/




























































































































































































































































































































































































































Текст
В. Н. АНТОНОВ, А. С. ЛАПИДУС
ПРОИЗВОДСТВО
АЦЕТИЛЕНА
ИЗДАТЕЛЬСТВО «ХИМИЯ»
Москва 1970
УДК 661.715.342
А 72
3-14-7
79-69
Антонов В. Н., Лап иду с А. С,
Производство ацетилена.
Книга является первой отечественной монографией
по технологии ацетилена, включающей сведения по всем
методам производства этого важнейшего сырья для
промышленности органического синтеза. Наибольшее
внимание уделено методам производства ацетилена из
нефтяного сырья: высокотемпературному пиролизу,
электрокрекингу и окислительному пиролизу. Детально разобраны
технологические схемы процессов, даны
технико-экономические сопоставления, подробно описана аппаратура;
имеется специальная глава по технике безопасности.
Монография предназначена для
инженерно-технических работников нефтехимической промышленности и
промышленности органического синтеза, а также может
быть использована в качестве пособия учащимися
химических вузов и техникумов.
В книге содержится 104 табл., 211 рис., 447 библ.
ссылок.
СОДЕРЖАНИЕ
Предисловие 7
Введение 9
Литература 12
Глава I. Свойства ацетилена и ацетиленовых углеводородов 13
Физические свойства ацетилена 13
Горючие и взрывные свойства ацетилена 17
Химические свойства ацетилена 26
Высшие ацетиленовые углеводороды 34
Литература 41
Глава II. Производство ацетилена из карбида кальция 43
Производство карбида кальция 43
Технологическая схема производства 46
Усовершенствования в производстве карбида кальция 49
Требования к сырью и качество получаемого карбида кальция ... 52
Получение ацетилена из карбида кальция 55
Ацетиленовые генераторы 56
Очистка ацетилена 59
Литература 63
Глава III. Получение ацетилена пиролизом углеводородов 64
Основные сведения о процессе 64
Термодинамика процесса 64
Механизм и кинетика процесса 71
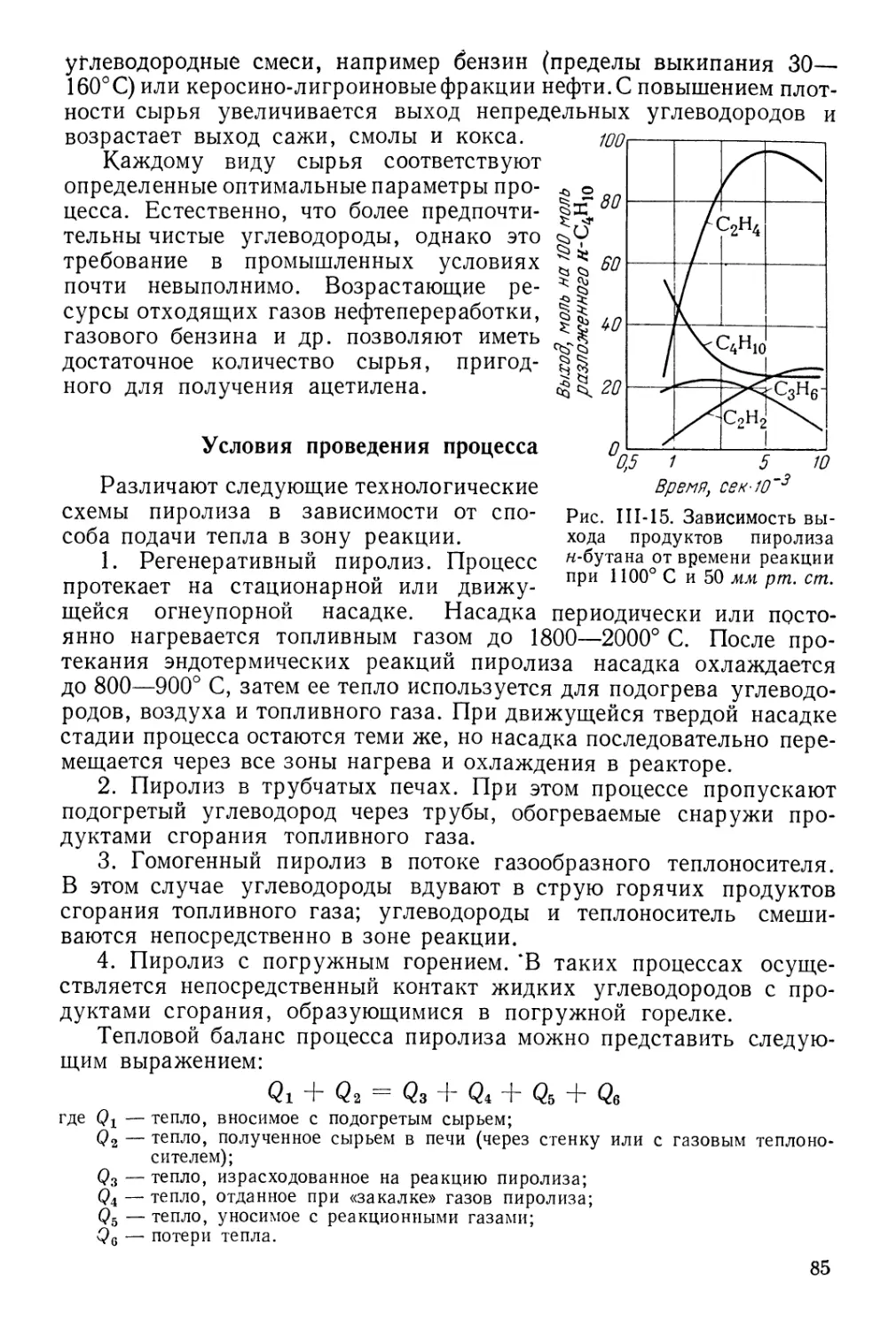
Выбор сырья для пиролиза 81
Условия проведения процесса 85
Добавки водяного пара 89
Регенеративные процессы 90
Пиролиз в регенеративных печах 90
Пиролиз в движущемся слое твердого теплоносителя 97
Пиролиз в трубчатых печах 98
Пиролиз с электронагревом 101
»' з
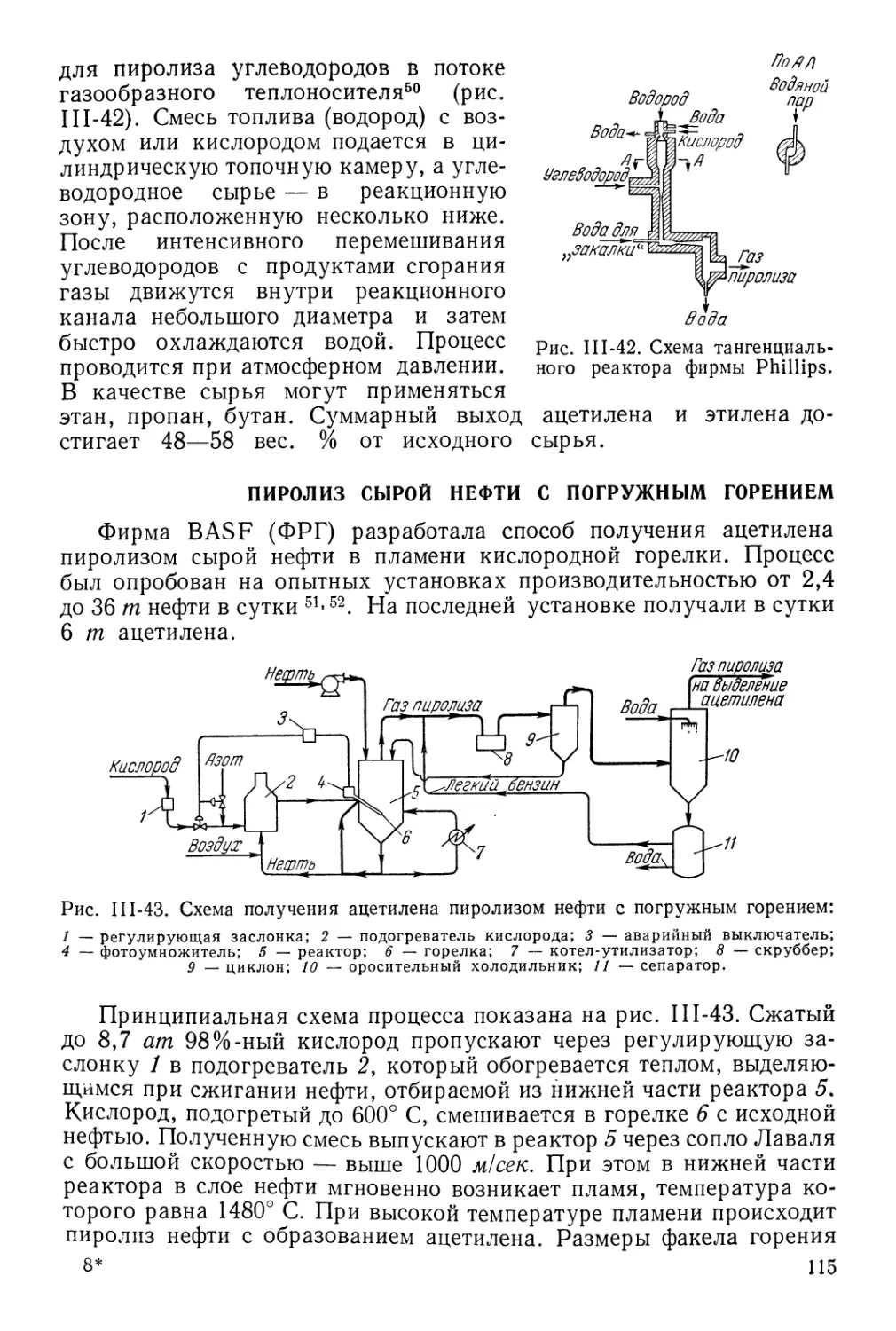
Гомогенный пиролиз в потоке газа-теплоносителя 102
Пиролиз сырой нефти с погружным горением 115
Сравнение различных способов пиролиза углеводородов 117
Литература 119
Глава IV. Получение ацетилена электрокрекингом углеводородов ... 121
Электрические разряды в газах 122
Реакции углеводородов в электрических разрядах 126
Методы получения ацетилена в электрических разрядах 131
Электрокрекинг газообразных углеводородов 134
Электрокрекинг жидких углеводородов 145
Крекинг углеводородов в плазменной струе 147
Литература 152
Глава V. Получение ацетилена окислительным пиролизом углеводородов 154
Основные сведения о процессе 154
Термодинамика и кинетика процессов неполного горения 155
Механизм образования и разложения ацетилена 157
Условия горения метано-кислородных смесей 160
Основные параметры процесса окислительного пиролиза 168
Материальный и тепловой балансы процесса 185
Реакторы окислительного пиролиза метана 187
Технологические схемы окислительного пиролиза ч. 195
Получение ацетилена при атмосферном давлении 195
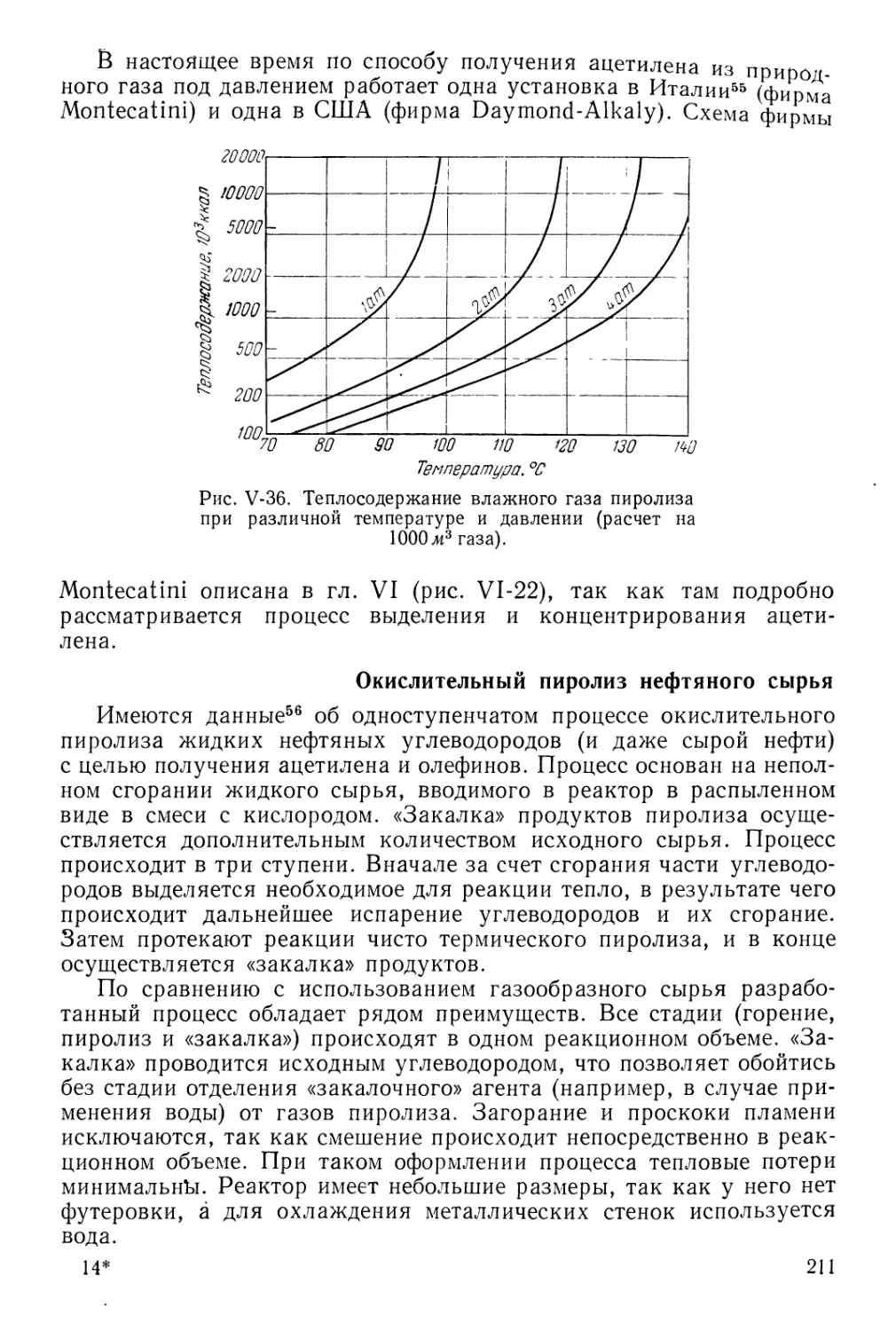
Получение ацетилена при повышенном давлении 209
Окислительный пиролиз нефтяного сырья 211
Литература 213
Глава VI. Методы разделения ацетилен содержащих газов и выделения
ацетилена 215
Общие сведения об абсорбционных процессах 217
Абсорбция ацетилена водой 227
Растворимость ацетилена в различных органических растворителях . . . 228
Абсорбция ацетилена селективными растворителями 231
Выделение ацетилена диметилформамидом 231
Выделение ацетилена N-метилпирролидоном 238
Выделение ацетилена у-бутиролактоном 246
Схемы установок концентрирования с селективными растворителями 246
Абсорбция ацетилена низкотемпературными растворителями 253
Выделение ацетилена жидким аммиаком 254
Выделение ацетилена метанолом 263
Выделение ацетилену ацетоном 268
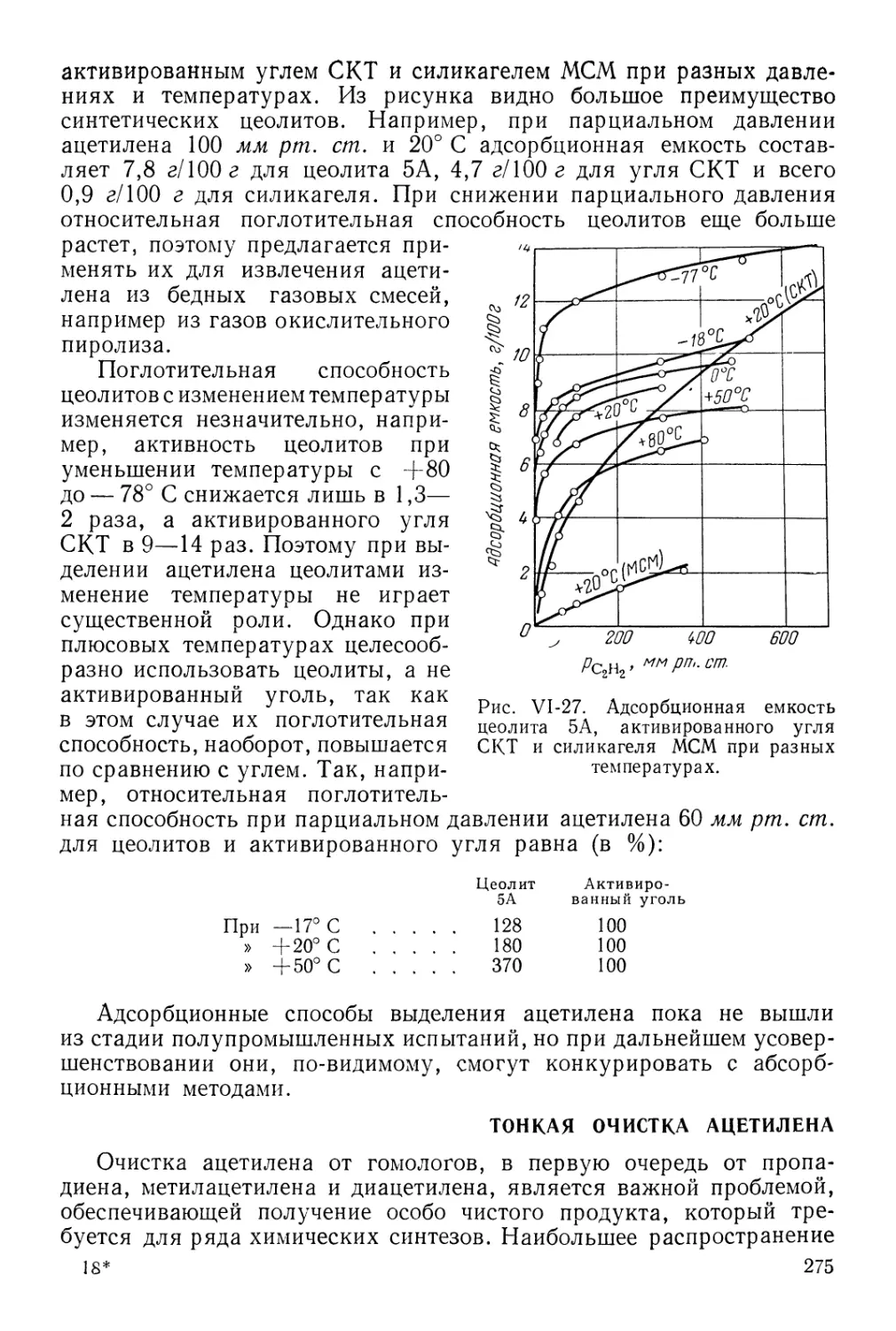
Адсорбция ацетилена 271
Тонкая очистка ацетилена 275
Литература 277
4
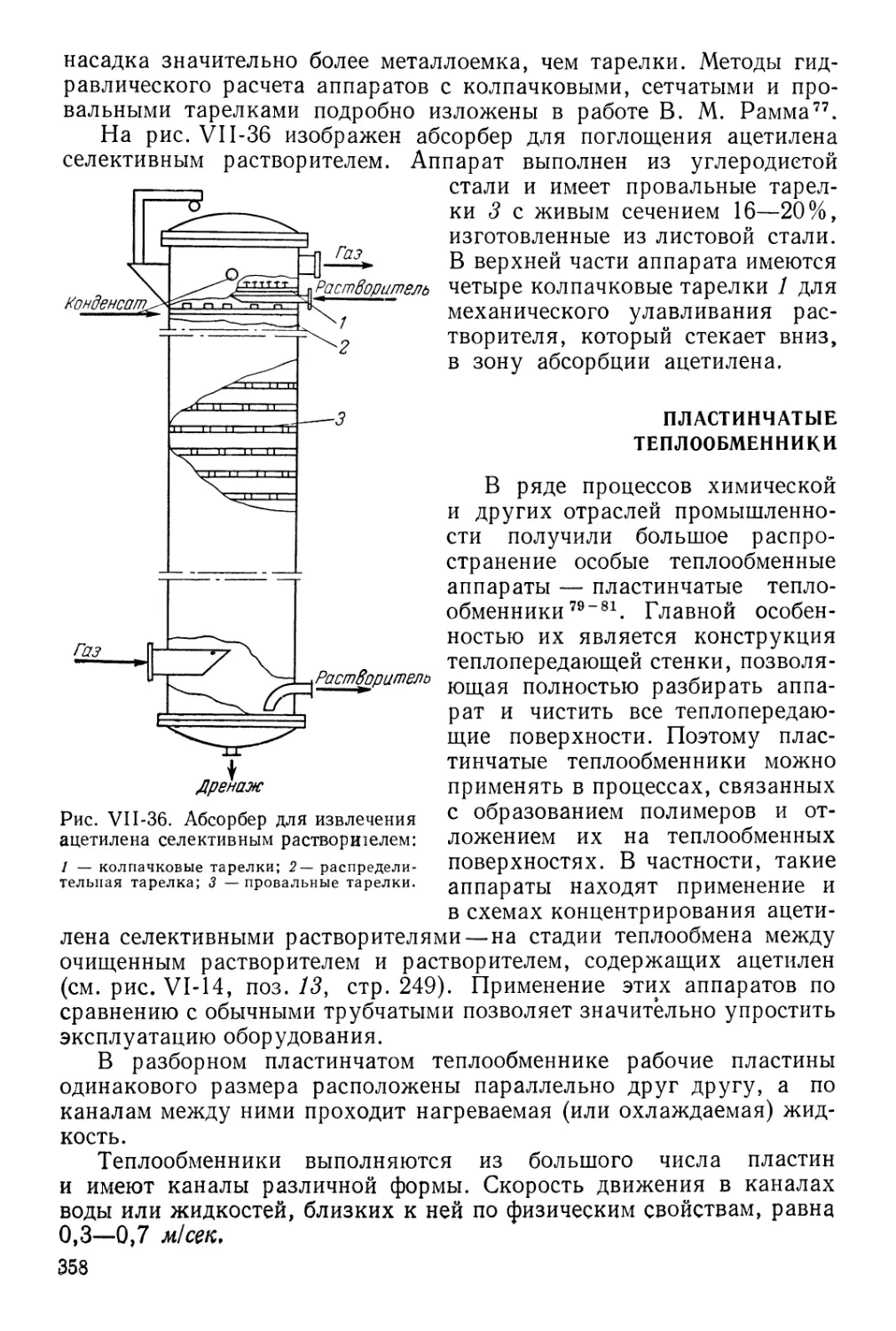
Глава VII Процессы и основные аппараты производства ацетилена
из углеводородов 279
Радиационно-конвективные подогреватели змеевикового типа 279
Горелки для сжигания топливного газа 283
Расчет подогревателей 285
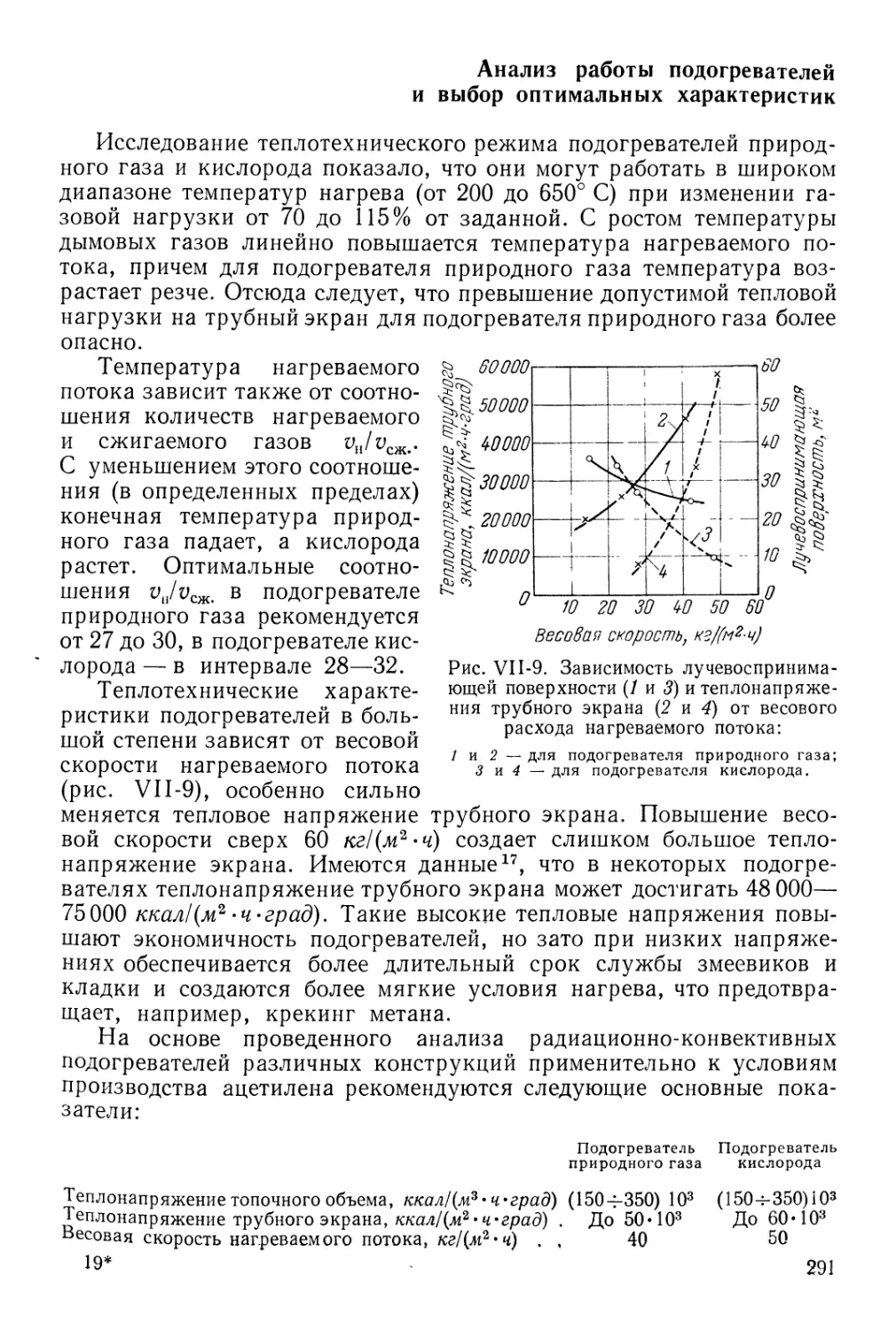
Анализ работы подогревателей и выбор оптимальных характеристик 291
Ацетиленовые реакторы 292
Смесительные устройства 297
Конструкции смесителей 297
Процесс смешения на основе теории турбулентных струй 298
Расчет смесителей для процесса окислительного пиролиза 302
Анализ смесителей различных конструкций и рекомендации по их
разработке 307
Расчет процесса «закалки» реакционных газов 310
Факторы, влияющие на скорость «закалки» 310
Выбор параметров процесса «закалки» 316
Выбор режима охлаждения газов после «закалки» 322
Аппараты сажеочистки и охлаждения газов 324
Полые скрубберы 325
Турбулентные газопромыватели 325
Электрофильтры 328
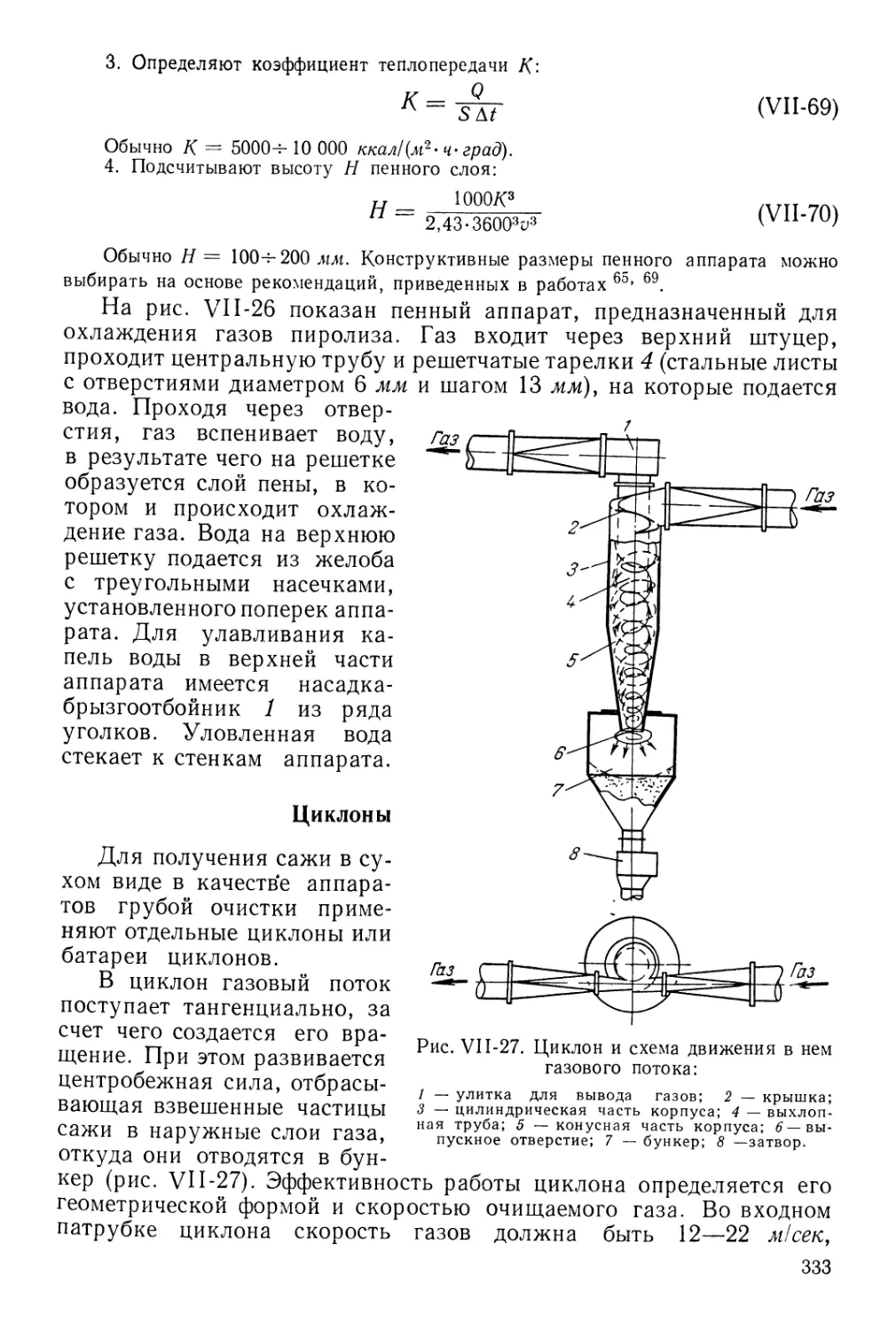
Пенные аппараты 332
Циклоны 333
Рукавные фильтры 335
Очистка воды от сажи 336
Компримирование ацетилена и ацетиленсодержащих газов 339
Условия сжатия 339
Типы компрессоров 343
Разделение многокомпонентных систем 349
Оборудование для абсорбционно-десорбционных процессов
разделения ацетиленсодержащих газов 357
Пластинчатые теплообменники 358
Литература 361
Глава VIII. Меры безопасности в производстве ацетилена 364
Факторы, влияющие на взрывоопасность ацетилена 364
Выбор габаритов ацетиленопроводов 367
Статическое электричество и скорость движения ацетилена по трубам 372
Защита оборудования 372
Системы транспортирования ацетилена 374
Защитные устройства 376
Ацетиленопроводы и арматура 384
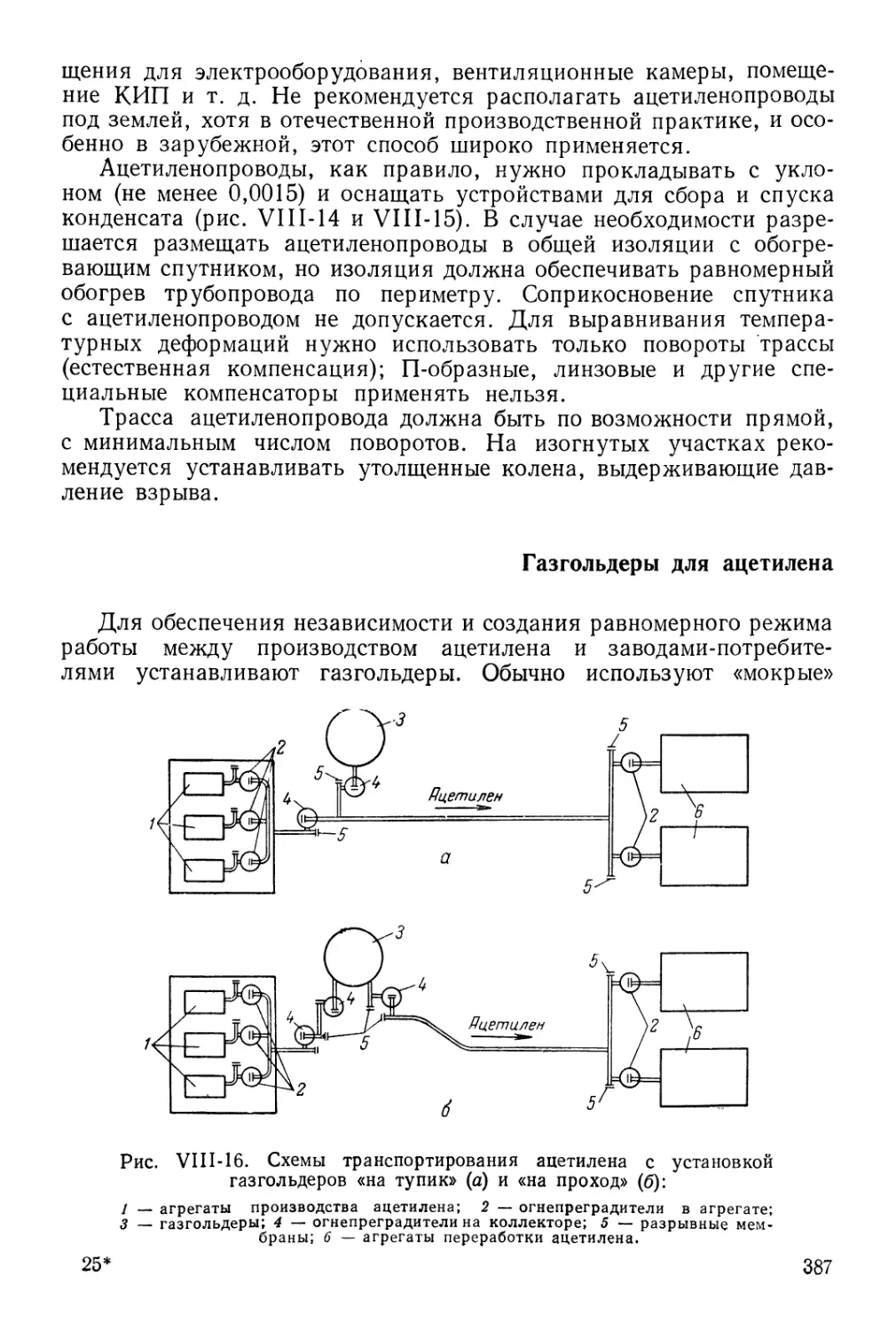
Газгольдеры для ацетилена 387
Подготовка аппаратов и коммуникаций к ремонту . 388
Конструкционные материалы 389
Наполнение баллонов «углеводородным» ацетиленом 390
Литература 391
5
Глава IX. Технико-экономическое сравнение методов производства
ацетилена 393
Получение ацетилена из карбида кальция 393
Получение ацетилена из углеводородного сырья 394
Сравнение методов производства ацетилена 401
Литература 407
Предметный указатель - 408
ПРЕДИСЛОВИЕ
Ацетилен является исходным сырьем для синтеза мономерных
веществ, из которых получают химические волокна, пластические
массы, каучук и другие важные продукты и материалы. К таким
мономерам относятся винилхлорид, винилацетат, акрилонитрил,
хлоропрен и т. д. В связи с большой потребностью в продуктах,
получаемых на основе ацетилена, планами развития народного хозяйства
предусмотрено увеличение производства ацетилена как из
углеводородного сырья, так и классическим способом — через карбид кальция.
В последнее время особенно широкбе распространение получили
методы производства ацетилена из углеводородного сырья, главным
образом из природного газа. В настоящей монографии рассмотрены
все способы производства ацетилена; при этом использованы
экспериментальные и расчетные данные, литературные материалы и опыт
эксплуатации отечественной ацетиленовой промышленности.
При описании методов получения ацетилена принят следующий
порядок: вначале карбидный метод, а затем — пиролиз
углеводородов, электрокрекинг и окислительный пиролиз. Такое изложение
совпадает с историей возникновения и развития этих методов в
промышленности, а также подчиняется определенной системе: сначала
приводятся теоретические и физико-химические основы чисто
термического расщепления углеводородов, а затем пиролиза под
действием окислителей. В книге изложено большинство используемых
в промышленной практике процессов получения ацетилена из
углеводородов, однако многие разновидности методов остались
неосвещенными, поскольку некоторые из них еще недостаточно
разработаны, а другие не нашли применения и потеряли свое значение.
В последней главе сосредоточены материалы по
технико-экономическим оценкам эффективности различных методов получения
ацетилена в Советском Союзе. В связи с различием экономических условий
районов страны выбор метода производства и переработки ацетилена
требует предварительного серьезного анализа и расчетов.
Авторы считают приятным долгом отметить большой вклад в
развитие отечественной ацетиленовой промышленности (особенно в части
7
переработки углеводородного сырья) работников Северодонецкого
химического комбината, Государственного института азотной
промышленности и продуктов органического синтеза (ГИАП),
Саратовского химического комбината, Гипрохлора, Ереванского
химического комбината им. С. М. Кирова, Гипрокаучука и других
организаций и предприятий.
Главы I—IV монографии написаны В. Н. Антоновым, главы V—
VIII — А. С. Лапидусом, введение и глава IX написаны совместно.
Монография по производству ацетилена различными методами
выпускается в нашей стране впервые, поэтому авторы, возможно,
неполностью осветили все разнообразные аспекты производства
ацетилена и с признательностью примут замечания и предложения
читателей.
В. Н. АНТОНОВ
Л. С. ЛАП ИДУ С
ВВЕДЕНИЕ
В послевоенные годы произошло существенное изменение
сырьевой базы химической промышленности. Если ранее основным
поставщиком сырья для органических синтезов являлись коксохимическая
промышленность и сельское хозяйство, то в настоящее время
ведущую роль играют неисчерпаемые и более дешевые ресурсы нефти
и природного газа. На основе переработки простейших
углеводородов нефти и природного газа современная промышленность
органического синтеза способна производить полимерные материалы,
азотные удобрения, химические средства защиты растений, моющие
средства и другие химикаты, количественным выпуском которых
определяется уровень индустриализации любого промышленно
развитого государства.
Одним из видов сравнительно дешевого сырья в промышленности
органического синтеза является ацетилен. Высокая реакционная
способность позволяет использовать его для синтеза различных
веществ, при переработке которых можно получать, например, поли-
винилхлорид, перхлорвиниловую смолу, синтетический хлоропре-
новый каучук, химические волокна и пленки типа «саран» и «винол»,
различные хлорорганические растворители, три- и перхлорэтилен
и другие продукты.
Ацетилен стал доступен в конце XIX в., после того как был
получен в промышленных условиях карбид кальция, явившийся
сырьем для производства ацетилена. Использование дешевого
природного газа и продуктов переработки нефти стало новым мощным
стимулом для получения ацетилена и последующего развития на его
основе крупной промышленности органического синтеза.
Предпочтительное использование методов получения ацетилена из
углеводородов или карбидного метода зависит главным образом от наличия
в данном районе страны нефтяного сырья, природного газа или кокса
и энергетических ресурсов. Из новых способов получения ацетилена
чаще применяются окислительный пиролиз природного газа,
электрокрекинг углеводородов и пиролиз нефтяных фракций в потоке
высокотемпературных газов, образующихся в кислородной горелке.
Новые методы позволяют организовать производство ацетилена
по более простой технологической схеме, с меньшими капитальными
9
вложениями и снизить его себестоимость. На основе использования
отходящих газов, богатых водородом, создаются кооперированные
производства синтетического аммиака или метанола, что в свою
очередь снижает себестоимость продуктов, вырабатываемых на основе
ацетилена.
Несмотря на высокий уровень технологического оформления
используемых промышленных процессов производства ацетилена,
во многих странах появляются все новые предложения по
совершенствованию оборудования и технологии, что свидетельствует о
большом интересе к этой проблеме. Освоение новых способов позволило
накопить огромный опыт безопасной работы со взрывоопасными
газами, что само по себе явилось большим стимулом для развития
химической промышленности. Однако производство карбида кальция
и в настоящее время не утрачивает своего значения. Большие
количества этого химиката необходимы для получения ацетилена,
используемого при резке и сварке металлов. Иногда необходимость
строительства новых производств карбида кальция обусловливается
огромной территорией нашей страны и отсутствием в ряде
экономических районов нефтяных и газовых месторождений, а также
трубопроводов для транспортирования нефти и газа. Часто такое решение
экономически оправдано.
С развитием современной химической науки стало возможным
во многих случаях использовать в органическом синтезе вместо аце-,
тилена этилен и даже пропилен, что позволяет значительно снизить
потребность в ацетилене и ограничить ее теми производствами, где
подобная замена невыгодна, технически невозможна или равноценна.
Тем не менее, до последнего времени во всех странах производство
ацетилена продолжает расти (табл. 1).
За последние годы в Советском Союзе введены в действие
предприятия по получению ацетилена из природного газа и низкоокта-
ТАБЛИЦА 1
Рост производства ацетилена в некоторых странах1"20 (в тыс. т)
Страна
США . . .
ФРГ* . .
ГДР* . .
Япония .
Англия .
Франция
Италия .
Польша .
Румыния
1940 г.
всего
57
70
100
120
23**
55
20
9
9
из
углеводородов
3
—
—
—
—
—
—
1950 г.
всего
175
210
174
52
30
48
48
45
3
из
углеводородов
30
70
—
—
—
—
—
—
I960 г.
всего
420
373
277
285
96
146
186
95
40
из
углеводородов
136
79
—
—
>у ■ '
f "
1967 г.
всего
544
550
340
440
160
190
410
120
80
из
углеводородов
300
230
80
60
40
230
20
60
* Данные по 1940 г. для ФРГ и ГДР приведены
** На импортном карбиде кальция.
их современных границах.
10
нового бензина. В СССР освоены наиболее совершенные методы
производства ацетилена, поэтому в настоящее время нет технических
препятствий для выпуска любых видов химической продукции на
основе ацетилена.
Данные табл. 1 свидетельствуют о том, что в настоящее время все
промышленно развитые страны получают ацетилен и из карбида
кальция, и из нефтяного сырья и природного газа, причем доля
углеводородного ацетилена заметно растет. Производство различных
продуктов, для синтеза которых используется ацетилен, также из
года в год возрастает. В 1967 г. по основным видам промышленной
продукции, получаемой из ацетилена (винилхлорид, винилацетат,
хлоропрен, трихлорэтилен и акрилонитрил), объем производства
значительно превысил прежний уровень. Вероятно, общая
потребность в этих продуктах столь велика, что в переработку оказываются
вовлеченными известные и новые виды сырья (табл. 2).
ТАБЛИЦА 2
Производство некоторых химических продуктов в ряде стран1"20 в 1967 г.
(в тыс. га)
Продукт
Винилхлорид
общее производство
из ацетилена
Винилацетат
общее производство
из ацетилена
Хлоропрен
общее производство
из ацетилена
Трихлорэтилен
общее производство
из ацетилена
Акрилонитрил
общее производство
из ацетилена
США
1361
116
295
37
208
104
213
47
499
55
Япония
485*
172
190
54
29*
15
64
14
190
28
ФРГ
275*
102
70
26
50*
25
106*
23,5
120*
7
Италия
240*
78
50
18
74*
16,3
35*
14
* Данные на 1966 г.
Табл. 2 не может претендовать на полную точность в связи с
трудностью получения сведений, однако из этих данных видны основные
тенденции потребления ацетилена в некоторых странах.
Несмотря на разработку методов получения винилхлорида деги-
Дрохлорировани^м. и пиролизом дихлорэтана, т. е. через этилен,
расход ацетиле.. 'яя этой цели остается высоким и продолжает
увеличиваться, чтз связано главным образом с необходимостью
использования больших количеств хлористого водорода,
образующегося при производстве различных хлорорганических продуктов.
В ряде стран для очистки хлористого водорода от органических
примесей методом абсорбции и последующей десорбции из водных
растворов разработано специальное графитовое оборудование.
11
Увеличивается потребление ацетилена для производства хлоро-
прена (мономера для синтетического каучука) и винилацетата, а
также на синтез тетрахлорэтана, дегидрохлорированием которого
получают трихлорэтилен. Акрилонитрил в настоящее время производят
главным образом гидроцианированием ацетилена, однако в
последнее время внедряется метод совместного каталитического окисления
пропилена и аммиака, поэтому потребность в ацетилене для синтеза
акрилонитрила в ближайшем будущем может измениться. Большое
количество ацетилена подвергается гидратации в ацетальдегид, но за
последние годы разработаны методы производства ацетальдегида
окислением этилена, что позволяет уменьшить потребление ацетилена
для этой цели в странах, обеспеченных нефтяным сырьем.
В настоящее время в крупных промышленных странах
организовано производство N-метил- и N-винилпирролидона на основе
ацетилена, компримированного до 15—50 am. Созданы также
установки диенового синтеза для получения новых гербицидов,
используемых для уничтожения сорняков на полях бобовых растений и
зерновых культур.
Широкое применение найдет ацетилен в производстве различных
галоидпроизводных, в первую очередь винилфторида и винилбромида,
которые затем полимеризуют для получения ценных полимеров.
В частности, одним из перспективных полимеров ближайшего
будущего является поливинилфторид. Имеются и многие другие областд
использования ацетилена, о которых невозможно сказать в кратком
введении.
ЛИТЕРАТУРА
1. М о р о з Л. Н., Хим. пром., № 3, 66 (1961).
2. Chem. Techn., 12, № 7, 438 (1960).
3. Chem. Techn., 12, № 8, 501 (1960).
4. Chem. Techn., 13, № 6, 311 (1961).
5. К 1 oc her A., Chem. Ind., 11, № 12, 762 (1959).
6. Erdol u. Kohle, 12, № 9, 55 (1959).
7. Chem. Techn., 16, № 6, 7 (1964).
8. Chem. Ind., 16, № 12, 22 (1964).
9. Europ. Chem. News, № 3 (1965).
10. Europ. Chem. News, № 1 (1965).
П. Туров Ю. Я., Журн. ВХО им. Д. И. Менделеева, 9, № 1 (1964).
12. Chem. Eng. News, 42, № 1, 82 (1964).
13. Chem. Eng. News, 43, № 1, 43 (1965).
14. T у р о в Ю. Я., Хим. пром., № 3 (1962).
15. Т у р о в Ю. Я., Ю д о в и ч Е. А., Журн. ВХО им. Д. И. Менделеева, 7,
№ 1 (1962).
16. К у л и ч к и н Н. П., Яковлева Д. А., Обмен опытом в азотной
промышленности, Госхимиздат, № 3 (1962).
17. К о р с у н с к и й О. В., Хим. пром., № 3 (1962).
18. Т у р о в Ю. Я., Паршина Г. А., Фокина М. А., в сб.
«Сопоставительные обзоры по отдельным производствам химической промышленности
в СССР и за рубежом», вып. 1, изд. НИИТЭхим, 1967, стр. 34.
19. Т у р о в Ю. Я., Паршина Г. А., Химия и технология топлив и масел,
№ 12, 57 (1967).
20. Б а р и н о в а Р., Производство карбида кальция в капиталистических
странах, в сб. «Химическая промышленность за рубежом», вып. 10, изд.
НИИТЭхим, 1967, стр. 56
12
ГЛАВА
I
СВОЙСТВА АЦЕТИЛЕНА
И АЦЕТИЛЕНОВЫХ УГЛЕВОДОРОДОВ
Физические свойства ацетилена
Ацетилен СН^СН был открыт в 1836 г. английским химиком
Дэви при исследовании состава светильного газа. В ряду
непредельных алифатических углеводородов общей формулы CrtH2rt_2
ацетилен является первым представителем и имеет молекулярный вес
26,04.
Ацетилен при обычных условиях представляет собой бесцветный
газ со следующими физическими характеристиками1"3:
Температура, °С
кипения —83,8
плавления —81
возгонки —84,1
критическая 35,6
Критическое давление, am ... 61,6
Тройная точка ацетилена, соответствующая устойчивому
равновесию трех фаз, характеризуется следующими величинами3:
температура —80,6° С, давление 962 мм рт. ст.
Плотность жидкого ацетилена при критическом давлении и 0° С
равна 0,451 кг/л. Об изменении плотности, а также вязкости и
коэффициента сжимаемости ацетилена с температурой и давлением можно
судить по данным табл. 1-1, 1-2, 1-3, 1-4 и 1-5.
ТАБЛИЦА 1-1
Плотность ацетилена3"5
Температура
°С
0
10
20
30
40
Плотность, г/л
при 0,5 am
0,5833
0,5624
0,5430
0,5248
0,5079
при 1,0 am
1,1716
1,1290
1,0895
1,0528
1,0185
при 1,5 am
1,7650
1,7000
1,6398
1,5838
1,5318
при 2,0 am
2,3636
2,2753
2,1936
2,1180
2,0478
13
ТАБЛИЦА 1-2
Динамическая вязкость ацетилена3"0 (при 1 am)
Температура
°К
200
220
240
260
273,15
280
300
320
Динамическая
вязкость
мкпз
70,8
77,7
84,6
91,2
95,5
97,8
104,7
110,4
Температура
°К
340
360
380
400
450
500
550
600
Динамическая
вязкость
мкпз
115,9
122,4
128,1
133,8
147,5
160,4
172,7
184,5
ТАБЛИЦА 1-3
Динамическая вязкость ацетилена7
Температура
°с
20
60
100
150
200
250
при 1 am
103
115
128
142
155
172
Динамическая вязкость, мкпз
при 20 am
ИЗ
125
135
148
161
177
при 40 am
128
138
144
156
170
—
при 60 am
156
157
165
180
—
при 80 am
_
196
176
182
193
—
ТАБЛИЦА 1-4
Коэффициент сжимаемости ацетилена '8
Давление
am
0,5
1,0
2,0
4,0
Коэффициент
сжимаемости
при 0° С
1,0057
1,0000
0,9891
0,9708
при 25° С
1,0989
1,0937
1,0841
1,0684
Давление
am
6,0
8,0
10,0
12,0
Коэффициент
сжимаемости
при 0° С
0,9530
0,9360
0,9194
0,9026
при 25° С
1,0531
1,0385
1,0255
1,0139
В
0е
табл. 1-6.
G равен
Данные о теплоемкости ацетилена представлены
Коэффициент теплопроводности12 ацетилена при
15,8-10~3 ккал/(м-ч-град).
Давление паров ацетилена (табл. 1-7) изменяется с температурой
в соответствии с формулой13»14:
lgpc2H2 = 12,7718 -
14
ТАБЛИЦА 1-5
Температура
°С
0
20
40
60
80
100
150
200
250
Коэффициент
при
20 а т
0,779
0,846
0,891
0,925
0,949
0,961
0,975
0,983
0,988
сжимаемости ацетилена"
Коэффициент
при
40 am
_
0,620-
0,734
0,800
0,844
0,872
0,916
0,940
0,955
при
60 am
—
0,463
0,652
0,730
0,780
0,860
0,898
0,921
сжимаемости
при
80 am
—
0,146
0,443
0,618
0,700
0,822
0,873
0,902
при
100 am
—
—
0,323
0,517
0,629
0,787
0,852
0,891
при
120 am
—
—
0,328
0,439
0,562
0,752
0,828
0,876
Теплоемкость ацетилена6'10'11 (при 1 am)
ТАБЛИЦА 1-6
Температура
°С
0
100
200
300
400
500
600
Теплоемкость
ккал/(м3-град)
Ср
0,4466
0,5189
0,5670
0,6032
0,6331
0,6593
0,6828
cv
0,3578
0,4301
0,4783
0,5144
0,5443
0,5707
0,5942
Температура
°С
700
800
900
1000
1100
1200
Теплоемкость
ккал/(м3-град)
Ср
0,7044
0,7241
0,7420
0,7583
0,7729
0,7861
Су
0,6158
0,6355
0,6534
0,6697
0,6843
0,6975
ТАБЛИЦА 1-7
Давление паров ацетилена14' 15
Температура
°С
—90
—85
—81
—70
—60
—50
—40
—30
Давление
паров Рс2Н2
am
0,7
1,0
1,3
2,2
3,6
5,3
7,7
10,9
Температура
—20
—10
0
+ 10
+ 15
+ 20
+ 30
+35,6
(критическая)
Давление
паров Рс2Н2
am
14,9
20,0
26,3
33,9
37,9
43,1
54,1
61,6
(критическое)
Ацетилен хорошо растворяется во многих органических и
неорганических жидкостях, что позволяет выделять концентрированный
ацетилен из реакционных газов при производстве его из природного
15
газа или нефтяных углеводородов. Растворимость ацетилена в воде,
измеряемая числом его объемов, растворимых в 1 объеме воды при 0° С
и парциальном давлении ацетилена 760 мм рт. ст., характеризуется
следующими данными15'1б:
Температура
°С
0
5
10
15
20
Растворимость
М3/М3
1,73—1,77
1,49—1,52
1,31—1,33
1,16—1,17
1,03
Температура
°С
25
30
35
40
45
Растворимость
М3/М*
0,93
0,85
0,77
0,71
0,65
Температура Р
°С
50
60
70
80
90
'астворил
М3/М3
0,50
0,37
0,25
0,15
0,05
В табл. 1-8 приведены данные о растворимости ацетилена в
некоторых растворителях, а в табл. 1-9 перечислено несколько
растворителей, в которых ацетилен растворяется наиболее полно. При
повышении давления растворимость ацетилена возрастает в соответствии
с общими физическими законами.
ТАБЛИЦА 1-8
Растворитель
Аммиак (в м3/т)
Ацетон . .
Y-Бутиролактон
Диметилформамид
Метанол ....
N-Метилпирроли-
дон
Растворимость
ацетилена17"19 (при
1 am)
Растворимость ацетилена, м3/м3
при
—80° С
2000
462
при
-60° С
600
161
при
-40° С
300
141
74
при
—20° С
76
40
при
0° С
42
19,5
67,0
20
65
при
4-20° С
24
11,5
37,4
11,2
38,7
при
+40° С
6,5
6,6
21,4
6,0
23
при
+60° С
—
4,4
13,0
10
при
+80° С
—
8,0
7
Примечание. Все объемы приведены к нормальным условиям.
ТАБЛИЦА 1-9
Физические свойства некоторых растворителей ацетилена '
Растворитель
Молекулярный вес
Плотность
при 20° С
г/ смя
Температура, °С
кипения затвердевания
Гексаметилфосфамид
[(CH3)2N]3PO
Диметилформамид
HCON (СН3)2
N-Метилпирролидон
СН3—N—СН2СН2 СН2СО
у-Бутиролактон
СН2—СН2—СН2—GO
I о 1
179,2
73,1
99,1
86,1
1,024
0,950
1,030
1,130
153,0
202
204
4
—61
—24
—42
Горючие и взрывные свойства ацетилена
Ацетилен может гореть с выделением большого количества тепла.
Теплотворная способность его равна22 13 387 ккал/мг (высшая)
и 12 710 ккал/м3 (низшая). Благодаря такой высокой теплотворной
способности ацетилен находит применение для газопламенной резки
и сварки металлов. Ацетилен является эндотермическим
соединением, в определенных условиях способным к взрывному разложению
на простые вещества:
С2Н2
2С + Н2
АЯ = —54,2 ккал/моль
Температура при этом достигает 2800° С. При наличии источника
воспламенения ацетилен также способен взрываться. Взрывчатые
свойства его выше, чем у многих сильно взрывчатых веществ (табл.
1-10).
ТАБЛИЦА 1-10
Свойства некоторых взрывчатых веществ
Взрывчатые вещества
Ацетилен
Нитроглицерин ....
Нитрат целлюлозы . .
Нитротолуол
Нитрат аммония . . .
Тепло,
выделяемое
при взрыве
ккал/кг
2070
1455
1025
950
347
Объем газов,
образующихся
при взрыве 1 кг
вещества
л
860
715
765
690
980
Как показали исследования, в составе продуктов разложения
ацетилена основными компонентами являются углерод и водород,
кроме того, обычно присутствует и метан. Механизм разложения
заключается в первоначальной полимеризации ацетилена и
последующей полной дегидрогенизации. Сажа, образующаяся при
взрывном разложении ацетилена, является высококачественной — имеет
хорошую дисперсность и высокую активность. В технике для
получения сажи обычно проводят взрывное разложение электрической
искрой при 10 am.
Ацетилен способен к самопроизвольному разложению нескольких
видов36"42 : при горении, взрыве, детонации и каскадном разложении
(нестационарное горение). В зависимости от характера разложения
может создаваться небольшое или очень высокое конечное давление
газов.
При горении скорость распространения пламени составляет лишь
несколько сантиметров в секунду, а конечное давление увеличивается
незначительно. Обычно о таком разложении свидетельствуют следы
сажи на стенках сосуда. Взрыв идет со значительно большей
скоростью распространения пламени, достигающей нескольких десятков
метров в секунду* Конечное давление взрыва в 8—12 раз превышает
начальное и является функцией температуры.
2 В. Н. Антонов 17
Расширение газов при горении приводит к образованию ударной
волны, распространяющейся перед фронтом пламени. Сжатие газа
и его нагревание в ударной волне протекает тем сильнее, чем больше
скорость движения расширяющихся газов, которая в свою очередь
определяется скоростью горения. При быстром горении нагревание
смеси в ударной волне может быть настолько значительным, что
смесь воспламенится перед фронтом пламени. Возникает режим
горения, называемый детонацией. При детонационном горении образуется
комплекс из ударной волны и следующей за ним зоны сжатой и
нагретой реагирующей смеси.
Детонация возможна при поджигании горючей смеси взрывчатым
веществом или при достаточной скорости пламени, например в
длинных трубах, где происходит турбулизация горящей смеси. Давление
в детонационной волне до ее отражения от стенки (а также от торца,
изгиба и т. д.) может превышать начальное в 30 раз, а в отраженной
волне — в 50—100 раз. Явление детонации в основном изучено для
трубопроводов, хотя известны случаи сходных по характеру взрывов
в помещениях. Например, в помещении объемом 100 м3 при
воспламенении ацетилено-воздушной смеси (10 объемн. % С2Н2),
находящейся под давлением 1 am, конечное давление достигало
80 am.
Для ацетилена известно так называемое нестационарное горение
(каскадное разложение), при котором часть газа сгорает, а остальное
количество сжимается перед фронтом пламени и детонирует уже в
сжатом состоянии. Давление, развивающееся при таком разложении,
может превысить давление в отраженной детонационной волне и в 500—
600 раз превышать начальное.
Все перечисленные виды самопроизвольного разложения
ацетилена отмечались на практике. Например, в длинных трубопроводах
диаметром до 200 мм (толщина стенки 12 мм) при введении
раскаленной проволоки или в струе горячих газов происходила детонация
ацетилена42. При начальном давлении 1,7—5 am давление взрыва
возрастало в 100—200 раз. При увеличении диаметра труб до 380 мм
и начальном давлении 1,05 am самопроизвольный распад ацетилена
не наблюдался; при инициировании искрой происходил взрыв,
но давление не превышало 25 am. В трубопроводах диаметром более
200 мм при давлении выше 1,4 am в случае самопроизвольного
разложения ацетилена у торца трубы возникали чрезвычайно высокие
давления (в 340—660 раз больше начального). В этом случае
происходило каскадное разложение ацетилена.
Скорость распространения пламени. При горении чистого
ацетилена скорость видимого распространения пламени сильно меняется в
зависимости от размеров и формы труб (или сосудов) и примерно
составляет15'23~25 0,03—0,9 м/сек при 1— 2 am и 0,033—0,015 м/сек при
2—7 am. Скорость распространения пламени при детонационном
разложении ацетилена в длинных трубопроводах неодинакова на
разных участках и возрастает с перемещением ударной волны по
трубопроводу. Например, в трубопроводе длиной 20 м скорость распро-
18
странения пламени увеличивается следующим образом42:
Расстояние от конца трубопровода, м . . . 2,5—2,0 2,0—1,5 1,5—1,0 1,0—0,5
Скорость, м/сек 732 1280 1700 1980
Для обеспечения полного сгорания ацетилено-воздушной смеси
в ней должно быть не более 7,75 объемн. % С2Н2. При горении
ацетилено-воздушной смеси, содержащей более 17,37 объемн. % С2Н2,
выделяется свободный углерод (сажа). Максимальная температура
горения ацетилено-воздушных смесей15 2350° С; пределы детонации
для них составляют от 4,2 до 50 объемн. % С2Н2. На рис. 1-1
показано изменение скорости видимого распространения пламени
ацетилено-воздушных смесей в зависимости от их состава и диаметра
трубок (при малых диаметрах)26. Значения нормальной скорости *
распространения пламени при горении ацетилена в смеси с воздухом
приведены ниже27:
Содержание С2Н2,
объемн. % 7,12 7,54 7,84 8,41 8,83 9,35 9,7 10,2 11,43
Нормальная скорость
горения, м1сек .... 1,46 1,53 1,61 1,64 1,67 1,69 1,64 1,60 1,50
При горении ацетилено-кислородных смесей развивается более
высокая температура, например максимальная температура
горения27 смеси с 45 объемн. % С2Н2 равна около 3150° С. Пределы
детонации для смесей ацетилена с кислородом меняются от 3,5 до
93 объемн. % С2Н2, а скорость распространения детонационной
волны достигает 1800—3300 м/сек. Нормальная скорость горения
в зависимости от содержания ацетилена составляет28:
Содержание С2Н2, объемн. % 15 20 25 30 35 40 45 50
Нормальная скорость горения,
м/сек 10,0 11,8 13,3 13,1 11,3 9,3 7,8 6,7
Температура самовоспламенения ацетилена и его смесей.
Температура самовоспламенения чистого ацетилена меняется с
давлением следующим образом (см. также рис. I-2)29:
Давление, am 1 2 3—11 21
Температура
самовоспламенения, °С 635 540—570 475—530 350
В присутствии газов-разбавителей, например СО, ацетилен может
воспламениться и при 250—300° С. Некоторые твердые вещества
также понижают температуру самовоспламенения ацетилена в 1,5—
2 раза30, т. е. действуют как катализаторы (табл. 1-11).
* Скорость перемещения пламени в неподвижной смеси по нормали к его
поверхности называют нормальной скоростью горения. Эта скорость минимальна для
данной горючей смеси и является ее физико-химической характеристикой.
Практически наблюдаемые скорости горения зависят от степени турбулентности горючей
смеси и значительно отличаются от нормальной.
2* 19
ТАБЛИЦА 1-И
Температура самовоспламенения ацетилена при 5 am
Катализаторы
— 2 ^
V о СТО
Дч Н со о
&5S со
й а я я
с о а) о
Ьи2н
Катализаторы
Без катализатора .
Железные опилки
Медные опилки
Латунные опилки
Ржавчина . . . .
Окись меди . . .
Перекись марганца
Окись алюминия .
5—6
12
15
17
12
17
17
20
505—530
520
460
500—520
280—300
240
310
490
Пемза
Силикагель . ,
Активный уголь
Карбид кальция ,
Окись железа
То же
12
10
20
15
5
— 1
300
500
470
400
500
280
325
340
(при 4 am)
Температура самовоспламенения ацетилена в смеси с воздухом
или кислородом зависит от состава смеси. Для ацетилено-воздушных
смесей температура самовоспламенения приведена ниже31'32:
Содержание С
си, объемн.
Температура
пламенения,
9Н2 в сме-
% ....
самовос-
°С . . .500*
10
и 387 **
20
400 *
30
374 **
45—55
335 *
50
353 **
* Смесь подавали через трубку при определенной температуре.
** Смесь сгорала в закрытом сосуде при определенной температуре.
Температура самовоспламенения ацетилено-кислородных смесей
(70—90 объемн. % О2) составляет примерно 300° С при 1 am. С
повышением давления эта величина снижается33, что видно на примере
ацетилено-кислородной (1:1) смеси: 245° С при 5 am и 230° С
при 10 am.
700
$ 650
й
к1^ 550
500
О
г
1
1
1
\ф90мм
W
\
Ч
\
\
Облает
\
V^
ь Взрыва
1,0 2,0
Давление, am
3ft
10 20
Содержание С2Н2, %
Рис. 1-2. Зависимость температуры
самовоспламенения ацетилена от
давления.
Рис. 1-1. Зависимость скорости видимого распространения пламени при
горении ацетилено-воздушных смесей от их состава и диаметра трубок.
20
Пределы взрываемости ацетилена. Концентрационные пределы
взрываемости смесей ацетилена с воздухом зависят от температуры
и давления смеси 34, например при 0° С меняются от 2,53—78 объемн.
% С2Н2 (при 1 am) до 2,53—100 объемн. % С2Н2 (при 5 am). О
расширении пределов взрываемости с ростом температуры можно судить
по следующим данным (при 1 am):
Температура, °С 17 50 100 150 200 250 300
Ппелелы взрываемости *,
объемн. % С2Н2 2,9-55 2,83-59 2,68—65 2,52-73 2,39—81 2,3-100 2,19-100
* В опытах использовали закрытую трубу диаметром 25 мм и длиной 2500 мм.
Изучение взрывных свойств ацетилена в смеси с воздухом при
разрежении и 20—25° С показало, что область взрывоопасных
концентраций сужается с увеличением глубины вакуума. При этом
верхний предел взрываемости снижается непропорционально уменьшению
давления35 (78 и 39 объемн. % С2Н2 при остаточных давлениях
соответственно 740 и 150 мм рт. ст.).
В процессе исследования смеси ацетилена, ацетальдегида и
воздуха (остаточное давление 740 мм рт. ст., температура 20—25° С)
установлено, что такая смесь может взрываться даже тогда, когда
содержание любого горючего компонента в ней меньше нижнего
предела взрываемости этого компонента в смеси с воздухом.
Следовательно, взрываемость смеси нельзя характеризовать
концентрацией только одного компонента. Нужно учитывать также, что
верхний предел взрываемости ацетилена в отличие от нижнего не зависит
от давления и температуры (не соблюдается принцип Ле-Шателье).
Условия взрываемости различных бинарных смесей ацетилена
видны из следующих данных35:
С формальдегидом при 150° С
С ацетальдегидом при 100° С
С метанолом при 150° С . .
объемн. %
С2Н2
75
57
55
85
72
57
80
52
30
Давление
am
2,5
4,5
6,0
2,5
3,5
6,0
3,0
6,0
10,0
Начальное давление распада ацетилена. Минимальное давление,
при котором возможен взрывной распад ацетилена, зависит от его
начальной температуры36 (рис. 1-3), а также от температуры
поджигающей проволоки, если она используется в качестве инициатора
(рис. 1-4). При давлении ниже 1,4 am даже при 2500° С (температура
плавления молибденовой проволоки) не происходит взрывное
разложение ацетилена, т. е. чистый ацетилен при 20° С и 1,4 am
практически не взрывается (при условии, что энергия источника зажигания
не превышает 5 дж). Однако Когарко с сотр. показали37"39, что при
очень больших импульсах взрывное разложение ацетилена может
21
Наступить и при давлении 0,65 am, но для инициирований
разложения необходима энергия, равная 1200 дж. С возрастанием
начального давления ацетилена требуемая энергия инициирования
уменьшается (рис. 1-5).
На рис. 1-6 показано
изменение энергии, необходимой для
инициирования разложения аце-
«
ьо,
1
(
к
I
г
г
^ о
о
200
\
\
РЬ
Al Cu I
! 1:
<ePt
4°
SO WO 150
Температура, °С
Рис. 1-3. Зависимость
минимального давления взрывного распада
ацетилена от его начальной
температуры.
2500
500 1000 1500 2000
Температура, °С
Рис. 1-4. Зависимость минимального
давления взрывного распада
ацетилена от температуры поджигающей
проволоки (на оси абсцисс указана
температура плавления некоторых
металлов).
тилена, в присутствии различных добавок при начальном давлении
1 am. Как следует из приведенных данных, при энергии
инициирования выше 5 дж верхних концентрационных пределов взрываемое™
2000
I"
1000
|
1
I
I
500
Искра о
т
катушии
Румкорсра
V
и 0,50,65 1,0 1,5
Давление, am
Рис. 1-5. Зависимость энергии
инициирования взрывного
распада ацетилена от начального
давления.
400
1 300
I
$. 200
1
1 100
I
0 10 20 30 40
Содержание добавки, с/о
Рис. 1-6. Изменение энергии
инициирования взрывного
распада ацетилена в зависимости от
добавок азота (1), воздуха (2)
и кислорода (3).
А
'/
lf\
/
V
\
Искра от \
\ катушки \
\ Румкорсра \
смесей ацетилена с кислородом и воздухом не существует*'.
Наиболее взрывоопасны смеси, содержащие до 8 и выше 33 объемн. %
* Известные значения верхних пределов взрываемости смесей ацетилена с
воздухом и кислородом установлены при небольших значениях энергии инициирования,
обычно достигаемых при зажигании ацетилена и его смесей (плавление и
пережигание проволочек).
22
воздуха (кривая 2). Перегиб кривой 2 объясняется тем, что
на начальном участке разбавляющее действие имеющегося в
воздухе азота превалирует над действием кислорода, облегчающего
воспламенение. Для смеси ацетилена с кислородом (кривая 3)
энергия поджигающей искры меньше, чем для чистого ацетилена,
и при увеличении содержания кислорода эта энергия
уменьшается. Из данных рис. 1-6 можно сделать важное заключение
1100
1000
, 300 >
[ 800
X 700
% 300
^200]
wo
-0,65am-
м
А
/i
08-
Л
f 0,9
/1
flam-
]
\0j8~
Л
\\
\\
1\\
\\\
\4\
чШ
о том, что небольшие добавки
кислорода к ацетилену делают смесь более
опасной по сравнению с чистым
ацетиленом, а небольшие добавки
воздуха понижают взрывоопасность
ацетилена.
10 20 30 ЬС
Содержание воздуха,°/о
20 W 60 80 37,5'100
Содержание Воздуха, %
Рис. 1-7. Зависимость энергии Рис. 1-8. Зависимость минимального
инициирования взрывного
распада ацетилена в смеси с
воздухом от состава смеси при
различном начальном давлении.
начального давления распространения
пламени в смеси ацетилена и воздуха
от состава смеси при 18° С.
Взрывоопасность системы в большой степени характеризуется
зависимостью энергии зажигания (инициирования) смеси ацетилена
с воздухом от начального давления смеси: при уменьшении
начального давления энергия инициирования взрывного распада ацетилена
значительно возрастает (рис. 1-7).
При горении ацетилена пламя начинает распространяться лишь
при давлении выше определенной величины.
На рис. 1-8 показана зависимость минимального давления, при
котором начинается распространение пламени, от состава смеси.
Заштрихованная область называется областью негорючих составов
смеси. Из графика видно, что при содержании воздуха около
17 объемн. % смесь наименее взрывоопасна, а в пределах от^67 до
93 объемн. % — наиболее взрывоопасна.
Флегматизация ацетилена. Взрывоопасность газообразного
ацетилена можно понизить, если добавить к нему газы-разбавители,
например предельные углеводороды, инертные газы, водород, аммиак,
азот, водяной пар и др. 40~46. Этим часто пользуются для безопасного
транспортирования ацетилена, Механизм действия таких флегматизи-
рующих добавок еще не выяснен окончательно. Предполагается,
что в их присутствии понижается температура горения, что
обусловлено в свою очередь величиной теплоемкости разбавителя и его
способностью к эндотермическому разложению. В свете этого азот менее
эффективный флегматизатор, чем, например, насыщенные
углеводороды, так как у последних теплоемкость выше.
Изучение влияния добавок различных предельных углеводородов
на стабильность газообразного ацетилена показало, что взрыв
предотвращается при следующем содержании разбавителя (в зависимости
от давления)43:
Давление
смеси
am
6,8
9,2
33,8
% сн4
15
21
36
С3Н8
10
13
24
% с4н10
11
19
20
X
I
|
60
Т
w
\ 4V
\
\
\
со
/
Область Взрыва
'"о
/Н2
100
В опытах с добавками азота или гелия было найдено, что для
предотвращения взрыва их количество должно быть в 2 раза больше,
например, количества
пропана или бутана.
Начальные давления,
при которых может
начаться разложение
смесей ацетилена с
различными газами, выше, чем
при разложении чистого
ацетилена. На рис. 1-9
показано влияние
различных разбавителей на
начальное давление, при
котором может произойти
взрывной распад
ацетилена36. Видно, что добавки
разбавителей до 20 объемн.
% не оказывают
практически никакого влияния
на начальное давление
распада. Взрывчатые свойства
I
^
4
я w зо ■
начальное давление распада, am
Рис. 1-9. Зависимость начального давления
взрывного распада различных ацетиленовых
смесей от содержания разбавителей при 15° С.
ацетилена начинают
заметно ослабевать лишь
при содержании
разбавителей более 20 объемн. %.
Наиболее эффективным разбавителем является метан.
Различное влияние разбавителей видно также из данных49
рис. 1-10. Если примем, что давление смеси, при котором начнется
взрывной распад при 15° С, составляет 10 am, то для смеси ацетилен—
водород этим условиям соответствует содержание 32% ацетилена,
а для смеси ацетилен —метан — 58%, т. е. при равном давлении
24
взрыв смеси ацетилена с метаном произойдет при большем
содержании ацетилена, чем, например, смеси с водородом. С повышением
температуры указанная закономерность сохраняется, хотя величина
минимального давления смеси, при котором начнется взрывной
распад, понижается.
Эффективным флегматизатором ацетилена является аммиак44.
Газообразные смеси ацетилена с аммиаком и растворы ацетилена
ДаВление смеси, am
Рис. 1-10. Зависимость предельжГдопустимых парциального давления и
концентрации ацетилена в смесях от'температуры и давления (пунктир —
изменение концентрации С2Н2).
в жидком аммиаке находят применение в промышленности, например
для выделения ацетилена из газов пиролиза. Пределы концентраций,
при которых происходит взрыв таких смесей, в жидкой фазе
значительно шире, чем в газообразной.
В табл. 1-12 приведены данные опытов по исследованию горения
газообразных смесей ацетилена с аммиаком в воздухе при действии
ТАБЛИЦА 1-1
Содержание
ацетилена
в смеси
с аммиаком
объемн. %
1,2
1,2
3,1
- 3,8
Горение смесей ацетилена с аммиаком
Избыток
воздуха
по отношению
к
теоретическому
%
1,2
10
1J
1,5
Количество
сгоревших
газов
% от общего
объема
28
24
54
55
Содержание
ацетилена
в смеси
с аммиаком
объемн. %
7,6
7,7
16,7
в воздухе47
Избыток
воздуха
по отношению
к
теоретическому
%
3
10
6,3
Количество
сгоревших
газов
% от общего
объема
80
65
100
25
3,0
w
о
0 ■
—J
«=^
r^
1
y\
3 -
_tl/
2,5
2,0- -
1,5
80 |
1
'SO it
X
4/7 V
0
-20
0
WO
1
25 50 75
Температура, °C
Рис. I-11. Зависимость начального
давления взрывного распада смеси
ацетилена с водяным паром от
температуры:
/ — предельное содержание ацетилена в
смеси; 2 — начальное давление распада;
3 — парциальное критическое давление
ацетилена в смеси.
Электрических искр. Из этих данных видно, что уже й% ацетилена
способны создать в системе взрывной распад, но при этом разложение
не идет до конца. В отходящих газах в основном содержится аммиак,
количество сгоревших газов
увеличивается пропорционально
содержанию ацетилена. Избыток
воздуха при одном и том же
количестве ацетилена оказывает флегма- ,
тизирующее действие.
Исследовалась также взрывае-
мость растворов ацетилена в
жидком аммиаке при расплавлении
проволоки под действием тока
50 а. При температуре ниже 50° С
смеси невзрывоопасны до
содержания ацетилена в жидкой фазе
околр 30 вес. %, а выше 30%
взрываются с определенным
индукционным периодом. Конечное
давление приблизительно в 5—6 раз
выше начального. При добавлении
воздуха смесь может взорваться
(нижний предел взрываемое™
около 23 вес. % ацетилена). Смесь горит характерным желтым
пламенем, которое быстро гаснет.
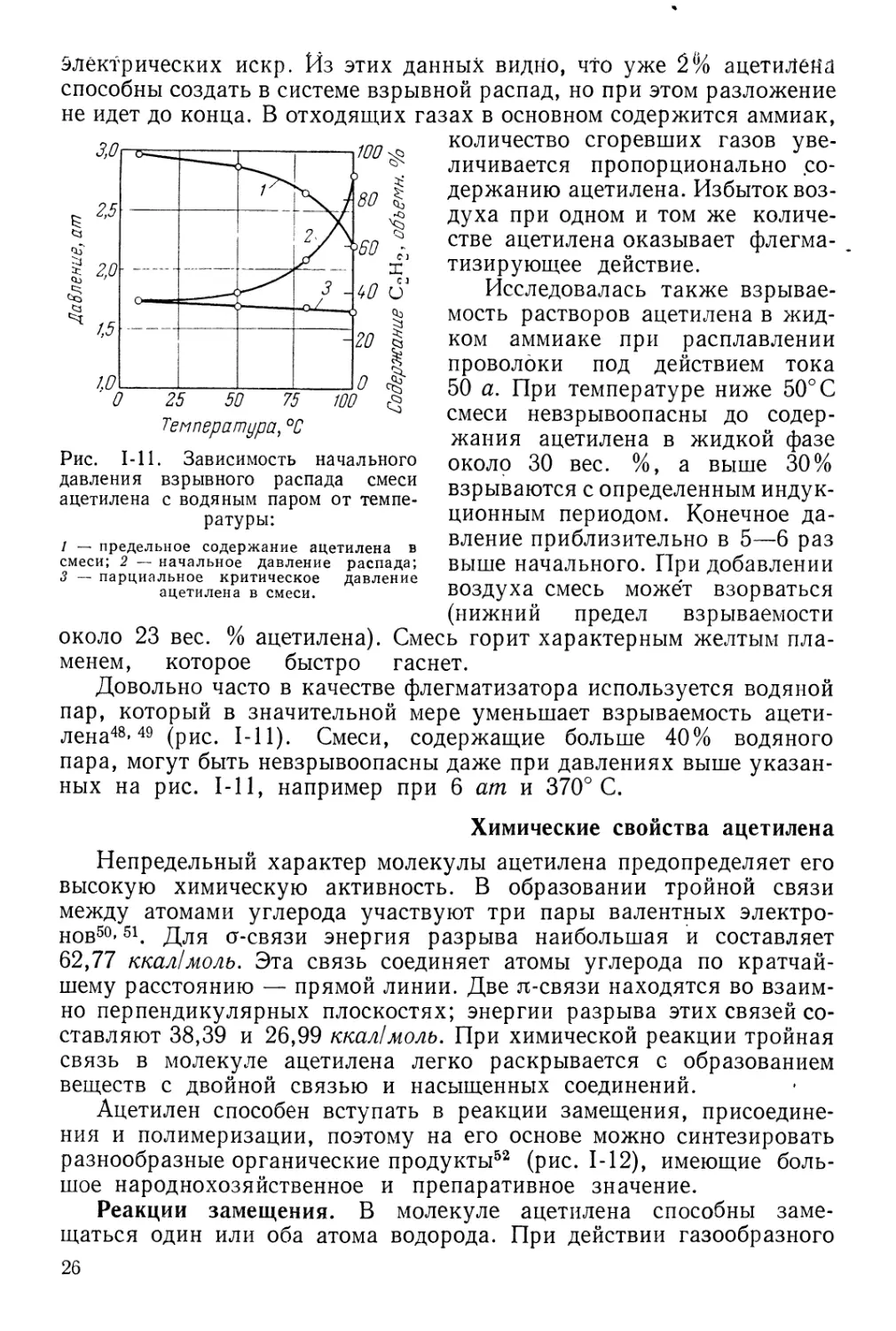
Довольно часто в качестве флегматизатора используется водяной
пар, который в значительной мере уменьшает взрываемость
ацетилена48» 49 (рис. 1-11). Смеси, содержащие больше 40% водяного
пара, могут быть невзрывоопасны даже при давлениях выше
указанных на рис. 1-11, например при 6 am и 370° С.
Химические свойства ацетилена
Непредельный характер молекулы ацетилена предопределяет его
высокую химическую активность. В образовании тройной связи
между атомами углерода участвуют три пары валентных
электронов50» 51. Для а-связи энергия разрыва наибольшая и составляет
62,77 ккал/моль. Эта связь соединяет атомы углерода по
кратчайшему расстоянию — прямой линии. Две я-связи находятся во
взаимно перпендикулярных плоскостях; энергии разрыва этих связей
составляют 38,39 и 26,99 ккал/моль. При химической реакции тройная
связь в молекуле ацетилена легко раскрывается с образованием
веществ с двойной связью и насыщенных соединений.
Ацетилен способен вступать в реакции замещения,
присоединения и полимеризации, поэтому на его основе можно синтезировать
разнообразные органические продукты52 (рис. 1-12), имеющие
большое народнохозяйственное и препаративное значение.
Реакции замещения. В молекуле ацетилена способны
замещаться один или оба атома водорода. При действии газообразного
26
Ацетилен —
Винилхлорид
Дихлорзтилен
ПолиВинил-
хлорид
Сополимеры
сакрилатами,
Винилиденщ-
ридом ит.о.
Трихлорэтан
>
| Г]
Пластические]
массы
ЛерхлорВини-
лобая смола
Винилиден-
хлорид
^Гетрахлорэтан
Трихлорэтилен
Монохлоруксус-
ная кислота
Дцетальдегид
Уксусная
кислота
Уксусный
ангидрид
Лактонитрил
—
Винилаиешат
Лцетат
целлюлозы
Дирилонитрил
г*
1
г*
1
Сополимеры
L диниЛХЛО-
ридом, ахрило-
нитриломит.д
Фреоны
Карбоксиметил-
целлюлоза
Гербициды
ПолиВинил-
ацетали
спирт
Сополимеры с
Винилхлоридом
Полиакрило-
нитрил
Сополимеры
сакрилатами
Бутадиен-нит-
рильный каучук
Гексаметилен-
диамин
Соль ДГ
Пдликапрозмид
ВинилоцетилЕн
Хлоропрен
ДиВинил-
ацетилен
Полихлоропре-
ноВыи каучук
Полимеры
[лаки)
Лцетон
Синтезы на
основе ацетона
Нетвн
Рис. 1-12. Синтезы на основе ацетилена,
ацетилена на щелочные металлы, медь, серебро, никель, ртуть,
кобальт, цинк и др. образуются ацетилениды *, часто очень
взрывоопасные, особенно в сухом состоянии:
СН^СН + 2 Си -> Си—С^С—Си + Н2
При пропускании ацетилена через аммиачный раствор
гидроокиси меди также выпадает осадок ацетиленида меди. Эта реакция
используется51 для качественного определения ацетиленовых
соединений с атомом водорода у углерода с тройной связью.
С металлическим натрием реакция замещения протекает53 в среде
жидкого аммиака с образованием моно- и дизамещенных продуктов
NaHC2 и Na2C2.
Ацетилен и его производные с атомом водорода у углерода с
тройной связью способны реагировать с метилмагнийиодидом с
образованием магнийорганических соединений:
RC^CH + CH3MgI -> RC^CMgl + СН4
Реакции присоединения. Гидрирование ацетилена с целью
получения этилена протекает над палладиевым катализатором при 1 am
и 250—300° С:
Pd
СН^СН + Н2 — СН2-СН2 Д#=—41,7 ккал/моль
Процесс гидрирования ацетилена в этилен ранее использовался
в Германии в связи с недостатком нефтяного сырья. В настоящее
время эту реакцию применяют для очистки этиленовых фракций от
примесей ацетилена.
При действии воды на ацетилен в присутствии катализаторов
образуется ацетальдегид:
СН^СН -f Н2О -> СН3—СНО ЛЯ - —38,8 ккал/моль
Если этот процесс протекает в присутствии солей ртути, то он
называется реакцией Кучерова54. Реакция Кучерова проводится
в промышленности при избыточном давлении 1,7 am и 75—100° С.
В зависимости от состояния катализатора конверсия ацетилена
протекает на 30—60%, в связи с чем часть газа подвергают
рециркуляции. Обычно в промышленности выход ацетальдегида составляет
около 90% по ацетилену. В настоящее время для гидратации
ацетилена в ацетальдегид используются также другие, нертутные
катализаторы. Указанный метод получения ацетальдегида до сих пор
является в промышленности основным, однако за последнее время в
связи с открытием реакции прямого окисления этилена до
ацетальдегида этот новый, более перспективный метод внедряется все шире.
* Ацетилениды металлов образуются также при сплавлении окислов металлов
с углем. Например, при сплавлении угля с окисью кальция образуется ацетиленид
(карбид) кальция, при взаимодействии которого с водой выделяется ацетилен.
28
При действии водяных паров на ацетилен в присутствии окисей
цинка и железа как катализаторов при 360—450° С образуется
ацетон (выход 85% по ацетилену):
2СН^СН+ЗН2О — СН3—СО—СН3+СО2+2Н2
Эта реакция не используется в промышленности, но применялась
ранее на опытных установках, где переработка разбавленного
ацетилена велась для научных целей.
Со спиртами ацетилен реагирует с образованием простых
виниловых эфиров (А. Е. Фаворский, 1888 г.)55. Ацетилен и
соответствующий спирт взаимодействуют в присутствии едкого кали как
катализатора при 150—160° С и 4—20 am:
СН^СН + ROH -> CH2-CHOR
Выход эфира достигает 95% по ацетилену. Конверсия ацетилена
за один проход составляет 20—40%. Перед компримированием
ацетилен разбавляют азотом (10—20 объемн. % N2) для снижения взры-
воопасности процесса. Обычно эта реакция используется для
получения виниловых эфиров низших насыщенных спиртов.
С сероводородом ацетилен реагирует при 120° С с образованием
целого ряда веществ, содержащих серу56:
сн-сн + h2s —► ch2=chsh +"f > c2h5sh
винил меркаптан этилмеркаптан
| |
н2с—сн2 c2h5-s-ch==ch2
с этилвинилсульфид
лена
I t
тиоокись
этилена
I
C2H5-S-(CH2)2-S-C2H5
Тиоколовые L ъ v '
полимеры этилендиэтилсульфид
При действии ацетилена на меркаптан аналогично простым
виниловым эфирам получаются виниловые тиоэфиры:
CH^CH + RSH -, CH2-CHSR
Ацетилен легко присоединяет хлор, образуя тетрахлорэтан53:
СН^СН + 2С12 — СНС12—СНС12
Для получения тетрахлорэтана в реактор, заполненный жидким
тетрахлорэтаном, вводят раздельно хлор и ацетилен в стехиометри-
ческом соотношении. Температура реакции 80—90° С. При реакции
выделяется тепло, расходуемое на испарение части тетрахлорэтана.
Для регулирования испарения в реакционной зоне поддерживается
вакуум. Поскольку хлор и ацетилен могут реагировать со взрывом,
в реакторе не должно быть незаполненного жидкостью объема.
Тетрахлорэтан производится в промышленности в больших количе-
29
ствах, главным образом для получения трихлорэтилена, применяемого
как эффективный растворитель и для переработки в монохлоруксус-
ную кислоту.
Таким же образом ацетилен присоединяет другие галоиды с
образованием тетрабромэтана и тетраиодэтана. Фтор реагирует с
ацетиленом довольно бурно, поэтому для получения фторпроизводных
этана обычно используют реакции обмена хлорпроизводных с
фтористым водородом.
Присоединением одной молекулы галоида к ацетилену могут
быть получены дигалоидные производные этилена:
СН^СН + Вг2 -> СНВг = СНВг
Эта реакция проще протекает в случае присоединения брома
и иода, а для получения, например, симметричного дихлорэтилена
используют действие цинковой пыли на тетрахлорэтан:
СНС12—СНС12 + Zn -> CHCl = CHCl+ZnCl2
Гидрохлорированием ацетилена в присутствии катализатора
получают хлористый винил:
+ — СНа=СНС1
Процесс осуществлен в промышленности в жидкой и в паровой
фазах. В первом случае катализатором является однохлористая
медь Си2С12, во втором — хлорная ртуть HgCl2. Реакция протекает
с высоким выходом.
Аналогично присоединяется серная кислота:
СНееСН + H2SO4 -> CH2=CH-OSO3H
С азотной кислотой ацетилен реагирует с образованием тетранит-
рометана со степенью превращения 60% по ацетилену:
СН = СН + 6HNO3 -> С (NOa)4 + СО2 + 2NO2 + 4Н2О
Гидроцианирование57 ацетилена протекает при небольшом
избыточном давлении и 80° С в присутствии хлористой меди:
СНееСН + HCN - СН2=СН—CN
акрилонитрил
Акрилонитрил получают, пропуская смесь ацетилена й циани-
хтого водорода в соотношении 10:1 через насыщенный раствор
катализатора. После адсорбции образовавшегося акрилонитрила водой
избыток ацетилена возвращается в цикл. Давление в реакторе обычно
определяется высотой столба раствора и гидравлическим
сопротивлением системы. Обычное давление 1,2—1,4 am,
30
С аммиаком ацетилен реагирует при 100° Си 16 am с
образованием 2-метил-5-этилпиридина57
с2н5
Н3С
,5-метил-2-этил пиридин
Для реакции необходим трехкратный избыток аммиака.
При действии ацетилена на некоторые гетероциклические
соединения, содержащие пиррольный цикл (пиррол, индол, карбазол,
имидазол), образуются N-винильные производные. Так, например,
с карбазолом при 180° С и 15 am в присутствии некоторых
катализаторов (окиси кадмия и цинка, органические соли этих металлов и
смеси солей) реакция винилирования идет относительно легко57"60:
\ /ч //\ /ч
NH N—СН=СН2
карбазол N-пинилкарбазол
Выход N-винилкарбазола, перекристаллизованного из метилового
спирта, составляет 75% по ацетилену. N-Винилкарбазол хорошо поли-
меризуется в поливинилкарбазол, применяемый в промышленности
в качестве электроизоляционного материала.
Винилирование алифатических и циклических амидов протекает
в присутствии едкого кали57"60 при 140—160° С и 15 am:
кон
СН = СН + СН8—СО—NHCH8 -> СН3— СО—N—СН = СН2
I
сн3
С а-пирролидоном при 30—40° С и 22—25 am образуется N-ви-
нилпирролидон:
НоС—СНо НоС—СН2
СН = СН + | I >~ "I I
НоС СО Н2С СО
" \н N-CH=CM2
N-Винилпирролидон полимеризуется с образованием поливинил-
пирролидона, применяемого в медицине как заменитель плазмы крови.
В лабораторной практике используется реакция присоединения
гидрида бора к алкинам для последующего получения альдегидов
или кетонов посредством окисления аддукта щелочным раствором
перекиси водорода61:
+н2о2
RC=CH + ВН3 — RCH=CHBH2 -> RCHO
31
При карбонилировании ацетилена (Реппе, 1939 г.) получается
акриловая кислота57:
СН = СН + СО+ Н2О -» СН3=СН—СООН
АН = —50,6 ккал/моль
Реакция осуществляется в присутствии карбонила никеля,
являющегося переносчиком окиси углерода, при 150—200° С и 30 am.
Соотношение окиси углерода и ацетилена 1:1.
Со спиртами в тех же условиях образуются эфиры акриловой
кислоты
CHeeCH + CO+ROH -> CHa=CH—COOR
а с меркаптанами — тиоэфиры акриловой кислоты:
CHseCH + CO+RSH -> СН2=СН—COSR
С аммиаком получается амид акриловой кислоты:
+ CO+NH8 -> СН2-СН—CONH2
Если вводятся насыщенные карбоновые кислоты, то образуются
смешанные ангидриды насыщенной и акриловой кислот57:
CH = CH + CO+RCOOH -> CH2-CH—COO-COR
Ацетилен присоединяет альдегиды с образованием ацетиленовых
гликолей (синтез Реппе)67:
+ 2RCHO -^2HOCH—C = C—CHOH
Катализаторами таких реакций являются ацетилениды
некоторых тяжелых металлов, главным образом меди, осажденные на сили-
кагеле. Для предотвращения полимеризации ацетилена добавляют
25% окиси висмута. Так же, как и при реакции со спиртами,
ацетилен разбавляют азотом (10—20 объемн. % N2).
Для получения бутиндиола57 реакцию нужно проводить при 90—
95° С и 4—6 am. Выход бутиндиола-1,4 составляет 90—92%,
образование его протекает через пропаргиловый спирт:
СН = СН + НСНО -> СН = С—СН 2ОН АН = — 33 ккал/моль
пропаргиловый спирт
сн=с—сн„он + нсно -> носн2—с^с—сн2он
бутиндиол-1,4
32
Пропаргиловый спирт и бутиндиол используются для получения
аллилового спирта, бутандиола и других растворителей.
При взаимодействии ацетилена с алифатическими кетонами также
образуются ацетиленовые гликоли (синтез Реппе)49»57:
—CO-R
ОН ОН
Реакция протекает в тех же условиях, что и с
альдегидами.
При реакциях ацетилена с гетероциклическими кетонами
(пиранового, тиопиранового и пиперидинового рядов) можно
синтезировать гетероциклические третичные ацетиленовые
спирты62"64:
сн^сн
Ч. ЛЧ>"\\^11'{»7 *^ _-
2,2-диметил-4-
этинилтетрагидро-
пиранол-4
=сн
2,2-диметил-4-
этинилтетрагидро-
тиапиранол-4
сн^сн
NH
2,5-диметил-4-
этинилпиперидол-
Реакции протекают в абсолютном диэтиловом эфире при 5—10° С
в присутствии сухого порошка едкого кали с 30—60%-ным
выходом.
С органическими кислотами ацетилен образует в присутствии
катализаторов сложные виниловые эфиры59"'61'66:
CHeeCH + RCOOH -> CH2=CH—OCOR
С уксусной кислотой реакция в паровой фазе протекает при
170—210° С в присутствии ацетата цинка, нанесенного на
активированный уголь (в жидкофазном процессе при 60—65° С используют
соли ртути). Выход винилацетата составляет 95% по ацетилену и
3 В. Н. Антонов 33
97—99% по уксусной кислоте. Степень однократной конверсии
ацетилена 20%, что вызывает необходимость его рециркуляции. Винил-
ацетат вырабатывают во многих странах для получения поливинил-
ацетата, поливинилового спирта и винилита (сополимер с винил-
хлоридом).
Реакции полимеризации. В присутствии порошкообразной меди
при 200—300° С ацетилен полимеризуется в купрен:
пСН ее СН -> (СН)2П
Не растворимый в воде и почти ни в каких органических
растворителях купрен имеет большой молекулярный вес и низкую
теплопроводность. Купрен может применяться^ в качестве изоляционного
материала.
При пропускании ацетилена через насыщенный раствор однохло-
ристой меди происходит димеризация с образованием винилацети-
63
2СН = СН С"-2 СН2-СН-С =
Реакция осуществляется при 80° С и небольшом избыточном
давлении. Степень конверсии ацетилена за один проход — до 15%.
Выход винилацетилена составляет 80% по ацетилену. Винилацетилен
является исходным полупродуктом для получения хлоропрена и в
больших количествах производится во многих странах.
При пропускании ацетилена через раствор соединений никеля
образуется тетрамер ацетилена — циклооктатетраен57:
нет хн
I I
up PLI
C4CH_CH^CH
По данным Реппе, реакция протекает с промежуточным
образованием ацетиленида никеля.
При температуре красного каления (около 1000° С) или в более
мягких условиях над активированным углем ацетилен полимери-
зуется в бензол (Н. Д. Зелинский).
Высшие ацетиленовые углеводороды
При производстве ацетилена из углеводородов в качестве'
примесей образуются высшие ацетиленовые и другие непредельные
углеводороды, наличие которых в продуктах пиролиза осложняет процесс
выделения ацетилена из реакционных газов, его очистку и
переработку. Некоторые из них (диацетилен и триацетилен) значительно
более взрывоопасны, чем ацетилен. В ацетилене, полученном из
34
карбида кальция, также имеются примеси ацетиленовых
углеводородов, но в гораздо меньшем количестве.
физические свойства ацетиленовых углеводородов — алкинов —
закономерно меняются в гомологическом ряду. Некоторые
показатели ацетиленовых и других непредельных углеводородов, обычно
присутствующих в газах пиролиза углеводородов, приведены в
табл. 1-13, 1-14 и 1-15.
ТАБЛИЦА 1-13
Физические свойства некоторых ненасыщенных углеводородов10' 65' 70
Углеводород
Метилацетилен
Этилацетилен
н-Пропилацетилен . . .
Диметилацетилен ....
Метилэтилацетилен . . .
Диацетилен
Метилдиацетилен ....
Этилдиацетилен.
Триацетилен
Пропадиен (аллен) . . .
Бутадиен-1,3
Винилацетилен
Формула
СН3-С=СН
с2н5-с=сн
с3н7-с=сн
СН3—С=С—СН3
СН з—С=С—&2 Н 5
сн=с—с=сн
СН3-С=С-С=СН
с2н5-с=с—с=сн
сн=с-с=с-с=сн
СН2=С=СН2
СН2=СН-СН=СН2
СН2=СН-С=СН
Молекулярный
вес
40,065
54,0.2
68,119
54,092
68,119
50,060
64,087
78,114
80,082
40,065
54,092
52,074
Температура
кипения, °С
—23,2
8,1
40,2
28
56,1
9,5
55
87
70-80
-34,5
—4,5
5,5
Температура
плавления, °С
—103
—126
—106
-32,8
—109
—36
-80
-80
—20
—136
—108,9
Относительная
плотность
(жидкостей) d4
0,550
0,625
0,60
0,650
0,711
0,736
0,595
0,624
0,686
Теплота
сгорания, ккал/моль
463,1
620,6
778,0
620,6
774,3
1646
830
720
940
550
602,8
670
ТАБЛИЦА 1-14
Теплоемкость ацетиленовых углеводородов6'10'65'70 [в ккал/(м3 -град)]
Температура
°С
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
Метилацетилен
ср
0,617
0,741
0,853
0,949
1,031
1,103
1,166
1,221
1,271
1,313
1,350
1,383
1,412
cv
0,522
0,652
0,764
0,860
0,942
1,014
1,077
1,133
1,182
1,225
1,262
1,295
1,324
Этилацетилен
ср
0,812
1,015
1,189
1,338
1,466
1,576
1,672
1,756
1,829
1,892
1,948
1,997
2,040
cv
0,723
0,927
1,101
1,250
1,377
1,487
1,583
1,667
1,740
1,804
1,859
1,908
1,951
Диметилацетилен
Ср
0,767
0,963
1,132
1,285
1,420
1,538
1,641
1,730
1,808
1,876
1,934
1,986
2,030
cv
0,678
0,874
1,043
1Д96
1,331
1,449
1,552
1,641
1,719
1,787
1,846
1,897
1,942
Примечание. Для винилацетилена
630 ккал/(м*. град) при 1200° С.
3*
= 0,716 ккал/{м3-град) при 0° С и
35
ТАБЛИЦА I -15
Давление насыщенных паров ацетиленовых углеводородов10'68 (в am)
Темпера-
4g*
—40
—20
—10
— 5
0
5
Метил-
ацетилен
0,49
1,34
1,83
2,25
2,67
3,24
Этилаце-
тилен
0,10
0,30
0,49
0,59
0,70
0,91
Диметил-
ацетилен
0,03
0,13
0,21
0,28
0,35
0,42
Темпера-
тур*
10
20
30
40
60
Метил-
ацетилен
3,87
5,28
7,03
9,13
15,46
Этилацетилен
1,13
1,55
2,11
2,95
5,27
Диметил-
ацетилен
0,52
0,78
1,13
1,69
2,99
Кривые изменения скрытой теплоты испарения10»70 и плотности69-70
некоторых ацетиленовых углеводородов в зависимости от
температуры приведены на рис. 1-13 и 1-14. Видно, что с ростом температуры
~ скрытая теплота
испарения и плотность
уменьшаются.
Следует иметь в виду,
что физические свойства
рассматриваемых
углеводородов исследованы
недостаточно подробно и
многие представленные цифры
не являются полностью
воспроизводимыми
результатами измерений. Раство-
I
—1 —
у!
fes^-
^ч
Г
V
-80
О 80
Температура, °С
160
Рис. 1-13. Зависимость скрытой теплоты
испарения некоторых ацетиленовых углеводородов
от температуры:
1 — ацетилен; 2 — диметилацетилен; 3 — этилаце-
тилен; 4 — метилацетилен.
римость ацетиленовых
углеводородов исследована
относительно хорошо,
количественные показатели
приведены
растворимости
в гл. VI.
Метилацетилен36 СН3—С = СН получается вместе с алленом
(пропадиеном) при действии воды на карбид магния:
2Mg2C3 + 8Н2О - СН3-С=СН + СН2=С=СН2 + 4Mg (OH)2
Поскольку обычно в товарном карбиде кальция присутствует
карбид магния, в ацетилене, полученном из карбида кальция, всегда
имеются незначительные примеси аллена и метилацетилена..
Концентрированный метилацетилен способен к взрывному
разложению71-73 при давлении выше 4,4 am. Концентрационные пределы
взрываемости метилацетилена в смеси с воздухом 2,2—13 объемн. %.
Добавки азота, метана, этана и некоторых других веществ оказывают
на метилацетилен флегматизирующее действие. При добавлении,
например, 30 объемн. % азота (или 26 объемн. % метана, или
36
0,6
I
15 объемн. % этана) допустимое безопасное давление возрастает
до 7,8 am. При дальнейшем увеличении количества добавок
предельное безопасное давление можно еще больше повысить.
Мети л ацетилен, имея в молекуле тройную связь между атомами
углерода, способен вступать во все реакции замещения,
присоединения и полимеризации, описанные для ацетилена. В технике метилаце-
тилен начинают применять для газопламенной обработки металлов
в связи с меньшей опасностью взрыва. По теплотворной
способности метилацетилен превышает ацетилен в 1,5 раза.
Винилацетилен74 СН2—СН—С = СН присутствует в газах
пиролиза при получении ацетилена из углеводородов в количестве до
1 объемн. %.
Впервые винилацетилен
был получен Ньюлендом в
1931 г. путем димеризации
ацетилена в присутствии
однохлористой меди. В
настоящее время этот способ
применяется в
промышленности. В лабораторной
практике для синтеза винилацети-
лена используют методы деги-
дрохлорирования галоидпро-
изводных углеводородов или
дегидратации ацетиленовых
спиртов.
Жидкий винилацетилен в
открытом сосуде горит
сильным коптящим пламенем.
Винилацетилен склонен к
взрывному распаду. Он более взрывоопасен, чем ацетилен, так как при
транспортировании по трубам малого диаметра начальное давление
взрывного распада винилацетилена повышается в меньшей степени,
чем ацетилена. Предельное безопасное давление винилацетилена
1,32—1,38 am. Пределы взрываемости в смеси с воздухом67
2,13—22,9 объемн. % и с кислородом 1,7—73 объемн. %.
Газообразный винилацетилен не окисляется кислородом воздуха,
но в жидкой фазе легко образует перекиси, которые остаются после
испарения винилацетилена в виде желтой взрывчатой массы.
Винилацетилен способен полимеризоваться со взрывом; кислород
инициирует реакцию полимеризации, а вода замедляет этот процесс.
Поскольку в молекуле винилацетилена имеются двойная и тройная
связи между атомами углерода, он легко вступает во все химические
реакции, свойственные ацетилену и этилену. Следует отметить, что
гидрирование винилацетилена проходит через стадию образования
бутадиена:
•ч
V
N
\
\
-^
\ ^
\
-80
160
О 80
Температура, °С
Рис. 1-14. Зависимость плотности
некоторых ацетиленовых углеводородов в жидком
состоянии от температуры:
/ — ацетилен; 2 — метилацетилен; 3 — этилаце-
тилен; 4 — диметилацетилен.
снг=сн—с=сн-}-н3 -> снг=сн—сн=сна
37
Гидрохлорирование винилацетилена при 50—110° С приводит
к получению хлоропрена:
GU2°l2; NH4°> CH2-CH-CC1=:CH2
Винилацетилен получается в промышленности в больших
количествах для синтеза хлоропренового каучука. Хлоропреновый
каучук является ценным полупродуктом для получения маслобензо-
стойких резин.
Диацетилен СН = С—С = СН обычно -присутствует в относительно
большом количестве (до 1 объемн. %) в газах электрокрекинга и
пиролиза метана. Имеется он также и в газах пиролиза других
углеводородов. В процессе выделения ацетилена в продувочных газах иногда
содержится до 5—8% диацетилена. Обычно эти газы сжигают или
возвращают в реактор (при электрокрекинге метана).
В лабораторных условиях диацетилен можно получить
окислительной димеризацией моноацетиленидов меди или натрия в
присутствии катализатора — окиси железа (таким путем диацетилен был
впервые получен Байером):
2CH = CNa ±2 СН-С—C = CH+Na2O
Можно также получить диацетилен щелочным омылением
1,4-дихлорбутадиена:
СНС1-СН—СН-СНС1 jg? СН = С-С = СН
Диацетилен очень взрывоопасный газ, поэтому обращение с ним
требует особой осторожности. Согласно литературным данным,
взрывной распад диацетилена происходит уже при давлении 0,05—
0,2 am (в зависимости от импульса)35. Взрывоопасные концентрации*
его в смесях с воздухом лежат в пределах 1,5—100 объемн. %, т. е.
диацетилен не имеет верхнего предела взрываемости (взрыв
происходит в отсутствие кислорода). Взрыв диацетилена под действием
электрической искры сопровождается яркой вспышкой, выделением
сажи, водорода и метана. Минимальное давление в автоклавах после
взрыва превышает начальное более чем в 14—15 раз. При
атмосферном давлении газовые смеси, содержащие более 12% диацетилена,
взрываются от удара, искры и даже от падения кусочков полимера53'57.
Флегматизаторы (пропан, бутан, пентан, пропилен, бутилен,
двуокись углерода и водород) позволяют значительно повысить
безопасность диацетилена35. Так, например, при нагревании чистого
жидкого диацетилена до 60° С происходит сильный взрыв (давление
паров 6,5 am). При флегматизации жидким бутаном (1 : 1) смесь
можно нагревать до 220° С без взрыва. Давление паров диацетилена
при этой температуре 160 am.
Кривые изменения минимального давления, при котором
начинается взрывной распад диацетилена, в зависимости от концентрации
флегматизирующих добавок представлены на рис. 1-15. С
понижением давления область взрыва уменьшается. Смеси диацетилена
38
невзрыворпасны, если содержание разбавителя превышает
определенный минимум.
Взрывы диацетилена часто связаны с его полимеризацией,
начинающейся при температуре немного выше 0° С. Полимеризация
ускоряется при соприкосновении газа со стеклом, сталью и особенно
с ржавчиной. Диацетилен, как и другие ацетиленовые углеводороды,
легче полимеризуется в растворителях — в диметилформамиде, N-ме-
тилпирролидоне и др. В нейтральном водном растворе в темноте
диацетилен сравнительно
стабилен, однако следы щелочи
или соды ускоряют
образование хлопьев полимера67.
| Для предотвращения
полимеризации были
предложены различные
ингибиторы76-78: пиридин, уротропин,
морфолин и др. Добавление
0,1—3,0% (от количества
растворителя) уротропина или
метиленового голубого
препятствует образованию
полимера или приводит к получе-
I
>§ 80
съ
I 60
1
I
1
20
I
\сг
YV
\
Область
Взрыва
V.
>^
/
/
•■»——р.—
800
200 400 600
Давление смеси, мм рт. ст.
Рис. 1-15. Зависимость начального давления
взрывного распада диацетилена от влияния
разбавителей и общего давления смеси:
/ — с метаном; 2 — с азотом; 3-е водородом.
нию веществ, растворимых в
диметилформамиде или в
N-метилпирролидоне.
Иногда для очистки
ацетилена от диацетилена
последний специально подвергают
полимеризации в присутствии
солей металлов79. Полимеры, выделенные из растворителей в сухом
виде (из диметилформамида или N-метилпирролидона),
нерастворимы в воде и органических растворителях. Такие полимеры могут
самопроизвольно воспламеняться на воздухе, поэтому все операции,
связанные с ними, рекомендуется проводить в атмосфере азота.
По химическим свойствам диацетилен во многом сходен с
ацетиленом. Он также склонен вступать в реакции замещения одного или
двух атомов водорода в молекуле и в реакции присоединения. При
гидрировании диацетилена образуется бутадиен, затем я-бутан.
Многие реакции присоединения приводят к образованию замещенных
диацетиленовых углеводородов. Так, например, с ацетоном
получается дииндиол
(СН3)2 С-С = С-С = С-С (СН3)2
он он
а с водой — диацетил СН3—СО—СО—СН3. Диацетилен пока не
используется в технике.
Триацетилен СН = С—С = С—С = СН впервые был обнаружен
в газах электрокрекинга метана Бауманом в 1948 г.80.
39
Триацетилен крайне нестабилен и способен быстро пблимеризб-
ваться с образованием взрывоопасных продуктов; 20%-ный раствор
триацетилена в этиловом спирте самопроизвольно разлагается со
взрывом81. Триацетилен полимеризуется при температуре
плавления (—20° С) в течение 1 ч, при комнатной температуре — в течение
нескольких минут с образованием коричневой массы, которая при
соприкосновении со стеклянной поверхностью взрывается с
образованием сажи82.
\ Л
Область
взрыва
V \
80 60 40 20
~с4н2
б
80 60 UO 20
-с4н4
2
Рис. 1-16. Концентрационные пределы взрываемости различных смесей высших
ацетиленовых углеводородов с воздухом:
а — диацетилен С4Н2—винилацетилен С4Н4—воздух; б — диацетилен С4Н2— метилацети-
лен С3Н4—воздух; в — диацетилен С4Н2— метан СН4—воздух; г — винилацетилен С4Н4 —
метилацетилен С3Н4—воздух (точки — воспламенение; кружки — нет воспламенения).
Смеси ацетиленовых углеводородов83'84, например смеси ацетилена
с диацетиленом, метил- и винилацетиленом, при комнатной
температуре и 700 мм рт. ст. не взрываются, если содержание в них
диацетилена составляет 18—32 объемн. %. Следует отметить, что
минимальная концентрация диацетилена, при которой происходит
взрывной распад его смесей с ацетиленом, винилацетиленом и метилацети-
леном, с повышением давления уменьшается. Нижние пределы
взрываемости смесей диацетилена и винилацетилена и смесей метилаце-
тилена и винилацетилена с достаточной точностью подчиняются
уравнению Ле-Шателье. Максимальное давление газов после взрыва
40
смесей ацетилена с метил- и винилацетиленом превышает начальное
давление в 8—13 раз. Концентрационные пределы взрываемое™
различных смесей ацетиленовых углеводородов с воздухом показаны
на рис. 1-16.
ЛИТЕРАТУРА
1. Справочник химика, т. I, Изд. «Химия», 1966.
2. Brans ky E., Acetilen, Bratislava, 1958.
3 Miller S. A., Acetylene, its Properties, Manufacture and Uses, v. 1, London,
' 1965.
4 Bottom ley G., Rewes C. G., Seiflow G., J. Appl. Chem., 9,
№ 10, 517 (1959).
5. В г о m 1 e у L. A., W i 1 с е С R.f Ind. Eng. Chem., 43, 1641 (1951).
6. Вукалович М. П., Кирилин В. А., Ремизов С. А. и др.,
Термодинамические свойства газов, Госнаучтехиздат, 1953.
7. К i У a m a R., M a k i t a, Rev. Phys. Chem. Japan, 12, 49 (1952).
8. M а к i t a, Mem. Fac. Ind. Arts, Kioto Techn. Univ., 4, № 19 (1955).
9. Kiyama R., J. Chem. Soc. Japan, 61, 17 (1958).
10. Thermodynamic Function of Gases, v. 2, London, 1956.
11. He use W., Ann. Phys., 59, 86 (1919).
12. Landolt — Bornstein (Red.), Physikalisch-chemishe Tabellen, Springer,
Berlin, 1927, S. 1305.
13. J. Chem. Phys., 21, 264 (1953).
14. Kordes E., Z. Elektrochem., 58, № 1, 76 (1954).
15. С т р и ж е в с к и й И. И., Гузов С. Г., Ковальский В. А.,
Ацетиленовые станции, Машгиз, 1959.
16. Hiraoka, Rev. Phys. Chem. Japan, 24, № 13, 221 (1954).
17. H о n t i G., Mag. kern, lap., 13, № 5/6 (1958).
18. M с К i n n i s A., Ind. Eng. Chem., 47, 850 (1955).
19. H о 1 e m a n n P., Hasselmann R., Forschungsber. Wirtschafts- und
Verkehrsministeriums Nordrhein Westfalen, 1954, S. 109.
20. Chem. Ing. Techn., 25, 466, 735 (1953).
21. К i у a m a R., Hiraoka, Rev. Phys. Chem. Japan, 25, № 16, 52 (1955).
22. Дубовкин Н. Ф., Справочник по углеводородным топливам и их
продуктам, Госэнергоиздат, 1962.
23. И в а н о в Б. А., К о г а р к о СМ., ПМТФ, 3, 59 (1963).
24. Prout W., Anderson R., Fuel, 33, 125(1954).
25. Иванов Б. А., Ко г ар ко СМ., ДАН СССР, 150, 1300(1963).
26. Волков А. Е., Лапидус А. С, Техника безопасности в производстве
ацетилена из природного газа, Изд. «Химия», 1964.
27. L u r i e, Sherman, Ind. Eng. Chem., 25, 404 (1933).
28. В a r t о 1 о m e, Z. Elektrochem., 53, 191 (1949).
29. Miller S. A., Chem. Eng. Proc. Ind., 32, 476 (1951).
30. Rimarski W., Konschak M., Autogene Metallbearbeitung, 13, 211
(1930).
31. Koesche-Kris cher В., Wagner H. G., Brennst. Chem., 39, №3/4,
15 (1958).
32. H i г о s h i, T e r a n i s h i H., Rev. Phys. Chem. Japan, 25, № 2, 52 (1955).
33. К i у a m a R., О s u g i D., T e r a n i s h i H., Rev. Phys. Chem. Japan,
u23, 43 (1953).
34. While A. C, J. Chem. Soc, 127, 672 (1925).
35. M у ш и й Р. Я., Г л и к и н М. А., Вестник НИИТЭхим, № 7, 28 (1963).
36 R е р р е W., Chemie und Technik der Acetylen-Druck-Reactionen, 2 Aufl.,
Verlag-Chemie, Weinheim, 1952, S. 34.
37. К о г а р к о СМ., Иванов Б. А., ДАН СССР, 140, № 1, 165 (1961).
38. К о г а р к о СМ., Иванов Б. А., ДАН СССР, 142, № 3, 637 (1962).
39. Иванов Б. А., К о г а р к о СМ., Журн. ВХО им. Д. И. Менделеева,
9, № 3, 280 (1964).
41
40. S а г g e n t H., Chem. Eng., 64, № 2, 250 (1957).
41. Стрижевский И.И., Бородулин А.А., К онер Б.А., Охрана
химических предприятий от пожаров и взрывов, изд. НИИТЭхим, вып. 172
(1961).
42. Когарко С. М., Бородулин А. А. и др., Хим. пром., № 7 (1962).
43. J о n e s С, Kennedy R., S р о 1 е n J., U. S. Bur. Mines Rept. Invest.,
№ 4196 (1948).
44. F г е у а с q u e s M., Chim. et ind., 46, № 5, 579 (1941).
45. С т р и ж е в с к и й И. И., Зайцева В. П., Труды ВНИИавтоген, вып. V,
1961.
46. Clark A., Z. Schweisstechnik, 42, № 4, 80 (1952).
47. Д е л а г о Ю. В., в сб. «Газопламенная обработка металлов», Труды
Всесоюзной научно-технической конференции», 1956, стр. 144.
48. Jahresber. Chem. Techn. Reichsanstatt, 8, 43 (1930).
49. P e п п е В., в сб. «Химия ацетилена», под ред. А. Д. Петрова, Издатинлит,
1954.
50. И н г о л ь д К- К., Механизм реакций и строение органических соединений,
Издатинлит, 1959.
51. Роберте С, К а с е р и о М., Основы органической химии, Изд. «Мир»,
1968.
52. Антонов В. Н., Производство ацетилена, Госхимиздат, 1959.
53. Н ь ю л э н д Ю., Фогт Р., Химия ацетилена, Издатинлит, 1947.
54. Кучеров М. Г., ЖРФХО, 20, 418 (1888).
55. Фаворский А. Е., Сборник избранных трудов, Химтеорет, 1934.
56. Copenhaver J., Bigelow M., Acetylene and Carbon Monoxide
Chemistry, Reinhond Publ. Corp., New York, 1949, p. 32.
57. Химия ацетилена, под ред. Петрова А. Д., Издатинлит, 1954.
58. Назаров И. Н., Химия ацетилена, 1942.
59. Шостаковский М. Ф., Простые виниловые эфиры, Изд. АН СССР, 1954.
60. Шостаковский М. Ф., Ацетилен в химической промышленности, Изд.
«Правда», 1942.
61. Химия ацетилена, под ред. Шостаковского М. Ф., Изд. «Наука», 1968.
62. Сарбаев Т. С, докт. дисс, Алма-Ата, 1968.
63. Маканов У., канд. дисс, Алма-Ата, 1967.
64. А з е р б а е в И. Н., Сарбаев Т. С, Маканов У., Труды химико-
металлургического института АН Каз. ССР, т. 2, 1967.
65. Физико-химические свойства индивидуальных углеводородов, под ред. Та-
тевского В. М., Госнефтеиздат, 1960.
66. Шостаковский М. Ф., Богданова А. В.,
Красильщиков Б. К., Усп. хим., 28, 1052 (1959).
67. Strauss F., Ко 11 en L., Ber., B59, 1664(1926).
68. S t u I I D. В., Ind. Eng. Chem., 39, 517 (1947).
69 Flynn L. W., T ho das G., J. Chem. Eng. Data, 6, 457 (1961).
70. G a 1 1 a n t R. W., Hydrocarb. Proc. a. Petrol. Ref., 44, № 9, 225 (1965).
71. F i t z g e r a 1 d F., Nature, 186, 386 (1960).
72. F i t z g e r a 1 d F., Fuel, 40, 275 (1961).
73. Nelson H., Combustion, Reinhold Publ. Corp., New York, 1957, p. 823.
74. Петров А. А., Усп. хим., 29, № 9, 1049 (1960).
75. Z о b e 1 F., Angew. Chem., 20, 260 (1948).
76. Муший Р. Я., Хим. пром., № 2 (1963).
77. N e 1 s о n H., пат. США 2715101, 2715105 и 2861041, 1955 г.
78. S a r a b i а О., пат. США 2915138 и 2915142, 1959 г.
79. О d u k a H у d о d у, яп. пат. 3864, 1961 г.
80. В a u m a n P., Angew. Chem., 20, 257 (1948).
81. Н u n s m a n n W., Angew. Chem. 62, 219 (1950).
82. С о о к С, Jones E., W h i h i n g M., J. Chem. Soc, 1952, 2883.
83. M о ш к о в и ч Ф. Б., Муший Р. Я., Крайнюк Б. П., Хим. пром.,
№ 2 (1965).
84. Мошкович Ф. Б., Р о з л о в с к и й А. И., Муший Р. Я., Хим.
пром., № 6, 433 (1969).
42
ГЛАВА
II
ПРОИЗВОДСТВО АЦЕТИЛЕНА
ИЗ КАРБИДА КАЛЬЦИЯ
ПРОИЗВОДСТВО КАРБИДА КАЛЬЦИЯ
Первые попытки1 получения карбида кальция сплавлением
обожженной извести и угля в электрической дуге были сделаны еще
в 1839 г., однако лишь в 1892 г. Муассан (Франция) и Вильсон
(Канада) предложили конструкцию электродуговой печи, пригодной
для промышленного использования. Сначала во Франции и Канаде
в 1896 г., а вскоре и в других странах этот способ получил широкое
распространение.
В России производство карбида кальция было организовано
в 1908 г. До революции объем его производства в нашей стране был
очень небольшой, и только с развитием промышленности в годы
первых пятилеток началось ускоренное строительство новых заводов
и отдельных крупных цехов. В настоящее время СССР имеет развитое
производство карбида кальция.
Вначале карбид кальция использовался для получения ацетилена
в небольших переносных или стационарных генераторах — только
для целей сварки или резки металла. В машиностроении и в
ремонтных службах заводов и в настоящее время применяется карбид
кальция, причем во все возрастающих количествах. Но, как отмечалось
ранее, теперь большая часть карбида кальция используется
химической промышленностью для производства продуктов органического
синтеза. Некоторая доля карбида кальция все еще потребляется
в производстве цианамида. Однако расход карбида на эти цели не
превышает 3—5% от его общего производства, и это — единственная
область применения собственно карбида кальция.
Метод производства ацетилена из карбида кальция имеет ряд
технологических достоинств. Ацетилен получается более чистым и очень
высокой концентрации, что исключает необходимость выделения его
из реакционных газов и специальной очистки. Карбид кальция можно
легко транспортировать, поэтому районы его производства можно
отдалить от мест потребления ацетилена.
Производство ацетилена из карбида кальция делится на две
стадии: 1) получение карбида кальция в электродуговых печах и 2)
разложение карбида кальция водой с образованием ацетилена.
Как было указано выше, карбид кальция получается сплавлением
обожженной извести и угля (смеси кокса и антрацита):
СаО + ЗС -> СаС2 + СО АН = + 108 ккал/моль
43
И
II
2400
2000
1600
v]
/
У
/
/
При этом получается окись углерода, и таким образом осуществляется
восстановление обожженной извести.
Обожженная известь сплавляется с углеродом лишь на 70—80%,
поэтому в товарном продукте всегда присутствует 12—25% СаО.
Последнее обстоятельство очень важно, так как для облегчения
выпуска расплава из печи необходимо максимально снижать
температуру плавления карбида кальция, а наличие извести в сплаве
как раз и понижает температуру его плавления. Чистый карбид
кальция плавится при 2300° С, а обожженная известь — при 2585° С.
При сплавлении карбида кальция с обожженной известью
температура плавления смеси снижается в зависимости от содержания
компонентов (рис. П-1). Наиболее
низкая температура
плавления (1750° С) у эвтектичной
смеси состава 69,4% СаС2 и
30,6% СаО.
Присутствие различных
химических и механических
примесей в обожженной
извести и в золе кокса также
отражается на температуре
плавления получаемого
карбида кальция3. В нем обычно
имеются окись магния, фосфорит, остатки известняка, соединения
серы, кремния и аммония, окиси металлов (железа, алюминия,
ванадия и др.). Все эти примеси в условиях высокой температуры
реагируют с углем или окисью кальция, образуя различные соединения.
Например, окись магния образует карбид магния:
MgO + 3C -> MgC2 + CO
Фосфорит восстанавливается до фосфида кальция:
Са3(РО4)2 + 8С -* Са3Р2 + 8СО
Остатки известняка вначале обжигаются, а затем образуется
карбид кальция:
СаСО, -^ СаО ^ СаС2
wo
о
60 Ь0 20
20 ЬО 60 80
Содержание, %
О СаС2
W0 СаО"
Рис. Н-1. Диаграмма плавкости смеси
карбида кальция и обожженной извести.
Окись железа сплавляется с кремнистыми соединениями с
образованием ферросилиция:
2Fe2O3 + 3SiO2+12C -> Fe4Si3+12CO
Сульфат кальция превращается в сульфид кальция:
CaSO4 + 4C — CaS + 4CO
Окись алюминия сплавляется с углеродом с образованием
карбида алюминия:
2А12О3
А14С3
44
Таким же путем образуются соединения кальция с мышьяком
и некоторые другие вещества.
При обработке карбида кальция водой многие примеси
разлагаются с выделением различных газов. Например, фосфид кальция
выделяет фосфин:
Са3Р2+6Н2О -> ЗСа(ОН)2 + 2РН3
Из соединений мышьяка образуется мышьяковистый водород:
Ca3As2 + 6H2O — 2AsH3 + 3Ca(OH)2
Карбид алюминия разлагается водой с выделением метана:
А14С3+12Н2О — ЗСН4 + 4А1(ОН)3
В соответствии с теоретическими подсчетами для образования
1 т СаС2 в электродуговых печах требуется затратить 1965 квт-ч
электроэнергии, однако практически расходуется от 2800 до
3500 квт-ч. Электроэнергия затрачивается на разогрев и
расплавление шихты, на проведение основной эндотермической реакции,
а также на побочные химические реакции и потери в окружающую
среду. Кроме того, некоторое количество электроэнергии расходуется
на подогрев и расплавление избыточной (20%) обожженной извести,
загружаемой в печь вместе с шихтой для снижения температуры
плавления карбида кальцкя.
Создание высокопроизводительных карбидных печей позволяет
более эффективно использовать электроэнергию, так как тепловые
потери в окружающую среду в таких печах значительно ниже, чем
в маломощных печах. Если в малопроизводительных печах расход
электроэнергии составляет 3500 квт-ч на 1 т СаС2, то в крупном
производстве эта величина снижается до 2800 квт-ч.
Высокопроизводительные печи с большими ваннами для накопления плава имеют
еще одно достоинство — высокую тепловую инертность, что
позволяет снижать потребляемую мощность на 40—50% в часы перегрузки
обслуживающей энергосистемы и тем самым способствует
стабильности печи. По этой же причине для крупных печей часто
предоставляется льготный тариф на электроэнергию, вследствие чего
себестоимость продукции снижается.
Плав карбида кальция можно получать также окситермическим
способом в шахтных печах по методу И. С. Голынкера (СССР)2.
Необходимое тепло выделяется в результате сгорания части кокса
при продувке шихты воздухом, обогащенным кислородом (до 70%).
В связи с высокой температурой в шахте (2200—2400° С) возникают
особые трудности в подборе огнеупорных материалов для футеровки
печи. При образовании 1 т СаС2 в шахтной печи образуется 5100 м3
газов, что в 6,5 раз больше, чем в электродуговых печах. По окситер-
мическому способу была построена опытная установка фирмы BASF
производительностью 70—100 т/сутки 80%-ного карбида кальция4.
При этом процессе получается большое количество отходящих газов,
45
содержащих СО (95%), Н2, N2, CO2 и О2. Эти газы можно
использовать для различных химических синтезов.
Однако окситермический способ производства карбида кальция
промышленного развития не получил, так как наряду с кислородом
приходится расходовать в несколько раз больше металлургического
кокса, чем при электротермическом методе. Это связано с тем, что
около 70% кокса при окситермическом способе расходуется на
получение необходимого тепла. Электротермический метод использует
электроэнергию, полученную из различных топлив (природный
газ), или гидроэлектроэнергию и является экономически более
выгодным.
Технологическая схема производства
Производство карбида кальция состоит из следующих стадий:
обжига известняка, приготовления шихты, получения карбидного
плава, дробления или гранулирования карбида кальция.
Для обжига известняка применяют пересыпные шахтные печи,
в которые засыпают известняк вместе с коксом или антрацитом, или
шахтные печи с выносной топкой. В обожженной извести,
получаемой в пересыпных печах, содержится много вредных примесей из золы
топлива. Для удаления золы, а вместе с ней и части извести,
измельчившейся при обжиге и пересыпке, применяется отсев. В печах с
выносной топкой используется тепло от сжигания природного газа или
отходящих газов карбидных печей. Известь, получаемая в таких печах,
чище,однако в данном случае необходимо сооружать несколько печей,
так как диаметр печи с выносной топкой меньше, поскольку топочным
газам трудно проникать через слой обожженной извести и
известняка. Применяются также вращающиеся печи с газовым обогревом.
Обожженная известь после отсева от золы и пыли поступает
на дробление, где измельчается до кусков размером менее 50 мм
в поперечнике. После отсева от мелочи и пыли дробленая известь
направляется посредством транспортеров и элеваторов в приемные
бункеры карбидных печей для приготовления шихты.
Технологическая схема крупного производства карбида кальция
приведена на рис. П-2. Исходное сырье (обожженная известь, кокс
и антрацчт) заготавливается в отдельных приемных бункерах /, 2 и 3,
из которых специальными автоматическими весами дозируется в
смеситель 4. Кокс и антрацит направляются на составление шихты в виде
кусков размером 10—40 мм.
Из смесителя приготовленная шихта ковшовым элеватором 5
подается в загрузочный напорный бункер 6, расположенный над кар- .
бидной печью 8. Периодически, по мере срабатывания шихты; через
специальную систему неподвижных и качающихся рукавов в ванну
(к электродам) подается дополнительное количество шихты из
напорного бункера, и печь загружается до уровня газосборников.
Под действием электрического тока известь и углеродное сырье
нагреваются до температуры реакции. При их взаимодействии в рас-
46
плаве образуются карбид кальция и окись углерода, которая отса
сывается из-под газосборников вентилятором для последующего"
использования при обжиге известняка. В окиси углерода всегда
присутствует водород, образующийся при разложении воды,
поступающей в ванну печи в небольшом количестве вместе с шихтой. Несмотря
на наличие газосборников, сверху печи через неплотности около
электродов, а также при загрузке шихты всегда выделяется много
пыли и газов, для отсоса которых устанавливается специальный
вентилятор. За вентилятором имеется промывочный скруббер для
Антрацит 5 улавливания пыли из дымо-
ИзВесть
Честь Лнтрса
I т I
/
2
3
вых газов, выходящих в
атмосферу (на рисунке не
показан).
Дымовые^
газы
Рис. П-2. Технологическая схема производства карбида кальция:
1, 2, 3 — приемные бункеры; 4 — смеситель; 5 — ковшовый элеватор; 6 —
загрузочный напорный бункер; 7 — вентилятор; 8 — карбидная печь; 9 — сливное корыто;
10 — барабан; 11 — магнитный сепаратор.
Печной газ кроме окиси углерода (70—75%) содержит до 15%
водорода, 1—3% кислорода и 10—12% азота. Калорийность такого
газа сравнительно невелика (до 1800 ккал/мг). Химическое
использование его затруднено наличием воздуха и других
примесей.
Для питания карбидных печей переменным током применяются
трансформаторы высокой мощности. Например, при производстве
карбида кальция с одной печи в количестве 100 тыс. т в год и более
используются трансформаторы мощностью 64 тыс. кет на ток до
120 тыс. а с 24 ступенями напряжения от 140 до 240 в. При переходе
с одной ступени напряжения на другую производительность печи
увеличивается; полная мощность достигается при 240 в. Сравнительно
невысокое напряжение на электродах позволяет уменьшить утечку
тока из зоны реакции на кожух печи и тем самым увеличить срок
безремонтного пробега ванны.
В карбидном производстве применяются печи с тремя электродами
(по числу фаз питающего тока). Электроды располагают по одной
прямой или под углом в виде треугольника. Печи обоих типов имеют
равноценные технико-экономические показатели, однако
расположение электродов по одной прямой несколько удобнее для подачи
47
шихты в ванну. Кроме того, при таком расположении электродов
легче выпускать из ванны карбидный плав, а следовательно, для этой
печи снижаются требования к качеству шихты. Крупная карбидная
печь с электродами, расположенными по одной прямой, имеет
футерованную ванну 8x6 ж глубиной 2 м. В качестве футеровочного
материала используется высококачественный корунд. Под печи
выкладывается угольными и графитовыми блоками.
В настоящее время во всех карбидных печах применяют
самоспекающиеся угольные электроды. В мощных печах высота таких
электродов достигает 10 м, сечение — 2,8x0,65 м. На электродах
имеется стальной кожух, в который со специальной изолированной
площадки периодически подается готовая электродная масса. Весь
электрод висит в ванне на нескольких стальных лентах,
закрепленных на лебедке, управляемой автоматически. При работе печи
нижняя часть электрода срабатывается, поскольку углерод электрода
частично расходуется на образование карбида кальция. При этом
увеличивается напряжение между электродом и подом печи, на
управление лебедки поступает сигнал и электрод автоматически
опускается на нужную глубину для восстановления прежней силы
тока.
Ток к электродам подводится через гибкие шины и токоподво-
дящие кольца, охлаждаемые водой. При каждом опускании
электродов токоподводящие кольца переставляются выше.
Обычно самоспекающиеся электроды подпитывают электродной
массой, изготовляемой непосредственно в помещении карбидного
производства на специальном оборудовании. Для приготовления
электродной массы расходуются кокс, особо обработанный антрацит
и смолопек. Куски готовой электродной массы закладывают в
кожухи электродов. При опускании электродов в зону высоких
температур вначале происходит расплавление и образование монолитного
электрода, а затем коксование и частичная графитизация. В зоне
реакции ток протекает уже по твердому электроду.
Карбидный плав обычно выпускают из ванны периодически,
по мере накопления. Предварительно расплавляют корку твердого
карбида в летке, для чего устанавливают вспомогательные
переносные или чаще подвесные графитовые электроды, питаемые током от
небольших специально предназначенных для этой цели
трансформаторов. Из леток плав направляется в специальные изложницы или
во вращающийся охлаждаемый барабан.
В случае использования изложниц для приема плава устраивают
летки под каждым электродом, что позволяет полнее опорожнять
ванну. После охлаждения карбид кальция извлекают из изложниц'
и направляют на дробление и упаковку.
При наличии вращающихся барабанов плав обычно выпускают
через одно отверстие в поде печи. Барабаны изготовляют из
жаростойких сталей. В печах производительностью 100—120 тыс. т СаС2
в год устанавливают барабаны диаметром 1,2 м и длиной 50 м. В
барабанах карбид кальция получается в гранулированном виде. После
48
отсева от пыли и мелочи гранулированный продукт готов к
переработке в ацетилен.
В настоящее время в производстве используются как изложницы,
так и вращающиеся охлаждаемые барабаны. Использование
барабанов является одним из видов механизации карбидного производства,
однако, как показала практика, в этом случае до 40% карбида
кальция получается в виде пыли и мелочи. При непосредственной
переработке на этом же предприятии карбида кальция в ацетилен эту мелочь
и пыль можно использовать. Но перевозка мелочи и пыли для
использования на другом заводе затруднительна в связи с выделением
ацетилена, так как мелкий карбид кальция, имеющий большую
поверхность, легче адсорбирует атмосферную влагу.
Усовершенствования в производстве карбида кальция
Основным направлением в развитии производства карбида
кальция является создание крупных карбидных заводов. В 1954 г. в СССР
были введены в эксплуатацию механизированные пересыпные шахт-
Рис. П-3. Схема закрытой карбидной печи с вращающейся ванной:
/ — сливные изложницы; 2 — предохранительный сливной экран; 3 — зонд отсоса газов из
летки печи; 4 — трубы для отвода реакционных газов; 5 — гибкие шины; 6 — трубы для
шихты; 7 — однофазные трансформаторы; 8 — бункеры для шихты; 9 — гидравлические
устройства для перемещения электродов; 10 — механизм перепуска электродов; 11 —
самоспекающиеся электроды; 12 — закрытый свод с водяным охлаждением; 13 — устройство для
прожига летки; 14 — механизм вращения ванны; 15 — опорная плита ванны; 16 —
контактные плиты.
В. Н. Антонов
49
ные печи для обжига известняка производительностью 100—130 т
обожженной извести в сутки3. Одновременно начали применять
полузакрытые карбидные печи мощностью 40 Мва с механизированным
сливом и охлаждением, имеющие специальные устройства для
грануляции карбида кальция. Эти печи оборудованы установками для
улавливания и очистки реакционных газов и устройствами для
регулирования потребляемой электроэнергии в процессе работы.
За последнее десятилетие достигнуты значительные успехи в
области усовершенствования конструкций карбидных печей3"5. Так,
на некоторых заводах ФРГ, Норвегии и США разработаны карбидные
печи с более высокой степенью механизации и автоматизации
управления. Особое внимание обращено на улучшение санитарных
условий труда. На карбидном заводе фирмы Demag (ФРГ, г. Тросберг)
в 1952 г. была построена полностью закрытая печь мощностью до
20 Мва, снабженная вращающейся ванной (рис. П-3). Позднее была
построена вторая печь мощностью 42 Мва производительностью 260 т
карбида кальция в сутки5.
На карбидном заводе фирмы Knapsack-Griesheim (ФРГ) работают
две закрытые печи конструкции фирмы Demag мощностью 47,7 Мва,
построенные в 1955—1958 гг.6»7. При работе этих печей
осуществляется автоматический непрерывный слив карбидного плава в
изложницы и достигается практически полное улавливание окиси
углерода (в открытых печах она сгорает). В реакционных газах из
закрытых печей содержится 85—87% СО и 6—12% Н2 (остальное СО2
и N2). После надлежащей очистки эти газы можно использовать
для органических синтезов.
Сравнительные данные о работе печей разных конструкций приве-'
дены ниже:
Закрытая Печь с
газопечь сборниками
Литраж * карбида кальция, л/кг 308 300
Расход на 1 т СаС2 (условный литраж
250 л/кг):
известь, кг 916 930—950
(96% СаО) (95% СаО)
кокс, кг 650 580—600
(91—92% С) (85% С)
электроэнергия, квт-ч 2940 2800—2900
* Литраж — число литров ацетилена (при 20° С и 760 мм рт. ст.),
образующегося при полном разложении водой 1 кг карбида кальция.
Как видно из приведенных технико-экономических показателей,
усовершенствованные закрытые печи по расходным показателям
мало отличаются от обычных крупных печей, поскольку схема
использования электрической энергии и, следовательно, баланс тепла
практически не изменились. Однако применение
усовершенствованных печей позволяет полнее автоматизировать процесс получения
карбида кальция и создать наиболее безопасные условия
эксплуатации. Обслуживание усовершенствованных печей сводится к
прожигу леток с помощью специального электрода и к дистанционному
50
управлению технологическими аппаратами. За счет улучшения
санитарных условий и повышения производительности труда уменьшаются
эксплуатационные расходы на 10—15%, что приводит к снижению
себестоимости карбида кальция.
При сравнительной оценке достигнутых технологических
показателей следует иметь в виду более жесткие требования к качеству
используемых в усовершенствованных печах сырьевых материалов.
В частности, в таких печах применяются зерна малого размера (кокс
3—40 мм и известь 3—80 мм).
Производительность усовершенствованных печей может
изменяться в зависимости от диапазона потребления активной
мощности, создаваемого без отключения печи, путем регулирования
напряжения на понижающей стороне трансформатора. Это позволяет
не отключать трансформатор при опускании самоспекающихся
электродов.
На 1 т СаС2 в закрытых печах получается от 350 до 380 м3
отходящих газов (содержание карбидной пыли в них 80—250 г/м3).
Газы очищают от пыли при повышенной температуре в пористых
жестких фильтрах, которые периодически продувают очищенным газом.
Часть отходящего газа удаляется для дальнейшего использования,
а остальное количество компримируется и возвращается в цикл для
очистки фильтров. Установка работает автоматически и
обеспечивает практически полную очистку газов от пыли (конечное
содержание пыли до 5 мг/м8). Очищенные реакционные газы используют для
синтеза аммиака (до 150 кг NH3 на 1 т СаС2).
Электродуговые карбидные печи типа Demag построены и в
других странах, например в Японии8 (мощность печи 27 Мва).
В США9'10 на заводе фирмы Air Reduction Со. (г. Калверт-Сити)
в 1955 г. были пущены трехфазные электрические печи мощностью
39 Мва с вращающимися ваннами (конструкция фирмы Elektrokemisk,
Норвегия). Каждая печь питается током от трех однофазных
трансформаторов мощностью по 13 Мва. Диаметр ванны 9 м. Ванна
окружена металлическим кожухом и футерована огнеупорным
кирпичом.
Она расположена на широкой круговой площадке, снабженной
катками, которые движутся по круговому монорельсу,
укрепленному на специальной опоре. Электроды перемещают электрической
лебедкой.
Суточная выработка трехфазной печи составляет в среднем 225 т
карбида кальция. Загрузка сырья производится автоматически. После
смешения кокса и извести в соответствующих пропорциях шихта
поступает через питатель в ряд ковшей, движущихся с помощью
бесконечной цепи на катках по рельсовому пути. Каждый ковш
автоматически разгружается в момент прохождения
соответствующей точки ванны. Вокруг ванны расположено девять загрузочных
отверстий. Пустые ковши автоматически возвращаются к питателям.
Выпуск и розлив карбида по изложницам также происходит
автоматически. Карбидный плав застывает, образуя слитки 20 х50 см
4* ' 51
и толщиной 5 см. Для транспортирования слитков к месту
дробления и охлаждения используют специальные конвейеры.
В закрытых печах с механизированными загрузкой шихты,
сливом и транспортированием карбидного плава можно
автоматизировать процесс и облегчить условия эксплуатации.
Основные показатели карбидного производства в г. Калверт-
Сити следующие:
Средняя активная мощность каждой печи,
Мва 30
Литраж карбида кальция, л/кг 270—285
Расход на 1 т СаС2 (условный литраж
250 л/кг):
электроэнергия, квт-ч 2900—3000
электродная масса, кг 22—24
Требования к сырью и качество
получаемого карбида кальция
Количество примесей в извести и углеродистом материале
довольно жестко лимитируется. Особенно ограничивается в извести
наличие фосфора (не более 0,01% в пересчете на Р) и магния (не
более 1,8% в пересчете на MgO). При наличии воды соединения фосфора
способствуют образованию фосфористого водорода, вызывающего
самовозгорание ацетилена, а окись магния резко повышает
температуру плавления шихты.
Обычный состав извести, применяемой в производстве карбида
кальция, следующий (в %):
СаО 94,0 MgO 0,77
СаСО3 2,61 Другие окислы ме-
SiO2 1,19 таллов .... 0,74
S 0,68 Р 0,01
Содержание примесей в извести ограничивается еще и потому,
что они вызывают дополнительный расход электроэнергии и кокса
при получении карбида кальция, что видно из следующих данных
(расчет на 1 кг примесей в 1 т извести):
Расход Расход
электро- Расход электро- Расход
энергии кокса, кг энергии кокса, кг
кет • ч кет • ч
MgO 12 0,4 Fe2O3 2 0,3
А12О3 .... 8 0,5 СО2 1,5 0,27
SiO2 .... 7 0,5 Н2О 1,0 0,5
Состав углеродистого материала (кокса и антрацита) колеблется
в зависимости от месторождения угля. Вредной примесью,
неблагоприятно отражающейся на протекании реакции образования
карбида кальция, является зола. В коксе ее имеется от 9 до 13%.
Зольность антрацита меняется от 5 до 13%. Углеродсодержащие
материалы с большей зольностью обычно в карбидном производстве не
используются.
52
При повышении зольности й влажности кокса увеличивается
расход электроэнергии3» п. Так, при увеличении зольности кокса на 1 %
расход электроэнергии на 1 тчистого углерода возрастает на 57кет-ч,
а для уменьшения влажности на 1 % требуется дополнительно
затратить 19 квт-ч. Присутствие золы в коксе способствует образованию
сплава железа с кремнием — ферросилиция, что приводит к более
частым остановкам карбидной печи на ремонт.
Лучшим углеродистым материалом для производства карбида
кальция является нефтяной кокс, образующийся при глубоком
крекинге тяжелых-нефтяных остатков. Нефтяной кокс является
малозольным материалом (не более 0,8%) и содержит незначительное
количество влаги и других примесей. В США на ряде карбидных заводов
используется нефтяной кокс. При работе на нефтяном коксе удается
осуществить непрерывный слив карбидного плава и улучшить
показатели работы печей. В качестве углеродистого материала
используется также коксовый «орешек», который на 40% дешевле, чем
металлургический кокс. Довольно эффективным способом снижения
энергоемкости карбидного производства может явиться
использование мелкого кокса и кокса с большим электрическим
сопротивлением, так как при этом уменьшается электрическое сопротивление
ванны печи.
В готовом блочном карбиде кальция обычно присутствуют все
примеси, содержащиеся в обожженной извести и в золе
углеродистого материала. Однако состав соединений, в которые входят эти
примеси, в значительной степени меняется под влиянием высокой
температуры и восстановительной среды в ванне карбидной печи.
Примерный состав блочного карбида кальция приведен ниже (в %):
СаС2 73,9 MgO 0,27
СаО 17,92 Другие окислы ме-
Свободный углерод 1,34 таллов 2,67
o.~ о -г П2 U,UO
SiO2 3,75 рНз Следы
В готовом карбиде кальция присутствуют карбиды и окислы
металлов и кремния, а также ферросилиций, который отстаивается
в карбидном плаве и периодически сливается из ванны печи по мере
накопления. В случае соприкосновения расплавленного
ферросилиция с водой происходят хлопки и выбросы, поэтому при сливе плава
из печи во вращающийся барабан принимаю! меры предосторожности.
Нельзя также допускать попадания охлаждающей воды в барабан.
Часто в блочном карбиде кальция присутствуют соединения фосфора
(0,02—0,03% в пересчете на фосфин).
Как показывает практика отечественных и зарубежных заводов,
наименьшие расходные коэффициенты достигаются при получении
карбида высокой степени чистоты и большого литража. Согласно
приведенным в литературе данным3» п-14, наиболее экономично
получать 85%-ный карбид кальция. Уже при получении 80%-ного СаС2
вместо 70%-ного (в результате экономии сырья и электроэнергии)
53
себестоимость получаемого продукта снижается на 7,4%. В
результате повышения литража карбида кальция на 25 л/кг расход извести
сокращается на 10%, расход электроэнергии — на 1%. Для
производства высоколитражного карбида кальций необходимо повышать
чистоту используемого сырья и уменьшать его гранулометрический
состав.
Материальный баланс карбидной печи приведен в табл. П-1.
ТАБЛИЦА П-1
Примерный материальный баланс карбидной печи
(на 1 т товарного продукта)
Приход
Известь
Известняк ....
Окись кремния (с
золой кокса и
известью) . .•
Окись магния . . .
Окислы других
металлов
Сера
Фосфор
Углерод (с коксом
и электродной массой)
Влага (с коксом и
известью)
Всего ...
кг
863,0
23,0
31,8
2,0
10,2
8,0
0,08
478,6
23,2
1439,88
%
59,93
1,60
2,21
0,14
0,71
0,55
0,01
33,24
1,61
100
Расход
Карбидный плав
в том числе
карбид кальция
карбид магния
карбиды
других металлов
окись магния
окись кальция
сульфид
кальция
фосфид кальция
ферросилиций
свободный
углерод ....
Газы
в том числе
окись углерода
водород ....
Пыль
в том числе
окись кальция
карбид кальция
Всего ...
кг
1000
757,72
2,4
16,6
31,2
161,8
18,1
0,24
2,04
10,0
398,6
396,0
2,6
41,28
18,48
22,8
1439,88
%
]
J
1
J
69,45
27,68
- 2,87
100
Расходные показатели крупного производства карбида кальция
в настоящее время достигают следующего уровня (расчет на 1 т,
карбида кальция при литраже 250 л/кг)12:
Электроэнергия, квт*ч
в том числе
на производство СаС2
на привод механизмов и вентиляцию
Обожженная известь (95% СаО), т
Кокс и антрацит (в пересчете на условное топливо), т
Электродная масса, кг
Вода, м3
2935
2750
185
0,875
0,5,2
22—31
70
Поскольку производство карбида кальция освоено сравнительно
давно, дальнейшее снижение расходных показателей протекает
относительно медленно. Все усовершенствования процесса направлены
54
в основном на улучшение условий труда, автоматизацию и
механизацию производства.
Для снижения стоимости карбида кальция целесообразно
кооперировать его производство с содовым производством, а также
использовать отходы извести в строительной, металлургической и других
отраслях промышленности. В этом случае известь сначала
расходуется в производстве карбида кальция, а затем после дегазации
может быть передана для дальнейшего использования.
ПОЛУЧЕНИЕ АЦЕТИЛЕНА ИЗ КАРБИДА КАЛЬЦИЯ
При действии воды на карбид кальция выделяется ацетилен и
образуется гашеная известь:
СаС2 +2Н2О -> С2Н2 +Са (ОН)2 АН = —30,4 ккал/моль
Тепло, выделяющееся при разложении технического карбида
кальция, представляет собой сумму количеств тепла, выделяемого
при взаимодействии с водой карбида кальция и содержащейся в
техническом карбиде негашеной извести. Реакция извести с водой
протекает по уравнению:
СаО + Н2О — Са (ОН)2 А Я - —15,2 ккал/моль
Теоретически для разложения 1 кг химически чистого карбида
кальция нужно затратить 0,562 кг воды; при этом образуется 1,156 кг
гашеной извести и 0,406 кг ацетилена. Таким образом, теоретический
выход сухого ацетилена из химически чистого карбида кальция при
0° С и 760 мм рт. ст. составляет 14'19 372,3 л/кг. Фактический выход
ацетилена из технического карбида кальция вследствие наличия
в последнем примесей и разложения его влагой воздуха колеблется
от 230 до 310 л/кг.
Ниже приведены данные о выходе сухого ацетилена при 20° С
и 760 мм рт. ст. из технического карбида с различным содержанием
СаС2:
Содержание СаС2, % .... 80 75 70 65 60 55
Выход С2Н2, л/кг 298 279 260 242 223 204
Нормы выхода ацетилена при величине кусков карбида кальция
от 2 до 80 мм регламентируются ГОСТ 1460—56. С повышением
размеров выход ацетилена растет, так как мелкие куски сильнее
притягивают атмосферную влагу и вследствие этого теряют ацетилен
во время транспортирования. Значительный тепловой эффект
реакции разложения карбида кальция и нежелательность перегрева
ацетилена приводит к тому, что в практических условиях процесс
разложения ведут с большим избытком воды против теоретического
количества.
Процесс образования ацетилена часто затрудняется из-за
появления корки на кусках карбида кальция, возникающей при
контакте их с гашеной известью, в которую превращается СаС2 при
действии воды. Это приводит к заиливанию и перегреву генератора.
55
Для нормального протекания реакции корку извести
необходимо удалять.
При погружении кусков карбида кальция в воду процесс
разложения протекает непрерывно. Реакция начинается очень бурно,
с большим выделением ацетилена, *затем скорость ее постепенно
снижается. Это тоже связано с образованием корки извести,
препятствующей свободному доступу воды.
При перемешивании реакционной
массы^в генераторе разложение
карбида кальция протекает" быстрее и
более равномерно. С уменьшением раз-
19
I 17
. 15
О
$350
б
О
1
Y,
I
у^
/-"
^-2
^-
3
1
t
5 8 13 17
Время, мин
Рис. П-4. Выход ацетилена при
разложении карбида кальция в
зависимости от его состава и
времени реакции:
/ — 93%-ный СаС2; 2 — 83%-ный
СаС2; 3 — 72%-ный СаС2.
А
—о—
сг
&>
О—<
о-
А
А
0-^
(=и
8==»
/
■о—
te=
Л
1
2
3
Л
-J-C
5\
6
10 20 30 40 50
Температура, °С
Рис. Н-5. Скорость
разложения карбида кальция
в зависимости от
температуры воды и размеров
кусков:
/ — от 2 до 4 мм; 2 — от 4
до 8 мм; 3 — от 8 до 15 мм;
4 — от 15 до 25 мм; 5 — от
25 до 50 мм; 6 — от 50 до
80 мм.
меров кусков возрастает общая поверхность соприкосновения фаз
и, следовательно, скорость разложения повышается.
Графически зависимости скорости разложения карбида кальция от
его состава, времени реакции, температуры воды и размеров кусков
представлены1'13»18 на рис. П-4 и Н-5. На практике процесс
производства ацетилена оценивают по времени, в течение которого выделяется
98% общего количества ацетилена, образующегося при разложении
данного количества карбида кальция. Остаток карбида разлагается
очень медленно, вследствие чего это время практически не
характеризует процесс в условиях работы ацетиленовых генераторов.
Ацетиленовые генераторы
Ацетиленовые генераторы классифицируют по
производительности, предельному давлению вырабатываемого ацетилена и главное —
по способу разложения карбида кальция водой.
Производительность генераторов по ацетилену может быть от 0,8
до 2000 мв/ч и более. Для сварочных работ используют небольшие
56
передвижные генераторы — преимущественно до 80 м3/ч, в
химической промышленности применяют генераторы на 500—2000 м3/ч.
По предельному давлению вырабатываемого ацетилена
генераторы разделяют14-17 на генераторы низкого давления—до 0,1 am
(герметизация с помощью гидравлического затвора), генераторы
среднего давления — от 0,1 до 1,5 am (закрытые аппараты) и
генераторы высокого давления — свыше 1,5 am (только закрытые
аппараты).
По способу разложения карбида кальция водой генераторы
подразделяют на три основные системы:
1. Генераторы системы «карбид в воду» (аппараты непрерывного
действия). Куски карбида сбрасывают в воду, находящуюся в газо-
образователе, и там происходит разложение. Выделяющееся тепло
расходуется в основном на нагревание воды. Образующаяся при
реакции гидроокись кальция удаляется вместе с водой в виде
суспензии;
2. Генераторы системы «вода на карбид». Воду периодически (или
непрерывно) подают на куски карбида, загруженные в газообразо-
ватель. Регулируют процесс разложения, изменяя количество
подаваемой воды. В этих аппаратах можно вести «мокрый» или «сухой»
процесс газообразования. В генераторах «мокрого» типа воду подают
на неподвижный карбид кальция, который полностью разлагается
в присутствии значительного избытка воды. В «сухих» генераторах
вода поступает на непрерывно движущиеся куски карбида кальция,
который находится в избытке по отношению к количеству
подаваемой воды. Отходом является порошкообразная гидроокись кальция
(известь-пушонка);
3. Контактные генераторы. Взаимодействие карбида кальция
с залитой в аппарат водой осуществляется периодически, по мере
расходования ацетилена. Такие генераторы бывают только малой
производительности (использование ацетилена для сварки и резки
металлов).
В генераторах системы «карбид в воду» происходит наиболее
полное разложение карбида кальция с минимальными потерями
ацетилена; в них можно перерабатывать куски любых размеров,
включая мелочь, а также регулировать производительность. Недостатком
этих генераторов являются относительная сложность и громоздкость
загрузочных устройств, высокий расход воды (10 м3 на 1 т С2Н2)
и вследствие этого большое количество отходов (жидкого ила).
Генераторы системы «карбид в воду» применяются преимущественно как
стационарные аппараты большой производительности и находят
самое широкое применение в химической промышленности.
На рис. П-6 показан генератор системы «карбид в воду»
производительностью до 500 м3/ч. Аппарат заполнен водой на 3/4 высоты.
Гранулированный карбид кальция (размер кусков 50—80 мм) из
загрузочных бункеров 1 и 2 поступает в генератор через шахту 4У
нижняя часть которой погружена в воду. Подача карбида
регулируется секторным питателем 3, вращающимся со скоростью 16—18 об/ч.
57
Посредством установленного под шахтой конуса 5 карбид равномерно
распределяется по сечению генератора. Для удаления корки извести
с кусков карбида имеется перемешивающее устройство — вращаю-
Карбид кальция
Ацетилен
т i
Известковое молоко
Рис. П-6. Ацетиленовый генератор системы «карбид в воду»:
/ _ верхний загрузочный бункер; 2 — нижний загрузочный бункер; 3 — секторный
питатель; 4 — шахта; 5 — распределительный конус; 6 — корпус аппарата; 7 — гребки;
8 — полки; 9 — вращающийся вал; 10 — шлюзовой затвор; // — уравнительна^ труба;
12 — гидравлический затвор.
щийся (до 120 об/ч) вертикальный вал 9 с лопастями. На верхней
сетчатой полке 8 куски карбида перемещаются от центра к периферии
и попадают на нижнюю полку, имеющую уклон к центру. При
трении кусков карбида о полки корка извести ссыпается. Гашеная из-
58
весть удаляется из генератора в виде известкового молока по
сифонной трубе 11, являющейся регулятором уровня воды. Тяжелые
примеси карбида кальция (ферросилиций и др.) собираются в нижнем
бункере (шлюзовой затвор 10)у откуда их периодически удаляют.
Более рациональны генераторы системы «вода на карбид» с
«сухим» процессом разложения карбида кальция. Получаемая в них
сухая известь-пушонка (5% влаги) является хорошим строительным
материалом и легко транспортируется. Этим упрощается
использование отходов. Небольшой избыток воды при работе генератора
практически полностью испаряется, что способствует созданию
большей безопасности при эксплуатации аппарата. На испарение
расходуется часть реакционного тепла, поэтому получаемый ацетилен не
перегревается. Он насыщен водяными парами, вследствие чего
значительно менее взрывоопасен. Это является одним из важных
преимуществ генераторов системы «вода на карбид».
В «сухом» генераторе карбид кальция через секторный питатель
и горизонтальный шнек непрерывно поступает на верхнюю полку,
куда разбрызгивающими устройствами подается вода. Карбид
взаимодействует с водой на полках генератора. При большом числе полок
достигается почти полное разложение карбида (на 98%). Сухая
известь выводится из генератора через конусную часть и попадает в
нижний цилиндр. Чтобы предотвратить «зависание» извести, конусная
часть снабжается механическими ворошителями. Температура должна
поддерживаться 50—60° С, для чего в генератор подают холодную
воду. Загрузочные и нижние выгрузные бункеры устроены по
принципу шлюзовых затворов и продуваются азотом. Постоянное
избыточное давление в генераторе не должно превышать 300—400 мм
рт. ст. В случае повышения давления избыточный ацетилен через
гидравлический затвор отводят в атмосферу. Расход воды в «сухих»
генераторах почти в 5 раз меньше, чем в «мокрых».
На рис. Н-7 изображен «сухой» ацетиленовый генератор, в
котором разложение карбида кальция проводят при небольшом избытке
воды. В нижнем цилиндре поддерживается определенный уровень
извести, который препятствует прониканию ацетилена в нижний
шнек. Для окончательной подсушки пушонки в конус и нижний
цилиндр подают пар по специальным рубашкам. Ацетилен отводится
из генератора по трубе, расположенной в конусной части аппарата.
Очистка ацетилена
Неочищенный ацетилен-сырец непригоден для химической
переработки, так как в нем обычно содержатся следующие примеси (в %):
РН3 .
NH3 .
SiH4 .
AsH3
СН4 .
0,01—0,03
. , , % 0,02—0,06
.... 0,01
.... 0,002
0,03—0,5
Н, . . .
О»
N2 . . .
СО . .
со2 . .
. . . 0,1
0,1
. . . 0,4
. . . 0,1
. . . 0,03
Некоторые из указанных веществ (РН3, SiH4, AsH3 и СН4)
образуются при действии воды на примеси, имеющиеся в карбидном
59
Карбид кальция
Азот
'шонка
Рис. И-7. «Сухой» ацетиленовый генератор системы «вода на карбид»:
/ — гребки; 2 — полки; 3 — ворошитель пушонки; 4 — устройство для замера
уровня пушонки; 5 — рубашки; 6 — вал; 7 — стальной корпус,
18
плаве (стр. 53); водород, окислы углерода и аммиак десорбируют из
карбида кальция; кислород и азот вносятся с водой. Кроме того,
в ацетилене могут присутствовать ацетиленовые углеводороды"
(0,001—0,01%), хлорпроизводные ацетиленов (образующиеся в
процессе очистки ацетилена) и сероводород (попадающий в газ при
промывке его серной кислотой).
Карбид кальция
Вода
| СаОС1:
Пушонка ^»
Рис. П-8. Схема очистки ацетилена:
/ — «сухой» ацетиленовый генератор; 2 — скруббер водной промывки; 3 —
очистной скруббер; 4 — щелочной скруббер; 5 — скруббер для осушки ацетилена
серной кислотой; 6 — сборники.
Для удаления наиболее вредных примесей, оказывающих влияние
на последующие химические синтезы с участием ацетилена,
применяют очистные составы и осуществляется промывка ацетилена водой
и щелочными растворами. Обработку ацетилена обычно проводят
в следующем порядке:
а) промывка водой для удаления аммиака;
б) обработка раствором гипохлорита натрия для окисления фос-
фина, сероводорода и мышьяковистого водорода
РН3 + 4NaOCl — Н3РО4 + 4NaCl
H2S +4NaOCl — H2SO4 + 4NaCl
AsH3 + 4NaOCl — H3As04 + 4NaCl
в) промывка щелочным раствором для нейтрализации следов
кислот;
г) осушка серной кислотой или силикагелем.
Иногда для очистки ацетилена используют различные составы
(смеси хлорной извести, солей железа и других соединений). Обычно
применения гипохлорита натрия или кальция достаточно для
получения ацетилена требуемого качества.
На рис. П-8 представлена технологическая схема очистки
ацетилена. Ацетилен из генератора 1 поступает в скруббер 2, где промы-
61
вается теплой оборотной водой для удаления аммиака и части
сероводорода. По мере накопления аммиачных солей оборотную воду
заменяют свежей. В скруббере 3 ацетилен промывается гипохлоритом
натрия или кальция для удаления фосфористого и мышьяковистого
водорода и кремниевых соединений. В скруббере 4 из ацетилена
раствором щелочи вымываются остатки соединений кислотного
характера. В случае необходимости ацетилен в скруббере 5 осушают
серной кислотой или обрабатывают в специальном аппарате силика-
гелем (на рисунке не показано). Очищенный ацетилен направляют
на переработку или собирают в газгольдере.
При «мокром» способе производства ацетилена образуется в
больших количествах карбидный ил. При разложении 1 кг карбида
кальция получается до 2 л тестообразного карбидного ила, содержание
воды в котором может колебаться от 35 до 55 вес. %. Сухой
карбидный ил имеет следующий состав (в вес. %):
Са (ОН)2 . \ . 83—88 CaS 0,6—0,9
СаСО3 .... 4—8 Fe2O3 .... 0,2—0,3
А12О3 .... 2—3 Влага .... 1,5—3,0
Прочие . . . 2—4
* При длительном соприкосновении с воздухом часть
гидроокиси кальция взаимодействует с содержащейся в воздухе
двуокисью углерода с образованием карбоната кальция.
Карбидный ил отводится по трубам или каналам в специальные
ямы, где отстаивается и уплотняется. Заполняют иловые ямы и
сливают из них осветленную воду периодически. Слив осветленной
воды в канализацию недопустим, так как в ней может содержаться^
до 20% ацетилена, уходящего вместе с карбидным илом. Иловые
ямы представляют собой железобетонные резервуары, закрытые
сверху плитами. Ил удаляют оттуда центробежными, диафрагменными
или пульсационнымй насосами (выдавливание сжатым воздухом).
Время отстаивания карбидного ила зависит от количества воды,
затраченной на разложение карбида кальция. В воде, полученной
при отстаивании ила, содержится растворенный ацетилен; кроме
того, часть ацетилена адсорбируется мелкодисперсными частицами
ила. При хранении ила количество содержащегося в нем ацетилена
постепенно уменьшается. Потери ацетилена с карбидным илом
являются основными при эксплуатации генераторов.
Карбидный ил может быть использован в тех случаях, когда
требуется применение гашеной извести, но переработка ила осложняется
наличием в нем большого количества воды. На крупных производствах
ацетилена, территориально связанных с карбидными заводами, ил
можно частично использовать в качестве известкового сырья для
получения карбида кальция (он должен быть предварительно обезвожен).
Карбидный ил используется для строительных работ и в
производстве строительных материалов, в качестве химического реагента в
различных отраслях промышленности, а также в сельском хозяйстве
(для известкования почв, обмазывания стволов фруктовых деревьев,
приготовления защитных растворов медного купороса).
62
При получении ацетилена и его очистке от вредных примесей
дополнительно расходуются электроэнергия и дорогие химические
реагенты (гипохлорит натрия, едкие щелочи и серная кислота).
Увеличиваются также затраты на амортизацию дополнительного
оборудования и оплату обслуживающего персонала.
Суммарные расходные показатели на 1 т очищенного ацетилена
составляют12:
Известняк, т 6,6—7,6
Углеродистое сырье (включая расход на электродную
массу и производство извести), т 2,7—3,0
Карбид кальция (в пересчете на литраж 250 л/кг), т 3,7
Вода, ж3 500—600
Шламовая известь, т 4,4—4,5
Отходящие газы (калорийность 1800 ккал/м3), ж3 ... 1000
Электроэнергия (включая расход на производство
извести), квт-ч 11 000—11 500
ЛИТЕРАТУРА
1. Кузнецов Л. А., Производство карбида кальция, Госхимиздат, 1950.
2. Г о л ы н к е р И. С, Кислород, № 5 (1945).
3. Конюх Г. Д., Вестник НИИТЭхим, № 1 (1959).
4. Chem. Age, 85, № 2172, 327 (1961); Hummel Т., Eeastern Metals Rev., № 29,
333 (1960).
5. Sevin P., J. electrigue, № 6, 191 (1961).
6. H e h 1 e n H., Chem. Techn., 13, № 7/8, 386 (1961).
7. К u ess F., Vogel E., Chem. Ing. Techn., 28, № 12, 759 (1956).
8. Chem. Ing. Techn., 31, № 7, 492 (1959).
9. J. electrigue, № 5, 163 (1958).
10. T у р о в Ю. Я., Ю д о в и ч Е. А., Журн. ВХО им. Д. И. Менделеева, 7,
№ 1 (1962).
11. U dy M. Z., J. Electrochem. Soc, 101, № 12, 622 (1954).
12. Туров Ю. Я., Паршина Г. А., Фомина М. А., в сб.
«Сопоставительные обзоры по отдельным производствам химической промышленности
в СССР и за рубежом», вып. 1, изд. НИИТЭхим, 1967, стр. 34.
13. Т а р а с о в Г. Г., ЖПХ, № 7, (1964).
14. Стрижевский И. И., Гузов С. Г., Ковальский В. А.,
Ацетиленовые станции, Машгиз, 1959.
15. Юкельсон И. И., Технология основного органического синтеза, Изд.
«Химия», 1968.
16. Е р ш о в В. А., А л ь п е р о в и ч И. Г., Хим. пром., № 3 (1967).
17. А й р а п е т я н ц Э. П., Ершов В. А., К а г а н о в а А. К., ЖПХ,
40, № 2, 443 (1967). -
18. Miller S. A., Angew. Chem., 62, № 7, 166 (1950).
19. С т р и ж е в с к и й И. И., Технологические основы и безопасность
производства газообразного и растворенного ацетилена, Изд. «Химия», 1968.
ГЛАВА
HI
ПОЛУЧЕНИЕ АЦЕТИЛЕНА
ПИРОЛИЗОМ УГЛЕВОДОРОДОВ
До второй мировой войны карбид кальция являлся практически
единственным источником получения ацетилена для промышленных
целей. Отсутствие разработанных методов не позволяло
использовать для производства ацетилена большие ресурсы углеводородов
нефти и природного газа, хотя в лабораториях
научно-исследовательских институтов многих стран уже велись обширные исследования
по определению условий превращения низших парафинов в ацетилен.
Между тем пиролиз углеводородов для получения олефинов (этилена
и пропилена), а также термический крекинг углеводородов уже давно
получили промышленное развитие. Постепенное накопление
теоретических и практических сведений позволило создать первые
полупроизводственные установки, а затем и крупное промышленное
производство ацетилена на основе высокотемпературного пиролиза
углеводородного сырья.
Применение высоких температур для расщепления
углеводородов, несмотря на кажущуюся простоту, потребовало разработки
новых способов теплопередачи и использования специальных
огнеупорных материалов. Прямой пиролиз углеводородов реализован в
промышленности сравнительно недавно (1955—1960 гг.) — позже
электрокрекинга метана и карбидного способа производства ацетилена.
Впервые пиролиз углеводородов с целью получения ацетилена
был применен фирмой Wulf Process (США) для расщепления пропана
в регенеративной печи (процесс Вульфа). Дальнейшее развитие
получил метод гомогенного пиролиза, когда в качестве теплоносителя
используют топочные газы, полученные сжиганием углеводородного
сырья в кислороде. По этому способу построены установки в США,
Франции, Италии, ФРГ. В Советском Союзе также реализован метод
гомогенного пиролиза и проведены исследования процесса в
трубчатой печи.
ОСНОВНЫЕ СВЕДЕНИЯ О ПРОЦЕССЕ
Термодинамика процесса
Пиролиз заключается в разложении и различных превращениях
исходных углеводородов при температурах выше 1000° С в
адиабатических условиях за время 0,0005—0,02 сек. В течение этого вре-
64
ыени нужно осуществить нагрев сырья, саму реакцию и охлаждение
образовавшихся продуктов до 200° С. Процесс является1»2 суммой
последовательных и параллельных реакций, и анализ его состоит
главным образом в установлении возможности протекания этих
реакций и их относительной движущей силы.
Как известно, показателем возможности протекания реакции
является изменение свободной энергии (изобарный термодинамический
потенциал):
AG° - ЛЯ° — *
где AG° — свободная энергия системы при постоянном давлении;
АЯ° — изменение энтальпии в результате протекания реакции при
постоянном давлении (теплота реакции);
А5° — изменение энтропии.
Данные расчетов изменения свободной энергии позволяют
установить термодинамическую возможность протекания различных
реакций и относительную стабильность углеводородов в широком
интервале температур. В табл. III-1 приведены температурные
зависимости энергии образования углеводородов, с которыми
приходится наиболее часто встречаться в процессах получения ацетилена3'4.
ТАБЛИЦА III-1
Температурная зависимость энергии образования
некоторых углеводородов
Углеводороды
Метан
Этан
Другие парафины СЛН2/г+2
Этилен
Другие олефины CrtH2rt . .
Ацетилен
Энергия образования
—21 470 + 26,0Г
—24 900+ 51,0Г
—11 260+ 6440/г +
—9 100 + 19,0Г
— 18 940—6440я—33,
+ 53 350—12,7Г
AG, кал/моль
25,6Г
ST + 25,6/гГ
На рис. III-1 эти же зависимости показаны графически. Из
рисунка видно, что относительная стабильность ацетилена, в отличие
от других приведенных углеводородов, с повышением температуры
увеличивается, причем выше 1400° К свободная энергия
образования ацетилена меньше, чем пропана или этана. Это свидетельствует
о возможности образования ацетилена непосредственно из этих
углеводородов в условиях высокой температуры. Например, для
получения ацетилена из метана необходима температура не менее 1500° К.
Высокотемпературный пиролиз углеводородов до ацетилена можно
рассматривать как ряд последовательных обратимых реакций,
первичные продукты которых не связаны непосредственно с конечными
равновесными продуктами. В табл. II1-2 представлены возможные
высокотемпературные реакции образования ацетилена из
различных углеводородов и выражения для их констант равновесия4.
Все эти реакции идут с увеличением объема, поэтому уменьшение
давления (путем снижения общего давления или парциальных давле-
5 в- Н. Антонов 65
ний компонентов при добавлении разбавителей) способствует
разложению углеводородов на промежуточные продукты и ацетилен.
На рис. II1-2 показаны зависимости lg К от температуры для
реакций, приведенных в табл. II1-2. Повышение температуры, как видно
из графиков, способствует разложению исходных углеводородов
(метана, этана, пропана и др.) до промежуточных продуктов, а
последних — до ацетилена. Значения константы равновесия реакции
образования ацетилена из метана
С о Н 9 -\
2СН
4 ч—
ЗНо
гЗ,5
в зависимости от температуры
представлены в табл. III-3. Изпри-
35000
25000
15000
5с О
-2
+5000^
-5000
300 500 700 900 1/00 1300 1500 1700
Температура, °К
Рис. III-1. Температурная зависимость
энергии образования различных
углеводородов из простых веществ (расчет
на 1 атом углерода).
t^
^f
8^
у^-з
4s
-—
^^
<^>
у
1 У
/)
/'
*.—
И
V
^^
-7/
/X
J-
•р
У/
13
Рис. II1-2. Температурная зависимость
констант равновесия реакций,
указанных в табл. II1-2 (цифры на кривых
совпадают с номером реакции в табл.
Ш-2).
веденных данных следует, что ацетилен с повышением температуры
становится термодинамически более устойчивым.
Степень равновесного превращения метана в ацетилен резко
возрастает (от 0 до 99,66%) в интервале температур 500—1800° К,
а при дальнейшем повышении температуры (до 3000° К) меняется
мало; давление в интервале 0,1—1,0 am влияет на состав продуктов
реакции лишь до 1600° К, а при более высоких температурах степень
превращения метана от давления не зависит3 (табл. II1-4).
Аналогичные выводы были сделаны при изучении термического
распада этана и других парафинов. С повышением температуры
процесса и глубины превращения углеводорода непрерывно
увеличивается количество образующегося ацетилена3.
Результаты6 термодинамического расчета реакций разложения
метана и этана до ацетилена и этилена приведены в табл. II1-5.
Из этих данных видно, что, например, теоретически 25%-ное
содержание ацетилена можно получить при пиролизе этана при 902° К,
а при пиролизе метана — лишь при 1407° К. Что же касается
этилена, то при пиролизе метана максимального содержания С2Н4 (33%)
66
ТАБЛИЦА Ш-2
Возможные превращения углеводородов в процессе получения
ацетилена и выражения для констант равновесия этих реакций
№
п/п
Реакция
Константа равновесия
10
2СН4 ^± С2Н2 + ЗН2
с2нб ^ с2н4 + н2
СН4
Н2
С4Н10 ^_1 С2Н4 + С2Н6
С4Н10 +2. С3Нб + СН4
С4Н10 ^zt С4Н8 + Н2
С2Н4 < > С2Н2 + Н2
С2Н2 + СН4
Рсн4
к =
к = -
is ^С2Н4
тг ' ^>3*"
^с4н10
К= °4Н8 Н2
к=^Рс2и2Рн2
„ Рс2и2Рсн4
ТАБЛИЦА III-3
Константы равновесия реакции образования ацетилена
из метана
Температура
500
1000
1200
1400
1600
1800
\gK
—27,905
—7,030
—3,474
—0,888
+0,981
+2,504
Температура
°К
2000
2200
2400
2800
3000
\gK
+ 3,645
+4,620
+5,416
+6,093
+7,181
67
ТАБЛИЦА III-4
Степень превращения метана в ацетилен
Температура
°К
500
1000
1200
1400
1600
1800
Степень превращения *
%
при 0,1 am
0,000
4,840
35,200
85,450
97,970
99,660
при 1 am
0,000
1,530
11,800
46,200
84,000
96,540
Температура
°К
2000
2200
2400
2600
2800
3000
Степень
превращения *
при 0,1 am
99,900
99,968
99,987
99,994
99,997
99,998
, %
при 1 am
99,030
99,680
99,870
99,963
99,971
99,983
* При стехиометрическом соотношении компонентов.
ТАБЛИЦА III-5
Результаты термодинамического расчета реакций образования ацетилена
и этилена при 1 am
Температура, °К
2cpj —>. С Н 4-
+зн2
806
853
893
913
940
1017
1107
1214
- 1407
1407
—
с2нв^->с2н2 +
+ 2Н2
656
681
705
723
737
783
827
864
895
902
956
1064
—
2СН4 —>C2H4-t-
+ 2Н2
703
747
792
823
837
948
1052
1162
1267
1297
1527
1527
—
с2н6 ^-> с2н4 +
+н2
448
475
493
510
523
567
607
642
669
671
698
724
735
777
841
902
1055
Содержание
ацетилена
(или этилена)
в равновесной
смеси, мол. %
2
3
4
5
6
10
15
20
24
25 .
30
33
35
40
45
47,5
49,5
можно добиться только при 1527° К; при использовании этана такое
количество этилена отмечается уже при 724° К. Следовательно,
с точки зрения термодинамики для получения ацетилена этан более
выгодное сырье, чем метан. Практический выход ацетилена
значительно меньше, чем это следует из термодинамических расчетов, из-за
протекания различных побочных реакций.
Расчетные кривые изменения равновесного- состава продуктов
пиролиза этана, пропана и бутана в зависимости от избыточного
давления и температуры представлены7 на рис. II1-3. Анализ этих
кривых показывает, что при 900° К главными продуктами распада
68
C2H2
2n4
2н6
0,5 am
1,0am
Рис. 111-3. Расчетный равновесный
состав продуктов пиролиза бутана (а),
пропана (б) и этана (в).
900 1100 1300
Т, °К
а
69
этана являются метан (70%) и этилен (30%), а ацетилена в смеси
имеется всего около 2%; при 1300° К содержание ацетилена
достигает 17%. В случае разложения пропана и бутана при 1300° К
наряду с основными продуктами (метаном, этиленом, ацетиленом и
водородом) в смеси имеются пропилен, бутилен, бутадиен и метилаце-
тилен С3Н4.
Кроме реакций, указанных в табл. 111-2, при пиролизе могут
протекать реакции образования этана3 и ароматических углеводородов,
например бензола:
2СН4 ^ С2Н6 + Н2
6СН4 ^ С6Н6 +9Н2
Образуются также высшие ацетиленовые углеводороды, но
термодинамические условия образования этих веществ еще мало
исследованы.
При пиролизе большое значение имеют реакции крекинга
исходных углеводородов на простые вещества5:
СН4 ^ С +2Н2
С2Н6 ^ 2С +ЗН2
С2Н4 ^ 2С +2Н2
Константы равновесия этих реакций приведены5 в табл. II1-6,
откуда видно, что термодинамически более устойчив этилен. Вообще
в ряду парафиновых углеводородов склонность к крекингу с
выделением углерода повышается с увеличением их молекулярного веса.
ТАБЛИЦА III-6
Константы равновесия реакций крекинга углеводородов на простые
вещества
Температура, °к
500
600
700
800
900
1000
1100
1200
1300
1400
1500
lg*
СН4^С+2Н2
—3,684
—3,088
—2,655
—2,325
—2,062
— 1,849
—1,673
—1,525
—1,398
—1,289
—1,195
/~» т_т ^. of"» | от_т
—6,783
-. —2,722
— 1,839
— 1,171
—0,650
—0,232
+ 0,112
+0,397
+ 0,649
+0,845
+ 1,025
С,Н4^2С+2Н2
— 17,981
— 14,433
— 11,888
—9,974
—8,482
—7,287
—6,310
—5,494
—4,816
—4,213
—3,702
Получающийся в процессе пиролиза ацетилен при высокой
температуре также способен к разложению на простые вещества:
C2HS
2С +Н3
70
Это следует иметь в виду при выборе оптимальных условий
пиролиза. Давление на реакцию разложения ацетилена не оказывает
влияния, поскольку процесс идет без изменения объема, а повышение
температуры препятствует этой реакции; поэтому, чем выше
температура пиролиза, тем больше ацетилена можно получить в
равновесной смеси за счет уменьшения его разложения3 (табл. III-7).
ТАБЛИЦА II1-7
Равновесные концентрации ацетилена и значения lg К при реакции
•С2Н2 ^ 2С + Н2
Температура
°С
500
1000
1200
1400
1600
1800
2000
2200
lg- Л'
—20,899
—8,993
—7,025
—5,637
—4,594
—3,763
—3,142
—2,613
Концентрация с2н2
объемы. %
—
—
0,003
0,02
0,08
0,24
Температура
°С
2400
2600
2800
3000
3500
4000
4500
5000
lg К
—2,186
— 1,824
— 1,515
— 1,260
—0,741
—0,368
—.
+ 0,136
Концентрация с2н2
объемн. %
0,65
1,47
3,0
5,2
15,4
30,0
45,0
57,8
Механизм и кинетика процесса
Механизм процесса пиролиза. Термический крекинг
углеводородов является сложным многостадийным процессом, при котором
протекают молекулярные и радикально-цепные реакции. Наиболее полно
исследован процесс низкотемпературного крекинга углеводородов
(в интервале 400—700° С). С точки зрения современных
представлений низкотемпературный крекинг является радикально-цепным
процессом, в котором молекулярные реакции имеют подчиненное
значение. В пользу цепного механизма крекинга при нормальном или
пониженном давлении, по крайней мере для простейших парафинов,
свидетельствует уменьшение константы скорости мономолекулярной
реакции разложения с увеличением глубины превращения. Скорость
суммарного распада парафинов описывается уравнением Динцеса и
Фроста г:
4
где к — константа скорости;
т — время реакции;
х — количество распавшегося углеводорода;
|3 — константа, характеризующая торможение реакции крекинга;
Такое самоторможение объясняется обрывом реакционных цепей
некоторыми продуктами реакции. Изучение зависимости состава
продуктов крекинга газообразных парафинов от температуры,
давления и глубины превращения также позволяет сделать вывод о
самотормозящемся характере распада и его радикально-цепном механизме.
В отличие от обычных мономолекулярных реакций константа ско-
71
рости крекинга газообразных углеводородов несколько увеличивается
с ростом парциального давления, что также можно объяснить
цепным характером реакций.
При небольшой глубине превращения парафинов первичные
реакции их термического распада носят молекулярный характер и
описываются следующими стехиометрическими уравнениями (пит —
целые числа):
f-2 —
12П1
(п+т)
Возможные направления распада парафинов, число которых
возрастает с увеличением пит, определяются либо экспериментально,
либо расчетным путем на основании известных термодинамических
ТАБЛИЦА I1I-8
Кинетические данные по распаду парафинов
Углеводород
Метан ....
Этан
Пропан . . .
я-Бутан . . .
Изобутан . . .
я-Пентан . . .
я-Гексан . . .
Интервал
температур
°С
1249—2046
600—700
587—679
600—650
600—650
425—575
525—565
Предэкспо-
нента А
12,00
15,12
13,44
14,14
14,20
13,40
14,22
Энергия
активации Е
ккал/моль
79,4
73,2
62,0
65,0
65,0
61,3
64,5
соотношений. В табл. II1-8 приведены кинетические
характеристики2 — предэкспонента А и энергия активации £, входящие в
уравнение Аррениуса для константы скорости k реакции распада
парафинов:
k = Ae~EIRT
Механизм высокотемпературного разложения углеводородов
изучен недостаточно, поэтому отсутствует единая теория образования
ацетилена при термическом разложении углеводородов в интервале
1300—1500° С. Однако существующие исследования позволяют
высказать предположения об изменении механизма крекинга при переходе
к более высоким температурам: замедляются реакции развития цепей
и ускоряются процессы молекулярной деструкции1»2. Вторичные
реакции образования конденсированных продуктов и кокса протекают
с участием радикалов при 900—1000° С. Однако при еще более
высоких температурах наблюдаются молекулярные реакции распада
с образованием водорода, сажи и ацетилена, ускоряемые
кристаллическими зародышами углерода. Так, при исследовании распада
метана над раскаленной до 1500—1700° С угольной нитью
(образующиеся продукты немедленно выводили из сферы реакции) основным
72
первичным продуктом превращения являлся этан, что согласуется
с предложенным Касселем механизмом этого процесса:
СН4 ■—> 'СН2' -{- Н2
•СН2- + СН4 —> С2Нб
С2Н6 —> С2Н4 -f- Н2
С2Н4 -> С2Н2 +Н2
С2Н2 -> 2С +Н2
Кинетика разложения исходных углеводородов. Выход ацетилена
в процессе пиролиза углеводородов определяется кинетикой
протекающих реакций: скорость
образования ацетилена должна превышать
скорость его разложения на простые
вещества. Поэтому для получения высоких
выходов ацетилена необходимо
подавлять нежелательные реакции,
например путем резкого снижения
температуры образующихся газов.
Разложение метана является,
согласно данным Касселя и др., реакцией
юоо
500
300
200
100
50
30
20
^ 10
СП
~ 5
3
2
0,5
0,3
0,2
0,1
I
\фП,230^Ц\
\
I
I
II
V/
//
I
\\
//
.ft
f
1800
% то
1000,
Метан
Углерод
Бензол
т ш , к.
0,01 0,10
Время, сек
Рис. 111-4. Температурная
зависимость константы скорости
реакции разложения метана.
Рис. 111-5. Образование различных
продуктов при пиролизе метана.
первого порядка6»8, скорость которой может быть представлена
следующим уравнением, справедливым до 1500° К:
f-
dx
или
Q
In ■—- = k (т2 —
^2
где Сг и С2 — концентрации метана на входе в зону реакции и на выходе из нее,
мольные доли;
(т2 — хг) — время реакции, сек.
Зависимость константы скорости этой реакции от температуры
описывается следующими уравнениями (рис. Ш-4):
=. 11,864 —
17 352
\gk= 11,230 —
15700
73
Последнее уравнение выведено4 на основе анализа многочисленных
данных и справедливо для разложения метана в большом интервале
температур, давлений и концентраций.
На рис. 111-5 схематически показаны области образования
различных продуктов пиролиза метана в зависимости от температуры и
времени контакта8. При 1500° С и времени реакции 0,01 сек,
происходит в основном образование ацетилена, с увеличением времени
реакции ацетилен распадается с выделением углерода. При более
низких температурах и более продолжительном нагреве возможно
появление бензола.
В результате пиролиза этана при 650—700° С и времени реакции
более 0,25 сек в основном получаются водород, метан и жидкие
продукты. Ацетилен образуется в малых количествах. Начиная с 1000° С
ацетилен становится основным компонентом, этан отсутствует и
обнаруживается только при очень малом времени контакта. Например,
при 1400° С и времени контакта 0,56 мсек концентрация этана
незначительна.
Уравнение скорости разложения этана может быть представлено
в виде4:
dCt
с2нв
dx
= «ьсянв
Значения k при разных температурах приведены4 в табл. III-9.
ТАБЛИЦА 111-9
Константы скорости пиролиза этана
Время
реакции
сек
Константа
скорости
сек'1
Время
реакции
сек
Константа
скорости
сек'1
При 750° С и 750 мм рт. ст.
0,38
2,90
13,10
1,74
0,50
0,13
При 1400° С и 50 мм рт. ст.
0,28-Ю-3
0,41-Ю-3
0,88-Ю-3
7800
9550
5850
Термическое разложение этилена в интервале до 750° С и при
низких давлениях (до 25 мм рт. ст.) протекает по реакции первого
порядка, а при более высоких давлениях — по реакции второго
порядка. До 600° С ацетилен не присутствует в продуктах реакции,
а начиная с 800° С является уже основным продуктом разложения
этилена. С увеличением температуры повышается скорость
разложения этилена и степень его конверсии до ацетилена. При температуре
выше 1000° С время контакта измеряется миллисекундами и
наблюдается интенсивное сажеобразование. Уменьшение давления и
разбавители замедляют скорость разложения и полимеризации этилена
и повышают выход ацетилена.
Пиролиз пропана протекает по реакции первого порядка 4
dCr м
й-х
74
и значения константы скорости, рассчитанные по уравнению4
-13.46
соответствуют экспериментальным данным, определенным для
температуры выше 700° С.
Разложение пропана идет в двух основных направлениях:
С3Н8 ~^_ С2Н4 + СН4
С3Н8 Tit С3Н6 + Н2
Представляется вероятным, что образующийся пропилен в
условиях реакции разлагается на этилен и метан. Заметное влияние на
состав продуктов оказывает температура: при 600—700° С
получаются этилен и пропилен в соотношении примерно 1:1; выше 1000° С
достигается большой выход ацетилена в результате разложения
этилена и пропилена. Ацетилен при пиролизе пропана получается в
результате расщепления первичного продукта реакции — этилена и,
возможно, пропилена.
Изменение давления от 1 до 7 am мало влияет на состав конечных
продуктов пиролиза парафинов, но при давлении ниже атмосферного
состав продуктов начинает зависеть от начального парциального
давления парафинов. Поэтому при пиролизе метана, этана и пропана
условия для получения высоких выходов ацетилена одинаковы:
низкое парциальное давление углеводорода, высокая температура
и небольшое время пребывания в зоне высоких температур.
Пиролиз бутанов протекает в тех же условиях, что и пиролиз
пропана. Продуктами разложения н-бутана являются бутен-1, бу-
тен-2, водород, метан, этан, этилен и пропилен. Температура выше
700° С способствует образованию этилена; при более низкой
температуре получается пропилен.
В промышленных условиях широко применяется пиролиз смесей
газообразных углеводородов: этана и пропана, пропана и
пропилена, бутана и бутилена и др. Кинетика пиролиза таких смесей
исследована недостаточно, однако можно предположить, что
разложение любого углеводорода в смеси зависит от присутствия других
компонентов.
Отмечалось как инициирующее, так и ингибирующее действие
одного углеводорода на другой9. Так, в опытах по пиролизу этана
с примесью пропилена было установлено, что в присутствии 9% С3Нб
константа скорости разложения этана уменьшается в 10 раз при
700° С и в 3 раза при 775° С по сравнению с процессом в отсутствие
пропилена. Кроме того, при добавке пропилена уменьшается
скорость образования бутана.
Кинетика образования и разложения ацетилена. Образование
ацетилена при пиролизе метана начинается при температуре выше
1200° С, причем время реакции составляет несколько десятков
миллисекунд. Однако для достижения больших степеней превращения
75
требуются более высокая температура (около 1700 С) и
время реакции (порядка десятых и сотых долей миллисекунд):
температурам 1700—1800° С и времени реакции 0,1—0,5 мсек
соответствует степень превращения 60—80%. Выход ацетилена
возрастает с увеличением температуры и
уменьшением времени реакции10 (рис. III-6).
Имеется определенный оптимум времени
реакции, который соответствует
максимальному выходу ацетилена для данной
температуры. С отклонением от этого
оптимума выход ацетилена уменьшается.
Образование ацетилена при пиролизе
метана является реакцией первого
порядка9"12. Если начальная концентрация
метана составляет Со, то концентрация его
-J в момент т равна С:
1200 /400 1600
Температура, °С
i i i i i
О 0,2 0,2 0,0050,00k
Время, се/с
С =
2)
и г- =
Рис. II1-6. Зависимость
степени конверсии метана в
ацетилен от оптимального
времени реакции при разной
температуре.
где k — константа скорости
образования ацетилена, a k2 — константа
скорости разложения метана на простые
вещества.
Количество метана, превратившегося в ацетилен, равно:
С =±(С0-
Фактически количество ацетилена меньше получаемого из метана
вследствие частичного распада ацетилена:
dC I r>i
С = Ce-k
dx
Отсюда
° - 2(*1 + *а) L
Где fc3 — константа скорости разложения ацетилена.
Максимальная концентрация ацетилена Смгкс при оптимальном
времени реакции равна:
1 1.. ki + k2 + k3
^макс —
In
Величина полезного крекинга т), т. е. степень превращения
метана до ацетилена (стр. 81)
2С" h
. e-ksT
при т —» 0 стремится к 100%.
76
Имеется ряд эмпирических формул, связывающих скорость
реакции с парциальным давлением метана, общим давлением смеси и др.,
на основе которых делались попытки определить оптимальные
кинетические условия образования ацетилена3.
Кинетика образования ацетилена из других низших парафинов
и олефинов в основном подчиняется тем же законам, но с поправкой
на влияние температуры при увеличении молекулярного веса
углеводородов.
При температурах ниже 600° С ацетилен преимущественно поли-
меризуется с образованием жидких и твердых веществ.
Полимеризация ацетилена является гомогенной реакцией второго порядка.
При повышении температуры выше 600° С одновременно с
полимеризацией происходит разложение ацетилена на простые вещества:
С2Н2 ^ 2С + Н2
На начальной стадии при 400—500° С разложение ацетилена
является относительно медленной реакцией, затем скорость ее
довольно быстро возрастает, пока не достигнет максимума.
Максимальная скорость обычно в 3—5 раз больше начальной и устанавливается
только после того, как в реакцию вступит 1—2% ацетилена. В
течение короткого промежутка времени скорость максимальна и почти
постоянна, а затем по мере расходования оставшегося ацетилена
снова медленно снижается.
Разложение ацетилена в широком интервале температур и
давлений является реакцией второго порядка. Константу скорости этой
реакции можно подсчитать по уравнению11'12:
\ и л ал 6540
lg Л = 4,64 y~
На начальных стадиях реакции в основном образуются полимеры,
а также бензол, этилен и метан. При высоких степенях разложения
количество полимеров и бензола резко уменьшается и значительно
увеличивается количество метана. Спектрографические исследования
показывают присутствие фенантрена, хризена, пирена и 1,2-бензпи-
рена.
Изучение термического разложения ацетилена, меченного
углеродом14 С, в смеси с этиленом при 800—1000° С и 100 мм рт. ст.
показало, что ацетилен в исследованных условиях нестабилен и
разлагается с образованием различных продуктов. Основными из них
являются водород, метан, этилен, бутадиен, бензол и кокс. В
значительно меньших количествах получаются этан, пропилен, пропадиен,
метил- и винилацетилен, толуол. Причем большая часть
радиоактивного углерода (от 0,5 до 0,75 от общего количества) переходит
в кокс, бензол, винилацетилен, этилен и метан. Это свидетельствует
о весьма важной роли ацетилена в реакциях образования кокса при
высокой температуре2.
Добавки азота, водяного пара и бензола влияют на ход реакции
незначительно. Добавление окиси азота уменьшает скорость разло-
77
жения ацетилена. Аналогичный эффект оказывают й галоидсодержа-
щие соединения, причем действие их зависит как от природы, так и
от количества добавки (табл. Ш-10). Между влиянием окиси азота и
галоидсодержащих соединений, однако, имеется существенное
различие. В присутствии окиси азота состав продуктов разложения
ацетилена не меняется, а при введении галоидсодержащих соединений
(особенно бромистого этила) образование бензола и полимеров
ацетилена почти полностью прекращается и в основном появляются
этилен и метан.
ТАБЛИЦА 111-10
Влияние галоидсодержащих соединений на скорость
разложения ацетилена при 413° С и 250—400 мм рт. ст.
Добавка
Без добавки . . .
Хлористый водород
Хлористый этил . .
Хлористый я-пропил
Бромистый водород
Бромистый этил . .
Концентрация
добавки
объемн. %
0,2
0,4
1,5
2,0
1,0
2,0
2,5
2,0
3,8
0,2
1,6
2,0
Константа
скорости
разложения
ацетилена,сек-1
0,90
0,62
0,57
0,40
0,30
0,83
0,69
0,58
0,86
0,81
0,53
0,05
0,38
При 1200—1500° С скорость разложения ацетилена значительно
меньше скорости его образования, что является основным
кинетическим фактором, позволяющим получать ацетилен. При
температурах выше 2000° С разложение ацетилена на углерод и водород
замедляется, так как в этих условиях достигается термодинамическое
равновесие. Соотношение скоростей образования и разложения
ацетилена можно регулировать изменением общего давления; с
уменьшением давления это соотношение увеличивается (образование
ацетилена ускоряется, а разложение его замедляется). Так, например,
при снижении давления со 150 до 35 мм рт. ст. соотношение
скоростей возрастает в 3 раза.
Процесс пиролиза метана до ацетилена можно представить как
совокупность следующих реакций разложения метана, этана,
этилена и
ацетилена:
1)
2)
3)
4)
2СН4 Zl
с2нв ^
с2н4 zi
С9Н2 ~1
с2
с2
с2
2С
:Нв
н4
н2
+
+ н2
+ н2
;+Н2
н2
кг
k2
k3
k 4
= 4
= 9
С\
= 1
,5-
• 1С
,57
,7-
JQ13e—91 000//?Г
|13в-69 000/ЯГ
. l08^~40000/jRr
106^"3000/jRr
78
Кривые температурных зависимостей констант скоростей
указанных реакций приведены на рис. II1-7. Анализ графика13 объясняет
различные экспериментальные факты. Например, концентрация этана
при термическом разложении метана всегда чрезвычайно мала, а
при высоких температурах в продуктах реакции его совсем не
бывает. Это объясняется тем, что константа скорости разложения этана
k2 в интервале 1500—2500° К почти на три порядка превышает
константу скорости его образования, т. е. константу скорости
разложения метана kx. Из графика видно также, что этилен может присутст-
\
>
ч
ч
\
\
К'"
\
\S
Ui
\
NN
ч N
ч
ч
WOr
гШТооото 1500 /яо/ооо
Рис. III-7. Температурная
зависимость констант
скоростей разложения метана klt
этана k2, этилена k3 и
ацетилена /г4; пунктир
соответствует данным, полученным
экстраполяцией.
I75
or
% 50
r
г
1
к
/ 2 3 A 5
Время реакции £ мсек
Рис. Ш-8. Зависимость степени
конверсии е метана в ацетилен
при 1900° К от времени реакции.
вовать в продуктах термического
разложения метана при
температурах около 1500° К, причем при
более высоких температурах его количество возрастает, так как
константа скорости разложения этилена k3 растет с температурой
меньше, чем константа скорости его образования k2. Максимальное
превращение метана в ацетилен происходит при температуре выше
2000° К, так как в этих условиях отношение констант скоростей
реакций разложения метана и ацетилена kx\ &4 больше 10.
Результаты проведенных расчетов степени конверсии е метана
в ацетилен и степени разложения ацетилена по реакциям
2СН4
С2Н2 4" ЗН
2С+Н,
для 1900° К графически показаны на рис. Ш-8. Зависимость степени
превращения е от константы скорости и времени реакции т
выражается уравнением:
8 =
(e
—kxX
S)
Преобразованием этого уравнения можно получить следующие
выражения для максимальной степени превращения метана в аце-
79
тилен емакс. и времени топт., за которое она достигается:
е = г-!— \е~г 1п %1(Г-Х) - е~]п х^г-1)]
__ \пг
Топг. —
(r—\)k
где г = &4 : &х;
/г — константа скорости превращения метана в ацетилен.
В процессах получения ацетилена осуществляется «закалка»
продуктов реакции — резкое охлаждение их большим количеством
специально подаваемой воды. В связи с этим определенный интерес
представляет взаимодействие ацетилена с водяным паром
С2Н2 + 2Н2О ^ 2СО + ЗН2
которое может снижать количество образовавшегося ацетилена. Эта
реакция имеет первый порядок и ее константа скорости выражается
уравнением14:
7П 37 700
= 7,0 ^у-
Скорость взаимодействия ацетилена с водяным паром
относительно невысока, а при резком падении температуры при «закалке»
эта реакция может совсем прекратиться.
Ацетилен и высшие ацетиленовые углеводороды, образующиеся
при пиролизе метана, в условиях процесса могут не только
разлагаться на углерод и водород, но и образовывать различные
ненасыщенные соединения. Было исследовано15 разложение ацетилена, ви-
нилацетилена и диацетилена в интервале 500—800° С. Углеводород,
разбавленный гелием (25% от объема углеводорода), пропускали
через проточный реактор. Оказалось, что по способности к
разложению рассмотренные углеводороды располагаются в следующий
ряд: диацетилен >> винилацетилен >> ацетилен.
Степень конверсии углеводородов изменяется в зависимости от
температуры:
Температура, °С 400 500 600 700 800
Степень конверсии, %
ацетилен 0 22 48 73 92
винилацетилен 0 28 66 97 100
диацетилен 4 68 96 100 100
В продуктах пиролиза ацетилена найдены водород, метан,
этилен, винилацетилен и бензол. В продуктах разложения винилацети-
лена содержание бензола меньше, но имеются пропилен и бутадиен.
Пиролиз диацетилена характеризуется еще меньшим образованием
бензола и газообразных веществ. Количество водорода и метана
повышается с температурой, а наибольший их выход наблюдается при
пиролизе ацетилена. С повышением температуры содержание
бензола в отходящих газах проходит через максимум. Таким же образом
влияет температура на выход винилацетидена при пиролизе ацети-
80
лена и на выход ацетилена при пиролизе винилацетилена и диацети-
лена. Добавки водорода повышают выход метана и этилена и
уменьшают образование углерода.
Выбор сырья для пиролиза
В качестве сырья для получения ацетилена практически могут
быть использованы любые углеводороды и их смеси — газообразные
и жидкие. Однако химический состав сырья является одним из
факторов, влияющих на выход целевых продуктов пиролиза (ацетилена и
сопутствующего ему этилена). Известно, что лучшие результаты
получаются при пиролизе парафинов нормального строения, причем
энергетика процесса находится в прямой зависимости от
молекулярного веса углеводорода:
АЯ298 °К>
ккал/моль
2СН4 ^> СН2 + ЗН2 —89,97
С2Нб ^ С2Н2+2Н2 -71,4
2С3Н8 ^ ЗС2Н2 + 5Н2 —70,9
С4Н10 ^ 2С2Н2+ЗН2 -69,3
Пиролиз изопарафинов и нафтенов характеризуется значительно
меньшими выходами целевого продукта; наименее пригодным сырьем
оказываются ароматические углеводороды, при разложении которых
образование ацетилена незначительно, а кокса и смол очень велико.
Исследовалась также возможность применения различных
нефтяных фракций, при этом в отдельных случаях получался суммарный
выход этилена и ацетилена 40—55%.
Для оценки выхода ацетилена при пиролизе обычно используют
две величины: общий крекинг А (общая степень конверсии) и
полезный крекинг г\ (степень конверсии до ацетилена)
* число молей ацетилена -f- число молей других непредельных
общее число молей углеводородов в сырье
число молей ацетилена
общее число молей углеводородов в сырье
причем полезный крекинг можно рассчитать по отношению к
прореагировавшему или к исходному углеводороду.
Пиролиз характеризуется также глубиной разложения исходного
сырья до простейших углеводородов или до углерода и водорода.
Часто о глубине разложения судят по плотности получаемого газа,
которая находится в обратной зависимости от глубины разложения.
Состав сырья определяет температуру и давление процесса: чем
выше молекулярный вес углеводородов, тем ниже оптимальная
температура14 (рис. II1-9). Аналогичная зависимость наблюдается и для
давления: чем больше молекулярный вес пиролизуемого
углеводорода, тем неблагоприятнее повышение давления.
6 В. Н. Ahtqhojj §1
Метанпиролизуетсяпринаиболеевысокойтемпературе(>1400°С),
что обусловливает очень малое время пребывания его в зоне
реакции — менее 0,01 сек. В существующих технологических схемах, за
исключением разложения метана в потоке газа-теплоносителя, это
условие с сохранением приемлемых выходов ацетилена обеспечить
невозможно. Разложение метана при остаточном давлении 0,1—0,2 am
происходит с глубиной превращения 30%, концентрация ацетилена
в газах пиролиза достигает 8—10 объемн. %.
На рис. II1-10 показана зависимость выхода ацетилена от глубины
разложения метана. В трубке диаметром 1,5 мм при 1350° С и
глубине разложения 10% за время
3,8-10~3шс образуется 2,1
объемн. % ацетилена и 17 объемн. %
юо
//
//
F
ft /
/У
У
/
80
160
140
I
^ 20
700 900 1100 1300 1500
Температура, °С
Рис. II1-9. Зависимость
равновесного выхода ацетилена при
пиролизе углеводородов от температуры:
/ — бутан; 2 — этан и пропан; 3—
метан.
12
у^
1
у^
X
м
и
1
В 14 22 30 38
Глубина разложения СН4,%
Рис. 111-10. Зависимость выхода
ацетилена от глубины
разложения метана:
/, 2, З—в трубке диаметром 1,5 мм
при 1500° С (У), при 1450°-С (2) и
при 1350° С (3); 4 — в трубке
диаметром 5 мм при 1350° С.
водорода16. При увеличении глубины разложения метана до 25%
резко возрастает выход жидких продуктов. При концентрации
7,5 объемн. % ацетилена и 52 объемн. % водорода в реакционных
газах появляется сажа.
Этан является более подходящим сырьем для получения
ацетилена. В промышленности накоплен большой опыт по пиролизу этана
для получения этилена. Процесс обычно проводится в трубчатых
печах высокой производительности. Обогрев этана до 850—900° С
осуществляется через металлическую стенку. Для получения
ацетилена необходима более высокая температура (1100—1200° С). В связи
с трудностью подбора конструкционных жаростойких сплавов
пиролиз этана до ацетилена в печах очень сложен, процесс легче
протекает при разложении этана в потоке газа-теплоносителя.
В лабораторных условиях17"19 пиролиз этана в трубчатом
реакторе исследован достаточно полно. Например, при 986° С и времени
пребывания в зоне нагрева 0,022 сек степень конверсии этана
составляет 83%, выходы ацетилена и этилена соответственно 3,8 и 74 вес. %
S2
(рис. III-11). При этом максимальная концентрация ацетилена в газе
пиролиза составляет 3 объемн. %, этилена — 37 объемн. %. При
увеличении температуры выше 1000°С выход и концентрация ацетилена
резко возрастают за счет уменьшения образования этилена. Мак-
70
x S°
О 50
1
\
\
<s
C2H
S^
C2H
i6
\ ^T
^N^CO _
%%_
0,1
I
I
w
a 30
^t
20
-0,01
4
1
10
0,001
°0
<j
Л
/
го
с2н4
/
\
\
ч
ч
У
>
А
\
\
>
СгН2
ч
\
~^>о
^^
°0,2 0,3
0,5 0,7 1,0 1,5 2,0 3,0
Время контакта, моек
5,0 7,0 10,0
Рис. II1-12. Зависимость выхода продуктов
пиролиза этана от времени реакции при 1400° С
и 50 мм рт. ст.
700 800 900 WOO 1100
Температура, °С
Рис. 111-11. Зависимость
выхода продуктов пиролиза этана
от температуры.
симальный выход ацетилена из этана достигнут4 при 1400° С, 50 мм
рт. ст. и времени контакта около 5 мсек (рис. II1-12) и составляет
50 вес. %. Это соответствует 17 объемн. % ацетилена в получаемом
газе.
Окись и до у окись
Сажа
и смолистые 8ещест8а -
100
углерода
800
900
1000 1100 1200 1300
Температура, °С
Рис. II1-13. Распределение углерода в продуктах
пиролиза пропана при времени реакции 0,11 сек
и разбавлении сырья водяным паром (1 : 2).
Пропан, бутан и более тяжелые парафины используются в
производственных условиях для получения ацетилена и этилена
пиролизом в трубчатых печах при 1000—1100° С. При времени пребывания
б* 83
в зоне пиролиза 0,14 сек и разбавлений углеводородов водяным
паром в соотношении углеводород: пар = 1:2 получается газ,
состоящий из 12—13 объемн. % ацетилена, 14—15 объемн. % этилена,
25—27 объемн. % метана и 0,25 объемн. % гомологов ацетилена
(40 объемн. % составляют водород и другие газы)20'22.
Распределение углерода в продуктах пиролиза гомологов метана зависит от
температуры процесса (рис. III-13) и мало изменяется с ростом
молекулярного веса углеводорода.
1
20
10
О.
\
\l
\
с2н2 +
\
2н2 N
>»,
\с2н
\
с2н4
\
Ч
0,1 0,2 0,3
Время, сек
а
0,5 1 3 10 30 100
Время, сек-10 ~3
б
80
г
<
-^СгН,,
N
хГ"
с3н6
И'!
ч
0,3 0,5
1,0 2 4
Время, сен to'3
R
Рис. II1-14. Зависимость выхода
продуктов пиролиза пропана от
времени реакции, температуры и
давления:
а —при 1100° С и 1 am; б—при 1100° С
и 50 мм рт. ст.', в — при 1400°С и 50 мм
рт. ст.
Кривые, приведенные на рис. III-14, иллюстрируют влияние
времени реакции на выход продуктов пиролиза пропана. При 1100° С и
1 am максимальный выход ацетилена отвечает времени реакции 0,2 сек
(рис. III-14, а); при снижении давления21'22 до 50 мм рт. ст.
оптимальное время реакции сокращается до 0,05 сек (рис. III-14, б), а
при 1400° С и 50 мм рт. ст. максимум ацетилена достигается уже при
0,005 сек (рис. Ш-14, в). Аналогичная зависимость в отношении
времени пиролиза существует для я-бутана (рис. Ш-15)22~26.
При пиролизе этилена и пропилена выход ацетилена растет с
температурой процесса и меньше зависит от времени реакции.
В промышленных условиях используются газообразные смеси
углеводородов, состоящие из низших парафинов, иногда с примесью
олефинов27"30. Кроме газообразных смесей применяются жидкие
84
углеводородные смеси, например бензин (пределы выкипания 30—
160°С) или керосино-лигроиновые фракции нефти. С повышением
плотности сырья увеличивается выход непредельных углеводородов и
возрастает выход сажи, смолы и кокса.
Каждому виду сырья соответствуют
определенные оптимальные параметры
процесса. Естественно, что более
предпочтительны чистые углеводороды, однако это
требование в промышленных условиях
почти невыполнимо. Возрастающие ре- ^ cS
сурсы отходящих газов нефтепереработки, I ^
газового бензина и др. позволяют иметь ,Л|
достаточное количество сырья, пригод- §g>
ного для получения ацетилена. <§ ч
юо
о
Г80
ВО
Условия проведения процесса
1
у
1
/
:2н4
-4Н10
/^
С2Н2
"^Ч
^^
ЛН6"
^ч
0,5
1
5
10
~3
Время, сен-Ю~
Рис. II1-15. Зависимость
выхода продуктов пиролиза
я-бутана от времени реакции
при 1100° С и 50 мм рт. ст.
Различают следующие технологические
схемы пиролиза в зависимости от
способа подачи тепла в зону реакции.
1. Регенеративный пиролиз. Процесс
протекает на стационарной или
движущейся огнеупорной насадке. Насадка периодически или
постоянно нагревается топливным газом до 1800—2000° С. После
протекания эндотермических реакций пиролиза насадка охлаждается
до 800—900° С, затем ее тепло используется для подогрева
углеводородов, воздуха и топливного газа. При движущейся твердой насадке
стадии процесса остаются теми же, но насадка последовательно
перемещается через все зоны нагрева и охлаждения в реакторе.
2. Пиролиз в трубчатых печах. При этом процессе пропускают
подогретый углеводород через трубы, обогреваемые снаружи
продуктами сгорания топливного газа.
3. Гомогенный пиролиз в потоке газообразного теплоносителя.
В этом случае углеводороды вдувают в струю горячих продуктов
сгорания топливного газа; углеводороды и теплоноситель
смешиваются непосредственно в зоне реакции.
4. Пиролиз с погружным горением. *В таких процессах
осуществляется непосредственный контакт жидких углеводородов с
продуктами сгорания, образующимися в погружной горелке.
Тепловой баланс процесса пиролиза можно представить
следующим выражением:
Qi + Q2 = Qs + Q4 + Q5 + Qe
где (?! — тепло, вносимое с подогретым сырьем;
Q2 — тепло, полученное сырьем в печи (через стенку или с газовым
теплоносителем);
Q3 — тепло, израсходованное на реакцию пиролиза;
Q4 — тепло, отданное при «закалке» газов пиролиза;
(?5 — тепло, уносимое с реакционными газами;
QG — потери тепла.
85
Основной составляющей теплового баланса является величина Q2,
определяющая тепловой потенциал процесса. При использовании
в качестве теплоносителя стационарной или движущейся твердой
насадки Q2 зависит от теплоемкости материала насадки и определяет
цикличность процесса. При пиролизе с газовым теплоносителем Q2
изменяется с теплотворной способностью топливного газа: применяя
топливо повышенной калорийности, можно создать более высокую
температуру в печи.
I*
^$5
II
Я
/
/J
ъ
W
V
\
I
-so
t
4
If
fa
200
Р
Г
Время, мксек
Рис. 111-16. Кинетические кривые процесса
образования дисперсного углерода при самовоспламенении
ацетилена:
/ — скорость образования частиц; 2 — суммарная
поверхность частиц; 3 — степень разложения ацетилена. *
На условия пиролиза оказывает влияние поверхность теплообмен-
ных труб и твердого теплоносителя. Некоторые материалы ускоряют
распад получаемых углеводородов на простые вещества, тем самым
снижая выход целевых продуктов. К таким материалам относятся
кобальт,- платина, палладий и в большей степени никель31. Значение
твердой поверхности уменьшается с ростом температуры. Несмотря
на многие попытки, не удалось определить влияние различных
веществ, входящих в состав материала стенки, на образование
ацетилена. Не найдены до сих пор и катализаторы, ускоряющие этот
процесс.
Большое влияние на пиролиз оказывает углерод, осаждающийся
на неуглеродных поверхностях (на стенке), который в свою очередь
увеличивает дальнейшее сажеобразование. При пиролизе
углеводородов сажеобразование протекает в две стадии31'32. Вначале при
достижении определенной критической температуры (в интервале 500—
800° С) образуются зародыши — сажевые частицы; затем за счет
распада ацетилена на их поверхности количество частиц быстро
растет. На рис. II1-16 приведены расчетные кинетические кривые
процесса сажеобразования при самовоспламенении ацетилена. Из
общей продолжительности процесса 7 мксек индукционный период
составляет 1,5 мксек, а время до начала быстрого роста частиц —
2 мксек. После достижения максимума скорость сажеобразования
86
25
Й
ч*
г
!
I
—
4
\
\
начинает быстро падать при еще высокой концентрации ацетилена.
Максимальная скорость образования сажевых частиц
соответствует степени разложения ацетилена 100%. Энергия активации
процесса сажеобразования составляет 60 ккал/моль\ высокое значение
энергии активации указывает на цепной механизм образования
углерода.
Сажа может образовываться не только из ацетилена, но и из
различных ароматических соединений, содержащихся в смолистых
веществах. Однако при исследовании
диффузионного факела горения природного
газа было установлено32, что зоны
образования сажи и смол не совпадают:
сажа обнаружена в светящейся зоне
пламени —в тонком слое газа,
прилегающем непосредственно к фронту
горения, а смолы появляются
преимущественно во внутренней темной зоне
пламени. Максимальный выход сажи для
условий опыта составлял 1,7% от
общего содержания углерода в
природном газе и соответствовал
относительной высоте факела 62—64%.
Наибольшее содержание смолистых веществ
(0,7%) соответствует начальным
участкам (на высоте 25%). Процесс
сажеобразования в диффузионном
факеле осложняется тем, что
образование дисперсной фазы совмещается
с процессами диффузионного перемешивания и горения.
Для начала образования сажевых частиц при термическом
разложении углеводородов, разбавленных азотом,требуется некоторая
критическая (пороговая) концентрация углеводорода в исходной смеси,
причем, чем ниже температура разложения газовой смеси, тем выше
этот концентрационный порог (рис. III-17). Для смесей
углеводородов критическая концентрация компонентов ниже, чем каждого
из этих компонентов в отдельности. Например, при 1100° С сажеобра-
зование в смеси метана и ацетилена, разбавленной азотом, начинается
при 0,47 объемн. % С2Н2 и 11,7 объемы. % СН4. Сажеобразование
в смеси метана с азотом при 1100° С начинается, как это видно из
рис. 111-17, при 21 объемн. % СН4.
Выяснение особенностей процесса сажеобразования особенно
важно при использовании нефтяных фракций в качестве сырья для
получения ацетилена. Одним из возможных путей снижения доли
реакций конденсации и выхода кокса в процессе пиролиза является
систематическое изучение кинетики и химизма сажеобразования при
пиролизе индивидуальных углеводородов и использование
полученных закономерностей для установления оптимальных
параметров,
87
о
950 1000 1050 1100 1150 1200
Температура, "С
Рис. III-17. Температурная
зависимость критической
концентрации углеводородов,
разбавленных азотом, при образовании
сажи:
/ —бензол; 2—ацетилен; 3 — метан
(-90% СН4).
Скорость процесса сажеобразования оценивается по скорости
образования карбоидов — коксообразных веществ, не растворяющихся
в горячем бензоле г. Между скоростью крекинга и скоростью
сажеобразования наблюдается определенное соответствие: чем выше
константа скорости крекинга углеводорода, тем больше скорость
образования карбоидов. Наибольшая скорость образования карбоидов
наблюдается при крекинге циклических непредельных углеводородов
Парафины
±
Парасрины
1
Циклические
непредельные
соединения
Длкилароматцческие
соединения
Высокомолекулярные
конденсированные
угле8одороды
JL
Дсазальтены
±
Карбоиды
±
Кокс, сажа
| Оленины I
—L
I
Оленины | | Диены| | Нафтены\ | Парафины
| Олефин
\Парафины
Рис. III-18. Схема образования
карбоидов при крекинге
парафинов.
типа индена. Далее следуют ароматические углеводороды с боковыми
цепями (алкилбензолы, дибензил) и полициклические
конденсированные соединения (например, антрацен, пирен, аценафтен). По
кинетике коксообразования олефины располагаются между
ароматическими углеводородами и парафинами. Медленнее всего образуются
карбоиды при крекинге ароматических углеводородов без боковых
цепей. Именно для последней группы углеводородов механизм
образования карбоидов изучен наиболее полно: молекулы ароматических
углеводородов конденсируются с выделением водорода и
образованием полициклических ядер, обедненных водородом. Наиболее
характерным для процессов коксообразования при разложении
углеводородов остальных групп является наличие стадии полимеризации
или конденсации. Поэтому при крекинге парафинов, олефинов и наф-
тенов предварительно должны образоваться циклические углеводен
88
роды, после чего начинаются процессы коксообразования. На рис.
II1-18 приведена упрощенная схема термического превращения
углеводородов, заканчивающегося образованием кокса1.
При исследовании сажеобразования в процессе пиролиза метана
в смесях с предельными и непредельными углеводородами было
установлено, что выход сажи тем больше, чем выше молекулярный вес
углеводорода32. При этом для бинарных смесей соблюдается правило
аддитивности, т. е. образование сажевых частиц из каждого
углеводорода происходит так же, как из индивидуальных углеводородов.
Это, по-видимому, связано с тем, что механизм образования сажевых
частиц одинаков для всех ациклических углеводородов и различие
заключается только в скорости процесса. Добавки к метану
небольших количеств ароматических углеводородов вызывают резкое
возрастание выхода сажи, объясняющееся большей скоростью
образования сажевых зародышей. Примеси нафталина тоже сильно
увеличивают количество образующейся сажи; можно предположить, что
в процессе пиролиза происходят реакции алкилирования нафталина,
а алкилнафталины характеризуются высокой скоростью
сажеобразования.
50
I
Добавки водяного пара
Пиролиз углеводородов (газообразных и жидких) ведут при
различном разбавлении их водяным паром. Добавки пара позволяют
регулировать температурный режим процесса и подавляют
образование смол и кокса.
Механизм действия водяного пара
в процессе пиролиза изучен
недостаточно. Установлено, что с увеличением
молекулярного веса сырья повышение
соотношения пар: сырье благоприятно
сказывается на выходе целевых
продуктов и уменьшает выход смол и
кокса. При пиролизе этановой
фракции оптимальное количество водяного
пара составляет29'30 10 объемн. %. При
таком разбавлении и температуре 900° С
концентрация этилена в газах пиролиза
доходит до 36—37 объемн. %, а
ацетилена получается не более 1,0—1,2
объемн. %. При пиролизе пропана
суммарный выход ацетилена и этилена с
уве/^
^—-^
L_
^2^2 ~^~ 2 4
АН;
м—
с2н4
>
10О 2 4 6 8
Пар-сырье
Рис. II1-19. Зависимость выхода
продуктов пиролиза пропана
от соотношения пар : сырье при
времени
1100° С и
контакта
0,15 сек.
личением весового соотношения пар : сырье повышается, хотя
количество ацетилена возрастает, а этилена падает22 (рис. III-19).
При различных технологических методах пиролиза влияние
водяного пара неодинаково. При регенеративных процессах и пиролизе
в трубчатых печах добавки водяного пара особенно важны, так как
изменением подачи пара практически можно регулировать темпера-
89
турный режим процесса. При гомогенном пиролизе с
газом-теплоносителем влияние соотношения пар : сырье сказывается меньше, так
как в этих условиях водяной пар необходим только для уменьшения
глубины разложения сырья до сажи и смол, а регулирование
температуры достигается изменением соотношения топливного газа и
воздуха (или кислорода).
РЕГЕНЕРАТИВНЫЕ ПРОЦЕССЫ
Пиролиз в регенеративных печах
Пиролиз метана с целевым выходом ацетилена, как было
показано, осуществим при температурах выше 1200° С и парциальном
давлении метана 0,05—0,2 am. Технологическое оформление процесса
крайне затруднено в связи с необходимостью подвода большого
количества тепла при высоких температурах и проведения процесса
в вакууме. Поэтому, несмотря на большое число патентов,
лабораторных работ и даже опытных установок, промышленного
применения этот способ не нашел.
Рассмотрим результаты пиролиза метана на опытной установке
в печи регенеративного типа33, имеющей насадку в виде цилиндров
диаметром 60 мм из белого корракса *. Цикл работы печи составляют
следующие процессы: разогрев, продувка водяным паром, создание
вакуума, пиролиз, гашение вакуума.
На установке изучалось влияние разбавления метана водяным
паром и воздухом, а также действие инициирующих добавок пропана
и бутана. Добавки водяного пара позволяют несколько уменьшить
содержание сажи и водорода в газах пиролиза за счет подавления
распада метана на простые вещества. Добавки пропана и бутана
не повышают выход ацетилена и вызывают заметное- сажеобразова-
ние. Применение воздуха в качестве разбавителя затрудняет
проведение процесса в вакууме. Наиболее благоприятные результаты
получены при температуре 1500° С, остаточном давлении 0,2—0,4 am
и времени реакции 0,007—0,01 сек. Газ, получаемый при пиролизе
чистого метана, имел следующий состав (в объемн. %):
С2НО 5,0 СН4 10,2
QH4 0,3 СО 6,0
Н2 78,0 СО2 0,5
Результаты отдельных опытов33 приведены в табл. III-11.
Процесс Вульфа. В 1930—1940 гг. был разработан процесс
получения ацетилена в регенеративных печах по методу Вульфа. Первая
установка мощностью 500 т/год была пущена в США3--37. В 1962 г.
фирма Union Carbide (США) пустила в эксплуатацию установку
производительностью 4500 т ацетилена в год36, а в 1965 г. в
Англии^ началось производство ацетилена по методу Вульфа на
* Корракс (88% А12О3) огнеупорен до 1890° С.
90
ТАБЛИЦА III-ll
Результаты пиролиза метана в регенеративной печи при 1500° С
Показатели
Остаточное давление в зоне
реакции, am
Время реакции, сек
Выход ацетилена на 100 ж3 метана,
м*
исходного
превращенного
% от теоретического
Расход метана на 1 кг ацетилена, кг
Пиролиз метана
0,3
0,012—0,016
11,3
18,0
36,0
7,6
Пиролиз метано-
паровой смеси (1 : 1)
0,2—0,4
0,009—0,012
12,8
18,3
36,6
6,8
заводской установке годовой производительностью по ацетилену
30 000 т. В настоящее время имеются две установки в ФРГ, три —
в США, две —в Англии.
Пиролиз по методу Вульфа протекает при высокой температуре
(1100—1300° С) и низком общем давлении (0,4—0,5 am). Для
снижения парциального давления исходное сырье разбавляют водяным
паром или возвратными газами; можно также проводить процесс
в вакууме.
Одной из особенностей метода является одновременное
образование ацетилена и этилена из любого углеводородного сырья, кроме
метана. Соотношение ацетилена и этилена в газе пиролиза зависит
от режима работы печи. Этилен можно выделять в виде готового
продукта или возвращать в цикл. В последнем случае выход
ацетилена увеличивается примерно в 2 раза.
Основным аппаратом процесса Вульфа является регенеративная
печь, представляющая собой горизонтальный аппарат
прямоугольного сечения, состоящий из двух камер-регенераторов, разделенных
топкой. Аппарат имеет стальную обечайку и внутри футерован
огнеупорным кирпичом. С обоих концов печи имеются устройства для
подвода и вывода газа. Для нагрева печи и проведения реакции
используется тепло, выделяющееся при сгорании топливного газа,
подаваемого в центральную часть топки. •
Каждая камера-регенератор имеет огнеупорную насадку, которая
поглощает тепло в период нагрева и отдает его в течение рабочего
цикла крекинга. Насадка выполнена в виде плоских прямоугольных
пластин, имеющих продольные канавки. При укладке пластин по
всей длине регенератора образуются цилиндрические каналы
диаметром 6,4 мм. Насадка и прилегающие к ней слои футеровки
изготовлены из 99%-ной окиси алюминия.
На рис. II1-20 показана принципиальная схема процесса. Сырье
(жидкий пропан) испаряется в испарителе 1 за счет использования
отходящего тепла со стадии выделения ацетилена. Далее пары
пропана поступают в смеситель 2, где пропан разбавляется водяным
паром и рециркулирующим газом со стадии выделения ацетилена.
91
<^
j-S!
11-
ь*
С&-
^
-С)
С- C\j"""
С:
V
пиролиза
N
Ш
Пропан
г
.пар
Дымовые
i
Топливный
Соотношение количеств пропана, водяного пара и рециркулирующего
газа составляет 1:6:2.
Процесс ведут в вакууме (при остаточном давлении 370 мм рт. ст.).
Перепад давления в печи не превышает 25 мм рт. ст. Подачу
пропана и рециркулирующего газа
газы ,—, воздух прекращают за 3 сек до
окончания рабочего цикла и в
течение этого времени печь
продувают водяным паром. Перед
поступлением в топку воздух
фильтруется и нагревается в
одной из печей 3 до 1100° С.
Дымовые газы выходят из цикла
пиролиза нагрева при 300° С, охлажда-
7 ются в холодильнике и
отсасываются из него ротационным
вакуум-насосом, чем достигает-
газ
л
Дымовые
газы
П
Топливный
1
газ
Воздух
газы
Газы
Ш
Проспан
Пропан
Ш
Ш
Газы
пиролиза
Воздух
.Пар
Дымовые
газы
Е
Топливный
газ
Воздух
Пропан
Ш
ся в печи требуемое
разрежение, и выбрасываются в
атмосферу (на схеме не показано).
Пиролиз происходит в
нагретой печи <?, затем газы
охлаждаются до 300° С в другой печи 3.
1200
1000
800
600
ЬОО
IF
Пазы
газы
пиролиза
- //
•* Длина
\ч
Х\
г
печи *-
Рис. II1-21. Цикл работы регенеративных
печей Вульфа:
1, 2, 3, 4 — различные положения двух
параллельно работающих печей; / — подогрев
воздуха; // — нагрев насадки дымовыми газами;
/// — пиролиз; IV — нагрев насадки газами
пиролиза.
Рис. III-22. График изменения
температуры по длине регенеративной печи
Вульфа.
Средняя скорость газа во время
рабочего цикла примерно
55 м/сек. Газы пиролиза выходят
из печи при 300° С, подвергаются «закалке» водой в аппарате 4,
после смолоотделителя 5 дополнительно охлаждаются в
холодильнике 6, затем поступают в газгольдер 11 и на стадию выделения
ацетилена диметилформамидом (ДМФ).
Непрерывность процесса пиролиза достигается установкой
сдвоенных печей. Печь Вульфа работает по четырехтактному циклу в
такой последовательности (рис. 111-21): такт пиролиза и такт нагрева
93
при одном направлении газового потока, затем такт пиролиза и такт
нагрева при противоположном направлении газового потока.
Каждый такт длится около 1 мин, в результате на весь цикл требуется
около 4 мин.
На рис. II1-22 показано изменение температуры печи по ее длине.
Эта зависимость получена при ведении процесса с максимальным
выходом ацетилена и максимальным суммарным выходом ацетилена и
этилена. При переключении с прямого потока газов на обратный
колебания температуры в любой точке печи не превышают 15° С.
Максимальная температура зависит от характера сырья и требуемого
состава газа и может быть изменена при изменении любого из этих
параметров. Конечная температура печи является функцией ее длины
. и пропускной способности.
При пиролизе образуются смолы и масла; для предотвращения их
оседания на насадке и стенах камеры температура на выходе из печи
должна быть выше температуры конденсации этих соединений.
Образующаяся при пиролизе сажа отлагается на поверхности насадки;
ее удаляют в процессе разогрева за счет сжигания вместе с
топливным газом. При этом улучшается тепловая эффективность печи. При
работе на различном сырье установлено, что в газах, выходящих
из печи, сажа практически не содержится.
Цикличный характер процесса и многообразие протекающих
реакций затрудняют анализ работы и расчет регенеративной печи.
Кроме того, сложнее сохранять заданное время реакции, поскольку
здесь нет «закалки» реакционных газов. Однако отсутствие «закалки»
позволяет использовать тепло и увеличить к. п. д. печи до 70%.
На рис. II1-23 приведены данные о пиролизе пропана с целевым
получением суммы ацетилена и этилена и раздельным получением
этих продуктов. Условия опытов: давление 0,5—1,2 am, весовое
соотношение пар : сырье ^ 1 : 3. Максимальный суммарный выход
соответствует 1060° С и равен 55 вес. %, максимальный выход ацетилена
около 30 вес. % при 1150° С. Выход ацетилена зависит также от
парциального давления пропана — возрастает с понижением давления
(рис. II1-24). На выход этилена изменение парциального давления
пропана существенно не влияет.
Состав газов пиролиза пропана приведен ниже (в объемн. %):
Ацетилен 10,0 Метил ацетилен ... 0,2
Этилен 3,8 Винилацетилен ... 0,16
Метан 15,0 Диацетилен 0,04
Этан 0,1 Водород 55,98
Пропан 0,2 Кислород 0,7
Пропилен Следы Окись углерода ... 6,9
Бензол 0,14 Двуокись углерода . . 1,6
Азот 5,2
В регенеративных печах Вульфа можно подвергать пиролизу не
только -индивидуальные углеводороды (пропан, бутан), но и
различные нефтяные дистилляты. Например, при пиролизе газойля
(пределы выкипания 220—312° С) в присутствии водяного пара при дав-
94
ленйи 0,49 am (рис. II1-25) максимальный суммарный выход р
дельных углеводородов составляет ~45%, а максимальный выход
ацетилена равен 22% (при 1100° С).
900 /000 1100 1200 1300
Температура, °С
Рис. II1-23. Выход продуктов
пиролиза пропана в
зависимости от температуры.
0,07 О,)Ч
Парциальное да8ление,ат
Рис. II1-24. Зависимость
выхода ацетилена от
парциального давления пропана:
/ — при 1090° С; 2 —при 1090 —
1125° С; 3— при 1140 —1200° С.
На рис. II1-26 представлены результаты пиролиза легкого
лигроина при атмосферном и пониженном давлении; видно, что выход
800 SOO 1000 /100 f200
Температура, °С
Рис. III-25. Температурная
зависимость выхода продуктов
пиролиза газойля при 0,49 am.
S00 1000 1100 1200
Температура°С
Рис. II1-26. Температурная
зависимость выхода продуктов
пиролиза легкого лигроина при
атмосферном и пониженном давлении.
ацетилена при атмосферном давлении довольно медленно возрастает
с повышением температуры до 1000° С и резко увеличивается при
разрежении и дальнейшем повышении температуры (1000—1200° С).
95
Топливный
газ
Газ
Установка фирмы Roppers-Hasche (США). Фирмой Koppers-
Hasche в 1954 г. в г. Вероне (США) была сооружена опытная
установка с регенеративной печью.
Печь системы Koppers-Hasche является разновидностью печи
Вульфа. Особенностью процесса фирмы Koppers-Hasche является его
цикличный характер: через печь (сдвоенную или одинарную)
попеременно пропускают сначала воздух и
топливный газ, а затем крекируемые
углеводороды (рис. III-27)38.
При нормальной работе печи
максимальная температура в регенераторах
создается в областях, примыкающих к
камере сгорания 2, причем
температура снижается почти линейно по на-
правлению к обоим концам печи. В
зоне нагрева 1 воздух подогревается
примерно от 150° С до 815—1040° С в
зависимости от установленной
температуры реакции, в зоне пиролиза 3
температура углеводородного сырья
возрастает от 150° С до максимальной (870—
1425° С), при которой и протекает
процесс пиролиза.
Исходным сырьем может служить
пропан, бутан или газовый бензин. В
результате пиролиза пропана (табл.
III-12) можно получать в качестве целевых продуктов ацетилен,
этилен или их совместно в зависимости от выбранного режима.
Выход ацетилена возрастает при разбавлении сырья паром и при
ТАБЛИЦА 1Г1-12
Результаты пиролиза на установке фирмы Koppers-Hasche
Рис. II1-27. Схема
регенеративной печи системы Koppers-
Hasche:
1,3— регенеративные части; 2 —
камера сгорания топливного газа;
4 — насадка.
Показатели
Целевые продукты
Температура, °С
Давление, am
Количество водяного пара,
объем/объем
Общая степень конверсии,%
Состав * газа пиролиза, объ-
емн. %
ацетилен
этилен
метан
Пиролиз
природного газа
(95О/О СН4)
с2н2
1338
0,7
2,2
57,2
5,4
0,8
18,9
Пиролиз
зтана
С2Н4
954
1,05
0
83,9
2,6
30,0
10,8
Пиролиз пропана (94,2% С3Н8)
с2н2+
СоН4
1343
0,63
6,2
98—100
11,7
5,2
15,6
с2н4
1010
1,05
0
87,8
1,7
25,6
30,6
С2Н2+
С2Н4
1240
1,05
4,4
99—100
8,9
16,8
26,0
* В расчете на безвоздушный газ.
96
Продол ж. табл. III -12
Показатели
Пиролиз
природного газа
(95% СН4)
Пиролиз
этана
Пиролиз пропана (94,2% С3Н8)
Состав * газа пиролиза,
объемы. %
этан
пропан
пропилен
другие углеводороды С3
углеводороды С4
углеводороды С5
водород
окись углерода
двуокись углерода . . . .
азот
Выход, % от сырья
ацетилен
этилен
пропилен
0,2
57,2
9,1
2,5
5,9
20
8,9
3,0
42,5
0,3
0,5
4,1
50,7
0,9
1,5
1,5
44,8
6,8
2,0
10,0
28,3
13,4
1,9
5,8
4,2
1,1
0,9
25,8
0,3
0,3
1,8
2,1
34,3
8,4
1,0
40,0
3,6
1,4
2,3
16,1
32,6
снижении давления. Максимальный выход ацетилена 27—28% (от
сырья), причем в этом случае одновременно получается до 13%
этилена. Суммарный выход ацетилена и этилена при пиролизе,
например, пропана может составить 41—48%, в том числе 16—28%
ацетилена.
Промышленного применения для производства ацетилена печь
фирмы Koppers-Hasche не нашла.
Пиролиз в движущемся слое твердого теплоносителя
Фирмой Phillips (США) разработана опытно-промышленная
установка для пиролиза углеводородного сырья в движущемся слое
твердого теплоносителя 35>38 (рис. III-28).
Гранулированный теплоноситель (размер зерен 3—5 мм)
нагревается топочными газами до 1100—1200° С в камере 3 и затем
пересыпается в реакционную камеру 5, куда поступают этан, пропан или
более тяжелые углеводороды. Насадка-теплоноситель в процессе
пиролиза охлаждается, отдавая тепло реакционным газам, и
загрязняется сажей, которая в следующей зоне выжигается. Чистая
насадка передается элеватором или пневматически в нагревательную
камеру 3 и затем вновь поступает в реакционную камеру 5.
Для обеспечения интенсивной теплопередачи поддерживается
высокая разность температур между гранулами теплоносителя и газом
путем повышения кратности циркуляции теплоносителя. В связи
с этим даже при относительно низкой температуре исходного
газообразного сырья на входе в реакционную камеру (90° С) температура
выходящих из нее гранул достаточно высока (500° С).
Предварительное нагревание сырья перед подачей в реакционную зону
нежелательно, так как при этом повышается температура теплоносителя
и усложняется работа пневмотранспорта.
7 В. Н. Антонов 97
При пиролизе этан-пропановой фракции, содержащей 63,1
объемы. % этана, 4,6 объемы. % пропана, 32,2 объемы. % метана
и 0,1 объемы. % двуокиси углерода, общая степень конверсии
составила 96,8% и полученный газ имел следующий состав:
Ацетилен 5,4
Этилен 20,1
Метан 25,8
Этан 1,1
Пропан 0,1
Пропилен . . . : 0,3
Бутадиен 0,4
Высшие ацетиленовые
углеводороды 0,4
Бензол 0,8
Толуол 0,2
Водород 43,8
Окись углерода 1,2
Двуокись углерода .... 0,3
Прочие 0,1
Выход целевых продуктов
составил 14,5% по ацетилену и 53,4% по
этилену (в расчете на пропущенное
сырье). Количество этилена
оказалось больше, чем, например, в
трубчатых реакторах. Выход ацетилена,
однако, был очень мал, поэтому,
а также из-за сложности установки и
высоких требований к
гранулированному теплоносителю, процесс не
нашел промышленного применения для
совместного получения ацетилена и
этилена.
Описанные установки
регенеративного пиролиза сложны в
эксплуатации, так как для повышения
выхода целевых продуктов требуются вакуум, относительно высокие
температуры и жаростойкие материалы (окись алюминия).
Пиролиз в трубчатых печах
Ацетилен получают в трубчатых печах при тех же основных
условиях, которые требуются для регенеративных процессов: высокая
температура, низкое парциальное давление исходного углеводорода
и кратковременный нагрев реакционной смеси. В трубчатых
реакторах теплообмен осуществляется через стенку, поэтому необходимо
следить за равномерностью обогрева труб (за отсутствием местных
перегревов) и применять металлы повышенной жаростойкости и
прочности.
98
■ Топливный
газ
Рис. II1-28. Схема пиролиза
углеводородного сырья в движущемся
слое твердого теплоносителя:
1 — пылеотделитель; 2 — сепаратор;
3 —нагревательная камера; 4
—топочная камера; 5 — реакционная камера;
6 — подогреватель воздуха.
Процесс в трубчатых печах имеет большие преимущества по
сравнению с регенеративными установками: возможность создания
непрерывного процесса и получения более концентрированных газов,
не разбавленных продуктами сгорания. На протекание процесса
влияет материал труб, причем в данных условиях олефины более
чувствительны к каталитическому действию металлов, чем парафины.
В присутствии кобальта, железа, меди и особенно никеля ускоряется
разложение сырья до углерода и водорода. Наличие хрома снижает
Вода
Газ
пиролиза
Вода
Рис. 111-29. Схема опытной установки фирмы Koppers-Hasche с трубчатым
реактором:
1 — подогреватели; 2 — пароперегреватель; 3 — зона смешения; 4 — реакционная трубка;
5 — сердечник из карбида кремния; 6 — нагревательные элементы; 7 — зона «закалки»;
8 — вакуум-насос; 9 — сепаратор.
действие никеля, поэтому целесообразно применять сплавы,
содержащие оба эти компонента.
На опытной установке37, построенной фирмой Koppers-Hasche
(рис. II1-29), было исследовано влияние на процесс пиролиза
температуры и времени контакта. Реактором служила трубка длиной 1,73 м
и диаметром 100 мм из карбида кремния с сердечником диаметром
75 или 89 мм из того же материала. В трубку подавали смесь
углеводородного сырья (пропан, бутан, бензин, реактивное топливо) и
водяного пара, нагретую до 900—1100° С. Условия проведения
опытов следующие; температура стенки трубки 1160—1480° С, давление
0,23—1 am, количество водяного пара 2—8 кг на 1 кг сырья, время
контакта 0,018—0,14 сек.
Во всех случаях выход газа возрастает с повышением
температуры, причем выход этилена уменьшается и при 1100° С этилен
практически не образуется. В газе пиролиза пропана и бутана при 870° С
содержится примерно 40% этилена. При пиролизе пропана, бутана
и бензина (общее давление 0,5 am) максимальный выход ацетилена
достигает 35% и соответствует температуре 980—1040° С. С ростом
давления до 1 am оптимальная температура повышается (до 1200° С),
но по абсолютной величине выход ацетилена оказывается меньшим,
чем при пониженном давлении. При уменьшении времени контакта
выход ацетилена и этилена возрастает, однако, если одновременно
повышается общее давление, увеличивается выход только этилена.
Выход сажи и смол возрастает с увеличением молекулярного веса
7* 99
сырья, а при пиролизе бензина и керосина, кроме того, и с
повышением давления и температуры.
В нашей стране на опытно-промышленных установках в течение
ряда лет ведутся опыты с целью создания отечественного метода
получения ацетилена в многопоточной трубчатой печи41'42.
Основным аппаратом является печь с беспламенными панельными
горелками. Трубы изготовлены из железо-хромо-алюминиевого сплава,
пригодного для работы в условиях температур порядка 1200° С и
обладающего удовлетворительными механическими свойствами.
«Закалка» газов пиролиза осуществляется в специальном аппарате
коллекторного типа, куда через восемь форсунок подается вода. Аппарат
установлен на двух Катковых опорах, которые вместе с П-образным
компенсатором воспринимают изменения длины труб при перепаде
температуры в печи.
Процесс проводится с периодическими остановками для прожига
кокса, постепенно накапливающегося в трубках. В период прожига
в трубки подают воздух. Длительность работы печного агрегата между
прожигами зависит от вида сырья (например, на бутане агрегат может
работать беспрерывно 250—350 ч). Время, расходуемое на прожиг,
зависит от температуры и составляет 10—20% рабочего цикла.
Температура газов прожига достигает 1050° С. Концентрация окислов
углерода в газах прожига 1,6—3 объемн. % при работе на бутане
и 4—8 объемн. % — при работе на пропане, если в нем высоко
содержание непредельных углеводородов (пропан-пропиленовая
фракция).
Типичный состав газов и выход компонентов при пиролизе бутана
в трубчатой печи (1050° С, сырье : пар = 1:2) приведен ниже:
Ацетилен
Этилен
Метан
Этан
Пропилен
Ароматические углеводороды
Гомологи ацетилена ....
Водород
Окись углерода
Смола и кокс
Технологические показатели работы трубчатой печи42 приведены
в табл. II1-13.
По основным показателям процесс в трубчатой печи мало
отличается от установок регенеративного пиролиза. Сложность
конструирования подобных печей и отсутствие достаточно жаростойких
материалов для изготовления труб затрудняет дальнейшее развитие этого
способа, поэтому он пока не вышел за масштабы
опытно-промышленных установок. Однако следует заметить, что развитие в химической
промышленности различных высокотемпературных процессов, осу-
100
Состав
объемн. %
12,80
14,60
26,10
1,00
0,50
0,25
1,70
40,60
2,15
0,30
Выход
вес. %
20,75
25,50
26,03
1,87
1,37
0,76
8,25
5,03
3,73
1,31
ТАБЛИЦА 111-13
Результаты пиролиза пропана и бутана в трубчатой печи
Показатели
Нагрузка по сырью, кг/ч ....
Весовое соотношение сырье : пар
Скорость потока в реакционных
трубках, м/сек . . .
Время контакта, сек
Температура, °С
исходной смеси
на входе в зону подогрева . .
на выходе из зоны подогрева
под сводом реакционной печи . .
газа пиролиза
после реакционной зоны . . .
после зоны «закалки» ....
Избыточное давление, am
на входе в зону подогрева . . .
после реакционной зоны ....
Плотность газа пиролиза, г/л . ,
Содержание непредельных
углеводородов в газе пиролиза, объемн.
%
Тепловой к. п. д. печного
агрегата, %
Пиролиз пропана
1200
1 : 3
97,7
0,138
140
716
1124
828
345
0,7
0,17
1,97
33,2
46,0
Пиролиз бутана
1400
1 : 2,57
89,6
0,134
140
729
1193
877
349
0,75
0,21
2,365
29,2
44,9
ществляемых в аппаратах трубчатого типа, а также появление
жаростойких высокопрочных материалов может привести к дальнейшему
совершенствованию этого перспективного метода пиролиза.
Пиролиз с электронагревом
Кинетические исследования процесса пиролиза углеводородов
показали, что если проводить процесс в вакууме в изотермических
условиях, то можно достичь высокого выхода ацетилена (90%). В
идеальных условиях (при оптимальном времени контакта и исключении
зоны промежуточных температур, в которой возможно протекание
побочных реакций) можно получать газ, состоящий только из
ацетилена и водорода.
Такой процесс с использованием электронагрева для создания
изотермической зоны был разработан в США Отмером39'40 на
лабораторных и пилотных установках с применением различного
углеводородного сырья (от метана до дистиллятов нефти). Реактор
представляет собой сосуд с концентрически расположенными трубками.
Собственно реакционная трубка из алюминия помещена в графитовый'
элемент сопротивления. Последний обеспечивает необходимый
электронагрев, для чего используется напряжение 3 кв. Для
осуществления селективного пиролиза метана до ацетилена и водорода
необходимо точно установить границы реакционной зоны. За ее начало при-
101
нимается точка, температура которой примерно на 250° С ниже
максимально достигаемой температуры процесса. Разница между
температурой газа в реакционной зоне и температурой стенки реактора
составляет примерно 100° С. Таким образОхМ создаются почти
изотермические условия.
Углеводород (метан) вводится в реактор при нормальной
температуре, а в реакционную зону поступает при температуре выше 1500° С;
получающиеся газы непрерывно выводятся из нее при 600—800° С.
Время реакции 0,05 сек, процесс проводится при остаточном
давлении ~100 мм рт. ст. Из реакционной зоны газ пиролиза охлаждается
сначала в камере с водяной рубашкой, а затем происходит его
быстрое охлаждение до 300—600° С в отдельном холодильнике путем
впрыскивания рециркулирующего охлажденного газа, благодаря
чему предотвращается разложение, гидрирование и полимеризация
образовавшегося ацетилена. Охлажденные газы очищаются от сажи
в специальном фильтре.
Степень конверсии метана 90% за один проход. В газе пиролиза
содержатся (в объемн. %):
Ацетилен 21,1 Высшие ацетиленовые
Этилен 0—0,1 углеводороды . . 0,1
Метан 4,9 Водород 74,0
Водород, получающийся в этом процессе после выделения
ацетилена, можно непосредственно использовать для синтеза аммиака,
что существенно улучшает технико-экономические показатели метода.
ГОМОГЕННЫЙ ПИРОЛИЗ В ПОТОКЕ ГАЗА-ТЕПЛОНОСИТЕЛЯ
В последнее время появились процессы совместного получения
ацетилена и этилена пиролизом различного сырья (от пропана до
легкого бензина) в потоке высокотемпературного газа-теплоносителя.
К таким процессам относятся методы фирм Eastman-Kodak (США),
SBA-Kellog (США), Hoechst (ФРГ), Montecatini (Италия), Phillips
(США)43-44 и др. Теплоносителем при гомогенном пиролизе являются
топочные газы, получаемые при сгорании топливного газа в
атмосфере воздуха или кислорода. Тепловое напряжение в камерах
сгорания и в реакционной зоне очень высоко, что позволяет создавать
относительно малогабаритные реакторы при высокой
производительности по сырью.
При гомогенном пиролизе в газовой фазе осуществляется
непосредственный контакт пиролизуемого сырья с газом-теплоносителем,
поэтому реакторы можно изготовлять из металла с минимальным
использованием огнеупорных материалов (футерованы только стенки
реакционной камеры). Охлаждение стенок осуществляется водой или
паром, создающим завесу. Применение металла значительно
упростило конструкцию и уменьшило капитальные затраты на основное
оборудование. При таком конструктивном решении облегчается
«закалка» реакционных газов и снижаются выделение сажи и отложе-
102
ние кокса. В отличие от регенеративных процессов, в реакторе
создается более высокая температура, благодаря чему выход ацетилена
и этилена при гомогенном пиролизе выше, однако общий тепловой
к. п. д. гомогенных процессов несколько меньше.
Процесс фирмы Eastman-Kodak43(США). Реактор, разработанный
фирмой Eastman-Kodak (рис. 111-30), испытан в масштабе
полузаводской установки. Топочные газы, образующиеся в камере 2 при
сжигании природного газа или пропана, поступают в смеситель 3,
куда подается пиролизуемое сырье (пропан или бензин),
разбавленное паром. Требуемая температура достигается регулированием
количества пара.
Реакционная камера футерована
окисью циркония,
выдерживающей температуру до
2500° С.
Пиролиз протекает при
давлении, несколько
превышающем атмосферное, и
при 2000° С. По окончании
процесса температура
снижается до 900° С.
Образовавшаяся газовая смесь
проходит «закалку» водой
и направляется на
охлаждение, очистку от сажи и
далее на установку выделения ацетилена и этилена. Объемное
соотношение ацетилена и этилена в реакционных газах может изменяться
от 0,3 : 1 до 4 : 1 в зависимости от состава пиролизуемого сырья и
температуры процесса. Например, при сжигании части пропана
(около 20%) в атмосфере кислорода температура топочных газов
достигла 1985° С. Затем эти газы смешивались с водяным паром и
основным потоком пропана, подогретого до 600° С. В результате
пиролиза получался газ следующего состава (в объемн. %):
Сырье
Газ пиролиза
Рис. II1-30. Реактор фирмы Eastman-Kodak для
пиролиза углеводородов в потоке
газа-теплоносителя:
/ — горелка; 2 — топочная камера; 3 — смеситель;
4— реакционная камера; 5—смотровое окно; 6— зона
«закалки».
Ацетилен 9—10
Этилен 16—18
Метан 17—18
Пропилен 0,04
Водород 32—35
Окись углерода . . 17-—18
Двуокись углерода . ' 4—5
Азот 0,4—0,5
При этом выход целевых продуктов составлял (в вес. % от сырья):
*
Ацетилен 18,6
Этилен 36,7
Пропилен 3,2
При повышении температуры процесса до 2000—2100° С
концентрация целевых продуктов в газе пиролиза значительно снижалась:
3—8 объемн. % ацетилена и 4,5—5 объемн. % этилена. Выходы
продуктов, соответствующие интервалу 1985—2100° С, приведены
в табл. IIM4.
103
Таблица nl-i4
Выход продуктов при пиролизе пропана и газового бензина по методу фирмы
Eastman-Kodak
Продукты пиролиза
Ацетилен
Этилен
Пропилен •
Всего
при
18,6
36,7
3,2
58,5
Выход i
1родуктов, вес. %
пиролизе пропана *
25,0
29,9
1,7
56,6
26,3
30,1
1,0
57,4
45,6
9,6
0,0
55,2
при пиролизе газового
бензина **
18,7
40,3
3,4
62,4
26,6
31,3
1,8
59,7
42,6
10,4
0,0
53,0
* Теплоноситель — продукты сгорания пропана в кислороде.
** Теплоноситель — продукты сгорания природного газа в воздухе.
При выходах, указанных в табл. III-14, в газе пиролиза
получается низкое соотношение С2Н2 : С2Н4 (примерно 0,3 : 1). При таких
соотношениях выход целевых продуктов пиролиза бензина выше,
чем при пиролизе пропана. При более высоком соотношении С2Н2 :
: С2Н4 (например 4 : 1) выход продуктов из газового бензина меньше,
чем из пропана. Во всех случаях суммарный выход.целевых
продуктов превышал 50%. Наряду с этим получались ароматические
углеводороды (2—3 вес. % от исходного пропана): 65% бензола, 19%
толуола, 16% ксилола и других тяжелых углеводородов. При пиролизе
газового бензина выход ароматических углеводородов иногда
достигал 25 вес. %.
Способ фирмы Eastman-Kodak не получил промышленного
применения из-за сложной конструкции реактора и высокого расхода
сырья.
Процесс фирмы SBA-Kellog44'45. Этот процесс осуществляется
путем впрыскивания смеси парообразного углеводородного сырья
(например, лигроина) и водяного пара в раскаленные топочные газы.
В качестве топливного газа можно применять, например, отходящий
газ после выделения ацетилена и этилена, коксовый газ или водород;
в период пуска можно также использовать легкие бензины. Процесс
позволяет получить ацетилен и этилен с суммарным выходом 52—
70 вес. % от исходного сырья, при этом остальная часть сырья
превращается в синтез-газ (СО + Н2), пригодный для химической
переработки.
На рис. 111-31 показан реактор, применяющийся для пиролиза.
Кислород и топливный газ, предварительно подогретые, проходят
через многочисленные каналы, расположенные вокруг верхней части
камеры сгорания /, поджигаются и образуют устойчивое пламя.
Камера сгорания / выполнена из металла и ограничена огнеупорным
кольцом 2 с водяным охлаждением. Кроме того, она защищается
завесой из водяного пара, впрыскиваемого параллельно стенкам по
104
Топливный
Кислород
периметру. При необходимости карборундовое кольцо можно
заменять. Предварительно подогретое сырье (лигроин; пределы
выкипания 30—150° С) смешивается с небольшим количеством водяного
пара, испаряется в подогревателе и через каналы, окружающие
камеру сгорания, равномерно поступает в реакционную камеру 3. По
металлической стенке последней
непрерывно стекает тонкая
водяная завеса, препятствующая
отложению сажи на стенках.
В нижней части реакционной
камеры предусмотрена «закалка»
газов пиролиза водой. Длину
реакционной камеры, а
следовательно, время ацетиленообразо-
вания можно изменять с помощью
специального устройства 6.
Производительность таких
реакторов достигает 10 000 ml год по
ацетилену и этилену (суммарно).
Существенное значение для мая вода
проведения процесса имеет
количество водяного пара. Мини- Soda на
мальный расход пара
определяется его количеством,
добавляемым к топливному газу, и
расходом на охлаждение стенок
камеры сгорания. Увеличение
подачи пара позволяет снизить
температуру топочных газов и
изменить соотношение
восстановителей (СО и Н2) в газах
пиролиза. Оптимальное количество
пара зависит от соотношения
между паром, лигроином и
топливным газом и от заданного
соотношения СО : Н2. Например,
при необходимости уменьшения
количества синтез-газа нужно
добавлять больше пара, при
этом получается больше
ацетилена и этилена. При снижении подачи пара выход синтез-газа
возрастает (этого можно добиться также, увеличив соотношение
топливного газа и кислорода в зоне сгорания).
В табл. II1-15 приведены показатели пиролиза лигроина по
методу SBA-Kellog45.
Распределение углеводородов в продуктах пиролиза лигроина
при разном мольном соотношении С2Н4 : С2Н2 графически показано
на рис. II1-32. Кроме углеводородов образуются также жидкая
105
на очистку
от сажи
Распылен-
Рис. II1-31. Реактор фирмы SB A-Kellogg
для пиролиза углеводородов в потоке
газа-теплоносителя:
/ — камера сгорания; 2 — карборундовое
кольцо; 3 — реакционная камера; 4 — зона
«закалки»; 5 — термопара; 6—устройство для
регулирования границ реакционной зоны.
ТАБЛИЦА 111-15
Показатели пиролиза лигроина по методу SBA-Kellog*
Показатели
Мольное соотношение С2Н4 : С2Н2
0,44
0,50
374
272
250
60
270
600
2680
962
9,3
4,6
8,0
0,1
0,3
0,6
0,2
43,7
25,1
5,3
2,8
28,0
15,0
1,68
510
250
232
60
112
500
2630
979
7,8
11,9
10,4
0,8
2,5
1,1
0,1
35,4
24,1
4,8
1,8
15,8
28,6
1,69
373
203
178
542
133
580
1880
640
8,5
14,4
13,5
0,7
2,8
1,6
0,4
32,5
12,3
10,0
3,3
17,3
31,6
1,94
Расход
лигроин, кг/ч
топливо (коксовый газ),
м3/ч
кислород (92% О2), м3/ч
водяной пар, кг/ч
в зону горения . . . .
на процесс
Температура, °С
подогрева лигроина . . .
в зоне горения (расчетная)
Количество газа пиролиза,
м3/ч
Состав газа пиролиза, объемн.
%
ацетилен
этилен
метан
другие парафины . . . .
высшие олефины . . . .
гомологи ацетилена . . .
ароматические
углеводороды
водород
окись углерода
двуокись углерода . . . ,
азот
Выход целевых продуктов в
расчете на превращенный
лигроин, вес. %
ацетилен
этилен
230
199
187
462
185
500
2030
584
10,0
4,4
8,9
0,2
0,5
0,7
0,2
42,4
17,8
10,5
4,3
30,3
14,4
360
245
188
545
127
580
1380
650
7,7
14,9
14,5
0,4
3,1
1,8
0,2
33,3
10,9
9,5
3,6
16,2
33,6
* Данные опытной установки производительностью 900 т ацетилена в год.
смола и сажа, причем соотношение сажи и смол увеличивается при
уменьшении соотношения С2Н4 : С2Н2.
Выход и соотношение целевых продуктов в газе пиролиза влияют
на количество тепла, необходимого для процесса (его определяют по
температуре топливного газа на выходе из горелки), и на расходные
показатели (рис. 111-33 и Ш-34). При изменении соотношения С2Н4 :
: С2Н2 от 0,1 до 2 выход меняется следующим образом (в вес. %):
Ацетилен ....
Этилен + ацетилен
От 16 до 36
От 40 до 50
Процесс фирмы Hoechst. Фирмой Hoechst (ФРГ) разработан
своеобразный процесс гомогенного пиролиза углеводородного
106
сырья48 : в настоящее время по этому способу работает установка
мощностью по ацетилену 50 000 т/год в г. Франкфурте.
Принципиальная схема реактора изображена на рис. II1-35.
В верхнюю часть камеры сгорания / подается топливный газ с высо-
4/7
1
35
SO
25
8
О
6
2
3
О
0,8
О
0,8
•О
'\
0,5 1,0 1,5 2,0
/4\
2,5 3,0 0,5 1,0 1,5 2,0
Мольное соотношение
2,5 3,0 0,5
С2Н4 : C2H2
1,0 1,5 2,0 2,5 3,0
Рис. II1-32. Выход продуктов пиролиза лигроина в потоке газа-теплоносителя при
разном мольном соотношении этилена и ацетилена:
/ — этилен; 2 — пропилен; 3 — бутадиен; 4 — бутилен; 5 — пентадиен; 6 — пентен; 7 —
ацетилен; 8 — метилацетилен; 9 — диацетилен; 10 — винилацетилен; // — циклопентадиен;
12 — гдета,н; 13 — этан; 14 — пентан; 15 — гексан; 16 — бензол.
65
I
|55
О
§ 35
L
^ 700 900 1100 1300
^° Температура,°С
Рис. II1-33. Зависимость
между температурой
топливного газа на выходе
из горелки и содержанием
ацетилена в смеси с
этиленом.
/
/
/
/
1
6000
то
§
ч
1
;
2
и=
^3
2000
°0,25 0}5 1 1,5 2 2,5
Весобое соотношение С2Н4:С2Н2
Рис. II1-34. Расходные коэффициенты
на 1 т суммы этилена и ацетилена при
пиролизе лигроина в зависимости от
соотношения С2Н4 : С2Н2 в газе
пиролиза:
/ — пар; 2 — лигроин; 3 — кислород.
ким содержанием водорода, который сгорает в кислороде с
образованием так называемого вторичного пара. Обычно в качестве
топливного газа используется газ, отходящий с производства ацетилена.
Тепловыделение в камере 1 превышает 8,9 • 106 ккал/(м3-ч), для
регулирования температуры туда подается водяной пар. Камера изготов-
107
лена из металла и охлаждается водой, при этом потери тепла очень
невелики. Отсутствие керамических деталей позволяет производить
быстрый пуск и остановку реактора.
Газообразный теплоноситель, имеющий высокую температуру,
смешивается с предварительно нагретым сырьем (бензин, лигроин)
и вместе с ним входит в реакционную камеру 2, где протекают
основные реакции разложения углеводородов. Температура газов при
этом непрерывно снижается. Время реакции около 0,001 сек. Для
сохранения образовавшегося
ацетилена газовая смесь
подвергается «закалке» водой в зоне 3.
Для улавливания сажи и смол,
имеющихся в газе пиролиза,
в нижнюю часть реактора
подают тяжелую нефтяную
ароматическую фракцию (на рисунке
не показано), основными
компонентами которой являются
декалин, дифенил и др.
Выходящий из реактора газ
охлаждается в
котле-утилизаторе 2 (рис. II1-36) и после
промывки в сепараторе (на рис. не
500 W00 1500 2000 2500 3000 П0Казан) поступает во фракцио-
Температура, °с
Вода
Газ
пиролиза
Рис. III-35. Схема реактора фирмы Hoechst
для пиролиза углеводородного сырья
в потоке газа-теплоносителя (сбоку
показана кривая распределения температур):
У—камера сгорания; 2— реакционная камера;
3 —зона «закалки»; Гн—начальная
температура; Тк — конечная температура.
нирующую колонну 3, где от
него отделяются в первую
очередь наиболее тяжелые
компоненты. Для удаления более
легких компонентов (на рисунке
не показано) газ охлаждается;
при этом легкая фракция,
состоящая в основном из бензола,
декалина, нафталина и др., конденсируется. Затем она поступает на
орошение колонны 3. Окончательно газ охлаждается в
холодильнике 4. Охлажденный газ сжимается компрессором 6 и поступает
в систему очистки. Вначале в аппарате 7 путем промывки щелочью
или другими реагентами от газа отделяют сероводород и двуокись
углерода. Затем удаляют ацетиленовые и другие нежелательные
углеводороды трехступенчатой промывкой исходным
углеводородным сырьем, например бензином (на рис. не показано).
После этого газ проходит на установку 8 для выделения
ацетилена селективным растворителем. Выделенный ацетилен осушается
и направляется на переработку. Газ, содержащий этилен, сжимается
и поступает на низкотемпературную установку 9 для выделения
этилена. Остающийся газ, обогащенный окисью углерода и
водородом, возвращается в качестве топлива в реактор, метан расходуется
на производство аммиака, а этилен — на производство ацетальдегида
и других продуктов.
108
Выход продуктов пиролиза зависит от химического состава сырья
и температурного режима реактора (рис. II1-37). Повышение
температуры газа на выходе из реакционной камеры способствует увеличению
выхода ацетилена, однако при этом возрастают потери тепла с
отходящими газами. Например, при пиролизе легкого бензина, когда
температура на выходе из реакционной камеры равна 900° С, выход
ацетилена и этилена составляет соответственно 3 и 25 вес. %. При
Метан Этилен
Дцетилен
углерода
Рис. II1-36. Схема установки фирмы Hoechst для
совместного получения ацетилена и этилена пиролизом
углеводородного сырья:
/ — реактор; 2 — котел-утилизатор; 3— фракционирующая
колонна; 4—газовый холодильник; 5—газгольдер; 6—компрессор;
7 — аппарат для очистки от H2S и СО2; 8 —установка для
выделения ацетилена; 9 — установка для выделения этилена.
повышении температуры до 1400° С выход ацетилена возрастает почти
до 40 вес. %, а выход этилена снижается практически до нуля.
Поэтому на промышленной установке путем изменения
технологического режима можно изменять соотношение ацетилена и этилена от
4 : 1 до 1 : 2,2.
Газ пиролиза легкого бензина, полученный на установке фирмы
Hoechst, имел следующий состав (в объемн. %):
Ацетилен 10,70
Этилен 15,70
Метан \ 13,20
Этан 0,40
Пропан + двуокись
углерода 8,10
н-Бутан 0,01
Пентаны 0,15
Пропилен + изобутан 0,70
Бутилены 0,04
Бутадиен 0,67—1,3
Другие диены 0,60
Гомологи ацетилена . . 1,90
Бензол 1,50
Толуол 0,04
Водород 31,8—33,0
Окись углерода .... 11,90
Кислород 0,29
Азот 0,11
Вода 0,64
109
Исследования влияния качества исходного сырья на выход
ацетилена и этилена показали47, что состав газов пиролиза
определяется главным образом соотношением сумм парафинов, изопарафи-
нов, нафтенов и ароматических углеводородов в сырье. Наименьший
выход целевых продуктов и наибольшее смолообразование
наблюдаются при крекинге ароматических углеводородов.
Процесс фирмы Montecatini. Фирмой Montecatini (Италия)
разработаны способы получения ацетилена и этилена на основе
сочетания пиролиза низкооктанового бензина с окислительным пиролизом
метана 48. Поскольку
пиролиз бензина идет при более кислород
низкой температуре, на его
Метан
Температура, °С
800 1000 /200 МО 1600
Газ
гшро-
лиза
500 1000 1500 2000 2500
Энергия реакции, мм ал/кг
Рис. II1-37. Выход продуктов
пиролиза легкого бензина и степень его
разложения в зависимости от
энергии и конечной температуры
реакции:
/ — степень разложения; 2 — С2Н4;
3— С3Н8; 4 — СН4; 5—(СО+СО2+Н2);
6 — С4Н8; 7 - С2Н2.
О 500 1000 1500
Температура,°С
Рис. II1-38. Реактор фирмы Montecatini для
пиролиза метана и другого углеводородного
сырья до ацетилена (справа показана кривая
распределения температур):
/ — эжекционный смеситель; 2 — диффузор;
3 — горелочная плита; 4 — зона пиролиза метана;
5 — зона пиролиза бензина; 6 — зона «закалки».
расщепление можно расходовать часть тепла нагретых газов
неполного окисления метана.
На рис. II1-38 изображена схема реактора для совместного
получения ацетилена и этилена. Кислород и метан, предварительно
увлажненные и нагретые, смешиваются в эжекционном смесителе / и,
пройдя диффузор 2, поступают на горелочную плиту 3 с большим
числом отверстий. В реакционной зоне 4 происходит пиролиз метана
ПО
25°
О
до ацетилена при избыточном давлении 4 am и температуре около
1500° С. Ниже зоны 4 поступают пары бензина, нагретого до 500° С.
За счет пиролиза бензина в зоне 5 (1500—800° С) дополнительно
образуются ацетилен, этилен и синтез-газ. Получаемая газовая смесь
резко охлаждается водой, которая улавливает часть сажи и выходит
с низа реактора. Выход ацетилена и этилена зависит от ряда
факторов и в первую очередь от свойств бензина и температуры
предварительного подогрева сырья. На рис. II1-39 показана зависимость
выхода продуктов пиролиза от количества бензина, вводимого в
реакционную зону (в расчете
на 1000 м3 метана).
Фирмой Montecatini
разработан также процесс
получения ацетилена и
этилена из жидких
углеводородов 48, аналогичный про-
цессу фирмы SBA-Kellog,
но проводимый при
давлении около 10 am. В
камере сгорания реактора
можно сжигать как
газообразные, так и жидкие
углеводороды. Для
поддержания эффективного горения
соотношение топливного
газа и кислорода
принимают очень близким к
стехиометрическому.
Температуру регулируют,
изменяя подачу водяного пара.
Кислород и топливный газ компрессорами 11 и 8 подаются в
увлажнители 1 и 2 и после предварительного нагрева в подогревателе 3
вводятся в реактор (рис. II1-40). В камере сгорания 4 происходит
процесс полного горения. Сюда же поступают пары бензина. Ацетилен
образуется в основном в верхней реакционной зоне 5 при 1200—
1400 °С. В нижнюю реакционную зону 6 впрыскивается
дополнительное количество бензина, в результате чего температура там
снижается до 800° С и преобладает образование олефинов. Ниже
проводится окончательная «закалка» газов водой. Вода с сажей и смолой
уходит снизу аппарата, а газ пиролиза направляется в колонну 12,
где конденсируется водяной пар. Горячая вода, скопившаяся в кубе
колонны, нагнетается насосами в увлажнители 1 и 2.
По этому методу можно в широких пределах изменять
соотношение этилена и ацетилена в газах пиролиза путем изменения
соотношения водяного пара, кислорода и топливного газа, а также
количества первичного и вторичного бензина. Соотношение С2Н4 : С2Н2
в получаемом газе определяет расходные и тепловые показатели
процесса (рис. 111-41).
?П
50 WO 150 200
Количество бензина, кг
Рис. II1-39. Выход продуктов пиролиза в
зависимости от количества бензина, поданного
в реактор.
Данные рисунка относятся к бензину, имеющему следующие
характеристики:
Пределы выкипания, °С *. . 30—160
Групповой состав, вес. %
парафины 86,2
нафтены •....' 9,6
ароматические углеводороды 3,8
другие ненасыщенные углеводороды 0,4
В качестве исходного сырья в рассматриваемом процессе можно
применять различные бензины. Лучшие выходы получаются при
большем содержании парафинов в бензине. Присутствие нафтеновых
и ароматических углеводородов усиливает образование смол. Вообще
Рис. II1-40. Схема полупромышленной установки фирмы Моп-
tecatini для получения ацетилена и этилена пиролизом жидких
углеводородов:
jt 2 — увлажнители; 3 — подогреватель; 4 — камера сгорания;
5, 6— зоны пиролиза; 7—насос; 9, 10 — газодувки; 8, 11 —
компрессоры; 12 — фракционирующая колонна; 13 — сепаратор.
говоря, при использовании высококипящих жидкостей выход
целевых продуктов снижается.
Пиролиз бензина в потоке перегретого водяного пара. Интересен
разработанный в АрмНИИхимпроекте 49 метод пиролиза газового
бензина в токе перегретого водяного пара, получаемого от сжигания
водорода в избыточном кислороде, с частичным окислением сырья
в реакционной камере. При этом наряду с ацетиленом получается
большое количество этилена, пропилена и синтез-газа. В качестве
сырья был использован газовый бензин Бакинского месторождения
следующих характеристик;
112
Пределы выкипания, °С 28—148
Групповой состав, вес. %
парафины 54,48
нафтены 39,64
ароматические углеводороды
5,88
|
1500
W
1,2 1Л Ь6
Соотношение С2Н4:
1,8
\
- -
I—
\
\
3
11
_ /U
I —
Схема процесса заключается в следующем. Смесь водорода
(топливный газ) и кислорода при избыточном давлении 1,2—1,4 am
поступает в горелку реакционной печи. Газовый бензин испаряется
за счет сжигания топливного газа, перегревается до 200—250° С и
проходит в смеситель реакционной печи, где смешивается с горячими
продуктами сгорания (2400—2500°С), а затем попадает в реакционную
камеру. За счет тепла дымовых газов бензин пиролизуется.
Образующийся газ, температура которого составляет 1000—1400° С в
зависимости от принятого режима,
подвергается «закалке» водой. 2500г
Печь состоит из трех основ- Д W0Q
ных частей: топочной камеры,
смесительной камеры и
реакционной зоны. В топке с
диффузионным горелочным
устройством происходит
сжигание водорода. Для
охлаждения металлических стен и
уменьшения диссоциации
топочных газов при
случайном повышении температуры
в рубашку топочной камеры
подается вода. При
использовании водорода в качестве
топливного газа в камере
могут протекать реакции
горения водорода в кислороде
и частичной диссоциации
продуктов сгорания.
В смесительной камере продукты сгорания (в основном
перегретый, частично диссоциированный водяной пар) и избыточный
кислород смешиваются с перегретыми парами бензина. При смешении
более холодного потока бензина с горячими газами продукты
сгорания резко охлаждаются, вследствие чего протекают реакции
рекомбинации диссоциированных компонентов. При этом смесь паров
бензина и продуктов сгорания в основном состоит из Н2О, СО2 и Н2.
В реакционной зоне с устройством для «закалки» протекают
следующие процессы: 1) Окисление части бензина избыточным
кислородом до СО2 и Н2О с выделением тепла (—11 000 ккал/кг),
расходуемого на эндотермические реакции пиролиза основного количества
бензина. 2) Конверсия части бензина перегретым водяным паром
с образованием СО и Н2. 3) Пиролиз бензина с получением целевых
продуктов и «закалка» образующихся газов.
8 В. Н. Антонов ЦЗ
1,2 1,Ь 1,6 1,8
Соотношение С2Н4 : С2Н2
2,0
Рис. III-41. Влияние соотношения С2Н4:С2Н3
в газах пиролиза на показатели процесса:
/ — расход бензина; 2 — расход кислорода;
3— теплосодержание остаточного газа; 4— тепло,
рекуперируемое из охлаждающей воды при 135° С.
Для определения оптимального режима процесса и выхода
целевых продуктов проводили опыты при расходе газового бензина 28—
42 кг/ч и объемном соотношении топливного газа и окислителя
(Н2 : О2) в интервале от 1,59 : 1 до 2,14 : 1 (табл. II1-16).
ТАБЛИЦА 111-16
Выход ацетилена и этилена при пиролизе газового бензина
Расход бензина
кг/ч
Выход, кг/ч
С2Н2
с2н4
Выход, «рее. % от
количества бензина
С2Н2
с2н4
Н2: О2= 1,59
28
36
42
28
36
42
28
36
42
36
42
28
36
5,1
6,9
6,1
6,4
7,5
7,4
Н2
6,8
7,2
6,4
Н2
7,2
6,2
Н2
6,5
7,0
4,2
7,0
7,3
,= 1J2 : 1
5,5
8,3
8,1
= 1,86: 1
6,9
8,2
7,2
О2 = 2,0 : 1
8,1
7,9
О2= 2,14 : 1
6,5
7,9
17,8
19,0
16,9
23,0
20,7
17,6
24,4
19,9
15,2
20,0
14,8
23,5
19,3
15,0
19,5
20,2
19,7
23,0
19,3
24,1
22,8
17,1
22,5
18,7
23,2
22,0
Эксперименты показали, что для принятой конструкции аппарата
оптимальными являются следующие условия: расход сырья 36 кг/ч
и температура 1150—1200° С. При этом на 1 кг бензина расходуется
1,375 м3 водорода и 0,805 м3 кислорода. Состав получаемого газа
такой (в объемн. %):
С2Н2 . . . 9—10 С3Н6 .... 2—2,7 СО . . . .' 13—15
С2Н4 . . .9,1—10 Н2 . . . .45,5—48,2 СО, . . . . 6,5—7
СН4 ... 4—6 О2 0,6—0,8 N2 .... 2,5—3,5
С2Н6 . . . 1-1,5
На основе лабораторных данных были подсчитаны примерные
расходные показатели процесса:
u л На 1 т
rl a 1 /72 /"• т_г 1
Г тт ^2«2 -Г
<-2н2 + с2н4
Газовый бензин, кг 4850 2300
Кислород, кг 5600 2650
Электроэнергия, квт-ч 700 350
Установка фирмы Phillips. Фирмой Phillips (США) разработан
и испытан в полупромышленном масштабе тангенциальный реактор
114
Водород
Водяной
пар
„закалкии
Газ
^пиролиза
для пиролиза углеводородов в потоке
газообразного теплоносителя50 (рис.
II1-42). Смесь топлива (водород) с
воздухом или кислородом подается в
цилиндрическую топочную камеру, а
углеводородное сырье — в реакционную
зону, расположенную несколько ниже.
После интенсивного перемешивания
углеводородов с продуктами сгорания
газы движутся внутри реакционного
канала небольшого диаметра и затем
быстро охлаждаются водой. Процесс
проводится при атмосферном давлении.
В качестве сырья могут применяться
этан, пропан, бутан. Суммарный выход ацетилена и этилена
достигает 48—58 вес. % от исходного сырья.
ПИРОЛИЗ СЫРОЙ НЕФТИ С ПОГРУЖНЫМ ГОРЕНИЕМ
Фирма BASF (ФРГ) разработала способ получения ацетилена
пиролизом сырой нефти в пламени кислородной горелки. Процесс
был опробован на опытных установках производительностью от 2,4
до 36 т нефти в сутки 51«52. На последней установке получали в сутки
6 т ацетилена.
вода
Рис. II1-42. Схема
тангенциального реактора фирмы Phillips.
Рис. II1-43. Схема получения ацетилена пиролизом нефти с погружным горением:
/ — регулирующая заслонка; 2 — подогреватель кислорода; 3 — аварийный выключатель;
4 — фотоумножитель; 5 — реактор; 6 — горелка; 7 — котел-утилизатор; 8 — скруббер;
9 — циклон; 10 — оросительный холодильник; // — сепаратор.
Принципиальная схема процесса показана на рис. II1-43. Сжатый
до 8,7 am 98%-ный кислород пропускают через регулирующую
заслонку 1 в подогреватель 2, который обогревается теплом,
выделяющимся при сжигании нефти, отбираемой из нижней части реактора 5.
Кислород, подогретый до 600° С, смешивается в горелке 6 с исходной
нефтью. Полученную смесь выпускают в реактор 5 через сопло Лаваля
с большой скоростью — выше 1000 м/сек. При этом в нижней части
реактора в слое нефти мгновенно возникает пламя, температура
которого равна 1480° С. При высокой температуре пламени происходит
пиролиз нефти с образованием ацетилена. Размеры факела горения
8* 115
ограничиваются окружающей его нефтью, температура которой
специально поддерживается 200 —260° С. Резкое снижение температуры
по периферии факела предотвращает разложение ацетилена.
Поскольку реакции пиролиза осуществляются при высоких
скоростях потока, процесс можно вести при повышенном давлении без
существенного уменьшения выхода ацетилена. Проведение процесса
под давлением исключает последующее компримирование газа
пиролиза, что очень удобно, поскольку в нем имеется большое количество
легко полимеризующихся веществ, которые затрудняют сжатие.
Горячий газ пиролиза, насыщенный низкокипящими
углеводородами, отмывается в скруббере 8 легким бензином от сажи и нефтяного
битума. Эмульсия сажи в бензине отделяется от газового потока
в циклоне 9 и возвращается в реактор. Окончательная конденсация
паров бензина происходит в орошаемом водой холодильнике 10. Бензин
после отделения от воды в сепараторе 11 возвращают в скруббер 8.
Если в исходной нефти имеются сернистые соединения, то они
переходят в газ пиролиза в виде сероводорода, сероокиси углерода
и сероуглерода. Для отделения сернистых соединений газ промывают
сырой нефтью (при этом удаляются также углеводороды С4). В
отсутствие серы в исходном сырье крекинг-газ направляют
непосредственно на стадию выделения ацетилена N-метилпирролидоном. После
этой стадии из отходящих газов можно выделить этилен и пропилен
методом низкотемпературной ректификации.
В реакторе необходимо поддерживать определенную постоянную
концентрацию сажи, образующейся при сжигании и пиролизе нефти.
Это достигается сжиганием сажи в горелке 6 при циркуляции через
нее некоторого количества нефти из нижней части реактора. Часть
сажи вместе с нефтью сжигается в подогревателе 2.
Для поддержания в нижней части реактора температуры 200—
260° С некоторое количество нефти непрерывно отводят из него и
охлаждают водой в теплообменнике (на рисунке не показан), в
результате чего образуется пар низкого давления, который используется
на стадии выделения ацетилена. При этом удается на 1 т нефти
получить около 1 т пара.
Установка снабжена автоматической защитой — при погасании
пламени (фиксируется фотоэлементом) прекращается подача
кислорода и система продувается азотом. Это исключает возможность
попадания углеводородов через горелку в трубопровод для кислорода
и образования в нем взрывоопасной смеси.
Состав газов пиролиза (в объемн. %) мало зависит от природы
сырья и давления процесса:
При При При При
1,5 am 8,7 am 1,5 am 8,7 am
Ацетилен 8,0 7,0 Гомологи ацетилена 1,0 0,9
Этилен 6,5 6,6 Водород 23,0 27,0
Метан 4,0 4,0 Окись углерода . .42,1 45,0
Пропилен 1,2 1,2 Двуокись углерода 9,0 6,0
Углеводороды Q , 2,0 1,0 Азот и др 3,2 1,3
116
70
I.
Для получения 1000 м* газа пиролиза расходуется 0,5—0,8 т
нефти и 370 м3 кислорода. В ацетилен превращается ~16 вес. %
исходного сырья.
СРАВНЕНИЕ РАЗЛИЧНЫХ СПОСОБОВ
ПИРОЛИЗА УГЛЕВОДОРОДОВ
Одним из основных преимуществ получения ацетилена пиролизом
углеводородного сырья является возможность использования для
этих целей самых разных углеводородов, что позволяет строить
установки пиролиза в районах с практически любыми углеводородными
ресурсами. В основном выбор сырья определяется
производительностью установки и заданным соотношением ацетилена и этилена
в получаемом газе.
На рис. II1-44 приведена зависимость выхода ацетилена и этилена
от вида сырья для случая, когда установка предназначена для работы
на индивидуальном сырье. Максимальный выход целевых
продуктов получается при пиролизе этана
(углеродный показатель* 2). Выход
ацетилена сначала уменьшается (при
увеличении углеродного показателя
до 4), а затем остается постоянным.
Суммарный выход ацетилена и
этилена с увеличением молекулярного
веса сырья постепенно снижается.
Практически достигаемые выходы
определяются температурой,
временем контакта и общим давлением
процесса, независимо от применяемого
технологического оформления метода
ПИрОЛИЗа35. 37,49,52, 53,
В табл. Ш-17 приведены
сравнительные данные о различных
процессах пиролиза с учетом выхода целевых компонентов и
технологических параметров. Как видно из таблицы, при регенеративных
процессах и процессах в трубчатых печах можно получать большие
концентрации ацетилена, чем при процессах с газовым
теплоносителем. Однако регенеративные процессы, несмотря на преимущества
по расходным показателям, особенно в части выделения ацетилена,
связаны со сложными конструктивными решениями и требуют
применения жаростойких материалов. Наиболее существенный
недостаток процесса — периодичность. Перспективность развития
процессов в трубчатых печах целиком обусловлена изготовлением
жаростойких труб.
Наибольшее распространение получили способы пиролиза в
потоке газа-теплоносителя. Они отличаются гибкостью (возможность
50
1
«8 to
V
V.
,С2Н2+
/С2Н2
С2Н4
5 9 13
Углеродный показатель
17
Рис. II1-44. Выход ацетилена и
этилена при пиролизе различного
углеводородного сырья.
* Углеродный показатель (С-число, С-индекс) означает среднее число
атомов углерода, приходящееся на 1 моль исходной углеводородной смеси.
117
ts;
to
b-.
дии
я о о
о х <у
citf
О) О) О
н о о.
U и и
3 к
3 О) CQ
s з о
О д Я
О-, я 03
gg tS
,6 ,6 .6
3 >» g Э?^^ 3 >>s
3 CD 5 3 рн CQ 3 d) cQ
S3O ^ л О S3O
О я Я ОдЯ О я Я
О, w й ^ Я °3 Сч w сз
Е £ н CoJh tZlS^
18
С Н
и
00
СМ
CD
ю
ю
ю
ю
со"
^s
ftco ^
О эт
и
ж
о
г^
^-Г ^
и
1^.
0,7
ю
(N
о
о
X
я
PQ
СО.
ю
ю
со
ю
"чГ
J,
'cD
ю
со
CD
LO
о"
О
3
s
X
ев
Bt
0)
S
ua
s
Е
Cfl
ej
О.
Йи
а°
0)
Sft
я 3
§
и
Сырь
ю
о
8
СО
О
8
О
ю
о
8
О
см
о
00
сС
о
о
о
о
Tf
О
00
OvS
глеСЗ
5 з |
*§!
си Он2
3 g си
^ о S
9go
я
СО
S
я"
сз
С
О
Он
с
I.U
Е-
>.
\О
опан, i
Он
t—t
бен
ропан,
С
ЗИН
я
ы
Лигро
я
Бензи
ккщ.
cgs
а ° 2?
s g"04
со V «
Я я со
CU со CQ
с я
СО
Он
сЗ
я я
сз О
с о,
as
роцесс
с
■§-
ч
>>
CQ
Hasche
00
!_i
CL)
Он
Он
о
1тых пе
ЕГ1
VO
>■»
Он
CQ
-Kodak
с
со
"с/5
со
W
log
О)
<
DQ
СО
3
я
£s
>-> си
Он Я
о5
С Он
ииС
# * #
изменения мощностей по ацетилену и этилену) и внедрены в
промышленность в ФРГ, США, Японии и СССР. Удачное конструктивное
решение узлов сжигания топливного газа и смешения продуктов
сгорания с парообразными углеводородами привело к созданию
удобных в эксплуатации аппаратов. Проведение процесса под давлением
позволяет улучшить технико-экономические показатели, но
расходные коэффициенты различных способов с газовым теплоносителем
зависят от многих факторов и в первую очередь от соотношения
ацетилена и этилена в газах пиролиза, поэтому сравнение их в
сопоставимых условиях затруднено. В последнее время в Англии, Франции и
Голландии получили также распространение регенеративные
процессы.
При пиролизе с преимущественным получением ацетилена общий
выход целевых продуктов уменьшается, поэтому такое направление
пиролиза экономически невыгодно. Пиролиз целесообразно вести
с целевым выходом ацетилена и этилена и даже с получением
большего количества этилена. Отсюда ясна также необходимость
кооперирования производств ацетилена и этилена с организацией их
переработки.
ЛИТЕРАТУРА
1. Тиличеев М. Д., Химия крекинга, Гостоптехиздат, 1941.
2. Степухович А. Д., Кинетика и механизм термического крекинга алканов,
Изд. Саратовского университета, 1965.
3. Ф р о с т А. В., в сб. «Труды завода «Химгаз», № 2, 98 (1934).
4. Брукс, Бурд, КурЦ, Шмерлинг, Химия углеводородов нефти,
т. 2, Гостоптехиздат, 1958.
5. Л а в р о в Н. В., Коробов В. В., Филиппова В. И.,
Термодинамика реакций газификации и синтеза из газов, Изд. АН СССР, 1960.
6. Введенский А. А., Термодинамические расчеты нефтехимических
процессов, Гостоптехиздат, 1960.
7. Р i e t s с h H., Erdol u. К о h 1 е, 10, № Ю, 666 (1957).
8. Фастовский В. Г., Метан, Гостоптехиздат, 1947.
9. М у х и н а Т. Н., Барабанов Н. Л., Майоров В. И. и др., в сб.
«Труды НИИСС», № 1, Госхимиздат, 1959.
10. F i с h e r F., P i с h I e r H., Brennst. Chem., 11, № 24, 501 (1930); 13, № 20,
281 (1932).
П. Гриненко Б. С, Зелизный А. М., Производство ацетилена из
природного газа, Гостехиздат, Киев, 1963.
12. Знаменский Н. Н., Франк-Каменецкий Д. А., ЖФХ, 20,
№ 11 (1946).
13. Козлов Г. И., Кнорре В. Г., Газ. пром., № 1 (1963).
14. L ер г i псе P., Genie Chimique, 89, № 5, 137 (1963).
15. Нои К. С, Anderson К. С, J. Phys. Chem., 67, № 8, 15797 (1963).
16. Полякова М. М., Рафалькес И. С., Рабинович Е. Я., Тес-
нер П. А., в сб. «Труды ВНИИгаз», вып. И, Гостоптехиздат, 1959.
17. Frolich R. К., Wiezevich P. I., Ind. Eng. Chem., 27, 1055(1935).
18. С о г г i g a n Т. Е., К о b e К. A., Unit processes in organic synthesis, New
York, 1958.
19. H e p p H. I. et al., Ind. Eng. Chem., 41, № 11, 2531 (1949).
20. Schmidt H., Brennst. Chem., 33, № 9/10, 144 (1956).
21. Tropsch H., Egloff G., Ind. Eng. Chem., 27, 1963(1935).
Mop и на И. Н., в сб. «Химическая переработка нефтяных углеводородов»,
23. F а г n s w о г t h J.,' Ind. Eng. Chem., 47, 1517 (1955).
119
24. К i 1 p a t r i с к М. О. et al., Petrol. Ref., 33, № 4, 29 (1954).
25. Новейшие достижения нефтехимии и нефтепереработки, под ред. К. А. Кобе
и Д. Д. Мак-Кета, т. 2, Гостоптехиздат, 1961, стр. 98.
26. К i n n е у R. Е., С г о w 1 у D. I., Ind. Eng. Chem., 46, № 2, 258 (1954).
27. Л у к ь я н о в П. И., Б а с и с т о в А. Г., Пиролиз нефтяного сырья,
Гостоптехиздат, 1962.
28. Д а л и н М. А., Мухина Т. Н., Прокофьева Т. В.,
Талисман Л. В., в сб. «Химическая переработка нефтяных углеводородов», Изд.
АН СССР, 1956, стр. 79.
29. Коган П. С, Потоловский Л. А., Химия и технология топлив
и масел, № 12, 22 (1958).
30. Е b г е у G., Е п g e I d e r G., Ind. Eng. Chem., 41, № 11, 2531 (1949).
31. Тес н ер П. А., в сб. «Химическая переработка нефтяных углеводородов»,
Изд. АН СССР, 1956.
32. Переработка природного газа (ред. П. А. Теснер), Гостоптехиздат, 1961,
стр. 3—102.
33. Б е р к о в и ч В. Б., Гордон С. А., Газ. пром., № 2 (1962).
34. Химия ацетилена, под ред. А. Д. Петрова, Издатинлит, 1954.
35. Антонов В. Н., Производство ацетилена, Госхимиздат, 1959.
36. Chem. Proc, 9, № 1, 28 (1963).
37. В i x 1 е г G., С о b е г 1 е у G., Ind. Eng. Chem., 45, № 12, 2596 (1953).
38. Chem. Ing. Techn., 33, № 4, 248 (1961).
39. Othmer D., Chem. Age India, 16, № 2, 101, 157 (1965).
40. Chem. Week, 95, № 21, 89 (1964).
41. 3 у б к о в а К. А., Л а п и ц к а я О. И., Газ. пром., № 2, 24 (1964).
42. 3 у б к о в а К. А., Л а п и ц к а я О. И., Нефтепереработка и нефтехимия,
№ 1 (1963).
43. А с i n С. A., Reid Т. F., S с h r a d e r, Chem. Eng. Progr., 54, № 1, 41
(1958).
44. Pat ton, Grubb, S t e p h e n s о n, Petrol. Ref., 37, № 11, 80 (1958).
45. В arr у M. I. et al., Chem. Eng. Progr., 56, № 1, 39 (1960).
46. К r e k e 1 e r H., W e r t z R., P e с h t о 1 d N., Erdol u. Kohle, 12, № 5,
358 (1959).
47. Schneck E., Brennst. Chem., 44, № 11, 354 (1963).
48. Fa user G., Chim. e ind., 42, № 2, 150 (1960).
49. Хачатурян Т. А., Джанцмов А. Н., Нефтепереработка и
нефтехимия, № 9 (1964).
50. Petrol. Ref., 38, № И, 204 (1959).
51. Chem. Eng., 70, № 21, 92 (1963).
52. Кг о per Н., Platz R., 6th World Petroleum Congress, Francfurt/Main,
1963.
53. Friz H., Chem. Ing. Techn., 40, № 20, 992 (1968).
ГЛАВА
IV
ПОЛУЧЕНИЕ АЦЕТИЛЕНА
ЭЛЕКТРОКРЕКИНГОМ УГЛЕВОДОРОДОВ
При осуществлении электрического разряда в среде газообразных
углеводородов происходит местный разогрев газа и расщепление его
молекул на радикалы, рекомбинация которых приводит к
образованию ацетилена, его гомологов, сажи, водорода и некоторого
количества этилена. Такое действие электрического тока на
углеводородные газы впервые наблюдалось французским химиком Бертло1»2 еще
в 1860 г., однако в связи с недостаточным развитием электротехники
того времени это явление не нашло практического применения.
Только в тридцатых годах текущего столетия немецкой
фирмой BASF и советскими исследователями Н. И. Кобозевым, С. С.
Васильевым и Н. П. Божко были начаты подробные исследования
электрических разрядов в среде метана и других углеводородов. В
Германии эти исследования завершились созданием на заводе в г. Хюльсе
промышленной установки по получению ацетилена в реакторах,
использующих линейную электрическую дугу постоянного тока.
Установки подобного типа созданы в послевоенные годы в СССР и
Румынии.
В 1962 г. фирмой Du Pont (США) был разработан новый тип
реактора с коаксиальным расположением электродов для электрокрекинга
углеводородов. Несколько позднее на заводе в г. Монтегью построили
промышленную установку с таким реактором мощностью 25 тыс. т
ацетилена в год. В последнее время метод одноступенчатого
электрокрекинга усовершенствован; в реакционную смесь после выхода ее
из зоны электрической дуги добавляют новые порции углеводородов,
благодаря чему температура реакционных газов понижается с 1500—
1600 до 800—1000° С. Использование на этой стадии процесса теплоты
реакционных газов приводит к увеличению выхода ацетилена и
этилена на единицу количества затраченной электроэнергии. Этот двух-
стадийный (двухступенчатый) процесс внедрен в промышленность
фирмами Chemishe WerKe Hiils (ФРГ) и Du Pont. В 1942 г. Шох (США)
разработал аппарат роторного типа для электрокрекинга метана
в дуге переменного тока. Реактор прошел испытание на пилотной
установке, но практического применения метод Шоха не нашел.
В СССР в 1932 г., в США —в 1938 г. и в СССР в 1960 г.
(Н. С. Печуро) были оформлены патенты на получение ацетилена
электрокрекингом жидких углеводородов. При таком способе
121
электрическая дуга образуется между кусочками графита,
погруженными в керосин или мазут. Однако этот метод не применяется
в промышленности.
Дальнейшим усовершенствованием процесса электрокрекинга для
получения ацетилена (Л. С. Полак и Г. В. Гуляев, СССР) явилось
использование низкотемпературной плазмы водорода или аргона35,
что позволило на 25—30% снизить расход электроэнергии на единицу
количества товарного ацетилена. В ФРГ также исследуется крекинг
бензина в низкотемпературной водородной плазме с целью
одновременного получения ацетилена и этилена. Работы доведены до стадии
опытных установок.
ЭЛЕКТРИЧЕСКИЕ РАЗРЯДЫ В ГАЗАХ
Если в газе имеются заряженные частицы (ионы, электроны), то
он является проводником электричества. В электрических разрядах
всех видов одновременно протекают различные элементарные
процессы: возникновение, движение и уничтожение заряженных частиц3.
Несамостоятельные
разряды
Объемная
ионизация
Поверхностная
ионизация
Самостоятельные
разряды
Темный
разряд
Тлеющий
разряд
Дуговой
разряд
Промежуточная область
(темно-тлею-
I щий разряд)
Промежуточная область
[тлеюще-дуго-
8ой разряд)
Рис. IV-1. Основные типы стационарных электрических разрядов.
Типы стационарных разрядов представлены на рис. IV-1. К
стационарным формам относятся колеблющиеся во времени разряды,
в которых изменения происходят настолько медленно, что
предыдущие состояния не влияют на состояние в данный момент.
Стационарные разряды делятся на несамостоятельные и самостоятельные.
В несамостоятельных разрядах ионизация происходит в
результате некоторых внешних воздействий; разряды исчезают при
прекращении подачи ионов извне.
122
ю~
п-В
Значительно бсЗльшую роль играют самостоятельные
(самоподдерживающиеся) разряды, сами производящие все ионы, необходимые
для переноса тока. Во всех стационарных самостоятельных разрядах
протекают ионизация газа в объеме при электронном ударе и
образование заряженных частиц на поверхности электрода под действием
положительных ионов. Эти элементарные процессы взаимно
поддерживают друг друга: при действии ионизирующих электронов в газе
образуются положительные и отрицательные ионы; положительные
ионы при попадании на электрод (катод) «выбивают» из него
электроны, в свою очередь ионизирующие газ, и т. д. При достаточной силе
тока эти процессы протекают
одновременно и параллельно,
что обеспечивает однородный
разрядный ток.
Существует общее правило
ионизации при элементарных
процессах, справедливое для
всех стационарных
самостоятельных разрядов: каждая
заряженная частица должна
в среднем ионизовать
настолько интенсивно, что при учете
всех потерь вместо
первоначальной частицы должна
появиться подобная ей
заряженная частица. Стационарные
самостоятельные разряды
представляют собой равновесные процессы: несмотря на
непрерывный отвод и уничтожение отдельных частиц вследствие диффузии,
рекомбинации и т. д., эта убыль постоянно возмещается генерацией
точно таких же заряженных частиц.
Характеристика трех главных типов самостоятельного разряда
(темный, тлеющий и дуговой разряды) определяется соотношением
напряженности электрического поля и силы протекающего тока. На
рис. IV-2 приведена зависимость 4 потенциалов горения V всех этих
разрядов от силы тока /.
Область темного разряда соответствует силе тока 10~6—10~8 а
при потенциале выше 1000 в.
Область тлеющего разряда представляет собой светящееся
пространство вблизи катода, отделенное от него темной полосой. При
тлеющем разряде катод испускает электроны вследствие
бомбардировки его заряженными частицами и световыми квантами. Поле
У катода создается в основном положительным пространственным
зарядом. Тепловые явления при тлеющем разряде отсутствуют и не
являются необходимым условием его существования. Виды тлеющего
разряда определяются свойствами и давлением газа, размерами
разрядного сосуда, формой, размерами и материалом электродов, а также
межэлектродным расстоянием. Как видно из рис. IV-2, область нор-
123
1О'
Сила тока /7 а
Рис. IV-2. Потенциалы горения трех
главных типов самостоятельных разрядов (Vn —
нормальное катодное падение потенциала).
, я it .L \ t - ti светящиеся
Катодный слоМтрицательное Положишь- Днодное\ 3QHbt
! сбечение ныи столб сбечениех \
I
мального тлеющего разряда (горизонтальный участок кривой)
соответствует силе тока от 10~5 до 10~2 а при потенциале 100 в.
Для объяснения особенностей тлеющего разряда рассмотрим распределение
электрического поля 4, схематически показанное на рис. IV-3. Перенос тока в
тлеющем разряде осуществляется за счет движения электронов и положительных ионов
от одного электрода к другому. Электрон начинает свой путь от катода с очень малой
начальной энергией (порядка 1 эв) и возбуждает молекулы только на определенном
расстоянии от катода, эквивалентном 5—10 эв, что соответствует астонову темному
пространству. В области катодного слоя электрон приобретает энергию, соответ-
о о а ствующую максимуму функции воз-
Дстонобо Катодное Форадеево Лнодное Темные «ужения
II Г У пространства суждения.
▼ т т т Электроны, поступающие в зону
отрицательного свечения, можно
разделить по меньшей мере на две
группы. Первую группу составляют
быстрые электроны, появившиеся
на катоде или вблизи него и не
успевшие потерять энергию при
столкновениях в астоновом темном
пространстве. Вторая, большая группа
состоит из медленных электронов,
имеющихся в астоновом темном
пространстве и испытавших много
неупругих столкновений. Энергия
медленных электронов меньше
величины, соответствующей максимуму
ионизации, но больше или близка
к энергии, отвечающей максимуму
функции возбуждения. Поэтому
электроны испытывают много
столкновений с возбужденными
молекулами (отрицательное свечение),
после чего их энергия становится
настолько малой, что они легко
рекомбинируют с положительными
ионами. Рекомбинация, вероятно,
протекает в зоне отрицательного
свечения и за ней, так как
концентрации ионов и электронов в этой
области велики, а напряженность
электрического поля незначительна. Однако рекомбинационное излучение имеет
в общем малую интенсивность. С удалением от границы свечения количество
быстрых электронов уменьшается и интенсивность свечения падает.
Последующее медленное увеличение напряженности поля приводит к тому,
что вероятность рекомбинации уменьшается и появляется фарадеево^ темное
пространство, свойства которого являются промежуточными между свойствами зоны
отрицательного свечения и зоны положительного свечения (положительный столб),
расположенной за фарадеевым пространством. Так как напряженность поля
возрастает в направлении к положительному столбу, то в первую очередь появляются
спектральные линии, для которых максимумы вероятности возбуждения лежат
в области малых энергий.
В однородном положительном столбе продольная составляющая
электрического поля одинакова по всей длине столба. Отсюда следует, что результирующий
пространственный заряд в положительном столбе равен нулю, т. е. концентрации
электронов и положительных ионов в любой точке равны (рис. IV-3). Вследствие
малой подвижности положительных ионов практически весь ток разряда переносится
электронами, а положительные ионы лишь компенсируют пространственный заряд
электронов. Величина напряженности поля в положительном столбе на несколько
порядков меньше, чем в катодном темном пространстве. Это обстоятельство так же,
124
Рис. IV-3. Схема расположения темных и
светящихся зон в тлеющем разряде и
изменение характеристик электрического поля
(напряженности X, потенциала У, плотности
пространственного заряда р+ и р~ и
плотности тока i+ и /") в зависимости от
расстояния х между электродами.
как и однородность положительного столба, указывает, что ионизация в этой области
осуществляется главным образом благодаря высоким скоростям электронов,
приобретенным ими в результате большого числа упругих столкновений в электрическом
поле.
В примыкающей к аноду области положительного столба электроны
притягиваются анодом, а положительные ионы отталкиваются. В результате перед анодом
возникает отрицательный пространственный заряд. Это вызывает увеличение
напряженности электрического поля и резкое изменение (анодное падение) потенциала.
Электроны, вышедшие из положительного столба, вступают в область анодного
падения потенциала с малой начальной скоростью. Здесь они ускоряются в направлении
анода и после прохождения анодного темного пространства приобретают скорость,
достаточную для возбуждения и ионизации газа в пространстве перед анодом.
Поэтому вокруг анода образуется светящийся слой (анодное свечение), который иногда
распадается на отдельные светящиеся пятна.
Одной из своеобразных форм
тлеющего разряда является коронный
разряд, наблюдающийся в пространстве
около острия или проволоки при
наличии на них высокого потенциала
относительно окружающей среды. Коронный
разряд возникает в сильно
неоднородном электрическом поле; в зависимости
от того, какой заряд несет острие или
проволока (отрицательный или
положительный), в коронном разряде
отсутствует заряженная зона
противоположного знака. Разряд этого типа широко
применяется для получения
заряженных частиц в газах, например в элек-
wo
80
60
i
s
О
\
ч
ч^/
х=о,
-—.J
х=0,<
6 см
?сни
Г*
\__
-*h
\__
10 15 20 25 30
Плотность тока о & дуге, а
Рис. IV-4. Зависимость
напряжения дуги от силы тока между
двумя угольными электродами
в воздухе при 1 am.
трофильтрах, а также в озонаторах для стерилизации воды. Крекинг
углеводородов в коронном разряде не происходит.
Луговой разряд можно вызвать при соприкосновении и
последующем разведении двух металлических или угольных электродов,
находящихся в цепи, по которой протекает ток не менее нескольких
ампер. Этот разряд наблюдается как при атмосферном давлении, так
и при более низких и высоких давлениях в воздухе и различных газах
и парах. О характере дугового разряда удобно судить, сравнивая его
катодную область с катодной областью тлеющего разряда. В
тлеющем разряде катодному падению потенциала 100—400 в соответствует
малая сила и соответственно низкая плотность тока (см. рис. IV-2,
стр. 123); эмиссия катода при тлеющем разряде не сопровождается
тепловым эффектом. В дуге катодное падение потенциала
невелико — около 10 в, однако плотность тока очень высока; при
дуговом разряде весьма существенен и температурный режим катода,
а излучение соответствует спектру материала катода.
Основные электрические свойства дуги иллюстрируются 4
рис. IV-4. Для заданного расстояния х между электродами (т. е. для
заданной длины дуги) напряжение дуги Е уменьшается при
увеличении плотности тока i\ кривые, отражающие эту зависимость,
располагаются тем выше, чем больше х. Для угольных электродов Е резко
падает в точке h («шипение» дуги), что связано с явлениями на аноде.
125
Другой характеристикой дугового разряда является распределение
температуры * газа, потенциала и плотности тока в зависимости от
расстояния между электродами4 (рис. IV-5).
С помощью калориметрических измерений
установлено, что в коротких дугах между
угольными электродами в воздухе при токе 5 а около
42% всей электроэнергии выделяется на катоде,
около 37% —на аноде, остальное теряется за
счет излучения, конвекции, химических
реакций и т. д. Таким образом, около 80% энергии
расходуется на обоих электродах. В длинных
дугах на электродах выделяется не более 15—
20% всей электроэнергии. Баланс изменится,
если воздух заменить азотом, так как при этом
угольный анод не окисляется.
В положительном столбе дугового разряда
газ находится в состоянии изотермической
плазмы, при котором электроны и ионы
находятся в термодинамическом равновесии.
Вследствие высокой температуры, достигающей
18 000° С в центре дуги, а также большой
плотности тока и возможности варьирования
давления в дуговом разряде создаются весьма
благоприятные условия для проведения
высокотемпературных химических процессов 23.
РЕАКЦИИ УГЛЕВОДОРОДОВ
В ЭЛЕКТРИЧЕСКИХ РАЗРЯДАХ
Реакции расщепления и превращения углеводородов
наблюдаются в электрических разрядах всех типов.
В темном и тлеющем разрядах образование и рекомбинация
радикалов и ионов в основном протекают под действием электрического
разряда. Заметную роль в этих процессах играют атомы водорода,
образующиеся при расщеплении молекул углеводородов. В зонах
темного и тлеющего разрядов развиваются невысокие температуры
(60—150° С), что исключает реакции термического распада, однако
последующее повышение температуры приводит к увеличению выхода
продуктов превращения. Большое влияние на расщепление
углеводородов оказывает давление, от которого часто зависит характер
разряда.
Выход и состав продуктов реакции в темном и тлеющем разрядах
определяются, естественно, структурой исходного углеводорода,
однако в течении процесса расщепления существуют некоторые
закономерности. Наряду с реакциями дегидрогенизации (разрыв связи
Рис. IV-5. Зависимость
температуры газа Г,
потенциала V и
плотности тока i от
расстояния между
электродами х для
длинной дуги (VK и Va —
соответственно
катодное и анодное падение
потенциала).
* Действительная температура в п- 103 раз больше отложенной на графике,
где п — коэффициент, зависящий от материала электродов, плотности тока и
природы газа и равный 5-=- 50.
126
С—Н) и разрывом связей С—С с образованием ацетилена, этилена,
метана и элементарного углерода (сажи) протекает также
присоединение молекул непредельных углеводородов к предельным и
получаются более сложные алканы и полимеры. С ростом температуры
количество продуктов превращения увеличивается.
По убыванию стойкости * к действию тлеющего разряда
углеводороды можно расположить в следующем порядке5: ароматические
углеводороды >> нафтены > алканы > алкены > бициклоалкены.
В каждом гомологическом ряду по мере увеличения молекулярного
веса углеводорода снижается его стойкость к действию разряда,
уменьшается относительный расход электроэнергии на расщепление
и растет выход конечных продуктов реакции.
Изучение кинетики и механизма превращения метана в тлеющем
разряде при 28—36 мм рт. ст. показало, что основными продуктами
реакции являются ацетилен, этилен и водород. Количество
образующегося ацетилена и его последующий распад зависят от приложенной
удельной электроэнергии, определяемой по формуле 6:
q = KU/v
где q — удельная электроэнергия, вт* ч1л\
К — коэффициент, зависящий от природы исходного углеводорода;
U — расход электроэнергии, вт\
v — объемная скорость газа, л/ч.
В опытах при раздельном пропускании ацетилена, этилена и этана
через зону тлеющего разряда было установлено 6, что ацетилен
распадается с образованием водорода, гомологов и полимеров ацетилена
и сажи, а этилен и этан в этих условиях разлагаются главным образом
на ацетилен и водород. При U/v = 8,5 вт-ч/л распадается половина
исходного ацетилена, а при 20 вт-ч/л происходит почти полное его
разложение, но только 9% ацетилена превращается в газообразные
моно- и диацетиленовые производные, а основными продуктами
являются полимеры коричневого цвета. Количества этилена и этана
незначительны (0,1%). Разбавление водородом тормозит разложение
ацетилена — уменьшается скорость реакции, которая в зависимости
от условий может быть первого или второго порядка.
Ацетилен и другие продукты реакции могут получаться из метана
по схеме, приведенной на рис. IV-6 (см. стр. 128). Предполагается,
что образование ацетилена протекает одновременно по четырем
направлениям (реакции образования метана из ацетилена, этана
и этилена не учитываются ввиду их малых скоростей):
1) непосредственно из метана (константа скорости &5);
2) через этан, минуя этилен (k2 и ke)\
ih чеРез этилен, получающийся непосредственно из метана
\кх и /г7);
/и К4ePef этан и этилен, т. е. по схеме, предложенной Касселем7
(я2> #з и It,).
K действию разряда определяется величиной, обратной скорости
газов в зоне разряда.
127
Изучение поведения метана, ацетилена, этана и этилена в
тлеющем разряде, а также расчет скоростей и степени превращения этих
углеводородов показали, что основная часть ацетилена получается
по первому направлению — непосредственно из метана 6. Несколько
меньшая роль принадлежит второму и третьему направлениям, и
только 10% ацетилена образуется по схеме Касселя.
Мр
пан
4,
4
Этан
k
%з
в
►
Этилен |—
Дцетилен
Полимеры, гомологи
ацетилена, сажа,
пропан, пропилен
*е
Рис. IV-6. Возможные пути образования ацетилена из метана.
Разложение метана в тлеющем разряде протекает по следующему
радикальному механизму 8"11:
сн4 -> сн3- +н-
СН3
-сна
Н*
-сн
Все эти радикалы были обнаружены в газах крекинга
спектроскопическим путем 12. Ацетилен образуется при димеризации метин-
ных радикалов -СН- и в результате присоединения атомарного
углерода к метиленовым радикалам «CH^. Присутствие атомарного
углерода также было установлено спектроскопически. Исследование
спектров поглощения показало, кроме того, наличие в газах
крекинга диацетилена и высших ацетиленов. Рассчитанные энергии
активации для процессов в тлеющем разряде приближаются к
энергии активации реакций, протекающих при термических процессах13»17.
Механизм активации в тлеющем разряде сводится к образованию
ионов посредством соударения электронов с молекулами
углеводородов.
При получении ацетилена из метана и других углеводородов
в тлеющем разряде было установлено 12>14^16, что с увеличением
расходуемой мощности на единицу объема исходного метана растет
содержание ацетилена в продуктах реакции (табл. IV-1). Понижение
давления в зоне разряда также приводит к увеличению выхода
ацетилена. Минимальные затраты энергии на образование ацетилена из
метана в условиях тлеющего разряда составляют 9,6 квт*ч на 1 кг
С2Н2 при оптимальном давлении 100 мм рт. ст. Оптимальная
удельная нагрузка 1,2—1,8 квт-ч на 1 м3 СН4. Вследствие низкой общей
мощности тлеющего разряда он не нашел промышленного применения.
В дуговом разряде применяются высокие плотности тока (до
1000 а/см2), что приводит к нагреву пропускаемого газа до высокой
температуры и создает благоприятные условия для протекания эндо-
128
ТАБЛИЦА IV-1
Крекинг метана до ацетилена в тлеющем разряде
Скорость
подачи метана
л/ч
15
58
750
1080
Содержание
ацетилена в
газе крекинга
объемн.'%
16,0
8,2
12,4
6,9
Общая
мощность, кет
0,79
0,72
2,40
1,40
Расход
электроэнергии на 1 кг
ацетилена, квт-ч
122
94
12
10,5
термических процессов; к ним относятся большинство реакций,
входящих в схему рис. IV-6.
Термодинамические и кинетические условия образования
ацетилена в электрической дуге в основном такие же, как при термических
процессах. Наложение
электрического поля и соприкосновение
исходного углеводорода и конечных
продуктов реакции с узким
потоком газа, находящегося в
состоянии изотермической плазмы,
инициирует термический крекинг; при
этом из реактора выходят газы
с более высоким, чем при чисто
термических процессах, содержанием
ацетилена и продуктов его
полимеризации и деструкции.
В процессе электрокрекинга
метана при температуре 1600° С
протекает несколько конкурирующих
100
| 50
I 25
N
У
\
/
/
\
N
/ 2
N
ч
V/
Л
f\
I
V
с
о д ю~8
2
/Z7"y 10'° 1O'f Ю'° Ю'° 10'" 1O'J 10'
Время, сен
Рис. IV-7. Кинетика образования
различных продуктов электрокрекинга
метана при 1600° С.
2СН4
СН4
С2Н2
С2Н2 + ЗН2
С + 2Н2
2С + Н2
между собой реакций:
к = + 91 ккал/моль
Л#шз° к = + 15,4 ккал/моль
к — — 26,9 ккал/моль
В зоне высокого нагрева электрической дуги имеется достаточно
энергии для осуществления всех этих процессов и, следовательно,
суммарное направление крекинга определяется главным образом
кинетикой составляющих его стадий. На рис. IV-7 показаны
кинетические условия различных реакций при электрокрекинге метана9.
при установлении оптимального режима (температура 1500—1600° С,
продолжительность реакции 0,5-10"3-Ы0~3 сек) можно добиться
максимального выхода ацетилена и подавления побочных реакций.
пряЖ* электР°дУГ0ВЫХ процессах необходимая для химических
rTenZv нИЯ/°ЛуЧаеТСЯ За СЧет пРевРаЩения электроэнергии
тепловую. На образование 1 кг ацетилена при 1600° С теоретически
в- Н. Антонов ing
требуется затратить следующее количество сырья (при 100%-ном
его превращении) и энергии:
Расход Расход энергии
сырья
кг
ккал квт-ч
Метан 1,23 3760 4,37
Этан 1,15 1870 2,17
Пропан 1,13 1250 1,45
Бутан 1,11 1345 1,56
Этилен 1,07 1775 2,06
Пропилен 1,07 933 1,08
Из приведенных данных видно, что с увеличением
молекулярного веса несколько уменьшаются затраты энергии, и поэтому для
получения ацетилена в электрической дуге выгоднее использовать
гомологи метана. Для них оптимальная температура крекинга
составляет 1000—1400° С.
При электрокрекинге углеводородов энергия расходуется на
подогрев исходного газа до температуры реакции и на образование
ацетилена и побочных продуктов. Полезный расход энергии на 1 кг
ацетилена составит а квт-ч. Если обозначить степень превращения
углеводорода в ацетилен г), степень общего превращения А, общий расход
электроэнергии U и объем углеводорода 1/,то удельный расход энергии
на единицу объема исходного углеводорода составит6 U/V.
Благодаря предварительному подогреву углеводорода перед подачей в зону
электрокрекинга и использованию тепла реакционных газов
значительно снижается как полезный расход энергии а, так и общий
расход U.
При электрокрекинге различных углеводородных газов,
например т и п, в оптимальных условиях (площадь разрядного канала,
размеры реакционной зоны и конструкция реактора) соблюдается
постоянство соотношений 6»18:
Ат (К'+КГ)т
An {K'+K")n
Цт <2/г ^т
% ~~~ Ыт ~~ К'
х/г
где /('—энергетическая эффективность разряда при образовании ацетилена,
м3/квт- ч\
К" — энергетическая эффективность разряда при распаде ацетилена на
простые вещества, м3/квт-ч.
Процесс крекинга углеводородов в электрической дуге является
примером очень быстро протекающих химических реакций:
исходный газ вводится в аппарат при относительно низкой температуре,
за 0,5-10~3-^-2-10~3 сек успевает нагреться в среднем до 1600° С
и затем быстро охлаждается водой до температуры, при которой
активные химические реакции прекращаются. На характер химических
процессов, происходящих в дуге, наряду с температурой влияют
давление, скорость и режим газового потока.
130
Электрическая дуга может гореть при низких и при высоких
давлениях. При пониженном давлении ионы, и в особенности
электроны, под действием электрического поля приобретают большую
кинетическую энергию, поскольку свободные пробеги велики и
соударения ионов с молекулами редки. Ионизованные молекулы газа
также приобретают значительный запас кинетической энергии и,
следовательно, высокую температуру. Нейтральные молекулы,
наоборот, имеют малые запасы кинетической энергии и соответственно
низкую температуру. Поэтому при небольших давлениях много
энергии расходуется на нагрев газа и образование ионов, но степень
разложения молекул невелика, поскольку взаимные соударения редки.
С повышением давления учащаются столкновения молекул газа,
вследствие чего быстрее достигается термодинамическое равновесие.
Важное значение для получения равномерного температурного
поля имеет турбулизация газового потока, проходящего через дугу.
Для того чтобы создать наименьшее сопротивление, обычно придают
потоку газов и дуге вращательное движение, в результате чего под
действием центробежной силы образуется поток с меньшим
статическим давлением, соответственно уменьшается плотность газа и
возрастают длина и устойчивость дуги. Стационарные дуги
используются только в экспериментальных целях — для изучения
кратковременного действия дуги на испытуемый газ.
При технологическом оформлении процесса электрокрекинга
наибольшее распространение получили трубчатые аппараты с линейным
или коаксиальным расположением электродов. В таких реакторах
поток газа непрерывно проходит через зону высоких температур.
Трубчатые катод и анод охлаждаются проточной водой, что
предотвращает их выгорание. Газообразный углеводород, проходящий через
дугу, при линейном ее расположении нагревается неравномерно.
В центральной «нити» дуги в зоне нижнего электрода газ имеет
наиболее высокую температуру, при которой в основном протекают
процессы деструкции и образуется сажа («горячая нить» дуги). Около
этой «нити» происходит термический крекинг углеводорода с
образованием ацетилена. По мере удаления от «нити» температура
углеводорода снижается; у стенки электрода газ имеет самую низкую
температуру и охлаждает электрод, нагретый до 700° С излучением
электрической дуги. В описанных реакторах дуга перемещается
с большой скоростью.
МЕТОДЫ ПОЛУЧЕНИЯ АЦЕТИЛЕНА
В ЭЛЕКТРИЧЕСКИХ РАЗРЯДАХ
Перед тем как приступить к описанию технологических схем
электрокрекинга углеводородов, приведем на примере метана расчет
необходимых затрат электроэнергии (в кет -ч на 1 м3 исходного
углеводорода). Электроэнергия дуги превращается в тепловую энергию,
расходуемую на подогрев газа до оптимальной температуры
реакции (q±)9 на осуществление эндотермических реакций образования
9* 131
ацетилена и побочных продуктов (q2) и на возмещение потерь в
окружающую среду (q3).
Оптимальная удельная электроэнергия (в квт-ч/м?) определится
из уравнения19"20:
„ _ <7i + 92 + <7з
К
где К — коэффициент пересчета тепловой энергии (в ккал) в
электрическую (в квт-ч), равный 860,28.
Количество тепла ql9 расходуемого на подогрев метана * до
оптимальной температуры реакции, можно рассчитать по формуле:
1000^5(1600 — 25) cD
Яг = Шм " = 1300 ™<*л1м8
где ^5 — плотность метана при 25° С, равная 0,656 г/л;
ср — средняя мольная теплоемкость метана в интервале 25—1600° С, равная
20,4 кал/(моль-град);
М —вес 1 моль метана, равный 16,043 г.
Количество тепла q2, затрачиваемого на осуществление основной
и побочных реакций при электрокрекинге метана, можно подсчитать,
исходя из состава реакционных газов и теплового эффекта
реакций21' 22:
?а = S AHini
В этой формуле ДЯ/ft/ — произведение теплового эффекта при
1600° С и мольной доли продуктов реакции (ацетилена, этилена,
диацетилена, метилацетилена, пропадиена, винилацетилена, триаце-
тилена, фенилацетилена, бензола, нафталина и сажи). Для
упрощения подсчетов принято, что все указанные вещества образуются из
метана. Метан присутствует в газах крекинга вследствие его
неполного превращения, а появление водорода определяется реакциями,
происходящими в электрической дуге.
Теплоты реакций образования ацетилена и других продуктов (АЯ)
определены из известных термодинамических соотношений. На
основе приведенного в табл. IV-2 материального баланса
электрокрекинга метана (данные производственных установок) определены
количества продуктов реакции nh полученных превращением 1 мъ
100%-ного метана. Исходя из приведенных величин, рассчитываем q2'
q2 = 800 ккал/м3
Из материального баланса следует, что в процессе
электрокрекинга метана образуется 32,41 вес. % ацетилена, т. е. в ацетилен
превращается 4^2% метана. Общее превращение метана в ацетилен и
побочные продукты составляет 55,5%, что свидетельствует о расходе
13,5% метана на побочные реакции.
* Для удобства расчета принимаем 100%-ный метан.
132
ТАБЛИЦА IV-2
Материальный баланс электрокрекинга метана при 1600°С (расчет на 1 м? СН4)
Продукты реакции
Количество
вес. %
Объем при 0° С и 1 am
объемы. %
Ацетилен . .
Этилен ....
Метан ....
Водород . . .
Сажа ....
Диацетилен
Винилацетилен
Метилацетилен
Триацетилен .
Фенилацетилен
Пропадиен . .
Бензол . . .
Нафталин . .
Всего
232,7
19,2
318,0
77,4
24,0
16,7
8,7
5,4
1,7
1,0
2,7
7,8
1,5
32,41
2,68
44,40
10,80
3,36
2,33
1,21
0,75
0,24
0,14
0,37
1,10
0,21
201,0
15,4
445,0
865,0
7,5
3,8
3,0
0,5
0,2
1,5
2,3
0,3
13,0
1,00
28,8
56,02
0,48
0,21
0,19
0,03
0,01
0,10
0,14
0,02
716,8
100
1545,4
100
Если принять потери тепла в окружающую среду равными
50 ккал/м3, то можно определить оптимальную удельную
электроэнергию:
„ _ Яг + Яг + <7з _ 1300 + 800 + 50 _ ~ -
а Щ ~ 25
860,28
Щ28
На непосредственное превращение метана в ацетилен расходуется
31 % тепла электрической дуги, а на образование побочных продуктов
реакции — 6,3%; 60% тепла затрачивается на подогрев метана до
оптимальной температуры реакции и, вероятно, все это тепло уходит
с газами крекинга. В случае охлаждения продуктов реакции тепло
подогрева метана полностью воспринимается водой, впрыскиваемой
на выходе газов из зоны электрической дуги. Использование тепла
реакционных газов является резервом для снижения удельной
электроэнергии до 2 квт-ч на 1 м3 СН4, однако это можно осуществить
только при добавлении низших гомологов метана (без учета
образования этилена)10'18-37.38.
Дальнейшее использование тепла реакционных газов на
производство пара или для других целей не приводит к снижению удельной
энергии (в расчете на 1 кг С2Н2), поскольку в этом случае количество
образующегося ацетилена не меняется.
Следует иметь в виду, что при расчете реальных энергетических
затрат необходимо учитывать наличие в природном газе 3—7% азота,
имеющего значительно более низкую теплоемкость по сравнению
с метаном. На каждый 1% азота в 1 м* природного газа необходимо
уменьшать удельную электроэнергию на 0,01 квт-ч.
133
Электрокрекинг газообразных углеводородов
с Вода на
'охлаждение
углеводороды
Энергию дугового электрического разряда для крекинга
углеводородов впервые начали применять в Германии на заводе в г. Хюль-
се2з-25# д0 1945 г. ацетилен получали из газов, образующихся при
синтезе моторного топлива по методу Фишера—Тропша. Затем
длительное время для этой цели использовали природный газ и
метановую фракцию коксового газа. В
настоящее время сырьем для
производства ацетилена служат
фракции углеводородов нефти. При
мощности завода в г. Хюльсе
(ФРГ) свыше 100 тыс. т С2Н2
в год к 1968 г. было выработано
более 1,5 млн. т ацетилена25.
Электрокрекинг углеводородов
осуществляется в реакторе
постоянного тока мощностью 8200 кет
(рис. IV-8). Стальной реактор
состоит из верхнего колоколообраз-
ного катода / диаметром 150 мм,
нижнего трубчатого анода 5
длиной 1500 мм и диаметром 100 мм,
вихревой камеры 3 между
электродами и фарфорового изолятора 2,
отделяющего катод от электрически
соединенных вместе и заземленных
вихревой камеры и анода. Сбоку
на вихревой камере имеется
вспомогательный пусковой электрод 6.
При пуске реактора
вспомогательный электрод пневматическим
устройством накоротко
замыкается с катодом и затем отводится
в крайнее нижнее положение. При
этом возникает электрическая дуга,
которая как бы оттягивается от
катода и вихревым потоком газа
увлекается в анодное
пространство. Напряжение дуги 7000 в,
сила тока 1150 a, cos ср = 0,7ч-0,75.
Общая длина свободно висящей дуги устанавливается около 1000 мм
(соответственно ее электрическим характеристикам). Электроды
охлаждаются водой через специальные рубашки. Все части реактора
изготавливаются из стали.
В вихревую камеру через специальные отверстия тангенциально
вводят газообразные углеводороды (метан) при нормальной
температуре. В камере создается вращательное движение газа, который
134
l ^ Углеводороды
Вода на
\закапку"
Крекинг-газ
Рис. IV-8. Реактор фирмы Chemische
Werke Htils для электрокрекинга
метана:
1 — катод; 2 — фарфоровый изолятор;
3 — вихревая камера; 4 — заземление;
5 — анод; 6 — пусковой электрод.
проходит в трубчатый анод. Там под действием электрической дуги
газ нагревается в среднем до 1600° С. Электрическая дуга под
действием вращающегося газового потока также вращается вокруг оси
нижнего электрода, при этом точки примыкания дуги к электродам
все время перемещаются. Это позволяет избежать выгорания
материала электродов.
На 20 см ниже точки примыкания электрической дуги к аноду
в анодное пространство вводится дополнительное количество
углеводородов со средним углеродным показателем ^ 2 (обычно
используется смесь углеводородов С3—С4). За счет тепла реакционных газов
крехинг-газ
No+CO
Рис. IV-9. Технологическая схема двухступенчатого электрокрекинга -метана на
установке фирмы Chemische Werke Hiils:
/ — реактор; 2 — циклон; 3,8 — водяные скрубберы; 4, 5,6,7— масляные скрубберы;
9 — газгольдеры; 10 — компрессоры; // — водный абсорбер; 12 — осушитель газов; 13,
14 — колонны для выделения фракций Сх и С2; 15 — колонна для выделения водорода; 16 —
сажеотделитель; 17 — насосы; 18 — колонна регенерации масла; 19 — колонна выделения
синильной кислоты; 20 -т колонна регенерации воды; 21, 25, 26 — аппаратура выделения
ацетилена; 22, 23, 24 — аппаратура выделения этилена; 27 — газодувка; 28, 29 — колонны
регенерации растворителей.
эти углеводороды расщепляются с,образованием новых количеств
ацетилена, этилена, метана и водорода; температура смеси при этом
понижается до 800° С. В пространство под анодом через специальные
сопла вводится вода, за счет чего газы охлаждаются до 180—200° С.
Поскольку температура выходящих газов составляет 150—180° С,
значительная часть сажи (более 60%) улавливается в сухих
циклонах 2 (рис. IV-9) и после грануляции выдается в товарном виде.
1 юсле циклонов реакционные газы промываются водой в скруббере 3,
а затем маслом в скрубберах 4У 5, 6 и 7 для удаления сажи, синильной
кислоты, нафталина и высших ацетиленовых углеводородов.
Очищенный газ собирается в газгольдере 9. После очистки в 1 м* газа
содержится менее 3 мг сажи. При выделении ацетилена водой газы ком-
примируются до 18 am. Стадия выделения концентрированного
ацетилена и этилена подробнее описана в гл. VI (стр. 246).
135
При использовании для электродугового процесса природного
газа, содержащего 92,3 объемы. % метана, 1,4 объемы. % этана,
0,5 объемы. % пропана, 0,4 объемн. % бутана и 5,4 объемн. % азота,
крекинг-газ имеет следующий состав (в объемн. %)25:
Ацетилен 14,5 Метан 16,3
Метилацетилен . . 0,4 Этан 0,03
Диацетилен .... 0,6 Пропан 0,02
Винилацетилен ... 0,1 Бензол 0,3
Диметилацетилен . . 0,01 Толуол 0,02
Метилвинилацетилен 0,04 Водород 63,46
Этилен 0,9 Окись углерода . . 0,6
Пропилен 0,02 Азот 2,7
Бутилен 0,02
В реакторе, показанном на рис. IV-8, часовой расход природного
газа составлял 2400 м3. Из 2400 м3 природного газа указанного
состава получено 680 кг ацетилена, 46 кг этилена, 1900 м3 водорода
и 145 /сгсажи, что свидетельствует о 46%-ном превращении исходных
углеводородов (44,2% превратилось в ацетилен). Наряду с
ацетиленом образовались его гомологи (1,15 объемн. % или 12,3 вес. %).
Выход сажи составил 9,4 вес. % от количества исходных
углеводородов. Водород используется для синтеза аммиака, этилен — для
получения полиэтилена, этилбензола и окиси этилена.
Природный газ для увеличения С-числа обогащается отходящими
углеводородами, полученными на установке концентрирования
ацетилена. Поэтому в исходной смеси содержится часть ацетилена и
водорода и почти все образующиеся ацетиленовые углеводороды. При
работе на обогащенном природном газе (С-число = 1,8) и с
использованием углеводородов С3—С4 для дополнительного крекинга
получают газ следующего состава (в объемн. %):
Ис- Ис-
ход- Кре- ход. Кре-
ный кинг" ный кинг"
газ газ газ газ
Ацетилен 1,2 15,9 Метан 53,8 17,0
Метилацетилен .... 1,0 1,0 Этан 10,3 1,2
Диацетилен ...... 0,8 0,5 Пропан 7,9 0,8
Винилацетилен 0,7 0,5 Бутан 12,6 2,1
Диметилацетилен .... 0,4 0,3 Бензол 0,4 0,4
Метилвинилацетилен . . 0,2 0,2 Толуол — 0,1
Этилен 1,7 7,1 Водород 2,8 50,2
Пропилен 2,3 0,9 Окись углерода .... 0,8 0,7
Бутилен 0,4 0,3 Азот 2,7 0,8
Расходные показатели процесса в расчете на 1 т ацетилена
составляют25;
Исходные углеводороды (с учетом рециркуляции и добавки *
углеводородов С3—С4), кг 2900
Электроэнергия, квт*ч 13 200*
Водяной пар на выделение ацетилена, кг 1500
* Непосредственно в реакторе расходуется 10 300 квт-ч.
136
Метан
Полезное использование электроэнергии в процессе крекинга
составляет 43%. Удельный расход электроэнергии на 1 мъ
природного газа и отходящих углеводородов равен 2,5 квт-ч (без учета
дополнительно введенных 900 кг/ч углеводородов С3—С4). В сажу
превращается 10% сырья.
На заводе в Хюльсе
(ФРГ) установлены 34
спаренных реактора, из
которых 17 находятся в работе.
В реакторе стальные
электроды служат
ограниченное время (анод заменяют
через 500 ч работы,
катод— через 150 ч), поэтому
для обеспечения
бесперебойной работы
устанавливают по два реактора на
каждой линии,
работающие попеременно.
В целях использования
колебаний мощности в
энергосистеме производство
ацетилена работает по
ступенчатому графику: при
перегрузке системы
сокращается число работающих
реакторов, а в часы
минимального расходования
энергии производство
работает с максимальной
нагрузкой, чем достигается Вода,
снижение стоимости
электроэнергии.
В 1960 г. на
Саратовском химическом
комбинате2^ в 1963 г. на
химическом комбинате в г. Бор-
зешть в Румынии были
введены в действие
производства ацетилена
электрокрекингом природного
газа. В основу
технологических схем обоих заводов положены конструктивные разработки
электродугового реактора мощностью 7200 кет румынских
инженеров Петреску и Ионеску (рис. IV-10).
В отличие от реактора в г. Хюльсе, в румынском реакторе (позже
усовершенствованном совместно с советскими специалистами) основ-
вде элементы анода 6, вихревой камеры 3 и катода 2 рассчитаны на
137
Бодана
\охшждениь
Реакционные
газы
Рис.'ПМО. Реактор для электрокрекинга метана:
/ — высоковольтный ввод; 2 — катод; 3 — вихревая
камера; 4 — гетинаксовая плита; 5 — заземление;
6 — анод; 7 — пусковой электрод.
более оптимальный газогидродинамический режим, что позволяет
уменьшить сажеобразование в анодном пространстве и увеличить
срок службы деталей реактора. Катод изолирован от вихревой
камеры гетинаксовой плитой 4. Вихревая камера и анод заземлены,
чем достигается безопасность работы. Природный газ поступает
в камеру 3 по одному тангенциальному вводу. Часть газа (до 10%)
подается в колоколообразный катод, что позволяет несколько
регулировать напряжение во время работы реактора.
Так же, как и в реакторе фирмы Chemische WerKe Htils, в
вихревой камере устанавливается вращательное движение газа.
Вращающийся поток газа устремляется в нижний трубчатый электрод—
анод, в центре которого создается относительное разрежение,
стабилизирующее электрическую дугу.
Линейная дуга протягивается между катодом и анодом на
расстояние 800—1000 лш..Она питается постоянным током от ртутного
выпрямителя. Напряжение в дуге зависит от силы тока / и расхода
газа Q:
Чем выше сила тока, тем сильнее прогревается газ и тем большая
часть его переходит в плазменное состояние, что увеличивает токо-
проводимость среды и уменьшает напряжение дуги. При увеличении
расхода газ охлаждается и ионизация уменьшается, что приводит
к обратному эффекту — увеличению напряжения между
электродами.
При пуске реактора в момент подачи тока между
вспомогательным электродом и катодом осуществляется короткое замыкание.
Затем вспомогательный электрод отводится при помощи
пневматического устройства в нижнее крайнее положение, а образовавшаяся
дуга потоком газа увлекается в анодное пространство. Дуга работает
более устойчиво, когда рабочее напряжение не превышает 2/3
максимально допустимого напряжения на клеммах трансформатора. Для
поддержания стабильности дуги в цепи постоянного тока установлены
мощные индукционные катушки. Стальные электроды приходится
часто заменять (анод — через 500 ч работы, катод — через 800—
1000 ч)у поэтому на каждой технологической линии установлено по
два реактора. Переключение с одного реактора на другой происходит
за 5—7 мин.
Технологическая схема24 получения ацетилена
электрокрекингом природного газа на Саратовском химическом комбинате несколько
отличается от схемы фирмы Chemische WerKe Hiils. Исходным сырьем
является природный газ Саратовского месторождения, не
обогащенный гомологами метана и имеющий после очистки следующий состав
(в объемн. %):
Метан 89,2—93,6
Этан 1,5—1,7
Пропан 0,4—0,7
Бутан 0,15—0,49
Азот 3,3—7,3
138
Для выделения ацетилена применяется селективный абсорбент —
диметилформамид (ДМФ) при избыточном давлении до 10,5 am.
Природный газ подвергается очистке этаноламином от
сероводорода и двуокиси углерода, осушается и после редуцирования до
избыточного давления 1,5 am поступает в сепаратор 1 (рис. IV-11)
для улавливания возможных брызг этаноламина, а затем
направляется в реактор 2. В каждый реактор газ поступает в количестве
2800—3000 м3/н (10% подают через верх колоколообразного катода).
Анод и катод непрерывно охлаждаются водой, циркулирующей через
окружающие их рубашки. Температура воды в рубашках
автоматически поддерживается не выше 42° С. Реактор автоматически
выключается при повышении сверх нормы температуры охлаждающей воды
в рубашках электродов, температуре реакционных газов более 250° С,
давлении газов в реакторе выше 0,65 am, снижении давления воды и
азота, появлении метана в помещении, где установлен реактор, и др.
Около 42—43% углеводородов природного газа превращается
в ацетилен, 12—14% расходуется на образование побочных
продуктов и сажи, 2—2,5% исходного сырья расщепляется с образованием
сажи (содержание ее в газе пиролиза 14—20 г!мъ). Примерный
состав газов крекинга приведен ниже (в объемн. %):
Ацетилен .... 11—13,0 Метан ...... 28—30
Бензол ..... 0,14
Нафталин .... 0,02
Водород .... 56—58
Цианистый
водород 0,1—0,2
Азот 3,2—7,0
Газы крекинга в реакторе охлаждаются водой с 1600 до 250° С,
затем проходят последовательно четыре циклона 3, где улавливается
до 70—80% сухой сажи. Сажа из циклонов через промежуточные
бункеры пневматически транспортируется в соседний корпус для
грануляции и упаковки. Полное удаление сажи и примесей цианистого
водорода происходит на стадиях мокрой и масляной очистки.
Предварительно газы охлаждаются до 50° С в полом скруббере 4, где водой
вымывается 90% оставшейся сажи. В крекинг-газе содержится
цианистый водород, большая часть которого попадает в промывные
воды. Поэтому после отдувки воздухом в башне 16 сточные воды
поступают на очистку, а удаленный из них цианистый водород
поглощается щелочью в специальном скруббере (на рисунке не
показан). Насыщенная цианистым водородом щелочь направляется на
утилизацию синильной кислоты.
Крекинг-газ, очищенный от цианистого водорода, через
сепаратор ь направляется в систему очистки I ступени, состоящую из скруб-
оеров 6 и 7, орошаемых дизельным топливом (масляная очистка),
и пенного аппарата 8 (мокрая очистка). Назначение I ступени очи-
ки отделение от газа части оставшейся сажи, нафталина и наи-
139
Метилацетилен
Винилацетилен . .
Фенилацетилен . .
Диацетилен . . .
Триацетилен . .
Этилен ...
Пропадиен ....
0,19
0,21
0,01
0,48
0,03
1,0
0,1—0,2
более тяжелых ацетиленовых углеводородов. Скрубберы 6 и 7
снабжены смесителями типа трубы Вентури. При последующей мокрой
очистке водой в пенном аппарате, также снабженном смесителем
типа трубы Вентури, из газа удаляются остатки сажи. После
промывки 4%-ным раствором щелочи в аппарате 9 для удаления
остатков цианистого водорода крекинг-газы направляются на II ступень
в скрубберы 10 и 11, также орошаемые дизельным топливом, для
удаления оставшегося нафталина. Поскольку температура газов
во II ступени близка к нормальной, очистка здесь протекает полнее.
Дизельное
Вода топливо Вода
Природный^
~г
Крекинг-газ
на компрессию
Раствор гомологов
ацетилена в ДМФ
На утилизацию
' HCN
Рис. IV-11. Технологическая схема электрокрекинга углеводородов на
Саратовском химическом комбинате 24:
1,5 — сепараторы; 2 — реактор; 3 — циклон; 4, 13 — промывные скрубберы; tf, 7, 10,
11—скрубберы, орошаемые дизельным топливом; 8—пенный аппарат; 9 —нейтрализатор;
12 — абсорбционная колонна; 14 — газгольдер; 15 — насос; 16— башня для отдувки HCN.
Окончательная очистка газов происходит в колонне 12, орошаемой
диметилформамидом. Количество диметилформамида рассчитывается
на проведение полной очистки газа.
Очищенный крекинг-газ идет через промывной скруббер 13 в
газгольдер 14 и после компримирования поступает на установку
выделения ацетилена диметилформамидом. Технологический процесс
выделения ацетилена диметилформамидом описан ниже (стр. 246).
Концентрация ацетилена после системы выделения достигает 99,9%,
коэффициент извлечения более 95%.
Дизельное топливо из скрубберов 10 и 11 возвращается в
скрубберы 6 и 7, а затем для регенерации продувается отходящими или
природным газами, направляемыми далее в котлы для производства
пара. Загрязненный диметилформамид из колонны 12 продувается
рециркулирующими газами, возвращаемыми на очистку для
извлечения из них ацетилена, и регенерируется.
140
В процессе получения ацетилена электрокрекингом метана на
Саратовском химическом комбинате на 1 т ацетилена расходуются:
Природный газ, м3 4500
Дизельное топливо, кг 100—200
Щелочь (92%-ная), кг 150
Электроэнергия (включая расход на
выделение С2Н2), кет- ч 13 500—14 000
Пар, Гкал 5—7
Сажа, образующаяся в производстве ацетилена, по специальному
пневмопроводу поступает в классифайер 4 (рис. IV-12), одновременно
Газ на дальнейшее
охлаждение и
очистку
В атмосферу
Вода
Этиловый
^jf
ТоплиВнь/й
газ
Сажа
Рис. IV-12. Технологическая схема получения гранулированной сажи:
Л 5 — циклоны; 2, 9 — затворы; 3 — промежуточные бункеры; 4, 18 — вентиляторы; 6 —
измельчитель; 7 — элеватор; 8 — усреднитель; 10 — гранулятор; // — мерник; 12 —
смеситель; 13 — насос; 14 — сушильный барабан; 15 — элеватор; 16 — бункер; 17 — печь;
19 — фильтр.
являющийся вентилятором. Там от нее отделяются посторонние
примеси, кокс, грит и смолы, присутствующие в количестве до 2 вес. %.
Затем сажа попадает в циклон 5, где освобождается от инертного газа,
и направляется в измельчитель 6 и в усреднитель 5, снабженный
мешалкой. Усреднитель является одновременно напорным бункером.
После усреднителя сажа, имеющая более равномерную кажущуюся
плотность, через шлюзовой затвор 9 пересыпается в гранулятор 10.
нем она перемешивается с распыляемой водой (соотношение 1:4),
содержащей 4% этилового спирта. Раствор спирта в воде готовится
в отдельном смесителе 12. Сырую (до 80% влаги) гранулированную
сажу сушат топорными газами в сушильном барабане 14. Исполь-
Г^16 топо™ые газы вентилятором выбрасываются через мешоч-
фильтры 19 в атмосферу.
141
Для получения 1 т гранулированной сажи расходуются:
1оплибный газ, м3
Конденсат, кг ....
Этиловый спирт, кг
Электроэнергия, кет-я
1000
3800
152
240
Гранулированная высушенная сажа представляет собой комки
размером 0,8—2 мм (кажущаяся плотность 0,02—0,025 г/см3,
гигроскопичность 1—2%). При использовании комки рассыпаются, что
облегчает применение. Сажа, получаемая в процессе
электрокрекинга, более дисперсна, чем при взрывном разложении ацетилена
(табл. IV-3). Такая сажа может использоваться для крашения в массе
полимеров, перерабатываемых в синтетические волокна, а также в
полиграфической и резиновой промышленности. Использование сажи
как побочного товарного продукта снижает себестоимость ацетилена
на 8—10%.
ТАБЛИЦА IV-3
Свойства сажи, полученной при производстве ацетилена
Процесс
Взрывное разложение
ацетилена (сажа сорта II-1250) . .
Процесс фирмы Chemische
Werke Htils
Процесс Саратовского
химического комбината
Истинная
плотность
г/см3
1,72
1,21
1,39
Кажущаяся
плотность
г/см3
0,03
0,03
0,025
г/л
150 *
150*
140*
Гигроскопичность, г/г
0,8
2,8
2,8
Масляное
число, г/г
2,05
2,5
2,5
Удельная
поверхность
м2/г
120
180
200
* Гранулированная сажа.
Фирма Du Pont в 1963 г. на заводе в г. Монтегью (США) начала
производство ацетилена электрокрекингом природного газа26*27
с использованием тепла реакционных газов для пиролиза
дополнительных количеств углеводородов (гомологов метана). Процесс
фирмы Du Pont мало освещен в технической литературе, и многие
подробности технологической схемы неизвестны.
Процесс ведется в реакторе коаксиального типа (рис. IV-13)
с электромагнитной стабилизацией электрической дуги26»27. Дуга
устанавливается между центральным графитовым катодом 2 и
окружающим его цилиндрическим стальным охлаждаемым анодом 3.
Под действием электромагнита 5 дуга вращается по внутренней
поверхности анода со скоростью до 7000 об/сек, чем достигается ее
устойчивость и относительно долгий срок работы анода. К катоду
дуга примыкает главным образом в его нижней части.
Для компенсации выгорания графита катод периодически
опускается на определенную величину. При этом дуга касается все новых
142
участков поверхности анода и катода, что обеспечивает
непрерывность работы реактора. Поскольку под действием газового потока
точки примыкания дуги к аноду перемещаются вниз, при работе
реактора образуется электродуговой конус, верхней точкой которого
является нижний конец графитового катода, основанием —
обегающая линия примыкания дуги к аноду, а боковой поверхностью —
сама электрическая дуга.
Преимуществом коаксиального
реактора является малое время
соприкосновения горячей «нити» дуги с потоком
газа, вследствие чего можно свести до УглёШ
минимума процессы деструкции углево- -0/Р \ \
дорода. В таком реакторе за счет умень- \
шения образования сажи и побочных
продуктов полнее используется тепло,
развиваемое электрической дугой, а это
приводит к повышению степени крекин- водана-
га углеводорода. В реакторе фирмы ""** ~
Du Pont концентрация ацетилена в
получаемом газе значительно выше, чем
в реакторах линейного типа, и
составляет 16—18 объемн. %.
В коаксиальном реакторе одной из \
трудностей является необходимость pa- Вч°п3,м,,«
J- J _, ' о „JUKLUIKy
боты с токами относительно большой
СИЛЫ, ПОСКОЛЬКУ Протяженность Дуги Рис. IV-13. Коаксиальный реак-
сравнительно невелика. Применяя ко- ^рокрекинга1*
■Углеводороды
крекинг-газ
/ — обойма катода; 2 — катод;
3 —анод; 4— охлаждающая
рубашка; 5 — электромагнит; 6
—сепаратор; 7 — скреперное кольцо.
р
нусообразную форму дугового
пространства, удается увеличить напряжение
между электродами. Мощность реактора
фирмы Du Pont составляет 3000 кет
при напряжении между катодам и анодом 750 е\ сила тока 4000 а.
Состав газов крекинга (в объемн. %):
Ацетилен 16—18
Ацетиленовые углеводороды . . . 0,8—1,2
Этилен 5—7
Метан 15—18
Водород 45—50
Сажа 8—12 г/си3
Выделение ацетилена из газов крекинга осуществляется
несколько проще, поскольку при использовании коаксиального
реактора в реакционных газах содержится меньше сажи, смол и
ацетиленовых углеводородов. Очистку газов от следов сажи, синильной
кислоты, ацетиленовых углеводородов и смол проводят по обычной,
но несколько сокращенной схеме. Абсорбцию ацетилена
осуществляют с использованием селективного абсорбента.
Другой принцип электрокрекинга газообразных и испаренных
жидких углеводородов разработан Щохом28. Исследования, начатые
143
в 1942 г., закончились в 1950 г. созданием при Техасском
университете (США) полупромышленной установки мощностью до 200 кг/ч
ацетилена. На установке имеются три основных и один резервный
реактор. В каждом из основных реакторов, последовательно
соединенных по ходу газов крекинга с остальными, создается дуговой
разряд мощностью 815 кет. Таким образом, общая мощность
работающей установки составляет около 2500 кет.
Разложение углеводорода протекает последовательно в трех
реакторах (рис. IV-14). В центре каждого реактора установлен ротор 2
с закрепленными на нем тремя
вращающимися электродами /. К этим
электродам подведены три
стационарных высоковольтных электрода 3,
расположенных радиально по
отношению к оси камеры и
изолированных друг от друга и от ротора 2.
Питание высоковольтных (154 ё)
электродов осуществляется переменным
током от трех однофазных
трансформаторов, подводимым к ротору. Ротор
является «нулем» трехфазной системы
и заземлен. Для пуска реактора
приводят во вращение ротор, затем
приближают к нему стационарные
электроды и после включения тока
отводят их в рабочее положение.
Газообразные углеводороды
непрерывно вводятся и выводятся из
реактора через два патрубка,
установленные на кожухе реактора. При вращении ротора, служащего
одновременно газодувкой, газ из камеры проталкивается в щели
между стационарными и вращающимися электродами, где возникает
дуговой разряд и происходит процесс электрокрекинга. Для
охлаждения крекинг-газы смешивают со свежими порциями исходных
углеводородов. На линиях перехода газа из одного реактора в
другой установлены трубчатые теплообменники, где тепло снимается
водой. Температура в реакторах поддерживается 290° С.
Из последнего реактора отводится газ с содержанием 10—
14объемн. % ацетилена, 1—Зобъемн. % этилена, 0,2—0,3объемн. %
метилацетилена, 0,4—0,6объемн. % диацетилена и 0,2—0,Зобъемн. %
винилацетилена. При использовании газообразных углеводородов
(метана или его гомологов) в реакционных газах содержится 30—
50 объем». % исходного углеводорода. Содержание сажи в продуктах
электрокрекинга составляет 10—12 вес. % от количества исходного
углеводорода. При переходе от метана к бутану расход
электроэнергии уменьшается с 10 700 до 7300 кет-ч на 1 т ацетилена (без учета
стадии выделения ацетилена). Содержание ацетилена в реакционных
газах можно повысить до 12—14%, пропуская меньшее количество
144
Рис. IV-14. Схема реактора Шоха:
/—вращающийся электрод; 2—ротор;
3— высоковольтный электрод; 4 —
корпус реактора; 5 — ввод высокого
напряжения; 6— станина; 7 —фундамент.
газа через реакторы, однако при этом расход электроэнергии на 1 т
ацетилена увеличивается на 15—20%. Ацетилен выделяют из
реакционных газов селективным абсорбентом — диметокситетраэтилен-
гликолем.
Для использования в процессе Шоха природный газ необходимо
предварительно полностью освободить от сернистых соединений;
содержание двуокиси углерода (или азота) допускается не более
2 объемн. %, водорода — не более 10 объемн. %.
Процесс Шоха не нашел промышленного применения по
следующим причинам. Реакционные газы аналогичны газам,
полученным при линейном электрокрекинге: в них практически
одинаково соотношение между гомологами ацетилена и ацетиленом (1 : 10
по объему) и такое же количество углеводорода превращается в сажу
(10—12 вес. %). Процесс Шоха не имеет преимуществ и в части
расхода электроэнергии на 1 т получаемого ацетилена. Поскольку
крекинг углеводородов осуществляется в условиях высокой
температуры электрической дуги, использование тепла реакционных газов
с целью подогрева вводимого в камеру углеводорода до 290° С,
т. е. до температуры, поддерживаемой в камере, не дает ощутимой
экономии энергетических затрат. Основное тепло процесса снимается
водой в промежуточных теплообменниках.
Однако этот процесс интересен для специалистов, исследующих
методы получения ацетилена из углеводородов и, в частности, в связи
с использованием оригинального приема «закалки» реакционных
г,азов — передача тепла при перемешивании. Другие элементы
процесса Шоха также представляют интерес, поэтому целесообразность
их использования в процессе получения ацетилена необходимо
изучать.
Электрокрекинг жидких углеводородов
До последнего времени для получения ацетилена в дуговом
разряде использовали газообразные углеводороды, в основном метан.
Будучи самым дешевым сырьем, метан, однако, наименее выгоден
термодинамически, так как для его разложения требуется
значительно больше электроэнергии, чем, например, для крекинга жидких
углеводородов. Кроме того, содержание ацетилена в газе,
получаемом при крекинге метана, мало из-за недостаточной степени
превращения метана.
В настоящее время в разных странах исследуется возможность
получения ацетилена электрокрекингом жидких углеводородов.
Первые работы в этом направлении32 были проведены в СССР в период
1932—1934 гг. Они закончились созданием небольшой пилотной
установки по получению ацетилена из различных нефтяных
фракций. Аналогичные работы были проведены во Франции2^—31
фирмой Air Liquide, которая в 1935 г. разработала и построила
небольшую установку по получению ацетилена электрокрекингом
низкосортного жидкого сырья. В последние годы к этому методу
неоднократно возвращались многие исследователи,
10 В. Н. Ahtqhqb И5
t
тт
крекинга
а
1
В нашей стране этой проблеме уделяет большое внимание Н. С. Пе-
чуро с сотр. Предложенный ими32-33 лабораторный реактор для
электрокрекинга жидких углеводородов представляет собой
герметичный аппарат, в котором параллельными рядами размещаются
графитовые электроды (рис. IV-15). В пространство между
электродами загружаются графитовые шарики диаметром 5—12 мм. Через
нижний штуцер в реактор подают жидкие углеводороды (нефть,
мазут или другое нефтяное сырье), которые проходят через аппарат
снизу вверх, заполняют все свободное пространство между
графитовыми шариками и вытекают
через два боковых штуцера.
Между каждой парой
электродов через графитовые
шарики пропускается
переменный ток напряжением от 60 до
п Жидкие 300 в. При этом в среде жид-
^углеводороды КОго углеводорода образуется
множество коротких электри-
^5 ческих дуг, положение
которых непрерывно меняется под
действием потока сырья.
Вблизи электрических дуг
углеводороды разогреваются
до температуры термического
крекинга и образуются аце-
тиленсодержащиегазы. Часть
углеводородов, по-видимому,
непосредственно в местах
прохождения дуг
подвергается деструктивному
разложению с образованием свободного углерода — сажи. Крекинг-газы
выводятся из аппарата через верхние штуцеры. Постепенно за счет
нагрева всей массы нефтяного сырья в аппарате устанавливается
температура 100—120° С, которая в дальнейшем поддерживается
заданным потоком жидкости. В образующемся крекинг-газе
содержатся ацетилен и его гомологи, этилен, метан и его гомологи и
водород (в объемн. %)34:
Ацетилен и его гомологи (метилацетилен,
диацетилен, винилацетилен) 29—32
Олефины (этилен, пропилен) 8—11
Парафины (метан, этан) 2,6
Водород 52—58
При наличии в нефтяном сырье кислородсодержащих
органических соединений в крекинг-газе содержатся окись и двуокись
углерода. В отличие от реакционных газов других процессов, в газе
практически отсутствует сажа, так как она оседает на графитовых
стержнях и шариках и смывается с них потоком жидких углеводо-
146
Жидкие
углеводороды
Рис. IV-15. Схема реактора для
электрокрекинга жидких углеводородов:
/—улавливающий козырек; 2, 4 —изолированные
реакционные камеры; 3— электроды; 5 —шарики
из электропроводящего материала.
0^0Bt Полученный газ промывают исходным нефтяным сырьем;
получаемый ацетилен выделяют абсорбцией селективными
растворителями. Промытый крекинг-газ можно непосредственно
использовать для сварки и резки металла.
Из 1 кг нефтяного сырья образуется 1,25—1,35 м3 реакционных
газов. Полезный крекинг до ацетилена составляет 48%. Выход
сажи 0,15—0,25/сг на 1 /сгС2Н2. Расход электроэнергии 9,5—10 квт-ч
на 1 кг С2Н2 (без учета стадии выделения ацетилена).
Жидкие углеводороды, выходящие из реактора, необходимо
фильтровать для отделения сажи. Кроме того, нужно строго следить за
циркуляцией нефтяного сырья через аппарат во избежание
накопления сажи в отдельных местах и короткого замыкания через
образующиеся сажевые «мостики». Сильный местный перегрев может
привести к взрыву.
Наряду с нефтяным сырьем в реакторах подобного типа
исследовался электрокрекинг обводненных жидких углеводородов.
Установлено, что при подаче стойких в данных температурных условиях
водно-органических эмульсий процесс электрокрекинга протекает
таким же путем, но с некоторым снижением выхода ацетилена за
счет образования окиси и двуокиси углерода.
Несмотря на кажущуюся простоту, метод получения ацетилена
электрокрекингом жидких углеводородов пока не нашел
практического применения. Это объясняется, вероятно, трудностями создания
аппаратуры большой мощности, а также отсутствием значительных
преимуществ в сравнении другими методами, уже нашедшими
широкое распространение в промышленности.
Крекинг углеводородов в плазменной струе
При соприкосновении потока углеводородов в реакторе с
горячей «нитью» электрической дуги некоторое количество исходного
сырья, как уже указывалось, подвергается деструкции под действием
высокой температуры (18 000° К), что обусловливает
непроизводительный расход части энергии. При деструкции образуются сажа,
осмол и ацетиленовые углеводороды. Наличие этих веществ в
реакционных газах осложняет и удорожает технологический процесс
выделения ацетилена.
Поэтому представляет интерес проведение пиролиза углеводородов
в среде теплоносителя, например водорода или аргона, нагретых
электрической дугой до 4000—5000° К, т. е. до частично
ионизованного состояния (низкотемпературная плазма). Поскольку при
соприкосновении водорода и аргона с горячей «нитью» дуги никаких
побочных продуктов, осложняющих выделение ацетилена, не
образуется, в струе такой низкотемпературной плазмы можно осуществить
направленный пиролиз углеводородов с целью получения ацетилена
при малом количестве побочных продуктов и сажи. Степень
превращения углеводородов и выход ацетилена и этилена в этих условиях
10* 147
>
]
^ 9
I
п _
-
1Г—!
,/
У
-
300 1000
2000 3000
Температура, °К
О
5000
ШО
2000
О
выше36, чем при линейном электрокрекинге. В настоящее время
плазменные процессы исследуют во многих странах36'40"42.
В основу химических процессов, протекающих в струе
низкотемпературной плазмы, положена способность некоторых газов
диссоциировать на атомы, ионы и электроны при сравнительно невысокой
температуре. Так, например, степень диссоциации водорода по
реакции
Н2 -> 2Н АН = +102,48 ккал/моль
в зависимости от температуры36 изменяется следующим образом:
Температура, °К 2500 3000 3500 4000 5000
Степень диссоциации, % 1,36 8,39 29,8 64,2 98,8
Вследствие высокой степени диссоциации значительно
увеличивается энтальпия водорода (рис. IV-16), благодаря чему он является
эффективным теплоносите-
пппп% лем для плазменных про-
g4 цессов. Поскольку при
охлаждении газа степень его
диссоциации уменьшается,
водород возвращается в
молекулярное состояние и
отдает большую часть тепла
(>>90%), полученного от
электрической дуги. Это
тепло расходуется на
пиролиз углеводорода.
Легкость диссоциации
водорода при высоких температурах способствует стабилизации
электрической дуги в плазматроне, что облегчает управление реактором.
Особенно сильно увеличивается токопроводимость в интервале
3000—4500° К.
В качестве теплоносителя в лабораторных условиях используется
также аргон, всегда находящийся в атомарном состоянии. Энтальпия
аргона возрастает с повышением температуры медленнее и
равномернее, чем энтальпия водорода (см. рис. IV-16). На рисунке видно
также, насколько выше энтальпия водорода в условиях высоких
температур.
Пиролиз углеводородов в струе низкотемпературной плазмы
подчиняется обычным закономерностям термических превращений.
Выход ацетилена и непредельных углеводородов зависит от
степени нагрева углеводорода и времени его пребывания в зоне высоких
температур. В случае подачи метана при достаточном перемешивании
его с водородной или аргонной плазмой реакция расщепления
протекает с хорошими выходами по ацетилену (табл. IV-4). Как видно
из таблицы, общее превращение метана и конверсия его в ацетилен
значительно выше, чем при линейном электрокрекинге (стр. 132),
однако и в данном случае образуются побочные продукты и сажа,
хотя и в меньшем количестве.
148
Рис. IV-16. Изменение энтальпии водорода и
аргона в зависимости от температуры.
ТАБЛИЦА IV-4
Экспериментальные данные о процессе превращения метана
в струе водородной плазмы35
>едняя тем
ратура, °К
4100
4140
4450
4600
5400
о
К
IN
>о2Х
о8Й
1,54
1,34
1,72
1,72
2,15
X
79,3
81,5
76,0
76,1
76,0
Состав реакционных газов, объемн. %
X
и
4,8
2,2
5,8
6,0
7,2
К
и
0,4
0,3
0,5
0,4
0,6
£
и
13,1
13,7
15,5
15,5
13,8
СЗ
-Е^
nJo я
Дч cu<U
о 55
—
0,01
0,02
0,02
"я"
1 (U
.53
д&|
OwW
0,20
0,10
0,16
0,10
0,20
д
и
0,20
0,30
0,09
0,07
0,10
д
о
0,60
0,50
0,36
0,27
0,52
1.
£ Я 2
со Ч <У
2-££
*£о
1,52
1,54
1,54
1,54
1,56
Степень
превращения
метана, %
общая
88
94
86
86
84
в
ацетилен
66
73
76
76
63
Механизм разложения метана в струе низкотемпературной
водородной плазмы подчиняется схеме, предложенной Касселем7:
СН4-> -СН2. + Н2
-СН2
С2Н6
Н2
С2Н4—>С2Н2 + Н2
С2Н2
2С + Н
Время реакции, сек
2800
10
1600
Кинетические и термодинамические условия разложения метана
в этом процессе35 графически представлены на рис. IV-17.
Максимум концентрации этилена (кривая 2) достигается при времени
реакции 10"6—10~5 сек, т. е.
раньше, чем максимум
концентрации ацетилена,
соответствующий времени 10~4—
10"3 сек (кривая 5).
Температура плазменной струи
(кривая 6) в начале процесса % 2000
очень быстро падает за счет
передачи тепла введенному
метану и протекания
эндотермических реакций его
расщепления. В конце процесса
температура несколько
повышается в связи с выделением
тепла при начинающемся
распаде ацетилена на углерод
и водород. Путем сравнения
кривых 3 и 4 ясно видно, что
свободный углерод в данном
случае образуется вследствие
распада ацетилена.
'2
10~3 W'2 W 1 Ю1 Wz
Протяженность реакционной зоны, см
Рис. IV-17. Кинетические и
термодинамические условия пиролиза метана в струе
водородной плазмы:
/, 2, 3, 4, 5—изменение концентрации метана (У),
этилена (2), ацетилена (3), сажи {4) к водорода (5)
в зависимости от времени реакции; 6,7—
изменение температуры смеси (6) и скорости потока (7)
в зависимости от протяженности реакционной
зоны.
149
При пиролизе низших гомологов метана и низкооктанового
бензина в струе водородной плазмы процесс протекает с соблюдением
тех же закономерностей, что и при подаче метана, однако в
реакционных газах кроме ацетилена появляются в значительном количестве
этилен и пропилен, доля которых зависит от конечной температуры
газовой смеси. При подогреве
исходного сырья
увеличивается выход ацетилена и
непредельных углеводородов
и сокращаются затраты
электроэнергии35. При
использовании аргона в
качестве теплоносителя пиролиз
бензина протекает так же,
как и метана, но
концентрация ацетилена и
непредельных углеводородов
получается значительно ниже в связи
с меньшим теплосодержанием
аргона. Следовательно, в
данном случае необходимо брать
теплоноситель в большем
количестве.
Фирма Knapsack - Gries-
heim (ФРГ) в 1963 г. провела
испытания полузаводского
реактора для пиролиза
различных углеводородов в струе
водорода, нагретого до
состояния плазмы36. Мощность
реактора составляла 2500 и
4000 кет на 1 т бензина в 1 ч.
В реакторе (рис. IV-18)
между двумя графитовыми
электродами / диаметром по
70 мм устанавливается
электрическая дуга. В дуговое
пространство 2 подается
водород в количестве 700—
820 м3/ч. Теплонапряжение
дугового пространства дости-
Маслона
„закалм
Газы пиролиза
Рис. IV-18. Электродуговой плазменный
реактор фирмы Knapsack-Griesheim мощностью
2500 кет на 1 т бензина в 1 ч:
/—электроды; 2— дуговое пространство; 3— зона
пиролиза.
гает при этом 2 млн. кет •
В зону пиролиза 3 поступает низкооктановый бензин. Часть бензина
нагретым водородом поднимается вверх до нижней границы дугового
пространства, а остальное количество увлекается вниз, обеспечивая
тем самым хорошее перемешивание газов. В конце реакционной зоны
устанавливается температура 900—1100° С, которая затем резко
снижается до 200° С вспрыскиванием масла. С потоком масла из
150
реакционной зоны удаляются сажа, наиболее тяжелые углеводороды
и смола. Выходящие из реактора газы промываются маслом в
скруббере, охлаждаются там до 30° С и направляются на компрессию,
а затем на выделение ацетилена и этилена. Тепло нагретого масла
используется для получения водяного пара.
Результаты пиролиза различных углеводородов в струе
водородной плазмы * представлены в табл. IV-5. Из этих данных видно,
что количество сажи и смол колеблется от 0,7 до 7 вес. % в
зависимости от свойств углеводородного сырья. Наименьшие количества
этих веществ образуются при пиролизе метана и циклогексана,
относительно большой выход сажи и смол наблюдается при
переработке бензола. Выход ацетиленовых углеводородов также зависит
от состава сырья. Средняя концентрация целевых продуктов в газе
пиролиза составляет: 12—Нобъемн. % ацетилена и 6—10 объемн. %
этилена.
ТАБЛИЦА IV-5
Результаты пиролиза различных углеводородов в струе водородной плазмы 36
Компоненты
Выход при пиролизе различного сырья, вес. %
Ацетилен
Этилен
Метан
Ацетиленовые
углеводороды
Бензол
Этан и остальные
углеводороды
Водород *
Двуокись углерода . .
Сажа и смола . . . .
Всего
Примерный суммарный
выход ацетилена
и этилена
Примерное
соотношение С2Н2 : С2Н4 . . .
65,58
3,57
9,80
4,50
0,93
16,81
0,70
36,04
8,11
8,96
6,67
1,12
33,42
2,77
0,22
2,10
39,12
11,96
13,49
2,57
19,41
2,66
0,27
1,40
53,94
12,23
5,88
7,89
3,92
12,05
1,45
0,30
1,50
41,20
11,81
9,60
9,59
9,54
14,41
0,36
0,23
3,00
40,93
7,71
5,98
11,41
4,19
22,04
3,89
1,40
2,20
40,34
8,10
3,37
3,76
1,68
36,60
3,26
1,30
0,65
50,96
4,10
1,66
4,34
26,80
0,99
0,33
2,79
7,30
99,89
67
16: 1
99,41
44
5: 1
99,72
51
4 : 1
99,22
66
4,5 : 1
99,74
53
4: 1
99,75
48
5: 1
99,06
49
5: 1
99,27
55
Бгз учета водорода, введенного с плазмой.
Расходные показатели процесса пиролиза углеводородов в струе
водородной плазмы по данным36 фирмы Knapsack-Qriesheim состав-
Процесс осуществлен в полузаводском масштабе.
151
ляют в расчете на 1 т ацетилена (без учета стадии выделения
ацетилена):
Углеводороды, т 1,6—2,5
Водород, ж3 1750—2250
Электроэнергия, квт-ч
общий расход 7000—9000
на 1 ж3 Н2 3—4
на 1 т С2Н2 + С2Н4 4600
Переработка углеводородов в плазменной струе с целью
получения ацетилена еще не получила практического применения, пока
лишь продолжаются научно-исследовательские и конструкторские
работы по созданию аппаратуры для плазменного пиролиза. Метод
пиролиза в струе водородной плазмы, его перспективы, а также
разработка и внедрение в заводскую практику больше зависят от общей
конъюнктуры в отношении использования ацетилена для
промышленного органического синтеза и перспективного снижения стоимости
электроэнергии.
* *
*
Из всего сказанного в гл. IV следует, что электрическая дуга для
расщепления углеводородов до ацетилена используется в
промышленности довольно широко. В настоящее время методом
электрокрекинга производится во всех странах более 180 тыс. т
ацетилена в год. Ведется разработка методов пиролиза углеводородов
в струе водородной плазмы и методов электрокрекинга жидких
углеводородов, усовершенствуется аппаратура получения и выделения
ацетилена37"39.
Процесс электрокрекинга в настоящее время характеризуется
отработанностью технологической схемы, высокой степенью
механизации и автоматизации управления. Концентрация ацетилена
в газе, полученном этим способом, выше, чем при других методах.
Предварительный подогрев углеводородов еще более улучшит техно-
экономические показатели электрокрекинга. Применение
коаксиального реактора и проведение пиролиза в струе водородной плазмы
позволят получить дополнительные экономические выгоды.
ЛИТЕРАТУРА
1. Ньюлэнд Ю. А., Фогт Р. Р., Химия ацетилена, Издатинлит, 1947.
2. Фогель И. Г., Ацетилен, его свойства, изготовление и применение, Гос-
химтехиздат, 1934.
3. Энгель А., Штен.бек М., Физика и техника электрического разряда
в газах, т. 2, ОНТИ НКТГ СССР, 1936.
4. Э н г е л ь А., Ионизированные газы, Физматгиз, 1959.
5. Андреев Д. Н., Органический синтез в электрических разрядах, Изд.
АН СССР 1953
6. Борисова Е. Н., Еремин Е. Н., ЖФХТ, 36, 1261, 2234 (1962).
7. К a s s e I L. I., J. Am. Chem. Soc, 54, 3949 (1932).
8. В а с и л ь е в С. С, Кобозев Н. И., Еремин Е. Н., ЖФХ, 7, № 5,
619 (1936).
152
9 Кондратьев В. Н., Кинетика химических газовых реакций, Изд. АН
' СССР, 1960.
Ю Ильин Д. Т., Еремин Е. Н., ЖПХТ, 35, вып. 9 (1962).
11 Э й д у с Я. Т., Изв. АН СССР, сер. хим., № 3, 737 (1938).
12. Peters К., Wagner О. Н., Z. physik. Chem., 153, 161 (1931).
13. Б о ж к о Н. П., Оршанский Д. Л., Химия твердого топлива, № 7 (1936).
14. Колер Д. К., ЖПХ, 13, № 1, 102 (1940).
15. Колер Д. К., Химия твердого топлива, № 8 (1936); № 1, 8, 9 (1937).
16. Peters К. Meyer К., Brennst., Chem., 10, № 16 (1929).
17. Кобозев Н. И., Васильев И. И., Еремин Е. Н., ЖФХ, 10,
543 (1937). j|
18. Е р е м и н Е. Н.,^ Ильин А. Т., Хим. пром., № 6, 408 (1962).
19. Ц е н ц и п е р й А. Б., Еремин Е. Н., Кобозев Н. И., ЖФХ, 37,
№ 4 (1963).
20. А н т о н о в В. Н., Производство ацетилена, Госхимиздат, 1959.
21. Физико-химические свойства индивидуальных углеводородов, Гостоптехиздат,
1960.
22. Кей Д. Ж., Л оби Т., Таблицы физических и химических постоянных,
Физматгиз, 1962.
23. Glad is ch M., Hydrocarb. Proc. a. Petrol. Ref., June, 1962.
24. Химия ацетилена, под ред. М. Ф. Шостаковского, Изд. «Наука», 1968.
25. G 1 a d i s h M., Chim. et ind., 88, № 5, 471 (1962).
26. Chem. Eng., 71, № 2, 67 (1964).
27. Пат. США 3073769; канад. пат. 573701; англ. пат. 938823.
28. The University of Texas Publication, June 1, 5011 (1950).
29. Andrusow L., Chim. et ind., 79, № 4 (1958).
30. Пат. США 2632731, 1936 г.
31. Пат. ФРГ 925891, 1955 г.
32. Печуро Н. С, Меркурьев А. Н., Проблемы электрической
обработки материалов, Изд. АН СССР, 1962.
33. П е ч у р о Н. С, Г р о д з и н с к и й Э. А., П е с и н О. Б., Газ. пром.,
47, № 2 (1963).
34. Химические реакции органических продуктов в электрических разрядах, Изд.
«Наука», 1966, стр. 84, 93, 111, 129, 137, 181.
35. Кинетика и термодинамика химических реакций в низкотемпературной плазме,
Изд. «Наука», 1965 (ред. Л. С. Полак), стр. 90.
36. Semuwald К-, Schablus E., Pohf F., Chem. Ing. Techn., 35,
№ 1, 1 (1963).
37. Ильин Д. Т., канд. дисс, 1963, ИНХС АН СССР.
38. L e u t n e r H., Sokes Ch., Ind. Eng. Chem., 53, № 5, 341 (1961).
39. Ильин Д. Т., Еремин Е. Н., ЖПХ, 38, № 12, 2876 (1965).
40. КобзевЮ., Козлов Г., Худяков А., Электр, обраб. матер., № 1
(1968).
41. Полак Л. С, Эндюськин П. А., Углев В. Б., Химия высоких
энергий, 3, № 2 (1968).
42. Gladish H., Chem. Ing. Gechn., 41, №4 (1969).
ГЛАВА
V
ПОЛУЧЕНИЕ АЦЕТИЛЕНА
ОКИСЛИТЕЛЬНЫМ ПИРОЛИЗОМ УГЛЕВОДОРОДОВ
Первые опыты по получению ацетилена частичным окислением
метана кислородом были проведены в Германии в 30-х годах, а
в начале 40-х годов там же были осуществлены полупромышленные
испытания процесса и разработан реактор мощностью 2500 т
ацетилена в год. В 50-х годах метод окислительного пиролиза
природного газа до ацетилена начали использовать в промышленном
масштабе в ФРГ (фирма BASF), Бельгии (фирма SB А) и Италии (фирма
Montecatini). В настоящее время работают заводские установки
также в Румынии, СССР, Франции, Японии, Бельгии, Польше,
Чехословакии и других странах.
В СССР исследования процесса окислительного пиролиза метана
начались с 1950 г. при участии П. А. Теснера, Б. С. Гриненко,
В. У. Шевчука, Я. С. Казарновского, В. П. Семенова, И. М. Рома-
нюка, В. В. Харламова, Ф. П. Ватлаева, Н. Е. Алипова, С. А. Аст-
вацатряна и др. В результате исследовательских и опытных работ
были сконструированы отечественные ацетиленовые реакторы
большой производительности, а также аппаратура систем сажеочистки
и др. Крупные исследования процесса получения ацетилена
окислительным пиролизом природного газа проводились в Венгрии и
Румынии (А. Ласло, М. Немет, М. Молдован), где также были
сооружены опытно-промышленные установки.
В настоящее время в промышленности разных стран процесс
получения ацетилена окислительным пиролизом имеет наибольшее
применение по сравнению с другими методами его получения из
углеводородного сырья. За последние годы созданы крупные реакторы
производительностью 5000 —8500 т ацетилена в год и продолжается
разработка еще более мощных агрегатов.
ОСНОВНЫЕ СВЕДЕНИЯ О ПРОЦЕССЕ
При окислительном пиролизе углеводородов тепло, необходимое
для проведения эндотермической реакции образования ацетилена,
получается в результате сжигания части исходного сырья в
атмосфере кислорода. Процессу неполного горения присущи особые
факторы (скорость горения, пределы взрываемое™, индукционный
период самовоспламенения горючих смесей и т. п.), которые
существенно влияют на образование ацетилена.
154
Термодинамика и кинетика процессов неполного горения
Взаимодействие метана с кислородом может протекать в
нескольких направлениях в зависимости от содержания кислорода
в смеси и режима процесса (температуры, давления и времени
пребывания продуктов в зоне реакции):
СН4 + 0,5 О2 ^ СО + 2Н2 АН = —8,5 ккал/моль (1)
СН4 + О2 ^ СО + Н2 + НаО АЯ = —66,4 ккал/моль (2)
СН4 + 2О2 ^ СО2 + 2Н2О Д# = —191,7 ккал/моль
(3)
Например, при содержании кислорода в исходной смеси до 35—
40% и температуре выше 1400° К можно получить газовую смесь,
состоящую в основном из окиси углерода и водорода. Расчетные
значения констант равновесия1 реакций (1) и (2) приведены в табл. V-1.
ТАБЛИЦА V-1
Расчетные константы равновесия реакций неполного горения метана
Температура
°К
600
700
800
900
1000
Константа равновесия К
СН4+0,5 О2 Z
?СО+2Н2
реакция (1)
2,1691 • 1012
1,0296-1012
6,0475-1011
4,1081-Ю11
3,0557-1011
СН4+О2 t
tco+H2+
+н2о
реакция (2)
I I I I I
Температура
°К
1200
1400
1600
1700
1800
Константа равновесия /С
СН4+0,5 О2 t-
£СО+2Н2
реакция (1)
1,9574-1011
1,4236-1011
1,0281-1011
СН4+О2 t
?СО+Н2+
+н2о
реакция (2)
1,17-Ю57
0,28-1056
0,79-1055
0,25-1055
Реакция (3) полного окисления метана практически необратима и
в дальнейшем не рассматривается. Из данных табл. V-1 видно, что
при 1400° К и выше термодинамически более вероятна реакция (2),
так как константа равновесия ее значительно выше. Из таблицы
следует также, что константа равновесия реакции (1)
„ РсоРн2
уменьшается с повышением температуры, т. е. при этом возрастает
количество метана, не вступившего в реакцию. Опытные данные,
однако, показывают, что доля остаточного метана при повышении
температуры, наоборот, уменьшается. Это противоречие исчезает,
если предположить, что метан взаимодействует с кислородом в две
стадии. На первой стадии весь кислород расходуется на полное
сгорание части метана по реакции (3), а на второй — образовавшиеся
155
двуокись углерода и водяной пар взаимодействуют с остальным
количеством метана по эндотермическим реакциям:
СН4 + Н2О ^ СО + ЗН2 ДЯ = +49,3 ккал/моль (4)
СН4 + СО2 ^ 2СО + 2Н2 ДЯ = +59,1 ккал/моль (5)
Результаты расчета по этим уравнениям хорошо согласуются
с экспериментальными данными. Таким образом, можно считать,
что 25% метана, взаимодействующего с кислородом, расходуется
по реакции (3), а 75% — по реакциям (4) и (5), причем последние
реакции протекают достаточно быстро, а в некоторых условиях
и мгновенно 2. Высказанные здесь представления в основном
относятся к взаимодействию метана и кислорода при низких
температурах (процесс конверсии метана при 1073—1173° К).
При повышении температуры, как видно из данных табл. V-1,
в условиях неполного горения (окислительный пиролиз) более
вероятной становится реакция (2):
СН4 + О2 ^ СО + Н2 + Н2О АЯ = —66,4 ккал/моль
Выделяющегося при этом тепла достаточно для образования
ацетилена в условиях окислительного пиролиза:
2СН4 ^± С2Н2 + ЗН2 АЯ - +91 ккал/моль (6)
Равновесный состав продуктов, получающихся в процессе
неполного сгорания метана, по данным П. А. Теснера 3 и Говарда 4,
отвечает так называемой реакции водяного газа
СО + Н2О^СО2 + Н2 (7)
причем равновесие этой реакции устанавливается в конце процесса,
в «бескислородной» зоне 3 при 1300—1450° С. Убедительным
доказательством этого служит совпадение температур — измеренной
термопарой и рассчитанной Я. С. Казарновским и др.5 по константе
равновесия:
Например, если измеренная температура составляла 1300° С,
то рассчитанная величина 5 находилась в пределах 1287—1322° С.
Путем эффективной «закалки» реакционных газов можно добиться
равновесия при любой заданной температуре в реакционной зоне.
В обстоятельной работе Леру и Матье 6, посвященной
термодинамике и кинетике процесса окислительного пиролиза, наоборот,
показано, что равновесие реакции водяного газа практически не
достигается: опытные данные реакции водяного газа значительно
отклоняются от расчетных.
По мнению авторов 7, наиболее рационально представить процесс
окислительного пиролиза следующей схемой. Часть метана сгорает
по реакции (2), причем образующиеся продукты вступают в реак-
155
иЮ (7), равновесием которой определяется общий состав продуктов
сгорания. Остальной метан превращается по реакции (6) в ацетилен
и водород.
Интересно сравнить окислительный пиролиз метана до ацетилена
и широко распространенную высокотемпературную конверсию
метана с образованием синтез-газа, так как состав реагирующих
компонентов и температура процесса одинаковы. Если представить
образование ацетилена в виде промежуточной реакции при
высокотемпературной конверсии метана, оба процесса можно описать
следующей схемой:
СН4 + 2О2 ^ СО2 + 2Н2О
2СН4^С2Н2 + ЗН2
Кроме того, образуется сажа
С2Н2^2С+Н2 (8)
которая при температуре процесса частично подвергается
газификации:
С+Н2О^СО+Н2 (9)
В процессе высокотемпературной- конверсии метана
продолжительность реакции не является лимитирующим фактором, а состав
получаемых продуктов соответствует равновесию реакции (7)
водяного газа, что подтверждается многочисленными данными. В процессе
окислительного пиролиза метана продолжительность реакции,
наоборот, очень важна, так как состав получаемых продуктов
фиксируется на определенной промежуточной стадии, что и позволяет
получать ацетилен. При этом также достигается равновесие реакции
водяного газа, но фактический состав продуктов отклоняется от
расчетного на некоторую величину.
Механизм образования и разложения ацетилена
Окислительный пиролиз метана можно представить как процесс,
состоящий из трех этапов: горения метана, образования ацетилена
и разложения ацетилена8"10. Б. С. Гриненко и А. М. Зелизный 10
предлагают следующую схему окислительного пиролиза:
СН4 + О2 i± СО + Н2О + Н2 АН = —66,4 ккал/моль
СН4 + 2О2^-СО2 + 2Н2О АН = —191,7 ккал/моль
2СН4^С2Н2 + ЗН2 АН = +91 ккал/моль
С2Н2 т± 2С + Н2 АН = —10,5 ккал/моль
В табл. V-2 приведены данные, характеризующие долю указанных
Реакций в расходовании исходных метана и кислорода, а также в теп-
л°вом балансе процесса. Как видно из таблицы, всего в реакциях уча-
157
ствует примерно 90% метана и 99% кислорода. До 10% метана и 1%
кислорода переходят в газы пиролиза в неизмененном состоянии.
ТАБЛИЦА V-2
Доля отдельных реакций в процессе окислительного пиролиза
Показатели
Степень участия исходных
компонентов, %
метан
кислород
Доля в общем тепловом
балансе, %
сн4+о8 Z со+
+Н2+Н2О
50—52
78—79
71—72
СН4+2О2?СО2 +
+2Н2О
5 7
20—22
28—29
2СН4^С2Н2 +
+зн2
До 33
32—33
В отличие от чисто термического пиролиза, где реакция крекинга
метана лимитируется стадией зарождения первичных радикалов,
при окислительном пиролизе возможно образование метиленовых
•СН2- и метальных СН3- радикалов при горении части метана.
Поэтому процесс может протекать по двум направлениям8"10:
1.СН4 + О —
•сн2. +о2
нсно ->
и. сн4+ -он
СН3- +О2 -»
нсно
.СНа- +Н2О
> нсно + о
со + н2
— сн3- +н2о
НСНО+ -ОН
со + н2
В обоих случаях процесс горения части метана можно выразить
общим уравнением (2):
СН4 + О2 -СО + Н2О + Н2
Леру и Матье полагают 6, что несгоревший метан подвергается
крекингу с образованием ацетилена:
OHL
С2Н6 —> С2Н4 —> С2Н2
Подтверждением этой схемы является совпадение
экспериментальных и расчетных данных по температуре и распределению
углерода в продуктах окислительного пиролиза метана (табл. V-3).
Расчет вели по константам скоростей для отдельных стадий
процесса.
3. В. Иевлевой и П. А. Теснером 3 были исследованы условия
образования ацетилена в процессе неполного горения в лабораторной
горелке при различных соотношениях метана и кислорода в исходной
смеси, а также проведены опыты с добавкой пропана. Большая часть
метана взаимодействует с кислородом на очень коротком участке
по вертикальной оси пламени горелки, и реакция неполного горения,
158
ТАБЛИЦА V-3
Результаты исследования окислительного пиролиза метана
Показатели
Температура, С
Содержание углерода, %
в метане
в этилене
в ацетилене
в саже и тяжелых углеводородах
в окиси углерода
н
2
с
О
1840
18,4
• 3,1
65,7
9,6
3,2
н
0)
о
а
1845
18,3
—
69,8
7,2
1,7
А
С
О
1800
20,1
2,1
66,2
9,7
0,8
н
(L)
о
0-
1799
20,2
66,6
8,6
1,9
1
2
с
О
1670
28,9
53,1
10,5
4,2
1670
27,3
55,3
13,2
2,2
сопровождаемая значительным образованием ацетилена,
заканчивается в основном в кислородной зоне шириной в несколько десятых
миллиметра.
Содержание ацетилена за кислородной зоной увеличивается до
максимума, соответствующего расстоянию 0,3—0,4 мм от конца зоны,
а затем начинает уменьшаться при дальнейшем движении газовой
смеси по оси горелки. Было установлено, что в начале кислородной
зоны образуются только водяной пар и окислы углерода, а ацетилен
и этилен появляются позднее. Количество водорода в газе к концу
кислородной зоны возрастает. Это, вероятно, можно объяснить
протеканием реакции водяного газа.
В кислородной зоне температура по оси пламени быстро
возрастает, а по достижении примерно 1850° С остается почти постоянной.
Это еще раз подтверждает, что образование ацетилена в процессе
окислительного пиролиза носит чисто термический характер и
происходит при расщеплении молекул метана и последующих
превращениях соответствующих углеводородных радикалов. Необходимая
для этого высокая температура создается за счет сгорания части
метана до СО, Н2О и СО2.
По Д. П. Алейнову и Я. С. Казарновскому, наиболее вероятна
следующая схема образования и последующего разложения части:
ацетилена при окислительном пиролизе метана11:
Метан->Этан-^Этилен->Ацетилен —
| Ф+н2о
~> Окись углерода
Ди ацетилен
i
Сажа и водород
t
Винилацетилен
Видно, что разложение ацетилена протекает гомогенно до
газообразных ацетиленовых углеводородов и гетерогенно — на простые
вещества. Экспериментально установлено11'12, что при атмосферном
Давлении и высоких температурах ацетилен разлагается
преимущественно до диацетилена, а при повышении давления и низких
температурах главным продуктом является винилацетилен. Это перерас-
159>
пределение углерода обусловлено тем, что на образование диаце-
тилена
2С2Н2 —> С4Н2 -f- Н2
повышение давления влияет мало, а образование винилацетилена
при этом заметно ускоряется:
2С2Н2 —* С4Н4
В продуктах пиролиза замечены также метил ацетилен,
бутадиен-1,3 и пропадиен, которые, по-видимому, также образуются
при распаде ацетилена11. Суммарная концентрация ацетиленовых
углеводородов не зависит от давления и пропорциональна
концентрации ацетилена в газе пиролиза. Считается, что разложению
подвергается около 5% образовавшегося ацетилена.
С. С. Абаджев и В. У. Шевчук12 изучали разложение ацетилена,
разбавленного инертным газом, в интервале 1300—1750° К. По их
представлению, ацетилен распадается гомогенно — с образованием
в основном винилацетилена и диацетилена и гетерогенно — на С
и Н2 без промежуточных стабильных продуктов (например, доля
гомогенной реакции при 1000° К составляет 35%). При температурах
около 1300° К преобладает разложение ацетилена через винилаце-
тилен, при ~ 1700° К процесс в основном идет через диацетилен,
хотя при этом также появляются С и Н2.
Условия горения метано-кислородных смесей
Процесс неполного горения метана определяется главным образом
концентрационными пределами взрываемости смеси его с
кислородом. В первую очередь рассмотрим влияние температуры на верхний
предел взрываемости. Из рис. V-1 видно13, что с ростом температуры
верхний предел взрываемости углеводородов в смеси с кислородом
(по кислороду) понижается, причем для метана медленнее, чем,
например, для этана.
Эта особенность метана позволяет допускать высокие температуры
предварительного подогрева, что, как будет показано ниже, очень
существенно для проведения процесса пиролиза. Примеси этана и
пропана в природном газе значительно расширяют пределы
воспламенения и делают эти смеси более взрывоопасными по сравнению
с метано-кислородной.
Как известно, повышение давления расширяет концентрационные
пределы взрываемости горючих газов и уменьшает предельно
допустимую концентрацию кислорода. Поэтому с ростом давления взры-
воопасность смеси метана и кислорода увеличивается. Из данных
рис. V-2 видно14, что при повышении давления с 1 до 4 am предельно
допустимая концентрация кислорода при 0° С уменьшается с 48 до
43 объемн. %. С ростом температуры предельно допустимая
концентрация кислорода тоже уменьшается. Изменение верхнего
предела взрываемости метана в смеси с кислородом в зависимости от
160
давления выражается следующей эмпирической формулой15
(графически зависимость показана на рис. V-3):
С = 56,0 (Р - 0,9)0'04
где С — концентрация метана в смеси, объемн. %;
Р — общее давление смеси, am.
Добавки водяного пара уменьшают взрывоопасность метано-
кислородных смесей. Например, при 2 объемн. % Н2О (см. рис. V-2)
предельно допустимая концентрация кислорода при 0° С и
избыточном давлении 4 am возрастает с 43 до 47 объемн. %.
50
•
ч
р
^ й: 45
it
^ ^ /,п
100 200 300 400 500
* Температура, °С
Рис. V-1. Температурная
зависимость верхнего предела взры-
ваемости смесей метана (/) и
этана (2) с кислородом (площадь
над кривыми — область взрыва).
100 200 300 400 500
Темг/ература, °С
Рис. V-2. Изменение предельно допустимой
концентрации кислорода в смеси с метаном
в зависимости от температуры и давления
(площадь над кривыми — область взрыва):
/ — при 4 am; 2 — при 1 am; 3 — при 4 am
(в смеси содержится 2 объемн. % Н2О).
Начало взаимодействия метана с кислородом определяется
периодом индукции — временем, необходимым для развития цепного
процесса горения. Период индукции, или время, по истечении
0,25*
50 70100
Рис. V-3. Изменение верхнего
предела взрываемости метана
в смеси с кислородом в
зависимости от давления.
860 900 9Ь0
Температура, Ч
980 W00
Рис. V-4. Температурная зависимость
периода индукции метано-кислородной
смеси (37 объемн. % О2) при 1 am.
которого начинается самовоспламенение газов, определяется
температурой, давлением и составом горючей смеси. На рис. V-4
показано изменение периода индукции метано-кислородной смеси
В. II. Антонов
(37 объемы. %О2) в зависимости от температуры13 при 1 am,
например при 1000° С период индукции составляет около 0,01 сек. На
практике обычная температура подогрева метано-кислородной смеси
600—700° С, при этом если проэкстраполировать данные рис. V-4,
период индукции составляет несколько секунд.
Изменение периода индукции метано-кислородной смеси в
зависимости от температуры и давления (при высоких давлениях)
16
12
10
Ь
f
Iе
I
{
if:
\
Л
:• Ф
^f^i^
1
1'
!
\
1
\
^V^f
350 400 U50 500
Температура, °С
а
20 40
Избыточное давление, am
6
Рис. V-5. Изменение периода индукции метано-кислородной
смеси (33 объемн. % О2) в зависимости от температуры при
избыточном давлении 10 am (а) и в зависимости от давления
при 420° С (б).
показано17 на рис. V-5. Видно, что он довольно сильно зависит от
начальных параметров, причем наибольшее изменение наблюдается
в области меньших температур и давлений. Зависимость периода
индукции ти от давления Р и температуры Т выражается следующим
уравнением:
Pne~VITxll = const
где п и v — постоянные величины. Например, при 420° С в интервале
давлений 5—30 am эта формула приобретает вид:
э2'7ти = const
162
Высокое значение п — 2,7 указывает на необычайно сильное
влияние давления на период индукции данной смеси. Зависимость
периода индукции от температуры в интервале 360—510° С при
давлении 10 am выражается уравнением:
тиб-4900°/г = const
Период индукции метано-кислородной смеси существенно меняется
с изменением ее состава: чем богаче кислородом смесь, тем меньше
ее период индукции (табл. V-4). Небольшие добавки тяжелых
углеводородов (до 4 объемн. %) не оказывают влияния на период
индукции. Если количество тяжелых углеводородов (например, пропана
или бутана) превышает 4 объемн. %, то период индукции метано-
кислородных смесей заметно уменьшается и температура их
воспламенения снижается16 на 150 — 200° С (табл. V-5).
ТАБЛИЦА V-4
Период индукции метано-кислородных смесей
Содержание
кислорода
объемн. %
50
60
33
50
66,6 '
Избыточное
давление
am
5
5
10
10
10
Температура
°С
310
310
380
380
380
Период
индукции, сек
4-8
0,1—2
5,3
0,1—0,4
<0,02
ТАБЛИЦА V-5
Температура самовоспламенения и период индукции метано-кислородных смесей
сн4
61,5
61,5
61,5
52,0
52,0
52,0
52,0
51,0
51,0
51,0
51,0
Состав смеси,
С3Н8
—
4,3
4,3
4,3
4,3
1,4
1,4
1,4
1,4
С4н10
—
—
3,6
3,6
3,6
3,6
эбъемн. %
о2
37,0
37,0
37,0
41,7
41,7
41,7
41,7
42,0
42,0
42,0
42,0
N2
1,5
1,5
1,5
2,0
2,0
2,0
2,0*
2,0
2,0
2,0
2,0
Температура
самовоспламенения
°С
600
650
700
450
500
550
600
400
450
500
550
Период
индукции
сек
>15
2,4
1,18
>15
13,85
3,12
0,59
>15
14,0
4,16
0,96
Изучалось также действие добавок17 N2, CO2, Н2О, СН3С1 и СС14.
Добавки азота, даже в больших количествах, не оказывали влияния
на период индукции. Причина этого явления, однако, не выяснена.
Другие примеси в малых количествах не влияют на период индукции,
11* 163
но при увеличении их концентрации период индукции
изменяется. Например, период индукции метано-кислородной смеси
(80 объемн. %О2) при 5 am и 290—360° С составляет 0,02 сек\ при
добавлении 7,4 объемн. %СО2 эта величина не изменилась, а при
15 объемн. %СО2 возросла до 0,3 сек. Следовательно, СО2 действует
как флегматизатор.
Режим горения
определяется многими параметрами.
Для характеристики
процесса получения ацетилена рас-
400 Ш Ш 1000 смотрим скорость горения и
Температура,°с длину факела.
Рис. V-6. Температурная зависимость нор- Горящую смесь характе-
мальной скорости горения
метано-кислородной смеси (37 объемн. % О2).
200
ризует нормальная скорость
горения, т. е. минимальная
скорость распространения
пламени по нормали к его поверхности в неподвижной смеси
данного состава. Эта скорость зависит от температуры, например для
метано-кислородной смеси (37 объемн. %О2) нормальная скорость
горения возрастает примерно в 2,5 раза с повышением температуры
в 10 раз —от 100 до 1000° С (рис.
V-6). Практически наблюдаемые
скорости горения значительно
отличаются от нормальной скорости.
Они зависят от многих факторов
(степени турбулизации, диаметра
отверстия горелки и др.).
Устойчивость горения метана в воздухе
0х—
5 10 15
Диаметр, мм
20
Рис. V-7. Зависимость скорости
гашения для смесей углеводородов с
воздухом от диаметра сопла горелки.
Рис. V-8. Схема факела горения
одиночной турбулентной струи.
и кислороде характеризуется также скоростью гашения, при которой
пламя отрывается от горелки. Скорость гашения изменяется в
зависимости от диаметра сопла горелки13 (рис. V-7) и особенно сильно —
при малых диаметрах (до 5 мм). Для пропана характерна
меньшая скорость гашения, чем для метана.
Схема факела горения одиночной турбулентной струи показана
на рис. V-8. Он состоит из трех основных зон18: зоны
воспламенения LB, фронта горения (собственно пламени) аг и зоны
догорания ЬЛ.
164
Общая длина факела Ьф равна:
Длина зоны воспламенения (ядро факела) LB в первом
приближении может быть рассчитана по формуле:
и
Толщина фронта горения ат описывается выражением:
Зона догорания в процессе образования ацетилена не играет
существенной роли. Длина зоны догорания практически зависит от
кинетических свойств горючей смеси и в общем виде может быть
выражена уравнением:
1Д % К3®
Величины, входящие в эти уравнения, означают следующее:
Ki и /С3 — экспериментальные константы;
со — скорость истечения горючей смеси из отверстия (кратера) горелки;
d — диаметр отверстия горелки;
и — скорость распространения пламени;
/С2 — константа, зависящая от характера горения (ламинарное или
турбулентное).
Теоретический расчет длины факела Ьф на основании данных
о составе смеси, скорости горения и и скорости истечения горючей
смеси со практически невозможен, поскольку длина факела зависит
еще и от других параметров, которые можно определить только
экспериментально18'19.
Ламинарное горение (простейший режим горения)
характеризуется резко очерченными границами факела. При ламинарном
горении толщина фронта пламени ат практически составляет доли
миллиметра, поэтому общая длина факела определяется только
длиной зон воспламенения и догорания:
Поверхность зоны воспламенения для ламинарного горения можно
принять равной поверхности конуса, основанием которого является
отверстие горелки. Под действием определенных факторов эта
поверхность может сильно изменяться. В процессе получения
ацетилена при ламинарном горений зона наивысшей температуры
расположена непосредственно за зоной воспламенения, поэтому процесс
горения и соответственно ацетиленообразование заканчивается на
очень малом участке.
На практике горение метано-кислородной смеси обычно протекает
турбулентно. По поводу механизма турбулентного горения
существует несколько теорий, которые пока еще не дают однозначного
решения. Предполагают, что турбулентное горение происходит на
165
поверхности газовой смеси. Состояние турбулентного потока в каждой
отдельной зоне непрерывно меняется: изменяется величина
отдельных объездов газа, а также скорость и направление относительного
движения потоков.
Различают два типа турбулентности: мелкомасштабную (с
большими колебаниями скорости горения) и крупномасштабную (с малыми
колебаниями скорости горения). Это обстоятельство приводит к
различным закономерностям распространения пламени. При
мелкомасштабной турбулентности скорость турбулентного горения ит
зависит от нормальной скорости горения в ламинарном потоке ил:
щ
Ил
I/Re
При крупномасштабной турбулентности выброс языков пламени
перед фронтом горения настолько значителен и общая поверхность
воспламенения так велика, что практически скорость
распространения пламени не зависит от нормальной скорости горения и
определяется лишь величиной турбулентности:
С точки зрения механизма образования факела следует
различать два предельных процесса: первый — возникновение факела
в заранее перемешанной смеси горючего газа и окислителя
(кислорода); второй — образование факела при раздельной подаче горючего
газа и кислорода — диффузионный факел. Природа поверхности
воспламенения (ядра факела) при этих двух процессах
принципиально различна. Ядро факела первого типа связано с
распространением фронта пламени в струе, как, например, в обычной бунзе-
новской горелке и в туннельных горелках. В большинстве
ацетиленовых реакторов происходит именно такое горение. Факел второго
типа связан с процессом перемешивания горючего газа с кислородом
в струе. В ацетиленовых реакторах некоторых конструкций
применяется, правда не в полной мере, и этот тип горения.
Необходимым условием для устойчивого режима горения при
отсутствии посторонних импульсов является равенство скоростей
истечения со и горения и. При ламинарном горении это условие
соблюдается в нижней периферийной части конуса, так как при
истечении смеси у края отверстия образуется область медленных
скоростей потока, в которую проникает горячий газ из основной струи.
При наличии такого устойчивого участка воспламеняется основная
масса газа. В обычных условиях (турбулентное горение) пламя не
может существовать самостоятельно, поэтому в реакторе всегда
предусматриваются соответствующие поджигающие устройства.
При нарушении равенства со = и режим горения меняется:
с уменьшением скорости истечения ниже скорости горения (со << и)
происходит проскок пламени внутрь горелки, с увеличением
скорости истечения выше скорости горения (со > и) наблюдается отрыв
пламени от горелки. Скорости со и и в различных участках сечения
166
сопла меняются по параболическому закону (рис. V-9): скорость со
у края отверстия всегда меньше, чем в центре. Увеличивая скорость со
вблизи стенки, можно препятствовать проскоку пламени. В
большинстве горелок для этого используют конфузор — небольшое
сужение на конце сопла.
Воспламенение горючей смеси цри факельном горении может быть
двояким: с периферии струи и центральное — с оси потока (последнее
возможно только для заранее перемешанных смесей)20'21.
Периферийное воспламенение горючей смеси осуществляется по
внешней оболочке пламени путем подсоса горячих продуктов
сгорания к корню факела, что обеспечивает аутостабилизацию процесса
горения. Такое горение может
происходить либо в пространстве,
заполненном горячими продуктами
сгорания, либо в узком туннеле.
При попадании охлажденных
продуктов сгорания в туннель факел
гаснет (если он не заполняет
туннель целиком).
Центральное воспламенение
достигается путем изменения
аэродинамики газового потока
(использование закрученных потоков или
различных стабилизаторов). При
этом получается более развитая поверхность воспламенения, что
позволяет уменьшить зону воспламенения LB и, как следствие,
длину факела О
Рис. V-9. Условия отрыва (а) и
проскока (б) пламени при истечении
горючей смеси из отверстия горелки.
поверхность
у воспламенения LB
$ Отсюда следует, что центральное воспламенение
более выгодно, поскольку уменьшать длину факела L$ можно путем
увеличения поверхности зажигания без изменения диаметра сопла d.
Суммируя изложенное, можно отметить, что для изменения
величины LB, являющейся основной составляющей величины L$,
имеются следующие пути:
а) изменение диаметра кратера;
б) изменение скорости вылета струи со;
в) изменение условий воспламенения смеси, приводящие к
увеличению поверхности воспламенения (закрученные потоки,
стабилизаторы).
Для интенсификации горения, т. е. уменьшения дальнобойности
факела, наиболее часто прибегают к изменениям условий
воспламенения горючей смеси (создание закрученных потоков и
использование стабилизаторов).Образование закрученных потоков достигается
путем вставок различных завихрителей в отверстия для истечения
горючей смеси. Закрученная струя характеризуется большим углом
расширения газового потока при истечении и уменьшенной
дальнобойностью факела, т. е. уменьшением L$, но главной особенностью
закрученных струй является повышенная эжекционная способность,
что в свою очередь приводит к увеличению поверхности
воспламенения21.
167
Аэродинамика закрученных струй и их основные особенности
обстоятельно изложены в работе Д. Н. Ляховского и др.21. В
зависимости от вида завихрителя в центре закрученных струй образуются
области с отрицательным значением осевой скорости, т. е.
наблюдается обратное движение газа. Как показали опыты с
лопаточными и тангенциальными завихрителями, интенсивность падения
скорости вдоль сильно закрученной струи больше, чем вдоль слабо
закрученной21. Поэтому несмотря на относительно большие скорости
закрутки в устье, дальнобойность сильно закрученных струй меньше,
чем слабо закрученных. Следовательно, для уменьшения длины
факела целесообразна большая степень закрутки.
И. М. Романюк, А. М. Зелизный и В. У. Шевчук изучали влияние
завихрения потока на длину факела при получении ацетилена
окислительным пиролизом22'23. При одной и той же длине реакционного
канала в закрученном потоке степень ацетиленообразования заметно
возрастала по сравнению с незакрученным потоком, причем наиболее
эффективным оказался угол наклона лопастей завихрителя в
пределах 0—45°. Основным критерием для установления угла наклона
является допустимое гидравлическое сопротивление горелки.
Например, увеличение угла наклона с 0 до 60° вызвало возрастание
перепада давления с 0,04 до 0,3 am. Оптимальный угол наклона
лопастей завихрителя составил 45° — при этом достигнута мощная
интенсификация процесса неполного горения при относительно низких
потерях напора в.завихрителе.
Анализировалось также влияние скорости потока и ширины щели
(т. е. диаметра кратера) кольцевой горелки на длину образующегося
факела. При увеличении ширины щели с 14 до 32 мм относительное
удлинение факела составило 54%, при повышении скорости в 2 раза
относительная длина факела возросла только на 25%.
Следовательно, для условий опыта (скорость вылета 120—60 м/сек и ширина
щели 14—32 мм) факел сильнее удлиняется при расширении щели,
чем при увеличении скорости газового потока.
Основные параметры процесса окислительного пиролиза
Окислительный пиролиз метана представляет собой сумму
параллельных и последовательных реакций и в общем виде при
оптимальном времени образования ацетилена выражается следующим
суммарным уравнением8' 13> 16> 24:
45СН4 + 30,25О2 --> 3,5СО2 + 8С2Н2 + 25,5СО + 28Н2О + 54Н2
Значения коэффициентов в этом эмпирическом уравнении зависят
от условий подогрева, соотношения О2 : СН4, состава пиролизуемого
сырья и кислорода, конструктивных особенностей реактора, потерь
тепла в зоне «закалки» реакционных газов, степени конверсии метана
и разложения ацетилена, степени сажеобразования и др.
Как было показано ранее, степень превращения метана в
ацетилен определяется соотношением скоростей образования ацетилена и
168
разложения его на простые вещества. Наиболее интенсивно ацетилен
образуется тогда, когда основное количество кислорода переходит
з продукты полного и неполного окисления метана, при этом
парциальное давление метана составляет25 0,2 am.
Соотношение О2 : СН4. При окислительном пиролизе это
соотношение играет очень значительную роль, определяя термодинамику
и кинетику процесса.
Прежде всего от соотношения О2 : СН4 существенно зависит
температура процесса (рис. V-10), которая определяет выход ацетилена.
Это не значит, однако, что чем
больше величина О2 : СН4, тем
выше температура реакции и
соответственно выход ацетилена. Как
уже говорилось, процесс
окисли150д0,50 0,55 0,60
Соотношение О2: СНд
Рис. V-10. Зависимость расчетной
температуры пиролиза от
соотношения О2: СН4 при различной
температуре предварительного
подогрева:
/—при 700° К; 2 — при 800° К; 3— при
900° К-
0,2 0,4 0,6
Соотношение О2: СН4
Рис. V-11. Области окисления
метана и крекинга метана до
ацетилена в зависимости от состава
метано-кислородной смеси:
/ — метан не реагирует; 2 —
окисление метана; 3 — крекинг метана
до ацетилена.
тельного пиролиза определяется не только подводом тепла в систему,
но и необходимым парциальным давлением СН4.
Направленность процесса неполного окисления метана в
зависимости от соотношения О2 : СН4 показана на рис. V-11. Видно,
что оптимальное количество ацетилена образуется при О2 : СН4 =
= 0,55^0,65. Дальнейшее увеличение соотношения способствует
развитию реакций окисления метана.
В зависимости от значения О2 : СН4 меняется распределение тепла
в процессе (рис. V-12). Тепловой баланс окислительного пиролиза
без предварительного подогрева исходной метано-кислородной смеси
замыкается при О2 : СН4 = 0,65 (точка А), а с подогревом до
660° С — при О2 : СН4 = 0,58 (точка Б). В интервале значений
0,58—0,65 окислительный пиролиз носит аутотермический характер.
Зависимость концентрации ацетилена в газах пиролиза от
соотношения О2 : СН4 показана26 на рис. V-13. Максимальная
концентрация (8,5 объемн. %С2Н2) получается при О2 : СН4 = 0,65.
169
Представляет интерес зависимость полезного крекинга метана до
ацетилена от величины О2:СН4 (рис. V-14). Максимальный
полезный крекинг получен при О2 : СН4 = 0,65 и составляет 36,5% в
расчете на прореагировавший
углеводород (33% в расчете на исходный
углеводород).
и оо
Oi
и
!
300
200 —
а
-
Б
|
д
о,б
Соотношение О2:СН4
Рис. V-12. Изменение
тепловыделения при окислительном
пиролизе метана:
/ — необходимое тепло без
подогрева метана и кислорода; 2 — то
же, но при подогреве до 600° С;
3 — суммарное тепловыделение
реакций окисления.
зГ о.
\
0,80
и0,50 0,60 0,70
Соотношение О^ СН4
Рис. V-13. Изменение
концентрации ацетилена в газах
пиролиза метана в зависимости от
соотношения О2 : СН4 в
реакционной смеси.
Состав пиролизуемого сырья.. Для пиролиза с получением
ацетилена можно использовать не только метан, но и другие
газовые смеси, например содержащие кроме метана этан и этилен27.
Такие смеси могут быть получены на нефтеперерабатывающих заво-
^ 35
Iй
90
*
/V
2>
К'
\
Ю
0,U 0,6 0,8
Соотношение
7
Рис. V-14. Изменение полез -
ного крекинга метана до
ацетилена в зависимости
от соотношения О2 : СН4:
1 —в расчете на
прореагировавший метан; 2 — в расчете
на исходный метан.
70,9 1,1 1,3 1,5 1,7
Углеродный показатель 1С7
Рис. V-15. Изменение
концентрации ацетилена в газах пиролиза
в зависимости от углеродного
показателя сырья (температура
предварительного подогрева
550° С).
дах, а также в качестве побочных продуктов при разделении
коксового газа методом глубокого охлаждения.
С ростом углеродного показателя (стр. 117) увеличивается
степень превращения углеводородов и, следовательно, содержание аце-
170
?илена в газах пиролиза (рис. V-15). Поэтому для пиролиза до
ацетилена целесообразно использовать природные газы, содержащие
небольшое количество этана. Переработка углеводородов с
углеродным показателем выше 2, т. е. с очень большим содержанием этана,
дает менее благоприятные результаты вследствие образования
больших количеств сажи. В табл. V-6 приведены значения полезного
крекинга метана, этана и пропана (из расчета полного превращения
сырья в ацетилен) при оптимальной температуре процесса8, а
в табл. V-7 собраны сведения о распределении тепла при
окислительном пиролизе этих же углеводородов, полученные расчетным
путем 8. Для расчетов были приняты следующие допущения:
1) углеводород и кислород подогревают до 400° С;
2) 85% углерода из метана, участвующего в горении, окисляется
до СО и 15% —до СО2;
3) около 50% участвующего в горении водорода окисляется
до Н2О.
ТАБЛИЦА V-6
Полезный крекинг углеводородов до ацетилена при оптимальной температуре *
Показатели
Оптимальная температура, °С . . . .
Полезный крекинг до ацетилена, %
теоретический
фактический
Пиролиз
метана
1450
81
50—68
Пиролиз
этана
1050
87
47—49
Пиролиз
пропана
1050
89
38-45
* Данные усредненные, приведены для разных процессов пиролиза (чисто термический
и окислительный) и для разных типов реакторов.
ТАБЛИЦА V-7
Использование тепла при пиролизе углеводородов до ацетилена
(расчетные данные)
Показатели
Пиролиз
метана
Пиролиз
этана
Пиролиз
пропана
Температура реакции, °С . . . .
Количество тепла, ккал
необходимое для образования 1 кг
С2Н2
расходуемое на нагрев реагентов
(в расчете на 1 кг С2Н2) . . .
выделяющееся при сгорании 1 кг
сырья, ккал
Выход ацетилена, вес % от
количества сырья
1650
3889
8612
4834
55
1370
3111
5112
4723
48
1370
2945
4889
4611
40
Из табл. V-7 видно, что с ростом молекулярного веса исходного
углеводорода уменьшается количество тепла, выделяющееся при его
сгорании, однако расход тепла на нагрев реагирующих веществ
171
и образование ацетилена также становится меньше. Это приводит
к уменьшению количества топлива при использовании сырья с
большим молекулярным весом и к образованию меньшего количества
водорода, а следовательно, к снижению количества побочных
продуктов.
В результате для получения ацетилена из тяжелых углеводородов
создаются наиболее благоприятные термодинамические условия,
однако меньшее количество продуктов сгорания при одном и том же
давлении ведет к возрастанию парциального давления побочных
продуктов (например, водорода). При рассмотрении суммарных
реакций получения ацетилена можно от-
метить, что в результате пиролиза >
<* 8
i
С
г
и
Л
/А
1 ■
V
"0,30 ОАО 0,50
Соотношение О2:2'С1
0,60
Рис. V-16. Изменение концентрации
ацетилена в газах пиролиза я-бутана в
зависимости от соотношения О2 : JjCi-
I
1
^ 20
0,30 0,^0 0,50 0,60
Соотношение O2'ZCX
Рис. V-17. Зависимость
полезного крекинга я-бутана до
ацетилена от соотношения
Метана На 1 Объем ацетилена ПОЛучаЮТ /-расчет на прореагировавший
^ J углеводород; 2 — расчет на
3 Объема ВОДОрОДа, При ИСПОЛЬЗОВаНИИ исходный углеводород.
этана — 2 объема, при пиролизе
пропана — 1,65 объема. С повышением молекулярного веса исходного
углеводорода уменьшается расход кислорода на процесс. Если
принять количество кислорода для пиролиза метана в условиях,
указанных в табл. V-7, за 100%, то для этана эта величина снижается
до 50%, а для пропана—до 40%.
При окислительном пиролизе я-бутана оптимальное отношение
О2 : SQl составляет примерно 0,49; этому соответствует
концентрация ацетилена И объемн. % (рис. V-16). При О2 : 2Сг выше 0,64
процесс затрудняется из-за пульсаций в реакционной зоне26.
Максимальный полезный крекинг при О2 : 2Q = 0,49 составляет 34,5%,
считая на прореагировавший я-бутан, и 30,0%, считая на исходный
я-бутан (рис. V-17). Для окислительного пиролиза я-бутана в
оптимальных условиях можно составить следующее эмпирическое
уравнение (без учета образования сажи) 26:
28,6О2 -> 3,7СО2 + 8,7С2Н2 +
29,80СО + 32,7 Н2 + 5,0СН4 + 18,80 Н2О
14,32С4Н10
0,7СаН4 + 0,6О2
При пиролизе смеси н-бутана с метаном с возрастанием
концентрации любого компонента полезный крекинг сначала
уменьшается, а затем, пройдя минимум, стремится к значению выхода для
172
соответствующего чистого компонента. Концентрация ацетилена
в продуктах пиролиза составляет 8—9 объемн. %. Зависимость
полезного крекинга от углеродного показателя для смесей метана
и «-бутана 26 показана на рис. V-18 (линия 1). На том же рисунке
(линия 2) показано изме- ^
нение расхода кислорода *S 5000p
(лабораторные данные).
Видно, что уменьшение
выхода ацетилена
(полезный крекинг)
сопровождается повышением расхода
кислорода.
С ростом углеродного
показателя сырья резко
уменьшается расход
углеводородов и кислорода *
при окислительном
пиролизе в расчете, например26, на 1 т С2Н2 (рис. V-19 и V-20). Это
говорит о том, что выгоднее работать на смесях, содержащих гомологи
метана («жирные» газы), так как можно значительно сократить расход
углеводорода. Однако при большом количестве гомологов метана
2 з
Углеродный показатель 2С j
Рис. V-18. Зависимость параметров
окислительного пиролиза смесей метана и «-бутана от
углеродного показателя:
/ — полезный крекинг; 2 — расход кислорода на
1 т С2Н2.
6000
5000
\
\
/
X
и
1
ЗШ0,9 1,1 1,3 1,5 у,
Углеродный показатель Z С j
Рис. V-19. Изменение расхода пи-
рол изуемых углеводородов и
кислорода на 1 т С2Н2 в зависимости
от углеродного показателя сырья
при разной температуре
предварительного подогрева:
/ — расход углеводородов, м3 (550° С);
2 — расход кислорода, кг (550° С);'
3 — расход углеводородов, м3 (200° С).
\ '7500
U 5000\
* 2500
%
i °.
ч
<
2 3 4
Углеродный показатель ZQ1
Рис. V-20. Зависимость расхода
углеводородов на 1 т С2Н2 от
углеродного показателя сырья
(предварительный подогрев до 200° С):
/ — экспериментальные данные по
пиролизу метана, «-бутана и их смесей;
2 — расчетные данные при условии,
что полезный крекинг не зависит от
природы углеводорода и составляет
31,5%.
может снизиться допустимая температура предварительного
подогрева и тем самым увеличиться фактический расход сырья. На
практике максимально допустимое суммарное содержание гомологов
* Кривые 3 (рис. V-19) и 1 (рис. V-20) построены по одним и тем же
экспериментальным данным.
173
метана составляет 2—4 объемы. %. Если содержание гомологов
выше 2—4 объемы. %, то приходится принимать специальные меры
во избежание преждевременного воспламенения углеводородо-кис-
лородной смеси в реакторе.
Следует отметить, что влияние гомологов неодинаково и расход
углеводорода уменьшается в первую очередь при возрастании
содержания этана и пропана. Бутан, пентан и особенно гексан менее
желательны, так как они склонны к интенсивному сажеобразованию.
Концентрация кислорода. При увеличении количества инертных
примесей в кислороде, как и в природном газе, уменьшается доля
тепла, используемого на проведение пиролиза углеводородного сырья,
и вследствие этого снижается выход ацетилена. Отсюда можно было бы
сделать вывод, что наиболее выгодна концентрация кислорода 100%,
однако это нецелесообразно с экономической точки зрения.
Практически используемая концентрация кислорода составляет 95—98%
и зависит в основном от типа аппаратов для разделения воздуха*.
В промышленной практике особенно важно сохранять
постоянной кондицию кислорода для обеспечения надежной и безаварийной
работы ацетиленовых реакторов. Именно об этом, а не об абсолютном
содержании О2 в газе-окислителе следует заботиться в первую
очередь.
Давление. Процесс получения ацетилена неполным сжиганием
газообразных углеводородов можно проводить при пониженном
(0,2—0,6 am) и при повышенном (3—5 am) давлении.
Процесс при пониженном давлении. Поскольку
окислительный пиролиз протекает с увеличением объема
образующихся продуктов, то, согласно термодинамическим расчетам,
понижение давления должно ускорять процесс. Однако практически
процесс при пониженном давлении протекает менее эффективно, так
как уменьшается количество тепла, выделившегося при неполном
горении части метана, и поэтому нехватает тепла на проведение
эндотермической реакции разложения метана до ацетилена.
В табл. V-8 приведены лабораторные данные по пиролизу метана
при 0,24 и 0,33 am на насадке 28. Видно, что изменение скорости
подачи метана при постоянном объеме реактора мало влияет на ход
реакции. При возрастании скорости горение было неустойчивым,
т. е. происходило затухание факела с понижением температуры
насадки. Поэтому увеличение скорости подачи метана возможно до
определенного предела. Минимальная скорость, при которой
наблюдалось затухание, при пониженном давлении была
приблизительно такой же, как и в опытах при нормальном давлении.
В опытах при пониженном давлении наблюдалось значительное
сажеобразование, причем количество сажи возрастало с повышением
молекулярного веса углеводорода и с понижением содержания кис-
* При использовании кислорода, содержащего менее 95% О2, в газах пиролиза
повышается содержание аргона, что может отрицательно влиять, например, на
пэреработку в аммиак синтез-газа, получаемого после выделения ацетилена.
174
§
з
X
о
с
s
си
с
X
н
о
т
ч
о
о.
Выход,
мол. %
Состав сухого газа пиролиза,
объемн. %
•*
и
Д
и
о
и
X
и
о
о
£
д
и
£
и
Количество
газа
пиролиза
М3/Ч
Максимальная
температура, °С
Площадь
поперечного
сечения
реактора
см2
8S*
яКЕ
sgu
<L> Я .
& о
Sg°
Скорость подачи,
м*/ч
д
о
о
CD
00
16,
О
СМ
1С
16,
iC^ CO CD О ОО
—* ~ —" ~ О"
оо
О
СМ
CD
О
со со
^ см"
CD
о4
Is- 1С Tf
со аГ о"
*—' Th1 СП 1С rf CO
cd" —* —Г —? -^ CD~
СМ О СМ -^ СЮ 1С
00 СП СП *—< 1С ^
—I — г-, СМ —•
§
СО
СО
О
S
S
к
CQ
ч
S
Оч
С
00
см
см
о
СП
со
1—1
00
06
1С
1С
см
1С
см
со
1С
см
о
57,
1С
о
СП
со
1С
00
о
со
1С
см
а
см
о
S
к
CQ
S
Оч
с
о
см
см
см
1С
со
о
см
см
§
CD
1
1
CD
СП
см
о
СП
см
о
со
—'
1С
СО
см
_
22
со
со
о
|
со
о
CD
У-4
о
СО
§5
СП
см
t
о
см
см
,30
Tf1
о
см
о
со
см
тН
a
00
45,
со
о
о
со
,90
о
см
со
см
см
см
S
см
о
СО
см
о
1С
1
1
CD
CD
1С
ю
1С
о
о
СП
о
ю
§
00
о
см
CD
о
1С
8
CD
О
1С
СМ
|><
о
1С
см
со
1С
о
^t4
СП
о
ю
о
СП
CD
о
Tf
СП
о
CD
СО
1С
CD
О
CD
00
CN
*■*■
00
175
лорода в газовой смеси. В качестве сырья использовали я-гексан,
смесь я-гексана с 10 вес. % бензина 28, а также высококипящие
углеводороды, содержащие большое количество ароматических
соединений. Для подавления сажеобразования в реактор добавляют азот,
однако это не дало положительных результатов и выход ацетилена
снижался с увеличением количества сажи. Следовательно,
окислительный пиролиз при пониженном давлении можно считать
малоперспективным.
Процесс при повышенном давлении. С точки
зрения термодинамики повышение давления неблагоприятно,
поэтому не следует ожидать увеличения выхода ацетилена. Но так как
с ростом давления разложение метана ускоряется, можно было бы
предположить, что концентрация ацетилена в получаемом газе
снижаться не будет. Однако экспериментально установлено, что
концентрация ацетилена в газах пиролиза с повышением давления
уменьшается п:
Давление, am 1 4 6
Время реакции, мсек 3 3 3
Концентрация ацетилена в газе
пиролиза, объемн. % 8,0 6,6 5,5
Вероятно, с ростом давления скорость разложения ацетилена
увеличивается в большей степени, чем скорость его образования.
Разложение ацетилена сопровождается значительным сажеобразо-
ванием, поэтому, как следует из приведенных выше данных, время
реакции нужно уменьшать. Оптимальное время реакции при разном
давлении установлено опытным путем и:
Давление, am 1 4 6 10
Оптимальное время реакции, мсек . . 2—6 1—3 0,6—2 0,5—1,5
Повышение избыточного давления сверх 5 am приводит к
уменьшению концентрации ацетилена. Например, при 10 am в
лабораторных условиях концентрация ацетилена в газах пиролиза метана
достигала только 3,5 объемн. %. То же самое получалось при
окислительном пиролизе я-бутана: с повышением давления уменьшалась
концентрация ацетилена с 9,8—10 до 7—8,5 объемн. % с
одновременным ростом содержания этилена, причем сумма этих продуктов
оставалась постоянной.
Для пиролиза метана при повышенном давлении оптимальное
отношение О2 : СН4 практически такое же, как и при 1 am и в
интервале 1—14 am составляет 0,56—0,6. С повышением давления
уменьшается концентрация ацетилена и увеличивается концентрация
этилена:
Давление, am 1 6 10
Содержание ацетилена, объемн. % 8 6 3,5
Содержание этилена, объемн. % 0,4 1,5 2
176
В интервале давлений 1—14 am уменьшается суммарный выход
непредельных углеводородов и увеличивается концентрация Н2 и
СО. Возможно, что это происходит вследствие взаимодействия
ацетилена с еодяным паром
СоН,
2НХ>
2СО + ЗН,
поскольку эта реакция второго порядка, а разложение ацетилена —
реакция первого порядка.
Весьма важным показателем является расход кислорода в
процессе. Если рассматривать в качестве целевых продуктов
окислительного пиролиза метана сумму С2Н2 + С2Н4, то в интервале 1—14 am
расходные коэффициенты по метану и кислороду практически
постоянны.
Следует, однако, указать что
пиролиз при повышенном давлении
изучен недостаточно и о нем имеются
противоречивые данные. Например,
по данным Ричи29 при пиролизе
метана под давлением выход ацетилена
не снижается, а наоборот, несколько
повышается: 16ж3С2Н2 на 100 ж3СН4
при 1 am и подогреве реагентов до
400° С (полезный крекинг 32%) и
17 м3 С2Н2 на 100 м3 СН4 при 5 am и
той же температуре подогрева
(полезный крекинг 34%). Расход
кислорода в обоих случаях одинаков.
'1500 1600 1700 1800 Щ0
Температура реакции, °К
Рис. V-21. Изменение полезного
крекинга (в пересчете на исходный
метан) в зависимости от
температуры реакции при разном
предварительном подогреве.
В промышленности проводят окислительный пиролиз природного
газа при давлении 4—5 am и получают газ с содержанием ацетилена
8—8,5 объемн. %. Преимуществами процесса под давлением являются
возможность теплоиспользования газов пиролиза и упрощение са-
жеочистки, так как смачиваемость сажи повышается с ростом
давления.
Температура предварительного подогрева. С ростом температуры
предварительного подогрева исходной смеси уменьшается доля
сгоревшего углеводорода и, следовательно, увеличивается количество
углеводорода, разлагающегося до ацетилена, т. е. повышается
полезный крекинг процесса. Из графика рис. V-21 видно, что с
повышением температуры предварительного подогрева теоретический
полезный крекинг увеличивается. Однако теоретически возможный
полезный крекинг 37,5% не был достигнут даже в лабораторных
условиях.
Практически по литературным данным известен максимальный
полезный крекинг 33%.
Ниже приведены данные Браконье о полезном крекинге (в
пересчете на исходный метан) при разной температуре
предварительного подогрева исходной смеси, а также о соответствующем расходе
!2 В. Н. Антонов 177
метана (96% СН4) и кислорода (98% О2) на 1 т С2Н2 (с учетом
стадии выделения ацетилена)27:
500 600 700
Температура предварительного подогрева, СС . .
Полезный крекинг (в пересчете на исходный
метан),
27,1 29,0 31,2
Расход, т
метан 6,4 6,0 5,3
кислород 5,5 5,1 4,6
Время реакции. Оптимальное время пребывания веществ в зоне
реакции определяется условиями проведения процесса (составом
исходного сырья, температурой, давлением и др.), а также
конкретной конструкцией реактора. Для каждого реактора и заданных
условий процесса необходимо определить оптимальное время реакции.
Время ацетилен*.
'я, сек-Ю~
Рис. V-22. Изменение степени превращения метана в продукты пиролиза в
зависимости от времени ацетиленообразования при 1400 и 1600° С.
На рис. V-22 показана зависимость оптимального времени
реакции от температуры процесса20: чем выше температура, тем меньше
время образования ацетилена. Поскольку температура в первую
очередь определяется соотношением О2 : СН4, время реакции также
должно зависеть от него.
По практическим данным время пребывания газов в реакционной
зоне составляет 0,003—0,008 сек. Более длительное время, как
правило, в промышленных реакторах не выдерживается.
Оптимальное время реакции определяется также температурой
(рис. V-23). Из рисунка видно, что при высокой температуре и
соответствующем соотношении О2 : СН4 количество углерода,
перешедшее в ацетилен, растет и оптимум соответствует 0,001 сек. Этому
условно отвечает температура ~1780° С.
Оптимальное время ацетиленообразования уменьшается с ростом
углеродного показателя пиролизуемого сырья. На рис. V-24 показана
зависимость концентрации ацетилена в газах пиролиза я-бутана от
178
времени реакции (лабораторная установка, реактор с внешним
обогревом26). Оптимальное время реакции составляет около 0,002 сек, что
примерно в 2 раза меньше, чем при пиролизе метана.
Об изменении оптимального времени реакции при повышении
давления процесса говорилось ранее (стр. 176).
Влияние добавок. Добавки различных газов (например,
водорода, окиси углерода) существенно изменяют тепловой баланс
окислительного пиролиза. При взаимодействии с метаном кислород
распределяется между углеродом и
2300
-2200
водородом в основном в
соотношении 1:1, что соответствует
реакции:
СН4
О2 -> СО + Н2О
Н2
i
10
- 2100 °
2000
1900
1800
Щ1700
2
При добавлении водорода
распределение кислорода
меняется в зависимости от
кинетики окисления метана и
водорода в интервале температур
Время реакции, сен
Рис. V-23. Изменение состава продуктов
пиролиза метана и температуры в
зависимости от времени реакции (соотношение
О2 : СН4 = 0,48; температура
предварительного подогрева смеси 610° С):
/ — температура реакции; 2 —метан; 3—
ацетилен; 4— этилен; 5—сажа; 6— окись углерода.
6
0,001 0,003 0,005 0,007 0,009
Время реакции, сек
Рис. V-24. Зависимость концентрации
ацетилена в газах пиролиза я-бутана
от времени реакции.
пиролиза (1200—1500° С)31. Это отражается на материальном и
тепловом балансах процесса, так как водород вступает в реакцию с
кислородом. При этом увеличивается доля метана, подвергающегося
крекингу, и выделяется дополнительное количество тепла,
затрачиваемое на полезную реакцию пиролиза. Лабораторные данные по
пиролизу метана.в присутствии водорода32 представлены в табл. V-9.
При введении водорода количество метана, превращенного в
различные продукты, меняется следующим образом (в %):
Без водорода С водородом
В продукты окисления . . . —60 До 42
в СО 50—52 32—37
в СО2 8—10 3—5
В СоНо и С,Н4 33—35 До 40
(при 1300— (при 1530Х)
1500° С)
12* 179
ТАБЛИЦА V-9
Окислительный пиролиз метана с добавкой водорода (при 1 am)
Темпера-
Tffa
°С
1200
1200
1300
1300
1400
1400
1530
Время
реакции
сек
0,001
0,01
0,001
0,01
0,001
0,004
0,002
с2н2
0,8
3,9
3,6
4,9
6,0
5,3
7,2
с2н4
2,4
1,2
1,6
0,4
0,8
0,9
0,4
Состав газов пиролиза, объемн. %
СН4
29,2
19,7
13,6
14,8
19,8
12,6
6,5
н2
47,2
55,8
62,0
61,2
55,0
62,1
67,3
О2
7,0
0,3
4,2
0,4
1,8
0,4
0,2
со
11,8
16,8
13,2
15,8
14,6
16,5
16,0
СО2
-
0,8
1,4
0,8
1,7
1,0
1,2
1,4
N2
0,8
0,9
1,0
0,8
1,0
1,0
1,0
Примечание. Состав исходной смеси (в объемн. %):
СН4
Н2 .
. 37
.36,9
О2
N2
.25,1
. 1,0
Кислород (общее количество) распределяется между углеродом
и водородом в соотношении от 1 : 2 до 1 : 2,2. В реакцию с
кислородом вступает 45—50% дополнительно введенного водорода.
Максимальный выход ацетилена получен при пиролизе смеси, в которой
соотношение СН4 : О2 : Н2 = 2 : 1 : 1.
Аналогичные данные получены для процесса при пониженном
давлении28 (табл. V-10). В результате добавки водорода повышалась
температура пиролиза и улучшались условия зажигания горючей
смеси. При соотношении Н2 : СН4 ^ 1 : 1 с понижением скорости
подачи газов в отдельных случаях происходил проскок пламени.
ТАБЛИЦА V-JO
Окислительный пиролиз метана при пониженном давлении с добавкой водорода28
Объемное
соотношение
Н2:СН4
Скорость
подачи, м3/ч
о2
Н2 +
+ СН4
Температура
реакции
°С
Количество
газов
пиролиза, м3/ч
Состав газов пиролиза, объемн. %
С2Н2
с2н4
СН4
нг
со
со2
03 q
га аи
180
При давлении 0,24 am
: 2,5
: 2
: 1,5
: 1
2,0
2,0
2,0
2,0
4,0
4,0
4,0
4,0
—
1365
1496
1447
4,57
5,25
5,40
4,90
2,5
6,7
3,6
2,3
3,1
0,7
1,0
0,9
19,7
4,9
2,1
1,8
43,2
56,6
60,4
62,5
19,3
25,3
27,7
27,2
12,2
5,8
5,2
5,3
8,0
25,5
16,2
10,0
При давлении 0,33 am
1
2
2
3
,8
,1
,6
,0
3,7
4,34
5,25
6,0
1420
1440
1445
1447
7,30
8,15
9,35
10,70
6,4
5,6
6,0
5,5
0
0
0
0
,7
,4
,6
,7
2,2
5,9
6,1
3,9
58,
57,
56,
57,
6
2
2
7
25,6
24,8
24,9
26,4
6
6
6
5
,5
,1
,2
,8
26,9
24,3
25,4
24,0
9,9
2,7
4,5
3,9
3,2
1,9
2,4
3,0
Наибольший выход ацетилена достигнут при объемном соотношении
Н^ : СН4 - 1 : 2.
При добавлении окиси углерода в исходную метано-кислородную
смесь также следовало ожидать изменения материального и теплового
балансов процесса, так как в интервале 1200—1500° С окись углерода
окисляется до двуокиси углерода. Однако при лабораторных опытах
в фарфоровых трубах, обогреваемых в электропечи, было
установлено32, что с введением окиси углерода в метано-кислородную смесь
(О2 : СН4 = 0,6) содержание двуокиси углерода в реакционных
газах повышалось незначительно. Вероятно, при температурах
пиролиза окисление окиси углерода тормозится метаном, который в
интервале 1200—1300° С окисляется гораздо быстрее окиси углерода.
Скорость окисления окиси углерода обычно значительно
возрастает при добавлении водяных паров и водорода, которые способствуют
образованию радикалов -ОН, однако в присутствии метана влияние
этих добавок не сказывалось. Следовательно, можно считать, что
окись углерода, вводимая в метано-кислородную смесь, является
только разбавителем.
Изучение влияния добавок водорода и окиси углерода на
процесс получения ацетилена представляет практический интерес, так
как при этом устанавливается возможность использования не только
метана, но и коксового газа, а также газов, остающихся после
выделения ацетилена. Впервые экспериментально это было проверено
Фишером и Пихлером24, которые проводили окислительный пиролиз
коксового газа в трубках с внешним обогревом. Превращение метана,
содержащегося в коксовом газе, в ацетилен достигало 50%, однако
вследствие разбавления водородом и окисью углерода концентрация
ацетилена-в газах пиролиза не превышала 5,6 объемн. %.
Впоследствии Я. С. Казарновским и др.33 были проведены опыты
по получению ацетилена из коксового газа и его фракции,
обогащенной метаном («богатый» газ), на модельных установках в односопло-
вом реакторе производительностью около 20 м3/ч по исходной
газовой смеси. Средний состав исходной смеси в реакторе (в объемн. %):
сн4 . .
с2н4 .
н, . .
. . 39—41
. . 3,5—4,5
. . 1,5—2,5
о2 . .
СО . .
N* . .
. . 34—36
. . 7,5—8,5
. . 8-12
Температура в реакционной зоне достигала 1450° С. При этом
общий крекинг (в расчете на метан) составлял 86,8—93%, а
полезный крекинг в среднем 31% и иногда даже 32,5%. Оптимальное
отношение О2:(СН4 + С2Н4) в исходной смеси 0,65—0,75.
Получаемый газ пиролиза имел следующий состав (в объемн. %):
С2Н2
с2н4 .
сн4 .
н2 . .
. . 6,4—7,0
. . 0,1—0,5
. . 2—7
. . 38—43
о2 . .
СО . .
ох . .
N2 . .
. . 0,3—1,0
. . 30—33
. . 4,8—5,6
. . 8—11,9
181
Повышенное содержание окиси углерода связано с наличием ее
в исходной газовой смеси. Помимо газообразных продуктов
образуется свободный углерод (сажа) в количестве до 2,5 г на 1 м3
газов пиролиза.
Проводились опыты по окислительному пиролизу коксового газа,
предварительно очищенного от серы и имеющего следующий средний
состав (в объемн. %):
СН4 ... 25,3 СО .... 6,5
С2Н4 ... 2,3 СО2 .... 1,7
Но .... 62,0 N2 .... 1,4
О2 .... 0,3
Концентрация применяемого кислорода составляла, как и в
случае «богатого» коксового газа, 89—93%. Температура
предварительного подогрева исходных компонентов была 200° С, температура
пиролиза 1400—1450° С. Соотношение О2 : (СН4 + С2Н4) колебалось
в пределах 0,95—1,06. Общий крекинг доходил до 84%, а полезный
крекинг составлял 39—44%. В связи с малым содержанием метана
в коксовом газе по сравнению с «богатым» газом концентрация
ацетилена, естественно, была меньше — примерно 5 объемн. %.
Средний состав газа пиролиза такой (в объемн. %):
С2Н2 . . . 5,0—5,5 О, .... 0,4—0,9
С2Н4 . . . 0—0,2 СО .... 18,2—20,8
СН4 . . . 3,1—4,6 СО2 . . . . 3,3—5,7
Н2 .... 60—65 N .... 3,0—4,8
Вследствие высокой концентрации водорода в коксовом газе
должен понижаться расход кислорода и повышаться выход
восстановителей (СО + Н2) на единицу количества получаемого ацетилена
по сравнению с соответствующими данными для природного газа.
Расчеты, проведенные по экспериментальным данным для условий
модельной установки, показали, что при переходе от «богатого»
газа к коксовому количество восстановителей повышается примерно
на 20%.
В табл. V-11 приведены расчетные показатели окислительного
пиролиза коксового и природного газов33 при следующих условиях:
температура предварительного подогрева исходной смеси 550° С,
температура процесса 1560° С, общий крекинг (в расчете на метан) 90%,
полезный крекинг —до 48%. Из данных таблицы следует, что
расход кислорода при пиролизе коксового газа на 17% меньше, чем при
использовании природного газа.
Промышленное производство ацетилена из коксового газа в
настоящее время отсутствует, но приведенные выше данные показывают,
что в отдельных случаях, когда нет достаточных ресурсов
природного газа, он может быть заменен коксовым газом. Следует, однако,
указать, что из-за меньшего содержания ацетилена в газах
пиролиза расходы по его выделению соответственно возрастут
(гл. X).
182
ТАБЛИЦА V-ll
Некоторые показатели окислительного пиролиза коксового
и природного газов
Показатели
Концентрация С2Н2 в газе
пиролиза, объемн. %
Расход на 1 т С2Н2, м3
сырья
кислорода (93% О2)
Выход восстановителей (СО + Н2)
на 1 т С>Н2, ж3
Пиролиз
коксового газа
5,9
13 320
2 965
12 670
Пиролиз
природного
газа
8,3
5880
3560
8340
Влияние твердой поверхности. Степень воздействия твердой
поверхности на процесс окислительного пиролиза определяется
отношением поверхности F реактора к его объему V и условиями
контакта газа со стенкой реактора. Влияние поверхности реактора
изучал В. У. Шевчук34 (смесь кислорода и метана в отношении 0,66
пропускали через кварцевые трубки диаметром 4,8 мм, помещенные
в электропечь). Результаты опытов показали, что скорость
образования ацетилена существенно зависит от интенсивности переноса
(скорости потока) реагентов и продуктов горения в определенных
условиях. С увеличением скорости потока до 3 м/сек степень
превращения метана в ацетилен заметно возрастает, а дальнейшее
повышение скорости (до 10 м/сек) не оказывает на процесс ацетиленообразо-
вания значительного влияния. В интервале температур 1200—•
1400° С влияние скорости потока до 3 м!сек значительно, при
повышении температуры оно менее ощутимо, а при 1500° С процесс ацетилено-
образования от скорости почти не зависит.
При умеренных температурах газового потока загорается смесь
у стенки реактора и интенсивность горения зависит от отношения
поверхности реакционной зоны к объему. Поэтому в определенном
диапазоне времени контакта с увеличением диаметра реакционной
трубки возрастает количество непрореагировавших метана и
кислорода и снижается выход ацетилена, вероятно, потому, что при
гетерогенном горении тормозится гомогенная реакция образования
ацетилена. Особенно это заметно в случае использования насадки,
причем с уменьшением размеров насадки увеличивается количество
сгоревшего метана и уменьшается скорость образования ацетилена.
По данным В. У. Шевчука, тормозящее действие стенки
проявляется при очень малой скорости газов в реакторе и при большом
отношении поверхности к объему (F : V ^ 25), что соответствует
диаметру трубки 1,6 мм. Практически в ацетиленовых реакторах это
соотношение значительно меньше, поэтому действие стенки не так
заметно.
Интересны исследования окислительного пиролиза метана в
присутствии водорода в реакторе с различной насадкой — кварцевые
183
трубки диаметром 5, 7 и 9 мм и мелкозернистая алундовая насадка31
(табл. V-12). Из таблицы видно, что выход ацетилена значительно
уменьшается при замене трубок насадкой, а при изменении
эквивалентного диаметра трубки концентрация ацетилена меняется мало.
ТАБЛИЦА V-12
Влияние твердой поверхности на состав газов пиролиза метана в присутствии водорода
(время реакции 0,015 сек)
Эквивалентный
диаметр *
трубки
мм
2
4
6
Насадка
2
6
Насадка
Отношение
поверхности
к объему
см'1
20
10
6,6
50
20
6,6
50
Температура
°С
1230
1230
1230
1230
1300
1300
1300
Состав газа пиролиза, объемн. %
С2Н2
4,4
4,4
4,0
1,4
4,8
4,0
1,0
с2н4
1,0
0,9
1,3
0,7
0,3
0,8
0,2
сн4
15,4
14,0
12,6
16,7
12,5
9,6
11,9
н2
59,2
59,8
63,2
59,0
62,7
65,0
61,3
О2
0,3
0,3
0,2
0,1
0,3
0,8
0,2
со
17,1
18,0
16,1
19,9
17,3
17,4
23,1
со2
1,6
1,6
1,6
1,4
1,6
1,4
1,3
N2
1,0
1,0
1,0
0,8
0,5
1,0
1,0
* Эквивалентный диаметр трубки равен ^экв = 4//Т7 (/ — площадь сечения трубки,
заполненной газом, П — периметр трубки).
Сажеобразование. Под сажеобразованием следует понимать
появление частиц сажи (углерода) и различных смолистых веществ.
Это процесс сложный и недостаточно изученный. Известно, что
характер сажи и смолистых веществ зависит от условий их
образования: времени, соотношения О2 : СН4, аэродинамики процесса
неполного горения (температурная неоднородность факела в реакторах
разных конструкций). Чем больше время реакции (в пределах
допустимого), тем эффективнее идет образование сажи, причем при
увеличении времени даже на 10% свойства сажи резко меняются. С
повышением соотношения О2 : СН4 сажа становится более гидрофильной.
Это, по-видимому связано с большим образованием смолистых веществ.
П. А. Теснер35 высказывает предположение, что в условиях
окислительного пиролиза метана сажа почти исключительно образуется
при разложении ацетилена. Непосредственно из метана сажа
практически не получается из-за менее благоприятных кинетических
условий его разложения.
Сажа, полученная при окислительном пиролизе природного газа,
подобна стандартной ацетиленовой саже, но отличается от нее
большей гигроскопичностью (табл. V-13). Следует отметить, что выход
сажи при окислительном пиролизе такой же, как в целевых
процессах ее получения.
Возможность технического использования сажи определяется
в основном ее пористостью. Исследования сажи, полученной в
процессе окислительного пиролиза, показали ее пригодность для
изготовления резино-технических изделий, однако в промышленности
184
ТАБЛИЦА V-13
Свойства сажи, полученной
Процессы
получения сажи
Окислительный
пиролиз
природного газа ....
Электрокрекинг
природного газа
Взрывное
разложение
ацетилена
Гигроскопичность
0/
/о
0,8—1
—
0,05
Зольность
0/
%
7—10
—
0,04
Содержание
углерода
%
85—95
99
99
разными методами
Плотность, г /см3
истинная
1,5—1,65
1,21 — 1,39
1,75
кажущаяся
0,05-01
0,04—0,03
0,03
Удельная
поверхность
м2/г
16—35
180—200
120
эта возможность пока не реализована (в основном из-за высокой
зольности сажи).
Материальный и тепловой балансы процесса
В табл. V-14 приведен материальный баланс процесса,
рассчитанный при средних реальных условиях, типичных для процесса
при атмосферном давлении (отношение О2 : СН4 = 0,6, температура
подогрева 600° С, состав кислорода: 95%02h5%N2 + Аг). Из
таблицы видно, что в результате реакции образуется более 27 объемн. %
пирогенетической влаги. Общий крекинг при этом составляет 89%,
а полезный крекинг в расчете на прореагировавший метан — около
28%. В продуктах окисления содержится 54% углерода от общего
количества.
Количество влажного газа пиролиза, необходимое для
получения 1 т С2Н2, составляет (с учетом потерь):
1381-1000-92,9
83,0
15 500 м3/т
где 92,9 и 83,0 — количество ацетилена (в кг)у содержащееся в 1000 м3
газов пиролиза после реактора и после выделения ацетилена.
Количество сухого газа пиролиза равно:
15 500-0,722 = 11 200 м3/т
Методика расчета материального баланса связана с трудностями,
поскольку степень превращения метана в ацетилен меняется в
зависимости от ряда факторов и должна быть определена опытным путем.
Для иллюстрации влияния режима процесса на выход продуктов
ниже приведены экспериментальные данные6:
Отношение О2 : СН4 0,48—0,57
Температура предварительного подогрева,
°С 400—610
185
Выход продуктов пиролиза, % от прореагировавшего метана:
С2Н2
С2Н4
сн44 .
с3нб
с3н4 ;
С4Н2 .
. . 28,9—25,2
. . 2,4—3,2
. . 0,43—0,52
. . 0,1—0,2
. . 1,1—1,2
. . 2,8—3,0
С4Н4 . . .
Сажа . . .
СО ....
СО2 . . . .
0,9
1,6—2,2
3,4—4,7
53,1—60,1
0,9—4,2
Общее уравнение теплового баланса процесса окислительного
пиролиза можно записать в следующем виде:
где Qx — тепло, вносимое с подогретой исходной смесью;
Q2 —тепло, выделяющееся при неполном сгорании метана;
<?з — тепло, расходуемое на пиролиз;
(?4 — тепло, уносимое реакционными газами;
Q5 — потери тепла.
Анализ теплового баланса позволяет выявить влияние отдельных
факторов, изменяющих направление процесса. Например, увели-
ТА БЛИЦА V-14
Материальный баланс окислительного пиролиза
(расчет на 1000 ж3 сухого газа пиролиза)
Приход
Природный газ
в том числе
метан ....
этан ....
пропан . . .
бутан ....
другие
углеводороды. .
Кислород . . .
в том числе
кислород . .
азот ....
Всего..
м*
557,4
518
21,1
8,3
5,0
5,0
334,4
317,7
16,7
891,8
Объемы.
%
62,5
92,9
3,8
1,5
0,9
0,9
37,5
95,0
5,0
100
кг
445,5
370
28,2
16,25
12,8
18,25
473,9
453
20,9
919,4
Расход
Газ пиролиза
(сухой) * . .
в том числе
ацетилен . .
этилен . . .
водород . . .
окись
углерода . . .
метан . . .
двуокись
углерода . .
азот ....
кислород . .
бутадиен +
+ бензол..
диацетилен. .
метилацетилен
винилацеги-
лен . . .
Влага пироге-
нетическая. .
Всего. .
ж3
1000
80
3
547,3
258,4
50
40
16,7
2
1
1
0,5
0,1
381
1381
Объемн.
%
72,2
8,0
0,3 ■
54,73
25,84
5,0
4,0
1,67
0,2
0,1
0.1
0,05
0,01
27,8
100
кг
612,1
92,9
2,1
49,4
324
35,8
78,5
20,9
2,8
2,4
2,2
0,9
0,2
307,3
919,4
* Содержанием сажи и смолы пренебрегаем.
186
чение Qx должно повышать выход ацетилена. При возрастании Q2
выход снижается, поскольку в системе остается меньше исходного
сырья, которое может пиролизоваться до ацетилена. Вследствие
сложности процесса неполного горения невозможно определять
однозначно влияние тех или иных факторов, поэтому результаты анализа
являются в некоторой степени условными.
В табл. V-15 приведен примерный тепловой баланс процесса,
рассчитанный в соответствии с составом газов пиролиза, указанным
в табл. V-14. Из данных баланса видно, что основное тепло
выделяется при образовании водяных паров. Доля тепла, вносимого
за счет предварительного подогрева исходных газов, составляет
около 15%, и, как будет показано далее, играет существенную роль
в снижении расходных коэффициентов. В статье расхода 50%
принадлежит количеству тепла, уносимому с газами пиролиза. Это
потенциальный источник использования тепла, но он в настоящее
время используется только в процессах под давлением.
ТАБЛИЦА V-15
Тепловой баланс процесса окислительного пиролиза
(расчет на 1 м3 исходного метана)
Приход
Тепло, вносимое с
подогретой исходной смесью
Тепло, выделяющееся
при образовании
продуктов сгорания метана
окиси углерода и
водорода
двуокиси углерода
водяных паров . .
Всего...
ккал
340
134
216
1510
2200
%
15,4
6,1
9,8
68,7
100
Расход
Тепло, затрачиваемое
на образовалие
ацетилена и других продуктов
пиролиза
Тепло, уносимое с
реакционными газами . .
Потери тепла в
окружающую среду ....
Всего ...
ккал
900
1100
200
2200
%
40,9
50,0
9,1
100
РЕАКТОРЫ ОКИСЛИТЕЛЬНОГО ПИРОЛИЗА МЕТАНА
При промышленном получении ацетилена методом неполного
окисления метана приходится осуществлять три взаимосвязанных
процесса: получение гомогенной метано-кислородной смеси,
неполное горение этой смеси с образованием ацетилена и «закалку»
реакционных газов.
На стадии получения ацетилена основным аппаратом является
реактор, который должен быть высокопроизводительным и
надежным в эксплуатации. Продолжительность существования ацетилена
при высоких температурах пиролиза составляет всего 0,01—0,008 сек,
поэтому время неполного горения метана в реакторе должно быть
187
очень небольшим во избежание разложения образующегося
ацетилена.
Одним из важнейших условий нормальной работы реактора
является достаточное смешение исходных компонентов до получения
гомогенной смеси. При горении предварительно перемешанных
смесей пламя более однородно по температуре, что благоприятно
сказывается на процессе получения ацетилена. Однако в этих условиях
часто возможен проскок пламени.
Во избежание проскока можно подавать в зону горения метано-
кислородную смесь, в которой концентрация метана близка к
верхнему пределу взрываемое™ или даже превышает его. В этом случае
необходима существенная стабилизация пламени, например путем
подачи кислорода и создания повышенной местной концентрации
окислителя. При таких условиях можно осуществлять интенсивное
горение с факелом малой дальнобойности, т. е. вести процесс в
условиях, оптимальных для образования ацетилена.
При сжигании раздельно подаваемых углеводородов и
кислорода возникает диффузионный факел, при котором можно избежать
проскока пламени. В этом случае главное — организовать
стабилизацию горения так, чтобы провести процесс за время, допустимое
для получения ацетилена. Такие конструкции уже запатентованы.
Предлагалось также проводить горение в прерывистом потоке
окислителя за счет тепла полного сгорания части метана с
кислородом (остальное количество метана в отсутствие кислорода
крекируется с большим выходом ацетилена), т. е. была сделана попытка
изменить тепловой баланс неполного горения. Однако наибольшее
распространение в промышленном масштабе получили реакторы,
работающие на предварительно подготовленных смесях природного
газа и кислорода.
Процессы горения, в том числе неполного, трудно моделировать,
поэтому данные о длине факела, способе стабилизации пламени,
возможности избежать проскока и отрыва пламени и др., полученные
на установках малой производительности, при увеличении масштаба
принимают совершенно иной характер. Следовательно, при
разработке новых конструкций ацетиленовых реакторов и при увеличении
производительности уже известных и отработанных аппаратов в
первую очередь необходима их опытная проверка.
Значительное разнообразие процессов горения, а также путей его
интенсификации обусловило создание большого числа конструкций
реакторов, подробно описанных в литературе. Все они имеют
примерно одинаковые показатели и различаются в основном условиями
эксплуатации (предотвращением проскока и отрыва пламени и т. п.),
а также решением ряда вспомогательных операций, например
способом удаления образующейся сажи.
Ацетиленовые реакторы * можно разделить на две категории 36~43:
* Методы расчета реакторов и выбора конструктивных размеров изложены
в гл. VII (стр. 292—296).
188
1) односопловые (одноканальные, с одноствольным факелом),
Б ряде конструкций для обеспечения высокоинтенсивного горения
предусматривается разделение основного потока на секторы или
наличие кольцевого зазора (одного или нескольких) определенной
ширины (кольцевые реакторы);
2) многосопловые (многоканальные, с многоствольным факелом);
диаметр отверстий сопел обычно не превышает 30 мм.
Такое деление во многом условно, так как в некоторых
конструкциях сочетаются оба
принципа.
На рис. V-25 изображен однока-
нальный реактор конструкции
Б. С. Гриненко36. Природный газ при
избыточном давлении 2 am в
смесителе / смешивается с кислородом.
Полученная смесь поступает в сопло 2
и истекает из него с критической или
сверхкритической скоростью (300—
500 м/сек) в реакционную зону 4,
стенки которой выложены
огнеупорным кирпичом.
Для стабилизации горения,
происходящего при таких больших
скоростях, применяется дежурное пламя,
создаваемое оригинальным
способом — часть метана с кислородом
сжигают в отдельной боковой
горелке 6, образующиеся при этом
высокотемпературные дымовые газы
стабилизируют основной факел. Горение метано-кислородной смеси
происходит в реакционном канале, где создается необходимая для
процесса температура (1500° С). Скорость движения газов в канале
150 м/сек. На выходе из реакционной зоны они проходят «закалку»
водой и направляются на переработку.
При работе этого реактора на крупной лабораторной установке
получены положительные результаты, но испытания в
полупромышленном масштабе прошли не столь удачно. Основной недостаток
конструкции — большие скорости потока в реакционной зоне и
высокая температура стабилизующих горячих газов. Все это приводит
к быстрому разрушению футеровки реакционной зоны и к
оплавлению сопла. Кроме того, этот реактор трудно моделировать, так как
определенное в лабораторных условиях оптимальное соотношение
между длиной реакционного канала и диаметром сопла не
подтвердилось при испытании в полупромышленном масштабе. Тем не менее
развиваемые Б. С. Гриненко представления о необходимости
получения ацетилена в режиме высокотурбулентного горения были
использованы при создании ряда других, более рациональных
конструкций реакторов22»39-40,
189
^Реакционные
Рис. V-25. Высокоскоростной одно-
канальный реактор:
/—смеситель; 2— сопло; 3— гляделка;
4— реакционная зона; 5—зона
«закалки»; 6 — боковая горелка.
На рис. V-26 показан одноканальный реактор, в котором процесс
горения стабилизуется керамической вставкой, расположенной в
реакционной зоне37-33. Испытания реактора на крупной лабораторной
установке показали, что при такой стабилизации обеспечивается нор.
мальное горение с факелом малой длины. Однако эту конструкцию
трудно моделировать — при разработке реакторов промышленной
мощности потребовалось бы
создание очень большого
туннеля с громоздкой
керамической вставкой.
Более интересны
конструкции одноканальных
реакторов, в которых для
сокращения длины факела и
стабилизации горения при
Вода на
\закалку"
Реащион\
ные газы
Рис. V-26. Туннельный
одноканальный реактор со стабилизатором:
/ — инжекционный трубчатый
смеситель; 2 —переходный патрубок; 3—
реакционный канал; 4 — керамический
стабилизатор; / — зона горения; // —
зона «закалки».
Газ пиролиза
Рис. V-27. Схема
кольцевого одноканального
реактора:
1 — сопло; 2 —завихри-
тели; 3 — футеровка;
4 — реакционная зона;
5 — зона «закалки».
больших скоростях истечения искусственно разделяют
одноствольный поток метано-кислородной смеси и подают его в реакционную
зону по кольцевому зазору либо через отверстия со вставленными
в них завихрителями.
На рис. V-27 изображен одноканальный кольцевой реактор 22«40.
Метано-кислородная смесь из смесителя поступает в кольцевой
зазор, разделенный на несколько секторов, и истекает со скоростью
150—200 м/сек в реакционную зону. Для стабилизации горения пс
центру горелки подают небольшой струей кислород. Основное
преимущество этой конструкции — интенсификация горения путем со-
190
чдапия кольцевого закрученного потока с центральным
воспламенением. При этом значительно увеличивается поверхность
воспламенения, сокращается дальнобойность факела и горение завершается
за малый промежуток времени. Этот реактор, в отличие от
описанных выше, возможно моделировать, что позволяет увеличивать его
производительность.
Исследованиями В. У. Шевчука, И. М. Романюка и др.
установлено, что с увеличением диаметра газовой струи при постоянной
скорости потока длина факела возрастает 39
увеличении диаметра туннеля диаметр
и площадь кольцевого зазора увеличу
ваются в соответствии с уравнениями.
" з_ __ „nnct _^кз
Ft
в степени 0,5—0,6. При
сн
= const
= const
Газ пиролиза
Рис. V-28. Одноканальный
реактор со шнековым завихрите-
лем:
/ — завихритель; 2, 4—-термопары;
3 — слой шамотного кирпича; 5 —
шнековая форсунка.
При испытаниях конструкции в
полупромышленном и промышленном
масштабах были достигнуты хорошие
результаты.
В изображенном на рис. V-28
реакторе со шнековыми завихрителями,
вставленными в отдельные отверстия,
путем создания закрученных потоков
можно создать короткий факел41.
Испытания такого реактора показали его
работоспособность, но из-за сложностей
при изготовлении завихрителей
(сварные, охлаждаемые изнутри водой) эту
конструкцию нельзя считать удачной.
Общей особенностью всех
рассмотренных одноканальных реакторов
является относительно большая длина
реакционной зоны по сравнению, например, с многоканальными
реакторами. Поэтому реакционную зону приходится футеровать
огнеупорными керамическими материалами, способными выдержать большие
скорости газов и высокую температуру.
Рассмотрим многоканальные ацетиленовые реакторы с так
называемыми многосопловыми горелками, выполненными в виде
горелочных плит, иногда охлаждаемых изнутри водой, с большим
числом отверстий относительно малого диаметра. Элементы горелоч-
ной плиты можно набирать в требуемом количестве в зависимости
не от условий неполного горения метано-кислородной смеси, а от
возможности ее равномерного распределения по значительному числу
малых отверстий.
Согласно закономерностям горения при малых диаметрах
отверстий (d = 25 -f- 30 мм) и относительно низкой скорости истечения
газов (100—120 м/сек) длина факела не превышает 8—10 d. Это
обеспечивает более равномерную, чем в одноканальных реакторах, тем-
191
пературу в реакционной зоне. Наличие большого числа маленьких
факелов создает возможность взаимной стабилизации одного пламени
другим. Для стабилизации горения при небольших скоростях
истечения газов подают несколькими потоками кислород между
основными отверстиями и по периферии горелочной плиты.
СН
Вода
CYL
Вода
Вода
еИЬ
"вода
пиролиза
Рис. V-29. Схемы многоканальных реактороз:
а) реактор с эжекционным смесителем (/ — смеситель; 2 — диффузор; 3 —
футеровка; 4 — горелочная плита; 5 — форсунки; 6 — устройство для
удаления кокса; 7 — предохранительная мембрана; 8 — термопара);
б) реактор с водяной завесой (7 —смеситель; 2 —горелочная плита; 3 —
реакционная зона; 4— зона «закалки»)..
На рис. V-29 изображены многоканальные реакторы, в которых
метано-кислородная смесь получается в отдельных смесителях
различной конструкции. Приготовленная смесь распределяется по
отверстиям горелочной плиты, на выходе из них поджигается и поступает
в реакционную зону. Зажигание смеси при работающей горелке
происходит за счет подсоса горячих продуктов горения к корням
факелов (аутостабилизация горения).
192
Основной недостаток этих аппаратов заключается в том, что в
случае проскока пламени за горелочную плиту в предреакционном
пространстве начинается горение. Проскок пламени в многоканальных
реакторах возможен чаще, чем в одноканальных, из-за меньших
скоростей потока метано-кислородной смеси. Для прекращения
горения реактор оснащается специальными автоматическими
приборами — блокировками, которые прерывают подачу кислорода или
подают азот, разбавляющий метано-кислородную смесь.
Наряду с этими конструкциями разработан ацетиленовый
реактор, в котором многоэжекционный смеситель совмещен с горелочной
плитой, имеющей большое число реакционных каналов. Это
позволяет улучшить распределение газа по отдельным отверстиям и
уменьшает случаи проскока пламени в предреакционную зону. Возможные
проскоки локализуются в отдельных каналах и не распространяются
по всей плите. С помощью термопары, помещенной в горелочную
плиту, можно отмечать повышение температуры в случае проскока
пламени.
В процессе работы на стенках реакционной зоны отлагается сажа,
которую периодически приходится удалять. Для этой цели
применяются разнообразные механические приспособления. Сажеочистные
устройства, расположенные в зоне высоких температур, необходимо
охлаждать водой, что усложняет их конструкцию и создает трудности
при эксплуатации. В некоторых реакторах для предотвращения
отложения сажи подается вода, стекающая тонкой пленкой по
наружным стенкам реакционной зоны и частично испаряющаяся.
Механические устройства в этом случае не нужны, поскольку они заменены
движущимся водяным экраном.
Различные ацетиленовые реакторы с многосопловыми горелками
широко распространены в промышленности.
Основные сравнительные характеристики реакторов приведены
в табл. V-16 (данные о промышленных реакторах и результаты
испытаний отдельных полупромышленных конструкций).
Наиболее интересно сопоставить одно- и многоканальные
реакторы по величине соотношения w1 : до2. В многоканальных реакторах
оно практически равно 1. Следовательно, метано-кислородная смесь
и поток реакционных газов движутся там с одинаковыми скоростями
и поэтому в меньшей степени возможен обратный ток горячих (1500° С)
реакционных газов. При этом горелочная плита как бы защищается
экраном из относительно холодной метано-кислородной смеси (зона
воспламенения в турбулентном факеле), еще не вступившей в
реакцию.
В одноканальных реакторах, наоборот, создаются условия для
эжектирования горячих реакционных газов метано-кислородной
смесью. Во всех одноканальных реакторах имеются в той или иной
степени закрученные потоки, в которых наблюдается обратный ток
газов к горелке — в область с пониженным давлением, а площадь
живого сечения горелки невелика (20—25%), поэтому создаются
условия для омывания горелки горячими газами. При этом горелка,
13 В. Н, Антонов 193
ТАБЛИЦА V-16
Основные показатели ацетиленовых реакторов
Показатели
Скорость вылета мета-
но-кислородной смеси
(wi) м/сек
Скорость газов в
реакционной зоне (w2),
м/сек
Отношение скоростей
10) ' ТО)
Отношение длины
реакционной зоны (Я)
к диаметру горелки
(D) или отверстия
в горелочной плите (d)
Площадь живого
сечения горелки, % ....
Метод стабилизации
основного факела ....
Одноканальные реакторы
системы
Гриненко
300—500
150
2—3,3
Н : D =
= 6-г-8
100
Отдельной
горелкой
туннельный, со
стабилизатором
200
100
2
—
Аутоста-
билиза-
ция
кольцевой
150—200
50—160
1,3—3
Н : D = 2
25
Многоканальные реакторы
со шне-
ковыми
завихри-
телями
120
50
2,4
Н : d = 0,5
20
с
отдельным
смесителем
85—^0
80
0,8—1,0
Н : d = 0,5
35
с
совмещенными
горелкой
и
смесителем
100
so
0,9
Н : d =
= (0,5^-0,7)
40
Подача кислорода
даже охлаждаемая водой, работает в жестких условиях и чаще
прогорает.
Из данных табл. V-16 видно также, что с возрастанием скорости
вылета метано-кислородной струи (w±) резко повышается
соотношение Н : D (или Н : d). Это усложняет конструирование сопел
горелок и подбор материала реакционной зоны для проведения процесса
в сверхтурбулентном режиме.
Существенным отличием многоканальных реакторов является
значительно меньшее соотношение Н : d, т. е. реакционная зона имеет
гораздо меньшие размеры. Поэтому ее можно выполнять из металла
с охлаждаемыми водой стенками. Теплопотери через боковую
поверхность реакционной зоны в этом случае должны быть меньше,
чем в одноканальных реакторах. Однако, по-видимому, правильно
предположение, что общие потери тепла из реакционной зоны в
многоканальных реакторах несколько выше за счет большей отдачи тепла
реакционных газов в зону «закалки» (площадь горелочной плиты
в многоканальных реакторах больше, чем в одноканальных).
Поэтому в одноканальных реакторах выход ацетилена должен быть
несколько выше.
Одним из основных вопросов при выборе ацетиленового реактора
является его производительность. Для первых промышленных
реакторов она составляла 150—200 т ацетилена в год, в дальнейшем
(в 50-х годах) на ряде заводов были установлены реакторы
мощностью 2500 т/год. Повышение производительности тормозилось
техническими трудностями при разработке конструкций аппаратов.
Лишь в последние годы в СССР и за границей были созданы реакторы
194
на 5000—8500 т ацетилена в год, а сейчас разрабатываются реакторы
годовой производительностью до 10 000 т ацетилена. В части
повышения производительности одноканальные кольцевые реакторы
имеют бесспорные преимущества.
Необходимо, однако, остановиться на допустимом и
целесообразном пределе повышения производительности ацетиленового
реактора. При атмосферном давлении увеличение производительности
выше 10 000 т ацетилена в год, по-видимому, технологически
невыгодно. В этом случае для установок средней мощности (около
35 000 т ацетилена в год) потребуется два-три реактора, а для такого
чувствительного и малоинерционного процесса, как пламенный, это
создаст трудности в эксплуатации и не обеспечит надежную работу
цехов, перерабатывающих ацетилен. Повышение производительности
одного реактора также нецелесообразно экономически, поскольку
создание реактора обходится гораздо дешевле, чем остальных
аппаратов. Стоимость же этих аппаратов (подогреватели, аппараты саже-
очистки и др.) с увеличением их пропускной способности снижается
очень мало. В связи с этим повышение производительности
ацетиленового реактора целесообразно сочетать с интенсификацией
сопутствующих ему процессов (нагрев, очистка газа пиролиза от сажи).
Одним из возможных путей интенсификации окислительного
пиролиза является повышение давления. При этом можно увеличить
производительность реактора в 2—4 раза; одновременно
интенсифицируются остальные аппараты, что в сумме дает ощутимый
экономический эффект. Известно, что на промышленных установках при
повышенном давлении мощность ацетиленовых реакторов составляет
10—20 тыс. т С2Н2 в год.
Конструкции реакторов, предназначенных для процесса под
давлением, принципиально не отличаются от описанных выше.
Поскольку свойства горючей метано-кислородной смеси (скорость
горения, период индукции и пр.) с ростом давления изменяются, это нужно
учитывать при разработке конструкции реактора. В промышленности
для процесса под давлением используют многоканальные реакторы.
Другой экономически эффективный путь интенсификации
процесса пиролиза — применение высокоподогретых (до 800—900° С)
природного газа и кислорода. При этом значительно сокращаются
расходные показатели.
ТЕХНОЛОГИЧЕСКИЕ СХЕМЫ ОКИСЛИТЕЛЬНОГО ПИРОЛИЗА
Получение ацетилена при атмосферном давлении
В промышленности для получения ацетилена окислительным
пиролизом наиболее широко распространен процесс при атмосферном
давлении. Для этой основной схемы имеется несколько различных
вариантов, связанных с конструкцией ацетиленового реактора и
типом систем очистки газов пиролиза и сточных вод от сажи. Процесс
получения ацетилена включает следующие стадии: компримирование
кислорода, подогрев природного газа и кислорода и процесс пиро-
13* 195
лиза, очистка газов пиролиза от сажи, смол и высших ацетиленов.
В производстве ацетилена потребляется кислород с 95—98 объемн.
% О2. Кондиция кислорода и постоянное содержание О2 в нем
являются обязательным и неотъемлемым условием нормальной и
безопасной работы ацетиленового реактора. Допустимые колебания
концентрации О2 составляют ±0,5 объемн. %. Сохранение постоянной
концентрации кислорода в агрегатах для разделения воздуха
связано с определенными трудностями, поэтому целесообразно
устанавливать кислородный газгольдер «на проход» — при этом
обеспечивается постоянное давление кислорода и облегчается
перемешивание газа с различным содержанием О2. При установке газгольдера
«на тупик» эти задачи выполнить труднее, хотя сопротивление линии
при этом меньше.
В зависимости от конструкции реактора требуется избыточное
давление кислорода от 2 до 3,5 am. Для компримирования кислорода
обычно используют высокопроизводительные центробежные машины.
После каждой машины имеется холодильник, через который
пропускают все количество кислорода. Иногда кислород не охлаждают,
и тогда холодильник переключают на обводную линию. Естественно,
что чем больше требуется давление кислорода, тем менее выгоден
процесс экономически, поскольку при других равных параметрах
возрастает расход электроэнергии на сжатие кислорода.
Для сокращения расходных показателей по природному газу и
кислороду необходим их подогрев до максимально возможной
температуры (600—750° С). Температура подогрева природного газа
определяется его составом и условиями разложения содержащихся
в нем примесей (этан, пропан, пентан, гексан и др.) до углерода и
водорода. Для окисления гомологов метана добавляют в газ
кислород или пар.
Температура подогрева кислорода практически ограничена
только жаропрочностью применяющихся легированных сталей.
Исходные газы подогревают в специальных радиационно-конвективных
подогревателях за счет тепла топливного газа (обычно природный
газ). Иногда для этой цели применяют газы, отходящие со стадии
концентрирования ацетилена, так называемую фракцию высших
ацетиленовых углеводородов. Дымовые газы с температурой 200—
300° С не используются и выбрасываются в атмосферу.
Природный газ поступает на производство из общезаводской сети
при избыточном давлении 3—4 am и подвергается специальной очистке
от имеющихся в нем примесей (ржавчина, пыль и др.), способных
понижать температуру самовоспламенения метано-кислородной смеси.
Например, окалина Fe2O3 при 400—600° С под действием
природного газа восстанавливается до свободного железа. Восстановленное
железо в зоне смешения метана с кислородом окисляется:
3 Fe + 2 О2 — Fe3O4 Д# = — 195 ккал/моль
Значительный тепловой эффект этой реакции способствует
самовоспламенению метано-кислородной смеси до поступления в зону
горения, поэтому на промышленных установках предусматривается
специальная очистка природного газа от окалины.
Другим источником воспламенения может быть сажа,
образующаяся при крекинге метана. Вместе с другими механическими
примесями она понижает температуру самовоспламенения метано-кис-
лородной смеси с 650 до 340° С. От этих примесей природный газ
очищают в фильтрах из керамических трубок (паролит и другие
материалы) или из нескольких слоев мелкой металлической сетки.
При длинных коммуникациях кислорода и возможности его
загрязнения или в некоторых других случаях аналогичные фильтры
применяют и для очистки кислорода. Обычно же очистка кислорода
не так необходима, поскольку имеющиеся в нем примеси в стадии
нагрева должны сгореть.
Вследствие сложности окислительного пиролиза (пламенный
процесс), его малой инерционности и высокой чувствительности к
колебаниям нагрузок, давлений и пр. для стадий получения ацетилена
принята агрегатная схема. В немалой степени это связано с
возможностями применяемых регулирующих и блокирующих приборов.
Агрегаты получения ацетилена состоят из подогревателей природного
газа и кислорода, ацетиленового реактора и аппаратов сажеочистки.
Дальнейшая стадия переработки полученных газов пиролиза —
компрессия — организована по коллекторной схеме.
На рис. V-30 показана технологическая схема производства
ацетилена окислительным пиролизом природного газа, разработанная
в СССР (ГИАП)57.
Природный газ из сети при избыточном давлении 2,5 am
поступает в радиационно-конвективный подогреватель 3. Вначале газ
проходит конвективную зону, где нагревается до 350—400° С, затем
фильтр 4 для очистки от механических примесей и поступает в
радиационную зону, после которой температура его повышается до 650° С.
Подогрев производится за счет тепла дымовых газов, получаемых
в топочной камере подогревателя при сжигании природного газа.
Дымовые газы в радиационной зоне охлаждаются с 1200 до 800° С,
а в конвективной — с 800 до 300° С. Для нормальной работы в
подогревателе поддерживается постоянное количество сжигаемого газа
с коррекцией по температуре. В топке подогревателя установлена
постоянно горящая дежурная горелка (с сигнализацией о погасании
пламени) для поджигания исходной газовой смеси, выходящей из
рабочей горелки. Кислород, сжатый турбогазодувкой 1 до
избыточного давления 1,5 am, в подогревателе 2 нагревается до 650° С. По
конструкции этот подогреватель аналогичен подогревателю 3.
Нагретые газы в смесителе 5 образуют метано-кислородную смесь,
которая поступает в кольцевую горелку реактора б и на выходе из
горелки загорается за счет аутостабилизации. Для стабилизации
горения в-реактор отдельно подают дополнительное количество
кислорода (5—6% от основного потока).
Ацетиленовый реактор снабжен рядом регулирующих устройств
и защитных блокировок. Главным для нормальной работы реактора
197
является поддержание постоянного отношения О2 : СН4. С этой
целью на линии природного газа предусмотрен регулятор количества,
а по расходу природного газа с помощью регулятора соотношения
устанавливается необходимое количество кислорода.
Возникающие в процессе пиролиза неисправности связаны в
первую очередь с возможностью проскока или отрыва пламени; оба
обстоятельства могут привести к аварии: в первом случае — к
прогоранию горелки, во втором — к попаданию взрывоопасной горячей
метано-кислородной смеси в газопровод и далее в остальные аппараты.
В случае колебания концентрации кислорода более чем на ±1%
при одинаковом объемном соотношении О2 : СН4 изменяется
фактическое содержание кислорода в смеси. Поскольку концентрация
метана в горячей метано-кислородной смеси близка к верхнему пределу
взрываемости, то с повышением количества кислорода смесь либо
приобретает другую скорость горения, либо вообще выходит за
пределы взрываемости. Поэтому резкие колебания в составе кислорода
могут вызывать проскок или отрыв пламени. В том и другом
случаях реактор необходимо остановить. Остановка реактора и
связанных с ним аппаратов производится автоматически.
Защитные блокировки реактора срабатывают в следующих
случаях: при повышении или понижении давления метано-кислородной
смеси перед горелкой реактора; при возрастании температуры в
смесителе или в отдельных отверстиях горелки (в случае
многоканального реактора); при понижении температуры реакционных газов после
«закалки» (это свидетельствует о затухании горения); при уменьшении
давления природного газа или кислорода; при падении давления воды,
поступающей на «закалку»; при понижении давления конденсата,
расходуемого на охлаждение горелки.
Во всех указанных случаях в реактор в первую очередь подается
азот для разбавления метано-кислородной смеси (изменение
концентрационных пределов) и понижения ее температуры, что также
уменьшает взрываемость смеси. Азот поступает непосредственно
в смеситель в течение определенного времени (несколько секунд).
Если за это время защитные блокировки не вернутся в
первоначальное состояние, то подача кислорода прекращается совсем.
Образующиеся в этот период в реакторе некондиционные газы
пиролиза сбрасывают на факел 11 с постоянно горящей дежурной
горелкой. В это время прекращается поток газа в систему сажеочистки
и отключаются основные горелки в подогревателях природного газа
и кислорода. При пуске реактора также получаются некондиционные
газы, содержащие метан, кислород и ацетилен. Эти газы нельзя
сбрасывать в атмосферу, поэтому их тоже направляют на факел 57.
При нормальной работе реактора газы пиролиза имеют
следующий состав (в объемн. %, в пересчете на сухой газ):
СоН2 7,5-8,2 Н2 51,7—£6,0
СН4 4,0—3,0 О2 До 0,2
Высшие ацетилены и СО 26,0—27.0
ароматические уг- СО2 3,0—4,0
леводэроды .... 0,3-0,4 N.," 1,6
199
Кроме того, в газе содержится пирогенетическая влага, смолы,
ароматические углеводороды, нафталин, антрацен (до 3—5 мг/м*
в сумме) и сажа (3—5 г/мп).
На выходе из реакционной зоны газы, имеющие температуру
— 1500° С, резко охлаждаются (до 100° С) большим количеством воды;
на «закалку» подается горячая (50° С) и холодная (25—27° С) вода.
При этом примерно 45% сажи вымывается водой и уходит с ней через
гидравлический затвор 7. Частично очищенный газ направляется
на окончательное охлаждение и доочистку от сажи, смол и
ароматических углеводородов.
В схеме, изображенной на рис. V-30, система сажеочистки состоит
из двух аппаратов: полого скруббера 8, орошаемого водой, и мокро-
пленочного электрофильтра 9. Газы вначале поступают в скруббер 8,
где происходит конденсация водяных паров и улавливание сажи за
счет смачивания ее мелкораспыленной горячей водой. При
использовании горячей воды (до 80° С) смачиваемость сажи улучшается.
Поэтому в нижнюю часть аппарата подается горячая вода, а в
верхнюю — более холодная, чтобы обеспечить температуру газа на входе
в электрофильтр примерно 60° С.
. За счет высокого напряжения, создаваемого в электрофильтре 9,
происходит ионизация сажевых частиц, они приобретают
отрицательный заряд (катод) и оседают на коронирующих электродах
(аноды). Для удаления отложившейся сажи Янады постоянно
орошаются тонкой пленкой воды. Сажа способна агломерировать и
образовывать большие наросты на электродах, что может привести к
пробою электрофильтра. Поэтому предусматривается периодическая
промывка электрофильтра большим количеством воды. Для
обеспечения безопасной работы электрофильтра на линии входа газов
пиролиза установлен автоматический газоанализатор на кислород,
включенный в t систему аварийной блокировки электрофильтра.
При повышении содержания кислорода более 0,3—0,8 объемн. %
газы пиролиза автоматически направляются на сжигание.
Очищенные от сажи (до нескольких миллиграммов на 1 м3) газы
пиролиза при температуре около 60° С поступают на окончательное
охлаждение в пенный аппарат 12. Там газ и вода проходят
противотоком, в результате чего температура газа понижается до 30—35° С,
а вода нагревается до 50° С.
Охлажденные в пенном аппарате 12 газы пиролиза перед стадией
компримирования и выделения ацетилена очищают от
ароматических углеводородов и нафталина. Очистка (форабсорбция)
производится тем же селективным растворителем (N-метилпирролидон или
диметилформамид), который используется на стадии выделения и
концентрирования ацетилена. Для этой цели применяют тарельчатые
или барботажные аппараты 13, в которых снизу вверх проходит газ
пиролиза, а противотоком течет абсорбент. Система форабсорбции
выполнена с замкнутым циклом абсорбента, циркуляция которого
осуществляется насосом 14. Количество абсорбента выбирают с
таким расчетом, чтобы концентрация влаги, уловленной из газов пи-
200
ролиза, не превышала 10 вес. %. Кратность орошения 20 м3 на 1000 м3
газа.
За счет тепла, выделяющегося при растворении воды, температура
абсорбента и соответственно давление его паров повышаются,
поэтому абсорбент охлаждают в холодильнике 15. Для улавливания
брызг жидкого растворителя, а также его паров несколько верхних
тарелок в аппарате 13 орошаются водяным конденсатом. Полученная
на тарелках смесь воды и абсорбента смешивается со всей массой
абсорбента. Очищенные и охлажденные газы направляются в
газгольдер и далее на выделение ацетилена.
Вода, отходящая из реактора, аппаратов сажеочистки и
охлаждения газов пиролиза, загрязнена сажей, смолой и различными
водорастворимыми газами. Примерный состав примесей такой (в г/м3):
Нафталин
Аценафтилен
Антрацен
Фенантрен .
. 0,1—0,2
0,3
. 0,25
. 0,1
Коронен . . .
Пирен ....
Фенол и др. . .
Сажа ....
0,2
0,2
1,6
2-3
Некоторые из этих примесей токсичны, поэтому воду,
содержащую их, не сбрасывают в обычную канализацию, а направляют в
специальный цикл загрязненных вод. Для поддержания баланса солей
жесткости замкнутая система водооборота пополняется конденсатом,
а иногда устанавливается специальная стабилизационная установка
для отделения от воды временных солей жесткости. В условиях про-,
изводства ацетилена целесообразна обработка воды соляной кислотой
или другим реагентом.
Из аппаратов 8, 9 и 12 вода через соответствующие гидрозатворы
направляется в сажеотстойник 18 для очистки от сажи и смол. Са-
жеотстойник представляет собой бассейн, через который вода
проходит со скоростью около 0,1 м/сек в течение 20—30 мин. Сажа и смолы
являются гидрофобными веществами и поэтому всплывают на
поверхность воды. Шлам (смесь сажи, смол и воды) с помощью
скребков, насаженных на цепной транспортер 17, собирается на
поверхности воды и подается в емкость 16 с мешалкой. Часть сажи (3—5%)
не успевает всплыть и оседает на дно сажеотстойника, поэтому его
необходимо периодически опорожнять и очищать. Вода из
сажеотстойника направляется на охлаждение в водооборотный цикл
загрязненных вод, а небольшое ее количество (20—25%) при
температуре около 80° С направляется в реактор для «закалки» газов
пиролиза. Пульпа сажи и смолы с водой (до 95% воды) подается на
установку для сжигания отходов.
Наряду с описанной схемой, в которой для сажеочистки
используются мокропленочные электрофильтры, распространены схемы
с так называемыми коксовыми фильтрами, разработанные фирмой
BASF13 (рис. V-31). Природный газ и кислород раздельно
подогреваются в подогревателях ) и 2 до 650° С и поступают в многоканальный
ацетиленовый реактор 3. Газ, охлажденный после «закалки» примерно
201
до 80° С, проходит в полый скруббер-охладитель 4, где температуру
его снижается до 35° С.
Тонкая очистка газов пиролиза от сажи проводится в фильтре 7}
заполненном мелкозернистым прочным коксом. Газы поступают
в низ аппарата и проходят противотоком к медленно движущемуся
сверху вниз коксу, при этом сажа из газов оседает на смоченной
поверхности кокса. Содержащий сажу кокс спускается в бункер 10.
На (ракел
Вода
Топливный
газ
Конденсат
Вода для
■3 Топливный
газ
-^ ^Стабилизу -
ющий О2
7-
Ш
OOOvx
ЛлД/у
т
Лиетилен
на
концентрирование
вода для
транспортирования кокса
Рис. V-31. Схема установки с коксовыми фильтрами для получения
ацетилена по методу фирмы BASF:
/ — подогреватель природного газа; 2 — подогреватель кислорода; 3 — реактор;
4 —скруббер-охладитель; 5 —коксовый питатель; 6—предохранительный затвор;
7 — коксовый фильтр; 8 -циклон; 9 — сажепромыватель; 10— бункер для кокса;
// — сажеотстойник; 12 — насос.
Там кокс отмывается от сажи большим количеством воды и водяным
подъемником подается в коксовый питатель 5, а затем в коксовый
фильтр 7.
Вода из фильтра, содержащая сажу, через циклон 8 и
сажепромыватель 9 направляется в сажеотстойник 11. Туда же поступает
загрязненная вода из реактора 3 и скруббера 4.
Ацетиленовый реактор и газовые подогреватели оснащены
системой защитных блокировок, аналогичной описанной на стр. 197,
но с отличиями, присущими данной схеме, Некондиционные газы
пиролиза сбрасывают на факел.
2)2
Ниже приведены полученные автором сравнительные данные
о схемах сажеочистки с мокропленочными электрофильтрами и
с коксовыми фильтрами (расчет на 1 т ацетилена):
Схема Схема
с электро- с коксовыми
фильтрами фильтрами
Энерго-материальные затраты,
% 100 80
Капитальные затраты, % . . 100 120—130
Видно, что схема с электрофильтрами характеризуется
несколько большими энерго-материальными затратами. Это связано в
первую очередь с потреблением электроэнергии — примерно 1 квт-ч
на 1000 м3 газов пиролиза. Капитальные затраты, наоборот, выше
при схеме с коксовыми фильтрами (в расчете на их разовое
заполнение). Однако, несмотря на увеличенные энерго-материальные
затраты, способ очистки газов в электрофильтрах следует считать
перспективным, так как при этом получается более чистый газ
(содержание сажи не превышает 5 мг/м3). Кроме того, процесс в
электрофильтрах значительно автоматизирован и не связан с необходимостью
периодических чисток.
Бельгийской фирмой SBA разработана установка для получения
ацетилена при высоком предварительном подогреве природного газа
и кислорода (до 650° С) с последующей очисткой газа пиролиза в
электрофильтрах27. По этой схеме (рис. V-32) природный газ и кислород
подогреваются до 650°С в совмещенном змеевиковом подогревателей
за счет тепла дымовых газов и поступают в многоканальный реактор 3
со смесителем. Газы пиролиза проходят «закалку» водой и при 100° С
направляются на дальнейшее охлаждение в полый скруббер 4,
орошаемый горячей и холодной водой, а затем в мокропленочный
электрофильтр 5 прямоугольного сечения. Очищенные от сажи газы
окончательно охлаждаются в скруббере-охладителе 6 (до 30—35° С) и
поступают на стадию выделения и концентрирования ацетилена.
Загрязненные воды из реактора и аппаратов сажеочистки
направляются на выделение сажи. Из-за высокого предварительного
подогрева исходных компонентов и специфической конструкции
реактора (удаление сажи с помощью водяной пленки) образующаяся сажа
обладает малой гидрофобностью, по-видимому, потому, что она
низкопористая и довольно хорошо смачивается водой. Для очистки от сажи
воду фильтруют в вакуум-фильтрах. Полученная сажевая пульпа
направляется для сжигания н& специальные установки, а
очищенная вода поступает на градирню для охлаждения (часть воды
возвращается в реактор для «закалки» газов пиролиза).
На рис. V-33 изображена схема работающей в Румынии44'45
опытно-промышленной установки по получению ацетилена
окислительным пиролизом метана. Производительность установки по
исходному метану примерно 2000 м3/ч. По этой схеме очищенный от
механических примесей природный газ нагревается в радиационно-
конвективном подогревателе 7 до 420° С. Кислород поступает на
203
EX. -< TO w
^-ftS
1 ФИ
I 3fl&;
о ft
О 2 a ] ^
> ~ Г
я
s со г
°-2
2Г
g^
О S
>> Я
11
gb
g
I g
Ig
о, н
?s
О го
Я I
e^
.^ I I
о
установку из газгольдера 1, сжимается газодувкой 4 до избыточного
давления 0,8 am, проходит фильтр 5 для очистки от окалины и
поступает в подогреватель 6, где температура его повышается до 420° С.
Метан и кислород в определенном соотношении, поддерживаемом
специальным регулятором, поступают в многоканальный
ацетиленовый реактор 10. Температура в диффузорном смесителе реактора
достигает 380° С.
Газы пиролиза проходят «закалку» водой, охлаждаясь до 70° С,
и направляются на сажеочистку; давление газов после реактора 80—
160 мм вод. ст. Сажеочистка осуществляется в
скруббере-охладителе 11 и мокропленочном электрофильтре 12. Для улавливания сажи
в верхнюю часть скруббера подается горячая вода, а в нижнюю его
часть и в электрофильтр — холодная, которая предварительно
очищена от механических примесей. Около 60% сажи удаляется в зоне
«закалки» реактора и в скруббере, остальное количество — в
электрофильтре. Очищенные газы направляются непосредственно на
использование.
Загрязненные воды из реактора, скруббера и электрофильтра
собираются в сажеотстойнике 15. Образующаяся там пульпа (смесь
сажи с водой) откачивается насосом 14. Осветленная горячая вода
проходит фильтры 16 и 8 и возвращается в реактор для «закалки»
газов пиролиза.
Отношение О2 : СН4 на установке поддерживается 0,665, при этом
общий крекинг метана достигает 93—94%, а полезный крекинг
в пересчете на прореагировавший метан — 32,5%. Получаемый газ
имеет следующий состав (в объемн. %):
С2Н2 7,5 О2 0,2
С2Н4 + высшие СО 26,0
ацетилены . . 0,5 СО2 4,0
СН4 3,4 N2 2,0
Н2 56,4
Ниже приведены расходные показатели (в расчете на 1 т
ацетилена):
Метан (на процесс), м3 6000
Кислород, м? 4100
Электроэнергия (без учета кислородного цеха и
стадии выделения ацетилена), квт-ч 95
В последнее время появились сообщения о разработанном
фирмой Hydrocarbon Research Process (США) процессе производства
ацетилена из природного газа и кислорода при высоком подогреве
готовой метано-кислородной смеси (до 800° С)46"49. При этом возможно
снижение расхода природного газа и кислорода на 15—20%. Процесс
отработан в масштабе опытно-промышленной установки.
В метано-кислородной смеси соотношение О2 : СН4 выбирают
таким, что при нормальной температуре смесь находится за верхним
205
Кислород
i^-УглеВодород
ДымоВые
газы ~"~
Инертный
газ ~~*~
Вода
II II \-^Jlbi>
S 2 ST Й
Дымовые
газы
пределом взрываемости. Подогрев смеси расширяет пределы ее
взрываемое™ и она становится взрывоопасной, поэтому подогрев следует
проводить за очень малый промежуток времени — меньше периода
индукции для смеси данного состава. Поскольку в описываемом
процессе соотношение О2 : СН4 берется ниже, чем обычно при
атмосферном давлении, то соответственно требуется и более высокая
температура предварительного подогрева. Кроме того, при совместном
подогреве метана и кислорода создаются условия для лучшего их
перемешивания, что повышает выход
ацетилена.
На рис. V-34 показана конструкция
реактора49 для получения ацетилена
способом фирмы Hydrocarbon Research
Process при подогреве реакционной
смеси до 750—800° С. Кислород и
углеводород смешиваются в конической
верхней части реактора при температуре
ниже 315° С и затем попадают в узкие
трубки, где нагреваются дымовыми
газами, проходящими по межтрубному
пространству, до 750° С. Метано-кисло-
родная смесь находится в трубках очень
короткое время, меньше периода
индукции для этой смеси.
При описываемом способе
необходимо поддерживать скорость смеси
в трубках более 30 м/сек при
температуре выше 537° С, практически же
скорость равна 90—120 м/сек. Выходящая
из трубок горячая газовая смесь через
небольшое паровое пространство и
пористую керамическую плиту поступает в реакционную камеру.
Перепад давления в пористой плите 0,014—0,35 am, скорость газа
на выходе из нее 3—9 м/сек. Температура процесса при атмосферном
давлении около 1540° С. На выходе из реакционной зоны газы
подвергают «закалке» водой, при этом их температура снижается до
80° С. Затем газы промывают водой для улавливания сажи и
направляют на выделение ацетилена.
Содержание ацетилена в газе пиролиза и расход кислорода
зависят от температуры подогрева метано-кислородной смеси48:
Температура, °С 482 705
Содержание С2Н2, объемн. % 7,0 9,0
Расход О2 на 1 кг С2Н2, кг ч 5,6 4,0
На полупромышленной установке фирмы National Research
Development (США)50'51 природный газ и кислород подогревали до еще
более высокой температуры (850—900° С) в подогревателях, пред-
206
Газы пиролиза
Рис. V-34. Реактор для
получения ацетилена при
высокотемпературном подогреве исходных
компонентов.
ставляющих собой виток спирали небольшого размера. В газах,
выходящих из подогревателей, содержались пылевидные частицы
сажи, пирофорного железа и меди, что приводило к
преждевременному загоранию газовой смеси уже при 400° С. Поэтому природный
газ и кислород предварительно пропускали через фильтры из
стеклянной ваты.
Ацетиленовый реактор состоит из камеры смешения и
реакционной зоны. В камере смешения имеется семь трубок из нержавеющей
стали диаметром 8,5 мм и длиной 380 мм. Метан и кислород
распределяются по ним, а окончательно смешиваются в трубе Вентури,
расположенной в нижней части камеры. Труба Вентури
отрегулирована таким образом, чтобы скорость и состав газовой смеси на
выходе из трубок были одинаковы (допустимые колебания
концентрации метана ±0,5 объемн. %).
Уже при 850°С в камере смешения начинаются реакции окисления
с образованием побочных продуктов (в потоке газов были, например,
обнаружены значительные количества формальдегида). Поэтому время
пребывания газов в камере должно быть сведено к минимуму; в ряде
опытов оно составляло 1,11 мсек. Для предотвращения
преждевременного загорания газов в камере смешения в каждую трубку вблизи
точки смешения метана и кислорода помещали небольшой
предохранитель, соединенный с реле, с помощью которого при загорании
смеси отключалась подача кислорода и начиналась подача азота.
Однако это оказалось неудобным, так как для замены предохранителя
нужно было останавливать установку. Оптимальным оказалось
применение термопар, которые реагируют на повышение
температуры стенок камеры и с помощью усилителя включают защитную
систему.
Выходящая из камеры смешения метано-кислородная смесь
поступает в реакционную зону, представляющую собой цилиндр из
нержавеющей стали диаметром 180 мм, футерованный изнутри окисью
алюминия. Для предотвращения эрозии футеровка закрыта платино-
родиевой сеткой. Через пористую футеровку продувается пар,
перегретый до 450° С. Пористость подбирается такой, чтобы перепад
давления между паровой коробкой нагревательного цилиндра и
внутренней поверхностью стенок горелки был положительным.
Температура стенок реакционной зоны составляет 1100° С. Образующаяся
сажа в этих условиях ссыпается со стенок.
Направление потока газов, выходящих из реакционной зоны,
изменяют на 90° с помощью отбойной перегородки и затем пропускают
газы через пять концентрических кольцеобразных разбрызгивателей
воды* для «закалки».
В ходе испытаний использовались несколько систем
стабилизации пламени в реакционной зоне, состоящих из стабилизаторов
различных конструкций и дежурной горелки, в которой сжигали смесь
метана и кислорода (или водорода и кислорода). Наиболее
удовлетворительным вариантом была подача кислорода по кольцу вокруг
входа каждой трубки смешения в реакционную зону. Оптимальное
207
количество стабилизующего кислорода составляло около 5% от
основного потока.
При пиролизе наблюдалась прямая зависимость концентрации
получающегося ацетилена от температуры подогрева, а в случае
разбавления исходного метана азотом концентрация ацетилена падала
прямо пропорционально степени разбавления. Охлаждение
реакционных газов водой в зоне «закалки» мало влияет на концентрацию
получаемого ацетилена (в пределах ±0,2 объемн. %) в широком
интервале изменения количества воды и скорости охлаждения
(подробнее см. стр. 310).
На установке имелась газовая турбина для использования тепла
продуктов реакции. Газ из реакционной зоны поступал
непосредственно в рекуперационную турбину, где частично охлаждался и лишь
потом подвергался «закалке». Однако турбина позволяет снизить
стоимость ацетилена всего на 2—3%, поэтому ее применение
малоперспективно.
В лабораторном масштабе изучались ацетиленовые реакторы
поверхностного горения52: горение происходит на поверхности насадки
без видимого пламени. В таких реакторах практически исключается
возможность проскока пламени, при этом достигается высокая
газовая нагрузка на единицу поверхности насадки (0,3—0,8 м3/м2).
Насадкой служили кусочки шамота диаметром 2—3 мм и длиной 30—
40 мм. Для воспламенения метано-кислородно'й смеси применялся
платиновый катализатор, который позволял зажигать смесь при
300° С. Катализатор в значительной степени улучшает стабильность
горения и увеличивает возможную газовую нагрузку на реактор
до 1—1,5 мъ метана в 1 ч на 1 см2 насадки. Перепад давления в
реакторе при такой нагрузке не превышал 200 мм рт. ст. При
поддержании оптимальных параметров образования ацетилена (величина
насадки, скорость подачи смеси, соотношение О2 : СН4) процесс
шел без образования сажи. При этом были получены такие основные
показатели:
Расход О2 на 1 кг разбавленного С2Н2, кг 4,2—4,4
Полезный крекинг, %
на исходный метан 31,5
на прореагировавший .метан 36,6
Коэффициент увеличения объема газов . . 1,16—1,18
Показатели процесса. Во всех способах получения ацетилена
при атмосферном давлении, за исключением метода фирмы National
Research Development с высоким подогревом метано-кислородной
смеси, выход ацетилена и другие показатели примерно одинаковы.
Они изменяются в зависимости от состава природного газа и
кислорода, температуры их предварительного подогрева и конструкции
реактора.
Следует отметить, что доля расходов на стадии получения ацетилена
в общей себестоимости продукции составляет 60—70%; это в первую
очередь связано со стоимостью природного газа и кислорода. Поэтому
208
снижение расходных показателей по сырью даже на 5% существенно
сказывается на стоимости получаемого ацетилена. Ниже приведены
расходные показатели на стадии получения ацетилена (в расчете
на 1 т С2Н2)13'27»57:58 (см. также стр. 398):
Схема
Природный
газ (100% СН4)
ГИАП
BASF
SBA
6300*
6220 **
6200**
Кислород
м3
3570 (95% О2)
3600 (99% О2)
3670(100% Оо)
С учетом расхода в подогревателях.
Только на пиролиз.
%Г20]
t
I"
I
WO
1QITI
Jg£
122-
^
^
^-—■—-
—' /
~j
~j
-j—
Получение ацетилена при повышенном давлении
За последние годы построены установки по получению ацетилена
под давлением 3—5 am. Проведение процесса под давлением, как было
показано выше, не дает значительного сокращения расходных
показателей, но обусловливает возможность снижения капитальных
затрат. При пиролизе под давлением в первую очередь значительно
интенсифицируются сопутствующие процессы, например
теплопередача при нагреве исходных
компонентов, при охлаждении
получаемых газов и пр. Вследствие этого
габариты оборудования резко со;
кращаются, что дает заметный
экономический эффект.
При пиролизе под давлением
можно полезно использовать тепло
реакционных газов. На выходе из
реакционной зоны газы обычно
имеют температуру 1600° С, однако
основное количество тепла
снимается охлаждающей водой на
стадии «закалки». Речь может идти,
следовательно, только об
использовании тепла газов после «закалки».
При этом процесс теплопередачи
уже не лимитируется временем,
а зависит только от конечной температуры газов после «закалки»
(рис. V-35). Повышение давления, как видно из рисунка, позволяет
значительно повысить количество тепла, которое можно
рекуперировать.
Наиболее целесообразно затрачивать тепло газов пиролиза на
получение горячей воды, которую можно использовать на стадии
выделения ацетилена. Минимальная температура получаемой воды
определяется точкой росы водяных паров, зависящей от давления.
При 1 am парциальное давление водяного пара 0,5 am и точка росы
80° С — эта температура слишком низка для эффективной рекупе-
14^ Э.1 Н. Антонов 209
800 ~ 200 400 600 800
Количество тепла, к к ал на 100 м3 газа
Рис. V-35. Изменение количества
тепла, которое можно рекуперировать,
в зависимости от температуры газов
после «закалки» (пунктир соединяет
точки максимальной температуры
газов после «закалки» для
соответствующего давления).
рации тепла. При увеличении давления точка росы водяных паров
будет выше 100° С, например при 4 am в результате рекуперации
тепла можно получать горячую воду, имеющую температуру 142° С
(рис. V-36).
В газах пиролиза содержится большое количество сажи — около
20 г/м3, поэтому рекомендуется осуществлять их непосредственный
контакт с водой. В этом случае сажеочистка эффективно сочетается
с процессом теплоиспользования: во-первых, при высокой
температуре улучшается сажеулавливание, во-вторых, получается большое
количество горячей (120° С) воды, которую можно использовать на
стадии-концентрирования ацетилена низкотемпературными
абсорбентами, например метанолом (стр. 267).
Еще одним важным преимуществом процесса под давлением
является возможность использования для тонкой сажеочистки более
простых и не требующих высоких энергозатрат аппаратов — труб
Вентури. В трубе Вентури частицы сажи под действием центробежной
силы, получаемой за счет использования динамического напора
газового потока, отбрасываются на периферию к стенкам трубы.
Стенки все время орошаются небольшим количеством воды, которая
и увлекает с собой частицы сажи. При повышенном давлении
эффективность труб Вентури в несколько раз больше, чем при
атмосферном, поскольку допустимый перепад давлений в трубе значительно
выше.
Проведение процесса под давлением выгодно еще и потому, что
уменьшаются энергозатраты на последующее сжатие газов до
давления, которое поддерживается на стадии концентрирования ацетилена
(около 10 am). Если проводить пиролиз, например, при 5 am, то
затраты энергии по сравнению с процессом при атмосферном
давлении будут примерно вдвое меньше. Это видно из следующих данных
о расходе энергии в расчете на 1 т С2Н2 (в квт-ч):
При 1 am При 5 am
На сжатие кислорода с 1 до " На сжатие кислорода с 1 до 5 am 325
2 am 115
На сжатие газов пиролиза с 1 На сжатие газов пиролиза с 5
до 10 am 1350 до 10 am 430
Итого 1465 Итого 755
Из приведенных данных видно, что значительная доля
электроэнергии тратится на сжатие кислорода. Поэтому появились
предложения подавать кислород в реактор с помощью эжектора (в
процессах при атмосферном, и особенно при повышенном
давлении) 53.
При проверке54 лабораторного реактора с эжектором при
избыточном давлении природного газа 17 am кислород сжимался от 1
до 2,5—2,7 ату следовательно, становилось возможным вести
процесс под давлением,
210
В настоящее время по способу получения ацетилена из
природного газа под давлением работает одна установка в Италии55 (фирма
Montecatini) и одна в США (фирма Daymond-Alkaly). Схема фирмы
20000
§ 10000
^ 5000
| 2000
I'»
I»
^ #70
too.
I
i
У
/
/
/ /
70 80
90
ЮО 110 Щ 130 НО
Температура. °С
Рис. V-36. Теплосодержание влажного газа пиролиза
при различной температуре и давлении (расчет на
1000ж3 газа).
Montecatini описана в гл. VI (рис. VI-22), так как там подробно
рассматривается процесс выделения и концентрирования
ацетилена.
Окислительный пиролиз нефтяного сырья
Имеются данные56 об одноступенчатом процессе окислительного
пиролиза жидких нефтяных углеводородов (и даже сырой нефти)
с целью получения ацетилена и олефинов. Процесс основан на
неполном сгорании жидкого сырья, вводимого в реактор в распыленном
виде в смеси с кислородом. «Закалка» продуктов пиролиза
осуществляется дополнительным количеством исходного сырья. Процесс
происходит в три ступени. Вначале за счет сгорания части
углеводородов выделяется необходимое для реакции тепло, в результате чего
происходит дальнейшее испарение углеводородов и их сгорание.
Затем протекают реакции чисто термического пиролиза, и в конце
осуществляется «закалка» продуктов.
По сравнению с использованием газообразного сырья
разработанный процесс обладает рядом преимуществ. Все стадии (горение,
пиролиз и «закалка») происходят в одном реакционном объеме.
«Закалка» проводится исходным углеводородом, что позволяет обойтись
без стадии отделения «закалочного» агента (например, в случае
применения воды) от газов пиролиза. Загорание и проскоки пламени
исключаются, так как смешение происходит непосредственно в
реакционном объеме. При таком оформлении процесса тепловые потери
минимальны. Реактор имеет небольшие размеры, так как у него нет
футеровки, а для охлаждения металлических стенок используется
вода.
14* 211
Нефть
Газы пиролиза
Принципиальная схема установки окислительного пиролиза
сырой нефти показана на рис. V-37. В верхней части реактора 4,
имеющего водяную рубашку, расположено сопло 2> через которое в
реактор впрыскивается смешанная с кислородом нефть (для лучшего
распыления нефти необходимо оснащать реактор несколькими соплами).
Смесь поджигается дежурной горелкой 3, работающей на топливном
газе и кислороде. Газы после «закалки» поступают в циклон 5, где
от них отделяются капли нефти, и затем — на переработку. Жидкие
кислород углеводороды, собранные в
циклоне и нижней части
реактора, насосом 6 вновь
подаются на процесс.
Около 20% образующейся
сажи возвращается с
жидкими углеводородами в
реактор, а 80% уносится с
газами пиролиза. Содержание
сажи в рециркулируемых
продуктах допускается 100 г
на 1 кг, излишнее количество
выводится из системы.
Начальная концентрация сажи
в газе пиролиза 10—15 г на
1 м3 газа или примерно 65 г
на 1 кг смеси ацетилена и
этилена. Для отделения сажи
газ промывают нефтяными
фракциями.
Для производства 20 т суммы ацетилена и этилена в сутки
достаточно реакционного объема 1 м3, что свидетельствует о
допустимых высоких тепловых напряжениях в зоне реакции.
Соотношение ацетилена и этилена в получаемом газе может изменяться
от 2 : 1 до 1 : 2. Характерный состав газов следующий (в объемн.
Сажа и кокс
Рис. V-37. Принципиальная схема
окислительного пиролиза сырой нефти:
1 — теплообменник; 2 — сопло; 3 — дежурная
горелка; 4 — реактор; 5 — циклон; 6 — насос.
С2Н2
с2н4
сн4
с2н6
Он
C4HS
6,7
6,4
4,4
0,35
0,06
1,2
0,95
Гомологи ацети-
лена 6,97
СбН6
Н2
О2
СО
N2
0,1
28,3
0,8
42,27
1,5
Расходные показатели при использовании сырой нефти составляют
(в расчете на 1 кг С2Н2) 6,7 кг нефти и 3,9 кг кислорода. Примерно
45% введенной нефти сжигается для достижения необходимой
температуры процесса, остальное количество подвергается пиролизу.
Расход нефти на «закалку» реакционных газов примерно вдвое больше,
чем на стадиях горения и пиролиза.
212
ЛИТЕРАТУРА
1. Иоффе Б. В., Основы производства водорода, Гостоптехиздат, I960.
2. Падовани С., Сальви Г., Фиамура А., Доклад на IV
Международном нефтяном конгрессе в Риме, Гостоптехиздат, 1955.
3. И е в л е в а 3. В., Т е с н е р П. А., ДАН СССР, 115, № 3 (1957).
4. Howard W., Wood В., Kaltenbacher E., Chem. Eng. Progr.,
57, № 11, 50 (1961).
5. Солнцева Л. Н., Ирлин А. Л., Казарновский Я. С, Труды
ГИАП, вып. 12, Госхимиздат, 1961.
6. L е г о и х P. J., M a t h i e u P. H., Chem. Eng. Progr., 57, № 11, 54 (1961).
7. Ватлаев Ф. П., Вайнштейн В. Б., Л an иду с А. С.,
Бюллетень по обмену опытом в азотной промышленности, № 10, Госхимиздат, 1958.
8. Lock wood D. С, Petrol. Ref., 39, № 11, 223 (I960).
9. В e n e d e k P., L a s 1 о A., Mag. kern, fol., № 5, 372 (1951).
10. Г р и н е н к о Б. С, 3 е л и з н ы й А. М., Производство ацетилена из
природного газа, Гостехиздат, Киев, 1963.
И. АлейновД. П., Казарновский Я. С, Хим. пром., № 5, 332 (1964);
№ 6, 422 (1964).
12. А б а д ж е в С. С, Шевчук В. У., Газ. пром., № 8, 33 (1965).
13. S а с h s s e H., Bartolome E., Dehema. Monog. В., 24, № 283—290,
9, 39 (1954).
14. Fa user G., Chim. e ind., 42, № 2, 150 (1960).
15. L a s 1 о A., N e m e t h A., Mag. kern, fol., Mb 7, 254 (1960).
16. Ш е в ч у к В. У., Газ. пром., № 3, 36 (1962).
17. А н и с о н я н А. А., Е в л а н о в С. Ф., Труды ВНИИГАЗ, вып. 12,
Гостоптехиздат, 1961, стр. 103.
18. X и т р и н Л. Н., Физика горения и взрыва, Изд. МГУ, 1957.
19. Спейшер В. А., Сжигание газа на электростанциях и в промышленности,
Госэнергоиздат, 1960.
20. Иванов Ю. В., Основы расчета и проектирования газовых горелок,
Гостоптехиздат, 1963.
21. Л я х о в с к и й Д. Н. и др., Труды ЦКТИ, кн. 2, вып. 1, 1947.
22. Р о м а н ю к И. М., Зелизный А. М., Шевчук В. У., Газ. пром.,
№ 10, 34 (1964).
23. Зелизный A.M., Рома ню к И. М., Шевчук В. У., Хим. пром.,
№ 12 (1964).
24. F i s с h e r F., P i с h I e r H., Brennst. Chem., 11, № 24, 501 (1930).
25. Зелизный А. М., канд. дисс, 1955.
26. А л е й н о в а Л. Н., Работы молодых специалистов ГИАП, изд. ГИАП, 1961.
27. Br aconnier F., Ind. chim. beige (XXXI Congress Intern, de Chimie indu-
striell, Liege), № 11, 167 (1958).
28. Цусими Г., Нихон кагаку дзасси, 63, № 2, 197 (I960),
29. R i g h i A., La Rivista del Combastibill, 13, № 7/8, 523 (1959).
30. Fischer F., Pichler H., Brennst. Chem., 13, №20, 281 (1932).
31. Ш е в ч у к В. У., Газ. пром., № 7 (1957).
32. Ш е в ч у к В. У., Газ. пром., № 8, 44 (1960).
33. Казарновский Я. С и др., Газ. пром., № 7, 19 (1960).
34. Шевчук В. У., канд. дисс, 1958.
35. Тес н ер П. А., Труды ВНИИГаз, вып. 6 (14), Гостоптехиздат, 1959.
36. Гриненко Б. С, Химия и технология топлива, № 10 (1956); Г р и -
н е н-к о Б. С, в сб. «Химическая переработка нефтяных углеводородов»,
Изд. АН СССР, 1956, стр. 106.
37. А н т о н о в В. Н., Производство ацетилена, Госхимиздат, 1959.
38. Зелизный А. М., Романюк И. М., Шевчук В. У., Хим. пром.,
№ 12 (1964).
39. Балыта И. М., Зелизный А. М., Романюк И. М.,
Шевчук В. У., Газ. пром., № 9, 36(1959).
40. Р о м а н ю к И. М., Балыта В. И., Б е р к о в и ч В. Б. и др., авт.
свид. СССР 126105.
213
41. Казарновский Я. С., Семенов В. П. и др., Хим. пром., № I,
11, (1961).
42. С е м е н о в В. П., Казарновский Я. С, Ивановский Ф. П.,
Новости нефтяной и газовой техники, Газовое дело, № 10, 42 (1961).
43. Sachsse H., Kosbahn Т., Lehrer А., пат. США 2715648, 16/VIII
1955 г.
44. Las 1 о A., Nemeth A., Erdol. u. Kohle, № 12, 105(1959); Mag. kern,
lap., № 10—12, 384 (1958).
45. Л ас л о А., Немет А., Газ. пром., № 10, 39 (1960).
46. Chem. Age, 83, № 2122, 459 (1960).
47. Petrol. Ref., 40, № 11, 210 (1961).
48. S i t t i n g M., Petrol. Ref., 41, № 3, 177 (1962).
49. Пат. США 2765358, 2/X 1956 г.
50. H у g h e s E., H о w 1 a n d E. et al., Chem. a. Ind., № 5, 189 (1963).
51. Chem. Age, 87, № 2237, 858 (1962).
52. Freund M., Nemeth A., Chem. Techn., № 4, 194 (1962).
53. В а с и л ь е в С. С, Мосин Б. Г., Труды ИГИ АН СССР, 16, Изд. АН
СССР, 1961, стр. 16.
54. Г л е б о в А. А., X о л и н Б. Г., Хим. маш., № 3, 4 (1964).
55. Z u n d e I M. I., Chim. et ind., 91, № 2, 99 (1964).
56. S с h m i d t A., Schmidt H., I a h n e n W., Erdol u. Kohle, 16, № 9,
940 (1963); Chem. Week, 94, № 9, 61 (1964); Chem. Eng., 71, № 6, 83 (19э4).
57. Волков А. Е., Лапидус А. С, Техника безопасности в производстве
ацетилена из природного газа, Изд. «Химия», 1964.
58. Dumbcen G., Neumayr F., Chem. Ing. Techn., № 20, 1004 (1958).
ГЛАВА
VI
МЕТОДЫ РАЗДЕЛЕНИЯ АЦЕТИЛ ЕНСОДЕРЖАЩИХ
ГАЗОВ И ВЫДЕЛЕНИЯ АЦЕТИЛЕНА
При производстве ацетилена из углеводородного сырья
различными методами содержание его в реакционных газах колеблется от 8
до 30 объемы. %. В этих газах кроме ацетилена содержатся также
водород, метан, двуокись углерода, этилен, окись углерода и в
небольших количествах имеются ацетиленовые и другие ненасыщенные
углеводороды. Поэтому очень важной представляется проблема
выделения и концентрирования ацетилена. Процессы выделения и
концентрирования пиролизного ацетилена достаточно полно
исследованы в СССР (Ф. П. Ивановский, Е. Р. Шендерей, С. П. Сергеев,
В. В. Дильман, И. Л. Лейтес, Г. Е. Брауде, С. Ф. Шахова, И. Г. Дрей-
цер, Н. А. Кочергин и др.) и за границей (Заксе, Бартоломе, Хассель-
ман, Холлеман, Фаузер и др.).
Большой интерес представляет возможность использования
разбавленного ацетилена. На первой установке фирмы BASF
получаемый разбавленный ацетилен использовали для производства ацетона7.
Аналогично работает установка в Бучумени (Румыния). Кроме того,
сообщалось о возможности получения акрилонитрила
непосредственно из газов окислительного пиролиза 8, а также об
использовании разбавленного ацетилена для синтеза винилхлорида и, что
более предпочтительно, винилацетата9. Однако в настоящее время
разбавленный ацетилен в промышленном масштабе используется
только для получения ацетона и винилхлорида.
Для разделения ацетиленсодержащих газов в производстве
ацетилена нашли применение различные методы: абсорбция водой,
селективными растворителями (диметилформамидом, N-метилпирроли-
доном и др.), низкотемпературными растворителями * (метанолом,
аммиаком, керосином, ацетоном и др.)» активированным углем
в стационарном или движущемся слое (гиперсорбция) и др.
Более чем на 70% всех промышленных установок используется
метод абсорбции ацетилена селективными растворителями. Второе
место принадлежит низкотемпературным растворителям, далее
следуют методы адсорбции ацетилена активированным углем, хотя
все они не вышли за рамки опытно-промышленных установок.
i- Низко температурными раствори 1сли называются потому, что процесс с
их использованием идет при минусовых температурах.
215
t-Q
I
0
3
o^
Я
i
vo
о
я
0)
s
сЗ
oa
О
i
X
Z
ci x
x +
о
i
6
и
Д
и
£
и
a?
о
«ч
н
о|
я§
д
и
я
Абсорбе
и
о
я
о
др.
"зГ
О)
я
а дет
0)
о
о"
S
о
о
т—1
о
о
СО
о4
см
о
о
я амид
)ро-
л я
Димети лфо
N-Метилп
со
Н
j2l
к
ная
§£
OCQ
о
о"
°\
со
о^о^
2 I
о '
^ I
о '
^ |
о !
ю
сч
о"
о
о
СО
о"
о о
<М СО^
СП СП
СП СП
\шиак
• оЗ
ЛИДОН . .
Керосин,
<
CQ
со
^ ^ ч
"l^"X
I
I
i
I
i
I
i
|
g
СП
СП
СП
Метанол
'с
?cati
"с
<
э
S
I
S
I
I
о
S
1
I
VO
бъемн.
о
СЗ
<D
Ч
Я
н
сЗ
М
сз
Сост
О
и
о
д
и
£
и "
д
и
| Я
^г^щ
U s о;
СЗ
^^
Д^ о 0)
£ч
д
и
н
\£
|
О.
с
о>
туча
>-»
о
Ю СО
ю
о" о* о"
,-н — О —
о" о" х о"
СЗ
о я
о" х
я
003
,01
е й м
эолее
,03
О О о <ц °
си к
СО S
о
О
О, О^
о с о
CD
о о £ Ч^
о ft °
СО О
ЦЯи go1
о о о-
со
ю
00 <М
"S
cn~cn^$7
СП СП СП С
СП
п
я . •
о.
о • •
«=:
X • •
Я * ' С,
-^ 7
Я ь-
я!; «
Я с? "«
ё£| |
IIS 1
СЗ О Й С .
Ч Ч § °
Я Я н л
Я Оч CD С)
С
S3
Q<
Й Я"
t,< >
<
Один из ранних методов — абсорбция ацетилена водой, примененная
на заводе фирмы Hiils (ФРГ), из-за сложности технологической схемы
и высокой себестоимости не получил дальнейшего распространения.
Для химических синтезов возможно применение ацетилена с
различным содержанием примесей. Состав ацетилена-концентрата
зависит от способа концентрирования: при использовании
низкотемпературных растворителей можно получить более чистый ацетилен.
В табл. VI-1 указан состав ацетилена-концентрата, полученного
различными способами6. Из приведенных данных видно, что при
абсорбции селективными растворителями (диметилформамидом и N-метил-
пирролидоном) ацетилен имеет примерно одинаковый состав; при
использовании аммиака и керосина удается полностью очистить
ацетилен от диацетилена и винилацетилена. Наиболее сложна очистка
от метилацетилена, который гораздо больше, чем диацетилен и винил-
ацетилен, растворяется в селективных растворителях (в диметил-
формамиде в 3,5 раза, в N-метилпирролидоне в 50 раз) и приближается
по этим свойствам к ацетилену. Тонкую очистку от метилацетилена,
если это требуется, можно проводить активированным углем.
Ацетилен указанного в табл. VI-1 состава можно использовать
в производстве акрилонитрила, синтетического каучука, ацетальде-
гида и др. Требования к ацетилену, используемому для некоторых
производств, приведены6 в табл. VI-2. Приведенные в таблице данные
получены на опытных и промышленных установках. Следует
отметить, что допустимое содержание примесей в ацетилене пока точно
не установлено.
ОБЩИЕ СВЕДЕНИЯ ОБ АБСОРБЦИОННЫХ ПРОЦЕССАХ
Растворимость газов в жидкостях зависит от свойств тех и
других, а также от давления, температуры и состава газовой смеси. При
некотором давлении и температуре мольные концентрации газа У и
X соответственно в газовой G и в жидкой L фазах находятся в
определенном соотношении, соответствующем равновесию между фазами:
У = ф (X)
Идеальные растворы подчиняются закону Рауля:
. Р = РКХ (VI-1)
где р — парциальное давление компонента над раствором;
рк — давление насыщенного пара этого компонента.
Это соотношение применимо к растворам газов, критическая
температура которых выше температуры раствора и которые поэтому
способны конденсироваться при температуре раствора. Если же
температура газа в данных условиях выше критической, то растворы
подчиняются закону Генри:
р = КХ (VI-2)
(здесь К — коэффициент Генри). Так как X — величина
безразмерная, то коэффициент К имеет размерность давления. Как правило,
217
коэффициент Генри с ростом температуры увеличивается, т. е.
растворимость газа уменьшается. Зависимость коэффициента Генри от
температуры может быть с некоторым приближением выражена
уравнением:
где Н — теплота растворения газа, ккал/кмоль;
R — газовая постоянная, равная 1,99 ккал!(кмоль-град);
С — постоянная, определяемая опытным путем.
Если состав газовой фазы выражен в мольных долях, формула
(VI-2) принимает вид:
Y = -j-X или Y = mX (VI-4)
где Р — общее давление;
т — константа фазового равновесия или коэффициент распределения (эта
величина безразмерная и не зависит от состава раствора); т = KIP.
В тех случаях, когда состав обеих фаз выражен в относительных
мольных соотношениях (кмоль/кмоль), можно привести условие
равновесия (VI-4) к следующему виду:
У __ тХ А/т гч
1-1- Y ~~ \+Х (VK>)
Отсюда
Y = — (VI-6)
Закон Генри применим лишь к разбавленным растворам, по-
скольку он точно соблюдается только для идеальных растворов,
а при сильном разбавлении любой раствор приближается к
идеальному и тем ближе, чем больше разбавление. Этот закон точно
характеризует равновесие для плохо растворимых газов, а для сравнительно
хорошо растворимых — только при низких концентрациях. При
более высоких концентрациях растворимость газов обычно ниже,
чем это должно быть по закону Генри1"3. Для растворов, сильно
отклоняющихся от закона Генри, можно пользоваться уравнением
(VI-4), принимая, что коэффициент т зависит от концентрации
растворенного газа.
При повышенных давлениях вследствие значительных отклонений
свойств реальных газов от идеальных вместо давления компонентов
пользуются их летучестью. Для практических целей летучесть
можно выражать через коэффициент активности а, связывающий
летучесть чистого газа /0 с давлением Р:
/о = аР (VI-7)
Коэффициент активности а обычно определяют по уже
составленным графикам3 в зависимости от приведенных температурыЭ и
давления л:
т р
i кр Лф
218
где Ткр и Ркр — критические температура и давление. Летучесть /
каждого компонента газовой смеси определяется из уравнения:
Летучесть /' компонента в жидкости при температуре Т и давлении
его насыщенного пара рк равна летучести насыщенного пара /п,
а при давлении Р определяется по формуле:
где v0 — мольный объем чистой жидкости, м3/кмоль;
R — газовая постоянная, равная 0,082 м3-ami (кмоль - град).
Мольный объем определяется по формуле:
где М — масса 1 моль компонента, г;
у — плотность компонента в жидком состоянии, г/см3.
При замене давления на летучесть закон Рауля и выражение
для константы фазового равновесия m будут иметь следующий вид:
"»=х = 7Г (VM2)
Закон Генрй выражается через летучесть следующим образом
/ - КХ (VI-13)
а константа фазового равновесия имеет вид:
/п = -£=-£- (VI-14)
А /о
При высоких давлениях вычисление по уравнению (VI-14) дает
заметные отклонения от экспериментальных данных даже при
небольших значениях X. И. Р. Кричевским и А. А. Ильинской
предложена для высоких давлений следующая формулировка закона Генри
Нгпх=г-о ~лт = К
где X — мольная доля растворенного вещества в жидкой фазе,
причем К является функцией температуры и давления. Для расчета
фазового равновесия в этих случаях можно пользоваться
уравнением Кричевского—Ильинской, записанным в следующем виде:
ln-£ = In /С + ^~Ро) (VI-15)
где v — парциальный мольный объем — увеличение объема жидкости при
растворении 1 кмоль газа, м31кмоль\
Ро — давление пара чистого растворителя, am.
219
Для расчетов систем с относительно небольшим давлением, где
возможно применение парциального давления, можно пользоваться
преобразованным уравнением Кричевского — Ильинской:
где р2 — парциальное давление газа; (5 — A/2,3Q3RT (А — коэффициент,
зависящий от природы растворителя); Х±— мольная доля растворителя в
жидкой фазе.
Растворимость газов в воде значительно понижается при наличии
электролитов и неэлектролитов, склонных к гидратации. Это
явление было изучено И. М. Сеченовым (1892 г.), установившим
следующее соотношение:
ln-^ = А'Х3 или X = е~А>х* (VI-17)
где /Со — коэффициент Генри для чистой воды;
К — коэффициент Генри для раствора;
А' — коэффициент, зависящий от природы добавочного компонента и
растворителя;
Х3 — концентрация добавочного компонента в растворе.
Для числового выражения растворимости пользуются
коэффициентом растворимости а, определяющим количество объемов газа,
растворимых в 1 объеме (или в 1 г) растворителя. Иногда эту
величину приводят к 760 мм рт. ст. и получают коэффициент
приведенной растворимости апр:
«nP = f 760
Селективные растворители характеризуются определенным
коэффициентом селективности
/Сс = -^- (VI-18)
(здесь ааи аь — растворимость соответственно первого и второго
компонентов в одинаковых условиях). В общем случае
селективность является сложной функцией, зависящей от состава
растворителя, давления, температуры и других факторов, влияющих на
скорость абсорбции. Поэтому, изменяя условия абсорбционного
процесса, можно изменять и селективность растворителя. При этом ее
удобнее выражать как соотношение коэффициентов Генри для
первого и второго компонентов:
По современным представлениям, передача вещества между
фазами определяется так называемой пленочной теорией абсорбции.
Предполагается, что с обеих сторон поверхности соприкосновения
фаз образуются неподвижные или ламинарно движущиеся пленки.
22Q
На границе обе фазы всегда находятся в равновесии; оно
устанавливается за очень малый отрезок времени, в течение которого
концентрация вещества меняется лишь в пленках, а не в основной массе
фазы. По достижении равновесия концентрация вещества в пленках
остается постоянной, а в основной массе фазы меняется за счет
молекулярной диффузии.
Различают два основных вида массопередачи:
между жидкостью и газом (паром) или между двумя несмешиваю-
щимися жидкостями;
между твердым телом и жидкостью, паром или газом (см. раздел
«Адсорбция ацетилена», стр. 271).
Перенос вещества из одной фазы в другую в первом случае можно
представить следующим образом. Концентрация распределяемого
вещества в фазе G выше равновесной, и вещество переходит из фазы G
в фазу L. В фазе G оно перемещается к поверхности раздела фаз,
а в фазе L — от этой поверхности. Перенос вещества в обеих фазах
осуществляется путем молекулярной диффузии (движущимися
частицами носителя и распределяемого вещества).
В каждой фазе различают две области: ядро (основная масса)
и пограничный слой (диффузионная пленка). Перенос
распределяемого вещества в ядре фазы осуществляется преимущественно путем
конвективной диффузии (диффузия в движущейся среде). Вследствие
интенсивного перемешивания в ядре концентрация распределяемого
вещества в любом' сечении системы почти постоянна. Перенос
вещества в пограничном слое осуществляется путем конвективной
и молекулярной диффузий, причем по мере приближения к
поверхности раздела фаз конвективные потоки ослабевают и возрастает
роль молекулярной диффузии.
В простейшем случае молекулярная диффузия в одном
направлении выражается уравнением:
CA = -DF -§-r (VI-20)
где бд — количество диффундирующего вещества, кмоль/ч;
D — коэффициент пропорциональности или коэффициент диффузии, м21ч\
F — поверхность раздела фаз, ж2;
dCldz — градиент концентрации, выражающий изменение концентрации С
(в кмоль/м3) на единицу длины г (в м) в единицу времени и
являющийся движущей силой процесса диффузии;
т — время, ч.
Знак минус в выражении (VI-20) показывает, что диффузия
происходит в направлении уменьшения концентрации. Значения
коэффициента D могут быть определены экспериментально и расчетным
путем.
При переносе вещества через поверхность раздела фаз количество
вещества Сд, переносимое в единицу времени из фазы, отдающей
вещество, к поверхности раздела (или от поверхности раздела в фазу,
воспринимающую это вещество), пропорционально поверхности F
221
и разности концентраций Сг—С2 распределяемого вещества у
поверхности раздела фаз:
Од = $'F (Сг — С2) (VI-21)
где |3' — коэффициент переноса массы через поверхностное
сопротивление (коэффициент поверхностного сопротивления).
Существует следующая зависимость между коэффициентом массо-
передачи Км и коэффициентами |3j и $'2 для двух фаз:
-^ - -4- + Л- (VI-22)
При контакте между газом и жидкостью величина 1/pj
определяет сопротивление газовой пленки, величина 1/$'2 —
сопротивление жидкостной пленки. Если коэффициент |3j велик, то член 1/|3{ мал
и коэффициент Км определяется сопротивлением жидкостной пленки
(плохо растворимые газы). При большом $'2 член l/ft'2 мал и
величина Км определяется сопротивлением газовой пленки (хорошо
растворимые газы).
В некоторый момент скорости перехода вещества из одной фазы
в другую станут одинаковыми и между фазами установится
равновесие, при котором явного переноса вещества не будет. В состоянии
равновесия существует определенная зависимость между
концентрациями распределяемого вещества в обеих фазах, выражаемая
уравнением (VI-4). Условие равновесия, выраженное этим уравнением,
позволяет определить направление процесса. Выражение указанной
зависимости в координатах Y—X называется линией равновесия.
Уравнение материального баланса между фазами G и L может
быть записано в следующем виде:
а (V V \ — Т (X X ) (V] 9Т*
Соотношение количеств растворителя в обеих фазах выражается
уравнением:
Величина I представляет собой удельное количество поглотителя
(в кг/кг). Если в произвольном сечении абсорбера составы фаз
соответственно равны Y и X, то уравнение материального баланса для
части аппарата, расположенной выше этого сечения, имеет вид:
GY + LX2 - GY2 + LX
Отсюда можно получить так называемое уравнение рабочей линии:
Y = Y2 + ±-(X-X2) = Y2 + l(X— X2) (VI-25)
Как следует из этого уравнения, на диаграмме Y—X рабочая
линия представляет собой прямую с наклоном к оси абсцисс под
углом, тангенс которого равен /.
222
При абсорбции содержание компонента всегда выше равновесного,
поэтому рабочая линия располагается над линией равновесия. При
подстановке в уравнение (VI-25) значения / после простейших
преобразований получим:
— Y2 _ Х — Х2
У± — У 2 Xi — Х2
(VI-26)
Расчет абсорбционных аппаратов состоит в определении
необходимой поверхности F соприкосновения фаз по уравнению:
G = ^.V^-Cs) (VI-27)
Средняя движущая сила Дср. определяется по формуле
Аср. = -^^Г (VI"28)
2,3 lg А-
^2
в которой Ах и А2 — движущие силы соответственно на входе и на
выходе из абсорбера. Среднелогарифмическое значение движущей
силы точно лишь в том случае, если линия равновесия и рабочая
линия являются прямыми и коэффициент массопередачи сохраняется
постоянным по всей длине аппарата.
Необходимая высота абсорбера Н в общем виде выражается
уравнением:
Н = Cl~C2 »^Д- (VI-29)
Аср. Ам^
где F — поверхность раздела фаз, равная срП (ср — поверхность раздела фаз в
единице объема абсорбера; П — площадь поперечного сечения аппарата).
Первый член в этом уравнении выражает изменение рабочих
концентраций на единицу движущей силы и называется числом единиц
переноса (одна единица переноса соответствует участку абсорбера,
на котором изменение рабочих концентраций равно средней
движущей силе на данном участке):
п = Cl~°2 (VI-30)
АСр.
Второй член представляет собой высоту участка,
соответствующего одной единице переноса, и называется высотой единицы
переноса:
н = -кЖ <VI"31)
Следовательно, рабочая высота Н равна произведению числа
единиц переноса п на высоту единицы переноса h. Высоту аппарата
определяют графическим методом.
Для расчета абсорбционных аппаратов можно использовать
метод теоретической тарелки и метод единиц переноса1'4.
Метод теоретической тарелки применяется преимущественно для
расчета тарельчатых аппаратов. Основным недостатком этого метода
223
является условие равновесия йа каждой тарелке; при этом не
принимается во внимание кинетика процесса массопередачи. Так как
на практике равновесие на тарелке не достигается, то для
определения действительного числа тарелок нужно разделить число
теоретических тарелок на так называемый к. п. д. тарелок, под которым
понимают отношение числа теоретических тарелок пт к практически
необходимому числу. Величина к. п. д. лежит в пределах 0,2—0,6.
За последнее время при расчете абсорбционных процессов
появилась тенденция рассматривать процесс массопередачи как с точки
зрения статики (определение констант равновесия и состава фаз),
так и с точки зрения кинетики (определение коэффициентов скорости
или коэффициентов массопередачи)1'5. Следует указать, что понятие
к. п. д. тарелок не учитывает кинетику процесса и не отражает тех
закономерностей, которые характеризуют работу тарелки, в
частности поля концентрации на тарелке.
С учетом движения жидкости по тарелке абсорбционные аппараты
подразделяют на аппараты полного вытеснения, полного смешения
и промежуточные. По этой классификации число реальных тарелок
зависит от интенсивности перемешивания и выбранного типа
аппарата. Поэтому при использовании метода теоретической тарелки для
определения к. п. д. целесообразно принимать экспериментальные
данные, относящиеся к определенному виду тарелки: к. п. д. зависит
от относительного направления движения газа и жидкости на
тарелках и вдоль абсорбера, характера массопередачи на тарелке
(отсутствие равновесия в практических условиях), степени уноса капель
жидкости с газом и от других факторов.
Число ступеней (или теоретических тарелок) пт можно определить
и аналитически, если линия равновесия прямая:
где Лф — абсорбционный фактор
аф = -kr
L — количество жидкости;
V — количество газа;
m — константа фазового равновесия;
(vi-34)
Метод единиц переноса (метод единичных объемов) сходен с
методом теоретической тарелки, но в первом случае абсорбционная
колонна разбивается на ряд элементов (единиц переноса) с последующим
определением их числа и эквивалентной высоты каждого. Оба метода
имеют тот недостаток, что при расчете нельзя получить в явном виде
зависимость необходимой поверхности абсорбции от заданной
степени извлечения.
Эффективность работы абсорбционных аппаратов характеризуется
коэффициентами извлечения. Полнота извлечения зависит не только
224
от размеров поверхности абсорбера, но и от условий равновесия.
В зависимости от удельного расхода поглотителя / равновесие при
бесконечно большой поверхности может быть достигнуто или на
стороне выхода газа, или на стороне входа. Поэтому максимальное
значение коэффициента извлечения определяется не только величиной
поверхности, но и достаточным удельным расходом поглотителя, при
котором равновесие достигается на стороне выхода газа.
Коэффициентом извлечения е называется отношение количества
поглощенного компонента к его содержанию в исходной газовой
смеси:
е = Fl~F2 (VI-35)
При полном извлечении компонента 8 = 1; практически е << 1.
Отношение количества поглощенного компонента к количеству
компонента, которое было бы извлечено при бесконечно большой
поверхности абсорбции (при определенном удельном расходе
поглотителя), называется коэффициентом извлечения Е:
^ YYlyY- (VI-36)
* 1 * 2 МИН
bF=o
Указанные уравнения применимы, если равновесная линия
прямая и справедливо соотношение (VI-4).
Относительным коэффициентом извлечения Ео называется
степень приближения коэффициента Е к состоянию равновесия.
Пользуясь введенными коэффициентами, можно установить зависимость
между степенью поглощения компонента и размерами абсорбера.
Относительный коэффициент извлечения Ео связан следующей
зависимостью с числом теоретических тарелок (Лф — абсорбционный
фактор):
п +1
Ео= Л%+г~Лф ' (VI-37)
А
Отсюда число теоретических тарелок равно:
Лф — Ео
J
-l (VI-38)
Приняв Лф — 1 и раскрыв неопределенность, получим число
теоретических тарелок
пт = EJ(l -Eo).
Зависимости Ео и Е от Лф и пт приведены в работе В. М. Рамма1.
Аналогичные формулы имеются и для расчета теоретических тарелок
методом единиц переноса.
В процессах выделения ацетилена практически происходит
абсорбция многокомпонентных систем, причем присутствие других
компонентов влияет на равновесие и на скорость диффузии каждого
компонента. Влияние других компонентов на скорость диффузии опре-
15 В. Н. Антонов 225
Делит .ложно, но оно обычно невелико, поэтому при практических
расчетах в большинстве случаев пренебрегают действием других
компонентов и применяют обычные уравнения абсорбции и равновесия
к каждому компоненту. При этом один из компонентов выбирают
в качестве ключевого, задаются для него коэффициентом извлечения
и определяют размеры аппарата, необходимые для достижения
заданной степени извлечения. Затем рассчитывают коэффициенты
извлечения для остальных компонентов и определяют состав уходящего
газа. Абсорбцию многокомпонентной системы можно рассчитывать
любым из описанных методов, при этом требуется определить
положение рабочей линии для всех компонентов (кроме ключевого)
путем подбора при заданном числе тарелок.
Наиболее прост расчет через абсорбционный фактор, который
можно подсчитать для всех компонентов, и с его помощью установить
коэффициенты извлечения. Затем по коэффициентам извлечения
определяют число теоретических тарелок в абсорбере (расчет процесса
выделения и концентрирования ацетилена методом абсорбционного
фактора см. в гл. VII, стр. 352).
Десорбция — процесс, противоположный абсорбции,
заключающийся в выделении из растворителя растворенного в нем газа.
Практически процесс десорбции сочетается с очисткой (отгонкой), в
результате чего получаются абсорбированный газ и растворитель
в чистом виде; последний можно направлять на повторное
использование. Существуют три основных вида десорбции: отгонка или от-
дувка в токе инертного газа, отгонка путем повышения температуры
и отгонка путем снижения давления или создания вакуума.
Уравнения массопередачи (VI-27) и (VI-28) остаются в силе и для
процесса десорбции. Однако при десорбции движущая сила, а
следовательно, количество десорбируемого компонента приобретают
отрицательные значения. Поэтому в левой части этих уравнений следует
ставить знак минус. Тогда движущая сила будет выражаться
величиной (А2 — Ai), и, следовательно, для процесса десорбции рабочая
линия будет всегда располагаться ниже линии равновесия.
Относительный коэффициент извлечения Ео (или степень от-
дувки) для процесса десорбции определяют по уравнению
ь° - х, - тх;
а число теоретических тарелок пт — по формуле:
где V — количество инертного газа;
L — количество растворителя;
Л о — фактор отдувки
(v — удельный расход инертного газа, равный V/L).
226
АБСОРБЦИЯ АЦЕТИЛЕНА ВОДОЙ
Ацетилен обладает значительной растворимостью в воде
(стр. 16). Это свойство было использовано для разработки
промышленного способа выделения ацетилена из газов электрокрекинга
на заводе фирмы Hills13-14.
Схема выделения ацетилена водой и его тонкой очистки от высших
ацетиленовых углеводородов заключается в следующем (рис. VI-1).
Газ предварительно очищают от сажи, бензола, нафталина, частично
от высших ацетиленов и сероводорода. Очищенную смесь нагнетают
Непоглощенные газы
[на разделение
0,5%-ный NaOH
H2SO4
т
Рис. VI-1. Схема выделения ацетилена водой:
/, 10 — компрессоры; 2, 12 — холодильники; 3 — абсорбционная башня; 4, 5, 6,
7 — сепараторы; 8 — дроссельные вентили; 9 — буферный сборни-к; // — насос на
20 ат\ 13, 14, 15 — скрубберы.
компрессором 1 под давлением около 20 am в абсорбционную башню 5,
орошаемую водой. Для растворения 1 кг ацетилена при 18° С
подают около 400 л воды. Непоглощенные газы (Н2, СН4, С2Н4, N2
и др.), содержащие менее 0,1% ацетилена, направляют на
разделение методом глубокого охлаждения. Из них выделяют
концентрированные водород и этилен, смесь метана и этана, а также смесь азота
с окисью углерода. Водород и этилен передают потребителям, а метан-
этановую фракцию возвращают на пиролиз.
Водный раствор ацетилена, вытекающий из нижней части
абсорбционной башни 5, разделяют путем последовательного
четырехступенчатого снижения давления. В первой ступени давление при
помощи дроссельного вентиля понижают до 2 am, в результате чего
из раствора выделяются ацетилен и другие растворенные в воде газы;
отделенный в сепараторе 4 первой ступени 45%-ный ацетилен
возвращают во всасывающий трубопровод компрессора / и вместе с
продуктами электрокрекинга вновь направляют на абсорбцию. Раствор
из сепаратора 4 первой ступени через дроссельный вентиль, в котором
15* 227
давление снижается до 1 am, поступает в сепаратор 5 второй ступени,
где выделяется 92%-ный ацетилен. В сепараторах 6 и 7 третьей и
четвертой ступеней при давлении соответственно 0,15 и 0,05 am
выделяется 94—96%-ный ацетилен. Из сепараторов 5, 6 и 7 ацетилен
(примерно 95%-ный) компрессором подают в три последовательно
установленных скруббера. В скруббере 13, орошаемом минеральным
маслом, и в скруббере 14, орошаемом серной кислотой, ацетилен
очищается от диацетилена и других ненасыщенных углеводородов
С3—С4. Двуокись углерода связывается 0,5%-ным раствором щелочи
в скруббере 15, из которого выходит 98%-ный ацетилен, содержащий
до 1 % СО2 и 1 % инертных газов.
В результате водной абсорбции удавалось получить 99%-ный
ацетилен с содержанием наиболее взрывоопасного компонента —
диацетилена не более 1 г/ж3. Способ выделения ацетилена водой оказался
экономически нецелесообразным и технически очень сложным и
дальнейшего промышленного применения не получил. Следует указать,
что на отдельных опытных установках этот способ применялся с
некоторыми изменениями, в частности высшие ацетиленовые
углеводороды улавливались селективными растворителями. Отдельные узлы
описанной схемы были использованы при разработке схем с метанолом.
РАСТВОРИМОСТЬ АЦЕТИЛЕНА
В РАЗЛИЧНЫХ ОРГАНИЧЕСКИХ РАСТВОРИТЕЛЯХ
Большая растворимость ацетилена во многих органических
растворителях объясняется тем, что молекула ацетилена образует комплекс
с молекулой растворителя (вероятнее всего, между атомом водорода
молекулы ацетилена и электроотрицательным атомом растворителя
возникает водородная связь). Из этого предположения вытекает, что
растворимость ацетилена должна быть больше, если водородная
связь прочнее и если растворитель неассоциирован, т. е. если в
молекуле растворителя нет других атомов, способных к образованию
водородной связи с электроотрицательным атомом.
Механизм растворимости ацетилена в различных растворителях
еще полностью невыяснен. Их условно можно разделить на
растворители с низким давлением паров при 0° С и 1 am (например, N-метил-
пирролидон, диметилформамид) и растворители с высоким давлением
паров (например, тетрагидрофуран, диоксан). Данные 10~12 о
растворимости ацетилена в различных растворителях приведены
в табл. VI-3 и графически на рис. VI-2, VI-3 и VI-4.
Данные10 об увеличении объема раствора вследствие растворения
ацетилена приведены в табл. VI-4. С повышением температуры объем
раствора также заметно увеличивается — практически в линейной
зависимости от температуры. Исключение составляет только раствор
ацетилена в диметилформамиде, для которого максимальное
увеличение объема отмечается в интервале 25—40° С.
Указанные на рисунках и в таблице растворители можно разбить
на две группы. К первой относятся N-метилпирролидон, диметилформ-
228
ТАБЛИЦА
Растворимость ацетилена в различных растворителях
V1-3
Растворитель
Температура, °С
Максимальное
парциальное
давление рс2н
мм рпг. ст.
Растворимость С2Н2
в 1 г
растворителя
мг
Растворители с низким давлением паров
N-Метилпирролидон *
Диметилформамид
Тетраметилмочевина
Диметилацетамид
у- Бути рол а ктон
Этиловый эфир ацетоуксусной кислоты
Бутанол
Этиленгликоль
15
25
40
15
25
40
15
25
40
25
25
25
25
25
275
438
457
376
598
341
530
730
720
679
730
734
877
624
Растворители с высоким давлением паров
Тетрагидрофуран
Этиловый эфир уксусной кислоты . .
Диоксан
Метанол
Аллиловый спирт
Дихлорэтан
15
25
15
25
25
25
583
594
609
587
778
770
65,0
41,6
27,2
50,6
37,7
24,7
37,0
27,9
20,4
41,9
14,9
12,2
6,3
3,5
25,3
19,3
16,6
16,4
7,3
5,6
* Указанные цифры относятся к условиям, при которых растворимость ацетилена
подчиняется закону Генри.
** Понижение парциального давления этих растворителей из-за растворения
ацетилена вычислялось, исходя из предположения, что действителен закон Рауля.
ТАБЛИЦА
Удельное увеличение объема при растворении ацетилена
(Рс2н2 = 760 мм рт. ст.}
VI-4
Растворитель
N-Метилпирро-
лидон
Диметилформ-
амид
о
°.
СЗ
Си
ь^
грат;
Темп«
, 15
25
40
15
15
25
40
СЗ
&§.
ю о
н в «*
ш
1,93
2,12
2,39
2,01
1,88
2,13
3,07
Растворитель
Диметилацетамид
у-Бутиролактон
Тетраметилмоче-
вина
и
_
о.
ерат;
с
S
<У
н
25
40
15
25
25
40
•
о £*
ag
н в ^
1ш
1,94
2,05
1,95
1,70
2,07
2,18
амид и диметилацетамид. Растворимость ацетилена в них
относительно высока и сильно понижается с повышением парциального
давления ацетилена, а при давлении от 15 до 20 am приближается
к предельному значению, особенно у N-метилпирролидона. Для
30
11
Р ю
р
0
Рис. VI-2. Растворимость
ацетилена при 25° С в диметилацет-
амиде ()),тетраметилмочевине (2)t
Y-бутиролактоне (З) и бутано-
ле (4).\
—у
/
/
гу*
200 Ш 600 800
мг/г
X
О
1
три
20
15
Ю
5
0
/
/v.
//
/
'2j/ i
/зУ
"5
200 № 600 800
Рис. VI-3. Растворимость
ацетилена при 25° С в тетрагидрофу-
ране(/), сольвеноне (2), диокса-
не (5), аллиловом спирте (4) и
дихлорэтане (5).
диметилформамида, по-видимому, следует ожидать повышения
коэффициента растворимости с увеличением давления. Растворители
второй группы — у-бутиролактон, эфиры уксусной и ацетоуксусной
кислот — обладают при низком давлении относительно малой
растворяющей способностью,
которая меньше зависит от
парциального давления ацетилена.
Из приведенных данных
видно, что в указанных на рис. VI-4
растворителях при нормальной
температуре и низких давлениях
(до 2—3 am) ацетилен
растворяется лучше, чем, например,
в ацетоне.
Однако с повышением
давления ацетилена растворимость его
уменьшается. Поэтому в
настоящее время ацетон является
лучшим растворителем ацетилена
в области высоких давлений
(15—25 am) и замена его,
например на N-метилпирролидон,
нецелесообразна. Наоборот,
применение селективных
растворителей для выделения
ацетилена из газовых смесей
рационально и технически обоснованно.
\
о
I
*5
20
.
10
о
0>^—
о
Ч|
-о——'
—о—
■о
и*"
«2^
5
^Г
о»
-"■"
0-
г ^т
о
10
15
.am
20
Рис. VI-4. Растворимость ацетилена при
25° С в диметилформамиде (/), диметил-
ацетамиде (2), N-метилпирролидоне (5),
этиловом эфире уксусной кислоты (4),
7-бутиролактоне (5) и этиловом эфире
ацетоуксусной кислоты (6).
230
АБСОРБЦИЯ АЦЕТИЛЕНА СЕЛЕКТИВНЫМИ РАСТВОРИТЕЛЯМИ
Существует большая группа органических растворителей,
обладающих значительной избирательной растворимостью
(селективностью) по отношению к ацетилену. Однако промышленное
применение для процессов концентрирования ацетилена могут найти лишь
те растворители, которые характеризуются следующими качествами:
высокой селективностью по отношению к ацетилену; достаточно
хорошей поглотительной способностью; малыми вязкостью и
плотностью; низкой температурой затвердевания и высокой температурой
кипения; стабильностью при высокой температуре и обводнении;
доступностью и дешевизной; малой токсичностью.
С учетом этих требований промышленное применение нашли диме-
тилформамид, у-бутиролактон и главным образом N-метилпирроли-
дон.
Выделение ацетилена диметилформамидом
Диметилформамид получается в промышленных масштабах при
взаимодействии диметиламина с метилформиатом:
NH(CH3)2 + HCOOCHg — HCON(CH3)2 + CH3OH
Полученный диметилформамид остается в жидкой фазе, а метанол
и другие побочные продукты — в газовой. Известен также метод
получения диметилформамида взаимодействием синильной кислоты
и диметиламина в водном растворе метанола при 20° С.
При нормальных условиях диметилформамид представляет собой
бесцветную жидкость. Выше 177° С он может разлагаться на диметил-
амин и окись углерода. Важнейшие константы диметилформамида
приведены ниже:
Относительная плотность d^ 0,95
Вязкость, спз:
при 20° С 0,93
» 90° С 0,453
» 150° С 0,311
Температура, СС
кипения (при 1 am) 153
затвердевания —61
вспышки 50—87
Показатель преломления rt^ 1,427
Поверхностное натяжение при 25° С, дин/см 35,2
Теплота парообразования, ккал/кг 188
Пределы взрываемости в смеси с воздухом,
объемн/% 4,93—13,6
На организм человека диметилформамид оказывает
общеядовитое и раздражающее действие, на кожу действует аналогично
бензолу. Для нервной системы наибольшую опасность представляет
ЗДыхание его паров. Предельно допустимая концентрация
диметилформамида 10 мг/м3.
231
При нормальной температуре диметилформамид не корродирует
сталь, латунь, медь, но выше 100° С и при обводнении (4—5% Н2О) он
гидролизуется с образованием муравьиной кислоты, которая
характеризуется значительной коррозионной активностью. В качестве
уплотнителей в аппаратуре могут применяться паронит, тефлон,
полиэтилен, асбест.
В промышленности диметилформамид широко используется15
при переработке синтетических смол, в производстве лаков, красок
и пигментов, для очистки газов от серы. В смеси с другими
растворителями диметилформамид является хорошим растворителем
синтетических и естественных смол, мягчителей и пр. (полиакрилонитрил,
поливинилхлорид, поливинилацетат, полиамиды, кумароновые
смолы, бутадиен-нитрильный каучук и др.).
Диметилформамид хорошо смешивается с эфирами, спиртами,
кетонами, ароматическими углеводородами; парафиновые
углеводороды растворяются в нем ограниченно. Легко дает комплексы с
фтористым бором, хлористым водородом, серным ангидридом и др.
Наибольшее сродство к диметилформамиду имеют синильная
кислота, хлористый водород, сероводород, ацетилен; наименьшее —
предельные углеводороды, олефины, окись углерода, азот.
Данные16 о равновесии между жидкостью и паром в системе
диметилформамид — вода приведены в табл. VI-5.
Растворимость некоторых газов в диметилформамиде при их
парциальном давлении 760 мм рт. ст. и 20° С приведена ниже
(в см3 на 100 г растворителя):
Диацетилен .... 30 100 Цианистый водород 103 000 *
Винилацетилен ... 29 700 Сернистый ангидрид 42 860 **
Метилацетилен ... 8 800 Хлор . 39 700 ***
Ацетилен 4 650 Сероводород .... 3 690 **
Бутадиен-1,3 .... 4650 Аммиак 3290**
Пропадиен 3 500 Двуокись углерода 520
Бутен-1 2 900 Кислород 11,4
я-Бутан 1 800 Окись углерода . . 11
Пропилен 884 Азот 6,0
Пропан 425 Водород 5,0
Этилен 230
ддртя„ 1O7 * При 500 мм рт. ст. и 25° С.
метан 16/ ** При 25° С.
-** При 0° С.
Ацетилен и высшие ацетиленовые углеводороды растворяются
в безводном диметилформамиде и в его водных растворах19""22.
Теплота растворения Д# этих газов и двуокиси углерода в безводном
диметилформамиде при 25° С составляет:
— ля
кал/моль
Ацетилен 4600—5500
и 6550 *
Метилацетилен 5700
Винилацетилен 6500
Диацетилен 7200
Двуокись углерода 3300—3890
* При —55° С.
232
ТАБЛИЦА VI-5
Равновесие между жидкостью и паром в системе диметилформамид — вода
Содержание диметилформ-
амида,
в жидкости
вес. %
в парах
При 200 мм рт
10,3
19,2
20,2
32,1
40,2
50,3
60,4
70,3
80,0
90,4
97,4
100
2,4
4,7
5,0
8,2
11,6
16,0
23,6
32,0
44,6
63,5
85,2
100
Температура
w txTTci и lt a
мл. rlllcri И. И
смеси, °С
. ст.
67,35
67,60
67,85
68,60
69,35
70,70
72,35
75,20
79,05
86,70
98,15
109,5
Содержание
амида,
в жидкости
циметилформ-
вес. %
в парах
При 760 мм рт
10,1
19,1
20,1
32,7
40,1
50,0
61,3
70,9
81,0
91,0
97,4
100
3,2
6,0
6,2
9,6
12,4
16,6
24,1
31,5
44,1
59,1
82,8
100
Температура
v \л гтр*ы ы а
гЧ. И Не о И л
смеси, °С
. ст.
100,50
100,90
100,90
101,80
102,65
104,00
106,0
108,95
113,65
123,50
138,20
153
Г. Е. Брауде, И. Л. Лейтес и И. В. Дедова19 получили данные
о растворимости двуокиси углерода, ацетилена, метил-, винил-
и диацетилена в диметилформамиде, содержащем до 15% (а в
отдельных случаях до 23%) воды, в широком интервале температур (25—
140° С) и парциальных давлений указанных углеводородов
(ацетилена — до 760 мм рт. ст., его гомологов и двуокиси углерода —
при 100—140 мм рт. ст.). Результаты опытов показали, что
растворимость ацетилена, двуокиси углерода, метилацетилена и винил-
ацетилена в указанном интервале температур и давлений подчиняется
закону Генри. Значения коэффициента Генри для этих газов
приведены в табл. VI-6.
Растворимость диацетилена подчиняется закону Генри лишь при
высоких температурах и большем содержании воды в растворителе
(т. е. когда мольная доля диацетилена в растворе не превышает 0,2).
Данные, полученные для диацетилена, хорошо согласуются с
преобразованным уравнением Кричевского — Ильинской (VI-16).
В табл. VI-7 приведены значения коэффициентов К и (3, входящих
в это уравнение, для системы диацетилен — диметилформамид —
вода.
На рис. VI-5 показана экспоненциальная зависимость
растворимости (в мольных долях) ацетиленовых углеводородов в
диметилформамиде от температуры. Как видно из рисунка, для метилацетилена
при высоких температурах указанная зависимость несколько
отклоняется от прямолинейной.
Растворимость ацетиленовых углеводородов и двуокиси углерода
зависит также от обводнения растворителя (рис. VI-6). Для
метилацетилена наблюдается прямолинейная зависимость растворимости
233
ТАБЛИЦА VI-С
Коэффициенты Генри для разных систем с диметилформамидом
Содержание
воды
вес. %
Коэффициент Генри /С-10 3, мм рт. ст.
при 25е С
при 40° С
при 60е С
при
80° С
при
100° С
при
120° С
Ацетилен — диметилформамид — вода
0,5
4,0
12,0
15,3
Me
0,0"
3,7
12,3
14,8
В и
0,0
3,7
12,3
22,9
Д в у о
0,03
3,7
8,0
12,3
15,0
7,7
9,9
17,5
21,3
11,6
14,3
25,5
29,0
тилацетилен — д
3,69
4,81
10,47
13,32
н и л а ц е т i
1,055
1,445
3,45
7,05
к и с ь у г л
47,0
60,2
87,2
117,5
136,0
5,86
7,70
15,90
20,1
1лен — д
1,78
2,41
5,14
9,62
е р о д а -
58,5
80,4
109,0
145,8
177,5
—
—
—
—
и м е т и л ф
9,47
13,11
23,5
29,8
и м е т и л ф
3,38
4,31
8,85
16,10
- д и м е т и
86,0
113,0
150,0
182,5
209,0
26,6
33,1
53,4
67,9
о р м а м и д -
14,5
20,1
35,1
39,4
о р м а м и д -
5,15
7,10
13,75
28,80
лформами
107,0
135,5
174,0
212,0
•248,0
37,4
44,4
68,1
79,6
— вод
21,55
26,6
42,7
53,0
— вод
8,40
11,90
20,60
35,60
45,8
68,0
—
—
а
25,9
30,8
—
—
а
10,95
16,60
—
д — в од а
—
—
—
—
—
—
—
—
Примечание. Мольные объемы v высших ацетиленовых углеводородов
рассчитаны по их критическим параметрам с помощью уравнения Ван-дер-Ваальса и составляют
(в л/моль): 22,04 — для метилацетилена; 21,9 — для винилацетилена; 21,9 — для диаце-
тилена.
ТАБЛИЦА VI-7
Коэффициенты /Сир для системы диацетилен—диметилформамид — вода
Коэффициент
При
25° С
При
40°. С
При
60° С
При
80° С
При
100° С
При
120° С
Без воды
К у мм рт. ст.
р
/С, мм рт. ст.
р
/С, мм рт. ст.
р
/С, мм рт. ст.
Р
0,55
1,30
2,24
0,61
4,30
0,60
С 3,7 в е с. % воды
1,20
0,98
2,70
0,75
8,10
0,46
3,50
0,60
С 12,3 вес. °/о воды
6,50
0,55
С 22,9 вес. % в о д ы
13,3
0
10,5
0
13,9
0
27,0
0
15,4
0
21,7
0
39,4
0
24,0
0
28,2
0
7,55
0
14,3
0
25,6
0
42,4
0
58,7
0
от мольного содержания воды в растворе (рис. VI-6, д). Для
остальных веществ эта зависимость экспоненциальная (графики VI-6, а—г
построены в полулогарифмических координатах), т. е. растворимость
to
1,5
. 2,0
V$
W^
26 28 30 32 34
2,5
/v
s<s
э^
p^
<si
^r
'и24 26 28
30 32
°K
0,5
2,0
Й
26 30 32
Рис. VI-5. Растворимость
метилацетилена (а),винил-
ацетилена (б) и диацети-
лена (в) в диметилформ-
амиде * при парциальном
давлении углеводорода
100 мм рт. ст.:
1 — без воды; 2 —с 3,7 вес.
% Н2О; 3— с 12,3 вес. % Н2О;
4 - с 14,8 вес. % Н2О;
5 —с 22,9 вес. % Н2О.
изменяется более резко. До Хн2о = 0,5 ее изменение
удовлетворительно описывается уравнением Сеченова (VI-17):
С понижением температуры растворимость ацетилена в диметил-
формамиде повышается. В табл. VI-8 приведены данные Е. Р. Шенде-
рея и Ф. П. Ивановского23 по растворимости ацетилена в диметил-
ТА БЛИЦА \ 1-8
ое да-
етиле-
о
<и
и
о
X
СО
О,
\ъ
<и
м х
100
300
500
760
S
сх
5*
35
Растворимость ацетилена
При
25° С
8
4,65
13,35
21,40
31,30
X
и
><
0,015
0,042
0,036
0,093
При
0°С
8
10,35
28,70
44,75
64,20
X
и
0,033
0,086
0,128
0,174
При
—10° С
8
15,32
41,20
62,60
88,10
X
и
0,048
0,119
0,170
0,222
8
23,
57,
83,
Ш,
В
п
_2
10
30
50
60
д и мет ил формам и де
ри
0°С
X
и
0,074
0,158
0,217
0,275
При
—30° С
8
34
80
111
157
,70
,70
,70
,00
X
и
0,102
0,209
0,277
0,341
8
53
106
160
209
При
—40° С
70
70
50
00
X
и
н
0,148
0,272
0,345
0,407
с
93
183
252
336
При
—55°С
5
,00
,50
,50
,00
X
V
0,233
0,374
0,453
0,525
Примечание. В таблице a — растворимость ацетилена, см*/г\
Доля ацетилена в растворе.
~ м°льная
235
-W
5> -ZO
(-^-—
^—.
"°*^ч
"° ,
-°-—.
■^22r*^°^
§0
■ZSg;
—*°"H
o.
X)
0 0,1 0,2 0,3 0,4 0,5
^HoO > мольные доли
a
"О 0,1 0,2 0,3 0,4 0,5
xHi o, мольные доли
-0,5г
-3,0
0,030\
0,1 0,2 0,3 0,4 0,5
^HoO» мольные доли
0,2 0,3 0,4 0,5 0,6
2о> мольные доли
г
0,1 0,2 0,3 0,4 0,5
), мольные доли
д
Рис. VI-6. Изотермы растворимости ацетилена.,
его гомологов и двуокиси углерода в диметил-
формамиде при разном содержании воды:
а — при PCq2 ^380 мм рт. ст.\ б — при
Рс2н2 = 760 *MM Рт- ст-'> в "" ПРИ Рс4н4 ^
= 100 мм рт. ст.; г — при Рс4н2" ^ мм Рт- ст"»
д — при рСзн = Ю0 мм рт. ст.
0,1
0,2 0,3 0,4 0,5 0,6
мольные доли
с2н2
Рис. VI-7. Изотермы растворимости
ацетилена в диметилформамиде:
/ — при 25° С; 2 — при 0° С; 3 — при
— 10° С; 4— при —20° С; 5—при —30° С;
6 —при —40° С; 7 — при —55° С.
236
1750
§ 1500
%1250
V юоо
750,
'25 0 -25 -50 -75
Температурйу °С
Рис. VI-8. Зависимость
коэффициента А" от температуры
для системы ацетилен—диме-
тилформамид.
формамиде при минусовых температурах, на рис. VI-7 показаны
изотермы растворимости ацетилена в диметилформамиде. При
пониженных температурах растворимость ацетилена в
диметилформамиде не подчиняется закону Генри. Коэффициент растворимости апр.
уменьшается по мере роста равновесного давления ацетилена, а
фактическая растворимость Хф превышает идеальную растворимость Хи,
равную:
Хи = If- (VI-42)
где /р — летучесть при равновесном давлении;
/—летучесть чистого газа при данной температуре.
Растворимость ацетилена в интервале концентраций от 0,015 до
0,52 (в мольных долях) можно определять по уравнению:
A"lRT (l~x$
Рс,н, = KXe
(VI-43)
где Х2 — мольная доля растворителя в растворе;
К и Л" — коэффициенты, значения которых даны в табл. VI-9 и на рис. VI-8.
В пределах температур от +25 до —50° С коэффициент Генри
при различном содержании воды в растворе можно вычислять по
уравнению Сеченова (VI-17) или по следующему уравнению23:
lg/(-lg/Co-4,44X3 (VI-44)
При выделении ацетилена диметилформамидом очень важно
создать наиболее благоприятные условия для разделения пары
С2Н2 — СО?, потому что двуокись углерода также растворяется
в этом абсорбенте. Значения коэффициента селективности Кс для
ТАБЛИЦА VI-9
Коэффициент Генри для систем ацетилена и диметилформамида или его водных
растворов при пониженных температурах
Содержание воды
вес. %
0,0
2,0
5,0
10,0
при
25° С
6,39
Коэффициент 1
при
0°С
2,75
при
—10° С
1,77
2,15
2,73
4,54
"енри /С-Ю 3, мм рт.
при
-20° С
1,15
1,38
1,69
2,97
при
—30° С
0,71
0,82
1,03
1,87
ст.
при
—40° С
0,40
0,47
0,59
1,09
при
—50° С
0,16
пары С2Н2 — СО2 при растворении в диметилформамиде при
различных температуре и парциальном давлении ацетилена приведены
в табл. VI-10.
Из данных таблицы видно, что при снижении температуры
селективность повышается только при давлении 100 мм рт. ст.; при более
высоких давлениях селективность вначале растет, а затем снижается.
237
ТАБЛИЦА VI-10
Коэффициент селективности Кс для пары С2Н2 — С02 при растворении
в диметилформамиде
Равновесное
давление Рс2Н2
мм рт. ст.
100'
300
500
760
Коэффициент Кс
при +25° С
8,45
8,05
7,75
7,35
при —20° С
12,30
10,00
9,05
7,83
при —40° С
14,50
9,40
8,30
6,80
при —55° С
14,05
8,80
6,97
5,69
•Следовательно, концентрирование ацетилена целесообразно
проводить при небольших парциальных давлениях. Однако отсутствие
данных о растворимости в диметилформамиде других газовых
компонентов (С2Н4, Н2) не позволяет однозначно определить характер
изменения селективности растворителя при абсорбции газовой смеси
сложного состава.
Выделение ацетилена N-метилпирролидоном
N-Метилпирролидон получают из у-бутиролактона и метиламина:
их- сн2 н2с—сн2
г\ I + CH3NH2 >- I I
Н2С СО Н2СЧ /СО
О N —СН3
При нормальных условиях он представляет собой бесцветную
горючую жидкость со слабым специфическим запахом. Важнейшие
константы N-метилпирролидона приведены ниже:
Относительная плотность, df* 1,03
Вязкость при 20° С, спз 1,7—1,85
Температура, °С
кипения
при 1 мм рт. cm 60—65
» 12 мм рт. cm 71—72
» 24 мм рт. cm 105—ПО
» 760 мм рт. cm 200—202
плавления —24
вспышки 85—95,6
самовоспламенения 348
Давление паров, мм рт. ст.
при 15° С 5
» 25° С 6,5
» 40° С 8,2
Теплота парообразования, ккал/кг 118
При высоких концентрациях N-метилпирролидон действует
раздражающе на кожу и слизистую оболочку. Предельно допустимая
концентрация аэрозоля 100 мг/м3.
238
N-Метилпирролидон коррозионноактивен, так как при большой
степени обводнения (50% Н2О) и температуре выше 50° С разлагается
с образованием муравьиной кислоты.
Он полностью смешивается с водой и с большинством
органических растворителей, применяется в качестве растворителя для поли-
акрилонитрила, поливинилхлорида, полиакрилатов, найлона и
ароматических полиамидов17. Его применяют также как растворитель
при перекристаллизации терефталевой кислоты и для получения
прядильного раствора синтетических волокон.
N-Метилпирролидон обладает свойствами слабого основания.
В нейтральном растворе он очень стабилен, однако может гидроли-
зоваться 4%-ным водным раствором едкого натра. С добавлением
небольшого количества воды оказывается селективным
растворителем ароматических углеводородов. Если, например, обработать смесь
углеводородов, содержащих 25% ароматических соединений,
раствором N-метилпирролидона с 5% воды, то отделяется фаза,
содержащая почти все ароматические углеводороды. N-Метилпирролидон
является эффективным растворителем для ртутных
дезинфицирующих веществ и таких инсектицидов, как ДДТ, гексахлоран и др. Он
обладает высокой смачивающей и диспергирующей способностью,
поэтому его вводят в состав органических и неорганических
пигментов для повышения стойкости красителей.
Данные о растворимости некоторых газов в N-метилпирролидоне
при 1 am и 20° С представлены ниже (в см3/см3):
Диацетилен 4870 Сероокись углерода . 11,4
Винилацетилен 298,5 Двуокись углерода . 3,95
Метил ацетилен 86,0 Кислород 0,055
Ацетилен . . 38,7 Окись углерода . . . 0,055
Пропадиен . . 30,4 Водород 0,050
Пропилен . . . 8,62 Азот 0,050
Этилен . . . . 2,24
Теплота растворения ацетилена в N-метилпирролидоне составляет
— (5000-f-7000) кал/моль в зависимости от содержания воды. Система
ацетилен — N-метилпирролидон не подчиняется закону Генри 24:
коэффициент Генри при любой температуре значительно меньше
летучести чистого ацетилена (наблюдаются отрицательные
отклонения от законов для идеальных растворов). Наоборот, растворимость
двуокиси углерода и этилена в N-метилпирролидоне и его водных
растворах подчиняется закону Генри. Данные по растворимости
ацетилена в N-метилпирролидоне и его водных растворах приведены
в табл. VI-11.
Изменение растворимости ацетилена и двуокиси углерода в
водных растворах N-метилпирролидона в зависимости от давления
подчиняется уравнению
ГДе Р =
X — мольная доля газа в растворе,
239
Os
тел
ex
о
m
CTl
сх
<v>
а
з$
<j
03
X
о
fc3
я
н
аце
н
о
о
сх
о
ю
н
и
КЗ
а,
ьное
аце- |
циал
ение
Пар
давл
и
сх
с
и
?о
S
сх
и
о
ю
а
сх
с
С
LO
СМ
а
сх
с
и
ю
при
и
о
а
сх
с
и
о
ю
а
сх
с
и
о
о
а
сх
с
£g
«Ё
н
00 СО"Ь- Tt*
о" —<" с^Г со"
—< CM h- 00
О5 Is* Ю 00
мм
CD CM Tt- CO^
—1 СМ СО
ююою
LO О О LO
CM CD СО СМ
00 LO О LO
~* lo" сгГ со
см о о о
^ с-- со t-.
-н Ю О О
LO —• СМ СО
с;
о
S
о"
и
МОЛ
э^: м
—< 00 О О
ОО^Г^СО^ОО^
юхлхпю
»—1 СМ СО
МОЛ
и
I I I
мм
^юоо
t^. CO >—• 00
со" о" h-T ^"
*-* —4 СМ
Ь- О — LO
• СО СО t"- rf
5§
*о
2о
ао
0)0
£Г—
>> I
00 ОО О
t>- ^н Tf 00
00
CM
LO CM LO
CD LO CO
Ю^СОСО
~ CM CO
LO ОО О
СО^^СМ^О^
см"—Too'lo"
—• СО ^f CO
LO О О О
<J5 *-н СО С^
OLO О О
LO СО "*• СО
. о»
за
СО О- t>- 00
^ СМ СО
о о о
О О О С
-ч СО LO t
о ооо
О О О СО
1—I CO LO «^
о о о о
о о о со
—* СО LO О
ТАБЛИЦА VI-12
Коэффициенты Генри для различных систем с N-метилпирролидоном
Содержание
воды
мол. %
Коэффициент Генри /С-10 3, мм рт. ст.
при
0°С
при
5° С
при
10° С
при
15° С
при
25° С
при
35° С
при
45° С
[А ц е т и лен — N-м етилпирролидон — вода
0,0 1,59 1,99 2,42 3,06 4,37 6,17 8,64
10,4
24,1
37,8
55,7
Двуокись углерода — N-м етилпирролидон — вода
1,59
2,24
3,02
4,90
16,60
1,99
2,59
3,57
5,93
21,40
2,42
3,16
4,60
7,29
25,20
3,06
3,83
5,32
8,56
30,22
4,37
—
—
6,17
—
—
0,0
10,1
21,6
35,6
26,65
—
—
—
—
—
—
32,20
42,60
85,58
36,60
47,80
65,00
96,75
42,80
—
—
54,55
—
—
61,9
Значения коэффициента Генри К для рассматриваемых систем,
вычисленные по этому уравнению, представлены в табл. VI-12.
Величину коэффициента А можно рассчитать по эмпирическому
уравнению
А = 238 000 — 5927 (VI-45)
В табл. VI-13 приведены значения этого коэффициента,
определенные опытным путем.
ТАБЛИЦА VI-13
Коэффициент А для системы ацетилен — N-метилпирролидон — вода
Содержание
воды
мол %
0,0
10,4
24,1
37,8
55,7
п
0
75
77
82
82
79
ри
ЭС
000
500
500
500
000
при
5° С
73 500
78 500
84 000
72 000
74 000
Коэффициент A, am
при
10° С
72 000
80 000
78 500
74 000
74 000
при
15° С
67 000
76 000
76 000
76 000
75 000
•см3/моль
при
25° С
61 823
—
—
—
при
35° С
56 144
—
—
—
—
при
45° С
49 976
—
—
—
—
На рис. VI-9 представлены кривые изменения коэффициента
Генри для системы ацетилен — N-метилпирролидон при разном
содержании воды и различных температурах. Из этих данных
следует, что с ростом температуры растворимость ацетилена меняется
заметнее, чем при увеличении степени обводнения N-метилпирроли-
дона. Поэтому представляют интерес приведенные на рис.УЫО
16 в. Н. Антонов 241
3,8
изотермы растворимости ацетилена при его различных парциальных
давлениях. Видно, что увеличение растворимости с температурой тем
заметнее, чем выше равновесное давление ацетилена над раствором.
Изменение растворимости ацетилена в
зависимости от его парциального давления
необходимо знать для выбора оптимального
давления в процессе выделения ацетилена.
Из рис. VI-11 видно, что коэффициент
растворимости апр. для системы
ацетилен—N-метилпирролидон возрастает с понижением
парциального давления ацетилена. Так,
например, при 0° С и 100 мм рт. ст. апр# = 96,2сж3/г,
а при той же температуре и 760 мм рт. ст.
этот коэффициент составляет только 65 см3/г.
Поэтому при промышленной абсорбции
ацетилена N-метилпирролидоном с повышением
давления в 7,6 раза абсорбционная емкость
растворителя возрастает всего в 5 раз.
Изменение содержания воды в
N-метилпирролидоне также сказывается на коэффициенте
3,2,
%
Y
Ys
N
W
V
'3,1 3,3
г,5
Рис. VI-9. Коэффициент
Генри для систем
ацетилен —N-метилпирролидон
при разных температурах:
J —без воды; 2-е 10,4 мол. %
Н2О; 3— с 24,1 мол. % Н2О;
4 — с 37,8 мол. % Н2О.
апр.. Если при 0° С и увеличении
равновесного давления от 100 до 760 мм рт. ст. в
чистом растворителе величина апр# уменьшается, как уже говорилось,
с 96,2 до 65 см3/гу то в системе с 20 вес. % воды апр. снижается с 23,2
до 22 см3/г. 8СЮ
800
^ 600
I
| 400
Ъ 200
ч
и 0,05 0,10 0,15 0,20 0,25
^с2н2> мольные доли
Рис. VI-10. Изотермы растворимости
ацетилена в N-метилпирролидоне при разном
давлении ацетилена.
45'
*
0
^0^35/
U 4
щ
f 1
25/ 15ч
У Г
А//
<у
У5/
Р г/
{/
¥
Рис. VI-11. Изотермы коэффициента
растворимости апр. для системы
ацетилен — N-метилпирролидон при
разном давлении ацетилена.
Для количественной оценки зависимости растворимости
различных газов в N-метилпирролидоне от содержания третьего компонента
(в данном случае — воды) можно пользоваться уравнением Сеченова
(VI-17):
4
или Х =
242
Экспериментальные значения коэффициента А\ входящего в это
уравнение, приведены в табл. VI-14.
ТАБЛИЦА VI-14
Предельное
содержание воды, до
которого справедливо
значение А'
мол. %
Коэффициент
А в уравнении
Сеченова
Коэффициент А'
при 0° С
при 5° С
при 10° С
при 15° С
Ацетилен — N-м етилпирролидон — вода
30 | 1,25 | 1,13 | 1,20 | 1,10
Двуокись углерода— N-м етилпирролидон — вода
40 1,25 1,25 1,25 1,25
Растворимость ацетилена в водном N-метилпирролидоне
описывается уравнением Сеченова только до концентрации воды 20 мол. %,
а при большем ее количестве растворимость во всех случаях выше,
чем следует по этому уравнению.
Растворимость двуокиси углерода в N-метилпирролидоне также
изменяется с температурой, давлением и содержанием воды в
растворителе. Эту зависимость можно описать эмпирическим уравнением
Е. Р. Шендерея24:
lga = ^-
fср.
(VI-46)
где a — растворимость;
Х3 — мольная доля воды;
Мср. — средний молекулярный вес растворителя; Мср. = M1XS -f- M2X2
(М1 и М2 — молекулярные веса воды и N-метилпирролидона).
С уменьшением температуры растворимость двуокиси углерода
так же, как и ацетилена, заметно возрастает (рис. VI-12). Поэтому
особенно важно исследовать условия изменения селективности
растворителя по отношению к ацетилену. Лабораторные исследования 24
показывают, что селективность N-метилпирролидона по отношению
к ацетилену для пары С2Н2 — СО2 возрастает с понижением
температуры и с уменьшением давления (табл. VI-15). Селективность
увеличивается, кроме того, с увеличением обводненности
растворителя.
Высшие ацетиленовые углеводороды растворяются в
N-метилпирролидоне и его водных растворах в разной степени (табл. VI-16
и рис. VI-13). Теплота растворения —ЛЯ составляет (в кал/моль):
Метилацетилен .... 4800
Винилацетилен 6700
Диацетилен 9900
16*
243
ТАБЛИЦА VI-15
Селективность N-метилпирролидона и его водных растворов по отношению к ацетилену
(для пары С2Н2 — СО2)
Содержание воды
мол. %
0—10
10—20
20—40
40—60
при
и
h
Я
о.
с
9,85
10,05
11,50
11,50
760 мм рт.
и
о
к
Си
К
9,10
9,40
10,50
10,60
Коэффициент
ст.
и
ю
я
о,
К
8,85
9,40
10,50
10,50
селективности Кс
i
а.
я^
&2
11,80
12,50
13,45
12,00
я
а
с
11
11
12
11
при 10°
к
а*
00
00
25
50
с
я
а,
с
ю,
ю,
11,
11,
§
5.
Si
3
00
03
55
00
S
1
Si
Я^
g-8
9,10
9,40
10,70
10,60
ТАБЛИЦА VI-16
Растворимость ацетиленовых углеводородов в N-метилпирролидоне
При 25° С
Р X
При 40° С
Р X
При 60° С
Р X
При 90° С
Р X
При 120° С
Р X
100
200
300
400
500
100
200
300
400
500
600
10
20
30
40
50
0,032
0,062
0,091
0,117
0,142
0,145
0,254
0,339
0,410
0,476
0,536
0,167
0,222
0,264
0,296
0,326
100
200
300
400
500
100
200
300
400
500
600
50
100
150
200
250
Метилацетилен
0,019
0,038
0,057
0,075
0,094
100
200
300
400
500
Винил ацетилен
0,075
0,136
0,193
0,244
0,290
0,330
100
200
300
400
500
600
0,040
0,078
0,114
0,145
0,175
0,202
Диацетилен
0,212
0,295
0,364
0,387
0,416
200
200
300
400
0,207
0,299
0,363
0,408
100
200
300
400
500
600
25
50
75
100
125
0,006
0,013
0,019
0,025
0,032
0,018
0,036
0,054
0,072
0,090
0,106
0,031
0,058
0,084
0,107
0,130
100
200
300
400
500
100
200
300
400
500
600
25
50
75
100
150
0,004
0,008
0,012
0,016
0,020
0,010
0,020
0,030
0,040
0,050
0,060
0,021
0,025
0,037
0,050
0,075
Примечания. 1. В таблице р — парциальное давление углеводорода, мм рт. ст.;
X — мольная доля углеводорода в жидкой фазе.
2. Для винилацетилена и диацетилена данные приведены не при 25° С, а при 20° С.
При исследовании25 растворимости метил-, винил- и диацетилена
в N-метилпирролидоне и его водных растворах (до 4,5 мол. % воды)
в интервале температур 20—120° С было установлено, что растворы
подчиняются закону Генри только до мольной доли углеводорода
в растворе 0,1. При больших концентрациях растворимость
описывается уравнением Кричевского — Ильинской (VI-16). Изменение
800
Xя
I 400
U200
4S°L
л
/72
р/
к
у*
0,005 0,010 0,0/5 0,020 0,025 0,030
ХС02, мольные доли
Рис. VI-12. Изотермы растворимости
двуокиси углерода в
N-метилпирролидоне при разном давлении двуокиси
углерода.
150
!
\т
% 50
I
7гу
* •—'
f
U 5
62Х-
100
200 300
Ру/*м рт. cm
400
Рис. VI-13. Изотермы растворимости
диацетилена (У, 2, 3) и винилацетилена (4У 5)
в N-метилпирролидоне при разном
давлении углеводорода.
растворимости ацетиленовых углеводородов в зависимости от
содержания воды в N-метилпирролидоне подчиняется уравнению
Сеченова (VI-17). Значения коэффициента Генри для исследованных
систем приведены в табл. VI-17.
ТАБЛИЦА VI-17
Коэффициент
Температура
°с
Генри
Iff К
0
К для систем ацетиленовых
с N-метилпирролидоном
при разном содержании воды (в
0,05 | 0,10
углеводородов
мольных долях)
0,15
0,20
20
40
90
120
20
40
90
20
40
90
120
Метилацетилен
3,50
3,70
4,20
4,40
2,70
3,10
3,70
1,40
1,90
2,90
3,20
3,58
3,75
4,25
4,45
3,60
3,80
4,30
4,49
Винилацетилен
2,72
3,20
3,75
2,75
3,25
3,80
Диацетилен
1,45 1,50
1,95 2,00
3,00 3,05
3,25
3,28
3,70
3,85
4,35
4,50
2,80
3,30
3,85
1,70
2,10
3,10
3,30
3,75
3,90
4,40
4,52
2,95
3,38
3,90
1,80
2,15
3,20
3,35
245
Выделение ацетилена у-бутиролактоном
у-Бутиролактон синтезируют путем неполного окисления бутан-
диола-1,4 на медном катализаторе при 200—250° С. По Реппе
механизм реакции следующий:
СНр СНо — СНр СНп +Оо
I I —2->
ОН ОН "Н2°
бутандиол-1,4
сн2—сн2-сн2—со
ОН ОН
н^сн,
у-бутиролакгон
В промышленных условиях его получают из ацетилена и
формальдегида. у-Бутиролактон образуется также в качестве
промежуточного продукта при получении N-метилпирролидона.
Важнейшие физико-химические константы у-бутиролактона:
Относительная плотность d^° 1,12
Вязкость, спз 1,7
Температура, °С
кипения 203—204
плавления От —41
до —44
вспышки 98,3
Показатель преломления п2^ 1,4366
у-Бутиролактон полностью смешивается с водой, метанолом,
ацетоном, четыреххлористым углеродом, диэтиловым эфиром и др. Он,
как и N-метилпирролидон, применяется в качестве растворителя для
поливинилхлорида, полиэфиров и др.18 у-Бутиролактон химически
и термически стабилен и, что главное, в малой степени токсичен.
Он является полупродуктом для синтеза гербицидов,
пластификаторов и фармацевтических препаратов, а также применяется как
растворитель ацетилен.
О растворимости ацетилена в у-бутиролактоне можно судить
по данным табл. 1-8 (стр. 16) и рис. VI-2 и VI-4 (стр. 230).
Схемы установок концентрирования
с селективными растворителями
В схемах с селективными растворителями, в которых возможно
применение диметилформамида, N-метилпирролидона или у-бутиро-
лактона, выделение ацетилена обычно осуществляется при
повышенном давлении и плюсовых температурах. При создании установок
246
особенно важно определить оптимальное давление процесса. Как
видно из данных рис. VI-4 (стр. 230), растворимость ацетилена почти
во всех селективных растворителях с повышением его
парциального давления более 2 am понижается. Отсюда ясно, что повышение
парциального давления сверх этой величины нецелесообразно. Кроме
того, следует учитывать и допустимые пределы сжатия ацетилен-
содержащих газов (стр. 339).
Сравним расходные и энергетические показатели процесса выделения
ацетилена, например, диметилформамидом при 10 и 5 am. При 10 am, 25° С и степени
обводнения растворителя 3% (на стадии абсорбции) относительный расход его
составляет 4,75 л на 1 ж3 газа. Необходимое количество циркулирующего раствора (на
1000 м3 газа в час) составит
1000-4,75= 4750 л/м*
При 5 am и прочих равных условиях растворимость ацетилена в диметилформ-
амиде возрастет в 2,5 раза и расход растворителя соответственно повысится до
4,75-2,5= 11,9 л/м3
Этому отвечает необходимое количество циркулирующего раствора (на 1000 м3
газа в час), равное:
1000-11,9= 11 900 л/ж3.
Расчетные энергетические затраты на 1 т С2Н2 на стадии концентрирования
представлены ниже (в кг условного топлива):
При 10 am При 5 am
Расход пара на подогрев растворителя 230 400
Расход электроэнергии
на сжатие газов пиролиза . . . 660 210
на перекачивание растворителя 140 350
Итого . . . . 1030 960
Энергетические затраты на процесс абсорбции при 5 am несколько ниже, чем
при 10 am. Однако при 5 am возрастают потери растворителя с отходящими газами,
пропорциональные давлению при одной и той же степени улавливания растворителя.
Кроме того, при 5 am значительно повышаются капиталовложения в связи с
увеличением габаритов оборудования на стадии абсорбции. Указанные факторы
нивелируют разницу в энергетических затратах. Поэтому в промышленности применяют
давление около 10 am. Более высокое давление для селективных растворителей не
используется.
Схемы концентрирования ацетилена селективными
растворителями обычно строятся поагрегатно. Агрегат включает следующие
основные аппараты: абсорбер, десорбер I ступени и десорбер II
ступени со вспомогательной колонной, а также теплообменной и другой
аппаратурой.
Газы пиролиза, сжатые до 9—10 am, поступают в цех
концентрирования, где путем избирательной абсорбции разделяются на три
фракции. Первая фракция (синтез-газ) содержит газы, мало
растворимые в селективном растворителе (водород, окись и двуокись
углерода, метан, азот). Вторая фракция (товарный ацетилен) состоит
в основном из ацетилена с небольшими примесями двуокиси
углерода, пропадиена, метилацетилена, диацетилена и ароматических
247
углеводородов. И, наконец, третьей фракцией (гомологи ацетилена)
является смесь метил-, ди-, винилацетилена и др. с примесью
ацетилена.
На рис. VI-14 показана схема выделения* ацетилена диметилформ-
амидом или N-метилпирролидоном при 36—46° С и 10 am. Смесь
газа пиролиза и возвратного газа поступает в нижнюю часть
абсорбера 2, а селективный растворитель (рабочий раствор, содержащий
2% воды и менее 1% полимеризатов), из расходного бака 3 подается
насосом 4 в верхнюю часть абсорбера 2. Растворитель абсорбирует
углеводороды в количествах, соответствующих их растворимости
и парциальному давлению.
Непоглощенные газовые компоненты (синтез-газ) в верхней части
абсорбера проходят через промыватель, состоящий из четырех кол-
пачковых тарелок, где промываются конденсатом для улавливания
паров и брызг растворителя. Осушенный синтез-газ направляется
в коллектор и затем на переработку. В случае прекращения приема
газа потребителем или сокращения потребляемого количества синтез-
газ можно направлять на факел 5. Отходящая из промывателя смесь
растворителя с водой стекает в низ абсорбера и смешивается с
основным потоком растворителя.
Рабочий раствор с растворенными в нем газами из куба абсорбера
подается в верхнюю часть десорбера I ступени 8, где дегазируется.
При этом за счет снижения избыточного давления с 9,0 до 0,23—
0,20 am из раствора выделяется большая часть плохо растворимых
газов вместе с двуокисью углерода (возвратные газы). Затем рабочий
раствор через тарельчатую верхнюю и среднюю зоны попадает в куб
десорбера 8. Температура в аппарате поддерживается 40° С.
Из средней части десорбера 8 отводится товарный ацетилен. Он
отмывается конденсатом от паров растворителя в промывателе 7,
представляющем собой аппарат с колпачковыми тарелками.
Вытекающий из промывателя обводненный растворитель направляется
в качестве флегмы в десорбер 8, а ацетилен-концентрат через
предохранительный скруббер-огнепреградитель 6 поступает в коллектор
товарного ацетилена и далее в газгольдер. В случае понижения
давления на выходе с установки концентрирования ацетилен автоматически
направляется через другой предохранительный скруббер 6. на факел 5
с постоянной горящей дежурной горелкой. Оставшаяся часть газа,
которая не выводится в качестве товарного продукта, из средней
части десорбера 8 направляется в верхнюю его часть (на рисунке не
показано) для отдувки двуокиси углерода из рабочего раствора.
Рабочий раствор из десорбера 8 проходит через пластинчатые
теплообменники 13, в которых нагревается с 45 до 95° С за счет тепла
раствора, выходящего из куба десорбера 12 II ступени. После
теплообменников раствор подогревается в паровом подогревателе до 100—
* Для выравнивания колебаний производительности на линии между
производством ацетилена и стадией концентрирования (перед компримированием)
устанавливают газгольдеры.
248
105° С (на рисунке не показан) и поступает в верхнюю часть десор-
бера 12. Здесь поддерживается избыточное давление около 0,30 am
и температура 100° С, за счет которой выделяется большая часть
растворенного газа. Затем раствор стекает вниз противотоком к газу,
нагнетаемому вакуум-компрессором 19. При этом улавливается
дополнительное количество высших ацетиленовых углеводородов
и вся газовая смесь, состоящая главным образом из ацетилена с
незначительными примесями его гомологов (сырой газ), направляется
в нижнюю часть десорбера 8. Рабочий раствор из куба десорбера 12
через регулирующий клапан возвращается для выделения ацетилена
в среднюю часть десорбера 12, где поддерживается остаточное
давление 600 мм рт. ст. Вместе с ацетиленом частично уходят и высшие
ацетиленовые углеводороды.
Газ из зоны выделения ацетилена охлаждается, проходя через
ацетиленовый конденсатор 9 и сепаратор 10, сжимается вакуум-
компрессором 19 до избыточного давления 0,3 am и возвращается
в верхнюю часть десорбера 12. Если избыточное давление на
нагнетании вакуум-компрессора превысит 0,4 am, то срабатывает
предохранительный гидрозатвор 20 и газ устремляется на факел 5.
Газообразные высшие ацетиленовые углеводороды отсасываются
из средней части десорбера 12 вакуум-эжекционной установкой 18
через вспомогательную колонну 15 и конденсатор 16 и после
обязательного разбавления синтез-газом для флегматизации
взрывоопасных смесей направляются на сжигание. Вместе с высшими
ацетиленовыми углеводородами из десорбера 12 уходит большое количество
паров воды и растворителя. Поэтому во вспомогательной колонне 15
паро-газовая смесь промывается конденсатом для отделени-я паров
растворителя *. Смесь водяного пара и высших ацетиленов
охлаждается в конденсаторе 16, а вода уходит через гидравлический
затвор 17 в цикл загрязненной воды.
Рабочий раствор из средней части десорбера 12 после отделения
ацетилена и высших ацетиленовых углеводородов стекает в куб **
десорбера 12, а оттуда поступает в кипятильник И. Здесь из раствора
выпаривается почти вся вода (до 2 вес. % Н2О), а остальная жидкость
охлаждается в теплообменниках 13, где отдает свое тепло раствору
из десорбера 8, и в водяных холодильниках 14, а затем насосом (на
рисунке не показан) перекачивается в емкость 3. Растворитель из
емкости 3 насосом 4 направляется в абсорбер 2. Подпитка свежим
* Практически происходит процесс ректификации, при этом колонна 15
представляет собой укрепляющую часть, а низ десорбера 12 — исчерпывающую часть
ректификационной колонны. Подаваемый конденсат является флегмой, а с верха
колонны 15 отходит продукт (пары воды), который конденсируется в
барометрическом конденсаторе 16.
** В этой части аппарата при пониженном давлении и повышенной температуре
ацетиленовые углеводороды отдувают водяным паром, который по отношению к ним
является инертным газом. Отсюда ясно, что на предшествующих стадиях
желательно обводнять растворитель, чтобы улучшать процесс отдувки за счет добавочного
количества пара.
250
растворителем с общезаводского склада производится периодически,
по мере надобности.
Используемый на установке концентрирования раствор
содержит 97 вес. % селективного растворителя и. 2 вес. % воды. Кроме
того, в нем присутствуют так называемые смолы — вещества,
образующиеся за счет полимеризации высших ацетиленовых
углеводородов. До определенного содержания полимеры не влияют заметным
образом на физико-химические свойства циркулирующего раствора
(адсорбционную емкость, вязкость и др.), вследствие чего
концентрация полимеров не должна быть выше 1 вес. %. Во избежание
накопления полимеров часть циркулирующего раствора (около 1 объемн. %)
после десорбера 12 отбирается на регенерацию, которую ведут в две
стадии.
На первой стадии (упарка) при 30 мм рт. ст. (0,05 am) и 120° С
отгоняют 90 вес. % растворителя (при этом концентрация полимеров
в циркулирующем растворе повышается примерно до 10 вес. %).
Полученная паро-газовая смесь проходит сепаратор и конденсатор
(где охлаждается до 50° С), а затем поступает на трехступенчатую
вакуум-эжекционную установку. Там очищенный растворитель
конденсируется, а пары его направляются на стадию концентрирования.
Периодически с первой стадии регенерации осмоленный раствор
отводят на вторую стадию (давление 45 мм рт. ст., температура
до 100° С). За счет постоянной температуры на второй стадии
регенерации резко снижаются потери растворителя с полимерами.
Остаток после регенерации состоит из сухих тонкодисперсных
полимеров, легко воспламеняющихся на воздухе. После охлаждения
полимеры замешиваются в шлам небольшим количеством воды в
атмосфере азота и направляются на установку сжигания отходов.
Отдельно рассмотрим вопрос об уменьшении содержания примесей
в ацетилене-концентрате. Максимальное содержание гомологов
ацетилена в товарном ацетилене практически определяется парциальным
давлением их над раствором. Для того чтобы содержание высших
ацетиленовых углеводородов не превышало допустимой величины,
необходимо полностью отделять их в десорбере II ступени. В
противном случае концентрация гомологов ацетилена в товарном ацетилене
будет возрастать и может достичь концентрации их в газе пиролиза,
поступающем на установку концентрирования.
Как было показано ранее (стр. 237), поглотительная способность
диметилформамида с понижением температуры значительно
повышается. На основе этого предложена схема выделения ацетилена
диметилформамидом при пониженной температуре и атмосферном
или повышенном давлении с последующей отдувкой высших
ацетиленовых углеводородов инертным газом (например, синтез-газом)26.
При такой схеме можно значительно снизить энерго-материальные
затраты (против обычных схем с селективными растворителями):
1) снижается расход электроэнергии на 30% за счет исключения
стадии компрессии поступающих на концентрирование газов и
использования захоложенного диметилформамида;
251
2) уменьшается расход пара на 40%, так как растворитель не
обводняется, а отдувку высших ацетиленовых углеводородов
проводят синтез-газом.
Процесс при атмосферном давлении заключается в следующем.
Газы пиролиза, пройдя форабсорбер, газодувкой подаются на стадию
концентрирования при 35° С. Вначале они охлаждаются в
теплообменнике до —10° С (хладоагент—синтез-газ, отходящий из абсорбера)
и в холодильнике до —30° С (хладоагент — аммиак), а затем
поступают в абсорбер. Туда же подается диметилформамид, предварительно
охлажденный в теплообменнике до —10° С (хладоагент—раствор,
выходящий из абсорбера) и в аммиачном холодильнике до —30° С.
В абсорбере при —25° С и 1,3—1,4 am ацетилен и его гомологи
поглощаются диметилформамидом. Непоглощенные газы (синтез-газ)
после охлаждения в теплообменнике и промывки водой можно
направлять на производство аммиака или метанола. Выходящий из
абсорбера раствор с поглощенными газами сначала поступает в
теплообменник, где отдает свой холод свежему диметилформамиду,
а затем подвергается трехступенчатой десорбции.
Ацетилен выделяется на первых двух ступенях. В первом десор-
бере процесс осуществляется при 1,2—1,3 am и 15° С. Выделившиеся
возвратные газы направляются во второй десорбер. Предварительно
от них отдувают двуокись углерода товарным
ацетиленом-концентратом. Во втором десорбере ацетилен выделяется при 80° С и 0,2 am
за счет подогрева паром. Выделившийся ацетилен проходит
конденсатор, где конденсируется диметилформамид, и отсасывается вакуум-
насосом.
На третьей ступени десорбции путем продувки синтез-газом при
пониженном давлении из растворителя выделяются высшие
ацетиленовые углеводороды. Пары растворителя, выходящие из третьего
десорбера, конденсируются в конденсаторе, а смесь продувочных
газов и высших ацетиленовых углеводородов промывается водой для
улавливания оставшихся паров растворителя. Все промывные воды,
получаемые на установке, направляются в дистилляционную
колонну, где происходит разгонка смеси в вакууме. Регенерированный
диметилформамид возвращают в цикл, а воду можно использовать
для. промывки газовых потоков.
Для процессов выделения ацетилена существенное значение имеет
расход растворителя, который определяется потерями с полимерами
на стадии регенерации и уносом с газовыми потоками. С целью
сокращения потерь растворителя, как уже говорилось, необходим строгий
температурный режим на последней стадии регенерации. Для
уменьшения потерь с отходящими газами необходимо, чтобы промыватели
синтез-газа и ацетилена-концентрата работали в оптимальных
гидродинамическом и технологическом режиме. При этом в верхнюю
часть промывных аппаратов будет практически поступать чистая
вода, что- повысит к. п. д. тарелок.
Форабсорбция углеводородов. В ряде схем с выделением
ацетилена селективными растворителями (в первую очередь это относится
252
к системам разделения газов электрокрекинга) перед компрессией
предусматривается форабсорбция. Ее целью является удаление
основной массы высших ацетиленов, а также ароматических
углеводородов и нафталина, которые в противном случае будут интенсивно
выделяться при компримировании газа и оседать в машине и
межступенчатых холодильниках.
Цикл форабсорбции может быть осуществлен с использованием,
например, масла, в котором достаточно хорошо растворяются
ароматические углеводороды и нафталин. Отработанное масло для
регенерации продувают природным газом или азотом, после чего подогревают
до 150—250° С для удаления оставшихся поглощенных
углеводородов. Часть масла периодически выводится либо на регенерацию, либо
на сжигание, если смолообразных веществ (полимеров) образуется
слишком много. В систему при этом добавляют соответствующее
количество свежего масла. Особенно часто масляная форабсорбция
предусматривается для разделения газовой смеси с большим
количеством ароматических углеводородов и смол, т. е. для газов
пиролиза, например в схемах пиролиза легкого бензина (фирмы Hoechst
и SBA).
Форабсорбцию можно проводить тем же селективным
растворителем, который применяют в основном цикле абсорбции. Такую схему
принимают при наличии в исходном газе большого количества
ацетиленовых углеводородов, склонных к полимеризации. При этом
облегчаются основной цикл абсорбции — десорбции и получение
ацетилена требуемого качества. Основным недостатком такого
процесса являются безвозвратные потери ацетилена на стадии
форабсорбции. Для сокращения потерь количество растворителя должно быть
минимальным, т. е. определяться условиями поглощения ключевого
компонента — диацетилена. Параметры форабсорбции такие же, как
и основного цикла абсорбции — десорбции.
АБСОРБЦИЯ АЦЕТИЛЕНА
НИЗКОТЕМПЕРАТУРНЫМИ РАСТВОРИТЕЛЯМИ
Применение низкотемпературных растворителей (метанол,
ацетон, жидкий аммиак) для целей выделения ацетилена обусловлено
в первую очередь их доступностью и дешевизной по сравнению
с селективными растворителями. Кроме того, вследствие
относительно низкой температуры кипения метанола (64,7° С), ацетона
(56° С) и др. для регенерации этих растворителей можно использовать
отходы производства, имеющие низкую температуру (80—90° С),
например, горячую воду, получаемую на стадии пиролиза.
Метанол и ацетон обладают определенным коэффициентом
селективности по отношению к двуокиси углерода, а аммиак химически
связывается с ней. В связи с этим разделение пары С2Н2—СО2
метанолом или ацетоном принципиально возможно, однако практически
связано с образованием больших количеств возвратных газов после
отдувки двуокиси углерода. Например, при разделении этой пары
253
метанолом количество возвратных газов почти в 4 раза больше, чем
в случае использования диметилформамида в этих же условиях: при
760 мм рт. ст. и 30° С коэффициент селективности в диметилформ-
амиде равен 8 ,а в метаноле только 2,2. Поэтому для разделения пары
С2Н2 — СО2 в промышленных схемах обычно применяют другие
растворители, например аммиачную воду или раствор едкого натра.
При этом можно удалять двуокись углерода либо до стадии
абсорбции ацетилена низкотемпературным растворителем —
промывкой ацетиленсодержащих газов щелочью и аммиачной водой,
либо выделять двуокись углерода из ацетилена-концентрата. В
схемах с аммиаком проводится только предварительное удаление
двуокиси углерода, для схем с метанолом возможно двоякое решение.
Положение стадии очистки от двуокиси углерода в общей схеме
выделения ацетилена следует определять в зависимости от параметров
процесса концентрирования и выбирать способ очистки после
соответствующего технико-экономического анализа.
Выделение ацетилена метанолом или аммиаком из смеси его с
ацетиленовыми углеводородами затруднено из-за высокой
растворимости ацетиленовых углеводородов в этих растворителях; при
фракционной десорбции требуются высокие температуры и глубокий
вакуум. Поэтому предпочтительнее стадии удаления ацетиленовых
углеводородов и абсорбции — десорбции ацетилена осуществлять
раздельно.
В цикле очистки ацетиленсодержащих газов от высших
ацетиленовых углеводородов возможно применение как метанола, так и
других растворителей (например, керосина), что определяется
выбранной схемой разделения ацетиленсодержащих газов. В случае
применения метанола (на стадии очистки от ацетиленовых углеводородов
и на стадии выделения и концентрирования ацетилена) не удается
полностью очистить газ от метилацетилена. Для этой цели ацетилен-
сырец подвергают дополнительной обработке, например
активированным углем. При такой системе очистки двуокись углерода
целесообразно выделять в конечной стадии процесса концентрирования.
Выделение ацетилена жидким аммиаком
Давление паров смеси аммиака и ацетилена при разных
концентрациях и температурах можно определять по номограмме27
рис. VI-15. Система отклоняется от закона Рауля: при одинаковом
составе жидкости давление и содержание ацетилена в парах ниже, чем
получается по расчету, причем для одного и того же состава жидкости
содержание ацетилена в газовой фазе уменьшается при повышении
температуры. Опытные данные показывают, что выше критической
температуры ацетилена давление смеси возрастает незначительно,
что свидетельствует об очень большой растворимости ацетилена
в жидком аммиаке.
В табл. VI-18 приведены данные о растворимости ацетилена в
жидком аммиаке в зависимости от общего давления системы и темпера-
254
30
у/
у25
*/Г
55-L
\
\
\
\
\
\
\
\
\
ч
ч
ч
vv
с 0
15 11^(Ш
20^<цГр20/0°
25 ^^5(кА35
У 15 Ч}&Р
^20 M-t43^^^^.
В57^5О
™+во
1г
9о\
-85
\
\
\
^*-*^ \
*** *J ^2 2
100
%NH3)
<5Г
t
fi
<
1
\
Г70
\~60
[-50
ыо
ho
[о
1-10
[-20
--30
- -4/7
-50
-60
70-
60-
50-
ио-
зо-
20-
15-
/Z7-
9-
^ 8~
^з 7 —
^ъ 5 —
3-
2-
1,5-
1-
0,9-
0,8-
0,7-
0,6-
0,5-
о,и-
0,3-
0,2-
t
\
i
-300
-200
-150
-100
-90
-80
-70
-60
-50
-40
-30
-20
-15
Рис. VI-15. Давление паров смеси аммиака и ацетилена при разных концентрациях
и температурах:
/ — содержание С2Н2 в жидкости, вес. %; 2 — то же, мол. %; 3 — содержание С2Н2 в газе,
вес. %; 4 — то же, мол. %.
туры; из этих данных видно, что с понижением температуры
растворимость заметно повышается28. На рис. VI-16 представлены кривые
равновесия в системе ацетилен — жидкий аммиак.
Для растворов ацетилена в жидком аммиаке в области изученных
температур (от —40 до —80° С) справедливо уравнение Кричевского—
Ильинской (VI-15), записанное в следующем виде:
А
RT In К = #7Чп/'
где /' — летучесть компонента в жидкости.
(VI-47)
255
Растворимость ацетилена в жидком аммиаке
ТАБЛИЦА VI-18
При —42,4° С
Робщ.
546,0
570,0
575,0
577,0
577,5
639,0
651,0
656,0
712,0
716,0
X
0,032
0,031
0,031
0,031
0,031
0,059
0,058
0,059
0,075
0,074
При -49,0° С
Робщ.
415,5
422,0
431,5
528,5
540,0
580,0
587,0
589,0
679,0
715,5
X
0,032
0,032
0,039
0,075
0,075
0,091
0,091
0,100
0,112
0,143
При —58,5° С
Робщ.
346,0
397,5
405,5
453,0
477,5
486,0
534,5
607,0
625,0
788,0
X
0,092
0,101
0,114
0,134
0,154
0,153
0,179
0,214
0,214
0,299
При —64,1° С
Робщ.
264,0
355,5
360,5
362,0
367,0
407,5
428,0
455,0
537,0
583,0
X
0,097
0,142
0,163
0,145
0,154
0,179
0,215
0,198
0,263
0,299
При —79,9° С
р
*общ.
114,0
176,0
228,0
252,0
305,0
411,0
446,5
473,0
X
0,053
0,077
0,139
0,146
0,190
0,236
0,263
0,300
Примечание.
растворе.
— общее давление, мм рт. ст.; X — мольная доля ацетилена
По этому уравнению можно рассчитывать растворимость
ацетилена в жидком аммиаке в широкой области концентраций, используя
приведенные ниже значения:
t, °с
Г л
мм рт* cm* кал/моль
/ -А
мм рт. ст. кал/моль
—42,4
—49,0
—58,5
4790
3660
2500
120
55
60
—64,1
—72,9
—76,0
1980
1350
ИЗО
70
13
25
Теплота растворения ацетилена в жидком аммиаке составляет
3300 кал/моль. Мольный объем растворов ацетилена равен: 24,6—
26,0 смЧмоль при —42,4° С и X = 0—0,075; 23,3—27,4 см3/моль
при —76° С и X = 0—0,252 смЧмоль.
О растворимости высших ацетиленовых углеводородов в жидком
аммиаке можно судить по данным29 рис. VI-17. Сравнительные
данные о растворимости этих газов в жидком аммиаке приведены ниже
(в мольных долях):
При 100 мм рт. ст. При 200 мм рт. ст.
Метилацетилен .... 0,43 (при —70,5° С) 0,80 (при —56,7° С)
Винилацетилен 0,95 (при —44,4° С) 0,28 (при —55° С)
Диацетилен 0,70 (при —44° С) 0,20 (при —55° С)
Видно, что при парциальном давлении 200 мм рт. ст. и
одинаковой температуре (около —55° С) наибольшей растворимостью в
жидком аммиаке обладает метилацетилен, а наименьшей — диацетилен
(эта зависимость сохраняется и для других давлений). Отсюда
следует, что в качестве ключевого компонента для расчета стадии
выделения ацетиленовых углеводородов нужно выбирать диацетилен.
В системах аммиак — метилацетилен и аммиак — винилацетилен
имеются положительные отклонения от закона Рауля; в системе
аммиак —диацетилен, наоборот, отклонения отрицательные.
256
юоо
0,5 1,0
Хс н , мольные доли
Рис. VI-16. Равновесие
жидкость—газ в системе ацетилен —
жидкий аммиак (прямые
линии— в жидкой фазе, кривые—
в газовой; пунктир — данные,
полученные экстраполяцией):
У—при —42,4° С; 2—при —49,0° С;
3— при —58,5° С; 4— при —64,1° С;
5—при —72,9° С; 6— при —76,0° С.
700
600
500
К 200
' 100
О
500\
к 300
-J00
х
\\
0,2 ОЛ 0,6 0,8 1,0
j мольные доли
а
п
р*
\yOG~
^S
-70
,5
\
ft
~п
0,2 0,k 0,6 0,8 1,0
q,y\z, мольные доли
б
0,2 ОЛ 0,6 0,8 1,0'
, мольные i
17 В. Н. Антонов
Рис. VI-17. Изотермы
растворимости ацетиленовых углеводородов
в жидком аммиаке при разном
парциальном давлении углеводорода:
а — винилацетилен; б — диацетилен;
в — мети лацети лен.
В табл. VI-19 приведены данные о растворимости метилацетилена
в жидком аммиаке и мольном объеме этих растворов при разной
мольной доле метилацетилена и температуре. Аналогичные мольные
объемы имеют растворы диацетилена и винилацетилена.
ТАБЛИЦА VI-19
Растворимость метилацетилена в жидком аммиаке
При —43,9° С
^общ.
435
467
488
493
419
390
343
284
X
0,000
0,042
0,106
0,368
0,760
0,839
0,916
1,000
V
24,5
24,3
27,9
37,0
48,7
51,1
53,3
57,5
При —56,7° С
^общ.
203
222
246
246
245
209
188
145
X
0,000
0,045
0,270
0,355
0,406
0,777
0,870
1,000
и
24,0
24,6
31,7
34,8
37,0
47,6
51,1
56,4
При —70,5° С
робщ.
79
90
77
X
0,000
0,120
0,813
V
23,5
26,2
46,3
Примечание. Р0$щ — общее давление, мм рт. ст.; X
ацетилена в растворе; v — мольный объем раствора, см3/моль.
мольная доля метил-
Теплота растворения — Д# ацетиленовых углеводородов в жидком
аммиаке равна (в кал!моль):
Метилацетилен
Ди ацетилен
Винилацетилен
6 000
6 000
11 000
Выбор растворителя на стадии очистки от ацетиленовых
углеводородов. Способ выделения ацетилена аммиаком основан на
применении органических растворителей (для очистки газов пиролиза
от высших ацетиленовых углеводородов) и аммиака (для выделения
ацетилена). Этот процесс можно проводить либо при атмосферном
давлении, либо при 7—15 am (выбор давления на стадии выделения
ацетилена определяется принятым способом его получения). В
процессах при атмосферном давлении для очистки от высших
ацетиленовых углеводородов используют метанол, а при повышенном
давлении — керосин30.
Характеристика применяемого керосина:
Плотность, г/ж3 0,79
Температура затвердевания, °С . . . —34
Пределы выкипания, °С 190—250
Групповой состав, вес. %
парафины 99,15
олефины 0,57
ароматические углеводороды . . 0,28
И метанол, и керосин отвечают всем требованиям, которые
предъявляются к растворителям на стадии очистки: они способствуют
258
минимальному образованию полимеров, мало растворяют ацетилен
в условиях очистки, не вызывают коррозии и дешевы. Следует
отметить, что метанол применяется для очистки и при повышенном
давлении, но в этом случае требуются минусовые температуры.
Растворимость углеводородов в керосине и метаноле различна
(рис. VI-18). Среди высших ацетиленов наибольшей растворимостью
в керосине обладает винилацетилен, а наименьшей — метилацети-
лен. При абсорбции метанолом наивысшей растворимостью характе-
100
80
60
2о\
п
L?
h2
fflp
f
1A
I ////
11 ///
A-o
9
—JL---
W
2±=
180
I
%В0\
1 20
8 16
Содержание, обьемн.%
а
О
Ш7
\\ I 5 j
W/
W
"I&
Г
1
10-у/
/ 1
/О /
/° ^
9
8 16
Содержание, объемн. %
б
Рис. VI-18. Количество углеводородов, растворяющихся
в керосине (а) и в метаноле (б) при 7 am к —30° С, в
зависимости от их содержания в газе:
1 —бутадиен; 2—винилацетилен; 3— бутилен; 4— бутан; 5 — про-
падиен; 6— пропан; 7—метилацетилен; 8— пропилен; 9 — этилен;
10 — ацетилен.
ризуется винилацетилен. Ацетилен, как видно из рис. VI-18,
растворяется в метаноле в 10 раз более, чем в керосине. Например, при
10 объемн. % С2Н2, 7 am и —30° С растворимость ацетилена в
керосине составляет 3 объем/объем, а в метаноле — 35 объем/объем.
Отсюда следует, что метанол можно применять только для очистки
ацетиленсодержащих газов во избежание больших потерь ацетилена
и чтобы исключить дополнительную установку регенерации
растворенного ацетилена.
Применение аммиака для концентрирования ацетилена в
сочетании с использованием керосина для предварительной очистки газов
пиролиза позволяет получать очень чистый ацетилен-концентрат.
Температура кипения аммиака находится в интервале между
температурами кипения ацетилена и других углеводородов,
присутствующих в ацетиленсодержащих газах, что обусловливает значительную
разницу в растворимости и, следовательно, возможность полного раз-
17* 259
деления. Благодаря этому относительно легко осуществима
регенерация аммиака, которая обычно проводится под давлением.
В табл. VI-20 приведены значения коэффициентов селективности
керосина и безводного жидкого аммиака по отношению к ацетилену
и другим углеводородам30. Из этих данных следует, что абсорбция
этилена и пропилена аммиаком очень незначительна, поэтому их
можно извлекать из аммиака путем отдувки. Наоборот, высшие
ацетиленовые углеводороды обладают гораздо большей растворимостью
в аммиаке, чем в керосине, и их необходимо удалять до абсорбции
ацетилена аммиаком.
ТАБЛИЦА VI-20
Коэффициенты селективности Кс керосина и безводного жидкого аммиака
Углеводород
Ацетилен ....
Этилен
Метил ацетилен . .
Пропадиен . . .
ас2н2
керосин
1
0,8
0,01
0,01
жидкий
аммиак
1
50
0,5
2
Углеводород
Пропилен . . .
Бутадиен . . .
Бутилен . . ,
аС2Н2
^с аг
керосин
0,01
0,01
0,01
жидкий
аммиак
15
2
4
Важной особенностью жидкого аммиака является способность
к самоохлаждению путем испарения некоторого его количества за
счет тепла газа, поступающего на абсорбцию. Это дополнительное
охлаждение дает возможность улавливать ацетилен при значительно
меньшем давлении. Испарившийся аммиак конденсируют и
используют в абсорбере в качестве орошения. Еще одним преимуществом
способа выделения ацетилена аммиаком является возможность
использования некоторых отходов производства (горячей воды) на
стадии десорбции ацетилена из аммиака.
Возможность относительно простого извлечения аммиака из смеси
его с ацетиленом путем промывки водой с последующей регенерацией
получаемой аммиачной воды позволяет создавать безопасные условия
при компримировании и транспортировании ацетилена. Например,
из сжатой ацетилен-аммиачной смеси после отмывки аммиака можно
получить ацетилен, находящийся под давлением около 5 am. Кроме
того, эту смесь, являющуюся взрывобезопасной из-за флегматизи-
рующего действия аммиака, можно транспортировать на
завод-потребитель ацетилена и извлекать аммиак там. При этом ацетилен после
отмывки аммиака можно получать либо в сжатом состоянии, либо,
если это не требуется, при атмосферном давлении (после
дросселирования).
Технологические схемы выделения ацетилена жидким аммиаком.
В зависимости от метода получения ацетилена и давления процесса
применяют схемы выделения ацетилена аммиаком при атмосферном
260
и повышенном давлениях (стр. 209). Процесс абсорбции под
давлением наиболее рационально сочетать с пиролизом под давлением.
На рис. VI-19 показана схема концентрирования в процессе
совместного получения ацетилена и этилена из легких фракций нефти
(нафта)31. Газы пиролиза, полученные в реакторе 4 и очищенные от
Ацетилен
Этилен
Остаточный газ
(СО + Н2)
Рис. VI-19. (^хема концентрирования в процессе совместного получения ацетилена
и этилена из углеводородов:
1,2— газодувки; 3 — подогреватель; 4 — реактор; 5, 25, 26 — колонны промывки водой;
6 — колонна промывки маслом; 7 — смолоотделитель; 8 — насосы; 9 — газгольдер;
10 — компрессор; // — колонна промывки аммиачной водой; 12 — колонна для
регенерации аммиачной воды; 13 — колонна промывки раствором едкого натра; 14, 16, 17, 21 —
теплообменники; 15 — колонна промывки керосином; 18 — аппарат для регенерации керосина;
19 — сборник аммиака; 20 — абсорбер ацетилена; 22 — разделительная установка; 23,
24 — десорберы ацетилена; 27, 28 — ректификационные колонны.
сажи и смол, направляются в газгольдер 9. После этого они
сжимаются компрессором 10 до 3 am и направляются для отмывки
двуокиси углерода аммиачной водой в колонну И. Насыщенная
двуокисью углерода аммиачная вода выходит с низа колонны, проходит
теплообменник, где подогревается до 45° С за счет тепла
регенерированного раствора аммиачной воды, затем дросселируется до
атмосферного давления и направляется в колонну 12 для регенерации аммиач-
261
ной воды паром низкого давления; регенерированная аммиачная вода
возвращается в колонну 11.
Освобожденные от двуокиси углерода газы из колонны 11
поступают в колонну 13, где из них раствором щелочи улавливается
оставшееся количество двуокиси углерода. Затем газы охлаждаются до
—25° С в теплообменнике 14 (хладоагент — газы, выходящие из
абсорбера 20) и поступают на очистку керосином в колонну 15.
Керосин предварительно также охлаждают до —25° С в
теплообменнике 16. В результате промывки керосином из газа улавливаются
высшие ацетиленовые углеводороды.
Для регенерации керосина сбрасывают давление почти до
атмосферного, нагревают керосин до 110° С в теплообменнике 17 и
продувают острым паром в аппарате 18. Десорбированные из керосина
углеводороды возвращают на пиролиз, благодаря чему
утилизируются отходы и предотвращается попадание в атмосферу горючих
и взрывоопасных веществ.
Очищенный и охлажденный газ поступает в абсорбер 20, где
ацетилен растворяется в безводном жидком аммиаке. Тепловое
равновесие в колонне достигается за счет дополнительного охлаждения
аммиака путем его испарения. С верха абсорбера отбирается газ,
насыщенный аммиаком. Он проходит теплообменник 14, где отдает свой
холод газовому потоку, идущему на очистку керосином, и
направляется на водную отмывку от аммиака в колонну 25. Отделенный от
аммиака газ направляется на установку 22 для выделения этилена.
Раствор аммиака, насыщенный ацетиленом и этиленом, собирается
в нижней части абсорбера 20, отдает свой холод регенерированному
аммиаку в теплообменнике 21 и поступает в десорбер 23, где за счет
сброса давления и нагревания весь растворенный этилен десорби-
руется и возвращается в газгольдер 9. Жидкий аммиак, содержащий
только ацетилен, поступает в десорбер 24, где при повышенном
давлении за счет нагрева горячей водой, полученной на стадии пиролиза,
происходит десорбция смеси ацетилена с аммиаком. Отделенная
газовая смесь направляется в колонну 26 для улавливания аммиака
водой; с верха колонны выходит очищенный ацетилен-концентрат.
Степень извлечения ацетилена из газовой смеси достигает 97%,
а чистота его не менее 99,6%.
Безводный жидкий аммиак, отобранный в нижней части десор-
бера 24, после очистки керосином содержит небольшое количество
примесей (высшие ацетиленовые углеводороды). Часть его рециркули-
рует в абсорбер 20, а остальное количество поступает в
ректификационную колонну 27. Раствор аммиака из колонн 25 и 26 перегоняют
под давлением в колонне 28, обогреваемой паром высокого давления.
Очищенный аммиак, отбираемый с верха колонн 27 и 28, возвращают
в цикл абсорбции. Вода, не содержащая аммиак, возвращается в
промывные колонны 25 и 26. Кубовый раствор из колонны 27 сжигают.
Схема выделения ацетилена из газов окислительного пиролиза
аммиаком, аналогичная описанной выше, приведена в гл, V (см.
рис. V-32, стр. 204).
262
Этилен в газах пиролиза присутствует в небольшом количестве,
поэтому в схеме концентрирования нет аппарата для выделения
этилена — он поглощается совместно с ацетиленом в абсорбере 20.
Затем газовую смесь разделяют методом глубокого охлаждения на
установке 22. Степень извлечения этилена этим методом не ниже
95%, а чистота его 99,9%.
Выделение ацетилена метанолом
Растворимость ацетилена в метаноле возрастает с понижением
температуры11' 12 (рис. VI-20 и табл. VI-21). Система не подчиняется
закону Генри32; но приведенные данные согласуются с уравнением
(VI-16) Кричевского — Ильинской. По этому уравнению можно
рассчитывать растворимость ацетилена в широком интервале
концентраций. Значения коэффициентов /С, р и А приведены в табл. VI-22.
Высшие ацетиленовые углеводороды также растворяются в
метаноле. На их растворимость оказывают влияние парциальное давление
углеводорода и температура (рис. VI-21 и табл. VI-23). Изменение
растворимости диацетилена подчиняется уравнению Кричевского —
Ильинской, а винилацетилена и метилацетилена — уравнению Генри.
ТАБЛИЦА VI-21
Растворимость ацетилена в системе метанол — вода
При
РС2Н2
—10° С
а
При
РС2Н2
—25° С
♦ а
При -40° С
Рс2н2
а
При —55° С
РС2Н2
а
При -
РС2Н2
-70° С
а
При
РС2Н2
—78
С
а
С 0,05 вес. % воды
106 3,5 136 8,6 93 11,1 189 40,3 192 85,4 125 92,7
318 12,1 291 18,5 311 33,3 403 77,5 409 162,1 280 181,0
554 20,3 536 33,0 545 56,6 594 108,7 587 223,0 486 316,0
762 28,2 747 45,4 696 70,9 714 126,6 709 261,0 687 435,0
С 4,4 вес.
6,4
12,2
16,9
25,0
143
352
575
720
7,3
17,6
28,4
36,0
101
260
496
723
8,9
22,7
42,7
61,0
151
367
599
743
27,8
63,7
98,6
120,7
С 9,5 вес.
ВОДЫ
190
371
512
749
146
293
482
792
Примечание: Рс2Н2 ~ парциальное давление ацетилена, мм рт. ст.; а —
отношение объема растворенного ацетилена (при нормальных условиях) к объему метанола
пРн указанной температуре.
253
3,9
8,1
13,7
20,3
132
307
501
750
5,8
13,4
21,4
31,5
161
301
543
761
11,5
21,0
36,8
50,1
147
275
481
684
22,1
39,3
67,1
93,5
\
<
\ ч
я
^3
3
тгод этичной '
И
I
nwog Э1яняиои'
тгод
о
/
1
/
tl
I
, J
/
/
/
1
/
/
с
/
/
V»/
f
/
/
1
1
-(.
1
1
/
1
1
/
/
'1
-■(.
1
I
/
/
/
/
f
1
1
I
1
1
/
I
_J
/
1
1
1
1
1
i.
vf 1
ц£
I
f
/ 1
1
1
J
J
fl
/
I
и
I
I
I
I
n.
X CX,^4
ПЗ ПЗ Й
IcS
s я s
-g-l
я v~' ^
m 3 ^
U^S
2 с о
S 5 n
||S
S н g
Й ой
£fi
о 9 «=t
^is
>§s
g«s
а. я
ТАБЛИЦА VI-22
Значения коэффициентов К, Р
Коэффициенты
При
—10° С
и А для системы ацетилен—метанол — вода *
При
—25° С
При
—40° С
При
-55° С
При
—70° С
При
—78° С
С 0,05 вес. % воды
К, мм рт. ст.
А, см3-am/моль
/С, мм рт. ст.
Л, см3-am/моль
К, мм рт. ст.
А, см3-amiмоль
* В интервале температур от —10 до —78° С теплота растворения ацетилена в метаноле
составляет —АН = 4600 кал/моль.
ТАБЛИЦА VI-23
Растворимость ацетиленовых углеводородов в метаноле
15 470
0,251
12 480
С
18 520
0
0
С
23 180
0
0
8930
0,473
22 180
4,4 вес.
12 920
0
0
Э,5 вес.
15 ПО
0
0
4800
0,634
27 920
% воды
6730
0,420
18 500
% воды
9650
0
0
2535
0,631
26 000
3130
0,619
25 390
4335
0,422
17 390
1122
0,598
22 950
—
716
0,571
21 050
Е
—
При 20° С
р а
При 5° С
р а
При —10° С
р а
При —25° С
р а
При —40° С
р а
19,00
66,0
123,5
189,5
12,4
37,7
62,9
91,7
14,0
49,0
82,0
155,0
Д
19,5
52,2
78,3
133,0
и а ц е т
10,0
39,0
58,0
103,0
и л е н
26,4
68,8
99,4
161,0
15,0
42,5
66,5
85,0
67,8
153
231
298
9,5
12,5
44,5
50,0
46,5
116,5
165,5
209
37,0
97,0
158,0
216,0
Метилацетилен
2,3
6,0
8,2
10,3
30,5
73,0
102,0
136,0
2,3
5,3
7,6
10,4
31,5
77,0
128,0
179,0
4,4
8,7
16,2
24,3
43,5
78,5
148,5
193,5
11,1
20,6
37,8
52,5
29,0
68,5
94,0
126,0
Винилацетилен
5,4
15,1
24,4
34,9
23,0
85,0
116,0
181,0
6,2
24,0
32,7
54,7
35,5
101,5
142,5
178,0
17,8
54,4
80,5
104,0
34,5
70,5
120,5
140,5
38,2
77,0
140
183
18,0
37,5
53,0
86,5
175
341
395
35,2
95,1
124,0
200
51,1
102
152
Примечания. 1. В таблицер — парциальное давление углеводорода, мм рт. ст.;
а — отношение объема растворенного углеводорода (при нормальных условиях) к объему
метанола при указанной температуре.
2. Для метилацетилена приведены данные не при —40° С, а при —55° С
Коэффициенты /С, Р и А для системы диацетилен — метанол при
разных температурах составляют:
Температура, °С 20 5 —10 —25 —40
К, мм рт. cm 470 440 220 103 65,4
Р 0,810 0,667 0,759 0,657 0,500
Л, см3-ami'моль 1088 847 912 745 532
В системах метилацетилен — метанол и винилацетилен —
метанол коэффициент Генри меняется в зависимости от температуры
следующим образом:
Температура, °С 20 5 —10 —25 —40 —55 —70
Коэффициент Генри К, мм рт. ст.
С3Н4 — СН3ОН 11200 7400 4440 2250 — 500 190
С4Н4 —СН3ОН 4 170 2050 1150 580 254 — —
Из приведенных цифр видно, что наибольшей растворимостью
в метаноле обладает диацетилен, затем следуют винилацетилен,
метилацетилен и ацетилен.
Данные о теплоте растворения указанных углеводородов —АН
(в кал/моль) следующие:
Диацетилен 6700
Винилацетилен 6200
Метилацетилен .... 5400
Ацетилен 4600
Технологические параметры выделения ацетилена метанолом.
Растворимость ацетилена в метаноле, как уже указывалось, при
пониженной температуре очень велика: в 1000 кг метанола при —70°-С
и 1 am растворяется ~300 м3 ацетилена. Однако имеются данные,
что сверхнасыщенные ацетиленом растворы метанола взрывоопасны,
поэтому насыщение не должно превышать 350 м3 ацетилена на 1000 кг
метанола. Вследствие такой большой растворимости ацетилена
коэффициент его извлечения метанолом может быть очень высок.
Правильный выбор температуры позволяет резко сократить потери
растворителя с отходящим газом. Так, например, если парциальное
давление метанола при —70° С составляет 0,1 мм рт. ст., то в 1 м3
газа после выделения ацетилена содержится 0,13 г метанола. Однако
применение относительно низких температур связано с высокими
затратами энергии.
Селективное действие метанола особенно заметно проявляется
в отношении диаце.тилена: при —10° С растворимость его в
метаноле в 100 раз больше, чем ацетилена. Вследствие высокой
поглотительной способности метанола по отношению к высшим
ацетиленовым углеводородам, их можно выделять на первой ступени
охлаждения газа; при этом требуется очень небольшое количество
растворителя, и следовательно, малые габариты оборудования.
Метанол хорошо растворим в воде, что позволяет легко
улавливать его из газовых потоков водой. Промывные воды подвергают
266
ректификации, а регенерированный метанол возвращают в цикл.
Вследствие низкой температуры кипения метанола (64,7° С) на его
регенерацию достаточно затратить относительно небольшое
количество тепла. Для этой цели можно использовать, например,
горячую воду, получающуюся при отмывке газа пиролиза от сажи.
Эту воду целесообразно использовать также на абсорбционных
холодильных установках.
Например, на установке производительностью 1 т ацетилена
в 1 чдля его выделения требуется 1700 тыс. ккал/ч холода при —30° С.
Это количество можно получить на абсорбционной холодильной
установке путем рекуперации 3700 тыс. ккал/ч тепла, выделяющегося
при охлаждении газа пиролиза (за счет конденсации водяного пара,
содержащегося в нем) от 125 до 115° С. При дальнейшем
охлаждении воды (от 115 до 85° С) образуется дополнительное количество
тепла (3300 тыс. ккал/ч), которое можно использовать на стадии
регенерации аммиачной воды после извлечения двуокиси углерода
и на стадии регенерации метанола. Таким образом, тепло
рекуперируется полностью и тепловой баланс стадии выделения ацетилена
замыкается.
Растворимость ацетилена и двуокиси углерода в метаноле
выражается величинами примерно одного порядка (см. рис. VI-20),
однако коэффициент селективности для пары С2Н2— СО2 в рабочем
интервале температур (от +30 до — 70° С) равен примерно 3,
поэтому двуокись углерода можно удалять из газовой смеси до
абсорбции ацетилена, или на стадии доочистки выделенного ацетилена.
Обычно для отделения СО2 в отличие от схем с селективными
растворителями используют дополнительные абсорбенты, в первую очередь
щелочь и аммиачную воду (процесс хемосорбции). Известны схемы
с разделением С2Н2 и СО2 фракционной десорбцией, основанной
на достаточно высоком коэффициенте селективности для этой пары.
В промышленности для выделения ацетилена метанолом широко
распространены процессы при повышенном давлении* (рис. VI-22).
Газ пиролиза 31, полученный в реакторе 4, после охлаждения
и очистки поступает в абсорбер 14, где небольшим количеством
метанола поглощаются высшие ацетиленовые углеводороды. Очищенный
ацетиленсодержащий газ компрессором 12 дожимается до 13—16 am
и поступает на абсорбцию ацетилена метанолом в аппарат 15.
Отходящий из аппарата 15 газ (Н2, СО) промывается водой в
скруббере 16 и направляется на конверсию СО и далее на промывку
жидким азотом и на разделение глубоким холодом.
Метанол из абсорбера 15, насыщенный ацетиленом и двуокисью
углерода, дросселируется до атмосферного давления и поступает
на стадию десорбции ацетилена в аппараты 17 и 18. Десорбция
осуществляется при повышенной температуре (теплоноситель —
горячая вода, получаемая на стадии пиролиза). Десорбированный поток
газов (С2Н2 и СО2) проходит циклон-сепаратор 19 и скруббер вод-
Установки пиролиза при повышенном давлении описаны в гл. V (стр. 209).
267
ной промывки 21, после чего поступает на очистку от СО2 щелочью
в аппарат 22. Ацетилен-концентрат из этого аппарата подвергается
доочистке от метилацетилена активированным углем в аппарате 23
и направляется к потребителю. Раствор метанола из абсорбера 14,
насыщенный ацетиленовыми углеводородами, поступает на
установку регенерации, состоящую из аппаратов 24 и 25, где продувается
синтез-газом. Продувочный газ вместе с десорбированными
компонентами возвращается на стадию выделения ацетилена.
Ацетиленовые углеводороды из скруббера 26 сжигают в огневых
подогревателях 2 и 3.
Промывные воды (смесь метанола с водой) разгоняются в
ректификационной колонне 27\ метанол возвращается в цикл, а вода
направляется на отмывку газовых потоков. В качестве холодильного
блока применяется абсорбционная установка, где используется тепло
горячей воды, получаемой на стадии пиролиза.
Имеются и другие схемы выделения ацетилена метанолом,
отличающиеся в основном положением стадии выделения двуокиси
углерода (может быть в начале процесса) и схемой регенерации
холода (так, в частности, для первоначального охлаждения газа пиро-
,лиза используют фракции СН4 и СО + Н2 с установки
газоразделения). Существует схема, по которой сначала удаляют высшие
ацетиленовые углеводороды, а затем ацетилен при плюсовой
температуре и 15 am. Широкого распространения процесс выделения
ацетилена метанолом не получил, он применяется в промышленном
масштабе только на установках фирмы Montecatini.
Выделение ацетилена ацетоном
Применение ацетона для выделения ацетилена из газовых
смесей не нашло столь широкого распространения, как можно было бы
ожидать исходя из того, что ацетон в настоящее время является
наилучшим растворителем, используемым для наполнения
баллонов ацетиленом. Объясняется это тем, что при давлении 1,0—1,2 am,
соответствующем парциальному давлению ацетилена в разделяемой
газовой смеси, растворимость ацетилена в ацетоне меньше, чем в
других растворителях*.
При низких температурах в ацетоне растворяется
значительное количество ацетилена (рис. VI-23), причем с уменьшением
температуры растворимость резко возрастает (рис. VI-24)33'34.
Повышение давления также способствует увеличению растворимости,
как это следует из приведенных данных, полученных при —80° С
экстраполяцией:
Рс2н2> мм Рт- ст 776 889 1040 1240 1530
X-2102, мольные доли 77,6 80,0 82,7 85,8 90,0
* При наполнении ацетиленом баллонов ацетон является не только
растворителем, но и флегматизатором взрывного распада ацетилена.
268
Растворимость ацетилена в ацетоне при низких температурах
хорошо описывается уравнением (VI-16) Кричевского—Ильинской.
Значения коэффициентов К, Р и А для ацетилена, растворенного
в ацетоне, приведены ниже:
Температура, °С
/С, мм рт. cm
—40 —50 —60
2160 1390 950
0,031 0,140 0,201
]4, см3-ат1моль 1360 5900 8060
—70 —80
580 250
0,336 0,625
12 900 22 800
Теплота растворения газообразного ацетилена в ацетоне
составляет —ДЯ=4700 кал/моль.
300
200
100
80
——
\
\;-
"V
NJ
\
ч
о
Nx
1000
t
I
I
800
600
20\— -
10
8
6
200
It
\k
w
t /
//
Л
\ л
-jy.
ГО
OfV"*1
-80^*>
30 50
-70 -50 -30 -10 10
Температура, °С
Рис. VI-23. Растворимость ацетилена
и двуокиси углерода в ацетоне при
разных температурах и парциальном
давлении газов 760 мм рт. ст.
200 ЫЮ 600 800 1000
Растборимость, мг/г
1200
Рис. VI-24. Изотермы растворимости аце-
чилена в ацетоне при разном давлении
ацетилена.
Объем раствора превышает первоначальный объем ацетона и
значительно увеличивается с повышением концентрации ацетилена.
Так, при —80° С и 60 вес. % ацетилена объем раствора в 3 раза больше
объема взятого ацетона (табл. VI-24).
ТАБЛИЦА VI-24
Увеличение объема ацетона при растворении в нем ацетилена (при 1 am)
Содержание
ацетилена
в растворе
вес. %
5
10
15
20
25
30
40
50
60
Отношение объема раствора к объему ацетона
При —40° С
1,090
1,190
1,282
1,388
1,490
При —50° С
,080
1,175
1,272
1,253
1,498
1,520
1,930
При —60° С .
1,079
1,069
1,268
1,369
1,557
1,605
2,230
При —70° С
1,085
1,176
1,267
1,376
1,495
1,620
1,940
2,478
При —80° С
,075
1,165
1,255
1,368
1,482
1,615
1,960
2,4Ю
3,070
259
Такое увеличение объема сравнимо с увеличением объема при
смешении ацетона с жидким ацетиленом. Например, при
атмосферном давлении и —80° С в 1 г ацетона растворяется 1,3 г жидкого
ацетилена. Удельный объем ацетона при —80° С равен 1,12 см3/г,
удельный объем жидкого ацетилена при —83° С равен 1,77 см3/г.
Следовательно, общий объем раствора равен 3,42 см3/г и
увеличение объема составляет 3,42 : 1,12 =3,05. Исходя из этих данных,
можно ожидать, что при —80° С и давлении выше 1 am жидкий
ацетилен будет смешиваться с ацетоном во всех отношениях.
Из всего сказанного следует, что охлажденный до низких
температур ацетон можно использовать как растворитель ацетилена
значительно более эффективно. На этом основано предложение Ю. В. Де-
лаго о транспортировании ацетилена в танках с низкоохлажденным
ацетоном, так как это более экономично, чем в баллонах при обычной
температуре.
Один из вариантов схемы выделения ацетилена ацетоном описан
ниже*. Газовую смесь, сжатую компрессорами до 10 am, очищают
маслом от высших ацетиленовых углеводородов, а затем — шелочью
от двуокиси углерода. Для регенерации масло подогревают глухим
паром и продувают природным газом при 1 ат\ отдувочные газы
(смесь природного газа и ацетиленовых углеводородов) направляют
в качестве топливного газа в подогреватели стадии пиролиза. Для
регенерации щелочи нагревают раствор образовавшегося
карбоната.
Ацетилен извлекают ацетоном при низких температурах и
давлении около 10 am в скруббере, снабженном внутренними
холодильниками для отвода теплоты абсорбции. Отходящий из скруббера
газ промывается водой для улавливания паров ацетона.
С целью выделения ацетилена рабочий раствор из скруббера
направляют последовательно в два десорбера. В первом десорбере
после дросселирования давления до атмосферного и продувки
раствора небольшим количеством азота выделяется часть ацетилена,
а также другие газы, растворимость которых в ацетоне меньше,
чем ацетилена. Во втором десорбере раствор нагревают водяным
паром, при этом выделяется основное количество ацетилена, который
затем отмывается водой от паров ацетона. Образующиеся
промывные воды перегоняют в дистилляционной колонне; ацетон
возвращают в цикл, а очищенную воду направляют на промывку газовых
потоков.
Во всех рассмотренных схемах выделения ацетилена
низкотемпературными растворителями применяются по меньшей мере
два растворителя, а иногда (например, при использовании метанола)
проводится, кроме того, доочистка ацетилена от метил ацетилена
* Подобный способ выделения ацетилена осуществлен в промышленном масштабе
в Италии (завод в г. Равенне) 35.
270
добавочным растворителем. Это осложняет технологические^ схемы
и обусловливает значительные капитальные затраты.
Низкотемпературные растворители (метанол, ацетон) так же, как и селективные,
обладают сильно токсичными свойствами. Вследствие этого
низкотемпературные растворители не получили широкого распространения
для выделения ацетилена, несмотря на их доступность и дешевизну.
Однако их важным преимуществом является возможность
использования тепловых отходов производства с относительно низкой
температурой, например горячей воды, полученной на стадии пиролиза,
для их регенерации. Особенно выгоден в этом отношении способ
с применением метанола.
Ниже приведены сравнительные данные по способам выделения
ацетилена селективным (N-метилпирролидон) и
низкотемпературным (метанол) растворителями:
Себестоимость*
Капитальные стадии выде-
затраты, % ления, %
Выделение N-метилпирролидоном 100 100
Выделение метанолом
при 10 am и —35° С 110—120 105
при 16 am и +35° С 106—110 96—97
* Включая стадию компрессии.
Видно, что выделение ацетилена метанолом (при 16 am) более
выгодно, чем N-метилпирролидоном, главным образом потому, что
в первом случае используются тепловые отходы (горячая вода).
Капитальные затраты в схемах с метанолом несколько больше,
однако нужно отметить, что необходимое для процесса количество
метанола можно получать при имеющихся мощностях, а на
производство селективных растворителей требуются значительные
капитальные затраты, которые в приведенных данных не учтены.
Более подробное технико-экономическое сравнение различных
способов выделения и концентрирования ацетилена приведено
в гл. X (стр. 405). Стоит указать, что выборочное сравнение методов
выделения носит условный характер, поскольку в большинстве
случаев имеется определенная зависимость между стадиями пиролиза
и выделения: так, при пиролизе под давлением получающаяся
горячая вода используется на стадии выделения (это в первую очередь
относится к схемам выделения метанолом). Схемы выделения
ацетилена и соответственно расходные показатели меняются, например,
даже при небольшом изменении содержания высших ацетиленовых
углеводородов в газе пиролиза. Поэтому процесс получения
ацетилена целесообразно характеризовать общими расходными
показателями, учитывающими все стадии производства.
АДСОРБЦИЯ АЦЕТИЛЕНА
Для выделения ацетилена из газовых смесей используют
адсорбционные процессы с применением активированного угля в
стационарном или движущемся слое. Наибольшее распространение нашел
V\
способ с применением движущегося слоя угля—гиперсорбция.
Поскольку ацетиленовые углеводороды на активированном угле склонны
к полимеризации с образованием взрывоопасных веществ,
высказывалось мнение, что адсорбционный способ неприменим без
тщательного предварительного удаления ацетиленовых углеводородов.
Первая установка подобного типа была опробована в США 35.
При очистке исходного газа (9,2 объемы. % С2Н2),
пропускаемого противотоком к слою угля, с низа гиперсорбера выводили
ацетилен-концентрат, содержащий 82 объемн. % С2Н2, а в качестве
головного продукта получали смесь, которую можно использовать
как синтез-газ (табл. VI-25). Основной примесью в
ацетилене-концентрате являлась двуокись углерода, ее предполагалось удалять
промывкой щелочью.
ТАБЛИЦА VI-25
Состав газов, полученных на установке гиперсорбции (США)
Компоненты
Ацетилен
Гомологи ацетилена . . .
Метан
Водород
Окись углерода
Двуокись углерода ....
Состав, объемн. %
исходная газовая
смесь
9,2
0,1
6,0
48,4
32,3
4,0
ацетилен-концентрат
82,8
0,9
0,1
16,2
[синтез -газ
0,1
6,7
54,4
36,3
2,5
Интересные данные получены на пилотной установке по
гиперсорбции, созданной в Венгерской Народной Республике37
(рис. VI-25). Установка работала на искусственной смеси газов,
не содержащих гомологов ацетилена (смесь водорода, окиси и
двуокиси углерода и ацетилена), при избыточном давлении 1 — 1,5 am.
Основной аппарат — гиперсорбер 2 состоял из четырех частей
(сверху вниз): теплообменника, адсорбционной зоны,
ректификационной колонны и десорбционной зоны. Диаметр адсорбционной зоны
25 см, ректификационной колонны — 20 см. Эффективная высота
аппарата 25 м.
Подлежащая обработке газовая смесь подавалась в нижнюю
часть адсорбционной зоны и двигалась вверх, противотоком к слою
адсорбента (уголь). Газ, выходящий с верха адсорбционной зоны
(синтез-газ), содержал лишь следы ацетилена. В этой зоне частично
поглощалась и двуокись углерода, так как адсорбционные изотермы
С2Н2 и СО2 близки. В ректификационной колонне выделялась
двуокись углерода вследствие подачи ацетилена из десорбционной зоны,
расположенной ниже. Адсорбент, содержащий ацетилен, опускался
в десорбционную зону. Там при нагревании ацетилен десорбировался,
часть его выводили, а остальное количество возвращали в
ректификационную колонну.
272
В. опытах при 1 am десорбция осуществлялась при глухом
обогреве даутермом; при избыточном давлении 1,5 am полная
десорбция ацетилена не достигалась даже при 300° С, поэтому для от-
дувки ацетилена в десорбционную зону подавали острый пар ( на 1 кг
проходящего ацетилена 0,6—0,7 кг пара). При этом учитывался
нагрев угля в десорбционной зоне теплоносителем—даутермом.
Уголь
На 1 кг ацетилена расходовалось
20—25 г угля. Активность угля
(адсорбционная емкость его по
отношению к ацетилену)
практически не изменялась при работе
установки в течение месяца. На
описанной установке удалось
получить непосредственно ацетилен-
концентрат без дополнительной
очистки от двуокиси углерода
(табл. VI-26).
При разработке метода
выделения ацетилена гиперсобцией не
был решен вопрос
предварительной очистки газовой смеси от
гомологов ацетилена, а также о
разделении парыС2Н2 — С2Н4,
адсорбционные изотермы компонентов
которой практически равны.
Поэтому для промышленного
внедрения описанного способа требуются
дополнительные
исследовательские работы.
Н. Н. Кельцев и А. Л. Халиф38
проводили лабораторные опыты
по гиперсорбции ацетилена активированным углем различных
марок. Ниже приведены данные об адсорбционной емкости
отечественных углей АГ-2 и АР-3:
Рис. Vl-25. Схема установки для
выделения ацетилена методом
гиперсорбции:
/, 4,5,6— сепараторы; 2 — гиперсорбер;
3 — питатели; 7, 8 — теплообменники;
9— газодувка.
Давление ацетилена, мм рт. cm
Адсорбционная емкость, г С2Н2 на 100 г угля:
АГ-2
АР-3
50 700
1,4
2,6
6,3
7,8
В результате опытов была выявлена возможность достаточно
полного выделения ацетилена из газов окислительного пиролиза.
Вначале из газового потока адсорбировали активированным углем
ацетилен и двуокись углерода, которые затем разделяли хромато-
графическим способом. Надо, однако, отметить, что при таком
разделении содержание двуокиси углерода в ацетилене-концентрате было
довольно высоким — 20 объемн. %.
В работе 39 описывается гиперсорбционная установка
полупромышленного типа для выделения ацетилена из газов пиролиза уГЛе-
iS в. Н. Ahtohqi? 273
ТАБЛИЦА У1-26
Состав газов, полученных на установке гиперсорбции (Венгрия)
Компоненты
Ацетилен
Водород
Окись углерода
Двуокись углерода ....
Состав, объемы. %
исходная газовая
смесь
7,0
63,3
25,5
4,2
ацетилен-концентрат
99,1
0,9
синтез-газ
68,0
27,5
4,5
водородов. Первоначально в лабораторных условиях изучалась
адсорбция в подвижном слое активированного угля. Испытания
показали, что при полном удалении гомологов ацетилена отложений
смолистых веществ на активированном угле почти не было. Однако
в связи с осложнениями при увеличении
скорости газового потока подвижный слой
оказался неприемлемым, поэтому была
опробована система с псевдоожиженным
слоем (рис. VI-26), которая оказалась
более эффективной.
Исходная газовая смесь газодувкой 7
подавалась в зону адсорбции 2. С верха
этой зоны выходили газы, часть которых
с помощью газодувки 8 использовали для
транспортирования угля.
Ацетилен-концентрат выходил из зоны десорбции 5.
Уголь, уловленный из газовых потоков,
возвращался в систему через бункер 3.
Для целей выделения ацетилена и его
очистки исследовалась возможность
использования цеолитов (молекулярных сит).
Синтетические цеолиты типа А
представляют собой щелочные алюмосиликаты
общей формулы МеО-А12О3-2SiO2-xH2O.
Полученные осаждением из водного раствора
алюмосиликаты кристаллизуют, отделяют
от маточного раствора, сушат и
прокаливают для удаления влаги.
Гранулированные цеолиты имеют поры,
соизмеримые по размерам с молекулами
адсорбируемых веществ, благодаря чему цеолиты
могут играть роль молекулярных сит. Они хорошо адсорбируют
полярные и ненасыщенные соединения.
На рис. VI-27 представлены сравнительные экспериментальные
данные 40 по адсорбции ацетилена цеолитом 5А (размер пор 5 А),
274
Рис. VI-26. Схема установки
для выделения ацетилена
в псевдоожиженном слое
активированного угля:
/ — дезинтегратор; 2 — зона
адсорбции; 3 — бункер; 4 — зона
концентрирования; 5, 6 —зоны
десорбции; 7,8— газодувки.
активированным углем СКТ и силикагелем МСМ при разных
давлениях и температурах. Из рисунка видно большое преимущество
синтетических цеолитов. Например, при парциальном давлении
ацетилена 100 мм рт. ст. и 20° С адсорбционная емкость
составляет 7,8 г/100 г для цеолита 5А, 4,7 г/100 г для угля СКТ и всего
0,9 г/100 г для силикагеля. При снижении парциального давления
относительная поглотительная способность цеолитов еще больше
растет, поэтому предлагается
применять их для извлечения
ацетилена из бедных газовых смесей,
например из газов окислительного
пиролиза.
Поглотительная способность
цеолитов с изменением температуры
изменяется незначительно,
например, активность цеолитов при
уменьшении температуры с +80
до — 78° С снижается лишь в 1,3—
2 раза, а активированного угля
СКТ в 9—14 раз. Поэтому при
выделении ацетилена цеолитами
изменение температуры не играет
существенной роли. Однако при
плюсовых температурах
целесообразно использовать цеолиты, а не
активированный уголь, так как
в этом случае их поглотительная
ММ /7/7/. CIV.
Рис. VI-27. Адсорбционная емкость
цеолита 5А, активированного угля
Способность, наоборот, повышается СКТ и силикагеля МСМ при разных
по сравнению с углем. Так, напри- температурах.
мер, относительная
поглотительная способность при парциальном давлении ацетилена 60 мм рт. ст.
для цеолитов и активированного угля равна (в %):
При —17° С
» +20° С
» +50° С
Цеолит
5А
128
180
370
Активированный уголь
100
100
100
Адсорбционные способы выделения ацетилена пока не вышли
из стадии полупромышленных испытаний, но при дальнейшем
усовершенствовании они, по-видимому, смогут конкурировать с
абсорбционными методами.
ТОНКАЯ ОЧИСТКА АЦЕТИЛЕНА
Очистка ацетилена от гомологов, в первую очередь от пропа-
диена, метилацетилена и диацетилена, является важной проблемой,
обеспечивающей получение особо чистого продукта, который
требуется для ряда химических синтезов. Наибольшее распространение
18* 275
получил метод очистки активированным углем*. При этом
обязательно предварительное удаление диацетилена, например,
промывкой селективным растворителем. В результате обработки ацетилена
активированным углем можно снизить количество ацетиленовых
углеводородов в 10—20 раз и добиться содержания диацетилена не
более 0,003 объемн. % (столько же остается суммарно пропадиена
и метил ацетилена). Очистку проводят при 1 am и 20° С,
регенерацию использованного угля — при температуре выше 250° С.
Процесс периодический, с относительно большим расходом пара.
Для очистки ацетилена-концентрата, например, от винил- и
дивинил ацетилена можно использовать активированный уголь40, сили-
кагель, а также цеолиты 41. С этой целью изучали адсорбцию винил-
ацетилена на различных цеолитах и других сорбентах
(активированный уголь и мелкопористый силикагель) при 20° С (табл. VI-27).
Из данных таблицы видно, что наилучшими сорбентами являются
цеолит 13Х и активированный уголь СКТ. Адсорбционная
способность цеолитов особенно ярко проявляется при малых парциальных
давлениях ацетилена. При давлении более 10 мм рт. ст. для
цеолитов адсорбционная способность практически не изменяется, а для
углей значительно возрастает. Коэффициент селективности
цеолитов в 2 раза выше,чем угля, хотя адсорбционная емкость их примерно
одинакова. Условия регенерации цеолитов такие же, как при очистке
углем.
ТАБЛИЦА VI-27
Результаты адсорбции винилацетилена на различных сорбентах при 20° С
Давление
мм рт. ст.
5
10
25
100
300
700
ЗА
0,2
0,3
0,6
2,5
4,0
Количество адсорбированного
4 А
4,8
5,0
6,0
12,4
12,5
13,0
на цеолитах
5Л
6,7
8,5
10,0
13,5
13,5
15,0
юх
6,8
7,0
10,0
13,9
—
винилацетилена,
13Х
8,2
10,0
11,0
15,0
15,0
17,0
г на 100 г
сорбента
на активированном
угле
АГ-2
2,1
7,0
10,0
—
22,0
—
скт
4,7
10,0
19,0
31,4
37,0
39,0
на с ил ii-
it а геле
МСМ
0,8
1,5
4,5
10,8
16,0
24,0
Для очистки ацетилена применяется также серная кислота
концентрацией не ниже 95%. При 25—40° С почти полностью
улавливаются метил ацетилен, пропадиен, бутадиен, винилацетилен и
дивинилацетилен (наиболее затруднительно отделение пропадиена
и метил ацетилена). Очистка улучшается при добавлении, например,
0,001 вес. % сульфата алюминия или 0,5 вес. % нитрата меди. Ди-
ацетилен в этих условиях выделяется лишь на 30—50%, поэтому
* Для этих целей возможно применение углей марок СД и СДО.
276
сначала приходится обрабатывать ацетилен селективным
растворителем для выделения диацетилена, а затем серной кислотой —
для очистки от других ацетиленовых углеводородов. Очищенный
ацетилен отмывается щелочью от брызг кислоты. Этот метод
применяется и для очистки карбидного ацетилена, а также в процессах
осушки ацетилена. Основным недостатком является трудность
использования отработанной кислоты, так как расход ее относительно
велик (50—100 кг на 1 т С2Н2) и в процессе очистки она
обводняется.
Возможна тонкая очистка ацетилена керосином, имеющим пределы
выкипания 220—250° С. Растворимость ацетиленовых углеводородов
в керосине при 760 мм рт. ст. и 20° С равна (в объем!объем):
Метиладетилен .... 9,2
Винилацетилен 30
Пропадиен 15
Бутадиен 30
Диацетилен керосином поглощается в значительно меньшей
степени, чем указанные углеводороды, поэтому его требуется
отделять предварительно. Очистка керосином проводится при
нормальной температуре и соотношении газ : жидкость от 2 : 1 до 2,5 : 1.
Степень очистки в этих условиях достигает 100%. Для регенерации
керосин нагревают выше 100° С, подавая острый пар. Этот способ
применяется на промышленной установке для тонкой очистки
ацетилена, полученного электрокрекингом. Преимуществом очистки
керосином является небольшое количество отходов; правда, часть
керосина нужно выводить из цикла и уничтожать, так как в нем
накапливаются взрывоопасные полимеры.
ЛИТЕРАТУРА
1. Р амм В. М., Абсорбционные процессы в химической промышленности, Гос-
химиздат, 1951, Ра мм В. М., Адсорбция газов, изд. «Химия», 1966.
2. Кричевский И. Р., Фазовые равновесия в растворах при высоких
давлениях, Госхимиздат, 1952.
3. Карапетянц М. X., Химическая термодинамика, Госхимиздат, 1953.
4. Плановский А.Н., Рамм В.М., Коган С. 3., Процессы и аппараты
химической технологии, Госхимиздат, 1955.
5. К а с а.т к и н А. Г., Плановский А. Н., Чехов О. С, Расчет
тарельчатых, ректификационных и абсорбционных аппаратов, Стандартгиз,
1961.
6. Ивановский Ф. П., Д р е й ц е р И. Г. и др., Труды ГИАП, вып. 15,
Госхимиздат, 1963, стр. 117.
7. Sachsse H., Chim. Ing. Techn., 22, № 1/21 (1949); № 7/8, 129 (1949).
8. Chem. Age, 81, № 2106, 396 (1959).
9. Chem. Technik, № 2, 123 (1958).
10. Holemann P., Hasselmann R., Forschungsber. Wirtschafts- und
Verkehrsministenums Nordrhein Westfalen, № 109 (1954).
11. Ha i degger E., Schebeny I., Szekely A., Mag. kern, fol., № 10,
365 (1958).
12. Fa user G., Chim. e ind., 42, № 2, 150 (1960).
277
13. Химия ацетилена, под ред. А. Д. Петрова, Издатинлит, 1954.
14. Ф е д о р е н к о Н. П., Методы получения ацетилена, Изд. ВИНИТИ, 1958-
15. Chem. Ztg., 84, № 8, 239—249 (I960).
16. Су cap ев М. П., ЖПХ, 34, вып. 3, 412 (1961).
17. Chem. Week, 89, № 18, 154 (1961).
18. Remond I., Rev. prod, chim., 65 (1962).
19. Б р а у д е Г. Е., Лейтес И. Л., Дедова И. В., Хим. пром., № 4,
232 (1961).
20. Howard W., S с h о с h E., Petrol. Ref., 33, № 1, 143 (1954).
21. F r i e d L., A d 1 e r S., Chem. Eng. Progr., 53, № 9, 452 (1957).
22. S t a n t о п W. H., Petrol. Ref., 38, № 3, 209 (1959).
23. Шендерей Е. Р., Ивановский Ф. П., Газ. пром., № 7, 33 (1962).
24. Ш е н д е р е й Е. Р., Ивановский Ф. П., Хим. пром., № 2, 91 (1963).
25. Б р а у д е Г. Е., Шахова С. Ф., Хим. пром., № 3, 26 (1965).
26. И в а н о в с к и й Ф. П., Шендерей Е. Р., Лап иду с А. С, авт.
свид. СССР 149529, 1961 г., Бюлл. изобр., № 16 (1962).
27. Fregaiguess,M., Chim. et ind., 46, № 5, 579 (1941).
28. X о д е е в а СМ., ЖФХ, 35, № 3, 629 (1961).
29. Ш а х о в а С. Ф., Б р а у д е Г. Е., Хим. пром., № 7, 30 (1963).
30. В г а с о n i e r F., Ind. chim. beige (XXXI Congress Intern, de Chimie indu-
striell, Liege), № 11, 167(1958).
31. Z u n d e 1 M. I., Chim. et ind., 91, № 2, 99 (1964).
32. Б р а у д е Г. Е., Шахова С. Ф., Хим. пром., № 3, 177 (1961).
33. Ц и к л и с Д. С, К о ф м а н Л. Н., Шендерей Л. И., ЖПХ, 32,
№ 9, 2012 (1959).
34. Ш е н д е р е й Е. Р., Ивановский Ф. П., ЖПХ, 37, № 7, 1557 (1964).
35. Ерофеев Н., Талызин Н., Тес н ер П. А., Газ. пром., № 6,
45 (1958).
36. Scherwood P., Erdol u. Kohle, № 12 (1954).
37. Benedek P., Szepesy L., Erdol u. Kohle, № 9 (1956).
38. Кельцев Н. Н., Халиф А. Л., Химия и технология топлив, № 12
(1956).
39. Chem. Eng. Progr., 57, № 11, 43(1961).
40. Тороче ш ников Н. С, Кольцов Н. В., Д ж а р ы л к а н о в Ж- А.,
Журн. ВХО им. Д. И. Менделеева, 5, № 6, 710 (1960).
41. Николинский А., Известия химического института Болгарской АН,
№ 3, 255 (1955).
ГЛАВА
VII
ПРОЦЕССЫ И ОСНОВНЫЕ АППАРАТЫ
ПРОИЗВОДСТВА АЦЕТИЛЕНА ИЗ УГЛЕВОДОРОДОВ
РАДИАЦИОННО-КОНВЕКТИВНЫЕ ПОДОГРЕВАТЕЛИ
ЗМЕЕВИКОВОГО ТИПА
Для высокотемпературного подогрева газовых компонентов
(до 600—650° С) в производстве ацетилена широко применяются
радиационно-конвективные подогреватели1"5. В соответствии с
технологической схемой окислительного пиролиза эти подогреватели
входят в состав агрегатов получения ацетилена, что определяет их
необходимую тепловую нагрузку, равную 0,7-106 ккал/ч.
Для этих условий нецелесообразно применять подогреватели
типа трубчатых печей, оснащенных факельными или панельными
горелками, так как при указанных тепловых нагрузках они
громоздки и неудобны. Например, при одинаковой лучевоспринимаю-
щей поверхности в радиационной зоне (26 м2) общая боковая
поверхность стен составит в змеевиковом подогревателе 30 ж2, а в
трубчатой печи 65 м2 (с учетом площади, занятой панельными горелками).
В соответствии с этим для печи с панельными горелками вес
футеровки и кожуха радиационной зоны примерно в 2 раза, а площадь
застройки в 1,5 раза больше, чем для змеевикового подогревателя.
На рис. VI1-1 показаны возможные тепловые схемы радиационно-
конвективных подогревателей. Наиболее рациональна схема а.
Противоток в конвективной (верхней) зоне обеспечивает высокую
среднюю температуру, что приводит к уменьшению
необходимых поверхностей нагрева. Прямоточная схема греющего и
нагреваемого газовых потоков в радиационной (нижней) зоне позволяет
снизить температуру стенок труб. Схема б, основанная на противо-
точном режиме, менее благоприятна; в радиационной зоне стенки
трубок сильно нагреты, так как температуры греющего и
нагреваемого потоков максимальны. При схемах виг, где применяются
смешанные режимы, можно получить по зонам более низкие температуры
стенок, что особенно важно, если трубки изготовлены из
углеродистой стали. Иногда подогреватели разделяют на секции с
нагревом газа до определенной температуры в каждой из них: нагретый
поток выводится из аппарата и после проведения технологической
операции возвращается в другую секцию.
Использование тепла радиации для высокотемпературного
нагрева газов интенсифицирует теплопередачу, позволяет осуществлять
нагрев со значительно меньшими теплопередающими поверхностями.
Кроме того, за счет больших коэффициентов теплоотдачи со стороны
279
топочных газов при высоких скоростях движения нагреваемого
потока можно снизить температуру стенок труб по сравнению с
конвективными подогревателями. Например1, для подогрева воздуха
до 700° С при температуре дымовых газов 1380° С при радиацион-
♦ ♦ ♦
Рис. VI1-1. Тепловые схемы радиационно-конвективных
подогревателей (сплошная линия — нагреваемый поток, пунктир —
греющий поток).
ном подогреве и 1100° С при конвективном подогреве минимальная
температура металлической стенки в радиационном подогревателе
760° С, а в конвективном — 900° G. Максимально допустимая
температура стенок труб в радиационной зоне
определяется материалом труб и характером
нагреваемого газа (кислород, природный газ
и т. д.).
При высоких скоростях потоков достигается
относительно низкая температура стенок труб
(рис. VI1-2), что удовлетворяет технологическим
и конструктивным требованиям. Наиболее
целесообразна скорость природного газа от 50 до
200 м/сек, при этом обеспечиваются высокие
коэффициенты теплоотдачи, снижается
температура стенки и облегчаются условия работы
металла. Нужно особо подчеркнуть, что высокие
скорости природного газа выгодны даже с
учетом расхода электроэнергии на создание
дополнительного напора, поскольку при этом
обеспечивается более длительная работа металла.
Вообще же скорость йужно выбирать в каждом
отдельном случае в зависимости от природы
нагреваемого компонента, допустимого
гидравлического сопротивления аппарата и свойств
металла.
Радиационно-конвективные подогреватели змеевикового типа
можно классифицировать по следующим теплотехническим
характеристикам2'3: по удельной тепловой нагрузке топочного объема
радиационной зоны; распределению тепловой нагрузки между радиационной
280
и0 100 200 300
Скорость, м/сек
Рис. VII-2.
Зависимость средней
температуры стенки змеевика
от скорости движения
нагреваемого потока.
и конвективной зонами; удельной тепловой нагрузке поверхности
радиационных труб.
Удельная тепловая нагрузка топочного объема радиационной
зоны. Правильным выбором этой величины определяется создание
высокоэффективной и наиболее экономичной конструкции
подогревателя.
На рис. VI1-3 показана зависимость поверхностей
радиационной Fp и конвективной FK зон от тепловой нагрузки в радиационной
зоне. Видно, что наибольшее изменение претерпевает величина Fp.
Оптимальная нагрузка составляет от 150-103 до 550» 103 ккал/(м3 • ч).
При уменьшении тепловой нагрузки ниже 150• 103 ккал1(м3-ч)
резко увеличивается суммарная тепловоспринимающая
поверхность Fp -f- FK, особенно Fp. С другой стороны, с увеличением
нагрузки выше 550-103 ккал1(м3 -ч) суммарная
тепловоспринимающая поверхность меняется мало.
Известно, что явления теплопроводности и конвекции
определяются главным образом разностью температур, а лучистый
теплообмен — температурным уровнем. Поэтому при низких
температурах теплопроводность и конвекция играют основную роль в
общей теплопередаче, а при высоких температурах основным видом
теплопередачи является излучение. Как уже говорилось, в радиа-
ционно-конвективных подогревателях температурный уровень
ограничен допустимой температурой стенки змеевиков (практически
металл труб, используемых в радиационно-конвективных
подогревателях, позволяет держать среднюю температуру стенки примерно
650° С).
Поэтому на рис. VI1-3 показана также зависимость средней
температуры стенки змеевиков радиационной tp и конвективной tK
зон подогревателя от тепловой нагрузки в радиационной зоне.
Из графика видно, что при нижнем пределе тепловой нагрузки
150-103 ккал1(м3-ч) можно иметь достаточно низкую температуру
труб в радиационной и конвективной зонах. Верхний предел
тепловой нагрузки в радиационной зоне в соответствии с
допустимой температурой стенки змеевиков понизится и составит всего
350-103 ккал/(м3-ч). Таким образом, тепловая нагрузка
радиационной зоны с учетом применяемых материалов для труб должна
быть от 150• 103 до 350-103 ккал/(м3-ч).
Распределение тепловой нагрузки по зонам подогревателя.
Распределение тепла также зависит от тепловой нагрузки
радиационной зоны (рис. VI1-4). Из графика видно, что тепловая нагрузка
выше 350 • 103 ккал/(м3'Ч) не оказывает значительного влияния1 на
распределение тепла между радиационной и конвективной зонами
подогревателя, а ниже, например, 150• 103 ккал/(м3-ч) позволяет
увеличить Qp — долю тепла, воспринимаемого в радиационной зоне.
Однако снижение тепловой нагрузки приводит к уменьшению
температурного уровня в подогревателе, что значительно влияет на
теплопередачу излучением. Отсюда следует, что оптимальной по-
прежнему остается тепловая нагрузка от 150 • 103 до 350 • 103ккал(мя • ч),
281
соответствующая количеству тепла, воспринимаемого в
радиационной зоне, 25—45% от общего, т. е. 0,25Q < Qp^0,45Q.
Удельная тепловая нагрузка поверхности радиационных труб.
Предельно допустимое теплонапряжение определяется максимально
допустимой температурой стенок труб. Это соответствует такому
распределению тепла по высоте радиационной зоны, при котором
температура внутренних стен труб сохраняется постоянной
(практически ±10% от оптимальной)
и равной максимально
допустимой температуре для
конкретных условий.
100 200300 Ш 500 600 7006
Тепловая нагрузка q</D3,
ккал/(м*-ч)
Рис. VI1-3. Зависимость поверхностей
нагрева радиационной и конвективной зон
и средних температур стенок в этих зонах
от тепловой нагрузки q в радиационной
зоне подогревателя (заштрихованный
участок соответствует оптимальному
интервалу q с учетом применяемых
материалов).
80
70
60
,50
^30
20
Ю
О 100 200 300 WO 500 600 700 800
Тепловая нагрузка q-Ю3
ккал1(мъ*ч)
Рис. VI1-4. Зависимость количества
тепла, воспринимаемого в
радиационной зоне, от тепловой нагрузки q
в этой зоне (заштрихованный участок
соответствует оптимальному
интервалу q).
Максимально допустимая удельная тепловая нагрузка
поверхности радиационных труб qTi доп. с учетом неравномерности нагрева
по диаметру и длине труб может быть определена по формуле3'4:
, доп.
— Ф1Ф2 (едоп. — О
(VIM)
где cpv и ф2 — коэффициенты, учитывающие степень неравномерности нагрева
по диаметру и длине трубы и определяемые из условий освещенности
труб (принимаем фх = 0,55 и ф2 = 0,80);
Одоп. — максимально допустимая температура стенки, °С;
t — температура нагреваемого потока, °С;
6Т — толщина стенки трубы, м\
а — коэффициент теплоотдачи от стенки к нагреваемому потоку,
ккал/(м2- ч- град);
X — коэффициент теплопроводности, ккал/(м> ч* град).
282
Далее приведены максимально допустимые расчетные удельные
тепловые напряжения для нижнего и верхнего витков
радиационной зоны подогревателя:
Нижний Верхний
виток виток
Температура, °С 425 650
Коэффициент теплоотдачи,
ккал!(м2-ч-град) 278 288
Максимально допустимое теплонапря-
жение, ккал/(м2-ч-град) 33 000 6200
Горелки для сжигания топливного газа
Для обеспечения равномерной температуры металла змеевиков
по высоте радиационной зоны подогревателя необходимо, чтобы
на каждом участке нагрева была соответствующая тепловая нагрузка.
В этой связи целесообразно рассмотреть существующие типы
горелок и способы сжигания топливного газа для условий радиационно-
конвективных подогревателей. Виды горения и характеристика
факелов, образующихся в различных горелках, широко описаны в
литературе 6~10.
При сжигании топливного газа в беспламенных (туннельных)
горелках конструкции «Стальпроект»6»7 удельное тепловое
напряжение на начальных участках радиационной зоны повышается, а
затем примерно на 1/5 высоты уменьшается на 40—50%. Последнее
происходит потому, что при беспламенном сжигании газов все тепло
выделяется в пределах туннеля горения, т. е. там процесс
горения практически заканчивается; следовательно, нельзя
регулировать удельную тепловую нагрузку по высоте радиационной зоны.
Поэтому беспламенное сжигание топливного газа в радиационно-
конвективных подогревателях вызывает определенные
технологические затруднения и рекомендуется применять факельные горелки.
Как известно, излучение любого тела, в том числе и факела,
может быть выражено формулой:
Е = аЬТ* (VI1-2)
где Е — количество тепла, излучаемого \ м2 поверхности тела, ккал1(ч*м2);
а — степень черноты тела;
6 — постоянная излучения, равная 4,9« 103 ккал1'(м2• ч- град*)\
Т — температура излучающего тела, °К.
. Для условий радиационно-конвективных подогревателей рост
температуры факела ограничивается допустимым тепловым
напряжением в топке и в случае нагрева природного газа или кислорода
температура факела может быть от 1100 до 1500° С.
Экспериментальные данные показывают6, что при сжигании газа в диффузионном
потоке со светящимся факелом удельная теплоотдача незначительно
изменяется только на начальном участке горения, а потом на
расстоянии до 70% высоты реакционной зоны остается постоянной.
При факельном сжигании газовой смеси, предварительно подготов-
283
Дымовые газы
ленной в стехиометрическом соотношении, максимальная
температура создается на границе зон воспламенения и догорания, а
положение этой границы по высоте факела определяется типом горелки9-10.
Факельные горелки, например, характеризуются тем, что
максимальная температура не может произвольно перемещаться по
высоте факела при постоянной производительности горелки.
Из всех возможных вариантов факельного сжигания газа в
турбулентном потоке для радиационно-конвективных подогревателей
представляют интерес горелки частично
завершенного смешения. Они устроены
таким образом, что при изменении
соотношения количеств основного и
дополнительного воздуха можно получать
различные конфигурации факела, а это
дает возможность регулировать
удельную теплоотдачу от факела к тепловос-
принимающей поверхности по высоте
радиационной зоны. Кроме того, в таких
горелках можно достичь повышенной
степени черноты факела, что увеличит
теплоотдачу излучением. Степень
черноты газового факела обусловливается
наличием трехатомных газов (СО2, Н2О)
и главным образом сажевых частиц.
Последние при прочих равных условиях
позволяют иметь в радиационной зоне
более низкую температуру продуктов
сгорания при постоянном радиационном
теплообмене.
Важную роль в подогревателях
играет характер движения дымовых
газов в сечении радиационной зоны, так
как образование холодных слоев газа
между факелом и трубным экраном
может существенно ослабить лучистый теплообмен. При этом
влияние слоя холодных газов между факелом и тепловоспринимающей
поверхностью тем больше, чем меньше степень черноты основного
факела и выше температура поверхности 10.
Обычно применяемые факельные горелки работают с
коэффициентом избытка воздуха а = 1,05—1,1. При таком количестве
воздуха нельзя получить температуру, допустимую для радиационно-
конвективных подогревателей, и необходима подача дополнительного
воздуха. Если его подают по периферии факела, то температура
дымовых газов меняется по диаметру подогревателя: в середине
аппарата находится слой более горячих газов, получаемых в горелке.
Эти горячие газы обтекаются холодным вторичным воздухом.
Кроме того, возможен обратный ход высокотемпературных газов,
приводящий к аутотермическому самовоспламенению свежей смеси,
284
Рис. VII-5. Схема бездиффузор-
ной инжекционной горелки:
/ — горелочный венчик с
отверстиями; 2 — смесительный канал;
3 — инжекционный смеситель; 4 —
регулирующая шайба; 5 — сопло.
Следовательно, аэродинамические условия в топочной зоне
необходимо выбирать такими, чтобы исключить указанные
нежелательные явления.
Чем выше турбулентность факела (т. е. чем больше диаметр
газовой струи), тем, вероятно, более равномерной будет температура
по его сечению, что улучшит условия теплообмена. В свою очередь,
повышение теплообмена соответствует расширению зоны горения.
Отмечается, что применение вихревых горелок значительно
повышает теплообмен в камере сгорания по сравнению с прямоточными
и тем больше, чем выше степень закручивания потока (максимально
на 70%)9.
Рассмотренные закономерности позволили сделать вывод о
целесообразности применения в радиационно-конвективных
подогревателях факельных горелок частично завершенного смешения,
обеспечивающих турбулизацию факела и равномерное распределение
его по сечению радиационной зоны.
На рис. VII-5 показана бездиффузорная инжекционная горелка,
принятая для радиационно-коивективных подогревателей с
нагрузкой 6500 м3 природного газа в 1 ч. Горение осуществляется при а —
= 0,8—0,9; подачей вторичного воздуха обеспечивается
окончательный коэффициент избытка воздуха в топке, равный 1,2—1,3.
Вторичный воздух поступает внутрь рассредоточенного факела и
способствует догоранию природного газа. Для более равномерного
заполнения шахты факелом истечение смеси газа и воздуха из горелки
осуществляется под некоторым углом к вертикали, что создает
закрученный поток. Описанная горелка позволяет иметь
равномерную по сечению и регулируемую по высоте тепловую нагрузку на
змеевики подогревателя.
Расчет подогревателей
Для проектирования радиационно-конвективных подогревателей
змеевикового типа предложена методика теплотехнического и
конструктивного расчета, в основу которой положены следующие
зависимости4- 5* п~16 :
1) зависимость теплового излучения, определяемая по закону
Стефана—Больцмана;
2) уравнение теплового баланса аппарата;
3) эмпирическая зависимость температуры излучающей среды
от максимальной температуры горения или от температуры
отходящих дымовых газов.
Для радиационно-конвективных подогревателей змеевикового типа
наиболее приемлема методика ВТИ — ЦКТИ 2« 3>п, принятая для
расчета топок котельных агрегатов, по следующим причинам:
в радиационно-конвективных подогревателях, как и в котельных
агрегатах, применяются факельные горелки;
по тепловой нагрузке в топочном объеме радиационно-конвектив-
ные подогреватели близки к котельным агрегатам;
285
конструктивные характеристики (температура стенок трубного
экрана, высокая степень экранирования лучевоспринимающей
поверхности и др.) радиационно-конвективных подогревателей
приближаются к аналогичным характеристикам пароперегревателей
котельных агрегатов.
Теплотехнический расчет. Количество тепла в радиационной
зоне можно определить следующим образом:
Qi = Q2 + Qs + Q,
где Q± — общее количество тепла, воспринятое трубным экраном в радиационной
зоне;
Q2 — количество тепла, переданное от факела;
Q3 — количество тепла, отданное кладкой с учетом обратного излучения;
(?4 — количество тепла, переданное конвекцией.
Основными составляющими являются Q2 и Q3.
Объем радиационной зоны рассчитывают по формуле:
V = 9р_±9к_ (VIb3)
<7топ. 7
где <7Т0П. — удельная тепловая нагрузка топочного объема. Как было
показано выше, для радиационно-конвективных подогревателей ^топ.
может составлять 150 • 103—350 • 103 ккал/(м3 *ч).
Лучевоспринимающая поверхность Нл (в м2) радиационной зоны
определяется по формуле2* п*12:
Н 0,79.10^
вых. + 273 )
где ^вх. и /вых. — температура греющего газа соответственно на входе в
радиационную зону и на выходе из нее;
ат — степень черноты топки при равномерном распределении трубного
экрана по ее стенкам;
g — условный коэффициент загрязнения лучевоспринимающей
поверхности.
Лучевоспринимающая поверхность связана с геометрическими
размерами радиационной зоны зависимостью:
Ял = XF3 (VII-5)
где X — угловой коэффициент экрана, зависящий от характера освещенности
экрана, от соотношения s/d и от / (s — шаг между трубками; d — диаметр
трубки; / — расстояние трубки от стены);
F9 — боковая экранированная поверхность, принимаемая в соответствии с
размерами радиационной зоны, м2.
В свою очередь боковая экранированная поверхность Рэ
связана с поверхностью змеевиков /^м.» которые могут быть
расположены на этой поверхности, уравнением:
F3 = KF3M. (VII-6)
где К — поверхностный коэффициент экрана, также зависящий от
соотношения s/d. Из уравнений (VI1-5) и (VI1-6) найдем:
/■зм. =ff (VII-7)
286
Правильный выбор значений К и X позволяет при одинаковой
расчетной величине Ял получить минимальную поверхность
змеевиков F3M.. На рис. VII-6 показана расчетная зависимость К и X
от величины sld для трубного экрана одностороннего освещения2.
Из графика видно, что для трубок диаметром rf = 100 мм при I =
= 0,8d наиболее оптимально отношение sld = 1,8; для трубок
меньших диаметров эта величина несколько уменьшается.
Конвективную зону подогревателя
можно рассчитывать обычным методом
в соответствии с [условиями работы
подогревателей на естественной тяге.' Обычно
принимают скорость движения дымовых
газов в свободном сечении конвективной
зоны 6—10 м/сек, температуру дымовых
газов на выходе из подогревателя 250—
350° С.
Коэффициент теплопередачи
конвективной поверхности можно определять по
формуле Н.16:
1,00
0,95
Кк =
(VI1-8
где осд — общий коэффициент теплоотдачи
от дымовых газов конвекцией и
радиацией; -Ш^
ат — коэффициент теплоотдачи от
стенки к нагреваемому
компоненту;
6А — термическое сопротивление
стенки;
гт — термическое сопротивление
загрязнений.
Общий коэффициент теплоотдачи от
дымовых газов конвекцией и радиацией
равен:
Рис. VI1-6. График определения
оптимальных характеристик однорядного
гладкотрубного экрана радиационно-
конвективных подогревателей:
/ — с учетом излучения футеровки при
/ = l,4d; 2 — при / = 0,8d; 3 — при / =
= 0,5d; 4 — при / = 0; 5 — без учета
излучения футеровки при / ^ 0,5^.
(VI1-9)
Частный коэффициент теплоотдачи конвекцией вычисляем по формуле:
где Л — коэффициент, зависящий от физико-химических свойств нагреваемого
компонента;
G — весовой расход дымовых газов.
Частный коэффициент теплоотдачи радиацией составляет:
ap = a'Ka'rG' (VIM1)
где а.к — коэффициент теплоотдачи излучением !:;
сс^ — степень черноты незапыленного потока *;
G' — поправка на незапыленный объем \
* Значения этих величин в зависимости от температуры дымовых газов могут
быть найдены в «Теплотехническом справочнике», Госэнергоиздат, 1957.
287
Коэффициент теплоотдачи от стенки к нагреваемому компоненту равен!
(VI1-12)
где а — коэффициент теплоотдачи для прямой трубы, равный
(здесь А' — коэффициент, зависящий от физико-химических свойств
нагреваемого компонента);
d — диаметр трубки;
R — радиус закругления среднего змеевика.
После расчета поверхностей проверяется средняя температура стенок змеевика:
а) в радиационной зоне
0,20
0,16
0,12
0,10
0,08
0,06
0,05
ОМ
0,03
Д02
Щ
\^
\
\ 1
ш
ж
\ч
\
г
К
щ
щ
\
и
^5
\
к
У'
[в
\
щ
tcp., р = 'сР.,г + -7- (VII-13)
ат
где ^ср., г — средняя температура
нагреваемого газа,
б) в конвективной зоне
''ср., к^
■t2) (VII-14)
3 5 7
2 3
5 7
2 3
Рис. VII-7. Зависимость местного ко-
эффициента трения А, от числа Re и
соотношения Rid:
l-R/d = 3,l; 2-R/d = 3,9; 3—R/d=5,\;
4 — R/d = 6,9; 5—Rid = 12; 6—R/d=2l;
7 _ R/d = 43; 8 — R/d = loo.
где tx и /2 — средняя температура
дымовых газов и нагреваемого
компонента.
Конструктивный расчет. В ра-
диационно-конвективных
подогревателях особое значение имеет
выбор конструктивных размеров
аппарата (диаметр D и высота Н).
Поскольку расчет лучевосприни-
мающей поверхности ведется через
боковую экранированную
поверхность F3 радиационный зоны, то
в зависимости от соотношения
HID величина F9 будет меняться2. Например, при объеме
радиационной зоны 10 ж3:
HID .... 12,7 7,4 4,7 3,1 2,1 1,6
F9, м2 . . . 40,0 33,4 28,6 25,0 22,0 20,0
С конструктивной точки зрения наиболее целесообразно
отношение HID =6-M0. Этот интервал хорошо согласуется с
теплотехническими и конструктивными требованиями на подогреватели
теплопроизводительностью до МО6 ккал/ч, но при
производительности выше 1-106 ккал/ч это соотношение целесообразно уменьшить
до 3-ь 6. В противном сучае получится громоздкая конструкция,
в* которой очень трудно обеспечить равномерный нагрев по высоте
зоны. Например/ для подогревателя теплопроизводительностью
288
750 000 ккал/ч при диаметре 1200 мм необходима высота
радиационной зоны 11 м.
Уменьшение величины HID вызовет отклонение от оптимальных
теплотехнических условий, но в то же время позволит работать
с меньшими колебаниями локальных тепловых напряжений на
трубчатке. Уменьшить отношение HID можно либо сократив высоту
радиационной зоны, что благоприятно сказывается на регулировании
теплоотдачи от дымовых газов к нагреваемому компоненту, либо
увеличив диаметр радиационной зоны. Последнее усложняет
возможность получения равномерной температуры по сечению, что
существенно влияет на теплоотдачу излучением. Поэтому при диаметрах
2—4 м целесообразна установка нескольких горелок. При больших
значениях HID трудно обеспечить оптимальную теплоотдачу на
верхние змеевики радиационной зоны. Для этого целесообразно
применять вторичные излучатели в виде футерованных сводов и т. п.4.
В большинстве радиационно-конвективных подогревателей тепловосприни-
мающие радиационные поверхности представляют собой змеевики. Такое
расположение наряду с достоинствами (можно создать любой шаг навивки) имеет
существенный недостаток — трудности при изготовлении и ремонте в случае прогорания
змеевиков. Поэтому тепловоспринимающую поверхность можно выполнять в виде
вертикальных трубок, соединенных сварными калачами. Такое расположение трубок
наряду с конструктивными достоинствами улучшает теплопередачу, так как
образуются смешанные тепловые потоки. Однако при этом расстояние между трубами
составляет не менее двух диаметров трубки, что не является оптимальным.
Промышленное применение в радиационно-конвективных подогревателях нашли
трубки диаметром 50—100 мм. В этом случае, как показали расчеты, применение
вертикальных трубок в радиационной зоне снижает степень экранирования на 5%
по сравнению со змеевиками. Поэтому способ расположения трубок следует
выбирать в каждом конкретном случае отдельно.
Сопротивление змеевиков радиационной и конвективной зон газовых
подогревателей можно рассчитывать обычными методами. Местный коэффициент трения X
в зависимости от числа Re и соотношения радиуса закругления змеевика R к
диаметру трубки d может быть определен по графику, приведенному на рис. VI1-7.
На рис. VII-8 показана одна из конструкций2 подогревателя,
предназначенного для нагревания природного газа с 30 до 650° С.
Природный газ поступает в конвективную зону, где нагревается
до 400° С. Дальнейший нагрев до 600—650°С протекает в
радиационной зоне. Топливный газ сжигается в бесфакельных туннельных
горелках при а = 1,05. Подогреватель работает на естественной
тяге. Путем подсоса вторичного воздуха температура греющего газа
на входе в радиационную зону снижается до 1300° С, температура
на перевале между радиационной и конвективной зонами сохраняется
800—850° С. Температура природного газа на выходе из
подогревателя поддерживается регулятором количества топливного газа.
Скорость движения газа в радиационной зоне 70 м!секу средняя
расчетная температура стенки труб в радиационной зоне 650° С,
суммарное гидравлическое сопротивление по тракту газа 0,3—0,4 am.
Материал змеевиков в радиационной зоне — сталь 1Х18Н9Т, в
конвективной зоне — Ст.З. Подогреватель в нижней части футерован
высокоглиноземистым шамотом, легковесным шамотом и диатомовым
кирпичом.
]9 В. Н. Антонов 289
Дымоиые
газы
Природный
*тття 203
реактор
ТопливныйА '
газ-^\
Рис. VI1-8. Радиационно-конвективный подогреватель:
/ — змеевики конвективной зоны; 2—коллектор; 3 — змеевики
радиационной зоны; 4 — горелка.
Анализ работы подогревателей
и выбор оптимальных характеристик
Исследование теплотехнического режима подогревателей
природного газа и кислорода показало, что они могут работать в широком
диапазоне температур нагрева (от 200 до 650° С) при изменении
газовой нагрузки от 70 до 115% от заданной. С ростом температуры
дымовых газов линейно повышается температура нагреваемого
потока, причем для подогревателя природного газа температура
возрастает резче. Отсюда следует, что превышение допустимой тепловой
нагрузки на трубный экран для подогревателя природного газа более
опасно.
Температура нагреваемого & 6ОООО\ 1 ■ i :—z—\60
потока зависит также от
соотношения количеств нагреваемого
& 60000
"%Д 50000
Ш^
%зоооо-
\ 20 000-
\ 10000 -
о
—
>
I
I гУ
— -
d-
-Ч^
/
'X*
\
//'
_1_
—
1
-30
}
ц
Щ
О
10 20 30 4/7 50 60
Весовая скорость, кг/(м2-ч)
Рис. VI1-9. Зависимость лучевосприыима-
ющей поверхности (1 и 3) и теплонапряже-
ния трубного экрана (2 и 4) от весового
расхода нагреваемого потока:
/ и 2 — для подогревателя природного газа;
3 и 4 —• для подогревателя кислорода.
и сжигаемого газов vn/vc_
С уменьшением этого
соотношения (в определенных пределах)
конечная температура
природного газа падает, а кислорода
растет. Оптимальные
соотношения vjvcx, в подогревателе
природного газа рекомендуется
от 27 до 30, в подогревателе
кислорода — в интервале 28—32.
Теплотехнические
характеристики подогревателей в
большой степени зависят от весовой
скорости нагреваемого потока
(рис. VII-9), особенно сильно
меняется тепловое напряжение трубного экрана. Повышение
весовой скорости сверх 60 кг/(м2-ч) создает слишком большое
теплонапряжение экрана. Имеются данные17, что в некоторых
подогревателях теплонапряжение трубного экрана может достигать 48 000—
75000 ккал/(м2 -ч-град). Такие высокие тепловые напряжения
повышают экономичность подогревателей, но зато при низких
напряжениях обеспечивается более длительный срок службы змеевиков и
кладки и создаются более мягкие условия нагрева, что
предотвращает, например, крекинг метана.
На основе проведенного анализа радиационно-конвективных
подогревателей различных конструкций применительно к условиям
производства ацетилена рекомендуются следующие основные
показатели:
Подогреватель Подогреватель
природного газа кислорода
Теплонапряжение топочного объема, ккал/(м3-ч-град) (150-^350) 103 (150-ь350)103
Теплонапряжение трубного экрана, ккал/(м2-ч-град) . До 50• 103 До 60• 103
Весовая скорость нагреваемого потока, кг/(м2- ч) . , 40 50
19* 291
Сравнительная характеристика различных радиационно-конвек-
тивных подогревателей природного газа приведена в табл. VII-1.
Видно, что подогреватели с бездиффузорными горелками значительно
экономичнее за счет снижения расхода материалов.
ТАБЛИЦА VI1-1
Сравнительная характеристика радиационно-конвективных подогревателей природного
газа
Показатели
Теплонапряжение топочного
объема, ккал/(м3-ч-град)
Теплонапряжение трубного экрана,
ккал/(м2-ч-град)
Лучевоспринимающая поверхность,
Объем радиационной зоны, м3 . .
Степень экранирования
Поверхность змеевиков, м1 ...
Средняя температура стенки труб
в радиационной зоне, °С
Весовая скорость газа в
радиационной зоне, кг/(м2-ч)
Расход материалов, %
нержавеющая и
низколегированная сталь
углеродистая сталь
огнеупоры
Подогреватель
с бесфакельной
горелкой
конструкции
«Стальпроект»
375-Ю3
31,4-103
32,2
13,6
0,99
80
635
31,6
100
100
100
Подогреватель
с бездиффузорной
инжекционной
горелкой
275-103
47-Ю3
28,33
11,0
0,88
64,6
696
45,8
91
83
80
АЦЕТИЛЕНОВЫЕ РЕАКТОРЫ
Реакторы для получения ацетилена из углеводородного сырья
могут быть следующих видов:
а) регенеративные и трубчатые печи;
б) реакторы с полным горением части углеводородов и
получением гомогенной газовой смеси теплоносителя и пиролизуемого
углеводорода;
в) реакторы с электрической дугой (одно- и двухступенчатый
электрокрекинг);
г) реакторы неполного горения углеводородов (газообразных или
жидких) в атмосфере кислорода.
Общими условиями работы всех аппаратов являются высокая
температура реакционной зоны, кратковременное пребывание в ней
пиролизуемого сырья и последующее быстрое охлаждение
продуктов пиролиза с целью сохранения образовавшегося ацетилена.
В реакторах почти всех указанных конструкций имеются: зона под-
292
готовки сырья (смесители, горелки)*, реакционная зона, создаваемая
за счет теплообмена между твердым или газообразным теплоносителем
и исходными газами или путем аутотермического горения
углеводородов в кислороде, и зона быстрого охлаждения продуктов реакции
(«закалка»).
Назначение горелки — дополнительное перемешивание
углеводорода и кислорода и подача рабочей смеси в реакционную зону
одним или несколькими потоками различной конфигурации. Горелки
рассчитывают, исходя из заданной производительности, типа
реактора и принятой скорости истечения газовой смеси. Ниже
приведены формулы скорости истечения W при допустимом перепаде
давления для реакторов неполного окисления. Если давление газа перед
соплом Р2 больше 1000 мм вод. ст. и соотношение давлений газа после
и до сопла меньше критического, т. е. PJPX < vKp (ДЛя метано-
кислородной смеси vKp. = 0,5), расчет можно вести по формулам
адиабатического истечения:
W =
где /ск. — коэффициент скорости, принимаемый 0,6-^0,9;
g — ускорение силы тяжести;
к — показатель адиабаты;
R — газовая постоянная;
7\ — абсолютная температура смеси перед соплом;
Рг и Р2 — начальное и конечное давление.
При соотношении давлений больше критического весовая скорость
газа равна:
A (VIM6)
\f к 7 2 \2/<к-1)
где а=|/2()
/х — площадь сечения сопла Лаваля;
v1 — объемный расход газа перед соплом.
Отсюда можно найти необходимое сечение сопла Лаваля и
подобрать скорость истечения. Диаметр отверстий или ширину кольца
горелки обычно выбирают на основе экспериментальных данных
и проверяют опытным путем. Толщина горелки зависит от метода
охлаждения горелки и от принятого материала (см. стр. 187).
В реакционной зоне любого реактора должны быть соблюдены
условия быстрого прогрева пиролизуемого сырья и поддержания
изотермического режима. Как известно, при этом в меньшей степени
будут протекать побочные процессы (например, при пиролизе с
электронагревом и в плазменных процессах, где практически осуществлен
* В регенеративных и трубчатых печах и в реакторах электрокрекинга зона
подготовки сырья отсутствует (сырье поступает непосредственно в реакционную
3°ну). Для реакторов гомогенного пиролиза и неполного окисления зона
подготовки сырья играет очень существенную роль.
293
изотермический процесс). В этих условиях уменьшаются отложения
сажи и кокса, что облегчает разработку конструкции реакторов.
Кроме того, реакционная зона должна отвечать оптимальному режиму
ацетиленообразования.
Объем реакционной зоны V можно определить по формуле:
/-\ _^
V =
3600
где Q — производительность по пиролизуемому углеводороду, мъ1ч\
т — время ацетиленообразования, сек.
Для создания изотермического процесса необходимо свести до
минимума теплопотери из реакционной зоны. Это связано с
подбором огнеупорных материалов или с созданием металлических
конструкций, охлаждаемых водой. В большинстве реакторов
применяется водяное охлаждение, например в реакторах гомогенного
пиролиза фирм Hoescht и Montecatini (рис. II1-35 и 111-38), а также
в реакторах неполного окисления (рис. V-29). Подбор
материалов, стойких к эрозии и высокой температуре, затруднен. Правда,
известны реакторы, которые полностью или частично футерованы.
Скорость движения пиролизуемого углеводорода в реакционной
зоне определяется требованиями теплопередачи и допустимым
сопротивлением аппарата. В регенеративных и трубчатых печах, где
теплообмен не является достаточно эффективным, скорости газовых
потоков составляют до 50 м/сек. Эти аппараты имеют сильно развитую
поверхность для обеспечения достаточной теплопередачи.
Коэффициент теплоотдачи от дымовых газов к стенке (нагрев
регенеративной печи) и от стенки к пиролизуемому углеводороду
(нагрев и пиролиз сырья в регенеративной и трубчатой печи) можно
определять из следующего выражения:
Р = ак + аЯЗл. (VII-17)
где р — суммарный коэффициент теплоотдачи, ккал/'(м2• ч- град);
ак — коэффициент теплоотдачи конвекцией, определяемый по известным
формулам, ккал1(м2*Ч'град)\
аизл. — коэффициент теплоотдачи излучением между стенкой и газом,
ккал!(м1 - ч- град).
Коэффициент аизл. является сложной функцией, зависящей от
излучающей способности газа и стенки, их температуры, толщины
газового слоя, размеров топки и состава газа. Формулы для этих
величин приведены в специальной литературе11-16 (см. также стр. 285).
Толщина прогреваемого слоя насадки в печах для пиролиза
углеводородов выбирается, исходя из требований быстрой отдачи
тепла, накопленного насадкой, углеводороду. При эффективной работе
регенеративной печи отдача тепла конвекцией должна быть
соизмерима со скоростью подвода тепла из глубины насадки, т. е. с ее
теплопроводностью.
В регенеративных печах Вульфа и на установке Koppers-Hasche
90% всей массы насадки составляют слои, удаленные от поверхности
294
теплообмена не более чем на 5 мм. Аппараты регенеративного типа
имеют насадку с высоким содержанием А12О3, выдерживающую
быстрый нагрев. При всех этих процессах происходит плавное
распределение температуры по длине реактора, причем максимальная
температура достигается на середине аппарата (см. рис. II1-22,
стр. 93). В трубчатых реакторах распределение температуры
регулируют панельными горелками, обеспечивающими более равномерный
нагрев трубного экрана.
Реакторы электрокрекинга разрабатываются с учетом
требований устойчивого горения электрической дуги необходимых размеров
и по возможности с уменьшенными поверхностью и сечением
реакционной зоны. Это необходимо для снижения влияния охлаждающих
стенок на процесс сажеобразования. При уменьшенном поперечном
сечении можно более эффективно увлекать газовым потоком сажу
и кокс со стенок реакционной зоны. Для процессов
одноступенчатого электрокрекинга целесообразно увеличивать скорость подачи
газа в реактор и следить за равномерностью его поступления
в зону дуги с целью повышения стабильности горения дуги.
Исследования показали, что оптимальная линейная скорость подачи газа
в разрядную камеру близка к 300 м/сек.
Один из реакторов электрокрекинга описан ниже. Он состоит
из следующих основных деталей: разрядной камеры, верхнего
высоковольтного электрода (катода), нижнего электрода (анода), пускового
электрода и устройства для «закалки». Катод и анод изготовлены
из углеродистой стали и охлаждаются водой. Катод изолирован
от корпуса гетинаксовой крышкой. В реакторе осуществлена
вихревая стабилизация дуги путем подачи газа через несколько
тангенциальных отверстий в корпусе. Равномерное распределение газа
в разрядном пространстве достигается с помощью экрана в виде
цилиндра, установленного коаксиально с корпусом реактора. Между
верхним торцом экрана и гетинаксовой крышкой есть зазор, через
который вращающийся поток газа поступает в зону дуги. В катоде
имеется цилиндрическая полость П-образного сечения диаметром
126 мм. Часть газа (2—10% от общего количества) подается в эту
полость, что позволяет регулировать напряжение и способствует
равномерному износу электрода. В аноде имеется цилиндрический
канал диаметром 95 мм и длиной 1 ж, являющийся реакционной
зоной.
Расстояние между электродами 156 мм. «Закалка» осуществляется
водой, подаваемой по трубе через 50 отверстий диаметром 3 мм,
расположенных в один ряд по окружности трубы.
Объем реакционной зоны в реакторах гомогенного пиролиза
и неполного окисления углеводородов также определяется
допустимым временем реакции. В реакторах гомогенного пиролиза
необходимо учитывать смешение двух потоков (теплоносителя и
углеводородного сырья), протекающее при большой средней разности
температур (1500—2000° С) и при высоких скоростях (100 м/сек и более),
что обеспечивает очень эффективную теплопередачу. Поэтому в реак-
. 295
торах гомогенного пиролиза рабочие условия ближе к изотермическим
чем, например, в регенеративных печах.
Габариты камеры горения определяют по допустимой величине
теплового напряжения с целью обеспечения полного горения
(практически тепловое напряжение превышает 106 ккал/(м3-ч-град).
Кислород
\
Метан'\
Длина L и диаметр D
реакционной зоны в реакторах
неполного окисления
определяются следующими выражениями:
Т __
3600 D*- 0,785
(VII-18)
где
Вода
Газ
пиролиза
w — скорость газов в реакционной
зоне, м/сек;
тац. — время ацегиленообразования,
сек;
Q — производительность по
метано-кислородной смеси, м*/ч;
К — коэффициент увеличения
объема газов пиролиза (до
«закалки»), определенный с
учетом температуры и давления
(К = 9,5 при 1 am и К =
= 2,6 при 4 am);
D — диаметр реакционной зоны, м.
На рис. VII-10 изображен
реактор с кольцевой горелкой
из нержавеющей стали,
имеющий водяное охлаждение. Для
футеровки реакционной зоны
применялся корунд и
высокоглиноземистый шамот.
Во всех процессах пиролиза
необходима «закалка»
реакционных газов, осуществляемая
водой, различными маслами или
рециркулирующим
охлажденным газом пиролиза.
Наибольшее распространение в
промышленности получила «закалка»
водой; даже при
двухступенчатом процессе (гомогенный пиролиз, электрокрекинг по методу фирмы
Hiils) в конечной стадии подают охлаждающую воду.
Ацетиленовые реакторы плохо моделируются по различным зонам,
поэтому требуется отработка принятой конструкции на
полупромышленных установках. Смесители и устройства для «закалки» газов
водой, имеющие большое значение для реакторов различной
конструкции, рассмотрены ниже более лодробно.
еа—Ьэ АВодп
\
Рис. VII-10.
Вода
Реактор с
кой:
кольцевой горел-
/ —смеситель; 2—горелочная плита; 3—зави-
хрители; 4 — реакционный канал; 5 —
форсунка; 6 —сажеочистной механизм;
7—взрывная мембрана; 8 — термопара.
296
СМЕСИТЕЛЬНЫЕ УСТРОЙСТВА
Конструкции смесителей
Смешение двух газовых потоков может происходить в
следующих случаях: за счет диффузии одного газа в другой (диффузионное
смешение); при движении двух потоков по одной оси
(концентрические потоки) или под углом (поперечные потоки); путем
завихрения одного потока относительно другого (закрученные струи);
вследствие эжектирования одного потока другим за счет разности
давлений между ними.
В общем виде необходимый путь движения газов для
осуществления перемешивания можно выразить следующей функциональной
зависимостью.
Hn = f(wl9 wu, а, а, d)
где w\ и w\\ —линейные скорости потоков / и //;
а — соотношение линейных скоростей потоков;
а — угол встречи потоков;
d — диаметр отверстия для истечения смеси.
При отношении скоростей wulwY > 1 перемешивание
значительно улучшается, что можно объяснить увеличением
турбулентности потока 37. Из этого следует, что целесообразно в допустимых
пределах повышать скорости потоков, поскольку это приводит
к сокращению длины смесительных устройств. Равномерность
перемешивания двух параллельных потоков, движущихся с разными
скоростями, сдвигается в сторону потока с меньшей скоростью.
Перемешивание потоков, движущихся под углом, происходит тем
лучше, чем больше угол встречи потоков а. При а = 0°
целесообразно одному из потоков придавать большую скорость.
Схемы наиболее распространенных смесителей, применяемых
в производстве ацетилена, изображены на рис. VII-11 - Частым
случаем является смешение двух концентрических спутных (а = 0°)
потоков, движущихся с разными скоростями (рис. VI1-11, а). При
этом перемешивание улучшается, если облегающий (внешний)
поток // имеет большую скорость, что можно объяснить сжатием струи
центрального потока /, т. е. уменьшением его диаметра и,
следовательно, пути перемешивания. Поэтому при концентрических
потоках целесообразно иметь соотношение линейных скоростей wn /wY =
= 2-г-З. Более высокое соотношение приведет к нерациональному
соотношению диаметров, поскольку с конструктивной точки зрения
диаметр центрального потока будет мал.
Выбор скоростей зависит от допустимого перепада давлений
и в пределе скорость может иметь значение критической. Для
упрощения конструкции трубчатых смесителей рационально применять
скорости облегающего потока wu = 100—150 м/сек. Для
окислительного пиролиза выбирают в качестве центрального потока
природный газ, так как потери его давления несущественны из-за
наличия избыточного давления газа в трубопроводе. С учетом соот-
297
ношения wn fwi = 2-^3 для центрального потока обычно выбирают
скорость W[ -= 40-^50 м/сек.
На рис. VII-11,6 изображен щелевой смеситель. При такой
конструкции перемешивание улучшается с ростом угла встречи
потоков а. Однако эти смесители неудобны, так как при высоких
газовых нагрузках щель должна быть очень длинной, что
неконструктивно.
Л I Л
/
л
J
I „ А
W/
В\\\П
\У\\
ч>
4
S
Рис. VII-11. Схемы смесителей для газов:
а — концентрическиII; б -- щелевой, в — поперечный угловой; г, д — поперечные струнные
В смесителе, изображенном на рис. VI1-11, в, один поток входит
в другой через щель или через отверстия под некоторым углом.
Для лучшего перемешивания целесообразно придать
облегающему потоку закрученное движение с выходом его навстречу
центральному потоку или обеспечить вход одного потока в другой отдельными
струйками. В последнем случае происходит так называемое
смешение с поперечным потоком (рис. VI1-11, г, д). Такие смесители
широко применяются при высокотемпературном и окислительном
пиролизе.
Процесс смешения на основе теории
турбулентных струй
Для расчета смесительных устройств необходимо рассмотреть
основы процесса смешения газов18'21-40. При развитом
изотермическом турбулентном движении сила сопротивления определяется
не молекулярной вязкостью, а турбулентным переносом и
перемешивание потоков не зависит от числа Re.
Как известно40, для неизотермического истечения определяющим
является критерий Архимеда:
(VII-19)
т<
где g — ускорение силы тяжести;
Л Г — изменение температуры при истечении;
d — диаметр отверстия;
Т — температура потока;
wn — скорость истечения потока.
В смесителях ацетиленовых реакторов вследствие больших
скоростей истечения критерий Аг мал и влияние его на траекторию
298
струи практически ничтожно. Процесс смешения зависит от
конструктивных размеров аппарата (диаметр и форма отверстия, шаг между
отверстиями) и от принятых скоростей потоков.
С точки зрения скоростных показателей смесительные
устройства принято характеризовать по степени турбулентности потока
и струи и по наиболее важному показателю — гидродинамическому
параметру40, т. е. по соотношению объемных расходов струи и
потока v\yjv\yn.
Интенсификация процессов смешения идет в основном двумя
путями: дроблением потока на более мелкие (переход к струям)
и повышением интенсивности и турбулентности потока (закрученные
потоки, эжектирование с закруткой и др.)29»40. По интенсивности
смешения струй различные потоки можно расположить в такой
последовательности 37»40: спутные (а = 0°), поперечные (0° < а ^ 90°),
встречные (90° < а < 180°).
Турбулентная струя характеризуется беспорядочным движением
вихревых масс. Особенностью свободной турбулентной струи является
то, что поперечные скорости в любом ее сечении малы по сравнению
с осевой скоростью, поэтому все основные характеристики струи
связаны с осевой скоростью. Закон изменения пути смешения Нп
по длине смесителя устанавливается на основании подобия
скоростных полей пограничных слоев в различных поперечных сечениях
свободного потока.
Подобие скоростных полей предопределяет подобие полей
температуры (в нагретых и охлажденных струях), полей концентрации
(в струях с примесями) и вообще всех физических характеристик,
связанных с переносом вещества струи. Поскольку изменение этих
характеристик (например, концентрации) в пограничном слое
происходит за счет перемешивания струи с окружающей средой, то об
изменении рассматриваемой характеристики судят по ее избытку
над величиной в окружающей среде.
Характер распределения избыточных значений температуры в
свободной струе аналогичен характеру распределения значений скорости,
поэтому для установления закона распределения температуры вдоль
оси основного участка струи нужно лишь вместо постоянства
количества движения использовать постоянство теплосодержания струи.
При определении теплосодержания струи по избыточным
температурам считают, что газ, подсасываемый струей из окружающей среды,
не является теплоносителем, так как его избыточная температура
равна нулю. Иначе говоря, избыточное теплосодержание всей массы
газа, протекающей через произвольное сечение струи, равно
избыточному теплосодержанию первоначальной массы, вытекающей из
сопла за равный промежуток времени.
Избыточная концентрация (разность между локальной
концентрацией примеси в струе и койцентрацией вне струи) оказывается
одинаковой в различных поперечных сечениях струи точно так же, как
сохраняется неизмененным теплосодержание(температура)21'22.
Принцип постоянства избыточной концентрации примеси дает возмож-
4 299
ность найти законы распределения избыточных концентраций в
поперечных сечениях и вдоль оси струи, причем эти законы точно
совпадают с законами распределения избыточных температур. В этом
смысле изменение концентрации примеси вдоль оси струи круглого
сечения описывается выражением:
где АСт и АС0 — значения концентраций, относящихся к сечению струи на
расстоянии Нп и к устью;
а — экспериментальная константа;
Ro — радиус устья.
Это соотношение подтверждает, что путь смешения при
турбулентной стр уе (в пределах Re до 107) не зависит от скорости и определяется
выбранным диаметром струи и ее геометрической характеристикой
(плоская, осесимметричная) 26-28.
Анализ работ 24« 26-28> 5з показывает, что вопрос расчета
турбулентных изотермических и неизотермических струй переменного
состава (поперечных и спутных) до настоящего времени не имеет
исчерпывающего решения. По-видимому, сложность задачи
объясняется тем, что изменение плотности в поперечных сечениях струи
обусловлено изменением скорости, температуры и концентрации
вещества.
При истечении турбулентных изотермических и неизотермических
струй имеется подобие скоростных, температурных и
концентрационных полей. Различие скоростных полей турбулентной струи,
втекающей в движущийся поток, и турбулентной струи, втекающей в
неподвижный поток, в первую очередь связано с определением
экспериментальной константы а, величина которой колеблется 26>28 в
пределах 0,07—0,11. Поэтому можно предположить принцип подобия
и для турбулентных струй, втекающих в подвижный поток, и как
следствие этого — сохранение закона изменения скоростей,
температур и концентраций по длине смесителя по закону турбулентной
струи, втекающей в неподвижный поток. Наличие движущегося
потока способствует лучшему перемешиванию, и путь смешения будет
в действительности короче, чем это можно рассчитать по теории
турбулентных струй.
Смешение с поперечным потоком. Большое значение для
процесса смешения имеет механизм взаимодействия струи с поперечным
потоком.
При исследовании истечения круглой струи в поперечный поток
в широком интервале диаметров (5—22 мм), температур потоков
(300—575° К) и струй (300—860° К), углов встречи (30—150°) и
скоростей истечения (wjwc = 3-^20) было установлено39"42, что
характер изгиба струи не зависит от температуры потока и струи, если
обработку результатов проводить по гидродинамическому параметру.
Вся аэродинамика струй различной формы, развивающихся
в поперечном потоке, сводится к закономерностям развития экви-
300
валентной свободной круглой струи 41. Из условия теор-ии подобия
следует, что относительный шаг между струями входит в условия
однозначности и может в некоторых пределах стать определяющим
параметром. Для определения влияния шага были проведены опыты
по развитию струй в поперечном потоке, ограниченном стенками
(канал). Опыты с соплами диаметром 5,1, 10, 25 и 20 мм показали,
что уравнение оси струи, развивающейся в свободном поперечном
потоке, можно с некоторыми условиями распространить на струю,
развивающуюся в ограниченном поперечном потоке.
Относительную глубину проникания струй в поперечный поток
можно определить по формуле 40:
где h — абсолютная глубина проникания струи;
d — диаметр отверстия для истечения струи;
Ks — безразмерный эмпирический коэффициент (при проникании
единичной струи в поперечный поток Ks = 2,2; в случае нескольких
струй Ks зависит от относительного шага sid между струями в ряду 40,
например при s/d = 5-М5 Ks = l,6-f-l,8);
wc и wn — линейные скорости соответственно струи и потока;
Тс и уп — удельный вес соответственно струи и потока.
Наоборот, если задаться величиной А, то по формуле можно
определить диаметр отверстия d для истечения струи.
При расчете струй различного сечения было определено, что
наибольшей проникающей способностью обладает круглая струя.
Ю. В. Иванов40-41, изучавший проникание струй в поперечный
поток, определял закономерности равномерного распределения струй
в потоке, приняв, что при этом будет обеспечено достаточное смешение.
Он показал, что падение скорости и температуры по оси струи,
развивающейся в поперечном движущемся потоке (так же, как и в
неподвижном потоке), является функцией диаметра d ее устья. При
этом скорость изменяется вдоль оси струи, развивающейся в
свободном поперечном потоке, тем интенсивнее, чем меньше отношение
скоростей wc и wn в устье. Полное смешение фиксируется
определенной величиной HJd равной 4—10 (для d = 20,8 мм). Эта величина
достигается при Тс/Тп — 1 и wjwn = 4.
Все приведенные выше данные справедливы для тех случаев,
когда скорость истечения струи невелика либо ее направление
совпадает с направлением потока.
При истечении газа в неподвижную среду той же плотности,
находящуюся в канале, направление и величину скорости можно
приближенно получить 42»43 геометрическим сложением осевой
скорости истечения и ее составляющей, нормальной к плоскости
отверстия. Поэтому при расчете дальнобойности струй, для которых
направление скорости совпадает с направлением потока, нужно
учитывать изменение угла их истечения в поток. При этом формула (VI1-21)
примет вид:
301
При расчете равномерного распределения струй в потоке
представляет интерес определение диаметра струй, принявших направление
потока. При исследовании струи, втекающей в поперечный поток,
экспериментально было установлено, что отношение диаметра
струи dC) измеренного на расстоянии h от устья, к этому расстоянию
оказалось постоянной величиной 43:
-^ = 0,75 (VII-23)
Ю. В. Иванов установил следующую зависимость между
диаметром и глубиной проникания двух струй, размещенных в ряду
с одинаковым относительным шагом и вытекающих в поперечный
поток при одинаковых условиях:
Как видно из соотношения, при увеличении диаметра струи,
например, вдвое при прочих равных условиях увеличится в 2 раза
и глубина проникания этой струи в поток.
Расчет смесителей для процесса
окислительного пиролиза
Правильный выбор конструкции и режима работы смесителя
особенно важен для процессов окислительного пиролиза — здесь один
из газовых потоков является окислителем, а это обусловливает
особые требования с точки зрения техники безопасности.
Конструкция смесителя должна обеспечивать оптимальную степень
смешения * компонентов и исключать возможность образования
взрывоопасных смесей: полученная в смесителе гомогенная газовая смесь
должна поступить в реакционную зону раньше, чем закончится
период индукции 19»20.
Расчет смесителя должен включать определение геометрических
размеров аппарата, пути и времени перемешивания, а также
установление условий безопасной работы с получаемой
мешано-кислородной смесью.
Перемешивание газов, имеющих разные удельные веса и
температуры, является довольно сложным процессом. Поэтому представим
механизм перемешивания в виде двух стадий. Начальной стадией
является проникание струй в поток. Для данного случая можно
ограничиться рассмотрением условий проникания всей струи (если
принимается принцип струйного распределения) либо невозмущенного
ядра струи (если струя проникает в поток на малую глубину).
Следующая стадия — перемешивание образовавшихся струй в
поперечном потоке до получения заданной степени смешения.
* Степень смешения — отношение фактической концентрации заданного
компонента (при окислительном пиролизе — кислорода) в смеси к расчетной величине.
302
В смесительных устройствах кроме турбулентного переноса
наблюдается явление молекулярного переноса. При расчете
смесителей нужно было бы учитывать оба механизма переноса, а также
влияние на них температуры. Однако для упрощения исключим
возможность молекулярного переноса и рассмотрим только
турбулентный перенос в соответствии с теорией турбулентных струй.
Распределение струй. Первую стадию процесса перемешивания
газов (проникание струй в поток) можно с достаточным приближением
рассчитать с помощью зависимостей, /##
характеризующих поведение струи
в поперечном потоке [формулы
(VII-21) h(VII-22)]. Для условийсме- I 80
шения метана и кислорода в процессе
окислительного пиролиза при одина-
1
ковой температуре подогрева компо- §
нентов формула (VI1-21) примет вид: §
фру
а) струя — метан
Wn
(VII-25)
б) струя — кислород
$ 20
£ (VII-26) о
f
л
А
т
tffi^i
/
.—-о
w
При одинаковых диаметрах d
глубина проникания будет тем больше,
Соотношение
Рис. VI1-12. Зависимость глубины
проникания струй в поперечный
чем больше скорость wc. Поскольку поток от диаметра отверстий и от
величина h прямо пропорциональна соотношения скоростей струи и по-
плотности газа в струе, целесообраз- тока ПРИ s/d= 3 : 5 (сплошные ли-
нии — подача кислорода струями:
НО подавать струями кислород как пунктир-подача метана струями)!
более тяжелый газ.
На рис. VI1-12 показана расчетная зависимость глубины
проникания струй, истекающих из отверстий различных диаметров, от
соотношения скоростей wjwn. Для расчета взято отношение О2 :
: СН4 = 0,6 и величина /Cs = 1,5 при s/d = Зч-5. Из графика видно,
что проникание при одинаковом соотношении скоростей сильно
зависит от диаметра отверстия d.
Для упрощения расчета за начало поперечных струй,
распространяющихся с углом раскрытия 22—24°, можно принять границу
потока. Пренебрежение участком, где струи распространяются как
затопленные, при расчете дальнобойности дает погрешность, не
превышающую +5%.
Диаметр и скорость струй при встречных потоках выбирают
такими, чтобы при самой низкой скорости потока дальнобойность
струи при отсутствии столкновения на оси смесителя составляла бы
h/Dc = 0,5-^-0*8 (Dc —диаметр смесителя). Если желательно свести
к минимуму смешение струй в точке столкновения, то диаметр струй
Должен определяться условием DJd ^ 6—8, основанным на резуль-
3Q3
татах исследования скорости турбулентной диффузии при
взаимодействии струй 21.
Определение пути перемешивания. Путь перемешивания можно
с достаточной для инженерных расчетов точностью определять на
основе теории турбулентных струй с учетом некоторых
экспериментальных данных 21"24. По теории турбулентных струй можно найти
минимальное расстояние, при котором количество вещества в струе
(концентрация Сс) будет равно количеству этого вещества в потоке
(концентрация Сс) при достижении заданной степени смешения.
В условиях окислительного пиролиза (для О2) Сс = 100%, а
Сс = 37%. Изменение концентрации примеси вдоль оси струи
круглого сечения описывается выражением (VI1-20).
Методы определения пути перемешивания Яп, при котором в
потоке будет достигнута средняя концентрация вещества струи, для
смесителей поперечного смешения и эжекционных смесителей в
соответствии с теорией турбулентных струй несколько различны.
В случае поперечного смешения струя входит в поток и ось струи
принимает направление его оси. Для упрощения примем, что диаметр
струи, принявшей направление потока, равен диаметру отверстия d>
т. е. dc = d. В этом случае, имея фактический запас по значению Яп,
определение пути перемешивания можно значительно упростить.
Для эжекционных смесителей это упрощение не требуется, поскольку
в них спутные потоки. На основе формулы (VI1-20) можно вывести
условие для определения пути перемешивания в зависимости от
диаметра отверстия. Подставляя значение экспериментально
определяемой константы а и значение Ro = d/2, после несложных
преобразований получим условие смешения:
-^-^ 7,5 ч-11,5 (VII-27)
Определение времени смешения. Для определения времени смешения можно
рассмотреть безразмерное отношение 35 пути перемешивания Нп к диаметру
осевого потока D. Использование величины D в качестве определяющего параметра
нельзя достаточно строго обосновать из-за отсутствия данных о структуре
внутреннего потока, однако при таком методе анализа можно сравнивать различные
конструкции аппаратов. Если поток имеет сложную конфигурацию (например, кольцо
или сумма отверстий), то более правильно определять безразмерное соотношение
с учетом ^экв. потока. Безразмерное отношение HJD по разным данным 35>4°
составляет 2,5—4.
Время смешения можно определить по формуле 30:
Тс-. =4=- (VI1-28)
где w — средняя линейная скорость, полученная при взаимодействии обоих
потоков.
Удобнее выражать тсм. через объемные расходы газов в струе и потоке соответственно
(vc и vu) и скорость потока на входе в смеситель wn- Для упрощения примем, что
в точке входа поперечных струй поток сохраняет свою начальную скорость, а струи
теряют скорость. В действительности поток приобретает некоторую скорость за счет
количества движения в поперечных струях, но пренебрежение этой величиной дает
достаточно большой запас времени, что позволяет считать предлагаемый метод
надежным.
304
В конце процесса смешения оба потока приобретают одинаковую скорость wKOH
Определим w как среднее арифметическое в соответствии с условием 35 постоянства
импульсов т:
"-ъ-"
где q = jx YY
Конечная скорость суммарного потока равна: доко„ = (Щ\ + w)/2. После
преобразований получим
где Р = (1 + <?)/(! + <?/2).
Диаметр потока в начальном сечении для смесителей поперечного смешения
в условиях окислительного пиролиза (600° С, 1,5 am, поток—метан) равен:
D = УЧтШ-шъг = °'0028 Vlt (VII"29)
При окислительном пиролизе время смешения тсм> должно отвечать условию:
^-^Тсм.^Тинд. (VII-30)
По экспериментальным данным для смесителей поперечного смешения#пД) =
= 2-=-4 при wn^> 100 м/сек. При wn = 100 м/сек примем для дальнейших расчетов
максимальную величину HJD — 4. Тогда
W W Wn г
Подставляя в формулу (VI1-31) значения £>, получим:
j-j- (VII-32)
При изменении q от О до оо величина р изменяется от 1 до 2, т. е. Р мало влияет
на время смешения.
Из формулы (VI1-32) можно определить время смешения для
процесса окислительного пиролиза (Р = 1):
тсм. = 0,011 л[-\ (VII-33)
При скорости потока (метан) wn = 100 м/сек и его объемном
расходе vn = 6000 д3/</ (что применяется, например, в промышленных
аппаратах) время смешения равно 0,7-10"3 сек. Практически, как
уже говорилось, период индукции метано-кислородной смеси равен
около 0,51 сек, т. е. имеется запас времени примерно в 725 раз.
Следовательно, для процесса смешения с поперечным потоком
выдерживается условие:
7,25.102tcm.<W
На основе изложенного и проведенных автором исследований
можно сформулировать следующие оптимальные условия работы
смесителей поперечного смешения:
а) безразмерное соотношение HJd ^ (30—100), т. е. в*4—10 раз
больше теоретического (см. формулу VI1-27).
20 В. Н. Антонов 3Q5
СН4
2ot
б) величина HJD ^ 4;
9 9
в) гидродинамический параметр ocyc/vnyn ^ (1 —10).
Расчет эжекционных смесителей. Расчет эжекционных
смесителей основан на теории эжекции, разработанной для паро-воздуш-
ных эжекторов и водоструйных насосов 40» 44~47. При окислительном
пиролизе в отличие от полного горения эффективность эжекции
должна быть незначительной: исходные метан и кислород поступают
в смеситель под давлением и смешиваются в основном за счет своей
кинетической энергии, поэтому явление собственно
эжекции играет небольшую роль.
Главными частями эжекционного смесителя
(рис. VI1-13) являются камера смешения У,
горловина 2, диффузор 3 и так называемый кратер 5.
Камера смешения обеспечивает частичное
выравнивание скорости и концентрации потоков, а
основное выравнивание концентрационных полей
происходит в диффузоре. Конфузор 4 (небольшое
сужение после диффузора) служит для
выравнивания скорости суммарного потока по всему сечению
после диффузора, поскольку имеется определенное
торможение у стенок (пристеночный эффект),
обусловливающее заметное снижение скорости потока
в этих местах.
В качестве наиболее приемлемой для условий
окислительного пиролиза принята методика
расчета эжекционных смесителей, предложенная
В. А. Спейшером 48. По этому способу
соотношение площади горелки /5 к площади сопла (или
суммы сопел) fx (см. рис. VI1-13) определяется
следующей формулой:
Рис. VII-13. Эжек-
ционный смеситель:
/—камера смешения;
2 — горловина; 3 —
диффузор; 4—
конфузор; 5 — кратер.
h.
h
где
(VII-34)
: Wvo2;
i —соотношение удельных весов метана и кислорода, i-
ти — коэффициент инжекции; тн = уо2^О./иСН4^сн4'
VO2 и исн4 — объемные расходы кислорода и метана;
I — суммарный коэффициент местного сопротивления смесителя
и горелки.
Для условий производства ацетилена при одинаковой температуре
подогрева исходных компонентов имеем ти = 1,2 и i = 0,52. С
учетом этих величин выражение (VI1-34) может быть преобразовано:
£ = 3,60(1+4-)
(VII-35)
Задаваясь значениями £ и /5, можно найти величину /х. Другие
конструктивные размеры смесителя можно определять по
нижеследующим формулам. Угол раскрытия диффузора 3 необходимо
выбирать таким, чтобы исключить возможность пристеночного отрыва
306
потока44"47. Расстояние tv от выходного сечения сопла до конца
горловины можно подсчитать по величине необходимого
оптимального угла 2а (угол вписывания газовой струи в ограниченный
стенками объем):
Ll 2tga
Опытным путем установлено, что оптимальный угол вписывания газовой струи
в цилиндрическую горловину равен 2а и составляет примерно'15°. Длину
горловины /2 можно определить в зависимости от отклонения Да от оптимальной
величины а (отклонение вызвано существующим на практике расширением газовой
струи в горловине):
/ Sin
/2 - 2 sin2 а ^ — ai)
где Да = 1,5°. Наиболее важным участком является длина диффузора /3, зависящая
от угла его раскрытия:
Путем сопоставления фактических и расчетных данных можно установить, что
для эжекционных смесителей в ацетиленовых реакторах соотношение fjf1 равно
200—215. Для этих смесителей отношение пути перемешивания к диаметру
отверстия (в данном случае струя—метан) HJd = 55; отношение пути перемешивания
к диаметру потока HJD = 30. В этом случае время смешения составляет:
30D
Поскольку диаметр потока согласно формуле (VI1-29) равен
D = 0,0028]/^ (VII-36)
то время смешения определится из уравнения:
тсм. = 0,034 l/^i- (VII-37)
Практически wn & 450 м/сек и тсм# = 3- 10"2 сек. При этом выдерживается
условие смешения:
^см. ' ^ U ^^^ ^инд.
Анализ смесителей различных конструкций
и рекомендации по их разработке
Из всего сказанного выше можно установить факторы,
влияющие на эффективность работы смесителя. Основное условие
смешения, которое должно соблюдаться в смесителе любой конструкции,
выражается уравнением (VI1-30):
20* * 307
Время смешения тсм. для поперечных и эжекционных смесителей
при wn = (100-7-450) м/сек можно приближенно оценивать по
формуле:
f- (VII-38)
В этом уравнении А —коэффициент, зависящий от типа смесителя
и равный 0,011 для поперечного смешения и 0,084 — для эжекцион-
сн4 Т4 с.н*
а 6 6
Рис. VI1-14. Схемы промышленных смесителей:
а — поперечный смеситель с мелкими струями;
б — то же, с крупными струями; в — эжекционный
смеситель.
ного смешения. В указанном диапазоне скорости wn время смешения
составляет:
тсм.<(10-2--10-3) сек
Приведенные нами данные хорошо совпадают с
литературными 35-36. Например, по А. М. Цирлину 35, время смешения тсм# ^
^ (10~2-т-10~3) сек, по данным С. П. Гориславца 36 для смесителей
с поперечным смешением при закрученных потоках тсм. (3,8-10"2^-
-т-6' 10"2) сек. Как показывают расчеты, величина wn мало влияет на
время смешения, что должно учитываться при разработке новых
конструкций аппаратов. При ^п^100 м/сек (эта скорость
поддерживается на практике в смесителях различных конструкций)
выдерживается условие смешения:
тсм. (102-М03)<тинд.
Это хорошо подтверждается многими данными, поскольку
изменение газовой нагрузки в пределах 40—60% от номинальной не
влияет на выход ацетилена.
Другой важной характеристикой смесителей является
безразмерное соотношение пути перемешивания Яп и диаметра отверстия ' d.
Для окислительного пиролиза HJd ^ (30^-100).
308
Это условие выдерживается во всех рассмотренных конструкциях
смесителей.
Еще одной важной характеристикой является гидродинамический
параметр, который в соответствии с характером процесса при
окислительном пиролизе должен быть больше или равен 1:
1
На рис. VI1-14 изображены схемы некоторых смесителей,
используемых в промышленности, а в табл. VI1-2 приведена их
сравнительная характеристика. Из данных таблицы видно, что достаточная
степень смешения (HJd = 30 ч-100) достигается в широком диапазоне
размеров (при d3KB. = 0,1-^0,0035) и отношений скоростей метана и
кислорода (гидродинамический параметр от 0,85 до 12,9).
Следовательно, для процессов окислительного пиролиза возможно
применение смесителей различных конструкций.
ТАБЛИЦА VII-2
Сравнительная характеристика смесителей ацетиленовых реакторов
Показатели
Отношение Hn/d
Эквивалентный диаметр
отверстия d3KB.
Отношение Hn/d3KQm . . .
Гидродинамический
параметр v2cyjvlyn
Время пребывания метана
и кислорода в смесителе,
сек
Поперечный смеситель*
(струя—кислород)
с мелкими
струями
90—180
0,104
10,6
6,7
с крупными
струями
27
0,069
16
0,85
0,012
Трубчатый
смеситель с
поперечным
смешением
(струя—кислород)
100
• 0,0035
70
3,2
0,004
Эжекционный
смеситель
(струя-
метан)
55
0,044
100
12,9
0,02
1 В поперечном смесителе с крупными струями выход ацетилена был наименьшим.
Ниже обобщены основные технологические требования, которые
нужно учитывать при выборе конструкции смесителя для реакторов
окислительного пиролиза19:
1. Смеситель должен иметь по возможности малое сопротивление.
2. Смешение необходимо проводить в кратчайшее время, которое
должно быть меньше периода индукции.
3. В смесителе должна быть предотвращена возможность
проскока пламени из реакционной зоны, а в случае проскока —
обеспечена возможность его быстрейшего обнаружения.
Самыми надежными с точки зрения безопасности можно считать
устройства, совмещенные с горелочной плитой реактора и имеющие
конструкцию эжекционного смесителя. Число отдельных элементов
309
смесителя можно определять из условия допустимых скоростей
истечения метана и кислорода. К эжекционным можно отнести и
многосопловые смесители, при этом расстояние от смесителя до горелочной
плиты должно быть таким, чтобы обеспечить равномерное
распределение газового потока по отверстиям плиты. В идеальном случае число
элементов такого смесителя должно соответствовать числу элементов
в горелочной плите (в кольцевом реакторе это необязательно).
Эжекционный смеситель достаточно эффективен, однако его основным
недостатком являются довольно большие габариты и, как следствие
этого, длительное пребывание метано-кислородной смеси во
взрывоопасных условиях.
Поперечному смесителю свойственны недостатки эжекционного
смесителя — он также имеет большие габариты. Преимуществом
поперечного смесителя по сравнению с эжекционным является
меньшее гидравлическое сопротивление.
РАСЧЕТ ПРОЦЕССА «ЗАКАЛКИ»
РЕАКЦИОННЫХ ГАЗОВ
Факторы, влияющие на скорость «закалки»
«Закалка» реакционных газов проводится при двух возможных
типах химических реакций. В первом случае равновесная
концентрация целевого продукта в конечной смеси достаточно велика и задача
«закалки» состоит в том, чтобы при быстром охлаждении газов
сохранить возможно большее количество целевого продукта. Во втором
случае равновесная концентрация целевого продукта при высокой
температуре мала и для его сохранения необходима принудительная
«закалка», чтобы остановить реакцию на определенной ступени.
К таким процессам относится производство ацетилена.
Для процессов второго типа дать обоснованные рекомендации
по выбору наиболее целесообразных параметров «закалки»
значительно труднее. В этом случае необходима последовательная теория
математической оптимизации процесса. Из-за отсутствия такой
теории находят относительно простые соотношения, позволяющие
оценить скорости «закалки» в тех или иных конкретных условиях
и выяснить факторы, способные обеспечить необходимую скорость
охлаждения реакционных газов 49~51.
При различных способах получения ацетилена (окислительный
пиролиз, электрокрекинг, пиролиз) общее время процесса равно
сумме времени реакции и времени «закалки». Время реакции по
производственным и опытным данным колеблется от 0,002 до 0,01 сек.
Время «закалки» обусловлено контактом горячих реакционных
газов с охлаждающей водой и определяется конструктивными
особенностями зоны «закалки». Практически оно составляет 10—20%
от времени реакции.
Для сохранения целевого продукта необходимо так организовать
процесс, чтобы температура реакционных газов достаточно быстро
310
упала от значения ТА, при котором проходила реакция, до
величины Tf, ниже которой целевой продукт (в данном случае ацетилен)
может сохраняться без разложения достаточно долго 51~53.
Необходимая скорость понижения температуры dTldx, зависящая от типа
вещества и вида реакции, может быть оценена по формуле:
Если принять, что dTldx = const, то за время Ат <т (ТА)
температура реакционных газов снизится настолько, что ацетилен
сможет быть сохранен достаточно долго. Оценка скорости «закалки»
по формуле (VI1-39) является завышенной, так как по мере снижения
температуры Еремя т (ТА) будет быстро возрастать, вследствие чего
может снизиться скорость «закалки».
Стадии «закалки». В зоне «закалки» происходят следующие
процессы: торможение реакции разложения ацетилена; нагревание
охлаждающей жидкости от начальной температуры на входе до
температуры ее испарения; испарение жидкости; нагревание паров.
Необходимо учесть ряд гидродинамических факторов, в первую
очередь влияющих на величину эффективной поверхности воды, рас-
пыливаемой форсунками. Струя воды, попадающая в поток газа,
подвергается динамическому воздействию со стороны этого потока
и при относительно больших скоростях газа и воды распыляется на
крупные и мелкие капли. Согласно данным В. Г. Левича 54, средний
радиус L капель можно оценить по следующим формулам:
L = —?Ц- L ъ (3 -f- 5) б' (VII-40)
(мелкие капли) (крупные капли)
где а — поверхностное натяжение жидкости;
рг — плотность газа;
wr — скорость газа;
6'/2 — радиус кривизны капли.
Для подсчета поверхности распыленной воды воспользуемся
методикой, разработанной Ю. Л. Хаитом51. Число мелких капель 7г,
образующихся в реакторе в единицу времени, равно:
dn n dVB
d,T 4 dT cc3
ТГЯ
где ц — доля распыленной воды;
—~- 3iL3 — объем капли;
dVB/dT — объем воды, подаваемой в реактор в единицу времени: в
=
CLT 4
б — диаметр отверстия;
b — скорость воды,
311
За время Ат образуется число капель Дя, которые распределяются
в объеме газа Д1/г:
где DT — диаметр газового потока. Предполагая, что капли не
успевают испариться, среднее число капель в единице объема Д1/г можно
определить так:
_^LJ dn
ь^г Dp»r dx
Подставив значение dnidx, получим:
nd2w о3оу5
п = 0,2т| f^- (VI1-42)
Г
С учетом значения L поверхность F мелких капель равна:
F = 4nL*n = 2,5л n6%frPr (VII-43)
г
Из этой формулы видно, что поверхность распыленной воды
пропорциональна общему количеству воды, подаваемой со скоростью шв
через п отверстий диаметром 6.
Скорость «закалки». Для рассмотрения основного параметра
процесса «закалки» — скорости — можно оценить скорость
охлаждения, не прибегая к детальному рассмотрению всех протекающих
реакций, и рассчитать лишь общую суммарную энергию, отдаваемую
потоком горячего газа при контакте с каплями жидкости. Л. С. Полак
и Ю. Л. Хаит, рассматривая процесс «закалки» в плазмохимическом
реакторе, показали 49-5i? что роль достаточно интенсивных стоксов
(отрицательных источников теплового переноса) играют жидкость,
поглощающая тепло из газа, и пар, образующийся при ее испарении.
Напишем уравнение теплового переноса для процесса «закалки» в ацетиленовом
реакторе окислительного пиролиза (в цилиндрической системе координат):
сР рг Vr-^-= — Qz (VII-44)
где Vr — объем газа;
Qz — энергия, идущая на испарение жидкости Qx и нагрев пара Q2(Qz = Qi + Q2)-
Для определения Qx и Q2 воспользуемся следующими уравнениями 51. Энергия,
передаваемая жидкости в единицу времени при соударении молекул газа с
поверхностью мелких капель, находящихся в объеме газа, равна:
dE^zdF (VI1-45)
где 8 — коэффициент, учитывающий долю кинетической энергии, передаваемой
молекулами при ударе:
г==р у _i^_ (VII-46)
(здесь К — постоянная Больцмана; Т — температура, °К; m — масса
молекулы);
312
dF — поверхность мелких капель, равная:
dF = 4я!2/г dVB = FdVB (VI1-47)
F — площадь поверхности мелких капель, находящихся в единице объема
газа dV:
F = 25 n№w^ (VII-48)
Dzra
Величина Qx равна dE/dV, ее можно определить по уравнению (множители
порядка единицы опущены):
dE - гр -тип п62(°в^гРгР -\f~KT
^гРгР -\f~KT am AQ\
2a у -ffr (VH-49)
Найдем величину энергии, поглощаемой при нагревании пара, образовавшегося
при испарении капель:
(VII-50)
где Спар. — теплоемкость пара;
г — теплота парообразования.
Следовательно
[г»
1 +-J-J cnap.
Ti
.(T)dT
(VII-51)
Введя величину со' (безразмерный кинетический коэффициент передачи энергии)
и подставляя в выражение Q2, получим:
со' = (о
= © [ 1 + С-^Р (Tt - Г,)] (VII-52)
Т1<Т<Т2.
Тогда
6
КТ
^- -(vn-53)
Из уравнения (VI1-44) при стационарном режиме работы ацетиленового
реактора найдем постоянный во времени градиент температуры:
Отсюда можно найти скорость изменения температуры dTldi элемента газа,
движущегося со скоростью wr через зону «закалки»:
у (V.,-55)
313
Формула (VII-55) связывает скорость «закалки» с величинами,
характеризующими газовую фазу (давление Р, температура Г, масса молекул газа т, удельная
теплоемкость газа (ср), жидкость (поверхностное натяжение а), режим работы
реактора (шг и wa), взаимодействие газа с жидкостью (величина г], определяющая долю
распыливаемой жидкости, величина со, определяющая долю энергии, которая
передается жидкости молекулами газа).
Для приближенных оценок можно принять т]0) & 1, поскольку т] -> 1, так как
струйки жидкости подвергаются интенсивному разрушению, а крупные капли —
дальнейшему дроблению; со -> 1, если учитывать высокую температуру газа 51.
Полное изменение температуры для зоны «закалки» длиной / равно:
/
«■,-/■£*-(■£■)•«
О
где выражение в правой части получено на основании теоремы о среднем интеграле,
а величина (дТ/дт*) согласно этой теореме представляет промежуточное значение
дТ/дт в некоторой точке с координатой т*, лежащей внутри зоны /.
Можно определить длину зоны «закалки» /, положив, что А/ (Т) = Та — Tf.
Если на отрезке / температура монотонно падает, то величину / можно определить
из следующих соотношений:
/
J -g- dx = TA - Tf или / = fjffL (VII-57)
Зависимость дТ/дх от т можно приближенно определить по формуле (VI1-54)
для величин, отвечающих условию 0 ^ т* ^ /.
Преобразуем уравнение (VI1-55), введя для удобства расчетов весовые расходы
воды GB и газа Gr. .Если примем, что
Bn62 = Усек. =
и отношение wrID2r подставим в виде
WBn62 = Усек. = —2-
Y в
wT yrwrwr _ Угшг
~ ~ ~^
то окончательная формула будет такой:
дт
(VII-58)
Для расчета по формуле (VI1-58) можно пользоваться следующими числовыми
значениями:
ср = 3,7 ккал!(кг* град) — по водороду;
а = 59 дин/см — по воде;
уг/ув = 0,01» Юг2 (по газу пиролиза при 1 am с учетом температуры).
По формуле (VI1-58) можно подсчитать изменение градиента
температуры в условиях окислительного пиролиза:
-J^- = 0,0166 -§*- w2r (VII-59)
Из этой формулы видно, что скорость снижения температуры
прямо пропорциональна скорости газа wr. Поэтому в
высокоскоростных реакторах (туннельные реакторы с кольцевой горелкой) темпе-
314 *
ратура должна снижаться быстрее, чем, например, в многоканальных
реакторах, где объемный расход реакционных газов меньше.
Рассмотрим влияние отношения GJGr на скорость «закалки»
При окислительном пиролизе это отношение равно:
GB __ 5-Ю3 _ q
~<57 "" 0,56-103 ~~
где 5» 103 — весовой расход воды на «закалку», кг на 1000 ж3 реакционных газов;
0,56- 103 — вес 1000 м3 газа, кг.
При изменении GB/Gr в 1,5 раза скорость «закалки» меняется очень
незначительно, например при ^г = 2-104 см/сек:
-^- = 1,66-10"2.9(2-104)2= 6,0-107 град/сек
dT
dx
= 1,66-10"2-9-1,5 (2-104)2 = 9,0-107 град/сек
Практически время «закалки» остается без изменения при
значительном увеличении расхода воды. Опытные данные показывают, что
время нахождения газа пиролиза в зоне «закалки» в различных
конструкциях ацетиленовых реакторов составляет около 10% от времени
ацетиленообразования:
Ат = 0,1 Атац. = 0,003 • 0,1 = 3 • 10"4 сек
Для процесса окислительного пиролиза Т = 1500° С. Отсюда
можно найти скорость «закалки» в промышленных условиях:
-|^- = 0,5-107 град/сек
Минимальное отношение GB/Gr для обеспечения указанной
скорости «закалки» можно определить по формуле (VI1-59):
дТ/дх 0,5-107 П7г
V Gr Дин. ~ 1,66- 1(Г2а£ 1,66-Ю-2 (2-104)2 ~ U'/0
что составляет примерно 0,42 м3 воды на 1000 м3 газа пиролиза.
Указанные теоретические данные по скорости «закалки» хорошо
согласуются с фактическими. При широком изменении расхода воды
(до 100%) на «закалку» в процессе окислительного пиролиза
концентрация ацетилена в реакционном газе практически не меняется, но
изменение величины GJGr даже на 20% дает изменение расхода воды
на 10 мд на 1 m ацетилена (5—10%). В табл. VII-3 приведена
сравнительная оценка зоны «закалки» в различных реакторах
окислительного пиролиза. В промышленных реакторах удельные нагрузки по
воде меняются почти в 2 раза *, но выход ацетилена примерно
одинаков.
* На некоторых установках расход воды доходил до 15—17 м3 на 1000 ж3 газа
пиролиза,
315
ТАБЛИЦА VI1-3
Сравнительная характеристика зоны «закалки» в различных реакторах окислительного
пиролиза
Показатели
Вариант I
Вариант II
Вариант III
Вариант IV
Объем зоны «закалки»,
м*
Сечение зоны «закалки»,
м2
Температура, °С
газа после «закалки»
воды на «закалку»
горячей
холодной
воды после «закалки»
Удельная нагрузка по
воде, м3 на 1000 м3 газа . .
Плотность орошения,
м3/(м2-ч)
Число форсунок
общее
на 1 м2 сечения . .
0,045
0,245
80—86
27—36
50—60
5—7
200—285
32
* 130
0,045
0,245
80—86
27—36
50—60
10
350
192
334
0,038
80
70-75
30-35
75-80
9,2—11,0
510
90
720
90
80
40—50
80
7
175
120
580
Из сказанного следует, что оптимальные условия «закалки»
(расход и температура воды) точно не определены. Это приводит
к излишним затратам, связанным с подачей и последующим
охлаждением больших количеств воды, а также создает трудности при
конструировании зоны «закалки».
Выбор параметров процесса «закалки»
В зоне «закалки» наряду с торможением разложения ацетилена
происходит частичное улавливание образовавшейся в процессе сажи.
Поэтому для «закалки» целесообразно использовать горячую воду,
так как при повышенной температуре повышается смачиваемость
сажевых частиц и, следовательно, улучшается процесс сажеулавли-
вания.
При выборе температуры воды необходимо учитывать тот факт55,
что растворимость ацетилена в воде с ростом температуры понижается.
Поэтому для уменьшения потерь ацетилена с водой температура ее
должна быть максимально высокой, практически не менее 70—80° С.
Повышение температуры, например, с 60 до 80° С позволяет
уменьшить относительные потери ацетилена более чем в 2 раза. Однако
высокая температура воды на входе в зону «закалки» приведет к
малому перепаду температур и, следовательно, большому расходу воды
на «закалку». Оптимально допустимая температура отходящей воды
может быть не выше 80° С, что определяется возможностями
замкнутых водооборотных циклов.
Зона «закалки» должна обеспечивать большой теплосъем и поэтому
характеризуется очень интенсивным теплообменом между газом и
316
водой. Например, в условиях «закалки» средние объемные
коэффициенты теплоотдачи достигают (0,5^-1,0) • 106 ккал/(м3 -ч-град). Такие
высокие коэффициенты обеспечиваются следующими факторами-
средняя скорость газа в зоне «закалки» 20—50 м/сек, средняя
логарифмическая разность температур газа и воды в условиях прямотока
в реакторе составляет 400° С (а для процесса электрокрекинга —
порядка 900° С). Например, при охлаждении газа в испарительных
скрубберах (процесс, в какой-то мере подобный «закалке»)
коэффициенты теплопередачи 56~58 составляют только 200—
400 ккал/(м2 • ч • град).
Выбор параметра оптимизации. Как было ранее показано,
изменение расхода воды в 1,5—2 раза практически не влияет на скорость
«закалки». Поэтому для определения оптимальных условий «закалки»
(минимального расхода воды) нужно ориентироваться на другие
показатели, а именно: на тепловой баланс системы и наибольшую
степень очистки газа от сажи. Вторая величина при достаточно
совершенной системе очистки (например, в мокропленочных
электрофильтрах) практически не влияет на режим «закалки», поэтому
главным является оптимальный тепловой режим стадии «закалки»
и дальнейшего охлаждения газа. Тепловой баланс процесса
«закалки» может быть представлен выражением:
Q, =Q2 + Q3
где Qx — тепло, получаемое водой от газа;
(?2 — тепло, расходуемое на испарение воды; Q2 = GBcpr (r — теплота
испарения);
<?з — тепло, расходуемое на нагрев пара; Q3 = GB (I — ф)
С технологической точки зрения интересно определить
соотношение между Q2 и Q3. В процессе теплосъема теплота
парообразования г больше величины At&c. Следовательно, чтобы Q2 было больше
Q3, целесообразно при «закалке» вести охлаждение за счет испарения
воды. В этом случае упрощается конструкция зоны «закалки»
(уменьшаются габариты, сокращается число форсунок, облегчается их
расположение и т. п.) и снижается расход воды.
Тепловой баланс дальнейшего охлаждения газа после «закалки»
может быть выражен уравнением:
Qi = Qs + Qe
где <?4 — общее количество тепла, выделяемое при охлаждении;
<?5 — количество тепла, выделяемое при охлаждении водяного пара;
Q6 — количество тепла, выделяемое при охлаждении газа пиролиза.
Чем больше водяного пара получится в процессе «закалки»,
тем больше воды потребуется на его конденсацию. Введя понятие
«влагосодержание газа пиролиза», характеризующее как стадию
«закалки», так и стадию охлаждения, можно определить оптимальное
влагосодержание d (или коэффициент ф испарения воды) на стадии
«закалки», при котором расход воды на общее охлаждение газа будет
наименьшим.
317
Необходимо также рассмотреть режим «закалки» с точки зрения
теплопередачи. Теплообмен в процессе «закалки» может быть
выражен уравнением:
Q = Fa M (VI1-60)
где F — поверхность, образующаяся при распылении воды:
а — коэффициент теплопередачи.
При постоянной разности температур А/ интенсивность
теплообмена будет зависеть от F и а.
Согласно формуле (VI1-43) суммарная поверхность капель воды
пропорциональна количеству воды и в общем виде может быть
представлена формулой 59> 60:
F = lnGB
В ней /п — коэффициент пропорциональности:
/„ = /(<р, А/, АФ)
где ф — коэффициент испарения воды;
А* — температурный потенциал процесса;
АФ — характеристика применяемой форсунки.
При А^ = const и АФ = const поверхность капель
пропорциональна коэффициенту испарения ср. Этот коэффициент связан в
первую очередь с величиной А/, что является отличием
высокотемпературных процессов, и с характеристикой форсунки АФ. При
постоянном расходе воды GB поверхность F может меняться в зависимости
от коэффициента пропорциональности /п.
Коэффициент теплопередачи, отнесенный к 1 кг распыляемой
воды (а/п), может быть представлен выражением 60:
Усек,р) (VII-62)
где Я — плотность орошения, кг/(м2-ч);
^сек.Р — весовой расход воды, кг/ч (иСек. — секундный расход; р — плотность
воды).
Экспериментально установлено, что величина а/п уменьшается
с ростом плотности орошения и возрастает с увеличением весового
расхода воды. Отсюда следует, что процесс теплообмена при
охлаждении газа распыленной водой через форсунки можно
интенсифицировать либо путем повышения поверхности F (т. е. за счет увеличения
подачи воды), либо путем увеличения коэффициента теплопередачи а/п,
г. е. фактически за счет уменьшения количества воды.
Из приведенного анализа можно заключить, что оптимизация
процесса «закалки» с точки зрения как теплового баланса, так и
процесса теплопередачи при распылении воды должна сводиться к
определению минимального количества воды, подаваемой на «закалку»,
в зависимости от влагосодержания газа пиролиза d после «закалки»
(или к определению коэффициента испарения <р).
Определение оптимального режима «закалки». В процессе
«закалки» охлаждается ненасыщенный газ, поэтому вначале
охлаждение газа идет при его постоянном теплосодержании за счет испарения
318
воды. Влагосодержание газа растет, температура воды на этой стадий
остается постоянной и практически равной температуре воды на входе
в зону «закалки». По достижении точки росы начинается конденсация
содержащихся в газе водйных паров, его влаго- и теплосодержание
уменьшаются, а вода нагревается до конечной температуры на
выходе из аппарата.
Из этого следует, что «закалку» целесообразно осуществлять
в две стадии. Вначале необходимо подавать горячую воду, что
исключает период нагрева воды и интенсифицирует процесс испарения.
После насыщения газа для его дальнейшего охлаждения нужно
подавать по возможности холодную воду. Поскольку вода на выходе
из зоны «закалки» должна иметь высокую температуру (70—80° С),
то чем больше разность температур газа и воды, тем интенсивнее
будет теплообмен в зоне конденсации.
Количество тепла, которое отдает газ, равно:
где V — объем газа (сухого), ж3;
сг и сп — теплоемкость газа и пара, ккал/(град- м3);
Ad — изменение влагосодержания газа в процессе (Ad = d± — d2), кг/м3;
Atr — изменение температуры газа (А^г = tx — /2), °С.
При заданном влагосодержании газа количество воды (в кг1ч),
расходуемое на «закалку», можно определить по формуле 57' 59:
e V(cr + Ad)Atr
q>('-'i) + (i-q>)(<2-<i)
где i — теплосодержание водяного пара при температуре газа t2;
t[ и t'2 — начальная и конечная температура воды.
Величина ф (i — t\) показывает долю тепла, которое можно
снять за счет испарения воды, а величина (1 — ф) (/2 — h) —
долю тепла, снимаемого за счет нагрева воды.
Исследуем зависимость GB от ф. При ф —■* 0
G,
Практически t2 < 100° Си/к 30° С, тогда GB = Q/70.
При ф —> 1
Г Q = Q
в , _ ^ 620
где f ^s 650. Следовательно, при «закалке» расход воды может
колебаться приблизительно в 10 раз:
70 ^=^в^= 620
На рис. VI1-15 показано изменение расхода воды, необходимой
Для «закалки» в процессе окислительного пиролиза, в зависимости
319
от коэффициента испарения ср. Изменение начальной температуры
воды существенно влияет на ее расход только в области значений
Ф ^> 0,5. Следовательно, при большом значении <р расход воды на
«закалку» не зависит от ее начальной температуры. Это означает, что
неиспарившуюся после «закалки» воду можно использовать вновь.
Из рис. VI1-15 видно, что расход воды стремится к пределу,
соответствующему минимальному количеству воды (1,5 м31ч), если
в процессе «закалки» она полностью испаряется, т. е. ф = 1.
Следовательно, 1,5 м31ч —
минимальный расход горячей воды в так
называемой зоне испарения. Из
графика можно также
определить расход воды в зоне
конденсации. Например, при ф = 0,5
и средней температуре воды 50° С
расход горячей воды (75° С)
в зоне испарения составит, как
уже говорилось, 1,5 м31ч, а
расход холодной воды (25° С) в зону
конденсации будет равен 2,7—
— 1,5 - 1,2 м3/ч.
Изменение конечного влаго-
содержания газа после «закалки»
в зависимости от коэффициента
испарения воды при разных
температурах показано на рис.
VII-16.
Исследования процесса
«закалки» газов окислительного
пиролиза в широком интервале отношения GB/Gr=5-M2 (табл. VII-4)
показали, что в этих условиях концентрация ацетилена в
реакционных газах остается постоянной (около 7 объемн. %).
Коэффициент ф в процессе меняется от 0,1 до 0,4, а количество испаряемой
воды колеблется от 1,2 до 0,7 м3 на 1000 м3 газа.
ТАБЛИЦА VI1-4
Режим «закалки» в туннельных реакторах окислительного пиролиза
2 4 6
Расход Воды, м3/ч
10
Рис. VI1-15. Зависимость расхода воды
(на 1000 м3 таза пиролиза) на «закалку»
от коэффициента испарения воды и
начальной температуры при 1,07 am.
Расход воды
на «закалку»
м3 на 1000 м3
газа
3,0
4,0
4,5
5,0
5,5
6,0
6,5
7,0
Отношение
°в/вг
5,4
7,2
8,0
8,6
9,9
10,8
11,6
12,5
Температура, ° С
газа после
«закалки»
_
80
82
80
83
83
81
82
воды при
«закалке»
33—65
33—62
33—61
33—61
33—58
33—55
33—51
33—49
Коэффициент
испарения
воды ф
0,40
0,30
0,25
0,20
0,20
0,18
0,15
0,10
Концентрация
ацетилена в
газе пиролиза
объемн. %
7,2
7,2
7,3
7,4
7,2
7,1
7,3
7,2
320
Процесс теплосъема при малых расходах воды
несколько меньшей суммарной поверхности капель F ^^четСумПрИ
шения расхода воды GB и увеличения коэффициента испарений"™)
и одновременно с некоторым увеличением коэффициента
теплопередачи а/п за счет понижения плотности орошения Я в 1,75 раза.
Фактический расход воды GvB = Зч-7 м3/ч превышал расчетный (0,4 м'3/ч)
в 7—15 раз. Указанное обстоятельство объясняет, почему при
Дф = const, несмотря на изменение подачи воды, поверхность
распыляемой жидкости обеспечивала достаточный контакт и
практически неизменный градиент температуры
дТ/дт. Уменьшение фактического расхода
воды на «закалку» ниже 3—7 м31ч нецелесо-
Фбмм
ЬО
0,8
0,6
о,и
0,2
1
I
1
0,6 0,8 1,0 1,2 1,4 1,6 1,8
Общее благосодержание газа d, кг/м3
Рис. VI1-16. Зависимость влагосодержания газа
от коэффициента испарения воды при разной
начальной температуре.
Рис. VII-17. Схема
форсунки для
тонкого распыления
воды:
/ — направляющий
конус; 2— четырехза-
ходный шнек; 3 —
входной патрубок.
образно, так как при этом ухудшается очистка газа от сажи в зоне
«закалки».
На основании сказанного выше можно рекомендовать для
«закалки» режим с большими коэффициентами испарения (ср ^ 0,5),
т. е. процесс теплосъема при увеличенных коэффициентах
теплопередачи за счет относительно малой плотности орошения в зоне
«закалки», например порядка 250 м3/(м2-ч). Это обстоятельство очень
существенно при разработке реакторов большой производительности
(свыше 10 000 m ацетилена в год). С учетом опыта работы различных
ацетиленовых реакторов можно рекомендовать следующие
оптимальные величины для проведения «закалки»:
1) объемный коэффициент теплоотдачи
(0,5-М).106 ккал/(м3-ч-град)]
2) скорость движения газового потока 20—50 м/сек;
3) угол раскрытия форсунки 10—15°;
4) плотность орошения 250—300 м3/(м2 -ч).
21 В. Н. Антонов 321
Процесс «закалки» должен проходить в две стадии: орошение
горячей водой и орошение холодной водой.
Конструкции форсунок, их число и расположение нужно
выбирать отдельно для каждого конкретного случая. Форсунки
целесообразно устанавливать таким образом, чтобы их факелы
перекрывали друг друга, а поток воды был направлен под некоторым углом
к потоку газа. Методы расчета и выбора форсунок подробно описаны
в специальной литературе 57> 61. Схема одной из промышленных
форсунок показана на рис. VI1-17.
Выбор режима охлаждения газа после «закалки»
Эффективность процесса охлаждения газа после «закалки»
в скрубберах и количества необходимой воды определяются влаго-
содержанием и температурой поступающего газа. Поэтому режим
«закалки» нужно выбирать с учетом последующего охлаждения
газа в скрубберах. Параметры газа влияют на условия его последу-
Газы на
Вода *_
на^акал-
W
Вода ..- п
охлаждение
Рис. VI1-18. Схема ацетиленового реактора со
скруббером для охлаждения и очистки газа от сажи:
/ — реактор; 2 — зона «закалки»; 3 — скруббер.
ющей очистки от сажи. При сухой очистке (батарея циклонов и др.)
' температура газа после «закалки» должна быть 200—250° С, т. е.
выше точки росы для данных условий. При улавливании сажи
мокрыми способами (скрубберы с горячей водой, электрофильтры)
целесообразна температура газов после «закалки» около 100° С.
На рис. VI1-18 показаны реактор окислительного пиролиза и
скруббер для охлаждения реакционных газов. Рассмотрим параметры
совместной работы обоих аппаратов.
Расход воды на охлаждение насыщенного или ненасыщенного
газа в скруббере определяется по формуле 57:
322
где Сохл. — расход воды, кг/ч;
V — объем газа, м*;
сГ и сп — теплоемкость газа и пара, ккал/(м3-град);
Atr — изменение температуры газа в скруббере (Л/г — t\ — t'2), °C;
Л<2 — изменение влагосодержания газа в скруббере, кг/м3;
iH — начальное теплосодержание водяного пара при температуре t'u
ккал/м3;
tH и tK — начальная и конечная температура воды в скруббере, °С.
Конечная температура воды tK должна быть, как и в процессе
«закалки», максимально высокой, чтобы уменьшить потери
ацетилена.
На рис. VII-19показано
изменение расчетных
расходов воды на «закалку» и
охлаждение газа в
производстве ацетилена
окислительным пиролизом в
зависимости от
влагосодержания газа, т. е. от конечной
температуры газа в зоне
«закалки». Из графика
видно, что в зависимости
от начальной температуры
воды, подаваемой на
«закалку» и охлаждение,
меняется оптимальное влаго-
содержание,
соответствующее минимальному
суммарному расходу воды (при
одинаковой температуре
воды в зонах «закалки» и
охлаждения точки
пересечения сплошных и
пунктирных линий отвечают
минимальным расходам
воды в соответствующих
«стадиях»).
0 ОА 0,8 1,2 1,6
Влагосодержание d газа после реактора, кг/м3
Рис. VI1-19. Зависимость расхода воды на
«закалку» (сплошные линии) и на охлаждение
(пунктир) от влагосодержания газа после
«закалки» при давлении 1,03 am и разной
начальной температуре воды (расчет на 1000 ж3 газа).
)
При разной температуре воды, подаваемой на «закалку» и
охлаждение газа, что обычно наблюдается в производственной практике,
по этому графику также можно выбрать оптимальное влагосодержа-
ние d. Например, при средней температуре горячей и холодной воды,
подаваемой на «закалку», 50° С, температуре воды на охлаждение
газа 36° С и влагосодержании газа 0,8 кг/м3 суммарный расход воды
на обе стадии составит 24,8 м3 на 1000 м3 газа. В этих же условиях,
но при влагосодержании газа после «закалки» 1,5 кг/м3 общий расход
воды будет 21,4 м3. Снижение составит 15%, причем в основном за
счет уменьшения расхода воды на «закалку». Габариты аппаратуры
Для охлаждения газа практически не увеличатся, поскольку повыше-
21* 323
ние начального влагосодержания паро-газовой смеси (с 0,8 до
1,5 кг/м3) не скажется на теплообмене.
Ранее говорилось (см. рис. VII-15, стр. 320), что наименьший
расход воды на «закалку» отвечает высоким значениям коэффициента
испарения ер ^ 0,5. Влагосодержание газа при этом должно быть
выше 1,4 кг/м3 (см. рис. VII-16, стр. 321), а суммарный расход воды
на «закалку» и охлаждение, как следует из данных рис. VII-19,
будет меньше, чем, например, при режиме «закалки» с ф^ 0,5.
АППАРАТЫ САЖЕОЧИСТКИ И ОХЛАЖДЕНИЯ ГАЗОВ
Процесс очистки реакционных газов от сажи делится на две
ступени — грубая очистка при большом содержании сажи и тонкая
доочистка. На первой стадии применяют аппараты скрубберного
типа, иногда с насадкой в виде уголков, а обычно без насадки,
поскольку она сильно забивается частицами сажи. Для тонкой до-
очистки используют трубы Вентури, мокропленочные электрофильтры
и коксовые фильтры. Очищенный газ подвергается охлаждению в на-
садочных или пенных скрубберах (последние более эффективны).
В некоторых схемах электрокрекинга метана применяют аппараты
для улавливания сажи в сухом виде — батареи циклонов,
рукавные фильтры, однако эксплуатация рукавных фильтров связана
с большими затруднениями: они быстро забиваются полимеризу-
ющимися на ткани соединениями.
В табл. VI1-5 показаны аппараты, применяемые для очистки
реакционных газов от сажи и смол при различных способах
производства ацетилена.
ТАБЛИЦА VI1-5
Аппараты очистки реакционных газов от сажи при производстве ацетилена
различными способами 65~70
Способ производства
ацетилена
Аппаратура
для грубой очистки
для тонкой доочистки
Пиролиз
Электрокрекинг
Окислительный
пиролиз
при атмосферном
давлении
•при повышенном
давлении
324
Орошаемые
скрубберы
полые
Циклоны или батареи
циклонов. Полые
скрубберы
Полые скрубберы или
скрубберы с насадкой из
уголков
Полые скрубберы
Турбулентные промыва-
тели. Насадочные
скрубберы, орошаемые маслом
Турбулентные
газопромыватели (промывка водой
или маслом)
Мокропленочный
электрофильтр. Коксовые
фильтры
Турбулентные
газопромыватели. Насадочные
скрубберы, орошаемые
горячей водой
Полые скрубберы
Полый скруббер (рис. VI1-20) представляет собой цилиндрическую
башню, в которой газ проходит сверху вниз, а вода распыляется
через форсунки, размещаемые поодиночке или группами таким
образом, чтобы все поперечное сечение скруббера было перекрыто струями
воды. Избыточное давление воды на форсунки не менее 5—6 am,
скорость движения газа в поперечном сечении скруббера 1—1,5 м/сек.
Расчет полого скруббера рекомендуется вести
в следующем порядке65> 66.
1. Определяют количество тепла, которое
требуется отвести от газа (все объемы взяты при 0° С
и 1 am):
d1cn)(t1-t2) (VII-64)
где
Q — количество отводимого тепла, ккал/ч;
V — объем сухого газа, м3/ч;
с — теплоемкость газа, ккал/(м3- град);
dx — начальное влагосодержание газа (в
данном случае — после «закалки»), кг/м3;
сп — теплоемкость водяного пара,
ккал/(кг- град),;
t\ и ^2 — начальная и конечная температура
газа, °С.
2. Определяют среднюю разность температур
в скруббере:
М = *i ~ h ,
ОО U *1 «"2
Z,6 lg —
(VII-65)
В этой формуле tK—точка росы (температура, выше
которой жидкость при соприкосновении с газами не
нагревается). Точка росы в основном определяется
начальной температурой газа и его влагосодержа-
нием.
3. Рассчитывают полезный рабочий объем
скруббера:
Q
V =
(VII-66)
Рис. VI1-20. Полый скруббер:
/—корпус; 2—коллектор с
форсунками; 3 — гидравлический
затвор.
Здесь К — объемный коэффициент теплопередачи. В полых скрубберах этот
показатель зависит от условий распыления воды и коэффициента ее испарения,
температурного режима и многих других факторов и колеблется от200до 400/с/сал/(л13- ч- град).
Известны аппараты с насадкой из уголков, где коэффициент К достигает
10 000 ккал1(м3-ч-град).
4. Определяют диаметр скруббера, принимая, что скорость газа на выходе не
должна превышать 1 — \,Ь м/сек.
Турбулентные газопромыватели
Принцип действия турбулентных газопромывателей основан на
использовании энергии динамического газового потока для
распыления вводимой в него воды. Например, при скорости газа 100 м/сек
струя воды, поступающая через мелкие отверстия, дробится на
325
капли размером около 10 мк\ при скорости газа 150 м/сек
средний размер капель составляет несколько микрон. По данным
В. Н. Ужова 65« 66 число капель, образующихся в 1 см3 газа при
скорости 100 м/сек, равно 106, а их суммарная поверхность в 1 м3 газа
составляет 500 м2. Раздробленная на мельчайшие капли вода,
сталкиваясь со взвешенными в газе частицами сажи и смол, способствует
агломерации этих частиц. Под действием центробежной силы,
возникающей за счет подачи газа, укрупненные частицы сажи оседают
в последующем циклоне-каплеуловителе.
На рис. VII-21 показана схема
установки турбулентного
газопромывателя (трубы Вентури).
Очищаемый газ поступает в трубу Вентури /.
В горловину б через сопла диаметром
5—8 мм подается распыленная вода,
и там образуется сплошная завеса
из водяных капель. Газ, проходя
через горловину со скоростью 90—
120 м/сек (скорость выбирают исходя
из характеристик сажи),
сталкивается с завесой жидкости и разрывает ее
на капли. Величина образующихся
капель обратно пропорциональна
скорости газового потока. При большой
скорости газа капли соударяются
с частицами взвешенной в газе сажи
и друг с другом. После уменьшения
скорости потока в диффузоре в
образуются капли диаметром 10—20 мк, легко улавливаемые в
циклоне-каплеуловителе 2. Вместе с водой там из газа удаляются
остатки сажи и смол.
В литературе описано использование турбулентных промывателей
для очистки газа, получающегося в процессе электрокрекинга
метана. На рис. VI1-22 приведена схема очистки газов
электрокрекинга на заводе фирмы Htils 67.
Газы, содержащие около 30 г/ж3 сажи, проходят последовательно
через циклоны 1 и 2, в которых улавливается основное ее количество.
В скруббере 3 газ охлаждается водой до 47° С, при этом содержание
сажи снижается до 350 мг/м3. В трубе Вентури 4 газы промываются
маслом при 52° С и вводятся тангенциально в колонну 5, куда
противотоком поступает масло (30 т/ч) при 52° С. Количество масла
составляет примерно 0,75 л на 1 ж3 газа, сопротивление трубы 4
450 мм вод. ст. Из колонны 5 масло, содержащее сажу, стекает
в сборник 10, откуда через холодильник 9 возвращается обратно
в колонну 5. В систему циркуляции вводится дополнительно 0,25 т
в 1 ч чистого масла и такое же количество выводится на регенерацию.
Тонкая очистка газов водой проводится в трубе Вентури 6 до
содержания сзжи 2—2,5 мг/м3. Окончательно газ охлаждается в колонне 7,
326
Вода с I
саже и \
Рис. VI1-21. Схема турбулентного
газопромывателя:
1 — труба Вентури (а — конфузор;
б — горловина; в—диффузор); 2—цик-
лон-каплеуловитель.
Для нормального протекания процесса сажеочистки важен
строгий температурный режим в аппаратах 3, 5 и 7, так как при
уменьшении температуры газа ниже точки росы конденсируется вода и
образуются стойкие эмульсии масло—вода—сажа. В первой трубе 4
при 52° С (это на 2—3° С выше точки росы) давление паров масла
незначительно, но все же достаточно, чтобы покрыть частицы сажи
масляной пленкой.
Для процессов, сопровождающихся образованием больших
количеств пирогенетической влаги, схема и технологический режим
очистки несколько иные. Например, при получении непредельных газов
Вода но охлаждение
Рис. VI1-22. Схема очистки газа в процессе
электрокрекинга:
1,2 — циклоны; 3 — скруббер-охладитель; 4 — труба Вентури
с впрыском масла; 5—колонна для промывки маслом; 6— труба
Вентури с впрыском воды; 7 — колонна водной промывки;
8 — насосы; 9 — холодильник; 10 — сборник.
из жидких углеводородов (процесс неполного окисления) 95%
образующейся сажи должно быть выделено при температуре выше
точки росы. Поэтому газ сначала охлаждается в трубчатом
холодильнике, после чего в трубе Вентури выделяется сажа. В этом случае
возможно образование эмульсии масла в воде и выделившийся
водный конденсат можно легко отделить от масла. При использовании
скруббера-охладителя, как, например, для процесса
электрокрекинга, из-за большого количества воды образуется, наоборот,
эмульсия воды в масле, которую отделить очень трудно.
На рис. VII-23 показана схема установки трубы Вентури для
очистки газа, содержащего большое количество водяных паров67.
Из реактора / газ поступает в первую трубу Вентури 2, куда
впрыскивается масло, которое затем вместе с сажей отделяется в циклоне 3.
Дальнейшее охлаждение газа происходит в трубчатом
холодильнике 7. Тонкая очистка проводится во второй трубе Вентури 8
и в скруббере-промывателе 9. Масло возвращается в систему, а часть
его выводится с основным количеством сажи.
327
При сочетании трубы Вентури, например, с мокропленочным
электрофильтром или пенным аппаратом (в последнем случае циклон
не требуется) обеспечивается высокая степень очистки (99%) и
остаточное содержание сажи в газе не превышает 2—3 мг/м3.
Эффективность очистки возрастает за счет конденсации водяных паров,
происходящей в горловине трубы и после нее вследствие разности
статических давлений.
Газ
Исходный газ
масло
с сажей,
Рис. VI1-23. Схема очистки газа с
повышенным содержанием водяных паров:
/ — реактор; 2, 8 — трубы Вентури; 3 — циклон;
4 — промежуточный сборник; 5, 7 —
холодильники; 6— фильтр; 9— скуббер; 10 — насосы;
// —фильтр; 12 — сепаратор.
Трубы Вентури
изготовляют из листовой стали или
отливают из чугуна.
Горловина трубы в большинстве
случаев выполняется
круглого, а иногда
прямоугольного сечения. Диаметр входного
отверстия (конфузор) трубы
рассчитывают, исходя из
скорости газов перед трубой
10—12 м/сек; угол сужения
конфузора обычно равен 25°.
Длина горловины трубы
составляет 0,15 ее диаметра.
Диаметр выходного отверстия
(диффузора) рассчитывают,
исходя из скорости газов
20 м/сек; угол расширения
диффузора принимают равным 6—8°. Вода в трубу Вентури подается
через радиальные сопла под давлением 3—4 am.
Гидравлическое сопротивление турбулентного газопромывателя
равно 500—1200 мм вод. ст. Расход воды на 1 м3 очищаемого газа
составляет 0,25—1,25 л. В производстве ацетилена турбулентные
газопромыватели наиболее широко применяются при окислительном
пиролизе под давлением и электрокрекинге. Методика расчета
турбулентных газопромывателей разработана институтами НИИОгаз
и Гипрогазоочистка 65.
Электрофильтры
Действие электрофильтров основано на явлении ионизации
газов 65' 66. Как известно, любой газ представляет собой скопление
беспорядочно движущихся частиц, большая часть которых
(молекулы) обычно нейтральна, т. е. лишена заряда. Вместе с тем в газе
содержится также некоторое количество заряженных частиц — ионов
и свободных электронов. При создании электрического поля ионы
и электроны начинают двигаться по силовым линиям поля, причем
направление движения определяется знаком заряда, а скорость
движения — напряженностью электрического поля. При достаточно
большой напряженности поля движущаяся заряженная частица
приобретает столь высокую энергию, что при столкновении с ней-
328
тральной молекулой выбивает из нее свободный электрон, превращая
молекулу в положительный ион. Вновь образующиеся заряженные
частицы также приходят в движение под действием поля и
способствуют дальнейшей ионизации газа (ударная ионизация), благодаря
чему создаются условия для электрического разряда.
В зависимости от конфигурации электродов, напряженности поля
и других факторов разряды в газах могут принимать различные
формы. Наиболее распространены и важны для очистки газов
искровой, дуговой и особенно коронный разряды. Коронный разряд
возникает только в неоднородном электрическом поле и носит
незавершенный характер: вследствие неоднородности поля начавшаяся
у одного электрода ударная ионизация не распространяется до
другого электрода. В зависимости от знака на проводе коронный разряд
(или корона) может быть положительным или отрицательным.
Под действием короны в газовом пространстве между электродами
происходят следующие явления. В области, непосредственно
примыкающей к коронирующему проводу, в результате ударной ионизации
возникают ионы обоих знаков и свободные электроны. В
электрическом поле положительные ионы начинают двигаться к
коронирующему проводу (если корона отрицательная) и нейтрализуются на
нем. Свободные электроны образуют с газовыми молекулами
отрицательные ионы, которые перемещаются к положительному электроду
и также нейтрализуются на нем. При этом в пространстве между
электродами возникает электрический ток.
Установка с электрофильтром состоит из двух частей: собственно
электрофильтра, через который проходит очищаемый газ, и
высоковольтной аппаратуры, устанавливаемой в преобразовательной
подстанции и предназначенной для питания электрофильтра
выпрямленным током высокого напряжения. Питающий электроагрегат
(рис. VII-24) состоит из регулятора напряжения 8 и повышающего
трансформатора 7, преобразующего переменный ток 220—380 в
в ток напряжением до 100 000 в (поддержание постоянного
напряжения очень важно для нормальной работы электрофильтра).
Механический высоковольтный выпрямитель * 5 преобразует переменный
ток в постоянный, последний по высоковольтному кабелю или через
шины подается на электроды 2 и 3 электрофильтра 4-.
Для питания электрофильтров выпрямленным током высокого
напряжения отечественная промышленность выпускает повысительно-
выпрямительные электроагрегаты АФА-90-200 и АФАП-80-225.
В электроагрегат АФА-90-200, выпускаемый в трех модификациях
(модели А, Б и Д), входят следующие основные элементы:
однофазный масляный трансформатор, повышающий напряжение заводской
* В последнее время вместо механических выпрямителей находят применение
полупроводниковые, которые обходятся дешевле, имеют малые габариты и
обеспечивают более стабильную работу электрической системы электрофильтра. В
качестве полупроводниковых выпрямителей применяются кремниевые и селеновые
выпрямители, которые обладают свойством пропускать в одном направлении
значительно больший ток, чем в другом.
329
сети; механический выпрямитель, преобразующий переменный ток
высокого напряжения в постоянный ток; щит управления с
пусковыми, регулирующими и контрольно-измерительными приборами;
высоковольтный переключатель с приводом.
От сета
380(220)6
Рис. VI1-24. Принципиальная электрическая схема
питания электрофильтра:
/ — изолятор; 2 — осадительный электрод; 3 — коронирующий
электрод; 4 —электрофильтр; 5—высоковольтный выпрямитель;
6 — заземление; 7 — повышающий трансформатор; 8 —
регулятор напряжения; 9 — рубильник; 10 — предохранитель.
Ниже приведена техническая характеристика электроагрегата
АФА-90-200:
Модель Модель Модель
А Б Д
Напряжение питающей сети, в 380 220 500
Номинальная мощность на стороне
выпрямленного напряжения, ква 18 18 18
Номинальное выпрямленное (амплитудное)
напряжение, кв 90 90 90
Номинальный выпрямленный ток, ма 200 200 200
В осадительной части электрофильтра смонтированы осадитель-
ные и коронирующие электроды. Осадительные электроды могут
быть пластинчатыми (из листовой стали, из пластин уголкового
профиля и пр.) или трубчатыми (круглого или прямоугольного
сечения); коронирующие электроды делают из круглой или другой
профилированной проволоки. Осадительные электроды соединены
с положительным контактом механического выпрямителя и
заземлены, коронирующие электроды соединены с отрицательным
контактом выпрямителя и изолированы. При подаче высокого напряжения
в пространстве между электродами возникает электрическое поле,
напряженность которого можно менять, регулируя подаваемое
напряжение. При увеличении напряжения до определенной величины
образуется коронный разряд, в результате чего начинается
направленное движение заряженных частиц к электродам, т, е. в
пространстве между электродами возникает ток,
330
форсунки
При пропускании через электродное пространство газа,
содержащего взвешенные частицы сажи, происходит их заряжение
движущимися ионами. Заряженные частицы под действием электрического
поля движутся к электродам и оседают на них. Очищенный газ,
пройдя электрическое поле, направляется на дальнейшую
переработку, а основная масса взвешенных частиц осаждается наосадитель-
ных электродах. Для удаления сажи электроды приходится
периодически встряхивать, обстукивать или непрерывно промывать водой.
Работа электрофильтра в
значительной степени зависит от влагосодержания
поступающего газа: при увеличении
влагосодержания (повышение
плотности газа) потребление тока уменьшается.
Даже небольшое повышение влажности
сажи резко уменьшает электрическое
сопротивление слоя, оседаемого на
электродах, а частицы, абсорбировавшие
пары воды, укрупняются, что
способствует улучшению работы
электрофильтра. Однако во избежание налипания
сажи на электроды температура
очищаемого газа не должна быть ниже точки
росы, в противном случае дальнейшее
улавливание сажи невозможно.
На рис. VI1-25 изображен мокропле-
ночный пластинчатый электрофильтр,
применяемый в производстве ацетилена.
Газ при 60° С входит в нижнюю часть
фильтра, затем поступает в осадитель-
ную камеру прямоугольного сечения и
движется в пространстве между
электродами со скоростью около 0,5 м/сек.
Коронирующие электроды 3
представляют собой нихромовые провода
диаметром 2 мм, подвешенные к верхней раме. Нижняя рама корони-
рующих электродов фиксирует их положение и предотвращает
раскачивание. Осадительные электроды 4 изготовлены из свободно
подвешенных стальных листов толщиной 3 мм. При работе
электрофильтра разность потенциалов составляет 45—55 тыс. в.
Гидравлическое сопротивление аппарата равно около 20 мм вод. ст.
Для увлажнения газов и удаления осевшей сажи и смолы с оса-
дительных и коронирующих электродов в верхнюю часть
электрофильтра через форсунки тонкого распыления непрерывно подается
вода при избыточном давлении 5 am. Кроме того, предусматривается
периодическая промывка электродов большим количеством воды,
подаваемой через специальную поворотную систему форсунок.
На время промывки подача тока прекращается, а газ нужно очищать
в другой секции электрофильтра или осуществить дополнительную
331
Вода £
сажей
Рис. VI1-25. Мокропленочный
пластинчатый электрофильтр
СПМ-8:
/ — изоляторная коробка; 2—
корпус; 3 — коронирующий электрод;
4 — осадительный электрод;
5—гидрозатвор.
очистку в следующем технологическом аппарате. При нарушениях
технологического режима и при понижении температуры в коробках
изоляторов питание электрофильтров током высокого напряжения
также автоматически прекращается. Очищенный газ выходит с верха
электрофильтра и направляется на дальнейшую переработку, а вода,
содержащая сажу, из нижней части аппарата через гидрозатвор
уходит в систему очистки сточных вод.
В настоящее время мокропленочные электрофильтры нашли
наиболее широкое применение, особенно в схемах окислительного
пиролиза. Однако электрофильтры можно использовать только в
процессах при атмосферном давлении, так как изготовление изоляторов,
работающих при повышенном давлении, связано со значительными
трудностями.
Пенные аппараты
Принцип действия пенного аппарата заключается в следующем.
Жидкость поступает на горизонтальные решетки сверху вниз и
вспенивается тонкими струями газа, проходящего через отверстия
решеток снизу вверх. Образование пенистого слоя, имеющего развитую
поверхность, позволяет значительно
интенсифицировать процессы теплообмена между
газом и жидкостью. В пенных аппаратах
одновременно с охлаждением * происходит
очистка газа от сажи и смол.
Расчет пенных аппаратов теплообменного типа
ведут по уравнению теплообмена 69:
Q = KSM (VII-67)
где Q — количество передаваемого тепла, ккал/ч;
К — коэффициент теплопередачи, ккал/(м2*Ч'град);
S — сечение аппарата, ж2;
А^ — разность температур, °С.
^с fc М. Е. Позин, И. П. Мухленов, Э. Я. Тарат б9
рекомендуют следующий порядок расчета пенных
теплообменников.
1. Определяют количество отводимого тепла:
Q = 3600St> (iH — iK)
где v — расчетная скорость газа в полном
сечении аппарата, м/сек;
*н и **к — начальное и конечное теплосодержание
газа, ккал/м3.
2. Рассчитывают разность температур для
процесса теплопередачи между газом и жидкостью (без
учета испарения и конденсации):
л 4 \Рн — * н) — (^н — tK) _ ^н — ^
Рис. VI1-26. Пенный
аппарат с решетчатыми
тарелками:
/ — брызгоотбойник;
2—корпус; 3—центральная труба;
4 — решетчатые тарелки;
5 —гидрозатвор.
In
вн — ;
(VII-68)
где 0Н и 0К — температура
tH и tK — температура
газа до и после решетки, °С;
жидкости до и после решетки, °С.
* В пенных аппаратах нельзя охладить газ до точки росы и тем более — ниже
этой температуры.
332
3. Определяют коэффициент теплопередачи К:
*- Q
Обычно К = 5000-М0 000 ккал/(м2-ч-град).
4. Подсчитывают высоту Н пенного слоя:
10(Ж3
Н =
2,43-36003и3
(VII-69)
(VII-70)
Газ
Обычно Я = 100-^200 мм. Конструктивные размеры пенного аппарата можно
выбирать на основе рекомендаций, приведенных в работах ' .
На рис. VII-26 показан пенный аппарат, предназначенный для
охлаждения газов пиролиза. Газ входит через верхний штуцер,
проходит центральную трубу и решетчатые тарелки 4 (стальные листы
с отверстиями диаметром 6 мм и шагом 13 мм), на которые подается
вода. Проходя через
отверстия, газ вспенивает воду,
в результате чего на решетке
образуется слой пены, в
котором и происходит
охлаждение газа. Вода на верхнюю
решетку подается из желоба
с треугольными насечками,
установленного поперек
аппарата. Для улавливания
капель воды в верхней части
аппарата имеется насадка-
брызгоотбойник 1 из ряда
уголков. Уловленная вода
стекает к стенкам аппарата.
Циклоны
Для получения сажи в
сухом виде в качестве
аппаратов грубой очистки
применяют отдельные циклоны или
батареи циклонов.
В циклон газовый поток
поступает тангенциально, за
счет чего создается его
вращение. При этом развивается
центробежная сила,
отбрасывающая взвешенные частицы
сажи в наружные слои газа,
откуда они отводятся в
бункер (рис. VI1-27). Эффективность работы циклона определяется его
геометрической формой и скоростью очищаемого газа. Во входном
патрубке циклона скорость газов должна быть 12—22 м/сек,
333
Газ
Рис. VI1-27. Циклон и схема движения в нем
газового потока:
/ — улитка для вывода газов; 2 — крышка;
3 — цилиндрическая часть корпуса; 4 —
выхлопная труба; 5 —конусная часть корпуса; 6—
выпускное отверстие; 7 — бункер; 8 —затвор.
а в условном сечении циклона (по среднему диаметру
цилиндрической части) — от 2,5 до 4,5 м/сек. Существует большое число
циклонов для различного вида частиц, но наиболее распространены
циклоны конструкции НИИОгаз. В каждом отдельном случае
необходимый типоразмер циклона нужно подбирать по нормалям или
каталогам.
Газ
(вариант)
Рис. VI1-28. Батарейный циклон (а) и схемы движения газа в элементах
батарейного циклона с завихрителями «винт» (б) и «розетка» (в):
1 — конфузоры; 2 — диффузор; 3 — завихрители; 4 — корпус циклонного элемента; 5 —
набивка; 6 — крышка; 7 — верхняя опорная решетка; 8 — выхлопная труба; 9 —
циклонный элемент; 10 — кожух; // — нижняя опорная решетка; 12 — опорный пояс; / — камера
очищенных газов; // — бункер для сажи; /// — газораспределительная камера.
В батарейных циклонах (мультициклонах) газовый поток
приводится во вращение с помощью завихрителей, в качестве которых
применяют элементы «винт» и «розетка» (рис. VI1-28). Завихритель
«винт» имеет две лопасти, наклоненные под углом 25°, завихритель
«розетка» снабжен восемью лопатками, расположенными под углом
25 или 30°. Наиболее распространены батарейные циклоны
конструкции «Гипрогазоочистка» 65. Нормализованные диаметры цикло-
334
нов составляют 250, 150 и 100 мм. Расчет батарейных циклонов можно
проводить по каталогам и литературным данным.
Для обеспечения нормальной работы циклонов и
мультициклонов выгрузные устройства должны быть герметичными, при этом
система выгрузки уловленного продукта
может быть периодической или непрерывной.
Рукавные фильтры
Рукавные (мешочные) фильтры состоят из
нескольких тканевых рукавов, подвешенных
в металлической камере; верхняя часть
рукавов обычно заглушена (рис. VI1-29).
Очищаемый газ поступает в нижнюю часть
аппарата и проходит через ткань, на которой
оседает сажа. Обязательным условием
эффективной очистки газов в тканевых фильтрах
является поддержание температуры выше
точки росы, иначе сажа увлажняется,
вследствие чего ткань забивается и растет
гидравлическое сопротивление фильтра.
Процесс фильтрации зависит от характера
сажи и типа ткани. В производстве
ацетилена приходится фильтровать горячие газы,
следовательно, в этих условиях необходимы
материалы, выдерживающие высокую
температуру. Известны, например, синтетические
ткани из волокна нитрон, выдерживающие
температуру до 130° С, и стеклянные
ткани — до 700° С.
Рис.
VI1-29. Рукавный
фильтр:
/ — дроссель; 2 — рукава;
3 — корпус; 4 — сажа;
5 — шнек; 6 — затвор.
* *
*
Описаны результаты исследования в масштабах опытной
установки (70—120 м3 газа в 1 ч) двух систем для очистки газов пиролиза
от сажи и смолистых веществ. Очистке подвергался газ, выходящий
из скруббера, установленного непосредственно после реактора70.
Первая система состояла из двух последовательных пенных
аппаратов и коксового фильтра. Сопротивление каждого пенного
аппарата не превышало 17—20 мм рт. ст. Скорость газа в
сечении аппарата составляла 1,0—1,5 м/секу а в отверстиях решетки
7,5—11,2 м/сек. При таких условиях температура газа в первом
пенном аппарате снижалась с 55 до 25—30° С, во втором — до
20—22° С. Температура охлаждающей воды 15—18° С. Частично
очищенный газ после второго пенного аппарата направлялся в
коксовый фильтр, а сажа и смола сбрасывались в канализацию. При
использовании в качестве насадки двух слоев коксовых гранул
размером 15—20 мм конечное содержание сажи и смолистых веществ
335
в газовой смеси составляло 76—100 мг/м3. При работе на
активированном угле (гранулы 3—5 мм) содержание примесей в газе снизилось
до 10 мг/м3, однако при этом слой угля быстро забивался и
сопротивление фильтра повышалось в 2—2,5 раза.
Некоторые данные об очистке газа с начальным содержанием
сажи 2,5—3,1 мг/м3 приведены ниже:
Содержание Степень очист-
сажи в газе, ки, %
мг/м3
После пенного аппарата I .... 170—210 30—31
» » » II .... 130—150 —38
» фильтра 10—30 80—92
Степень очистки в пенных аппаратах невысока, и лишь за счет
установки коксовых фильтров удалось получить общий к. п. д.
системы, равный 88—97%. Следовательно, для очистки
рекомендуется устанавливать коксовые фильтры.
Во второй системе очистки применялся мокропленочный
электрофильтр, установленный непосредственно после скруббера. Скорость
движения газа в зоне фильтрации 0,3 м/сек. Коронирующие электроды
были изготовлены из нихромовой проволоки диаметром 0,7 мм и
периодически обмывались водой. Кроме того, имелось специальное
обдувочное устройство, с помощью которого на электроды подавали
природный газ при избыточном давлении 1,5—2 am. Во избежание
взрыва газовой смеси в активной зоне электрофильтра на
ацетиленовом реакторе был установлен специальный фотоэлектрический
прибор, отключающий электрофильтр в случае погасания горелки
реактора. Очистка газов в электрофильтрах по описанной схеме
была очень эффективной: остаточное содержание сажи и смолистых
веществ не превышало 2—3 мг/м3, к. п. д. электрофильтра
достигал 99%.
Очистка воды от сажи
Промывные воды, образующиеся при мокрой очистке газа
пиролиза, содержат значительное количество сажи и смол. В процессах
производства ацетилена вся вода находится в замкнутом водообо-
ротном цикле, так как в ней содержатся различные органические
вещества из газа пиролиза. Замкнутый цикл выдвигает жесткие
требования по очистке воды от сажи и смол. Промышленной
практикой установлено максимально допустимое содержание
механических примесей в циркулирующей воде 100—200 мг/л.
Очистка воды от сажи и смол проводится различными способами:
отстаиванием, фильтрацией и др. Технико-экономическое сравнение
показало, что отстаивание значительно экономичнее, чем
фильтрация. При методе отстаивания загрязненную воду пропускают через
специальный аппарат — сажеотстойник. Вследствие создания в
аппарате определенных условий частицы сажи всплывают на поверхность
воды.
336
ч
/
\
I
1
Механизм всплывания сажи заключается в следующем 62-64ш
Истинная плотность сажи в 1,5—2 раза больше, чем плотность воды,
потому сажа должна была бы оседать на дно. Однако благодаря
большой пористости в саже оказывается много газа, кроме того, каждая
частица сажи вследствие ее высокой адсорбционной способности
окружена газовыми пузырьками. В этих условиях фактическая
плотность сажи меньше, чем воды. Проявляется эффект флотации,
и частицы сажи, увеличенные осевшими на них пузырьками газа,
всплывают.
Степень всплывания сажи зависит
от ее пористости, температуры воды
и гидравлического режима работы
сажеотстойника. При высокой
пористости сажа хуже смачивается,
поскольку слишком большая
поверхность ее частиц окружена газовыми
пузырьками. В этом случае
получается так называемая гидрофобная сажа.
При меньшей пористости сажа лучше
смачивается, т. е. становится
гидрофильной. С повышением температуры
воды растворимость в ней газовых
пузырьков, окружающих сажевые
частицы, уменьшается, благодаря чему
сажа всплывает быстрее. Однако
повышение температуры в сажеотстойнике ограничено возможностью
охлаждения горячей воды. Практически увеличить температуру
сверх 80—85° С трудно, поэтому обычно она колеблется от 50 до
80° С.
Проведенные автором исследования показали, что степень
очистки воды связана с физико-химическими свойствами сажи,
главным образом с ее смачивающей способностью (всплываемостью).
Характеристикой всплываемости предложено считать количество
сажи, отбираемой в различных местах сажеотстойника (на^ входе
или на выходе), по отношению к общему объему загрязненной воды.
Эта величина зависит от соотношения природного газа и кислорода
и температуры их предварительного подогрева, а также от физико-
химических свойств сажи, причем пока точно не установлено, какие
из всех этих параметров являются определяющими.
Данные об изменении количества сажи в зависимости от
соотношения О2 : СН4 представлены на рис. VII-30. Из графика видно,
что при О2 : СН4 > 0,60 начинается резкое осаждение части сажи
с одновременным ее всплыванием, следовательно, образуются два
вида сажи — гидрофильная и гидрофобная.
Кроме всплываемости сажи для работы сажеотстойника важно
качество сажевой «шапки», собирающейся на поверхности. В
некоторых случаях она состоит из легкой гидрофобной сажи, которая
сильно пылит, что мешает работе, Это отмечалось, например, при
22 в. И. Антонов ^37
8
55 6
Ь
ь.
CJq О
2Oj5 WO "0,65 0,70 \
Соотношение О2: СН4
Рис. VI1-30. Зависимость выхода
сажи от соотношения О2 : СН4
(время работы сажеотстойника 5 мин):
при температуре > 450°С:
/ _ всплывающая сажа; 2 —
оседающая сажа.
температуре предварительного подогрева 400—420° С и отношении
О2 : СН4 = 0,61-5-0,62.
Для более глубокого понимания зависимости физико-химических
свойств сажи от показателей пиролиза необходимы дальнейшие
исследования, однако уже сейчас можно сказать, что для нормальной
работы сажеотстойников и облегчения транспортирования сажевой
пульпы необходимо строго поддерживать заданный режим пиролиза,
например для окислительного пиролиза отношение О2: СН4 = 0,585-г-
-ь0,595 и температуру предварительного подогрева 450—600° С.
В этих условиях получается максимальный выход ацетилена и
минимальный выход сажи при высокой степени всплываемости (плохое
смачивание).
Очищенная
вода
Рис. VI1-31. Схема противоточного сажеотстойника:
/ — сажеотстойник; 2 — скребки; 3 — емкость с мешалкой.
Сажеотстойник представляет собой железобетонный резервуар
(открытый или закрытый), с одного конца которого подается под
уровень загрязненная вода, а с противоположного конца выводится
осветленная вода (рис. VI1-31).* На основании экспериментальных
данных можно принять скорость загрязненной воды 0,01—0,02 м1сек,
а время отстаивания 10—30* мин. Отношение длины и ширины
сажеотстойника может быть от 5 до 8. Для отвода всплывающей сажи
(пульпа сажи с водой) необходимо правильно подобрать скребковый
транспортер. Скорость его движения и число скребков определяют
по количеству пульпы. Часть сажи оседает на дно, поэтому
сажеотстойник необходимо периодически чистить.
Материальный баланс сажеотстойника складывается из
следующих количеств сажи: Gx — поступает с водой из водооборотного
цикла, G2 — поступает в сажеотстойник из аппаратов сажеочистки;
G3 — отводится с пульпой, G4 — уходит с очищенной водой.
Работу сажеотстойника можно характеризовать относительной А
и абсолютной Б степенями очистки воды (с учетом количества сажи,
которое может накапливаться в водооборотном цикле):
_ g2 — G4 1ПЛ „ _ G2 — Gj
100
100
Относительная степень очистки при нормальном качестве сажи
достигает 78—80%, при ухудшении показателей сажи эта величина
Широко применяются и прямоточные аппараты.
338
снижается до 55—60%. Абсолютная степень очистки составляет
80—90%. При таких условиях часть сажи накапливается в системе
водооборота, поэтому следует периодически очищать приемные
резервуары градирен водооборотного цикла. Однако при нормальном
качестве сажи, наоборот, часть ее, накопившаяся в цикле,
возвращается в сажеотстойник, отстаивается и выводится из системы.
Практически при условии нормальной работы в системе сажеотстой-
ники — водооборотный цикл устанавливается подвижное
равновесие. Полученную сажевую пульпу сжигают.
При удалении сажи методом фильтрации применяют барабанные
вакуум-фильтры. Полученную пасту после подсушивания можно
сжигать в специальных печах. Этот способ, хотя и обеспечивает
лучшую очистку, является более громоздким и дорогим (например,
расход электроэнергии в 5 раз больше, чем при использовании саже-
отстойников).
КОМПРИМИРОВАНИЕ АЦЕТИЛЕНА
И АЦЕТИЛЕНСОДЕРЖАЩИХ ГАЗОВ
Условия сжатия
Процессы концентрирования и выделения ацетилена связаны
с необходимостью сжатия ацетиленсодержащих газов и чистого
ацетилена.
В табл. VI1-6 приведены составы различных реакционных газов,
подлежащих сжатию в производстве ацетилена. Содержание
ацетилена в них составляет 8—16 объемн. %, а основными
разбавителями являются водород, этилен, окись углерода и метан. Как было
показано в главе I, эти примеси существенно увеличивают предельно
допустимое давление ацетилена и тем самым делают смесь менее
взрывоопасной. Если рассмотреть, например, смесь ацетилена
(20 объемн. %) и водорода, то минимальное давление распада
ацетилена в этой смеси при 15° С равно 20 ат> а минимальное давление
взрывного распада чистого ацетилена в этих условиях составляет
1,4 am.
ТАБЛИЦА VII-6
Состав газов, подлежащих сжатию в производстве ацетилена
Компоненты
Ацетилен
Этилен
Высшие ацетиленовые и
ароматические углеводороды
Метан
Водород
Окись углерода
Двуокись углерода
Состав газа, объемн. %
пиролиз
10—16
15—18
2—3
15
30—35
электро-
крекинг
12—15
1—4
1,5
25—27
49—55
окислительный
пиролиз
8,0
0,3
0,5
5—6
50—56
26—27
4—5
22*
339
Учитывая склонность ацетилена к взрывному распаду, при его
сжатии необходимо соблюдать соответствующий температурный
режим. Допустимая температура при сжатии ацетилена и
ацетиленовых смесей пока точно не определена. В промышленной практике
при сжатии ацетилена максимально допустимая температура
принимается 100—110° С. Поэтому компрессоры для ацетилена имеют
малую степень сжатия (не выше 2—2,4), чтобы при адиабатическом
сжатии температура не поднималась выше указанной. Величина
100—110° С выбрана, вероятно, с учетом зависимости условий
взрывного распада ацетилена от температуры. Как известно, при
наполнении баллонов ацетилен сжимают до 20—25 am. При этом, естественно,
превышается предельно допустимое давление, вследствие чего здесь
особенно тщательно нужно соблюдать условия, исключающие
возможность взрывного распада.
Температура начала взрывного распада ацетилена также зависит
от его давления: при 8 am температура, при которой начинается
распад ацетилена, превышает 500° С, а при повышении давления
до 20 am она снизится примерно до 250° С, что можно определить
путем интерполирования данных рис. 1-4 (стр. 22). Вообще же рост
температуры ацетилена вызывает уменьшение давления, при котором
может начаться его распад; снижается в этом случае и температура
самовоспламенения ацетилена. Следовательно, при 20 am и
увеличении температуры сжатия распад ацетилена начнется при температуре
ниже 250° С.
При установлении максимально допустимой температуры сжатия
ацетилена необходимо учитывать также возможность его
полимеризации при относительно низких температурах (250° С). На основе
всего выше сказанного можно установить максимальную
температуру сжатия ацетилена до 20 am не более 100° С. Особенно строго
нужно следить за температурой на последних ступенях сжатия.
При определении допустимой температуры ацетилена, сжимаемого
до более низких давлений, например до 1,4 ату применяется тот же
принцип, хотя, по-видимому, в этих условиях ограничения могут
быть менее жесткими.
На рис. 1-9 (стр. 24) показано изменение предельно
допустимого давления ацетилена в присутствии разбавителей (водород, окись
углерода, метан и др.). Видно, что смеси, содержащие до 15объемн. %
ацетилена, можно безопасно сжимать до 30 am, а смеси, содержащие
до 65 объемн. % ацетилена,— лишь до 2,5 am. Отсюда ясно, что
сжатие ацетиленовых смесей менее опасно, чем
ацетилена-концентрата. В настоящее время температура сжатия ацетиленовых смесей
принимается 100—110° С, т. е. как и для чистого ацетилена. Это,
по-видимому, не совсем правильно, так как в присутствии
разбавителей можно допускать повышение температуры сжатия. Однако
нужно принимать во внимание склонность ацетилена, особенно в
присутствии таких разбавителей, как СО, полимеризоваться при 250—
300° С.
340
С целью изучения условий компримирования была проведена работа по
определению минимальных давлений распада ацетилена в смеси с водородом при 20,
100 и 300° С в масштабе опытной установки п. Опыты велись в стальной бомбе!
Ацетилен нагревали до требуемой температуры и инициировали взрыв,
расплавляя током 127 в нихромовую проволоку длиной 10 мм и диаметром 0,15 мм.
При большом содержании ацетилена в смеси флегматизирующее действие
водорода оказывалось незначительным (рис. VI1-32). С уменьшением
концентрации ацетилена влияние водорода на предельно допустимое давление резко
повышалось. Например, взрывной распад ацетилена в смеси с водородом
10 15 20 25 30 35
Начальное давление взрывного распада, am
Рис. VI1-32. Начальное давление взрывного распада ацетилена в смеси с водородом:
7 — при 20° С; 2 — при 100° С; 3 — при 300° С.
(80 объемн. % Н2) при 300° С начинался при давлении 10 am, а при 100° С — при
17 am. В смеси состава С2Н2 : Н2 =1:1 при 300° С взрывное разложение
наблюдалось при 2,5 am.
Полученные экспериментальные данные с известным приближением можно
использовать для определения условий компримирования газовых смесей,
образующихся в различных производствах ацетилена, но при этом следует
предусмотреть некоторый запас надежности. В ацетилено-водородных смесях, содержащих
менее 10 объемн. % С2Н2 и имеющих температуру 300° С, взрывной распад
начинается при 30 am и выше. Следовательно, если температура газовых смесей не
превышает 100° С, то при сжатии их до 30 am будет соблюден достаточный запас
надежности. И, наоборот, если режим сжатия ограничивается 10 am, допустимая
температура сжатия для ацетиленовых смесей, содержащих менее 10 объемн. % С2Н2,
может быть повышена до 150—200° С.
Указанный температурный режим сжатия ацетилена до низких
давлений (1,4 am) и ацетиленовых смесей до 10—20 am принят
согласно существующей практике компримирования ацетилена при
наполнении баллонов (давление 20 am); по-видимому, в обоих
случаях допустимые температуры неоправданно занижены, однако этот
вопрос должен быть изучен более подробно.
341
При сжатии следует избегать местных перегревов газа, которые
могут привести к полимеризации ацетилена. С этой целью в
поршневых машинах ограничивают скорость движения поршня, обычно
ее принимают не более 0,7—1 м/сек. В машинах не допускается
искрообразование, которое может явиться импульсом взрывного
распада ацетилена. Для поддержания требуемого по условиям
безопасности температурного режима в компрессоре для ацетилена
или ацетиленовых смесей должно быть предусмотрено охлаждение
сжимаемого газа в корпусе или в межступенчатых выносных
холодильниках.
Расчет процесса сжатия. Процессы компримирования ацетилена и его смесей
связаны с расчетом степени сжатия, которая определяется допустимой
температурой на каждой ступени сжатия. В состав газовой смеси входят компоненты с
различными физическими и термодинамическими свойствами (например, метан и
водород). Повышение температуры Т[ каждого компонента i в процессе его
адиабатического сжатия определяется соотношением конечного и начального давлений и
показателем адиабаты ki для этого компонента. При сжатии для каждого
компонента соблюдается уравнение 72f 73:
■HtJ (V"-71)
где т\ и т\ — температуры компонента i в начале и в конце сжатия, °К;
Pi и Pi — давление компонента i в начале и в конце сжатия, am;
ki — показатель адиабаты для компонента i.
Более точно температуру смеси или ее отдельных компонентов в конце
адиабатического сжатия, а также показатели адиабаты для любого компонента можно
определять по диаграммам состояния этого компонента. При графическом
изображении полного процесса адиабатического сжатия компонента (от начального до
конечного парциального давления) можно определять температуру Т'\ в конце
процесса и удельные объемы в начале и в конце сжатия (v. и v^). Зная эти величины,
можно определить и действительное значение показателя адиабаты:
kt = —%■ (VII-72)
4
При адиабатическом сжатии смеси сложного состава показатели адиабаты для
отдельных компонентов изменяются по-разному, однако температура всех
компонентов в каждый момент времени одинакова вследствие непрерывного теплообмена:
одни вещества (метан, ацетилен) в результате сжатия нагреваются, а другие (водород)
отдают тепло.
Показатель адиабаты для смеси kCM. можно вычислить по формуле:
или kcN. = S SfiP g48 (VII-73)
где ср и Су — теплоемкость смеси;
gi — мольная доля компонента;
ср — теплоемкость компонента.
342
Поскольку теплоемкость ср изменяется с давлением, для более точного
определения /гсм. можно пользоваться формулой:
FW~ (VII'74)
1 g —-г -г 1 g У gfip, i—\g gjCp
Pi L^ \ Рем. /
Например, для реакционных газов, получающихся в процессе окислительного
пиролиза, /гсм. = 1,357.
В действительности сжатие каждого компонента смеси протекает по политропе
с внутренним относительным коэффициентом полезного действия
Ч* —
-^по
где Lafl. — работа адиабатического сжатия, а Ьполн. — полная работа сжатия.
Для компенсации внутренних потерь к 1 кг газа, кроме тепла, выделяющегося при
адиабатическом сжатии, подводится дополнительное тепло <7под.:
(VII-75)
Действительная температура смеси в конце сжатия может быть найдена по
уравнению:
Тем. = ГСМ. -?Ч (VII-76)
В этой формуле тсм. — показатель политропы, определяемый следующим
уравнением:
*«.
(VII-77)
Уже говорилось, что при сжатии ацетиленсодержащих газов, особенно чистого
ацетилена, необходим строгий температурный режим компрессора, что
обусловливает относительно низкую степень сжатия газа. Для каждой ступени работа сжатия
может быть определена так:
= -V-r^n^M.TCM. -S4 -И (VH-78)
Л/
В этой формуле /?см. = 2 giRi (gi— мольная доля компонента, R[—
газовая постоянная).
Типы компрессоров
Компрессоры для ацетиленсодержащих газов. Для сжатия
ацетиленсодержащих газов до 10—30 am применяют поршневые
компрессоры и турбокомпрессоры. Тип компрессора выбирают с учетом про-
343
изводительносГи установки, конечного давления, состава газовой
смеси и прочих факторов. Как известно, турбокомпрессоры обладают
лучшими технико-экономическими показателями, меньшими
энергозатратами и капиталовложениями и большим к. п. д., чем
поршневые машины, только при определенной производительности — не
менее 15—20 тыс. м3/ч. При меньшей производительности
преимущества турбокомпрессоров не столь значительны.
Ниже приведены сравнительные эксплуатационные данные для
поршневых компрессоров и турбокомпрессоров:
Поршневой Турбоком-
компрессор прессор
Производительность, м3/ч 10 000 25 000
Вес, т 88,4 86,5
Удельные капитальные затраты, % .... 100 58,3
Себестоимость 1 т ацетилена по стадии
компрессии газа, % 100 76 или 69 *
* С учетом установки рекуперационной турбины.
На выбор установок компримирования существенное значение
оказывает тип привода, который определяется стоимостью
электроэнергии и топлива в конкретном районе. В качестве привода в
настоящее время применяют электродвигатели, паровые и газовые турбины,
а также газомоторы в непосредственном исполнении с компрессорами.
На паровых турбинах, работающих с противодавлением,
используется отработанный водяной пар или пар, отбираемый на
промежуточных ступенях паротурбогенераторов ТЭЦ. Затем этот пар можно
использовать как теплоноситель.
На ряде установок, где давление отходящих газов рекуперируется
в газовой турбине, смонтированной на одном валу с компрессором,
электродвигатель компрессора выбирают таким, чтобы при
отключенной турбине компрессор мог действовать на полную мощность. На
установках пиролиза, работающих при 3—6 am, применяются
дожимающие компрессоры (без первой ступени), которые могут создать
конечное давление 12—16 am. При совместном получении ацетилена
и этилена требуются более мощные компрессоры, чтобы газовую
смесь после выделения ацетилена сжать до 30—40 am. Для сжатия
ацетиленсодержащих газов применяют поршневые компрессоры
производительностью 5000—10 000 м3/чу обеспечивающие конечное
давление 10—12 am. Создание более крупных машин затруднено, поскольку
при малой степени сжатия цилиндры, особенно на первых ступенях,
будут очень большими и неконструктивными. Указанные
компрессоры имеют четыре ступени с выносными промежуточными
холодильниками и циклонами для сбора капель влаги и растворителя,
отделяющихся при сжатии газа.
Для лучшего удаления сажи и смолистых веществ, оседающих
в цилиндрах, производится обильная смазка машин фильтрованным
маслом (т. вспышки выше 215° С). Повторное использование
отработанного масла в этих компрессорах запрещается, оно подлежит реге-
344
нерации и последующему использованию в менее ответственных
машинах. В отработанном масле могут растворяться ацетиленовые
углеводороды, поэтому регенерацию его нужно проводить на
отдельной установке. Там масло продувают инертным газом и одновременно
подогревают паром.
Турбокомпрессоры,
используемые для ацетилен-
содержащих газов, обычно
имеют производительность
25 000лг7</ и рассчитаны на
конечное давление 10—
12 am. У них бывает от
пяти до шести ступеней.
Газ после каждой ступени
охлаждается в выносных
промежуточных
холодильниках, при этом часть
содержащихся в нем
водяных паров конденсируется
и отделяется в циклонах,
установленных после хо-
а
Рис. VI1-33. Турбокомпрессор для сжатия ацетиленсодержащих газов:
а — общий вид; б — разрез по / и // ступеням (низкое давление).
лодильников. При компримировании из газа, выделяются также
полимеры, которые отлагаются в промежуточных холодильниках.
Поэтому газ обычно направляют по трубному пространству
холодильников для облегчения их чистки.
На рис. VH-33 показан турбокомпрессор для газов пиролиза
производительностью 25 000 м*1ч, совмещенный с регенеративной
345
газовой турбиной п. Машина имеет два корпуса — низкого и
высокого давления; в корпусе высокого давления размещена турбина.
Конечное давление, создаваемое компрессором, равно 9,2 am.
Важным фактором при выборе режима компримирования аце-
тиленсодержащих газов является схема работы компрессоров:
коллекторная или агрегатная. С учетом характера работы пиролизных
установок и их производительности компрессорные установки всегда
коллектируют по всасывающей линии, на которой устанавливают
газгольдер для выравнивания колебаний производительности и
обеспечения времени, необходимого для срабатывания аварийных
блокировок. На нагнетательной линии компрессоры обычно работают по-
агрегатно, что обеспечивает более гибкую работу установок
выделения ацетилена. Иногда для нагнетательных линий имеются и
коллекторные схемы, однако при этом приходится дополнительно
регулировать распределение газа по отдельным установкам выделения.
Из-за сложности коллекторной схемы, а также вследствие
трудности поддержания одинакового давления в агрегатах
концентрирования на нагнетательной линии чаще применяют агрегатные схемы.
Это обеспечивает независимую работу установок выделения и
возможность регулирования нагрузки как по газу, так и по жидкости.
Каждая машина снабжена аварийными блокировками на
всасывающей линии, позволяющими остановить ее при уменьшении давления
ниже допустимого.
На любой машине (поршневой компрессор или турбокомпрессор)
имеется обводная линия для газа, используемая при пуске и
регулировании, причем эта линия может быть короткой (непосредственно
с нагнетательного на всасывающий трубопровод) или длинной (через
технологическое оборудование). На турбокомпрессорах, кроме того,
имеется противопомпажное устройство, автоматически
переключающее газ на обводную линию.
Производительность компрессоров регулируют различными
способами в зависимости от типа привода. К преимуществам
газомоторного, паротурбинного и газотурбинного приводов относится
возможность регулирования производительности и давления путем
изменения числа оборотов, что особенно важно для турбокомпрессоров.
При электроприводе производительность регулируют энергетически
малорентабельным способом: перепускают газ с нагнетательного на
всасывающий трубопровод и дросселируют газ на всасывающей линии,
а для поршневых машин изменяют объем вредного пространства.
Чаще всего производительность поршневых компрессоров и
турбокомпрессоров регулируют, дросселируя газ на всасывающей
линии. При этом предел изменения производительности составляет
в имеющихся конструкциях 25—30%, т. е. производительность
можно снизить до 70% от номинальной. Дросселирование
осуществляется на второй ступени для того, чтобы не создавать вакуум
на всасывающей линии (первая ступень).
Компрессоры для ацетилена. Для компримирования ацетилена
применяются поршневые, центробежные и водокольцевые компрес-
346
соры. Условия сжатия ацетилена в части максимально допустимой
температуры и соответственно степени сжатия более жесткие, чем
ацетиленсодержащих газов. На установках выделения ацетилен
сжимают до 1,5—2,0 am, при наполнении баллонов и в процессе
переработки давление ацетилена от 5 до 50 am. Более высокие
давления в промышленных условиях не применяются.
В процессах компримирования ацетилена обычно стремятся
уменьшить производительность компрессоров при высоких давлениях
в целях безопасности: компрессоры на 25 am имеют
производительность 25—100 мъ\ч\ при производительности 300 мъЫ и давлении
50 am применяются ротационные компрессоры с водяным затвором;
при малых давлениях (1,5—2,0 am) используют компрессоры
производительностью до 2000 м3/ч.
При сжатии чистого ацетилена температура не превышает 100° С,
при росте температуры сверх этой величины машина должна быть
остановлена. Корпус машины рассчитывается на 12—13-кратное
давление против начального. Во всех компрессорах предусматривается
надежная система охлаждения ацетилена во встроенных или
промежуточных холодильниках. Известны случаи, когда компрессор
для сжатия ацетилена помещали в бассейн с водой.
Для наполнения ацетилена в баллоны выпускают поршневые
компрессоры производительностью до 25 м3/ч и давлением 25 am.
Показатели некоторых типов таких компрессоров приведены в табл.
VI1-7. Для охлаждения газа имеются выносные холодильники,
а цилиндр компрессора охлаждается водой, циркулирующей по
специальной рубашке.
ТАБЛИЦА VI1-7
Основные показатели поршневых компрессоров для заполнения баллонов
ацетиленом
Показатели
Производительность, м3/ч
Максимальное давление, am . . .
Число ступеней
Скорость вращения вала после
редуктора, об/мин
Мощность электродвигателя, кет
Компрессор
АКВ-3
20
20
3
175
7—10
Компрессор
КЛ-5
5—7
22
2
150/195
2,8
При сжатии ацетилена до 2 am можно применять водокольцевые
компрессоры типа РМК или ВГ. Промышленностью выпускаются
машины производительностью от 50 до 500 мг/ч. Преимуществом
водоко^ьцевых компрессоров является большая безопасность сжатия
ацетилена вледствие наличия воды, обеспечивающей интенсивный
теплоотвод от газа, нагревающегося в процессе сжатия. Но эти
машины имеют и существенный недостаток: из-за высокой
растворимости ацетилена запорная вода насыщается газом, поэтому сброс ее
в канализацию недопустим — для этой цели применяется замкнутый
цикл циркулирующей воды.
347
Поскольку в процессе сжатия газа вода нагревается примерно
на 10° С, в системе устанавливают холодильники для ее охлаждения
и насосы для циркуляции. Это значительно удорожает компримиро-
вание и создает трудности при эксплуатации — необходимость
установки резервных циркуляционных насосов или блокировки водо-
кольцевого насоса с водяным насосом. Кроме того, газ насыщается
водяными парами за счет повышенной температуры (30—45° С)
и большого механического уноса. Вода может попасть в абсорбент
на стадии выделения ацетилена, что вызовет повышение расхода
пара на концентрирование (упаривание) абсорбента. В связи с этим для
сжатия ацетилена до небольших давлений (порядка 1,5 am)
применяют центробежные компрессоры производительностью до2000ж3/^.
Для соблюдения строгого температурного режима в машине имеются
встроенные холодильники (обычно — это охлаждаемые изнутри
диффузоры между колесами машины), которые охлаждают
циркулирующим конденсатом или обессоленной водой.
При выделении концентрированного ацетилена приходится иногда
работать в вакууме (остаточное давление до 0,15—0,2 am) на чистом
ацетилене и затем сжимать его до 1,2—1,3 am. Для этой цели можно
применять водокольцевые и центробежные компрессоры. Хорошо
зарекомендовали себя в промышленных установках
вакуум-компрессоры, кцторые обеспечивают и вакуум до 80% (остаточное давление
0,2 am), и давление 1,3 am. В таких компрессорах газ охлаждается
непосредственно в корпусе машины.
Рекуперационные турбины на синтез-газе. В тех установках, где
для последующей переработки синтез-газа возможно атмосферное
давление, целесообразно применять рекуперационные турбины75.
Использование энергии сжатого газа в турбине связано с
процессом изоэнтропийного расширения, количество энергии
определяется разностью теплосодержаний газа в начальном и конечном
состояниях:
Q = cP(T1-T2)
где ср — теплоемкость газа;
Тг yl Т2 — начальная и конечная температура газа.
Температура Т2 в результате расширения (снижение давления
от Р1 до Р2) определяется следующим выражением (к — показатель
адиабаты):
(А)(«-1)/К (VII-79)
Найдя величины Г2 и Q, молено определить мощность рекупера-
ционной турбины:
д
102
где Усек — секундный расход газа, кг/сек;
г\е — эффгктивный к. п. д. турбины; це — г|мг| (здесь г|м — механический
к. п. д. турбины, т|м = 0,98; ц — внутренний к. п. д. турбины, обычно
г| = 0,9). Принимают це = 0,88—0,85.
348
Из приведенных формул видно, что полезная мощность реку-
перационной турбины зависит от разности теплосодержаний сжатого
и расширенного синтез-газа: чем выше подогрет газ, поступающий
в турбину, тем больше можно использовать конечное давление
сжатого газа. Подогрев синтез-газа до высоких температур, например
выше 250э С (в этом случае требуются огневые подогреватели),
нежелателен из-за наличия в газе следов ацетилена и его
гомологов и, следовательно, из-за опасности взрыва. Чаще всего газ
подогревают в паровых подогревателях до 120—130° С. В этих условиях
полезная мощность турбины составляет около 450 квт*ч на 1 т
ацетилена.
РАЗДЕЛЕНИЕ МНОГОКОМПОНЕНТНЫХ СИСТЕМ
В процессах выделения ацетилена приходится иметь дело с
многокомпонентными системами, в которых только один или два компонента
являются целевыми продуктами, а остальные вещества практически
представляют балласт.
Для упрощения представим многокомпонентную газовую смесь
состоящей из трех основных компонентов: А — ацетилена, В — дву-
ВЧ
ч
Q+Д + В*/
Р2
MJ
Рис. VI1-34. Принципиальная схема разделения
трехкомпонентной смеси селективным растворителем:
/ — абсорбер; 2 — отдувочная колонна; 3,4— десорберы.
окиси углерода, / — инертных газов (водород, окись углерода,
метан). Разделение такой смеси возможно, поскольку растворимость
ацетилена и двуокиси углерода, например, в диметилформамиде,
N-метилпирролидоне и метаноле достаточно велика и примерно
одинакова, а водорода и окиси углерода — в несколько раз меньше.
По растворимости компоненты смеси можно расположить в
следующий ряд:
А>В>1
Из системы А + В + / в производстве ацетилена в первую
очередь необходимо выделить компонент А (ацетилен); разделение
других компонентов практически не требуется. Процесс с
использованием селективного растворителя идет в соответствии со следующей
принципиальной схемой (рис. VI1-34). Смесь Л + В + /входитснизу
в абсорбер У, орошаемый растворителем Q. Компонент А поглощается,
349
сверху аппарата выходит смесь компонентов В | /. Часть
компонентов В и I в соответствии с их малой растворимостью тоже поглощается
растворителем Q. Затем их выделяют в колонне 2 (обычно этот
аппарат называют отпарной или отдувочной колонной) — за счет
отдувки компонентом Л, имеющим большую растворимость. В этом
случае компонент Л можно рассматривать как инертный газ по
отношению к компонентам В + /. Учитывая разницу растворимостей,
можно, вообще говоря, определить необходимое соотношение
давлений PJP'2, при котором происходит полное выделение компонента В,
и, следовательно, компонента /. Правильно выбранное соотношение
давлений позволяет определить расход растворителя на процесс
абсорбции.
Компонент А можно выделять из растворителя путем сброса
давления или создания вакуума, а также за счет подогрева и отдувки
его инертными газами (например, азотом) или смесью В + /. При этом
предъявляется ряд дополнительных требований, связанных с взры-
ваемостью ацетилена: давление газовых смесей, содержащих
более 65 объемн. % С2Н2, не должно быть выше 1,4—2,0 am. Этой
величиной ограничивается допустимое давление десорбции. Повышать
температуру или проводить отдувку инертным газом можно в одну
или в две ступени, что зависит от требований чистоты конечного
ацетилена. Обычно в десорбере 3 выделяется чистый компонент А в виде
готовой продукции, а часть его направляется в колонну 2 для
отдувки из растворителя компонентов В + /.
В действительности компонент А является смесью А + А1 (Аг —
высшие ацетиленовые углеводороды), причем растворимость А' в
растворителе Q значительно больше, чем А. Компонент А' выделяется
из растворителя в десорбере 4 путем понижения давления или
отдувки инертным газом. Для упрощения примем, что нагрев и отдувка
инертным газом оказывают на абсорбцию такое же влияние, как
понижение давления. Поэтому можно считать, что давление в
аппаратах, например, 2 и 3 связано выражением: Р2 = КР3, где К —
коэффициент пропорциональности. Для аппаратов, указанных на рис.
VII-34, существует определенное соотношение Р1: Р 2: Р3: Р4>
которое при заданном количестве растворителя будет
обеспечивать требуемую степень извлечения.
Е. Р. Шендерей76 предложил интересный метод расчета абсорбции
многокомпонентной смеси для условий выделения ацетилена
селективным растворителем. По этому методу можно подсчитать, например,
соотношение PJP^
Предварительно введем следующие условные обозначения:
VH, VCr, ^вг — количества исходного, синтез-газа, возвратного газа;
Vor — количество компонента А в отдувочном газе;
х'л> *в» x'i —мольные доли компонентов Л, В и / в исходном газе;
хА, хв, Xj —то же, в синтез-газе;
ХА* хв> Х1 — т0 же» в возвратном газе;
Рх, Р2 — давления в аппаратах / и 2\
35Q
им, olvb, ct,/— растворимость компонентов А, В и / в селективном раствори-
теле в аппарате 1\
L — количество селективного абсорбента;
Ф — степень насыщения.
Пользуясь этими обозначениями, можно записать уравнения,
материального баланса для аппарата 1:
1) по компоненту А:
VuxA =
2) по компоненту В:
Уих'в = VcrxB
3) по компоненту /:
Напишем аналогичные уравнения для аппарата 2:
1) по компоненту А:
L = VBrx'A
2) по компоненту В:
P2a1BL (l—xB) = VBr(l —xa)
Равновесие в аппарате 2 достигается при равенстве количеств
компонента А — поглощенного в аппарате 1 и содержащегося в газе,
отходящем из аппарата 2: Р\Ха^> = Pix'a- Тогда соотношение Р\1Рч будет
равно:
[(а\А — а\в) х'в + (фа1Л — а\в) ха]
Здесь Xj = (1 — хА — хв).
Приведенный расчет абсорбции многокомпонентной системы очень
удобен и позволяет определять работу отдельных аппаратов (анализ
состава газа при различных давлениях) и всей системы в целом*.
Аналитическое сравнение того или иного способа разделения
многокомпонентной системы необходимо для выбора параметров
процесса с их последующим технико-экономическим сравнением.
После оценки оптимальных условий разделения
многокомпонентной системы необходимо рассчитать газовый баланс и соответственно
аппаратуру методом абсорбционного фактора. Для рассмотренной
на рис. VI1-34 принципиальной схемы выделения ацетилена
селективным растворителем (диметилформамидом) при условиях, указанных
в табл. VI1-8, рассчитаны отдельные стадии процесса: абсорбции аце-
* В дальнейшем этот метод был применен С. П. Сергеевым для расчета
технологических схем получения пиролизного ацетилена высокой чистоты [Хим. пром.,
№ 3 (1967)].
351
ТАБЛИЦА VI1-8
Основные зоны разделения ацетилен содержащих газов
Зона
Абсорбция ацетилена
Десорбция двуокиси
углерода
Десорбция ацетилена
Десорбция ацетиленовых
углеводородов
Абсорбция ацетиленовых
углеводородов
Выделяемый
компонент
Ацетилен
Двуокись
углерода
Ацетилен
Ди ацетилен
Метилацетилен
Коэффициент
извлечения
0,98
0,98
0,99
0,99
0,7
Параметры
10 am,
25° С
1,2 am,
25—30° С
0,2 am,
RO f\^° Г
yj\J—DO V-i
0,2 am,
100° С
1,2 am,
35° С
(в среднем)
Степень
обводнения
%
3
5
5
2
5
тилена, десорбции двуокиси углерода, абсорбции высших
ацетиленов, тепловой и вакуумной десорбции ацетилена и выделения высших
ацетиленов. В табл. VI1-9 приведен для примера расчет абсорбции
ацетилена. Аналогичным способом рассчитаны составы газов по
отдельным зонам схемы. Данные расчета сведены в табл. VI1-10.
Из табл. VI1-10 видно, что в процессе имеется два рециркулирую-
щих газовых потока: возвратный газ и сырой ацетилен, содержащий
высшие ацетиленовые углеводороды. Путем изменения количеств этих
потоков можно регулировать степень очистки ацетилена от двуокиси
углерода и высших ацетиленовых углеводородов. Необходимо
учитывать циркуляцию сырого ацетилена для правильного выбора
производительности вакуум-компрессора.
После расчета газового баланса отдельных стадий процесса определяется число
ступеней разделения в соответствии с принятой схемой на стадиях абсорбции и
десорбции. Число ступеней разделения в зоне абсорбции ацетилена рассчитываем
по формуле (стр. 224):
п =
— 1
Лф — абсорбционный фактор; Лф = L/Vm.
Эти величины равны:
Y± = 9,78%
У 2 = 0,2о/0
т = 0,00417
L/V = 0,132
0,132
ф 0,00417
B= 1 + (1,25 — 1)
= 1,25
= 12,65
Отсюда п = 10,3.
352
-« О
СМ СО
о" о"
с©
00~
I I
I I I I
2 S
о о4
о"
2 S
^ ^ о
хнэипиффеоМ
8
о" — *-н
S
CN
8
..88..
^ ~-< о~ о" о" о" о" о
о д
СМ С
и
оЗ w
Е ^
So
S о
н о
03 сз
* х
ы
О,
ю
о
СО
о
CD Ю СО
Ю -н ^ ^
СО СМ СО СМ
о о о о
о о^ о^ о
о" о" о о
киээаонавс! олоз
-оевф
о
см
СО
ел см см
00 — ^ -ч
О —• СО СМ СО
d
HdH9J
ю
о"
О СО О
ел о см
о о о
—Г о" о"
8 §
00 ~н ^
^. со см
о
о
СО
см
о"
ю
см
t^ ^^ 1-ч Ю О О
00 СО О О *-< -н
ел о о о о о
2 S 2
2
S |
cf s
СЭ ^ ^
о" о" о"
ю
см
о см
00 СМ
2 S 8 8
О" О" "-ч" -н"
s
- со ел оо
СМ у—| Ю Tf1
см ю
о
00
о"
см
см
о"
I I I I
СП
t^i
о"
S S
cuj
ю о о
СО"
"■*
оо"
см
и и и и и и1
нС
и
О
и
о
и
23 в. Н. Антонов
К JL.
х U
о
а. —1
s
е-з
Is
X 03
О «
к о
■ I
ОТ,
о
X
3
ю
03
(-
те
g
о
5 л
3 я
о.
^ S сЗ 0) ^>
«[«С-с; °
к
о
К со
is
Си Н
<L> Я
ч a
5^
о |
5 *- s
3SS
3*1
vogi -
О. а) те
О ч s
о н
Газ
ЦИ1
диме
*
8
CQ
те
со
рОЛР
ой)
у
со —'
те
енты
мпон
о
vO
■"
чо
VP
V»
а?
п/и5Я
S
96
о
78,
см
о
о
см
СО
00
X
00
^
00
CD
80
100
8
о
00
8
О
О
00
о
80
X
и
|
1
о
СО
О
о
со
<л
о
см
см
о
со
о
22
СО
64
о
см
см
о
со
о
о
СО
J
см
см
о
^_
о
1
08
о
о
о
о
о
о
о
о
о
_
о
X
J
СС
см
СО
о
to
о
1
1
44
о
S
о
ю
о
о
S
о
05
о
ю
о
X
те
О
Tt
со
см
о
1
1
87
о
8
'—'
О
О
о
о
1
1
о
о
о
с*
X
J
ю
со
(М
"-*
о
1
1
87
о
S
о
о
00
1
|
о
о
о
(О
Х^
СО
|
1
СО
Ю
О
о
69
о
(Л
о
см
си
79
8
56
см
СП
о
8
LO
О
S
СН,
О
см
о
со
СО
00
75
00
98
СП
СО
00
78
49
о
а
00
о>
00
о
8
00
|
1
82
т~>
h*.
16,
8
О
8
О
О
LO
*'~н
73
16
00
О
о
со
о
о
67
'—'
со
(М
<Л
|
I
21
о
о
см
01
о
о
о
<л
о
01
см
§
о
о
о
а
о
о
см
о
о
о
28
Tt<
in
CM
s
о
74
о
о
ю
см
259
о
СМ
t4^
о
S
см
258
8
i
i
8
59
со
tp
ю
02
*-*
1~н
см
о
S
47
548
СО
'—1
т~н
СО
LO
СО
547
<м
см
а
00
80
о
см
19
(Л
8
34
114
о
о
S
1033
8
тр
Ю
S
8
О
оз
со со
^1
О Он
О S
—' С
03 оз
51 со
со
S
1
о
о
со
о
ю
т"""1
ю
ь*
СО
о
X
и
оз
X
2
354
III
Оста
В ДИМ
форм
S3
3%
Вые
ацети
ржится
метил-
мамиде
еред
лением
сших
иленов
Соде
в ди
фор
п
выде
вы
ацет
§33§
• § н g
III*
<*si
f-i JjJ
Ч д °
Я* q Q
Содержится
в диметил-
формамиде
перед
десорбером
II ступени
. в .|, g
?.|1«|а
о» 5. f* о s s
s г>ф е g р{
* К ^
■-
Si
с»
а?
*
ее
^5
99
мпоненты
о
u/l
чКГ
|
31,3
000
31,3
1,00
99,22
^
72
98,
о»
95,
96,58
о>
96,
S
8
©
8
00
£
и
1
1
3,08
960
©
3,08
0,10
i
о
0,0
' ©"
©
©
0,13
со
©
©"
8
©
X X
и J
с
ч со
1
3,62
115
©
3,62
0,12
| 0,52
00
0,3
(М
СО
©
©
со
©
0,72
©
СО
(Л
21
©
3?
им
т
*
о
0,01
31,30
990
©
31,3
1,00
1
03
©
СО
©
©
1,03
СО
о
'—*
©
СО
(М
о
©
Х^
30,70
976
©
30,7
0,97
см
0,0
38
©
со
СО
©
1,34
34
'—|
со
СО
33
©
£
и
СО
1
1 1
0,26
I
CN
©
55
о"
CN
©
0,21
см ,
о 1
СО
©
©
о
о
1
©
5 8 г" о 8
!>• 00 <Л ©
-н
о
о
*~*
0,01
100
оо
со
100
3,19
100
СО
77,
1 8
1 ^
1—1
97
100
CN
СО
100,
о
т^
52
О
се
со 2
о|
£ 8|
'X S
u
с
ч со
1
©
1
5,5
СО
1
35,7
1
г^.
1
СО
1
о
1
1
00
(М
d:
о
X
23*
Я «
я я
s~
US
о s
Is
за
«&
«В
Я Ч
-S
S* о
г
я
о.
С
355
Аналогичным образом могут быть рассчитаны остальные стадии
процесса разделения ацетиленсодержащей газовой смеси. Для
перехода от ступеней разделения к реальным аппаратам вводятся
значения единиц переноса, полученные на основе экспериментальных
данных. Метод расчета ступеней разделения с помощью
абсорбционного фактора можно применять в том случае, если растворимость
компонентов подчиняется закону Генри. Если закон Генри не
соблюдается, применяют обычные
методы, но с тем отличием, что
расчет ведут по ключевому
компоненту для каждой стадии
процесса.
Представляет интерес
определить зависимость числа
ступеней разделения и
абсорбционного фактора от количества
абсорбента для различных стадий
процесса. При этом изменение
абсорбционного фактора связано
с соотношением газ : жидкость.
На рис. VI1-35 представлена
зависимость абсорбционного
фактора (или фактора отдувки),
а также числа ступеней
разделения для различных стадий
разделения газовой смеси от
количества абсорбента (диметилформ-
амида).
Из графика видно, что
изменение количества абсорбента
вызывает незначительное
колебание абсорбционного фактора на
стадии абсорбции ацетилена и
десорбции двуокиси углерода,
а также на стадии десорбции
высших ацетиленов (кривые 5,
7, 8). Наоборот, число ступеней
в зависимости от количества
растворителя изменяется довольно резко. Незначительное
увеличение количества растворителя сильно изменяет число ступеней
на стадиях абсорбции ацетилена и десорбции двуокиси углерода
(кривые 1 и 2) и меньше сказывается на стадии десорбции
ацетилена и его гомологов (кривые 4 и 6).
Следовательно, при оптимальном количестве растворителя на
стадии абсорбции ацетилена практически будет создано
оптимальное количество растворителя на остальных стадиях процесса. В
частности, для рассматриваемых условий наиболее целесообразно
количество абсорбента 105 м61ч. Можно отметить, что действительное
'356
Количество абсорбента, м3/ч
Рис. VI1-35. Зависимость показателей
абсорбции от количества абсорбента:
/ — число ступеней в зоне абсорбции С2Н
2 — число ступеней в зоне десорбции СО
3 — фактор отдувки в зоне десорбции С2Н
4 — число ступеней в зоне десорбции С2Н_
5 — фактор отдувки в зоне десорбции высших
ацетиленов; 6 — число ступеней в зоне
десорбции высших ацетиленов; 7 — абсорбционный
фактор в зоне абсорбции С2Н2; 8 — фактор
отдувки в зоне десорбции СО2.
число ступеней зависит от абсорбционного фактора, поэтому
фактическая высота аппаратов также в какой-то мере определяется
количеством растворителя.
Оборудование для абсорбционно-десорбционных процессов
разделения ацетиленсодержащих газов
В процессах получения концентрированного ацетилена с
использованием селективных растворителей нашли применение аппараты
насадочного типа с насадкой из колец Рашига и Пааля и аппараты
с провальными тарелками. В схемах выделения ацетилена
низкотемпературными растворителями применяются абсорберы
насадочного типа и с колпачковыми тарелками.
Для процесса выделения ацетилена диметилформамддом при
режиме, указанном в табл. VI1-11, было проведено сравнение
различных абсорбционно-десорбционных аппаратов. Сравнивались аппараты
с двумя типами колпачковых тарелок (площадь колпачков 0,713 и
0,918 м2), с насадкой из колец Рашига и с провальными тарелками.
Расчеты показали, что для указанных в табл. VI1-11 соотношений газ:
жидкость колпачковые тарелки работают в неустойчивой
гидродинамической области. Аппараты с насадкой и с провальными тарелками
имеют лучшие показатели при данном гидродинамическом режиме,
причем сопротивление провальных тарелок примерно такое же, как
насадки77'78.
ТАБЛИЦА VII-Л
Основные параметры выделения ацетилена диметилформамидом
Аппарат
Избыточное
давление
am
Темпера-
тура
Весовое
отношение
газ :
жидкость
Абсорбер
Десорбер I ступени (зона выделения
СО2)
Десорбер II ступени
зона выделения С2Н2
зона выделения высших ацетиленов
10
1,25
0,2
0,2
25
30
80
100
0,13
0,02
0,04
0,07
При одинаковой производительности по газу диаметры аппаратов
следующие (в м):
д Аппараты с
Аппараты провальными
с насадкой тарелками
Абсорбер 1,8 1,6
Десорбер I ступени 1,6 1,4
Десорбер II ступени
зона выделения С2Н2 1,8 1,8
зона выделения высших ацетиленов .... 2,2 2,0
Аппараты с провальными тарелками имеют меньшие габариты,
поэтому они предпочтительнее. Кроме того, нужно учитывать, что
357
Газ
насадка значительно более металлоемка, чем тарелки. Методы
гидравлического расчета аппаратов с колпачковыми, сетчатыми и
провальными тарелками подробно изложены в работе В. М. Рамма77.
На рис. VI1-36 изображен абсорбер для поглощения ацетилена
селективным растворителем. Аппарат выполнен из углеродистой
стали и имеет провальные
тарелки 3 с живым сечением 16—20%,
изготовленные из листовой стали.
В верхней части аппарата имеются
^Растворитель четыре колпачковые тарелки 1 для
механического улавливания
растворителя, который стекает вниз,
в зону абсорбции ацетилена.
ПЛАСТИНЧАТЫЕ
ТЕПЛООБМЕННИКИ
В ряде процессов химической
и других отраслей
промышленности получили большое
распространение особые теплообменные
аппараты — пластинчатые
теплообменники 79"81. Главной
особенностью их является конструкция
теплопередающей стенки,
позволяющая полностью разбирать
аппарат и чистить все теплопередаю-
щие поверхности. Поэтому
пластинчатые теплообменники можно
применять в процессах, связанных
с образованием полимеров и
отложением их на теплообменных
поверхностях. В частности, такие
аппараты находят применение и
в схемах концентрирования
ацетилена селективными растворителями —на стадии теплообмена между
очищенным растворителем и растворителем, содержащих ацетилен
(см. рис. VI-14, поз. 13, стр. 249). Применение этих аппаратов по
сравнению с обычными трубчатыми позволяет значительно упростить
эксплуатацию оборудования.
В разборном пластинчатом теплообменнике рабочие пластины
одинакового размера расположены параллельно друг другу, а по
каналам между ними проходит нагреваемая (или охлаждаемая)
жидкость.
Теплообменники выполняются из большого числа пластин
и имеют каналы различной формы. Скорость движения в каналах
воды или жидкостей, близких к ней по физическим свойствам, равна
0,3—0,7 м/сек.
358
Дренаж
Рис. VI1-36. Абсорбер для извлечения
ацетилена селективным растворшелем:
/ — колпачковые тарелки; 2 —
распределительная тарелка; 3 — провальные тарелки.
На рис. VI1-37 показана схема теплообменника, состоящего из
группы теплообменных пластин 11, подвешенных на горизонтальных
штангах 5, концы которых заделаны в стойки 1 и 7. Пластины можно
изготовлять из любого металла, подвергающегося штамповке. Обычно
применяют листовую углеродистую или нержавеющую сталь
толщиной 1 мм. При помощи нажимной плиты 6 и винта 8 пластины в
собранном виде сжимаются в один пакет. Каждая пластина имеет две
резиновые прокладки: одна прокладка ограничивает на лицевой
стороне пластины канал для соответствующего потока жидкости
и имеет два отверстия (с одной стороны или по диагонали) — для под-
Рис. VI1-37. Схема пластинчатого теплообменника:
/ — передняя стойка; 2 — верхнее угловое отверстие; 3 — кольцевая
резиновая прокладка; 4— граничная пластина; 5—штанга; 6 —
нажимная плита; 7 — задняя стойка; 8 — винт; 9 —большая резиновая
прокладка; 10 — нижнее угловое отверстие; 11 — теплообмениая пластина;
lull— потоки.
вода и стока жидкости, другая прокладка изолирует входные
отверстия и создает проход для второго потока жидкости.
Пластинчатый теплообменник построен так, что после сборки
и сжатия пластин в нем образуются две системы каналов: одна —
для нагреваемой (или охлаждаеймо) жидкости, другая — для
теплоносителя. Первая система состоит из каналов с нечетными номерами,
вторая — с четными, благодаря чему потоки нагреваемой (или
охлаждаемой) жидкости и теплоносителя чередуются. Обе системы каналов
кончаются соответствующими штуцерами для входа и выхода
жидкостей. При параллельной расстановке пластин с малыми
промежутками между ними можно разместить рабочую поверхность
теплообменника наиболее компактно, что приводит к значительному
уменьшению габаритов пластинчатых аппаратов по сравнению с другими
конструкциями.
Сравним пластинчатые теплообменники, например, с трубчатыми.
В пластинчатых аппаратах коэффициент компактности (величина
рабочей поверхности в 1 мв пространства) равен 6,5. Практически это
означает, что для создания трубчатых аппаратов с такими же
габаритами рабочей зоны и для той же тепловой нагрузки необходимо под-
359
держиватьв них перепад температур или коэффициент
теплопередачи в 6,5 раза выше по сравнению с пластинчатыми. Если
коэффициент теплопередачи в современных пластинчатых аппаратах при
умеренной скорости потока достигает 3000—3500 ккал/(м2'4>град),
то для выравнивания коэффициентов компактности в трубчатых
аппаратах необходимо увеличить коэффициент теплопередачи в 6,5
раза, что неосуществимо.
В современных теплообменниках пластины снабжены
специальными элементами — гофрами, вызывающими искусственную турбу-
лизацию потока с целью
повышения эффективности
теплопередачи. Гофры имеют
в сечении профиль
равнобедренных треугольников и
расположены под углом к
продольной оси симметрии
пластины, образуя форму «елки».
Существуют пластины с
гофрами другой формы. По
данным УкрНИИхиммаш81 на
изготовление теплообменника
из пластин с гофрами в «елку»
требуются сравнительно
меньшие затраты, чем с гоф-
100
80
60
20
10
/
У
--
У
-1
у
/
У
УА
—
Ч
1
у
/
/
/
/l"
/у
у/
/
/
К _|_
2-W2 5-Ю2
Ю3
Re
2-Ю*
5-Ю3
рами других типов. При
гофрах в «елку» обеспечивается
хорошая искусственная тур-
булизация рабочих потоков
(при Re > 200). Это
способствует улучшению
теплопередачи и сокращению эксплуатационных расходов.
В результате исследований УкрНИИхиммаш получена
следующая зависимость81 теплоотдачи в каналах при Re ^ 200:
Рис. VI1-38. Теплопередача в различных
теплообменниках:
/ — пластинчатые теплообменники с гофрами
в «елку»; 2 —то же, с горизонтальными гофрами;
3 — труба круглого сечения (режим движения
потока переходный).
Nu = CRe°.rapfo.43
(VII-81)
В этой формуле С — коэффициент пропорциональности. Для
пластин с гофрами в «елку» он равен 0,135, а с горизонтальными
гофрами — всего 0,097.
На рис. VII-38 представлены графики теплоотдачи в различных
теплообменниках. Видно, что при одном и том же значении
критерия Re теплопередача в пластинчатых теплообменниках значительно
интенсивнее, чем в трубчатых. Еще одним преимуществом
пластинчатых аппаратов с узкими каналами является возможность обработки
потоков в тонком слое и при малой разности температур, что
обусловливает мягкий обогрев, без местных перегревов.
■ Пластинчатые теплообменники имеют и крупные недостатки,
связанные с наличием большого количества прокладок, которые
360
трудно уплотнять. Кроме того, при высоких давлениях (более 10—
12 am) и температуре выше 120—150° С затруднен подбор
прокладочных материалов. Применение синтетических каучуков новых типов
(силиконовых и др.) позволит поднять температурный предел до
300° С.
Сравнительные результаты замены трубчатых
теплообменников пластинчатыми на стадии концентрирования ацетилена
селективными растворителями приведены ниже80 (теплообменники
двухсекционные):
Пластинчатый Трубчатый
теплообмен-
теплообменник ник
Нагрузка, м3/ч 200 200
Температурный режим, °С
основной поток 116—36 116—36
I хладоагент 50—97 50—97
II хладоагент 27—40 27—40
Количество аппаратов 7 13
Поверхность теплообмена, м2 520 1066
Общий вес, т 17 28
Применение пластинчатых теплообменников на стадии
концентрирования ацетилена позволяет интенсифицировать процесс
теплопередачи примерно в 2 раза, сократить расход металла примерно
в 1,5 раза и уменьшить занимаемую производственную площадь.
Нужно, однако, отметить, что в пластинчатых теплообменниках
расход электроэнергии на перекачивание потоков несколько выше,
чем в трубчатых. В настоящее время для схем с селективными
растворителями повсеместно применяются пластинчатые теплообменники.
ЛИТЕРАТУРА
1. Тебеньков Б. П., Рекуператоры для промышленных печей, Металлург-
издат, 1958.
2. Л а п и д у с А. С, А л и п о в Н. Е., Хим. маш., № 2 (1962).
3. А л и п о в Н. Е., Лапидус А. С, Газ. пром., № 5 (1965).
4. Б а х ш и я н Ц. А., Трубчатые печи с излучающими стенками топки,
ГОСИНТИ, 1960.
5. Бахшиян Ц. А., Баклашов Б. Е., Хим. маш., № 6 (1960).
6. Ш о р и н С. Н., П р а в о в е р о в К. Н., Изв. АН СССР, ОТН, № 8 (1953).
7. М и х е е в В. П., Сжигание природного газа в промышленных установках,
Гостоптехиздат, 1962.
8. Д р у с к и н Л. И., Сжигание газа в промышленных печах и котлах,
Гостоптехиздат, 1962.
9. С у х о в В. И., Шорин С. Н., Газ. пром., № 9 (1965).
10. 3 а х а р и к о в Н. А., Сталь/ № 10 (1965).
11. Нормы теплового расчета котельного агрегата, под ред. А. М. Гурвича и
Н. В. Кузнецова, Госэнергоиздат, 1957.
12. Теплотехнический справочник, Госэнергоиздат, 1957.
13. Филимонов С. С, Хрусталев Б. А., Андрианов,
Конвективный и лучистый теплообмен, Изд. АН СССР, 1960.
14. Р а б и н о в и ч О. М., Котельные агрегаты, Машгиз, 1963.
361
15. Л е з и н В. И., Л и п о в Ю. М., Селезнев М. А. и др.,
Пароперегреватели котельных агрегатов, Изд. «Энергия», 1965.
16. Михеев М. А., Основы теплопередачи, Госэнергоиздат, 1949.
17. Ellwood P., Danatos S., Chem. Eng., № 8, 151 (1966).
18. Las 1 о A., Nemeth A., Mag. kern, fol., 66, № 1, 25 (1960).
19. Волков А. Е., Лапидус А. С, Техника безопасности в производстве
ацетилена из природного газа, Изд. «Химия», 1964.
20. Л а п и д у с А. С., Безопасность труда в промышленности, № 11 (1965).
21. Абрамович Г. Н., Прикладная газодинамика, Гостехтеоретиздат, 1951.
22. Абрамович Г. Н., Теория турбулентных струй, Физматгиз, 1960.
23. Хинце И. О., Турбулентность, Физматгиз, 1963.
24. В у л и с А. А., Струйные задачи прикладной газодинамики, в сб.
«Исследование физических основ рабочего процесса топок и печей», Изд. АН КазССР,
Алма-Ата, 1957.
25. Исследование турбулентных струй воздуха, плазмы и реального газа, сб. под
ред. Г. Н. Абрамовича, Изд. «Машиностроение», 1967, стр. 182.
26. Г и н е в с к и й А. С, в сб. трудов ЦАГИ им. Жуковского «Промышленная
аэродинамика», вып. 23, Оборонгиз, 1962.
27. Гроздовский Г. Л., там же.
28. Г и н е в с к и й А. С, в сб. трудов ЦАГИ им. Жуковского «Промышленная
аэродинамика», вып. 27, Изд. «Машиностроение», 1966.
29. Л я х о в с к и й Д. П., Теплопередача и аэрогидродинамика, кн. 12, вып. 3,
Госэнергоиздат, 1942, стр. 82.
30. И в а н о в Ю. В., Изв. АН СССР, ОТН, № 8, 37 (1954).
31. Хитри н Л. Н., Физика горения и взрыва, Изд. МГУ, 1957.
32. Михеев В. П., Промышленное сжигание природного газа, Куйбышевское
книжное изд., 1959.
33. И о н и н А. А., Горелки для сжигания газа, изд. Министерства
коммунального хозяйства РСФСР, 1951.
34. Brit. Chem. Eng., 10, № 5, 292 (1965).
35. Ц и р л и н А. М., Теорет. основы хим. технол. 1, № 4 (1967).
36. Г о р и с л а в е ц С. П. и др., Нефтепереработка и нефтехимия, № 3 (1967);
№ 5 (1968).
37. Арсеев А. В., Сжигание газа, Металл у ргиздат, 1952.
38. А н т о н о в Г. С, в сб. «Труды совещания по прикладной газовой динамике»,
Изд. АН КазССР, Алма-Ата, 1959.
39. Иванов Ю. В., Газ. пром., № 9, 1958.
40. Иванов Ю. В., Основы расчета и проектирования газовых горелок, Гостоп-
техиздат, 1963.
41. Иванов Ю. В., Эффективное сжигание подслойных горючих газов в топках,
Эстгосиздат, 1959.
42. Ш а н д о р о в Г. С, Истечение из канала в неподвижную и движущуюся
среду, ЖТФ, 26, № 1 (1957).
43. Чернобыльский И. И., Щеголев Г. М., в сб. «Труды института
теплоэнергетики АН УССР», № 7, 1952.
44. П о п л а в с к и й А. Н., Газ. пром., № 9 (1961).
45. Г р о д с к и й Я. С, К а р м и н с к и й В. Д., Газ. пром., № 2 (1959).
46. Л е в и н А. М., Брюханов О. Н., Газ. пром., № 9 (1962).
47. Привалова К. А., в сб. «Теория и практика сжигания газа», Гостоптех-
издат, 1958.
48. Спейшер В. А., Сжигание газа на электростанциях и в промышленности,
Изд. «Энергия», 1967.
49. Полак Л. С, Хаит Ю. Л., ДАН СССР, 156, 920 (1964).
50. X а ит Ю. Л., Приклад, механ. и техн. физ., № 3, 44 (1965).
51. Хаит Ю. Л., в сб. «Кинетика и термодинамика химических реакций в
низкотемпературной плазме», Изд. «Наука», 1965, стр. 167.
52. Л а н д а у Л. Д., Л и ф ш и ц Е. М., Механика сплошных сред, Гостехиз-
дат, 1957.
53. Э м а н у э л ь Н. М., Кнорре Д. Г., Курс химической кинетики, Изд.
«Высшая школа», 1964.
362
54. Л е в и ч В. Г., Физико-химйЧеская гидродинамика, Физматгиз, 1959.
55. С т р и ж е в с к и й И. И., Г у з о в С. Г., Ковальский В. А.,
Ацетиленовые станции, Машгиз, 1959.
56. Кафаров В. В., Основы массопередачи, Изд. «Высшая школа, 1962.
57. Е г о р о в Н. Н., Охлаждение газа в скрубберах, Госхимиздат, 1954.
58. И р а к А. Ю., Газ. пром., № 1, 34 (1958).
59. Л а п и д у с А. С, Нефтепереработка и нефтехимия, № 11 (1963).
60. Зусманович Л. М., Водоснабж. и сан. тех., № 7, 2 (1961).
61. Головачевский Ю. А., Оросители и форсунки в химической
промышленности, Машгиз,- 1967.
62. Веселовский К. В., Гамбург Д. Ю., Т у р к и н а Т. И,, Труды
ГИАП, вып. 17, 1966, стр. 181.
63. Теснер П. А., Рафалькес И. С, Исследование образования сажи
при термическом разложении углеводородов, Труды ВНИИгаз, вып. 3, Гос-
топтехиздат, 1958.
64. 3 у е в В. П., Михайлов В. В., Производство сажи, Изд. «Химия», 1965.
65. У ж о в В. Н., Борьба с пылью в промышленности, Госхимиздат, 1962.
66. У ж о в В. Н., Очистка промышленных газов электрофильтрами,
Госхимиздат, 1962.
67. В a u m H., Erdol u. Kohle, 15, № 11, 914 (1962).
68. В а л ь д б е р г А. Ю., Зайцев М. М., Сидоров В. И., Вестник техн.
и экон. информ., изд. НИИТЭхим, № 4 (1964).
69. Позин М. Е., Мухленов И. П., Тарат Э. Я-, Пенный способ
очистки газов от пыли, дыма, тумана, Госхимиздат, 1953.
70. К о р н е в М. А., М е л и к а т я н С. А., Вестник техн. и экон. информ.,
изд. НИИТЭхим, № 12 (1963).
71.' Б е г р о в В. В., Стрижевский Н. И., Вестник техн. и экон. информ.,
изд. НИИТЭхим, № 10 (1963).
72. Киселев В. И., Насосы, компрессоры и вентиляторы, Металл у ргиздат,
1959.
73. Рис В. Ф., Центробежные компрессорные машины, Машгиз, 1951.
74. Heide P., GWF, 99, № 49, 1266 (1958).
75. Кириллов И. И., Газовые турбины и газотурбинные установки, т. 1 и 2,
Машгиз, 1956.
76. Ш е н д е р е й Е. Р., Ивановский Ф. П., Хим. пром., № 9, 10 (1963).
77. Р а м м В. М., Абсорбция газов, Изд. «Химия», 1966.
78. Д и л ь м а н В. В. и др., Хим. пром., № 3, 156 (1956).
79. Барановский Н. В., Пластинчатые теплообменники в пищевой
промышленности, Машгиз, 1962.
80. Коклюшкин Г. Ф., в сб. «Химия и технология азотных удобрений и
продуктов органического синтеза», Изд. ГИАП, 1963.
81. К о в а л е н к о Л. М., Хим. маш., № 2 (1961).
ГЛАВА
VIII
МЕРЫ БЕЗОПАСНОСТИ В ПРОИЗВОДСТВЕ
АЦЕТИЛЕНА
Производство ацетилена из углеводородного сырья и из карбида
кальция является пожаро- и взрывоопасным, так как в этом процессе
приходится иметь дело с горючими газами и жидкостями.
Как было указано в гл. I, ацетилен и высшие ацетиленовые
углеводороды способны к самопроизвольному разложению в условиях,
близких к параметрам технологических процессов. Поэтому
технология производства ацетилена по возможности строится таким
образом, чтобы избежать критических условий, способных вызвать
взрыв. Дополнительные мероприятия (установка специальных огне-
преградителей, повышение запаса прочности аппаратуры и
коммуникаций и т. п.) также позволяют повысить безопасность процесса.
Вопросы, связанные с разработкой строительной части и
санитарной техники, электрооборудования, а также общие требования
техники безопасности здесь не рассматриваются, так как они описаны
во «Временных нормах по технике безопасности в производстве
ацетилена из природного газа» и других официальных изданиях1-2.
Факторы, влияющие на взрывоопасность ацетилена
Взрывные свойства ацетилена были подробно рассмотрены в гл. I
(стр. 17). Здесь следует подчеркнуть, что под техническим
ацетиленом в производственной практике подразумеваются газовые смеси,
содержащие не менее 95% С2Н2. Обычно в этих смесях имеются
небольшие примеси высших ацетиленов, двуокиси углерода и этилена,
а также следы растворителей (при получении ацетилена из
углеводородов). При более значительном разбавлении ацетилена другими
газами взрывчатость его существенно понижается, так как заметно
возрастает минимальное давление, при котором возможен взрывной
распад ацетилена (для чистого С2Н2 эта величина составляет 0,65—
1,4 am в зависимости от энергии инициирования).
Важным показателем, характеризующим взрывчатость ацетилена
является его начальная температура (см. рис. 1-3, стр. 22): чем
она выше, тем ниже допустимое предельное давление ацетилена.
Поэтому температура ацетилена должна быть возможно меньше.
Аналогичным образом повышенная температура влияет на
разложение ацетиленсодержащих газов (см. рис. VII-32, стр. 341). Это об-
364
стоятельство учитывается при выборе режима их компримирования
(стр. 340).
Температура самовоспламенения чистого ацетилена в разных
условиях составляет 300—625° С, однако под действием различных
веществ она может снижаться (особенно опасна ржавчина). В
присутствии некоторых газов-разбавителей (например, СО) температура
самовоспламенения также понижается.
Смеси ацетилена с кислородом и воздухом по возрастанию
взрывоопасное™ можно расположить в такой ряд3:
С2Н2 + О2 > С2Н2 > С2Н2 + воздух
Отсюда можно сделать существенно важный практический
вывод — чистый ацетилен (поскольку смеси его с кислородом в
процессах производства ацетилена маловероятны) более опасен, чем
.в смеси с воздухом. Поэтому при работе с чистым ацетиленом
необходимо применять защитные средства, выбор которых зависит от
давления и температуры ацетилена, степени разбавления его другими
газами, скорости движения по трубопроводам и др. Пределы взрывае-
мости ацетилена с воздухом определены путем многочисленных
экспериментов и составляют 2,5—78,0 объемн. %.
.Для ацетиленсодержащих газов допустимые пределы давления
и концентрации точно не установлены, но ориентировочно пределы
взрываемости этих смесей можно рассчитывать по формуле:
a -f- b + с + d
п =
В ^ С ' D
где П — нижний (или верхний) предел взрываемости газовой смеси
сложного состава, объемн. %;
а, Ь, с, d — содержание горючих компонентов в смеси, объемные доли;
Л, В, С, D — нижние (или верхние) пределы взрываемости горючих
компонентов, объемн. %.
. Предельно допустимые концентрации ацетилена в тройной смеси
С2Н2—О2—N2 при атмосферном давлении показаны на рис. VII1-1.
Из рисунка видно, что эта смесь характеризуется очень большой
областью взрыва, например смесь, содержащая около 10 объемн. % О2,
становится невзрывоопасной лишь при содержании N2 более
80 объемн. %. Для чистого ацетилена при 1 am возможно применение
технического азота, содержащего 97 объемн. % N2. Это допускается,
так как максимальное содержание кислорода, поступающего с таким
азотом, в смеси будет 1,5 объемн. % (при разбавлении С2Н2 : N2 =
= = 1 : 1), а нижний предел взрываемости ацетилена в смеси с
кислородом составляет примерно 2 объемн. %. При повышенном давлении
требования к чистоте азота повышаются. Так, например, при сжатии
ацетилена до 5 am применяется азот чистотой не менее 99,999%.
В производстве ацетилена из углеводородного сырья большое
значение имеют смеси, в которых присутствуют ацетилен и водород.
По данным рис. VII1-2 можно хотя бы ориентировочно судить о
взрываемости газа пиролиза при различных способах получения
365
ацетилена4. Например, взрыв ацетилено-водородной смеси,
содержащей 8 объемы. % С2Н2, может наступить при добавке 5 объемы. % О2,
поэтому в смесях С2Н2 + Н2 содержание кислорода не должно
превышать указанной величины. Это требование особенно важно
соблюдать при окислительном пиролизе, так как в реакционных газах
может остаться непрореагировавший кислород.
Присутствие кислорода очень опасно при очистке газа в
электрофильтрах. Напряженность поля в электрофильтре очень высока.
При этом энергия может достигать 10 000 дж и быть достаточной для
инициирования поджигания смеси. Для безопасности содержание
ОМ
О 20 W 60 80 100
с2н2 *-
Рис. VII1-1. Пределы взрываемости
смеси С2Н2—О2—N2 при 1 am.
Рис. VII1-2. Пределы взрываемости
смесей ацетилена с кислородом при
разбавлении их азотом, метаном и
водородом (х — содержание разбавителя,
объемн. %).
кислорода в газовой смеси, поступающей в электрофильтр, не должно
превышать 0,5—0,8 объемн. %.
Из всех параметров, обусловливающих стабильность ацетилена,
давление является одним из основных: с увеличением давления
стабильность резко снижается. Все прочие величины (скорость движе
ния ацетилена по трубам, диаметр и длина трубопровода, наличие
статического электричества и др.), определяющие возможность,
характер и давление взрыва ацетилена, являются вторичными
факторами, зависящими в основном от давления. Поэтому оптимальное
давление при работе с ацетиленом следует выбирать с учетом
конкретных условий. Например, для производства ацетилена из
карбида кальция и углеводородов допустимо давление до 1,4 am; при
переработке ацетилена в акрилонитрил, хлоропрен, винилацетат и
N-метилпирролидон давление достигает 3—6 am и более.
Возможность ведения процесса при таком давлении обеспечивается строгим
соблюдением соответствующих условий и применением
специальных защитных средств (см. ниже), которые по мере увеличения
давления должны быть все более жесткими. Вообще же по соображениям
безопасности давление ацетилена рекомендуется принимать
минимально возможным.
366
При производстве ацетилена из углеводородного сырья в целевом
продукте иногда допускается до 2,5 объемн. % высших ацетиленов,
отдельные представители которых (диацетилен, триацетилен, винил-
ацетилен) значительно более взрывоопасны, чем ацетилен. В
сравнительно небольших количествах эти вещества не могут повлиять на
характер взрывного распада ацетилена. Но наличие их представляет
значительную опасность, так как они легко полимеризуются с
образованием твердых или жидких веществ, которые скапливаются на
поверхности аппаратов и труб и в любой момент при
соответствующих условиях (удар, падение капель или частиц, повышение
температуры и т. д.) могут вызвать взрывное разложение ацетилена.
Если по ацетилену в настоящее время накоплен сравнительно
большой экспериментальный и производственный опыт,
позволяющий определять основные условия безопасной работы, то по высшим
ацетиленовым углеводородам таких сведений имеется значительно
меньше. Отдельные представители высших ацетиленов
характеризуются чрезвычайно низким начальным давлением взрывного
распада, например для диацетилена оно равно 0,04—0,2 am. Поэтому
безопасное обращение с ними в условиях производства возможно
практически только при разбавлении инертным газом или водяным
паром.
Условия разбавления смесей, содержащих ацетиленовые
углеводороды (например, диацетилен), можно выбирать, исходя из
пределов взрываемое™ диацетилена (рис. 1-16, стр. 40). Минимальная
кратность разбавления по диацетилену равна 1:1, т. е. при
атмосферном давлении количество разбавителя должно быть равно количеству
диацетилена. При окислительном пиролизе и электрокрекинге
фракция высших ацетиленов содержит около 30% диацетилена и
находится при избыточном давлении 1,1—1,2 am. Разбавление в этом
случае должно быть не менее 3—5-кратного.
Выбор габаритов ацетиленопроводов
Взрывной" распад ацетилена проходит ряд стадий,
характеризующихся разными конечными давлениями. Характер взрывного
распада и возможность его возникновения помимо других факторов
зависят от длины и диаметра ацетиленопроводов, а также от
соотношения этих величин5.
Длина ацетиленопроводов. Для перехода взрывного распада
ацетилена в детонацию требуется определенное время — преддето-
национный периоду который при соответствующей скорости пламени
обусловливается преддетонационным расстоянием. Преддетонацион-
ным расстоянием называют длину участка трубопровода, по которому
должен переместиться фронт начавшегося взрывного разложения
ацетилена до момента детонации.
На основании различных экспериментальных данных была
установлена зависимость преддетонационного расстояния от начального
давления ацетилена5 (рис. VIII-3). Как видно из рисунка, с увеличе-
367
нием начального давления преддетонационное расстояние
сокращается. Так, переход к детонации от начала взрывного распада при
давлении порядка 4,8 am совершается в пределах 0,5—10 м, а при
1,4 am — лишь на расстоянии 15—40 м. Преддетонационное
расстояние существенно зависит также от состояния внутренней поверхности
трубопровода (наличие шероховатостей, сужений и пр.), поэтому
с точки зрения безопасности целесообразно применять для ацетилена
трубопроводы с гладкой внутренней поверхностью.
С увеличением диаметра труб минимальное давление, при
котором возможен детонационный распад ацетилена, уменьшается.
1
I
7,0
5,6
%
X
!
ьоо
300
cj 200
•х.
^ 100
x 0,3050,61 1,221,833,05 6,1 12,218,330,5 61 122
Преддегпонационное расстояние, м
Рис. VII1-3. Зависимость преддетонацион-
ного расстояния от начального давления
ацетилена (заштрихованная область означает
интервал экспериментальных данных).
0 ~ 0,2 ОА 0,6 0,8 1,0
К" Ld/Lmp.
Рис. VII1-4. Зависимость
отношения конечного давления
взрыва к начальному давлению
ацетилена от отношения преддетона-
ционного расстояния к длине
трубы.
Например, для трубы диаметром около 40 мм минимальное давление
детонационного распада равно 2,8 am, а для трубы диаметром около
115 мм оно составит 1,4 am.
•На рис. VIII-4 графически изображено изменение отношения
конечного давления РК9 возникающего при взрыве ацетилена, к
начальному давлению Рн в зависимости от величины К — отношения
преддетонационного расстояния ЬД к длине трубы LTp. На основе
анализа рис. VIII-4 можно установить возможность детонации и
оценить давление, которое при этом развивается.
Об опасности распада ацетилена в трубопроводах можно судить
по величине К следующим образом: при К <С 1 детонация возможна;
максимальное давление, возникающее при детонационном распаде,
будет тем больше, чем ближе величина К к единице. Особенно резко
оно возрастает в пределах значений К от 0,6 до 1. При К > 1
детонация невозможна; в этом случае может быть только взрывной
распад ацетилена.
Рассмотрим возможные варианты развития взрывного распада ацетилена в
трубопроводах различной длины. Примем условно начальное давление взрывного
распада 1,4 am. Преддетонационное расстояние, соответствующее этому давлению,
как видно из рис. VII1-3, находится в пределах от 15 до 40 м.
368
Вариант 1 — длина трубопровода значительно превосходит преддетона-
ционное расстояние (например, L тр.= 200 м). В этом случае детонация возможна,
так как К <С 1:
Давления, возникающие при распаде ацетилена, могут быть такими (см.
рис. VIII-4):
при К = 0,075 Рк/Рн = 60 и Рк = 60- 1,4 = 84 am
при К = 0,2 РК!РН - 68 и Рк - 68- 1,4 - 95,2 am.
Вариант 2 — длина трубопровода меньше минимального преддетонаци-
онного расстояния (LTp> = 10 м). В этом случае /(> 1:
Поэтому может протекать только взрывной распад ацетилена.
При взрывном распаде, как известно, возможно максимум
13-кратное увеличение давления против начального: 1,4 «13= 18,2 am.
Наиболее опасно каскадное разложение, возникающее на
сравнительно коротких участках ацетиленопровода. При каскадном
разложении конечное давление может превосходить начальное более
чем в 500—600 раз.
Отдельно стоит рассмотреть влияние поворотов, торцов и других
преград в системе транспортирования ацетилена на характер его
разложения. В этом случае детонационная волна, дойдя до преграды,
отражается и повышает давление в ударной волне, что приводит
к увеличению конечного давления. Поэтому все указанные преграды
нежелательны, так как они повышают опасность взрыва.
Что же касается прочности трубопроводов, то в отдельных
случаях после оценки возможной опасности сооружаемой системы
транспортирования и возникающих в ней давлений, по-видимому, можно
использовать трубы, выдерживающие давление детонационного
распада ацетилена. Промышленные трубопроводы обычно трудно
принимать только по этому критерию, так как небольшие по диаметру
трубопроводы обычно еще достаточно стойки против детонации, а
в трубопроводах больших диаметров давление детонации может
превышать предел прочности трубы.
Диаметр ацетиленолроводов. Для создания безопасных условий
транспортирования ацетилена необходимо правильно выбрать
диаметр трубопровода, так как от этой величины зависит возможность
возникновения взрывного разложения ацетилена и распространение
уже возникшего очага распада. На рис. VIII-5 цоказаны
минимальные давления, при которых может быть инициировано взрывное и
детонационное разложение ацетилена, и соответствующие им
внутренние диаметры трубопроводов. Видно, что при давлении до 1,4 am
взрывной распад ацетилена практически невозможен в трубах
любого диаметра, если импульс не превышает некоторой величины.
На рис. VII1-6 показано минимальное начальное давление
ацетилена, при котором возможен взрывной и детонационный распад
ацетилена даже при давлении ниже 1,4 am (т. е. меньше минималь-
24 В. Н. Антонов 369
10
ного давления начала распада), и соответствующий ему внутренний
диаметр трубопровода. С увеличением давления безопасный диаметр
трубопровода уменьшается.
Диаметр, ниже которого взрывное разложение ацетилена
практически невозможно, называется критическим. Так, например,
критическим диаметром при
избыточном давлении ацетилена доТД am
является 100 мм, а при 5 am —
12,0 мм. Однако следует иметь
в виду, что понятие критического
диаметра условно, так как при
значительной энергии воспламенения
и большом объеме горючей смеси
взрывное разложение ацетилена
может произойти в трубах
диаметром меньше критического. На
современных производствах
приходится транспортировать
ацетилен в больших количествах,
поэтому диаметр ацетиленопровода,
соответствующий условиям
наибольшей безопасности, нужно
выбирать с учетом изложенных выше
сведений и конкретных условий транспортирования ацетилена2'5.
Рассмотрим на примерах несколько возможных вариантов транспортирования
ацетилена по трубам2. Пусть транспортирование некоторого количества ацетилена,
осуществляемое под давлением газгольдера (1,05—1,06 am) по трубе диаметром 400 мм
со скоростью 15 м/сек, будет основным вариантом для сравнения.
1
Ц//
\V 2
Ч
1
I
°l 100 200 300
Внутренний диаметр трубопровода, мм
Рис. VII1-5. Зависимость
минимального давления взрывного (/) и
детонационного (2) разложения ацетилена от
внутреннего диаметра трубопровода
(длина трубопровода 30 м).
1
28,0
2,8
* 0,7
ОА
ч
ч
/7
".^
4
х^
5?
——
ч V.
% , fi 25b 25W
Внутренний диаметр трубопровода, мм
Рис. VII1-6. Зависимость минимального давления
взрывного (1) и детонационного (2) распада ацетилена от
внутреннего диаметра трубопровода при малых давлениях
(пунктир означает данные, полученные экстраполяцией).
Вариант 1. Примем диаметр ацетиленопровода Dx = 150 мм, т. е. меньше
критического диаметра, соответствующего данным условиям (при Р = 1,05 am
D = 170 мм, см. рис. VIII-6). В этом случае для транспортирования того же
количества ацетилена и с той же скоростью, которая была принята для трубы
диаметром 400 мм, потребовались бы три отдельные параллельные трубы. Разумеется,
это практически неприемлемо,
370
Вариант 2. С целью сокращения диаметра ацетиленопровода примем
скорость ацетилена 30 м/сек. В этом случае для транспортирования принятого
количества ацетилена диаметр трубы должен быть равен:
£>2 = Т/'5;°'42 = 0,28 м
где 15 и 30 м/сек — скорость ацетилена при первом и втором вариантах;
0,4 м — диаметр ацетиленопровода в основном варианте.
Определим необходимое увеличение давления ацетилена вследствие возрастания
сопротивления трубы в рассматриваемых вариантах и сравним с основным.
Увеличение сопротивления АРХ и АР2 в первом и втором вариантах равно:
ДР2_А,яу
где %± и Я2 — местное сопротивление в первом и втором вариантах (к1 = 0,0233;
К2 = 0,0253);
w-l и w2 — скорость газа в первом и втором вариантах (wL = 15 м/сек; w2 =
= 30 м/сек).
Сопротивление трубы во втором случае в 15 раз больше, чем в первом:
ЛР2 _ 0,0253» 0,282-302_ «г
АРХ ~~ 0,0233-0,42.152 ^
Следовательно, второй вариант нерационален.
Из рассмотренных примеров можно сделать вывод, что
предпочтительнее ацетиленопроводы меньшего диаметра, однако тенденция
к их применению должна быть ограничена разумными пределами
(нельзя создавать системы с высоким давлением ацетилена, так как
это опасно). В СССР практически ацетилен транспортируют по
трубам большого диаметра (> 500 мм) при скоростях газа 10—15 м/сек.
Безопасность транспортирования ацетилена под давлением свыше
1,4 am в большей мере зависит от диаметра ацетиленопровода, чем
при атмосферном давлении. С увеличением давления ацетилена
диаметр трубопровода должен уменьшаться, но скорость движения
должна быть невысокой. Критический диаметр, выше которого
возможен взрывной или детонационный распад ацетилена, можно
рассчитать по уравнениям (на основе кривых рис. VIII-6):
D= 157Р-1'82 (взрывной распад)
D = 240Р~1>82 (детонационный распад)
Для давления Р = 3 am критический диаметр равен ~25 мм,
а для 11 am — около 4 мм. Поэтому можно предотвратить
взрывной распад ацетилена, если выбрать диаметр трубопровода с
учетом критического. Практически малый диаметр неприемлем,
поэтому выбирают трубопроводы большего диаметра, но
выдерживающие давление взрывного или детонационного распада.
24* 371
Статическое электричество и скорость движения
ацетилена по трубам
Важнейшими источниками возникновения статического заряда
являются движение жидкости или газа (пара), а также падение
жидкости с большой высоты, сопровождающееся ударом о стенки сосуда.
Электризация усиливается в среде сухого нагретого малоионизован-
ного газа и при неоднородностях металла, из которого выполнены
трубопроводы.
Искра статического разряда может быть инициатором взрывного
разложения ацетилена. Минимальная энергия искры при
статическом разряде, необходимая для воспламенения ацетилена при 2 am,
составляет7 10,5 дж, причем по мере повышения давления эта
величина резко снижается. Так, при 5 am она равна 18-10"3 дж, при
10 am снижается до 2,7 • 10"3 дж, а при 15 am составляет всего лишь
0,56-10"3 дж.
На основе определения энергии искры при статическом разряде
и минимальной энергии воспламенения ацетилена установлено, что
для инициирования взрывного разложения ацетилена, находящегося
под давлением не выше 5 am, требуется больше энергии, чем
выделяется при образовании искры статического разряда. Таким образом,
при транспортировании ацетилена по трубопроводам под давлением до
5 am опасность взрыва за счет статического электричества
практически исключается. Для большей безопасности в обращении с
ацетиленом независимо от его первоначального давления необходимо
избегать возникновения статического электричества и отводить
возникающий заряд от коммуникаций, аппаратов и машин. Особенно
тщательно за этим нужно следить при давлении ацетилена выше 5 am.
Одним из факторов, стимулирующих возникновение
статического электричества, является скорость движения ацетилена, поэтому
ее нужно устанавливать с учетом давления ацетилена. Скорость
движения ацетилена по трубам ограничивается, кроме того, величиной
сопротивления и для давления 5 am и менее на промышленных
установках принимается 10—15 м1сек. При более высоком давлении
ацетилена скорость должна быть ниже указанной. Для ацетиленона-
полнительных станций при 20 am она не превышает 2—4 м/сек.
Защита оборудования
Одним из основных условий взрывного или детонационного
разложения ацетилена является определенное соотношение размеров
системы, в данном случае габаритов аппаратов или машин. Для
возникновения взрыва в аппаратах имеется больше возможностей, чем
в коммуникациях: там чаще повышается температура, больше
снижается содержание газов-разбавителей и т. п. Однако развитие взрыва
и переход его в детонацию в аппаратах менее вероятны, чем в
трубопроводах, поскольку диаметр аппарата сопоставим с его длиной.
372
Технологические аппараты, в которых находится ацетилен,
с точки зрения взрывоопасное™ надо классифицировать по
давлению ацетилена. При 1,4 am можно считать, что система не
отличается от обычных взрывоопасных газовых систем (например,
водород, метан и т. п.), поэтому пробное давление аппаратов следует
принимать, как обычно, в 1,25—1,5 раза более рабочего (согласно
нормам Гостехнадзора). При давлении более 1,4 am следует повышать
требования к прочности аппаратов. При расчете оборудования
необходимо, однако, учитывать температуру, наличие катализаторов и
растворителей, влажность и т. д. и принимать запас прочности не
менее 9—13-кратного по отношению к начальному давлению в
аппарате при давлении в системе до 4—5 am. Например, аппараты, в
которых давление чистого ацетилена составляет 7 am, нужно
рассчитывать на 64—100 am, т. е. на 9—13-кратное давление против рабочего8.
Были проведены специальные опыты по изучению влияния
разрывных мембран на возможность снижения запаса прочности
реакторов9"11. В сосуде диаметром 114 мм и длиной 381 мм была
установлена разрыв-ная мембрана диаметром 51 мм, рассчитанная на
давление 32—33 am. Взрывной распад ацетилена инициировался
расплавленной платиновой проволокой, а также пламенем. При
начальном давлении 10—20 am конечное давление составляло 50—
140 am (инициатор — проволока) и 70—150 am (инициатор — пламя).
Следовательно, даже относительно большая по сравнению с
диаметром сосуда разрывная мембрана не может защитить от превышения
давления в аппарате.
Безопасность синтезов на основе ацетилена можно повысить
при наличии жидкой фазы, поскольку раствор ацетилена в жидкостях
обычно весьма устойчив. Если же процесс необходимо проводить
в газовой фазе и по условиям процесса требуется высокое
парциальное давление ацетилена, то свободное пространство аппарата
заполняют насадкой и применяют газоподводящие трубки узкого
диаметра. В тех случаях, когда недостаточно малого парциального
давления, ацетилен разбавляют (флегматизируют) соответствующими
газами или парами до получения невзрывоопасных смесей.
При конструировании и эксплуатации аппаратов с ацетиленом
по соображениям безопасности необходимо предусматривать
следующее:
1) возможно минимальные размеры аппаратов;
2) отсутствие в аппаратах «мертвого» пространства во избежание
застоя ацетилена;
3) обязательную продувку азотом периодически действующих
аппаратов;
4) соответствующие предохранительные устройства (гидрозатворы
на линиях низкого давления и предохранительные клапаны на
линиях высокого давления) для поддержания давления в аппаратах
не выше заданного во избежание выбросов избытка ацетилена;
5) герметичность аппаратов, в особенности при работе в вакууме,
во избежание подсоса воздуха;
373
6) при работе с чистым ацетиленом и избыточном давлении >7 am
заполнение всего свободного пространства аппарата металлическими
кольцами Рашига соответствующих размеров.
Особым случаем является компримирование ацетилена. При этом
повышение давления сопровождается нагреванием газа, что делает
систему еще более взрывоопасной. Поэтому максимальную
температуру компрессора ограничивают и рассчитывают его на 5—10-
кратное давление против начального; при давлении сжатого
ацетилена 10—20 am расчетное давление принимают до 100 am.
Системы транспортирования ацетилена
С целью наибольшей безопасности ацетилен нужно
транспортировать при минимально возможном давлении, без учета давления,
которое требуется на производстве, потребляющем ацетилен. В связи
с этим промышленные системы транспортирования ацетилена
целесообразно классифицировать в зависимости от избыточного
давления 2- 13~15:
1. Системы низкого давления (до 0,07 am). К ним можно отнести
системы транспортирования ацетилена, находящегося под давлением
1 Ацетилен
потребителям
Ацетилен
Рис. VIII-7. Система транспортирования ацетилена низкого давления:
/ — газгольдер; 2 — устройства для отвода конденсата; 3 — газодувка; 4 — огнепрегради-
тель; 5 — река; 6 — дренажное устройство; 7 — контрольная трубка; 8 — железная дорога;
9 — предохранительный водяной затвор.
газгольдера, т. е. при избыточном давлении 0,02—0,07 am. Обычно
при таком давлении транспортируют ацетилен в процессах его
получения.
2. Системы среднего давления (до 2 am). Это — системы
транспортирования ацетилена в ряде процессов его переработки:
производство акрилонитрила, хлоропрена и др. (включая межцеховые и
внутрицеховые коммуникации).
3. Системы высокого давления (до 25 am). Так транспортируют
сжатый ацетилен при наполнении его в баллоны. Обычно это —
короткие линии от компрессора до ацетиленонаполнительной рампы.
Для каждой из указанных систем меры безопасности различны.
Системы низкого давления (рис. VIII-7). Относительно малое
давление ацетилена делает эту систему не столь взрывоопасной.
Однако в этих случаях применяют ацетиленопроводы больших
диаметров (до 1000 мм), длиной I—2 км и более, что увеличивает опас-
374
ность пожара и взрыва. Как уже говорилось, скорость перемещения
ацетилена в этих условиях согласно промышленной практике
составляет 10—15 м/сек и определяется главным образом допустимым
сопротивлением системы; особых ограничений к скорости не
предъявляется. В системах низкого давления можно применять «сухие» и
«мокрые» огнепреградители с разрывными мембранами. В схеме,
показанной на рисунке, у газгольдера огнепреградители не
установлены, вероятно, потому, что в этом месте система считается
наиболее безопасной, но, по мнению автора, огнепреградители здесь
лучше ставить.
Лцетилен^д
Лцетилен
потребител!
Рис. VII1-8. Система транспортирования ацетилена среднего давления:
/ — огнепреградитель; 2 — дренажные устройства; 3 — контрольная трубка; 4 — шоссе;
5 — трубная рубашка; 6 — река; 7 — железная дорога; 8 — предохранительный водяной
затвор.
Системы среднего давления (рис. VIII-8). В этих системах для
транспортирования ацетилена целесообразны меньшая скорость —
порядка 4—8 м/сек и более жесткие требования в отношении
защитных мероприятий. В частности, рекомендуется применять «мокрые»
огнепреградители (стр. 380), так как «сухие» огнепреградители в этих
условиях недостаточно надежны. В системах среднего давления
широко применяют также гидравлические обратные клапаны.
С целью уменьшения взрывоопасное™ систем низкого и среднего
давления в отдельных случаях, умеренно повышая температуру,
можно увеличивать влажность ацетилена. Однако при чрезмерном
увлажнении происходит конденсация водяных паров, что затрудняет
отвод конденсата. Оптимальная влажность ацетилена соответствует
температуре около 50° С.
Системы высокого давления. В этих системах необходимо строго
следить за скоростью движения ацетилена: чем выше давление, тем
меньше должна быть допустимая скорость. Кроме того,
ограничиваются диаметры трубопроводов, например до 50 мм на ацетилено-
375
наполнительных станциях. При транспортировании больших
количеств ацетилена трубопроводы, особенно изогнутые участки, нужно
заполнять трубками малого диаметра (менее критического для
данного давления).
В настоящее время в связи с расширением транспортирования
ацетилена под давлением и на большие расстояния много внимания
уделяется вопросам безопасности. На большие расстояния
(например, до 80 км) ацетилен предлагается транспортировать в виде
растворов в диметилформамиде под давлением 10 am, а процесс
десорбции осуществлять на месте потребления16. Процесс может быть
непрерывным. В качестве растворителя используют, например,
жидкий аммиак17 и перемещают ацетилен при давлении до 10 am.
Последующее выделение ацетилена путем промывки раствора водой
осуществляют на заводе-потребителе.
Заслуживает внимания метод18 транспортирования ацетилена
под давлением, когда в потоке газа суспендированы инертные
твердые частички размером 10—800 мку имеющие высокий коэффициент
теплопроводности (например, Fe, Mg, Al, A12O3 и SiO2). Скорость
перемещения взвеси рекомендуется от 0,6 до 3 м1сек. Можно также
диспергировать в ацетилене жидкий растворитель, например
метанол или ацетон19. При размерах частиц аэрозоля 10—100 мк полная
стабильность ацетилена, сжатого до 34—60 am, достигается при
10—35° С.
Практический интерес представляют рекомендации по
предотвращению распада ацетилена введением в его поток^ распыленного
масла20 (т. кип. 250—350° С): добавление около 1 м3 масла в час
обеспечивает безопасное транспортирование 20 000 м3 смеси,
содержащей до 35% ацетилена, под давлением 15 am.
Защитные устройства
Огнепреградители. Эти приспособления препятствуют
распространению взрывного и детонационного распада, если он начался
из-за нарушения нормальных условий процесса или по другим
причинам. В основу работы огнепреградителей положена известная
закономерность о том, что фронт пламени не может распространяться,
если теплоотвод от него становится прогрессирующим. На этом
принципе построены различные огнепреградители (впрыск воды, пучки
труб, сетки, насадка)21"24.
Действие насадочных огнепреградителей основано на так
называемом явлении гашения пламени в узких каналах. При горении
в трубках и каналах теплоотдача к стенкам возрастает с уменьшением
их диаметра. В широких трубках теплоотдача незначительна и
затухание пламени может происходить только за счет потерь тепла при
излучении; в узких трубках (каналах) теплоотдача приводит к
уменьшению скорости горения, что содействует затуханию пламени.
Условия гашения пламени в трубках зависят только от их
диаметра, материал трубок влияния не оказывает. Такая особенность
376
обусловлена различием плотностей газа и твердого тела. При
охлаждении газа, граничащего со стенкой канала, поверхностный слой
стенки прогревается незначительно и независимо от материала.
Условия гашения пламени в узких параллельных каналах мало
меняются с изменением их длины и числа, большее значение имеет
форма каналов и характер движения в них горячих газов.
По устаревшим представлениям Холма21 гашение пламени в узких
каналах объясняется теплопередачей из зоны пламени к холодной
исходной смеси. При этом с уменьшением диаметра канала
сокращается радиус сферического фронта пламени,
распространяющегося внутри канала, и возрастает теплоотдача к исходной смеси.
Однако зависимость между скоростью пламени и критическим
диаметром канала, полученная Холмом, верна.
Как было показано Я. Б. Зельдовичем22, концепция Холма,
несмотря на правильный конечный результат, ошибочна. Теплоотдача
от пламени к несгоревшему газу не является потерей тепла, а
представляет одну из стадий нормального процесса распространения
пламени. Согласно теории Я. Б. Зельдовича, в момент гашения
пламени в узком канале достигается определенное критическое значение
безразмерного критерия Пекле Рекр:
где иг — нормальная скорость горения смеси, см/сек;
^кр. — критический диаметр канала, см (dKp. = 0,36d, где d — диаметр
шариков);
ср — теплоемкость холодной смеси, кал/(моль • град);
р — плотность холодной смеси, г/см3;
Ркр. — критическое давление, am;
R — газовая постоянная, ат*смъ1(град\моль)\
То — начальная температура смеси, °К;
Я — теплопроводность смеси, кал/(см-сек* град);
По этой формуле, если известна величина иГ, можно
приблизительно оценить критический диаметр канала dKpm, при котором пламя
гаснет. Результаты опытов и теоретических расчетов подтверждают25,
что Рекр. составляет около 70. Однако в некоторых случаях Рекр.
оказывается намного большим и зависит от диаметра канала или
гранул насадки. При увеличении давления прохождение пламени через
трубки облегчается, так как температуропроводность %1ср газа
обратно пропорциональна давлению. Поэтому с ростом давления
критический диаметр трубок должен уменьшаться.
Поданным И. И. Стрижевского и В. Ф. Заказнова25, полученные
расчетом значения критерия Ре должны быть меньше Рекр., равного
65—70. Если найденное значение Ре больше Рекр>, огнепреградитель
не задержит распространение пламени данной горючей смеси. При
этом «запас надежности» огнепреградителя, который находят из
отношения Рекр. к вычисленному значению Ре, должен составлять
не менее 2:
Ре
377
Для оценки огнепреградителей необходимо знать нормальные
скорости распространения пламени горючих смесей и критический
диаметр для данного огнепреградителя. Величину dKp. для
различных горючих газов можно вычислить по приведенному выше
уравнению. Результаты такие:
н2 ....
со ....
сн4 . . .
с2н2 . . .
"Г
см/сек
... 280
... 45
... 35
... 2
-кр
см
0,8
30
4,0
26
Из этих данных видно, что ацетилен в отличие от других газов
имеет очень малую скорость распространения пламени и большое
значение dK?m. Поэтому в огнепреградителе необходимо применять
крупную насадку (35x35 мм или 50X50 мм).
Независимо от принятого метода расчета и достоверности
получаемых результатов огнепреградители, как правило, подлежат
проверке. Они могут быть использованы в промышленности, если при
испытании не было проскоков пламени.
С целью изыскания наиболее эффективных конструкций
огнепреградителей было проведено большое число экспериментальных работ.
В отдельных опытах в качестве теплоотводящей среды
использовалась вода как наиболее доступная жидкость, имеющая высокую
теплоту парообразования. Огнепреградитель (орошаемый штрек)
представлял собой небольшой участок трубы (около 3 м), в которую
интенсивно впрыскивалась вода. Он был опробован при взрывном
и детонационном распаде ацетилена: детонационный распад
протекал по штреку без заметного затухания, взрывное разложение
иногда задерживалось. Характерно, что работа огнепреградителя
в данном случае не зависела от начального давления ацетилена.
На основе исследований был сделан вывод, что и более длинный штрек
вряд ли может быть достаточно надежной преградой для
начавшегося взрывного разложения.
В качестве огнепреградительного устройства был также испытан2
пучок узких трубок диаметром 10 мм, который помещали в
трубопровод диаметром 100 мм и длиной 30 м. Возникавшее взрывное
разложение ацетилена при давлении до 5,5 am не распространялось
на весь трубопровод, а при 8,5 am взрыв не переходил в детонацию.
Аналогичные испытания проводились при низком давлении24: испы-
тывались трубки диаметром 10 мм, вмонтированные в трубопровод
диаметром 300 мм. В большинстве случаев узкие трубки
задерживали распад ацетилена. Минимальное начальное давление взрывного
распада ацетилена существенно зависит от внутреннего диаметра
этих трубок27: при определенных диаметрах и давлении взрывной
распад ацетилена не может распространяться (рис. VII1-9).
При работе с ацетиленом под давлением трубопроводы диаметром
25 мм и выше, особенно изогнутые участки, рекомендуется заполнять
378
А-
Взрывной,
распад
Ц
' О U Ч- i/
Внутренний диаметр трубой, мм
Рис. VIII-9. Зависимость
начального давления взрывного
распада ацетилена от
внутреннего диаметра трубок.
пучком более узких трубок, причем диаметр отдельной трубки
должен соответствовать критическому диаметру для начального
давления и не должен превышать 12,5 мм. Это целесообразно только при
повышенном давлении из-за относительной сложности и
значительного увеличения стоимости установки. Вообще использование
короткого пучка труб в качестве защитного средства при низких
давлениях требует дальнейшего изучения.
Наиболее надежной преградой против распада ацетилена, хорошо
зарекомендовавшей себя в условиях полевых испытаний, явились
огнепреградители в виде башен с насадкой из колец Рашига. Для
ацетилена при 1 —1,4 am исследовались
орошаемые и неорошаемые башни
(«сухие» и «мокрые» огнепреградители)22'26.
Испытывал ась орошаемая башня
диаметром 500 мм с высотой насадки 1 м
из колец Рашига 35x35 мм или 50 X
х50 мм при подаче 1000 л воды в час
(циркуляция с помощью насоса).
Исследования проводились и при
циркуляции ацетилена в башне со скоростью
0,6—0,7 м/сек с помощью вентилятора.
Было установлено, что орошаемая
башня локализует взрывной и
детонационный распад ацетилена.
В неорошаемой башне газ находился в покое или циркулировал.
Опыты показали, что неорошаемая башня тоже является хорошей
преградой для взрывного и детонационного распада, но в этом
случае необходима несколько большая высота насадки. И. И. Стри-
жевский и др.28 на опытной установке испытывали неорошаемую
башню диаметром 400 мм и высотой 5^ с насадкой из колец
Рашига размером 30X37 мм. В трубопроводе за колонной
признаков распада ацетилена (отложения сажи, разрушение мембраны)
ни в одном из 9 проведенных опытов не наблюдалось.
Оптимальные размер и высота слоя насадки в огнепреградителях
в настоящее время точно не определены. По-видимому, высота
насадки определяется величиной ее общей развитой поверхности,
необходимой для эффективного теплосъема с целью охлаждения
продуктов взрыва ниже температуры воспламенения. В противном
случае ацетилен после огнепреградителя может загореться при
соприкосновении с продуктами взрыва, имеющими высокую температуру,
особенно если эти вещества, не имея выхода наружу, проходят через
огнепреградитель в течение продолжительного времени. С этой точки
зрения целесообразна установка разрывных мембран.
На основе опытных и промышленных прозерок огнепреградите-
лей для ацетилена под давлением не выше 1,4 am даются следующие
рекомендации по их проектированию, установке и эксплуатации.
Скорость ацетилена-концентрата в огнепреградителе должна быть
не выше 0,7 м/сек, считая на свободный объем аппарата. Скорость
379
может быть и больше (1,5—2 м/сек), но тогда значительно
повысится сопротивление слоя насадки. По величине скорости и
нагрузки по газу рассчитывают диаметр огнепреградителя, но
принимают его не менее 500 мм.
В качестве насадки используют
металлические кольца Рашига обычно
размером 35x35 мм и 50х50лш, но
в последнем случае высота слоя
должна быть увеличена не менее чем на
40%, чтобы сохранить одинаковую
поверхность теплосъема. Высота слоя
для «сухих» огнепреградителей
должна быть не ниже 4 м, а для «мок-
Дцетилен
^=
Дцетилен
Вода „, ^=г-
Рис. VIII-10. Схема
«мокрого» огнепреградителя:
1 — насадка (кольца Рашига);
2 — влагоотделитель (кольца
Рашига).
\
Ацетилен
Рис. VI11-11. Схема «сухого»
огнепреградителя с
разрывными мембранами:
/ — насадка (кольца Рашига);
2—люк; 3 — разрывные мембраны.
рых» — не менее 2 м. Применение металлических колец обусловлено
их большей механической прочностью по сравнению, например,
с керамическими. Кольца Рашига нужно располагать равномерно,
чтобы незаполненный объем был минимальным.
При расчете корпуса огнепреградителя на прочность расчетное
давление принимают не менее 12-кратного по отношению к
абсолютному рабочему давлению в аппарате. Обычно пробное давление
равно 25 am, но некоторые исследователи не считают это
обязательным. 12
Конструкции огнепреградителей изображены на рис. VII1-10
и VI11-11. «Мокрые» огнепреградители равномерно по сечению
орошаются водой, плотность орошения 2,5—5 м3/(м2-ч). Во избежание
380
потерь ацетилена воду следует рециркулировать. При этом нужно
следить, чтобы через неплотности сальников насоса в систему не
попадал воздух. Ацетилен-концентрат может проходить в аппарат
прямотоком или противотоком по отношению к воде. Вход и выход
газа должны быть осуществлены под углом 90° по отношению друг
к другу и выполнены в виде тройника (см. рис. VIII-11), на тройнике
перпендикулярно движению газа установлена разрывная торцевая
мембрана.
Как было отмечено, орошаемый водой огнепреградитель является
более надежным, чем «сухой»14. Однако тип аппарата нужно выбирать
в зависимости от конкретных условий производства и
транспортирования ацетилена (степень взрывоопасное™, давление, скорость,
температура и т. д.). Огнепреградители устанавливают на внутри-
и межцеховых коммуникациях ацетилена-концентрата, например
в следующих местах: на выходе из каждого агрегата (при агрегатной
схеме производства); на коллекторе ацетилена-концентрата; на входе
в газгольдер, а также на выходе газа из него при установке
газгольдера «на проход»; на входе в цех или на агрегате переработки
ацетилена; на трубопроводе перед местом вывода некондиционного
ацетилена на факел.
Место огнепреградителя в производстве ацетилена или при его
переработке выбирают с учетом конкретных условий таким, чтобы
локализовать взрывной распад ацетилена на возможно меньшем
участке.
Отсекатели и гидравлические обратные клапаны. Для
предупреждения взрывного распада ацетилена в опытных условиях были
опробованы отсекатели — быстродействующие запорные устройства13.
Принципиально работа отсекателя состоит в том, что возникающий
импульс взрывного распада освобождает сжатую пружину, под
действием которой тарельчатый клапан прижимается к седлу и
перекрывает трубопровод. В результате этого распад ацетилена тормозится.
Отсекатель приходит в действие автоматически — под влиянием
давления взрыва. Время срабатывания отсекателя должно быть
возможно минимальным, но практически оно всегда значительно
превосходит требуемое. Поэтому для торможения распада ацетилена
до отсекателя целесоооразно предусмотреть замедляющий штрек
(башню с кольцами Рашига). Кроме того, штрек является хорошей
защитой для самого отсекателя при детонационном распаде ацетилена.
Инерционность штрековых отсекателей составляет 0,02—0,04 сек.
Существенными недостатками известных в настоящее время
отсекателей являются односторонность их действия, сложность
конструкции и сравнительно высокая стоимость, в особенности для
трубопроводов большого диаметра. В качестве примера на рис. VIII-12
показан гидравлический отсекатель, отличающийся от описанного
тем, что практически является безынерционным.
Гидравлические обратные клапаны предназначены для
прекращения взрыва или детонации в ацетиленопроводах. Большинство
гидравлических клапанов применяется в системах транспортирова-
381
среднего давления. При нормальной работе ацетилен проходит
через клапан и входит в центральную трубу, где барботирует через
находящуюся там жидкость. Часть жидкости при этом увлекается
потоком газа. В центральной трубе поток газа изменяет направление,
при этом жидкость отделяется и собирается внизу, откуда поступает
на циркуляцию. В качестве гидравлической жидкости часто
применяют воду, но при необходимости иметь сухой ацетилен можно
использовать другие жидкости. Потери жидкости нужно периодически
восполнять. Гидравлический клапан также включает
предохранительный пружинный клапан, назначение которого — регулировать
давление, при котором может начаться разложение ацетилена.
Гидравлические клапаны обычно устанавливают перед цехом
переработки ацетилена, чтобы предотвратить повреждение
трубопроводов и защитить оборудование. Их также можно устанавливать
в конце цикла производства.
Разъединительные клапаны. Для быстрого отключения установки
(или ее частей) с помощью разъединительных клапанов применяется
система затопления трубопровода, показанная на рис. VIII-13. При
нормальной работе ацетилен по трубопроводу 4 проходит вниз
под перегородкой 7. При срабатывании автоматического клапана 2
вода из верхнего резервуара перетекает в нижний и закрывает путь
газу. Обычно это устройство применяется в системах низкого
давления для трубопроводов большого диаметра.
Разрывные мембраны. Они не являются средством
предупреждения или замедления взрывного и тем более детонационного распада
ацетилена, так как скорость распространения ударной волны
настолько велика, что мембрана при любой толщине разрывной
пластины не успевает сработать. Кроме того, даже если большая часть
энергии ударной волны и потеряется при срабатывании мембраны,
то за первой волной набегают следующие и распад ацетилена будет
продолжаться. Поэтому мембраны находят применение только в
сочетании с огнепреградителями. Однако польза разрывных мембран
состоит в том, что они, воспринимая на себя основную энергию
ударной волны, уменьшают ее воздействие на аппараты или
трубопроводы и обеспечивают выход наружу горячих продуктов взрыва.
.Схема установки разрывных мембран на огнепреградителе
показана на рис. VIIЫ1 (стр. 380). Места их на трубопроводах у огне-
преградителей обусловлены характером распространения
детонационной волны. Установлено, что разрушению подвергается
разрывная мембрана, которая расположена перпендикулярно фронту
детонационной волны, в то время как другая мембрана, установленная
параллельно фронту волны, только деформируется. Исходя из этих
соображений, огнепреградитель располагают под углом 90° к
трубопроводу, чтобы вся кинетическая энергия набегающей
детонационной волны была направлена на разрывную мембрану, тогда
ослабится удар на огнепреградитель.
Давление разрыва мембраны, установленной на трубопроводе
у огнепреградителя, определяется конкретными условиями произ-
383
водстЁа, например при давлений ацетилена 1,4 am разрывное
давление мембраны принимают 2—2,5 am. При давлении ацетилена выше
1,4 am разрывное давление мембраны соответственно повышают.
Мембраны обычно изготавливают из листов алюминия или стали.
Диаметр их не должен быть меньше внутреннего диаметра
трубопровода, а толщину можно рассчитать по формуле6:
б - KPD -КГ3
где б — толщина мембраны, см;
К — коэффициент, зависящий от материала (для алюминия /(—-0,33—0,38);
Р — разрывное давление, кгс/см2;
D — диаметр мембраны, см.
Необходимо защищать мембраны снаружи от механических
повреждений, а на расстоянии 2—3 м перед ними устанавливать
предохранительные щиты. Независимо от результатов расчета партию
изготовленных мембран следует подвергать контрольному испытанию.
В настоящее время нет единой точки зрения по вопросу установки
взрывных мембран на трубопроводах с горючими газами. Некоторые
исследователи26 считают, что после разрыва мембраны в результате
подсоса воздуха в трубопровод может начаться вторичный взрыв,
по силе значительно превосходящий первичный. Для предупреждения
вторичного взрыва можно применять малоинерционные затворы или
другую специальную арматуру, чтобы, перекрыть трубопровод после
разрыва мембраны, а также подавать азот для устранения вакуума,
который может образоваться в результате взрыва. С целью
подавления вторичного взрыва рекомендуется помещать во взрывоопасную
зону бумажные пакеты с инертным порошком (твердый флегмати-
затор).
Металлокерамические вставки. Для гашения пламени можно
использовать металлокерамические вставки, которые изготовляют
путем спекания металлических шариков. Средние размеры пор этих
вставок составляют 40—200 ммк и зависят от размеров частиц
исходного материала и технологии изготовления. Гасящее действие
пористого материала обусловлено наличием узких лабиринтных
каналов.
- Для локализации распада ацетилена, сжатого до 25 am,
рекомендуют различные конструкции металлокерамических огнепрегра-
дителей9: набор нескольких металлокерамических элементов
двухстороннего действия применяется для защиты компрессоров и
технологических аппаратов, одиночные элементы используются в
манометрах.
Ацетиленопроводы и арматура
В соответствии с действующими «Временными нормами и
правилами по технике безопасности в производстве ацетилена из
природного газа» межцеховые ацетиленопроводы и установленную на них
384
арматуру в системах низкого давления следует рассчитывать на
давление, равное 13-кратному абсолютному давлению в системе
транспортирования. Для систем среднего давления предлагается
применять трубопроводы, выдерживающие давление, которое может
возникать при распаде ацетилена13. Так, например, трубопроводы
диаметром 150 мм рассчитывают на давление 100 am и более.
Трубопроводы для ацетилена изготовляют из стальных бесшовных
труб и соединяют сварными швами. Сварка может быть электрической
(ручной или автоматической под слоем флюса) и газопрессовой.
Газовая сварка допускается для трубопроводов диаметром не более
150 ^ж.
При ручной электросварке можно применять только
высококачественные электроды. Присадочная проволока для
автоматической сварки под слоем флюса и для газовой сварки должна
удовлетворять соответствующим требованиям. Все сварные продольные
швы после укладки трубопроводов следует располагать только
сверху, так как при конденсации влаги по шву может начаться
интенсивная коррозия.
Устанавливать на ацетиленопроводе чугунную арматуру
вследствие ее сравнительно низкой прочности разрешается с
соответствующими ограничениями, предусмотренными нормами. Применение
чугунной арматуры на трубопроводах, подвергающихся вибрации,
запрещается. Арматура на резьбе рекомендуется для трубопроводов
диаметром до 76 мм'. Фланцевые соединения допускаются только
в местах присоединения к оборудованию, арматуре, контрольно-
измерительным приборам и для возможности монтажа там, где сварка
невыполнима. Для изготовления прокладок, выдерживающих
высокое давление взрыва, предлагается использовать красную резину
или асбест. Хорошо прошли испытания кольцевых прокладок
толщиной около 1,6 мм из смеси резины с асбестом.
Транспортировать ацетилен и ацетиленсодержащие газы
рекомендуется по отдельным трубопроводам для каждого потребителя.
По территории завода ацетиленопроводы прокладывают по
эстакаде — самостоятельной или используемой для трубопроводов с
другими газами. В последнем случае ацетиленопроводы следует
располагать в верхнем ярусе эстакады, отдельно от трубопроводов для
кислорода, водорода, взрыво- и огнеопасных веществ и
корродирующих жидкостей. Ацетиленопроводы можно размещать на одной
эстакаде с паро- и теплопроводами, но при этом необходимо исключать
возможность местного перегрева ацетилена. Высказывалось мнение,
что ацетиленопроводы должны иметь антикоррозионное покрытие.
Однако практика работы отечественных заводов показывает
возможность эксплуатации и незащищенных трубопроводов.
Прокладка ацетиленопроводов в непосредственной близости от
воздушных линий электропередачи (включая линии связи) или
пересечение ацетиленопроводами этих линий воспрещается. Не
допускается прокладка ацетиленопроводов через бытовые, подсобные и
административно-хозяйственные помещения, а также через поме-
25 в. н. Антонов 385
Вода
Рис. VIII-14. Устройство для дренирования линий с ацетиленом
в системах низкого давления:
/ —воздушник; 2 —устройство для дренирования; 3 — закрытый сосуд.
ДцетиленС
Q
Диетилен
Рис. VIII-15. Устройство для дренирования линий с ацетиленом
в системах среднего давления при подземной прокладке:
/ — вентили; 2 — сальник; 3 — сифон; 4— закрытый сосуд.
щения для электрооборудования, вентиляционные камеры,
помещение КИП и т. д. Не рекомендуется располагать ацетиленопроводы
под землей, хотя в отечественной производственной практике, и
особенно в зарубежной, этот способ широко применяется.
Ацетиленопроводы, как правило, нужно прокладывать с
уклоном (не менее 0,0015) и оснащать устройствами для сбора и спуска
конденсата (рис. VIII-14 и VIII-15). В случае необходимости
разрешается размещать ацетиленопроводы в общей изоляции с
обогревающим спутником, но изоляция должна обеспечивать равномерный
обогрев трубопровода по периметру. Соприкосновение спутника
с ацетиленопроводом не допускается. Для выравнивания
температурных деформаций нужно использовать только повороты трассы
(естественная компенсация); П-образные, линзовые и другие
специальные компенсаторы применять нельзя.
Трасса ацетиленопровода должна быть по возможности прямой,
с минимальным числом поворотов. На изогнутых участках
рекомендуется устанавливать утолщенные колена, выдерживающие
давление взрыва.
Газгольдеры для ацетилена
Для обеспечения независимости и создания равномерного режима
работы между производством ацетилена и
заводами-потребителями устанавливают газгольдеры. Обычно используют «мокрые»
Рис. VIII-16. Схемы транспортирования ацетилена с установкой
газгольдеров «на тупик» (а) и «на проход» (б):
/ — агрегаты производства ацетилена; 2 — огнепреградители в агрегате;
3 — газгольдеры; 4 — огнепреградители на коллекторе; 5 — разрывные
мембраны; 6 — агрегаты переработки ацетилена.
25*
387
газгольдеры, которые, как правило, можно применять только при
давлении ацетилена до 1,04—1,05 am на входе. Ацетилен
сравнительно хорошо растворяется в воде, поэтому для уменьшения его
потерь в «мокром» газгольдере зеркало воды заливают слоем масла
(4—5 см) или добавляют в воду поваренную соль.
В производстве ацетилена газгольдеры можно устанавливать
«на проход» или «на тупик» (рис. VIII-16). С точки зрения техники
безопасности оба способа примерно равноценны, но некоторое
предпочтение все же необходимо отдать проходному газгольдеру. В нем,
в отличие от тупикового, благодаря постоянному обновлению газа
затрудняются процессы полимеризации и сглаживаются возможные
кратковременные изменения состава ацетилена, обусловленные
нарушением режима работы какого-либо агрегата. С другой стороны, при
установке газгольдера «на проход» в большинстве случаев требуются
более длинные коммуникации и дополнительный огнепреградитель
на выходе газа. Кроме того, этот газгольдер имеет повышенное
сопротивление.
Подготовка аппаратов и коммуникаций
к ремонту
В производстве ацетилена для продувки аппаратов и
трубопроводов, содержащих горючие или токсичные газы или пары,
используют азот. В азоте не должно быть горючих газов, а содержание
кислорода не должно превышать 3,0 объемн. %. Продувку аппаратов
и трубопроводов можно начинать только после их отключения от
работающей системы или перед пуском. Азот подают через
специальные съемные трубы и гибкие шланги с вентилями или задвижками.
После окончания продувки их снимают и ставят на это место
заглушки.
Если азот необходимо подавать в работающую систему (для
устранения проскока пламени в горелке реактора, защиты змеевиков
подогревателей, устранения подсоса воздуха в систему, работающую
в вакууме, и др.), то его направляют по стационарным
трубопроводам, соблюдая соответствующие правила, предусмотренные
нормами.
На трубопроводе для ацетилена должно быть не менее одного
штуцера и одного воздушника для продувки линии азотом перед
ремонтом и перед включением ее в работу. Предусматривается также
подвод азота для периодических продувок аварийных или дренажных
емкостей растворителя и для поддержания постоянной атмосферы
азота в расходных баках и в емкостях.
Большое значение для безопасной эксплуатации производства
имеет правильная организация и тщательная подготовка аппаратов
и трубопроводов к ремонтным работам. Уже говорилось, что
полимеры, как и ацетилен, сорбируемый в продуктах коррозии, при
продувке аппаратуры удаляются очень медленно. Известны случаи
388
взрыва ацетилена при сварочных работах даже в тщательно
продутых трубопроводах. Поэтому перед ремонтом аппараты и
трубопроводы, находившиеся в атмосфере ацетилена, необходимо продуть
азотом, длительно пропарить и вновь продуть азотом до отсутствия
в нем горючих.
Конструкционные материалы
Ацетилен может вступать во взаимодействие со многими
металлами.
Наиболее взрывоопасными являются ацетилениды ртути,
серебра, цинка, кадмия и особенно меди29-31. В зависимости от
условий реакции образуются ацетилениды меди общей формулы31
Си2С2-Н2О, но различных модификаций и с разными взрывными
свойствами (разной температурой разложения), зависящими от
содержания Си в ацетилениде (температура разложения может меняться
от 20 до 185 и даже до 200—250° С).
При контакте ацетилена со сплавами, содержащими менее 70% Си,
пленка ацетиленида (если она вообще образуется) очень тонка и
плотно прилегает к металлу; в этом случае даже искры не вызывают
взрывного разложения29. Если ацетилен соприкасается со сплавами,
содержащими более 70% Си, образуются более толстые отложения
ацетиленида. Его чешуйки можно отделить от металла, они
взрываются при трении и контакте с проволокой, нагретой до
150° С.
Были изучены31 свойства ацетиленидов меди, образующихся на
поверхности латуни (70% Си), фосфористой бронзы (93% Си) и
мельхиора (62% Си). Для этого осадки, собранные с пружинных трубок
манометров, помещали в атмосферу ацетилена. Ацетилениды,
образовавшиеся на поверхности фосфористой бронзы, показали признаки
взрывного распада при ударных нагрузках 0,5 кгс (в 50 испытаниях
из 100 образовались искры).
Осадки с мельхиоровых и латунных трубок при ударных
нагрузках 0,5 и 13,5 кгс оказались стабильными.
На поверхности металлической меди и латуни (до содержания
50% Си) взрывчатый ацетиленид меди образуется при следующих
условиях30: во влажной атмосфере ацетилена и аммиака, сильнее —
в присутствии воздуха и особенно СО2; после соприкосновения с
известковым шламом в атмосфере влажного ацетилена; после
погружения в кислоту (соляную, азотную, серную, а в определенных
условиях и в уксусную) или в щелочь в атмосфере влажного или сухого
ацетилена.
Количество и взрывоопасность отложений уменьшаются с
понижением содержания меди в сплаве, поэтому в некоторых опытах
латунь (50% Си) не дает ацетиленидов. Например, при автогенной
сварке (латунная горелка) медные детали соприкасаются с
ацетиленом (неочищенным или плохо очищенным) и при этом не возникает
каких-либо осложнений. На основании исследований ацетиленидов
389
меди и их превращения во влажном воздухе можно заключить, что
они могут образоваться в различных производственных условиях.
Это необходимо учитывать при выборе конструкционных материалов
(особенно если ацетилен влажный или при наличии аммиака или
кислот). •
Ацетилениды других металлов (серебра, ртути и др.) взрываются
лишь при трении и нагревании.
В связи с этим в производстве ацетилена должно быть обращено
особое внимание на материалы для изготовления аппаратов и
деталей, непосредственно соприкасающихся с ацетиленом или его
гомологами. Запрещается применять для этих целей медь и медные
сплавы, а также серебро и серебряные припои. В виде исключения
для изготовления пробоотборников и измерительных приборов
разрешается использовать сплавы, содержащие не более 70% Си. Это
относится также к оборудованию, в котором находятся
растворители или вода, содержащие ацетилен, в частности оборудование,
устанавливаемое в насосных водооборотного цикла, не должно иметь
медных частей. Не рекомендуется также применять цинк
(оцинкованные трубы), кадмий или сплавы с высоким содержанием этих
металлов.
При переработке синтез-газа, содержащего до 0,2 объемн. %
С2Н2, на оборудовании отделений конверсии метана и окиси
углерода (например, насосы в цикле сатурации и др.) не должно быть
деталей из меди и ее сплавов. Применение серебра и серебряных
припоев также запрещается.
Особо следует оговорить использование ртути, которая сама не
взаимодействует с ацетиленом, но ее окислы достаточно химически
активны по отношению к ацетилену. Поэтому приборы с ртутным
заполнением обязательно должны иметь защитный слой из жидкости,
в которой ацетилен плохо растворяется (например, 30%-ный
раствор хлористого кальция).
Наполнение баллонов «углеводородным»
ацетиленом
Наиболее опасной примесью в углеводородном ацетилене при
наполнении им баллонов является диацетилен. Согласно
официальным нормам32, принятым в ФРГ, содержание диацетилена в
ацетилене-концентрате, предназначенном для наполнения баллонов, не
должно превышать 0,05 объемн. %.
И. И. Стрижевский, Е. И. Кордыш и др.33»34 исследовали
полимеризацию диацетилена в ацетоне и взрываемость системы
диацетилен—ацетон—активный уголь. Было установлено, что полимеры
диацетилена, образующиеся в ацетоне, не являются взрывчатыми
и становятся опасными лишь тогда, когда количество диацетилена
в общей массе составит 50%. На практике при длительной
эксплуатации баллона (25 лет) содержание диацетилена в газовой смеси
не превышает 1%, т. е. не имеет практического значения. Поэтому
390
углеЁодородйым ацетиленом наполняют баллоны без
дополнительной его очистки.
Углеводородный ацетилен, подаваемый в баллоны, после
наполнительной рампы должен иметь следующий состав * (в объемн. %,
в расчете на сухой газ):
Ацетилен, не менее 98,4
Мети л ацетилен, не более 0,15
Диацетилен, не более 0,01
Кислород, не более 0,1
Интенсивность запаха углеводородного и карбидного ацетилена
одинакова, хотя обусловлена разными причинами: основным
компонентом, придающим запах углеводородному ацетилену, является
мети л ацетилен, а в карбидном ацетилене эту роль играет фосфористый
водород (0,01 объемн. %). Утечки ацетилена легко обнаружить орга-
нолептически.
ЛИТЕРАТУРА
1. Временные правила и нормы техники безопасности и промышленной санитарии
для проектирования, строительства и эксплуатации производств ацетилена
термоокислительным пиролизом и электрокрекингом метана, Госхимиздат, 1962.
2. Волков А. Е., Лапидус А. С., Техника безопасности в производстве
ацетилена из природного газа, Изд. «Химия», 1964.
3. К о г а р к о СМ., Иванов Б. А., Журн. ВХО им. Д. И. Менделеева,
9, № 3 (1964).
4. Las 1 о A., Nemeth A., Mag. kern, fol., 66, № 7, 254 (1960).
5. S a r g e n t H., Chem. Eng., 64, № 2, 250 (1957).
6. P о й з е н И. С, Вестник техн. и экон. информ., изд. НИИТЭхим, № 12,
47 (1961).
7. F u d s i с а к у, J. Chem. Soc. Japan, Ind. Chem. Sec, 61, № 5, 515 (1958);
РЖхим, 28217 (1960).
8. R ер p e W., Chem. Eng. Techn., № 13/14, 273 (1950).
9. С т р и ж е в с к и й И. И., Журн. ВХО им. Д. И. Менделеева, 7, № 6, 632 (1962).
10. Miller S. A., Penny Е., Simposium on Chemical Process Hazard, Inst.
Chem. Eng., 1960, p. 87.
11. P err a u den R., Cuivre, laitons, alliages, 43, 33 (1958).
12. К о rap ко С. M., Хим. пром., № 5, 368 (1969).
13. Acetylene Transmission for Chemical Synthesis. Recomended Minimum Safe
Practices for Piping System. International Acetylene Association, New York, 1960.
14. Л а п и д у с А. С., Безопасность труда в промышленности, № 11 (1965).
15. Chem. Eng., 67, № 18, 131 (1960).
16. Н е n d e r s о n E., пат. США 2930682, 1960 г.
17. Pat ton I., Crubb G., Stephenson K., Petrol. Ref., 37, № 11,
180 (1958).
18. Kemp L., пат. США 2730438, 1956 г.; англ. пат. 610755, 1955 г.; пат. ФРГ
1040169, 1959 г.
19. К iy a ma L., Kosishita H., яп. пат. 610755, 1955 г.
20. Н a b е г 1 К., пат. ФРГ 849555, 1952 г.
21. Шаулов Ю. X., Распространение пламени через пористые среды, Изд.
АН АзССР, 1954.
22. Зельдович Я. Б., Теория горения и детонации газов, Изд. АН СССР, 1944.
23. С о л о в ь е в Н. В., Ермилов П. И., Стрельчук Н. А., Основы
техники безопасности и противопожарной техники в химической
промышленности, Госхимиздат, 1960.
* Содержание влаги не должно быть выше 0,8 г/ж3.
391
24. S с h m i d t G., H a b e r 1 K., Technische Oberwachung, 9, № 12, 7 (1955).
25. С т р и ж е в с к и й И. И., 3 а к а з н о в В. Ф., Журн. ВХО им. Д. И.
Менделеева, 7, № 3 (1964).
26. К о г а р к о С. М., Лямин А. Г., Михайлов В. А., Хим. пром.,
№ 8, 621, 1965; ДАН СССР, 162, № 4, 857 (1965).
27. Holemann P., Hasselmann P., Dix G., Forschungber. Wirtschafts-
und Verkehrsministeriums Nordrhein Westfalen, № 382 (1957).
28. Стрижевский И. И., Бородулин А. А., Канер Б. Л., Охрана
химических предприятий от пожаров и взрывов, изд. НИИТЭхим, 1961, стр. 172.
29. Loos F., Schwesstechnik Z., 48, № 11, 290 (1958); 48, № 12, 329 (1958); 49,
№ 19 (1959).
30. J. Soc. Chem. Ind., 66, 346 (1947).
31. Feitknecht W., Hugi-Carmes L., Schweiz. Arch, angew. Wiss.
u. Techn., 23, № 10, 328 (1957).
32. Freitag H., Arbeitschutz, № 1, 16 (1949).
33. Стрижевский И. И., Кордыш Е. И., Воронова Л. Я. и др.,
Журн. ВХО им. Д. И. Менделеева, № 1 (1965).
34. Стрижевский И. И., Кордыш Е. И., Воронов Л. Я. и др.,
XiMi4Ha промисловють. Информ. науково-техн. 36., № 1 (1965).
ГЛАВА
IX
ТЕХНИКО-ЭКОНОМИЧЕСКОЕ СРАВНЕНИЕ МЕТОДОВ
ПРОИЗВОДСТВА АЦЕТИЛЕНА
Получение ацетилена из карбида кальция
Наиболее распространенным методом производства ацетилена
остается получение его из карбида кальция 1~5» 7>13»14. Во многих
странах строятся новые установки по производству карбида кальция.
Технологическая схема и оборудование современных промышленных
установок позволяют получать значительное количество карбида
кальция с одного агрегата. Высокий уровень механизации и
автоматизации технологического процесса обеспечивает низкую
себестоимость продукта.
Ацетилен, получаемый карбидным методом, имеет высокую
концентрацию после очистки от примесей (99,9%) и пригоден для любых
целей органического синтеза. До сих пор еще имеются утверждения,
что ацетилен из карбида кальция наиболее чистый и для ряда про1
цессов органического синтеза не может быть заменен ацетиленом,
получаемым из углеводородного сырья.
Потребляемая мощность одной новой карбидной печи достигает
65 000 кет и можно ожидать дальнейшего укрупнения агрегатов.
Такие крупные мощности печей обеспечивают лучшее использование
тепла в процессе, при этом снижаются относительные потери
продукта и отходящего газа, что также улучшает технико-экономические
показатели метода.
Мощности отдельных карбидных заводов достигли 200, 400 и
даже 600 тыс. т в год. Такие крупные заводы имеются в СССР, ГДР,
США, ФРГ и Японии, а также в других странах. Самый крупный
завод мощностью 600 тыс. т карбида кальция в год построен в ГДР
(в г. Шкопау). Крупнейшим заводом США является предприятие
фирмы Air Production в г. Луисвилле годовой мощностью 250 тыс. т
карбида кальция.
Обычно на крупных карбидных заводах используется известняк,
добываемый в местных механизированных карьерах. При этом
затраты на транспортирование известняка и обожженной извести
до места производства минимальные. В случае потребления
привозного карбида стоимость ацетилена возрастает в связи с необходи-
393
мостью перевозки и укупорки карбида кальция — в СССР * на 50%,
в США — на 25%. Однако возможность перевозки карбида кальция
является одним из достоинств карбидного метода, так как
транспортировать ацетилен по трубопроводам на сколько-нибудь большие
расстояния невозможно, а перевозка в баллонах больших количеств
ацетилена также неприемлема.
Большим достоинством карбидного метода является возможность
регулировать нагрузку энергетической системы в часы пик; в
частности можно без особого ущерба на 5—7 ч в сутки сокращать на 50%
расход электроэнергии. В крупных карбидных печах тепло в зоне
расплава сохраняется достаточно долго, поэтому за несколько часов
шихта не застывает и электропроводность ее не снижается. При
увеличении нагрузки или включении печи после непродолжительного
простоя работа возобновляется без особых затруднений.
Технико-экономические показатели современных крупных
производств карбида кальция, включая затраты на разложение его водой
для получения ацетилена и на обжиг извести, находятся на
следующем уровне (расчет на 1 т С2Н2):
Расход
известняк, т 7,6
кокс и другие углеродистые материалы, т . . . . 3,0
карбид кальция (литраж 250 л/кг), т 3,7
электроэнергия, квт-ч 11 500
вода, м3 600
пар, мГкал 0,4
Количество отходящих продуктов
шламовая известь, т 4,5
топливный газ, ж3 800—1000
Однако производство карбида кальция, несмотря на постоянное
совершенствование технологического процесса, остается громоздким
и тяжелым по условиям труда. Серьезным ограничением для
развития этого процесса остаются трудности в обеспечении известнякрм,
удовлетворяющим но качеству технологическим требованиям
(стр. 52), а также высококачественным коксом. Многие крупные
месторождения известняка, пригодного для переработки, не могут
использоваться, так как перевозка известняка на дальние
расстояния невыгодна. Все указанные обстоятельства часто служат
серьезным аргументом в пользу выбора других методов производства
ацетилена9- п- 12.
Получение ацетилена из углеводородного сырья
В настоящее время из всех известных методов получения
ацетилена из углеводородного сырья промышленное использование нашли
три: гомогенный пиролиз жидкого углеводородного сырья, электро-
* Несколько больший относительный рост стоимости ацетилена в Советском
Союзе объясняется небольшими масштабами контейнерной перевозки карбида
кальция в нашей стране.
394
крекинг природного газа и окислительный пиролиз природного
газа. Имеется много разновидностей каждого метода, отличающихся
конструкцией основного аппарата, технологической схемой и
параметрами производства15-22.
Гомогенный пиролиз жидкого углеводородного сырья. В
заводской практике используются три способа — фирмы Montecatini
(при повышенном давлении), и фирм SBA-Kellog и Hoescht (при 1 am).
Мощность одного реактора гомогенного пиролиза достигает 10 тыс. т
ацетилена в год.
Ниже дан перечень установок гомогенного пиролиза в
различных странах15"23:
Италия
ЧССР .
ПНР .
Япония
ФРГ .
Страна
Метод
Montecatini
Montecatini
, . Montecatini
Montecatini
SBA
. . Hoescht
Мощность no
ацетилену
тыс. т в год
25
10
10
40
9
50
Для этих способов опубликованы1» 3» 4 основные сравнительные
технико-экономические показатели, достигнутые в производстве
(табл. IX-1).
ТАБЛИЦА IX-1
Технико-экономические показатели * получения ацетилена гомогенным пиролизом
жидкого углеводородного сырья
(расчет на 1 т ацетилена)
Показатели
Схема
SBA-Kellog
Hoescht Montecatini
Расход
природный газ, мъ
бензин, т
пар, мГкал
вода, м3
электроэнергия, квт-ч
Количество получающихся продуктов
этилен, т
отходящие газы, м3
водород (общий после конверсии
метана), ж3
аммиак, т
Состав отходящих газов, объемн. %
водород
этилен
метан
2000
2,75
4,9
500
2775
До 0,5
3000—5000
4500
2,0
42
16
20
2200
2,7
4,9
875
2775
2,0
4500
4500
2,0
40
22
20
Предполагается, что кислород поступает со стороны.
3,2—5,0
6,0
3000
5,0
3000
4000
До 2,2
40
20
19
395
В процессе, проводимом под давлением 10 am *, исключается
необходимость последующего компримирования для выделения
ацетилена и облегчается выделение сажи, смол и гомологов ацетилена.
Сырьем в этом процессе могут быть любые нефтяные углеводороды,
но обычно стремятся использовать фракции с меньшим содержанием
ароматических соединений, которые значительно увеличивают
образование смолообразных продуктов.
Следует отметить, что использование нефтяного сырья для
получения ацетилена менее благоприятно по сравнению с применением
природного газа, для работы с которым практически не требуется
каких-либо специальных устройств и установок (часто оказывается
достаточным проложить трубопровод к месту потребления газа).
Кроме того, жидкие углеводороды более дефицитны, что также
ограничивает распространение гомогенного пиролиза в заводской
практике.
Особую группу составляют методы пиролиза в трубах, в
регенеративных печах (табл. IX-2) и пиролиз с движущимся
теплоносителем. Эти методы, несмотря на длительную разработку, до сих пор
еще не внедрены в промышленность. Наиболее перспективным
может оказаться пиролиз паров жидкого нефтяного сырья (пропан-
бутановая фракция, бензин) в трубах; после окончательной
разработки процесса и подбора необходимых жаростойких сталей он,
видимо, найдет широкое применение.
ТАБЛИЦА IX-2
Технико-экономические показатели некоторых способов получения
ацетилена
(расчет на 1 т ацетилена)
Показатели
Расход
бензин (или пропан-бутановая
фракция), т
топливный газ, м3
электроэнергия, квт-ч
Количество получающихся
продуктов
метано-водородная фракция
(35% СН4), т
смола и кокс, т
Пиролиз
в трубчатых
печах
5,20
2000
2000
збао
0,25
Пиролиз
в
регенеративных печах
5,55
3000
2750
4000
0,24
В регенеративных печах сырье используется менее эффективно,
чем в трубчатых, из-за необходимости чередовать периоды нагрева,
пропарки и пиролиза. Преимуществом регенеративных печей
является отсутствие жаростойких труб, для изготовления которых
Процесс пока полностью не освоен.
396
требуются дорогие сплавы. Однако регенеративные печи также
нуждаются в качественных дорогостоящих огнеупорных материалах.
Эти обстоятельства и периодичность процесса препятствуют
широкому использованию указанных методов. Преимуществом
гомогенного пиролиза является возможность применения любого
углеводородного сырья.
Электрокрекинг4'6. Этот метод производства ацетилена применен
в Румынии, США, ФРГ и СССР. В 1966 г. общая мощность установок
по получению ацетилена электрокрекингом достигла около 180 тыс. т.
В процессах электрокрекинга затрачиваемое тепло используется
более целесообразно, так как для получения 1 т ацетилена
требуется нагревать меньшее количество газов. Достоинством
электрокрекинга является возможность получения газов с меньшим
количеством примесей и с более высоким содержанием ацетилена. При
этом процессе источник тепла является наиболее дорогим, но зато
возможно кратковременное отключение реакторов для
регулирования нагрузок электрических систем в часы пик (если допускаются
перерывы в обеспечении потребителя ацетиленом). Сырьем для
электрокрекинга могут быть газообразные углеводороды (метан)
и жидкие (бензин).
Технико-экономические показатели метода можно улучшить
внедрением плазменного процесса. По данным фирмы Knapsack-Grie-
sheim применение водородной плазмы повышает эффективность
крекинга метана — .выход ацетилена составляет до 63,5 вес. % на
исходный метан, количество сажи сокращается до 0,7%, а расход
электроэнергии снижается до 9 тыс. квт-ч на 1 т ацетилена. В
случае пиролиза паров бензина в водороде, нагретом до состояния
плазмы, расход электроэнергии составляет 8,5 тыс. квт-ч на 1 т
С2Н2 (или 4,5 тыс. квт-ч на 1 т С2Н2 + С2Н4). При плазменном
процессе реакционные газы не содержат кислородсодержащих
соединений, а в зоне высоких температур образуется значительно меньше
сажи и смол, чем при обычном электрокрекинге.
Очень мало материалов имеется по методу электрокрекинга
жидких углеводородов. По данным Н. С. Печуро, расход
электроэнергии при крекинге бензина составляет 7,5 квт-ч на 1 т ацетилена (без
учета расхода на очистку и концентрирование). Для этого метода
характерно образование большого количества сажи и смол (до
30 вес. % от исходного бензина), однако большая часть сажи удаг
ляется на самой стадии пиролиза.
В табл. IX-3 приведены для сравнения показатели различных
способов электрокрекинга метана и жидких углеводородов
(принимается степень извлечения ацетилена 95% от количества,
содержащегося в реакционных газах). Как видно из приведенных данных,
для метана наиболее выгоден двухступенчатый электрокрекинг:
несколько снижается расход электроэнергии и значительно
сокращается количество отходящих газов.
При использовании бензина расход электроэнергии на 1 т С2Н2
резко снижается, однако в реакционных газах образуется значи-
397
"ТАБЛИЦА IX-S
Техн и ко- экономические показатели различных способов электрокрекинга
(расчет на 1 т ацетилена)
Показатели
Электрокрекинг
метана
одноступенчатый
двухступенчатый
Пиролиз в плазме
водорода
бензин
Расход
природный газ, м*
водород, м*
бензин, т
Концентрация ацетилена в
реакционных газах, объемн. % . . . .
Количество отходящих газов (после
выделения С2Н2), м3
Состав отходящих газов, объемн. %
водород
метан
этилен
Количество водорода, м3
в отходящих газах
общее после конверсии метана
Количество этилена, т . . . . . .
Количество сажи, т
Расход электроэнергии на 1 т
С2Н2, кв/П'Ч (учитывая стадию
выделения С2Н2)
4600
13,0
5900
62
32
1,2
3800
8200
0,1
12 500
3000
0,9
15,9
3980
50
17
7,5
2500
6000
0,5
0,3
10 300
2700
3000
16,5
7320
76,0
3,0
0,7
7050*
4500
И 000
3000
2,4
14,0
3740
60
9,0
10,5
3110*
920
0,7
0,07
(со смолой)
8500
* 3000 ж3 отходящих газов возвращают в процесс.
тельное количество бензола (до 230 кг на 1 т С2Н2) и этилена.
Поэтому необходимо организовать выделение этих веществ из ацетилен-
содержащих газов. Кроме того, при пиролизе бензина в плазме
сильно сокращается количество отходящих газов и, следовательно,
количество синтез-газа, а также аммиака, получаемого на его основе.
Серьезным препятствием для широкого внедрения
электрокрекинга является высокий расход электроэнергии по сравнению с ее
расходом при пиролизе бензина и окислительном пиролизе метана.
Необходимы дальнейшие усовершенствования метода в целях
снижения энергозатрат, например, путем подогрева исходного газа и
бензина перед подачей в реактор II ступени. Однако это мероприятие
возможно лишь после создания жаростойких электроизоляционных
материалов и после значительного усовершенствования конструкции
реактора.
Окислительный пиролиз природного газа6» 8' 10. Этот метод
получения ацетилена в настоящее время наиболее распространен и
используется в США, Италии, Франции, ФРГ, СССР и других странах.
Общая мощность заводов, использующих этот метод, превышает
600 тыс. т в год (с учетом СССР) и продолжает расти. Наиболее
распространены схемы ГИАП (СССР), фирм BASF (ФРГ) и SBA
(Бельгия). Однако заметной разницы в технико-экономических показа-
398
телях методов не имеется, поскольку температура подогрева
исходных газов одинакова и расходы газа на подогрев и на процесс
практически одни и те же (табл. IX-4). Отдельные способы отличаются
конструкцией реакционной печи и схемой выделения ацетилена (преиму*-
щественное распространение получили процессы выделения
ацетилена селективными растворителями).
ТАБЛИЦА IX-4
Технико-экономические показатели различных способов окислительного пиролиза
(расчет на 1 т ацетилена)
Показатели
Схема
ГИАП *
Схема
BASF
Схема
SBA
Природный газ, ж3
Кислород, ж3
Селективный растворитель, кг, ****
Керосин, кг
Аммиак, кг
Щелочь, кг
Кокс, кг
Поглотитель для сероочистки, кг
Синтез-газ, мъ
Электроэнергия, квт-ч
Вода, м3
Пар, т
6300 **
3570
(95% О2)
5
1,0
10 200
1900***
770
7,8
6220
3598
(98% О2)
о
10 150
2010***
790
7,5
6200
3720
(100% О2)
100
24
34
10 000
2350 ***
1250
6,8 (при 25 am)
2,5 (при 8 am)
0,05 (при 3 am)
* С рекуперационными турбинами.
** С учетом расхода в подогревателях.
*** Расход на производство кислорода, равный около 2000 кет» ч> не учитывается.
**** Даны расходы, близкие к теоретическим.
Указанные схемы отличаются в основном энергетическими
затратами. Путем сравнения методов BASF и SBA выявлено снижение
себестоимости ацетилена на 10% при использовании первой схемы.
В схеме SB А капитальные вложения О1носительно больше, что
связано в первую очередь с применением очень сложного и
металлоемкого оборудования при выделении ацетилена несколькими
растворителями (керосин, щелочь, аммиачная вода и аммиак).
Существенное значение для технико-экономических показателей
методов окислительного пиролиза имеет расход сырья, поэтому
в последнее Бремя на ряде установок для уменьшения расхода
углеводородного сырья и кислорода осуществляют более высокий
нагрев. Кроме того, исследуются возможности снижения сырьевых
затрат, наиболее рационального использования отходов и,
следовательно, удешевления стадии выделения ацетилена. После выделения
получается так называемая фракция высших ацетиленовых
углеводородов (около 5—10 вес. % на 1 т ацетилена). Квалифицированное
использование этих углеводородов, например для получения
гербицидов, позволит существенно улучшить показатели метода в целом.
399
Таким же ценным, но не утилизируемым на сегодняшний день
ресурсом является сажа, составляющая до 5 вес. % на 1 т ацетилена.
Необходимым условием рентабельности является также
использование отходящих газов после выделения ацетиленовых
углеводородов (синтез-газ) для получения аммиака или метанола. В синтез-
газе содержится большое количество водорода и окиси углерода
(26—28% СО, 58—60% Н2, 4—6% СН4). Количество синтез-газа
достигает 10 000 м3 на 1 т. После конверсии окиси углерода общий
выход водорода составит 9600—9800 л*3, что позволит получить до
4 т аммиака или 3,3 т метанола. При таком использовании синтез-
газа можно снизить себестоимость ацетилена почти на 30%.
В производстве метанола или аммиака расход синтез-газа обычно
оценивают по его теплотворной способности, т. е. по степени
энергетического использования. При такой оценке можно снизить
себестоимость метанола или аммиака, получаемого из синтез-газа, на
3—8% за счет сокращения расхода кислорода по сравнению с
получением их из природного газа (в зависимости от цен на
электроэнергию и природный газ).
При сочетании окислительного пиролиза природного газа до
ацетилена с производством метанола (или аммиака) достигается
экономия сырьевых ресурсов, как это видно из данных табл. 1Х-5: причем
более эффективно направлять синтез-газ на производство метанола.
ТАБЛИЦА JX-5
Расходные показатели производства ацетилена и метанола или аммиака
(расчет на 1 т ацетилена)
Показатели
Расход природного газа, м2
на ацетилен
на метанол *
Итого
на ацетилен
на аммиак **
Итого
Расход кислорода, м3
на ацетилен
на метанол *
Итого
на ацетилен
на аммиак **
Итого
Раздельное
получение
6300
3366
9666
6300
3200
9500
3570
1980
5550
3570
1800
5370
Сочетание
производств
6300
6300
6300
6300
3570
600
4170
3570
600
4170
Экономия
%
34
33
23
22,8
* На 3,3 т метанола.
** На 4 т аммиака,
400
Это доказывается следующим сопоставлением. При переработке
синтез-газа в метанол снижение расхода кислорода позволяет
сократить удельные капитальные затраты на 1 т метанола на 13,7%
(включая производство кислорода). При отпускной стоимости природного
газа 20 руб. за 1000 м3 и стоимости электроэнергии 1,4 коп. за
1 квт-ч, снижение себестоимости метанола из синтез-газа составит
около 8%. При сочетании производств ацетилена и аммиака экономия
удельных капитальных затрат и снижение себестоимости (считая
на 1 т аммиака) соответственно составят 9,8 и 4,7%, т. е. они будут
несколько ниже, чем при получении метанола. Добавим, что расход
сырья и энергозатраты (в пересчете на условное топливо) при
переработке синтез-газа в метанол на 8% ниже, чем при производстве
метанола из природного газа (расчет на 1 т метанола-ректификата).
Сравнение методов производства ацетилена
Исторически получение ацетилена развилось на базе
производства его из карбида кальция, но внедрение углеводородного сырья
(природный газ, различные нефтепродукты) в различные отрасли
химической промышленности привело за последние годы к
разработке новых способов производства ацетилена3» 4» 10.
В США все крупные карбидные производства, сооруженные еще
в годы второй мировой войны, размещены в районах с
высокоразвитой промышленностью и относительно дешевой гидроэнергией.
Развитие карбидной промышленности идет в основном за счет
реконструкции действующих заводов, на которых устанавливают
модернизированное высокопроизводительное оборудование. Установки
для получения углеводородного ацетилена, получившие развитие
в последние 15 лет, созданы в районах с большими запасами газа и
нефти и высокой концентрацией предприятий нефтехимической
промышленности, и их становится все больше (за последнее десятилетие
производство ацетилена из углеводородного сырья выросло более
чем в 6 раз).
Аналогичная картина наблюдалась и в Японии, где сначала
(1950—1960 гг.) развивалось производство ацетилена из карбида
кальция, однако из-за сезонности в работе гидроэлектростанций и
вследствие общего недостатка электроэнергии карбидные установки
в Японии используются лишь на 50—60%. Быстрое развитие
нефтехимической промышленности привело к появлению производства
ацетилена из углеводородов: в настоящее время в этой стране
работают две такие установки, заканчивается строительство еще трех
и, кроме того, имеется несколько опытных установок по разработке
новых методов.
Производство ацетилена из углеводородов различными методами
в крупнейших капиталистических странах распределяется
следующим образом (в тыс. т/год)24* 25:
ФРГ . . 310—330 Италия . . 210—230 Япония . . 51
США . .280—320 - Англия . . * 150 Франция. . 48—50
26 р. н. Антонов 401
Сравнение всех существующих методов производства ацетилена
является трудной задачей х~5. Для уточнения соотношения
показателей пришлось бы приводить громоздкие данные, из которых трудно
усвоить основные преимущества и недостатки в свете технико-
экономической оценки. Поэтому анализу подвергаются наиболее
известные методы, с использованием которых в настоящее время
производится основное количество ацетилена в промышленности.
Если распределить общее количество^ вырабатываемого ацетилена
по методам, применяющимся в промышленности, то на 1968 г.
картина такая (в тыс. т, без учета мелких установок)1» 2» 18:
Из карбида кальция ^2800
Окислительный пиролиз природного газа .... 600
Гомогенный пиролиз природного газа и
нефтепродуктов 180
Электрокрекинг природного газа 180
Регенеративный пиролиз 120
Всего 3880
Таким образом, в объеме всего мирового производства доля
ацетилена, получаемого на основе углеводородов, достигла 30% и
продолжает расти. Причем эти методы применяются в основном в про-
мышленно развитых странах, что свидетельствует об их высокой
перспективности. Однако нельзя не указать на сложность
технологического процесса, требующего средств развитого машиностроения
и автоматизации. На основе природного газа в настоящее время
производится более 800 тыс. т ацетилена или около 20% от общего
количества.
Для правильного представления о том или ином методе
получения ацетилена следует определить степень использования тепла
в процессе — так называемый энергетический к. п. д. Именно этот
показатель является главным при оценке методов. При его
определении принимаются во внимание все реагенты и энергетические
ресурсы, затрачиваемые в процессе: природный и топливный газ,
углеводородное сырье, кокс, уголь, пар, электроэнергия. Сравнительные
данные по степени теплоиспользования всех промышленных методов
производства ацетилена приведены в табл. IX-6. Из таблицы видно,
что в зависимости от условий получения электроэнергии
энергетический к. п. д. процессов различен; наиболее выгодны
окислительный пиролиз и гомогенный пиролиз нефтепродуктов.
При определении себестоимости ацетилена учитываются все
технико-экономические показатели, включая амортизацию зданий и
оборудования, обслуживание и другие расходы. Существенное
значение приобретает стоимость сырья и энергетических средств.
В табл. IX-7 приведены данные3 о себестоимости ацетилена для
условий США. Из этих данных видно, что если не принимать во внимание
Вульф-процесс, который хотя и имеет лучшие показатели по себе-
402
4s
I
са
ных
s
ч
В
3
S
про
оа
s
U
О.
Я
си
етил
sf
СО
'~н
СЗ
я
Е-
О>
D4
О
<Я
Он
со
Я
Ч CQ
оо
* о
3 о.
к о
If
о
и
со
Я
с
[иро.
l_i
=я
3
я
гслител
О
«и
н§
«j 5
»• * я
gg-S
>.«
ы
я*
л
СО
КЗ
я
"а
03
СО
S
о
о *
*11
CQ О К
«S
Ё Я
Ss
2 я
о со
ЙЯ8
* CQ С?
м ч о
я §
•Р
О
кг
>ВНОГ
лива
w § §
^Н
g«
Is
2с
^ CQ Ч
и | §
>.Н
Я S
1'
s
s
13
■3
<
D
С
ю
СО
о
-t
8
3
<М
о
см
\
ео
газ, м
эЯ
"Я
ЮДН1
X Он
с£
ю
"-'
ю
1^
сч
1
1
1
о
см
см
О5 00 О
•^f
О"
СО t^
0
о4
55
S
со
S
г-7
о
о
О)
**
§
о
ю
юо"
о
о
СО
|О9
со
со
о
о
5д
CQ
:ооктаз
со
я
Я
3*
90
гия, к
нер
троэ
бенз
кокс
элек
о"
пар,
о оо
СО
о.
00
00
00
00
О5
S
СО
со
СО
о4
СП
00
t^.
о
си
о
CQ
к
о
щих
Q
олуча]
с
§
1
«
кал/
(1800
со
СЗ
U
go
tr C:
§
Он
с
ОС
' °
ю
О
1
1
1
—«00
<=>" О
^нЮ
о"о
1
О '
800
те
1
cJ
о
• я
смол
этил
1
ие
(2900
п
с_
о
V-"
CN
8
S
ю
"^
000
о
°i-
о ^
оо
'is
' со
* ео
'If *
' О •
• °°
. см
^^ео
со' ^ *
CQ S
rh
СО"
LO
"^
1
1
О)
см"
1
1
см
о"
1
о
а
DQ
ю
а>
со"
3
-*
ю
СО
СО"
см
^h"
6
СИ
я
тепла
|
•—•
ю"
СМ
ю"
ь."
аз
г-
о .
^е^
зщии
\Л 7TOU
б 1
;
5
j
я*
С8Ч
' ca с;
ns
s«
g Я
is
° X
s со
; о 5
a с »* о
V О CQ
«SS.3
EBse
N1!
Ё
| О)
I Ч
O-^hX
ssgs
ЙЙ8А
26*
Ч
s О ffl
|
( я
Я О СП
403
ТАБЛИЦА 1Х-?
Сводные данные по себестоимости ацетилена, полученного различными методами
Способы производства
ацетилена
Себестоимость, %, при разной
мощности (тыс. т/год)
10
25
50
Из карбида кальция
Карбид собственного производства
Привозной карбид
100
102
84
95
73
93
Из газообразного углеводородного сырья
Huls (сырье—метан)
Двухступенчатый электрокрекинг
(сырье—метан)
Вульф-процесс (сырье—этан) . . . . .
BASF, SBA-Kellog (сырье—метан) . .
Phillips (сырье—пропан)
73,5
48
46
57
62
55
40
36
51
47
Из жидкого углеводородного сырья
Вульф-процесс
SBA-Kellog . .
Hoescht . . .
62
74
82,5
47
38
58
46
35
30,5
45
40
40
51
51
Примечание. Кислород поступает со стороны.
стоимости, однако пока не получил значительного распространения,
наименьшая себестоимость ацетилена оказалась при окислительном
пиролизе метана (BASF, SBA-Kellog) и двухступенчатом
электрокрекинге. Дороже всего обходится производство ацетилена из
карбида кальция.
При оценке удельных капитальных затрат следует учитывать,
что при окислительном пиролизе метана и гомогенном пиролизе
жидких углеводородов затрачивается кислород, а получение ацетилена
из карбида кальция и электрокрекинг метана связаны с большими
затратами электроэнергии. Следовательно, при расчете удельных
капитальных затрат на производство ацетилена необходимо
учитывать затраты на сопряженные производства (табл. IX-8). В СССР
решающую роль при сравнении различных методов играют
отпускные цены на сырье и электроэнергию, которые сильно различаются
по отдельным экономическим районам. Поэтому метод нужно
выбирать с учетом конкретных географических условий.
Поскольку производство ацетилена из углеводородного сырья
в нашей стране практически только начало развиваться, сравнение
эксплуатационных показателей может быть сделано только путем
расчетов и анализа проектных данных. На основе работ ГИАП и Лен-
гипрохим можно провести сравнительную оценку методов получения
ацетилена для различных географических районов нашей страны1'10> 14
(табл. IX-9).
404
ТАБЛИЦА IX-S
Технико-экономические показатели различных методов получения ацетилена в СССР
Методы производства
Из карбида кальция . .
Окислительный пиролиз
Электрокрекинг ....
Гомогенный пиролиз
жидких углеводородов ....
Удельные
на
производственные
и
вспомогательные
объекты
100
110—135
120—135
100—120
капитальные затраты, %
на
сопряженные
отрасли
100
75—85
85—90
70—80
всего
100 <
80—90
95—105
70—80
Себестоимость
ацетилена
%
100
75—85
88—95
70—80
Примечание. Для карбидного способа расчет вели на мощность по ацетилену
50 тыс. m/год, по остальным способам — на 35 тыс. т/год.
ТАБЛИЦА IX-9
Относительные капиталовложения и себестоимость ацетилена в СССР
(расчет на 1 т ацетилена)
Методы производства
ацетилена
Из карбида кальция . •
Пиролиз жидкого сырья*
Окислительный пиролиз
природного газа
Электрокрекинг
природного газа
Относительные
капиталовложения
%
100
80—100
80
90—100
Себестоимость ацетилена, %
Поволжье,
Урал
100
70—75
75
80
Сибирь
110
95
115
85
Центр,
Запад
100
70—80
90—95
130
Юг
100
70—80
70
95
* Приведены проектные показатели.
По опубликованным в зарубежной печати технико-экономическим
анализам методов производства ацетилена в большинстве случаев
отдается предпочтение получению ацетилена из углеводородов1'5.
Однако в ряде случаев карбидный ацетилен оказывается более
дешевым. Это объясняется, во-первых, конъюнктурой мирового рынка,
а также относительно дорогим природным газом и дешевой
гидроэнергией (США) или отсутствием собственной углеводородной базы
(ФРГ).
При производстве ацетилена из углеводородов преимущества
какого-либо одного метода не являются столь очевидными; во
многих странах развиваются все известные методы, а выбор одного из
них определяется конкретными условиями расположения
предприятия, наличием сырья и энергетических ресурсов. Очень важно
располагать эффективными методами, основанными на природном газе,
что позволяет вовлечь в химическую переработку огромные ресурсы
этого дешевого сырья и вырабатывать ацетилен в различных районах
страны.
405
Для условий СССР, по-видимому, необходимо одновременное
развитие обоих главных методов промышленного получения
ацетилена (из карбида кальция и из углеводородов) в силу экономических
преимуществ каждого способа и значительных резервов их
технического совершенствования. Поэтому необходимы
технико-экономические исследования перспектив производства и применения
ацетилена в 1971 —1980 гг. с учетом структурных изменений,
происходящих в сырьевой базе и в технологии промышленности органического
синтеза.
Отдельно стоит рассмотреть стадию выделения ацетилена из
реакционных газов, стоимость которой определяется концентрацией
ацетилена и выбранным абсорбентом. При одинаковом содержании
ацетилена энергоматериальные затраты для различных способов
выделения составляют 6»8 (в %):
Абсорбция селективными растворителями 100
Абсорбция низкотемпературными
растворителями
аммиаком 140—180
метанолом 80—100
ацетоном 120—160
Следует отметить, что в производстве продуктов органического
синтеза сильными конкурентами ацетилена являются низшие оле-
фины — этилен, пропилен, бутилен. Методы производства олефинов
значительно экономичнее существующих методов получения
ацетилена по следующим основным причинам:
1) энергетические затраты на получение олефинов
(температура 700—900° С) намного меньше, чем для ацетилена (температура
1500—2000° С).
2) концентрация олефинов в реакционных газах в 2—3 раза выше
по сравнению с ацетиленом, что удешевляет стадию их выделения.
3) процессы получения ацетилена связаны с большим количеством
отходящих газов, для использования которых требуются
значительные дополнительные капиталовложения.
В связи с этим стоимость низших олефинов меньше, чем
ацетилена: в СССР стоимость этилена в 2—2,5 раза меньше стоимости
ацетилена, аналогичное положение и за границей. Поэтому
представляется целесообразным исследовать возможность рациональной
замены ацетилена низшими олефинами при производстве некоторых
мономеров и полупродуктов (акрилонитрила, винилхлорида, ацет-
альдегида и др.) в определенных районах страны. В настоящее
время такие работы ведутся как за рубежом, так и в нашей стране.
Тем не менее, производство ацетилена развивается и будет
совершенствоваться. Это вызывается более выгодными условиями
получения ряда продуктов органического синтеза на основе ацетилена
(винилацетат, хлоропрен и т. д.). Увеличивается использование
ацетилена в связи с ростом потребности в виниловых эфирах, N-метил-
пирролидоне, N-винилпирролидоне и других продуктах, для
получения которых нельзя применить этилен.
406
Будущее производства ацетилена в первую очередь связано
с совершенствованием и развитием методов его переработки и с
получением новых синтетических продуктов, т. е. с новыми успехами
химии ацетилена.
ЛИТЕРАТУРА
1. Туров Ю. Я., Паршина Г. А., Фокина М. Б., в сб.
«Сопоставительные обзоры по отдельным производствам химической промышленности
СССР и за рубежом», вып. 1, изд. НИИТЭхим, 1967, стр. 34.
2. Т у р о в Ю. Я., Паршина Г. А., Химия и технология топлив и масел,
№ 12, 57 (1967).
3. Lob о W., Chem. Eng. Progr., 11, 35 (1961).
4. Z u n d e 1 M. I., Chim. et ind., 91, № 2, 99 (1964).
5. Scherwood P., World Petrol., 35, № 8, 61 (1964).
6. Антонов В. Н., Производство ацетилена, Госхимиздат, 1959.
7. Т у р о в Ю. Я., Ю д о в и ч Е. А., Хим. пром., № 7, 481 (1964).
8. Левитан Л. Б., Лапидус А. С, Вестник техн. и экон. информ., изд.
НИИТЭхим, № 2 (1960).
9. Туров Ю. Я., Вестник техн. и экон. информ., изд. НИИТЭхим, № 1 (1963).
10. Б о р и с о в и ч Г. Ф., Хохряков П. А., Родина Р. А., Вестник
техн. и экон. информ., изд. НИИТЭхим, № 2 (1963).
И. Chem. a. Ind., № 17, 551 (1968).
12. Chem. Age, № 2542, 98 (1968).
13. Б а р и н о в а Р., в сб. Химическая промышленность за рубежом, изд.
НИИТЭхим, № 10, 56 (1967).
14. Б р а г и н с к и й О., Щукин Е., в сб. «Экономика, организация и
управление в нефтеперерабатывающей и нефтехимической промышленности», № 4,
изд. ЦНИИТЭнефтехим, 1967, стр. 7—13.
15. Chem. Ind., 20, № 6, 400 (1968).
16. Chem. Ind., 19, № 5, 274 (1967).
17. Hydrocarbon Proc, 47, № 4, 188 (1968).
18. Chem. Ind., 19, №1,1 (1967).
19. Chim. et ind., 97, № 9, 1406 (1967).
20. Chem. Ind., 19, № 4, 181 (1967).
21. Ind. chim., 53, № 589, 173 (1966).
22. Chem. Age, 96, № 2471, 941 (1967).
23. Miller S. A., Acetylen, London, 1965, p. 474.
24. Chem. Week, 103, № 9, 63 (1968).
25. Chem. a. Ind., № 45, 1534 (1968).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорберы ацетилена 224, 357, 358
расчет 223 ел.
Абсорбционный фактор 224, 351 ел., 356
Абсорбция 217 ел.
ацетилена аммиаком 254 ел.
— ацетоном 268 ел.
— Y-бутиролактоном 246
— водой 227 ел.
— диметилформамидом 231 ел.,
351 ел., 357, 358
— метанолом 263 ел.
— N-метилпирролидоном 238 ел.
— низкотемпературными
растворителями 253 ел.
— селективными растворителями
231 ел., 349 ел.
высших ацетиленов аммиаком 256 ел.
— — керосином 258 ел., 277
— — метанолом 258, 263 ел.
— — N-метилпирролидоном 243 ел.
серной кислотой 276
двуокиси углерода ацетоном 269
метанолом 264
N-метилпирролидоном 241,
243 ел.
низкотемпературными
растворителями 253, 254
многокомпонентной смеси 349 ел.
Адсорбенты ацетилена 271 ел., 274, 275
Адсорберы ацетилена 272, 273
Адсорбционная емкость
(Поглотительная способность) 273, 274
цеолитов 276
Адсорбция
ацетилена 271 ел.
— активированным углем 272 ел.
— силикагелем 275
— цеолитами 274, 275
винилацетилена 276
Акриловая кислота 32
Акрилонитрил 30
Аллен см. Пропадиен
Аллиловый спирт 229, 230
Алкины (Высшие ацетилены) 34 ел.
Аммиак
абсорбция высших ацетиленов 256 ел.
как растворитель ацетилена 16,
254 ел.
как флегматизатор ацетилена 25
Ацетилен
абсорбция водой 227 ел.
адсорбция 271 ел.
в баллонах 268, 390, 391
взрывное разложение 17 ел., 21 ел.,
24, 340 ел.,- 364 ел., 367 ел.,
372, 379
выделение 227 ел., 231 ел., 233 ел.,
238 ел., 241 ел., 246 ел., 253 ел.,
263 ел., 268 ел., 349 ел.
горючие свойства 17 ел.
детонация 18, 19, 367 ел.
«закалка» водой 80
карбидный 55, 59 ел., 392, 393
компримирование (сжатие) 339 ел.
концентрат 251
обнаружение 391
образование из углеводородов 66 ел.,
75 ел., 158, 159
очистка см. Очистка ацетилена
пиролиз 80
полимеризация 34, 77
пределы взрываемости 20, 21, 365
производство 64 ел., 154 ел., 131 ел.
разложение 21, 70, 71, 77 ел., 80, 159,
372
растворители см. Растворимость
ацетилена
самовоспламенение 19, 20, 86, 365
себестоимость 403, 404
смеси с аммиаком 25, 26 '
— с ацетальдегидом 21
— С воздухом 19 ел,
Ацетилен
смеси с водородом 365 ел.
— с двуокисью углерода 234 ел.,
243, 244
— с кислородом 19, 20, 365 ел.
— с метанолом 21
— с формальдегидом 21
транспортирование 374 ел., 387
«углеводородный» 216, 393 ел.
физические свойства 13 ел., 36, 37
химические свойства см. Реакции
ацетилена
Ацетилениды (Карбиды) металлов 28,
38, 389
Ацетиленовые
гликоли 32, 33
спирты 32, 33
Ацетиленопроводы 384 ел.
арматура 384 ел.
диаметр 369 ел.
длина 367 ел.
критический диаметр 370, 371
скорость газа 372
статическое электричество 372
Ацетон 253, 254, 268 ел.
Ацетоуксусной кислоты этиловый эфир
229, 230
Бутадиен-1,3 35, 37
Бутанол 229, 230
Бутиндиол-1,4 32
у-Бутиролактон
как растворитель ацетилена 16, 229,
230, 246 ел.
получение 246
физические свойства 16, 246
Взрывное разложение
ацетилена 17 ел., 21 ел., 24, 340 ел.,
364 ел Г, 367 ел., 372, 379
— инициаторы 21, 22, 372
— начальное давление распада 21,
26, 368 ел., 379
— энергия инициирования 22 ел.
винилацетилена 36, 37, 367
высших ацетиленов 40, 41, 367
диацетилена 38, 39, 367, 390
метилацетилена 36
триацетилена 40, 367
Винилацетат 34
Винилацетилен 37 ел.
адсорбция 276
взрывное разложение 36, 37, 367
гидрирование 38
образование при окислительном
пиролизе 159
пиролиз 80
растворимость в аммиаке 256 ел.
— в диметилформамиде 233 ел.
Винилацетилен 37 ел.
растворимость в керосине 259, 277
— в метаноле 259, 264 ел.
— в N-метилпирролидоне 239, 243 ел.
синтез 34
Винилирование
амидов 31
карбазола 31
кислот органических 33, 34
меркаптана 29
а-пирролидона 31
спиртов 29
уксусной кислоты 34
Винилит 34
N-Винилкарбазол 31
Винилмеркаптан 29
Виниловые эфиры
простые 29
сложные 33
Виниловые тиоэфиры 29
N-Винилпирролидон 31
Винилхлорид 34
Водяного газа реакция 156
Воспламенение газов 167 ел.
Вульф-процесс 90 ел., 118
Газгольдеры ацетилена 387 ел.
Газопромыватели турбулентные 325 ел.
Гашение пламени в трубках 376, 377
Гексаметилфосфамид 16
Генераторы ацетиленовые 56 ел.
«вода на карбид» 56, 59, 60
«карбид в воду» 56, 58
контактные 57
«мокрые» 56
«сухие» 56, 60
Гидравлические клапаны 381 ел.
Гидродинамический параметр в
смесителях 299, 304, 309
Гиперсорбция ацетилена 272 ел.
Гомогенный пиролиз 85, 102 ел., 395 ел.
бензина 104, 109, ПО ел., 112 ел.
лигроина 105, 106
параметры 103, 106
пропана 104
расходные коэффициенты 106, 107,
395 ел.
реактор 103, 105, 108, ПО, 114
Горелки
бездиффузорные 284, 292
беспламенные 283
бесфакельные 292
в реакторах 293 ел.
в подогревателях 283 ел.
инжекционные 284, 292
кольцевые 296
скорость истечения смеси 293
туннельные 283
факельные 284
409
Горение
ацетилена нестационарное
(Каскадное разложение) 17, 18, 369
— с аммиаком 25
— с воздухом 19
— с кислородом 19
гашение 166
ламинарное 165
метано-кислородных смесей 160 ел.
нормальная скорость 19, 164
пламя 166, 167, 188, 193
скорость 166, 167
турбулентное 164 ел., 166
углеводородов неполное 155 ел.
факел 164 ел., 188, 283 ел.
фронт 165
Дальнобойность факела горения 167 ел.,
188
Детонационное разложение ацетилена
18, 19, 367 ел.
Диацетил 39
Диацетилен
взрывное разложение 38, 39, 367, 390
гидрирование 39
образование при окислительном
пиролизе 159
пиролиз 80
полимеризация 39, 390
растворимость в аммиаке 256 ел.
— в диметилформамиде 233 ел.,
352 ел.
— в метаноле 264 ел.
— в N-метилпирролидоне 239, 243 ел.
физические свойства 35
Дииндиол 39
Диметилацетамид 229, 230
Диметилацетилен 35, 36
Диметилформамид 231 ел.
как растворитель ацетилена 16, 229,
230, 351 ел., 357, 358
высших ацетиленов 233 ел.,
352 ел.
предельно допустимая концентрация
231
физические свойства 16, 231
Диоксан 229, 230
Дихлорэтан 229, 230
Дуговой разряд в газах 125, 127, 134, 329
Единичных объемов метод расчета
абсорберов 224
«Закалка» реакционных газов 80, 293,
296, 310 ел.
время 311
оптимальный режим 317 ел., 321
расход воды 319 ел., 323
скорость 312 ел.
стадии 311 ел.
410
Избыточные концентраций газов при
смешении 299, 300
Известь в карбидном производстве 52
Известь-пушонка 57, 59
Извлечения коэффициент при
абсорбции 225, 226
С-Индекс (Углеродный показатель,
С-Число) 117, 173
Инициаторы взрывного разложения
ацетилена 21, 22, 372
Ионизация газов
в плазме 147
в электрофильтрах 328, 329
при электрокрекинге 122 ел.
Искровой разряд в газах 329
Каскадное разложение ацетилена
(Нестационарное горение) 17, 18, 369
Касселя схема электрокрекинга метана
127, 128, 149
Карбазол 31
Карбид кальция
литраж 50
плавление 44
получение 43 ел., 52 ел.
примеси 44, 45, 53
разложение водой 55, 56
технический 52 ел.
Карбидный ил 61
Карбидный плав 44, 48
Карбиды (Ацетилениды) металлов 28, 38.
389
Карбоиды 88
Катализаторы
винилирования 31, 34
гидрирования ацетилена 28, 29
гидрохлорирования ацетилена 30
димеризации ацетилена 34
карбонилирования ацетилена 32
полимеризации ацетилена 34
разложения углеводородов 86
синтеза ацетиленовых гликолей 32
— винилацетилена 37
— диацетилена 38
Кинетика
взаимодействия ацетилена с водой 80
неполного горения углеводородов
155 ел.
образования ацетилена 75 ел.
— сажи 86, 88
пиролиза метана 73, 79
— парафинов 71
— пропана 74
— углеводородов 73
— этана 74
разложения ацетилена 77 ел.
— карбида кальция 56
— метана в плазме 149
электрокрекинга метана 129
Кокс в карбидном производстве 53
Коксовый газ, пиролиз до ацетилена
181 ел.
Компрессоры
для ацетилена 346 ел.
для ацетиленсодержащих газов
343 ел.
Компримирование см. Сжатие ацетилена
Коронный разряд в газах 125, 329
Коэффициенты
извлечения при абсорбции 225, 226
селективности 220, 237, 238
— аммиака 260
— керосина 260
— низкотемпературных
растворителей 253
Крекинг углеводородов
общий 81
полезный до ацетилена 76, 81, 170 ел.,
177
Критический диаметр ацетиленопрово-
дов 370, 371
Купрен 34
Ламинарное горение 165
Литраж карбида кальция 50
Материальный баланс
абсорберов 351
карбидной печи 54
окислительного пиролиза 185, 186
отделения концентрирования 354, 355
сажеотстойника 338
электрокрекинга метана 132, 133
Мембраны разрывные 383 ел.
Метанол как растворитель
ацетилена 16, 229, 253, 254, 263 ел.
гомологов ацетилена 258 ел., 264 ел.
Метилацетилен 36, 37
взрывное разложение 36
образование 36
получение 36
растворимость в аммиаке 256 ел.
— в диметилформамиде 233 ел.,
352 ел.
— в керосине 259, 277
— в метаноле 259, 264 ел.
— в N-метилпирролидоне 239, 243 ел.
физические свойства 35, 36, 37
N-Метилпирролидон 238 ел.
как растворитель ацетилена 16, 229,
230, 238 ел., 241 ел.
— — гомологов ацетилена 239,
243 ел.
предельно допустимая концентрация
238
физические свойства 16, 238
Метилэтилацетилен 35
5-Метил-2-этилпиридин 31
Механизм
образования ацетилена 127, 128, 159
— карбоидов 88
— сажи 86, 89
окислительного пиролиза метана
157 ел.
пиролиза метана 78
— углеводородов 71 ел.
разложения ацетилена 159, 160
— метана в плазме 149
электрокрекинга метана 127, 128
Молекулярные сита (Цеолиты) 274, 275,
276
Начальное давление взрывного распада
340, 341
ацетилена 21, 26, 368 ел., 379
диацетилена 38
Нестационарное горение ацетилена
(Каскадное разложение) 17, 18, 369
Низкотемпературные растворители
ацетилена 215, 253 ел.
Нормальная скорость горения 19, 164
ацетилена 19
Огнепреградители 376 ел.
Окислительный пиролиз
бутана 172
время реакции 176, 178
давление 174 ел.
добавки водорода 179 ел.
жидких нефтепродуктов 211 ел.
коксового газа 181 ел.
концентрация кислорода 174
метана ПО ел., 170 ел., 175 ел.,
180, 184, 255 ел.
нефтяного сырья 211 ел.
параметры 168 ел.
природного газа 168 ел., 397 ел.
пропана 171
расходные соотношения 173, 208, 209
реакторы 187 ел.
роль твердой поверхности 183
сажеобразование 184, 185
соотношение реагентов 169 ел.
сырье 169 ел.
температура 169*
— подогрева сырья 177 ел.
технико-экономические показатели
398 ел.
углеводородов 154 ел.
этана 171
Отмера пиролиз с электронагревом
101 ел.
Отсекатели 381
Очистка
ацетилена адсорбцией 272 ел., 276
— активированным углем 276
— карбидного 59 ел.
— керосином 258 ел., 277
411
Очистка
ацетилена метанолом 258 ел.
— от гомологов 258 ел., 275 ел.
— от метилацетилена 270, 271
— серной кислотой 276, 277
— тонкая 275 ел.
воды от сажи 336 ел.
газов от сажи 324 ел., 335, 336
Пенные аппараты 332, 333
Период
индукции 305, 307, 309
— при горении метана 161 ел.
преддетонационный 367, 368
Печи
карбидные 45—47, 49 ел.
многопоточные 100
регенеративные 90 ел., 96
трубчатые 98 ел.
шахтные 45—47, 49 ел.
Пиролиз 64 ел.
ацетилена
бензина 104, 109, ПО ел., 112 ел.
бутанов 75, 99 ел., 172
в плазме 151
в трубчатых печах 85, 88 ел., 396
газойля 94
гомогенный см. Гомогенный пиролиз
глубина разложения сырья 81
диацетилена 80
, жидких углеводородов 85, 94, 95,
111, 115 ел., 211 ел.
кинетика 73 ел.
лигроина 95, 105, 106
метана 66 ел., 73 ел., 76, 78 ел., 82,
90 ел., 96, 102
механизм 71 ел.
на насадке 86
окислительный см. Окислительный
пиролиз
парафинов 71 ел., 81
пропана 68, 69, 74, 75, 84, 89, 94 ел.,
99, 101, 104
регенеративный см. Регенеративный
пиролиз
с газом-теплоносителем 86, 102 ел.
с погружным горением 85, 115 ел.
с твердым теплоносителем 97, 98
с электронагревом 101 ел.
смесей углеводородов 84
состав продуктов 83
сырой нефти 115 ел.
сырье 81 ел., 84, 85, 94 ел.
температура 68, 81 ел.
термодинамика 66, 67, 70
технико-экономические показатели
394 ел.
условия процесса 85
этана 66 ел., 74, 82, 83, 96
этилена 74
412
а-Пирролидон 31
Плазменный крекинг углеводородов
147 ел.
Плазмообразователи 148
Пламя в газах 17, 18 ел., 167 ел.
отрыв 166, 167
проскок 166, 167, 188, 193
Пластинчатые теплообменники 358 ел.
Поглотительная способность
(Адсорбционная емкость) 273, 274
цеолитов 276
Подогреватели радиационно-конвектив-
ные 279 ел.
конвективная зона 287
конструкции 289, 290
показатели работы 291, 292
радиационная зона 286
расчет конструктивный 288 ел.
— теплотехнический 285 ел.
скорость газа 280, 281
соотношение HID 288, 289
температура стенки 280
тепловая нагрузка 281 ел.
тепловые схемы 279, 280
Полимеризация
ацетилена 34, 77
винилацетилена 37
диацетилена 39, 390
триацетилена 40
Пороговая концентрация при сажсобра-
зовании 87
Преддетонационное расстояние 367, 368
Преддетонационный период 367, 368
Предельно допустимые концентрации
диметилформамида 231
N-метилпирролидона 238
Пределы взрываемости
ацетилена 20, 21, 365
винилацетилена 37
высших ацетиленов 40, 41
диацетилена 38
метана 160, 161
Пропадиен (Аллен)
растворимость в диметилформамиде
233
— в керосине 259, 277
— в N-метилпирролидоне 239
физические свойства 35
Пропаргиловый спирт 32
я-Пропилацетилен 35
Проскок пламени при горении 166, 167,
188, 193
Разбавители см. Флегматизаторы
Разряды в газах
дуговой 125, 127, 134, 329
искровой 329
коронный 125, 139
темный 123, 129
тлеющий 123, 127, 129
Растворимость ацетилена 16, 215, 228 ел.
в аммиаке жидком 16, 254 ел.
в ацетоне 253, 254, 268, ел.
в у-бутиролактоне 246 ел.
в диметилформамиде 233 ел.
в метаноле 253, 254, 263 ел.
в N-метилпирролидоне 238 ел., 242 ел.
Расходные показатели 395 ел.
выделения ацетилена 247
гомогенного пиролиза 395 ел.
карбидного производства 50, 54, 394
пиролиза в трубчатых печах 396
— окислительного 205, 212, 399
— плазменного 152
— регенеративного 396
производства карбидного ацетилена
63, 394
электрокрекинга метана 136, 141, 397,
398
Расчеты
абсорберов 223 ел.
абсорбции многокомпонентной смеси
350 ел.
ацетиленопроводов 368 ел.
методом абсорбционного фактора
351 ел.
— единичных объемов 224
огнепреградителей 376, 378
пенных аппаратов 332, 333
поверхности распыленной воды при
«закалке» 311 ел.
подогревателей 285 ел.
полого скруббера 325
пределов взрываемости ацетилена 365
процесса «закалки» реакционных
газов 310 ел., 312 ел.
— сжатия газов 342, 343
— смешения газов 298 ел.
расхода воды на «закалку» 319 ел.
на охлаждение газов после
«закалки» 322 ел.
рекуперационной турбины 348
скорости «закалки» 312 ел.
смесителей 302 ел.
тепловой нагрузки в подогревателях
282
электрокрекинга 131 ел.
Реакторы ацетиленовые 101, 103, 293 ел.
гомогенного пиролиза 103, 105, 108,
114, 295
горелки 293 ел.
многосопловые (многоканальные) 189,
190, 193, 194
объем реакционной зоны 294, 295
односопловые (одноканальные) 189,
193, 194
окислиельного пиролиза 187 ел., 206
плазменные 150
поверхностного горения 208
с кольцевой горелкой 296
Реакторы ацетиленовые
соотношение HID 194
электрокрекинга 143, 295
— жидких углеводородов 146
— метана 134, 137, 138, 144
Реакции ацетилена 26 ел.
винилирование 29, 31, 33, 34
галоидирование 29, 30
гидрирование 28 ел.
гидрохлорирование 30
гидроцианирование 30
димеризация 34
замещение 26, 27 ел.
карбонилирование 32
полимеризация 34, 77
с альдегидами 32
с аммиаком 31
с водой 28, 29, 80
с гетероциклами 31, 33
с гидридом бора 32
с карбазолом 31
с кислотами минеральными 30
— органическими 33, 34
с меркаптаном 29
с сероводородом 29
со спиртами 29
Регенеративный пиролиз 90 ел., 118, 397
в печах 90 ел.
в твердом теплоносителе 97 ел.
Вульф-процесс 90 ел., 118
метана 90, 91, 96
параметры 85, 91, 95, 96
пропана 94, 95
технико-экономические показатели
396
этана 96
Ре пае синтез ацетиленовых гл и колей 32,
33
Сажа 141 ел.
всплывание в сажеотстойниках 337
газификация 157
образование 86 ел., 159, 184
— механизм 86, 89
пороговая концентрация 87
физические свойства 185, 337
Сажеотстойники 338
Самовоспламенение
ацетилена 19, 20, 86, 365
метана 161 ел.
Селективности коэффициент 220, 237,
238, 253, 260
Селективные растворители ацетилена
220, 231 ел., 246 ел., 349 ел.
Сжатие (Компримирование) ацетилена
339 ел.
параметры 340, 341
расчет 342 ел.
степень сжатия 342 ел.
условия 339 ел.
413
Скрубберы 324 ел.
расчет 325
Смесители в реакторах 297 ел.
время смешения 304 ел., 308
гидродинамический параметр 299, 304,
309
конструкции 297, 298, 308
концентрический 298
поперечные 298, 308, 309
путь перемешивания 304, 309
распределение струй 303 ел.
расчет 302 ел.
смешение с поперечным потоком
300 ел.
соотношение HID 305, 309
степень смешения 302
трубчатые 308, 309
турбулентные струи 298 ел.
условие смешения 307, 308
щелевые 298
эжекционные 306, 307, 309
Темный разряд 123, 126
Термодинамика
неполного горения углеводородов
155 ел.
пиролиза метана до ацетилена 79
— углеводородов 64 ел.
разложения метана в плазме 149
Теоретические тарелки в абсорбере
233 ел.
Тепловой баланс
окислительного пиролиза 169, 170,
186, 187, 209
пиролиза 85, 86
подогревателей 286
стадии «закалки» 317
электрокрекинга метана 132
Тетрагидрофуран 229, 230
Тетраметилмочевина 229, 230
Тетранитрометан 30
Тетрахлорэтан 29
Техника безопасности
в производстве ацетилена 364 ел.
при монтаже оборудования 373
при окислительном пиролизе 193,
199, 205
при пиролизе сырой нефти 116
при работе электрофильтров 366
при сжатии ацетилена 340 ел.
при транспортировании ацетилена 376
Технико-экономические показатели
гомогенного пиролиза жидких
углеводородов 395 ел.
окислительного пиролиза
природного газа 398 ел.
пиролиза в трубчатых печах 396
регенеративного пиролиза 396
электрокрекинга 398
414
Технологические параметры
выделения ацетилена аммиаком 262
— — диметилформамидом 357
метанолом 266 ел.
селективными растворителями
248 ел.
гомогенного пиролиза 103, 106, 109,
118
«закалки» 316 ел.
карбидного производства 45
окислительного горения 168 ел.
— пиролиза 175, 180
пиролиза в газе-теплоносителе 103
— в твердом теплоносителе 97
— в трубчатых печах 99, 101, 118
— с погружным горением 118
регенеративного пиролиза 90 ел., 96
сжатия ацетилена 340, 341
систем транспортирования
ацетилена 374 ел.
форабсорбции высших ацетиленов 253
электрокрекинга метана 134, 135
Технологические схемы
адсорбции ацетилена 273, 274
Вульф-процесса 92
выделения ацетилена аммиаком 261
ацетоном 270
водой 227
метанолом 267
селективными растворителями
249 ел.
гомогенного пиролиза 109, 112
окислительного пиролиза 198, 202,
204, 212
очистки газа после
электрокрекинга 327, 328
— карбидного ацетилена 61
пиролиза 92
— в твердом теплоносителе 98
— в трубчатых печах 99
— сырой нефти с погружным
горением 115
производства карбида кальция 47
— сажи 141
транспортирования ацетилена 374,375
установки компрессоров 346
форабсорбции гомологов ацетилена
253
электрокрекинга метана 135, 140
Тиоокись этилена 29
Тлеющий разряд в газах 123, 126, 127
Транспортирование ацетилена 374 ел.,
387
Триацетилен 39, 40
взрывное разложение 40, 367
полимеризация 40
физические свойства 35
Трубопроводы для ацетилена см.
Ацетил енопр оводы
Труба Вентури 326, 328
Турбины рекуперационные 348 ел.
Турбокомпрессоры для ацетиленсодер-
жащих газов 344 ел.
Турбулентное горение 164 ел., 166
Турбулентные струи при смешении
газов 298 ел.
газопромыватели 325 ел.
Углеродный показатель (С-Индекс,
С-Число) 117
при окислительном пиролизе 173
Факел горения 164 ел.
в горелках подогревателей 283 ел.
дальнобойность 167 ел., 188
диффузионный 188
Фильтры рукавные 335
Флегматизаторы (Разбавители)
ацетилена 19 ел., 23 ел., 26, 268, 340,
341, 364 ел., 376
диацетилена 38, 39
метана 163, 164
метилацетилена 36
Форабсорбция высших ацетиленов 252,
253
Фронт горения 165
Хлористый винил (Винилхлорид) 30
Хлоропрен 38
Цеолиты (Молекулярные сита) 274, 275,
276
Циклоны 324, 333 ел.
Циклооктатетраен 34
С-Число (Углеродный показатель, С-Ин-
декс) 117, 173
Шахтные карбидные печи 45—47, 49 ел.
Шоха реактор электрокрекинга метана
144
Экономика производства ацетилена
398 ел.
Электрические разряды в газах 122 ел.,
328, 329
Электрокрекинг 126 ел., 397
в плазме 147 ел.
газообразных углеводородов 134 ел.
жидких углеводородов 145 ел.
ионизация газов 122 ел., 328
метана 127 ел., 134 ел.
очистка реакционных газов 327
реакторы 143, 295
технико-экономические показатели
398
Электрофильтры 328 ел.
мокропленочные 331
Виктор Никитич Антонов,
Александр Семенович Лапидус
ПРОИЗВОДСТВО АЦЕТИЛЕНА
М., Издательство «Химия» 1970 г.
416 с. УДК 661.715.342
Редактор Н. И. Урывалова
Технический редактор В. М. Скитина
Художник М. Ф. Ольшевский
Корректоры Н. П. Шигина, Г. Е. Потапова
Т-14780 Подписано к печати 12/ХП 1969 г.
Формат бумаги 60x90Vie Печ. л. 26+1 вкл. Уч.-изд. л. 28,15
Тираж 4500 экз. Темплан 1969 г.. № 79 Тип. бум. № 1.
Цена 1 р. 59 к. Заказ 169
Ленинградская типография № 6 Главполиграфпрома
Комитета по печати при Совете Министров СССР
Ленинград, ул. Моисеенко, 10
01 on Чистый
■ ^ аиетжн
сн4
Рис. VI-22. Схема производства ацетилена окислительным пиролизом природного газа под давлением и выделения ацетилена метанолом:
/f 12 — компрессоры; 2, 3 — подогреватели природного газа и кислорода; 4 — реактор; 5 — сажеотделитель*,|6 — сборник горячей воды; 7—расширитель; 5—скруббер I ступени; 9 — скруббер II ступени;
Ю— скруббер-охладитель; // — газгольдер; 13, 20 — холодильники; 14, 15 — абсорберы; 16, 21 — промывател^; 17, 18 — десорберы ацетилена; 19 —сепаратор; 22, 23 — аппараты для очистки ацетилена;
24, 25 —десорберы ацетиленовых углеводородов; 26 —скруббер Для отдувки ацетиленовых углеводородов; 27'— ректификационная колонна; 28 — установка газоразделения; 29— холодильная установка.
В. Н. Антонов