/


















































Автор: Ластовский Р.П. Поспелов А.М.
Теги: органическая химия химия химическая промышленность химические реакции
Год: 1964




Текст
ГОСУДАРСТВЕННЫЙ КОМИТЕТ
ХИМИЧЕСКОЙ ПРОМЫШЛЕННОСТИ ПРИ ГОСПЛАНЕ ССС
МЕТОДЫ ПОЛУЧЕНИЯ
ХИМИЧЕСКИХ РЕАКТИВОВ
И ПРЕПАРАТОВ
Выпуск 10
ВСЕСОЮЗНЫЙ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ BEIHEiCT 5
М О С К В А — 1 9 6 4
Редакционная коллегия:
Проф. Р. П. Ластовский (гл. редактор),
А. М. Поспелов (зам. гл. редактора),
В. Г. Брудзь, А. В. Бромберг, Е. А. Божеиольнов,
В. М. Дзиомко, Г. А. Певцов
СОДЕРЖАНИЕ
3-Аминодифениламин. А. Н. Гриценко, С. В. Журавлев, В. А. Ско-
родумов ...........................
Ангидро-бис-индандион-1,3. Р. А. Жагат, В.НЗелмен, Г.Я.Ва-
наг ..................................................•
Анилиндиуксусная кислота. В. Г. Синявский, Л. П. Кошечкина,
Е. Г. Белкина ............. ...........
Ацетил- и бензоилапетоны. В. Т. Скрипкина, Н. Н. Дыханов . . .
п-трет.. Бутилпирокатехин. И. Г. Гах. Л. Н. Асланова, М. К. Ми-
трохина, Г. И. Митрюкова.............. ....
Высшие простые эфиры. Н. Н. Дыханов, В. Р. Шилов........
Диацетамид. А. Ф. Нагорный, II. Ф. Семенова, Т. И. Комаренко,
Г. А. Любченко .........................................
Лиа цетил. Н. Н. Дыханов, Л. М. Егупова ............
Дибеизиламин. Л. Л. Саввина, Е. Я. Яровенко ..........
Дибензилдитиокарбамат цинка. Л. Н. Саввина, Е. Я- Яровенко . .
Дибензилцианамид. Л. Н. Саввина, Е. Я. Яровенко.........
1,4-Ди-(1,5-дифенил-д а-пиразолипил-3) бензол. В. Г. Тищенко . . .
2,5-Диметилацетофенон. В. Т. Бурмистров.................
л.я'-Диоксибензолсульфимид. Н. II. Дыханов, Г. Н. Никитенко .
2-Дифенилацетилиидандиои-1.3(дифенацин) и его гидразон (дифе-
зон). В. Н. Зелмен, 3. Н. Пристынь, Г. Я. Ванаг ........
2,7-Дихлорхромотроповая кислота. В. И. Кузнецов, Н. И. Басар-
гин .................................'..................
Р, р',-Дициандиэтиловый эфир. Н. Семенов, С. Бельченко,
И. Хвостов .............................................
Диэтилентриаминпентаацетат меди. И. А. Селиверстова, Л. В. Горо-
хова, Н. М. Дятлова.....................................
Изобутилнитрит. Н, Н. Дыханов, Л. М. Егупова.........
Йодистый метил. М. Н. Баисова, Н. Н. Дыханов............
л-Метоксифенол. В. М. Савоськин, И. П. Гаркуша-Божко,
Р. Н. Логвиненко, Н. С. Двойных, Л. В. Брюханова........
Монобензоильное производное дифенилолпропана. И. В. Хвостов .
Монооксим днацетила. Н. Н. Дыханов, Л. М. Егуповп............ 62
п-Нитробензилхлорид. И. Н. Дыханов, Л. М. Егупова............ оо
З-Нитродифеииламин. С. В. Журавлев, В. М. Светмева .... 67
7-Нитро-2- и 4-метокснакридоны-9 и 6- и 7-нитро-2- и 1-метокси-
10-метилакридоны-9. В. И. Кихтева, Н. И. Дыханов........... 70
4-Нитро-2'-, 3'- и 4'-метоксидифениламин-2-карбоновые кислоты.
В. И. Кихтева, Н. Н. Дыханов............................... 71
7-Нитро-2- и 4-метокси-9-хлоракридины. В. И. Кихтева,
Н. II. Дыханов............................................. 78
Орто- и пара-Нитрофепилгидразины. В. Т. Скрипкина, Н. /7. Ды-
ханов ..................................................... 61
о-Оксифснилвинилкетои. А. И. Турбина, В. Г. Синявский........ 86
Нормальные пере, октиловые, нониловые и дециловые эфиры изомер-
ных нитробепзойиых кислот. II. Н. Дыханов, В. Р. Шилов . . 90
Полистироламииоуксусная кислота — комплексообразующий кати-
онит. В. Г. Синявский, Л. П. Кошечкина, М. Я. Романкевич . 93
Функциональные производные 1,3,5- трифенилниразолина - Да.
В. Г. Тищенко .................. • ... 97
1,4-Эндометилен-1,2,3,4-тетрагидроантрахинон-9,10. Н. Н. Дыханов,
В. И. Кихтева ............... ........................... 101
2-Этипилпиридин. Н. Н. Дыханов, Т. С. Рыжкова............... 104
Перечень соединений, описанных в настоящем сборнике . . . 107
УДК 547.553.2.07
3-АМИНОДИФЕНИЛАМИН
N-Фенил- м -фенилендиамин
А. Н. ГРИЦЕНКО, С. В. ЖУРАВЛЕВ, В. А. СКОРОДУМОВ
Н
N NH,
// \ / \ / /
I II II I
C]aHi5N2 . М. в. 184,23
Из литературы известен метод получения 3-аминодифенил-
амина, основанный на восстановлении 3-нитродифениламина
оловом в соляной кислоте [Ц. Имеется также упоминание о
восстановлении 3-нитродифениламина гидрированием над ни-
келем Ренея, но без приведения каких-либо подробностей 121.
Ввиду отсутствия точного описания методов синтеза 3-амино-
дифениламина и его хлоргидрата, нами разработаны два' ме-
тода, основанные а) — на каталитическом восстановлении
3-нитродифениламина в присутствии скелетного никелевого
катализатора и б) на взаимодействии того же нитросоедине-
ния с гидратом гидразина в присутствии никеля Ренея [3].
3-Аминодифениламин был предложен нами для синтеза неко-
торых производных фентиазина [4].
Ввиду неустойчивости основания 3-аминодифепиламина
рекомендуется хранить его в виде хлоргидрата и переводить
в основание по мере надобности.
СХЕМА СИНТЕЗА 3-АМИНОДИФЕНИЛАМИНА
н
I
N N02
2 I 11 |l | +3N2HrH2O -
4/ \//
н
-L7H2O + 3N2
Характеристика основного сырья
3-Нитродифениламин, т. пл. 111,5—112,5°.
Никель Ренея, активный.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Водород из газометра.
Гидразин-гидрат, ч., ГОСТ 5832—51.
Условия получения
Гидрирование. В склянку прибора для гидрирования,
установленную в аппарате для встряхивания, помещают су-
спензию из 50 г 3-нитродифениламина в 350 мл спирта и 30 а
(^ 18 мл) никеля Ренея (см. примечание 1) и пропускают че-
рез смесь, при взбалтывании, газообразный водород из газо-
метра. З-Нитродифениламип по мере восстановления раство-
ряется, образуя прозрачный раствор темного цвета. При этом
происходит значительное саморазогревание. За 2—3 часа по-
глощается почти теоретическое количество водорода. Катали-
затор отфильтровывают (см. примечание 2), от фильтрата от-
гоняют спирт. Сиропообразный остаток растворяют в доста-
точном количестве 10—15%-ной соляной кислоты и охлажда-
ют. Выделившийся осадок отфильтровывают, промывают хо-
лодной водой и высушивают. Получают 48 г хлоргидрата
3-аминодифеииламина с т. пл. 223—225°, что составляет 83%
от теоретического выхода. После перекристаллизации из
спирта т. пл. продукта 235—236°. По внешнему виду это
игольчатые бесцветные блестящие кристаллы, хорошо раст-
воримые в горячей воде и спирте.
Восстановление гидратом гидразина. К 21 г 3-питродифе-
ниламина и 10 г (^-6 мл) влажного никелевого катализатора
в 200 мл этилового спирта добавляют при размешивании, в
течение 1 часа по каплям 15 г гидрата гидразина. Реакция
сопровождается саморазогреванием. После прибавления гид-
б
рата гидразина реакционную массу размешивают 1 час при
кипячении и катализатор отфильтровывают. Затем из филь-
трата отгоняют спирт, остаток растворяют при нагревании в
200 мл 3—5 %-ной соляной кислоты, очищают его нагревани-
ем с углем, фильтруют и охлаждают. По охлаждении из
фильтрата выпадает хлоргидрат 3-аминодифениламина в ви-
де осадка белого цвета. Выход продукта — 15 г, что состав-
ляет 82% от теоретического; т. пл. 234—235°.
Основание 3-аминодифениламина выделяют из раствора
хлоргидрата в горячей воде прибавлением 20%-ного раствора
едкого натра до сильнощелочной реакции. По охлаждении
осадок отсасывают и перекристаллизовывают из спирта, раз-
бавленного водой. Выход продукта почти количественный,
т. пл. 72—74°; по литературным данным, т. пл. 3-аминодифе-
ниламина 76—77э [1].
Примечания: 1. Никель Ренея после активации ввиду его пиро-
форности (самовоспламеняемости) хранят под спиртом и при загрузке в
реакционную колбу его количество рассчитывают по объему.
2. Осадок катализатора при высушивании может самовоспламениться.
Поэтому его еще во влажном состоянии следует немедленно залить
10%-ной соляной кислотой для растворения никеля.
ЛИТЕРАТУРА
1. Н. W1 е 1 a n d, W. R h е i п h е I m е г. Liebigs Ann. Chem., 423, 28
(1921).
2. Р. О г a m m a t i с a k i s. Bull. Soc. chim. Fiance, 21, 99 (1954).
3. D. В a 1 с о m, A. Furst. J. Amer. Chem. Soc., 75, 4334 (1953).
4. С. В. Журавлев, В. А. Скородумов. ЖОХ, 31, 3129 (1961).
Поступила в декабре 1963 г. Институт фармакологии и химиотерапии
АМН СССР
УДК 547.7.07
АНГИДРО-БИС-ИНДАНД ИОН-1,3
Биндон
Р. А. ЖАГАТ, В. Н. ЗЕЛМЕН, Г. Я. ВАНАГ
С18Н10Оз М. в. 274, 27
Биндон применяется в качестве аналитического реактива
на первичные амины [1, 2], нитросоединения [3] и гидразин
[4], а в последнее время и для определения малых количеств
аминов при флотации калиевых солей [5, 6].
Биндон является также исходным продуктом при получе-
нии метилбиндона, бромметилбиндона (реагенты на различ-
ные амины) [7—9].
Впервые биндон получен Вислиценусом в 1887 году само-
конденсацией индандиона-1,3 [10]. Нами показано, что для
приготовления биндона нет необходимости употреблять гото-
вый индандион-1,3. При его получении можно исходить из
сырой Na-соли эфира индандион-1,3-карбоновой-2 кисло-
ты [11].
Синтез биндона делится на две стадии:
1) получение Na-соли эфира индандионкарбоновой кисло-
ты;
2)разложение этой соли кислотой и самоконденсация об-
разовавшегося индандиона-1,3.
В настоящей работе дан удобный метод получения на-
триевой соли метилового эфира индандион-1,3-карбоновой-2
кислоты, являющейся промежуточным продуктом для по-
лучения индандиона-1,3.
8
Этот метод может быть применен в укрупненных установ-
ках.
СХЕМА СИНТЕЗА Na-СОЛИ МЕТИЛОВОГО ЭФИРА
ИНДАНДИОН-СЗ-КАРБОНОВОЙ-2 КИСЛОТЫ
{ 4 COOCI|::
L J-СООСНз
СН3СООС.,Н,; _
- - сн,он
о
11
\ — с ч
-* I с z-ccooc2H5
ONa
переэсте-
рификация
о
// \__С X
! ус ^ссооснз
ONa
Характеристика основного сырья
Диметилфталат, ч., ГОСТ 3985—53.
Этилацетат, абсолютный, СТ ГОСТ 27—1839.
Этиловый спирт, техн., ГОСТ 5962—51.
Толуол, ч., ГОСТ 5789—51.
Натрий, ГОСТ 3273-55.
Условия получения
Получение Na-соли метилового эфира индандион-1,3-кар-
боновой-2 кислоты. В литровую трехгорлую колбу, снабжен-
ную обратным холодильником с хлоркальциевой трубкой,
капельной воронкой и мешалкой из нержавеющей стали, за-
гружают 100 г сухого толуола и 40 г металлического натрия.
Колбу нагревают на песочной бане до начала кипения раст-
ворителя, затем при помощи механической мешалки превра-
щают расплавленный натрин в пылеобразное состояние (см.
примечание 1) и прекращают нагревание. Когда температура
в песочной бане понизится до 110°, прибавляют 200 г диме-
тилфталата и 30—50 мл этилацетата, повышают температу-
ру в песочной бане до 130—140° и поддерживают эту темпе-
ратуру до конца реакции. Если реакция не начинается,, то
прибавляют 10—15 мл абсолютного этилового спирта. Во
время реакции прибавляют по каплям остальную часть этил-
ацетата (всего 200 мл). Прибавление смеси регулируют так,
чтобы содержимое колбы слабо кипело. При очень бурной
реакции ускоряют приливание этилацетата и прекращают на-
гревание. Продолжительность реакции 4 -5 часов. К концу
реакции образуется густая желтая масса — натриевая соль
9
метилового эфира индандионкарбоновой кислоты, которую
трудно перемешивать.
По окончании реакции в колбу вливают 300 мл техничес-
кого этилового спирта для удаления избытка натрия и нагре-
вают колб)' около получаса. Остывшую густую желтую массу
отсасывают, промывают на воронке четыре раза по 50 мл
техническим этиловым спиртом и сушат при 80—100° около
6 часов.
Выход натриевой соли метилового эфира индандион-1,3-
карбоновой-2 кислоты равен 200—225 г, что составляет 88,5—
99,5% от теоретического. Продукт является достаточно чис-
тым для получения биндона.
СХЕМА СИНТЕЗА АНГИДРО-БИС-ИНДАНДИОНА-1,3
Характеристика основного сырья
Кислота серная, ч., ГОСТ 4204—48.
Калий углекислый, ч., ГОСТ 4221—53.
Кислота уксусная, ледяная, ГОСТ 61—51.
Условия получения
Получение ангидро-бис-индандиона-1,3. 80 г полученной
натриевой соли метилового эфира индандион-1,3-карбоно-
вой-2 кислоты суспендируют в 500 мл разбавленной серной
кислоты (1:1) и нагревают на водяной бане 2 часа. При этом
выделяется углекислый газ, желтый раствор темнеет и выпа-
дает осадок песочного или оранжево-коричневого цвета. По
окончании реакции добавляют ~ 1 л горячей воды и отсасы-
вают. Осадок на фильтре промывают горячей водой. Выход
технического биндона, содержащего смесь биндона и изобин-
дона, составляет 26,5 г.
Способ очистки. 20 г технического биндона, 300 мл воды
и 6 г карбоната калия кипятят несколько минут, до полного
растворения биндона. Фиолетово-красный раствор фильтру-
10
ют. Остаток на фильтре, представляющий собой калиевую
соль изобиндона, промывают 200 мл воды. Оба фильтрата
соединяют, выдерживают 10—12 часов, после чего отфильтро-
вывают дополнительно выпавшее небольшое количество ка-
лиевой соли изобиндона.
Полученный фильтрат медленно подкисляют соляной кис-
лотой до исчезновения фиолетово-красной окраски (см. при-
мечание 2), выпавший буровато-желтый осадок биндона от-
сасывают, промывают водой до тех пор, пока промывная вода
не начнет окрашиваться в красный цвет, и перекристаллизо-
вывают из ледяной уксусной кислоты. Выход ангидро-бис-
индандиона-1,3 равен 12 г, что составляет 28—33% в пересче-
те на диметилфталат. По внешнему виду это желтые кристал-
лы с т. пл. 208—209°; капилляр при определении послед-
ней не окрашивается в зеленый цвет, как это наблюдается с
бипдопом, содержащим изобиндон.
По литературным данным, т. пл. биндона 209 [12].
Примечания: 1. Выход продукта отчасти зависит от степени из-
мельчения расплавленного натрия.
2. Прибавлять соляную кислоту следует очень осторожно, так как при
этом происходит бурное выделение углекислоты.
Л И 'Г Е Р А Т У Р А
1. G. Wanag. Z. analyt. Chem., 113, 21 (1938); 119, 412 (1940); 122,
119 (1941).
2. К. Бауер. Анализ органических соединении, М., ИЛ, 1953,
стр. 142.
3. G. Wanag. Z, analyt. Chem., 126, 21 (1943).
4. Г. Я В а на г, Р. А. Ж агат. Докл. АН СССР, 133, 362 (I960),
5. A. Singewald. Z. analyt. Chem., 164, 219 (1953).
6. A. Singewald. Kali tlnd Steisalz, 2, 9, 281 (1959).
7. G. Wanag. Z. analyt. Chem., 123, 292 (1942).
8. P. А, Ж агат, Г. Я. В а н а г. Изв. АН Латв. СсР, сер. хим., 1961,
стр. 175.
9. Р. А. Ж а г а т, Г. Я. В а н а г. Изв. АН Лати. ССР, сер. хим., 1962,
стр. 35.
10. W. W i s 1 i с е n u s. Вег., 20, 594 (1887).
11. Р. А. Ж а г а т, В. 3 е л м е н, Г. Я. В а н а г. Изв. АН Латв. ССР,
сер. хнм.. 1961, стр. 71.
12. G. Wanag. Вег., 66, 1678 (1933).
Поступила в марте 1964 г.
Институт органического синтеза
АН Латв. ССР
УДК 547.551.44.07
АНИЛИНДИУКСУСНАЯ КИСЛОТА
В. Г. СИНЯВСКИЙ, Л. П. КОШЕЧКИНА, Е. Г. БЕЛКИНА
у—. /СН,СООН
< X
v= СН2СООН
C10HnNO4 М. в. 209, 20
Как известно из литературы, анилинднуксусная кислота
применяется в качестве комплексообразователя [1], а также
для синтеза селективного катионита путем азосочетания ее с
диазотированным полиаминостиролом [2].
Несмотря на довольно частое применение и неоднократ-
ные попытки получения анилиндиуксусной кислоты, в литера-
туре до сих пор не приведен удовлетворительный метод син-
теза этого препарата.
По приведенным в литературе данным анилиндиуксусную
кислоту в большинстве случаев получают путем двустадий-
ной конденсации анилина с монохлоруксусной кислотой, вы-
деляя в качестве промежуточного продукта фенилглицин и
конденсируя его далее с монохлоруксусной кислотой в при-
сутствии соды или ацетата натрия. Реактив получают срав-
нительно с невысокими выходами и недостаточно чис-
тым [3—6].
Нами проверено несколько методов синтеза анилипдиук-
сусной кислоты. Варьировали порядок смешения реагентов,
количество воды, температуру; анилин и монохлоруксусную
кислоту вводили в реакцию в виде основания и кислоты и в
виде солей — солянокислого анилина и натриевой соли моно-
хлоруксусной кислоты.
Как оказалось, наиболее эффективным является односта-
дийный метод получения анилиндиуксусной кислоты, согласно
которому к смеси анилина и водного раствора монохлорук-
сусной кислоты при нагревании дабавляется раствор едкого
натра до прекращения понижения pH реакционной среды.
12
СХЕМА СИНТЕЗА АНИЛИНДИУКСУСНОЙ КИСЛОТЫ
//\
| II 4- 2С1СН2СООН -г 2NaOH --
ч/
I
NH,
П -I- 2NaCl -Г 2Н2О
ч/
I
N(CH2CUOH)2
Характеристика основного сырья
Анилин, ч., ГОСТ 5819—51.
Монохлоруксусная кислота, ч., ГОСТ 5836—51.
Натр едкий, ч., ГОСТ 4328—48.
Условия получения
В реактор, снабженный обратным холодильником, мешал-
кой и капельной воронкой, помещают 60 г (0,65 М) свежепе-
регнанного анилина и раствор 183 г (1,98 М) монохлоруксус-
ной кислоты в 800 мл воды. Смесь нагревают на водяной ба-
не при температуре воды 80—90°, поддерживая pH в пре-
делах 10—12 прибавлением %-ного водного раствора едко-
го натра (см. примечание 1).
После того как pH перестанет понижаться (обычно после
прибавления— 700 мл раствора едкого натра), реакционную
смесь нагревают еще 2—3 часа. Затем горячий раствор обра-
батывают активированным углем, фильтруют и подкисляют
концентрированной соляной кислотой до pH 2—1. В даль-
нейшем смесь охлаждают, выпавшие белые кристаллы от-
фильтровывают, промывают ледяной дистиллированной водой
до отрицательной реакции на хлор-ион в промывных водах.
Полученный продукт высушивают в сушильном шкафу при
70—75°.
Выход анилиндиуксусной кислоты равен 105—115 г, что
составляет 85—90% от теоретического. Методом потенцио-
метрического титрования было установлено, что продукт со-
держит основного вещества 99,8% (см. примечания 2,3).
Найдено, %: N—6,61; 6,64.
CWHUNO4. Вычислено, %: N—6,7.
Примечания: 1. Уменьшение количества е ”Ы (а также увеличе-
ние концентрации щелочи до 40%) с целью повышения выхода не. при-
вело к положительным результатам. При этом продукт получается недос-
таточно чистым, желтеет при высушивании.
1.3
2. Следует отметить, что продукт с содержанием основного вещества
ниже 90—95% плохо кристаллизуется из воды (образуется маслообраз-
ное вещество). При высушивании и хранении желтеет. Поэтому такого
сорта продукты рекомендуется сушить в вакууме при комнатной темпе-
ратуре.
3. Анилиндиуксусная кислота с содержанием основного вещества вы-
ше 95% оч)ень хорошо кристаллизуется из воды.
Не рекомендуется нагревать водные растворы анилиндиуксусной кис-
лоты до кипения во избежание декарбоксилирования [3].
ЛИТЕРАТУРА
1. Р. II р ш и б и л. Комплексоны в химическом анализе, М., ИЛ, 1955,
стр. 25.
2. М. Я. Романкевич, В. Г. Синявский, Н. П. Цыганкова.
Укр. хим. ж., 9, 1096 (1962).
3. A. HausdOrfer. Вег., 22, 1798 (1889).
4. A. Mouilpied. J. Chem. Soc„ 87, 438 (1905).
5. Т. Johnson, R. В e n g i s. J. Amer. Chem. Soc„ 33, 745 (1911).
6. D. Vorlander, В. M u m m e. Ber., 34, 1617 (1901).
Поступила в марте 1964 г.
Институт химии высокомолекулярных соединений
АН УССР
УДК 547.572.3.07
АЦЕТИЛ- И БЕНЗОИЛАЦЕТОНЫ
В. Т. СКРИПКИНА, Н. Н. ДЫХАНОВ
Ацетилацетон Бензоилацетон
СН8-С-СН2-С-СН3 С6Н5-С-СН2-С-СН3
II 1| II II
0 0 0 0
С6Н8О2 М. в. 100, И С10Н1(А М. в. 162, 18
Р-Дикетоны, в том числе ацетилацетон и бензоилацетон,
находят широкое применение в разнообразных органических
синтезах. Для их получения предложено много методов
[1—2], из которых в отечественной промышленности исполь-
зуется конденсация этилацетата с ацетоном или, соответст-
венно, с ацетофеноном под действием этилата натрия, не со-
держащего сольватационного спирта.
В данной методике реакцию этилацетата с ацетоном и
ацетофеноном впервые предлагается осуществлять в присутст-
вии сухого технического бутилата натрия [3]:
СХЕМА СИНТЕЗА АЦЕТИЛ- И БЕНЗОИЛАЦЕТОНОВ
СН3-С = О -1- СН3—С—R + C4H9ONa ->
II
ОС2Н5 о
- СН3—С—СН=С—R 4- С2Н6ОН + С4Н9ОН
II I
О ONa
1 +СН3СООН
СНз-С-СНг—C-R 4- CH3COONa
где R = —СН3 или — С6НБ.
15
Характеристика основного сырья
Бутилат натрия, техн., содержание основного вещества не
менее 88 %
Этилацетат, абсолютный, Ст. ГОХП 27—1839.
Ацетон, ч. д. а., ГОСТ 2603—51 (или ацетофенон, ч., ТУ
ОРУ 28—55).
Метилен хлористый, ТУ МХП 3105—52.
Спирт метиловый, синтетический, ч., ГОСТ 6995—54.
Кислота уксусная, ГОСТ 7077—54, сорт 1.
Получение ацетилацетона. К сухому техническому бутила-
ту натрия, приготовленному из 22' г 95%-ного едкого натра
[3], при энергичном перемешивании приливают 88 г абсолют-
ного этилацетата и сразу же начинают постепенное прибав-
ление 29 г ацетона, поддерживая реакционную массу в сос-
тоянии легкого кипения.
По прибавлении ацетона массу кипятят еще в течение
3 часов с обратным холодильником, затем оставляют на
12 часов при комнатной температуре. За это время из реак-
ционного раствора выделяются кристаллы натриевого произ-
водного ацетилацетона.
Не отделяя кристаллов, к реакционной массе прилива-
ют 600 мл воды, размешивают до образования гомогенного
раствора, который экстрагируют хлористым метиленом
(дважды по 150 мл).
К освобожденному от нейтральных органических приме-
сей водному раствору натриевого производного ацетилацето-
на прибавляют охлажденную до 0° 20%-ную серную кислоту
до pH 6—7 (по универсальному индикатору) и технический
ацетилацетои экстрагируют из водного раствора хлористым
метиленом 3 раза по 200 мл. Объединённые метиленхлорид-
ные вытяжки сушат над 25 г прокаленного сульфата натрия,
фильтруют, к фильтрату прибавляют 3 г сухого бикарбоната
натрия и отгоняют растворитель (39—43°), который использу-
ют в следующем опыте для той же цели.
Остаток фракционируют при атмосферном давлении с
елочным дефлегматором (высота 50 см), собирая фракцию,
кипящую в пределах 125—1453 (25—30 г). Эту фракцию вы-
сушивают ещё раз над прокаленным сульфатом натрия (5—
6 г) и подвергают повторному фракционированию с тем же
дефлегматором.
Ацетилацетои квалификации «чистый» собирают при 135—
137° в количестве 20—22 г, что составляет 40—45% от теоре-
тического; продукт отвечает требованиям ВТУ МХП
3349—52.
Получение бензоилацетона. Реакцию 88 г абсолютного
этилацетата с 60 г ацетофенона ведут в присутствии сухого
технического бутилата натрия, приготовленного из 22 г
16
95%-ного едкого натра [3], так же, как и в синтезе ацетил-
ацетона, с тою лишь разницей, что реакционную массу не на-
гревают до кипения, а выдерживают в течение 12 часов при
комнатной температуре.
Освобожденный от нейтральных органических примесей
раствор натриевого производного бензоилацетона нейтрали-
зуют 50%-ной уксусной кислотой, выпавший осадок техничес-
кого бензоилацетона отфильтровывают, промывают ледяной
водой и дважды перекристаллизовывают из метанола (1 :4) с
охлаждением раствора до 5—8°.
Получают 48—50 г бензоилацетона с т. пл. 57,8е (при
скорости нагревания капилляра 0,5°/лшн); свойства продукта
отвечают литературным данным [4].
ЛИТЕРАТУРА
1. Синтезы органических препаратов, сб. 3, М., ИЛ. 1952, стр. 93.
2. Органические реакции, сб. 8, М., ИЛ, 1956, стр. 184.
3. Н. Н. Д ы х а н о в, В. Т. С к р и п к и н а. Методы получения хи-
мических реактивов и препаратов, М„ ИРЕ/\, № 9, 1964, стр. 28.
4. L, С 1 a i s е п. Вег., 695 (1905).
Поступила н сентябре 1963 г. ВНИИ Монокристаллов
2 Зак. 613
УДК 547.978.4.07
п-ТРЕТ. БУТИЛПИРОКАТЕХИН
И. Г. ГЛХ, Л. Н. АСЛАНОВА, М. К. МИТРОХИНА, Г. И. МИТРЮКОВА
ОН
/к / он
\ //
СН;1- с-сн8
сн3
С10Н)4О2 м. в. 166, 22
п-трет. Бутилпирокатехин получают алкилированием пи-
рокатехина трет, бутиловым спиртом [1, 2], изобутиловым
спиртом [3] или изобутиленом [4, 5, 6] в присутствии серной
кислоты [2, 4, 5], фосфорной кислоты [2, 3], активированной
японской кислой глины [1} и катионнообменной смолы [6].
Из других методов известны следующие: нагревание 1-
окси-2-бром-трет. бутилбензола в щелочном растворе в при-
сутствии СнаО [7] и окисление п-трет. бутилфенола персуль-
фатом по Элбсу [8].
Мы применили метод алкилирования пирокатехина в при-
сутствии катионита КУ-2. В качестве алкилирующего агента
использовали изобутилен и изобутиловый спирт.
СХЕМА СИНТЕЗА n-tnpem. БУТИЛ ПИРОКАТЕХИНА
ОН он
I
сн3-с-сн3
I
сн3
18
он
I
сн3.
;сн-сн8он
сн3х
он
КУ-2
СН3-С-СНз
сн,
Характеристика основного сырья
Пирокатехин, ч., ВТУ МХП 2792—51.
Изобутилен, Ефремовского завода СК.
Изобутиловый спирт, ч.д.а.,ГОСТ 6016—51.
Ксилол, без. сернистых соединений, ч., ВТУ У 279а—51.
Катионит КУ-2, ГОСТ 5695—53, в Н-форме, обезвоженный,
имеющий статическую обменную емкость 4,9 мг-экв/г.
Условия получения
Алкилирование пирокатехина на катионите КУ-2 изобути-
ловым спиртом. В четырехгорлую колбу емкостью 1 л,
снабженную механической мешалкой, термометром, капель-
ной воронкой и водоотделителем, соединенным с обратным
холодильником, помещают 55 а пирокатехина, 20 г катиони-
та КУ-2 и 200 мл ксилола (см. примечание 1). Колбу нагре-
вают на масляной бане до 140° и при энергичном перемешива-
нии, из капельной воронки, в течение 5—6 часов, прибавляют
59 г изобутилового спирта, разбавленного 200 мл ксилола. 13
водоотделителе собирается вода, выделяющаяся в результате
реакции. По окончании прибавления спирта реакционную
смесь выдерживают при интенсивном перемешивании и тем-
пературе 140° примерно в течение часа до прекращения вы-
деления воды.
Алкилат осторожно декантируют с катализатора, который
может быть использован для повторного синтеза. Затем при
атмосферном давлении отгоняют растворитель (ксилол), а
остаток перегоняют под вакуумом при 130—160720 мм соби-
рают невступивший в реакцию пирокатехин, а при 160—
175720 мм— п-трет. бутилпирокатехин (сырой продукт с т.
пл. 45—49°).
Выход этого продукта равен 73,8 а, что составляет 88,9%
от теоретического.
Для очистки препарат перегоняют еще раз, отбирая фрак-
цию при 168—173720 мм, которая составляет 50% от взятого
количества сырого продукта. Температура плавления чистого
п-трет. бутилпирокатехина 54°.
2* 19
По литературным данным, т. пл продукта имеет различ-
ные значения: 47—48= [7]; 35, 5—36° [1]; 53—55° [2]; 55е
[6] и 62° [4] (см. примечание 2).
Алкилирование пирокатехина на катионите КУ-2 изобути-
леном. В четырёхгорлую колбу емкостью 1 л, снабженную
механической мешалкой, термометром, обратным холодиль-
ником и барботером для подвода газа, загружают 110 г пиро-
катехина, 40 г катионита КУ-2 и 400 мл ксилола (см. приме-
чание 1). Затем при перемешивании и температуре 90° про-
пускают 30 л газообразного изобутилена со скоростью 3—
5 л/час. После прекращения подачи газа реакционную массу
охлаждают и декантируют алкилат. Дальнейшую обработку
алкилата проводят так же, как описано выше.
Выход п-трет. бутилпирокатехина равен 120 г, что состав-
ляет 72%, считая на взятый пирокатехин.
Примечания: 1. В качестве растворителя можно использо-
вать и другие ароматические или алифатические соединения, температура
кипения которых близка к указанной температуре проведения реакции.
2. Сырой продукт можно очистить и перекристаллизацией. Для этого
100 г сырого продукта растворяют при слабом нагревании в 100 мл пет-
релейного эфира или бензина (т. кип. 80—120°). Затем раствор охлажда-
ют при интенсивном перемешивании. Через 20—30 минут выпадают бе-
лые кристаллы, которые отфильтровывают па воронке Бюхиера, сушат в
вакуум-эксикаторе и взвешивают. Выход продукта после перекристаллиза
ции равен 50—70 г; т. пл. 51—53°.
ЛИТЕРАТУРА
1. К у ват а, Куметика. РЖхим, 7, 22929 (1957).
2. Хэ Б и н-л нн, Ма Гу й-л ань, Ц з я н ь Т и н-б а о. Гао феицза
туньсинь, 4—6, 190 (1959).
3. В. Д. Тамбовцев а. Труды САГУ, кв. 33, 1953, стр. 69
4. J. F. Jelinek. Chem. Prumvsl, 9, 8, 398 (1959).
5. Пат. ГДР 11619; РЖхим, 8, 26312 п (1958).
6. Л. Н. Кириченко, В. И. И с агу ля нц. Кинетика, катализ и
нефтехимия, Труды МИНХиГП им. Губкина, (2), вып. 37, 133 (1962).
7. L. Mills, В. Fayerweathe. Пат. США 1942827 (1934).
8. S. М. S et h n a. Chem. Rev., 49, 91 (1951).
Поступила в марте 1964 г.
донецкий филиал ПРЕД
УДК 547.27.05
ВЫСШИЕ ПРОСТЫЕ ЭФИРЫ
Н. Н. ДЫХАНОВ, В. Р. ШИЛОВ
R-0-R,
где R = — н.СвН18, — н.С7Н15, — н,С8Н17, —п.СэН13 и
н.С10Н2|.
Высшие простые эфиры, применяемые в качестве специфи-
ческих высококипящих растворителей в гриньяровских синте-
зах, могут быть получены нагреванием спиртов с концентри-
рованной серной кислотой. Однако достигаемые при этом вы-
ходы простых эфиров весьма невелики, так как первоначаль-
но образующиеся алкилсерные кислоты легко распадаются до
олефинов; одновременно протекают реакции полимеризации,
окисления и др. [1].
В значительно меньшей степени те же побочные процессы
имеют место при проведении этерификации высших спиртов
в присутствии безводных арилсульфокислот [2—4], но работа
с последними, особенно в больших масштабах, осложняется
их высокой гигроскопичностью.
Авторами данной работы найдено [5], что эффективными
катализаторами этерификации спиртов с шестью и более ато-
мами углерода являются дешевые и доступные арилсульфо-
хлориды. В их присутствии процесс протекает через стадию
промежуточного образования сложных эфиров арилсульфо-
кислот, которые затем переэтерифицируются исходными спир-
тами с образованием простого эфира и арилсульфокислоты, а
последняя реагирует далее с новой молекулой спирта, обра-
зуя алкиларилсульфонат и воду. При непрерывном удалении
воды из сферы реакции выходы простых эфиров близки к
количественным.
21
СХЕМА СИНТЕЗА ВЫСШИХ ПРОСТЫХ ЭФИРОВ
Ar—SO2—С1 + R-OH Ar—SO2—OR + НС1;
Ar—SO2—OR R-OH --- R-O-R + Ar-SO„-OH;
Ar—SO2—OH + R-OH Ar—S0.2 —OR |-H2O,
где Ar—ароматический, a R —алифатический радикалы.
Характеристика основного сырья
Гексиловый спирт первичный, ч. д. а., ВТУ РУ 1299—57.
Гептиловый спирт первичный, ч. д. а., СТУ 12—10—61.
Октиловый спирт первичный, ч. д. а., ВТУ РУ 1301—57.
Нониловый спирт первичный, ч. д. а., ВТУ МХП РУ
1302—57.
Дециловый спирт первичный, ч., ВТУ РУ 1303—57.
n-Толуолсульфохлорид, ч., ВТУ ГХК 1577—61.
n-Хлорбензолсульфохлорид, т. пл. 53,5°.
Условия получения
В колбу, снабженную мешалкой с каучуковым обтюрато-
ром и автоматическим водоуловителем (насадка типа Дина-
Старка), верхний конец которого соединен с обратным холо-
дильником, помещают 1 г-моль нормального первичного гек-
силового (102,2 г), гептилового (116,2 г), октилового
(130,2 г), нонилового (144,2 г) или децилового (158,3 г)
спирта и 0,01 г-моля д-толуолсульфохлорида (19 г) или
п-хлорбензолсульфохлорида (21,1 г) *. Образовавшийся
раствор кипятят до полного прекращения накопления воды
в водоуловителе (~9 мл), на что требуется не более 3 часов.
Охлажденную до комнатной температуры реакционную
массу встряхивают в делительной воронке с примерно рав-
ным объёмом 2%-кого водного раствора едкого натра. Орга-
нический слой отделяют, промывают водой до нейтральной
реакции, фильтруют через слой прокаленного сульфата нат-
рия и фракционируют при обычном или уменьшенном давле-
нии. Получают простые эфиры с выходами 95—97% от теоре-
тических; их температуры кипения, удельные веса и резуль-
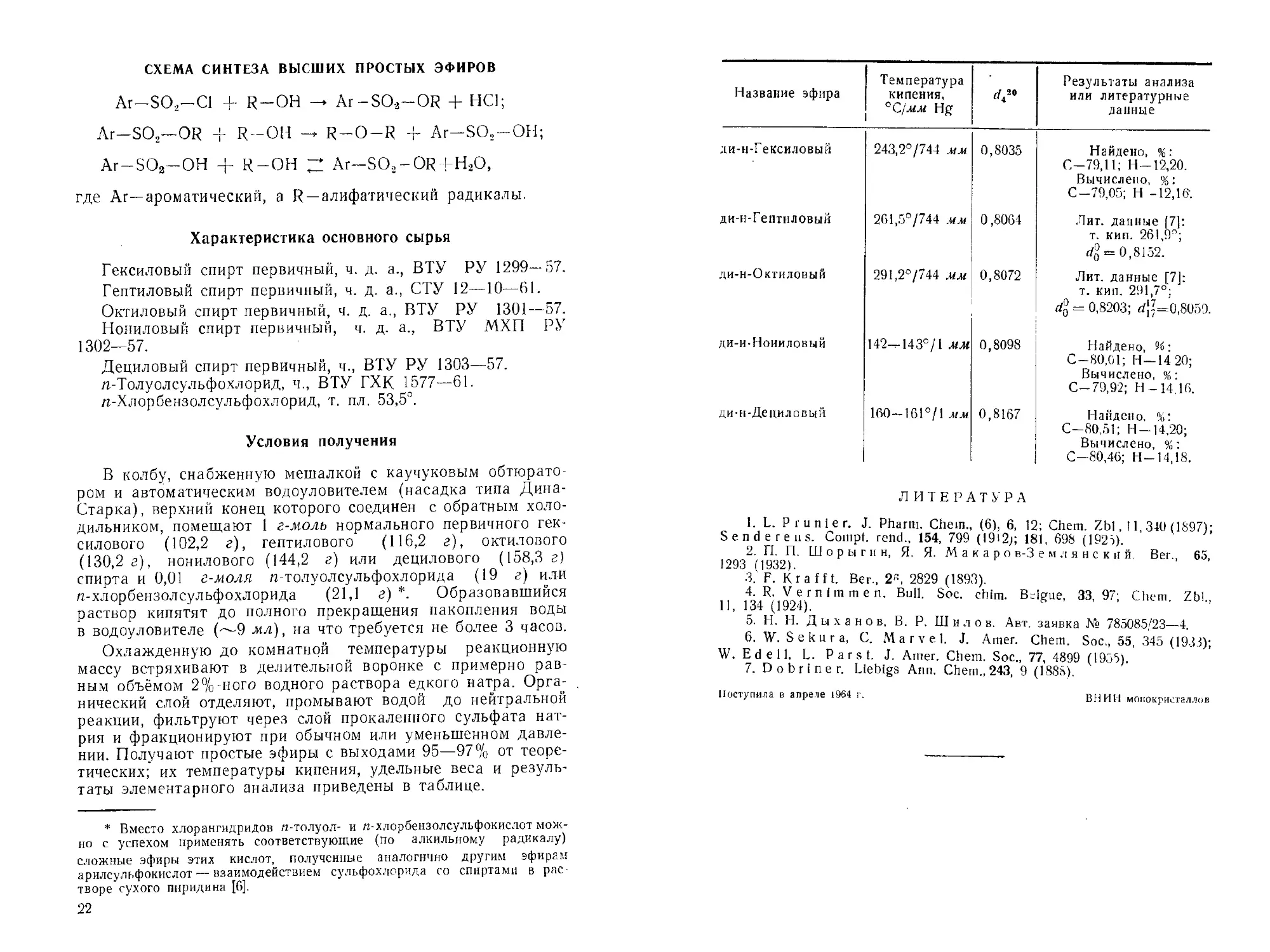
таты элементарного анализа приведены в таблице.
* Вместо хлорангидридов /г-толуол- и п-хлорбензолсульфокислот мож-
но с успехом применять соответствующие (по алкильному радикалу)
сложные эфиры этих кислот, полученные аналогично другим эфирам
арилсульфокислот — взаимодействием сульфохлорида со спиртами в рас-
творе сухого пиридина [6].
22
Название эфира Температура кипения, °С/лглг Hg <742’ Результаты анализа или литературные данные
ди-н-Гексиловый 243,2°/744 мм 0,8035 Найдено, %: С-79,11; Н—12,20. Вычислено, %: С-79,05; Н -12,16;
ди-н-Гептиловый 261,5°/744 мм 0,8064 Лит. данные |7): т. кип. 261,9°; 4 = 0,8152.
ди-н-Охтиловый 291,2°/744 мм 0,8072 Лит. данные [7]: т. кип. 291,7°; = 0,8203; d{?=0,8059.
ди-и-Ноииловый 142—143°/1 мм 0,8098 Найдено, 96: С-80,01; Н —14 20; Вычислено, %: С-79,92; Н-14.16.
ди-н-Дециловый 160-161°/! мм 0,8167 Найдено, %; С-80,.51; Н-14,20; Вычислено, %: С-80,46; Н-14,18.
ЛИТЕРАТУРА
1. L. Р г u n 1 е г. J. Pharm. Chem., (6), 6, 12; Chem. Zbl, 11,340 (1897);
Send ere us. Compt. rend., 154, 799 (19i2>; 181, 698 (1925).
2. П. П. Шорыгин, Я. Я. Макаров-Землянский, Вег., 65,
1293 (1932).
.3. F. Krafft. Вег., 2Л 2829 (1893).
4. R. Vernlmmen. Bull. Soc. chim. Belgue, 33,97; Chem Zbl.,
11, 134 (1924).
5. H. H. Дыханов, В. P. Шилов, Авт. заявка № 785085/23—4.
6. W. Sekura, C. Marvel. J. Amer. Chem. Soc., 55, 345 (1933);
W. Edell, L. P a r s t. J. Amer. Chem. Soc., 77, 4899 (1935).
7. Dobriner. Liebigs Ann. Chem., 243, 9 (1888).
Поступила в апреле 1964 г.
ВНИИ монокристаллов
УДК 547.298.1.07
ДИАЦЕТАМИД
N - Ацетилацетамид
А. Ф. НАГОРНЫЙ, Н. Ф. СЕМЕНОВА, Т. И. КОМАРЕНКО,
Г. А. ЛЮБЧЕНКО
СН3—СО—NH—СО—СН3
C4H7O2N М. в. 101,10
Диацетамид может быть получен;
1) взаимодействием ацетамида с хлористым ацетилом в
бензоле [1, 2];
2) взаимодействием ацетамида с хлористым ацетилом и
пиридином в дибутиловом эфире [3];
3) ацетилированием ацетамида уксусным ангидридом
[4, 5] в присутствии соляной кислоты [6], бромистого ацетила
или бромистого водорода [7];
4) из ацетамида и изопропенилацетата с т. кип. 220—222°
{8] и т. пл. 81—82° [9];
5) ацетилированием ацетамида кетеном [10].
Нами проверен и уточнен метод получения диацетамида
ацетилированием ацетамида хлористым ацетилом в бензоль-
ной среде с последующей перекристаллизацией продукта из
смеси бензола и петролейного эфира.
СХЕМА СИНТЕЗА ДИАЦЕТАМИДА
CH3CONH, 4-СН3СОС1 CH3CONHCOCH3 4-на
Характеристика основного сырья
Ацетамид, ч. д. а., ГОСТ 684—51.
Ацетил хлористый, ч., ГОСТ 5829—51.
Бензол, ч. д. а., ГОСТ 5955—51.
Петролейный эфир, ч., МХТУ 279—59.
24
Условия получения
В двух- или трехгорлую колбу, снабженную обратным хо-
лодильником и стеклянной мешалкой с глицериновым или
ртутным затвором, помещают 273 мл хлористого ацетила и
675 мл бензола. При постоянном энергичном перемешивании
добавляют небольшими порциями 423 г расплавленного аце-
тамида.
По прибавлении всего ацетамида смесь нагревают 3 часа
для полного завершения реакции, затем охлаждают и отфиль-
тровывают образовавшийся осадок нерастворимого в бензоле
хлоргидрата ацетамида.
Фильтрат переносят в колбу Вюрца и отгоняют бензол
при температуре 78—80°. Остаток перегоняют с воздушным
холодильником, собирая фракцию при температуре 205—220°.
Продукт быстро закристаллизовывается. Его перекристалли-
зовывают из 140 мл смеси бензола и петролейного эфи-
ра (1:1).
Выход диацетамида равен 22 г, что составляет 17%, счи-
тая на хлористый ацетил. Диацетамид представляет собой
белый мелкокристаллический порошок с температурой плав-
ления 76—79°.
ЛИТЕРАТУРА
1. A, W. Titherley. J. Chem. Soc., 412 (1901).
2. С. D. Hurd, M. F. Dull. J. Amer. Chem. Soc., 54, 2436 (1932).
3. V. R. Olson, H. B. Feldman. J, Amer. Chem. Soc., 59, 2004
(1937).
4. W. Hentschel. Ber„ 23 II, 2395 (1890).
5. D. Davidson, H. Skovronek. J. Amer. Soc., 80, 376 (1938).
6. J. B. Polya, P. L. Tardrew. J. Chem. Soc., 1081 (1948).
7. K. Shishido, H. M ozaki, M. Mato. C. A„ 89751 (1951).
8. H. J. Hagemeyer. C. A., 5522 (1955).
9. H. K. Hall, M. K. Brandt, R. M. Mason. J. Amer. Chem. Soc.,
80, 6420 (1958).
10. R. F. Dunbar, G. C. White. J. Organ. Client., 23, 915 (1958).
Поступила в декабре 1963 г.
ИРЕА, ЦЗЛ
УДК 547.442,2.05
ДИАЦЕТИЛ
Н. И. ДЫХАНОВ, Л. М. ЕГУПОВА
Н3С-С-С-СН3
II II
о о
С4НаО2 М. в. 86,09
Диацетил при сильном разведении напоминает по запаху
свежее масло и в связи с этим находит применение в произ-
водстве искусственных жиров.
Наиболее общераспространенным способом получения
диацетила является гидролиз изонитрозометилэтилкетона
(изомерная форма монооксима диацетила) разбавленной сер-
ной [1] или азотной [2] кислотой. Синтез исходного для этой
цели изонитрозометилэтилкетона описан в статье данного
сборника [3]. Ниже приводится описание гидролиза изонит-
розометилэтилкетона, полученного по этой методике, без вы-
деления его из реакционной массы.
СХЕМА СИНТЕЗА ДИАЦЕТИЛА
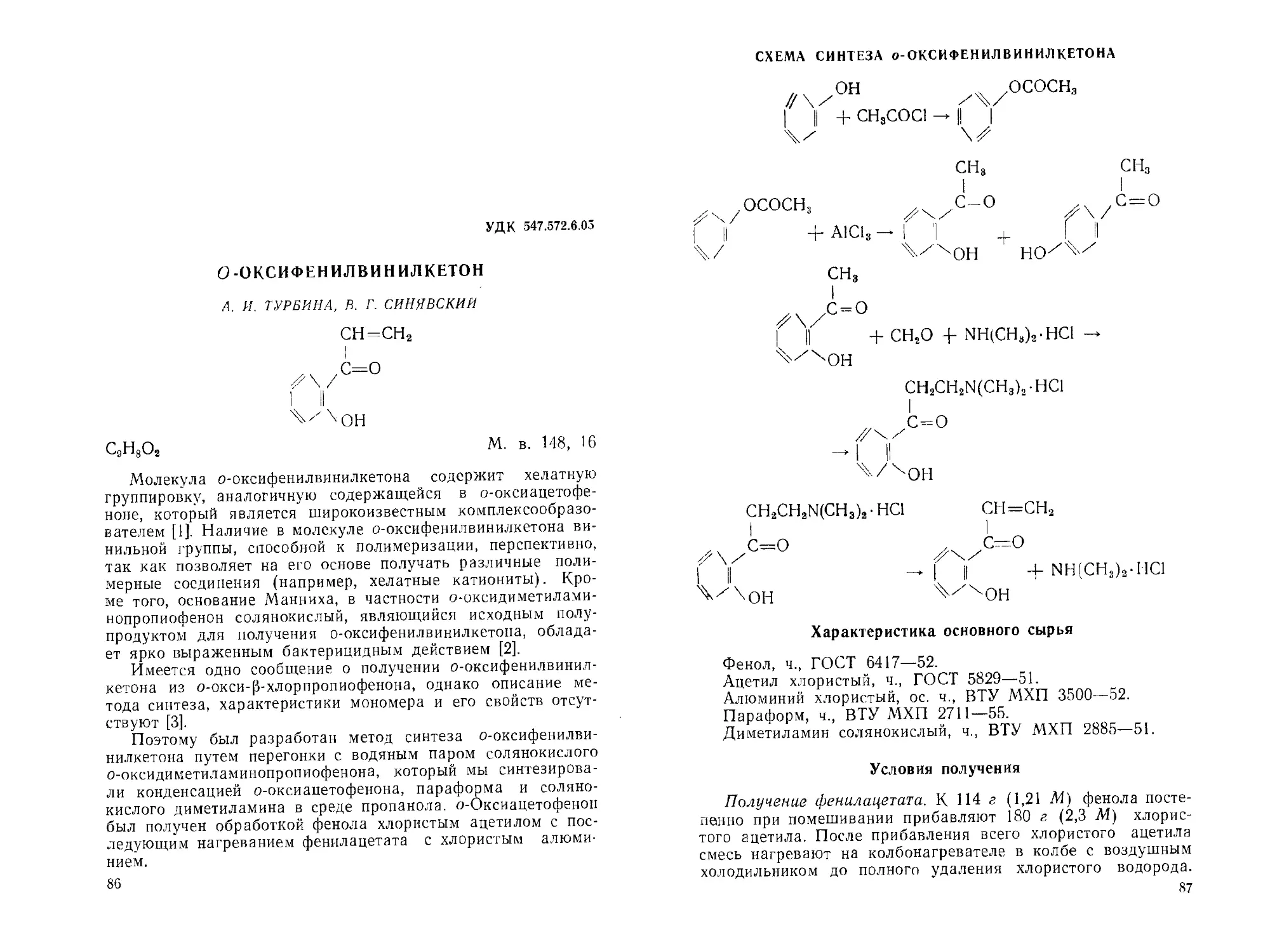
Н
I
Н3С-С-С-СН3 + 1-C4H9-O-N = O
11 1 н
он у
-> Н3С-С-С-СН3 + 1-С4НэОН
II I
О N=O
1 НА н+
Н2С-С-С-СН3 + nh2oh
26
Характеристика основного сырья
Метилэтилкетон, очищенный, ОСТ НКЛес 7606/90.
Изобутилнитрит, свежеприготовленный, т. кип. 65—70°.
Кислота соляная, техн., синтетическая, ГОСТ 857—57.
Натрий азотистокислый, техн., ГОСТ 6194—52, сорт 1.
Кислота серная, контактная, улучшенная, ГОСТ 2184—59.
Натрий хлористый, техн,, ГОСТ 4233—48.
Кальций хлористый, безводный, ГОСТ 450—58, сорт 1.
Условия получения
Смесь 43 мл метилэтилкетона и 2 мл концентрированной
соляной кислоты нитрозируют 53 мл свежеперегнанного изо-
бутилнитрита, как описано в синтезе [3]. Реакционную
массу переносят в трехлитровую круглодонную кол-
бу, снабженную капельной воронкой и воздушным холодиль-
ником, прибавляют раствор 141 г нитрита натрия в 240 мл
воды и затем прикапывают смесь 120 мл концентрированной
серной кислоты и 340 мл воды с такой скоростью, чтобы тем-
пература в массе не превышала 15°.
После того как в реакционной смеси перестанет обнару-
живаться свободная азотистая кислота (15—20 часов), при-
бавляют 200 г поваренной соли, воздушный холодильник за-
меняют на нисходящий прямой холодильник и отгоняют азе-
отроп диацетила с водой до тех пор, пока проба дистиллата
не перестанет темнеть от прибавления избытка 20%-ного вод-
ного раствора едкого натра.
Дистиллат переносят в делительную воронку, дают отсто-
яться и отделяют верхний слой сырого диацетила. Нижний
водный слой насыщают поваренной солью и вновь произво-
дят отгонку сырого диацетила, который отделяют и присо-
единяют к предыдущей порции, а нижний слой подвергают
ещё раз такой же обработке.
В результате трехкратной отгонки получают 18—20 г сы-
рого диацетила, который сушат в течение 2 дней над 2 г про-
каленного хлористого кальция и перегоняют, собирая чистый
диацетил в пределах 86—90°. Выход его равен 15—16 г, что
составляет около 35% от теоретического.
ЛИТЕРАТУРА
1. С. Schramm. Вег., 16, 177 (1883); Н. Р е с h in а п п. Вег., 20,
3113 (1887); 21, 1411 (1888).
2. S. С. J. Olivier. Bull. Soc. chim. France, (4), 51, 99 (1932).
3. H. H. Д ы x а и о в, Л. M. E г у п о в а. См. статью Монооксим диа-
цетила в этом сборнике.
Поступила в сентябре 1963 г.
ВНИИ монокристаллов
УДК 547.554.05
ДИБЕНЗИЛА.МИН
Л. Н. САВВИНА, Е. Я. ЯРОВЕНКО
(С6Н6СН2)2 NH
C14H15N м. в. 197, 28
По литературным данным, дибензиламин может быть по-
лучен взаимодействием спиртового раствора аммиака
[1, 2, 3, 4] или жидкого аммиака [5, 6] с хлористым бензилом
из цианамида кальция и хлористого бензила [7], а также вос-
становительным алкилированием аммиака бензальдегидом
под давлением [8], восстановлением альдазинов цинком в
уксусной среде [9], каталитическим гидрированием бензил-
амина [10, 11], бензальдоксима.
Нами дибензиламип получен гидролизом дибензилциан-
амида 40%-ной серной кислотой, при этом были найдены
оптимальные условия проведения реакции и выделения про-
дукта.
СХЕМА СИНТЕЗА ДИБЕНЗИЛАМИНА
Haso4
(C6H5CH2)2NCN 4-2Н2О-----> (C8H6CH2)2NH + СО2 4-NH3
Характеристика основного сырья
Дибензилцианамид, т. пл. 47—50°.
Кислота серная, ч., ГОСТ 4204—48.
Натр едкий, ч., ГОСТ 4328—-48.
Условия получения
В трехгорлую колбу емкостью 0,25 л, снабженную обрат-
ным холодильником, мешалкой, термометром и капельной во-
28
ронкой, загружают 68 мл 40%-ной серной кислоты и при
размешивании прибавляют 22,2 г (0,Ш) дибензилцианамида.
Смесь нагревают в течение 5 часов на глицериновой бане
при слабом кипении.
Затем, охладив содержимое колбы до 30—40°, добавляют
еще 26 мл 40%-ной серной кислоты (общая загрузка серной
кислоты составляет 0,5 М), снова нагревают до температуры
слабого кипения и поддерживают его в течение 3 часов.
Далее, реакционную массу снова охлаждают до комнат-
ной температуры и постепенно, по каплям, прибавляют око-
ло 70 ты (1 А4) 40%-ного раствора едкого натра до слабоще-
лочной реакции по универсальной индикаторной бумажке.
Образовавшийся дибензиламин всплывает в виде масла,
которое отделяют в делительной воронке. Для более полного
выделения дибензиламина к водному щелочному раствору
прибавляют хлороформ и трижды экстрагируют остатки ди-
бензиламина. Хлороформные экстракты сливают к основному
продукту и сушат плавленным едким натром.
Разгонку проводят в колбе Кляйзена. Вначале, при атмос-
ферном давлении, отгоняют хлороформ, затем, осторожно
включая вакуум, перегоняют дибензиламин при температуре
181 —183° и 20 мм.
Выход продукта с т. кип. 298—300° при 760 мм равен
13,0—13,8 г, что составляет 66—70%, считая на дибензилциа-
намид.
По литературным данным, т. кип. продукта 300° при
760 мм [13].
ЛИТЕРАТУРА
1. Н. Limpricht. Liebigs Ann. Chem., 144, 305 (1867).
2. S. Cannizzaro. Bull. Soc. Chim , (2>, 2, 126 (1861).
3. A. Mason. J. Chem. Soc,, 63, 1311 (1893).
4. S. Cannizzaro. Liebigs Ann. Chem., 134, 128 (1865).
5. S. Hubert, M. V i n z e n z. Чехосл. nar. 89637; РЖхим, 9 л. 169
(1961).
6. R. A s a m i, M. S h 1 m о. Япон. пат. 6556, С. A., 11921e (1958).
7. W. Traub e, A. E n g e I h a r d t. Ber., 44, 3152 (1911).
8. F. Winans. J. Amer. Chem. Soc., 61, 3566 (1939).
9. Th. C u r 11 u s, H. Franzen. Ber., 34, 552 (1901).
10. K. Kindler, W. P e s c h k e. Liebigs Ann. Chem., 644, 23 (1961).
11. C. Ainsworth. J. Amer. Chem. Soc., 78, 1635 (1956).
12. W. H. Hartung, j. Amer. Chem. Soc, 50, 3370 (1928).
13. Д P. Стэлл. Таблицы давления паров индивидуальных веществ,
М.,ИЛ, 1949.
Поступила в декабре 1963 г.
И РЕ А
УДК 547.496.3.07
ДИБЕНЗИЛДИТИОКАРБАМАТ ЦИНКА
Л. Н. САВВИНА, Е. Я. ЯРОВЕНКО
-С,Н5СН2
;n-c-s-
с6н6сн/ II
L S
C3oH28N2S4Zn
М. в. 610,19
Дибензилдитиокарбамат цинка находит применение для
аналитических целей при определении малых количеств меди
и в качестве ускорителя вулканизации.
По литературным данным, общим методом получения со-
лей диалкилдитиокарбаминовых кислот является взаимодей-
ствие диалкиламипов с сероуглеродом в присутствии едкого
натра с последующим обменным разложением диалкилдитио-
карбаматов натрия сернокислыми солями [1, 2].
Этот метод нами проверен и уточнен для получения дибсп-
зилдитиокарбамата цинка.
СХЕМА СИНТЕЗА ДИБЕНЗИЛДИТИОКАРБАМАТА ЦИНКА
С6Н6СН2 ч
'N—Н 4- C=-S 4 NaOH
с6н5сн2х II
S
с6н5сн2,
— \N-C-S-Na +Н2о
С6Н5СН/ II
S
С6н6сн3
2 )N-C -S-Na 4- ZnSO4'7H,O
С6Н5СН/ ||
S
30
ГС6Н5СН2
;n-c-s
сен6сн/ ||
L S
Zn + Na2SO4
2
Характеристика основного сырья
Дибензиламин, ч., СТУ 79—381—X—60.
Сероуглерод, ч., ГОСТ 1541—42.
Цинк сернокислый, ч., ГОСТ 4174—48.
Условия получения
В трехгорлую колбу емкостью 0,5 л, снабженную обрат-
ным холодильником, механической мешалкой, капельной во-
ронкой и термометром, загружают 19 мл (0,1 Л4) дибензил-
амина и, при энергичном перемешивании, прибавляют 6,1 мл
(0,Ш) 40%-ного раствора едкого натра, после чего переме-
шивание продолжают еще в течение 30 минут при комнатной
температуре.
К образовавшейся густой массе дибензилдитиокарбамата
натрия прибавляют 250 мл 1%-ного раствора уксуснокислого
натрия и перемешивают в течение 15—20 минут до полного
растворения массы.
Затем к полученному раствору прибавляют 72 мл (0,05 М)
20%-пого водного раствора сернокислого цинка, при этом не-
медленно начинают выпадать белые мелкие кристаллы дибен-
зилдитиокарбамата цинка. Для полноты проведения реакции
смесь, продолжая размешивать, выдерживают 1,5—2 часа при
комнатной температуре, после чего образовавшийся осадок
дибснзилдитиокарбамата цинка отфильтровывают, промыва-
ют дистиллированной водой до отрицательной реакции на
ион SO4" и сушат при температуре 60—70°.
Выход дибензилдитиокарбамата цинка с т. пл. 183—185°
равен 24,5—25 г, что соответствует 80% от теоретического.
По литературным данным, т. пл. продукта, перекристалли-
зованного из бензола, 185—186° [3].
ЛИТЕРАТУРА
1. A. Clifford, J. L i с h t i. J. Amer. Soc., <54, IIG3 (1932).
2. Польск. пат. 39141; РЖхим, Зл59 (1961).
3. L. Compin. Bull. Soc. chim. France. (4), 27, 468 (1920).
Поступила в декабре 1963 г. ПРЕД
УДК 547.554.07
ДИБЕНЗИЛ ЦИАНАМИД
Л. Н. САВВИНА, Е. Я. ЯРОВЕНЕО
едсн2.
;n—cn
C(iH5CH/
CI6H14N2 М. в. 222, 29
Дибензилцианамид является исходным продуктом в син-
тезе дибензиламина.
По литературным данным, дибензилцианамид получают из
цианамида кальция (или цианамида натрия) и хлористого
бензила при 12—24-часовом кипячении в 50%-ном спирте [1],
при введении хлорциана в спиртовой раствор дибензилами-
на [2, 3].
В основу нижеприведенной методики нами положен синтез
дибензилцианамида из цианамида кальция и хлористого бен-
зила, при этом были найдены оптимальные условия проведе-
ния реакции и растворитель для экстракции дибензнлциана-
мида.
СХЕМА СИНТЕЗА ДИБЕНЗИЛДИТИОКАРБАМАТА ЦИНКА
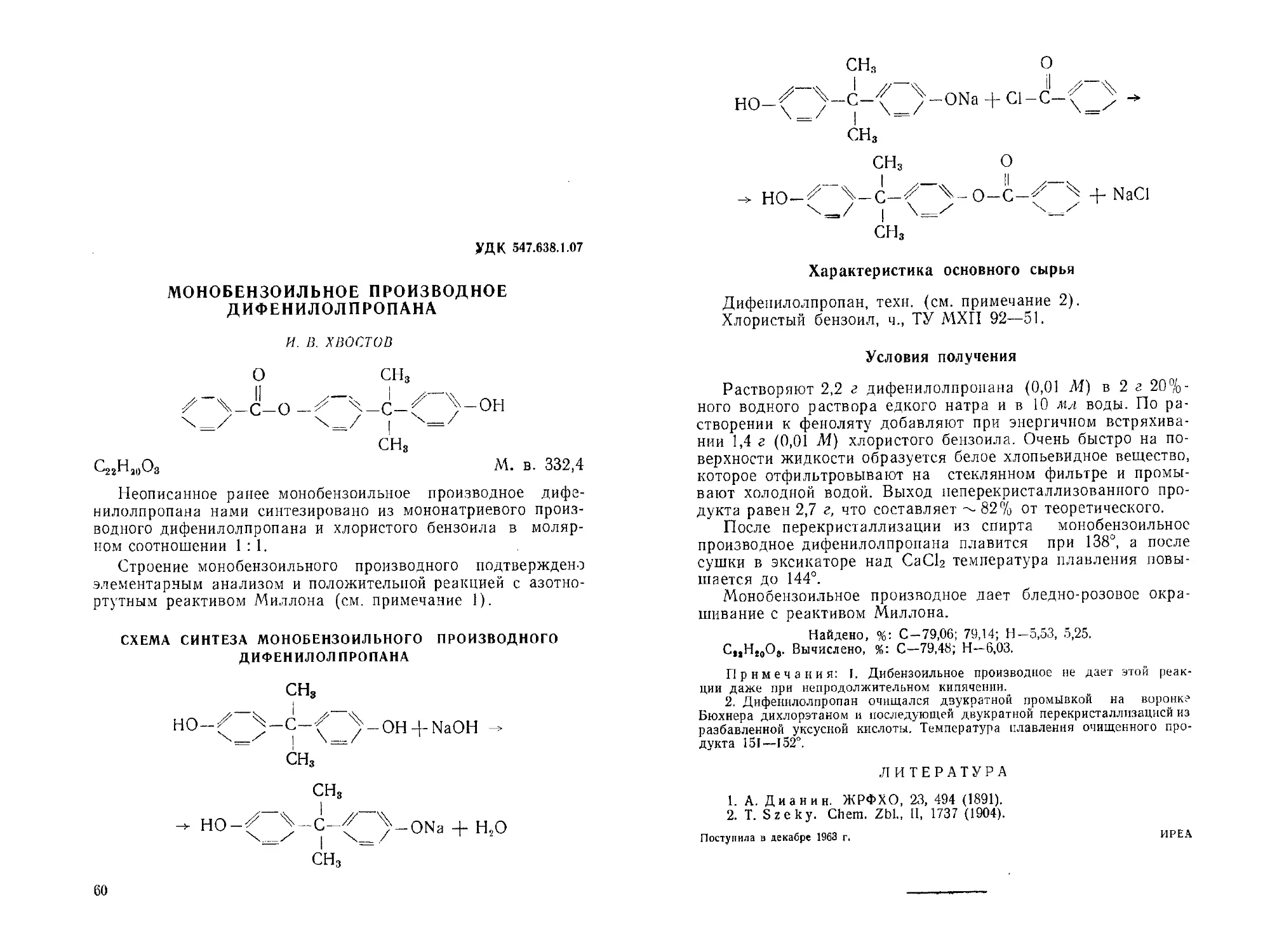
CaNCN + 2NaOH — Na2NCN Д- Ca(OH)2
2С2Н5СНаС1 + Na2NCN (CeH5CH2)2NCN + 2NaCl
Характеристика основного сырья
Хлористый бензил, ч., ТУ МХП 50—49.
Цианамид кальция, техн., ГОСТ 1780—56.
Условия получения
В трехгорлую колбу емкостью 0,25 л, снабженную обрат-
ным холодильником, мешалкой и капельной воронкой, загру-
32
жаюг 42 мл дистиллированной воды, 10 г измельченного льда
и при энергичном перемешивании вносят небольшими порция-
ми 14,6 г (0,1Л4) цианамида кальция и суспендируют 30 ми-
нут. Затем осторожно приливают 11,6 мл (0,2Л4) 40%-ного
водного раствора едкого натра, следя за тем, чтобы темпера-
тура при этом не поднималась выше 25° (см. примечание) и
перемешивают 1 час. К приготовленной таким образом вод-
ной суспензии цианамида натрия прибавляют 23 мл (25,3 г,
0,2/И) хлористого бензила, предварительно растворенного в
50 мл этилового спирта.
Реакционную смесь нагревают до слабого кипения и под-
держивают его 3 часа, после чего производят отгонку с водя-
ным паром непрореагировавшего хлористого бензила, спирта
и побочного маслянистого продукта. Затем содержимое колбы
охлаждают до комнатной температуры, фильтруют и осадок
сушат па воздухе до постоянного веса.
Из сухого осадка выделяют дибензилцианамид экстракци-
ей хлороформом в аппарате Сокслета. Из хлороформных вы-
тяжек отгоняют хлороформ (регенерируется около 80%).
Оставшуюся густую массу темно-желтого цвета в горячем
состоянии переливают в фарфоровую чашку и, при размеши-
вании, дают ей закристаллизоваться. Образовавшиеся крис-
таллы дибензилцианамида сушат на воздухе.
Выход продукта с т. пл. 47—50° равен 15—16,3 г, что сос-
тавляет 67,7—73,5% от теоретического.
Полученный дибензилцианамид без дополнительной очист-
ки используется для получения дибензиламина.
По литературным данным, т. пл. перекристаллизованного
продукта равна 53,5° [1].
Примечание. Если температура будет выше 25°, то часть циа-
намида полимеризуется в дициандиамид.
ЛИТЕРАТУРА
1. W. Trail be, A. Engelhardt. Вег., 44, 3151 (1911).
2. Н. Limpricht. Liebigs Ann. Chem., 144,313 (1867).
3. О. Wallach. Ber., 32, 1873 (1894).
Поступила в декабре 1963 г. И PEA
3 Зак. 613
УДК 547.7.07
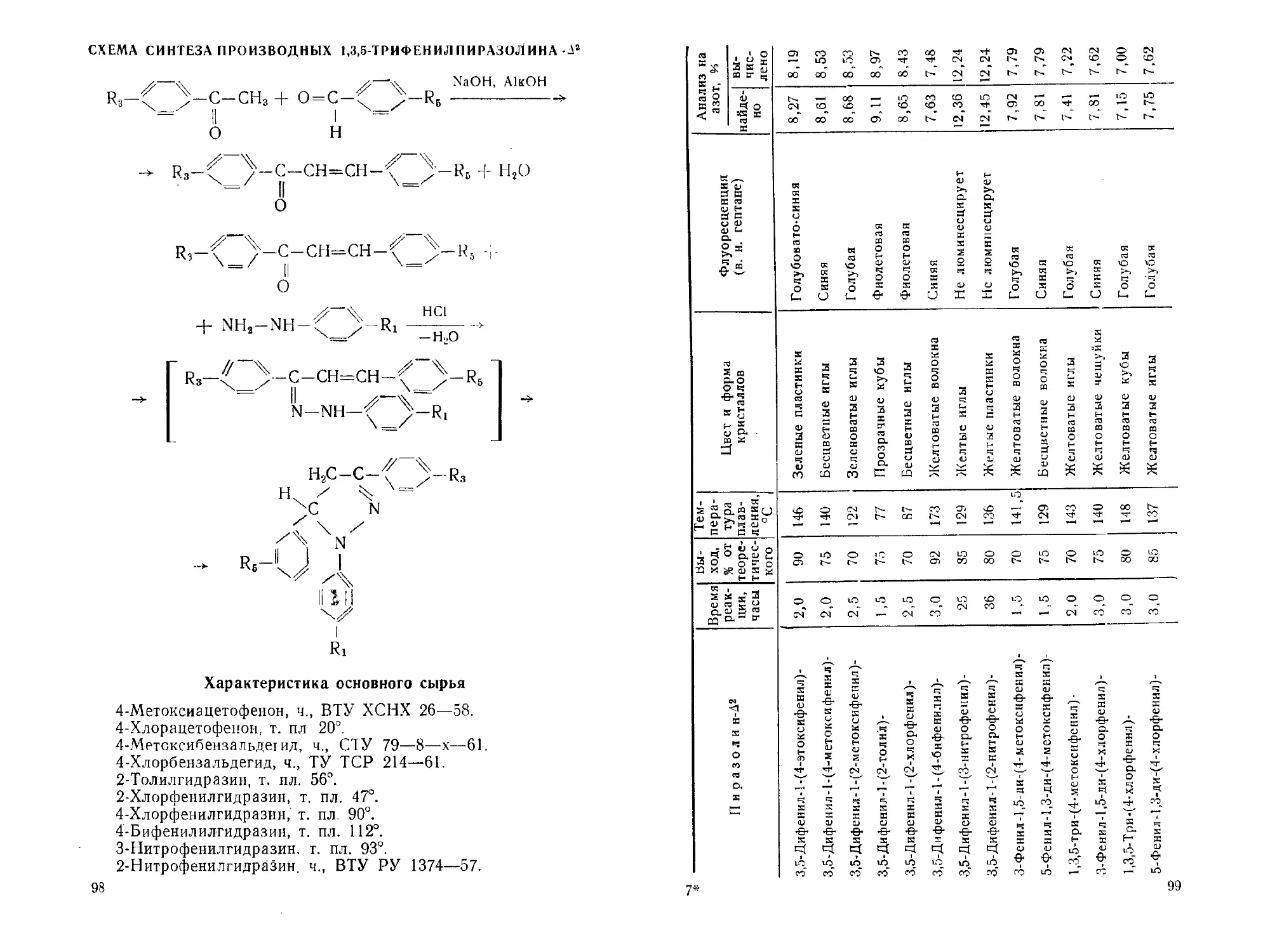
1,4-ДИ-(1,5-ДИФЕНИЛ-А2-ПИРА30ЛИНИЛ-3)-БЕН30Л
В. г. ТИЩЕНКО
^-С-СН2
C!6H3„N4
М. в. 518, 66
1,4-Ди- (1,5-дифенил-А2-пиразолинил-3) -бензол применяет-
ся в качестве сместителя спектра жидких сцинтилляторов в
области 500—510 ммк [1]. Его синтез впервые осуществлен
автором данной статьи по нижеприведенной методике.
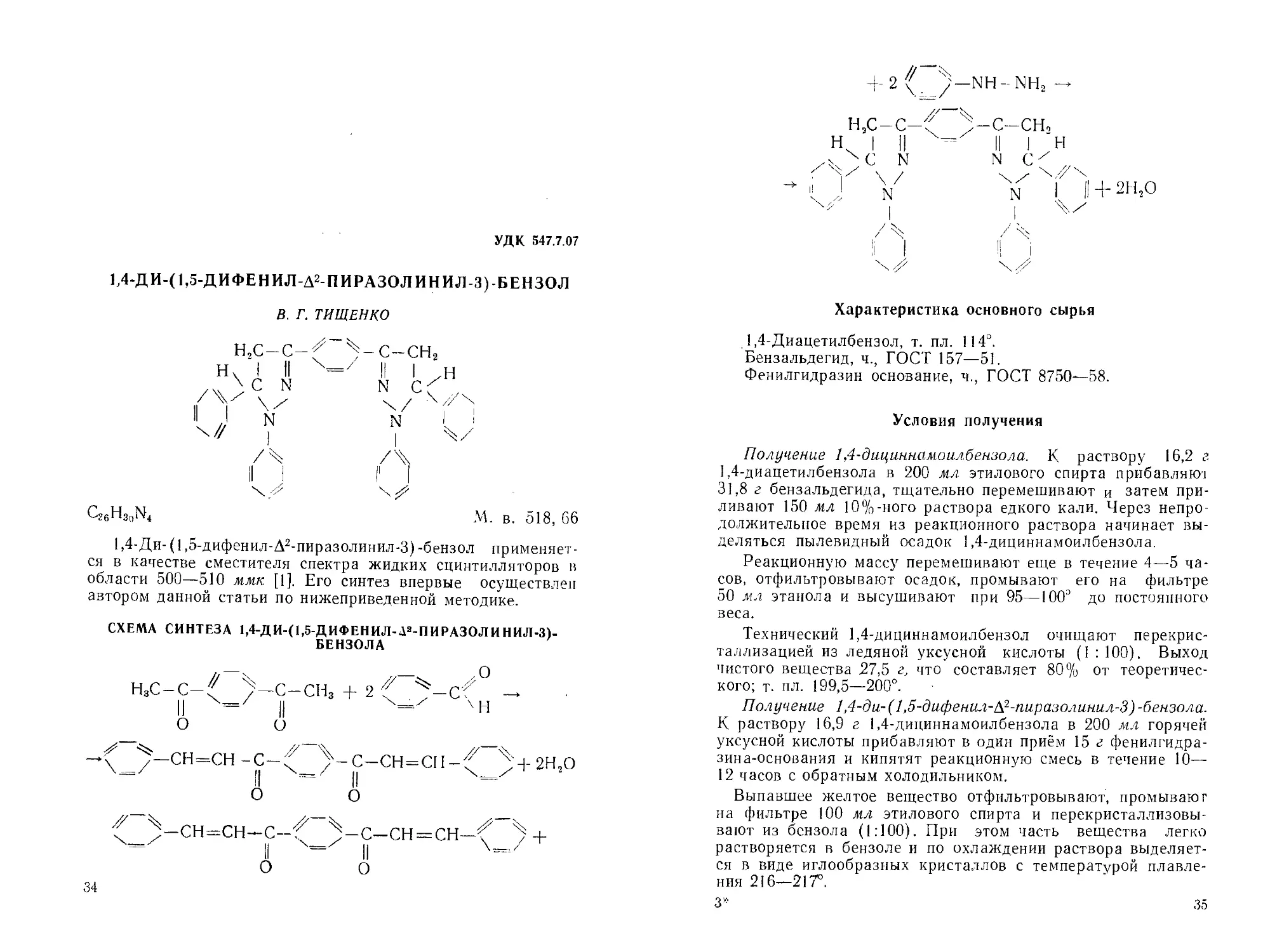
СХЕМА СИНТЕЗА 1,4-ДИ-(1,5-ДИФЕНИЛ-Д2-ПИРАЗОЛ ИНИЛ-3)-
БЕНЗОЛА
4-2 /'-NH-NH2 —
Н5С-С-^ ^-с-сн,
Н I II --- II I н
x'JjC N N С\4/"
1; J n | Н- 2Н2О
I I
Характеристика основного сырья
1,4-Диацетилбензол, т. пл. 114°.
Бензальдегид, ч., ГОСТ 157—51.
Фенилгидразин основание, ч., ГОСТ 8750—58.
Условия получения
Получение 1 А-дициннамоилбензола. К раствору 16,2 г
1,4-диацетилбензола в 200 мл этилового спирта прибавляю!
31,8 г бензальдегида, тщательно перемешивают и затем при-
ливают 150 мл 10%-ного раствора едкого кали. Через непро-
должительное время из реакционного раствора начинает вы-
деляться пылевидный осадок 1,4-дициннамоилбензола.
Реакционную масс}' перемешивают еще в течение 4—5 ча-
сов, отфильтровывают осадок, промывают его на фильтре
50 мл этанола и высушивают при 95—100э до постоянного
веса.
Технический 1,4-дициннамоилбензол очищают перекрис-
таллизацией из ледяной уксусной кислоты (1 :100). Выход
чистого вещества 27,5 г, что составляет 80% от теоретичес-
кого; т. пл. 199,5—200°.
Получение 1,4-ди-(1,5-дифен1иг-/^-пиразолинил-3)-бензола.
К раствору 16,9 г 1,4-дициннамоилбензола в 200 мл горячей
уксусной кислоты прибавляют в один приём 15 г фенилгидра-
зина-основания и кипятят реакционную смесь в течение 10—
12 часов с обратным холодильником.
Выпавшее желтое вещество отфильтровывают, промывают
на фильтре 100 мл этилового спирта и перекристаллизовы-
вают из бензола (1:100). При этом часть вещества легко
растворяется в бензоле и по охлаждении раствора выделяет-
ся в виде иглообразных кристаллов с температурой плавле-
ния 216—21 Г.
3 х 35
Другая часть вещества растворяется с трудом в большом
объёме бензола и выделяется при охлаждении в виде мелких
пластинок с температурой плавления 249—250°.
При длительном кипячении бензольного раствора первое
вещество переходит во второе и, по-видимому, является пово-
ротным изомером 1,4-ди-(1,5-дифенил-А2-пиразолииил-3)-беп
зола. Оба вещества имеют одинаковые сцинтилляционные ха-
рактеристики и элементарный состав.
Найдено, %: N—11,0.
СзвН3о^4. Вычислено, %: N-10,8.
ЛИТЕРАТУРА
1. В. Ф. Подужайло, В. Г. Тищенко. Авт. заявка № 832659/23-4.
Поступила в апреле 1964 г.
ВНИИ монокристаллов
УДК 547.572.1.07
2,5-ДИМЕТИЛАЦЕТОФЕНОН
В. Т. БУРМИСТРОВ
С10Н1аО
М. в. 148, 20
По литературным данным, 2,5-диметилацетофенон получа-
ют по реакции Густавсона-Фриделя-Крафтса при действии
ацетилхлорида на n-ксилол в присутствии хлористого алюми-
ния в растворе сероуглерода с выходом 53% [1, 2, 3, 4, 5].
Предлагаемая нами видоизмененная методика позволяет
получать 2,5-диметилацетофенон без применения сероуглеро-
да с выходом 80% от теоретически возможного.
СХЕМА СИНТЕЗА 2,5-ДИМЕТИЛАЦЕТОФЕНОНА
сн, сн3
Airi %'\ _ г_ СН
I II +- сНзСОС) | II V 3,
Ч/ х/ О
СН3 СН3
Характеристика основного сырья
п-Ксилол, ч., ТУ МХП 3601-53.
Хлористый ацетил, ч., ГОСТ 5829—51.
Хлористый алюминий, технический.
37
Условия получения
В сухую четырехгорлую колбу емкостью 1 л, снабженную
механической мешалкой, капельной воронкой, затвором и
термометром, вносят 173,3 г (1,3 М) хлористого алюминия и
42 г л-ксилола (см. примечание 1). Затем, при охлаждении
и интенсивном перемешивании, прикапывают в течение 30 ми-
нут 78,5 г (1 М) хлористого ацетила, при этом температура в
колбе не должна превышать 14—15°.
По окончании прикапывания ацетилхлорида температуру
реакционной смеси поднимают до 20’ и поддерживают ее еще
1 час. Затем реакционную массу выливают на смесь 60 г
измельченного льда и 5 мл концентрированной соляной ки-
слоты (см. примечание 2). Образовавшийся желто-коричне-
вый органический слой отделяют, а водный слой экстрагиру-
ют 2—3 раза эфиром. Экстракт соединяют с основной массой
кетона, сушат сульфатом магния (см. примечание 3) и фрак-
ционируют из колбы Клайзена, снабженной небольшим елоч-
ным дефлегматором. Сначала отгоняют эфир и н-ксилол (см.
примечание 4), а затем при 104—104,8°/9 мм— 2,5-диметнл-
ацетофенон с константами: di20— 0,9960 и —1,5300, что
точно соответствует литературным данным [3].
Выход продукта равен 118,56 г, что составляет 80% от
теоретического.
2, 5-Диметилацетофенон представляет интерес как проме-
жуточный продукт для синтеза третичных ароматических
спиртов, арилкетоспиртов и других соединений.
Примечания: 1. Холодильник и воронку закрывают хлоркаль-
циевыми трубками.
2. При недостаточном количестве льда (менее ЗЛ4 на Ш хлористого
алюминия) образуется значительное количество хлопьев, из которых 2,5-
диметнлацетофенон можно выделить обработкой концентрированной
серной кислотой и последующим разбавлением водой.
3. Сульфат магния необходимо проэкстрагировать двумя небольши-
ми порциями эфира, так как он адсорбирует значительные количества
2,5-диметилацетофенона.
4. Регенерированный и-ксилол может быть вновь использован.
ЛИТЕРАТУРА
1. М. G 1 а и s. Вег., 18, 230 (1885).
2. A. D. G 1 a u s, R. W о I 1 п с г. Вег., 18, 1856 (1885).
3. М. Freund, К. F 1 е 1 с h е г. Liebigs. Ann. Chem., 414, 5 (1918).
4. М. G u е b е г t. Compt. Rend., С XXV, 34 ('897).
5. V, D. Hightingale, B. Howard, J. Mucker. J. Organ. Chem.,
18, N 7-11, 1529 (1953).
Поступила в декабре 1963 г. Институт химии высокомолекулярных соединений
АН УССР
УДК 547.239.2.07
п, п' -ДИОКСИБЕНЗОЛ СУЛЬФИМИД
В. И. ДЫХАНОВ, Г. Н. НИКИТЕНКО
о о
НО—S— N—S—J^-OH
о н о
C12HnOeNS2 М. в. 329, 35
п, п'-Диоксибензолсульфимид рекомендован в качестве ис-
ходного материала для синтеза дубителей, красителей, капил-
лярно-активных веществ и инсектофунгисидов [1]. В том
же патенте его предложено получать путем 12-часового на-
гревания п, п'-дихлорбензолсульфимида с избытком 20%-ного
водного раствора едкого натра в присутствии каталитических
количеств медного порошка при температуре 210—220° и дав-
лении около 25 атмосфер. Выход и физико-химические кон-
станты п, n'-диоксибензолсульфимида не приведены [1].
Исходя из данных по аммонолизу п, п'-дихлорбензолсуль-
фимида [2], можно было сделать вывод, что соли закиси меди
или смесь эквивалентных количеств солей окиси меди и мед-
ного порошка будут более эффективными катализаторами и
в реакции гидролиза п, n'-дихлорбензолсульфимида. На осно-
вании экспериментальной проверки такого вывода разработа-
на нижеописываемая оптимальная методика получения п, ri-
диоксибензолсульфимида щелочным гидролизом п, п'-дихлор-
бензолсульфимида в присутствии каталитических количеств
смеси медного купороса и медного порошка. При температу-
ре 165—170° и давлении не более 6 атмосфер процесс обмена
обоих атомов хлора на гидроксильные группы полностью за-
канчивается за 7 часов, а выход п, п'-диоксибензолсульфими-
да составляет около 75% от теоретического.
39
СХЕМА СИНТЕЗА л,л'-ДИОКСИБЕНЗОЛСУЛЬФИМИДА
Cl-^_^-SO2-NH-SO2-^_^-CI 4-5NaOH^^4-*
— NaO-^_^-SO2-N-SO2— J-ONa 4-2NaCl + ЗНаО
Na
_ 1 + 3HC1
HO-SO2-NH-SO,-\J/-OH 4- 3NaCl
Характеристика основного сырья
n, n'-Дихлорбепзолсульфимид, очищенный, т. пл. 206—2073.
Натр едкий, жидкий, ГОСТ 2263—59, марка «Д».
Купорос медный, техн., ГОСТ 2142—58, сорт 1.
Медный порошок, свежеосажденный из медного купороса.
Кислота соляная, техн., синтетич., ГОСТ 857—57.
Условия получения
В нержавеющий стальной вращающийся автоклав емко-
стью 1 л помещают 106,1 г чистого п, п'-дихлорбензолсульфи-
мида, 800 мл 20%-ного водного раствора едкого натра, 1,87 г
медного порошка и 7,5 г медного купороса. Содержимое за-
крытого автоклава нагревают в течение 7 часов при темпера-
туре 165—170е; максимальное давление при этом достигает
6 атмосфер.
По окончании гидролиза автоклав охлаждают до комнат-
ной температуры и открывают. Реакционная масса представ-
ляет собой взвесь кристаллов тринатриевой соли п, п'-диок-
сибензолсульфимида и хлористого натрия в щелочном маточ-
нике. Кристаллы технической тринатриевой соли отфильтро-
вывают, растворяют при нагревании в двукратном количест-
ве воды, прибавляют 5 г активированного угля, перемешива-
ют в течение 10—15 минут и фильтруют в горячем состоянии.
К энергично перемешиваемому фильтрату при температуре
60—65° приливают равный объем концентрированной соляной
кислоты и оставляют на 1,5—2 часа для кристаллизации.
Осадок технического п, п'-диоксибензолсульфимида от-
фильтровывают на пористой пластинке, тщательно отсасывают
от маточника (вес воздушно-сухого продукта около 90 г) и
перекристаллизовывают из 10%-ной соляной кислоты в соот-
ношении 1:12. Получают п, n'-диоксибензолсульфимид с тем-
пературой плавления 209—2113 в количестве 70—75 г, что со-
ответствует 73—78% от теоретического выхода.
40
Вещество хорошо растворимо в воде, спирте, ацетоне,
очень мало растворимо в ароматических углеводородах и
эфире; кристаллизуется из диоксана (1:5) в виде мелких бес-
цветных игл; т. пл. 2 12—212,5°.
Найдено, %: С-43, 79; Н-3, 40; N-4, 31.
C12HnO6NS2. Вычислено, %: С -43, 76; Н—3, 36; N —4, 25.
ЛИТЕРАТУРА
1. Герм, пат, 3289; РЖхим,, № 1, реф. 1154 П (1955).
2. А. М. Григоровский, Н. Н. Д ы х а н о в. Ж. прикл. химии, 30,
1352 (1957).
Поступила в сентябре 1963 г. ВНИИ .монокристаллов
УДК 547.638.4.07
2-ДИФЕН ИЛ АЦЕТИЛ И НДАНДИОН-1,3 (ДИФЕНАЦИИ)
И ЕГО ГИДРАЗОН (ДИФЕЗОН)
В. Н. ЗЕЛ МЕН, 3. Н. КРАСТЬ! НЬ, Г. Я. ВАН АГ
/СвН5 //х ZCBH6
I II ^Х)СНСОСН I || "СНСОСН
X s х.> к_/ / \ «ч. > ' к
ХС6Н5 || \с,н5
n-nh2
М. в. 340, 37 CagHigOaNa М. в. 354, 40
2-Дифенилацетилиндандион-1,3 был получен конденсацией
диалкилфталата с дифенилацетоном в присутствии метилата
натрия [1, 2. 8]. После подкисления соляной кислотой и кри-
сталлизации получают желтое кристаллическое вещество с
т. пл. 146°.
2-Дифенилацетилиндандион-1,3 является антикоагулянтом
крови и зооцидом [3, 4].
Гидразон 2-дифенилацетилиндандиона-1,3 является реа-
гентом на карбонильные группы. Он легко конденсируется с
альдегидами и кетонами и дает соответствующие азины с до-
статочно острыми температурами плавления. Гидразон дифе-
нилацетилиндандиона может быть приготовлен по двум мето-
дам, описанным в статьях [5, 6]. Однако разработанный на-
ми способ его синтеза является более удобным и простым, так
как вместо безводного гидразина применяется его гидрат, со-
кращается время реакции и продукт получается вполне при-
годным по своей чистоте для дальнейших работ без перекри-
сталлизации.
СХЕМА СИНТЕЗА 2-ДИФЕНИЛАЦЕТИЛИНДАНДИОНА-1,3
^\.-COOR
L J—COOR
+ СНдСОСН
хс6н5
NaOCH,
42
/ \ > /С“Н6
-» Г Г?°ХСНСОСН + 2R0H
\ /—СО/ \
ч/ ХСвН6
Характеристика основного сырья
(см. примечание 1)
Голуол, ч. д. а., ГОСТ 5789—51.
Бензол, ч. д. а., ГОСТ 5955—51.
Натрий металлический, ч., ГОСТ 3273—55.
Метиловый спирт, безводный, ГОСТ 6995—54.
Димстилфталат, ч., ГОСТ 9657—61.
Дифенилацетон (см. примечание 2).
Этиловый спирт, ректифицированный, ГОСТ 5962—51.
Соляная кислота, ч., ГОСТ 3118—46.
Условия получения
В литровую трехгорлую колбу со шлифами, снабженную
механической мешалкой, капельной воронкой и обратным
холодильником, с которым соединен холодильник Либиха для
перегонки, помещают 50 мл безводного толуола и 6,9 г ме-
таллического натрия. Колбу нагревают на масляной бане
при 130—140°. Когда натрий расплавится, нагрев прекращают
и интенсивно перемешивают содержимое колбы, чтобы полу-
чить порошкообразный натрий. Затем через капельную ворон-
ку к смеси приливают 200 мл безводного бензола и медленно,
по каплям, 70 мл безводного метанола. После этого, не пре-
кращая перемешивания,колбу нагревают 1 —1,5 часа на мас-
ляной бане до полного растворения натрия и образования
метилата натрия. Используя обратный холодильник как деф-
легматор, отгоняют растворители, пока в колбе останется
только сухой метилат, к которому приливают 250 мл безвод-
ного бензола и 38 мл безводного диметилфталата. К этой
массе, в течение 1,5 часа, по каплям, добавляют раствор
21 г дифенилацетона в 100 мл безводного бензола при темпе-
ратуре в масляной бане НО—125°. Во время этого добавле-
ния производят равномерно отгонку 100 мл смеси бензола с
метанолом, образующимся в процессе реакции. Затем, в те-
чение 1,5 часа, прибавляют по каплям раствор 21 г дифенил-
ацетона и 25 мл диметилфталата в 100 мл безводного бензо-
ла. За это же время равномерно отгоняют еще 100 мл смеси
бензола с метанолом. После этого реакционную массу, при
интенсивном перемешивании, продолжают нагревать в тече-
ние 1 часа при температуре 140° (в бане). Затем температуру7
понижают до 90э, к образовавшейся темной вязкой массе при-
43
ливают 150 мл этанола и нагревают, при перемешивании, до
60—70°, пока масса станет гомогенной. Спирт и бензол отго-
няют на водяной бане под небольшим вакуумом. Отгонку пре-
кращают, когда в колбе остается ~ ЮОлл темной массы. Ее
охлаждают до 25° и постепенно, при сильном встряхивании,
приливают 50 мл концентрированной соляной кислоты. Масса
постепенно становится темно-оранжевой, густеет и из нее
выделяются белые кристаллы хлористого натрия.
Полученную смесь оставляют на 1 час в холодильном
шкафу, потом промывают тремя порциями воды по 200 мл,
каждый раз осторожно сливая водный раствор. Остаток в
колбе обливают 400 мл этанола и темно-оранжевая масса
при размешивании закристаллизовывается. Колбу оставля-
ют па 2—3 часа в холодильном шкафу, спирт отсасывают, а
осадок снова промывают спиртом. Выход дифенилацетилин-
дандиона равен 36—42 г, что составляет 54—62% от теоре-
тического. После перекристаллизации из спирта получают
желтые кристаллы с т. пл. 146—147°.
СХЕМА СИНТЕЗА ГИДРАЗОНА
2-ДИФЕН ИЛАЦЕТИЛ ИН ДАН ДИОНА-1,3 (ДИФЕЗОНА)
/СбН*
| ||_CQ/CHCOCH 4-HaN-NH3.HaO -
w ЧС6Н5
//X
-Ч II р/СНСОСН + 2НаО
у ХС6Н6
N-NHa
Характеристика основного сырья
2-Дифенилацетилиндандион-1,3, т. пл. 146—1477
Гидразин-гидрат, ч. д. а., ГОСТ 5832—51.
Этиловый спирт, ГОСТ 5962—51.
Условия получения
Загружают в колбу 5 г (0,014 М) 2-дифенилацетилиндан-
диона-1,3, 1,4 г (0,028 М) гидрата гидразина и 20 мл этилово-
го спирта и кипятят на водяной бане 1 час. Из оранжевого
раствора постепенно выпадают оранжевые кристаллы гидра-
зона 2-дифенил ацетилиндандиона-1,3.
Выход равен 4,4 г, что составляет 84,4% от теоретичес-
кого; продукт разлагается около 253—258°.
44
Конденсация гидразона 2-дифенилацетилиндандиона-1,3
с карбонильными соединениями.
Эквимолекулярные количества карбонильного соединения и
гидразона 2-дифенилацетилиндандиона-1,3 в хлороформе с,
добавлением одной капли концентрированной соляной кисло-
ты кипятят 5—10 минут на водяной бане. Полученный жел-
тый или оранжевый раствор фильтруют, осаждают метило-
вым спиртом или упаривают в вакууме и остаток кристалли-
зуют из уксусной кислоты или этилового спирта.
Продукты конденсации гидразона 2-дифенилацетилиндандиона-1,3
с альдегидами и кетонами (дифезины)
Карбониль- ное соедине- ние Т. пл. азина, °C Суммарная формула Содержание азота, %:
найдено вычислено
Ацетон .... 224 -225 226-227* ^26^22^1^2 6,92 7,10
Бензальдегид 243—244 239-240* C80H22O8N2 6,27 6,33
Акролеин , . 82-83 85,5-86,5х' ^26^20^2^2 7,01 7,14
* По литературным данным [5].
Примечания: 1. Использовались высушенные растворители и
реагенты.
2. Дифенилацетон приготовлен по прописи [7].
ЛИТЕРАТУРА
1. В. Н. Витол, Г. Я. Ванаг. Изв. АН ЛССР, 9, 111 (1955).
2. Chem. Abstrs., 3264а (1955); (U. S., 2672, 483 (1954)..
3. J. В. Field, М. S. Goldfarb, A. G. Ware, G С. Griffith.
Proc. Soc. Exptl. Biol. a. Med., 81, 678 (1952).
4. Г. Я. В а н а г, Б. Ю. Фалькенштейн, Л. В. Егорова,
В. Н. Зел мен. Изв. АН ЛССР, 5, 99 (1959).
5. R. A. Braun, W. А. М о s с h е г. J. Amer. Chem. Soc., 80, 3048
(1958).
6. В. Н. 3 е л м е н, 3. Н. К р а с т ы н ь, Г. Я. В а и а г. Изв. АН ЛССР,
№ 1, 77 (1961).
7. Синтезы органических препаратов, Сб. 4, 219 (1953).
8. Chem. Abstrs, 16325е (1958); (U. S., 2. 827, 489 (1958).
Поступила в апреле 1964 г.
Институт органического синтеза АН Латв. ССР
УДК 547.58.07
2,7 ДИХЛОРХРОМОТРОПОВАЯ КИСЛОТА
1,8-диокси-2,7-дихлорнафталин-3,6-дисульфокислота,
динатриевая соль, дигидрат
В. И. КУЗНЕЦОВ, Н. Н. БАСАРГИН
НО ОН
ciw'\A/a
J у -2Н.0
NaO3Sz XSO3Na
Cl0H4O8Cl2S2Na2-2H2O M. b. 469, 18
Дихлорхромотроповая кислота применяется в аналитичес-
кой практике в качестве чувствительного и избирательного
реактива для фотометрического определения микрограммовых
количеств титана [1]. С использованием этого реактива разра-
ботаны методы определения титана в сталях [2], уране [3],
алюминиевых сплавах [4], бериллии [5], минералах и породах
[6] и в других объектах.
Изучены комплексные соединения титана (IV) [7] и желе
за (III) с 2,7-дихлорхромотроповой кислотой.
Моно- и дигалоидозаметенные хромотроповой кислоты до
настоящего времени не были получены.
Обычные способы галоидирования в применении к хро-
мотроповой кислоте нам не давали положительных результа-
тов. При хлорировании ее в водной среде газообразным хло-
ром, смесью NaOCI с НС! или КСЮ3 с НС1 всегда наблюда-
лось очень сильное окисление.
Синтез галоидозамещенных хромотроповой кислоты, и в
том числе 2,7-дихлорхромотроповой кислоты, был осущест-
влен лишь при проведении галоидирования в среде 80--
100%-ной серной кислоты, где процесс окисления оказался
полностью подавленным [8, 9].
46
СХЕМА СИНТЕЗА 2,7-ДИХЛОРХРОМОТРОПОВОЙ КИСЛОТЫ
НО он
2Clg (газ)
A A A H2SO4
NaO3Sx /z SO3Na
НО ОН
с1\ AA/C1
'l II I + 2НС1
NaO3S X^/44^XSOsNa
Характеристика основного сырья
Хромотроповой кислоты динатриевая соль, ч., ВТУ МХП
4045—53, содержание основного вещества 98%.
Серная кислота, х. ч., ГОСТ 4204—48.
Хлор (из баллона).
Едкий натр, х. ч., ГОСТ 4328—48.
Условия получения
В реакционном сосуде емкостью 2—2,5 л, снабженном ме-
ханической мешалкой и рубашкой для охлаждения, смешива-
ют 180 г (0,45 Л1) хорошо измельченной динатриевой соли хро-
мотроповой кислоты с 1200 мл серной кислоты уд. веса 1,83.
Полученную смесь перемешивают в течение ~ 30—40 ми-
нут и хлорируют при температуре 15—20°, медленно пропу-
ская хлор из баллона в течение 15—20 часов до полного раст-
ворения суспензии хромотроповой кислоты. Хлор подводят ко
дну сосуда по стеклянной трубке при хорошем перемешива-
нии реакционной смеси. Затем подачу хлора прекращают и в
реакционный раствор медленно, небольшими порциями, при
продолжающемся перемешивании, вводят 240 г мелкого льда.
Смесь при этом слегка нагревается. Температуру реакционно-
го раствора понижают до комнатной внешним охлаждением и
снова пропускают хлор в течение 15—20 часов, не прекращая
перемешивания (см. примечание 1).
После этого реакционную смесь медленно выливают на
700—800 г льда при продолжающемся перемешивании и ох-
лаждают в ледяной бане до 5—7°. Осадок дихлорхромотропо-
вой кислоты отфильтровывают через воронку со стеклянным
фильтром № 2 (см. примечание 2) и хорошо отжимают. По-
лученный плотный светло-серый осадок удаляют с фильтра,
смешивают с 400—500 мл воды и снимают избыток серной
кислоты, добавляя порциями по 10—15 мл (всего 100—
47
150 мл) 30%-ный раствор едкого натра и хорошо перемеши-
вая всю реакционную массу, которая при этом несколько за-
густевает. Температура ее пе должна подниматься выше 50—
60°. После охлаждения до комнатной температуры осадок от-
фильтровывают, хорошо отжимают и промывают 50—60 мл
ледяной воды (порциями по — 10 мл), отсасывают и сушат
вначале на воздухе, затем при температуре 60—80° в сушиль-
ном шкафу.
Выход продукта в виде динатриевой соли 2,7-дихлорхро-
мотроповой кислоты равен 120—130 г, что составляет 57—
61,6% от теоретического.
Для получения более чистого препарата 50 г продукта
растворяют в 140—150 лл воды при нагревании. К раствору
добавляют 0,1—0,2 г животного угля, не содержащего желе-
за, и раствор быстро фильтруют через воронку, обогреваемую
паром. Фильтрат при охлаждении выделяет осадок динатрие-
вой соли дихлорхромотроповой кислоты в виде бесцветных
тонких иголочек.
Выход чистого препарата составляет 35—37 г (см. приме-
чание 3).
Найдено, %: С1-15.1; 14,9; S —14,1; 13,8;
Н2О-7,9; 8,1.
C10H4O8S2Cl2Na2-2H2O. Вычислено, %: CI-15,1; S—13,6;
Н2О - 7,7.
Примечания: 1. При стоянии после хлорирования в течение
20—30 часов смесь выделяет — 30—40% продукта в виде кристалличес-
кого осадка 2,7-дихлорхромотроповой кислоты.
2. Из полученного фильтрата можно при нейтрализации его сухим
Na2CO3 до слабокислой реакции (pH ~ 2—3) выделить 7—10 г менее чи-
стого вещества.
3. Реактив следует хранить в склянке из темного стекла, избегая пря-
мого солнечного света.
ЛИТЕРАТУРА
1. В. И. Кузнецов, И. Н. Басаргин. Ж. аналит. химии, 16, 573
(1961); Авт. свид. 148584; Бюлл. изобр. № 13 (1962).
2. Н. Н. Басаргин, А. Н. Ткаченко, Л. Р. Ступа, Л. Н. Б о-
родаевская. Заводск. лаборатория, 28, 1311 (1962).
3. В. И. Кузнецов, Н. Н. Басаргин, Т. Н. Кукишева. Ж.
аналит. химии, 17, 457 (1962).
4. Л. М. Буданова, С. Н. П и н а е в а. Заводск. лаборатория, 29,
149 (1963).
5. Н. Н. Басаргин, Т. Н. Кукишева, Н. В. Соловьева. Ж-
аналит. химии, 19, 553 (1964).
6. Н. С. Классов а, Л. Л. Леонова. Ж. аналит. химии, 19, 131
(1964).
7. Н. Н. Басаргин, Т. В. Петрова. Ж. аналит. химии, 19, 835
(1964).
8. В. И. Кузнецов, Н. Н. Басаргин. Ж. общ. химии, 34 (1964).
9. II. Н. Басаргин. Диссертация, М., 1962, стр. 203.
Поступила в апреле 1964 г. ГЕОХИ АН СССР
48
УДК 547.37.07
р, р'-ДИЦИАНДИЭТИЛОВЫЙ ЭФИР
Р, Р'-Пропиоиитрилоксид
Н. СЕМЕНОВА, С. БЕЛЬЧЕНКО, И. ХВОСТОВ
/CHa-CH2-CN
О(
ХСН3-СН3-CN
C6HsN2O М. в. 124,14
Описанные в литературе методы получения Р, р'-дициан-
диэтилового эфира основаны на реакции цианэтилирования
воды.
Так, Брассон и Райнер действовали на акрилнитрил едким
натром в диоксане [1]. Шостаковский, Шур и Филимонов
реакцию цианэтилирования проводили также в среде диок-
сана в присутствии 10°/о-ного едкого натра. В зависимости
от времени нагревания выход р, р'-дициандиэтилового эфира
составлял 43—53% [2].
Имеются указания на то, что цианэтилирование воды мо-
жет происходить в присутствии гидроокисей щелочноземель-
ных элементов и некоторых карбонатов, ацетатов и фосфа-
тов [3].
Терентьев и Кост получили р, Р'-дициандиэтиловый эфир
из этиленциангидрина и акрилнитрила с хорошим выхо-
дом [4].
Применяется р, р'-дициандиэтиловый эфир в качестве чис-
той стационарной фазы в газовой хроматографии [5], при
синтезе циапэтиловых эфиров крахмала и целлюлозы [6], а
также в качестве экстрагирующего агента при выделении
нафталина из смесей, содержащих его алкильные дериваты и
алкилбензолы, получаемые при гидроформинге [7].
Нами р, р'-дициандиэтиловый эфир получен методом циан-
этилирования воды в присутствии 10%-ного водного раствора
едкого натра в среде диоксана, при этом было установлено,
4 Зак. 613 49
что длительная выдержка щелочного реакционного раствора
сверх установленного времени снижает выход конечного про-
дукта.
СХЕМА СИНТЕЗА ₽, -ДИЦИАНДИЭТИЛОВОГО ЭФИРА
CH2-CH-CN + H2o -> НО-СНг-СНа--CN
HO-CH2-CH2-CN + ch2=ch-cn -
-> NC-CH2-CH2-O-CH2-CH3-CN
Характеристика основного сырья
Акрилонитрил, ч., ТУ У 797—53.
Диоксан, ч., ВТУ МХП 3111—52.
Натр едкий, ч., ГОСТ 4328—48.
Кислота соляная, ч., ГОСТ 3118—46.
Условия получения
В колбу, снабженную мешалкой и термометром, загружа-
ют 106 г (~2Л1) акрилонитрила, 100 г диоксана и 20 г
10%-пого раствора едкого натра и перемешивают при темпе-
ратуре 45э. По истечении 10 часов смесь охлаждают до ком-
натной температуры и фильтруют. К фильтрату прибавляют
8—10 мл концентрированной соляной кислоты до кислой ре-
акции по Конго, после чего реакционную массу разгоняют под
вакуумом. Сначала отгоняют диоксан, а затем, при 210—2157
25—30 мм, р, р'-дициандиэтиловый эфир в количестве 51 г, что
составляет -^-41,3% теоретически возможного выхода.
По внешнему виду продукт представляет собой прозрач-
ную, светло-желтую жидкость, хорошо растворимую в арома-
тических углеводородах, о'420— 1,046, «р — 1,440.
По литературным данным, rf420 — 1,050 — 1,055 [8], —
— 1,442 - 1,444 [2].
ЛИТЕРАТУРА
1. В г и son, Riener. J. Amer. Chem. Soc., 65, 23 (1943).
2. M. В. Ш о с г а к о в с к и й, А. Н. Шур, Б. Ф. Филимонов.
Ж. прикл. химии, 30, 971 (1957).
3. Пат. США 2816130; С. Zbl., 4626 (1959).
4. А. Т е р е н т ь е в, А. Кост. Реакции и методы исследов. орга-
иич. соединений, М., Госхнмиздат, 2, 1952, стр. 108.
5. Е. Bayer. Gas chromatographie, Springer Verlag, Berlin, Gottingen
Heidelberg 1959.
6. Пат. США 2842541; Zbl., 10763 (1959).
7. Пат. США 2812372; Zbl., 3649(1959).
8. Information ilber neu aufgenomrnenen Praparate Merk, № 10, 9 (1960).
Поступила в декабре 1963 г. ИРЕА, ЦЗЛ
УДК 547.Г 13.05
ДИЭТИЛЕНТРИАМИНПЕНТААЦЕТАТ МЕДИ
(Водородная форма)
И. А. СЕЛИВЕРСТОВА, Л. В. ГОРОХОВА, Н. М. ДЯТЛОВА
зн<-
сн2соо-
Н,С- N-CH,
Н2С сн2
оосн2с\ । ; сн2соо-
)N N<
сн/х / хсн2
/Си |
ОС —О7 'О-СО
Н2О
C14H21O10N3Cu H3O М. в. 472,89
В последнее время твердые комплексонаты находят широ-
кое применение в различных областях науки, техники и сель-
ского хозяйства.
В данной работе приведена методика получения одного из
них — диэтилентриаминпента ацетата меди.
Получение этого комплексоната представляло интерес с
точки зрения испытания его применения в аналитической хи-
мии, а также для устранения медного хлороза в сельском хо-
зяйстве.
Диэтилентриаминпентаацетат меди получен нами взаимо-
действием диэтилентриаминпентауксусной кислоты со свеже-
приготовленным гидратом окиси меди в соответствии с указа-
ниями, имеющимися в работе Сайверса и Бейлара [1], при
этом были уточнены условия синтеза и выделения комплек-
соната.
4* 51
СХЕМА СИНТЕЗА СОЕДИНЕНИЯ
H3[Hs(CuH18N3O10)] + Cu(OH)3
-> H8[Cu(CuH18N8O10)].H2() + н,о.
Характеристика основного сырья
Диэтилентриаминпентауксусная кислота (ДТПУ), ч., РТУ
ОЭЗ, ИРЕА, № 10—63.
Гидрат окиси меди, свежеприготовленный.
Условия получения
В трехгорлой колбе, снабженной обратным холодильни-
ком, мешалкой и термометром, растворяют 19,65 г (0,05А4)
ДТПУ в 800 мл дистиллированной воды при нагревании до
70—80°. К раствору добавляют 4,9 г (0,05Л4) свежеприготов-
ленной гидроокиси меди (см. примечание 1) и выдерживают
реакционную массу до полного растворения Сп(ОН)2.
Полученный синий раствор упаривают па водяной бане
при 70э до объема 60 мл, после чего осторожным прибавлени
ем 35 мл этилового спирта осаждают комплекс (см. приме-
чание 2), выпадающий при охлаждении в нормальных усло-
виях в виде синих кристаллов. Их отфильтровывают, промы-
вают спиртом и сушат на воздухе между листами фильтро-
вальной бумаги.
Выход диэтилентриаминпентаацетата меди равен 18 г, что
составляет 76% от теоретического.
Найдено, %: Си-13,28; С-36,28; Н - 5,35; N-8,63.
CuHjjNjOnCti. Вычислено, %: Си—13,43; С—35,5; Н—4,86; N-8,87.
Примечания: 1. Для получения пасты Си(ОН)2 растворяют
12,4 г медного купороса (CuSCh 5IhO) в 180 мл воды и фильтруют.
Гидрат окиси меди осаждают профильтрованным раствором 4—5 г едко-
го натра в 2—3 мл воды. Осажденную гидроокись фильтруют и промы-
вают до отсутствия реакции на ион SO4'' с раствором хлористого бария [2].
Рекомендованное авторами прибавление глицерина при промывании гидра-
та окиси в нашем случае является нежелательным.
2. Наиболее сложным моментом данного синтеза является выделение
комплексоната. Этанол необходимо прибавлять медленно, при перемеши-
вании, строго поддерживая температуру 70°.
ЛИТЕРАТУРА'
1. R. Е. Sievers, J. С. В a i 1 а г. Inorg, Chem., 1, 1, 174 (1962).
2. Ю. В. Карякин, И. И. Ангелов. Чистые химические реакти-
вы, М„ Госхимиздат, 1955, стр. 346.
Поступила в марте 1964 г. ИРЕА
УДК 547.26’117.07
ИЗОБУТИЛНИТРИТ
Н. Н. ДЫХАНОВ. Л. М. ЕГУПОВА
Н
Н3с-С—CH3-0-N-0
сн3
C4H9O2N М. в. 103,13
Из четырех изомерных бутилнитритов в литературе не
описан только пере, изобутилнитрит, вероятно, по той причи-
не, что исходный для его синтеза изобутиловый спирт (2-ме-
тилпропанол-1) трудно доступен в чистом состоянии.
Поскольку алкилнитриты применяются в органическом
синтезе в основном как нитрозирующие средства, то их инди-
видуальность для этой цели не имеет существенного значения.
В связи с этим для нитрозирования, например, карбонильных
соединений вместо дорогостоящего н.амилнитрита [I] весьма
целесообразно использовать изобутилнитрит, получаемый из
дешевого и доступного технического изобутанола. Методика
его синтеза приводится ниже.
СХЕМА СИНТЕЗА ИЗОБУТИЛ НИТРИТА
2СН3-СН-СН2ОН + 2NaNO2+HsSO4 ->
СН3
2СН3—СН —СН2—О—N=O + Na2SO4 4- 2Н3О.
I
СН3
Характеристика основного сырья
Спирт изобутиловый, техн., ТУ МХП 1704—47.
Натрий азотистокислый, техн., ГОСТ 6194—52, сорт 1.
53
Натрий двууглекислый, для мед. целей, ГОСТ 6194—52.
Натрий сернокислый, природный, прокаленный, ГОСТ
6318—52, 1 сорт.
Натрий хлористый, техн., ГОСТ 4233—48.
Кислота серная, контактная, улучшенная, ГОСТ 2184—59.
Условия получения
В круглодонную трехгорлую колбу емкостью' 350 мл, снаб-
женную мешалкой, капельной воронкой и термометром, до-
стигающим почти до дна колбы, помещают раствор 38 г нит-
рита натрия в 150 мл воды. Содержимое колбы охлаждают в
смеси льда с солью до 0° и прикапывают к нему смесь 15 мл
воды, 25 г концентрированной серной кислоты и 52 г изобути-
лового спирта с такой скоростью, чтобы температура в колбе
не превышала 2°.
Затем реакционную массу оставляют на 15 часов при ком-
натной температуре, после чего отфильтровывают выделив-
шийся осадок сульфата натрия, а фильтрат переносят в дели-
тельную воронку для расслаивания.
Верхний- слой сырого изобутилнитрита отделяют, промы-
вают раствором бикарбоната натрия (1 г в 20 мл воды), за-
тем раствором поваренной соли (5 г в 50 мл воды), сушат
над прокаленным сульфатом натрия (3—5 г), фильтруют и
фракционируют с елочным дефлегматором (Н = 15 см) при
атмосферном давлении, собирая изобутилнитрит в пределах
65—70°.
Выход его равен 45 — 46 г, что составляет около 62% от
теоретического.
ЛИТЕРАТУРА
1. Р е х t е г s. Bull. Soc. chim. Belg., 1906, стр. 796.
Поступила в сентябре 1963 г. ВНИИ монокристаллов
УДК 547.224.0~i
ЙОДИСТЫЙ МЕТИЛ
М. И. ВАИСОВА, И. И. ДЫХАНОВ
н
H-C-J
н
CH3J М. в. 141,93
По литературным данным, йодистый метил может быть
получен действием диметилсульфата на водный раствор йоди-
стого калия [1], медленной перегонкой смеси метилового спир-
та с большим избытком постоянно кипящей йодистоводород-
ной кислоты [2], электролизом водного раствора уксуснокис-
лого калия в присутствии йода или йодистого калия [3], дей-
ствием метилового эфира n-толуолсульфокислоты на водный
раствор йодистого калия [4] и действием метилового спирта
на раствор пятийодистого фосфора в йодистом метиле [5].
Наиболее широко распространенным способом получения
метилйодида является взаимодействие метилового спирта с
трехйодистым фосфором или со смесью йода и фосфора (без-
различно красного или желтого или их смесью) [6]. Однако
этот способ весьма огневзрывоопасен и громоздок в аппара-
турном отношении.
Ниже приводится методика получения йодистого метила
действием технического диметилсульфата на водный раствор
йодистого натрия. Гарантированный выход метилйодида по
этой методике составляет 75% от теоретического, считая на
йодистый натрий.
СХЕМА СИНТЕЗА ЙОДИСТОГО МЕТИЛА
NaJ + (CH3)2SO4
CH3J + СН3О- SOnONa
55
Характеристика основного сырья
Натрий йодистый с примесью таллия йодистого — отход от
производства монокристаллов.
Диметилсульфат, техн., ТУ МХП 3180—52.
Условия получения
Реакция проводится в полулитровой круглодонной колбе,
снабженной мешалкой, термометром и нисходящим прямым
холодильником, аллонж которого опущен под поверхность ле-
дяной воды, налитой в приемную колбу.
В реакционную колбу помещают раствор 150 г йодистого
натрия в 150 мл воды (см. примечание) и при работающей
мешалке к нему приливают 100 мл диметилсульфата. Реакция
образования метилйодида начинается самопроизвольно и при
температуре 24° (в парах) отгоняется его азеотроп с водой.
Как только реакция замедлится, колбу помещают в горячую
водяную баню (50—60°) и ведут процесс до прекращения от-
гонки метилйодида, который собирается на дне приемной кол-
бы под слоем воды в виде бесцветного маслянистого слоя.
Сырой метилйодид отделяют при помощи делительной во-
ронки, тщательно высушивают над прокаленным хлористым
кальцием, сливают в сухую перегонную колбу и перегоняют
на водяной бане. Чистый йодистый метил собирают при 42° в
количестве 107—108 г или около 75% от теоретического, счи-
тая на йодистый натрий.
Примечание. Из того же количества йодистого натрия и 172 мл
технического диэтилсульфата в аналогичных условиях получается 126,5—
127 г (81%) йодистого этила, азеотроп которого с водой отгоняется при
48—50°.
ЛИТЕРАТУРА
1. Wei nl and, Schmid. Ber., 38, 2327 (1905); Герм. пат. 175209
Frdl., 8, 17.
2. Norris. J. Amer. Chem., 38, 639 (1907).
3. F. К a u f 1 e г, С. H e r z о g. Ber., 42, 3860 (1909).
4. Peacock, Menon. Quart. Journ. Indian Chem. Soc., 2, 240 (1925);
В. M. Родионов. Bull. Soc. chim. France, (4), 39, 323 (1926).
5. J. W. W a 1 k e r, F. M. J о h n s о n. J. Chem. Soc., 87, 1595 (1905).
6. D umas, P e 11 g о t. Liebigs. Ann. Chem., 15,20 (1835); ЖРФХО, 27,
1, 364 (1895); Ber., 29 (R), 90 (1896); 17, 652 (1884); J. pract. Chem., (2),
53, 275 (1896); J. Chem, Soc., 87, 1592 (1905); 121, 1911 (1922).
Поступила в сентябре 1963 г. ВНИИ монокристаллов
УДК 547.565.2.07
л-МЕТОКСИФЕНОЛ
Монометиловый эфир гидрохинона
В. /И. САВОСЬКИН, и. И. Г АРКУША-БОЖКО, Р. Н. ЛОГВИНЕНКО,
Н. С. ДВОЙНЫХ, Л. В. БРЮХАНОВА
НО-^-^-ОСН3
С7Н8О2 М. в. 124,14
n-Метоксифенол получают метилированием гидрохинона
диметилсульфатом [1—5], метилкалийсульфатом [6], йодистым
метилом [7—8], метанолом или диметиловым эфиром над
окисноалюминиевыми или борофосфатными катализаторами
[9—10] и деметилированием диметилового эфира гидрохинона
в присутствии хлористого алюминия [11].
Монометиловый эфир гидрохинона применяется в качест-
ве стабилизатора метилакрилата и акрилонитрила.
Предлагаемый нами способ получения п-метоксифенола
заключается в диазотировании л-анизидина с последующей
заменой диазогруппы на гидроксил.
СХЕМА СИНТЕЗА п МЕТОКСИФЕНОЛА
СН3О—^~^-nh2 + H2SO4 CH3O-^~>-NH3HSO4
18-20°
СН3О-^ ^-NH3HSO4 + NaNO2 + HaSO4---->
—> СН8О-^~^-N2HSO4 + NaHSO4 + 2H2O
57
/—.. 85-86°
СНзО-f ^-N,HSO4 + H20-------------->
'=/ толуол,
CuSO4
CH3O-^ J-OH + N3 + H2SO4
Характеристика основного сырья
(См, примечание 1)
/i-Анизидин, ч., ТУ МХП 1485—51,
Кислота серная, ч., ГОСТ 4204—48.
Нитрит натрия, ч., ГОСТ 4197—48.
Медный купорос, ч., ГОСТ 4165—48.
Толуол, ч.д.а., ГОСТ 5789—51.
Натр едкий, ч., ГОСТ 4328—48.
Кислота соляная, ч., ГОСТ 3118—46.
Условия получения
Диазотирование п-анизидина. В трсхгорлую колбу ем-
костью 3 л, снабженную мешалкой, капельной воронкой, но-
сик которой доходит почти до дна колбы, термометром и воз-
душным холодильником, загружают 630 мл воды, 70 г
(0,71 М) серной кислоты и 62 а (0,5 Л!) измельченного п-ани-
зидина. Смесь нагревают при перемешивании до 60—65°, по-
ка л-анизидин не растворится полностью. Затем раствор ох-
лаждают до 18°; при этом выпадают кристаллы сернокислого
n-анизидина. К полученной суспензии при этой же темпера-
туре прибавляют в течение 40 минут под слой жидкости раст-
вор 34,5 г, (0,5 нитрита натрия в 100 мл воды. По оконча-
нии диазотирования раствор должен быть кислым по конго
и давать в течение 10 минут положительную реакцию на азо-
тистую кислоту по йодкрахмальной бумаге. В случае отсутст-
вия положительной реакции следует добавить необходимое
количество раствора нитрита натрия. Избыток азотистой кис-
лоты можно нейтрализовать добавлением сульфата п-анизи-
дина.
Замещение диазогруппы на гидроксил и выделение п-мет-
оксифенола. В трехгорлую колбу емкостью 5 л, снабженную
эффективной мешалкой, капельной воронкой и обратным хо-
лодильником, загружают 140 мл воды и 140 г (0,56 М) мед-
ного купороса. Смесь подкисляют концентрированной серной
кислотой до pH 1—2 и прибавляют 400 мл толуола. Содержи-
мое колбы доводят до кипения и в кипящую массу при пере-
мешивании в течение двух часов прикапывают раствор диазо-
соединения (см. примечание 2). По окончании прибавления
58
диазораствора смесь кипятят еще 10—15 минут и охлаждают
до комнатной температуры. Затем содержимое колбы перено-
сят в делительную воронку, отделяют верхний толуольный
слой, а водный слой обрабатывают тремя порциями толуола
по 30—40 мл. Объединенные толуольные вытяжки три раза
обрабатывают 2н. раствором едкого натра (см. примечание 3).
Первая обработка производится 200 мл щелочи, две пос-
ледующих— по 50 мл. Щелочные вытяжки подкисляют кон-
центрированной соляной кислотой до кислой реакции по кон-
го и охлаждают до комнатной температуры. я-Метоксифенол,
выделившийся в виде тяжелого масла, экстрагируют тремя
порциями толуола по 30—40 мл каждая. Отгоняют с дефлег-
матором толуол, а затем /г-метоксифенол при 132715 мм или
при 141 —141,5724 мм. Расплавленный n-метоксифенол выли-
вают в фарфоровую ступку и растирают. Выход равен 37—
40 г, что составляет 60—65% от теоретического. Полученный
продукт представляет собой белый порошок, плавящийся при
54—55°.
Под действием света и воздуха продукт изменяет окраску.
По литературным данным, л-метоксифенол плавится при
53-547
Примечания: 1. При применении техническою сырья и увеличении
загрузок в 10—100 раз против указанных в методике получают н-мстоксн-
фено.т с выходом 50—64% от теоретического.
2. Капельную воронку необходимо вставлять так, чтобы носик ее ло
возможности меньше обогревался парами внутри колбы и капли диазора-
створа не попадали на стенки колбы.
3. Раствор едкого натра лучше готовить на дистиллированной воде.
На водопроводной воде раствоп готовят заранее и перед употреблением
осюрожио декантируют его с осадка.
ЛИТЕРАТУРА
1. F. U И m а п п. Liebigs Ann. Chem., 327, 104 (190.3),
2. М. К о h и, L. W. Guttman n. Monatsh. Chem., 45, 581 (1924).
3. R о b i n s о п, Smith.! Chem. Soc., 393 (1926).
4. H. Bred e reck, Heidenheim, Brenz. Angew. Chem., № 3,
59 (1948).
5. H. В r e d e r e c k, J. H e n n 1 g, W. Ran. Ber., 86, 9, 1085 (1953).
6. H. И Iasi we tz, J. Habermann. Liebigs. Ann. Chem., 177, ,334
(1875).
7. Hesse. Liebigs Ann. Chem., 200, 232 (1880).
8 F. T i e m a n n, W. H. M ii 11 e r. Ber., 14, 1939 (1831).
9. Швейц, пат. 277986; Zbl., 5487 (1952).
10. Пат. ФРГ 827803; Zbl. 5187 (1952).
11. Merck D. R. P. 94852; Frdl , 4, 123
Поступила в 1963 г.
Ловецкий филиал ИРЕА
УДК 547.638.1.07
МОНОБЕНЗОИЛЬНОЕ ПРОИЗВОДНОЕ
ДИФЕНИЛОЛПРОПАНА
и. в. хвостов
О СН3
/'xJ-O-^-С-^ ^-ОН
СН8
С22На0О3 М. в. 332,4
Неописанное ранее монобензоильное производное дифе-
нилолпропана нами синтезировано из мононатриевого произ-
водного дифенилолпропана и хлористого бензоила в моляр-
ном соотношении 1:1.
Строение монобензоильного производного подтверждено
элементарным анализом и положительной реакцией с азотно-
ртутным реактивом Миллона (см. примечание 1).
СХЕМА СИНТЕЗА МОНОБЕНЗОИЛЬНОГО ПРОИЗВОДНОГО
ДИФЕНИЛОЛПРОПАНА
СН3
н°-/ Ч-C-Z y0H + Na0H
СНз
сн8
-4- НО-Ч Х 4_0Na + Н2О
СНз
60
сн3 о
^-С-^ у"—ONa + С1-С-^_^
сн3
СН3 О
НО-^"^-С-^_ Ч-О-С-^~ + NaCl
\ = / ।
СНз
Характеристика основного сырья
Дифенилолпропан, техн. (см. примечание 2).
Хлористый бензоил, ч., ТУ МХП 92—51.
Условия получения
Растворяют 2,2 г дифенилолпропана (0,01 М) в 2 г 20%-
ного водного раствора едкого натра и в 10 жл воды. По ра-
створении к феноляту добавляют при энергичном встряхива-
нии 1,4 г (0,01 М) хлористого бензоила. Очень быстро на по-
верхности жидкости образуется белое хлопьевидное вещество,
которое отфильтровывают на стеклянном фильтре и промы-
вают холодной водой. Выход пеперекристаллизованного про-
дукта равен 2,7 г, что составляет --82% от теоретического.
После перекристаллизации из спирта монобензоильное
производное дифенилолпропана плавится при 138°, а после
сушки в эксикаторе над СаС12 температура плавления повы-
шается до 144°.
Монобензоильное производное дает бледно-розовое окра-
шивание с реактивом Миллона.
Найдено, %: С-79,06; 79,14; Н-5,53, 5,25.
CjjHjuO,. Вычислено, %: С—79,48; Н—6,03.
Примечания: I. Дибензоильное производное не дает этой реак-
ции даже при непродолжительном кипячении.
2. Дифенилолпропан очищался двукратной промывкой на воронке
Бюхнера дихлорэтаном и последующей двукратной перекристаллизацией из
разбавленной уксусной кислоты. Температура плавления очищенного про-
дукта 151—152°.
ЛИТЕРАТУРА
1. А. Дианин. ЖРФХО, 23, 494 (1891).
2. Т. Szeky. Chem. Zbl., II, 1737 (1904).
Поступила в декабре 1963 г. ИРЕА
УДК 547.442.2.07
МОНООКСИМ ДИАЦЕТИЛА
И. Д. ДЫХАНОВ, Л. Л1. ЕГУПОВА
Н3С-С-С-СН,
II II
О N-OH
C4H7OaN М. в. 101, 10
Монооксим диацетила, являющийся полу продуктом синте-
за диацетила из метнлэтилкетона [1], рекомендовано получать
действием н. амилнитрита на метилэтилкетон с последующей
изомеризацией продукта этой реакции в слабощелочной сре-
де [2]. Однако этот способ малоэффективен для получения
значительных количеств вещества по причине дефицитности и
сравнительно высокой стоимости н. амилового спирта.
В нижеописанной методике синтеза монооксима диацетила
в качестве нитрозирующего средства предложен более деше-
вый и доступный изобутилнитрит [3].
СХЕМА СИНТЕЗА МОНООКСИМА ДИАЦЕТИЛА
Н
Н8С-С-С-СН3 4- 1-С4Нэ—О—N=O -
II I
О н
н
I
Н3С-С-С-СН3 + 1-С4Н6ОН
II I
О N = O
1 он-
HSC-C-C-CHS
II II
О N—ОН
62
Характеристика основного сырья
Метилэтилкетон, очищенный. ОСТ НКЛес 7606/90.
Изобутилнитрит, свежеприготовленный, т. кип. 65—70°.
Кислота соляная, техн., синтетическая, ГОСТ 857—57,
Натр едкий, жидкий, ГОСТ 2263—59, марка «Д».
Кислота серная, контактная, улучшенная, ГОСТ 2184—59.
Условия получения
В трехгорлую круглодонную колбу емкостью 500 мл, снаб-
женную мешалкой, термометром, капельной воронкой и воз-
душным холодильником, помещают 43 мл метнлэтилкетона
(см. примечание 1) и 2 мл концентрированной соляной кисло-
ты и подогревают до 48—50°.
К реакционной смеси при указанной температуре и энер-
гичном перемешивании прикапывают 41 мл свежеперегнанно-
го изобутилнитрита; весь процесс занимает не более 2 часов.
По прибавлении изобутилнитрита реакционную массу охла-
ждают до комнатной температуры и прибавляют к ней в те-
чение 10—15 минут смесь 40 г льда и 40 г 33%-ного водного
раствора едкого натра; температура в массе при этом должна
удерживаться в пределах 20—25°. Реакционную массу пере-
мешивают еще в течение 30 минут, дают разделиться слоям и
отделяют верхний слой изобутилового спирта, который сушат,
перегоняют и возвращают в процесс.
Водный слой охлаждают до 5е и к нему при энергичном
перемешивании прикапывают разбавленную (1:1) серную
кислоту до pH 6,5; при этом температура в массе не должна
превышать 10°, а длительность процесса нейтрализации и пос-
ледующей выдержки реакционной массы должна быть не бо-
лее 2 часов (см. примечание 2).
Выпавшие кристаллы монооксима диацетила отфильтро-
вывают, а маточник упаривают до начала кристаллизации и
охлаждают до 5°. Вновь отфильтровывают кристаллы моно-
оксима, присоединяя их к первоначальному осадку.
Получают сырой моноокенм диацетила в количестве 18 г,
который перекристаллизовывают из воды (1:3) или из петро-
лейного эфира (1 :20).
Выход чистого продукта с т. пл. 75—76° составляет 13,0 г,
или 40% от теоретического.
Примечания: 1. Содержание основного вещества должно быть не
ниже 98%.
2. При большей длительности процесса, особенно при меньшем значе-
нии pH, имеет место частичный гидролиз монооксима до свободного диаце-
тила, а последний в кислой среде подвергается бимолекулярной дегидрата-
ции с образованием ксилохинона, от которого весьма трудно избавиться
при последующей перекристаллизации технического моиооксима как из
води, так и из петролейного эфира.
63
ЛИТЕРАТУРА
1. Н. Н. Дыханов, Л. М. Е г у п о в а. См. статью Диацетил в этом
сборнике.
2. О. D lets и др. Вег., 35, 3292 (1902); 43, 4338 (1907).
3. Н. Н. Дыханов, Л. М. Е г у п о в а. См. статью Изобутилнитрит в
этом сборнике.
Поступила в сентябре 1963 г. ВНИИ монокристаллов
УДК 547.548.07
л-Н ИТРОБЕНЗИЛХЛОРИД
И. Н. ДЫХАНОВ, Л. М. ЕГУПОВА
СН2С1
NO3
C7H6O2NC1 М. в. 171,58
n-Нитробензилхлорид является полупродуктом синтеза
«-нитробензальдегида [1]. Его получение непосредственным
хлорированием n-нитротолуола в боковую цепь (в отличие от
п-нитробензилбромида [2]) осуществить не удается, так как
одновременно с введением хлора в метильную группу проис-
ходит замещение нитрогруппы на хлор[3]. В связи с чтим из-
вестные способы получения n-нитробензилхлорида сводятся к
различным приемам нитрования бензилхлорида. Наиболее эф-
фективным из них является описываемый ниже способ нитро-
вания бензилхлорида нитратом калия в серной кислоте [1].
СХЕМА СИНТЕЗА л- НИТРОБЕНЗИЛХЛОРИДА
z_... (-4°)-(-7°)
^_^-СН8С1 + KNO3 + H2SO4 -----
O2N-СНаС1 4- KHSO4 + Н2О
Характеристика основного сырья
Бензил хлористый, бензольный, ТУ МХП 588—49, сорт 1.
Калий азотнокислый, ч., ГОСТ 4144—48.
5 Зак. 613 65
Кислота серная, ч., концентрированная, ГОСТ 4204—48.
Спирт метиловый, синтетический, ГОСТ 2222—54.
Уголь активированный, древесный, ГОСТ 4453—48, щелоч-
ной марки «А».
Условия получения
В трехгорлую круглодонную колбу емкостью 500 мл, снаб-
женную мешалкой, термометром и капельной воронкой, поме-
щают ПО г нитрата калия (1,09 М) и 400 мл концентрирован-
ной серной кислоты и перемешивают до растворения соли.
К охлажденному до минус 6—7° раствору прикапывают
из воронки 114 мл хлористого бензила (1 А4) с такой ско-
ростью, чтобы температура в массе не превышала минус 4°.
Уже по прибавлении половины исходного объема бензилхло-
рида начинается выделение кристаллов п-нитробензилхлори-
да.
После прибавления всего бензилхлорида реакционную
массу размешивают при минус 4—7° еще в течение 1 часа,
затем выливают на 500 г льда.
Осадок п-питробензилхлорида тщательно отсасывают на
стеклянной пористой пластинке, промывают 500 мл воды.
Маслообразный фильтрат охлаждают в стакане до 0—5° и
выдерживают в течение 2—3 часов при периодическом поти-
рании палочкой по стенке стакана. При этом- выкристалли-
зовывается дополнительное количество л-нитробензилхлори-
да, который отсасывают, промывают водой и присоединяют к
основному осадку.
Сырой технический п-нитробензилхлорид очищают пере-
кристаллизацией из метанола (1:2,5) с добавлением активи-
рованного угля (5% от веса технического продукта).
Выход чистого n-нитробензилхлорида с т. пл. 70—71° со-
ставляет 74—75 г или ^-43% от теоретического.
ЛИТЕРАТУРА
1. Органические реакции. Сб. 8, М, ИЛ, 1952, стр. 276
2. С a vll 1. J. Amer. Chem. Soc., 72, 2804 (1950).
3. Е. К. У г л е ц к а я. Авт. свид. 57353; Л. С. С о л о д а р ь, В. В. М а р-
кии. Успехи химии, 16, 102 (1947).
4. Alway. J. Amer. Chem. Soc., 24, 1062 (1902).
Поступила в сентябре 1S63 г.
ВНИИ MOHOKfметаллов
УДК 547.551.2.07
3-НИТРОДИФЕНИЛАМИН
С. В. ЖУРАВЛЕВ, В. М. СВЕТЛАЕВА
NO2
C12H10N2O2 “ = М. в. 214,22
Синтез 3-нитродифениламина конденсацией бромбензолас
ж-нитроацетанилидом предложен уже давно [1]. Однако,
вследствие образования в реакции побочных продуктов вы-
деление 3-нитродифениламина связано со значительными
трудностями и при высоком выходе сырого продукта [2], часто
после кристаллизации получают небольшие количества ана-
литически чистого вещества [3].
В связи с необходимостью получения 3-нитродифенилами-
на для дальнейшего синтеза 2-аминофентиазина нами был
разработан метод очистки промежуточно образующегося
М-ацетил-З-нитродифениламина колоночной фильтрацией хло-
роформного раствора на А12О3 II степени активности [4]. Очи-
стка этого промежуточного продукта от образующихся смол
позволила нам надежно получать 3-нитродифениламин с вы-
ходом 80% и выше, причем вещество после однократной пере-
кристаллизации представляло собой аналитически чистый
препарат.
СХЕМА СИНТЕЗА 3-НИТРОДИФЕНИЛАМИНА
no2
_ _/ К2СО8
^-Br + HN-^
/ j \/ СIJ j 012
СОСНз
5*
67
no2
СОСНз
NOa
_ _Х HC1
// X — N—// 'Ч------->
\=-_/ | \ = /
СОСН3
Характеристика основного сырья
Бромбензол, ч., ТУ МХП 2753—51, высушенный над
СаСЬ и перегнанный; т. кип. 155—157°.
.и-Нитроапетанилид, ч., ВТУ 513—60, перекристаллизован-
ный, т. пл. 150—151°.
Калий углекислый, ч., ГОСТ 4221—51, прокаленный.
Медь бромистая (Си2Вг2), свежеприготовленная [5].
Алюминий окись, д. хр., II степени активности, ТУ МХП
2962—54.
Хлороформ, ч., ГОСТ 3160—51.
Спирт этиловый, ректифицированный, ГОСТ 5962—51.
Кислота соляная, ч., ГОСТ 3118—46.
Условия получения
В колбу емкостью 0,5 л загружают 54 г (0,3 А1) л-иитро-
ацетанилида, 99,2 г (0,6 /И) бромбензола, 14 г безводного,
растертого в порошок, углекислого калия и 0,63 г бромистой
меди (см. примечание 1); эту смесь нагревают с обратным
холодильником на масляной бане 24 часа при температуре
бани. 160—180° (см. примечание 2). Затем добавляют еще 15 г
углекислого калия (общее количество К2СО3 0,21 А1) и про-
должают нагревание при 180—200° еще 24 часа (см. примеча-
ние 3).
По окончании нагрева от реакционной массы отгоняют
бромбензол, а маслянистый остаток N-ацетил-З-нитродифе-
ниламина и частично 3-нитродифепиламина экстрагируют
~800 мл хлороформа (см. примечание 4). Полученный раст-
вор высушивают хлористым кальцием и пропускают через ко-
лонку с 200 г А120з II степени активности при полностью от-
крытом кране (отношение диаметра колонки к высоте столба
адсорбента~1 :7).
Колонку промывают хлороформом, который отгоняют по
мере пропускания раствора через колонку, в результате чего
68
последнюю промывку до получения почти бесцветного элюата
проводят регенерированным хлороформом.
После отгонки растворителя (конец отгонки для удаления
следов растворителя проводят в вакууме водоструйного на-
соса) к остатку добавляют 80 мл концентрированной соляной
кислоты в 100 мл этилового спирта и в течение 5 часов нагре-
вают раствор на глицериновой бане при перемешивании и
температуре 100—110°. Затем раствор охлаждают, не прекра-
щая при этом перемешивания, чтобы избежать образования
комков. После охлаждения ледяной водой полученный кри-
сталлический осадок отсасывают.
Выход 3-нитродифениламина равен 55,9 г, что составляет
86,9% от теоретического; т. пл. продукта 108—109°.
Вещество однократно кристаллизуют из раствора в горя-
чем этиловом спирте с добавлением горячей воды (на 1 г ве-
щества берется 5 мл спирта и 2,5 мл воды); т. пл. перекри-
сталлизованного 3-нитродифениламина 111,5—112,5°.
По литературным данным, т. пл. продукта 110—112° [6],
ИЗ—114° [7]. Выход к взятому для перекристаллизации ве-
ществу равен 85,4%. Кроме того, из маточного раствора мо-
жет быть выделено дополнительное количество вещества.
Найдено, %: С-67,07; 66,99; И-4,75; 4,80; N—13,05; 13,18
С1,Нц>М4О4. Вычислено, %: С 67,23; Н—4,70; N- 13,07.
Примечания: 1. Реакцию можно проводить в присутствии порош-
кообразной меди (ПМ ТУ 4451—54) с добавлением или без добавления
KJ.
2. При проведении реакции с большими загрузками, необходимо пе-
ремешивание.
3. При необходимости время нагрева может быть сокращено до 24 ча-
сов: 12 часов при' 160—180° и 12 часов при 180—200° с незначительным
при этом уменьшением выхода З-нитродифениламииа.
4. Экстракция продукта меньшим количеством хлороформа ведет к
получению более концентрированного хлороформного раствора, что за-
трудняет в последующем очистку вещества на колонке.
ЛИТЕРАТУРА
1. J. Goldberg. Вег., 40, 4541 (1907).
2. С. В. Ж у р а в л е в, В. А. С к о р о ду м о в. ЖОХ, 31, 3129 (1961).
3. Р. G г am m a 11 с a k i s. Bull. Soc. chim. France, 21, 99 (1954).
4. В. M. С в e г л a e в а, С. В. Журавлев. Ж. нрикл. химии, 35,
1857 (1962).
5. Синтезы органических препаратов, 1, М., ИЛ, 1949, стр. 137;
Ю. В. К а р я к и н, И. Н. Ангелов. Чистые химические реактивы, М.,
Госхимиздат, 1955, стр. 344.
6. Т. L. Dawis, A. A. A s h d о wп. J. Amer. Chem. Soc., 46, 1053
(1924).
7. M., J. S. Dewar, D. S. Urch. J. Chem. Soc., 3079 (1958).
Поступила в марте 1964 г. Институт фармакологии и химио-
терапии АМН СССР
УДК 547.835.07
7-НИТРО-2- И РО-2- И 4-МЕТОКС ИАКРИДОНЫ-9 И 6- И 7-НИТ- 4-МЕТОКСИ-10-МЕТИЛАКРИДОНЫ-9 В. И. К.ИХТЕВА, И. И. ДЫХАНОВ
О 8 II OaN-Z V? 61 II 10 Ч/v 5 . N О • II II -|-ОСНз O.N+ II II +ОСН3 N |
1 Н сн8
7-Нитро 2- и 4-метокси- 6- и 7-Нитро-2- и 4-метокси-
акридоны-9 -10-метилакридоны-9
CuHI0O4N3 М. в. 270,24 C]6H]2O4N2 М. в. 284,27
Из 16 теоретически возможных питрометоксиакридонов-9 с
нитро- и метоксигруппами в разных «бензольных» ядрах опи-
сан только один * нерастворимый в обычных органических ра-
створителях и неплавящийся до 300° 2-метокси-7-нитроакри-
дон-9, полученный нагреванием 4-нитро-4'-метоксидифенил-
амин-2-карбоновой кислоты с эквивалентным количеством
хлорокиси фосфора [1]. N-Метильные производные нитромето-
ксиакридонов-9 до сих пор не были описаны.
На основании литературных данных по растворимости
других замещенных акридонов-9 и их N-метильных производ-
ных [2] можно было ожидать, что метилированные при азоте
нитромстоксиакридоны будут значительно более растворимы-
ми в органических растворителях, и в связи с этим окажется
возможным их получение в достаточно чистом состоянии.
В 1963 году А. К Сухомлинов и В. П. Максимец [3] синтезировал:!
изомерные 6-нитро-1-, 2-, 3- и 4-метоксизамсщеиные акридона-9 путем гид-
ролиза соответственно замещенных 9-хлоракридинов.
70
Последнее обстоятельство весьма важно для оценки донорно-
акцепторного эффекта нитро- и метоксигрупп в трехкольцевой
гетероциклической системе и для исследования физических
характеристик подобных соединений.
В данной работе осуществлен синтез 2-метокси-7-нитро-
акридона-9 и его 4-метокси-изомера путем гидролиза соответ-
ственно замещенных 9-хлоракридинов [4] разбавленной соля-
ной кислотой. Оба эти вещества лишь с большим трудом ра-
створяются в ледяной уксусной кислоте и не растворяются в
других обычных органических растворителях. Очищенные пе-
рекристаллизацией из уксусной кислоты, они не плавятся до
300°, в связи с чем общепринятый способ идентификации ор-
ганических соединений по температуре плавления к ним не
применим. ,
Значительно более растворимыми и низкоплавкими оказа-
лись N-метильные производные 7-нитро-2- и 4-метоксиакридо-
нов-9, впервые полученные авторами данной статьи обработ-
кой названных акридонов метиловым эфиром бензолсульфо-
кислоты в присутствии едкой щелочи в кипящем ацетоновом
растворе.
Аналогичным путем впервые синтезированы N-метильные
производные 6-нитро-2- и 4-метоксиакридонов-9, полученных
по А. К- Сухомлинову и В. П. Максимцу [3].
СХЕМА СИНТЕЗА 6- И 7-НИТРО-2- И 4-МЕТОКСИАКРИДОНОВ-9
И ИХ N-МЕТИЛЬНЫХ ПРОИЗВОДНЫХ
С1
нао,н-
O2N+ II | +ОСН3-------------►
N
О
II
/Сч/х/х c,h,so,ch,
o3N+ II II +ОСН3 —
Ч/x/W
N
II
о
II
-> OgN-f- || || -f-OCHj
Характеристика основного сырья
2-Метокси-7-нитро-9-хлоракридин, т. пл. 226°.
4-Метокси-7-нитро-9-хлоракридин, т. пл. 267—267,5°.
2-Метокси-6-нитро-9-хлоракридин, т. пл. 219,5—220,5°.
4-Метокси-6-нитро-9-хлоракридип, т. пл. 224°.
Метиловый эфир бензолсульфокислоты, ч., РТУ 138—57.
Ацетон, ч.д.а., ГОСТ 2603—51.
Кислота соляная, ч., ГОСТ 3118—46.
Условия получения
Получение 6- и 7-нитро-2- и 4-ме.токсиакридонов-9. В трех-
горлую круглодонную'колбу с пришлифованным обратным
холодильником помещают 5,5 г 7-нитро-2- или 4-метокси-9-
хлоракридина и 370 мл 12 %-ной соляной кислоты и кипятят в
течение 7 часов.
Осадок нитрометоксиакридона отфильтровывают, промы-
вают на фильтре дистиллированной водой до отсутствия в
промывке ионного хлора (проба с азотнокислым серебром) и
высушивают при 100— 105° до постоянного веса. Технические
нитрометоксиакридоны перекристаллизовывают из ледяной
уксусной кислоты (1 : 1000) с осветлением уксуснокислых ра-
створов активированным углем (5% по весу) и получают очи-
щенные вещества с выходами 92—95% от теоретических. Цвет
и форма их кристаллов, а также результаты количественного
определения азота приведены в таблице.
Получение N-метильных производных 6- и 7-нитро-2- и 4-
метоксиакридонов-9. В круглодонную колбу, снабженную ме-
шалкой, термометром и капельной воронкой, помещают 12 г
нитрометоксиакридона-9, 4 г тонкоизмельченного едкого кали
и 200 мл ацетона, нагревают в течение 30 минут при 55—60°
и приливают 38,2 г метилового эфира бензолсульфокислоты.
Реакционную смесь кипятят в течение 1,5 часа, охлаждают
до комнатной температуры, отфильтровывают осадок N-ме-
тилакридона, промывают его на фильтре небольшим количе-
ством метанола (для удаления смолистых примесей), затем
водой до нейтральной реакции на универсальный индикатор и
сушат при 95—100° до постоянного веса. Получают техниче-
ские N-метилнитрометоксиакридоны с выходом около 70% от
теоретического, считая на исходные акридоны.
Чистые 6- и 7-нитро-2- и 4-метокси-10-метилакридоны-9
получают перекристаллизацией технических продуктов из
бензола или дихлорэтана. Их температуры плавления, цвет и
форма кристаллов, а также результаты количественного опре-
деления азота приведены в таблице.
72
Акридоны-9 Температу- ра плавле- ния, °C Цвет н форма кристал? лов, растворитель* Найдено азота, %**
7-Нитро-2-мегокси- > 300° Лимонно-желтый аморф- ный порошок из 1 12,98
7-Нитро-2-метокси-10- метил- 267- 267,5° Желтые пластинки из III 9,79
7-Нитро-4-метокси- >300° Желтые иглы из I 13,00
7-Нитро-4-метокси-10- метил- 235° Светло-желтые иглы из 11 9,81
6-Нитро-2-метокси- > 300° Оранжево-красный аморфный порошок из I 13,04
6-Нитро-2-метокси-10’ -метил- 314—315° Светло-красные иглы из III 9,86
6-Нитро-4-метокси- > 300° Оранжево-красный аморфный порошок из 1 13,01
6-Нитро-4-метокси-10- метил- 223-224° Светло-красные иглы из II 9,82
* I —Ледяная уксусная кислота, II—бензол, 111—дихлорэтан.
** Вычислено азота: у незамещенных при азоте акридонов— 13,02 %,
у N-метилакридонов-9,85 %.
ЛИТЕРАТУРА
1. W. Lesnianski. Chem. Zbl., 1, 3105 (1929); Н. С. Дроздов,
ЖОХ, 5. 691 (19 <5).
2 Н. С. Дроздов. ЖОХ, 7, 2292 (1937).
3. V К. Сухомлинов, В. П. Махсвмец. ЖОХ (в печати).
4. В. И. К и х т е в а, К. Н. Дых анов. См. статью 7-Нитро-2- и
4-метокси-9-хлоракридины в этом сборнике.
Поступила в апреле 1964 г.
ВНИИ монокристаллов
УДК 547.58.07
4-НИТРО-2 -, 3 - И 4-МЕТОКСИДИФЕН ИЛАМИН-2-
КАРБОНОВЫЕ КИСЛОТЫ
В. И. КИХТЕВА, Н. И. ДЫХАНОВ
3 СО.ОН 5' ,
°2N-Y \\? Р| |-ОСН3
YVZ
Н
4-Нитро-Х'-метоксидифениламин-2-карбоиовые кислоты
C14Hl2O5N2 М. в. 288, 26
Замещенные дифениламин-2-карбоновые кислоты являют-
ся основными исходными веществами в синтезе разнообраз-
ных производных акридина [1] и дифениламина [2]. Их получа-
ют, обычно, по методу Ульманна [3]—конденсацией орто-гало-
идбензойных кислот с анилинами в присутствии медного по-
рошка (катализатор) и поташа (средство для связывания га-
лоидоводорода) в высококипящих спиртах [4] или в концентри-
рованном водном растворе [5].
В ряду изомерных нитрометоксидифепиламин-2-карбоно-
вых кислот с нитро- и метоксигруппами в разных бензольных
ядрах описана только одна* 4-нитро-4/-метоксидифениламин-
2-карбоновая кислота, полученная нагреванием 2-хлор-5-нит-
робензойной кислоты с n-анизидином в растворе глицерина
при 117—120° в присутствии поташа и медного порошка [4].
В данной работе осуществлен синтез 4-нитро-4'-метокси-
дифениламин-2-карбоновой кислоты взаимодействием 2-хлор-
В 1963 году А. К- Сухомлинов и В. П. Максимец [6] синтезировали
б-нитро-а'-.З7- и 4/-метоксидифениламин-2-карбоновые кислоты взаимодей-
ствием 2-хлор-4-иитрэбензойной кислоты с изомерными анизидинами по
упомянутому методу Ульманна [3].
74
5-нитробензойной кислоты с n-анизидином в концентрирован-
ном водном растворе поташа (т. кип. 105—106°) в отсутствие
медного катализатора, а ее ранее неизвестные 2'- и З'-мето-
ксиизомеры получены нагреванием при 115—120° той же
хлорнитробензойной кислоты с о- и л-анизидинами в присут-
ствии медного порошка и поташа в среде н.амилового спирта.
Возможность проведения реакции между 2-хлор-5-нитро-
бензойной кислотой и я-апизидипом в отсутствие медного ка-
тализатора обусловлена, очевидно, двумя причинами: активи-
рующим действием на галоид двух о- и «-расположенных
электроноакцепгорных заместителей и более высокой основ-
ностью я-анизидина по сравнению с его о- и /«-изомерами [7].
Пример некаталитической реакции между 2,4-дихлор-5-нитро-
бензойной кислотой и я-анизидином был зафиксирован од-
ним из авторов данной статьи еще в 1959 году [8].
СХЕМА СИНТЕЗА 4-НИТРО-2'-, 3Z- и 4/-МЕТОКСИДИФЕНИЛАМИН-2-
КАРБОНОВЫХ КИСЛОТ
,СООН
O,N—
2 I + н м II +ОСН3 + к3со3 -*
Ч/хС1
COOK ч
ОЦ_/\/ /X
2 | II II +ОСН8 + КС1 + н2о + со2
N
I
н
I + НС1
7/ соон ..
O,N —
2 I II II +ОСН3 + КС1
Ч/\ N
н
Характеристика основного сырья
2-Хлор-5-нитробензойная кислота, ч., ТУ МХП 2169—49.
о-Анизидин, ч., ТУ МХП 2356—50.
л-Анизидин, ч., ВТУ РУ 865—53.
я-Анизидин, ч„ ТУ МХП 1485 51.
Калий углекислый, ч„ ГОСТ 4221—57.
Условия получения
Получение 4-нитро-4'-метоксидифениламин-2-карбоновой
кислоты. В трехгорлой круглодонной колбе, снабженной ме-
75
шалкой, термометром и обратным холодильником, имеющим
переключение на нисходящий прямой холодильник, готовят
раствор 41 г поташа в 130 яд воды (см. примечание 1) и при-
бавляют к нему небольшими порциями (во избежание силь-
ного вспенивания) 60 г 2-хлор-5-нитробензойной кислоты.
В образовавшийся раствор калиевой соли 2-хлор-5-нитро-
бензойной кислоты вносят в один прием 36 г н-анизидина, пе-
ремешивают реакционную смесь в течение 7 часов при энер-
гичном кипении (см. примечание 2) и охлаждают до комнат-
ной температуры.
Осадок калиевой соли 4-нитро-4'-метоксидифениламин-2-
карбоновой кислоты отфильтровывают, промывают метанолом
до бесцветного фильтрата (для удаления смолистых приме-
сей) и растворяют в достаточном объеме воды. Кипящий вод-
ный раствор перемешивают с 5 г активированного угля, филь-
труют и к фильтрату при 95—100° осторожно (во избежание
выброса!) прибавляют 15%-ную соляную кислоту до pH 4,5.
Выпавший аморфный осадок технической 4-HHTpo-4z-MeT-
оксидифениламин-2-карбоновой кислоты отфильтровывают,
промывают на фильтре кипящей дистиллированной водой до
отсутствия ионного хлора (проба с азотнокислым серебром),
высушивают при 95—100° до постоянного веса (52—55 е) и
перекристаллизовывают из 96%-ного этилового спирта (1:17)
с осветлением спиртового раствора активированным углем
(5% по весу).
Чистая 4-нитро-4'-метоксидифениламин-2-карбоновая кис-
лота (выход 58—60% от теоретического) имеет температуру
плавления 230—231°; смешанная проба с образцом этой кис-
лоты, полученным по Леснянскому [4], плавится без депрессии.
Примечания: 1. Для растворения поташа в указанном объеме
воды массу необходимо подогреть до 50—60°.
2. Если температура в кипящей массе будет ниже 105°, то необходи-
мо обратный холодильник переключить на прямой и отогнать некоторое
количество воды до достижения указанной температуры.
Получение 4-нитро-2'- и 3'-метоксидифениламин-2-карбо-
новых кислот. В колбу прибора, применявшегося для получе-
ния 4-нитро-4,-метоксидифениламин-2-карбоновой кислоты,
помещают 60 г 2-хлор-5-нитробензойной кислоты, 45 г пота-
ша, 170 мл н. амилового спирта и 1,2 г медного порошка. Со-
держимое колбы нагревают до 60°, прибавляют к нему в один
прием 55 г о-анизидина и перемешивают реакционную массу
ь течение 3 часов при температуре 115—120°.
По окончании реакции из реакционной массы отгоняют
н. амиловый спирт с водяным паром, а к остатку добавляют
горячую воду до полного растворения осадка образовавшейся
калиевой соли 4-нитро-2/-метоксидифениламин-2-карбоновой
кислоты. Раствор соли кипятят с 5 г активированного угля,
фильтруют в горячем состоянии и к кипящему фильтрату
76
прибавляют 15%-ную соляную кислоту до рН~5 по универ-
сальному индикатору.
Осадок технической 4-нитро-2'-метоксидифениламин-2-кар-
боновой кислоты отфильтровывают, промывают кипящей ди-
стиллированной водой до отсутствия ионного хлора (проба с
азотнокислым серебром), высушивают при 95—100° до посто-
янного веса (48—50 г) и перекристаллизовывают из этилово-
го спирта (1:15) с осветлением спиртового раствора активи-
рованным углем (5% по весу).
Чистая 4-питро-2/-метоксидифениламин-2-карбоновая кис-
лота кристаллизуется в виде ярко-желтых игл с температурой
плавления 215°. Выход ее составляет около 50%, считая на
2-хлор-5-нитробензойную кислоту.
Найдено, %: N—9, 66.
C14HlaOsN?. Вычислено, %: N—9, 72.
Аналогичным образом из 71,6 г 2-хлор-5-нитробензойной
кислоты,, 82 г, ж-анизидина, 50 г поташа и 1,2 г медного по-
рошка в 250 мл н. амилового спирта получают 60 г 4-нитро-
3'-метоксидифениламин-2-карбоновой кислоты с температурой
плавления 253—254°; выход 74,2% от теоретического.
Найдено, %: N—9, 69.
C14HiaO5Na. Вычислено, %: N-9, 72.
ЛИТЕРАТУРА
1. А. М. Григоровский. Успехи химии, 21, 615 (1952).
2. А. Ф. Б е х л и, А. М. Григоровский, Н. В. С а в и ц к а я,
Е. Ф. Тарасевич, Ю. С. Ц ы з и н, М. Н. Щукина. Авт. свид. 105356;
Бюлл. изобр. № 12 (1957).
3. U liman п. Вег., 36, 2382 (1903); 37, 853 (1904); 38, 2211 (1905);
Liebigs Ann. Chem., 355 312 (1907).
v ,
4. W. L е s n i a n s k i. Chem. Zbl., L 3105 (1929); H.C. Дроздов.
ЖОХ, 5, 691 (1935).
5. H. H. Дыханов, Г. А. Горлач, В. П. Серговская. Меди-
цинская промышленность СССР, № 4, 26 (1960).
6. А. К. Сухомлинов, В. П. Ма ксимец. ЖОХ (в печати).
7. И. Н. Ворожцов. Основы синтеза полупродуктов и красителей,
М., Госхимиздат, 1955, стр. 338.
8. Н. Н. Дыханов. Авт. свид. 126497 (1959).
Поступила в апреле 1964 г. ВНИИ монокристаллов
УДК 547.835.3.07
7-НИТРО-2- И 4-МЕТОКСИ-9-ХЛОРАКРИДИНЫ
В. И. КИХТЕВА, Н. И. ДЫХАНОВ
c]4han2ci
7-Нитро-2-и 4-метокси-9-хлоракр идины
М. в. 288, 69
Благодаря большой подвижности хлора, замещенные
9-хлоракридипы находят широкое применение в качестве по-
лупродуктов синтеза различных других производных акриди-
на [1]. Наиболее распространенным способом получения
9-хлоракридинов является циклизация дифениламин-2-карбо-
новых кислот [2] или их щелочных солей [3] под действием
хлорокиси фосфора.
Из четырех изомерных по положению метоксигруппы
7-нитро-метокси-9-хлоракридинов (с нитро- и метоксигруппа-
ми в разных «бензольных» ядрах) в литературе описан толь-
ко один * 7-нитро-2-метокси-9-хлоракридин [4].
В данной работе воспроизведена методика получения этого
соединения [4] и аналогичным путем, исходя из 4-нитро-2'-мет-
окси-дифениламин-2-карбоновой кислоты [6], синтезирован
его ранее неизвестный 4-метокси-изомер.
СХЕМА СИНТЕЗА 7-НИТРО-2- и 4-МЕТОКСИ-9-ХЛ ОРАКРИДИНОВ
соон
f^+OCHs
Н
* В 19G3 году А, К. Сухомлинов и В, П. Максиме^ [5] получили че-
тыре изомерных 6-нитро-Ь, 2-, 3- и 4-метокси-9-хлоракридина.
I
I
l_
o2n-
Cl
/А/ЧА
I II I +OCH3
N
H
Характеристика основного сырья
4-Нитро-4/-метоксидифени ламин-2-карбоновая кислота,
т. пл. 230°.
4-Нитро-2'-метоксидифениламин-2-карбоновая кислота,
т. пл. 215°.
Хлорокись фосфора, ч., ВТУ У 166—51.
w-Ксилол, ч., ТУ МХП 3601—53.
Бензол, ч.д.а., ГОСТ 5955—51.
Условия получения
В трехгорлую круглодонную колбу, снабженную мешал-
кой с каучуковым абтюратором, термометром и обратным хо-
лодильником, помещают 29 а сухой гонкоизмельченной 4-нит-
ро-4'-метоксидифениламин-2-карбоновой кислоты, 32 г хлор-
окиси фосфора и 200 мл сухого «-ксилола. Содержимое кол-
бы нагревают при размешивании до слабого кипения н вы-
держивают в течение 3 часов.
По охлаждении до комнатной температуры реакционная
масса разделяется на два слоя. Верхний .(ксилольный) слой
отделяют декантацией, а нижний маслообразный коричневый
слой выливают на 250—300 г льда, смоченного 70—75 мл
10%-ной аммиачной воды.
Образовавшийся желтый аморфный осадок отфильтровы-
вают, промывают на фильтре 150 мл метанола и сушат при
70—75° до постоянного веса. Получают 27,4 г технического
2-метокси-7-питро-9-хлоракридина, который перекристаллизо-
вывают из бензола (1 :25) с осветлением бензольного раство-
ра активированным углем (5% по весу).
В соответствии с литературными данными [4], чистый 2-
метокси-7-нитро-9-хлоракридин плавится при 226°; выход его
составляет ~ 65% от теоретического.
79
Аналогичным путем из 22,5 г 4-нитро-2/-метоксидифенил-
амин-2-карбоновой кислоты, 50 г хлорокиси фосфора и 180 мл
«-ксилола получают 23,4 г чистого 4-метокси-7-нитро-9-хлор-
акридина с температурой плавления 267—267,5°.
Найдено, %: N-9, 73; С1-12, 25.
CI4H9OSN2C1. Вычислено, %: N—9, 70; С1—12, 28.
ЛИТЕРАТУРА
1. А. М. Григоровский. Успехи химии, 21, 615 (1952).
2. О. Ю. Магидсон, А. М. Григоровский. ЖОХ, 3, 615
(1933); Вег., 66, 866 (1933).
3. Н. Н. Дыханов, Г. А. Горлач, В. П. С е р г о в с к а я. Меди-
цинская промышленность СССР, № 4, 26 (1960).
4. W. Lesnianskii. Chem. Zbl., 1, 3105 (1929); Н. С. Дроздов.
ЖОХ, 5, 691(1935).
5. А. К. Сухомлинов, В. П. М а к си м е ц. ЖОХ (в печати).
6. В. И. Кихтева, Н. Н. Дыханов. См. статью 4-Натро-2'-, 3'- и
4'-метоксидифенилалшн-2-карбоновые кислоты в этом сборнике.
Поступила в апреле 1964 г. ВНИИ монокристаллов
УДК 547.556.8.05
ОРТО- И ПАРА-НИТРОФЕНИЛГИДРАЗИНЫ
В. Т. СКРИПКИНА, Н. Н. ДЫХАНОВ
NH—NH2
CeH,O2N3
М. в. 153, 14
о-Нитрофенилгидразин был впервые получен в 1889 г.
Бишлером восстановлением хлористого о-нитрофенилдиазония
хлористым оловом в соляной кислоте [1], а в 1896 г. Бамбер-
гер и Краусс, восстанавливая хлористый я-нитрофенилдиазо-
ний сульфитом натрия, получили п-нитрофенилгидразин [2].
В промышленном синтезе арилгидразинов наибольшее
применение нашел способ восстановления арилдиазонийхло-
ридов сульфитно-бисульфитной смесью [3]. Ниже описывается
оптимальная методика синтеза этим путем о- и п-нитрофенил-
гидразинов, состоящая из 8 стадий.
I. ^-NH2 НС1 —> ( NHa-НС1;
no2 no2
II. ^-NH3-HC1 + NaNOa + HC1
NO2
^-N = N- Cl + NaCl + H2O;
no2
б Зак. 613
81
Ш. 2NaHSO3 + Na2CO3 — 2 Na2SOs + H20 -f CO3;
IV.
N-—N—Cl + Na2SO3
+^-N = N-SO3Na + NaCl;
NO2
V. ^-N=N-SO8Na + NaHSO3 -
NOa
SO3Na
==x I
^.-N-NH-SO3Na;
NO2
SO3Na
VI. ^-N-NH-SO3Na + H2O —-
NO2
-» ^-NH-NH-SO3Na-|-NaHSO4;
no2
VII. ^+^-“NH-NH-SO3Na + H2O + HC1
NO2
—NH—NHa-HCl + NaHSO4;
no2
VIII. ^.-NH-NH3-HC1 + CHgCOONa —
NO2
— ^+^-NH-NHa + CH3COOH + NaCl.
NO2
82
Характеристика основного сырья
о-Нитроанилин, ч., ТУ МХП 2639—51, или
n-Нитроанилин, ч., ТУ МХП 1461—46.
Кислота соляная, техн., синтетич., ГОСТ 857—57.
Бисульфит натрия, техн, (водный раствор), ГОСТ 902—41,.
Сода кальцинированная, ГОСТ 5110—49.
Натрий азотистокислый, техн., 6194—52, сорт I.
Условия получения
I. Получение хлоргидратов о- и п-нитроанилинов. В ста-
кан емкостью 250 мл, хорошо закрепленный на штативе и
снабженный механической мешалкой и термометром, поме-
щают 13,8 г (0,1 М) чистого о- или п-нитроанилииа и 70 мл
разведенной (1:1) соляной кислоты.
Содержимое стакана при энергичном перемешивании на-
гревают на водяной бане до 80—90° и выдерживают до пол-
ного растворения осадка.
II. Получение хлоридов о- и п-нитрофенилдиазониев. Син-
тезированные, как описано выше, растворы хлоргидратов
о- или n-нитроанилинов охлаждают в смеси льда с солью до
минус 2—3° и, не прекращая перемешивания, постепенно при-
бавляют 8,3 г сухого нитрита натрия с такой скоростью, что-
бы температура в массе не превышала 0°.
В процессе прибавления нитрита натрия осадок хлоргид-
рата исходного нитроанилина превращается в хлорид соответ-
ствующего нитрофенилдиазония и масса становится гомоген-
ной. Окончание процесса определяют по наличию некоторого
избытка свободной азотистой кислоты (легкое потемнение
йодкрахмальной бумажки), причем реакционная масса долж-
на быть слабокислой на конго.
III. Приготовление сульфитно-бисульфитной смеси. В
стакан, снабженный мешалкой и хорошо закрепленный на
штативе, помещают 52 мл продажного раствора бисульфита
натрия с концентрацией 505,9 г/л и к нему при энергичном
размешивании прикапывают из воронки заранее приготовлен-
ный раствор 13,8 г кальцинированной соды в 40 мл горячей
воды.
IV—VI. Получение о- или п-нитрофенилгидразинсульфона-
тов натрия. В свежеприготовленную и охлажденную до 3—5°
сульфитно-бисульфитную смесь при энергичном размешива-
нии прикапывают из воронки холодный (и также свежеприго-
товленный) раствор хлористого о- или п-нитрофенилдиазония
с такой скоростью, чтобы температура в массе удерживалась
в пределах 3—5° (понижение или повышение температуры
против указанных пределов приводит к осмолению реакцион-
ной массы).
6*' 83
По прибавлении диазораствора перемешивание реакцион-
ной массы при той же температуре продолжают еще в тече-
ние 30 минут, после чего приливают соляную кислоту до кис-
лой реакции на конго (~30 мл), нагревают до 40—50° и
оставляют самопроизвольно охлаждаться на 12—16 часов. За
это время выпадает осадок о- или п-нитрофенилгидразинсуль-
фоната натрия, который отфильтровывают и подвергают гид-
ролизу, как описано ниже. Не следует подкисленный раствор
фепилгидразинсульфонатов натрия нагревать выше 50°, так
как наступает их гидролиз.
VII. Получение хлоргидратов о- или п-нитрофенилгидрази-
нов. Хорошо отжатую пасту о- или п-нитрофенилгидразин-
сульфонатов натрия помещают в стакан, снабженный мешал-
кой и термометром, прибавляют 60 мл концентрированной со-
ляной кислоты и размешивают при 80—90° в течение 1,5—
2 часов. За это время гидролиз сульфонатов полностью за-
канчивается.
Реакционную массу по возможности быстрее охлаждают
во льду с солью до 0° и отфильтровывают выпавший осадок
хлоргидрата о- или н-нитрофенилгидразина.
VIII. Получение оснований о- или п-нитрофенилгидрази-
нов. Тщательно отжатый осадок хлоргидрата о- или п-нит-
рофенилгидразина растворяют в 100—150 мл горячей (^90°)
воды, прибавляют 3 г активированного угля, размешивают в
течение 5 минут и фильтруют в горячем состоянии. К еще
теплому (50—60°) фильтрату приливают при размешивании
30%-ный водный раствор уксуснокислого натрия (50—70 мл).
При этом немедленно выделяется осадок основания о- или п-
нитрофенилгидразина.
Реакционную массу охлаждают до 10°, осадок питрофенил-
гидразина отфильтровывают, дважды промывают холодной
дистиллированной водой (по 20 мл каждый раз), а фильтрат
проверяют на полноту осаждения нитрофенилгидразина при-
бавлением к его пробе избытка водного раствора ацетата
натрия, прн этом не должно выпадать осадка.
Выход технического о-нитрофенилгидразина составляет
около 20 г, а его п-изомера — 20—25 г.
Очистку технических оснований о- и п-нитрофепилгидра-
зинов осуществляют перекристаллизацией из воды (1:20) с
применением в качестве осветляющего средства активирован-
ного угля (-10% от веса технического продукта).
Выходы чистого о-нитрофенилгидразина составляют
4—4,5 г (~28% от теоретического), а его пара-изомера —
5—6 г (—36% от теоретического). Их качество удовлетво-
ряет требованиям соответственно ВТУ П—118—57 и
ТУ НКХП 378—41.
84
ЛИТЕРАТУРА
1. А. В i s с h 1 е г. Вег., 22, 280 (1889).
2. Е. Bamberger, Е. Krauss. Вег., 29, 1834 (1896).
3. Н. Pechman. Вег., 28, 863 (1895); Е. Bamberger, А. Mayen-
berg. Вег., 30, 374 (1897).
Поступила в сентябре 1963 г. ВНИИ монокристаллов
УДК 547.572.6.05
О -ОКСИФЕН ИЛ ВИ НИЛ КЕТОН
/1. И. ТУРБИНА, И. Г. СИНЯВСКИМ
сн=сн2
I
/с=0
С9Н8О2 М. в. 148, 16
Молекула о-оксифенилвинилкетона содержит хелатную
группировку, аналогичную содержащейся в о-оксиацетофе-
ноне, который является широкоизвестным комплексообразо-
вателем [1]. Наличие в молекуле о-оксифенилвинилкетона ви-
нильной группы, способной к полимеризации, перспективно,
так как позволяет на его основе получать различные поли-
мерные соединения (например, хелатные катиониты). Кро-
ме того, основание Манниха, в частности о-оксидиметилами-
нопропиофенон солянокислый, являющийся исходным полу-
продуктом для получения о-оксифенилвинилкетопа, облада-
ет ярко выраженным бактерицидным действием [2].
Имеется одно сообщение о получении о-оксифенилвинил-
кетона из о-окси-р-хлорпропиофенона, однако описание ме-
тода синтеза, характеристики мономера и его свойств отсут-
ствуют [3].
Поэтому был разработан метод синтеза о-оксифенилви-
нилкетона путем перегонки с водяным паром солянокислого
о-оксидиметиламинопропиофенона, который мы синтезирова-
ли конденсацией о-оксиацетофенона, параформа и соляно-
кислого диметиламина в среде пропанола. о-Оксиацетофеноп
был получен обработкой фенола хлористым ацетилом с пос-
ледующим нагреванием фенилацетата с хлористым алюми-
нием.
8G
СХЕМА СИНТЕЗА о-ОКСИФЕНИЛВИНИЛКЕТОНА
//\/он /w0C0CH3
| || + СН3СОС1 - || |
W
СНз сн3
„ ,ОСОСН3 С-0 /. . с=о
I II + А1С1з - I 1 , I II
X/ ;./х0Н НО/^
сн3
I II + СН5О 4-NH(CH3)3-HC1 —
CH2CH2N(CH3),-HC1
с=о
CH2CH2N(CH3)2-HC1
I II
'WXqH
сн=сн2
с—о
| II + NH(CH3)2.J IC1
'С/ХОН
Характеристика основного сырья
Фенол, ч., ГОСТ 6417—52.
Ацетил хлористый, ч., ГОСТ 5829—51.
Алюминий хлористый, ос. ч., ВТУ МХП 3500—52.
Параформ, ч., ВТУ МХП 2711—55.
Диметиламин солянокислый, ч., ВТУ МХП 2885—51.
Условия получения
Получение фенилацетата. К 114 г (1,21 А1) фенола посте-
пенно при помешивании прибавляют 180 г (2,3 М) хлорис-
того ацетила. После прибавления всего хлористого ацетила
смесь нагревают на колбонагревателе в колбе с воздушным
холодильником до полного удаления хлористого водорода.
87
Полученный фенилацетат перегоняют, собирая фракцию с
т. кип. 195—196°. Выход фенилацетата равен 148 г, что со-
ставляет 91 % от теоретического.
Получение о-оксиацетофенона. Тщательно перемешанную
смесь 75 г (0,55 М) фенилацетата и 88 г (0,66 Л4) хлористо-
го алюминия нагревают в колбе с воздушным холодильни-
ком на масляной бане. Температуру бани медленно поднима-
ют до 120—130° (см. примечание 1) и реакционную массу вы-
держивают при этой температуре 30 минут. После охлажде-
ния к смеси прибавляют лед и концентрированную соляную
кислоту. Выпавшие кристаллы n-оксиацетофенона отфильтро-
вывают, а фильтрат подвергают перегонке с водяным паром
(см. примечание 2). Получают светло-желтое (почти белое)
масло, которое очишают перегонкой в вакууме, собирая фрак-
цию с т. кип. 93 —94°/7 мм.
Выход о-оксиацетофенона 31,5 г, что составляет 42% от
теоретического; /г® = 1,5580, что совпадает с литературны-
ми данными [4].
Получение о-оксидиметиламинопропиофенона солянокис-
лого. Смесь 6,8 г (0,05 М) о-оксиацетофенона, 5,6 г (0,068 М)
солянокислого диметиламина, 2,0 г (0,067 М) параформа и не-
сколько капель концентрированной соляной кислоты (см.при-
мечание 3) нагревают с 10 мл пропанола (см. примечание 4)
в колбе с обратным холодильником на колбонагревателе. Пос-
ле одночасового кипячения смесь охлаждают, фильтруют,
промывают ацетоном. Получают 8,2 г о-оксидиметиламино-
пропиофенона солянокислого, что составляет 72% от теорети-
ческого; т пл. 162°.
По литературным данным, продукт плавится при 156° [2}.
Получение о-оксифенилвинилкетона. 11,6 г (0,051 М) со-
лянокислого о-оксидиметиламинопропиофенона перегоняют с
водяным паром (см. примечание 5). Отгон экстрагируют эфи-
ром, сушат сульфатом натрия, фильтруют, эфир отгоняют.
о-Оксифенилвинилкетон перегоняют в вакууме. После фрак-
ционной разгонки собирают фракцию с т. кип. 67—6871 — •
2 мм или 93—9473—4 мм.
Выход о-оксифенилвинилкетона равен 3,2 г, что составляет
42,7% от теоретического;
пв° = 1,5783 ; £>20° = 1,3004 г/см3-,
МПнайд = 37,79; MRem = 38,23.
Найдено, %: С-72. 43; 72. 51; Н-5,73; .5, 81.
CSH8O2. Вычислено, %; С—72, 90; Н -5,4.
Продукт присоединения НС1 имеет т. пл. 73—74°.
Найдено, %: CI—19, 12; 19, 20.
CgHgOgCl. Вычислено, %: CI—19, 30.
88
Процентное содержание двойных связей, определенное ме-
тодом гидрирования на Pd/CaCO3, составляет 99,2%.
Кетон легко полимеризуется по радикальному механизму.
Полимеры получены путем блочной и гранульной полимери-
зации. (М. в. 120 000).
Примечания: 1. Соотношение образующихся изомеров, по лите-
ратурным данным [5]. зависит от температуры реакции. Однако наши мно-
гочисленные опыты показали, что при температурах в интервале 100—
160° выход о-нзомера не превышает 42%.
2. После отгона о-изомера в перегонной колбе остается дополнитель-
ное количество н-оксиацетофенона, так что общий выход составляет 50%.
3. pH среды доводят до 2—3.
4. Проведение конденсации в других растворителях (диоксан, диме-
тилформамнд, бутанол) приводит к снижению выхода (58—65%).
5. Для предотвращения полимеризации образующегося виннлкетона в
перегонную колбу помещают небольшое количество ингибитора N-фенил-
0 -нафтнламипа.
ЛИТЕРАТУРА
1. Pfeifer. Liebigs Ann. Chem , 398, 137 (1913).
2. L. Nobles, J. Burckhalter. J. Amer. Pharm. Assoc., Sei, Ed.,
47, 77 (I158).
3. W. B. Geiger. Archive of Bfocliem., 16, 423 (1948).
4. H. Pauly, K. Lockemann. Ber., 48, 30 (1915).
5. Rosenmund. Schnur. Liebigs Ann. Chem., 460, 56 (1928).
Поступила в марте 1964 г.
Институт химии высокомолекулярных
соединений АН УССР
УДК 547.582.2.05
НОРМАЛЬНЫЕ пере. ОКТИЛОВЫЕ, НОНИЛОВЫЕ
И ДЕЦИЛОВЫЕ ЭФИРЫ ИЗОМЕРНЫХ НИТРОБЕНЗОЙ-
ных кислот
И. И. ДЫХАНОВ, В. Р. ШИЛОВ
COOR
COOR
OjN—\ / O2N-^ O2N-^ ^-COOR
Алкил-о-нитро-
бензоаты
Алкил-.м-нитро-
бензоаты
Алкил-л-нитро-
бензоаты
R = -h.C8Hi7, -н. CgHjD или -н. С1(|Н21.
В 1963 году авторами данной статьи, совместно с Авери-
ным и Дорошко [1], было установлено, что высшие эфиры изо-
мерных нитробеизойных кислот могут быть использованы в
качестве селективных «неподвижных фаз» при разделении сме-
сей близкокипящих органических вешеств методом газо-жид-
костной хроматографии [2]. Синтез высших алкилнитробензоа-
тов до сих пор описан не был.
Общеизвестный способ этерификации ароматических кар-
боновых кислот спиртами в присутствии концентрированной
серной кислоты неприемлем для получения высших алкил-
нитробензоатов по той причине, что высшие спирты под дей-
ствием серной кислоты легко окисляются и дегидратируют-
ся [3]. В связи с этим сложные эфиры нитробеизойных кислот
получают обычно взаимодействием хлорангидридов этих кис-
лот с абсолютными спиртами. Эта реакция протекает настоль-
ко легко и гладко, что рекомендована для идентификации
спиртов [4], однако применение ее для промышленного полу-
чения алкилнитробензоатов осложняется ядовитостью и срав-
нительно высокой стоимостью тионилхлорида, необходимого
для синтеза исходных нитробензоилхлоридов.
В данной работе найдена возможность непосредственной
этерификации нитробеизойных кислот высшими спиртами в
присутствии каталитических количеств легкодоступных и де-
шевых арилсульфохлоридов. Роль последних в этом процессе,
как и в случае получения простых эфиров из высших спиртов
[5], заключается в образовании алкиларилсульфонатов, кото-
рые затем переэтерифицируются нитробензойпыми кислотами
легче, чем исходными спиртами. Для достижения максималь-
ного выхода алкилнитробензоатов образующуюся в процессе
реакции воду необходимо удалять из сферы реакции с избыт-
ком исходного спирта; количество удаленной воды не должно
быть больше теоретического (по отношению к взятой кислоте),
в противном случае происходит нежелательное превращение
избыточного спирта в простой эфир [5].
СХЕМА СИНТЕЗА ВЫСШИХ АЛ КИЛ НИТРОБЕНЗОАТОВ
Ar-SO2-CI + R—ОН — Ar—SOa—OR + HC1;
O2N-C6H4-COOH + Ar—SO2—OR —
-> O2N-CeH4-COOR +Ar-SO2-OH;
Ar—SO2—OH +- R-OH Ar—SO2—OR Д- H2O,
где Ar — ароматический, a R — алифатический радикалы.
Характеристика основного сырья
о-Нитробепзойная кислота, ч., ВТУ РУ 1192—55.
.м-Нитробензойная кислота, ч., ВТУ У 82—51.
n-Нитробензойная кислота, ч., ВТУ ОРУ 8—54.
Октиловый спирт первичный, ч., ВТУ РУ 1301—57.
Нониловый спирт первичный, ч., ВТУ МХП РУ 1302—57.
Дециловый спирт первичный, ч., ВТУ РУ 1303—57.
n-Толуолсульфохлорид. ч., ВТУ ГХК 1577—61.
Условия получения
Раствор 1 г-моля о-, м- или n-нитробензойной кислоты
(148 г) в 3 гучалях октилового (390 е), нонилового (432 г)
или децилового (474 г) спирта кипятят в присутствии
0,01 г-моля п-толуолсульфохлорида (1,9 г) в колбе с автома-
тическим водоуловителем (насадка типа Дина-Старка) до от-
деления 18—18,5 мл воды (см. примечание). После этого во-
доуловитель заменяют клайзеновской насадкой с нисходящим
прямым холодильником, создают в системе остаточное дав-
ление в пределах 10 —15 мм и отгоняют избыточный спирт,
который используют в следующем опыте этерификации.
Остаток после отгонки спирта фракционируют при более
глубоком разрежении и получают высшие алкилнитробензоа-
91
1ы с выходами 90—95% от теоретических, считая на исходные
кислоты. Их физико-химические характеристики приведены в
таблице.
Примечание. Если процесс не остановить при указанном объеме
выделившейся воды, то происходит нежелательное превращение избыточ-
ного спирта в простой эфир.
Эфиры Температура кипения, °С/мм Hg d™
Октил-о-нитробензоат .... 168-169°/! мм 1,1453 1,4970
Октил-л-нитробеизоат .... 170—172°/2 мм 1,2104 1,5010
Октнл-п-нитробепзоат .... 178—179°/1 мм 1,0277 1,4980
Ноннл о-нитробензоаг .... 175 177°/4 мм 1,0787 1,4580
Ноннл-зг-нитробензоат .... 166-167°/1 мм 1,1319 1,5050
Нонил-п-нитробензоат .... 196-198°/1 мм 1,0517 1,5005
Децил-о-нитробензоат .... 186 —188°/3 мм 1,0183* 1,4983*
Децил-л/-нитробензоат .... 200—202°/3 мм 1,0435* 1,5021*
Децил-п-иитробензоат .... 203 - 205°/3 мм 0,9999* 1,4941*
’ Измерения проведены при 30°.
ЛИТЕРАТУРА
1. Н. Н. Д ы х а н о в, В. Р. Шилов, В. А. А в е р и н, Э. И. Дорош-
к о. Авт. заявка № 791379/23—4.
2. А. Кейл ем а нс. Хроматография газов, М_, ИЛ, 1959; Э. Байэр.
Хроматография газов, М., ИЛ, 1961.
3. L. Р г ц n 1 е г. J. Pharm. Chem., (6), 6, 12; Chem. Zbl., 11, 340 (1897);
A. Senderens. Compt. rend., 154,779(1912); 181,698 (1925).
4. Д. H. Прянишников. Практикум по органической химии, М„
Госхнмиздат, 1952, стр. 73; Р. Шрайнер, Р. Фьсон. Систематический
качественный анализ органических соединений, М., ПЛ, 1950, стр. 164, 168.
5. Н. Н. Дыханов, В. Р. Шилов. Авт. заявка № 785085/23—24.
Поступила в апреле 1064 г.
ВНИИ монокристаллов
УДК 547.466.22
ПОЛ ИСТИРОЛАМИНОУКСУСНАЯ КИСЛОТА — КОМП-
ЛЕКСООБРАЗУЮЩИЙ КАТИОНИТ
В. Г. СИНЯВСКИЙ, Л. И. КОШЕЧКИ НА, ЛГ Я. РО МАНКЕ ВИЧ
-СН-СН2-
-СН-СП8-
I
-СН2-СН-
NHCH2COOH
п
В последнее время комплексообразующие катиониты нахо-
дят применение при извлечении тяжелых металлов, очистке ре-
активов, разделении различных катионов и т. д. [1—3].
Одним из типов таких катионитов являются смолы, содер-
жащие в своем составе аминоацетатные хелатные группиров-
ки, аналогичные содержащимся в известных комплексообра-
зователях — глицине, фенилглицине и этилендиаминтетраук-
сусной кислоте (ЭДТА).
Известно несколько способов введения таких групп в по-
лимер. Так, например, хлорметилированный полистирол обра-
батывают этилендиамином, а затем хлоруксусной кислотой [4].
По другому методу хлоруксусную кислоту конденсируют с
.м-фенилепдиаминформальдегидной смолой [5].
Катионит «Dowex А-1» получают путем конденсации хлор-
метилированного полистирола с аминодиуксусной кислотой
[6, 7]. Известны также катиониты аналогичного строения КТ-1
и КТ-2, однако методы их синтеза не описаны [8].
Указанные методы обладают рядом недостатков: много-
стадийность. невысокая степень превращения на каждой ста-
дии и, как следствие, непостоянный состав и низкая обмен-
ная емкость.
Нами разработан довольно простой и удобный метод син-
теза комплексообразующего амфотерного катионита. По пред-
93
варительным данным (потенциометрическое титрование, СОЕ,
элементарный анализ) можно заключить, что в составе ка-
тионита наряду с аминоацетатными содержатся также амино-
диацетатные хелатные группировки.
Исходный аминополимер готовили путем:
а) гранульной сополимеризации ч-аминостирола с диви-
нилбензолом (5—15%), размер гранул 0,25—1,0 мм [9];
б) блочной сополимеризации /г-аминостирола с дивипил-
бензолом (5—20%), размер частиц после дробления 0,15—
1,0 мм [9];
в) восстановления нитрованных гранул сополимера стиро-
ла с дивинилбензолом (2—8%) [10]
Как известно, для облегчения полимераналогичных прев-
ращений на гранулах их предварительно выдерживают в
инертных растворителях для набухания. В данном случае
наиболее подходящим растворителем оказался одномолярный
раствор соляной кислоты. Солянокислый сополимер вносится
в раствор монохлоруксусной кислоты и при нагревании смесь
постепенно подщелачивается до тех пор, пока не прекратится
уменьшение pH реакционной смеси.
После окончания реакции катионит в натровой форме пе-
реносится в колонку и отмывается водой.
В зависимости от содержания аминогрупп и дивинилбен-
зола в исходном полимере колеблется как время и легкость
протекания реакции, так и количество хелатных групп в ка-
тионите, а также его способность к набуханию.
Статическая обменная емкость по Na+ и Си2+ максималь-
на у катионита, полученного на основе сополимеров а) и б),
и составляет соответственно 4,8 и 5,0 мг-экв/г. Катионит в
Na- и Cu-формах легко превращается в Н-форму при обра-
ботке его 1—3 М соляной кислотой.
СХЕМА СИНТЕЗА КОМПЛЕКСООБРАЗУЮЩЕГО КАТИОНИТА
-СН-СН2-
_-СН-СН2-"
nCICH.COOH
и NaOH
-сн2-сн-
NHgCl
-сн-сн,-
--СН--СН,--
-сн2-сн-
NHCH2COONa
n
94
Характеристика основного сырья
Сополимер аминостирола с дивипилбензолом (5%), СОЕ
по 0,1 н. НО — 6,8—6,9 мг-экв/г, размер гранул 0,25—1,0 мм.
Кислота монохлоруксусная, ч., ГОСТ 5836,—51.
Натр едкий, ч., ГОСТ 4328—48.
Условия получения
20 г гранульного сополимера аминостирола с дивинил-
бензолом в течение 2 часов (см. примечание 1) выдерживают
для набухания в 500 мл 1н. соляной кислоты, затем отфиль-
тровывают и сразу же помещают в реактор, снабженный ме-
шалкой, обратным холодильником и капельной воронкой. Ту-
да же загружают 80 г монохлоруксусной кислоты (пятикрат-
ный избыток) и 600 мл воды. Смесь нагревают на кипящей
водяной бане и постепенно прибавляют 20%-пый водный ра-
створ едкого натра по мере уменьшения pH реакционной сме-
си (см. примечание 2), поддерживая его в пределах 10—12.
После окончания процесса конденсации (см. примечание 3)
смесь при указанном pH нагревают еще 2—3 часа. Грану-
лы катионита переносят в колонку и промывают водой до ней-
тральной реакции (см. примечание 4). Катионит высушивают
на воздухе и получают 32,4 г воздушно-сухого продукта (см.
примечание 5).
Основные показатели катионита
Внешний вид — прозрачные, чуть желтоватые гранулы.
Размер частиц — 0,25—1,0 мм.
Влажность воздушно- 1 в Н-форме-- 18,8%;
сухого катионита / в Na-форме—24,5%,
„ , 1 в Н-форме — 24,6%;
Набухаемость -1 в Ма.форме _ 72,8 о/о
СОЕ по 0,1 н. NaOH 4,8 жг-экв/г;
СОЕ по Си2+ (в буфере с pH 4,2) — 5,0 мг-экв!г.
Примечания: 1. Солянокислый аминосополимер, полученный пу-
тем восстановления нитросополимера по методике [10] предварительно вы-
держивают для набухания в воде или растворе монохлоруксусной кисло-
ты, в котором затем проводят конденсацию. Для набухания сополимеров
с содержанием дивинилбензола выше 5—7% необходим больший проме-
жуток времени (до суток).
2. Контроль pH реакционной смеси осуществляли по универсальной
индикаторной бумаге; pH при этом колеблется в пределах 4—12.
3, Процесс, оканчивается обычно за 10—20 часов. Однако на сильно-
сшитых сополимерах процесс протекает намного медленнее (до 4—5 дней).
4, В результате реакции получают катионит в натровой форме, при-
годный для многих целей.
Катионит легко перевести в Н-форму, пропустив через колонку с ка-
тионитом 1н. соляную кислоту.
95
В случае работы в буферных растворах Н- или Na-форму обрабаты-
вают буфером с нужным pH.
Катионит амфотерен. Поэтому в силыюкислых средах образует соля-
ную форму по аминогруппе.
5. СОЕ, внешний вид, цвет, зависят, в основном, от качества исходно-
1 о полимера, т. е. от процентного содержания аминогрупп, дивинилбензо-
ла, цвета, формы и размера гранул.
В процессе реакции указанные качеств! почти не изменяются.
ЛИТЕРАТУРА
1. С. Е, Б р е с л е р. Успехи химии, 29, 993 (1960).
2. J. R. Millar. Chem. and. Ind., 20, 606 (1957).
3. А. А. Берлин, H. Г. Матвеева Успехи химии, 29, 277 (1969).
4. D. К. H ol e, S. L. Т h о m a s, К. W. P e p p e г. Англ. пат. 767821;
РЖхим, 17, 62874 (1959).
5. E. В 1 a s i u s, G. О 1 b r i c h. Z. Analyt. Chem., 151, 81 (1956).
6. L. R. Morris, R. A. Mock и др. J. Amer. Chem. Soc., 81,377 (1959).
7. Dow. Chemical Co. Chem. a. Eng. News, 4, 49 (1959).
8. E. Б. T p о с т я п с к а я, А. С. T e в л и п а. Заводск. лаборатория,.
23, 1042 (1957).
9. В. Г. Синявский, А. И. Турбина, М. Я. Романкевич.
Методы получения химических реактивов и препаратов, вып. 9, М..ИРЕА,
1963, стр. 17.
10. В. Г. Синявский, М. Я. Романкевич, Н. П. Цыганкова
Там же, стр. 21.
Поступила в марте 1964 г.
Институт химии высокомолекулярных
соединений АН УССЕ
УДК 547.772.2.07
ФУНКЦИОНАЛЬНЫЕ ПРОИЗВОДНЫЕ 1,3,5-ТРИФЕНИЛ-
ПИРАЗОЛИНА-Д2
В. Г. ТИЩЕНКО
Н2С- с-^
н-с I
Rs-fr ( N
W I
il I
Ri
R^/z-OCaHj; я-СеНБ; я-ОСН3; л-ОСН3;
о-ОСН3; о-СН3; О-Cl; o-NO2; .«-NO2;
R3=k-OCH3; «-С1;
R5 = «-OCH3; я-Cl.
Производные 1,3,5-трифенилпиразолина-Д2 успешно при-
меняются в качестве активаторов и сместителей спектров жид-
ких и пластмассовых сцинтилляторов [1, 2], а также в качест-
ве люминофоров.
Наиболее общим методом получения соединений этого ря-
да является циклизация соответственно замещенных гидразо-
нов а,р-ненасыщенных карбонильных соединений. Однако в
литературе нет сведений об оптимальных условиях протека-
ния этой реакции. i
В данной работе проверены и уточнены имеющиеся в ли-
тературе методы синтеза производных 1,3,5-трифенилпиразо-
лина-Д2 и разработана общая методика их синтеза, приводя-
щая к высоким выходам подобных соединений.
7 Зак. 613 97
СХЕМА СИНТЕЗА ПРОИЗВОДНЫХ 1,3,5-ТРИФЕН ИЛ ПИРАЗОЛ ИНА-Д8
R3-\ 'J-C-CHa + О=С—/-RB
= II I -
о н
NaOH, А1кОН
------------->
с-сн=сн
НС1
-Н2О
Rs-ч ^-С-СН=СН-^ Vr5
= 'I
N-NH—^-Rt
Характеристика основного сырья
4-Метоксиацетофенон, ч., ВТУ ХСНХ 26—58.
4-Хлорацетофенон, т. пл 20°.
4-Метсксибензальде1ид, ч., СТУ 79—8—х—61.
4-Хлорбензальдегид, ч., ТУ TCP 214—61.
2-Толилгидразин, т. пл. 56°.
2-Хлорфенилгидразин, т. пл. 47°.
4-Хлорфенилгидразин, т. пл. 90°.
4-Бифенилилгидразин, т. пл. 112°.
З-Нитрофенилгидразин. т. пл. 93°.
2-Нитрофенилгидразин. ч., ВТУ РУ 1374—57.
98
Флуоресценция (в. н. гептане) Голу бое ато-синяя Синяя Голубая Фиолетовая Фиолетовая Синяя Не люминесцирует Не люмнпесцирует Голубая Синяя Голубая 1 Синяя 1 Голубая Голубая
Цвет и форма кристаллов Зеленые пластинки Бесцветные иглы 1 о 1 । Зеленоватые иглы Прозрачные кубы г 1 Бесцветные иглы ‘ГУ 1 Желтоватые волокна । Желтые иглы Желтые пластинки Желтоватые волокна Бесцветные волокна 1 Желтоватые иглы I Желтоватые чешуйки Желтоватые кубы Желтоватые иглы
Тем- | пера- тура плав- ления, °C 146 140 122 £ сс 173 661 136 141,5 129 I 143 140 148 137
3 32 ход, % от теоре- тичес- кого О Ю о £ о CN О ей о £ О К ю 1С GO
Время реак- ции, часы 2,0 2,0 2,5 1 । LO Ю сч 3,0 ю CN 36 Ю ю 2,0 1 3,0 о 3,0
<1
к
s
4
о
co
ra
a.
x
t:
99
Условия получения
Получение производных бензальацетофенона. К раствору
эквимолекулярных количеств производных ацетофенона и
бензальдегида в этиловом спирте при перемешивании прили-
вают 10%-ный водный раствор едкого натра, количество ко-
торого должно составлять 1,5 г-моля на 1 г-моль карбониль-
ного соединения. Количество этилового спирта должно быть
таким, чтобы по окончании прибавления водного раствора
щелочи реакционная масса оставалась гомогенной.
Реакционную массу перемешивают в течение 2—3 часов
при комнатной температуре и затем оставляют на 10—12 ча-
сов без перемешивания. Выпавшие кристаллы замещенных
бензальацетофенонов отфильтровывают и очищают перекри-
сталлизацией из этилового спирта. Выход чистых веществ ко-
леблется в пределах 70—80% от теоретического.
Получение производных 1,3,5-трифенилпиразолина-\2. Эк-
вимолекулярные количества соответствующего производного
бензальацетофенона и солянокислого фенилгидразина (или
его производного) растворяют при кипении в этиловом спирте
и кипячение продолжают до начала помутнения реакционной
массы. Пробу реакционного раствора растирают в холодном
этиловом спирте до начала кристаллизации и ..полученную «за-
травку» вносят в реакционный раствор, который слегка ох-
лаждают. Немедленно начинается выделение кристаллов за-
мещенного 1,3,5-трифенилпиразолина, которое продолжается
в течение 10—12 часов.
Кристаллы замещенного 1,3,5-трифенилпиразолина-Д2 от-
фильтровывают и перекристаллизовывают из этилового спир-
та или смеси его с ацетоном.
Вышеописанным способом был получен ряд функциональ-
ных производных 1,3,5-трифенилпиразолина-Д2, характеристи-
ки которых представлены в таблице. Все перечисленные в
этой таблице вещества получены впервые.
ЛИТЕРАТУРА
1. Н. Willey, Н. Hayes, С. J а г Ь о е. J. Organ. Chem., 23,732 (1958).
2. Л. Л. Нагорная, А. П. Килимов, Б. Г. Ди ст а но в,
Л. М. Подгорная. Приборы и техника эксперимента, I960, стр. 48
Поступила в апреле 1964 г.
ВНИИ монокристаллов
УДК 547.673.07
1,4-ЭНДОМЕТИЛ ЕН-1,2,3,4-ТЕТРАГИДРОАНТРАХИ-
НОН-9,10
Я. Я. ДЫХАНОВ, В. И. КИХТЕВА
О
//\/\/1\
I il ||С11> I
о
C16HI2O2 М. в. 224,25
1,4-Эндометилен-1,2,3,4-тетрагидроантрахинон-9,10 предло-
жено получать (с близким к количественному выходом) изо-
меризацией гидрированного аддукта 1,4-нафтохинона с цикло-
пентадиепом-1,3 и дальнейшим окислением продукта изомери-
зации броматом калия в смеси уксусной и серной кислот [1].
На выход и качество 1,4-эндометилен-1,2,3,4-тетрагидро-
антра.хинона-9,10 по этому методу существенное влияние ока-
зывают чистота гидрированного аддукта и полнота его прев-
ращения в соответствующий антрагидрохинон. При наличии
в гидрированном аддукте негидрированного исходного веще-
ства или при неполной изомеризации гидрированного аддук-
та имеет место обильное смолообразование, в результате ко-
торого выделение 1,4-эндометилен-1,2,3,4-тетрагидроантрахи-
нона-9,10 в чистом виде чрезвычайно затрудняется, а выход
его составляет не более 15—20% от теоретического. Кроме
того, изменение цвета реакционного раствора при окислении
изомеризата броматом калия в кислой среде не может слу-
жить надежным критерием полноты протекания процесса.
Ниже приводится методика окисления гидрированного и
изомеризованного аддукта 1,4-нафтохинона с циклопентадие-
ном-1,3 посредством уксуснокислого раствора бихромата нат-
101
рия, позволяющая получать 1,4-эндометилен-1,2,3,4-тетрагид-
роантрахинон-9,10 даже из технического гидрированного ад-
дукта (т. пл. 110—118°) с выходом не менее 40% от теорети-
ческого [2].
СХЕМА СИНТЕЗА 1,4-ЭНДОМЕТИЛЕН-1,2,3,4-ТЕТРАГИДРОАНТРАХИ-
НО НА-9,10
NaaCr2O7
СН.СООН
Характеристика основного сырья
Гидрированный аддукт 1,4-нафтохинона с циклопентадие-
ном-1,3; т. пл. 110—118°.
Кислота уксусная, синтетическая, ГОСТ 7077—54, сорт 1.
Ацетон техн., ГОСТ 2768—60, марка «А».
Условия получения
Раствор 11,3 г гидрированного аддукта 1,4-нафтохинона с
циклопентадиеном-1,3 в 25 мл уксусной кислоты кипятят в
течение 1 часа с обратным холодильником, охлаждают до
35—40° и приливают к нему в один прием раствор 14,9 г дву-
водного бихромата натрия в 100 мл той же кислоты. Темпера-
тура в реакционной массе при этом сначала понижается, за-
тем вновь возрастает до 40—45° и удерживается в указанных
пределах в течение 25—30 минут.
Когда температура в массе снизится до 20—25°, ее медлен-
но, при перемешивании, выливают на 2 л холодной воды и
оставляют на 10—12 часов для формирования осадка. Техни-
ческий 1,4-эндометилен-1,2,3,4-тетрагидроантрахинон-9,10 от-
фильтровывают, тщательно промывают водой (вес сухого 8- -
9 ;г; т. пл. не ниже 85°) и, не подвергая высушиванию, пере-
кристаллизовывают из смеси 1 объема ацетона и 3 объемов
102
воды в соотношении 1:10. Перекристаллизованный продукт
сушат при 50—60° до постоянного веса.
Получают 1,4-эидометилен-1,2,3,4-тетрагидроантрахинон-
9,10 в виде зеленовато-оранжевого кристаллического порош-
ка; т. пл. 133—134°.
Выход его равен 4,5—5 г, что составляет 40—45% от тео-
ретического.
ЛИТЕРАТУРА
1. А. Н. Гринев, Н. К. В е н е в ц е в а, В. И. Фра н ч у к, А П. Т е-
р е и т ь е в. ЖОХ, 30, 1911 (1960).
2. Н. Н. Д ы х а в о в, В. И. Кихтева. Авт. свид. по заявке
№765157/23-4 (1962).
Поступила в сентябре 1963 г. ВНИИ монокристаллов
УДК 547.821,2.07
2-ЭТИНИЛПИРИДИН
(а-Пиридил ацетилен)
И. Н. ДЫХАНОВ, Т. С. РЫЖКОВА
V-c = c-H
N
CjH6N М. в. 103,16
Общепринятый способ синтеза этинилпиридинов многоста-
диен и сложен в технологическом отношении. Он заключает-
ся в обработке ацетилпиридинов пятихлористым фосфором и
дальнейшем бис-дегидрохлорировании образующихся при
этом а,|3-дихлорэтилпиридинов спиртовым раствором едкой
щелочи [1]. Исходные ацетилпиридины получают из эфироз
пиридин-карбоновых кислот и этилацетата по методу Клай-
зена [2].
В связи с разработкой в последние годы экономически вы-
годных способов получения изомерных винилпиридинов [3].
последние стали привлекать внимание исследователей как ис-
ходные вещества для синтеза соответствующих этинилпириди-
нов. В частности, исходя из 2-метил-5-винилпиридина, Кост и
сотрудники [4] получили с вполне удовлетворительным выхо-
дом 2-метил-5-этинилпиридин,
Ниже приводится простая и надежная методика получе-
ния 2-этинилпиридина из 2-винилпиридина [5].
СХЕМА СИНТЕЗА 2-ЭТИНИЛПИРИДИНА
2 Вг2
сн=сн2-------------->
Н—С—N(CHa)s
I!
О
104
Вг Br
I 1 1
|1-СН-СНа
N
KOH
CHgOH
Br2
Характеристика основного сырья
2-Винилпиридин, свежеперегнанный, = 1,545.
Диметилформамид, техн., ВТУ ЕУ—53—54.
Бром, техн., ГОСТ 54—41.
Спирт метиловый, синтетический, ГОСТ 2222—54.
Калий едкий, твердый, техн., ГОСТ 9285—59, марка «А».
Эфир петролейный, ТУ МХП 1867—48.
Магний сернокислый, прокаленный, ГОСТ 4523—48.
Условия получения
В колбу, снабженную мешалкой, термометром и капельной
воронкой, помещают 70 г свежеперегнанного 2-винилпириди-
на и 250 мл диметилформамнда. К полученному раствору при
энергичном перемешивании и температуре минус 5—0° прика-
пывают 84 мл брома.
Реакционную смесь перемешивают при указанной темпе-
ратуре еще в течение 1 часа, затем прибавляют раствор 153 г
едкого кали в 190 мл метанола с такой скоростью, чтобы тем-
пература в массе не превышала 10° (см. примечание 1). Пос-
ле этого массу кипятят в течение 2 часов с обратным холо-
дильником, охлаждают до комнатной температуры и отфиль-
тровывают осадок бромистого калия.
Из фильтрата при остаточном давлении 30—40 мм отго-
няют максимально возможное количество метанола (его ис-
пользуют в следующем опыте), а остаток выливают в 500 мл
воды. Водный раствор трижды экстрагируют петролейным
эфиром (по 150 мл каждый раз). Эфирные вытяжки объеди-
няют, сушат над 5 г безводного сульфата магния, фильтруют
и фракционируют в вакууме при температуре бани не выше
100° (см. примечание 2).
Чистый 2-этинилпиридин собирают при 75°/2 мм Hg в при-
емник, защищенный от солнечного света (см. примечание 3).
Выход его равен 17-—18 г, что составляет 25—26% от теорети-
ческого. В атмосфере азота, с добавкой 0,5% гидрохинона,
продукт сохраняется без заметных изменений в течение не-
дели.
Примечания: 1. Прибавление к пербромиду 2-(«, ₽-дибромэтил) -
пиридина метанольною раствора едкого кали при более высокой темпера-
туре приводит к сильному оСмолёиию реакционной массы.
105
2. При более высокой температуре бани 2-этипилпиридин полимери-
зуется в перегонной колбе. Продукт весьма чувствителен к кислороду воз-
духа (быстро темнеет), поэтому капилляр перегонной колбы следует сое-
динять с газометром, заполненным азотом или аргоном.
3. В качестве приемника удобно пользоваться ампулой с краном; в
этой ампуле, обернутой черной бумагой, следует затем хранит^ продукт,
ЛИТЕРАТУРА
1. U. Haug, G. Fiirst. Chem. Ber., 93, 593 (I960).
2. A. H. T e p e н т ь e в и др. Ж. всесоюзн. хим. об-ва им. Менделеева,
6, 116 (1961).
3. Г. С. К о л е с н и к о в. Синтезы винильных производных арома-
тических и гетероциклических соединений, М., АН СССР, 1960, стр. 219.
4. А. Н. Кост и др. Докл. АН СССР, 130,326 (1960).
5. Н. Н. Дыханов, Т. С. Рыжков а. Авт. заявка № 822972/23—4
(1963).
Поступила в сентябре 1963 г. ВНИИ монокристаллов
ПЕРЕЧЕНЬ СОЕДИНЕНИИ,
ОПИСАННЫХ В НАСТОЯЩЕМ СБОРНИКЕ
Алкил-о-иитробензоаты.......................................... 90
Алкил-лг-иитробензоаты......................................... 90
Алкил-п-нитробензоаты.......................................... 90
З-Амииодифеииламина хлоргидрат.................................. 5
З-Аминодифениламипа основание................................... 5
Ангидро-бис-йндандион-1,3....................................... 8
Анилнндиуксусная кислота....................................... 12
Ацетилацетон................................................... 15
Бензоилацетон.................................................. 15
п-трет. Бутилпирокатехин ..........•.......................... 18
Высшие простые эфиры (гексиловый, гептиловый, октиловый, нони-
ловый, дециловый) . . ...............................' . . . . 21
Гидразон 2-дифенилацетилиидандиона-1,3 (дифезон)............... 14
Дециловые нормальные пере, эфиры изомерных нитробеизойных
кислот......................................................... 92
Диацетамид..................................................... 24
Диацетил....................................................... 26
Дибензиламнн.............................................. . 28
Дибензилдитиокарбамат циика ................................... 30
Дибензилцианамид.............................................. 32
1,4-Ди-(1,5-дифенил-Да-пиразолинилил-3)-бензол................. 34
2,5-Диметилацетофеион.......................................... 37
и.п-Диоксибензолсульфимид...................................... 39
2-Дифеиилацетилиндандион-1,3(дифенацин)........................ 42
2,7-Дихлорхромотроповая кислота................................ 46
рф-Дициандиэтиловый эфир....................................... 49
1,4-Дициннамонлбензол.......................................... 35
Диэтилеитриаминпентаацетат медн ...........•................... 51
Изобутнлнитрит................................................ 53
Йодистый метил................................................. 55
п-Метоксифенол................................................. 57
Монобензоильное производное дифенилолпропана................... 66
Монооксим диацетила............................................ 62
Натриевая соль метилового эфира нндандиОн-1,3-карбоновой-2-кис-
логы............................................................ 9
о-Нитроанилина :хлоргидрат................................... 83
107
л-Нитроанилина хлоргидрат..........................I......... 83
я-Нитробензилхлорид..............................i............. 65
3-Нитродифениламин............................................ 67
7-Нитро-2-метоксиакридон-9.......................i............. 70
7-Нитро-4-метоксиакридон-9.................................... 70
6-Нитро-2-метокси-10-метилакридон-9..............|............. 70
6-Нитро-4-метокси-10-метилакридои-9..............i............. 70
7-Нитро-2-метокси-10-метилакридон-9 .............1............. 70
7-Нитро-4-метокси-10-метилакридон-9..............1............. 70
4-Нитро-2'-метоксидифеннламин-2-карбоновая кислота............ 76
4-Нитро-3'-метоксидифениламин-2-карбоновая кислота............ 76
4-Нитро-4'-метоксидифениламин-2-карбоновая кислота............ 75
7-Нитро-2-метокси-9-хлоракридин................................ 78
7-Нитро-4-метоксн-9-хлоракридин................................ 78
о-Нитрофенилгидразнн........................................... 81
и-Нитрофенилгидразии......................................... 81
о-Нитрофеннлгидразина хлоргидрат............................. 84
я-Нитрофенилгидразина хлоргидрат.................;............. 84
о-Нитрофенилгидразинсульфонат натрия........................... 83
л-Нитрофенилгидразинсульфоиат натрия.................. 83
о-Нитрофеннлдиазоиия хлорид......................' ........ 83
n-Ннтрофеннлдиазоння хлорид......................;............. 83
Нониловые нормальные пере, эфиры изомерных нитробеизойных
кислот ..........................................j ..... . 92
о-Оксндиметиламинопропиофенон солянокислый . . -J.......... 88
о-Оксиацетофенон ................................j............. 88
о-Оксифенилвинилкетон................... ... \................ 86
Октиловые нормальные пере, эфиры изомерных нитробеизойных
кислот . . . . •.................................• ;........... 92
Полистироламиноуксусная кислота............................... 93
Сульфитно-бнсульфитная смесь................................. 83
1,3,5-Трнфеиилпнразолина-Д2 функциональные производные .... 99
Фенилацетат..................................................... 87
1,4-Эндометилеи-1,2,3,4-тетрагидроаитрахинон-9,10.................. 101
2-Этинилпиридин.................................................... 104
Редактор Б. Г. Козлов
Техн, редактор As И; Пирожкова
Корректоры: Е. Л. Каждая, Э. С. Сысоева
Сдано в нроизв. 26. V. 1964 г. Подписано к печ. 8. X. 1964 г.
Формат бум. 6Ох9О'/и Печ. л. 6.75 Уч.-над. л. 5
Л-126023 Зак. 613 Тираж 2000 экз. Цена 40 коп.
Типография ВАХЗ
Замеченные опечатки
Страница Строка Напечатано Следует читать
50 20 сверху по Конго ПО КОНГО
57 12 сверху деметилированием диметилироваиием
80 6 снизу W. Lesnianskii W. Lesnianski
92 1 снизу 1064 г. 1964 г.
1964 Зак. 61;