/



































































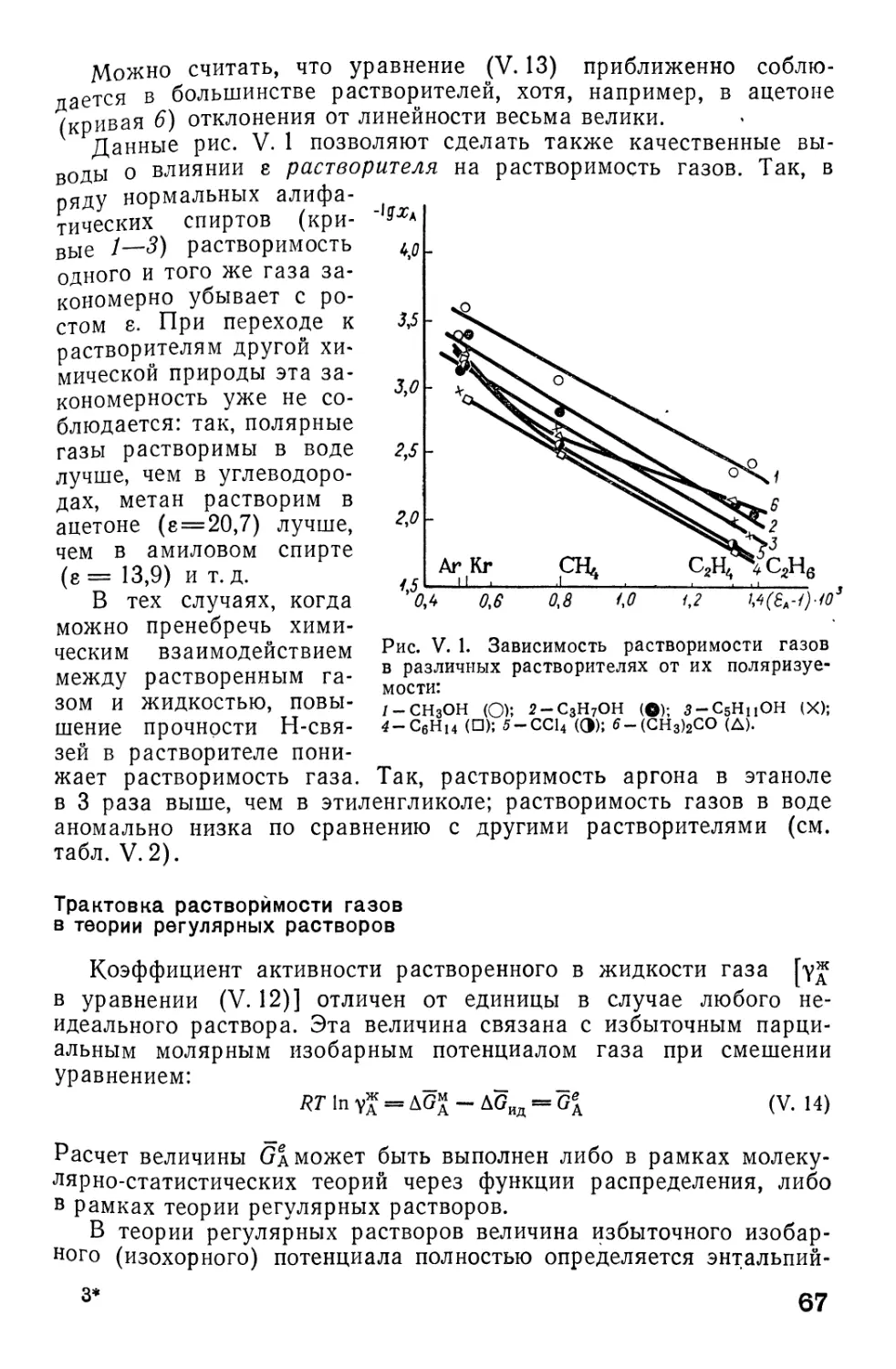





















































































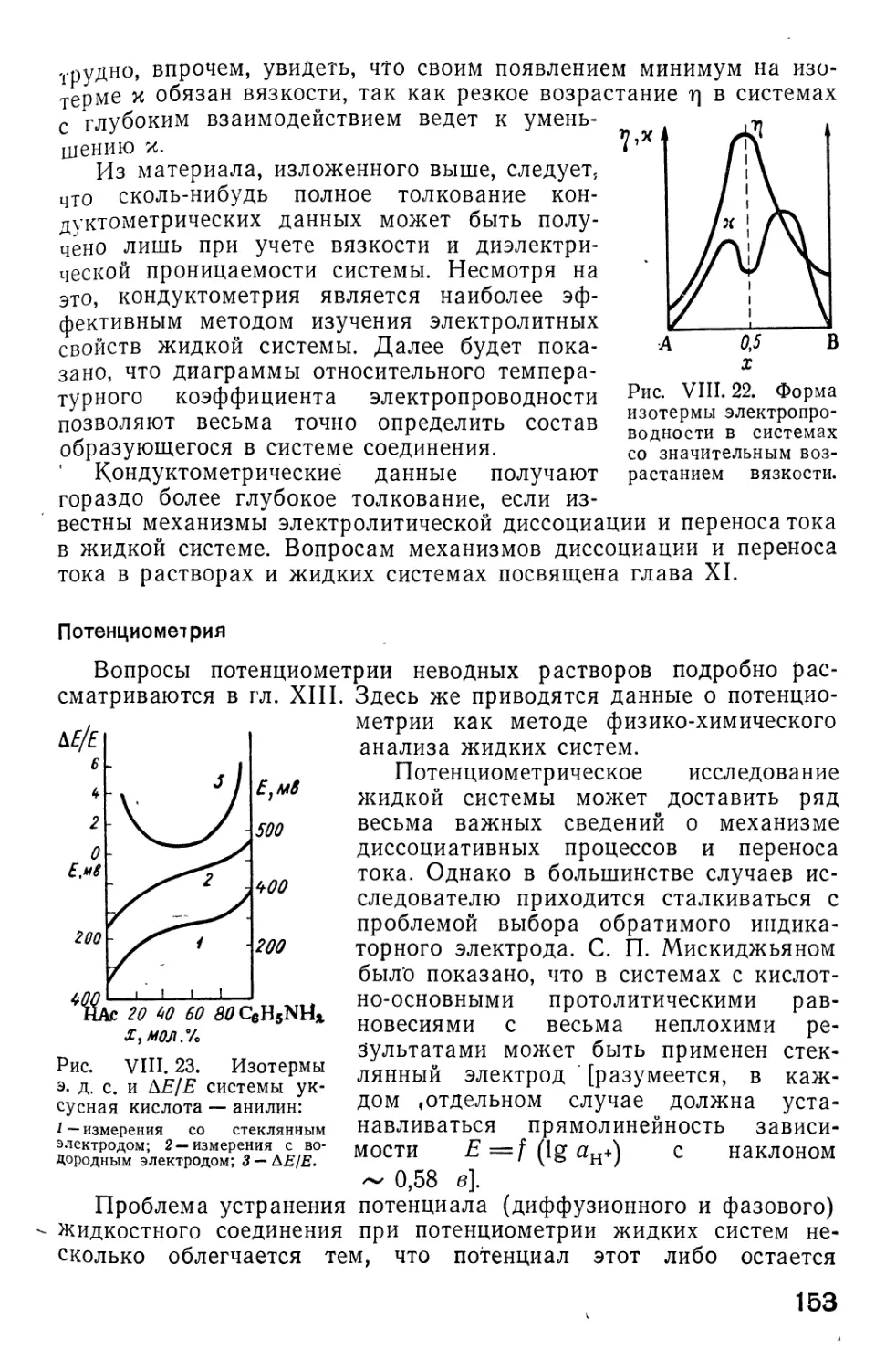





























































































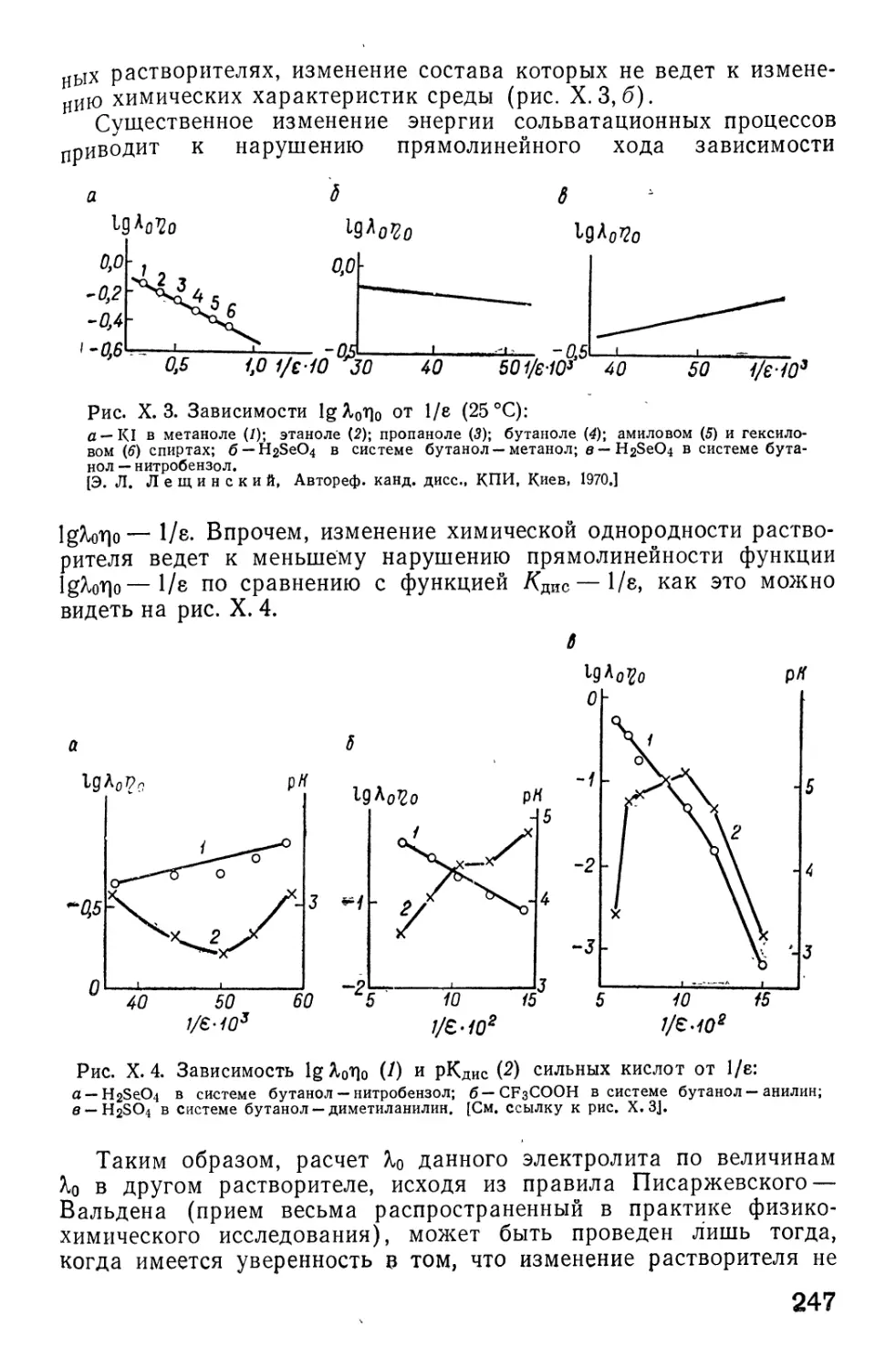

































































































































Автор: Фиалков Ю.Я. Житомирский А.Н. Тарасенко Ю.А.
Теги: состояние вещества химия физика физическая химия
Год: 1973




Текст
Ю.Я.ФИАЛКОВ
А.Н.ЖИТОМИРСКИЙ
Ю.А.ТАРАСЕНКО
ФИЗИЧЕСКАЯ
ХИМИЯ
НЕВОДНЫХ
РАСТВОРОВ
ЛЕНИНГРАД
ИЗДАТЕЛЬСТВО „ХИМИЯ"
Ленинградское отделение • 19 7 3
УДК 54-145.3/.4:541
Ф48
Фиалков Ю. Я., Житомирский А. Н., Тара-
сенко Ю. А.
Ф48 Физическая химия неводных растворов. Л.,
«Химия», 1973.
376 стр., 59 табл., 87 рис., список литературы 205 ссылок.
В книге рассмотрены основные аспекты физической химии
неводных растворов, классификация растворителей, строение неводных
растворов и связь между физическими и химическими свойствами
растворов. Отдельные разделы книги посвящены термодинамике,
равновесиям в неводных растворах, электролитической диссоциации
и электродным процессам, а также кинетике реакций в неводных
средах.
Книга предназначена для физико-химиков, аналитиков,
нефтехимиков и органиков, занимающихся электросинтезом. Книга
представляет также интерес для студентов старших курсов и аспирантов,
специализирующихся в области физической химии,
Издательство «Химия», 1973
ф_0254-152_ 13-73
050 (01)-73
ПРЕДИСЛОВИЕ
Исследование неводных растворов имеет большое значение в
становлении многих разделов общей теории растворов. За
последние годы резко возросло и практическое значение неводных
растворов как среды для проведения разнообразных синтезов и
электрохимических процессов, а также аналитических определений.
В этом причина повышенного интереса к проблеме неводных
растворов со стороны не только химиков, но и представителей многих
разделов естествознания. Отражением этого интереса является
рост числа публикаций, посвященных неводным растворам, рост,
интенсивность которого необычна даже на фоне резкого прироста
объема информации в области химии. Однако число обобщений —
монографий или обзорных статей — в области химии неводных
растворов еще не отвечает обилию накопившихся сведений. Это
обстоятельство — одна из причин, побудивших авторов к
написанию данной монографии.
Неопределенность границ между естественными науками в
полной мере свойственна и области, которой посвящена эта книга.
Действительно, весьма трудно разграничить физику жидкого
состояния и физическую химию растворов. Также нелегко
оконтурить рубеж между неводными и водными растворами. Хотя
впечатляющие различия между последними (особенно для
электролитных растворов) бросаются в глаза при первой же попытке
сопоставить эти системы, все же между ними гораздо больше
общего, чем это принято обычно считать. Тем не менее, совершенно
уникальные свойства воды обуславливают выделение физической
химии неводных растворов в отдельный раздел общей теории
растворов.
Объем книги побудил авторов ограничиться лишь гомогенными
системами, тем более, что на русском языке изданы монографии,
посвященные равновесиям жидкость — пар (В. Б. Коган, «Гете-
оогенные равновесия»), жидкость — жидкость (А. Френсис,
«Равновесие жидкость — жидкость») и жидкость — твердое тело
(В. Я. Аносов, С. А. Погодин, «Введение в
физико-химический анализ»); книг же по физической химии гомогенных
равновесий в неводных растворах не выходило ни в
отечественных, ни в зарубежных издательствах.
Применительно к неводным системам книга охватывает:
термодинамику, статику (равновесия), электрохимию (равновесную и
необратамую) и кинетику реакций в неводных средах. В каждом
разделе основное внимание уделяется влиянию растворителя на
течение химического процесса.
Поскольку книга посвящена физической химии именно
неводных растворов, читатель не встретит в ней многих традиционных
разделов, входящих в курсы физической химии растворов.
Естественно, что здесь рассматриваются лишь жидкие растворы, так
как проблема твердых растворов является специфической и
частной по отношению к вопросам, затронутым в книге. Не
рассматриваются также металлические и ионные (солевые) расплавы.
Авторы не ставили целью дать исчерпывающее изложение
каждого затронутого раздела физической химии неводных
растворов. Поэтому книга скорее является введением в физическую
химию неводных растворов, предназначенным для первоначального
знакомства с вопросом, которое предполагает осведомленность
читателя в объеме общего курса физической химии. По этой же
причине авторы свели к минимуму библиографию. [Напомним, что
библиография С. И. Якубсон «Электролитные неводные растворы»
(Киев, 1967), посвященная сравнительно частному по отношению
к кругу проблем, рассматриваемых в этой книге, вопросу, и
охватывающая литературу всего за период 1941 —1960 гг., имеет объем
свыше 60 п. л.]. Для более глубокого знакомства с излагаемыми
вопросами читатель может воспользоваться заключительным
разделом книги, где приведен перечень важнейших монографий и
обзорных статей.
Главы I—III, VII—IX написаны доктором хим. наук, проф.
Ю. Я. Фиалковым, главы IV—VI, XI, XIV, XV и Приложение —
канд. хим. наук А. Н. Житомирским и главы X, XII и XIII, а
также разделы VIII.8—VIII.10 — канд. хим. наук Ю. А. Тарасенко.
Обитая редакция книги осуществлена Ю. Я. Фиалковым.
Авторы выражают глубокую признательность Л. И. Антропову,
В. Д. Безуглому, И. А. Коппелю, Г. А. Крестову, Г. И.
Лихтенштейну, Л. Л. Спивак, Н. Е. Хомутову, и особенно А. И. Шатен-
штейну и Н. Л. Ярым-Агаеву, которые прочитали отдельные
разделы книги и сделали ценные замечания, а также рецензенту
книги А. Г. Морачевскому.
Мы будем признательны всем читателям, которые пожелают
сообщить авторам свою оценку книги*
г. Киев, 1973
ОБЩИЕ ПОЛОЖЕНИЯ
Глава I
ИСТОРИЯ РАЗВИТИЯ ИССЛЕДОВАНИЙ
В ОБЛАСТИ ФИЗИЧЕСКОЙ ХИМИИ
НЕВОДНЫХ РАСТВОРОВ
Предлагаемая краткая историческая справка не претендует на
сколько-нибудь подробный перечень имен и характеристику
ученых, которые внесли вклад в развитие физической химии неводных
растворов. Именно поэтому здесь и не будут рассмотрены
исследования в интересующей нас области, относящиеся к начальным
(т. е. доатомно-молекулярным) периодам развития химии — какие
бы славные имена, подобно М. В. Ломоносову и А. Лавуазье, не
были при этом упущены. Несомненно также и то, что работы этих
ученых в области неводных растворов, как и исследования
классиков естествознания Бургаве, Ловица, Фарадея, Гей-Люссака и
некоторых других, еще ждут своего историографа. Особенный
интерес в этом плане представляют совершенно недостаточно
оцененные историками химии работы Гротгуса, деятельность
которого, приходившаяся на первые десятилетия прошлого века,
предвосхитила многие идеи классиков физической химии, но, к
сожалению, в силу причин исторических, не стала их предтечей.
Развитие физической химии неводных растворов можно
разбить на несколько периодов. Первый из них характеризуется
накоплением— часто довольно бессистемным—данных по различным
свойствам электролитных и неэлектролитных неводных растворов.
Начало этого периода относится к 50-м годам прошлого столетия
(предшествующие, в общем, весьма немногочисленные
исследования относятся скорее к общей химии); именно в эти годы и
произошло выделение физической химии неводных растворов в
самостоятельные разделы физической химии и теории растворов.
Наиболее частыми в этот период являются исследования
электрохимических свойств неводных растворов. В 1853—1859 гг. Гит-
торф определял электропроводность галогенидов цинка и кадмия
в некоторых спиртах и этиловом эфире. Несколько позже Эффенди
и Блэкроди изучали электропроводность индивидуальных
жидкостей и растворов в них различных кислот и их ангидридов.
В 1870—1880 гг. Ленд получил данные по электропроводности
многих солей и пикриновой кислоты в "смешанных водно-
5
спиртовых растворителях. В конце 80-х годов Фитцпатрик провел
изучение в широком концентрационном интервале
электропроводности растворов ряда солей в метаноле и этаноле. В это же время
Гартвиг исследовал электропроводность растворов алифатических
карбоиовых кислот в различных спиртах.
Вообще следует отметить, что 80-е годы прошлого века
отличаются резким повышением интенсивности исследований в
области физической химии неводных растворов. Помимо только что
перечисленных электрохимических исследований, в это время
появляются классические работы Рауля по эбулиоскопии и
криоскопии растворов во многих органических растворителях — бензоле,
эфире, нитробензоле, спиртах, кислотах, бромэтиле. Общеизвестно
огромное влияние результатов этих исследований на последующее
развитие многих разделов физической химии.
В конце 80-х годов появляется цикл работ Н. А. Меншуткина
по влиянию растворителя на равновесие реакций этерификации и
образования четвертичных алкилзамещенных аммонийных солей.
По новизне подхода и тщательности экспериментального
выполнения работы эти с полным правом считаются классическими.
Наконец, в 1887 г. завершает свое обширное исследование
плотности водно-спиртовых растворов Д. И. Менделеев.
Выдающаяся работа великого химика явилась свидетельством его
глубокого интереса на протяжении всей научной деятельности к
растворам. Эти исследования положили начало обширному циклу работ,
выполненных учениками и последователями Д. И. Менделеева, и
легли в основу развивавшегося им учения об образовании в
растворах соединений определенного состава.
Нельзя не вспомнить в связи с этим, что уже тогда, на заре
развития химии неводных растворов, Д. И. Менделеев высказал
обоснованное мнение, что создание общей теории растворов может
быть достигнуто «только при умножении исследований ... с
другими растворителями, чем вода».
К сожалению, значительная часть экспериментальных данных,
полученных исследователями в этот первый период развития
физической химии неводных растворов, особенно данных,
относящихся к электропроводности, из-за недостаточного внимания к
очистке компонентов раствора, довольно противоречива. Это
привело к появлению якобы аргументированных экспериментами
весьма разнообразных точек зрения, и, как следствию этого,—
к значительной путанице во взглядах на природу
электропроводности в. неводных растворах, устранение которой потребовало от
следующего поколения исследователей значительных усилий.
Второй период развития физической химии неводных растворов
характеризуется возникновением и весьма быстрым развитием
двух основных направлений в теории растворов — физического и
химического. Интересно, что представители каждого направления,
аргументируя свои положения, черпали большей частью эту
аргументацию из изучения неводных растворов, Вот почему беспри-
6
мерный по протяженности научный спор между сторонниками
физической и химической теорий растворов послужил на пользу
физической химии неводных растворов — не только потому, что в
результате этого спора родилась истина, но и потому, что
экспериментаторами было получено много новых прецизионных данных.
Вряд ли стоит здесь характеризовать сколь-нибудь подробно
научные платформы, на которых стояли сторонники обоих
направлений, это общеизвестно и достаточно подробно освещено во
многих исторических и специальных работах. Однако краткий обзор
этих направлений здесь будет несомненно уместен.
физическое направление в теории растворов возглавляли
С. Аррениус и Я- Вант-Гофф— ученые, с именами которых
связаны наиболее выдающиеся достижения физической химии XIX в.
Являясь основоположниками современной теории растворов,
Аррениус и Вант-Гофф большее внимание уделяли водным растворам,
однако, их научные интересы распространялись и на иеводные
растворы.
Наиболее видными представителями физической теории начала -
второго периода развития физической химии неводных растворов
следует считать Нернста и Томсона, которые в 1893 г. связали
столь неопределенное понятие как «диссоциирующая сила
растворителя» с диэлектрической проницаемостью. С середины 90-х
годов Каррара начинает систематические и весьма точные
исследования электропроводности растворов галогенидов щелочных
металлов, тетраалкиламмония и триалкилтиония, а также некоторых
кислот в некоторых органических растворителях.
Наиболее существенный вклад в изучение электролитных
неводных растворов, который привел к существенному расширению
сведений о концентрационной, а в ряде случаев и температурной
зависимости электропроводности неводных растворов, был сделан
П. И. Вальденом, с именем которого связан обширный и весьма
плодотворный период развития физической химии неводных
растворов. Экспериментальные данные по электропроводности очень
большого числа электролитов в чрезвычайно обширном круге
растворителей, полученные П. И. Вальденом за период почти
тридцатилетней активной деятельности в этом направлении, и сейчас
используются многими исследователями для обоснования
выдвигаемых ими теоретических положений.
Основная заслуга школы П. И. Вальдена — установление
первых количественных соотношений в физической химии
электролитных неводных растворов. П. И. Вальден предпринял попытку
установить связь между диэлектрической проницаемостью,
концентрацией и степенью диссоциации электролита. Впоследствии из
этого правила было найдено множество исключений, однако и
сегодня оно не потеряло своего значения. П. И. Вальденом было
выведено эмпирическое соотношение между величинами электро*
проводности при бесконечном разбавлении
(Ко),электропроводности при данном разбавлении, вязкостью (г]),электропроводностью
и концентрацией раствора. Это приближенное уравнение нередко
используют для оценки константы диссоциации электролитов в
неводных растворах и в настоящее время.
Полученные экспериментальные данные позволили П. И. Валь-
дену сформулировать правило постоянства исправленной на
вязкость растворителя (г)0) предельной электропроводности. Впрочем,
несколько ранее эта же закономерность была установлена
Л. В. Писаржевским и поэтому закон коЦо = const часто и
совершенно справедливо называют правилом Писаржевского — Валь-
дена *.
Развитие физического направления в физической химии
неводных растворов в России в значительной мере связано также с
именем А. Н. Саханова, основные работы которого приходятся на
второе десятилетие нашего века. Он первым пытался установить
количественную зависимость электропроводности от концентрации
для неводных растворов в довольно широком концентрационном
интервале. При этом А. П. Саханов прибегнул к представлениям
об ассоциации электролита в растворе, что позволило весьма
логично объяснить экстремумы на кривых зависимости
электропроводности от состава.
Объективно к физическому направлению в теории растворов
должны быть отнесены весьма успешно начавшиеся в последней
четверти XIX в. и получившие значительное развитие
исследования по термодинамике растворов.
В первую очередь здесь должны быть названы классические
работы Дж. Гиббса. Созданный Гиббсом фундамент современной
химической термодинамики оказал решающее влияние на
физическую химию в целом и физическую химию растворов в
особенности. Гиббс является основателем статистической термодинамики,
на базе которой развивается теория растворов. В начале нашего
века Бозе сформулировал первую теорию теплот смешения и
теплопроводности двойных жидких смесей. Существенный вклад в
общую теорию растворов, в том числе и неводных, был сделан
М. Планком. Развитый им метод учета влияния физических
свойств растворителя на свойства растворов впоследствии лег в
основу большого раздела физики жидкого состояния. Наконец,
существенный вклад в термодинамику растворов, в особенности
концентрированных, был сделан Ван-дер-Ваальсом и Ван-Лааром,
работы которых в области растворов значительно способствовали
развитию химической термодинамики.
Следует, впрочем, отметить, что исследователи в области
термодинамики жидкого состояния большей частью стояли в стороне
от спора, который велся между сторонниками физической и
химической теорий растворов, а поскольку основным предметом дискус-
* В литературе были нашедшие сочувственный отклик предложения назы-
'вать коррегированную электропроводность Pwnrj0 числом Писаржевского.
8
бычи электролитные свойства, изучавшиеся с помощью элек-
поводности, термодинамические исследования не могли стать
пешающим аргументом в этой полемике.
Бопьшинство сторонников химической теории растворов на
рубеже "двух столетий сосредоточилось в России. Несомненно это
бьпо вызвано влиянием могучей личности Д. И. Менделеева, кото-
оый как известно, был одним из энергичных проповедников
химической теории растворов. Неприятие теории электролитической
диссоциации отнюдь не было проявлением научной
недальновидности великого ученого как об этом говорится в некоторых даже
весьма серьезных жизнеописаниях его. Д. И. Менделеев со
свойственной ему проницательностью усмотрел то, чего не замечали
многие апологеты теории электролитической диссоциации и что
обходили создатели этой теории: отсутствие указаний на источник
энергии, необходимой для разрыва связи в ионных соединениях
в процессе их растворения. Разумеется, это было достаточно
веским аргументом для отрицания теории электролитической
диссоциации.
Одним из самых видных представителей химической теории
растворов, работы которого посвящены в основном неводным
средам, является Д. П. Коновалов. Поскольку данная книга
рассматривает гомогенные равновесия, мы не будем останавливаться на
работах Д. П. Коновалова, посвященных равновесиям жидкость —
пар, хотя эти исследования, приведшие к формулировке законов,
носящих его имя, несомненно являются главным результатом
научной деятельности этого ученого. Напомним также, что
Д. П. Коновалов установил факт, определивший на несколько
десятилетий развитие химической теории электролитных
растворов: образование электролитного раствора при смешении не
проводящих в индивидуальном состоянии ток компонентов.
Исследовав ряд систем, образованных аминами и карбоновыми кислотами,
Д. П. Коновалов связал экстремальные точки на изотермах
электропроводности с определенным стехиометрическим составом
образующихся в системе соединений. Пусть теоретические
представления Д. П. Коновалова об особых точках на кривых
электропроводности впоследствии потребовали значительного уточнения,
а в ряде случаев и существенного изменения, но основной тезис
химической теории растворов —основные особенности раствора
должны быть связаны с конкретным химическим взаимодействием
строгой стехиометрии — был сформулирован в этих работах с
полной определенностью.
Существенный вклад в химическую теорию растворов был
сделан В. Ф. Тимофеевым, который в 90-х годах прошлого века
установил тесную связь между растворимостью различных
органических соединений в неводных растворителях и химическими
свойствами растворителя. В. Ф. Тимофеев одним из первых начал
систематическое исследование теплот смешения неводных
растворов.
9
Последовательным сторонником химического направления в
физической химии неводных растворов был Е. В. Бирон, который
установил параллелизм между степенью химического
взаимодействия и отступлениями от идеальности в растворах.
Весьма значительные достижения химической теории растворов
вообще и неводных растворов, в частности, связаны с работами
Н. С. Курнакова — основоположника того раздела общей и
физической химии, который получил наименование физико-химического
анализа. Учение о связи особых точек на диаграммах
концентрационных зависимостей различных свойств с определенными
химическими процессами, протекающими в системе, получило в
работах Н. С. Курнакова глубокое теоретическое обоснование. Он ввел
в практику исследования растворов ряд принципиально новых
методов, разработал классификацию диаграмм различных свойств и
предложил методы определения стехиометрии протекающих в
химических системах взаимодействий. Работы Н. С. Курнакова
способствовали значительному прогрессу в исследовании
концентрированных растворов.
За рубежом были также сторонники химической теории
растворов, внесшие вклад в изучение неводных сред.
Последовательно отстаивали химическую теорию растворов
Армстронг, Кромптон и Пикеринг, которые в подтверждение
тезиса об образовании соединений между растворенным веществом
и растворителем провели ряд экспериментов с неводными
растворами. С несколько иных позиций критиковал теорию
электролитической диссоциации Фитцпатрик.
Наиболее видным представителем химической теории растворов
за рубежом следует считать Ганча, об исследованиях которого
в области теории кислот и оснований пойдет речь далее (гл. II).
Интересно, что еще в 1899 г. Ганч высказал представления о
механизме электролитической диссоциации, весьма близко
примыкающие к современным взглядам на этот процесс. Он считал, что
взаимодействие растворенного вещества с растворителем
предшествует процессу электролитической диссоциации, а образующиеся
при этом ионы сохраняют свою связь с молекулами растворителя.
Объективно способствовали развитию химической теории
растворов школы Франклина и Крауса, которые начали и очень
интенсивно развили исследования растворов в жидком аммиаке.
Детальное исследование растворов в этом растворителе позволило
существенно расширить представления о роли химического
взаимодействия в образовании электролитного раствора.
Второй период развития физической химии неводных
растворов, завершение которого приходится на 20-е годы, принес много
ценных экспериментальных фактов и теоретических обобщений.
Этот период позволил достаточно определенно выявить позиции
физической и химической теорий растворов, их достижения и
слабости. Достижения эти, которые в случае физической теории
растворов сводились к бесспорно доказанной роли диэлектрической
10
проницаемости и вязкости среды в определении электролитных
свойств раствора, а в случае химической теории растворов —
к столь же бесспорному доказательству существования стехиомет-
рических соединений в растворах, были достаточно
впечатляющими и позволяли приверженцам каждого из этих направлений
весьма последовательно отстаивать свои позиции.
Однако даже в начале второго периода развития физической
химии неводных растворов находилось немало исследователей,
настроенных достаточно критически, чтобы ясно видеть недостатки
каждой теории.
В случае физической теории растворов эти недостатки
сводились к игнорированию энергетики процессов растворения и
электролитической диссоциации, а также к отрицанию необходимости
взаимодействия между растворенным веществом и растворителем
как фактора, приводящего к электролитической диссоциации.
Индифферентность растворителя не только не отрицалась
сторонниками физической теории растворов, но всячески ими
подчеркивалась, особенно в исследованиях по эбулиоскопии, криоскопии,
осмотическому давлению и т. п. '
К недостаткам химической теории растворов относился отказ
обсуждать механизм электропроводности (естественно, что это
обсуждение должно было неизбежно привести к признанию
существования в растворе ионов как носителей электрического заряда),
очевидным недостатком химической теории было нежелание
учитывать строгие экспериментальные доказательства существования
ионов в электролитных растворах.
Поэтому уже в конце XIX в. появились исследователи, которые
независимо от направления стремились к синтезу физической и
химической теорий растворов.
Одним из первых исследователей этого плана был И, А.
Каблуков, который еще в 1889 г. начал систематические исследования
электропроводности растворов хлористого водорода в
углеводородах, спиртах и этиловом эфире. Обнаруженный И. А. Каблуковым
факт уменьшения молекулярной электропроводности с
уменьшением концентрации раствора привел к пересмотру ряда основных
положений теории электролитической диссоциации и, прежде
всего, положения о полной диссоциации любого электролита при
бесконечном разбавлении. Явление аномальной
электропроводности И. А. Каблуков объяснял тем, что «нельзя смотреть на
растворитель как на среду, индифферентную к растворенному телу, а
нужно принять некоторое химическое взаимодействие между
растворенным телом и растворителем».
Новый этап в исследовании неводных растворов связан с
именем В. А. Плотникова, который своими исследованиями, начатыми
в 1900 г., положил начало «Киевской электрохимической школе»,
внесшей весьма существенный вклад в физическую химию невод-
кых растворов. Исследуя ряд остроумно подобранных систем,
В. А. Плотников доказал, что в любом растворителе, независимо
11
от его диэлектрической проницаемости, может быть получен
электролитный раствор. Действительно, подбирая растворенные
вещества так, чтобы они образовывали друг с другом ионогенные
соединения, В. А. Плотников доказал возможность получения
электролитных растворов даже в таких характеризующихся малой
величиной диэлектрической проницаемости растворителях, как
бензол или бромэтан. Для своего времени весьма прогрессивным
оказался выдвинутый В. А. Плотниковым принцип
«электрохимического соответствия», позже названный им «электрохимическим
резонансом». Согласно последнему для образования
электролитного раствора растворенное вещество и растворитель должны
вступать друг с другом в «резонанс». Этим самым В. А.
Плотников вплотную подвел физическую химию неводных растворов к
признанию взаимодействия между растворенным веществом и
растворителем как необходимого условия возникновения
электролитного раствора.
К тому направлению, представители которого стремились
объединить достижения физической и химической теории растворов,
принадлежат исследования Л. В. Писаржевского по влиянию
растворителя на равновесие реакций комплексообразования и
обменного солевого разложения, выполненные в первое десятилетие
нашего века.
Нельзя не отметить также и то, что хотя Планк считал себя
последовательным представителем физической теории растворов,
выполненный им термодинамический анализ свойств разбавленных
растворов привел его к необходимости признать обязательное'
взаимодействие молекул растворенного вещества и растворителя.
Подытоживая достижения, которыми характеризовался второй
период развития физической химии неводных растворов, можно
отметить, что все же между физической и химической теориями
растворов было гораздо больше общего, чем считали тогда, в пылу
дискуссии, представители этих направлений. Каждое направление
числило за собой как выдающихся теоретиков, так и незаурядных
экспериментаторов. Получая новые достоверные
экспериментальные данные, представители каждой теории объективно
способствовали развитию физической химии неводных растворов. И, наконец,
немаловажным обстоятельством в развитии каждой теории и
дискуссии вокруг нее явилось то обстоятельство, что направления эти
возглавлялись большими учеными, для которых успех «своего»
направления был, разумеется, весьма дорог, но еще дороже была
истина.
Третий период развития физической химии неводных растворов
характеризуется разработкой современных теоретических
положений в этой области. Для данного периода характерно резкое
сокращение числа работ, в которых фиксировалась качественная
сторона явлений в пользу работ ;с количественными обобщениями.
Хотя с началом этого периода дискуссии между сторонниками
физической и химической теорий растворов не прекратились, но
12
паботы в которых высказывались «крайние» точки зрения, стали
сточь редкими, что характеризуя этот период развития физической
химии неводных растворов, вряд ли стоит придерживаться
принципа использованного при описании предыдущего периода:
рассмотрение каждого направления в отдельности. Кроме того,
развитие исследований в этом направлении стало таким интенсивным,
что дальнейшее изложение материала этой главы будет
представлять скорее перечисление имен, чем подробную характеристику
вклада каждого упомянутого исследователя в физическую химию
неводных растворов.
Создание теории сильных электролитов явилось важным
событием в общей теории растворов, которое не могло не оказать
определяющего влияния на физическую химию неводных
растворов. Возникновению теории сильных электролитов предшествовал
ряд важных исследований, которые существенно пополнили
сведения и состояния ионов в растворах. Здесь следует прежде всего
назвать формулировку закона ионной силы Льюиса — Рендалла
(1921 г.) и вывод Борном уравнения для энергии гидратации
иона (1920 г.). Последнее уравнение связывает величину энергии
гидратации с ионным радиусом и диэлектрической
проницаемостью раствора, и с некоторыми допущениями распространяется на
неводные растворы. Существенным достижением явился также
вывод Бьеррумом уравнения, которое связывало коэффициент
электропроводности с осмотическим коэффициентом, активностью
растворенного электролита и диэлектрической проницаемостью
(1918 г.).
Возникновение и развитие теории сильных электролитов
достаточно подробно освещено во многих книгах, а также учебниках и
учебных пособиях по физической химии и электрохимии, поэтому
позволено будет авторам не останавливаться на истории этого
вопроса с той обстоятельностью, которой эта теория заслуживает.
Упомянем лишь, что возникновение этой теории связано с именами
Дебая, Хюккеля, Онзагера, Вина, Фалькенгагена, которым
современная физическая химия обязана созданием и обоснованием
одного из самых фундаментальных своих разделов.
Середина и конец 20-х годов характеризуется появлением
весьма важных исследований именно в области физической химии
неводных растворов.
Количественные закономерности протолитического равновесия
были разработаны Бренстедом (1922—1929 гг.), которому
принадлежит первая количественная теория диссоциации кислот в
неводных растворах *.
Вопросы рефрактометрии электролитных неводных растворов
и поляризации ионов в неводных растворах стали предметом
обширных исследований Фаянса (1927—1931 гг.).
Подробнее о теории протолитического равновесия Бренстеда см. в гл. II
и IX.
13
Поведению ионов в неводных растворах были посвящены
исследования Фуосса и Крауса (1933 г.), которые выдвинули и
экспериментально доказали ассоциацию ионов в ионные тройники —
явление, весьма распространенное в неводных растворах. Фуосс и
Краус предложили наиболее употребительный в настоящее время
метод расчета констант электролитической диссоциации,
впоследствии усовершенствованный и дополненный Шедловским и Онза-
гером.
Значительный интерес для физической химии неводных
растворов представляло перенесение на эти объекты выводов Бернала и
Фаулера, исследовавших строение жидкой воды и электролитных
водных растворов (1933 г.). Разработанная Берналом и Фаулером
теория эстафетной протонной проводимости в воде стала основой
для построения подобной теории в приложении к неводным
растворам.
На протяжении нескольких десятилетий проводил
плодотворные исследования в области физической химии концентрированных
неводных растворов Гильдебранд. Им была создана весьма
стройная теория регулярных растворов, разработаны теоретические
аспекты растворимости газов в неводных растворителях, а также
сформулированы физические условия подчинения смеси двух
жидкостей закону Рауля.
Начиная с 30-х годов, значительные достижения в области
физической химии неводных растворов все чаще связываются с
именами советских исследователей. Впрочем, весьма важные работы
в этом направлении были выполнены еще в 20-х годах.
Глубокий теоретический анализ электрохимических
характеристик неводных растворов был проведен А. И. Бродским (1926—
1934 гг.), которому принадлежит разработка ряда важных
вопросов определения коэффициентов активности электролитов в
неводных растворах и первая теория связи стандартных э.д. с. со
свойствами растворителя.
Энергетика образования ионных ассоциатов в растворах была
подробно рассмотрена В. К. Семенченко (1924—1929 гг.).
Впоследствии В. К. Семенченко разработал теорию растворимости в
неводных растворителях, связав эту характеристику с некоторыми
физическими свойствами и, в частности, с диэлектрической
проницаемостью.
Основополагающие исследования по рентгенографии жидкости
были выполнены в 30-х годах В. И. Даниловым. Эти исследования
позволили конкретизировать и подвести прочные
экспериментальные критерии под такие понятия как ближний и дальний порядок
в жидкости.
В это же время у нас в стране начинается весьма
интенсивное развитие исследований растворов в сжиженных газах,
связанное в основном с работами А. И. Шатенштейна, который получил
ряд важных физико-химических характеристик электролитных и
иеэлектролитных растворов в сжиженных газах. А. И. Шатен-
14
ейну принадлежат также фундаментальные разработки в обла-
Ш \ кислотно-основного взаимодействия (см. гл. II) и кинетики
изотопного обмена в неводных средах (см. гл. XV).
Исследованиями в сжиженных газах успешно занимался так-
В. А. Плесков, которому принадлежат и исследования э. д. с.
гальванических цепей в неводных растворах; большое значение
для практики эксперимента в неводных растворах имеет
предложенный В. А. Плесковым метод стандартного иона.
Начиная с конца 20-х годов, вопросами физической химии
неводных растворов интенсивно занимается М. И. Усанович, работы
которого оказали значительное влияние на развитие этой области.
М. И. Усановичу принадлежит заслуга окончательной
формулировки и экспериментального доказательства положения, что
возникновение электролитного раствора необходимо связано с
химическим взаимодействием компонентов. Большая часть работ
М. И. Усановича' связана с развитием выдвинутой им теории
кислот и оснований (см. гл. II), значительный вклад внесен им в
изучение концентрированных растворов, в частности, в методологию
и практику физико-химического анализа.
Основные достижения физической химии неводных растворов
последних десятилетий не только у нас в стране, но во всем мире
связаны с деятельностью выдающегося физико-химика Н. А.
Измайлова. К основным достижениям Н. А. Измайлова и его школы
(А. М. Шкодин, В. Д. Безуглый, В. А. Александров, Л. Л. Спивак,
Е. Ф. Иванова и др.) относится: разработка единой
количественной теории диссоциации электролитов, теоретическое обоснование
дифференцирующего и нивелирующего действия растворителей на
силу растворенных в них электролитов, создание общей схемы
равновесий в растворах, разработка теории поведения электродов
в неводных растворах, разработка теории и обоснование ряда
методов исследования взаимодействия между компонентами
раствора. Кроме того, Н. А. Измайлову принадлежат важные
разработки в области термодинамики неводных растворов, титрования
в неводных растворителях, теории и практики ионного обмена в
неводных средах и т. п. Основные результаты исследований
Н. А. Измайлова подытожены им в монографии «Электрохимия
растворов».
Общие разработки в области термодинамики растворов
электролитов в неводных растворах принадлежат К. П. Мищенко, чьи
исследования, начавшиеся в начале 30-х годов, существенно
обогатили этот важнейший раздел общей теории растворов. В
работах К. П. Мищенко был предложен получивший большое
распространение метод вычисления энергий сольватации, а также метод
разделения энергии сольватации на- ионные составляющие.
Вопросы термодинамики неводных растворов в последнее
десятилетие получили развитие в работах Г. А. Крестова, который
предложил метод разделения энтропии процесса растворения на
составляющие и получил весьма важные для термодинамики
15
неводных растворов данные по растворимости инертных газов в
ряде неводных растворителей.
Весьма сложные проблемы ионных равновесий в растворителях
с низкой диэлектрической проницаемостью были исследованы
A. М. Сухотиным, который предложил ряд оригинальных
уравнений для расчета констант электролитической диссоциации в таких
средах.
Термодинамика концентрированных растворов получила
существенное развитие в работах А. В. Сторонкина и его сотрудников
(А. Г. Морачевский, М. П. Сусарев, Е. В. Комаров, Н. А.
Смирнова), которые провели подробный анализ границ применимости
законов Гиббса — Коновалова к многокомпонентным жидким
смесям.
В последние годы интенсивными исследованиями
электрохимических свойств неводных растворов занимается Н. Е. Хомутов,
которому принадлежит оригинальный метод расчета э.д. с. неводных
гальванических цепей.
Кинетика реакций в неводных средах за последнее десятилетие
получила развитие в работах В. М. Литвиненко, М. И. Винника и
B. А. Пальма, исследующих влияние состава растворителей на
кинетику и механизм различных органических реакций.
Ряд гомогенных жидких систем, имеющих большое значение
для экстракции, был исследован А. В. Николаевым.
Существенный вклад в физическую химию неводных растворов
был внесен в 50—60 годах физиками, из которых прежде всего
следует назвать М. И. Шахпаронова. Развитая им теория
флуктуации свойств позволяет описать закономерности
концентрационного изменения многих важных свойств.
В. М. Чулановский, М. Ф. Вукс, Г. С. Денисов и другие
выполнили важные работы по спектроскопии неводных растворов.
Весьма важный раздел физической химии неводных
растворов— физико-химический анализ жидких систем преимущественно
развивался советскими исследователями.
Школа Н. С. Курнакова представлена работами Н. И.
Степанова, известного своими исследованиями метрики равновесной
химической диаграммы, и В. Я. Аносова, в работах которого
вопросы геометрии химической диаграммы нашли глубокое и
всестороннее рассмотрение. К этой же школе относятся работы
C. Ф. Жемчужного, Н. Н. Ефремова и Н. К. Воскресенской.
Второе направление в физико-химическом анализе жидких
систем связано с именем Н. А. Трифонова, который разработал ряд
методологических вопросов физико-химического анализа жидких
систем. К школе Н. А. Трифонова принадлежат Р. В. Мерцлин
(исследование поверхностного натяжения), О. А. Осипов
(исследования в области диэлькометрии и полярных свойств жидких систем),
С. П. Мискиджьян (исследования в области механизма
электропроводности систем, образованных непроводящими тока
компонентами, а также потенциометрия неводных систем), Н. Л. Ярым-
Д ев (исследования в области геометрии диаграмм и
термодинамики жидких систем).
Советскими исследователями были выполнены работы по обос-
ванию и развитию новых методов физико-химического анализа
Н лких систем. В. В. Удовенко рассмотрел и проанализировал
функциональные зависимости молекулярной массы от состава
жидкой фазы. Т. Н. Сумарокова провела методологические
исследования в области криоскопии жидких систем и предложила
классификацию криоскопических диаграмм. Е. Н. Гурьяновой принадлежит
метод диэлькометрического титрования.
За рубежом в настоящее время физическая химия неводных
растворов интенсивно развивается в исследованиях школы Н.
Пущина (СФРЮ —Тутунджич, Косанович, Лилер и др.), Джиллеспи
(Канада), Фалькенгагена (ГДР), Барроу (Великобритания) и др.
физическая химия неводных растворов относится к числу
наиболее быстро развивающихся разделов химии, вообще, в
физической химии, в особенности. Значительный интерес, который
представляют процессы в неводных средах для химической технологии,
является залогом дальнейшего развития физической химии
неводных растворов.
Глава II
ХИМИЧЕСКИЕ РЕАКЦИИ
В НЕВОДНЫХ РАСТВОРИТЕЛЯХ
Подавляющее большинство реакций, протекающих в неводных
растворах, а также реакций между растворенным веществом и
растворителем могут быть описаны с привлечением положений
теорий кислот и оснований. По этой причине до изложения
вопросов физической химии неводных растворов желательно
рассмотреть некоторые закономерности кислотно-основного
взаимодействия.
Ранее уже отмечалось, что характер протекания реакций в
неводных растворах является предметом рассмотрения
неорганической или органической химии (в зависимости от природы
участников реакции). Однако физико-химическая характеристика этих
реакций предполагает, естественно, знакомство с самими
реакциями. Поэтому в этой главе будут рассмотрены кратко также
основные закономерности протекания химических реакций в неводных
растворителях.
II. I. Теории кислотно-основного взаимодействия
Различные теории кислот и оснований мы рассмотрим лишь в
объеме, необходимом для уяснения материала следующих глав.
При этом авторы будут максимально кратки, так как данной
проблеме посвящена превосходная книга А. И. Шатенштейна «Теории
кислот и оснований», которая хотя и вышла более 20 лет назад
(1949 г.), но полностью сохранила свое значение.
Общей теории кислот и оснований, по отношению к которой
все остальные являлись бы безусловно частными, нет, да и
вряд ли она возможна, так как рассматривая такой широкий круг
явлений как кислотно-основные превращения, эта теория,
выиграв, быть может, в общности, несомненно проиграла бы в
конкретности.
Авторы хотели бы также предупредить естественный вопрос:
какая из существующих теорий кислот и оснований лучше?
Поскольку мы не считаем возможным посягать на время и труд
читателя, обременяя его рассмотрением заведомо ошибочных
положений, на поставленный вопрос мы с полным основанием можем
ответить: ни одна из излагаемых далее теорий кислот и оснований
не может быть категорически предпочтена иной. Все дело в том,
чтобы правильно очертить тот круг веществ и реакций, к которым
та или иная теория применима.
Авторы не останавливаются также на положениях теории
кислот и оснований Аррениуса — Оствальда, считая их
общеизвестными.
18
Химические теории
Возникновение химических теорий кислотно-основного взаимо-
- (ствия стало неизбежным после того, как было установлено, что
^новной продукт этого взаимодействия — соль — может быть
получен без участия воды — и как среды для проведения реакций и как
химического составляющего кислоты и основания. Действительно,
уже реакция НС1 + NH3 <=* NH4C1 — типичный пример солеобразо-
вания, протекающего без участия воды. Вот почему Вернер еще
в начале века ввел понятия «ангидрокислота» и «ангидрооснова-
ние», которые в результате кислотно-основного взаимодействия
образуют соль. Ниже приведены некоторые ангидрокислоты, ангид-
рооснования и соли, образующиеся в результате реакций
нейтрализации:
Ангидрокислота Ангидрооснование Соль
SO3 Na2O Na2SO4
As2O6 K2O K3As04
As2S5 K2S K3AsS4
PtCl4 KC1 K2PtCl6
К химическим принадлежит также теория Ганча, обоснованию
которой он посвятил ряд работ, публиковавшихся с начала века
в течение почти трех десятилетий. Подобно Вернеру, Ганч в.
качестве основного признака кислотно-основного взаимодействия
выдвигал солеобразование. При этом любые достаточно прочные
продукты присоединения к кислотам могут рассматриваться как соли.
Так, аналогично тому, как соединение NH3-HC1C>4 является
перхлоратом аммония NHtClOI, соединение HfeO-HClO^ по Ганчу,—
перхлоратом гидроксония Н3О+СЮ7. Соединения типа НК+А~,
образующиеся в результате кислотно-основного взаимодействия,
Ганч назвал ониевыми солями — термин, который сохранился в
химии и применяется весьма часто.
Развитие химической теории Ганча привело к теоретическому
представлению и экспериментальному обоснованию амфотерности
водородных кислот. Так, СН3СООН по отношению к воде является
кислотой и образует ониевую соль Н3О+СН3СОО~, но по
отношению к хлорной кислоте СН3СООН — основание образует ониевую
соль СН3СООН2СЮ7 — перхлорат ацилония.
Амфотерность водородных кислот была впоследствии изучена
методами физико-химического анализа на весьма большом числе
объектов. Значительный вклад в эту область исследований был
сделан школой М. И. Усановича, который исследовал двойные
системы с водородными и апротонными кислотами.
Развитие исследований в области амфотерности Н-кислот
показало, что нет практически такого соединения, которое при
подходящих условиях не могло бы присоединять протон, т. е. не могло
бы быть основанием. Так, в системе CF3SO3H—H2SO4 серная
кислота проявляет протоноакцепторную функцию:
CF3SO3H + HaSO4 *==* H2S(VCF3SO3H *=* H3SO+ + CF3SO3
19
Обе химических теории — Вернера и Ганча — отличались
существенной новизной и научной смелостью: впервые термины
«кислота» и «основание» были отнесены не к водным растворам, а к
индивидуальным веществам. Эти теории и особенно воззрения Ганча
обладали значительной предсказательностью, позволяя оценить
степень взаимодействия в той или иной системе.
Теории сольвосистем
Теория сольвосистем распространяет положение теории
электролитической диссоциации Аррениуса — Оствальда, согласно
которому все вещества, отщепляющие в воде катион Н+ (Н3О+),
являются кислотами, а отщепляющие анион ОН" — основаниями —
на все растворители, способные к автоионизации: 2S з=* S| + SI.
По теории сольвосистем вещество, отщепляющее при растворении
в данном растворителе тот же катион, который образуется при
автоионизации растворителя (Si), будет в этом растворителе
кислотой. Аналогично, вещество, отщепляющее тот же анион,
который образуется при автоионизации растворителя (Sj), будет
основанием (Кэди, Элсей, 1929). Согласно теории сольвосистем,
аналогия с кислотно-основным взаимодействием в воде простирается
также и на механизм этого взаимодействия: подобно тому как в
воде реакция нейтрализации ведет к образованию соли и воды,
в растворителе S при взаимодействии кислот и оснований в данной
сольвосистеме должны образовываться молекулы соли и
растворителя S. Некоторые сольвоеистемы представлены в табл. II. 1.
Таблица ИЛ
Сольвоеистемы растворителей
Растворитель
H2SO4
CH3COOH
н2о
С2Н5ОН
NH3
so2
I2
SbCl5
Катион
H3sot
CH3COOH+
н3о+
с,нбон+
NH+
so2+
I+
SbCl*
Анион
hso;
СНзСОСГ
OH"
с2н5сг
NHJ
SO32-
г
sbci;
P*I
3,1
12,6
14,0
19;5
32,7
—
—
Кислота
h3so;.cf3so3
CHgCOOHj. HSO;
Н3О+СГ
C2H6OHJCF
NH4C1
SOC12
IC1
SbCl4F
Основание
NaHSO4
CH3COONa
NaOH
NaOC2H5
NaNH2
Na2SO3
Nal
NaSbCl6
Продукт
нейтрализации (соль]
NaCF3SO3
NaHSO4
NaCl
NaCl
NaCl
NaCl
NaCl
NaF
20
Достоинства теории сольвосистем очевидны: закономерности
исаотно-основного равновесия распространяются на весьма
больше число растворителей, по отношению к которым вода занимает
Устное и отнюдь не исключительное положение. Теория эта
позволила дать четкий прогноз относительно поведения веществ в
определенных растворителях, вследствие чего оказалось
возможным осуществить ряд важных синтезов (см. раздел II. 2). Вместе
с тем, теории сольвосистем свойственны и очевидные недостатки:
сведение кислотно-основного взаимодействия исключительно к
одному механизму, а также невозможность ни предсказать, ни
даже объяснить проявление кислотных или основных свойств и
протекание кислотно-основного взаимодействия в растворителях,
не способных к автопротолизу. Несомненным недостатком этой
теории является также исключение из круга кислотно-основного
взаимодействия реакций, протекающих не в «своем» растворителе, как,
например, взаимодействие хлорного олова и пиридина в растворе
уксусной кислоты.
Протолитическая теория
Возникновение протолитической теории кислотно-основного
взаимодействия связано с именами Бренстеда, Лоури и Льюиса,
которые высказали ее почти одновременно в 1923 г. Однако
поскольку первый из них сделал это более обоснованно и
впоследствии затратил много увенчавшихся успехом усилий на развитие
этой теории, ее справедливо называют теорией Бренстеда.
Кислотные свойства, по Бренстеду, связаны со способностью
вещества отдавать протон партнеру, который в данном случае
выступает в роли основания. При этом безразлично, является ли
донор протона нейтральной молекулой, либо заряженной; точно
так же может быть нейтральна или заряжена молекула
основания— акцептора протона. В соответствии с этим возможны
нейтральные, катионные и анионные кислоты и основания, примеры
которых приводятся в табл. II. 2.
К существенным отличиям теории протолитического равновесия
от иных теорий кислотно-основного взаимодействия относится
положение, согласно которому продуктами этого взаимодействия
являются не соль, а новая кислота и новое основание:
Кислота I Основание II Основание I Кислота II
СНзСООН + Н2О ^=± СН3СОО" + Н3О+
нсю4 + сн3соон ^=± сю; + сн3соон£
НС1 + NH3 ^=± СГ + NH+
Н2О + NH3 ^z± ОН" + NH+ (II. 1)
Н2О + CN" ^=± ОН" + HCN
hso; +он~ ^z± sol" +Н2°
АН + В ^=± А" + ВН+
21
Таблица И.2
Нейтральные и заряженные кислоты и основания
Зарядный тип
Нейтральный
Катионный
Анионный
Кислота
нсю4
H2SO4
СНзСООН
н3о+
NH+
СН3СООН+
HSOJ
НРО42"
Основание
NH3
C6H5NH2
NH2-NH2
NH2-NH+
сю;, soj-,
СГ, ОН"
Таким образом, при кислотно-основном взаимодействии кислота
превращается в основание I, а основание II — в кислоту II. Эти
пары называются корреспондирующими (или сопряженными).
Согласно схеме (II. 1), основание А~ является корреспондирующим
кислоте НА, а кислота ВН+ сопряжена с основанием В. Очевидно
также, что участники реакции (II. 1) могут иметь произвольные
заряды, либо быть нейтральными — в зависимости оттого,
являются ли основания (кислоты) и кбрреспондирующие им кислоты
(основания) нейтральными, катионными или анионными.
Теория протолитического равновесия получила очень
значительное распространение и поныне является едва ли не самой
популярной теорией кислотно-основного взаимодействия, что вполне
естественно, так как все кислотно-основные процессы, связанные
с участием донора (кислоты) и акцептора протона (основания)
закономерно и без каких-либо исключений укладываются в рамки
теории Бренстеда. Из этой теории естественно вытекает ряд
положений, которые прежде были предметом весьма оживленной
дискуссии (см. гл. I). Так, представляется несомненным
возникновение электролитного раствора при кислотно-основном
взаимодействии; очевидным является взаимодействие кислот и оснований с
растворителем; весьма логично вытекает понятие амфотерности
и т. п.
Но все же самым большим достоинством теории Бренстеда
является возможность вывода уравнений, которые позволяют
рассчитывать весьма важные характеристики, как, например, константы
электролитической диссоциации кислот, а также зависимость
констант электролитической диссоциации от диэлектрической
проницаемости растворителя (об уравнениях количественной теории
Бренстеда и следствиях из них см. в разделе IX. 4).
К недостаткам теории Бренстеда следует отнести то
обстоятельство, что реакции с участием апротонных кислот исключаются
22
из круга кислотно-основных равновесий. Впрочем, Бренстед,
формулируя основы своей теории и развивая ее, неоднократно
подчеркивал, что теория эта относится лишь к реакциям с участием Н-
кислот, т. е. сам четко ограничивал рамки ее применения *.
Электронная теория
Электронная теория кислот и оснований была сформулирована
в 20-х годах Льюисом, который стремился охватить понятиями
«кислота» и «основание» более широкий круг веществ, чем это
позволяла сделать теория протолитического равновесия.
Последовательно развивая электронные представления, Льюис дал
следующие определения основным понятиям своей теории:
«Основание — вещество, обладающее свободной парой
электронов, которая может быть использована для образования
устойчивости электронной группировки другого атома. Обычно такой
группировкой является октет электронов. Кислота — вещество, которое
может использовать свободную пару электронов атома другой
молекулы для образования устойчивой электронной группировки
одного из своих атомов».
В соответствии с электронной теорией серный ангидрид, который
в виду дефицита электронов на атоме серы является кислотой (L-
кислотой), взаимодействует с основанием — водой, на атомах
кислорода которого имеются вакантные электронные пары:
ОН ОН
O:S + :O: —► O:S:O:
ОН ОН
В рамках теории Льюиса получает объяснение ряд фактов,
которые не могли быть объяснены изложенными выше теориями.
Так, становится очевидным сродство, которое испытывает L-кисло-
та А1Вгз к основанию — пиридину:
Вг Вг
Вг : А1 + : NC6H5 —> Вг: Aj: NC5H5
Вг Вг
Н-Кислоты являются частным случаем L-кислот, так как
протон, испытывая сильнейший дефицит электронов, проявляет сильно
выраженные кислые свойства. Справедливость включения Н-кис-
лот в более широкий круг L-кислот Льюис логично обосновал
также тем, что L-кислотам свойственны многие закономерности,
хорошо известные для Н-кислот, в частности, быстрота протекания
реакций с основаниями (реакций нейтрализации), вытеснение
более слабых кислот более сильными, изменение цвета
индикаторов, каталитическое действие на ряд химических реакций.
Точно так же не может быть отнесено к числу недостатков теории Брен-
стеда отрицание ею солеобразования как признака кислотноосновного
взаимодействия — положение, которое нельзя отвергать, не отвергая теорию в целом.
23
Льюисом был сделан еще один шаг в сторону развития теории
Бренстеда. Согласно теории протолитического равновесия,
основаниями являются все анионы (или, во всяком случае то
большинство из них, которое отвечает существующим водородным
кислотам), но кислые свойства обуславливаются лишь одним
катионом— Н+. Однако согласно трактовке Льюиса, катионы,
характеризующиеся дефицитом электронов, способны взаимодействовать
с анионами — основаниями, и, следовательно, являются кислотами.
Правда, кислые свойства Льюис приписывал не всем катионам,
а лишь тем, которые «способны присоединять пару электронов
основания».
Теория Льюиса, несмотря на внешнюю простоту и логичность,
обладает рядом недостатков, которые ее значительно
обесценивают. Так, поясняя факт взаимодействия кислоты НС1 с
основанием NH3, необходимо прибегнуть к предположению, что один из
атомов водорода в образовавшемся (но еще не продиссоциировав-
шем) продукте присоединения двухковалентен:
Н
: С1: Н : N: Н
Н
С большим трудом, лишь прибегая к допущениям, иногда
весьма произвольным, теория Льюиса поясняет факты амфотерности
(например, проявление водой основных и кислых свойств); в
некоторых случаях амфотерность теорий Льюиса вообще не может
быть объяснена.
Кислотно-основные превращения логично объясняются
электронной теорией лишь в случае простых соединений. Поэтому,
сталкиваясь со случаями, когда то или иное вещество, не являясь в
соответствии с начертанием электронной формулы, например
кислотой, тем не менее проявляет отчетливо выраженные кислые
свойства, Льюис прибегнул к представлениям о вторичных
кислотах и основаниях. Последние приобретают электронное строение,
отвечающее L-кислотам или L-основаниям в момент реакции, при
возбуждении; этим Льюис объясняет относительно большие
значения энергии активации реакций, в которых принимают участие
вторичные кислоты и основания.
Иногда теория Льюиса приводит к прямому противоречию: так,
взаимодействие AsCl3 с пиридином, носящее четко выраженный
кислотно-основной характер, не может быть объяснено в рамках
этой теории, поскольку формальные электронные формулы
показывают, что на атомах и мышьяка и азота имеются вакантные
электронные пары *.
* В действительности на атоме мышьяка из-за сильного электрофильного
действия трех атомов хлора наблюдается не избыток, а, напротив, дефицит
электронов; однако необходимость учета подобных факторов, конечно, сильно
обесценивает теорию»
24
Теория Усановича
Наиболее общая теория кислот и оснований была
сформулирована М. И. Усановичем (1939 г.). Согласно этой теории «кислота —
частица, которая может отщеплять катионы, включая протон, или
присоединять анионы, включая электрон; основание — частица,
которая может присоединять протон и другие катионы или отдавать
электрон и другие анионы» (формулировка 1964 г.).
В качестве основного признака кислотно-основного
взаимодействия М. И. Усанович выдвигает солеобразование. Таким образом,
в реакции SO3 + 2Н2О <=* H3O+ + HSO7 вода, отдавая анион О2",
является основанием, a SO3, присоединяя этот анион — кислотой;
аналогично в реакции SnCl4 + 2KCl^K2SnCl6 тетрахлорид олова,
присоединяющий анионы хлора, выступает в роли кислоты.
Приведенная выше формулировка понятий «кислота» и
«основание» позволяет включить в круг кислотно-основных равновесий
окислительно-восстановительные реакции. Действительно, в
реакции Zn -f- Br2-*ZnBr2, приводящей к образованию соли (признак
кислотно-основного взаимодействия!), бром, присоединяя
электроны, по Усановичу, выступает в роли кислоты, а цинк, отдавая
электроны, — в роли основания. Включение
окислительно-восстановительных реакций в круг кислотно-основных превращений не
покажется искусственным, если вспомнить, что металлический цинк
в реакции Zn + 2HBr ->ZnBr2 + H2 нейтрализует кислоту, образуя
при этом соль, т. е. в этой окислительно-восстановительной реакции
соблюдаются все традиционные признаки кислотно-основного
взаимодействия.
Иногда, основываясь на некоторых сходных чертах, считают
теорию Усановича развитием теории Льюиса, что неверно, так как
теория Усановича базируется на иных предпосылках: в основе
понятий «кислота» или «основание» лежит знак заряда, а не строение
электронной оболочки. Вот почему, вопреки*электронной теории
Усанович считает, что любой катион обладает кислыми свойствами,
а любой анион — основными. Поскольку кислотные и основные
свойства в рамках этой теории определяются лишь знаком заряда,
можно говорить о более и менее кислых катионах и более и менее
основных анионах. Очевидно, что кислотность катионов либо
основность анионов (при одинаковом строении внешних электронных
оболочек и одинаковом заряде) будет возрастать по мере
уменьшения ионного радиуса. Так, в ряду катионов щелочных металлов
кислотность падает от Li+ к Cs+, а в ряду галоген-анионов
основность падает от F" к 1~. Такой, на первый взгляд, не совсем
обычный подход к проблеме кислотности и основности позволяет
объяснить ряд закономерностей электролитической диссоциации солей
в неводных растворах (см. раздел IX. 4).
Понятие о корреспондирующих кислотах и основаниях
сохраняется в теории Усановича, однако предпосылки этой теории
позволяют сделать существенное уточнение, согласно которому
25
сильная кислота соответствует слабому основанию, например, в
реакции Вг2 + 2е -> 2Вг", сильная кислота Вг2 отвечает слабому
основанию Вг"; сильному основанию К отвечает слабая кислота К+
(К-*К+ + е-).
Основной упрек теории М. И. Усановича, который обычно
встречается в литературе, заключается в слишком большой общности
охватываемого этой теорией круга реакций. Действительно,
включение в круг кислотно-основных превращений
окислительно-восстановительных реакций приводит к, казалось бы, неоправданному
расширению понятия «кислотно-основное взаимодействие» и, как
следствие — к потере значительной доли конкретности этого
понятия. Авторы не склонны разделять эту критику, так как общность
любой теории вряд ли следует считать ее недостатком. Что же
касается конкретности, то, естественно, чем меньшее число признаков
кладется в основу классификации, тем менее она будет
конкретной. Можно считать, что в теории М. И. Усановича соотношение
между общностью рассматриваемых явлений и конкретностью
подхода к кислотно-основному взаимодействию ближе к
оптимальному, чем в каких-либо иных теориях.
К действительным недостаткам теории М. И. Усановича
относится недостаточно четкая определенность приведенной выше
формулировки понятий «кислота» и «основание», поскольку любая
«частица», отщепляющая катион (анион), неизбежно при этом
отщепляет и анион (катион). Вот почему, следуя М. И. Усановичу,
точнее было бы определить кислоту как частицу, которая может
отщеплять катион, присоединяющийся к основанию; соответственно
должно быть уточнено понятие «основание».
К недостаткам теории М. И. Усановича следует отнести также
и то обстоятельство, что она не описывает кислотно-основные
превращения, которые не носят ионогенный характер. Наконец,
теория М. И. Усановича много выиграла, если бы она, подобно
теории Бренстеда, позволяла делать количественные предсказания.
Теория жестких и мягких кислот и оснований
Своеобразным развитием положений теории М. И. Усановича
является теория жестких и мягких кислот и оснований,
сформулированная Р. Пирсоном (1963 г.). По Пирсону, к жестким
основаниям относятся донорные частицы, обладающие высокой
электроотрицательностью, низкой поляризуемостью и трудно
окисляющиеся (ОН", F", SO7, С1О7, NH3, RNH2); донорные частицы с низкой
электроотрицательностью и высокой поляризуемостью относятся
к мягким основаниям (I~, SCN~, R3P, СбН6). К жестким кислотам
относятся акцепторные частицы с низкой поляризуемостью (№",
Li+, Na+, BF3, SnCU, AICI3), а к мягким акцепторные частицы с
высокой поляризуемостью (Ag4", GaCl3, I+, J2). Чисто формальный
признак, по которому произведена первичная классификация в
рамках этой теории, заключается в том, что жесткие кислоты реа-
26
гируют лишь с жесткими основаниями, а мягкие — с мягкими
основаниями. При этом так же, как в теории Усановича, кислыми и
основными свойствами могут обладать и нейтральные, и
заряженные частицы.
Предложен количественный критерий деления кислот и
оснований на жесткие и мягкие (К. Б. Ядимирский, 1970 г.). В качестве
меры жесткости основания выбрана разность между р/С для
равновесия реакции данного основания с протоном (самая жесткая
кислота *) и реакции этого же основания с СНзН^ (одна из
самых мягких кислот): б (р/Св) = рКн+ — pKCH3Hg+ • Аналогично в
качестве меры жесткости кислоты выбраны разность 6(р/Сд) =
=-g-p#oH- ~~ Р^сг ~~ 2» где ^он- и #сг ~ константы устойчивости
комплексов Mez+OH~ (ОН" — одно из наиболее жестких
оснований) и MeCl*-1 (Cl~ — одно из наиболее мягких оснований). В
табл. II. 3 приведены показатели жесткости некоторых кислот и
оснований.
Несмотря на отдельные несогласованности с опытом (так,
анилин имеет показатель жесткости, заметно более высокий, чем
пиридин, хотя основные свойства последнего выражены более
отчетливо), показатели жесткости позволяют в большинстве случаев
весьма точно прогнозировать поведение тех или иных ионов в
кислотно-основных (в широком смысле этого определения)
равновесиях.
Таблица И.З
Показатели жесткости кислот и оснований (по К. Б. Яцимирскому)
Кислоты
частица
н+
Zr4+
Сг3+
F*e3+
In3+
Cu2+
Fe2+
6 (рКА)
9
4,9
3,8
2,7
1,2
1,0
0,6
частица
Ni2+
Zn2+
Pb2+
Cd2+
CH3Hg+
Ag+
Hg2+
б (picA)
0,2
0,2
—0,3
-1,0
-2,8
-3,5
—4,0
Основания
частица
ОН"
C6H5NH2
NH3
F"
NO7
CH3COO"
C5H5N
б (pKb)
6,3
2,0
1,8
1,7
0,9
0,8
0,5
частица
SO42"
H2O
CN"
S2"
СГ
Br™
Г
б (pKb)
0,5
0,0
-4,9
—8,4
-9,3
-13,7
-18,7
Приведенная шкала составлена для водных растворов. Каждый
неводный растворитель характеризуется своими определенными
значениями б (р/С). Хотя достоверных данных о константах
нестойкости и протонному сродству в неводных растворителях
ь настоящее время существует немного для составления шкал,
* Строго говоря, самой жесткой кислотой является позитрон, а самым
жестким основанием — электрон,
27
подобных приведенной в табл. П.З, можно предполагать, что в
большинстве случаев последовательность рядов жесткости кислот
и оснований будет такой же, как и в водных растворах.
Концепция жестких и мягких кислот и оснований свободна от
недостатка, упрек в котором адресовался теории М. И. Усановича:
в рамках этой концепции можно предсказать кислотно-основное
взаимодействие, не связанное с передачей заряженной частицы.
Развитие теории кислотно-основного взаимодействия
в работах А. И. Шатенштейна
Усматривая значительную аналогию между реакциями с
участием Н- и L-кислот, А. И. Шатенштейн (1939 г.) подчеркивает
необходимость выделения Н-кислот в отдельный класс.
Обусловлено это резким отличием катиона Н+ от всех иных катионов,
связанным с ничтожно малым радиусом и, как следствием этого,
значительно большей энергией сольватации этого катиона по
сравнению со всеми другими *.
Значительную роль в развитии представлений о кислотах и
основаниях сыграли работы А. И. Шатенштейна по изучению
кислотно-основных свойств углеводородов, для которых было
установлено, что в сильнокислых средах они ведут себя как основания,
а в сильноосновных — как кислоты (Н-кислоты). Действительно,
было установлено наличие дейтерообмена между бензолом и
жидким аммиаком и бензолом и жидким фтористым водородом:
С6Н6 + ND3 *=* СвН- • ND3H+ *=* C6H5D + ND2H (II. 2)
СбНб + D2F2 =*=* C6H6D+ . DF" *=* C6H5D + HDF (II. 3)
В реакции (II. 2) бензол проявляет свойства С—Н-кислоты, в
реакции (П.З)—свойства основания**.
Эти эксперименты позволили включить в круг кислот и
оснований еще один весьма значительный класс соединений.
Оставаясь в рамках теории протолитического равновесия,
А. И. Шатенштейн предлагает следующие существенно уточненные
определения: кислота — электроноакцепторный агент, атом
водорода которого участвует в равновесной реакции с основанием;
основание— электронодонорный агент, обладающий сродством к
протону. Апротонные кислоты А. И. Шатенштейн предлагает
называть кислотоподобными веществами.
Приведенные определения позволяют включить в круг
кислотно-основных равновесий весьма обширный тип реакций, которые
не приводят к образованию ионогенных продуктов. Действительно,
* Н. А. Измайлов высказывает интересную мысль, что а-частица должна
обладать в силу большего заряда значительно более кислыми свойствами, чем
протон. Однако кислоты на основе а частиц неустойчивы из-за сильнейшего
ионизирующего действия последних.
** Механизм дейтерообмена реакциями (II. 2) и (П.З) передается упрощенно.
28
например, в двойных жидких системах, образованных H2SO4 с
CF3COOH, ССЬСООН, CHCI2COOH, CH2CICOOH и СНзСООН, во
всех случаях констатировано взаимодействие между
компонентами через Н-связь. Однако в первых двух системах это
взаимодействие не приводит к появлению ионогенных продуктов, и
ограничивается лишь образованием продуктов присоединения H2SO4 •
. RCOOH, в остальных же системах этот комплекс подвергается
электролитической диссоциации на ионы HSOJ и RCOOH<J\
Очевидно, что исключение взаимодействия в первых двух системах из
круга кислотно-основных равновесий было бы весьма
искусственным.
Нами были рассмотрены лишь основные теории кислот и
оснований. На теориях, представляющих частный интерес, либо
уточняющих уже сформулированные положения, как, например, на
теориях Эберта и Кнопика (1949 г.) и Гутмана и Лундквиста
(1954) мы не останавливались.
Отметим также то, что определения «кислота» и «основания»
по своему химическому смыслу есть понятия динамические, а не
статические. Индивидуальное вещество не может проявлять ни
кислотных, ни основных свойств, потому что свойства эти
проявляются лишь во взаимодействии. Вот поэтому теории кислот и
оснований, дающие априорные определения этих понятий,
основываются на химически (да и логически) неверных предпосылках.
II. 2. Реакции синтеза в неводных растворах
Неводные растворители используют либо для проведения
реакций препаративного синтеза, либо электрохимического. Последний
аспект применения неводных растворителей будет рассмотрен
в гл. XIII.
Возможны следующие причины, которые побуждают
исследователя или технолога прибегнуть к проведению реакции синтеза
в неводных растворах:
синтез данного вещества по тем или иным обстоятельствам не
может быть осуществлен в водном растворе;
необходимость повысить выход продукта реакции вследствие
существенного сдвига равновесия в данном неводном
растворителе;
необходимость изменить скорость реакции.
Вопросы влияния растворителя на равновесие и скорость
процесса являются сугубо физико-химическими и будут рассмотрены,
в гл. IX и XV.
Более благоприятные условия проведения синтеза в данном
неводном растворителе по сравнению с водным раствором могут
быть обусловлены лучшей растворимостью исходных компонентов,
и повышением их реакционной способности; отсутствием реакции
сольволиза и нежелательных или, напротив, наличием
желательных таутомерных форм и т. п. Наконец, растворитель сам может
29
принимать участие в реакции, приводя к образованию
необходимого соединения.
Классификация протолитических равновесий, в которых
принимает участие растворитель, была дана Клагесом (1933 г.). В
соответствии с этой классификацией в реакции принимают участие
либо нейтральные молекулы растворителя, либо ионы
растворителя, образовавшиеся в результате процесса автопротолиза.
В* первом случае Кла^ес различает два варианта:
1. Переход протона от растворенного вещества к растворителю
CHgCOOH + NH3 *=* СНдСОСГ + NH+ (электролитическая диссоциация)
2. Переход протона от растворителя к иону растворенного
вещества —
CN" + H2SO4 —> HCN + HSOJ (реакция сольволиза)
Во втором случае возможны три варианта:
1. Реакции, в которых катион растворителя St
взаимодействует с анионом растворителя SJ—NHj + NHJ +=* 2NH3 (реакция
нейтрализации);
2. Переход протона от катиона растворителя к основанию либо от
кислоты к аниону растворителя—H3SOt + СН3СООН —► H2SO4+
+ СН3СООН2 и СбН5ОН + NH7 —> С6НбО" + NH3
3. Обмен между ионами растворителя и растворенного
вещества — Н3О+ + СОГ —> НСОз" + Н2О
Каждый из перечисленных вариантов взаимодействия
растворенного вещества с растворителем может быть положен в основу
методов синтеза в неводных растворах.
В качестве примеров синтеза в неводных растворителях
рассмотрим примеры протекания реакций в некоторых, кстати, весьма
часто применяющихся в препаративных целях, растворителях.
Реакции в серной кислоте
Благодаря чрезвычайно сильно выраженным кислотным
свойствам серная кислота «заставляет» выступать в роли основания
инертные или даже кислые (в других растворителях) соединения
и является, таким образом, весьма мощным средством повышения
реакционной способности.
Лишь весьма небольшое число веществ в серной кислоте
способно проявлять кислые свойства (НСЮ4, CF3SO3H, FSO3H). В
соответствии с положениями теории сольвосистем сильным
основанием в серной кислоте является KHSO4.
Поведение многих неорганических и органических соединений
в серной кислоте, а также свойства возникающих при растворении
этих веществ в H2SO4 соединений были предметом обстоятельных
исследований, среди которых выделяются работы Джиллесци,
Взаимодействие органических соединений с серной кислотой
может протекать по двум основным схемам — присоединения и
обмена:
- ROH + H2SO4 *=* ROH • H2SO4
ROH + 2H2SO4 *=* ROSO3H + H3O+ + HSOJ
ROH + 2H2SO4 *=* R+ + H3O+ + 2HSO7 (II. 4a)
RCOOH + H2SO4 *=* RCOOH • H2SO4 =F=fc RCOOHj + HSOJ
2RCOOH + H2SO4 *=* RCOOH++ RCOOH • HSOJ
RCOOH + 2H2SO4 <=* RCO+ + H3O+ + 2HSOJ (II. 46)
RCOORi + H2SO4 <=* RCOOR! + H2SO4
RCOOR, + 3H2SO4 *=* RCO+ + RjHSO4 + H3O+ + 2HSOJ
RONO2 + 3H2SO4 *=* N0+ + RQSO3H + H30+ + 2HS0J (II. 4в)
(RCO)2O + 2H2SO4 =f=* RCO+ + RCOOH+ + 2HS0J (II. 4r)
Как видно из приведенных реакций, специфическое действие
серной кислоты обусловлено также ее дегидратационными
свойствами. Высокая диэлектрическая проницаемость серной кислоты
(^100) обуславливает ионный механизм протекающих в ней
реакций.
Реакции в уксусной кислоте
Кислые свойства уксусной кислоты выражены гораздо слабее,
чем серной, однако они достаточно отчетливы, чтобы значительно
повышать силу растворенных "в ней оснований и еще более
значительно понижать силу кислот (подробнее о силе электролитов в
уксусной кислоте см. в разделе IX. 5).
Характер растворимости неорганических солей в уксусной
кислоте существенно иной, чем в воде, поэтому реакции с участием
этих соединений обладают значительным своеобразием. Так, в
уксусной кислоте можно проводить многие реакции, обратимые либо
вообще не протекающие в воде, поскольку удается сместить
равновесие этих реакций из-за малой растворимости какого-либо из
продуктов реакции, например C2H5COONa + HC1 -> С2Н5СООН +
+ NaClj. Уксусную кислоту широко используют в неорганическом
синтезе для получения веществ кислотного характера, которые в
воде разлагаются (например, 1СЬ).
В органической химии абсолютная либо весьма
концентрированная («ледяная») уксусная кислота является одним из наиболее
Распространенных неводных растворителей. Обстоятельный обзор
органических реакций в уксусной кислоте приведен в книгах
Л. Одрита и Дж. Клейнберга, а также в Г. Шарло и Б. Тремилло.
31
Реакции в гидразине
В качестве примера реакций в основных растворителях
рассмотрим реакции в гидразине (реакции в сжиженном аммиаке так
часто описываются, что вряд ли их стоит еще раз воспроизводить).
Высокая диэлектрическая проницаемость гидразина (е = 51,7)
обуславливает хорошую растворимость в нем многих
неорганических соединений. При этом можно установить соответствие между
химическими свойствами растворенного вещества и его
растворимостью. Так, обладающий гораздо более кислым характером LiCl
(см. предыдущий раздел) растворим в высокоосновном гидразине
вдвое лучше, чем NaCl (15,8 и 7,87 г в 100 г N2H4). Растворы
многих солей в гидразине характеризуются столь же высокой
электропроводностью как и в воде. Это позволяет с успехом
осуществлять в этом растворителе многие ионные реакции, связанные с
образованием веществ основного характера, которые в воде под-
' вергались бы далеко идущему гидролизу.
Сильноосновные свойства гидразина обуславливают течение
многих своеобразных органических реакций, например,
RCOOR, + N2H^ *=* RCONHNH2 + R,OH
RCOC1 + 2N2H4 =*=* RCONHNH2 + N2H4 • HCi (II. 5)
Многие аммонийные соли в гидразине подвергаются сольволизу
CH3COONH4 + N2H4 =*=* CH3CONHNH2 + NH3 + Н2О
либо переходят в соответствующие соли гидразина
NH4C1 + N2H4 —> N2H4- HCI + NH3
что свидетельствует о более высокой основности гидразина по
сравнению с аммиаком.
Глава III
КЛАССИФИКАЦИЯ РАСТВОРИТЕЛЕЙ
В основу любой системы классификации положен какой-либо
один признак или какая-либо группа признаков. Вот почему
каждая такая система, выдвигая на первый план лишь определенные
качественные либо количественные признаки, не может быть
универсальной и одинаково пригодной для всех случаев.
Классификация растворителей в этом смысле не представляет исключения.
Однако свойства, на которых основывается классификация
растворителей, можно разбить на две основные группы: физические и
химические.
Ш.1. Системы классификации, основанные
на физических свойствах растворителей *
К важнейшим физическим свойствам, в значительной степени
обуславливающим особенности неводного раствора, относятся
диэлектрическая проницаемость и вязкость.
По диэлектрической проницаемости (е) растворители
подразделяются на растворители с низкой (1,9—12), средней (12—50) и
высокой (>50) величиной е **.
Нижний предел первой группы определен значением е гекса-
на — одного из наиболее часто применяемых растворителей с
низким значением диэлектрической проницаемости; верхний же
предел определен тем, что именно в этой области чаще всего
наблюдаются аномалии на зависимостях констант
электролитической диссоциации (см. гл. IX) и предельной электропроводности,
исправленной на вязкость (см. гл. X), от 1/г. Отметим также, что
точность расчета констант диссоциации по данным
электропроводности в растворителях с е < 12 значительно меньше по
сравнению с таковой для растворителей с более высокими
значениями 8.
Верхний предел второй группы определен тем, что в
растворителях с е > 50 электролиты часто ведут себя как сильные, в то
время как в растворителях со средними значениями 8 (и тем
более в растворителях с низким е) практически никогда не
происходит полной электролитической диссоциации.
Классификацию растворителей, основанную на дипольных
моментах (ji), вряд ли можно считать рациональной. Связано это
с тем, что из-за очень распространенного в растворителях явления
* Сводку основных физических свойств неводных растворителей см. в
Приложении (стр. 357).
** Часто применяемые по отношению к этим группам термины
«низкополярные», «высокополярные» и т. п. представляются неудачными, так как
определение «полярность» правильнее относить не к е, а к дипольному моменту.
2 Зак, 500 ОО
ассоциации (см. гл. VII) истинный дипольный момент молекулы
ассоциата может значительно отличаться (чаще всего, в меньшую
сторону) от \х одиночной молекулы. Так, значение ji=l,7
присуще одиночным молекулам целого ряда растворителей в широком
диапазоне диэлектрических проницаемостей — от этанола (е = 24,3)
до тетрагидрофурана (е = 7,35). Вода — растворитель с весьма вы-
соким значением е— имеет сравнительно низкую величину jj, (1,8),
а характеризующийся заметно более низкой величиной 8 пропи-
ленкарбонат (65,1) имеет ji = 5,0*.
Следует отметить также, что на измеряемые значения ц
оказывает существенное влияние растворитель.
Вязкость (т|) растворителей может изменяться в весьма
широких пределах — от нескольких десятых долей до тысяч сантипуаз.
Впрочем, вязкость наиболее распространенных неводных
растворителей при 25 °С редко превышает 30 спз (очевидным исключением
является глицерин). В соответствии с этим принято различать
низковязкие растворители (<2 спз), растворители,
характеризующиеся средней величиной вязкости (2—10 спз), и высоковязкие
(> 10 спз).
По температуре кипения (при 760 мм рт. ст.) растворителя
подразделяются на низкокипящие (<100°С), среднекипящие (100—
150 °С) и высококипящие (> 150 °С).
Для классификации растворителей по их способности к
испарению предложена относительная шкала, в основе которой лежат
теплоты испарения жидкостей. По этой шкале летучесть этилового
эфира при 20°С и относительной влажности, равной
65±5%,принята за 1. В соответствии с этим различают легколетучие
(относительная летучесть <10), среднелетучие (10—35) и труднолетучие
(>35) растворители.
III. 2. Системы классификации, основанные
на химических свойствах растворителей
В практической деятельности растворители чаще всего
классифицируют в соответствии с рациональной системой классификации
химических соединений (углеводороды, галогеналкилы, спирты,
кислоты и т. д.). Классификация эта оправдывается также тем, что
в большинстве случаев растворы одних и тех же соединений в
растворителях одного типа проявляют весьма сходные свойства (за
исключением растворов в первых членах гомологического ряда
каждого из классов органических растворителей — метаноле,
формальдегиде, муравьиной кислоте и т. д.).
Для специальных целей вводят классификацию, основанную на
определенных химических свойствах. При этом качественные
признаки выбирают чаще, чем количественные. Так, часто подразде-
* Более подробно о корреляции между е и \i, как и вообще о корреляциях
между различивши физическими двойствами растворителей, см. в гл. IV.
34
ляют растворители на донорные (реагирующие с акцепторами
электронной пары) и акцепторные (реагирующие с донорами
электронной пары).
Нередко (а при выборе растворителя для проведения неводного
титрования — практически всегда) классифицируют растворители,
исходя из их способности принимать участие в кислотно-основных
равновесиях, т. е. вводят классификацию, основанную на кислотно-
основных свойствах растворителей.
Если рассматривать кислотно-основное взаимодействие в
рамках представлений Бренстеда, то в зависимости от участия в про-
толитическом кислотно-основном равновесии растворители
подразделяют на протолитические и апротонные.
К протолитическим относятся растворители, проявляющие про-
тонодонорную или протоноакцепторную функцию по отношению
к растворенному веществу. В зависимости от последней
протолитические растворители бывают: протогенными (кислыми), прото-
фильными (основными) и амфипротонными, т. е. такими, которые
приблизительно в одинаковой степени проявляют и кислотную
и основную функции. Примерами амфипротонных протолити-
ческих растворителей могут служить спирты и фенолы
(первые из них чаще проявляют основные свойства, а вторые —
кислотные) .
Апротонные растворители не способны вступать в кислотноОс-
новное взаимодействие, связанное с переносом протона. К этому
классу растворителей относятся углеводороды, галогеналкилы и
галогенарилы, четыреххлористый углерод и т. д.
Поскольку в подавляющем большинстве случаев амфотерность
присуща всем химическим соединениям, постольку приведенная
классификация весьма условна. Так, апротонные растворители,
даже такие, как бензол или гексан, в экстремальных условиях
способны проявлять протонодонорные или протоноакцепторные
свойства. Еще более условно подразделение на классы протолитиче-
ских растворителей, поскольку практически любой из
растворителей в определенных условиях способен проявлять и кислую, и
основную функцию.
В рассматриваемой системе классификации весьма
неопределенное место занимают апротонные растворители. Так, в
соответствии с введенным определением к этому классу растворителей
Должен быть отнесен, например, тетрахлорид олова, хотя взятый
в качестве растворителя он будет оказывать самое существенное
влияние на течение кислотно-основного протолитического
равновесия. Отсутствие четкого критерия для предварительной оценки
степени участия данного растворителя в протолитическом
равновесии приводит к тому, что в отдельных работах к апротонным
относят самые разнообразные растворители — нитробензол (в H2SO4
проявляет четкую основную функцию), кетоны (весьма легко
проявляют основные свойства и нередко, выступая в энольной форме,
и кислотные) и даже такие растворители как триэтиламин и
* 35
пиридин. Очень многие растворители способны к взаимодействию
посредством образования водородной связи (Н-связь). В
соответствии с этим разработана классификация растворителей по их
способности к образованию Н-связей (Е. Well, H. А. Измайлов).
В этой системе классификации растворители делятся на пять
классов.
К первому классу относятся жидкости, способные к
образованию объемной трехмерной сетки Н-связей. Растворители этого
класса (вода, муравьиная кислота, гликоли и т. п.) имеют, как
правило, весьма высокую диэлектрическую проницаемость и хорошо
растворяются друг в друге.
Второй класс — растворители, в которых возникает двухмерная
сетка Н-связей. Это определяется менее сильным стремлением к
образованию подобных связей по сравнению с растворителями
первого класса. Как правило, растворители этого класса содержат
одну ОН-группу. К этому классу по классификации Н, А.
Измайлова относятся фенолы, одноосновные низшие карбоновые кислоты
(за исключением муравьиной), одноатомные спирты.
Третий класс объединяет жидкости, молекулы которых имеют
в своем составе атомы азота, кислорода, серы, фтора и некоторые
другие, способные к образованию Н-связи с протоном партнера
(эфиры, амины, кетоны, альдегиды и вещества других классов
органических соединений).
В четвертый класс входят жидкости, молекулы которых имеют
атом водорода, способный к участию в Н-связи, но не имеют
атомов, которые могли бы быть акцептором протона. К этой группе
жидкостей относятся хлороформ, дихлорэтан и т. п.
К пятой группе относятся растворители, молекулы которых при
обычных условиях не способны к образованию Н-связей — ни в
качестве доноров, ни в качестве акцепторов протонов. В эту группу
входят углеводороды, четыреххлористый углерод, пергалогенугле-
водороды и т. п.
К предыдущей системе классификации близко примыкает
классификация Паркера (1962 г.), в основу которой положена
способность растворителя сольватировать ионы. Согласно Паркеру,
растворители делятся на следующие группы:
аполярные апротонные растворители — жидкости с
низкой диэлектрической проницаемостью (< 15) и небольшим диполь-
ным моментом (0—2D). В эту группу входят углеводороды и их
галогензамещенные производные, сероуглерод, третичные амины
и т. п.;
диполярные апротонные растворители — жидкости со
сравнительно высокими диэлектрическими проницаемостями (>15)
и дипольными моментами (>2,5). Если молекулы этих
растворителей и содержат водород, он все равно не способен образовывать
Н-связь; в эту группу входят такие растворители как сернистый
ангидрид, нитробензол, нитрометан, ацетонитрил, пропиленкарбо-
нат, диметилформамид и др.;
36
протонные растворители содержат группы, в которых атом
водорода соединен с электроотрицательным атомом, что
обуславливает его способность вступать в Н-связь. Диэлектрическая
проницаемость этой группы растворителей, как правило, высока.
Сольватационная способность растворителей повышается от
первой группы к третьей. Соответственно этому и константы
диссоциации растворов электролитов увеличиваются в этом же
направлении (на величины констант помимо сольватационной
способности растворителя оказывает существенное влияние еще ряд
факторов, см. гл. IX).
Бренстед предложил систему классификации, сочетающую
диэлектрическую проницаемость растворителя и его протогенность
или протофильность. В соответствии с этими признаками Бренстед
разделил растворители на 8 групп, отличительные признаки
которых приведены в табл. III. 1.
Таблица III-1
Классификация растворителей по Бренстеду
Свойство
Диэлектрическая
проницаемость
Протогенность
Протофильность
Тип растворителя
I
J
II
±
III
±
IV
±
V
+
VI
±
VII
VIII
—
Примечание. Знаки «+» и «—» в первой строке отвечают высокой и
низкой диэлектрической проницаемости; а в остальных строках — наличию или
отсутствию данного свойства.
Примерами растворителей отдельных типов могут служить: I — вода;
II — серная кислота, фтористый водород; III — гидразин; IV — пропиленкарбо-
нат, нитрометан, нитробензол, ацетонитрил *; V — бутиловый спирт, ж-крезол;
VI — жидкий бромистый водород и йодистый водород; VII — анилин, пиридин;
VIII — гексан, бензол.
Классификация растворителей по Бренстеду позволяет
предсказать проявление кислотно-основных свойств растворенных
веществ в растворителях каждого из типов. Так, в растворителе
типа II сила оснований заметно выше, чем в растворителе типа III;
сила кислот в растворителях типа III в общем случае выше, чем в
растворителях типа VII и т. д. (подробнее о соотносительном
влиянии диэлектрической проницаемости и энергии сродства к протону
на силу кислот и оснований см. гл. IX).
Строго говоря, перечисленные растворители IV типа обладают большей
или меньшей протофильностью, растворителей же, которые обладая высокой 8,
характеризовались бы полной инертностью в протолитических равновесиях, не
37
Весьма строгим критерием классификации растворителей,
который позволял бы точно предсказывать характер
кислотно-основного взаимодействия в этом растворителе, являлось бы протонное
сродство. Однако энергия сродства к протону с большей или
меньшей достоверностью определена лишь для весьма незначительного
круга нейтральных молекул.
Вот почему в качестве количественного критерия, который
позволил бы оценить степень участия растворителя в кислотно-основном
протолитическом равновесии, можно применить константу автопро-
толиза растворителя: /Ct=[stjfSi] (например, /Сн2о=[Нз0+] [ОН~];
/CNH8 = [NHt][NHj]; /Ссн4 = [CHt][CHl]). Хотя последние
определены для многих веществ также с недостаточной точностью
(нередко в литературе приводятся данные, различающиеся на 2—4,
а иногда и на 5—6 порядков), в большинстве случаев можно
составить правильное представление о последовательности изменения
этой величины в растворителях одного химического типа
(табл. III. 2).
Таблица Ш.2
Величины pKi некоторых растворителей
Растворитель
сн4
C6H5CH(CH3)2
С6Н5СН3
(С6Н5)2СН2
(С6Н5)3СН
С2Н2
С2Н4С12
ркг
50
37
37
35
33
26
10,1
Растворитель
NH3
C6H5NH2
(C6H5)2NH
С2Н5ОН
СН3ОН
НоО
(СН3)2СО
рК{
32,3
27
23
19,5
16,7
14,0
25,9
Растворитель
СН3СОС2Н5
СН3СОС3Н7
СН3СОС4Н9
СН3СООСбНи
СН3СООС4Н9
СН3СООС2Н5
СН3СООСНз
рк(
25,6
25,3
24,2
24,2
23,3
29,8
22,5
Растворитель
СНдСООН
нсоон
H2SO4
vKt
12,6
6,2
3,1
Среди растворителей одного типа кислотность увеличивается с
уменьшением pKi. Так, дифениламин (р/Сг = 23) обладает по
сравнению с анилином (pKi = 27) настолько более отчетливо
выраженными кислыми свойствами, что будучи смешанным с ним,
проявляет легко регистрируемую протонодонорную функцию.
Аналогично, по мере усиления кислых свойств уменьшаются величины
р/Сг в ряду соединений CH3COOR (от 24,2 для уксусноамилового
эфира до 12,6 для уксусной кислоты).
Величина pKi в аналитической химии часто определяет шкалу
(интервал) кислотности растворителя, так как чем выше для
данного растворителя величина рКи тем больше скачки титрования,
наблюдающиеся при титровании в этом растворителе. Поскольку
определение достоверной величины pKi является непростой
задачей, для составления относительной шкалы кислотности
растворителей можно пользоваться величиной Es — числом милливольт. Эта
величина получается вычитанием потенциала полунейтрализации
38
в данном растворителе (C2H5)4NOH (одно из наиболее сильных
оснований) из потенциала нейтрализации НСЮ4 (одна из наиболее
сильных кислот): £ls = £i/2Ft4NOH — £i/2hoci4 [А. П. Крешков,
Л Н. К а з а р я н, 1967 г.]. В табл. III. 3 приведены величины Е3
некоторых растворителей. Критерием основности растворителя
может служить величина химический сдвиг спектров ЯМР по
отношению к одному эталону и к определенной концентрации (чаще
всего экстраполированный на бесконечное разбавление).
Таблица Ш.З
Величина Es некоторых растворителей
Растворитель
растворитель
Вода
Этиленгликоль
Метанол . . «
Пиридин . . .
Этанол . . . .
Пропанол . «
610
620
660
710
740
760
Амиловый спирт .
Бутанол
Нитробензол . . .
Нитрометан . . .
Диметилформамид
Ацетон
800
830
1070
1140
1150
1350
В табл. III. 4 приведены величины Абоо для ряда соединений в
хлороформенном растворе при бесконечном разбавлении [D.
Norman t, 1967 г.]; величина Абоо растет по мере роста основности
растворителя.
Таблица Ш.4 -
Величины Абоо для некоторых растворителей
Растворитель
Растворитель
Циклогексан
Диоксан
Гексаметилтриамидофосфит
Этиловый эфир
Тетрагидрофуран
Ацетон
0
0,64
0,68
0,70
0,80
0,94
Циклогексанон
Трибутилфосфат
Диметилформамид ...
Диметилсульфоксид ...
Тетраметилмочевина . .
Триамидофосфат («гексаме-
танол»)
1,06
1,28
1,30
1,34
1,37
2,03
Глава IV
МОЛЕКУЛЯРНОЕ СТРОЕНИЕ
И ФИЗИЧЕСКИЕ СВОЙСТВА
НЕВОДНЫХ РАСТВОРИТЕЛЕЙ
Изучение физических свойств жидкостей в широком интервале
давлений и температур создает экспериментальный фундамент для
решения одной из самых общих проблем современной физической
химии —установления связи между строением и свойствами
вещества. Данные о физических свойствах растворителей
необходимы для уяснения механизма влияния растворителя на процессы,
протекающие в растворах, и, что особенно важно практически,
являются базой для расчетов аппаратуры и технологических
режимов многих химических и нефтехимических производств. Однако
накопление таких экспериментальных данных, как правило,
отстает от потребностей науки и техники, в связи с чем существует
настоятельная необходимость в разработке методов расчета
физических свойств растворителей.
Среди существующих методов можно выделить теоретические,
основанные на физически обоснованных корреляциях между
молекулярными и макроскопическими параметрами, эмпирические, в
которых применяется подбор математических зависимостей между
совокупностями значений правильно выбранных свойств, и
полуэмпирические. В последних устанавливаются такие соотношения
между параметрами, которые могут быть обоснованы
теоретически, но для согласования с экспериментом нуждаются во
введении дополнительных эмпирических коэффициентов.
Литература, посвященная упомянутым проблемам, весьма
обширна. Вопросы молекулярного строения жидкостей и оценка
(в простейших случаях) отдельных физических параметров по
данным о свойствах молекул рассмотрены в фундаментальной
монографии Гиршфельдера и др. (1961). Проблемам
молекулярного строения простых жидкостей и растворов посвящены
монографии М. И. Шахпаронова (1956), И. 3. Фишера (1961),
Э. Гуггенгейма (1952), И. Пригожина (1957), Дж. Роулинсона
(1969).
Эмпирические и полуэмпирические методы расчета ряда
физических свойств жидкостей являются предметом книг С. Бретшнай-
дера (1966), Р. Рида и Т. Шервуда (1971).
В этой главе будут даны элементарные сведения из теории
строения жидкостей, необходимые для понимания принципов
современных теоретических методов расчета физических и
термодинамических параметров, а также кратко изложены наиболее
употребительные эмпирические и полуэмпирические методы.
40
|V.1. Молекулярное строение жидкостей
и статистический расчет их свойств
общие представления о строении жидкостей
Большинство современных теорий строения жидкостей
основывается на представлениях о наличии в жидкостях определенной
регулярности в расположении молекул, характеризуемой терминами
«квазикристалличности» и «ближнего порядка». Эти представления
хорошо согласуются с результатами рентгеноструктурных
исследований жидкостей и данными по молекулярному (релеевскому)
рассеянию света. Мысль Дж. Бернала (1961), что жидкость
представляет собой «...существенно нерегулярно построенное скопление
молекул, не содержащее никаких кристаллических участков или
дырок...», не была воспринята большинством исследователей и не
послужила основой для разработки количественных теорий.
Представления о сходстве строения жидкостей и кристаллов
наиболее последовательно развиваются в ячеечной модели
жидкостей. Квазикристалличность здесь понимается в том смысле, что
колебания атомов или молекул жидкости происходят только в
определенном объеме («клетке»), предоставленном частице ее
ближайшими соседями. Этот объем, который формально можно
отождествить с узлом кристаллической решетки, превышает
собственный объем частицы на величину так называемого «свободного
объема». Явление самодиффузии (см. гл. V) в этой модели
объясняется смещением центра колебаний частицы вместе со
смещениями соседних частиц в процессе теплового движения. Многие
исследователи отмечают, что клеточная модель приписывает
жидкостям значительно большую степень упорядоченности по
сравнению с реально существующей. Тем не менее количественные
соотношения, вытекающие из этой модели, во многих случаях
удовлетворительно согласуются с экспериментом. С повышением
температуры, когда ослабляется упорядоченность структуры и
возрастает вероятность перескока частицы из одной ячейки в другую,
расхождение эксперимента с предсказаниями теории существенно
возрастает.
Дырочная модель, наиболее последовательно развитая
Я. И. Френкелем и Г. Эйрингом, основывается на допущении о
существовании в жидкостях свободных полостей («дырок»),
достаточно больших, чтобы молекула могла внедриться в них. При этом
суммарная энергия процесса перемещения молекулы складывается
из энергии образования полости (т. е. энергии, необходимой для
Удаления молекулы, находившейся в месте полости) и энергии
перемещения частицы в образовавшуюся полость. На основе
дырочной модели Г. Эйрингом дано теоретическое обоснование
температурных зависимостей вязкости, электропроводности и
коэффициентов диффузии в водных растворах.
Упомянутые модели строения жидкостей в принципе применимы
как для водных, так и для неводных растворов. Однако здесь
41
представляется уместным лишний раз подчеркнуть уникальные
особенности строения воды, оправдывающие рассмотрение свойств
неводных растворов в отдельной книге.
Вода занимает особое положение в ряду растворителей,
обусловленное созданием за счет Н-связей структуры высокой степени
упорядоченности. Согласно О. Я. Самойлову (1965), четко
выраженный ближний порядок в воде обусловлен тем, что каждая
молекула воды может участвовать в четырех Н-связях, а расположение
атомов в молекуле Н2О энергетически и геометрически
благоприятно для создания тетраэдрического каркаса. При этом тепловое
движение молекул может происходить внутри пустот этого
каркаса, без нарушения его структуры и, следовательно, без
заметного снижения стецени упорядоченности. Такое уникальное
свойство воды как повышение плотности с температурой в интервале
О—4°С также связано со структурной особенностью — переходом
открытой льдоподобной структуры в более упакованную структуру.
Как пишут К. П. Мищенко и Г. М. Полторацкий (1968), «в
делении на водные и неводные растворы ведущими факторами
являются неповторимая структура воды и специфика сил ближнего
действия в ней». Детальное описание особенностей строения воды
имеется в упомянутой книге К. П. Мищенко и Г. М. Полторацкого,
книге Р. А. Робинсона и Р. X. Стокса «Растворы электролитов» и
в специальной монографии [D. Eisenberg, D. Kauzmann,
1969].
Статистический подход к расчету свойств жидкостей
Основная задача статистического описания жидкого
состояния— представление структуры жидкости и расчет ее макросвойств
через молекулярные параметры, т. е. величины, характеризующие
размеры и форму молекул, а также энергию межмолекулярного
взаимодействия. Наибольших успехов теория достигла в расчете
свойств простых жидкостей, состоящих из частиц с шаровой
симметрией и нулевым дипольным моментом.
Простую однородную жидкость можно представить как систему,
состоящую из очень большого числа N одинаковых частиц
(атомов или молекул), распределенных в данном объеме. Если
допустить, что известны координаты центра инерции (#;) и
соответствующие импульсы (pi) частицы (/ — число степеней свободы),то
функция Гамильтона H(p,q), равная сумме кинетической и
потенциальной энергии системы, определяется уравнением:
Н (</, р) = 2 {P2il2m) + un M (IV. l)
где m —масса частицы; Un— потенциальная энергия.
Практически, разумеется, невозможно определить координаты
и импульсы каждой отдельной частицы, но задача сводится лишь
к оценке вероятности dW(qy p) наличия у частцц системы коорди-
42
нат и импульсов, близких к заданным. Эта вероятность дается так
называемым каноническим распределением Гиббса:
dW (q, р) = dq dpZ^h-™ exp [- Я (р, q)/kT] (IV. 2)
Величина Zn называется статистическим интегралом системы;
из этой величины может быть вычислена свободная энергия
F = — kT\nZN (IV. 3)
и следовательно, другие термодинамические параметры системы.
В статистической механике показано, что общее каноническое
распределение Гиббса можно разделить на два независимых
распределения— по координатам и по импульсам. В распределение по
координатам входит конфигурационный интеграл QNj связанный
со статистическим интегралом соотношением:
qn = ZNN\ (h2l2nmkTfNl2 (IV. 4)
Вместо гамильтониана H(q,p) вводится потенциальная
энергия, являющаяся функцией только координат. Таким образом,
задача расчета термодинамических функций сводится (при условии,
что известна масса частиц) к нахождению вероятности их
распределения в заданном объеме.
В случае раствора, состоящего из п компонентов, химический
потенциал каждого из которых \ц, вероятность распределения
частиц описывается с помощью так называемого большого
канонического ансамбля Гиббса
NQ\ Z
exp
- H fa>
kT
(IV. 5)
и задача расчета термодинамических функций опять-таки сводится
к вычислению статистического интеграла Z^.
Расчет статистического и конфигурационного интегралов
является весьма сложным и требует введения определенных
упрощающих допущений:
потенциальная энергия может быть представлена в виде
аддитивной суммы потенциалов взаимодействия пар частиц;
взаимодействие каждой пары частиц зависит только от
расстояний между их центрами.
Первое допущение достаточно хорошо оправдывается,
по-видимому, только для сжиженных благородных газов, хотя и в этом
случае, как следует из квантово-механических расчетов, в
конденсированной системе из большого числа частиц должен появиться
дополнительный «коллективный» член в энергии взаимодействия,
зависящий от взаимного расположения частиц. В системе же,
состоящей из дипольных частиц, вследствие индуктивного влияния
полей соседних диполей на диполь частицы отклонения от
аддитивности энергии должны возрастать весьма существенно.
.43
Второе допущение может быть оформлено математически в виде
выражения для энергии межмолекулярного взаимодействия.
Сводка различных формул для расчета этой энергии дана в
упомянутой монографии Гиршфельдера и др.; для неполярных молекул
наиболее известно уравнение
Леннард— Джонса:
В этом уравнении & —
постоянная, имеющая размерность энергии
и определяющая глубину
потенциальной ямы на диаграмме Ur(r)
(рис. IV. 1); а—эффективный
поперечник частицы, который может
быть определен по величине Гщщ
(см. рис. IV. 1) как a=rmin-2-1/e.
Величина Slk называется силовой
константой Леннард — Джонса;
значения ее измерены для ряда веществ
в газообразном состоянии. Первый
член в уравнении (IV. 6) характеризует отталкивание, второй —
притяжение молекул.
Для описания взаимодействия полярных молекул используют
потенциал Штокмайера:
Рис. IV. f. Графическое
представление потенциала Леннард—
Джонса.
[()()]
X [2 cos 8A cos 9В — sin 0A sin 9B cos (фв — фА)] (IV. 7)
Здесь 6а, Эв — углы между осями диполей молекул А и В и
линией АВ, соединяющей центры молекул; фл и фв — углы поворота
соответствующих диполей относительно координатной оси,
перпендикулярной к АВ.
Математические основы современной теории расчета
молекулярных функций распределения сформулированы Кирквудом,
Н. Н. Боголюбовым, Борном и Грином. В этих теориях прямое
суммирование с целью вычисления QN заменяется расчетом
вероятности конфигураций групп из двух, трех и более частиц, а
физической основой теории служит решеточная модель жидкости.
Вероятность обнаружения некоторой частицы на расстоянии
от г до г + dr до другой частицы в объеме V определяется
соотношением
dW (г) = g (r) Am2V~l dr (IV. 8)
где g(r)—так называемая радиальная функция распределения.
Важной особенностью функции g(r) является то, что она
может быть определена экспериментально из рентгеновских, нейтро-
44
и электронографических исследований, а также из данных по
оеаеевскому рассеянию света. Пики на кривой g(r) указывают на
наиболее вероятные расстояния между ближайшими частицами в
жидкости; по положению и интенсивности этих пиков могут быть
определены средние координационные числа.
В статистической теории жидкостей получены выражения,
связывающие g(r) с термодинамическими свойствами. Например,
изотермическая сжимаемость жидкости рг = — у у— -щ^)т
определяется уравнением:
[
$ +
l
Величину рт часто применяют для определения характера и
стехиометрии межмолекулярного взаимодействия в растворах.
В настоящее время развитие методов математической
статистики и применение ЭВМ позволяет рассчитывать функции
распределения не только для простых жидкостей, но и для растворов.
Однако вычисленные на этой основе физические и
термодинамические параметры существенно расходятся с экспериментальными.
По-видимому, причина этого состоит не в недостаточности
математического аппарата, а в неадэкватности решеточной модели
истинной физической структуры жидкостей. В этом смысле
важным шагом на пути приближения к реальной модели является
учет отклонений поведения жидкостей от средневероятностного
распределения: флуктуации плотности и ориентации в
индивидуальных жидкостях и флуктуации концентрации в растворах
[М. И. Шахпаронов, 1956]. Упомянутый выше коэффициент
изотермической сжимаемости может быть определен через
флуктуации плотности (Дй) с помощью относительно простого
соотношения:
h.<^ „V.,0,
Оценка величин флуктуации и анализ с их помощью
молекулярного строения растворов рассмотрены в работах М. И. Шахпа-
ронова (1956, 1963).
Сказанное выше позволяет заключить, что расчет макрофизи-
ческих свойств жидкостей на основании данных о свойствах
молекул не дает в подавляющем большинстве случаев
удовлетворительной сходимости с экспериментом. И. 3. Фишер (1961),
упоминая о возможности расчета коэффициентов вязкости, диффузии и
теплопроводности жидкостей из молекулярных функций
распределения, не приводит в своей монографии соответствующих
количественных соотношений. Расчетам поверхностного натяжения
^идкостей посвящена специальная монография С. Оно и С. Кондо
(1963), где удовлетворительная сходимость теории с
экспериментом продемонстрирована лишь для сжиженных благородных газов.
45
Некоторые эмпирические соотношения между параметрами урав-
нения (IV. 6) и макрофизическими свойствами жидкости
(критические температуры, давление и объем, а также мольный объем при
температуре кипения) приведены в книге С. Бретшнайдера (1966).
IV. 2t Полуэмпирические методы расчета
физико-химических свойств
Большинство используемых в настоящее время растворителей
являются органическими веществами, и расчет их свойств может
быть осуществлен, если известны соответствующие данные для
соединений— аналогов, принадлежащих к одному гомологическому
ряду. В основе расчетов лежит принцип химического подобия
веществ, сформулированный В. А. Киреевым., Общие принципы
сравнительных расчетов подробно описаны в монографии М.Х. Ка-
рапетьянца (1965).
Полуэмпирические методы расчета базируются на принципах:
химического сходства в рассматриваемом ряду соединений,
аддитивности и конститутивности изучаемого свойства. Последнее
означает, что величину свойства можно представить для данного
вещества в виде суммы инкрементов (составляющих), присущих
отдельным атомам или атомным группам и связям. Строго
аддитивной величиной является, например, молекулярная масса.
Мольный объем является аддитивной и конститутивной величиной, т. е.
он зависит от строения молекул.
Применительно к алканам методы расчета физико-химических
свойств в гомологических рядах были рассхмотрены В. М. Татев-
ским (1953, 1960).
Согласно первому методу, величину свойств представляют в
виде суммы инкрементов связей, имеющихся в молекуле данного
вещества. Величина же инкремента связи С—С или С—Н зависит
от природы окружающих связь соседних атомов или атомных
групп. Иными словами, инкремент связи С—Н в концевой группе
углеводорода —СН3 отличен от инкремента для групп —СН2—
или —СН— точно так же, как, отличаются инкременты связей
С—С в зависимости от того, к какой группе (метильной, метиле-
новой, метановой) относится данный углеродный атом.
Зная структуру углеводорода, можно определить число всех
возможных связей различных типов и, суммируя их вклады
(вычисленные ранее из сравнения свойств соответствующим образом
подобранных веществ), найти величину аддитивного свойства для
данного вещества.
Описанный метод расчета применяют для вычисления таких
свойств углеводородов, как мольный объем и мольная рефракция,
мольная теплота испарения (AHV) для различных температурных
интервалов, теплоты образования соединений из свободных
атомов, теплоты и свободные энергии образования из элементов,
теплоты сгорания.
46
Второй метод расчета по Татевскому заключается в
рассмотрении изменения свойства в ряду однотипных соединений в
зависимости от числа п углеродных атомов. Это изменение описывается
линейным уравнением вида
(IV. И)
где ат — постоянная величина для т-и группы алканов, а р
одинаково для всех групп алканов. Величины свойств у,
удовлетворяющие расчету по первому методу, пригодны для использования
и в уравнении (IV. И).
Третий метод расчета заключается в сопоставлении значений
различных физических свойств одной и той же гомологической
группы соединений. Линейная зависимость соблюдается обычно
при сопоставлении свойств, каждое из которых в отдельности
подчиняется уравнению (IV. И).
В работах В. М. Татевского показано, что при расчете
физических свойств даже таких простых соединений, как насыщенные
углеводороды, необходимо учитывать детали структуры.
Значительно сложнее оценить значения физических свойств полярных
жидкостей. Заметный прогресс в этой проблеме достигнут в
работах А. Е. Луцкого, который рассмотрел некоторые зависимости
между макрофизическими свойствами жидкостей и свойствами
микрочастиц. По А. Е. Луцкому и Е. М. Обуховой (1961), в
гомологических рядах, начиная с третьего или четвертого члена,
остаются практически постоянными дипольный момент молекул и
фактор Ф, характеризующий форму молекул, а изменяются
масса т, геометрический размер г и поляризуемость а молекул.
Рассмотрение зависимости макрофизического свойства от одного из
перечисленных выше молекулярных свойств приводит к ценным
расчетным соотношениям. Например, для нескольких гомологиче-
ских рядов мольная рефракция R = —■$ , взятая в
некоторой ^дробной степени (0,8-f-1,7), линейно зависит от функций
Ц Ум , (е — 1)(г • 1024)3/2 и уг2(з (г — диаметр молекулы).
Каждая такая зависимость характеризуется определенной величиной
показателя степени при /?, который может также измениться при
переходе к другому классу соединений.
Методика расчета, разработанная А. Е. Луцким, не требует
постулирования аддитивности и конститутивности исследуемого
физического свойства и, следовательно, данных об инкрементах
свойства, отнесенных к атомным группам и связям. Необходимо лишь
знать значения свойства (Уг)р,т,ц,ф двух членов гомологического
ряда, начиная с третьего или четвертого, чтобы затем определить
экстраполяцией свойство у г для других членов этого же ряда (для
первых членов ряда экстраполяция обычно ненадежна). В работах
А. Е. Луцкого не вводится количественное выражение для
фактора Ф, учитывающего форму молекул,
47
Удобный способ одновременного рассмотрения размеров и
формы молекул с привлечением понятия о среднеквадратичном
радиусе г2 молекулы предложен Альтенбургом [К. Altenburg, 1961].
Если в молекуле имеется N материальных точек (атомных групп)
с п\ числом одинаковых расстояний между группами, то
где z — число связей в молекуле; а\ — величины расстояний.
Например, для 2,3-диметилбутана
5
1
1
1
2
о
6
л
4
можно найти число кратчайших расстояний между группами
щ = 5 (1—2; 2—3; 3—4; 2—5; 3—6), число расстояний между
группами, разделенными одной группой п2 = 6 (1—3; 1—5; 2—4;
3—5; 4—6); число наиболее длинных возможных расстояний п3=4
(1—4; 1—6; 5—4; 5—6). Для этого случая уравнение (IV. 12)
принимает вид:
г2 = (БД? + Ьа\ + 4<2з)/36 (IV. 12а)
В работах Альтенбурга приведены эмпирические соотношения,
связывающие г2 в гомологических рядах соединений с мольной
рефракцией, температурой кипения, поверхностным натяжением
и другими свойствами жидкостей.
IV. 3. Аддитивные и конститутивные свойства жидкостей
Из аддитивных и конститутивных свойств наиболее широко
известны: мольный объем VM = M/d; парахор Ру = Му1/4/(^ж—dn)
и мольная рефракция /?м = M(n2D— \)jd{n2D + 2) [или M/(nD— l)dl
В литературе встречаются также:
ортохор — Or = M (dTlr\Tx — ^г2т1г2)/(лл — т)г2) [Н. Д. Литвинов,
1961];
реохор — Rh = M(l(h\)4*/(dm + 2dn) [J. N. F r i e n d, 1942];
константа Джоши — RT = MnD/d In Tmm [D. P. Josh i, 1962];
мольная скорость звука — Ru = ul/3M/dm [M. R. R a o, 1940] и др.
Инкременты для расчета большинства из них приводятся в
книге С. Бретшнайдера.
Указанные величины можно использовать либо для
установления строения молекул вещества, либо для расчета физических па-
48
раметров (например, поверхностного натяжения по парахору,
вязкости по реохору и т. д.). Вопрос о том, для какой величины
лучше соблюдается структурная аддитивность, обсуждался в
работах Экснера [О. Ехпег, 1967], который на основании
статистического анализа экспериментальных данных пришел к выводу,
что не найдено ни одной функции, для которой аддитивность
соблюдалась бы лучше, нежели для мольного объема. Для ортхора
и реохора эта аддитивность выполняется хуже.
Что же касается точности расчетов физических свойств по
структурно-аддитивным константам, то она в большинстве
случаев будет заведомо низкой, особенно если изучаемый параметр
входит в формулу в дробной степени (вязкость — реохор и т. п.).
Определение плотности по мольному объему, рассчитанному из
инкрементов, по-видимому, может быть выполнено с
удовлетворительной точностью; величины инкрементов приведены в табл. IV. 1.
Таблица IV.1
Аддитивные инкременты мольного объема при 20 °С
[О. Ехпег, Coll. Czech. Chem. Comm., 32, 1 (1967)]
Атомы, группы, связи
Доля FM,
смЗ/моль
Атомы, группы, связи
Доля VM,
смЗ/моль
сн2
СНз
н2с=сн
нс^с
сбн5
F )
С1 '
gj; } первичные
I J
ОН (первичный спирт)
Связь:
двойная
тройная
16,58
31,48
42,48
33,22
74,65
15,11
22,96
26,19
32,93
40,25
24,22
44,76
О (эфир)
СО (кетон)
соон
н
— NH2
NH
\N—(третичный амин) .
-CN
--Ш2
Шестичленное алицикличе-
ское кольцо .....
6,74
10,21
27,24
14,90
17,67
7,30
5,09
22,67
24,51
9,34
IV. 4. Эмпирические способы расчета
физико-химических свойств жидкостей
Здесь рассматриваются лишь те физические свойства
растворителей, для которых теоретически установлена взаимосвязь с
параметрами процессов, протекающих в растворах. Так, известно,
что скорость ионных и ион-дипольных реакций зависит от
диэлектрической проницаемости, а скорость реакций, контролируемых
Диффузией, от вязкости растворителя. Теория растворов
неэлектролитов использует параметры, вычисляемые из теплот
испарения или поверхностного натяжения (см. гл. V). Важнейшие
термодинамические функции растворов вычисляются по данным
49
о давлении насыщенного пара над ними. Такие свойства как
плотность (мольный объем), вязкость, электропроводность,
показатель преломления и др. используются при изучении равновесий
в растворах.
Представляют интерес как эмпирические соотношения для
вычисления значений свойства по данным для других свойств, так
я зависимости свойства от температуры и давления в области
существования жидкого состояния.
Плотность
Измерения плотности легко осуществимы экспериментально, и
практически для всех жидкостей известны значения плотности
хотя бы при одной температуре. Температурный коэффициент
плотности ad = [{d\/d2)—1]/(^2— ^i) незначительно увеличивается
с ростом температуры, причем для неполярных жидкостей в
определенном температурном интервале соблюдается приблизительно
линейная зависимость а от /. Для большого числа жидкостей в
широком интервале температур данные по плотности могут быть
с достаточной точностью аппроксимированы степенным
уравнением
df = do-a'*-p'/2- (IV. 13)
где а! и р7 — эмпирические коэффициенты.
По зависимости плотности (точнее, связанного с ней мольного
объема) от давления определяют коэффициенты сжимаемости:
_, о г» 1 / dVu \ / Г» \
адиабатический ps = — -tj— —^— ) (о—энтропия);
Ум \ OP /S
«о I
изотермический рг = — y-lj
С коэффициентом ps и скоростью звука, в жидкости и
плотность связана соотношением
d = 1/«2р5 (IV. 14)
Определение координационных чисел в жидкостях на основе
данных по плотности рассмотрено в работах Г. М. Панченкова и сотр.
(1964).
Показатель преломления
Расчетные методы определения показателя преломления nD
обычно не имеют большого значения, поскольку эта величина
легко определяется экспериментально. Отметим, однако, что этот
параметр может быть найден из значений плотности для одного
и того же вещества при различных температурах по уравнению
мольной рефракции (см. выше) или (более точно) по формуле
Эйкмана
• (4~0/И + °>4И = с (IV. 15)
где С — константа для данного веществу.
ьо
Показатель преломления, как и плотность, уменьшается с
ростом температуры. Для большинства жидкостей в интервале тем-
пеоатур, близких к комнатной (15—30 °С), величина темпе-
пятуоного коэффициента показателя преломления составляет
Вязкость
В качестве меры «вязкости» (динамической) обычно
используют коэффициент вязкости (внутреннего трения) ц в уравнении
F = i\S-42- (IV. 16)
связывающим силу F, которую надо приложить к слоям
жидкости площадью S, чтобы сообщить им относительно друг друга
скорость сдвига с градиентом dz/dl. Уравнение справедливо в
области ламинарного течения для большого числа органических и
неорганических жидкостей. [Для так называемых
структурированных жидкостей, в частности, растворов и расплавов полимеров,
уравнения (IV. 16) не применимо].
Наряду с динамической вязкостью используют такие
модификации, как текучесть ср = 1/г), кинематические вязкость v = r\/d
и текучесть d/ц.
Известны способы расчета вязкости жидкостей с помощью
структурно-аддитивных констант: упомянутого выше реохора,
величины lgr]*, константы В в уравнении (IV. 17) [L. H. Thomas,
1946]:
\g ц = 0,5 lg d + В (ГКр - Т)/Т - 0,933 (IV. 17)
Точность этих методов невысока и применимость весьма
ограничена.
Вязкость почти всех известных жидкостей уменьшается с
ростом температуры (к одному из очень немногих исключений
относится жидкая сера) и, за немногими исключениями, возрастает
с повышением давления, причем при давлениях ниже 40 атм
влияние давления на вязкость практически не сказывается. Вязкость
большинства органических растворителей находится в пределах
(0,5—10) -Ю-3 (н-сек)/м2 (0,5—10сп).
В ряде монографий, посвященных теориям вязкости (Э. Гат-
ч е к, 1935; Г. Б а р р, 1938; Г. М. П а н ч е н к о в, 1947 и др.], даются
сводки уравнений, описывающих зависимость вязкости от
температуры. (Среди позднейших сводок следует упомянуть обзор
Браша [St. G. Brush, 1962]). Эти уравнения можно подразделить
на чисто эмпирические, имеющие вид полиномов с
несколькими коэффициентами, и уравнения экспоненциального вида,
Применимо для неразветвленных соединений, не содержащих гидро-
ксильных групп [О. Е х п е г, 1967].
51
полученные на основе представлений дырочной теории жидкостей.
Зависимость
Л = А ехр (B/RT) (IV. 18)
найденная де-Гусманом (1913) как эмпирическая, была
обоснована в работах Я. И. Френкеля, Г. Эйринга, Г. М. Панченкова
и др., так что коэффициент В в уравнении (IV. 18) можно
трактовать как энергию активации вязкого течения Ё1.
Эйринг показал, что предэкспоненциальный множитель А
зависит от температуры и может быть выражен через мольный
объем и энергию испарения
з yuV2T8/2
Л = 1,09- 10"" ,, (IV. 19)
V*hEv
причем величина VM выражена в см?/моль. Ev— в кал/моль.
Величина £а может быть оценена для большого числа жидкостей/
исходя из допущения Эйринга, что Ev\El« 2,5. Для полярных
жидкостей с сильными водородными связями это допущение не
соблюдается.
Наличие определенной зависимости между Еа и структурно-
аддитивной величиной — мольным объемом — приводит к
естественному предположению, что величина Е& также может быть
представлена в виде суммы инкрементов отдельных групп и
связей. Подробно этот вопрос рассмотрен в работе А. А. Абрамзона
и М. В. Островского (1968).
Поверхностное натяжение
Величина поверхностного натяжения у, выражаемая в н/м,
численно совпадает с величиной поверхностной энергии (в дж/м2),
т. е. энергии, которую необходимо сообщить жидкости для
увеличения ее поверхности на единицу. Учитывая, что поверхность
сферы пропорциональна ее объему в степени 2/з, иногда
используют понятие о мольной поверхностной энергии уУ2^\ имеющее
вполне определенный физический смысл при условии, что
молекулы имеют сферическую форму. Установлены закономерности
изменения у^м3 в гомологических рядах [А. Е. Луцкий,
Е. М. Обухова, 1961] и эмпирическое соотношение между yV2^
и давлением насыщенного пара жидкости [С. А. Войткевич,
1964] .
lg p = ayV^3 + Ь (IV. 20)
где а и Ъ — постоянные в пределах гомологического ряда.
Поверхностное натяжение жидкости уменьшается с
температурой, падая до нуля при критической температуре. Среди
соотношений, описывающих эту зависимость, наиболее известны:
52
уравнение Этвеша
K{TT)jV^ (IV. 21)
уравнение Ван-дер-Ваальса:
у = уо(1-Т/Ткр)п (IV. 22)
Константы уо и п уравнения (IV. 22) сохраняют постоянство
в пределах одного класса соединений.
Из уравнений (IV. 21) и (IV. 22) может быть получено
уравнение Фергюсона
0 ~
где />ф==2,1 для неассоциированных жидкостей. В области
температур, далеких от критической, достаточно точно соблюдается
линейная зависимость у = f{T).
Среди эмпирических соотношений, связывающих у с другими
свойствами жидкостей, наиболее известен парахор (см. выше).
Установлены также корреляции у с вязкостью [Н. Schonhorn,
1967], коэффициентом изотермической сжимаемости [С. А. Ми-
хайленко, В. В. Пашков, 1970] и другими свойствами.
Давление пара и теплота испарения
Рассматриваемые величины имеют первостепенное значение
для расчета термодинамических свойств жидких
неэлектролитов. Методы измерения этих величин описаны, например, в
известной книге А. Вайсбергера «Физические методы органической
химии».
Теплоту испарения жидкости находят либо из
калориметрических данных [для большого числа жидкостей такие измерения в
последнее десятилетие выполнены Вадсо [Wadso], либо по
температурной зависимости давления насыщенного пара,
описываемой известным уравнением Клапейрона — Клаузиуса:
In р = — &HV/RT + const (IV. 24)
Уравнение (IV. 24) выведено в предположении, что теплота
испарения не зависит от температуры, пар подчиняется законам
идеальных газов, а объемом жидкости можно пренебречь в
сравнении с объемом газа. Несоблюдение указанных условий
приводит к отклонению от предсказываемой уравнением (IV. 24)
линейной зависимости \gp (1/7"). Более точно температурная
зависимость давления пара описывается уравнением Антуана:
lg р = А - В/(С + Т) (IV. 25)
Константа С для большого числа соединений близка к 230,.
константы А и В характерны для каждого данного соединения.
Поэтому уравнения (IV. 24) и (IV. 25) непригодны для расчета
Давления паров при отсутствии экспериментальных данных.
53
Уравнение (IV. 20) может быть использовано для вычисления р пс
плотности и поверхностному натяжению какого-либо соединения
если известны все параметры этого уравнения хотя бы для дву^
соединений-аналогов.
При отсутствии данных о давлении пара теплоты испарения
могут быть вычислены по эмпирическим уравнениям через тем.
пературу кипения. Для неполярных веществ AHV определяют пс
формулам:
Кистяковского —
ЛЯ^кип = 36'ЗГ*ип + 19>14Гкип 1* Гкип (IV. 26)
Скотта —
ДЯ *98 = 99,23ГКИП + 0,0837Г2ип - 12 350 (IV. 27)
[Коэффициенты уравнений (IV. 26) и (IV. 27) даны для
вычисления AHV в дж/моль].
Известен также способ расчета АНт с помощью
структурно-аддитивной константы — лиопарахора:
Инкременты для вычисления лиопарахора приводятся в
монографии Рида и Шервуда.
Наряду с теплотой (энтальпией) испарения часто используют
энергию испарения Ev. Эти величины связаны соотношением:
-pAV = AH*- RT
Для неполярных жидкостей (алканов) установлено простое
соотношение между энергией испарения и плотностью:
Ev = k(d- dKP) (IV. 28)
Здесь k — постоянная для данного соединения; dliV — плотность
его при критической температуре [A. A. Miller, 1964].
В обзоре Голда и Огла [Ph. I. Gold, G. J. Ogle, 1969] дана
сводка уравнений для расчета AHV по другим свойствам
(температура кипения, критические константы), а также для расчета
AHV при данной температуре по значениям АНтк . Все эти
уравнения включают эмпирические коэффициенты, постоянство
которых сохраняется лишь для, отдельных групп соединений.
Достоверно известно лишь то, что AHV уменьшается с ростом
температуры и обращается в нуль при критической температуре.
Диэлектрическая проницаемость и дипольный момент
Сведения о диэлектрических проницаемостях и дипольных
моментах неводных растворителей весьма существенны для понима-<
ния влияния растворителей на равновесие и скорость
протекающих в них процессов. Детальное обсуждение понятия о дипольных
моментах в газовой фазе и в растворе, а также методов их изме-
54
пения приводится в монографии В. И. Минкина, О. А. Осипова и
{п А Жданова (1968). Вопросы измерений диэлектрической про-
тшаемости также освещены достаточно, например в монографиях
Смайса [С. P. Smyth, 1955] и Ф. Эме (1967).
Обычно приводимые в справочниках значения диэлектрической
проницаемости представляют собой величины, найденные путем
измерения емкости конденсатора, заполненного исследуемым
веществом, на частоте 106—10s гц. Это — так называемая
статическая, или низкочастотная диэлектрическая проницаемость es-Связь
между es и дипольным моментом детально рассмотрена в
литературе.
Для полярных жидкостей наиболее обоснованным считается
уравнение Кирквуда с уточнениями Харриса и Алдера
(е5 - l)/(es + 2) = -| nNoa + 4nN0gn2Es/kT (2es + 1) (es + 2) (IV. 29)
где а — поляризуемость; No — число Авогадро; g — фактор
корреляции; k — постоянная Больцмана, а также уравнение Сыркина —
Осипова:
[flM (й)2] (IV-30)
Г* Эйрингом и сотрудниками (1966, 1967) получено уравнение,
пригодное для жидкостей с Н-связями:
^^] <■*•■>
где Ум — мольный объем «твердоподобной» структуры жидкости,
/ — фактор, близкий к единице. Дифференцированием этого
уравнения по температуре получено [J. Fexa и др., 1971] выражение
для температурного коэффициента es через температурные
коэффициенты плотности и показателя преломления.
Зависимость es от температуры описывается рядом
эмпирических уравнений, из которых наиболее часто используется
экспоненциальная форма
Bs = а ехр (— ЬТ) (IV. 32)
где а и Ь — величины постоянные для данной жидкости.
Для подавляющего большинства жидкостей es уменьшается
с температурой и лишь в отдельных случаях (например, для
масляной кислоты) найден положительный температурный
коэффициент 8S.
Достаточно надежные корреляции между es и другими
свойствами отсутствуют. Известно, что es не зависит непосредственно
°т массы молекул, растет с числом молекул в единице объема и
с увеличением поляризуемости а. В рядах соединений, молекулы
которых обладают примерно равной a, es обратно
пропорционально V% [А. Е. Луцкий, Е. М. Обухова, 1961),
55
Дипольный момент любого вещества измеряют либо в газовой
фазе (|ыо), либо в разбавленном растворе этого вещества в каком-
либо растворителе (jli3), по возможности химически инертном
относительно исследуемого вещества. Найдено [А. Е. Луцкий,
В. А. Г р а н ж а н, 1968], что и при отсутствии специфического
взаимодействия эффективный дипольный момент исследуемого
вещества изменяется от одного растворителя к другому. Интересно,
что в неполярном растворителе (например, декалине) \хэ
нитробензола практически не изменяется с температурой, а в полярных
растворителях (хлорбензоле, ацетофеноне) возрастает с
температурой в интервале 25—100°С примерно на 15%.
Электропроводность
Данные по удельной электропроводности (%) чистых неводных
растворителей весьма скудны и часто недостоверны. Это
объясняется, отчасти, тем, что подавляющее большинство
растворителей обладает весьма низкой электропроводностью (к <<
< Ю~7 ом-1см~1)у которую не всегда удается измерить с
достаточной точностью. Гораздо важнее, однако, то обстоятельство, что
электропроводность чрезвычайно чувствительна к примесям,
содержание которых столь мало, что не влияет на значения других
физических констант вещества. Это обусловливает относительную
неточность и ненадежность большинства приводимых в
литературе данных по электропроводности чистых растворителей, за
исключением, по-видимому, воды и обладающих заметной
проводимостью (10~2—10~4 омгх • см~1) некоторых минеральных кислот. На
основе качественных соображений о способности соединений
данного гомологического ряда к отрыву протона в процессе
автоионизации можно объяснить, например уменьшение
электропроводности в рядах алифатических спиртов и карбоновых кислот. Поиски
количественных корреляций пока не имеют реального физического
обоснования.
Зависимость электропроводности от температуры в общем
случае носит сложный характер. Для чистых жидкостей
температурный коэффициент электропроводности является обычно
положительной величиной, однако в ряде случаев возможны и отрица-
тельные значения -~г (см. стр. 158). Теория абсолютных
скоростей реакции по Эйрингу обосновывает экспоненциальную
зависимость электропроводности от температуры:
х = а' ехр (— Е1\КГ) (IV. 33)
Поскольку отрицательное значение энергии активации
электропроводности не имеет физического смысла, теория Эйринга не
предусматривает возможность отрицательного температурного
коэффициента электропроводности.
ТЕРМОДИНАМИКА НЕВОДНЫХ РАСТВОРОВ
Глава V
ТЕРМОДИНАМИКА
РАСТВОРОВ НЕЭЛЕКТРОЛИТОВ
В числе проблем, разрабатываемых термодинамикой растворов
вообще, для неводных растворов специфический интерес
представляет определение термодинамических функций смесей на
основании значений молекулярных параметров или макрофизических
свойств исходных компонентов, а также установление корреляции
между термодинамическими характеристиками процесса
растворения и природой растворителя. Последний вопрос, строго говоря,
не должен был бы рассматриваться в книге, посвященной
процессам, протекающим в гомогенных системах. Однако в данном
случае, по мнению авторов, исключение оправдывается как
специфичностью, так и практической важностью проблемы.
V. I. Классификация неэлектролитов.
Основные определения
Существующие системы классификации индивидуальных
неэлектролитных жидкостей учитывают прежде всего форму и
полярность составляющих их молекул (другие системы
классификации см. в гл. III). Роулинсон (1969) выделяет пять основных
классов неэлектролитов:
1. Сжиженные благородные газы, обладающие одноатомными
сферически симметричными молекулами;
2. Вещества, состоящие из гомоатомных двухатомных молекул
(Н2, О2, N2, Гал2) и гетероатомных двухатомных молекул с
пренебрежимо малыми дипольными моментами (СО);
3. Низшие углеводороды и другие простые неполярные
вещества (ССЦ);
4. Простые неполярные жидкости (SO2, НГал);
5. Жидкости с молекулами большой полярности и
электрической асимметрии (вода, спирты, кетоны, амины, нитрилы, аммиак).
Смеси, образованные веществами первых трех классов или
одного гомологического ряда для всех классов, характеризуются,
как правило, весьма слабым межмолекулярным взаимодействием.
67
Взаимодействие молекул одного сорта, характеризующее
склонность вещества к ассоциации, проявляется значительно лишь для
жидкостей пятого класса.
Межмолекулярное взаимодействие в растворах может быть
охарактеризовано с помощью величин, определяющих изменение
энтальпии и энтропии в результате смешения компонентов.
Достоверное определение этих величин методами статистической теории
жидкостей осуществимо пока лишь в случае простейших смесей
(например, образованных веществами первого класса). Для
оценки энергии взаимодействия из экспериментальных данных
необходимо проводить сопоставление с эталоном, в качестве которого
принят так называемый идеальный раствор.
Как известно, условием идеальности раствора является
подчинение его закону Рауля: fs =/°(1 — *а)[/* п fs~~ фугативности
(термодинамические летучести) растворителя S, содержащего
растворенное вещество А и не содержащего его; хА — молярная доля].
Характерными признаками идеального раствора являются
равенство нулю теплоты смешения и равенство общего объема смеси
сумме объемов компонентов при той же температуре.
Изменения изобарного потенциала AGCM и энтропии Д5СМ при
смешении компонентов, образующих идеальный раствор, даются
выражениями
HG^RT^ntlnxt (V.I)
i
Д5вд' *2*iln*l (V.2)
где щ — число молей i-ro компонента в растворе.
Для самопроизвольного процесса, каким является идеальное
смешение, всегда Дбид < 0 и AS«a > О-
Разность между значением термодинамической функции 2СМ
для исследуемого и для идеального раствора называют
избыточной термодинамической функцией Ze = ZCM — Z^. В идеальном
растворе по определению величины Ge, He9 Se и Vt равны нулю.
Гильдебранд выделил класс регулярных растворов, при
образовании которых Se = 0, a Ge и Не отличны от нуля. Определению
регулярных растворов удовлетворяют многие реальные смеси
жидкостей с неполярными молекулами. Это обусловило
применимость развитой Гильдебрандом, Скэтчардом, Скоттом и другими
исследователями теории регулярных растворов для многих
практических расчетов.
Атермальными растворами называют такие, для которых
Не = 0, a Ge и Se отличны от нуля. Эти смеси, наряду с
регулярными, занимают промежуточное положение между идеальными и
реальными растворами; в последних все перечисленные
избыточные функции отличны от нуля*
D8
V 2 Теория неэлектролитных растворов
Молекулярно-статистические теории
Расчеты термодинамических функций неэлектролитных смесей
методами статистической теории жидкостей выполнялись многими
исследователями. Здесь следует прежде всего упомянуть
фундаментальные работы Е. Гуггенгейма, И. Пригожина, Дж. Роулин-
сона. В этих расчетах постулирована ячеечная модель жидкостей
и универсальная форма потенциала межмолекулярного
взаимодействия: wt7===^/(|)(riy/r*/)> где ^ — расстояние между центрами
молекул i и /, <S]. и r*tj—характеристические параметры этой
пары молекул, а Ф^^/^/)— универсальная функция.
Выбор конкретного вида универсальной функции (сводку этих
функций см., например, в монографии Гиршфельдера и др. 1961)
далеко не очевиден. Часто используют уравнение
Леннард—Джонса [уравнение (IV. 6), стр. 44], принимая, что для молекул
компонентов 1 И 2 012 = (tfl + 02) /2 И <^12 = (<^l<^2) Va. РоуЛИНСОН
[J. S. Rowlinson, 1967] считает, однако, что лучшие результаты
достигаются с применением ^потенциала Перкуса — Йевика
[J. К. Р е г с u s, G. J. Y e v i с к, 1958].
Молекулярно-статистическая теория позволяет оценить с
привлечением данных о размерах молекул компонентов вероятность
распределения молекул в узлах квазирешетки. В ряде работ с
помощью современной вычислительной техники выполнены расчеты
функции распределения и конфигурационного интеграла (раздел
IV. 1), что позволяет определить термодинамические функции
смесей [N. A. Gokcen, E. Т. Chang, 1971]. Найденные в этой
работе расчетные и экспериментальные значения Не расходятся
весьма существенно, составляя, например, для системы СбН6+ССЦ
80,4 и 110,1 дж/моль (х = 0,5; Т = 298,15 °К).
Задача статистического расчета термодинамических функций
еще более усложняется, если по крайней мере один из
компонентов смеси склонен к образованию ассоциатов. В этом случае
помимо данных о размерах и форме молекул исходных компонентов
необходимо располагать сведениями о размерах и форме
ассоциативных комплексов в растворе. Примеры расчетов
термодинамических функций для систем такого рода (спирты — четыреххлори-
стый углерод) имеются в работах Е. В. Комарова и сотр.
[Л. Г. Кар до - С ы с о ев а, Е. В. Комаров, 1971], причем для
анализа формы ассоциатов используются данные ИК-спектроско-
пии и диэлькометрии.
Лучшее согласие с экспериментом достигается в тех молекуляр-
но-статистических теориях, где наряду с молекулярными
используются отдельные макрофизические параметры.
Флори [P. J. Flo г у, 1965] рассмотрел выражение для
конфигурационного интеграла системы, в который вместо собственных
Размеров частиц и свободных промежутков, определяющих число
69
возможных конфигураций, введены связанные с этими размерами
величины собственного объема частиц v* и доступного объема и.
Для оценки энергии межмолекулярного взаимодействия принято,
что каждую молекулу можно представить в виде набора
сегментов (частей, обладающих одинаковым количеством внешних
степеней свободы), причем на каждый сегмент приходится
определенное число мест контакта и взаимодействие пары соседних мест
характеризуется постоянным энергетическим параметром. Этот
энергетический параметр является единственной величиной,
которая не может быть предвычислена, исходя из физических свойств
компонентов.
Основное уравнение для избыточной энтальпии смешения в
бинарных системах имеет вид
Н* = NlPlvx (^ - у) + N2P2v2 (_ - т) + -i±i. Xl2
(V. 3)
где Ni — число частиц каждого компонента в системе; величина v*
связана с приведенным объемом v соотношением v* = v/v;
v, в свою очередь, определяется через коэффициент
термического расширения а
аТ = 3 (vl> - 1)/[1 - 3 (б1/з - 1)] (V. 4)
р* — характеристическое давление, определяемое через
термический коэффициент давления и приведенный объем
(V.5)
8; — доля контактирующих участков /-го типа, 02 = 1 — 6i;
Х12—-параметр, характеризующий энергию взаимодействия.
Для расчетов по методу Флори необходимо иметь хотя бы одно
экспериментальное значение энтальпии смешения А#см при любом
соотношении компонентов, чтобы рассчитать параметр Xi2, а затем
уже Д#см при других концентрациях. Было показано [A. Abe,
P. J. Flory, 1965], что для смесей неполярных жидкостей
наблюдается удовлетворительное согласие расчетных и
экспериментальных значений Яе, причем теория позволяет вычислить также
значения избыточного объема и энтропии.
Теория регулярных растворов
С точки зрения практики представляет интерес прежде всего
то обстоятельство, что теория регулярных растворов оперирует не
трудно определимыми молекулярными параметрами, а макрофизи-
ческими константами жидкостей, прежде всего энергией испарения
Ev и мольным объемом VM- Теория позволяет с
удовлетворительной точностью (в случае неполярных компонентов) предсказать
растворимость газов и твердых тел в жидкостях, условия
смешения жидкостей (критическую температуру смешения и соответ-
60
сТвующий состав), оценить энергию смешения. Разработанная
около полувека назад, эта теория с рядом уточнений и
дополнений при использовании несравненно более простого
математического аппарата позволяет достигнуть во многих случаях лучшего
согласия с экспериментом, чем молекулярно-статистические
теории. Подробно о преимуществах использования величин Ev и VM
по сравнению с молекулярными параметрами а и 8 говорится в
одной из последних работ создателя теории Дж. Гильдебранда
(1969).
Значение энергии испарения и мольного объема позволяет
вычислить основной параметр теории регулярных растворов —
плотность энергии когезии или параметр растворимости б. С учетом
связи между энергией и теплотой испарения (гл. IV) выражение
для параметра растворимости может быть записано в виде:
Обычно параметр растворимости вычисляют при стандартной
температуре (298 °К), а Д#298 в случае отсутствия
экспериментальных данных рассчитывают по формуле (IV. 27).
Величина б, согласно Гильдебранду и Скотту (1950), может
быть вычислена также по формуле
где а — коэффициент термического расширения; Рг — коэффициент
изотермической сжимаемости.
Выражение для б посредством поверхностного натяжения
имеет вид:
lg б = 0,6128 + 0,43 lg у — 0,143 lg Vu (V. 8) *
Значения параметров растворимости бб и 6g, вычисленные по
формулам (V. 6) и (V. 8), сведены в табл. V. 1 **. Наибольшие
расхождения между этими величинами наблюдаются для
жидкостей с Н-связями. Исходные физические константы жидкостей,
необходимые для расчета, даны в Приложении.
Основное уравнение для расчета свободной энергии смешения
&Ки (при постоянном объеме) в теории регулярных растворов
имеет вид
А/*" = (xaVUa + xBVM^ (бА - 6В)2 VAVB + RT (xA In хА + хв In *B) (V. 9)
где xAi хв — мольные, a VA, Vb — объемные доли компонентов.
* Числовые коэффициенты отвечают выражению величин через следующие
единицы: у —дин/см; ]/и — смъ1моль\ б— (кал/см3)У2.
** Приводимые обычно в литературе значения б в кал?2- см~^2 аля перевода
а систему СИ (дж1/г м'*1*) надо умножить на 2046.
61
Таблица V.I
Параметры растворимости некоторых жидкостей (в кдж^2м"Ъ12) при 25 °С
Растворитель
Растворитель
Гексан
Октан
Декан
Бутилацетат . . .
Декалин
Толуол
Этилацетат ....
Бензол
Уксусная кислота .
Циклогексанон . .
Уксусный ангидрид^
14,85
15,45
15,80
16,94
17,72 !
18,23
18,50
18,72
18,91
18,95
19,13
14,45
15,08
15,30
16,51
17,62
18,01
16,78
18,50
19,31
19,72
19,01
Хлорбензол . .
Анизол . . .
Ацетон ....
1,4-Диоксан .
1,2- Дихлорэтан
Анилин ....
Спирты
бутиловый
пропиловый
этиловый
метиловый
Вода ....
19,44
19,78*
19,87
19,91
20,28
24,14
23,16
24,43
26,07
29,26
47,94
19,40
19,80
17,31
19,95
19,77
22,10
17,27
17,51
18,50
18,72
55,35
Первый член правой части уравнения описывает энтальпийный
эффект, второй (с обратным знаком)—соответствует
энтропийному эффекту и совпадает, в соответствии с основным постулатом
теории регулярных растворов, с выражением (V. 2) для энтропии
идеального смешения. Из уравнения (V. 9), в частности, следует,
что компоненты с равными параметрами растворимости образуют
идеальный раствор.
Используя параметры растворимости для многих систем,
образованных неполярными жидкостями, удалось рассчитать энергию
смешения, а также сделать согласующиеся с, экспериментом
предсказания растворимости в различных растворителях (подробнее
об этом будет сказано ниже). Весьма заманчивым представляется
распространение теории на смеси полярных жидкостей, хотя бы
с помощью эмпирических параметров, характеризующих
полярность. В последнее время предложены методы разделения
величины б на составляющие, которые отражают соответственно
вклады диполь-дипольного взаимодействия, дисперсионных сил и
Н-связей [Ch. М. Н а п s e п, 1969].
Современные модификации теории регулярных растворов
За 20 с лишним лет с момента выхода третьего издания
фундаментальной монографии Гильдебранда и Скотта (1950)** основные
представления теории регулярных растворов не подвергались
существенному пересмотру. С целью же расширения круга объектов,
описываемых теорией, и улучшения согласованности теоретических
предсказаний с экспериментом, в ряде работ вводились дополни-
* Значения теплот испарения вычислены по уравнению (IV. 27).
** Эта книга в 1964 г, переиздана без изменений.
62
тельные допущения. Здесь рассматриваются вкратце основные по-
ложения теории Хаггинса [М. L. H u g g i n s, 1970].
Так же, как и Флори (см. выше), Хаггинс использовал
представление о том, что межмолекулярное взаимодействие
определяется природой и величиной поверхности частей молекулы —
«сегментов». Поверхность молекулы аддитивно складывается из
поверхностей сегментов: например, для молекулы алкана — из
2СНз-сегментов и (п — 2)СН2-сегментов. Исходные постулаты
теории формулируются Хаггинсом следующим образом:
1. Для молекул, где внутримолекулярные взаимодействия
сегментов отсутствуют, можно допустить, что средняя площадь
межмолекулярного контакта для данного типа сегментов при
постоянной температуре не зависит от типа и числа других сегментов;
2. Для каждого вида контактов сегмент-сегмент средняя
контактная энергия на единицу площади контакта не зависит при
данной температуре от типов и площадей других контактов;
3. Величины относительных поверхностей контакта различных
сегментов определяются условием минимизации свободной
энергии системы, причем средние площади молекулярных контактов
для различных типов контактов (между однородными
молекулами— 0аа, <*вв, между разнородными — (7Ав) связаны определенной
константой равновесия —К, = сгдВ/4стАА<гвв.
Величина К экспоненциально зависит от средней энергии
молекулярного -контакта А^, которая определяется как: А<? =
= 2(?f ав — <^аа — ^вв-
Полная величина энергии межмолекулярных взаимодействий
в расчете на 1 моль вычисляется, с одной стороны, как сумма
произведений <*if, с другой — согласуется с энергией когезии в теории
регулярных растворов (с обратным знаком), отличаясь от нее на
величину 3RT/2:
% = — AHv + pAv-YRT (V. 10)
Последний член правой части уравнения представляет собой
разность между суммарной энергией поступательного и
вращательного движения всех молекул в газовом состоянии и энергией
колебания и скручивания (в случае длинноцепочечных молекул7
в жидком состоянии.
Хаггинс отождествил_энтальпию смешения с энергией
межмолекулярного взаимодействия и получил таким образом выражение
Для избыточной энтальпии смешения:
H'^ff-lx&i + x&i) (V.11)
Исходя из данных о геометрических размерах молекул, Хаг-
гкнс рассчитал теплоты смешения для ряда простых систем (с
Участием ССЦ, С6Н6, цикло-СвН12, SiCU, SnCU, TiCl4).
Полученные результаты хорошо согласуются с экспериментом,
63
Можно считать, что в настоящее время применение теории
регулярных растворов и ее модификаций для расчетов
термодинамических функций смесей более оправданно с практической точки
зрения, нежели молекулярно-статистических теорий.
V.3. Растворимость газов в неводных растворителях
Растворимость газа в жидкости обусловлена в общем случае
физическими и химическими свойствами компонентов и внешними
условиями: температурой и давлением. Примером системы с
неограниченной растворимостью может служить жидкость,
находящаяся в равновесии с собственным паром. Это равновесие при
постоянном внешнем давлении описывается уравнением
Клапейрона — Клаузиуса.
Общей теории, количественно описывающей влияние природы
растворителя на растворимость веществ, находящихся в
различных агрегатных состояниях, не существует. Можно отметить, что
химическое взаимодействие между компонентами, как правило *,
Таблица V.2
Растворимость некоторых газов в воде и неводных растворителях
(р= 1 бар, * = 20°С)
[W. Gerrard. Chem. a. Ind., № 31, 884 (1971)]
Газ
Растворимость (моль газа/моль растворителя)«104
идеальная
2,5
5,1
5,5
16,0
15,5
17,0
18
36
38
180
200
240
250
500
600
1800
1800
1400
2 200
2 200
4 000
в воде
0,07
0,084
0,135
0,12
0,19
0,27
0,23
0,282
0,47
7,0
5,0
8,0
3 600
4 450
20
19
3 400
5 000
300(18°)
19
60
в неводных
растворителях (интервал
наблюдаемых
экспериментальных значений)
0,7-2,5
1,0-3,5
2,6-7,8
2,2-14
5-17
6,2-25
8,1-19
12-31
68-80
20—280
54,320
190-3 380
190-14 000
330—15 300
190—1200
2 200-2 400
2 700-12 000
850-13 100 (0°С)
1 300-6 300
1 600—3 400
1000-15 000
Гелий
Неон
Водород
Азот
Окись углерода
Аргон ,
Кислород
Метан
Криптон
Двуокись углерода
Закись азота . . .
Ацетилен
Хлористый водород
Бромистый водород
Сероводород . . .
Хлор
Йодистый водород
Аммиак
Метиловый эфир .
Хлористый метил .
Двуокись серы . .
* Для газов неизвестны исключения из этого правила, в других случаях —
возможно образование нерастворимых комплексов,
64
ведет к повышению растворимости. Растворимость газов в воде в
подавляющем большинстве случаев во много раз ниже, чем в
обычных неводных растворителях (табл. V. 2). Подробная
библиография работ по определению растворимости газов в различных
жидкостях (включая расплавленные металлы и соли) дана в
обзоре [R. Battino, H. L. Clever, 1966]; там же обсуждаются
методы измерений и современные теории растворимости.
Коэффициенты растворимости
Одним из наиболее употребляемых коэффициентов
растворимости газов является константа kH в известном уравнении Генри
pA = kKxAi где хА— мольная доля газа в растворе. Численное
значение ku зависит от способа выражения концентрации.
Коэффициент Оствальда (L) представляет собой отношение
объема поглощенного газа к объему абсорбирующей жидкости,
причем объемы приведены к одинаковой температуре.
Коэффициент Бунзена (а) характеризует объем газа
(приведенный к нормальным условиям), который растворяется при
температуре опыта в \ л растворителя при условии, что парциальное
давление газа рА составляет 760 мм рт. ст. Если рА отличается от
указанной величины, то а дается выражением: а =
= 273,15 vApa/760 Tvs. Коэффициент Куэнена отличается от
коэффициента Бунзена тем, что количество растворителя выражено в
граммах. Сводка соотношений между коэффициентами
растворимости приведена в упомянутом обзоре Баттино и Клевера.
Идеальная растворимость
Из общего термодинамического условия фазового равновесия
(равенство химических потенциалов газа А в газообразной
На и жидкой [xj фазах) выводится уравнение растворимости в
виде [Т. М. Reed, 1970]
где /а — парциальная летучесть (фугативность) газа в газовой
фазе, совпадающая с парциальным давлением рА при рА—>0;
/а — фугативность чистого компонента A; yf — коэффициент
активности частиц растворенного газа в жидкой фазе. Последняя
величина отражает совокупность эффектов растворителя.
Если система газ — растворитель ведет себя как идеальная в
газообразной и в жидкой фазах, то Ya = 1» фугативность при этом
можно заменить давлениями, и тогда уравнение (V. 12)
преобразуется в уравнение Рауля, записанное для растворенного
вещества, т. е. в уравнение растворимости идеального газа. Если
концентрация выражена в мольных долях, то идеальная
растворимость данного газа при" определенном парциальном давлении и
3 Зак. 500 gR
температуре одинакова в любом растворителе; при выражении
концентрации в моль/л численное значение идеальной растворимо
сти зависит еще и от мольного объема растворителя. Величины
идеальной растворимости газов сведены в табл. V. 2.
Качественные закономерности,
характеризующие растворимость газов
Говоря о влиянии природы растворяющегося газа на
растворимость, следует прежде всего отметить одно достаточно общее
правило [J. H. Hi I debr a n d, R. L. Scott, 1950]: в данном
растворителе лучше растворяется газ, имеющий более высокую
критическую температуру и температуру кипения. Применимость этого
правила для воды может быть проиллюстрирована данными
табл. V. 2 (газы расположены в порядке возрастания температур
кипения); исключения из правила обусловлены химическим
взаимодействием. Более четко указанная закономерность выявляется
в ряду подобных растворителей, например спиртов (табл. V.3).
Таблица V.3
Растворимость некоторых газов в спиртах'при 25 °С и 1 атм
[F. L. Воуег, L. J. В i г с h е г, J. Phys. Chem.. 64, 1330 (I960)].
Газ
Температура, °К
критическая
Растворимость,
(моль газа/моль растворителя)»\04
СНзОН
С2НЬОН
С3Н7ОН C8Hi7OH
Азот .
Аргон
Метан
Этилен
Этан .
126,2
150,7
191,1
283,1
305,5
77,4
87,1
111,6
169,5
184,7
2,55
4,15
8,67
43,9
40,5
3,48
6,18
12,8
59,7
68,6
4,18
7,82
16,1
74,6
90,0
6,47
13,7
28,0
126,8
173
В. К. Семенченко (1941), по-видимому, впервые попытался
дать полуколичественную трактовку растворимости на основе
данных о свойствах молекул компонентов. Им выведено основное
уравнение растворимости, включающее в качестве параметров
объемные концентрации молекул в различных фазах и
потенциальные энергии этих молекул. В уравнении Семенченко имеется
ряд параметров, не поддающихся точному расчету, однако
введение ряда упрощающих допущений приводит к соотношению
1пхА = а + р(еА-1) (V. 13)
где 8д — диэлектрическая проницаемость газа; аир — постоянные
для совокупности газов-аналогов в одном и том же растворителе.
Применимость уравнения (V. 13) для обработки
экспериментальных данных иллюстрируется рис. V, 1,
66
Можно считать, что уравнение (V. 13) приближенно
соблюдается в большинстве растворителей, хотя, например, в ацетоне
(кривая 6) отклонения от линейности весьма велики.
Данные рис. V. 1 позволяют сделать также качественные
выводы о влиянии е растворителя на растворимость газов. Так, в
ряду нормальных
алифатических спиртов (кри-
вые 1—3) растворимость
одного и того же газа
закономерно убывает с
ростом е. При переходе к
растворителям другой
химической природы эта
закономерность уже не
соблюдается: так, полярные
газы растворимы в воде
лучше, чем в
углеводородах, метан растворим в
ацетоне (s=20,7) лучше,
чем в амиловом спирте
(8= 13,9) и т.д.
В тех случаях, когда
б
3,0
2,5
2,0
Рис. V. 1. Зависимость растворимости газов
в различных растворителях от их
поляризуемости:
/-СНзОН (О); 2-С3Н7ОН (О); з-С5НпОН (X);
4-c6Hi4 (□); 5-ссц О); s-(ch3)2co (A).
0,6 0,8 1%0 <t2
можно пренебречь
химическим взаимодействием
между растворенным
газом и жидкостью,
повышение прочности Н-свя-
зей в растворителе
понижает растворимость газа. Так, растворимость аргона в этаноле
в 3 раза выше, чем в этиленгликоле; растворимость газов в воде
аномально низка по сравнению с другими растворителями (см.
табл. V.2).
Трактовка растворимости газов
в теории регулярных растворов
Коэффициент активности растворенного в жидкости газа \у™
в уравнении (V. 12)] отличен от единицы в случае любого
неидеального раствора. Эта величина связана с избыточным
парциальным молярным изобарным потенциалом газа при смешении
Уравнением:
Расчет величины G| может быть выполнен либо в рамках молеку-
лярно-статистических теорий через функции распределения, либо
в рамках теории регулярных растворов.
В теории регулярных растворов величина избыточного
изобарного (изохорного) потенциала полностью определяется энтальпий-
3*
67
ным эффектом, который, в свою очередь, может быть выражен
через параметры растворимости б [уравнение (V.9)]. Поэтому
окончательное выражение для растворимости газов в случае
образования регулярного раствора также включает величины б:
0,4343FM.
(V. 15)
Здесь FMA — парциальный мольный объем газа в растворе.
Для растворов, где молекулы газа и растворителя существенно
отличаются размерами, используется модификация этого
уравнения, предложенная Флори и Хаггинсом:
- lg VA = -
+ 0,4343 (1 -
0,4343 VM
(6S - 6A)2 (V. 16)
Практическое использование этих уравнений для расчета
растворимости связано с решением двух вопросов: при каких
условиях должен быть определен параметр растворимости газа и как
определить парциальный мольный объем растворенного газа.
Параметры растворимости жидкостей приводятся обычно к
стандартной температуре 298,15°К (табл. V. 1), т. е. при условиях,
когда теплоты испарения сжиженных газов экспериментально не
могут быть определены, а экстраполяция на эту температуру не
имеет физического смысла. Гильдебранд показал, что
экспериментальные данные для многих газов, растворенных в неполярных
растворителях, удовлетворительно описываются уравнением
(V. 16), если параметр растворимости газа взят при температуре
кипения,-а величина FMA заменена мольным объемом сжиженного
газа УмА. Параметры растворимости ряда газов при температуре
кипения приведены в табл. V. 4.
Таблица V.4
Параметры растворимости газов при температуре кипения
[j. H. H i I d e b г a n d, R. L. S с о 11, The Solubility of Non-electrolytes, N. Y., 1950, p. 435 ]
Газ
Водород
Гелий
Неон
Аргон
Криптон
Ксенон'
Радон
Т, °К
20,4
4,2
27
87
121
165
211
б,
кдж1!2.м-312
5,11
1,02
10,03
14,32
16,35
16,37
17,19
Газ
Фтор
Хлор
Кислород ....
Азот
Окись углерода
Двуокись
углерода
Окись азота . .
т, °к
85
239
90
77
82
223
121
б,
кдж112.м-312
12,89
20,05
14,73
12,07
12,48
18,21
23,33
68
В последнее время выполнены исследования, посвященные
методам оценки К„ растворенных газов. В частности,
предпринимаюсь попытки связать эту величину с мольным объемом
гипотетической жидкости УмА, которая могла бы образоваться из данного
газа при тех условиях (Т и /?), в которых происходит растворение.
Теория регулярных растворов предлагает для этого соотношение
[Е. В. Smith, J. Walkley, 1962]
где ps^ f j —изотермическая сжимаемость чистого раство-
/ dEv \ i( Ev\ *
рителя; п==[~~^~) /\~7Г)» для большинства неполярных
жидкостей эта величина равна 1.
Значение VM вычисляют по данным о диаметрах молекул
А
газа а по формуле VM = Af0cr3, где No — число Авогадро.
Ю. В. Гуриков (1969) показал, что рассчитанные таким путем
значения FM даже в случае неполярных жидкостей (бензол,
сероуглерод) примерно в два раза превышают экспериментальные. Он
же предложил статистико-термодинамический метод расчета VM ,
в котором используются значения свободной энергии сольватации.
Поскольку статистические методы весьма громоздки и трудоемки,
для практических целей удобнее пользоваться все же методами
теории регулярных растворов, скорректированными с помощью
дополнительных допущений. Так, на основе предположения о
пропорциональности VM критическому объему газа получено
простое соотношение [W. Y. N g, J. W a 1 k 1 е у, 1969]
где а — коэффициент пропорциональности между Vm и
критическим объемом газа (VM =Q>VMV\\ A — величина постоянная
V А А/
Для данного растворителя.
Теория регулярных растворов позволяет сформулировать
следующие положения, характеризующие зависимость растворимости
от свойств исходных компонентов [J. E. Jolly, J. H. H i I d e-
brand, 1958]:
1. Для одного и того же газа, растворенного в ряду
растворителей, растворимость при одинаковых внешних условиях
уменьшается с ростом б растворителя;
2. В одном и том же растворителе величина \gxA
увеличивается линейно с ростом силовой постоянной Леннард — Джонса
е/£ газа;
69
3. Энтропии растворения различных газов в том же
растворителе изменяются линейно с величиной R\t\Xa (это используют для
оценки температурных коэффициентов растворимости, см. ниже);
4. Парциальная молярная энтропия растворения любого газа
при постоянном давлении для раствора той же концентрации
приблизительно одинакова во всех растворителях (во фторуглеродах
несколько выше) и растет в любом растворителе при переходе
к газам с меньшей силовой константой.
Закономерности растворения газов,
вытекающие из решеточной модели
Согласно представлениям решеточной модели жидкостей
процесс растворения газа можно расчленить на стадии: образования
полостей в жидкости, необходимых для внедрения молекулы газа,
и собственно внедрения молекулы в эту полость. Растворимость
газа, характеризуемая константой Генри kH, связана с величинами
изменения парциального молярного изобарного потенциала на
этих стадиях уравнением [R. A. P i e г о 11 i, 1965]:
In kH «-^r + -gr + In (RT/V^) (V. 19)
Величина &с, характеризующая стадию образования полости,
рассчитывается методами статистической механики [Н. Reiss a. oth,
1959] и определяется в конечном итоге в зависимости от
плотности, температуры, давления и диаметра полости для жидкости.
Величина Gi, характеризующая взаимодействие молекул газа и
жидкости, вычисляется через межмолекулярный потенциал с
учетом индукционных взаимодействий; последний требует знания ди-
польного момента растворителя и его поляризуемости.
Молекулярно-статистическая теория дает соотношения,
применимые в принципе как к воде, так и к неводным растворителям.
Различие между растворами в воде и в неассоциированных
органических растворителях проявляется в том, что для водных
растворов наблюдаются большие отрицательные значения теплоты и
энтропии растворения и положительные мольные теплоемкости
растворения. Расчеты с помощью молекулярно-статистической
теории для растворов, где нет химического взаимодействия, дают
результаты, удовлетворительно согласующиеся с экспериментом.
Исходя из представлений решеточной модели, была
предпринята попытка [Н. Н. Uhlig, 1937] связать растворимость газа с
поверхностным натяжением растворителя. Предполагая, что
энергия образования полости в жидкости равна произведению
площади поверхности полости на поверхностное натяжение жидкости,
Улиг получил соотношение, из которого вытекает линейная
зависимость между igL (коэффициент Оствальда) и у. Позднее было
показано, что это соотношение имеет ограниченную применимость
и, в частности не соблюдается для полярных жидкостей,
70
поирода растворителя
и температурный коэффициент растворимости газов
Общее выражение, связывающее температурный коэффициент
растворимости газов с термодинамическими характеристиками
процесса растворения, можно получить на основе известного
уравнения Вант-Гоффа для температурной зависимости константы
равновесия. Рассматривая в качестве такой константы равновесия
коэффициент Генри kH, находим
АСР lg Т Л#о
Ч**+(У20)
где Д#о — теплота перехода 1 моль газа в раствор при
стандартной температуре: АСР — разность теплоемкостей газа и раствора;
д — постоянная для данной пары газ — жидкость.
В зависимости от природы растворителя температурный
коэффициент растворимости может быть положительным или
отрицательным. Так, растворимость благородных газов в метаноле,
этаноле и бензоле возрастает, а в воде убывает при повышении
температуры [Г. А. Крестов, К. М. Пацация, 1969], причем при
33 и 50 °С для водных растворов гелия и неона происходит
перемена знака температурного коэффициента растворимости.
Если газ и растворитель образуют, идеальный раствор, то в
этом случае растворимость газа должна уменьшаться с
увеличением температуры. В теории регулярных растворов установлена
связь между температурным коэффициентом растворимости и
энтропией растворения, которая дает возможность приближенно
предсказывать растворимость при различных температурах.
В современной интерпретации [W. Hayduk, W. D. Buckley,
1971] метод оценки температурных коэффициентов растворимости
в рамках теории регулярных растворов заключается в следующем.
Установлено, что растворимости всех газов в данном растворителе
достигают одной и той же постоянной величины, когда
температура раствора совпадает с критической температурой
растворителя. Эта постоянная величина х0 соответствует растворимости
газа, который в данном растворителе обладает нулевой энтропией
растворения и, соответственно, нулевым температурным
коэффициентом растворимости. Между лг0 и параметром растворимости б
растворителя на основании обработки большого числа
экспериментальных данных найдена зависимость вида lgx0 = а — 66, где
a yl b — эмпирические коэффициенты. Зная х0 и критическую
температуру растворителя, а также растворимость при одной
температуре, можно найти растворимость при любой другой
температуре с помощью уравнения:
Надежнее определять x0 не по параметрам растворимости, а путем
графического анализа кривой температурной зависимости
растворимости в координатах \gx-\gT (экстраполяция на Гкр),
71
Более трудоемкий (но не более точный) метод определения
температурной зависимости константы Генри дает молекулярно-
статистическая теория [D. A. J о n a h, M. В. К i n g, 1971].
Выражение для расчета d In kJdT включает такие параметры
растворителя как теплота испарения, коэффициент термического
расширения и давление насыщенного пара, а также молекулярные
параметры Леннард —Джонса.
Растворение газов в смесях неэлектролитов
Обширные исследования растворимости газов в
водно-неводных смесях выполнили за последнее десятилетие Бен-Найм
[А. В en-Nairn, 1964, 1965, 1968] с сотрудниками. Полученные
данные рассмотрены с учетом влияния растворенных газов на
структуру воды. В частности, было отмечено, что в системе вода—
этанол кривая растворимости аргона проходит через максимум, а
в системе вода —диоксан максимум не обнаруживается.
В отдельных смесях неполярных растворителей обнаружилась
линейная зависимость растворимости газа от состава смешанного
растворителя (водород в смеси бензол — циклогексан). Это
связано,, по-видимому, с тем, что взаимодействие этой пары
растворителей пренебрежимо мало. В общем случае было предложено
[J. Р. (УСonnell, J. M. Prausnitz, 1964] уравнение,
учитывающее межмолекулярные взаимодействия в смешанном растворителе
1П kH, CM==XllnkH,l+ Х2 ln kH, 2 - «12*1*2 ( V' 22)
где ai2 — параметр, характеризующий взаимодействие
растворителей 1 и 2 и определяемый из данных по равновесию жидкость —
пар в смешанном растворителе.
Теория регулярных растворов во многих случаях
удовлетворительно описывает растворимость газов в смешанных
растворителях, если параметр растворимости смешанного растворителя
вычисляется из параметров растворимости исходных жидкостей как
объемно-аддитивная величина [J. H. Hil d eb r a n d, R. L. Scott,
1962].
V. 4. Растворимость жидких и твердых неэлектролитов
В предыдущем разделе говорилось, что при отсутствии
заметного химического взаимодействия между компонентами
полуколичественная оценка растворимости может базироваться на теории
В. К- Семенченко, учитывающей полярность компонентов, либо на
теории регулярных растворов, рассматривающей разность
внутренних давлений или параметров растворимости. Каждая из этих
теорий оправдывается для определенного круга объектов.
Сказанное относится в равной степени к взаимной
растворимости жидкостей и к растворимости твердых неэлектролитов, при-
72
чем эти вопросы разработаны в меньшей степени, чем в случае
растворимости газов. Поэтому они будут рассмотрены здесь
вкратце и в самом общем виде.
Взаимная растворимость жидкостей
В. К. Семенченко (1941) предложил полуколичественную
трактовку растворимости, основанную на сопоставлении величин
«молекулярных силовых полей» растворяющихся компонентов. При
дальнейшей разработке этой теории М. И. Шахпароновым (1956)
получено уравнение, связывающее растворимость со средним
координационным числом молекул в растворе и коэффициентом,
характеризующим различие молекулярных полей частиц
компонентов и способность этих частиц к взаимодействию. Трудности, свя-.
занные с вычислением коэффициентов, входящих в это уравнение,
побуждают ограничиться изложением только качественных
выводов этой теории, которые применимы, разумеется, и в случае
растворения твердых веществ.
Полагая, что интенсивность силовых полей молекул
компонентов коррелирует в первом приближении с макроскопической
диэлектрической проницаемостью е, Семенченко и Шахпаронов
рассмотрели зависимости растворимости (х) веществ от 8
растворителей и классифицировали растворяемые вещества по виду
кривых х(г).
В общем случае такая кривая должна иметь максимум. Если
молекулы растворенного вещества обладают молекулярным полем
средней силы, то можно подобрать растворители,
характеризующиеся как более слабым, так и более сильным молекулярным
полем. Максимум растворимости достигается в том растворителе,
молекулярное поле которого наиболее близко к молекулярному
полю растворенного вещества. Если растворяемое вещество
обладает слабым молекулярным полем, у кривой х(г) реализуется
только нисходящая ветвь, а для высокополярных соединений на
этой кривой наблюдается лишь восходящая ветвь. Семенченко и
Шахпаронов (1948) привели большое число данных,
иллюстрирующих эти зависимости для твердых веществ (см. ниже).
Теория регулярных, растворов позволяет определить условие
оптимального смешения двух химически не взаимодействующих
неполярных жидкостей на основе уравнения (V. 9). Минимальное
значение изменения свободной энергии при смешении в системе
достигается, как отмечалось выше, в случае равенства параметров
растворимости компонентов. Чем меньше разность параметров
растворимости, тем больше вероятность полного
взаиморастворения компонентов.
Критическая температура смешения жидкостей в теории
регулярных растворов определяется из соотношения:
) (V. 23)
73
При этом мольная доля одного из компонентов смеси,
соответствующая критической температуре смешения, определяется как:
kT - ч]
-24)
Для жидкостей с равными мольными объемами при Ха=^в = 0,5
получается упрощенное соотношение:
(V.25)
Последнее выражение может быть использовано для растворов
с невзаимодействующими компонентами, имеющими близкие
мольные объемы, причем величина VM в этом случае вычисляется как
среднее арифметическое из мольных объемов компонентов.
Уравнения (V. 24) — (V. 25) неприменимы, если хотя бы один из
компонентов является полярным соединением.
Растворимость твердых неэлектролитов
Говоря об общих закономерностях, характеризующих процессы
растворения твердых тел, наряду с известным правилом
повышения растворимости их с температурой, следует упомянуть
зависимость, установленную Лавуазье: твердое тело с более высокой
температурой плавления при данной температуре менее
растворимо, чем тело с низкой температурой плавления. Это
эмпирическое правило может быть объяснено на основании
термодинамического уравнения Шредера — Ле-Шателье для идеальной
растворимости, полученного примерно через 100 лет после Лавуазье
__ АЯПЛ (Уд, - Т) АСр (Гпл - Т) ЛСр _7пл
lg*A"~ 2,303#Гпл^ 2.303ЯГ + R g T ( 6)
где АСр —разность между мольной теплоемкостью растворенного
вещества в чистом виде и парциальной мольной теплоемкостью
его в растворе при температуре Т и мольной доле хА. Уравнение
(V. 26) выведено в предположении, что процесс растворения
твердого тела можно условно расчленить на две стадии: плавление
твердого тела и смешение получившейся жидкости с
растворителем, причем образуется идеальный раствор. Это уравнение не
учитывает природу растворителя.
Теория регулярных растворов дает для расчета растворимости
твердых неэлектролитов выражение, аналогичное (V. 16):
(V. 27)
Для расчета параметра растворимости твердого вещества 6а
могут быть использованы значения теплоты испарения и мольного
объема этого компонента при температуре плавления в жидком
состоянии. На практике чаще используется другой путь, основан-
74
ный на анализе величин растворимости данного вещества в
растворителях с известными 6s-
Для качественного предсказания растворимости твердых
неэлектролитов в случае, когда хотя бы один из компонентов
раствора является полярным веществом, используют классификацию
Семенченко, основанную на подразделении веществ по мере
увеличения их полярности (слабой, средней и сильной). Здесь
возникают затруднения, связанные с оценкой полярности твердых
веществ. Если принять в качестве такой меры «обобщенный момент»
по Семенченко, равный отношению дипольного момента к длине
связи, то для сложных органических молекул не всегда удается
оценить эту величину. В работах Семенченко и Шахпаронова
(1948) классификация базируется на анализе кривых х(&) (см.
выше); при этом, в частности, наблюдаются такие аномалии, когда
в группе веществ средней полярности фумаровая кислота
оказывается рядом с перхлоратами лития и кальция, а сахар и винная
кислота попадают в группу веществ высшей полярности.
Растворимость полимеров
Проблемы растворимости полимеров, так же как и
термодинамические свойства растворов полимеров в неводных растворителях
подробно рассматриваются в специальных монографиях Ч. Тен-
форда (1965), Г. Моравеца (1967), С. Е. Бреслера и Б. А. Еруса-
лимского (1965) и др. Единственной теорией, позволяющей
установить хотя бы качественную связь между растворимостью
полимера и природой растворителя, является теория регулярных
растворов. При этом параметр растворимости для полимера
определяется, согласно Смоллу [P. A. Small, 1953], по уравнению
6 = 2w^ <v-28>
где U — постоянная для данной функциональной группы или связи
мономера; d — плотность мономера; М — молекулярная масса.
Опыт показывает, однако, что растворимость одного и того же
полимера может существенно изменяться в зависимости от его
молекулярной массы и степени кристалличности. Поэтому трудно
ожидать успехов в применении теории регулярных растворов к
описанию растворимости полимеров даже при условии введения
Дополнительных параметров, характеризующих полярность (см.
раздел V. 2).
V.5. Термодинамические свойства
жидких неэлектролитных смесей
Важнейшая задача теории растворов неэлектролитов — расчет
термодинамических функций смешения на основе
характеристических параметров исходных компонентов. В общем виде эта задача
Решена только для идеальных и регулярных растворов. Реальные
75
растворы характеризуются величиной отклонения от идеальности,
выраженной через коэффициенты активности, которые в свою
очередь связаны известным термодинамическим соотношением с
избыточной свободной энергией (изобарным потенциалом) системы.
Принципы расчета избыточных термодинамических функций
растворов неэлектролитов в рамках молекулярно-статистической
теории и теории регулярных растворов были кратко рассмотрены
в разделе V. 2. Обзор современных достижений теории и
результатов экспериментальных определений термодинамических функций,
содержащий подробную -библиографию, опубликован Скоттом и
Фенби [R. L. Scott, D. V. Fend у, 1969].
Определение термодинамических параметров
разбавленных растворов неэлектролитов
Закон Рауля, характеризующий поведение компонента в
идеальном растворе, строго говоря, неприменим в случае реального
раствора, даже если концентрация одного из компонентов очень мала.
Для смеси компонентов А и S, где количество «растворенного
вещества» А мало по сравнению с количеством «растворителя» S,
химический потенциал компонента S выражается соотношением
Us = Us + RT In xs + Л (1 — *s)2 (V. 29)
где \x°s — химический потенциал компонента в стандартном
состоянии (чистое вещество при тех же внешних условиях);
А—эмпирический коэффициент, характерный для данной пары
растворитель— растворенное вещество. Сравнивая уравнение (V. 29) с
известным выражением для химического потенциала /-го компонента
в реальном растворе
I*/ = \i°i + RT In xt + RT In v, (V. 30)
видим, что уравнение (V. 29) по существу представляет собой
концентрационную зависимость коэффициентов активности. Другие
термодинамические параметры, например, парциальный мольный
объем, также могут быть представлены как концентрационные
зависимости
Уравнения (V. 30) и (V. 31) показывают, что в разбавленных
растворах отклонения термодинамических величин от найденных
в предположении идеальности растворов несущественны для
растворителя. В случае же растворенного вещества поправка на
неидеальность дается уже членом, линейно зависящим от
концентрации. Согласно Пригожину, зависимость коэффициентов
активности от концентрации в наиболее общем виде выражается
степенными функциями.
76
для растворителя —
- RT In ys = А2х\ + Л3х\ + А,х\+ ... (V. 32)
и, в соответствии с уравнением Гиббса — Дюгема,
для растворенного веще^ва (A. M. Розен, 1969) —
RT In YA = - 2Л2*А + (а2 - уА,) х\ + ( Л3 - у Л4) 4 + ... (V. 32а)
Отбрасывая в этих уравнениях члены высших порядков, можно
получить приближенные соотношения, справедливые для
разбавленных растворов. Методы оценки коэффициентов, входящих в эти
соотношения, а также определение концентрационных границ их
применимости содержатся в работах А. М. Розена (1969).
Описано вычисление коэффициентов активности в предельно
разбавленных растворах на основе усовершенствованной теории Гильде-
бранда, распространенной на смеси полярных веществ [J. Q. Hep-
pi ns til 1, М. Van Winkle, 1968].
Экспериментальное определение термодинамических функций
Для определения трех важнейших термодинамических функций
смешения — изобарного потенциала AGCM, энтальпии Д#см и
энтропии ASCM из экспериментальных данных используют следующие
приемы:
1. Нахождение AGCM из коэффициентов активности (чаще
всего, на основании тензиметрических измерений) и Д#см из
калориметрических измерений;
2. Нахождение AGCM при нескольких температурах с
последующим расчетом ASCM и Д#см на основании известных
термодинамических соотношений
М
V оТ р
Сопоставление этих приемов при условии, что коэффициенты
активности определяются по давлениям паров, проведено В. П. Бе-
лоусовым и А. Г. Морачевским (1964). Проведенный ими анализ
Уравнения для расчета коэффициента активности, выведенного
с учетом отклонения поведения паров от законов идеальных газов,
показал, что при расчете Д#см по второму способу поправки на
неидеальность могут изменять значение этой величины на 20—
30%. Для вычисления поправок на неидеальность необходимо
знать вириальные коэффициенты в уравнении состояния для паров
каждого данного компонента и их температурные зависимости.
Из-за недостатка необходимых данных второй метод применяется
8 ограниченном числе случаев.
В последнее время выполнены измерения избыточных
термодинамических свойств во всем интервале концентраций для боль-
шого числа двойных жидких систем, Сводка экспериментальных
77
данных по теплотам смешения дана в книге В. П. Белоусова и
А. Г. Морачевского (1970). Химические диаграммы теплот
смешения рассматриваются в разделе VIII. 2. Реже встречаются работы,
где были бы одновременно приведены значения Ge, Нв и Se.
Зависимость избыточных термодинамических функций от
состава представляют чаще всего в форме полуэмпирических
уравнений, анализ которых дан, например, в монографии В. Б. Когана
(1968). Эти зависимости обычно имеют вид кривых с максимумом,
который для неполярных смесей в большинстве случаев
приходится на эквимолекулярный состав. Некоторые примеры даны в
табл. V. 5.
Таблица V.5
Максимальные значения избыточных термодинамических
функций для некоторых систем при 25 °С
[R. L. Scott, J. Phys, Chem., 64, 1241 (I960)]
Система
Vе,
смЪ/моль
0,24
0,01
—0,19
1,07
4,89
Ge,
дж/моль
25,9
81,6
-556
1046
1364
Не,
дж/моль
60,7
109,2
-1828
1460
2159
Se,
дж/мольХ
Хград
С6Нб-1,2-С2Н4С12 0,24 25,9 60,7 0,116
С6Н6—СС14 0,01 81,6 109,2 0,092
(СН3)2СО—СНС13 —0,19 —556 -1828 4,265
(СН3)2СО—CS2 1,07 1046 1460 1,389
изо-С8Н18—C7F16 4,89 1364 2159 2,708
Избыточные термодинамические функции
и физические свойства жидких смесей
Величины избыточных термодинамических функций в жидких
системах характеризуют степень отклонения этих систем от
идеальности и, следовательно, глубину химического взаимодействия.
Та же задача ставится при изучении диаграмм свойство — состав,
подробно рассматриваемых в гл. VIII. К сожалению, работы, в
которых рассматривались бы вопросы корреляции между
термодинамическими и физическими свойствами, весьма немногочисленны.
В качестве примера можно указать на уравнение,
связывающее избыточные значения энтальпии, изобарного потенциала и
вязкости [М. R о t h e r, H.-J. В i 11 r i с h, 1967]:
2Не = Ge + 3RT In (l/rtf*
(V. 33)
В этом выражении используется понятие об «избыточной
текучести» (1/т])в, определяемой как разность между
экспериментальным значением текучести и вычисленным в предположении
мольной аддитивности текучести для системы с невзаимодействующими
компонентами. Корреляцию между Ge и диэлектрической
проницаемостью смеси рассмотрели Хериг и Биттрих [H.-J. Но rig,
H.-J. Bittrich, 1967].
78
Говоря о применимости этих соотношений, следует,
по-видимому, учитывать, насколько физически обосновано уравнение
аддитивности свойства для системы с невзаимодействующими
компонентами (см. гл. VIII). В частности, публикуемые в отдельных
работах расчеты коэффициентов активности по «избыточному
поверхностному натяжению» уе = усм — Ya#a — Yb#b имеют
определенный смысл лишь для ограниченного круга систем.
V.6. Взаимодействие неэлектролита с растворителем
В наиболее широком смысле явление взаимодействия частиц
растворенного вещества с растворителем обозначают термином
«сольватация» [К. П. Мищенко, Г. М. Полторацкий, 1968].
Мы термину «сольватация» придаем более узкий смысл (см.
гл. VII), поэтому понятие, часто обозначаемое в литературе
термином «сольватация неэлектролитов», в заголовке этого
параграфа названо общим термином «взаимодействие».
Изменение термодинамических функций в процессе
сольватации электролита определяют из цикла, включающего образование
газообразных ионов из кристаллической решетки соли и переход
этих ионов в раствор с образованием сольватов (см. подробнее
в гл. VI). Аналогичный цикл можно составить для растворения
твердого неэлектролита, с той разницей, что вместо образования
газообразных ионов из кристаллической решетки (процесс,
характеризуемый энергией решетки) здесь необходимо рассмотреть
образование газообразных молекул из твердого неэлектролита
(процесс, характеризуемый энергией сублимации).
Соответствующее термодинамическое соотношение может быть
записано в виде:
АЯС0ЛЬ^ = ДЯраств + £суб (V. 34)
Так как различие между энтальпией и изобарным потенциалом
для конденсированных систем невелико, можно согласно Н. А.
Измайлову (1959), записать аналогичное соотношение, используя
вместо энтальпии сольватации химическую энергию сольватации:
<4сольв = —Абсольв — изменение изобарного потенциала при
взаимодействии молекулы растворенного вещества с растворителем без
учета энергии, затрачиваемой на преодоление границы раствора.
Изменение изобарного потенциала при растворении можно
связать с активностью растворенного вещества в насыщенном
растворе а\ соотношением:
-AGpacTB = RT In a\ (V.35)
Из уравнений (V. 34) и (V. 35) следует:
* 4сольв £суб
Можно рассматривать процесс сольватации, как это делает
И. А. Измайлов, независимо от фазового состояния исходных ком-
79
понентов. В этом случае энергия сольватации определяется
изменением изобарного потенциала растворенного вещества при
переносе его из вакуума в среду растворителя или из одной среды в
другую. Это изменение изобарного потенциала, как упоминалось
выше, эквивалентно разности химических энергий сольватации
исследуемого вещества в соответствующих средах, или работе
переноса. Эти величины (и связанные с ними коэффициенты
активности) можно определить на основании измерений ряда свойств
растворов: давление паров, понижение точки замерзания,
повышение точки кипения, осмотическое давление, а также по данным
о растворимости.
Удобно в качестве стандарта принять бесконечно разбавленный
раствор вещества в воде и рассматривать работу переноса этого
вещества в исследуемый неводный растворитель. Обозначив
коэффициент активности, соответствующий этому стандартному
состоянию, через y° (no H. А. Измайлову), находим:
AGnep = — RT In y° (V. 37)
где величина AGnep равна разности химических энергий
сольватации в воде и в неводном растворе.
Значения у° могут быть рассчитаны, например, из данных по
растворимости веществ в Н2О и в растворителе S (разумеется,
если растворимость имеет конечную величину). Некоторые
величины, полученные таким образом, приведены в табл. V. 6.
Таблица V.6
Единые нулевые коэффициенты активности некоторых
органических кислот в метаноле и ацетоне
[Н. А. Измайлов, Избранные труды, Изд. «Наукова Думка»,
1967, стр. 76.]
Растворенное вещество
Бензойная кислота ....
Салициловая кислота . . .
2,6-Динитрофенол
2,4-Динитрофенол
ч
Метанол
4,67
3,16
1,41
1,02
Y°.1O3
Ацетон
6,46
3,55
0,013
0,20
V.7. Диффузия в растворах неэлектролитов
Диффузия является необратимым процессом и детально
рассматривается в курсах неравновесной термодинамики. Здесь этот
вопрос будет затронут лишь постольку, поскольку исследование
диффузии дает информацию о межмолекулярном взаимодействии
в растворах. Методы определения коэффициентов диффузии как
неэлектролитов, так и электролитов подробно описаны в
монографии Робинсона и Стокса,
80
При рассмотрении процессов диффузии выделяют обычно са-
модиффузию и взаимную диффузию. Первая характеризует меру
подвижности молекул данного сорта. Коэффициенты диффузии
(самодиффузии) чистых жидкостей изменяются с температурой по
экспоненциальному закону в соответствии с теорией Эйринга;
энергия активации этого процесса связана с энергией испарения.
Для многокомпонентных систем существует столько
коэффициентов самодиффузии, сколько компонентов имеется в системе.
Коэффициент взаимной диффузии определяет скорость
движения смеси жидкостей в направлении выравнивания градиента
концентраций. Для идеальных двойных систем коэффициент взаимной
диффузии Diz дается соотношением
D12 = Dxx2 + D2Xi (V. 38)
а для реальных систем — уравнением Даркена:
Dl2 = (Di*2 + D2xx) [1 + (д In Yi/a In Xl)TtP] (V. 39)
Для систем, близких к идеальным, величина (DiX2 + D2Xi)
является линейной функцией плотности [J. С. S h i e h, P. A. Lyons,
1969]. Показано [Р. М. Васенин, 1969], что для таких систем
отношение коэффициентов самодиффузии компонентов постоянно и
равно обратному (отношению мольных ^объемов компонентов.
Изучение зависимости коэффициентов диффузии от состава
жидких систем дает, наряду со сведениями о других
термодинамических и физических свойствах, весьма ценную информацию об
ассоциации молекул и межмолекулярном взаимодействии.
Совместное изучение коэффициентов диффузии и активности для
различных неэлектролитных бинарных систем показало, что
уравнение (V. 39) удовлетворительно согласуется с экспериментом.
Неучет коэффициентов активности [уравнение (V. 38)] приводит
к расхождению с экспериментом даже для систем, весьма близких
к идеальным. Если же взаимодействие проявляется отчетливо
(система ацетон — вода), на концентрационных зависимостях
величин Di, D2 и Da обнаруживается глубокий минимум. По
наблюдениям Васенина для систем, характеризующихся отрицательными
отклонениями от закона Рауля, концентрационные зависимости D
выпуклы от оси состава, а в системах с положительными
отклонениями — выпуклы к оси состава.
В последнее время большое число измерений диффузии
выполнено импульсным методом ядерного магнитного резонанса («спин-
эхо»). Эти измерения позволяют оценить молекулярную массу
протононосителя и, следовательно, делать выводы о степени
ассоциации молекул (см. гл. VII).
Глава VI
ТЕРМОДИНАМИКА РАСТВОРОВ
ЭЛЕКТРОЛИТОВ
VI. 1. Растворимость солей в неводных растворителях
В литературе накоплен обширный экспериментальный материал
по растворимости солей в неводных растворителях,
систематизированный в «Справочнике по растворимости» [сост. В. Б. Коган
и др., 1961 —1963] и справочнике Сейделла [A. Seidell,
W. F. Linke, 1959—1966]. Многими исследователями
предпринимались попытки вывести общие закономерности, связывающие
величину растворимости с определенными параметрами,
характеризующими как растворитель, так и растворенное вещество.
Качественные предсказания растворимости с большей
вероятностью могут быть сделаны на основании представлений о
«химическом подобии» компонентов, нежели по величинам е
растворителей. Соли, как правило, плохо растворимы в углеводородах и их
галопенпроизводных (одно из исключений —перхлорат серебра в
бензбле и толуоле), лучше — в спиртах, амидах, сульфоксидах, ке-
тонах, нитропроизводных и пр. Соли щелочных и
щелочноземельных металлов обычно растворимы хуже в органических
растворителях, нежели аналогичные соли элементов, склонных к
образованию координационных соединений с растворителем. В последнее
время нашли применение некоторые апротонные растворители,
обеспечивающие довольно высокую растворимость многих
неорганических солей: пропиленкарбонат, N-метилацетамид, N-метилпир-
ролидон, гексаметилфосфортриамид и др.
Н. А. Измайлов с сотрудниками (1950) получил выражение,
связывающее растворимость полярной соли х\ в двух
растворителях I и II с отношением коэффициентов активности соли в этих
растворителях и величинами е растворителей:
■. {'УЛ„) - ■. (А/О+^ 2 7 (i - v) «vi ■>
Здесь г — среднее межионное расстояние для данной соли в
растворе, а символ * означает, что коэффициенты активности
определены по отношению к бесконечно разбавленному раствору в
данном растворителе.
Выражение VI. 1 учитывает изменение активности ионов только
за счет электростатического взаимодействия и, следовательно,
справедливо только при рассмотрении пары растворителей одной
химической., природы.
При введении ряда упрощающих допущений уравнение (VI. 1)
сводится к-виду:
lg xA = const + J5/e (VI. 2)
82
нвон
0,8
0,6
HCON(CH3)2
Применимость последнего уравнения к растворителям одной хими-
еской природы проиллюстрирована рис. VI. 1. Из того же рисунка
идно, что данные по растворимости LiCl в ацетоне и диметил-
(Ьормамиде резко выпадают из общей закономерности для
гомологического ряда спиртов.
Качественно можно на многих примерах показать, что
растворимость солей повышается с увеличением диэлектрической
проницаемости [В в уравнении
(VI.2)—отрицательная величина]. Тем не менее, на- \цхА-ю*
блюдаются многочисленные отклонения
от этого правила. Н. А. Измайлов
установил, что коэффициент В по абсолютной
величине тем больше, чем меньше ди-
польный момент растворителя, и
подтвердил это экспериментальными
данными по растворимости солей в спиртах
(^=1,65—1,75) и кетонах (^=2,7—2,9).
Можно сказать, следовательно, что
близость дипольных моментов растворителей
одной химической природы
обусловливает постоянство величины В, благодаря
чему выполняется уравнение (VI. 2).
В работах школы Н. А. Измайлова
рассматривалась также применимость
уравнения (VI.2) для описания
растворимости малополярных соединений с
органическими анионами, а также органит
ческих кислот. В частности, В. С.
Черный и Е. И. Вайль (1968) подтвердили
соблюдение этого уравнения для бензо-
ата, салицилата и хлорида серебра, а
также бензойной и салициловой кислот
в метаноле, этаноле и спирто-водных смесях. Эти же авторы
рассмотрели влияние электростатических взаимодействий ион —
растворитель на температурные коэффициенты растворимости
электролитов.
Используя выражение типа (VI. 1) и полагая, что
температурная зависимость е описывается уравнением (IV. 32), В. С. Черный
и Е. И. Вайль получили уравнение зависимости растворимости от
температуры, включающее температурный коэффициент
диэлектрической проницаемости и параметры, характеризующие межион-
ное взаимодействие и взаимодействие ион — растворитель. Из
уравнения следует, что если межионное взаимодействие сильнее, чем
ион-дипольное, то растворимость должна падать с повышением
температуры, и наоборот. Действительно, для хлоридов натрия и
Серебра растворимость растет с повышением температуры в воде
и падает — в метаноле (в последнем случае диссоциация и, значит,
межионное взаимодействие слабее),
0,02
JCH5)2CO ,
0,06 0,08
Рис. VI. 1. Растворимость
LiCl в различных
растворителях при 25 °С.
83
Учет взаимодействия ионов, а также малополярных радикалов
(в случае солей органических кислот) с растворителем позволяет
объяснить также изменение знака коэффициента В в уравнении
(VI.2) [Н. А. Измайлов, В. С. Черный, 1958], что согласуется
с теорией Семенченко, предусматривающей возможность уменьше-
ния растворимости с ростом е.
VI. 2. Термодинамические характеристики
процесса растворения электролитов
в неводных растворителях
Определение термодинамических параметров при растворении
Процесс образования электролитного раствора, как известно,*
можно представить в виде совокупности последовательно
протекающих стадий: образование соединения электролит —
растворитель; ионизация этого соединения и сольватация образующихся
ионов. В задачу термодинамического описания процесса входит
расчет изменения термодинамических параметров для каждой из
перечисленных стадий в отдельности, однако, это далеко не всегда
осуществимо.
Экспериментально может быть определен тепловой эффект
растворения электролита А в растворителе S. Отнесенный к одному
молю вещества при постоянных температуре и давлении, этот
эффект называется интегральной теплотой растворения АНт.
Величина Д#т зависит от концентрации А в S; экстраполяция этой
величины к состоянию, отвечающему бесконечно разбавленному
раствору, дает так называемую первую теплоту растворения Д#о,
которая зависит от природы и растворенного вещества, и
растворителя. Это относится, собственно говоря, и ко всем остальным
термодинамическим функциям: невозможно достоверно определить
долю параметра, характеризующую изменение термодинамического
свойства только одного компонента. В этом смысле, как показал
Г. А. Крестов (1973), использование парциальных молярных
величин не решает проблему.
Изменение изобарного потенциала системы соль —растворитель
может быть найдено из данных по растворимости с помощью
уравнения
AGp = — vRT in а± = — RT In ПР (VI. 3)
где у — число ионов, образующих молекулу соли; ПР —
произведение растворимости; а± — средняя активность ионов в насыщенном
растворе соли.
Дифференцированием уравнения (VI. 3) по температуре
находят изменение энтропии при образовании насыщенного раствора
/ д In ПР \
*|п пр+RT НН (VL 4)
84
сле чего соответствующее изменение энтальпии определяют на
основании известного термодинамического соотношения: Д#р =
С + TAS
Теплоемкость растворения, определяемая как изменение
теплоемкости при переходе соли из кристаллического состояния в
раствор, может быть найдена из калориметрических измерений, либо
методом сравнительного расчета. Согласно Г. А. Крестову,
теплоемкость растворения линейно связана с энтропией кристаллической
решетки для однотипных солей
(ДС)Р = т А5Реш + п (VI. 5)
где т и п — постоянные для данной группы солей.
Концентрационные зависимости теплот растворения
Изучение термодинамических свойств разбавленных растворов
электролитов связано с развитием теории Дебая — Хюккеля — Он-
загера. Эта теория позволяет рассчитать по результатам
термохимических измерений в разбавленных
растворах значения теплоты растворения
при бесконечном разведении Д#о и
величину наклона кривой ДЯт(т).
Подробное рассмотрение методики и примеры
расчета для неводных и водно-неводных
растворителей даны в статье Г. А. Кре-
стова и И. В. Егоровой (1968).
Детальное изучение термохимических
свойств неводных растворов в широком
интервале концентраций начато
сравнительно недавно К. П. Мищенко И Г. А. Рис. VI. 2. Различные типы
Крестовым. Основные закономерности из°теРм теплот растворе-
г ^ нйя электролитов (по Ми-
концентрационных изменении термодина- щеНко).
мических свойств растворов с ростом [к. п. мищенко, г.м.
Полконцентрации электролита сформулиро- ^и^Г^
Ваны В МОНОГрафиИ К. П. МиЩеНКО И неводных растворов электроли-
Г. М. Полторацкого (1968)} они различа- тов'<<Химия>>- л" 1968' стр' 122]'
ют три типа изотерм.
I. С непрерывно убывающей экзотермичностью;
- П. С непрерывно возрастающей экзотермичностью;
III. Проходящие через максимум экзотермичности (рис. VI. 2).
Известные до сих пор концентрационные зависимости Д#т в
неводных растворителях относятся к первому типу. Те
электролиты, которые в воде дают изотермы второго типа (например, Nal)
характеризуются изотермами / типа во всех изученных
органических растворителях: ацетоне, этиленгликоле, метаноле, этаноле,
ацетонитриле, метилэтилкетоне, пиридине, пиперидине. При этом
Уменьшение экзотермичности с концентрацией Д(Д#ш)/Дт для
одних и тех же солей в неводных растворах происходит гораздо бо-
Лее резко, нежели в водных (табл. VI. I),
85
Таблица VI. 1
Наклоны кривых Д#т (т) в различных растворителях
при 25 °С и т = (0,04—0,06) моль/кг
[К. П. Мищенко, Г. М. Полторацкий, Вопросы
термодинамики и строения водных и иеводных растворов электролитов.
Изд. «Химия», 1968, стр. 148].
Соль
NaBr
Nal
Растворитель
Вода
Метанол . . .
Вода
Метанол . . .
Ацетон ....
Пиридин . . .
Л (Л#т)/Дт, кдж/моль2
2,09
35,56
2,09
48,12
66,94
41,84
Первые интегральные теплоты растворения
и изменения теплоемкости
при растворении в различных растворителях
При рассмотрении зависимости теплот растворения солей от
свойств растворителя естественно в качество стандартного
состояния принять бесконечное разведение. Именно поэтому большой
интерес представляет накопление данных по первым интегральным
теплотам растворения одних и тех же солей в различных
растворителях (табл. VI. 2).
Таблица VI.2
Первые теплоты растворения (в кдж/моль) солей в различных растворителях
(-АЯ°т, 25 °С)
[см. К. П. М и щ е н к о, Г. М. Полторацкий].
Растворитель
Соль
NaBr
Nal
NaCNS
KI
Вода
Пиридин ....
Метилэтилкетон
Ацетон ....
Аллиловый спирт
Этанол ....
Метанол ....
Этиленгликоль .
Формамид . . .
0,602
—
—
17,15
18,41
7,53
55,65
40,58
43,93
46,02
24,52
32,05
33,89
30,96
-6,82
25,10
—20,33
13,39
7,95
4,18
4,18
Данные табл. VI. 2 не позволяют установить какую бы то ни
было взаимосвязь между диэлектрической проницаемостью
растворителей и термодинамическими параметрами растворения. Не
86
•о
1
§
^г
1
0 0,2
8
10
12о
б
-
W*°
0,2
0,4
а
г
1
ОЛ
0,6
-HftO
*£г^
i
0,6
0,8
***
1
0,8
U0
|
f,o
лючено, однако, что определенные закономерности могут
выбиться при сопоставлении термохимических параметров для групп
яВствОрителей одной химической природы, где вклад «химического
фактора» в энергию взаимодействия
соль — растворитель будет
приблизительно постоянным. Тогда различие
термохимических параметров может
определяться различием физических свойств
растворителей и, в первую очередь, их
полярности.
В то же время данные табл. VI. 2
подтверждают вывод, что вода стоит
особняком в ряду растворителей. Так,
первые теплоты растворения во всех
изученных неводных растворителях гораздо
более экзотермичны, чем в воде.
Весьма специфический интерес для
теории растворов представляет
исследование термодинамики растворения в
смешанных растворителях. Данные Г. А.
Крестова показывают, что величины Д#о
в смешанном растворителе изменяются
с составом растворителя по сложной
кривой, экспериментальные точки которой
можно связать с изменениями
структуры растворителя. Данные для
растворов нитрата кальция в различных
смешанных растворителях приведены на
рис. IV. 3.
Для растворов некоторых солей в
смешанных растворителях найдены
изменения теплоемкости при растворении,
экстраполированные на бесконечное
разведение. Данные для нитрата натрия в
водно-спиртовых растворах различного
состава показаны на рис. VI. 4.
Величины теплоемкости, измеренные с достаточной точностью, также
могут служить весьма чувствительным индикатором структурных
изменений в растворе.
VI.3. Сольватация ионов
Термодинамический цикл и теплоты сольватации
Термодинамически растворение электролита можно рассматри-
Вать как конечный результат возгонки и диссоциации на ионы
твердого кристалла и введения ионов в растворитель. Переход за-
Ряженного иона из газообразного состояния в раствор можно
87
О 0,2 0,4 0,6 0,8 1,0
Рис. VI. 3.
Концентрационные зависимости первой
теплоты растворения Ca(NO3)2
при 25°С[Г. А. Крестов,
В. И. Клопов, 1968].
Раствооители:
а-СНзОН—Н2О;
б - СНзОН—С2Н5ОН;
в - С2Н5ОН—Н2О.
представить схемой [Г. А. Крестов, 1973]:
/ // /// IV
K? + mS« = {[(K«|sn)|sp]|s,;|
/ // /'// IV
Ч
Обозначения К? и Ks относятся к и.ону с зарядом q в со
стоянии одноатомного идеального газа и в неводном растворителе
В состав сольватированного иона входит (га — k) молекул
растворителя. В непосредственном контакте с сольватированным ионом
0,25 0,5 0,75
• ^CHgOH » ХС2Н5ОН
О 0,005 0,01
Рис. VI. 4. Теплоемкости растворов NaNO3 в водно-спиртовых
смесях при бесконечном разведении и температуре 25°С. '
[Г. А. К Р е с т о в, В. И. К л о п о в. Тр. I коиф. по аналитич. химии
неводных растворов. Ч. II. МХТИ, М„ 1968].
находится п молекул растворителя и соответствующая гранидз
раздела / — / отвечает границе ближней сольватации. Этой
границе и числу п отвечает так называемое число сольватации, либо
координационное число иона в растворе *. Граница IV—IV
отвечает границе дальней сольватации, за которой молекулы
растворителя уже не изменяют своих термодинамических свойств под
действием иона.
При бесконечном разведении взаимодействие ионов в растворе
между собой исключается. Поэтому термодинамические параметры
* Подробнее об этом см. в гл. VIII, стр. 186,
льватации соли можно аддитивно разложить на соответствующие
с аметры для отдельных ионов. При этом, например, энергия
Поаьватации иона представляет собой изменение энергии при
переходе иона из газовой фазы в раствор. Изменение энергии при пере-
-оде ионов в газовую фазу из твердой соли представляет собой
энергию кристаллической решетки. Поэтому растворение
кристаллической соли в жидкости можно описать термодинамическим
соотношением:
Д/4 = A^bB + АЯ-ЛЬВ - Ереш (VI. 6)
где ДЯ^ольв и ДЯ^льв — энтальпии сольватации катиона и
аниона при бесконечном разведении.
Способы вычисления энергии решетки из термохимического
цикла или с помощью полуэмпирических уравнений Борна, Капу-
стинского и др. описаны в курсах физической химии. Очевидно, что
энергия решетки должна быть определена при той же температуре,
что и теплота растворения. Обычно это требование не влияет
существенно на точность расчета. Различие между величинами Е и Ео
(при 0°К) для галогенидов щелочных металлов не превышает
4—7 кдж/моль, т. е. того же порядка, что отличия в энергиях
решетки, найденных различными методами. Во многих случаях и
теплота растворения соли оказывается величиной того же порядка,
что и погрешность в определении энергии решетки *. Это,
естественно, сильно снижает надежность оценки энтальпий
сольватации. Для раствора вещества с ковалентной связью величину
энергии решетки Evem заменяют на энергию диссоциации Е при
образовании газообразных ионов из молекул. Определение теплот
сольватации по разности двух величин, которые сами известны
недостаточно надежно, существенно ограничивает возможности
использования их для оценки, например, влияния растворителя на
энергию сольватации.
Значения энтальпий растворения и сольватации для ряда солей
в различных растворителях приведены в табл. VI. 3, из которой
следует, что значения суммарных теплот сольватации ионов в
различных" растворителях отличаются очень мало. Имеется весьма
обширная литература, посвященная задаче разделения суммарной
теплоты сольватации соли на ионные составляющие. Задача эта
по существу является столь же формальной, как в кристаллохимии
Деление межатомных расстояний на ионные радиусы. В
действительности ион не существует в растворе сам по себе, в отсутствие
иона противоположного знака, и, следовательно, энтальпия
сольватации отдельного иона не отвечает реальному изменению энер-
гии, которое может быть зафиксировано экспериментально. Тем не
Менее, аддитивная схема расчета энергий сольватации оказалась
^ьже.полезной, как и концепция ионных радиусов, поскольку
* Энергии решетки для галогенидов щелочных металлов составляют от не-
ольких Десятков до 4000 кдж/моль, а энтальпии растворения их в воде и не-
°Дных растворах не превышают 40 кдж/моль,
89
Таблица VI.3
Энтальпии растворения и сольватация (кдж/моль) солей щелочных металлов
в различных растворителях при бесконечном разведении и 25 °С
Соль
LiCl
NaBr
Nal
NaNO3
KBr
KI
KNO3
Вода
о S
1
370,3
0,59
7,53
-20,50
-19,87
-20,33
-34,89
сольв
1
879
740
699
736
657
615
653
Метанол
о S
аз
1
51,9
-17,15
32,05
-2,09
-3,77
-0,71
—
сольв
1
895
757
711
757
674
636
—
Эганол
о S .
1
54,1
11,09
24,52
-2,93
—
—
—
и
А
8
1
908
749
707
632
—
—
—
Формамид
о S
1
39,37
18,41
30,96
—
-0,96
4,18
—
сольв
1
<
1
883
761
724
—
678
640
—
Диметилформами-
о S
1
51,05
29,71
56,90
10,04
9,62
32,22
—0,21
сольв 1
а;
1
895
770
749
757
678
669
686
она позволяет на основе сравнительно небольшого набора
исходных экспериментальных данных рассчитывать суммарные теплоты
сольватации с удовлетворительной точностью. Точность же таких
расчетов зависит и от физической обоснованности исходных
предпосылок разделения и от точности экспериментального
определения исходных данных. Различные методы разделения теплот
сольватации, описанные ^з литературе, дают в общем достаточно
хорошо согласующиеся между собой результаты.
Методы разделения термодинамических параметров
сольватации на ионные составляющие
Рассмотрим вначале метод, основанный на теории Борна —
Бьеррума. Уравнение Борна — Бьеррума для энтальпии
сольватации отдельного иона ДЯ1'
сольв
N.z.e2 [ 1 Тйг\
° 1 11 I
2г. \ е e2dTJ
(VI. 7)
(N0 — число Авогадро; е — заряд электрона) показывает, что
теплоты сольватации ионов с равными и противоположными по знаку
эффективными зарядами' z и равными радиусами г морут
совпадать по величине. Основываясь на этом допущении, Ланге и
Мищенко приравняли энтальпии гидратации цезия и иода, а Бернал
и Фаулер — калия и фтора. В настоящее время предпочтительным
считается деление по цезий-иодидной шкале. Для водных
растворов расчеты, основанные на этой шкале, дают результаты, хорошо
согласующиеся с экспериментом. Для перехода к неводным
растворам можно воспользоваться правилом Мищенко,
постулирующим постоянство отношений энтальпий сольватации катионов и
анионов в различных растворителях:
ЛЯС+ОЛЬВ/Д#С-ОЛЬВ = const (VI. 8)
90
~& &шь в)
Из правила Мищенко следует, в частности, что в любом рас-
орителе Д# Cs+= A#j-. Этот вывод является опорным пунктом
для вычисления величин АЯСОльв отдельных ионов в различных
астворителях. С правилом постоянства отношений энтальпий
сольватации связано и так называемое правило постоянства разностей.
Оно гласит, что для пар солей, содержащих одноименные катионы
анионы, разности энтальпий сольватации при бесконечном
разведении постоянны и не зависят
от растворителя.- Это правило
подтверждено
экспериментальными измерениями первых теплот
растворения для всех галогени-
дов щелочных металлов в воде и
нескольких неводных
растворителях. Некоторые отклонения для
растворов в жидком аммиаке, со- дйщЦв
гласно Мищенко и
Полторацкому, могут быть отнесены за счет
недостаточной точности
экспериментальных данных. Правило
постоянства разностей
иллюстрируется табл. VI. 4.
Метод Измайлова относится к
разделению изобарных потенциа-
ттгш \Gl гппьпятяттии* R пс Рис* VI-5* Схема определения энер-
лов А^сольв сольватации . ь ос- гии сольватацИИ иона водорода по
нове его лежат допущения: вели- методу Измайлова.
ишя Д fti гтинртшл QiinwpHT lH-А- Измайлов, Электрохимия раство-
ЧИНа АСгсольв ЛИНеИНО ЗаВИСИТ ^ов# Изд. ХГУ, Харьков, 1959, стр. 325].
от величины, обратной радиусу
иона, с некоторой поправкой 1/(Гг±р); при 1/гг —>0 ДО£Ольв->0."
Исходя из этого, изобарный потенциал сольватации, например Н+,
может быть определен следующим образом. Строят график
зависимости суммарных величин ДОСОльв для галогеноводородных
кислот от 1/(ггал ± Р), а затем производят экстраполяцию на величину
1/(г±Р) = О (рис. VI. 5). Сравнивая реальные изобарные
потенциалы соответствующих кислот и солей с одним и тем же анионом,
тт+ д»е+
можно вычислить разность Абсольв — Абсольв. Откладывая эту
разность как функцию от 1/(/*Ме+ ± Р7), получаем при экстраполяции
На ^(гме+ ± РО== 0 величину Аб"льв. Совпадение графиков при
экстраполяции служит критерием правильности расчета. Помимо
Этого, для нахождения предела может быть. использована
Точнее, химических энергий сольватации Ах, отличающихся от изобарных
отенциалов на величину энергии, затрачиваемой на преодоление границы рас-
*ВОрЦ
91
Таблица VI.4
Усредненные значения разностей энтальпий сольватации
катионов и анионов при бесконечном разведении и 25°
Ионы
Na+—Li+
К+—Li+
Rb+—Li+
Cs+—Li+
Ca2+—Li+
K+—Na+
Rb+—Na+
Cs+—Na+
Rb+—K+
Cs+—Rb+
Br-—СГ
Г—СГ
Г—ВГ
NO"—СГ
Вода
109
197
217
255
— 1075
84
109
142
21
38
29
71
^ 38
29
Метанол
109
197
222
255
84
113
146
25
33
29
63
29
29
Этанол
117
197
_
—
79
—
-—
—
—
38
71
29
—
Формамид
100
184
209
243
—
84
109
142
25
33
33
59
29
—
некоторых пар
С (в кдж/г-ион)
N-Метил-
формамид
121
197
222
268
—
75
100
146
25
46
21
50
29
Диметил-
формамид
' 113
197
217
251
-1251
84
105
138
21
33
8
29
21
8
вспомогательная линия, описываемая уравнением
Me+
причем
= f[l/(rcp±p")] (VI. 9)
(VI. 10)
Эмпирическая поправка |р| = |р'| для водных растворов
составляет 0,28 А. Н. А. Измайлов предположил, что эта величина
сохраняется и в неводных растворителях и произвел разделение
изобарных потенциалов сольватации в ряде растворителей.
Интересно, что несмотря на очевидную нестрогость исходных
допущений, разделение по Измайлову приводит к значениям энергий
сольватации отдельных ионов, весьма близким к найденным другими
методами. ' .
Позднее Н. А. Измайлов и Ю. А. Кругляк (1960) отметили
линейную зависимость изобарных потенциалов и энтальпий
сольватации от 1/я2, где п — главное квантовое число внешней
электронной оболочки ионизированного атома *. На основании этой
зависимости Измайлов разработал метод определения энергий
сольватации отдельных ионов, основанный на том, что разность величин
энергий сольватации изоэлектронных ионов галогенов и щелочных
металлов стремится к нулю, когда 1/д2->0.
* О квантово-механическом описании процесса сольватации методом молеку*
лярных орбит см. стр. 200.
92
Как и в предыдущем методе Измайлова, исходные значения
vmm и разностей энергии сольватации могут быть получены
непосредственно из электрохимических данных: по ЭДС
гальванических цепей без переноса рассчитывают суммы изобарных
потенциалов сольватации, а по э.д. с. цепей с переносом — их разности.
Значения энтальпий сольватации, как упоминалось выше, опреде-
т1яют на основе данных о теплотах растворения электролитов и
энергиях решетки.
Указанный метод дает величины, достаточно хорошо
согласующиеся с рассчитанными по методу Мищенко и по первому методу
Измайлова [экстраполяция
от 1/(г±Р)], причем Н. А.
Измайлов считал
экстраполяцию от 1/п2 более
надежной и обоснованной.
Обзор теоретических
расчетов термодинамических
параметров сольватации,
основанных на
электростатических моделях, дан в
монографии К. П. Мищенко и
Г. М. Полторацкого
(стр. 83—88). Мы здесь ИЗ-
ложим только не вошедший
Рис. VI. 6. Геометрическая схема взаимо-
действия катиона с молекулой раствори-
теля ПРИ сольватации (по Крестову и Зве-
. о РевУ)-
[В' А' Звере в' Ав2Й'«&? дисс> ИХ™' Ива'
в этот обзор метод, разра-
ботанный Г. А. Крестовым и
В. А. Зверевым (1969).
В основе метода лежит расчет электростатического
взаимодействия ионов с центрами зарядов диполей с учетом распределения
этих зарядов, основных характеристик ионов и постоянства их
координационных чисел. Для расчета необходимо знать размеры
молекул растворителя, длину диполя,> положение диполя по
отношению к центру молекулы и размеры ионов. Эффективный радиус
молекул растворителя вычисляют по уравнению
.{Г
(VI. 11)
где #M = -i? .Асимметрию диполя растворителя определяют из
межатомных расстояний, причем для кислородсодержащих
растворителей центр атома кислорода можно считать центром
отрицательного заряда диполя.
Обозначим (рис. VI. 6): Ак—расстояние от периферии молекулы
растворителя до центра отрицательного заряда диполя, Да —
расстояние до положительного центра диполя, / — длина диполя. Тог-
^а разделение суммарной энтальпии сольватации соли ДЯСОльв на
Теплоты сольватации катиона ДЯсольв и аниона ДЯ?ОЛЬв может
93
быть произведено по формуле:
дя!
(VI.
Геометрические характеристики молекул растворителей, рас.
считанные Г. А. Крестовым и В. А. Зверевым, приведены в табл
Таблица VI.5
Параметры молекул в уравнении (VI.12)
Растворитель
г, А
ДК,А
Вода 1,86 1,45 1,39
Метанол 1,68 1,89 1,22
Этанол 1,67 2,19 0,99
Аммиак 0,93 1,70 1,63
Формамид 3,37 2,06 0,89
N-Метилформамид 3,82 2,32 0,80
Диметилформамид 3,82 2,53 0,60
Таблица VI.6
Разделение энтальпий сольватации ионов в различных
растворителях при 25 °С, кдж/(г-ион) *
0,93
0,41
1,16
1,39
1,54
1,93
2,55
0,58
2,15
2,23
0,38
1,69
1,91
1,91
Растворитель
Ион
LP
NaT
С1"
Вг"
Вода
[М]
[К]
Аммиак
[М]
[К]
Метанол
[М]
[К]
Этанол
[М]
[К]
Формамид
[М]
[К] .....
N-Метилформамид
[М]
[К]
Диметилформамид
[М]
[К]
506
535,5
510
448
523
485
623
674
724
427
423
435
385
414
485
412
523
552
611
335
339
343
397,5
310
326
4Р6
326
439
477
527
349
347
393
406
372
376,5
259,5
226
167
320
318
360
368
343
264
343
238,5
205
159
284,5
276
318
280
322
310
230
303
201
176
138
* [М] и [К] — разделения по методам К. П. Мищенко (1968) и Г. А. Кре'
стова (1969).
94
VI 5. Рассчитанные на основании этих данных энтальпии
сольватации сопоставлены в табл. VI. 6 с величинами, рассчитанными
другими способами. Сопоставление изобарных потенциалов ионов,
найденных различными способами, дано в табл. VI. 7.
Таблица VI.7
Разделение изобарных потенциалов сольватации ионов в различных
растворителях при 25 °С, кдж/(г-ион) *
растворитель
Вода
1 . ....
2
3
4
Метанол
1
2
3
4 ....
Этанол
1
2
3
4
Муравьиная
кислота
1
2
3
4
Ацетонитрил
4
Ион
Н+
1063
1077
1065
1090
1050
1056
1054
1114
1048
1052
1050
1112
1033
1033
1017
1062
1079
Li+
483
490
490
511
477
485
481
539
469
481
473
532
481
485
475
520
507
Na+
389
402
393
411
379
389
385
435
368
377
372
510
418
416
383
428
413
К+
314
326
322
337
307
318
314
360
299
305
303
363
307
305
305
351
347
Rb+
301
310
303
_
—
—
289
—
—
278
307
306
293
__
-—
Cs+
259
268
263
—
253
249
—
—
—
245
«___
274
270
253
—■
СГ
318
310
312
296
306
300
305
249
301
298
303
244
324
327
330
285
233
Вг"
296
284
289
271
291
280
282
227
284
276
280
222
298
252
227
I"
259
248
253
239
256
249
253
199
251
245
245
195
__
—
247
212
207
* Разделения: 1 — по Мищенко {^О^олъв//^G^0JlbB = const); 2 и 3 — по
Измайлову [1/(г±Р)] и (1/я2); 4 —по В. Case, R. Parsons [Trans. Faraday Soc,
63, 1224 (1967)].
Как видно из табл. VI. 6 и VI. 7, различие в теплотах и энергиях
сольватации ионов, найденных различными методами, в одном и
том же растворителе, часто значительно превышает разницу в
термодинамических параметрах сольватации одного и. того же иона
в различных растворителях. Из этого можно было бы заключить,
что энергия сольватации ионов мало зависит от природы
растворителя, и вода не является уникальным растворителем.
Действительно, такого рода представления развиваются и в настоящее
время [С. И. Дракин и др., 1968], хотя их невозможно
согласовать с обширным экспериментальным материалом, показывающим
специфичность воды как растворителя, Это противоречие может
95
быть разрешено только на основании более глубокого анализа
механизма сольватации и установления относительного вклада
различных взаимодействий в этот процесс.
Построение детальной модели процесса сольватации
необходимо для учета вкладов различных составляющих эффектов в
энергетику процесса. Полный расчет процесса гидратации
выполнен К. П. Мищенко и А. М. Сухотиным; для неводных растворов
расчеты такого рода не производились, по-видимому, из-за
недостатка необходимых исходных данных. Выражение для теплоты
гидратации содержит следующие восемь членов:
произведение «внутренней» теплоты испарения растворителя *
Ev = AHV — RT на координационное число п\
экзоэффект а взаимодействия иона с жесткими диполями
растворителя;
экзоэффект £ПолГ обусловленный ориентационной и
деформационной поляризацией диполей растворителя в поле иона;
экзоэффект £дисп, обусловленный дисперсионным
взаимодействием между ионом и молекулами растворителя;
эндоэффект С, возникающий вследствие взаимного
отталкивания диполей растворителя в сольватной сфере;
эндоэффект Е0Ту вызванный отталкиванием иона и диполей соль-
ватного слоя при взаимопроникновении их электронных оболочек:
экзоэффект 5, обусловленный поляризацией молекул
растворителя, окружающих сольватный комплекс;
экзоэффект G, характеризующий взаимодействие между
«свободными» молекулами растворителя и входящими в сольватный
комплекс.
Расчеты показывают, что для большинства ионов все эффекты,
кроме первого, компенсируют друг друга и, таким образом,
энтальпия сольватации ионов определяется прежде всего этим эффектом
первичной сольватации. Можно было предположить, что такой
результат расчета получается лишь для водных растворов. Однако
Н. А. Измайлов показал, что величины теплот сольватации ионов
щелочных металлов и галогенов с одними и теми же вакантными
электронными уровнями совпадают между собой как для водных,
так и для неводных растворов [Н. А. Измайлов, Ю. А.
Кругляк, 1960]. Поскольку в углеводородах, за редким исключением,
не образуются электролитные растворы, сольватация ионов в
неводных растворителях связана обычно с поставкой недостающих
электронов донорными атомами кислорода, азота, фтора молекулы
растворителя. Так как во всех случаях электроны, будут переходить
с одного и того же электронного уровня, следует ожидать что
теплоты сольватации ионов будут весьма.близки.
Изложенные здесь представления о механизме сольватации
позволяют истолковать кажущуюся независимость энергий и теплот
сольватации от свойств растворителя. Малое различие энергий и
* Эквивалентна энергии испарения (см. гл. IV).
96
теплот сольватации ионов в различных растворителях объясняется
тем, что практически во всех исследованных растворителях
главную часть полной энергии сольватации составляет энергия
первичной сольватации Л<^ольв (перв)>которая, как было показано выше,
очень мало зависит от свойств растворителя. Вклад же энергии
вторичной сольватации AG*0JIbB (втор), зависящей от диэлектрической
проницаемости, дипольного момента и мольного объема
растворителя, обычно весьма невелик.
Из всего сказанного выше следует, что энергии и энтальпии
сольватации не дают достаточной информации о специфике
поведения растворителя. Значительно больший интерес с этой точки
зрения представляют энтропийные характеристики растворов,
поскольку энтропия, характеризующая степень упорядоченности
системы, должна весьма чутко реагировать на структурные
изменения в растворе.
Энтропия сольватации ионов
Суммарная величина изменения энтропии при сольватации
электролита легко может быть найдена из термодинамических
параметров кристаллической решетки и термодинамических
параметров растворения
ДЗсольв = ^раств ~ ЛЗреш (VI. 13)
либо на основе значений изобарного потенциала и энтальпии
сольватации
либо по температурной зависимости изобарного потенциала
сольватации
Г д (А(?сольв) 1 /ут 1,-.
I gj; J IV1. ID;
Затруднения возникают при выделении в суммарной величине
изменения энтропии составляющих, которые отражают изменение
энтропии электролита и растворителя в отдельности. Для такого
разделения используют метод относительных парциальных моляль-
ных величин и метод, основанный на определении реальных
термодинамических характеристик составных частей раствора.
В методе парциальных моляльных величин, о котором речь уже
шла выше, разделение суммарного изменения энтропии
производят формально, путем простого дифференцирования
термодинамической функции по числу молей компонентов. Как и в случае
растворов неэлектролитов, значительный интерес здесь представляют
избыточные парциальные моляльные функции, получаемые путем
вычитания из наблюдаемого значения функции значения ее для
идеальных растворов. Избыточная парциальная моляльная
4 Зак, 500 Q7
энтропия растворителя определяется выражением:
Щ - (ля5 - RT in -JV г (VI. i6)
При этом мольную долю растворителя xs вычисляют по формуле:
1000/Ms
*s = 1000/Ms + vm (VL17)
Здесь m — моляльность электролита, v — число ионов, на которые
электролит распадается при растворении.
Данные по концентрационной зависимости избыточных
относительных парциальных моляльных энтропии в некоторых неводных
растворах получены в лаборатории К. П. Мищенко. Оказалось, что
для всех изученных неводных растворов, в отличие от водных,
наблюдаются отрицательные значения S| во всем интервале
концентраций. Это свидетельствует об упорядочивающем влиянии
сольватационных процессов на структуру жидкости. Вода же
насколько превосходит другие жидкости по степени упорядоченности,
что разупорядочивающее действие ионов на ее структуру
превалирует над упорядочивающими процессами.
Иной подход к оценке энтропийных характеристик сольватации,
приводящий качественно к аналогичным выводам, развит
Г. А. Крестовым (1966). Следуя О. Я. Самойлову, он
рассматривает изменение энтропии в процессе сольватации как сумму двух
составляющих
Ассоль»-A^ + AS^ (VI. 18)
где AS/— изменение энтропии при образовании сольватирован-
ного иона из газообразного; AS//— изменение энтропии,
связанное со структурными изменениями растворителя. Величина AS/J
согласно Крестову, может быть представлена как разность
энтропии поступательного движения иона в поле молекул растворителя
и в газе:
AS] = S'.p -5j (VI. 19)
Энтропию иона в газовой фазе вычисляют по уравнению Заккура—
Тетроде
S[ «|- R In Mt + 25,7 (VI. 20)
где Mi — атомная масса иона.
Теоретический расчет величины Sp.p затруднен. Для водных
растворов и, с оговорками, для неводных растворов принимают ее
равной 0,385 S[. Следовательно, величина AS// может быть
определена из уравнения;
A^-AS^ + 0,616^ (VI. 21)
98
В свою очередь, величина ASJ/ может быть представлена в виде
суммы двух величин, одна из которых связана с образованием
полостей в структуре растворителя — ASA, а другая — с действием
иона на структуру растворителя Д5С:
A5j7 = Д5Л + Д5С (VI. 22)
Величина AS а имеет тот же физический смысл, что и для процесса
растворения изоэлектронйых с данным ионом благородных газов
(см. гл. V). Из эмпирических соотношений, найденных Г. А.
Крестовым, следует, что
^SA = o,345Sr
ASC =■
— 0,3455г
(VI. 23)
(VI. 24)
Разделение энтропии сольватации соли на составляющие,
относящиеся к катиону и аниону, может быть проведено, исходя из
предположения, что правило постоянства отношений
термодинамических функций отдельных ионов, выведенное Мищенко для теплот
сольватации, может быть распространено и на энтропии
сольватации, т. е.
Р/Д5Г-ИДР = ASc+QJIbB/ASc-0JIbB (VI. 25)
Значения энтропии сольватации некоторых ионов, и
составляющие энтропии представлены в табл. VI. 8. Приведенные данные
свидетельствуют, что изменения энтропии, связанные со структурой
растворителя, в спиртах намного более отрицательны, чем в воде,
причем различие между водой и метанолом намного больше, чем
между метанолом и этанолом. Суммарное уменьшение энтропии
обусловлено упорядочиванием структуры раствора под действием
ионов. В то же время, внедрение ионов в первичную структуру
Таблица VI.8
Составляющие энтропии [в дж/(град • моль)] некоторых ионов в воде
и спиртах при 25 °С
Г. А. Крестов, Ж. структ. хим., 3, 516 (1962)].
Ион
Li+
Na+
к+
сг
Вг-
г
Вода
гидр
со
1
127,6
96,6
61,1
89,1
74,1
51,5
со
-45,6
—5,4
33,9
5,0
26,8
52,7
Метанол
m
л
8
со°
1
198,3
152,7
—
162,3
139,7
121,7
к.
со*
1
116,7
61,9
—
68,2
39,3
17,6
с
со
<з
1
162,3
113,0
—
120.9
95,8
76,1
со
45,6
51,0
—
52,7
56,5
58,6
Этанол
ю
л
со°
1
208,4
172,0
140,2
167,8
139,3
136,4
со*
1
126,8
81,2
44,8
73,6
38,9
32,2
2
i
172,4
132,2
136,0
130,5
95,4
90,8
<
%
45,6
51,0
53,6
56,9
56,5
58,6
99
чистого растворителя должно вызывать разупорядочивание
структуры и повышение энтропии, что и наблюдается в воде. Какой из
этих эффектов будет преобладать, зависит от степени
упорядоченности структуры чистого растворителя. На основании энтропийных
измерений Г. А. Крестов расположил растворители в ряд по
уменьшению компактности структуры:
Н2О > СН3ОН > С2Н5ОН > С6Н5С1 > СН3СООСН3 > (СН3)2СО >
> СС14 > QH6 (и другие углеводороды).
Этот ряд согласуется с представлениями, базирующимися на
результатах измерений физических свойств растворителей.
Корреляция между составляющей ионной энтропии, характеризующей
чистый растворитель, и физическими свойствами растворителя,
рассмотрена в работе Крисса и др. [С. М. Criss, L. P. Held,
Е. Luksha, 1968].
Весьма интересные данные о процессе сольватации могут быть
получены на основании измерений теплоемкости растворов, однако
в литературе имеются сведения в основном о водных и
водно-неводных растворах, ^поэтому анализ связи этих величин со
структурой растворителя не может пока быть осуществлен.
VI. 4. Коэффициенты активности электролитов
в неводных растворах
Отличия в определении коэффициентов активности
электролитов и неэлектролитов обусловлены в первую очередь выбором
стандартного состояния. Так, если для неэлектролитов в качестве
стандартных состояний выбираются обычно чистые компоненты, то
для определения активности соли в растворе нецелесообразно в
качестве стандартного состояния рассматривать кристаллическую
соль, поскольку это исключает возможность сопоставления
растворов различных солей в различных растворителях. Гораздо удобнее
выбрать в качестве стандартного состояния гипотетический
раствор, в котором отсутствует межионное взаимодействие (т. е.
состояние ионов такое же, как в бесконечно разбавленном растворе),
а концентрация ионов в принятой шкале равна единице *.
Парциальный _изоба_рный потенциал_растворенного вещества в таком
растворе Ga = G°a + RT In a == Ga, to есть он равен стандартному
изобарному потенциалу, а коэффициент активности у = 1.
Численные значения коэффициентов активности зависят от
выбора шкалы концентраций:
в шкале моляльности уьт = ailmi\
в шкале молярности — у1с = а.[с.;
в шкале мольной
* В дальнейшем такой раствор для упрощения будем называть бесконечно
разбавленным.
100
Величины у1т, Yc и Y* называют моляльным, молярным и
рациональным коэффициентами активности. В разбавленных растворах
(при выборе упомянутого выше стандартного состояния)
коэффициенты активности в разных шкалах численно весьма близки, по
мере увеличения концентрации различие между ними возрастает.
В растворах неэлектролитов обычно используются рациональные
коэффициенты активности. Индекс I относят к компонентам
системы, которые могут присутствовать в молекулярной или иной
форме.
Если электролит pKq+qLp~ диссоциирует по схеме: pKg+qLp~7^.
^pKq+ + qLp~, то константа равновесия этого процесса
определяется из соотношения:
(VL26)
При определении коэффициентов активности растворов
электролитов, например по давлению пара или из криоскопических
измерений, не удается рассчитать в отдельности коэффициенты
активности, относящиеся к ионам и к недиссоциированному
электролиту; для такого расчета необходимы данные по
электропроводности или по э. д. с. Величины же активностей отдельно катионов и
анионов экспериментально вообще не могут быть раздельно
определены.'Поэтому для оценки активности ионов в растворе вводят
понятие о средней ионной активности:
Величины средних ионных активностей зависят в общем случае
от изменения: количества частиц в растворе — в связи с
диссоциацией, ассоциацией и сольватацией — и энергии частиц в растворе
в результате межионных и ион-дипольных взаимодействий.
Действие первого фактора связано с концентрацией раствора, что
обуславливает концентрационную зависимость коэффициентов
активности. Изменение энергии частиц в растворе зависит от природы
компонентов, образующих электролитный раствор.
В принципе возможно расчленить действие этих факторов,
выбирая при изучении растворов различные стандартные состояния.
Например, исследуя термодинамические свойства растворов иоди-
Да натрия в этиленгликоле, можно выбрать в качестве
стандартного состояния либо бесконечно разбавленный раствор его в этом
растворителе, либо бесконечно разбавленный раствор в воде.
В первом случае величины коэффициентов активности отражают
только изменение состояния раствора в связи с изменением его
концентрации («концентрационные» коэффициенты активности),
во втором случае с помощью коэффициентов активности
учитывается и специфика растворов этиленгликоля в сравнении с водой.
В качестве единого стандарта выбирают обычно водный
Раствор. Это означает, что в бесконечно разбавленном водном
101
растворе коэффициент активности исследуемого вещества
принимается равным единице, и активность вещества при любой
концентрации в любом другом растворителе отсчитывается от этого
стандарта. Вычисленные таким образом коэффициенты
активности называются едиными. Концентрационные коэффициенты
активности обозначаются, соответственно, у*т, у*с и у*х. Специфику
растворителя отражают единые нулевые коэффициенты
активности у°т, у°с и у°х. Они связаны с изменением химического
потенциала при переносе вещества из бесконечно разбавленного
водного раствора в бесконечно разбавленный раствор в неводном
растворителе
Yo - ехр [(|4 - \1и2о)1Щ (VI. 27)
где \xos — химический потенциал растворенного вещества в
неводном растворителе S.
Можно установить соотношение между названными выше
коэффициентами активности, если учесть, что обратимая работа
переноса вещества из бесконечно разбавленного водного раствора в
раствор конечной концентрации в растворителе S может быть
представлена в виде суммы работ переноса вещества: из
бесконечно разбавленного водного раствора в бесконечно
разбавленный раствор в растворителе S и из бесконечно разбавленного
раствора в растворителе S в раствор конечной концентрации в этом
же растворителе. Тогда:
\i *~ \х° + \х* (VI. 28)
RT In y = RT In Yo + RF In Y* (VI. 29)
Y = YoY' (VI. 30)
Величина yo отлична от единицы в том случае, когда раствор в
стандартном растворителе (воде) и в исследуемом растворителе
S характеризуются различными отклонениями от идеальности. На
практике это наблюдается почти всегда.
Выбор водного раствора в качестве стандартного обусловлен
только наибольшей изученностью водных растворов. Если же
учесть специфичность воды как растворителя, то такой выбор
представляется не совсем удачным. Тем не менее, поиски иного
стандарта среди неводных растворителей вряд ли были бы
целесообразны, поскольку, как показывает опыт, в принципе
невозможно подобрать растворитель, который со всеми электролитами
давал бы идеальные растворы, либо растворы с одинаковыми
отклонениями от идеальности.
Определение коэффициентов активности компонентов
растворов электролитов может быть выполнено прямыми либо
косвенными методами. Если изучается давление пара над раствором
соли, то метод является прямым по отношению к растворителю и
косвенным по отношению к растворенному веществу. Наоборот,
метод э. д. с, язляется прямым по отношению к растворенному
102
еществу. В основе расчетов по косвенному методу лежит
уравнение Гиббса —Дюгема:
d In п2 = d In a\
x2
Наиболее широко применяют методы изучения активности
растворителя, основанные на измерениях давления пара и
понижения температуры замерзания раствора, а также методы изучения
активности растворенного вещества по измерениям э. д. с.
гальванических цепей и давлению пара растворенного вещества. Реже
используются измерения растворимости, осмотического давления
и коэффициентов распределения растворенного вещества между
двумя растворителями. Подробная теория и описание техники
эксперимента указанных методов даны в монографиях Н. А.
Измайлова (1959), Робинсона и Стокса (1963).
Коэффициенты активности могут изменяться с концентрацией
в чрезвычайно широких пределах. Высокие значения их
приписывают обычно сольватации ионов, умеренно низкие — объясняют
образованием ионных пар и очень низкие — образованием
комплексных ионов. Как в водных, так и в неводных растворах для
многих электролитов кривая зависимости коэффициента
активности от концентрации имеет минимум, обусловленный сочетанием
указанных факторов. Однако, например, для нитратов щелочных
металлов такой минимум отсутствует как в водных, так и в форм-
амидных растворах.
При одинаковых концентрациях соли коэффициент активности
убывает в ряду: LiCl > NaCl > КС1 > RbCl > CsCl как для
водных, так и для формамидных растворов. Для одного и того же
электролита коэффициенты активности уменьшаются с
уменьшением е среды (табл. VI. 9).
Таблица VI.9
Концентрационные коэффициенты активности НС1 в различных
растворителях при 25 °С и т = 0,10
Растворитель
н2о
СНзОН
с2н5он
*-с3н7он
8
78,3
32,6
24,3
18,3
0,796
0,440
0,314
0,180
Растворитель
С4Н9ОН
*-С4Н9ОН
/-СБН„ОН
СНзСООН
;е
17,7
17,7
14,7
6,1
?•
0,125
0,121
0,100
0,033
VI. 5. Термодинамические функции отдельных ионов
Все существующие методы разделения термодинамических
Функций электролитов на ионные составляющие относятся к бес-
Конечно большому разведению электролита, т, е. к такому
103
состоянию, в котором практически можно пренебречь межионньщ
взаимодействием. В реальных растворах эти методы неприложимы.
Поясним это примером.
Пусть известны коэффициенты активности солей KiLi, К2Ц и
K1L2 в растворителе S при нескольких концентрациях, а
экстраполяцией найдены их значения при бесконечном разведении. Пред.
положим, что парциальные молярные изобарные потенциалы
электролитов, связанные с коэффициентами активности, можно
аддитивно разделить на ионные составляющие так же, как и
соответствующие термодинамические параметры сольватации. Если
это предположение справедливо, то найденный на основании
указанных выше данных коэффициент активности электролита K2L2
в том же растворителе должен совпадать с экспериментальным
значением. Действительно, оперируя справочными данными для
водных растворов NaBr, КВг и NaCl, находим для КС1 при
бесконечном разведении значение коэффициента активности,
совпадающее с табличным. Однако при концентрации электролитов
/п = 0,1 расхождение между экспериментальным и рассчитанным
значением составляет 0,010, и с ростом концентрации это
расхождение возрастает.
Разделение термодинамических параметров процессов
сольватации на ионные составляющие было рассмотрено в разделе VI. 3.
Здесь мы коснемся вопроса о вкладах отдельных ионов в теплоту
растворения электролита. Поскольку теплота сольватации,
теплота растворения и энергия кристаллической решетки солей
связаны уравнением (VI.6), а методы разделения теплот
сольватации известны, решение поставленной задачи сводится к
разделению энергии кристаллической решетки по вкладам аниона и
катиона. Такой расчет для ряда солей был выполнен Г. И. Микули-
ным (1966). Им выведена формула для расчета разности энергий
Таблица VI.10
Значения (Ек — Еа) для галогенидов щелочных металлов, кдж/г-ион
Соль
LiF
LiCl
LiBr
Lil
*K-a
-2,9
2,9
0
-2,9
Соль
NaF
NaCl
NaBr
Nal
*к-а
-5,0
-5,9
—7,1
-0,8
Соль
KF
KCl
KBr
KI
-5,9
-10,9
-7,1
-8,8
катиона и аниона в кристаллической решетке (Ек — £а),
включающая члены, связанные с силами диполь-дипольного и диполь-
квадрупольного взаимодействия, а также члены, обусловленные
квантовомеханическими силами отталкивания электронных
оболочек ионов (табл. VI. 10).
104
Теплоты растворения катиона и аниона вычисляются по
формулам:
Л„к __ АНт . Ек-"Еа (— А#£ольв) - (— А#сольв) ,VT on
а т— 2 2 2
а г/а АЯт Ек~Еа , ("" АЯсольв) "" (—АЯсольв) ,„. q~
A^m^—2 2 ' 2 (VI. dJ)
Раздельное определение теплот растворения по этим
формулам было произведено Г. И. Микулиным для водных растворов;
для неводных сред такое разделение отсутствует.
Определенный интерес для термодинамического описания
растворов представляет нахождение теплоемкости отдельных ионов
при бесконечном разведении. Для разделения теплоемкостей
электролитов при бесконечном разведении на ионные составляющие
необходимо либо задаться определенной величиной С°р для
какого-либо иона, либо разделить пополам теплоемкость какого-
либо электролита, полагая равными теплоемкости катиона и
аниона. При использовании первого метода обычно приравнивают
к нулю парциальную мольную теплоемкость иона водорода.
Второй метод подробно рассмотрен в монографии К. П. Мищенко и
Г. М. Полторацкого, где авторы, сравнивая различные шкалы
ионных теплоемкостей, считают наиболее целесообразным принять
допущение С°р ,=ѰР_ = — 27,6 дж1(г-ион-град) при 25° С для
NH. С1
4
водных растворов. К сожалению, для неводных растворов данные
по теплоемкости электролитов практически отсутствуют. Это
лишает нас возможности судить о применимости той или иной
шкалы теплоемкостей для неводных растворов, а также о наличии
или отсутствии специфики теплоемкостных характеристик
электролитов в различных неводных растворителях.
РАВНОВЕСИЯ В НЕВОДНЫХ РАСТВОРАХ
Глава VII
ОБЩАЯ СХЕМА РАВНОВЕСИЙ
В РАСТВОРАХ
Процессы, сопряженные с образованием раствора, особенно
электролитного, неизменно находились в центре внимания общей
теории растворов. Именно вокруг вопросов, связанных с числом
и характером стадий, предшествующих возникновению
электролитного раствора, велись дискуссии между сторонниками различных
теорий вопроса (см. гл. I). Синтез этих двух основных
направлений в учении о растворах в значительной мере был связан именно
с установлением общей схемы равновесий в растворах.
Представления о процессах, приводящих к возникновению
электролитного раствора, развивались рядом исследователей
[Хальбан, 1948; Смит и Эллиот, 1950; Корней-, 1963; Ш а-
тенштейн, 1955; Кольтгоф и Брукенштейн, 1956]. В
наиболее общем виде схема равновесий, учитывающая как
химические взаимодействия в системе, так и энергетику равновесий
процессов была разработана Н. А. Измайловым (1955—1959 гг.).
VII. I. Ассоциативно-диссоциативные процессы
Ассоциированная молекула растворенного вещества Ах при
растворении в растворителе S может претерпевать молекулярную
диссоциацию:
А* *=* ... *=* хк (VII. 1)
Последняя может быть связана с физическим влиянием
растворителя (например, ослаблением межмолекулярного диполь-ди-
польного взаимодействия в растворителях с высокой диэлектриче-
екой проницаемостью). Однако большей частью энергия, нужная
для разрыва связей в ассоциате, черпается из химического
взаимодействия между Ах и S:
Ах + mxS *=* xASm (VIL 2)
Если молекулы растворителя ассоциированы, то образование
раствора приводит к изменению этого состояния тем больше, чем
больше концентрация раствора. Так же, как и в условии (VII. 2)*
106
молекулярная диссоциация растворителя может быть вызвана
химическим взаимодействием между А и S:
nyA + Sy *=* ... *=* yAnS (VII. 3)
Определение степени ассоциации растворенного вещества в
растворе относится к числу наиболее распространенных в
практике физико-химического исследования экспериментов. В случае
разбавленных растворов для решения этой задачи успешно
применяют группу методов, основанных на законе Рауля — эбулио-
скопию и, особенно, криоскопию. Большинство сведений о
молекулярном состоянии растворенных веществ в разбавленных
растворах, которыми располагает в настоящее время физическая
химия, получены с помощью последнего метода. К недостаткам
криоскопии и эбулиоскопии следует отнести сильное влияние на
точность определения флуктуации концентрации в растворе
(обстоятельство, которое не всегда учитывается исследователями),
а также приложимость этих методов лишь к весьма
разбавленным растворам.
Весьма полная информация о молекулярном состоянии
растворенного вещества, особенно в тех распространенных случаях,
когда образование ассоциатов обусловлено Н-связью, может быть
получено с помощью ИК (КРС)-спектроскопии. Уменьшение
степени ассоциации, сопряженное с разрывом Н-связей, приводит
к сдвигу частот валентных колебаний ОН в длинноволновую
область. Во многих случаях установлена весьма определенная
корреляция между сдвигом максимума полосы поглощения и
степенью ассоциации. Впрочем, так же, как и в случае методов,
основанных на законе Рауля, сколь-нибудь исчерпывающая
количественная информация о степени ассоциации по
спектроскопическим "данным может быть получена в разбавленных растворах.
Ассоциативно-диссоциативные процессы в растворах с
несколько более высокой концентрацией могут быть исследованы с
помощью ЯМР-спектроскопии: разрыв ассоциатов через Н-связь
приводит к сдвигу сигнала протонного магнитного резонанса в
сторону более сильного поля. (В системах, где компоненты содержат
арильное ядро, это явление не проявляется с необходимой для
точного расчета степени ассоциации четкостью).
С повышением концентрации раствора точность методов
определения степени ассоциации быстро падает. Связано это не только
с несовершенством экспериментальных и расчетных методик,
которые основаны на недостаточном теоретическом обосновании
методов, но прежде всего на возрастании по мере роста
концентрации раствора степени неопределенности понятия «ассоциация».
Действительно, в предельном случае — в индивидуальной
жидкости— далеко не всегда бывает очевидным, какой критерий следует
положить в основу понятия «степень ассоциации».
Определенность, с которой физика вводит это понятие через представления
107
о ближнем и дальнем рорядках в жидкости, лимитируется отсут.
ствием четкой границы между «порядками».
Для определения степени ассоциации индивидуальной жидко-
сти предложено несколько методов. Весьма часто для этой цели
применяют уравнение Рамзая — Шильдса, использующее данные
по поверхностному натяжению у:
где х — степень ассоциации; Гкр — критическая температура; Г —
температура жидкости; А — константа, равная приблизительно
279 °К; d — плотность; М — формульная молекулярная масса.
Близко по характеру входящих в него величин к уравнению
(VII. 4) примыкает уравнение Беннета — Митчелла
(у-Т ||г) MV2£ = const (VII. 5)
где VM — мольный объем; а М — истинная молекулярная масса.
Для оценки степени ассоциации индивидуальной жидкости
может быть использована энергия активации вязкого течения El
[см. уравнение (IV. 18)]. Установлено существование
функциональной зависимости между Ё1 и VM для отдельных групп химически
сходных веществ [Tiuzzio, 1953]. Построив такую зависимость
для заведомо неассоциированных жидкостей (например, алканов),
можно определить условный мол>ный объем исследуемой
жидкости в предположении отсутствия ассоциации и, сопоставляя эту
величину с экспериментальным значением Ум, оценить степень
ассоциации.
Следует, впрочем, подчеркнуть, что методы, основанные на
применении уравнений (VII. 4) и (VII. 5), а также метод Тиуццио,
неточны и зачастую приводят к противоречивым результатам.
Поэтому в настоящее время их можно рекомендовать лишь для
качественного описания ассоциации индивидуальной жидкости.
Весьма точная оценка ассоциативного состояния
индивидуальной жидкости может быть получена из экспериментальных
определений коэффициентов самодиффузии. Для водородсодержащих
веществ весьма действенным методом является «спиновое эхо» —
вариант метода ЯМР, который позволяет определить коэффициент
самодиффузии. Найденное значение сравнивают со значением
коэффициента самодиффузии Z)t, рассчитанным по уравнению Хауг-
тона: Dx = RTcpl2d/6r\M (ф — объем, занимаемый одной молекулой,
включая свободный объем). Сравнение экспериментального
значения Dx с теоретическим позволяет рассчитать отношение
Дэксп/Мгеор и, таким образом, определить степень ассоциации.
Уравнение Хаугтона вытекает из развиваемой им модели
кубических ячеек,, которая принимает жидкость как квазикристалл,
обладающий лишь ближним порядком. Диффузию при этом рас-
108
сматривают как непрерывный стохастический процесс, при котором
в движении участвует не одна молекула, а кубическая ячейка,
содержащая определенное число молекул, располагающихся около
центральной молекулы.
Большей частью уравнение Хаугтона пригодно лишь для оценки
величины коэффициента самодиффузии. Одна из причин этого
состоит в том, что в уравнении используют величины
макровязкости, характеризующие силы трения в непрерывной среде, в то
время как движение молекулы происходит в дискретной среде.
Для исследования молекулярного состояния индивидуальной
жидкости с весьма хорошими результатами применяют группу
методов, основанную на релеевском рассеянии света на флуктуациях
ориентации молекул [М. И. Шахпаронов, 1963, 1970].
Тепловое движение молекул жидкости приводит к случайным
локальным упорядочениям ориентации молекул — флуктуациям
ориентации, которые являются одной из причин релеевского
рассеяния света в жидкостях.
Коэффициент рассеяния света на флуктуациях ориентации ра~
вен
_ 13 / г2 А /лпт ..
*°р = — ТГТГ-Т+д- ^ (VIL6)
Здесь /0 — интенсивность монохроматического, неполяризован-
ного параллельного пучка лучей, падающего на кювету с
жидкостью; / — интенсивность релеевской линии света, рассеянного
под углом 90°; г — расстояние от центра рассеивающего объема
до точки наблюдения; V — объем жидкости, рассеивающей свет;
Д — степень деполяризации линии.
Установлена связь между #Ор и анизотропией поляризуемости
молекул или молекулярных ассоциатов:
где Я — длина волны света; п — показатель преломления; Л^ —
число Авогадро; d — плотность; М — молекулярная масса; g —
средняя статистическая анизотропия поляризуемости.
Развитие этого метода позволяет определить не только число,
но и геометрию молекул в ассоциате. Метод обладает высокой
чувствительностью. Установлено, например, что молекулы бензола
образуют я-комплексы, в которых оси (С6) этих молекул взаимно-
перпендикулярны, а энергия связи составляет ~8 кдж/моль
(2 ккал/моль).
В жидких системах глубокий распад ассоциированных
молекул происходит лишь в случаях значительного преобладания
одного из партнеров (при этом, разумеется, протекает в основном
Диссоциация компонента, концентрация которого мала). Следует
считать неподтвердившимися предположения авторов многих
работ, в которых постулировался глубокий распад ассоциированных
109
компонентов даже при образовании концентрированных
растворов.
Действительно, на ИК-спектрах растворов ассоциированного
компонента в неассоциированном заметный сдвиг частоты
валентных колебаний, отвечающих Н-связи, проявляется лишь при
больших разбавлениях. К аналогичным выводам приводит анализ
спектров ЯМР систем типа «ассоциированный компонент — неас-
социированный компонент». Термодинамические характеристики
таких систем объясняют, почему заметная молекулярная
диссоциация протекает лишь в достаточно разбавленных растворах.
Так, ДЯСМ эквимолекулярных количеств бутанола и гексана
составляет всего 0,84 кдж (0,2 ккал) на 1, моль спирта, в то время
как образование 1% раствора спирта в гексане ведет к тепловому
эффекту Д#=+154 кдж (+39 ккал) на 1 моль спирта, что
вполне достаточно для образования мономеров спирта, так как
соизмеримо с энергией Н-связи в ассоциатах бутанола.
VII. 2. Образование продуктов присоединения (сольватация)
Переходя к рассмотрению следующей стадии равновесий в
растворах, связанной со взаимодействием между компонентами
раствора, необходимо, хотя бы кратко, остановиться на определении
понятия «химическое взаимодействие в растворе».
Ответ на вопрос, что следует считать необходимым и
достаточным признаком для суждения об отсутствии химического
взаимодействия между компонентами раствора с самого начала был
связан с трудностями методологического характера. Как известно, не
может быть дано сколь-нибудь строгого определения негативного
понятия, а отсутствие взаимодействия несомненно принадлежит
к числу таковых.
Далее, если считать признаком возникновения химической
связи взаимодействие электронных облаков атомов химических
элементов, то строго говоря, поскольку плотность электронного
облака на любом конечном расстоянии от атомного ядра не равна
нулю, следует признать наличие химического взаимодействия
между любыми двумя атомами, находящимися друг от друга на
любом конечном расстоянии. Но, очевидно также и то, что
исповедование этого положения лишило бы химию сколь-нибудь четких
критериев в определении самого ее фундаментального понятия.
Поэтому в определении понятия химическое взаимодействие (так
же, как и атомный радиус) всегда лежит какое-либо
предварительно оговоренное условие, а само оно всецело зависит от этого
условия.
По-видимому, наиболее удобным критерием, который мог бы
служить для суждения о наличии или отсутствии
химического взаимодействия в химической системе, является энергия
взаимодействия. Можно было бы выбрать такую минимальную ее
величину, которая могла бы быть отнесена к химическому про-
110
цессу. Однако несовершенство и значительная сложность методов
определения энергии взаимодействия не позволяет в настоящее
время считать такой выбор удачным.
Тип связи также не может служить критерием, который лег
gbI в основу суждения о наличии или отсутствии взаимодействия.
3 самом деле, между крайними случаями — образованием чисто
ковалентной или ионной связи и слабым вандерваальсовским
взаимодействием — существует множество промежуточных типов,
одна классификация которых подчас вызывает значительные
затруднения.
Строго говоря, процесс молекулярной ассоциации
(диссоциации) должен быть отнесен к химическому взаимодействию.
Действительно, если, например, ассоциат (АН)* через Н-связь
подвергается молекулярной диссоциации, то это сопряжено с
разрывом Н-связи, т. е. по всем формальным признакам (в число
которых включается и весьма значительная величина энергии
связи) подходит под химическое взаимодействие. Однако
ассоциативно-диссоциативные процессы не принято относить к
химическому взаимодействию. В противном случае пришлось бы
разграничивать химическое взаимодействие, относящееся к первой стадии
равновесий в растворах и межмолекулярное взаимодействие
между компонентами раствора:
(АН)* + (SH)j, 5F=fc ... *=* АН ... SH (VII. 8)
(предполагается для простоты, что молекулы растворителя также
ассоциированы через Н-связь).
Впрочем, вряд ли было бы целесообразным считать любое
межмолекулярное взаимодействие типа (VII. 8) химическим. Так,
если АН и SH — соединения тождественной химической природы,
стоящие к тому же близко в гомологическом ряду (например,
пропанол и бутанол, изомеры крезола, валериановая и масляная
кислоты и т. п.), то образование новых межмолекулярных связей
(перекрестных ассоциатов) не может быть отнесено к
химическому взаимодействию, хотя бы потому, что это взаимодействие
практически не сопряжено с таким изменением энергетических
характеристик системы, которое могло бы быть с уверенностью
зафиксировано.
Выбор критерия, которым следует пользоваться для суждения
о взаимодействии в химической системе, в каждый данный период
развития химии зависит от развития теории, а также методов
исследования химических систем. Так, в настоящее время благодаря
интенсивному развитию ИК- и радиоспектроскопии, появляется
возможность получения гораздо более интимных сведений о
химической системе, чем два-три десятилетия назад. И несомненно,
что спустя определенный промежуток времени появятся методы
исследования, которые углубят степень познания процессов,
протекающих в системе, столь же значительно, как это сделали
упомянутые методы. Вот почему всегда, гозоря о наличии или
111
отсутствии химического взаимодействия, следует оговаривать, 0
какой степени приближения идет речь, так как очевидно, что
универсального определения, одинаково пригодного для всех случаев,
быть не может.
Хотя современные методы исследования позволяют
зафиксировать весьма слабые химические взаимодействия с энергией
порядка десятой дол-и ккал/моль, в подавляющем большинстве
случаев в растворах они не могут быть с уверенностью обнаружены.
Дело в том, что энергетические эффекты смешения соизмеримы,
а зачастую значительно превышают указанную величину. Вот
почему слабая энергетика химического взаимодействия в растворах
не может быть выделена на фоне значительных тепловых
эффектов смешения, а также теплового движения молекул.
В этих случаях предпочитают говорить об отсутствии
химического взаимодействия между компонентами, образующими
раствор. Такая точка зрения является приближением, весьма
пригодным для рассмотрения процессов, протекающих в растворах.
Таким образом, процесс химического взаимодействия между
растворенным веществом АК и растворителем S
nS *=* (AK)mS« (VII. 9)
с точки зрения представлений, которые будут соблюдаться и
в этой книге, протекает не всегда. Он необходим лишь тогда,
когда образуется электролитный раствор. В литературе
химическое соединение (AK)mSn часто называют сольватом, а сам
процесс (VII. 9) — сольватацией. Прежде понятие «сольват» очень
часто отождествляли с понятием «соединение неопределенного
состава». В тех же случаях, когда стехиометрия взаимодействия
между АК и S была точно известна, употребляли термин «продукт
присоединения». По мере усовершенствования методов
исследования растворов смысловое различие между этими двумя понятиями
постепенно исчезает. В этой книге мы будем считать эти понятия
синонимами, применяя, следуя традиции, укоренившейся в
литературе, термин.«сольватация» для обозначения процессов
присоединения молекул растворителя к ионам, а термин «продукт
присоединения»— для сольватов, образующихся в жидких системах.
В случае ионных сольватов принято различать первичную и
вторичную сольватные оболочки. При этом в первичную оболочку
входит то сравнительно небольшое количество молекул
растворителя, которые непосредственно связаны с ионом (причем энергия
связи ион — молекула растворителя соизмерима с энергией
химической связи) и которые перемещаются в растворе вместе с ионом.
Вторичная сольватная оболочка включает значительно большее
число молекул растворителя. Границы этой оболочки
определяются изменением физико-химических характеристик
растворителя под влиянием иона.
Исследование стехиометрии и глубины взаимодействия в
растворах, т» е. изучение стехиометрии и константы равновесия про*
112
цесса (VII. 9) является предметом обширнейшего раздела теории
растворов. Относительно подробному рассмотрению этих^ методов
будет посвящена следующая глава.
VII. 3. Ионизация
Энергетический анализ процесса электролитической
диссоциации показывает, что сольват (AK)mSn не может непосредственно
перейти в диссоциированные на ионы соединения. Так, продукт
присоединения амина к кислоте RCOOH • NR£ не может
непосредственно дать ионы RCOO- и [NR3H]+. Для этого потребовалось бы
затратить весьма значительную энергию, необходимую для
разрыва связи —О—Н. Поэтому стадии электролитической
диссоциации продукта присоединения амина к кислоте предшествует
стадия ионизации продукта присоединения, заключающаяся в
отторжении протона от гидроксильной группы с переходом его на
атом азота, причем образуется ионизированный комплекс
RCOO-.[NR3H]+.
В общем виде процесс ионизации передается схемой:
(АК)« S* ^=* pKq+Sn-y • qbpSy (VII. 10)
Нередко взаимодействие между компонентами раствора
может ограничиваться стадией ионизации. Особенно часто это
встречается в растворах с низкой диэлектрической проницаемостью.
Например, при сливании разбавленных бензоловых растворов
трихлоруксусной кислоты и триэтиламина образуется
практически нацело ионизованный продукт присоединения,, однако
электропроводность раствора, очень низка. Кондуктометрические и
криоскопические исследования такого раствора показывают, что
ионизованный комплекс СС1зСОО~*[Е1з]МН]+ подвергается
электролитической диссоциации в ничтожной степени.
Наиболее удобным методом изучения стадии ионизации в
растворах является исследование колебательных спектров. Однако
константа ионизации при этом может быть рассчитана лишь тогда,
когда электролитическая диссоциация протекает в малой степени,
так как в подавляющем большинстве случаев на ИК-спектрах
полосы поглощения, отвечающие ионам, находящимся и в свобод-
ком состоянии и в ионизованном комплексе, неразличимы.
В жидких системах информация о стадии ионизации может
быть получена на основании кондуктометрических измерений.
Анализ кондуктометрических диаграмм применительно к отдельным
стадиям равновесий в растворах будет приведен в гл. VIII.
В тех случаях, когда ионизованный комплекс не ассоциирован,
т. е. процесс
t[pKq+Sn-yqAp-Sy] *=* [pKq+Sn-yqAP-Sy]t (VII. 11)
113
не протекает, данные о константе ионизации могут быть получены
из криоскопических измерений.
В растворителях с высокими диэлектрическими проницаемо-
стями электролитическая диссоциация может протекать полностью
и равновесная концентрация ионизованного комплекса будет
пренебрежимо мала. Однако это не означает, что электролитный
раствор мог образоваться, минуя стадию ионизации. Развитие методов
изучения быстрых реакций показывает, что даже в растворителях
с весьма высокими диэлектрическими проницаемостями (вода,
серная кислота) возникновению свободных ионов неизбежно
предшествует стадия ионизации.
VII. 4. Электролитическая диссоциация
Ионизация сольвата является необходимым, но еще не
обязательным условием образования электролитного раствора.
Обязательным же условием диссоциации ионизированного комплекса
на ионы является достаточно высокая диэлектрическая
проницаемость раствора. Лишь в этом случае ионизованный комплекс
распадается на ионы:
PK'+S^A'-S, *=* ... =f=* рК20+льв + ^А?-ьв (VII. 12)
Несмотря на то, что полная ионизация протекает достаточно
часто, полная электролитическая диссоциация (сильные
электролиты) наблюдается значительно реже. Абсолютно подавляющее
большинство изученных электролитных неводных растворов
образовано слабыми электролитами, реже —электролитами средней
силы. (Подробнее о закономерностях, определяющих силу
электролитов, см. в гл. IX).
Основные методы определения константы электролитической
диссоциации обычно сводятся к кондукто- либо потенциометриче-
ским методикам и приложимы лишь к весьма разбавленным
растворам (см. гл. X). Другие методы определения констант
диссоциации (например, спектрофотометрические) имеют ограниченное
применение.
Общая схема равновесий в растворах объединяет все
рассмотренные стадии:
/
mKA + nS ^ (КА)да Sn Л* PKQ+Sn-yqAp-Sy ^
V*
Приведенная схема может обрываться на любой из стадий,
т.е. компоненты раствора могут це образовывать химического qq-
114
единения (обрыв на стадии /); продукт присоединения не
обязательно подвергается ионизации (обрыв на стадии II) и, наконец,
ионизованный комплекс не обязательно подвергается
электролитической диссоциации (обрыв на стадии III).
Современный арсенал методов исследования растворов
позволяет почти всегда уверенно определять число стадий схемы
(VII- 13) для каждого конкретного объекта.
В последующих главах будет рассмотрено соотносительное
влияние химических и физических факторов на константу
равновесия каждой из стадий этой общей схемы равновесий в растворах.
Глава VIII
МЕТОДЫ ИССЛЕДОВАНИЯ ХИМИЧЕСКОГО
ВЗАИМОДЕЙСТВИЯ МЕЖДУ КОМПОНЕНТАМИ
РАСТВОРА
Природа и степень взаимодействия между компонентами
системы, образующей жидкий раствор, оказывают решающее
влияние на свойства последнего. Ни один раствор не может быть
охарактеризован сколько-нибудь полно, если не известна
стехиометрия, а часто и степень взаимодействия растворенного вещества
с растворителем.
В этой главе будут рассмотрены методы исследования процесса
образования продуктов присоединения (сольватации) в неводных-
растворах, которыми располагает современная теория растворов.
Число этих методов весьма обширно. Велико и разнообразие
теоретических предпосылок, положенных в основу этих методов.
Методы исследования разбавленных и концентрированных
растворов разнятся настолько, что нередко выявить общие черты в
них можно не без значительного труда.
Современная теория электролитных растворов позволяет строго
количественно описывать лишь разбавленное состояние с верхним
концентрационным пределом, лишь редко превышающем 0,01 М.
Даже усовершенствования классической теории электролитных
растворов позволяют лишь незначительно углубиться в область
более высоких концентраций. Кроме того, подавляющее
большинство теоретических работ в этой области посвящено сильным
электролитам, т.е. ограничено применимы к неводным растворам, где
случаи, отвечающие сильным электролитам, встречаются весьма
редко.
Ассортимент строгих методов для изучения концентрированных
растворов довольно узок. Црежде всего, к таким растворам
неприложим круг методов, основанный на законе Рауля. Нехимические
взаимодействия между молекулами растворенного вещества в
концентрированных растворах настолько значительны, что вклад их
в изменение свойств раствора становится весьма существенным, а
подчас и решающим.
Основные успехи в области исследования концентрированного
состояния связаны с физико-химическим анализом, т. е. с
построением и анализом химических диаграмм. Установление связи между
геометрией химических диаграмм, с одной стороны, и
стехиометрией и степенью взаимодействия между компонентами раствора,
с другой стороны, позволяет получить многие из тех
характеристик, которые необходимы для построения полной схемы
равновесий в растворах.
Существуют также различия в теории и методах исследования
растворов, образованных твердым (в индивидуальном состоянии)
116
электролитом, и жидкими системами, компоненты которых во всем
температурном интервале исследования системы являются
жидкостями. В свою очередь, методы подхода к анализу диаграмм свой-
ств0 — состав различны для двухкомпонентных (двойных) систем,
где концентрация компонентов изменяется в самых широких
пределах, для двухкомпонентных систем в инертном растворителе,
когда концентрация реагирующих компонентов значительно ниже
концентрации растворителя, для тройных и многокомпонентных
систем и т. п. Вот почему целесообразно рассматривать методы
исследования химического взаимодействия между компонентами
раствора отдельно для каждой из названных выше градаций.
ФИЗИКО-ХИМИЧЕСКИЙ АНАЛИЗ ДВОЙНЫХ ЖИДКИХ СИСТЕМ
Двойная система — наименьшая по числу компонентов
химическая система, из которых может быть образован раствор. Хотя
любой раствор-вещества А в растворителе S сколь-угодно малой
концентрации представляет собою двойную систему, в литературе
укоренилась традиция относить к двойным жидким системам
системы, образованные жидкими компонентами, концентрация
которых изменяется в весьма широких пределах (по возможности от
одного компонента до другого).
VIII. 1. Общие свойства химических диаграмм двойных
жидких систем
Способы выражения состава двойной системы
Наиболее распространенный способ выражения концентрации
двойной системы — долевой, при котором доля, например,
компонента А выражается так:
сА = —х— (VIII. 1)
А сА + с .
Если с — масса компонента А (/?А), отнесенная к сумме масс
компонентов, то концентрация выражается через массовые доли
(Ра)', если с — объе'м компонента А (£>а), отнесенный к сумме
исходных объемов компонентов, то концентрация выражается через
объемные доли (VA)\ наконец, если с — число молей компонента А
в смеси (тА), отнесенное к суммарному числу молей, то
концентрация выражается через мольные доли (хА).
Поскольку концентрация в физико-химическом анализе почти
всегда выражается через доли исходных компонентов, то незави-,
симо от того протекает или отсутствует химическое взаимодействие
117
в системе сумма долей равна 1, то есть:
Доли, рассчитываемые по соотношению (VIII. 2), называются
аналитическими. Если же при химическом взаимодействии в
системе учитывать равновесное количество компонента и относить
его к сумме равновесных количеств компонентов системы и
образующихся в ней соединений, то получаемые при таком расчете доли
будут истинными. Так, если в системе одновременно протекают
реакции: А + В^АВ и АВ + В ^ АВ2, то истинная мольная
доля, например компонента А, вычисляется по уравнению
тА
а истинная мольная доля продукта присоединения АВ2 — по
уравнению:
N e 5^—г (VIII. 4)
Будем далее обозначать аналитическую мольную долю через ху
а истинную мольную долю через N. Очевидно, что в системах с
химически невзаимодействующими компонентами N = х.
При расчете как х, так и N для определения количества молей
компонентов всегда берут формульную молекулярную массу —
ассоциативное состояние компонентов при этом не учитывают.
Переход от одного способа выражения состава к другому
может быть сделан без труда:
от массовой доли Р к объемной V
PJdA
v*=PA/dA + (l~PA)ldB <VIIL5>
от массовой доли Р к мольной х
*А= p.im. мп—рАумв - (VIII. 6)
от объемной доли V к мольной х
VTA" (VIII. 7)
В пересчетных формулах (VIII. 5) — (VIII. 7): d — плотность;
М — молекулярная масса; 9 — мольный объем.
Свойство называется аддитивным, если в идеальной системе
величина свойства У является прямолинейной функцией аналити-
* Часто вместо долевого используют процентный способ выражения
концентраций, при котором ^Р=^У=^х = 100*
118
ческого состава смеси:
: Уаса
(VIII. 8)
Свойство относится также к аддитивным, если уравнением
прямой связана не величина исходного свойства, а какая-либо его
функция f(Y):
Так, нередко бывает аддитивен логарифм свойства:
te Y в са 18 У а + св lg У в
(VIII. 10)
Неаддитивными называются свойства, которые не подчиняются
зависимостям (VIII. 9) и (VIII. 10) ни при одном из способов
выражения концентрации. у
Разность между экспериментальной
величиной свойства и его аддитивной величиной
называется отклонением от аддитивности:
№ = ^эксп - ^ад = Кэксп - {УасА + Увсв) (УШ- И)
Диаграммы отклонения от аддитивности
чрезвычайно часто применяются в
физико-химическом анализе.
Основные типы и геометрические элементы
химических диаграмм двойных систем
На рис. VIII. 1 приведены основные типы д
изотерм свойств и отвечающие им диаграммы ри
отклонения от аддитивности. Аналитико-гео-
метрическая характеристика этих типов
кривых элементарна и вряд ли на ней стоит
останавливаться.
Особое значение на диаграммах
химических систем приобретают экстремальные
точки. На первом этапе развития
физико-химичеб
В
. 1.
Основные типы изотерм
сзойство — состав:
/ — аддитивная; 2 —
монотонно выпуклая от оси
состава (положительная);
3— монотонно выпуклая
к оси состава
(отрицательная); 4—с
максимумом; 5 — с минимумом*.
5 —с сингулярным макси-
ского анализа жидких систем любой экстре- мумом; г-э-образная.
мальной точке приписывалось образование
соединения определенного состава. В настоящее время известно,
что связь между положением экстремальной точки относительно
оси состава и стехиометрией взаимодействия в жидкой системе
далеко не однозначная. Далее в этой главе вопросы соответствия
отдельных геометрических элементов диаграммы свойство — состав
определенным химическим образом будут рассмотрены достаточно
подробно.
* Здесь и далее величина свойства У, приведенная без индекса, относится
к свойству смеси., . . _ .
119
Основные типы реакций, протекающих в жидких системах
Наиболее распространенный тип реакций, которые могут
протекать в жидких системах, это уже неоднократно упоминавшиеся
здесь реакции присоединения (сольватации) *
тА + пВ *=* АтВ„ (VIII. 12)
Значительно реже встречаются реакции соединения:
тА + пВ *=* рС (VIII. 13)
Реакции соединения, примером которых может служить
взаимодействие (СНзСО^О + НзСГ—>2СН3СООН, как правило,
сопряжены с более или менее глубокой перегруппировкой связей в
молекулах реагирующих веществ. Это обстоятельство в подавляющем
большинстве случаев обусловливает протекание реакций
соединения до конца.
Относительно распространены в жидкой фазе двойных систем
реакции обменного взаимодействия:
пгА + пВ *=* рС + qD (VIII. 14)
Примерами реакций обменного взаимодействия являются эте-
рификация, ацидолиз, либо гидролиз сложных эфиров и т.д.
Непременным условием проведения физико-химического
анализа жидкой системы является предварительное определение типа
взаимодействия, протекающего в системе.
Преобразование координат химической диаграммы
Для решения вопроса о стехиометрии и степени взаимодействия
в жидкой системе на основании данных физико-химического
анализа далеко небезразлично, каков способ выражения состава
системы. Дело в том, что каждое данное свойство имеет лишь один
физически обоснованный способ выражения концентраций. Так,
плотность идеальной системы аддитивна при выражении состава
в объемных долях, а мольный объем — при выражении состава в
мольных долях.
Это обстоятельство необходимо иметь в виду при.переходе от
одного способа выражения концентрации к другому.
Изменение геометрии изотерм химических диаграмм при
переходе от одного способа выражения концентрации к другому
подробно рассмотрено В. Я. Аносовым (1959 г.). Аналитико-геомет-
рический анализ диаграмм химических двойных систем
показывает, что если данное свойство при одном способе выражения
* Реакции присоединения нередко называют реакциями аддатационного
взаимодействия; этот термин будет применяться и в этой книге, поскольку адда-
тационное взаимодействие в растворах встречается наиболее часто. И если не
будет следовать специальных оговорок, под термином «взаимодействие» будем
понимать именно аддатационное взаимодействие.
120
концентрации аддитивно, то при переходе к другому способу
выражения концентрации прямая превращается в гиперболу.
Действительно, если свойство аддитивно при мольно-долевом
способе выражения состава
(VIII. 15)
то при переходе, например к массовым долям [см. уравнение
(VIH. 6)], уравнение прямой (VIII. 15) преобразуется в уравнение
гиперболы:
МА + МВ /уА J/b_
Y- мр + мр[мр^м
В физико-химическом анализе нередко прибегают к
логарифмическим единицам. Если при этом исходная изотерма была
аддитивна, то есть описывалась уравнением (VIII. 8), то уравнение
In У = In (уАсА + увсв) (VIII. 17)
будет описывать кривую, монотонно выпуклую от оси состава.
Напротив, если аддитивен логарифм свойства [см. уравнение
(VIII. 10)], то изотерма исходного свойства •
(VIII. 18)
будет монотонно выпукла к оси состава.
В ряде случаев представляется целесообразным рассматривать
диаграммы так называемых обратных свойств: Z = 1/У. При этом,
если прямое свойство аддитивно, то обратное свойство будет при
том же способе выражения концентраций описываться уравнением
гиперболы:
г =4- * . „ (VIII. 19)
Напротив, если обратное свойство аддитивно, то изотерма
исходного (прямого) свойства — гипербола.
Очень часто для суждения о характере и глубине протекающего
в системе взаимодействия рассматривают не изотерму исходного
свойства, а изотерму отклонения этого свойства от аддитивности.
(Это рассмотрение лишь в том случае имеет физический смысл,
если данное свойство в идеальной системе аддитивно). Анализ
уравнения (VIII. 11) показывает, что если на кривой уЭксп имеется
экстремум, то он сохраняется и на кривой Ау, причем в общем
случае положения экстремумов на этих кривых не будет совпадать.
Мольные свойства
Мольными называют свойства, отнесенные к 1 моль смеси.
Естественно, что для расчета мольного свойства необходимо
задаться значением молекулярной массы смеси. Эту величину
обычно рассчитывают как аддитивную при выражении состава в
121
аналитических мольных долях величин из молекулярных масс ком»
понентов:
(VIII. 20)
Очевидно, впрочем, что в системах с взаимодействием
молекулярная масса смеси должна определяться выражением, которое
учитывает истинные мольные доли компонентов системы и
продуктов их взаимодействия. Так, если в системе протекает
взаимодействие (VIII. 12), то:
^ист - MANA + MBNB + Mkn?fNknfin (VIII. 21)
Мольные свойства, рассчитываемые с введением истинной
молекулярной массы [выражение (VIII. 21)], называют истинно
мольными, а мольные свойства, рассчитываемые с введением
аддитивной молекулярной массы — псевдомольными.
Между истинными мольными и псевдомольными свойствами
имеется различие, которое является принципиальным с точки
зрения применения этих свойств в физико-химическом анализе.
Различие это выявляется при анализе уравнений связи * каждой из
разновидностей мольных свойств.
Уравнения связи различны для каждого из случаев
стехиометрии взаимодействия в жидкой фазе. Рассмотрим уравнения связи
отклонения псевдомольных свойств от аддитивности.
Если в системе образуется лишь эквимолекулярный продукт
присоединения
А + В 38=* АВ (VIII. 22)
то связь между отклонением свойства равновесной смеси от
аддитивности (Д*/^) с величинами свойств компонентов (уа,Ув) и
образующегося соединения (#ав), аналитической мольной долей
() и константой равновесия процесса (VIII. 22)
передается уравнением:
-0,5(yAB-yA-yB) I-"!/ 1- ^ *"Ч (VIII. 24)
Анализ уравнения (VIII. 24) показывает, что экстремум ^$
всегда отвечает стехиометрии процесса (VIII. 22), т. е. приходится
на абсциссу с хА = 0,5— независимо от величины К (за
исключением, разумеется, случая, когда при К = 0 Дудв = 0)-
Уменьшение К приводит лишь к уменьшению абсолютной величины
* Уравнениями связи называют уравнения, связывающие свойства смеси и
компонентов с содержанием их в смеси и константой равновесия процесса.
122
не влияя на положение экстремума. Изотерма Ayjfg, как это видно
из рис. VIII. 2 [на котором приведены изотермы Az/^g для
рассматриваемого случая взаимодействия (VIII.22), рассчитанные для
условных величин констант равновесия, причем К\ < /G < Кг <
< /С»], является сингулярной лишь в случае, когда взаимодействие
протекает до конца.
tyS
KooJ
Ad
if
/
1 //4s
F/ K<
*L~-~—
В - i
I l >Л
!\\
J \_ >\
\ ^^V^
1 ^*
9.5 A
в
Рис. VIII. 2. Изменение гео-
метрии изотерм Лг/дв с изме"
нением степени взаимодей-
ствия.
44
Рис. VIII. 3. Изменение геометрии
изотерм Л#ав(а#ав) с
изменением степени взаимодействия.
Если в системе протекает только реакция
А + 2В <=* АВ2
(VIII. 25)
уравнение связи для отклонения псевдомольного свойства для
этого случая передается уже более сложным уравнением
(VIII. 26)
где
W =
К+1
(27Кх* - 27Кх2 + 9КХ-К-1)
1
- (729К2*4~1458/С2*3 + 972К2х2 - 270К2х + 27К2 - 54/С* + 27К)
Анализ уравнения (VIII. 26) показывает, что экстремум
независимо от величины константы равновесия
^АВ2 *
К
АВ2:
(VIII. 27)
приходится на точку стехиометрии процесса (VIII. 25), то есть на
*а = 0,33.
123
К абсолютно идентичным выводам приводит анализ уравнений
отклонений от аддитивности псевдомольного свойства для реакций
соединения (VIII. 13) и обменного взаимодействия (VIII. 14).
Экстремум же уравнения отклонения от аддитивности
истинномольного свойства, как показывает анализ, приходится на состав
соединения лишь в тех случаях, когда взаимодействие прошло до
конца, т. е. /С—>оо; при конечных же значениях К экстремум ДУ(^)
истинномольного свойства тем больше отклоняется от точки
стехиометрии, чем меньше /С
Анализ уравнений отклонения от аддитивности
объемно-аддитивных— АУ<У) или массово-аддитивных ДУ(р) свойств, например
~ х)К
] [8АВ8ад (уАВ - Уад) + 6А9В (уА - ув) (2*-!)]
(8АВ - еА - ев) + 29ад] еад
(VIII. 28)
показывает, что точно так же, как в случае истинномольного
свойства экстремум ДУ(^) (или ДУ<р)) приходится на состав
образующегося в системе соединения лишь в том случае, если
взаимодействие прошло до конца. Во всех иных случаях, когда К конечно,
экстремум ДУ<Г> (ДУ<р)) тем больше сдвигается от точки
стехиометрии, чем меньше К (рис. VIII. 3).
Итак, для строгого суждения о стехиометрии взаимодействия
в жидкой системе необходимо пользоваться псевдомольными
свойствами (точнее, отклонениями этих свойств от аддитивности).
Лишь в этом случае строгое определение стехиометрии может быть
проведено без учета точного значения константы равновесия.
Впрочем, мольноаддитивные свойства, применяемые в
физико-химическом анализе, за редким исключением обычно являются
псевдомольными, поскольку нетрудно увидеть, что для определения
истинной молекулярной массы жидкой смеси (VIII. 21) необходимо
располагать данными о равновесной концентрации компонентов и
продукта (продуктов) присоединения, а определение этих величин
в двойных жидких системах, как будет показано далее,
представляет весьма сложную задачу.
Преимущественное положение псевдомольных свойств * в
физико-химическом анализе вызывает необходимость перевода
свойств, аддитивных при выражении состава в объемных или
массовых долях, в их модификации, которые были бы аддитивны в
мольных долях. Применяемые для этого приемы4 достаточно
элементарны.
Если свойство аддитивно при выражении состава в массовых
долях, т. е.
УР=*Ъу{ьР>Р1 (VIII. 29)
* Если не будут приведены необходимые оговорки, далее под термином
«мольные свойства» будем понимать именно псевдомольные свойства.
124
путем несложных преобразований можно придти к равенству:
~ ' '"ч ч , (VIII. 30)
где Мад определено уравнением (VIII. 20).
В случае объемноаддитивного свойства
Yv = ^yfVi (VIII. 29а)
справедливо равенство:
(у)2(^)^ (VIII. 31)
Итак, для перевода в мольноаддитивную модификацию
необходимо величину весовоаддитивного свойства умножить на
соответствующую величину молекулярной массы, а величину
объемноаддитивного свойства — на значение мольного объема (точнее —
псевдомольного объема, так как значение свойства смеси
перемножается на величину Мад/й, а не M^a/d).
Из сказанного следует, что выбор координат при построении
химической диаграммы не может быть произвольным. Так,
координатой состава при построении диаграммы объемноаддитивного
свойства должна быть именно объемная доля, т.~ е._ диаграмма
должна строиться в координатах У<^)—V; диаграмма же мольноад-
дитивной модификации этого свойства должна строиться в
координатах YWQ — х. Аналогично, диаграмма массовоаддитивного
свойства имеет физический смысл лишь в координатах У(р) — Р и т. д.
Рациональные и иррациональные химические диаграммы
В практике физико-химического анализа диаграммы,
экстремальные элементы которых приходятся на состав образующегося
в системе соединения, принято называть рациональными;
диаграммы же, экстремальные элементы которых отклоняются от точки
стехиометрии, называются иррациональными.
Одна из причин, приводящих к иррациональности химической
диаграммы, очевидна. Так, в случаях, когда взаимодействие не
прошло до конца, диаграммы любых свойств, за исключением
псевдомольных, будут иррациональны. Такой вид
иррациональности назовем геометрическим, поскольку при переводе диаграмм
У{Р) или У(У) в их мольноаддитивные модификации можно придти
к рациональной диаграмме.
Однако рассмотрение обширного экспериментального
материала по диаграммам двойных жидких систем показывает, что и
диаграммы псевдомольных свойств весьма часто, бывают рациональны,
т- е. экстремум FW или ДУ(ДТ) приходится на состав, который
заведомо не может быть приписан никакому вероятному продукту
присоединения. Такой вид диаграммы псевдомольного свойства
будет реализовываться во всех случаях, когда в равновесной смеси
одновременно будут находиться два или более различающихся
по составу продуктов присоединения, т. е. одновременно будут
пРотекать"несколько процессов [например, (VIII. 22) и (VIII. 25)].
125
В этом случае экстремум отклонения псевдомольного свойства от
аддитивности (A{/(iV)) будет приходиться на состав, расположенный
между значениями Ха = 0,33 и 0,5.
Очевидно, что никакие геометрические пребразования диа.
грамм такой системы не приведут к рациональной диаграмме. По.
добный вид иррациональности будем называть химическим.
Помимо отмеченной причины появления химической иррадио.
нальности, в разделе VIII. 4 будет разобрана еще одна.
VIII. 2. Основные методы физико-химического анализа
двойных жидких систем
Для изучения равновесий в гомогенных жидких системах
применяют методы, основанные на исследовании следующих групп
свойств: механические — плотность, вязкость, давление истечения;
поверхностные — поверхностное натяжение; оптические —
показатель преломления; спектральные — оптическая плотность или
интегральная интенсивность полос поглощения в различных областях
спектра (главным образом, в ИК, видимой и УФ); поглощение в
области радиочастот (резонансная спектроскопия); акустические —
скорость распространения звука (адиабатическая сжимаемость);
тепловые — теплоты смешения, теплопроводность; электрические
(магнитные) — электропроводность, доли переноса тока,
электропотенциалы, магнитная восприимчивость, диэлектрическая
проницаемость.
На измерении и анализе концентрационной зависимости
перечисленных свойств основан ряд методов физико-химического
анализа жидких систем, которые, впрочем, разработаны в различной
степени. Кроме того, эффективность их для исследования природы
равновесий в жидких системах также различна. Вот почему в
предлагаемом читателю обзоре основных методов
физико-химического анализа жидких систем эти методы будут характеризоваться
не с одинаковой степенью обстоятельности.
Одной из возможных систем классифицирования методов
физико-химического анализа является форма изотермы свойства
в идеальной системе. Исходя из этого можно все свойства
подразделить на три группы:
I. Свойства смеси аддитивно слагаются из свойств компонентов
(при определенном способе выражения состава);
II. Свойства смеси в более или менее явной форме
рассчитываются исходя из свойств компонентов;
III. Свойства смеси не находятся в какой-либо связи с
величинами свойств компонентов.
К группе I относятся плотность, показатель преломления,
оптическая плотность, магнитная восприимчивость и др.; примером
свойств, относящихся ко II группе, могут служить вязкость и дй*
электрическая проницаемость; к III группе относятся теплота
смешения и электропроводность,
126
Теоретическое обоснование каждого из методов II группы
начинается с решения вопроса о закономерностях, которым
подчиняется форма изотермы идеальной системы (очевидно, что в
идеальных системах изотерма свойства, относящегося к группе I,
будет прямой; для свойств III группы вопрос о форме изотермы
идеальной системы вообще лишен смысла так, например, теплоты
смешения в идеальной системе по определению равны 0).
Вторым этапом теоретического обоснования соответствующего
метода является решение вопроса о закономерностях, которым
подчиняется форма изотермы системы с химически
невзаимодействующими компонентами. Последнее определение
предусматривает необходимость учета влияния на форму изотермы свойства
ряда явлений, которые объединяются под общим названием
«нехимические взаимодействия».
Обоснованное суждение о том, имеет ли место химическое
взаимодействие в системе, какова его стехиометрия и степень, может
быть получено лишь при сравнении экспериментальной изотермы
свойства с изотермой, рассчитанной в предположении отсутствия
взаимодействия (т. е. изотермой, относящейся к системе с
химически невзаимодействующими компонентами),
Волюмометрия
В физико-химическом анализе жидких систем строят
диаграммы плотности (d) и ряда функций, производных от этого свойства.
К последним относятся: удельный объем—Va = 1/d; мольный
объем — VM = M/d *; атомная концентрация [И. И. Заславский,
1941]—Ad= 1000 ^fNQ, где No— число Авогадро, а п — «среднее
число атомов в молекуле бинарной смеси», рассчитываемое по
соотношению аддитивности п = пАхА + пвхв; физический смысл
«атомной концентрации» — среднее число атомов в 1 л смеси.
Плотность и атомная концентрация являются объемноаддитив-
ными свойствами;'
d=dAVA + dBVB (VIII. 32)
Ad = AdAVA + AdBVB (VIII. 33)
Удельный объем — массовоаддитивное свойство
^-Va + Wb (VIII. 34)
* В соответствии с обозначениями, введенными в предыдущем разделе, бу-
У, MtNi
Дем различать истинный мольный объем смеси—*7М = ^ и псевдомоль»
2 Mixi
ный объем —6 = -г. ; очевидно, что в системах с химически
невзаимодействующими компонентами FM == 9*
127
а мольный объем — мольноаддитивное свойство:
^м^м.Л^У WB (VIII. 35)
А В
Анализ большого числа систем с химически
невзаимодействующими компонентами показывает, что нехимические взаимодействия
приводят к отклонениям от аддитивности, не превышающим 1%,
причем эти отклонения могут быть как положительными, так и
отрицательными. При этом в каждой данной системе мера
отклонения от аддитивности любого из перечисленных свойств одина-
Ad AAd AVd AVU *
кова, т. е. — — -^- — -^ — ТТ" '
В абсолютно подавляющем большинстве случаев аддатацион-
ное взаимодействие приводит к сжатию, то есть к положительным
отклонениям от аддитивности d и Ad и отрицательным Va и Ум.
Традиционные представления о малой чувствительности
плотности и производных от нее свойств к химическому
взаимодействию в жидкой системе в значительной степени несправедливы.
В системах с взаимодействием нередки величины сжатия,
достигающие 10%, а встречаются даже системы (например, изотиоциа-
наты — амины), где величины сжатия, обусловленные
взаимодействием, достигают 15%. Таким образом, объемные эффекты,
вызванные нехимическими взаимодействиями, могут составлять
относительно небольшую долю общего изменения объема.
Как следует из предыдущего раздела, преимущественное
значение для целей физико-химического анализа имеют мольноаддитив-
ные свойства. Мольный объем относится к весьма ограниченному
кругу мольноаддитивных свойств, которые применяют в физико-
химическом анализе. Поэтому построение и анализ диаграмм
мольного объема (или отклонения его от аддитивности) является
весьма желательным этапом анализа процессов, протекающих в
жидкой системе. Тут следует заметить, что измерение плотности
жидкой системы является, весьма несложной задачей; если же
учесть, что вычисление ряда свойств (вязкость, мольные рефракция
и поляризация и т. д.) не может быть проведено без данных о
плотности, то весьма широкое распространение волюмометрии как
метода физико-химического анализа становится понятным.
Полезной для практического применения является
классификация изотерм отклонения от аддитивности мольного объема (А9),
основанная на геометрических признаках (рис. VIII. 4). Две
основных разновидности изотерм относятся к системам с
невзаимодействующими (/) и взаимодействующими (//) компонентами
(напоминаем, что обоснованное суждение о взаимодействии при
реализации диаграмм типа // может быть сделано лишь в тех случаях,
когда величина сжатия превышает 1%). Разновидность // делится
* При этом, как и ранее, обозначения без индекса отвечают аддитивной
величине свойства смеси, причем аддитивность рассчитывается при физическом
обоснованном для данного свойства способе выражения концентрации,
128
на три основных типа: // — / — сингулярные изотермы
(взаимодействие прошло до конца); // — 2— рациональные изотермы (в
системе образуется лишь одно соединение) и // — 5
—иррациональные изотермы (в системе образуется несколько продуктов
присоединения).
-Д0 4
Рис. VIII 4. Классификация изотерм отклонения
мольного объема от аддитивности.
Тип //—2 можно подразделить на два подтипа II—2^
—симметричные изотермы и //—22 —несимметричные изотермы; как бу-
Дег показано далее, симметричность и асимметричность диаграмм
Д8 (также, как и любого иного свойства ДУОТ) вызвана
причинами, имеющими четкое химическое толкование (см. раздел VIII. 4).
Рефрактометрия
Показатель преломления {п) — одно из наиболее часто
определяемых свойств жидкостей. В физической химии нередко
применяются различные функции показателя преломления; удельная
Зак? 500
129
рефракция —
г = -^П (VIII. 36)
мольная рефракция —
RM*=rM (VIII. 37)
[Как и в случае иных мольных свойств, рефракция смеси
является истинным — если вводится величина истинной
молекулярной массы (VIII. 21) — и псевдомольным свойством, если вводится
величина аддитивной 'молекулярной массы (VIII. 20). При этом
функция f(n) вводится по Гладстону — Далю (я—1), либо по
Лорентцу —Лоренцу (^^)]
)
Установлено, что в жидких системах, приближающихся по
своему поведению к идеальным, весьма хорошо соблюдается правило
аддитивности (в массовых долях) удельных рефракций:
r-rAPA + rBPB (VIII. 38)
Из уравнения (VIII. 38) путем несложных преобразований типа
Р— V можно придти к уравнению, из которого следует, что f(n)
является объемноаддитивным свойством:
f (*) = / Ы ^а + / Ы ^в (VIII. 39)
[Анализ соответствующих функций показывает, что мера
отклонения от аддитивности —— наименьшая, когда f(n) вводится по
Гладстону — Далю].
Из уравнения (VIII.39), подставляя в него f(n) = п— I,
можно вывести уравнение объемнодолевой аддитивности п [J.
Wallot, 1903].
n^nAVAfnBVB (VIII. 40)
Из уравнения (VIII. 31) следует, что мольноаддитивной
модификацией показателя преломления будет функция Fn = я8:
^-V'a + 'Vb (VIII. 41)
А В
Анализ отношений — и —^ показывает, что при расшире-
нии в системе (Ad < 0) правилу аддитивности лучше
удовлетворяет показатель преломления; при сжатии (Ad > 0) правилу
аддитивности лучше удовлетворяет функция Fп.
На изотермах показателя преломления экстремум встречается
нечасто. Поэтому о характере протекающих в системе процессов
следует судить по диаграммам отклонения показателя
преломления от аддитивности (An),
130
Анализ метрического уравнения отклонения показателя
преломления от аддитивности (для случая А + В ^
j ) [еАве (Яав - «) + еАев („А - „в) (2* - D]
[(1-У1"1£1ТТМ)(еАв-ел-ез) + 2е]е
(VIII. 42)
показывает, что в соответствии с выводами, приведенными в
предыдущем разделе, максимум Д/z приходится на состав
образующегося в системе соединения лишь в тех случаях, когда
взаимодействие протекает до конца.
В очень большом числе систем химическое взаимодействие
приводит к положительным отклонениям показателя преломления
от аддитивности. Впрочем, анализ уравнения, связывающего Д/г
с отклонениями от аддитивности удельной рефракции
(напоминаем, что удельная рефракция является
массово-аддитивным свойством): o,ooi
An = (d + Ad)Ar + rAd (VIII. 43) ~'
-0,001
показывает, что возможны случаи, когда _0f002
и в системах с взаимодействием Д/i бу- р003
дет отрицательно. Так, например, если нсоон f
выполняется условие |d Дг| > |r Ad| при v
Дг < О, ТО Д/г < О [Б. В. И О ф ф е, Рис. VIII. 5. Изотермы
1959 г 1 (25 С) Д/г в системе му-
о *J* „„ равьиная кислота — диме-
Следует учесть, что в одной и той же ТИЛанилин для линий С, D
системе вид изотерм Д/г может суще- и F.
ственно изменяться при переходе от
красной к фиолетовой частям спектра (рис. VIII. 5). (В подавляющем
большинстве случаев измерение показателя преломления проводят
для линии D).
Рефрактометрия не относится к чувствительным методам
физико-химического анализа. Нехимические взаимодействия могут
приводить к относительно большим отклонениям от аддитивности,
достигающим 5-Ю"3, что всего на порядок меньше величин Д/г,
появляющихся в системах с очень глубоким взаимодействием.
Впрочем, самым большим недостатком рефрактометрии, как
метода физико-химического анализа являются особенности функции
Fn — мольноаддитивной модификации показателя преломления.
Действительно:
dkn nkd /ллттт лл\
<VIIL44>
Если же рассчитывать отклонение от аддитивности мольной
рефракции [при /(/г) = п — 1], то
ко хп .. d An —Ad (п — i) /лт-ттт л*\
АЛ,-1 = Л*м-Л* ща^гщ ' (VIII. 45)
б* 131
В системах с химически невзаимодействующими компонентами
величины An и Ad весьма малы, и поэтому величины AFn- так же,
как и ARM одинаково мало отличны от 0. Но и в системах с
взаимодействием, поскольку в подавляющем большинстве случаев Л/г
и Ad однозначны и близки по абсолютной величине, AFn и ARM
близки к 0, т. е^ функции Fn и RM независимо от глубины
взаимодействия, протекающего в системе, весьма близки к аддитивным.
К достоинствам рефрактометрии следует отнести весьма малое
время, необходимое для проведения измерения этого свойства,
а также предельную несложность выполнения эксперимента. Этот
метод может оказаться небесполезным для предварительного
заключения о том, взаимодействуют ли компоненты жидкой
системы.
Спектрофотометрия
Изучение спектров поглощения, столь распространенное при
изучении разбавленных растворов, при исследовании жидких
систем .получает относительное распространение лишь в последние
годы. Связано это и с отсутствием до последнего времени
теоретических разработок в области спектрофотометрии как метода
физико-химического анализа двойных жидких систем, и со
значительными экспериментальными трудностями, с которыми
сопряжена работа с концентрированными растворами.
Из не имеющего концентрационных ограничений закона
Ламберта— Беера, следует, что в идеальных системах оптическая
плотность (D) и интегральная интенсивность (В) полос
поглощения являются объемноаддитивными функциями:
Д^А^А + ^В (VIII. 46)
B = BAVA + BBVB (VIII. 47)
Если же, как это нередко бывает, компоненты жидкой системы
поглощают в разных областях спектра, то D (В) при какой-то
данной длине волны зависит лишь от концентрации
поглощающего компонента
D = DAVA (VIII. 46а)
B = BAVA ' (VIII. 47а)'
Неидеальность систем с химически невзаимодействующими
компонентами приводит к большим относительным отклонениям
от аддитийности, чем это наблюдается в случае иных объемно-
аддитивных свойств — плотности и показатели преломления. Так,
если в случае d величина относительного отклонения от
аддитивности G = Ау/у не превышает 0,01, а в случае п составляет
~ 0,005, то для изотерм D относительное отклонение от
аддитивности может достигать 0,1. Во всех случаях, где удалось провести
сопоставление, изотермы интегральной интенсивности В характе-
132
ризуются меньшими величинами G, чем изотермы оптической
плотности. Объемнодолевая аддитивность оптической плотности в
системах с невзаимодействующими компонентами соблюдается и
для спектров комбинационного рассеяния света (КРС).
Анализ спектрофотометрических данных в системах с
химическим взаимодействием проводится так же, как и в случае иных
аддитивных свойств, по изотермам отклонения от аддитивности.
Взаимодействие может приводить к исчезновению полос
поглощения, отвечающих какому-либо из компонентов; в этом
случае величины AD (AB) будут отрицательны. В качестве примера
ЗООнм
1ft
0}5
с3н7сно
0,5
х
0.5
C5H5N
Рис. VIII. 6. Оптическая плотность (D) и ^отклонение
оптической плотности от аддитивности (Д/)) в системах:.
а) масляныГ альдегид—этанол (УФ); б) дифениламин — пиридин
(ИК, ^ = 3280 см"1).
на рис. VIII. 6, а приводятся изотермы для ряда длин волн
системы масляный альдегид — этанол, где протекает
взаимодействие, связанное с исчезновением связи С = О (образование полу-
ацеталя).
Взаимодействие может быть также сопряжено с
возникновением новой связи, которой на спектрах поглощения будет
отвечать новая полоса. Так, в системе дифениламин — пиридин, где
образуется продукт присоединения через Н-связь, можно
наблюдать появление новой полосы поглощения 3280 слс1, максимум
оптической плотности которой приходится на область стехиометрии
образующегося соединения (рис. VIII. 6, б).
Мольноаддитивная модификация оптической плотности
Fd = Z)0 (мольное поглощение) является мольным свойством,
особенности которого делают применение его для целей
физико-химического анализа весьма целесообразным. Поскольку в системах
с взаимодействием величины Ad и AD не обязательно однозначны
и, кроме того, сильно различаются по абсолютной величине, то,
как следует из уравнения
d i\D - D i
(d + Ad) <
* (VIII. 48)
133
CeH5NH
отклонение мольного поглощения от аддитивности, в отличие от
мольной рефракции, будет существенно отличаться от нуля, и
характеризуется всеми качествами псевдомольного свойства,
которые были разобраны в предыдущем разделе.
В качестве примера на рис. VIII. 7 приводятся изотермы AFD
для различных полос поглощения в системе анилин — хлороформ,
в которой образуется эквимолекулярный продукт присоединения!
Как видно из рис. VIII. 7, во всех
случаях максимум AFD приходится
на состав продукта присоединения.
Эта особенность функции FDy a
также весьма четкая идентифика-
СНС1 ция факта взаимодействия в
случае появления новых полос
поглощения, позволяют отнести спектро-
фотометрию к чувствительным
методам физико-химического
анализа.
Нередко встречающееся в
литературе мнение, что в системах,
образованных одним или двумя
ассоциированными компонентами,
возникновение смеси сопряжено с
сильным сдвигом максимума
полосы поглощения, ответственной за
связь, вызывающую ассоциацию,
механически перенесено из практики исследования разбавленных
растворов —на концентрированные и является неверным. В
подавляющем большинстве случаев в области концентраций 1—0,1 мол.
долей ассоциированного компонента этот сдвиг не превышает в
ИК области 10 см'1; лишь при весьма сильных разбавлениях
наблюдается значительный сдвиг, который обычно и используют для
изучения. Н-связи. Это отвечает высказывавшимся выше
положениям о природе ассоциативно-диссоциативных процессов в
двойных жидких системах.
Спектроскопия, особенно ИК (КРС), доставляет весьма
ценные сведения при изучении кислотно-основных равновесий в
жидких системах.
В случае протолитического кислотно-основного взаимодействия
кислоты А—Н с основанием В схема равновесий (VII. 13) (в
предположении, что продукт присоединения и ионизованный
комплекс имеют простейший состав) объединяет стадии:
А"ВН+ =*=*= А" + ВН+ (VIII. 49)
'0,1
Рис. VIII. 7. Изотермы ДР^ в
системе анилин — хлороформ:
/—для полосы, отвечающей валентным
колебаниям N—Н (3370 см"1)'* 2— для
полосы, отвечающей связи С—H»»»N
(З331см~1) в продуктах присоединения.
АН + В
АН
В
Обычно на ИК спектрах сохраняются полосы поглощения
компонентов, отвечающие не вступившим во взаимодействие их
молекулам; часто можно найти полосу поглощения, отвечающую
продукту присоединения АНВ. И практически всегда возникновение
134
ионов сопряжено с появлением легко идентифицируемых новых
полос поглощения, что позволяет рассчитать суммарную
константу ионизации и электролитической диссоциации.
К специфическим особенностям спектрофотометрии как метода
физико-химического анализа жидких систем следует отнести
возможность получения прямых сведений о строении продуктов
взаимодействия и о механизме реакций в жидкой фазе. Так, именно
с помощью ИК-спектроскопии школой Ингольда были получены
данные о механизме реакций нитрования органических
соединений.
радиоспектроскопия
Несмотря на значительное развитие радиоспектральных
методов, исследования двойных жидких систем выполнены
практически лишь методом ЯМР-спектроскопии, причем почти всегда на
ядрах водорода, значительно
реже — на ядрах фтора и
лишь в единичных случаях —
на ядрах фосфора.
ЯМР-Спектроскопия —
весьма удобный и наглядный
метод изучения ассоциативно-
диссоциативных процессов в
системах с химически
невзаимодействующими
компонентами. Приведенные на рис.
VIII. 8, а изменения
химических сдвигов протонных
сигналов ЯМР гидроксильной
группы (относительно значений,
отвечающих чистым спиртам) в
системах алифатические
спирты — ССЦ показывают, что в
широком интервале
концентраций эти изменения
незначительны; лишь при весьма
больших разбавлениях величины
начинают резко возрастать.
При этом в растворах
одинаковых концентраций Дб
уменьшается от бутанола к
метанолу. Поскольку величина Дб
симбатна глубине
диссоциативного распада спирта, подобное изменение Дб отвечает
уменьшению прочности ассоциатов с возрастанием углеводородного
Радикала.
Для характеристики процессов, протекающих при образовании
Раствора, немаловажную информацию могут нести данные о
135
Рис. VIII. 8 Концентрационная
зависимость относительных изменений
химических сдвигов протонных сигналов:
а—сигнал 'от протонов группы ОН в
системах ССЦ с метанолом (/), "этанолом (2), пропа-
нолом (3) и бутанолом (4).
б —сигналы от протонов различных
молекулярных групп в системе БЬС1з — бутанол.
[Сб. «Ядерный магнитный резонанс», Изд. ЛГУ
1969, стр. 75, 73].
химическом сдвиге сигналов от протонов, принадлежащим разным
молекулярным группам компонентов, которые образуют жидкую
систему. На рис. VIII. 8, б приведена концентрационная
зависимость Аб химических сдвигов протонных сигналов ЯМР для
различных групп протонов спирта в системе SbCb—С4Н9ОН;
изотермы Аб — состав показывают,
р-Ю*смгсек'1 что кислотно-основное
взаимодействие приводит к
наибольшему изменению электронной
плотности у гидроксильной группы.
Эти изменения последовательно
уменьшаются с удалением
соответствующего атома водорода от
гидроксильной группы.
Связь Экстремумов на
диаграммах химический сдвиг —
состав со стехиометрией протекаю-
Рис. VIII. 9. Концентрационная
зависимость химического сдвига в
системе Н2О—C5D5N.
щих в системе процессов изучена
недостаточно. Примером такой
связи может служить диаграмма
для б, приведенная на рис.
VIII. 9. Максимум на изотермах б (прерывистые линии)
приходится на соотношение компонентов 1:1, что соответствует составу
продукта присоединения, установленному другими методами
физико-химического анализа.
Химический сдвиг может отражать и процессы
электролитической диссоциации, поскольку разрыв ионной пары ведет к
изменению электронного
состояния атомов, входящих в
состав ионов. Как следует из
рис. VIII. 10, химический
сдвиг сигнала ЯМР ядер %sos
F19 концентрированных
растворов трифторуксусной
кислоты незначителен; лишь
508
501,
0,6 0,8
CFsCOOH
Рис. VIII. 10. Концентрационная
зависимость химического сдвига сигналов
ЯМР F19 в системе CF3COOH—Н2О.
[Сб. «Ядерный магнитный резонанс», Изд. ЛГУ,
1969, стр. 79].
0,2 0A
при больших разбавлениях, Н*О V
когда раствор приобретает
высокую
электропроводность, свидетельствующую
о значительной
концентрации ионов, происходит
заметное изменение
величины б.
Образованию продукта присоединения отвечает более или
менее значительное увеличение массы диффундирующей молекулы.
Это соответствует уменьшению коэффициента самодиффузии,
минимум которого должен приходиться на состав продукта
присоединения. Примером диаграмм коэффициент самодиффузии — со-
136
став может служить изотерма этого свойства в системе C5D5N —
Ц2О, приведенная на рис. VIII. 9 (о расчете коэффициента само-
диффузии по данным ЯМР см. в разделе VII. 1.
диэлькометрия
Диэлектрическая проницаемость — одно из макроскопических
свойств жидкости, наиболее тесно связанных со строением
образующих эту жидкость молекул. Практически все существующие и
наиболее достоверные методы экспериментального определения
дипольнрго момента молекулы основаны на первичных данных по
диэлектрической проницаемости раствора. Методы эти достаточно
общеизвестны (см. гл. \у).
Отметим только, что достаточно строгие соотношения,
связывающие е с дипольным моментом \i, выведены лишь для неассо-
циированных жидкостей. Примерами таких связей могут служить
уравнения (IV. 29) —(IV. 31).
Одним из следствий теории Онзагера является
объемно-долевая аддитивность''диэлектрической проницаемости в идеальных
системах:
(VIII. 50)
Учет неидеальности в системах с химически
невзаимодействующими компонентами привел М. И. Шахпаронова к следующей
связи е раствора со значениями диэлектрической проницаемости
компонентов
(еА-ев)2(Аср)2
8 = 8ад + е .2 + 6 ) (VIIL 5l)
где (Аф)2 — средний квадрат флуктуации концентрации, а бе =
дёэксп
dV I dV #
Рассмотрение экспериментального материала показывает, что
уравнение Шахпаронова позволяет весьма удовлетворительно
рассчитывать е в системах с химически невзаимодействующими, Re-
ассоциированными компонентами *. Расхождение
экспериментальных данных с рассчитанными по уравнению (VIII. 51) не
превышает 0,2—0,3 единицы е для систем, образованных
высокополярными компонентами, причем расхождение это е не превышает по-
погрешностей эксперимента.
Теория жидких смесей, образованных одним ассоциированным
компонентом с другим, неассоциированным, а тем более теория
смесей двух химически невзаимодействующих ассоциированных
* Можно принять, что для равнообъемных смесей химически
невзаимодействующих иеассоциированных жидкостей (Аф)2 = 0,13; с незначительной
погрешностью можно считать, что для этих же смесей б8= 1. Таким образом, от-
пломение 8 от объемно-долевой аддитивности в точке Va = 0,5 можно
рассчитывать по следующей формуле:
(е д — eR)2-
As = 0,043 i^ -;-
бдд
137
компонентов разработана в столь малой степени, что до
настоящего времени не существует уравнения, которое позволяло бы
рассчитывать е таких смесей со сколь-нибудь значительной
точностью. Заметим, однако, что в системах, образованных одним и
тем же ассоциированным компонентом с рядом
невзаимодействующих с ним неассоциированных компонентов, соблюдается
постоянство отклонений эксперименатальных значений от значений,
рассчитанных по уравнению (VIII. 51). При этом соблюдается
постоянство не только абсолютной величины этих отклонений ДеХар =
= 8ад— ефлукт, но и геометрия изотерм ДеХар-
Поскольку не соблюдается объемно-долевая аддитивность ё,
постольку не будет соблюдаться аддитивность (ни при одном из
способов выражения состава) ни мольной поляризации *, (р) ни
квадратов средних дипольных моментов, рассчитываемых по
уравнению (IV. 31). Можно показать, что всегда —5- >— (за исклю-
чением систем, характеризующихся весьма малыми значениями е)
d(\x2) dp d& **
и, аналогично, —J-rL>—— > —.
\Х р Е
Взаимодействие между компонентами системы может
приводить как к возрастанию, так и к понижению 8 смеси по сравнению
со значениями, отвечающими смеси с невзаимодействующими
компонентами. Связано это с тем, что \i продукта присоединения
может быть выше \i компонентов (в случае цепочечной или
параллельной ориентации молекул компонентов), либо ниже \х
компонентов (в случае циклизации, либо антипараллельной ориентации
молекул компонентов).
В общем случае экстремумы на изотермах е приходятся на
состав продукта присоединения лишь в тех случаях, когда
взаимодействие прошло достаточно далеко.
В качестве примеров изотерм е систем с достаточно четко
выраженным взаимодействием на рис. VIII. 11 приводятся диаграммы
систем уксусная кислота — хинолин и изоамиловый спирт — бенз-
альдегид. В первой из них образуется весьма прочный продукт
присоединения C9H7N(RCOOH)2. Присоединение кислоты к амину
сопряжено с разрывом кольцевого димера молекулы кислоты и
превращением его в цепочечный; это ведет к знaчиteльнoмy
возрастанию |i, а, следовательно, и е, что находит отражение в
рациональности изотерм этой системы. Выпуклость ветви изотермы 8,
лежащей в области кислоты, объясняется установленной иными
методами тенденцией соединения 1:2 присоединять некоторое
количество кислоты. Поскольку энергия этого дополнительного
взаимодействия невелика, оно не ведет к разрыву димеров, и
величина диэлектрической проницаемости уменьшается. В системе
* Мольно-долевая аддитивность поляризации Р = 2 Pixv как это
вытекает из теории Дебая, соблюдается лишь в весьма разбавленных растворах.
** Если аддитивна 8(У), то аддитивен — в мольных долях — не ц,, а ц2.
138
язоамиловый спирт — бензальдегид весьма глубокое обменное
взаимодействие, приводящее к образованию ацеталя и воды
СбН5СНО + 2С5Нц0Н -> С6Н5СН(ОС5Нц)2 + 2Н2О, обусловливает
локализацию экстремума на диаграммах е в точке стехиометрии
реакции.
Измерение 8 многих систем с взаимодействующими
компонентами затруднено возникновением электропроводящих растворов.
Впрочем, в настоящее время разработано несколько относительно
простых схем [например, Ю. Я. Боровиков, Ю. Я. Фиалков,
6
20
10
-
/IV
/ ! \
I ! »
CH3COOH 0,33 0,5 Cj!
/5 -
/^ -
V од
у
ч /^
I t
,OH 0,5 C6H
5CHO
Рис. VIII. 11. Изотермы (при 25 °С) диэлектрической
проницаемости систем: уксусная кислота — хинолин (а) и изоами-
ловый спирт — бензальдегид (б).
1965], с помощью которых можно производить измерение
диэлектрической проницаемости растворов, электропроводность
которых достигает 10"2 ом-1-см-1. Это существенно расширяет
возможности диэлькометрии как метода физико-химического анализа.
К специфическим особенностям диэлькометрии следует отнести
возможность определения структуры соединений, образующихся
в системе. Как уже было отмечено, уже сама форма изотермы
может нести некоторую информацию о строении продукта
присоединения. Правда, точный расчет \i соединения, образующегося
в двойной жидкой системе, затруднен, но в разбавленных
растворах определение дипольного момента растворенного соединения,
особенно в тех случаях, когда растворитель имеет малую е,
является одним из традиционных методов исследования в химии.
Определенность, с которой могут трактоваться данные диэлько-
метрического исследования жидкой системы, зависит в
значительной степени от ассоциативного состояния компонентов. В случае
системы, образованной неассоциированными компонентами,
экспериментальная изотерма диэлектрической проницаемости может
быть сопоставлена с изотермой, рассчитанной в предположении
отсутствия взаимодействия. В системах же, где ассоциирован один
или тем более два компонента — подобное сопоставление
произведено быть не может из-за отсутствия сколь-нибудь достоверных
методов расчета е смеси невзаимодействующих компонентов.
139
Вот почему диэлькометрия в приложении к системам первого
класса может считаться весьма чувствительным методом физико-
химического анализа, в то время, как в случае иных классов
двойных систем диэлькометрия вряд ли может дать определенные
сведения о стехиометрии взаимодействия.
Вискозиметрия
Как отмечалось ранее, мерой вязкости является коэффициент
внутреннего трения ц в уравнении (IV. 16). Многочисленные
попытки найти простую функцию типа т] = f (r]A, т]в, *a), которая
описывала бы вязкость смесей с химически
невзаимодействующими компонентами, оказались безуспешными. Одна из причин,
обусловливающих эту неудачу, заключается в изменении
геометрии изотерм вязкости с изменением отношения вязкости
компонентов S^ =—- (здесь и далее будем выбирать обозначения
компонентов так, чтобы всегда ца > )
Рассмотрение обширного экспериментального материала по
вязкости двойных систем с химически невзаимодействующими
компонентами показывает, что изотермы вязкости таких систем
представляют собою кривые, монотонно выпуклые к оси состава. Был
предложен [Ю. Я. Фи ал ко в, 1963] полуэмпирический метод
расчета вязкости систем с химически невзаимодействующими
компонентами, основанный на связи между S. и L= 7]эксп . Последняя
Лад
величина является отношением экспериментального (или, что в
данном случае одно и то же, расчетного) значения вязкости
к аддитивному значению, рассчитываемому по уравнению г]ад ==
= 11аУа + Лв^в (при этом аддитивному значению вязкости не
придается никакого физического смысла).
Для смесей, содержащих 0,25 или 0,50 объемн. долей более
вязкого компонента
о
Lo,26; 0.50 = -5 + 7 (VIII. 52а)
для смеси, содержащей 0,75 объемн. долей более вязкого
компонента
5 1
^0,75 = ^0,25; 0,50 + 5S + 20 (VIII. 526)
Определяя L по уравнениям (VIII. 52), можно вычислить
значения вязкости, отвечающие смесям с содержанием 0,25; 0,50; 0,75
объемн. долей более вязкого компонента. Дальнейшее построение
изотермы вязкости по пяти точкам (3 значения вязкости,
отвечающие смесям, и 2 значения вязкости, отвечающие исходным
компонентам) не представляет труда.
140
Для расчетов изотерм вязкости в предположении отсутствия
химического взаимодействия в системах, вязкость компонентов
которых разнится не очень значительно (до S^6), можно
использовать уравнение
КЖ) (VIII. 53)
которое обычно (не совсем верно) называют уравнением Арре-
ниуса.
Помимо рассмотренных соотношений типа г\ = /(т)а, Цв, *а)
для расчета вязкости существует значительное количество
уравнений, куда помимо значений вязкости компонентов и содержания
их в смеси входят различные иные параметры, определение
которых в подавляющем большинстве случаев производят
экспериментально. Для целей физико-химического анализа, т. е. для расчета
изотерм вязкости в предположении отсутствия химического
взаимодействия, подобные уравнения не подходят, так как в качестве
исходных данных для такого расчета могут вводиться лишь
величины т|а, г)в и х. Однако для решения ряда специфических задач
такие уравнения оказываются несомненно полезными. В качестве
примера уравнений этого типа приведем уравнение Г. М. Панчен-
кова:
W (| ) (VIII. 54)
где А = 3 ]Л)/? у ~- M~5/e (w — собственный объем молекул в
1 моль), а <§ГСм — усредненная энергия связи между молекулами
смеси: <ГСМ = &ккх\ + 2&ABxAxB + <^Bb*b (<^аа, ^ав и <2?вв —
энергия связи между молекулами А, А и В, В).
Рассмотрение механизма вязкого течения в жидкой фазе
позволяет сделать вывод, что аддатационное взаимодействие
обязательно приводит к возрастанию вязкости смеси по сравнению со
значениями, отвечающими смесям с невзаимодействующими
компонентами. Поэтому форма изотермы вязкости двойной системы
в значительной степени является функцией глубины
взаимодействия в этой системе; именно степень взаимодействия лежит в
основе приводимой ниже классификации по геометрическому
признаку изотерм вязкости подобных систем.
Геометрическая хистема классификации изотеру ц состоит из
четырех основных типов (рис. VIII. 12).
К типу / относятся изотермы вязкости, монотонно выпуклые
d2r\
к оси состава (во всем интервале концентрации -т-j- > 0, в то
время как ни в одной точке -А^Окт. е. по форме эти
изотермы не отличаются от изотерм с химически невзаимодействующими
компонентами. Взаимодействие на изотермах вязкости типа /
проявляется в том, что они характеризуются положительными
141
отклонениями от изотерм, рассчитанных в предположении
отсутствия взаимодействия (рис. VIII, 13, а). При этом максимум изотерм
Дг) = г]эксп — %ас приходится на область стехиометрии
взаимодействия. Таким образом, построение диаграмм Дт] дает возможность
не только установить факт взаимодействия (что не может быть
сделано по самим
изотермам вязкости), но и состав
продукта присоединения.
К типу // принадлежат
S-образные изотермы
вязкости (в точке перегиба -гЛ =
= 0, в то время как на всем
интервале концентраций
А
IV
Рис. VIII. 12. Классификация изотерм
вязкости двойных систем с взаимодействием
по геометрическому признаку.
d2r\
двойной системы — ¥=
S-образные изотермы
вязкости реализуются в тех
системах, компоненты
которых значительно
различаются по вязкости, а
количества образующегося
продукта присоединения
недостаточно, чтобы превысить
вязкость более вязкого
компонента.
Общим признаком
изотерм типа /// является
положительные отклонения от
аддитивных значений
вязкости (еще раз напоминаем,
что при этом аддитивной
вязкости не придается
никакого физического смы-
сла), то есть выполнение условия -г"г< 0- Тип /// делится на два
подтипа. Подтип ///-/ составляют изотермы, монотонно
выпуклые от оси состава, то есть изотермы, у которых ни в одной точке
~-^-¥= 0. Принцип установления состава соединений в системах,
характеризующихся изотермами вязкости подтипа ///-/, тот же,
что для изотерм предыдущих типов: построение диаграмм Дт],
максимум которых приходится на область стехиометрии
взаимодействия в системе (рис. VIII. 13,6).
Подтип изотерм, вязкости Ш-2 характеризуется наличием
максимума. Изотермы вязкости этого подтипа свойственны
большинству систем с химическим взаимодействием и поэтому,
естественно, подтип III-2 характеризуется большим разнообразием, что
вызывает необходимость введения ряда подразделов.
142
К подразделу III-2{ относятся рациональные изотермы, к
подразделу ///-22 — иррациональные. Для установления состава
соединений, образующихся в системах, которые характеризуются
изотермами вязкости подраздела ///-22, прибегают к обычному
приему: сравнению экспериментальных изотерм вязкости с
изотермами, рассчитанными в предположении отсутствия взаимодей-
0,5
х
В
0,5
х
В
Рис. VIII. 13. Экспериментальные (/) и рассчитанные
в предположении отсутствия взаимодействия (2)
изотермы вязкости и отклонения изотерм 1 от изотерм 2
(АЛ).
ствия. При этом часто изотермы Дт}, в отличив от исходных
изотерм т), становятся рациональными (рис. VIII. 13, в). В свою
очередь, подраздел иррациональных изотерм делится на
разновидности: Н1'22,а — изотермы, положение максимума которых
относительно оси состава с изменением температуры не изменяется и-
Ш-22,6 — изотермы У], максимум которых сдвигается с
изменением температуры.
0,5
х
0,5
X
Рис. VIII. 14. Типы изотерм вязкости систем с
обменным взаимодействием:
/ — изотерма вязкости системы с отсутствием взаимодействия;
степень взаимодействия увеличивается от изотермы 2 к
изотерме 5.
К типу IV относятся сингулярные изотермы вязкости, которыми
характеризуются системы, где взаимодействие прошло до конца.
В системах с обменным взаимодействием геометрия изотерм
вязкости отличается от приведенных выше для систем с аддата-
Ционным взаимодействием (рис. VIII. 14). Тип изотермы опреде-
143
ляется величиной вязкости стехиометрической смеси продуктов
реакции (VIII. 4) рС + <?D. Эта величина может быть больше
вязкости компонентов, т. е. Црс+дв > tia > Цв (рис. VIII. 14, а);
может располагаться между значениями вязкости компонентов, т. е.
г)в > Црс+qD > т]а (рис. VIII. 14, б) и, наконец, может быть меньше
вязкости компонентов, т. е. т]в > т]а > ^pc+gD (рис. VIII. 14,в).
Примером систем с изотермами вязкости первого типа может
служить система SiCl4—CH2(OR)2, в которой протекает реакция,
приводящая к образованию SiCl2(OR)2 и CH2CIOR в
соотношении 1 :2. К системам с изотермами вязкости второго типа относится
система (СН3СО)2О—С6Н5ОН (стехиометрическая смесь
продуктов реакции — СН3СООС6Н5 и СН3СООН). Изотермами вязкости
третьего типа характеризуется система С2Н5ОН—СН3СООН, где
протекает равновесная реакция этерификации.
Вискозиметрия относится к числу наиболее чувствительных
методов физико-химического анализа. Этот метод позволяет не
только фиксировать слабые взаимодействия в двойных жидких
системах, но и, что значительно важнее, определять стехиометрию
этого взаимодействия. Далее будет показано, что к весьма
определенным выводам о процессах, протекающих в системе, приводит и
анализ изотерм относительного температурного коэффициента
вязкости.
Поверхностное натяжение двойных систем
Многочисленные попытки вывести уравнение, которое
связывало бы поверхностное натяжение у даже идеального раствора
только со значениями у компонентов, увенчались лишь
относительным успехом.
К одним из наиболее обоснованных относится уравнение >
А. А. Жуховицкого (1944 г.). Это уравнение связывает у раствора
со значениями у компонентов, с мольной долей v, а также с
удельными числами молей п компонентов в поверхностном слое
раствора:
• Y = Yava + Yb^b + nAvART ln-JT + nBvBRT In-~ (VIII. 55)
Впрочем, от уравнения (VIII. 55) можно перейти к уравнению,
где у будет представлено как функция состава системы в объеме
(однако, по-прежнему, будут входить величины удельных чисел
молей компонентов в поверхностном слое):
Y = Yb - n*RT In (1 - NA + NAK) (VIII. 56)
где /г* — среднее число молей на 1 см2 поверхности жидкости, а
Так как парахор Ру = yV40 весьма часто применяли в
органической химии для приблизительного решения вопроса о строении
144
соединения, то неоднократно предпринимали попытки проследить
концентрационную зависимость этой функции в двойных системах.
Нетрудно увидеть, что парахор аддитивен
(VIII. 57)
постольку, поскольку можно считать, что функция y1/4 не зависит
(либо зависит весьма незначительно) от состава. Действительно,
если принять, что в системах с химически не
взаимодействующими компонентами мольный
объем является мольно-аддитивной функцией
состава, то Ру может описываться прямой
лишь в том случае; если yiji не зависит от
состава. Это условие действительно
выполняется, если отношение величин поверхностного
натяжения не очень велико. Так, при
Ya Ya
Sv = — = 1,5, S i/4 = —тг = 1,1, т. е. функция
Y Yb- y Yb
по форме напоминает прямую, весьма мало
зависящую от состава. (Разумеется, факт ма- А
лого изменения ^1/4 с составом никак не
может быть распространен на самое у).
В литературе описано весьма много систем верхностногТ^натя-
с химическим взаимодействием, изученных по Жения в системах с
поверхностному натяжению. К сожалению, не взаимодействием:
все эти данные обладают одинаковой
достоверностью. Дело в том, что несмотря на
внешнюю простоту, метод измерения у позволяет
получить точные данные лишь при
соблюдении ряда условий.
Трем возможным случаям отношений
между величинами поверхностного натяжения хлораль.
компонентов и продукта (продуктов)
взаимодействия отвечают три основных типа изотерм поверхностного
натяжения [Н. А. Трифонов, 1940] (рис. VIII. 15). Примерами
указанных типов могут служить изотермы в системах фенол —
пиперидин, вода — уксусная кислота и вода — хлораль.
ъ
Рис- VIII. 15. Основ-
V^A>vB:
Системы: 1 — фенол —
пиперидин; 2 — вода —
уксусная кислота; 3 — вода —
Скорость звука в двойных системах
Скорость и прохождения звуковых волн относится к одному из
наиболее характерных свойств каждой жидкости. Естественно,
поэтому, что был предпринят ряд попыток применения и, а также
адиабатической сжимаемости $s = -^j- для целей
физико-химического анализа.
Обработка экспериментального материала показывает, что в
системах с химически невзаимодействующими компонентами, где
145
нехимические взаимодействия не очень сильны, соблюдается мае-
сово-долевая аддитивность величины и2 [Б. Б. К у др я в цев, 1956];
u2 = u\PA + ulPB (VIII. 58)
В ряде работ утверждается, что адиабатическая сжимаемость
в таких системах — объемно-аддитивная функция состава. Однако,
если принять справедливость уравнения (VIII. 58), то (3 ф 2 &iVt.
Лучше согласуется с уравнением (VIII. 58) уравнение
аддитивности мольной скорости звука (см. IV. 10)
Ru = Ru *А + \*В (VIII. 59)
часто называемое эмпирической зависимостью Рао.
Возможности применения величин ц, ps и Ru в
физико-химическом анализе требует дальнейших исследований.
Калориметрия
Калориметрия принадлежит к числу методов
физико-химического анализа, особенность которых состоит в том, что величина
свойства смеси заведомо не может быть связана с величинами
свойств компонентов. Более того, понятие «теплота смешения»
вообще лишено смысла в приложении к чистому компоненту.
Наиболее часто под теплотой смешения понимают тепловой
эффект образования раствора, отнесенный к 1 моль раствора. При
выборе термодинамической системы знаков тепловой эффект
смешения будет энтальпией этого процесса, а экзотермическим
эффектам будут отвечать отрицательные значения —Д#см.
Для систем, где протекают реакции соединения типа (VII. 13)
или иные типы взаимодействия, проходящие до конца, правильнее
применять термин не теплоты смешения, а тепловой эффект
реакции. Однако, поскольку число таких систем относительно невелико,
в практике исследования жидких систем бытует первый из этих
терминов.
Теплота смешения, как и многие иные свойства жидкой фазы,
отражает суммарный эффект ряда процессов, которые
сопровождают образование жидкой смеси. Даже в тех случаях, когда
известны все процессы, вносящие вклад в величину суммарного
эффекта теплоты смешения, не удается провести разделение
энергетических эффектов, относящихся к каждому из этих процессов.
В случае калориметрии этот общий недостаток физико-химического
анализа жидких систем становится особенно чувствительным, так
как тепловые эффекты нехимических взаимодействий по
абсолютной величине (а нередко и по знаку) близки к тепловым эффектам
химического взаимодействия, особенно в тех случаях, когда
смешение ведет к разрыву Н-связей между молекулами каждого из
компонентов и к образованию новой Н-связи.
Геометрически изотермы Д# подразделяются на три основных
типа (см. рис. VIII. 16). Большей частью (однако далеко не всег-
да) эндотермические изотермы свойственны системам с химически
^взаимодействующими компонентами, а экзотермические и S-об-
разные —к системам с химическим взаимодействием. S-образный
ход изотермы АН вызван наложением эндотермического эффекта,
обусловленного нехимическими взаимодействиями, и
экзотермическими эффектами, связанными с взаимодействием. Существенно,
что при этом в системах с взаимодействием существует довольно
Значительная область концентраций, где
д# близко или даже равно 0. 4,
В. П. Белоусов и А. Г. Морачевский
(1970 г.) предлагают рассматривать
теплоты смешения раздельно в системах
без водородных связей и в системах с
таковыми.
Системы без Н-связей могут быть
образованы компонентами: неполярными;
неполярным и полярным и полярными.
Эффекты, наблюдающиеся в
системах первой Группы, как правило, Неве- ри-с. VIII. 16. Основные ти-
лики и эндотермичны (Д#>'0). Изотер- пы изотерм теплот смеше-
мы АН в этих системах симметричны, ния:
р
т. е. хорошо описываются теорией регу- £-j^
ЛЯрНЫХ раСТВОрОВ (СМ. ГЛ. V). В СИСТемаХ термический); 3 -S-образный.
второй группы теплоты смешения в
значительной степени определяются изменениями энергии ориентаци-
онного взаимодействия, сопровождающими образование раствора.
Большей частью эффекты в таких системах эндотермичны.
В системах первых двух групп, в отличие от систем полярных
компонентов, химическое взаимодействие отсутствует. Поэтому в
системах последней группы, в которых большей частью протекает
взаимодействие, могут наблюдаться как положительные, так и
отрицательные величины АН.
Системы А—В с Н-связями подразделяют на четыре группы:
о) через Н-связь ассоциирован только компонент А; б) Н-связь
образуется только между молекулами компонентов А и В; в)
компонент А ассоциирован через Н-связь и между А и В образуется
Н-связь; г) присутствуют Н-связи всех типов.
В системах группы а в подавляющем большинстве случаев
взаимодействие отсутствует; в остальных группах системам
присуще химическое взаимодействие.
Изотермы АН группы а положительны и характеризуются
максимумом, который большей частью сдвинут в сторону неассоцииро-
ванного компонента, так как именно в этой области концентраций
относительная доля молекул компонента А, у которых произошел
разрыв Н-связи, максимальна (рис. VIII. 17,а).
В системах группы б взаимодействие относится к типу
незавершенного кислотно-основного, а тепловые эффекты практически
всегда экзотермичны. В большей части систем экстремум на
147
изотерме ДЯ точно или почти точно приходится на состав обра.
зующегося продукта присоединения (рис. VIII. 17, б).
, В системах группы в образование смеси сопровождается
разрывом ассоциатов А и образованием продукта присоединения Ав_
Наложение тепловых эффектов, отвечающих этим двум процесса^
приводит к S-образности изотерм (рис. VIII. 17,в).
Разнообразие и самих процессов и их энергетических эффектов
в системах группы г приводит к возможной реализации изотерм Д//
всех трех геометрических
типов. На рис. VIII. 17, г
приведены изотермы ДЯдля
систем, в которых вода
образует более " или менее
прочные продукты
присоединения.
Исследование теплот
смешения при нескольких
температурах позволяет
почерпнуть весьма ценную
дополнительную информацию
из калориметрических
измерений. Политермические
сведения об изотермах АН
позволяют рассчитать
величины избыточных теплоем-
костей смеси: —Ся.
С1
Кроме того, изменение
температуры может вести к
изменению, и иногда весьма
значительному, не только
знака, но и геометрии
изотермы ДЯ, как это можно
видеть, например, из рис. VIII.17-е. Сопоставление изотерм ДЯ,
относящихся к различным температурам, позволяет оценить
соотносительный вклад в общую величину ДЯ диссоциативного и
химического эффектов.
Экспериментальное определение теплот смешения, особенно в
тех системах, где величины ДЯ невелики, представляет собою
непростую задачу. Точное определение этих величин возможно лишь
при учете ряда факторов, среди которых важными являются
поправки на испарение и изменение давления.
Несмотря на ряд ограничений, которые свойственны
калориметрии как методу физико-химического анализа жидких систем,
обращение к нему желательно уже хотя бы потому, что он
единственный среди всего арсенала методов исследования жидких систем,
который доставляет прямые термодинамические сведения о
системе.
Рис. VIII. 17. Изотермы теплот смещения
в системах:
а — пропанол — бензол; б — хлороформ — ацетон;
в — вода — ацетон; г — вода —• ацетонитрил (/),
пиридин (2), диоксан {3).
148
К группе термодинамических свойств, характеризующих
молекулярные превращения в жидкой фазе, относятся также
теплоемкость (С) и теплопроводность (X). Отличие этих свойств от теплот
смешения с точки зрения вопросов, рассматриваемых в этой главе,
заключается прежде всего в том, что существует принципиальная
возможность вывода уравнений, связывающих величины С или I
компонентов с величинами свойств смесей. Впрочем, для
теплоемкости такое уравнение не выведено, хотя анализ имеющегося
экспериментального материала показывает, что С аддитивно при
выражении состава в массовых долях (как любое удельное
свойство).
Уравнение для теплопроводности, выведенное и обоснованное
Ю. Л. Расторгуевым и Ю. А. Ганиевым (1968 г.), показывает, что
в системах с химически невзаимодействующими компонентами это
свойство также аддитивно при выражении состава в массовых
долях.
Кондуктометрия
Кондуктометрия относится к наиболее распространенным
методам исследования растворов, вообще, и жидких систем, в
частности. В этом разделе будут рассматриваться вопросы приложения
кондуктометрии для решения основных задач физико-химического
анализа.
В отличие от свойств жидкой фазы, разбиравшихся прежде,
понятие «электропроводность» лишено конкретности. Большей частью
отнесение данного раствора к категории «электропроводного», либо
«неэлектропроводного» является следствием совершенства
аппаратуры и измерительных схем, которые имеются в распоряжении
исследователя. Точно так же отсутствует и вряд ли может
появиться вообще единый критерий для введения понятий «плохо
проводящий раствор», «умеренно проводящий раствор» и т. д.
В настоящее время в литературе, посвященной кондуктометрии
жидких систем, принято считать проводящими индивидуальные
жидкости и растворы с х порядка 10~7 ом~1-см~1 и выше; умеренно
проводящими — с 10~7—10"11 олг*"СМ~*; ниже 10~и —
непроводящими. Условность этой классификации очевидна.
Более определенной является система классификации,
основанная на механизме электропроводности (см. гл. XI).
Электропроводность относится к той, сравнительно небольшой
группе свойств жидкой фазы, величина которых для смеси не
может быть ни в явной, ни в неявной форме связана с величинами
свойств компонентов. Действительно, большая часть
электропроводных жидких систем образована непроводящими ток
компонентами.
В физико-химическом анализе принято пользоваться
диаграммами удельная электропроводность % — состав. Диаграммы
молекулярная электропроводность К — состав распространены в го-
149
раздо меньшей степени. Действительно, как уже упоминалось, в
весьма большом числе случаев электропроводность жидкой смеси
обусловлена ионогенностью продукта взаимодействия неионоген-
ных компонентов. Концентрация же продукта взаимодействия,
необходимая для расчета изотермы X двойной жидкой системы,
большей частью неизвестна, либо определяется весьма приблизительно.
В соответствии с общей схемой (VII. 13) равновесий в
растворах электропроводность в системах с химически
невзаимодействующими компонентами может наблюдаться лишь в тех случаях, когда
хотя бы один из компонентов жидкой системы — электролит в ин-
21
А х В
Рис. VIII. 18.
Основные типы изотерм
электропроводности
в системах с
химически
невзаимодействующими
компонентами.
0,5
X
О 1,0
0,5
X
О 1,0
0,5
X
Рис. VIII. 19. Изотермы удельной
электропроводности систем, образованных
ацетатами пиридиния (а), анилиния (б) и нико-
тиния (в) с бензолом.
давидуальном состоянии. Поскольку практически невозможно
подобрать два электролита, которые не взаимодействовали бы друг
с другом, закономерности изменения электропроводности в
системах без взаимодействия могут быть прослежены на системах типа
«электролит — индифферентный растворитель».
Возможны два основных типа изотерм % в системах с химически
невзаимодействующими компонентами (рис. VIII. 18): монотонно
выпуклые к оси состава (/) и с максимумом (2). Принято
связывать форму изотермы вязкости в таких системах с влиянием
вязкости второго (неэлектролитного) компонента (далее будем
считать, что А — электролитный, а В—неэлектролитный компоненты):
если т]а < Лв, то прибавление более вязкого компонента к
электролитному компоненту ведет к понижению х; при цА > г|в на
изотермах % появляется максимум. Впрочем, эксперимент показывает,
что для появления максимума на изотермах к необходимо, чтобы
в системе происхбдило весьма значительное снижение вязкости. На
рис. VIII. 19 приводятся изотермы удельной электропроводности
двойных систем, образованных весьма близкими по значениям х,
но сильно различающихся по вязкости ацетатами пиридиния,
анилиния и никотиния с бензолом. Из приведенных диаграмм видно,
150
что даже при 5^ = —— = 21 на изотермах к еще не появляется
™с6не
максимум.
Влияние вязкости на электропроводность может быть
нивелировано весьма популярным в физико-химическом анализе и в теории
растворов электролитов вообще приемом исправления
электропроводности на вязкость. Как показывает обработка
экспериментального материала, в системах с невзаимодействующими
компонентами, либо в системах с взаимодействием, где образующиеся
продукты реакции неэлектролитны, изотермы и всегда монотонно
выпуклы, к оси состава, в отличие от систем с взаимодействием, где
образуются электролитные продукты, когда исправление
электропроводности на вязкость не приводит к исчезновению максимума.
Это положение, называемое правилом М. И. Усановича, позволяет
в ряде случаев определять число стадий в общей схеме
равновесий (VIII. 13) *. Так, в системах ряда H2Se04 — карбоновые
кислоты электролитные продукты присоединения образуются в
системах с уксусной и монохлоруксусной кислотами; в системе же с
дихлоруксусной кислотой общая схема взаимодействий в этом ряду
. i и
H2Se04 + RCOOH ^=± H2Se04 • RCOOH ;~±'
ш
^Z± HSeOJ • RCOOHJ ^± HSeO" + RCOOHj
обрывается на первой стадии. Соответственно этому, изотермы
щ — а первых двух систем имеют максимум; изотерма же а
последней системы монотонно выпукла к оси состава (рис. VIII. 20).
На ход изотерм х значительное влияние оказывает также
диэлектрическая проницаемость. При прочих равных условиях
электропроводность раствора данного электролитного соединения будет
тем выше, чем выше диэлектрическая проницаемость. Зависимость
электропроводности от е находит отражение в прямолинейном
ходе lga как функции 1/е, соблюдающемся во всем
концентрационном интервале двойных систем с химически
невзаимодействующими компонентами. Эта особенность электропроводности в
концентрированных растворах, объединяющая ее.с электропроводностью
в разбавленных растворах (см. гл. X) является одним из немногих
мостов, наведенных в настоящее время между теориями
концентрированных и разбавленных электролитных растворов.
Зависимость lga от 1/е может быть иллюстрирована системами,
образованными комплексом [Н3О-ЗТБФ]+Т1С14" с
индифферентными растворителями (рис.. VIII. 21) **.
* Геометрический анализ уравнений изотермы электропроводности
показывает, что при малых S^ даже в системах без взаимодействия на изотермах a
может сохраняться максимум; однако, как следует из сказанного ранее, в таких
системах появление изотермы к с максимумом маловероятно.
** ТБФ — трибутилфталат.
151
Прямолинейность функции lga(l/e) позволяет вывести
уравнение электропроводности в системах с химически
невзаимодействующими компонентами [Ю. Я. Фиалков, Ю. А. Тарасенко, 1966]
к = — exp In aA — L -г — —■
L
(VIII. 60)
где L = а
8А8В
(а — коэффициент пропорциональности).
Представляя 8 и ц в виде соответствующих функциональных
зависимостей от состава [например, (VIII.51) и (VIII.53)], можно
уравнение к представить в более общем виде:
и = w l v ехр ( In аА - L Г—4—г —1 \
f(*b*) 1 А L*(e»x) 8AjJ
(VIII. 61)
Определенность, с которой могут быть истолкованы
диаграммы х, в значительной степени зависит от природы составляющих
систему компонентов. Если система
(5, спз-ом' -см'
образована непроводящими ток
компонентами, то уже сам факт
возникновения электропроводности
при образовании смеси является
2,0
A5
нсоон
Рис. VIII. 20. Изотермы a = щ
(25 °С) для систем,
образованных селеновой кислотой с
уксусной (У), монохлор- (2) и
дихлоруксусной (3) кислотами.
Рис. VIII. 21. Изотермы lgir— 1/е
систем, образованных:
[Н3О-ЗТБФ]+Т1С14 с ж-ксилолом (7),
хлорбензолом (2), ацетофеноном (3)
и нитробензолом (4).
бесспорным признаком химического взаимодействия, приводящего
к образованию электролитных соединений. Экстремальные точки
на изотермах к никак не связаны со стехиометрией
межмолекулярного взаимодействия в жидкой системе. Действительно,
электропроводность, являющаяся результатом протекания стадии III
общей схемы (VII. 13), не может быть сколь-нибудь
непосредственно связана со стехиометрией стадии I. Лишь в тех системах, где
взаимодействие протекает весьма глубоко, на изотермах
электропроводности появляется минимум, расположенный между двумя
максимумами (рис. VIII. 22). Локализация минимума относительно
оси концентрации отвечает составу продукта присоединения. Не-
152
7,*
трудно, впрочем, увидеть, что своим появлением минимум на
изотерме х обязан вязкости, так как резкое возрастание г) в системах
с глубоким взаимодействием ведет к
уменьшению х.
Из материала, изложенного выше, следует,
qTo сколь-нибудь полное толкование кон-
дуктометрических данных может быть
получено лишь при учете вязкости и
диэлектрической проницаемости системы. Несмотря на
это, кондуктометрия является наиболее
эффективным методом изучения электролитных
свойств жидкой системы. Далее будет
показано, что диаграммы относительного
температурного коэффициента электропроводности
позволяют весьма точно определить состав
образующегося в системе соединения.
Кондуктометрическиё данные получают
гораздо более глубокое толкование, если
известны механизмы электролитической диссоциации и переноса тока
в жидкой системе. Вопросам механизмов диссоциации и переноса
тока в растворах и жидких системах посвящена глава XI.
В
0,5
х
Рис. VIII. 22. Форма
изотермы
электропроводности в системах
со значительным
возрастанием вязкости.
Потенциометрия
Вопросы потенциометрии неводных растворов подробно
рассматриваются в гл. XIII. Здесь же приводятся данные о
потенциометрии как методе физико-химического
анализа жидких систем.
Потенциометрическое исследование
жидкой системы может доставить ряд
весьма важных сведений о механизме
диссоциативных процессов и переноса
тока. Однако в большинстве случаев
исследователю приходится сталкиваться с
проблемой выбора обратимого
индикаторного электрода. С. П. Мискиджьяном
было показано, что в системах с
кислотно-основными протолитическими
равновесиями с весьма неплохими
результатами может быть применен стек-
200
20 40 60 Я0СвН5Шг
z', мол.%
?Ид:с.Гд£/1'™ыРуМкЫ лянный электрод [разумеется, в каж-
сусная кислота — анилин:
I — измерения со стеклянным
электродом; 2 — измерения с
водородным электродом; 3 — АЕ/Е.
дом отдельном случае должна
устанавливаться прямолинейность
зависимости E=f(lgaH+} с наклоном
~ 0,58 в].
Проблема устранения потенциала (диффузионного и фазового)
Жидкостного соединения при потенциометрии жидких систем
несколько облегчается тем, что потенциал этот либо остается
153
постоянным, либо изменяется весьма незначительно на большей
части концентрационного интервала двойной системы и, таким
образом, не может оказать существенного влияния на, геометрию
изотерм Е = f(x).
В качестве примера диаграмм э.д. с. двойных систем на
рис. VIII. 23 приводятся данные по потенциометрии системы
уксусная кислота — анилин. Экстремум АЕ/Е в этой системе приходится
на состав СбН5ЫН2-2СНзСООН; соединение такого состава
обнаружено при исследовании этой системы иными методами физико-
химического анализа.
Температурные коэффициенты свойств двойных жидких систем
Политермическое исследование жидкой системы в большинстве
случаев оказывается более эффективным, чем изотермическое:
сопоставление ряда изотерм данного свойства одной и той же
системы в возможно более широком температурном интервале
позволяет получить дополнительную и, нередко, весьма ценную
информацию о процессах, протекающих в системе. Особенно
эффективным оказывается политермическое изучение вязкости,
диэлектрической проницаемости и электропроводности.
Обычно в физико-химическом анализе политермическое
исследование системы сводят к построению диаграмм температурного
коэффициента соответствующего свойства:
абсолютного
YT - YT АГ dY
<v- ъ-ъ-w или «Vе-зг (VIIL62)
относительного
aY 1 dY
^У = ~ или рк = т— (VIII. 63)
Рассмотрение экспериментального материала по вязкости
двойных систем показывает, что для смеси жидкостей справедливо
уравнение (IV. 8) температурной зависимости цу выведенное для
индивидуальной жидкости.
Аналитико-геометрический анализ изотерм ц, а также кривых
ссц и Рп приводит к некоторым выводам, представляющим интерес
для физико-химического анализа.
Кривые сц и рл в системах без взаимодействия могут
рассчитываться по тем же соотношениям, что и изотермы ц [для чего в
уравнениях (VIII. 52) или (VIII. 53) следует г] заменить на с^
или PJ.
Геометрия кривых ач всегда повторяет геометрию исходных
кривых г]. Напротив, концентрационная зависимость рл отличается
значительным своеобразием. В системах с взаимодействием,
изотермы вязкости которых монотонно выпуклы к оси состава (тип /,
см. рис. VIII. 12), кривые Рп характеризуются S-образным ходом.
Это обстоятельство может служить дополнительным способом уста-
164
яовления факта взаимодействия по вискозиметрическим
данным в системах, характеризующихся подобным ходом изотерм
вязкости.
Диаграммы Досп и Дрп (отклонение экспериментальных
величин температурных коэффициентов от значений этих свойств,
рассчитанных в предположении отсутствия взаимодействия) в
системах с взаимодействием характеризуются экстремумами,
приходящимися на область стехиометрии реакции, что, естественно, также
может быть использовано для целей
физико-химического анализа.
В качестве примера соотношения
между кривыми ц типа /, ач, рл, Лац
и Дрл на Рис- VIII.24 приводятся
данные по системе CF3COOH—
—СН2С1СООН, где образуется
продукт присоединения
эквимолекулярного состава.
Аналитико-геометрический анализ
политерм вязкости в системах,
характеризующихся максимумом вязкости
(тип III-2), показывает, что
максимумы c&rj и рл приходятся не всегда на
тот же состав, что и исходные изотер- сн^асоон 0,5
мы т). В этом смысле кривые ал и рл jc
не несут дополнительной информации .Рис< уш 24# Вязкость и тем.
по сравнению с изотермами ц. В слу- пературные коэффициенты вяз-
чаях же, когда положение максимума кости системы трифторуксус-
изотерм г] относительно оси состава ная кислота — монохлоруксус-
с изменением температуры изменяется ная кислота-
(тип Ш'22,б), максимумы на кривых
с^ и р^ лежат ближе к положению максимума на изотерме г],
отвечающей наиболее низкой из рассматриваемых температур.
Можно установить связь между рл и усредненной энергией
связи между молекулами бинарной смеси 8, входящей в
уравнение (VIII. 54). Дифференцируя уравнение (VIII. 54) по Г в
предположении, что Л и а не зависят от Т (действительно, эти величины
изменяются с Г на 1—2 порядка медленнее, чем вязкость) и деля
полученное выражение на исходное уравнение, получают
выражение
где
dT T \RT
1 + ехр (— &IRT)
1 - ехр (~ &IRT)
(VIII. 64)
Анализ экспериментального материала показывает, что
безразлично от наличия или отсутствия в системах взаимодействия,
155
L весьма мало изменяется с концентрацией. Поэтому в
соответствии с уравнением (VIII. 64) геометрия рл полностью определяется
геометрией &, как это можно видеть из диаграмм рис. VIII. 25, где
сопоставлены кривые рл и & для систем без взаимодействия (а),
а также в системах с взаимодействием (бив).
0,15 -
CgH0 0,5
X
3,0
СНСУСООН 0yS HC1O*
в
0,2
0,1
40
20
ft
10
Рис. VIII. 25. Вязкость,
относительный .температурный
коэффициент вязкости, энергия
активации вязкого течения и
энергия связи (по VIII.54) в
системах:
а — бензол — этилбензол; б — дихлор-
уксусная кислота —хлорная кислота;
в — хлораль — амиловый спирт.
СНзСНО 0,5 С5Н„ОН
X
В свою очередь, сопоставлением уравнений (IV. 18) и (VIII.54),
можно установить весьма простую аналитическую связь между
энергией активации вязкого течения Е^ к &
El = H + k%2 (VIII. 65)
где Н и k — постоянные для данного интервала температур Т2—Т\ч
равные
£i
Анализ уравнения (VIII. 65) показывает, что геометрия кривой
£а — состав определяется геометрией кривой & — состав. Расчет
показывает, что обычно величины коэффициентов Н и k таковы,
156
qTo El,' повторяя ход &, отличается от последней не более, чем на
Ю%- Так, например, в системе SnCl4—C6H5NO2 при jcc6h5no2 = 0,4
й тj == 293°, Г2 = 353°К, £? = 3,12 ккал/моль, а величина *?,
рассчитанная по уравнению (VIII. 54), равна 2,93 ккал/моль.
Аналогия в ходе кривых Е^ и & сохраняется независимо от того,
взаимодействуют ли или не взаимодействует компоненты системы, как
это можно видеть из рис. VIII. 25.
Из-за ограниченности экспериментального материала по
диэлектрической проницаемости двойных жидких систем, изученных
в более или менее широком температурном интервале, сколь-ни-
будь определенные теоретические обобщения по закономерностям
концентрационного хода кривых абсолютного (ае) и
относительного (р8) температурных коэффициентов диэлектрической
проницаемости сделаны быть не могут.
Можно утверждать, что с
достаточной степенью точности в
относительно нешироком
температурном интервале' температурная
зависимость е передается три- W
виальным выражением
[уравнение (IV.32) описывает более ши- сп
рокии температурный интервал]:
6 = -^ + 6 (VIII. 66) 20_
0уз
Из соотношений (VIII.62) вы-СЛО* *.*Ь*ЪР с^о, o,5o,sr
текает, что кривые ав по необхо- Рис> vill. 26. Свойства системы диок-
димости должны повторять гео- сан — вода,
метрию исходных кривых е.
Так же, как и в случае вязкости, кривые относительного
температурного коэффициента |Зе вч системах с взаимодействием
обладают экстремумом, который приходится на состав образующегося
в системе соединения (рис. VIII. 26).
Политермы электропроводности большинства двойных жидких
систем хорошо описываются уравнением.(IV.33), из которого еле-.
Дует, что так.же, как и в случае вязкости, относительный
температурный коэффициент определяется энергией активации
электропроводности Е*:
1 d%
RT2
(VIII. 67)
Хотя электропроводность связана с электролитной стадией об-
й схемы равновесий в растзорах, кривые рк, как правило,
хорошо отражают стехиометрию продукта присоединения —
независимо от геометрии и положения экстремумов на исходных
изотермах, как это можно видеть из примеров, приведенных на
157
рис. VIII. 27. Это кажущееся противоречие может быть пояснено
анализом уравнения (VIII.61) электропроводности, дифференвд.
руя которое по Г и разделяя переменные, легко придти к
выражению
ИЛИ
где
8А
(VIII. 68)
(VIII. 68а)
Таким образом, из уравнения (VIII. 68а) следует, что ход ри
определяется ходом кривых р^ и р8— тех свойств, которые, как
2,5-10
2 10'1 -
H2SO* 0,5 CF3COOH UzSOk 0,5 CH3COOQH5
xx
0,5 C3H5SCN
x
Рис. VIII. 27. Удельная электропроводность, абсолютный (<хи) и
относительный (рк) температурные коэффициенты электропроводности в системах:
а —серная кислота —трифторуксусная кислота; б —серная кислота — уксусноэтиловый эфир;
в — аллиловое горчичное масло — пиперидин.
было только что показано, весьма хорошо регистрируют
стехиометрию процесса образования продукта присоединения.
Итак, построение и анализ диаграмм относительных
температурных коэффициентов свойств, в частности, рл и р8 и рк является
весьма желательным этапом физико-химического анализа
жидкой системы. Отсюда вытекает естественная рекомендация
изучать жидкую систему в возможно более широком интервале тем*
ператур,
158
VIII. 3. Сравнительная характеристика методов
физико-химического анализа двойных жидких систем
Рациональный выбор методов физико-химического анализа для
каждой конкретной системы зависит от тех задач, которые ставит
перед собой исследователь; чаще всего эти задачи сводятся к
установлению факта и стехиометрии взаимодействия.
Установление факта взаимодействия, как очевидно, необходимо
лишь при изучении систем с малой энергией взаимодействия,
поскольку в иных системах факт взаимодействия очевиден уже хотя
бы по феноменологическим свойствам компонентов. Можно
считать, что одним из наиболее чувствительных для данных целей
методов является вискозиметрия. В тех случаях, когда система
образована неассодиированными компонентами, весьма
определенные сведения о факте взаимодействия могут быть почерпнуты из
диэлькометрических данных.
Установление состава соединений, как уже отмечалось,
является основной задачей физико-химического анализа. В разделе VIII. 1
было показано, что преимущественное значение для определения
состава соединений, образующихся в жидкой фазе двойной
системы, имеют мольно-аддитивные свойства (точнее, псевдомольные
свойства). Сопоставление относящегося к различным методам
физико-химического анализа материала, приведенного в предыдущем
разделе, показывает, что, во-первых, далеко не каждое из свойств
жидкой фазы может быть представлено в виде мольноаддитивной
модификации, а, во-вторых, не каждое из мольноаддитивных
свойств в одинаковой степени пригодно для целей
физико-химического анализа. Так, мольная рефракция RM, а также отклонение
мольной рефракции от аддитивности Д/?м чрезвычайно
малочувствительны к аддатационному взаимодействию [см.
уравнение (VIII. 45) и анализ этого уравнения].
Для целей физико-химического анализа весьма удобным
псевдомольным свойством является мольный объем. Как показывает
сопоставление диаграмм ДУ<*) различных мольных свойств, степень
определенности, с которой можно судить о составе соединения^
в случае диаграмм Д8 большей частью не меньшая, чем в случае
отклонения от аддитивности остальных псевдомольных свойств.
Построение диаграмм Д0 часто не требует дополнительного
эксперимента, так как данные по плотности необходимы для расчета или
обработки диаграмм многих иных свойств жидкой фазы. Из-за
соотношения величин AD и Дй [уравнение (VIII.48)] весьма
определенные сведения о составе образующегося в системе продукта
присоединения дает оптическая плотность, вернее, ее мольно-аддитив-
ная модификация FD. Как будет показано в следующем разделе,
Диаграммы Д8 и AFD являются наиболее удобными и для
суждения о степени взаимодействия в системе.
Сведения об электрохимических свойствах системы могут быть
получены из измерений электропроводности, чисел переноса,
159
а также потенциометрии). Помимо этой группы методов, ценные
сведения о природе возникающих при электролитической
диссоциации продуктов присоединения ионов могут быть получены при
помощи ИК- и КРС-спектроскопии. Последний метод может также
доставить весьма определенные сведения о строении продуктов
присоединения; сведения о строении могут быть получены также
исследованием диэлектрической проницаемости и анализом диаграмм
ряда расчетных свойств, основанных на диэлектрической
проницаемости.
VIII. 4. Количественный физико-химический анализ
двойных жидких систем
Связь элементов химической диаграммы
со степенью взаимодействия
Методы определения выхода продуктов реакции в растворе,
относительно подробно разработанные для взаимодействия между
двумя веществами, растворенными в индифферентном
растворителе (см. разделы VIII. 5 и VIII. 6), принципиально не распростра-
нимы на двойные системы, поскольку эти методы основаны именно
на изменении свойств растворителя (например, криоскопия). По
этой причине количественный физико-химический анализ двойных
систем обладает рядом специфических особенностей, некоторые из
которых и будут рассмотрены в этом разделе.
Ранее отмечалось, что геометрия изотерм свойство — состав
связана со степенью взаимодействия между компонентами системы.
Особенно наглядно эта связь выявляется при сопоставлении
экстремальных элементов диаграмм рядов подобных систем, т. е. систем,
образованных одним и тем же компонентом А с рядом близких по
химической и классификационной характеристике компонентов Вг-.
В табл. VIII. 1 сопоставляются максимальные отклонения от
аддитивности мольного объема А0, показателя преломления An,
а также максимальные величины относительной вязкости т]Отн =
= Цтах/цкА в системах, образованных уксусной кислотой (НАс)
с различными кислотами (НА). Кислоты расположены по
убыванию их силы, т. е. в порядке увеличения показателя константы
электролитической диссоциации в уксусной кислоте рК^ис'.
В табл. VIII. 1 также приведены значения /Ср реакций образования
продукта присоединения НАс + НА ^ НАс • НА, определенные
методом, о котором будет идти речь ниже.
Таким образом, кислотно-основное взаимодействие, в котором
уксусная кислота проявляет основную функцию, должно
уменьшаться от системы с хлорной кислотой к системе с пропионовой
кислотой, где взаимодействие вообще не протекает. Из табл. VIII. 1
видно, что экстремальные элементы химических диаграмм
действительно монотонно уменьшаются в данном ряду, и следовательно,
наблюдается симбатность изменения степени взаимодействия и
величин экстремальных элементов диаграмм свойство — состав.
160
Таблица VIII. 1
Экстремальные элементы химических диаграмм систем
уксусная кислота — кислоты (25 °С)
Кислоты
„НАс
^дисс
-де
Д/ЫО*
Хлорная
Пиросерная . . .
Серная
Селеновая ....
Ортофосфорная .
Азотная
Трифторуксусная
Трихлоруксусная .
Дихлоруксусная .
Монохлоруксусная
Пропионовая . .
2,7
3,8
4,3
4,4
5,1
5,4
11,6
9,1
7,1
5,6
4,5
1,6
0,6
6,06
5,91
4,20
2,84
1,76
0,72
0,70
0,68
0,40
0,25
-0,40
52,80
13,70
4,75
2,25
2,29
1,64
441
384
156
93
45*
35*
25*
10
При 50 °С.
Помимо рассмотренных в табл. VIII. 1 свойств, связь геометрии
изотерм со степенью взаимодействия обнаруживает ряд других
свойств. Так величины электропроводности, исправленной на
вязкость а = щ> закономерно увеличиваются в ряду подобных систем
с увеличением степени взаимодействия.
Определение констант равновесия реакций
образования продуктов присоединения
Аналитическая связь геометрии изотерм свойство — состав с
константой равновесия процесса образования продукта
присоединения была установлена ранее [см. уравнения (VIII.23), (VIII.24),
(VIII. 26) и (VIII. 28)].
Каждое метрическое уравнение двойной системы, где протекает
реакция типа (VIII. 12), имеет вид
У = !(Уа>Ув'Уа в >*>К) (VIII. 69)
т. е. связывает величину свойства равновесной смеси У с
аналитическим составом смеси х, величинами свойств компонентов z/a и
Ув и продукта присоединения г/А в , а также с константой
равновесия образования продукта присоединения:
/С = !1LJL (VIII. 70)
A TlTL \ Tib N '
Определение константы равновесия решением относительно К
Уравнений связи типа (VIII. 69) не может быть проведено, так как
в подавляющем большинстве случаев величина свойства продукта
присоединения уА в неизвестна. Эта величина не может быть
6 Зак, 500
161
определена экспериментально, так как в случае, если К конечна
т.е. взаимодействие (VIII. 12) не проходит до конца, AmBn не
может быть выделено из равновесной смеси. Кроме того, не
существует сколь-нибудь достоверных способов и теоретического расчета
величины уа в (об оценочных способах расчета этой величины
применительно к некоторым свойствам см. ниже).
Исключение у к в может быть достигнуто лишь одновремен-
т ti
ным решением двух уравнений, относящихся к различным
составам х = Х\ и х = х%. Так, если в системе образуется
эквимолекулярный продукт присоединения, то определение величины К может
быть произведено совместным решением двух метрических
уравнений [(VIII.24) или (VIII.26)], относящихся к этому типу
взаимодействия.
Обозначим отношение отклонений от аддитивности
псевдомольного свойства в точках состава х = Х\ и х = х2 через в, т. е. 0 =?
== -г-^- (выбираем точки так, чтобы &уХх > куХ2, т. е. @ > 1); а =
&Ух2
= х(1—х). Тогда, решая совместно два уравнения (VIII.24) для
х = xi и х = хг> придем к выражению *
(а1_а202)2
v
Метод расчета константы равновесия реакции аддатационного
взаимодействия, основанный на уравнениях типа (VIII. 71),
предполагает, что: отклонения от аддитивности обусловлены лишь
химическим взаимодействием, константа равновесия которого
может быть рассчитана; в системе во всем интервале концентраций
протекает лишь один процесс, например, только А + В^АВ или
только А + 2В^АВ2; во всем интервале концентраций
сохраняется постоянство константы равновесия.
Проследим, как несоблюдение этих условий отражается на
точности расчета /С.
Поскольку в реальных системах отклонения свойств от
аддитивности сопряжены как с химическими, так и с нехимическими
взаимодействиями, расчет К сопряжен с ошибкой, которая в ряде
случаев может стать настолько значительной, что речь должна
идти лишь об оценке константы равновесия. Так (впрочем,
приводимый пример относится к наиболее неблагоприятному случаю),
при значениях 0, близких к 5, изменение 0 на 1% приводит к
изменению К на 48%. Вот почему точность расчета К может быть
повышена лишь в тех случаях, когда удается определить вклад
нехимических взаимодействий в общую величину Дг/М.
* В тех случаях, когда в системе образуется соединение АВ2, расчет /Сдв2
производится совместным -решением двух уравнений (VIII. 26). Тогда
аналитическая связь 0 с /С, т. е. в = f(K), будет передаваться выражением, отличным
от (VIII. 71).
162
Метод расчета /С, основанный на уравнении (VIII. 71), не
приложим к иррациональным системам, где одновременно образуются
несколько соединений. Впрочем, если константа равновесия
одного из равновесий мала по сравнению с константой равновесия
второго-, например, /Cab > /Сав2, экстремум Д#(*) будет
приходиться на точку стехиометрии основного равновесия; побочное же
равновесие будет приводить лишь к асимметричности изотермы
Ду(*), как это можно видеть из рис. VIII. 28. При этом расчет
константы равновесия должен быть проведен по неискаженной
побочным взаимодействием ветви изотермы
Так как при исследовании двойной
системы изменение концентрации
компонентов происходит в весьма широких
пределах, постоянной является лишь
термодинамическая константа равновесия процесса
образования продукта присоединения, т. е.
1АВ
■•Ка
Уав
(VIII. 72)
Ц5
X
В
где у — коэффициент активности. Рис. VIII. 28. Изотерма
Так как методы определения коэффи- ^y{x) в иррациональной
циентов активности в двойных жидких си- системе (КАВ > Яав2)-
стемах с взаимодействием в большинстве
случаев редко являются надежными, в физико-химическом анализе
обычно оперируют концентрационной частью константы
равновесия, которая тождественна введенному ранее выражению (VIII.70)
и которую далее будем называть функцией равновесия.
' Из выражения (VIII. 71) видно, что нет оснований
предполагать неизменность /СФ во всем интервале концентраций двойной
системы. А поскольку величины Ау№ и Ау№ [см., например,
уравнения (VIII. 24), (VIII. 26) и (VIII. 28)] определяются именно
значениями /Сф, то непостоянство этой функции системы может стать
дополнительной причиной, обуславливающей геометрическую
иррациональность диаграмм этих свойств (как и исходных свойств
у(х) и y(v)y
Из сказанного вытекает, что определение константы
равновесия по уравнению (VIII. 71) может быть произведено лишь в том
приближении, с которой эту величину можно считать неизменной.
Некоторые специальные методы
определения степени взаимодействия
в двойных системах с аддатационным взаимодействием
Нередко взаимодействие между компонентами двойной
системы приводит к появлению новых полос поглощения на ИК- или
КРС-спектрах. Иногда появление новых полос поглощения может
происходить в УФ и в видимой области спектра. В этих случаях
определение степени взаимодействия может быть проведено по
6* 163
концентрационной зависимости этой новой полосы поглощения.
Равновесная концентрация продукта присоединения Сктъп (в
данном случае С выражается в моль/л) определяется по соотношению
Г — Апах (VIII 7Ч\
где D — оптическая плотность; сэ — показатель поглощения в
максимуме полосы; / — толщина поглощающего слоя.
Величина о определяется как тангенс угла наклона
касательной, проведенной к кривой D-C в области концентраций,
отвечающей избытку одного из компонентов; при этом предполагается,
что в этой области второй компонент нацело связан в комплекс.
б
'50
20
10
0,5
N
C5H5SN
Рис. VIII. 29. Функция равновесия в системах
дифениламин — пиридин (а) и масляный альдегид —
этанол (б).
Ошибка, связанная с этим предположением, составляет лишь
незначительную часть общей погрешности определения функции
равновесия в двойной системе. Зная равновесную концентрацию
продукта присоединения (а, следовательно, и равновесные
концентрации компонентов), можно рассчитать величину функции
равновесия для реакции (VIII. 12) образования продукта
присоединения.
Расчет равновесной концентрации продукта присоединения
может быть проведен также по убыванию оптической плотности
одного из компонентов. В этом случае экспериментальная
изотерма оптической плотности полосы поглощения сопоставляется
с аддитивной прямой, отвечающей отсутствию взаимодействия;
это позволяет определить равновесную концентрацию
поглощающего компонента. По найденным данным, зная стехиометрию
взаимодействия, нетрудно рассчитать равновесные концентрации
второго компонента и продукта присоединения, а следовательно, К$.
На рис. VIII. 29 в качестве примера приводятся изотермы
функции равновесия в системах дифениламин — пиридин [реакция типа
(VIII. 12)] и масляный альдегид — этанол [реакция типа (VIII, 14)].
164
Константы равновесия реакций образования продуктов в
системах кислоты — вода [М. И. В и н н и к, Н. Г. 3 а р а х а н и,
1963—1964] были определены в предположении, что молярные
коэффициенты поглощения анионов, образующихся при
взаимодействии воды с кислотами, в растворах кислот и
соответствующих солей одинаковы. Это предположение позволяет определять
равновесную концентрацию аниона; по оптической плотности
полосы поглощения, отвечающей недиссоциированной молекуле
кислоты, можно рассчитать равновесную концентрацию продукта
присоединения воды к кислоте. Таким образом, спектральное
исследование позволяет определить равновесные концентрации всех
стадий общей схемы (VII. 13) взаимодействия между
компонентами двойной системы вода — кислота.
В некоторых случаях представляется возможность рассчитать
дипольный момент продукта присоединения, образующегося в
системе. Подставляя вычисленное значение \ia в в одно из
уравнений, связывающих \х с е [например, (IV. 30)], находят величину
диэлектрической проницаемости продукта присоединения ед в .
Далее, зная величину е равновесной смеси, определяют
равновесную концентрацию продукта присоединения.
Несколько более общим является метод расчета констант
равновесия процесса образования продукта присоединения по данным
поляризации [О. А. Осипов, И. К. Шеломов, 1959]. Так, если
в системе образуется продукт присоединения AwBn, то степень
его" диссоциации связана с поляризациями компонентов и
соединения:
для области диаграммы A —AmBn
(хв/п)РА в + pf[iB]
::— <vin-r4*)
для области диаграммы AmBn — В
Отсюда константа равновесия вычисляется по уравнениям:
для области диаграммы А — AmBn
для области диаграммы AmBn — В
l1 ?«-)] (VIIL756)
165
Количественный физико-химический анализ систем
С обменным взаимодействием
Уравнения связи для систем, в которых протекают реакции
обменного взаимодействия (VIII. 14), представляют собою
выражения типа:
Y = f (Уа> Ув> У о У& х, К) (VIII. 76)
В отличие от уравнений связи для систем с аддатационным
взаимодействием (VIII. 69) величины свойств как компонентов,
СН3СООСзН7 0,5
х
CF3COOH
Рис. VIII. 30. Свойства системы уксуснопропиловый эфир —
трифторуксусная кислота.
Индексы:
«исх» характеризует смесь компонентов до начала реакции (величину
свойства находят расчетом; при условии медленного течения реакции-»"
экспериментально);
«равн» —равновесная смесь;
«кон» —смесь компонентов, прореагировавших до конца (расчет).
так и продуктов реакции могут быть определены
экспериментально. Поэтому константа равновесия реакции обменного
взаимодействия может быть найдена с большей точностью, чем
константа равновесия реакции аддатационного взаимодействия.
Расчет степени взаимодействия в системах с реакциями
рассматриваемого типа основан на определении содержания
продуктов реакции в равновесной смеси, исходя из аналитической связи
между величинами свойств: равновесной смеси У, той же смеси
в предположении отсутствия взаимодействия и при
взаимодействии, прошедшем до конца,
166
в
Так, если в системе протекает реакция А + В^±С + D, то
уравнение связи мольноаддитивного свойства представляет собою
следующее выражение:
у - УаХ + Ув и -,) + (2,см - Уа - Ув) ^
(VIII. 77)
где Уем — величина свойства эквимолекулярной смеси С + D,
которую можно определить
экспериментально, либо расчетом по
соответствующему уравнению
аддитивности.
На рис. VIII.30 в качестве
примера приведены диаграммы свойств
системы СН3СООС3Н7—CF3COOH,
в которой протекает реакция аци-
долиза, приводящая к образованию
CF3COOC3H7 и СНзСООН. Из
диаграмм видно, что
экстремальные элементы изотерм точно
приходятся на стехиометрию взаимо-
действия (Д — означает в данном
случае разность между значениями Рис. VIII. 31. Сечения тройной
величин свойств равновесной смеси системы А—В—С.
и смеси до конца
прореагировавших компонентов). Как и следовало ожидать функция равновесия
/Сф изменяется с изменением аналитического состава смеси.
ФИЗИКО-ХИМИЧЕСКИЙ АНАЛИЗ ДВОЙНЫХ СИСТЕМ
В РАСТВОРИТЕЛЕ (КВАЗИДВОЙНЫЕ СИСТЕМЫ)
Один из наиболее распространенных типов химических
реакций—это взаимодействие двух веществ А и В в растворителе S.
Большей частью при этом растворитель считают
индифферентным, хотя эта предпосылка соблюдается относительно строго лишь
в тех случаях, когда энергия взаимодействия между А и В
значительно превышает энергию взаимодействия каждого из этих
веществ с растворителем. Здесь нелишним будет отметить, что
вследствие специфических особенностей воды последнее условие
в водных растворах соблюдается весьма редко. Поэтому
исследование взаимодействия двух веществ в индифферентном
растворителе может быть (да и то с известной степенью приближенности)
реализовано лишь в случае неводных растворов.
Н. А. Измайлов (1951) отмечает следующие преимущества
физико-химического анализа двухкомпонентных систем в
растворителе по сравнению с методами исследования в отсутствии
растворителя:
I. В этом варианте физико-химического анализа появляется
возможность судить о характере взаимодействия между компо-,
167
нентами А и В по изменению свойств растворителя S. Связь
между свойствами исследуемых веществ и растворителя может
быть найдена из уравнения Гиббса—Дюгема — Маргулеса
Nkd In aA + NBd In aQ + Nsd In as = 0 (VIII. 78)
(a — активность).
К числу свойств растворителя, измерение которых наиболее
распространено в практике физико-химического исследования,
относятся давление пара, повышение температуры кипения и
понижение температуры замерзания.
II. Учет изменения ассоциативного состояния компонентов при
^образовании раствора в присутствии растворителя может быть
проведен весьма полно. Это обстоятельство позволяет, сравнивая
экспериментальную изотерму свойства с изотермой, отвечающей
отсутствию взаимодействия (т. е. с изотермой, характеризующей
свойства растворов А и В после смешения, но до реакции), весьма
точно определять как стехиометрию, так и степень
взаимодействия даже в случае значительно разбавленных растворов и при
малой энергии (степени) взаимодействия. ,
III. Наличие растворителя позволяет рассчитать парциальные
молярные свойства растворенных веществ, которые большей
частью весьма чувствительны к наличию взаимодействия и весьма
точно регистрируют его стехиометрию.
IV. Применение растворителя позволяет уменьшить различие
между свойствами компонентов, так как величины свойств
растворов А и В в S, как правило, характеризуются значительно
меньшим различием, чем сами А и В. Это дает возможность
более точно рассчитывать изменение величины свойства раствора,
вызванное химическим взаимодействием.
К перечисленным особенностям физико-химического анализа
двойных систем в растворителе можно добавить еще несколько:
V. Рассматриваемый вариант физико-химического анализа
охватывает значительно более широкий круг систем по
сравнению с физико-химическим анализом двойных жидких систем.
VI. Возможность, подбор соответствующей концентрации
позволяет оперировать с растворами компонентов,
характеризующихся постоянной степенью ассоциации. В этом случае картина
взаимодействия не искажается ассоциативно-диссоциативными
процессами.
VII. Расчет констант равновесия различных процессов может
быть проведен достаточно точно даже в тех случаях, когда не
известны величины коэффициентов активности, для чего
необходимо проводить исследование в достаточно- разбавленных
растворах.
Впрочем, исследование взаимодействия между двумя
веществами в растворах нельзя считать универсальным вариантом
физико-химического анализа, так как в случае слабых
взаимодействий оно не может выявить химизма в системе. Кроме того, не-
168
редки случаи, когда введение растворителя приводит к изменению
стехиометрии процесса взаимодействия.
Двухкомпонентная система А — В в растворителе S является,
разумеется, трехкомпонентной (рис. VIII. 31). Однако во всех
вариантах физико-химического анализа предпочитают вести
исследование при таких условиях, когда фиксацией концентрации
какого-либо из компонентов (чаще всего компонента S) система
может быть сведена к квазидвойной.
Наиболее распространенным является метод изомолярных
серий, часто называемый также методом Остромысленского — Жоба.
Условием исследования по этому методу
является сохранение постоянства
концентрации растворителя, то есть изучение
растворов, лежащих на сечениях Ai—Вь
Аг—Вг .. • Обычно исследование в
подобных случаях проводят, сливая изоконцент-
рационные (чаще всего, изомолярные)
растворы компонентов в различных
соотношениях, но так, что сумма исходных объемов
растворов компонентов остается
постоянной. Следует, впрочем, подчеркнуть, что
соблюдение изомолярности растворителя
(то есть «движение» по прямым, параллель- Рис. VIII. 32. Схема крио-
ным стороне АВ) в этом случае достаточно скопического метода
v * ' J ^ физико-химического ана-
точно соблюдается лишь при работе с £
б
разбавленными растворами, когда можно
пренебречь различием мольной доли растворителя в исходных
растворах компонентов *.
Исследование системы может быть проведено при постоянном
мольном соотношении компонентов А и В, например, при А: В =
= т:п, но с изменяющейся концентрацией растворителя, то есть
изучение растворов ведется по лучу AmBn — S.
Наконец, можно, прибавляя к одному и тому же количеству
одного компонента все возрастающие количества раствора
второго компонента, вести изучение растворов, отвечающих,
например, лучевым сечениям Ai — В, либо В2—А.
VIII.5. Методы физико-химического анализа квазидвойных
систем, основанные на изменении свойств растворителя
Общие положения
Если определить среднюю активность компонентов
квазидвойной системы, как
аА аВ
(VIII. 79)
* Действительно, два изомолярных раствора, характеризующиеся
равенством С а = С в (в моль/л), характеризуются различными значениями мольных
лх>лей хА и^в; различие это тем меньше, чем меньше С.
1Q9
то, учитывая, что
<-d ln (П <
уравнение (VIII. 78) принимает вид:
xsd In as + (хА + хв) d In acp = О (VIII. 80)
Из уравнения (VIII. 80) следует, что:
d In acp = - ** rf In as (VIII. 80a)
В случае метода изомолярных серий —~— = const, т. е.
*A + *В
dlnaCp = —constdlnas. Таким образом, изменение активности А и В
определяется лишь изменением активности растворителя. Вот
почему при постоянной сумме хА + хв изменение, например,
давления пара растворителя связано лишь с изменением суммарного
числа молекул А и В в результате взаимодействия (здесь и
далее подразумевается, что при смешении растворов А и В не
происходит изменение' ассоциативного состояния молекул
компонентов).
Комбинация уравнений Клапейрона — Клаузиуса и Рауля, дает
выражение
^(Г-Го) (VIII. 81)
связывающее активность растворителя as с мольной теплотой
агрегатного перехода (жидкости в пар либо кристаллизации
растворителя)— Д№ и разностью температур агрегатного перехода
чистого растворителя То и раствора Г.
Комбинирование уравнений (VIH.80a) и (VIII.81) позволяет
связать аср с понижением температуры замерзания или
повышением температуры кипения раствора, т. е. из криоскопических либо
эбуллиоскопических данных получать данные об изменении числа
частиц в растворе в результате взаимодействия.
Криоскопия
Криоскопические измерения более распространены в практике
физико-химического исследования, чем эбуллиоскопические или
измерения давления пара. Причина этого лежит прежде всего
в большей простоте криоскопического эксперимента по сравнению
с определением температуры кипения или давления пара раствора.
Разработано несколько вариантов криоскопического метода
физико-химического анализа. Первый из них [Н. А. Измайлов,
1951 —1953] состоит в сопоставлении экспериментальной величины
170
f—T0 = hT с величиной, полученной расчетом в предположении,
что при смешении компонентов уже произошло изменение
ассоциативного состояния, но еще не произошло взаимодействия.
При этом расчет производится следующим образом. Пусть
суммарная концентрация компонентов СА-\- Св = С. Тогда
исследованию системы предшествует изучение понижения температур
растворов каждого из компонентов в растворителе, т. е. систем
A — S и В — Sot концентраций А и В, равных С, до бесконечного
разбавления. Полученные данные изображаются на криоскопиче-
ской диаграмме кривыми I и II (рис. VIII. 32). Из этих кривых,
учитывающих изменение ассоциативного состояния А и В при
изменении концентрации каждого из них, строится расчетная
кривая /Я, соответствующая смеси непрореагировавших компонентов.
Экстремум разности (кривая V) между экспериментальной
кривой IV и кривой ///, т. е. значений 6ДГ, указывает на состав
образующегося в системе соединения.
Полученные криоскопические данные когут быть пересчитаны
на молекулярную массу. В простейшем случае при отсутствии
взаимодействия молекулярная масса раствора смеси компонентов
аддитивно слагается из молекулярных масс компонентов [см.
уравнение (VIII. 20)]. Если компоненты вступают в протекающее до
конца взаимодействие с образованием продукта присоединения
AmBn, то в точке стехиометрии молекулярная масса будет равна
(тМА + пМв).
Таким образом, диаграммы молекулярная масса — состав
[В. В. Удовенко, 1941] могут непосредственно указывать на
состав образующегося в системе продукта присоединения, а
также давать данные о степени взаимодействия. При этом возможны
следующие варианты (рис. VIII. 33, а—е):
а. Неассоциированные компоненты А и В не вступают во
взаимодействие;
б. Ассоциированный компонент А не вступает во
взаимодействие с неассоциированным компонентом В;
в. Ассоциированные компоненты А и В не вступают во
взаимодействие, причем степень ассоциации в перекрестных ассоциа-
тах АВ не отличается от степени ассоциации компонентов;
г. Компоненты А и _В образуют продукт присоединения,
причем взаимодействие протекает до конца;
д. Компоненты А и В вступают во взаимодействие,
характеризующееся конечной величиной константы равновесия;
е. Продукт присоединения AmBn обладает тенденцией к
присоединению компонента А.
Криоскопическое исследование может быть проведено в
варианте криоскопического титрования [М. И. У с а н о в и ч,
Т. Н. Сумарокова, 1954]. Особые точки на диаграммах ДГ —
состав связаны со стехиометрией взаимодействия. При этом в круг
реакций, которые исследуются с привлечением криоскопической
методики, могут быть включены и реакции осаждения, т. е. реакции,
171
"О 20 40 60 80 100
Этил бензол Толуол
Х,шл.%
100 80 60 40 20
сндон
х,мол%
C9hLOH
100 80 60 40 20 О
С2Н5ОН СН3ООН
х,мол.%
сбн5сн3
100 80 60 ЬО 20 О
C5HnN CeH5NCS
е %
220
М
100 80 60 W 20 О
СН3ОН СС23СНО
Рис. VIII. 33. Диаграммы молекулярная масса — состав
в системах А—В — бензол:
i, 2, 3, 4— изоконцентрат ы для 0 (экстраполяция на бесконечное
разбавление); 0,5- 1,0 и 1,5 моль/ШОе бензола.
приводящие к образованию соединений, нерастворимых в данном
криоскопическом растворителе.
Методика криоскопического титрования заключается в
определении AT растворов, образующихся при приливании к
определенному объему раствора А определенных порций раствора В.
Так, в случае реакций осаждения величина ДГ по мере
прибавления второго компонента будет уменьшаться, достигая либо
минимального значения (если образующееся соединение ограниченно
растворимо в данном растворителе), либо 0 (если образующееся
соединение совершенно нерастворимо в данном растворителе) при
соотношении компонентов, отвечающем стехиометрии
образующегося & системе соединения.
Наиболее чувствителен к установлению и факта
взаимодействия, и состава образующегося соединения первых из
рассмотренных вариантов криоскопического метода физико-химического
анализа.
Криоскопические данные позволяют определять не только
стехиометрию, но и ассоциативное состояние продукта
присоединения, образующегося в изучаемой системе.
Следует отметить, что иногда в тех случаях, когда в растворе
наблюдаются значительные флуктуации концентраций,
криоскопические данные могут привести к неправильным выводам. Вот
почему условием проведения криоскопических измерений
является отсутствие сколь-нибудь значительных флуктуации
концентрации. Существующая в настоящее время теория этого
явления позволяет в большинстве случаев произвести довольно
точную оценку вклада флуктуации концентраций в общую величину
понижения температуры замерзания.
Отклонение величин свойств от значений,
рассчитанных в предположении отсутствия взаимодействия
Метод сопоставления изотермы свойства, относящейся к
образовавшейся смеси непрореагировавших компонентов, с
экспериментальной изотермой свойства, описанный выше на примере
расчета понижения температуры замерзания, может быть
распространен на любые иные свойства жидкой фазы квазидвойной системы.
Н. А. Измайлов, разработавший этот метод, предложил называть
величины Уэксп—Уневз отклонениями от «аддитивности».
Очевидно, что этот термин не является строгим, так как без каких-либо
оговорок он может быть приложен лишь к свойствам, которые
в идеальной системе являются аддитивными, т. е. описываются
линейным уравнением Y = 2 У&1 (в случае двухкомпонентных
систем в растворителе tji и х%— величины свойств и мольных
долей компонентов А и В и растворителя). Поскольку термин
«отклонение от аддитивности» более удобен, чем приведенный в
подзаголовке, мы, следуя Н. А. Измайлову и традиции,
укоренившейся в литературе, будем пользоваться им в дальнейшем, помня,
173
шпз
0,6
0,5
А,
0,06
0,03
0,00
что в случае неаддитивных свойств это обозначение является
нестрогим.
Рассмотрим более подробно методику расчета отклонений от
аддитивности на примере вязкости квазидвойной системы
уксусная кислота — ацетон в бензоле при изоконцентрате
растворителя 0,5 мол. долей. На рис. VIII. 34 изотерма / характеризует
вязкость системы уксусная кислота — бензол от точки,
отвечающей чистому бензолу (в данном случае эта
точка лежит на ординате ацетона), до точки
с эквимолекулярным соотношением
уксусной кислоты и бензола.
Изотерма // отвечает вязкости системы
ацетон—бензол, причем здесь вязкость
изменяется от эквимолекулярного раствора
ацетона в бензоле (точка, отвечающая
бензолу, для данного случая лежит на
ординате уксусной кислоты). Изотерма ///,
полученная графическим суммированием изо-
СН СООНД5 СН СОСН теРм ^ и ' отвечает вязкости растворов
3 г з з системы в бензоле в предположении
отсутствия взаимодействия. Изотерма IV —
экспериментальная изотерма вязкости;
изотерма V — отвечает разности между
изотермами IV и ///; максимум на этой
изотерме соответствует составу образующегося
продукта присоединения СНзСООЩСНзЬСО. Из приведенной
диаграммы видной что в то время, как взаимодействие между
компонентами системы весьма слабо, отражается на
экспериментальной изотерме IV, на изотерме Дг) весьма четко отражается и
сам факт взаимодействия и его стехиометрия.
Описанная методика расчета отклонений от аддитивности
проводится в приближении, что концентрация ассоциированных и не-
ассоциированных молекул компонентов в «непрореагировавшей»
смеси такая же, как и в прореагировавшей. При не очень больших
константах равновесия это приближение является достаточным
даже в тех случаях, когда на основании этих зависимостей
рассчитывают выход реакции (см. ниже); для определения же
стехиометрии взаимодействия это приближение оказывается
достаточным почти всегда.
Рис. VIII. 34. Вязкость
системы уксусная
кислота — ацетон при
изоконцентрате бензола
0,5 мол. долей.
Диаграммы парциальных молярных свойств
Сопоставление изотерм парциальных молярных свойств,
относящихся к смесям с взаимодействием и к смесям, где после
смешения произошло изменение ассоциативного состояния
компонентов, но компоненты не вступили еще в химическое взаимодействие,
является весьма чувствительным методом установления факта и
стехиометрии взаимодействия в квазидвойной системе.
174
Общая теория применения парциальных (дифференциальных)
молярных свойств для целей физико-химического анализа развита
{-{. А. Измайловым (1951 г.). Эта теория связывает величину
свойства равновесной смеси с величинами свойств и истинными
мольными долями всех ассоциативных форм компонентов и продукта
присоединения. Так, если в системе А — В компонент А частично
димеризован и находится в растворе в виде молекул Аг, то
У = £а^а + # А + Ув^в + Уав^ав + h^s (VIII. 82)
где уi — величины парциальных молярных свойств, а N{ —
истинные мольные доли, которые могут быть найдены, например, из
криоскопических измерений.
Совпадение экспериментальных данных с рассчитанными по
уравнению (VIII. 82) показывает, полностью ли учтены все формы
отдельных молекул в растворе
и правильно ли определены их
истинные мольные доли.
Понятие «парциальное мо- ~m
лярное свойство» может быть /'
применено к любому
экстенсивному свойству раствора,
независимо от того, является ли f<°
данное свойство аддитивной
функцией мольных долей.
Например, вязкость
раствора выражается уравнением
n~2VV, (VIII. 83)
где
(VIII. 84)
0,5
СвНв 0,5
х
Рис. VIII. 35. Вязкость системы уксус-^
ная кислота — ацетон — бензол:
а —при различных изоконцентратах бензола;
б—парциальная молярная вязкость.
Смеси, в мол. долях: / —СН3СООН—С
//-0,4(СНз)2СО-0,бСН3СООН в бензоле;
///-0,7(СНз)2СО-0,ЗСН3СООН в бензоле;
Используем приведенный /v - ссвдгсб - с6нб;
выше пример квазидвойной си- 7и7Гчет; 7/-°пыт: "'-""■«*"• »«w '
стемы уксусная кислота —
ацетон в бензоле для рассмотрения принципа построения диаграмм
парциальных молярных свойств (рис. VIII.35,a).
Если вести расчет для системы при изоконцентрате
растворителя, например, 0,5 мол. долей, то величины парциальных
молярных свойств находятся из точек пересечения касательных к
изотермам вязкости в точке 0,5 с ординатами компонентов. При
этом на ординате бензола касательная отсекает величину
парциального молярного свойства растворителя.
Расчетная величина парциальной молярной вязкости смеси
компонентов находится суммированием величин Г компонентов
(рис. VIII. 35, б).
175
VIII. 6. Методы физико-химического анализа квазидвойных
систем, основанные на измерении свойств образующегося
в системе соединения, либо на измерении свойств раствора
Особенности- группы методов физико-химического анализа,
основанные на измерении величины свойства образующегося
в системе соединения, рассмотрим на примере спектрофотомет-
рии — одного из наиболее распространенных в практике
химических исследований метода.
Классификация диаграмм оптической плотности в
квазидвойных системах [А. К- Б а б к о, 1955], относящаяся к методу изо-
молярных серий и к определенной длине волны, предусматривает
два основных типа изотерм (рис. VIII.36), первый из которых
D
АтЪп В
АпВ„ В Л
лтъп в
А
В
Рис. VIII. 36. Основные типы изотерм оптической плотности в
квазидвойных системах:
1 — экспериментальная изотерма; 2 — изотерма, отвечающая отсутствию взаимодействия
(аддитивная); #—изотерма отклонения от аддитивности.
(а) характеризует поглощение окрашенного продукта
присоединения, которому присуща бесконечно большая константа
равновесия (сингулярная изотерма); второй тип изотерм (б) отвечает
случаю, когда реакция образования продукта присоединения
протекает не до конца. В последнем случае величина оптической
плотности Da в может быть найдена по точке пересечения
касательных, проведенных к кривой D в самых начальных ее участках.
Тогда, как следует из элементарных предпосылок, степень
диссоциации продукта присоединения связана с оптической
плотностью простым соотношением
D,
(VIII. 85)
из которого легко может быть вычислена константа равновесия
процесса образования продукта присоединения.
Все принципы определения состава соединения и константы
равновесия реакции его образования, основанные на спектрофо-
тометрии, могут быть распространены на случаи, когда
соединение при данной длине волны не поглощает, а поглощающим
является один из компонентов (например В). Принтом основные
17©
типы изотерм, как и в предыдущем случае, отвечают системам,
в которых взаимодействие прошло до конца (рис. VIII. 36, в) и
где константа равновесия образования продукта присоединения
конечна (рис. VIII. 36,г).
Существует далеко идущая аналогия между относящимися
к различным концентрациям изоконцентратам свойств
квазидвойной системы и соответствующим .различным температурам
изотермам свойств двойной системы. В .ряде случаев изменение
концентрации растворителя в квазидвойных системах оказывает на
равновесие в системе такое же действие, как изменение температуры
А 0,5
На
0,5 0,33 В
Рис. VIII. 37. Рациональные (а) и иррациональные (б)
изоконцентраты в квазидвойных системах.
в двойных смесях. Так, в квазидвойных системах встречаются
рациональные изотермы поглощения (рис. VIII. 37, а),
характеризующие поглощение в системах, где образуется лишь один
компонент, и иррациональные изотермы (рис. VIII. 37, б), отвечающие
системам, где одновременно образуется несколько продуктов
присоединения (изотермы относятся к изоконцентратам с различным
содержанием растворителя Cs, причем Cfs > Cs > С$" ...). При
этом так же, как и в случае двойных систем, при одновременном
образовании, например, продуктов присоединения АВ и АВг
максимум поглощения (или максимум отклонения от аддитивности,
если рассматривается случай, изображенный на рис. VIII. 37, б)
лежит между абсциссами, соответствующими хА = 0,33 и 0,5.
Связь между константами равновесия реакций образования
различных продуктов присоединения и положением экстремума
на кривых D или AD может быть установлена анализом
соответствующих метрических уравнений [Н. П. К о м а р ь, В. Н. То л-
мачев, 1950—1955].
Спектрофотометрия как метод определения состава
образующегося в системе соединений может быть проведена при
постоянной концентрации одного из компонентов (рис. VIII. 38).
Все, что было здесь описано для оптической плотности, без
каких-либо ограничений может быть распространено на любое
Иное аддитивное свойство в квазидвойных системах.
177
Методы физико-химического анализа, основанные на
измерении свойств образующегося в системе соединения (соединений),
имеют более ограниченную область применений, чем методы,
описанные в предыдущем разделе. Впрочем, точность определения
констант равновесия с помощью этой группы методов, как
правило, выше, чем с помощью группы методов, основанной на
измерении свойств растворителя. ■
8
D J
АЪ А*В СА
ЛВ
СА
Рис. VIII. 38. Основные типы изотерм оптической плотности на
диаграммах, отвечающих постоянной концентрации одного из компонентов (В):
а —в системе образуется один прочный продукт присоединения; б) образующийся
продукт присоединения заметно диссоциирован на компоненты; в — в системе образуется
несколько продуктов присоединения; г — аналогично случаю «в», но Dj^2^ < Ь
К числу методов физико-химического анализа квазидвойных
систем, основанных на измерении свойств образующегося в
системе соединения относится калориметрия. Предложено
[И. П. Гол{>дштейн, Е. Н. Гурьянова, И. Р. Карпович,
-Д//
ЛтВп
Рис. VIII. 39. Диаграмма калориметрического титрования.
1965] проводить определение состава и константы равновесия
продукта присоединения в растворе методом калориметрического
титрования, который основан на измерении количества тепл£ как
функции количества небольших количеств компонента В,
прибавляемого к определенному количеству раствора компонента А.
При отсутствии взаимодействия будет наблюдаться
прямолинейная зависимость между энтальпией смешения и концентрацией
прибавляемого компонента СА (VIII. 39, а). Если образующееся
соединение AmBn недиссоциировано (К—юо), то его составу на
диаграммах калориметрического титрования соответствует точка
пересечения двух прямых (рис. VIII.39,б). При конечной кон-
178
станте равновесия процесса образования продукта
присоединения кривая калориметрического титрования будет монотонна
(рис. VIII. 39, в). Состав соединения в этом случае определяется
по точке пересечения прямых, экстраполированных из
прямолинейных участков калориметрической диаграммы; точка
пересечения (—А#оо) отвечает тепловому эффекту образования продукта
присоединения в предположении прошедшего до конца
взаимодействия. Сравнивая —Д#оо с экспериментальной, легко
рассчитать степень диссоциации продукта присоединения, а
следовательно, и константу нестойкости соединения AmBn з данном
растворителе.
Примером метода физико-химического анализа квазидвойной
системы, основанного на измерении свойств раствора, может
служить диэлькометрия. С помощью кривых диэлькометрического
титрования [Е. Н. Гурьянова, 1964] по принципу, аналогичному
калориметрическому титрованию, можно определять стехиометрию
и константу равновесия процесса взаимодействия. Данные по ди-
элькометрической проницаемости раствора могут быть
использованы для расчета поляризации.
Диаграммы диэлектрической поляризации [И. А. IIIека, 1957]
позволяют более точно определить константу равновесия процесса
комплексообразования.
VIII. 7. Метрика двухкомпонентной химической системы
в растворителе
Как и в случае двойных систем, метрические уравнения
равновесной химической диаграммы двойной системы в растворителе
связывают величину свойства равновесной смеси У с величинами
свойств компонентов и растворителя, их содержанием в растворе
и константой равновесия химического процесса, протекающего
в системе:
У = / (Уа> У* УАтвп> *а. *в> К) (VIII. 86)
В случае квазидвойных систем при постоянной мольной доле
растворителя метрические уравнения можно выводить лишь с
одной переменной по составу (считая Ха + *в=1), и уравнение
(VIII. 86) сводится к уравнению (VIII. 69).
Метрические уравнения квазидвойных систем различаются для
систем, образованных неассоциированными и ассоциированными
компонентами. Поэтому здесь эти два случая будут
рассматриваться раздельно.
Метрика квазидвойных систем,
образованных неассоциированными компонентами
Как и прежде, рассмотрим метрику систем, где протекают
реакции присоединения типа (VIII. 12).
Обозначим количества молей компонентов в равновесной смеси
через а и 6, количество молей продукта присоединения AmBn —
через г, количество молей растворителя — через s.
179
Уравнение связи для этого случая представляет собою выра-
жение [Н. И. С т е п а н о в, 1924—1933]
г [1 - г (т + я - 1) + s]m+n"1 - К (х - mz)m (1 - * - яг)" = О (VIII. 87а)
где т и п—стехиометрические коэффициенты в уравнении VIII. 12.
При постоянном содержании растворителя (изоконцентрата по
растворителю) уравнение связи модифицируется в
г [1 - г (т + п - [)]т+п~1 -К (х- тг)т - (1 - * - пг)п = О (VIII. 876)
Уравнение (VIII. 87,а) относится к типу (VIII. 86), а
(VIII. 876)—к типу (VIII. 69).
Количество молей продукта присоединения z в 1 моль
равновесной смеси принято называть выходом химической реакции.
Диаграмма, изображающая зависимость выхода реакции от
константы равновесия (изотермы выхода), представлена на
рис. VIII. 40, а. Как обычно, сингулярная изотерма отвечает взаи-
п
ZI
в
в
Рис. VIII. 40. Диаграмма выхода в квазидвойной
системе (Коо > Къ>К2> Ki):
а— система, образованная неассоциированным компонентом; б
—система, один из компонентов которой ассоциирован.
модействию, прошедшему доконца (/С—>оо); поскольку выход
реакции может быть (с известными оговорками) отнесен к
псевдомольному свойству, изменение константы равновесия не приводит
' к иррациональности изотерм выхода.
Практически единственным способом экспериментального
приложения уравнений связи являются методы, основанные на
измерении свойств растворителя в квазидвойной системе. Так,
Н. И. Степанов при выводе уравнения изотермы растворимости
предложил исходить из закона постоянства изотермической
растворимости, гласящего, что при данной температуре отношение молей
растворенного вещества к числу молей растворителя в
насыщенном растворе постоянно. При этом состав растворителя может быть
сколь-угодно сложным; тогда при расчетах, основываясь на
постоянстве состава растворителя, состав его можно передавать
одной переменной —так же, как и в случае индивидуального
растворителя.
180
Если сумма молей компонентов смеси и образовавшегося
соединения равна ^т = а-\-Ь-\-2>а в соответствии с законом
изотермической растворимости -=— =К, то уравнение растворимости
7\ tn
передается выражением [Е. Е. Ч е р к а ш и н, 1958]
(с + cs - s)(т + п - 1)^+я-1^+я-1 _ к [сх (т + п _ {) _
- т (с + cs - s)]m [с (т - 1) - ns (с - 1) - сх (т + п - 1)]п = О (VIII. 88)
Метрика квазидвойных систем,
образованных ассоциированными компонентами
Пусть в системе А—В компонент В ассоциирован, т. е. в
растворах может существовать равновесие /В *± Bt, константа
которого с учетом принятых выше обозначений запишется
bt }— (VIII. 89)
где р — равновесное количество молей В*.
Тогда уравнение изотермы выхода реакции будет передаваться
следующим выражением [Е. Е. Ч е р к а ш и н, 1958 г.]:
гъ (К2 + К- Kac) + Z2 (2KacS + KacX + Кае - К2Х - Кх - К2 ~ К -
+ г (2К2х + Кх + Kxs - К2х2) + К2х2 (х - 1) = О (VIII. 90)
На рис. VIII. 40, б приводятся изотермы выхода для
рассматриваемого случая. Сингулярная изотерма, максимум которой точно
соответствует составу продукта присоединения, характеризует лишь
прошедшее до конца взаимодействие: чем меньше константа
равновесия процесса комплексообразования, тем больше экстремум
выхода сдвигается от точки стехиометрии.
Величина константы ассоциации /Сас также влияет на
положение экстремума изотермы выхода: чем выше /Сас, тем более
при прочих равных условиях максимум сдвигается от точки
стехиометрии.
Для системы А—В с обоими ассоциированными компонентами
уравнение изотермы выхода слишком сложно, чтобы пользоваться
им в общем виде. Некоторые частные формы этого уравнения
рассмотрены в монографии Е. Е. Черкашина.
Определение выхода реакций образования продуктов
присоединения в растворах
Расчет выхода реакций присоединения рассмотрим на примере
криоскопии, которая позволяет наиболее полно учесть изменение
количества частиц в растворе, вызванное не только
взаимодействием, но и изменением ассоциативного состояния компонентов,
а также образовавшегося продукта присоединения.
181
Криоскопический метод определения выхода продукта
реакции [Н. А. Измайлов, 1953] состоит из следующих
последовательных операций:
1. Из криоскопических данных для растворов А в S и В в S
различной концентрации, рассчитывают концентрационную
зависимость фактора ассоциации /= Л7,эксп для каждого из компо-
ЛУ теор
нентов исследуемой двойной системы в растворителе;
2. Определяют АГ растворов с различным соотношением А: В,
но с постоянным содержанием S;
3. Из криоскопических данных для растворов А в S и В в S
рассчитывают теоретическую величину АГтеОр в предположении
отсутствия взаимодействия: АГтеОр = А7"а + А7в. Диаграмма 6ДГ =
= ДГТеор — АГЭКсп как функция состава квазидвойной системы
указывает на состав образующегося в растворе соединения.
Первые три этапа исследования системы совпадают с
описанным в разделе VIII. 5 методом исследования квазидвойных
систем;
4. Расчет выхода продукта реакции производят методом
последовательных приближений. Вначале по уравнению S\c =—±г-
жшшА tK
(Ек — криоскопическая постоянная для данного растворителя)
находят изменение количества частиц в растворе в результате реакции;
у с
5. По уравнению Сдв = f , f __ 1 находят в первом
приближении равновесную концентрацию продукта присоединения;
6. Второе приближение учитывает ассоциацию компонентов
в данном растворителе:
где С* — равновесные концентрации компонентов, рассчитанные
с учетом уменьшения исходной концентрации в результате
образования продукта присоединения.
Приближения производят до тех пор* пока не достигают
постоянства величины Сдв *.
* Расчеты по пунктам 5 и 6 приведены для системы, в которой образуется
эквимолекулярный продукт присоединения; в случае образования, например
соединения состава АгВ, расчет ведут по формулам:
первое приближение
2 2'А "г 'В
второе приближение
>.C-Cf.-
2fA-fB-l
182
В литературе можно найти лишь единичные работы,
посвященные исследованию гомогенных жидкофазных равновесий
в тройных системах, все компоненты которых взаимодействовали
а
40 60
Мол.% - - Шл%
Рис. VIII. 41. Система H2SO4—Н3РО4—Н20 при 25 °С:
а —изотермы вязкости нулевых разрезов; б —изотермы электропроводности нулевых разрезов;
в — проекции кривых изовязкостных растворов; г — проекции кривых изоэлектропроводных
растворов.
бы друг с другом (в тех случаях, когда один из компонентов не
вступает во взаимодействие с двумя другими, система может быть
сведена к квазидвойной).
В качестве одного из немногочисленных примеров такой
тройной системы приведем диаграммы вязкости и электропроводности
183
'системы H2SO4—Н3РО4—Н20. В каждой из двойных систем этой
тройной системы протекает отчетливое взаимодействие: в
система** H2SO4—Н2О и Н3РО4—Н2О образуется ряд гидратов; в
системе H2SO4—H3PO4 протекает кислотно-основное взаимодействие
с образованием продукта присоединения H2SO4-H3PO4, в
котором Н3РО4 проявляет основную функцию. На рис. VIII.41
приводятся данные по вязкости (а) и удельной
электропроводности (б) лучевых сечений, идущих от воды к растворам с
различным фиксированным соотношением кислот, а также проекции
кривых изовязкостных (в) и изоэлектропроводных (г) растворов
для температур 25 °С.
В настоящее время теория гомогенных жидкофазных тройных
систем не разработана в степени, достаточной для того, чтобы
на основании анализа тройных диаграмм можно было бы
достаточно достоверно судить об образовании в системе тройных
соединений AmBnCp.
Ввиду исключительной громоздкости эксперимента в
литературе нет данных по исследованию гомогенных жидкофазных
равновесий ни в одной полной системе с числом компонентов,
большим трех.
МЕТОДЫ ИССЛЕДОВАНИЯ СОЛЬВАТАЦИИ ИОНОВ
Материал, изложенный в гл. VI, был посвящен
преимущественно анализу термодинамических характеристик процесса
сольватации. В этом разделе будут рассмотрены некоторые
физические методы исследования процесса сольватации. Ряд из них
описан достаточно подробно в обзорных статьях [Б. Е. К о н у э й,
Дж. О'М. Б окр й с, 1958; Ж. Денуайе, К. Жоликер,
1971 г.], монографической [Н. А. Измайлов, 1966] и учебной
[Л. И. Антропов, 1969] литературе, в то время как другие
методы стали интенсивно развиваться (особенно применительно
к неводным растворам) лишь в последние годы.
Как и в исследовании структуры растворителей, пониманию
процесса сольватации ионов способствует огромное число
физических методов и свойств, например таких, как вязкость,
диэлектрическая проницаемость и время релаксации, диффузия ионов
и самодиффузия молекул растворителя в ионных растворах,
поглощение ультразвука, поверхностное натяжение, дифракция
рентгеновских лучей и ЯМР, инфракрасная и рамановская
спектроскопия.
Естественно, что ни один из методов не обладает достаточной
универсальностью, чтобы дать полное описание такого сложного
процесса как сольватация. Достаточно сказать, что отсутствие
однозначных методов определения четкой границы между
первичной и вторичной сольватными оболочками, отмечавшееся
в гл. VI, приводит к значительному расхождению данных о чис-
184
лах сольватации одного и того же иона, получаемых различными
методами.
Такое положение вполне понятно, поскольку почти во всех
случаях связь изучаемых свойств с сольватацией является лишь
косвенной и при нахождении чисел сольватации делаются
различные дополнительные, зачастую произвольные, допущения.
Как было показано ранее (см. гл. VI), иногда удается
рассчитывать термодинамические функции сольватации с
использованием определенной модели сольватированного комплекса,
описываемой некоторыми параметрами процесса сольватации (энергия
сольватации, радиус сольватированного иона, объема сольвата-
ционной оболочки, число сольватации). Среди них наибольшее
распространение получили числа сольватации.
Кроме перечисленных выше методов, количественные
характеристики процесса сольватации ионов находятся также из данных
по плотности, диализу, термохимических и электрохимических
свойств растворов.
В данном разделе основное внимание будет сосредоточено на
методах исследования сольватации, основанных на измерении
подвижности ионов и спектральных характеристиках процесса
сольватации.
VIII.8. Методы исследования сольватации, основанные на
измерении подвижности ионов
Рассмотрим, как сказывается сольватация на движении ионов.
Этот эффект может быть оценен из данных по числам переноса,
либо на основе определения размера сольватированного иона.
Число молекул растворителя, совершающих вместе с ионом
движение под действием электрического поля, предложено называть
трансляционным числом -сольватации [А. М. Сухотин,
А. Ф. К а з а н к и н а, 1970].
Числа переноса
Классическим методом исследования сольватации является
изучение переноса ионов. Числа переноса ионов, определяемые
на основе модифицированного Уошборном метода Гитторфа по
изменению ионных концентраций в приэлектродных пространствах
в процессе электролиза, будут истинными числами переноса, если
концентрационные изменения связаны только с перемещением
ионов. Однако ионы в своем движении уносят некоторое
количество связанных с ними молекул растворителя. Это обстоятельство
также будет причиной изменения концентрации в приэлектродных
областях.
В соответствии с этим вводятся понятия об истинных (w) и
кажущихся (п) числах переноса, что позволяет учесть количество
связанного растворителя и определить числа сольватации ионов.
185
При прохождении IF электричества анионы переносят в сторону
анода (1 —шк)Ла моль, а катионы в сторону катода wKnK моль
растворителя. Всего из анодного в катодное пространство будет
перенесено т = (\—аук)яа — wKnK моль растворителя.
Число переноса п равно кажущемуся числу эквивалентов
электролита, a w — истинному числу эквивалентов электролита,
перенесенных из анодного пространства в катодное. Разность этих
двух величин определяется числом эквивалентов электролита,
которое было бы растворено в т моль растворителя и дает
значение чисел сольватации.
"Весьма распространенным приемом определения чисел
сольватации из данных по переносу ионов является использование
какого-либо инертного неэлектролита, например, углевода,
который, как предполагают, неподвижен в электрическом поле. Тогда,
если определить изменение концентрации электролита и вещества
сравнения, можно установить количество растворителя,
переносимого ионами. Поскольку изменение коцентрации неэлектролита
является результатом сольватации ионов обоих знаков, этот метод
дает только разность между числами сольватации катиона и
аниона.
Впрочем, для определения чисел сольватации отдельных
ионов стараются подобрать такие пары катион—анион, в которых
сольватацией одного из ионов можно пренебречь, или же для
которого точно известно число сольватации. Недостаток метода
состоит в трудности подбора инертного вещества, которое не
вступало бы во взаимодействие с ионом и не переносилось бы вместе
с ним. Низкая точность метода может быть обусловлена также
изменением концентрации вещества сравнения вблизи ионов из-за
эффекта высаливания.
Перспективным для исследования процесса сольватации путем
измерения чисел переноса является радиоизотопный вариант
метода Гитторфа (см. гл. XI).
Электропроводность
Этот метод исследования процесса сольватации ионов основан
на законе Стокса — Эйнштейна.
Рассматривая движение сольватированного иона во внешнем
электростатическом поле как единого комплекса, можно
рассчитать радиус гс такой движущейся частицы по уравнению Стокса
r = JZJF[ (VIII. 91)
с бяА^оЯо х
где \z\— абсолютный заряд иона; Яо — предельная эквивалентная
ионная электропроводность; щ — вязкость растворителя; А^о —
число Авогадро; F — число Фарадея,
186
После введения числовых коэффициентов
(VIII. 91) можно переписать в виде:
г _ 0,82 1 z 1
уравнение
(VIII. 92)
Вывод уравнения Стокса, как известно, основан на применении
законов классической гидродинамики для случая движения
сферической макроскопической частицы в идеальной непрерывной
среде. Отсюда необходимо учитывать недостаточную
обоснованность в применении такого приближения к описанию поведения
сольватированного иона.
Действительно, значения гс, рассчитанные по уравнению
(VIII. 91), обычно получаются заниженными. Поэтому для
вычисления истинных радиусов сольватированных ионов гист было
предложено видоизмененное уравнение Стокса
0,821 z | гкр
т-1 — (VIII. 93)
/"ист = "
в котором гс — стоксовское значение радиуса сольватированного
иона, рассчитанное на основании предельных подвижностей; гкр —
кристаллографический радиус. Фактор корреляции -^- был вве-
гс
ден Р. Робинсоном и Р. Стоксом (1963) как поправочный
коэффициент для закона Стокса.
В табл. VIII. 2 приведены значения стоксовских и
коррелированных радиусов ионов в различных растворителях. Из таблицы
видно, что анионы характеризуются уменьшением гист от СГ
Таблица VIII. 2
Значения радиусов (в А) ионов, полученные из уравнения Стокса (гс),
и коррелированные радиусы ионов гкор в различных растворителях
1д е л л а, Моника, Сенаторе, 1970]
Ион
Li+
Na+
К+
Rb+
Cs+
NH+
СГ
Br-
Г
сю;
NO"
Метанол
ГС
3,77
3,32
2,86
2,62
2,40
2,59
2,86
2,65
2,39
2,11
2,80
гкор
4,74
4,46
4,16
4,02
3,88
3,99
4,16
4,08
3,88
3,70
4,14
Формамид
'С
2,92
2,46
1,96
1,94
,84
,59
/45
,44
1,45
1,45
,44
гкор
4,36
3,99
3,58
3,57
3,48
3,30
3,14
3,14
3,21
3,14
3,14
К,М-диметил-
формамид
ГС
4,12
3,45
3,34
3,18
2,99
2,66
1,87
1,92
1,97
1.97
1,80
гкор
5,10
4,46
4,36
4,22
4,04
3,70
3,00
3,08
3,09
3,08
2,92
N, N-диметил-
ацетамид
ГС
3,49
3,54
—
—
2,06
2,13
2,08
1,92
гкор
4,60
4,65
—
—
—
3,51
3,56
3,54
3,40
Нитробензол
ГС
2,78
2,54
—
2,46
2,04
2,10
2,22
2,17
2,00
гкор
3,99
3,77
—
—
3,70
3,32
3,39
3,49
3,46
3,30
Ацетонит-
рил
ГС
2,98
3,10
2,86
—
2,44
—
2,54
2,35
2,38
2,31
2,24
гкор
4,13
4,24
4,04
—
3,68
—
3,76
3,60
3,56
3,55
3,48
Сульфолан
ГС
1,92
2,30
2,05
2,00
1,95
1,67
0,89
0,93
1,15
1,24
—
гкор
3,74
4,23
3,92
3,85
3,78
3,36
1,98
2,06
2,47
2,64
—
187
к СЮ7 в метаноле и ацетонитриле; в формамиде, диметилформ-
амиде и диметилацетамиде величина гист почти постоянна, тогда
как в сульфолане гист увеличивается от СГ к СЮ'. Радиус
катионов уменьшается от лития к цезию.
Поправочный коэффициент рассчитывался на основании
допущения, что большие тетраалкиламмониевые ионы не гидрати-
руются в водных растворах. Справедливость этого утверждения
в свете представлений о структуре водных растворов тетраалкил-
аммониевых солей [И. В. Кудрявцева, Б. С. Крумгальз,
К. П. Мищенко, 1970] оказывается весьма условной. Кроме того,
для расчета поправочного коэффициента необходимо знать
собственные размеры ионов. Расчет их радиусов по Робинсону и
Стоксу также основан на ряде не вполне строгих допущений.
Критический анализ экспериментальных данных по
определению радиусов сольватированных ионов [Б. С. Крумгальз,
К. П. Мищенко, Д. Г. Трабер, 1971] показал возможность
использования для расчета радиусов сольватированных ионов
непосредственно уравнения (VIII. 91) без учета поправок.
Радиусы сольватированных ионов можно использовать для
определения чисел сольватации ионов яСольв. Объем сольватной
оболочки рассчитывают по формуле
Гсольв-Тя(^-'кр) (VIII. 94)
причем величина г может быть найдена из уравнений (VIII. 92)
или (VIII. 93).
Число сольватации иона определяется выражением
«сольв = ~^- (VIII. 95)
1 L
где Vl — объем, занимаемый одной молекулой растворителя,
может быть оценен на основе ее пространственной структуры.
Считая, что молекулы растворителя вблизи иона должны
характеризоваться высокой упорядоченностью, можно предположить, что
структура сольватной оболочки отвечает плотнейшей упаковке.
С другой стороны, предельное сжатие молекул растворителя
в поле иона должно уменьшать объем растворителя до
величины, которая определяется средним расстоянием между
атомами. (Найденные из таких предпосылок объемы молекул
растворителей ^составляют, например, для воды 12,2 А3, для этанола
28,9 А3).
В табл. VIII. 3 приведены литературные данные по
трансляционным числам сольватации ионов при бесконечном разбавлении
в различных растворителях, полученные из данных по
электропроводности. Они показывают, что числа сольватации катионов
довольно близки для ряда растворителей; для анионов
наблюдаются некоторые различия, причем в алифатических спиртах
получаются более высокие значения, чем в других растворителях.
188
Таблица VIII.3
Трансляционные числа сольватации ионов в различных растворителях
Ион
и+
Na+
К+
с\~
вг
г
no;
сю~
Вода
4,6
1,8
-0,1
-1,4
-2,0
-2,9
—
-3,7
Этанол
11,0
7,1
4,3
4,9
3,0
1,6
3
0,4
Фенол
_
6
4
2
1,5
0,5
0
—
Ацетон
5
5
4
1
—
0
—
—
Ацето
нитрил
6
8
4
3
2,5
0,5
1,5
—
Диметил-
формамид
_
2,9
2,3
—
—
1
—
—
Сульфо-
лан
1,4
2,0
1,5
0
0
0
—
0
В ряду катионов, обладающих изоэлектронной структурой,
числа сольватации, как правило, уменьшаются с увеличением
ионного радиуса — в соответствии с изменением -напряженности поля,
создаваемого ионом. Однако на величины чисел сольватации
оказывают значительное влияние химические свойства растворителя.
Так, растворители, молекулы которых обладают приблизительно
одинаковым размером, будут сольватировать данный ион в тем
большей степени, чем выше химическое сродство иона к
растворителю (подробнее о химическом сродстве ионов к растворителю
как факторе, обуславливающем электролитные свойства раствора,
см. в разделе IX. 5). Размеры молекулы играют весьма
значительную роль в числах сольватации ионов в этом растворителе. Так,
в сульфолане из-за значительного объема занимаемого молекулой
этого соединения числа сольватации малы, либо вообще равны
нулю.
Отрицательные значения чисел гидратации ионов К+, СГ, Вг~
и I- соответствуют представлениям О. Я. Самойлова о структуре
водных растворов электролитов.
Диффузия
Аналогичный методу электропроводности способ оценки
сольватации ионов основан на определении скоростей диффузии.
В классической теории диффузии коэффициент диффузии D
определяется уравнением:
(VIIL96)
Уравнение (VIII. 96) строго соблюдается лишь для частиц,
имеющих большие размеры по сравнению с частицами
окружающей среды. С некоторыми же ограничениями его можно применить
Для расчета радиусов сольватированных ионов, а из них — чисел
сольватации.
189
Расчет коэффициентов диффузии ионов может быть проведен
с использованием предположения, что величины D ионов при бес-
конечном разведении будут пропорциональны их эквивалентным
электропроводностям. Кроме кондуктометрических данных, для
нахождения коэффициентов диффузии пригодны метод меченых
атомов, полярография, оптические методы.
Не меньшее значение представляет изучение влияния ионов
на самодиффузию молекул растворителя, хотя исследованию
этого вопроса посвящено очень небольшое число работ. Между
тем, такие исследования помогают выяснению характера
взаимодействия ионов с растворителем, дают информацию об изменении
структуры растворителя под влиянием электролита и т. д.
Кроме традиционного метода изотопных индикаторов
[J. H. Wang, 1954] в последние годы находит широкое
применение для определения коэффициентов самодиффузии растворителя
в ионных растворах метод спинового эха [К. А. Валиев и др
1964].
Сопоставление чисел сольватации ионов, полученных из
данных по электропроводности, с рассчитанными значениями яСольв
из величин D растворителей показывает, что метод спинового эха
приводит всегда к более высоким значениям Ясольв- Такое
расхождение можно объяснить довольно слабым взаимодействием
ионов с молекулами растворителя: при измерении подвижности
ионы под действием электрического поля легче покидают
молекулы растворителя, а в методе спинового эха электрическое поле
на них не действует.
VIII. 9. Спектроскопические методы исследования
сольватации
В современных работах по сольватации спектральным
методам уделяется много внимания, поскольку они дают большой
объем информации, непосредственно касающейся взаимодействия
ионов с растворителем. Данные по электронным спектрам
позволяют оценить энергетические характеристики связей,
инфракрасная и рамановская спектроскопия отражают изменения в
колебательных и либрационных движениях молекул растворителя,
а сдвиги ЯМР и времена релаксации дают сведения об изменении
распределения электронов вокруг ядер в молекулах растворителя
и о средней скорости обмена молекул растворителя в
координационной оболочке.
Электронные спектры
Данные об электронных спектрах часто используют для
выводов о строении электролитных веществ в растворах. Так как
положение полосы поглощения иона в растворе характеризует
энергию, необходимую для переноса электрона между ионом и моле-
190
пулами растворителя в сольватной оболочке, то смещение этой
полосы при переходе от одного растворителя к другому может
служить мерой различия энергии связи между ионом и
молекулами растворителя в сольватной сфере.
Исследованиями спектров поглощения в видимой области
растворов солей многих переходных металлов в различных
растворителях установлено существенное изменение оптических свойств
ионов при переходе от одного растворителя к другому. Например,
полосы поглощения сольватированных ионов Си2+ и Со2+ в
метаноле и этаноле значительно смещены в длинноволновую
область по сравнению со спектрами поглощения этих ионов в
водных растворах [П. А. Загорец, С. А. Скобелев, 1967].
Анализ спектров поглощения в видимой области позволяет
получить информацию о геометрии сольватных оболочек ионов
в растворах, т. е. судить о структуре раствора.
Так, установлено, что ионы Си2+ образуют в спиртовых
растворах сольваты с конфигурацией тетрагональной бипирамиды,
а сольваты ионов Со2+ — тетраэдра. Исследования электронных
спектров позволяют оценить прочность связей ионов с сольвати-
рующими молекулами растворителя. Из спектральных данных
для хлоридов и бромидов Со2+ в спиртовых растворах [И. С. По-
минов, Е. К. Макарова, 1964] установлено, что прочность
связи иона с молекулами спирта понижается в
последовательности: метанол > этанол > пропанол > бутанол.
На основании изучения сольватации иона Со2+ с помощью
электронных спектров поглощения в спиртах гомологического
ряда [И. И. Антипова-Каратаева, Н. Н. Ржевская,
1972] был построен следующий ряд снижения прочности связи
Со2+ со спиртом:
СН3ОН > С2Н5ОН~СзН7ОН~мзо-СзН7ОН~
~С4Н9ОН~ «зо-С4Н9ОН~ мзо-С5Н11ОН>С5Н11ОН-'
~С6Н13ОН > С7Н15ОН > С8Н17ОН > трет-С4Н9ОН
С помощью электронных спектров ионов металлов в неводных
растворителях можно не только обнаруживать сольватацию, но
и определять типы присутствующих частиц в разных диапазонах
концентраций и структуру таких сложных сольватных комплексов.
При исследовании УФ-спектров поглощения растворов
перхлората железа(III) в 1М,1М-диметилацетамиде [R. S. Drago, 1961,
1963] в зависимости от прибавления иона хлора было обнаружено,
что по мере увеличения соотношения [С1~]: [Fe3+] наблюдается
исчезновение максимума поглощения, относящегося к иону
[Fe(flMA)6]3+, а взамен появляются полосы поглощения ионов
FeCl2 и FeCl^, сольватированных различным количеством
молекул диметилацетамида. Аналогичное поведение ионов железа(III)
было обнаружено в растворах N-метилацетамида и диметилсульф-
оксида. Однако в N-метилацетамиде, который обладает кислотным
191
Протоном, вследствие высокой анионной сольватации УФ-спекгр
FeCl3 указывает на присутствие только ионов [Fe(NMA)6pV
и С1-.
С помощью электронных спектров можно изучать процессы
перестройки сольватных сфер в смешанных растворителях в
зависимости от их состава. Добавление воды к спиртовым
растворам ионов меди и кобальта сдвигает край полосы поглощения
этих ионов в коротковолновую область. Особенности зависимости
длинноволновой границы спектров Си2+ и Со2+ от содержания
воды в спиртах объясняется образованием смешанных сольватов,
содержащих в координационной сфере молекулы воды и спирта
[N. J. Friedman, R. A. Plane, 1963].
Колебательные спектры
Поскольку координация, возникающая в растворе при
образовании сольватного комплекса, влияет на распределение
электронов в молекуле растворителя, то обязательно будут
изменяться силовые постоянные из связей, а следовательно, и
колебательные частоты. Таким образом, ИК-спектр растворителя
можно использовать для обнаружения координации молекул
растворителя вокруг ионов в растворе и в ряде случаев проследить
ион-молекулярное взаимодействие в растворах электролитов.
Так, при изучении ИК-спектров растворов иодидов и
перхлоратов лития и натрия в ацетоне [И. С. Перелыгин, 1964] было
найдено смещение полосы поглощения валентного колебания
группы С = О в сторону меньших частот (для Na+ на 3 см~х и
для Li+ на 8 слг1).
Независимость величин сдвига от аниона растворенной соли
позволяет приписать эти сдвиги влиянию катионов, которое
проявляется тем в большей степени, чем выше электроно-акцептор-
ные свойства катиона. При этом сосуществование в спектре,
наряду со смещенными полосами, полос поглощения чистого ацетона
свидетельствует о том, что влияние катиона сказывается лишь на
тех молекулах растворителя, которые непосредственно связаны
с катионом.
Аналогичная картина получена при ИК-спектроскопическом
исследовании ионов щелочных металлов в пиридине (V. J. Me К i n-
ney, A. I. Popov, 1970]. Ионы щелочных металлов по-разному
сольватированы молекулами этого растворителя. Как видно из
табл. VIII. 4, величина сдвига скелетных колебаний молекулы
пиридина ^соответствует степени кислотности катиона (см. гл. II).
При исследовании колебательных спектров солей аммония и
щелочных металлов с различными неорганическими анионами
(СГ, Вг", I", N07, C10J", SCN") в диметилсульфоксиде
[В. W. Maxey, A. I. Popov, 1967] обнаружено, что в спектрах
появляется при растворении новая полоса поглощения. В спектрах
солей с одним и тем же катионом она не зависит от природы и
192
Таблица VIII. 4
Сдвиги скелетных колебаний связей
в молекуле пиридина для растворов солей
щелочных металлов (в см~1)
Катион
Li+
Na+
NH+
AVC=N
12
9
11
Avc-h
И
4
7
Avc-c
17
8
И
массы, аниона и обусловлена взаимодействием ,катиона с
молекулами растворителя (Li+ — 429; Na+—199 — 206; NHl —214; К+—
— 153; Rb+— 123—129 см~1). Различие максимумов поглощения
свидетельствует о том, что ионы по-разному сольватированы
молекулами диметилсульфоксида.
Изменения частот в ИК-спектрах молекул растворителя могут
указывать на то, какой атом участвует в качестве донора в ион-
молекулярном взаимодействии. Например, было показано, что ди-
метилацетамид координирован с ионами металлов через кислород
[W. E. Bull, S. К. М a d a n, J. E. Willis, 1963]. Через кислород
происходит также координация при сольватации ионов
переходных металлов диметилсульфоксидом [F. А. С о 11 о n, R. Francis,
1961].
На основании данных по ИК-спектрам, как и из электронных
спектров, может быть Оценена прочность связи растворителя
с ионом. При спектрофотометрическом изучении СоС12 в
спиртовых растворах был построен следующий ряд
изо-С3Н7ОН > г/?ег-С4Н9ОН > С2Н5ОН > С3Н7ОН~
~С4Н9ОН > M30-C4H90H~tt30-C5Hn0H > С5НПОН >
> С7Н15ОН > С6Н13ОН > С8Н17ОН
характеризующий постепенное понижение ковалентности связи
Со2+ — спирт [И. И. Антипов а-Кар ата ева, Н. Н.
Ржевская, 1972].
Радиоспектроскопия
Высокая чувствительность ядерного магнитного резонанса
позволяет регистрировать слабые межмолекулярные взаимодействия,
что открывает перед этим методом возможности для изучения
процесса сольватации. Методом ЯМР можно изучать как процессы
релаксации, так и химические сдвиги в растворах.
Химический сдвиг позволяет по-разному изучать взаимодействия
ионов с растворителем. Можно определять резонанс ядер атомов
как в молекуле растворителя, например *Н или 17О, так я ядер
растворенных электролитов, например 23Na, 35C11 или 19F.. В заданном
7 Зак, 500. 193
магнитном поле резонансная частота ядер зависит от распреде,
ления электронов вокруг этих ядер. Любое поле, искажающе
электронное облако, будет изменять магнитное экранирование ядер
электронами и, таким образом, влиять на его резонансную частоту,
т. е. вызывать химический сдвиг.
При цзучении сольватации методом ЯМР весьма желательно
иметь возможность наблюдения отдельных резонансных линий от
свободных молекул растворителя и от молекул, находящихся в
первой координационной сфере иона. Связывая химический сдвиг
веществ в растворах с явлением сольватации, необходимо
учитывать все возможные вклады в величины сдвигов, отражающие
взаимодействие ионов с растворителем. Суммарный химический
сдвиг является следствием следующих процессов: разрыв связей
между молекулами растворителя; поляризация среды,
взаимодействие ион-растворитель в первой сольватной оболочке, а также
неэлектростатическое взаимодействие растворителя с ионом. Таким
образом, интерпретация получаемых результатов по химическим
сдвигам весьма сложна.
Парамагнитные и диамагнитные вещества сильно отличаются
друг от друга по характеру влияния на параметры спектров ЯМР.
Парамагнитные ионы (например, Cu2+, Co2+, Fe2+) влияют на
спектры ЯМР органических растворителей из-за неспаренного
электрона, возмущающего электронную структуру молекул
растворителя. Величина химического сдвига б растворителя в
растворах парамагнитных ионов выражается формулой Бломбергена
6__С
otl
где ^(О)!2 — вероятность пребывания неспаренных электронов
около протона растворителя; С — концентрация молекул
растворителя; п — константа, учитывающая координационное число иона.
Сольватацию диамагнитных ионов можно изучать по
химическому сдвигу магнитных ядер в молекулах растворителей. Для
ионов в смешанных растворителях по величие химического сдвига
можно судить о силе взаимодействия с различными компонентами
смешанного растворителя, избирательной сольватации,
разрушающем влияние ионов на структуру смешанного растворителя.
Работы по изучению сольватации ионов в неводных
растворителях методом химического сдвига ЯМР еще сравнительно
малочисленны. Достаточно указать, что первые систематические
исследования химических сдвигов, вызываемых ионами в водно-неводных
смесях и органических растворителях стали появляться менее
десяти лет назад [A. F г a t i е 11 о, D. С. Douglass, 1963; R. L. В и с-
kson, S. G. Smith, 1964]. Тем не менее изучение химических
сдвигов весьма перспективно для решения проблем теории
растворов (характеристика избирательной сольватации, константы
ассоциации электролитов, коэффициенты диффузии, кинетика
реакций и т. д.),
194
Второй параметр, изучаемый при помощи ЯМР, это время ре-
паксации возбужденных ядер. Механизм электронной релаксации
в электролитных растворах (N. Bloembergen, V. Morgan,
1961) связан с тем, что флуктуация растворителя около сольвати-
рованного иона модулирует окружающее магнитное поле в
результате деформации и искажения сольватного комплекса. Эти
йскажения через спин-орбитальную связь вызывают релаксацию
электронного спина, что дает возможность при изучении жидких
веществ методом ЯМР определять структуру раствора (взаимное
расположение частиц, расстояние наибольшего сближения,
молекулярное движение).
При исследовании растворов электролитов задача сводится к
анализу поведения молекул растворителя, находящихся в составе
данного сольвата и взаимодействующих как с ионом, так и с
остальной массой растворителя, изменяя характер ее движения и
координацию молекул растворителя около ионов.
Релаксационные измерения в растворах позволяют определять
координационные числа сольватации и кинетику обмена молекул
растворителя между сольватной оболочкой и основной массой
растворителя.
При исследовании связанного со временами релаксации
относительного уширения линий в спектрах ЯМР для растворов
электролитов в смешанных растворителях могут быть решены вопросы
избирательной сольватации ионов и оценены специфические
эффекты взаимодействия иона с растворителем.
Проиллюстрируем применение метода ЯМР для изучения
сольватации ионов в неводных растворителях некоторыми примерами.
Исследование сольватации ионов Li+ и Na+ по химическому сдвигу
протонов в диметилсульфоксиде [В. W. Maxey, A. I. Popov,
1968] показало, что ионы натрия менее сольватированы этим
растворителем, чем ионы лития. Это подтверждают другие свойства
растворов солей Li+ и Na+ в диметилсульфоксиде.
При изучении поведения иона Na+ в четырнадцати различных
кислород- или азотсодержащих растворителях [Е. G. В loo г,
R. G. Kidd, 1968] измерена концентрационная зависимость
химического сдвига 23Na в иодиде натрия. Установлено, что между
величинами химических сдвигов при бесконечном разбавлении и
константами диссоциации иодида натрия в соответствующих
растворителях существует прямолинейная зависимость.
При изучении растворов солей алюминия в ДМФ [W. G. Мо-
vius, N. A. Matwiyoff, 1968] при сопоставлении резонансных
линий протонов, характеризующих свободные молекулы ДМФ,
одинаковые для растворов любой соли, и линий протонов,
характеризующие координированные молекулы ДМФ, была определена
природа комплекрообразования в этих системах.
Аналогичная картина наблюдается в спектрах ЯМР раствора
Со(СЮ4)2 в метаноле при — 60°С [Z. Luz, S. Meiboom, 1964],
где присутствуют линии, соответствующие по своему положению
7* 195
свободному метанолу, а также линии групп ОН и СН3, сильно
смещенные в слабое поле, отвечающие молекулам метанола в первой
координационной сфере иона Со2+.
Из температурной и концентрационной зависимостей б и
ширины линий Av сигналов ПМР растворителя в растворах солей
Со2+ и Ni2+ в ацетонитриле [N. Ma t w iy of f, S. Hooker, 1967]
найдено, что в области температур (—45 °С)— (— 79°С) в растворе
присутствуют только комплексы состава [Me(CH3CN)6]2+.
Существенный интерес представляют данные ЯМР
электролитов в смешанных растворителях. Для водных смесей растворителей
(ацетона, Г\[,]М-диметилформамида, диметилсульфоксида, диоксана,
тетрагидрофурана и тетраметилмочевины) было измерено
координационное число А13+ в широкой области концентраций компонентов
смешанных растворителей [A. Fratiello, R. Lee, V. N i s-
h i d a, R. Schuster, 1967]. Координационное число А13+
рассчитывали интегрированием сигналов, соответствующих протонам
свободной воды и воды в сольватной сфере иона алюминия. Кроме
того, показано, что в водных смесях ацетона, тетраметилмочевины
и в небольшом интервале концентраций диоксана и
тетрагидрофурана А13+ сольватирован только молекулами воды, тогда как
вводных растворах диметилформамида и диметилсульфоксида в
координации участвуют молекулы обоих компонентов растворителя.
При установлении координационного числа Mg2f в смесях
метанол— вода различного мольного соотношения снимали спектры
ПМР перхлората магния [J. H. Swinehart, H. T a u b е, 1962].
При температуре —75 °С удалось наблюдать отдельные сигналы от
молекул спирта и воды — как связанных, так и свободных;
найденные числа сольватации иона магния для метанола и воды
-равны, соответственно, 5,0 и 0,7.
Концентрационные зависимости б-сигналов воды в смешанных
водно-неводных растворителях позволяют оценить степень участия
каждого из компонентов растворителя в комплексообразовании с
растворенным электролитом. Так, ЯМР-спектроскопия
диамагнитных растворов ТЮЦ и парамагнитных растворов C0CI2 [A.
Fratiello et al., 1967], позволила оценить способность органических
молекул к сольватации. Найдено, что степень взаимодействия с
ионами убывает в последовательности:
диметилсульфоксид > амиды > спирты > тетраметилмочевина,
гетрагидрофуран > ацетонитрил, диоксан.
Найденная последовательность коррелирует со значениями
основности этих органических соединений.
Исследование времени релаксации позволяет установить
скорость обмена молекулами растворителя между сольватной
оболочкой иона и окружающей средой.
Определение константы скорости обмена координированное и
свободного диметилсульфоксида при различных температурах и
концентрациях перхлоратов Ni2+ и Со2+ [S. Thomas, W. L.
Reynolds, 1967] позволило оценить энтальпию и энтропию активации
196
Рис. VIII. 42.
Концентрационная зависимость релаксации
в растворах NaC104 при 25°.
(Ti — время релаксации в растворе
исследуемой концентрации; Tt° — время
релаксации чистого растворителя);
/ — метанол; 2 — ацетонитрил; 3 —
ацетон, 4 — вода.
[Ж. общ. хим. 41, 1971, 13].
этого процесса, а также связать величины скорости обмена со
строением комплексов.
Из релаксационных данных можно получать сведения о
структурных изменениях в растворах электролитов. На рис. VIII. 42
приведены зависимости времени
спин-решеточной релаксации протонов для
растворов перхлората натрия разной
концентрации. Уменьшение времени
релаксации с концентрацией
свидетельствует о том, что в растворах
происходит упорядочение структуры,
причем в неводных растворителях
упорядочение примерно одинаковое; для
водных растворов упорядочение более
слабое.
Измерение концентрационной ' и
температурной зависимости времени
спин-решеточной релаксации х\ в
растворах иодидов натрия и калия в
гидразине [А. А. Б о р и ч е в, К. П. М и-
щенко, В. В. Кущенко, 1971]
показало, что скорость протонной
релаксации растет с увеличением
концентрации электролита. Для водных растворов при тех же условиях
наблюдается уменьшение релаксации, обусловленное разрушением
структуры растворителя, в то время как в гидразине
упорядочивающее влияние ионного поля преобладает над разрушающим
действием ионов на структуру растворителя. Стабилизирующее
влияние Nal на структуру гидразина проявляется значительно
сильнее, чем KI.
VIII. 10. Некоторые другие методы исследования
сольватации
Сжимаемость
Поскольку входящие в состав сольватной оболочки молекулы
растворителя сжаты силовым полем иона, при наложении внешнего
давления они будут подвергаться сжатию в меньшей степени, чем
молекулы, не связанные с ионом. Вследствие этого раствор будет
характеризоваться меньшей сжимаемостью по сравнению с чистым
растворителем.
На основании наблюдаемых объемных изменений свойств
растворителя, вызванных присутствием ионов, из данных по
сжимаемости при бесконечном разбавлении можно определить числа
сольватации по формуле [Б. К о ну эй, Дж. О'М. Б окр и с, 1958]
п —
Лсольв — —
-7
(VIII. 98)
197
где фк и Ро кажущаяся (моляльная) изотермическая сжимаемость
электролита и чистого растворителя; V°M — молярный объем
растворителя. Получаемые значения чисел сольватации относятся при
этом к ионным парам.
Сольватационные эффекты в растворах мож'но оценивать,
например, из скорости распространения ультразвука. Расчет чисел
сольватации на основании ультраакустических измерений
производят по формуле [И. Г. М и х а й л о в, В. А. Соловьев, Ю. П.
Сырников, 1964]
d — ^гольв "Z
(VIII. 99)
Мсл
где d — плотность; р— адиабатическая сжимаемость [см.
уравнение (IV. 14)]; С — концентрация электролита (обозначения с
индексом 0 относятся к растворителю, обозначения без индекса — к
раствору; Мо— в данном случае молекулярная масса электролита).
Таблица VIII. 5
Числа сольватации электролитов в метаноле при 25 °С
[Г. П. Р о щ и н а, А. С. К а у р о в а, И. Д. К о ш е л е в а, 1968]
Электролит
LiCl
NaCl
CsCl
Концентрация, н.
0,09
0,09
0,09
лсольв
5,8
5,0
3,0
Электролит
NaCl
NaBr
Nal
Концентрация, н.
0,1
0,1
0,1
^сольв
4,9
5,3
4,5
Приведенные в табл. VIII. 5 значения чисел сольватации для
электролитов с ионами различной поляризующей способности в
метаноле, найденные из данных по сжимаемости [Г. П. Рощи на,
А. С. К а у р о в а, И. Д. Ко шел ев а, 1968] показывают, что с
уменьшением поляризующей способности катионов и ослаблением
общего взаимодействия (включающего как донорно-акцепторное,
так и ион-дипольное) между ионами и молекулами растворителя
число сольватации уменьшается. Замена аниона С1~ на Вг~ или I"
практически не сказывается на числе сольватации.
Магнетохимический метод
Этот метод исследования сольватации ионов в растворах
предполагает, что разница между величинами магнитной
восприимчивости в кристаллическом и растворенном состояниях обусловлена
в основном изменением ванфлековского парамагнетизма
ближайших к иону молекул растворителя по сравнению с его
значением для молекул растворителя в объеме раствора [Я. Г. Дорф-
ман, 1961].
198
Значения магнитной восприимчивости солей щелочных
металлов в воде и спиртах приведены в табл. VIII. 6.
Таблица VIII. 6
Молярные магнитные восприимчивости (X • 106 ед. CGSM)
солей при 20 °С
[Ю. В. Е р г и н, Л. И. К о с т р о в а, 1970
Соль
LiCl
LiBr
NaBr
Nal
KI
хкр
24,4
34,7
40,6
57,0
63,5
XH2O
24,0
35,8
41,8
59,6
67,9
хсн3он
23,0
26,6
31,0
49,1
57,6
хс2н5он
22,8
22,4
—
46,9
54,2
Разделение магнитной восприимчивости солей на катионную и
анионную составляющие, проводят в предположении, что
независимо от природы растворителя справедливо соотношение:
(Хсольв)к = (Хгидрк (VIH 1(Ю)
(Хсольв)а (Хгидр)а
На рис. VIII. 43 представлены ионные значения магнитной
восприимчивости Д% для воды, метанола и этанола как функция кри-
сталлохимического радиуса ионов. лх
В воде величины Д% переходят от
положительных значений (Li+, Na+)
к отрицательным (К+, Rb+), в
растворах спиртов величины Д% только
положительны.
Таким образом, явление
отрицательной сольватации ионов не
обнаруживается в спиртах, т. е.
упорядоченность спиртов выражена
менее отчетливо, чем у воды. w 0,5
Рентгено- и электронография растворов
электролитов
Рассеяние рентгеновских лучей
и электронов, а в некоторых
случаях и нейтронов, относится к числу
наиболее распространенных
методов структурного анализа жидкости
[А. Ф. С крыш ев с кий, 1971].
Эта группа методов позволяет определить изменения в структуре
раствора, происходящие по сравнению со структурой исходного
растворителя. Кроме этого, указанные методы являются весьма
Рис. VIII. 43. Зависимость
магнитной восприимчивости ионов от
их кристаллографического
радиуса в различных растворителях:
7-Н2О; 2-СН3ОН; 3 - С2Н5ОН.
[Ж. структ. хим., 11, 1970, 762].
199
действенными при изучении структурных изменений, происходя,
щих в результате изменения концентрации раствора.
Так, сопоставление кривых атомно-электронной плотности рас-
творов галогенидов различных металлов в низших алифатических
спиртах между собой, а также с соответствующими кривыми для
водных растворов [А. Ф. С крыше в с кий, А. К. Дорош, 1971]
позволило установить кратчайшие расстояния между сольватиро-
ванными ионами, расстояния Me—HORb а также числа
сольватации.
Описание процесса сольватации
методом молекулярных орбиталей
Вследствие общепризнанной в настоящее время
недостаточности электростатических моделей для описания явления
сольватации, весьма плодотворным оказалось рассмотрение сольватации
иона в растворе как процесса комплексообразования. Наиболее
подходящим приближением при этом для описания связей в соль-
ватных комплексах является метод молекулярных орбиталей
[Н. А. Измайлов, Ю. At Кругляк, 1960], который охватывает
все типы связей от ковалентной до ионной, а электростатические
представления включает в себя как частный случай.
Для объяснения характера связей в сольватных комплексах
вводится понятие о донорно-акцепторных связях между ионом и
растворителем. Модель взаимодействия основана на
представлениях о том, что в сольватах донорами электронов служат атомы
в молекулах растворителя, имеющие свободные неподеленные
электронные пары (кислород, азот, фтор), акцепторами
электронов— ионы, имеющие вакантные орбиты — орбиты ближайшие по
энергии к заполненным в соответствии с порядком расположения
электронных уровней в атомах.
Волновая функция связывающих электронов в такой модели
является результатом линейных комбинаций из волновых функций
вакантных орбит ионов и орбит электронов в атомах-донорах
молекул растворителей.
Рассмотрение симметрии полученного набора молекулярных
орбит в образовании связей позволяет предсказать геометрию
образующихся сольватов, т. е. указывает числа сольватации и
пространственную конфигурацию сольватного комплекса.
Предполагаемый Н. А. Измайловым и Ю. А. Кругляком
механизм процесса сольватации в рамках метода молекулярных орбит
позволил разъяснить ряд закономерностей сольватации ионов.
Поскольку в сольватах, как и в комплексных соединениях
степень ковалентности связей уменьшается с увеличением радиусов
ионов, процесс сольватации для них можно рассматривать как
ион-дипольное взаимодействие. Для малых ионов (в особенности
это касается Н+) механизм сольватации и значение энергии
отличаются от этих величин для других ионов вследствие
специфического взаимодействия с растворителем,
200
Квантово-механический расчет протонного сродства [Н. А.
Измайлов и др., 1961] позволяет дать количественное описание ряда
процессов кислотно-основного взаимодействия.
Нужно отметить, что этот метод в настоящее время
ограничивается описанием в основном процесса гидратации. Так, на основе
ковалентной модели гидратных комплексов ионов Na+ и К+
удалось объяснить их различное поведение в водных растворах
[Ю- А. Кругляк, 1967].
Ю. П. Сырников (1967) методом, аналогичным методу МО,
вычислил энергию взаимодействия ионов щелочных металлов и
ионов галогенов с молекулами воды и показал, что изменение
энергии электронов при гидратации уменьшается в ряду щелочных
металлов при переходе от лития к цезию.
Описанием процессов гидратации ионов методом МО
[Н. В. Gray, 1965; R. E. Burton, J. Daly, 1970] удается
объяснить устойчивость ионов в водном растворе, структуру гидратного
комплекса, цветность и т. д.
Перспективы применения этого метода для описания процесса
сольватации, кислотно-основного взаимодействия и других
характеристик неводных растворов несомненны,
Глава IX
ВЛИЯНИЕ РАСТВОРИТЕЛЯ НА РАВНОВЕСИЕ
В ХИМИЧЕСКИХ СИСТЕМАХ
Проблема влияния растворителя на равновесие процессов, про
текающих в химических системах, сводится в первую очередь к
рассмотрению двух вопросов — как изменяется равновесие
процесса при переходе из газовой фазы в данный растворитель и от
одного растворителя к другому. Наиболее важным в этом разделе
химической статики является раздельная оценка влияния
физических и химических свойств растворителя на химическое
равновесие. Впрочем, объем экспериментального материала и степень
разработки расчетных методов явно недостаточны для обоснованного
ответа на первый из поставленных вопросов. Однако, несмотря на
относительное обилие экспериментальных данных, относящихся ко
второй проблеме, разграничение влияния физических (физические
свойства) и химических (природа и энергия взаимодействия
растворенного вещества с растворителем) факторов на химическое
равновесие при переходе от одного растворителя к другому также
может быть проведено лишь с относительной строгостью. Дело в
том, что изменение физических свойств среды при переходе от
одного растворителя к другому неизбежно сопряжено с
изменением химических свойств, и наоборот.
В этой главе будет рассмотрено влияние растворителя на
стадии общей схемы равновесий в растворах. Несмотря на
значительное разнообразие процессов, объединяемых этой схемой,
можно усмотреть много общего во влиянии растворителя на каждую
из стадий этой схемы.
Характер изменения констант равновесия при переходе из
газовой фазы в растворитель следует из уравнения, связывающего
тепловой эффект реакции в газовой фазе Д#г с тепловым
эффектом реакции в данном растворителе A#s и суммой изменения
энтальпий при переходе каждого из компонентов из раствора с
единичной концентрацией в газовую фазу, где парциальное
давление каждого из этих компонентов также равно 1:
Изменение констант равновесия реакции при переходе из
одной фазы в другую рассчитываются из данных по Д#, привлекая
известные соотношения.
Влияние физических свойств растворителя на константы
равновесия процессов в химических системах может быть объяснено с
электростатических позиций, если учесть, что в значительном числе
случаев в первом приближении химические процессы сводятся к
электростатическому взаимодействию, константа равновесия кото-
202
оГо описывается уравнением Борна:
in к = const + е(/1Г2* )kT (IX. 2)
Из уравнения (IX. 2) следует, что из физических свойств
растворителя, определяющих константу равновесия, основным
является диэлектрическая проницаемость. Действительно, как будет
показано в этой главе, почти во всех случаях можно установить
весьма простую корреляцию между константами равновесий
разнообразных процессов и диэлектрической проницаемостью.
Влияние диэлектрической проницаемости на равновесие в
химических системах может быть установлено и в случае
взаимодействия двух диполей. Энергия этого взаимодействия передается
выражением
rv» а —9 »» —3 /"TV Q\
<gf = Ar — Br (IX- 6)
где r — расстояние между диполями; А—константа сил отталки-
4 В3
вания, равная ^ (во— энергия взаимодействия в наиболее
г 27 щ
устойчивом состоянии); В — константа сил притяжения равная
IX. I. Влияние растворителя
на молекулярные ассоциативно-диссоциативные процессы
Как отмечалось в гл. VII, во многих случаях молекулярная
диссоциация ассоциированных компонентов, образующих жидкий
раствор, обусловлена химическим взаимодействием между ними,
т. е. описывается схемами (VII. 2) и (VII. 3). В соответствии с
этими схемами можно" установить тесную связь между
ассоциативным состоянием и химическими свойствами компонентов,
образующих раствор.
В табл. IX.1 приведены значения констант равновесия про-
ГНАс!2
цесса распада димеров уксусной кислоты /Смон = "г7иТ~тт в паРе
и различных растворителях. Несмотря на то, что таблица
составлена по данным различных авторов и данные эти относятся к
довольно широкому интервалу температур, можно установить
четкую корреляцию между ассоциированным состоянием уксусной
кислоты и химическими свойствами растворителя. В паре
молекулы уксусной кислоты практически полностью являются димерны-
ми. Во всех растворителях уксусная кислота находится в виде
равновесной смеси димерных и мономерных молекул. Нетрудно
заметить, что на положение равновесия (НАс)2 =е^2НАс
диэлектрическая проницаемость * оказывает гораздо меньшее влияние,
* Данные по диэлектрической проницаемости (е) растворителей сведены в
Приложении,
203
Т а б л и ца IX. 1
Константы равновесия и теплоты процесса (в кдж/моль)
диссоциации димеров уксусной кислоты в различных растворителях
Растворитель
Пар*
СеН14 *
cs2
СС14*
свн6*
«мои'103
0,07
1,9
2,8
3,1
12,4
-дя,
кдж/моль
60,7
37,7
29,5
31,8
34,3
Растворитель
с4н8о2 **
о-С6Н4С12 3*
СНС133*
СбН5С1 *
C6H5NO2
Кмон'108
8,00* 103
1,4
7,75
9,4
20
-АН,
кдж/моль
20,9
22,0
* Спектральные данные при 35 °С [А л лен, Гэлдин, 1953].
** Криоскопические данные [Л. И. Деревянно, 1971].
8* Спектральные данные при 25 °С [В. В. В а щи некая, 1971].
чем химические свойства растворителей. Так, в растворителях,
характеризующихся весьма близкими значениями е — гексане,
fmm сероуглероде, четыреххлори-
стом углероде, бензоле и диок-
сане, величины /Смон
различается весьма существенно.
В химически инертных по
отношению к уксусной кислоте
растворителях равновесие
практически смещено в
сторону димера. Величина /Смон в
к бензоле приблизительно на по-
0,5
0,2
ОА
Ж
0,8 т
рядок выше, чем в остальных
инертных растворителях, что
обусловлено взаимодействием
(впрочем, довольно слабым)
кислоты с бензолом по я-свя-
зям последнего. В диоксане
же, который является
растворителем с отчетливо выраженными основными свойствами,
равновесие уже значительно смещено в сторону мономера.
Влияние химических свойств растворителя на процесс распада
димерных молекул иллюстрируется также рис. IX. 1, где
приведены полученные по криоскопическим данным факторы ассоциа-
Рис. IX. 1 Факторы ассоциации
уксусной кислоты в различных
растворителях:
/ — бензол; 2 — нитробензол; 3 — диоксан.
ции fac =
Ммон
и Мэксп — формульная и
экспериментальная молекулярные массы) уксусной кислоты в различных
растворителях. Из рисунка видно, что значительно большее значение 8
нитробензола по сравнению с бензолом приводит лишь к
незначительному повышению fac; в то же время величины /ас в диоксано-
вых растворах либо равны 1, либо весьма близко к ней.
204
Химические свойства растворенного вещества также в
значительной степени определяют его ассоциативное состояние. Так,
величины Кмое растворов уксусной, монохлоруксусной и трихлор-
уксусной кислот в диоксане, рассчитанные по криоскопическим
данным [М. Н. Цареве к а я, Л. И. Деревянко, 1971],
составляют 0,8; 24 и оо, т. е. увеличиваются по мере увеличения степени
взаимодействия кислот с растворителем.
Причиной, обусловливающей распад ассоциированных молекул
многих веществ, является в большинстве случаев образование
-дНмон, ни ал/моль
Ш
10
ч,о
0,5
Рис. IX. 2. Зависимость рКмои уксусной Рис. IX 3. Зависимость р/С
е + 2 /г. . . /7.ч А тт конформерного равновесия мо-
кислоты от -^- (/) и 1/е (//); и Д#мон от Лекул 1,2-дибромэтана от 1/е.
1/е (///):
1-пар; 2-С6Нн; 3-СС14: 4-CS2; 5-С6НбС1;
5-G6H6NO2.
Н-связи между компонентами раствора. Как известно, мерой
прочности Н-связи (а, соответственно, и степени распада ассоциатов)
является сдвиг максимума полосы поглощения, относящейся к
группировке, которая участвует в образовании Н-связи.
Спектроскопическая литература изобилует примерами, иллюстрирующими
эту закономерность.
Влияние растворителя на ассоциативное состояние
растворенного вещества в соответствии с уравнением (IX. 3) связано прежде
всего с е. Как отмечалось выше, это влияние отчетливо
сказывается лишь в тех растворителях, энергией взаимодействия
которых с растворенным веществом можно пренебречь по сравнению
с энергией связи молекул в ассоциате. В таких растворителях
можно установить неплохо соблюдающуюся прямолинейную зави-
стоимость р/Смон от -^—р- (прямая /, рис. IX. 2) и 1/е (прямая //).
Поскольку величина AS в процессах мономеризации в
различных растворителях весьма близки, можно считать, что различия
P^moh(= —lg Kmoh=AGMoh) однозначно определяются величинами
205
А//мон. Таким образом, должна соблюдаться линейная
зависимость и для ДЯмон =/(1/е) (рис. IX. 2, прямая ///).
Прямолинейный характер зависимости —Д#мон от 1/е выявляется значительно
отчетливее, если учесть зависимость величины —ДЯмон от тепло-
вого эффекта разрыва димеров в газовой фазе (—Д#г), темпера-
- , 1 /dine г \ *
туры и температурного коэффициента lge I дт = — L\ :
АЯМ0Н= l~LT ДЯГ (IX. 4)
о
Можно установить влияние растворителя и на константу
равновесия конформационных превращений молекул в растворах.
Поскольку последние сопряжены с изменением дипольного
момента, молекулы, в соответствии с уравнением (IX. 3) следует
ожидать влияния е на константу равновесия этого процесса.
Действительно, из рис. IX. 3 видно, что наблюдается прямолинейная
зависимость между логарифмом константы превращений
трансформа +*? гош-форма молекул 1,2-дибромэтана(р/Спр =— lg »> гош )
V -'"транс /
и 1/е для ряда инертных растворителей [J. Leffer, E. Grun-
wald, 1963].
В литературе имеются весьма скудные данные по влиянию
растворителя на константу равновесия процессов образования
продуктов присоединения mk + nB ^AmBn. Исследования в этом
направлении [например, Т. Jasinsky с соавторами, 1967—1970]
охватывают ограниченный круг растворителей в небольшом
интервале диэлектрических проницаемостей; кроме того, не всегда
удается разграничить влияние физических и химических факторов,
IX. 2. Влияние растворителя
на константы устойчивости комплексных соединений
Рассмотрим вопросы, связанные с изменением константы
равновесия (константы устойчивости) /Ск процессов комплексообразо-
вания, т. е. процессов присоединения к иону металла иона (в
частном случае нейтральной молекулы) лиганда
Mem+ + pLl~ *=* [МеЦ](т-р/)+ (IX. 5)
при переходе от одного растворителя к другому, либо с
изменением состава смешанного'растворителя.
Многочисленные литературные данные свидетельствуют, что при
переходе от одного растворителя к другому состав комплексного
иона может изменяться — вследствие внедрения молекул
растворителя во внутреннюю сферу комплекса
sS *=* [МеЦ-^Г"-' <*-*>+ + tLp~ (IX. 6а)
* Величины L приведены в табл, XIV. 2 (стр. 331). .
206
иЛи замены внедрившихся молекул одного растворителя
молекулами другого
Равновесия (IX. 6) (последнее из них часто и не совсем точно
называют пересольватацией) в этой книге не рассматриваются *.
Изменение констант устойчивости комплексных соединений с
изменением растворителя (так далее будет называться переход от
одного растворителя к другому, либо изменение состава двойного
смешанного растворителя) обусловлено изменением как
физических, так и химических характеристик среды. Кроме того, в случае
комплексных соединений добавляется еще один специфический
фактор, играющий большую роль в определении степени, с
которой растворитель влияет на изменение Кк — это характер связи
между центральным атомом и лигандом.
Для объяснения влияния физических свойств растворителя и,
прежде всего, диэлектрической проницаемости на прочность
комплекса в первом, а в большинстве случаев достаточно хорошем,
приближении можно ограничиться представлениями о том, что
взаимодействие типа (IX. 5) подчиняется закономерностям
электростатического взаимодействия [см. уравнение (IX. 2)]. Однако в
рассматриваемом случае постоянная const в уравнении (IX. 2)
равна логарифму константы устойчивости комплекса, принятой
для выбранного стандартного состояния. В соответствии с
уравнением (IX. 2) In Kk должен быть линейной функцией 1/е.
Более подробное уравнение [Я. И. Турьян, 1955] связывает
константу устойчивости с величиной е растворителя и стехиометри-
ческими коэффициентами уравнения (IX. 5):
Если принять, что с изменением растворителя величины г*
остаются постоянными (это предположение оправдывается в
близких по химической природе растворителях Si, S2, S3 либо в
двойных смешанных растворителях Si — S2, где Si и S2 близки по
природе и энергии сольватационных процессов), то
р/Ск = const + — - (IX. 7а)
£
т. е. зависимость р/Ск — 1/е прямолинейна, так как В не зависит
от растворителя.
В литературе имеется обильное количество примеров,
подтверждающих справедливость уравнения (IX.7а). В качестве примера
такого рода зависимости на рис. IX. 4 приводятся данные по
* Весьма обширный материал по процессам комплексообразования в
системах Mew+ — Sj, Mem+ — S2 обобщен в монографии В. Гутмана (1968).
207
рЯ,
Р«г
w
/ 2 3 1/е-ю2
Рис. IX. 4. Зависимость рКк
роданидных комплексов
кадмия от 1/е:
1 - Cd SCN+; 2 - Cd(SCN)2;
S-Cd(SCN)~.
[Я. Н, Турьян, Н. Н. Во н-
д а р е н к о, 1959].
константам устойчивости роданидных комплексов в смешанном
растворителе вода — метанол [Я. Н. Т у р ь я н, Н. Н. Бондарен,
ко, 1959].
Уравнение (IX. 7а) соблюдается достаточно хорошо лишь в
разбавленных растворах — там, где можно пренебречь изменением е
раствора по сравнению с е исходного растворителя.
Рассмотрение экспериментального материала по изменению
констант устойчивости с изменением растворителя показывает, что
в соответствии с условиями, принятыми
при выводе этого уравнения,
прямолинейная зависимость рКк от 1/е
соблюдается лишь в растворителях, близких
по химической природе. Тогда же, когда
растворители существенно различны по
химической природе, отсутствует не
только подобная корреляция — нельзя
усмотреть даже приблизительного
соответствия между Кк и е.
Приведенные в табл. IX. 2 данные для
системы Ph3CCl—SbCls свидетельствуют,
что^ химическая природа растворителя
оказывает решающее влияние на
константы устойчивости. Действительно, в
почти изодиэлектрических дихлорэтане
и пиридине константы устойчивости
различаются более, чем на 7 порядков.
Связано это с тем, что один из компонентов
рассматриваемой реакции — SbCls —
будучи сильной апротонной кислотой,
энергично взаимодействует с сильно основным пиридином, в то
время, как взаимодействие SbCls с дихлорэтаном протекает в
несравненно меньшей степени. Столь же различаются константы
равновесия в весьма близких по величинам е ацетонитрилеи ДМФ.
Напротив, в разнящихся по величине е почти вдвое PhPOF2 и
ГРОС13 константы устойчивости практически одинаковы. Все эти
различия связаны с различием в сродстве (энергии сольватации) одного
из компонентов реакции комплексообразования к растворителю.
Влияние сольватационной способности растворителя на
величины Кк хорошо иллюстрируются примерами растворов
роданидных комплексов металлов [А. М. Голуб, В. М. С а мой лен ко,
1963—1965]. Константы устойчивости этих комплексов в весьма
близких по величинам е растворителях (метанол, ДМФ и ацето-
нитрил) различаются на много порядков. Кроме того прочность
комплексов в ацетонитриле значительно выше, чем в метаноле и
ДМФ — в соответствии с гораздо меньшей сольватационной
способностью первого по сравнению с последними.
Иногда изменение растворителя ведет к существенному
изменению природы лиганда. Так, например, глицин, который в диок-
208
Таблица IX.2
Степень взаимодействия в системе Ph3CCl—SbCl5
и константы устойчивости комплекса PhSbClg • (Ph)3C+
в различных растворителях
[V.Gutmann, E. W у с h e г а, 1966]
Растворитель
SO2C12
SOC12
1,2-С4Н4С12
C6H5N
POCI3
(СНзО)зРО
СбН5СОС1
СбН5РОС12
C6H5POF2
CH3CN
Диметилформамид
Диметилсульфамид
Пропиленкарбонат
е(25°С)
9,1
9,2
10,4
. 12,3
13,9 (22 °С)
20,6
23,0
26,0
27,9
36,0
36,7
46,6
65,1
а
1,0
1,0
1,0
0
0.061
0,011
1,0
0,036
0,088
0,98
0,001
0,001
0,13
>105
>105
>ю5
<10"2
1,1. 102
2,5
>105
3,9-10
1,3- 102
105
5,0- 10-^
4,0-10"2
3,4-102
сане находится в молекулярной форме, в воде находится в форме
+
H3NCH2COO". Поэтому константа устойчивости комплекса никеля
с глицином в смешанном растворителе вода — диоксан изменяется
чрезвычайно сильно, в то время как константы устойчивости
комплексов никеля с лигандами, молекулярное состояние которых не
зависит от растворителя, в этом смешанном растворителе
изменяются гораздо меньше.
В ряде работ рассматриваются зависимость константы
устойчивости от состава двойного смешанного растворителя Si—S2. Во
многих случаях установлена линейная зависимость lg/CK от
мольной доли *s одного из компонентов смешанного растворителя.
Г. Ирвинг и Г. Росотти (1956) предложили уравнение,
связывающее величины констант устойчивости KV в данном неводном
растворителе и в воде К^2° с мольной долей первого л^ в
смешанном растворителе Н2О—Si и с коэффициентом активности
соответствующих ионов:
«н2о н2о
VMe VL
YMeLp
(IX. 8)
(/ —постоянные для ионов металла, лиганда и комплексного
иона).
Из уравнения (IX. 8) следует, что \gKl] будет линейно
зависеть от xs в тех случаях, если отношение коэффициентов
209
активности в смешанном растворителе и в воде будет близко к 1
или пропорционально xS\. Кроме того, можно заключить, что коль
скоро соблюдается* прямолинейная зависимость \gKK—1/е, то
прямолинейность в координатах \gKK — xs будет соблюдаться в
случае, если 1/е есть также прямолинейная функция состава xs.
В первом приближении это условие выполняется для многих
систем, изотермы е которых монотонно выпуклы к оси состава (см.
гл. VIII, стр. 137). В качестве примера характерного хода
зависимости lg Кк — #s на рис. IX. 5 приводятся данные по константам
устойчивости ацетатных комплексов
лантана в смешанном растворителе
вода — этанол.
Природа связи металла с лиган-
дом играет существенную роль
0.1 0,2 0,3
х
0,4
-J/
28
24
20
16
12
8
1,нк ал/моль
-
-
-
1/
у/г
i |
2 4
6 у
К5
6
8 10
9
У
f
1
12 р/Гк
Рис. IX. 5. Величины р/Ск
ацетатных комплексов
лантана как функция мольной
доли спирта в смешанном
растворителе вода —
этанол;
1 - LaAc2+; 2 - LaAc+; 3 - LaAc3.
[П. К. М и г а л ь, Н. Г. Ч е б о-
т а р ь, 1967].
Рис. IX. 6. Зависимость — АН от
р/Ск комплексов SbCl5 • S:
/ —РОС13; 2 — CH3GN; 3 — пропиленкар-
бонат; 4 —ацетон; 5 —вода; 6—(СгВДгО;
7 — (СНзСО)зРО; 8 — диметилацетамид;
9 — диметилсульфид.
IV. G u t m а п, 1966].
в определении степени влияния растворителя на константу
устойчивости. Влияние это, по-видимому, достаточно непосредственное,
так как в ряде случаев существует прямолинейная зависимость
между тепловым эффектом реакции комплексообразования и
температурной зависимостью lg/CK образовавшегося комплексного
соединения. Примером подобной зависимости могут служить
тепловые эффекты реакций комплексообразования SbCls с различными
растворителями SbCl5 + S +±- SbCl5-S (рис. IX. 6).
Тесная зависимость характера изменения константы
устойчивости комплекса от растворителя вытекает уже из следующей
элементарной предпосылки: чем выше степень ионности связи, тем
влияние е должно сказываться сильнее. Действительно, величины
210
констант устойчивости [CdClnp~nb изменяются с изменением
состава смешанных растворителей вода — спирты гораздо более
резко, чем константы устойчивости [Cdlnp-n>- [О. И. X о ц я н о в с к и й,
1956—1957]. Это соответствует большей степени ионности связи
металл — лиганд у первого комплекса по сравнению со вторым.
Диалогичная зависимость наблюдается для комплексов,
образованных разными металлами с одним и тем же лигандом.
В общем случае изменение устойчивости комплексов со связью
jVie- • -О с изменением е более резкое, чем в комплексах со связью
Д1е- • -N. Иногда уменьшение е приводит — вопреки уравнению
(IX. 7, а) — к уменьшению устойчивости комплексов со связью
]Vie---N — явление, которое не свойственно в общем случае
комплексам со связью Ме---О. Это обусловлено не только большей
степенью ионности связи в комплексах последнего типа, а также,
в соответствии с интерпретацией, данной Ирвингом и Россоти
(1956 г.), тем, что уменьшение е растворителя приводит к
усилению ионного взаимодействия Мет+ с донорным атомом лиганда;
ион-дипольное взаимодействие Мёт+ с растворителем в то же
время усиливается в гораздо меньшей степени.
IX. 3. Влияние растворителя
на таутомерные равновесия1
Рассмотрим влияние растворителя на константу равновесия
таутомерного превращения Ai ч=* к%
/Ст = -^- (IX. 9)
или для разбавленных растворов:
=~[ХТГ
(1Х*9а)
В подавляющем большинстве случаев исследуются кето-эноль-
ные превращения К—*Э; тогда константа таутомерного
равновесия представляет собою отношение активностей (в случае
разбавленных растворов — концентраций) энольной и кетонной форм:
(IX. 10)
Кт=[Э]/[К] (IX Юа)
Значительный экспериментальный материал по влиянию
растворителя на кето-энольное равновесие был накоплен работами
К. Мейера (1912—1914) и О. Димрота (1910—1913). Общая
теория вопроса разработана М. И. Кабачником (1952—1955 г.),
выводы из работ которого главным образом положены в основу этого
раздела. -
211
Общая схема равновесий в системе AiH—A2H—S
/ // /// iv
АН -I-9 > Д Н . <n >" Д~.Н<ч + > Д И . <ч > Д Н JL Q /TY П\
«П ^ О ^ rljO • о ^ /ij • П.О ^ •**-2 ^ 2 ^ \1Л. i [\
+ I
I ///.а
U
А" + HS+
объединяет стадии:
/ — взаимодействие AiH с растворителем, приводящее к
образованию продукта присоединения AiH«S (разумеется, состав
продукта присоединения может быть более сложным);
// — ионизация сольвата с образованием ионной пары Af-HS*,
которая может подвергаться электролитической диссоциации
(стадия ///а);
/// — переход ионизованного комплекса в продукт
присоединения A2H-S с последующим образованием свободной таутомерной
формы А2Н (стадия IV).
В наиболее часто встречающемся случае кето-энольного
равновесия С = О—>С—ОН схема (IX. 11) имеет вид:
—С=О _c=o-.-S —С—О"
—С—Н < —С—Н < — C-HS+ *~
I I I
и
А" + HS+
—С—OH-..S —С—ОН
1± || ._ (IX. Па)
—С — С + S
I I
Обращает на себя внимание далеко идущая аналогия между
приведенной схемой (IX. 11) и общей схемой равновесий в
растворах (VII. 13). Таким образом, для таутомерных превращений
должны быть справедливыми многие из тех закономерностей по
влиянию растворителя на равновесие, которые выполняются для
обычных равновесий в растворах. Даже формальное рассмотрение
схемы (IX. 11) показывает, что растворитель должен оказывать
решающее влияние на величину /(т, причем протофильность
растворителя, точнее, энергия сродства молекулы S к протону, играет
здесь решающую роль.
Исследование таутомерных равновесий в растворах привело к
установлению двух основных эмпирических закономерностей.
Первая из них связывает Кт с характерной для каждого данного
вещества константой энолизации Е [К. Мей ер, 1912]
Кт = ЕЛ (IX. 12)
где Л — «константа растворителя», численно равная константе
таутомеризации ацетоуксусного эфира в данном растворителе
(табл. IX. 3),
212
Таблица 1Х.З
Константы энолизации
Соединение
Метилдибензоилметан . .
Этилацетоуксусный эфир .
Метилбензоилацетон . . .
Ацетоуксусный эфир . . .
Бензоилуксусный эфир . .
2-Метилиндандион ....
Индандион-1,3
Е
0,02
0 23
0,6
1,00
2,3
2,75
3,75
7,0
Соединение
Трибензоилметан ....
З-Бромацетилацетон • • .
Дибензоилметан
Димедон .
Е
10,0
12
22,5
33
120
350
5075
Вторая закономерность, эмпирически установленная Димротом,
учитывает растворимости LK и L3 кетонной и энольной форм в
данном растворителе:
tfT = G-~- (IX. 13)
где G—постоянная величина, независимая от природы
растворителя. В качестве примера в табл. IX. 4 приведены входящие в
уравнение (IX. 13) величины, которые характеризуют
кето-энольное равновесие 3-бензоилкамфоры в различных растворителях.
Из табл. IX. 4 видно, что величина G остается постоянной с
удовлетворительной точностью (до 5%).
Таблица IX.4
Кето-энольное равновесие 3-бензоилкамфоры (О °С)
Растворитель
Этиловый эфир . . ,
Уксусноэтиловый эфир
Этанол
Метанол •
Ацетон
*т
6,81
1,98
1,67
0,87
0,85
'Ч
6,39
1,81
1,57
0,75
0,80
1,06
1,09
1,06
1,15
1,06
Уравнения (IX. 12) и (IX. 13) обоснованы теоретически
[М. И. Кабачник, 1952]. Если рассматривать кето-энольное
равновесие как кислотно-основное, то для описания его применимо
следствие, вытекающее из основного уравнения Бренстеда *. В
приложении к кето-энольному равновесию это следствие запишется:
р/ф e pKs2 + const (1Х и)
(здесь /С?1 и Кт2 —■ константы таутомерного равновесия в
растворителях Si и S2).
* Об уравнении Бренстеда и следствиях из него см. в разделе IX. 4.
213
Уравнение (IX. 14) хорошо подтверждается
экспериментальными данными, как это, например, можно видеть из данных,
приведенных на рис. IX. 7.
Из уравнения (IX. 14) следует, что
К.
•s2
= const
или
*?;/*?;=^J^i ='... = (const)1 =:
Следовательно, в любом растворителе
(IX. 15)
Кг
■ = Е
(IX. 16)
On
' JL/S «Л
Л
У
/
-/ 0 1 *"'т
Рис. IX. 7. Зависимость
pKj1 — р/Ст2 Для паР
растворителей:
1 — метанол — муравьиная кислота;
// — метанол — уксусная кислота;
///— метанол — этанол; /У —
метанол— этиловый эфир.
/ —дибензолметан; 2 — бензоилаце-
тон; 3 — ацетилдибензоилметан; 4 —
ацетилацетон; 5—бензоилкамфора;
6 — ацетилкамфора; 7 — бензоилуксус-
ный эфир (метиловый); 8 — ацето-
ацетанилид; 9 — ацетоуксусный эфир.
Если, согласно К. Мейеру, принять
Кт2 за стандартное состояние
(ацетоуксусный эфир), то есть /Ст2 = Л, то
уравнение (IX. 16) переходит в
(IX. 12).
Константы таутомерного
равновесия в растворах со значительными
концентрациями, согласно уравнению
(IX. 10), передаются выражениями:
А
■Kt
*?■■
* о ~~~ 1\ 4 о ' ^1/V. 1 /)
где у — коэффициент активности, а
/Ст — термодинамическая константа
таутомерного равновесия, которая не
зависит от природы растворителя.
Из уравнения (IX. 17) следует, что:
vSl vSa
гк тэ
vSl vS2
Уэ »к
(IX. 18)
Последнее соотношение справедливо для растворов любой
концентрации, в том числе и для насыщенных. Так как Lsy^ = const
(Ys — коэффициент активности вещества в насыщенном растворе),
то для кетонной и энольной форм будут справедливы
соотношения:
Vk
Ls2
ьк
LSl
(IX. 19)
Подставляя выражения (IX. 15) в (IX. 16), можно придти к
уравнению
Кт'^т2^'^ (IX. 20)
LT
Если считать растворитель S2 стандартным, т. е. К^2 —|- = G,
то выражение (IX. 20) переходит в уравнение Димрота (IX. 13).
В следующем разделе будет показано, что в тех случаях, когда
изменение растворителя не ведет к сколь-нибудь значительному
изменению химической характеристики среды, р/СДИс является
прямолинейной функцией 1/е. Приложимость
к кето-энольному равновесию общих
закономерностей диссоциации
электролитов, а также тот факт, что различные
таутомерные формы характеризуются -*- /
различным дипольным моментом,
позволяет предположить существование такой
же зависимости от диэлектрической про- о4—~*—-г—^**Ц. 1/£-10Ё
ницаемости и величин р/Ст (рис. IX. 8).
IX. 4. Влияние растворителя
на константы электролитической
диссоциации
Рис. IX. 8. Зависимость
констант таутомерного
превращения ацетоуксусного
эфира от диэлектрической
проницаемости:
/—метанол; 2 — этанол; 3— про-
панол; 4 — амиловый спирт.
В проблеме, которой посвящена
настоящая глава, вопрос, составляющий
предмет этого раздела, может считаться наиболее исследованным.
В литературе имеется обширный экспериментальный материал по
константам электролитической диссоциации (Ктс) кислот,
оснований и солей в различных классах растворителей. Ряд весьма
представительных научных школ, среди которых имеются такие,
как школы П. Вальдена и Р. Фуосса, почти полностью посвятили
свои исследования электролитической диссоциации в неводных
растворах.
Наиболее полная разработка теории влияния растворителя на
процесс электролитической диссоциации принадлежит Н. А.
Измайлову. Эта проблема, являющаяся центральной в научном
наследии выдающегося исследователя в области теории растворов,
рассмотрена Н. А. Измайловым весьма полно. Вопросы единой
количественной теории электролитической диссоциации подробно
изложены в монографии Н. А. Измайлова «Электрохимия
растворов». Вот почему в этой книге вопросы влияния растворителя на
Кдис будут рассмотрены лишь в той мере, которая обеспечит
целостность и естественную последовательность излагаемого
материала. Теоретический материал этого раздела в значительной
степени основан на работах Н. А. Измайлова.
215
Рассмотрим связь между величиной /СДИс и константами
равновесия процессов, предшествующих стадии электролитической
диссоциации *. Для удобства представим общую схему равновесий в
растворе (VII. 13) в следующем частном виде:
^нест ^дис ^ас
KA + pS ^± KASp +=> К^ольв + А~ольв ^=> КС+ОЛЬВА^ОЛЬВ (IX. 21)
где индекс «сольв» означает сольватированную частицу.
В схеме (IX. 21) величины констант равновесия отдельных
стадий передаются выражениями:
; в разбавленных растворах ^ест =
aKASp
Q> + a a + a
^сольв -^сольв ^— 1 ^сольв Асольв
v лсольвлсольв
vnp а
(IX. 22)
KASp j
Между величинами /Сдис» Кае и /СпР существует простая
аналитическая связь:
'^дис *^ас '* пр \1Л.. zo)
Если константа электролитической диссоциации определяется
методами, дающими возможность вычислить активность ионов, то
находимая таким образом константа является общей, т. е.
а + a
V Ксольв Асольв
Аоб = (IX. 24)
"недис
ГДе ^недис = СнедисУрР == f^КА + CkAS + С + _ YyCp
\ v лсольв сольв J
В разбавленном растворе величины у всех частиц при
соответствующем выборе стандартного состояния «1, тогда
Аналитическая связь между Коб и константами равновесий
отдельных стадий схемы (IX. 21) передается выражениями:
К- . ^Сдис 1 1 "Ь ^Снест ^^ v
Аоб = t . у -т-Тг— и 1?— 1? ' Г Аас
1 "1" Анест ~Г Апр Аоб Адис
* Здесь пропущена стадия ионизации [К+А~]Сольв, учет которой не играет
особой роли в рассмотрении разбираемых далее соотношений.
216
Рассматривая работу кругового процесса, состоящего из стадий
переноса электролита КА из растворителя S в вакуум,
диссоциации КА на ионы К+ и А" и их сольватации при переносе из
вакуума в растворитель, Н. А. Измайлов вывел уравнение,
связывающее величины /(об, /Сдис(в) (константа диссоциации КА на ионы
в вакууме) с работой переноса ионов из вакуума в растворитель
2^сольв и энергией £/мол образования продукта присоединения
AKSP:
Ков - Кдис (об) ехр (2^-С/мол j {lx 2?)
Величина UM0Jl рассчитывается по уравнению:
kaso
f/мол = - RT In • р (IX. 28)
1 -Г Анест "Г Апр
Из уравнения (IX. 27) следует, что изменение констант Ков при
переходе от одного растворителя к другому определяется только
разностью в химических энергиях сольватации ионов и молекул:
А 2л ^сольв (ион) ~ A 2j ^сольв (мол)
Д In Доб = -Kf (1л. 29)
Н. А. Измайловым выведено полное уравнение для /СОб,
связывающее эту величину с диэлектрической проницаемостью среды и
физическими характеристиками ионов, образующихся при
диссоциации:
In Ко6 = In /Сдис (в) + In YKASp - In (1 + Д-нест + кпр) +
Анализ величин, входящих в уравнение (IX. 30) показывает,
что р/Соб должна быть линейной функцией от 1/е. Впрочем, эта
линейность будет соблюдаться лишь в растворителях,
характеризующихся одинаковой химической и классификационной
характеристикой (у которых величина £/Сольв не изменяется сколь-нибудь
существенно при переходе от одного растворителя к другому),
либо в тех смешанных растворителях, изменение состава которых
не ведет к сколь-нибудь существенному изменению сольватацион-
ной характеристики среды.
Рассмотренные зависимости являются общими и приложимы к
электролитической диссоциации трех основных групп
электролитов — солей, кислот и оснований. Впрочем, для каждой из этих
групп электролитов существуют и специфические
феноменологические выражения, обусловленные особенностями этих веществ.
Связи констант диссоциации кислот с физическими
характеристиками среды и, прежде всего, с г посвящены классические
исследования Бренстеда (1922—1934 гг.), В соответствии с общей
217
схемой кислотно-основного равновесия по Бренстеду (см. гл. II)
начальная и конечная стадии этого процесса передаются схемой:
НА + S ^=± SH+ + A" (IX. 31)
Константа равновесия процесса (IX. 31) равна:
ах. 32)
Здесь а—активности, отнесенные к бесконечно разбавленному
раствору в той же среде (стандартное состояние), т. е. к раствору
с нулевой ионной силой.
Константа протолитического равновесия /СДИс представляет
собою отношение двух констант кислотности:
незаряженной кислоты НА — /СДИс* на =
aH+V
"НА
и катионной кислоты SH+ — /Сдис SH+ = ^
Нетрудно увидеть
„ ^дис, НА /TV ооч
*\jmc==:~7f \i\» 66)
дис, SH+
Решение вопроса so влиянии диэлектрической проницаемости на
константы электролитической диссоциации кислот в рамках
теории Бренстеда достигается сравнением величин /Сдис в данной
среде с величинами констант электролитической диссоциации /Сдис в
среде с бесконечно большой е:
%+ад- . 4%+
_ _
%ис, НА * » ^дис, SH+ — *
aHA aSH+
(a* — абсолютные активности, отнесенные для стандартизации
к среде с бесконечно большой е). В отличие от /Сдис величины
К дис не зависят от среды (точнее, от физических характеристик
среды). Величины /С*ис Бренстед назвал константами собственной
кислотности. Обратные им величины
ьр^,8 7
^дис.НА ^дис, SH+
называются константами собственной основности.
Учет зависимости коэффициентов активности от е и величин
радиусов аниона кислоты гА- и молекулы растворителя rs привел
Бренстеда к следующему общему уравнению: ^
1 г*
С| НА - lg/СДИС|
(IX. 34)
218
Поскольку ЛТдис, на и /С*ИС) SH+ не зависят от е, и приняв в
первом приближении, что величины гА- и rs также не зависят от е,
можно зависимость /СДИс от е представить следующим уравнением
lgKRnc = A + — (IX. 35)
о
где константа А связана только со свойствами данной кислоты, а
константа В — со свойствами растворителя лишь постольку,
поскольку можно считать, что величины гА~ и rs изменяются при
переходе от одного растворителя к другому.
Из уравнения (IX. 35) следует, что в общем случае между
^Кдис и 1/е должна соблюдаться прямолинейная зависимость.
Таким образом, выводы, полученные при рассмотрении уравнения
(IX. 30) для /(об, полностью согласуются с выводами, полученными
Бренстедом для частного случая электролитической диссоциации
кислот.
Приложение уравнения (IX. 34) к частному случаю катионных
кислот приводит к выводу, что поскольку у них rA « rSH+,
величина /Сдис не должна зависеть от е, что подтверждается
экспериментальными данными.
Общая константа электролитической диссоциации и константы
собственной кислотности, по Н. А. Измайлову, связаны
уравнением:
егНА *\цис, НА 2ш1 ^сольв, Н+ *•* "сольв, А~^мол /TV оа\
^об =—* ехР ™ (IX. db}
*Wc,SH+ Ki
ИЛИ
-НА ± ^—J ^сольв, Н+ "•" ^сольв, А" , ^мол /TV о« ч
= const -3^ -^ + 1Ш (IX. 36а)
В соответствии с общей теорией сольватации
f
сольв, *+ ^ А") " ~ ^f S
" Н*+ ^, А") " ~
2 (
(формула Борна — Уэббса)
Подставляя уравнение (IX. 37) в (IX. 36) находим, что:
(ix-38)
Если считать, что величины С/Мол и г\ остаются постоянными *
при переходе от одного растворителя к другому либо при
изменении состава смешанного растворителя, то очевидно, что уравнение
(IX. 38) переходит в уравнение (IX. 35). Таким образом,
ограничениями, накладываемыми на условие прямолинейной зависимости
рКдис от 1/е, является постоянство энергий сольватации и ионных
радиусов с изменением растворителя — обстоятельства, которые не
* О строгости предположения о постоянстве г< см. раздел X. 2.
219
всегда учитываются при обсуждении этого следствия из общего
уравнения Бренстеда.
К аналогичным выводам о прямолинейной зависимости р/СДИс
от 1/е (при соблюдении названных условий) приводит теория
электрической диссоциации оснований.
Исходя из некоторых положений статистической
термодинамики, А. М. Сухотин (1957) вывел уравнение, из которого также
следует экспоненциальная зависимость /Сдис от 1/е
Кцис = ( т 1 ехР( 1 (IX. 39)
Af0 [totrfaTj Ч rBkT) '
где го — минимальное расстояние между ионами в ионном ассо-
циате; п — постоянная, равная « И.
Ценность уравнения (IX. 39) заключается в том, что в ряде
случаев оно дает хорошие результаты в растворителях с низкими
диэлектрическими проницаемостями, там, где иные уравнения
приводят к недостоверным значениям Ктс- Уравнение (IX. 39)
позволяет также описывать зависимость /Сдис от температуры.
В литературе можно встретить множество подтверждений
хорошо соблюдающейся прямолинейной зависимости р/(ДИс от 1/е.
Однако не меньшее количество данных свидетельствует о
неподчинении величин р/СДис одного и того же электролита в разных
растворителях уравнению (IX. 30) [и, следовательно (IX. 35)].
Соответственно этому можно встретить различные суждения о функции
р Кдпс — 1/е — от категорического утверждения о необходимости
прямолинейности этой зависимости до столь же категорического
признания, что встречающиеся случаи такой прямолинейности
являются случайным совпадением.
Рассмотренный выше перечень ограничений, накладываемых на
уравнения (IX. 30) и (IX. 35), показывает, в каких случаях можно
ожидать соблюдения прямолинейности функции- р/СДИс—1/е. На
рис. IX. 9 приведены зависимости р/СДИс — 1/е для ряда
электролитов в некоторых смешанных растворителях. На диаграммах а и б
растворитель однороден (постоянство либо близость энергий
сольватации). И как следствие этого зависимости р/СДИс — 1/е являются
прямолинейными. На диаграмме в изменение е сопряжено со
значительным изменением природы и энергии сольватационных
процессов, т. е. не соблюдаются ограничения, накладывавшиеся при
выводе уравнений (IX. 30) и (IX. 35), вследствие чего зависимость
рКдис — 1/е в этой системе нелинейна.
Особенности хода зависимости р/Сдис—1/е в системах, где не
выполняются ограничения, наложенные при выводе уравнений
(IX. 30) и (IX. 34), могут быть достаточно хорошо объяснены. Так,
весьма быстрое падение е при добавлении диметиланилина к бу-
танолу вызывает соответственно стремительный рост р/СДИс серной
кислоты (рис. IX.9,б), однако при значительном содержании
диметиланилина в смеси сольватации осуществляется преимуще-
220
ственно этим компонентом, имеющим энергию сольватации боль-
шую, чем бутанол, соответственно чему величины р/Сдис начинают
падать.
Влияние члена с [/мол (в уравнении (IX. 38) на
прямолинейность функции р/Сдис — 1/е особенно четко выявляется в
смешанных растворителях, один из компонентов которых обладает
значительно большей энергией сольватации по сравнению с другим. Это
р/с
EUNI
1
рН
2,5
б
7,5
H2SO4
6
-1/В'Ю2
4,5
1/е-ю2
Рис. IX. 9. Зависимость р/СДИс от 1/е для некоторых электролитов:
а — в алифатических нормальных спиртах [А. М. Ш к о д и н, Л. П. Садовничая,
С. Г. Р о с е н к о, 1971]; б —в бутанол-метанольном растворителе; в —в бутанол-диме-
тиланилиновом растворителе [Ю. Я. Фиалков, Э. Л. Лещинский, 1970];
г— в бутанол-гексановом растворителе [Е. М. Рыжков, А. М. Сухотин, 1961].
позволяет считать, что в широком интервале концентрацией
подобного растворителя сольватации осуществляется лишь одним
компонентом, а роль второго сводится лишь к изменению величины е.
Соответственно, в этом интервале концентраций зависимость
Р^дис — 1/е будет прямолинейной, как это можно видеть на
рис. IX. 9, г.
Из уравнения (IX. 34) следует, что различие между
величинами р/Сдис, на Двух кислот HAi и НА2 в одном и том же
растворителе S р/Сдис, на, — рДдис, на2 = Др^Сди? равно:
, НА,
с, НА2
А2
(IX. 40)
221
Отсюда следует, что в случае незаряженных кислот (г = 0)
ИЛИ
4$eRT
л' л. в'
-А + —
(IX. 40а)
Этот вывод, который нетрудно распространить на все типы
незаряженных электролитов, соблюдается гораздо более строго, чем
уравнение (IX. 35), так как не требует наложения тех
ограничений, которые вводились при
обосновании последнего уравнения.
В качестве примера выполнения
уравнения (IX. 36, а) на рис. IX. 10
приводится зависимость р/С£™ —
1/е для уксусной кислоты
(стандарт— бензойная кислота).
• б
Рис. IX. 10. Зависимость рК°™ от 1/е
для уксусной кислоты:
1 — вода; 2 — диоксан — вода (е=45); 3—ацето-
нитрил; 4 — этанол и диоксан — вода (е=25);
5 —бутанол; 6 — диоксан — вода (е=15);
7— ж-крезол.
[Н. А. Измайлов. Электрохимия
растворов, «Химия», 1966.].
IX. 5. Дифференцирующее и
нивелирующее действие
растворителей на силу
электролитов
Рассмотрим связь между
величинами констант
электролитической диссоциации одного и того же электролита в двух разных
растворителях Si и S2.
Из уравнения (IX. 36) с учетом уравнения (IX. 38) следует, что
с. НА
Разность между величинами р/Сдис кислоты НА в
растворителях Si и S2 будет равна:
Р^дис, НА "" Р^дис, НА =* (рЯдис, S2H+ "~ Р^дис, S^*) +
е'Ко \( 1 , 1
Первый и последний члены правой части этого уравнения для
каждой пары растворителей являются постоянными величинами,
поэтому уравнение (IX. 42) может быть переписано в виде:
e2N0
Р^дис, НА ~" Р^дис, НА
■ const +
43RT
L-_!___L\_f_! L_\l
. rs3H+ 8s2/ VSih+ rssH+/J
(ix.42a)
222
Если следуя Н. А. Измайлову, предположить, что rs н+ *» ^s
то разность р/Сдис, на —
нием:
pJ&e. НА " Р^ис, НА = const + ^gr [(^ + ^ j (^- -±-) j (IX. 426)
на выразится следующим
уравнеа для каж-
Для каждой данной кислоты сумма [ Ь -—).
V SH+ A /
дой данной пары растворителей разность ( ] — величины
V es, es2 /
постоянные, поэтому
(IX. 43)
10
Таким образом, зависимость р/Сд1ис от р/Сд2ис является
прямолинейной. Этот вывод без труда может быть обобщен на любые хипы
электролитов и относится к числу
наиболее важных следствий теории элек-
тролитической диссоциации в
неводных растворах. Действительно, в
соответствии с уравнением (IX. 43), если
известны величины р/СДИс
электролитов KiAi, K2A2, К3А3 ... в растворителе
Si, то, зная величины р/СДИс хотя бы
двух электролитов KiAi и К2А2 в
растворителе S2, можно определить
величины р/Сдис всех остальных
электролитов в этом растворителе. К подобным
расчетам приходится прибегать в тех
весьма распространенных случаях,
когда электролиты К3А3, K4A4 ... в
растворителе S2 являются слабыми,
вследствие чего точность методов
определения константы
электролитической диссоциации весьма низка.
В качестве примера выполнения
уравнения (IX. 43) на рис. IX. И
приводится зависимость величин р/СДИс различных кислот в бутаноле
от р/Сдис тех же кислот в метаноле.
В соответствии с уравнением (IX. 43) зависимости р/Сдис — р/^дис
графически всегда должны располагаться под углом 45°. Этот
вывод действительно соблюдается в весьма большом числе случаев
(один из которых изображен на рис. IX. 11), когда оправдываются
приближения, принятые при выводе уравнения (IX. 43). В тех же
случаях, когда нельзя считать, что разность между энергиями
сольватации для любых электролитов, растворенных в данной паре
растворителей Si и S2 постоянна, угол наклона прямой р/Сдис—р/(дис
может отличаться от 45° в большую или меньшую сторону,
Рис. IX. И. Зависимость рДдИС
кислот в бутаноле от р/Сдис
тех же кислот в метаноле:
I — пикриновая; 2 — трихлоруксусная:
3 — дихлоруксусная; 4 — монохлор-
уксусная; 5 — о-нитробензойная;
6—салициловая; 7 — гс-нитробензойная;
8 — ж-нитробензойная; 9 — бензойная;
10— уксусная.
223
Таким образом, возможны три основных типа зависимостей
р/Сдис—р^дис (рис. IX. 12). Прямая / отвечает тем системам, где
наблюдается полное подчинение уравнению (IX. 43). Прямая //
соответствует случаю, когда сила электролитов в растворителе S2
дифференцируется по сравнению с силой электролитов в
растворителе Si. Действительно, величины Ар/СДИс = рДдис, к,а, — рДдис, к2а2 в
растворителе S2 значительно больше, чем в растворителе Si.
Прямая /// отвечает нивелированию силы электролитов в растворителе
S2 по сравнению с силой электролитов в растворителе Si,
соответственно чему величины Ар/(ДИс в
растворителе S2 меньше, чем в
растворителе Si.
Пояснение причин
дифференцирующего или нивелирующего действия
растворителей на силу растворенных
в них электролитов может быть
найдено из общей схемы (VII. 13)
равновесий в растворах.
Е а иу.^--—^г^ [ Как уже неоднократно отмечалось,
^^^^ \ \ v nflf2 электролитическая диссоциация обус-
К2А2 К^4 ис ловлена как взаимодействием (соль-
_ тлг ю т ватацией) растворенного вещества с
Рис. 1ХЛ2^ Типы зависимо- paCTBopH^eM, так и распадом возни-
стеи рддис р/\дис. кающих при этом взаимодействии
ионизированных комплексов (ионных
ассоциатов). В соответствии со схемой (VII. 13) растворители,
обладающие противоположной химической функцией по отношению
к растворенному веществу, должны в общем случае способствовать
повышению силы электролита. Это положение, очевидное в случае
протонных или льюиСовских кислот и оснований распространяется
и на электролитическую диссоциацию солей, поскольку этот
процесс в данном случае можно рассматривать как следствие
кислотно-основного взаимодействия между катионом (кислота) и
растворителем (основание), либо между анионом (основание) и
растворителем (кислота).
Даже это первое приближение позволяет, исходя из общей
схемы (VII. 13), давать качественный прогноз относительно силы
электролитов в растворителях определенных классов.
Естественно, что число кислот в кислых растворителях и число
оснований в основных растворителях значительно меньше числа
кислот и оснований в амфотерном растворителе — воде. Так,
кислоты, сильные в воде, в кислотных растворителях являются
слабыми. При этом (в растворителях с не очень разнящимися
значениями г) сила кислоты в кислом растворителе будет тем меньше,
чем более кислым является растворитель.
Например, H2SO4 в уксусной (в = 6,15) и трихлоруксусной
(е = 4,36 при 50 °С) кислотах характеризуется значениями р/Сдис»
равными 4,30 и 8,47,
224
Подавляющее большинство аминов и амидов, будучи
растворенными даже в сильноосновных (т. е. характеризующихся
высоким значением энергии сродства к протону) растворителях
(жидкий аммиак, этилендиамин, гидразин, хинолин, пиридин),
являются неэлектролитами. Так, анилин в гидразине (е = 51,7)
образует непроводящие ток растворы, в то время как в близкой по
значению 8 (56,2) муравьиной кислоте он' является электролитом
средней силы (р/С = 0,28).
Если, следуя М. И. Усановичу (гл. II), делить соли на более
или менее «кислые» или, соответственно, на более или менее
«основные», то в большинстве случаев можно предсказать качественно
поведение соли в том или ином растворителе. Так, обладающий
весьма «кислым» катионом пикрат лития LiPi диссоциирован в
сильноосновном пиридине (р/Сдис = 4,08; см. табл. IX. 6)
значительно лучше, чем в обладающем значительно большей величиной
8 нитробензоле (р/Сдис = 7,22). Увеличение размера катиона при
переходе от Li+ к Na+ должно было бы вести к повышению силы
электролита из-за ослабления энергии электростатического
взаимодействия с повышением размера катиона. Но, будучи менее
«кислым», чем LiPi, пикрат натрия диссоциирован в пиридине
хуже (рКдис = 4,37). Напротив, в нитробензоле
последовательность в изменении силы этих солей находится в соответствии с
размерами катионов (p/CNapP2 = 4,55).
Итак, растворители, обладающие противоположной по
отношению к электролиту химической функцией, в общем случае должны
способствовать нивелированию, а растворители, обладающие
сходной химической функцией — дифференцированию силы
электролитов.
Однако обоснованный прогноз относительно нивелирующего
либо дифференцирующего действия растворителя может.быть
сделан лишь с "'учетом второго из основных факторов, влияющих на
процесс электролитической диссоциации, — диэлектрической
проницаемости.
Сколь бы не разнились химические функции растворенного
вещества и растворителя, при низких величинах s образовавшийся
ионный ассоциат будет диссоциировать в малой степени, либо не
будет диссоциировать вовсе. Напротив, даже при незначительном
сродстве растворенного вещества к растворителю при высокой е
раствора образующийся ионный ассоциат будет диссоциировать в
заметной степени, приводя к сдвигу вправо равновесий
предшествующих электролитической диссоциации стадий общей схемы
(VII. 13).
Нетрудно проследить, что в растворителях с высокими е сдвиг
равновесий всех стадий схемы (VII. 13) происходит в той степени,
которая обуславливается равновесной концентрацией ионов;
последняя же величина в таких растворителях почти полностью
зависит именно от е. Вот почему степень электролитической
диссоциации в таких растворах в основном зависит от растворителя, а
8 Зак, 500 225
высокое значение е растворителя ведет к нивелированию силы
электролитов.
Напротив, в растворах с низкими е на первый план выступают
особенности, связанные с образованием продуктов присоединения
(сольватов) в растворах, так как е не «диктует» сдвиг равновесий
стадии комплексообразования и в значительной степени — стадии
образования ионного ассоциата. Поскольку энергия образования
продукта присоединения (сольвата) растворенного вещества к
растворителю, а, следовательно, и константа равновесия этого
процесса являются весьма специфической характеристикой каждой
пары растворенное вещество — растворитель, в растворителях с
низкой е степень электролитической диссоциации даже
однотипных электролитов, заметно разнится, а низкая величина s
способствует дифференцированию силы электролитов.
Рассмотрим некоторые аспекты проблемы нивелирующего и
дифференцирующего действия растворителей на силу
растворенных в них электролитов на примере различных классов
растворителей. При этом в качестве очевидного стандартного растворителя
выберем воду.
Кислые растворители, как следует из сказанного, в общем
случае должны дифференцировать силу кислот и нивелировать силу
оснований. При этом дифференцирующее действие кислого
растворителя проявляется тем более отчетливо, чем ниже е; в свою
очередь, нивелирующее действие растворителя выступает с
очевидностью при высоких е (табл. IX. 5).
Следует отметить, что вследствие экспериментальных
трудностей, возникающих при определении констант электролитической
диссоциации слабых электролитов в неводных растворах, различие
в значениях констант, определенных различными методами, часто
достигает 1—2, а иногда и большего числа порядков. По-видимому,
по этой же причине значения констант диссоциации, определенные
одним и тем же методом, но различными авторами, также разнятся
нередко весьма значительно.
Немалый разнобой в приводимых в литературе значениях
констант электролитической диссоциации вносит недостаточная
разработка проблем, возникающих при таком расчете. Вот почему
нередко обработка разными авторами экспериментальных данных,
сообщаемых в какой-либо одной работе, приводит к существенно
разнящимся значениям констант диссоциации. Так, А. М. Шкодин
(1961 г.), обработав литературные данные по электропроводности
ацетатов щелочных металлов, (J. М. Коlthoff, A. Wilman,
1934), нашел, что константы электролитической диссоциации этих
солей на 2—3 порядка выше значений, рассчитанных прежде
[S. Bruckenstein, J. M. Kolthoff, 1956].
Как и следовало ожидать, сила кислот в кислых растворителях,
сильно понижена по сравнению с водой. Все сильные в воде
минеральные кислоты являются слабыми в уксусной. При этом
вследствие малой величины е в уксусной кислоте происходит резкое
226
Таблица 1Х.5
Сила электролитов в кислых растворителях
Электролит
Растворитель
Н2-0
СНзСООН СН2С1СООН
нсоон
нсю4 . . .
CF3SO3H . .
h2s2o7 . . .
H2SO4 . . .
H2Se04 . . .
Н3РО4 . . .
HNO3 . . .
CF3COOH .
CCI3COOH .
Пиперидин .
Диэтиламин
Триэтиламин
Пиридин . .
Анилин . .
Кодеин . . .
Мочевина .
Теобромин .
NaCl....
КС1 ....
LiCOOCH3 .
NaCOOCH3
КСООСН3 .
LiCOOH . .
NaCOOH . .
КСООН . .
Et4NPi . . .
О
О
О
О
1,80
0
0,23
0,7
2,80
2,90
3,25
8,69
9,40
13,39
13,82
13,89
0
0
2,70
3,3
3,8
4,30
4,40
5,10
5,40
11,60
4,35
4,33
4,20
4,45
7,05
7,30
6,20
3,78
5,43
5,03
4,50
7,09
5,79
1,10
2,4
1,6
1,94
0,56
0,94
4,01
3,62
0,58
0,27
0,28
0,60
одо-
0,51
1,35
1,43
0,28
0,10
-0,01
р*й!?С1СООН
дифференцирование силы кислот. Так, НС1О4 и СС13СООН,
различающиеся в воде приблизительно на порядок, в уксусной кислоте
различаются по силе почти в миллиард
раз.
Даже не очень значительное
изменение сродства к протону у кислого
растворителя сказывается на проявлении
электролитных свойств у растворенных в
кислом растворителе кислот. Например,
СН2С1СООН и СНС12СООН, которые в
уксусной кислоте являются
неэлектролитами, в растворах ж-крезола проявляют
хотя и слабые, но отчетливо
фиксируемые электролитные свойства (р/СДИс
равны 8,7 и 7,3).
Повышение г растворителя в
соответствии с высказанными выше
положениями ведет к нивелированию силы кислот. На рис. IX. 13
приводится зависимость величин р/Сдио некоторых кислот
_J 1 1— CH3COOH
2 3 4-5 Q^duc
Рис. IX. 13. Зависимость
Р^Сдис кислот в СН2С1СООН
от р/Сдис тех же кислот
в СНзСООН.
227
в монохлоруксусной кислоте, 8 которой приблизительно
вчетверо больше е уксусной кислоты, от величин р/СДИс тех же кислот
в уксусной кислоте. Из рисунка видно, что СН2С1СООН оказывает
заметное нивелирующее действие на силу кислот: разности р/СДИс
(Ар/Сдис) в СН3СООН, равной 1, отвечает в СН2С1СООН величина
Др/Сдис = 0,55.
Рассмотрение соотносительного влияния на константы
электролитической диссоциации химического фактора и диэлектрической
проницаемости позволяет утверждать, что последняя оказывает на
изменение величин /Сдис гораздо более существенное влияние.
Действительно, хотя энергия сродства к протону у
монохлоруксусной и муравьиной кислот ниже, чем у уксусной, кислоты в
первых растворителях диссоциированы в большей степени, чем в
последнем, причем константы диссоциации в НСООН на 1— 3
порядка выше, чем в СН2С1СООН — соответственно большей
величине е муравьиной кислоты.
Соотносительное влияние химического и физического факторов
особенно отчетливо проявляется в величинах констант
диссоциации (точнее, протонизации) оснований в кислых растворителях.
Вследствие того, что величина е уксусной кислоты значительно
меньше, чем у воды, амины в этом растворителе в ряде случаев
(пиридин, диэтиламин и др.) слабее, чем в воде, хотя сродство
к протону у воды выше, чем у уксусной кислоты. Впрочем, в случае
очень слабых (в воде) аминов происходит повышение их силы
в уксусной кислоте по сравнению с водой.
Поскольку химические функции аминов и кислого растворителя
противоположны, в этих растворителях происходит нивелирование
силы оснований — тем большее, чем выше значение е растворителя
(сравни величины р/СДИс аминов в уксусной и муравьиной
кислотах). Сравнение силы аминов в двух растворителях, обладающих
высокой е — воде и муравьиной кислоте — служит весьма
наглядной иллюстрацией влияния химических факторов на
нивелирование и дифференцирование силы электролитов: в значительно более
кислой по сравнению с водой муравьиной кислоте амины
нивелированы в значительно большей степени.
Роль кислотно-основного взаимодействия достаточно четко
проявляется и в диссоциации солей в кислых растворителях. Ацетаты
и формиаты щелочных металлов, будучи растворенными, в
уксусной и муравьиной кислотах, должны в согласии с положениями
теории сольвосистем быть в этих растворителях основаниями, сила
которых увеличивается от LiA к КА, что и наблюдается (табл.
IX. 5). Причину повышения силы солей при переходе от Li+ к К+
можно в данном случае трактовать, полагая, что: в этом ряду
ослабляются кислые свойства солей и, следовательно, повышается
степень диссоциации в кислых растворителях.
К числу основных растворителей, в которых относительно
хорошо исследованы константы диссоциации электролитов,
относятся жидкий аммиак, пиридин, этилендиамин, диметилформамид и др.
228
Основные растворители должны в общем случае нивелировать
силу кислот и дифференцировать силу оснований. Это положение
действительно оправдывается, как можно видеть из данных,
приводимых в табл. IX. 6. Эти данные, как и в случае кислых
растворителей, свидетельствуют о главенствующей роли е.
Действительно, все растворители, данные по которым сведены в табл. IX. 6,
обладают значительно большей энергией сродства к протону по
сравнению с водой; однако величины г этих растворителей меньше,
чем у воды — соответственно и кислоты в этих растворителях
являются слабыми, хотя в воде они сильные электролиты.
Таблица IX.6
Сила электролитов в основных растворителях
Электролит
Растворитель
NH3
C2H4(NH2)2
C5H5N
PhNEt2
нсю4 . . .
HI
HBr ....
HC1 ....
HF
HNO3 . . .
HCOOH ..
CH3COOH .
C6H5COOH .
CF3COOH .
CHC12COOH
CH2C1COOH
N1
Bt4NI . . .
Bt4NNO3 . .
AgNO3 . . .
LiPi ....
NaPi ....
KPi ....
Bt4NCH3COO
AgC104 . .
2,27
2,62
2,89
2,37
3,47
4,12
3,82
3,16
3,27
3,29
3,24
2,23
2,53
4,0
5,4
8,53
4,3
9,8
3,43
3,39
3,43
3,03
4,08
4,37
4,00
3,77
2,72
4,8
2,56
3,12
3,82
Если же 8 основного растворителя достаточно велико, то
происходит существенное повышение силы кислот. Так, п-хлорфенол,
фенол и окрезол, которые в воде имеют р/СДИс равные 9,4; 9,9 и
10,2, в гидразине (е = 52) характеризуются величинами р/Сдис
равными 2,2; 2,3 и 3,4.
Даже относительно небольшого повышения диэлектрической
проницаемости оказывается достаточно, чтобы начало проявляться
нивелирующее действие основного растворителя. На рис. IX. 14
приведены величины р/СДИс кислот в аммиаке (е = 17) как
функция р/Сдис величин тех же кислот в пиридине (е = 12,9). Как видно
из рисунка, аммиак по сравнению с пиридином оказывает на
229
кислоты отчетливо выраженное нивелирующее действие: величины
Др/Сдис = 1 в пиридине, в аммиаке отвечает Др/Сдис = 0,25.
Фактом, который несомненно заслуживает того, чтобы обратить
на него внимание, является четкая прямолинейная корреляция
между величинами р/Сдис кислот в пиридине и энергией сродства
соответствующих анионов к протону, как это можно видеть из
данных,-приведенных на рис. IX. 15 [величины энергии сродства к
протону взяты из работ К. Б. Ядимирского (1951—1957 гг.)].
Выше уже отмечалось, что практически ни в одном случае не
удается с уверенностью зафиксировать электролитные свойства
аминов в основных
растворителях. Л
Рассмотрение общей схемы
(VIII. 13) возникновения
электролитного раствора с точки зрения
кислотно-основного взаимодей-
w
PhCOOH
Z'S
5,0
&
300
325
350 375
Ен+,ннал/г-ион
Рис. IX. 14. Зависимость р/Сдис
кислот в жидком аммиаке от р/СДИс
тех Же кислот в пиридине.
Рис. IX. 15. Зависимость силы кислот
в пиридине от энергии сродства
анионов кислот к протону.
ствия в случае солей позволяет объяснить ряд закономерностей
относительно силы солей в основных растворителях. Так,
сопоставление электролитных свойств растворов солей в двух
практически изодиэлектрических растворителях пиридине и этилендиа-
мине показывает, что соли в последнем диссоциированы в большей
степени — в соответствии с большей основностью этилендиамина.
При этом при переходе от пиридина 'к этилендиамину сила более
«кислого» Nal повышается в большей степени (Др/(ДИс = 0,27), чем
у менее «кислого» Bt4NI (Др/(ДИс = 0,12).
В обладающих общим анионом ряду солей с катионами Li+—
— Na+—К+ сила солей должна возрастать в соответствии с ростом
ионного радиуса. Однако, сопоставляя силу пикратов этих
металлов в пиридине, можно заметить в закономерности изменения
величин р/Сдис этих солей борьбу двух противоположных тенденций;
ослабление «кислых» свойств катионов при переходе от Li+ к К+ и
связанное с этим понижение степени взаимодействия соли с
растворителем, и возрастание степени диссоциации, связанное с
ростом ионного радиуса катионов.
Из двух солей — Bt4NNO3 и Bt4NCH3COO — последняя является
более «основной», следовательно, в основном растворителе^-пи-
230
ридине она должна быть хуже диссоциирована, что имеет место
в действительности (см. табл. IX. 6). В то же время сила Bt4NI и
Bt4NNO3 практически одинакова — соответственно близости
основных свойств анионов этих солей (энергия сродства к протону у
этих анионов равна, соответственно, 1306 и 1360 кдж/г-ион).
Аналогично, более «кислый» AgC104 диссоциирован в пиридине
лучше, чем менее «кислый» AgNO3 (величина энергии сродства к
протону с С1О4 равна 4140 кдж/г-ион).
Прежде, чем перейти к рассмотрению некоторых
закономерностей электролитической диссоциации в амфотерных растворителях,
надо напомнить читателю об относительности понятия «амфотер-
ность» (подробнее об этом см. в гл. II). Классическим примером
амфотерного растворителя может служить вода.
По установившейся в физической химии растворов традиции
к амфотерным растворителям относят спирты; некоторые авторы
включают в эту группу также кетоны, что оправдывается
возможностью проявления последними как протоноакцепторной, так и
протонодонорной (энолы) функций.
Таблица IX.7
Сила электролитов в амфотерных растворителях
Электролит
сн3он
3,17
4,9
6,3
7,8
9,7
6,00
6,20
5,20
5,54
1,44
—
-7
1,47
1,42
1,42
1,42
—
—
1,19
'
1,05
1,15
с2нбон
3>57
5,7
7,1
8,5
10,4
5,70
5,90
4,40
4,27
—
—
—
—
—
—
—
—
—
2,09
—
—
1,01
—
1,83
1,85
Растворител1
с4н9он
6,30
7,30
8,5
10,35
—
—
—
—
—
—
—
—
—
3,19
—
—
—
2,96
3,32
э
(СН3)2СО
5,9
4,9
4,5
2,99
2,87 '
2,46
2,95
1,95
1,76
—
1,65
2,64
2,48
2,28
2,26
2,02
2,75
2,10
2,10
—
СН3СОС2Нв
—-
6,6
—
___
7,5
3,71
3,47
3,04
—
2,42
2,36
—
—
__.
—
—
—
3,44
2,72
—
HNO3 ....
CCI3COOH .
СНС12СООН
СН2С1СООН
СНзСООН .
C6H5NH2 . .
/z-CH3C6H4NH2
C6H5N(CH3)2
C5H5N . . .
LiPi
NaPi ....
KPi
NH4Pi ....
(CH3)4NPi . .
Et4NPi . . .
Pr4NPi . . .
Bt4NPi . . .
Bt4NCl . . .
Bt4NBr . . .
Bt4NI ....
Bt4NNO3 . .
Bt4NC104 . .
Lil . . , . .
Nal
KI
Csl
231
Данные по величинам р/Сдис некоторых электролитов и амфо-
терных растворителей сведены в табл. IX. 7.
Сила кислот в спиртах резко понижена по сравнению с водой.
Связано это прежде всего с тем, что величины 8 у спиртов
значительно меньше, чем у воды, так как энергия сродства к протону у
воды и спиртов разнится не очень значительно: на 10—15%
(707 кдж/моль для воды и 753 кдж/моль для метанола, по оценке
Н. А. Измайлова).
Определяющее влияние диэлектрической проницаемости на
силу кислот проявляется также в монотонном падении силы кислот
в гомологическом ряду спиртов — в соответствии с падением е, но
вопреки росту энергии сродства к протону.
/Mt2CO
рд
2,5
9
H
ROH
d
о
300 350
Ен+,ккал/г-ион
Рис. IX. 16. Величины р/СДИс кислот
в диметиланилине от р/СДИс тех же
кислот в метаноле (/) или
этаноле (//):
/ - СНС12СООН; 2 - СН2С1СООН;
3-СНзСООН.
Рис. IX. 17. Зависимость
Р^Сдис солей тетрабутил-
аммония в ацетоновых
растворах от энергии
сродства анионов к
протону.
Роль химических свойств растворителя в определении силы
кислот выразительно проявляется при сопоставлении данных по
константам диссоциации кислот в основных амфотерных
растворителях. На рис. IX. 16 приводятся зависимости величин р/СДИс
некоторых кислот в диметиламилине от величин рН этих же кислот
в метаноле и этаноле. Из прямых, выражающих эту зависимость,
следует, что несмотря на меньшую величину е диметиланилин в
соответствии с большим по сравнению со спиртами сродству к
протону оказывает на кислоты нивелирующее действие величине Др/Сдис
в спирте, равной 1, отвечает в диметиланилине р/СДИс = 0,45.
Амфотерйые растворители понижают по сравнению с водой и
силу оснований [А. М. Ш ко дин и др., 1961; Н. А. Измайлов и
Т. В. Можарова, 1962]. Естественно также, что сила оснований
в спиртовых растворителях значительно ниже, чем в кислых.
Спирты по сравнению с кислыми растворителями должны
дифференцировать силу оснований. Действительно, различие между
величинами рКдис пиридина и а-пиколина, составляющее в уксусной
кислоте 0,05, в этаноле равно 1,24,
232
Все исследованные в амфотерных растворителях соли
являются слабыми электролитами. Закономерности, определяющие
силу солей в подобных растворителях, пока что изучены слабо.
Влияние диэлектрической проницаемости с несомненностью
выявляется и здесь. Если считать, что мерой ионности связи в солях
является энергия сродства аниона к протону, то представляется
естественной прямолинейная зависимость величин рКдис солей с
одноименным катионом в ацетоне от величины энергии сродства
соответствующего аниона к протону (рис. IX. 17). Обращает на
себя внимание также нивелирующее действие метанола на силу
растворенных в нем солей.
IX. 6. Кислотность неводных растворов
Проблема количественной характеристики кислотности
неводных растворов состоит из двух аспектов. Первый посвящен
разработке методов сопоставления кислотности растворов в одном
растворителе, а второй — в разных растворителях.
Распространение понятия «рН» на неводные растворы
сталкивается с рядом трудностей. Прежде всего отметим, что почти во
всех неводных растворителях кислоты являются слабыми, и
поэтому даже в весьма разбавленных растворах не представляется
возможным рассчитать концентрацию Н+, не располагая данными
по константе диссоциации данной кислоты в данном растворителе.
Более строгий подход к проблеме кислотности в неводных
растворах вызывает необходимость учесть, что кислотность любого
раствора характеризуется, собственно, не концентрацией, Н+, а
концентрацией ионов лиония Sp образующихся в результате процесса
автоионизации растворителя 2S *=* S* + S2. Впрочем, в протоли-
тических неводных растворителях, так же, как и в воде, отнесение
показателя кислотности раствора не к концентрации Sf, а к
концентрации Н+ не приводит к сколь-нибудь существенным
погрешностям, поскольку можно считать, что протоны, введенные в
систему, практически полностью присоединяются к молекулам
растворителя, повышая концентрацию ионов лиония в полном
соответствии с повышением концентрации Н+. Однако в апротон-
ных растворителях, особенно растворителях, обладающих
достаточно ярко выраженным стремлением к автоионизации, таких,
например, как SbCis, определение кислотности сопряжено с
необходимостью прямого определения концентрации ионов лиония (в
приведенном примере SbClt).
Подобно водным растворам, в каждом неводном растворителе,
точка нейтральности совпадает с концентрацией ионов лиония,
равной YKi *• Таким образом, точка нейтральности pKi в жидком
* Разумеется, в том абсолютно подавляющем большинстве случаев, когда
процесс автоионизации приводит к возникновению двух ионов.
233
аммиаке соответствует значению 16,15 {[NHj]=(lO 32'3) '}, в этаноле
9,75{[С2Н5ОН2]=(10"19>5)0'5}, в уксусной кислоте 6,3 {[СНзСООН,]^
= (Ю""12'6)' } (см. табл. III.2). Приведенные примеры обос-
новывают высказанное в разделе III. 2 утверждение, что
кислотность растворителей увеличивается с увеличением величины Кг.
Действительно, чем выше Ки тем большая концентрация ионов
лиония должна быть создана для достижения точки
нейтральности.
Из сказанного следует, что каждый растворитель будет
характеризоваться своей шкалой рН. Предложено отмечать показатель
рН для каждого растворителя индексом, равным точке
нейтральности. Так, для аммиака показатель будет отмечен индексом
«16, 15», то есть pHie is, для этанола рН9 75, или в общем виде:
рНр.
Итак, в неводных растворах показатель кислотности pSf равен
pS+ = - lg ag+ = - lg Cs+Ys+ (IX. 44)
В частном (хотя и весьма распространенном) случае
растворителей с протолитическим равновесием
pSf = PHS = - lg <£+ = - lg Ch+yh+ <ix- 45)
Поскольку из уравнения (IX. 44) следует, что два раствора в
различных растворителях, характеризующиеся одинаковым
значением pSi", будут обладать различной кислотностью, очевидно, что
из двух перечисленных выше аспектов проблемы кислотности в
неводных растворах более сложным является сравнение кислотности
растворов в различных растворителях. Поэтому было предложено
несколько критериев, которые могут лечь в основу сравнения
кислотности растворов в различных растворителях.
Бренстед (1934) предложил, в качестве критерия сравнения
химический потенциал протона в данной среде;
\хи+ = RT In aH+ (1Х. 46)
Величина \хн+ была названа Бренстедом потенциалом
кислотности, а величина 1/м-н+ называется потенциалом основности. Из
материала, изложенного в разделе IX. 4, с очевидностью следует, что
\iH+ связан с константой собственной кислотности простым
соотношением
Впрочем, практическое использование уравнения (IX.47)
сопряжено с известными затруднениями. Для расчета химического
потенциала необходимо располагать данными о константе
собственной кислотности кислоты и энергиях переноса кислоты
(основания) из вакуума в соответствующий растворитель. Однако и эти
234
величины не могут быть определены в прямом эксперименте и
в подавляющем большинстве случаев находятся с помощью
косвенных методов, причем степень приближенности, с которой
определяются эти величины, часто оказывается совершенно
недостаточной для сравнения кислотности растворов в различных
растворителях.
К числу весьма распространенных методов сравнения
кислотности в различных растворителях (как, впрочем, и сопоставления
кислотности растворов в одном растворителе) относится метод
Гаммета (1932). В основе этого метода лежит положение о том,
что константы кислотности оснований и катионных кислот не
зависят от растворителя (см. раздел IX. 4). Если какой-либо индикатор
является по отношению к раствору основанием, то степень лревра-
щения индикатора из молекулярной в конную форму может
служить мерой кислотности среды. Если выразить активности
молекулярной и ионной форм индикатора через ав и авн+, то
кислотность среды выразится функцией
Н0 = Р^вн + 18-^~ (IX. 48)
авн+
где Квн+ — константа электролитической диссоциации ионной
ан+ав
формы основания в воде, т. е. Квн+ = Если концентрация
вн+
индикатора невелика, то функция Гаммета может быть выражена
через концентрации:
^H^
вн+
Если индикатор ионизован наполовину, то есть Св = Свн+, то;
Можно путем несложных преобразований показать, что в
разбавленных водных растворах Но = рН. *
Широкое распространение метода Гаммета обусловлено
относительной простотой определения Св и Свн+, производимого
большей частью спектрофотометрическими методами.
В основе сравнения кислотности растворов в различных
растворителях, как уже упоминалось, лежит представление о том, что
Квн+ не изменяется при переходе от одного растворителя к
другому. Однако это положение оправдывается с той точностью, с
которой можно считать, что константы кислотности оснований могут
быть выражены через абсолютные активности, отнесенные к
водным растворам; последние в данном случае принимаются за
стандартные. Н. А. Измайлов показал, что Но отличается от lgaH+ на
величину lg °ВН , где yo — коэффициенты активности, отнесенные
Yob
23Б
к бесконечно разбавленному водному раствору. Вот почему метод
Гаммета может служить лишь для приблизительного сравнения
кислотности в различных растворителях.
Тем не менее, функция Гаммета может служить весьма
неплохим полуколичественным методом сравнения кислотности
различных растворов, причем в широком интервале концентраций. На
рис. IX. 18 приводятся значения Но для растворов H2SO4 в воде и
уксусной кислоте. Из рисунка видно, что растворы серной кислоты
в уксусной значительно (на 102»65, т. е. в
450 раз) превышают кислотность
растворов той же концентрации в воде, что
вполне естественно, так как вода по
отношению к серной кислоте является
гораздо более основным растворителем,
чем уксусная кислота.
Предложенные в развитие метода
Гаммета аналогичные функции
кислотности [Б ей с, Шварценбах, 1955;
Вестгеймер, Караш, 1946] не нашли
широкого распространения, так как, с
одной стороны, они относятся к более
частным случаям, чем функция Гаммета,
а с другой стороны, они обладают тем же
недостатком, что и Но, так как величины
этих функций определяются не только
кислотностью раствора, но и
активностью ионной и молекулярной форм
индикатора.
В значительной степени лишен недостатков, присущих
перечисленным выше методам, метод «стандартного иона» [В. А.
Плесков, 1947], который относительно подробно будет охарактеризован
в гл. XII. Однако общим этот метод быть не может, так как
приложим без каких-либо ограничений лишь к весьма узкому кругу
растворителей с высокой диэлектрической проницаемостью.
Н. А. Измайлов (1959) предложил проводить сравнение
кислотности неводных растворов, приняв в качестве стандартной
величины состояние протонов в бесконечно разбавленном водном
растворе. При этом показатель единой кислотности рА определяется
выражением
Рис. IX. 18. Функция
кислотности растворов серной
кислоты в воде (2) и
уксусной кислоте (1).
[А. И. Ш а т е н ш т е й н. Теория
кислот и оснований. Госхимиздат,
1949.]
рА = - lg а_
= -lgaSH+-lg0H+
= pEL - \g у
0H+
(IX. 50)
В уравнении (IX. 50) — ан+— активность, а уон+ — единый
нулевой коэффициент активности протонов. Обе эти величины
отнесены к бесконечно разбавленному водному раствору. Величина
Y0H+> по Измайлову, определяется изменением энергии протонов
при перенесении из бесконечно разбавленного неводного раствора
236
в бесконечно разбавленный водный раствор, т. е. выражением:
*J • /U ГЛ\ ГТТ /П\ Т Т "Г
(IX. 51)
Н+(Н2О)
г0Н+
2,3/?*
2,3Rt
где Лн+ — химические энергии сольватации протонов в воде и
данном неводном растворителе. Величины Yoh+ для некоторых
растворителей сведены в табл. IX. 8 (Н. А. Из м а й л о в, 1963).
Таблица IX.8
Изменение протонного сродства (Yoh+) и интервал шкалы рА
для некоторых растворителей
Растворитель
Аммиак . . .
Гидразин . . .
Вода
Метанол . . .
Этанол ....
Бутанол . . .
Y0H+
-15,8
— 13,2
0
3,1
3,9
4,2
Интервал
шкалы
15,8-48,1
13,2-37,9
0 -14,0
-3,1-13,6
-3,9-15,6
Растворитель
Пентанол . .
Ацетон ....
Ацетонитрил .
Муравьиная
кислота . .
W
4,2
3,3
5,5
10,0
Интервал
шкалы
-3,3-22,6
— 10,0—(—3,8)
Величина y0 h+ является надежным теоретическим критерием
для разработки единой меры изменения кислотности при переходе
от одного растворителя к другому. Следует, впрочем, учесть, что
если обозначить начало шкалы кислотности для воды через 0, то
поскольку например, для метанола Yoh+ = 3>1> a абсолютная
протяженность шкалы 16,7 (см. табл. III. 2), то шкала разместится в
интервале от —3,1 до 13,6. Положения шкалы рН для каждого
растворителя приведены в табл. IX. 8.
Величины yOh+ Для различных кислот в данном растворителе
могут быть найдены сопоставлением с раствором НС1, для
которого достаточно достоверно определены величины у0 во многих
неводных растворителях. Однако, поскольку неизвестно, какая
доля yohci в неводных растворителях приходится на Н+ (SH+), а
какая на С1", принимают, что lg у0 н+ = lg Yo cr-
Глава X
ЭЛЕКТРОПРОВОДНОСТЬ
НЕВОДНЫХ РАСТВОРОВ
Пожалуй, ни одно из свойств растворов не зависит так сильно
от растворителя и не изменяется с изменением его столь
существенно, как электропроводность. Именно на различии
электропроводности одного и того же электролита в разных растворителях
основывались первые теоретические обобщения физической химии
неводных растворов. В то же время электропроводность наиболее
непосредственно связана с электролитной стадией общей схемы
(VII. 13) равновесий в растворах и поэтому кондуктометрия
является наиболее действенным методом изучения этой стадии.
В проблеме электропроводности неводных растворов можно
выделить два основных аспекта: зависимость электропроводности
от концентрации раствора и влияние на электропроводность
физических и химических свойств растворов.
X. I. Зависимость электропроводности
от концентрации растворов
Количественные соотношения между электропроводностью и
концентрацией, известные в настоящее время, почти во всех
случаях описывают разбавленные и весьма разбавленные растворы.
При этом практически всегда используют величины эквивалентной
электропроводности А, в то время как для характеристик
электропроводности концентрированных растворов и жидких систем (см.
гл. VIII) большей частью применяют величины удельной
электропроводности и. Связано это с тем, что, как уже отмечалось, в
концентрированных растворах и жидких системах, далеко не всегда
бывает известна истинная концентрация электролитного
соединения, ответственного за электропроводность раствора.
Именно неправильным подсчетом концентраций электролитного
компонента объясняется явление так называемой «аномальной»
электропроводности. Состоит оно в' том, что во многих неводных
растворителях, в отличие от воды, X с уменьшением
концентрации растворенного вещества падает, вместо того, чтобы
повышаться, как этого требует классическая теория электролитов Арре-
ниуса — Оствальда. Действительно, кривая Я, «аномальная» при
обычном способе выражения состава, часто превращается в
нормальную, если концентрация растворителя подсчитана
правильно— прием, названный «обращением» кривых X [А. И.
Рабинович, 1921]*.
* Подробнее о закономерностях концентрационного хода кривых в жидких
системах см. далее.
238
Лишь одно уравнение
(4 ) (Х-О
являющееся основным соотношением классической теории Аррени-
уса — Оствальда, непосредственно связывает электропроводность
ус с концентрацией С (а — степень диссоциации, Xt и Хо—
подвижности, или электролитические проводимости ионов). Все
остальные уравнения относятся к типу
* = f(4> V»C av <*2> •••) (Х.2)
где аи я2, ... — постоянные величины для данного электролита в
данном растворителе, либо для данной группы электролитов в
данной группе растворителей. В зависимости от степени физической и
математической обоснованности параметры аи а2, ... в уравнениях
типа (Х.2) являются либо эмпирическими константами, либо
выражаются через различные свойства растворителя и среды (е, т|,
Г, Р и т. п.) с той или иной степенью приближенности.
Многие годы развития физической химии электролитных
растворов сводились к усовершенствованию и более глубокому
физическому обоснованию способов расчета ab а2, ..., а также к
расширению верхнего предела концентрации раствора, описываемого
уравнениями типа (Х.2).
Общее уравнение Кольрауша (1900)
Яс = Я0-а/с" (Х.З)
в подавляющем большинстве случаев хорошо описывает
концентрационную зависимость X как водных, так и неводных растворов
в пределах концентраций 10~5—10~3 моль/л*. В растворителях с
высокой 8 верхний концентрационный предел может быть повышен
на 1 —1,5 порядка. Например, было найдено, что X перхлоратов
щелочных металлов в смешанном растворителе ацетон —
хлороформ (во всем концентрационном интервале смешанного
растворителя) хорошо описывается уравнением (Х.З), причем постоянство
величины а для каждой соли в растворителе определенного состава
соблюдается с точностью порядка 0,5% [А. М. Ш ко дин,
И. А. Сергеева, 1971].
Ряд уравнений, генетически связанных с уравнением (Х.З),
более точно передают связь X = f(C) и охватывают более широкий
интервал концентрации. Среди них основными являются
уравнения:
Онзагера (1927)
%с = Яо - (а{Х0 + а2) VC (X. 4а)
в котором
2801106lU __ 41,25(1 g|| + |gal)
п
* Данные по электропроводности растворов с более низкой концентрацией,
как правило, весьма неточны,
239
(г заряды ионов; е и г)— диэлектрическая проницаемость и
вязкость растворителя; q — определяется зарядами и подвижностям.и
ионов);
Шедловского (1932 и 1938)
Яс = Ко - (а{Х0 + а2) VC + а3С (l - а{ VT) (X. 46)
где п\ и а2 — те же, что и в уравнении (X. 4а); аъ — эмпирический
коэффициент, выбираемый так, чтобы наблюдалось согласие с
опытом;
Фуосса-Онзагера (1952)
Яс = Яо - (а{Х0 + а2) VC + а3С In С + аАС (X. 4в)
(константы а\ и а2 — обычны, аъ — определяется теми же
параметрами, что #i и #2, #4 — величина, зависящая от размера иона);
Фалькенгагена (1952) и Робинсона и Стокса (1954)
с ° 1 + хя
в котором делитель 1+%а вводится для учета конечности размера
иона.
Все уравнения типа (Х.4) выведены на основе теории сильных
электролитов. В неводных растворителях, в особенности тех,
которые характеризуются более низкой, чем у воды диэлектрической
проницаемостью, следует ожидать образования значительного
количества ионных пар даже в растворах низких концентраций, что
обязательно следует учитывать при использовании разлдчных
форм уравнения (Х.4).
Так, электропроводность галогенидов щелочных металлов в
разбавленных метаноловых растворах (е = 32,6) хорошо
согласуется с наиболее часто используемым предельным законом Онза-
гера [уравйение (Х.4а)]. В частности, для NaCl в метаноле при
концентрации 0,001 н. по расчету из этой формулы получается
% =z 89,89, в то время как эксперимент дает величину X = 89,87.
При исследовании электропроводности ряда солей щелочных
металлов в жидком цианистом водороде найдена (почти для всех
солей) линейная зависимость между X и ]/С вплоть до 0,003 н
растворов; расчет вели по уравнению (X. 4а), в котором для
данного случая п\ = 0,1271, #2 = 233 (при 18°С). Значения п\ и а2 в
уравнении (X. 4а) для различных растворителей представлены в
табл. X. 1.
Весьма интересны измерения электропроводности солей в
цианистом водороде, проведенные с целью проверки приложимости
уравнения (X. 4г), в котором учитывается поправка на конечность
размера ионов. Согласие опытных, и расчетных данных
наблюдается вплоть до концентрации 0,02 н.
Большинство обычно используемых неводных растворителей
характеризуются низкими значениями е, поэтому ассоциация еу-
240
Таблица X.I
Значения коэффициентов а.\ и а2 в уравнении Онзагера
для I—I-электролитов в различных растворителях при 25 °С
Растворитель
Вода
Метанол
Этанол
Пропанол
Бутанол
Ацетон
Пиридин
Нитрометан ....
Нитробензол . . .
а\
0,230
0,856
1,330
1,768
2,140
1,692
3,694
0,664
0,776
60,56
153,3
89,80
53,21
46,10
347,2
154,0
124,1
44,37
Растворитель
Ацетонитрил . . .
Формамид
N-Метилформамид .
Диметилформамид .
Диметилсульфоксид
Пропиленкарбон ат
Глицерин
Уксусная кислота .
N-метилпирролидон
«,
0,738
0,139
0,065
0,717
0,501
0,303
0,575
10,32
0,841
О-2
233,2
13,81
21,40
98,95
35,66
23,84
127,3
169,6
202,6
щественно влияет на электропроводность и отклонения от
уравнений (X. 4) наблюдаются уже при гораздо более низких
концентрациях, чем в воде.
Закономерности изменения электропроводности
концентрированных растворов и жидких систем описываются уравнением
(VIII. 61), выведенным для систем типа электролит — неэлектролит
в предположении отсутствия химического взаимодействия между
ними. Количественных обобщений, описывающих
электропроводность концентрированного раствора или жидкой системы с ярко
выраженным взаимодействием нет и трудно надеяться на
появление таких обобщений в ближайшем будущем. Действительно,
такое уравнение должно представлять электропроводность как
функцию ряда физических свойств компонентов раствора, равновесных
концентраций компонентов системы и продуктов взаимодействия,
энергии взаимодействия и т. д. Многие из этих величин для
неводных растворов не могут быть ни определены, ни даже оценены.
Вопрос о правильном способе подсчета концентраций раствора,
как уже отмечалось, приобретает особое значение при построении
диаграмм К — состав. Часто кривая X «аномальная» в том случае,
если считать растворителем компонент А, становится нормальной,
если за растворитель принять компонент В. Примером может
служить система AsBr3—(С2Н5)2О, изотермы в зависимости от
разбавления ф для которой представлены на рис. X. 1. Как видно из
рис. X. 1,а изотерма «аномальна», если считать растворителем
(С2Н5)2О, но становится нормальной, если за растворитель
принять AsBr3 (рис. X. 1,6).
Рассматривая наиболее распространенный тип жидких систем,
в которых электролитом является образующееся из компонентов
соединение, М. И. Усанович (1936) получил зависимость X от
состава системы во всем интервале концентрации и объяснил
причины появления так называемой «аномальной»
электропроводности.
241
Анализ причин, приводящих к аномальной электропроводности
в разбавленных растворах, был дан А. М. Сухотиным (1952) на
основании общих свойств ионных растворов без привлечения пред.
ставлений о ионных тройниках. Это предположение основано на
анализе выражения для
термодинамической константы равновесия между
ионами и ионными парами
• 10 ? ом'-1 см1- мольU
2*2
f
(Х.5)
где а — степень диссоциации ионных
пар на ионы; f+ — средний ионный
коэффициент активности.
Для случая а<С1, уравнение (Х.5)
дает:
При очень малой ионной
концентрации, когда f± » 1, изменение
степени диссоциации практически
полностью определяется стоящим в
знаменателе множителей ]/С. Это ведет
<р к уменьшению а с ростом С. При уве-
1 2 личении концентрации ионный коэф-
Рис. X. 1. Электропроводность фициент активности уменьшается, по-
в системе трехбромистый мы- ' ? лГ~п
шьяк — этиловый эфир (18 °С): этому величина произведения J±y L
а-пересчет на AsBr3; б-пересчет МОЖеТ Начать уменьшаться, ЧТО ПОВе-
?мшенн^й1?каИлЛеЙВЫА Плотникову" ДеТ К ВОЗраСТаНИЮ СТепеНИ ДИССОЦИа-
священный аКад. хэ, а. Плотникову. * fl^
Киев, 1936.J ции с ростом концентрации. Таким
образом, уравнение (X. 5)
предусматривает возможность появления максимума на кривой а—С, а
следовательно, и на изотерме электропроводности.
Для экспериментальной проверки этих предположений были
проведены исследования электропроводности ряда солей в
неводных растворителях [А. М. Сухотин, 3. Н. Тимофеева, 1959]
и сделана оценка применимости высказанных положений к
электропроводности растворов в растворителях с низкой
диэлектрической проницаемостью.
X. 2. Зависимость электропроводности
от физических свойств среды
Зависимость электропроводности от вязкости .и
диэлектрической проницаемости следует уже из уравнений (X. 4), где значения
г) и е входят в коэффициенты аи а2, .. В случае жидких систем
это влияние становится определяющим, как это следует из
уравнения (VIII. 61).
242
влияние вязкости на электропроводность
Предположение об обратно-пропорциональной зависимости
электропроводности от вязкости были высказаны еще на заре
развития исследования электролитных растворов, когда считалось,
что электропроводность однозначно связана лишь с миграцией
ионов в среде с определенной вязкостью. Именно тогда было
сформулировано широко известное правило Писаржевского —
Вальдена
Яот]о = const (X. 7)
сыгравшее весьма значительную роль в развитии теории
электролитных неводных растворов.
Влияние вязкости на удельную электропроводность к в жидких
системах было рассмотрено в разделе VIII. 2. В дополнение к
приведенному там анализу следует отметить, что учет влияния
вязкости на электропроводность в жидких системах позволяет
объяснить ряд особенностей концентрационной зависимости х. Так,
М. И. Усанович (1940) положив, что зависимость а = щ в
системе А—В с электролитным компонентом А выражается
уравнением
а = оАхп (X. 8)
где п — целое число, зависящее от природы электролита, показал,
что максимум на изотермах и будет появляться всегда при ?^~>еп.
Несмотря на очевидную приближенность этого вывода
подобное рассмотрение позволило сформулировать разбиравшееся в
гл. VIII правило о связи геометрии изотерм а с электролитными
свойствами продукта присоединения.
Зависимость электропроводности
от диэлектрической проницаемости
Зависимость электропроводности от е в разбавленных
растворах следует из уравнений (IX. 34) — (IX. 35). Действительно, эта
связь становится явной, если учесть, что электропроводность в
первом приближении зависит от ионной концентрации, а
последняя, в свою очередь, определяется величиной /Сдис. *
К числу первых количественных соотношений между X и 8
относится известное эмпирическое правило Вальдена (1913)
3
8 "Кфт1п = Const « 30 (X. 9)
/
где фт1п — разведение в точке минимума электропроводности.
Более обоснованная связь концентрации в точке минимума
Cmin кривой К с е [А. М. Ш ко дин, 1960] показывает, что поскольку
Сщш связано с /Сдис прямой пропорциональной зависимостью (см.
раздел X. 3), а /Сдис является экспоненциальной функцией от 1/г
243
[см. уравнения (IX. 30) и (IX. 39)], то Cmin должно быть также
экспоненциальной функцией от 1/е:
Уравнение (X. 10) соблюдается более точно, если разведение
выражать не в литрах, а в количестве молей растворителя,
приходящихся на 1 моль электролита; оно приложимо к растворам од-
ного электролита в растворителях
различной химической природы
(рис. Х.2).
Нахождение вида зависимости X
от е лежит в основе практически
всех теорий ионной ассоциации в
растворах. В. К. Семенченко и
Н. Бьеррум основывались на том,
что в растворах электролитов (в
концентрационных
Ю i2
1/ечо*
ДЛЯ
проводности qpmin от
AgNO3 (О) и KI (О):
i —вода при 18 °С (8=80); 2—вода при
100 °С (е=47,5); 3 — ацетонитрил; 4—
пиридин; 5 —метиламин; 6 — анилин (е=7,5).
Разведение выражено в молях
растворителях на моль электролита.
определенных
Рис. Х.2. Зависимость разведе- интервалах, обусловленных величи-
ния в точке минимума электро- ной е растворителя) возможно об-
разование ионных пар или ионных
двойников М+А~.
В. К. Семенченко (1923, 1924)
определил ионную пару как
агрегацию двух противоположно
заряженных ионов, сблизившихся в
результате действия сил кулоновского взаимодействия на
расстояние, меньшее некоторой критической величины (г0). При этом
выполняется условие равенства электростатической энергии
взаимодействия ионов и их кинетической энергии:
(X. И)
Такая связь приводит к появлению квазимолекул, в которых
ионы ведут себя как незаряженные частицы.
Главными обстоятельствами, от которых зависит ионная
ассоциация, являются концентрация раствора и диэлектрическая
проницаемость. Чем выше концентрация, тем больше вероятность
ассоциации; чем ниже е, тем больше сила кулоновского
взаимодействия.
Степень ассоциации Семенченко определяет как отношение
суммы объемов сфер радиуса rQ к объему всего раствора.
Основные предпосылки теории Бьеррума (1926, 1927)
базируются на тех же положениях, что и теория Семенченко, однако
здесь принято во внимание больцмановское распределение ионов
по энергиям.
Анализируя величину 1/у= у ^пе2, называемую радиусом
ионной сферы, Бьеррум показал, что вероятность Р нахождения
244
она на расстоянии г от заданного противоположно заряженного
11 на дается выражением
()
число ионов данного сорта), причем функция Р(г) проходит
минимум при значении
г . __£l£2fl fX 13ч
При уменьшении суммы радиусов ионов до величины /mm сте-
пень ассоциации резко увеличивается, а при г < Гщщ ионы
полностью находятся в состоянии «недиссоциированных» ионных пар
внутри сферы радиуса rmin; при г > rmin ионные пары не
образуются.
Весьма подробно рассмотрели вопрос об образовании ионных
пар в растворе Фуосс и Краус. Ими показано, что ассоциация
происходит не только в средах с низкими значениями е, но и в
растворителях с любыми (в том числе и очень высокими) значениями е.
Уравнения теории Фуосса — Крауса лежат в основе методов
расчета констант электролитической диссоциации по кондуктометри-
ческим данным (см. раздел X. 3).
Обобщая существующие теории ионной ассоциации, Н. А.
Измайлов получил соотношение между /СДИс и /(ас (см. раздел IX. 4).
Оригинальный подход к построению теории ассоциации,
основанной на статистических представлениях, был предложен
А. М. Сухотиным. Он учитывает лишь два крайних состояния
системы, которые характеризуются наличием свободных и
соприкасающихся ионов. Последнее позволило устранить затруднения с
разграничением ассоциированных и неассоциированных частиц.
Именно в результате развития этих представлений было выведено
уравнение (IX. 39).
Изолированный анализ влияния е на Я в большинстве случаев
не может быть произведен, поскольку (даже в случае
индифферентных растворителей) изменение растворителя, ведущее к
изменению е, неизбежно ведет и к изменению вязкости. Вот почему
обычно рассматривают влияние е не на Я, а на \г\.
Особенности функции ког\о
Правило Писаржевского — Вальдена (X. 7) обычно
обосновывают зависимостью, легко получаемой из закона Стокса:
Необходимо учесть, что одним из условий применения закона
Стокса является отсутствие взаимодействия между движущейся
частицей и вязкой средой. Кроме того, из уравнения (X. 14)
следует, что правило Писаржевского — Вальдена может соблюдаться
лишь в том случае, если радиус мигрирующей в электрическом
245
поле частицы остается постоянным. Совершенно очевидно, что н,.
первое, ни второе условия в растворах электролитов не собд1г|
даются: ионы образуют с молекулами растворителя более или щ
нее прочные сольваты, размер которых изменяется при переход,
от одного растворителя к другому.
Отметим, что Вальден, обосновывая правило (X. 7),
использовал экспериментальные данные по электропроводности растворов
тетраалкилзамещенных солей аммония, катионы которых из-за
большого размера сольватируются в малой степени (либо не соль^
ватируются вовсе), а изменение эффективного радиуса которых
при переходе от одного растворителя к другому относительно не-
велико.
В литературе имеется большое, количество данных, которые
указывают на несоблюдение правила Писаржевского —Вальдена
при рассмотрении поведения одного электролита в широком круге
растворителей. Поскольку эффективный радиус ионов должен за-
висеть от е растворителя (а в пределах группы растворителей,
обладающих близкой химической природой, число сольватации
можно считать неизменным), то естественно предполжить, что
произведение ЯоТ]о должно быть функцией е.
Фуосс (1959) выразил зависимость эффективного радиуса иона
г± от е эмпирическим уравнением:
г± = г0 + В-\/е (X. 15)
А. М. Шкодин (1961) предложил общее уравнение
Хопо = Л ехр (- В/г) * (X. 16)
качественное обоснование которого следует из совместного
решения уравнений типов K = f(K) и /С = *ф(1/в). Однако при этом
строгий вывод уравнения (X. 16), к сожалению, невозможен, так
как для этого требуется знать аналитическую связь Ко с
концентрацией. Как следует из раздела X. 1, в общем виде получить такую
зависимость т представляется возможным.
Можно привести ряд примеров, свидетельствующих о хорошем
выполнении уравнения (X. 16) (рис. X. 3). Анализ
экспериментального материала ло электропроводности неводных растворов
показывает, что в соответствии с выведенными предпосылками
прямолинейность lgЛ,ог|о от 1/е, вытекающая из уравнения (X. 16), так
же как и прямолинейность р/Сдис от 1/е (раздел IX. 4) соблюдается
лишь в растворителях тождественной химической природы. По-
следние характеризуются близостью энергетических характеристик
сольватационных процессов. Последнее,- например, присуще
растворам солей щелочных металлов в низших алифатических
спиртах* (рис. X.3,а). Аналогичная картина наблюдается в смешан-
* Прямолинейность функции lgXor)o— 1/е для растворов в высших
алифатических спиртах (начиная с гептилового) нарушается в соответствии с
уменьшающейся энергией сольватации [А. М. Шкодин, Л. П. Садовничая, С. Г. Росенко,
1971].
246
Hbix растворителях, изменение состава которых не ведет к измене-
нИю химических характеристик среды (рис. X. 3, б).
Существенное изменение энергии сольватационных процессов
приводит к нарушению прямолинейного хода зависимости
Рис. X. 3. Зависимости lg Яо^о от 1/е (25 °С):
a — KI в метаноле (/); этаноле (2); пропаноле (3); бутаноле {4)\ амиловом (5) и гексило-
вом (6) спиртах; б — H2SeO4 в системе бутанол —метанол; в — H2SeO4 в системе бута-
нол — нитробензол.
[Э. Л. Л е щ и н с к и й, Автореф. канд. дисс, КПИ, Киев, 1970.]
lg^oT]o — I/8- Впрочем, изменение химической однородности
растворителя ведет к меньшему нарушению прямолинейности функции
lgVno—I/8 по сравнению с функцией /СДИс — 1/е, как это можно
видеть на рис. X. 4.
40
p/f
50 60
3 Ч
у
10 15
5 10 f5
i/e.w8
Рис. X. 4. Зависимость
(1) и рКдис (2) сильных кислот от 1/е:
в системе бутанол — нитробензол; б—CF3COOH в системе бутанол —анилин;
в — H2SO4 в системе бутанол — диметиланилин. [См. ссылку к рис. X. 3J.
Таким образом, расчет Ко данного электролита по величинам
Ко в другом растворителе, исходя из правила Писаржевского —
Вальдена (прием весьма распространенный в практике физико-
химического исследования), может быть проведен лишь тогда,
когда имеется уверенность в том, что изменение растворителя не
247
приводит к изменению величины эффективного радиуса, одним из
условий чего является постоянство химической характеристики
среды. Многочисленные работы, где подобный расчет проводился
без учета отмеченных факторов, должны быть подвергнуты реви*
зии, в основу которой следует положить уравнение (X. 16).
Влияние диэлектрической проницаемости на коррегированную
удельную электропроводность а = щ было рассмотрено в
разделе III. 2.
Влияние температуры на электропроводность
Во многих руководствах по физической химии считается, что
в подавляющем большинстве случаев электропроводность
растворов растет с температурой. Остальные случаи характеризуются в
этих руководствах как «аномальные». Однако экспериментальный
материал показывает, что в достаточно широком температурном
интервале функция l = f(T) почти всегда характеризуется
максимумом *. Таким образом, растворы с отрицательным
температурным коэффициентом электропроводности должны встречаться
сравнительно нередко.
Даже полуколичественное рассмотрение закономерностей
изменения электропроводности в неводных растворах позволяет
сформулировать условия, при которых температурный коэффициент
отрицателен. Так, считая, что изменение % с температурой
обусловлено лишь уменьшением вязкости раствора, М. И. Усанович
(1939, 1941) пришел к выводу, что «температурный коэффициент
удельной электропроводности может быть отрицательным лишь
при условии, что l/r\-dif]/dT по абсолютной величине меньше
1/o-do/dT, причем последняя величина отрицательна».
Условия появления отрицательного температурного
коэффициента, учитывающие помимо изменения вязкости раствора, также
и изменение е могут быть получены при анализе уравнения
(VIII. 68, а). Действительно, в тех случаях, когда |Pn| >(pe/e—Ci),
относительный температурный коэффициент электропроводности
будет отрицателен.
Зависимость электропроводности от температуры в
разбавленных растворах может быть объяснена на основе формальной связи
/Сдис слабых электролитов с T[N. van Meurs, 1958]; для любых
растворителей справедливо соотношение
1п/СДис = ~-ф" + Л2~АгТ ' (х-17)
где Ai и Л3 > 0, тогда как Л2 — может быть положительной или
отрицательной.
* При анализе экспериментального материала, имеющегося ^-литературе,
необходимо учесть, что авторы этих работ, измеряя электросопротивление растворов
с изменением температуры и используя эти данные для расчета X, почти никогда
не вводят поправку на изменение концентрации электролита вследствие
изменения объема раствора с изменением температуры.
248
Сочетание уравнения (X. 17) с уравнениями связи между
ионными размерами и подвижностями, вязкостью растворителя и
степенью ионизации электролита, позволили Ван Меурсу придти
к соотношению между хиГ
Л,/2 + EllR А3
In и « -—y^— + Л4 - -у- Г (X. 18)
где Еа — энергия активации вязкого течения; Л4 — константа,
независящая от температуры.
Уравнение (X. 18) описывает кривую, проходящую через
максимум, т.е. позволяет объяснить наличие максимума на кривой
/0О
/О
Данные об изменении /СДИс с температурой, необходимые для
объяснения и прогноза изменения К от Г, могут быть получены из
сопоставления уравнений, которые, с одной стороны, связывают
/(Дис с Я, а, с другой, — Кдис с Т [примером уравнений последнего
типа может служить уравнение (IX. 39)].
Некоторые вопросы термодинамики и температурные
зависимости X будут рассмотрены в разделе XI. 1. Применение
температурных коэффициентов электропроводности для решения
вопросов о характере взаимодействия между компонентами раствора
было рассмотрено в разделе VIII. 2.
X. 3. Определение констант диссоциации
из данных по электропроводности
Константы диссоциации электролитов в растворах определяют
из данных по низкочастотной электропроводности в основном
двумя методами: Крауса — Брея и Фуосса — Крауса.
Закон разведения Оствальда
*дис== ЫЛо-Л) (ХЛ9)
применим только к растворам слабых электролитов в средах с
высокой 8.
В средах с низкой е сильные электролиты находятся в
ассоциированном состоянии, которое сохраняется и при больших
разбавлениях.
Краус и Брей предложили графический метод определения
констант диссоциации, используя другую форму закону
разведения Оствальда (X. 19):
(х-2°)
В этом уравнении Ктс и Ко являются постоянными величинами,
поэтому в координатах 1Д— (ХС) зависимость (X. 20) будет
передаваться уравнением прямой сугловым коэффициентом 1/Ддис • Яо.
249
Поскольку уравнение (X. 19) не учитывает возможность
ния на электропроводность образования комплексных ионов й
взаимное притяжение между ионами, находящимися на
значительных расстояниях, Фуосс и Краус предложили для определения
констант диссоциации видоизмененное уравнение:
F(z) ^ 1 [ 1 ^ CW (Х21)
^ ^0 ^дисАо ^(г)
При расчете /СДИс по методу Фуосса—Крауса сначала находят
приближенное значение Ко, по которому рассчитывают г и по
интерполяционным таблицам Фуосса находят F(z):
(X.22)
По величинам F(z) определяют степень диссоциации а и
коэффициент активности f. Построением графика в координатах
F (z) CKf2
* —F7T нах°Дят более точное значение Ко. Расчет
повторяется до тех пор, пока на графике не получится прямая линия.
По найденному значению Ко вычисляется константа
диссоциации
(Х23>
В качестве примера расчета по методу Фуосса — Крауса
можно привести расчет второго приближения при определении
константы диссоциации селеновой кислоты.
Ниже приведены исходные данные для расчета Ко и /Сдис в
системе бутанол — нитробензол с содержанием 20% вес. бутанола
(г] = 0,0165 /13, в = 27,1) [Э. Л. Лещи не кий, 1970]:
С • 104 . . . 31,62 15,81 6,324 3,794 2,530 1,138 0,7589 0,5692
К 9,950 12,39 16,42 19,25 20,75 23,73 24,50 25,52
Построением графика l/K — f(KC) определяют приближенное
значение величины предельной электропроводности; Ко = 27,77.
Для расчета z применяют выражение
z = sKfIiVKC' (X.24)
в котором 5 вычисляют по формуле Онзагера
s = сцХ0 + а2 (X. 25)
250
де щ и йч — коэффициенты*, зависящие от свойств растворителя
/6 и л) и температуры.
Для температуры 25 °С ai и а2 находят по уравнениям:
a1==163,45/88/j (X.26)
!/ (X.27)
2,0
Для выбранного растворителя а^ = 1,158; пг = 56,08, откуда
QQ 0^
Величины У КС и ЯС определяют отдельно для каждой точки;
умножение У КС на значение sK^'2 = 0,06032 приводит к
значению z—и далее из интерполяционных
таблиц—к F(z). Например, для первой точки
2=0,1070, a Fz = 0,88634.
Степень диссоциации а определяют из
отношения it~ftt'> 0Ha равна 0,4043; эф-
фективную концентрацию находят из
произведения аС : 0,4043-0,00005692 = 0,001278.
Из выражения
— lg f2 = 2р (аС)1/з (X. 28)
где 2|3 — множитель, учитывающий влияние
8 на коэффициент активности; ;он равен
2В = —т,— = 5,153, находим квадрат ко-
е'2
эффициента активности /2.
Произведение 2р(а1)!/з для выбранной
точки равно 0,1842; откуда: f2 = 0,6543.
Определяем значение F (z) /К = 0,08908 и CXf2/F(z) = 0,02322.
Вычислив таким образом значения F(z)/K и CXf2/F(z) для всех
точек, строим кривую; пересечение ее с ординатой дает
значение 1До.
Далее по Ко и по тангенсу угла наклона получаем по формуле
(X. 23) значение /Сдис.
В выбранном примере величина р/СДИс = 3,29.
Для облегчения трудоемких расчетов по уравнению Фуосса —
Онзагера может быть составлена сравнительно простая
программа, позволяющая вести расчеты на ЭВМ [В. М. Центовский,
В. С Центовская, 1971]; при этом в память машины вводятся
Данные по Я, С, е, т].
Один из методов определения /Сдис по кондуктометрическим
Данным основывается на определении концентрации,
соответствующей минимальному значению К, которое приходится на область
Умеренных концентраций.
* В общем виде эти коэффициенты входят в уравнение предельного закона
^ электропроводности (X. 4),
2 3456
Рис. X. 5. Зависимость
концентрации минимума
электропроводности Cmin
от силы кислот в
уксусной кислоте:
/-НСЮ4; 2-
3-H2S2<
dj; I-
251
Из уравнения (X. 10) и (IX. 35) следует, что lg Стщ являете,
линейной функцией р/СДИс [А. М. Ш к о д и н, 1960]. к
с (X. 25
Полуэмпирическое уравнение (X. 29) можно использовать дл,
определения р/СДИс электролитов сходной химической природы *
одном растворителе, если хотя бы для двух электролитов этогг
ряда известны величины р/СДИс, установленные независимым путем'
необходимые для определения коэффициентов тип уравнен^
(X. 29). Примером соблюдения уравнения (X. 29) может служить
рис. X. 5.
Глава XI
МЕХАНИЗМЫ ЭЛЕКТРОЛИТИЧЕСКОЙ
ДИССОЦИАЦИИ И ПЕРЕНОСА ТОКА
Изучение механизма электропроводности в неводных средах
связано прежде всего с выяснением природы носителей заряда.
Исходя из этого, можно выделить три основных типа
проводимости в растворах: ионный, протонный и электронный. При
исследовании электролитных растворов чаще всего имеют дело с первым
типом. Протонный механизм проводимости связан с передачей
протона по цепи Н-связей, причем в этой передаче участвуют
протонсодержащие ионы и молекулы растворителя. По этому
механизму могут проводить и хорошо проводящие электролиты
(например, серная кислота: к = 1-Ю"2 ом^-см-1) и плохо проводящие
(метанол, к= 1,5-10~9).
Электронный механизм проводимости наблюдается в
органических полупроводниках — в основном, ароматических
углеводородах, способных к передаче электрона по сопряженным связям.
Электропроводность, обусловленная таким механизмом, весьма
низка (10~7 —10~13 ом^-слг1). Гораздо большей проводимостью
обладают растворы, содержащие сольватированные электроны.
К ним относятся растворы щелочных металлов в жидком аммиаке,
этилендиамине, гексаметилфосфортриамиде и других аминах,
амидах и эфирах. Установлено, что подвижность сольватированного
электрона в NH3 примерно в 7 раз превышает таковую иона
щелочного металла. Сольватированные электроны могут возникать
также при облучении растворов а-, р- и улучами, а также
ультрафиолетовым излучением.
Ниже будут рассматриваться только ионный и протонный
механизмы проводимости, как наиболее распространенные в
электролитных растворах.
Исчерпывающий анализ механизма электропроводности в
растворе может быть сделан тогда, когда установлен состав ионов, а
также измерены их подвижности и концентрации. Практически это
осуществимо в настоящее время лишь для разбавленных
растворов, где применимы уравнения теории Дебая — Хюккеля—
Фуосса — Онзагера и соблюдается закон независимости движения
ионов Кольрауша. В этом случае справедливо соотношение
t
x=10-32<V^ . (XL 1)
где Ci — концентрация /-го иона, г-экв/л; U — число переноса этого
иона.
Для концентрированных растворов, особенно неводных,
возникают затруднения при определении истинной ионной концентрации
253
Сг, поскольку не удается достоверно вычислить степень диссоциа-
ции электролита. Независимые же определения Сг, например из
данных по спектрам комбинационного рассеяния^ до сих пор
крайне малочисленны. Серьезные проблемы возникают также и
при интерпретации чисел переноса в концентрированных
растворах, о чем подробнее будет сказано ниже.
Поэтому в настоящее время применительно к
концентрированным неводным растворам не может идти речь о количественном
описании изотермы электропроводности. Более того, во многих
случаях спорными являются вопросы о составе ионов и о схемах
электролитической диссоциации.
Наиболее распространенным методом изучения
электролитической диссоциации в неводных средах является электролиз.
Анализируя продукты электролиза и изменения концентрации
компонентов в приэлектродных пространствах, можно судить о направлении
электромиграции и составе ионов (разумеется, с учетом
сольватации). Наряду с этим, обработка данных об изменении состава
электролита по методу Гитторфа позволяет (с помощью
найденных таким образом кажущихся чисел переноса) оценить вклад
в суммарную электропроводность различных механизмов переноса
тока.
Для анализа механизма переноса ионов в растворах
используются также данные о зависимости ионных подвижностей от
температуры и давления. Теория, объясняющая эту зависимость,
разработана Г. Эйрингом и развита позднее Бокрисом, Хиллсом и
другими.
XI. I. Перенос и подвижность ионов
в разбавленных растворах
Если электролитный раствор в неводном растворителе в
определенном интервале концентраций подчиняется закономерностям
теории сильных электролитов, то числа переноса и, соответственно,
подвижность ионов могут быть найдены с помощью' обычных
методов: электролиза по Гитторфу, движущейся границы и э.д. с.
(Все эти методы достаточно подробно описаны в общих курсах
теоретической электрохимии.) Применение метода Гитторфа не
приводит к особым затруднениям, помимо обычных мер
предосторожности при работе с неводными растворами и выбора
подходящей аналитической методики. При использовании метода
движущейся границы часто нелегко бывает подобрать подходящий
индикаторный электролит, поэтому таким методом в неводных
растворах выполнено очень мало исследований.
В качестве примера можно сослаться на работу Кэя и др.
[Ch. H. Springer, J. F. Coetzee, R. L. Kay, 1969], где при
измерениях переноса (^зо-С5Нц)4 NC1O4 в ацетонитриле в
качестве индикаторного электролита был использован (CeHsbAsClO^
При измерениях чисел переноса методом э.д.с. необходимо подо-
254
брать обратимый электрод, устойчиво работающий в данном
растворителе, а также независимо определить коэффициенты
активности.
Экстраполяция концентрационной зависимости чисел переноса
на бесконечное разведение может быть выполнена, как и в случае
водных растворов, с помощью линейной зависимости гг(Ус) по
Кольраушу или более сложных соотношений, данных в известной
монографии Харнеда и Оуэна (1952). Найденные таким путем
значения $ позволяют вычислить ионные подвижности. При этом
необходимо определить значения ti хотя бы для одного
электролита в данном растворителе; после этого можно последовательно
подбирать электролиты, имеющие один общий ион с изученным
ранее, находить для этих электролитов Ко и затем вычислять Л*
с помощью правила Кольрауша: Яо = 2 А,?. Приведенные в табл.
XI. 1 значения Я?, указывают на отсутствие корреляции между
l| и 8 растворителя: наименьшие значения Я? обнаруживаются
в N-метилацетамиде — растворителе, значение е которого примерно
вдвое больше, чем у воды. Легко показать также с помощью этих
данных несоблюдение правила Писаржевского — Вальдена о
сохранении постоянства величины Я?т]о в различных растворителях.
Таблица XI. 1
Ионные подвижности в различных растворителях при 25 °С
( 1 2 !)
(в ом"1* см2
Ион
н+
Li+
Na+ '
к+
NH+
N(CH3)J
N(C4H9)t
СГ
Br-
Г
NOg
сю;
Пикрат
sac
• г-элв"
Растворитель
г
X
349,8
38,68
50,1
73,5
73,55
44,92
19,47
76,35
78,14
78,84
71,46
67,36
30,4
66,5
X
О
X
и
146,1
39,8
45,2
52,4
57,9
69,0
39,4
52,4
56,5
62,7
—
71,0
49,2
62,2
ю
и
59,5
17,05
20,3
23,5
19,3
29,65
19,67
21,87
23,88
27,00
—
—
—
27,4
1
со
и
88
72,8
78,4
80,6
94,5
102,5
—
105,2
115,9
ПО
120,1
117
85,3
123
о
ео
X
и
_
69,3
76,9
83,7
—
94,5
61,4
100,7
102,4
—
103,7
77,7
""—
о
ю
д
о
23
—
16,3
17,8
18,4
—
11,9
22,2
21,6
20,4
22,6
20,9
16,0
"—
X
и
О
и
X
25,0
29,9
30,8
38,Г
38,9
26,2
55,1
53,6
52,3
57,3
52,4
31,5
59,6
о
3
со
_
—
13,1
13,9
—
—
11,2
36,5
24,7
24,3
27,0
25,2
17,3
29,9
X
8
X
10,8
8,5
10,1
12,7
15,6
12,5
6,8
17,1
17,2
16,6
17,4
—
—
—~
СО
и
|?
Х%
9,2
6,6
8,1
8,2
9,5
12,0
73
11,5
12,9
14,7
14,5
16,8
12,0
16,25
256
Некоторые авторы склонны объяснить такие результаты тем
что экстраполяция данных электропроводности на бесконечное раз'
ведение не позволяет определить истинные значения Аю, свобод,
ные от искажений, вносимых явлениями сольватации и ассоциации
ионов. Другие исследователи полагают это следствием несовер.
шенства гидродинамической теории движения ионов, из которой
вытекает правило Писаржевского — Вальдена. Адаме и Лейдлер
[W. A. A d a m s, К. J. L a i d 1 е г, 1968] показали, что для
ацетоновых растворов зависимость Я?г]о от радиуса ионов проходит через
максимум, причем значение г*, отвечающее этому максимуму,
совпадает с радиусом молекулы. Исходя из этого, можно рассмотреть
различие в поведении ионов при переносе, обусловленные
различием во взаимодействии ионов с диполями растворителя: на одной
ветви кривой оказываются катионы с малыми радиусами,
склонные к сольватации, а по другую сторону максимума — большие
катионы типа алкил- и арилзамещенных катионов аммония, арсо-
ния и т. п.
Гидродинамическая теория не позволяет количественно описать
изменение произведения Я?т]о с температурой и давлением. В то
же время теория переходного состояния Эйринга,
модифицированная Бруммером и Хиллсом, позволяет предсказать зависимость Я?
от этих параметров, хорошо согласующуюся с экспериментом.
В отличие от гидродинамической модели, рассматривающей
движение иона как жесткой сферы в среде растворителя, теория
переходного состояния представляет движение иона как
совокупность скачков на расстояние / между равновесными положениями
иона в исходном и в активированном (переходном) состоянии.
Переход в активированное состояние связан с затратой энергии на
образование полости в среде растворителя и на перенос
заряженной частицы в эту полость. Подвижность иона Я? связана с
изменением парциального изобарного потенциала моля ионов при этом
переходе соотношением [S. В. В г u m m e r, G. J. H i 11 s, 1961]:
п zfeFl2 , _
^="V"ехр (- AG
Величина / может быть вычислена по данным о тепловом
расширении системы (фактически, растворителя) с помощью
уравнения
где а — коэффициент термического расширения. Используя
известные термодинамические соотношения между изобарным
потенциалом, энтальпией и энтропией обратимого процесса, можно
рассчитать раздельно изменение энтальпии и энтропии при образовании
256
переходного состояния
2 (^) 4 RT*a (XL 4)
(XI. 5)
причем AG(f в первом приближении вычисляется из графика
In Я? (1/Г), а более точно [если по зависимости а (Г) найдено /]—
из графика (inЯ?-2\пl) = f(1/Г).
Ценный материал для суждения о механизме проводимости
дают расчеты параметров процесса активации при постоянном
объеме, выполненные на основании данных о зависимости Я? от
давления. Для неводных растворов такие данные получены в
последнее время Хиллсом, Бруммером, Лейдлером и другими
исследователями.
XI. 2. Особенности экспериментального определения
переноса ионов в концентрированных неводных растворах
Величины, определяемые на практике при помощи обычных
методов измерения чисел переноса в условиях неполной
диссоциации, представляют собой по существу количества
грамм-эквивалентов данного иона, переносимые через электролит одним фарадеем
тока. Эти величины, согласно Бокрису (1962), целесообразно
называть долямц переноса тока *. Последние совпадают с числами
переноса лишь при условии, что искомый заряд связан лишь с теми
ионами вещества, перемещение которых изучается. Это условие
соблюдается лишь для полностью диссоциированных
электролитов. Если же исследовать, например, раствор CdBr2, где металл
может входить в состав катионов Cd2+, CdBr+ и анионов CdBrJ»
CdBr24~, то определить число переноса Cd2+ по изменению
концентрации кадмия в приэлектродных пространствах в этом случае
невозможно. В неводных растворах, не содержащих ионы
металлов, определение чисел переноса также осложнено сольватацией.
Для исследований концентрированных растворов наиболее
удобен метод Гитторфа. Поскольку с увеличением концентрации
вещества возрастает количество электричества, которое необходимо
пропустить через раствор, чтобы достичь ощутимых
концентрационных изменений, возникает необходимость в повышении
чувствительности метода. Для этого применяют его радиоизотопный
вариант.
* Керкер [М. Кегкег, 1957] предлагал называть эти величины стехиометри-
ческими числами переноса.
9 Зак. 500 267
В этом варианте радиоизотоп вводится только в одно (среднее)
отделение трехкамерного электролизера, разделенного пористыми
диафрагмами. Это позволяет определить изменение концентрации
компонента не по разности аналитических определений до и после
электролиза, а непосредственно по величине радиоактивности после
электролиза в приэлектродном отделении.
Удобство применения этого метода для исследования
концентрированных неводных растворов определяется еще и тем
обстоятельством, что вследствие комплексообразования в таких объектах
весьма часты случаи, когда определяемый элемент входит в состав
и катиона и аниона. При обычном анализе на основании
увеличения концентрации анализируемого элемента, например в
катодном пространстве, может быть сделан вывод, что элемент входит
только в состав катиона, хотя тот же экспериментальный результат
получается, когда элемент входит в состав и катиона и аниона,
но подвижность катиона выше. Изотопный же метод
позволяет непосредственно определить направление электромиграции
ионов.
Для количественной трактовки данных по переносу в
рассматриваемом" методе можно воспользоваться упрощенной схемой
[А. Н. Житомирский, О. К. Кудра, Ю. Я. Фи ал ков, 1962].
Согласно этой схеме, в результате электролиза Ар г-экв
исследуемого иона перемещается из одного из приэлектродных отделений
в среднее, а из среднего в другое приэлектродное отделение
переходит такое же количество этих ионов, в котором содержится
определенная доля радиоактивных атомов. Доля переноса ионов
(г-экв/фарадей) по этой схеме может быть рассчитана из
уравнения:
*± = 2РК <«/, (а)/<? ('с + 1'с- ^к (а)) (XI- 6)
где t+(t-)—доля переноса катиона (аниона), /?коо— количество
исследуемых ионов в катодной (анодной) части до электролиза
(в г-экв), /К(а) — относительная скорость счета пробы, взятой из ка-
толита (анолита); 1°с—скорость счета пробы из средней части
до начала электролиза; /с — то же после электролиза; q —
количество пропущенного через раствор электричества (фара-
деев).
Несмотря на очевидную упрощенность этой схемы, значения
долей переноса, полученные радиоизотопным методом, для
разбавленных водных растворов удовлетворительно согласуются с
данными, найденными классическими методами.
Поскольку изменение концентрации вещества находится не по
разности двух аналитических определений, а непосредственно по
величине активности в приэлектродном пространстве, описанный
метод позволяет исследовать практически любые концентрации
растворов при пропускании относительно небольших количеств
электричества,
258
XI. 3. Перенос ионов
и электролитическая диссоциация
в концентрированных растворах
с протогенными компонентами
Общие представления
о прототропном механизме проводимости
Рассматривая механизм проводимости в системах с
протогенными компонентами, следует прежде всего обратить внимание на
тот факт, что некоторые вещества способны в индивидуальном
состоянии передавать электрический ток путем перемещения
протонов по цепи Н-связей (так называемый прототропный, или
эстафетный механизм). Эти вещества характеризуются относительно
высокими значениями электропроводности и констант автопрото-
лиза. Установлено, что аномально высокие подвижности имеют
сольватированные ионы водорода в Н2О, СН3ОН, С2Н5ОН, H2SO4,
H2SeO4, H3PO4 и анионы в воде и названных кислотах, а также
H2F2. В жидком NH3, HCN, НСООН, СН3СООН и других прото-
генных веществах подвижности ионов, образующихся при авто-
протолизе, — величины того же порядка, что и подвижность других
ионов в этих растворителях.
Механизм, обуславливающий способность данного протон-
содержащего иона переносить ток по эстафетному механизму,
в настоящее время недостаточно ясен. Наиболее детально
обсуждался механизм аномальной проводимости в воде, но и в этом
случае единое мнение не выработано. В обзоре Конуэя (1967)
детально рассматриваются различные теории протонного переноса,
в которых в качестве лимитирующих стадий постулируются
следующие процессы:
1. Частота протонного перескока через потенциальный
энергетический барьер;
2. Квантово-механический туннельный проход через барьер;
3. Первичная реориентация иона и молекулы растворителя в
положения, благоприятствующие протонному переносу;
4. Поворот молекулы после переноса протона в положение,
благоприятствующее дальнейшему переносу.
Каждая из этих теорий согласуется с представлением, что
перенос по цепи Н-связей требует меньшей энергии активации,
нежели перенос ионной частицы. Интересно отметить, что в чистых
жидкостях, где обнаружен эстафетный механизм переноса (H2SO4,
Н3РО4, D3PO4, H2Se04) всегда кажущаяся энергия активации
электропроводности меньше кажущейся энергии активации
вязкого течения (£* < £а). Чистая вода, для которой £*,
вычисленная по данным Кольрауша для воды с предельно низкой
электропроводностью, равна 36,2 кдж/моль, а Е& — 16,9 кдж/моль, и
здесь составляет исключение.
9* 259
Теории, рассматривающие ориентацию ионов и молекул
растворителя в качестве лимитирующих стадий процесса, не объясняют
сохранение аномально высокой подвижности ионов с повышением
температуры, когда вероятность благоприятной для переноса
ориентации частиц должна была бы уменьшиться. Ни одна из
перечисленных теорий не дает согласующегося с экспериментом
отношения подвижностей ионов водорода и дейтерия ^h3o+/^d3o+ ~
= 14
По мнению М. И. Шахпаронова (1964), эти противоречия
устраняются, если допустить возможность существования в жидкости
свободных протонов. Используя представления Дармуа (1950)
о существовании в водных растворах протонного газа, Шахпаро-
нов рассмотрел уравнения для расчета подвижности протона
в обычной и тяжелой воде и получил отношение подвижностей
протона и дейтерона, равное 1,4, что согласуется с опытом. Из данных
о температурной зависимости е, в жидкой и твердой фазах им
был сделан вывод об отсутствии влияния температуры на
переориентацию диполей. Это позволяет объяснить упомянутое выше
сохранение аномальной подвижности при изменении температуры.
Наиболее серьезным аргументом против гипотезы протонного газа
мог бы служить общеизвестный факт высокой энергии сольватации
протонов, однако Шахпаронов считает, что это не противоречит
возможности перемещения свободных протонов, поскольку
энергия, необходимая для перемещения протона в конденсированной
фазе, гораздо меньше энергии, требующейся для его десольвата-
ции. Можно добавить к этому, что высокая энергия сольватации
протонов не противоречит и сверхбыстрому обмену протонов между
частицами в конденсированной фазе.
Теория протонных полупроводников получает в последнее время
все более широкое распространение [В. Н. М а слов, Ю. А.
Зотов, 1968], поскольку на базе этой теории удается выяснить
многие аспекты, связанные с применением растворов электролитов
в электронных устройствах («химотроника»). Однако неясно
сейчас, удастся ли с помощью этой теории объяснить, в каких
случаях и почему создаются благоприятные условия для эстафетного
переноса протона.
Анализ механизма проводимости
в концентрированных растворах
В системах с протогенными компонентами изучение долей
переноса тока позволяет установить относительный вклад ионного и
прототропного (эстафетного) механизма проводимости в общую
электропроводность.
На основании этих исследований системы, содержащие прото-
генные компоненты, могут быть разделены в соответствии с
преобладанием того или иного механизма проводимости.
260
В системах с преобладанием ионного механизма проводимости
взаимодействие между компонентами выражено, как правило,
достаточно отчетливо. Значения долей переноса ионов велики по
абсолютной величине и в сумме могут быть больше единицы
(вследствие сольватации). Возрастание суммы долей переноса ионов
связано с увеличением концентрации ионов в системе (если
подвижности существенно не различаются). В системах с
взаимодействующими компонентами состав, отвечающий максимальной
концентрации комплекса, часто отвечает ^ о
и его максимальной диссоциации.
Поэтому максимум суммы долей переноса
ионов может приходиться на стехиомет-
рический состав. Примером могут
служить системы, образованные диэтилани-
лином с уксусной и монохлоруксусной
кислотами [Ю. Я. Фиал ко в, С. Н. Хо-
лодникова, 1968].
0,8
^-> " ." " ~ •»- V») •» -' ~— j.
Если один из компонентов системы в СН3СООН
8,0
6,0
4,0
2.0
OJS
av
Рис. XI. 1. Доли переноса
тока в системе
H2Se04—CH3COOH (25 °С):
1 —t_ для HSeOjJ*; 2 — i+ для
СН3СО2н£; 3 —удельная
электропроводность; 4 — энергия
активации электропроводности.
[Ю. Я. Ф и а л к о в, А. В. Я к о-
в л е в а, 1971].
чистом виде обладает эстафетной
проводимостью, то по крайней мере на одном
из концентрационных интервалов
системы (вблизи этого компонента) сумма
долей переноса тока будет существенно
меньше единицы. Комплексообразование
и диссоциация комплекса на ионы, как и
в предыдущем случае могут приводить
к возникновению максимума на изотерме
сумм долей переноса (t+-\-tJ). Этот
случай реализуется в системах: H2SO4—
Н3РО4, H2SO4— CH2C1COOH, H2Se04—НзРО4 и H2Se04—
СН2С1СООН. Степень кислотно-основного взаимодействия в этих
системах относительно невелика.
Если же кислотно-основное взаимодействие протекает более
интенсивно, в различных концентрационных интервалах системы
отчетливо прослеживается переход от чисто эстафетного к чисто
ионному механизму проводимости. Этот случай целесообразно
рассмотреть более подробно на примере системы H2Se04 — CH3COOH
(рис. XI.1). В области, богатой уксусной кислотой (содержание
H2Se04 0,1—0,3 мол. долей), на 1 г-экв иона HSeOT,
перенесенного к аноду, приходится 2 г-экв СН3СООН2, перенесенных к
катоду (наиболее точно это соотношение соблюдается при мольном
отношении компонентов СН3СООН : H2Se04 = 2:1). На
основании этих данных можно представить схему диссоциации:
2СН3СО2Н. H2Se04 ^z± (CH3CO2H. CH3CO2H2)++ HSeOJ
С дальнейшим ростом концентрации селеновой кислоты
отношение t+/t~ уменьшается и при эквимолекулярном составе смеси
261
достигает единицы, что отвечает схеме диссоциации:
СН3СО2Н • H2Se04 ^z± CH3CO2Hj + HSeO^
Сумма долей переноса в этой точке снижается уже до 0,3, что
свидетельствует о значительном возрастании вклада эстафетного
механизма в общую проводимость. Наконец, при содержании H2Se04
0,88 мол. долей, соответствующем максимальным значениям к
(кривая 5), доля переноса аниона достигает практически нулевых
значений (значения t+ также весьма малы).
Таким образом, в области, соответствующей максимальному
значению электропроводности двойной системы, перенос тока
практически осуществляется только по эстафетному механизму.
Кривая 4 наглядно показывает, что этот процесс требует
значительно меньшей энергии активации, нежели миграция ионов (спад
кривой 4 в области, богатой уксусной кислотой, объясняется
уменьшением общей концентрации ионов).
Если оба компонента системы в индивидуальном состоянии
обладают эстафетной проводимостью, то (t+-\-t-) при любом
составе смеси значительно меньше единицы. Здесь также возможен
случай, когда кислотно-основное взаимодействие выражено
достаточно отчетливо (системы Н3РО4—H2SO4, Н3РО4 — H2Se04); при
этом ионный механизм вносит определенный вклад в общую
проводимость, и положение максимума изотермы (t+-\-L) совпадает
со стехиометрическим составом комплекса. Если же кислотно-
основное взаимодействие выражено очень слабо (система H2SO4 —
FbSeC^), то обнаруживается незначительная миграция только
одного иона (H3Se0t) на небольшом интервале концентраций, а
на большей части концентрационного интервала системы перенос
тока осуществляется практически целиком по эстафетному
механизму.
XI. 4. Механизм электролитической диссоциации
в апротонных системах
Изучению механизма электролитической диссоциации в
системах, содержащих апротонные кислоты и основания, посвящено
большое количество исследований. В подавляющем большинстве
этих работ рассматриваются системы, включающие галогениды
металлов. Отдельные работы посвящены исследованию систем, где
одним из компонентов является чистый галоген (иод, бром) или
межгалоидное соединение.
Большое количество исследования выполнено в 20—40-х гг.
киевской школой электрохимиков во главе с В. А. Плотниковым.
Для выяснения состава ионов и механизма электролитической
диссоциации в этих работах проводился электролиз исследуемых
объектов с последующим анализом электролита в приэлектродных
пространствах. В одних случаях выводы основывались на данных
262
о качественном составе продуктов электролиза, в других — на
определении чисел переноса по Гитторфу. Определение чисел переноса
во многих случаях давало величины, большие единицы. Такие
результаты были получены для растворов пятибромистого фосфора
в броме [В. С. Ф и нкел ьштей н, 1927], пиридина в броме
[В. А. Плотников, В. И. Михайловская, 1940], растворов
галогенидов металлов в межгалоидных соединениях [Я. А. Фиал-
ков, 1946, 1954]. Аномальные значения чисел переноса относились,
как правило, за счет сольватации и полимеризации.
Следует отметить, что определение состава ионов по анализу
продуктов электролиза не может считаться достоверным. Можно
для сравнения указать, что в водных растворах галогенидов ряда
металлов (кадмия, железа и др.) с несомненностью доказано
присутствие металла в комплексном анионе, и, тем не менее,
металл разряжается на катоде.
Вызывает также сомнение обоснованность схем диссоциации,
составленных по результатам анализа изменения концентрации
лишь одного из компонентов в приэлектродных пространствах.
Наконец, во многих работах электролиз производился в ячейках,
разделенных пористыми диафрагмами, причем далеко не всегда
учитывалась возможность искажений за счет электроосмотического
переноса вещества.
Не удивительно поэтому, что для одних и тех же
электролитных систем в литературе предложены противоречащие друг другу
схемы диссоциации; согласно одним — рассматриваемый элемент
входит в состав только аниона, согласно другим — в состав
катиона. Это затрудняет составление обобщенной схемы механизма
диссоциации даже для однотипных систем.
Для систем, образованных галогенидами металлов с
органическими соединениями, можно выделить, по крайней мере, четыре
основных типа схем электролитической диссоциации.
Ионизация галогенида металла. По этой схеме предполагается,
что галогенид металла диссоциирует на простые либо
комплексные ионы, причем молекула органического основания сольватирует
либо оба иона, либо только катион:
ft + aB ^=± [MeXft-mBfl]m+ + mX'
2МеХ„ + (а + Ъ) В +
Ни для одной из приведенных схем вхождение элемента-комп-
лексообразователя в состав катиона не было подтверждено прямым
экспериментом с применением меченых атомов.
Ионизация органического соединения. В соответствии с этой
схемой элемент-комплексообразователь и галоген входят только
в состав аниона, например:
МеХ„ + RCOOR' ^=± [MeX^ORT + RCO+
Такая схема была предложена М. И. Усановичем при
исследовании растворов галогенидов металлов в спиртах и карбоновых
263
кислотах. Аналогичной схемой пользовался Р. Ч. Пауль с сотруд.
никами для объяснения механизма электропроводности растворов
А1С13, FeCl3, SnCl4, TiCl4, ZrCl4, SiCl4, M0CI5, BF3 и SbCl5 в этил-
ацетате [R. С. P a u 1, К. С. M a 1 h о t г а, 1963].
Реакции сольволиза. Взаимодействие галогенидов элементов
III—VIII групп периодической системы с органическими
основаниями может протекать необратимо с отщеплением атома водорода
от органической молекулы и выделением галогеноводорода. Обычно
такие процессы протекают в системах, где в качестве
органического компонента взят спирт или кислота:
МеХ„ + aROH —► MeXrt-a(OR)a + aHX
Электропроводность системы в этом случае обусловлена
диссоциацией растворенного галогеноводорода.
Схема с образованием карбкатиона и аниона типа МеХ^-Гт.
Эта схема отличается от второй схемы тем, что образующийся
анион не содержит углеводородного радикала. Примером может
служить взаимодействие хлорида алюминия с хлористым ацетил-
хлоридом, приводящее [F. Fairbrether, 1937] к образованию
ионов СН3СО+ и А1СЦ":
СН3СОС1 + А1С13 —> СН3СО+ + А1С17
В настоящее время затруднительно, по-видимому,
сформулировать общие положения, позволяющие предпочесть ту или иную
схему в каждом конкретном случае. Вполне очевидно, однако, что
для суждения о составе ионов и механизме переноса тока
необходимо иметь данные о направлениях электролитической миграции
всех элементов, входящих в состав компонентов смеси, в возможно
более широком концентрационном интервале. Исследования,
выполненные на кафедре физической химии Киевского
политехнического института, позволяют заключить, что лучшим методом в этом
случае является радиоизотопный вариант метода Гитторфа.
Разнообразный ход изотерм долей переноса различных ионов в
изученных системах наводит на мысль, что в подавляющем большинстве
случаев механизм электролитической диссоциации в данной
конкретной системе не может быть описан одной схемой, а связан
с одновременным протеканием нескольких процессов, причем в
определенном интервале концентраций какой-то из этих процессов
может быть преобладающим.
В системах, образованных апротонными кислотами и
содержащими подвижный протон, и органическими основаниями (спирты,
карбоновые кислоты) возможен эстафетный механизм передачи тока
протоном- по цепи Н-связей. Некоторые авторы допускают
возможность передачи тока по эстафетному механизму и для чисто
апротонных систем, вдоль полимерной цепи атомов галоида. Гут-
манн и Гиммль [V. Gutmann, R. Himml, 1955] провели
электролиз по Гитгорфу смесей хлорокиси фосфора с А1С13 и SbCb-
264
Для ионов А1С1Г и SbC17 найдены аномально низкие числа
переноса, соответственно, 0,04 и 0,05. Аномально высокое число
переноса иона POClt, по их мнению, можно объяснить переносом С1~
от РОС13 к POClg по механизму, аналогичному передаче
протона по цепи Н-связей. Такое же предположение было
использовано Циглером [К. Ziegler, H. Lehmkuhl, 1956] для
объяснения аномально высокой проводимости металлоорганического
комплекса NaF-2Al(C2H5)3, а также Е. Я. Горенбейном и-А. Е. Го-
ренбейном (1967) при обсуждении данных по проводимости PBrs
в жидком броме. Однако физическая картина такого процесса
еще не разработана на том уровне, на каком это было сделано
для переноса протонов.
ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ
В НЕВОДНЫХ РАСТВОРАХ
Глава XII
ЭЛЕКТРОДНОЕ РАВНОВЕСИЕ
Величина э.д. с. цепи связана с состоянием электролитов в
растворе, поэтому измерение электродных потенциалов и э. д. с. как
функции природы растворителя либо состава смешанного раствора
применяют при исследовании свойств электролитов. Широкое
применение также находит измерение э.д.с. в аналитической практике.
В этой главе будет рассмотрена зависимость э.д.с. и
электродных потенциалов от свойств растворителя и растворенного
электролита, поскольку интенсивное развитие электрохимического синтеза,
гальванотехники и других отраслей электрохимии вызывает
необходимость определений электродных потенциалов в различных
(в том числе и неводных) средах. Кроме того, данные по
электродным потенциалам в неводных растворителях позволяют проследить
изменение энергии ионов с изменением свойств растворителя и тем
самым получать ценные сведения о состоянии электролитов в
растворах.
XII. 1. Природа электродных потенциалов
Проблема возникновения электродных потенциалов и э. д. с.
относится к числу важнейших в современной теоретической
электрохимии. Одним из основных вопросов при этом является влияние
свойств растворителя и состояние растворенного электролита на
величины электродных потенциалов.
Если рассматривать общий случай электрохимической цепи
(рис. XII. 1, а) с двумя неодинаковыми металлическими
электродами Mei и Мег, погруженными в различные растворы
электролитов Li и L2, то ее э. д. с. (Е) представляет собою алгебраическую
сумму отдельных скачков потенциалов ср на межфазных границах:
Е =
В цепи (рис. XII. 1,6), составленной из исследуемого электрода
и некоторого выбранного электрода сравнения, разность
потенциалов между концами металла Me называется электродным потен-
266
циалом. Как и э.д. с, электродный потенциал, представляющий
собой сумму скачков потенциала ф на границах раздела
соответствующих фаз, обозначается через Е.
Следовательно, для выяснения роли растворителя в процессе
возникновения э.д. с. и электродных потенциалов необходимо про-
Металл
Ме2
Металл
МеА
Раствор
Раствор
L2
Металл
Ме2
6
Ме2
Ме1
Электролит
ЗлентроЬ
сравнения
в
Ме2
М&1
L
Ме2
Ме2
Рис. XII. 1. Схемы электрохимических цепей.
анализировать выражение (XII. 1) и установить связь отдельных
скачков потенциала со свойствами металлов и растворов
электролитов.
Контактный потенциал
Контактный потенциал на границе двух металлов вызывается
тем,^ что при их соприкосновении часть электронов одного металла
с большим запасом свободной энергии переходит к другому
металлу, в котором этот запас меньше. Если предположить, что до
соприкосновения уровень Ферми в первом металле лежит выше,
чем во втором, то при их контакте электроны с самых высоких
уровней в первом металле станут переходить на более низкие
свободные уровни второго металла. Такой процесс будет
продолжаться до тех пор, пока общая энергия в обоих металлах не
уравняется.
В статистической физике доказывается, что условием
равновесия между соприкасающимися металлами является
расположение уровней Ферми на одной высоте. При наличии между
металлическими фазами электронного равновесия, справедливо
термодинамическое условие равновесия: равенство
электрохимических потенциалов jl электронов в обоих металлах Mei и Ме2
(XII. 2)
267
Возникший при установлении динамического равновесия скачок
потенциалов определяется разностью свободных энергий, которые
необходимо дополнительно сообщить находящимся на уровнях
Ферми электронам для выхода их через поверхность металлов
в окружающую среду, например, в вакуум. Эта разность
потенциалов устанавливается между точками, лежащими вне металлов
в непосредственной близости к их поверхности, и называется
внешней контактной разностью потенциалов или вольта-потенциалом.
Величина вольта-потенциала ФМв1ме2 определяется разностью
работ выхода электронов из каждого металла в вакуум со^е
. 3)
и колеблется для различных пар металлов от десятых долей до
нескольких вольт.
Поскольку величина со^е доступна непосредственному
определению, контактный потенциал между двумя металлами можно
рассчитать по формуле (XII. 3), пользуясь табличными данными
для работ выхода электронов из различных металлов. Например,
для пары медь — цинк она равна 0,3 в.
Таким образом, скачок потенциала между двумя металлами
зависит не только от природы обоих металлов; зачастую он
составляет заметный вклад в общую величину э.д.с. обратимых
электрохимических цепей.
Жидкостный потенциал
Жидкостный потенциал qpLL возникает в пограничной зоне
между двумя разными соприкасающимися электролитами. При
измерении э.д.с. такая ситуация возникает, когда измерительная
ячейка содержит два раствора.
Здесь следует выделить два возможных случая. Если
электролиты растворены в несмешивающихся между собой растворителях,
то возникает фазовый жидкостный потенциал. Тогда же, когда
имеются растворы двух различных электролитов в одном
растворителе, либо эти растворы отличаются только концентрацией
электролита, возникающий при этом потенциал называется
диффузионным щ.
Причина появления скачка потенциала на границе раздела
растворов кроется в неодинаковой подвижности или различии
коэффициентов диффузии ионов электролита. Если на границе
растворов имеется градиент активностей (в разбавленных
растворах— концентрации), то это порождает диффузию. Подвижности
ионов зависят от особенностей их сольватации молекулами
различных растворителей, т.е. определяется свойствами иона и
растворителя.
268
При отсутствии внешнего тока в электрохимической цепи
разность потенциалов на границе растворов принимает стационарное
значение, соответствующее равенству скоростей перехода через
границу раздела положительных и отрицательных ионов.
Результирующий ток на границе раздела становится равным нулю, а
разность потенциалов будет соответствовать величине щ.
Такой квазиравновесный процесс допускает трактовку
диффузионного потенциала в рамках термодинамической теории, что
приводит к следующему общему выражению для величины щ между
двумя растворами
и
JS^1"^ (XIL4)
где t{ — доля тока, переносимая данным сортом ионов: й\ —
активности отдельных ионов.
Уравнение (XII. 4) содержит термодинамически неопределимые
активности отдельных ионов аи поэтому вычисление величины щ
возможно на основе определенных упрощающих предположений
о строении переходного слоя, а также о характере изменений в нем
активностей и подвижностей ионов. Оценка диффузионного
потенциала для простейших электролитов может быть сделана с
помощью приближенных уравнений Гендерсона, Льюиса, Сарджента
и др.
Существует метод, позволяющий настолько снизить величину
cpd, что им можно практически пренебречь. Для уменьшения
диффузионных потенциалов электролиты обоих электродов соединяют
через так называемые «солевые мостики» или электролитические
ключи, заполненные раствором электролита, состоящим из ионов
с близкими подвижностями.
Диффузионный потенциал может быть полностью устранен
путем использования электрохимической ячейки, содержащей только
один электролит (рис. XII. 1,в). Тогда из выражения (XII. 1а) для
э. д. с. и электродного потенциала выпадает слагаемое <pLiL2.#
Е =
Если в практических условиях во многих случаях величинами
диффузионных потенциалов можно пренебречь, то при выяснении
влияния природы растворителя на стандартные электродные
потенциалы межжидкостной потенциал должен быть обязательно учтен.
Потенциал на границе металл — раствор
N
Впервые теоретические представления о важной роли
растворителя в процессах возникновения электродных потенциалов были
высказаны в 1912—1914 гг. Л. В. Писаржевским. В соответствии
с его воззрениями переход ионов на границе металл — раствор
269
совершается за счет взаимодействия металла с молекулами
растворителя. Для объяснения природы электролитической упругости
растворения металлического электрода Л. В. Писаржевским была
выдвинута гипотеза о наличии в металле диссоциации атомов на
ионы и электроны, степень диссоциации при этом должна зависеть
от свойств атомов структуры кристаллической решетки.
Согласно предложенной схеме, возникновение скачка
потенциала на границе металл — раствор обусловлено двумя
процессами. Первый из них, определяемый природой металлического
состояния, сводится к ионизации металла электрода; второй
процесс— это взаимодействие растворителя S с ионами металла
(сольватация). Общая реакция, обуславливающая появление
скачка потенциала на границе металл — раствор, по Л. В. Писар-
жевскому, может быть записана в следующем виде:
Me + *S ^Z± №Z+SX + ze (XII. 5)
Процесс (XII. 5) подчиняется закону действующих масс и
характеризуется константой равновесия, зависящей от свойств
металла и растворителя. Таким образом, переход ионов из металла
в раствор совершается в результате взаимодействия его с
молекулами растворителя с образованием сольватированных ионов,
свойства которых зависят от природы растворителя. Это позволило
объяснить не только причину возникновения скачка потенциала на
границе металл — раствор, но и подойти к вопросу о влиянии
растворителя на электродные потенциалы. В частности, Писаржевский
подчеркивал, что величина электродного потенциала должна
определяться суммой некоторой постоянной величины, зависящей от
свойства металла и величины, изменяющейся при переходе от
одного растворителя к другому.
Здесь необходимо отметить, что работы Писаржевскогсг по
теории возникновения электродных потенциалов представляли собой
первую попытку конкретной физико-химической интерпретации
процессов на электродах с точки зрения электронных
представлений и в этом их большое значение для электрохимии. Идеи Писар-
жевского нашли дальнейшее развитие в двух направлениях. Одно
из них характеризовалось термодинамическим подходом к
описанию процесса возникновения электродных потенциалов (Н. А. Из-
гарышев, А. И. Бродский, В. А. Плесков, Н. А. Измайлов, Н. Е.
Хомутов) .
Другое направление привело к созданию кинетической теории
электродных потенциалов. В 1924 г. Батлер для описания
процессов перехода ионов на границе металл — раствор предложил метод
потенциальных кривых. Используя основное условие равновесия —
равенство скоростей перехода ионов из металла в раствор и
обратно,—Батлер вывел уравнение, в котором потенциал электрода
связывался с потенциальными энергиями иона в металле ^ме и
растворе Wl (рис. XII. 2). Но поскольку в работах Батлера не
270
были предложены методы расчета потенциальных энергий ионов,
этот подход нельзя было использовать для непосредственного
вычисления электродных потенциалов.
В 1932 г. Герни применил кинетический подход к расчету
равновесия на границе металл — раствор, использовав метод
потенциальных кривых Батлера. Герни использовал также законы
квантовой статистики для расчета распределений ионов по
энергетическим уровням в металле и в растворе. Из кинетических
условий равновесия, т. е. равенства скоростей перехода иона в металл
и в обратном направлении, было выведено уравнение для
электродного потенциала:
Tf
(XII. 6)
Уравнение (XII. 6) формально совпадает с уравнением Нернста,
но оно раскрывает физический смысл электродного потенциала.
При яМе2+=1
zF
(XII. 6а)
Разностью WL — №ме
учитывается изменение
электродного потенциала,
вследствие зависимости энергии
сольватации иона от свойств
растворителя.
Для вольта-потенциала
на границе металл—
раствор уравнение (XII. 6а) может быть записано в виде [Л. И.
Антропов, 1946]:
-Граничный слой '
Металл Me Раствор S
Рис. XII. 2. Потенциальный барьер на
границе между металлом и раствором.
— со
Ме
zF
(XII. 66)
Здесь с величиной фьме связаны работы выхода иона в газовую
фазу из металла и из раствора.
Величины, входящие в уравнение (XII. 66) имеют вполне
определенный физический смысл.
Пользуясь так называемым методом цикла, процессы, которые
происходят на границе раздела металлический электрод — раствор
электролита, можно представить в виде ряда отдельных стадий и
попытаться вычислить энергетический эффект каждой из них. Для
электрода «металл — ионы металла в растворе» величина
электродного потенциала будет определяться суммой энергетических
эффектов трех стадий: сублимация металла; ионизация газообразного
атомного металла и сольватация ионов металла молекулами
271
растворителя. Эти процессы можно изобразить в виде следующего
цикла
(Me)+2I
При непосредственном переводе 1 г-ион металла из металла
в вакуум затрачивается энергия, равная работе выхода со^|2+-
Осуществить такой мысленный эксперимент можно также и в две
стадии: сперва сублимировать 1 г-ат металла, затратив на это
энергию сублимации £Суб, а затем ионизовать газообразные атомы
металла в ионы с валентностью г. Расход энергии при этом равен
сумме потенциалов ионизации 2 /• Освобожденное количество
электронов г возвращается в металл с выделением «химической
энергии», равной работе выхода электрона из металла со^е.
Следовательно, работу выхода иона в вакуум можно выразить
следующим образом
причем все входящие в уравнение величины могут быть определены
экспериментально. В представленном цикле ионы металла
переводятся в электролит с освобождением энергии сольватации £С0Льв.
Следует отметить, что в уравнении (XII. 7) только величина
энергии сольватации зависит от растворителя.
Величина со^е2+ в уравнении (XII. 66) представляет собой
реальную энергию сольватации Лр, взятую с обратным знаком
„М.6 __ Л /VTT О\
©L «- Лр (XII. 8)
но поскольку катионы металла при переходе в раствор не
пересекают границу раздела вакуум — жидкость, то фх,ме в различных
растворителях определяется лишь химической энергией
сольватации Ах, а не реальной энергией сольватации.
XII. 2. Зависимость э. д. с, электрохимических цепей
от свойств растворителя и природы электролита
Из рассмотрения природы возникновения всех скачков
потенциалов, из которых складывается э. д. с. электрохимической цепи и
электродного потенциала [уравнение (XII. 1а)], следует, что qpMeiMe2
определяется только природой металла, в то время как величины
Фьме зависят еще и от свойств растворителя^
272
В 1947 г. В. А. Плесковым была выведена количественная
зависимость величины электродного потенциала от свойств
растворителя и электрода. Основной предпосылкой при этом было
положение о том, что в ряде случаев влияние растворителя сказывается
не менее существенно, чем зависимость потенциала от природы
самого электрода. Электродный потенциал при этом определяется
величиной изменения свободной энергии перехода иона из электрода
в раствор, следовательно, он зависит от величины энергии
сольватации.
Наибольший вклад в разработку теории электродных
потенциалов в неводных растворах внесен работами Н. А. Измайлова,
основывавшихся на теории А. Н. Фрумкина, согласно которой э. д. с.
цепи без переноса состоит из разности скачков потенциалов на
границах двух металлов с раствором и из вольта-потенциалов между
металлами.
Уравнение Измайлова для э. д. с.
различных электрохимических цепей
При выводе основного уравнения зависимости э.д. с. цепи от
свойств металла и растворителя Н. А. Измайлов воспользовался
уравнением (XII. 1а) для э.д.с, которое справедливо для цепи
Mej | Mef" 11 Mef+ | Me2 (XII. 9)
без жидкостной границы или с элиминированным диффузионным
потенциалом. Вольта-потенциал на границе контакта двух
металлов определяется уравнением (XII. 3). Вольта-потенциалы на
границе металл — раствор для металлов Mei и Ме2 запишутся с
учетом уравнений (XII. 7) и (XII. 8):
--ЛР1 - Есубх - S 7i
^р
■ гейм*
^ (XII. 11)
Подставляя в уравнение (XII. 1а) все вольта-потенциалы, а
также полагая, что активности участников реакции равны 1,
получаем уравнение для стандартной величины э.д. с. Е°
гальванической цепи (XII. 9):
е Ме2
е2+
Ме2 °>
. °>Ме2 Лр2 . ®Мех \ (уи
1 jp 1 jp (All.
Учитывая, что Лр = Ах ± zF%L 0 (%ь о — скачок потенциала на
границе раствор — вакуум), уравнение для Е° можно переписать
в следующем виде
g +
273
где первое слагаемое определяется только свойствами электродов,
а второе — свойствами потенииалопределяющих ионов и природой
растворителя.
Поскольку для данной цепи из двух определенных металлов
величины энергий сублимации и ионизации не зависят от природы
растворителя и (для стандартных условий) от концентрации
раствора, уравнение (XII. 13) может быть представлено в общем виде
yj д
Е° = const + Xl Хг (XII. 14)
откуда следует, что э. д. с. цепей и стандартные электродные
потенциалы зависят от природы растворителя.
Если уравнение (XII. 13) привести к виду
ЕО = Дсу^+ал+х _ £СУб2+2/2+л,2 (XIL 15)
то становится ясно, что э.д. с. цепи является алгебраической
суммой, каждый член которой относится к одному из электродов,
образующих цепь. При этом полное изменение энергии для каждого
из металлов определяется энергией превращения металла в ион
и энергией сольватации иона металла.
Запишем гальваническую цепь с переносом, включающую
водородный электрод, следующим образом:
Pt(H2)| H+ ||Mez+|Me (XII. 16)
Пренебрегая диффузионным потенциалом, к ,цепи (XII. 16)
можно применить видоизмененное уравнение (XII. 1а)
Е° = ФМеР! + <Pl Me "" 4>L Pt (XIL 16)
В такой системе (с учетом газообразной природы водорода)
величина со"* может быть найдена из следующего цикла
(Н) < т Н2
-"а
1, на
адс
где приняты следующие обозначения:
DHz —энергия диссоциации молекулы водорода; £/адс — энергия
адсорбции атомов водорода на Pt; /H+ — потенциал ионизации
атомов водорода; co£t — работа выхода электрона из платины.
Из цикла следует, что
<=yOH2+/H+-cDePt (XII. 17)
274
Подставляя граничные скачки вольта-потенциалов в уравнение
(ХП- 16) и выполняя соответствующие преобразования, получим
выражение для э. д. с. элемента (XII. 16)
Р = — : -F " — + гр т (XII. 18)
или
Е° = const + Н zF MeZ+ (XII. 19)
Уравнение (XII. 19) можно использовать для подсчетов
разности энергий сольватации ионов водорода и ионов металлов.
Если цепь без переноса содержит электрод второго рода,
например
Pt(H2)|H+r-|| Agr|Ag (XII. 20)
то уравнение для э. д. с. в этом случае запишется следующим
образом:
И1-ад
Изменение энергии при растворении в насыщенном растворе
равно нулю, поэтому можно записать
Ах +АХ =АХ (XII. 22)
Ag+ Г" АдГ
откуда
Ах =АХ —Ах (XII. 22а)
Ag+ Agr г-
Тогда выражение для э. д. с. элемента (XII. 20) будет:
_ + -
(XII. 23)
Для цепи без переноса типа
Pt (H2) | НГ | (Г2) Pt (XII. 24)
в которую входит галоидный газовый электрод, уравнение для Е°
записывается в виде
(XII.25)
275
Для цепи Na|NaCl||AgCl|Ag выражение для Е° можно
представить в виде:
_ ( У)( у Л8С1)
£ _ 1
F
(XII. 26)
Из полученных уравнений видно, что для всех гальванических
элементов с электродами, обратимыми относительно катиона или
аниона, выражение э. д. с. можно представить в общем виде
£° = const + J^p± (XII. 27)
т. е. стандартная э. д. с. цепи без переноса определяется двумя
слагаемыми, первое из которых зависит только от свойств металла и
соли, второе — зависит от свойств растворителя и ионов, точнее,
от суммы энергии сольватации катиона и аниона.
Влияние диэлектрической проницаемости среды
на величины э. д. с. цепей и условные электродные потенциалы
Приведенные выше уравнения для э. д. с. цепей и электродных
потенциалов могут быть использованы для объяснения зависимости
Е° электрохимических систем от свойств растворителя. В этом
случае с изменением природы растворителя изменяется только второе
слагаемое уравнения (XII. 27). Получаемые таким образом э.д. с.
и электродные потенциалы могут быть названы условными
величинами Е°, поскольку в уравнении (XII. 1 а) при переходе от
одного растворителя к другому будут изменяться оба скачка
потенциала фьме на границе металл — раствор.
Если рассматривать раствор с активностью растворенного
вещества, равной единице, то химическая свободная энергия
сольватации ионов молекулами растворителя приближенно может быть
описана выражением
z2e2Nл tioiiezN л
л (1-1/е) + — - (XII. 28)
х- 2г w ч~, г r
где ns — число молекул растворителя, сольватирующих ион; \х —
дипольный момент молекул растворителя; г — радиус сольватиро-
ванного иона.
Первое слагаемое этого выражения учитывает борновские
вклады в энергию сольватации, а второе суммирует энергию всех
видов взаимодействия ионов с дипольными молекулами
растворителя. Поэтому величина э.д. с. цепи и стандартного электродного
потенциала одной и той же цепи без переноса должна быть
линейной функцией от 1/е растворителя; этот вывод справедлив с
точностью, с которой можно считать, что ионные радиусы не
изменяются при переходе от одного растворителя к другому.
276
Таблица ХП.1
Стандартные э. д. с. цепей Li(Hg)^| LiCl | AgCl, Ag и Na(Hg)y | Nal J Agl> Ag
в смешанном растворителе метанол—дециловый спирт при 25 °С
Вес. доля
C10H21OH
0,10
0,20
0,30
0,40
0,50
8
30,0
27,0
25,1
22,5
19,9
£LiCl' в
2,0520
2,0260
2,0120
1,9825
1,9600
%al' в
1,5060
1,4900
1,4660
1,4550
1,4250
Вес. доля
С10Н21ОН
0,60
0,70
0,80
0,90
0,95
8
17,7
15,3
12,7
10,4
9,6
Еис\> в
1,9080
1,8750
1,8200
1,7560
1,6950
о
ENaV в
1,4000
1,3575
1,3000
1,2380
1,1860
Таблица XII.2
Стандартные э. д. с. цепей Li(Hg)^ | УС11 AgCl, Ag
и Na(Hg)jc I Nal | Agl, Ag в некоторых спиртах при 25 °С
Спирт
СН3ОН
С2Н5ОН
С3Н7ОН
С4Н9ОН
изо-СвНцОН
^LiCl' в
2,040
1,9105
1,8525
1,7930
1,7280
ENaI' в
1,5690
1,4230
1,3065
1,2460
Спирт
с6н13он
с7н15он
с8н17он
С10Н21ОН
о
£ЫС1' в
1,6940
1,6795
1,6640
1,6300
о
%аГ в
1,1501
1,0902
1,0400
0,9480
Имеющиеся литературные данные (табл. ХП.1 и XII. 2)
показывают, что э.д. с. и электродный потенциал тем выше, чем выше е,
но величины условных стандартных электродных потенциалов не
являются линейной функцией от 1/е. Выведенная Бродским (1927)
теоретически — на основе анализа уравнения Борна — линейная
зависимость Е° от 1/е строго соблюдается, по-видимому, лишь
в узком интервале е и в растворителях с близкой химической
природой. Так, в смешанном растворителе этанол —вода, где
изменение состава растворителя сопряжено с существенным изменением
химической характеристики среды, рассматриваемая зависимость
нелинейна (рис. XII. 3).
Исследования зависимости Е° от 1/е растворителя в ряду
растворителей одинаковой химической природы (спирты жирного
ряда), данные по которым приведены в табл. XII. 2, показали, что
отклонение от линейности начинается в области е= 14—12. Эго
связывается с изменением молекулярных свойств и структурных
различий самих спиртов при переходе от низших спиртов к высшим
и, прежде всего, поляризуемости.
В табл. XII. 1 приведены данные по стандартным потенциалам
в смешанном растворителе метанол — дециловый спирт. Из
таблицы следует, что на зависимости Е°—1/е излом приходится на
соотношение компонентов смешанного растворителя ~ 1 :2.
Приведенные данные показывают, что при интерпретации результатов
277
влияния растворителей на величины э. д. с. необходимо кроме их
природы учитывать также и структурные факторы.
Зависимость э.д. с. цепей без переноса, содержащих водород,
ные электроды от 1/е, нелинейна вследствие особого механизма
сольватации иона водорода и большой энергии этого процесса.
Нелинейность в цепи Pt(H2) | НС11 AgCl, Ag подтверждается данными
для некоторых спиртов (рис. XII. 3,6).
а д
*.*
0.091
0/7
««■V >
0,13
4"
рСН3ОН
7Н15ОН
ofii o,O2 0,03i/e
Рис. XII. 3. Зависимость Е от 1/е.
a — /—AgCl I AgBr; 2-Hg2Cl2| Hg2Br2; в смесях воды с этанолом;
б- Pt(H2) | НС1 | AgCl, Ag в ряду спиртов.
Такие результаты следует считать вполне закономерными,
поскольку борновским слагаемым уравнения (XII. 28) не
исчерпывается вся энергия сольватации, которая включает в себя также
специфические силы взаимодействия. Кроме того, величина е,
используемая в уравнении Борна, относится ко всему объему
растворителя, тогда как е вблизи иона может принимать совершенно
другое значение; радиусы ионов также изменяются причререходе от
растворителя к растворителю.
XII. 3. Электродный потенциал
и химическая природа растворителя
Непосредственное сравнение потенциалов одного и того же
электрода в различных растворителях наталкивается на
существенные трудности, поскольку любая гальваническая цепь из двух
одинаковых электродов с различными растворителями должна
иметь границу раздела между двумя жидкостями.
В настоящее время не существует достаточно надежного метода
для экспериментального определения скачков потенциала на
границе двух жидкостей. Для теоретического расчета с помощью
приближенных уравнений типа (XII. 4) необходимо располагать
278
такими данными, как активность ионов и коэффициенты
распределения ионов между двумя растворителями, достоверные методы
определения которых, как правило, отсутствуют.
Проблема выбора электрода сравнения
Для сравнения электродных потенциалов в различных
растворителях нужно относить все потенциалы к одному электроду
сравнения. В водных растворах последним обычно является водородный
электрод, стандартный потенциал которого в воде условно принят
за нуль при всех температурах. Такой выбор не может считаться
удачным для неводных сред, поскольку протон характеризуется
весьма специфическим взаимодействием с растворителем и
потенциал водородного электрода будет наиболее сильно изменяться при
переходе от одного растворителя к другому. Кроме того, возникает
дополнительная трудность, заключающаяся в необходимости
оперировать с активностью ионов Н+ в неводных растворах, значения
которой во многих растворителях оценить можно лишь с большей
или меньшей степенью неопределенности (см. раздел IX. 6).
Наиболее правильным было бы отнесение всех электродных
потенциалов к такому электроду сравнения, для которого энергия
сольватации ионов была бы одинаковой или изменялась весьма
незначительно для различных растворителей. Очевидно, что такие
условия могут быть реализованы в тех случаях, когда энергия
сольватации ионов будет наименьшей.
Существует несколько способов преодоления трудностей,
возникающих при сравнении электродных потенциалов в различных
растворителях.
В качестве одного из вариантов решения проблемы
стандартизации электродных потенциалов В. А. Плесковым был предложен
относительно простой метод выбора подходящего иона сравнения
для вспомогательного электрода, стандартный потенциал которого
не зависит от растворителя. Идеальный ион сравнения должен
иметь в любом растворителе одинаковую свободную энергию
сольватации и, следовательно, свободную энергию переноса, равную
нулю. Такой ион, разумеется, не существует, поэтому, согласно
В. А. Плескову, наилучшим приближением реального иона
к идеальному иону сравнения является то, для которого
соблюдаются следующие условия: свободная энергия сольватации в
любом растворителе по возможности минимальна; отсутствует
специфическое взаимодействие (например, сильное комплексообразо-
вание) с растворителем; ион слабо поляризуем.
Поскольку лучше всего такой совокупности требований
отвечал ион рубидия, В. А. Плесков предложил, чтобы в качестве
независящего от свойств растворителя электрода был принят
рубидиевый электрод сравнения. Иными словами, в первом
приближении, свободная энергия переноса иона Rb+ должна быть близка
к нулю. (Ион Cs+ не мог быть выбран Плесковым в качестве
279
электрода сравнения, так как <в то время было недостаточно на*
дежных данных о свойствах этого электрода. Полученные в
последние годы результаты показали, что при сопоставлении свойств
рубидиевого и цезиевого электродов разница между их
стандартными потенциалами очень мало зависит от свойств растворителя).
При использовании метода Плескова следует учесть, что даже
в случае отсутствия значительной сольватации энергия переноса
иона будет зависеть от диэлектрической проницаемости обоих
растворителей.
Если, пользуясь приближением Плескова, построить ряд
напряжений металлов относительно рубидиевого электрода сравнения,
то выполняются все качественные предсказания о свободной
энергии переноса других ионов из одного растворителя в другой. Так,
найдено, что водородный электрод имеет более отрицательный
потенциал в основных растворителях, но более положительный -—
в кислых.
Такой подход позволил привести в общую систему
значительный экспериментальный материал по электродным потенциалам
в различных растворителях, поэтому метод Плескова следует
рассматривать в качестве, по крайней мере, первого шага к
разрешению проблемы.
Действительно, при учете только электростатического
взаимодействия иона Rb+ с молекулами растворителя, характеризующихся
различными е, можно ожидать лишь незначительную энергию
переноса. Произвести же полный теоретический расчет величины
энергии переноса иона с помощью существующих теорий сольватации
трудно, так как многие физические явления в растворах могут быть
оценены только приблизительно (например, диэлектрическое
насыщение, электрострикция, изменение структуры растворителя вблизи
ионов).
Работами Штрелова (Н. Strehlow, 1952, 1958, 1962] было
показано, что среди окислительно-восстановительных систем можно
подобрать значительно более подходящий электрод сравнения, чем
рубидиевый. Причем такой электрод будет удобен как в
экспериментальном отношении, так и с точки зрения учета его сольвата-
ционных характеристик. Было исследовано значительное
количество окислительно-восстановительных систем, из которых выбраны
системы: ферроцен-феррициниевый и кобальтоцен-кобальтициние-
вый ионы.
Хотя кобальтоцен до некоторой степени нестабилен во всех
растворителях, ионы его почти обратимо восстанавливаются на
ртутно-капельном электроде. Это позволяет удобно сравнивать
ряды напряжений в различных растворителях путем
полярографического измерения потенциала полуволны кобальтоцена.
Разница между E°RedOx для °беих указанных систем равна
1,33 в и в пределах точности эксперимента не зависит от раствори;
теля. В исследованных растворителях оказалась очень стабильной
также разница Я;ерроцен - £°Rb+<
280
Метод Н. А. Измайлова основан на возможности расчета или
измерения энергии сольватации отдельных ионов, в том числе и
протонов в различных растворителях. Такой подход исключает
необходимость выбора единого электрода сравнения, поскольку
изменение потенциала любого электрода при переходе из одной среды
в ДРУГУЮ может быть рассчитано, если принять потенциал
нормального водородного электрода в воде равным нулю.
Подсчет энергии сольватации протонов и других ионов обычно
проводится на основании измерения э. д. с. соответствующих
электрохимических цепей. Данные по энергии сольватации протона
в различных растворителях позволяют определять разность теплот
сольватации при переносе протона между любой парой
растворителей и вычислить разницу в стандартных потенциалах
водородного электрода в изучаемых системах.
Используя значения Е°н+ j Hj> можно создать единую шкалу
потенциалов, отнесенных к потенциалу водородного электрода,
значение которого для водного раствора принимается за стандартное
и полагается равным нулю. Такой метод позволяет составлять ряды
напряжений и выявить влияние растворителя на электродные
потенциалы.
Электрохимические ряды металлов
в неводных растворителях
Наиболее подробные исследования электродных потенциалов
в неводных растворителях принадлежат В. А. Плескову. Сводка
надежных данных, составленная В. А. Плесковым в 1947 г., лишь
с очень небольшими дополнениями и изменениями воспроизведена
в табл. XII. 3.
Из табл. XII.3 следует, что при расположении электродов враз-
личных растворителях в порядке возрастания их электродных
потенциалов образуются ряды, в которых места отдельных
электродов зависят от природы растворителя.
Потенциалы металлов в спиртах мало отличаются от их
потенциалов в воде. В сильноосновных жидком аммиаке и гидразине
потенциалы щелочных металлов изменяются сравнительно мало, а
ярко выраженная склонность аминов к присоединению протона
сдвигает потенциал водородного электрода в отрицательную
сторону относительно их значений в водных растворах.
Действительно, в аммиаке и в гидразине потенциал водородного электрода
отрицательнее, чем даже в воде, которая также проявляет
значительное сродство к протону за счет образования катиона гидр-
оксония.
У безводной муравьиной кислоты и ацетонитрила склонность
к присоединению протона весьма незначительна, вследствие этого
потенциал водородного электрода в них оказывается значительно
положительнее. Таким образом, водородный ион в ацетонитриле
является более сильным окислителем, чем в воде, а в воде более
281
Таблица ХИ.З
Стандартные электродные потенциалы (в в) в воде
и различных растворителях при 25 °С [Е0^ = 0J
Н. S t г е h 1 о ^
v. in: The Chemistry о
Solvents, ed. by J. J. Lagowski, N. Y.
Электрод
Cs, Cs+
Rb, Rb+
K, K+
Na, Na+
Li, Li+
Ca, Ca2+
Zn, Zn2+
Cd, Cd2+
Tl, Tl+
Pb, Pb2+
Ag, AgBr
H2, H+
Ag, AgCl
Hg, Hg2Cl2
Cu, Cu2+
Cu, Cu+
Hg, Hgf
Ag, Ag+
H2O
-2,923
-2,923
-2,923
-2,714
-3,045
-2,87
-0,763
-0,483
-0,316
-0,126
-0,085
0
0,222
0,263
0,337
0,521
0,798
0,799
CH3OH
-2,912
-2,921
-2,728
-3,095
—
-0,74
-0,43
-0,38
—
-0,138
0
-0,010
—
0,490
—
0,764
Non-Aqueous
1966.
C2H5OH
—
—
—2,657
-3,042
—
—
-0,34
—
-0,132
0
-0,088
—
—
—
—
CH3CN
-3,16
-3,17
-3,16
-2,87
-3,23
-2,75
—0,74
-0,47
-0,12
—
0
—
—
-0,28
-0,38
—
0,23
HCOOH
-3,44
-3,45
-3,36
-3,42
-3,48
-3,20
— 1,05
-0,75
-0,72
—
0
—
—
-0,14
—
0,18
0,17
HCONH2
-2,855
-2,872
—
—
—
-0,757
-0,408
-0,344
-0,193
0
, 0,198
0,245
0,279
—
—
—
N2H4
-2,01
-2,02
-1,83
—2,20
-1,91
-0,41
-0,10
0,35
—
0
—
0,22
0,77
—
NH3
-1,95
-1,93
-1,98
-1,85
-2,24
-1,74
-0,53
-0,20
0,32
—
0
—
—
0,43
0,41
—
0,83
сильным, чем в жидком аммиаке. В муравьиной кислоте, типично
кислом растворителе, потенциалы Cu, Ag, Zn и Cd смещены в
положительную сторону.
Ацетонитрил является растворителем с нерезко выраженными
основными свойствами и значительной склонностью к комплексо-
образованию с ионами тяжелых металлов; этим объясняется сдвиг
потенциалов меди и серебра в этом растворителе в отрицательную
сторону, в то время как в других растворителях (аммиак,
гидразин, муравьиная кислота) их потенциалы по сравнению с водой
практически неизменны.
Приведенные в табл. XII. 3 данные показывают существенное
влияние химической природы растворителя на величину
электродного потенциала. Иногда это влияние оказывается столь сильно,
что для рядов электродных потенциалов в разных растворителях
может изменяться даже последовательность расположения
металлов. Так, в водных растворах медь положительнее водорода, тогда
как в ацетонитриле наблюдается обратная картина. Это связано
с тем, что ацетонитрил проявляет малую склонности к
присоединению протона, но образует прочные комплексы с медью. Поэтому
в водных растворах медь не может окисляться ионом водорода, а
в растворах ацетонитрила медь окисляет Н+, вытесняя водород из
растворов кислот.
282
XII. 4. Расчет э. д. с. и электродных потенциалов
Зависимость э. д. с. цепи от концентрации ионов, применимость
уравнения Гельмгольца — Гиббса и т. д. остаются неизменными,
независимо от того, какой растворитель, образующий раствор,
будет при этом использован. В настоящее время экспериментальному
определению наиболее доступны значения химической энергии
сольватации электролитов, поэтому задача термодинамической
теории электродных потенциалов должна предусматривать
установление прямой связи величин электродных потенциалов с
характеристиками сольватации ионов.
Метод Н. А. Изгарышева
Н. А. Изгарышевым (1926—1928) была предложена теория
электродных потенциалов, основанная на количественном учете
сольватации ионов. Поскольку взаимодействие ионов с молекулами
растворителя определяется энергией сольватации, а способность
металла электрода к ионизации определяется энергией отрыва
электронов, то энергия zFE перехода иона в раствор будет
зависеть от обеих указанных величин:
= /(o), С/С0ЛЬр) (XII. 31)
Такое представление о механизме электродных процессов
позволило Н. А. Изгарышеву выбрать величины, на основании которых
можно вычислить э. д. с. электрохимической цепи.
Таблица XII.4
Энергетические величины для расчета э. д. с. цепи
Zn | ZnSO41 H2SO41 H2, Pt по Н. А. Изгарышеву
Энергетическая величина
Обозначение
-hJ+
-ян+
xi 2
Н2
/2н
Энергия, к/едл
525
-666
-250 • 2 = -500
-2-40,5 =-81
2 • 50 = 100
2-311=622
Энергия гидратации
образующихся ионов
Энергия ионизации паров
металла
Энергия дегидратации
выделяемого Н+ на электроде .
Теплота испарения водорода,
выделяемого в газообразном
состоянии
Теплота образования
молекул Н'2 из атомов
Энергия, выделяемая в
процессе разряда
В табл. XII. 4 сведены энергетические величины, необходимые
для теоретического вычисления э. д. с. цепи:
Zn | ZnSO411 H2SO41 H2, Pt (XII. 32)
283
Если просуммировать все эти величины, то они будут
соответствовать тепловому эффекту реакции Q, протекающей в цепи
(XII 32). Теоретические расчеты значений Q и найденные из них
по уравнению Гельмгольца — Гиббса значения э.д.с. и электрод,
ных потенциалов показали довольно близкое совпадение с
экспериментальными величинами. Нужно отметить, что расчеты Н. А. Из-
гарышева относились к электродам первого рода, причем основная
идея сводилась к установлению связи между величинами
электродных потенциалов и энергией сольватации ионов. Однако эта
правильная точка зрения не получила законченного оформления, так
как использовались энергетические эффекты вместо строгих
термодинамических величин — свободных энтальпий (изобарных
потенциалов) сольватации, значения которых в то время отсутствовали
в литературе.
Методы Н. Е. Хомутова (1962-1970)
В одном из методов Н. Е. Хомутова для расчета величин Е°
применяются термодинамические уравнения связи стандартных
электродных потенциалов с изобарными потенциалами
сольватации ионов, образования ионов в газовой фазе и другими
термодинамическими характеристиками участников электродных
реакций.
Для электродов первого рода стандартный электродный
потенциал равен э. д. с. электрохимической цепи:
Pt | Me | МеАЦНА (aR+ = 1) | Pt, Н2 (рНг = lam) (XII. 33)
Реакция, протекающая в этой системе
Ме2+ (ж) + */2Н2 = Me + zU+ (ж.)
может быть представлена как совокупность стадий:
Ме2+ (г.) + zH+ + ze —> Me (г.) + zH+ (г.)
f
zU
t
Me2+ (ж.) + */2H2 > Me (тв.) + zU (ж.)
Mez+ (ж.) = Ме2+ + ж. + kG°2
Ме2+ (г.) + ze = Me (г.) + AG3
Me (г.) = Me (тв.) + AG^
*/2Н2 (г.) = zH (г.) +• Д<?5
zU (г.) = zH+ (г.) + ze + AGg
гН+ (г,) + ж. = *Н+ (ж.) + AG°7
284
(XII. 34)
(XII. 35)
(XII. 36)
(XII. 37)
(XII. 38)
(XII. 39)
(XII. 40)
Величина AG° реакции (XII. 34)
bG\ = -zFE* (XII. 41)
связана с AG реакций (XII. 35) —(XII.40) соотношением
AG° = ^GO2 + AG° + AG4 + AG5 + AG°6 + AG^ (XII. 42)
Величины изобарных потенциалов отдельных стадий процесса
(XII. 34) обозначены AG%, AG3, AG4, Те же величины, но с
обратным знаком относятся к сольватации ионов Ме2+, ионизации
атомов Me и сублимации твердого или испарению жидкого
металла; — Дбб, —Абб, —AG?—изобарные потенциалы
диссоциации молекулярного водорода, ионизации атомарного водорода и
сольватации протонов. Величины — AG5 и — AG? представляют
сумму химических изобарных потенциалов сольватации АОсольв и
величин zF%L0—изменения энтальпий при прохождении ионами
скачка потенциала на границе газ — жидкость (поскольку zF%LtQ
входит в уравнение для электродного потенциала дважды с
противоположными знаками эта величина уничтожается). Величины
£ и AGq вычисляют при помощи соотношений
/ AG3 = А#з - Т kS°3 (XII. 43)
° (XII. 44)
В КОТОрЫХ Д#з = — 2 ^Ме^» А#6 =ZIH+ (2 /ме*+ ~~ СУММа И0НИ"
зационных потенциалов атомов Me; zIH+ — ионизационный
потенциал водорода); ASJ и А5б — изменение энтропии в процессах
нейтрализации ионов Мег+ и ионизации атомов водорода.
Если пренебречь разницей между энтропией ионов и атомов.,
то величины AG? и AG? в уравнениях можно заменить на
величины Д#з и АЯе.
После введения перечисленных упрощений уравнение (XII. 42)
может быть выражено в форме
Е° = Ч?° + А (XII. 45)
где 4го — сумма стандартных изобарных потенциалов образования
1 г-экв газообразных ионов и химической энергии сольватации
ионов, выраженных в электрических единицах; А — такая же сумма
для водородных ионов.
Величины 4го и А для случая участия одновалентных ионов
вычисляют из уравнений
уе _ Ме2+ сольв Mez+ (XII. 46)
л— AGT н + AG н + AG°0JIbB H+
Л — — "р \Л11. 4/)
где AGax н, AGS н, АОсольв н+ — изобарные потенциалы атомиза-
Дии, ионизации водорода и сольватации протонов.
285
При постоянной температуре для определенного растворителя
величина А будет сохраняться постоянной.
Уравнение (XII. 45) справедливо для любых типов обратимых
электродов, т. е. аналогично выражениям (XII. 46) и (XII. 47)
можно написать для электродов 2-го рода и
окислительно-восстановительных электродов.
Так, для электродов второго рода Pt, Х2|МеХ и Me, МеХ|Х
И
для окислительно-восстановительных электродов
xp-o _. сольв Me сольв Me zx-z2 ,„,, _,
где zi и z2 — заряды окисленной и восстановленной форм вещества
(z = zx — z2); AGSi-zj — изобарный потенциал реакции ионизации
металла в газовой фазе.
Рассчитанные по уравнениям первого метода величины
стандартных электродных потенциалов приведены в табл. XII. 5 и
обозначены Ей
Второй сравнительный метод термодинамического расчета
стандартных электродных потенциалов для трех основных типов
электродов базируется на связи стандартных потенциалов в неводных
растворителях £s со стандартными потенциалами в воде £н2о
и изобарными потенциалами гидратации AGh2o и сольватации
Абсольв ИОНОВ.
Основное уравнение сравнительного метода расчета может быть
получено вычитанием из уравнения (XII. 45), составленного для
неводного растворителя, такого же уравнения, но для воды
4 = £°н2о + ^s - ^°н2о + ^s - ^н2о = Я°н2о + № + ДЛ (XII. 51)
где AW0 = ^s — Ч'нгО и АЛ = As — Лн2о, а индекс S относится
к неводному растворителю.
После подстановки значений W° из уравнений (XII. 46), (XII. 48)
и (XII. 50) получаем уравнения для различных типов электродов
AG° л, z+ - AG° z+
E°s = £°НгО + сольвМе^ ^~ + АЛ (XII. 52)
AG%- - AG° Yz-
4 = £°н2о + — zFC0JlhBX + ^ (XIL 53)
AG° лл z,+ — AG° Zl+ — AG° лд г2+ + AG° г2+
o о сольв Me Me сольв Me Me , A -
^s = £h2o H Jf г дл
(XII. 54)
286
К
X
о
tt
к
■ S
I
са S
g « S3
e я si
а я w
«S x*
о р. $
X
яз
Sf
а
и
g о
5 S
нС
О оо
«т
If
М II
4
«1
о II
LO CD CO" <D N N
^ CO CO CO 00^ ^ (» S CO N Tt S i2 § £ I ^ S S
со" см cm" cm" cm" cm" cm" cn" cm" о" о cp" о о" о" о" ' о" о" о"
со см см см см см см см см о с
I I I I II I I I I I
OOCMNOOOCOOOOCMIOLOOO ООСПсО со N О)
OOCOCOOfO-'OCO-'CON^ COo~-< CDCiin
*"1 Ю« ^ °°* °°ж ^ °°- °°« °1 ^ ^L °Я. О ^ N | СМ СО СЛ
со" см" см" см" см" см" см" см" см" о" о" о" о о" о" о" ' о" о* о"
О5 СО О Ю
*- СО СО СО
С^ _ (^ _
«—* rf CO 00
00
о
сосмсмсмсмсмсмсмсмоооооооооо-^
О5 Ю N
О СО 00
111
О) lo i>» оо
^ 00 Ю -н CD ^ О N ^
со^ сол . ^ смл о.^ t>-^ со^ ь.л ол
о" о" I о о" о" о" о" о" о" -н
NCOCO-hN^OCMOLOCMCO
NCMLOCDCOCONO^rfNCOCO
со" см" см" см" см" см" см" см" со" о" о* о" о
!$ ОС> СО —I CM LO
СМ СМ 00 rf О) rf
Tf 00 N CM N eD
о" о" о" о" о" -<"
^чСМ~»'"Ф—<СМОСМЮ00ОЮ
^NWNWCN^CDfOOgW
СГ5 сО О^ 00 t4** ^^ CD С^ ^3 О^ *^* 1-О
см" см" см" см" см" см" со" со" со" о" о" о" о
t^ о —I а>
S3 2 £ ё?
о" о" о" о*
со см ь- ю
" "
со
со" см" см" см" со"
s to >-• союсосоюсоо
I I ^ ^ ^ (N 00 NN 00 00 О
' ' о" о* о" о о" о" о" о"4 о" <э *-Г
О0)ОЮ00М
СООСМЮОО —
—н оо о СГ5 оол Юл
" " " " " "
-^ CONCOCOCM00-H
*—| у—| О5 ^ 00 00 Ю -н
СОСМСОСМСМСМСМСМСО'-^ООООООООО»-^4
00 СМ О
см о со
о оо о
O)
CO
—^ см со оо
оо смл о^ о>
со"смсо"смсмсмсмсосооооооо I ооо"о"
ю^ююсососооососососм «о N оо О5 со к о
т+<—.OJCMCMCOCOOOOCOO^ W ОО 00 О) СО 00 ^
О NO) СО О>ж СО^ 00^ 00л О^ N^ rj^ 00^ —1 СОл N Ь-^ Юл Ол СО^
со" см" см" см" см" см" см" см" см" о" сГ о" о о о" о" о" о" *-Г ~
ъ Н $ ь Щ11 Н+к I
287
Записанные уравнения позволяют использовать не абсолютные
изобарные потенциалы сольватации ионов, а их относительные
величины, что устраняет неточности, связанные с нахождением
абсолютных изобарных потенциалов сольватации ионов.
Величины Es в спиртах (метиловом, этиловом, бутиловом и
амиловом), ацетоне и в муравьиной кислоте рассчитаны по второму
методу с применением второго набора формул; полученные данные
приведены в табл. XII. 5 и обозначены El.
Для сравнения в табл. XII. 5 приведены также имеющиеся
экспериментальные данные по значениям потенциалов различных
электродов в выбранных растворителях; для большинства
электродов расхождения опытных и расчетных данных не превышает 0,1 в.
Для многих электродов (табл. XII. 5) величины Ё° рассчитаны
впервые. Таким образом, метод Н. Е. Хомутова позволяет
существенно пополнить таблицы стандартных электродных потенциалов
для неводных растворителей.
Необходимые для расчетов электродных потенциалов по
методам Н. Е. Хомутова значения AG0 для многих реакций в неводных
растворах могут быть определены экспериментальным путем или
же рассчитаны теоретически. Точность методов определяется
надежностью имеющихся данных для изобарных потенциалов ионов
в газовой фазе и изобарных потенциалов сольватации ионов.
Расчетные уравнения Н. Е. Хомутова не содержат величин
реальной энергии сольватации. Устранение необходимости учета
скачков потенциалов на границе жидкость — газ достигается путем
проведения реакций ионизации водорода и нейтрализации атомов
металла в газовой фазе, благодаря чему отсутствуют стадии ввода
электронов в металл и удаления электронов из металла.
Метод А. И. Бродского
Значительный практический интерес представляет косвенное
вычисление электродных потенциалов по данным о растворимости.
Возможность эта основывается на том, что э. д. с. цепи с
насыщенным раствором соли МеХ, например
Hg I Hg2Cl2 • КС11 KBr • Hg2Br21 Hg (XII. 55)
электроды которой обратимы по отношению к ионам Ме+ и Х~,
должна быть независима от свойства растворителя, а зависеть
только от растворимости КС1 и КВг. Такой вывод основан на том
очевидном факте, что э. д. с. подобной цепи определяется свободной
энергией образования твердой соли, не зависящей от свойств
растворителя, поскольку максимальная работа реакции в насыщенных
растворах та же, что и при отсутствии растворителя.
Очевидно, что э. д. с. такой цепи, равная разности электродных
потенциалов Me и X, выражается уравнением
288
или
== ЕМе ~ Е
In й
(XII. 57)
где а — активности ионов и соли в насыщенном растворе.
Составив подобные выражения для двух растворителей и
приравняв их (поскольку Е от растворителя не зависит), получаем
выражение
'Me
■ In -
(XII. 58)
(CHac — концентрация насыщенного раствора; у± — средний
коэффициент активности) дающее, например, возможность вычисления
изменения стандартного потенциала галогена X, если известно
соответствующее изменение потенциала металла Me,
растворимости и коэффициенты активности соли в обоих растворителях.
Поскольку для труднорастворимых солей у±=1, то этого
оказывается вполне достаточно для расчета.
Очевидно, что приведенные уравнения должны рассматриваться
лишь как вспомогательные, так как для их использования
необходимы данные по разности стандартных потенциалов металла Me
в обоих растворителях.
XII. 5. Особенности потенциометрических измерений
в неводных растворителях
Экспериментальные измерения величин э. д. с. цепей в неводных
растворителях связаны со значительными трудностями, которые
прежде всего обусловлены низкой растворимостью солей и слабой
диссоциацией электролитов;
необходимо также тщательно
обезвоживать растворители и
соли, а также предохранять
растворы от попадания вла-
ги, значительно влияющей как
на величины потенциалов, так
и на воспроизводимость изме- "
рений.
Воспроизводимость данных
по э. д. с. для водно-неводных
систем обычно лучше, чем для
чистых неводных
растворителей из-за всегда имеющихся в
Рис. XII. 4. Зависимость £° хлорсере-
бряного электрода в цепи
Pt(H2) | НС11 AgCl, Ag от содержания
воды в этаноле.
чистом растворителе трудноустранимых следов воды. На рис. XII. 4
представлены данные по величинам Е° хлорсеребряного
электрода в водно-спиртовых смесях, полученные для различных
составов смесей: добавление 1% воды к этанолу снижает э.д. с.
цепи Pt(H2)|HCl||AgCl, Ag больше, чем на 0,1 в при общем
изменении э.д.с. при переходе от воды к спирту на 0,3 0&
Ю Зак. 500
289
В аппаратурном оформлении возникает трудность подбора
электродов сравнения, поскольку для многих из них невозможно
получать цепи без жидкостных контактов. Из-за этого при
измерении э. д с. в неводных растворах часто приходится электроды
цепи соединять с помощью солевых мостиков.
Наиболее широкое применение при измерениях электродных
потенциалов получили электроды второго рода на ртутной основе,
благодаря доступности ртути высокой степени чистоты. Такие
электроды характеризуются высокой воспроизводимостью и легкостью
приготовления.
В качестве электрода сравнения при исследовании
электролитических цепей в неводных средах и в смесях воды с органическими
растворителями широко используют каломельный электрод.
Стандартные моляльные потенциалы каломельного электрода при 25 °С
в различных смешанных растворителях приведены в табл. XII. 6.
Таблица XII.6
Стандартные электродные потенциалы
каломельного электрода, полученные
из измерений э. д. с. цепей Pt(H2) | HCI | Hg2Cl2, Hg
в смешанных растворителях вода — органический
компонент при 25 °С
Растворитель
Диоксан
Этанол
Метанол
Вес. доля Р
0,90
0,20
0,45 .
0,70
0,82
0,20
0,40
0,65
0,90
0,98
0,20
0,43
0,68
0,97
6
78,30
60,79
38,48
17,69
9,53
66,99
54,89
40,62
28,13
25,15
69,8
59,4
' 47,9
34,2
о
Ет'°
0,2683
0,2501
0,2104
0,1126
-0,0014
0,2539
0,2406
0,2111
0,1376 ,
0,0638
0,2545
0,2415
0,2173
0,1127
Для хлорсеребряных электродов стандартные потенциалы
определены во многих безводных растворителях; их величины приведены
в табл. XII. 7.
Некоторые растворители могут вступать в химическое
взаимодействие с материалом электрода, не исключено «отравление»
электродов растворителем; в окислительно-восстановительных системах
растворители могут взаимодействовать с окисленной или
восстановленной формами участников электродной реакции,
290
Таблица XII.7
Стандартные потенциалы электрода Ag, AgCl | HC1
в безводных органических растворителях при 25 °С
Растворитель
СНзОН
С2Н5ОН
«зо-С3Н7ОН
С4Н9ОН
0,099
0,08131
0,109
0,132
Растворитель
изо-С4Н9ОН
С6Н5СН2ОН
НСООН
СНзСООН
-Е°, в
0,134
0,163
1,200
0,618
Недостаточным учетом всех перечисленных обстоятельств
объясняется расхождение величин э. д. с! одинаковых цепей в работах
различных авторов.
Рассчитываемые из эксперимента величины стандартных
электродных потенциалов зависят от способа выражения концентрации
(наиболее часто используется моляльность).
Зависимость электродных потенциалов от концентрации
необходима для нахождения стандартных электродных потенциалов,
которые определяются преимущественно с помощью цепей без
жидкостных контактов. Для этого составляют цепь из электрода
второго рода и электрода первого рода с общим электролитом.
Определение Е° наталкивается на затруднение, обусловленное
недостаточной воспроизводимостью результатов измерений э. д. с.
в сильно разбавленных растворах и медленным установлением
равновесия между электродом и раствором. В концентрированных
растворах равновесие обычно устанавливается быстро и достаточно
хорошо воспроизводятся результаты, но зависимость э. д. с. от
концентрации весьма сложна. Поэтому в настоящее время для
нахождения Е° используются измерения э.д. с. в растворах малых и
умеренных концентраций. Процедура экстраполяции для
определения значений Е° описана в главе VI.
10*
Глава XIII
КИНЕТИКА ЭЛЕКТРОДНЫХ ПРОЦЕССОВ
В НЕВОДНЫХ РАСТВОРАХ
ПОЛЯРОГРАФИЯ
Поскольку большинство органических соединений нерастворимо
либо плохо растворимо в воде, полярографическое изучение этих
веществ можно проводить лишь в неводных растворах. На первой
стадии развития неводной полярографии основное внимание
уделялось подбору подходящих растворителей, в которых исследуемое
вещество давало бы хорошо выраженные волны, позволяющие
производить количественные определения.
Применение неводных растворителей в полной мере
способствовало раскрытию нового аспекта полярографии: оказалось, что
по сравнению с водными растворами существенно изменяется
характер восстановления растворенных соединений. Это расширило
возможности полярографического метода: были выявлены
специфические особенности влияния неводных растворителей на
протекание электродного процесса, а также на некоторые
полярографические характеристики. Было установлено, что свойства
растворителя, как правило, существенно влияют на величины диффузионных
токов и значения потенциалов полуволн деполяризаторов; такое
влияние обусловлено, прежде всего, изменением сольватации
деполяризаторов и их коэффициентов диффузии. Эти результаты
дают ценную информацию о поведении данного вещества на
электроде и его состоянии в растворе, кроме того, они имеют важные
практические применения, прежде всего, раздельное определение
нескольких веществ при их совместном присутствии, получение
отчетливых волн таких ионов, для которых в водных растворах
невозможно измерить их предельные токи и т. п.
Таким образом, неводные растворители можно считать не
вспомогательным средством, обеспечивающим, благодаря высокой
растворяющей способности, возможность проведения
полярографического исследования различных органических соединений в
растворе, а фактором, существенно влияющим на^ течение
электрохимической реакции и ее результаты.
Указанные обстоятельства поясняют, почему проблеме
использования неводных растворителей в полярографии для физико-
химических, аналитических и других приложений уделяется
в последнее время такое большое внимание.
XIII. I. Влияние природы растворителя
на основные полярографические характеристики
Изменения основных полярографических характеристик
деполяризаторов: предельного тока id (или высоты волны) и потенциала
полуволны £у2, которые наблюдаются при переходе от воды к орга-
292
ническому растворителю или замене одного неводного растворителя
другим, как правило, весьма существенны. Поэтому проблема
использования неводных растворителей в полярографии охватывает
широкий круг вопросов, главными из которых являются: каково
влияние неводных растворителей на предельные токи и форму
полярографических волн и какова зависимость между свойствами
растворителя и потенциалами полуволн.
Закономерности, которым подчиняются изменения id и £у2 под
влиянием природы растворителя, могут быть выявлены путем
анализа уравнений, лежащих в основе полярографических измерений.
Влияние растворителя на диффузионные токи
Наиболее часто используемым в полярографии рабочим
электродом является ртутно-капельный *. Проблема тока,
ограниченного скоростью диффузии частиц к сферическому электроду, была
решена Д. Ильковичем (1934). Рассматривая зависимость величины
тока от параметров образующейся ртутной капли и концентрации
частиц деполяризатора в растворе, он вывел уравнение для
среднего значения предельного тока id за период жизни капли
^ = 605zD1/2m2/3TI/6C0 (XIIL 1)
где z — число переходящих электронов; D — коэффициент
диффузии деполяризатора, см2/сек; т—скорость истечения ртути, мг/сек;
х — период жизни одной капли, сек; Со — концентрация
деполяризатора в глубине раствора, моль/л, а ток измеряется в мка.
В полярографической литературе усредненный по времени
жизни ртутной капли ток обычно называют диффузионным током,
а выражение (XIII. 1) —уравнением Ильковича.
Изменение величины предельного тока в неводных
растворителях по сравнению с водой рассматривали многие авторы. Так,
И. М. Кольтгоф и Дж. Дж. Лингейн в своей монографии (1948)
сделали общий вывод о том, что изменение id при переходе от воды
к другим растворителям определяется исключительно изменением
коэффициента диффузии. Они связывали это с изменением
коэффициента диффузии деполяризаторов в неводных средах как за
счет изменения вязкости, так и вследствие различной степени
сольватации ионов, определяющей их размеры.
Первая попытка количественно объяснить зависимость высоты
волны при переходе от одного растворителя к другому с учетом
всех специфических особенностей неводных растворителей была
сделана в работах Н. А. Измайлова и В. Д. Безуглого (1949).
Сопоставляя имеющиеся литературные данные и результаты
собственных исследований по применению неводных растворителей в
полярографии, Н. А. Измайлов и В. Д. Безуглый подвергли анализу
* Все дальнейшее изложение будет относиться именно к этому типу
электрода, хотя основные положения останутся, по-видимому, справедливыми и для
других типов применяемых в полярографии рабочих электродов.
293
основное уравнение диффузионного тока (XIII. I) с точки зрения
изменения его параметров пбд влиянием свойств растворителя.
Анализ уравнения (XIII. 1) показал, что замена растворителя
может приводить к изменениям id за счет всех входящих в него
величин.
Однако опыт показывает, что масса ртути, вытекающая за 1 сек,
практически не зависит от природы растворителя, а время
образования одной капли, хотя и изменяется, но это изменение
сказывается мало, поскольку т входит в уравнение Ильковича в степени
7б- Величина D зависит от степени сольватации электролита и
вязкости среды, уменьшаясь с ростом этих факторов. Число
электронов г, участвующих в электродной реакции, также может
изменяться с переменой растворителя; причинами могут быть:
химическое взаимодействие восстанавливающегося вещества с
растворителем; изменение рН, так как его значение влияет на
механизм электродного процесса и на химические реакции
восстановления; изменение адсорбируемости поляризатора, влияющее на
механизм процесса.
Переменной величиной в уравнении (XIII. 1), зивисящей от
свойств растворителя, является также величина Со — так
называемая «активная» концентрация восстанавливающегося вещества.
Она может изменяться под влиянием различных причин, главными
иЗ которых являются: изменение степени ассоциации;
взаимодействие восстанавливающегося вещества с фоном или
растворителем; переход молекул деполяризатора под влиянием растворителя
в другую форму; изменение адсорбируемости деполяризатора,
связанное с изменением растворителя.
Итак, анализ уравнения (XIII. 1) показывает, что природа
растворителя влияет не только на коэффициент диффузии, но также и
на количество электронов, принимающих участие в восстановлении
одной молекулы деполяризатора, и на активную концентрацию
восстанавливающегося вещества. В общем случае изменение
высоты волны при переходе от одного растворителя к другому
является результатом сложного сочетания вышеперечисленных
факторов.
Однако, в некоторых случаях можно выделить определяющее
действие одного или двух из указанных выше факторов, при
незначительном изменении остальных. Так, если фактор вязкости
является определяющим, то при неизменных z, m, т и Со можно, как
частный случай, получить предложенную И. М. Кольтгофом и
Дж. Дж. Лингейном зависимость между id и вязкостью ц
const (XIII. 2)
Влияние растворителя на потенциалы полуволн
Изменение природы или состава растворителя оказывает
большое влияние на кинетику электрохимических реакций, что в случае
полярографии на ртутно-капельном электроде сказывается на
величинах потенциалов полуволн деполяризаторов.
294
Форма катодной полярографической волны при протекании
восстановления растворенного вещества с учетом коэффициентов
активности передается уравнением (Я. Гейровский, Д. Иль-
кович, 1935)
где vox, YRed, Dox и Z?Red — коэффициенты активности и
коэффициенты диффузии окисленной и восстановленной форм
деполяризатора; id — диффузионный ток, определяемый уравнением Илько-
вича; i— ток при любом данном потенциале Е.
Когда t = V2 ^d, последнее слагаемое в уравнении (XIII. 3)
равно нулю. Потенциал, при котором ток равен половине
предельного тока, известен как потенциал полуволны £i/2; он равен:
Еч =E°~—lnl™- I/^5- (XIII. 4)
k ZF Yox V DRed
Величина Eif2 отвечает точке перегиба i— ^-кривой и, как видно
из уравнения (XIII. 4), не зависит от характеристики капельного
электрода.
Уравнение (XIII. 4) можно переписать в виде:
£v=£°~ —!nl/^ + -^-in^L (XIII. 4а)
к zF V #Red zF YRed
Из этого уравнения следует, что при изменении природы
растворителя или состава раствора могут изменяться величины всех
трех слагаемых правой части, влияя тем самым на Еу2. Во многих
Случаях Dox И D^ed Обычно Не СИЛЬНО ОТЛИЧаЮТСЯ; Т. е. Ьох/^Red» 1.
Поэтому с достаточной степенью приближения уравнение
(XIII. 4а) можно упростить
RT vox
ЧЕ 1^ <XIIL46)
Поскольку в уравнение (XIII. 46) входит величина
стандартного электродного потенциала, то значение Еч2 должно зависеть от
природы разряжающихся частиц. Отсюда очевидно, что влияние
растворителя на потенциалы полуволн будет сказываться как через
изменения £°, так и за счет изменения коэффициентов активности
YOx И YRed.
Характер влияния природы растворителя на величины
электродных потенциалов был подробно рассмотрен в гл. XII.
Коэффициенты активности ионов, как правило, сильно возрастают при
уменьшении диэлектрической проницаемости среды, причем
являются линейной функцией от 1/е (см. гл. VI).
Из ура^чения XIII. 46 следует, что, если при замене
растворителя (в ряду однотипных растворителей) изменяется лишь е, то
295
изменение Еу2 обусловлено влиянием е на величину электродного
потенциала. Сдвиги Еу2 могут быть обусловлены и другими
причинами: в одних случаях это связано с образованием комплексов
между ионом и растворителем, в других — с явлением сольватации;
сдвиг потенциала полуволны может определяться и влиянием
фонового проводящего электролита. Эта последняя зависимость
объясняется тем, что при образовании комплексов
деполяризаторов с электролитом фона они стабилизируются органическим
растворителем и £у2 становится более отрицательным.
В общем случае на сдвиг Еу2 оказывают влияние оба
слагаемых уравнения (XIII. 46). Здесь сказываются, по крайней мере,
два противоположно действующих эффекта. Во-первых,
присутствие органических растворителей может повышать активность
иона деполяризатора, а это сдвигает Еу2 в область более
положительных значений. Во-вторых, молекулы растворителя при
возрастающем содержании их в растворе могут вытеснять ионы
деполяризатора из граничного слоя, затрудняя тем самым их разряд
и смещая Еу2 к отрицательным значениям.
На сдвиг потенциалов полуволн оказывают влияние еще и1
другие факторы. Например, растворители с низкой 8 могут
благоприятствовать образованию ионных пар и затруднять процесс
восстановления.
В случае органических деполяризаторов действие растворителя,
вызывающее сдвиг £у2 в отрицательную сторону, будет
проявляться за счет адсорбции его на электроде; при весьма сильной
адсорбции самого деполяризатора величина Еу2У как правило, не
будет зависеть от свойств растворителя. Следовательно, при
оценке сдвига Ei/2 для органических деполяризаторов под влиянием
растворителя необходимо обязательно учитывать адсорбционное
действие растворителя.
XIII. 2. Полярография различных соединений
Полярографическое поведение иона водорода
Исследование поведения иона водорода по своему
теоретическому значению занимает в полярографии одно из центральных
мест. Обусловлено это, помимо чисто аналитического интереса, той
важностью, которую занимает процесс разряда водорода в теории
водородного перенапряжения и некоторых других вопросах.
Изучение полярографического поведения ионов водорода в ме-
танольных растворах безводной НС1 на фоне индифферентных
электролитов (KCNS и NaC104) показало, что существует
пропорциональность между id и Сн+ и возрастание диффузионного тока
с увеличением концентрации НС1 (рис. XIII.1,а), в то же время
величины Еу2 не зависят отСн+. Аналогичные результаты
получены и для этанола (рис. XIII.1,6).
Полярографические данные о поведении растворов сильных
кислот в спиртах [А. VI сек, 1955] показывают, что из растворов
НС1О4 и НС1 в этаноле водород выделяется при потенциале, более
296
положительном (на 0,27 в), чем из водных растворов, и с
пониженным на 0,1 в перенапряжением по сравнению с водой. Это
объясняется тем, что протон в этоксониевом ионе (С2Н5ОН^)
слабее связан, чем в гидроксонии: образование иона Н3О+
сопровождается ростом перенапряжения.
При изучении полярографического поведения ряда Н-кислот
в безводном ацетонитриле [J. F. Coetzee, I. M. Kolthof f, 1957]
было установлено, что различие их £\/2 связано со значительным
дифференцирующим действием ацетонитрила на силу кислот:
Кислота
-Ей
Хлорная 0,70
Бромистоводородная . . 0,90
Хлористоводородная . . 1,06
д-Толуолсульфоновая . 1,20
Серная 1,20
Кислота —E\j , в
Фторвалериановая ... 1,50
Щавелевая 1,55
Фосфорная 1,75
Бензойная 2,1
Уксусная 2,3
20
Волна выделения водорода из растворов НС1О4 и НС1 в
насыщенном растворе (CH3)4NI в ацетонитриле [А. VI сек, 1954]
лежит при значительно более
высоких потенциалах, чем
волна его выделения из водных
растворов. В безводном CH3CN
волна начинается от
потенциала растворения ртути. В
99,8%-ном CH3CN высота вол- ю
ны водорода растет линейно
с концентрацией кислоты.
Величина Ei/2 постоянна и
равна —0,60 в относительно н. к. э.
Представляют интерес дан-
ф
Ю 20 зо 40
Сц*,МГ-ЫОН//1
р р
ные о полярографическом по- £ ^ Н#?Ге£
ведении органических КИСЛОТ нольных (а) и этанольных (б) раство-
В невоДНЫХ растворителях pax при 25 °С на фоне 1 М индифферент-
(табл. XIII. 1). Показано, что ных электролитов:
гттриттиятткт nnnvRnrm ппгяни- l -KCNS; 2- NaClOt; 3-LiCl: 4-NaCI (0,1 М).
ПОТеНЦИаЛЫ ПОЛУВОЛН ОргаНИ- [Г. К. Мига л ь, Я. И. Т урьян, Н. И. Бон-
ЧеСКИХ КИСЛОТ ОПредеЛЯЮТСЯ даре нко, 1956].
их силой. Установленная ли- ;
нейная зависимость между £у2 и р/С кислот позволяет
использовать полярографический метод для оценки силы кислот в
различных растворителях, а также для оценки
дифференцирующего действия различных органических растворителей по
отношению к кислотам.
Полярографическое поведение неорганических ионов в неводных
растворителях
Изучение поведения целого ряда ионов в органических
растворителях показало применимость уравнения (XIII. 2), т. е.
вязкость является во многих случаях основным фактором,
влияющим на коэффициент диффузии (рис. XIII. 2). Однако в таких
297
Т аб лица XIII.1
Величины Еул (в в) некоторых органических кислот
[Л. Я. Хейфец, Н. М. Пржиялговская, Л. И. Дмитриевская, 1971]
Кислота
Муравьиная . . .
Уксусная . .
Хлоруксусная
Бензойная . .
Салициловая ,
1-Нафтойная
2-Нафтойная
, #
Метанол
2,03
2,05
2,00
2,03
1,66
1,63
1,70
ДМФА
2,11
2,21
2,06
2,15
2,02
1,86
1,91
Кислота
2,1 -Оксинафтойная
3 1-Оксинафтойная
З-Хлор-1 -нафтойная
1,2-Оксинафтойная
2,3-Оксинафтойная
1,3-Оксинафтойная
1,3- Диокси-2-нафтой-
ная
Метанол
1,57
1,59
1,47
1,56
1,56
1,64
1,55
ДМФА
1,76
1,85
1,70
1,75
' 1,70
1,86
1,30
растворителях, как спирты, происходит смещение указанной
зависимости в сторону меньших значений id (рис. XIII. 3).
Таким образом, наряду с влиянием вязкости на предельные
токи в различных растворителях, значение имеют и сольватные
оболочки, размеры которых могут
меняться при замене одного
растворителя другим.
На величины потенциалов
полуволн ионов влияют следующие
О 0,5 1
Объемная доля этиленгликоля
Рис. XIII. 2. Зависимость
произведения idVy\ для иона Т1+ от
состава растворителя в смеси
этиленгликоль — вода.
02-
Н2О 0,2 0,4 0,8
Объемная доля спирта
Рис. XIII. 3. Диффузионные
токи кадмия в метаноле (У) и
этаноле (2).
[Я. И. Т у р ь я н, Б. П. Ш а т а л а й,
1960].
факторы: свободная энергия сольватации иона; степень, комплек-
сообразования иона с другими (кроме растворителя) веществами;
возможность реакции ассоциированного иона с электролитом
фона.
Величины Et/2 неорганических ионов в неводных растворах
отличаются от соответствующих величин в воде (см. табл. XIII.2,
XIII.3, XIII.4),
При переходе от воды к спиртам (табл. XIII.5) потенциалы
полуволн изменяются мало, вследствие близости свойств этих
растворителей и воды. Однако в таком основном растворителе как
аммиак найденВ! значительные изменения потенциалов полуволн
298
(табл. XIII.4), что совпадает с характером влияния аммиака на
электродные потенциалы этих металлов.
Таблица XIII.2
Величины Еу2 (в в) катионов щелочных металлов
в различных растворителях при 25 °С
Ион
Li+
Na+
К+
Rb+
Cs+
Вода
2,34
2,10
2,12
2,11
2,09
Диметил-
формамид
1,81
1,53
1,55
Пропилен-
карбонат
2,07
1,92
2,02
2,04
2,03
Этилендиамин
2,41
1,97
2,00
1,86
1,82
Пиридин
2,17
1,89
1,91
1,91
1,89
Таблица ХШ.З
Величины £у2 (в в) ионов в воде и нитрилах
по отношению к «рубидиевому нулю» в том же растворителе
Ион
Li+
Na+
к+
Rb+
Cs+
Zn2+
Cd2+
Ag+
Вода
-0,20
0,01
—0,01
0
-0,04
1,13
1,55
Ацето-
нитрил
'0,013
0,13
0,02
0
0,01
1,36
1,72
2,40
Пропил-
нитрил
0,01
0,11
0
1,35
1,69
2,39
Изобутил-
нитрил
-0,02
0,11
0,02
0
1,35
2,37
Бензо-
нитрил
0,06
0,14
0
1,35
1,71
2,42
Фенилацето
нитри л
0,06
0,12
0
1,38
1,71
2,41
Таблица ХШ.4
Величины — Ей (в в) ионов в воде и аммиаке
Ион
Са2+
Sr2+
Ва2+ . . '
Вода
2,22
1,94
1,94
Аммиак
1,96
1,67
1,54
При исследовании полярографического поведения ряда ионов
металлов в безводных нитрилах на фоне солей тетраалкиламмо-
ния (табл. XIII.4) найдено, что большинство катионов в этих
растворителях вследствие меньших по сравнению с водой значений
энергии сольватации восстанавливаются при более
положительных потенциалах, чем в воде. Различие между потенциалами
299
Таблица XIII.5
Величины Еу2 (в 0) ионов в воде и спиртах
Ион
Cd2+
Sn2+
Pb2+
Ni2+
Mn2+
Al3+
Ba2+
Вода
0,64
0,47
0,46
1,09
1,54
1,70
1,97
Метанол
0,67
1,13
1,54
1,70
2,00
Этанол
0,66
1,14
1,58
1,73
2,03
Глицерин
0,70
0,44
1,54
1,54
полуволн катионов в ацетонитриле и в воде больше для катионов
с меньшим радиусом, которые сильнее гидратированы в водных
растворах. Однако серебро и медь из-за сильного их комплексооб-
разования с нитрилами восстанавливаются в них труднее, чем в
воде. При введении .в нитрилы небольших количеств воды волны
катионов сдвигаются ib сторону отрицательных потенциалов,
благодаря образованию более прочных гидратов, в то время как в
нитрилах они сольватированы очень слабо.
Полярографическое поведение органических веществ
в неводных растворителях
Исследование влияния неводных растворителей на высоты
полярографических волн различных органических соединений,
особенно тех, которые восстанавливаются в молекулярной форме,
показало, что в этом случае изменение id связано только с
изменением коэффициентов диффузии. Это обусловлено, прежде всего,
тем, что молекулы органических деполяризаторов, как правило,
меньше сольватированы по сравнению с неорганическими ионами
и степень сольватации их изменяется незначительно с переменой
растворителя.
Во всех случаях наблюдается линейная зависимость
диффузионного тока от концентрации деполяризатора. Приведенная на
рис. XIII. 4 весьма характерная зависимость id от концентрации для
процесса восстановления бензальдегида показывает, что
диффузионный ток линейно зависит от концентрации*
Если же при заданной концентрации деполяризатора
варьируется вязкость путем замены растворителя или приготовлением
смесей растворителей с изменяющейся вязкостью, то всегда
выполняется соотношение (XIII. 2).
Однако далеко не всегда изменение диффузионного тока
обусловлено изменением вязкости среды — существенное влияние
может оказывать также изменение активной концентрации деполя-
300
ризаторов за счет взаимодействия его с растворителем. Так, при
восстановлении формальдегида было показано, что при переходе
от чистой воды к ее смесям с диоксаном наблюдается увеличение
высоты волны, обусловленное возрастанием скорости
дегидратации этого альдегида. В смесях же воды с метанолом
полярографическая волна формальдегида снижается, что связано с
образованием из альдегида и спирта полуацеталя,
который является весьма устойчивым
соединением, не восстанавливающимся на ртутно-ка-
пельном электроде.
Значительно уменьшаются высоты волн
восстановления различных алкалоидов по
мере добавления спирта к воде за счет
уменьшения силы алкалоидов как оснований. При
добавлении спирта происходит смещение
равновесия в сторону увеличения концентрации
невосстанавливающейся молекулярной
формы алкалоида по отношению к
восстанавливаемой— ионной.
Существенно влияет на
полярографические характеристики органических
деполяризаторов в неводных растворителях, в
частности, на потенциалы полуволн, природа и
концентрация сопутствующего электролита
(фона). Влияние это обусловлено
дифференцирующим действием таких фонов; особенно
сильно оно проявляется в полярографии
различных ароматических альдегидов, кетонов, хинонов и некоторых
других классов органических соединений.
Приводимая ниже зависимость A£\/2 = i?^22 —£у2 для антра-
хинона от состава фона показывает, что дифференцирующее
действие зависит как от природы катиона, так и от природы аниона
и уменьшается для растворов в диметилформалиде в следующем
порядке:
(C2H5)4NC1O4 > (C2H5)4NI >LiC104 >LiCl
О 0t2 0A 0,6
С,ммоль/л
Рис. XIII. 4.
Зависимость диффузионного
тока восстановления
бензальдегида от его
молярной
концентрации в безводном
глицерине на фоне 0,1 М
LiCl.
[Th. D e V г i e s,
D. В. В г u s s, 1953].
Фон (0,1 M)
LiCl
LiClCV
)
AEij2. в
0,27
0,34
V
(C2H5)4NI 0,60
(CH)C1O 075
(25)4
(C2H5)4NC1O4
0,75
Анионы в приведенном выше ряду оказывают значительно
меньшее влияние, чем катионы.
Имеющиеся в настоящее время экспериментальные работы
являются недостаточными для исчерпывающего описания
возможного влияния неводных растворителей на полярографические
характеристики различных веществ. Тем не менее, такие
исследования, кроме ценных сведений о количественных закономерностях
301
в полярографии, могут дать существенную информацию о
состоянии деполяризатора в растворе и его изменениях под влиянием
растворителя.
На основании уже полученных теоретических и
экспериментальных данных, можно провести последовательный (хотя пока
качественный) учет влияния неводных растворителей на сдвиги
потенциалов полуволн различных деполяризаторов и на высоты волн.
XIII. 3. Некоторые особенности эксперимента
при использовании неводных растворителей
в полярографии
Несмотря на то, что полярографии в неводных средах
посвящено большое число исследований, многие результаты получены
в различных условиях, поэтому зачастую их невозможно
сравнивать. Это обстоятельство выступает наиболее отчетливо, когда на
полярографическое поведение деполяризаторов влияет
трудноустранимые следы воды и недостаточная чистота применяемого
растворителя.
Важнейшим из условий полярографирования в неводных
растворителях при работе с капельным ртутным электродом является
соблюдение требований, предъявляемых к подбору подходящего
неводного растворителя и индифферентного электролита (фона).
Для успешной съемки полярографических кривых в неводных
растворителях существенным является выбор ячейки, электрода
сравнения и конструкционных особенностей применяемой аппаратуры.
Выбор электрода сравнения
При выполнении полярографических исследований в неводных
растворителях в первую очередь возникает проблема выбора
электрода сравнения для измерения потенциала полуволны.
В качестве электрода сравнения рекомендуют применять
хлорсеребряный электрод в растворе хлористого тетраметиламмо-
ния или каломельный электрод в растворе ацетона, насыщенном
хлоридом лития. Такой электрод обратим, неполяризуем,
характеризуется высокой устойчивостью и воспроизводимостью данных.
Зачастую применяют выносные электроды сравнения с
водными растворами, например, насыщенный каломельный электрод.
При этом, однако, нужно учитывать, что значение потенциала
полуволны будет содержать неустранимую величину диффузионного
потенциала, определяемую ионами индифферентного электролита
на границе раздела двух сред. Следует, правда, отметить, что при
большой концентрации фона величина диффузионного
потенциала почти постоянна, а это дает возможность проводить
относительное сравнение значений потенциалов полуволн различных
деполяризаторов в данной среде. В ряде работ для отделения неводного
растворителя от электродов сравнения, содержащего воду, реко-
302
мендуют применять специальную мембрану: пористую стеклянную
перегородку, заполненную жидким стеклом и имеющую
незначительное омическое сопротивление. Такая перегородка в известной
мере препятствует заметному смешению водной и неводной
фаз.
Методы нахождения значений стандартных.электродных
потенциалов, не содержащих ошибок за счет появления диффузионного
потенциала, описаны в гл. XII. Влчек [А. V 1 с е к, 1951]
распространил представления о практическом совпадении нормальных
потенциалов ионов рубидия и цезия во всех неводных
растворителях на область, полярографических исследований. Однако,
поскольку во многих средах очень трудно определить значение
потенциалов полуволн ионов рубидия и цезия (вследствие
электрохимической неустойчивости растворителей), то в качестве
потенциала сравнения Влчек предложил использовать величину
потенциала полуволны восстановления иона калия, число сольватации
которого в большинстве растворителей мало.
Позднее, когда было доказано, что это положение для
указанных катионов щелочных металлов не соблюдается достаточно
строго, Штрелов [Н. S f r e h 1 о w с сотрудниками, 1960] предложил
использовать в качестве электрода сравнения вместо рубидия
подходящую окислительно-восстановительную систему. В частности,
оказалось удобным использовать систему Fe2+ — Fe3+.
Значительно точнее, чем в случае щелочных катионов,
постоянство стандартных потенциалов в различных растворителях
выполняется для одновалентных ионов с большим радиусом и
равноценными связями (типа NRt). Такие ионы, правда, не могут быть
использованы в качестве «нулевой точки», так как они
разряжаются при очень отрицательных потенциалах и не дают
полярографическую волну.
Влияние электрического сопротивления неводного раствора
При расшифровке полярографических кривых, получаемых в
неводных растворителях, нужно учитывать влияние падения
потенциала в растворе, которое в этом случае проявляется в
значительно большей степени, чем в водных растворах, так как даже
в присутствии избытка фона электрическое сопротивление
раствора довольно высоко (от 2000 до 20 000 ом). Для нормального
полярографирования в неводных средах, как показывает опыт,
внутреннее сопротивление системы электрод — ячейка не должно
превышать 8000 ом.
Капельный ртутный электрод устанавливают так, чтобы
нижний конец возвышался над поверхностью донной ртути не менее,
чем на £—6 см; уменьшение этого расстояния может вести к
появлению ложных полярограмм. За счет изменения расстояния от
капельного электрода до электрода сравнения достигается
значительное снижение внутреннего сопротивления (до 10 раз).
303
При полярографировании в неводных средах из-за большого
сопротивления рабочих растворов могут появляться дополнительные
полярографические максимумы I рода, которые препятствуют
точному измерению полярограмм. Необходимо иметь в виду, что
некоторые поверхностно-активные вещества,< применяемые для
подавления таких максимумов, могут значительно сдвигать
потенциал полуволны и искажать прямолинейные зависимости между
высотой волны и концентрацией деполяризатора. Необходимое
количество подавителя выбирается в зависимости от растворителя,
концентрации проводящей соли и деполяризатора.
Для уменьшения сопротивления рабочих растворов к ним
добавляются полярные соли (фон), которые имеют достаточную
растворимость в неводном растворителе с одновременной хорошей
проводимостью и более отрицательным потенциалом выделения
катионов фона по сравнению с анализируемым веществом.
Для компенсации эффекта сопротивления ячейки предложены
различные схемы. Лучшим методом [Т. Дамокош, 1967]
является наложение асимметричной поляризации на ртутный электрод.
Электрод сравнения должен быть расположен близко к
капающему электроду.
Выбор индифферентного электролита
Весьма существенным для работы в неводных средах является
выбор подходящего индифферентного электролита, который
должен достаточно хорошо растворяться в данном растворителе.
В большинстве случаев для этого можно использовать соли
лития. При аналитических определениях в органических
растворителях часто используются четырехзамещенные аммониевые соли;
так как разряд.этих электролитов происходит только при очень
высоких отрицательных потенциалах, можно использовать
относительно широкую область напряжений. Если проводящая соль
совершенно нерастворима в «выбранном неводном растворителе, то
для достижения необходимой растворимости используют смеси
растворителей. При выборе проводящей соли фона учитывают
также устойчивость ее в данном растворителе.
Выбор фона оказывает большое влияние на форму
полярографической кривой; подбором фона можно изменять и величину £\/2,
что особенно важно для анализа многокомпонентных систем*.
Наиболее часто используемые неводные растворители
В настоящее время для полярографирования используется
большое количество неводных растворителей и многие из них
обеспечивают получение воспроизводимых полярограмм.
В литературе описаны достаточно надежные способы
получения и контроля чистоты неводных растворителей и приготовления
растворов для электрохимических исследований,
304
При выборе растворителя должны быть учтены: его
устойчивость во времени; трудность восстановления на ртутном катоде и
отсутствие взаимодействия ртути с растворителем; склонность к
образованию максимумов и легкость их подавления;
растворимость проводящих солей в безводном растворителе,
обеспечивающая достаточную величину диффузионного тока.
Наиболее широко распространенными растворителями в
полярографии являются спирты и их смеси с водой. Благодаря хорошей
растворимости многих органических веществ в СхМесях спиртов с
бензолом, водой или диоксаном оказалось возможным исследовать
полярографическое поведение многих классов органических
соединений и проводить в указанных смесях их аналитическое
определение, а также исследовать кинетику и механизм многих
органических реакций .в неводных средах.
Найденные при исследовании поведения катионов щелочных
металлов в этаноле значения их диффузионных токов меньше, чем
в воде.
При использовании в качестве растворителей карбоновых
кислот наиболее подходящим индифферентным электролитом
являются их аммонийные соли. Отрицательная область потенциалов для
уксусной кислоты при этом ограничивается величиной —1,7 в; при
этом потенциале происходит выделение водорода. Последнее
обстоятельство делает невозможным определение таких катионов,
как А13+ или Сг3+.
Ледяная уксусная кислота является удобной средой для
исследования полярографического поведения соединений с хиноидной
структурой, а из неорганических ионов — главным образом ионов
таллия, кадмия, меди, уранила и палладия. Значения
диффузионных токов ионов Cd2+, Pb2+, Zn2+ в уксусной кислоте составляют
примерно 2/з величины предельных токов этих ионов для водных
растворов. В случае ионов, восстанавливающихся при очень
положительных потенциалах (например, Си2+) на полярограммах
образуются острые максимумы, которые можно подавлять некоторыми
поверхностно-активными веществами. В муравьиной кислоте
потенциалы полуволн катионов более положительны по сравнению
с водой, а максимумы I рода в значительной степени подавлены
поверхностно-активными свойствами молекул самой муравьиной
кислоты.
Достаточно распространенными растворителями являются
жидкий аммиак, амины и нитрилы. В жидком аммиаке щелочные
металлы разряжаются обратимо, их предельные токи
описываются уравнением Ильковича, а приведенные В. А. Плесковым
(табл. XII. 5) потенциалы полуволн, рассчитанные из значений
стандартных потенциалов, совпадают с экспериментальными.
Диффузионные токи щелочных металлов в аммиаке при одних и тех
Же концентрациях деполяризатора в 2—3 раза выше, чем в
водных растворах при 25°С. Ряд потенциалов полуволн щелочно-
305
земельных металлов в этих условиях имеет ту же
последовательность, что и в водных растворах.
Очень удобен в качестве растворителя этилендиамин,
поскольку в нем достаточно хорошо растворимы многие соли. Если
электродный процесс обратим, то по разности значений потенциалов
полуволн катионов в воде и в этилендиамине можно оценить
величину константы равновесия соответствующего этилендиамино-
вого комплекса в воде.
В ацетонитриле энергия сольватации катионов меньше, чем в
воде, поэтому большинство катионов восстанавливается в нем при
более положительных потенциалах по сравнению с водой.
Добавлением же к ацетонитрилу небольших количеств воды (от 0,4
до 1 вес.%) вызывает образование гидратированных катионов и
потенциалы полуволн сдвигаются к более отрицательным
значениям.
Помимо неорганических деполяризаторов в ацетонитриле
исследовалось поведение многих органических веществ. Было,
например, показано, что восстановление хинонов протекает через
образование семихинонов, а восстановление некоторых
углеводородов, например, стильбена и др. —с образованием устойчивых
анион-радикалов.
Из других растворителей в полярографии применяются
ацетон, уксусный ангидрид, хлорбензол, диметилформамид, диметил-
сульфоксид.
ЭЛЕКТРОХИМИЧЕСКОЕ ВЫДЕЛЕНИЕ МЕТАЛЛОВ
XIII. 4. Стадии электроосаждения
и перенапряжение процесса
Разряд катионов металла из раствора становится возможным,
когда выполняется условие
Е<Е° + ^\па^г+ (XIII. 5)
где Е — потенциал электрода, на котором разряжаются ионы
металла; правая часть неравенства выражает равновесный
потенциал металла в растворе его ионов.
При восстановлении ионов металла на катоде происходит ряд
последовательных процессов: подход сольватированных ионов
Mez+ • S* к электроду; деформация сольватной оболочки с
одновременным вытеснением чужеродных частиц с поверхности
электрода; освобождение ионов от сольватной оболочки; адсорбция и
миграция их к активным участкам электрода; разряд ионов и
включение образующихся атомов металла в кристаллическую
решетку.
В общем виде процесс разряда катионов металла можно
выразить следующим образом:
[Ме*+ • SjpacTI| + ze —► [МеЦ + xS (XIII. 6)
306
Уже это элементарное описание процесса разряда показывает,
что природа сольватной оболочки, а следовательно, природа
растворителя должны оказывать сильное влияние на процесс
электроосаждения металла. Кроме того, процесс электрохимического
выделения металла зависит от природы металла и условий
электролиза и может протекать различными путями, поэтому
последовательность стадий катодной реакции не является вполне
определенной. Например, неясно, во всех ли случаях адсорбция
восстанавливающихся ионов предшествует акту разряда,
образуются ли атомы металла на поверхности кристалла, или разряд
ионов происходит в момент их вторжения в кристаллическую
решетку. Впрочем, этот вопрос в большей степени касается общей
теории электроосаждения, чем специфических проблем влияния
растворителя на электролитическое выделение металлов.
Термин «перенапряжение выделения металлов» характеризует
те затруднения, которые возникают на различных' стадиях
процесса катодного осаждения металлов. Перенапряжение ц равно
разности между потенциалом электрода под током Е и
равновесным потенциалом Е°.
Общая теория перенапряжения подробно рассматривается во
всех курсах электрохимии и поэтому здесь на ней мы
останавливаться не будем.
Миграция ионов в электрическом поле уже рассматривалась в
связи с определением чисел сольватации ионов (гл. VIII),
описанием закономерностей электропроводности растворов (гл. X) и
изучением механизма переноса ионов (гл. XI). Закономерности
диффузии рассмотрены в гл. VI и частично в гл. XII (в связи
с возникновением диффузионных потенциалов). При этом было
показано, что скорость перемещения ионов зависит от свойств
растворителя.
Теория конвективного переноса на основе гидродинамических
представлений развита в работах В. Г. Левича (1944—1950).
Изменение заряда частицы на электрохимической стадии
приводит к изменению ее физико-химического и энергетического
состояния. Прежде чем перейти в кристаллическую решетку, ион
должен освободиться от связывающей его сольватной оболочки;
природа растворителя при этом оказывает значительное влияние,
так как обуславливает величину затрат энергии на процесс де-
сольватации иона. Кроме того, при пропускании тока через
электролит происходит изменение состава ионов в приэлектродном
слое по сравнению с составом ионов в объеме раствора, что
связано с концентрационными изменениями вблизи электрода.
Величина энергетического барьера (или энергия активации)
при осаждении металла представляет сумму трех слагаемых
UssaUtoe + UL + Ux (XIII. 7)
где f/ме — химическое сродство разряжающегося иона к металлу;
UL — сродство иона к раствору ; Ux — фактор, обусловленный
307
внешним электрическим полем и полями молекул, ионов и атомов
в двойном слое.
В ходе процесса электроосаждения металла происходит непре.
рывное изменение состояния поверхности катода, изменяется
соотношение между активной поверхностью электрода и пассивными
участками (покрытыми чужеродными адсорбированными части-
цами); поверхность катода ингибируется. Ингибирующее перена.
пряжение возникает в том случае, когда замедленность
электрохимической реакции обусловлена прочной адсорбцией
посторонних частиц на поверхности металла. Этот вид перенапряжения
связан с состоянием поверхности электрода и часто может
определять характер и кинетику электровыделения металлов вслед,
ствие влияния примесей поверхностно-активных веществ,
присутствующих в растворе.
Обычно для выяснения механизма электроосаждения металла
и определения основных закономерностей этого процесса изучают
зависимость перенапряжения процесса от токовых характеристик
реакции.
При катодном разряде величина перенапряжения г), когда
можно пренебречь влиянием структуры двойного слоя, запишется
в виде:
bn/K (XIIL8)
где а — коэффициент переноса; iK — катодный ток; 10 — ток
обмена.
Учет характера строения двойного слоя приводит к
видоизмененному выражению для перенапряжения:
*--Ш*пг*--ШгыЬ + (1-£)ъ <XIIL8a>
Третье слагаемое появляется потому, что поверхностная
концентрация разряжающихся частиц не равна их объемной
концентрации. Таким образом перенапряжение является функцией
строения двойного электрического слоя и зависит от g — потенциала
двойного слоя.
Уравнение (XIII. 8) было впервые предложено Фольмером
(1930), уравнение (XIII. 8 а) выведено А. Н. Фрумкиным. Теория
электрохимического перенапряжения получила дальнейшее
развитие в работах В. Г. Левича и его сотрудников.
Из анализа уравнений (XIII. 8) и (XIII. 8а), описывающих
явление электрохимического перенапряжения, следует, что
наиболее важными характеристиками перенапряжения являются
величины токов обмена i0 и коэффициентов переноса ос.
Токи обмена обычно относят к единице поверхности раздела
электрод — раствор; они служат кинетической характеристикой
равновесия между электродом и раствором при равновесном
значении электродного потенциала £°.
308
Если скорость медленной катодной электрохимической реакции
выразить при помощи уравнения Фольмера
i = ioC exp (— azFEJRT) (XIII. 9)
где С — ионная концентрация; Еи — потенциал поляризации, то из
него можно получить выражение для стандартного тока обмена i°0
i°o = k exp ( - azFE°lRT) (XIII. 10)
где k — константа скорости электродной реакции.
Таким образом, ток обмена зависит от природы
электрохимической реакции, материала электрода и состава раствора
(природы растворителя и концентрации). При одном и том же
отклонении потенциала электрода от равновесного значения скорость
процесса восстановления будет тем больше, чем выше ток обмена.
Коэффициент переноса а, введенный Эрдей-Грузом и Фоль-
мером в 1930 г., имеет величину в интервале от 0 до 1; в неявном
виде он появился уже в работах Батлера (1924 г.) при
рассмотрении влияния потенциала на скорости прямой и обратной реакции
(см. гл. XII).
Множитель а в уравнениях (XIII. 8) и (XIII. 8а)
характеризует степень влияния электрического поля электрода на энергию
активации электрохимической стадии и определяет симметричность
формы потенциальных кривых Батлера. При а = 0,5 ветви
потенциальной кривой симметричны, при отклонении от этого значения
симметричность нарушается. Для данной электродной реакции
коэффициент переноса сравнительно мало меняется при переходе
от одного электродного материала к другому.
Отличительной чертой реакции разряда на катоде
металлических ионов является также возникновение новой твердой фазы,
поэтому здесь необходимо учитывать перенапряжение, связанное
с построением кристаллической решетки. Эта стадия может играть
решающее значение в кинетике осаждения и обязательно должна
учитываться при осуществлении электролиза на твердых катодах.
Скорость процесса осаждения на различных его стадиях
зависит от свойств ионов, природы растворителя, условий
кристаллизации и других факторов, поэтому лимитирующими могут быть
различные стадии процесса.
Х1И. 5. Электроосаждение металлов
Общие положения
Из более семидесяти металлов периодической системы, лишь
около тридцати могут быть выделены электроосаждением из
водных растворов. В связи с этим в зависимости от природы металла
все металлы можно разделить на две группы. \ К первой из них
относятся те металлы, которые осаждаются на катодах при столь
отрицательных потенциалах, что достижение их в водных растворах
309
невозможно из-за выделения водорода (даже при использовании
электродов из металлов с высоким перенапряжением водорода).
Другую группу образуют все остальные металлы, которые в
принципе могут осаждаться из водных растворов.
Применение неводных растворителей представляет
значительный интерес особенно для получения тех металлов, которые
принципиально не могут быть выделены в свободном состоянии из
водных растворов.
На основании общих соображений могут быть сформулированы
весьма важные общие принципы, позволяющие решить вопрос о
выборе соответствующего растворителя для проведения
электролиза с целью выделения металла [Н. П. Федотьев, Р. Н. Кин-
кульская, 1935].
Прежде всего, растворитель должен обладать значительной
растворяющей и ионизирующей способностью^ позволяющей
получить высокую концентрацию и электропроводность раствора
электролита. Растворитель не должен химически реагировать с
продуктами электролиза и материалом электродов. Выделяющийся
металл не должен служить катализатором для реакций
полимеризации и конденсации растворителя. В крайнем случае он может
служить катализатором для таких превращений растворителя, в
результате которых получались бы вещества, не затрудняющие
процесс электролиза и не представляющие опасность из-за
легкой взрываемости или сильной ядовитости.
Растворитель должен быть электрохимически устойчивым и
позволять получать гомогенный раствор при сравнительно низких
температурах.
Вопросы растворимости и ионизирующей способности
растворителей рассматривались в гл. IX, где было показано, что только
учет физических свойств и химической природы растворителя, а
также структуры и свойств соли, позволяет делать обоснованный
прогноз о том, будет ли раствор являться проводником тока.
Однако, если образующиеся сольваты будут слишком
устойчивыми, то процесс осаждения будет затруднен.
Весьма важным обстоятельством при электроосаждении
металлов из неводных растворов является электрохимическая
устойчивость растворителей. Поскольку сами растворители имеют, как
правило, очень низкую электропроводность, то наименее
изученной областью в электрохимических исследованиях является
электролиз абсолютно чистых растворителей. В основном электролиз
проводился при внесении добавок, способствующих повышению
электропроводности. Так, электролиз безводного сернистого
ангидрида путем пропускания в течение шести месяцев малых токов
не привел к химическим изменениям; введение добавок иодида
калия приводило к анодному образованию 'иода на аноде и
образованию соответствующих иодидов с металлами анодов (Zn, Си).
Электролиз жидкого аммиака в присутствии различных
органических соединений приводит к образованию водорода и азота в сте-
310
хиометрическом отношении. Электролиз безводной
фтористоводородной кислоты в присутствии добавки KF приводит к
образованию водорода и фтора.
В' процессе электролиза хлорокиси фосфора,
электропроводность которой обусловлена автоионизацией, на катоде происходит
образование полимерной моноокиси фосфора: ЗРОС124+Зе—>
—* РО + 2РОС13. Аналогично протекает электролиз тионилхлорида.
При электролизе органических растворителей обычно
протекают сложные электродные процессы. Часто такие процессы
осуществляют с целью получения продуктов электроорганического
синтеза; этим вопросам посвящена специальная монография по
электрохимии органических соединений А. П. Томилова, С. Г. Май-
рановского, М. Я. Фиошина, В. А. Смирнова (1968). Здесь можно
привести лишь некоторые примеры. При электролизе формамида
образуется циануровая кислота с выходом по току до 80%; из
других амидов также получен ряд сложных продуктов
органического синтеза. Электролиз этилового эфира, содержащего А1С)3
и LiCl, приводит к образованию на катоде этилена.
Если ведение процесса имеет конечной целью
электровыделение металлов, процессы электроразложения растворителя неже-'
лательны, что накладывает дополнительные ограничения на
рецептуру электролитической ванны.
Осаждение металлов ,,неводной" группы
К этой группе относятся такие металлы, которые за редким
исключением не могут быть выделены электролизом водных
растворов их солей: щелочные и щелочноземельные металлы;
элементы III группы Периодической системы; элементы подгруппы
титана и ряд других.
Для щелочных металлов в литературе содержатся данные по
осаждению из неводных растворителей только лития, натрия и
калия. Электролиз простых солей лития в индивидуальных
растворителях (спирт, ацетон, пиридин) обычно приводит к
образованию катодных осадков, содержащих продукты внедрения
растворителя в осаждаемый металл, при этом процессы характеризуются
низкими выходами по току [R. Millie г, 1924]. Такая же картина
характерна для выделения натрия и калия. Однако введение
добавок веществ, способствующих образованию центров
кристаллизации может оказать существеБное влияние на скорость процесса
и качество осадка. Так, добавление ничтожных количеств солей
меди к растворам хлористого лития в диметилформамид
позволило в десятки раз увеличить плотность тока [Н. А. Зотов,
К. Б. X л ы с т о в а, 1966].
Намного лучше результаты получаются при осаждении
щелочных металлов из комплексных солей типа МеХ • А1Х3 (или
подобных) из органических растворителей. Работы В. А. Плотникова и
его школы показали, что хорошие осадки лития, натрия и калия
получаются при электролизе галогенидов (хлоридов и бромидов)
311
алюминия и щелочного металла в различных неводчых
растворителях. Особенно хорошие осадки и высокие выходы по току
характерны для смешанных растворителей* (нитробензол — ксилол).
Аналогично, при электролизе системы LiCl — TiCl4 — нитрометан
на платиновом катоде происходит осаждение металлического лития
[И. М. Подорван, И. И. Пен кал о, 1967].
Выделение щелочных металлов из неводных растворов является
перспективным в отношении замены существующих
высокотемпературных способов получения этих металлов путем электролиза
расплавов их солей.
Бериллиевые покрытия отличаются высокой коррозионной
стойкостью при повышенных температурах; этим объясняется высокий
интерес к электролитическому осаждению из неводных сред
бериллия и его сплавов. При электролизе смеси хлорида бериллия и
деметилбериллия в этиловом эфире Вуд и Бреннер [G. В. Wood,
A. Brenner, 1957] получили плотные осадки, содержащие не
более 95% Be. Наиболее чистые осадки бериллия получались из
раствора ВеСЬ в этиловом эфире при плотности тока 1 а/дм2
[Н. В. Коровин, 1962]. Для получения блестящих бериллиевых
покрытий может быть использован электролит, содержащий
раствор KF-2Be(C2H5)2 в этиловом эфире; процесс необходимо вести
при температуре 85 °С и катодной плотности иона 0,6 а/дм2. Сплав
Be —A1, содержащий 57% Be, мож^о осаждать из раствора
Ве(А1Н4Ь и ВеСЬ в эфире при температуре 18—20°С и плотности
тока 0,5 а/дм2. Плотные блестящие покрытия сплавом Be — В,
содержащим 70% бериллия, получаются из эфирных растворов при
плотности тока 0,6 а/дм2. При электролизе ВеС12 в безводном
аммиаке на электродах обнаруживается только газовыделение
[J. M e n z i e s, D. L. H i 11, L. W. О w e п, 1959].
При осаждении магния из неводных растворителей (пиридин,
этилендиамин, моноэтаноламин) на твердых катодах осадки, как
правило, получаются плохого качества. Выделение магния из
эфирных растворов( галоидов, гидрида, борогидрида или реактивов
Гриньяра позволяет получать сплавы магния с алюминием и
бором [J. H. С о n n о г, W. E. R e i d, G. В. W о о d, 1957].
Кальций,, стронций и барий не осаждаются из неводных
растворов на твердых катодах [А. Р. Stuart, 1957], но могут быть
осаждены из растворов в амине на ртутном катоде с
образованием амальгам соответствующих металлов [М. Hai'sinsky, 1937].
Электрохимическое осаждение алюминия из неводных
растворов привлекает особое внимание, связанное как с разработкой
метода получения металлического алюминия, так и с широким
использованием процесса алюминирования.
Электролиз системы бромид алюминия — этиловый эфир при
15—75 °С и катодной плотности тока 1—2 а/дм2 приводит к осаж-
* Применение смешанных неводных растворителей позволяет значительно
улучшить условия осаждения щелочных металлов и из простых солей.
312
дению алюминия; использование титановых или серебряных
анодов приводит к образованию соответствующих сплавов с
алюминием [А. Л. Левинскас, Ч. В. Ге р а с и м а в и ч у с, Я- Ю. С и-
нюс, 1967]. Значительный интерес вызывают в настоящее время
эфирно-гидридные электролиты, из которых при катодной
плотности тока 2—3 а/дм2 получается качественный катодный осадок
(белый электролитический алюминий) при использовании
алюминиевого анода [F. J. S с h m i d t, I. J. H e s s, 1966; А. Л. Левин-
c к а с, Я. Ю. С и н ю с, А. И. Ч е с н а в и ч у с, 1969].
В работах К. Циглера [К. Ziegler, 1956—1959] было
показано, что исключительно хорошими электролитами для
электрохимического выделения алюминия высокой чистоты (> 99,999)
с высокими выходами по току являются жидкие комплексные
соединения NaF.2Al(C2H5)3 и Ыа[А1(С2Н5)зР]-А1(С2Н5)з. Процесс
характеризуется малыми затратами энергии и пригоден как для
рафинирования алюминия, так и для нанесения покрытий.
Индий обычно осаждают из растворов на основе глицерина при
очень высоких плотностях тока (2000—5000 а/дм2). В этих
условиях индий или сплавы осаждаются в жидком состоянии за счет
локального повышения температуры на 50—80 °С, что исключает
возможность образования дендритов, рыхлых осадков, губки.
Выход по току составляет приблизительно 20—25%; скорость
осаждения металла достигает нескольких миллиметров в минуту.
Этот же способ используется при необходимости осаждения
сплавов на основе индия с кадмием, галлием, оловом, серебром
и т. д. [G. L. S с к о п о b I e, 1961].
Электрохимическое осаждение таллия можно осуществить из
его метаноловых перхлоратных растворов [F. J. Schmidt,
I. J. Hess, 1967]. При электролизе растворов Т1Вг3—А1Вг3 в
бромистом этилене с платиновым катодом и таллиевым анодом на
катоде образуется сплав А1: Т1 = 2 : 1 [В. Г. П р и х о д ч е н к о, 1953].
Германиевые покрытия обладают высокой коррозийной
стойкостью и высоким коэффициентом отражения, что дает
возможность использовать их в качестве рефлекторов. Электроосаждение
германиевых покрытий может применяться в технологии
изготовления монокристаллических пленок, которые повторяют структуру
подложки.
Несмотря на то, что потенциал германия не слишком
отрицателен, германий не удается выделить из водных растворов: обычно
осаждаются тонкие пленки, после чего процесс выделения
металла прекращается и выделяется лишь водород. Причина этого
явления возможно связана с более высоким перенапряжением
выделения германия на германии по сравнению с перенапряжением
на нем водорода или с возможностью образования GeH4*.
Блестящие плотные осадки германия толщиной до 25 мк
получаются из 5—7% раствора хлорида германия в этилен- или
* То же самое наблюдается для вольфрама и молибдена.
. 313
пропиленгликоле при температуре электролиза 50—60 °С и
плотности тока 0,4 а/дм2 [В. В. Остроумов, Т. В. Ананьева, 1964].
Для электроосаждения титана можно использовать растворы
его галогенидов в спиртах, эфирах или их смесях с небольшими
добавками толуола и смол; процесс ведется с графитовыми
анодами при комнатной температуре, и плотности тока 200 а/дм2.
При использовании ванн осаждения титана и циркония с
добавками борогидрида или гидрида алюминия получаются осадки,
содержащие только до 7% титана и до 40% циркония [W. E. Reid,
Jr. J. М. В i s h, А. В г e n n e r, 1957].
При электроосаждении молибдена и вольфрама использовались
растворы молибдата или вольфрамата натрия в формамиде
[А. Л. Левинскас, 1965]. Без применения добавок при
электролизе молибдата в формамиде при плотности тока 0,02—0,08 а/дм2
за 30—60 мин на катоде выделяется смесь соединений молибдена и
растворителя. Введение ионов SOT и ЭДТА позволяет получить
на катоде чистую поверхность молибдена со средним выходом по
току; значительно тормозится образование побочных продуктов
также введением добавок ацетамида, сульфаниловой кислоты,
бенаолсульфамида. При электролизе вольфрамата на катоде идет
образование вольфрамовой сини, поверхность электрода остается
чистой. Однако при добавлении к раствору Ы,1М-диметилформамида
под слоем вольфрамовой сини выделяется серебристый тонкий
слой металлического вольфрама.
Осаждение металлов „водной" группы
Электроосаждение меди может производиться с
использованием большого числа растворителей, таких, как пиридин,
фурфурол, жидкий аммиак, формамид, метанол, этиленгликоль, ацетон.
В многочисленных исследованиях было показано, что
электролиты с низкой растворимостью соли меди, как правило, дают
порошкообразные, плохо сцепленные с основой покрытия. В то же время
в электролите, обладающем высокой электропроводностью,
получаются крупнозернистые пористые осадки, которые не могут
служить покрытиями.
Пригодный для получения качественных медных покрытий
электролит содержит ацетат меди, растворенный в смеси уксусная
кислота — пиридин [I. A.Menzies, T. Broughton, 1965]. При
25 °С и плотности тока ~ 0,1 а/дм2 осаждаются зеркальноблестя-
щие, пластичные, прочно сцепленные с подложкой покрытия.
Хотя1 с увеличением плотности тока выход по току возрастает,
однако прочность сцепления уменьшается, а поверхностные
неровности увеличиваются. Режимы процесса и качество осадка зависят
от состава растворителя. Оптимальным режимом будет
ведение процесса при 50 °С, плотности тока 0,65 а/дм2 и объемной
концентрации пиридина в смеси с уксусной кислотой — 20%,
314
Блестящий зеркальный осадок серебра может быть выделен из
растворов его соли в жидкости Диверса (NH4NO3- n NH3); при
0°С и плотности тока 0,4 ма/см2 выход по току составляет около
65% [W. H u b i с k i, J. К о w a 1 с z у к, 1958].
Катодное выделение золота происходит з растворах НАиСЦ в
ацетамиде; при отсутствии избытка ионов С1~ в электролите
наблюдается склонность к образованию губчатых осадков
[И. Г. Е р у с а л и м ч и к, Е. А. Е ф и м о в, 1969].
Осаждение цинка можно осуществить путем электролиза
системы ZnCb — А1С13 — ксилол; при этом на катоде получаются
мелкокристаллические осадки [П. Ф. Кал южна я, Д. П. Зоси-
м о вич, 1955].
В литературе описаны методы электроосаждения кадмия из
растворов его солей в различных органических и водноорганиче-
ских растворителях. При осаждении кадмия из насыщенных
растворов Cdl2 в ацетоне при 20 °С, плотности тока 0,4 а/дм2 за 1—2 ч
осаждался слой плотного кадмиевого покрытия, не содержащего
посторонних включений, толщиной 15—20 мк [I. A. Menzies,
P. J. Morel and, S. R. О u 1 s n a m, 1967].
В растворах хлорида кадмия в ацетамиде на катоде происходит
осаждение металлического кадмия, причем в интервале
концентраций CdCl2 0,05—0,4 М выход по току близок 100% [И. Г. Ер уса-
л и м ч и к, Е. А. Ефимов, 1969].
Олово и свинец могут быть выделены из растворов их солей
в различных неводных растворителях; можно подвергать
электролизу системы SnCl2 — А1С13 — ксилол и РЬС12 — А1С1з — ксилол
[П. Ф. К а л ю ж н а я, Д. П. 3 о с и м о в и ч, 1955].
АНОДНОЕ ПОВЕДЕНИЕ МЕТАЛЛОВ
Все многообразие процессов, происходящих при
электрохимическом растворении металлов, можно разделить на две основные
группы: принудительное растворение металла с помощью
внешнего тока (анодное растворение) и самопроизвольное
растворение в результате химического воздействия окружающей среды
(коррозия.) Обе эти группы процессов являются реакциями
электрохимического окисления и. обладают как общими, так и
специфическими особенностями, причем часто происходит совместное
протекание обоих процессов.
Самопроизвольное растворение металлов в кислых растворах
в общем случае для двухвалентного металла протекает по схеме:
«Me + 2Н+ —> Ме2+ + Н2 (XIII. 11)
На такой процесс может влиять кислотно-основное равновесие:
НА ^z± H+ + А" (XIII. 12)
Существенную роль может играть и взаимодействие растворителя
г продуктами коррозии. Например, образование защитного слоя,
315
блокирующего поверхность металла по реакции
Me + 2HA —> МеА2 + Н2 (XIII. 13)
тормозит коррозию. Однако, если растворитель вступает в
реакцию с продуктами коррозии, образуя растворимый комплекс
МеА2 + xS —> Ме2+ • S* + 2А~ (XIII. И)
это приводит к ускорению процесса коррозии.
Вышеупомянутые процессы, как было показано в предыдущих
главах, зависят от сродства молекул растворителя к протону, их
полярности и диэлектрической проницаемости, а также от свойств
металла и образующихся соединений.
Все реакции анодного растворения, протекающие с участием
металла и раствора электролита, во многих чертах являются
обращением катодного выделения металлов. Общие реакции
анодного растворения металла, когда образуются простые или
комплексные сольватированные ионы, можно представить в виде
уравнений:
Me + xS —> Ме2+ ♦ S* + ze (XIII. 15)
Me + xS + yk" —> Жъкг-у -Sx + ze (XIII. 16)
Влияние растворителя на анодные процессы может
сказываться также в результате его электроокисления.
Анодное растворение металлов происходит при более
положительных потенциалах, чем соответствующие равновесные значения;
это вызвано анодной поляризацией, которая связана с
замедленностью стадий транспортировки ионов, разрушения твердой фазы
или ионизации атомов металла.
Так как свойства растворителя влияют на ток обмена и
коэффициент переноса, то они влияют и на скорость анодной реакции,
определяемую через плотность анодного тока /а:
/а = /0 exp (azFr\JRT) , (XIII. 17)
Величина анодного перенапряжения растворения металла г]а
выражается уравнением:
т)а = 2,303/?Г (Ig /а - Ig io)/azF (XIII. 18)
При изучении анодных процессов нужно учитывать также
влияние скорости диффузии и температуры на коррозионные
процессы в различных средах.
XIII. 6. Электрохимическое растворение металлов
В качестве растворимых анодов обычно используются металлы,
которые при наложении внешнего тока начинают переходить в
раствор в виде простых сольватированных или комплексных ионов.
316
Закономерно, что те металлы, которые характеризуются
малым перенапряжением при катодном выделении (большой ток
обмена), обладают малой анодной поляризацией; металлы,
характеризующиеся при катодном восстановлении малыми токами
обмена, имеют большую анодную поляризацию. Адсорбционный слой,
тормозящий разряд металлических ионов, должен тормозить и
обратную реакцию ионизации, причем каждое
поверхностно-активное вещество должно по-разному влиять на
электрокристаллизацию и анодное растворение металла. Такой вывод вполне
понятен, поскольку адсорбция поверхностно-активных веществ, а,
значит, и состояние адсорбционного слоя, как правило, зависят от
потенциала ионного слоя.
Приведем некоторые примеры анодного растворения
различных металлов.
Титан является одним из немногих металлов, анодно
растворяющихся в эфирно-гидридных электролитах. При растворении
титана в электролите LiH — А1С13 — этиловый эфир [А. Л. Л е-
винскас, Я. Я. Юстинович, 1969] раствор приобретает
коричневый оттенок, интенсивность которого пропорциональна
количеству растворенного титана. Одновременно с растворением на
аноде идет газовыделение; оба процесса протекают в широком
интервале плотностей тока — от 0,02 до 13 а/дм2. При высоких
плотностях тока на поверхности анода наблюдаются коричневые
диффузионные зоны растворяющегося металла. Такая ванна
позволяет осуществить процесс полирования поверхности титана до
зеркального блеска. Накопление титановых соединений в
электролите ведет к интенсификации анодного растворения титана.
Изучение электрохимического поведения кадмиевого анода в
расплавленном ацетамиде [И. Г. Ерусалимчик, Е. А.
Ефимов, Н. В. М о тыл ев а, 1969] показало, что растворение
кадмия происходит стадийно: Cd — е-> Cd+ и Cd+ — е -> Cd2+, причем
наиболее замедленной является вторая стадия. При высоких
плотностях тока на поверхности кадмиевого анода появляется белый
налет, что свидетельствует, вероятно, об образовании основных
хлоридов кадмия или комплексных соединений с ацетамидом.
Торможение процесса анодного растворения кадмия резко
увеличивается в присутствии добавок нейтральных солей и несколько
уменьшается при увеличении концентрации CdCl2 в электролите.
Сходные процессы наблюдаются при изучении анодного
поведения индия, золота и сурьмы в ацетамидных электролитах
[И. Г. Ерус ал имчик, Е. А. Е ф и м о в, Т. И. П р и л е п и н а,
Т. В. Гериш, 1969]. Исследование анодного растворения индия
в ацетамидных растворах 1пС13 показало, что процесс протекает в
три стадии. При низких плотностях тока самой медленной стадией
является стадия отщепления последнего электрона. При высоких
плотностях тока наиболее медленной стадией является
единственный электрохимический процесс — образование одновалентного
индия. Сульфаты и хлориды тормозят процесс растворения.
317
Анодный процесс растворения сурьмы в растворах SbF3 в ацет-
амиде протекает в две стадии, причем замедленной является от-
щепление двух электронов от иона одновалентной сурьмы;
соединений Sb5+ при электролизе не образуется.
Весьма интересными и перспективными являются процессы
электрохимического синтеза металлорганических соединений. Так,
в 1959 г. Циглер показал, что если в качестве электролита
использовать комплекс типа NaAl(C2H5)4, то при его электролизе с
алюминиевым анодом на аноде образуется триэтилалюминий. Позже
аналогичным путем Циглером [К. Ziegler, 1963] был
синтезирован тетраэтилсвинец.
При электролизе меркаптанов с ртутным анодом образуются
соединения типа R—SHg [R. В г dick a, 1942].
Имеются сведения о приготовлении электрохимическим путем-
комплексных соединений марганца, оловоорганических соединений
и других элементоорганических соединений, которые зачастую не
могут быть получены традиционным органическим синтезом
[А. П. Т о м и л о в, И. Н. Б р а г о, 1969].
При осуществлении процессов анодного растворения следует
учитывать возможность пассивации анодов за счет образования
окисных пленок, труднорастворимых солей и т. п.
Потенциал равновесного процесса образования окисной пленки
лежит положительнее потенциала основной реакции растворения,
поэтому именно в процессе растворения наиболее резко
проявляется влияние поверхностных окислов. Труднорастворимые соли
или комплексы также могут блокировать поверхность анода, тогда
растворение частично или полностью прекращается.
Так, Колотыркин и Коссый (1965) нашли, что в метаноловых
растворах соляной кислоты скорость растворения хрома зависит
от содержания воды в растворе только при наложении высоких
анодных потенциалов. В безводных электролитах хром не
пассивировался. Оказалось, что в изученных растворах при
определенном потенциале, который находится положительнее потенциала
пассивации и зависит от содержания воды, пассивирующий слой
на хроме разрушается ионами хлора. Эти результаты объясняются
зависящей от потенциала конкурирующей адсорбцией
пассивирующих молекул воды и активирующих ионов хлора.
При исследовании влияния воды и природы электролита на
анодное активирование титана в метаноловых растворах было
найдено, что потенциал активирования не зависел от плотности
тока, природы катиона и кислотности среды. Однако с увеличением
содержания воды и с изменением природы аниона в ряду
СГ < Вг~ < Г < СЮ7 < N07 потенциал активирования
сдвигался к более положительным значениям, что также объясняется
конкурирующей адсорбцией пассивирующих кислородсодержащих
групп и активирующих анионов [А. И. Цинман, В. С. Кузуб.
А. Н. Катревич, 1966].
318
Из приведенных данных следует, что помимо учета влияния
природы растворителя и добавок воды на механизм анодного
растворения металлов требуется учет конкурирующей адсорбции
молекул растворителя и ионов.
XIII. 7. Коррозия металлов
в неводных растворах
Коррозионное поведение металлов и сплавов в неводных
растворах, кроме чисто теоретического интереса о влиянии природы
растворителя на кинетику процесса, имеет-большое значение для
нефтяной и химической промышленности.
При растворении металла, например, в кислом растворе, на
скорость процесса может существенно влиять кислотно-основное
равновесие в системе растворителя. Может также сказываться
возможность взаимодействия растворителя с продуктами
коррозии: на поверхности может образоваться защитный слой,
тормозящий коррозию. Возможность применения таких процессов
зависит от свойств металла, химической природы растворителя,
кислотности среды и характеристик образующихся соединений. Кроме
того, нужно учитывать, что скорость диффузии может по-разному
влиять на коррозионные процессы в различных средах.
Исследование коррозии цинка и железа в растворах
муравьиной кислоты в органических растворителях [К. Schwa be, 1952]
показало, что при отсутствии кислорода и окислов протекает
лишь незначительное химическое растворение металлов, причем
ингибиторы действуют менее эффективно, чем в водных средах.
Коррозия цинка в безводных растворах НС1 в спиртах,
ацетоне, эфире сильнее, чем в водных растворах с той же
концентрацией кислоты [A. Bukowiecki, 1959], причем в случае спиртов
коррозия протекает с образованием алкоголятов, что препятствует
образованию защитной пленки.
Интересны данные по влиянию содержания воды на коррозию
в неводных средах [A. Bukowiecki, 1948]: при воздействии
спирта на магний, свинец или алюминий активное растворение
протекает лишь в безводной среде; добавка менее 1 % воды
пассивирует эти металлы.
Особое значение для практики имеет коррозия в смазочных
материалах, нефти и различных хлор- и сераорганических
соединениях Коррозия в этих средах в присутствии воды, как
правило, ускоряется: часто это выражается в возникновении питтин-
гов. Окислитель может также значительно повышать скорость
коррозии, действуя как катодный деполяризатор.
КИНЕТИКА РЕАКЦИЙ В НЕВОДНЫХ РАСТВОРАХ
По определению Н. М. Эмануэля «химическая кинетика — это
учение о химическом процессе, закономерностях его протекания во
времени и механизме». Теоретические основы химической
кинетики подробно излагаются в общих курсах физической химии и в
специальных монографиях по химической кинетике, в большом
числе опубликованных за последнее десятилетие.
В этой книге будет затронута лишь одна из многочисленных
проблем химической кинетики: особенности протекания
химических реакций в неводных средах. Решение этой проблемы в
общем виде предусматривает выяснение влияния растворителя как
на скорость, так и на механизм реакции.
Определить влияние растворителя на механизм реакции —
означает установить на молекулярном уровне, какие стадии
химического процесса протекают с участием растворителя, образуются
ли при этом промежуточные комплексы с растворителем, либо
выяснить, как физические свойства растворителя (чаще всего,
полярность молекул и диэлектрическая проницаемость) влияют на
особенности молекулярных превращений.
Известно, что многие органические реакции могут протекать
в различных направлениях, т. е. приводить к образованию разных
продуктов в зависимости от природы растворителя. Обсуждение
этого вопроса требует привлечения обширного материала из
курса теоретической,органической химии и нецелесообразно в рамках
этой книги, тем более, что этой проблеме посвящен ряд
специальных монографий (К. Ингольд, Л. Гамметт, Б. Чубар,
Р. Бреслоу и др.).
Качественное обсуждение проблемы влияния растворителя на
скорость реакций впервые' дано в классических исследованиях
Н. А. Меншуткина (1890). За истекший период этой проблеме
посвящены тысячи работ, где рассматриваются различного рода
корреляции между физическими свойствами растворителей и
параметрами протекающих в них реакций: порядком реакции,
константой скорости, квазитермодинамическими параметрами
процесса активации, вычисляемыми из температурной зависимости
320
константы скорости. Наибольшего успеха здесь достигла пока
теория, учитывающая электростатические взаимодействия. В
работах Кирквуда, Лейдлера, Амиса и других исследователей
получены количественные соотношения между константой скорости
реакции в данном . растворителе и диэлектрической
проницаемостью растворителя.
В тех же многочисленных случаях, когда влияние растворителя
не сводится к роли среды в электростатических взаимодействиях,
особенно при наличии специфической сальватации реагентов
растворителем, строго обоснованных количественных соотношений
найти не удается. Здесь количественные соотношения получаются
в частных случаях, с использованием эмпирических параметров.
Для выбора этих параметров используются так называемые
экстратермодинамические допущения (принцип «линейности
свободных энергий»), постулированные и развитые в работах Л. Гамме-
та, Р. Тафта, В. А. Пальма и многих других исследователей.
Вопрос о роли растворителя в кинетике химических реакций в
настоящее время не выяснен еще настолько, чтобы можно было
изложить весь накопленный экспериментальный материал в
рамках единой теории. Здесь будет лишь сделана попытка дать
введение в проблему и обсудить вкратце границы применимости
отдельных теорий.
1/3Ц Зак. 500
Глава XIV
ОБЩИЕ ВОПРОСЫ ВЛИЯНИЯ
СРЕДЫ НА СКОРОСТЬ РЕАКЦИЙ
Прежде чем переходить к обсуждению кинетических
характеристик реакций в различных растворителях, необходимо вначале
хотя бы качественно уяснить, какие же параметры могут
изменяться при переходе от одного растворителя к другому. Наиболее
чувствительна к перемене растворителя константа скорости
реакции; в отдельных случаях может измениться порядок реакции.
Замена растворителя сказывается также на величине параметров
уравнения, описывающего зависимость константы скорости
реакции от температуры.
XIV. I. Качественное обсуждение влияния среды
на константу скорости и энергию активации реакций
В общих курсах химической кинетики подробно обсуждаются
температурные зависимости скорости реакции [A. A. Frost,
R. G. Pearson, 1961] и отмечается, что, хотя эта зависимость
для сложных реакций может быть весьма разнообразной,
истинная константа скорости элементарного химического процесса
связана с температурой уравнением вида:
/г = Л ехр ( - £а/7?Г) (XIV. 1)
Предэкспоненциальный множитель А в классической теории
Аррениуса определяется как некоторый числовой коэффициент,
размерность которого совпадает с размерностью константы
скорости; Е& — энергия, необходимая для перевода молекулы в так
называемое «активированное состояние» (энергия активации).
В молекулярно-кинетической ■ теории столкновений
соответствующая зависимость представлена в виде
k = Z'f1* exp ( - EJRT) (XIV. 2)
где Z'— коэффициент, не зависящий от температуры и
определяемый (в случае бимолекулярной реакции) выражением:
Z' - 1<Г3Л0 (гА + гв)2 \8*R [ маМв)\ (XIV. 3)
(гА и гв— радиусы молекул; МА и Мв — молекулярные массы).
Наконец, теория переходного состояния (абсолютных скоростей
реакций) также приводит к экспоненциальному выражению
температурной зависимости скорости реакции
(XIV. 4)
It, \ S.\J. f 4, \ i.\* / \ Л\ /
322
где % — трансмиссионный коэффициент, учитывающий вероятность
перехода активированных комплексов в продукты реакции; W —
постоянная Больцмана; AGcT, Д#<Г и AS<f — изменения
изобарного потенциала, энтальпии и энтропии системы при образовании
активированного комплекса в стандартных условиях, т. е. при
концентрации исходных веществ и активированных комплексов
1 моль/л.
Квазитермодинамические параметры процесса активации в
уравнениях (XIV. 1), (XIV.2) и (XIV.4) связаны между собой
соотношениями
Ea = Ec + ±RT (XIV. 5)
Еа = Д#<f + RT (XIV. 5а)
а предэкспоненгу А уравнения (XIV. 1) можно выразить через
энтропию активации:
In A = In (%k'Tlh) + AS^/tf (XIV. 6)
С помощью уравнений (XIV. 1), (XIV. 4) и (XIV. 5а) могут быть
раздельно определены A Go*, A#<f и ASo*.
Как следует из уравнений (XIV. 5) и (XIV. 5а), энергетические
параметры процесса активации в классической и молекулярно-ки-
нетической теориях, а также в теории переходного состояния
отличаются сравнительно мало при обычных температурах.
Действительно, величина энергии активации реакций колеблется обычно
в пределах 40—100 кдж/моль, а величина RT при комнатной
температуре составляет около 2,5 кдж/моль, что близко к
погрешности определения. В современной литературе для обработки
экспериментальных- данных чаще всего используют уравнение
(XIV. 4), как наиболее обоснованное теоретически.
Количественная трактовка проблемы влияния растворителя на
кинетику реакций должна включать, по-видимому, определенные
соотношения между квазитермодинамическими параметрами
процесса образования активированного комплекса и
характеристиками растворителя. В общем виде эта задача не решена, однако
само наличие такой связи сейчас не вызывает сомнений.
Количественный учет влияния растворителя на кинетику
процесса мог бы быть выполнен весьма наглядно, если бы имелась
возможность раздельно оценить энергию всех взаимодействий в
растворе [реагентов — между собой, реагентов, промежуточного
состояния (активированного комплекса) и продуктов реакции —
с растворителем]. Тем самым можно было бы выделить вклад
растворителя в величину свободной энергии (изобарного
потенциала) активации реакции и уже на основании этих данных
установить зависимость от природы растворителя величины пред-
экспоненты Л, связанной уравнением (XIV. 6) с энтропией
активации.
V.ii* 323
Один из путей решения этой проблемы заключается в
представлении изобарного потенциала активации реакции в виде
суммы [О. А. Ершов и др., 1960]
AG* = AGf + ДС^ЛЬВ + ДССОЛЬВ (XIV. 7)
где AGcf — изобарный потенциал активации данной реакции в
вакууме, AG^bB и Абсольв— изменения изобарного потенциала
при сольватации активированного комплекса и реагентов.
О. А. Ершов (1961) предположил, что ДОс^льв мало зависит от
растворителя и, следовательно, вклад растворителя в свободную
энергию активации определяется главным .образом сольватацией
реагентов. Из этого, в частности, следует зависимость AG^ от е
растворителя для ионных и ионо-дипольных реакций,
аналогичная такой же зависимости для энергии сольватации (ом. гл. VI).
При рассмотрении реакций между дипольными молекулами
можно использовать аналогичный подход, учитывая вместо
энергии сольватации энергию взаимодействия реагента с
растворителем *. Термодинамический цикл для расчета энергии активации
реакции через теплоты смешения реагентов с растворителем
рассмотрен в работе Н. К. Воробьева и Л. В. Курицына (1968) на
примере реакции анилина с хлористым бензоилом в различных
растворителях. Точнее, в этой работе определяется не энергия
активации, а энтальпия образования активированного комплекса в
жидкой фазе Д#<ж)» выражаемая соотношением
д# (*} = д#£} + 6 (дя ?) - 6 (дя°°) (XIV. 8)
где Д#(£) — энтальпия образования активированного комплекса в
газовой фазе; б(Д#?)—алгебраическая сумма теплот испарения
реагентов и активированного комплекса [б (АН°) = Д#Х + А#в —
—AHf) и б(ДЯГ)—аналогичная сумма парциальных молярных
теплот смешения каждого из реагентов и активированного комплекса
с растворителем при бесконечном разведении.
Г. И. Лихтенштейн (1970) представил эффективные значения
энергии и энтропии активации в виде суммы двух членов, один из
которых отражает вклад собственно химического акта, другой —
вклад процесса обратимой перестройки растворителя,
обусловленной кооперативными эффектами взаимодействия молекул
растворителя за пределами первой координационной сферы
растворенного вещества. Точная количественная оценка этих параметров
представляется в настоящее время весьма проблематичной.
Более подробное обсуждение зависимости
квазитермодинамических параметров реакции от природы растворителя будет дано
ниже, при рассмотрении отдельных типов реакций.
* Разница между этими понятиями по существу терминологическая (см.
гл. V, VII).
324
XIV. 2. Основные отличия реакций,
протекающих в газовой фазе и в растворе
Различия в условиях протекания реакций в газообразной и
жидкой фазах определяются прежде всего отклонением в
поведении реагирующих молекул в жидкой фазе от законов,
описывающих газовое состояние. В общем виде учет этого отклонения
можно осуществить с помощью коэффициентов активности,
отражающих всю совокупность химических и нехимических
взаимодействий молекул реагентов с молекулами среды. Для более
детального анализа желательно разделить эти типы
взаимодействий и учесть их относительные вклады. Если бы последнее
удалось, то можно было бы выяснить, какие именно свойства
растворителя в каждом конкретном случае определяют скорость и
механизм реакций, протекающих в растворе.
Удобнее всего было бы решать такую задачу путем
сопоставления параметров одной и той же реакции в газовой фазе и в
различных растворителях. Однако на практике так поступают весьма
редко. Бенсон (1964) отмечает, что «из многих тысяч реакций,
изученных в растворах, лишь менее 20 могут быть изучены в газовой
фазе». Основная сложность заключается в несоизмеримости
скоростей, с которыми протекает реакция, в каждой из фаз.
Если же удается подобрать растворитель, константа скорости
реакции в котором близка к таковой в газовой фазе, то возникает
вопрос, обусловлено ли это близким совпадением и предэкспонен-
циального и энергетического члена, либо компенсационным
изменением обоих этих факторов. Физическая сущность такой
постановки вопроса заключается в следующем. Если растворитель
заведомо не взаимодействует ни с исходными веществами, ни с
активированными комплексами, ни с продуктами реакции, то
свободная энергия активации реакции в нем должна быть такой же,
как и в газовой фазе. Тогда различие в константах скорости
определится различием чисел столкновений (по молекулярнокине-
тической теории) или трансмиссионных коэффициентов (по теории
переходного состояния).
Мельвин-Хьюз предположил ранее (1933 г.), что можно
подобрать инертный растворитель, скорость реакции в котором
равнялась бы скорости реакции в газовой фазе. В этом случае
было бы одинаковым число столкновений, испытываемых
реагирующими молекулами в той и в другой системе. Базируясь на
данных, полученных в таком инертном (обычно неполярном)
растворителе, можно было бы проводить сопоставления с кинетикой
реакций в других растворителях.
Однако представление о возможности равенства числа
столкновений в газовой фазе и в инертном растворителе не
подтвердилось при более строгом теоретическом анализе. Рабинович (1937)
и Белл (1937), исходя из различных допущений, показали, что
11 Зак, 500 325
частота столкновений в растворе, даже при условии отсутствия
специфических взаимодействий, примерно втрое выше, чем в
газовой фазе. Естественно отличаются и скорости реакций.
Увеличение числа столкновений в жидкой фазе может быть
объяснено так называемым «клеточным эффектом». Этот эффект
обусловлен наличием определенной структуры ближнего порядка
в жидкости. Реагирующие молекулы попадают в элемент
структуры — «клетку» из молекул растворителя, вследствие чего они не
могут сразу разойтись после столкновения, и прежде чем хотя бы
одна из столкнувшихся частиц успевает вырваться из «клетки»,
между ее и другими частицами могут происходить
дополнительные соударения. Особенно существенное значение имеет
клеточный эффект для некоторых реакций с участием свободных
радикалов, когда энергия активации относительно мала или близка к
нулю.
Количественное сопоставление констант скоростей реакций в
газовой фазе и в растворе удобно провести в рамках теории
переходного состояния. При этом целесообразно использовать
условное понятие о константе равновесия между реагирующими
частицами и активированным комплексом. Это равновесие неадэкват-
но обычному химическому равновесию, поскольку активированный
комплекс представляет собой не индивидуальное химическое
соединение (хотя бы неустойчивое), а лишь промежуточную
конфигурацию атомов в ходе реакции. Однако введение понятия о
«квазиравновесии» приводит к правильным количественным
результатам, поскольку скорость реакции пропорциональна концентрации
активированных комплексов, а последние находятся с исходными
молекулами в статистическом равновесии [М. И. Темкин, 1958].
Связь между константой скорости и условной константой
равновесия выражается для реакций в газовой фазе соотношением
kg = t^K£ (XIV. 9)
а для реакции в жидкой фазе:
ks = t¥l~Kf (XIV. Ю)
Пренебрегая различием трансмиссионных коэффициентов в
обеих фазах, находим:
ksjkg = KflKf (XI V.I 1)
Для идеальных газов и идеальных растворов справедливы,
соответственно, следующие соотношения:
TI (XIV. 12)
К? =*s?7*a*b (XIV. 13)
326
Для приведения к одной размерности констант уравнений
(XIV. 12) и (XIV. 13) могут ^быть использованы известные из
термодинамики соотношения:
(XIV. 14)
n (XIV. 15)
где А/г — изменение числа молей в результате реакции; VM —
мольный объем растворителя (раствор настолько разбавлен, что
изменением объема за счет растворенных веществ можно
пренебречь).
Используя два последних равенства и закон Рауля РА = Р°АхА,
находим:
оо
kslkg в Щ^ (i?WM)M (XIV. 16)
Рф
Соотношение (XIV. 16) справедливо в случае идеальных
растворов. Трудность его практического использования заключается в
том, что оно требует данных о свойствах активированных
комплексов. Величину же р°ф можно определить с помощью уравнения
(XIV. 9), если только определено значение трансмиссионного
коэффициента (что далеко не всегда удается осуществить независимо)
и известна концентрация активированного комплекса хф.
Поэтому целесообразнее с помощью уравнения (XIV. 16)
рассчитывать величину р°ф для данной реакции и затем производить
вычисления для других идеальных растворителей.
Таким образом, вопрос о сопоставлении скоростей реакций в
газовой фазе и в идеальном растворителе может считаться в
принципе решенным, если только можно с достоверностью
утверждать, что выбранный растворитель является идеальным.
Сопоставление скоростей реакций в идеальном растворе и в
реальных растворах также можно осуществить, исходя из
термодинамических соображений. Для этого достаточно ввести в уравнение
(XIV. 16) отношение коэффициентов активностей реагентов и
активированного комплекса в данном растворителе:
* А - Т
В упрощенной форме это соотношение впервые было выведено
Бренстедом (1925).
Однако введение коэффициентов активности не раскрывает
физической сущности действия растворителя. В общем случае это
действие может быть обусловлено:
неспецифическими взаимодействиями за счет вандерваальсовых
и дисперсионных сил;
И* 327
электростатическими взаимодействиями между ионами и
между диполями;
явлениями специфической сольватации молекул исходных
веществ, активированных комплексов и продуктов реакции
растворителем.
Разумеется, каждый из перечисленных эффектов нелегко
выделить в чистом виде, и потому дальнейшее обсуждение будет
носить в определенной степени условный характер. В зависимости от
преобладающей роли того или иного эффекта в одном случае
будут существенными такие свойства растворителя, как вязкость и
энергия когезии, в другом — диэлектрическая проницаемость и
дипольный момент, в третьем — его химические свойства. Те или
иные свойства растворителя приобретают определяющее значение
в зависимости от типа и (или) механизма реакции.
XIV. 3. Реакции между неполярными молекулами
в растворах электролитов
Оценка влияния растворителя на реакции, протекающие в
неэлектролитных регулярных растворах, может быть выполнена на
базе теории Гильдебранда (см. гл. V), в которой коэффициент
активности растворенного вещества определяется как функция
разности плотностей энергии когезии (параметров растворимости)
растворенного вещества и растворителя.
Для разбавленных растворов справедливо соотношение
RT\nyi^VUi(6i-6sy (XIV. 18)
где индекс i относится к частицам растворенных веществ.
Обозначим разность параметров растворимости между реагентами А, В
и активированным комплексом, с одной стороны, и
растворителем — с другой, соответственно, через Ал, Аб и А^. Запишем
уравнение (XIV. 17) в упрощенной форме (по Бренстеду), исключив
все коэффициенты, введенные для соблюдения размерностей.
Тогда
ln (kJkg) =ln Ya + In YB - ln Уф (XIV. 19)
Подставляя в это уравнение значения коэффициентов активности
из уравнения (XIV. 18), находим:
1п (W " ~W 0ЧАа + У«в^ ~ V"A*) (XIV'20)
Для полуколичественной оценки величин с помощью этого
уравнения необходимо ввести упрощающие допущения. В качестве
первого такого допущения предположим, что разности параметров
растворимости близки для реагентов и активированного
комплекса, т. е. ДА — Ав — А^ = Д. Тогда
ln W - w (*Ч+Ч - v») (XIV-
328
Второй путь упрощения уравнения (XIV. 20) заключается в
предположении равенства мольных объемов реагентов и
активированного комплекса. В этом случае:
ln (KIK) - "fe К + 4 - А*) (XIV. 22)
Учитывая вариации параметров растворимости и мольных
объемов для неполярных соединений (см. гл. V), можно оценить
интервал изменений константы скорости реакции в растворе по
сравнению с газовой фазой, обусловленный неспецифической
сольватацией. Отношение ks/kgf исходя из этих данных, может
изменяться в пределах от 0,2 до 25.
Рассмотрим качественно основные положения,
характеризующие влияние параметров растворимости на скорость реакции.
Если близки параметры растворимости растворителя и
активированного комплекса, то скорость реакции близка к скорости в
идеальном растворе. Если же, напротив, близки параметры
растворимости реагентов и растворителя, а величина 6^ существенно
отличается от них, то и скорость реакции существенно отличается
от скорости в идеальном растворе. Определение свойств
активированного комплекса представляет собой весьма сложную задачу,
поэтому до сих пор сохраняют значение эмпирические правила,
сформулированные Ричардсоном и Сопером (1929), независимо
от теории регулярных растворов:
1. Если продукты реакции обладают большей когезией,
нежели реагенты, то реакция ускоряется растворителем с большей
когезией;
2. Если продукты реакции обладают меньшей когезией, нежели
реагенты, то растворители с высокой когезией тормозят реакцию;
3. Если когезии реагентов и продуктов близки, то влияние
растворителя незначительно.
Таблица XIV.1
Константы скорости бимолекулярной реакции конденсации
малеинового ангидрида с 1,3-бутадиеном в различных
растворителях при 25°С
[К. F.Wong, С. А. Е с k e r t, Trans. Faraday Soc, 66, 2313, (1970)].
Растворитель
Нитрометан .......
Дихлорметан
Бензол
Сероуглерод
Четыреххлористый углерод
Уксусноэтиловый эфир . .
Этиловый эфир
Параметр
растворимости, кдж1ум1*
24,76
20,25
18,72
20,46
17,60
18,50
15,75
k,
л/(моль-ч)
0,18
0,13
0,12
0,11
0,11
0,06
0,03
329
Данные, иллюстрирующие влияние плотности энергии когезии
(внутреннего давления) растворителей на скорость реакции,
приводятся в табл. XIV. 1. Они свидетельствуют о приближенной
корреляции между константой скорости рассматриваемой реакции и
параметрами растворимости растворителей. Соблюдение этой
корреляции можно отнести за счет отсутствия заметного химического
взаимодействия между реагентами и растворителями и
образования сравнительно малополярного переходного соединения. В
случае реакции Меншуткина такая корреляция качественно не
соблюдается.
Описание поведения реакции усложняется в случае, когда
реакция протекает в смешанном растворителе. Растворенные
вещества могут образовывать регулярные растворы с компонентами
растворителя, либо давать продукты присоединения с одним
растворителем. Анализ этих данных на основе теории растворов дан,
в частности в работах Кондо и сотрудников [Y. Коп do, N. То-
kuraa. oth., 1964—1969].
XIV. 4. Влияние диэлектрической проницаемости среды
на скорость реакций с участием ионов
и полярных молекул
Реакции между ионами
В случае реакций между ионами влияние растворителя
сказывается прежде всего на изменении электростатических сил,
зависящих от зарядов ионов, межионных расстояний и
диэлектрической проницаемости растворителя. Последняя, согласно закону
Кулона, влияет на величину электростатических сил межионного
притяжения.
Количественное описание этих эффектов может быть дано на
основе теории Дебая — Хюккеля — Онзагера как для водных
растворов, так и для неводных растворов с достаточно высокой
диэлектрической проницаемостью. Для растворов с малой е
соответственно снижается верхняя граница концентрационного
интервала, в котором применимы теории сильных электролитов
(см. гл. X).
Применение теории сильных электролитов к расчету скоростей
реакций между ионами может быть .сведено к определению
активностей ионов и промежуточных комплексов и подстановке
полученных данных в уравнение (XIV. 17). Скэтчард, основываясь на
теории Дебая — Хюккеля, и Лейдлер, рассчитывая изменение
свободной энергии иона за счет электростатического
взаимодействия, пришли к аналогичным выводам. Выражение для константы
скорости реакции имеет вид
in к = in kQ - zAzBe2R/erAQNQT (XIV. 23)
330
где k0 — константа скорости реакции в среде с бесконечно
большой диэлектрической проницаемостью; гА и гв — заряды ионов;
Гав — расстояние максимального сближения между ионами.
Проверка этого уравнения производилась в большинстве
случаев не в различных растворителях, отличающихся значениями е,
а в водно-неводных смесях, причем неводный компонент
выбирался со сравнительно низкой е. Например, получены линейные
зависимости 1п£(1/е) для реакции между ионами NHt и CNO" в
водно-метанольной и водно-гликолевой смесях. Почти во всех
случаях прямолинейная зависимость сохраняется только в
определенном интервале концентраций смешанного растворителя и
претерпевает излом в области малых значений е. Используя значения
тангенсов углов наклона прямолинейных участков, можно
рассчитать величины гАв для различных ионных реакций.
Важно также оценить влияние растворителя на величины
квазитермодинамических параметров процесса активации.
Электростатический вклад в изобарный потенциал процесса активации
можно выразить соотношением:
^Gti = N0e2zAzBlerAE (XIV. 24)
Дифференцируя это выражение по температуре, определяем
электростатический вклад в энтропию активации:
Noe zAzB I dine
8/*AB
дГ jp
(XIV. 25)
Электростатический вклад в энтальпию активации может быть
определен с помощью обычного термодинамического соотношения:
АЯ^ = Д6Й +T&Sm. Значения (д In г/дТ) для различных
растворителей приведены в табл. XIV. 2.
Таблица XIV.2
Температурные коэффициенты диэлектрической проницаемости
некоторых растворителей
Растворитель
Гексан
Четыреххлористый
углерод
Бензол . ;
Толуол
Анизол
Хлороформ
Хлорбензол
(д In e\
\ дТ )Р
0,71. Ю-3
0,843
0,876
0,673
5,2
3,33
2,89
Растворитель
Бромэтил
Ацетофенон
Ацетон
Этанол
Метанол
Нитробензол
Вода
(д In e\
V дТ )р
4,91
4,10
4,63
6,02
5,39
5,21
4,63
331
Поскольку коэффициенты активности ионов, согласно теории
сильных электролитов, зависят от ионной силы раствора, можно
рассмотреть также влияние этого параметра на скорость реакции
между ионами. Основное уравнение, полученное на базе теории
Дебая-Хюккеля, имеет вид
\g k = lg k0 - 2QzAzB VT (XIV. 26)
где / — ионная сила раствора, a Q определяется из соотношения:
Q = А#2е3 (2я8/2)/2,303 (10е2ЯГ)3/2 (XIV. 27)
Это уравнение обычно хорошо соблюдается в разбавленных
водных растворах. Наблюдаемая в неводных растворах ассоциация
ионов обусловливает отклонения от уравнения (XIV. 27)
Реакции между ионами и диполями
Выражение для скорости реакции между ионом и дипольной
молекулой, полученное Амисом, аналогично уравнению (XIV.23)
In k = In kQ + zenR/er2ABNQT ' (XIV. 28)
где (li — эффективный дипольный момент молекулы (по Онзагеру).
Анализ применимости этого уравнения, так же как и уравнения
(XIV. 23), практически тоже сводится к построению зависимости
In k—1/е при постоянной температуре. Амис показал, что
уравнение (XIV. 28) хорошо описывает экспериментальные данные по
кислотному и щелочному гидролизу сложных эфиров в
водно-органических смесях и приводит к разумным значениям расстояния
наибольшего сближения между реагирующими частицами.
Реакции между дипольными молекулами
Свободная. энергия диполя в среде с диэлектрической
проницаемостью е при условии учета только кулоновских сил
определяется, согласно Кирквуду, выражением
Д(/Эл = - \х2 (е - 1)/г3 (2е + 1) (XIV. 29)
где |ы — дипольный момент; г — радиус молекулы.
Используя уравнение теории переходного состояния,
связывающее константу скорости с изменением свободной энергии
активации, а также уравнение (XIV. 29), Лейдлер и Эйринг
получили следующее выражение:
In k = In k0 \-~- + -f- - -zr + —- > Ф/ (XIV. 30)
RT 2e + 1 - "
В этом уравнении принято, что диэлектрические проницаемости
разбавленных растворов и чистого растворителя равны. Член
332
-1,0 ■■
-2,0-
0,38 Q42 0,46
е-1
* огшсывает вклад некулоновских взаимодействий в
свободную энергию активации реакции. Если этот вклад относительно
невелик либо может считаться постоянным, то из уравнения
(XIV. 30) следует линейная зависимость \g k от (е—1)/(2е+1).
Большое количество
экспериментальных работ
посвящено проверке
соблюдения этой зависимости
в средах с широко
варьирующимися значениями е.
Удобными моделями
для исследования
влияния е растворителя на
скорость реакций служат
смешанные
водно-неводные растворители;
большое количество
экспериментальных данных для
таких систем получено, в
частности, Томмила [Е.
То mm И а, 1967]. Мы
ограничимся здесь
примером реакции
каталитического разложения
гидроперекиси трет-бутила в
смесях уксусной кислоты
с водой [Э. А.
Кузьмина, М. К. Щенникова,
1964].
Для этого случая
линейная зависимость
от функции
0,38 0,42 0,46
£-1
2S-H
0,46 0,48
еЧ
ТёТГ
Рис. XIV. 1. Зависимость кинетических пара-
К пкт/ття метРов реакции разложения гидроперекиси
- - - трет-бутила. в смеси уксусной кислоты с во-
(е— 1)/(е+ 1) СООЛЮда- дой От функции Кирквуда:
еТСЯ Не ТОЛЬКО ДЛЯ ЛОГа- а —зависимость константы скорости; б —зависимость
рифма КОНСТаНТЫ СКОрОС- ^Н^ и AS^; в -зависимость предэкспоненты А.
ти, но и для логарифма
предэкспоненты Л уравнения (XIV. 1) и для квазитермодинамических
параметров активации Д#^, AS^=. Это иллюстрируется рис. XIV. 1.
Значительно сложнее обстоит дело в случае смесей
растворителей, образованных компонентами различной химической
природы. Некоторые примеры показаны на рис. XIV. 2.
Линейная зависимость \gk от функции Кирквуда, как
выяснилось, хорошо описывает данные по реакциям окисления
полярных органических веществ в различных растворителях, включая
аполярные. Г. Е. Заиков и 3. К. Майзус (1969) измерили
константы скорости реакций продолжения и обрыва цепей при
окислении метилэтилкетона в растворах декана, /г-дихлорбензола и
33?
четыреххлористого углерода и, пользуясь упрощенной формой
уравнения (XIV. 30), вычислили значения дипольных моментов
(я^ активированных комплексов, образующихся в указанных
реакциях. Аналогичные результаты получены в бензоловых и
уксуснокислых растворах. Однако для растворов в хлорбензоле
найдены существенные отклонения от линейной зависимости
\gk — (е—1)/(2е + 1), которые объяснены
авторами с помощью предположения о
специфической сольватации в хлорбензоле
радикалов, образующихся при окислении.
Более простое уравнение для констант
скорости диполь-дипольных реакций
получено Амисом, также с учетом только
электростатических взаимодействий, для
случая, когда дипольные моменты реагентов
имеют одинаковое направление и
расположены вдоль одной прямой:
1дк
2,0
3,0
4,0
5,0
(XIV. 31)
0,44 0,46 0,48 £-1
2е+1
метилформ амид; 8 — диметил-
сульфоксид
[И. А. К о п п е л ь,
В. А. Пальм, Реакц.
способ,, орт. соед., 4, № 4, 892,
1967].
Уравнение Амиса, как и в случае ион-
ных реакций, позволяет рассчитать гАв—
R реакции сольвол^а расстояние, на которое должны сблизиться
урег-бутилхлорида от диполи, чтобы прореагировать. Как указы-
функции Кирквуда для вает сам Амис, полученные значения гАв
некоторых бинарных сме- нельзя считать достоверными. Однако не-
/С!ЙводРаИ (0,40 мол. долей); сомненное достоинство уравнения (XIV. 31)
2- метанол; з - этанол; заключается в том, что оно, будучи более
TT6lt простым, с не меньшей точностью
описывает экспериментальные данные, нежели
уравнение (XIV. 30).
Вполне естественно ожидать, что ни
уравнение (XIV.30),hh уравнение (XIV.31)
не будут удовлетворительно описывать
изменение константы скорости реакции в случаях, где возможна
специфическая сольватация реагентов. Это подтверждается
рассмотрением кинетических данных для реакции Меншуткина и
реакции сольволиза гр£г-бутилхлорида, (рис. XIV. 3). Интересно, что
даже для растворителей одной химической природы —
алифатических спиртов точки, соответствующие метанолу и этанолу,
не укладываются на одну прямую с высшими гомологами
(рис. XIV. 3,6).
Таким образом, раздельное рассмотрение реакций,
протекающих в электролитных и неэлектролитных растворах, позволило до
сих пор выявить роль только двух факторов: энергии когезии
(параметра растворимости) растворителя (качественно) и
диэлектрической проницаемости растворителя (полуколичественно).
Теперь можно перейти к описанию свойств растворителя, имеющих
334
существенное значение как для электролитов, так и для
неэлектролитов. Сюда относятся химические свойства растворителя, от-
а
ЦЬ+4
1.В
1,2
0.8
ОА
8
с
J
о
2
о
0,35
ОАО
о9
0А5 е-1
о2
4°
оЗ
0,1 0,2 0,3 ОА 0,5
Ю 2еТТ
Рис. XIV. 3. Несоблюдение уравнения Кирквуда:
а —Реакция Меншуткина в различных растворителях при 30 °С:
/ — хлороформ; 2 — хлорбензол; 2 — ацетон; 4 — циклогексанон; 5— пропионитрил; 6 — бензонйтрил;
7—нитробензол; 5 —1,2-дихлорэтан; 9 — хлорциклогексан; 10 — бутанон 2 [Н. Hartraann,
А. Р. S с h m i d t, Z. phys. Chem. (NF), 66, 183 [1969)]
б— Сольволиз трег-бутилхлорида в различных растворителях при 120 °С:
2 —гептан; 2 — бензол; 3— этиловый эфир; 4 — хлорбензол; 5—ацетон; 6 — нитробензол; 7
—метанол; 8 — этанол; 9— пропанол; 10 — бутанол; // — гексанол; 72 — октанол; 13— фенол
[И. А. Коппель, В. А. П а л ь м, Реакцион. способ, орг. соед., 4, 862 (1967)].
ветственные за специфическую сольватацию реагентов, и
некоторые физические свойства: вязкость, сжимаемость и др.
XIV. 5. Специфическая сольватация реагентов
и скорость реакций
В огромном числе реакций, особенно органических,
растворитель выступает не как инертная среда, а как вещество
определенной химической природы, оказывающее влияние на скорость и
механизм реакции путем специфической сольватации исходных
веществ, активированных комплексов и продуктов реакции.
Специфическая сольватация обусловлена обычно образованием
водородных или донорно-акцепторных связей.
Если активированный комплекс прочнее сольватирован
растворителем, нежели реагенты, разность энергий активированного
комплекса и реагентов меньше, чем при отсутствии сольватации,
и, следовательно, реакция протекает с меньшей свободной
энергией активации. Наоборот, в случае большей сольватации
335
реагентов по сравнению с активированным комплексом энергия
активации реакции возрастает.
Для оценки способности растворителя к специфической
сольватации необходимо найти определенную меру электронодонорной
или электроноакцепторной способности веществ. В качестве такой
меры предложены различные параметры, например, донорное
число по Гутманну, растворимость НС1 (мера кислотности), теплота
смешения с хлороформом (мера основности) и ряд других
параметров полярности.
Влияние специфической сольватации реагентов на механизм
реакции рассматривается в общих руководствах по органической
химии и в специальных монографиях по механизмам органических
реакций, поэтому здесь нет смысла обращаться к этим вопросам,
тем более, что для этого требуется привлечение обширного
материала по органической химии.
Влияние водородной связи
Влияние Н-связи на кинетику химических реакций может
проявляться как в реакциях с переходом протона, так и с участием
сильно электроотрицательных анионов и гидроксильных
растворителей, где возможно, например, образование связи О—Н---С1.
Реакции со специфической сольватацией анионов будут
рассмотрены ниже.
Для реакций, связанных с переходом протона по Н-связи,
Н. Д. Соколов (1947) определил энергию активации процесса
X—H---Y—► Х-—H+---Y как простую линейную функцию от
изменения частоты колебаний Ду по связи Н—X в комплексе
относительно частоты колебаний по изолированной связи
E = a-bkv (XIV. 32)
где а и b — коэффициенты, принимающие определенное значение
для данной изолированной связи X—Н.
Зная энергию активации, можно в принципе рассчитать и
константу скорости, определив предэкспоненциальный множитель с
помощью уравнений теории переходного состояния.
Интересно рассмотреть также влияние внутримолекулярной
водородной связи. Например, реакция меламина с салициловым
альдегидом протекает в этаноле примерно в 80 раз быстрее, чем
с n-оксибензальдегидом. Этот эффект может быть приписан
различию в прочности внутримолекулярной водородной связи в
указанных альдегидах. Отношение скоростей реакции меламина с
обоими альдегидами неодинаково в различных растворителях.
В общем, можно считать, что эта величина убывает по мере
ослабления способности молекул растворителя к образованию
водородной связи: С2Н5ОН > С6Н5СН2ОН > 1,4-диоксан >
> С6Н5Ш2 > (СбН5СНз + 3% С2Н5ОН) > СНС13.
336
Избыточная энтальпия смешения молекул растворителя
с реагентами и скорость реакции
Трудности количественного учета специфической сольватации
в рамках молекулярной теории растворов обусловливают
целесообразность отыскания такой количественной характеристики
сольватации, которую можно было бы определенным образом
связать со скоростью реакции. Основываясь на уравнении (XIV. 19),
можно сопоставлять константы скорости в различных средах,
выбрав в качестве стандартного состояния не газовую фазу, а
какой-либо инертный (для данной реакции) растворитель. Тогда
мерой изменения скорости реакции при переходе от одного
растворителя к другому будут служить относительные коэффициенты
активности (см. гл. VI), или соответствующие им стандартные
свободные энергии переноса.
Учитывая, что в растворах часто наблюдаются линейные
соотношения между энтальпиями и энтропиями процессов, можно в
первом приближении выразить константу скорости реакции как
функцию энтальпии смешения реагента с растворителем [С. Г. Э н-
телис, Р. П. Тигер, 1973]. Рассматривая константы скорости
реакций и энтальпии смешения в ряду растворителей, необходимо
обратить особое внимание на тот реагент, сольватация которого
должна более существенно изменяться при переходе от
растворителя к растворителю.
Так, для реакции ацилирования анилина хлористым бензоилом
(Н. К. Воробьев, Л. В. Курицын, см. выше) была установлена
приближенная линейная зависимость энергии активации и
логарифма константы скорости реакции от теплоты смешения анилина
с растворителями. Для теплот смешения Д#°° бензоилхлорида с
теми же растворителями линейная корреляция с \g k отсутствует.
Из этого можно заключить, во-первых, что упомянутая
зависимость может иметь лишь весьма ограниченное применение и, во-
вторых, что следует с большой осторожностью относиться к
попыткам" интерпретировать поведение смешанных растворителей на
основании данных об изменении скорости какой-либо реакции в
зависимости от состава раствора. Иными словами, если
избыточную энтальпию смешения можно рассматривать как характерное
свойство в физико-химическом анализе жидких систем, то
константа скорости, не связанная однозначно с энтальпией, таким
свойством служить не может.
• Тем не менее, накопление кинетических данных для реакций
в смешанных растворителях представляет значительный интерес.
Смешанный растворитель представляет собой удобную модель для
кинетических исследований влияния растворителя. Можно
ожидать, что развитие общей теории влияния растворителя на
кинетику реакции во многом будет зависеть от успехов в изучении
таких моделей. Данные по кинетике реакций в смешанных
растворителях приведены, в частности, в монографии В. А. Пальма (1967).
337
Сольватация ионбв в диполярных растворителях
и скорость реакций
Этот вопрос детально изучался главным образом в последние
десять — пятнадцать лет, в особенности, в работах А. Паркера
(A. Parker) и его сотрудников (1960—1970). Для книги, где столь
сжато излагаются основные проблемы кинетики реакций в
растворах вообще, такой вопрос может показаться частным. Однако эти
примеры весьма удобны, чтобы показать, как изменение
растворителя может в сотни тысяч раз и более изменить скорость реакции.
Анионы обычно сольватируются как в протонных, так и в
диполярных апротонных растворителях, причем в последних — в
значительно меньшей степени. Наоборот, большинство катионов
сильно сольватируется в высокополярных растворителях, у
которых отрицательный заряд локализован на атоме кислорода (ди-
метилсульфоксид, тетраметилсульфон, диметилацетамид и др.)-
Бимолекулярные реакции замещения, в которых не участвуют
анионы, протекают в диполярных апротонных растворителях так
же, как и в протонных. Например, пиридин и бромбутил
реагируют в диметилформамиде примерно с такой же скоростью, как
и в изодиэлектрических смесях метанол —- вода при той же
ионной силе.
Для реакции же*с участием ионов различие в скоростях
реакций, обусловленное специфической сольватацией, проявляется очень
сильно, Диполярные апротонные растворители, как правило, ос-
новны, т. е. являются донорами электронов. Поэтому их
взаимодействие с анионами имеет чисто электростатическую природу
(отталкивание), в то время как с катионами они образуют
комплексы. Помимо этого, диполярные апротонные растворители обладают
достаточно высокой среди органических растворителей
диэлектрической проницаемостью (30—40), что способствует диссоциации
ионных пар. Сочетание всех этих эффектов приводит к тому, что
анион становится как бы «свободным» и обладает повышенной
реакционной способностью. Наоборот, катионные реакции в таких
растворителях вследствие сильной сольватации катиона идут
медленно. В гидроксильных растворителях степень сольватации анио-
Та блица XIV.3
Относительная скорость анионных реакций
в различных растворителях
[A. J. Р а г к е г, Quart. Rev., 16, 163 (1962)]
CH3I
—
338
Реакция
1 /"* 1 — ртт
-J- L.1 ► ^Г1з
«-NO2C6H4N3
ci + i-
+ F"
CH3OH
1
1
HCONH2
12,5
5,6
HCONCH3
45,3
15,7
HCON(CH3)2
1,2-106
2,4-104
CH3CON(CH3)2
7,4.106
8,8* 104
на возрастет, и реакционная способность его существенно падает.
Примеры, характеризующие изменение скорости анионных реакций
в зависимости от растворителя, сведены в табл." XIV. 3.
Использование диполярных апротонных растворителей
позволило провести ряд органических реакций, которые протекают в
других растворителях чрезвычайно медленно. Особенно высокие
скорости анионных реакций найдены в гексаметилфосфортриамиде
и 1М-метил-2-пирролидоне — растворителях с очень высокой
ионизирующей способностью (по Гутману).
XIV. 6. Скорость реакций и принцип линейности
свободных энергий (ЛСЭ)
Приведенные выше данные о влиянии различных свойств
растворителя на скорость химических реакций наглядно иллюстрируют
сложность теоретического расчета скоростей реакций в жидкой
фазе с количественным учетом электростатических, вандерваальсо-
вых и других, в особенности донорно-акцепторных, взаимодействий.
Возможность совершенствования теоретических расчетов на основе
более детального и углубленного познания физической сущности
процессов межмолекулярного взаимодействия, несомненно, сулит
большие перспективы в будущем, однако приложение
современных физических теорий для расчета скоростей реакций в
конкретных системах часто наталкивается на непреодолимые
математические трудности.
Другой путь априорной оценки параметров, характеризующих
химические соединения и процессы, основывается на допущениях,
часто интуитивных, о наличии скрытой взаимосвязи между какими-
то параметрами в определенных условиях. Эти допущения обычно
невозможно строго обосновать с помощью представлений
молекулярной физики или термодинамики, поэтому они называются
экстратермодинамическими допущениями [J. E. L e f f 1 е г, Е. Grun-
v a I d, 1963]. В качестве примеров таких. корреляций можно
привести зависимость между шириной запрещенной зоны в
полупроводниках и энергией их атомизации, и многие другие
соотношения, рассмотренные в известной монографии М. X. Карапетьян-
ца «Методы сравнительного расчета физико-химических свойств»
(1965). Общей чертой соотношений такого рода является их
линейность для ряда подобных соединений или процессов. Этот путь,
несмотря на отсутствие достаточно строгих теоретических
обоснований, часто позволяет предсказывать результаты эксперимента с
удовлетворительной точностью, там где строгий теоретический
подход отступает перед трудностями конкретных решений.
Применительно к рассматриваемой проблеме задача состоит в
том, чтобы отыскать пригодные хотя бы для одного типа реакций
и серии химически подобных реагентов эмпирические соотношения
между кинетическими параметрами и параметрами,
характеризующими химическую природу реагентов. Для отыскания таких
339
корреляций может быть использован принцип линейности
свободных энергий (ЛСЭ).
Формулировка принципа ЛСЭ была впервые дана Гамметом
(1935) на основании рассмотрения серии реакций м- и я-замещен-
ных бензола. Была обнаружена линейная зависимость между
логарифмом константы скорости или константы равновесия реакции
некоторых замещенных и логарифмом константы диссоциации в
воде соответствующей ж- или n-замещенной бензойной кислоты.
Это соотношение может быть записано в виде
lg6 = lg£0 + plg-^- (XIV. 33)
где k и ko — константы скоростей реакций для замещенного и
незамещенного производных бензола, а К/ и /Со—константы
диссоциации замещенной и незамещенной бензойной кислоты в воде.
Коэффициент р называется константой реакции; он изменяется в
зависимости от типа реакции и внешних условий. Коэффициент а —
константа заместителя — может быть условно приравнен величине
lg(/C7/Co). т.е.
\gk = \gk0 + G9 (XIV. 34)
Величина константы скорости связана с энергией активации
процесса. Используя уравнение (XIV. 4) и обозначая через A Go* и
AG^ изобарные потенциалы активации незамещенного и
замещенного соединения, получим соотношение
AG# = AGf - RTop (XIV. 35)
дающее линейную зависимость между изобарными потенциалами
активации для двух различных гомологических серий реакций.
Выяснилось, что принцип ЛСЭ соблюдается с удовлетворительной
точностью и для многих других типов реакций. Подробное
рассмотрение принципа ЛСЭ и его обоснование дается в монографиях
Леффлера и Грюнвальда (1963) и В. А. Пальма (1967).
По аналогии с уравнением Гаммета, где константа скорости
изменяется в зависимости от типа реакции и заместителя, были
предложены аналогичные соотношения, учитывающие вариацию
растворителя. В качестве примера может быть рассмотрено
уравнение (XIV. 36) [G. С. Swain, D. С. Dittmer, 1953]
lg (k/ko)A - lg (k/k0)Ao = ab (XIV. 36)
описывающее сольволиз органических хлоридов и бромидов.
Здесь А — произвольно выбранное соединение; А° — стандартное
соединение (CH3Br); k — константа скорости сольволиза
вещества А или А° в любом растворителе; k0 — то же в стандартном
растворителе; а — константа соединения (для стандартного
соединения СНзВг а = 0); Ь — константа растворителя (в стандартном
растворителе — 80% объемн. этанола в воде — Ь = 0).
Уравнение такого типа пригодно для описания реакций, протекающих по
340
одинаковому механизму, хотя иногда наблюдаются отклонения от
линейной зависимости. В цитированной работе не обсуждался
физический смысл параметра растворителя Ьу который определялся
эмпирически, путем приравнивания этой величины нулю в
произвольно выбранном стандартном растворителе.
• В более поздних публикациях было показано существование
линейной зависимости между величиной Ъ и частотой максимума
поглощения излучения какого-либо высокополярного соединения в
данном растворителе. Исходя из этого, можно полагать, что
параметр Ь является некоторой функцией диэлектрических и
специфических эффектов растворителя.
Для распространения принципа ЛСЭ на более широкий круг
объектов И. А. Коппель и В. А. Пальм (1971) предложили общее
уравнение, учитывающее как полярные, так и сольватационные
эффекты среды:
А = Ло + yY + рР + еЕ + ЬВ (XIV. 37)
Здесь А и Ло — характеристики процесса в исследуемом
растворителе и в стандартной среде; У — параметр полярности
растворителя; Р — параметр поляризуемости; Е и В — обобщенные
кислотность и основность растворителя; коэффициенты у, /?, е, b
характеризуют чувствительность процесса к действию каждого из
перечисленных факторов. Параметр Y выражают обычно через
функцию (е—1)/(2е+1)>* Р приравнивают к (n2D — l)[(n2D + 2).
В качестве меры обобщенной кислотности или основности могут
быть использованы параметры, упомянутые в гл. III, а также
параметры Димрота, Гутманна, Козоуэра и др., методы определения
которых описаны, например, в монографии В. А. Пальма (1967) и
Рейхардта [Chr. R e i с h а г d t, 1969].
Для расчета коэффициентов, входящих в уравнение (XIV, 37)
используются методы статистической обработки
экспериментальных данных. Конкретные примеры применения корреляционных
уравнений типа (XIV. 37) имеются в работах И. А. Коппеля и
В. А. Пальма, опубликованных в последние годы в журнале
«Реакционная способность органических соединений».
XIV. 7. Скорость реакции
и вязкость растворов
Учитывая, что для взаимодействия в растворе реагирующие
молекулы должны перемещаться в жидкой среде, и вязкость
оказывает сопротивление этому движению, уместно обсудить^ в
отдельности вопрос о влиянии вязкости раствора на скорость
реакции в жидкой фазе.
Мелвин-Хьюз отметил, что кинетическая теория газов приводит
к соотношениям, в которых число столкновений Z может либо
прямо либо обратно пропорционально зависеть от вязкости смеси
Z = 4RTn2/N0Kr\ = 4л2о4цп2/т (XIV. 38)
341
где о — диаметр молекулы реагента; т — ее масса; п — число
молекул. Часто, используя прямую пропорциональную зависимость
между Z и п, записывают уравнение Аррениуса в виде:
k = const rj ехр ( — E[JRT) (XIV. 39)
Энергия активации £а в этом случае определяется из графика
зависимости \g{klr\) — 1/7', где k/ц — константа скорости,
«исправленная» на вязкость. Очевидно, Е'а отличается от Еа,
рассчитанной из обычного уравнения Аррениуса, но основное затруднение
заключается не в согласовании величин энергий активации, а в
том, что экспериментальные данные часто не укладываются на
кривую, описываемую уравнением (XIV. 39).
Недавно была предпринята попытка [С. F. Well s, 1970]
интерпретировать изменение скорости некоторых
окислительно-восстановительных реакций в водно-органических смесях различного
состава на основании данных об изменении вязкости растворителя.
Анализ кинетических данных по реакции окисления изопропилово-
го спирта фотовозбужденным антрахинон-2-сульфонат анионом в
смесях трет-бутапол — вода и ацетон — вода показал, что
зависимость константы скорости реакции от вязкости среды описывается
с помощью уравнения теории столкновений только в определенном
интервале концентраций. При этом экспериментальные точки для
системы ацетон — вода не укладываются в зависимость для
системы трет-бутанол — вода, а максимум на диаграмме вязкость —
состав в последней системе не находит отражения в кинетических
данных.
Можно заключить, что попытки интерпретации кинетических
данных, с учетом одной только вязкости растворителя не
оправданы даже в простых случаях. Удовлетворительное совпадение
теории с экспериментом наблюдается при учете вязкости лишь для
реакций, где лимитирующей стадией является диффузия.
Подробнее об этом будет сказано ниже, при рассмотрении быстрых
реакций (гл. XV).
XIV. 8. Влияние давления на скорость реакций
в различных растворителях
Общее решение вопроса о влиянии давления на скорость
реакции базируется на известном термодинамическом соотношении,
выражающем зависимость изменения термодинамического
потенциала системы от давления:
С учетом уравнения (XIV. 4) можно получить
(iiLij^-Д^/яг (XIV. 40)
342
где Av^ — изменение объема при переходе реагентов в
активированное состояние. Эта величина называется объемом активации.
Изменение скорости реакции с давлением обычно невелико,
поэтому для определения объема активации исследуются широкие
интервалы давлений. Интегрирование уравнения (XIV. 40) дает
In (kJk0) = - -^ ^ф (XIV. 41)
где kP — константа скорости при данном давлении, а ко— при
давлении, равном нулю (когда Ауф = 0). Величина k0 практически
совпадает со значением к при атмосферном давлении.
Согласно уравнению (XIV. 41), величину Аи^ можно вычислить
из наклона прямой \g(kP/ki) — Р. Это уравнение описывает
экспериментальные данные для большого числа реакций в широком
интервале давлений: от атмосферного до 1500 атм. Для более
точных расчетов [Н. Н а г t m а п n, A. P. Schmidt, 1969]
необходимо учесть сжимаемость жидкости р:
(XIV. 42)
Перрин (1938) показал наличие линейной зависимости между
объемом и энтропией активации для большого числа реакций.
Энтропийный фактор как мера упорядоченности системы может
быть связан со структурными эффектами, причем надо учитывать
как собственные структурные изменения молекулы реагента при
переходе в активированное состояние, так и перестройку структуры
растворителя. При этом для реакций с участием ионов или
диполей растворитель влияет на объем и, соответственно, на энтропию
активации в большей степени, нежели перестройка самой молекулы
реагента.
Влияние давления на скорость реакции в различных
растворителях можно проиллюстрировать на примере детально
изученной реакции Меншуткина (триэтиламин + йодистый этил).
В табл. XIV. 4 приведены отношения констант скорости этой
реакции при давлении 1500 атм и 1 атм и объемы активации.
Таблица XIV.4
Влияние давления на скорость реакции Меншуткина
в различных растворителях при 50 °С
[Н. Hartmann, H. D. В г a u e r, G. Rink, Z. phys. Chem. (NF), 61, 47 (1968)].
Растворитель
Гексан
Бензол
Метанол
Хлорбензол . . .
&1500
ki
' 7,05
6,15
3,26
4,81
-Ло8^,
мл/моль
58,2
50,2
58,0
45,1
Растворитель
Ацетон
Нитробензол ....
/г-Ксилол (45 °С) . .
&1500
kl
6,07
3,39
7,90
- Ди^,
мл/моль
53,8
30,3
49,3
343
Г л а в а XV
НЕКОТОРЫЕ СПЕЦИАЛЬНЫЕ ВОПРОСЫ
КИНЕТИКИ
В этой главе рассматриваются особенности протекания в
неводных средах некоторых специальных типов реакций,
представляющих большой практический и теоретический интерес. Это
прежде всего гомогенные жидкофазные каталитические реакции.
В присутствии катализаторов протекают также многие реакции
изотопного обмена, которые заслуживают отдельного
рассмотрения, поскольку изучение их весьма ценно не только с точки
зрения кинетической теории, но и для решения одной из центральных
проблем современной химии — связи между строением и
реакционной способностью. Наконец, очень интересны в теоретическом
и прикладном отношении быстрые реакции в растворах, изучение
которых осуществлялось довольно интенсивно в последние два
десятилетия.
XV. I. Каталитические реакции
в жидкой фазе
Для уточнения терминологии необходимо с самого начала
оговориться, что термин «катализ» здесь относится не к действию
растворителя, а только к действию той добавки, которая вводится
в раствор помимо реагентов и изменяет кинетические
характеристики реакции *. Экспериментально измеряемая константа
скорости зависит и от катализатора и от растворителя. Поэтому в
выражении
k = k0 + kK [катализатор]" (XV. 1)
величины ko — (константа скорости некатализируемой реакции) и
kK (каталитический коэффициент) сохраняют определенное
значение только в данном растворителе. Это наглядно иллюстрируется
данными для реакции 2,4-динитрохлорбензола (ДНХБ) с аминами,
приведенными в табл. XV. 1. Эта реакция имеет второй порядок по
ДНХБ и описывается уравнением (XV. 1), причем п = 1.
Измерение константы скорости при постоянной концентрации ДНХБ и
переменной концентрации амина (катализатора) позволяет
определить константы k0 и kK.
Из табл. XV. 1 достоверно'следует, что в растворителях,
способных к образованию Н-связей, «некатализируемая» реакция
протекает значительно быстрее, т. е. эффект растворителя
перекрывает эффект реагента-катализатора.
* Или к действию одного из основных реагентов, если константа скорости
реакции зависит от его концентрации.
344
Т а блица XV.1
Влияние растворителя на константы скорости каталитической
реакции ДНХБ с аминами при 24,8 °С
[С. Д. Росс
1967, стр. 33].
Амин
Аллиламин
Бутиламин
, сб. «Современные проблемы
Растворитель
Хлороформ
Хлороформ — бензол
[1: 1 (объемы.)] . . .
Этанол
Диоксан — вода [1 : 1
(объемы.)]
физической органической химии», Изд. «Мир»,
fco-105,
л/{молЬ'Свк)
3,00
4,8
13,5
20
90
220
*К.Ю5.
л21 {мо ль2-сек)
13,8
16
12
51,7 -
67
63
W
л1моль
4,60
3,3
0,89
2,59
0,74
0,29
Анализируя механизм действия растворителя в каталитических
реакциях, необходимо прежде всего выяснить, какого рода
взаимодействия — дисперсионные, кулоновские или донорно-акцептор-
ные — будут преобладать в растворе и между какими
компонентами они наиболее отчетливо выражены: реагентами и
растворителем, реагентами и катализатором, активированным комплексом
и растворителем и т, д. Помимо перечисленных выше
взаимодействий, определяющих энергию образования различных комплексов
в растворе,, необходимо учитывать и энтропийный фактор.
Обычно образование комплексов связано с упорядочением частиц, то
есть с уменьшением энтропии, но в некоторых случаях возможно
такое комплексообразование с растворителем, которое будет
сопровождаться возрастанием энтропии. Например, если
растворитель обладает прочной сеткой Н-связей и структурой высокой
степени упорядоченности, то взаимодействие его с реагентами может
происходить за счет разрыва связей, обусловливающих
собственную структуру растворителя, причем нарушение этих связей может
вызвать такое разупорядочение структуры растворителя, что
общее изменение энтропии окажется положительным.
Влияние химической природы и физических свойств
растворителя на кинетику гомогенных каталитических реакций можно
проследить на конкретных примерах. В неводных средах наиболее
широко изучены кислотно-основные каталитические процессы.
Кислотно-основной катализ
Реакции, катализируемые водородными или гидроксильнымй
ионами, изучены в водных растворах значительно лучше, чем вне-
водных. Одним из наглядных примеров каталитического действия
неводного растворителя является каталитическая мутаротация
тетраметилглюкозы. Этот процесс не происходит в чистых пиридине
12 Зак, 500
345
и крезоле, но в смеси этих растворителей протекает быстрее,
чем в воде [Е. А. М о е 1 w у п - Н u g h e s, 1947].
Общее уравнение для кислотно-основного катализа дано Брён-
стедом в виде:
^b = Gb(/Cb)3 (XV. 26)
Здесь ^на и &в — каталитические коэффициенты [см. уравнение
(XV. 1)] для кислоты НА и основания В; Кяа и Kb — константы
диссоциации. Параметры GA, GB, аир сохраняют определенное
значение, для данной реакции, катализируемой серией кислот или
оснований. Уравнение Брёнстеда может рассматриваться как
частный случай принципа линейности свободных энергий (см. гл. XIV).
Из теории Брёнстеда вытекает линейная зависимость р/Сдис
кислоты (основания) от 1/е (гл. IX). Логарифмируя уравнения
(XV. 2), легко показать, что для группы объектов с постоянными
G и а такая же зависимость должна наблюдаться и для
каталитических коэффициентов. Естественно, что и отклонения от этой
зависимости обнаружатся при сопоставлении растворителей
различной химической природы. По-видимому, можно, считать, что
соотношение (XV. 2) будет удовлетворительно выполняться
в водно-неводных растворителях при не очень низкой
концентрации воды. Данные по каталитическим коэффициентам кислотно-
основных реакций в чистых неводных растворителях пока крайне
малочисленны. Однако для характеристики реакционной
способности веществ в реакциях кислотно-основного катализа можно
использовать значения констант скоростей изотопного обмена
водорода в неводных растворах, изученные довольно широко за
последние десятилетия (прежде всего, в работах А. И. Шатен-
штейна и сотрудников, см. ниже).
Другие гомогенно-каталитические реакции
Помимо кислотно-основных, в последнее время изучено
большое число реакций, протекающих в неводных средах в
присутствии растворенного катализатора. Значение этих исследований
для теории и практики далеко не соответствует тому скромному
объему, который может быть выделен для них в этой книге. Здесь
будут вкратце рассмотрены на конкретных примерах реакции
каталитической полимеризации и радикального распада.
Колкло и Дэйнтон [R. О. С о 1 с 1 о u g h, F. S. D a i n t о п, 1958]
исследовали каталитическую полимеризацию стирола в
присутствии SnCU в неводных растворителях: ССЦ, l,2-C2H4Cl2, C6H5NO2
и их смесях. Они обнаружили, что в ССЦ реакция сильно
ускоряется добавкой воды; это позволяет, по их мнению, приписать
истинному катализатору состав 5пС1зОН-Н2О. Реакция также
сильно ускоряется при добавлении к ССЦ дихлорэтана. Авторы
346
убедительно показали, что ускорение реакции не связано с
изменением полярности растворителей. Так, в дихлорэтане
полимеризация идет быстрее, чем в малополярном ССЦ и в более полярном
нитробензоле. В данном случае дихлорэтан, как и другие алкил-
галогениды, является сокатализатором реакции.
В растворах могут протекать реакции с участием свободных
радикалов, образование которых инициируется как физическим
воздействием (различные виды облучения, электронный удар
и т. д.), так и химическими реагентами. Если растворитель не
вступает в реакцию с радикалами, последние попадают в «клетку»
из молекул растворителя, где они быстро рекомбинируют. В
случае же взаимодействия растворителя с радикалами с передачей
свободной валентности молекуле растворителя реакции могут
протекать с высокой скоростью, в несколько десятков раз
превышающую скорость диффузии [Н. Н. Семенов, 1958]. Взаимодействие
радикала с растворителем было доказано, например, для реакции
[Ф. С. Дьячковский, Н. Е. Хрущ, А. Е. Шилов, 1967]
C5H5VOC12 + В -* С5Н;+ VOC12-B
где В—донор электронов (органическое основание). Радикал
С5Н^ отрывает водородный атом от метильной группы
растворителя толуола, при этом образуется циклопентадиен. Поскольку
ионизации в данном случае не происходит, константа скорости
реакции практически не зависит от е растворителя (исследованы
смеси толуола с тетрагидрофураном).
Роль растворителя в жидкофазных реакциях,
протекающих при участии твердого катализатора
Реакции такого рода изучались достаточно детально и
подробно описаны, например, в монографии Д. В. Сокольского (1962).
Из всего обширного круга проблем, возникающих при изучении
этих реакций, мы рассмотрим лишь вкратце влияние растворителя
на скорость процесса и состав продуктов.
Установлено, что ацетон в присутствии платинового
катализатора в водном растворе подвергается гидрогенизации с
образованием изопропилового спирта, а чистый ацетон при тех же
условиях превращается в пропан.
В гетерогенных каталитических реакциях направление и
скорость процесса в большой мере зависят от адсорбции реагента
на катализаторе. Растворитель может взаимодействовать с
реагентом, образуя ассоциаты различной прочности, либо сам
адсорбироваться на поверхности катализатора. В том и в другом случае
растворитель влияет на селективность процесса а, выражаемую
соотношением
l (XV. 3)
где ki и k2 — константы скорости образования соответствующих
продуктов, bi и Ъ%—их коэффициенты адсорбции на металличе-
13* 347
ском катализаторе. Так, для реакции совместного каталитического
гидрирования циклогексена и ацетона в циклогексане и диоксане
на скелетном никелевом катализаторе селективность гидрирования
а = 1гцЬп/1гаЬа (ц — циклогексен, а — ацетон) возрастает в 10 раз
при переходе от циклогексана к диоксану. Этот факт можно
объяснить, если рассмотреть смешиваемость реагентов с указанными
растворителями. Циклогексен хорошо смешивается с циклогекса-
ном и плохо — с диоксаном; с ацетоном наблюдается обратная
картина. Можно заключить, что вещество, более прочно
связанное в растворе, обладает меньшим сродством к катализатору, и,
следовательно, меньшей реакционной способностью в процессе
гидрирования.
В отдельных случаях влияние растворителя определяется
растворимостью в нем реагента. В процессе гидрирования хинона на
платине, никеле и палладии скорость
реакции убывает в ряду растворителей:
бензол, диоксан, этанол, вода и
уксусная кислота. В таком же порядке
изменяется в названных растворителях
растворимость хинона.
При изучении скорости гидрирования
в смешанных растворителях изотермы
констант скорости во многих случаях
Рис. XV. 1. Изменение рас- обнаруживают минимум в зависимости
хода водорода при гидри- QT состава Такие МИНИмУМЫ обнаружи-
ровании гексена-1 на РЮ2
в этанол-гептановом
растворителе [Д. В.
Сокольский, 1962.]
ваются, например, при исследовании
гидрирования диметилэтилкарбинола в
смесях диоксана с водой и со спиртом.
По мнению Д. В. Сокольского, наличие
минимумов на изотермах константа скорости каталитического
гидрирования — состав смешанного растворителя свидетельствует
о межмолекулярном взаимодействии в смешанном растворителе,
а наличие максимумов — об отсутствии такого взаимодействия.
Глубина минимумов и высота максимумов зависят от природы
катализатора и непредельного соединения. Эффекты проявляются
более отчетливо, если исследуемое вещество само сольватировано
растворителем.
По-видимому, можно прийти к общему выводу, что влияние
растворителя на скорость гетерогенной каталитической реакции
гидрирования обусловлено прежде всего участием растворителя
в создании оптимальных концентраций реагирующих веществ на
поверхности катализатора. Очевидно, это участие нельзя описать,
используя только одно какое-либо свойство растворителя, и,
следовательно, ход изотермы константа скорости — состав
растворителя в общем виде вряд ли возможно интерпретировать с учетом
только межмолекулярного взаимодействия в смешанном
растворителе. Это может быть проиллюстрировано на примере
каталитического гидрирования гексена-1 на РЮ2 в системе этанол —
348
гептан (рис. XV. 1 схематически иллюстрирует изменение расхода
водорода при гидрировании указанного соединения в зависимости
от состава растворителя).
Очевидно, что перегибы на кривых никак не могут быть
приписаны эффектам межмолекулярного взаимодействия в системе
этанол — гептан, которое в этой системе исключительно слабое.
XV. 2. Кинетика изотопного обмена
в неводных средах
При исследовании реакций с применением изотопов возможны
следующие варианты:
меченый атом принимает участие в той связи, которая
непосредственно деформируется или разрывается в результате реакции;
реакция протекает без образования или разрыва связи с
меченым атомом.
В принципе рассмотрение роли растворителя в кинетике таких
реакций не должно было бы отличаться какими-либо
особенностями, однако, следует учесть изменение скорости реакции в
зависимости от изменения массы изотопа — так называемый
кинетический изотопный эффект.
Кинетический изотопный эффект наиболее отчетливо
проявляется в первом варианте — в особенности, когда изменение массы
значительно (изотопы водорода). Во втором варианте этот эффект
проявляется весьма слабо и называется вторичным эффектом
(подробно этот вопрос обсуждается в обзоре Е. А. Халеви).
Теория кинетического изотопного эффекта основывается на
расчете изменений относительной энергии связей для различных
изотопов ^следствие различия масс. Детальное количественное
рассмотрение проблемы содержится в монографии Л. Меландера
(1964) и в обзоре Саундерса [W. H. S a under s, Jr. 1961]. Здесь
дается более простой анализ согласно Ф. С. Якушину (1962). »
Для расчета констант скоростей реакций с участием различных
изотопов необходимо учесть влияние массы изотопа на
трансмиссионные коэффициенты и энергии активации в соответствии
с уравнением
2 DT Ч <XV-4)
где индексы 1 и 2 относятся к различным изотопам одного
элемента. Различие в энергетических членах, по Якушину, можно
свести к разности колебательной энергии на нулевых уровнях
замещенного и незамещенного соединения в исходном и
переходном состоянии. Колебательная энергия нулевого уровня
связана с частотой колебания связи
2!v' (XV>5)
349
и может быть найдена из спектроскопических измерений. Для
переходного состояния прямое определение Ео затруднительно, но
можно оценить граничные значения этой величины, полагая, что
связь в активированном комплексе либо такая же, как в исходном
соединении, либо полностью разорвана. Отсюда, пренебрегая
различием в трансмиссионных коэффициентах, можно найти
возможный интервал изменения отношений констант скоростей,
например, kH/kD = 29-7- 1,4, kn/kT = 125 -f-1,7).
В предыдущей главе говорилось о том, как растворитель влияет
на изменение констант скоростей и энергий активации различных
реакций. Возникает вопрос, могут ли проявиться какие-то отличия
во влиянии растворителя при изучении совершенно одинаковых
по химической природе реагентов и по механизму реакций,
каковыми являются реакции с участием веществ, содержащих разные
изотопы одного и того же элемента. Для ответа на этот вопрос
надо знать, во-первых, насколько введение изотопа изменяет
физические свойства молекулы реагента, прежде всего, дипольный
момент, и, во-вторых, происходит ли сольватация реагента
молекулами растворителя с участием той связи, в которую включен
рассматриваемый атом. Кроме того, для реакций изотопного
обмена по С—Н-связи, которые, как правило, протекают с участием
катализатора, необходимо учитывать возможность изменения
состояния катализатора в различных растворителях.
Влияние изменения дипольного момента, по-видимому, можно
считать пренебрежимо малым. Действительно, дипольные моменты
обычной и тяжелой воды разнятся всего на 0,01 D (Н2О—1,86;
D2O— 1,87). Для более полярных соединений различие может быть
несколько большим, но вряд ли превысит 1—2%. Подстановка
этих измененных значений [i в уравнение (XIV. 30) приведет к
изменению константы скорости, не превышающему погрешности, с
которой электростатическая теория описывает экспериментальные
данные. Таким образом, задача о влиянии растворителя на
кинетический изотопный эффект в случае водорода практически сводится
к рассмотрению эффектов специфической сольватации
растворителем реагентов и катализаторов реакции.
Весьма интересны данные наблюдений кинетического изотопного
эффекта самого растворителя. В качестве примера могут быть
взяты реакции бимолекулярного нуклеофильного замещения,
проведенные в этаноле и дейтерированном этаноле C2H5OD,
(табл. XV. 2).
Полученные результаты могут быть объяснены специфической
сольватацией реагентов этанолом с участием Н-связи. Реакции
такого типа обычно протекают медленнее в гидроксилсодержащих
растворителях, нежели в диполярных апротонных. Как видно,
замещение водорода дейтерием ослабляет сольватацию по Н-связи.
Примеры таких исследований в неводных растворах пока
малочисленны. Отсутствуют достаточно надежные данные по
температурной зависимости кинетического изотопного эффекта среды, что
360
Таблица XV.2
Влияние изотопного замещения в растворителе на скорость реакций
[Р. В е 11 r a m e a. oth., J. Chem Soc, В, 1970, № 3. 453.]
Реакция
2,4-(NO2)2C6H3Cl +
+ С2Н5О- —* 2,4-
-(NO2)2C6H3OC2H5 + Cl-
2,4-(NO2)2C6H3Cl +
+ C5H5N —> 2,4-
-(NO2)2C6H3N+C5H6C1-
Растворитель
C2H5OH
C2H5OD
C2H5OH
C2H5OD
t, °c
25
90
fe.104,
лЦмоль'сек)
842 ±32
1645 ±44
1,178 ±0,038
1,530 ±0,065
^C2H5OD
^C2H5OH
1,84
1,30
не позволяет с достоверностью оценить изменение энергии
активации и предэкспоненты уравнения Аррениуса под влиянием
изотопного замещения в растворителе.
Выше уже упоминалось, что скорость водородного обмена
(кинетическая кислотность) может служить мерой способности
веществ вступать в реакции по связям, содержащим
обменивающийся водород. Для выяснения роли растворителя в этом процессе
необходимо хотя бы вкратце обсудить механизм обмена.
А. И. Бродский (1954) отметил, что обмен в связях N—Н,
О—Н, С1—Н, S—Н для большинства соединений, обменивающихся
с кислотноосновными донорами дейтерия (D2O, C2H5OD),
протекает практически мгновенно при любой температуре без
катализаторов (см. ниже).
Медленный обмен характерен для связей типа С—Н в
органических соединениях, Р—Н в фосфинах и т. д. Скорость обмена
дейтерия определяется строением и свойствами обеих
обменивающихся молекул (акцептора и донора дейтерия), а также
свойствами среды. Медленный обмен обычно идет с измеримой
скоростью в присутствии катализаторов протолитических реакций.
Скорость такого обмена зависит от температуры; из этого следует,
что процесс характеризуется определенной энергией активации.
Быстрый или медленный тип обмена, согласно А. И. Бродскому,
определяется наличием или отсутствием свободных электронных
пар около атома, непосредственно связанного с атомом водорода
дейтерия. Это обусловлено легкостью присоединения протона (дей-
терона) к свободной электронной паре. Энергия активации этого
процесса близка к нулю. Но при отсутствии свободной электронной
пары (например, в связи С—Н) присоединение дейтерона к этой
связи может происходить лишь после удаления протона, что
требует значительной энергии активации.
351
Упрощенная схема процесса обмена может быть представлена
в виде:
X—Н
X—H + Y—D —> ' i —> X—D + Y—Н
t ••
D--Y
причем быстрый обмен отличается тем, что процесс перехода
атомов Н и D в комплексе идет синхронно.
Здесь будет вкратце рассмотрен только катализируемый
медленный обмен для связей С—Н, протекающий в неводных средах,
поскольку роль растворителя в этом случае проявляется наиболее
наглядно.
Обмен в связях С—Н может идти по ионизационному или
ассоциативному механизмам. Ионизационный обмен протекает через
промежуточную стадию образования ионов в соответствии со
схемами [А. И. Бродский, 1957]:
I !
RH + AD ^± W + HAD+ т=± RD + НА
t I
-± RHD+ + A" ;p± RD + НА
| t
В случае ионизационного механизма важное значение имеют
диэлектрические свойства растворителя. По данным А. И. Шатен-
штейна, обмен водорода соответственно в метиленовой и в мети-
новой группах дифенилметана и трифенилметана с дейтерогидра-
зином протекает быстрее, чем с дейтероаммиаком, что можно
объяснить большей диэлектрической проницаемостью гидразина.
Особенно эффективно протекает ионизация углеводородов с
образованием карбониевых ионов в сильнокислых высокополярных
растворителях, например, смесях SbF5 с HF и HSO3F [F. А. 01 ah,
1964, 1965].
Ассоциативный механизм (через ассоциированные молекулы),
согласно А. И. Бродскому, характерен для быстрого обмена. Для
медленного обмена возможен как ионизационный, так и
ассоциативный механизмы. А. И. Шатенштейн показал, что для одного и
того же вещества (бензола) можно провести водородный обмен,
переходя от ассоциативного (в присутствии DBr) к
ионизационному механизму (смесь HF + BF3) через промежуточные стадии
с образованием комплексов различной полярности (добавки кDBr
бромидов титана, алюминия, бора).
Приведенные выше примеры характеризуют непосредственное
участие растворителя в реакциях водородного обмена. Раствори*
тель может влиять на реакцию обмена и косвенно, путем измене-
352
ния силы электростатического взаимодействия или специфической
сольватации реагентов через донорно-акцепторное взаимодействие
(как это описано в общем случае в гл. XIV). Например, скорость
дейтерообмена между инденом и флуореном, катализируемая
грет-бутил атом лития, последовательно уменьшается в ряду эфи-
ров: 1,2-диметоксиэтан > тетрагидрофуран > 1,2-метоксиэтокси-
этан > 1,2-диэтоксиэтан > 1,4-диоксан, причем этот порядок
согласуется с изменением относительной сольватирующей
способности эфиров [А. И. Шатенштейн и др., 1967].
Специфическая сольватация растворителем реагентов,
участвующих в водородном обмене, не обязательно должна быть
сопряжена с наличием протолитического растворителя. И. П. Али-
ханов и А. И. Шатенштейн (1968), изучая дейтерообмен CF3COOH
с пентаметилбензолом, содержащим дейтерий в бензольном кольце,
обнаружили изменение порядка реакции по кислоте в разных апро-
тонных растворителях от 2,3 до 2,9 и изменение констант скорости
обмена (в скобках даны значения &-106, сек-1): C6Hi4(l,7),
ССЦ (2,0), С6Н6(8,5), 1,2-С2Н4С12(21), С6Н5С1(69).
Большую, нежели в ССЦ, скорость обмена в бензоле можно
объяснить более высокой протоноакцепторной способностью
я-электронного облака бензола по сравнению с атомом хлора
в ССЦ (диэлектрические проницаемости С6Н6 и ССЦ примерно
равны), Эффект растворителя в этом случае можно рассматривать
как «протолитическое взаимодействие на уровне образования Н-
связи» (А. И. Шатенштейн).
Рассмотрим еще роль сольватационной способности
растворителя на примере исследования реакции изотопного обмена иода
[Ch. A. Marcopoulos, 1967]. Реакция изотопного обмена иода
между KI и 1-1-2,4-динитронафталином имеет первый порядок по
иодиду в ацетоне, нулевой — в метаноле и дробный — в смесях
этих растворителей (0 < п < 1). Для смесей ацетон —вода и
ацетон—метанол с одинаковой е порядок реакции составляет,
соответственно, 0,86 и 0,58. Этот пример, как и предыдущие,
подтверждает, что для реакций изотопного обмена донорно-акцептор-
ные свойства обычно более существенны, чем полярные.
XV. 3. Быстрые реакции в растворах
К быстрым реакциям в настоящее время относят такие, для
которых время полупревращения реагентов не превышает
нескольких секунд. Начало современных исследований быстрых реакций
было положено работой Хартриджа и Роутона (1923),
применивших метод непрерывной струи для изучения реакций со временем
полупревращения порядка тысячных долей секунды. В настоящее
время наиболее чувствительные методы позволяют исследовать
реакции с периодом полупревращения ~ 10~10 сек. Эти методы
связаны с применением специальной аппаратуры для смешения
реагентов и современных физических приборов для регистрации
353
концентрационных изменений. Так, при исследовании быстрых
реакций широко используют полярографию, фотоэлектроколори-
метрию, измерение флуоресценции, электронный парамагнитный
резонанс (если реакции идут с образованием свободных
радикалов), ядерный магнитный резонанс (особенно, импульсный метод).
Методика исследования быстрых реакций описана в монографии
Колдина (1964) и в специальных обзорах.
Поведение растворителя в быстрых реакциях
Многие быстрые реакции, если отвлечься от специфики
методов наблюдения, не имеют каких-либо существенных отличий от
обычных реакций с точки зрения формальной кинетики. Однако
некоторые реакции, протекающие с участием ионов водорода,
имеют константы скорости ~1010 — 1011 сект*. При этом доля
активных столкновений от общего числа столкновений молекул,
вычисляемого по молекулярно-кинетической теории, близка к
единице. Это означает, что взаимодействие между молекулами
происходит практически при каждом столкновении, и энергия
активации реакции будет определяться энергией активации
процесса диффузии. Поскольку скорость реакции определяется
скоростью диффузии, то из свойств растворителя наиболее важным
в данном случае окажется вязкость. Соответствующая зависимость
получена Дебаем. Полагая, что движение молекул в жидкости
описывается уравнением Стокса — Эйнштейна, Дебай нашел (для
реакции второго порядка):
£ = -!' — • 10~3 (л-моль*1 .сек~1) (XV.6)
о г\
Из этого уравнения, в частности, следует, что температурные
коэффициенты скорости таких реакций будут определяться
температурными коэффициентами вязкости растворов и, значит, будут
намного ниже, чем для обычных реакций, описываемых
уравнением Аррениуса. Уравнение (XV. 6) приближенно описывает
экспериментальные результаты для некоторых очень быстрых реакций
в растворах, хотя, разумеется, оно не может оправдываться в
общем случае, поскольку игнорирует как полярные свойства
растворителя, так и его химическую природу. Близость значений
константы скорости к вычисленным по уравнению (XV. 6) может
служить лишь указанием на то, что скорость реакции
лимитируется диффузией.
Наиболее характерными примерами очень быстрых реакций
в неводных растворах могут служить реакции переноса протонов.
Обмен протонами может происходить как между частицами,
образующимися в результате самодиссоциации (автопротолиза) чистых
прототропных растворителей, так и вследствие кислотно-основного
взаимодействия кислоты НА и основания В по схеме:
НА + В 5=£ НАВ ^=£ ВН+ + А"*
364
В водных растворах скорость реакции
Н3О+ + ОН" ^=± 2Н2о
в интервале температур 5—30 °С изменяется в пределах
(1,0—1,7)X Ю"11 л/(моль• сек). [Э. Грюнвальд, 1969]. Механизм
процесса перемещения протона вдоль водородных связей,
обеспечивающий чрезвычайно большую скорость реакций водородного
обмена, аналогичен для воды, гидроксилсодержащих
растворителей и ряда протонных кислот. Следует ожидать поэтому, что
скорость передачи протона в таких неводных растворах будет близка
по порядку к упомянутой величине.
Однако экспериментальные данные для неводных растворов
крайне малочисленны. Теоретически скорости протолитических
реакций могут быть рассчитаны по данным о подвижности
водородных ионов в соответствующих растворителях. Связь между
удельной константой скорости и подвижностями ионов А и В
дается уравнением Дебая
(XV. 7)
""" \0Q0eF2 {ехр (zAzBNQe2/eoRT) - 1}
где е — заряд электрона, гА, z& — заряды ионов; а —среднее
расстояние между центрами молекул в комплексе А---В; Яа и Kb—
эквивалентные электропроводности. Поэтому вместо рассмотрения
корреляций между константами скорости переноса протона и
свойствами растворителей можно проанализировать изменение
подвижности протона в этих растворителях.
В табл. XV. 3 величина Яп, характеризующая по Грюнвальду
долю подвижности, обусловленную эстафетным перескоком
протона, сопоставлена с величинами вязкости и временем
диэлектрической релаксации td растворителей. Как и следовало ожидать,
величины Ки не коррелируют с вязкостью, поскольку движение прог
токов не описывается законом Стокса (см. гл. XI). Три
экспериментальные точки, представленные в таблице, укладываются на
прямую в координатах \gKu — td, из чего Грюнвальд делает
вывод, что диэлектрическая релаксация определяет лимитирующую
стадию переноса протона.
Таблица XV.3
Подвижность протона и свойства растворителей при 25° С
[Э. Грюнвальд, сб. «Новые проблемы физической органической химии»,
Изд «Мир», 1969].
Растворитель
Вода
Метанол
Этанол
V104.
см2/(в'сек)
31
10
4,1
ть спз
0,894
0,545
1,087
1/тд.Ю-10,
сек"1
12
2,0
0,64
355
Известны еще некоторые примеры реакций, связанных с
переносом протона в неводных растворителях. Так, методом
ультразвуковой релаксации была исследована димеризация бензойной
кислоты в четыреххлористом углероде и толуоле. Константы
скорости ассоциации в этих растворителях составляют 4,7-109 и
1,6-109 л/(моль-сек), а энтальпии активации—2,9 и 3,0 ккал]моль.
Наблюдаемое различие в конбтантах скорости может быть
объяснено большей степенью сольватации молекул в толуоле.
[Согласно уравнению (XV. 7), скорость реакции должна быть большей
в толуоле, обладающем меньшей вязкостью].
Детальному изучению процессов переноса протона в растворах
посвящен специальный выпуск дискуссий Фарадеевского общества
(1965). В работе Колдина и Каспарян [Е. F. Caldin, M. К as-
р а г j a n], опубликованной в этом выпуске, показано, как
проявляется влияние растворителя в очень быстрых, реакциях. Кван-
тово-механическим путем вычислена ширина энергетического
барьера для нескольких реакций с переносом протона и показано, что
в случае замены растворителя (С2Н5ОН вместо Н2О и D2O)
изменение ширины барьера значительно больше, чем при изменении
температурного интервала исследований и даже для различных
типов реакций (перенос протона в связи СН.. .О или в связи
СН...). Таким образом, отчетливые доказательства роли сольвата-
ционных процессов могут быть найдены даже в случае очень
быстрых реакций.
cq 5 2
й^ £.
h
SO |
Oho '
Сю P
з^ь?
a'o-
4 cokS
OS (Уи
go
X^ Л a)
"* cu 3 ,
го м a I
£ c 2 -
S I pgggg1
ao^cup^Ha^,
, IS I
a '
i я
-S3 w
B^S1
*.У я я
со v
« я
« cu ё
к 5 ^
о
я
Ш s s i .
51 я S l£
gC^l» к.
i
к С О)
R о з
ф
ство
05
а
х
15
а
о
ения
авл
1=2
С
"А
к
ч
fcf
к
S
я
CU
fcj
ё<§
>>2
6 <DS
И « И
СГ о
-г Я си >
^ Я S
' М <1>О
S Я Ю О
Я Я СМ Я
И
Cl
2 сЗ
К Он
1
Sc^
ш
Л
ш
О
Q.
С
ffi
О
>s
о
ш
о
з:
о
О)
3я
в
5 н
со
il
Ч о
8 §
к й У
§ --
&^§
IS-
in
cu £ ■
H ca
^. Я <
q
« cu
а/ и * ~
g В S a o
н 5 « о в
н . . -■
оз и р
я «J й
lfs
Я
g
5 w
в я
Л Он
s s
Sq
o
X Н
о^
cu
IS
ч ^5
о с
CQ Ou
s о
я ж
Я ' О
ч<1>в
О) S
нх
Ц
со
gsj
"У
о о
t0.2 2 odd- c?-
00 CD - '> -t^ t^ О — Ю
о
°
CD
00
oo/cd аэо
аГсо'>со''^Г
'-н CD СМ СО
, ю
JCXJlO^CDrf^OOO
) t-h (N t^. CX) —н Ю —^ CD
С S S
S-M ч -a
o
UU
о о ю
СО CD СО
<м ^
СТ5 ю "^f ^t1 l
Ь- Ь- t4- f~ '
t^
^ ~ О CM CO
QO I
— ю N. а> о со !>■
оо см ooioq^co
'Г^
q^q
сосо'сгГ-^oootCcM''o4
т—i оо "^ | '—* С4- сп
357
358
I ^
uu
° °
C4 (NOOJ
CM О ^-CM «
N-
О
• si
*7<b
af
(О CO O5
* *
N СО
^^©tCio^
^ сч со <м с^ со
U
оГ ctToo'nTio oo oo
оососмсооосчсп
O ,
^ I
о
* CM
КО
© © CO —* CO О © О —н »— —i LO© — »-* О ~ CM СО О 00©rf©LO
© 0Ол Ю LO О 00 00 N- CM CJ5 N^ CD J "^ 00^ C\ 00^ ^ ^ CO 00 G^
см см со см lo oo lo со со со со со lo oo Tt4 со см ^ Ю lo oo
Sco
uu
о о
CD
O
N
* * * * *
CD
OOCMOOOCMCM—'COCMOOOO—'
CDN^COOOCDCMCMCrJN
OOCMOOO
CD'——^N-^
со ww© ^-.
^^'Ю
CM Tf
Ю ^
CD CM
© см oo —< ю
N CM Ю ЮСО
tO N —< 00 N.
rh CO ^00 Tt<
7 Д
uu и и
о о о о
§2 §.-S?KE^fe
^^ 00 ~* ОО 1^
oo J^ tjh oooi ©
^^°°-Soo-
) чф CD CD 00
~ ~ ~ j; ©*
S 00 LO LOCM
^ЮО5 00
00C
CD ■<
CM CO О ^©00 Ю© — Oi Ю CO G5 N. Tt«NC0
»—i © CD^LO O^N^ "^CM_CM_CD ©^ LO "^ —^N^C7^C0_LO N^00_C^ N^ ^ ^O
nT© юстГоо со
Mi
CM сОл
cd ^
L000
0lO00O00©
^н _н ^ ^, CM
с ^
NC0
^CMC
со сою
N- О Tf< ©00
« CMCOCD^CD-
СОСЧт^^ООГчГю , ^^Ч ОоСгЧ^^ЧЧ^ « CMCOCD^CD
NN©ЮN.C0 I Ю ЮО) CD 00 LO LOCM © 00 00O5 00 LO N-LO ^ © 00
I i*-«i i i I ' a> Tt< ~ч см см —• ^со»-н оо©аэ аяосл^
1'I''' II II 11 11 I
I
i i
II III
117
1
I
s.
J
&
44J
2§§ Ё
|«f I
:
S
" -н " Z-* и^СОЮСО ° N^ NOW CM СМ*т^
^Т) *-н ^—^ jlO с«и* f о [s^ £s^ ф} ^j* ^^ ^sj^ СИ) CO CO 00 CO CO CO СЭ 4|sf* O^ ^^ I4*4-» ^-* ОС CO CO O^
^^^S cxTw ccT cb*of схГо^сГстГ^см N*~^of iT^of of of looo~n~ to **£ об оо ~
CMOI CO TP CO —* — CM •— Г4! CO CO 00 —. — CO LOCOCOCM
csT
8S?
l
I
О
* СО 00
CM N "^Р *~н "^ Ю N 00 I*4» N N СМ *—*
СО СМ СМ СО "tf Оа OJ СЧ СО О1 CD OJ rf
•^ о
00 О^ СО
^б"
О^об 1Гсд
СО СМ тр СО^
со"'-*^>
СОСОО!
ю tj<
—' Ю
t< 00 О
i4
) CO O5
СОЮСОЮЮ
СЛ Ю CM CO 00
и
— OCN
CO 00 CO CO Ю
oaT — — осэо
СО
СО
00 O5 тр « ^— О CM CO N C0N-^x
^оОСОООСОтР СО Ю .ОЮ CM Ol (M CO —i
N N Ю СО Ю ^-^Ю CD CO I^CO ~ — _ _ _
f— — d^do"o"o о'оЪ*со о ю d ~f —
CO - g
CO*
. СО О
^1«
N. ^ ^» СО П« N.
c»ois^cq
00 N CO.О — Ю О О Ю СО
00 Tf СО СМ СО СО СО ^ ТР СО
о*
ю
N
со
Ю. СО '""*"*
SSS
о
5 О1
О5 СЛС^СОЮ
СО СО О ~-* гр
N OJC^CN-^
оз
SCD
U О
о о
о о со
_. Э OI СО СО СО
СО СМ 00 О ^^"^ "—'СО
£$Ч^оо?£со
- — — --rf^CM—•
и
о
f^> >***' f f) I CJT5 C^ O^ с j t^O
со i^nI coionoo
"11 ^ ^Я ^ ^ ^ °^ °?.
^ o- -----
Ю
^ CM CO
■■ "5O1
S3
do"
и
00 O5 1
и
о
COS CO Ю05 000)1ООС0ЮЮ* С5ЙЮ О *-ч COION CONOJO)
_ _ £ф »Г 1^ O^ ^^ CO ^iQ t4** lO CO C7^ t^** *^t^ ^^ ^^ j^i-CO t4^* ^O O^ w^ *■•** CO 00 1™™< o0
^ ^^ ^t* CO ^w^ ^^ C4^ ^^ CO ^^ *""* ^O l-O O4^ ^^ lO 00 ^^ л "***~ ~~~ "**
DNO O5(N _Г С5ОС0~чО1—^CT5OOC7)CO0OCON О
5*O- o*^ o---*-*-*OO*-^*O*go -
щ
г-н 00 00 *
ЮФ'-» —HQ5COCOC-
О N- О СМ —н —. О СО
^ ~—.^н —о
О)
оо
СМ Ю —< 00 СО СП О О 1-н
СО С^ СО "^ СО 00 СО СО Ь^ — ^ 1Л
^*'ГГГГГ*
J ^ N 00 00 О CO
'К g°82 "
—■ ЮСО О5
00 rnrf Ю (ММЛ N
«0>lO ОсО 00 *—«'—'СОСОЮОСОСО'—i-^NON СО СО ЮСОО ООЮОООСО
l OJ CO OJ <N —i CO — О О5 СО —• ^ О —• *-• ^f СМ СЛ *-• 1 00 «0 ^ CM O5 Ю
I I I
I I
I
03
о
Он
g
С» н
Й й § ИНН
§
a
SS^!
• S
о
g 2 g
о н к &, t
со <v О >> <
Е-" Н Н н
s s s т
хххо
359
W
«* at
о «
1
€%
^1
ti
со*4 О
1 ^
О "-"
*■* со
"ii
з© .
и
0
В
Растворитель
О5
00
ю
со
-
3
о
о
СМ
Чх"
00
ю
29,4
1
С?
о
о
со
СП
00
о
со
CD
о
СП
1
иперидин . . .
с
Cvf
36,48 *
826
°"
40
1,5067
о
97
о
58
ю
00
_
1
иридин .....
с
ео
СП
36,9*
1
1
3
о
16 (15
ю
и
о
8
00
О5
8?
О
8
О
СО
ю
оо
7
иррол
ропиленкарбо-
in
со
1
ОО
СМ
1
1
1,4195
а?
СП
"-1
__
rh
см
см
СП
I
нат
г-Г
см
23,3*
004
см
см
со
47,
1,3835
ю
N
О
L0
^,
СМ
СО
т
ропанол ....
пп
21,0*
S
см
§3
1,3747
о
оо
о
о
см
ОО
ю
СП
оо
1
ч
о
я
СЗ
я
о
о.
с
6
со
2,625
31,52
998
о
о
N
СМ
,6397
СО
СМ
*-*
8
с©
57
__
7
ероуглерод . .
и
1
3
о
О
СО
9,87
1
1
о
17 (30
и
о
О
СО
ю
261
—«
со
00
см
СО
00
оо
см
ульфолан . . .
и
етрагидронаф-
Н
,771
см
34,54 *
003
см
в
к
1,5392
см
СП
о
со
о
см
оо
)П
со
талин
,39
1
СМ
см
о
см
со
3
о
)50 (20
00
о
ю
СО
)П
CD
Си
•е-
о
&.
н
ь
1,3,3-Тетраме-
^Г
ю
"^
1
о
в
К
^,
16,9
1,4659
СО
со
о
I
ю
о>
ю
00
N
тилгуанидии . .
,379
см
27,92
СО
551
о
8
N
СО
1,49414
228
8
о
см
СО
о
8
СП
1
1
,05
00
27,2*
8
со
в
S
,42
СО
ft
о
26 (20
СО
97
о
СП
00
см
<->
00
рибутилфосфат
Н
эй фтору ксусная
Н
,26
00
1
со
О)
о
в
к
,73
со
3
о
о
см
см
см
ю
см
ю
т
кислота ....
,41
со
28,8
532
о
в
а
>w
,47
со
1,4750
97*
ю
•~н
СП
N
00
^
CD
00
эихлорэтилен
Н
,42
см
20,14*
со
о
1
1
о
)10 (20
ч
*
nI
о
S3
СП
00
■^f
7
риэтиламин . .
Н
риэтиленгли-
Н
1
45,13*
гг
о
О
СМ
оо
со
см
1
S
см
""-■
со
00
сч
ю
1
КОЛЬ
CD
26,88
со
с§
1,36995
S
о
N.
63
СО
ксусная кислота
360
и
о
ю
см
о
со"
U
S
см
см
U
, | з s? § s
— ю см" ^"" ю
rf СМ СМ СО
О N О О « О5
Ю "^ W Ю об" -^
CM
ON- О СМ
СО СО О) СО СМ
CM" ^ CM" N* СМ" ^ т£
CD « - «
CM
CM
3
см
CD CT) О
о" см N."
ТГ СО СО
^ I
CD т£ CO rf
СМ СМ СО СО СМ
— CO CD О CM —
53 £ 8 83 3 :? S3 £
I
I
о
* oo
%.-
о -Г
e>
со ю I
со о
О 00 * CD
t O5 О —
ю oo ^^tl
d 6 о -" о
"'Sf
N*
о со со о
О О CD^ t-~
d -* о
CM
rt4 o> см
LO О CM
со -^ - o"
О •*•» О) О CD "*-»
CXD CO ^ « Ю -и "^
-*• см со — о со
- • - - -^^'1Г:}1ООСЧЮ
cmco^"oo"cm^co ' —Г со" см" oT oo"
■и Ю <N (N W ^ (N CO 00 CD W (M
°°* oo" сч" со* ю
Ю
см"
—
CM N. CD CM
fe"
r %
$ -
g
00
о
00
о
rf * CD
CMCOcOO^TfOOOCM
^^(МСЙСОЮКЮ
00N.CDOO500OCM
^:_:o"-"c" ooo
со*
СМ
со о> ю ю ^ о ю со
COOOcDNcO^S^CDfN
ю
OON^
о о - о t" »нй « -и" со о> ^* о ^* ю о>*
ThN-00—'OOCOCDCON.lOCOOOCDlO^f
СМ см н
O> Ю CM
1— —< 00 О CO
N- CO CO
со*
N-
О^ ^ЮС^
" Г
Ю-н!^^
* "
сослосмсмюсою — смсосоюсосо
N-CM^ t 00 CO 'Ф Ю I CD CM —' C5
I I
Mill
I
rf CM" -" 'Ф CD" I
О —* —« —« »-'
1 ' T"
О СП
к
a
Я
3
д
о 1*
Si 10
• § §
как
gig
Я jt; щ
2 *о
S a.
Q, О
о н
s§ §
СЗ
fl
CU
CU
С
CU
S >>
р. ^
а я
4S5S5«
fc[ t-
s о
и со
СиХ
OCU
Ч О
X Ч
я a
с н
Е-
« СП
о
Г5 -* ч
ДЛП
CQWCU
О О Д
Ч Ч Ч
s я s
н н н
361
ОСНОВНАЯ ЛИТЕРАТУРА
Как было оговорено в предисловии, в нижеследующем перечне приводится
лишь основная литература по материалу соответствующих глав. В большинстве
случаев даются указания на монографии и обзорные статьи. Кроме того, в ряде
случаев в основном тексте приводятся (на языке оригинала) фамилии авторов
оригинальных статей, которые могут быть отысканы читателем по авторским
указателям к реферативным журналам. В тех разделах, по материалу которых
в последние годы опубликованы монографии, ссылки на журнальные статьи не
даются.
Глава I
1. Ю. Н. Соловьев, История учения о растворах, Изд. АН СССР, 1959.
2. Я. И. Турченко, Основные пути развития общей, неорганической и
физической химии на Украине, Киев, 1957.
3. Успехи физической химии в СССР, Изд. АН СССР, 1967.
Глава II
1. А. П. Крешков, Л. Н. Быкова, Н. А. Казарян, Кислотно-основное
титрование в неводных растворах, Изд. «Химия», 1967.
2. М. И. У с а н о в и ч, Исследования в области теории растворов и теории кислот
и оснований, Алма-Ата, 1970.
3. А. И. Шатен штейн, Теория кислот и оснований, Госхимиздат, 1949.
4. Л. Ф. О д р и т, Я. К л е й н б е р г, Неводные растворители, ИЛ, 1955.
5. G. Chariot, В. Tremillon, Les reactions chimigues dans les solvants et
les sels fondus, Paris, 1963.
6. И.Денеш, Титрование в неводных средах, Изд. «Мир», 1971.
7. А. К. Holliday, A. G. Massey, Inorganic Chemistry in Non-aqueous
Solvents, Oxford, 1965.
8. Т. Ваддингтон, Неводные растворители, Изд. «Химия», 1971.
9. R. G. Pearson, J. Chem. Educ, 45, 581, 643 (1968); Усп. хим., № 7 (1971).
Глава III
1. А. Вайсбергер, Э. Проскауэр, Дж. Р и д д и к, Э. Т у п с, Органические
растворители, ИЛ, 1958.
2. М. М. Davis, Acid-base behavior in Aprotic Organic, Solvents, Washington,
1968.
3. J. T i m m e r m a n s, Physico-chemical Constants of Pure Organic Compounds.
Vol. I, II, Amsterdam, 1950, 1965,
362
Глава IV
1. С. Бретшнайдер, Свойства газов и жидкостей, Изд. «Химия», 1966.
2. Дж. Гиршфельдер и др., Молекулярная теория газов и жидкостей, ИЛ,
1961.
3. С. Глесстон, К. Лейдлер, Г. Эйринг, Теория абсолютных скоростей
реакций, ИЛ, 1948.
4. В. И. М и н к и н, О. А. Осипов, Ю. А. Жданов, Дипольные моменты
в органической химии, Изд. «Химия», 1968.
5. С. Оно, С. К о н д о, Молекулярная теория поверхностного натяжения в
жидкостях, ИЛ, 1963.
6. О. А. Осипов, В. И. М и н к и н, А. Д. Г а р н о в с к и й, Справочник по ди-
польным моментам, Изд. «Высшая школа», 1971.
7. Г. М. Панченков, Теория вязкости жидкостей, Гостоптехиздат, 1947.
8. Р. Рид, Т. Шервуд, Свойства газов и жидкостей, Изд. «Химия», 1971.
9. В. М. Т а т е в с к и й, Химическое строение углеводородов и закономерности
в их физико-химических свойствах, Изд. МГУ, 1953.
10. В. М. Т а т е в с к и й, В. А. Б е н д е р с к и й, С. С. Яровой, Методы
расчета физико-химических свойств парафиновых углеводородов, Гостоптехиздат,
I960.
11. Физические методы органической химии, под ред. А. Вайсбергера, т. I, ИЛ,
1950.
12. И. 3. Фишер, Статистическая теория жидкостей, Физматгиз, 1961.
13. М. И. Ш а х п а р о н о в, Введение в молекулярную теорию растворов, Гостех-
теоретиздат, 1956.
14. Ф. Э м е, Диэлектрические измерения, Изд. «Химия», 1967.
15. J. R. P a r t i n g t о n, An Advanced Treatise on Physical Chemistry, Vol. 2,
Properties of Liquids, London, 1951.
16. I. Prigogine, The Molecular Theory of Solutions, Amsterdam, 1957.
17. С P. Smyth, Dielectric Behavior and Structure, N. Y., 1955.
18. Discuss. Faraday, Soc, № 43 (1967).
19. J. Wiemann, Y. Pascal, J. С huсhe, Relations entre la structure et les
properties physiques, Paris, 1965.
20. A. E. Л у цк и й, Усп. хим., 23, 479 (1954). Влияние Н-связи на свойства
органических соединений (обзор).
21. St. G. Brush, Chem. Rev., 62, 513 (1962). Обзор теорий вязкости.
22. О. Exner, Coll. Czech. Chem. Comm., 31, 3222 (1966); 32, 24, 4327 (1967).
Статистический анализ структурно-аддитивных констант жидкостей.
См. также литературу к гл. III.
Глава V
1. В. П. Белоусов, А. Г. Морачевский, Теплоты смешения жидкостей,
Изд. «Химия», 1970.
2. Н. А. Измайлов, Электрохимия растворов, Изд. «Химия», 1966.
3. Н. А. Измайлов, Избранные труды, Изд. «Наукова думка», Киев, 1967.
4. Г. А. Крестов, Термодинамика ионных процессов в растворах, Изд.
«Химия», 1973.
5. И. Пригожий, Р. Д е ф э й, Химическая термодинамика, СО Изд. «Наука»,
Новосибирск, 1966.
6. В. К. Семенченко, Физическая-теория растворов, ГИТТЛ, 1941.
7. Сб. «Химия и термодинамика растворов», вып. 1, Изд. ЛГУ, 1964; вып. 2,
1968.
8. D. Dreisbach, Liquids and Solutions, Boston, 1966.
9. J. H. H i 1 d e b r a n d, R. L. S с о 11, The Solubility of Non-electrolytes, N, Y.,
1950.
10. J. H. Hildebrand, R. L. Scott, Regular Solutions, N. Y., 1962.
11. J. S. Ro wlinson, Liquids and Liquid Mixtures, London, 1969.
12. H. C. V a n Ness, Classical Thermodynamics of Non-electrolyte Solutions,
Oxford, 1964.
363
13. A. G. Williamson, An. Introduction to Non-electrolyte Solutions, Oxford
1967.
14. P. M. В асе нин, ЖФХ, 43, 1201 (1969). Диффузия в бинарных жидких
системах (неэлектролиты).
15. R. Battino, H. L. Clever, Chem. Rev., 66, 395 (1966). Растворимость газов
в жидкостях (обзор).
16. P. J. Flory, J. Am. Chem. Soc, 87, 1833 (1965). Статистическая
термодинамика жидких смесей.
17. Н. G. Hertz, Ber. Bunsenges. Phys. Chem, 75, 183 (1971). Самодиффузия
в жидкостях (обзор).
18. М. L. Huggins, J. Phys. Chem., 74, 371 (1970). Развитие методов расчета
термодинамических свойств жидкостей и растворов.
19. D, A. Jonah, М. В. King, Proc. Roy. Soc. (London), 323A, 361 (1971). Mo-
лекулярно-статистический расчет температурных коэффициентов
растворимости газов.
20. J. S. Rowlinson, Ind. Eng. Chem., 59, № 12, 28 (1967). Молекулярные
теории жидкостей и смесей (обзор).
21. R. L. Scott, D. V. Fenby, Ann. Rev. Phys. Chem., 20, 111 (1969).
Растворы неэлектролитов. (Современные достижения теории и обзор работ за
1966—1968 гг.) См. также к гл. IV [2, 12, 13, 16, 18].
Глава VI
1. К. П. Мищенко, Г. М. Полторацкий, Вопросы термодинамики и
строения водных и неводных растворов электролитов. Изд. «Химия», 1968.
2. Р. Робинсон, Р. Стоке, Растворы электролитов, ИЛ, 1963.
3. О. Я. Самойлов, Структура водных растворов электролитов и гидратация
ионов, Изд. АН СССР, 1957.
4. Труды I Конференции по аналитической химии неводных растворов и их
физико-химическим свойствам, ч. II, изд. МХТИ, 1968.
5. Труды Совещания по влиянию растворителей на свойства электролитов, Изд.
ХГУ, Харьков, 1960.
6. Электролиты в воде и неводных средах. Труды I Менделеевской ^дискуссии,
Изд. «Химия», 1970.
7. Б. Е. К о н у э й, Дж. О'М. Б о к р и с, сб. «Новые проблемы современной
электрохимии», ИЛ, 1954, стр. 63. Сольватация ионов.
8. Г. А. Крестов, Автореф. докт. дисс, МХТИ, 1966.
9. Н. S. Н а г n e d, В. В. Owen, The Physical Chemistry of Electrolytic
Solutions, N. Y., 1963.
10. С R. Allen, M. R. Wright, P. G. Wright, J. Chem. Soc, A, № 6, 892
(1967). Сводка энергий сольватации.
11. С. М. Criss, R. P. Held, E. Luksha, J. Phys. Chem., 72, 2970 (1968).
Ионные энтропии: их оценки и взаимосвязь со структурой растворов
электролитов.
См. также гл. V. [2, 3, 6, 7], гл. IV [13].
Глава VII
См. гл. V [2, 3, 8].
Глава VIII
1. В. Я. Аносов, Геометрия химических диаграмм двойных систем, Изд. АН
СССР, 1959.
2. В. Я. Аносов, С. А. Погодин, Основные начала физико-химического
анализа, Изд. АН СССР, 1948.
3. Б. В. Иоффе, Рефрактометрические методы химии, Госхимиздат, 1960.
4. Н. С. Кур на ков, Введение в физико-химический анализ, Изд. АН СССР,
1940.
364
б. С. П. Мискиджьян, Электрохимия неводных ионогенных систем,
образованных из электролитов, Изд. «Наукова думка», Киев, 1970.
6. Ю. Я. Фиал ко в, Двойные жидкие системы, Изд. «Техшка», Киев, 1969.
7. J. Tim mer mans, The Physico-chemical Constants of Binary Systems in
Concentrated Solutions, N. Y., 1957—1959, Pt. I—IV.
8. А. К. Б а б к о, Физико-химический анализ комплексных соединений в
растворах, Изд. АН УССР, Киев, 1955.
9. Е. Е. Черкашин, Метрика равновесной химической диаграммы систем
с ассоциированными компонентами, Львов, 1958.
10. И. А. Шека, сб. «Работы по химии комплексных соединений», Изд. АН
УССР, Киев, 1959, стр. 163. Физико-химический анализ растворов, основанный
на их диэлектрических свойствах.
11. Н. А. Измайлов, ЖФХ, 25, 1070 (1951). Физико-химический анализ двух-
компонентных систем в растворах.
См. также гл. V [2, 3].
12. А. И. Бродский, Современная теория электролитов, Госхимиздат, 1934.
13. Б. И. И о н и н, Б. А. Е р ш о в, ЯМР-спектроскопия в органической химии,
Изд. «Химия», 1967.
14. А. Ф. Скрышевский, Рентгенография жидкостей, Изд. КГУ, Киев, 1966.
Структурный анализ жидкостей, Изд. «Высшая школа», 1971.
15. Н. А. Измайлов, Ю. А. Кругляк, ДАН СССР, 134, 1390 (1960).
Описание процесса сольватации методом молекулярных орбиталей.
16. N. A. Ismaylov et al., Acta phys. Hung., 13, 203 (1961). Квантово-меха-
нический расчет протонного сродства.
17. И. С. Перелыгин, Оптика и спектроскопия, 16, 40 (1964). Исследование
сольватации методом ИК-спектроскопии.
18. J. Burgess, М. С. R. Symons, Quart. Rev. Chem. Soc, 22, 276 (1968).
ЯМР — исследование взаимодействия ион — ион и ион — растворитель.
19. М. Delia Monica, L. Sen a tore, J. Phys. Chem., 74, 205 (1970).
Радиусы сольватированных ионов в неводных растворителях.
20. Дж. Десноерс, С. Джоликер, Современные проблемы электрохимии,
Изд. «Мир», 1971, стр. 11.
21. G. E. Ewing, Accounts Chem. Res., 2, 168 (1969). Спектроскопическое
исследование движения молекул в жидкостях
22. Н. G. Hertz, Angew. Chem., 82, 91 (1970). Структура сольватной оболочки
растворенных частиц.
23. J. F. Hint on, E. S. Amis, Chem. Rev., 67, 367 (1967). Изучение ионов в
индивидуальных и смешанных растворителях методом ЯМР.
24. J. F. Hi n ton, E. S. Amis, Chem. Rev., 71, 627 (1971). Числа сольватации
ионов. Обзор.
25. S. Mine, Wiadom. Chem., 21, 621 (1967). Исследование сольватации ионов
методом ЯМР.
26. Т. Zeegers-Huyskens, Ind. chim. Belg., 33, 525 (1968). Применение
ИК-спектроскопии для исследования молекулярных взаимодействий, в том
числе сольватации.
См. также гл. V [2, 3, 18]; гл. VI [1—3, 7, 8].
Глава IX
1. Устойчивость смешанных комплексных соединений в растворах, под ред.
Я. Д. Фридмана, Фрунзе, 1971, гл. VI.
2. Р. Бейтс, Определение рН, Изд. «Химия», 1972.
3. Т. К. Blackburn, Equilibrium. Chemistry of Solution, N. Y., 1969.
4. G. M. Fleck, Equilibria in Solution, N. Y., 1967.
5. R. W. G u r n e y, Ions in Solution, N. Y., 1962.
6. В. Г у Т м ан, Химия координационных соединений в неводных растворах,
Изд. «Мир», 1971.
7. J. L e f f I e r, E. G г u n w a I d, Rates and Equilibria of Organic Reactions,
N, Y., London, 1963.
365
8. М. И. Кабачник, Изв. АН СССР, ОХН, 1955, 98. К теории таутомерного
равновесия в растворах.
9. М. И. Кабачник, ДАН СССР, 83, 859 (1952). О влиянии растворителя на
таутомерное равновесие и кислотность таутомерных форм.
10. Я. И. Турьян, ДАН СССР, 102, 295 (1955). Влияние растворителя на кон-
станту нестойкости комплексного иона.
11. V. G u t m a n n, Rev. chim. miner., 3, 941 (1966). Влияние растворителя на
координационные реакции в неводных растворах.
12. R. M. F u о s s, Ch. А. К г a u s, Обширный цикл статей по константам
диссоциации электролитов в растворителях различных классов, опубликованных в
основном в J. Am. Chem. Soc.
Весьма обширный материал по теме гл. IX содержится в работах Н. А.
Измайлова (см. лит. к гл. V [2, 3]).
Глава X
1. Г. Фалькенгаген, Электролиты, ОНТИ Химтеорет., 1935.
2. Г. Фалькенгаген, Г. Кельбг, Новые проблемы современной
электрохимии, ИЛ, 1962, стр. 11. Современное состояние электролитических
растворов.
3. А. М. Сухотин, Е. М. Рыжков, ЖФХ, 34, 762 (1960). Об особенностях
изотерм электропроводности 1 — 1-валентных солей в растворах с низкой
диэлектрической проницаемостью.
4. А. М. Шкодин, Укр. хим. ж., 26, 565 (1960). Аномальная
электропроводность и сольватация ионов.
5. А. М. Шкодин, Изв. вузов. Хим. и хим. технол., 1961, 941. О влиянии
растворителей на предельную подвижность ионов.
6. J. В art hell, Angew. Chem., 80, 253 (1968). Электропроводность растворов
электролитов. (Обзор)\
7. W. Ebeling, Z. phys. Chem. (DDR), 238, 400 (1968). О теории Бьеррума
ассоциации ионов в электролитах.
8. W. D. Кг a eft, W. Ebeling, Z. phys. Chem. (DDR), 240, 141 (1969). О
теории электропроводности ассоциированных электролитов.
9. R. Fernandez-Prini, Trans. Faraday Soc, 65, 3311 (1969).
Электропроводность растворов электролитов. Модифицированное выражение для
зависимости электропроводности от концентрации.
10. W. D. Kraeft, R. S a n d i g, J. chim. phys. e phys.-chim. biol., 67, 1265
(1970). Уравнение электропроводности для ассоциированных электролитов.
11. R. Paul, J. Chem. Phys., 50, 1491 (1969). Вывод обобщенного правила Валь-
дена методами статистической механики.
12. М. Szwarc, Accounts Chem. Res., 2, № 3, 87 (1969). Ионы и ионные пары.
(Обзор).
См. также гл. V [2, 3]; гл. VI [2, 9]; гл. VIII—В [1].
Глава XI
1. Л. И. Антропов, Теоретическая электрохимия, Изд. «Высшая школа», М.,
1969.
2. Зб1рник, црисвячений 35-р1ччю науково1 д1яльноси академжа В. О. Плотш-
кова, Вид. АН УРСР, Кшв, 1936. Работы киевской школы электрохимиков по
исследованию механизма проводимости неводных растворов.
3. Е. А. К а й м а к о в, Н. Л. В а р ш а в с к а я, Усп. хим., 35, 2, 201 (1966).
Измерение чисел переноса в водных растворах электролитов.
4. Б. Е. К о н у э й, сб. «Современные аспекты электрохимии», № 3, Изд. «Мир»,
1967, гл. II. Процессы переноса и сольватации протона в растворах.
5. М. И. Ш а х п а р о н о в, ЖФХ, 38, 2, 252 (1965). Вещества с межмолекулярной
водородной связью как протонные полупроводники.
366
6. G. J. Hills, in «Physico-chemical Processes in Mixed Aqueous Solvents,
London, 1967, p. 91. Рассмотрение механизма переноса ионов в водных и неводных
растворах по температурным зависимостям ионных подвижностей.
7. М. Spiro, J. Chem. Educ, 33, № 9, 464 (1956). О физическом смысле понятия
«число переноса».
См. также гл. IV [4], гл. VIII [6].
Глава XII
1. О. I. Бррдський, Досл1ди з термодинам1ки та електрох1м11 розчин1в, Тех-
видав УРСР, Дн1пропетровськ, 1931.
2. Н. А. И з г а р ы ш е в, С. В. Горбачев, Курс теоретической электрохимии,
Госхимиздат, 1951.
3. Р. П а р с о н с, сб. «Некоторые проблемы современной электрохимии», ИЛ,
1958, стр. 125.
4. В. А. Плесков, Усп. хим., 16, 254 (1947). Электродные потенциалы и
энергия сольватации ионов.
5. М. А. Рязанов, Электрохимия, 6, 211 (1970). К теории диффузионного
потенциала.
6. А. Н. Фрумкин, Усп. хим. 15, 385 (1946). Механизм возникновения
потенциалов.
7. Н. Е. Хомутов, ЖФХ, 42, 2229 (1968). Термодинамические методы расчета
стандартных электродных потенциалов в неводных и смешанных
растворителях.
8. Н. Е. Хомутов, Итоги науки. Электрохимия. 1967. изд. ВИНИТИ, 1969,
стр. 95. Электроды сравнения в растворах.
9. J. Brenet, J. chim. phys. et phys, et phys.-chim. biol., 66, 1057 (1969).
Теоретические основы общей проблемы электрода сравнения в неводных средах.
10. J. N. Butler, Advances of Electrochemistry and Electrochemical Engineering
vol. 7, N. Y., 1970, p. 77. Обратимые электроды в апротонных органических
растворителях.
11. Н. Strehlow, in «The Chemistry oi Non-aqueous Solvents, Vol. 1, N. Y.,
1966, p. 129. Электродные потенциалы в неводных растворителях.
См. также гл. V [2], гл. XI [1].
Глава XIII
1. Г. В. Акимов, Теория и методы исследования коррозии металлов, Изд. АН
СССР, 1959.
2. Дж. О'М. Б о к р и с, сб. «Некоторые проблемы современной электрохимии»,
ИЛ, 1958, стр. 209. Кинетика электродных процессов.
3. Дж. О'М. Бокрис, А. Дамьянович, сб. «Современные аспекты
электрохимии», Изд. «Мир», 1967, стр. 259. Механизм электроосаждения металлов.
4. Я. Гей р о веки й, Я. Кута, Основы полярографии, Изд. «Мир», 1965.
5. П. Д е л а х е й, Двойной электрический слой и кинетика электродных
процессов, Изд. «Мир», 1967.
6. Р. Р. Д о г о н а д з е, А. М. Кузнецов, сб. «Итоги науки. Электрохимия».
1967, изд. ВИНИТИ, 1969, стр. 5. Современное состояние теории электродных
процессов.
7. Б. Н. Кабанов, Электрохимия металлов и адсорбция, Изд. «Наука», 1966.
8. Л. I. К а д а н е р, Електроосаждения благородних i рцгдасних метал!в, «Тех-
Н1ка», Кшв, 1968.
9. С. Г. Майрановский, Каталитические и кинетические волны в
полярографии, Изд. «Наука», 1966.
10. П. Л. Перрин, сб. «Новые проблемы физической органической химии», Изд.
«Мир», 1969, стр. 95. Механизмы органических полярографических реакций.
11. В. А. Плотников, сб. «Работы по химии растворов и комплексных
соединений», Изд. АН УССР, 1959, стр. 3. Электрохимические свойства галогенидов
алюминия в неводных растворах.
367
12. К. Ф е т т е р, Электрохимическая кинетика, Изд. «Мир», 1967.
13. А. Н. Ф р у м к и н, В. С. Б а г о ц к и й, 3. А. И о ф а, Б. Н. Кабанов,
Кинетика электродных процессов, Изд. МГУ, 1952.
14. Т. П. Хор, сб. «Новые проблемы современной электрохимии», ИЛ, 1962,
стр. 284. Анодное поведение металлов.
15. Т. Дамокош, Венгерское исследовательское оборудование, 1967, ноябрь,
стр. 32. Некоторые вопросы полярографии систем с высоким сопротивлением.
16. Н. А. Измайлов, В. Д. Б е з у г л ы й, Труды комиссии по аналитической
химии, т. IV, Изд. АН СССР, 1952, стр. 29. Влияние растворителя на поляро.
графические характеристики.
17. Л. Я. Хейфец, В. Д. Безуглый, ЖОХ, 41, 514 (1947). О влиянии
неводных растворителей на потенциалы полуволн некоторых органических
деполяризаторов.
18. В. Шмидт, Защита металлов, 5, 608 (1969). О коррозии металлов и сплавов
в органических средах.
19. A. Brenner, Advances of Electrochemistry and Electrochemical Engineering,
Vol. 5, N. Y., 1967, p. 205. Электролиз неводных систем.
20. P. Т. Elving, Pure a, Appl. Chem., 15, 297 (1967). Современные
направления в органической полярографии.
21. Н. Schneider, H. Strehlow, J. Electroanalyt. Chem., 12, 530 (1966).
Сравнение потенциалов полуволн в различных растворителях.
22. К. Schwabe, Progress in Polarography, Vol. 1, London, 1962.
Полярография в неводных растворителях.
См. также гл. XI [1].
Глава XIV
1. Э. Амис, Влияние растворителя на скорость и механизм химических
реакций, Изд. «Мир», 1966.
2. С. Бен с он, Основы химической кинетики, Изд. «Мир», 1964, гл. XV, XVI.
3. М. С. Захарьевский, Кинетика химических реакций, Изд. ЛГУ им.
А. А. Жданова, 1969.
4. К. Л ей д л ер, Кинетика органических реакций, Изд. «Мир», 1966.
5. Е. А. М е л в и н - X ь ю з, Кинетика реакций в растворах, ГОНТИ, 1938.
6. Е. А. Мелвин-Хьюз, Физическая химия, ч. II, ИЛ, 1962, гл. XXIV.
7. В. А. Пальм, Основы количественной теории органических реакций, Изд.
«Химия», 1967.
8. Г. М. Панченко, В. П. Лебедев, Химическая кинетика и катализ, Изд.
МГУ, 1961.
9. Н. В. С а п о ж н и к о в а, Кинетика химических реакций в растворах, изд.
У ПИ, Свердловск, 1963.
10. Н. Н. Семенов, О некоторых проблемах химической кинетики и
реакционной способности, Изд. АН СССР, 1958.
11. Н. М. Эма ну эль, Д. Г. Кнорре, Курс химической кинетики (Гомогенные
реакции), Изд. «Высшая школа», 1969.
12. Е. S. Amis, Kinetics of Chemical Change in Solution, N. Y., 1949.
13. H. Eyring, E M. E u r i n g, Modern Chemical Kinetics, in Selected Topics
of Modern Chemistry, N. Y., 1963.
14. S. L. Friess, E. S. Lewis, A. Weissberger, Investigations of Rates and
Mechanisms of Reactions, in «Technique ob Organic Chemistry», Vol. VIII,
Ch. I, II, XI, N. Y. — London, 1961.
15. A. A. Fro s't, R. G. Pearson, Kinetics and Mechanism. A. Study of
Homogeneous Chemical Reactions, N. Y., 1961.
16. G. M. H a r r i s, Chemical Kinetics, Boston, 1966.
17. E. A Moelwyn-Hughes, The Kinetics of Reactions in Solution, Oxford,
1947/
18. A. M. North, The Collision Theory of Chemical Reactions in Liquids,
London, 1964.
19. Chr. Reichardt, Losungsmittel — Effekte in der organischen Chemie, Wein-
heim, 1969.
368
20. M. Ritchie, Chemical Kinetics in Homogeneous Systems, Edinburgh, 1966.
21. E. T о m m i 1 a, The Influence of Solvent, on Substituent Effect in Chemical
Reactions, Helsinki, 1967.
22. Д. Г. Кнорре, Н.М.Эма н у э л ь, Усп. хим., 24, 3, 275 (1955). Водородная
связь в кинетике химических реакций.
23. Б. Ч у б а р, Усп. хим., 34, 7, 1227 (1965). Некоторые аспекты роли
растворителя в органической химии.
24. A. J. Parker, Quart. Rev., 16, 2, 163 (1962). Влияние сольватации на
свойства анионов в диполярных апротонных растворителях.
25. A. J. Parker, Chem. Rev., 69, 1 (1969). Влияние протонных и диполярных
апротонных растворителей на скорости бимолекулярных реакций.
См. также гл. IV [4], гл. IX [2].
Глава XV
1. Д. Бете л, В. Голд, Карбониевые ионы, Изд. «Мир», 1970.
2. А. И. Бродский, Химия изотопов, Изд. АН СССР, 1957, гл. 6, 7.
3. Сб. «Гомогенный катализ», под ред. Д. В. Сокольского, Изд. «Наука», Алма-
Ата, 1970.
4. Катализ, стереохимия и механизмы органических реакций, пер. под ред.
А. А. Баландина, А. М. Рубинштейна, Изд. «Мир», 1965.
5. Сб. «Каталитические реакции в жидкой фазе», Изд. «Наука», Алма-Ата, 1967.
6. Е. Ко л дин, Быстрые реакции в растворе, Изд. «Мир», 1966.
7. Л. Мела н дер, Изотопные эффекты в скоростях реакций, Изд. «Мир», 1966,
8. Д. В. Сокольский, Гидрирование в растворах, Изд. АН КазССР, Алма-
Ата, 1962. -
9. А. И. Ш а т е н ш т е й н, Изотопный обмен и замещение водорода в
органических соединениях, Изд. АН СССР, 1960.
10. А. И. Бродский, ЖОХ, 24, 413 (1964). Механизм водородного обмена в
некоторых протолитических реакций в растворах
11. Э. Г р ю н в а л ь д, сб. «Новые проблемы физической органической химии»,
Изд. «Мир», 1969, стр. 207.
12. Е. А. Халеви, сб. «Современные проблемы физической органической
химии», Изд. «Мир», 1967, стр. 95.
13. А. И. Ш а тен ш тей н, Кинетика и катализ, 8, 1056 (1967). Измерение
кинетики реакций изотопного обмена водорода в неводных растворах.
14. Ф. С. Якушин, Усп. хим., 31, 241 (1962). Исследования кинетического
изотопного эффекта трития.
15. S. A. Bernard, in «Investigations of Rates and Mechanisms of Reactions»,
Ed. by S. L. Friess a. oth., Vol. VIII, Pt I, N. Y,,' 1961, p. 579, Гомогенный
катализ.
16. E. F. Caldin, Chem. Rev., 69, 135 (1969). Туннельный эффект в реакциях
переноса протона в растворе.
17. Discuss. Faraday Soc, № 39, 278 (1965). Дискуссия по проблемам кинетики
переноса протона.
18. М. Eigen (См. [15], р. II, N. Y., 1963, р. 793) Очень быстрые реакции в
растворе. См. там же, р. 799. Методы изучения быстрых реакций.
19. Е. М. tyring, in «Survey of Progress in Chemistry», Ed by A. F. Scott,
Vol. 2, N. Y., 1964. p. 57. Быстрые реакции.
20. W. H. Saunders, Jr. (См. [15], p. 389). Кинетические изотопные эффекты.
21. F. J. W. Roughton, B. Chance (см. [18], p. 703). Быстрые реакции. См.
также гл. XIV [2, 4, 6, 8, 11].
УКАЗАТЕЛЬ
Автоионизация 20, 233, 259, 311
Аддитивность 118 и ел.
отклонение от нее 119, 121 и ел., 124, 128,
130, 132, 137, 160, 173
Ассоциация 8, 14, 59, 106 и ел., 111 и ел.,
171, 181, 203, 205, 225, 256, 296, 298
влияние на
обмен 352 '
состояние компонентов 139
среднюю ионную активность 101
зависимость от
возникновения водородных связей 205
и ел.
диэлектрической проницаемости 203 л
ел.
константа 181, 194
перекрестные ассоциаты 111
связь с процессами диффузии 81
степень 107 и ел., 205, 244 и ел., 294
фактор 182, 204
Водородная связь, влияние на
кинетику 336, 345
переход протона 336
распад ассоциатов 205
теплоты смешения 147
электропроводность 259
Водородная связь, образование 36, 55, 67, 107,
111
Диаграммы состояния двойных жидких
систем
иррациональные 125
определение констант равновесия 161
определение функции при наличии
обменного взаимодействия 166
основные типы 119
рациональные 125
связь элементов со степенью
взаимодействия 160
способы выражения состава 117
Доли
аналитические 118
истинные 118
переноса тока 257
Инкременты физико-химических свойств 46,
48, 52, 54
Ион лиония 233
Ионность связи 211
Кислоты и основания
апротонные 19, 22, 28, 208
вторичные 24
заряженные 22
как среда для проведения реакций 29,
31
льисовские 23, 224
нейтральные 22
сопряженные 22, 25
Кислотно-основное взаимодействие 18 и ел.
влияние
на природу механизма
электропроводности 261
на силу электролита 224
Константа
автопротолиза 38, 259 и ел.
нестабильности 216
общая 216
собственных
кислотности 218, 234
основности 218
таутомеризации 212
условная 326
Константа Джоши 48
Коэффициент активности
единый 102
концентрационный 101
моляльный 101
молярный 101
нулевой 102
рациональный 101
Лиопарахор 54
Межмолекулярное взаимодействие
в регулярных растворах 63
как основа классификации
неэлектролитов 57 и ел
Методы выбора стандартного состояния 100
Методы расчета свойств жидкостей 46 и ел.
в гомологических рядах по
Луцкому 47
Татевскому 46
основанные на принципе химического
подобия (по Кирееву) 46
с использованием понятия среднеквадра»
тичный радиус (по Альтенбургу) 48
статистические 42, 59
Ортохор 48
Отклонение от идеальности 77, 78, 102
Катализ
кислотно-основной 345
реакций
гидрирования 348
полимеризации 346
радикального распада 347
Квазидвойные системы, см физ.-хим.
анализ двойных систем в растворителе
Кинетическая кислотность 351
Парахор 48, 53, 144
Показатель кислотности
единый 235
по Бренстеду 234
по Гам мету 234
Полярография, выбор
индифферентного электролита 304
неводного растворителя 304
электрода сравнения 300
370
Порядок реакции 320, 322
Потенциал
кислотности 234
основности 234
химический протона 234
Правило
Вальдена 8, 243
Гильдебранда — Скотта 66, 69
Лавуазье 74
Мищенко 90, 99
Писаржевского—Вальдена 243, 245, 255
Ричардсона—Сопера 329
Усановича 248
Протолитическое равновесие 30
Растворение
изменение теплоемкости 86
определение термодинамических
параметров 84
теплота 85
Растворимость газов
в смесях 72
идеальная 65
коэффициенты 65
объяснение при помощи
решеточной модели 70
теории регулярных растворов 67
температурный коэффициент 71
Растворимость жидких неэлектролитов,
взаимная 73
Растворители
влияние на
диффузионные токи 293
потенциал полуволны 294
классификация по
Бренстеду 37
вязкости 34
дипольным моментам 33
диэлектрическим проницаемостям 33
температурам кипения 34
химическим свойствам 34
применение в полярографии 304
типы
акцепторные 35
амфипротонные 35
амфотерные 231
аполярные 36
апротонные 35, 82, 262, 264, 338
донорные 35
протогенные 35, 37
протофильные 35, 37, 338
способные к образованию
водородных связей 36
способные сольватировать ионы 36
Растворы
атермальные 58
идеальные 58, 74, 75
регулярные 14, 60, 62, 68, 69, 71, 73 74
75, 329 '
Реохор 48
Сила электролитов 224, 225, 227, 228 и ел.
Скорость реакции, влияние
водородной связи 336
вязкости 341
давления 342
избыточной энтальпии смешения 337
сольватационных процессов 335, 338
Сольватация
влияние на электропроводность 256
граница 88
дифференцирующее действие. 224
допущение равенства теплот 90
иона 279
методы определения по
диффузии 189
Сольватация, методы определения по
магнетохимическим данным 198
рентгено- и электронографическим
данным 199
сжимаемости 197
спектральным данным 190, 192, 193
числам переноса 185
электропроводности 186
механизм 70
модель 88, 96
неспецифическая 329
нивелирующее действие 224
оболочки 112
вторичная 188
объем 188, 298
первичная 188
структура 188
описание
методом МО 200
с использованием величин рКдис 219
отрицательная 199
первая координационная сфера 194
пересольватация 207
протона 260, 285
радиус иона 136
разделение термодинамических
параметров на составляющие 91, 93, 104
разрушение структуры растворителя 194,
197
специфическая 334 и ел., 350, 353
способность 36
теплота 87, 90, 95 - .
электрона 253
энергия 69, 79
энтальпия 90
энтропия 97
Сродство к протону 38, 212, 225, 227, 281, 316
Структура воды 42 и ел.
Структура жидкостей 40 и ел., 45, 108
влияние на кинетику 345
изменение под влиянием кооперативного
характера взаимодействия молекул 324
компактность 100
модели
дырочная 41
решетчатая 44, 45, 59, 70
ячеечная (клеточная) 41, 59, 108, 326
образование водородных связей 36, 67,
107, 111 и ел.
порядок
ближний 14, 326
дальний 14
разрушение 194, 197
Теория
активированного комплекса 322 и ел.
ассоциативно-диссоциативных процессов
106
влияние растворителя на процесс
электролитической диссоциации 215, 219
кислот и оснований
Бренстеда (протолитическая) 21, 218
Вернера 19
Ганча 19
Льюиса (электронная) 23, 224
Пирсона 26
сольвосистем 20
Усановича 25, 225
молекулярно-статистическая жидкого
состояния 42 и ел.
переноса тока 253, 259 и ел.
переходного состояния 322 и ел.
расчета молекулярных функций
распределения 44 и ел.
регулярных растворов 58 и ел.
Теория
Батлера 270
Борна^-Бьеррума 90 ч
871
Теория
Бренстеда 346 и ел.
Бродского 288
Бьеррума 244
Гильдебранда 56 и ел.
Дебая—Хюккеля—Онзагера 330
Левича 307
Писаржевского 269
Семенченко 244
Семенченко — Шахпаронова 73
Фрумкина 273
Фуосса — Крауса 245, 250
Хаггинса 63
Эйринга 81, 254, 256
Точка нейтрализации 233
Уравнение
Альтенбурга 48
Амиса 332
Антуана 53
Беннета — Митчелла 108
Бломбергена 194
Борна 203
Борна — Бьеррума 90
Борна — Уэббса 219
Бренстеда 213, 218, 328, 346
Бродского 289
Бруммера — Хиллса 256
Ван-дер-Ваальса 53
Ван-Меурса 248, 249
Войткевича 52
Гаммета 235
Гейровского — Ильковича 295
Гильдебранда 61
Даркена 81
Дебая 354
Де-Гусмана 52
Жуховицкого 144
За кура — Тетроде 98
Измайлова 82, 92, 217, 219, 223, 236, 273
Ильковича 293
Ирвинга — Росотти 209
Кирквуда 332
Кирквуда — Харриса — Алдера 55
Кистяковского 54
Конуэя — Бокриса 197
Крестова 85
Лейдлера — Эйринга 332
Ленард — Джонса 44
Мелвин-Хьюза 341
Микулина 105
Миллера 54
О'Коннела — Праузнитца 72
Онзагера 239
Пальма — Коппеля 341
Панченкова 141
Пригожина 77
Пьеротти 70
Рамзая — Шильдса 108
' Рао 146
Рида 65
Робинсона — Стокса 240
Розен а 77
- Розера — Биттриха 78
Семенченко 66
Скотта 54
Смита — Уолкли 69
Смолла 75
Стокса 187
Сухотина 220
Сыркина — Осипова 55
Томаса 51
Ту рьян а 207
Фалькенгагена 240
Фергюсона 53
Фиалкова 152
Уравнение
Флори 60, 68
Фольмера 308
Фрумкина 308
Фуосса 246
Фуосса — Онзагера 240
Хаггинса 63. 68
Хаугтона 108
Хомутова 284
Шахпаронова 137
Шедловского 240
Шкодииа 246
Шредера — Ле-Шателье 74
Эйкмана 50
Эйринга 52, 55
Этвеша 53
Физико-химический анализ
двойных систем
вискозиметрия 140
волюмометрия 127
диэлькометрия 137
калориметрия 146
кондуктометрия 149
поверхностное натяжение 144
потенциометрия 153
радиоспектроскопия 135
рефрактометрия 129
скорость звука 145
спектрофотометрия 132
сравнение методов 159
температурные коэффициенты
свойств 154
двойных систем в растворителе 167, 169
в предположении отсутствия
взаимодействия 173
криоскопия 170
с использованием парциальных
молярных свойств 174
Физико-химические свойства жидкостей
вязкость 51
давление пара 53
дипольный момент 54
диэлектрическая проницаемость 54
плотность 50
поверхностное натяжение 52
показатель преломления 50
теплота испарения 53
электропроводность 50
Характеристика поведения одного
электролита в другом 222, 224, 225
Числа переноса 185, 257
истинные 185
кажущиеся 185
трансляционные 188
Электродные потенциалы
жидкостной 268
контактный 267
на границе металл — раствор 269
условные 276
Электропроводность
аномальная 259
зависимость
от вязкости 243
от диэлектрической проницаемости 243
от температуры 248
механизмы 259, 264
Электрохимический ряд в неводных
растворах 281
ОГЛАВЛЕНИЕ
Предисловие 3
ОБЩИЕ ПОЛОЖЕНИЯ
Глава I. История развития исследований в области физической химии
неводных растворов 5
Глава II. Химические реакции в неводных растворителях 18
II. 1. Теории кислотно-основного взаимодействия 18
II. 2. Реакции синтеза в неводных растворах 29
Глава III. Классификация растворителей 33
III. 1. Системы классификации, основанные на физических свойствах
растворителей 33
III. 2. Системы классификации, основанные на химических свойствах
растворителей . 34
Глава IV. Молекулярное строение и физические свойства неводных
растворителей . . . 40
IV. 1. Молекулярное строение жидкостей и статистический расчет их
свойств . 41
IV. 2. Полуэмпирические методы расчета физико-химических свойств 46
IV. 3. Аддитивные и конститутивные свойства жидкостей 48
IV. 4. Эмпирические способы расчета физико-химических свойств
жидкостей 49
ТЕРМОДИНАМИКА НЕВОДНЫХ РАСТВОРОВ
Глава V. Термодинамика растворов неэлектролитов • • . 57
V. 1. Классификация неэлектролитов. Основные определения .... 57
V. 2. Теория неэлектролитных растворов 59
V. 3. Растворимость газов в неводных растворителях 64
V. 4. Растворимость жидких и твердых неэлектролитов ...... 72
V. 5. Термодинамические свойства жидких неэлектролитных смесей 75
V. 6. Взаимодействие неэлектролита с растворителем 79
V. 7. Диффузия в растворах неэлектролитов 80
373
Глава VI. Термодинамика растворов электролитов §2
VI. 1. Растворимость солей в неводных растворителях 82
VI. 2. Термодинамические характеристики процесса растворения
электролитов в неводных растворителях 84
VI. 3. Сольватация ионов 87
VI. 4. Коэффициенты активности электролитов в неводных растворах . Юо
VI. 5. Термодинамические функции отдельных ионов Юз
РАВНОВЕСИЯ В НЕВОДНЫХ РАСТВОРАХ
Глава VII. Общая схема равновесий в растворах . . «, 106
VII. 1. Ассоциативно-диссоциативные процессы 106
VII. 2. Образование продуктов присоединения (сольватация) . . . .110
VII. 3. Ионизация 113
VII. 4. Электролитическая диссоциация 114
Глава VIII. Методы исследования химического взаимодействия между
компонентами раствора 116
Физико-химический анализ двойных жидких систем 117
VIII. 1. Общие свойства химических диаграмм двойных жидких систем 117
VIII, 2. Основные методы физико-химического анализа двойных
жидких систем 126
VIII. 3. Сравнительная характеристика методов физико-химического
анализа двойных жидких систем 159
VIII. 4. Количественный физико-химический анализ двойных жидких
систем 160
Физико-химический анализ двойных систем в растворителе (квазидвойные
системы) - 167
VIII. 5. Методы физико-химического анализа квазидвойных систем,
основанные на изменении свойств растворителя 169
VIII. 6. Методы физико-химического анализа квазидвойных систем,
основанные на измерении свойств образующегося в системе
соединения, либо на измерении свойств раствора 176
VIII. 7. Метрика двухкомпонентной химической системы в растворителе 179
Методы исследования сольватации ионов • 184
VIII. 8. Методы исследования сольватации, основанные на измерении
подвижности ионов 185
VIII. 9. Спектроскопические методы исследования сольватации . • . 190
VIII. 10. Некоторые другие методы исследования сольватации . . ♦ . 197
Глава IX. Влияние растворителя на равновесие в химических системах 202
IX. 1. Влияние растворителя, на молекулярные
ассоциативно-диссоциативные процессы 203
IX. 2. Влияние растворителя на константы устойчивости комплексных
соединений 206
IX. 3. Влияние растворителя на таутомерные равновесия 2П
IX. 4. Влияние растворителя на константы электролитической
диссоциации . .... . . • 215
IX. 5. Дифференцирующее и нивелирующее действие растворителей на
силу электролитов 22^
IX. 6. Кислотность неводных растворов 2**
374
Глава X. Электропроводность неводных растворов 238
X. 1. Зависимость электропроводности от концентрации растворов . . 238
X. 2. Зависимость электропроводности от физических свойств среды . 242
X. 3. Определение констант диссоциации из данных по
электропроводности 249
Глава XI. Механизмы электролитической диссоциации и переноса тока . . 253
XI. 1. Перенос и подвижность ионов в разбавленных растворах ... 254
XI. 2. Особенности экспериментального определения переноса ионов в
концентрированных неводных растворах 257
XI. 3. Перенос ионов и электролитическая диссоциация в
концентрированных растворах с протогенными компонентами 259
XI. 4. Механизм электролитической диссоциации в апротонных системах 262
ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ В НЕВОДНЫХ РАСТВОРАХ
Глава XII. Электродное равновесие 266
XII. 1. Природа электродных потенциалов в 266
XII. 2. Зависимость э. д. с. электрохимических цепей от свойств
растворителя и природы электролита 272
XII. 3. Электродный потенциал и химическая природа растворителя . . 278
XII. 4. Расчет э. д. с. и электродных потенциалов 283
XII. 5. Особенности потенциометрических измерений в неводных
растворителях 289
Глава XIII. Кинетика электродных процессов в неводных растворах . . .
Полярография 292
XIII. 1 Влияние природы растворителя на основные полярографические
характеристики 292
XIII. 2. Полярография различных соединений 296
XIII. 3. Некоторые особенности эксперимента при использовании
неводных растворителей в полярографии 302
Электрохимическое выделение металлов 306
XIII. 4. Стадии электроосаждения и перенапряжение процесса . . . 306
XIII. 5. Электроосаждение металлов • • 309
Анодное поведение металлов 315
XIII. 6. Электрохимическое растворение металлов 316
XIII. 7. Коррозия металлов в неводных растворах 319
КИНЕТИКА РЕАКЦИЙ В НЕВОДНЫХ РАСТВОРАХ
Глава XIV. Общие вопросы влияния среды на скорость реакций .... 322
XIV. 1. Качественное обсуждение влияния среды на константу
скорости и энергию активации реакций 322
XIV. 2. Основные отличия реакций, протекающих в газовой фазе и в
растворе 325
XIV. 3. Реакции между неполярными молекулами в растворах
электролитов 328
XIV. 4. Влияние диэлектрической проницаемости среды на скорость
реакций с участием ионов и полярных молекул 330
XIV. 5. Специфическая сольватация реагентов и скорость реакций . . 335
XIV. 6. Скорость реакций и принцип линейности свободных энергий 339
375
XIV. 7. Скорость реакций и вязкость растворов 341
XIV. 8. Влияние давления на скорость реакций в различных
растворителях 342
Глава XV. Некоторые специальные вопросы кинетики 344
XV. 1. Каталитические реакции в жидкой фазе 344
XV. 2. Кинетика изотопного обмена в неводньГх средах .349
XV. 3. Быстрые реакции в растворах . . ! 353
Приложение 357
Основная литература 362
Указатель 370
Юрий Яковлевич Фиалков
Александр Натанович Житомирский
Юрий Александрович Тарасенко
ФИЗИЧЕСКАЯ ХИМИЯ
НЕВОДНЫХ РАСТВОРОВ
Редактор В. А. Станкевич
Технический редактор Е. М. Соболева
Переплет художника Л. А. Яценко
Корректор М. 3. Васина
М-36449. Сдано в набор 17/1 1973 г. Подписано к
печати 27/IV 1973 г. Формат бумаги 60X907i5. Бумага
типогр. № 2. Усл. печ. л. 23,5. Уч.-изд. л. 25,44.
Тираж 5400 экз. Заказ № 5U0. Изд. № 187. Цена 2 р,70 к.
Издательство «Химия». Ленинградское отделение,
191186, Ленинград Д-186, Невский пр., 28
Ордена Трудового Красного Знамени
Ленинградская типография № 2 имени Евгении Соколовой
«Союзполиграфпрома» при Государственном комитете
Совета Министров СССР по делам издательств,
полиграфии и книжной торговли,
198052, г, Ленинград, Измайловский проспект, 29