ОСН2СН,С1
о
Хлористый водород, всегда присутствующий в небольшом
количестве в треххлористом фосфоре, образует с окисью
этилена этиленхлоргидрип — побочный продукт этого
процесса.
108
РЕАКТИВЫ
Фосфор треххлористый, ч., ГОСТ 91—41.
Окись этилена, ГОСТ 7568—64.
Калий уксуснокислый, безводный, ГОСТ 5820—51.
Натрий уксуснокислый, безводный, МРТУ 6-09-2121—65.
Способ получения
Прибор для получения ди-р,р'-хлорэтилового эфира
винилфосфоновой кислоты состоит из круглодонной четы-
рехгорлой колбы (см. примечание 1), снабженной мешал-
кой с глухим затвором*, термометром, обратным холодиль-
ником с хлор кальциевой трубкой и барботером. В колбу
помещают 302 г (1 моль) треххлористого фосфора и при
сильном перемешивании пропускают около 320 г (3,3 моль)
окиси этилена (см. примечание 2), которая предвари-
тельно проходит через две колонки с твердой щелочью
для осушения (см. примечание 3) и достаточно большую
пустую промежуточную склянку (см. примечание 4).
Наружным охлаждением (минус 20—40 °C) и регулирова-
нием скорости подачи окиси этилена поддерживают тем-
пературу реакции в пределах 10—20 °C (см. примеча-
ние 5). Незадолго до прекращения подачи окиси этилена
температура реакционной смеси резко падает, что указы-
вает на завершение реакции. Затем удаляют барботер,
обратный холодильник заменяют на нисходящий, соеди-
ненный с приемником, и в колбу вводят около 600 г о-
или л-дихлорбензола или их смеси (см. примечание 6).
Из реакционной смеси отгоняют при перемешивании
избыток окиси этилена и образовавшийся этиленхлор-
гидрин до тех пор, пока температура жидкости не достиг-
нет 170 °C. Затем прямой холодильник заменяют на обрат-
ный, смесь кипятят при 170—175 °C и перемешивании
(см. примечание 7) 3—3,5 ч, после чего охлаждают до
80—90 °C и добавляют в колбу 225 г уксуснокислого калия
(или 189 г уксуснокислого натрия). Реакционную массу
снова перемешивают, выдерживают при 120—125 °C в те-
* Затвор мешалки состоит из стеклянной трубки, вставленной
в пробку, которая входит в горло колбы. На верхнюю часть стек-
лянной трубки надевается резиновая трубка, охватывающая также
и стержень мешалки. Свободное вращение мешалки и одновременно
герметичность затвора достигались смазыванием верхнего края
резиновой трубки каплей глицерина.
109
чение 3—3,5 ч, охлаждают до комнатной температуры
и фильтруют. Осадок растворяют в 10—15%-ном растворе
карбоната натрия. Образовавшийся органический слой
объединяют с фильтратом. Объединенный фильтрат нагре-
вают до 100 °C при 5—10 мм рт. ст. При этом отгоняется
уксусная кислота и основное количество дихлорбензола.
Остаток нейтрализуют 10—15%-ным раствором карбо-
нате натрия, органический слой отделяют и перегоняют,
собирая фракцию с т. кип. 100—150 °C при 2—3 мм рт. ст.
(см. примечание 8). После второй перегонки получают
250—265 г продукта; выход 49—52% (от теоретического),
считая на треххлористый фосфор; т. кип. 120—
122 °C при 2 мм рт. ст.\ п2° = 1,4?85, d%° = 1, 3205
По литературным данным4, т. кип. 131—132 °C при
3 мм рт. ст., = 1,4787, df = 1,3233.
Вещество представляет собой бесцветную прозрачную
жидкость, растворяется в бензоле, толуоле, эфире, диок-
сане, не растворяется в воде.
Примечания. 1. При проведении реакции в укрупненном
масштабе мы применяли 16-литровый цилиндрический реактор из
нержавеющей стали марки 1Х18Н9Т.
2. Треххлористый фосфор ядовит, разъедает кожу и слизистые
оболочки. На воздухе дымит, при этом выделяется хлористый водо^
род. Хлорокись фосфора, пятихлористый фосфор и хлорангидриды
кислот трехвалентного фосфора имеют аналогичные свойства. Рабо-
тать с этими соединениями необходимо в резиновых перчатках.
Окись этилена легко воспламеняется и образует с воздухом взрыв-
чатые смеси. Реакцию следует проводить в вытяжном шкафу при
отсутствии источников огня (включенные плитки, зажженные
горелки и пр.) Процесс экзотермичен; его следует вести осторожно,
имея наготове большое количество хладоагента: смеси сухого льда
и ацетона или сухого льда и четыреххлористого углерода.
3. По мере увлажнения щелочи в колонках ее необходимо заме-
нять свежей для предупреждения спонтанной полимеризации окиси
этилена.
4. При неравномерной подаче окиси этилена возможно заса-
сывание жидкости из реакционной колбы через барботер в колбу
с окисью этилена. Это может вызвать серьезную аварию вследствие
выделения большого количества тепла.
5. При температуре выше 30—35 °C реакция может стать
«неуправляемой».
6. Отсутствие растворителя может привести к взрыву. Другие
возможные растворители указаны в литературе2.
7. В случае прекращения работы мешалки необходимо немедлен-
но прекратить нагревание и перемешивать вручную, пока температу-
ра не снизится до 120—130 °C.
8. При таком давлении растворитель и вода отгоняются при
невысокой температуре, что препятствует гидролизу эфира.
110
Литература
1. Гефтер Е. Л., Мошкин П. А., Пласт, массы, № 4, 54
(1960).
2. Г е ф т е р Е. Л., Кабачник М. И., Усп. хим., 31, 285
(1962).
3. К а б а ч н и к М. И., Изв. АН СССР, ОХН, 1947, 233.
4. Гефтер Е. Л., ЖОХ, 28, 2500 (1958).
5. Leu р old Е. Zorn Н., Пат. ФРГ 1006414; Zbl, 1958,
7895.
ди-р, р'-ХЛОРЭТИЛОВЫЙ ЭФИР
ФЕНИЛФОСФОНИСТОЙ кислоты
Е. Л. ГЕФТЕР, И. А. РОГАЧЕВА
ZOCH.,CH,C1
СвН5Р<
ОСН2СН2С1
С18Н13С12О2Р Мол. вес 267,10
Ди-р,Р'-хлорэтиловый эфир фенилфосфонистой кис-
лоты сравнительно легко превращается в соответствую-
щие эфиры кислот пятивалентного фосфора при присоеди-
нении серы или кислорода, при взаимодействии с галоид-
ными алкилами, галоидоводородами и при действии повы-
шенных температур; переэтерифицируется спиртами и фе-
нолами г. Мы разработали метод получения указанного
эфира взаимодействием фенилдихлорфосфина с окисью
этилена с выходом 90—95%.
Схема синтеза
C6HSPC12 + 2СН2-СНг-----» С6Н6Р(ОСН2СН2С1)2
О
РЕАКТИВЫ
Фенилдихлорфосфин 2.
Окись этилена, ГОСТ 7568—64.
Бензол, ч., ГОСТ 8448—61.
Способ получения
В двухлитровую колбу, снабженную мешалкой, термо-
метром, длинной трубкой для ввода окиси этилена и обрат-
111
ным холодильником с хлоркальциевой трубкой, поме-
щают 200 г (1 моль) фенилдихлорфосфина и 1000 мл сухого
бензола. Перед началом реакции воздух в системе вытес-
няют сухим азотом. Реакцию проводят в атмосфере
азота. В интенсивно перемешиваемый бензольный раствор
фенилдихлорфосфина пропускают 295 г (6 моль) окиси'
этилена, высушенной предварительным пропусканием
через колонку с твердой щелочью. Реакция экзотерми-
ческая (см. примечание 1),. причем сначала наблюдается
индукционный период (от 20 до 40 мин), во время которого
температура смеси поднимается сравнительно медленно.
Затем происходит быстрый подъем, иногда до температуры
слабого кипения растворителя (см. примечание 2), после
чего температура поднимается равномерно. Температуру
реакции в пределах 30—45 °C поддерживают, регулируя
скорость подачи окиси этилена и охлаждая смесь в бане
с ледяной водой. Более высокая температура может
привести к снижению выхода продукта. В ходе реакции
раствор слегка мутнеет. Подачу окиси этилена закан-
чивают через 30 мин после прекращения экзотермиче-
ского процесса. За это время температура смеси снижается
до комнатной при прежнем охлаждении. Избыток окиси
этилена и бензол отгоняют вначале при атмосфер-
ном давлении, а затем при 30—10 мм рт. ст. и 40—50 °C.
Получают 280 г светло-желтого практически чистого
продукта, выход 94% (от теоретического). Из 5 г этого
эфира выделяют перегонкой в вакууме (см. примечание 3)
4,2 г совершенно чистого продукта; т. кип. 138—140 °C
при 3 мм рт . стл, выход 84% (от теоретического), п® =
= 1,5420, dl° = 1,2620. Найдено MRO = 66,74; вычис-
лено — 66,61. Аналогично получены соответст-
вующие эфиры других арилфосфинистых станты которых приведены ниже: кислот, кон- d2° 4
Ди-Р.р'-хлорэтиловые эфиры кислот Брутто- формула Т. кип. (°C) при давлении (Жм pin. ст.)
Анизи лфосфочистой* Ci 1^1 rftCUP СМ.** 1.5'62 1,2996
Хлорфенилфосфоиистой* С10Н12О2С13Р 158—165; 0,3-0.5 1,5534 1,3600
Толилфосфотис гой* СцН15О2С].2Р см.** 1,534 1.2311
Этилфенилфосфо. [истой* С12Н17О2С12Р см.** 1,5366 1,2203
♦Получены в основном н-нзомеры
♦♦Вещество частично изомеризуется в ходе перегонки при давлении 0,5—I мм
рт, ст.
11?
Примечания. 1. Взаимодействие окиси этилена с толил-
дихлорфосфином протекает гораздо более энергично, а с хлорфенил-
дихлорфосфином менее энергично, чем с фенилдихлорфосфином.
2. Для более плавного выхода на постоянный режим следует
в начале процесса пропускать окись этилена медленно и поддержи-
вать наружным охлаждением температуру реакционной смеси
25—30 °C.
3. Перегонку необходимо вести с предохранительным щитом
из органического стекла. При перегонке больших количеств возмож-
на изомеризация эфира, которая протекает чрезвычайно бурно.
Литература
1. Г е ф т е р Е. Л., Рогачева И. А., ЖОХ, 31, 949, 952
(1961); 32, 964, 3962 (1962); 33, 1177, 3548 (1963); 34, 88, 92 (1964).
2. Г е ф т е р Е. Л., ЖОХ, 28, 1338 (1958).
ДИ-0, р'-ХЛОРЭТИЛОВЫЙ ЭФИР
р ХЛОРЭТИЛФОСФОНОВОЙ кислоты
Е. Л. ГЕФТЕР, Л С. ЖУРАВЛЕВА
/ОСН,СН2С1
С1СНгСН2—Р<
|| ОСН2СН2С1
О
С6Н12С13О,Р Мол. вес 269,49
Ди-р,р'-хлорэтиловый эфир р-хлорэтилфосфоновой
кислоты является исходным веществом при получении
дихлорангидрида этой кислоты, а также ди-р,р'-хлор-
этилового эфира винилфосфоновой кислоты (мономера
для изготовления огнестойких полимерных материа-
лов). Впервые ди-р,р'-хлорэтиловый эфир р-хлорэтил-
фосфоновой кислоты был синтезирован в небольших
количествах термической изомеризацией три-р,р',Р"-хлор-
этилфосфита х. Метод опасен, особенно в укрупненном мас-
штабе, т. к. не исключена возможность взрыва реакцион-
ной смеси. Мы разработали способ 2, позволяющий спо-
койно получать большие количества этого эфира.
Схема синтеза
Р(ОСН2СНаС1)8---> С1СНаСН2РО(ОСН2СН3С1)9
8—287
113
РЕАКТИВЫ
Tpu-fi,^' ,^”-хлорэтилфосфит 3, получают и затем перемешивают
30 мин при 80—100 °C и 10—20 мм рт. ст.
Кумол, ч., МРТУ 6-09-2402-65.
Способ получения
Внимание! Во избежание взрыва реакция проводится
обязательно в присутствии растворителя и при интенсивном
перемешивании.
В бачок емкостью 2 л из нержавеющей стали (см. при-
мечание 1), снабженный мешалкой, термометром и обрат-
ным холодильником с хлоркальциевой трубкой, поме-
щают 910 а неперегнанного три-р,р',р"-хлорэтилфосфита
и 300—350 г (25—30% от общего веса смеси) кумола
(см. примечание 2). Смесь кипятят 12 ч при энергичном
перемешивании. Температура кипения смеси постепенно
понижается от 158—160 °C до 149—150 °C. Из полученной
жидкости желтого цвета отгоняют растворитель при
50—100 °C и 20—40 мм рт. ст., г остаток (850—870 а)
перегоняют на масляной бане при 1—4 мм рт. ст., при
этом вместо холодильника ставят форштос. Собирают
фракцию, начиная с т. кип. 130 °C при 1 мм рт. ст.
(или 145 °C при 4 мм рт. ст.).
Получают 610—620 а густой бесцветной жидкости
(см. примечание 3). После повторной вакуумной перегон-
ки получают 590—610 а продукта; выход 65—67% (от тео-
ретического). Вещество представляет собой бесцветную
прозрачную густую жидкость, постепенно кристаллизую-
щуюся при стоянии; т. кип. 161—163 °C при 4 мм рт. ст.,
п® = 1,4844, dl° = 1,3910.
По литературным данным х, т. кип. 167—171 °C при
4 мм рт. ст., ri^ = 1,4828, = 1,3892. Вещество
постепенно кристаллизуется при стоянии; растворяется
в ряде органических растворителей, не растворяется
в воде.
Примечания. 1. Реакцию можно вести также в колбе
соответствующих размеров.
2. Можно применять и другие растворители 2> 4.
3. В колбе остается 220—250 г сиропообразной жидкости желто-
го цвета.
Литература
1. Кабачник М. И., Российская П. А., Изв. АН
СССР, ОХН, 1946, 515.
114
2. Гефтер Е. Л., ЖОХ, 28, 1908 (1958).
3. Кабачник М. И., Российская П. А., Изв. АЙ
СССР, ОХН, 1946, 295.
4. Г е ф т е р Е. Л., М о ш к и и П. А., Пласт, массы, № 4
(1960).
ди-р, Р'-ХЛОРЭТИЛФОСФОРИСТАЯ КИСЛОТА
Е. Л. ГЕФТЕР, М. И. КАБАЧНИК
.ОСН„СН„С1
НР<
II ХОСН2СН2С1
о
C^HgClgOgP
Мол. вес 206,99
Ди-р,р'-хлорэтилфосфористая кислота является исход-
ным веществом при получении производных ди-р,-р'-
хлорэтилфосфорной и а-оксиалкилфосфоновых кислот,
а также полиэфиров. Ди-р,р'-хлорэтилфосфористая кис-
лота получена взаимодействием треххлористого фосфора
с этиленхлоргидрином 2, а также взаимодействием фос-
фористой кислоты с три-р,р',Р"-хлорэтилфосфитом 3 или
с этиленхлоргидрином4. Мы использовали первый из
указанных методов.
Схема синтеза
РС13 + ЗС1СН2СН2ОН-> НРО(ОСН2СН2С1)2+ С1СН2СН2С1 4- 2НС1
РЕАКТИВЫ
Фосфор треххлористый, ч., свежеперегнанный, ГОСТ 91—41.
Этиленхлоргидрин, СТ У 12-10222—62.
Способ получения
В круглодонную четырехгорлую колбу, снабженную
капельной воронкой, обратным холодильником с хлор-
кальциевыми трубками, мешалкой и термометром, поме-
щают 121 г этиленхлоргидрина. В колбу прибавляют
по каплям при перемешивании и температуре 5—10 °C
в течение 2 ч 69 г треххлористого фосфора (мольное соот-
ношение РС13 : С1С2Н4ОН = 1:3). Затем смесь пере-
мешивают при комнатной температуре еще час, отгоняют
8*
115
при давлении 15—30 мм рт. ст. легколетучую фракцию
и медленно поднимают температуру бани до 100 °C. Оста-
ток перегоняют в вакууме. Получают 76—80 г продукта,
т. кип. 119—120 °C при 3,5 мм. рт. ст.; выход 73—77%
(от теоретического), и® = 1,4708, = 1,4025. По лите-
ратурным данным2, т. кип. 129 °C при 0,6 мм рт. ст.
Вещество представляет собой бесцветную прозрачную
жидкость с неприятным запахом, не растворяется в воде,
растворяется в ряде органических растворителей.
Состав синтезированного продукта (С4Н9С12О3Р):
с н Р
Найдено, %.......................... 23,16 4.44 14.82
Вычислено, %........................ 23,20 4,38 14,98
Литература
1. Г е ф т е р Е. Л., Кабачник М. И., Изв. АН СССР,
ОХН, 1957, 194.
2. Cook Н. G., Saunderes В. С., Smith F. Е., J.
Chem. Soc., 1949, 635.
3. Walsh Е. N„ J. Am. Chem. Soc., 81, 3023 (1959).
4. П e т p о в К- А., Нифантьев Э. Е., Г о л ь ц о-
ва Р. Г., Белавенцев М. А., Корнеев С. М.,
ЖОХ, 32, 1277 (1932).
ДИЭТИЛАМИНОАЦЕТАЛЬ ПРОПИЛЕНГЛИКОЛЯ
2-ДИЭТИЛАМИНОМЕТИЛ-4-МЕТИЛ-
1,3-ДИОКСОЛАН
Б. Г. ЯСНИЦКИИ, С. А. САРКИСЯНЦ, Е. Г. ИВАНЮК
I >СН—СН2—N<
сн3-сн—О' ХА
CeHlaNOa Мол. вес 173,25
Диэтиламиноацеталь пропиленгликоля и ряд других
аминоацеталей получены нами впервые1 по методу,
основанному на взаимодействии соответствующих хлор-
ацеталей с диэтиламином под давлением в присутствии
солей меди.
116
Схема синтеза
СН2О
| >CH-CH2C1+HN(C2H6)2
CHg-СНО '
СН2О. /ОД
---» | >СН—СН,—N<
СН3—СНО/ хС2Нб
РЕАКТИВЫ
Диэтиламин, ч., ГОСТ 13279—67.
Медь однохлористая, ч., ГОСТ 4164—61.
Хлорацеталь пропиленгликоля (получение см. на стр. 215).
Способ получения
В автоклав емкостью 400 мл, снабженный термомет-
ром, штуцером для сброса давления и монометром, поме-
щают 50 г (0,36 моль) хлорацеталя пропиленгликоля,
53,6 г (0,73 моль) диэтиламина (необходим двукратный
избыток диэтиламина для связывания выделяющегося
хлористого водорода) и 0,2 г однохлористой меди. Реак-
ционную смесь выдерживают 6 ч при температуре 130—
140 °C и давлении 5 атм (см. примечание). После окон-
чания реакции выпавшие кристаллы солянокислого
д! этиламина отфильтровывают, а фильтрат перегоняют
в вакууме. Получают 49 г продукта; т. кип. 65—66° С
при 4 мм рт. ст. Выход 77,2% (от теоретического),
dl°= 0,9324, п*> = 1,4362.
Диэтиламиноэцеталь пропиленгликоля представляет
собой бесцветную жидкость со слабым запахом, характер-
ным для аминов, растворяется в спирте, эфире, ацетоне;
в воде не растворяется.
Так же получают другие диэтиламиноацетали, кон-
станты которых приведены ниже:
Т. кип, (°C) при
Название соединения Выход % давлении (мм рт.ст.) 20 nD .20 d4
Диэтиламиноацеталь
этиленгликоля 71,7 58-59; 4 1,4420 0,9640
Диэтиламиноэцеталь глицерина . 65,2 122-127; 7 1,4665 1,0719
Диэтиламиноацеталь диэгилен-
ГЛИКОЛЯ 32 97-99; 4 1,4550 1,0240
Диэтиламиноэцеталь диэтиламиио-
пропиленгликоля 44,5 118121; 5 1,4545 0,9428
Ди-(диэтиламиноацеталь)-пента-
эритрита 51.8 187-188;! 1,4725 1,0340
Три-(диэтиламиноацеталь)-маннита 61,5 156-157:0,004 1,48.0 1.1308
Три-(диэтнламиноацеталь)-сорбита 41 205-206; 0,07 1,4800 1,0935
117
Примечание. Автоклав нагревают медленно, так как
возможно самопроизвольное повышение температуры за счет резкого
увеличения скорости реакции, при этом давление быстро возрастает.
Следует работать с автоклавом, испытанным при 200 ат.
Литература
1. Я с н и ц к и й Б. Г., С а р к и с я н ц С. А., И в а-
ию к Е. Г., ЖОХ, 34, 1945 (1964).
1,3-ДИЭТИЛБЕНЗОЛ
А. В. СТРАШНЕНКО, Е. С. ЭЙДЕЛЬМАН
Мол. вес 134,22
1,3-Диэтилбензол образуется наряду с 1,4-диэтил-
бензолом при действии этилбромида 2> 3 или этилена4’ 5
на бензол в присутствии хлористого алюминия. 1,3-Диэтил-
бензол получают также при нагревании этилбензола с хло-
ристым алюминием 6. Разделение изомерных диэтилбен-
золов частичным сульфированием или фракционной
кристаллизацией бариевых солей диэтилбензолсульфо-
кислот не всегда приводит к чистому м-изомеру. В лабо-
раторных условиях более удобным является предлагаемый
способ получения индивидуального 1,3-диэтилбензола,
основанный на алкилировании 3-этилфенилмагнийбро-
мида диэтилсульфатом. Исходный 3-этилбромбензол
получают восстановлением легкодоступного 3-бромацето-
фенона 7.
Схема синтеза
118
РЕАКТИВЫ
3- БромацетофеноьЗ .
Гидразингидрат технический, ГОСТ 10729—64.
Этиленгликоль, ГОСТ 11033—64.
Диэтилсульфат, технический.
Способ получения
Синтез 3-бромэтилбензола. В 'трехгорлую колбу
емкостью 2 л, снабженную мешалкой, обратным холо-
дильником и термометром, помещают 1,4 кг этиленгли-
коля, 0,17 кг едкого кали, 0,2 кг (1 моль) 3-бромацетофено-
на и 170 мл гидразингидрат?. Содержимое колбы мед-
ленно нагревают до 130 °C и выдерживают при этой
температуре 2 ч, затем обратный холодильник заменяют
на нисходящий и отгоняют смесь воды и восстановлен-
ного продукта, постепенно повышая температуру в колбе
до 200 °C. Дистиллят и охлажденную реакционную смесь
последовательно обрабатывают эфиром (2 раза порциями
по 0,5 л). Эфирный экстракт промывают водой и сушат
безводным сульфатом натрия. Эфир отгоняют, а остаток
перегоняют в вакууме.
Получают 150 г продукта; т. кип. 85—86 °C при
20 мм рт. ст., выход 81% (от теоретического), =
= 1,3489, Пд — 1,5449. По литературным данным8,
3-бромэтилбензол представляет собой жидкость с т. кип.
200—208 °C, б/20 = 1,3493, = 1,5465.
Синтез 1,3-диэтилбензола. Реакцию проводят в токе
азота. В трехгорлую колбу емкостью 2 л, снабженную
мешалкой Гершберга, капельной воронкой и обратным
холодильником, защищенным хлор кальциевой трубкой,
помещают 24 г (1 моль) магниевых стружек и активируют
их иодом. В колбу вводят по каплям с такой скоростью,
чтобы эфир слабо кипел, сначала раствор 0,15 кг (0,81 моль)
3-бромэтилбензола в 400 мл сухого эфира, после чего
реакционную смесь кипятят 2 ч, затем раствор 0,24 кг
(1,7 моль) свежеперегнанного диэтилсульфата в 400 мл
сухого эфира и смесь кипятят еще 2 ч. Содержимое колбы
выливают в 400 мл охлажденного 10%-ного раствора
серной кислоты. Эфирный слой отделяют, промывают
10%-ным раствором серной кислоты, водой и сушат
сернокислым натрием. После отгонки эфира остаток
перегоняют в вакууме. Получают 65 г продукта: т. кип.
119
70—72 °C при 20 мм рт. ст., выход 60% (от теоретиче-
ского), dl° — 0,859, п% = 1,4946. По литературным дан-
ным С6, 1,3-диэтилбензол представляет собой бесцвет-
ную жидкость с т. кип. 181—182 °C, d^ = 0,8602 и =
= 1,4955.
Литература
1. Voswinkel А., Вег., 21, 2829 (1888).
2. А 1 1 е n R. Е., Underwood, Bl. [2], 40, 100 (1883).
3. Faurnier, Bl. [3], 7, 661 (1892).
4. В e г г у Т. М., Reid Е. Е., J. Am. Chem. Soc., 49, 3157
(1927).
5. Copenhaver J. E., Reid E. E., J. Am. Chem. Soc.,
49, 3157 (1927).
6. S t r a t f о r d R. E., Ann. Off Combust, lig., 4, 93, 321 (1928).
7. Синтезы органических препаратов, сб. 12, Изд. <'Мир», стр. 11.
8. Brown J. Н., Marvel С. S., J.Am. Chem. Soc., 59, 1176
(1937)
КОМПЛЕКСЫ
О-ОКСИФЕНИЛИМИНОДИУКСУСНОЙ кислоты
С МЕДЬЮ и свинцом
М. Н. РУСИНА, В. я. ТЕМКИНА, Н. М. ДЯТЛОВА
СН2СОО
......\>Н,0
ОН
СН.2СОО
N<\ch.2coo/
Pb
C10H9NO5Cu-H2O
Мол. вес 304,74
C10H9NO5Pb
Мол. вес 430,37
Нерастворимые в воде комплексы меди и свинца с отно-
сительно высоким содержанием металла представляют
интерес для решения ряда новых технических задач.
В связи с этим были синтезированы комплексы о-оксифе-
нилиминодиуксусной кислоты, обладающие значитель-
ной устойчивостью г. Синтез комплексов мы осуществили
взаимодействием дикалиевой соли комплексона с экви-
валентным количеством хлорида металла. Оптимальные
значения pH образования протонированных комплексог
рассчитывались из потенциометрических данных С
120
Схема синтеза
где
РЕАКТИВЫ
Кислота о-оксифенилимимдиуксусная, ч., ТУ 033-82—56.
Медь хлорная, ч. д. а., ГОСТ 4167-—61.
Свинец хлористый, ч. д. а., ГОСТ 4210—48.
Кали едкое, х. ч., ГОСТ 4203—65.
Способ получения
В двугорлой колбе емкостью 500 жл, снабженной
мешалкой, при комнатной температуре и перемешивании
растворяют 20,4 г (0,09 моль) о-оксифенилиминодиуксус-
ной кислоты в 200 мл 0,82 М раствора едкого кали. К полу-
ченному раствору при перемешивании добавляют соот-
ветственно 50 мл 2 М раствора хлорной меди (при pH 2)
или хлористого свинца (при pH 3) и выдерживают реак-
ционную массу 30 мин. Выпавший мелкокристаллический
осадок отфильтровывают, промывают водой до отрица-
тельной реакции па СГ-ион и высушивают до постоянного
веса при 115 °C. Получают 23,8 г комплексоната меди
(выход 87% от теоретического) и 32,8 г комплексоната
свинца (выход 84% от теоретического). Комплексонат
меди имеет зеленую окраску, комплексонат свинца —
светло-бежевую.
Состав синтезированного продукта (C10H9NO5Cu • Н2О):
с н N Си
Найдено, % .........39,8; 40,2 3,5; 3,7 3,8; 4,0 20,1; 20,0
Вычислено, % ....... 39,5 3,6 4,6 20,8
Состав синтезированного продукта (CloH0NO5Pb):
с н N рь
Найдено, % ..........27.9; 28,1 2,2; 2,4 3,2; 3,1 47,4; 47,6
Вычислено, % ........ 27,9 2,1 3,2 48,1
Литература
I. Ластовский Р. П., Дятлова Н. М., Темки-
н а В. Я., Русина М. Н., Жданов Б. В., Л а в -
о в а О. Ю., Химические реактивы и препараты, вып. 30,
РЕА, 1967, стр. 118.
121
П-КР ЕЗО Л-2-МЕТ И Л Е Н ИМ И НОД И УКСУСНАЯ
КИСЛОТА
В. я. ТЕМКИНА, Н. В. ЦИРУЛЬНИКОВА, Р. П. ЛАСТОВСКИЙ
0Н СН,СООН
CH2-N<
чСН2С00Н
CHS
ClaH16NO5
Мол. вес 253,28
«-Крезолметилениминодиуксуспая (4-метил-1-оксифе-
нил-2-метилениминодиуксусная) кислота образует ком-
плексы с рядом катионов.
Синтез этого не описанного в литературе соединения
мы осуществили взаимодействием «-крезола с формальде-
гидом и иминодиуксусной кислотой по реакции Манниха
в водно-щелочном растворе 2. При проведении реакции
в среде ледяной уксусной кислоты3 продукт не был
получен в чистом виде.
Схема синтеза
ОН
1 СН2СООН
СН2СООН
(к
CHjCOOH
х:н2соон
сн3
РЕАКТИВЫ
n-Крезол, ч., ВТУ МГ УХП 402—59.
Формальдегид, ч., ТУ МХП 2631—51.
Иминодиуксусная кислота, ч., ВТУ МГ УХП 190—58.
Натр едкий, ч., ГОСТ 4328—66.
Кислота соляная, ч., ГОСТ 3118—46.
Способ получения
В трехгорлую колбу, снабженную обратным холо-
дильником, механической мешалкой и капельной ворон-
кой, помещают) раствор 8,64 г (0,080 моль) «-крезола
122
в 12 мл дистиллированной воды и 8 мл 30%-ного раствора
едкого натра. К полученной смеси при комнатной темпе-
ратуре и размешивании прибавляют раствор 11,2 г
(0,084 моль) иминодиуксусной кислоты в 12 мл дистилли-
рованной воды, нейтрализованный 30%-ным раствором
едкого натра до pH 7—8. Затем при температуре 10° С
и размешивании добавляют по каплям 6,46 мл (0,088 моль)
37%-ного водного раствора формальдегида. Реакционную
массу размешивают 1 ч при комнатной температуре, 2 ч
при 60—70 °C и подкисляют (при охлаждении льдом)
концентрированной соляной кислотой до pH 2. При этом
выпадает смолообразный осадок. Реакционную массу
упаривают в вакууме при 60—70 °C досуха. Остаток
перемешивают в колбе с 50 мл дистиллированной воды
при 40 °C в течение 6 ч. Порошкообразный осадок отфиль-
тровывают, промывают водой и сушат двое суток в вакуум-
эксикаторе над хлористым кальцием. Получают 10 г
продукта; выход 45,8% (от теоретического). Комплексон
окрашен в бледно-розовый цвет, растворяется в щелочах,
плохо растворяется в воде, не растворяется в ацетоне,
бензоле и спирте.
Состав синтезированного продукта (C12H13NO5):
с н N
Найдено, % . . . . 56,24; 55,95 5,90; 6,15 5,61; 5,82
Вычислено, % . . . 56,91 5,96 5,53
Литература
1. An deregg G., F 1 a s h k а Н., S а 1 1 m a n n R.,
Schwarzenbach G., Helv. Chim. Acta, 37, 113 (1954).
2. Diehl H., E 1 1 i n g b о e J., Anal. Chem,, 28, 882 (1956).
3. Schwarzenbach G., A n d er egg G., S a 1 1-
m an n R., Helv. Chim. Acta, 35, 1785 (1952).
МЕТИЛИЗОЦИАНАТ
Ю. А. НАУМОВ, Л. Г. БАЖАНОВА, А. П. КНЯЗЕВА
CH3NCO
CjjH3ON Мол. вес 57,05
Метилизоцианат применяется в синтезе уретанов.
Он может быть получен взаимодействием циановокислого
калия с диметилсульфатом 1> 2, а также с помощью ряда
9* 123
других методов, общих для всех изоцианатов 3~5. При
систематической работе с метилизоцианатом, а также для
получения его в значительном количестве наиболее при-
годен метод синтеза этого продукта из N-метилкарбамино-
илхлорида. По литературным даннымв, при действии
пиридина на N-метилкарбаминоилхлорид образуется
метилизоцианат с выходом 88%. По нашим данным, при
обработке N-метилкарбаминоилхлорида эквимолярным
количеством диметиланилина выход метилизоцианата
93—95%.
Схема синтеза
CH3NHCOC1 4- CeH5N(CH3)a-- CH3NCO + C„H6N(CHS)2HC1
РЕАКТИВЫ
N-Метилкарбамиюилхлорид (получение см. на стр. 125).
У,У-Диметимнили.н, ч., ГОСТ 5855—51.
Углерод четыреххлористый, ч. д. а., ГОСТ 5827—51.
Способ получения .
В куб лабораторной ректификационной колонки, пред-
ставляющий собой трехгорлую колбу емкостью 250 мл,
помещают 67,6 г (0,725 моль) N-метилкарбаминоилхлорида
(см. примечания 1,2) и 50 мл четыреххлористого угле-
рода; смесь охлаждают до 10—15 °C и прибавляют по кап-
лям 87,8 г (0,725 моль) диметиланилина. После прибавле-
ния диметиланилина реакционную массу перегоняют
на лабораторной ректификационной колонке, отбирая
фракцию с т. кип. 35—40 °C. Получают 40,5 г продукта;
выход 98% (от теоретического). При повторной перегонке
отбирают предгон (около За) с т. кип. 35—37,8 °C и соби-
рают фракцию (т. кип. 37,8—38,0 °C), состоящую из чисто-
го метилизоцианата (см. примечание 3).
Получают 33,9 г продукта; выход 82% (от теоретиче-
ского); = 1,3632, d%° = 0,9620. По литературным
данным, т. кип. 37—39 °C ®, п77 = 1,3630 7, d\5 = 0,9670 ?.
Примечания. 1. Взвешивать N-метилкарбаминоилхло-
рид следует по возможности быстрее и обязательно под тягой.
2. Прибор для получения метилизоцианата должен быть защи-
щен от попадания влаги, так как вещество легко гидролизуется.
3. Метилизоднанат является сильным лакриматором. Хранить
его рекомендуете^ в холодильнике, в склянке с хорошо притертой
пробкой
124
Литература
1. W й г z A., Compt. rend., 27, 242 (1848); Ann., 71, 329 (1849).
2. S 1 о t t а К. H., T sc h esc he R., Ber., 60, 298 (1927).
3. Saunders J. H., Slocombe R. J., Chem. Rev., 43,
203 (1948).
4. Благонравова А. А., Левкович Г. А., Усп.
хим., 24, 93 (1955).
5. Ullmanns Enzyklopadie der technischen Chemie, 3Aufi., Bd. 9,
1957 S I
6. SI ocombe R. J., H a r d у E. E., S a u n d e r s J. H.,
Jenkins R. L., J. Am. Chem. Soc., 72, 1888 (1950).
7. Tolles W. M., Tichy J. R„ J. Phys. Chem., 62, 1606
(1958).
N-МЕТИЛ КАРБАМИНОИЛХЛОРИД
Ю. А. НАУМОВ, А. П. КНЯЗЕВА, Л. Г. БАЖАНОВА
C2H4C1ON
CH3NHCOC1
Мол. вес 93,51
N-Метилкарбаминоилхлорид используется для полу-
чения метилизоцианата и уретанов. По литературным
данным, N-метилкарбаминоилхлорид получают, пропу-
ская газообразную смесь фосгена и метиламина через
нагретую до 240—350 °C трубку. Прошедшую через
трубку газовую смесь конденсируют 1 или поглощают
нейтральным органическим растворителем2. Для лабо-
раторных целей удобнее иметь продукт в твердом виде.
Мы подобрали для этой реакции оптимальные условия,
позволяющие получить максимальпый выход N-метилкар-
баминоилхлорида и по возможности предотвратить побоч-
ную реакцию образования хлоргидрата метиламина.
Схема синтеза
CHSNH2 + СОС12--» CHgNHCOCl + НС1
РЕАКТИВЫ
Фосген, ГОСТ 2345—43.
Метиламин, газообразный, ВТУАИ 87-56—55.
Азот, газообразный, ос. ч., СТУ 36-13-748—61
125
Способ получения
Реакцию ведут в приборе для газофазного фосгениро-
вания, изображенном на рисунке. Поскольку N-метил-
карбаминоилхлорид на воздухе легко гидролизуется,
вся система должна быть защищена от попадания влаги.
Расход газов регулируют с помощью игольчатых венти-
лей и контролируют по капиллярным реометрам. При
Прибор для получения N-метилкарб-
аминоилхлорида:
1 — реактор с электрообмоткой; 2 — асбестовый
шнур; 3 — трубка для подачи фосгена; 4 — труб-
ка для подачи метиламина; 5 —термометр;
6 — трубка для отвода реакционных газов:
7 — ловушки; 8 — охлаждающая баня; 9 — отвод
к поглотительной системе.
(Размеры даны в мм).
градуировке реометров количество прошедшего газа
определяют по привесу, пропуская фосген в толуол при
—20 °C, а метиламин — в разбавленную серную кислоту.
Через общий вход, сделанный в виде сопла, фосген
и метиламин поступают в реактор (объем реактора около
50 см3), изготовленный из стекла пирекс и снабженный
термометром и электрообмоткой для нагревания. Для
термоизоляции реактор обматывают асбестовым шнуром.
Степень нагрева регулируют лабораторным автотранс-
форматором (ЛАТР). Реактор нагревают до 350 °C, про-
дувая систему слабым током сухого азота (азот пропу-
126
екают через склянку с 'Серной кислотой й вводя 1 В линию
подачи фосгена). Затем вентиль баллона с азотом закры-
вают и открывают вентиль баллона с фосгеном. Вслед
за фосгеном вводят метиламин. Для успешного протека-
ния процесса необходим некоторый избыток фосгена
(5—10%), в противном случае образуется большое коли-
чество хлоргидрата метиламина. Реакционные газы после
реактора попадают в специальную ловушку, состоящую
из двух последовательно соединенных широких (/-образ-
ных трубок, охлаждаемых смесью ацетона и сухого льда
до —40 °C. При этом N-метилкарбаминоилхлорид кон-
денсируется, а хлористый водород и избыток фосгена
поступают далее в поглотительные склянки с водой,
20%-ным раствором едкого натра и 25%-ным водным
аммиаком. По окончании работы в первую очередь пре-
кращают подачу метиламина, для того чтобы предотвра-
тить образование хлоргидрата метиламина. Затем пре-
кращают подачу фосгена, выключают электрообогрев
реактора и систему продувают 1,5 ч током сухого азота.
Твердый N-метилкарбаминоилхлорид расплавляют, осто-
рожно нагревая ловушку на водяной бане, и переливают
в сосуд. При расходе метиламина 0,4 л]мин, фосгена —
-0,44 л/мин и продолжительности процесса 45 мин полу-
чают 74,6 г N-метилкарбаминоилхлорида; выход 96,7%
(от теоретического), считая на метиламин, т. пл. продуд-
та ~80—85 °C. Так как N-метилкарбаминоилхлори к
легко гидролизуется на воздухе, температуру его плав-
ления нельзя определить точно в обычном приборе.
В литературе 3 температура плавления N-метилкарбами-
ноилхлорида также указана ориентировочно (около 90 °C).
По данным анализа, содержание основного вещества
в продукте не ниже 99%.
Определение проводят следующим образом. Навеску
N-метилкарбаминоилхлорида вводят в реакцию с более
чем двойным избытком дибутиламина, с которым он
образует метилдибутилмочевииу и хлоргидрат дибутил-
амина. Непрореагировавший дибутиламин титруют соля-
ной кислотой.
Литература
l.Slocombe R. J., Hardy Е. Е., Saunders J. Н.,
Jenkins R L., J. Аги. Chem. Soc., 72, 1888 (1950).
2 Фр. пат. 1344292 (1962); РЖХим., 8н77 (1965).
3. Gattermann L„ Ber., 20, 118 (1887); Ann., 244, 29 (1888).
127
N-МОРФОЛИЛ- И
N-ПИПЕРИДИЛИЗОПРОПИЛДИАЛКИЛФОСФАТЫ
N-Морфолилизопропилдиэтил фосфат
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
ChH24NOeP
ZCH2-CH2.
О . N—СН2-СН—О—Р</
\зн2—сн2/
ос2н5
сод
Мол. вес 281,29
Вследствие своей высокой физиологической активно-
сти большой интерес представляют фосфорорганические
соединения, являющиеся аналогами продуктов жизне-
деятельности различных организмов. К таким веществам
относятся фосфаты холинового ряда.
Со времени получения первых представителей этого
ряда 1 появилось много работ, посвященных изучению
данной группы соединений 2. Они могут быть использова-
ны в качестве пестицидных препаратов, физиологически
активных веществ, эстрогенных препаратов и пр. Фос-
фаты N-морфолил- и К-пиперидилпропаиола-2 в лите-
ратуре не описаны и получены нами впервые 3. Их син-
тез заключается во взаимодействии алкоголятов натрия
N-пиперидил- и Ы-морфолил'пропанола-2 с диалкилхлор-
фосфатами.
Схема синтеза
/СН2-СН2< /СН3 Na хн.-сн^ ,сн3
О N—СН2—СН » О N—СН2—СН
\сн2— СН2/ \он \сн2-сн/ \oNa
Ю
С1Р/
\ОС2Н»)2
РЕАКТИВЫ
,СН2— СН2ч /СН3
Э \-СНг-4?Н 0
\сн2—сн2/ \э-р/
Х(ООД)2
ТА-Морфолилпропанол-2, т. кип. 77—79 °C при 5 мм рт. ст. *,
1,019, п™ = 1,4648.
N-Пипериди.лпропанол-2, т. кип. 59—61 °C при 6 мм рт. ст. 4
d24° = 0,9263, = 1,4623.
128
10
Натрий металлический, ч., ТУ МХП 1664—50.
Диэтилхлорфосфат, свежеперегнанный, т. кип. 81—83 ®С при
мм рт. ст.
Бензол, ч. д. а., ГОСТ 5955—51.
Способ получения
В колбу с обратным холодильником помещают 14,52 г
(0,1 моль) Ь1-морфолилпропанола-2, прибавляют 50 мл
сухого бензола и 2,30 г (0,1 моль) металлического натрия
и при перемешивании нагревают реакционную смесь
до полного растворения натрия (60—70 °C). Охладив
полученный раствор алкоголята натрия до 5 °C, по кап-
лям добавляют 17,25 г (0,1 моль) диэтилхлорфосфата
с такой скоростью, чтобы температура реакционной
массы не поднималась выше 10 °C. Затем смесь нагре-
вают на кипящей водяной бане 30 мин, охлаждают до ком-
натной температуры и выпавший осадок хлористого
натрия отделяют центрифугированием. Из фильтрата
отгоняют бензол, а остаток перегоняют в вакууме. Полу-
чают 20 г продукта; т. кип. 151 —152 °C при 3 мм рт. ст.,
выход 71% (от теоретического), df = 1,113, п'д = 1,4459.
Содержание азота и фосфора в препарате (CUH24NO6P):
N Р
Найдено, % .... . 5,14 10,92
Вычислено, % . . . . 4,98 11,01
Так же получают другие N-замещенные изопропил-
диалкилфосфаты, константы которых приведены ниже:
Название соединения
Т. кип. (°C)
Выход, при давлении 20 j20
% {мм рт. cm.) nD а4
N-Морфолилизопропилдиизопро-
пилфосфат....................
N-Пиперидилизо пропилдиэтил-
фосфат ...
N-Пиперилилизопропилди-я-про-
пилфосфат.................
83 152—154; 3 1,4410 1,062
63 141—142; 3 1,1460 1,061
73 154—156; 3 1,4472 1,026
Литература
l. Qhosh R., Newman J. F., Chem. a. Ind., 1955, 1186.
2. Tamm elitl L. E., Acta chem. Scand., 11, 859 (1957).
Tammel in L. E., Ark. Kemi., 12, 30, 287 (1958).
3. Самуилов A. M., Дрегваль Г. Ф., авт. свид. СССР
188969; Билл, изобр., № 23 (1966),
4. Малиновский М. С., Окиси олефинов и их производные,
Госхимиздат, 1961, стр. 309.
9—287
129
1-НАФТИЛФОСФАТ
МОНО-а-НАФТИЛОВЫЙ ЭФИР ФОСФОРНОЙ
кислоты
В. М. ОСТРОВСКАЯ, т. А. МАРЬЯШКИНА
ОРО(ОН)8
Об
Сц|Н8О4Р Мол. вес 224,15
1-Нафтилфосфат применяют в гистохимии для
исследования гидролитических ферментов 1-3. Препарат
получают гидролизом 1-нафтилдихлорфосфата в воде4> s
или в вакуум-эксикаторе над раствором едкого кали в.
1-Нафтилдихлорфосфат получают при действии хлороки-
си фосфора на нафтол-1 (а-нафтол) 4-7. Мы проверили
и уточнили методику синтеза 1-нафтилдихлорфосфата,
сснованную на действии хлорокиси фосфора на раствор
нафтола-1 в бензоле в присутствии эквивалентного коли-
чества пиридина в. Гидролиз полученного 1-нафтилди-
хлорфосфата проведен в воде.
Схема синтеза
ОН ОРОС12 ОРО(ОН)2
РЕАКТИВЫ
Нафтол-1 (а-Нафтол), ч. д. а., ГОСТ 5838—51.
Хлорокись фосфора, ч., ВТУ 166—51.
Пиридин, ч., ГОСТ 2747-44.
Бензол, ч. д. а., ГОСТ 5955—51.
Вода, дистиллированная, ГОСТ 6709—53.
Способ получения
Синтез 1-нафтилдихлорфосфата. Смесь 100,4 г
(0,695 моль) нафтола-1, 111,5 г (0,695 моль) хлорокиси
фосфора и 360 мл бензола нагревают до 80 °C, перемеши-
130
вают до полного растворения нафтола-1, после чего при-
бавляют по каплям 54,8 мл (0,695 моль) пиридина и пере-
мешивают еще 15 мин. Реакционную массу охлаждают
до комнатной температуры и отфильтровывают осадок
солянокислого пиридина. Из фильтрата отгоняют бензол,
а остаток подвергают двукратной перегонке в вакууме.
При первой перегонке отбирают фракцию с т. кип. 163—
168 °C при 3 мм рт. ст.', получают 150 г продукта; выход
82% (от теоретического). После второй перегонки полу-
чают 135,6а продукта с т. кип. 167—168 °C при 3 мм
рт. ст.-, выход 74,6% (от теоретического), ng* = 1,5982.
Получение 1-нафтилфосфата. В 430 мл дистиллиро-
ванной воды вводят при перемешивании 135 г (0,530 моль)
1-нафтилдихлорфосфата, перемешивают 2 ч и оставляют
при комнатной температуре на ночь. Смесь расслаивается
и из нижнего слоя выкристаллизовывается 1-нафтилфос-
фат. Осадок отфильтровывают и сушат в вакуум-эксика-
торе над твердым едким кали до постоянного веса. Полу-
чают 100,4 а продукта в виде белых кристаллов; т. пл.
155,5—156 °C, выход 83,5% (от теоретического). По лите-
ратурным данным,4-5,М-10, т. пл. 1-нафтилфосфата 155—
159 °C.
Литература
1. Бер стон М., Гистохимия ферментов, Изд. «Мир», 1965,
стр. 213, 400.
2. В а г k а Т., Anderson Р. J., Histochemistry, theory,
practice, bibliography, New York, Evanston, London, 1963,
p. 383.
3. Пи p с Э., Гистохимия теоретическая и прикладная, Издат-
инлит, 1962, стр. 391, 799.
4. Kunz Р„ Вег., 27, 2559 (1894).
5. Маха J., Coll. Czech. Chem. Comm., 25, 603 (1960).
6. Friedman M., Seligman M., J. Am. Chem. Soc.,
72, 624 (1950).
7. Катышкина В. В., Крафт М. Я., ЖОХ, 26, 3060
(1956).
8. С h a n 1 е у J. D., Feageson Е., J. Am. Chem. Soc.,
77, № 15, 4002 (1955).
9. Light L. and Co. Ltd, Biochemicals, 1963—1964, p. 27.
10. Aldrich organic chemicals, USA, 1966, p. 268.
2-НАФТИЛФОСФАТ
МОНО-р-НАФТИЛОВЫЙ ЭФИР ФОСФОРНОЙ
кислоты
В. М. ОСТРОВСКАЯ, т. А. МАРЬЯШКИНА
ОРО(ОН)
CioHfOjP
Мол. вес 224,15
2-Нафтилфосфат применяют в гистохимии для
исследования гидролитических ферментов1-3. Препарат
получают гидролизом 2-нафтилдихлорфосфата в воде4
или в вакуум-эксикаторе над раствором едкого кали 8.
2-Нафтилдихлорфосфат получают при действии хлорокиси
фосфора на нафтол-2 (Р-нафтол) 4-6.
Мы проверили и уточнили методику синтеза 2-нафтил-
фосфата, основанную на действии хлорокиси фосфора
на раствор нафтола-2 в бензоле в присутствии эквивалент-
ного количества пиридина 5. Гидролиз полученного
2-нафтилдихлорфосфата проведен в воде.
Схема синтеза
РЕАКТИВЫ
Нафтол-2 (fi-нафтол), ч. д. а., ГОСТ 5835—51.
Хлорокись фосфора, ч., ВТУ 166—51.
Пиридин, ч., ГОСТ 2747—44.
Бензол, ч. д. а., ГОСТ 5955—51.
Вода дистиллированная, ГОСТ 6709—53.
Способ получения
Синтез 2-нафтилдихлорфосфата. Смесь 100,4 г
(0,695 .ноль) нафтола-2, 111,5 г (0,695 моль) хлорокиси фос-
фора и 360 мл бензола нагревают до 80 °C и размешивают
до полного растворения нафтола-2, после чего добавляют
по каплям 54,8 мг (0,695 моль) пиридина и перемешивают
еще 15 мин. Реакционную массу охлаждают до комнат-
132
ной температуры и отфильтровывают осадок солянокис-
лого пиридина. Из фильтрата отгоняют бензол, а остаток
подвергают двукратной перегонке в вакууме. При первой
перегонке отбирают фракцию с т. кип. 166—172 °C при
3 мм рт. ст.; получают 143 г продукта; выход 78,5%
(от теоретического). После второй перегонки получают
134,4 г продукта с т. кип. 169—171 °C при 3 мм рт. ст.;
т. пл. 39—40 °C, выход 74% (от теоретического).
Получение 2-нафтилфосфата. В 430 мл дистиллиро-
ванной воды вводят при 40—50 °C и перемешивании 134 г
(0,525 моль) 2-нафтилдихлорфосфата. Через 5—10 мин
реакционная масса сильно загустевает в результате
образования 2-нафтилфосфата. Реакционную массу раз-
мешивают 1 ч при комнатной температуре. Выпавший
осадок отфильтровывают и сушат его в вакуум-эксикаторе
над твердым едким кали до постоянного веса. Получают
113 г 2-нафтилфосфата в виде белых кристаллов; т. пл.
172—173°C, выход 97% (от теоретического). По литератур-
ным данным, т. пл. 167 °С\ 172—173 °C7, 176—177 °C 5,
177— 178 °C 8.
Литература
1. Берстон М., Гистохимия ферментов, Изд. «Мир», 1965,
стр. 155, 162.
2. П и р с Э., Гистохимия теоретическая и прикладная, Издат-
инлит, 1962, стр. 366, 801.
3. В а г k а Т., Anderson Р. J., Histochemistry,
theory, practice, bibliography, New York, Evanston, London,
1963, p. 231, 233, 244.
4. К u n z P., Ber., 27, 2559 (1894).
5. Friedman M., Seligman M., J. Am. Chem. Soc.
72, 624 (1950).
6. К а т ы ш к и н а В. В., Крафт M. Я., ЖОХ, 26, 3060
(1956).
7, A th ert on F. R., О p e n s h a u H. T., Todd A. R.,
J. Chem. Soc., 1945, 382.
8. C h a n I e у J. D., F e a g e s о n E., J. Am. Chem. Soc.,
77, 15, 4002 (1950).
1-НАФТИЛФОСФАТА ДИНАТРИЕВАЯ СОЛЬ
В. М. ОСТРОВСКАЯ, т. А. МАРЬЯШКИНА
/ONa
О-Р<
| || ONa
C10H,NaaO4P • 1,5Н2О Мол . вес 268,12 +27,02
Динатриевую соль 1-нафтилфосфата применяют в гисто-
химии для количественного и качественного определения
гидролитических ферментов. В иностранных каталогах
указана цена препарата методики получения этой соли
в литературе не найдено. Мы получили динатриевую
соль 1-нафтилфосфата действием метилата натрия на 1-наф-
тилфосфат.
Схема синтеза
ОРО(ОН)а OPO(ONa)2
I
+ CH3ONa
РЕАКТИВЫ
1-Нафтилмонофосфат (получение см. на стр. 139), т. пл.
155,5—156 °C.
Спирт метиловый, ч., ГОСТ 6995—54.
Натрий металлический, ч., ТУ МХП 1664—50.
Эфир диэтиловый, ч., ГХФП 25/IV—41 г.
Способ получения
К раствору метилата натрия в метаноле, полученному
при растворении 4,15 г (0,18 моль) металлического натрия
в 80 мл метилового спирта, приливают при размешивании
раствор 20 г (0,09 моль) 1-нафтилфосфата в 30 мл метило-
вого спирта (последний раствор можно отфильтро-
вать). После 15-минутиого размешивания реакционную
массу оставляют при комнатной температуре. Через
20—30 мин начинает выпадать динатриевая соль 1-наф-
тилфосфата в виде длинных прозрачных игл. Колбу выдер-
живают в холодильнике при 5 °C 1—1,5 ч. После этого
134
осадок отфильтровывают и промывают 10 мл эфира. Филь-
трат упаривают почти досуха и приливают к остатку
50 мл эфира. Выпавшую динатриевую соль отфильтро-
вывают и сушат в вакуум-эксикаторе над концентриро-
ванной серной кислотой. Получают 22,5 г продукта;
выход 88,5% (от теоретического).
Состав синтезированного продукта (C10H,Na2O4P х
х 1,5НаО):
С Н Р Na Н2О
Найдено, % 40,70; 40,723,62; 3,44 10,50; 10,54 15,75; 16,00 9,0
Вычислено,
% . . . . 40,67 3,11 10,50 15,60 9,15
Литература
1. Каталог ВДН, Laboratory chemikals catalogue, 1964, р. 171;
проспект ВДН, Enzyme substrates, 1966.
2-НАФТИЛФОСФАТА ДИНАТРИЕВАЯ СОЛЬ
В. М. ОСТРОВСКАЯ, Г. А. МАРЬЯШКИНА
ONa
C10H7Na2O4P0,7H2O Мол. вес 268,12+ 12,61
Динатриевая соль 2-нафтилфосфата применяется
в гистохимии для качественного и количественного опре-
деления гидролитических ферментов в тканях. В ино-
странных каталогах указана цена препарата \ методики
получения этой соли в литературе не' найдено. Мы полу-
чали динатриевую соль 2-нафтилфосфата действием мети-
лата натрия на 2-нафтилфосфат.
Схема синтеза
ОРО(ОН)2
+ CHgONa
OPO(ONa)2
135
РЕАКТИВЫ
2-Нафтилфосфат (получение см. на стр. 132), т. пл. 172—173 °C.
Спирт метиловый, ч., ГОСТ 6995—54.
Натрий металлический, ч., ТУ МХП 1664—50.
Эфир диэтиловый, ч., ГХФП 25/IV—41 г.
Способ получения
К раствору метилата натрия б метаноле, полученному
постепенным растворением 4,15 г (0,18 моль) металличе-
ского натрия в 80 мл метилового спирта, приливают при
размешивании раствор 20 г (0,09 моль} 2-нафтилфосфата
в-30 мл метилового спирта.
Выпавший осадок динатриевой соли 2-нафтилфос-
фата после 15-минутного размешивания реакционной
массы отфильтровывают, промывают диэтиловым эфиром
и сушат в вакуум-эксикаторе над концентрированной
серной кислотой до постоянного веса. Из метанольного
маточника добавлением диэтилового эфира (в объемном
отношении спирт: эфир =1:4) осаждают еще некоторое
количество динатриевой соли 2-нафтилфосфата. Полу-
чают 23 г продукта; выход 90% (от теоретического).
Динатриевая соль представляет собой белое кристалли-
ческое вещество, не плавящееся до 250 °C.
Состав синтезированного продукта (C1()H7Na2O4P х
X 0,7Н2О):
С н Р нао
Найдено, % . . . 43,46; 43,18 3,31; 3,29 11,10; 11,06 4,3
Вычислено, % . . 42,79 3,02 11,03 4,49
Литература
1. Aldrich organic chemicals, 1966, р. 268.
НИТРИЛ 2-ХЛОРПЕНТЕН-2-ОВОЙ КИСЛОТЫ-5
Е. Л. ГЕФТЕР
СНа—СС1 =СН—СНа—CN
C6HeClN Мол. вес 115,56
Нитрил 2-хлорпентен-2-овой кислоты-5 является про-
межуточным продуктом при синтезе (из 1,3-дихлорбуте-
136
на-2) самой кислоты и ее эфиров, а также левулиновой
кислоты и ее эфиров. Описан синтез нитрила 2-хлорпен-
тен-2-овой кислоты-5 из 1,3-дихлорбутена-2 и цианистого
натрия 1-3. Методика получения приведена только в одном
случае1, причем выходы продукта составляют 60% (в пере-
счете на прореагировавший) и 38% (в пересчете на загру-
женный) дихлорбутен. Используя цианистую медь вместо
цианистого натрия, н’ам удалось существенно улучшить
выход продукта.
Схема синтеза
СН3—СС1=СН—СН2С1 + CuCN---> СН3—СС1=СН—СН2—CN
РЕАКТИВЫ
1, 3-Дихлорбутен-2, свежеперегнанный, технический (отход при
получении хлоропрена), фракционирован на колонке эффектив-
ностью 8—10 тарелок; т. кип. 126—129 °C, Лд' = 1,4725, dj0 =
= 1,1595 (см. примечание 1).
Л4едб цианистая, сухая, ГОСТ 10018—62.
Способ получения
В круглодонную четырехгорлую колбу, снабженную
капельной воронкой с хлоркальциевой трубкой и обрат-
ным холодильником с хлоркальциевой трубкой, термо-
метром и мешалкой с глухим затвором *, позволяющим
проводить реакцию в вакууме, помещают 88 г цианистой
меди (см. примечания 1 и 2) и 30 г свежеперегнанного
1,3-дихлорбутена-2. Колбу нагревают на водяной бане,
время от времени перемешивая почти сухую массу Через
20—60 мин после установления температуры 90—95 °C
(в бане) смесь становится более жидкой, и постепенно
начинается экзотермическая реакция. Включают пере-
мешивание, удаляют баню и добавляют из капельной
воронки 82 г дихлорбутена (см. примечание 3) с такой
скоростью, чтобы температура реакционной смеси не под-
нималась выше 115—120 °C (в случае необходимости
колбу охлаждают водой). Через 10—15 мин после добав-
ления, дихлорбутена экзотермическая реакция прекра-
щается, после чего смесь нагревают при непрерывном
перемешивании до 95—100 °C еще 30 мин. Обратный
холодильник заменяют на прямой и отгоняют (см. при-
* Описание затвора приведено иа стр. 101 настоящего сборника.
137
мечание 4) при перемешивании и температуре бани 100—
120 °C 85—90 г чистого продукта. После вторичной пере-
гонки в вакууме получают 81—85 г нитрила с т. кип.
100—102 °C при 55 мм рт. ст.-, выход 78—82% (от тео-
ретического), п2£ 1,4618—1,423, d20 = 1,0705. По лите-
ратурным данным *, т. кип. вещества 102—105 °C при
55 мм рт. ст., п22 = 1,462, d22 = 1,07063.
Нитрил 2-хлорпентен-2-овой кислоты-5 представляет
собой прозрачную бесцветную жидкость (желтеющую
при стоянии) со своеобразным запахом, растворяется
в спирте, эфире, этилацетате и других органических
растворителях, но практически не растворяется в воде.
Примечания. 1. Внимание! Работать с цианистой медью
опасно (при ее подкислении выделяется синильная кислота) Необ-
ходимо надевать резиновые перчатки. В случае попадания цианистой
меди на стол следует залить ее раствором сернокислого железа,
и образовавшийся продукт синего цвета тщательно смыть в ракови-
ну.. 1,3-Дихлорбутен-2 является лакриматором. Работу следует
проводить в хорошо действующем вытяжном шкафу.
2. Цианистая медь должна быть совершенно сухой, так как
даже следы влаги увеличивают индукционный период реакции и
снижают выход продукта. При использовании несвежеперегнанно-
го дихлорбутена индукционный период реакции может продолжать-
ся 2 ч.
3. При быстром добавлении дихлорбутена энергичное выделе-
ние тепла может привести к выбросу реакционной массы и даже
к взрыву.
4. Если реакционная масса охлаждается (например, при прове-
дении перегонки на следующий день) и затвердевает, ее нужно
предварительно расплавить в бане с горячей водой.
Литература
1. Варданян С. А., ЖПХ, 25, 1322 (1952).
2. А з а т я н В. Д., Аракелян М. А., Научно-исследова-
тельские работы химических институтов и лабораторий АН
СССР, Изд. АН СССР 1941-1943, стр. 310.
3. Chem. Eng. News, 24, 1085 (1946).
ОКСИМЕТИЛФОСФОНОВАЯ КИСЛОТА
Е. Л. ГЕФТЕР, Н. И. БОНДАРЬ
ZOH
НОСН,Р<
II4 он
о
СН6О4Р
Мол. вес. 112,02
138
Оксиметилфосфоновэя кислота представляет собой
твердое гигроскопическое вещество. Неочищенная кислота
содержит небольшие количества формальдегида, чем
и объясняется ее специфический запах. При длительном
нагревании до 170—200 °C (особенно в вакууме) наблю-
дается самоконденсация кислоты, причем ее кислотное
число падает, а температура плавления возрастает. Окси-
метилфосфоновая кислота является специфическим ката-
лизатором отверждения метилолполиамидных смол, при-
дающим огнестойкость и ценные физико-механические
качества1. В литературе описано получение оксиметил-
фосфоновой кислоты взаимодействием треххлористого
фосфора и параформа в отсутствие растворителей 2 с после-
дующим гидролизом комплекса, что является опасной
и неприятной реакцией при получении значительных
количеств продукта; гидролизом под давлением хлорме-
тилфосфоновой кислоты 3, взаимодействием треххлори-
стого. фосфора с формалином4 (получение технической
кислоты).
Мы разработали безопасный метод получения чистой
оксиметилфосфоновой кислоты взаимодействием треххло-
ристого фосфора и параформа в инертных растворителях.
Схема синтеза
О
/ \ н2о
РС13 + ЗСН2О---> [Р(ОСН2С1)з-> СН2-Р(ОСН2С1)2]->
---> НОСН2РО(ОН)2
РЕАКТИВЫ
Фосфор треххлористый, ч., ГОСТ 91—41.
Параформ, ч., МРТУ 6-09-2128—65.
Хлороформ, ч., ГОСТ 3160—51.
Способ получения
В круглодонную четырехгорлую колбу емкостью
700 мл, снабженную капельной воронкой с хлоркальцие-
вой трубкой, нисходящим холодильником с приемником,
мешалкой и термометром, помещают 135 г (3 моль) пара-
форма и 180 мл сухого хлороформа (см. примечание 1).
139
Смесь перемешивают и нагревают на масляной бане
(см. примечание 2) до 55—68 °C, пока не отгонится около
30 мл хлороформа (см. примечание 3). Затем охлаждают
до 35—40 °C, заменяют нисходящий холодильник с прием-
ником на обратный холодильник с хлоркальциевой труб-
кой, вновь нагревают до 60—65 °C (до легкого кипения
хлороформа), прекращают нагревание и из капельной
воронки начинают прибавлять при энергичном перемеши-
вании тонкой струей или по каплям 207 г (1 моль) пере-
гнанного треххлористого фосфора (см. примечание 4).
Реакция протекает с выделением тепла. Скорость подачи
треххлористого фосфора регулируют так, чтобы смесь
непрерывно кипела (60—65 °C). Не следует допускать
снижения температуры реакции, так как накопление
в реакционной смеси непрореагировавшего треххлористого
фосфора может привести к аварии. После добавления треххло-
ристого фосфора и прекращения экзотермической реакции
содержимое колбы перемешивают еще 30 мин при 60—65 °C,
охлаждают в бане с ледяной водой до 5—10 °C и прибав-
ляют из капельной воронки 150 мл воды с такой скоростью,
чтобы температура смеси не поднималась выше 40—45 °C
(повышение температуры гидролиза сопровождается
уменьшением выхода оксиметилфосфоновой кислоты).
Затем заменяют обратный холодильник на прямой и пере-
ь сшивают реакционную смесь, постепенно нагревая ее до
температуры кипения водяной бани. Сначала при темпе-
ратуре 30—45 °C отгоняется хлористый водород, который
поглощается водой, затем при температуре 45—70 °C хло-
роформ. Оставшуюся жидкость упаривают на водяной бане,
подливая в чашку для выпаривания воду (2—3 раза
по 50 мл) для удаления соляной кислоты и образовавше-
гося при гидролизе формальдегида. Оставшаяся желтая
густая жидкость постепенно кристаллизуется в бело-
желтую массу с т. пл. 79—82 °C. После перекристалли-
зации из смеси этанола и уксусной кислоты получают
160—165 г продукта; т. пл. 84—85 °C; выход 96—98%
(от теоретического). По литературным данным2, т. пл.
85 °C.
Данные анализа синтезированного продукта (СН5О4Р):
Кислотное
Р число
Найдено . . 27,4% 985—990
Вычислено . 27,7% 1000
140
Примечания. 1. Можно применять и_другие инертные
растворители, например бензол, дихлорэтан, четыреххлористый
углерод. Однако лучшие результаты были получены с хлороформом.
2. Водяную баню не рекомендуется применять вплоть до стадии
гидролиза, так как в случае поломки колбы треххлористый фосфор
может попасть в воду, что вызовет серьезную аварию.
3. Технический параформ обычно недостаточно сухой, и отгонка
части растворителя сопровождается удалением воды из параформа.
4. Слабое перемешивание реакционной смеси может вызвать
местные перегревы и бурное вскипание. В случае прекращения
работы мешалки необходимо немедленно прекратить подачу трех-
хлористого фосфора и нагревание и некоторое время вращать мешал-
ку рукой, пока температура смеси не снизится на 10—12 °C.
Литература
1. Г е ф т е р Е. Л., Родивилова Л. А., авт. свид. СССР
114022; Бюлл. изобр., № 7 (1958).
2. Р a g е Н. J., J. Chem. Soc., 101, 423 (1912).
3. К а б а ч н и к М. И., Шепелева Е. С., Изв. АН СССР,
ОХН, 1951, 185.
4. Г е ф т е р Е. Л., Мошкин П. А., Перцов Д. Д.,
Пласт, массы, № 4, 62 (1961).
2-ОКСИ-6-МЕТИЛЦИНХОНИНОВАЯ КИСЛОТА
А. Л. ГЕРШУНС, А. А. ВЕРЕЗУБОВА
cuh9no3
Мол. вес 203,19
2-Окси-6-метилцинхониновая кислота может слу-
жить полупродуктом в синтезе различных производных
хинолина. В литературе это соединение не описано.
Мы получили 2-окси-6-метилцинхониновую кислоту по
методу Пфитцингера 1 с использованием в качестве исход-
ных веществ 5-метилизатина и малоновой кислоты.
Схема синтеза
СООН
с"-- ______с0 у0011 сн, <<.Л
I II (от — III
соон Vх х <^\он
ГЧН 14
141
РЕАКТИВЫ
5-Метилизатин 2, т. пл. 187 °C.
Малоновая кислота, ч., МРТУ 6-09-327—63.
Кислота уксусная, ч. д. а., ГОСТ 61—51.
Способ получения
Смесь 32,2 г (0,2 моль) 5-метилизатина, 31,2 г (0,3 моль)
малоновой кислоты и 50 мл ледяной уксусной кислоты
нагревают 2 ч в колбе с обратным холодильником на кипя-
щей водяной бане. Затем смесь кипятят на открытом
пламени газовой горелки (используя сетку) еще 6 ч,
охлаждают и фильтруют. Осадок промывают на фильтре
уксусной кислотой до получения бесцветного фильтрата.
После кристаллизации 31 г сырого продукта (из расчета
10 г вещества на 1 л воды) получают 23,5 г чистого про-
дукта в виде белого или с сероватым оттенком порошка;
т. пл. 307—310 °C; выход 58% (от теоретического). Веще-
ство хорошо растворяется в водных растворах щелочей
и в диметилформамиде; при нагревании растворяется
в спиртах и пиридине; плохо растворяется в эфире, бен-
золе и ацетоне 2.
Содержание азота в препарате (CnHeNO3):
Найдено, %......... 6,74, 6,93
Вычислено, %....... 6,89
Литература
1. Pfitzinger Н., J. Prakt. Chem,, 33, 100 (1886),
2. Синтезы органических препаратов, т. 1, Издатинлит, 1949,
стр. 216.
ОКСИФЕНИЛДИ-а-ТИЕНИЛМЕТАН
И ДИАРИЛ-а-ТИЕНИЛМЕТАНЫ
И. И. ЛАПКИН, М. И. БЕЛОНОВИЧ
При действии сложных эфиров муравьиной или щаве-
левой кислот на галоидмагнийди-а-тиенилкарбиноляты
(или галоидмагнийарил-а-тиенилкарбиноляты) образуются
ди-а-тиенилметилгалоидиды (или арил-а-тиенилметилга-
лоидиды). Галоидмагнийфеноляты в аналогичных усло-
142
виях не взаимодействуют с названными сложными эфи-
рами. Эти факты мы использовали для синтеза оксифенил-
ди-а-тиенилметана и диарил-а-тиенилметанов. Метод
заключается в том, что смесь ди-а-тиенилкарбинола (или
арил-а-тиенилкарбинола) и фенола после прибавления
соответствующего количества бромистого этилмагния
превращается в смесь броммагнийди-а-тиенилкарбинолята
(или броммагнийарил-а-тиенилкарбинолята) и фенолята.
Последующее прибавление к реакционной смеси сложного
эфира муравьиной или щавелевой кислот переводит бром-
магнийди-а-тиенилкарбинолят в ди-а-тиенилметилбромид
(или арил-а-тиенилметилбромид), который, взаимодей-
ствуя с неизменяющимся в этих условиях броммагний-
фенолятом, образуют оксифенилди-а-тиенилметан (или
монооксидиарил-а-тиенилметан).
Для получения названных веществ не обязательно
исходить из предварительно синтезированных карбино-
лов. Очень удобно эту реакцию сочетать с получением
магнийорганическим синтезом самого карбинола. Необ-
ходим только избыток реактива Гриньяра, чтобы его
хватило для последующего превращения фенола в галоид-
магнийфенолят.
4-ОКСИДИФЕНИЛ-а-ТИЕНИЛМЕТАН
C17H14OS
Мол. вес. 266.36
Схема синтеза
CeHsCHO
HCOOCjjHs
I К
\s/\MgI
СеНбОН
143
''OMgl
S CH
—Mgl2
РЕАКТИВЫ
Тиофен, ч., ВТУ МХП 3329—52.
Фенол, ч., ГОСТ 6417—52.
Бензальдегид синтетический, ч., ГОСТ 157—51.
Этилформиат, ч., ВТУ РУ 1285—56.
Этил бромистый, ч., ТУ МХП 80—47.
Способ получения
К раствору 21,2 г (0,2 моль) бензальдегида в 60 мл
абсолютного диэтилового эфира при постоянном пере-
мешивании и охлаждении прибавляют раствор 94 а
(0,4 моль) йодистого а-тиенилмагния в 200 мл абсолют-
ного эфира (см. примечания 1 и 2). Смесь нагревают
15 мин на водяной бане. К раствору, содержащему иод-
магнийфенил-а-тиенилкарбинолят и иодистый а-тиенил-
магний, прибавляют раствор 18,8 г (0,2 моль) фенола
в 60 мл эфира и снова нагревают 15 мин. К образовав-
шейся смеси эквимолярных количеств иодмагнийкарби-
нолята и фенолята добавляют раствор 15 г (0,2 моль)
этилформиата в 30 мл эфира. Реакционную массу нагре-
вают 3 ч на водяной бане и затем разлагают водой и 10%-ной
уксусной кислотой. Эфирный раствор отделяют, промы-
вают насыщенным раствором бикарбоната натрия, водой
и сушат прокаленным сернокислым натрием. Раствори-
тель отгоняют на водяной бане, и продукт реакции очи-
щают перекристаллизацией из смеси петролейный эфир —
бензол (4 : 1). Получают 38 г 4-оксидифенил-а-тиенилме-
тана; т. пл. 82 °C, выход 75% (от теоретического).
144
По аналогичной методике получают 39 г 5-метил-2-
оксидифенил-а-тиенилметана (используя вместо фенола
n-крезол); т. пл. 96 °C, выход 70% (от теоретического).
4-ОКСИФЕНИЛДИ-а-ТИЕНИЛМЕТАН
C15H12OS2
Мол. вес 272,38
Схема синтеза
Получение из ди- а-тиенилкарбинола
А.
CftMgBr
OMgBr
НСООС2Н6
С6Н5ОН
OMgBr
—MgBr2
10—287
145
Б.
Получение из <х-иодтиофена
НСООСгНб _ __
~~LAAJ
S CH S
I HCOOCjHs
I СвНбОН
Способ получения
А. К раствору 39,2 г (0,2 моль) ди-а-тиенилкарбинола
(см. примечание 3) и 18,8 г (0,2 моль) фенола в 180 мл
абсолютного диэтилового эфира при постоянном переме-
шивании и охлаждении прибавляют эфирный раствор
53,2 г (0,4 моль) бромистого этилмагния (см. примечание4).
Смесь нагревают 15 мин. К образовавшейся смеси бром-
магнийди-а-тиенилкарбинолята и фенолята добавляют
15 г (0,2 моль) этилформиата в 30 мл эфира и нагревают
на водяной бане 3 ч. Реакционную массу при охлаждении
гидролизуют водой и 10%-ной уксусной кислотой. Эфир-
ный слой отделяют, промывают насыщенным раствором
бикарбоната натрия, водой и сушат прокаленным серно-
кислым натрием. Растворитель отгоняют на водяной
бане, а продукт реакции перекристаллизовывают из
смеси петролейный эфир—бензол (4 : 1). Получают 38 г
продукта; т. пл. 81 °C, выход 70% (от теоретического).
146
Б. Получение 4-оксифенилди-а-тпенилметана можно
сочетать с получением магнийорганическим синтезом
исходного карбинола.
К раствору 140 г (0,6 моль) йодистого а-тиенилмагния
в 250 мл абсолютного диэтилового эфира (см. примеча-
ние 5) прибавляют раствор 15 г (0,2 моль) этилформиата
в 30 мл эфира. Смесь нагревают на водяной бане 1 ч.
Затем приливают раствор 18,8 г (0,2 моль) фенола в 60 мл
эфира, вновь нагревают на водяной бане 15 мин, прибав-
ляют еще 15 г (0,2 моль) этилформиата в 30 мл эфира
и нагревают на водяной бане 3 ч. Разложение реакционной
массы и выделение продукта проводят, как указывалось
выше. Получают 27 а продукта, выход 50% (от теорети-
ческого).
Примечания. 1. а-Иодтиофен получен по описанной
в литературе методике *.
2. Йодистый а-тиенилмагний синтезирован в обычных условиях
гриньяровских реакций из 10 а (0,4 моль) магния и 84 г (0,4 моль)
а-иодтиофена.
, 3. Ди-а-тиенилкарбинол синтезирован из 84 г (0,4 моль) а-ирд-
тиофена, 10 г (0,4 моль) магния и 15 г (0,2 моль) этилформиата;
т. пл. 57 °C.
4. Бромистый этилмагний получен из 10 г (0,4 моль) магния
и 44 г (0,4 моль) бромистого этила.
5. Йодистый а-тиенилмагний синтезирован в обычных услови-
ях гриньяровских реакций из 15 а (0,6 моль) магния и 126 а (0,6 моль)
а-иодтиофена.
Литература
1. Л а п к и н И. И., Лапкина О. М., ЖОХ, 25, 298 (1955).
2. Л а п к и н И. И., Лапкина О. М., Рыба но-
ва М. И., ЖОХ, 28, 391 (1958).
3. Л а п к и н И. И., Белонович М, И., ЖОХ, 28, 605,
(1958); 31, 3182, 1961.
4. Синтезы органических препаратов, т. 2. Издатинлит, 1949,
стр. 287.
8-ОКСИХИНАЛЬДАЗИН
АЗИН 8-ОКСИХИНАЛЬДИНОВОГО АЛЬДЕГИДА
В. м. ДЗИ0МК0, И. А. КРАСАВИН, Ю. П. РАДИН
Мол. вес 342,36
10*
147
; Об экстракции смешанных (при участии купферона)
хелатов 8-оксихинальдазина с редкоземельными эле-
ментами уже сообщалось 2, но способ получения-8-окси-
хинальдазина был лишь кратко упомянут 3. 8-Оксихи-
нальдазин образуется с 90%-ным выходом при взаимо-
действии 8-оксихинальдинового альдегида с гидразин-
гидратом в спиртовом растворе.
Схема синтеза
РЕАКТИВЫ
8-Оксихинальдиновый альдегид дважды перекристаллизованный
из я-гептана. т. пл. 98,5—99 °C.
Гидразингидрат, ч. д. а., ГОСТ 5832—65.
Способ получения
В круглодонной колбе емкостью 100 мл, снабженной
обратным холодильником, растворяют 3,46 г (0,02 моль)
8-оксихинальдинового альдегида в 20 мл горячего этило-
вого спирта, прибавляют. раствор 0,60 г (0,012 моль)
99,5%-ного гидразингидрата в 10 мл этилового спирта.
При этом выпадает ярко-желтый объемистый осадок. Сус-
пензию кипятят на водяной бане 1 ч, охлаждают в бане
со льдом и выдерживают 2—3 ч при 0° С. Выпавший оса-
док отсасывают, промывают на фильтре 5 мл этилового
спирта и сушат в вакуум-эксикаторе. Получают 3,1 г
продукта; т. пл. около 258—260 °C (с разложением),
выход 90,6% (от теоретического). После перекристалли-
зации из 400 мл диоксана получают 2,5 г продукта в виде
блестящих ярко-желтых пластинок; т. пл. 261° С (с раз-
ложением); выход 73,1% (от теоретического).
148
Состав
синтезированного
продукта (C20H14N4O2):
Найдено, %
Вычислено,
с
70,26; 70,57
70,17
н
4,59;4,41
4,12
N
16,03; 16,21
16,37
Литература
1. Дзи ом ко В. М., Красавин И. А., Марко-
вич И. С., Островская В. М, Зеличенок С Л.,
Совещание по методам концентрирования элементов в аналити-
ческой химии, Тезисы докладов, 1963, стр. 6.
2. Дзи ом ко В. М., Химические реактивы и препараты, вып.
27, ИРЕА, 1965, стр. 336.
3. Дзиомко В. М., К р а с а в и н И. А., Применение
органических реактивов в аналитической химии, Второе все-
союзное совещание, Тезисы докладов, Саратов, 1966, стр. 50.
4. Д з и о м к о В. М., Красавин И. А., Химические
реактивы и препараты, вып. 26, ИРЕА, 1964, стр. 29.
З-ПИ^ИДИЛ СУЛЬФАМИД
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
N
so2nh.
C5H6N2O.2S
Мол. вес 158,17
З-Пиридилсульфамид применяется для синтеза различ-
ных органических соединений и физиологически актив-
ных препаратов. В литературе имеется сообщение о син-
тезе 3-пиридилсульфамида J из 3-пиридилсульфохлорида
и аммиака в растворе бензола. Нами была проверена эта
методика; выход продукта составил 20%.
Предлагаемое изменение в методике получения 3-пири-
дилсульфамида заключается в том, что в качестве раство-
рителя применяется вода. При этом в несколько раз
увеличивается выход вещества и упрощается методика
проведения синтеза.
149
Схема синтеза
РЕАКТИВЫ
3-Пиридилсульфокислота, техн., т. пл. 332—340 °C.
Пятихлористый фосфор, ч., ТУ НКХП 386—41.
Аммиак водный, ГОСТ 3760—64.
Спирт метиловый, ГОСТ 6995—54.
Эфир этиловый, ГОСТ 6265—52.
Способ получения
Синтез 3-пиридилсульфохлорида. В колбу, снабжен-
ную нисходящим холодильником с хЛоркальциевой трубкой,
помещают 47,7 г (0,3 моль) 3-пиридилсульфокислоты
и 93,8 г (0,45 моль) пятихлористого фосфора (реактивы
тщательно измельчены). Реакционную смесь перемеши-
вают и нагревают на масляной бане. В течение 1 ч тем-
пературу в бане поднимают до 135 °C и выдерживают
смесь при этой температуре еще около часа (до полного
прекращения выделения хлористого водорода и хлороки-
си фосфора). Получают 92 г неочищенного продукта в виде
желто-красной жидкости.
Получение 3-пиридилсульфамида. В трехгорлую колбу,
снабженную механической мешалкой и капельной ворон-
кой, помещают 300 мл 25%-ного раствора аммиака,
к которому при перемешивании и охлаждении ледяной
водой прибавляют полученный ранее 3-пиридилсульфо-
хлорид. Затем реакционную массу упаривают на водяной
бане до загустения, охлаждают и экстрагируют метано-
лом. Спиртовой экстракт упаривают на водяной бане,
остаток высушивают и растворяют в абсолютном эфире.
После упаривания эфира получают 28,5 г амида в виде
желтых игл; т. пл. ПО—111 °C (после перекристаллиза-
150
ции из бензола), выход 60% (от теоретического). По лите-
ратурным данным х, т. пл. НО—112 °C.
Содержание азота в препарате (C6H6N2O2S):
Найдено, % .
Вычислено, %
18,08
17,71
Литература
1. G. Machek. Monatsch., 72, 77 (1938).
ПИКРАМИН С
2,7-БИС-(2'-0КСИ-3',5'-ДИНИТР0БЕН30ЛА30-1')-
1,8-ДИОКСИ-3,6-НАФТАЛИНДИСУЛЬФОКИСЛОТА
Л. А. ОХАНОВА, С. Б. САВВИН
Мол. вес 740,51
Пикрамин С впервые синтезирован в Институте гео-
химии и аналитической химии им. В. И. Вернадского
АН СССР и применялся для спектрофотометрического
и экстракционно-фотометрического определения Nb, Zr,
Al, Си и некоторых других элементов. Пикрамин С
получают азосочетанием концентрированных растворов
хромотроповой кислоты с избытком диазотированной
пикраминовой кислоты в присутствии гидроокиси каль-
ция и пиридина. Пиридин применяется для стабилизации
диазония пикраминовой кислоты и как катализатор
реакции азосочетания С В отсутствие пиридина сочетание
проходит только с образованием моноазокрасителя.
151
Схема синтеза
РЕАКТИВЫ
Пикраминовая кислота, ч., ГОСТ 881—53.
Хромотроповой' кислоты динатриевая соль, ч., ТУ МХП
4045—53.
Натрий азотистокислый, ч., ГОСТ 4197—66.
Кальция окись, ч., ГОСТ 8677—58.
Пиридин, ч., ГОСТ 2747—44.
Способ получения
Получение сульфата диазония пикраминовой кислоты 2.
Хорошо размешивают 12 г (0,06 моль) пикраминовой
кислоты в 40 мл воды, прибавляют к суспензии 2,4 г
едкого натра и перемешивают. Среда должна быть щелоч-
ной по фенолфталеиновой бумаге; при недостатке щелочи
добавляют по каплям 20%-ный раствор едкого натра.
Смесь нагревают до 50 °C, охлаждают в ледяной бане,
вводят 8 мл разбавленной (1:1) серной кислоты и затем
при перемешивании прибавляют по каплям 12 мл 5 М
раствора нитрита натрия. В ходе диазотирования среда
должна быть кислой (индикатор — бумага конго), в реак-
ционной смеси должен быть слабый избыток нитрита
(индикатор — иод-крахмальная бумага). После достиже-
ния устойчивого избытка нитрита перемешивание про-
должают еще 30 мин. Осадок соли диазония быстро отфиль-
тровывают на воронке Бюхнера, отжимают, промывают
152
холодной водой (2 раза порциями по 5 мл), переносят
на фильтровальную бумагу, измельчают в мокром состоя-
нии и сушат вдали от источников тепла. Получают 14,5 г
продукта.
Синтез пикрамина С. Смешивают 35 мл пиридина
с раствором 6,5 г окиси кальция в 40 мл воды и охлаждают
в ледяной бане. К суспензии прибавляют смесь 11,6 г
(0,045 моль) сухой соли диазония и 5,46 г (0,015 моль)
динатриевой соли хромотроповой кислоты, охлаждая реак-
ционную массу в ледяной бане. Смесь приобретает темно-
зеленую окраску. Через 6—8ч добавляют при охлаждении
по каплям 60 мл концентрированной соляной кислоты.
Осадок отфильтровывают и промывают 10—15 мл холод-
ной воды. Для очистки сырой продукт размешивают
в 200 мл воды, отфильтровывают от растворившейся
примеси моноазокрасителя и полученный осадок раство-
ряют при нагревании на водяной бане в смеси 350—
400 мл воды и 3 мл 20%-ного раствора едкого натра.
Раствор охлаждают и выделяют пикрамин С, добавляя
40 г мелко растертого хлористого натрия. На следующий
день осадок отфильтровывают и промывают 10—15 мл
холодной воды. Получают 6 г пикрамина С в виде тем-
ного, почти черного порошка.
Для определения содержания основного вещества
применяют метод спектрофотометрического титрования.
Аликвотную часть 0,1 %-ного раствора пикрамина С
титруют 0,3-10-3 М раствором ZrOCl2 в 6 н. растворе НО
так, чтобы кислотность в ходе титрования соответствовала
0,1 н. раствору НС1. Оптическую плотность измеряют
при 610 ммк. Мольную концентрацию пикрамина С рас-
считывают, имея в виду, что в состав образующегося
комплекса ZrR2 цирконий и реагент входят в отноше-
нии 1:2.
Литература
1. Цоллингер Г., Химия азокрасителей, Госхимиздат, 1960,
стр. 187.
2. Николенко Л. П., Лабораторный практикум по проме-
жуточным продуктам и красителям, 1961, стр. 181.
153
ПИРОМЕЛЛИТОВАЯ КИСЛОТА
БЕНЗОЛ-1,2,4,5-ТЕТРАКАРБОНОВАЯ
КИСЛОТА
М. Т. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Г. И КОРЕЛЬСКАЯ
НООС
Соон
СООН
СООН
С1„Н6О,
Мол. вес 254,15
Пиромеллитовая кислота применяется для синтеза
термостойких полимеров1 и органических полупровод-
никовых материалов 2. Мы уже сообщали ранее о получе-
нии этой кислоты окислением дуриловой кислоты и
дурола 3.
В данной статье предлагается другой, препаративный
метод получения пиромеллитовой кислоты окислением
ацетилпсевдокумола перманганатом калия.
Схема синтеза
сн3
COOK
на
СООН
коос^
СООН
соон
РЕАКТИВЫ
Ацетилпсевдокумол, ч., РТУ ОЭЗ ИРЕА 04—63.
Калий марганцовокислы., техн., ГОСТ 5777—51.
Натр едкий, ч., ГОСТ 4328—66.
Кислота соляная, ч., ГОСТ 3118—46.
Спирт этиловый, ректификованный, ГОСТ 5962—51,
154
Способ получения
В четырехгорл ую круглодонную колбу емкостью 3 л,
снабженную механической мешалкой с вазелиновым
затвором, обратным холодильником, термометром и ворон-
кой, вводят 1 л 6%-ного раствора едкого натра и 81 а
(0,5 моль) ацетилпсевдокумола. Смесь нагревают на мас-
ляной бане до 85—90 °C и при интенсивном перемешива-
нии прибавляют в течение 9 ч 650 г марганцовокислого
калия порциями по 15—20 г. Перед прибавлением новой
порции проверяют, раскислялась ли предыдущая: темпо-
малиновая окраска раствора должна перейти в бурую.
После прибавления первых 325 г марганцовокислого
калия в реакционную массу вводят 500 мл горячей воды
и заканчивают окисление прибавлением остального коли-
чества такими же порциями при 95 °C (в колбе). Окисле-
ние считается законченным, если вновь введенная порция
не реагирует в течение часа при 95—100 °C. Затем тем-
пературу реакционной массы снижают до 70 °C и прибав-
ляют 15—20 мл этилового спирта для разложения избыт-
ка марганцовокислого калия. Горячую смесь деканти-
руют и тут же отфильтровывают от осадка двуокиси
марганца. Осадок трижды порциями по 150 мл промывают
на фильтре горячей водой. Основной фильтрат (~1500 мл)
и промывные воды (450 мл) объединяют и упаривают
в вакууме на кипящей водяной бане до объема 700—
800 мл. Сконцентрированный фильтрат переливают
в двухлитровый химический стакан, охлаждают до 10 °C
ледяной водой и при перемешивании постепенно прибав-
ляют из капельной воронки 170 мл концентрированной
соляной кислоты до pH 1 по универсальной индикаторной
бумаге. Кислый раствор оставляют стоять в течение ночи
при комнатной температуре. Выпавший белый мелко-
кристаллический осадок отфильтровывают и тщательно
отжимают. Продукт в количестве 150 г перекристаллизо-
вывают из 1225 мл 10%-ного раствора соляной кислоты.
Полученный после перекристаллизации осадок отса-
сывают, промывают холодной водой до отрицательной
реакции промывных вод на СГ-ион и сушат при ПО—
120 °C в сушильном шкафу до постоянного веса. Полу-
чают 66 г продукта; т. пл. 273—275 °C, выход 52% (от тео-
ретического). Содержание основного вещества 99%. По
литературным данным 2, т. пл. продукта 273—275 °C.
155
Литература
1. В г е п n а п Р. J., Chem. Eng., 70, 68, 2 (1963); Chem. Week,
88, 46 (1961); Chem. Age, 88, 955, 2267 (1962).
2. Epstein A., W i 1 d у В. S., J. Chem. Phys., 32, 324 (1960).
3. Разумовская M. T., Белякова А. И., Его-
рова Л. А., Методы получения химических реактивов и пре-
паратов, вып. 12, ИРЕА, 1965, стр. 96.
N-ПРОПИЛАМИНОИЗОПРОПИЛДИАЛКИЛ-
ФОСФАТЫ
N-ПРОПИЛАМИНОИЗОПРОПИЛДИЭТИЛФОСФАТ
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
СН, О
I и /Оед
c3h7-nh-ch,-ch-o-p<
ХОС2Н6
С10Н24\’О4Р Мол. вес 253,28
N-Пропиламиноизопропилдиалкилфосфаты являются
фосфорилированными аналогами продуктов жизнедея-
тельности организмов. Подобные соединения применяют-
ся в качестве инсектицидов 1 и фармацевтических пре-
паратов 3. N - пропиламиноизопропилдиалкилфосфаты
могут быть получены при реакции диалкилхлорфосфатов
с алкоголятом натрия пропиламипопропанола-2 в ней-
тральных растворителях. Описанные нами соединения
получены впервые 3.
Схема синтеза
N-пропиламиноизопропилдиэтилфосфата
сн3
I Na
CsH7-NH-CH2-CH-OH------»
СН3
I юс2н6
--» CgHj-NH-CHj-CH-ONa---------->
CHS О
I II УОС2Н5
,—> СдН,—NH—СНа—СН—О—Р<
хосгнЕ
156
РЕАКТИВЫ
Пропиламинопропанол-2 л, т. кип. 169—170 °C, т. пл. 26 °C.
Натрий металлический, ч., ТУ МХП 1664—50.
Диэтилхлорфосфат, свежеперегнанный, т. кип. 81—83 °C при
10 мм рт. ст.
Бензол, сухой, ч. д. а., ГОСТ 5955—51.
Способ получения
В трехгорлую колбу, снабженную мешалкой и обрат-
ным холодильником, помещают 11,72 г (0,1 моль) про-
пиламинопропанола-2, 50 мл сухого бензола и прибав-
ляют 2,3 г (0,1 моль) металлического натрия (кусочками
по 0,2—0,3 г). Реакционную массу выдерживают, пере-
мешивая при температуре 60—70 °C, до полного раство-
рения натрия. К полученному раствору алкоголята
натрия, охлажденному ледяной водой до 5 °C, прибавляют
по каплям 17,25 г (0,1 моль) диэтилхлорфосфата, поддер-
живая температуру реакционной смеси 5—10 °C. Затем
смесь нагревают 15 мин на кипящей водяной бане, охлаж-
дают до комнатной температуры и выпавший осадок хло-
ристого натрия отделяют центрифугированием. Из филь-
трата отгоняют бензол в вакууме водоструйного насоса.
Из остатка удаляют непрореагировавшие исходные веще-
ства до постоянного веса (2—3 ч; 40 °C; 3—4 мм рт. ст.).
Получают 22 г продукта; выход 87% (от теоретиче-
ского), = 1,036, ng1 = 1,4354. При перегонке продукт
разлагается.
Состав синтезированного продукта
с
47,45
47,42
(C10H24NO4P):
н
9,54 :
9,55,
р
11,54
12,23
Найдено, % .
Вычислено, % .
Аналогично получают другие N-пропиламиноизопро-
пилдиалкилфосфаты, константы которых приведены ниже:
Название соединения Выход, % „20 nD d?
N- Пропиламиноизопропилдиизопро-
пилфосфат М-Пропиламиноизопропилди-я-про- 86 1,4308 0,9961
пилфосфат N-Пропиламиноизопропилди-н-бутил- 84 1,4380 1,0120
фосфат 88 1,4394 0,9913 157
Литература
1. Пат. ФРГ 1143512 (1957); РЖХим, Н426 (1965).
2. Фр. пат. 1072 (1962); РЖХим, 11128 (1963).
3. Дрегваль Г. Ф., Самуилов А. М., авт. свид. СССР
187021; Билл, изобр., № 20 (1966).
4. Малиновский М. С., Окиси олефинов и их производ-
ные, Госхимиздат, 1961, стр. 267.
СИММЕТРИЧНЫЕ ДИАЗОАМИНОСОЕДИНЕНИЯ
(ДИАРИЛТРИАЗЕНЫ)
О. М. СТАШКЕВИЧ, г. Т. ПИЛЮГИН, В. В. СТАШКЕВИЧ
Из обширного класса диазоаминосоединений (диарил-
триазенов) в настоящее время в качестве реактива исполь-
зуется лишь диазоаминобензол. Между тем производные
диазоаминобензол" могут найти применение как устой-
чивые формы диазосоединений или как исходные вещества
для синтеза амипоазосоединений.
Общий способ получения диарилтриазенов заключает-
ся в действии аминов на соли диазония. Для синтеза
производных диазоаминобензола, содержащих метильную
группу, алкоксигруппу и атомы галоида, мы использова-
ли методику с применением ацетата натрия \ уточнив
в процессе работы условия синтеза и очистки диазоамино-
соединений.
4,4'-ДИАЗОАМИНОТОЛУОЛ
НдС——N=N—NH СНд
Cj^HjgNg Мол. вес 225,14
4,4'-Диазоаминотолуол получают действием водного
раствора азотистой кислоты на раствор л-толуидина
в этиловом спирте2, а также действием л-толуидина
на раствор солянокислого n-толилдиазония 3> 4.
Схема синтеза
нс—^^-NHg + ^HgC—^2^,—N=n] СГ —
------- HgC-/“^-NH-N=N—
158
РЕАКТИВЫ
п-Толуидин, ч., ВТУ МХП 2965—54.
Кислота соляная, х. ч., пл. 1,19, ГОСТ 3118—46.
Натрий азотистокислый, ч. д. а., ГОСТ 4197—66.
Спирт этиловый, ректификованный, ГОСТ 5962—51.
Натрий уксуснокислый, ч. д. а., ГОСТ 199—52.
Способ получения
В термостойкий стакан емкостью 1 л помещают 54 г
(0,5 моль) п-толуидина, 120 мл концентрированной соля-
ной кислоты, 100 мл воды и кипятят смесь до растворе-
ния n-толуидина. Горячий раствор при перемешивании
быстро охлаждают в ледяной бане для получения воз-
можно мелких кристаллов солянокислого п-толуидина.
Суспензию охлаждают смесью льда и соли (внешнее
охлаждение) до —5 °C и диазотируют (при перемешива-
нии механической мешалкой) раствором 35 г азотистокис-
лого натрия в 100 мл воды. Конец диазотирования опре-
деляют с помощью иод-крахмальной бумажки. К полу-
ченному раствору соли диазония добавляют охлажден-
ную до 0 °C смесь 54 г n-толуидина в 40 мл соляной кис-
лоты, 40 мл воды и 100 мл этилового спирта.
Отдельно в батарейном стакане емкостью 3 л раство-
ряют 400 г уксуснокислого натрия в 1,5 л воды и прибав-
ляют 300 г истолченного льда. К раствору уксуснокислого
натрия при энергичном перемешивании тонкой струей
приливают раствор соли диазония и n-толуидина. При
этом выпадает желтый осадок 4,4'-диазоаминотолуолэ.
После прибавления раствора смесь перемешивают еще
10 мин, оставляют стоять 1 ч и фильтруют под вакуумом.
Осадок промывают на фильтре водой до исчезновения
запаха уксусной кислоты и сушат на воздухе.
Получают 95 г продукта, достаточно чистого для син-
тетических целей; т. пл. НО—111 °C, выход 80% (от тео-
ретического). При быстрой кристаллизации из спирта
(от длительного нагревания вещество разлагается) выпа-
дает продукт в виде желто-оранжевых игл с т. пл. 116—
117°С. По литературным данным, т. пл. 115—116 °C3
или 118 °C1.
159
2,2'-ДИАЗОАМИНОТОЛУОЛ;
2,2'- И 4,4'-ДИМЕТОКСИДИАЗОАМИНОБЕНЗОЛ;
4,4'-Д ИЭТОКСИД ИАЗОАМИНОБЕНЗОЛ;
4,4'-ДИХЛОРД ИАЗОАМИНОБЕНЗОЛ;
4,4'-ДИБРОМДИАЗОАМИНОБЕНЗОЛ
Схема синтеза
Rv=\ R^==\ + CH3COONa
0^ + O~n=n
R
R
где R: o-CH3; о- или n-OCH3; n-OC^Hj; n-Cl; n-Br.
РЕАКТИВЫ
о-Толуидин, ч., ТУ МХП 95—47.
о-Анизидин, ч., ТУ МХП 2356—50.
п-Анизидин, ч., ТУ МХП 1485—51.
п-Фенетидин, ч., ВТУ РУ 586-52.
п-Хлоранилин 5, т. пл. 71 °C.
п-Броманилин, ч., МРТУ 6-00-1227—64.
Кислота соляная, х. ч., ГОСТ 3118—46.
Натрий азотистокислый, ч. д. а., ГОСТ 4197—66.
Натрий уксуснокислый, ч. д. а., ГОСТ 199—52.
Спирт этиловый, ректификованный, ГОСТ 5962—51.
Бензол, ч. д. а., ГОСТ 5955—51.
Способ получения
Указанные диазоаминосоединения получены анало-
гично 4,4'-диазоаминотолуолу. Условия получения, кон-
станты и данные анализа для этих соединений приведены
в таблице.
Литература
1. Голодников Г. В., Низовкина Т. В., Рыс-
ка л ь ч у к А. Т., Практикум по органическому синтезу,
Изд. ЛГУ, 1957, стр. 156.
2. G г i е s s Р., Ann., 121, 277 (1862).
3. Bernthsen A., Goske А., Вег., 20, 924 (1887).
4. N о f f J., Ann., 311, 92 (1900).
5. Общий практикум по органической химии, Изд. «Мир», 1965,
стр. 513.
6. Tischer В., Wimmer Н., Вег., 20, 1581 (1887).
7. Meh пег D., J. Prakt. Chem., 65, 444 (1901).
8. Н u п t е г L., J. Chem. Soc., 1937, 320.
9. М е 1 d о 1 а К-, Streatfeild J., J. Chem. Soc., 53,
670 (1888).
10. H a n t z c h A., Perkin F. M., Ber., 30, 1412 (1897).
160
Условия получения, константы и данные анализа симметричных диазоаминосоединений
К <О СО Оо «о о>
| Данные анализа | % ‘N оиэ1гэныча О М* -чр 00 00 00 00 ср СО Xif LO -М
% м оиэгивн Сч£ C?S Сч£? со 2 ед ^2 ссГ^ X Ш ££ OJ
оэн унн -dKirXMairow со оэ о т со сч сч сч со —о LQ Ь- Ь- Ю <£> Ю СЧ LQ Ю 00 Ср LQ сч сч сч сч сч со
1 Брутто- . формула <n <м « . га -s £ °« 3 °- 5 < % Ч «Й 3 ®: % з [_) i-( ТМ тМ t V и< ООО ° и
Эо КИНЭ1ГЯВ1ГЦ BdKxBdanmj, СЧ 1 LQ 1 О | ь~ О СП СЧ ю ь~ 1 о о —«—< сч -ч- ю
ИИПВСИ1Г -ireiandM виг 4iroiHiioa±3Bd 5^5^ 5 5 О о. о о о о СО tt* СО СО СО СО В S S S к В 3 s 3 « й «
% Гохкд о о о сч сн ю Ь- Ь- 00 СО Ь- 00
i Реактивы 1 для азосочетания • - го . - CV -г го го го • ’ Ч «0 го го ГО • - ГО s го з- = о во хо го •* ж х SO So So х о So КО S.OO "oQ я я я ЕГ<мо к —ю s<»Q gUZ =U'Z. 5<JZ Sr Z сЛ z ?Г 2 £хо ?хо «ко аУ .о §9 .о °У..о счО с о е „ О •е--’ га о g ” га О ю я О S О §U Sj У V и ^3 К и Л 5 ш га т-S? гаЧт? ra ^S3 s т« с я-А1 с < s т" ю тН «о ?Ч to ?Ч го ш к гН <м о с гЧ го о с гЧ •хгО •чгО хг (-»О —- иО — оU ’ГГ сч сч ед со г- U0 о со со — —«
' для диазотиро- вания го г-: го гг го ~ s $ S У* gs? ж О о» X О S1 х U сч rx П ас о Fg Fg rg |ой S3® SjS S2 „ « es « 24Z й % & О z • .O SO io" 6o" ;o'" e..~-Z |CZ rag^Z га“Й »«XZ « -4- СЧ СЧ co CO b. ю <o <O CO —< —-
Название соедине- ния i Si8 SS 5i Si я i § so so u $y ^,3^ ra 2 5 2 5 “ о 0^8 So о ЬЯ P я О о go O° tn - О O 2 н® 5 Cx. ?• ra 5 £ ra Sra mS x s >os s P So so as ss s J Чч ЧйчЧячЧ§ч 4§ч ^ЧЗч ' О ._' S О „• S О ./ со О в,' со О к го О сч н с^ггл-^ссго^гого га со га со Сч сч” *«±* Tf”
И—287
161
СЛОЖНЫЕ ЭФИРЫ а-ТИЕНИЛГЛИОКСАЛЕВОЙ
И а-ТИЕНИЛГЛИКОЛЕВОЙ КИСЛОТ
И. И. ЛАПКИН, Н. А. КАРАВАНОВ, Ю. П. ДОРМИДОНТОВ
При взаимодействии сложных эфиров щавелевой кис-
лоты с эквимолярными количествами мэгнийорганиче-
ских соединений, содержащих арильные, алициклические
или алифатические радикалы, получаются сложные эфи-
ры вторичных а-оксикислот 1> 9. Изучение механизма
этой реакции показало, что образующееся вначале ком-
плексное соединение распадается по уравнению
OMgBr OMgBr
R'—С—COOCH2R----» R'—CH—COOCH2R + RCHO
icHaR
что приводит после гидролиза к образованию сложных
эфиров вторичных а-оксикислот.
Термическая стойкость комплексного соединения
зависит от характера входящих в него радикалов. Если
R' — арильный или алициклический радикал, то ком-
плексное соединение распадается при сравнительно низ-
кой температуре (40—42 °C); если же R' — алкильный
радикал, то для распада более стойкого в этом случае
комплексного соединения требуется и более высокая
температура (ПО—120 °C). Термическая стойкость ком-
плексного соединения, содержащего а-тиенильный ради-
кал, несмотря на его ярко выраженный ароматический
характер, также высока, поэтому в обычных условиях
проведения магнийорганических реакций комплексное
соединение не распадается, и после гидролиза вместо
сложных эфиров а-тиенилгликолевой кислоты (вторич-
ной а-оксикислоты) образуются с выходом от 30 до 50%
сложные эфиры а-тиенилглиоксалевой кислоты.
Термический распад комплексного соединения, содер-
жащего а-тиенильный радикал, происходит при темпе-
ратуре 100—110 °C. Поэтому проведение реакции между
сложными эфирами щавелевой кислоты и галоидным
а-тиенилмагнием при указанной температуре приводит
к образованию сложных эфиров а-тиенилгликолевой
кислоты. Практически синтез сложных эфиров а-тиенил-
162
гликолевой кислоты лучше проводить путем взаимодей-
ствия сложных эфиров а-тиенилглиоксалевой кислоты
с броммагнийалкоголятами при ПО °C. В этом случае
образуется комплексное соединение такого же строения,
как указывалось выше 3.
СЛОЖНЫЕ ЭФИРЫ а-ТИЕНИЛГЛИОКСАЛЕВОЙ КИСЛОТЫ
Схема синтеза изобутилового эфира
1| |1 -|- изо-С4Н9ООС—СООС4Н9-шо-»
Y^Mgi
____ OMgl ____
---* I\SJ-C-COOC4H9-U30----, \со -соос4не-изо
ОС4Н9-изо
РЕАКТИВЫ
Тиофен, ч., ВТУ МХП 3329—52.
Натрий двууглекислый, ч., ГОСТ 4201—66.
Бисульфит натрия, ч,, ТУ МХП 1944—49.
Натрий сернокислый, безводный, ч., ГОСТ 4166—66.
Диизобутиловый эфир щавелевой кислоты.
Кислота соляная, ч., ГОСТ 3118—46.
Эфир этиловый, ГОСТ 6265—52.
Йодистый а-тиенилмагний, получен из 5 г (0,2 моль) магния
и 42 г (0,2 моль) а-иодтиофена 4.
Способ получения
Диизобутиловый эфир щавелевой кислоты в количе-
стте 40,4 г (0,2 моль) растворяют в безводном этиловом
эфире в объемном соотношении 1:3. К полученному
раствору, охлажденному в бане со смесью льда и соли,
прибавляют при перемешивании механической мешалкой
эфирный раствор йодистого тиенилмагния, взятого
в эквимолярном количестве. Реакционную смесь кипятят
на водяной бане 2 ч и затем проводят гидролиз при охлаж-
дении в бане со льдом и перемешивании. Для этого в колбу
вводят 50 мл воды и 100 мл 10%-ной соляной кислоты.
Содержимое колбы переносят в делительную воронку;
эфирный слой отделяют, промывают 5%-ным раствором
бикарбоната натрия, 10%-ным раствором бисульфита
11* 163
натрия (для удаления свободного иода) и сушат сульфа-
том натрия (15 г). Эфир отгоняют и продукт реакции пере-
гоняют в вакууме. После двукратной перегонки полу-
чают 19 г изобутилового эфира а-тиенилглиоксалевой
кислоты; т. кип. 106—107 °C при 2,5 мм рт. ст., выход
45% (от теоретического), п2£ = 1,5240 (см. примечания
1 и 2).
1 По аналогичной методике получают следующие эфиры
а-тиенилглиоксалевой кислоты: этиловый эфир (т. кип.
101 °C при 3,5 мм рт. ст., выход 24%, а* = 1,2364,
/2^ = 1,5485); «-пропиловый эфир (т. кип. 97—99 °C
при 2 мм рт. ст., выход 25%, d’9 = 1,1809, = 1,5345);
изопропиловый эфир (т. кип. 97—98 °C при 2,5 мм рт. ст.,
выход 30%, dl° ~ 1,1819, ri® ~ 1,5230); н-бутиловый эфир
(т. кип. 110—112 °C при 2,5 мм рт. ст., выход 35%,
d'9 = 1,1679, п* = 1,5350).
СЛОЖНЫЕ ЭФИРЫ а-ТИЕНИЛГЛИКОЛЕВОЙ КИСЛОТЫ
Схема синтеза «-пропилового эфира
4- «-CgH7OMgBr
।—СООС3Н--н
____ OMgBr
У-^-СООСзН,.» -^-4
OMgBr
JI Н2о
-С -СООС3Н7-н -----»
S I
н
РЕАКТИВЫ
Пропил бромистый, ч., МРТУ 6-09-446—63.
н-Пропиловый спирт, СТ ГОПХ 15—2354.
Эфир этиловый, ГОСТ 6265—52.
Толуол, ч., ГОСТ 5789—51.
н-Пропиловый эфир а-тиенилглиоксалевой кислоты (получение
см. выше).
Натрий двууглекислый, ч., ГОСТ 4201—66.
Кислота соляная, ч., ГОСТ 3118—46.
164
Способ получения
В трехгорлую круглодонную колбу емкостью 0,5 л,
снабженную капельной воронкой, механической мешал-
кой с масляным затвором и обратным холодильником
с хлоркальциевой трубкой, помещают 2,4 г (0,1 моль)
магния в виде тонкой стружки и покрывают его слоем
безводного диэтилового эфира. В капельную воронку
помещают раствор 12,3 г (0,1 моль) бромистого «-пропила
в 40 мл безводного эфира. Для начала реакции приливают
1,5—2 мл этого раствора и слегка подогревают содержи-
мое колбы на водяной бане. Когда реакция начнется,
включают мешалку и по каплям прибавляют раствор
бромистого «-пропила с такой скоростью, чтобы смесь
в колбе равномерно кипела. После введения всего раство-
ра и прекращения кипения эфира реакционную массу
нагревают на водяной бане около 30 мин до почти полного
растворения магния, охлаждают и, продолжая перемеши-
вание, из капельной воронки прибавляют по каплям
6,2 г (0,1 моль) «-пропилового спирта, растворенного
в 15 мл безводного эфира. К образовавшемуся эфирному
раствору броммагнийпропилата приливают довольно
быстро из капельной воронки раствор 19,8 г (0,1 моль)
н-пропнлового эфира а-тиенилглиоксалевой кислоты
в 50 мл эфира, а затем 100 мл безводного толуола. Заменив
вертикальный холодильник на нисходящий, отгоняют
эфир и выдерживают реакционную смесь при температуре
кипения толуола 1 ч. Гидролиз реакционной массы про-
водят водой (30 мл) и 10%-ной соляной кислотой (50 мл).
Органический слой отделяют, промывают 10%-ным рас-
твором бикарбоната натрия, водой и сушат безводным
сульфатом натрия. После перегонки получают «-пропи-
ловый эфир а-тиенилгликолевой кислоты; т. кип. 103—
105 °C при 2 мм рт. ст., выход 78% (от теоретического).
По аналогичной методике получают следующие эфи-
ры а-тиенилгликолевой кислоты: изопропиловый эфир
(т. кип. 98—99 °C при 2 мм рт. ст., выход- 80%); «-бути-
ловый эфир (т. кип. 110—112 СС при 2 мм рт. ст., выход
82%); изобутиловый эфир (т. кип. 105—107 °C при 2 мм
рт. ст., выход 85%).
Примечания. 1. Сложные эфиры а-тиенилглиоксалевой
кислоты могут быть получены этерификацией. Для этого кислоту
растворяют во взятом для этерификации безводном спирте и про-
пускают в раствор сухой хлористый водород.
16S
2. В случае применения низкомолекулярных эфиров щавелевой
кислоты (этилового, пропилового) наблюдается образование неболь-
шого количества продуктов второй стадии реакции — эфиров
ди-а-тиенилгликолевой кислоты, которые имеют более высокую
температуру кипения и легко отделяются перегонкой от эфиров
а-тиенилглиоксалевой кислоты.
Литература
1. Л а п к и н И. И., ЖОХ, 16, 721 (1946); 17, 1332, 1339 (1947).
2. Л а п к и н И. И., Караванов Н. А., ЖОХ, 30, 1638
(I960); 30, 2677 (1960).
3. Beilst., 18, 407 (4 изд.).
4. Синтезы органических препаратов, т. 2, Издатинлит, 1949,
стр. 287.
СУЛЬФИДЫ ФУРАНОВОГО
И ТИОФЕНОВОГО РЯДОВ
И. И. ЛАПКИН, Н. В. БОГОСЛОВСКИЙ
Предлагаемый способ синтеза сульфидов гетероцикли-
ческого ряда основан на том, что при взаимодействии
галогенмагнийкарбинолятов ароматического или гетеро-
циклического ряда и сложных эфиров муравьиной или
щавелевой кислоты (взятых в эквимолярном количестве)
образуются соответствующие галоидиды1-4. Если эту
реакцию проводить в присутствии галогенмэгнийтиофепо-
лятов, то последние реагируют с галоидидами, что при-
водит к образованию сульфидов. Для получения сульфи-
дов не обязательно исходить из предварительно синтези-
рованных карбинолов. Очень удобно эту реакцию сочетать
с получением самого карбинола магнийорганическим
синтезом. При этом в реакционной смеси содержится
уже готовый галогенмагнийкарбинолят, необходим толь-
ко избыток реактива Гриньяра, чтобы его хватило для
превращения тиофенола в галогенмагнийтиофенолят.
ФЕНИЛФУРФУРИЛСУЛЬФИД
cnH10os
Мол. вес 190,26
166
Схема синтеза
VXCH2OH -------
C2H6MgBr----------»- (I IL
\CH2OMgBr
CoHaSH
A
^Z\SMgBr
COOCtHs
COOC2H5
OC2H5
I 0
C<( >Mg
0
COOC2H5
—МгВг2
u
о
РЕАКТИВЫ
Фурфуриловый спирт, ч., ТУ МХП 2623—51.
Тиофенол, ч., ВТУ ХСЦХ 87—58.
Диэтилонсалат, ч., ТУ ХЗР 101—60.
Этил бромистый, абсолютный, ч., ТУ МХП 80—47.
Магний, стружка.
Эфир этиловый, ГОСТ 6265—52.
Способ получения
В трехгорлую круглодонную колбу емкостью 0,5 л,
снабженную капельной воронкой, механической мешалкой
с масляным затвором и обратным холодильником с хлор-
кальциевой трубкой, помещают 4,8 г (0,2 моль) магния
в виде тонкой стружки и покрывают его слоем безводного
диэтилового эфира. В капельную воронку помещают
раствор 21,8 г (0,2 моль) бромистого этила в 70 мл абсо-
лютного эфира. Для начала реакции приливают 1,5—
2 мл этого раствора и слегка нагревают содержимое колбы
на водяной бане. После начала реакции включают мешал-
ку и по каплям прибавляют весь раствор бромистого этила
с такой скоростью, чтобы реакционная смесь в колбе
167
равномерно кипела. Мосле прибавления всего раствора,
на что требуется около 1,5 ч, и прекращения кипения
эфира реакционная масса нагревается на водяной бане
около 30 мин почти до полного растворения магния.
Если не весь магний растворился, необходимо прилить
1—2 мл бромистого этила и нагреть реакционную массу
до растворения магния. Колбу охлаждают в бане с водой
и льдом и, не прекращая охлаждения и перемешивания,
приливают по каплям раствор 9,8 г (0,1 моль) свежепере-
гнанного фурфурилового спирта и 11 г (0,1 моль) свеже-
перегнанного тиофенола в 50 мл абсолютного эфира.
Реакция протекает бурно. Затем реакционную массу
кипятят 15—20 мин на водяной бане. На дне колбы выде-
ляется серо-зеленое масло; в эфирном слое, в масле
и на стенках колбы образуется белый осадок. К этой
массе при перемешивании прибавляют по каплям, но
довольно быстро (10—15 мин) раствор 14,6 г (0,1 моль)
дпэтилоксалата в 30 мл диэтилового эфира; при этом
масса несколько желтеет. Перемешивание и нагревание
продолжают до загустения нижнего слоя. Затем мешалку
выключают, а нагревание продолжают еще 1 —1,5 ч. За
это время осадок обычно кристаллизуется. В случае,
если этого не произойдет, нагревание продолжают до
кристаллизации. После охлаждения осадок в колбе измель-
чают шпателем под слоем эфира. Для дальнейшего измель-
чения осадка продолжают нагревание и перемешивание
еще 1—2 ч. Затем реакционную массу выливают в банку
емкостью 1 л, содержащую 250 г колотого льда и 150 мл
насыщенного раствора хлористого аммония. Эфирный
слой отделяют, водный слой дважды экстрагируют эфиром
порциями по 50 мл, и эфирные вытяжки присоединяют
к основному эфирному слою. Эфирный раствор промы-
вают 50 мл воды, 10%-ным раствором едкого натра (два
раза порциями по 20 мл), водой до нейтральной реакции
(индикатор - метиловый оранжевый) и сушат 10—12 г
безводного сульфата натрия. Эфирный раствор фильтруют
через вату. Эфир отгоняют на водяной бане, э оставшееся
легкоподвижное коричневое масло перегоняют в вакууме.
Получают 12,2гфенилфурфурилсульфида; т. кип. 138—
140 °C при 8 мм рт. ст., выход 64% (от теоретического),
d™ = 1,1463, п% = 1,5952.
Аналогично получают п-толилфурфурилсульфид;
т. кип. 149—150 °C при 9 мм рт. ст., выход 68% (от тео-
168
ретического), tig = 1,5780, dg = 1,080, а также ж-толил-
фурфурилсульфид с т. кип. 151—153 °C при 10 мм рт. ст.
и выходом 53% (от теоретического); ng = 1,5837,
1,1171.
CisH18OS
n-ТОЛ ИЛ-(а-ФУРИЛ-/г-ТОЛ ИЛМЕТИЛ)-СУЛЬФИД
РЕАКТИВЫ
Фурфурол, ТУ ХЗР 27—1831.
п-Тиокрезол, ТУ ХЗР 79-514-Х—60.
Диэтилоксалат, ТУ ХЗР 101—60.
п-Бромтолуол, ВТУ 4-280а—51.
Способ получения
В трехгорлую круглодонную колбу емкостью 0,5 л,
снабженную капельной воронкой, механической мешал-
кой с масляным затвором и обратным холодильником
с хлор кальциевой трубкой, помещают 4,8 г (0,2 моль) маг-
ния в виде тонкой стружки и покрывают слоем абсолют-
ного эфира. В капельную воронку помещают раствор
34,2 г (0,2 моль) n-бромтолуола в 70 мл абсолютного
эфира. Для начала реакции в колбу вводят 2—2,5 мл
этого раствора и нагревают 10—15 мин на водяной бане
до кипения эфира. Затем включают мешалку и по каплям
прибавляют остальное количество бромида с такой ско-
ростью, чтобы раствор в колбе равномерно кипел без
подогревания. На эту стадию синтеза требуется около
1,5 ч. После прекращения кипения реакционную массу
нагревают на водяной бане 1—1,5 ч до почти полного
169
растворения магния, после чего колбу охлаждают в банё
с водой и льдом и, не прекращая охлаждения и переме-
шивания, прибавляют по каплям раствор 9,6 г (0,1 моль)
свежеперегнанного фурфурола в 10 мл абсолютного
эфира. Реакция протекает бурно, после ее окончания
реакционную массу нагревают на водяной бане 1,5 ч.
При этом образуется белый осадок.
Затем в колбу приливают по каплям раствор 12,4 г
(0,1 моль) п-тиокрезола в 25 мл абсолютного эфира,
нагревают 10—15 мин, после чего довольно быстро при-
бавляют по каплям раствор 14,6 г (0,1 моль) диэтилокса-
лата в 30 мл абсолютного эфира. После загустевания осадка
(см. примечание) нагревание продолжают без перемеши-
вания, а когда осадок начнет кристаллизоваться, его
размельчают шпателем и продолжают нагревать при меха-
ническом перемешивании 2,5—3 ч. Затем содержимое
колбы выливают в банку емкостью 1 л, содержащую 250 г
колотого льда и 150 мл насыщенного раствора хлористого
аммония. Массу перемешивают стеклянной палочкой
до полного растворения осадка и образования двух слоев.
Жидкость фильтруют через вату в делительную воронку.
Эфирный слой отделяют; водный слой экстрагируют два
раза эфиром. Эфирные вытяжки присоединяют к основ-
ному эфирному слою, промывают дважды 10%-ным рас-
твором едкого натра и четыре-пять раз водой до ней-
тральной реакции. Эфирный слой сушат 15 г безводного
сульфата натрия 12—15 ч. Эфир отгоняют на водяной бане
при температуре не выше 50 °C и получают красно-корич-
невый маслообразный осадок, который в вакуум-экси-
каторе кристаллизуется через сутки. После трехкратной
перекристаллизации из этилового спирта (один раз с акти-
вированным углем) получают 17 г п-толил-(а'-фурил-
п-толилметил)-сульфида в виде бесцветных кристаллов;
т. пл. 99 °C, выход 61% (от теоретического). Из маточных
спиртовых растворов можно выделить еще 1,5—1,7 г
сильно окрашенных кристаллов сульфида. Аналогичным
способом получают п-толил-(а-фурил-п-ксилилметил)-
сульфид, т. пл. 73,5 °C, выход 60,5% (от теоретического);
а также п-толил-(а-фурил-о-толилметил)-сульфид, т. пл.
49—50 °C, выход 58,2% (от теоретического).
Примечание. В процессе прибавления диэтилоксалата
осадок оседает па дно, уплотняется и становится настолько вязким,
что механическое перемешивание невозможно.
171
ФЕНИД-(а-ТЙЁНЙЛФЕНИЛМЕТЙЛ)-СУЛЬФИД
^17^14^2
Мол. вес 282,42
Схема синтеза
\MgBr
ОМсВг
| __
+ 0™---> J
C2H5MgBr + HS-^ --» ^A-S-MgBr
OMgBr
oJh-O +1°0СЛ —
\=/ v cooc2H5
РЕАКТИВЫ
Тиофен, ч., ВТУ МХП 3329—52.
Бензальдегид синтетический, ч., ГОСТ 157—51.
Этил бромистый абсолютный, ч., ТУ МХП 80—47.
Тиофенол, ч., ВТУ ХСНХ 87—58.
Диэтилоксалат, ч., ТУ ХЗО 101—60.
172
Способ получений
В трехгорлую круглодонную колбу емкостью 0,5 л,
снабженную капельной воронкой, механической мешал-
кой с масляным затвором и обратным холодильником
с хлоркальциевой трубкой, помещают 2,4 г (0,1 моль)
магния и покрывают слоем эфира. В капельную воронку
помещают раствор 16,3 г (0,1 моль) свежеперегнанного
2-бромтиофена (см. примечание 1) в 50 мл абсолютного
эфира. В колбу вводят 1 —1,5 мл этого раствора и нагре-
вают до начала реакции, згтем включают мешалку и по
каплям прибавляют остальное количество 2-бромтиофена
с такой скоростью, чтобы эфир равномерно кипел без
нагревания. Затем реакционную массу кипятят на водя-
ной бане 3 ч почти до полного растворения магния (неболь-
шой остаток магния в дальнейшем реакции не мешает).
Не прекращая перемешивания, к образовавшемуся
эфирному раствору броммзгний-2-тиенила при охлажде-
нии ледяной водой прибавляют по каплям раствор 10,6 г
(0,1 моль) свежеперегнанного бензойного альдегида.
Реакция протекает бурно, и образуется белый осадок.
По окончании прибавления раствора бензойного альде-
гида реакционную массу кипятят на водяной бане 1,5 ч.
Выделяющийся броммагний-2-тиенилфенилкарбинолят
образует суспензию.
В колбе емкостью 250 мл из 2,4 г (0,1 моль) магния
и раствора 10,9 г (0,1 моль) бромистого этила в 30 мл
абсолютного эфира обычным способом готовят этилмаг-
нийбромид. После растворения магния к полученному
магпийорганическому соединению по каплям прибав-
ляют раствор 11 г (0,1 моль) тиофенола в 25 мл абсолют-
ного эфира. Реакция идет бурно. После ее прекращения
реакционную массу кипятят при перемешивании на водя-
ной бане 15 мин (иногда выпадает белый осадок броммаг-
пийтиофеполята).
Полученную реакционную массу по возможности коли-
чественно переносят в колбу с броммагний-2-тиенилфенил-
карбинолятом, хорошо перемешивают, после чего по кап-
лям, не прекращая энергичного перемешивания, прибав-
ляют раствор 14,6 г (0,1 моль) диэтилоксалата в 30 мл
абсолютного эфира. При этом находящийся во взвешен-
ном состоянии осадок опускается на дно и густеет. Меха-
ническое перемешивание прекращают, а нагревание про-
173
должают. Реакционная масса окрашивается в красно-
коричневый цвет и в течение 1 —1,5 ч кристаллизуется
(см. примечание 2). Кристаллический осадок размель-
чают шпателем и продолжают нагревание с механическим
перемешиванием еще 1,5—2 ч. Затем содержимое колбы
переносят в банку емкостью 1 л, содержащую 250 г коло-
того льда, хорошо перемешивают стеклянной палочкой
и осадок магниевых солей растворяют, добавляя 50 мл
разбавленной (1 : 2) серной кислоты. Образуется два
слоя. Жидкость фильтруют через вату в делительную
воронку и эфирный слой отделяют. Водный слой дважды
экстрагируют эфиром порциями по 50 мл. Эфирные вытяж-
ки присоединяют к основному эфирному слою. Эфир
промывают один раз водой, два раза 10%-ным раствором
едкого натра и снова 4—5 раз водой до нейтральной
реакции. Эфирный раствор (красно-коричневого цвета)
сушат 15 г безводного сульфата натрия. Эфир отгоняют
на водяной бане, остаток перекристаллизовывают из эти-
лового спирта. После трехкратной перекристаллизации
получают 11,9 г фенил-(ос-тиенилфеиилметил)-сульфида
в виде бесцветных игольчатых кристаллов; т. пл. 46 °C,
выход 41% (от теоретического).
Примечания. 1. 2-Бромтиофен получают бромированием
тиофена в растворе четыреххлористого углерода при охлаждении
льдом ъ.
2. Если кристаллизация за это время не закончилась, переме-
шивание с нагреванием продолжают до окончания кристаллизации.
Литература
1. Л а п к и н И. И., Лапкина О. М., ЖОХ, 25, 298 (1955).
2. Л ап кив И. И., ЖОХ, 25, 536 (1955)
3. Лапкин И. И., Лапкина О. М., Рыбакова М. Н.,
ЖОХ, 28, 391 (1958).
4. Л а п к и н И. И., Панова Н. И., ЖОХ, 32, 795 (1962).
5. В eike F. F., Burckholter J. Н., J. Am. Chem. Soc.,
64, 473 (1942).
2,2,4,4 ТЕТРААЛКИЛОКСЕТАН-3-КАРБОНОВЫЕ
КИСЛОТЫ
И. к. КОРОБИЦЫНА, Л. Л. РОДИНА
R Н
R'-j-рСООН
о—j-R
R'
174
2,2,4,4-Тетраалкилоксетан-З-карбоновые кислоты до
сих пор не были известны. Мы установили 1, что 2,2,4,4-
тетраалкилоксетан-3-карбоновые кислоты образуются
с выходом 60—97% при облучении 4-диазо-2,2,5,5-тетра-
алкилфуранидонов-3 в водно-диоксановом растворе ртут-
ной лампой ПРК.-4 в течение 50—110 ч*. В этих условиях
4-диазо-2,2,5,5-тетраалкилфуранидоны-3 претерпевают
перегруппировку Вольфа с сужением цикла 2> 3.
Исходные 4-диазо-2,2,5,5-тетраалкилфуранидоны-3 мы
получали окислением моногидразонов 2,2,5,5-тетраалкил-
фуранидиндионов-3,4 желтой окисью ртути4. Выход
80—89%.
Схема синтеза
О N=N R н
УФ-облучение R/_i Г*СООН
” О—pR
R'
I п
где R, R' = СН3 (соединение a) R+R' = —(СН,)4— (соединение в)
R = СН3; R' = С2Н6 (соединение б) R+R' = —(СН2)5— (соединение г)
РЕАКТИВЫ
4-Диазо-2,2,5,5-тетраалкилфуранидоны-3 (см. стр. 60).
Диоксин, х. ч., ГОСТ 20455—-63.
Способ получения
Синтез 2,2,4,4-тетраметилоксетан-З-карбоновой кис-
лоты (Па). Раствор 2,2 г (0,013 моль) 4-диазо-2,2,5,5-
тетраметилфуранидона-3 (1а) в 84 мл диоксана и 6 мл
воды облучают в кварцевой колбочке ртутной лампой
ПРК.-4 в течение 50 ч. Растворитель отгоняют в вакууме
водоструйного насоса, выпавший кристаллический оса-
док отфильтровывают и перекристаллизовывают из смеси
спирта с водой. Остальные кислоты (соединения Пб,
Не, На) получают в тех же условиях. Выход кислот,
время проведения реакции, константы и данные элемен-
тарного анализа приведены в таблице.
* При использовании погружной лампы «Hanau-S-81» время облуче-
ния сокращается в п./гь раз.
175
Данные синтеза и анализа 2,2,4.4-тетраалкилоксетан-З-карбоновых
ки"ЛОТ
2,2.4,4-Тетраалкилоксетан- 3-карбоновые кислоты Выход % Всеми реакции ч • Темпера- тура плав- ления °C Найдено % Вычислено %
С н С н
2,2,4,4-Тетраметилоксетая- -3 карбоновая кислота (Ра) 97 50 114—115 60,90 60,64 9,03 9,20 60,73 8,92
2,4 Диметил-2,4-диэтилок- сетан-3-карбоновая кис- лота (I б) 92 68 105—106 64,29 64,24 9,84 9,99 64,48 9,74
2,4-Бис (тетраметилен)-ок- сетан-3 карбоновая кис- ло-а (Ре) 61 90 147,5— 148,5 68,88 68,76 8,77 8,81 68,54 8,62
2,4 -Бис- (пент аметилеи) -ок - сетан-З-карбоновая кис- лота (На) 60 ПО ИЗ—114 70,31 70,39 9,30 9,01 70,55 9,30
Литература
1. Коровины на И. К., Родина Л. Л., ЖОХ, 1, 932
(1965).
2. W о I f f L., Ann., 325, 144 (1902); Horner L., Spie-
ls ch k a E., Ber., 88, 934 (1955).
3. Родина Л. Л., Коробинына И. К., Усп. Хим., 36, 611
(1967).
4. К о Р о б и ц ы н а И. К-, Родина Л, Л., ЖОХ, 34,
2851 (1964).
ТЕТРАМИД 3,3',4,4'-ДИФЕНИЛОКСИДТЕТРА-
карбоновой кислоты
М. Т. РАЗУМОВСКАЯ, А. И. БЕЛЯКОВА, Л. А. ЕГОРОВА
CONH, CONH2
H2NOC—о—conh2
CieH14O3N4
Мол. вес 342,13
Тетрамид 3,3',4,4'-дифенилоксидтетра карбоновой кис-
лоты используется в синтезе органических полимеров,
обладающих полупроводниковыми свойствами. Тетрамид
получают обработкой 3,3',4,4'-тетракэрбоксидифенилок-
сида водным раствором аммиака по схеме: 3,3',4,4'-
17§
тетракарбоксидифенилоксид -»• диимид 3,3',4,4'-дифенил-
оксидтетра карбоновой кислоты тетрамид 3,3',4,4'-ди-
фенилоксидтетракарбоновой кислоты
Мы предлагаем менее сложный метод получения тетр-
амида, основанный на взаимодействии 3,3',4,4'-тетракарб-
оксидифенилоксида с мочевиной в среде хлорбензола;
при этом продолжительность синтеза сокращается до 6 ч.
Схема синтеза
СООН
соон
\ / zNH2
НООС—А>._О—/ СООН + со
~— Ан2
conh2 conh2
--> H2NOC—CONH2
РЕАКТИВЫ
3,3',4,4'-Тетракарбоксидифенилоксид 2, ч., т. пл. 227—229 °C.
Мочевина, ч., ГОСТ 6691—53.
Хлорбензол, ч„ ТУ МХП 2877—54.
Аммиак водный, ч. д. а., ГОСТ 3760—64.
Способ получения
В трехгорлую круглодонную колбу емкостью 1 л,
снабженную механической мешалкой, обратным холо-
дильником и термометром, помещают 34,6 г (0,1 моль)
3,3',4,4'-тетракарбоксидифенилоксида, 68 г (1,1 моль)
мочевины (реактивы тщательно растерты и перемешаны)
и 500 мл (550 г) хлорбензола. Смесь нагревают на масля-
ной бане до 130—135 °C и выдерживают при этой темпе-
ратуре 6 ч, затем охлаждают до комнатной температуры,
дают ей отстояться и декантируют. К остатку в колбе
прибавляют 300 мл 25%-ного водного раствора аммиака
и смесь перемешивают 4 ч до образования белой однород-
ной массы. Осадок отфильтровывают, промывают 12,5%-
ным водным раствором аммиака 2 раза порциями по 50 мл,
водой до нейтральной реакции промывных вод (индика-
тор— лакмус), ацетоном (25 мл), тщательно отжимают
и сушат в сушильном шкафу при 90—95 °C до постоян-
ного веса. Получают 28 г продукта; т. пл. выше 300 °C,
12-287 177
ыход 80% (от теоретического). По литературным дан-
ым \ т. пл. выше 300 °C.
Препарат представляет собой аморфный порошок
белого цвета, нерастворимый в органических раствори-
телях и воде.
Содержание азота в синтезированном продукте
(CleH14OgN4):
Найдено. %...........
Вычислено, % . . . .
16,50; 16,46
16,37
Литература
1, Marvel С. S., Martin М., J. Am. Chem. Soc., 80, 6600
(1958).
2. Разумовская М. Т., Белякова А. И., Его-
рова Л. А., Методы получения химических реактивов и пре-
паратов, вып. 15, ИРЕа, 1967, стр. 130.
1,1,4,4-ТЕТРАХЛОРБУТАН
10. М. ЗИНОВЬЕВ, в. Н. КУЛАКОВА, Л. 3. СОБОРОВСКИЙ
СНС12СН2СН2СНС12
С4Н6С14 Мол. вес 195,90
Галоидирование углеводородов с целью получения
индивидуальных, не содержащих изомеров полигалоид-
ных производных очень трудная и практически почти
неразрешимая задача. Между тем лишь индивидуальные
полигалоидные углеводороды оказываются пригодными
для дальнейших превращений.
В настоящей методике описан относительно простой
способ получения впервые синтезированного 1,1,4,4-тег-
рахлорбутана, практически не содержащего каких-либо
посторонних примесей Г 1,1,4,4-Тетрахлорбутап обра-
зуется при нагревании в автоклаве до 100—110 °C смеси
хлористого винила с пятихлористым фосфором. Можно
предполагать, что реакция протекает с образованием
промежуточного комплекса дихлорэтилтетрахлоридафос-
фора с пятихлористым фосфором, превращающегося далее
в 1,1,4,4-тетрахлорбутан с выделением хлоридов фосфора.
Возможно также, что промежуточным соединением в этой
реакции является комплекс, аналогичный комплексу,
178
возникающему при взаимодействии непредельных угле-
водородов с пятихлористым фосфором. Такое взаимодей-
ствие, в частности, подробно изучено на примере реакции
стирола с пятихлористым фосфором 2.
При работе следует иметь в виду, что в реакционной
массе содержатся агрессивные вещества: треххлористый
фосфор, пятихлористый фосфор, хлористый водород, по-
этому при выпуске газообразных продуктов из автоклава
необходимо соблюдать осторожность. Выходящие из авто-
клава газообразные продукты пропускают через раствор
щелочи.
Схема синтеза
СН2=СНС1 + РС15---» [С2Н3С12РС1з PCI']-> СНС12СН2СН2СНС12
РЕАКТИВЫ
Хлористый винил, ТУ МХП 3897—53.
Пятихлористый фосфор, ч. д. а., МРТУ 6-09-604—63.
Способ получения
В автоклав из специальной стали емкостью 0,5 л поме-
щают 208,3 г (1 моль) пятихлористого фосфора, охлаж-
дают его до минус 20—30 °C и конденсируют 100 г
(1,6 моль) хлористого винила. Автоклав нагревают до
ПО °C и выдерживают при этой температуре 12 ч. Дав-
ление сначала поднимается до 20 атм, а затем снижается
до 14 атм. После охлаждения из автоклава через вентиль
выпускают непрореагировавший газообразный хлористый
винил (27—30 г). Оставшуюся в автоклаве жидкость
(260—270 г) перегоняют. При 70—115 °C отгоняется
160—165 г смеси треххлористого фосфора и 1,1,2-трихлор-
этана (см. примечание). Остаток перегоняют в вакууме
и собирают фракцию (55—60 г) с т. кип. 30—90 °C при
1 мм рт. ст., содержащую наряду с 1,1,4,4-тетрахлор-
бутэнолом продукты побочных реакций — тетрахлор-
этан и пентахлорэтан. После повторной перегонки этой
фракции выделяют 35—40 г продукта; выход 35% (от тео-
ретического), т. кип. 200 °C при 745 мм рт. ст.-, 52—
54 °C при 1 мм рт. cm., dl° = 1,4477, — 1,4995.
Состав синтезированного продукта (С4Н6С14):
с на
Найдено, %. . 24,82; 25,14 3,57; 3,20 73,02; 72,16
Вычислено, % . 24,52 3,09 72,39
179
Примечание. Наряду с образованием 1,1,4,4-тетрахЛор-
бутана происходит хлорирование хлористого винила главным обра-
зом по кратной связи до 1,2,2-трихлорэтана: СН2=СНС1 4-
+ РС15 — СН2С1СНС12 + РС13.
Выход 1,2,2-трихлорэтана составляет более 30% (считая по
израсходованному хлористому винилу). Кроме того, в результате
хлорирования образуется некоторое количество тетрахлорэтана
и, возможно, пентахлорэтана.
Литература
1. Зиновьев Ю. М., Кулакова В. Н., Соборов-
с ки н Л. 3., ЖОХ, 27, 151 (1957).
2. Федорова Г. К., Кирсанова А. В., ЖОХ, 30,
4044 (1960).
1,1,3,3-ТЕТРАЦИАНПРОПАН
МЕТИЛ ЕН-БИС-МАЛОНОНИТРИЛ
И. М. РЫБКИНА, А. А. БЫЛИНКИНА, Г. И. МИХАИЛОВ
C7II4N4
CN CN
Н—С— СИ,—С—Н
I I
CN CN
Мол. вес 144,13
Синтез 1,1,3,3-тетрацианпропана из динитрила мало-
новой кислоты и формалина описан в ряде работ1-4.
Реакция не всегда протекает в одном направлении
и в некоторых случаях получается продукт с нечетко
выраженной температурой плавления. Мы показали, что
кислотность исходного динитрила малоновой кислоты
влияет на качество и выход 1,1,3,3-тетрацианпропана
и предложили применять динитрил малоновой кислоты
с минимальной кислотностью5. Установлено также влия-
ние катализаторов на скорость реакции6!7,8 и показано,
что в присутствии р-аланина7 синтез веществ проте-
кает 4 ч.
Схема синтеза
CN4 . CN
>сн2 + сн2о+ н2с/
CNZ 'CN
CN CN
---- НС—CH2—CH
I I
CN CN
180
РЕАКТИВЫ
Динитрил малоновой кислоты 8.
Формалин, технический, 37%-ный водный раствор, пл. 1,094,
ГОСТ 1625—61.
Спирт этиловый, технический (гидролизный), ГОСТ 8314—57.
^-Аланин, ч., МРТУ 6-09—62.
Способ получения
В круглодонную колбу емкостью 6 л с лопастной
мешалкой и термометром вводят 750 г (11,36 моль) свеже-
перегнанного динитрила малоновой кислоты (см. приме-
чание 1) и 3,75 л этилового спирта. Колбу помещают
в водяную баню, включают мешалку и перемешивают
смесь до полного растворения веществ. Затем прибавляют
430 мл (470 г; 5,68 моль) раствора формальдегида, 2,5 г
0-аланина и размешивают (см. примечание 2). Реакцион-
ную смесь нагревают до 26 °C и выдерживают при этой
температуре 50—60 мин. При этом начинается кристалли-
зация. Температуру в колбе постепенно повышают до
35—40 °C и перемешивают реакционную массу 3,5 ч,
считая с момента появления первых кристаллов. К концу
перемешивания температура реакционной массы сни-
жается до 23—25 °C. Выпавший осадок ’ ,1,3,3-тетрациан-
пропэна отфильтровывают и тщательно отжимают. Маточ-
ный раствор сохраняют. Через сутки из него выпадает
около 50—60 г продукта. Осадки объединяют, промывают
95%-ным этиловым спиртом (800 мл), дистиллированной
водой пять раз порциями по 600 мл, еще раз спиртом (1 л),
тщательно отсасывают и отжимают досуха. После высу-
шивания на воздухе продукт сушат в вакуум-сушильном
шкафу при остаточном давлении 5—10 мм рт. ст. (1 ч
при 20 °C и 3 ч при 95—100 °C). Получают 620—640 а
продукта; т. пл. 132—137 °C (см. примечание 3), выход
80% (от теоретического). По литературным данным4, т. пл.
130—135 °C.
Примечания. 1. Максимально допустимое количество
соляной кислоты 0,01%. В противном случае динитрил обрабатыва-
ют окисью магния и перегоняют в вакууме 5.
2. Лучше применять 40%-ный водный раствор формальдегида;
перед употреблением его нужно проанализировать.
3. Температуру плавления 1,1,3,3-тетрацианпропана опреде-
ляют в запаянном капилляре при быстром нагревании, так как при
температуре плавления может начаться пиролиз вещества.
181
Литература
1. Diels О,, Gartner Н., К a ack R., Вег., 55, 3439
(1922).
2. D i е 1 s О., Сепп В., Вег., 56, 2078 (1923).
3. Шервуд П., Химия и химическая технология, Издатинлит,
1958, стр. 64
4. Якубович А., Разумовский В., Розен-
штейн С., ЖОХ, 28, 2292 (1958).
5. Михайлов Г. И., Радле-Десятник С. С., авт.
свид. СССР 175947; Билл, изобр., № 21 (1965).
6. Михайлов Г. И., Богомолова Л. И., авт. свид.
СССР 168287; Билл, изобр., Ns 4 (1965).
7. Михайлов Г. И., Рыбкина Н. М., Былинки-
н а А. А., авт. свид. СССР 193486; Бюлл. изобр., Ns 7 (1967).
8. Rosenberg A., D е 1 v i п J. Р., Spectrochim. Acta, 2]
16114 (1965).
9. Радле-Десятник С. С., Розенфельд Н. И.,
Методы получения химических реактивов и препаратов, вып..13,
ИРЕА, 1965, стр. 40.
2-(2'-ТОЗИЛАМИНО-5'-БРОМФЕНИЛ)-
4Н-НАФТО [2,3-d]-[l,3]-OKCA3OH-4
М. В. ЛОСЕВА, В. Г. БРУДЗЬ, Б. М. БОЛОТИН
CjsHjjBrNjOjS
Мол. вес 521,39
2 - (2' - Тозиламино-5' - бромфенил) - 4Н - нафто[2,3 - d\ -
[1,31-оксазон-4 или ангидрид 2-(2'-тозиламино-5'-бром-
бензоиламино)-3-нафтойной кислоты в литературе не
описан. Однако известен общий метод получения подоб-
ных соединений путем циклодегидратации соответствую-
щих о-ациламинобензойных или нафталинкарбоновых
кислот 1i s. Все промежуточные продукты также не опи-
саны и были получены нами по аналогии с известными
182
методами. Ацилирование аминокислот проводилось
в среде ледяной уксусной кислоты, которая, как известно,
намного ускоряет этот процесс3. Последняя стадия син-
теза — циклодегидратация — может быть осуществлена
под действием различных агентов: уксусного ангидрида 4,
серной кислоты Б, хлористого тионила 6> \
Мы остановили свой выбор на хлористом тиониле,
так как в этом случае исключается возможность пере-
или деацилирования. Циклизация идет легко и с хоро-
шим выходом.
Схема синтеза
.NH.
Вг-
С ООН
Вг
NHSO3C6H4CH3
СООН
NHSO2C6H4CH3
/\^\соон
Вг/^/Ч-СОС1
II
__/Вг
NH—СО—
СООН nhso2c6h4ch3
N
Вг
\/ nhso2c6h4ch3
II
о
111
РЕАКТИВЫ
5-Бромантраниловая кислота, ч., РТУ 79-64—63.
п-Толуолсульфохлорид, ч., ВТУ ГКХ 1577—61.
Хлористый тионил, ч., ВТУ МХП 3591—52.
183
2-Амино-З-нафтойная кислота ’.
Бензол, ч., ГОСТ 5955—51.
Гептан, ч., МХТУ 286—60.
Кислота уксусная, ГОСТ 61—51.
Способ получения
Синтез 5-6poM-N-тозилантраниловой кислоты (I). Это
соединение получают тозилированием 5-бромантранило-
вой кислоты (методика аналогична указанной в литера-
туре 8). В стеклянный стакан емкостью 100 мл, снабжен-
ный механической мешалкой и термометром, достигаю-
щим почти до дна стакана, помещают 30 мл воды и 5,33 г
(0,05 моль) карбоната натрия. При нагревании к смеси
в 3 приема прибавляют 4,2 г (0,0205 моль) 5-бромантра-
ниловой кислоты. При температуре 60 °C, когда кислота
полностью растворится, к раствору добавляют в 5 приемов
за 20 мин 4,72 г (0,0248 моль) п-толуолсульфохлорида.
По окончании этой операции температуру реакционной
смеси (60—70 °C) поддерживают еще 20 мин, затем повы-
шают до 85 °C, осторожно вводят 0,3 г активированного
угля и все вместе фильтруют. Затем в стакан емкостью
100 мл, снабженный эффективной мешалкой, помещают
10,5 мл 6 н. раствора соляной кислоты. Охлажденный
до 50 °C фильтрат приливают небольшими порциями
к соляной кислоте. Выпавшую смолообразную массу
отфильтровывают и кипятят 1 ч с 40 мл воды. Осадок
полностью переходит в кристаллическое состояние; его
отфильтровывают и перекристаллизовывают из бензола.
Получают 1,5 г продукта; т. пл. 200—200,4 °C, выход
22% (от теоретического). При повторной перекристалли-
зации т. пл. не изменяется.
Содержание углерода и водорода в синтезированном
продукте (C14H12BrNO4S):
с н
Найдено, % ................. 45,91; 45,29 3,15; 3,03
Вычислено, %................... 45,42 3,26
Получение хлорангидрида 5-6poM-N-тозилантранило-
вой кислоты (II). Раствор 1,5 г (0,00445 моль) 5-бром-М-
тозилантраниловой кислоты и 5,3 г (0,0445 моль) хлори-
стого тионила в 45 мл бензола кипятят 2,5 ч с обратным
холодильником. Бензол и не вступивший в реакцию хло-
ристый тионил отгоняют в вакууме водоструйного насоса
184
при ЙО—60 °C. Твердый осадок перекристаЛлийовывайФ
из гептана. Получают 0,8 г продукта; т. пл. 123—123,4 °C,
выход 51% (от теоретического). При повторной перекри-
сталлизации т. пл. не изменяется.
Содержание углерода и водорода в синтезированном
продукте (Ci^HHBrClNOgS):
с н
Найдено, % ................ 43,36; 43,63 2,73; 2,83
Вычислено, %................... 43,26 2,85
Синтез 2-{2'-тозиламино-5-бромфенил)-4Н-нафто-
12, 3-d]-[] ,3]-оксазона-4 (III). Растворяют 0,37 г
(0,00198 моль) 2-амино-З-нафтойной кислоты в 16мл ледя-
ной уксусной кислоты. Раствор нагревают до кипения
и добавляют к нему 0,7 г (0,00198 моль) хлорангидрида
5-бромтозилантраниловой кислоты. Реакционную смесь
кипятят 2 ч с обратным холодильником и оставляют
на ночь. Выпавший осадок отфильтровывают и сушат.
Затем продукт растворяют в 5 мл (0,0445 моль) хлористого
тионила. Смесь кипятят 30 мин. Уже через 1—2 мин
начинает выпадать осадок. По охлаждении к смеси добав-
ляют 40 мл гептана. Осадок отфильтровывают и пере-
кристаллизовывают из ледяной уксусной кислоты. Полу-
чают 0,37 г продукта; т. пл. 286,5—287,5 °C, выход 65%
(от теоретического). При повторной перекристаллизации
т. пл. не изменяется.
Содержание углерода и водорода в синтезированном
продукте (C25H17BrN2O4S):
с п
Найдено, %.................
Вычислено, %..............
57,36; 57,68 3,00; 3,15
57,48 3,28
Литература
1 Sr hroeter Q., Е i s 1 е b О., Ann., 367, 1 (1909).
e c h W. F., Legg N., J. Chem. Soc., 1950, 961.
твиненко Л. M., Александрова Д. M.,
цкин Г. Д., Укр. хим. ж., 28, 77 (1962).
X u g g 1 i P., Leonhardt W., Helv. Chim. Acta, 7,
906 (1924).
о. Болотин Б. M., Рябокобылко Ю. С., Драп-
кина Д. А., Брудзь В. Г., Химические реактивы
и препараты, вып. 27, ИРЕА, 1965, стр. 289.
6. I о п е s с u М., Mantsch Н., G о 1 a J., Вег., 93, 2063
(1960).
185
t. Синтезы органических препаратов, т. 3, Издатинлит, 1949,
стр. 51.
8. Синтезы органических препаратов, т. 4, Издатинлит, 1949,
стр. 32.
2-(3'-тозиламино-2-нафтил)-
4Н-3.1-БЕН30КСА30Н-4
В. Г. БРУДЗЬ, Б. М. БОЛОТИН, М. В. ЛОСЕВА
C26H18N2O4S
Мол. вес 442,46
Это соединение получено так же, как и 2-(2'-тозил-
амино-5'-бромфенил)-4Н-нафто[2,3-с!]-1,3-оксазон-4.
Схема синтеза
186
о
NHSO2CeH4CH3
in
РЕАКТИВЫ
2-Амино-З-нафтойная кислота \ ч
п-Толуолсульфохлорид, ч_, ВТУ ГКХ 1577—61.
Хлористый тионил, ч., ВТУ МХП 3591—52.
Антраниловая кислота, ч., ТУ 79П ь86—61.
Кислота уксусная, ч., ГОСТ 61—51
Бензол, ч., ГОСТ 5955—51.
Гептан, ч., МХТУ 286—60.
Способ получения
Синтез 2-тозиламино-З-нафтойной кислоты (/). В стек-
лянный стакан емкостью 100 мл, снабженный механиче-
ской мешалкой и термометром, достигающим почти до
дна стакана, вводят 60 мл воды и 5,33 г (0,05 моль) карбо-
ната натрия. Смесь нагревают до 60 °C и прибавляют
в 3 приема 3,85 г (0,0205 моль) 2-аминонэфтойной кисло-
ты-3, а затем в 5 приемов в течение 20 мин 4,71 г
(0,0248 моль) n-толуолсульфохлорида. Реакционную
смесь выдерживают при этой температуре 20 мин и охлаж-
дают. Выпавший осадок натриевой соли кислоты отфиль-
тровывают и нейтрализуют 11 мл 6 н. раствора соляной
кислоты. Образовавшуюся свободную кислоту вновь
фильтруют и кристаллизуют последовательно из ледяной
уксусной кислоты и метанола. Получают 2,44 г продукта;
т. пл. 222—223 °C, выход 48% (от теоретического). По
литературным данным 2, т. пл. вещества 224 °C.
Получение хлорангидрида 2-тозиламино-З-нафтойной
кислоты (11). Смесь 2,44 г (0,0071 моль) 2-тозиламино-З-
нафтойной кислоты, 8,3 г (0,07 моль) хлористого тионила
и 50 мл бензола кипятят 3 ч с обратным холодильником.
Бензол и избыток хлористого тионила отгоняют в вакууме
водоструйного насоса при 50—60 °C. Твердый остаток
перекристаллизовывают из бензола. Получают 1,9 г
продукта; т. пл. 183—184 °C, выход 75% (от теоретиче-
187
ского). При повторной перекристаллизации т. пл. не из-
меняется.
Содержание углерода и водорода в синтезированном
продукте (C18H14C1NO3S):
с н
Найдено, %.................. 60,11; 60,23 3,89; 3,68
Вычислено, %................... 60,08 3,92
Синтез 2-(3'-тозиламино-2' -нафтил)-4Н-3,1 -бензокса-
зона-4 (III). К раствору 0,57 г (0,0042 моль) антранило-
вой кислоты в 15 мл ледяной уксусной кислоты добавляют
1,5 г (0,0042 моль) хлорангидрида 3-тозиламинонафтойной
кислоты-2 и полученную смесь кипятят с обратным холо-
дильником 3,5 ч. Охлажденную реакционную массу выли-
вают в 60 мл воды. Выпавший осадок отфильтровывают
и обрабатывают последовательно 60 мл 8%-ного раствора
соляной кислоты и 60 мл воды, вновь фильтруют и сушат.
Высушенный осадок растворяют в 7 мл (0,097 моль) хло-
ристого тионила и кипятят с обратным холодильником
1,5 ч. По охлаждении в реакционную смесь добавляют
30 мл гептана. Выпавший осадок отфильтровывают
и перекристаллизовывают из ледяной уксусной кислоты.
Получают 0,4 г продукта; т. пл. 252—252,5 °C; выход
75% (от теоретического). При повторной перекристалли-
зации т. пл. не изменяется.
Содержание углерода и водорода в синтезированном
продукте (C26H18N2O4S):
с н
Найдено, %.................. 67,48; 67,84 4,01; 4,11
Вычислено, %................... 67,86 4,10
Литература
1. Синтезы органических препаратов, Издатннлит, т. 3, 1949,
стр. 51.
2. В е е с h W. F., Legg N., J. Chem. Soc., 1950, 961.
8-(п-Т0ЛУ0ЛСУЛЬФ0НИЛ0КСИ)-КАРБ0СТИРИЛ
2-ОКСИ-8-(п-ТОЛУОЛСУЛЬФОНИЛОКСИ)-
хинолин
В. М. ДЗИОМКО, И. А. КРАСАВИН, И. В. РУБЦОВ
C16H13NO4S
Мол. вес 315,35
Синтез 8-(и-толуолсульфонилокси)-карбостирила опи-
сан в работе \ авторы которой получили его двумя неза-
висимыми методами: перегруппировкой 1-оксикарбостири-
ла под действием и-толуолсульфохлорида в пиридине
(выход 89,5%) и реакцией 8-оксикарбостирила (2,8-ди-
оксихинолина) с и-толуолсульфохлоридом в присутствии
углекислого калия (выход 61,3%). Проверка показала,
что первый из этих методов приводит к образованию про-
дукта, сильно загрязненного примесями и трудно под-
дающегося очистке. Напротив, второй метод дает вполне
удовлетворительные результаты.
РЕАКТИВЫ
8-Оксикарбостирил (2,8-диоксихинолин)2< 3 перекристаллизованный
из диметилформамида, т. пл. 288—289,с °C (с разл.).
п-Толуолсульфохлорид перекристаллизованный из петролейного
эфира, т. пл. 70—71 °C.
489
Схема синтеза
Способ получения
В колбу емкостью 3 л, снабженную эффективной
мешалкой, помещают 5,64 г (0,035 моль) 8-оксикарбости-
рила (2,8-диоксихинолина), 69,1 г (0,5 моль) углекислого
калия, 750 мл воды и добавляют 11,4 г (0,06 моль) п-толуол-
сульфохлорида и 750 мл хлороформа. Смесь энергично
размешивают 4—6 ч при комнатной температуре. Ниж-
ний слой отделяют, верхний слой дважды экстраги-
руют хлороформом порциями по 125 мл. Экстракты
объединяют, высушивают прокаленным сульфатом нат-
рия, фильтруют и упаривают досуха. Остаток перекри-
сталлизовывают из 400—460 мл этилового спирта с при-
менением активированного угля и получают 7,2—8,3 г
препарата в виде бесцветных кристаллов; т. пл. 212—
213 °C, выход 65,2—75,2% (от теоретического). По лите-
ратурным данным1, т. пл. 210—211 °C.
Литература
1. Ham ana М., Funakoshi К., Yakugaku Zasshi, 84,
28 (1964).
2. Д з и о м к о В. М., Красавин И. А., Радии ГО. П.,
Методы получения химических реактивов и препаратов, вып. 13,
ИРЕА, 1965, стр. 44.
3. Красавин И. А., Д з и о м к о В. М., Радин Ю. П.,
Методы получения химических реактивов и препаратов, вып. 15,
ЬРЕА, 1967, стр. 73.
190
2-ХЛОР-8-
(и-ТОЛУОЛСУЛЬФОНИЛОКСИ)ХИНОЛИН
И. А. КРАСАВИН, В. М. ДЗИОМКО, И. В. РУБЦОВ
GuHuCINOgS
Мол. вес 333,80
Единственный известный метод получения 2-хлор-8-(п-
толуолсульфонилокси)хинолина заключается в нагрева-
нии 8-(п-толуолсульфонилокси)-карбостирила с пятихло-
ристым фосфором и небольшим количеством хлорокиси
фосфора 1 в течение 10 ч при 140—160 °C (выход
90,6%). Как показала проверка, по этой методике
получается загрязненный примесями маслянистый про-
дукт, очистка которого сопряжена с' большими поте-
рями вещества. Мы установили, что применение одной
хлорокиси фосфора, взятой в избытке, не только позво-
ляет снизить температуру реакции и сократить продол-
жительность нагревания (с 10 ч до 15—30 мин), нои при-
водит к образованию с почти количественным выходом
очень чистого продукта. 2-Хлор-8-(п-толуолсульфонил-
окси) хинолин мы получили и другим путем: при взаи-
модействии 2-хлор-8-оксихинолина с п-толуолсульфохло-
ридом в среде пиридина; этот способ также дает достаточно
чистый продукт с 98—99%-ным выходом.
РЕАКТИВЫ
8-(п-Толуолсульфонилокси)-карбостирил (получение см. на
стр. 189), т. пл. 212—213 °C.
Хлорокись фосфора, ч., МРТУ 6 09 337—63.
п-Толуолсульфохлорид, перекристаллизованный из петролейкого
эфира, т. пл. 70—71 °C.
Пиридин, ГОСТ 13647—68, высушенный над едким кали и перегнан-
ный.
2-Хлор-8-оксихинодин2, очищенный перегонкой с водяным паром
и перекристаллизацией из 60%-ного спирта, т. пл. 82,5—83 °C.
191
Схема синтеза 2-хлор-8-
(п-толуолсульфонилокси)хинолина
из 8-(/г-толуолсульфонилокси)-карбостирила
Способ получения
В колбу емкостью 500 мл с пришлифованным обратным
холодильником, защищенным хлоркальциевой трубкой,
помещают 4,10 г (0,013 моль) 8-(п-толуолсульфопилокси)-
кгрбостирила и 20 г (12 мл; 0,13 моль) свежеперегнанной
хлорокиси фосфора. Колбу погружают в глицериновую
или масляную баню, нагретую до 125—128 °C, и выдержи-
вают при этой температуре 30 мин. Содержимое колбы
периодически перемешивают, причем через 2—3 мин
осадок полностью растворяется. Затем избыток хлор-
окиси фосфора отгоняют в вакууме; остаток охлаждают
в бане со льдом и добавляют к нему в один прием 100 г
дробленого льда. Когда выделившийся маслянистый про-
дукт закристаллизуется, содержимое колбы осторожно под-
щелачивают до pH 8 раствором 42,4 г (0,4 моль) безвод-
ного углекислого натрия в 170 мл воды. Осадок отфиль-
тровывают, тщательно промывают водой и сушат. Полу-
чают 4,3 г продукта; т. пл. 132,5—133 °C, выход 99%
(от теоретического). После перекристаллизации из 130—
140 мл этилового спирта получают 3,75 г продукта, т. пл.
при этом не изменяется; выход 86,4% (от теоретического).
По литературным данным1, т. пл. 132—134 °C.
192
Схема синтеза 2-хлор-8-
(и-толуолсульфонилокси)хинолина
из 2-хлор-8-оксихинолина
Способ получения
В конической колбе емкостью 25 мл растворяют 1,80 г
(0,01 моль) 2-хлор-8-оксихинолина в 5 мл пиридина,
добавляют при размешивании раствор 2,29 г (0,012 моль)
n-толуолсульфохлорида в 5 мл пиридина и оставляют
на 10—20 ч при комнатной температуре. Затем реакцион-
ную массу с выпавшим в ней кристаллическим осадком
переносят в колбу с 100 мл ледяной воды и перемешивают
при охлаждении в ледяной бане до тех пор, пока продукт
полностью пе закристаллизуется. Осадок отсасывают,
промывают водой и сушат. Получают 3,3 г вещества;
т. пл. 131,5—132,5 °C, выход 98,6% (от теоретического).
После перекристаллизации из 100 мл этанола получают
2,7 г препарата в виде бесцветных кристаллов; т. пл.
132,5—133 °C (см. примечание).
Примечание. Образны 2-хлор-8-(л-толуолсульфонилок-
си)хинолина, полученные двумя описанными выше методами, не
дали депрессии температуры плавления в пробе смешения.
Литература
1. Ham ana М., Funakoshi К., Yakugaku Zasshi, 84,
28 (1964)
13—287
193
2. Красавин И. А., Дзиомко В. М., Мй р ош
кина Н. И., Радин Ю. П., Методы получения химиче-
ских реактивов и препаратов, вып. 14, ИРЕА, 1966, стр. 150.
УРАМИЛДИУКСУСНАЯ КИСЛОТА
В. я. ТЕМКИНА, Н. А. ЕГОРУШКИНА, Р. П. ЛАСТОВСКИИ
NH—СО
| | ,СН,СООН
СО CH-N<f
| | ЧСН2СООН
NH—СО
C3HeN3O7
Мол. вес 259,18
Урамилдиуксусная кислота (Н3Ур) образует комплек-
сы с металлами, превосходящие для катионов Li, Na, К,
Tl, Be по прочности комплексы с ЭДТА L 2> 3. В литера-
туре описано получение урамилдиуксусной кислоты
взаимодействием урамила (5-амипобарбитуровой кислоты)
с монохлоруксусной кислотой L 2. В процессе синтеза
получают малоустойчивое соединение Н3Ур-МаН2Ур х
X Н2О. Перекристаллизация его из хлорной кислоты
приводит к получению комплексона в виде кислоты Н3Ур,
которая устойчива при хранении 2.
Мы проверили и уточнили методику 2 получения ура-
милдиуксусной кислоты. Исходный урамил получен вос-
становлением нитробарбитуровой кислоты губчатым оло-
вом в среде соляной кислоты 4. На этой стадии количество
соляной кислоты уменьшено вдвое по сравнению с коли-
чеством, предусмотренным методикой.
Схема синтеза
NH-CO NH- СО
| 1 Sn-f-HCI I | C1CH2COOH
СО CH-NO2-------► СО CH—NHa-------->
II II
NH-CO NH—СО
NH-CO
I | ,CH,COOH
CO CH—N<
I I VlLCOOH
NH—CO
194
РЕАКТИВЫ
Нитробарбитуровая кислота, ч_, ВТУ РУ 835—53.
Олово металлическое, губчатое, ч., ВТУ РУ 589—52.
Кислота соляная, ч., ГОСТ 3118—46.
Кислота монохлоруксусная, ч., ГОСТ 5836—51.
Кислота хлорная, ч., ТУ ОРУ 87—57.
Натр едкий, ч., ГОСТ 4328—66.
Способ получения
Синтез 5-аминобарбитуровой кислоты. В трехгорлую
колбу, снабженную мешалкой, обратным холодильником,
термометром и загрузочной воронкой, помещают 50 г
(0,3 моль) нитробарбитуроврй кислоты и 300 мл (9,7 моль)
концентрированной соляной кислоты. Смесь нагревают
на кипящей водяной бане, добавляют при 90 °C в течение
0,5 ч 125 г (1,05 г-атом) губчатого олова, затем 200 мл
(6,4 моль) концентрированной соляной кислоты и выдер-
живают при 90—95 °C и перемешивании до тех пор, пока
не исчезнет желтая окраска раствора. Затем добавляют
еще 1000 мл (32 моль) концентрированной соляной кисло-
ты, нагревают до полного растворения осадка, прибавляют
2—3 г активированного угля и фильтруют горячую
реакционную смесь через фильтр Шотта № 4. Фильтрат
упаривают в вакууме до объема 0,5 л и охлаждают смесью
соли и льда. Выпавший осадок фильтруют, грэмывают
1 л разбавленной (1 : 1) соляной кислоты, 1 л воды и сушат
в вакуум-эксикаторе сначала над серной кислотой, затем
над 40%-ным раствором едкого натра. Получают 18 г
продукта. Выход 47,5% (от теоретического), считая на
нитропроизводное.
Синтез урамилдиуксусной кислоты. В трехгорлую
колбу, снабженную мешалкой, холодильником и капель-
ной воронкой, помещают 10 г (0,07 моль) урамила и 14 мл
5 н. раствора едкого натра. К полученной суспензии
малинового цвета при работающей мешалке прибавляют
20 г (~0,21 моль) монохлоруксусной кислоты, предвари-
тельно нейтрализованной 46 мд5н. раствора едкого натра
до pH 8—10. Смесь нагревают до кипения, после чего
прибавляют по каплям 28 мл 5 н. раствора едкого натра
с такой скоростью, чтобы поддерживать pH смеси в преде-
лах от 8 до 10. В течение первых 15лы« суспензия почти
полностью растворяется. После прибавления всего коли-
чества щелочи цвет раствора становится оранжевым,
195
pH раствора больше не понижается. Кипячение прОдолИ
жают еще 10 мин, затем реакционную массу охлаждают
до 20 °C, фильтруют и фильтрат подкисляют концентри-
рованной соляной кислотой до pH 2. Выпавшую натрие-
вую соль комплексона (Н3Ур-НаНгУр-Н2О) фильтруют
и дважды перекристаллизовывают из воды. Получают
продукт серого цвета, розовеющий на воздухе. Вещество
переводят в Н+-форму без предварительного высушивания.
Для этого его растворяют в 65 мл 25%-ной хлорной кис-
лоты и раствор охлаждают смесью соли и льда. Выпавший
осадок фильтруют, промывают небольшим количеством
воды, затем 80%-ным этиловым спиртом и сушат в сушиль-
ном шкафу при 50 °C. Получают 9 г продукта. Выход
49,7% (от теоретического).
Содержание азота и углерода в препарате (C8HBN3O7):
N С
Найдено, % ................ 16,66; 16,62 37,04; 36,74
Вычислено, %.................... 16,2 37,1
Литература
1. Schwarzenbach G.; Helv. Chim. Acta, 29, 364 (1946).
2. Irving H., da Silva J. J. R. F., J. Chem. Soc., 1963,
448.
3. Irving H., da Silva J. J. R. F., J. Chem. Soc., 1963,
3309.
4. Хартман В., Шеппард О., в сб. «Синтезы органи-
ческих препаратов», т. 2, Издатинлит, 1949, стр. 491.
2-ФЕНИЛ-1,2,3-ТРИА30Л
Ю. А. НАУМОВ
Мол. вес 145,16
2-Фенил-1,2,3-триазо'’ образуется с 50%-ным выходом
при нагревании до 7'< .00 °C фенилозазона глиоксаля
с водным раствором медного купороса l> а. С незначитель-
196
ным выходом 2-фенил-1,2,3-триазол получают также при
сухой перегонке фенилозазона глиоксаля 3.
Мы установили, что 2-фенил-1,2,3-триазол можно
получить при нагревании до 180 °C фенилозазона глиокса-
ля с каталитическим количеством однохлористой меди.
Реакция протекает быстро и с хорошим выходом.
Схема синтеза
CH=N-NHCeHs
ii=N-Niic6H5
--->|| II + c6h6nh2
N N
CeH6
РЕАКТИВЫ
Фенилозазон глиоксаля1, т. пл. 169—170 °C.
Фенилозазон монофенилглиоксаля 5, т. пл. 148—149 °C.
Медь однохлористая, ч. д. а., ГОСТ 4164—61.
Кислота соляная, ч., ГОСТ 3118—46.
Калий углекислый, прокаленный, ч. д. а., ГОСТ 4221—65.
Способ получения
Нагревают до 180 °C 20 г фенилозазона глиоксаля
и 0,2 г однохлористой меди в колбе Вюрца на открытом
огне с сеткой. В результате начавшейся реакции темпера-
тура быстро поднимается до 210 °C и в приемник отго-
няется светло-желтая жидкость. Через несколько минут
в колбе остается лишь небольшое количество смолы.
Полученный дистиллят (15,6 г) растворяют в эфире; эфир-
ный раствор для удаления анилина встряхивают несколь-
ко раз с 2 н. соляной кислотой, промывают водой и сушат
прокаленным углекислым калием. После отгонки эфира
продукт перегоняют в вакууме. Получают 8 г 2-фенил-
1,2,3-триазола в виде бесцветной жидкости; т. кип. 104—
105 °C при 18жж рт. ст., выход 66,6% (от теоретического),
п”> = 1,5870, dg0 = 1,139. По литературным данным 2,
т. кип. продукта 115—118 °C при 22 жж рт. ст. и 80 °C
1ри 12 жж рт. ст.
Аналогичным образом из 9,4 г фенилозазона моно-
)енилглиоксаля получают 4,4 г 2,4-дифенил-1,2,3-три-
1зола в виде белых кристаллов; т. пл. 41—42 °C
197
(после перекристаллизации из спирта), выход 70%
(от теоретического). По литературным данным6, т. пл.
продукта 56—57 °C.
Несмотря на значительное отличие температуры плав-
ления от величины, указанной в литературе, данные
анализа вещества соответствуют 2,4-дифенил-1,2,3-три-
азолу.
Состав синтезированного продукта (CUHUN3):
с н
Найдено, % .............. . 75,79; 75,81 5,18; 5,25
Вычислено, %................... 76,00 5,02
Литература
1. R i eb s от er J. L., J. Org. Chem., 13, 815 (1948).
2. El-Khadem H., E 1 - S h a f e i Z. M., J. Chem. Soc
1958, 3117.
3. Pechmann H., Ann,, 262, 265 (1891).
4. F i s c h e r E., Ber., 17, 575 (1884).
5. L a u b m a n n H., Ann., 243, 247 (1888).
6. C h i g i E_, Pozzo-Balbi T., Qazz. chim. ital 71,
232 (1941); C.A., 36, 2862 (1942).
4-ФЕНИЛХИНАЛЬДИНОВЫЙ АЛЬДЕГИД
А. Л. ГЕРШУНС, И. A. PACT РЕП ИН A
C16HnNO
Мол. вес 233,27
4-Фенилхинальдиновый альдегид и его производные
являются полупродуктами в синтезе органических ком-
плексообразователей, используемых для фотометриче-
ского определения меди. Это соединение в литературе
не описано и впервые получено нами окислением 4-фе-
нилхинальдина 1 двуокисью селена в среде диоксана.
198
Схема синтеза
РЕАКТИВЫ
Диоксан, ч_, ТУ ГКХ 1558—61.
4-Фенилхинальдин1, т. пл. 98—99° С.
Спирт метиловый, ч., ГОСТ 6995—54.
Селена двуокись, ч., ТУ ГКХ 1502—61.
Способ получения
В круглодонную колбу емкостью 0,5 л, снабженную
термометром, мешалкой, обратным холодильником
и капельной воронкой, помещают раствор 11,1 г (0,1 моль)
свежеперегнанной двуокиси селена в смеси 130 мл диокса-
на и 6 мл воды, нагревают до 45 °C и при размешивании
в течение 0,5 ч прибавляют раствор 10,95 г (0,05 моль)
4-фенилхинальдина в 150 мл диоксана. Реакционную
массу кипятят 3 ч, горячий раствор сливают, отделяя
от осевшего на стенках металлического селена, отгоняют
150 мл диоксана и добавляют воду до помутнения раство-
ра. Затем раствор кипятят до тех пор, пока он не станет
прозрачным, и еще горячим фильтруют. Выделившийся
после нейтрализации раствором карбоната натрия белый
или розоватый осадок альдегида фильтруют, отжимают
на фильтре и сушат при комнатной температуре. Полу-
чают 3,2 г продукта; т. пл. 85—86 °C; выход 30% (от тео-
ретического). В таком виде альдегид вполне пригоден
для синтетических целей.
Для очистки продукт кристаллизуют из 70%-ного
метилового спирта. 4-Фенилхинальдиновый альдегид
в чистом виде представляет собой бесцветные игольчатые
кристаллы, хорошо растворимые в спиртах, ацетоне,
бензоле, хлороформе, эфире и плохо растворимые в воде.
Т. пл. 90 °C, выход 27% (от теоретического).
199
Содержание азота в препарате (Ci6HuNO):
Найдено, %........... 6,21 6,08
Вычислено, %......... 6,00
Литература
1. Fischer О., Scheibe G., Merkel Р., Miiller R.,
J. Prakt. Chem., 100, 91 (1920).
2-ФТОРЭТИЛОВЫЙ ЭФИР ЭТИЛЕНГЛИКОЛЯ
Ю. M. ЗИНОВЬЕВ, В. Н. КУЛАКОВА, Л. 3. СОБОРОВСКИИ
FCHaCH2OCHaCHaOH
CJ^OjF Мол. вес 108,11
В настоящей методике описан удобный в препаратив-
ном отношении синтез 2-фторэтилового эфира этиленгли-
коля из 2-фторэтанола \
Схема синтеза
Na FCHaCHaOH
FCH,CH20H---» FCH3CH2ONa-------»- FCH2CH2OCH,CH2OH
A A a A L L i i
РЕАКТИВЫ
2-Фторэтанол 2.
Натрий металлический, ТУ МХП 1664—50.
Способ получения
В колбу для перегонки емкостью 100 мл помещают
32 г (0,5 моль) 2-фторэтанола и постепенно вносят 2,3 з
(0,1 моль) металлического натрия. Колбу во время реак-
ции охлаждают водой со льдом. После растворения нат-
рия из реакционной массы при 100—105 °C отгоняют
непрореагировавший 2-фторэтанол (18,7 г), а затем соби-
рают фракцию с т. кип. 58—62 °C при 7 мм рт. ст. При
повторной перегонке получают 8 г продукта. Выход 71,5%
(считая по израсходованному 2-фторэтанолу) и 85%
200
(считая по натрию); т, кип. 169—171 СС, №> — 1,1216,
= 1,4070.
Содержание углерода и водорода в препарате (C4HBO2F):
с н
Найдено, % .................. 44,30; 44,41 8,92; 8,77
Вычислено, %.................. 44,43 8,39
Литература
[.Зиновьев Ю. М., Кулакова В. Н., Соборов-
с ки й Л. 3., ЖОХ, 27, 2858 (1957).
2. Н о f f m a n F., J. Am, Chem. Soc., 70, 2596 (1948).
2-ХИНОЛИЛ ГИДРАЗОН
8-ОКСИХИНАЛЬДИНОВОГО АЛЬДЕГИДА
В. М. ДЗИОМКО, И. А. КРАСАВИН, Н. И. ДУДАРЕВА
ОН
Q19H14N4O
Мол. вес 314,35
Первое сообщение 1 о комплексообразующих свойст-
вах 2-хинолилгидразонэ 8-оксихинальдиновОго альдегида
сделано в 1965 г., но метод получения реагент? не был
тогда опубликован. В следующем году появилась работа 2,
также посвященная исследованию свойств этого соедине-
ния, но синтез был описан очень кратко. Ниже приведена
разработанная нами методика получения 2-хинолилгид-
разона 8-оксихинальдинового альдегида (выход 95—98%).
реактивы
8-Оксихинальдиновый альдегид 4, дважды перекристаллизованный
из н-гептана, т. пл. 98,5—99 °C.
2-Гидразинохинолин? дважды перекристаллизованный из бензола,
т. пл. 142,5—143 °C.
2fil.
Схема синтеза
Способ получения
При нагревании” на водяной бане и перемешивании
растворяют 3,98 г (0,025 моль) 2-гидразинохинолина в
75 мл этилового спирта и добавляют к раствору 4,33 г
(0,025 моль) 8-оксихинальдинового альдегида в 50 мл
теплого этилового спирта. Выделяется светло-желтый
осадок. Суспензию кипятят с обратным холодильником
2—2,5 ч на водяной бане, охлаждают в бане со льдом
и выдерживают 1—2 ч при 0 °C. Осадок отсасывают, про-
мывают на фильтре 10 мл этилового спирта и сушат
в вакуум-эксикаторе. Получают 7,7 г гидразона; выход
98% (от теоретического). Продукт перекристаллизовы-
вают из 500—550 мл диоксана (не содержащего переки-
сей); выпавшие кристаллы промывают на фильтре спиртом.
Получают 6,2 г продукта; т. пл. 270—275 °C, выход 79%
(от теоретического). По литературным данным 2, т. пл.
274 °C.
Состав синтезированного продукта (Ci9H14N4O):
с н N
Найдено, % . . . 72,43; 72,35 4,72; 4,66 17,83
Вычислено, % . . 72,60 4,49 17,82
Литература
1. Дзиомко В. М., Красавин И. А., Кремен-
с к а я И. Н., XX Международный конгресс по теоретиче-
ской и прикладной химии, Тезисы докладов, Секция Е, 1965,
стр. 64.
2. Rudolph Т., Phillips J. Р., Anal. Chim. Acta, 34,
235 (1966).
3. Дзиомко В. М., Красавин И. А., Мирошки-
и а Н. И., Методы получения химических реактивов и препа-
ратов, вып. 12, ИРЕ А, 1965, стр. 50.
4. Дзиомко В. М., Красавин И. А., Химические
реактивы и препараты, нып. 26, ИРЕА, 1964, стр. 29.
202
8-ХИНОЛИЛГИДРАЗОН
8-ОКСИХИНАЛЬДИНОВОГО АЛЬДЕГИДА
И. А. КРАСАВИН, В. М. ДЗИОМКО, Б. В. ПАРУСНИКОВ,
Ю. П. РАДИН
C19H14N4O
Мол. вес 314,35
Об избирательной экстракции кальция 8-хинолилгид-
разоном 8-оксихинальдинового альдегида уже сообщалось
ранее1-3. Недавно предложен люминесцентный метод
определения кальция этим реагентом4, но способ его
получения до сих пор не был опубликован. Ниже приво-
дится методика синтеза 8-хинолилгидразона 8-оксихи-
нальдинового альдегида, основанная на взаимодействии
8-оксихинальдинового альдегида с 8-гидразинохинолином.
РЕАКТИВЫ
8-Оксихинальдиновый альдегид 6, дважды перекристаллизованный
из «гептана, т. пл. 98,5—99 °C.
8-Гидразинохинолин «, т. пл. 63,5—65° С.
Схема синтеза
203
Способ получения
В круглодонной колбе емкостью 100 мл, снабженной
обратным холодильником, растворяют 2,60 г (0,015 моль)
8-оксихинальдинового альдегида в 50 мл этилового спирта
и прибавляют раствор 2,40а (0,015 моль) 8-гидразинохино-
лина в 25 мл этилового спирта. Полученную суспензию
кипятят с обратным холодильником на водяной бане 2 ч,
охлаждают в бане со льдом и выдерживают 2—Зчпри 0°С.
Осадок отсасывают, промывают на фильтре 25 мл этилового
спирта и сушат в вакуум-эксикаторе. Получают 4,6 а
продукта; выход 97,5% (от теоретического). После пере-
кристаллизации из 300 мл бензола получают 3,9 г про-
дукта в виде ярко-желтых волокнистых игл; выход
82,6% (от теоретического). Т. пл. 196 и 223 °C; при даль-
нейшей очистке образуется вещество с т. пл. 201,5—202 °C
и 223—223,5 °C. (Гидразон имеет двойную температуру
плавления: первоначально образовавшийся расплав при
дальнейшем нагревании затвердевает и затем плавится
вновь). Образец для анализа был приготовлен трехкрат-
ной перекристаллизацией из бензола.
Состав синтезированного продукта (C19H14N4O):
с н N
Найдено, % . . 72,04: 72,13 4,53; 4,73 17,61; 17,80
Вычислено, % . 72,60 4,49 17,82
Литература
1. Дзиомио В. М., Красавин И. А., Марко-
вич И. С., Островская В. М., Зеличенок С. Л.,
Совещание по методам концентрирования элементов в аналити-
ческой химки, Тезисы докладов, 1963, стр. 6.
2. Дзиомио В. М., Химичесиие реаитивы и препараты,
вып. 27, ИРЕА,’1965, стр. 336.
3. Дзиомио В. М., Красавиц И. А., К р е м е н-
с и а я И. Н., XX Международный ионгресс по теоретичесиой
и прииладной химии, Тезисы доиладов, Сеиция Е, 1965, стр. 64.
4. Божевольнов Е. А., Дзиомио В. М., Федо-
рова Л. Ф., Красавин И, А., авт. свид. СССР 210468;
Бюлл. изобр., № 6 (1968).
5. Дзиомко В. М., Красавин И. А., Химические
реактивы и препараты, вып. 26, ИРЕА, 1964, стр. 29.
6. К р а с а в и н И. А., Парусников Б. В., Дзи о м-
ио В. М., Методы получения химических реактивов к препа-
ратов, вып. 7, ИРЕА, 1963, стр. 5.
204.
5-ХЛОР-2-АМИНОТОЛУОЛ
Н. В. ДОПИНГ, В. В. ПАНОВА
C,HBC1N
Мол. вес 141,60
5-Хлор-2-аминотолуол — промежуточный продукт
в синтезах диазоля красного ТР и фосфат-нафтол а ТР,
являющихся ценными гистохимическими препаратами,
В литературе описано получение 5-хлор-2-аминотолуола
хлорированием о-анетотолуидина хлоратом калия или
натрия и соляной кислотой 2, молекулярным хлором 3,
хлористым сульфурилом4. Мы проверили и уточнили
последний способ получения, применив в качестве раство-
рителя вместо сероуглерода хлороформ.
Схема синтеза
РЕАКТИВЫ
о-Ацетотолуидин, ч., МРТУ 6-09-423—63.
Хлороформ, ч., ГОСТ 3160—51.
Спирт зтиловый, ректификованный, высшей очксткк, ТУ
366—63.
Кислота соляная, ч., ГОСТ 3118—46.
Натр едкий, ч., ГОСТ 4328—48.
Сульфурил хлористый, ч., ТУ НКХП 45—40.
205
Способ получения
Синтез 5-хлор-о-ацетотолуидина. В трехгорлой колбе
емкостью 1 л, снабженной мешалкой, шариковым холо-
дильником, капельной воронкой и термометром, раство-
ряют при размешивании и комнатной температуре 60 а
(0,4 моль) о-ацетотолуидина в 220 мл хлороформа. Колбу
помещают в ледяную баню и в течение 2 ч прибавляют
по каплям 36 мл (60 а, 0,45 моль) хлористого сульфурила.
Реакционную смесь перемешивают 30 мин, кипятят
на водяной бане 1 ч и затем отгоняют хлороформ. Остав-
шийся в колбе коричневый маслообразный продукт при
охлаждении кристаллизуется в монолитную белую массу.
После высушивания при 75 °C получают 72,4 а техниче-
ского продукта;.т. пл. 127—128 °C, выход 98% (от теоре-
тического). Вещество перекристаллизовывают из 1400 мл
25%-ного этилового спирта и получают 50—51 а продукта
в виде кристаллов белого цвета; т. пл. 139—141 °C, выход
69% (от теоретического).
Получение 5-хлор-2-аминотолуола. В одногорлую колбу
емкостью 1 л, снабженную мешалкой и шариковым холо-
дильником, помещают 51 а 5-хлор-о-ацетотолуидина
и 850 мл разбавленной (1 : 1) соляной кислоты. Смесь
нагревают на открытом пламени с сеткой, при этом осадок
растворяется -и через 10—15 мин начинают выпадать
кристаллы в виде перламутровых пластинок. Реакционную
массу размешивают при слабом кипении 2 ч, охлаждают
и фильтруют. Осадок растворяют в 420 мл кипящей дистил-
лированной воды и приливают раствор едкого натра
до нейтральной реакции по универсальной индикаторной
бумаге (примерно 135 мл). Выпавший осадок отфильтро-
вывают и сушат в эксикаторе над едким натром. Продукт
очищают перегонкой в вакууме, собирая фракцию с т. кип.
118—120 °C при 18 мм рт. ст. Получают 28,5 г 5-хлор-
2-аминотолуола, представляющего собой белые гигро-
скопичные кристаллы с т. пл. 28—30 °C; выход 50%
(считая на о-анетотолуидин). По литературным данным,
т. пл. 29—30 °C з-4 и 30—31 °C в.
Литература
1. R е v е d г i n F., Стер i eu х Р., В е г, 33, 2499 (1900).
2. Магидсон О. Ю., Травин А. И., ЖОХ, 7, 844
(1937).
206
3.2 el Im an E., Klotz C., A n n., 231, 317 (1885).
4. Wynn W. P., J. Chem. Soc., 61, 1046 (1892).
5. Bamberger A., Ann, 441, 302 (1925).
ХЛОРАНГИДРИД ДИФЕНИЛФОСФОРНОЙ
кислоты
А. Г. ФАДЕИЧЕВА, Л. Г. РУДЕНКО, Т. Н. СДУРАТОВСКАЯ
C^HjoClOgP
Мол. вес 268,63
Хлорангидрид дифенилфосфорной кислоты исполь-
зуется как реагент для синтеза разнообразных фосфорор-
ганических соединений. Хлорангидрид дифенилфосфор-
ной кислоты получают при взаимодействии эквимоляр-
ных количеств фенола и хлорокиси фосфора х. Однако
известные условия реакции требуют значительной затра-
ты времени и выход вещества составляет только 20%,
при этом в качестве побочного продукта получается хлор-
ангидрид фенилфосфорной кислоты (выход 74%). В ли-
тературе есть ряд работ, посвященных изучению влияния
катализаторов,' в том числе хлористого алюминия, на
выход хлорангидридов арилфосфорных кислот 2. Мы изу-
чали условия получения хлорангидрида дифепилфосфор-
ной кислоты в зависимости от количества катализатора,
времени и температуры реакции и разработали метод
синтеза с выходом до 82—85%. Предложенный способ
получения хлорангидрида дифенилфосфорной кислоты
проверен в заводских условиях; продолжительность син-
теза 2,5—3 ч.
Схема синтеза
А1С13 ДО
свн5он + роа3 —, (CeH5o)2p<;ci
РЕАКТИВЫ
Фенол, ч., ГОСТ 6417—52.
Хлорокись фосфора, ч., перегнанная, ВТУ У-166—51
Алюминий хлористый, безводный, ч. д. а., ГОСТ 4452—66.
207
Способ получений
В трехгорлую колбу емкостью 1 л с боковым тубусом,
снабженную обратным холодильником с хлоркальциевой
трубкой, механической мешалкой и термометром, поме-
щают 235 г (2,5 жоль) сухого свежеперегнанного фенола.
Фенол расплавляют при 60—70 °C и через боковой тубус
при перемешивании прибавляют 3,25 г (0,025 лголь) сухого
хлористого алюминия и 192,3 г (1,25 моль) хлорокиси фос-
фора (см. примечание 1). Реакционную смесь нагревают
на глицериновой бане и выдерживают 2 ч при перемеши-
вании и температуре внутри колбы 125 ± 5 °C. Получен-
ную смесь перегоняют из колбы с елочным дефлегмато-
ром в вакууме при остаточном давлении 6—7 мм рт. ст.
и собирают первую фракцию с т. кип. около 170 °C. По-
лучают 30 г дихлорангидрида фенилфосфорной кислоты;
выход 8—9% (от теоретического), п2® = 1,5230 — 1,5520.
Вторая фракция с т. кип. 170—180 °C представляет собой
хлорангидрид дифенилфосфорной кислоты. Выход веще-
ства равен 286 г (см. примечание 2), что составляет 85—
83% (от теоретического); — 1,5520 — 1,5550. Полу-
ченный продукт, согласно данным анализа, содержит
не менее 99—99,5% основного вещества.
Примечания. 1. При проведении синтеза принципиально
важен порядок добавления реактивов. Кроме того, следует учиты-
вать, что реакции, катализируемые галоидными соединениями
металлов, чувствительны к влаге, поэтому фенол и хлорокись фосфо-
ра должны быть тщательно перегнаны. Плохо очищенная хлорокись
фосфора, содержащая продукты гидролиза н влажный фенол, сни-
жает скорость реакции и выход вещества.
2. Вакуум-разгонку нужно проводить быстро (1,5—2 ч). Дли-
тельный нагрев приводит к снижению выхода н образованию
3-фенилфосфата. Необходимо учесть, что в первых погонах могут
присутствовать следы не вступившей в реакцию хлорокиси фосфора,
а также хлористый водород, являющийся продуктом реакции,
поэтому рекомендуется в начале разгонки на 10—15 мин включить
водоструйный насос, а затем вакуум-насос. Для быстроты перегонки
перегонную колбу н дефлегматор необходимо хорошо термостатиро-
вать н не допускать их перегрева.
Литература
1. Jakobson G., Вег., 8, 1521 (1875).
2. П л е ц В. И., Органические соединения фосфора, Оборонгнз,
1940, стр. 276.
3. Катышкина В. В., Крафт М. Я., ЖОХ, 26,
3060 (1956); Orlof Н. D., W or г el G. J., Маг-
208
с 1 e i F. X., J. Am. Chem. Soc., §0, 726 (1958); Fried-
mann M., Seligman M., J. Am. Chem. Soc., 76, 655
(1954).
ХЛОРАНГИДРИД ФЕНИЛ-
р-ХЛОРЭТИЛФОСФИНОВОй кислоты
E. JI. ГЕФТЕР, И. А. РОГАЧЕВА
CcH5 0
>p<
ClHjCHjC/ XI
CBH9C12OP
Мол. вес 223,066
Хлорангидрид фенил-Р-хлорэтилфосфиновой кислоты
легко реагирует с водой, спиртами, аминами, являясь,
таким образом, исходным веществом для синтеза самой
кислоты, ее эфиров, амидов, а также хлорангидрида
фенилвинилфосфиновой кислоты. Указанный хлорангид-
рид получен нами при взаимодействии Р-хлорэтилового
эфира фенил-Р-хлорэтилфосфиновой кислоты с пятихлори-
стым фосфором х.
Схема синтеза
С6Н6 ,0 СЙП5 о
>р< >р\
ClHjCH^/ ХОСН2СН2С1 + РС16----» С1Н2СН2СХ ХС1
РЕАКТИВЫ
Хлор этиловый эфир фенил-^-хлорэтилфосфиновой кислоты,
перегнанный, получен нз ди-р,р'-хлорэтилового эфира фенилфосфи-
нистой кислоты термической изомеризацией
Пятихлористый фосфор, ч., МРТУ 6-09-604—63.
Способ получения
В четырехгорлую колбу, снабженную обратным холо-
дильником и капельной воронкой с хлоркальциевыми
трубками, мешалкой и термометром, помещают 80 г
(1,03 моль) пятихлористого фосфора и к нему медленно
при перемешивании прибавляют 100 г (1 моль) р-хлорэти-
лового эфира фенил-Р-хлорэтилфосфиновой кислоты.
Реакция сопровождается сильным разогреванием и раз-
14—287
209
жижением смеси. Температуру реакции поддерживают
в пределах 70—100 °C. После завершения экзотермиче-
ской реакции обратный холодильник заменяют прямым
и, нагревая смесь до 120 °C, отгоняют образовавшиеся
дихлорэта.1 и хлорокись фосфора. Остаток нагревают
до 150 °C и в течение 30 мин пропускают сухой хлористый
водород *. После перегонки в вакууме получают 62 г
продукта; т. кип. 150—153 °C при 2 мм рт. ст., выход
74% (от теоретического), п^1 = 1,5482, d?0 = 1,3409. Ве-
щество представляет собой бесцветную прозрачную жид-
кость; растворяется в бензоле, толуоле, эфире, хлориро-
ванных углеводородах.
Содержание хлора и фосфора в синтезированном про-
дукте (С8Н9С12ОР):
а
гидролизуемый Р
Найдено, %.................... 16,08 13,8
Вычислено, %......... .... 15,9 13,9
Литература
1. Гефтер Е. Л., ЖОХ, 31,952 (1961).
ХЛОРАНГИДРИД Р-ХЛОРЭТИЛОВОГО ЭФИРА
ВИНИЛФОСФОНОВОЙ кислоты
Е. Л. ГЕФТЕР, X. М. ЗИЯВУДДИНОВ
ZOCH,CH2C1
СН,=СН—р/
II хп
о
Мол. вес 188,97
В литературе описан метод получения хлорангидрида
0-хлорэтилового эфира винилфосфоновой кислоты, осно-
ванный на взаимодействии ди-Р,р'-хлорэтилового эфира
винилфосфоновой кислоты и пятихлористого фосфора С
Применив общий метод синтеза хлорангидридоэфиров фос-
* Если хлористый водород не пропускать, то хлорангидрид
имеет растянутую температуру кипения и перегоняется с некото-
рым разложением. Причина этого описана в статье х.
210
фоновых кислот2, мы проверили и улучшили описанный’
в литературе метод 1 и получили продукт с выходом около
85%.
Схема синтеза
,ОСНаСН2С1
СН2=СН—Р(ОСН2СН2С1)2 + РС15 -> СН2=СН—Р<
II II ЧС1
о о
РЕАКТИВЫ
Ди-$,р' -хлорэтиловый эфир винилфосфоновой кислоты, ч., (полу-
чение см. на стр. 10S).
Пятихлористый фосфор, ч., МРТУ 6-09-604—63.
Способ получения
Прибор для синтеза представляет собой четырехгор-
лую круглодонную колбу, снабженную термометром,
мешалкой и обратным холодильником с хлоркальциевой
трубкой. В колбу помещают 466 г (1 моль) ди-0,Р'-хлор-
этилового эфира винилфосфоновой кислоты и 700 мл
сухого бензола. При энергичном перемешивании смеси
вводят 417 г (1 моль) пяти хлор истого фосфора порциями
по 20—30 г. Каждую следующую порцию пятихлори-
стого фосфора прибавляют после полного растворения
предыдущей. В ходе реакции температура реакционной
смеси поднимается на 2—3 °C (колбу не охлаждают),
а сам раствор слегка желтеет. Смесь перемешивают при
комнатной температуре 1 ч и затем отгоняют образовав-
шиеся дихлорэтан и хлорокись фосфора при давлении
20—30 мм рт. ст., постепенно нагревая баню до 90—
100 °C. Остаток перегоняют, собирая фракцию с т. кип.
100—НО °C при 4 мм рт. ст. После повторной пере-
гонки получают 302—330 г продукта; выход 80—87% (от
теоретического); т. кип. ПО °C при 4 мм рт. ст., п® =
= 1,4764, d20 = 1,3578. По литературным данным1,
т. кип. 94—100 °C при 1 мм рт. ст. Вещество представ-
ляет собой бесцветную прозрачную жидкость, которая
растворяется в бензоле, толуоле, эфире. Содержание
хлора в синтезированном продукте (С4Н7С12О2Р):
Найдено. %................. 19.0
Вычислено, %............... 18,79
14*
211
Литература
I. Welch С. M., Ganroles Е. J., Gutrie J. D.,
J. Org. Chem., 26, 3270 (1961).
2. Разумов А. И., Маркович E. А., Мухаче-
ва O. А., Химия и применение фосфорорганических соедине-
ний. Труды 1-н конференции в Казани, Изд. АН СССР, 1957,
стр. 194.
ХЛОРАНГИДРИДЫ МЕТИЛ-
И ЭТИЛТРИХЛОРМЕТИЛФОСФИНОВЫХ кислот
С. 3. ИВИН, Л. Е. ДМИТРИЕВА, К. В. ПАРАВАНОВ
R\ -Cl
>Р<
C13CZ ^0
Хлорангидриды метил- и этилтрихлорметилфосфино-
вых кислот являются исходными продуктами в синтезе
различных фосфорорганических соединений. Описанный
ранее1 метод получения хлорангидрида фенилтрихлор-
метилфосфнновой кислоты основан на взаимодействиг
комплексного соединения фенилтетрахлорфосфина и хло
ристого алюминия со спиртом в четыреххлористом угле
роде; выход равен 50%.
Нами показано, что хлорангидридьГ метил- и этил-
три хлорметилфосфиновых кислот могут быть получены
взаимодействием комплексного соединения метил- и этил-
три-хлорметилтрихлорфосфина и хлористого алюминия
с водой. Реакцию проводят в хлористом метилене в при-
сутствии хлористого калия.
ХЛОРАНГИДРИД МЕТИЛТРИХЛОРМЕТИЛФОСФИНОВОЯ
кислоты
С2Н,С14ОР
Мол. вес 215,83
Схема синтеза
Н3С.
>РС1, • А1С1, + Н,0 + КС1-►
C1SCZ
Н3(\ -С1
—> \р/ _L А1С13-КС1 Н-2НС1
C19CZ X)
£12
РЕАКТИВЫ
Комплексное соединение метилтрихлорметилтрихлорфосфина
с хлористым алюминием 2.
Хлористый калий, х. ч., свежепрокаленный, ГОСТ 4234—65.
Способ получения
В фарфоровый стакан помещают 40,4 г (0,1 моль)
комплексного соединения метилтрихлорметилтрихлор-
фосфипа с хлористым алюминием и 7,5 г (0,1 моль) хлори-
стого калия в 100 мл хлористого метилена. К смеси при
—20 °C из капельной воронки постепенно прибавляют
2 г (0,1 моль) воды. Затем доводят температуру реакцион-
ной массы до комнатной, осадок комплекса А1С13-КС1
отфильтровывают, хлористый метилен отгоняют. После
перекристаллизации из хлористого метилена получают
18 г продукта; т. пл. 84 °C, выход 87% (от теоретического).
Аналогично получают хлорангидрид этилтрихлорме-
тилфосфиновой кислоты при действии воды на соответст-
вующее комплексное соединение; т. пл. 36 °C, выход
70% (от теоретического).
Литература
1. В i d d 1 е Р., Kennedy J., W i 1 1 а п s J. Z., Chem. a.
Ind., 45, 1481 (1957).
2. И в и н С. 3., Караванов К. В., Методы получения
химических реактивов и препаратов, вып. 12, ИРЕА, 1965,
стр. 76.
ХЛОРАЦЕТАЛЬ ЭТИЛЕНГЛИКОЛЯ
2-ХЛОРМЕТИЛ-1.3-ДИОКСОЛАН
Б. Г. ЯСНИЦКИИ, С. А. САРКИСЯИЦ, Е. Г. ИВАНЮК
Н2с-оч
| >СНСН2С1
H8C-OZ
С4Н7СЮ2
Мол. вес 122,55
Хлорацеталь этиленгликоля получают из диэтилхлор-
ацеталя и этиленгликоля переацетилированием в при-
сутствии минеральных кислот1’ 2, взаимодействием а,0,0'-
213
трихлорэтилового эфира или водного раствора хлорацет-
альдегида с этиленгликолем в присутствии ионообмен-
ных смол3> 4, восстановлением 2-хлорметилен-1,3-диоксо-
лана Б. Перечисленные методы весьма сложны и часто
дают низкие выходы.
Предлагаемый способ6 основан на взаимодействии
этиленгликоля с димергидрэтом хлорацетальдегида в при-
сутствии ионообменной смолы КУ-1. Метод прост в выпол-
нении и приводит к высокому выходу продукта.
Схема синтеза
Н2С— о.
2СН,ОН-СН,ОН + (С1СН,СН0)2-Н.,0 «тгд 2 | >СНСН2С14-ЗН„О
H2C-OZ
РЕАКТИВЫ
Этиленгликоль, ч., ГОСТ 10164—62.
Ионообменная смола КУ-/, ТУ МХП 2115—49.
Хлорацетальдегид, димергидрат 7.
Способ получения
В двухлитровую колбу, снабженную обратным холо-
дильником, термометром и механической мешалкой, поме-
щают 1000 г (16,1 моль) этиленгликоля, 352,5 г (2,01 моль)
димергидрата хлорацетальдегида (см. примечание 1)
и 200 г ионообменной смолы КУ-1 в Н+-форме. Реакцион-
ную массу нагревают на водяной бане 12 ч при 80—90 °C,
после чего определяют содержание хлорзцетальдегида8
(см. примечание 2). Если содержание хлорацетальдегида
в реакционной массе при дальнейшем нагревании в тече-
ние 2—3 ч практически не изменяется, то смесь образую-
щегося хлорацеталя и выделившейся воды отгоняют при
65—70 °C и 20 мм рт. ст. (1-й отгон). Затем в реакцион-
ную колбу добавляют еще 352,5 г (2,01 моль) димергидрата
хлорацетальдегида и выдерживают смесь при той же
температуре 10—12 ч. Определяют содержание хлорацет-
альдегида. При отсутствии хлорацетальдегида или при
наличии незначительных количеств (как и в первый раз)
отфильтровывают в горячем состоянии ионообменную
смолу КУ-1 (см. примечание 3) и фильтрат подвергают
разгонке. При 60—70 °C и 20 мм рт. ст. отгоняют смесь
хлорацеталя с водой (2-й отгон), при 70—80 °C и
214
и 20 мм рт. ст.— чистый продукт. Первый и второй от-
гоны смешивают, отделяют в делительной воронке воду
и остаток присоединяют к чистому продукту. Полученный
хлорацеталь этиленгликоля сушат сульфатом натрия
и перегоняют в вакууме. Получают 736 г продукта;
т. кип. 51—53 °C при 9 мм рт. ст., 157—158 °C при
760 мм рт. ст.-, выход 75% (от теоретического), —
= 1,2475; п™ = 1,4474. Хлорацеталь этиленгликоля —
бесцветная подвижная жидкость с легким запахом.
По этой методике можно получить и ряд других хлор-
ацеталей. Однако в отличие от описанного способа при
получении следующих ацеталей хлорацетэльдегид вносят
в реакционную массу в один прием. Ниже приведены
оптимальные мольные соотношения реагентов (мно-
гоатомного спирта и димергидрата хлорацетальдегида
Л4ГП : /ИХаа), время (/) проведения процесса при 90 °C
и физико-химические константы полученных соединений.
Хлорацеталь глицерина (смесь двух изомеров). /И|П : =
= 2 : 0,5; t = 12 ч; т. кип. 125—130 °C при 9 мм рт. cm.; dj1 =
— 1,3245, njj1 = 1,4750; выход 96%. По литературным данным2,
т. кип. 130—134 °C при 13 мм рт. ст.
Хлорацеталь пропиленгликоля. МсП : /Ихаа = 1 : 0,5; t = 19 ч;
т. кип. 45—46 °C при 5 мм рт. cm.; dlJ = 1,1597, n2D = 1,4420;
выход 65% .
Хлорацеталь a-хлоргидрина глицерина. Л7СП : Л1хаа —
= 1,5: 0,5; t = 12 ч; т. кип. 82—83 °C при 4 мм рт. cm.; di’1 =
= 1,3498, н3д = 1,4750; выход 77%.
Хлорацеталь диэтиленгликоля. Д4ГП ' Л4ХЯа — 1,5 : 0,5; 1 =
= 20 ч; т. кип. 115—118 °C при 5 мм рт. cm.; dj = 1,1670, п™ —
= 1,4562; выход 63%. По литературным данным3, т. кип. 152—
160 °C, т. пл. 45—46 °C.
Дихлорацеталь пентаэритрита. МСЛ : Л4хаа = 1 : 1; t = 24 ч;
т. пл. 92 °C; выход 86%. По литературным данным 8, т. пл. 91,8 °C.
Трйхлорацеталь маннита. Мсп ’• Л4хаа ~ 1 •’ 1,5; t = 22 ч;
т. кип. 195—200 °C при 0,1—0,2 мм рт. ст.; dla = 1,4473; п^ =
= 1,5048; выход 60 о.
Трйхлорацеталь сорбита. Л4СП : Afxaa = 1 : 1,5; <=19 ч;
т. кип. 219—220 °C при 0,05 мм рт. cm.; diJ = 1,4484; п*р — 1,5096;
выход 62 %.
Примечания. 1. Внимание! Димергидрат хлорацетальде-
гила является сильным лакриматором и при попадании на кожу вы-
зывает ожоги, поэтому работу следует проводить с предосторожностью,
в вытяжном шкафу.
2. Анализ на содержание хлорацетальдегида в реакционной
массе проводят следующим образом: навеску реакционной массы
(1—3 г) переносят в мерную колбу емкостью 100 мл, доводят обьем
215
Ьодой до метки и отбирают 5 мл раствора. К отобранной пробе
добавляют 5—10 мл (в зависимости от величины навески) 2,5%-ного
водного раствора гидразинсульфата и оставляют стоять в течение
10 мин. Затем вводят 10 мл титрованного раствора азотнокислого
серебра, 2 мл азотной кислоты, 2 мл индикатора (железоаммиачные
квасцы) и титруют 0,1 н. раствором роданида аммония долоявления
красной окраски раствора. Расчет ведут по формуле
(oftt-fr/C,) 0,00785-100-100
х 5с
где х — количество хлор ацетальдегида (%); а — количество 0,1 и-
раствора нитрата серебра (мл)-, — поправка на титр 0,1 н. раство.
ра нитрата серебра; b — количество 0,1 н. раствора роданида аммо-
ния (мл)-, К2 — поправка на титр 0,1 н. раствора роданида аммо-
ния; с — навеска реакционной массы 9.
3. Так как реакционная масса имеет вязкую консистенцию,
фильтрование следует проводить через четырехслойный фильтр из
марли.
Литература
1, D е 1 е ф 1 п е М., Bull. Soc. chim. France [3], 25, 582 (1901).
2. Hallonomis E. G., Hilbert H., Can. J. Res., 8,
29 (1936).
3. Фр. пат. 1113862; С.A. 84771958.
4. A s t 1 e M., Zaslowsky I., L a f i a t i s P., Ind. Eng.
Chem., 2, 46, 789 (1954).
5. Fa ass U., Hilgert H„ Ber., 87, 1345 (1954).
6. Ясницкий Б. Г., Саркисянц С. А., Ива-
нюк Е. Г„ ДАН УССР, 2, 229 (1964).
7. Ясницкий Б. Г., Дольберг Е. Б., Сатаиов-
с кая Ц. И., Мед. пром. СССР, № 1, 35 (1963).
8. М х и т а р я н В. Г„ ЖОХ, 9, 1923 (1 939).
9. Ясницкий Б. Г., Сатановская Ц. И., Мед. пром.
СССР, № II, 36 (1960).
2-ХЛОРПРОПИЛДИХЛОРТИОФОСФАТ
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
S С1
СЕ II j
>Р—О—СН2—СИ- сн3
си
C3HeCl3OPS Мол. вес 227,47
2-Хлорпропилдихлортиофосфат благодаря наличию
реакционноспособных атомов галоида может быть ис-
216
пользован в качестве промежуточного продукта при син-
тезе разнообразных фосфорорганических соединений.По
патентным данным \ 2-хлорпропилднхлортиофосфат по-
лучают из окиси пропилена и тиохлорокиси фосфора
в присутствии третичных аминов. Известен также способ
получения 2-хлорэтилдпхлортиофосфата2.
Мы предлагаем более простой и удобный способ полу-
чения 2-хлорпропилдихлортиофосфата из 2-хлорпропил-
дихлорфосфита и серы.
Схема синтеза
Ск I
>Р—О—СН2—СН—СН3 + S -
СИ
S С1
Cl.ll I
\ Р—О—СН2—сн—сн3
РЕАКТИВЫ
2-Хлорпропилдихлорфосфит3, т. кип. 55—56 °C при
7 мм рт. ст., == 1,4954.
Сера, черенковая, ч., ТУ МХП ОРУ 83—57.
Способ получения
В колбу с обратным холодильником помещают 19,54 г
(0,1 моль) 2-хлорпропилдихлорфосфита и 3,21 г
(0,1 моль) серы. Реакционную смесь выдерживают 8 ч
на масляной бане при температуре 150—160 °C, охлаж-
дают до комнатной температуры и отфильтровывают
непрореагировавшую серу. Образовавшийся 2-хлорпро-
пилдихлортиофосфат перегоняют в вакууме. Получают
6,15 г продукта; т. кип. 92—96 °C при 10 мм рт. ст.,
выход 27% (от теоретического), d® = 1,455, = 1,5302.
Состав синтезированного продукта (C3H6CI3OPS):
Найдено, % .
Вычислено, %
16,28
15,84
2,67 46,84 13,26 14,13
2,66 46,76 13,61 14,09
217
Литература
1. Пат. США 2866809; РЖХим, 81992 П (1960).
2. Malatesta Р., A t г i D., Farmaco (Pavia), 8, 398 (1953);
РЖХим, 25272 (195-1).
3. Самуилов А. М., Дрегваль Г. Ф., Методы получе-
ния химических реактивов и препаратов, вып. 18, ИРЕА, 1969,
стр. 219.
2-ХЛОРПРОПИЛДИХЛОРФОСФАТ
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
О С1
с\ II I
> Р -о-сна- СН-СН3
СИ
С3Н6С13ОаР
Мол. вес 211,41
2-Хлорпропилдихлорфосфат может быть использован
в качестве исходного продукта в синтезе различных произ-
водных пятивалентного фосфора. По патентным данным \
2-хлорпропилдихлорфосфат получают взаимодействием
хлорокиси фосфора с парами окиси пропилена в при-
сутствии третичных аминов в качестве катализаторов.
Процесс протекает при небольшом давлении. Малинов-
ский проводил взаимодействие хлорокиси фосфора
с окисью этилена 2.
По предлагаемой нами методике синтез протекает при
нормальном давлении без катализатора; окись пропи-
лена вводится в реакцию в жидкой фазе.
Схема синтеза
РОС13 + СН3—СН—СН3
О С1
>Р—О—СНа—СН—СН3
РЕАКТИВЫ
Хлорокись фосфора, свежеперегнанная, т. кип. 105—106 °C.
Окись пропилена, ч., свежеперегнанная, т. кип. 35 °C.
218
Способ получения
В трехгорлую колбу с обратным холодильником, тер-
мометром, капельной воронкой и механической мешалкой
помещают 15,3 г (0,1 моль) хлорокиси фосфора, к которой
при температуре минус 3—10 °C и перемешивании при-
бавляют 5,8 г (0,1 моль) окиси пропилена. Реакционную
смесь нагревают 3 ч на водяной бане до полного прекра-
щения кипения и затем перегоняют в вакууме. Получают
13,9 г продукта; т. кип. 84—86 СС при 7 мм рт. ст.,
выход 66% (от теоретического), d20 = 1,459, п2° = 1,4640.
Состав синтезированного продукта (С3Н6С13О2Р):
с и р
Найдено, %...................
Вычислено, %.................
17,03 2,96 14,58
17,04 2,86 14,66
Литература
1. Пат. США 2866809; РЖХпм, 81992 (1960).
2. М а л и н о в с к и й М. С., Окиси олефинов и их производ-
ные, Госхимиздат, 1961, стр. 212.
2-ХЛОРПРОПИЛДИХЛОРФОСФИТ
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
С1
С1 |
\Р-О-СН2—СН—сн3
СИ
CgHgClgOP Мол. вес 195,41
2-Хлорпропилдихлорфосфит является доступным
реагентом для синтеза разнообразных фосфорорганиче-
ских соединений. По литературным данным, 2-хлорпро-
пилдихлорфосфит получают из паров окиси пропилена
и треххлористого фосфора в присутствии третичных
аминов в качестве катализаторов. Процесс проводят при
небольшом давлении *. Аналогично получают другие
2-хлоралкилдихлорфосфиты 2.
По предлагаемой нами методике синтез протекает при
нормальном давлении без катализатора; окись пропи-
лена вводят в реакцию в жидкой фазе.
219
Схема синтеза
Ск
РС13 + СНа-СН -СН3 >Р—о—сна—сн—сн3
\/ си I
о а
РЕАКТИВЫ
Фосфор треххлористый, ч., ГОСТ 91—41.
Окись пропилена, ч., свежеперегнанная, т. кип. 35 °C.
Способ получения
В трехгорлую колбу, снабженную обратным холо-
дильником, термометром, капельной воронкой и механи-
ческой мешалкой, помещают 13,7 г (0,1 моль) треххлори-
стого фосфора, к которому постепенно при температуре
0—5 °C и перемешивании прибавляют 5,8 г (0,1 моль)
окиси пропилена. Реакционную смесь нагревают на кипя-
щей водяной бане 30 мин и образовавшийся продукт
перегоняют в вакууме. Получают 11,8 г вещества; т. кип.
55—56 °C при 7 мм рт. ст. Выход 60% (от теоретиче-
ского); df — 1,383, п*> = 1,4954.
Содержание хлора и фосфора в препарате (С3НвС13ОР):
а р
Найдено, % ....... 54,46 15,92
Вычислено, % . . . . 54,42 15,86
Литература
1. Пат. США 2866809; РЖХим, 81992П (1960).
2. Малиновский М. С., Окиси олефинов и их производ-
ные, Гослитиздат, 1961, стр. 210.
3-ЭТИЛ- И 3-и-БУТИЛАНИЛИН
А. В. СТРАШНЕНКО, А. И. АЛЕКСЕЕНКО, Р. К. СТРАШНЕНКО
СН2-СН2-СН3-СН3
сн.2-сн3
СвНцЫ
Мол. вес 121,18
C10HnN Мол. вес 149.23
220
З-Этиланилинполучают восстановлением 3-нитроэтил-
бензола железом в присутствии уксусной кислоты 1 или
восстановительным разложением гидразона 3-аминоаце-
тофенона в присутствии этилата натрия З-н-Бутилани-
лин получают восстановлением 3-нитро-н-бутилбензола
цинком в присутствии соляной кислоты ®. Оба соедине-
ния могут быть также получены каталитическим гидри-
рованием соответствующих 3-нитро-алкилфенилкетонов
в присутствии палладия 4 или восстановлением этих
кетонов сплавом Ренея в щелочном растворе 6. Разрабо-
танный нами способ получения 3-этил- и 3-н-бутиланили-
нов основан на одновременном восстановлении гидра-
зингидратом нитро- и карбонильной групп в 3-нитро-
анетофеноне и 3-нитробутирофеноне. Этот способ приме-
ним также для получения других 3-алкиланилинов.
Схема синтеза
где R: —СН3 или —СН2—СН2—СН3
РЕАКТИВЫ
3-Нитроацетофенон 6, т. пл. 68—72 °C.
3-Нитробутирофенона, т. пл. 60—61 °C.
Гидразингидрат, технический, ГОСТ 10729—64.
Этиленгликоль, ГОСТ 11033—64.
Способ получения
Синтез З-н-бутиланилина. В четырехгорлой колбе
емкостью 500 мл, снабженной мешалкой, термометром,
капельной воронкой и холодильником, растворяют 20 г
(0,11 моль) 3-нитробутирофенона в 180 мл этиленгликоля
и при температуре 40 °C в течение 1 ч вводят по каплям
при перемешивании 34 г гидразингидрата. Реакционную
массу перемешивают еще 1 ч при температуре 40 °C,
после чего медленно нагревают содержимое колбы до
80 °C, недолго выдерживают при этой температуре, ох-
лаждают до 40 "С и небольшими порциями прибавляют
18 г (0,32 моль) едкого кали, предварительно растертого
221
в порошок, с такой скоростью, чтобы температура реак-
ционной массы не поднималась выше 50 °C (см. примеча-
ние 1). Эта температура поддерживается за счет выделяю-
щегося тепла в течение 1 ч. Затем обратный холодильник
заменяют на нисходящий и температуру в колбе медленно
поднимают до 200 °C, при этом отгоняется вода и неболь-
шое количество 3-н-бутиланилина. Дальнейшую отгонку
ведут в вакууме водоструйного насоса при медленном
повышении температуры в колбе до 240 °C. Перегнанный
продукт растворяют в эфире, эфирный раствор промывают
водой и сушат едким кали. После отгонки эфира продукт
перегоняют в вакууме. Получают 8г 3-н-бутиланилина,
представляющего собой слегка желтоватое масло; т. кип.
110—112 °C при 8 мм рт. ст., выход (см. примечание 2)
50% (от теоретического), = 1,5228. По литературным
данным 5, 3-н-бутиланилин представляет собой жидкость
с желтоватым оттенком; т. кип. 132—134 °C при
19 мм рт. ст.
Синтез 3-этиланилина. З-Этиланилин получают вос-
становлением 3-нитроацетофенона в условиях, описанных
для 3-н-бутиланилина, исходя из 35 г (0,21 моль) 3-нит-
роацетофенона, 60 г гидразингидрата, 220 мл этиленгли-
коля и 35 г (0,62 моль) едкого кали. После перегонки в ва-
кууме получают 16 г 3-этиланилина; т. кип. 214—215 °C,
92 °C при 12 мм рт. ст., выход 68% (от теоретического),
«о = 1,5208. По литературным данным1, т. кип. 214—
215 °C.
Примечания. 1. Не следует температуру в реакторе
поднимать выше 50 °C, так как происходит очень быстрое разогрева-
ние и вспенивание реакционной массы, что может послужить причи-
ной ее выброса.
2. Применяя в качестве катализатора никель Ренея, можно
повысить выход анилина до 75%.
Литература
1. Behal J., Chody F., Bull. Soc. chim. France, 11, 211
(1894).
2. Ma u er F., E n g 1 i c h B., Ann., 417, 84 (1918).
3. Reilly B. J., Hickinbottom W. J., J. Chem.
Soc., 117, 109 (1920).
4. Oelschloger H., Ber., 89, 2025 (1956).
5. P a r a D., Schwenk E., Whitman B., J. Org.
Chem., 6, 587 (1942).
6. К о p с о н Б. Хазен P., Синтезы органических препара-
тов, т. 2, Издатинлит, 1949, стр. 361.
222
ЭТИЛЕНДИАМИН-N, N'-ДИУКСУСНАЯ КИСЛОТА
И. В. ЦИРУЛЬНИКОВА, В. я. ТЕМКИНА, Г. А. ГРОМОВА,
Р. И. ЛАСТОВСКИЙ
HOOC-H2C-HN-CH2-CH2-NH-CH2-COOH
C6H12N2O4 Мол. вес 176,17
Этилендиамин-М,М'-диуксусная кислота представляет
интерес как комплексообразователь и как промежуточный
продукт в синтезе различных окрашенных и флуоресци -
рующих комплексонов В литературе указано два способа
получения этого соединения: взаимодействие дихлорэтана
или дибромэтана с глицином 2 и цианметилирование эти-
лендиамина ®’ 4. Попытка воспроизвести синтез первым
из указанных способов не дала положительных резуль-
татов.
Мы проверили и уточнили методику получения эти-
лендиамин-Ы-ЬГ-диуксусной кислоты методом цианмети-
лирования.
Схема синтеза
Н,СО4
h2nch2ch2nh2 + СН2О + KCN —.
---> HOOC-HSC-HN-CH2-CH2-NH—СН2-СООН
РЕАКТИВЫ
Этилендиамин, ч., МРТУ 6-09-1238—64.
Формальдегид, ч., ТУ МХП 2631—51.
Цианистый калий, ч., ВТУ 3961—53.
Кислота серная, ч., ГОСТ 4204—48.
Натр едкий, ч., 4328—66.
Способ получения
В трехгорлую колбу, снабженную обратным холодиль-
ником, механической мешалкой и капельной воронкой,
помещают 6 г (0,1 моль) этилендиамина, прибавляют
20%-ный раствор едкого натра до pH 8,5—10,5 и затем
прибавляют 13 г (0,2 моль) цианистого калия и 100 мл
дистиллированной воды. Затем при 45—50 °C перемеши-
вая , вводят в 5 приемов 100 мл 7%-ного водного рас-
твора формальдегида. Каждую порцию формальдегида
вводят по каплям в течение 1 ч, после чего при той же
223
температуре в вакууме водоструйного насоса отгоняют
20 мл воды. Затем прибавляют следующую порцию форм-
альдегида и операция повторяется. После отгонки по-
следней порции воды реакционную массу выдерживают
1 ч при 45—50 °C, фильтруют (не охлаждая), нейтрали-
зуют при энергичном размешивании и охлаждении
30%-ным раствором серной кислоты до pH 6,5 и снова
фильтруют. Из фильтрата в вакууме при температуре до
70 °C порциями (в 4—5 приемов) отгоняют воду, каждый
раз отфильтровывая выпадающий осадок для отделения
основного количества сульфатов. Отгонку воды продол-
жают до получения вязкого раствора (объем около 20 мл).
Полученный раствор смешивают с равным объемом этило-
вого спирта и оставляют стоять 3—4 дня. Выпавшие кри-
сталлы отфильтровывают, растворяют при 50 °C в 100 мл
дистиллированной воды и осаждают 300 мл абсолютного
этилового спирта. Переосаждение повторяют 2—3 раза
до получения кислоты, свободной от сульфатов (отрица-
тельная проба с хлористым барием). После высушивания
при 50 °C получают 5,2 г продукта. Выход 30% (от теоре-
тического).
Состав синтезированного продукта (C6H12N2O4):
с н N
Найдено, % .
Вычислено, %
40,63: 40,71 6,76: 6,57 15,92: 15,88
40,90 6,81 15,90
Литература
1. Budesinsky В., Haas К-, Coll. Czech. Chem. Comm.,
29, 100, (1964).
2. A iken J. K, Chem. a. Ind., 45, 1334 (1956).
3. Герм. пат. 638071 (1936).
4. Ber s worth F. С., пат. США 2588923 (1951).
ЭТИЛ ЕНДИАМИНО-БИС-
ИЗОПРОПИЛФОСФОНОВАЯ КИСЛОТА, МЕЧЕНАЯ
ПО УГЛЕРОДУ
Т. Я. МЕДВЕДЬ, М. В. РУДОМИНО
НО. /ОН
\p_i4C-NH—СН.-СН,—NH—«С------Р< 2Н2О
НО-^Н /\ || 'ОН
О СН3 СН3 СН3 СН3 о
12C614C2H26N2O8P3 Мол. вес 344,25
224
Этилендиамино-бис-изопропилфосфоновая кислота,
являясь высокоэффективным комилексообразователем, се-
лективным в отношении ионов некоторых переходных
металлов, предложена в качестве профилактического
и терапевтического средства при лечении свинцовых
отравлений \ Для изучения механизма физиологического
действия, а также вопроса о путях выведения этилен-
диамино-бис-изопропилфосфоновой кислоты из организма
животных получен препарат кислоты, меченой по углероду
в изопропильной группе. В основу синтеза положен пред-
ложенный ранее метод получения а-аминоалкилфосфоно-
вых кислот конденсацией диалкилфосфонатов с аминами
и кетонами 2> 3. С целью повышения выхода этиленди-
амино-бис-изопропилфосфоновой кислоты метод был не-
сколько видоизменен и в этом варианте использован для
получения меченого продукта. В качестве исходного ак-
тивного реагента был выбран ацетон (меченый по С2),
который взаимодействовал с этилендиамином и диэтил-
фссфонатом.
Схема синтеза
2(С2Н5О)2РНО + 2СШ*СОСП3 + H3N—СН2—СН2—NH2----»
---> (C2H5O)2P-,4C-NH-CH2-CH2-NH-14C-----Р(ОС2Н5)2 -->
II /X /х II
о сн3 сн3 сн3 сн3 о
---» (HO)2P-14C-NII-CH2-CI12-NH-14C---Р(ОН)3
II /\ /X II
о сн3 сн3 сн3 сн3 о
РЕАКТИВЫ
Диэтилфосфонат, ТУТСР 1048р—63, т. кип. 68—70 °C при
10 мм рт. ст., = 1,4080.
Этилендиамин, безводный, т. кип. 116,5 °C, Пр = 1,4540
(см. примечание I).
Ацетон, меченый, удельная активность 3,96 мк/г.
Ацетон, ч. д. а., ГОСТ 2603—63.
Бензол, ч. д. а., ГОСТ 5955—51.
Натрий сернокислый, прокаленный, ч. д. а., ГОСТ 4166—66.
Кислота соляная, ч., ГОСТ 3118—46.
Способ получения
Синтез этилового эфира этилендиамино-бис-изопро-
пилфосфоновой кислоты. В четырехгорлую колбу, снаб-
женную механической мешалкой, термометром, двумя
15—287
225
капельными воронками с хлор кальциевыми трубками
и холодильником с хлоркальциевой трубкой, помещают
15 г (0,25 моль) этилендиамина, нагревают его до 60 °C
и при перемешивании прибавляют по каплям из двух
разных воронок одновременно 29 г (0,5 моль) ацетона (из
них 3,5 г меченого) и 69 г (0,5 моль) диэтилфосфоната с такой
скоростью, чтобы соблюдалось примерное соотношение:
1 капля ацетона — 2 капли диэтилфосфоната, а в реак-
ционной смеси поддерживалась температура 60 °C. Реак-
ционную смесь выдерживают 1 ч при 60 °C, охлаждают
и экстрагируют 500 мл бензола. Бензольный экстракт
промывают 40 мл насыщенного раствора поташа и сушат
сернокислым натрием. Бензол удаляют в вакууме. Полу-
чают 79,7 г продукта, представляющего собой густую
светлую жидкость, кристаллизующуюся при стоянии;
т. пл. 32—32,5 °C (см. примечание 2), выход 76,4% (от
теоретического).
Получение этилендиамино-бис-изопропилфосфоновой
кислоты. Кипятят 7 ч 79,7 г этилового эфира этиленди-
амино-оис-изопропилфосфоновой кислоты с 410 мл кон-
центрированной соляной кислоты. Избыток соляной кис-
лоты отгоняют в вакууме, выпавший осадок отфильтровы-
вают, промывают водой и сушат в вакуум-эксикаторе.
После перекристаллизации из разбавленной соляной кис-
лоты получают 34 г меченой этилендиампно-бис-изопро-
пилфосфоновой кислоты; т. пл. 235—236 °C, удельная
активность 0,14 мк!г, выход 40% (от теоретического).
Выход вещества по активности составляет 35% (от теоре-
тического). По литературным данным 3, т. пл. продукта
235—236 °C.
Примечания. 1. Безводный этилендиамин получен из
70%-ного водного раствора этилендиамина-основания (МРТУ 6-09-
1238—64) после удаления воды в вакууме водоструйного насоса
при 50 °C и последующей перегонки над натрием.
2. Для немеченого этилового эфира этилендиамино-бис-изопро-
пилфосфоновой кислоты с т. пл. 32—32,5 °C, синтезированного по
этой же методике, получены следующие данные анализа.
Состав синтезированного продукта (C16H38N2O6P2):
с н N Р
Найдено, % .
Вычислено, %
45,5: 45,7 9,2: 9,1 7,0; 7,0 14,7; 14,5
46,1 9,2 6,7 14,8
226
Литература
1. Архипова О. Г., Медведь Т. Я., Р у д о м и -
н о М. В., Кабачник М. И., Гигиена труда и проф-
заболевания, № 12, 33 (1963).
2. К а б а ч и и к М. И., Медведь Т. Я-, ДАН СССР, 84,
717 (1952).
3. Кабачник М. И., Медведь Т. Я., Козлова Г. К.,
Балабуха В. С., Сенявин М. М., Т и х о н о -
ваЛ. И., Изв. АН СССР, ОХН, 1958, 1070.
ЭТИЛ ЕНД И АМИ Н-М-(4-СУЛ ЬФО-2-НАФТОЛ)-
N, N', N-ТРИУКСУСНАЯ КИСЛОТА И
ЭТИЛЕНДИАМИН-М-(4,6-ДИСУЛЬФО-8-НАФТОЛ)-
N, N', N-ТРИУКСУСНАЯ КИСЛОТА
Г. Ф. ЯРОШЕНКО, В. я. ТЕМКИНА, Н. Е. ХАВЧЕНКО,
Р. И. ЛАСТОВСКИЙ
соон
CieHaoNgOtoS
HO3S—
<^н2
I
—N-CH2-CH2-N
он
/CHjCOOH
''СН2СООН
Мол. вес 456,39
СН2СООН
СН2СООН
Ci8H20N2O13S2
Мол. вес 536,43
Этилендиамин-М-(4 - сульфо-2-нафтол) - N,N',N' - три -
уксусная кислота (I) и этилендиамин-Й-(4,6-дисульфо-8-
нафтол)-М,М',М'-триуксусная кислота (II) являются не-
описанными в литературе флуоресцирующими комплек-
сонами — аналогами ЭДТА, содержащими нафталиновое
ядро в качестве сопряженной системы, ответственной за
флуоресценцию. Эти соединения образуют комплексы
15* 227
с рядом канонов, причем в Некоторых случаях комп-
лексообразование сопровождается гашением флуоресцен-
ции, что может быть использовано для флуориметриче-
ского определения этих катионов. Комплексоны получены
взаимодействием 1-аминоэтиламино-2-нафтол-4-сульфо-
кислоты и 1-аминоэтиламино-8-нафтол-4,6-дисульфокис-
лоты с цианистым калием и формальдегидом в щелочной
среде *.
Схема синтеза
R—NH—СН2—СН3—NHa + KCN СН2О -
ZCH соон
---- R-N-CH2-CH —n< '
| хсн,соон
сн3
I
соон
он
РЕАКТИВЫ
]-аминоэтиламино-2-нафтол-4-сульфокислота (получение см. на
стр. 28).
I-аминоэтилалино-в- нафтол-4,6-дисцлырокислота (получение
см. на стр. 28).
Цианистый калий, ч., ТУ МХП 3961—53
Формальдегид, ч., ТУ МХП 2631—51.
Натр едкий, ч. д. а., ГОСТ 4328—48.
Кислота соляная, ч., ГОСТ 3118—46.
Способ получения
Реакцию ведут в токе азота. В четырехгорлой
колбе, снабженной механической мешалкой, обратным
холодильником, капельной воронкой и термометром, рас-
творяют 8,5 а (0,03 моль) 1-аминоэтиламино-2-нафтол-4-
сульфокпслоты (при получении соединения I) или 10,8 г
(0,03 моль) !-аминоэтиламино-8-нафтол-4,6-дисульфокис-
лоты (при получении соединения II), 2 г едкого натра
и 6,5 г (0,1 моль) цианистого калия в 300 мл воды. Разбав-
228
ляют 8,2 г (0,1 моль) 37%-ного раствора формальдегида
водой до объема 100 мл и 20 мл этого раствора при 75 °C
добавляют к реакционной смеси в течение 1 ч. По окон-
чании добавления формальдегида отгоняют в вакууме
20 мл растворителя. Остаток формальдегида добавляют
к смеси в 4 приема: при 80, 85, 90 и 95 °C, каждый раз
в течение часа, и после каждой порции отгоняя в вакууме
по 20 мл растворителя. После добавления всего количе-
ства формальдегида реакционную массу выдерживают 2 ч
при 95 °C и перемешивании. Для выделения соединения
I реакционную массу охлаждают, добавляют 10 мл рас-
твора бисульфита натрия и подкисляют соляной кислотой
до pH 2. Выпавший осадок натриевой соли кристалли-
зуют из воды с добавлением активированного угля. Полу-
ченный после перекристаллизации осадок отфильтровы-
вают, отмывают 80%-ным этиловым спиртом до отрица-
тельной пробы на СГ-ионы и сушат в сушильном шкафу
при 70—80 °C. Получают 4,8 г натриевой соли соедине-
ния I; выход 33% (от теоретического). Для выделения
комплексона в виде кислоты 2 г натриевой соли раство-
ряют в 200 мл воды и пропускают ? через колонку с катио-
нитом КРС-5 (емкость 50 мл, диаметр 14 мм). Собирают
фракцию со значением pH 1—2, отгоняют воду в вакууме
в токе азота, в конце отгонки добавляют в несколько
приемов 100 мл абсолютного этанола и отгоняют досуха.
Полученный осадок сушат в сушильном шкафу при 50 °C.
Получают 1,8 г комплексона в виде кислоты.
Состав синтезированного продукта (C18H20N2O10S):
Найдено, %...........
Вычислено, % ... .
С Н N S
47,0; 46,9 4,5; 4,4 6,i; 5,8 7,4; 7,6
47,2 4,4 6,1 7,0
Для выделения соединения II из подкисленного до
pH 2 реакционного раствора отгоняют воду в вакууме
до образования густой сиропообразной массы, которая
кристаллизуется при обработке смесью ацетона и эти-
лового спирта (1 : 1). Осадок кислоты отделяют, промы-
вают 80%-ным этиловым спиртом до отрицательной пробы
на СГ-ион и сушат в сушильном шкафу при 60—70 °C.
Получают 9,3 г соединения II; выход 58% (от теоретиче-
ского).
229
Состав синтезированного продукта (ClgH20N2O13S2):
С Н N S
Найдено, % , . . 39,7; 39,6 4,3; 4,1 5,1; 5,0 12,2; 12,3
Вычислено, % . . 40,2 3,7 5,2 11,9
Литература
1, A i k е п J. К., Chem. a. Ind., 17, 1334 (1956).
2. Ластовский Р. П., Тростянская Е. Б., Тем-
кина В. Я., Колпакова И. Д., Макарова С. Б.,
Ярошенко Г. Ф., А п т о в а Т. А., авт. свид. СССР
178000; Бюлл. изобр., № 2 (1966).
N-ЭТИЛ ЕНИМИНОИЗОПРОПИЛДИАЛКИЛ-
ФОСФАТЫ
N-ЭТИЛ ЕНИМИНОИЗОПРОПИЛДИЭТИЛФОСФАТ
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
сн3 о
I II
-СНз-СН-О-Р^
СН2\
I
CH.Z
ОС2Н5
OC2HS
C8H20NO4P
Мол. вес 237,24
N-Этилениминоизопропилдиалкилфосфаты представ-
ляют интерес как физиологически активные соединения
и как исходные вещества для синтеза других производных
холииового ряда. Они могут быть получены при взаимо-
действии алкоголята натрия М-этилениминопропэнола-2
и диалкилхлорфосфатов. N-Этилениминоизопропилдиал-
килфосфаты в литературе не описаны и получены нами
впервые г.
Схема синтеза
СН2\
I N—
сн/
ОН
СН2-СН-СН3
Na
СН
ONa
!\ I
N—СН2—СН—СН3
230
о
II /OC^Hs
Cl-P(
хос2н5
сн2Х
I N—СН2-
сн3 о
I „ II /ОС2Н5
2П5
РЕАКТИВЫ
N-Этилениминопропанол 2 2, т. кип. 53—56 °C при 10 мм рт.
ст., п% = 1,4455.
Натрий металлический, ч., ТУ МХП 1664—50.
Диэтилхлорфосфат, свежеперегнанный, т. кип. 81—83 °C при
10 мм рт. ст.
Бензол, ч. д. а., ГОСТ 5955—51.
Способ получения
В трехгорлую колбу с обратным холодильником поме-
щают 10, 12г (0,1 моль) М-этилениминопропанола-2 и 50 мл
сухого бензола, вводят 2,3 г (0,1 моль) металлического
натрия (кусочками по 0,2—0,3 г) и выдерживают реак-
ционную массу, перемешивая, при 60—70 °C до полного
растворения натрия. Охладив полученный раствор алко-
голятэ натрия до 5 °C, добавляют по каплям 17,25 г
(0,1 моль) диэтилхлорфосфата, поддерживая температуру
5—10 °C. Затем смесь нагревают 30 мин на кипящей
водяной бане, охлаждают до комнатной температуры
и выпавший осадок хлористого натрия отделяют центри-
фугированием. Из фильтрата отгоняют бензол в вакууме
водоструйного насоса и продукт перегоняют в вакууме.
Получают 18,2 г вещества; т. кип. 117—119 °C при
4 мм рт. ст.; выход 76% (от теоретического), <й5 —
= 1,073, п* = 1,4327.
Содержание азота в препарате (C9H20NO4P):
Найдено, %.................... 5,71
Вычислено, %.................. 5,90
Так же получают другие N-этилениминоизопропил-
диалкилфосфаты, константы которых приведены ниже:
Название соединения Выход
N-Этилениминоизопропнлди
пропилфосфаг . . 35
N-Эгилениминоизопропилди-
изопропилфосфат . • 28
N-Эгилениминоизопропилци- н-
бутилфосфат.................. 24
Т. кип. (°C)
0. при давлении 25 j25
• '° (леле рт. cm.) nD а4
134—137; 4 1,4310 1,038
131—132; 4 1,4280 1,028
150—151; 4 1,4345 1,007
231
Литература
1. Дрегваль Г. Ф_, Самуилов А. М., авт. свид. СССР
188969; Бюлл. изобр., № 23 (1966).
2. Самуил ов А. М., Дрегваль Г. Ф., Методы полу-
чения химических реактивов и препаратов, вып. 18, ИРЕА,
1969, стр. 232.
N-ЭТИЛ ЕНИМИНОПРОПАНОЛ-2
А. М. САМУИЛОВ, Г. Ф. ДРЕГВАЛЬ
I <а'\ I
N-CH2-CM-CHS
сн/
CjHnNO Мол. вес 101,14
М-Этилениминопропанол-2 можно использовать как
промежуточный продукт для синтеза физиологически
активных веществ. В литературе описан способ получения
Н-этилениминопропанола-2 из окиси пропилена и этилен-
имина в присутствии воды с последующей обработкой
реакционной смеси щелочью и экстракцией бензолом,
из раствора которого М-этилениминопропанол-2 выделяют
перегонкой в вакууме (выход не приводится 2). Мы
предлагаем более простую и удобную методику синтеза,
исключающую обработку щелочью и экстракцию.
Схема синтеза
СН-
I NH + CH2-CH2-CH3
сн/ \/
РЕАКТИВЫ
Этиленимин, свежеперегнанный, т. кип. 54—57 °C.
Окись пропилена, ч., свежеперегнанпая, т. кип. 35 °C.
Способ получения
В трехгорлую колбу, снабженную обратным холодиль-
ником, термометром, капельной воронкой и механической
232
мешалкой, помещают 4,3 г (0,1 моль) этилениминг и при
О—10 °C, перемешивая, прибавляют 1,8 г (0,1 моль) воды,
а затем 5,8 г (0,1 моль) окиси пропилена. Реакционную
массу перемешивают 4 ч при 10—20 °C и 1 ч при 40—
50 °C. После перегонки в вакууме получают 3,3 г продукта;
т. кип. 53—56 °C при 10 мм рт. ст.\ выход 32% (от теоре-
тического), = 0,9487, ng = 1,4455. По литературным
данным2, т. кип. 161—163 °C, d™ = 0,941, ng = 1,4488.
Содержание азота в препарате (CsHllNO):
Найдено, %.................... 13,55
Вычислено, %.................. 13,85
Литература
1. Малиновский М. С., Окиси олефинов и их производные,
Госхимиздат, 1961, стр. 305.
2. Пат. США 2475068; С. А. 43, 7955 (1949).
ЭФИРЫ МАЛОНОВОЙ кислоты
И. П. РОДИОНОВ
ROOC-CH2-COOR
Эфиры малоновой кислоты обычно получают алкого-
лизом циануксусной кислоты или ее натриевой соли \
действием недокиси углерода С3О2 на соответствующие
спирты2 или переэтерификацией малонового эфира 3.
Концен-Кровет 4 получал кислые эфиры малоновой кис-
лоты действием расчетного количества спирта на малоно-
вую кислоту при нагревании, отгоняя воду в виде азео-
тропа со спиртом на ректификационной колонке. Этери-
фикация малоновой кислоты осложнена тем, что при
140 °C она декарбоксилируется, а в присутствии обыч-
ных катализаторов этерификации — сильных кислот —
разложение идет и при более низких температурах.
Поэтому мы проводили этерификацию без катализатора,
нагревая на водяной бане малоновую кислоту с избытком
спирта в присутствии четыреххлористого углерода. Для
выведения воды из сферы реакции отгоняющийся азеотроп
пропускался через патрон с водопоглощающим вещест-
вом (безводной сернокислой медью), а спирт возвращался
в колбу.
233
Физнко химические
к
S
X
о
со
<Я
к
Температура
кипения в за-
висимости от
давления
Название соединения
Брутто-
формула
Диметилмалочат . . ,
Ди-« пропилмалонат .
Ди-в/иор-бутилмалонат
Ди-н-гексилмалоиат .
Дициклогексилмалоиат
Ди-«-нонилмалонат . ,
132,12
188,22
216,27
272,38
268,35
356,55
—68,5
—71,5
—41,5
—24,0
4-4,0
72
88
105
157
177
214
10
4
7
4
8
5
1,1535
1,0111
0,97279
0,91697
1,0662
0,92143
Схема синтеза
НООС-СН2-СООН 4- 2ROH-> ROOC-CH2-COOR 4- 2НаО
РЕАКТИВЫ
Малоновая кислота, ч_, МРТУ 6-09-397—63,
Метиловый спирт, синтетический, ч., ГОСТ 2222—65.
н-П ропиловый спирт, ч., СТ ГОХП 15—2354.
н-Гексиловый спирт, ч., ВТУ РУ 1299—57.
Циклогексанол, ч., ВТУ МХП 3530—52.
втор-Битиловый спирт, ч.
н-Нониловый спирт, ч., СТУ 12-10-155—61.
Углерод четыреххлористый, ч., ГОСТ 4—65.
Медь сернокислая, безводная, ч., ГОСТ 897—41.
Способ получения
В круглодонную двугорлую колбу емкостью 500 мл,
снабженную насадкой Сокслета с обратным холодильни-
ком, помещают 20,8 г (0,2 моль) малоновой кислоты
и четыреххлористый углерод (количества этих реагентов
указаны в табл. 2). В насадку Сокслета вводят патрон
из фильтровальной бумаги, заполненный 0,3—0,5 моль
безводной сернокислой меди. Реакционную смесь кипя-
тят с обратной флегмой, проходящей через осушительный
патрон, на водяной бане (см. примечание 1), переносят
в колбу Клайзена, отгоняют четыреххлористый углерод,
234
свойства малоновых эфиров
Литературные данные
Таблица 1
i Молекулярная
nD рефракция
температура
кипения в за-
висимости от
давлення
g
е
cl
5*
5*
nD
1,4130
1,4202
1,4210
1,4343
1,4728
1,4438
1,4087
1,416
1,4165
1,4305
1,4689
1,4400
28,558
47,126
56,378
74,964
70,573
102,783
28,477
47,069
56,365
74,960
70,533
102,848
—699
—775
0,5—1,5’
1819 760
ИЗ5 13
118° 12
15110 1
167’ 0,18
1.41409
1,42066
1,4213’
1,473610
1,1544»
1,0097е
0,973s
0,9525s
1,0631»
d*>
4
взбыток спирта и остаток перегоняют в вакууме (см. при-
мечание 2). Выход малонатов составляет 45—84%. Усло-
вия реакций и физико-химические константы получен-
ных эфиров приведены в табл. 1 и 2.
Таблица 2
Условия этерификации малоновой кислоты
Название соединения Мольное соотношение реагентов Пподолжи- тельность реакции ч Выход %
малоновая кислота спирт четырех- хлопистын углерод
Диметилмалонат .... 1,о 30,0 16 68
Ди н-пропилмалонат . . . 1,0 2,5 1,87 8 84
Ди-втор-бутилмалолат . . 1,0 2,5 1 ,87 14 71
Ди-н-гексилмалонат . . . 1,0 2,5 1,87 8 83
Дицик логексилмалонат 1,0 2,5 2,39 15 45
Ди-н-нонилмалонат . . . 1,0 2,5 2,07 9 74,5
Синтезированные эфиры малоновой кислоты — про-
зрачные, бесцветные жидкости с характерным запахом,
нерастворимые в воде.
Примечания. 1.0 скорости реакции можно судить по
скорости окрашивания CuSO3 в синий цвет.
2. Чтобы в продукт не попала примесь моноэфира, смесь после
отгонки спирта нагревают до 180—190° С, при этом моноэфир декар-
обксилируегся и отгоняется из смеси.
235
Литература
1. R oss A. A., Bibbins F. E., Ind. Eng. Chem., 29, 1341
(1927); Препаративная органическая химия, Госхимиздат,
1959, стр. 370.
2. Дашкевич Л. G , ДАН СССР, 132, 1319 (1960).
3. Б о н д а р ь Л. С., Окунев Р. А., Родионов П. П.,
Пылина Н. В., Изв. АН СССР, Сер. хим., 10,. 1894 (1955).
4. Contzen-Crowet С., Bull. Soc. chim. Beiges, 35, 165
(1926).
5. Serwy H., Bull. Soc. chim. Beiges, 42, 483 (1933).
6. P a 1 о m a a M. H., Mikkila J., Chem. Ber., 75. 1659
(1942).
7. Staudinger H., Schwalenstoker H., Chem.
Ber., 68, 727 (1935).
8. Backer H. J., L о 1 k e m a J., Rec. trav. chim., 57,
11, 1234 (1938).
9. Справочник химика, т. II, Госхимиздат, 1963, стр. 762; т. IV,
1963, стр. 828.
10. F г a n k R. L., D a v i s W. R., (jr), DrakeS. S., M c-
Pherson J. R., (jr), J. Am. Chem. Soc., 66, 1509 (1944).
ЭФИРЫ САЛИЦИЛОВОЙ кислоты
И НОРМАЛЬНЫХ АЛИФАТИЧЕСКИХ СПИРТОВ
П. П. РОДИОНОВ, В. А. МАСЛЕННИКОВ
/ч/ОН
Cl
^^COOR
где R = С3Н7; С4Н9: C5Hn: С9Н19: Ci0H21
Алкилсалицилаты получают этерификацией салици-
ловой кислоты спиртами в присутствии различных ката-
лизаторов 1-5. Алкилсалицилаты с С4 — С4 в алкильном
остатке получают при действии диалкилфосфонатов в.
Описано также получение п-оксиметилбензоата при дей-
ствии диметилсульфата 7 и эфиров этиленгликоля при дей-
ствии хлорангидрида салициловой кислоты 8-9.
Нами предложен метод синтеза алкильных эфиров
салициловой кислоты с R С3 в алкильном остатке пере-
этерификацией метилсалицилата спиртами в присутствии
катализаторов основного характера — 5—10%-ных рас-
творов алкоголятов щелочных и щелочноземельных ме-
таллов. Серная кислота и л-толуолсульфохлорид (в коли-
честве 10 мол. %) не катализируют обмен алкоксильной
группы в салицилатах.
236
Схема синтеза
РЕАКТИВЫ
Метилсалицилат, ч., Л1РТУ 6-09-1239—64.
н-Пропиловый спирт, о. ч., ОРУ 53—56.
н-Бутиловый спирт, ч., ГОСТ 6006—51.
н-Амиловый спирт, ч., СТУ 10-158—61.
н-Нониловый спирт, ч., СТУ 12-10-155—61.
н-Децимиый спирт, ч., МРТУ 6-09-2476—65.
Hump едкий, х. ч., ГОСТ 4328—66.
Лития гидроокись, ч. а. а., МРТУ 6-09-1471—64.
Способ получения
В двугорлую колбу, снабженную термометром и елоч-
ным дефлегматором, соединенным с прямым холодильни-
Таблица 1
Условия переэтерификации
Названые соединения Мольное соотношение реагентов Пгсдолжи- дельность i реакции ч и ; MUH Выход алкил- салицилата %
исходный эфир алифати- ческий спирт катали- затор
«-Пропил салицила т Метилсалицилат 1,0 ОДОН 4,27 NaOH 0,112 4- 73,7
«-Бутилсалицилат Метилса лицила т 1,0 ОДОН 1,6 NaOH 0,1 2— 77,4
н-Амилсалицилат Метилсалицилат 1,0 С^цОН 1,6 Li ОН 0,04 10- 92
«-Нонилсалицилат Метилсалицилат 1,0 ОД8ОН 3,84 NaOH 0,07 о- 64
Бутилсалицилат 1,0 C,HWOH 2,0 NaOH 0,1 0— 74
н-Децилсалицилат Метилсалицилат 1,0 С10Н21ОН 3,84 NaOH 0,07 1- 65
Бутилсалицилат 1,0 CioH21OH 2,0 NaOH 0,05 о- 65
237
Физике химические константы алкилсалицилатов
ком и приемником, помещают 15,2 г (0,1 моль) метилен*
лицилата, 14,1 г (0,16 моль) амилового спирта, 0,1 г
(0,004 моль) гидроокиси лития (см. примечание 1). Смесь
нагревают, регулируя нагрев так, чтобы температура
паров не превышала 67—70 °C. Выделяющийся метанол
полностью отгоняют из реакционной смеси, после чего
реакция заканчивается. Реакционную массу охлаждают,
промывают 200 мл воды, отделяют органический слой,
водный слой экстрагируют 50 мл бензола, экстракт объ-
единяют с органическим слоем, отгоняют азеотроп бен-
зола с водой, спирт и остаток перегоняют в вакууме.
Получают 19,1 г амилсалицилата; т. кип. 125—133 °C
при 4—5 мм рт. ст., выход 92% (от теоретического).
Условия получения других алкилсалицилатов (см. при-
мечание 2) и их физико-химические константы приведены
в табл. 1 и 2.
Алкилсалицилаты с С! — СП| в алкильном остатке —
бесцветные прозрачные жидкости со специфическим запа-
хом, мало или почти нерастворимые в воде, при длитель-
ном хранении окрашиваются в желтый цвет. Для пре-
дотвращения появления окраски при хранении алкилса-
лицилатов рекомендуется прибавлять к ним 0,1 вес. %
фосфорной кислоты ,0.
Примечания. 1. В качестве катализаторов могут быть
использованы гидроокиси или окислы бария и кальция, а также
карбонат натрия.
2. Методом газо-жидкостной хроматографии на хроматографе
ХЛ-4 с детектором по теплопроводности показано отсутствие
в синтезированных алкилсалицилатах (R = С3 — С6) исходного
метилсалицилата, салициловой кислоты и соответствующего спирта.
Условия анализа: температура колонки 180—245 °C: диаметр колон-
ки 3,6 мм\ длина 1 м\ носитель — целит 545, фракции 0,14—12 мм',
неподвижная фаза — апиезон-Л4 (20% от веса носителя); газ-носи-
тель — гелий (3—4 л/>); ток детектора 92 ма.
Литература
1. Препаративная органическая химия, перев. с польского, Издат-
инлит, 1964, стр. 373—-374.
2. F а 1 t i n g s К., пат. ФРГ 957574; РЖХим, 40626П (1958).
3. Д ы х а и о в Н. Н., Шилов В. Р., Зорина Е. И.,
авт. свид. СССР, 162122; Бюлл. изобр., № 9 (1964).
4. Гельштейн Р. М., Кривошеева Н. Ф., Методы
получения химических реактивов и препаратов, вып. 13, ИРЕА,
1965, стр. 5.
5. Rahassa В а 1 1 а г a R., Rev. fac. cienc. quim., 27, 31
(1955; 1952); РЖХим, 66016 (1957).
239
6. H of f tn an F. W., Weiss H. t>., J . Am. Chem. Soc
79, № 17, 4759 (1957); РЖХим, 46.642 (1958).
7. Hirwe U. N., Thakor V. M., Jadhav V.,
Shah R. C., J. Univ. Bombay, 22, N° 5, 14 (1954); РЖХим,
11.624 (1955).
8. Kovac S., Chem. zvesti, 7, № 10, 659 (1953); РЖХим,
31580 (1955).
9. Англ. пат. 738178; РЖХим, 32, 117П (1957).
10. R a d u e R. W„ Пат. США 2918491; РЖХим, 16Л158 (1961).
МЕТОДЫ ПОЛУЧЕНИЯ ХИМИЧЕСКИХ РЕАКТИВОВ И ПРЕПАРАТОВ
выпуск 18
Издательство «Химия». М., 1969 г.
240 с. УДК [543 = 4 + 542 22]; 001.8.546/547.05
Редактор Л. Б. Демушкина
Технический редактор Л. С. Кочетова
Корректор М. С. Хрипунова, Е. П. Павлов
Т—14758 Подписано к печати 14/XI 1969 г.
Формат бумаги 84X1081/32 Печ. л. 7.5 Усл. печ. л. 12,6 Уч.-изд. л. 12.89
Типогп. 6vm. № 2 Тираж 3300 экз. Тем. план 1969 г.. № 97
Цена 75 коп. Зак. № 287
Типографии V? 21 ГтавттлчграЪпрэца Комитета по печати при
Совете Министров СССР- Москва, 88. Угрешская, 12