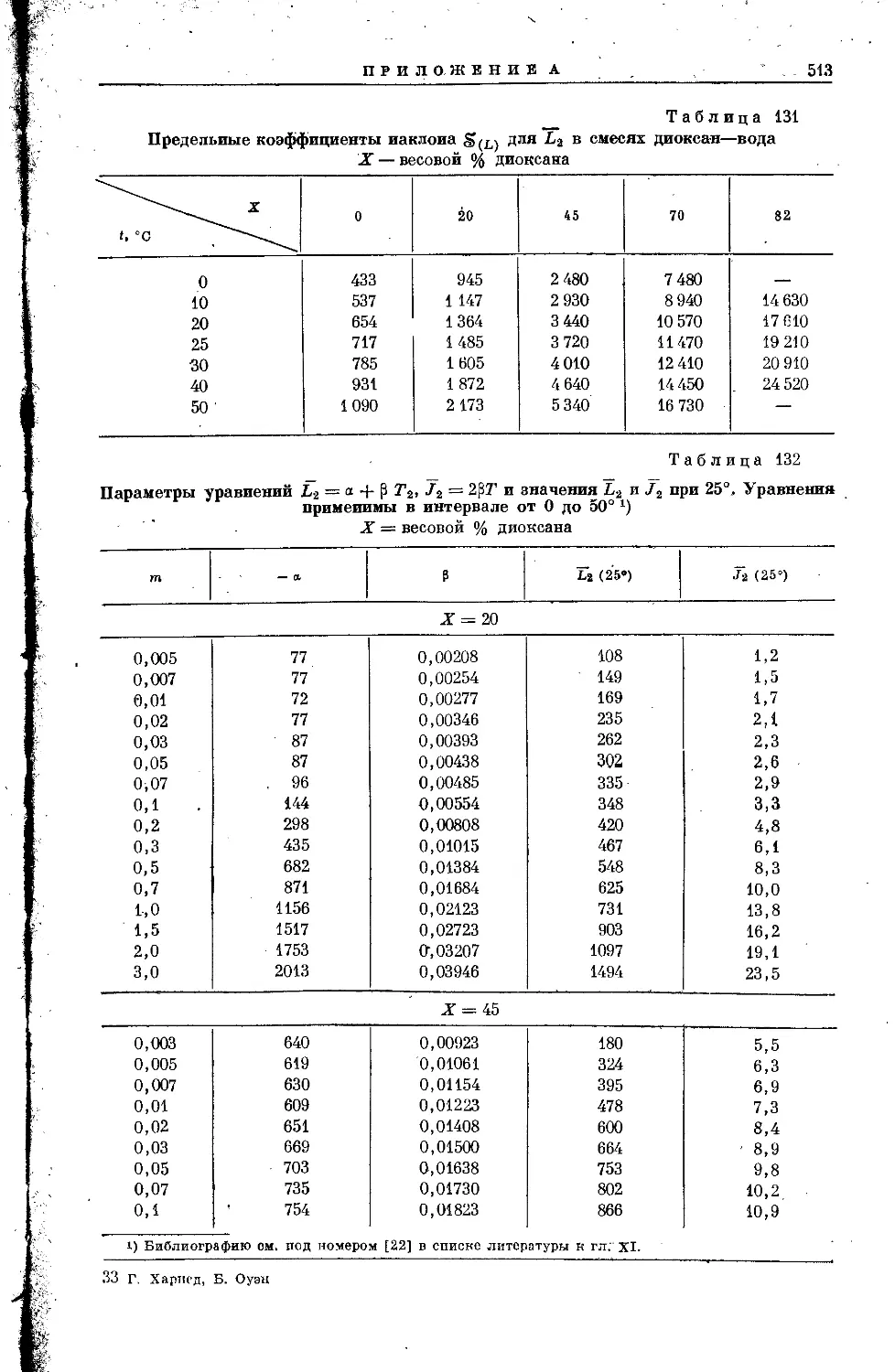
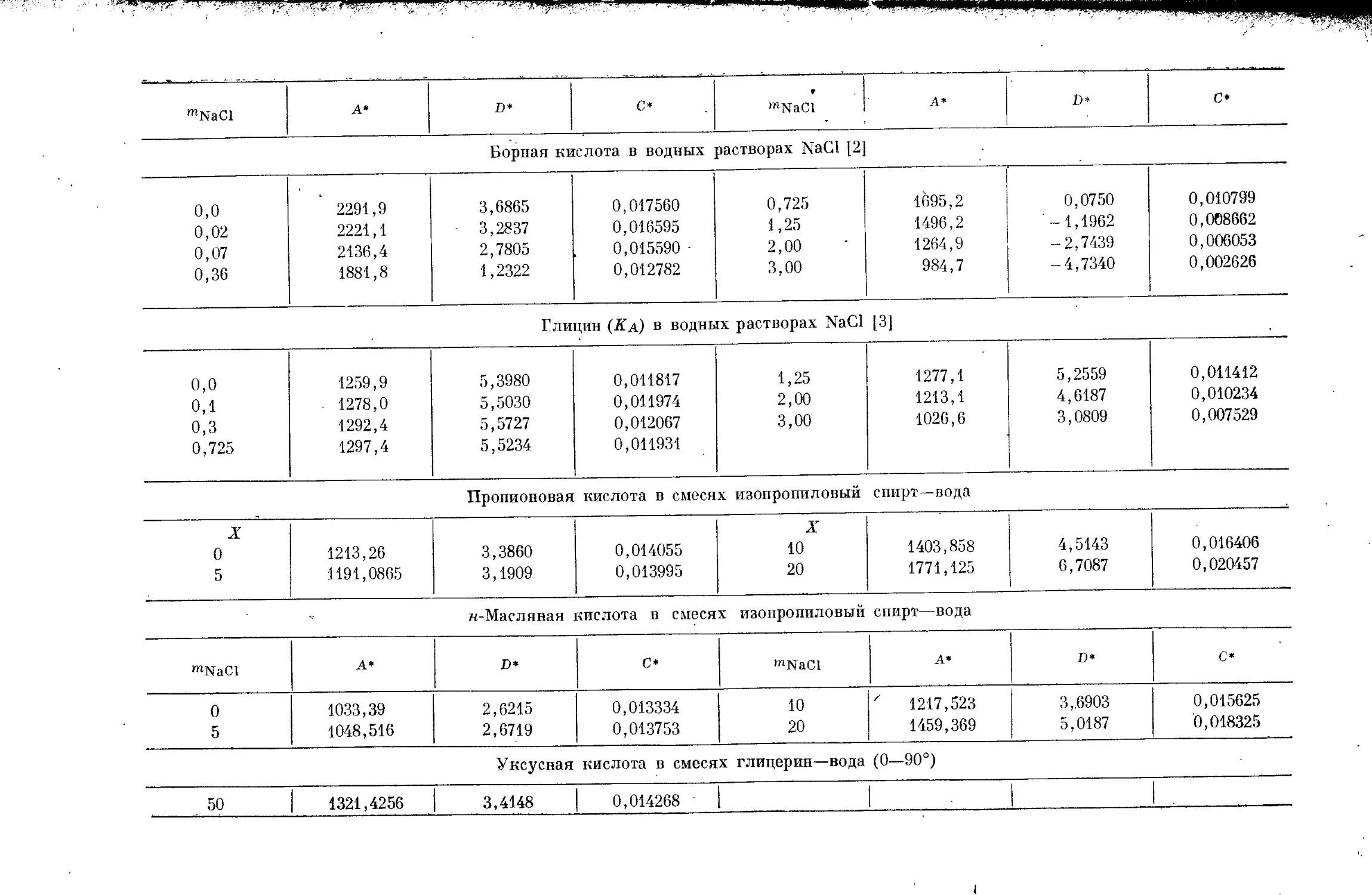
/










































































































































































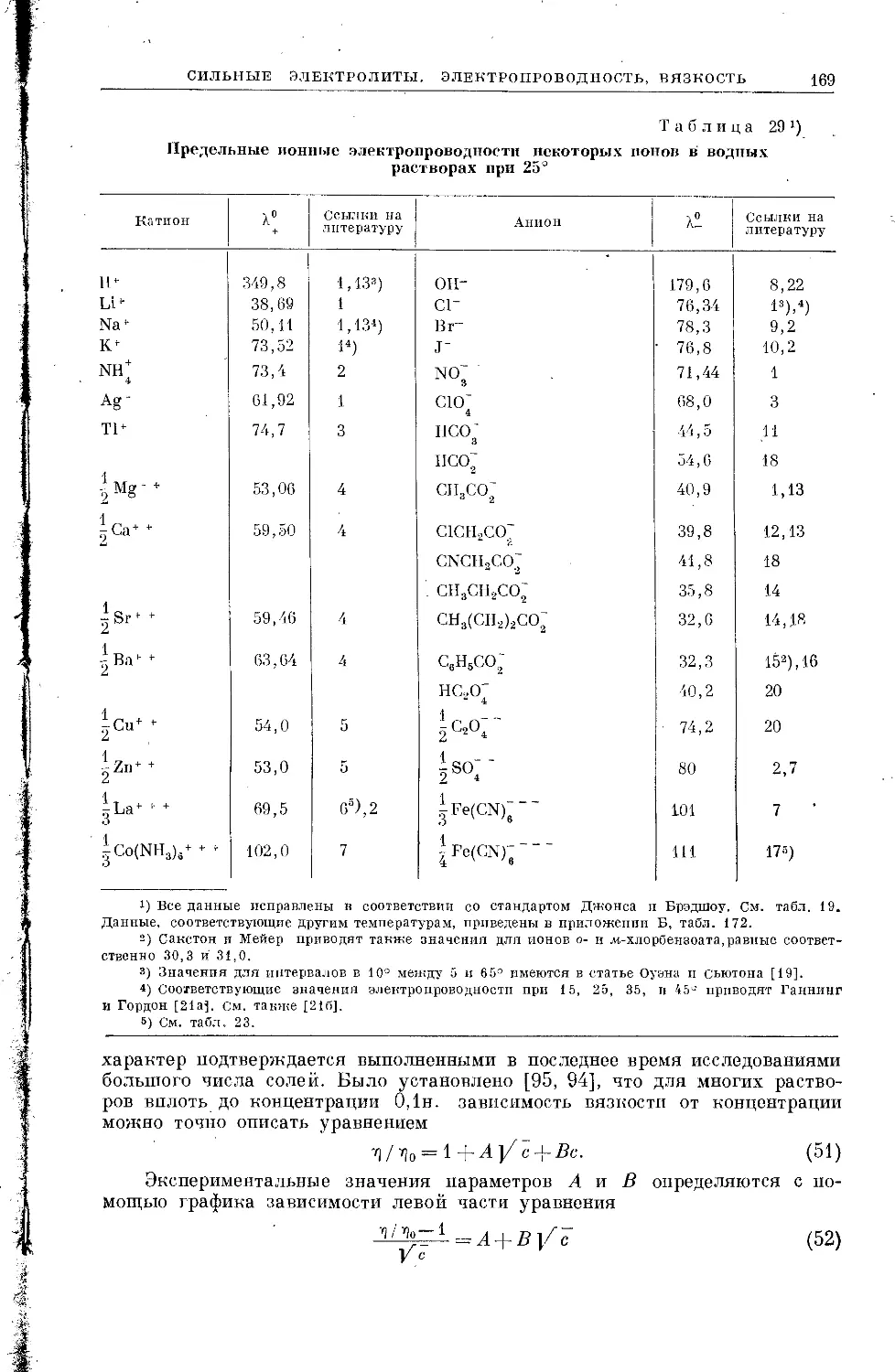






















































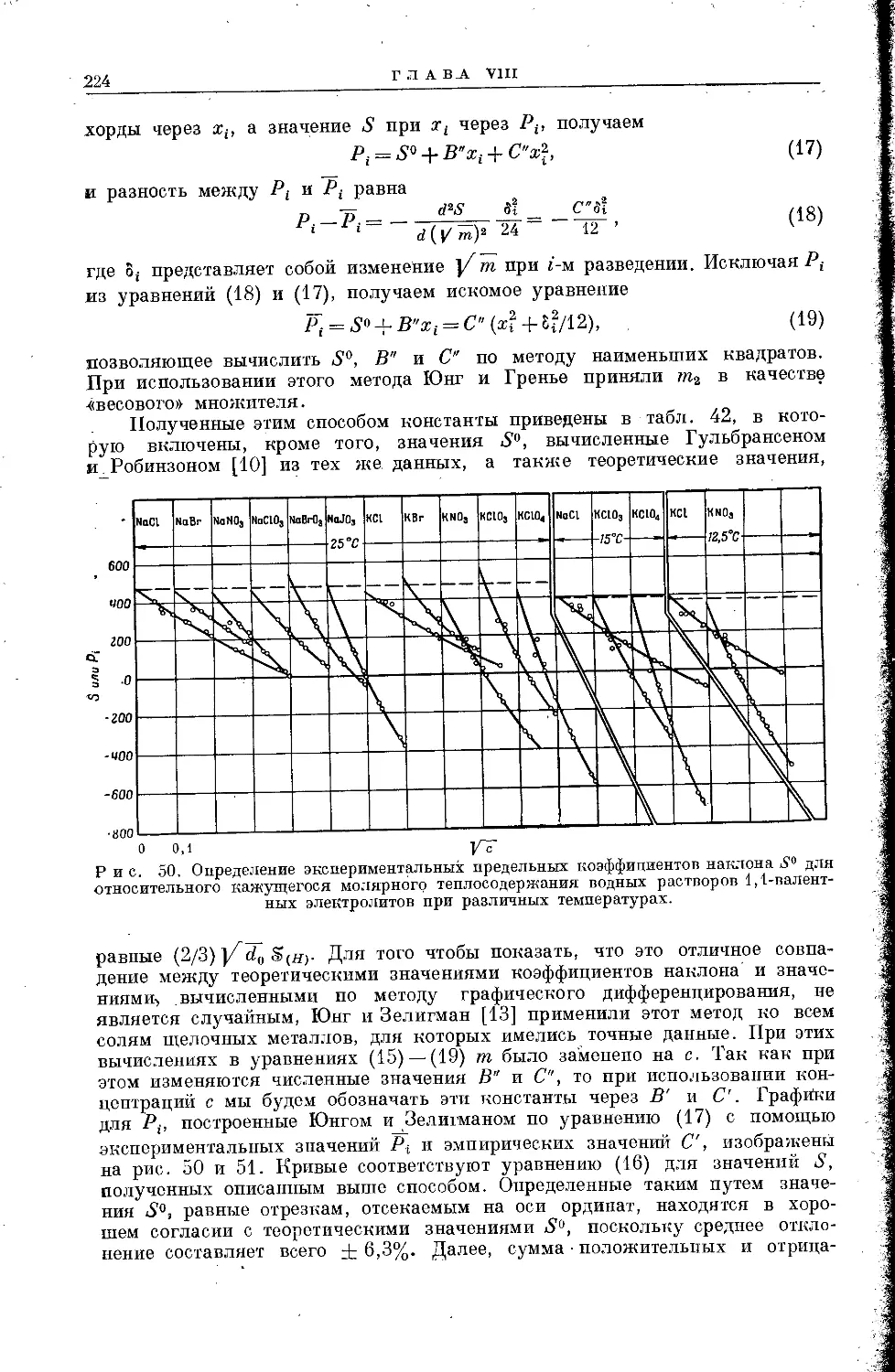







































































































































































































































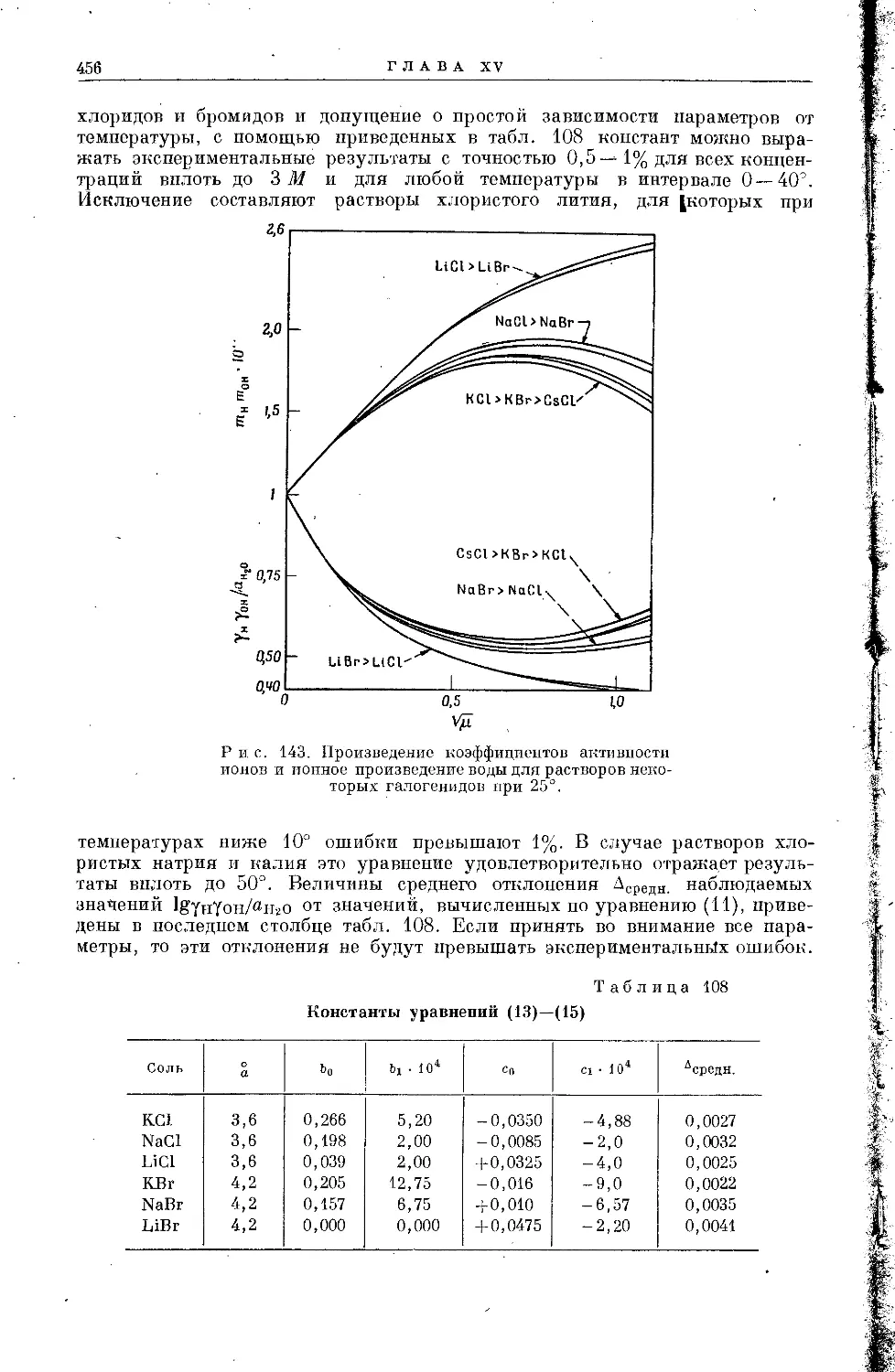
































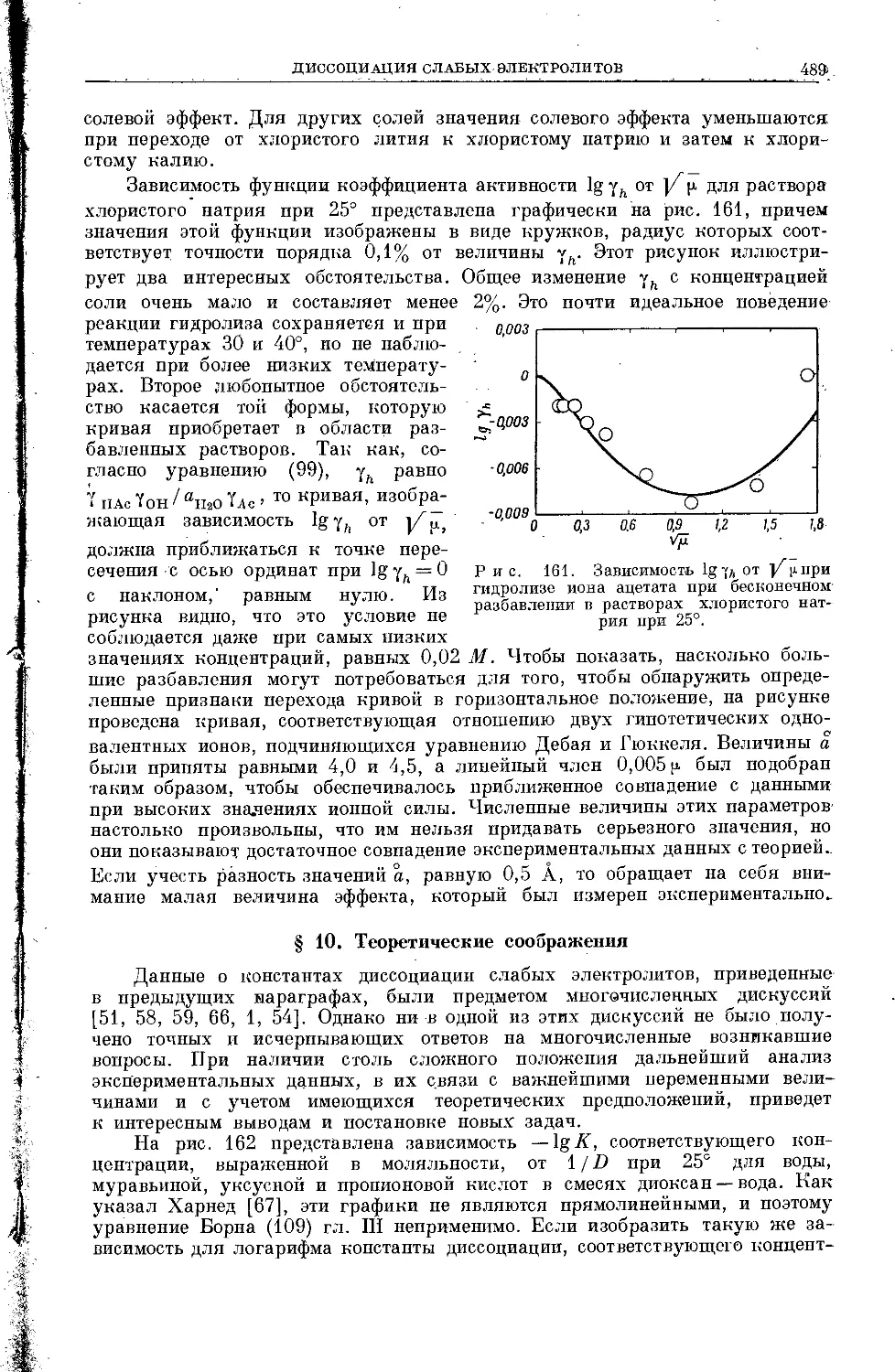














































































































































Текст
ХХарнед .Ь.Оуэн.
ФИЗИЧЕСКАЯ ХИМИЯ
РАСТВОРОВ
ЭЛЕКТРОЛИТОВ
Больше химической литературы на
vk.com/chemzone
More chemistry books you can find on
vk.com/chemzone
vk.com/chemzone
Г. ХАРНЕД, Б. ОУЭН
ФИЗИЧЕСКАЯ ХИМИЯ
РАСТВОРОВ
ЭЛЕКТРОЛИТОВ
Перевод с английского
И. И. ЛИПИЛИНОЙ ИМ. С. СТАХАНОВОЙ
Под редакцией
чл.-корр. АН СССР А. Ф. КАПУСТИПСКОГО
THE PHYSICAL CHEMISTRY
OF ELECTROLYTIC
SOLUTIONS
by
HERBERT S. HARNED
and
BENTON B. OWEN
Second edition, revised and enlarged
NEW YORK
.19 5 0
ПРЕДИСЛОВИЕ
В настоящее время некоторые разделы физической химии выросли
в обширные научные области, К числу таких областей относится теория
растворов, которая разрослась настолько, что отдельные ее части, в свою
очередь, имеют самостоятельное значение. Это может быть сказано о теории
растворов электролитов, которой посвящена монография американских
исследователей Харнеда и Оуэна. Эта книга представляет собой фундамен-
тальную работу, освещающую современное состояние физической теории
растворов электролитов.
Книга Харнеда и Оуэна состоит из пятнадцати глав и двух приложений
и может быть, по существу, разделена на три части. Первая часть (гл. I—V)
посвящена всестороннему изложению теории междуионного взаимодействия,
причем в гл. V даны краткое обобщение всего этого раздела и теоретические
уравнения, выраженные в форме, наиболее удобной для их эксперименталь-
ной проверки. Вторая часть (гл. VI—X) содержит изложение принципи-
альных основ экспериментальных методов исследования свойств растворов
электролитов (электропроводности, вязкости, диффузии, парциальных моляр-
ных величин, температур замерзания и кипения, упругости пара). Дан под-
. робный обзор экспериментальных результатов и методов их обработки, а также
сопоставление их с теоретическими. Наконец, третья часть (гл. XI—XV) по-
священа описанию термодинамических свойств растворов конкретных веществ
(соляной кислоты, 1,1-валентных и поливалентных электролитов, а также
смесрй сильных электролитов). Здесь же рассматриваются константы диссо-
циации отдельных слабых электролитов и их смесей. В приложении А даны
таблицы, иллюстрирующие экспериментальный материал, приведенный в
тексте книги. В приложении Б, введенном авторами во второе издание, даны
исправленные значения некоторых величин, а также сделаны краткие добав-
ления к первому изданию.
Последовательно применяя ставшие в настоящее время классическими
в области теории растворов термодинамические и статистические методы,
авторы дают строгое изложение физической теории растворов электролитов
в ее современной форме, обсуждая при этом и физический смысл различных
ее положений. Книга содержит также подробное изложение эксперименталь-
яых работ и эмпирических обобщений, большое число графических иллю-
страций, сводных таблиц и, наконец, обширную библиографию. Все это
делает ее весьма интересной для советского читателя.
Наряду с несомненными достоинствами книга обладает и сущесхвен-
иыми недостатками. Основным недостатком ее является то, что авторы из-
лагают теорию растворов электролитов в отрыве от общей теории растворов.
В тесной связи с этим стоит недооценка роли растворителя—его химических
свойств, недостаточное внимание к химическому взаимодействию компонен-
тов раствора, к явлениям сольватации, без учета которых исчерпывающее
понимание процессов, протекающих в растворах, невозможно.
4
ПРЕДИСЛОВИЕ
Русские ученые, начиная с М. В. Ломоносова, занимавшегося изуче-
нием солевых растворов, успешно работали над созданием общей теории
растворов, уделяя особое внимание химическим явлениям. Большую
роль сыграла гидратная (химическая) теория растворов, созданная
Д. И. Менделеевым и развитая далее в работах его учеников. По этой
теории растворы представляют собой переменные диссоциирующие химиче-
ские соединения растворенного тела и растворителя, называемые теперь
сольватами, а в случае водных растворов—гидратами. В. А. Кистяковский
и И. А. Каблуков ввели понятие о гидратации (сольватации) ионов
электролитов и этим положили начало объединения физической и хими-
ческой теорий растворов, которое характерно для русской и советской
школы физической химии растворов. Между тем в книге Харнеда и Оуэна,
как и почти во всей современной американской научной литературе, не
учитываются и даже не цитируются работы крупнейших русских ученых,
так же как и более поздние исследования советских ученых в области
теории растворов электролитов.
В связи с отмеченными недостатками книги при редактировании пере-
вода были даны в отдельных местах краткие примечания к тексту, содер-
жащие указания на некоторые важнейшие работы советских ученых, непо-
средственно относящиеся к вопросам, излагаемым авторами. Естественно,
что примечания не охватывают всего вклада, внесенного нашей наукой
в общую теорию растворов.- Поэтому советский читатель, знакомясь с моно-
графией Харнеда и Оуэна, должен иметь в виду, что невозможно составить
себе полное представление о физической химии растворов электролитов,
без изучения русских и советских работ в этой области1.
Книга Харнеда и Оуэна представляет большой интерес для науч-
ных работников — специалистов по термодинамике, теории растворов,
электрохимии и смежных областей, работающих по вопросам, связанным
с физической химией сильных и слабых электролитов, а также для
инженеров многих химических производств. Эта книга может быть исполь-
зована не только для глубокого ознакомления с теорией растворов электро-
литов, но и в повседневной исследовательской работе в качестве источника
для справок.
Перевод был сделан со второго издания книги, вышедшего в 1950 г.
Главы I—IX переведены И. И. Липилиной, а главы X—XV и приложения—
М. С. Стахановой.
Чл.-корр. АН СССР
А. Ф. Капустинский.
1 См., например, следующие книги: А. Ф. Капустинский, Очерки по исто-
рии развития неорганической и физической химии в России, Изд. АН СССР, М.—Л.,
1949; «Свойства растворов электролитов», отчет о V конференции по физико-хими-
ческим вопросам, НХТИ, Л., 1930; А. И. Бродский, Современная теория электроли-
тов, Л.,1934; В. К.Семенченко, Физическая теория растворов, ГИТТЛ, М.— Л., 1941.
Некоторые из разделов книги Харнеда и Оуэна трудны для понимания читате-
лей, впервые приступающих к изучению данной области; они требуют основательного
знания термодинамики и общей электрохимии. В этом отношении помощь читателям
могут оказать книги: М. X. Карапетьянц, Химическая термодинамика, Госхим-
издат, М.—Л., 1949, и Н. А. Изгарышев и С. В. Г о р б а ч е в, Курс теоретической
электрохимии, Госхимиздат, М.—Л., 1951.
УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
В связи со сложностью излагаемого в книге материала возникает необходимость
часто употреблять одно и то же обозначение для различных величин. Так, например,
буква А употребляется главным образом для обозначения работоспособности, но в не-
которых случаях эта же буква служит для обозначения площади, специальных функций
или эмпирического параметра. Это обстоятельство не должно служить источником ка-
ких-либо недоразумений, так как все специальные случаи применения обозначений
оговорены в тексте.
Согласно системе обозначений, предложенной Льюисом и Рендаллом, черта над
термодинамической величиной употребляется для того, чтобы обозначить ее парциальное
значение, т. е. производную по числу молей при постоянных давлении и температуре.
Так, F представляет собой парциальную молярную свободную энергию, V — парциаль-
ный молярный объем.
Кружок, расположенный непосредственно над буквой, обозначающей расстояние,
указывает, что оно выражено в ангстремах. Так, если через а обозначается расстояние
в сантиметрах, то через а обозначается расстояние в ангстремах.
Все векторные величины напечатаны жирным шрифтом, за исключением оператора у.
Для изображения произведения векторов употребляются и точка, и крестик. Из
скалярных величин жирным шрифтом напечатаны только три—Е, F и N, выражающие
электродвижущую силу, число Фарадея и число эквивалентов.
Для использования верхних и нижних индексов принято несколько правил.
Нижним индексом обозначаются: а) ионные или молекулярные составляющие; так,
например, +, —, г, / обозначают ионные составляющие, 1, 2, ... , с — молекулярные
компоненты, w обозначает воду, V—пар и т. д.; б) нижний индекс ;£ употребляется
для обозначения средней величины для ионов данного электролита; так, представ-
ляет собой средний моляльный коэффициент активности, т± — среднюю моляльность;
в) нижний индекс нуль употребляется для обозначения свойств чистого растворителя;
например, d0 и тд0 — плотность и вязкость растворителя. У
Верхний индекс нуль, за исключением тех специальных случаев, когда использу-
ется иное обозначение (например, звездочка), употребляется для обозначения величины
функции в стандартном состоянии. Под стандартным состоянием вещества понимается
такое состояние, когда его активность в водном растворе равна единице.
В одном случае употребление верхнего индекса нуль не подчиняется изложенному
выше правилу. Мы обозначаем эквивалентную электропроводность электролита и экви-
валентную электропроводность и число переноса иона при бесконечном разведении
соответственно через А°, X? и Ti. Здесь пришлось предпочесть верхние индексы нижним
потому, что иначе было бы трудно описать все явления электропроводности, не при-
бегая к двойным нижним индексам.
В системе обозначений имеется одно весьма важное нововведение. Многим предель-
ным теоретическим уравнениям придается простая форма, например:
lg/±=-^w/r=-S(;;/c”
^2 = &(£,) }^Г= S(L) Ус>
6
УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
и т. д. для многих свойств. В этих уравнениях §>(/) и т- Д- представляют
собой теоретические константы для данной среды при постоянных давлении и темпе-
ратуре. Нижний индекс обозначает рассматриваемое свойство. Часто приходится иметь
дело с графиками, на которых левая часть этих уравнений представлена в виде функ-
ции от корня квадратного из концентрации. Физический смысл упомянутых констант
состоит поэтому в том, что они равны коэффициентам наклона соответствующих кривых
и обычно так и называются. Мы надеемся, что при последовательном применении это-
го способа обозначения, если он станет общепринятым, удастся устранить путаницу в
обозначениях.
Л—работоспособность, отнесенная к одному молю.
А—парциальная молярная работоспособность.
Лх, Л2 и т. д., А/, Aj и т. д. —парциальная молярная работоспособность компонентов
и ионных составляющих, обозначенных нижними индексами.
Л — составляющая (когда впереди стоит Д): 1) ДЛ (эл.) —электрическая составляющая
работоспособности [уравнение (6) гл. III]; 2) ДЛ(8)—составляющая работоспособ-
ности, обусловленная добавлением электролита [уравнение (103) гл. III].
Aw—составляющая работоспособности, обусловленная учетом ко-объема (собственного
объема частиц) по Ван-дер-Ваальсу.
Л, Л'—характеристические параметры уравнений (9) и (И) гл. V, представляющие
собой важные функции. Следует учесть, что Лт = Л' y/^<Z0-
Л — площадь.
Л —специальная функция, выражаемая уравнением (237) гл. IV.
Л —специальная функция (гл. XII, § 7).
ЛДа — специальная функция в табл. 12, определяемая уравнением (19) гл. V.
Л, Ап, Лд-х, Л*_р Л', А* и т. д. — эмпирические коэффициенты.
а — активность.
alt а2 ... — активности указанных компонентов.
я+, a—, at, aj — условные активности указанных ионных составляющих.
я±—средняя активность ионов электролита [уравнение (31) гл. 1].
ав—активность компонента в стандартном растворе.
а„— активность воды.
аН> аон и т- Д- — активности указанных ионных составляющих раствора электролита.
алт, ат> ас —активность, когда концентрация выражена в единицах IV, т и с.
а* — активность твердого растворителя при Т'.
а®' — активность чистого жидкого растворителя при Т'.
а[— активность растворителя в растворе при Т'.
а'2—активность растворенного вещества в растворе при Т'.
av — активность паров растворителя при Т'.
а — среднее расстояние сближения ионов, определяемое уравнением (5) гл. XIV.
О
а — то же в ангстремах.
я,-, Ду — средние расстояния сближения указанных ионов,
о о
aj — то же в ангстремах.
а — расстояние сближения ионов в теории образования ионных пар (гл. III, § 7).
as — расстояние в уравнении теории образования ионных тройников [уравнение (65) гл. III].
я4 —расстояние в уравнении образования квадруполей [уравнение (72) гл. III].
я12, «хо, а20 — плотности энергии взаимного сцепления (гл. XII, § 7).
а* — составляющая скорости деформации (гл. IV, § 2).
а, а0, ах, а', а" и т. д.—коэффициенты эмпирических уравнений.
В2(ха), В* (ха), В3 (ха) — функции обобщенной теории [уравнение (44) гл. III].
В, В', В", BRI и т. д. —коэффициенты эмпирических уравнений.
УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
7
Ь— характеристический параметр в теории образования ионных пар Бьеррума [уравне-
ние (51) гл. III].
63—характеристический параметр в теории образования ионных тройников [уравнение
(65а) гл. III].
Ь—функция, употребляемая в теории влияния напряженности поля на диссоциацию
слабых электролитов; определяется уравнением (245) гл. IV.
bj—ионный радиус в теории солевых эффектов (гл. III, § 10).
Ъ, Ъо, Ъ1Г Ъ', Ъ", Ъс, Ът и т. д. — коэффициенты эмпирических уравнений.
Ср — молярная теплоемкость при постоянном давлении.
Ср3 — молярная теплоемкость твердого вещества.
СРу—молярная теплоемкость пара.
<7Р — молярная теплоемкость растворителя.
,Ср — парциальная молярная теплоемкость.
СР1, Ср2—парциальные молярные теплоемкости указанных компонентов.
Ср, Cpt, Ср2 — стандартные парциальные молярные теплоемкости.
Сп—функция, определяемая уравнением (9) гл. IX.
Сп— функция, определяемая уравнением (20) гл. IX.
С-2, C_lt Со, Clt С2 —коэффициенты в уравнении (30) гл. XI.
С', С* и т. д. — коэффициенты эмпирических уравнений.
ср, ср, ср и т. д. — удельная теплоемкость при постоянном давлении или объеме.
с—молярная концентрация в молях на 1000 мл раствора.
c+j с—, с,, cj—молярные концентрации указанных составляющих.
с± —средняя молярная концентрация [уравнение (52) гл. I].
с* — концентрация в эквивалентах на 1000 мл раствора.
си — молярная концентрация недиссоциированного электролита.
cj[—нормальность ионной составляющей А.
с^ — нормальность раствора индикатора.
с*'—исходная нормальность раствора индикатора.
с (х)— концентрация растворенного вещества на расстоянии х от границы раствора
(гл. III, § 11).
с—число компонентов в системе (по Гиббсу).
с0, Ci, с2, сп нт. д, — коэффициенты, определяемые уравнениями (105) и (106) гл. IV.
с, с0, с', с", с'", cj и т. д.—коэффициенты эмпирических уравнений.
D—диэлектрическая постоянная раствора.
Dj, jD2 и т- Д- — диэлектрические постоянные указанных компонентов.
—диэлектрическая постоянная растворителя.
^—коэффициент диффузии [уравнение (57) гл. VI].
—коэффициент диффузии при бесконечном разведении.
— вторые частные производные по координатам.
D', D* — коэффициенты эмпирических уравнений.
d— плотность раствора.
«70 — плотность растворителя.
d( [ — полный дифференциал.
dlt d«, d12, d+, d_, — диаметры указанных составляющих (гл. XII, § 8).
Е — электродвижущая сила гальванического элемента.
Е° — стандартная электродвижущая сила гальванического элемента.
Ejv, Em, Ес — стандартная электродвижущая сила гальванического элемента, когда
концентрация выражена в единицах m и с.
Е°* — стандартная электродвижущая сила гальванического элемента в смешанных или
неводных растворителях.
8
УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
Ei(T)> Е2 (Т) — электродвижущая сила цепей гальванических элементов с переносом.
Е12, ®2i и т. д.— электродвижущая сила цепей некоторых гальванических элементов
без переноса.
Е/—электродвижущая сила жидкостного соединения (диффузионный потенциал).
Едоп, — составляющая электродвижущей силы, обусловленная присутствием дополни-
тельных членов высшего порядка.
Е°н—постоянная в уравнении (77) гл. X.
Elt Е2—парциальные молярные расширяемости указанных компонентов.
Е°, Ё2—стандартные парциальные молярные расширяемости.
Е2—Е% — относительная парциальная молярная расширяемость компонента 2.
е—основание натуральных логарифмов.
е — электрический заряд.
«1. е2, ... , es, et, в/ — общий электрический заряд указанных ионов (со знаком).
I «11, | е2 |: , | es | , | е, | , | е}-1 — абсолютные величины зарядов ионов.
S — коэффициент поглощения света [уравнение (48а) гл. XV].
El ( ) — экспоненциальная интегральная функция.
F — число Фарадея.
F— молярная свободная энергия.
F°— стандартная молярная свободная энергия.
Fi, F2, ... , Fc — молярные свободные энергии указанных компонентов.
F°, F2, ... , F°— стандартные молярные свободные энергии указанных компонентов.
F\, F2, ... , Fc — парциальные молярные свободные энергии указанных компонентов.
F°, F2, • •• , —стандартные парциальные молярные свободные энергии указанных
компонентов.
Fi,Fj — условные парциальные молярные свободные энергии указанных ионов.
Ft, F°j — условные парциальные молярные свободные энергии указанных ионов в стан-
дартном состоянии.
F°n, Fm, Fc —стандартная парциальная молярная свободная энергии, когда концен-
трация выражена в единицах 7V, т и с.
Fw—составляющая свободной энергии, обусловленная учетом ко-объема по Ван-дер-
Ваальсу (гл. XII, § 8).
—сила.'
F (&) — специальная функция, определяемая уравнением (248) гл. IV.
F (Z) —специальная функция, определяемая уравнением (9) гл. VII.
Fji (Q) — число ионов, покидающих внутренний объем Q области 5 [уравнение (168)
гл. IV].
/ — рациональный коэффициент активности.
—рациональный коэффициент активности неэлектролита.
/+, ft, fj — условные коэффициенты активности указанных ионов.
— средний рациональный коэффициент активности [уравнение (48) гл. I].
/±(s) —рациональный коэффициент активности электролита в уравнении (106) гл. III.
/±(()— рациональный коэффициент активности электролита в уравнении для переноса
электролита [уравнение (109) гл. III].
iij (г2> Г12), fjt (ri> r2i)> /;ч, ftj — функции распределения согласно уравнению (4) гл. II.
/“i, fii — функции распределения для раствора в невозмущенном состоянии.
f'ji, /ij —функции распределения для раствора в возмущенном состоянии.
, /т — специальные функции, определяемые уравнением (55) гл. XII.
/ (х) — функция в теории эффекта Вина [уравнение (206) гл. IV и табл. 18].
Gji, Gq, G12, G21, Gu, G22, G (r)—функции, употребляемые в теории влияния частоты
и напряженности поля (эффект Вина) на электропроводность (гл. IV, § 5 и 6).
УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
9
g— рациональный осмотический коэффициент [уравнение (65) гл. I].
8 (®) — функция в теории эффекта Вина [уравнение (209) гл. IV и табл. 17].
g (с) —специальная функция в уравнении (32) гл. VII.
g— специальная функция (гл. IV, § 7).
Н — молярное теплосодержание.
HIt Н%, ... , Нс—молярные теплосодержания указанных компонентов.
Hit Нг,..., Нс — парциальные молярные теплосодержания указанных компонентов.
Hj, Н] — условные парциальные молярные теплосодержания указанных ионов.
Н° — стандартное молярное теплосодержание.
Н^, Н%, ... , Н° — стандартные молярные теплосодержания указанных компонентов,
Н°, Н°г, ... , Нс — стандартные парциальные молярные теплосодержания указанных
компонентов.
~ГГ° “Г7°
Hi, Hj—условные стандартные парциальные молярные теплосодержания ионов.
h — высота.
hji, hji—элементы матрицы (гл. IV, § 3).
Н (изменение величины Н): &HD—теплота разведения; \HD(ii, ^HD^ay —
теплоты разведевия соли, кислоты и основания; Д/7П —теплота нейтрализации;’
ХЯ* = &Нп — ^НD (s) (т -+ т'); Д//“— теплота диссоциации.
70—интенсивность света до прохождения через раствор.
I—интенсивность света после прохождения через раствор.
I (&3) — специальная функция в теории образования ионных тройников [уравнение
(69) гл. III и табл. 10].
i — сила электрического тока.
i — вектор силы электрического тока.
I—характеристическая постоянная в уравнении (127) гл. VIII.
J ( = Ср — Ср), относительная парциальная молярная теплоемкость.
J\, J2, ... , Jс — относительные парциальные молярные теплоемкости указанных ком-
понентов.
^2(Тд) = ,;Г2 ПРИ стандартной температуре TR.
J — поток ионов [уравнение (113) гл. IV].
Jj, Jj—поток г-ионов, /-ионов.
(Jpj/fJpjj, (Jj)f (%)—специальные функции в дебаевской теории солевых эффектов
[уравнение (140) —(145) гл. III].
J (Ь3, х)— специальная функция в теории образования ионных тройников [уравнение
(70) гл. III].
/— функция в теории понижения температуры замерзания [уравнение (51) гл. IX].
К1УК-_, ..., Кс — парциальные молярные сжимаемости указанных компонентов.
К,-, К;-—полные силы, действующие на ионы.
К — константа равновесия.
К л—константа диссоциации кислоты.
КR — константа диссоциации основания.
Ка — константа диссоциации воды.
— вторая константа диссоциации двухосновной кислоты.
К;, — константа равновесия реакции гидролиза.
Кц — максимальное значение константы диссоциации при температуре 0.
—специальные константы диссоциации, употребляемые при описании влияния
среды (гл. XV, § 7).
10
. УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
К3 — константа диссоциации тройных ионов.
Kt— константа диссоциации ионных агрегатов, образующих квадруполи.
К (X), К (0)—константы диссоциации при наличии и в отсутствие внешнего ноля.
Кг— специальная функция, определяемая уравнением (145) гл. IV.
Ж (и} — специальная функция, определяемая уравнением (80) гл. III.
Ж<уу— специальная функция, определяемая уравнением (90) гл. III.
Л —постоянная Больцмана.
к,-, к/—внешние силы, действующие на ионы.
к0 — сила, действующая на молекулы растворителя.
к, к0— объемное сжатие раствора и растворителя [уравнение (115) гл. VIII].
к(= 2,3026 BT/NF) [уравнение (3) гл. XI и^др.].
ki — константа в уравнении (18) гл. IV.
к1г к2 — кинетические постоянные в уравнении (3) гл. XIV.
Л„ кт, &v—коэффициенты высаливания при концентрации, выраженной в единицах
с, т и N.
( = твтов)
*А (= твта/тва).
= ^hr^ohMr).
Л'.4) к'оА~~специальные функции (гл. XV, § 7).
Llt Lz—относительные парциальные молярные теплосодержания указанных компонентов.
•^2(Тд) обозначает Л2 при стандартной температуре TR.
Ls—относительное молярное теплосодержание твердого вещества при Т'.
Ls (0)—относительное молярное теплосодержание твердого вещества при То.
Lv — теплота испарения при Т'.
(0) — теплота испарения при Уо.
L—удельная электропроводность раствора.
Lo— удельная электропроводность растворителя.
L*(=L — L0).
L— постоянная в уравнении (127) гл. VIII.
1 — длина.
I—длина волны [уравнение (32) гл. V].
М — молекулярный вес.
М1г М2 —молекулярный вес указанных компонентов.
Л7Х, MY—молекулярные веса компонентов в бинарных смесях растворителей.
Мхг—средний молекулярный вес [уравнение (60) гл. I].
Ф1, ДЯИ, SJiijt, W-ki, ДЭЛ', ДЯЛ"—функции в теории диффузии (гл. IV, § 4).
m—моляльность (концентрация в молях на 1000 г растворителя).
mR—стандартная моляльность.
т2, ... , тс — моляльности указанных компонентов.
mi, mj, m-|_, —моляльности указанных ионов.
т± — средняя моляльнос!ь ионов электролита [уравнение (51) гл. I). /
тв, тощ, tohsoi и т. д. — моляльности указанных ионов.
Мц и т. д.—кажущиеся'концентрации ионов водорода и т. д.
N — число эквивалентов.
IV—число Авогадро.
Nlt N2, ... , 7VC —молярные доли указанных компонентов.
Nw — молярная доля воды.
Л± —средняя молярная доля компонента-электролита [уравнение (47) гл. I].
IV?, №—молярные доли на большом расстоянии от иона (гл. III, § 10).
УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
11
«1, п2, ..., пс—числа молей указанных компонентов.
«!, па, ..., ns—числа ионов указанного вида в см3.
п, п'—числа молей неэлектролита и электролита (гл. III, § 10).
—число г-ионов в присутствии /-ионов.
—число /-ионов в присутствии г-ионов.
п’у—число г-ионов в присутствии /-ионов и наоборот (раствор в невозмущенном
состоянии).
ПЯ> п'ц—числ0 г'-ионов в присутствии /-ионов и наоборот (раствор в возмущенном
состоянии).
Р—общее внешнее давление.
Ро—исходное общее внешнее давление (гл. VIII, § 7).
Ре—эффективное давление (гл. VIII, § 7).
Рг—молекулярная поляризация [уравнение (44) гл. VII].
Pi—экспериментальный коэффициент наклона кривой в точке ж, [уравнение (17)
гл. VIII].
Р,—определяется уравнением (15) гл. VIII.
.^(= гп^тУу), где ш_|_, т_—моляльности ионов насыщающей соли.
(== —в чистом растворителе.
д>1{= —компонента 1 в растворе соли.
<^1(0) (= «3s)—компонента 1 в чистом растворителе.
Pi> Р2> • Рс—парциальные давления указанных компонентов.
pt)—давление пара растворителя.
рН(= Igl/ag-t).
р—постоянная в уравнении (67) гл. XV.
Q—количество тепла.
Q—специальная функция, определяемая уравнением (24) гл. V.
Q—специальная функция, определяемая уравнением (160) гл. IV.
Q (Ь)—функция в теории образования ионных пар [уравнение (54) гл. III и табл. 9].
<7—функция в теории ассоциации ионов .[уравнение (48) гл. III].
9*—функция в теории необратимых процессов [уравнение (87) гл. IV].
R—газовая постоянная на моль.
R&—электрическое сопротивление при частоте ш.
Rm—электрическое сопротивление при бесконечно большой частоте.
Я—характеристическое расстояние, определяемое уравнением (123) гл. III.
Rj— специальная функция, определяемая уравнением (134) гл. III.
R—специальная функция в теории влияния частоты поля на электропроводность,
определяемая уравнением (160) гл. IV.
R—расстояние в уравнении (65) гл. III.
г—расстояние.
г—вектор расстояния.
Г1, Та, г3> ••> г12, *21—векторы переменных расстояний.
?+, г-— ионные радиусы.
ГР ГР ^П)-фУнкЧии> определяемые уравнениями (107) и (108) гл. IV.
5—энтропия.
«У—растворимость нейтральных молекул в растворе соли.
—растворимость нейтральных молекул в чистом растворителе;
S'—экспериментальный коэффициент наклона кривой.
<У°—экспериментальный коэффициент наклона кривой при г, = 0 [уравнение (17) гл. VIII].
12
УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
5(7р1 Sv, S£, Sк—экспериментальные коэффициенты наклона для парциальной моляр-
ной теплоемкости, кажущихся молярного объема, молярной расширяемости
и молярной сжимаемости.
^(/)> S(L). S(J)> ^(л)’ S(T+) и т- Д-—теоретические предельные коэффициенты
наклона для указанных величин. Переменной является ]/с.
®(2)> ®*(Т+) и т- Д-—теоретические коэффициенты наклона для
указанных величин. Переменной является УI1.
Sxy, Syxt $тп> $пт и т. д.—составляющие тензора напряжения сдвига.
S*y, $ух> ^тп’ ^пт и т- Д-—электростатические составляющие напряжений.
«У® , S^n и т. д.—составляющие напряжения при взаимодействии нейтральных моле-
кул растворителя (гл. IV, § 2).
S(Z)—функция удельной электропроводности, определяемая уравнением (17) гл. VII.
Т—абсолютная температура.
Т'—температура замерзания или температура кипения раствора.
Ти—температура замерзания или температура кипения растворителя.
Уд—стандартная температура.
У1( Т-2, ..., Т+, Т-, Ti, Tj—числа переноса указанных составляющих.
У^_, у», T°j—предельные числа переноса указанных ионов.
Уд—число переноса [уравнение (29) гл. VI].
Т’А—кажущееся число переноса [уравнение (28) гл. VI].
Уд—число переноса при стандартной концентрации.
Т°А—функция числа переноса, определяемая уравнением (38) гл. VI.
t—температура в градусах Цельсия. .
t—время в секундах.
И, t-—время, в течение которого в элементе объема находится I- пли /-ион (гл. II, § 1).
гЪ'’ —время, в течение которого /- и г-ионы находятся соответственно в заданных
элементах объема (гл. II, § 1).
tji— элемент матрицы, определяемый уравнением (109) гл. IV.
U—энергия.
U0—стандартная энергия.
Е7^, —потенциальная энергия /-иона в присутствии i-иона и наоборот.
Uo—энергия ионного тройника при 6 = 0 [уравнение (61) гл. 111].
L7 (г)—функция в теории эффекта Вина [уравнение (188) гл. IV].
и—скорость звука [уравнение (129) гл. VIII].
и,-, и-—подвижности ионов (= г,- /X) при градиенте потенциала, равном единице
(в системе CGS).
Ui, uj—подвижности ионов (в практической системе единиц).
и?, uj—предельные подвижности ионов (в практической системе единиц).
V—общий объем.
V—молярный объем.
V—потенциал в в/см.
V i, V2, .—молярные объемы указанных компонентов.
V i, V2, —парциальные молярные объемы указанных компонентов.
V i, Vj—условные парциальные молярные объемы указанных понов.
V i, V2, . •., 7®, —парциальные молярные объемы в стандартном состоянии.
V*—критический объем разрушения (гл. VIII § 5).
V (= Vt + v2 V2) (гл. XII, § 7).
У(гг), V (г2)—скорости движения всего раствора.
УКАЗАТЕЛЬ ОБОЗНАЧЕНИИ
13
V—скорость.
v—вектор скорости.
V,-, V-—скорости указанных ионов.
v.{—скорость г-иона в присутствии /-иона.
vf-—скорость /-иона в присутствии г-иона.
vx, vz—составляющие скорости.
Си V2—объемные постоянные в теории солевых эффектов (гл. III, § 10).
т, Vo—удельные объемы раствора и чистого растворителя.
W—работа
JV (эл.)—электрическая работа.
IVji (г)—взаимный потенциал средних сил взаимодействия между ионами i и / [урав-
нение (221) гл. IV].
JF (ж)—работа, необходимая для переноса молекулы или иона из объема раствора
(с глубины, равной х) на поверхность [уравнение (146) гл. III].
W (п;), W (n), W (х)—выражения для электрической работы [уравнения (97) и (99) гл. III].
Wt—электрическая работа переноса [уравнение (108) гл. III].
(Н)—специальная функция, определяемая уравнением (79) гл. III.
специальная функция, определяемая уравнением (89) гл. III.
w—функция, определяемая уравнением (3)- гл. V.
w'—функция, определяемая уравнением (6) гл. V.
щ"—функция, определяемая уравнением (12) гл. V.
®*—функция, определяемая уравнением (25) гл. V.
X—электрическое поле.
X-—вектор электрического поля.
X —напряженность электрического поля при частоте ш.
\Хдействующее на ион / поле, обусловленное как ионной атмосферой, так и элек-
трофоретическим эффектом.
Х2т+1 (ха)> ^(х“)> Хв(ха), Хг (ха), X* (ха)—функции в обобщенной теории [уравне-
ния (42) и (44) гл. III и табл. 8].
X—представляет собой различные специальные функции, определяемые уравнениями
(65), (84) гл. III, (47) гл. IX, (29) гл. XI.
Хц—специальная функция, определяемая уравнением (29) гл. XI.
X, Y—весовые проценты компонентов смеси двух смешивающихся растворителей.
.rj, ylt ..., Xlt У1. х[, у'х, ..., Х{, У]—числа молей. Специальное значение
(гл. I, § 10).
х—различные функции, употребляемые в качестве переменных.
х—специальная функция, определяемая уравнением (204) гл. IV.
-^2т+1 (ха)’ 1% (ха)> У8 (ха)—функции в обобщенной теории [уравнение (42), (44) гл. III
и табл. 8].
У, Ус, Yf, У', У', У), у2—функции в теории поверхностного натяжения [уравнение
(159) и (160) гл. III и табл. 12].
У—переменная, определяемая уравнением (50) гл. III.
У (г)—функция, определяемая уравнением (179) гл. IV.
у—молярный коэффициент активности.
Ул., У1< Уг-i Уц и т- д-—молярные коэффициенты активности указанных составля-
ющих
—средний молярный коэффициент активности электролита [уравнение (50) гл. I].
уи—молярный коэффициент активности недиссоциировапных молекул.
у—различные функции, употребляемые в качестве переменных.
14
УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
Z—специальная функция, определяемая уравнением (10) гл. VII.
Zj—среднее число молекул, окружающих ион (гл. III, § 10).
ZJJ—число молекул компонента 2 в месте расположения иона (гл. III, § 10).
Z2—число молекул в растворе после прибавления электролита (гл. III, § 10).
2 —валентность.
zi, 2у—валентности указанных ионов (со знаком заряда).
zt\, | z. I, | 2+|, | z— |—абсолютные величины валентности указанных ионов.
z—функция, определяемая уравнением (23) гл. XII.
а—степень диссоциации в общем случае.
«о—степень диссоциации в отсутствие внешнего поля (гл. IV, § 7).
в—коэффициент расширения раствора (гл. VIII).
а0—коэффициент расширения растворителя (гл. VIII).
а*—коэффициент в уравнении электропроводности (22) гл. V.
а—эмпирический коэффициент в термохимических уравнениях.
«—специальная функция, определяемая уравнением (133) гл. III
«12, о21—эмпирические коэффициенты в уравнениях для смешанных' электролитов
(гл. XIV).
Р—коэффициент сжатия раствора (гл. VIII).
ft?—адиабатический коэффициент сжатия [уравнение (128) гл. VIII].
Ро~коэффициент сжатия растворителя (гл. VIII).
Р*—коэффициент в уравнении электропроводности [уравнение (23) гл. V].
[1—эмпирический коэффициент в термохимических уравнениях.
р, р' — эмпирические константы в теории солевых эффектов (гл. III, § 10).
Pi2, Р21 — эмпирические коэффициенты в уравнениях для смешанных электролитов
(гл. XIV).
р—специальная функция, определяемая уравнением (232) гл. IV.
Г — ионная концентрация.
S
Г2—число отрицательно адсорбированных молей растворенного вещества, приходящихся
на единицу прироста поверхности [уравнение (87) гл. I].
Г (г)—специальная функция в теории эффекта Вина [уравнение (185) гл. IV].
7 — моляльный коэффициент активности.
7+, 7—> 7г> 7; — условные коэффициенты активности указанных ионов.
7± — средний моляльный коэффициент активности электролита [уравнение (49) гл. 1]_
7Л — стандартный коэффициент активности при концентрации mR.
7U—коэффициент активности недиссоциированных молекул.
— коэффициент активности неэлектролита.
7^., 7± — коэффициенты активности при температурах Т’ и Т".
7н(Н)> 7ci(H)> 7н(К) и т- Д-— коэффициенты взаимодействия (гл. XIV, § 5).
7(Н+К)’ 7(С1) и т- Д-.— коэффициенты высаливания (гл. XIV, § 5).
7нС1(МС1) и т- д'—коэффициент активности НС1 в растворе MCI и т. д. для других:
смесей.
7о, 7°> 7о> 7*> 7а> 7оа> 7а> 7а> 7оа—специально определяемые коэффициенты активности,
употребляемые в теории влияния среды (гл. XV, § 7).
7(0) 1> 7(0)2’ 71(о)» 72(0) — специальные коэффициенты активности, употребляемые в тео-
рии смешанных электролитов [уравнения (16) и (17) гл. XIV].
7,—специальный коэффициент активности, употребляемый в гл. XI и XIII.
УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
15
А( )—конечное, приращение величины ( ).
V, Vi> V2 — векторный оператор.
W — оператор Лапласа.
А—«разница» в электропроводности (гл. VI, § 3).
А— отклонение.
Аср. — среднее отклонение.
д( ) —частный дифференциал.
8( )—вариация.
{1 ’ / = i 1
0: . J — обозначения Кронекера.
8—специальная функция, определяемая уравнением (15) гл. V.
8, —специальная функция, определяемая уравнением (18) гл. VIII.
е — заряд электрона.
специальный коэффициент активности а/п (гл. IV, § 4).
1)—вязкость раствора.
и]0—вязкость растворителя.
^♦—составляющая вязкости раствора, обусловленная взаимодействием ионов.
к]'—специальная функция, определяемая уравнением (196) гл. IV.
8 —понижение температуры замерзания.
8—эмпирическая константа [уравнение (97) гл. XV] — температура, при которой кон-
станта диссоциации в растворе соли имеет максимальное значение.
в —повышение температуры кипения.
9 — эмпирическая константа [уравнение (67) гл. XV]—температура, при которой кон-
станта диссоциации имеет максимальное значение.
9—переменный угол.
х — величина, обратная среднему радиусу ионной атмосферы; определяется уравнением
(22) гл. II.
xj(== j*x2)—по уравнению (86) гл. IV.
хр х„-функции из уравнения (150) гл. IV.
. А—эквивалентная электропроводность электролита.
А0—предельная эквивалентная электропроводность электролита при бесконечном раз-
ведении.
Ат— молярная электропроводность.
•^т—предельная молярная электропроводность.
А----молярная электропроводность при частоте ш.
А.---составляющая молярной электропроводности, обусловленная влиянием частоты
1ш
поля а.
Ап—составляющая молярной электропроводности, обусловленная электрофоретическим
эффектом.
Aj(0)—равно А^^, если ш = 0.
А0'— специальная функция электропроводности, выражаемая уравнением (12) гл. VI.
А'— специальная функция электропроводности, выражаемая уравнением (36) гл. VI.
Ajj — электропроводность в поле с напряженностью X.
10 УКАЗАТЕЛЬ ОБОЗНАЧЕНИИ
А+, Х_, \ — эквивалентные электропроводности указанных ионов.
к°_, Х®_, ХР, XJ, Х[>— предельные эквивалентные электропроводности указанных ионов
при бесконечном разведении.
X— молярное понижение температуры замерзания.
X*— молярное повышение температуры кипения.
X — длина волны света.
Х1; Х2, ).3, — специальные переменные, определяемые уравнениями (200) и (201) гл. IV,
X, Хь Х2— эмпирические константы в уравнениях (37) и (38) гл. XII.
ц—химический потенциал.
ц"— химический потенциал в стандартном состоянии.
|ij, |i2, ..., |лс — химические потенциалы указанных компонентов.
Hi, Ну— условные химические потенциалы указанных ионов.
p.j р-У, Р-с’ р4> Н® — химические потенциалы в стандартном состоянии.
[j.j, ..., — химические потенциалы компонента 1 в каждой из р фаз.
Р-2, P-L > рГ— химические потенциалы компонента 2 в каждой из р фаз.
Pi, Ру — электрохимические потенциалы [уравнение (71) гл. X].
|х—химический потенциал в эргах на моль (гл. IV, § 4).
Р—электрический момент диполя (гл. VII, § 4).
Р — составляющая (когда впереди стоит А): 1) — Др.у (эл.) — электрическая составляющая
химического потенциала, 2) — Др(.,у— составляющая химического потенциала не-
электролита, обусловленная прибавлением электролита [уравнение (104) гл. III].
Р mtzf^ — ионная сила.
S
р' — приближенная «реальная» ионная сила [уравнение (19) гл.- XIII и сл.].
р' (= eXikT) — специальное обозначение в теории эффекта Вина [уравнение (197) гл. IV].
р' (= Гу/Г) [уравнение (110) гл. IV].
v+, — числа катионов и анионов, возникающих при диссоциации одной молекулы
электролита.
( И + '*_')
— v стандартного растворенного вещества.
v-j—число молекул, которые диссоциируют (гл. IV, § 1 и 7).
<!, с2> Се — пространственные координаты.
Е2, ..., — соответствующие скорости.
(г) — специальная функция, определяемая уравнением (43) гл. IV.
11 — произведение.
-к ( = 3,1416).
ло — стандартный электродный потенциал.
р — плотность в общем случае.
pf, Ру — коэффициенты трения указанных ионов.
р, р2— функции, определяемые уравнениями (48) и (49) гл. IV
1/р — функция, определяемая уравнением (9) гл. IV.
S — сумма.
УКАЗАТЕЛЬ ОБОЗНАЧЕНИЙ
17
I3VIS1
о—в качестве нижнего индекса обозначает сумму.
а—поверхностное натяжение раствора.
во — поверхностное натяжение растворителя.
а—электропроводность, специальное употребление (гл. IV, § 1 и 7).
а—специальная функция, определяемая уравнением (130) гл. III.
а—специальная функция, определяемая уравнением (37) гл. IX.
°т — специальная функция, определяемая уравнением (41) гл. IX.
т—время релаксации.
т'—время запаздывания по Ланжевену (гл. IV, § 1 и 7).
т (= zq*) [уравнение (144) гл. IV].
Ф—полная свободная энергия в теории солевых эффектов [уравнение (ИЗ) гл. III].
?—практический осмотический коэффициент, определяемый уравнением (66) гл. I.
<р' —осмотический коэффициент при температуре замерзания или кипения.
—осмотический коэффициент стандартного раствора.
¥х> 72(0)’ 71 (о)-осмотические коэффициенты, употребляемые при описании смесей
(гл. XIV, § 5).
?н> 7l> 7cp> 7у> 7к и т- Д-— кажущиеся молярные значения указанных величин,
и т. д. — стандартные кажущиеся молярные величины при бесконечном раз-
ведении.
7н (а)’ ?я(Ь)-кажущиеся молярные теплосодержания соли, кислоты и основа-
ния [уравнение (31) гл. VIII].
Чн—кажущиеся молярные теплоты разведения при концентрациях т' и т".
?i> 7г — свободные энергии, рассчитанные на молекулу, в теории солевых эффектов
(гл. III, § 10).
?(za) — специальная функция, определяемая уравнением (76а) гл. IV.
?—специальная переменная в уравнении (212) гл. IV.
Ру — функция в уравнении (74) гл. VIII.
р — переменный угол.
ф*( = фу/7*)— функция, употребляемая в гл. VIII, § 5.
ф— электрический потенциал.
разность электрических потенциалов.
ф{, фу—электрические потенциалы ионов и их атмосфер.
фр ф®—электрические потенциалы ионов и их атмосфер для невозмущенного раствора,
ф’, ф' — электрические потенциалы ионов и их атмосфер для возмущенного раствора.
44 (о) = 4*1 (г=о)-
— потенциал г-иона в присутствии /-ионов.
фц — потенциал /-иона в присутствии г-ионов.
фу” — потенциалы г- и /-иона соответственно в присутствии /- и г-ионов для не-
возмущенного раствора.
ф{\, фу; — потенциалы г и /-иона соответственно в присутствии /- и г-ионов для воз-
мущенного раствора.
—потенциал, обусловленный ионной атмосферой. *
фь ф2 —кажущиеся удельные объемы растворителя и растворенного веществфЛ. раФйвбре;,;
[уравнение (120) гл. VIII]. ( '
ф (d)—специальная функция, определяемая уравнением (61) гл. JVJ,j-
2 г. Харнед, Б. Оуэн •
18
УКАЗАТЕЛЬ ОБОЗНАЧЕНИИ
/ (?*, “у) — функция, выражаемая уравнением (155) гл. IV.
у.д — функция, выражаемая уравнением (159) гл. IV.
Я, —функции, употребляемые й уравнениях электропроводности (гл. IV, § 5).
<о—подвижность в общем случае.
ш/, <i>. — подвижности ионов в общем случае (и>г-=1/р^ «>;-= 1/ру-).
ш— частота переменного электрического поля.
Г лава 1
ОБЩЕЕ ТЕРМОДИНАМИЧЕСКОЕ ВВЕДЕНИЕ
Свойства растворов электролитов представляется естественным разде-
лить на две группы: 1) свойства, определяемые путем изучения равновес-
ных систем, 2) свойства, определяемые путем изучения систем, находя-
щихся в неравновесных состояниях. К первой группе принадлежат те свой-
ства, которые могут быть найдены в результате определений понижения
давления пара, повышения температуры кипения, понижения температуры
замерзания, растворимости, теплоемкости, теплосодержания, потенциалов
гальванических элементов и поверхностного натяжения. Ко второй группе
относятся те свойства, которые могут быть найдены путем определений диф-
фузии, электропроводности и вязкости. Так как термодинамика представ-
ляет собой формальный метод изучения равновесных систем, то необходимо
кратко рассмотреть те общие термодинамические выводы, которые будут
использованы при дальнейшем изложении. В нашу задачу не входит под-
робное и строгое изложение термодинамических вопросов, мы ограничимся
введением и определением основных переменных величин, а также уста-
новлением обозначений и терминологии.
§ 1. Энергия, энтропия и химические потенциалы Гиббса
. Первый закон термодинамики можно выразить в достаточно общем
виде, если рассматривать энергию данной фазы V как функцию давле-
ния Р, объема V, температуры’ Т, электрического заряда е и числа молей
nX) и2, ...,пс каждого из с компонентов, находящихся в этой фазе. Все
другие переменные, как, например, величина поверхности, сила тяжести,
внешние поля и т. д., считаются в ходе нижеследующего изложения
постоянными. В дальнейшем будет специально разобрано термодинамиче-
ское уравнение, в которое входит в качестве переменной также величина
поверхности раздела. Энергия фазы выражается через перечисленные пере-
менные следующим образом:
U =.f (Р, V, Т, tii, п2, . . ., пс, е) + U°, (1)
где U° — энергия в некотором произвольно выбранном стандартном состоя-
нии. Так как любая из переменных может быть исключена с помощью1
уравнения состояния фазы, то число независимых переменных в этом выра-
жении равно с4-3. Первый закон термодинамики требует, чтобы увеличе-
ние энергии системы при постоянном составе и массе было равно сумме
тепла, поглощенного системой, и механической и электрической работы,
совершенной над системой окружающей средой. В последующих термоди-
намических выводах под механической работой подразумевается только1
изменение объема системы. Следовательно,
dU =dQ — PdV 4- гЛГ(эл.). (2)
2*
20
ГЛАВА I
(5)
(6)
(7)
образом:
(8)
(9)
(Ю)
Если ограничиться рассмотрением только обратимых процессов, то Р будет
равновесным давлением системы, а (эл.) — обратимой электрической
работой.
Согласно второму закону термодинамики, тепло, поглощенное систе-
мой при обратимом процессе, равно
dQ = TdS. (3)
Тем самым вводится понятие энтропии 5. Уравнения (2) и (3) могут быть
объединены и дополнены [1а] переменными, отражающими изменения co-
der dU
става системы, путем введения химических потенциалов рх = , ра =
и т. д., которые характеризуют изменение энергии при изменении числа
молей компонентов на единицу. Таким образом, обобщенное выражение
первого и второго законов для обратимых процессов имеет следующий вид:
dU = TdS — PdV 4- pjrfnx 4- ... 4- ?cdnc 4-de. (4)
Член — de представляет собой электрическую работу обратимого процесса,
выраженную через заряд как независимую переменную.
§ 2. Теплосодержание, работоспособность (максимальная работа
изотермического процесса) и свободная энергия
Теплосодержание Н, работоспособность А и свободная энергия1 F
определяются уравнениями
h=u+pv,
A = U-TS,
F— U — TS + PV — Н — TS.
Дифференциалы этих величин можно записать следующим
dH — TdS 4-7dP 4- ^dn^ 4- ... 4- pc<7nc 4- de,
dA — — SdT — PdV 4- ^idnx 4- ... 4- pcdnc 4- de,
dF = — SdT 4- VdP + Pidnr-V • • • 4* V-cdnc + ~ de,
причем определяется следующими соотношениями:
V, П2n„, e \dn1Js, P, na ,..., nc, e X^i/V, T, na.nc, ।
Kdi^/p, T, n2,..., ne, e • • •
Аналогичные уравнения можно написать для р2, ..., рс.
Рассматривая систему, состоящую из р фаз и находящуюся при по-
стоянных температуре, давлении и заряде, Гиббс получил с помощью
уравнения (4), справедливого для каждой фазы, следующие условия равно-
весия системы:
» " р
Pi = Н = • • • = Pi >
р2 = р2 = • • • = Р2 > (12)
' ” р
Рс — Рс — ••• - Рс >
1 Н, А и F представляют собой х, у и 5-функции Гиббса [1].
(И)
ОБЩЕЕ ТЕРМОДИНАМИЧЕСКОЕ ВВЕДЕНИЕ
21
где н, р[, •••> Р? представляют собой химические потенциалы первого
компонента, p'z, . ..,р2 — второго компонента и т. д. в каждой из р
фаз.
§ 3. Системы при постоянных составе и заряде
Так как U, Н, A, F и S всегда относятся к некоторому стандартному
состоянию, которое почти для всех случаев выбирается произвольно, то
нас больше всего интересуют изменения этих величин, и поэтому мы вво-
дим символ оператора А для обозначения указанных изменений. Таким
2
образом, 'AT*1 = dF, где интегрирование производится от состояния 1 до
1
состояния 2, причем AF, ДУ/ и т. д. можно пользоваться вместо F, Н
и т. д. в основных уравнениях.
При постоянных составе и заряде уравнение (4) принимает вид
dS = dU+^d- . (13)
Подставляя dS из этого уравнения в полный дифференциал уравнения (7),
получаем
(g)p=-5, или <“>
И
— V, или = AV. (15)
\дРут \др Jt ' '
Используя выражение (14), можно записать уравнение (7) так:
или
р(ДР/7’)1 _ ДЯ
<16>
§ 4. Системы при постоянных давлении, температуре и заряде.
Парциальные молярные величины
В данных условиях уравнение (10) принимает вид
С
dF = + р2(йг2 4-... + рс(йгс = 2 (17)
1
Так как это уравнение является однородным уравнением первой степени
относительно переменных пг, п2, . . ., пс, то, интегрируя его при постоян-
ных р1; р2, ..., рс, получаем [2]
С
F — 4~ ^2П2 + • * • + ” 2 (1®)
1
С
Если разделить это выражение на (W1 + «2+ ... 4-ис) = 2 получается
1
52
ГЛАВА I
уравнение
с
= Рх-^х + Рг^г + • • + Р-с^с — 2 (19)
1 1
где
= • . . (20)
2У1; N2, • • - , Na представляют собой молярные доли с компонентов, а F/^щ —
свободная энергия, отнесенная к одному молю фазы. Так как р1; р2, • • , Рс
являются частными производными свободной энергии по числу молей при
постоянных температуре и давлении, то они называются парциальными
молярными свободными энергиями и обозначаются Flt F2, ...,FC. Для
упрощения вида последующих уравнений мы будем вместо F/^ni всегда
писать F, поскольку обычно приходится иметь дело с молярными величи-
нами. В тех случаях, когда это условие не соблюдается, в, тексте даются
соответствующие пояснения Ч
Полный дифференциал уравнения (18) равняется
С с
dF — 2 v-idnt 4- 2(21)
1 1
и после подстановки dF из (17) получаем важное уравнение
с с
2 nid[4 = 0, или 2 Mdpx = 0, (22)
1 1
которое связывает химические потенциалы компонентов данной фазы при
постоянных температуре и давлении.
§ 5. Системы при постоянных температуре и давлении. Обратимый
гальванический элемент2
Рассмотрим элемент, состоящий из двух электродов, соединенных
раствором электролита. Пусть ф'— ф" представляет собой разность электри-
ческих потенциалов между двумя проводами из одного и того же металла,
присоединенными к этим двум электродам. Если этот гальванический элемент
обратимо заряжать при постоянных давлении и температуре, то электри-
ческая работа, которая совершается над системой, будет (ф'— ф") de, при-
чем она- равна общему увеличению свободной энергии. При постоянных
температуре, давлении и составе системы уравнение (10), выражающее
полное изменение свободной энергии р фаз, приобретает вид
«r-2(g),.T.w.......... *»*-«* (23)
1
Следует отметить, что разность потенциалов, определение которой было
дано выше, является величиной, которая поддается измерению, в то время
как разности потенциалов между каждым из двух электродов и раствором
не могут быть измерены отдельно3.
1 Эти обозначения соответствуют той системе обозначений в химической термо-
динамике, которая является наиболее распространенной в США.
2 Такой элемент, работающий обратимо, Гиббс назвал «идеальным электрохими-
ческим аппаратом» [16].
з См. [1в].
ОБЩЕЕ ТЕРМОДИНАМИЧЕСКОЕ ВВЕДЕНИЕ
23
При постоянных температуре и давлении из уравнения (10) получается
-следующее выражение для полного изменения свободной энергии р фаз:
dF = + 0/ — Ф’) й-е- (24)
1 1
Если все изменения состава происходят в одной фазе, тогда
С
dF =' 2 Pidni + (ф' — ф") de. (25)
1
Это уравнение можно проинтегрировать так же, как уравнение (17), при-
чем получается
С
^ = 2^{ + (ф'-ф")е. (26)
1
Полный дифференциал этого выражения равняется
dF = 2 ^i^ni + 2 —V') de + ed (ф' — ф"), (27)
i i
и после подстановки в это уравнение dF из (25) получаем
С
2 riid^i 4-ев?(ф' — Ф") = 0. (28)
1
Это выражение представляет собой уравнение концентрационной цепи,
с помощью которого при соответствующих условиях можно вычислить
электрическую работу обратимого процесса, если известны изменения
состава системы.
Важной особенностью этого уравнения является то обстоятельство,
что измерению поддается лишь суммарная электрическая работа, выражае-
мая соответствующим членом уравнения. Следовательно, измерив электро-
движущую силу концентрационной цепи, можно вычислить химические
потенциалы компонентов, определяемых по Гиббсу; однако с помощью
таких измерений нельзя вычислить потенциалы отдельных составляющих
компонентов, т. е., точнее говоря, потенциалы отдельных видов ионов1.
§ 6. Функция активности. Активность электролита
Приведенные в предыдущих параграфах уравнения содержат все пере-
менные и являются достаточно общими для того, чтобы служить термо-
динамической основой при рассмотрении всех систем, с которыми нам
придется иметь дело, за исключением тех случаев, когда необходимо учи-
тывать поверхностное натяжение. Пользуясь химическими потенциалами
и вводя соответствующие ограничения, можно вполне строго применить
термодинамику к растворам. При топ трактовке, которая является в настоя-
щее время наиболее употребительной, вводятся дополнительные функции
1 Харнед и Оуэн обходят молчанием дискуссию о возможности определения абсо-
лютных потенциалов, отраженную в советской периодической печати; они игнорируют
также весьма важные работы советских ученых, посвященные механизму возникновения
равновесного электродного потенциала и его зависимости от растворителя [Н. А. Изга-
рышев, ЖРФХО, 58, 1175 (1926); Z. Electrochem., 3, 128 (1928); А. И. Бродский,
Z. phys. Chem., 121, 1, 26 (1927)]. См. также обзоры: В. А. Плесков, Электродные
потенциалы и энергия сольватации ионов, Успехи химии, XVI, вып. 3, 254 (1947);
Б. В. Эршлер, Проблема абсолютного потенциала в электрохимии, Успехи химии,
21, вып. 2, 237 (1952). (Прим, ред.)
24
ГЛАВА I
более специфического характера. Разрабатывая термодинамику растворов,
Льюис [3] ввел две новые функции, а именно летучесть и активность.
Активность широко применяется в термодинамике растворов, и ниже будет
дано ее определение и приведены ее общие свойства. Ради простоты мы
совершенно не будем касаться понятия летучести1. Активность at чистого
химического вещества или одной из составных частей раствора в общем
виде определяется следующим уравнением:
^«ЯПп^+р?, (29)
где |ij—химический потенциал вещества в некотором условно выбранном
стандартном состоянии. Далее будет показано, что значение ц? зависит от
выбора единиц концентраций (моляльности, молярные доли и т. д.), в которых
выражено а, (§ 8).
Если в уравнение (29) вместо химических потенциалов ввести, согласно §4,
равнозначащие им переменные, то можно определить активность г-ой состав-
ной части раствора следующим образом:
Fi^RTXnai + Fl, (30)
причем Fi представляет собой парциальную молярную свободную энергию
данной составной части, a Fi — соответствующее значение в некотором
условно выбранном стандартном состоянии.
Поскольку в случае раствора электролита должно соблюдаться условие
электронейтральности, число молей ионов данного вида не может меняться
независимо. Поэтому следует рассматривать ионы данного вида как состав-
ную часть раствора, отличную от компонента, и тогда понятие компонента
может сохранить точное значение, данное ему Гиббсом, а именно: компонент
представляет собой такую составную часть раствора, которая является незави-
симой переменной. Так, в системе, состоящей из NaCl и Н2О, имеются два
компонента, химические потенциалы которых могут быть определены чисто
термодинамическим путем. Этими компонентами являются, конечно, NaCl
и НЯО. Хотя ионные составные части Na+ и С1_ имеют существенное значение
для определения поведения и свойств системы, их концентрации не являются
независимыми переменными. Термодинамика не дает возможности вычислять
химические потенциалы, свободные энергии, активности и т. д. ионов того
или иного вида. Несмотря на зто ограничение, удобно использовать во многих
термодинамических выводах гипотетические активности ионов, всегда имея
при этом в виду, что только некоторые произведения или отношения актив-
ностей ионов имеют реальное физическое значение2.
Другие удобные выражения для активности электролита можно
получить, исходя из формальных представлений о диссоциации электролита
в растворе3. Этот прием оказался весьма полезным при сопоставлении
свойств электролитов различных валентных типов. Так, если электролит
^v+-4v_ диссоциирует на v+ катионов и v_ анионов, согласно реакции
Cy+Av_—>v+C'-]-v_A, то его активность может быть выражена так:
v+ v- v /ом
а — а+ а_ = а±, (31)
где у = у+ + v_, а+ и а_ являются условными активностями отдельных
ионных составных частей и а± носит название средней активности ионов.
1 Подробное изложение общей термодинамики этих функций можно найти
в работах, приведенных под рубрикой [4].
2 Называя активности ионов гипотетическими, авторы допускают ошибку. Отсут-
ствие в настоящее время путей для вычисления активностей отдельных ионов не ли-
шает эти величины реального физического смысла. (Прим, ред.)
3 См. книгу Льюиса и Рендалла [4а].
ОБЩЕЕ ТЕРМОДИНАМИЧЕСКОЕ ВВЕДЕНИЕ
25
Таким образом, химический потенциал и парциальную молярную свободную-
энергию диссоциирующего на ионы компонента раствора можно выразить так:
pi - ио = RT In (a‘++al~) = vRT In а± (32)
И
F-F» = RT\n(a^a~)== vRTlna±. (33)
Следует отметить, что активность является менее общей функцией, чем:
химический потенциал или парциальная молярная свободная энергия,
поскольку при ее определении обычно приходится вводить специальное
стандартное состояние для каждой фазы. Очевидно, прежде чем приписать-
активности то или иное определенное численное значение, необходимо
совершенно точно установить стандартное состояние. В качестве примера
рассмотрим уравнение (12), согласно которому при постоянных температуре,
давлении и заряде химический потенциал данного компонента будет одина-
ков во всех фазах. Согласно определению активности, такого рода утверж-
дение в отношении активности данного компонента должно сопровождаться
соответствующей оговоркой. Так, активность данного компонента системы
при постоянных температуре, давлении и заряде будет одинаковой во всех,
фазах при том условии, что она определяется во всех фазах по отношению
к одному и тому же стандартному состоянию. Если иметь в виду эту
оговорку, не приходится испытывать каких-либо затруднений при изучении
многофазных систем с помощью методов определения растворимости, рас-
пределения, понижения температуры замерзания и других аналогичных
способов.
§ 7. Зависимость активности от температуры и давления
Согласно уравнению (19), относительная свободная энергия одного моля:
раствора составляет
F-F°:=N1{F1-Foi) + N2(F2-Fo2)+ ... (34)
Точно так же относительное теплосодержание раствора при расчете на моль
равно
+ ... (35)
Если ввести в первое из этих уравнений определение активности, данное
в предыдущем параграфе, получается выражение
F — F° = N]RT In aj + N2RT In а2 + . .. (36)
Подставляя полученные значения вместо kF и ДЯ в уравнение (16) и взяв,
..согласно этому уравнению, частную производную по температуре, получаем:
(Nid In я, + N2d In а2 + .. А (Н}-—Н®) + N2 (Н2 —Н®) + ...
< дТ )р~ ВТ* '
Это общее уравнение употребляется редко; обычно каждый компонент
изучают в отдельности. Таким образом,
_ -(Йг-н?)
< дт Jp вт* • I66/'
_ Обозначим относительное парциальное молярное теплосодержание
Нi — Hi компонента i через Lp.
L^Hi-Ht (39)
26 . . .. .. ... . . ГЛАВА I
Дифференцируя это уравнение по температуре при постоянном давлении,
можно определить такие важные величины, как парциальную молярную
теплоемкость Ср. и относительную парциальную молярную теплоемкость
Ср.— Ср. при постоянном давлении. Таким образом,
При постоянных составе, температуре и заряде
согласно уравнению (15), причем общий объем фазы V определяется
•выражением
V = (42)
1
где Vi и т. д. — парциальные молярные объемы компонентов. Сочетая эти
уравнения с уравнением (30), которое определяет активность, получаем
ZJVj д In In а2 + • • .\ JVi (Fj — V°) + JV2 (V2— T2) + • • /ло\
< др )t~ RT • \ 6)
Для каждого отдельного компонента
zain^ '
k dp Jt RT 1 >
§ 8. Зависимость активности от состава при постоянных температуре
и давлении. Определение коэффициентов активности
При сочетании уравнения (22) с (29) или (32) получаем при постоян-
ных температуре, давлении и заряде уравнение
2Mdlnai = 0. (45)
1
С помощью этого важного соотношения можно вычислять активность одного
из компонентов раствора, если известны активности остальных компонентов.
В случае бинарных растворов оно чаще всего употребляется для вычисле-
ния активности растворенного вещества на основании измерений так назы-
ваемых коллигативных свойств растворителя (т. е. таких свойств, которые
зависят лишь от числа молекул, а не от их специфической природы, как,
например, понижения температуры замерзания раствора и т. д.).
Как экспериментально так и на основании теоретических соображений
было показано [5], что в очень разбавленных растворах, когда можно
пренебречь взаимодействием частиц растворенного вещества между собой,
активности растворенных веществ становятся пропорциональными их кон-
центрациям, когда последние стремятся к нулю. Таким образом, в беско-
нечно разбавленном растворе любой электролит подчиняется следующему
предельному закону:
F-F^ = v7?7,lnAr±. (46)
1 См., например, [4 в].
ОБЩЕЕ ТЕРМОДИНАМИЧЕСКОЕ ВВЕДЕНИЕ
27
Это уравнение имеет тот же вид, что и уравнение (33), и вводит понятие
средней ионной молярной доли, определяемой уравнением
(47)
которое аналогично уравнению (31). Парциальная молярная свободная
энергия в стандартном состоянии обозначается через Fn для того, чтобы
показать, что активности выражены в молярных долях. Активности, выра-
женные в молярных долях, особенно важны в теоретическом отношении,
поскольку уравнение (46) служит для определения поведения гипотетиче-
ских «идеальных» распадающихся на ионы растворенных веществ при всех
концентрациях. Отклонения реальных растворенных веществ от идеального
состояния удобно выражать с помощью коэффициента
«Уд.
/±^^ = (/:+/Я1/А ~ (48)
называемого рациональным коэффициентом активности. Это название будет
сохранено, так как оно удобно ввиду его краткости, но более точное обо-
значение этой величины гласит — средний стехиометрический коэффициент
выраженной в молярных долях активности ионов.
Для многих целей оказывается удобным применять активности, выра-
женные через моляльности (число молей растворенного вещества в 1000 г
растворителя) или молярности (число молей в 1000 см3 раствора), и опре-
делять соответствующие коэффициенты активности следующим образом:
ат,
= (49)
И
y±^^ = (yv+j,v-)i/v. (50)
Средняя моляльность и средняя молярность ионов определяются таким образом:
т± = (m^+w^-)1/v = [(v+m)v+ (v_t^)v-]1/v = т (yj+v2_)1/v (51)
и
с± == (с^с»1/'’ = [(v+c)v+ (v_c)v-]Vv = c (Vv+Vv-)1/V. (52)
Средний стехиометрический моляльный коэффициент активности ионов у±
наиболее часто применяется на практике и обычно называется практическим
коэффициентом активности. Средний стехиометрический молярный коэффи-
циент активности ионов у± не нашел столь широкого применения, чтобы
получить особое сокращенное наименование. В последующих главах все три
коэффициента будут именоваться просто «коэффициентами активности».
В § 6 было показано, что величины р® или Fi для стандартного состо-
яния должны быть выбраны таким образом, чтобы значения [и или Fi для
данного компонента были независимы от единиц концентраций, в которых
выражается Принимая это во внимание, мы можем совместить уравне-
ния (30), (48 — 50) и выразить парциальную молярную свободную энергию
электролита следующим образом:
F = F.v 0- vRT In /±Л\ Fm + vRT In = F°c + ^RT In y±c±. (53)
При всех термодинамических исследованиях, за исключением тех, в кото-
рых применяются смешанные растворители, обычно выбирают стандартное
состояние так, чтобы при бесконечном разведении раствора /j. = у± = у+ = 1.
28
ГЛАВА I
Если придерживаться этого условия, можно ввести предельные значения:
NjJm± и с±/иг± при бесконечном разведении и получить из уравнения (53)
соотношение
^==^ + VjRTln^ = F“ + vJR71ln^^1 (54)
1YL j 1YL 1
Где Му — молекулярный вес растворителя, a d0 — его плотность. Появление-
d0 в последнем члене этого уравнения указывает на существенное разли-
чие между весовыми и объемными единицами концентраций. Концентрация.'
(состав) фазы в том смысле, в каком это выражение употребляется при
выводе основных термодинамических соотношений, является независимой
переменной. Когда процесс происходит при постоянном составе и соответ-
ственно при этом же условии производится частное дифференцирование
уравнений, то концентрации т, N и т. д., выраженные в весовых единицах,
остаются постоянными, в то время как концентрации с, выраженные-
в объемных единицах, могут меняться и на практике обычно меняются.
Для постоянства с обычно требуются два условия: постоянный состав
и постоянный объем. Следовательно, если F® (и другие термодинамические
функции, выраженные через концентрации с) нужно дифференцировать-
по Т или Р при постоянном составе, то нельзя забывать, что с является
переменной величиной. Те ошибки, которые могут произойти, если не учесть-
этого обстоятельства, делают нежелательным применение объемных единиц
концентрации, за исключением случаев, когда температура и давление-
постоянны.
Путем сочетания уравнений (53) и (54) получаем
ln/± = lnY±-|-ln^(^L) = lny± + ln^(1^?). (55)
Соотношение между концентрациями при любых разведениях дается общим
выражением
± vm+lOOO/THt w + (1000d—' r
в котором т и с представляют собой стехиометрическую моляльность и мо-
лярность электролита, М2 — его молекулярный вес, a d — плотность раствора.
Подставляя (56) в (55), получаем важные соотношения:
In /± = In Y± -[-In (1 + wvAfj/lOOO), (57)
In /± = In y± + In [d/dQ + c (v>! - >2)/1000 d0], (58)
In Y± = In y+ -f- In (d/d0 — cJ/2/1000 d0). (59)
Хотя зависимость уравнений (57 — 59) от (54) не представляет
сколько-нибудь существенного интереса при исследованиях, относящихся
только к изучению зависимости различных коэффициентов активности
от концентрации, она приобретает большое значение при изучении влияния
изменений состава растворителя на термодинамическое поведение раство-
ренного вещества. При такого рода исследованиях стали широко приме-
няться смешанные растворители, так как их свойства можно изменять
непрерывно и в желаемом направлении. Если Мх и Му — молекулярные
веса компонентов бинарной смеси растворителей, а X и Y отражают ее
состав в весовых процентах, то в предыдущие уравнения вместо Мj следует
подставить средний молекулярный вес смешанного растворителя:
М™= (X/Mx+Y/M^ •
ОБЩЕЕ ТЕРМОДИНАМИЧЕСКОЕ ВВЕДЕНИЕ
29
§ 9. Коэффициент активности и осмотический коэффициент
растворителя
Рациональный коэффициент активности растворителя определяется
уравнением _ _
F1 — Ft=RTlnfiNl, (61)
которое, вообще говоря, является одинаковым для любого компонента
раствора; однако индекс 1 будет всегда употребляться нами для обозначе-
ния растворителя. В разбавленных растворах, где количественное преобла-
дание растворителя над растворенным веществом проявляется очень сильно,
применение коэффициента активности растворителя при вычислениях
является практически неудобным. Для растворенного вещества, обозна-
чаемого индексом 2, в соответствии с (30) и согласно определению рацио-
нального коэффициента активности можно написать следующее уравнение:
= (62)
Дифференцируя эти два уравнения при постоянных температуре и дав-
лении и используя.уравнение (45), получаем
ЛГ1(/1п/Л1 + ^2с/1п/2^2 = 0. (63)
Так как +-/V2 = 1, т0 из (63) получается выражение
dln/1== —^dln/2, (64)
из которого следует, что в разбавленных растворах отклонение /j от еди-
ницы значительно меньше, чем соответствующее отклонение /2- Чтобы
получить более чувствительную меру отклонения растворов от идеального
поведения, выраженную через свойства растворителя, Бьеррум [6] ввел
понятие осмотического коэффициента. Рациональный осмотический коэф-
фициент g определяется уравнением
'Ft-F^gRTVaNi, (65)
а практический осмотический коэффициент ср —уравнением
Ft - Fl = - ср RT 2 miМ!/1 ООО, (66)
где Мх — молекулярный вес растворителя. Связь между этими коэффици-
ентами дается уравнением
<p = -s-glnjvi . (67)
2 тгЛЛ/ЮОО
В случае разбавленных растворов можно воспользоваться соотношением
8
— In Ni = In (1 -f- 2wi-^i/1000) и после разложения логарифмического члена
в ряд получить выражение
? = g [l-l^wtiMi/lOOO+^dj^i/lOOO)2-... ]• (68)
8
Множитель представляет собой результат суммирования по всем
растворенным веществам. Если в растворе имеется только один электролит,
8
диссоциирующий на v ионов, то = 'т- В этом случае уравнение
30 ГЛАВ а I
(66) приобретает следующий вид:
In <71= —®У?ИТ()о'б . (69)'
Дифференцируя уравнение (69) и подставляя cl In av в уравнение (45), напи-
санное в виде
Nx d In щ N2 d In a2 — 6, или d In a± + vmd In '{±m = 0, (70)
получаем важное соотношение, выведенное Бьеррумом, которое связывает
практический осмотический коэффициент с коэффициентом активности,
а именно:
d\т (1 — <р>)] J- md In у± = 0. (71)
В более общем случае получаем
d (1 — ?)] + In "(i -= 0. (72)
В интегральной форме эти уравнения имеют следующий вид:
о = 1 +1 / т md In v± (73)
и
4 = 1-j-1/2 m,. 2 m/cZlnYi • (74)
§ 10. Константа равновесия химической реакции
Рассмотрим какую-либо химическую реакцию, протекающую при по-
стоянных температуре, давлении и заряде, при которой жх, х2 .. . молей
Х2 . • и у1г у2 ... молей Ух, У2 • • реагируют с цбразованием ж), х'2 ...
молей Х'х, Х'2 . , . и у'х, у2 молей TJ, У'2 . . ., причем через X обозначены
чистые вещества, а через У — компоненты имеющихся в системе многоком-
понентных фаз. Тогда
xi Хх 4- х2 Х2 4- . . .71 Р1 //11-2 - • • • =
.<! .V । .<о А1 . 4-у'х Yt 4-у2Y'2 4- • •! (Tty
и по определению активности можно написать
(F— F°)Xi - Хх 7?7Чп a и т. д.,
(F — F°)Yi = г/х КТ In аУ1 и т. д.
и (76)
(F — F°)x^x'x RT In ах{ и т. д.,
(Т-?)гГу(ЯПпау,ит. д.
Общее изменение свободной энергии при этой реакции будет
(av')X1 (ау')х~ (с'-у'У11 ...
SF — SF11---RTln —Х1>-..{ х‘2>--LIT------, (77)
. («У1)У1Л • •
где
— (Fxx + Fх2 + • • • 4- F^x + Fy2 . ..) (78)
ОБЩЕЕ ТЕРМОДИНАМИЧЕСКОЕ ВВЕДЕНИЕ
3t
И
Д7го = {Fox, + Fox, + ... + F°y>+F°y, ...)-
+ + (79>
Очевидно, &F представляет собой изменение свободной энергии в общем
случае, &F0 — изменение свободной энергии в том сйучае, когда все реаги-
рующие вещества и продукты реакции находятся в их стандартных со-
стояниях.
Если система достигла равновесного состояния при постоянных темпе-
ратуре, давлении и заряде, тогда состав каждой фазы будет неизменным.
Из уравнения (10) следует, что при этом должно выполняться условие
Д7? = 0.
Следовательно, в этих условиях
Д/го = _ RT in ("V ('rv'• JM- • • •
(80>
Поскольку при постоянных температуре и давлении AF" является величи-
ной постоянной1,. то можно написать
(flx^1 (ах2)Х2 • • • • ’
(81)
где К — константа равновесия реакции. Это соотношение служит основой
при изучении ионных равновесий. В другой форме оно может быть запи-
сано так:
к (^(^...(ту^...Н7'
(«х/1 («х/2 ••• (mY1)yi ... Ну ’ . '
где Пу' и Пу — соответствующие произведения коэффициентов активности.
Если мы принимаем условие, что все коэффициенты активности чистых
фаз (веществ Xlt Х2, . ..) выбраны так, что их активности равны единице,
а коэффициенты активности всех компонентов многокомпонентных фаз
(веществ Ylt Y2, ...) равны единице при бесконечном разведении соответ-
ствующих компонентов, тогда при бесконечном разведении из уравнения (82)
получается классический закон действующих масс
_ ("h-;)81 •
(83)
Из уравнений (80) и (81) получается важное соотношение
AF° = -RTAnK. (84)
Поскольку зависимость свободной энергии от температуры и давления
выражается уравнениями (15) и (16), следовательно,
ZainFA _ Луо
< дР )т~ RT
(85)
1 Величина AF0 до некоторой степени произвольна, поскольку ее значение зави-
сит от выбора стандартных состояний компонентов и от тех единиц, в которых выра-
жаются активности. В том частном случае, когда реакция симметрична и протекает
лишь в одной фазе, AF° не зависит от этих произвольных условий.
32
Г. Л АВА I
/д1п£Л ДН»
\.-dT-)P = R^' <86>
Последнее соотношение называют иногда уравнением Вант-Гоффа.
§ И. Поверхностное натяжение. Уравнение адсорбции Гиббса
При последующем изучении зависимости поверхностного натяжения
от концентрации электролита мы будем пользоваться уравнением адсорб-
ции Гиббса в форме
5а 5а
5^“ ~ЯГ51пв2 '
(87)
Г2 представляет собой число «отрицательно» адсорбированных молей рас-
творенного вещества, приходящихся на единицу прироста поверхности,
.а а — поверхностное натяжение. Вывод и обсуждение этого уравнения
привели бы нас к слишком большому отклонению от основного предме-
та, и поэтому мы на них не останавливаемся [1г, 7].
ЛИТЕРАТУРА
1. Дж. В. Гиббс, Термодинамические работы, ГИТТЛ, М.—Л., 1950: а) стр. 104,
уравнение (12); стр. 430, уравнение (691); б) стр. 430, 431; в) 430—444;
г) стр. 288—310.
2. П. С. Эпштейн, Курс термодинамики, ОГИЗ, М.—Л., 1948.
3. G. N. Lewis, Proc. Am. Acad. Sci., 37, 45 (1901); 43, 259 (1907).
4. а) Г. H. Л ьюис, M. Рендалл, Химическая термодинамика, ОНТИ—Химтеорет.,
Л., 1936; 6)J.N. Bronsted, J. Am. Chem. Soc., 42, 76 (1920); N. В j e r r u m,
Z. physik. Chem., 104, 406 (1923); в) X. С. Тейлор, Физическая химия,
ОНТИ—Химтеорет., Л., 1935, гл. ХП.
5. Е. A. Guggenheim, Proc. Roy. Soc., A135, 181 (1932).
6. N. В j e r r u m, Z. Elektrochem., 24, 259 (1907); Proc. Internet. Congr. Appl. Chem.,
Sect. X, London, 1909.
7. J. W. Gibbs, Commentary on the Scientific Writings of J. W. Gibbs, Section
by James Rice, Vol. I, Yale University Press, New Haven, 1936, p. 504 ft;
E. A. Guggenheim, J. Chem. Phys., 4, 689 (1936).
Глава II
ОБЩИЕ ОСНОВЫ ТЕОРИИ МЕЖДУИОННОГО ПРИТЯЖЕНИЯ
И СВОЙСТВА ИОННЫХ АТМОСФЕР
Успешное развитие современной теории ионных растворов обусловлено
тем обстоятельством, что нам известен закон, которому подчиняются силы
притяжения, действующие между ионами. На основе этого закона и при-
менения основных представлений электростатики, гидродинамики и стати-
стической механики была развита точная теория, которая описывает свой-
ства электролитов в тех случаях, когда силы междуионного притяжения
являются преобладающими и когда можно пренебречь влиянием других
сил, как, например, междумолекулярных сил и сил взаимного отталкивания
ионов, проявляющихся при сближении ионов на очень малые расстояния.
Следовательно, построение теории должно начинаться с количественного
изучения влияния сил взаимодействия между ионами на все известные
свойства ионных растворов. Если этот первый шаг будет сделан правильно
и если полученные результаты будут подтверждены экспериментально при
соответствующих условиях, тогда можно с известной надеждой на успех
предпринять следующий шаг в деле выяснения свойств растворов электро-
литов на основании наблюдаемых отклонений от этих закономерностей.
К счастью, выводы теории междуионного притяжения могут быть про-’
верены непосредственно, так как существует класс сильных электролитов,
которые при умеренных концентрациях в водных растворах, невидимому,
полностью диссоциированы и растворы которых соответствуют простой
электростатической схеме заряженных ионов, находящихся в среде с данной
диэлектрической постоянной. Уже давно были высказаны предположения,
что поведение сильных электролитов в разбавленных растворах можно
объяснить на основании гипотезы полной диссоциации и соответствующего
учета влияния междуионного притяжения. Среди первых исследователей,
которые придерживались такой точки зрения, были Сэзерлэнд [1],
Нойес [2] и особенно Бьеррум [3]. Ван-Лаар [4] еще раньше указал на важ-
ность учета электростатических сил для понимания свойств ионных раство-
ров. Герц [5] и Гош [6] пытались дать математическую теорию влияния сил
междуионного притяжения, однако те основные положения, из которых они
исходили, оказались несостоятельными. Мильнер [7] дал успешный анализ
этого вопроса, но его математический подход был чрезвычайно сложен и не
привел к вполне удовлетворительным результатам.
Введенное Дебаем понятие «ионной атмосферы» и использование им
уравнения Пуассона позволили дать остроумную и простую математическую
трактовку, из которой вытекали точные соотношения, позволявшие количе-
ственно предсказывать свойства разбавленных растворов электролитов.
В своей первой работе по теории растворов Дебай и Гюккель [8]1 * 3 успешно
вычислили предельный закон для коэффициента активности, т. е. дали точ-
ное теоретическое выражение для характеристики этой величины в очень
разбавленных растворах.
1 Статистические основы этой теории исследовали Фоулер, Онзагер, Крэмер
и Кирвуд [9 ].
3 Г. харнед, Б. оуэн
34
ГЛАВА II
На основании своей теории Дебай и Гюккель [10] внесли также сущест-
венный вклад в теорию электропроводности электролитов. Несколько позже,
развивая общую теорию движения ионов, Онзагер [11] вывел предельный
закон для электропроводности электролитов. Впоследствии теория электро-
проводности Онзагера была расширена Дебаем и Фалькенгагеном [12],
которые учли влияние высокой частоты переменного тока на электропровод-
ность и диэлектрическую постоянную. Предельный закон для вязкости
растворов электролитов.вывел Фалькенгаген [13], а общие законы диффузии
электролитов были изучены Онзагером и Фуоссом [14]. Далее, Иоос и Блю-
ментрит [15] исследовали с теоретической точки зрения эффект Вина, т. е.
влияние сильных электрических полей на свойства растворов электролитов.
Позднее Вильсон [16] дал полное решение этого вопроса для случая электро-
литов, диссоциирующих на два иона. Очень интересная теория влияния силь-
ных полей на ионизацию слабых электролитов была развита Онзагером [17].
Этот краткий исторический обзор1 * показывает, что в настоящее время
теория междуионного притяжения достигла высокой ступени развития
и что она была успешно применена для описания большинства термодинами-
ческих свойств и необратимых процессов в растворах. В настоящей главе
будут разобраны основные положения теории и выведены общие уравнения
для ее приложения к различным явлениям. В гл. III и IV будут рассмотрены
обратимые и необратимые свойства.
Ионная атмосфера
При рассмотрении ионного раствора следует обратить внимание на два
фактора, имеющих существенное значение. Первым из них является рас-
пределение ионов в растворе по отношению друг к другу, а вторым—дейст-
вующие на ионы силы, обусловленные как наличием самих ионов, так и на-
ложенными извне полями. Эти факторы не являются взаимно независимыми,
так как силы влияют на распределение ионов, а распределение ионов в свою
очередь определяет силы. Первым шагом в построении теории будет опреде-
ление функций распределения, которые должны быть применимы к ионным
растворам в общем случае, независимо от того, находятся ли эти растворы
в равновесном или возмущенном состоянии, причем наличие последнего
обусловлено действием внешних возмущающих факторов, как, например,
наложенных извне электрических полей или движения всего раствора в целом.
Со времени появления первоначальной работы Дебая и Гюккеля теория
растворов, не подверженных действию внешних полей, неоднократно изла-
галась в упрощенном виде [18]. Хотя такой упрощенный подход пригоден
для случая равновесия, он является недостаточно общим для теории рас-
творов, находящихся под действием внешних сил, например для вычисления
электропроводности. Кроме того, если не уточнить систему обозначений,
то характер многих важных допущений теории междуионного притяжения
останется невыясненным. Этот недостаток можно исправить, если восполь-
зоваться более тщательно разработанной системой обозначений, предложен-
ной Онзагером и Фуоссом. Хотя применение этой системы обозначений
и создает вначале большие трудности, оно в конечном счете приводит
к более строгому и ясному пониманию теории.
а) Функции распределения. Рассмотрим раствор электролита, содержа-
щий в 1 см3 пг, п2, . . ns ионов различных видов, обозначенных соответствую-
1 Указания на наиболее важные исследования советских авторов, имеющие от-
ношение к отдельным вопросам, рассматриваемым в данной книге, приводятся ниже,
в подстрочных примечаниях редактора. (Прим, ред.)
ТЕОРИЯ МЕЖДУИ01Ш0Г0 ПРИТЯЖЕНИЯ
35,
щими индексами, причем заряды ионов равны соответственно е1. е2, . .., е8.
Поскольку любые два. иона, несущие заряды е, и , взаимодействуют
друг с другом, согласно закону Кулона, с силой, равной Dr-, то дви-
жение ионов не является вполне беспорядочным. Таким образом, присут-
ствие в данной точке раствора определенного иона влияет на простран-
ственное распределение других ионов, находящихся в непосредственной
близости от этого иона. Так, например, каждый положительный ион, вызы-
вая вблизи себя увеличение плотности отрицательных зарядов, будет окру-
жен «атмосферой», которая содержит больше отрицательных и меньше
положительных ионов, чем их содержится в соответствующем элементе
объема раствора в среднем. Подобным же образом отрицательный ион
будет окружен положительно заряженной атмосферой. Для решения вопроса о
распределении ионов в наиболее
общем виде, что необходимо для
создания теории, применимой не
только к равновесным состоя-
ниям, но и к необратимым про-
цессам (как, например, электро-
проводность электролитов, диф-
фузия и вязкость)*, требуется
функция, которая выражает
нероятность одновременного на-
хождения двух ионов в двух дан-
ных элементах объема раствора.
Для определения этой функ-
ции выделим два элемента объ-
ема (рис. 1), положение которых
определяется концами векторов
г, и г2, проведенных из какой-либо произвольной исходной точки. Расстояние
между этими элементами объема равно
Г21 = Г2-Г1= — Г12. (1)
Обозначим концентрации (число ионов н сл«3) ионов нида j и i соответ-
ственно через nj и пг. Далее, обозначим через п-,, среднюю концентрацию
за некоторый промежуток нремени Лионов в dV2 при нахождении вблизи
в элементе объема dV1 одного /-иона, а через и,;/ —среднюю концентрацию
за некоторый промежуток времени /-ионов в dVr, когда поблизости н эле-
менте объема dV2 находится один ион вида i.
В общем случае концентрации и,.; и n\j будут зависеть от нескольких
переменных:
1. Эти концентрации будут зависеть от расстояния г между /-ионом
и точкой, в которой требуется определить концентрацию Лиона, и обратно.
2. Если на ионы действуют внешние силы, в пространстве выделяется
особое направление, и n>i и пц будут зависеть не только от величины г,
но и от его направления. Так, например, если растнор находится под дей-
ствием ннешнего электрического поля, можно считать, что ионы двигаются
н определенном направлении, т. е. в направлении оси х. Таким образом,
в выражение для электропроводности будет входить не только расстояние
между ионами г, но и направление оси ж.
3. Если раствор в целом находится в движении, причем градиент ско-
рости этого движения является переменным, то зависит от расположения
/-иона, а — от расположения Лиона. Эти условия можно выразить с по-
мощью следующих функциональных зависимостей:
Пц = Пц (г1; r2J), = Пц (г2, г12). (2)
3*
36
ГЛАВА Ы
Эти общие соотношения будут использованы в теории вязкости.
Теперь необходимо найти функции распределения. Рассмотрим два
элемента объема dVг и dV 2, фиксированных в пространстве в течение не-
которого промежутка времени t, и два вида ионов j и i. Ту часть этого
промежутка времени t, в течение которой /-ион находится в dV\, обозначим
через tj, а ту часть промежутка времени, в течение которой г-ион нахо-
дится в dV2, обозначим через . Вероятность tj/t нахождения /-иона
в объеме dV\ и вероятность ti/t нахождения г-иона в объеме dV 2 связаны
с концентрациями гг7- и пг соотношениями1 *
4 = ~ = nidV2.
Далее, обозначим через время, в -течение которого /-ион и г-ион одно-
временно находятся соответственно в dVг и dV 2. Тогда
= bL^nadV..
Li li
Исключая из предыдущих уравнений tj и tjt получаем выражение
-2L = щпц dV1 dV2 = -~- = ni nij dV\dV2. (3)
Так как t^/t представляет собой вероятность одновременного нахожде-
ния /-иона в dV! и г-иона в dV2, то это соотношение можно использовать
для определения искомых функций распределения, а именно
tii (г1, Г21) == П) ПЦ (ГЬ Г21) =-- Щ Пц (г3, Г13) ~ fij (г2, г12). (4)
Отметим, что /ц представляет собой концентрацию (число ионов в см3)
i-ионов на расстоянии г21 от /-иона, умноженную на п}-, т. е. на число
ионов вида / в см3, находящихся в пространстве на расстоянии г2 от исход-
ной точки. Другими словами, fa равняется концентрации i-ионов в атмо-
сферах tij ионов вида /. Отметим также важное условие симметричности,
согласно которому /д должно равняться fa- Эта величина должна быть
известна для решения задачи о распределении ионов. Значения /г, и щ
известны, а значения пц^, г21) и П{Дг2, ri2) должны быть найдены с по-
мощью дальнейших статистических рассуждений. В случае равновесия,
когда нужно учитывать только кулоновские силы взаимодействия между
ионами, для определения этих значений требуется только применение закона
распределения Максвелла—Больцмана.
Удовлетворительное представление об «ионной атмосфере» можно полу-
чить, рассматривая положительный ион как фиксированный в dyr. Поскольку
между положительными и отрицательными ионами действуют силы при-
тяжения, то в dV2 (в среднем) будет находиться больше отрицательных
ионов, чем положительных. Такой избыток отрицательных ионов вокруг
данного положительного иона, или наоборот, создает ионную атмосферу.
В растворе электролита, на который не действуют внешние силы, поле
вокруг данного иона во всех направлениях одинаково, и поэтому сила этого-
поля зависит только от расстояния г, но не от направления. В этом случае
ионная атмосфера обладает сферической симметрией. Если же ионы нахо-
дятся под действием внешних сил, как, например, в случае раствора, на
который наложено электрическое поле, ионная атмосфера является несим-
метричной.
1 Здесь авторы допускают неточность, так как для согласования размерности
в правую часть этих уравнений нужно было бы еще ввести некоторый коэффициент.
Однако отсутствие такого коэффициента не вносит ошибки в дальнейшее изложение,
так как в ходе последующих подстановок он сокращается. {Прим, ред.}
ТЕОРИЯ МЕЖДУИОННОГО ПРИТЯЖЕНИЯ
37
б) Скорость изменения функции распределения во времени. Пусть
скорость i-иона вблизи у'-иона равна
V;i = Vyi(r1, Г21).
Аналогичным образом
vy = vy(r2, г12). (5)
Далее, обозначив через ...Д6 шесть пространственных координат,
которые определяют положение двух точек в растворе, можно одновременно
следить за положением dV\ и dV2. Компоненты скорости у-иона в dV х
равны $1; i2 и i3, а i-иона в dV2 — i4, и |6. Здесь точки над буквами,
как обычно, обозначают дифференцирование по времени. В этом шести-
мерном пространстве рассмотрим шестимерный элемент объема, положение
центра которого определяется координатами Si, ...,£6. Так как /ц пред-
ставляет собой концентрацию (в шестимерном пространстве) в этой точке,
то поток i-ионов Unyiib в координате входящий в элемент объема через
пятимерную поверхность di2...dig, перпендикулярную к оси равен
£1 fji di% ..• dig,
а поток, выходящий из этого объема в координате £1 + сД1, равен
|_ Si fa Н-d J di2 ... d^a
Таким образом, увеличение числа ионов в элементе объема в единицу вре-
мени, обусловленное движением ионов в направлении координаты соста-
вляет
ЧЫц) st
----дё~ ^£1 • • • dig»
Разделив сумму соответствующих разностей для всех шести координат на
величину элемента объема di1 . . . dig, получим скорость изменения кон-
центрации в элементе объема во времени
dfji _ _ /ИХ
dt & дсг * ' '
г«=1
Дифференциальный оператор справа представляет собой шестимерную
дивергенцию, которая может быть записана так:
6
Г = 1
где индексы 1 и 2 обозначают дифференцирование по компонентам гх и г2.
Поскольку //Дг!, г21) = fn (г2, г12), то уравнение (6) можно записать в сле-
дующем виде:
(Г1, г21) 5/{ (г2, г12)
----—gt-----= Un Vo) + V2 • Uii Vji) =-’-fr.- . (7)
Это выражение представляет собой уравнение непрерывности, записанное
в форме, подходящей для применения в теории движения ионов. В ста-
df-
ционарном состоянии -0 и, следовательно,
V1- (/vVy) + V2- (/дУд) = 0.
(8)
38 Г Л А В А II
"Эти- гидродинамические уравнения служат основой теории необратимых
процессов. Однако прежде чем их применять, необходимо определить как
функции распределения, так и скорости.
в) Уравнения движения. Ионы могут двигаться под влиянием трех
причин: 1) сил, действующих на ионы, 2) теплового (беспорядочного) дви-
жения и 3) движения всего раствора в целом. Действующие на ионы силы
могут быть внешними (внешнее электрическое поле) и внутренними (гра-
диенты концентраций и электростатические силы, обусловленные присут-
ствием самих ионов). Если подвижность i-иона равна еоу тогда действу-
ющая на него сила К, сообщит ему скорость Коэффициент трения
этого иона составляет р( = 1/<и;. Если считать константу диффузии иона
равной кТ& [19], то под влиянием градиента концентраций V/ возникает
-поток ионов — кТыЪ/. Поскольку этот поток равен v/, скорость диффузии
составляет — Zc7’o>V In/. Далее, обозначив через V (г2) скорость движения
раствора в целом в точке, определяемой вектором г2, можно найти суммар-
ную .скорость Vji в этой точке:
v;i = V(r2)+ <0г(Кд —ATValn/ji). (9)
Аналогичным образом:
уi) = V (гД + СО; (Ку - ЛТ?! In fij).
После подстановки эТих значений скоростей в уравнение (8) для стацио-
нарного состояния это- уравнение принимает следующий вид:
V1 [ fijV (ГД + М/ч Ку - кТ'^1 АД] +
+ V2. [А{У(г2) + ш{(/дКд-*7?2/д)] = 0. (10)
Чтобы применить это уравнение к теории необратимых процессов, следует
ввести представление о характере влияния междуионных сил. С этой целью
необходимо сначала хорошо изучить свойства растворов электролитов
в отсутствие внешних полей. Мы не остановились на разборе этого сравни-
тельно простого случая раньше, так как для объяснения некоторых важ-
нейших положений, на которых основана вся теория, требуется выраже-
ние для скоростей ионов, даваемое уравнением (9).
г) Основные уравнения для потенциалов иона и его атмосферы
в отсутствие внешних полей. Теория Дебая. Если электролит находится
в стационарном состоянии и на него не действуют внешние силы, ионные
атмосферы обладают сферической симметрией и функции распределения
зависят только от расстояния г = | r121 = | r2i |. Вертикальные черточки озна-
чают, что берется абсолютное значение данных величин. Для этого случая
функции распределения будут обозначаться верхними нулевыми индексами;
следовательно,
/?Дг) = п,пО (r)^„.no.(r) = yo.(r); (11)
Разберем подробно действие кулоновских сил и влияние теплового
движения ионов на функции распределения и потенциалы. Допустим, что
ионы являются заряженными точками, и не будем пока останавливаться
на эффектах, связанных с конечными размерами ионов. Электрический
потенциал иона, обладающего зарядом е7- и находящегося в жидкости
с диэлектрической постоянной D, на расстоянии г от этого иона равен
e^Dr. В растворе электролита образуются ионные атмосферы и происходит
наложение полей ионов, которые могут быть найдены из значений потен-
циалов. Обозначим потенциал, обусловленный ионом и его атмосферой,
ТЕОРИЯ 1ИЕЖДУИОННОГО ПРИТЯЖЕНИЯ ' 39
в общем случае через фДгх, г21), а в статическом (невозмущенном) состоя-
нии—через ф?(г).
Если среда содержит электрические заряды (или другие источники
полей), находящиеся под действием сил, которые обратно пропорциональны
квадрату расстояния, тогда соотношение между плотностью заряда р и
потенциалом дается в общем случае уравнением Пуассона
Согласно этому уравнению, в любой точке среды, определенной тремя
пространственными координатами (ж, у, z; г, в, <р), дивергенция градиента
потенциала, т. е. общий силовой поток из этой точки, пропорциональна
плотности заряда в этой точке. В растворе электролита плотность заряда
8
(заряд, приходящийся на 1 см3) на расстоянии г от /-попа равна У, пд в,,
i = l
где суммирование производится по всем видам ионов в растворе. Потенциал
в этой точке обусловлен /-ионом и его атмосферой. Таким образом, в общем
случае
8
V-V9y(ri, г21) = -^2 ппе'-- (12)
i=i
Для электролита, находящегося в невозмущенном состоянии,
s
у.уфо(г)=_^2^е4 (13)
г-1
и зависит только от г.
‘ 1
Принимая во внимание тепловое движение ионов, можно допустить,
что концентрация ионов данного вида вблизи определенного иона опреде-
ляется законом распределения Максвелла — Больцмана, представленным
в следующей форме:
где Uji—потенциальная энергия г-иона вблизи /-иона, а кТ— его кинети-
ческая энергия.
Сопоставление этих соотношений с уравнениями движения дает воз-
можность обнаружить важнейшие положения, лежащие в основе теории.
Случай равновесия характеризуется тем, что из уравнений исчезают сред-
ние величины скоростей v,i и Уц. Следовательно, уравнения (9) приобре-
тают такой вид:
Кц = АТПп/?{ = т1п/?,=К{,. (15)
Согласно уравнению (14), flt = пут^ц = — fl,, и тогда из уравнения (14)
получаем
Л -U^IkT —Uij/kT ,0
fji = П)П1в ]г = nttlje O' — .
Так как п,- и ni — постоянные концентрации, то после подстановки этих
соотношений в уравнение (15) получаем при постоянной температуре
кТ^ -VUif = kTV In fl,-. (16)
40
ГЛАВА II
Если предположить, что средняя сила, действующая на один ион,
определяется потенциалом другого иона, что эквивалентно допущению
о линейном наложении полей, то потенциальные энергии Uji и будут
равны соответственно фу (г) е, и фу (г) бу, и по уравнению (16)
#я = Ф°(г)ву==фу (r)ey = tfyy. (17)
Таким образом, мы допускаем, что потенциальная энергия i-иона на
расстоянии г от /-иона равна потенциалу, возникающему в этой точке
под влиянием /-иона и его атмосферы, умноженному на заряд i-иона,
причем эта потенциальная энергия равна работе заряжения i-иона в поле
потенциала фу (г). Согласно уравнению (17), потенциал иона и его атмо-
сферы пропорционален его заряду.
Необходимо учитывать, что при изложении теории междуионного при-
тяжения всегда делается допущение о линейном наложении полей. Так,
например, теоретическое вычисление термодинамических свойств (химических
потенциалов, коэффициентов активности и т. д,) основано на допущении
о существовании пропорциональности между фу (г) и бу. В ходе последую-
щего изложения будут рассмотрены условия, при которых зто соотношение
пропорциональности между потенциалом и зарядом совместимо с распреде-
лением Максвелла—Больцмана [20].
Путем сочетания уравнений (13) и (14) и подстановки фу (г) бу вместо
получаем
S
V • 7ф° (г) = - g 3 ехР [ - (r) (18)
г=1
Это уравнение не отвечает требованию, чтобы фу (г) было пропорциональное,,
так как левая часть уравнения, согласно сделанному выше предположению,
линейно зависит от еу, в то время как для правой части уравнения это
условие не соблюдается. Для устранения этого противоречия можно вос-
пользоваться приближенным выражением
ехр [ — фу (г) 6y/fcT]^l — фу (г)ецкТ, (19)
которое получается, если пренебречь членами ряда более высокого порядка,
чем первый. Это приближение допустимо лишь в том случае, если
ф® (г) еу мало или если фу (г) мало, причем зто условие осуществляется при
очень малых концентрациях. Для электролитов с высоким типом валент-
ности и для электролитов, растворенных в средах с низкой диэлектрической
постоянной, ф° (г) еу имеет более высокое значение, и зто приближение нельзя
использовать.
При подстановке приближенного значения из уравнения (19) в урав-
нение (18) получаем
V-ViOy(r) = -^3 Пубг2ф«(г), (20)
г=1
так как условие электронейтральности раствора в целом требует, чтобы
s
JSeyny = O. (21)
г=1
ТЕОРИЯ МЕЖДУИОННОГО ПРИТЯЖЕНИЯ
41
Пусть ef = zt s2, где Zj— валентность иона, a s —заряд электрона; опре-
делим следующим образом величину х:
»А2; (22)
i=l
тогда уравнение (20) принимает вид
У-Уф?(г) = х*ф?(г). (23)
В электролите, на который не действуют внешние силы, ионные атмосферы
обладают сферической симметрией и удобнее всего выразить V-Уфу (г)
в сферических координатах. Тогда
1 5 г 2а^(г) 1 0
а^-1=хфЛг)’ (24>
поскольку ф° (г) зависит только от г. Путем интегрирования этого урав-
нения получаем фу (г) как функцию от г и х, причем х являетсц функцией
диэлектрической постоянной среды, температуры и концентрации элек-
тролита .
Общее решение уравнения (24) имеет следующий вид:
,0, V Ае-" , А'е"
Ф/ (г) = —— + > (25>
где А и А' — постоянные интегрирования. Постоянная Л' должна равняться
нулю, так как фу (г) обращается в нуль, когда г стремится, к бесконеч-
ности, т. е. фу(со) = 0. Это уравнение можно разложить в ряд, и для
малых значений хг, пренебрегая членами более высокого порядка, полу-
чаем выражение
Ф?(6=4-Ах- <26>
В гл. 3, § 5, будет показано, что А не зависит от г и х. Поэтому
А/r зависит только от г, в то время как Лх зависит лишь от х и исчезает
при бесконечном разбавлении. Следовательно, А/r представляет собой по-
тенциал на расстоянии г от точечного заряда еу, величина которого равна
AD, а фу (г)—потенциал, соответствующий точечному заряду (иону) вели-
чиной AD, плюс потенциал, создаваемый пространственным зарядом атмо-
сферы. Таким образом Л можно заменить через ejlD. Из уравнений (13),
(23) и (25) следует, что
V • Уф? (г) = - J 2 = х2Ф> (И = *2 ’ (27>
1=1
и таким образом пространственный заряд г-иона в атмосфере /-иона на
расстоянии г от последнего равен 28
(28)
i=l
Из уравнения (25) получаются очень важные уравнения для значений
потенциалов после подстановки е(/D или Zjs/D вместо Л.
Так,
(29>
42
ГЛАВА II
или, в другой форме,
zis
Dr
V (1 —е
Dr
(30)
Первый член справа в этом уравнении представляет собой потенциал
на расстоянии г от отдельного иона в среде с диэлектрической постоянной D.
Второй член равен потенциалу ионной атмосферы. Первый член не входит
в выражения для зависимости термодинамических свойств электролитов от
концентрации ионов. Однако в некоторых случаях, когда нужно учитывать
влияние изменения растворителя, первый член сохраняется. Так как
(1—е~") приближается к величине хг при малых значениях хг, то потен-
циал иона и его атмосферы в этом случае становится равным
• 0 / к
'Ь (Н =
zr
Dr
zp-t.
(31)
D '
Ото уравнение вытекает также непосредственно из уравнения (26). Второй
член правой части уравнения равен потенциалу ионной атмосферы при
условии, что значения хг малы; это условие будет выполнено, если (при
конечной величине г) х приближается к 0, т. е. концентрация стремится
к нулю. Очевидно, величина 1/х в последнем члене уравнения (31)
л # Z/SX
ф, ---------—
(32)
аналогична расстоянию г в первом члене правой части этого уравнения и обла-
дает размерностью длины. Таким образом, 1/х представляет собой средний
радиус ионной атмосферы; благодаря этому обстоятельству х является величи-
ной, которая играет очень важную роль в теории междуионного притяжения.
Для того чтобы придать уравнению непрерывности (10) подходящую
форму для применения к специальным теориям необратимых процессов
в растворах электролитов, функции распределения /# и /г° необходимо вы-
разить через г. Искомые выражения можно получить из уравнений (11),
.(14), (17), (19), (29):
fa = П)Пц = ехр [ — Ф° (г) et/kT] = n^i £ 1 — 61 ] =
= • (33)
Три из вышеприведенных уравнений имеют важнейшее значение для
дальнейшего изложения теории междуионного притяжения. Соотношение
(32) является основным уравнением теории для вывода предельных зако-
нов для активности и осмотических коэффициентов, а также для вычисле-
ния парциальных молярных теплот разбавления и теплоемкостей (гл. III,
§ 1—4). Уравнение (29) служит основой для вывода уравнения Дебая —
Гюккеля для коэффициента активности, в которое входит величина сред-
него расстояния сближения ионов (гл. III, § 5). Последнее из этих урав-
нений (33) будет использовано в следующем параграфе для преобразова-
ния уравнения (10) с тем, чтобы это уравнение удовлетворяло требованиям
теории необратимых процессов в растворах электролитов.
д) Общие уравнения для ионных атмосфер в возмущенном состоянии.
Если электролит оказывается в возмущенном состоянии под действием
внешних сил, то как потенциалы, так и функции распределения приобре-
тают несимметричный вид. Тогда
ФДг,, г21)=ф)(Г1, г21) + ф“ (г)
/л(Г1, r2i) = j ji (Г1( Г21) + fji (г),
ТЕОРИЯ МЕЖДУИОННОГО ПРИТЯЖЕНИЯ
43
где члены со штрихом отражают влияние возмущений. Потенциалы и функ-
ции распределения связаны между собой согласно уравнению Пуассона
следующим образом:
ф-- (35)
• 4=1 1
Для простоты, мы часто пишем ф( вместо ф/(Гх, r2i) и /ц вместо г21).
Под действием возмущающих факторов в растворе идут необратимые
процессы, и распределение ионов уже не подчиняется уравнению Максвелла —
Больцмана. Поэтому мы должны обратиться к уравнению непрерывности
(10), поскольку средние значения скоростей v# и vy не равны нулю.
Общая сила Иц, действующая на г-ион, выражается уравнением
Кд = кг —e;V2<p-(0) —е^аОДгх, г21), (36)
где к; — приложенная внешняя сила. Второй член в правой части уравне-
ния представляет собой силу (градиент потенциала), действующую на г-ион
и обусловленную наличием атмосферы у этого иона. Следует иметь в виду,
что это будет потенциал в начале координат (г = 0) или в точке, где по-
мещается t-ион. Третий член —это действующая на г-ион сила, обусловлен-
ная потенциалом /-иона и его атмосферы. Важно помнить, что это вычис-
ление общей силы сделано при допущении, что общее силовое поле, обу-
словленное ионами и их атмосферами, может быть получено в результате
линейного наложения отдельных полей.
Подставляя К,{ из уравнения (36) в уравнение (9), умножая послед-
нее на fji и беря дивергенцию, получаем
• (filin') = ?2 • { Л'гV (г2) + [/якг — е(/лГ2ф( (0) — ег/дГ2ф; - Лг?Т2/д]}. (37)
После раскрытия скобок некоторые члены в правой части уравнения
исчезают. Во всех изучаемых нами случаях как скорость раствора в целом,
так и внешнее электрическое поле остаются постоянными, и поэтому два
члена исчезают, так как V2-V(r2) = 0 и Г2-кг = 0. Членом, содержащим
V2ty$ (0), можно пренебречь, так как порядок величины этого члена ef, в то
время как остальные члены — порядка е; (см. уравнение (29)). Далее,
согласно уравнению (33), разность /ц — имеет порядок величины et.
Следовательно, в выражении можно заменить на п,П1, и по-
скольку ^2nini равно нулю, то член V2 •(ei<i>(n7nfV2ty/) превращается в
е;<и;гг;ггг72‘После этих сокращений уравнение (37) принимает вид
2 ' (/jiVji) — (Г2) • V>]ji + (,,i (Ь{ ‘ 2/71)
— е^гг^ • V2 • — <ntkTV2 V2/;i. (38)
Подобное уравнение можно получить также для V (/i/V,,). Для того,
чтобы получить уравнение непрерывности в желаемой форме, значения
Vi’(Дуг,;,) и V2-(/jiVji) подставляют в уравнение (7) и производят некоторые
дополнительные преобразования. Вместо fjit fl} и ф,-, ф, подставляют их
значения из уравнений (34). Так как для состояния равновесия к, = 0,
к, = 0 при V;T = 0 и Vi/ = 0, то с помощью уравнений (9) и (36) находим, что
v'ikT~2 ~ winjrtiei^2
и
MjkT -= — • VpL?,
m, следовательно, четыре члена исчезают в результате сокращения. Урав-
нение непрерывности в окончательной форме, которая является достаточно
44
ГЛАВА II
общей для всего дальнейшего анализа необратимых процессов в растворах
электролитов, имеет вид
V (Г2),уг/д + V (ri)'Vi/y +
4- од (к{ • V2/3i) + Ш] (kj • Vj/ij) —
— eiwinjni^ 2 • V2tyy — ер>ргщ37 г • V — (39)
— инкТУ 2 • v 2//г — <s>jkT\ i • Vj/ty =
д1ц (П, r8i) 5/,--(r2, r12)
=--------di----~~--------di---- (Аля нестационарных полей),
= 0 (для стационарных полей).
Как правило, за исключением одного случая, а именно эффекта Вина
(гл. IV, § 5), V (гг), V (г2), kj и ку настолько малы, что в первых четы-
рех членах можно заменить / на /°.
е) Общий обзор поведения ионных атмосфер в возмущенных состояниях.
Уравнение (39) является достаточно общим для того, чтобы его можно было
применить к теории вязкости, электропроводности и диффузии электролитов.
Первые четыре члена содержат возмущающие факторы, а последние четыре —
асимметрические составляющие части потенциалов. Для получения диф-
ференциальных уравнений для потенциалов, с помощью которых можно
вычислять силы, вызывающие движение ионов, нужно исключить функции
распределения /'ц и с помощью уравнения Пуассона (35).
Для решения тех или иных частных задач уравнению (39) можно
придавать различный вид. Так, в случае приложения к явлениям вязко-
сти, члены, содержащие скорости движения всего раствора в целом V (г2)
и V (гД, сохраняются, в то время как члены, содержащие внешние воз-
мущающие силы kj и к,, исчезают. Можно показать, что составляющая
вязкости, обусловленная ионами, связана со значениями потенциалов, кото-
рые получаются из уравнения (39), и может быть найдена путем соответ-
ствующих математических вычислений.
При изучении таких вопросов, как электропроводность и диффузия,
могут быть опущены первые два члена уравнения (39), содержащие ско-
рости движения раствора в целом. В случае электропроводности возмуща-
ющими силами к, и к, являются наложенные внешние поля, а в случае
диффузии — градиенты термодинамических потенциалов1. При движении
иона в электрическом поле он увлекает за собой свою атмосферу, и в резуль-
тате его подвижность уменьшается. Величину этого эффекта можно найти,
вычисляя значения потенциалов по уравнению (39) и затем силы, действу-
ющие на ионы. Кроме того, имеется другой эффект, обусловленный движе-
нием растворителя по отношению к иону, называемый «электрофоретиче-
ским эффектом», который следует вычислять независимо и добавлять к
эффекту, связанному с асимметричностью ионных атмосфер.
Все рассмотренное относится к стационарным случаям постоянных
возмущающих полей, когда /ц и /,{ не меняются во времени. Однако имеются
некоторые явления, для которых необходимо исследовать нестационар-
ный случай; при этом левая часть уравнения (39) равняется не нулю, а
—д/МП, г21)
dt
Если ионная атмосфера сделалась асимметричной под влиянием возму-
щающего поля и если затем это поле исчезает, то атмосфера приобретает
свою первоначальную сферическую форму. Для этого восстановления исход-
1 Под термодинамическими потенциалами подразумеваются термодинамические
функции U, Н, А и F (см. гл. J, стр. 20). (Прим, ped.)
ТЕОРИЯ МЕЖДУИОННОГО ПРИТЯЖЕНИЯ
45
ной формы атмосферы требуется конечное время, которое называется «вре-
менем релаксации» атмосферы.
Если к раствору электролита приложено переменное электрическое
поле с частотой порядка этого времени релаксации, то атмосфера не успе-
вает сделаться асимметричной и изменение подвижности ионов, связанное
с асимметричностью их атмосфер, будет меньше, чем в стационарном случае.
Эта идея лежит в основе развитой Дебаем и Фалькенгагеном теории влияния
высоких частот на электропроводность ионных растворов. Отправным пунк-
том этой теории является уравнение (39) для нестационарного случая.
Имеется еще одно важное явление, которое будет нами изучено в даль-
нейшем. Если раствор электролита находится под действием сильного элек-
трического поля, центральные ионы будут извлекаться этим полем из своих
атмосфер. Это явление приводит к увеличению проводимости ионов, по-
скольку им уже не приходится «тянуть» вместе с собой ионную атмосферу.
Теория этого явления, известного под названием эффекта Вина, может быть
изучена с помощью уравнения (38).
Приложения уравнения (39) к явлениям вязкости, электропроводности
и диффузии будут рассмотрены в гл. IV. Более простые теории термодинами-
ческих свойств будут детально изложены в следующей главе.
Л ИТЕРАТУРА
1. W. Sutherland, Phil. Mag. [6], 3, 167 (1902).; 7, 1 (1906).
2. A. A. Noyes, Congress Arts ScL, St. Louis Exposition, 4, 317 (1904).
3. N. В j e r r u m, D. Kgl. Danske Vidensk. Selsk. Skrifter [7], 4, 1 (1906); Proc. 7th
Intern. Congr. Applied Chemistry, Spect. X, London (1909); 16 Skand. Naturforsk.
Forhandl., 226, 1916; Z. Electrochem., 24, 321 (198); Meddel. Kgl. Vet. Akad. Nobel
inst., 5, No 16 (1919); Z. anorg. Chem., 109, 275 (1920).
4. J. J. v a n L a a r, Z. physik. Chem., 15, 457 (1894); 17, 245 (1895); 19, 318 (1896);
Z. anorg. Chem., 139, 108 (1924).
5. P. Hertz, Ann. Physik. (4), 37, 1 (1912).
6. I. G. Ghosh, J. Chem. Soc., 113, 449, 627, 707, 790 (1918); Trans. Faraday Soc.,
15, 154 (1919); J. Chem. Soc., 117, 823, 1390 (1920); Z. Physik. Chem., 98, 211 (1921).
7. R. Milner, Phil. Mag., 23, 551 (1912); 25, 742 (1913).
8. P. Debye, E. Hiickel, Physik. Z., 24, 185 (1923).
9. R. H. Fowle r, Trans. Faraday Soc., 23, 434 (1927); L. О n s a g e r, Chem. Rev,,
13, 73 (1923); H. A. К r a m e r s, Proc.Royal Acad. Sci. Amsterdam, 30, 145 (1927);
J. G. Kirkwood,.!. Chem. Phys., 2, 767 (1934),
10. P. D e b у e, E. H ii c k e 1, Physik. Z., 24, 305 (1923).
ILL. О n s a g e r, Physik. Z., 28, 277 (1927).
12. P. D e b у e, H. F a 1 k e n h a g e n, Physik. Z., 29, 121, 401 (1928).
13. H. Falkenhagen, M. D о 1 e, Z. physik. Chem,, 6, 159 (1929); Physik. Z., 30,
611 (1929); H. Falkenhagen, Physik. Z., 32, 365, 745 (1931).
14. L. Onsager, R. M. F u о s s , J. Physical Chem., 34, 2689 (1932).
15. G. J о о s, M. В 1 u m e n t r i t t, Physik. Z., 28, 836 (1927).
16. W. S. Wilson, Диссертация, Yale University, June, 1936.
17. L. О n s a g e r, J. Chem. Phys., 2, 599 (1934).
18. A. A. N о у e s, J. Am. Chem. Soc.,46, 1080 (1924); X. С. Тейлор, Физическая химия,
т. I, ОНТИ—Химтеорет, Л., 1935, гл. XII, стр. 766; А. Эйкен, Курс химиче-
ской физики, вып. второй, ГХТИ, М.—Л., 1933; М. Дол, Основы теоретической
и экспериментальной электрохимии, ОНТИ—Главная редакция химической лите-
ратуры, М., 1937. D. А. М а с I n n е s, The Principles of Electrochemistry,
Reinhold Publishing Corporation, N. Y., 1939; W. M. Clark, The Determina-
tion of Hydrogen Ions, 3rd ed., Williams and Wilins Co., Baltimore, 1928.
19. W. Nernst, Z. physik. Chem., 2, 613 (1888); Theoretical Chemistry, Macmillan Co.,
London, 1911, p. 368—374; R. C. Tolman, Statistical Mechanics, Chemical
Catalog Co., (Reinhold Publishing Corp.), N. Y., 1927, p. 231, equation (535).
20. L. О n s a g e r, Chem. Rev., 13, 73 (1933).
Глава III
ТЕОРИЯ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ Р АСТВОРОВ
ЭЛЕКТРОЛИТОВ
В настоящей главе будет развита полная теория термодинамических
свойств разбавленных ионных растворов путем сочетания теоретических
уравнений, полученных для случая равновесия (гл. II, 8 4) с термодинами-
ческими зависимостями, приведенными в гл. I, а также путем дальнейшего
обобщения полученных результатов. Будут выведены предельные законы
зависимости коэффициента активности, осмотического коэффициента, отно-
сительного парциального молярного теплосодержания, теплоемкости, рас-
ширяемости и сжимаемости от концентрации. Теория будет распростра-
нена на тот случай, когда учитывается влияние конечных размеров ионов.
Дальнейшее расширение теории будет заключаться в устранении прибли-
женного характера математической трактовки, которая была обусловлена
отбрасыванием членов высших порядков при разложении в ряд экспонен-
циальной функции в уравнении (18) гл. II; кроме того, будет рассмотрена
также теория, учитывающая влияние ассоциации ионов. В сжатой форме
будет изложена теория влияния электростатических сил на поверхностное
натяжение раствора. Наконец будет рассмотрена теория влияния распа-
дающихся на ионы солей на растворимость нейтральных молекул.
§ 1. Свойства 1/х
При обсуждении распределения ионов предполагалось, что последние
эквивалентны точечным зарядам; при этом было развито представление
о том, что каждый ион окружен избытком ионов противоположного
знака, что приводит к возникновению электрического потенциала, кото-
рый для случая раствора электролита, не подверженного действию внеш-
ней силы, мы обозначали через Ф’*. Допуская, что распределение ионов
подчиняется закону Максвелла — Больцмана, и используя приближенное
условие, выраженное уравнением (19) гл. II, мы получаем первое, очень
важное приближенное соотношение теории Дебая—Гюккеля, которое
выражается уравнением (32) гл. II:
~ziгх
D
где -х определяется выражением (22) гл. II:
,2— 4д~2 у
— DkT Zj
i=l
Tli Zi.
Величина х обладает размерностью, обратной размерности расстоя-
ния. и таким образом 1/х связано с потенциалом ионнои атмосферы ,
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
47-
так же как г связано с потенциалом Ф" отдельной частицы с зарядом е;-,
находящейся в среде с диэлектрической постоянной D.
Концентрация С\ ионов г-го вида в молях на литр раствора связана с чис-
лом этих ионов в с,и3 Wj соотношением
где N — число Авогадро. Следовательно, по уравнению (22) гл. II
< 4tcs2A \’/2
* < ЮООЛАТ 1 ) ’
где через Г обозначена так называемая «иональная» концентрация
Г = ci zi- (3>
1
Подставляя численные значения констант, получаем в общем случае
4 = 2,811 • 10-/^ см, (4>
а в частном случае 1,1-электролита в водном растворе при 25°
1 3,041 10~8 ,5<
Таким образом, в нормальных растворах 1/х имеет величину порядка диа-
метра молекулы. Заметим, что поскольку это соотношение содержит корень
квадратный из с, то при стократном уменьшении концентрации 1/х увели-
чивается в 10 раз.
§ 2. Методы вычисления работоспособности и химического
потенциала
Электростатическая составляющая работоспособности ДА [уравне-
ние (6) гл. П] П/ ионов и их атмосфер дается уравнением
пз ез
W (эл) = ДА (эл) = 2 \ Ф (еД de. (6)-
7-1 0
Таким образом, если незаряженный ион при постоянном составе системы
обратимо заряжается в поле потенциала, то ф (еД зависит от заряда е
в данный момент. Процесс заряжения, согласно Дебаю, изображается
выражением
пз л=1
W (эл) = ДА (эл) = 2 е;-ф(кеД(/к, (7)'
7 = 1 Л=0
которое получается непосредственно из уравнения (6), если обозначить
величину заряда в данный момент времени через ке;-.
На основании определения химического потенциала р;-, данному в урав-
нении (И) гл. I, можно ввести представление о другом «процессе заря-
жения», которое непосредственно вытекает из уравнения (6), причем элек-
тростатическа'я составляющая химического потенциала в случае иона, как
48
ГЛАВА Ш
показал Гюнтельберг [1], будет равна
<эл)=(gAffn)) v~ S * de-
Если воспользоваться приближенным соотношением, которое дается урав-
нением (19) гл. II, и первым приближением для ф9*, согласно уравне-
нию (32) гл. II, то оба процесса заряжения приводят к одинаковому
результату. С другой стороны, если -при интегрировании уравнения Пуас-
сона-Больцмана (18) гл. II не пользоваться этим приближением, то про-
цессы заряжения по Дебаю и Гюнтельбергу приводят к несколько различ-
ным результатам. На этот факт указали Гронвол, Ла-Мер и Сэндвед [2],
которые произвели полное интегрирование, используя процесс заряжения
согласно Дебаю. Полученные ими результаты несколько отличаются от
результатов Мюллера [3], который воспользовался более простым методом
Гюнтельберга. На основании теоретического анализа этого затруднения
Онзагер [4] сделал вывод, что указанное расхождение обусловлено непри-
менимостью уравнения Пуассона — Больцмана при более высоких концентра-
циях, когда потенциалы ионных атмосфер уже нельзя считать аддитивными.
§ 3. Процесс заряжения и вычисление электростатической
составляющей химического потенциала
Из основного допущения о линейном наложении полей ионных атмо-
сфер следует, что потенциал ионной атмосферы пропорционален заряду:
«%°* = е/, (9)
где а — коэффициент пропорциональности. Воспользуемся процессом заряже-
ния, который выражается уравнением (7) гл. III, в этом случае электро-
статическая составляющая химического потенциала /-иона определяется вы-
ражением:
2 ,е
7 1 . О* , 0* > О*
г аф- ф- z-гфУ
Ар-/ (эл) = \ (е;) de, = \ aW* d^* = . (10)
о о
Подставляя сюда значение фр* из уравнения (32) гл. II, получаем
(z,. s)2 х
Др,-(эл)=—(И)
§ 4. Предельный закон для зависимости коэффициента
активности от концентрации
Согласно уравнению (53) гл. I
F = >RT In /± + vRT In N± + Fn-
F можно разбить на две части, а именно:
Fn=vRT In N± + F°n, (12)
~Ff = vRT\nf±. (13)
Первое из этих уравнений представляет собой предельный закон для раз-
бавленных растворов неионизированных растворенных веществ/ причем оно
было бы, вероятно, справедливым также для растворов электролитов,
если бы ионы не обладали электрическими зарядами.
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
49
Поэтому делается допущение, что в разбавленных растворах в случае
полностью ионизированных электролитов отклонение растворов электроли-
тов от идеального состояния может быть отнесено только за счет электро-
статических сил,-Действующих между ионами. Это допущение эквивалентно
отождествлению Ff с 7УДр(.эл.) для любого иона."Следовательно,
(z • s)2 X
Ди.3(эл.) = А7’In/у =---(!4)
Подставляя в это уравнение'значение х из (2), получаем
1п/у= -z) [ 1000(дЛГ)3 ] /2/f, (15)
предельное выражение для коэффициента активности иона (произвольного
вида /) в растворе, содержащем s видов ионов с концентрациями nlt
П.2, .. ., ns в см3.
Средний коэффициент активности электролита, диссоциирующего на р
видов ионов, согласно уравнению (48) гл. I, равен
р
ln/± = v3v>ln//> (16)
1
если при диссоциации одной молекулы электролита образуется всего
v ионов, из которых vy являются ионами вида j. Путем сочетания этого урав-
нения с уравнением (15) получаем
р
О’)
1
Для удобства переходим к десятичным логарифмам и подставляем числен-
ные значения констант; полученное уравнение записываем следующим
образом:
lg/±= - &(/)ГГ , (18)
где
р
= 1,283 . 10е. ' (19)
1
Символ с соответствующим индексом будет употребляться исключительно
для обозначения предельных коэффициентов наклона в наиболее общей
форме теоретических уравнений (при рассмотрении междуионного притяже-
ния). всегда применяется в сочетании с/Ги обычно относится к рас-
творам смешанных электролитов. В растворах, содержащих только один
электролит, Г пропорционально с, и в этом случае более удобно ввести
в теоретические уравнения jZс вместо |/Г. При этом возникает необходи-
мость в новом определении понятия предельного коэффициента наклона,
и для этой цели мы применяем в гл. V обозначение 5.
В очень важном, хотя и частном случае, когда р = 2, фактор валент-
ности в сводится к | ZiZa |, и мы имеем1
^(/) = |г122|(ПГ)-3/2 . 1,283 • 10е; р = 2. (19а)
1 Наличие вертикальных черточек в выражении | ZjZ2 | указывает на то, что берется
абсолютная величина произведения валентностей независимо от знаков валентностей.
4 Г. Харнед, Б. Оуэн
50
ГЛАВА III
Уравнения (17) —(19) представляют собой «предельный закон» Дебая —
Гюккеля для коэффициентов активности в наиболее общей форме1. Со-
гласно этому закону, в крайне разбавленных растворах логарифм коэффи-
циента активности электролита должен убывать линейно по мере возра-
стания величины корня квадратного из Г. Следует помнить, что Г является
функцией количества всех ионов 1......г, .... s в растворе независимо
от их источника, в то время как в суммирование производится только
по ионам 1, ... j, ... р, возникающим при диссоциации того электролита,
к которому относится /±. Так как мы оставили только два члена в ряде,
взятом для вычисления и допустили, что отклонение растворов элек-
тролитов от идеального поведения может быть приписано только наличию
кулоновских сил между ионами, то нельзя ожидать, чтобы эти уравнения
были строго справедливы для концентраций, поддающихся измерению.
Однако можно с полным правом ожидать, что эти уравнения будут точно
отражать предельное поведение электролитов в таких условиях, когда
иональная концентрация стремится к нулю.
Важной особенностью «предельного коэффициента наклона» яв-
ляется то, что он однозначно выражается через основные физические кон-
станты: абсолютную температуру, диэлектрическую постоянную раствори-
теля и валентный тип изучаемого электролита.
Прежде чем закончить этот параграф, необходимо представить предель-
ный закон в нескольких различных формах, которые будут использованы
в дальнейшем. Во многих случаях более удобно использовать у, т. е.
коэффициент активности, соответствующий моляльной концентрации (чис-
ло молей на килограмм растворителя), чем рациональный коэффициент
активности /. Из уравнений (57) гл. I и (18) получаем
lgY±= - &(/)/Г -lg(l + ^) , (20)
однако для тех разбавлений, при которых это уравнение становится спра-
ведливым, последний член в правой части уравнения2 может быть
опущен. Гак как с{ = 7пИ а—j , то Ci = mid0 при крайних разбавле-
ниях. Поэтому иональная концентрация I' связана с ионной силой
• S
i
следующим образом:
S S
Г = 2 CiZi=d0 2 miZi = 2(/op; с<1. (21а)
1 1
Следовательно,
lgT±=-&(/)/2^. (22)
1 Границы применимости теории Дебая и Гюккеля были указаны А. И. Брод-
ским (Труды V физико-химической конференции, 1930, стр. 52). См. также работу
Измайлова, который дал единую интерпретацию различных коэффициентов активности
и развил метод подсчета влияния основности среды на изменение коэффициента актив-
ности [Н. А. Измайлов, ЖФХ, 23, вып. № 5, 647 (1949)]. {Прим, ред.)
2 Этот член имеет такой вид для случая, когда раствор содержит один электро-
лит. Если в растворе имеется несколько электролитов с концентрациями mlt т2, ...,
этот член приобретает следующий общий вид:
/ S чтМ >
~lg<1+ 1000 J '
Термодинамические свойства растворов электролитов
51
Преобразуя уравнение (17) или непосредственно сочетая уравнения (14)
и (16), мы получаем в общем виде
1п /± = — Cv VjZj ) 2kDT ’
i
а для электролита, диссоциирующего только на два вида ионов,
С24)
Для применения предельных уравнений для вычисления парциальных мо-
лярных величин нам потребуется уравнение (17) в форме
, , 1 х1 2 2,457 • 1014 /оеч
In /± =---Л у Г. (25)
'а ч t-i J 1 R(DTylz ' ' '
При сочетании этого уравнения с уравнением (19) получаем полезное соот-
ношение
адОЗ^Й,,,»^,/2^!^. (26)
§ 5. Влияние кажущихся диаметров ионов.
Уравнения для коэффициента активности и осмотического коэффициента
Обсуждая в предыдущей главе применение уравнения Пуассона — Больц-
мана в теории Дебая и Гюккеля, мы рассматривали ионы как точечные
заряды. Ниже мы рассмотрим эту теорию в более общем виде с учетом
конечных размеров ионов и введем понятие «ионного параметра» а. Этот
параметр представляет собой минимальное среднее расстояние, на которое
могут сближаться как положительные, так и отрицательные ионы. Как
указывалось ранее [уравнение (22) гл. II], исходное уравнение Пуассона—
Больцмана имеет вид
где ф°(г)— потенциал иона и его атмосферы. Это уравнение можно пред-
ставить в такой форме:
Z.-S
Ъ» = ^ + Ф* (г).
(27)
где первый член в правой части уравнения представляет собой потенциал
на расстоянии г от иона с точечным зарядом z3-i, а ф* (г) — потенциал ионной
атмосферы на этом расстоянии.
Следовательно,
(28)
, * , . Ае~" zjs
ф* (г) =.------А- .
т ' ' г Dr
Для того чтобы выполнялось условие непрерывности поля при г, равном
среднему минимальному расстоянию ajt на которое могут сближаться ионы,
необходимо, чтобы суммарная величина напряженности поля иона и его атмо-
сферы дф“ (г)/дг равнялась величине напряженности поля одного иона
— Zj&lDr^ при г = цу. Это условие выполняется, если
Г5ф*(г)"| Ае ХО/ •• z,s
L dr Jr=a. 02
JDq j
4*
52
ГЛАВА III
Отсюда
Следовательно,
z.e /“/ А — -L е Д 1-|- -Mij (29)
„ • / ха. \ 1 ' ' Dr \1 + xaj J (30}
Z:£ X jL/ A л(4у (31)
и
Следует отметить, что это выражение отличается от значения ф®*, которое
дается уравнением (32), на коэффициент ха Если допустить, что потен-
циал пропорционален заряду, то, согласно уравнению (8) для процесеа
заряжения по Гюнтельбергу, получаем соотношения для химического потен-
циала
Л I \ ' х
(32)
и для коэффициента активности иона
(z,e)2 х
—<33)
С помощью тех же преобразований, которые были использованы для полу-
чения уравнения (18), получаем уравнение для 1g /± электролита, диссоци-
ирующего на два вида ионов:
1g /± ~ - ^</>У £ , (34)
8/± 1+Л/Т’ v ’
где ^(/) определяется уравнениями (19) и (19а), и
. ах 35,57 . 103а
А~—^ —------г,— ; « в сантиметрах,
/Г (DT)1^ г
о (35)
. а% 35,57-а
А~—-_==-----гг- ; а в ангстремах,
/г (ДГ)1/2 г
Следует отметить, что когда Г стремится к нулю, уравнение (34) переходит
в уравнение предельного закона, выведенное для точечных зарядов.
Теоретические уравнения для осмотического коэффициента можно полу-
чить путем сочетания любого из приведенных выше выражений для In /у с
уравнением (73) или (74) гл. I. Так, уравнение (74) гл. I можно написать в виде
? = 1 + S c>d ln fi’ (36)
2jcj j
поскольку Cj — mjd0 и = в очень разбавленных растворах. Если ввести
величину din/у, получаемую из уравнения (15), то после интегрирования
получаем общее предельное уравнение для ср (или g):
Г у/2(Ес;4)8/2
* 1 [ 1000 (ДАТ)8 J 3 3 ’ (
которое применимо к растворам, содержащим любое число ионизированных
растворенных веществ. Это уравнение употребляется редко. Для важного
частного случая раствора одного электролита, когда 2q = vc и 2c,zy =
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
53
= c^Vjzj = V, для приведения этого уравнения к более простому виду
можно воспользоваться уравнениями (17) и (19). При этом уравнение (37)
сводится к уравнению
? = 1+ |-In/± = 1 -4-2,303 (38)
Этот результат получается также путем сочетания уравнений (17) и (19)
непосредственно с уравнением (73) гл. I и последующего интегрирования.
• Более подробный вывод и обсуждение теоретических уравнений для
будут приведены в гл. IX, § 5, где будет рассмотрена зависимость <р от
параметра А в уравнении (31).
§ 6. Развитие теории Дебая и Гюккеля с учетом членов высших порядков
(Гронвол, Ла-Мер и Сэндвед)
Как было показано [уравнение (18) гл.II], основное уравнение теории
Дебая и Гюккеля имеет вид
8
V . 2 ni€i exp ( - ^ei/kT).
i — i
До сих пор мы пользовались приближенным выражением [уравнение (19)
гл. II]
ехр (— е^/кТ) 1 — е^/кТ,
которое получается, если взять первые два члена разложения в ряд экспо-
ненциальной функции. Рассмотрим электролит с концентрацией п молекул
в 1 см3, который диссоциирует на два вида ионов. Тогда
2 nvid ехр ( — е^/кТ) =
= п [Vj I в! I exp (— j 611 $/кТ) — v21 е21 ехр (| е21 (ЛТ)] = (39)
= nvi | 6i | [ехр (— | ei | $/кТ) — ехр (| е21 <$/кТ)],
так как Vi | ej ] = v21 е2 |.
Если 1611 равно |е2|, то выражение в скобках равно отрицательному
значению удвоенного гиперболического синуса угла | ег | ^/кТ, и после раз-
ложения последнего в ряд получаем
Отсюда видно, что для электролитов симметричного типа валентности мы
ранее пренебрегали влиянием третьего, пятого и других нечетных членов
более высокого порядка- Далее, очевидно также, что в случае электролитов
несимметричного валентного типа следует учитывать как нечетные, так
и четные члены. Полное решение этой проблемы имеется в работе Гронвола,
Ла-Мера и Сэндведа [2], которые вывели уравнение для электролитов сим-
метричного типа, а также в работе Ла-Мера, Гронвола и Грифа [5], которые
исследовали случай электролита несимметричного валентного типа. Ввиду
сложности математических выводов мы не будем излагать эти работы под-
робно и приведем лишь полученные этими авторами уравнения в окончатель-
ной форме, а также рассмотрим характер тех эффектов, которые обусловлены
включением членов высшего порядка.
54
ГЛАВА III
Для электролитов симметричного валентного типа (|Z]| = |z2|) обобщен-
ное уравнение для In /± имеет вид
оо
ln/±=-2WTTTa+ S X2m+i(*a)-2mY2m+i (ха)]. (41)
т= 1
Если взять при суммировании все члены разложения вплоть до членов пятого
порядка, получаем
'"А= +
+ (Ж.)5[1х‘(«>)-4к.М]. («)
Первый член справа идентичен выражениям (33) и (34) и представляет
собой первое приближение, полученное Дебаем и Гюккелем. Следующие
два члена уравнения соответствуют членам разложения третьего и пятого
порядков; Х3 (ко), Y3 (ха), Х5 (ха) и У5 (ха) являются сложными функциями ха,
значения которых были получены и представлены в виде таблицы Гронволом,
Ла-Мером и Сэндведом. Члены седьмого и более высокого порядков были
этими авторами опущены.
Характер коэффициентов перед членами в квадратных скобках указывает
на те условия, при которых проявляются отклонения от первоначальной
теории Дебая и Гюккеля. Следует ожидать такого рода отклонений для
электролитов с высокой валентностью (например, 2,2 и 3,3), а также для
электролитов, у которых значение а мало. Далее, значительные отклонения
от первоначальной приближенной теории должны наблюдаться в случае
растворителей с низкой диэлектрической постоянной.
В случае электролитов несимметричного валентного типа, как показали
Ла-Мер, Гронвол и Гриф, положение становится еще более сложным. Так,
ln -1। {(W(Z1 + Z2)2 B1 м ~
- (1^3 (z? - | zxz21 + z|) (Zj + z2)2 Вt (xa) - (43)
- aw(z' ~1 ZiZa' +‘Вз -•••}’
где
B2 (xa) = (j^y у X* (xa) — Y2 (xa) ] ,
^(ха) = (^)3Г|Л(ха)-2У3*(ха)] , (44)
у ик j. / l J
53 (xa) = (^У [IX3 (xa) - 2У3 (xa) ] .
Для численных значений членов, заключенных в квадратные скобки,
Ла-Мер, Гронвол и Гриф составили таблицы. На основании этого уравне-
ния следует ожидать больших отклонений от первоначальной приближен-
ной теории Дебая и Гюккеля для электролитов несимметричного типа более
высокой валентности даже в средах с большой диэлектрической постоянной,
как, например, в случае воды. Таблицы всех этих специальных функций
будут даны в гл. V, § 2.
§ 7А. Теория ассоциации ионов Бьеррума [6]
Вскоре после опубликования теории Дебая и Гюккеля Бьеррум, учи-
тывая математические трудности полного решения уравнения Пуассона —
Больцмана в форме, данной Гронволом, Ла-Мером и Сэндведом, предложил
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
55
значительно более простую теорию. В теории Бьеррума рассматриваются
факторы, определяющие степень ассоциации ионов или, точнее говоря, обра-
зование ионных пар под влиянием кулоновских сил1. Бьеррум принимает
простейшую модель ионных растворов — он рассматривает ионы как несжи-
маемые и неполяризующиеся сферические частицы с радиусом а, находя-
щиеся в среде с определенной макроскопической диэлектрической постоян-
ной. При этом совершенно не учитываются неполярные квантовые связи
между ионами, а также взаимодействие ионов с молекулами растворителя.
Вероятность того, что д-ион находится на расстоянии г от /-иона,
согласно закону распределения Максвелла — Больцмана, выражается уравне-
нием
Вероятность == eu/kT 4-/.2 ^г. (45)
где Nci/iQQQ — число г-ионов в см3 раствора, а 4w2<Zr—объем сферической
оболочки толщиной dr и радиуса г, окружающей /-ион, U — потенциальная
энергия, т. е. работа удаления г-иона от /-иона с расстояния г на беско-
нечно большое расстояние. Для заданных значений г потенциальную энер-
гию U можно заменить выражением, полученным на основании закона
Кулона:
U==_^L. (46)
Таким образом получаем
Вероятность = [ ехр ( ] 4тсг2 dr'
Эта функция обладает некоторыми интересными свойствами. Если заряды
ионов имеют одинаковые знаки, то вероятность их ассоциации очень мала.
Если же заряды ионов имеют противоположные знаки, то можно показать, что
вероятность имеет минимальное значение на некотором расстоянии д, так что
г (мин.) = 7 = ^|^. (48)
Отсюда следует, что д представляет собой расстояние, на котором энергия
удаления ионов друг от друга равна 2кТ. Для значений г, меньших чем д,
вероятность быстро возрастает с уменьшением г. При значениях г, больших
чем д, вероятность увеличивается медленно. Бьеррум делает допущение,
что два иона, находящиеся на расстоянии г < д, являются ассоциирован-
ными. Для 1,1-электролитов вводе при 18° д равно 3,52 А, и, следовательно,
в электролитах этого типа, для которых величина среднего расстояния
сближения ионов а меньше 3,5А, будут возникать короткие ионные пары2.
О о
Для 1,1-электролитов, обладающих значениями а > 3,5 А, справедлива тео-
рия Дебая — Гюккеля 3.
1 До Бьеррума представление об образовании «ассоциированных пар» в растворах'
электролитов было высказано Семенченко [В. К. С е м е н ч е н к о, Z. physik. Chem., 112,
128 (1924)]. (Прим, ped.)
2 Так обозначаются ионные пары, у которых г < q. (Прим, перев.)
3 «Длинные» ионные пары (т. е. такие, для которых г > q), которые не учи-
тывал Бьеррум, были исследованы Фуоссом в его работе, посвященной тщательному
анализу всей теории [7]. В случае «длинных» ионных пар потенциальная энергия
данного иона по отношению к ближайшему неспаренному иону очень мала по сравне-
нию с энергией теплового движения. С помощью теории Дебая и Гюккеля можно
показать, что общая потенциальная энергия складывается из большого числа малых
членов.
56
ГЛАВА III
На основании этих рассуждений можно сделать вывод, что два иона,
находящиеся на расстоянии г < q, будут вступать во взаимодействие друг
с другом и количество образующихся ионных пар будет очень быстро
возрастать с уменьшением г. Степень ассоциации, т. е. (1—а), будет
выражаться определенным интегралом
' 9 I Zl?2 I е2
, 4ti2Vc Г QrkT о , ,/п.
<1-a)=’iooo'}е r~dr' (49)
а
Расстояние между центрами двух ионов, образующих ионную пару при
их наибольшем сближении, равно а. Если ввести обозначения
(50)
Ь=-^ (54
и подставить эти значения в уравнение (49), получаем
ъ
<‘-“>=тет(1^Л,Ьгг’*‘,г= <52>
2
“Ж (7я77 <53>
где Q (Ь) определяется таким образом:
ь
,(? (6) == e^Y-idY. (54)
2
Из уравнений (81) и (82) гл, I получаем, что константа равновесия обра-
зования ионной пары К'1 равна
К-1 =2/12(1 ~а) . (55)
3/i3/2a2c х >
При уменьшении концентрации отношение коэффициентов активности стре-
мится к единице, а2 также приближается к единице, и поэтому
(l-a)=7f-1c. * (56)
Таким образом, величина, обратная константе ионизации ассоциированной
ионной пары, равна
<5’)
Другой вывод этого уравнения принадлежит Фуоссу и Краусу [8], которые
воспользовались несколько более сложным, хотя и более общим фазовым
интегралом.
Интеграл Q (5) сводится к выражению
(2(6)= ^еуу-^у = ±[е2 -£i(2) +^(&)~4(1+4 + ^-)] > (58)
2
где Ei (х) — интегральная экспоненциальная функция
Ei(x) = e-^dt. (59)
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
57
Бьеррум составил таблицу значений Q (6) в интервале 1 <6 <15 на осно-
вании известных таблиц функций [9]. Значения Q (Ь) для интервала
15<6<80 были получены Фуоссом и Краусом с помощью асимптотиче-
ского разложении функции Ei (ж) в ряд. Это разложение становится непра-
вильным при 6 = 15, но ошибка быстро уменьшается с увеличением Ъ. Зна-
чения Q(b) приведены в табл. 9.
Для того чтобы ясно представить себе различные стороны явлений
ассоциации ионов и электролитической диссоциации и определить условия, при
Которых будет справедлива теория Бьеррума, следует остановиться на неко-
торых общих положениях. Мы будем различать два вида образования свя-
зей, благодаря которым уменьшается число ионов в растворе в результате
возникновения нейтральных частиц. Для ионов с одним зарядом можно
представить себе ассоциацию таким образом:
С+ + А- ДГ [С+А-]° СА,
где через [С+А~]° обозначена ионная пара, а через СА — педиссоциированная
молекула с неполярной связью. Первая образуется только благодаря дей-
ствию кулоновских сил, а последняя — в результате возникновения электрон-
ной связи. Кроме того, возможны также последующие реакции, как, напри-
мер, образование ионного тройника:
[С+А-]° + А- [А-С+А-]-
' [С+А-]° + С+ Jt[C+A-C+]+,
а также более сложных ионных групп.
Теория Бьеррума основана полностью на допущении о применимости
закона Кулона, т. е. принимает, что потенциал равен ф (г) = , а также
на том допущении, что ионы ведут себя как несжимаемые тела1. Зави-
симость потенциала от г для этого случая изображена на рис. 2, а. Так как
произведение е1е2 отрицательно и имеет постоянное значение, то ф (г)
является величиной отрицательной и кривая представляет собой гиперболу.
При минимальном расстоянии, на которое могут сблизиться положительный
и отрицательный ионы, кривая функции ф (г) поднимается перпендикулярно
оси абсцисс, т. е. на этом расстоянии имеется бесконечно большой потен-
циальный барьер. Интервал расстояний, в пределах которого образуются
ионные пары, равен (<? —а).
Нельзя ожидать, что теория Бьеррума будет справедлива в том случае,
когда имеются недиссоциированные молекулы с неполярной связью. Так,
например, рассмотрим такой случай диссоциации:
НАс Д Н2О Н3О+ + Ас-.
В этом случае зависимость ф (г) от г при г < / определяется квантовыми
условиями и соответствующая кривая имеет вид, изображенный на рис. 2, б.
Система молекул в левой части вышеприведенной реакции должна обла-
дать энергией, достаточной для того, чтобы возникающие из этих молекул
ионы Н3О+ и Ас~ могли преодолеть максимум потенциальной энергии
в точке d.
1 Советскими исследователями были введены в теорию электролитов выражения,
дающие одновременный учет зарядов и радиусов [В. К. Семенченко, ЖФХ, 6,
вып. 3, 285 (1932): Z. physik. Chem., 129, 176 (1927); А. Ф. К а и у с т и н с к и й,
Z. physik. Chem., 144, 187 (1929); Ю. В. Хода к о в, Элементы электростатической
химии, ОНТИ, М.—Л., 1934]. Семенченко дал расширенную трактовку кулоновского
потенциала — , введя удачный термин «обобщенный ,момент» и указал, что эта величи-
Г
на является важной электростатической характеристикой иона. (Прим, ред.)
58
ГЛАВА III
Эти соображения приводят к выводу, что для слабых электролитов О (г)
является значительно более сложной функцией от г, чем та функция,
которая принимается в теории Бьеррума. С другой стороны, мы можем
ожидать, что эта теория будет справедлива для электролитов, имеющих
Р и с. 2. Кривые электрического потенциала для случая:
а—ассоциации ионов и б—образования неполярной связи.
достаточно большое значение а в средах с малой диэлектрической постоян-
ной. В ходе последующего изложения мы встретим многочисленные примеры
применимости этой теории.
§ 7 Б. Теория образования ионных тройников и квадруполей
по Фуоссу и Краусу [10]
Применяя метод Бьеррума для расчета ассоциации ионов, можно полу-
чить выражение для константы диссоциации К3 следующих реакций:
[А-С+А-]- А- + [А-С+]<>,
[С+А-С+]+ ;± с+ + [С+А-]°.
Рассмотрим ионный тройник, состоящий из двух отрицательных и одного
положительного ионов, и допустим, что ионы представляют собой заряжен-
ные частицы сферической формы. Из ряда возможных способов образова-
ния ионных тройников рассмотрим тот случай, когда отрицательный ион
приближается к ионной паре. Пусть расстояние между ионами, соединен-
ными в ионную пару, равно а. Обозначим через 0 угол между осью ионной
пары и прямой, проведенной к центру ближайшего отрицательного иона
из центра положительного иона пары, а через г —расстояние между
приближающимся отрицательным ионом и положительным ионом пары.
Тогда расстояние между приближающимся отрицательным ионом и отри-
цательным ионом пары будет равно (г2 -ф а2 + 2ar cos 6)1/2, а потенциальная
энергия U отрицательного иона будет
U = - 1 - —— 1 ___. (60)
\ т j/r2 4- а2 + 2аг cos 0/
Если рассматривать потенциальную энергию как функцию от 9, то оказы-
вается, что потенциальная энергия имеет минимальное значение при
6 = 0, так что
<61>
Так как минимум потенциальной энергии соответствует максимальной
устойчивости системы, то наиболее благоприятные условия для образования
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
59
ионного тройника будут в том случае, когда все три иона лежат на одной
прямой.
Вероятность нахождения отрицательного иона в dV2 на некотором
расстоянии от положительного иона ионной пары, находящейся в dV1, будет
Вероятность = ne~uikT dVx dV2 = ne~v°lkTe(Uo~U)lkT dV1 dV2, (62)
где n — число свободных ионов в единице объема. В полярных координатах
с началом крординат, расположенным в центре положительного иона,
это выражение переходит в следующее:
Вероятность = пе~и<‘!кГ e<'Uu~v'> kT 2№ sin 9 dG dr dV\. (63)
Функция r2e uo,hT имеет минимум при r = R, причем R является корнем
уравнения
1 2РкТ г е2 , г (г + а)2 (64)
Если подставить значения
Г Х=== , Х~ — , «3 (65)
е2 £>2 Ьз а3РкТ (65a)
4 2РкТ ’
в уравнение (64), то мы получим
1 + 2х • (66)
х (ж + I)2 ч ь3
Минимум указанной функции соответствует корню X этого уравнения.
Параметры а3 и 63 играют ту же роль в случае образования ионных
тройников, что а и b в теории ионных пар.
Аналогично тому, как это сделал Бьеррум в своей теории образования
ионных пар, мы допустим, что ионные тройники возникают при г <; R
и 6 = 0. Если г > R, ионный тройник диссоциирует на ионную пару и ион.
Принимая во внимание эти соображения и используя уравнение (63), можно
представить константу диссоциации К3 ионных тройников в виде следу-
ющего интеграла:
e^dr $ e(Uo-^Wsin6rf6.
аз 0
(67)
Введением переменной х, которая определяется уравнением (65), это
уравнение можно привести к форме, более подходящей для численных
расчетов. Таким образом,
2-xNal Г 2
______2 \ 'У4 О
1000 J
1
2тс/Уа| г / д \
(68)
(69)
Через J (Ь3, х) обозначен определенный интеграл с переменной 6:
+1
J(b3, х)=\ ехр Г2^(1- Ш, (70)
L ж т 1 \ у х2 -|- 1 -f- Ixz J
где z = cos 6. Фуосс и Краус графически определили J (Ь3, х) как функцию
от х и затем вычислили 1(Ь3). В табл. 10 представлены полученные ими
значения I (Ь3).
60
ГЛАВА III
Фуосс и Краус [И, 106] развили также теорию диссоциации ионных
скоплений по схеме
С+А-у
А-С+
2 [C+A-JO.
Вводя допущение, что подобный квадруполь представляет собой эллипсоид
с осями ал и Ха4 (где к < ]/ 2), содержащий в центре диполь с моментом р,
расположенный параллельно главной оси, они получили выражение
„—1 А / те \3/2 р2 e?y
Л 4 = 2000 J Т5Ю
(71)
причем у определяется
уравнением
Р2
У — (Xa4)3 DkT
(72)
уравнения для относительного парциального
§ 8. Предельные
молярного теплосодержания и относительной парциальной
молярной теплоемкости
Из уравнения (38) гл. I и определения коэффициента активности /
следует, что относительное парциальное молярное теплосодержание одного
моля электролита при данной температуре равно
L2 Н2 - /у ° = - )Р' N
Если увеличить интервал концентраций, охватываемый уравнением (34),
посредством добавления эмпирического линейного члена ВГ, то выражение
для In /± принимает вид
(73)
где
2,303 V Г
------+ 2,303 вг,
(74)
р
• 1,283 • 106
(75)
Ь /± =
и
A = a{DT)~1!i . 35,57,
(76)
согласно уравнениям (19) и (35). При дифференцировании уравнения (74)
по Т и подстановке полученного результата в уравнение (73) необходимо
учитывать зависимость от температуры величин D, Г, а и В, однако жела-
тельно, чтобы в конечный результат входили обычные величины и А.
Таким образом получаем
]/Г
+-
(77)
где
И
д In D
дТ
= 2,303 № &(/)А 1 (1 + +
'(/) у
(78)
Ж(Н) = - 2,303 vRT2B - а
2d In а
дТ
(79)
(80)
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
61
Численные значения указанных параметров для водных растворов при-
ведены в табл. 11. Член а представляет собой коэффициент термического
расширения раствора, который, согласно уравнению (83) гл. VIII, равен
— dlnl'/dT1. Полные уравнения, аналогичные выражению (77), были выве-
дены Харнедом и Элерсом [12] и Харнедом и Хеккером [13] *, которые
показали, что член, содержащий В, имеет существенное значение для уме-
ренно разбавленных растворов. В случае очень разбавленных растворов
уравнение (77) превращается в предельный закон
Z2=^(H)/T, (81)
который был впервые получен Бьеррумом [14]2 без учета зависимости Г
от температуры, однако позднее был исправлен с учетом этого фак-
тора [15, 16]. Знак ^(Ц) определяется членом dlnD/dT, который является
отрицательным и обычно превышает i/T на величину, большую чем а/3.
Поэтому для воды и для большинства (если не для всех) других раство-
рителей, согласно теории Дебая—Гюккеля, L2 должно быть прямо пропор-
ционально корню квадратному из иональной концентрации при бесконечном
разведении.. _ _
Относительная парциальная молярная теплоемкость Ср^ — Ср2 s J2
получается путем дифференцирования уравнения (73) или (77) по темпе-
ратуре при постоянных давлении и составе. Таким образом, в общем виде
J2~\dT Jp,N ПдТ\ ОТ ) ’
Чтобы не иметь дела с уравнениями, слишком громоздкими для практи-
ческих целей, мы рассмотрим только предельный закон
72 = ^(Ср)/Г, (82)
получаемый путем дифференцирования уравнения (18) или (81). Предель-
ный коэффициент наклона равен
&{Ср) = 2,303 vfl ^(/) (х2 - Т -1) , (83)
где
Х^1+|т’^Д + |Та. (84)
Это уравнение дано в форме, пригодной для вычисления ^(С ) Для боль-
шинства растворителей, поскольку было показано [17], что InZ линейно
зависит от Т в широком интервале температур. Член в скобках в урав-
нении (83) можно представить таким образом:
3 { \ л.‘)т dD дЕ‘У‘ 4-2Г2 dD дг 4- 2 т дГ л.
4 дТ1 + й k-D дт) "Г DV дТ 0Тг 3 V дТ +
, /ТйГу 2T2d2D 2T2d2V~\
+ \ДЭ дт) D дТ2 3 V дТ2] ‘
В такой форме это выражение обычно встречается в литературе [18, 19].
В этом выражении а заменено эквивалентными функциями от V, через
которое обозначают либо удельный, либо общий объем раствора.
1 Переменная у, которую указанные авторы определяют с помощью уравнения,
обозначенного в их статье (И), должна иметь в числителе величину а.
2 Формула Бьеррума в действительности была выведена ранее А. И. Брод-
ским [Z. physik. Chem., 121, 1, 26 (1926)]. {Прим, ред.}
62
ГЛАВА lit
§ 9. Предельные уравнения для относительного парциального
молярного объема, расширяемости и сжимаемости электролита
Согласно уравнению (41) гл. I, продифференцированному по п2, парциаль
ный молярный объем электролита в растворе равен
у2 _ у» = Г 1 = v RT
L дР J Т, N
д In /.
При сочетании этого выражения с уравнением (74) получается
уравнение
Уравнения (75) и (76) используют для нахождения параметров
V) = 2,303 vRT ± ,
-2,зозуЯГ^(/)л|(^-^-з)
& иг иг /
И
(86)
общее
(87)
(88)
(89)
^(V)=2,303y .
(90)
Коэффициент сжимаемости р = д In Y/dP известен для ряда растворителей,
но численные значения dh\D/dP обычно отсутствуют. Однако значение
dinD/dP для воды при комнатной температуре известно. До сих пор у нас
нет никаких данных о коэффициентах д In a/дР и д In B/дР, и поэтому
два последних члена уравнения (87) нельзя вычислить. Таким образом,
это уравнение может быть использовано лишь при крайних разведениях,
когда можно применять предельный закон
V2-V°2 = ^(V) У Г. (91)
Относительная парциальная молярная расширяемость электролита
в растворе определяется так:
(92)
Путем дифференцирования уравнения (91) по температуре получаем соот-
ветствующее предельное уравнение
В2-£1 = ^(£)/Г, (93)
в котором [20]
= - 2.303 , RT [ 1 + .) -
<94>
В настоящее время было бы совершенно бесполезным выводить более общее
выражение, подобное уравнению (87), потому что даже предельный коэф-
фициент наклона §fe(E) уже содержит неизвестный температурный коэф-
фициент члена dinD/dP. Гукер [20] определил порядок величины этого
члена и пришел к заключению, что S(e> должно равняться примерно
4 • 10~13 J] у у Zy при 20°.
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
63
Путем аналогичных рассуждений можно получить предельное выра-
жение для кажущейся молярной сжимаемости, которая определяется
следующим образом:
е К2 -К°2=- --Ц7 Г°а) = &(К) УТ\ (95)
Предельный коэффициент наклона [19] выражается уравнением
^(K) = 2,303 v/ГТ+ 15(Ц^)2-Jg*] • (96)
Гукер определил величины отдельных членов этого уравнения и получил
значение S>(x)=9,l • 10~4 J] v, zj см3 на атмосферу при 25°.
§ 10. Увеличение или уменьшение растворимости нейтральных
веществ и электролитов при добавлении соли к их растворам
Хорошо известно, что добавление соли к водному раствору, содер-
жащему нейтральные молекулы какого-либо вещества, часто приводит
к уменьшению его растворимости. Это явление соответствует увеличению
коэффициента активйости молекулы данного вещества. Подобное умень-
шение растворимости («высаливание») не является общим правилом,
и можно привести примеры противоположного эффекта увеличения рас-
творимости («всаливания»). Полная и надежная теория этих явлений отсут-
ствует, но можно показать, что порядок величины экспериментальных
результатов может быть вычислен теоретически, если рассматривать только
влияние кулоновских сил, не учитывая других факторов.
Дебай и Мак-Олэй [21] вычислили из обратимой электрической работы
парциальную свободную энергию молекулы по отношению к чистому рас-
творителю. При этом ионы рассматривались как несжимаемые сферические
частицы. Позднее Дебай [22] развил теорию, которая основана на влиянии
ионов на распределение молекул неэлектролита, и вывел выражение длц
концентрации этих молекул как функции расстояния от иона. Эта теория
была развита далее Гроссом [23], который учел влияние ионной атмосферы.
а) Теория Дебая и Мак-Олэя. Допустим, что ион представляет собой
шар с радиусом Ъ,. Тогда потенциал на его поверхности будет равен
ej/Dbj. Работа заряжения п, ионов такого рода может быть найдена
с помощью уравнения (7):
nj Л — 1
Ж(тгД = 2
1 /. = 0
«р. d/.
-Db~
2Dbj '
(97)
Рассмотрим раствор, содержащий пу . .. п, . . . ns ионов в см3, образовав-^
шихся при диссоциации п' молекул электролита, и вычислим электрическую
работу разряжения ионов при большом разведении в растворителе (воде)
с диэлектрической постоянной Do и работу их заряжения в среде с ди-
электрической постоянной D. Изменение диэлектрической постоянной от До
до D может быть вызвано прибавлением к раствору либо электролита,
либо неэлектролита. Общая электрическая работа, совершенная над систем
мой, будет равна
S „ S „
.... n niei кп ЩП
= <98>
1 у 1 J
ГЛАВА III
64
С помощью уравнения (32) гл. II можно вычислить добавочную электри-
ческую работу, совершенную против действия потенциала, обусловленного
ионными атмосферами:
s пу л =1 „ .
1 11-0
--22$ (")£—й 2•<* w
1 1 Л=0 1
Появление х>, в этом выражении обусловлено тем, что х пропорциональное.
Общая электрическая работа процесса заряжения, полученная путем сло-
жения уравнений (98) и (99), в первом приближении может быть приравнена
увеличению работоспособности системы. Таким образом,
л ,.е?
1
2Д0
3D 2 п$-
I 3
(100)
В первом приближении можно
смеси в следующем виде:
представить диэлектрическую
постоянную
D = Do (1 — fin — ₽'«'),
(101)
где п и п' — соответственно число молекул неэлектролита и электролита,
а Р и р'— эмпирические постоянные. Пренебрегая членами, содержащими
более высокие степени пип', получаем из двух последних уравнений
ди - ±1 У I 2Х У
- 27JO 21 Ь + 2/J0 2
1 3 1
2 S
n&i % v-i
~bT~~^Da 2 м-
* 1
(102)
Это уравнение переходит в предельный закон Дебая и Гюккеля при усло-
вии, что п' = 0 и р = О. В последующем изложении будут рассмотрены
только те случаи высаливания и всаливания электролитов и неэлектроли-
тов при крайних разведениях, которые обусловлены изменениями диэлек-
трической постоянной в соответствии с уравнением (101). В этих условиях
I _ Рп 5Л nje] . >?'п' ,fjeJ
W-2Z)0 2 b. I" 2D0 A bi
1 3 1 1
(103)
Это выражение соответствует «чистому» высаливанию (или всаливанию)
и не зависит от членов, содержащих х.
Из этого уравнения можно определить составляющую химического
потенциала неэлектролита (Др<8) на молекулу), обусловленную добавлением
электролита и, наоборот, уменьшением его концентрации. Так, для нейтраль-
ной молекулы
^) = ^ = i2^ = ^ln/(s) . (104)
1 3
или
•"/<«-^2^- б»)
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
65
Последйее выражение представляет собой уравнение Дебая и Мак-Олэя.
Как видно, при положительном |3 макроскопическая диэлектрическая посто-
янная среды уменьшается; при этом / увеличивается и происходит высали-
вание. Если р отрицательно, происходит веаливание. Поскольку мы приняли
упрощенную картину и при выводе сделали несколько приближенных
допущений, уравнение (105) является предельным и его можно рассматри-
вать лишь в качестве первого приближения.
Выражение для «солевого эффекта», оказываемого электролитом на рас-
твор того же самого электролита, можно получить из уравнения (103) путем
дифференцирования по п', При дифференцировании следует учесть, что
= Таким образом,
in/±(S)— ^kTD 2 ъ. (106)
1 3
Исключая $'п' с помощью (101), получаем
Ы , — (До—Д)£2 х? У)
если в растворе нет никаких неэлектролитов, кроме растворителя (воды).
Это уравнение описывает также солевой эффект одного электролита, оказы-
ваемый на раствор другого электролита при большом разведении. В этом
случае суммирование производится по всем ионам обоих электролитов.
Другие уравнения, описывающие солевые эффекты ионов, были также
выведены с помощью подобной упрощенной схемы. Батлер [24] получил
уравнение, идентичное (105), с той разницей, что в знаменателе вместо Do
находится D. Его уравнение для взаимного высаливания ионов отличается
от (107) тем, что в знаменатель вместо D2 входи^ D2.
Выражение для действия неэлектролита на коэффициент активности
электролита при крайних разведениях нетрудно получить из уравнения (98).
Так, электрическая работа Wt переноса зарядов п' молекул электролита
из чистой воды с диэлектрической постоянной Do в смесь вода—неэлектролит
с диэлектрической постоянной D равна
(10S)
• _ п'е2 5Л V? я'е2 чл ‘’у2/
t 2D Zj & 2Z>0 XJ & •
1 3 1 3
Приравнивая эту работу увеличению работоспособности и дифференцируя
по п', получаем 1 1 3
или = S4;- <‘09)
Это уравнение выведено Борном [25а] Ч Величина 1g /±(t> представляет собой
первичный эффект среды 1g /0, который будет рассмотрен в гл. XV, § 7.
б) Теория Дебая. Ниже будет получено выражение для зависимости
концентрации нейтральных молекул в присутствии ионов от их расстояния
1 См. также [256].
5 г. харнед, Б. оуэн
66
ГЛАВА III
от данного иона по методу Дебая [22]. Рассмотрим сферический ион радиуса Ь,
находящийся в среде, состоящей из двух смешивающихся неэлектролитов.
Для идеальной смеси этих веществ
И1«1 + ^2«2 = 1, (110)
где г?х и «2 — молярные объемы чистых компонентов, деленные на число
Авогадро, а пг и п2— число молекул каждого вида в 1 см3. Свободная
энергия элемента объема, согласно уравнению (61) гл. I, равна
[??i (<pi + йТЧп А\) Д- п2 (^>2 + кТ In TV2)] dV, (111)
где N-! и ZV2— молярные доли, a ^х и <р2 — стандартные свободные энергии,
приходящиеся на одну молекулу, когда концентрация выражена в моляр-
ных долях. Мы придерживаемся обозначений Дебая, согласно которым
Fly Fx и т. д. представляют собой свободные энергии и парциальные сво-
бодные энергии на один моль. Поскольку мы допустили, что растворы
являются идеальными, то коэффициенты активности равны единице. Для
определения общей свободной энергии раствора необходимо к свободной
энергии добавить составляющую, обусловленную полем, создаваемым ионом.
Согласно законам электростатики, эта величина равна энергии элемента
объема на расстоянии г от иона и выражается так:
(OX)2 ,1у Lrfr С г J iy е2 ,у П121
8-r.D aV 8nD dV ~ 8ъг*О aV ’
где X— напряженность электрического поля, равная . Общая сво-
бодная энергия системы равна, таким образом,
£ йх (?х + + ц2 (<р2 + AT1 In-/V2)J (ИЗ)
причем интегрирование распространяется на весь объем раствора. Пусть
мх и и2 изменились на Вих и 8и2, тогда
8Ф=$ + +
+ + (114)
Согласно условию равновесия, требуется, чтобы 8Ф = 0, т. е. чтобы Ф при-
нимало минимальное значение, при условии, что общее число молекул
остается неизменным или что nxdV и гг2dVимеют постоянное значение.
Из уравнения (110) следует, что
«х8их + С-ЛП2 = 0.
Положим
8их = v2bx; Ъп2 = — «х8ж,
где Zx — произвольная вариация х. После подстановки этих значений урав-
нение (114) приобретает вид
8Ф = ( dV 5 Д (Т1 + кТ 1» N, - -L. «Г) _
+ (115)
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
67
Так как
dV 4- Ъпг dV = («2 — ^i) 8^ dV = О,
то получаем, что
^Зжс/7 = 0. (116)
Согласно этому выражению, необходимо, чтобы при 5« = 0 и &Ф=0 член,
^стоящий в скобках под знаком интеграла в уравнении (115), был равен
постоянной величине, т. е.
-И1(?2+ЛГ 1п^2-g^A^)=const- (117)
Обозначим через 7VJ и молярные доли на большом расстоянии от иона
(/*= оо); тогда
const == у2 (<р2 + кТ In 2VJ) — (<р2 + кТ In 2Vg)
и
, i Nx , N3 e* f dD dD\ 1 .. .0V
y2 Ь yo vi111 - 8те/гТ£)2 (v2 9n^ V3 9n^ j ri . (118)
Отсюда видно, что если известна зависимость D от состава раствора, то
можно вычислить концентрации обоих компонентов на расстоянии г от иона.
Следует отметить, что эта теория не учитывает изменения D в зависимо-
сти от г.
Простой и полезный результат можно получить для случая смеси,
которая содержит небольшое количество компонента 2. Так как7У1 = 1—TV2
и 2VJ = 1 — №, то левая часть уравнения (118) при этом равна
, i — N3 , ТУ2
Мп 1 —дт» У11п7У§ ‘
Допустим, что компонент 2 «высаливается», вследствие чего N2/№ < 1, и до-
пустим, кроме того, что значение TV2 мало. При этих условиях у нас
останется только один член —v± In N2 /N%, и уравнение (118) приобретает вид
, Ж е2 1 / dD dD 4 1 (119)
TV" SnkT VjD2 Va 9n3 1 dn2 Jr1 ’
Пусть 1V e* 1 (v dD v, dD \ (120
Л 8ъкТъ1 Ds V2 дпг 1 dn3 ) ’
тогда -(-Г N3 = N%e , (121)
т. е. R представляет собой характеристическое расстояние, уравнения ь!ожно рассматривать как основной результат Если D является линейной функцией от N± и N2, т. е. Последние два теории Дебая.
+ (122)
то
Я — 8^RT DI ’ '
где Fi — объем одного моля компонента 1.
5*
68
ГЛАВА III
Отсюда видно, что эффект высаливания очень быстро увеличивается
при уменьшении г. В зависимости от того, окажется ли D1— D2<0 или
— Da>0, будет происходить либо всаливание, либо высаливание.
Из предыдущих уравнений могут быть получены важные уравнения
для солевых эффектов. Из уравнения (110) получаются в общем случае
выражения
1 П1 = = ’ (124)
которые при малом количестве компонента 2 приобретают вид
Рассмотрим насыщенный раствор компонента 2 в компоненте 1 в при-
сутствии ионов в единице объема. Тогда, согласно уравнению (121),.
среднее число молекул, окружающих данный ион, равно
£1= ^тг2йИ = ^Ц = (1 - ) dv] . (126)
Интегрирование производится по объему, в котором расположен ион. При
r=co,TV2 совпадает с и равно концентрации в любой точке, не содер-
жащей ионов. Число молекул Z2 компонента 2 в месте расположения дан-
ного иона до прибавления электролита будет, следовательно, равно
Zl^^-^dV. (127)
Отношение числа молекул, присутствующих в растворе после добавления
электролита Z2 к Z2, равно, таким образом,
р -И
z2 .
(128)
Интегрирование числителя производится по объему, в котором расположен ДД
данный иен. Поэтому ЯК
3nzJdF = l, (129) ®
где — общее число ионов в 1 см3. Д|
Пусть iK
°° _[«) w!
а = 4гс\(1 — е \rl)r2dr, (130) Ж
Ь
тогда
= f = (131) Я
где /2 и /2 — рациональные коэффициенты активности компонента 2 соот-
ветственно в присутствии и в отсутствие ионов. Так как при изучении
Солевых эффектов мы допускаем, что растворы неэлектролитов являются
идеальными, то /2 обычно принимается равным единице.
При обосновании и выводе уравнения (131) мы точно следовали
Дебаю [22], но для нахождения численной величины члена следует тВ
несколько видоизменить это уравнение с тем, чтобы определить различные
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
69
физические условия, при которых его можно применять. По уравнению (122)
= = П1С] , (132)
так как концентрация компонента 2 в разбавленном растворе равна
Удобно определить численную величину | а | следующим образом:
тогда
I’ln'-ir;''"
—. А А • 1000 _
Rj = Hr.HTIR । а ।
(133)
(134)
и знак члена зависит от того, будет ли Di больше или меньше D<>.
В первом случае (I), когда /Д > D2,
D = Dj (1 — | a | c) (135)
и мы вводим величину (/7)i ,
ОО /Ну\4
(J7)i = 5 (1-е '4 )r2dr. (136)
bi
Во втором случае (II), когда Dx < D2,
D = D1(l + \^\c) (137)
и мы вводим величину (J7)n ,
с 4-Г
(J7)n=^ (1-е
(138)
Таким образом, уравнение для солевого эффекта (131) принимает форму
“ 1 ~ 1000 2 Ji ei‘ (139)
Входящие в выражевия для J j интегралы определяются с помощью
рядов [26, 27]
(/,), = Й," [1,21 -1 (=()’• ]; Л, >*/. (КО)
(141>
+ «;<»/ (К2)
Гросс [28, 26а] дал дальнейшее развитие этой теории путем учета влияния
ионных атмосфер. Для учета этого влияния надо добавить к энергии эле-
мента объема, находящегося на расстоянии г от иона [см. уравнение (29)
гл. II и (112)], следующую величину.
е2 (1 + ха) е 2гг
8*r*D
(143)
I
70 Г л А В A III I
В результате добавления этой величины уравнения (140) и (141) принимают
вид
(Jy)I(z)=^[l>21-|^y-|zftJ.+ ...] ; Hj>a/ (144)
И
; Rj<aj. (145)
§ 11. Теоретический предельный закон для поверхностного натяжения
растворов электролитов
Вагнер [29], а также Онзагер и Сэмарас [30] развили теорию влияния
междуионного притяжения на поверхностное натяжение растворов электро-
литов. Для решения этого вопроса необходимо вычислить уменьшение коли-
чества растворенного вещества в поверхностном слое [уравнение (87) гл. I]:
„ да да
и затем увеличение поверхностного натяжения ® путем интегрирования
этого уравнения.
Благодаря силам притяжения, действующим между ионами внутри
раствора, йли, более точно, благодаря отталкиванию ионов от поверхности
под действием силы электрического изображения, количество растворенного
вещества вблизи поверхности меньше, чем в глубине раствора. Экраниру-
ющее влияние ионных атмосфер радиуса 1/х определяет предел расстояния,
на котором действуют эти силы. Теория Вагнера приводит к уравнениям,
решение которых очень затруднительно. Путем введения дальнейших упро-
щающих допущений в теорию Вагнера Онзагер и Сэмарас получили простое
решение, которое, как показывает анализ, может служить хорошим первым
приближением. ,
Поверхностный слой и адсорбционный потенциал. Концентрация рас-
творенного вещества с (я) на расстоянии х от поверхности раздела дается ii
уравнением Максвелла—Больцмана
—W (х)
с (х) = се кТ . (146) ‘
Здесь с представляет собой концентрацию растворенного вещества в объеме
раствора [с = с(оо)], a W (х) — работу переноса молекулы или иона рас-
творенного вещества из объема раствора в точку, находящуюся на расстоя- ;
нии х от поверхности. Величина W (я) называется «адсорбционным потен- .
циалом». Каждый вид ионов обладает своим собственным адсорбционным
потенциалом W( (х).
Для определения W (х) Вагнер воспользовался методом электрического
изображения. Рассмотрим ион с зарядом е, находящийся на расстоянии х
от фиксированной поверхности. Ион находится в среде с диэлектрической
постоянной D, причем эта среда соприкасается с воздухом, имеющим ди-
электрическую постоянную D', равную единице. Построим электрическое
отражение иона, как показано на рис. 3. Электростатический потенциал ф ;
в любой точке среды будет равен ;
_ ei Г (Д~ Д') ej _ е/ , (Д —1) е/ м 47')
“ Dr, (D Л Д') Дг2 ' Д^МДИ) Дг2 ’ k >
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
71
где тд — расстояние этой точки от иона, а г2 — ее расстояние от точки,
в которой расположен отраженный заряд1. Второй член в правой части
этого уравнения представляет собой потенциал, обусловленный наличием
поверхности раздела, который выражается через величину отраженного за-
ряда [(Z) — 1)/(Z) + 1)] ej- Этот член соответствует силе, действующей на ион:
oW(z). Г 1)е; Л _(£) — !) еу
дх L(jD+D Drl Jr2=2x ](Л + 1) 40ж2 ’
причем потенциал этой силы равен
. ч (D— 1) ej
IK ( х\ = :—тт“ 7'7,— •
1 ' (D + 1) ' IlDx
(148)
Вагнер показал, что такой адсорбционный потенциал должен был бы
привести к бесконечному увеличению поверхностного натяжения, так как
количество адсорбированного растворенного
вещества было бы при этом бесконечным:
оо W (ас)
— Г2 = с^(1 — е кГ )б(т=оо, (149)
о
что представляет собой, конечно, парадоксаль-
ный результат. В случае ионных растворов
экранирующее влияние ионной атмосферы
ограничивает эффективную область действия
электростатических сил расстоянием 1/х, и,
следовательно, это затруднение исчезает.
Рис. 3. Схема для вычисления
электростатического потенциала,
обусловленного зарядом е и его
изображением,
[(D-l)/ (D 4-1)1 е~е.
Как было показано для растворов электроли-
тов, потенциал подчиняется уравнению (23)
гл. II:
Г-7ф = х2ф. (150)
Далее, поскольку концентрация у поверхности отлична от концентрации
в объеме раствора, х2 является функцией z, если уравнение записано
в цилиндрических координатах, а именно:
<151)
Решение этого уравнения ограничивается условием г = 0 при z = X; если
z= — х, то также г = 0; если z > 0, x2(z) выражается уравнением (146)
и уравнением, определяющим х2 [уравнение (22) гл. II]; если z < 0, х2 (z) = 0.
На поверхности раздела z = 0, <|> непрерывно и dty/dz меняется на множи-
тель D.
Чтобы получить приближенное решение этого уравнения, Вагнер до-
пустил, что D' равно не единице, а нулю. Кроме того, он заменил x2(z)
через х2(ж). Онзагер и Сэмарас упростили задачу, введя дополнительное
допущение, что
х2 (z) ~х2 (со) = х2. (152)
При этих условиях W (х) определяется уравнением
W (ж)
Юе-2>ж
4Dx
(153)
1 Простой вывод уравнения (147) можно найти в книге Томсона [31].
72
ГЛАВА III
Как видно, это выражение отличается от уравнения (148) на множитель е~их-
при условии, если пренебречь членом D' ( = 1). Путем сочетания уравнений
(153) и (146) получаем для значения концентрации на расстоянии х от
поверхности выражение
с(я;) = сехр(^^ е-2«) = сехр , (154)
где
? = J ' <155>
представляет собой характеристическое расстояние, применяемое в теории
ассоциации ионов Бьеррума [уравнение (50)]. Это выражение является
простейшим предельным уравнением. Если ввести в него среднее расстояние
сближения ионов, то
РИ (Ж) = —Li- е-2хх (156)
4 ' 1 н- ха iDx ’ v '
(„.т
-Т+^Г -17") (157)
И
00 л
-Г2 = с^ [l-expf^-^——)] dx. (158)
Онзагеру и Сэмарасу удалось решить уравнение (158). Полученное таким
образом значение Г2 было затем подставлено в уравнение (146). После
интегрирования было получено выражение для увеличения поверхностного
натяжения Да, или а — <т0, где а0 — поверхностное натяжение растворителя.
Для предельного случая, когда не учитывается среднее расстояние
сближения ионов, эти авторы выразили полученный результат в следующем
виде:
= + = <159>
Аналогичным образом уравнение, которое содержит размеры ионов, имеет
вид
(160)
где Y и У' —сложные функции (гл. V, табл. 12).
Для 1,1-электролитов предельный закон приобретает вид
79,517 , 1,143 10~13(.ОГ)3
a-a0 = ^-clg----------. (161)
Если растворителем является вода при 25°, то
a-ao = l,O124clg^. (162)
ЛИТЕРАТУРА
1. Е. Giintelberg, Z. physik. Chem., 123, 199 (1926).
2. Т. Н. Gronwall, V. К. L а М е г, К. S a n d v е d, Physik. Z., 29, 358 (1929).
3. H. M U 1 1 о г, Physik. Z., 28, 324 (1927); 29, 78 (1928).
4. L. О n s a g e г, Chem. Rev., 13, 73 (1933).
5. V. K. L a M e r, T. H. Gronwall, L, J. G г i e f f, J. Physical Chemistry,
35, 2245 (1931).
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
73
6. N. Bjerrum, Kgl. Danske Vidensk. Selskab., 7, No 9 (1926).
7. R. M. Fuoss, Chem. Rev., 17, 27 (1935).
8. R.M. F u о s s, С. А. К r a u s, J. Am. Chem. Soc., 55, 1919 (1933).
9. E. Янке, Ф. E м д e, Таблицы функций с формулами и кривыми, ГТТИ, М.— Л.,
1949.
10. a) R. М. F и о s s, С. А. К г a u s, J, Am. Chem. Soc., 55, 2387 (1933); б) 57, 1
(1935).
11. R. М. F и о s s, J. Am. Chem. Soc., 56, 2017 (1934).
12. H. S. H a r n e d, R. W. Ehler s, J. Am. Chem. Soc., 55, 2179 (1933).
13. H. S. H a r n e d, J. С. H e c k e r, J. Am. Chem. Soc., 55, 4838 (1933).
14. N. В j e г г и m, Z. physik. Chem., 119, 145 (1926).
15. G. S c a t c h a r d, J. Am. Chem. Soc., 53, 2037 (1931).
16. 0. Gatty, Phil. Mag. [7], 11, 1082 (1931).
17. G. A k e r I 6 f, J. Am. Chem. Soc., 54, 4125 (1932).
18. V. K. L a M e r, I. A. Co w p er t h w a i t e, J, Am. Chem. Soc., 55, 1004 (1933).
19'. F. T. G и c k e r, Jr., Chem. Rev., 13, 111 (1933).
20. F. T. G и c k e r, Jr., J. Am. Chem. Soc., 56, 1017 (1934).
21. P. D e b у e, J. M с А и 1 a y, Physik. Z., 26, 22 (1925).
22. P. D eby e, Z. physik. Chem., 130, 56 (1927).
23. P. Gross, Monatsh. Chem., 53, 54 (1929).
24. J. A. V. В и t 1 e r, J. Phys. Chem., 33, 1015 (1929).
25. a) M. Born, Z. Physik., 1, 45 (1920); б) H. S. H a r n e d, N. N. T. Samara s,
J. Am. Chem. Soc., 54, 1 (1932).
26. P. Gross, K. Schwarz, a) Monatsh. Chem., 55, 287 (1930); 6) Sitz. Akad. Wiss.
Wien., 139, 179 (1930).
27. P. Gross, Sitz. Akad. Wiss. Wien., 138, 449 (1929).
28. P. Gros s, Monatsh. Chem., 53, 445 (1929).
29. C. Wagner, Physik. Z., 25, 474 (1924).
30. L. 0 n s a g e r, N. N. T. Samara s, J. Chem. Phys., 2, 528 (1934).
31. J. J. Thomson, Elements of Electricity and Magnetism, Fourth Edition, Cambrid-
, ge University Press, 1909, p. 169—171.
Глава IV
ТЕОРИЯ НЕОБРАТИМЫХ ПРОЦЕССОВ В РАСТВОРАХ
ЭЛЕКТРОЛИТОВ
В гл. II мы ознакомились с основными положениями теории, необхо-
димыми для изложения динамики ионных атмосфер. Используя для решения
этой проблемы общее уравнение непрерывности (39) гл. II, а также вводя
некоторые другие важные представления, можно вывести точные уравнения,
которые позволяют вычислить обусловленные кулоновскими силами элек-
тростатические составляющие вязкости, электропроводности и диффузии
разбавленных растворов электролитов. В создании и дальнейшем развитии
этой сложной теории участвовали Дебай и Гюккель, Фалькенгаген
и Онзагер. Так как для решения всех этих вопросов требуется применение
весьма специализированных математических методов, то мы не будем при-
водить полное изложение указанной теории. Нами будут рассмотрены прин-
ципиальные физические основы теории и изложены важнейшие этапы выво-
дов. Это облегчит читателю знакомство с литературой, к которой он может
обратиться, если пожелает получить более глубокие познания в этой области.
Вслед за теорией вязкости, электропроводности и диффузии будет рассмот-
рена теория влияния высокой частоты переменного тока и сильных электри-
ческих полей на электропроводность. В окончательном виде полученные тео-
ретические закономерности будут иметь форму, удобную для вычислений.
Связанные с теорией вопросы, более важные для практических вычислений,
подробно рассматриваются ниже, в гл. V, в которой приведены упрощенные
уравнения, а также таблицы соответствующих численных констант.
§ 1. Время запаздывания
а) Время релаксации ионной атмосферы (Дебай и Гюккель). Всякий
раз, когда ион электролита подвергается возмущению, например действию
электрического поля или градиента концентраций, центральный ион сдви-
гается по отношению к своей атмосфере и последняя утрачивает симмет-
ричную (сферическую) структуру. Если мгновенно удалить возмущающую
силу, то ионная атмосфера стремится возвратиться к своему нормальному
равновесному состоянию. Для этого перехода от состояния возмущения
к нормальному состоянию требуется некоторое конечное время.
Предположим, что появилась возмущающая сила, которая вызвала
изменение плотности заряда р. Если после этого источник возмущения
мгновенно удалить, то система будет возвращаться в первоначальное состоя-
ние со скоростью , пропорциональной р:
dt ‘ т “
Время t, которое мы назовем «временем релаксации», является величиной,
обратной константе скорости.
Рассмотрим возмущение, обусловленное электрическими зарядами, и
соответствующее изменение плотности заряда. Если ф — потенциал возму-
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
75
щающей силы, а а — электропроводность, то сила тока, согласно закону
Ома, равна
1=-а7ф. (1)
Скорость изменения плотности во времени дается уравнением непрерывности
и равна
g=-V.i = aV-V<p. (2)
Далее, по уравнению Пуассона,
(3)
а отсюда, в сочетании
После интегрирования
1
тр •
с уравнением (2), получаем
rfp _ 4irap
~di'~ +^D~
получаем
p(£) = p(°) е о =р(О)е
(4)
(5)
Отсюда видно, что
D
4ка
(6)
впервые получена Максвеллом.
Эта зависимость была
Пользуясь этими соотношениями, можно легко получить выражение для
времени релаксации ионной атмосферы. Так как сила —?ф;- заставляет
г-I C.-V-i;
г-ион двигаться со скоростью —оде, ф; или------и так как сила тока
равна скорости, умноженной на общий заряд, то
-2
Pl U
(7)
и, следовательно,
s
п>ег
УГ ’
i = l
причем и pt представляют собой подвижность и коэффициент
иона, которые были определены в гл. II, § 3. Пусть
S п
Г)
(Т
(8)
трения
V/
(9)
Тогда после введения величины
получаем
:2, согласно уравнению (22)
гл. II,
(Т
2 *4)kT T-
'i = •
(10)
2
S
Следовательно, согласно уравнению (6), время релаксации равно
1
т =-----— .
;.Ч-Т I/O
(И)
76
ГЛАВА IV
Если p1 = pa = p3 = pi, то 1/е = l/р, и
' = <12)
*
Это упрощенное уравнение будет вами использовано при предварительном
кТ
рассмотрении теории вязкости. —представляет собой коэффициент диффузии.
Р i
Чтобы определить время релаксации, выразим уравнение (И) для
электролита, который диссоциирует на два вида ионов, через известные
величины. Эквивалентная электропроводность иона Xj равняется силе тока
при градиенте потенциала в 1 в на 1 см, возникающей при прохождении
одного грамм-эквивалента ионов. Таким образом,
М = 96500и{ = 96500^ = 96500 | 6i | <о{ (13)
и, следовательно,
л / 300мI /л /\
= (14)
Здесь подвижность щ выражена в электростатических единицах, а и, отно-
сится к градиенту потенциалов в 1 в на 1 см.
Тогда
1 • 300Х,-
0)4 “ ?f — 96500 \zt I г •
Подставляя полученное значение pi в уравнение (9), мы найдем, что для
электролита симметричного типа, диссоциирующего на два вида ионов,
ту- 1 300 с Х1 + Ха \
1/1 е 96500 <|г1 1 + 1 \) ’
а отсюда с помощью уравнения (11) получаем
___(I zi I + I 1Л 15,34 • 10 8 ,, 7.
Ч 4 + ^2 ) кТ'л2 ’
причем z1 = |z1| —со знаком z±, a z2 = |z2| —со знаком z2.
Очевидно, что при данной температуре это уравнение принимает после
подстановки значения х2 простую форму
т %-у 10'10сек., (18)
где с — концентрация, выраженная в долях нормальности, к± — характери-
стическая постоянная для данного электролита в данном растворителе. Так,
для хлористого калия и соляной кислоты при 18° fcj равно соответственно
0,55 и 0,19. В 0,001 н. водных растворах электролитов т имеет величину
порядка 10~7--10_8 сек.
б) Время запаздывания процесса образования ионной группы. При
последующем рассмотрении скоростей диссоциации молекул и рекомбинации
ионов с образованием нейтральных молекул нам нужно будет знать кон-
станты скорости и время запаздывания этих реакций. Если щ и п, предо-
ставляют собой число ионов двух видов, которые рекомбинируют, a —
число диссоциирующих молекул, то уравнения скоростей химических реак-
ций будут иметь вид
-dS = An^-KA^’ <19>
НЕОБРАТИМЫЕ , ПРОЦЕССЫ В РАСТВОРАХ
77
где А и КА — соответственно константы скоростей рекомбинации и диссоциа-
ции, л—константа диссоциации, равная •, гДе а0 является степенью
диссоциации в невозмущенном состоянии. Если п — число молекул электро-
лита и а —степень диссоциации в общем случае, то
v;i=(l — а) п и = =
Из уравнения (19) и этих соотношений следует
л = . = dt. (20) Ап^-КА^ АК
После интегрирования получаем л 9~ао)- 1g г + с', (21) Ап (2— а0) “о Ь । “о ' a -f- 1 — “о
или - =се~^’, (22) “ + Г”2- 1—а0
где <23>
Таким образом, если равновесие диссоциации нарушается под действием
внешней силы и если эта возмущающая сила мгновенно исчезает, то
система возвращается в обычное невозмущенное состояние. Величина <с'
является мерой того времени, которое требуется для этого процесса, и
аналогична времени релаксации ионной атмосферы. Так как теория таких
процессов была впервые исследована Ланжевеном [1], мы будем называть
т' «временем запаздывания по Ланжевену». Приведенные выше основные
соотношения будут использованы нами при изучении влияния сильных
полей на диссоциацию слабых электролитов (см. § 7).
§ 2. Вязкость. Теория Фалькенгагена1
а) Предварительное обсуждение теории вязкости. Теория изменения
вязкости среды под влиянием кулоновских сил, действующих между ионами,
была впервые успешно развита Фалькенгагеном [2] для случая бинарных
электролитов. При изложении этой теории мы будем следовать методу
изложения Онзагера и Фуосса [За]. Им удалось получить общее решение,
пригодное как для отдельных электролитов, так и для их смесей. Так как
уравнение для потенциалов имеет такую же общую форму, но несколько
более простой вид, чем уравнение для вычисления электропроводности,
то теория вязкости будет изложена сначала.
Прежде всего будет дано упрощенное изложение вопроса для того,
чтобы показать, как электростатические силы, действующие между ионами,
влияют на вязкость, и примерно определить порядок величины этого
эффекта. Это послужит для. ознакомления с основными положениями тео-
рии динамики вязких жидкостей, которые необходимы для изложения
общей теории2.
1 Теория электропроводности, приведенная в § 3, является более простой по
сравнению с излагаемой здесь сложной теорией вязкости, и ее можно изучать неза-
висимо от последней. Данный порядок изложения совпадает с тем, который был при-
нят Онзагером и Фуоссом, исходя из логических соображений.
2 Исчерпывающее изложение этого вопроса можно найти в книге Пэйджа [4а].
78
ГЛАВА IV
Рассмотрим раствор, находящийся между двумя параллельными
плоскостями А и В, которые удалены друг от друга на h см, как пока-
зано на рие. 4. Пусть плоскость А неподвижна, а В движется с посто-
янной скоростью v' в направлении оси х. Допустим, что в направлении
оси у имеется постоянный градиент скорости, который определяется соот-
ношением
^ = 0, «г = 0. (24)
Коэффициент Sxy if-й составляющей напряжения сдвига [46] связан
с соответствующей составляющей скорости деформации [4в] сдвига уравнениемт
Р и с. 4. Схема для вычисления вязкости.
которое определяет вязкость vj, а именно:
= (2S>
В случае простого ламинарного течения
^« = 1^. (26)
Коэффициент вязкости ц представляет собой напряжение, отнесенное
к единице площади, которое передается от каждого слоя жидкости к со-
седнему слою при градиенте скорости, равном единице; иными словами,
это та сила, отнесенная к единице поверхности раздела, с которой часть
среды, находящаяся над данной плоскостью, действует на часть среды,
находящуюся под этой плоскостью.
В растворе электролита часть силы внутреннего трения обусловлена
деформацией ионной атмосферы. В невозмущенном растворе каждый ион
окружен атмосферой из ионов противоположного знака, находящихся
в среднем на расстоянии 1/х; как было показано (гл. II), это распределе-
ние обладает сферической симметрией. Под влиянием градиента скорости
в растворе эти атмосферы деформируются, превращаясь из сферических
в эллипсоидные. Электростатические силы и тепловое движение стремятся
восстановить сферическую форму ионных атмосфер. В результате влияния
этих двух противоположных тенденций, а также вследствие того, что время
релаксации т является конечной величиной, установится некоторая ста-
ционарная деформация. Если считать относительную скорость деформации
равной dvx/dy, тогда стационарная деформация будет tdvx/dy. Так как,
согласно уравнению (12), т = р{/хаА71, то деформация ионных атмосфер
^ОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
79
является величиной порядка
?i dPjc
*?кТ ду ’ ' '
Сила, действующая между двумя ионами с зарядами е, находящимися
на расстоянии 1/х, равна e2*2/D, а полная сила, действующая между ионом
и его атмосферой, равняется этой величине, умноженной на 1/х, т. е.
e2x/Z). Умножая это значение на величину смещения ионной атмосферы,
которая дается уравнением (27), получаем
ea?i dvx
ду ’
причем по порядку величины это значение должно быть равно количеству
движения, переданному от иона к его атмосфере. При подстановке значе-
ния х2 из уравнения (22) гл. II мы получаем выражение для электроста-
тической составляющей напряжения S*y
(28)
Это уравнение справедливо с точностью до коэффициента пропорциональ-
ности 1/480^, который может быть получен только при более общем рас-
смотрении величины деформации ионной атмосферы. Таким образом, для
1
случая, когда — = «ц = о>2 = ... = ws,
Напряжение, действующее между молекулами растворителя, определяется
уравнением
S°vx = ^, (30)
где т)0 — вязкость растворителя. Следовательно, составляющая вязкости,
обусловленная влиянием ионной атмосферы, равна
^* = £- <31>
Этот результат Фалькенгагена подтверждает вывод, сделанный Джонсом
и Долом [5] на основании экспериментальных данных, согласно которому
в разбавленном растворе пропорционально квадратному корню из кон-
центрации.
б) Краткое изложение общего решения проблемы вязкости. Общее
уравнение движения вязкой жидкости [4г]1 имеет вид
+ v-Vvd = FcZ — VP-f- у t]VV • v + • Vv, (32)
где F — сила на единицу объема и Р — давление. Согласно уравнению
непрерывности, если жидкость несжимаемая, то V.v = divv = 0. Если при-
нять плотность d равной единице и пренебречь вторым членом в левой части
уравнения для малых скоростей течения, поскольку в него входит квадрат
скорости v, то мы получим для постоянных скоростей течения выражение
•rJV.Vv = VZ5-F. (33)
Рассмотрим случай общего движения жидкости в точке, отмеченной век-
тором г, в которой скорость v обладает компонентами vx, vy, vz', Sxy—ком-
понент силы внутреннего трения по оси у, приходящийся на единицу поверх-
1 Следует отметить, что в этом частном случае через F обозначена общая сила.
80
ГЛАВА IV
ности плоскости, перпендикулярной к оси х,—выражается уравнением (25).
Существуют два фактора, под влиянием которых возникает напряжение.
Одним из них является взаимное трение молекул растворителя, а вторым —
электростатическое взаимодействие между ионами. В результате, если
обозначить через Smn общий коэффициент матрицы напряжения для пере-
дачи количества движения в чистом растворителе, а через Smn— ту же
величину для движения раствора, то
Smn — Smn— Smn, (34)
где Smn — составляющая напряжения, обусловленная взаимодействием
ионов. Далее,
71 —7’о = 71*> (35)
где »)* — увеличение вязкости, вызванное добавлением растворенного веще-
ства. После подстановки последнего выражения в уравнение (33) получаем
7ioV.Vv = VZ5-(F + 7]*p.vv). (36)
Этот важный результат показывает, что электростатические силы, дей-
ствующие между ионами, можно рассматривать как некоторое добавление
к объемной силе.'
Количество движения передается от ионов к растворителю посредством
трения, причем ионы движутся по отношению к растворителю. Если для
внутренних частей раствора V-Vv равно нулю, то не происходит передачи
количества движения между ионами и растворителем. В этом случае силы,
действующие на поверхностях раздела (Л и В, рис. 4), равны ± г,дох/ду,
а не ±’codvx/dy. Наличие разности т* дох/ду можно объяснить тем, что
ионы перемещаются относительно растворителя только вдоль поверхностей
раздела па расстояниях от последних, не превышающих 1/х.
Эти соображения, а также общее уравнение непрерывности (39) гл. II
содержат все основные физические предпосылки, необходимые для решения
проблемы вязкости. Математическая трактовка этой проблемы очень сложна,
и обычно при изложении метода Онзагера и Фуосса предполагается, что
читатели знакомы с линейными преобразованиями квадратичных форм, ь на-
шем изложении будут даны некоторые этапы вывода, из которых будет
видно, что для вычисления напряжения SyX, как объемной силы, должны
быть известны потенциалы ионных атмосфер, которые в случае необратимых
процессов могут быть определены только с помощью общего уравнения
непрерывности.
Найдем напряжение путем вычисления общей объемной силы1. С этой
целью рассмотрим снова рис. 4 и вычислим полное количество движения
ASyx, переданное в результате действия электростатической силы в напра-
влении х через поверхность А плоскости у — уа, а также предположим,
что градиент скорости одного из компонентов является постоянным:
где индексы 1 и 2 относятся к переменным х и у. Электростатическая
сила, действующая между любыми двумя ионами, равна
D rs ’
где г21 или г — расстояние между ионами. Составляющую напряжения дают
только те из ионов, которые находятся на противоположных сторонах
1 Здесь и на стр. 86 авторы применяют необычный термин «объемная сила».
О смысле этого термина можно судить из последующего вывода (стр. 80, 81). (Прим, ред.)
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
плоскости у — у0, поэтому мы учитываем лишь те пары ионов, которые
отвечают условию
У1 < Уо < Уг, (37)
причем через гг и г2 обозначено расположение соответствующих ионов.
Количество движения, перенесенное от /-иона, находящегося в dVlt к ионам,
находящимся в dV2, равно
7Г 2 ein/i(rai)
а полная величина количества движения для всех пар ионов в dV\ и dV2
равна
2 ¥dV± 2 ^2^=2 ¥’ fa (мdV*dV* ->=
j i ji
= -l^fij^dVidV^. (38)
ij
Таким образом, согласно уравнению (4) гл. II, в этом уравнении появи-
лась функция распределения /,;(г21). Интегрируя по всем элементам объема,
которые удовлетворяют условию (37), можно вычислить величину полного
напряжения. Таким образом, если A=^dxdz, то х— составляющая пе-
ренесенного количества движения—равна
- = $ Ш й 2 (r^) WdV* dV*> (39)
VI < VO < V2 ji
интегрирование по и zx дает коэффициент А, а интегрирование по уг
при постоянном у21 дает коэффициент уг1. После соответствующих преобра-
зований пределов интегрирования й опуская нижние индексы у величин
ж21, У21> dV\ и d,V2, получаем интеграл
-ф-ОО
(40)
—со ji
Исключая jji (г) с помощью уравнения Пуассона, можно ввести в уравне-
ние величину потенциала вблизи от /-ионов и получить интеграл
+ оо
= (41)
—оо ;
Для упрощения математических преобразований вводится функция (г),
которая связана с потенциалом. При этом интеграл принимает вид
+<»
£ Ш 2 (r) ] > dv- (42)
-оо j :
Здесь через а* обозначена составляющая скорости деформации сдвига,
а через — вторая частная производная по координатам. В рассматри-
ваемом случае й| является единственной составляющей напряжения сдвига
а*, которая не исчезает. Следовательно,
да
(г) = $/(/•)]. (43)
6 Г. Харнед, Б. Оуэн
82
ГЛАВА IV
Поэтому
-j-oo
—да з
При переходе к полярным координатам и интегрировании по переменным
значениям углов получаем
A* __al у Г d*y-xgj(r) ,
° и*- 30 п!&1 3 (г dr)2 а
3 о
и при интегрировании по частям получаем
J
Следовательно, электростатическая составляющая вязкости »]* равна
(44)
3
Из этих рассуждений видно, что для решения проблемы вязкости
требуется вычисление v-VE,(O), которое может быть осуществлено только
с помощью общего уравнения непрерывности (35) гл. II. Для того чтобы
получить из уравнения (35) гл. II дифференциальное уравнение для потен-
циалов в подходящей форме, необходимо прибегнуть к нескольким довольно
сложным заменам переменных.
Так как эта часть вывода сводится главным образом к математиче-
ским преобразованиям, мы остановимся на ней лишь в той мере, в какой
это необходимо для наших целей.
При изучении проблемы вязкости третий и четвертый члены общего
уравнения непрерывности (35) гл. II исчезают, так как при этом отсут-
ствуют внешние силы типа к, и к;. Чтобы придать уравнению (35) гл. II
более удобный вид, необходимо найти подходящие выражения для величин
скоростей. В данном случае, когда движется весь раствор, расстояния
между ионами r2i=—г12, и расстояния щ и г2 являются независимыми
переменными. Так как
Г2 — Г1 = Г21 ~ Г12>
то все члены можно преобразовать так, чтобы они были функциями от двух
из этих переменных (гп г21) или (г2, г12). Такое преобразование функций
было осуществлено. Оказывается, что при решении всех этих вопросов
более удобно употреблять потенциалы ф/ вместо функций распределения
так как при такой подстановке получаются s функций вместо —•
G помощью уравнения Пуассона /)г и заменяют потенциалами фу и ф',
а эти потенциалы в свою очередь заменяют на функции Еу и с помощью 'Л
соотношения
V.V^(r) = a^V-V^ (r).
После этих преобразований уравнение непрерывности (35) гл. II
мает следующий вид:
, 8
2 5
nie^i Ajr
(r) — /)/fy 2
' i=0
6^6,0) Л;
Wi + W: 1 4 '
4тг
(D/гГ)2
шг +
(45)
прини-
(46)
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
83.
Как будет показано в дальнейшем, левая часть этого уравнения идентична
левой части уравнения (80) для ф), с которым мы встретимся при изучении
проблемы электропроводности. Этим объясняется необходимость сложных
преобразований и введения Е/.
Общее решение некоторых сложных дифференциальных уравнений,
включающих суммирование, проще всего можно получить с помощью мат-
ричной алгебры1. Пользуясь матричным исчислением, Онзагер и Фуосс
получили общее решение уравнения (46) при соблюдении соответствующих
граничных условий задачи. Подробное изложение сложных алгебраи-
ческих методов, применяемых для решения этого и других дифференциаль-
ных уравнений, получающихся в теории необратимых процессов растворов
электролитов, потребовало бы много места и отвлекло бы наше внимание
от физической стороны вопроса. Подробности можно найти в работе Онза-
гера и Фуосса, а также в других работах, на которые мы будем ссылаться
в ходе изложения.
Для случая электролита, который диссоциирует на два вида ионов
(.5 = 2), решение уравнения (46) можно выразить в форме
Г_?___(q . __ .(^1 ?*) А ”1 //уч
v 2DkT |_4х2 V7 р / (1 +
где р, р3 и д* определяются уравнениями
е1 , е2
пх-----F л2 —
7 _ Ц>1 - а>2
nsel + nael
n^l + n2e|
(48)
(49)
(50)
n1efw1 + n2elw2
+ n2e2) + o>2)
Из уравнения (47) нетрудно получить электростатическую составляющую
вязкости т(*, которая определяется уравнением (44)3:
при условии, если воспользоваться уравнением (22), определяющим х, для
исключения 2 fy6;. Следует отметить, что если опустить второй член
в правой части уравнения (51), то получается выражение, идентичное урав-
нению (31), которое было выведено с помощью приближенных методов.
Второй член в скобках включает р3, т. е. вторую степень подвижности
или эквивалентной электропроводности. Как было указано, уравнение (51)
дано в форме, удобной для перехода к практическим единицам, и совпа-
дает с уравнением Фалькенгагена [7а]. Если Wj = <о2 — . .. = <os = <в, то второй
член в правой части уравнения исчезает; Онзагер и Фуосс преобразуют
в этом случае уравнение
* X
7)* — --------
480 тс ш
(52)
1 В гл. I книги Куранта и Гильберта [6] содержатся стандартные методы, необ-
ходимые для решения данного вопроса.
2 Уравнение (51) в статье Онзагера и Фуосса не приводится. Этот результат полу-
чен авторами при консультации профессора Онзагера.
6*
84
ГЛАВА IV
посредством подстановки значения коэффициента трения р из формулы
Стокса [уравнение (62)]:
1 и
— = Р = Ь'.-'пг
со 1 ’
где г —радиус иона. Путем сочетания этого соотношения с уравнением
(52) мы получаем
Таким образом, в первом приближении относительное понижение вязкости
пропорционально отношению радиуса иона к радиусу его атмосферы.
§ 3. Электропроводность. Теория Онзагера [3]
А. ОБЩИН СООБРАЖЕНИЯ И ПРЕДВАРИТЕЛЬНЫЙ ОБЗОР ТЕОРИИ
Чтобы рассмотреть проблемы электропроводности и диффузии с общей
точки зрения, Онзагер и Фуосс сначала развили общую теорию движения
ионов в гомогенном поле сил, обозначенных через к1; к2, . . ., ks (на ион),
действующих на различные виды ионов 1, . . ., я, и силы к0, действующей
на молекулы растворителя, которая уравновешивает остальные силы со-
гласно уравнению
иоко= — 2 «Зч- (54)
i-1
Такая система сил эквивалентна совместному действию электрического
поля Е = — \~ф, где ф — электрический потенциал, и градиентов концентра-
ций ..., Vns при условии, что сохраняется электропейтральность
согласно уравнению
S S
2 etVni^V У пге( = V (0) = 0. (55)
i-1 г=1
Правильность этого положения можно доказать с помощью следующих
термодинамических рассуждений.
При такой системе градиентов и электрических сил для сохранения
равновесия должны быть приложены силы, иные ио своей природе и про-
тивоположные по знаку, — к0, — к1( ..., — ks, так что
к,= -V(h +efO), (56)
где — обычный химический потенциал, а р,- — полный химический потен-
циал или «электрохимический потенциал» (гл. X, § 7). Можно принять, что
скорости, обусловленные различными причинами, вызывающими движение
ионов, можно суммировать. Следовательно, скорости, вызванные градиента-
ми — Vp{, должны быть равны скоростям, обусловленным силами к0, кх, . . ., к6,
потому что эти последние силы уравновешивают приложенные силы
— к0, — к1; . . ., —ks. Так как при постоянной температуре и давлении, согласно
уравнению Гиббса — Дюгема, 2 = то можно показать, что силы, опре-
i
деляемые уравнением (56), подчиняются условию равновесия, которое выра-
жается уравнением (54).
Рассмотрим важный раздел теории, относящийся к скоростям переноса
ионов. Если бы между ионами не действовали кулоновские силы или если
бы ионы были изолированы, то скорость иона по отношению к среде, в
которой он движется, была бы равна
Vi = *J = kiMi. (57)
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
85
Этот закон движения ионов был бы справедлив для идеального раствора,
в котором силы, возникающие между ионами, настолько малы, что
они не могут отразиться на применимости этого закона. При рассмот-
рении реальных растворов электролитов необходимо иметь в виду два
эффекта:
1. Непосредственное взаимодействие между ионами. Полный анализ
этого явления, при котором учитывается броуновское движение ионов,
принадлежит Онзагеру.
2. Электрофоретический эффект, который заключается в том, что ион
перемещается в среде, которая в свою очередь находится в движении.
Поскольку ион окружен ионной атмосферой, которая движется в противо-
положном направлении, то он перемещается по отношению к движущейся
среде. На наличие эффекта электрофореза впервые указали Дебай и Гюк-
кель.
а) Предварительное определение сил взаимодействия. Под действием
силы к ион движется со скоростью, приблизительно равной
1г .
= (58)
Когда приложена внешняя сила, ион стремится выйти из своей атмосферы,
однако смещенный ион будет все же притягивать свою отстающую атмосферу,
которая будет все время возникать вокруг него по мере его движения.
Если т —время релаксации, то ион будет опережать свою атмосферу на
расстояние порядка
к
= (59)
Отношение этого расстояния к толщине ионной атмосферы к,тх / р; служит
мерой относительной несимметричности атмосферы. Приближенное значе-
ние направленной силы Лк/, вызванной этой несимметричностью, получается
умножением этой величины на общую силу, действующую между ионом
и его атмосферой. Таким образом, после подстановки значения т из урав-
нения (12) получаем
Дку
D Р/
DkT '
(60)
Оказывается, что это значение Дк/ с точностью до некоторого численного
множителя хорошо согласуется с результатом, который получается при
использовании более точного общего метода.
Эта сила обусловлена несимметричностью атмосферы по отношению к
центральному иону. Если некоторые из ионов атмосферы перемещаются под
влиянием внешних сил, то центральный ион становится частью их атмо-
сфер. Связь этих сил становится взаимной, и для полного учета этой вза-
имосвязи нужно рассмотреть броуновское движение центрального иона,
так как оно способствует релаксации его собственной атмосферы. Этот
важный'эффект учитывается в основном уравнении Онзагера [уравнение
(39) гл. II].
б) Предварительное рассмотрение электрофоретического эффекта. Вы-
числение электрофоретического эффекта проще всего сделать для случая
электропроводности, когда имеется электрическое поле X и когда
4 = 0;
к, = еу-Х (/= 1, . . ., s). (61)
86 .
Г ЛАВ А IV
Ион с зарядом еу обладает ионной атмосферой с зарядом — еу, и эта атмо-
сфера находится под действием силы — Хеу. Эта сила стремится двигать
атмосферу и вместе с ней жидкость, содержащую эту атмосферу, в направ-
лении силы —Хеу. Центральный ион будет также переноситься средой
в направлении, противоположном его движению под действием силы—Хеу.
Скорость этого встречного движения центрального иона можно вычислить,
если принять, что весь заряд атмосферы —еу расположен на расстоянии 1/х
от центрального иона и распределен на сферической поверхности с радиу-
сом 1/х и что движение этой сферы подчиняется закону Стокса для дви-
жения шара в вязкой жидкости. Таким образом,
куХ Хе;.х
6лт| 6т1Т] ’
(62)
где дуу — скорость внутренней части сферической оболочки, а — вязкость
среды. Таким образом, мы приходим к выводу, что среда во внутренней
части оболочки движется с этой скоростью и что центральный ион будет
перемещаться против этого потока. Это уравнение справедливо для наибо-
лее существенной части эффекта электрофореза, хотя оно и не учитывает
некоторые тонкости этого явления.
Если бы центральный ион не обладал атмосферой, он двигался бы
со скоростью ку/^у, однако вследствие наличия атмосферы ион находится
под действием силы ку — Дку и движется по отношению к окружающей
его среде со скоростью (ку — Дку)/ру. Следовательно, окончательная ско-
рость vy равна
у. = Ду/+ЬГ^.. (63)
Ту
Для случая электропроводности получаем
Так как оба уравнения — для силы взаимодействия между ионами (60)
и для электрофоретического эффекта (62)—содержат первую степень от х,
то оба эти эффекта пропорциональны квадратному корню из концентра-
ции. Все сказанное выше служит введением к изложению физических
представлений, лежащих в основе общей теории.
Б. ОЧЕРК ОБЩЕЙ ТЕОРИИ ЭЛЕКТРОПРОВОДНОСТИ СОГЛАСНО ОНЗАГЕРУ
а) Электрофоретический эффект. Последующее изложение общей тео-
рии электропроводности и диффузии потребует более глубокой разработки
теории электрофоретического эффекта по сравнению с той, которая была
дана выше. Причиной электрофоретического эффекта является объемная
сила, действующая на среду, окружающую ион. Если Пу —средняя кон-
центрация /-ионов, то среднее значение внешней силы, действующей на
единицу объема, будет
8
У nyky==nsk„,
у = 1
где буква а, стоящая в индексе, обозначает суммирование. Эти силы пере-
даются молекулам растворителя, концентрация -которых составляет nQ
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
87
в единице объема. Все силы уравновешиваются согласно уравнению (54),
поэтому
4-поко = О. (65)
В элементе объема dV вблизи /-иона объемная сила равна n7-sk0 dV, так
как присутствие /-иона влияет на среднюю концентрацию ионов.в объеме dV.
Общая сила, действующая на элемент объема dV, равна
(n7Bks + noko)d7 = (n/s —ns) k0 dV. (66)
В сферических координатах сила, действующая на сферический слой, на-
ходящийся на расстоянии г от центрального иона, будет равна
4кг2 (n/s — ns) ks dr. (67)
Эта сила действует в направлении приложенной внешней силы и равно-
мерно распределяется по поверхности сферы. Эти условия аналогичны тем,
для которых применим закон Стокса *,
(68)
и, следовательно, можно применить этот закон в данном случае. Сила
заставляет йаходящиеся внутри сферы частицы двигаться со скоростью v.
Далее, необходимо выразить (n7i — п,) как функцию от г с помощью
закона распределения Максвелла — Больцмана, иными словами,
(69)
Онзагер и Фуосс сохранили второй член разложения экспоненциальной
функции в ряд, так как он представляет интерес для теории диффузии.
Для определения величины потенциала применяется второе приближение
теории Дебая и Гюккеля:
*/Г е™ е~хг~1
D L(1 + *а) г J
(70)
Это уравнение легко получить из уравнений (25) гл. II и (29) гл. Ш. Из
уравнений (67), (69) и (70) можно найти выражение для искомой силы,
действующей на сферический слой:
, „I eiea е*а e~xr 1 Г е, е„ "12 б>~2хг1
с/г = 4кг 0 + ха) — F J (14-ха) J 4 } Tl'k'dr, (71)
которое можно записать в более простой форме:
& dr = 4~ (— Л1ге-хг 4- Л2е~2хг) dr. (72)
Применяя уравнение Стокса, получаем для скорости выражение
(73)
Общую скорость получаем путем интегрирования по всем значениям г от а
до со, следовательно:
Ду,
9 Г л „~у-а Д
1 Ср. [8].
(74)
88
ГЛАВА IV
где Ei(2ха)— экспоненциальная интегральная функция:
00
Ei(x)= $ = —0,5772 —+ x — (74а)
X
е е„ ех-а "1 2
пак<; Ei (2ха).
(75)
После подстановки значений А± и Л2 окончательное выражение для эффекта
электрофореза принимает вид:
2 е/еаПок.
(1+za) З-Г] \_DkT (Ц-ха)
Для электрических сил, когда kj'=Xej, и при малых концентрациях, когда
можно пренебречь ха, первый член в правой части этого уравнения сов-
падает с правой частью уравнения (62), полученного при предварительном
рассмотрении теории электрофоретического эффекта. В этом можно убе-
диться путем подстановки в это уравнение значения х согласно уравне-
- нию (22) гл. II.
В случае диффузии электролита, который диссоциирует на два вида
ионов, s = 2 и к, -- Vy/шу. Если v —общая скорость диффузии, то уравне-
ние-(75) принимает вид
Av• = — — fП1в1 e/v 1 , I
1 Зт] \ «j "Г" o>2 J DkT x (1 za)2 ' 37
ve.
дат'Н™)’ (76)
где
. . е2хаEi(2ха) ,пс. ч
<Р (ха) =—. (76a)
1 х ' (1 + ха)2 v '
При последующем изложении теории диффузии нам нужно будет найти
составляющую, вызванную электрофорезом. Произведение числа
на электрофоретическую скорость равняется величине потока
числу ионов, проходящих в секунду через единицу поверхности,
кулярной к направлению диффузии.
Таким образом,
И0Н0В Пу
AJy, или
перпенди-
AT А Г ' 2 ni ei n° e°
AJy _ nyAv; - [ -
d\J.
Если AJy = A3JI/oka, то ,
ках в приведенном выше уравнении,
жение электрофоретического эффекта.
, ПЯ «а nj ef ?(“) 1 ,
Зт] (DkT)2 J ка’
(77)
т.
е. эта величина равна члену в скоб-
На этом мы заканчиваем общее изло-
1
б) . Общая теория ионных сил в применении к частному случаю
электропроводности и диффузии. В случае электропроводности и диффу-
зии исчезают первые два члена общего уравнения непрерывности (39) гл. II,
которые содержат скорости V (г2) и V (гД, относящиеся к движению всего
раствора в целом. Для изучаемых нами стационарных полей это уравне-
ние имеет вид
“{[кг ' V2 /ц (/•)] + *; [ку Vx/?y г)]—егЫЩЩу?2-?2фу(г21) —
— еуЫуПЩуУ 1 • (— г21) — 1щ7сТ72 • V2/л (r21) — uiyfcTVj • Vif'u (— r21) = 0. (78)
Принимая во внимание некоторые симметричные соотношения между пере-
менными, а также то обстоятельство, что к • V/д можно заменить на
<?/»
, не нарушая общности уравнения, последнее уравнение можно упро-
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
89
стить таким образом:
(Wikt — Wjkj) — е^тцп^ -V% +
+ ejWjniTijV • V tpi — (®i + <ь;-) kTV • yfji = 0. (79)
С помощью s уравнений Пуассона
i=l 1
из этого уравнения можно исключить функции распределения /д. Далее, со-
гласно уравнению (33) гл. II,
,0 (, eiei е-'-т\
1ц-П;Пг(^1 DkT r J,
и следовательно, можно исключить также /д. Произведя это преобразова-
ние и используя некоторые другие простые соотношения, в которые входят
6) и можно выразить окончательное дифференциальное уравнение для
потенциалов в .такой форме:
„ 4 7^ X 1 'I,"С; , Alt х 1 71-С-С .Ш-
г-1 3 г=1 г 3
= У п.е2е. £ ЛСГ Y (80)
(DkT)2 со{ + шу 1 1 3 дх \ т ) ' '
Как видно, левая часть этого уравнения совпадает по своей форме с левой
частью уравнения для потенциалов (46), выведенного при общем изложении
теории вязкости.
в) Предельный закон для электропроводности электролитов при слабых
электрических полях и при низких частотах [9]. Электролиты, диссоции-
рующие на два вида ионов. Прежде чем перейти к общему решению
проблемы электропроводности, справедливому для любого числа ионов s,
мы для большей ясности изложения рассмотрим более простой случай,
когда электролит диссоциирует на два вида ионов, т. о. когда s = 2. С этой
целью мы воспользуемся уравнением (80), которое в данном случае будет
иметь вид
(V. У)2 ф'. __ у. Уф'. ±L у. у.у,
' / • ।1 DkT а>! -р со2 '1 DkT «j + ш3 12
= 4тсТ е2°-'2 —п2е2ег д /'е~УГ \ ,g .,
DkT (Oj 4- а>2 DkT dx\rJ '
Заметим, что проводимости k, и k, заменены в этом уравнении на Хе2
и Лех, где через X обозначена составляющая внешнего электрического поля
в направлении оси х. Кроме того, в этом случае должно выполняться
условие
«161^1 + Паб-афг = 0 (82)
и поэтому должно быть справедливым равенство
^=<14 (83).
так как согласно условию электронейтральности
«1^1 + п2е2 = 0. (84).
30
ГЛАВА IV
После преобразования с учетом этих соотношений уравнение (81) приобре-
тает вид
_4<Г /е2<«2 —д (е~^Т\ /ок\
DkT к «>! + о>2 J DkT дх < г J ’ * ’
Введем следующие обозначения:'
х2 = 7*х2, (86)
4 ~~ (ei — е2) (ш1 + “2) (87),
о -^gle2 DkT * (88)
Тогда уравнение (85) приобретает вид
(V.W;-x*V.V^=Sx^(4^) . (89)
После интегрирования может быть выражено в виде степенного ряда
относительно г, а именно;
«’й.[-(Т)+(^)'-1 W
'Следовательно, величина поля ДХ1; вызванного возмущением атмосферы,
равна
“.“-'««'МпТ?)'11 (91)
Это и есть уравнение Онзагера [9] в его первоначальной форме.
Этот результат, а также результат, полученный для электрофоретиче-
ского эффекта, потребуются нам для вычисления подвижности, ионной элек-
тропроводности и эквивалентной электропроводности электролита. Общее
поле, действующее на изучаемый нами ион, равно В результате
электрофоретического эффекта скорость иона была меньше, чем величина
(X + &Xj). Учитывая все эти обстоятельства, можно найти результиру-
ющую скорость в направлении оси х:
„ / Д-У,- е,х \
Vj = X Qey ш, + —’ - gCJ . (92)
Последний член в этом уравнении представляет собой поправку на электро-
форетический эффект, которая вытекает из уравнений (64) или (75), если
пренебречь квадратичными членами. Подвижность представляет собой
скорость при разности потенциалов, равной единице, т. е. Uj — JVjI/X.
Таким образом, для величины подвижности, выраженной в практических
единицах, получаем
“у = зббОе/1 °’’4' т~1е»Н — ’ (93)
| с,-1 о>у/200 представляет собой предельную подвижность tty при бесконечном
разведении, а ку = 96500 Пу. После подстановки этих величин и значения х
-из уравнения (4) гл. III получаем
, , \Х: „ 96500 1е(| х
'-j х Ч 1800ТГГ]
(94)
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
91
или
28,98 |2,.|
х '} T[(Z)Z)1/2
Для частного случая, когда s = 2, вместо kXj/X можно подставить его
значение из уравнения (91), а вместо х —его значение из уравнения (4)
гл. III. Таким образом,
(95)
где
- 1,970-10®/ 9*' \. , - о , 28,98 | zy |
(Л)— (,О7’)3/2 \1 + }<^*^212г ' ‘ Э
Выражая подвижности ионов в уравнении (87) через соответствующие
электропроводности, получаем
__ I ziz21______М + Mj)
(I Z1 | + I z2 I) (I z2 | ^1 + I Z1 1 ^2)
Если написать уравнения (95) для обоих видов ионов данного электролита
и сложить их, получается уравнение
А = Л°-^(Д)Г1/2, (97)
где
„ 1,970- 10» / 9* , о t 28,98 (| Zj | + | z2 |)
s<«=)1 1 + —кот)1'- " ’ ( ’
так как, согласно закону Кольрауша,
до =(99)
Это выражение еще больше упрощается в случае электролита с сим-
метричным типом валентности, для которого |zi| = |za| = z, следовательно,
q* =1/i, g*/(1 4-}/g*) = 0,2929 и
_ 5,770 • 10s 2 . 0 57,96z
<Л)— (Z)Z)3/2 Z +r1(D7’)I/2‘
(100)
Оказалось, что это уравнение является очень важным, так как с его
помощью можно истолковать результаты многолетних экспериментальных
работ в области электропроводности сильных электролитов, а также опре-
делить константы диссоциации слабых электролитов со значительно боль-
шей точностью, чем это было возможно до создания теории междуионного
притяжения.
Прежде чем рассматривать в следующем параграфе применение урав-
нения (94) к наиболее общему случаю, разберем важный частный случай,
когда $=2 и | z2| = 2| zj. В этом случае
g* _ 0,2929 • 2,2761 f 101 'J
1 + /<Т*~1 + П + 0,8165 /1 + Л ’
если ввести число переноса Г® = >,?/(}.? +ко иона с меньшей валентностью zx.
Значение числителя, которое равно точно 2/3, представлено в виде произве-
дения двух множителей 0,2929 • 2,2761 для того, чтобы облегчить пользо-
вание табл. 13. После подстановки выражения (101) в уравнение (98)
92
ГЛАВА IV
получаем
5,770 • 105 2,276| z1Z.JA° 28,98 (| z, | + | z2 |)
(ДГ)3/2 (1+7’5+0,8165 + т|(7>7’)1/2
(102)
Электролиты, диссоциирующие на любое число ионов [За]. Чтобы опре-
делить ^Xj/X в уравнении (94) для общего случая электролита, диссоции-
рующего па s > 2 ионов, необходимо найти общее решение уравнения (80).
Эту задачу успешно решили Онзагер и Фуосс с помощью матричной алгебры
аналогично тому, как это было сделано в случае теории вязкости. Оконча-
тельное полное решение, которому можно придать формы, пригодные для
численных расчетов, имеет вид
ДА;
1,970 ЮЧ’1/2
(ДТ’)3-'2
п—0
(103)
Подставляя это выражение
получаем
в уравнение
для электропроводности ионов (94),
- } _ Г С970 • 1°6
~ L (Д7’)3/2
28,98 | Z; |
(ОТ)1/г
(104)
Сумму ряда 2 можно
вычислить с помощью следующих соотношений.
Коэффициенты сп равны
Со = |(2-/2),
(105)
причем значения ) представляют собой коэффициенты перед членами
ряда (1 + ж)1/2, т. е. — 1/2, Vg, —Vie, +5/i2s> —Vase Для соответственно
равного 1, 2, 3, 4, 5. Таким образом, первые шесть коэффициентов равны
с0= 0,2929,
Cj = — 0,3536,
с2 = 0,0884,
с3 = -0,0442,
с4= 0,0276,
с.= -0,0193.
Далее,
Г; = 40) = Zj 1
где tji — элемент матрицы, равный
tji-ojt 2j НйШ;г+ + +,
k
В этих уравнениях определяется выражением
Гг ”iei
Т ~~ Хп -в2 ’
(106)
(107)
(108)
(109)
(110)
__ ^V-tZi л
J
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
9»
а символ Кронекера S;i равен
8я=| 1 вол,, / = Л (111)
( 0, если ] =f=i.
Значение связано с элементом h,i матрицы Н [Онзагер и Фуосс, урав-
нения (224) и (225)] следующим образом:
tji = 2hji — ?jji = 2h[j — Ъ-ji. (112)
При использовании этих уравнений нужно следить за тем, чтобы
знак Zj совпадал со знаком соответствующего заряда иона. В простейшем
случае, когда s = 2 и zr = —z2, очевидно, что r® = zy. Нетрудно также
показать, что в этом случае г;п> = 0 и поэтому 2сп^п) превращается в c0Zj,
т. е. 0,2929 Zj, а уравнение (104) сводится к уравнению (94). Для случая
s = 2, | z± | = | z21 и <7* = 1 */г уравнение, эквивалентное (95), можно получить
из уравнений (104) —(109). Для случая смесей, когда s = 3 или больше,
указанный алгебраический метод становится чрезвычайно сложным. Мы
будем иметь возможность применить эти выражения при рассмотрении
некоторых, особенностей электропроводности смесей электролитов.
§ 4. Диффузия электролитов. Теория Онзагера [3,6]1
а) Некоторые общие положения. Коэффициент дуффузии 3) обычно
определяется законом Фика, т. е. соотношением
J=wv=— ЗЯп, (ИЗ)
где J —поток, V —скорость и п — число молекул растворенного вещества в см3.
Поток обычно определяют по отношению к некоторой плоскости, которую
считают неподвижной. В некоторых случаях это определение является
удобным, но оно не всегда удобно при изучении диффузии электролитов,
так как при смешении растворов происходит заметное изменение объема.
Мы будем определять поток любого растворенного вещества по отношению
к некоторому определенному участку, который движется вместе с раство-
рителем. Объемную скорость можно определить так:
(114)
где Fo, V\, . . ., Vs — парциальные объемы молекул растворителя и ионов
растворенного вещества !,...«.
Выраженный уравнением (ИЗ) закон можно написать в форме
J= -3Vn = -ЭПГр, (115)
где [i—химический потенциал; отсюда
э=(Хи®- <116>
Для идеального раствора
дч._ RT
дп, п ’
1 Германе [10а] предложил более общую теорию диффузии, которая не ограни-
чивается условием сферической симметрии ионной атмосферы. Мы не пытались рас-
смотреть его общие уравнения, поскольку при аналогичных условиях они сводятся
к уравнениям Онзагера. Хотя уравнения Германса могут быть применимы при несколь-
ко более высоких концентрациях, чем уравнения Онзагера, они вряд ли приложимы
к таким концентрациям, для которых имеются непосредственные экспериментальные
данные по диффузии. Германе основывает экспериментальную проверку своей теории
на измерениях диффузионных потенциалов. См. [106].
94 Г Л А В А IV
и, следовательно,
7?7, = ШЗ?.
В случае концентрированных растворов это уравнение неприменимо в связи
с отклонением этих растворов от идеального поведения и поэтому должен
быть введен коэффициент активности; при этом
= + , (117)
где С (= а/п) — коэффициент активности растворенного вещества, если
п выражено в молях на см3. Если можно было бы полностью выразить
зависимость S> от концентрации через отклонение раствора от идеального
поведения, то величина имела бы постоянное значение. Ни теорети-
ческие соображения, которые будут приведены в дальнейшем, ни фактиче-
ский материал не подтверждают этого допущения. представляет собой
подвижность, т. е. скорость при условии, что разность потенциалов, обус-
ловленная силой к =—Vp, равна единице.
Уравнению (115) можно придать общую форму:
S
(118>
k=i
если допустить, что имеется линейная зависимость между скоростью и гра-
диентом потенциалов растворенных веществ. Изучая соотношения между
переменными для необратимых процессов, Онзагер [11,За,в] показал на осно-
вании чрезвычайно общих соображений, включающих принцип микроско-
пической обратимости, что коэффициенты матрицы симметричны, т. е.
= (119)
Этот важный результат означает, что поток ионов i-го вида при действии
силы, равной единице, приходящейся на единицу количества ионов к-го вида,
равен потоку ионов к-го вида при действии силы, равной единице, приходя-
щейся на единицу количества ионов i-го вида.
б) Теория диффузии простого электролита. В случае равтвора электро-
лита, содержащего ионы двух видов, условием отсутствия электрического
тока является наличие одинаковой скорости как у катионов, так и у анио-
нов, т. е.
Vi = v2=v; J1 = «1v; J2 = n2v. (120)
При малых концентрациях можно пренебречь взаимодействием между
ионами, и в этом случае (120)
V — k1co1 = к2со2= — o^Vpj = — co2Vp2. (121)
Если р— химический потенциал молекулы, которая диссоциирует на Vj
анионов и v2 катионов, то мы получаем
k = Viki-E v2k2 = — Vp. (122)
Из последних двух уравнений следует, что
к’“(»к‘=(5)к-
kt =•. ~P1 Vp = -----------Vp
^1Р1 + ^2Р2 •Щ«1 + ч20,1
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
95-
И что
Если раствор идеальный, то
Vp = (vx 4- v2) RT Vn/n,
и, следовательно,
J = hv = - (V1 +-2) кТ^п. (123)
•*1“2 +
Это выражение представляет собой простой закон диффузии электролитов,
выведенный впервые Нернстом [12]. Если принять во внимание силы
междуионного взаимодействия, получаем
Vp = (vi + v2) кТ V In (:±тг),
где С± — коэффициент активности электролита.
То обстоятельство, что при диффузии как положительный, так и отри-
цательный ионы перемещаются с одинаковой скоростью, имеет существенное-
значение. Так как
= ^2®2 >
то нетрудно показать, что те члены в уравнении непрерывности (39) гл. II,
которые характеризуют возмущенное состояние, сокращаются. Вследствие
этого упрощения ионные атмосферы сохраняют симметричную форму, а это-
обстоятельство в значительной степени облегчает решение поставленной
задачи.
С другой стороны, некоторое влияние оказывает электрофоретический
эффект, так как он является результатом объемной силы, действующей
в ионной атмосфере. Величину поправки, учитывающей электрофоретиче-
ский эффект, можно получить из уравнения (76) путем подстановки 1/щ
вместо со;:
л Г 2 . ' е1
Д V> = V L “ 3^ (П161Р1 + + +
+^(n1eiei + n2e?2i>2) ?(*«)]• (134>
Это явление, которое влияет на скорость, изменяет силы к2 и к2 таким
образом, что
к; = р7 (v —Ду7), (/=1,2). (125)
Подставляя эти значения к;- в уравнение (122), получаем
— Vp = к = V1p! (V— Д¥х) 4- v2p2 (v — Av2) (126)
и, пользуясь уравнением (124) для исключения Ду2 и Ду2, находим
- Vp = v [ v1P1 + у2р2 + (Р1 - р2)« -
( ^2pi +^1рг V *“? (ха) 1 (127>
" к ъ + *2 ) 48^п J • к 7
Это выражение представляет собой силу, рассчитанную на молекулу раство-
ренного вещества при концентрации п — п1/у1 = молекул в 1 см3. Трети®
член в скобках в правой части уравнения соответствует эффекту «первого
порядка» (электрофоретическому), и он пропорционален квадратному коршо
96
ГЛАВА IV
--,----
из концентрации. Последний член имеет величину порядка п Ign и является
отрицательным.
Решая уравнение (127) относительно v и умножая на п, получаем J.
Если пренебречь членами, содержащими вторую и более высокие сте-
пени Avi и Av2, то получается
JE„V_ -S№= - (,Д+7,.,+Д№)Г|‘- <128>
причем ДЖ равно
дто = _( Y (А'ЖЛ______™_____l
+ W Vi + Спт] (1 + ха)
, О1м1 + ^амг)2 (*«) /л од')
' (V1 + 4 * * * * *s)2 (v.l0)2 + v2“l)2 48t.2V] ' ' '
Чтобы перейти к более привычным единицам, воспользуемся соотношениями
n — Nn, pN = р,.
где N — число Авогадро, п — концентрация в молях на см3 и и. — химиче-
ский потенциал в эргах на моль. Следовательно, величина потока, выра-
женная в молях на см3, равна
j=nv= (130)
Если исключить и <о2 из уравнения (129) с помощью соотношения
а также учесть, что A° = k?-|-k2, а с =1000 п, и ввести численные значения
универсальных постоянных согласно Бёрджу [13], то получаем
где Ж = Е074 • IO’20 —УХ2'| + дЖ' + АЖ", *1 1 ZJL | (131)
_ дт' = (1^?-Ы^)2 З’ш •..10~19 с Уг A»2|^2|(v1 + v2) 710 (РГ)1'2 (Ц-ха) (132)
и
(133)
Функция <р(ха) дается уравнением (76а).
Согласно уравнению (116), коэффициент диффузии равен
3) = Ж^ = ЮООЖ^ • (134)
дп и(-
Для бинарного электролита
+ (135)
и, следовательно,
sro / д In у . \
^ = 2000ДГу И + с . (136)
4
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
97
Это выражение представляет собой уравнение 1 для коэффициента диффузии
электролита, диссоциирующего только на два вида ионов. Значение R в этом
уравнении должно быть выражено в эргах на градус на моль.
§ 5. Зависимость электропроводности
и диэлектрической постоянной сильных электролитов от частоты.
Теория Дебая и Фалькенгагена
а) Уравнения для потенциалов и сил в переменном поле. Мы изложи-
ли теорию электропроводности электролитов и определили изменение подвиж-
ности ионов под влиянием несимметричности их ионных атмосфер. Так
как мы совсем не останавливались на влиянии частоты переменного тока,
а также сильных полей на электропроводность, то полученные результаты
справедливы для случая низких частот и слабых электрических полей.
Теория влияния высокой частоты на электропроводность и диэлектрическую
постоянную была успешно развита Дебаем и Фалькенгагеном [14, 76].
В переменном поле происходит колебательное движение ионов. При
низких частотах ионные атмосферы обладают асимметрией, обусловленной
действием внешнего поля. Если же частота настолько велика, что период
колебания центрального иона представляет собой величину того же порядка,
что и время релаксации атмосферы, то нарушение симметрии ионной атмо-
сферы становится менее вероятным. В результате по мере возрастания час-
тоты ионная атмосфера по своей форме все менее и менее отличается от атмо-
сферы, находящейся в невозмущенном состоянии и обладающей сферической
симметрией, и электропроводность раствора соответственно увеличивается.
Поскольку при наличии переменного внешнего поля возникает состо-
яние, которое не является стационарным, то мы должны использовать
для вывода уравнения для потенциалов нестационарную форму уравне-
ния непрерывности [(39) гл. II]. Таким образом,
- = шг [кг • V2f3i (г)] + со/ [к/ • VJ?, (г)] -
— (г) — е/со/^и/^. (— г) — (137)
Путем введения относительных координат 721 = —Qi и т- Д- или
т = х2 — х1, у = У2 — У1 и т. д. можно получить следующие’соотношения:
/Л(г) = —/?/(г),
?Х(*) = -V2/?y (r)=-<(г),
V2-V2 = v1-V1 = V-V,
fti(r)= -/G(r) = /0(-r).
Следовательно, так как направление k; и к/ совпадает с направлением
оси ж, то получается
к дх — ле; дх .
1 При сравнении
и [Фу осе а [Зг] следует
(4 — 13—19), содержится
коэффициент уменьшен в 1000 раз.
7 Г. Харнел, F. Оуэн
этого уравнения с окончательным результатом Онзагера
иметь в виду, что в уравнении, обозначенном в их статье
типографская ошибка, состоящая в том, что численный
ГЛАВА IV
Используя эти соотношения и исключая V2-Va<|»/ и с помощью соот-
ветствующих уравнений Пуассона, можно преобразовать уравнение (137)
и получить1
= кТ (ш; + ш,.) + (еуш,- - е;со;) Хд~—
-JT 2 f'jiei + eiMini 2 №>' ] ' (138>
i j
Допустим, что напряженность поля X мала, а частота равна ад, и что
Хй = Х(й=0)е{“г = Хе4йг. (139)
Далее, пусть
/л = /л + ^ = /и^г, (140)
где
.0 Л eiej • е ~*г\
fii — nini\l DokT г J'
Подставляя эти соотношения в уравнение (138), получаем2
(ш£ + ш;) кТ? VGit - i&Gfi - 2 G/A- -
4n7l;e;CO;
—2 Gi>ei=x it (141)
i
В случае электролита, диссоциирующего на ионы двух видов, для
изучаемой нами проблемы представляют интерес следующие условия:
G12 = —G21 и G11 = G22==0;
с учетом этих условий уравнение (141) принимает вид
(шх ш2) кТ V- VG12 — iwG12 — (wiejUh -f-Gi2 —
> d/?,
= - (eiah — е2ш2) X . (142)
Используем, далее, уравнения (86) и (87), а именно:
= дг*и2,
* — е1м1 —е2м2
4 (ei —е2) (“i + “2) '
Из этих соотношений и уравнения (11) находим величину времени
релаксации
’ЧтйОйЯ- (НЗ)
Введем обозначение
7*т = т (144)
1 Уравнение (138) соответствует уравнению (4—3'—6) статьи Онзагера и Фуосса,
если в последнее ввести уравнение Пуассона, с той лишь разницей, что используется
нестационарная форма. Это уравнение совпадает с уравнением (411) Фалькенгагена [7в].
2 Это соответствует уравнению (462) книги Фалькенгагена [7г]. Уравнение (142)
совпадает с уравнением (465) той же книги.
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
99
и определим комцлексную величину К2 с помощью уравнения
= (145)
При подстановке всех этих соотношений в уравнение (142) получаем
V VC1! - . (146)
Далее, согласно уравнению (12) гл. II, функция распределения /?2 равна
/о _ „ „ п^е^е-"
ъ—г - ,
а значения потенциалов асимметрии которые можно найти с помощью
выведенных ранее соотношений, определяются следующим выражением:
= (147)
Путем сочетания двух последних уравнений с уравнением (146) получается
дифференциальное уравнение для потенциала атмосферы вокруг иона (1).
Таким образом,
(V.V)2^=^2V-V^ = 2z?^(^)e«, (148)
где 2 определяется уравнением (88), когда w равно нулю. Отметим, что
уравнение (148) имеет ту же форму, что и уравнение (89), выведенное
Онзагером. К2 определяется уравнением (145) или выражением
= 2Р1Р. Й '
Р1 + Р2 (Pi + Ра) ZkT ' >
a xj и у-п определяются следующим образом:
х^ + х^^и^да^. (150)
Когда & равно нулю, уравнение (148) переходит в уравнение (89) и явля-
ется достаточно общим для вычисления эффекта изменения электропровод-
ности и диэлектрической постоянной в случае переменного тока высокой
частоты. ф( представляет собой потенциал асимметрии колеблющегося иона.
ф] и соответствующее значение ф2 можно найти путем интегрирования
уравнения (148) и с помощью основных соотношений
+ п2е2ф' =0,
~ ^2^2’
Асимметричная часть потенциала в непосредственной близости к ионам
определяется выражением
3"(x + g) e^[rcos6+ 0(7-2)], (151)
где 0 —угол между радиус-вектором г и направлением поля.
Из этого уравнения можно найти величину напряженности поля
в месте расположения иона .(1) при 0 = 0 с помощью метода, которым
было получено уравнение (91). Таким образом,
^ = -?«(^ = 0) = 5^(—+f^=), (1S2)
\1 + JZ g* |/ ! +
где v1eiwt — абсолютная скорость иона 1.
7*
100
ГЛАВА V
б) Уравнения для электропроводности и для уменьшения диэлектриче-
ской постоянной в переменном поле. Применим опять способ, аналогичный
тому, который был использован для вычисления электропррводности в слу-
чае отсутствия колебаний ионов. Нам нужно найти скорость y;ei<of. Если
видоизменить уравнение (92) таким образом, чтобы оно соответствовало
рассматриваемым условиям, то получается
г,/ег«( = Qxe у + е'~>. (153)
Используя уравнение (152) и заменяя pit>i на Хёу, получаем
v}e^ = X [е;%-+зд^ ’WZ(7*. G>T)_g.J е^, (154)
где
= —---------------• (155)
у 1 4- /шт + 1
Из написанного выше уравнения, для скорости можно путем соот-
ветствующих преобразований получить окончательные уравнения для мо-
лярной электропроводности Ат и для уменьшения диэлектрической посто-
янной D — Do. В практических единицах можно выразить полученный
результат следующим образом:
Д~ = Д^-Ай-Дп, (156)
где А~—суммарная молярная электропроводность при частоте G>, — мо-
лярная электропроводность при бесконечном разведении, — составляющая
Л~, обусловленная несимметричностью ионной атмосферы, и Дп — состав-
ляющая, обусловленная электрофоретическим эффектом. Дг~ и Лп опреде-
ляются уравнениями
~ д о б157>
и
_ v Иу^х Л]000Л 1
Z 67t7]0 V с ) 9 • 1011 ’ (158'
У
где т. е. действительную часть у, можно вычислить из уравнения
7.r = (i-Z^+йМ [(!- vr) (Я- 1//F) + SxQ], (159)
в котором
/? = 2-1/2[(1+“М1/24-1]1/2; Q = 2-1/2[(l 4-й2г2)1/2_1]1/2. (160)
Увеличение диэлектрической постоянной, обусловленное наличием иере-
. менного поля, равно
п Г> — 4те I file2 I „V-n Ли ____________
» — ^DakT *2jnieiw} — 1/g*)3 + <№] ‘
• [Q (1-1/9*)- Фт(Я-1//^)]. (161)
Уравнения (156) —(160), а также уравнение (161) представляют собой
основные выводы теории Дебая — Фалькенгагена относительно зависимости
электропроводности и диэлектрической; постоянной от высокой частоты.
(7*
Для стационарных полей (& = 0), становится равным :----------7/^’ чт0
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
101
согласуется с результатом теории электропроводности Онзагера. Упрощен-
ные уравнения и таблицы для практических вычислений будут приведены
в гл. V, § 3 *.
§ 6. Теория влияния сильных полей на свойства сильных
электролитов. Эффект Вина
При обсуждении свойств ионных атмосфер (гл. II) указывалось, что
при наложении сильных полей ионы выскакивают из своих атмосфер
и поэтому их движение уже не замедляется из-за того, что им приходится
тянуть за собой свою атмосферу. Это явление можно проиллюстрировать
с помощью простого расчета, принадлежащего Фалькенгагену [7д]. Согласно
уравнению (15),
Возьмем в качестве примера раствор хлористого калия в воде при 18°
и рассмотрим движение иона калия. Его подвижность равна 0,000675 см/сек,
и, следовательно, его абсолютная скорость в поле с напряженностью
100 000 в/см равна 67,5 см{сек. Так как XJ равно 65, то р,. равно 0,236 10-8.
Если принять концентрацию с равной 0,0001, то, согласно уравнению (12),
время релаксации составляет 0,276 10~6 сек. При этой концентрации
средняя толщина ивнной атмосферы 1/х равна 3,06 • 10-6 см. За время
релаксации ион пройдет расстояние 18,6 • 10~6 см, которое в 6 раз превышает
толщину атмосферы. Образование атмосферы в этих условиях является
невозможным.
Таким образом, при наличии поля ионы обладают большей подвижно-
стью, чем в отсутствие поля. Следовательно, при наложении сильных
полей электропроводность растворов увеличивается, и можно ожидать, что
закон Ома будет применим только в ограниченной области.
Поскольку теория электропроводности Дебая—Гюккеля—Онзагера была
развита для случая слабых полей, ее пришлось видоизменить таким обра-
зом, чтобы учесть влияние сильных полей. Первая попытка в этом направ-
лении была сделана Иоосом и Блюментритом [16]. Они получили следую-
щее уравнение:
-г^-= A*~AA=o = АХЦ1-ВХ2), (163)
лх-=о --хх=о
где Ад и Ад=0 представляют собой молярные электропроводности при
полях с потенциалами X и Х = 0. Параметры А и В являются функциями
Т, D п с, а также электропроводностей ионов и их валентностей. Этот
результат согласуется с опытом в случае сравнительно низких значений
потенциалов. Характер допущений, сделанных Иоосом и Блюментритом,
таков, что их теория носит лишь качественный характер.
Иной путь избрал Фалькенгаген [17], который развил качественную
теорию для полей, потенциал которых меняется в пределах от 0 до со.
На основе этой теории Фалькенгаген и Флейшер [18] исследовали случай
нестационарного поля и сделали расчет влияния частоты на эффект Вина.
Онзагер и Вильсон [19] развили более точную теорию эффекта Вина
для сильных электролитов, которая учитывает как гидродинамический,
1 Для дальнейшего развития теории влияния частоты тока на электропроводность
и диэлектрическую постоянную в применении к смесям сильных электролитов сыграли
роль работы Фалькенгагена и Фишера [15].
102
ГЛАВА IV
так и электрофоретический эффекты. Ниже будут изложены основные
положения и окончательные результаты этой теории.
Если за переменную принять г, то из уравнения (38) гл. II следует,
V • //» (г) [v7i (г) - vv ( - г)] = 0>i [k4 • V/Zi (г)] -
— 1к/ • (r) — nini [ei0)iV -УФ/ (г) + e^j V • V<h ( — г)] —
— АгТ (ю{ 4-.<oz) V. V/;i (г) = 0; (г,/=1,2, ...,s).
Рассмотрим только случай бинарного электролита. Выразим силы к2 и к2
следующим образом:
к, = Хе2; к2 — Хе2,
где X — компонент электрического поля в положительном направлении
оси х. Далее, для частного случая бинарного электролита
I ei i = I e3j = е,
П! = И2= П.
С учетом этих соотношений уравнения движения приобретают форму
п2е[Г-Г’(ф1(г) + ф1(-г))} + 2Л:717-Г/11(г) = 0, (164)
- n*e{V- V [ф2 (г) + ф2 ( - г)]} + 2kTV- (г) = 0, (165)
Хе (оу + <о2) — п2е {л1 V • ?ф2 (г) — <о2 7 • V<h (— г)} +
+ /cT1(®1 + a)2)V-V/12(r) = 0, (166)
— Хе (wj + <о2) — п2е {«.'j V.Гф2 (— г) — ш2 V • V (г)} +
+ кТ (ш2 + ®1)V-V/21 (г) = 0. (167)
а) Граничные условия. Поток. Число ионов Fp (S), покидающих
область 2, ограниченную поверхностью S, равно
-----\ 1а (г) [е„ • [Vji (г) - viZ ( - г)]} dS =
s
= j) V • {/Л (г) [vZi (г) - viy ( - г)]} dQ = 0, (168)
s
где еп —единичный вектор, нормальный к поверхности X. Закон сохра-
нения вещества требует, чтобы этот интеграл был равен нулю для неко-
торой области Q. Это основное условие обычно выражается в форме
lim fjt (г) {еп • [vyt(r) — viz( — r)]}c76’ = O, (169)
S (s) = 0 J
причем из него следует, что векторное поле потока /л (г) [vZ{ (г) — Vy (— г)]
не должно иметь источников.
Уравнения Пуассона. Согласно уравнениям (И) и (12) гл. II,
S S
V. 7ф. (Г) = - J Pi (Г) = - g 3 пр (г) ei = - g -L 2 jp (г) 6i. (170)
i=i 7ч=1
1 Отметим, что в этих уравнениях фигурирует величина / вместо /°, так как
эффекты возмущения велики.
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
103
В обобщенной форме эти уравнения имеют для бинарных систем следую-
щий вид:
™Ш = ЙНи(г) + Ш. (171)
v • =Й t (г)+(r)i • c172>
Ионные поля. Пространственный заряд внутри области Q равен
^/(г)^ = $Р/(ГИ2,
а й
причем рj, (г) можно интегрировать, за исключением того случая, когда
в начале координат находится точечный заряд. Правая часть этого урав-
нения стремится к нулю, когда Q стремится к нулю, если только в Q не
содержится начало координат; в этом случае .
lim \ р/ (г) dQ = ej.
9=°й3
С помощью этих
уравнений и пользуясь теоремой Гаусса, получаем
lim \
Гф, (г) t/S =
4ле.
(173)
где 8 равно единице, если начало координат совпадает с местом располо-
жения центрального иона, и 8 равно нулю, если начало координат нахо-
дится в другом месте.
На очень близком расстоянии от данного иона можно пренебречь экра-
нирующим влиянием ионной атмосферы, и тогда потенциал иона буден равен
ej/Dr, Потенциал иона и его атмосферы ф (г) будет отличаться от этого
значения потенциала на конечную величину, т. е.
Ф/ (r) — ei!Dr < 00 •
В бесконечности
ф/(г)=ф1(г)=О,
откуда для бинарного электролита
(г) — ex/Dr < оо,
ф2 (г) — e2!Dr < оо,
Ф1 (л) = ф2 (оо) = 0
(174)
(175)
(176)
Условия симметрии для потенциалов. Можно вывести два условия
симметрии для потенциалов: одно из них определяется тем обстоятельством,
что /п (г) и /22 (г) являются четными функциями, а другое зависит от нечет-
ной части функций распределения, для которых /12(г) =/21 (— г). Из этого
равенства и из уравнений Пуассона (171) и (172) получаем
V-V [ф2,(г) —ф2( —г)—ф^ + ф^ —г)] = 0. (177)
Так как разности ф(- (г) — ф; ( — г) являются конечными, то величина в скобках
всегда имеет конечное значение. Кроме того, эта величина является
постоянной, так как, согласно теории гармонических функций, любая
конечная функция, которая удовлетворяет уравнению Лапласа, имеет
постоянное значение. • Далее, поскольку ф> (со) = фг (оо) = 0, то
Фг (г) — *2 (- г) — 91 (г) + Ф1 (— г) = 0, (178)
104
ГЛАВА IV
ИЛИ
62 (г) — ф2 ( — г) = <]>! (г) — фх (— г) = 2Y (г), (179)
где Y (г) представляет собой нечетную часть потенциала.
Условия симметрии для четной части потенциала можно получить
следующим образом. Согласно уравнению (137), при i = j = l величина
потока становится равной
/и (г) [Vn (г) — VU (— г)] = wjV {— п2е [фх (г) + Ф1 (— г)] — 2kTfn (г)}. (180)
Согласно граничному условию для потока, его векторное поле не имеет
истоков. Величина градиента в правой части равна нулю, и член в скобках
является постоянным. Так как /;г(оо) = п2, а фг (оо) = ф, (оо) = 0, то отсюда
следует, что
/11(г) = /11(-г) = п2—^[^(гН'Ы-г)] (181)
и аналогичным образом
/22(r) = /23(-r) = n2 + g['b(r) + 62(-r)]. (182)
Суммируя уравнения Пуассона (171) и (172), подставляя/и (г) и /22(г), 01й°
раз суммируя и применяя основное уравнение (4) гл. II, получаем
(v • V - 4) № (г) + «М - г) + Ъ (г) + ф2 (- Г)] = 0, (183)
причем для бинарного электролита
„ 8ягее2
Х — DkT ’
Граничные условия требуют, чтобы член в скобках, содержащий сумму
потенциалов, имел конечное значение. Так как уравнение (183) не имеет
решения (кроме равного 0), которое было бы везде конечным и не обла-
дало особенностями, то, следовательно,
Ф1 (г) + (— г) + Ф-2 (г) + фг (— г) = 0,
Ф1 (г) + Ф1 (— г) = — [4*2 (г) + Фг (— г)] = 2Г (г), (184)
где Г (г) —четная часть потенциала.
При решении уравнений (179) и (184) получаются выражения
ф1(г)== — ф2( —г) = Г(г) + У(г), (185)
Ф1( — г)= — ф2(г) = Г (г) — Y (г), (186)
которые являются основными условиями симметрии ионных полей для
бинарного электролита. Сочетая эти выражения с уравнениями (181) и (182),
получаем
/п(г) = /й(г) = п2-$Г(г). (187)
б) Упрощение основных дифференциальных уравнений для функций
распределений и потенциалов. Дифференциальным уравнениям (166) и (167)
можно придать удобную форму с помощью условий симметрии (185) и (186).
Уравнения (164) и (165) содержатся в уравнении (187), и это можно
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
105
использовать для исключения /ц (г) и /22 (г)- Если обозначить четные
и нечетные члены функций распределения /12(f) через G(r) и U(г), так
чтобы
/12 (± г) = /21 (+ г) = G (г) — п2 ± и (г), (188)
где G (г) — G (— г) = U (r) + G (— г) = 0, то уравнения (166) и (167) приоб-
ретают вид
V. ГГ (Г) - V • VG (Г) = -g- , (189)
^-V.VK(r)-V.VG(r) = -^^i. (190)
кТ х ' v ' кТ дх 4
В эти формулы для стационарного состояния не входят.подвижности uij и ш2.
После введения граничных условий для потока и использования уравне-
ний Пуассона дифференциальные уравнения для функций распределения
и потенциалов можно выразить следующим образом:
(v-V-4-)G(r)=(191)
ЛД.д_2±у(г)_^4Г(г)=-1ь/^’ (192>
(193)
’w<r>= w'7(r)- (194)
Из уравнений (191) —(193) получаем
V.V(V.V_x2)G(r) = p'2^p, (195)
откуда можно вычислить G(r). Зная G(r), можно вычислить Г(г), К(г)
и U (г) из уравнений (188) и (190). В приведенных выше уравнениях
^' = е/ЛТ, (196)
?' = еХ/кТ. (197)
в) Решение системы дифференциальных уравнений. Системой
дифференциальных уравнений удобно оперировать, если представить ее
в цилиндрических координатах х, р, б, так чтобы она являлась инвариант-
ной по отношению к вращению вокруг оси х, вследствие чего угловую
переменную б можно опустить. Онзагер и Вильсон решают эти уравнения,
выражая G(r), U (г), Г (г) и У (г) с помощью преобразовании Фурье [20].
Получающиеся в результате интегралы для /12 (±г) и Wi1) имеют
следующий вид:
о k
• [(л2 — а2) Ко (Zip) — (Ц — а2) К0 (k2p)] cos (аж) da ±
СО
± 5 7l=^[(X2-a2)^0(M + (A22-a2)^0(M)--
о
— 2L_A’0(X3p) J sin(aa:)dal . (198).
106
ГЛАВА IV
(± г) = 4 5 [ (х2 - к0 (х1?) +
+ (к! - а2) Ко (Х2р) - Ко Х(3р) ] cos (аж) da±
± 5^Д^^о(Х1р)+^о(Хгр)-27Го(Х3р)]8ш(аж)С/а} , (199)
о
где К0 (Хр)—преобразованная функция Бесселя второго рода и нулевого
порядка, a Хь Х2 и Х3 определяются соотношениями
Х| = Х| = а2 + х2/2 ± ]А4/4-р.'2а2, (200)
Х2 = а2 + х2/2 и Х4 = а. (201)
Приведенные выше решения в интегральной форме можно успешно исполь-
зовать как для нахождения эффекта, обусловленного силами междуионного
взаимодействия, так и для определения электрофоретического эффекта.
г) Электрофоретический эффект. Влияние электрофоретического
эффекта на- скорость иона, движущегося под действием внешнего электри-
ческого поля с напряженностью X, можно найти посредством решения
гидродинамических уравнений
7)o[V X [V-v]]= -VP + F,
V-v = 0,
где v —скорость, Р — давление, т)0— вязкость, F—плотность силы, которая
возникает при действии внешнего поля на ионную атмосферу.
Используя результаты, полученные в предыдущем параграфе, находим
величину скорости в направлении поля:
(0. 0. «) - - 2^ ) 71=^- [ - >‘Ч Q) -
о
+ 4аУД + ^ ln(A)pa. (203)
Вводя переменную
х = р'/ъ, (204)
где р' — Хе/кТ, и выполняя интегрирование, получаем из уравнения (203)
МО, 0,9)=- —|Д-/(ж), (205)
6 у 2 лт|0
где / (ж) определяется уравнением
у (х\ = 1 _]-— Г 2ж2 arsh ж 4~ 1/ 2х — х\/~ 1-f-x2 —
4 у 2 ж3 L
— (1 4- 2х2) arctg ()/2 х) -j- (1 -|- 2х2) arctg ж J. (206)
Отметим, что при х = 0, / (х) = 2 и уравнение (2Q5) принимает вид
уравнения (62):
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
107
Этот результат был получен ранее [уравнение (92)]. Далее, в случае
бесконечно большой напряженности поля х = оо, j (х) — 1 и
^(0,0,9)=--^-, (207)
6 у 2
т. е. при бесконечно большой напряженности поля электрофоретический
эффект не исчезает.
д) Ионное поле. Как и в случае электропроводности при слабых
полях (§ 3), необходимо найти ионное поле, обусловленное возмущением
атмосферы. Для этого необходимо вычислить—Уф (г) при г = 0. Таким
образом, общая сила равна
ieAX (0) = ieAX (0, 0, 9) = lim [Гф7 (0)],
rr- р = 0
где i —единичный вектор, направленный по оси х. Вводя переменную
ж = /х и используя результат уравнения (199), получаем
ДХ(0, 0, ^Tgg^), (208)
где
g(x) = — 2М — ж]/1+ a:2 + arctg-^L=r+ У2х — arctg ж}, (209)
и поэтому
g(O) = 2~3^j И g(oo) = 0. (209а)
е) Функции f (х) и g (х) в форме, удобной для вычислений. Уравне-
ния (206) и (209) можно преобразовать в форму, удобную для вычислений,
с помощью следующих [подстановок:
tg" /2-1 = -^,
6 8 v ]/2 + 1
arctg s Q ct g -g J 4- arctg s Qtg - J = arctg ,
arctg s (ctg - arctgs (tg y) = arctg ,
где
s—i + )/TF^2’
Отсюда следует далее, что
arctg (l/ 2 x) — arctg — x
(тт?) =2aKte («
108
ГЛАВА IV
Используя эти выражения, можно записать уравнение (209) в следующем
виде:
, . / 1 —s2\ 1 1 . rt / 1 —S2 \
К 2~2 Т <~2~) ~
i
5 ‘ 7
(211).
Если заменить (1 — s2)/2 на 1/cp, то получим простое численное уравнение-
е{х} = _ ± {po.2071OHS_^0.MW«ssy [ 0,55228 - 0,0569 +
+0,00695(у)‘. (212),
Рис. 5. Функции / (х) и g (х) для вычисления эф-
фекта Вина для сильных электролитов.
Подобным же образом уравнение (206) принимает вид
1 , (stgi)4 1)
-<i+s ctgMtgЧ ГТЗ-----------37^~ + -^П^- ’ (213>
откуда получаем
/(0)=/2 и /(оо) = 1. (213а}
На рис. 5 изображены кривые для / (ж) и g (х) и показаны пределы,
к которым стремятся эти функции при сильных и слабых полях. Таблицы
этих функций приведены в гл. V, § 3.
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
109
ж) Уравнение электропроводности, преобразованное применительно
к эффекту Вина. Уравнение электропроводности сильных электролитов
в присутствии поля нетрудно вывести из предыдущих выражений. Если при-
нять во внимание уравнения (205) и (208) для электрофоретического эффекта
и влияния ионных полей, как и при выводе уравнения (92), то скорость
ионов можно выразить следующим образом:
2ВГ 8 ~ ’ (2И)
и уравнение (93). приобретает форму
Следовательно,
принимает вид
после подстановки в]Х/кТ вместо р/ уравнение для X,
превращается в
2— У"2
96 500 | е - ] х/ (г)
W ^8 -----• • (216)
уравнение (95) при да = 0, так как / (да) = ]/”2,
Это уравнение
a g(x)
С помощью рис. 5 можно
нения электропроводности при
величины / (да) и g (да) при увеличении силы поля уменьшаются и так как
оба эффекта — ионных полей и электрофоретический эффект [уравнение
(216)] — вычитаются из значения предельной электропроводности, то X; воз-
растает с увеличением силы поля. Однако при очень сильных полях Х7-
не стремится к XJ, так как / (да) равно единице при да = оо. Вид кривых
получить представление о характере изме-
увеличении напряженности поля. Поскольку
зависимости электропроводности от силы поля и количественная проверка
этой теории будут обсуждаться в гл. VII, § 8.
Фалькенгаген, Фрелих и Флейшер1 развили дальше предложенную
Дебаем и Фалькенгагеном (см. § 5) теорию влияния частоты на электро-
проводность и диэлектрическую постоянную в присутствии сильных полей.
Эта теория не является такой полной, как изложенная выше теория
Онзагера и Вильсона для нулевой частоты, так как в ней не учитывается
электрофоретический эффект. Уравнения для влияния частоты имеют сле-
л
дующий вид:
АХ=оо~~АХ _ е2ха у' _ ех2
АХ=оэ ~ 2DkT Iх' Х У
х2еу"
(217)
(218)
где р/ равняется 2DkTjXe. Для функций у' и у", представляющих собой
действительную и мнимую части комплексной функции у*, имеются таб-
лицы, которые содержат значения у' и у" в зависимости от р'/К DT/c и
от (от.
§ 7. Влияние сильных полей на диссоциацию слабых
электролитов. Теория Онзагера
В присутствии сильных полей для слабых электролитов наблюдаются
отклонения от закона Ома, которые во много раз превышают соответст-
вующие отклонения для сильных электролитов [21]. Электропроводность
1 О влиянии частоты на электропроводность см. [186, в]; о влиянии частоты на
диэлектрическую постоянную см. [18г].
110
ГЛАВА IV
пропорциональна абсолютной величине напряженности поля в большом
интервале изменения напряженности, причем предельное значение электро-
проводности соответствует полной диссоциации слабого электролита. Онза-
гер [16] дал объяснение этого явления на основании теории • междуион-
ного притяжения, причем это объяснение, повидимому, является очень
хорошим приближенным решением вопроса и на основании этого решения
можно сделать важные выводы. Теория Онзагера является результатом
тщательного изучения скоростей диссоциации электролитов и рекомбинаций
ионов. Условия ассоциации ионов получены из обобщенной теории Бьеррума
(гл. III, § 7). Далее, поскольку при наличии поля X закон распределения
Больцмана неприменим, то необходимо воспользоваться общими законами
броуновского движения ионов, которые для нестационарного случая выра-
жаются уравнением (39) гл. II. Для полного решения этого сложного вопроса,
касающегося движения ионов, требуется тщательное изучение необходимых
упрощающих допущений, а также вычисление интеграла, которое до сих
пор не удалось осуществить.
Онзагер принимает допущение Бьеррума о том, что пара ионов проти-
воположного знака, находящихся на расстоянии г < q, является ассоцииро-
ванной. Так как мы рассматриваем слабые электролиты, то концентрация
свободных ионов в растворе невелика и, следовательно, 1/х значительно
больше, чем «эффективное расстояние» между ионами q. Таким образом,
’ -/.</ = —xe1e2/2Z)A71 < 1, (219)
2__ (чде| 4- ч2е|) /9901
' “ DkT ’ ' >
где пг и и2— концентрации свободных ионов. Если в растворах сильных
электролитов v.q мало, как предполагается согласно уравнению (219),
влияние междуионных сил будет незначительным. В то же время в случае
слабых электролитов соединение ионов может происходить даже при ма-
лых значениях v.q. Бьеррум показал, что этот эффект подчиняется закону
действующих масс независимо от того, какие силы обусловливают образо-
вание связи между ионами. Прежде всего мы должны исследовать зави-
симость константы закона действующих масс от силы поля.
Остановимся на некоторых особенностях ионных атмосфер в условиях,
когда х/у < 1. В отсутствие внешнего поля концентрация /-ионов при нали-
чии /-ионов равна
Мд (/•) = »i ехр( — Wfi(r)/kT), (221)
где Wji (г) — взаимный потенциал средней силы, действующей между i-
и /-ионами. При г > q ионы свободны, а при г < q — ассоциированы. Для
случая r>q можно найти W-ц (г) из теории Дебая и Гюккеля; применяя
уравнение (30) гл. Ш для взаимного потенциала, получаем
(1 + z^)r ’
г > q-
(222)
Далее, согласно Бьерруму, работа, которую нужно затратить для разделе-
ния ионов, равна 1
^л(/-) = -^, ?<q.
(223)
1 Поправочным членом с величиной порядка xqkT мы гренебрегли, тай как
приняли допущение, выраженное уравнением (219).
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
111
Концентрация ассоциированных ионных пар равна
vyi = vij = П] пц (г) 4srr2 dr, (224)
а
где а —наименьшее расстояние сближения положительного и отрицатель-
ного ионов. Для слабых электролитов/ а также для любых электролитов
вереде с низкой диэлектрической постоянной, основной эффект имеет место
при расстоянии г, немного большем, чем а. Верхний предел q задан про-
извольно, и нетрудно показать, что его можно заменить на q/2 или 2q и это
не отразится существенным образом на результатах.
Некоторые важные положения, касающиеся пределов интегрирования,
можно получить из уравнений (222) и (223). В уравнении (222) величина
е~" характеризует «экранирование» поля иона его атмосферой, т. е.
пространственным зарядом, который окружает Ион. Если опустить этот
множитель в уравнении (222) и подставить получающееся при этом зна-
чение W}i\r) в уравнение (221), то можно показать, что этот простран-
ственный заряд является бесконечным, так как интеграл уравнения (221)
с верхним пределом, равным со, является расходящимся. Однако Онзагер
указал, что можно пренебречь членом е~хг, если принять, что уравнение (220)
применимо вплоть до некоторого расстояния г' > q, или, точнее говоря,
при условии, что
- q г' <£ 1/х. (225)
Исходя из этих соображений, можно применить уравнение (223) для слу-
чая расстояний г' > г, так что
FE/i (г) = ejedDr, r<r'. (226)
Это приближение является вполне точным в случае слабых электролитов,
так как неполная диссоциация зависит главным образом от внутренней
области поля ионов, в то время как поле за пределами г' не имеет суще-
ственного значения.
Для сильных электролитов уравнение (221) можно заменить, согласно
уравнению (19) гл. II, приближенным выражением
n^r^n^-W^/kT), (227)
которое хорошо оправдывается для больших значений г, а также при
Жд (r)/kТ < 1. Для малых значений г это выражение непригодно. С дру-
гой стороны, приближенные соотношения, выражаемые уравнениями (226)
и (224), применимы в случае малых расстояний, но приводят к абсурдным
результатам при г=оо. Однако Онгазер показал, что если увеличить г'
до бесконечности и пренебречь влиянием бесконечно большого общего
пространственного заряда на больших расстояниях, то можно получить
простое и важное решение.
Для вычисления величин эффектов, обусловленных внешним полем X,
необходимо использовать общие уравнения движения ионов. Так как общая
сила К равна сумме действующих на ионы внешней силы еХ и силы к,
т. е. среднему.значению силы, обусловленной ионной атмосферой, то мы
получаем из уравнения (9) гл. II
vyi (г) — Vy (- г) = — о», [eiX + kji (г) - kTV2 In (г)] —
-«/[еуХ + куД-^-Л^ЬДД-г)]. (228)
Далее, из уравнения (7) гл. II следует, что
• [hi (г2 ~ Ti) V/i (r2 - гД] - [fi3‘hi - r2) Vy (fj - r2)] =
= Да • {/о (r) [vy (- r) - v,4 (r)]} 0. (229)
112
ГЛАВА IV
Этому уравнению можно-придать удобную форму, если принять сделанные
ранее допущения: 1) при допущении, что v.q < 1, можно пренебречь вли-
янием ионной атмосферы; это будет вполне справедливо для сильных
полей, так как в этом случае атмосфера вокруг иона не образуется;
2) если в уравнении (226) считать г' = со, то можно заменить W через
кулоновский потенциал e,ei/Dr; 3) если выбрать внешнее поле X так,
чтобы оно было параллельным оси х и обладало потенциалом — Хх,
а также допустить, что Хе, > 0, Хе, < 0 и опустить индексы у /, то полу-
чается следующее выражение для потока:
/(r)v(r) - / (г) [vyi (г) - yi; (- г)] = .
. = A7,(<Oy4-U)i)[-v/ + /~(v + 2^)] ’ (23°)
где
5'=— U5kT ’ С231)
Условие стационарности, которое определяется уравнением (229), выра-
жается следующим образом:
Так как поле с потенциалом не имеет дивергенции, то
V-V0^-4-2£a?) = O
и
?.?/== +2^ . (233)'
Суммарная скорость, с которой пары ионов проходят через какую-либо
замкнутую поверхность S, может быть получена из уравнения (230). Таким
образом,
dV = кТ + од) 5 [ Гп/ - /Гп (А + 2^) ] dS, (234)
где Vn — градиент, нормальный к
Чтобы развить теорию неполной диссоциации на основании принятых
ранее допущений, необходимо решить уравнение (233) для определенных
граничных условий. Онзагеру удалось проинтегрировать это уравнение,
однако он не привел подробностей математического вывода в своей
работе, посвященной теории эффекта Вина1. Поскольку все основные
допущения были приведены выше, мы дадим лишь краткое описание
метода Онзагера и окончательные результаты.
Онзагер счел более удобным вычислять плотность / отдельно для
процессов соединения и диссоциации.. Скорость химического процесса
соединения определяется уравнением (19), или
<235>
где А и КА — соответственно константы скорости соединения и диссоциа-
ции, а К — константа диссоциации. При полной диссоциации распределе-
ние ионов дается уравнением (233) и п3п-[ — / (г) = const. Следовательно,
1 Решения даны Онзагером в его диссертации [22].
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ
113
скорость соединения ионов можно найти из уравнения (234), если восполь-
зоваться сферическими координатами г, 6 и <р:
Atijiii = кТ (о>; + о>г) njiii — 23 sin 0^г 2 sin 0 <20 с?<р =
= 8ttqkT (<о; + u>i) (236)
Для бинарного электролита ех= — е2 и
А = 471 (е^+е2ш2) (237)
Таким образом, поле не влияет на константу скорости рекомбинации
ионов. Если поле отсутствует и, следовательно, Р = 0, то из уравнений
(226), (221) и соотношения / (г) = п,пц можно найти, что число недиссоци-
ированных молекул определяется уравнением
/(r) = v;i7f(0)(e2^-l) = ?i;?ii(e29/'--l), (238)
где К (0) обозначена константа диссоциации в отсутствие поля. Это един-
ственное решение уравнения (233), удовлетворяющее граничным условиям,
согласно которым / = 0 при г — оо и поток из начала координат через
любой пространственный угол имеет конечную величину.
Если X Ф 0, решение уравнения (233) при этих граничных условиях
является сложным. Онзагеру удалось получить это решение в форме опре-
деленного интеграла, а именно:
s = 2q
j (г, 0) = ge-$r+$r cos 6 /0[/ — 4ps (1 + cos 0)] ехр , (239)
s=0 ...
где Jo — функция Бесселя нулевого порядка. Для малых значений г можно
заменить q через оо и получить хорошее приближение:
/ (г, 0) ge2qlre2?,rcos e. (240)
Если, как было упомянуто выше, не учитывать область r>q и, кроме
того, приравнять второй экспоненциальный член единице, то получается
/ (r) ~ ge2q!r, (241)
т. е. то же распределение, которое было получено для случая равновесия
при 1 < ехр (2g/r). Сравнивая это выражение с уравнением (238), полу-
чаем
v7i = g/^(O), (242)
т. е. искомое соотношение между g и K(Q),
Для вычисления скорости диссоциации согласно уравнению (239) необ-
ходимо подставить /(г, 0) в уравнение (234). Если разбить интеграл на
две части
2q оо со
0 0 2q
то оказывается, что только второй интеграл в правой части уравнения
дает некоторую составляющую. Можно показать (легче всего для частного
случая, когда г мало), что
. - К (X) Av}i = - 8r.qkT + ш{) g • (243)
8 Г. Харнед, Б. Оуэн
114
ГЛАВА IV
Из уравнений (236) и (243) явствует, что относительное увеличение кон-
станты диссоциации при действии поля равно
АК(Х) вд _J1(4/-^)_1 2„ (4^ (4р?Г ,
АК-(О) К(0) 2/ —р? "Г 17 21 3! ^314!
(244)
где А — функция Бесселя первого порядка. Напомним, что константа
скорости соединения ионов А оказывается независимой от поля.
Уравнение (244) представляет собой основной результат теории Онза-
гера. В настоящем кратком очерке этой теории могли быть освещены лишь
ее основные положения, а также подчеркнуты те значительные трудности,
с которыми она сталкивается. После изложения своей теории Онзагер дал
подробный критический разбор принятых допущений и вычислил порядок
величины ошибок, обусловленных этими допущениями. Он отметил, что
с целью упрощения не было учтено гидродинамическое взаимодействие,
и сделал попытку определить порядок величины гидродинамической по-
правки. Онзагер пришел к заключению, что при сильных полях этот
эффект может оказаться существенным, но все же он недостаточен для
того, чтобы изменить порядок величины основного результата.
Из уравнения (244) видно, что мерой относительного увеличения кон-
станты диссоциации является параметр 2fiq. Согласно уравнению (232),
этот параметр, обозначенный через Ь, равен
Б = 2^ = 1.21-Ь + К.Ь । । = + Y 9 636 V (245)
17 «>! + «>2 2.0/гт- I z2 I Ax + I z1 I A2 DT2 ' '
где zx и z2 — валентности ионов, и k2 —эквивалентные электропровод-
ности, e — заряд электрона и У — интенсивность поля в вольтах на санти-
метр. Для 1,1-электролитов это выражение приобретает простой вид:
6 = 9,636^. (246)
Функция в правой части уравнения (244) выражается степенным рядом
К (0) (6) — 1 + Ь + 3 + 18 + 180 + 2700 + 56700 + • ’ ' (247)
или для больших значений Ь асимптотическим разложением в ряд
^(i)^l/2^,(86)"s/4er^r 1---------------------------------- 1, (248)
r L 8 (8Ь)1/2 128(86) 1024 (86)3/2 J v
где при 6>3 остаток ряда меньше 1/13062, тЛе. меньше 10~3.
Теперь мы имеем возможность перечислить некоторые наиболее важ-
ные результаты теории.
1. Из уравнения (247) явствует, что первичное влияние поля обуслов-
ливает линейную зависимость К(Х)/К(0) от Ь, или, согласно урав-
нению (245), линейную зависимость этой величины от абсолютной величи-
чины силы поля X.
2. Из двух последних уравнений видно также, что зависимость К от
силы поля определяется двумя характерными свойствами электролита —
валентностями и подвижностями ионов.
3. С помощью закона действующих масс можно определить, какое
влияние оказывает поле на концентрацию свободных ионов Cj. Для бинар-
ных электролитов
са2
1—а
#(X) = tf(0)F(6)„
(249)
НЕОБРАТИМЫЕ ПРОЦЕССЫ В РАСТВОРАХ Й5
(250)
важный
измене-
а„.
где с — концентрация ЭйектроЛита, а —степень диссоциации, cj/c. Кроме
того,
Ад- Ct(X)_a
лх=о Ci (0) а0 ’
где Л — электропроводность.
Из закона, выражаемого уравнением (249), получается тот
результат, что изменение равновесия, соответствующее данному
нию К, зависит от первоначального значения степени диссоциации
4. Следует отметить, что а/а0 уменьшается при увеличении а0. Для
случая малых значений b и для малых изменений К
ДА ДА .(!-«) г Г25П
Ах=о “о (2-а)К(0) (2-а)и ’
если пренебречь в уравнении (247) всеми членами порядка Ь2 и выше. Че-
рез ДА обозначено увеличение электропроводности, вызванное присутствием
поля. При -а, близком к единице, изменение электропроводности, обуслов-
ленное присутствием поля, пропорционально величине (1—а). При а < 1
этот эффект достигает максимального значения, которое равно
'л^=.т-/т=,/р®-1 + 4';+г4г+--- t252)
5. Из уравнения (245) вытекает, что b пропорционально третьей степени
заряда электрона и обратно пропорционально диэлектрической постоянной.
Уравнение (252), справедливое для бинарных электролитов, является особен-
но важным для растворов с низкой диэлектрической постоянной, где, как
правило, диссоциация очень мала. Величина максимального эффекта для
растворов в воде (21 = 78,57) и в бензоле (D =2,28) в случае полей по-
рядка 5 • 105 в/см оказалась равной Ах/Ах=0=1>37 для воды и 121 для
бензола.
6. Мы показали, что время запаздывания в ионизационном равнове-
сии т' для бинарных электролитов можно вычислить с помощью уравне-
ния (23). Подставляя в это уравнение вместо А его значение из уравне-
ния (237), мы получаем важное соотношение
__ 1 ао ____ (1 %) В /9^4^
Ап (2—а0) “о (2 — а0) '
где а равно аоп (е^ + е|о>2)> т- е- удельной электропроводности раствора,
выраженной в электростатических единицах. Если а0 1, время замедле-
ния Ланжевена равно
• х’ = Л/8тса = D/8r. (9 • 10“£), (254)
где L — удельная электропроводность в практических единицах [уравне-
ние (2) гл. VI]. Сравнение с уравнением (6) показывает, что х' в точности
равно половине величины времени релаксации Максвелла, т. е. времени,
необходимому для экранирования зарядов.
В уравнении (253) а0 и а относятся к стационарному состоянию в поле
с напряженностью X. Поскольку, согласно изложенной теории, как а0,
так и а увеличиваются с возрастанием силы поля, то х' должно при этом
уменьшаться.
Рассмотрением влияния поля на диссоциацию мы заканчиваем краткий
обзор теории междуионного притяжения и ее приложений к обратимым
и необратимым явлениям в растворах электролитов. Полученные резуль-
8*
116
ГЛ АВА IV.
таты будут неоднократно использованы при последующем изложении, свойств
этих растворов.
Поскольку выведенным на основании теории законам еще не придана
форма, наиболее пригодная для вычислений, мы посвятим следующую
главу упрощению этих уравнений, чтобы сделать возможным их практи-
ческое применение.
ЛИТЕРАТУРА
1. а) Р. Langevin, Ann. Chim. Phys., 28, 433 (1903); 6) L. О n s a g e r, J. Chem.
Phys., 2, 599 (1934).
2. H. F a 1 k e n h a g e n, M. D о 1 e, Z. physik. Chem. Abt. B, 6, 159 (1929); Physik.
Z., 30, 611 (1929); H. Falkenhagen, там же, 30, 611 (1929); H. Falken-
‘ h a g e n, E. L. V e г n o’nr там же, 33, 140 (1932).
3. a) L. О n s a g e r, R. M. Fuoss, J. Phys. Chem., 36, 2689 (1932); б) там же,
p. 2759—2770; в) p. 2760—2762; г) p. 2767.
4. L. Pag e, Introduction to Theoretical Physics, Van Nostrand Co., N. Y., 1928:
a) p. 225—229; 6) Equation (43—13); в) Equation (71—3); r) p. 229, Equation (71—18).
5. G. J о n es, M. Dole, J. Am. Chem. Soc., 51, 2950 (1929).
6 P. Курант, Д. Гильберт, Методы математической физики, т. I, изд. 3-е,
ГИТТЛ, М.—Л., 1951.
7 Г Фалькенгаген, Электролиты, ОНТИ—Химтеорет, Л., 1935: а) уравнения
£580) и (581), стр. 302, 303; б) стр. 224—235; в) стр. 213; г) стр. 226, 227; д) стр. 283.
8. М. J. Polissar, J. Chem. Phys., 6, 833 (1938).
9. L. О n s a g e r, Physik. Z., 28,277 (1927).
10. a) J. J. Herman s,- Rec. trav. chim., 56, 635 (4.937); 6) 56, 658 (1937); 58, 1, 259
(1939).
11. L. O n sager, Physical Review, 37, 405 (1931).
12. W. N e г n s t, Z. physik. Chem., 2, 613 (1888).
13. R. T. Birge, Phys. Rev. Suppl., 1, 1 (1929).
14 P. Debye, H. Falkenhagen, Physik Z., 29, 121, 401 (1928); Z. Elektrochem.,
34, 562 (1928).
15. H. Falkenhagen, W. Fischer, Physik Z., 33, 941 (1932); 34, 593 (1933);
Nature, 130, 928 (1932).
16. G. J о о s, M. В 1 u m e n t r i t t, Physik. Z., 28, 836 (1927); M. Blumentritt,
Ann. Physik., 85, 812 (1928).
17. H. Falkenhagen, Physik. Z., 30, 163 (1929); 32, 353 (1931).
18. a) H. F a 1 k e n h a g e n, H. Fleischer, Physik. Z., 39, 305 (1938); б) H. Fa 1-
kenhagen, F. Frohlich, H. Fleischer, Naturwiss., 25, 446 (1937);
в) H. Fleischer, диссертация, Dresden, 1938; r) F. Frohlich, Physik. Z.,
40, 139 (1939).
19. W. S. Wilson, диссертация, Yale University, 1936; H. С. E о k s t г о m,
C. Schmelzer, Chem. Rev., 24, 367 (1939).
20. E. T. W h i t a k e r, G. N. W a t s о n, Modern Analysis, Fourth edition, Cambridge
University Press 1927, Chapter IX, p. 160 ff; P. К у p а и т, Д. Гильберт,
Методы математической физики, т. I и II, изд. 3-е, ГИТТЛ, М.—Л., 1951.
21. М. Wien, J. Schiele, Physik. Z.,, 32, 545 (1931).
22. L. О n s a g e г, диссертация, Mathieu functions of period 4^, Yale University, June
1935.
Глава V
ЗНАЧЕНИЯ ФИЗИЧЕСКИХ КОНСТАНТ, ХАРАКТЕРИСТИЧЕСКИХ
КОЭФФИЦИЕНТОВ НАКЛОНА И МАТЕМАТИЧЕСКИХ ФУНКЦИЙ
Принимая во внимание особенности настоящей монографии, мы собрали
в данной главе основные уравнения теории междуионного притяжения, при-
дав им форму, пригодную для их практического применения. Для соответ-
ствующих уравнений приведены таблицы численных значений предельных
и характеристических коэффициентов наклона, а также дана сводка часто
встречающихся физических констант и математических функций. Мы пола-
гаем, что тщательное изучение содержания этой главы принесет значитель-
ную пользу всем, занимающимся приложениями теории междуионного
притяжения, так как многие уравнения представлены здесь в более удобной
форме по сравнению с их формой, приведенной ранее, и, кроме того, в этой
главе дана сводка таблиц, публикуемых здесь впервые.
§ 1. Основные физические константы
В работах, которые ведутся в течение длительного периода и связаны
С вычислениями, необходимо придерживаться определенных значений физи-
ческих констант, а именно тех значений, которые приведены в «International
Critical Tables» [la] и воспроизведены в табл. 1. Среди этих величин
Таблица 1 *)
Основные физические константы, используемые в последующих таблицах.
Абсолютные электрические единицы
Обозначение Константа Значение
S Заряд электрона 4,774-10~10 абс. эл. ст. ед.
F Число Фарадея 9,6500-104 кулон-г-же-1
23060 кал15'в~1-г-экв~1 2,89365-1011 абс. эл. ст. ед. • е-экв~1
та Нормальный объем идеального газа Температура таяния льда 0° (1 атм) 22,4115 литр-.иолъ-1 273,1° К
R Газовая постоянная 8,316-107 эрг-град^ моль'1
N » » Число Авогадро 1,9869 кал15 град'1 моль'1 6,06.1-1023 лео.гь'1
к кал J5 Газовая постоянная, рассчитанная на одну молекулу Нормальная атмосфера Нормальная атмосфера (широта 45°) Литр Грамм-калория 1,372-10'16 эрг-г/икЭ-1 1,01325-10® ДИН-СЛ4-2 1,01320-10® дин-сл4~2 1000,027 елг> 4,185 джоуля
««л (средн.) 4,186 джоуля
1) Вновь исправленные значения этих констант приведены в табл. 168, см. приложение Б.
118
ГЛАВА V
наибольшим изменениям подверглись значения заряда электрона и числа
Авогадро, поскольку в более ранних вычислениях принималось ошибочное
значение вязкости воздуха. В последнем обзоре Берджа 12] для заряда
электрона и числа Авогадро приведены соответственно значения 4,8025-10-10
эл. ст. ед. и 6,0228-1023. Далее, было найдено, что температура замерзания
воды равна 273,16°. Менее значительные исправления могут быть внесены
в значения других констант. Эти поправки обычно меньше ошибок, прису-
щих физико-химическим экспериментальным данным, и поэтому замена
прежних значений констант на исправленные значения почти не отра-
жается на результатах вычислений.
В табл. 2 приведено несколько функций абсолютной температуры в интер-
вале температур от 0 до 100°, причем в некоторые из этих функций входят
значения газовой постоянной R и числа Фарадея F.
Таблица 2
Некоторые употребительные функции абсолютной температуры
(Т=273,1О+0
1, °C 1g т 1000 т ВТ In ю1) BI2 In 10 Ч !/'Юч 2) z-io5 3)
Р 105
0 2,43632 . 3,66166 0,054191 3,4122 6712 86
5 2,44420 3,59583 0,055183 3,5383 5273 54
10 2,45194 3,53232 0,056175 3,6667 3885 30
15 2,45954 3,47102 0,057168 3,7973 2545 13
18 2,46404 3,43525 0,057763 3,8769 1763 6
20 2,46702 3,41181 0,058160 3,9302 . 1251 3
25 2,47436 3,35458 0,059151 4,0655. 0 0
30 2,48159 3,29924 0,060143 4,2030 - 1210 3
35 2,48869 3,24570 0,061135 4,3428 — 2380 12
40 2,49568 3,19387 0,062128 4,4849 — 3513 26
45 2,50256 3,14367 0,063120 4,6293 — 4610 45
50 2,50934 3,09502 0,004112 4,7760 — 5674 69
55 . 2,51601 3,04785 0,065104 4,9250 — 6704 97
60 2,52257 3,00210 0,066096 5,0762 — 7704 130
70 2,53542 2,91460 0,068081 5,3857 — 9617 206
80 2,54790 2,83206 0,070066 5,7043 — 11421 296
90 2,56003 2,75406 0,072050 6,0320 —13126 398
100 2,57183 . 2,68025 0,074035 6,3688 —14740 511
1) in 10 = 2,3026, В in 10 = 4,575
_ 298,1-Т
' V 2,3026В • 298.1Т '
3) z = 298,11/-А ]g 298Д .
гС 1
а) Диэлектрические постоянные. С точки зрения теории междуионного
притяжения наиболее важным физическим свойством растворителя является
диэлектрическая постоянная. За последнее время выполнено несколько
обширных работ по исследованию диэлектрической постоянной воды. Изме-
рение высоких диэлектрических постоянных в жидкостях, обладающих
заметной электропроводностью, затруднительно. Различные методы не при-
ХАРАКТЕРИСТИЧЕСКИЕ КОЭФФИЦИЕНТЫ И МАТЕМАТИЧЕСКИЕ ФУНКЦИИ Ц9
водят к хорошо совпадающим результатам, в особенности в тех случаях,
когда измерения производятся при высоких температурах. Наиболее точные
данные были получены Дрейком, Пирсом и Дау [3] и Вайменом [4], которые
пользовались методами резонанса. Хотя для выбора той или иной группы
экспериментальных данных нет достаточных теоретических оснований, мы,
исходя из практических соображений, приняли значения Ваймена. Как
показал Акерлёф [5а]1, изменение результатов, полученных Вайменом,
в зависимости от колебаний температуры составляет для воды менее 0,2%.
Оба автора исследовали большое количество смесей неэлектролитов с водой.
Ваймен выразил полученные им в случае воды опытные данные с помощью
следующего уравнения:
D = 78,54 [1 - 0,00460 (t - 25) + 0,0000088 (t - 25)3], (1)
которое было использовано для вычисления значений, приведенных в табл.З.
Данные Акерлёфа для смесей вода—органический растворитель при 25°
приведены в табл. 4.
Таблица 3
Диэлектрическая постоянная воды и ее наиболее употребительные функции
t, °C I) 1g D lg DT dD dT TdP DdT
0 88,00 1,94450 4,38082 0,3958 1,228
5 86,04 1,93471 4,37891 0,3889 1,257
10 84,11 1,92487 4,37681 0,3820 1,286
15 82,22 1,91499 4,37453 0,3751 1,314
18 81,10 1,90904 4,37308 0,3710 1,332
20 80,36 1,90506 4,37208 0,3682 1,343
25 78,54 1,89509 4,36945 0,3613 1,371
30- 76,75 1,88508 4,36667 0,3544 1,400
35 75,00 1,87504 4,36373 0,3475 1,428
40 73,28 1,86496 4,36064 0,3406 1,455
45 71,59 1,85486 4,35742 0,3337 ' 1,483
50 . 69,94 1,84472 4,35406 0,3268 1,510
55 68,32 - 1,83457 4,35058 0,3199 1,536
60 66,74- 1,82440 4,34697 0,3130 1,562
70 63,68 1,80401 4,33943 0,2991 1,611
80 60,76 1,78362 4,33152 0,2853 1,658
90 57,98 1,76325 4,32328 0,2715 1,700
100 55,33 1,74297 4,31480 0,2576 1,737
Диоксан (1,4-окись диэтилена) обладает низкой диэлектрической посто-
янной, имеет нейтральный характер и смешивается с водой во всех отно-
шениях, поэтому он приобрел важное значение в качестве растворителя
в работах по изучению влияния изменения диэлектрической постоянной
на свойства ионных растворов. В связи с этим в приложении А приведены
специальные таблицы (124, 125 и 133), которые могут быть использованы
1 В более поздней работе Ваймен и Ингэлс [56] вычислили заново д£> / дТ.
120
Г Л А BAY
для термодинамических расчетов и для. определений электропроводности
-в. смесях вода—диоксан. Диэлектрические постоянные смесей вода—диоксан
можно вычислить для любой температуры между 0 и 80° по уравнению [6],
\gD = A-Bt, (2)
параметры которого приведены в табл. 5. ?
Таблица 4
Диэлектрические постоянные при 25° смесей воды и (1)—метилового спирта, (2)—
этилового спирта, (3)—н-пропилового спирта, (4)—изопропилового спирта, (5)—
трет.-бутилового спирта, (6)—этиленгликоля, (7)—глицерина, (8)—ацетона, (9)-г
маннита, (10)—сахарозы и (И)—диоксана
Вода, весовой % (1)1) (2) (3) (4) (5) (6) (7) (8) (9) (10) (Н)
90 74,1 72,8 71,8 71,4 70,0 75,6 75,5 73,0 77,1 76,3 69,69
80 69,2 67,0 64,9 64,1 61,3 72,8 72,9 67,0 75,5 73,6 60,79
70 64,3 61,1 57,7 56,9 52,6 69,8 70,0 61,0 70,9 51,90
60- 59,6 55,0 50,3 49,7 43,9 66,6 67,1 54,6 67,9 42,98
50 54,9 49,0 43,0 42,5 35,4 63,2 64,0 48,2 64,2 34,26
40 50,1 43,4 36,4 35,3 27,9 59,4 60,0 41,8 59,9 25,85
30- 45,0 38,0 30,7 28,7 21,4 54,7 55,6 35,7 54,2 17,69
20 40,1 32,8 26,1 23,7 16,5 49,3 50,6 29,6 10,71
10 35,7 28,1 22,7 20,3 12,4 43,7 45,5 24,0 • 5,605
0 31,5 24,3 20,1 18,0 9,9 37,7 40,1 19,1 2,101
1) Ср. [1].
Таблица 5
Параметры, необходимые для вычислении значений диэлектрической постоянной
смесей диоксан—вода с помощью уравнения (2)
Диоксан, весовой % А В Диоксан, весовой % А В
о1) 1,9461 0,00205 60 1,4747 0,00249
10 1,8969 . 0,00215 70 1,3090 0,00245
20 1,8398 0,00224 80 1,0860 0,00225
30 1,7734 0,00233 90 0,7896 0,00164
40 1,6935 0,00241 95 0,5923 0,00100
50 1,5965 0,00247 100 0,3234 0,00004
1) Результаты АкерлВфа для чистой воды помещены в таблице для сравнения. При последу-
ющих расчетах мы всегда будем пользоваться соответствующими значениями, приведенными
в табл. 3.
б) Ионные радиусы. В,табл. 6 приведены значения радиусов некото*-
рых простых ионов, взятые из обзора Паулинга [7а]1. В дальнейшем
изложении часто будут встречаться ссылки на эти величины.
См. также [76].
ХАРАКТЕРИСТИЧЕСКИЕ КОЭФФИЦИЕНТЫ И МАТЕМАТИЧЕСКИЕ ФУНКЦИИ .121
Таблица 6
Ионные радиусы, полученные из кристаллогра-
фических данных [1] (в ангстремах)1 * * * *
F- 1,341 С1- 1,806 Вг- 1,951 J- 2,168 Li* 0,607 Na* 0,958 К* 1,331 Си* 0,96 Rb* 1,484 Ag* 1,26 Cs* 1,656 Au* 1,37 Be** 0,31 Mg** 0,65 Ca** 0,99 Zn** 0,74 Sr** 1,13 Cd** 0,97 Ba* * 1,35 Hg** 1,10 In*** 0,81 La*** 1,15 Tl*** 0,95
§ 2. Уравнения, выведенные в гл. III для вычисления термодинамических
функций из электростатических свойств ионных атмосфер
В гл. III было показано, что предельное выражение для среднего рацио-
нального коэффициента активности сильного электролита, диссоцииру-
ющего на р видов ионов, имеет вид [уравнение (18) гл. III]
1g/±= -^(/)/г,
В этом выражении [согласно уравнению (19) гл. III]
1 -Д 2 1,283 • 106
(/) (DT)a^
и [см. уравнение '(3) гл. III]
Г = У CizX.
1
Значения теоретического предельного коэффициента наклона для вод-
ных растворов при разных температурах приведены в табл. 7. Значе-
ния DT, употреблявшиеся при вычислении этих коэффициентов, были взяты
из четвертого столбца табл. 3. Для упрощения вида таблицы фактор
1 Большинство советских исследователей пользуются не данными Паулинга, рас-
пространенными в американской литературе, а более достоверными, так называемыми
эмпирическими ионными радиусами Гольдшмидта (В. М. Гольдшмидт, Кристаллов
химия, 1942). В последнее время эти данные были расширены благодаря введению
так называемых термохимических радиусов [А. Ф. К а п у с т и н с к и й, Z. Kristallograpk.,
86, 359 (1933); ДАН СССР, 32, № 1, 59 (1941); А. Ф. К а п у с т и н с к и й, К. Б. Яци-
мирский, ЖОХ, 19, вып. 12, 2192 (1949). См. также Ю. В. X о д а к о в, Элементы
электростатической химии, 1934]. (Прим, ред.)
122
ГЛАВА V
Таблица 71)
Постоянные Дебая и Гюккеля для водных растворов2)
(, °C ~lg z± ®(Л- е -1g /± A_ _ x*10 8 2 КГ CO 1
0 0,3446 w 0,4870 w' 0,2294 0,3244w"
5 0,3466 w 0,4902 w' 0,2299 0,3251 w"
10 0,3492 w 0,4938 w' 0,2305 0,3260w"
15 0,3519 w 0,4977 w' 0,2311 0,3268 w"
18 0,3538 w 0,5002 w' 0,2315 0,3274 w"
20 0,3549 w 0,5019 w' 0,2318 . 0,3278w"
25 0,3582 w 0,5065 w' ’ 0,2325 0,3288 w"
30 0,3616 w 0,5114 w' 0,2332 0,3298w"
35 0,3653 w 0,5166 w' 0,2340 0,3309 w"
40 0,3692 w 0,5221 w' 0,2348 0,3321 w"
45 ' 0,3733 w 0,5280 w' 0,2357 0,3333 w"
50 0,3777 w 0,5341 w' 0,2366 0,3346 w”
55 0,3823 w 0,5406 w' 0,2376 0,3360 w*
60 . 0,3871 w 0,5474 w' 0,2386 0,3374 w"
70 0,3973 w 0,5619 w' 0,2407 0,3403w"
80 0,4083 w 0,5774 w' 0,2429 0,3434 w"
90 0,4200 w 0,5940 w' 0,2452 0,3467 w"
100 0,4325 w 0,6116 w' 0,2476 0,3501 w"
1) Факторы валентности w, wr и w" определяются уравнениями (3), (6) и (12).
2) Соответствующие предельные коэффициенты наклона для некоторых важных смесей
растворителей даны в табл. 125. Исправленные значения см. в табл. 169 (приложение Б).
валентности обозначен через w, причем
р
«’=72’4 с3 * *)
1
Если электролит диссоцирует только на два вида ионов (р = 2), то w
в общем случае равно ]z1z2], а для 1,1-валентных электролитов w ста-
новится равным единице.
Для важного частного случая раствора, содержащего только один
п
электролит, Г = с^ущ{, и предельный закон может быть записан следу-
1
ющим образом:
lg/±=-§(П)/с, - (4)
где
_ Vv 2у/21,283 • 106
= (Д^з/з- • (5)
ХАРАКТЕРИСТИЧЕСКИЕ КОЭФФИЦИЕНТЫ И МАТЕМАТИЧЕСКИЕ ФУНКЦИИ 123
Значения приведенные в табл. 7, выражены через фактор валентности
<6>
Коэффициент 2 введен в это выражение для того, чтобы для 1,1-ва-
лентных электролитов w' превращалось в единицу.
В очень разбавленных растворах, для которых выводится предельный
закон, отношение объемной концентрации к весовой становится равным d0,
т. е. плотности чистого растворителя. Таким образом, согласно уравнению
(21а) гл. III, в общем случае
lg/±=-/2^(/д/ь (7)
а для растворов, содержащих только один электролит,
1g/± = — j/Л
(8)
При умеренных разбавлениях, когда уже нельзя пренебрегать ионным
параметром а (средним расстоянием сближения ионов), логарифм коэффи-’
циента активности равен [уравнение (34) гл. III] •
1g /± =
1 + А / Г
Член А у/ Г равняется ха. Удобно выразить ионный параметр в ангстремах
и обозначить его в этом случае через а. Соответственно этому
л °
А = ак
10~8
/т
о 35,57
а------— .
(DT)1/2
(9)
Значения х10-8Г~1/2, вычисленные по уравнению (2) гл. III, приведены
в табл. 7.
Для растворов, содержащих только один электролит, можно исполь-
зовать уравнение (34) гл. III в упрощенном виде:
g(/> Ус_
1 + А' /с ’
где
1g /± =
,, °
А = а
ИО"8
V*
(Ю)
(И)
Значения хПГ8с-1/2, приведенные в табл. 7, выражены с помощью фактора
валентности
<12>
1
Следует отметить, что для бинарных электролитов, когда (| zr | = | z21 = z),
фактор валентности w" становится равным z.
Вычисления, при которых учитываются так называемые члены высших
порядков теории Дебая и Гюккеля, выполняются с помощью уравнений
Таблица 8
Члены высших порядков уравнения Дебая и Гюккеля
х=ха 10s (±х8-2У8) 105 (±Х5-4У5) 102 (-уХ2-У2 ) 103 (4Хз-2Гз*)
0 0,00000 0,00000 0,000000 0,000000
0,005 -0,00092 -0,00107 -0,001482 0,000009
0,01 -0,00351 -0,00416 -0,004797 0,000071
0,02 —0,01239 - 0,01487 -0,014784 0,000423
0,03 -0,02497 -0,02985 -0,027666 0,001264
0,04 -0,04014 -0,04725 -0,042363 0,002693
0,05 . -0,05711 -0,06564 -0,058209 0,004750
0,06 -0,07522 -0,08394 -0,074748 0,007457
0,07 — 0,09403 -0,10138 -0,091660 0,010809
0,08 . -0,11316 -0,11737 - 0,108703 0,014766
0,09 -0,13231 . -0,13153 -0,125699 0,019291
0,10 — 0,15130 - 0,14363 -0,142514 0,024335
0,11 -0,16992 . -0,15356 -0,15905 0,02986
0,12 '• -0,18802 -0,16126 - 0,17522 0,03580
0,13 ' -0,20355 -0,16680 -0,19097 0,04211
0,14 -0,22240 -0,17023 -0,20626 0,04874
0,15 -0,23853 -0,17166 -0,22105 0,05563
0,16 -0,25391 -0,17123 -0,23533 0,06274
0,17 -0,26851 -0,16910 -0,24908 0,07002
0,18 -0,28231 -0,16543 -0,26230 0,07743
0,19 . -0,29530 -0,16037 -0,27497 0,08493
0,20 -0,30750 - 0,15409 -0,28710 0,09248
0,21 ! -0,31892 - 0,14674 -0,29870 0,10005
0,22 -0,32953 -0,13847 -0,30977 0,10761
0,23 '-0,33943 -0,12942 -0,32032 0,11513
0,24 ' -0,34859 -0,11973 -0,33036 0,12258
0,25 -0,35703 -0,10953 -0,33991 0,12994
0,26 -0,36478 -0,09895 -0,34896 0,13720
0,27 -0,37187 -0,08804 -0,35755 0,14433
0,28 -0,37832 -0,07696 -0,36567 0,15132
0,29 -0,38416 -0,06577 . -0,37335 0,15816
0,30 -0,38942 -0,05453 -0,38060 0,16482
0,31 -0,39411 -0,04335 -0,38743 0,17132
0,32 -0,39827 -0,03222 -0,39386 0,17762
0,33 -0,40193 -0,02131 -0,39990 0,18374
0,34 -0,40510 -0,01053 -0,40557 0,18965
0,35 -0,40780 -0,00001 -0,41087 . 0,19536
0,36 -0,41007 +0,01027 -0,41583 0,20087
0,37 -0,41193 +0,02022 -0,42045 0,20617
0,38 -0,41339 +0,02986 -0,42475 0,21116
0,39 -0,41448 +0,03917 -0,42875 0,21613
0,40 -0,41525 +0,0481 -0,43244 0,22080
ХАРАКТЕРИСТИЧЕСКИЕ КОЭФФИЦИЕНТЫ И МАТЕМАТИЧЕСКИЕ ФУНКЦИИ 125
(42)—(44) гл. III. Эти уравнения содержат дополнительные члены, по-
являющиеся при учете высших членов разложения в ряД функции
ехр( — e^i/kT), которыми мы пренебрегали при выводе уравнения (18)
гл. III. Коэффициенты активности электролитов с симметричным типом
валентности вычисляются по уравнению (42) гл. III. Величины
10® [|хз(ха)-2У3 (ха)]
и
10® [|У5(ха)-4У5(ха)]
приведены в табл. 8 для значений ха, изменяющихся от 0,005 до 0,4 [8] У
Уравнения (43) и (44) гл. III применяются для электролитов с несимметрич-
ным типом валентности. Численные величины выражений
102 £-|х2(ха) — У2(ха)]
и.
10» [|Х5(ха)-2У1ха)]
также приводятся в табл. 8 для значений ха, изменяющихся от 0,005 до
0,4'[9]. Вычисление членов высших порядков осложняется необходимостью
применения последовательных арифметических приближений. Для определе-
ния этих функций должна быть известна величина а; для определения а
в свою очередь должны быть известны эти функции. Некоторые приложе-
ния этих величин будут детально рассмотрены ниже. С помощью приведен-
ных в двух последних столбцах табл. 7 отношений можно легко находить х,
если известно Г или с.
а) Функции теории ассоциации ионов Бьеррума [10]. Уравнение (57)
гл. Ill связывает константу диссоциации с диэлектрической постоянной
и функцией Q(b). Согласно этому уравнению,
К ~ 1000 Ч kDT J Q
где b равно [уравнение (51) гл. III]
, _ | Z1z2 |
aDkT ’
и Q(b), согласно уравнению (58) гл. III, равно
ь
Q (ь) = еГу-4 dY [е2 - Ei (2) + Ei(^ - т С1 + т+!)]’
2
и по уравнению (59) гл. III
—X
Ei (ж) = е~Ч~1 dt.
ОО
Значения Ei(b) до 5 = 15 были взяты Бьеррумом из таблиц [И]. Значе-
ния Ei(b) для b от 15 до 80 были получены Фуоссом и Краусом [12],
которые использовали асимптотическое разложение в ряд
+1+^+4^-+•••)• (13>
1 Отметим, что авторы принимают х/ — ха.
126
ГЛАВА V
Логарифмируя и пренебрегая членом -g- [е2 — Ei (2)] — 0,41, получаем урав-
нение
lg Q (6) 0,43436 — 41g 6+ 1g (1 + 5), (14)
где
л 4 . 4-5 , 4-5-6 ,
g = ¥ + ”^^-&з~+•••
(15)
Принимая это приближение, мы вплоть до значения Ь, равного 15, делаем
ошибку в величинах lg Q(b), но эта ошибка быстро уменьшается с увели-
чением Ь. Значения lg Q (b) в табл. 9 были получены с помощью приве-
денных выше выражений.
Таблица 9
Функция lg Q (&) теории Бьеррума
ь IgQ(b) ь lg Q (Ь) b lg Q (b)
2 _ Qf' 10 0,655 40 11,01
'• 2,5 - -0,728 12 1,125 45 12,99
3 -0,489 14 1,680 50 14,96
, 4 -0,260 16 2,275 55 16,95
5 -0,124 18 2,92 60 18,98
6 +0,016 20 3,59 65 21,02
7 0,152 25 5,35 70 23,05
8 0,300 30 7,19 75 25,01
9 0,470 35 9,08 80 27,15
б) Функции теории образования ионных тройников, развитой Фуос-
сом и Краусом. Мы показали (гл. III, § 7), что для диссоциации иовного
тройника, выражаемой уравнениями
I
[С+А-С+]+ [С+А“]° + С+,
[А~С+А~]- [С+А-]0 + А~,
константа диссоциации ионного тройника К3 может быть вычислена по фор-
муле (69) гл. III:
1 2-rc.ZV<z§ Г Г h \
Ks --Tooo^bs>’
где 63 [уравнение (51) гл. III] равно
л
°8 asDkT •
Табл. 10 содержит 7(63) и lg/(63) для значений Ь3 от 3,5 до 36,6. В по-
следнем столбце приведены значения X = R/a3, где Л,— характеристическое
расстояние, от величины которого зависит возможность образования ионного
тройника.
ХАРАКТЕРИСТИЧЕСКИЕ КОЭФФИЦИЕНТЫ И МАТЕМАТИЧЕСКИЕ ФУНКЦИИ 127
в) Предельные коэффициенты наклона для относительных парциаль-
ных молярных теплосодержания и теплоемкости в водных растворах.
Как было показано, относительное парциальное теплосодержание зависит
от концентрации, согласно уравнению (81) гл. III:
Г2=я2-я- = ^(Я)/Т,
а относительная парциальная молярная теплоемкость — согласно урав-
нению (82) гл. III:
J2 = Срз — Ср2 = &(СР) Г.
Согласно уравнению (78) гл. Ill, §г>(Я) является функцией D, Т и коэффи-
циента расширения раствора а.. представляет собой более сложную
функцию от тех же переменных и вычисляется по уравнениям (83) — (85)
Таблица 10
Л
Lg/(63), 7(63) и Х=— как функции &3
я3
1g Т(Ьз) 1g ЦЬз)-31gb3 •Г(Ь3) X
8/3 — СО — СО 0 1,0
3,5 0,096 —1,536 1,25 1,225
5 0,668 —1,429 4,66 1,56
10 1,534 —1,466 34,2 2,46
15 2,183 —1,345 152,4 3,17
20 2,894 — 1,009 784,0 3,76
25 3,732 —0,462 5,40-103 4,28
30 4,634 +0,203 4,30-Ю1 4,75
35 5,565 + 0,933 3,67-105 5,19
36,6 5,878 + 1,187 7,55-Ю5 5,33
I (Ьз)=0> когда так что кривая, выражающая зависимость 1g I (Ьз) от Ь3, асимптоти-
чески стремится к вертикальной прямой Ьз=8/3. Это означает, что ионные тройники образуются
при Ъ > 8/3 и являются неустойчивыми при 8/3.
гл. III. Природа этих функций такова, что величины &(Н) и > очень
чувствительны к экспериментальным погрешностям при измерений темпе-
ратурного коэффициента D. Поэтому между значениями коэффициентов
наклона, приводимыми в литературе, имеются большие расхождения, зави-
сящие от различий в значениях диэлектрической постоянной; эти различия
связаны с тем, что значения диэлектрической постоянной берутся из разных
источников. Коэффициенты наклона, приведенные в табл. 11, были вычис-
лены [13] с помощью значений диэлектрических постоянных, полученных
Вайменом и приведенных в табл. 3, и значения плотности воды, взятого
из «International Critical Tables» [16].
Для растворов, содержащих только один электролит, предельные урав-
нения удобно представить в следующем виде:
L2 = H2-H° = S(H)yc (16)
И
? = СР2-С°2= 5(Ср)]А. (17)
Значения 5(Я) и §(ср) приводятся в табл. 11. Для 1,1-валентных элек-
тролитов (у = 2) факторы валентности w и w' равны единице.
128
ГЛАВА V
Таблица И1)
Коэффициенты наклона, вычисленные с помощью теории
Дебая и Гюккеля для относительных парциальных молярных
теплосодержания и теплоемкости в водных растворах2).
(°C ^(H)=b2/Vf S(H) = L2/Kc ^>ср“^/Кг Scp=J2 / Ke
0 153vw 216vw' 3,40vw 4,81vw'
5 169vw 239vw' 3,67vw 5,19vw'
10 190vw 269vw' 3,96vw 5,60vw'
15 210vw 297vw' 4,22vw 5,97vw'
20 231 vw 327vw' 4,48v w 6,34vw'
25 254vw 359vw' 4,72vw 6,68vw'
30 278vw 393v w' 4,96vw 7,01vw'
35 303vw YUdvw' 5,19vw 7,34vw'
40 329vw 5,44vw 7,69vw'
45 357vw bOb'iw' 5,67vw 8,02vw'
50 385vw 5,89vw 8,33vw'
55 414v w 585vw' 6,08vw 8,60vw'
60 446v w 631vW' 6,30vw 8,91vw'
1) Факторы валентности w и w’ определяются по уравнениям (3)
п (6).
2) Соответствующие предельные коэффициенты наклона для неко-
торых важных смесей растворителей приведены в^табл. 133. Исправ-
ленные значения см. в табл. 169 (приложение Б). t
г) Предельные коэффициенты наклона для относительных пар-
циальных молярных объема, расширяемости и сжимаемости электролита.
Как видно из уравнений, приведенных в гл. III, § 9, S>(E> и
являются функциями как температурного коэффициента диэлектрической
постоянной растворителя, так и коэффициента, характеризующего зависи-
мость диэлектрической постоянной растворителя от давления. Величины
этих коэффициентов для случая воды можно вычислить по уравнениям,
приведенным в § 1 приложения Б. Экспериментальные значения приведены
в табл. 169.
д) Теоретические уравнения для поверхностного натяжения электро-
литов. Как было показано, предельный закон изменения поверхностного
натяжения воды при растворении в ней бинарных электролитов выражается
уравнением (159) гл. III
л № /V । v \ кТ v
Да = а -— ~Г5---5" (^С “Ь ^/) — Tfi-Г ’
и ТОЯ?2 ' с 11 IOkj2
где по уравнению (48) гл. III
_ S222
q~ 2ВкТ •
Если учесть среднее расстояние наибольшего сближения ионов, то по урав-
нению (160) гл. III
а кТ /v, . кТ v,
Да = а — ап = —j- (Yc +1/) = -тл—5- Y .
0 16л?2 v ‘' Ibrcj2
Пусть
, , » 1,746 • 1013 6 D
J/2 = %2?2 = .. zec = Bc
(18)
ХАРАКТЕРИСТИЧЕСКИЕ КОЭФФИЦИЕНТЫ И МАТЕМАТИЧЕСКИЕ ФУНКЦИИ 129
И
?/2 2/2 N**z2 \ CQf) J ’ v '
где Да — относительное увеличение поверхностного натяжения, рассчитанное
на моль растворенного вещества.
Таблица 12
Значения функций, применяемых для вычисления увеличения
поверхностного натяжения в растворах для 1,1-валентных электролитов при 25°
У2 Ч-/р2 Yf/v- А-а V2 г'/у2 Г^/У2 АДа
0 СО 0
0,0063 3,048 —0,076 2,972
0,0126 2,747 —0,097 2,650
0,0189 2,576 —0,111 2,465
0,0315 2,367 —0,134 2,233
0,0378 2,295 —0,141 2,154 0,0378 2,308 —0,107 2,201
0,0630 2,095 —0,163 1,932 0,0850 2,003 —0,125 1,878
0,0944 1,944 —0,184 1,760
0,1259 1,840 —0,203 1,637 0,1259 1,867 —0,132 1,735
0,1574 1,760 —0,217 1,543
0,1889 1,697 —0,228 1,469 0,1963 1,718 —0,139 1,579
0,2014 1,675 —0,232 1,443
0,2266 1,634 —0,241 1,394
Численные значения Yc, Yf, Y'c и Y'f для 1,1-валентных электролитов
были получены Онзагером и Сэмарасом [14]. Эти величины, деленные на у2,
приведены для соответствующих значений у2 в табл. 12. Значения ЛДа
также включены в таблицу. Отметим, что если при вычислениях учесть
среднее расстояние наибольшего сближения ионов, то это не окажет суще-
ственного влияния на результаты.
§ 3. Уравнения, выведенные в гл. IV
для необратимых процессов
а) Предельный закон для вязкости. Предельный закон для зависимости
увеличения вязкости раствора от количества добавленного электролита, дис-
социирующего на два вида ионов, имеет следующий вид [уравнение (51)
гл. IV]:
* х Г" , / ( 1—1
г,• = ,-,.=вф.Ц ,
где по уравнению (48) гл. IV
wIe|/o)I + n2eg/a>f
~ ще2 -р я2е| ’
по уравнению (49) гл. IV
и по уравнению (50) гл. IV
* _ 4- я2е|ч>2
* («lCl + »2e!) ('”1 +“2)
9 Г. Харнед, Б. Оуэн
130
ГЛАВА V
Следует помнить, что во всех этих соотношениях е± и е2 имеют соответ-
ствующий знак заряда. Таким образом, для бинарного электролита
61= — е2. Для того чтобы перейти к практическим единицам, заменим х
и o>i их значениями. По уравнению (2) гл. III
< 4®-^ V/2 лр _ 35,57 108 УТ
7 ~ кдОООЯАГ) И 1 —
п по уравнению (15) гл. IV
300/“
~ 96 500 I zf I е ’
где Г равно У] az?, к? представляет собой электропроводность данного вида
ионов при бесконечном разбавлении, а | Zj | — валентность. Далее, по усло-
вию электронейтральности
п1е1 = — п2е2, пг | zx | г = п21 г21 г,
где е —заряд электрона, а
__ CjN
ni — 1000 ’
Здесь с,— концентрация i-ионов в молях на литр раствора. Для бинар-
ных электролитов z?,j=re2 и |z1|=jz2|. Следовательно, д'* = 1/з> р =
= Va (l/«>i + 1/«>2) и р2 = Va (1/^1 + l/^i), чт0 значительно упрощает урав-
нение (51) гл. IV для данного частного случая.
б) Предельный коэффициент наклона для эквивалентной электро-
проводности сильного электролита. Вычисление предельного коэффициента
наклона для общего случая электропроводности смесей растворов электро-
литов является весьма сложным. Оно может быть выполнено при помощи
уравнений (104) —(109) гл. IV. Отдельные примеры разбираются в гл. VI
и VII.
Для наиболее важного частного случая одного электролита, диссоци-
ирующего только на 'два вида ионов, уравнение эквивалентной электропро-
водности [уравнение (97) гл. IV] можно написать так:
А = А»-§(Д)/ё, (20)
а предельный коэффициент наклона выражается в удобной форме
(21)
(22)
(23)
§(Д) = а*А°-|-р*.
Значения постоянных
* Г 1,970 10е • 0,2929 /2 I ,
L (_ог)3/2 J #
II
О* Г 28,98 21/2 1 *
--------------17— \ w*
L т](£)Г)/2 J
приведены в табл. 13. Значения вязкостей, применявшихся для расчета
величин р*, приводятся в последнем столбце этой таблицы. Величины
0,2929 /2 и 2 / 2 введены в скобки, и, таким образом, множители
Q =---------. (24}
4 0,2929 (! + /?*) 4
И
I Z1 N~’l 22 I v | г122 I
ХАРАКТЕРИСТИЧЕСКИЕ КОЭФФИЦИЕНТЫ И МАТЕМАТИЧЕСКИЕ ФУНКЦИИ 131
равны единице для 1,1-валентных электролитов. Фактор валентности w',
определяемый уравнением (6), для рассматриваемого случая имеет следующее
значение:
w' = |z1z2|(|z1z2|v/2)1/2. (26)
Параметр q*, который входит в выражение для Q, согласно уравнению (96)
гл. IV, равняется
п* _ ________I Z1Z2 I Cl + ^2)_
7 ( I Z1 I + | Z2 I ) ( | z2 И + I Z1 I Хз) ‘
Q можно также выразить через предельные числа переноса ионов раствора.
Тогда
<9 =_________З.'4141‘у2_____ (27)
где v = | z2| /1 Zi I и 52 = в/(1-j-и). При вычислении Q по этому уравнению
несущественно, обозначаем ли мы индексом 2 катион или анион. Следова-
тельно, Q имеет одинаковый вид для сернокислого натрия и хлористого
бария. Q равно единице для всех электролитов симметричного типа
Таблица 131)
Значения постоянных, входящих в выражение (21),
определяющее предельный коэффициент наклона*
кривых электропроводности в водных
растворах2)
(°C а* 103v)0 [П
0 0,2198 w’Q 29,47 w* 17,938
5 0,2205 w’Q 34,87 w* 15,188
10 0,2220 w’Q 40,56 w* 13,097
15 0,2237 w’Q 46,53 w* 11,447
. 18 0,2249 w’Q 50,31 w* 10,603
20 0,2257 w’Q 52,95 w* 10,087
25 0,2277 w’Q 59,86 w* 8,949
30 0,2299 w’Q 67,15 w* 8,004
35 0,2322 w’Q 74,81 w* 7,208
40 0,2348 w’Q 82,79 w* 6,536
45 0,2374 w’Q 90,99 w* 5,970
50 0,2401 w’Q 99,28 w* . 5,492
55 0,2431 w’Q 107,93 w* 5,072
60 0,2461 w’Q 116,98 w* 4,699
1) Факторы валентности w',Q и w* определяются урав-
нениями (24)—(26). Исправленные значения см. в табл. 169
(приложение Б).
2) Значения а* и ₽* для некоторых важных смесей
растворителей даны в табл. 124.
. Для эквивалентной электропроводности ионов одного вида в раство-
рах, содержащих только два вида ионов, уравнение (95) гл. IV имеет вид.
Х/ = Х°- 5(Л)/с, г (28)
где
+ ' = * 2' <28а)
132
ГЛАВА V
в) Теория диффузии. В гл. IV, § 4, было показано, что для коэффи-
циента диффузии электролита, диссоциирующего на ионы двух видов,
согласно теории диффузии Онзагера и Фуосса, получается выражение [урав-
нение (136) гл. IV]
S = 16,632 • 1010 Z7 —f 1 -i-c-^^O .
с \ ос J
представляет собой сложную функцию, которую можно вычислить
с помощью уравнений (131) —(133), (74а) и (76а) гл. IV. Для электроли-
тов симметричного типа эта функция может быть написана в следующей
форме:
(Ы\ 4020 -V 074 AV-Д 0,4373 Л°+-^°_> ха ,
W г V А" ) к Д° ) l + xa^
<29)
Такая форма удобна для вычислений. Значения (ха)2 • ® (ха) приведены
в табл. 14. В гл. VI, § 10, приводится эквивалентное выражение для
зависимости предельного коэффициента наклона 55 от ]/ с [см. уравнения
(58) и (63) гл. VI].
Таблица 14
Значения функций и входящих в уравнения
(133) гл. IV и (53) гл. VI
{ха) ¥(ха) (ха)2?(ха) (ха) ?(ха) (ха)2 ’Р(ха)
0 СО 0 0,5 0,2651 0,06628
0,010 3,3550 0,00034 0,6 0,2054 0,07394
0,025 2,4695 0,00154 0,7 0,16304 0,07989
0,05 1,8273 0,00457 0,8 0,13194 0,08444
0,10 1,2342 0,01234 0,9 0,10844 0,08784
0,15 0,9245 0,02080 1,0 0,09033 0,09033
0,20 0,7277 0,02911 1,2 0,06477 0,09327
0,25 0,5907 0,03692 1,4 0,04813 0,09433
0,30 0,4890 0,04409 1,6 0,03676 0,09411
0,35 0,4130 0,05059 1,8 0,02876 0,09318
0,40 0,3527 0,05643 2,0 0,02292 0,09168
0,45 0,3044 0,06164 2,5 0,01391 0,08694
0,50 0,2651 0,06628 3,0 0,00908 0,08172
г) Теория влияния частоты переменного тока на электропроводность.
Молярная электропроводность А. при наложении переменного напряжения
с частотой & может быть представлена, согласно уравнению (156) гл. IV,
-в виде суммы трех составляющих, а именно:
А~ = А^ — А ~ — Ац.
Здесь Am —молярная электропроводность при бесконечном разбавлении,
Ах~ — составляющая А~, обусловленная несимметричностью ионной атмо-
сферы, и Ап— составляющая, обусловленная электрофоретическим эффектом.
ХАРАКТЕРИСТИЧЕСКИЕ КОЭФФИЦИЕНТЫ И МАТЕМАТИЧЕСКИЕ ФУНКЦИИ 133-
Aj~ определяется уравнением (157) гл. IV и является сложной функ-
цией частоты переменного тока, времени релаксации и величины q*.
Aj (0) представляет собой вычисленную Онзагером составляющую молярной
электропроводности, обусловленную несимметричностью ионных атмосфер
при й, равном нулю. Численные значения Ат-/А1(-=0), полученные Фаль-
кенгагеном [15а] для разных йт и q*, приведены в табл. 15.
лца=о)
Таблица 15
d>T 0,5 0,45 0,40 0,35 0,30
0,0 1,0 1,0 1,0 1,0 1,0
0,2 0,997 0,997 0,997 0,997 0,997
0,5 0,980 0,981 0,981 0,981 0,981
0,75 0,956 0,959 0,962 0,965 0,968
1,0 0,930 0,934 0,938 0,942 0,946
1,25 0,904 0,909 0,913 0,918 0,922
1,5 0,876 0,882 0,888 0,894 0,900
2,0 0,826 0,833 0,841 0,848 0,856:
2,5 0,783 0,791 0,799 0,808 0,817
3,0 0,745 0,753 0,763 0,773 0,785
4,0 0,682 0,691 0,701 0,713 0,727
6,0 0,593 0,603 0,614 0,628 0,643
8,0 0,531 0,540 0,553 0,565 0,582
10,0 0,486 0,496 0,507 0,520 0,537
15,0 0,409 0,417 0,428 0,442 0,458
20,0 0,360 0,368 0,378 0,390 0,406
25,0 0,326 0,334 0,344 0,354 0,369
30,0 0,299 0,307 0,317 0,327 0,342
35,0 0,279 0,286 0,295 0,305 0,318
40,0 0,261 0,267 ’ 0,272 0,287 0,299
45,0 0,247 0,254 0,263 0,272 ©,288
50,0 0,235 0,242 0,250 0,259 0,271
75,0 0,193 0,199 0,206 0,214 0,223
100,0 0,168 0,173 0,179 0,186 0,195
150,0 0,138 0,142 0,147 0,153 0,160
200,0 0,1195 0,1232 0,1278 0,1328 0,1394
300,0 0,0983 0,1011 0,1046 0,1088 0,1145
500,0 0,0759 0,0783 0,0812 0,0844 0,0888
700,0 0,0643 0,0663 0,0688 0,0716 0,0751
1000,0 0,0538 0,0555 0,0576 0,0599 0,0629
5000,0 0,0241 0,0249 0,0258 0,0269 0,0282
10000,0 0,0171 0,01755 0,0182 0,0190 0,0199
Влияние частоты на диэлектрическую постоянную выражается уравне-
нием (160) гл. IV, которое определяет D& — Do как функцию от q*, fit,
Тис. Вычисленные Фалькенгагепом значения (Z)~ — Z)o) / (Л~= Q — Ло)
для разных шт и q* вошли в табл. 16 [156].
134
ГЛАВА V
(Dil-D0)/(D&=0-D0)
Таблица 16
Q* 0,5 0,45 0,4 0,35 0,3
0,0 1,0 1,0 1,0 1,0 1,0
0,1 0,999 0,999 0,999 0,999 0,999
0,2 0,989 0,989 0,989 0,9895 0,990
0,35 0,974 0,974 0,974 0,975 0,975
0,5 0,940 0,941 0,943 0,944 0,946
0,75 0,879 0,882 0,885 0,887 0,890
1,0 0,813 0,816 0,820 0,824 0,830
1,25 0,748 0,752 0,757 0,763 0,768
1,5 0,689 0,694 0,700 0,706 0,713
2,0 0,585 0,591 0,598 0,605 0,615
2,5 0,502 0,510 0,517 0,525 0,535-
3,0 0,437 0,444 0,452 0,406 0,471
4,0 0,342 0,349 0,356 0,365 0,375
6,0 0,231 0,237 0,244 0,251 0,261
8,0 0,1703 0,1753 0,1811 0,1877 0,1957
10,0 0,1326 0,1369 0,1419 0,1477 0,1546
15,0 0,0822 0,0853 0,0889 0,0931 0,0983
20,0 0,0576 0,0600 0,0627 0,0660 0,0700
25,0 0,0434 0,0453 0,0475 0,0501 0,0533
30,0 0,0343 0,0358 0,0376 0,0398 0,0425
35,0 0,0280 0,0293 0,0316 0,0327 0,0350
40,0 0,0235 0,0246 0,0259 0,0275 0,0298
45,0 0,0201 0,0210 0,0222 0,0236 0,0253
50,0 0,01741 0,01826 0,0193 0,0205 0,0220
75,0 0,01001 0,01053 0,01114 0,01190 0,01284
100,0 0,00672 0,00707 0,00750 0,00803 0,00869
150,0 0,00380 0,00401 0,00426 0,00457 0,00497
200,0 0,00252 0,00267 0,00284 0,00302 0,00332
300,0 0,001411 0,001493 0,001592 0,001714 0,001870
500,0 0,000674 0,000713 0,000762 0,000822 0,000899
700,0 0,000413 0,000437 0,000467 0,000505 0,000553
1000,0 0,000245 0,000259 0,000268 0,000300 0,000329
5000,0 0,0000227 0,0000241 0,0000258, 0,0000280 0,0000308
10000,0 0,00000808 0,00000859 0,00000922 0,00000999 0,0000110
Время релаксации, входящее в эти расчеты, можно вычислять по
простой формуле, получающейся из уравнения (17) гл. IV:
(30)
Здесь A0 = kj4-),2) с* — vi | zi Iс = v21 z21 c и — диэлектрическая постоянная
растворителя; q* определяется уравнением (96) гл. IV:
_ 8,85 • 10~и п
Д°с* °'
__________I ziza I (^i + ^2)_________
(I Z1 I + I z2 I) (I z2 I + I Z1 I M)
ХАРАКТЕРИСТИЧЕСКИЕ КОЭФФИЦИЕНТЫ И МАТЕМАТИЧЕСКИЕ ФУНКЦИИ 135
Для вычисления Aj~/AI(~=o)h А~, а также — Z)o)/(Z)~=O —Z>0) необ-
ходимо знать валентности, предельные электропроводности ионов при бес-
конечном разбавлении, концентрацию электролита, диэлектрическую по-
стоянную растворителя и температуру. Так как в этих расчетах иногда при-
меняется величина длины волны I, выраженная в метрах, то ниже приво-
дится уравнение для вычисления к.
, 2л 3 .. Ю1» _ 18,85 • 10s
Z “ Й100 ~ '
д) Влияние сильных полей на электропроводность электролитов.
Уравнение (216) гл. IV теории эффекта Вина, предложенной Онзагером
Вильсоном, переходит в случае бинарного электролита в следующее выра-
жение:
. n I е}’ I2 хЛ° , х 96500 к I е/I 2х х /зо\
Л=Л'-ТЙГ (3"’
где по уравнению (204) гл. IV
р.' Xz z
х —--= .
х кТх —
Таблица 171)
Функция g (х)'-}
' X g (х) X g (х) X g (%) X g (х)
0,0 0,19526 2,5 0,11977 10,0 0,04354 24,0 0,01965
0,1 0,19492 2,6 0,11721 10,5 0,04173 25,0 0,01891
0,2 0,19391 4,2 0,08656 11,0 0,04007 26,0 0,01822
0,3 0,19228 4,4 0,08376 11,5 0,03853 27,0 0,01758
0,4 0,19009 4,6 0,08113 12,0 0,03711 28,0 0,01698
0,5 0,18742 4,8 0,07865 12,5 0,03579 29,0 0,01643
0,6 0,18437 5,0 0,07632 13,0 0,03456 30,0 0,01590
0,7 0,18102 5,2 0,07411 13,5 0,03341 31,0 0,01541
0,8 0,17745 5,4 0,07203 14,0 0,03233 32,0 0,01495
0,9 0,17373 5,6 0,07005 14,5 0,03132 33,0 0,01452
1,0 0,16992 5,8 0,06818 15,0 0,03037 34,0 0,01413
1,1 0,16607 6,0 0,06640 15,5 0,02948 35,0 0,01372
1,2 0,16223 6,2 0,06472 16,0 0,02864 38,0 0,01269
1,3 0,15842 6,4 0,06311 16,5 0,02784 41,0 0,01178
1,4 0,15466 6,6 0,06158 17,0 0,02709 44,0 0,01101
1,5 0,15098 6,8 0,06012 17,5 0,02638 47,0 0,01032
1,6 0,14740 7,0 0,05873 18,0 0,02570 50,0 0;00972
1,7 0,14390 7,2 0,05740 18,5 0,02506 55,0 0,00886
1,8 0,14051 7,4 0,05613 19,0 0,02444 60,0 0,00814
1,9 0,13723 7,5 0,05551 19,5 0,02387 65,0 0,00753
2,0 0,13406 7,6 . 0,05491 20,0 0,02331 70,0 0,00700
2,1 0,13099 8,0 0,5262 20,5 0,02278 75,0 0,00654
2,2 0,12803 8,5 0,05002 21,0 0,02227 80,0 0,00614
2,3 0,12518 9,0 - 0,04766 22,0 0,02133 85,-0 0,00579
2,4 0,12242 9,5 0,04550 23,0 0,02045
1) Эти данные были сообщены авторам В. С. Вильсоном.
2) См. табл. 170.
136
ГЛАВА V
Для практических расчетов можно использовать значения и / (ж),
приведенные в табл. 17 и 18. Уравнение (32), выраженное с помощью
Таблица 18
Функция f (х)
X / (х) X / (*) X / (х)
0,0 1,4142 4,2 1,2895 15,5 . 1,1531
0,1 1,4139 4,4 1,2845 16,0 1,1502
0,2 1,4133 4;6 1,2797 16,5 1,1475
0,3 1,4121 4,8 1,2750 17,0 1,1449
0,4 1,4104 5,0 1,2705 17,5 . 1,1424
0,5 1,4085 5,2 1,2662 18,0 1,1399
0,6 1,4061 5,4 1,2622 18,5 1,1376
0,7 1,4035 5,6 1,2583 19,0 1,1354
0,8 1,4006 5,8 1,2545 19,5 1,1332
0,9 1,3975 6,0 1,2507 20,0 1,1312
1,0 1,3942 6,2 1,2471 20,5 1,1291
1,1 1,3907 6,4 1,2437 21,0 1,1272
1,2 1,3872 6,6 1,2403 22,0 1,1235
1,3 1,3836 6,8 1,2370 23,0 1,1201
1,4 1,3799 7,0 1,2338 24,0 1,1168
1,5 1,3762 7,2 1,2308 25,0 1,1138
1,6 1,3724 7,4 1,2278 26,0 1,1109
1,7 1,3687 7,5 1,2263 27,0 1,1082
1,8 1,3650 7,6 1,2249 28,0 1,1057
1,9 1,3613 8,0 1,2193 29,0 1,1032
' 2,0 1,3576 8,5 1,2129 30,0 1,1010
2,1 1,3539 9,0 1,2068 31,0 1,0988
2,2 1,3503 9,5 1,2011 32,0 1,0969
2,3 1,3468 10,0 1,1958 33,0 1,0947
2,4 1,3433 10,5 1,1908 34,0 1,0928
2,5 1,3398 11,0 1,1860 35,0 1,0910
2,6 1,3364 11,5 1,1815 38,0 1,0861
2,8 1,3298 12,0 1,1773 41,0 1,0816
3,0 1,3234 12,5 1,1733 44,0 1,0780
3,2 1,3172 13,0 1,1695 47,0 1,0742
3,4 1,3112 13,5 1,1659 50,0 1,0710
3,6 1,3055 14,0 1,1625 55,® 1,0664
3,8 1,2999 14,5 1,1592 60,0 1,0623
4,0 1,2946 15,0 1,1561 65,0 1,0588
постоянных а* и р*, значения которых приводятся в табл. 13, принимает
вид
А = Ло_(^до + _Г/(1)у;. (33)
е) Влияние силы поля на диссоциацию. Согласно теории Онзагера,
влияние сильных полей на константы диссоциации выражается уравне-
ХАРАКТЕРИСТИЧЕСКИЕ КОЭФФИЦИЕНТЫ И МАТЕМАТИЧЕСКИЕ ФУНКЦИИ
137
ниями (247) и (248) гл. IV. Таким образом, по уравнению (247) гл. IV
-^J = ^(6)= 1 +6 + -у+-]8 +18б+---
Для больших значений при асимптотическом разложении в ряд, согласно
уравнению (248) гл. IV,
F (b) (-V2 (86)-3/4 (1---------i-f------------------105_-з7- ... ) ,
v ' V71/ v ' I 8 (86)1/2 128(86) 1024 (86)3/2 J
где b определяется уравнением (245) гл. IV
I zjzj (Xt + Ха) 9,636 V
I z2 I ^-1 + I Z1 I ^-2 DT2
Для важного случая, когда а < 1, эквивалентная электропроводность, сте-
пень диссоциации и константа диссоциации в сильном поле могут быть,
вычислены с помощью выражения (252) гл. IV
Ах _ а
Х = 0 “о
где Ах, « и К (А) представляют собой, соответственно, электропроводность,
степень диссоциации и константу диссоциации при наличии поля с напря-
женностью V (в/см), а Ах=о, «о и К (0) — соответствующие величины
в отсутствие поля.
ЛИТЕРАТУРА
1. International Critical Tables, McGraw-Hill Book Co., N. Y., 1930—1931: a) Vol. I, p. 17;
6) Vol. Ill, p. 25.
2. R. T. Birge, Rev. Mod. Phys., 13, 233 (1941).
3. F. H. D r a k e, G. W. P i e г с e, M. T. Do w, Phys. Rev., 35, 613 (1930).
4. J.’ Wyman, Jr., Phys. Rev., 35, 623 (1930).
5. a) G. Akerlof, J. Am. Chem. Soc., 54, 4125 (1932); 6) J. Wyman, Jr., E. N. I n-
galls, J. Am. Chem. Soc., 60, 1182 (1938).
6. G. A k e r 1 6 f, O. A. Short, J. Am. Chem. Soc., 58, 1241 (1936).
7. а) Л. Пау линг, Природа химической связи, Госхимиздат, М.—Л., 1947, гл. X;
б) A. Lande, Z. Physik, 1, 191 (1920); J. A. Wasastjerna. Soc. Sci.
Fenn. Comm. Phys. Math., 38, 1 (1923); V. M. Goldschmidt, Geochemische
Verteilungsgesetze der Elemente, Skrifter det Norske Videnskaps, Acad., Oslo, I.
Matem.—Naturvid. Klasse, 1926.
8. T. H. G г о n w a 1 1, V. K. L a M e r, K. S a n d v e d, Physik. Z., 29, 358 (1928).
9. V. K. L a M e r, T. H. G г о n w a 1 1, L. J. G r i e f f, J. Phys. Chem., 35, 2245»
(1931).
10. N. В j e r r u m, Kgl. Danske Vidensk. Selskab., 7, No 9 (1926).
11. E. Я н к e, Ф. Эмд e, Таблицы функций с формулами и кривыми, ГТТИ, М.—Л.,
1949.
12. ' R. М. Fuoss, С. А. К г а u s, J. Am. Chem. Soc., 55, 1019 (1933).
13. H. S. H a r n e d, J. С. H e c k e r, J. Am. Chem. Soc., 35, 4838 (1933).
14. L. О n s a g e r, N. N. T. S a m a r a s, J. Chem. Phys., 2, 528 (1934).
15. Г. Фалькенгаген, Электролиты, ОНТИ—Химтеорет, Л., 1935, а) табл. 31.
стр. 253; б) табл. 37, стр. 262.
ЛИТЕРАТУРА К ТАБЛИЦАМ
К табл. 4
1. Т. Т. Jones, R. М. Davies, Phil. Mag. [7J, 28, 289, 307 (1939).
К табл. 6
1. Л. Па у л ин г, Природа химической связи, Госхимиздат, М.—Л., 1947, гл. X.
К табл. 13
1. International Critical Tables, McGraw-Hill Book Co., N. Y., 1931, Vol. V , p. 10.
Глава VI
ЭКСПЕРИМЕНТАЛЬНОЕ ИССЛЕДОВАНИЕ НЕОБРАТИМЫХ
ПРОЦЕССОВ В РАСТВОРАХ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ.
ЭЛЕКТРОПРОВОДНОСТЬ, ЧИСЛА ПЕРЕНОСА, ВЯЗКОСТЬ
И ДИФФУЗИЯ
В предшествующих главах были выведены и собраны воедино теорети-
ческие уравнения, необходимые для описания свойств разбавленных ионных
растворов. Начиная с этой главы мы будем излагать экспериментальные
результаты, полученные теми методами, которые оказались наиболее пло-
дотворными при исследовании данной области явлений. В первую очередь
мы рассмотрим измерения электропроводности (гл. VI и VII), а затем опреде-
ления термодинамических свойств (гл. VIII—X). Уже в самом начальном
периоде изучения свойств растворов электролитов измерения электропро-
водности оказались весьма ценными, в дальнейшем же их значение все больше
увеличивалось. Это объясняется тем, что метод электропроводности отличается
большой точностью и применим к исследованию растворов как сильных,
так и слабых электролитов в любой устойчивой среде, в которой они раство-
римы. Кроме того, с помощью этого метода можно изучать зависимость
электропроводности растворов электролитов от частоты и от градиента
потенциала внешнего электрического поля. Благодаря этому число перемен-
ных оказывается больше, чем число тех переменных, которые рассматриваются
при применении термодинамических методов.
§ 1. Эквивалентная электропроводность А.
Стандартные растворы
Эквивалентная электропроводность раствора электролита определяется
уравнением
д = 1ооил1 (1)
где с*—концентрация электролита в эквивалентах на литр и L*—та часть
удельной электропроводности раствора, которая обусловлена присутствием
электролита. В простейшем случае L* определяется вычитанием удель-
ной электропроводности чистого растворителя Lo из электропроводности
раствора L. Если Lo не очень мало по сравнению с L, то необходимо
учитывать влияние растворенного электролита на электропроводность
растворителй. Например, если большая часть электропроводности раство-
рителя обусловлена присутствием ионов, образовавшихся из растворенной
углекислоты, то очевидно, что эта часть электропроводности значительно
уменьшится в присутствии сильной кислоты, например такой, как соляная.
Таким образом, при вычислении L* из экспериментальных значений L
и Lo следует принимать во внимание как природу электролита, так и при-
роду растворителя. Подробности таких расчетов обсуждаются в ряде
работ [1].
Измерение электропроводности растворов электролитов производится с по-
мощью несколько видоизмененного мостика Уитстона. При всех измерениях
электропроводности растворов, за исключением очень редких случаев [2],
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
139
следует уменьшать поляризацию электродов. Для этого при таких изме-
рениях применяют переменный ток1, а также платинируют электроды2.
Даже при средних частотах применение переменных токов усложняет изме-
рение сопротивления, так как в результаты измерений входит емкостное
сопротивление сосуда для измерения электропроводности, а также индук-
тивное и емкостное сопротивления различных частей цепи мостика. Устра-
нение или точная компенсация этих эффектов требует значительной изобре-
тательности. С тех пор как Кольрауш [5] впервые применил мостик с исполь-
зованием переменного тока, в конструкцию мостика, нулевого прибора
и источника переменного тока было внесено много технических усовершен-
ствований3 4. Большая часть наиболее точных определений электропроводности
растворов была проделана с помощью измерительных схем Джонса
и Джозефса 17] и Шидловского [8а]1. В большинстве исследований сильно
разбавленных растворов электролитов ошибки, присущие наилучшим кон-
струкциям современных мостиков, значительно меньше ошибок, связанных
с наличием емкостного сопротивления ячеек для измерения электро-
проводности и неточностью при определении электропроводности раство-
рителя.
Джонс и Боллингер [9] и Шидловский [10] тщательно исследовали
вопрос о конструкции сосуда для измерения электропроводности. Емкостное
сопротивление сосуда обычно компенсируется переменным конденсатором,
находящимся в противоположном плече мостика, однако Джонс и Боллингер
показали, что если вводы электродов недостаточно далеко отведены от неко-
торых частей сосуда, заполненных раствором и обладающих полярностью
противоположного знака, то возникает такая побочная емкость, компенсация
которой является практически невозможной. Как было установлено, неучет
этого обстоятельства при конструировании сосуда обычно приводит к ошибке,
величина которой зависит от удельного сопротивления раствора5 6. Этим
объясняются небольшие расхождения между величинами постоянных сосуда
[12], наблюдающиеся для сосудов некоторых типов в том случае, когда изме-
рения постоянных производятся с помощью растворов с различной удельной
электропроводностью.
Соотношение между сопротивлением, измеренным на зажимах сосуда,
и удельной электропроводностью раствора зависит от относительного рас-
положения частей сосуда. В простом случае, когда электроды проходят через
параллельные стенки цилиндрического сосуда X, имеющего длину I и оди-
наковое по всей длине поперечное сечение а, удельная электропроводность
равна
L = KX/RX. (2)
Величина Кх, равная На, характеризует свойства данного сосуда и пазы-
1 Поляризация не устраняется при звуковых частотах, но необходимую поправку
можно внести, определив сопротивление при бесконечно большой частоте R с помощью
экстраполяции значений найденных при разных частотах, к бесконечно большой
частоте. Джонс и Христиан [3] показали, что экстраполяция будет линейной, если поль-
зоваться графическим изображением зависимости от !//«>.
2 Способы уменьшения поляризации с помощью платинирования электродов были
исследованы Джонсом и Боллингером [41.
3 Подробный обзор по этому вопросу приведен в книге Эгю [6].
4 Эти исследователи применяли способ заземления, подобный тому, которым поль-
зовался Вагнер [86]. Астин [8в] и Людер [8г] предложили балансную детекторную
схему, благодаря которой увеличивается чувствительность при измерении больших
сопротивлений
6 Ср. [11].
140
ГЛАВА VI
вается постоянной сосуда1. Вместо того чтобы всегда изготовлять сосуды
с одинаковыми и точно известными размерами, можно вычислять Кх с по-
мощью уравнения (2), измерив величину Rx для сосуда с неизвестными
размерами, заполненного стандартным раствором с известной удельной
электропроводностью L.Исходным стандартом для этой цели служит чистая
ртуть, так как она употребляется для изготовления эталона международного
ома. Удельная электропроводность чистой ртути весьма велика, и поэтому в
качестве вторичного стандарта при определении постоянных сосудов, предна-
значенных для изучения электролитов, употребляются растворы хлористого
калия. Кольрауш [13] впервые определил удельную электропроводность
стандартных растворов хлористого калия в абсолютных единицах, и резуль-
таты ого измерений использовались в течение многих лет, причем точность
найденных Кольраушем значений удельной электропроводности стандарт-
ных растворов не подвергалась сомнениям. Однако в сделанном Кольраушем
описании состава его стандартных растворов имелась некоторая неопределен-
ность. Паркер и Паркер [14] определили удельную электропроводность
растворов хлористого калия и устранили несогласованность в применении
единиц, введя понятие демалъного раствора (обозначается так: W) как такого
раствора, который содержит одну граммолекулу соли в одном кубическом
дециметре раствора при 0°. Данные Паркера и Паркера были приняты в ка-
честве стандартных при составлении таблиц электропроводности в «Interna-
tional Critical Tables». Однако этот выбор вскоре встретил возражения.
Джонс и Брэдшоу [15] сделали новые абсолютные измерения удельной
электропроводности растворов такого же состава, как и растворы, применяв-
шиеся в измерениях Паркера и Паркера, а Джонс и Прендергаст [16] по-
вторили исследования растворов, описанных Кольраушем. Результаты этих
новых измерений оказались более близкими к значениям Кольрауша, чем
к значениям Паркера и Паркера, но существенно отличаются как от тех,
так и от других. Данные Джонса в настоящее время обычно принимаются
в качестве наиболее надежных вторичных стандартов, применяемых при
вычислении электропроводности растворов. Все значения электропровод-
ности, которые нриводятся в тексте и таблицах этой книги, отнесены к стан-
дартам Джонса. Для того чтобы облегчить переход от одного с/андарта
к другому, в табл. 19 приведены значения удельной электропроводности
различных растворов хлористого калия по Кольраушу, Паркеру и Джонсу.
В нижней части этой таблицы концентрации выражены в демалях:
ID (1 демаль), 0,1 D (одна десятая демаля) и т. д. Джонс устранил все воз-
можные неясности в определении концентраций растворов, выражая состав
применявшихся им растворов на основании результатов взвешивания
в вакууме солей и соответствующих растворов.
§ 2. Уравнение электропроводности, выведенное Онзагером.
Сравнение с опытом
Теоретическое уравнение (20) гл. V для эквивалентной электропровод-
ности
А = А0 — S(A) ]/<Г = А0 — (а* А0-Д р*) ]/с (3)
было выведено для крайне разбавленных растворов полностью диссоци-
ированных электролитов. В этой главе излагаются результаты экспери-
1 Если поперечное сечение ячейки а неодинаково, но известна его зависимость от
расстояния до одного из электродов, то постоянная ячейки вычисляется по уравнению
—к-
о
Таблица 19 ’J
Удельная электропроводность стандартных растворов хлористого калия
в ол»'1 • СЛ4'1
Температура, СС Автор 0 18 20 25
1 N КС1 71,3828 г КС1 на 1 кг раствора в вакууме
Кольрауш 0,06541 0,09822 0,10207 0,11180
Паркер 0,065312 0,098116 0,11168,
Джонс 0,06543, 0,09820т 0,102024 0,11173з
- 0,1 N КС1 7,43344 0 КС1 та 1 кг раствора в вакууме
Кольрауш . 0,00715 0,01119 0,01167 0,01288
Паркер 0,007141, 0,011184, 0,012876,
Джонс 0,0071543 0,011191g 0,011667, 0,0128862
0,01 N КС1 0,746558 г на 1 кг раствора в вакууме
Кольрауш 0,000776 0,001225 0,001278 0,001413
Паркер 0,00077422. 0,0012223в 0,0014103,
Джонс 0,0007751а 0,0012226g 0,0012757а 0,0014114,
1 D КС1 71,1352 a КС1 ш 1 кг раствора в вакууме
Паркер . 0,065098 0,09779о 0,11132а
Джонс 0,06517, 2) 0,09783,2) 0,11134g
0,1 D КС1 7,41913 а на 1 кг раствора е вакууме
Паркер П, 007129, 0,011163, 0,0128524
Джонс 0,0071379 2) 0,011166,2) 0,012856о 2)
0,01 D КС1 0,745263 г на 1 кг раствора в вакууме
Паркер 0,00077284 0,00122023 0,0014078g
Джонс 0,00077364 2) 0,0012205о2) 0,0014087, 2)
!) На разницу между электропроводностями нормального и демального стандартных раство-
ров, равную нескольким сотым долей процента, указали Ганнинг и Гордон [1].
2) Эти значения Джонса и Брэдшоу применены в качестве стандартных для всех данных по
электропроводности, приведенных в этой книге.
142
ГЛАВА VI
ментальной проверки уравнения (3). Ниже будет показано, что теория
Дебая и Гюккеля приложима также к ионам неполностью диссоциированных
электролитов.
Онзаге'р [17] сравнил предельные теоретические коэффициенты накло-
на с многочисленными экспериментальными коэффициентами наклона,
вычисленными Дебаем и Гюккелем [18] по данным Кольрауша. Результаты,
полученные для растворов с концентрациями от 0,001 до 0,005 н., были
использованы для вычисления произвольных постоянных А нВ в уравнении
А = А0 — А \/ с + Вс (4)
по способу наименьших квадратов. Расхождение между экспериментальными
коэффициентами наклона1 А и теоретическими коэффициентами наклона
составляло 5—10% для 1,1-валентных электролитов и доходило до 13%
Рис. 6. Сравнение результатов, полученных с помо-
щью теоретического уравнения (3), с опытными данны-
ми для 1,1-валентных электролитов.
Прямые линии соответствуют теоретическому уравнению (3).
для 2,1-валентных электролитов. Для более строгий проверки теории мы
располагаем результатами более поздних весьма точных измерений. Обшир-
ный материал по проверке теории помещен в приложении Б.
Данные, полученные для некоторых 1,1-валентных электролитов, сопо-
ставлены на рис. 6 с теоретическими предельными функциями. Зависимость
коэффициента наклона этих кривых от температуры и от величины А0 выра-
жена очень отчетливо. Значение А0 для соляной кислоты, при 25° почти
в три раза больше, чем для хлористого калия при той же температуре.
1 Вальдсн [19] показал, что А= бб.Т/АЧв- воспользовавшись эмпирическим
правилом Оствальда—Вальдена—Бредига. С помощью этого соотношения получаются
значения того же порядка, что и для большого числа электролитов в различных
растворителях.
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
145
В выражении для теоретического коэффициента наклона температура в явном
виде входит в произведение DaT, которое для воды в интервале от 0 до 100°
уменьшается всего на 18%. Наиболее сильно влияние температуры прояв-
ляется в быстром уменьшении вязкости, что приводит к увеличению как Д°,
так и р*.
Влияние типа валентности электролита на электропроводность видно
из рис. 7. Данные, использованные для этого рисунка, были получены при 25°,
и значения А0 для различных солей являются приблизительно одинаковыми.
Все значения, за исключением данных для сернокислого цинка, прекрасно
согласуются с теорией. Во всем интервале концентраций, применявшихся
в этих исследованиях, с ростом]/с электропроводность сернокислого цинка
Рис. 7. Влияние валентности на эквивалентную
электропроводность водных растворов при 25°.
Прямые линии соответствуют теоретическому уравне-
нию (3). Следует отметить, что значения 5(д) для
LaCl3 и ZnSO4 при данном масштабе совпадают между
собой.
уменьшается сильнее, чем это соответствует теории. Такое поведение харак-
терно для слабых электролитов. Из рис. 7 видно, что в случае азотнокислого-
калия при умеренных концентрациях экспериментальные точки также лежат
ниже теоретической кривой, но здесь это расхождение менее заметно, чем
в предыдущем случае. Описанный эффект будет рассмотрен ниже.
Средние значения эквивалентных электропроводностей электролитов
различных типов валентности приведены в табл. 119 (приложение Б).
Для дальнейшей прямой проверки теории могут служить изображенные
на рис. 8 результаты исследований влияния диэлектрической постоянной
растворителя на электропроводность растворов пикрата тетраэтиламмония.
На этом рисунке изображена зависимость (А — А0) т)0 от ]/с для растворов пи-
крата тетраэтиламмония в нитрометане [20], метиловом спирте [21], этиловом
спирте [22] и ацетоне [23]. Во всех этих опытах температура поддерживалась
равной 25°. Включение множителя т]0 в функцию, отложенную по оси
ординат, устраняет влияние значительных различий, имеющих место между
144
ГЛАВА VI
величинами вязкости растворителей, так как и А°гои 5<д) т]0 не зависят от т]0,
если растворенное вещество, как в данном случае пикрат тетраэтиламмония,
подчиняется правилу Вальдена [уравнение (4) гл. VII]. Следовательно,
Рис. 8. Влияние диэлектрической посто-
янной на электропроводность пикрата те-
траэтиламмония при 25°:
1—нитрометан, D = 37, ц = 0,00627; 2—метиловый
спирт, D = 30,3, v)=0,00454; 3— этиловый спирт,
0=25, т) = 0,01078; 4-ацетон, D = 21, т, = 0,00316.
Точки пересечения двух соседних кривых
с осью ординат сдвинуты на 0,01.
различия в предельных коэффициен-
тах наклона и характере кривых, изо-
браженных на рис. 8, обусловлены
почти целиком различиями в диэлек-
трических постоянных. Специфиче-
ское действие растворителя сильно
сказывается на электропроводности
небольших ионов. При сделанном
нами выборе растворенного вещества
это действие растворителя сводится
к минимуму [24].
Предсказанные теорией законо-
мерности, как видно из рис. 8, под-
тверждаются экспериментальными
данными, полученными в условиях,
близких к теоретическим, а именно
для очень разбавленных растворов
полностью диссоциированных элек-
тролитов. Многие явления указывают
на то, что в средах с низкой диэлек-
трической постоянной диссоциация
бывает далеко не полной. По мере
уменьшения диэлектрической посто-
янной экспериментальные значения А
убывают по сравнению с теоретиче-
скими значениями. Это явление в
значительной мере связано с умень-
шением степени диссоциации. Эти от-
рицательные отклонения от предель-
ных уравнений разбираются в сле-
дующем параграфе и в гл. VII.
Общее уравнение (104) гл. IV
для эквивалентной электропроводности данного вида ионов, присутствующих
в растворе вместе с произвольной смесью других ионов, имеет вид
о
1,970 • 106
(Оо?7)3^
У с ,<"> ' 28,98V
“ п 3. м-ог)1/2
(5)
В этом уравнении множитель z,- У спг(,п) заменяет выражение | zrz21 </*/(! +
_______ о
+ Vвходящее в уравнение (95) гл. IV, которое было выведено только для
двух видов ионов. Различие между влиянием этих двух множителей на числен-
ное значение не является существенным во всех случаях и постепенно
исчезает, по мере того как уменьшается различие между величинами по-
движностей разных ионов одинакового заряда. Поэтому строгая эксперимен-
тальная проверка эффекта, обусловленного наличием добавок других элек-
тролитов, является более трудной, чем проверка уравнения (3). Легче всего
устанавливается наличие предсказываемых уравнением (3) значительных
отступлений от закона Кольрауша о независимости движения ионов в слу-
чае смесей, содержащих ионы одинакового знака, но весьма различных
подвижностей. В случае смеси растворов соляной кислоты и хлористого
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
145
калия, согласно теории, подвижность иона водорода должна быть меньше,
а подвижность иона калия больше, чем в отдельно взятых растворах этих
электролитов при той же ионной силе. Это явление отчетливо видно из кри-
вых, изображенных.на рис. 9 и 10. На этих рисунках по оси ординат отло-
жена разность (в процентах от а7) между электропроводностью катиона
в -смеси и в отдельно взятом растворе только одного электролита при той же
ионной силе. Сплошными линиями изображены теоретические кривые,
вычисленные для 25° Онзагером и Фуоссом [25]. Кружками обозначены
экспериментальные значения, полученные при той же температуре [26].
Крестиками обозначены независимые серии измерений [27] при 18°. Как
видно из рисунков, экспериментальные и теоретические величины, харак-
теризующие описываемые эффекты, совпадают по знаку и по порядку
Рис. 9. Изменение электропроводности
ионов водорода в смесях HCI—KCI.
Рис. 10. Изменение электропроводности
ионов калия в смесях HCI—KCI.
величины. Нельзя ожидать точного совпадения численных значений при
взятой концентрации (Г = 0,2). Результаты, полученные для более разба-
вленных растворов, лучше согласуются между собой, как видно из табл. 20.
Экспериментальные данные, по которым были рассчитаны величины, вошед-
шие в таблицу, не могут быть выражены через электропроводности отдель-
ных видов ионов, потому что для смесей хлористого натрия и соляной кис-
лоты неизвестны числа переноса. Поэтому для сравнения с теоретическими
данными были использованы значения удельных электропроводностей.
В третьем столбце помещена выраженная в процентах разность между наблю-
давшимися удельными электропроводностями смесей и значениями, вычислен-
ными по закону Кольрауша из измерений для растворов отдельных элек-
тролитов. В столбце четвертом содержатся соответствующие значения для
удельных электропроводностей, вычисленные по уравнению (5). Из данных,
помещенных в последнем столбце, видно, что разность между эксперимен-
тальным и вычисленным значениями эффекта, обусловленного наличием
добавок электролитов, исчезает в случае очень разбавленных растворов.
Аналогичные данные были получены Кригером и Кильпатриком [28а]1,
исследовавшими смеси растворов хлористых лития и калия.
Ренгольм [29] тщательно исследовал растворы смесей хлористого калия
с азотнокислым барием и сернокислой медью и обнаружил указанный эф-
фект для электропроводности очень разбавленных растворов (10~3—10~4).
Проверить уравнение (5) с помощью данных Ренгольма нельзя из-за непол-
ной диссоциации азотнокислого бария и сернокислой меди.
' 1 Исправленные значения см. [286].
10 Г. Хариед, Б. Оуэн
146
ГЛАВА VI
Таблица 20 Ч
Экспериментальные и вычисленные эффекты влияния
добавок электролитов на удельную электропроводность для
смесей НС1 и NaCl при 25°
cNaCl СПС1 Г % наблюд. % вычпел. д
1 0,400 1,99 3,31 1,32
1 0,080 1,02 1,38 .0,36
1 0,020 0,76 0,66 — 0,10
2 0,300 1,72 3,29 1,57
2 0,060 0,98 1,35 0,37
2 0,015 0,62 0,65 0,03
5 0,240 1,44 2,50 1,06
5 0,048 0,88 1,03 0,15
5 0,012 0,49 0,49 0,00
1) Приведенные в таблице значения взяты из статьи Онзагера
и Фуосса [1]. Измерения выполнены Брэем и Хантом [2].
§ 3. Расширение области применимости уравнения Онзагера.
Вычисление А0
Область концентраций, в которой применимо уравнение Онзагера,
может быть расширена с помощью трех различных методов. Первый метод
приложим только к таким растворам электролитов, электропроводность
которых меньше электропроводности, определяемой из данных, получен-
ных для разбавленных растворов с помощью предельного закона. Этот метод
основан на допущении о том, что «разница в электропроводности» обусловлена
конечной величиной константы диссоциации. В основе второго метода
лежит представление о полной диссоциации электролита, что соответствует
бесконечной величине константы диссоциации; при этом все отклонения
от предельного уравнения пытаются объяснить с помощью более тщатель-
ного теоретического исследования, при котором либо учитываются члены
высших порядков, либо в исходную физическую картину вводится пред-
ставление о «среднем (минимальном) расстоянии, па которое могут сбли-
жаться ионы». Третий метод состоит в чисто эмпирическом добавлении
членов, содержащих с в более высокой степени, чем1/2. Для ясности
изложения каждый из перечисленных методов будет рассмотрен отдельно.
а) Введение конечных констант диссоциации. Вопрос о том, каким обра-
зом по мере разбавления раствора происходит сближение экспериментальных
и вычисленных по предельному закону данных, имеет важное теоретиче-
ское значение. Онзагер [17а1 указал, что в случае полностью диссоцииро-
ванных электролитов экспериментальные кривые асимптотически прибли-
жаются к теоретическим со стороны больших значений электропроводности,
а в случае неполностью диссоциированных электролитов—со стороны мень-
ших значений. Наличие ассоциации ионов ограничивает область концентра-
ций, внутри которой можно пользоваться предельным коэффициентом на-
клона, но не влияет на его численную величину, потому что в очень разба-
вленных растворах ассоциация пропорциональна первой степени концентра-
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, ВЯЗКОСТЬ. U7
ции. Это легко показать путем сопоставления выражения для константы
диссоциации бинарного электролита
!.г/±«)2с
(1-а)
(6)
с уравнением (3). Для очень разбавленных растворов как а2, так и коэф-
фициент активности можно приравнять единице. Таким образом
1— а.^с/К. (7)
Так как уменьшение электропроводности, обусловленное ионной ассоциа-
цией, равно А (1—а) или, в пределе, Д°(1— а);" то можно написать урав-
нение
Л = А0 - (а*А0 4-₽*) Vc - с\°/К, (8)
которое эквивалентно уравнению (62), приведенному в статье Онзагера [17а].
Уравнение (8) представляет теоретический интерес, однако его использо-
вание для вычисления гюнстаит диссоциации связано с серьезными затруд-
нениями. С. помощью этого уравнения можно выразить электропроводность
частично диссоциированного электролита только в том интервале концентра-
ций, в котором уравнение (3) справедливо для полностью диссоциированных
электролитов. Как видно из рис. 6 и 7, экспериментальные кривые для
азотнокислого калия и сернокислого цинка заметно отклоняются вниз в той
области концентраций, в которой кривые для типичных сильных электро-
литов изгибаются вверх. Поэтому при любом определении степени диссо-
циации азотнокислого калия пли сернокислого цинка следует принимать
во внимание оба эти эффекта. Первый метод, с помощью которого учиты-
ваются эти факторы, поясняется ниже.
Онзагер допустил, что отклонение экспериментальной кривой для
хлористого калия от кривой, вычисленной по предельному закону, является
нормальным отклонением такого рода для сильных 1,1-валентных электро-
литов. При этом предположении разность А(Наблюд.) —^(пред. зав.)' для азот-
нокислого калия, вычтенная из соответствующей разности для хлористого
калия, полученной для той же концентрации, будет равна «разнице в элек-
тропроводности», обусловленной ассоциацией азотнокислого калия. Доля
ассоциированного электролита, выраженная с помощью этой разности
А, равна
1 а = А/А(Наблюд.)- (9)
Отсюда по уравнению (6) можно вычислить К, если известен коэффициент
активности. В связи с этим следует отметить, что стехиометрические коэф-
фициенты активности, вычисленные пз понижений .температуры замерзания
и из других коллнгативных свойств, фактически равны произведению
которое встречается, например, в уравнении (6). Коэффициент активности
недиссоциированного азотнокислого калия принимается равным единице.
В табл. 21 содержатся значения К и данные, необходимые для их вы-
числения. Как видно из таблицы, во всем рассматриваемом интервале кон-
центраций К сохраняет приблизительно постоянное значение. Такая неза-
висимость К от концентрации является необходимым, но не достаточным
условием для того, чтобы уменьшение экспериментальных значений элек-
тропроводности азотнокислого калия (в разбавленных растворах) по сравне-
нию со значениями, вычисленными по предельному закону, можно было
объяснить ассоциацией ионов. Это обстоятельство является весьма важным,
и мы еще вернемся к нему в этом параграфе.
1Q*
148
ГЛАВА VI
Таблица 21 *)
Вычисление константы диссоциации KN03 при 25° по методу Онзагера
с Данные, необходимые для вычисления 0 ,005 0,01 0 ,02 0 ,05 0,07 0,1
"^(наблюд.)’ K.NO3 138,48 135,82 132,41 126,31 123,55 120,39
^(наблюд.)~^(пред. зав.)’ 0,45 0,89 1,79 4,50 6,22 8,77
^(наблюд.)— ^(пред. aan.J’K-NOs " 0,08 0,14 0,56 2,08 3,13 4,77
«Разница» Д . : . 0,37 0,75 1,23 2,42 3,09 4,00
Степень ассоциации 1 — а . . 0,0027 0,0055 • 0,0093 0,0192 0,0250 0,0332
?/±а; KNO3 [2] 0,927 0,899 0,862 0,794 0,763 0,726
К . 1,59 1,47 1,60 1,64 1,63 1,59
1) Все приводимые значения электропроводностей являются средними значениями, получен-
ными из данных Шидловского [1], исправленными в соответствии со стандартом Джонса
и Брэдшоу.
Дэвис [30J1 произвел большое количество расчетов подобного рода2
и составил таблицы [31] констант диссоциации разнообразных солей, в осо-
бенности солей со сложными типами валентности. Некоторые типичные ре-
зультаты приведены в табл. 22. На основании значений К для многих из
этих солей можно предположить, что в растворах электролитов, состоящих
из ионов высокой валентности, диссоциация является далеко не полной.
Однако с таким выводом нельзя согласиться полностью и без оговорок,
потому что вычисление К зависит от произвольного выбора некоторых кри-
вых электропроводности, необходимых для того, чтобы изобразить поведе-
ние гипотетического полностью диссоциированного электролита. При наших
вычислениях мы воспользовались кривой для хлористого калия и полу-
чили очень хорошо согласующиеся между собой значения К для азотнокис-
лого калия. Если бы мы применили в качестве стандартной любую другую
экспериментальную кривую, то значения К сильнее менялись бы с изме-
нением концентрации. Эта трудность увеличивается в случае таких солей,,
как сернокислый цинк, потому что ни для одного 2,2-валентного электро-
лита еще не найдена кривая электропроводности, приближающаяся со сто-
роны больших значений к кривой, вычисленной по предельному закону.
Поэтому нам трудно решить вопрос о том, каково нормальное поведение
полностью диссоциированного 2,2-валентного электролита. До тех пор, пока
этот вопрос не будет достаточно надежно решен, физический смысл этих
значений К следует считать невыясненным.
Задача значительно облегчится, если мы, не вычисляя К в интервале
концентраций, применявшихся в этих исследованиях, будем одновременно
вычислять К и А0 путем соответствующей экстраполяции до бесконечного
1 Ссылки на более позднюю литературу приведены в табл. 22.
2 Если электропроводность соли значительно отличается от электропроводности,
вычисленной по предельному закону (как, например, для сернокислого цинка), то точное
значение Д° нельзя получить путем простой экстраполяции кривой, выражающей зави-
симость А от Ус. В этом случае Дэвис подбирает такое значение А0, которому в разба-
вленных растворах соответствуют наиболее постоянные значения К. Если точные значе-
ния коэффициентов активности неизвестны, то этот подбор не приводит к полному постоян-
ству значений А0 и К в пределах изучаемых интервалов концентраций и поэтому допу-
скает значительный произвол при вычислении каждой из этих констант.
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
149
Таблица 22 4)
Константы диссоциации солей и комплексных ионов в воде,
определенные из данных по электропроводности при 25 2) и 18°
Соль или ион к Ссылка на литературу Соль или ИОН 100 к Ссылка на литературу
КС1О3 1,4 1 (Са-ацстат)h 100* За4)
KNO-з 1,4 1 (СаОН) + 3,1* 4
AgNO3 1,2 1 (ВаОН) - 23* 4
T1NO3 0,52 1 MgSO4 0,78 5 5)
Т1С1 0,300 1 CaSO4 0,53 6
(CdNO3)>- 0,394 2а 3) CuSO4 0,50 5 5)
(CdClB 0,0101 2а з) ZnSO4 0,53 5 5)
(PbNO3)<- 0,0647 2 а.3) CdSO4 0,49 5 5)
(PbCI)1- 0,0304 2а 3) MgSO4 0,63* 6
(CaNO3)4 0,521 2а з) CoSO4 0,34* 6
(SrNO3)- 0,150 2а з) NiSO4 0,40* 6
(BaNO3)b 0,121 2а з) CvSO4 0,43* 7
(LiSO4)- 0,229 2а з) ZnSO4 0,49* 7
(NaSO4)~ 0,198 2а з) Си-малонат 0,00025* 6
(KSOJ- 0,151 2а з) Zn-малонат 0,021* 6
(AgSOJ- 0,05 2а з) Cd-малонат 0,051* 6
(T1SO4)- 0,0472 2а з) (KFe(CN)e~-- 0,55* 8
1) См. табл. 181.
2) Отмеченные звездочкой значения были определены при 25°.
з) Ср. [261.
4) Диссоциация уксуснокислого кальция исключительно велика. Значения
100 К для иона кальция, полученные для девяти других одноосновных органи-
ческих кислот, лежат между 2 и 30. Ассоциацию катионов щелочноземельных
металлов с радикалами карбоновых кислот исследовали с помощью потенциометри-
ческого метода Кэпнан и Кибрик [36] и Топп и Дэвис [Зв].
5) Ср. [1].
разбавления. Две аналогичные удобные для экстраполяции функции были
предложены Фуоссом [32] и Шидловским [33]. Эти функции будут подробно
рассмотрены в гл. VII, XI и XIII. Фуосс допускает, что гипотетический
полностью диссоциированный электролит подчиняется предельному закону
Онзагера при всех концентрациях. Шидловский пользуется уравнением
(15), согласно которому в интервале концентраций, применяющихся в иссле-
дованиях, допускаются небольшие отклонения вверх от кривой, вычислен-
ной по предельному закону. Для применения этих способов, так же как
и для применения способов Онзагера и Дэвиса, необходимо знать зависи-
мость коэффициента активности электролита от концентрации. При вычи-
слениях, относящихся к 1,1-валентным электролитам, во многих случаях
можно успешно применять предельный закон Дебая—Гюккеля, но для элек-
тролитов, состоящих из ионов с высокой валентностью, еще нет возможности
достаточно надежно вычислять необходимые для этой цели коэффициенты
активности с помощью одной только теории. Кроме того, эксперименталь-
ные данные о коэффициентах активности сложных электролитов не являются
столь точными и столь многочисленными, к£к в случае более простых солей.
Поэтому для сложных электролитов особенно желательно избегать введения
коэффициентов активности и вычислять А0 этих электролитов только по дан-
ным об их электропроводности. В следующем параграфе будет показано,
как могут быть осуществлены подобные вычисления с помощью дальнейшего
теоретического развития предельного закона [уравнение (10)].
150
ГЛАВА VI
б) Дальнейшее развитие теории, основанное на предположении о полной
диссоциации. В одной из своих ранних работ [17а], а позднее в совместной
работе с Фуоссом [25] Онзагер вычислил погрешность, получающуюся
вследствие математических упрощений, сделанных при выводе его предель-
ного уравнения. Отклонение от линейной зависимости А от с учитывается
путем введения двух новых членов. Таким образом, для разбавленных
растворов1
А = А0—£(д) | ' с 4- Ac 1g с 4- Вс. (10)
Это выражение считалось ранее полуэмпирическим из-за того, что числен-
ные значения постоянных А и В не могли быть вычислены теоретическим
путем. Фуосс [34], пренебрегая воздействием электрофоретического эффекта
и броуновским движением центрального иона, вывел выражения для по-
стоянных А и В для случая 1,1-валентных электролитов. Так как мы не рас-
полагаем количественной‘оценкой погрешности, связанной с допущениями
Фуосса, то, для того чтобы показать формальное согласие между уравне-
нием (10) и опытом, мы будем вычислять константы А и В с помощью данных
по электропроводности. Такая проверка для случая разбавленных растворов
1,1-валентных электролитов требует исключительно точных данных, но она
легко может быть выполнена для электролитов, состоящих из ионов с более
высокой валентностью, поскольку для таких электролитов вычисленные
теоретически [17а] и наблюдаемые отклонения от предельного коэффициента
наклона выражены более резко. Преобразовав уравнение (10) следующим
образом:
[л+Л,л,4,)^-Д’]-л18С + в,- (11)
мы можем вычислить постоянные А и В, построив график зависимости выра-
жения, заключенного в скобки, от 1g с. Очевидно, что если для А° выбрано
правильное значение, то зависимость выражения, заключенного в скобки,
от 1gс должна изображаться прямой линией с коэффициентом наклона А,
а отрезок, отсекаемый этой прямой на оси ординат, должен равняться В.
Правильное значение Л° можно легко найти путем подбора, так как выбор
того или иного значения А0 для вычисления члена а*А° не отражается суще-
ственным образом на форме кривой. Поэтому ориентировочное значение
А0 (и а*) получают посредством приближенной экстраполяции кривой, изо-
бражающей зависимость А от |/с, Эту величину используют для вычисления
значений Л + (а*А° + Р*) с, которые сохраняют неизменными во время про-
цесса подбора такого нового значения А0, которое приближенно удовлетворяет
графическому построению, сделанному по уравнению (И). Найденное таким
путем новое значение А0 в свою очередь используют для более точного вычи-
сления значений Л 4-(а*А0-[-£*) |/с и процесс повторяют до тех пор, пока
не будет найдено такое значение А0, при котором экспериментальные данные
в пределах вероятной ошибки опыта будут соответствовать уравнению (11)
вплоть до концентрации, равной 0,01 н. Кривые [35], полученные с помощью
этого способа для хлоридов калия, бария и лантана, изображены на рис. 11.
На этом рисунке мы выразили концентрацию с*, входящую в обе величины,
значения которых откладываются по осям координат, в эквивалентах на
литр. Из рисунка видно,что для разбавленных растворов уравнение (10)
1 Это уравнение применимо к полностью диссоциированным электролитам. Линей-
ный член уравнения Вс не следует смешивать с дополнительным членом —сАп/К, полу-
чающимся в результате наличия ассоциации ионов [ср. уравнение (8)].
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
151
очень хорошо согласуется с опытными данными. Диаметры кружков, изо-
бражающих опытные данные, соответствуют ошибке в 0,02 единицы при
определении величины Д° — А. На участке концентраций от 0,008 н.1 и ниже
все кривые, изображенные на рис. 11, имеют линейный характер и проходят
через максимум примерно при 0,03 н.
Величины А, В и А0, полученные с помощью графиков, изображенных
на рис. И, и аналогичных графиков для других электролитов, приведены
в табл. 23. Вследствие значительной величины2 параметров для последних
Рис. 11. Графическое определение параметров
А и В уравнения (10).
Радиусы кружков соответствуют ошибке опыта, со-
ставляющей 0,01 единицы электропроводности (А).
Рис. 12. Отклонения; диаметры
кружков равны 0,06 единицы элек-
тропроводности; расстояния между
кривыми составляют 0,6 единицы
электропроводности.
трех электролитов наилучшим средством для иллюстрации точности урав-
нения (10) будет использование этого уравнения для вычисления А0 из А, В
и экспериментальных значений А. На рис. 12 по оси ординат отложена
разность между значениями А0, вычисленными по этому способу, и зна-
чениями А0, использованными для графического определения А и В.
Так как диаметры кружков на рисунке соответствуют ошибке, равной
0,06 единицы электропроводности, то, следовательно, уравнение (10) согла-
суется с опытными данными для хлоридов калия, бария и лантана и для
сернокислого цинка с точностью около 0,02 единицы. Для двух других
электролитов эти отклонения в 5 —10 раз больше, причем эти отклонения
1 В случае хлористого бария для концентраций меньше 0,000985 и. имеются дан-
ные пяти определений, однако результаты не были использованы, так как ошибка при
определении А0 — А при этих концентрациях в несколько раз больше, чем при более
высоких концентрациях.
2 По причинам, которые будут обсуждены ниже, физический смысл численных
значений этих параметров представляется неясным.
152
ГЛАВА VI
не имеют сколько-нибудь закономерного характера. То обстоятельство,
что эти отклонения больше, чем в остальных случаях, в значительной сте-
пени объясняется тем, что данные для железистосинеродистого калия были
взяты из двух различных источников, а в данные для соляной кислоты вхо-
дят результаты измерений, проделанных в двух приготовленных независимо
друг от друга 70%-ных растворах диоксана, взятых в качестве раствори-
телей. Таким образом, уравнение (10) удовлетворительно согласуется с
результатами, полученными для очень разбавленных растворов электролитов
с весьма различными типами валентности. Уравнение (10) основано на допу-
щении о полной диссоциации, однако следует отметить, что электропровод-
ность некоторых из этих электролитов может быть выражена также через
Таблица 231)
Параметры уравнения (10) при 25°
Электролит A В A0
КС1 31,8 144 149,87
ВаС12 175 540 140,00
LaCl3 320 1030 145,8
K4Fe[CN]6 3345 6170 185,0
ZnSO4 5736 10627 132,92
HCI (70% диоксана) 4800 9800 93,1
1) При вычислении А и В по уравнению (10) концен-
трации в членах Ac*lgc*+Bc* были выражены в эквивален-
тах на литр. Все результаты согласованы с исходными
стандартами Джонса и Брэдшоу [1].
конечные константы диссоциации (табл. 22). Такая неопределенность воз-
никает из-за произвольного выбора стандартной кривой электропроводности
при вычислении К, а также вследствие чисто эмпирического способа вычи-
сления величин А и В. Поскольку А и В не вычислены теоретически, трудно
установить различие между влиянием непроводящих ионных пар, наличие
которых предполагается при определении К, и влиянием дополнительных
членов Aclgc и Вс в уравнении (10). Если бы теоретические значения А и
В были известны, то выбор стандартной кривой электропроводности для
гипотетического полностью диссоциированного электролита уже не был бы
произвольным и ассоциацию таких солей, как азотнокислый калий и серно-
кислый цинк, можно было бы изучать с большей степенью достоверности.
Были сделаны две независимые попытки истолковать особенности кри-
вых электропроводности путем учета различий в величинах ионных радиу-
сов или средних (минимальных) расстояний сближения иопов. Поскольку
последняя [36] из этих попыток не является дальнейшим развитием урав-
нения Онзагера, то она будет рассмотрена в следующем параграфе.
Канеко [37] вывел уравнение электропроводности, которое в случае
бесконечно разбавленных растворов переходит в предельный закон [уравне-
ние (3)]. Для умеренных концентраций это уравнение должно быть дополнено
эмпирическими константами; эта форма уравнения будет рассмотрена в сле-
дующей части данного параграфа. Одной из особенностей этого уравнения
является то, что в него входит множитель 1/(1 -f- ха), что является до некото-
рой степени произвольным.
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
153-
в) Чисто эмпирические способы использования уравнения Онзагера.
Изучая электропроводность 1,1-валентных электролитов, Шидловский [38]
заметил, что при непосредственном вычислении А0 по предельному урав-
нению (3) с помощью экспериментальных значений А разность между после-
довательными значениями А0 оказывается пропорциональной разности кон-
центраций. Значения А0, вычисленные таким образом, будут в дальнейшем
обозначаться через А0' в отличие от истинных значений предельных электро-
проводностей А0, полученных экстраполяцией на нулевую концентрацию.
В-математической форме можно выразить эту обнаруженную Шидловским
закономерность уравнением
дол_£с (12}
1 — a* |/ с
ИЛИ
А = А° — §(А) Ус + Вс - а*Вс3/2.
(13)
Положительная эмпирическая константа В зависит от свойств данного
электролита и имеет величину того же порядка, что и §(Д). Электропровод-
ность сильных 1,1-валентных
электролитов можно, как пра-
вило, выражать с помощью
этого уравнения вплоть до
концентрации, равной 0,1 н.,
с точностью до величины
ошибки опыта. Интересно от-
метить, что электропровод-
ность растворов хлористого
калия при малых концентра-
циях не подчиняется уравне-
нию Шидловского. Электро-
проводность электролитов,
СОСТОЯЩИХ 1ГЗ ионов с высо-
кой валентностью, не может
быть выражена этим уравне-
нием, но из рис. 13 видно,
что график зависимости А0'
от концентрации допускает
удовлетворительную экстра-
поляцию данных для хло-
ристых бария и лантана. Этот
Рис. 13. Зависимость А0' от концентрации (выра-
женной в эквивалентах на литр) для водных раство-
ров при 25°; А0 найдено путем экстраполяции.
рисунок иллюстрирует также небольшое,хотя и поддающееся измерению,
отклонение величин А0' для хлористого калия от линейной зависимости, вы-
ражаемой уравнением (12). Почти линейный характер отдельных участков
кривых объясняется наличием перегиба этих кривых. При больших концен-
трациях кривые сильно изгибаются вниз. С помощью этих кривых можно
довольно точно вычислять А0 несмотря на то, что при самых малых концен-
трациях отклонения от линейной зависимости весьма значительны. Для
электролитов, состоящих из ионов более высокой валентности, например
для железистосинеродистого калия и сернокислого цинка, экстраполяция
практически невозможна. Из рис. 14 видно, что кривые электропроводностей
растворов этих солей при приближении к оси ординат резко изгибаются.
Уменьшение А0' с ростом концентрации в очень разбавленных растворах,
которое, можно наблюдать на рис. 14, предсказывается теоретически урав-
154
Г Л А В A VI
нением (10) для электролитов всех типов валентности [35], поскольку при с,
стремящемся к нулю, из этого уравнения следует
ДА0' __ Л0' — Д° _ A 1g с — В _
de ~~ с ~ —
(14)
Это • обстоятельство не оказывает значительного влияния на величины Д°,
полученные при экстраполяции кривых, изображенных на рис. 13. Вычи-
сления Ло< для растворов хлоридов калия, бария и лантана с помощью вели-
чин. А и В, приведенных в табл. 23, показывают, что Л0' проходит через
довольно плоский минимум при концентрации, несколько более низкой,
чем нижняя граница исследованной области концентраций. Значение Л0',
соответствующее этому минимуму, лишь немногим меньше, чем значение Л°,
удовлетворяющее уравнению (10). Для хлоридов калия, бария и лантана
разности этих значений равняются соответственно 0,00015, 0,021 и 0,031 еди-
ницы электропроводности.
Это показывает, что для
данных солей можно по-
лучить удовлетворитель-
ные значения Л° с помощью
простого продолжения в
горизонтальном направле-
нии участков кривых на
рис. 13 в области наибо-
лее низко расположенных
экспериментальных точек.
Шидловский [33] пред-
ложил еще один способ
использования уравнения
Онзагера, с помощью ко-
торого легче производить
некоторые вычисления,.
Р и с. 14. Зависимость Д° от концентрации (выражен-
ной в эквивалентах на литр) для водных растворов
при 25°.
чем по уравнению (13). Он
показал, что вплоть до с 0,01 электропроводность типичных сильных
1,1-валентных электролитов в водных растворах можно весьма точно пред-
ставить уравнением
Л = Л° - (а*Л° + р*) сйг.
(15)
Согласно этому уравнению, коэффициент наклона при увеличении концен-
трации постепенно уменьшается. В приведенной форме это уравнение непри-
годно для интерполяции, но его можно представить в виде ряда
q* 2 е з
Л = до - §(Д)СЙ2 + е - 4^ с3/2 + • • •, (16)
который для данного числа членов формально совпадает с уравнением (13)
и обладает тем преимуществом, что в нем отсутствует эмпирический пара-
метр В.
Для экстраполяции уравнение (15) можно использовать в форме1
Д _Д, <^л° + з*\ 1/2
Л “ Л ° < Д02 Д
(17)
1 Лоренц [39] предложил аналогичное уравнение, имеющее вид 1/Л=1/Л° +
+ (Л.\.р-1)с1/2. Однако он обнаружил, что значение р является специфическим свой-
ством данного электролита при умеренных концентрациях (с 0,1); это является недостат-
ком указанного уравнения по сравнению с уравнением (15), для которого р = 1 в случае
очень разбавленных растворов.
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
155
причем 1/Д° можно получить экстраполяцией кривой, изображающей зави-
симость 1/Л от с1/2. Хотя такого рода зависимость и удобна для графического
изображения, однако она менее пригодна для экстраполяции, чем зависи-
мость, изображенная на рис. 13, потому что часть кривой, получающаяся
при экстраполяции по уравнению (17), длиннее и круче, чем в случае рис. 13.
§ 4. Другие уравнения электропроводности.
Критический обзор
А = А0 — А]/с_,
Среди многочисленных эмпирических уравнений, которые были пред-
ложены для экстраполяции данных по электропроводности сильных элек-
тролитов в водных растворах, наиболее полезным оказался так называемый
закон квадратного корня Кольрауша1
(18)
который в формальном отношении согласуется с теорией междуионного
притяжения. Это уравнение успешно применяется в случае крайне разба-
вленных растворов, и большинство интерполяционных уравнений, предло-
женных для более высоких концентраций, в пределе сводится к уравне-
нию (18). Простейшим из этих уравнений является соотношение
д~ до_ д /с+Вс, (19)
на которое мы уже ссылались в § 2. Вальден [41] предложил уравнение,
содержащее только две константы:
А =---.
1 + В / с
Оно значительно менее точно, чем уравнение (19), но охватывает ту же
область концентраций ( < 0,01 М). Лэтти [42] объединил оба эти уравнения,
предложив соотношение
Д = А°---
1+В^с
которое вполне пригодно для растворов с концентрациями ниже 0,1 н.
Джонс и Дол [43] показали, что соотношение, которое получается из урав-
нения (21) при добавлении линейного члена
Л = Л°----ЛТ-';_ + £>с, (22)
выражает полученные ими экспериментальные данные для хлористого бария
вплоть до концентрации 2 н. с точностью до весьма незначительной ошибки
•опыта. С равным успехом это уравнение можно применять для случая бро-
мистого калия [44] при 0 и 25°. Все предыдущие уравнения обладают одним
•общим свойством, состоящим в том, что
=-(const.). (23)
\а у с / с->о
Значения этой константы различны для каждого из уравнений и, как пра-
вило, отличаются от теоретического предельного коэффициента наклона,
соответствующего уравнению (3). Принимая во внимание, что уравнение
Онзагера является вполне правильным, не следует пользоваться для экстра-
поляции ни одним из приведенных выше эмпирических уравнений.
1 С подробным разбором
миться по книге Кольрауша
вопроса о применении этого уравнения можно познако-
[40].
156
ГЛАВА VI
Поскольку из теории междуионного притяжения следует, что к водным
растворам сильных электролитов формула [45] разведения Оствальда
до (До_Д)
(24>
неприменима, то многочисленные уравнения электропроводности, которые-
включают это соотношение1, вышли из употребления.
Несмотря на успешное применение уравнения Онзагера, в некоторых
случаях [48] все еще используется исследованное Лорепцом [39, 47а]2 3 общее
уравнение
А = А0 — Асп, (25)
в котором показатель степени п рассматривается как эмпирический пара-
метр, характеризующий данный электролит (или группу электролитов)-
Согласно уравнению (25), график зависимости 1g (А0 — А) от 1g с при Правиль-
ном выборе значения До должен представлять собой прямую линию с коэф-
фициентом наклона п. Фергюсон и Фогель [49] применили это уравнение
к полученным ими для большого числа электролитов данным и нашли, что-
как А, так и п являются специфическими величинами для каждого электро-
лита. Специфический характер величины А следует из теории, так как тео-
ретический коэффициент наклона a*A°-|-J3* содержит член А0. Дэвис [50а] *
указал на то, что, за немногими исключениями, изменение п, которое должна
наблюдаться при переходе от одного электролита к другому (0,4 < п < 0,6),.
является величиной того же порядка, что и различия между значениями п.
для повторных серий измерений, относящихся к одному и тому же электро-
литу (т. е. лежит в пределах ошибок опыта). Это обстоятельство свидетель-
ствует о том, что уравнение (25) непригодно для экстраполяции.
Для того чтобы показать, какие большие ошибки могут быть допущены
при использовании чисто эмпирических уравнений для экстраполяции,
мы применим приведенные выше уравнения к данным для типичного 2,1-ва-
лентного электролита при 25°. Джонс и Дол [43] нашли, что их данныа
по молярной электропроводности хлорисдого бария при больших разбавле-
ниях могут быть представлены с помощью следующих выражений:
Аш = 278,75-449,9/с, (18а)
Ал1 = 280,64-550,12 / Г + 1265,1 с, (19а)
А,л = 278,69/(1 + 1,715/7), (20а)
А,п = 281,22-602,8 /7/(1 + 4,069 /7), (21а)
Ат = 282,13-636,3 /7/(1 +4,628 /7) — 31,13 с, (22а)
Ат = 288,18 — 227,32 с °’3276. (25а)
С помощью табл. 24 можно определить ту обдасть концентраций, в кото-
рой каждое из этих уравнений правильно воспроизводит экспериментальные
данные. Цифры в скобках представляют собой величины предельных моляр-
ных электропроводностей А„, удовлетворяющие этим уравнениям. Разброс
этих значений свидетельствует о необходимости располагать точной теоре-
1 Большинство этих уравнений рассмотрено Краусом и Брэем [46а]. См. также
[466, в].
2 Теоретические выводы, которые сделали Герц [476] и Гош [47в], в значительной
мере способствовали исследованию уравнения Д = А° — Ас1^. Ср. [39, 47а, 47г].
3 Более подробную дискуссию о возможности применения уравнения (25) можно
найти в материалах по дискуссии между Фергюсоном и Фогелем [506] и Дэвисом, Мар-
тином и Портером [50в].
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
157
тической предельной функцией. Правильное значение1 Л™, несомненно,
близко к 280,00, т. е. к удвоенной величине эквивалентной электропровод-
ности ВаС12, приведенной в табл. 23. Отметим, что наибольшее отклонение
от этой величины (288,18) получается при вычислениях с помощью урав-
нения (25), в котором показатель степени с выбирается произвольно. Пока-
зательно также и то, что при расчетах по остальным уравнениям среднее
отклонение составляет 1,3 единицы, хотя все эти уравнения обладают общим
свойством, которое выражается уравнением (23). Применение уравнения (22),
•содержащего наибольшее число эмпирических постоянных, приводит
к ошибке в значении Д^, превышающей 2 единицы.
Таблица 24
-Значения молярной электропроводности хлористого бария при 25°, вычисленные
с помощью эмпирических уравнений
Лгн(наблюд.) Уравнение
(18а) (19а) (20а) (21а) (22а) (25а)
0 (278,75) Д280,64) (278,69) (281,22) (282,13) (288,18)
0,001 264,53 264,53 264,51 264,25 264,33 264,55 264,53
0,0025 • 256,25 256,25 256,30 256,65 256,16 256,22 256,25
0,005 248,11 246,94 248,07 248,56 248,10 248,03 248,11
0,01 238,27 233,76 238,28 237,89 238,37 238,32 237,89
0,025 223,25 207,61 225,29 219,24 223,22 223,26 220,29
0,05 210,65 220,89 201,44 210,64 210,65 202,99
0,1 197,36 233,19 180,69 197,86 197,36 181,27
0,25 178,39 181,90 178,35
0,5 161,20 171,28 161,26
1,00 137,96 137,94
Горин [36] вывел интересное уравнение электропроводности, которое
также не сводится к предельному уравнению Онзагера:
, Л° + (а_Х° 4 аД» ) %
Л ~ 14- (а+ + а_) х ’
При выводе этого уравнения для бинарных электролитов Горин воспользо-
вался теорией Дебая — Гюккеля для вычисления потенциала на поверхности
иона, допустив при этом, что наложенное внешнее поле не искажает суще-
ственным образом строение ионных атмосфер. При этом допущении не учи-
тывается потенциал асимметрии ф). Напомним, что наличие ф) в уравнении
•Онзагера является существенной особенностью этого уравнения, удовлетво-
рительно описывающего с помощью функции ф) влияние сильных полей
и высокой частоты. Уравнение Горина, несмотря на его недостаточную тео-
ретическую обоснованность, можно применять для вычисления электро-
проводности и чисел переноса в пределах значительного интервала концен-
1 Значения электропроводностей в пределах от 279,6 до 280,0 получаются из трех
различных кривых, построенных на основании уравнения Онзагера. Две из этих кри-
,вых показаны на рис. И и 13. Третья представляет собой зависимость Л от j/c, по-
строенную для нахождения теоретического предельного коэффициента наклона для
крайне разбавленных растворов. При использовании этой кривой необходима сравни-
•тельио далекая экстраполяция, приводящая к значению 279,8 с точностью до 0,2—0,3
единиц электропроводности.
158
ГЛАВА VI
траций. Это, несомненно, связано с введением двух дополнительных кон-
стант, а именно ионных радиусов а+ п а_.
Как и другие уравнения этого параграфа, уравнение (26) в предельном
случае сводится к уравнению (23). Предельный коэффициент наклона в этом
случае становится равным
(27)
если в качестве независимой переменной выбрано х. В табл. 25 приведены
результаты некоторых вычислений, выполненных с помощью уравнения (26)
на основании экспериментальных данных Шидловского [51] и Лонгс-
ворта [52]. Значения а+ довольно хорошо соответствуют величинам радиусов
^драгированных ионов, a aci практически не зависит от природы катиона.
Горин указал, что порядок величины а+ и ее изменение при переходе от одних
ионов к другим находятся в согласии с классической гидродинамической
теорией.
Таблица 25
Ионные радиусы (в ангстремах) при 25°, вычисленные по уравнению (26)
Электролит л" + л» dA dVc с-><) (Z.b (Z_
НС1 426,06 349,73 76,33 — 156,5 0,945 1,921
КС1 * 149,86 73,53 76,33 — 96,1 1,978 1,933
NaCl 126,45 50,12 76,33 — 90,6 2,562 1,937
1.IC1 115,03 38,70 76,33 — 88,7 3,095 1,976
§ 5. Определение чисел переноса1 по методу движущейся границы.
Основные уравнения
Лодж [54] показал, что можно непосредственно наблюдать движение
ионов, а Уэзем [55], Нернст [56], Мэзон [57] и особенно Денисон и Стил [58]
разработали метод, с помощью которого можно количественно определять
числа переноса по скорости передвижения границы между двумя раство-
рами. В дальнейших исследованиях Кэди [59], Смита [СО], Мак-Иннеса [61]
и Лонгсворта [52а]2 этот метод был значительно усовершенствован,
и в настоящее время он является весьма точным. В связи с важностью опре-
деления чисел переноса для проверки3 теории междуионного притяже-
ния, а также в связи с практическим применением чисел переноса в исследо-
ваниях электропроводности и электродвижущих сил ниже приводится
в общих чертах описание метода определения чисел переноса по скорости
перемещения границы между двумя растворами.
Обычно начинают с вычисления предварительного или кажущегося
числа переноса. При этом вычислении пренебрегают влиянием электропро-
водности растворителя и изменениями объема у электродов. Истинное число
переноса получают затем посредством введения соответствующих поправок,
учитывающих эти эффекты. На рис. 15 изображен вертикальный разрез
1 Обширная сводка литературы по общим вопросам, относящимся к числам пере- .
носа, имеется в статьях Ле-Роя [53а] и Мак-Вэна [536].
2 Подробный обзор теории и методических вопросов, а также результатов, полу-
ченных с помощью метода движущейся границы, приведен в статье Мак-Иннеса
и Лонгсворта [62].
3 За исключением отдельных случаев, числа переноса, определяемые с помощью
известного аналитического метода Гитторфа, как правило, бывают недостаточно точны
для указанной цели.
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
159-
трубки, в которой у х0 образуется граница между двумя растворами электро-
литов. Эту границу получают, наслаивая один из растворов AR на другой IR,
причем оба раствора имеют общий ион R. В течение t сек. пропускают
постоянный ток i, при этом граница переходит в положение xt. Если с& —
выраженная в нормальностях концентрация ионов А, а V—объем в литрах,.
равный произведению поперечного сечения трубки на путь, пройденный
границей от х0 до xt, то при перемещении ионов А вверх по трубке через
любое сечение трубки, расположенное выше xt, переносится FcJE кулонов.
Поэтому кажущееся число переноса ТА иона А равно
т, Fcav
1 А = —т;—
(28)
где произведение it представляет собой количество
электричества, протекшего через трубку за время t.
При очень малых концентрациях заметная часть
тока переносится ионами, возникающими при дис-
социации растворителя и следов примесей, напри
мер углекислоты. Лонгсворт [52а] обнаружил этот
эффект и вычислил его величину. Можно показать,
что для раствора 1,1-валентного электролита AR с
концентрацией с*, находящегося в трубке, попереч-
ное сечение которой равно а, истинное число перено-
са ТА — иА/ (Цд + мд) равно
Рис. 15. Схематическое-
изображение трубки и
ТА =
F«4 с* /1000 ,г L„
+ 1а т
границы между раство-
(29) рами.
Так как подвижности относятся к неподвижному растворителю, то иА
представляет- собой расстояние, проходимое ионами А через раствор в се-
кунду при напряженности электрического поля, равной единице. Если из-
менения объема, происходящие из-за электролиза и перемещения ионов,
вызывают движение растворителя по отношению к трубке, в которой опре-
деляется положение движущейся границы, то объем, входящий в уравнение
(28), выразится' следующим образом:
У = Тож + АУ- (30)
где ДЕ, выраженное в литрах, представляет собой приращение объема V,
обусловленное движением растворителя, которое добавляется к объему, обра-
зованному наблюдаемым перемещением границы в течение t сек. Сочетание
уравнений (28) и (30) с уравнением (29) приводит к выражению
(31)
На необходимость дополнения уравнения (28) поправочным членом,
содержащим ДЕ, указал Миллер [63]. ДЕ может быть либо измерено экспе-
риментально [64], либо вычислено из величин плотностей растворов и ком-
понентов электродов [65, 52а].
Если основной раствор (AR) и раствор индикатора (//?) выбраны правиль-
но, то диффузия через границу между двумя растворами невелика [26, 63, 66].
В описанном виде метод движущейся границы применим для определе-
ния чисел переноса таких ионов, для которых имеются подходящие (более
медленные) ионы-индикаторы. Однако он может быть применен также и для
непосредственного определения чисел переноса ионов-индикаторов. Кольрауш
[67] показал, что одним из условий сохранения отчетливой границы является
160
ГЛАВА VI
наличие пропорциональности между концентрация.ми и числами переноса
основного иона и иона-индикатора. Так как эти ионы должны у границы
двигаться с одинаковой скоростью, то объем, проходимый движущейся гра-
ницей при пропускании одного фарадея, равен не только Та/са, но также
равен и Ti/c*. Поэтому
Если исходная концентрация индикаторного раствора равняется с*', причем
с*' не очень отличается от с*, то концентрация в объеме, пройденном дви-
жущейся границей, должна автоматически приходить в соответствие с с*
согласно уравнению (32). Описанные изменения концентрации происходят
вблизи исходного положения движущейся границы, потому что значения
Tj при с*' весьма близки к значениям Тг при с*.
Если известна зависимость удельных электропроводностей растворов
от концентрации, то кольраушевскую концентрацию с* можно определять
кондуктометрически [68]. Хартли [69] предложил остроумный прибор с так
называемой «уравновешенной границей» и использовал уравнение (32) для
сравнения чисел переноса ионов водорода, калия и натрия в растворах
соответствующих хлоридов с числом переноса иона лития в растворе хло-
ристого лития, применявшемся в качестве индикаторного раствора. Расхо-
ждения между результатами, полученными Хартли, и данными Лонгсворта
[52а] не превышают 0,5%. Метод уравновешенной границы является прак-
тически важным, так как с его помощью можно непосредственно опреде-
лять числа переноса ионов с очень малой подвижностью. Этот метод был
применен для изучения солей, катионы которых содержали парафиновые
цепи с числом атомов углерода, доходившим до шестнадцати [70].
С помощью метода Хартли получены интересные экспериментальные
результаты, которые послужили основой для объяснения свойств коллоид-
ных электролитов [71].
Для определения чисел переноса по методу Гитторфа [72]1 необходимо
производить химический анализ, что весьма затрудняет определение чисел
переноса с большой степенью точности. Однако Мак-Иннес и Дол [73]
получили с помощью этого метода значения чисел переноса с точностью
примерно до 0,2%. Такова же величина расхождения между данными
Мак-Иннеса и Дола и соответствующими значениями, полученными
по методу движущейся границы. Джонс и Брэдшоу [74] измерили числа
переноса для хлористого лития, причем их результаты совпадают с дан-
ными, полученными по методу движущейся границы, с точностью до ± 0,7%.
Истинные числа переноса вычисляют из чисел переноса Гитторфа путем
введения поправок [75] на перенос &NW молей воды от анода к катоду
при прохождении одного фарадея. Таким образом,
Т+= Т+(Гитторф) + 0,018 A 7VWс*. (33)
Так как ДД% является величиной порядка единицы2, то эта поправка
делается существенной при умеренных концентрациях.
§ 6. Предельное уравнение Онзагера для чисел переноса.
Сравнение с экспериментальными данными
Для числа переноса ионов данного вида в растворе, содержащем
только один электролит, можно дать следующее определение:
T^j/A, = (34)
1 Обширную библиографию метода приводит Партингтон в книге Тейлора [46в].
2 Например, 1/4 < А 7%, < 3/2 для хлористого водорода и хлоридов щелочных
металлов.
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
161
Если ограничиться рассмотрением электролитов, состоящих только из двух
видов ионов (ф у), то значения /.у и А из уравнения (28) гл. V и из
уравнения (3) можно сочетать с их значениями по уравнению (34), что
приводит к предсказанному теорией Онзагера выражению
Т/ = ТН^(тр’Гс(А0/А'). (35)
В этом н последующих уравнениях значения эквивалентной электропровод-
ности, вычисленные по уравнению (3), обозначаются через А' в отличие
от определяемых опытным путем значе-
ний А. Таким образом,
Д'А0 —(а*Д° +Р*)]/с. (36)
Так как при больших разбавлениях от-
ношение Д°/А' близко к единице, пре-
дельное уравнение имеет вид
T^Tj + S^Vl (37)
где
S^)=L + —J Р • (37а)
Отметим, что выражение для коэффи-
циента наклона не содержит параметра
а*. Так как в это выражение входят
только абсолютные величины валентно-
стей, то индексом j можно по выбору
обозначать либо аннон, либо катион.
Как в том, так и в другом случае
условие Тj Т Ti = 1 выполняется при
^(Ту) = —
Некоторые из наиболее точных экспе-
риментальных значений, полученных с
помощью метода движущейся границы
при 25°, приведены в табл. 120. На
рис. 16 и 17 изображена полученная
опытным путем- зависимость числа пере-
носа Т+ от ]/с для водных растворов
электролитов при 25°. Предельная зави-
Р и с. 16. Числа переноса катионов
для водных растворов 1,1-валентных
электролитов при 25°.
симость числа переноса от Vс, соответствующая уравнению (37), изобра-
жается прямыми линиями, причем отрезки, отсекаемые этими прямыми
на оси ординат, равны Т°+. В случае хлористых солей эксперимен-
тальные значения прекрасно согласуются с теоретически вычисленными.
Такое поведение типично для таких 1,1-валентных электролитов, для кото-
рых в разбавленных растворах наблюдаются положительные отклонения
от предельного уравнения электропроводности (3). В случае таких солей
опытная кривая приближается к кривой предельного закона снизу, если §<т)
положительно, и сверху, если отрицательно. В случае азотнокислого
калия экспериментальные значения чисел переноса во всем интервале кон-
центраций, Применявшихся в этих исследованиях, не совпадают с теорети-
ческими значениями, и хотя для азотнокислого калия §<т+) положительно,
экспериментальная кривая приближается к кривой предельного закона
сверху. Как видно из рис. 7, отклонения экспериментальной кривой электро-
проводности этой соли от теоретической кривой, соответствующей уравне-
11 Г. Харнед, Б. Оуэп
162
ГЛАВА VI
Ри с. 17. Числа переноса катионов
электролитов несимметричного типа
(водные растворы) при 25°.
нию (3), имеют отрицательное значение. Числа переноса и электропровод-
ность для случая азотнокислого серебра во всех отношениях подобны числам
переноса и электропроводности азотнокислого калия.
На рис. 17 изображены экспериментальные данные для двух электро-
литов со сложным типом валентности—для хлоридов кальция и лантана.
Для растворов этих солей §(т+> отрицательно, и опытная кривая прибли-
жается к кривой предельного закона сверху, однако отклонения от теоре-
тических уравнений отчетливо выражены даже для наиболее разбавленных
растворов. С другой стороны, кривые электропроводности для этих электро-
литов, в согласии с теорией, приближаются к кривой предельного закона
сверху. Для этой аномалии еще не найдено
удовлетворительного объяснения. В связи
с этим было бы интересно теоретически
вычислить коэффициенты А и В членов
уравнения (10), содержащих с 1gс и с.
Для наименьших концентраций, применяв-
шихся в этих исследованиях, эти члены,
несомненно, сильно влияют на величины
чисел переноса для электролитов, состоя-
щих из ионов с высокой валентностью.
Предположение о том, что ассоциация
ионов является причиной аномалии чисел
переноса, дает возможность удовлетвори-
тельно объяснить экспериментальные ре-
зультаты, полученные при исследованиях
сернокислых солей щелочных металлов1 и
хлоридов щелочноземельных металлов
[77]. К сожалению, представление об ас-
социации ионов в растворах хлоридов
щелочноземельных металлов находится в
противоречии с данными по электропро-
водности, а данные по числам переноса
для азотнокислых калия и серебра не
подвергались количественной обработке
с учетом ассоциации.
Результаты проверки теоретического уравнения (37) можно суммиро-
вать следующим образом: коэффициент наклона экспериментальных кривых
зависимости числа переноса от с для большинства 1,1-валентных электро-
литов, исследованных по методу движущейся границы, близок к теорети-
ческому предельному коэффициенту наклона2. Сргласие между экспери-
ментальными и теоретическими значениями предельных коэффициентов
будет еще более убедительно показано в следующем параграфе, в котором
будут рассмотрены различные виды уравнений для чисел переноса. Экспе-
риментальные значения коэффициентов наклона для других электролитов
не обнаруживают никаких признаков совпадения с соответствующими
теоретическими значениями даже в случае наиболее разбавленных раство-
ров. В тех случаях, когда теоретические и экспериментальные значения’не
согласуются друг с другом, расхождение, повидимому, не связано каким-либо
простым соотношением со знаком теоретического коэффициента наклона
или с формой кривых электропроводности соответствующих электролитов.
1 Для того чтобы кривая для сернокислого натрия имела теоретический коэффи-
циент наклона, она должна обладать точкой перегиба при малых концентрациях [76а].
См. также [766].
2 Сэмис [78] показал, что хлористый водород и хлористый калий ведут себя при
40 и 50° так же. как и при 25°.
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ, ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
163
Измерения чисел переноса при температурах более высоких, чем 25°,
проведенные независимо друг от друга, не полностью согласуются между
собой. Однако некоторые данные свидетельствуют о том, что те электролиты,
которые подчиняются уравнению (37) при 25°, подчиняются ему и при дру-
гих температурах. В табл. 26 и 27 приведены значения чисел переноса
катионов в водных растворах [79а] хлоридов калия и натрия [796] при
Таблица 26 [1]
Числа переноса катионов в водных растворах хлористого калия
/,°с 0 А [2] т° Концентрация, моль/л
0,005 0,01 0,02 0,05 0,10
15 121,1 — 0,0027, 0,4928 0,4926 0,4925 0,4924 0,4923 0,4921
25 149,9 -0,0037э 0,4905 0,4903 0,4902 0,4901 0,4900 0,4900
35 180,5 -О,ОО46о 0,4889 0,4887 0,4886 0,4885 0,4885 0,4888
45 212,5 - 0,00543 0,4872 0,4869 0,4868 0,4868 0,4869 0,4873
Таблица 27 [1]
Числа переноса катионов в водных растворах хлористого натрия
ЩС Д°[2] S(T+) Т°+ Концентрация, моль/л
0,005 0,01 0,02 0,05 0,10
15 101,2 -0,04924 0,3929 0,3897 0,3885 0,3870 0,3846 0,3820
25 126,5 —0,04913 0,3962 0,3930 0,3918 0,3903 0,3878 0,3853
35 - 153,9 -0,04853 0,4002 0,3970 0,3958 0,3943 0,3919 0,3892
45 182,7 — 0,04785 0,4039 0,4008 0,3996 0,3982 0,3957 0,3932
различных температурах, а в табл. 133 содержатся подобные данные для
хлористоводородной кислоты1 в различных растворителях. В случае рас-
творов хлористого калия разность между Т + и увеличивается с темпера-
турой, что является исключением. Как правило [40], более высоким подвиж-
ностям ионов соответствуют более низкие температурные коэффициенты,
из-за чего числа переноса по мере увеличения температуры обычно сбли-
жаются [80, 79а].
§ 7. Уравнения для чисел переноса при умеренных
разбавлениях. Вычисление Т*
Для определения чисел переноса при разных концентрациях, начиная
от наименьших, применяющихся в исследованиях, и равных приблизи-
тельно 0,01 н., вплоть до 0,2 н. и выше, было предложено несколько полезных
уравнений. Три из этих уравнений сводятся при весьма малых концентрациях
к теоретическому предельному уравнению. Эти уравнения будут рассмотре-
ны в первую очередь.
1 Два из этих электролитов исследованы Сэмисом [78]. Полученные им данные
не приводятся, так как позднее (см. [79а]) было показано, что они могут содержать
ошибки, обусловленные особенностями поведения индикаторных растворов при высоких
температурах.
И* ш
164
ГЛАВА VI -
а) Лонгсворт [52а] обнаружил, что значения Т°+, вычисленные с по-
мощью уравнения (35) из экспериментальных значений Т+ для растворов
хлористых солей щелочных металлов и для соляной кислоты, линейно
зависят от с. Вычисленные таким образом величины обозначают через Т%. для
того, чтобы сохранить за Та+ его обычное значение. Полученную Лонгсвор-
том зависимость для 1,1-валентных электролитов можно представить в виде
=? г + вс.
+ Д'+ [1* у с +
(38)
Эмпирическая постоянная В является величиной, характерной для данного
электролита. Решая это уравнение относительно Т+, получаем
Т+ = Т*+ 4- §.<Т+) У с (А0/А')'+ В (1 + ₽* У с/ Д') с. (33)
Р и с. 18. Проверка уравнения (38) для
водных растворов 1,1 -валентных хло-
ридов при 25°.
Из рис. 18 видно, что уравнение (38) с большой степенью точности выра-
жает опытные данные в тех случаях,
когда экспериментальные значения при
экстраполяции к нулевой концентра-
ции совпадают со значениями, вычислен-
ными с помощью теоретического предель-
ного уравнения. Из данных, помещенных
в табл. 26 и 27, видно, что сделанное
заключение справедливо и при других
температурах. Значения Т\, получен-
ные путем экстраполяции зависимо-
стей, представленных на рис. 18, пол-
ностью соответствуют данным о предель-
ных электропроводностях электролитов
и согласуются с правилом о независи-
мости предельных подвижностей. Для
данных электролитов эти выводы под-
тверждают предсказания теории. С
другой стороны, уравнение (38) не-
применимо в случае растворов азотно-
кислых калия и серебра и электроли-
тов, состоящих из ионов более высокой
валентности.
б) Шидловский [81] предложил
общее эмпирическое уравнение
АУс-В*с, (40)
которое согласуется с экспериментальными данными, полученными для
различных электролитов при концентрациях, доходящих до 0,2 н. Для
таких электролитов, как хлористый кальций и азотнокислое серебро, оба
параметра, как А, так и В*, следует вычислять из данных по числам пере-
носа. В случае галогенидов щелочных металлов и других 1,1-валентных
электролитов можно пользоваться теоретическим значением А, находя-
щимся в соответствии с уравнением (37). При этом
Л=-§(Г.)(1/7’^. (41)
В этом случае в уравнение (40) входит только одна произвольная
постоянная В*. Поэтому можно использовать кривую, изобража-
ющую зависимость [1/У + £>(т/) (V^Pfc ] от с, для определения 1/Т^
с помощью экстраполяции с использованием метода последовательных
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
165-
приближений. Все предельные значения чисел переноса, полученные таким
образом, прекрасно согласуются с числами переноса, найденными с по-
мощью метода экстраполяции Лонгсворта. Если теоретическое значение А,
определяемое с помощью уравнения (41), является неприемлемым, то пра-
вильные значения Т° для хлористого кальция и азотнокислого серебра
можно получить из уравнения (40) по способу наименьших квадратов.
в) В случае разбавленных растворов 1,1-валентных электролитов, для
которых отсутствуют непосредственные экспериментальные данные, для
определения Т, было предложено уравнение [82]
7> = §(,-.)/с (AO/Д') [1 - (1 - а* Ус)У^с\ . (42)
Правая часть этого уравнения не содержит членов, которые нельзя было бы
определить из результатов измерений электропроводности с помощью
известного значения предельной ионной электропроводности. С помощью
уравнения (42) можно вычислять значения Т, при 25° с точностью до
0,2 — 0,3% в случае разбавленных растворов (с < 0,15) 1,1-валентных элек-
тролитов, для которых, в отличие от других электролитов, в предельном
случае справедливо уравнение (35). Наиболее интересной особенностью
уравнения (42) является то, что эмпирический член, заключенный в скоб-
ки, учитывает отклонения целой группы электролитов от предельного
уравнения. Эти отклонения более точно учитываются уравнениями (39)
и (40), так как последние содержат добавочный эмпирический параметр В
или В*, величина которого зависит от природы данного электролита. Зна-
чение В при 25° можно найти с помощью уравнений (39) и (42):
В=-]/25(Т). (43)
Имеющихся данных недостаточно для того, чтобы определить, в каком
интервале температур справедливо это соотношение; на основании некото-
рых данных можно полагать, что оно неприменимо при температурах,
превышающих 25°. Возможно, что коэффициент у 2, входящий в уравне-
ния (42) и (43), должен быть заменен параметром, зависящим от темпера-
туры (и от природы растворителя), для того чтобы эти уравнения имели
более общий характер.
г) Горин [36] вывел уравнение
(44)
Т_ Г» <l+a+x >
с помощью которого можно вычислять отношение чисел переноса электро-
лита с симметричным типом валентности. Вывод этого уравнения основан
на тех же допущениях, которые были сделаны при выводе уравнения (26).
Значения а+ и а_ можно вычислить с помощью уравнения (27), если из-
вестны экспериментальные предельные коэффициенты наклона кривых элек-
тропроводности и предельные подвижности ионов раствора. Следует отме-
тить, что, несмотря на наличие двух параметров а+ и а_, уравнение (44)
не будет выражать экспериментальные результаты в пределах столь же
широкого интервала концентраций, как уравнения (39), (40) и (42); сво-
дящиеся к теоретическому предельному закону.
д) Джонс и Дол [83] показали, что эмпирическое уравнение
А
-1
1 л^-.ву с
(45)
согласуется с полученными ими . экспериментальными данными для хло-
ристого бария при 25°, причем среднее отклонение вплоть до одномолярной
концентрации составляет лишь 0,0002. Благодаря столь высокой степени
166
ГЛАВА VI
точности и применимости в широком интервале концентраций это уравне-
ние с двумя константами удобно для интерполяции, однако оно неприме-
нимо для экстраполяции. Значение Т'} = А — 1,. полученное из данных для
хлористого бария, не согласуется с известными значениями предельных
электропроводностей ионов бария и хлора.
§ 8. Предельная ионная электропроводность. Закон Кольрауша
В своем классическом исследовании электропроводности растворов
электролитов Кольрауш [84] показал, что предельная эквивалентная электро-
проводность электролита является
суммой двух не зависящих друг от
друга величин, одва из которых ха-
рактеризует анион, а другая — ка-
тион. Это соотношение
Д° =
(46)
5
юо с
Р'и с. 19. Зависимость Х° от концентра-
ции при 25°.
известно под названием закона Коль-
рауша. Полная независимость пре-
дельных ионных электропроводностей
иллюстрируется рис. 19, на котором
изображены данные, полученные
Мак-Иннесом, Шидловским и Лонгс-
вортом [85] для некоторых 1,1-ва-
лентвых электролитов. Величина X®' ,
отложенная на этом рисунке по оси
ординат, отвечает «кажущемуся»
значению X® , вычисленному непо-
средственно из уравнения Онзагера
(28) гл. V путем подстановки в это
уравнение экспериментального зна-
чения ).j=TjA. При бесконечном
разведении значения Х“' равняются
истинным предельным электропро-
водностям XQ. Все изображенные на
рисунке зависимости практически ли-
нейны, за исключением кривой для
азотнокислого калия; следовательно,
при малых концентрациях X®' ли-
нейно зависит от с. Таким образом, зависимость
+ (47)
о
ю
для ионных электропроводностей в случае растворов 1,1-валентных
электролитов имеет тот же вид, что и уравнение (12) для эквивалентной
электропроводности. Эмпирическая постоянная b равняется коэффициенту
наклона соответствующих кривых.
Наиболее важным результатом, который можно получить на основании
анализа рис. 19, является возможность определения Х^ и Х^ из несколь-
ких согласующихся между собой, но независимых исследований. Все зна-
чения Х°, полученные с помощью экстраполяции отдельных кривых, согла-
суются друг с другом в пределах i и сумма средних значений
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
167
kOj= 76,34 и 7^ = 73,52 в точности равна Х°-С1 = 149,86, т. е. величине,
найденной путем прямого определения электропроводности растворов КО.
Таким образом, зная положение общей точки пересечения соответствующих
кривых с осью ординат, можно определять предельные электропроводности
ионов с точностью до 0,02%. Из рис. 19 видно также, что выраженная
уравнением (46) аддитивность весьма точно соблюдается вплоть до концент-
рации с = 0,02 для всех взятых электролитов, за исключением азотнокислого
калия. Этот результат имеет большое значение, так как благодаря приме-
нимости закона Кольрауша к растворам с конечными концентрациями этот
закон можно использовать для определения констант диссоциации слабых
электролитов по измерениям электропроводности. Следует, однако, помнить,
что на рис. 19 представлены только данные для 1,1-валентных электролитов.
Было установлено, что аддитивность электропроводностей одновалент-
ных ионов наблюдается также и в случае смешанных растворителей со
сравнительно высокими значениями диэлектрической постоянной. Из данных
Лонгсворта и Мак-Иннеса [86] , приведенных в табл. 28, видно, что
электропроводность иона хлора в растворах хлоридов натрия и лития
Таблица 28
в 0,05 н. растворах хлоридов натрия и лития; растворитель—смеси
воды и метилового спирта при 25°
Молярная ДОЛЯ спирта Раствор 0 0,1 0,2 0,4 0,6 0,8
NaCl 68,01 45,87 36,19 30,45 29,69 30,82
LiCl 67,99 45,85 36,24 30,46 29,77 30,82
в смесях метилового спирта с водой практически не зависит от природы
одновалентного катиона1.
Зависимость электропроводности ионов данного вида от валентности
другого иона, входящего в состав электролита, изображена на рис. 20 [87].
Теоретические предельные уравнения для электропроводности иона хлора
в трех растворах солей, выраженные через концентрацию с* (в эквива-
лентах на литр), имеют вид:
kciaC1 = 76,34—47,2/с*, (48)
Х^С12 = 76,34-74,0(49)
Х^С13 = 76,34-99,1 (50)
1 Начало изучению электропроводности неводных растворов было положено рабо-
той И. А. Каблукова [Об электропроводности хлористого водорода и серной
кислоты в различных растворителях, ЖРФХО, 22, отд. I, 79 (1890)]; затем оно про-
должалось в работах других советских ученых [А. И. Бродский, Ф. Трах-
тенберг, ДАН, 2, 490 (1934); В. А. Плесков, ЖФХ, 10, 601 (1938)]. В связи
с этим большое значение имели также работы В. А. Плотникова (Исследования по
электрохимии неводных растворов, Киев, 1908) и его учеников по изучению ионогенных
комплексных соединений, способных электролитически диссоциировать в данной системе
и обнаруживаемых методами физико-химического анализа [М. И. Усанович, Сбор-
ник, посвященный юбилею В. А. Плотникова, Киев, 1935; Я. А. Ф и а л к о в, Успехи
химии, 15, 485 (1947); Е. Я. Г о р е н б е й н, ЖФХ, 20, вып. 6, 547 (1946)]. (Прим, ред.)
168
ГЛАВ А VI
При таком выборе единицы концентраций кривые располагаются на ри- .
сунке, не сливаясь друг с другом. Гораздо ближе друг к другу кривые
располагаются, если по оси абсцисс откладывать значения ионной силы,
но и в этом случае они не налагаются друг на друга ни при каких зна-
чениях концентраций. Индивидуальные особенности этих кривых в случае
Р и с. 20. Зависимость ионной электропровод-
ности иона хлора от концентрации при 28°.
разбавленных растворов соот-
ветствуют предсказаниям тео-
рии; кроме того, следует отме-
тить, что отклонения экспери-
ментальных кривых от теорети-
ческих очень сильно зависят от
валентности второго иона. Отсю-
да, а также из данных, относя-
щихся к смесям электролитов
(§ 2), ясно видно, что принци-
пом независимости подвижно-
стей ионов можно пользоваться
только в качестве грубого при-
ближения при обычно приме-
няющихся в исследованиях кон-
центрациях электролитов, со-
стоящих из ионов высокой ва-
лентности или смешанных элек-
тролитов.
В табл. 29 приведены зна-
чения предельных электропро-
водностей некоторых ионов.
Значения электропроводностей
большинства одновалентных
ионов и щелочноземельных катионов определены с точностью, достигаю-
щей нескольких сотых процента. Некоторые из значений электропровод-
ностей многовалентных ионов являются весьма приближенными, о чем
можно судить по числу значащих цифр после запятой.
§ 9. Вязкость растворов сильных электролитов
и ее зависимость от концентрации
Со времени первых исследований Пуазейля [88] было опубликовано
очень много работ [89], посвященных вязкости и величине, обратной вяз-
кости, т. е. текучести растворов. С развитием теории междуионного при-
тяжения интересы исследователей были привлечены к задаче очень точного
определения [90] вязкости разбавленных растворов электролитов, а также
к теоретическому истолкованию результатов. Впервые весьма точные изме-
рения в области сильно разбавленных растворов были выполнены, невиди-
мому, Грюнайзеном [91], который, вопреки принятым ранее взглядам [92],
показал, что в разбавленных растворах вязкость не является линейной
функцией концентрации. Кроме того, отклонения от линейной зависимости
быстро увеличиваются по мере уменьшения концентрации. Такое поведение
электролитов является, невидимому, их общим свойством [93] и было на-
звано [94] эффектом Грюнайзена. Этот эффе'кт отсутствует в случае раство-
ров неэлектролитов.
Джонс и Дол [95, 94], а также Джой и Вольфенден [96] показали
экспериментальным путем, что вязкости очень разбавленных растворов
сильных электролитов линейно зависят от корня квадратного из концен-
трации растворов. Этот результат иллюстрируется рис. 21, а его общий
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ, ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
169
Таблица 29 х)
Предельные ионные электропроводности некоторых ионов в водных
растворах при 25°
Катион + Ссылки на литературу Анион /.1 Ссылки на литературу
н- 349,8 1,13я) OH- 179,6 8,22
Li- 38,69 1 Cl- 76,34 I3),4)
Na- 50,11 1,134) Br~ 78,3 9,2
К- 73,52 I4) J- • 76,8 10,2
NH+ 4 73,4 2 no; 71,44 1
Ag- 61,92 1 CIO’ 4 68,0 3
Tl- 74,7 3 HCO~ 3 44,5 11
neo; 54,6 18
1 2
-Mg - - 53,06 4 CHoCO 3 2 40,9 1,13
^Ca- + 59,50 4 C1CH2CO~ 39,8 12,13
cnch2co; 41,8 18
. ch3ch2co; 35,8 14
1 „ 59,46
2 Sr - - 4 CH3(CH2)2CO2 32,6 14,18
1 4 Ba- - 63,64 4 G6H5CO2 32,3 152), 16
HC2O' 2 4 40,2 20
1 ^Cu- " 54,0 5 h°;' • 74,2 20
Izn- 53,0 5 80 2,7
69,5 65),2 |Fe(CN);-' 101 7
|co(NH3V + " 102,0 7 ^Fe(CN);--' 111 175)
1) Все данные исправлены в соответствии со стандартом Джонса и Брэдшоу. См. табл. 19.
Данные, соответствующие другим температурам, приведены в приложении Б, табл. 172.
2) Сакстон и Мейер приводят также значения для ионов о- п .м-хлорбензоата,равные соответ-
ственно 30,3 и 31,0.
а) Значения для интервалов в 10° между 5 и 65° имеются в статье Оуэна п Сьютона [19].
4) Соответствующие значения электропроводности при 15, 25, 35, и 45° приводят Ганнинг
и Гордон [21а]. См. также [216].
5) См. табл. 23.
характер подтверждается выполненными в последнее время исследованиями
большого числа солей. Было установлено [95, 94], что для многих раство-
ров вплоть до концентрации 0,1н. зависимость вязкости от концентрации
можно точно описать уравнением
Г) / г10 = 1+А]/с-L-Bc. (51)
Экспериментальные значения параметров А и В определяются с по-
мощью графика зависимости левой части уравнения
= Л + В Vс
(52)
170
ГЛАВА VI
от у с. На рис. 22 и 23 изображены такие графики для различных элек-
тролитов. Параметры А и В равны соответственно численным значениям
Рис. 21. Относительная вязкость водных
растворов КС1 при 18, 25 и 35°.
Пунктирные прямые соответствуют теоретическому
предельному закону.
отрезков, отсекаемых прямыми на
оси ординат, и коэффициентов на-
клона этих прямых. Если зависи-
мости не являются линейными, то
значения параметров Л (см. рис. 23)
можно определить из соответствую-
щих кривых лишь тогда, когда
имеются данные для очень разбав-
ленных растворов, однако В в этом
случае, конечно, не является по-
стоянным при всех концентрациях.
Такие кривые являются необыч-
ными для водных растворов и
встречаются лишь в случае ча-
стично диссоциированных электро-
литов, а также электролитов, со-
стоящих из ионов высокой валент-
ности (в особенности электролитов
типа 2,2). Подобным же образом
ведут себя электролиты и в невод-
ных растворителях, однако в этом
случае интервал концентраций,
для которых справедливо уравнение
(51), ограничивается более раз-
бавленными растворами.
Конставта В является весьма специфической и представляет собой
в первом приближении аддитивное свойство ионов [97, 98]. Следует отме-
тить, что для азотнокислого цезия при 25° В имеет отрицательное значение.
Рпс. 22. Графики для определения предельных коэффи-
циентов наклона для относительной вязкости при 25°.
При низких температурах для многих солей, состоящих из больших, срав-
нительно мало гидратированных ионов, В также имеет отрицательное
значение. С другой стороны, температурные коэффициенты В всегда поло-
жительны, так что при высоких температурах вязкость растворов солей
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
171
должна увеличиваться во всем интервале концентраций, который охваты-
вается уравнением (51). Из рис. 21 видно, что в случае хлористого калия
значение В из отрицательного делается положительным между 25 и 35°.
До настоящего времени не найдено удовлетворительного теоретического
объяснения значения параметра В. Как было указано, В является в первом
приближении аддитивным свойством; это обстоятельство можно использо-
вать для создания эмпирических методов вычисления вязкости растворов,
содержащих один [97] или несколько электролитов [99]. Значения «теку-
чести» ионов также можно использовать для этой цели [100].
Рис. 23. Графики для определения предельных коэффи-
циентов наклона для относительной вязкости при 25°.
Хотя специфический характер параметра А менее резко выражен, чем
в случае В, и этот параметр не так сильно зависит от температуры, как В,
численные значения А представляют большой теоретический интерес. Так
как т] / т]0 = 1 + /j* / т]0, то теоретическое значение параметра А можно вы-
числить с помощью уравнения (51) гл. IV, которое применимо к растворам
одного электролита, диссоциирующего только на два вида ионов. Путем
сочетания уравнений (2) гл. III, а также (15) и (48) —(51) гл. IV можно
получить предельную зависимость для относительной вязкости;
7) /-го = 1 + °>(ч), (53)
где
е РН + ’Ф?
(ч)“ 80(|Z1! ГЫ)2 I
4 Г____________| z2 Pi — I zi ______Л21 (54)
L(A0)1/2 + (A’’+Xjp2/Zi| + X0|2l/22|)1/2j /
Это уравнение представляет собой немного видоизмененное соотношение
для предельного коэффициента наклона, впервые выведенное Фалькенгаге-
ном и Верноном [101]. Численные значения коэффициента Р* приведены
в табл. 13 и 124. Для бинарных электролитов
® - °'8863 (V02 ] <55>
а для частного случая, когда X® = Х§, это уравнение сводится к соотноше-
нию, выведенному Фалькенгагеном и Долом [102];
^ = пм-.- <5в>
172
ГЛАВА VI
В табл. 30 сопоставлены экспериментальные и теоретические предель-
ные коэффициенты наклона для водных растворов при 25°. Эксперименталь-
ные и теоретические величины весьма близки друг к другу; эти значения
Таблица 30
Параметры А и В уравнения вязкости (51) для водных растворов при 25°.
Предельные теоретические коэффициенты наклона
Растворенное вещество 1) Тип валент- ности S (Tj) 1°4 A • 10“ В Верхний предел концен- траций Ссылки на лите- ратуру
Сахароза 0 0 0 - 0,8786 0,02 8
Мочевина 0 0 0 0,0378 0,2 8
NH4C1 1,1 50 57 —0,0144 0,2 8
NaCl 1,1 60 ' 67 0,0244 0,2 4
KCI 1,1 50 52 —0,0140 • 0,2 8
KBr 1,1 49 47,4 —0,0480 0,1 7'
KNO3 1,1 52 50 —0,053 0,1 8
KC1O3 1,1 55 50 —0,031 0,1 8
KBrO3 1,1 58 58 —0,001 0,1 8
KMnO4 1,1 56 58 —0,066 0,1 6
CsJ 1,1 48 39 —0,118 0,2 6
CsNO3 1,1 51 43 —0,092- 0,02 8
AgNO3 1,1 56 63 0,045 0.1 9
KaSO4 1,2 131 140,6 0,194 0,1 2
K2CrO4 1,2 131 133 0,152 0,1 2
BaCla 2,1 147 201 0,207 o,t 9
LaCl3 3,1 284 304 0,567 0,1 3
K4Fe(CN), м 330 370 0,366 0,1 5
MgSO4 2,2 228 225 —• — 10
MnSO4 2,2 227 231 — — 10
CuSO4 2,2 228 230 0,540 0,01 10
ZnSO4 2,2 225 229 —. — 10
CdSO4 2,2 225 232 — — 10
Cra(SO4)3 3,2 507 495 — — 10
Ca3[Fe(CN)e]2 2,3 464 467 — — 10
CaaFe(CN)e 2,4 490 495 — — 10
1) Данные для других растворенных веществ см.
в статье Лоуренса и Вольфендена
[1].
так же хорошо согласуются в случае солей со сложным типом валентности,
как и для простых солей типа 1,1. Воспользовавшись значениями А и В
и уравнением (51), можно вычислить, в пределах точности опыта, относи-
тельную вязкость для верхнего предела концентраций, указанного в пред-
последнем столбце, а иногда и для более высоких концентраций. Для
иллюстрации того факта, что для растворенного вещества с незаряженными
частицами предельный коэффициент наклона равен нулю, в таблицу вклю-
чены неэлектролиты сахароза и мочевина. Установлено1, что влияние
1 Джонсом с сотрудниками были получены также для всех солей, приведенных
в табл. 30, соответствующие данные при 0°. Джой и Вольфенден [96] и Кокс п Воль-
фенден [97] исследовали ряд электролитов, в том числе азотную кислоту при 18 и 35°.
Данные, относящиеся к 35° и к более высоким температурам, приводят также Какра-
варти и Прасад [103а] и Ранад и Паранджпэ [1036]; кроме того, эти данные можно-
найти в обычных справочниках.
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, ВЯЗКОСТЬ 173
температуры на предельный коэффициент наклона находится в согласии
с теорией. Труднее было выяснить влияние диэлектрической постоянной
в случае неводных растворителей. Экспериментальные предельные коэффи-
циенты наклона могут быть ошибочны из-за случайной абсорбции влаги
гигроскопическими растворителями, а теоретические коэффициенты наклона
могут быть также неточны вследствие того, что неизвестны точные значе-
ния предельных ионных электропроводностей в этих средах.
В табл. 31 представлены некоторые типичные результаты, полученные
при исследованиях вязкости ацетоновых и спиртовых растворов солей. Для
Табл и ц а 3'1
Предельные коэффициенты наклона для вязкости
в неводных растворителях при 25°
Соль Растворитель А • 104 Sec'104 Ссылки на лите- ратуру
LiCl Ацетон 240 237 1
КС1 Метиловый спирт . . . 151 173 2
КВт — 142 165 2
KJ .— 159 158 2
NH4C1 — 183 165 2
NaJ Этиловый спирт .... 270 255 3
определения значений А, соответствующих этим растворам, необходимо рас-
полагать экспериментальными данными для очень малых концентраций.
Дэлиен и Бриско [104] показали, что при экстраполяции их данных, отно-
сящихся к сравнительно высоким концентрациям (с >0,04), для растворов
хлоридов никеля и алюминия в этиловом спирте получаются отрицатель-
ные значения А, а для растворов хлоридов кобальта, меди, железа, олова,
кадмия и ртути1 в этом же растворителе А равно нулю. На основании
рассмотрения рис. 23 можно предположить, что аномальные значения А,
полученные Дэлиеном и Бриско, не имеют физического смысла.
§ 10. Диффузия электролитов
Определение дифференциального коэффициента диффузии 3 для очень
разбавленных растворов связано со значительными экспериментальными
трудностями. Поэтому Онзагер и Фуосс [25] не смогли получить данные
для растворов с концентрациями ниже 0,05 н., необходимые для проверки
выведенного ими теоретического уравнения (136) гл. IV. С помощью усо-
вершенствованного концентрационного элемента Нортропа — Мак-Бэна [106],
в котором растворы двух различных концентраций разделяются горизон-
тальной диафрагмой из пористого стекла, можно сравнительно легко опре-
делять «интегральные» коэффициенты диффузии. К сожалению, определение
истинных дифференциальных коэффициентов диффузии очень разбавленных
растворов с помощью таких измерений практически неосуществимо. Поэтому
мы не можем надежно сравнивать величины, вычисленные по предельному
закону, с опытными данными. При рассмотрении данных, полученных при
1 Даже в разбавленных водных растворах двухлористая ртуть ведет себя как
типичный неэлектролит. [См. 105.]
174
ГЛАВА VI
более высоких концентрациях, будет показано, как используются уравне-
ния, а также будут1 приведены некоторые экспериментальные результаты.
Согласно уравнениям (136) и (131) гл. IV, дифференциальный коэф-
фициент диффузии равен
S = 16,632 • (57)
где у± представляет собой средний коэффициент активности, а подвиж-
ность Ж / с для 1,1-валентных электролитов выражается уравнением
/т . п2о = ! 074 ___Y-YY. YY +
’ К А° > y]0(DT)ll2\ А0 ) 1-гА’Ус
. 9,18 10’ ( /со.
+ Но(ДТ)2 г с)- (58)
В случае водных растворов коэффициенты 22 / т]0 (Л7’)1/2 и 9,18 • 107/тп0 (DT)*
соответственно равны 16,07 и 18,71 при 25° и 13,50 и 15,53 при 18°.
Первый член в правой части уравнения (58) представляет собой по-
стоянную величину, которая всегда превышает А9/4. Для многих солей
разность (Х+ — /._) настолько мала, что вторым членом можно пренебречь.
Из табл. 14 видно, что произведение ар (А']/с ), которое равно
(1/А')2 (ха)2®(ха), при больших концентрациях является практически по-
стоянной величиной и ни в одном случае не превышает 0,1. Следовательно,
изменение ЗЛ/с обычно настолько невелико, что зависимость коэффициента
диффузии от концентрации определяется только термодинамическим множи-
телем (1 + с с? In у±/дс). Этот множитель можно найти либо графически,
либо путем дифференцирования уравнения
le„t= - + , (59)
1 + A'fc 6 L J ' '
которое получается из уравнений (74) гл. III и (58) гл. I. В последнем
случае мы приходим к соотношению
/ д In и , \ 1,1514 Vс
+ +2'зозд2с--^(<;)- *6°)
Член
,Ь = dd/де+ 0,001 (2^-М2)
9(«)--- й+0,001с(2М1-М2) '
можно отбрасывать только в случае очень разбавленных растворов.
Путем сочетания уравнений (57), (58) и (60) (с учетом того, что зна-
чения функции сср(А'|/с) и ее первой производной по ]/ с приближаются
к нулю при с, стремящемся к нулю) получается предельное уравнение
S = S°-(62)
где
_ 3,732 • 10~2 ( WA \ , 3,659 10~~8 ( Xg \ 2
(S') дЗ/гуЛ/г \ A0 Z у-’/а \_ А» ) ’
И
S0 = 17,863 • 10~1от(^р) , (64)
Коэффициент диффузии бесконечно разбавленного раствора S0 был впервые
вычислен Нернстом [107]. Для водных растворов значение 17,863 • IO-10?7
составляет 5,325 • 10“7 при 25° и 5,200 • 10“7 при 18°. Множители
3,732 • 10-3/Z»3/a71/2 и 3,659 • 10~8 Tv~ / равны 0,03104 и 0,7965 • 10~5
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
175
при 25° и 0.02995 и 0,7781 • 10-5 при 18°. Приведенные выше три уравне-
ния можно получить из общего уравнения диффузии, выведенного Герман-
сом [108], если допустить, что ионные атмосферы имеют шаровую симмет-
рию, и отбросить члены, в которые концентрация с входит в более высокой
степени, чем
В качестве иллюстрации способа вычисления 3> для типичного элек-
тролита рассмотрим случай растворов хлористого натрия при 18°. Из дан-
ных Харнеда и Кука [109] следует, что при этой температуре А' = 1,310,
25 = 0,0502 и S(7) = 0,5002. Зависимость плотности [110] растворов от кон-
центрации можно выразить уравнением
d = 0,99862 + 0,0428 с - 0,00145 с3/+ (65)
Следовательно,
0 +°.1156—<66>
и
, . 0,0204 — 0,00217 /с
ф (d) =-------------------. (67)
0,9986 + 0,0204 с— 0,00145 с3'2
Подставляя соответствующие численные значения в уравнение (58) при 18°
и принимая предельные подвижности ионов1 равными 66,0 и 42,9, получаем
Ю2О = 27,92 - 0’6—% +15,53 с? (ха), (68)
< с J .1 + 1,31 /с п ' •
Сплошная линия на рис. 24 изображает зависимость 3) от "[/с, вычисленную
с помощью этих уравнений. Если при вычислении пренебречь членом ф (d),
то полученным значениям будет отвечать кривая 1. Кривая 2 получена
с помощью уравнения (58), в котором опущены два последних члена.
В данном случае влияние члена ф (d) оказывается более сильным, чем соот-
ветствующее теории изменение Ж/с с концентрацией. Экспериментальные
значения дифференциальных коэффициентов диффузии, полученные Кла-
ном [111]2, изображены кружками. При больших концентрациях экспери-
ментальные значения сильно отклоняются от теоретически вычисленных.
Хотя Онзагер и Фуосс [112] и выдвинули качественное объяснение такого
поведения, тем не менее не подлежит сомнению, что в рассматриваемом
интервале концентраций изложенная теория неприменима.
С другой стороны, при низких концентрациях экспериментальные зна-
чения, несомненно, приближаются к теоретически вычисленным3. В этой
области концентраций коэффициенты наклонов экспериментальных, кривых
совпадают с предельным коэффициентом наклона теоретической кривой4,
соответствующей уравнению (63). Полученные в последнее время данные
для растворов соляной кислоты [114] при 25° меньше отличаются от тео-
ретически вычисленных значений при концентрациях, превышающих 0,01 н.,
чем при меньших концентрациях, причем 3)°, найденное путем экстра-
поляции этих значений, примерно на 6% ниже значения, соответствующего
1 При 18°, согласно табл. 26, Л^аС1 =108,0; Д^С1=129,9 и 7’][=0,492.
2 Температура поддерживалась равной приблизительно 18,5°.
3 Данные для хлористого калия при 18° (Клак) и при 25° (Мак-Вен и Даусон.
в этом отношении не отличаются друг от друга.
4 Фридман и Карпентер [1131 обьаружили, что коэффициент диффузии неэлектро-
лита (глюкозы) в значительном интервале концентраций линейно зависит от }4С.
176
ГЛАВА VI
уравнению (64). Так как экспериментальные данные получены с помощью
элемента Мак-Бэна— Нортропа и их можно рассматривать [115] только как
известное приближение к истинным значениям w при средних значениях
концентраций, применявшихся в этих опытах, то результаты для наиболее
разбавленных растворов вряд ли можно использовать для проверки теории.
Онзагер и Фуосс показали, что при умеренных концентрациях измене-
ние вязкости, вызванное наличием в растворе попов, заметно отражается
на величине члена уравнения, который выражает подвижность, и что
в первом приближении этот эффект должен быть обратно пропорционален
макроскопической вязкости. На основании кривой 2 на рис. 24 можво
Р и с. 24 Л Коэффициенты диффузии хлористого
натрия в воде. Теоретические кривые относятся
к 18°.
Экспериментальные точки были получены при 18,5°.
судить о том влиянии, которое оказывает умножение величины ЯЛ / с,
входящей в уравнение (58), на т)0/т(. В результате умножения на эту ве-
личину расчетная кривая приближается к экспериментальным точкам
в области разбавленных растворов. Однако при более высоких концентра-
циях она значительно отходит от экспериментальной кривой. Ван-Риссель-
берг [116] подробно исследовал эту и другие аналогичные поправки.
Гордон [115] предложил полуэмпирическое уравнение
12) = 2000 1,074- (1+ , (69)
в котором множитель, выражающий подвижность, содержит как парциаль-
ный молярный объем растворителя, так и вязкость раствора. Это уравне-
ние в пределе не сводится к уравнению (62), однако с его помощью можно
получить значение ££>°, согласующееся со значением, вычисленным по
уравнению (64). Для ряда электролитов [115, 1146] уравнение Гордона
согласуется с экспериментальными данными с точностью до ошибки опыта
(см. рис. 25.). Следует отметить, что для растворов хлористого натрия
при 18° ч]о/т1 меньше единицы, тогда как для растворов хлористого и азотно-
кислого калия т]0/т] больше единицы для всех растворов, за исключением
наиболее разбавленных. Для электролитов,, состоящих из ионов более высо-
кой валентности, уравнение (69) менее пригодно, чем в описанных выше
случаях [117].
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, ВЯЗКОСТЬ 177
Другая попытка теоретически истолковать данные, полученные при
умеренных концентрациях, была предпринята Виноградом и Мак-Бэном [118].
Эти исследователи не учитывали коэффициенты активности и влияние
столкновений частиц при всех концентрациях, а также распространили
классическое соотношение Нернста [107, 119] на смеси электролитов.
В полученных ими уравнениях интегральный коэффициент диффузии любого
данного иона выражен через подвижности и градиенты концентраций каж-
дого из ионов. Непосредственное влияние градиентов концентраций в этих
уравнениях четко отделено от влияния диффузионного потенциала. Если
последний равен нулю, то ионы диффундируют со свойственной каждому
Р и с. 25. Сравнение значений, вычисленных с по-
мощью уравнения (69), с экспериментальными дан-
ными.
из них истинной скоростью; под влиянием диффузионного потенциала
движение быстрых ионов замедляется, а медленных ионов — ускоряется’.
Наличие такого ускорения и замедления подтверждается эксперименталь-
ными данными, полученными для смесей электролитов. Величины этих
эффектов хорошо согласуются с величинами,' вычисленными с помощью
соответствующих уравнений, если допустить, что градиенты концентрации
при диффузии в чистой воде являются линейными. Дальнейшее обсуждение
вопросов диффузии можно найти в приложении Б, § 3.
ЛИТЕРАТУРА
1. С. W. Davies, The Conductivity of Solutions, John Wiley and Sons, N. Y. 1930-
Trans. Faraday Soc., 28, 607 (1932). W. F. K. W у n n e-J о n e s, J. Phys. Chem ’
31, 1647 (1927).
2. J. N. Bronsted, R. F. Nielsen, Trans. Faraday Soc., 31, 1478 (1935);
R. M. F u о s s, С. A. К r a u s, J. Am. Chem. Soc., 55, 21, 3614 (1933); L. V. A n d-
rews, W. E. Martin, там же. 60, 871 (1938); H. E. Gunning A. R. Gor-
don, J. Chem. Phys., 10, 126 (1942).
3. G. J о n e s, S. M. Christian,!. Am. Chem. Soc., 57, 272 (1935).
4. G. J о n e s, D. M. В о 1 1 i n g e r, J. Am. Chem. Soc., 57, 280 (1935).
5. F. Ko h 1 r a u s c h, Z. physik. Chem., 2, 561 (1888).
6. B. Hague, A.C.Bridge Methods, Pitman, N. Y., 1938.
7. G. J о n e s, R. C. J о s e p h s, J. Am. Chem. Soc., 50, 1049 (1928); P. H. D i k e.
Rev. Sci. Instruments, 2, 379 (1931).
1 В оригинале сказано наоборот: движение быстрых ионов ускоряется, а медлен-
ных—замедляется. Это—явная ошибка авторов. (Прим. ред.).
12 Г. Харнед, Б. Оуэн
178
ГЛАВА VI
, 8. a) S h е d 1 о v s к у, J. Am. Chem. Soc., 52, 1793 (1930); б) К. W. Wagner,
Elektrotech. Z., 32, 1001 (1911); в) A. V. A s t i n, Bur. Standards, J. Research,
21, 425 (1938); r) W. F. L u d e r, J. Am. Chem. Soc., 62, 89 (1940).
9. G. J ones, G. M. В о 1 1 i n g e r , J. Am. Chem. Soc., 53, 411 (1931).
10. T. S h e d 1 о v s к y, J.Am. Chem. Soc., 52, 1806 (1930).
11. C. W. D a v i e s, J. Chem. Soc., 138, 432 (Д937).
12. H. C. Parker, J. Am. Chem. Soc.,'45, 1366, 2017 (1923).
13. F. К о h I r a u s c h, L. H о 1 b о r n, H. D i e s s e 1 h о r s t, Wien. Ann., 64,
417 (1898).
14. H. С. P а г к e r, E. W. Parker, J. Am. Chem. Soc., 46, 312 (1924).
15. G. J о n e s, В. C. Bradshaw, J. Am. Chem. Soc., 55, 1780 (1933).
16. G. Jones, M. J. Prendergast,!. Am. Chem. Soc., 59, 731 (1937).
17. a) L. О n sager, Physik. Z.,28, 277 (1927); 6) J. Lan g. e, Z. Physik. Chem., A188,
284 (1941).
18. P. D e b у e, E. H ii e к e 1, Physik. Z., 24, 305 (1923).
19. P. W a 1 d e n, Z. physik. Chem., 144, 297, 434 (1929); A147, 1 (1930).
20: С. P. W r i g h t, D. M. M u r r a y-R ust, H. Hartley, J. Chem. Soc., 134,
199 (1931).
21. A. U n m а с к, E. В ul lock, D. ,M. Murra y-R u s t, H. Hartley, Proc.
Roy. Soc., A132, 427 (1931).
22. M. В a г а к, H. Hartl e y, Z. physik. Chem., A165, 272 (1933).
23. P. W a 1 d e n, H. U 1 i c h, G. В u s c h, Z. physik. Chem., 123, 429 (1926).
24. H. U 1 i c h, Z. angew. Chem., 41, 1141 (1928).
25. L. О n s a g e r, R. M. F u о s s, J. Phys.-Chem., 36, 2689(1932).
26. L. G. Longsworth, J. Am. Chem. Soc., 52, 1897 (1930).
27. K. Bennewitz, C. Wagner, К. К ii c h I e r, Physik. Z., 30, 623 (1929),
28. a) K. A. Krieger, M. Kilpatrick,!. Am. Chem. Soc., 59, 1878 (1937);
б) там же, 60, 3601 (1938).
29. J. E. R e n h о 1 m, диссертация, Helsingfors, 1925.
30. C.. W. Davies, Trans. Faraday Soc., 23, 351 (1927).
31. C. W. D a v i e s, J. Chem. Soc., 140, 2093 (1938); W. H. В a n к s, E. C. Rig-
h e 1 1 a t о, C. W. Davies, Trans. Faraday Soc., 27, 621 (1931).
32. R. M. F u о s s, J. Am. Chem. Soc., 57, 488 (1935).
33. T. S h ed 1 о v s к y, J. Franklin Inst., 225, 739 (1938).
34. R. M. Fuoss, Physik. Z., 35, 59 (1934).
35. В. B. Owen, J. Am. Chem. Soc., 61, 1393 (1939).
36. M. H. G о r i n, J. Chem. Phys., 7, 405 (1939).
37. S. К aneko, J. Chem. Soc. Japan, 56, 793, 1320 (1935); 58, 985 (1937).
38. T. S h e d 1 о v s к y, J. Am. Chem. Soc., 54, 1405 (1932).
39. R. Lorenz, Z. anorg. Chem., 108, 191 (1919).
40. F. К о h 1 r a u s c h, L. H olborn, Das Leitvermogen der Elektrolyte, Teubner,
Leipzig, 1916.
41. P. Walden, Z. physik. Chem., 108, 341 (1924).
42. R. T. Latte y, Phil. Mag. [7], 4, 831 (1927).
43. G. J о n e s, M. D о I e , J. Am. Chem. Soc., 52, 2245 (1930).
44. G. J о n e s, C. F. Bickford, J. Am. Chem. Soc., 56, 602 (1934).
45. W. О s t w a 1 d, Z. physik, Chem., 2, 36 (1888).
46. a) C. A. Kraus, W. C. Bray, J. Am. Chem. Soc., 35,1315(1913);
б) E. W. W a s h b u r n, там же, 40, 150 (1918); в) X. С. Текло р, Физическая
химия, т.. I, ОНТИ—Химтеорет, Л., 1935, гл. XI, стр. 636—641.
47. a) R. Loren z, Z. anorg. Chem., 114, 209 (1920); б) P. Hertz, Ann. Phys. [4],
. 37, 37 (1912); в) I. C. Ghosh, J. Chem. Soc., 113, 449 и сл. (1918); г) R. Lo-
renz, P. Osswald, Z. anorg. Chem., 114, 209 (1920); R. Lorenz, W. M i -
c h a e 1, там же, 116, 161 (1921); P. Walden, H. U 1 i c h, Z. physik. Chem.,
106, 49 (1923).
48. A. Ferguson,!. Vogel, Phil. Mag. [4], 50, 971 (1925); [7], 4, I, 233, 300 (1927).
49. A. Ferguson, I. Vogel, Trans. Faraday Soc., 33, 404, 414 (1927); I. V о g e 1,
Phil. Mag. [7], 5, 199 (1928).
50. a) C. W. Davie s, The Conductivity of Solutions, Chapman and Hall, London,
1930, p. 84; 6) A. F e r g u s о n, I. Vogel, Trans. Faraday Soc., 27, 285 (1927);
в) C. W. Davie s, A. R. M a r t i n, A. W. Porte г, там же, 27, 547 (1927).
51. T. S h e d 1 о v s к у, J. Am. Chem. Soc., 54, 1411 (1932).
52. a) L. G. L о n g s w о r t h, J. Am. Chem. Soc., 54, 2741 (1932); 6) D. A. Mac I n-
n e s, T. Shedlovsky, L. G. Longsworth, там же, 54, 2758 (1932).
53. a) D. J. L e R о y, Univ. Toronto Studies, Papers Chem. Lab., No 156 (1939);
6) J. W. M с В a i n, там же, No 67 (1907).
54. О. Lodge, Brit. Assn. Advancement Sci., Birmingham, 1886, p. 389.
55. W. C. D. Whetham, Phil. Trans., A184, 337 (1893); Z. physik. Chem., 11, 220
(1893).
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
179
56. W. Nernst, Z. Elektrochem., 3, 308 (1897).
57. D. О. М a s s о n, Phil. Trans., А192, 331 (1899).
58. R. В. D е n i s о п, В. D. S t с e 1 e, J. Chem. Soc., 89, 999 (1906).
59. E. C. F r a n k 1 i n, H. P. C a d y, J. Am. Chem. Soc., 26, 499 (1904); H. P. C a-
d y, L. G. Longsworth, там же, 51, 1656 (1929).
60. E. R. S m i t h, D. A. M a с I n n e s, J. Am. Chem. Soc., 46, 1398 (1924); там же,
47, 1009 (1925); E. R. Smith, Bureau of Standards J. Research, 6, 917 (1931).
61. D. A. Mac Innes, E. R. Smith, J. Am. Chem. Soc., 45, 2246 (1923);
D. A. M a с I n n e s, I. A. Cowperthwai te, T. C. Huang, там же,
49, 1710 (1927); L. G. Longsworth, D. A. Maclnnes, Rev. Sci. Inst.,
19, 50 (1929).
62. D. A. M ac Inn es, L. G. Long swo rth, Chem. Rev., 11, 171 (1932).
63. W. -L. M i 1 1 e r, Z. physik. Chem., 69, 436 (1909).
64. E. R. Smith, Bur. Standards J. Research., 8, 457 (1932).
65. G. N. Lewis, J. Am. Chem. Soc., 32, 862 (1910).
66. D. A. M a с I n n e s, I. A. Cowperthwaite, Proc. Nat. Acad. Sci., 15, 18
(1929).
67. F. К ohlr ausch, Ann. Physik., 62, 209 (1897).
68. H. P. C a d y, L. G. L о n g s w о r t h, J. Am. Chem. Soc., 51, 1656 (1929).
69. G. S. Hartle y, Trans. Faraday Soc., 30, 648 (1934); E. D rev, G. S. Har-
tley, там же, p. 653; В. С о 1 1 i e, G. S. Hartley, там же, p. 657.
70. G. S. Hartley, В. С о 1 1 i e, C. S. Samis, Trans. Faraday Soc., 32, 795
(1936).
71. J. С. M oidli et, C. R. С о 1'1 i e, G. S. Hartley, Trans. Faraday Soc., 31,
120 (1935).
72. W. H i t t о r f, Ann. Physik., 89, 177 (1853); 98, 1(1856); 103, 1 (1858); 106, 337, 513
(1859).
73. D. A. M a с I n n e s, M. Dole, J. Am. Chem. Soc., 53, 1357 (1931).
74. G. J о n e s, В. C. Bradshaw,!. Am. Chem. Soc., 54, 138 (1932).
75. E. W. Washburn, Principles of Physical Chemistry, McGraw-Hill Book Co.,
N. Y., 1921, p. 277; J. Am. Chem. Soc., 31,322(1909); A. "A. N о у e s, K. G. F a Ik,
там же, 33, 1436 (1911).
76. a) L.. G. L о n g s w о r t h, J. Am. Chem. Soc., 57, 1185 (1935); 6) G. S. H a r t-
1 e y, G. W. Donaldson, Trans. Faraday Soc., 33, 457 (1937).
77. E. C. R i g h e 1 1 a t о, C. W. Davies, Trans. Faraday Soc., 26, 592 (1930).
78. C. S. Samis, Trans. Faraday Soc., 33, 469 (1937).
79. a) R. W. Allgood, D. J. L e R о y, A. R. Gordon, J. Chem. Phys., 8,
418 (1940); 6) R. W. A 1 1 g о о d, A. R. Gordon, там же, 10, 124 (1942).
80. H. S. Taylor, J. Chem. Phys., 6, 331 (1938).
81. T. S h e d 1 о v s k y, J. Chem. Phys., 6, 845 (1938).
82. В. B. Owen, J. Am. Chem. Soc., 57, 2441 (1935).
83. G. Jone s, M. Doi e, J. Am. Chem. Soc., 51, 1073 (1929).
84. F. Kohlrausch, Ann. Physik., 50, 385 (1893); 66, 785 (1898).
85. D. A. Maclnnes, T. Shed lo vsky, L. G. Longsworth, J. Am. Chem.
Soc., 54, 2758 (1932); Chem. Rev., 13, 29 (1933).
86. L. G. L о n g s w о r t h, D. A. M a с I n n e s, J. Phys. Chem., 43, 239 (1939).
87. L. G. L о n g s w о r t h, D. A. Maclnnes,!. Am. Chem. Soc., 60, 3070 (1938).
88. J. L. M. P о i s e u i 1 1 e, Ann. chim. phys. [3], 21, 76'(1847). •
89. E. C. Bingham, Fluidity and Plasticity, McGraw-Hill Book Co., N. Y., 1922;
Э. Гатчек, Вязкость жидкостей, ОНТИ, М.—Л., 1935.
90. Е. Griineisen, Wiss. Abh. phys. tech. Reichsanstalt, 4, 159, 241 (1904);
E. W. W a s h b u r n, G. V. Williams, I. Am. Chem. Soc., 35, 737 (1913);
E. С. В i n g h a m, R. F. Jackson, Bull. Bureau of Standards, 14, 59 (1918—
1919); G. J о n e s, S. K. Talley, Physics, 4, 215 (1933); G. Jones,,
H. J. F о r n w a 1 t, J. Am. Chem. Soc., 60, 1683 (1938); G. J о n e s, R. E. Sta-
uffer, там же, 59, 1630 (1937).
91. E. Griineisen, Wiss. Abh. phys. techn. Reichsanstalt, 4, 151, 237 (1905).
92. A. Sprung, Pogg, Ann. Phys. Chem., 159, 1 (1876); S. Arrhenius, Z. phy-
sik. Chem., 1, 285 (1887).
93. M. P. Appelbey, J. Chem. Soc., 97, 2000 (1910); T. R. Merton, там же,
97, 2454 (1910).
94. G. J о n e s, S. К. T a 1 1 e y, J. Am. Chem. Soc., 55, 624 (1933).
95. G. J о n e s, M. D о 1 e, J. Am. Chem. Soc., 51, 2950 (1929).
96. W. E. J о y, J. H. W о 1 f e n d e n, Proc. Roy. Soc., A134, 413 (1931).
.97. W. M. С о x, J. H. W о 1 f e n d e n, Proc. Roy. Soc., A145, 475 (1934).
98. V. D. Laurence, J. H. W о 1 f e n d e n, J. Chem. Soc., 136, 1144 (1934).
99. H. Tollert, Z. physik. Chem., A172, 129 (1935); A174, 239 (1935); A180, 383
100. E. С. В i n g h a m, J. Phys. Chem., 45, 885 (1941).
12*
180
ГЛАВА VI
101. Н. Falkenhagen, Е. L. Vernon, Physik. Z., 33, 140 (1932); Phil. Mag [7] >
14, 537 (1932); H. Falkenhagen, Nature, 127, 439 (1931);. Physik. Z., 32‘
745 (1931); Z. physik. Chem., B6, 159 (1931).
102. H. Falkenhagen, M, Dole, Physik, Z., 30, 611 (1929); Z. physik. Chem.,
B6, 159 (1929).
103. a) A. S. Chakravarti, B. Prasad, Trans. Faraday Soc., 35, 1466 (1939,);
6) J. D. R a n a d e, G. R. P a r a n j p e, J. Univ. Bombay, 7, 41 (1938).
104. F. E.D alien, H. T. В r i s с о e, J. Phys. Chem., 41, 1129 (1937).
105. F. Prasad, A. S. Chakravarti, B. Prasad, J. Indian Chem. Soc,, 15,
301 (1938).
106. J. H. N о r t h г о p, M. L. Anson, J. Gen. Physiol., 12, 543 (1929);
J. W. M с В a i n, T. H. L i u, J. Am. Chem. Soc., 53, 59 (1931); J. W. McB a i n,
C. R. Dawson, там же, 56, 52, 1021 (1934); Proc. Roy. Soc., A148, 32 (1935).
107. W. N e r n s t, Z. physik. Chem., 2, 613 (1888).
108. J. J. Hermans, диссертация, Leiden, 1937, p. 27; Rec. trav. chim., 56, 635,
658 (1937).
109. H. S. H a r n e d, M. А. С о о k, J. Am. Chem. Soc., 61, 495 (1939).
110. Landolt-Bornstein, Physikalisch-Chemische Tabellen, Egr. Ill-a, 1935,
p. 378/
111. B. W. Clack, Proc. Phys. Soc., 36, 313 (1924).
112. L. О n s a g e r, R. M. F u о s s, J. Phys. Chem., 36, 2769 (1932).
113. L. Friedman, P. G. Carpenter, J. Am. Chem. Soc., 61, 1745 (1939).
114. a) W. A. J ames, E. A. H о 1 1 i n g s h e a d, A. R. G о r d о n, J. Chem.
Phys., 7, 89 (1939); 6) W. A. J a m e s, A. R. Gordon, там же, 7, 963 (1939).
115. A. R. Gordon, J. Chem. Phys., 5, 522 (1937).
116. P. Van Rysselberghe, J. Am. Chem. Soc., 60, 2326 (1938).
117. E. A. H о 1 1 i n g s h e a d, A. R. Gordon,!. Chem. Phys., 9, 152 (1941).
118. J. R. V i n о g r a d, J. W. McBain, J. Am. Chem. Soc., 63, 2008 (1941).
119. S. A r r h e n i u s, Z. physik. Chem., 10, 51 (1892).
ЛИТЕРАТУРА К ТАБЛИЦАМ
К табл. 19
1. И. Е. Gunning, A. R. G о г d о n, J. Chem. Phys., 10, 130 (1942).
К табл. 20
1. L. Onsager, R. М. F и о s s, J. Phys. Chem., 36, 2689 (1932).
2. W. С. В r ay, F. L. Hun t, J. Am. Chem. Soc., 33, 781 (1911).
К табл. 21
1. T. S h e d 1 о v s к у, J, Am. Chem. Soc., 54, 1411 (1932).
2. G. Sc at c hard, S. S. P r e n t i s s, P. T. J о n e s, J. Am. Chem. Soc., 54, 2690
(1932).
К табл. 22
1. C. W. Davies, Trans. Faraday Soc., 23, 351 (1927).
2. a) E. C. R i g h e 1 1 a t о, C. W. D a v i e s, Trans. Faraday Soc., 26, 592 (1930);
6) J. Z i г к 1 e r, Z. physik. Chem:, A163, 1 (1932).
3. a) C. W. Davies, J. Chem. Soc. 140, 277 (1938); 6) R. К. С a n n a n, A. К i b-
r i c k, J. Am. Chem. Soc., 60, 2314 (1938); в) N. E. T о p p, C. W. D a v i es,
J. Chem. Soc., 142, 87 (1940).
4. C. W. Davies, J. Chem. Soc., 141, 349 (1939).
5. C. W. D a vies, J. Chem. Soc., 140, 2093 (1938).
6. R. W. M о n e у, C. W. Davies, Trans. Faraday Soc., 28, 609 (1932).
7. В. В. О w e n, R. W. Gurry, J. Am. Chem. Soc., 60, 3074 (1938).
8. C. W. D a v i e s, J. Am. Chem. Soc., 59, 1760 (1937).
К табл. 23
1. G. J о n e s, В. C. Bradshaw,!. Am. Chem. Soc., 55, 1780 (1933).
К табл. 26
J. R. W. A 1 1 g о о d, D. J. L e R о y, A. R. Gordon, J. Chem. Phys., 8, 418
(1940).
2. H. E. G и n n i n g, A. R. Gordon, J. Chem. Phys., 10, 126 (1942).
СИЛЬНЫЕ ЭЛЕКТРОЛИТЫ. ЭЛЕКТРОПРОВОДНОСТЬ, вязкость
181
К табл. 27
1.
2.
1.
2.
3.
4. T.
5. В.
6. G.
7.
8.
9.
10.
11.
12.
R. W.. Allgood, A. R. G о r d о n, J. Chem. Phys., 10, 124 (1942).
H. E. Gunning, A. R. Gor ’ ’ ™
К табл. 29
D. A. Maclnnes, T. Shedlo
Soc., 54, 2758 (1932). '
A. Maclnnes, J. Franklin
A. R о b i n s о n, C. W. D a v
S h e d 1 о v s k y, A. S. В г о w n, J. Am. Chem. Soc., 56, '1066 (1934).
В. О w e n, R. "r
J ones, C. F.
S. Hartley,
S i v e r t z, R.
1379 (1940).
G. J о n e s, C.
P. ' ’
T. Shedlovsky, D. A. Maclnnes, J. Am. Chem. Soc., 57, 1705 (1935).
T. Shedlovsky, A. S. " ~ ‘ ™
Soc., 66, 165 (1935).
S a x t о n, T. W. L a n g e r, J. Am. Chem. Soc., 55, 3638 (1933).
Belcher, J. Am. Chem. Soc., 60, 2744 (1938).
S a x t о n, H. F. M e i e r, J. Am. Chem. Soc., 56, 1918 (1934).
G. Brockman, M. Kilpatrick, J. Am. Chem. Soc., 56, 1483 (1934).
J о n e s, F. C. J e 1 e n, J. Am. Chem. Soc., 58, 2561 (1936); C. W. Davies,
J. Am. Chem. Soc., 59, 1760 (1937).
B. Saxton, L. S. Darken, J. Am. Chem. Soc., 62, 846 (1940).
В. B. Owen, F. H. S w e e t о n, J. Am. Chem. Soc., 63, 2811 (1941).
L. S. Darken,!. Am. Chem. Soc., 63, 1007 (1941).
a) H. E. Gunning, A. R. Gordon, J. Chem. Phys., 10, 126 (1942); 6) N. C. Li,
W. В r ii 1 1, J. "" " ‘ ~
64, 2517 (1942).
S. Darken,
don, J. Chem. Phys., 10, 126 (1942).
D.
R.
G.
13.
14. D.
15. B.
16. F.
17.
18.
19.
20.
21.
В.
G.
22. L.
F.
A. L a s s e 1 1 e.
К табл. 30
vsky, L. G. Longsworth, J. Am. Chem.
Inst.-, 225, 661 (1938).
i e s, J. Chem., Soc., 139, 574 (1937).
W. G urry, J. Am. Chem. Soc., 60, 3074 (1938)'.
Bickford, J. Am. Chem. Soc., 56, 602 (1934).
G. W. Donaldson, Trans. Faraday Soc., 33, 457 (1937).
E. R e i t m e i e r, H. V. T a r t a r, J. Am. Chem. Soc., 62,
E.
Bickford, J. Am. Chem. Soc., 56, 602 (1934).
J. G. A s t о n, J. Am. Chem. Soc., 55, 3067 (1933).
Brown, D. A. M a с I n n es, Trans. Electrochem.
Am.
H.
Chem. Soc., 64, 1635 (1942); A. R. Gordon, там же.
F.
Meier, J. Am. Chem. Soc., 64, 621 (1942).
1. V. D. Laurence, J. H. Wolf f enden, J. Chem. Soc., 136, 1144 (1934).
2. G. J о n e s, J. H. Colvin, J. Am. Chem. Soc., 62, 338 (1940).
3. G. J о n e s, R. E. S t a u f f e r, J. Am. Chem. Soc., 62, 335 (1940).
4. G. J о n e s, S. M. Christian, J. Am. Chem. Soc., 59, 484 (1937).
5. G. J о n e s, R. E. S t a u f f e r, J. Am. Chem. Soc., 58, 2548 (1936).
6. G. J о n e s, H. J. F о r n w a 1 t, J. Am. Chem. Soc., 58, 619 (1936).
7. G. J о n e s, S. К. T a 1 1 e y, J. Am. Chem. Soc., 55, 4124 (1933).
8. G. J о n e s, S. К. T a 11 e y, J. Am. Chem. Soc., 55, 624 (1933).
9. G. J о n e s, M. D о 1 e, J. Am. Chem. Soc., 51, 2950 (1929).
10. E. A s m u s, Ann. Physik, 35, 1 (1939).
К табл. 31
1. G. R. Hood, L. P. H о h 1 f e 1 d e r, J. Phys. Chem., 38, 979 (1934).
2. G. J о n e s, H. J. F о r n w a 1 t, J. Am. Chem. Soc., 57, 2041 (1935).
3. W. M. С о x, J. H. W о 1 f e n d e n, Proc. Roy. Soc., A145, 475 (1934).
Глава VII
КУЛОНОВСКИЕ СИЛЫ И АССОЦИАЦИЯ ИОНОВ.
СЛАБЫЕ ЭЛЕКТРОЛИТЫ. ВЛИЯНИЕ ЧАСТОТЫ И СИЛЫ ПОЛЯ
В предыдущей главе был подробно рассмотрен вопрос об электропро-
водности очень, сильно диссоциированных электролитов в средах с высокой
диэлектрической постоянной. С помощью уравнения электропроводности
Онзагера можно показать, что поведение большей части таких растворов
свидетельствует о полной диссоциации растворенного вещества при малых
концентрациях. В неводных средах, согласно теории ассоциации ионов
Бьеррума (гл. III, § 7)„ по мере уменьшения диэлектрической постоянной
растворителя увеличивается стремление ионов всех электролитов к ассоциа-
ции. Экспериментальные результаты, которые будут рассмотрены ниже,
подтверждают представления Бьеррума. В соответствии с этими представле-
ниями в средах с малой диэлектрической постоянной все электролиты
являются частично ассоциированными или «слабыми» и деление электроли-
тов на сильные и слабые становится до некоторой степени условным. Тем
не менее деление электролитов на сильные и слабые в соответствии с тем,
являются ли они сильно или слабо диссоциированными в водных растворах,
обычно сохраняется при описании свойств электролитов независимо от того,
какая среда выбрана в качестве растворителя.
§ 1. Закон Стокса и правило Вальдена 1 * * * * *
При растворении сильных электролитов в растворителях с очень низ-
кой диэлектрической постоянной под влиянием простых кулоновских сил
происходит ассоциация ионов даже при крайне малых концентрациях
ионов. В этом случае ионы, не участвующие в образовании ассоциирован-
ных ионных пар или более сложных агрегатов, настолько удалены друг
от друга, что можно пренебречь силами взаимного отталкивания ионов,
действующими лишь на коротких расстояниях; влияние ионных атмосфер
будет при этом также очень мало. Так, например, азотнокислый тетраизо-
амиламмоний в водном растворе является сильным электролитом; в 10-3 н.
растворе этой соли в смеси диоксан—вода, имеющей диэлектрическую
постоянную 2,3(0,343% Н2О), концентрация ионов тетраизоамиламмония
составляет весьма небольшую величину, порядка 10~8. Несмотря на
большие значения предельного коэффициента наклона 5(д), обусловлен-
ные низкой диэлектрической постоянной, наличие ионных атмосфер
изменяет А менее чем на 0,2%, и этой небольшой величиной можно пре-
небречь.
1 Харнед и Оуэн допускают ошибку. Закономерность, связывающая электро-
проводность и вязкость при бесконечном разведении, несправедливо приписы-
ваемая авторами Вальдену, была в действительности установлена Писаржевским
[Л. В. Писаржевский, Н. Лемке, ЖРФХО, 37, 492 (1905)] на год раньше Валь-
дена. Таким образом, это правило следует именовать правилом Писаржевского.
(Прим, ред.)
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
183
Вследствие чрезвычайно малой диссоциации, приводящей к образованию
ионов, определение Д° посредством прямой экстраполяции эксперименталь-
ных данных неосуществимо. В случае водных растворов слабых электроли-
тов Д° вычисляется на основе закона Кольрауша, т. е. косвенным путем из
данных для соответствующих сильных электролитов. Очевидно, этот способ
неприменим к растворителям, диэлектрическая постоянная которых настоль-
ко мала, что все электролиты, растворенные в них, сильно ассоциированы.
В этом случае обычно применяется приближенное соотношение, известное
под названием правила Вальдена. Это правило было установлено опытным
путем [1], однако можно.показать, что оно является прямым следствием при-
менения закона Стокса [уравнение (62) гл. IV] к движению ионов. Так
как правило Вальдена наиболее строго выполняется для систем, удовле-
творяющих тем условиям, которые принимаются при выводе закона Стокса,
то следует .рассмотреть оба эти соотношения одновременно.
Если электрическая сила действует на сферический ион радиуса а,,
находящийся в гомогенной среде с вязкостью т]0, то, согласно закону Стокса,
этот ион приобретает постоянную скорость1:
Часто форма ионов сильно отличается от сферической, и поскольку ионы
имеют величину того же порядка, что и молекулы растворителя, то вряд
ли можно точно описать тормозящее влияние последних с помощью пред-
ставления о макроскопической вязкости. В первом приближении можно
считать, что условия применимости уравнения (1) соблюдаются для боль-
ших сферических ионов. Сочетая это уравнение с уравнением (15) гл. IV
и соотношением оу- — Vt/Xzjs, получаем
л 96500 I ^, | s 0,8147 • 10~8 [ г,-/
^° = ^8ОоЦ. =----------«7^-- (2 * * * * *)
Таким образом, если А0 — предельная эквивалентная электропроводность
электролита, диссоциирующего на р видов ионов, то, следовательно,
АЧ = 0,8147.10-8 2 (3)
7=1 1
Радиусы простых ионов, вычисленные с помощью уравнения (2), имеют
величину порядка 1 —ЗА, что согласуется с величинами радиусов2 ионов
(сольватированных), вычисленными из других данных [4]. Так как нали-
чие сольватации ионов подтверждается при исследовании различных явлений,
то можно ожидать, что а, (а следовательно, и л'}7]0) в известной мере
должно зависеть от природы растворителя и в меньшей степени — от темпе-
1 Германе [2] учел взаимодействие диполей растворителя и вывел более точное
уравнение для скорости иона.
2 Величина радиуса иона водорода, вычисленная таким образом, представляет
собой весьма интересное исключение, так как она составляет для водных растворов
около &-• Поскольку независимым путем было показано, что высокая электропро-
водность растворов, содержащих эти ионы, обусловлена главным образом последователь-
ным перемещением иона водорода от одной молекулы растворителя к другой, то в этом •
случае закон Стокса неприменим. В связи с этим особый интерес представляют опыты
Бэкера и Ла-Мера [За]; см. также [36].
184
ГЛАВА VII
ратуры. Этот вопрос был подробно изучен Вальденом1, который показал,
что произведения Х’-7]0 и практически не зависят от температуры [6] 2
и изменяются меньше чем в 2 раза при переходе от одного растворителя
к другому. Эти приближенные соотношения
Т°~ I const- (4)
А /]0 — J
были установлены экспериментально [7а]3 и известны под названием пра-
вила Вальдена. Очень большие сферические ионы почти не сольватируются
из-за. малой плотности зарядов на их поверхности, и величины радиусов
этих ионо? не зависят от природы растворителя. Такие ионы лучше всего
Таблица 32 [1]
Влияние температуры на Х°7]0 и А%о
Ион Растворитель
Н + Вода 3,99 (0°) 3,14 (25°) 1,81 (100°)
Na+ » 0,466 (0°) 0,459 (25°) 0,43 (100°)
ci- » ...... 0,741 (0°) 0,682 (25°) 0,59 (100°)
(C2H5)4N + ъ 0,287 (0°) 0,295 (25°) .0,293 (100°)
Пикрат- » 0,269- (0°) 0,274 (25°) 0,27 (100°)
4 sof ~ » 0,73 (0°) 0,724 (25°) —
Электролит
NaJ Этиловый спирт 0,529 (0°) 0,524 (25°) 0,527 (50°)
(CH3)4NC1 » » 0,524 (0°) 0,549 (25°) 0,555 (56°)
(С2Н.Э4М-пикрат » » 0,567 (0°) 0,559 (25°) 0,564 (56°)
(С2Н6)4М-пикрат Ацетон 0,568 (0°) 0,568 (25°) 0,560 (50°)
NaJ » 0,557 (0°) 0,578 (25°) 0,570 (40°)
NaJ Изобутиловый
спирт 0,489 (0°) 0,488 (25°) 0,471 (50°)
NaJ Пиридин .... 0,638 (0°) 0,637 (20°) 0,640 (50°)
KJ Нитрометан . . . 1 0,765 (0°) 0,77 (25°) 0,75 (55°)
соответствуют той модели, для которой справедлив закон Стокса, т. е.
сферическим частицам, движущимся в гомогенной среде. Следует ожидать,
что отклонения от правила Вальдена будут больше всего в случае ионов
малых радиусов, несимметричной формы или с несимметричным распреде-
лением заряда, причем эти отклонения будут особенно велики для раство-
рителей, которые сильно различаются по значениям поляризуемостей
и молекулярных объемов. На основании данных, приведенных в табл. 32
и 33, можно составить представление о том, насколько значительным яв-
ляется влияние этих факторов.
1 Обширный материал, относящийся к этому вопросу, содержится в книге Валь
дена [5а]. В дальнейшем этот вопрос обсуждался в работах, указанных под номером [56].
Ср. [5в].
2 Наиболее значительные изменения этой величины с температурой наблюдаются
для растворов, содержащих ионы водорода, а также в случае сильно ассоциированных
пли очень вязких растворителей.
3 Ср. [76].
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
185
Таблица 33 [I]1)
Произведение для некоторых ионов в различных растворителях при 25°
Ион Растворитель К+ Na+ J- CNS“ (C2H5)4N+ Пикрат-
Вода . 0,668 0,459 0,686 0,598 0,295 0,269
Метиловый спирт .... 0,292 0,251 0,332 0,327 0,294 0,267
Этиловый » .... 0,267 0,238 0,285 0,28 0,295 0,263
Ацетон . . • 0,222 0,217 0,366 0,40 0,294 0,266
ФУРФУРОЛ 0,215 0,190 0,424 — 0,293 —
Ацетонитрил ........ . 0,31 0,28 0,38 0,44 0,294 —
Пиридин 0,27 0,22 0,42 0,45 0,294 —
1) См. также [2].
§ 2. Образование ионных пар. Вычисление констант диссоциации
Закон разведения' Оствальда
неприменим к водным растворам сильных электролитов, однако разбавлен-
ные растворы слабых электролитов в воде и растворы сильных электроли-
тов в средах с низкой диэлектрической постоянной хорошо подчиняются
этому закону. В растворителях с низкой диэлектрической постоянной все-
сильные электролиты очень сильно ассоциированы даже при самых малых
концентрациях, доступных экспериментальному изучению. Поэтому следует
определять А0 и К одновременно из одних и тех же данных. Это может
быть сделано путем последовательного подбора значений А0, который про-
должается до тех пор, пока К, вычисленное по уравнению (5), не перестанет-
зависеть от концентрации разбавленного раствора; более удовлетворитель-
ными являются графические методы. Краус и Брэй [8] представили урав-
нение (5) в следующей форме:
1 1 . сА
А — Л° К (А0)2 ’
где 1/А° равно отрезку, отсекаемому на оси ординат кривой, выражающей
зависимость 1/А от сА, а \/К (А0)2 — коэффициент наклона этой кривой.
Эти авторы построили кривые такого рода для большого числа растворов
солей (как органических, так и неорганических) в жидком аммиаке,
жидкой двуокиси серы и во многих органических растворителях. Они рас-
смотрели данные, приведенные в литературе, и пришли к выводу, что-
все растворы электролитов, в которых концентрация ионов меньше 10-3-
или 10~4 н., подчиняются закону действующих масс [уравнение (5)]. Они
указали также на то, что отклонения от этого закона, которые возрастают
по мере увеличения концентрации растворов, зависят не столько от общей
концентрации растворенного вещества1, сколько от концентрации ионов.
1 Основываясь на недостаточно точных измерениях чисел переноса, Краус и Брэй
отвергли предположение Саханова [9] о том, что присутствие комплексных ионов
и молекул, образующихся в ходе реакций
(СА)2^2(СА), (I)-
(CA)2j±C++CA7, (11>
СА^±С+ + А~, (Ш>
могло бы объяснить отклонение от закона действующих масс, справедливого для про-
186
ГЛАВА VII
С помощью точных измерений, а также теоретического анализа было
показано, что уравнение (5) не является точным даже при такой низкой
концентрации, как 10~4 н., поскольку оно .не учитывает влияния взаимного
притяжения ионов, находящихся на сравнительно далеких расстояниях друг
от друга, на электропроводность и активности ионов. Для учета этого
влияния Фуосс и Краус [10] предложили графический метод, состоящий
из ряда последовательных приближений. Применение этого метода облег-
чается с помощью таблиц, составленных в дальнейшем Фуоссом [И], по-
этому мы не описываем этого метода в его первоначальном виде.
В случае полностью диссоциированного электролита электропроводность
очень разбавленных растворов равняется А0—S (Д)]/с [уравнение (20)
гл. V]. Если доля электролита, присутствующего в виде ионов, равна а,
то средняя концентрация ионов составляет ас и эквивалентная электропро-
водность будет равна
Д = а (А°—5(Д) |/ас), (7)
а степень диссоциации выражается соотношением
а = —-----. (8)
А (1 —£(д) J^ac/A0)
В пределе это уравнение сводится к выражению а = А/А°, на котором
основан вывод уравнения (5). G помощью последовательных подстановок
значений а из уравнения (8) в выражение поправочного члена этого же
уравнения 1 — §(Д> |/ ас/А° последний может быть заменен непрерывной
дробью
F(Z)^1-Z{1-Z[1-Z (и. т. (9)
в которой
Z=5(i)]/S(A'j-:G (10)
Фуосс [И]* 1 составил таблицу численных значений F (Z) для 0<Z<0,209,
в которой промежутки между последовательными значениями Z равняются
0,001, так что при известном Д° можно вычислять а по уравнению
Для вычисления А0 обычно необходимо небольшое число последовательных
приближений, причем за исходное значение Д° принимается то значение А0,
которое получается путем очень приближенной экстраполяции данных
по электропроводности или на основании правила Вальдена. С помощью этого
исходного значения А0 получают приближенные значения F (Z) и а. Полу-
ченные значения а используют для определения коэффициента активности
по предельному закону2
lg?/± = — (12)
или по уравнению (10) гл. V. Согласно выражению
А А0 К (А0)2 '
стой диссоциации, выражаемой уравнением (II). Однако позже Краус, и Фуосс приме-
нили рассмотрение этих равновесий для интерпретации, кривых электропроводности-
•солей в растворителях с низкой диэлектрической постоянной.
1 F(Z) = |- cos2 -i-arccos (~3 3Z). Ср. [12].
2 Ср. уравнения (4) гл. V и (58) гл. I.
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
187
которое получается при сочетании термодинамического уравнения для кон-
станты диссоциации
" ПУГ
(14)
Р и с. 26. Определение К и А0 по уравне-
нию (13). Гваяколат натрия в растворах
гваякола при 25°.
и уравнения (11), i/K (А0)2 должно равняться коэффициенту наклона кри-
вой1, выражающей зависимость F (Z)/A. от c&y±/F(Z), а 1/А° — отрезку,
который эта кривая отсекает на оси ординат при экстраполяции. С помощью
подобной экстраполяции, основанной на приближенных значениях F (Z)
и у±, находят А0, которое используют для определения более точных зна-
чений F (Z) и ‘у±, по которым строится новая кривая. Описанный прием
повторяют? до тех пор, пока не
находят значение А0, удовлетво-
ряющее всем уравнениям — (11),
(12) и (13),—а затем по графику,
построенному с использованием это-
го значения А0, определяют К.
Рис. 26 иллюстрирует приме-
нение уравнения (13). Для построе-
ния этого графика использованы
значения а, вычисленные Шидлов-
ским и У лихом [13] из их данных
по электропроводности гваяколата
натрия, растворенного во влажном
гваяколе при 25°. Эксперименталь-
ные данные изображаются прямой
линией, которая отсекает на оси
ординат отрезок, равный 0,143, и
имеет коэффициент наклона 416.
Эти значения соответствуют А0 =
= 7,5 и К = 4,27 • 10’5. Линей-
ный характер кривой убедительно
подтверждает правильность описанного метода. Независимо от этого Шид-
ловский. и Улих подтвердили справедливость уравнения (8), показав,
что зависимость а от концентрации согласуется с их опытами по опре-
делению распределения [14].
В ряде случаев описанный метод является вполне удовлетворительным,
однако возможность его использования ограничена тем, что приходится
применять уравнение предельной электропроводности (20) гл. V, а также
практической необходимостью располагать таблицами Фуосса для F(Z).
Если для выражения электропроводности гипотетического полностью дис-
социированного электролита пользуются уравнением (59) гл. VI, то при
этом интервал концентраций значительно превосходит область, в которой
применимо предельное уравнение, и Шидловский [15] показал, что в этом
случае F (Z) можно заменить более простой функцией. Таким образом,
если принять, что наблюдаемая электропроводность равна степени диссо-
циации, умноженной на электропроводность, определяемую уравнением (15)
гл. VI, то получается квадратное уравнение
1 Фуосс пришел к выводу, что при концентрациях меньших, чем 3 • 10-7 D, этот
график должен носить линейный характер.
188
ГЛАВА VII
вместо уравнения третьей степени (8). Если решить уравнение (15) отно-
сительно а, используя переменную 1, определяемую уравнением (10), то
получается
<1в>
Для малых значений Z выражение в скобках можно представить в виде
ряда
^(Z) = i + z+4 + 4+•••
Обычно при вычислении 5 (Z) нет необходимости пользоваться членами
более высокого порядка, чем Z2.
Из уравнений (16) и (14) получается выражение
1 [eA^±S(Z)l -
AS (Z) + К(Д»)2 ’ V '
соответствующее уравнению (13). Для того чтобы сравнить уравнения (13)
и (18) с точки зрения их применения для определения констант диссо-
циации, представим эти уравнения в следующем виде:
A/^(Z) = A0-(ca2y2)A0/^, (19)
AS (Z) = Л° - (Са2у2 ) А°/К. (20)
На рис. 27 изображены кривые, построенные по результатам измере-
ний электропроводности водного раствора сернокислого цинка [16] при 25°.
Рис. 27. Определение Л° и К для водного раствора сер-
нокислого цинка при 25°.
1—вычислено по уравнению (19); 2—по уравнению (20). Про-
изведение ар± приравнено стехиометрическому коэффициенту
активности (табл. 93). Дальнейшее обсуждение этой экстрапо-
ляции см. в приложении Б, § 2.
Для построения нижней кривой использовано уравнение (20), и поскольку
эта кривая очень близка к прямой, то при ее экстраполяции получаются
более правильные результаты, чем при использовании верхней кривой,
соответствующей уравнению (19). Отрезок, отсекаемый нижней кривой на
оси ординат, и ее предельный коэффициент наклона соответствуют значе-
ниям А0 =132,8 и А = 0,0049. Значения К, вычисленные из этих же дан-
ных с помощью метода Дэвиса [17] (гл. VI, § 2), лежат в пределах от
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
189
0,0044 до 0,0047. Таким образом, уравнение (20) является, повидимому,
наиболее пригодным для ' определения констант диссоциации из данных
одной серии измерений электропроводности [18а,б ].
§ 3. Проверка теории междуионного притяжения Бьеррума.
Влияние диэлектрической постоянной на образование ионных пар
Уравнение Бьеррума [(57) гл. III]
К"1 = — ( |Z1Z2Is2Y Q (Ь)
1000 < DkT ) ’
было выведено на основании учета влияния кулоновских сид на образо-
вание ионных пар (гл. III, § 7). К представляет собой константу равно-
весия процесса диссоциации ионной пары
[С+А-]°.Х-С+ + А",
а о — параметр, равный: о —
Численные значения функции ()(Ь) приведены в табл. 9.
Своими исследованиями электропроводности Фуосс и Краус [19[ под-
твердили правильность теории Бьеррума и справедливость уравнения (47)
гл. III. Исследования Фуосса и Крауса оказались особенно важными потому,
что на основании их результатов была показана применимость теории
междуионного притяжения к растворам в самых различных средах от воды
(£) = 78,54) до диоксана (£>=2,1). Для этой цели Фуосс и Краус исполь-
зовали свой обширный опытный материал по электропроводности азотно-
кислого тетраизоамиламмония в смесях диоксан — вода. В растворе, содер-
жащем 53% Н2О (£> = 38 при 25°), ассоциация ионов заметна, но она выра-
жена не очень отчетливо. Поэтому можно было экстраполировать получен-
ные данные с помощью приближенного уравнения Онзагера (8) гл. VI для
определения Д° и приближенной оценки К. Более точное значение К было
затем получено на основании учета коэффициентов активности в нескольких
разбавленных растворах. В случае смесей растворителей с диэлектрическими
постоянными, равными 11,9 и 8,5 (20,2% и 14,95% Н2О), А0 и К вычи-
слялись с помощью графического метода, описанного в предыдущем пара-
графе. Данные Фуосса и Крауса относятся к таким растворам, степень раз-
бавления которых недостаточна для того, чтобы можно было использовать
этот метод в случае сред с низкими диэлектрическими постоянными. Поэтому
значения А° для растворов в смесях с диэлектрической постоянной, равной
5,84 и меньше, Фуосс и Краус находили путем внесения поправок на между-
ионное притяжение к приближенным значениям Аи, полученным с помощью
правила Вальдена (4). Вычисленные таким образом значения К следует
экстраполировать к нулевой концентрации, так как они возрастают с уве-
личением концентрации из-за образования ионных тройников.
Эти значения К можно использовать для вычисления параметра а
по уравнениям (57) и (51) гл. III и с помощью кривой, выражающей зави-
симость lg(6) от lg b (табл. 9). При подстановке численных значений
констант в эти уравнения получаем (для 25°)
-lg/C = 6,120-31g£> + lg(>(6) (21)
и
- lg b = 5,254 4- lg D + 1g а. (22)
Результаты вычислений Фуосса и Крауса приведены в табл. 34. Несмотря
too
ГЛАВА VII
Таблица 34
Значения констант для раствора азотнокислого
тетраизоамиламмония в смесях диоксан—вода при 25°
Вода, % вес. о1). -1g К К а 108
0,60 2,38 15,7 2 • 10-и 6,01
1,24 2,56 14,0 1 • Ю-k 6,23
2,35 • 2,90 12,0 1 Ю-!2 6,36
4,01 3,48 9,6 2,5 • 10-ю 6,57
6,37 4,42 7,53 3,0 Ю-s 6,65
9,50 5,82 5,78 1,65 • Ю-e 6,45
14,95 8,50 4,00 1,00 • Ю“4 6,50
20,2 11,9 3,08 9,0 Ю-4 6,70
53,0 38,0 0,60 2,5 ю-1 6,15
1) эти значения D, использованные Фуоссом и Краусом, являются
несколько более высокими, чем значения, приведенные в табл. 4.
на трудности определения К в случае растворителей е наиболее низкой
диэлектрической постоянной, изменение К в зависимости от D в этих средах
выражено настолько отчетливо,
Рис. 28. Зависимость диссоциации
ионных пар от диэлектрической по-
стоянной.
Азотнокислый тетраизоамиламмонин в
емесях диоксан—вода при 25°.
что можно надежно проверить теорию.
Как видно, вычисленные значения а посто-
янны для всех применявшихся смесей. Ма-
ксимальная разность между любыми двумя
значениями составляет 11%,'и среднее зна-
чение а (равное 6,4 • 10-8), невидимому,
хорошо согласуется со всеми опытными
данными с точностью до общей ошибки
вычислений и опытов. Фуосс и Краус ука-
зали, что во всех смесях растворителей,
включенных в таблицу, молярная концен-
трация воды всегда значительно больше,
чем молярная концентрация растворенно-
го вещества. Поскольку дипольный мо-
мент воды (1,85) велик ио сравнению с ди-
польным моментом диоксана (< 0,4), то
можно ожидать, что относительная гидра-
тация ионов растворенного вещества, а сле-
довательно, и их эффективный диаметр а
будут практически постоянными.
Поэтому при вычислении К для раз-
личных значений D с помощью уравнений
(21) и (22) применялось только одно зна-
чение а (равное 6,4-10~8). Вычисленные
таким образом значения К представлены
графически на рис. 28 в виде сплошной кривой. Как видно из рисунка,
экспериментальные точки, взятые из табл. 34, прекрасно ложатся на
эту кривую в пределах значений К, изменяющихся на пятнадцать поряд-
ков. Кривая имеет вертикальную асимптоту, соответствующую значению
£> = 43,6. Это означает, что в растворе азотнокислого тетраизоамиламмония
в смесях диоксан—вода не могут существовать устойчивые ионные пары,
если диэлектрическая постоянная этих смесей равна этому критическому
значению или превышает его.
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
191
§ 4. Ассоциация с образованием сложных агрегатов.
Ионные тройники, квадруполи и минимумы электропроводности1
Как видно из рис. 29, на кривых электропроводности разбавленных
растворов наблюдается минимум, если диэлектрическия постоянная раство-
рителя близка к десяти или меньше
диэлектрической постоянной минимум
становится более отчетливым и сдви-
гается в сторону меньших концентра-
ций. Согласно предположению Фуосса
и Крауса [20], увеличение числа ионов,
отнесенное к одному молю, нри возраста-
нии концентрации растворенного веще-
ства компенсируется тем, что ионы соеди-
няются с ионными парами и образуются
ионные тройники по следующей схеме:
[А+В-А+]+^ А+ + [А+В-]°, (23)
[В-А+В~]~ -t В- + [А+В-]°. (24)
Фуосс и Краус указали, что ионные
тройники должны быть устойчивыми,
поскольку избыток потенциальной энер-
гии, связанный с присоединением треть-
его иона, в несколько раз больше,
чем средняя тепловая энергия ионов
в растворителе с низкой диэлектриче-
ской постоянной.
Если простые ионы обладают сфе-
рической формой и одинаковыми раз-
мерами, то ионные тройники должны
возникать в равной степени по обоим
уравнениям и, следовательно 2,
(АВ) (В~) (АВ) (А*) _ „
(ВАВ*) (АВА*) — Лз- (^)
этой величины. При уменьшении
Рис. 29. Влияние диэлектрической
постоянной на зависимость электропро-
водности от концентрации.
Азотнокислый тетраизоамиламмоний в сме-
сях диоксан—вода при 25°. Соответствую-
шие значения диэлектрических постоянных
приведены с правой стороны графика.
Экспериментальные кривые построены по
данным Фуосса и Крауса.
Рассмотрим простое равновесие
[А*В-]о^А+ + В-, 2/2 1^2= #
(26>
и введем обозначения а = (А+)/с и а3 = (АВА+)/с; рассмотрим все равновесия
одновременно, причем концентрации простых, двойных и тройных ионов
будут равны соответственно са, с (1 — а — За3) и са3. Подставляя эти вели-
чины в приведенные выше уравнения и решая эти уравнения относительно-
1 Первые исследования в данной области были проведены И. Каблуковым,
установившим в своей диссертации [«Современные теории растворов в связи с учением-
о химическом равновесии», Москва, 1891] наличие экстремумов молекулярной электро-
проводности при разбавлении. Исследованиями отдельных вопросов электрохимии
неводных растворов в дальнейшем много занимались также Н. Зелинский, С. Крапи-
вин, В. Плотников, Л. Писаржевский, А. Думанский, А. Дорошевский и др. (Прим, ред.)'
2 Допущение об одинаковых размерах ионов делается для того, чтобы избежать-
необходимость учета двух различных констант ионного тройника, которые не могут быть,
вычислены независимо друг от друга. Каким бы приближением мы ни воспользовались,
указанное допущение проявляется в среднем размере иона, вычисленном из Ks. См. [21К
•192
ГЛАВА VII
а и а3, получаем
а3 = АТ (1-а-За3) = (]/Г7Ж3)(1-а-За3р у!1 - (25а)
лз
а=/^7’с(1-а-За3)1/2 у-1. (26а)
Если обозначить сумму предельных электропроводностей ионных тройников
обоих видов через Д3, то функция
. Д = (яА° + Я3Л3) (1 — S(A) с (я а3) /Л ) (27)
будет приближенно выражать электропроводность разбавленных растворов.
Согласно уравнению (12),
= ехр (2,303S(/) }/с(я + а3)). (28)
Для того чтобы объединить эти четыре уравнения, необходимо ввести до-
полнительное допущение. Фуосс и Краус предположили, что
с (а + а3) сД/Д° (29)
и
(1 — а — За3) (1 — Д/А°), (30)
и получили следующее окончательное уравнение, преобразованное таким
образом, что его можно использовать для экстраполяции:
д У с . g (с) = у к Л° + (/£л£//с3) (I - Л/Л°) с, (31)
где
_ exp (—2,303 §>(/) V сА/Л°)
(1—5(Д) /cA/A°/A°) (1 —А/А0)1/2
Так как с и Д/Д° обычно значительно меньше единицы, то можно поль-
зоваться приближенной формой уравнения (32) (см. [22,5]):
• 1 ! \ ~ Г °>4343^(Л) — S(/)A°1 1/-Г , 0,2171А - ,09 ч
lg 8 (с) L----(A0//2 ' J ^сЛ + • <32а)
Смысл члена g (с) состоит в том, что он отражает влияние ионных атмосфер
в выражении закона действующих масс и их влияние на электропроводность.
Если минимум электропроводности наблюдается при концентрации большей,
чем 1Сг3 н., то принятые при выводе уравнения (31) допущения становятся
слишком грубыми для количественной обработки данных, и эти допущения
можно использовать для указанной цели лишь в том случае, если измере-
ния продолжить до области очень малых концентраций.
Из уравнения (31) видно, что график, изображающий зависимость
Л}/ eg (с) от (1 — Д/Д0) с, представляет собой прямую линию, отсекающую
на оси ординат отрезок Л° У К', коэффициент наклона этой прямой равен
Д3 |/ KfK^. Такая прямая изображена на рис. 30. Для вычисления от-
дельных значений К и К3 должны быть известны Д° и Д3. В. растворите-
лях с очень низкой диэлектрической постоянной нельзя опредэлить Д°-
прямым путем, однако можно получить приближенное значение Д° с по-
мощью правила Вальдена (4). Значение произведения Л°т]0 можно опреде-
лить для растворителей, диэлектрическая постоянная которых достаточно
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
193
велика для того, чтобы можно было пользоваться экстраполяцией. Значе-
А 0
ние А3 нельзя определить непосредственно ни для какой среды, однако
отношение А3/А° можно найти, зная температурные коэффициенты электро-
проводности [23, 7, 24], если допустить, что А3/А° не зависит от Т. Так
как зависимость А3 от температуры и диэлектрической постоянной пред-
ставляет больший теоретический интерес, чем абсолютная величина А3,
то в большинстве случаев достаточно принять отношение А3/А° во всех
средах и при всех температурах [25]1 приближенно равным некоторой
достоянной величине, например 1/3 или 1/2.
Более просто, но несколько менее точно можно выразить Аа черев
А°/А3 с помощью численных величин
соответствующих минимуму элект-
ропроводности. Если пренебречь чле-
ном g (с) и множителем 1 — А/А0 в
уравнении (31), продифференцировать
это уравнение и использовать усло-
вие минимума, то получается соотно-
шение
А3 = смин А®/А°. (33)
При подстановке этого значения А3
в уравнение (31) получается выраже-
ние
"I /~ А^МИН. / Q \
V К = 2АГ~ • (34)
Так как графическое определе-
ние смин. и АМин. недостаточно точно,
а поправочным членом g (с) мы пре-
небрегли при выводе уравнения (33),
то более точно К3 определяют с по-
электропроводности и концентрации,
Рис. 30. Кривая, отвечающая уравнении
(31), причем g (с) определено из урав-
нения (32а).
Азотнокислый тетраизоамиламмоний в раство-
рах диоксана, содержащих 1,24% воды (1=25°.
Ц = 2,56).
МОЩЬЮ графиков, подобных тому, ко-
торый изображен на рис. 30; К3 находят из значений коэффициентов накло-
на кривых *и отрезков, отсекаемых ими на оси ординат. Эксперименталь-
ные значения — 1g А3, приведенные в табл. 35, были получены этим спо-
собом. Отметим, что К3 и концентрация, соответствующая минимуму, быстро
возрастают по мере увеличения диэлектрической постоянной. '
Теперь можно применить теорию образования ионных тройников,
изложенную в гл. III, § 7-. Согласно этой теории, К3 можно вычислить по
уравнению
А^1
__2пАа|
~ 1000
ЦЬЭ),
(35)
где
, I z+z_ I s2
= asDkT '
В табл. 10 приведены значения I (Ь3) для различных Ь3. Рассмотрение гра-
ничных условий показывает, что ионные тройники неустойчивы (К3 = со)
по отношению к тепловому движению при Ь3 < 8/3. Поэтому, согласно
уравнению (36), в растворителях, для которых
т-. . 31 Z+Z I S2
1 Коэффициент dk^jldT для большинства крупных ионов жмее® прантичеекж «дижа-
ковое значение. . • ,
13 Г. Харнед, Б. Оуэж
194
ГЛАВА VII
ионные тройники не образуются. О наличии такого критического значе-
ния D свидетельствует рис. 29, из которого видно, что минимум электро-
проводности исчезает при увеличении диэлектрической постоянной.
Для того чтобы уравнение (35) удовлетворяло данным, полученным
для растворов азотнокислого тетраизоамиламмония в смесях диоксан—вода,
Фуосс и Краус [20] выбрали для а3 значение 9 • 10~8 см. Цифры, помещен-
ные в последнем столбце табл. 35, представляют собой значения —1g К3,
вычисленные при а3 = 9- 10-8. Эти значения очень близки к эксперимен-
тальным значениям —lgKs. Как видно из табл. 34, для описания про-
цесса образования ионных пар в тех же растворах необходимо было вы-
брать значение а £^6,4 • НУ8 см. На рис. 31 кружками обозначены значе-
ния lg^3(onbiTH.), приведенные в табл. 35, а сплошная кривая построена
по уравнению (35) при а3=9 • 10~8 см. Согласно уравнению (37), при таком
значении а3 образование ионных тройников должно прекратиться при! ди-
электрической постоянной, равной 23,2.
Таблица 351)
Значения констант для растворов азотнокислого
тетраизоамиламмония в смесях диоксан—вода при 25°.
Вода, % вес. D “”18 смин. —lg Кз
опыты. вычисл.
0,60 2,38 4,10 4,68 4,34
1,24 2,56 3,60 4,12 4,03
2,35 2,90 3,15 3,50 3,56
4,01 3,48 2,60 3,00 3,02
6,37 4,42 2,30 2,5 2,50
9,50 5,84 2,05 2,0 2,06
14,95 8,50 1,60 — 1,56
*
1) Составлена по данным табл. III, приведенной в статье Фуосса
и Крауса [1].
С помощью уравнений (33) и (36) можно придать уравнению (35) сле-
дующий вид:
Г A«2rcNse \ 1 (Ь8)
смин. “<Л01000№ ) ы •
(38)
Величина отношения I (б3)/б3 в области, близкой к точке перегиба (рис. 31)* 1 *,
мало меняется при изменении Ь3. Поэтому Фуосс и Краус считают, что
с помощью соотношения (38) можно истолковать правило 7)3/смин. =
= const (^3 • 10*), эмпирически установленное Вальденом [26].
1 После подстановки численных значений констант уравнение (38) принимает для
25’ следующую форму:
смин. А Ь'3
Для больших ионов, например при а3 = 10~’’см и для Л = 3 или 4, 7 (6а)/Ь| по порядку
величины составляет 0,1—0,5. Вальденом найдено, что -£>3/смин ~3 ’ откуда следу-
1
ет, что значение A“/A° лежит в пределах между у и 1. Этот результат представляется
физически вполне вероятным.
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
195
В случае растворителей с очень низкой диэлектрической постоянной?
следует ожидать образования более сложных агрегатов, чем ионные трой-
ники. Известные успехи достигнуты в отношении количественной трак-
товки процессов образования квадруполей из ионных пар. О существова-
нии квадруполей свидетельствует наличие точек перегиба, расположенных
немного выше минимума на кривых электропроводность — концентрация для
растворов алкиламмониевых солей в бензоле [27] и диоксане [28], а также
величины кажущихся молекулярных весов растворенных веществ, полу-
ченные из криоскопических данных [29]. Кроме того, наблюдаемые значе-*
ния электропроводности настолько малы, что в качестве первого прибли-
жения можно пренебречь концентрацией простых ионов, ионных тройников
и других заряженных групп по сравнению с концентрацией нейтральных
ионных пар и квадруполей. В ограниченной области концентраций, для
которой справедливо такое допущение, можно
учитывать только один равновесный процесс
[А+ВД° [А+В~]° 2 [А+В']°, (39)
которому соответствует константа
к (^в)2
4 ([АВ] [АВ]) •
Сделав допущение, что квадруполь представляет
собой эллипсоид (с осями а и ).а), содержащий
в центре точечный диполь с моментом р, парал-
лельным главной оси, Фуосс и Краус [30] полу-
чили выражение
дг-1 _ N / Л \3/2 |12
4 — 2000<3/ DkTX
х4^[1/(2Х*)-1]-Ч (41)
уже приведенное выше [уравнение (71) гл. III].
Величина у определяется следующим выраже-
нием:
Рис. 31, Влияние диэлек-
трической постоянной на дис-
социацию ионных тройников.
Азотнокислый тетраизоамиламмо-
ний в смесях диоксан—вода при
25°.
= 112
У (Xa)3 DkT •
(42)
Данные по измерениям точек замерзания [29а] растворов пикрата триизо-
амиламмония в бензоле можно удовлетворительно выразить с помощью
уравнения (40), причем получается зависимость [30]
с(1—2/)2
4-----------
(43)
7
где для недиссоциированного вещества j определяется уравнением (51)
гл. IX при v = l. С помощью графического метода было найдено, что
Kt = 0,105 моль/л при 5,5°. При подстановке этого значения в уравнение (41)
получается физически реальная величина Ха = 5,54 • 10-8.- В этом случае
данные, полученные для интервала концентраций от 0,002 до 0,03 н.,
повидимому, подтверждают справедливость допущений, принятых при
выводе уравнения (43).
Пользуясь той же эллиптической моделью, которая применялась при
выводе уравнения (41), Фуосс [31] показал, что уравнение для предельного
коэффициента наклона кривой, выражающей зависимость молекулярной
13*
196
ГЛАВА VII
Поляризации Р2 растворенного вещества от концентрации, должно иметь
вид
_ 4к/к\»/2/2Уиу / 1___\~’/2 {44.
{de Д<3> \’UtJ 1000 у’/з <2X2 ' 1
Сочетая это уравнение с уравнением (41), можно найти зависимость
ОТ Рг, однако на численные значения К± слишком сильно влияют ошибки
при определении р и dP2jdc, и поэтому эти значения не представляют
иначительного интереса. Результаты, полученные при некоторых исследова-
ниях диэлектрической постоянной бензольных растворов, приведены в табл. 36.
Предельные значения молекулярной поляризации растворенных веществ Р*
и соответствующие дипольные моменты (в дебаях) содержатся во втором и
третьем столбцах. Предельные значения коэффициентов наклона dP2/de были
получены графическим путем. Для вычисления длины малой оси ка было
допущено, что л = 1/2. Значения а, полученные из, данных по изучению
влияния диэлектрической постоянной и из уравнения (44), приведены в пред-
последнем столбце. Они имеют правильный порядок величины и хорошо
согласуются с двумя значениями, полученными с помощью уравнений (43)
Таблица 36 [1]
Характеристические величины для эллипсоидальной модели диполя
Электролит Pg, СЛ13 (^) -• 10-6 ' de zc-*0 а 108
(D) (т. замерз.)
Тетра-к-бутиламмониевая соль окситрифенилбор- ной кислоты 8270 19,7 -14,30 6,37
Пикрат тетраизоамилам- мония 7090 18,3 -2,86 6,28 —
Пикрат тетра-н-бутиламмо- ния 6740 17,8 -1,52 6,28 —
Пикрат триизоамиламмо- ния 3830 13,3 -1,12 5,05 5,54 [81
Пикрат три-н-бутиламмо- ния 3670 13,1 -0,71 5,02 5,59 [3]
Ацетат тетра-н-бутиламмо- ния . t 2690 . 11,2 -0,27 4,59 —
и (41) из данных по температурам замерзания. К сожалению, представление
0 равновесии ионных пар и квадруполей до настоящего времени не применя-
лось для количественной интерпретации данных по электропроводности.
Фуосс [23] вывел приближенное уравнение
<1пЛ_ rflng, 1 /. rf In Д > /, . _j4\_
d Jn Т d In T "* 2 <1 "* d In T ) < b J
— (1+к3д"/л3ос) (1+disF)(63“4“ зь;)
для зависимости электропроводности сильных электролитов от температурвд
в средах с низкой диэлектрической постоянной. Это уравнение выведено
с помощью уравнения (31) и поэтому справедливо только при а < 1 и при
таких значениях с, которые меньше концентрации, соответствующей мини-
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
197
мальному значению А. Уравнение (45) может применяться только к систе-
мам, для которых 63 > 10, потому что При выводе использовано асимпто-
тическое разложение уравнения (35). Кроме того, при выводе этого урав-
нения приняты следующие три допущения:
d (А°>|0) _ d (AgT|0) „ / z g\
dT dT ’ ' '
do- _da3_(/Т7\
dT dT ' ’
• d (Ap/Ag) q (4g)
Для того чтобы применить уравнение (45) к данным по электропровод-
ности, необходимо построить ряд кривых и выполнить несколько последо-
вательных приближений, а также воспользоваться правилом Вальдена (4)
для определения А0. Подробности этих вычислений приводятся в работе
Бьена, Крауса и Фубсса [24], которые проверили уравнение (45) с помощью
данных, полученных при исследовании электропроводности растворов нит-
рата и пикрата тетрабутиламмония в анизоле (.0=4,29 при 25°) при
температурах между — 33 и 95°. Они представили полученные ими данные
в графической форме, подобно тому как это сделано на рис. 30, и нашли
значения А»/К/К3 и А0 / К из величин коэффициентов наклона полу-
ченной кривой и отрезков, отсекаемых ею на оси ординат. Для случая
нитрата тетрабутиламмония эти величины приведены в табл. 37. Для
вычисления А0 было использовано соотношение А°т)0 = 0,508, полученное
путем измерений в хлористом этилене [32] при 25°. Значения К были
вычислены из данных, приведенных во втором и четвертом столбцах
таблицы, и соответствующих значений а, вычисленных с помощью уравне-
ний (57) и (51) гл. III. Постоянство значений а свидетельствует о пра-
вильном определении величин А0.
Для вычисления К3 сначала определялось из опытных данных отно-
шение А°/А°, которое считалось не зависящим от температуры. Затем из
всех экспериментальных величин А3А°/А§ на основании среднего значения
А3/А° (равного 0,82) вычислялись значения К3. Эти значения К3, а также
вычисленные из них значения а3 приведены в последних двух столбцах
табл. 37. Как видно из таблицы, постоянство значений а3 соблюдается
удовлетворительно.
Таблица 370
Константы для раствора азотнокислого тетрабутиламмония в анизоле
i, °C л° К к ю* л/А 3 к3 А» К 1011 а 108 к3 . 104 а3 108
-33 0,650 0,115 14,22 2,08 4,88 4,61 5,79
0 2,34 0,324 31,8 5,42 4,95 5,89 5,79
25 4,75 0,550 49,5 9,20 4,96 7,05 5,84
61,3 10,50 0,950 82,1 16,30 4,91 9,04 5,82
80,2 14,70 1,145 102,1 20,65 4,89 10,47 5,88
95,1 18,10 1,288 118,9 23,2 4,85 11,48 5,88
1) Составлена по данным табл.
III, приведенной в статье Бьена, Крауса и Фуосса [1].
Бьен, Краус и Фуосс аналитически определили члены d Inr^/dT и
d In D/d In T с помощью эмпирических уравнений, удовлетворяющих полу-
198
ГЛАВА VII
ленным ими данным по текучести и молекулярной поляризации анизола.
Пользуясь средними значениями, а и а3, они с помощью уравнения (45) вычи-
слили d \nA/dT и сравнили результаты этих вычислений с эксперименталь-
ными значениями, которые определялись непосредственно как коэффициенты
Р if с. 32. Влияние температуры на
электропроводность раствора азотнокис-
лого тетр абу тиламмония в анизоле.
наклона хорд на графиках зависимости
1пЛотТ. Экспериментальные значения
сопоставлены с теоретической кривой 1
на рис. 32. Теоретическая кривая лишь
приближенно соответствует экспери-
ментальным значениям, но ничего
лучшего нельзя было ожидать, если
принять во внимание сложность вы-
числений и сделанные приближения.
Наклон теоретической кривой опре-
деляется в основном членом d In f^/dT,
и поэтому можно полагать, что наблю-
даемые расхождения между теорети-
ческими и экспериментальными зна-
чениями связаны с отклонениями от
условия, выраженного уравнением (46).
Путем сочетания уравнения (45)
с уравнениями (33) и (34) можно срав-
нить теоретические значения 1g смин.
и 1g Амин. с соответствующими зна-
чениями, найденными из логариф-
мических кривых, построенных до
экспериментальным данным для раз-
личных температур. Кривые 2 и 3 на
рис. 32 построены с помощью указанных уравнений, а опытные значения
1g сМин. и 1gАмин, обозначены соответственно точками и кружками.
§ 5. Влияние размеров и строения ионов
на электропроводность и ассоциацию
Так как степень ассоциации ионов зависит от потенциальной энергии
ионной пары (или более сложной группы), а потенциальная энергия в свою
очередь зависит от того расстояния, на которое могут сблизиться
заряженные ионы, то размер ионов является фактором, очень сильно влияю-
щим на величину константы диссоциации. Влияние параметра а количест-
венно учитывается в уравнении Бьеррума [(57) гл. III], а также в урав-
нении Фуосса и Крауса (35).
К сожалению, невозможно проверить эти уравнения посредством точ-
ных и независимых определений а, и эту последнюю величину нельзя точно
выразить через действительные размеры ионов и структурные параметры. При
выводе уравнений под а понимают расстояние между центрами (зарядами)
соприкасающихся между собой заряженных шаров, причем заряды этих
шаров распределены равномерно по их поверхности. Вообще говоря, ионы,
в особенности когда они соприкасаются друг с другом, не обладают сфе-
рической формой и не являются равномерно заряженными. Сложные во-
просы сольватации еще не получили удовлетворительного разрешения, так же
как и вопрос об истинной величине диэлектрической постоянной
в непосредственной близости от иона и между ионами, входящими в состав
ионной пары. Тем не менее можно сделать некоторые качественные выводы
на основании сравнения величины параметров а, вычисленных по уравне-
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
199 -
ниям (57) гл. III и (35), с результатами, полученными при исследовании
таких независимых свойств, как, например, постоянная кристаллической
решетки, подвижность ионов, дипольный момент и понижение температуры
замерзания.
Рассматривая вопрос только с качественной стороны, можно полагать,
что такая крайне упрощенная физическая картина будет наиболее близка
к реальной для тех систем, которые построены из больших ионов, обладаю-
щих сферической симметрией и находящихся в средах со слабой поляри-
зуемостью. Системы такого типа были исследованы Фуоссом и Краусом
[27а], которые определяли электропроводность растворов галоидных солей
тетраизоамиламмония в бензоле. Определяя положение минимума элек-
тропроводности графическим методом, с помощью уравнений (34) и (33)
они нашли значения К (А0)2 * * и AT8A0/Ag. Так как степень диссоциации элек-
тролитов в бензоле весьма мала, то А0 и А® нельзя определить эксперимен-
тально. Данные, полученные для галоидных солей, а также для тиоцианата
Таблица 38
Константы для растворов солей в бензоле при 25°
Соль -lg K(A°)2 -IgKi) a 10s * * -lg^° 4 -lgK32> аз • 10 8
(С6НП)4Ш 13,30 17,30 5,67 4,90 5,40 7,36
(C6Hn)4NBr 13,45 17,45 5,62 4,80 5,30 7,52
(C6H„)4NC1 13,90 17,90 5,47 4,70 . 5,20 7,67
(C6Hu)4NF 14,20 18,20 5,25 4,60 5,10 7,85
(C6Hn)4NSCN 13,25 17,25 5,68 4,65 5,15 7,76
(C6H11)4NOCeH2(NO2)3 13,05 17,05 5,75 4,35 4,85 8,34
(C6H11)3HNOCeH2(NO2)3 .... 16,60 20,60 4,74 2,90 3,40 16,3
1) А° принято равным 100.
2) Л°/Л® принято равным 3.
и двух пикратов [28], приведены в табл. 38. В неполярных растворителях,
подобных бензолу, в которых, как предполагается, отсутствует сольвата-
ция, размеры ионов галоидных солей в растворе изменяются в зависимости
от атомного номера в той же последовательности, что и ионные радиусы,
вычисленные на основании рентгенографического изучения соответствую-
щих твердых веществ (табл. 6). Из табл. 38 видно, что К (А0)2 уменьшается
при переходе от иодидов к фторидам, и это может служить доказательством
того, что К уменьшается в том же направлении, поскольку А° должно
несколько увеличиваться при переходе от большого иона иода к малому
(а следовательно, и более подвижному) иону фтора. Так как при вычис-
лении приведенных в таблице значений 1g АГ сделано предположение, что
Л°=100 для всех солей, наблюдаемые изменения К при переходе от одного
электролита к другому несомненно слишком малы.
Отношение А°/А|, вероятно, мало изменяется для любого ряда подобных
солей. Поэтому величина К3 должна изменяться так же, как отношение
АГ3А°/А®, а это последнее, как видно из табл. 38, увеличивается с уменьше-
нием атомного номера иона галоида. Этого слодовало ожидать, поскольку
связи между ионами больших размеров, входящими в состав ионных тройни-
ков, должны быть менее прочными вследствие меньшей потенциальной
энергии и, следовательно, величина К3 для таких ионных тройников дол-
жна быть меньше. Следует отметить, что значения а3 отличаются от значе-
ний а как по величине, так и по степени изменения при варьировании
'20®
ГЛАВА VII
состава электролита. Подобные различия иллюстрируются также табл. 37,
в которой приведены данные, относящиеся к одной системе при различных
температурах. Так как физическая картина образования ионных тройни-
ков, на которой основан вывод уравнения (35), является упрощенной, то
вряд ли следует придавать сколько-нибудь серьезное значение «наблюдае-
мым» величинам а3 и лучше ограничиться утверждением,'что размеры по-
рядка 5—10А являются физически приемлемыми для данных ионов. Конечно,
теория в ее современной форме недостаточна для объяснения, например,
такого факта [24], как увеличение а3 для нормального пикрата тетрабутил-
аммония на 1А между —33 и 95°.
Значения а практически не зависят от температуры, однако они, несо-
мненно, зависят от природы растворителя и растворенного вещества. Как
видно из табл. 39, а меняется при переходе от одного растворителя к дру-
гому. Это изменение является одним из примеров того, что взаимодействие
между ионом и растворителем [33] следует принимать во внимание в любой
теории, относящейся к полярным растворителям. Специфическое влияние
растворителя обычно выражено более отчетливо в тех случаях, когда раство-
ренным веществом является неполностью замещенная соль аммония. Значе-
ние а для пикрата к-трибутиламмония равно 4,66А, если в качестве рас-
творителя применяется трикрезилфосфат [34], и составляет всего 2,4 А,
когда растворителем служит хлористый этилен [35].
Таблица 39
Константы диссоциации и значения параметра а для растворов пикрата
«-тетрабутиламмония в различных растворителях
Растворитель t D л» к а 108
Хлористый этилен [1] 25 10,23 57,40 2,28 • 10~4 5,75
Трикрезилфосфат [2] 40 6,92 1,92 1,014 • IO'5 5,80
Хлорбензол [3] 25 5,63 60 1,88 10~8 4,75
Анизол [4] 25 4,29 43 1,16 • io-» 5,61
Дифениловый эфир [5] 50 3,53 24,5 2,70 • 10"11 5,33
Целый ряд работ [36] посвящен вопросу о влиянии строения ионов на
электролитическую диссоциацию та’кого типа. Некоторые указания о влия-
нии строения ионов можно получить из данных, приведенных в табл. 38,
если рассматривать замещение одного из галоидных ионов более крупным
и менее симметричным пикрат-ионом. Локализация отрицательного заряда
у атома кислорода на одном конце пикрат-иона противодействует образо-
ванию ионной пары вследствие эффекта несимметричного экранирования
тринитробензольной группы. С другой стороны, та же локализация заряда
придавала бы дополнительную устойчивость уже образованным ионным
парам, так как заряды могли бы располагаться ближе друг к другу, чем
в том случае, когда они симметрично распределены на поверхности ионов.
Не всегда можно точно предсказать, как отразятся на величине К оба эти
эффекта, действующие в противоположных направлениях, однако поскольку
эффект экранирования более сильно влияет на процесс образования ионных
тройников и поскольку он уменьшает составляющую потенциальной энер-
гии, обусловленную добавлением третьего иона к ионной паре, то следует
ожидать значительного увеличения К3 при появлении резко выраженной
несимметричности иона. Переход от четырехзамещенного иодида аммония
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
201
к пикрату сопровождается почти четырехкратным увеличением К3 и дву-
кратным увеличением К. При замене четырехзамещенного пикрата аммония
трехзамещенным оба иона становятся более несимметричными, в результате
чего А3 увеличивается в 30 раз, в то время как К возрастает в 3500 раз.
Значительное увеличение устойчивости, которое наблюдается при замеще-
нии большого изоамилового радикала водородом, можно приписать отчасти
способности заряженных атомов (О и N) подходить друг к другу на очень
близкое расстояние и отчасти взаимодействию протонов с молекулами
растворителя [37].
Абсолютная величина а представляет больший интерес, чем абсолютная
величина а3 , потому что а связано с более простым физическим процессом
и может быть найдено с помощью независимых экспериментальных методов;
Так, для раствора пикрата триизоамиламмония в бензоле из измерений
электропроводности было получено значение а < 108 = 4,74, из исследований
по влиянию диэлектрической постоянной—5,05 и из измерений температуры
замерзания—5,54 (табл. 38 и 36). Как видно из табл. 33, константа Вальдена
составляет 0,265 для пикрат-иона и около 0,295 для иона триизоамиламмония
(если считать, что электропроводность этого иона равна электропроводности
иона тетраэтиламмония). При подстановке этих величин в уравнение (2)
для радиусов отдельных ионов получаются значения 3,1 и 2,7 А, так как
вычисленное из подвижностей значение а равно 5,8 А. Поскольку физи-
ческий смысл величины а является довольно неопределенным и так как при
различных определениях этой величины приходится принимать некоторые-
допущения и учитывать ошибки опытов, то расхождения между этими неза-
висимо полученными значениями нельзя считать слишком большими. При
обсуждении влияния размеров и строения ионов на величину а мы не оста-
навливались на этой неопределенности в определении величины а, поскольку
наблюдаемые при изменении размеров и строения ионов эффекты больше,,
чем относительная точность определения а по методу электропроводности.
(49)-
’ § 6. Диссоциация слабых электролитов
При переходе от термодинамического выражения закона действующих
масс для диссоциации слабого бинарного электролита
у2, а2с
К==-~т-—
Уи(1 —
к закону разведения Оствальда [уравнение (5)] делаются допущения о том,,
что у±/уц = 1 и а=А/Л°, т. е. в этом уравнении не учитывается взаимо-
действие ионов. В случае очень разбавленных растворов оба эти допущения
приводят к небольшим ошибкам, которые по порядку величины равны
ДРУг другу, а по знаку противоположны. . Поэтому результаты ранних
измерений электропроводности слабых электролитов настолько хорошо-
подчинялись закону разведения Оствальда, что это согласие между теорией
и опытом рассматривалось, как триумф теории электролитической диссо-
циации Аррениуса [38]. Оствальд [39] заметил, что значения А, вычислен-
ные с помощью его уравнения, хотя и незначительно, но все же несомненно-
меняются при изменении с. Он указал, что допущение о постоянстве-
подвижностей ионов, на котором основано условие а=Л/Л°, следует уточ-
нить путем учета измёнения вязкости раствора с концентрацией. Иногда
можно добиться постоянства значений А в широком интервале концен-
траций, принимая а равным Лт]/А07]0, однако этот способ оказывался
пригодным лишь в отдельных частных случаях. До возникновения теории
междуионного притяжения был предложен также ряд эмпирических зави-
симостей, представляющих собой видоизменения первоначальной формы
202
ГЛАВА VII
закона разведения1, но ни одно из этих выражений не сыграло существенной
роли в дальнейшем развитии теории электролитической диссоциации.
При любом точном определении констант диссоциации слабых элек-
тролитов с помощью измерений электропроводности необходимо учитывать
зависимость коэффициентов активности и подвижностей от концентрации.
Это было сделано впервые Шерриллом и Нойесом [41], а также Мак-Инне-
-сом (42а)2. Они вычислили К для разбавленных растворов, применив пре-
дельный закон Дебая и Гюккеля для у± и определив степень диссоциации
следующим образом:
а == А/А„ (50)
где Л6 представляет собой эквивалентную электропроводность гипотетиче-
ского полностью диссоциированного электролита при концентрации ионов3
соответствующей экспериментальному значению эквивалентной электро-
проводности А, причем
Ci = ae. (50а)
“Определение As основано на допущении о том [44]4, что в изучаемой
области концентраций подвижности ионов являются аддитивными. Для
слабой кислоты HR, диссоциирующей по уравнению
HR^H+ + R",
величину ЛЕ можно определить из значений электропроводностей сильных
электролитов HCI, NaR и NaCl при концентрации, равной (\. Вычисления
производятся по уравнению
Аенц = Диш +-4-NaR~ A.NaCi. (51)
Определение концентрации ионов с, производится с помощью небольшого
числа последовательных приближений. В качестве первого приближенного
значения выбирают с[ = сА/А° и вычисляют величину А( из уравнения (51)
при этой концентрации. Вторым приближением будет значение с” = сЛ/А'е,
с помощью которого в свою очередь вычисляют А". Так как Ае не очень
сильно меняется в зависимости от cj, то для получения правильных зна-
чений С{ и Ае, лежащих в пределах ошибок опыта наилучших измерений,
редко приходится прибегать к третьему и четвертому приближениям.
Для удобства вычисления Мак-Иннес выразил электропроводности
сильных электролитов с помощью уравнения (13) гл. VI. Таким образом,
Ани = Лдс! - Shci Vd + (1 - а* ]/ct), (52)
^NaR — -4-NaR— ^NaR Ci + -^NaRci (1 — a* Cj), (53)
-4-NaCl = A^aCl — & NaCl ci + T?NaClci (1 — a*]/^i)- (54)
Параметры уравнения
AEhr = A'hr — §>hr p^Ci + BhrCi (1 — a* p^c’i) (55)
можно определить путем линейного сочетания этих уравнений согласно
1 Подробнее описание этих «законов разведения» приводится в книге Тейлора [40].
2 См. также [426].
3 Представление о гипотетической электропроводности подобного рода для Л° было
впервые использовано Кольраушем [43], который определил растворимость хлористого
серебра из соотношения с= 1000 х/Ае , вместо того чтобы вычислять ее с помощью соот-
ношения с=1000 х/Л0. Здесь х представляет собой удельную электропроводность насы-
щенного раствора, а с—растворимость.
4 См. гл. VI, § 8.
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
203
уравнению (51). Это аналитическое выражение для AShr широко приме-
няется и, невидимому, приводит к удовлетворительным результатам1.
Прежде чем закончить рассмотрение вопроса о вычислении а, следует
отметить, что хотя величина А/Де и представляет собой лучшее приближен-
ное значение для вычисления истинной концентрации ионов, чем А/А,
однако и эта величина не является вполне точной. Это объясняется тем,
что величина А измеряется для растворов, содержащих сц = с —<ц молей
недиссоциированного слабого электролита на литр, тогда как Ав вычис-
ляется для чистой воды. На пер-
вый взгляд может показаться,
что влияние такого изменения сре-
ды можно учесть, приняв а рав-
ной Дт]/Л0»]0. Поскольку отклоне-
ния от закона Стокса проявляются
наиболее отчетливо в случае рас-
твора, содержащего ионы водорода,
а вязкость си нормального рас-
твора слабого электролита, свобод-
ного от ионов, не может быть из-
мерена, то измерения электропро-
водности приводят к кажущимся
значениям а. Однако, экстраполи-
руя зти значения, можно получить
точные значения К а- Следует
отметить, что использование отно-
шения вязкостей в одних случаях
(45] оказывается весьма полезным,
/00 Vc]
Рис. 33. Графическое определение констан-
ты диссоциации уксусной кислоты в воде при
25° с помощью уравнения (57).
а в других случаях является излишним. Для применения этого отношения
требуются дополнительные экспериментальные данные, и поэтому его обычно
избегают.
Для вычисления К а из ряда значений с и а можно пользоваться
несколькими достаточно удобными способами. Если имеются эксперимен-
тальные данные для очень разбавленных растворов, то достаточно вычис-
лить значения кА для различных значений с{, согласно уравнению
и экстраполировать кривую зависимости 1g кА от ]/ а на нулевую концен-
трацию. На рис. 33 изображена подобная кривая, использованная Мак-Инне-
сом и Шидловским [46] для определения константы диссоциации уксусной
кислоты при 25°. Уравнение прямой, проведенной через эксперименталь-
ные точки, полученные для очень разбавленных растворов, имеет вид
lgкА = 1gКА +1,013]/Ci .
(57)
Из сравнения этого уравнения с уравнениями (49) и (56) видно, что
21g у± — 1g уи = — 1,013 |/7i .
(58)
1 В предыдущей главе было показано, что уравнение (13) не является строго справед-
ливым для крайне разбавленных растворов. Это обстоятельство может вызвать сомнения
в применимости этого уравнения в данном случае. Однако такое возражение не имеет
практического значения, поскольку уравнение (13) удовлетворительно описывает резуль-
таты для 1,1-валентных электролитов в интервале концентраций, применяемых для опре-
деления констант диссоциации. Поэтому описанный способ является не менее точным
и, повидимому, более удобным, чем аналитический способ.
204
ГЛАВА VII
Коэффициент активности нейтрального растворенного вещества для очень
разбавленных растворов настолько близок к единице [47], что членом lg уи,
несомненно, можно пренебречь. При этом численное значение предельного
коэффициента наклона кривой в точности совпадает со значением предель-
ного коэффициента наклона, соответствующим уравнению Дебая и Гюккеля
для водных растворов при 25°.
Отклонения экспериментальных точек от предельной прямой в области
более концентрированных • растворов нельзя учесть посредством найденных
опытным путем значений 1g уи
или с помощью введения в вы-
ражение для 1g у± величин ра-
диусов ионов Мак-Иннес и
Шидловский вычислили . 1g у±
по уравнению (34) гл. 111 и 1g уи
по данным измерений температур
замерзания, допустив, что для
диссоциированной уксусной кис-
лоты а =4. График зависимости
1g Ад от ]/Ci, построенный на ос-
новании таких вычислений, про-
ходит немного ниже пунктирной
линии на рис. 33, но сильно
отклоняется от эксперименталь-
ной кривой при более высоких
концентрациях.
Причиной некоторых рас-
мощью уравнения (60).
смотренных выше отклонений экспериментальных значений от теорети-
ческой кривой являются неприменимость закона Кольрауша и отсутствие
точного физического определения величины t’i при этих концентрациях.
Существенное значение имеет также «эффект среды», т. е. влияние недис-
социированных молекул уксусной кислоты на величину 1g у± ее ионов, которое
не учитывается в приведенных выше расчетах. [Количественные данные,
характеризующие этот эффект, отсутствуют. Вероятно, для данного случая
он не особенно отличается от влияния молекул уксусной кислоты на значение
1g у± соляной кислоты, которое составляет [48]1 примерно 0,106си. Не касаясь
вопроса о численной величине эффекта, мы можем допустить, что он про-
порционален си, т. е, написать, что
1g (Л/Уи) = - 1,013 V Ci + ₽cu. (59)
При этом предполагается, что зависимость 1g уи от си также носит линейный
характер. При сочетании этого уравнения с уравнением (49) получается
выражение [49, 50]
lgfcA-l,013/^ = lg^A-₽Cu. (60)
На рис. 34 изображена зависимость левой части этого уравнения от си
по данным Мак-Иннеса и Шидловского для концентраций, превышающих
0,0005 н. Величины радиусов кружков соответствуют точности в 0,1% от
значений Ад, причем эта точность, повидимому, совпадает с эксперименталь-
1 В условиях этих измерений диссоциация уксусной кислоты при рассматриваемых
концентрациях настолько подавлена из-за присутствия НС1, что ее стехиометрическую
концентрацию можно принять равной си. Величина 0,106сц представляет собой произве-
дение молярной доли уксусной кислоты на 5,87.
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
205
цой воспроизводимостью величины кА. Как видно, во всем интервале кон-
центраций, применявшихся в исследовании, зависимость носит линейный
характер в соответствии с уравнением (60), причем значение р равняется
примерно 0,134, т. е. по порядку, величины совпадает с теоретическим
значением 3.
Экстраполированное значение КА, полученное с помощью этой кривой,
равно 1,750 • 10~в, в то время как в случае кривой, изображенной на рис. 33,
цолучается значение 1,753- 10-в. Разность между этими значениями состав-
ляет 0,17%, т. е. она почти вдвое больше разброса экспериментальных
данных. Однако эта разность по своей величине не превышает расхождений
между значениями КА, полученными в наиболее точных работах различными
исследователями, а также при использовании различных методов. Выбор
одного из двух описанных способов экстраполяции определяется главным
образом не их точностью, а экспериментальными возможностями; исходя
из этого, безусловно следует предпочесть метод, отвечающий рис. 34. Как
видно из графиков, интервал концентраций, в котором изображенные зави-
симости носят линейный характер, расположен ниже 0,003 на рис. 33 и вы-
ше 0,005 на рис. 34; кроме того, в последнем случае экстраполяция является
гораздо более короткой.
Таблица 40
Константы диссоциации слабых электролитов в водных
растворах при 25°, определенные по методу
электропроводности
Электролит К (при концентра- ции, выражен- ной в долях моляльности) Ссылки на лите- ратуру
Муравьиная кислота 1,830 • 10-* 1
Уксусная кислота 1,758 • IO'5 2
к-Пропионовая кислота 1,347 • IO-3 3
н-Масляная кислота 1,512 • 10’5 3,1
Хлоруксусная кислота ...... 1,400 - 10“3 4,5
Циануксусная » . 3,370 - 10~3 1
Молочная кислота 1,391 • 10’4 61)
Серйая кислота (К2А) 1,18 • 10-’- 7
Фосфорная кислота (К1А,18°) 8,31 - 103 - 7
а-Кротоновая кислота 1,981 • 10~5 8
Бензойная кислота 6,313 • 10~5 9,10
о-Хлорбензойная кислота 1,200 • 10~3 9,10
.м-Хлорбензойная » 1,510 • 10-" 9,10
п-Хлорбензойная » 1,043 -10’4 9,10
Угольная кислота (А’1д) 4,323 • 10~7 11
Гидроокись моноэтаноламмония . . 3,19 • 10~5 12
Щавелевая кислота (.К%А) 5,376 • 10-« 13
1) От-0 до 50°.
В табл. 40 приведены значения констант диссоциации, полученные ив
данных по измерению электропроводности с помощью методов, описанных
в этом параграфе.
§ 7. Экспериментальное исследование влияния частоты переменного
тока на электропроводность и диэлектрическую постоянную
Зависимость электропроводности от частоты переменного тока была
обнаружена Вином [51]. Первые достаточно точные данные для высоких
частот получил Зак [52], показавший, что увеличение электропроводности
206
ГЛАВА VII
с частотой по порядку величины согласуется с предсказаниями теорий.
Зак [53], Цан [54], Дейбнер [55], Вин [56] и Малый [57] пользовались
различными экспериментальными методами. С помощью метода Вина опре-
деляется влияние высокой частоты на электропроводность или на диэлектри-
ческую постоянную.
Теория Дебая и Фалъкенгагена [58] подтверждается опытными данными
для многих электролитов. В качестве наиболее подходящего объекта для
исследования Фалькенгаген избрал водный раствор сернокислого магния.
Рис. 35. Влияние частоты переменного тока на
электропроводность растворов сернокислого магния
при 18°.
ЮОАпо)
Сплошная ливня------или 1= со; пунктирная ли-
Д’»
т
100АТ,..
1(<о)
ния-------5-----, теоретическая кривая, отвечающая
1 = 10; кружками показаны экспериментальные значения
для 1=10.
На рис. 35 изображена зависимость величины IGOA^/A^ от ]/с*, где с* —
концентрация, выраженная в эквивалентах на литр, а А„ — молярная электро-
проводность при <5 = 0. Согласно уравнению (156) гл. IV,
A~=A^-A~-An. . (61)
На рис. 35 сплошной линией изображен график зависимости отношения
1ООДк®=о)/^т от У с * для стационарного случая, полученный с помощью
уравнения Онзагера. Пунктирной линией изображена зависимость, отвечаю-
щая теории Дебая и Фалъкенгагена. Как видно, последняя зависимость
хорошо согласуется с экспериментальными данными Вина, представлен-
ными на рисунке кружками.
Рис. 36 иллюстрирует влияние частоты на электропроводность 0,001 н.
раствора сернокислого магния. Сплошной линией изображена зависимость,
эффекта дисперсии электропроводности, выраженного в процентах, от длины
волны в метрах. Эта сплошная линия соответствует вычисленным на основе;
теории значениям эффекта, а точки йредставляЮт собой экспериментальные
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
207
данные различных исследователей. Этот график не оставляет сомнений-
в справедливости теории Дебая— Фалькенгагена.
Теория уменьшения диэлектрической постоянной, развитая Дебаем
и Фалькенгагеном, интересна в том отношении, что она представляет собой.
Рис. 36. Влияние частоты переменного тока на эле-
ктропроводность растворов сернокислого магния
с*~001; сплошной линией показана теоретическая кривая;
кружками—экспериментальные значения.
Рис. 37. Влияние частоты переменного тока на
диэлектрическую постоянную растворов сернокисло-
го магния при 18°.
Сплошная линия—100 —По)/£>о> пунктирная ли-
ния — 100 (Ид—£>о)/Оо, теоретическая кривая, отвечающая
1 = 10; кружки—экспериментальные значения для ! = 10.
шаг вперед по сравнению с теорией электропроводности Онзагера и пред-
еказывает изменение диэлектрической постоянной с конпАнтряпией при
нулевой частоте. Для частного случая, когда & — 0, уравнение (161) гл. IV
можно свести к выражению
D~ — Da — + (aW/, ,fi?v
i®-0) 2Л^Г3/а(1 + 1//^)2 У > • (62>
208
ГЛАВА VII
Уравнением (96) гл. IV определяется значение д* и c* = v1|z1| c = va|z2[c.
Для простоты уравнение (62) можно записать в следующем виде:
Фалькенгаген нашел, что для водных растворов хлоридов калия, магния
и лантана и сернокислого магния при 18° S<d) равно соответственно 3,79;
10,9; 13,8 и 30,3.
Теория хорошо подтверждается также данными, полученными Вином
при определении диэлектрической постоянной разбавленных растворов серно-
кислого магния. На рис. 37 изображен график зависимости 100(Z>~ — D0),/Da
от j/c*. Сплошная линия отвечает эффекту при нулевой частоте, а пунктир-
ная линия — значениям, полученным с помощью табл. 16 для длины волны,
равной 10 м. Кружками изображены экспериментальные данные Вина.
Если принять во внимайие трудности измерения диэлектрической постоянной
разбавленных растворов солей, то согласие между экспериментальными и
теоретическими значениями следует признать удовлетворительным.
§ 8. Влияние сильного электрического поля
на электропроводность растворов электролитов
Вин [59] обнаружил, что электропроводность растворов электролитов
всегда увеличивается с увеличением напряженности электрического поля.
Он показал, что это увеличение является функцией концентрации, валент-
ности, а также природы раствора. Кроме того, эквивалентная электропро-
водность растворов сильных электролитов при очень сильных полях, по-
видимому, асимптотически приближается к постоянному предельному зна-
чению. Оказалось, что относительное увеличение электропроводности при
увеличении напряженности поля в растворах слабых электролитов [60]
во много раз больше, чем в случае сильных электролитов. Такое же
явление наблюдал Гемант [61] для растворителей с очень низкой диэлек-
трической постоянной (Л^ 3). Вин дал правильное объяснение этого эффекта,
считая, что под влиянием поля возрастает диссоциация электролита.
Так как сильные поля вызывают значительные тепловые эффекты, то
обычный метод измерения электропроводности должен быть заменен им-
пульсными методами, при которых поле накладывается на очень короткие
промежутки времени (^10~5 — 10~7 сек.). Трудности, связанные со сложностью
самих измерений, были в значительной степени устранены Вином и его
сотрудниками, которые получили большое количество точных эксперимен-
тальных данных. Обзор экспериментальных методов приведен в работе
Экштрома и Шмельцера [62].
а) Эффект Вина для случая сильных электролитов. На рис. 38 при-
ведены кривые, изображающие зависимость относительного увеличения
эквивалентной электропроводности различных сильных-электролитов от силы
поля, выраженной в кв/см [59а]. Концентрации растворов, к которым
относятся эти кривые, выбраны таким образом, чтобы значение Д(х=о) было
одинаковым для всех кривых. Относительное увеличение эквивалентной
электропроводности приблизительно пропорционально (z+z.)2 и при очень
сильных полях, повидимому, стремится к постоянному значению.
Влияние концентрации и общий характер явления иллюстрируется
рис. 39, на котором изображены теоретические кривые, полученные с по-
мощью уравнения (217) гл. IV для водного раствора гипотетического
сильного 2,2-валентного электролита. При слабых полях эффект Вина
исчезающе мал, так как при X = 0 кривые располагаются параллельно оси
абсцисс. По мере увеличения силы поля электропроводность все более
•
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
209
быстро возрастает, проходит через точку перегиба и, наконец, асимптоти-
чески приближается к предельному значению. Это предельное значение
Д(х=а) меньше, чем Д°, т. е. меньше, чем предельная электропроводность
при бесконечном разбавлении, из-за того, что разрушение ионной атмосферы
в сильных полях не сопрово-
ждается полным исчезновением
электрофоретического эффекта.
Теоретическое уравнение (33)
гл. V для электропроводности
симметричных электролитов при
напряженности поля, равной X,
можно представить в виде
?'=А'-[тиг'г(’) +
, 96500s (| z, | + | z2 j) /(x) ] .р/л
+ 6^300 У 2 J ’ ( }
а так как g(oo) = 0 и / (oo) = 1
(гл. IV, § 6), то разность меж-
ду Д°иДх равна
Рис. 38. Эффект Вина для сильных электро-
литов. Х(Х=0)=4,5 • 10~6.
Черные и белые кружки относятся к результатам, по-
лученным с помощью двух различных ячеек.
Д°—Д(Х=со) =
96500s (| Z1 | + | z2|) r
- 6лтп0 300 p42
(65)
Для слабых полей уравнение (64) можно свести к квадратичной форме,
предложенной впервые Иоосом и Блюментритом [63]. Так, Экштром и
Рис. 39. Общая характеристика эффекта Вина.
Кривые 1, 2 и 3 представляют эффект для растворов с тре-
мя различными концентрациями электролита, сз > С2 > ci.
Шмельцер [62] получили для х2 < 1, пренебрегая более высокими^ степе-
нями х, следующее выражение:
АХ — А(Х = 0) Г (0,0172) (zs)2A°x , (0,0243) (643,34)1 ze 1хД ,
' А(Х_О) = L.....XX_0YDkT + ’ <66>
(л —и) (Л = 0) ^(Х^О)0 V J
которое формально соответствует первому члену уравнения 1 Иооса и Блю-
ментрита [63] [уравнение (163) гл. IV]
АЛ—А(Х^0) АХ2 _ ВХ2у
Л(Х = 0) ' х
1 Численные ксэффициенты их уравнения равны 0,0133 и 0,0973 вместо значений
0,0172 и 0,0243 в случае уравнения (66).
14 г. Харпед, F. Оуэн
210
ГЛАВА VII
Уравнение (66) применимо только к бинарным электролитам, и поэтому
данные Вина [596, г] для сернокислого магния представляют собой единствен-
ные экспериментальные результаты для электролитов высокого валентного
типа, позволяющие сравнивать теорию с экспериментом. Экштром и Шмель-
цер сделали необходимые вычисления как для сильных, так и для слабых
полей. Для сильных полей они пользовались значениями постоянных с —
= 3,7 • 10~4 и А(х=0)= 105,5, а для слабых полей с = 1,22 • 10-3 и Д(х=о> =
= 99,0, кроме того, они приняли 7’ = 291, D = 81, т]0 = 0,0105 и А0 = 114.
Необходимые для расчетов значения g(x) и / (х), которые вычислил Вильсон,
приведены в табл. 17 и 18. На рис. 40 сопоставлены теоретические и экс-
периментальные данные: кружками представлены экспериментальные данные,
полученные для слабых полей, а точками — для сильных полей. Сплошные
кривые, построенные с помощью уравнений (64) и (66), прекрасно согласуются
Рис. 40. Эффект Вина для растворов сернокис-
лого магния.
Сплошные линии—теоретические кривые; белые круж-
ки—экспериментальные значения для слабых полей» чер-
ные кружки—то же для сильных полей.
с опытными данными при значениях напряженности поля, меньших чем
50 кв/см.
Фалькенгаген, Фрёлих и Флейшер [64] показали, что их теория влияния
частоты на электропроводность при сильных полях качественно согласуется
с данными Майкелса [65]. Так как экспериментальные данные в этой
области весьма немногочисленны, то необходимы дальнейшие исследования.
б) Эффект Вина для случая слабых электролитов, или влияние
силы поля на диссоциацию. Теория влияния сильных полей на диссоциацию
электролитов, развитая Онзагером, приводит к количественному соотноше-
нию, выражаемому уравнением (249) гл. IV:
Т(б) =1+Ь + ^3~ + Т8+180+"-’ (68>
где К (X) и К (0) представляют собой константы диссоциации соответственно
при наличии и в отсутствие внешнего поля. Переменная b для 1,1-валентных
электролитов определяется уравнением
Ь= 9,6361'77) Г2, (69)
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
211
причем V выражено в вольтах на сантиметр. Если степень диссоциации
мала (а < 1), то, согласно уравнению 252 гл. IV,
А(Л = оГ “о Г Л(О)-14' 2 6 + 246 +•••
(70)
С помощью данных, полученных для сред как с высокой, так и с низкой
диэлектрической постоянной, Онзагер [66] показал справедливость этих
уравнений.
На рис. 41 (согласно Онзагеру) точками представлены эксперименталь-
ные данные Шиле [67а] для уксусной и хлоруксусной кислот в воде
(Р= 78,57). Прямые линии соответствуют вычисленным теоретическим зна-
чениям относительного изменения электропроводности. Величина эффекта
в большой степени зависит от значения а, причем эффект тем сильнее,
чем слабее кислота. Для полей напряженностью свыше 50 кв/см совпадение
между экспериментальными и вычисленными значениями является пре-
восходным. Расхождения при более слабых полях объясняются тем фактом,
Рис. 41. Эффект Вина для уксусной и
хлоруксусной кислот; линии отвечают тео-
ретическим кривым.
эфире (D = 3,53) при 50°, причем сипа
что теория не учитывает влияния
ионных атмосфер, что является спра-
ведливым лишь в случае сильных
полей, при которых ионные атмо-
сферы разрушаются. Направление
этих отклонений подтверждает вы-
сказанное соображение, однако тео-
ретический расчет этого явления весь-
ма затруднителен.
Онзагер показал, что для слу-
чая сред с низкой диэлектрической
постоянной его теория подтверждает-
ся экспериментальными данными Ге-
манта [676], который исследовал,
влияние сильных полей на электро-
проводность раствора пикриновой
кислоты в смеси, содержащей 5%
этилового спирта и 1% минерально-
го масла в бензоле (£) = 2,7).
Мид и Фуосс [68а]1 измерили
электропроводность растворов пикра-
та тетрабутиламмония в дифениловом
поля менялась от 0 до 20 кв/см, а частота равнялась 60 периодам. Эти
измерения были проведены для ряда растворов с различными концентра-
циями, причем число этих растворов было достаточно для того, чтобы
с помощью способов, описанных в § 2 и 4, можно было вычислить кон-
станты диссоциации как [С+Л_]0, так и [С+Л-С+]+ для К иК3. Мид и Фуосс
получили соответственно значения 2,70 • 10~и и 1,00 • 10~3.
Влияние поля на электропроводность при очень малой и при несколько
более высокой концентрациях показано на рис. 42 и 43, на которых изо-
бражены графики зависимости удельной электропроводности от напряжен-
ности поля. При очень малых концентрациях растворенного вещества эта
зависимость является почти прямолинейной; при более высоких концентра-
циях начальная ветвь кривой (см. рис. 43) несколько изогнута, причем
она становится прямолинейной при возрастании силы поля. Как отмеча-
лось выше, это отклонение от прямолинейности обусловлено влиянием
ионных атмосфер, которые исчезают при более сильных полях. Путем
1 Уточненные данные см. [686].
14*
212
ГЛАВА VII
экстраполяции линейных участков кривых на нулевую напряженность
поля можно определить значения электропроводности растворов при малых
полях и при коэффициенте активности, равном единице (пунктирная линия).
Отношение электропроводности, полученной из обычных опытных данных
Р и с. 42. Эффект Вина для раствора ма-
лой концентрации. Пикрат тетрабутилам-
мония в дифениловом эфире.- 77=3,53;
£=50°; с=1,03 • 10~6»’
Рис. 43. Эффект Вина для раствора бо-
лее высокой концентрации. Пикрат тетра-
бутиламмония в дифениловом эфире.
Л=3,53; £=50°; с=9,73 . 10"4.
Рис. 44. Коэффициент активности
пикрата тетрабутиламмония в ди-
фениловом эфире, определенный из
данных для эффекта Вина.
Рис. 45. Влияние частоты перемен
ного тока на эффект Вина.
Бромистый тетрабутиламмоиий в дифени-
ловом эфире при 50°; с = 1,5 • 10-4.
при напряженности поля, равной нулю, к значению, найденному с помощью
линейной экстраполяции, является мерой коэффициента активности. Мид
и Фуосс определили таким путем коэффициент активности пикрата тетрабу-
тиламмония; вычисленные ими данные представлены на рис. 44. Пунктирная
линия соответствует предельной функции ’ lg/± — — 33,8с*/г, а эксперимен-
тальные значения обозначены кружками. Правильный порядок величины,
полученный этими исследователями, свидетельствует о том, что предложенное
Онзагером объяснение кривизны в области более слабых полей (рис. 43)
является справедливым. •
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
213
Согласно уравнению (70),
Лх/А(Х=0) = 1 +4 И- ... - 1 + 0.0132 (Ю-з V) + ..., (71)
если D = 3,53 и Z = 323. Мид и Фуосс нашли, что предельные значения
полученных ими коэффициентов - наклона при концентрации электролита,
равной нулю, точно совпадают с теоретическими.
Было показано, что коэффициенты наклона кривых зависимости
электропроводности от напряженности поля уменьшаются при увеличении
концентрации электролита. Это отклонение от хода кривой, соответствую-
щего теоретической зависимости,
происходит при той концентрации,
при которой зависимость IgA от
1g с начинает отклоняться от прямо-
линейной из-за образования ионных
тройников. Теория не учитывает
влияния поля на образование ион-
ных тройников. Некоторые измере-
ния, выполненные при частоте
500—1500 периодов, показывают,
что коэффициенты наклона кривых
зависимости электропроводности от
напряженности поля значительно
уменьшаются при увеличении ча-
стоты. Это явление подробно иссле-
довали Мид и Фуосс [22].
На рис. 45 изображено изме-
нение коэффициента наклона с
частотой для одной концентрации.
По оси ординат отложено отно-
шение электропроводности при
высокой напряженности поля к
электропроводности при слабом
поле1 для одной и той же ча-
стоты. По оси абсцисс отложены
значения напряженности поля
в кв!см. Влияние частоты2 наибо-
Р и с. 46. Влияние частоты переменного то-
ка и концентрации растворенного вещества
на эффект Вина.
Бромистый тетрабутиламмоний в дифениловом
эфире при 50°. Кривые для каждой последующей
концентрации смещены по оси ординат по 0,02 еди-
ницы. Цифры, приведенные в правой части гра-
фика, представляют собой с - 10*.
лее отчетливо выражено для очень разбавленных растворов и постепенно
исчезает по мере возрастания концентрации. Влияние концентрации иллю-
стрируется рис. 46, на котором изображены графики зависимости коэффи-
циента наклона (Дх/А<х=о)—l)/(10~3V) от частоты. Теоретическое значение
коэффициента наклона (0,0132) для каждой концентрации обозначено гори-
зонтальной линией со стрелкой в левой части рисунка.
При низких частотах экспериментальные значения коэффициентов
наклона 3 прекрасно согласуются с теоретическими; для самых высоких
концентраций это совпадение сохраняется вплоть до частоты, равной
1000 пер/сек. Эту экспериментальную зависимость можно заранее предсказать
на основании рассмотрения времени запаздывания Ланжевена [уравне-
ние (254) гл. IV]. Для наиболее разбавленных растворов (с = 10“*) т' состав-
1 Напряженность поля настолько мала, что ее увеличение в 2 раза не вызывает
заметного изменения электропроводности.
2 Следует отметить, что при низких напряженностях электропроводность пере-
стает зависеть от частоты; это указывает на то, что величина поляризации очень
незначительна.
3 Коэффициенты наклона на рис. 46 были умножены на 3/4 для того, чтобы учесть
влияние фильтра и усилителя, входивших в измерительную цепь. См. [686].
-214
ГЛАВА VII
ляет примерно 5-104 сек., и происходящая под влиянием поля реакция
диссоциации уже не успевает проходить полностью, так как направление
поля меняется через промежутки времени, близкие по порядку величины
ко времени т' (1000 периодов). Поэтому влияние поля резко уменьшается
с возрастанием частоты. При более высоких концентрациях время запазды-
вания меньше и распределение ионов может более точно соответствовать
меняющемуся полю. При с = 5 • ICh4, т'^8 • 10-5, и нормальные значения
эффекта Вина при низкой частоте приблизительно сохраняются до частоты,
равной 1000 периодам.
ЛИТЕРАТУРА
1. Р. Walden, Z. physik. Chem., 55, 207, 246 (1906).
2. J. J. H ermans, Z. physik., 97, 681 (1935).
3. a) L. E. В a k er, V. K. La M e r, J. Chem. Phys., 3, 406 (1935); 6) L. G. Longs-
worth, D. A. Maclnnes, J. Am. Chem. Soc., 59, 1666 (1937).
4. H. U 1 i c h, Z. Elektrochem., 36, 497 (1930).
5. a) P. Walden, Elektrochemie nicht-wasseriger Losungen, Barth, Leipzig, 1924;
б) P. W a 1 d e n, H. U 1 i c h, Z. phys. Chem., 114, 297 (1925); P. Walden,
H. U 1 i c h, G. Busch, там же, ИЗ, 429 (1926); H. U 1 i с h, Fortschritte Chem.,
18, No 10 (1926); H. S c h m i c k, Z. physik, 24, 56 (1924); R. T. L a t t e y. Phil.
Mag., 6, 258 (1928); в) M. Born, Z. Physik., 1, 221 (1920).
6. P. Wald en, H. U 1 i c h, Z. physik. Chem., 107, 219 (1923); H. U 1 i c h, Fort-
schritte Chem., 18, No 10 (1926).
7. a) P. W a 1 d e n, Z. physik. Chem., 55, 207, 249 (1906); 6) G. Angel, там же, A170,
81 (1934).
8. C. A. Kraus, W. C. Bra y, J. Am. Chem. Soc., 35, 1315 (1913).
9. A. S c h aji о v, Z. physik. Chem., 83, 129 (1913).
10. R. M. F u о s s, С. A. К r a u s, J. Am. Chem. Soc., 55, 476 (1933).
11. R. M. F u о s s, J. Am. Chem. Soc., 57, 488 (1935).
12. D. J. Mead, R. M. F u о s s, C. A. Kraus, Trans. Faraday Soc., 32, 594 (1936).
13. T. S h e d 1 о v s k у, H. H. U h 1 i g, J. Gen. Physiol., 17, 549 (1934).
14. T. S h e d 1 о v s k у, H. H. U h 1 i g, J. Gen. Physiol., 17, 570 (1934).
15. T. S h e d 1 о v s k y, J: Franklin Inst;, 225, 739 (1938).
16. В. B. Owen, R. W. G u r r y, J. Am. Chem. Soc., 60, 3074 (1938).
17. В. B. Owen, J. Am. Chem. Soc., 61, 1393 (1939).
18. a) D. Belcher, J. Am. Chem. Soc., 60, 2745 (1938); б) M. L. Kilpatrick,
J. Chem. Phys., 8, 306 (1940).
19. R. M. F u о s s, С. A. К r a u s, J. Am. Chem. Soc., 55, 1019 (1933).
20. R. M. F u о s s, C. A. Kraus, J. Am. Chem. Soc., 55, 2387 (1933).
21. M. Doi e, Trans. Electrochem. Soc., 77, 385 (1940); С. B. Wooster, J. Am. Chem.
Soc.,-60, 1609 (1938).
22. D. J. Mead, R. M. F u о s s, J. Am. Chem. Soc., 62, 1720 (1940).
23. R. M. F u о s s, J. Am. Chem. Soc., 56, 1857 (1934).
24. G. S. Bien, С. A. К r a u s, R. M. F u о s s, J. Am.. Chem. Soc., 56, 1860 (1934).
25. M. Born, Z. Physik., 1, 221 (1920); H. S c h m i с k, там же, 24, 56 (1924);
R. T. L a t t e y, Phil. Mag., 6, 258 (1928).
26. P. Walden, Z. physik. Chem., A147, 1 (1930).
27. a) R. M. F u о s s, С. A. К r a u s, J. Am. Chem. Soc., 55, 3614 (1933); 6) W. F. L u-
d e r, C. A. Kraus, там же, 58, 255 (1936).
28. R. M. F u о s s, C. A. Kraus, J. Am. Chem. Soc., 55, 21 (1933).
29. a)_ F. M. В a t s о n, С. A. К r a u s, J. Am. Chem. Soc., 56, 2017 (1934); б) C. A
К r a u s, R. A. V i n g e e, там же, 56, 511 (1934).
30. R. M. F u о s s, C. A. Kraus, J. Am. Chem. Soc., 57, 1 (1935).
31. R. M. F u о s s, J. Am. Chem. Soc., 56, 1031 (1934).
32. N. L. С о x, С. A. К r a u s, R. M. Fuoss, Trans. Faraday Soc., 31, 749 (1935).
33. R. L. M с I n t о s h, D. J. M e a d, R. M. Fuoss, J. Am. Chem. Soc., 62, 506
(1940); E. Swift, там же, 60, 2611 (1938); W. F. L u d e r, P. B. Kraus,
С. A. К r a u s, R. M. F u о s s, там же, 58, 255 (1936).
34. M. A. E 1 1 i о t t, R. M. F u о s s, J. Am. Chem. Soc., 61, 294 (1939).
35. D. J. M e a d, С. A. К r a u s, R. M. F u o s s, J. Am. Chem. Soc., 61, 3257 (1939).
36. С. A. К r a u s, J. Franklin Inst., 225, 687 (1938); С. A. К r a u s, E. G. John-
son, J. Am. Chem. Soc., 55, 3542 (1933); С. A. К r a u s, W. W. II awe s, там
же, 55, 2776 (1933); D. L. F о w 1 e r, C. A. Kraus, там же, 62, 2237 (1940).
37. C. A. Kraus, Science, 90, 281 (1939).
АССОЦИАЦИЯ ИОНОВ. СЛАБЫЕ ЭЛЕКТРОЛИТЫ
215
38. S. Arrhen ius, Z. physik. Chem., 1, 631 (1887); M. Planck, там же, 1, 577
(1887).
39. W. О s t w a 1 d, Z. physik. Chem., 2, 270 (1888).
40. X. С. Тейлор, Физическая химия, т. I, ОНТИ—Химтеорет, Л., 1935, гл. XII.
41, М. S. S h е г г i 1 1, A. A. Noyes, J. Am. Chem. Soc., 48, 1861 (1926).
42. a) D. A. M a с I n n e s, J. Am. Chem. Soc., 48, 2068 (1926); б) C. W. Davies,
J. Phys. Chem., 29, 977 (1925).
43. F. Kohlrausch, Z. physik. Chem., 64, 129 (1908).
44. F. К о h 1 г a u s c h, Wied. Ann., 6, 1 (1879); 26, 161 (1885).
45. C. W. Davie s, J. Am. Chem. Soc., 54, 3776 (1932).
46. D. A. M a с I n n e s, T. S h e d 1 о v s k y, J. Am. Chem. Soc., 54, 1429 (1932).
47. W. D. L a r s о n, W. J. T о m s i c e k, J. Am. Chem. Soc.,61, 65 (1939).
48. В. B. Owen, J. Am. Chem. Soc., 54, 1758 (1932).
49. ,B. S a x t о n, T. W. Langer, J. Am. Chem. Soc., 55, 3638 (1933).
50. B. S a x t о n, H. F. Meier, J. Am. Chem. Soc., 56, 1918 (1934); B. Saxton,
L. S. Darken, там же, 62, 846 (1940).
51. M. W i e n, Ann. Phys. [4], 83, 840 (1927).
52. H. Sac k, Physik. Z., 29, 627 (1928).
53. B. Brendel, О. M i t t e 1 s t a e d t, H. Sack, Physik. Z., 30, 576 (1929);
H. S a c k, B. Brendel, там же, 31, 345 (1930); В. Brendel, там же, 32,
327 (1931).
54. Н. Z ahn, Z. Physik., 51, 350 (1928); Н. R i е с k п о f f, Н. Zahn, там же, 53,
619 (1929).
55. A. D e u b n e r, Physik. Z., 30, 946 (1929); Ann. Physik. [5], 5, 305 (1930); Physik. Z.,
33, 223 (1932).
56. M. Wien, Physik. Z., 31, 793 (1930); 32, 183 (1931); Ann. Physik. [5], 11, 429 (1931).
57. J. M also h, Physik. Z., 33, 19 (1932); Ann. Physik [5], 12, 865 (1932).
58. P. D e b у e, H. Falkenhagen, Physik. Z., 29, 121, 401 (1928); Z. Elektroche-
mie, 34, 562 (1928).
59. a) M. Wien, Ann. Physik. [4], 83, 327 (1927); б) там же, 85, 795 (1928); в) Physik. Z.,
28, 834 (1927); г) там же, 29, 751 (1928).
60. М. W i е n, J. S с h i е 1 е, Physik. Z., 32, 545 (1931).
61. A. Gemant, Physik. Z., 29, 289 (1928).
62. H. C. Eckstrom, C. Schmelzer, Chem. Rev., 24, 367 (1939).
63. G. J о о s, M. Blumentritt, Physik. Z., 28, 836 (1927); M. В 1 u m e n t r i t t,
Ann. Physik., 85, 812 (1928).
64. H. Falkenhagen, F. F г б 1 i c h, H. Fleischer, Naturwiss., 25, 446 (1937).
65. F. Michels, Ann. Physik., 22, 735 (1935).
66. L. 0 n s a g e r, J. Chem. Phys., 2, 599 (1934).
67. a) S c h i e 1 s, Ann. Physik. [5], 13, 811 (1932); 6) A. G e m a n t, Electrophysik der
Isolierstoffe, Springer, Berlin, 1930, p. 78—80; Physik. Z., 29, 289 (1928).
68. a) D. J. M e a d, R. M. F u о s s, J. Am. Chem. Soc., 61, 2047 (1939); б) там же,
61, 3589 (1939).
ЛИТЕРАТУРА К ТАБЛИЦАМ
/7 табл. 32 «...
1. И. U 1 i с h, Fortschritte Chem., 18, No 10 (1926).
I{ табл. 33
1. H. U 1 i c h, E. R. Bit r, Z. Angew. Chem., 41, 443 (1928).
2. H. U 1 i c h, Trans. Faraday Soc., 23, 388 (1927).
К табл. 35
1. R. M. F u о s s, С. A. К r a u s, J. Am. Chem. Soc., 55, 2387 (1933).
77 табл. 36
1. J. A. G e d d e s, C. A. Kraus, Trans. Faraday Soc., 32, 585 (1936); G. S. H o-
o p e г, С. A. К r a u s, J. Am. Chem. Soc., 56, 2265 (1934); Proc. Nat. Acad. Sci.,
19, 939 (1933).
2. R. M. F u о s s, С. A. К r a u s, J. Am. Chem. Soc., 57, 1 (1935).
3. D. A. R о t h г о c k, С. A. К r a u s, J. Am. Chem. Soc., 59, 1699 (1937).
77 табл. 37
1. G. S. В i e n, С. A. К r a u s, R. M. F u о s s, J. Am. Chem. Soc., 56, 1860 (1934)»
216
ГЛАВА VII
Л табл. 39
1. С. A. Kraus, J. Franklin Inst., 225, 687 (1938).
2. М. А. Е 1 1 i о t t, R. М. F и о s s, J. Am. Chem. Soc., 61, 294 (1939).
3. L. R. M с I n t о s h, D. J. M e a d, R. M. F u о s s, J. Am. Chem. Soc., 62,
506 (1940).
4. N. L. С о x, С. A. К r a u s, R. M. F u о s s, Trans. Faraday Soc., 31, 749 (1935).
5. D. J. M e a d, R. M. F u о s s, J. Am. Chem. Soc., 61, 2047 (1939).
К табл. 40
1. В. S a x t о n, L. S. D a г к e n, J. Am. Chem. Soc., 62, 846 (1940).
2. D. A. Mac Innes, T. Shedlovsky, J. Am. Chem. Soc., 54, 1429 (1932).
3. D. Belcher, J. Am. Chem. Soc., 60, 2744 (1938).
4. T. S h e d 1 о v s к y, A. S. В г о w n, D. A. Maclnnes, Trans. Electrochem,
Soc., 66, 165 (1935).
5. B. S a x t о n, T. W. Langer, J. Am. Chem. Soc., 55, 3638 (1933).
6. A. W. M a r t i n, H. V. T a r t a r, J. Am. Chem. Soc., 59, 2672 (1937).
7. M. S. S h e r r i 1 1, A. A. N о у e s, J. Am. Chem. Soc., 48, 1861 (1926).
8. B. S a x t о n, G. W. W a t e r s, J. Am. Chem. Soc., 59, 1048 (1937).
9. B. S a x t о n, H. F. M e i e r, J. Am. Chem. Soc., 56, 1918 (1934).
10. F. G. В г о с к m a n, M. Kilpatrick, J. Am. Chem. Soc., 56, 1483 (1934).
11. T. S h e d 1 о v s к y, D. A. M a с I n n e s, J. Am. Chem. Soc., 57, 1705 (1935).,
12. V. S i v e r t z, R. E. R ei tmei er, H. A. T a r t a r, J. Am. Chem. Soc., 62,
1379 (1940).
13. L. S. Darke n, J. Am. Chem. Soc., 63, 1007 (1941).
Глава VIII
ТЕРМОХИМИЧЕСКИЕ ВЕЛИЧИНЫ, ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ
ОБЪЕМЫ И КОЭФФИЦИЕНТЫ РАСШИРЯЕМОСТИ И СЖИМАЕМОСТИ
§ 1. Введение
Начиная с этой главы, мы приступаем к систематическому рассмотрению
термодинамики растворов электролитов. Как видно из гл. I, содержащей
формальную трактовку термодинамики растворов электролитов, для изло-
жения этого вопроса необходимо знать все парциальные молярные величины
компонентов растворов. Парциальные величины можно разделить на две'
группы. В первую группу входят те величины, которые можно определить,
измеряя коэффициенты, характеризующие зависимость относительной пар-
циальной молярной свободной энергии или активности от давления и тем-
пературы. К таким величинам принадлежат относительные парциальный
молярные теплосодержание, теплоемкость и объем [уравнения (38), (40)
и (44) гл. I]. Эти величины можно измерить, не зная парциальной молярной
свободной энергии, однако последнюю нельзя определить из этих величин,
не располагая дополнительными данными. Ко второй группе относится
парциальная молярная свободная энергия растворенного вещества и раство-
рителя.
В целях удобства изложения вопрос о свободной энергии будет рас
смотрен в следующих главах, а в этой главе мы рассмотрим те величины,
которые могут быть получены из данных о свободной энергии, хотя такой
расположение материала и может показаться несколько произвольным.
Вначале будет рассмотрена наиболее важная из этих величин—относитель-
ное парциальное молярное теплосодержание и соответствующая ей инте-
гральная величина—относительное кажущееся молярное теплосодержание
(теплота разведения). Затем будет весьма подробно разобран вопрос о пар-
циальной молярной теплоемкости, абсолютное значение которой может
быть вычислено. Далее будет изложен также вопрос о теплоте нейтрализа-
ции, и таким образом все эти калориметрические величины можно будет
рассматривать в непосредственной связи друг с другом. Последние пара-
графы будут посвящены парциальным молярным объемам, расширяемости
и сжимаемости.
§ 2. Относительное кажущееся молярное теплосодержание, теплота
разведения и относительное парциальное молярное теплосодержание
Термохимические измерения всегда представляли большой интерес для
физико-химиков, но только с помощью современных многоспайных термо-
пар и постепенного усовершенствования методики адиабатической тепловой
компенсации удалось удовлетворительно измерить те небольшие тепловые
эффекты, которые имеют место в разбавленных растворах. Во многих слу-
чаях необходимо знать теплосодержания компонентов раствора по сравне-
нию с соответствующими значениями при бесконечном разбавлении. Для
определения этих величин необходимо прибегать к экстраполяции экспери-
218
ГЛАВА VIII
ментальных данных, полученных для очень разбавленных растворов.
С помощью уравнения Дебая—Гюккеля было получено выражение для теоре-
тического коэффициента наклона кривой [уравнение (16) гл. II], благодаря
чему эта экстраполяция стала более надежной. С другой стороны, возмож-
ность проверки значения предельного коэффициента наклона увеличивает
ценность измерений, произведенных при очень больших разбавлениях.
Прежде чем описывать и обсуждать эти данные, необходимо дополнить изло-
женные в гл. I представления о парциальных молярных величинах путем
определения некоторых новых величин.
Из общего уравнения (35) гл. I для случая двух компонентов полу-
чается выражение
+ (1)
с помощью которого общее теплосодержание раствора, состоящего из пг
молей растворителя и п2 молей растворенного вещества, определяется через
парциальные молярные теплосодержания его компонентов. Подобно этому
уравнение
Н = ПуН г-}-п2ун (2)
определяет теплосодержание через парциальное молярное теплосодержание
растворителя при бесконечном разбавлении /7° и через кажущееся молярное
теплосодержание растворенного вещества <рн. Как видно из этих двух
уравнений, при бесконечном разбавлении
Ти = М (3)
Так как абсолютные значения теплосодержания нельзя определить экспе-
риментально, то приходится использовать соответствующие относительные
величины, которые (в случае отсутствия специальных оговорок) будут отне-
сены к бесконечному разбавлению. Тогда из уравнений (1) и (2), пользуясь
уравнением (39) гл. I, можно получись выражение
Н _ = nr (Н, - И») + п2 (Н2-Н§ = п2 (?н - <&). (4)
Вводя обозначения L = H — Н°, — Н°1г L2~H2 — и =срн—
получаем уравнение
L = + n2L2 = n2'jfL. (5)
Относительное кажущееся молярное теплосодержание фн — равно по вели-
чине и противоположно по знаку теплоте разведения А7/D для изотерми-
ческого и одновременно изобарического добавления бесконечно большого
количества чистого растворителя к раствору, содержащему один моль
растворенного вещества в п1/п2 молях растворителя. Следовательно,
n2&HD = —njLi — n2L2 = — • (6)
Согласно этому определению, D положительно, если процесс растворения
сопровождается поглощением тепла. Такое условие относительно знаков
противоположно принятому Ланге и другими европейскими авторами, на
которых мы будем в дальнейшем ссылаться.
При дифференцировании первого и последнего членов уравнения (5)
по п2 при постоянных температуре, давлении и щ получается
^2 = + п2 , (7)
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
' 219
dL т гл
так как при этих условиях равно ь2- Это выражение представляет
собой главное уравнение, на котором основано калориметрическое опреде-
ление L2. Величина не может быть измерена непосредственно, но ее
легко определить, измеряя тепловые эффекты, сопровождающие последо-
вательные конечные разбавления, и экстраполируя данные, получающиеся
для наименьших концентраций, на нулевую концентрацию. Так, при раз-
бавлении бинарного раствора от концентрации растворенного вещества т
(в молях на килограмм растворителя) до т' и от т' до т" и т. д. полу-
чаются следующие тепловые эффекты:
— 'рд- tfjj, (8)
= <рд <рд (9)
ит. д. для последовательных разбавлений. Линейная комбинация этих
уравнений дает значения <ря—<рн, —®н и т> пУтем экстра-
поляции этих значений на нулевую концентрацию можно получить величину
'Ря—Далее, вычитая ч'н~~ <ря, и т- Д- 113 значения фя —
получают соответствующие теплоты разбавления для т', т" и т. д., т. е. для
всего изучаемого интервала концентраций. Пример такого вычисления при-
водится в табл. 41. Опытные данные относятся к водным растворам хлори-
стого натрия при 25°; первые три столбца таблицы взяты из работы Робин-
зона [1].
Таблица 41
Теплота разведения хлористого натрия при 25°
с9 моли/л. -Нр(спсх<-* сконечн.)> нал /'моль Конечная концентрация дн (0,1->с) ДНО
исходная конечная с Ус-
0,1 0,00308 -61,5 0,05 0,2236 -12,8 -70,2
0,1 0,00605 -52,9 0,025 0,1581 -27,8 -55,2
0,05 0,00154 -54,9 0,0125 0,1118 -42,9 -40,1
0,05 0,00302 -48,7 0,00605 0,0778 -52,9 -30,1
0,025 0,000770 -45,6 0,00305 ’) 0,0552 -61,5 -21,5
0,025 0,001515 -39,9 0,00153 0,0391 -67,4 -15,6
0,0125 0,000385 -33,1 0,00076 0,0276 -73,1 - 9,9
0,0125 0,000754 -30,5 0,00039 0,0197 -75,7 - 7,3
0 0 (-83,0) 0
1) Тепловой эффект при изменении концентрации от с=0,00308 до с=0,00302 очень незначи-
телен; поэтому вместо обеих концентраций принимается их среднее значение; то же проделано
и для других концентраций (меньших чем 0,00305), приведенных в таблице.
Из рис. 47 видно, как с помощью- экстраполяции определяется значе-
ние ДЯ(о,1->о), в данном случае равное—83,0. Теоретический предельный
коэффициент наклона, согласно уравнению (16) гл. II, равняется
Г2=5(Н)/Б = /^5(Н)Кот. (10)
Это выражение применимо к сильным 1,1-валентным электролитам при
таких больших разбавлениях, когда с можно заменить на dtim. Сочетая это
220
ГЛАВА VIII
уравнение с уравнением (7), получаем
= (2/3) УХ) S(H) К™’ (11)
или в более простой форме
ДЯО= _(2/3)§(Н)Ус. (12)
График зависимости Д#о от ]/с должен приближаться к прямой, соот-
ветствующей этому предельному выражению, по мере уменьшения с.
Рис. 47. Теплоты разведения водных растворов хлори-
стого натрия при 25°.
Черные кружки (0,1-> с), белые кружки—АН . (Ср.
рис. 170.) D
Следовательно, предельным уравнением для экспериментальных величин
Д7/(о,1->С) является выражение
=A7Z(o,i-»o) +(2/3) <5(Н) с, (13)
которое можно получить путем вычитания <рн(о,о из обеих частей урав-
нения (И).
Описанный метод экстраполяции предложен Ланге1 2. Ошибка, возни-
кающая при экстраполяции, оценивается им в + 2 кал/моль, что обычно соот-
ветствует действительности, однако значения «экспериментальных» пре-
дельных коэффициентов наклона, которые Ланге определял из данных для
наиболее разбавленных растворов, сильно отклоняются от теоретических
значений. Такое расхождение между теорией и опытом не возникает при
употреблении более современных методов расчета. Прежде чем приступить
к обсуждению 2 этих методов, следует вкратце описать экспериментальную
методику, применявшуюся при этих измерениях.
Первое точное измерение теплот разделения очень разбавленных раство-
ров было произведено Нернстом и Ортманом [2]; современный в высшей
1 Табл. 121.
2 Описание этой методики приводят Ланге и Робинзон [За], а полную библио-
графию более ранних методов исследования можно найти в статье Ричардса и Гу-
кера [36].
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ 221
Р и с. 48. Кажущиеся относитель-
ные молярные теплосодержания
водных растворов сернокислого каль-
ция и сернокислого магния при 25°.
Белые кружки—данные Ланге и Мон-
гейма; черные кружки—данные Ланге и
Стрека. П. 3.—предельный теоретиче-
ский закон.
степени точный метод разработан главным образом Ланге\ который
внес ряд остроумных усовершенствований в устройство двойного калори-
метра Джоуля—Пфаундлера. Наиболее замечательным нововведением яви-
лось употребление плоского термостолбика в качестве перегородки, разде-
ляющей калориметр на две части [4]. Это устройство состоит из 1000—1500
спаев железо-константановых термопар и позволяет измерять малые разно-
сти температур с точностью около 2 • 10~7 градуса. Так как теплопроводность
термопар препятствует измерению разностей температур, превышающих
10~3 градуса в разбавленных растворах, то возрастание температуры,
сопровождающее разбавление раствора в одной половине калориметра,
должно быть в значительной мере уравновешено посредством электриче-
ского нагревания чистого растворителя в другой половине. Назначение
термопар состоит в том, чтобы определять разность между теплотой раз-
бавления и точно измеренной теплотой,
выделяющейся при электрическом нагре-
вании.. В идеальном случае термопары
должны служить в качестве нуль-инстру-
мента. Пользуясь гальванометром Пашена
и специально сконструированным потен-
циометром, Гукер, Пикард и Планк [5]
понизили теплопроводность, применяя
медь-константановые термопары из по-
следовательно соединенных 60 спаев, и
благодаря этому смогли лучше изоли-
ровать друг от друга обе части калоримет-
рической системы. С помощью точной ре-
гулировки температуры термостата раз-
ность температур между калориметром
и окружающей средой уменьшается до
10~* градуса, а так как эта величина соиз-
мерима с изменениями температуры, про-
исходящими при разбавлении растворов с
малыми концентрациями, то измерения
можно рассматривать как адиабатические
и изотермические. О чувствительности та-
ких измерений и воспроизводимости ре-
зультатов можно судить на основании
сравнения данных, полученных для очень
разбавленных растворов сернокислого
кальция Ланге и Стреком [6], Ланге и
Монгеймом [7]. Из рис. 48 видно, что
данные двух независимых серий измерени)
телями, укладываются на одну и ту же кривую. Кривая для сернокис-
лого магния заметно отклоняется от кривой для сернокислого кальция
уже при столь малой концентрации, как 0,0005 М.
Специфические особенности графиков зависимости теплот разведения
от \/ с, проявляющиеся уже при самых малых концентрациях, для которых
доступны измерения, представляют собой общее явление, наблюдаемое.для
электролитов с различными типами валентности. На основании рис. 49
можно судить о поведении некоторых типичных 1,1-валентных электроли-
, проделанных этими исследова-
1 Детальное обсуждение точности различных методов термохимического иссле-
дования разбавленных растворов дано в монографии В. Свентославского (W. S w i е п-
toslawsky, Microcalorimetry, 1947). (Прим, ред.)
222
ГЛАВА VIII
тов, изученных ЛангеВ связи с этими данными возникают два важных
вопроса: действительно ли все кривые приобретают теоретический коэффи-
циент наклона при бесконечном разбавлении, и почему индивидуальные'
особенности солей, присутствующих в растворах в малых концентрациях,
гораздо отчетливее проявляются в случае термохимических данных,
чем в случае результатов, относящихся к коэффициентам активности. Ответ
на последний вопрос можно получить, дифференцируя полное уравнение
Дебая —Гюккеля [(74) гл. III]:
18/*--лЙ7Т-+-вг- <14>
В результате дифференцирования этого уравнения получается выражение,,
из которого видно, что температурные коэффициенты величин А и В в случае
очень разбавленных растворов1 2
имеют существенное значение.
Более ранние попытки3 объяс-
нить специфический характер
данных по теплотам разведения
посредством учета влияния толь-
ко параметра А либо в форме,
представленной выше, либо с
помощью уравнений (42) и (43)
гл. III, содержащих члены выс-
ших порядков, приводили к
некоторым любопытным ано-
мальным значениям ионного
параметра а. Было обнаруже-
но [9, За], что если ряд электро-
литов с одним общим ионом рас-
положить в порядке возраста-
ния величины а, найденной из
теплот разведения, то этот поря-
док обычно противоположен
Рис. 49. Относительные кажущиеся моляр-
ные теплосодержания водных растворов солей
калия при 25°; II. 3.—теоретический предельный
закон.
тому, который получается в том случае, когда величины а вычислены
из данных о коэффициентах активности. Кроме того, важную роль
играет при этом природа общего иона, и при замене данного общего
иона на другой в некоторых случаях этот порядок может изменяться на
обратный. Так, например, <iMgSo1 больше aCaso4> в т0 время как aMgCi2 мень-
ше acacia- Эти факты вызвали некоторые сомнения в справедливости урав-
нения Дебая —Гюккеля и показали, что необходимо точно определять,
приближаются ли экспериментальные значения коэффициентов наклона
к теоретическим предельным значениям. Подробное обсуждение этого вопроса
содержится в приложении Б, § 4.
Описанные расхождения между значениями экспериментальных и теоре-
тических предельных коэффициентов наклона в значительной степени объя-
сняются трудностями точного определения предельного коэффициента
1 Библиография экспериментальных работ приводится_в этой главе и в прило-
жении А—в ссылках на литературу к таблицам для АНD и Ь2.
2 См. гл. XI и XII.
3 Бонино и Ваглио [8] пытались отразить специфический характер кривых для
теплот разведения с помощью полуэмпирических выражений.
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
223-
наклона кривых, подобных изображенным на рис. 47. Графики такого рода,
в пределах точности опытных данных, являются прямолинейными в значи-
тельном интервале концентраций, но поскольку при бдльших концентрациях
появляются значительные отклонения от прямолинейности (рис. 49), то
значения экспериментального предельного коэффициента наклона оказы-
ваются зависящими от того интервала концентраций, в котором они
определялись. Гульбрансен и Робинзон [10] на основании полученных
ими данных для растворов хлористого натрия при 10, 15, 20 и 25° при-
шли к выводу, что теплота разведения является линейной функцией от р'Ъг
вплоть до]/т = 0,1. С другой стороны, эти же авторы утверждают, что
«имеются некоторые данные, свидетельствующие о том, что коэффициент
наклона продолжает изменяться вплоть до т = 0, однако тепловые эффекты
при значениях т меньше 0,1 слишком малы, чтобы окончательно ре-
шить этот вопрос». Согласно данным Гульбрансена и Робинзона, экспери-
ментальные значения предельных коэффициентов наклона меньше теорети-
ческих при 10° на 33% и при 25° на 12%. Позже с помощью аналитиче-
ского видоизменения метода графического дифференцирования [11] Юнг
и Гренье [12] показали, что из тех же данных получаются значения коэф-
фициентов наклона, отличающиеся от теоретических не больше чем на 5%.
Согласно этому методу, хорда
^(Ш1 -» тг)
|/т2— / тх
(15)
представляет собой среднюю величину коэффициента наклона S графика
зависимости ДЯС от |/ и в, интервале концентраций от Д° ]A»i. Со-
гласно уравнению (77) гл. III, для разбавленных растворов можно предста-
вить S в следующем виде:
S = 5° 4- В" т -j- С"т (16)
и путем предварительного построения кривых можно показать, что вклю-
чение членов, содержащих высшие степени ф' щ, при значениях ]/т < 0,64
является излишним. S0, В" и С" представляют собой эмпирические кон-
станты, которые следует определять из экспериментальных данных после-
того, как S будет выражено через 1\. Если опытные данные согласуются
с теоретическими, то полученное таким образом значение хУ0 должно
равняться (2/3) §(н). Обозначая величину (1/2) (]/т2 + p' ^i) для
Таблица 42
Значения констант для хлористого натрия
Температура, оС Метод графического дифференцирования Теоретиче- ское значение 8Э Значение 8°, найденное Гульбрансе- ном и Робин- зоном
- В" С" 8°
25 1452,2 730,4 476,1 477 (478)1) 418
20 1585,7 894,8 451,3 434 (435) 370
15 1650,8 924,5 413,6 393 (396) 340
10 1531,5 591,9 355,7 355 (358) 239
1) Значения, заключенные в скобки, вычислены из значений , при-
' ч* (хз )
веденных в табл. 11. Все остальные значения принадлежат Юнгу и Гренье.
224
Г Л A B A VIII
хорды через xt, а значение 6' при через Pt, получаем
Pi = S'i + B''xi + C"xl,
и разность между Pt и Pt равна
р _Р — _ А = _ £21
1 ' d (У т)2 14
(17)
(18)
где о; представляет собой изменение ]/т при г-м разведении. Исключая Р,
из уравнений (18) и (17), получаем искомое уравнение
Pi = S° + B"xl=:C,'(rf + tf/12), (19)
позволяющее вычислить S0, В" и С" по методу наименьших квадратов.
При использовании этого метода Юнг и Гренье приняли т2 в качестве
«весового» множителя.
Полученные этим способом константы приведены в табл. 42, в кото-
рую включены, кроме того, значения S0, вычисленные Гульбрансеном
Рис. 50. Определение экспериментальных предельных коэффициентов наклона .5'° для
относительного кажущегося молярного теплосодержания водных растворов 1,1-валент-
ных электролитов при различных температурах.
равные (2/3) / d0 SiH). Для того чтобы показать, что это отличное совпа-
дение между теоретическими значениями коэффициентов наклона и значе-
ниями-, вычисленными по методу графического дифференцирования, не
является случайным, Юнг и Зелигман [13] применили этот метод ко всем
солям щелочных металлов, для которых имелись точные данные. При этих
вычислениях в уравнениях (15) —(19) т было заменено на с. Так как при
этом изменяются численные значения В" и С", то при использовании кон-
центраций с мы будем обозначать эти константы через В' и С'. Графйки
для Pt, построенные Юнгом и Зелигманом по уравнению (17) с помощью
экспериментальных значений Р^ и эмпирических значений С, изображены
на рис. 50 и 51. Кривые соответствуют уравнению (16) для значений S,
полученных описанным выше способом. Определенные таким путем значе-
ния равные отрезкам, отсекаемым на оси ординат, находятся в хоро-
шем согласии с теоретическими значениями S0, поскольку среднее откло-
нение составляет всего ±6,3%. Далее, сумма • положительных и отрица-
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
225
Р и с. 51. Определение эксперименталь-
ных предельных коэффициентов накло-
на 5° для относительного кажущегося
молярного теплосодержания водных
растворов 1,2-валентных электролитов
при 25°.
тельных отклонений для 21 разной соли составляет только + 10,8%. Зна-
чения отдельных отклонений Д = 100 [№эк — £(теорет.)]/£(теорет.) приведены
в шестом столбце табл. 43. В связи с наблюдающимся согласием между
теорией и опытом Юнг и Зелигман считают, что теория полностью под-
тверждается в пределах вероятной суммарной ошибки опытных данных
и метода экстраполяции. Поэтому, воспользовавшись для всех случаев
теоретическим значением №, они вычислили заново константы В' и С
по методу наименьших квадратов. Полученные таким образом величины
приведены в табл. 43. При подстановке этих параметров в выражение
?, == - _ № /с 4- (1/2) В'с + (1/3) С'Сз/2 (20)
получаются точные значения относительного кажущегося молярного тепло-
содержания (или теплоты разведения) вплоть до |/ с —. 0,2 в общем слу-
чае и до ]/с = 0,4 для случая хлористых
представленных в табл. 43, в соответст-
вии с теорией предельный коэффициент
наклона выражается уравнением
№ = (2/3) §(Н). (21)
Что касается остальных солей, для ко-
торых имеются данные, относящиеся
к очень разбавленным растворам, то
Юнг и Зелигман нашли, что резуль-
таты измерений для случая растворов
хлористого [14] и фтористого калия [15]
согласуются с уравнением (21), хотя ко-
личество этих данных недостаточно для
применения метода наименьших квад-
ратов. Они указали также, что «при
исследовании солей щелочноземельных
металлов с типом валентности 2,1 об-
наруживается такое же согласие, по
крайней мере при 25°. Результаты экст-
раполяции представляются менее точ-
ными, и они не так определенно под-
тверждают теорию Дебая — Гюккеля,
однако нет никаких данных, которые
указывали бы на неправильность тео-
рии. Из имеющихся данных для солей
с типом валентности 2,2 при их обра-
ботке по описанному выше методу, не-
сомненно, нельзя получить предельные
значения, согласующиеся с теорией» [13,16]. Поскольку имеются доказательст-
ва того, что соли с типом валентности 2,2 неполностью диссоциированы в раство-
рах (табл. 22), то поведение этих солей нельзя рассматривать как серьез-
ное исключение из наблюдаемого в общем случае согласия данных о тепло-
тах разведения с теорией.
Вычисление относительного парциального молярного теплосодержания £2
из данных о теплотах разведения производится с помощью уравнения (7),
применяемого в следующей форме:
7“ л и д\нв
7^2 — ДЯ ц — и2---------------------
Агг 1 1 г— дЬНп
-я-— — ДЯВ----9 V т
Л дут
В случае разбавленных растворов, для которых т пропорционально с,
15 г. Харнед, Б. Оуэн
(22)
226 ГЛАВА VIII
путем сочетания этого уравнения с уравнением (20) получается выражение
Г2 = (3/2)5°/с + В,с + (5/6)С'с* 3 */2. _ (23)
Это выражение можно использовать для вычисления L2 вплоть до кон-
центраций порядка 0,05н.
При решении практических задач, в особенности при приведении
результатов определения точек замерзания к стандартной температуре,
часто приходится вычислять Lr. Общее выражение для этой величины, кото-
рое получается из уравнений (6) и (7), имеет вид
£ = пг dHHD^Mim3l2d^HD
1 • дп2 2000 д У т ‘ ' '
В достаточно разбавленных растворах с можно заменить через mdQ, причем
получается выражение
Л = - WW (5°с3/2 + в'с2 + С'с5/2)’ (25)
если исключить с помощью уравнения (20).
В табл. 121 и 122 собраны значения ерь и L2, полученные из данных
по калориметрическим измерениям для сильных электролитов при 25°. До-
полнительные значения, полученные с помощью измерений электро-
движущих сил, приводятся в гл. XI и XII. Подробные таблицы для
других температур, в особенности для 18°, можно найти в справоч-
никах и в монографии Биховского и Россини [16а]1.
Так как при экстраполяции большей части данных, использованных
для составления табл. 121, не был использован теоретический предельный
коэффициент наклона, то мы заново произвели экстраполяцию для всех
солей таким образом, чтобы кривые, изображающие зависимость <pL от ]/ т,
Имели близ начала координат коэффициент наклона, равный теоретическому,
т. е. (2/3) |/dQ £>(Н). Этот коэффициент наклона больше коэффициентов, при-
менявшихся ранее, поэтому обычно приходилось проводить кривые более
круто по мере их приближения к оси ординат. Полученные кривые смещались
в вертикальном направлении до тех пор, пока они не проходили через начало
координат, и затем определялись величины и dyijd ~\Ут при округленных
значениях концентраций. С помощью этих величин по уравнению (22) вычи-
слялись значения L2, которые усреднялись графическим путем, для того
чтобы устранить неточности, связанные с трудностью ©пределения d^tjd ]/т.
Часть I табл. 122 содержит эти пересчитанные и усредненные значения Л2-.
Не все данные, приведенные в части II этой таблицы, были пересчитаны
заново, но они в каждом случае подбирались таким образом, чтобы
соответствующие им точки хорошо укладывались на продолжениях кривых,
построенных по значениям, взятым из части I той же таблицы.
Значения ерь и Ь2 для разбавленных растворов, полученные авторами
этой книги, хорошо согласуются с величинами, вычисленными по урав-
нениям (20) и (23) с помощью констант, приведенных в табл. 43.
1 В указанной Харнедом и Оуэном книге Биховского и Россини собраны данные,
опубликованные до 1934 г. Советский читатель может пользоваться более полным источ-
ником, содержащим материал, опубликованный до 1941 г.; см. сборник, составленный
Э. Брицке, А. Капустинским, Б. Веселовским, Л. Шамовским,
Л. Ченцовой, Б. Анваером, Термические константы неорганических веществ.
Изд. АН СССР, 1949. (Прим, ред.)
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
227
Параметры уравнения (20)
Таблица 43
Соль t, °C 8° 1) - B' C' Ссылки на литературу
NaCl2) 25 477 1 532 1154 -0,2 1
15 393 1 411 335 +5,3 1
NaBr 25 477 1888 1 770 -1,5 2
NaNO3 25 ‘ 477 2 961 2 834 -4,8 3
NaC103 25 477 2 708 2 701 -3,1 3
NaBrO3 25 477 3 562 4 831 + 13,2 3
NaJOg 25 477 5 635 6 913 + 5,0 3
KC1 25 477 1893 2 144 -6,9 4
12,5 374 1569 273 +5,3 4
KBr 25 477 2 541 3 902 + 7,8 2
KNOg 25 477 5 006 6 079 -14,5 5
12,5 374 5 753 7 522 -6,4 5
KClOg 25 477 4 527 5 334 + 19,1 6
. 15 393 4 681 4 602 +4,1 6
KC1O4 25 477 7 492 10 510 -7,8 6
15 393 8 086 11 on +3,6 6
Li2S01 25 2 481 6 022 2 305 +2,7 7
Na2SO4 25 2 481 15 576 21482 -2,0 7
K2SO4 25 2 481 16 022 25 599 -3,3 7
Rb2SO4 . 25 2 481 17 896 31 381 +5,3 7
Cs2SO4 25 2 481 22 169 44 570 -10,0 7
!) 8° = (2/3)§(Я).
2) Параметры для NaCl относятся к моляльным концентрациям. Параметры для всех осталь-
ных солей относятся к концентрациям, выраженным в молях на литр раствора.
В случае концентрированных растворов наши значения обычно оказы-
ваются более высокими, чем значения, полученные1 с помощью первоначаль-
ной экстраполяции. Разности (Д) между прежними и новыми значениями
помещены в последнем столбце табл. 121, причем положительная величина Д
соответствует смещению кривых вверх, которое необходимо для того,
чтобы кривые, полученные по новому способу экстраполяции, проходили
через начало координат.
Из табл. 121 видно, что за исключением данных для хлористого лития
и 2,2-валентных солей, Д не превышает 7 кал. Значительное расхождение
1 Некоторые термодинамические величины, содержащиеся в таблицах, приводи-
мых в различных работах немецких авторов (Ланге и его сотрудников), нетрудно
привести в соответствие с системой обозначений, принятой в Америке и используе-
мой в этой главе, с помощью следующих соотношений
?L= Д7? д = —Рт>
т . . 55,51
1-2 — До — Дщ — Pm + Z7T- ?т>
Т/Ь
Lr~ ?т, i,2(s)==^'0 = До-
Здесь через —Lm обозначается суммарная (интегральная) теплота растворения, —Дт—
парциальная (дифференциальная) теплота растворения и Z,2(S)— относительное пар-
циальное теплосодержание твердого растворенного вещества. Более полное сопостав-
ление американских и немецких условных обозначений можно найти в книгах Льюи-
са и Рендалла [17а] и Шоттке [17б].
15*
228
ГЛАВА VIII
в случае хлористого лития объясняется отсутствием данных для концентра-
ций меньше 0,139 М. Применяемый нами способ экстраполяции основан
на близком сходстве кривых для умеренно концентрированных растворов
хлористого и бромистого лития, и хотя он гораздо менее надежен, чем
экстраполяции для других 1,1-валентных солей, его следует предпочесть
способу Ланге и Дюрра [18].
При экстраполяции данных по теплотам разведения для 2,2-валентных
солей получаются весьма ненадежные результаты, так как коэффициенты
наклона экспериментальных кривых при самых малых концентрациях
в 2—3 раза больше теоретических коэффициентов наклона. При попытке
придать экстраполированным кривым теоретический предельный наклон
приходится настолько увеличивать их кривизну1 при концентрациях,
лежащих за пределами экспериментальной области концентраций, что опре-
деление Д является в лучшем случае грубо приближенным. Кроме того,
экспериментальные данные для этих гидролизующихся солей нуждаются
в таких поправках, которые могут серьезно повлиять на результаты экстра-
поляции.
Дэльман п Ланге [20] указали, что происходящее при процессе разведе-
ния смещение ионного равновесия сопровождается заметными тепловыми
эффектами. В .случае разбавленных растворов нейтральных солей тепловой
эффект, связанный с изменением степени диссоциации воды, очень мал
(< 2 кал на моль соли), однако тепловые эффекты, вызываемые сдвигом
равновесия гидролиза или диссоциации таких солей, как, например, серно-
кислый цинк, неизвестны и могут оказаться весьма значительными. Для
полностью диссоциированного хлористого аммония поправка, связанная
с гидролизом, имеет тот же порядок величины, что и смещение (Д = 7) при
новой экстраполяции. Возможно, что тепловые эффекты, связанные с гидро-
лизом фторидов калия и рубидия, лишь частично компенсируются смещени-
ями Д, приведенными в табл. 121 для этих солей. В качестве примера край-
ней неопределенности, которая может встретиться при экстраполяции дан-
ных по теплотам разведения, зависящих от сдвига ионного равновесия,
в гл. XIII, § 13, приведены значения L2 для серной кислоты. В этом случае
смещение, необходимое для совмещения калориметрических значений L2
с'теоретическими, составляет около —450 кал. Значения L2 для серной кислоты
можно вычислять для различных температур с помощью параметров, приве-
денных в табл. 159.
Ланге и Робинзон [21] измеряли теплоты разведения хлористого калия
в водных растворах сахара и мочевины, чтобы выяснить, наблюдается ли при
изменении диэлектрической постоянной, обусловленном присутствием неэлек-
тролитов, тот эффект, который предсказывается теорией. Данные, получен-
ные этими исследователями, а также соответствующие теоретические пре-
дельные прямые представлены на рис. 52. При вычислении коэффициентов
наклона этих предельных прямых были использованы значения диэлектри-
ческих постоянных, полученные Вайменом для воды [22] и раствора моче-
вины [23]. Ваймен не опубликовал данных для растворов сахарозы, но из
результатов, полученных Акерлефом [24], можно заключить, что данные
Кокеля для этого случая согласуются с другими измерениями Ваймена.
Поэтому при вычислении теоретических коэффициентов наклона для рас-
твора сахара были использованы данные Кокеля [25]. Так как значение
dV/dT для растворов мочевины точно не установлено, то при вычислении
всех трех коэффициентов наклона не использовался член, в который входит
dV/dT. Из рис. 52 видно, что экспериментальные коэффициенты наклона
1 Экспериментальное доказательство такой кривизны приводится в расчетах Робин-
зона и Уоллеса [19].
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
229
примерно на 25% меньше теоретических, но этой простой закономерности
нельзя придавать серьезного значения, поскольку dD/dT определено недо-
статочно точно Г Отметим, что экспериментальные предельные коэффици-
енты наклона, найденные таким образом для растворов 1,1-валентных элек-
тролитов в чистой воде, обычно оказывались на 10—50% меньше теорети-
ческих. Поскольку с помощью метода графического дифференцирования
удалось добиться согласия между экспериментальными и теоретическими
величинами, этот метод, повидимому, можно распространить также на рас-
Р и с. 52. Относительное кажущееся
молярное теплосодержание КС1 в
сметанных растворителях при 25°.
Прямыми линиями изображены гра-
фики, соответствующие предельному
теоретическому закону.
творы сахара и мочевины.
Для разбавленных растворов некоторых неэлектролитов были опреде-
лены теплоты разведения и относительные парциальные молярные тепло-
содержания. Так как молекулы неэлек-
тролитов не заряжены, то в случае неэлек-
тролита в выражении для теплоты раз-
ведения должен исчезнуть член, содер-
жащий квадратный корень из концентра-
ции. В настоящее время мы не имеем точ-
ной теории, с помощью которой можно
было бы предсказать численные величины <f>L
коэффициентов при членах, содержащих
концентрацию в первой и более высоких
степенях. По этой причине калориметри-
ческие измерения в случае растворов не-
электролитов не привлекли к себе большо-
го внимания, однако для системы саха-
роза—вода имеются данные такой же сте-
пени точности, какая была достигнута в
случае растворов электролитов. Гукер,
Пикард и Планк 1 2 измерили теплоту разве-
дения очень разбавленных растворов саха-
розы в воде при 20 и 30°, а Ноде [27]
сделал подобные измерения при 18°. Дан-
ные для более высоких концентраций при-
водятся в работах Портера и Вуда3,
Пратта [29], а также Валлендера и Пер-
мена [30]. Гукер [26] экспериментальным
путем определил величины хорд ЬН/Ьт. и обработал полученные им резуль-
таты с помощью видоизмененного метода Юнга и Фогеля [11]. Он обна-
ружил, что между т = 0,002 и т = 0,2 в пределах точности опыта &Н/&т
имеет постоянное значение, причем средняя величина отклонения от сред-
него значения соответствует примерно 3 • 10~6 градуса. Предельные урав-
нения имеют следующий вид:
— &HD = — 128,9m; при 20°,
— ДЯд =<pL= 140,2m; при 30°.
(26)
(27)
1 При нагревании растворов мочевины о» 30 до 35° их электропроводность и диэлек-
трическая постоянная медленно и необратимо меняются; только путем очень быстрых
измерений Ваймену удалось получить воспроизводимые значения температурных коэф-
фициентов. Значение dD/dT, полученное ранее [25], в действительности соответствует
коэффициенту наклона противоположного знака по сравнению с коэффициентами наклона
кривых, изображенных на рис. 52.
2 См. также работу Гукера и Пикарда [26], посвященную исследованию растворов
мочевины.
3 Эти данные приведены в статье Портера [28].
230
ГЛАВА VIII
Для того чтобы из имеющихся данных для более высоких концентраций
получить значение оказалось необходимым дополнить аналитическое
выражение для членом, содержащим т2, однако коэффициент при линей-
ном члене (содержащем т) сохранял при этом практически то же значение,
что и при вычислении из данных, относящихся только к очень разбав-
ленным растворам. Уравнения (26) и (27) были дополнены членами, содер-
жащими т2, и, кромо того, были подобраны коэффициенты, позволяющие
удовлетворительно отразить все опытные данные. В окончательном виде
уравнения имеют следующий вид:
- = <pL= 128,9m — 6,917m2 при 20° (28)
вплоть до т = 5,9 и
- l\HD = <?L = 140,2m - 7,08m2
при 30°
(29)
Р[и’с. 53. Относительное кажущееся молярное
теплосодержание водных растворов сахарозы:
/—при 20 и 2—при 30°. Графин для 30° смещен вверх
га 200 кал.
вплоть до т = 2,2. На рис. 53 сплошными линиями представлены значения,
вычисленные Гукером, Пикардом и Планком [5] с помощью этих уравнений,
а точками изображены экспери-
ментальные данные. Пунктир-
ная кривая соответствует дан-
ным для 20°, причем по оси
абсцисс вместо т отложено с.
Такая замена единиц концентра-
ции мало отражается на кри-
визне в области концентриро-
ванных растворов.
§ 3. Теплота нейтрализации .
и теплота диссоциации
При рассмотрении вопроса
о теплоте нейтрализации различ-
ных кислот и оснований мы бу-
дем иметь дело'как с сильными,
так и со слабыми электролита-
ми, причем последним посвя-
щается § 6 гл. XV. Основное вни-
мание будет уделено определению предельных значений, к которым стремятся
теплоты нейтрализации при бесконечном разбавлении. Для случая теплоты
нейтрализации сильных кислот сильными основаниями таким предельным
значением является теплота образования воды из ее ионов или взятая с обрат-
ным знаком теплота диссоциации воды на ионы, определяемая по темпера-
турному коэффициенту константы диссоциации воды. Ниже будет показано,
как пользуются калориметрическими данными для вычисления теплоты
диссоциации воды, а также будут приведены наиболее надежные значения,
полученные этим способом. Эти последние значения будут сопоставлены
со значениями, полученными из данных по определению констант диссоциа-
ции методом электродвижу!цих сил. *
Реакция между сильной кислотой и сильным основанием, концентрации
которых равны и конечны, выражается уравнением
НВ • Н12О + МОН • яН2О = МВ • (2х + 1)Н2О, (30)
где х — 55,5/т. Это уравнение отвечает образованию одного моля воды из ее
ионов при исходной концентрации т — 55,5/х, а также разведению одного
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ 231
моля ионов М+ и одного моля ионов В- от концентрации т до
т' = 55,5/(2х + 1). Увеличение теплосодержания при этом, процессе соста-
вляет
— ?н (S) + ^н2О — 'Рн (а) ~ ?Н (ЬЦ
Нижние индексы обозначают соответственно соль, воду, кислоту и осно-
вание, а штрих над указывает на то, что концентрация соли равна
т'. Если мы напишем такое же уравнение для бесконечно разбавленного
раствора и вычтем его из уравнения (31), то получится выражение
дя„ = - ДЯЬ (S) + ДЯц (а)+ДЯц (Ь). (32)
Так как при бесконечном разбавлении ионы соли не влияют на ионы
водорода и гидроксила, то можно написать
-ДЯ°=Д/7°, (33)
где Д/71—теплота, диссоциации воды па ионы. Эту величину легко полу-
чить из значения теплоты нейтрализации, если известны соответствующие
теплоты разНедения. Ричардс и Холл .[31] нашли, что для реакции между
НС1 100 Н2О и NaOH-100H2O при 20° п = — 13 924. Подставляя значения
теплот разведения, которые они считают наиболее надежными, в уравне-
ние (32), Ричардс и Холл получили — 13 924 =—Д#? + 15 — 266 — 20,
откуда Д/£? = 13 653. Россини [32] произвел тщательный пересчет 52 зна-
чений теплот нейтрализации, полученных Ричардсом и Роу [33], и 7 зна-
чений, найденных Джиллеспай, Ламбертом и Гибсоном [34]. Сочетая их со
значениями Д77ц и Ср , которые он получил путем критического изучения
всех имеющихся данных [35], Россини нашел, что «наиболее точное» зна-
чение Д/ft при 18° составляет 13 721. Среднее отклонение от этого значе-
ния не превышает 20 кал. Согласно Россини, при 20° ДЯ? равно 13 606.
Такое совпадение результатов, полученных из большого числа опытов
с разными реагирующими веществами, является убедительным доказа-
тельством точности этих результатов. Однако некоторая неопределенность
вносится в расчеты из-за использования индивидуальных теплот разведения,
для вычисления каждой из которых необходима экстраполяция на нулевую
концентрацию. Данные, на которых основаны наши вычисления ДЯС для
кислот и оснований, либо относятся к концентрациям выше 0,1 М, либо,
за исключением нескольких отдельных случаев, являются настолько не-
точными при малых концентрациях, что их нельзя использовать для
надежной экстраполяции.
Весьма разбавленные растворы серной кислоты были изучены при 25°
с помощью калориметрических методов Ланге, Монгеймом и Робинзоном
[36]. Данные этих исследователей хорошо согласуются с данными измерений
Харнеда и Гамера [37] по методу электродвижущих сил. Соляная кислота
[38] и гидрат окиси натрия [39] были изучены с помощью метода электро-
движущих сил в широком интервале температур.
можно вычислять из упомянутых выше данных Ричардса и Роу
по теплотам нейтрализации с помощью метода, при котором используются
только непосредственно измеряемые промежуточные значения теплот разве-
дения и производится лишь одна экстраполяция на нулевую
концентрацию. Если значение теплоты разведения, соответствующее
предельному закону [уравнение (12)], подставить в уравнение (32),
а также учесть, что в разбавленном растворе т’= 0,5 т, то получается
232
ГЛАВА VIII
следующее выражение:
ЬНп = -ДЙ°- (2—[/6)5) [(2/3) |/ d„S(H)] \/т. (34)
Таким образом, располагая рядом экспериментальных {иди вычисленных)
значений &Нп для различных концентраций, можно построить график
зависимости от ]/т, который в области очень малых т должен иметь
прямолинейный характер, причем отрезок оси ординат должен равняться
— Д//?. Можно уменьшить наклон этой кривой и несколько изменить ее кри-
визну, пользуясь при построении графика экспериментальным значением
промежуточной теплоты разведения раствора соли от концентрации т до т'.
Ламберт и Джиллеспай [40] вычислили значения Д/£п—A//d<s)для
5 концентраций и построили, изображенные на рис. 54, графики зависи-
мости этих значений от ]/ т. Кривизна полученных ими графиков не-
велика, и они приближаются к общей касательной, которая определяется
уравнением
ДЯ£==[ДЯ„ — ДЯц(8)(т-ип’)] = — Д#?~ [(2/3)]AZ0S(H)J Yт. (35)
Через ДЯ* обозначено выражение, заключенное в скобки, для которого
Ламберт и Джиллеспай предло-
жили название «теплота нейтра-
лизации при постоянной концен-
трации». По величине отрезка,
отсекаемого на оси ординат
кривыми, изображенными на
рис. 54, они определили значение
ДЯ? = 13 650 при 20°. Это значе-
ние на 44 кал больше величины,
полученной из тех же данных с
помощью экстраполяции теплот
разведения, и на 42 кал меньше
величины, полученной из данных
по электродвижущим силам
(гл. XV, § 3). Индивидуальные
особенности кривых сохраняют-
ся при концентрациях, меньших
чем 0,1 М, в соответствии с ха-
рактером изменения теплот раз-
ведения .
Теплота диссоциации чистой воды равна — ДЯ^ при бесконечном раз-
бавлении, но для конечных концентраций — ДЯ)[ не имеет ясного физиче-
ского смысла, так как при замене выражения фщв) + ?Н(М)- — срн<в) — ?н<м>
через ДЯц(8)(т_т') молчаливо допускается, что <рн(МВ) в нейтральном рас-
творе соли равно сумме членов фщм» и ?Н(В), относящихся соответственно
к растворам основания и кислоты. Такое же допущение делается и в отно-
шении срц(Н) и 'рн(он). Мы лишены возможности проверить справедливость
этого предположения, поскольку термодинамическим путем теплосодержания
отдельных видов ионов не могут быть определены.
§ 4. Парциальная молярная теплоемкость при постоянном давлении
Основные уравнения, связывающие общую теплоемкость Ср бинарного
раствора с парциальными молярными теплоемкостями обоих компонентов Ср
п СР2, а также с молярной теплоемкостью чистого растворителя и кажу-
/6000
15000
4НП*
14000
1SOOO
0 0,4 0,8 1,2 1,6
Vrn
Р и с. 54. Экстраполяция теплот нейтрализации
растворов с постоянной концентрацией для опре-
деления теплоты диссоциации воды при 20°.
Для каждой кривой указана соль, образующаяся
при нейтрализации.
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
233
щейся молярной теплоемкостью растворенного вещества, аналогичны
тем уравнениям, которые были рассмотрены в § 2 и могут быть получены
из этих уравнений дифференцированием по температуре при постоянном
давлении. Таким образом, согласно уравнениям (1) и (2), для теплоемкости
раствора получаются выражения
Ср — П1СР1 + n2CVi, (36)
Ср = п1С°р -\-п2ус . ' (37)
р
Очевидно, что при бесконечном разбавлении
(38)
Зависимость между соответствующими относительными величинами выра-
жается уравнением
Ср — С* = п1 (С₽1 — ) + п2 (С₽2 — ) = п2 (срс — срО ), (39)
1 2 р р
которое получается из уравнений (36) и (37) или путем дифференцирова-
ния уравнения (2). Абсолютные значения Ср, Ср и Ср2 могут быть опре-
делены из имеющихся данных по удельным теплоемкостям, а из данных
об изменении активности с температурой можно получить только относи-
тельные значения этих величин. Для этих величин мы используем следу-
ющие обозначения: J= Ср — С%; = Ср —С° ;J2 = CP —С° и®г = срг —
Р 1 Рр 2 Р2 ‘ J Ь? Ьр •
С помощью этих обозначений уравнение (39) может быть представлено
в виде:
J = nxJl +n2J — n^j. (40)
J, J\ и J2 представляю!' собой частные производные (dL / дТ)р, (дЬ±/дТ^
и (дЬ2/ дТ)р соответствующих относительных теплосодержаний [урав-
нение (40) гл. 1]. Определение этих величин из относительных моляр-
ных теплосодержаний является настолько простым, что не нуждается
в пояснении, на определении же абсолютных значений Ср , СРп и <рс сле-
дует остановиться.
Согласно уравнению (37), кажущаяся молярная теплоемкость раство-
ренного вехцества равна
_ (1000 + тМ2) ср -1000.^
т ’ ( )
где т__моляльность растворенного вещества, молекулярный вес которого
обозначен через М2, а ср и ср— удельные теплоемкости раствора и чистого
растворителя.
Вычисление Ср и Ср из <?с настолько похоже на вычисления, при-
веденные в § 2, что мы ограничимся только перечислением основных
уравнений:
' д*ср д,?СР
СР=^р + п^ = ^ср^т-^’ (42)
ИЛИ
= ?ср
, т 3?Ср
2 д j/zn
(43)
234
ГЛАВА VIII
<1
/С] !'С'р гп- 1 Ср
nL дп2 1000/Л/г дт ’
(44)
ПЛИ
Ji —
т Ут ‘Ср
~Ж^М1
(45)
Поскольку в выражении для <рс
как и для других кажущихся моляр-
р
ных величин, часто предпочитают употреблять с вместо т, то можно
пользоваться следующими уравнениями [41]:
и
7 г 1000-с?у -|
р2 tgp ' L 2000 + с3/25?г/5/с J 3/с
= г° _ М± Г_________?_______ 1 1/о ^Ср
! d0 12000 + с^д^/дУс J а/г’
(46)
(47)
Эти уравнения можно применять для. определения L2 и других парциаль-
ных молярных величин. Дифференциальные коэффициенты обычно опреде-
ляются графически с помощью графиков зависимости <рс от т (для неэлек-
тролитов) или от рт (для электролитов). В последнем случае предельные
коэффициенты наклона кривых получают из уравнения (17) гл. V, которое
для 1,1-валентных электролитов имеет следующий вид:
/2 = S<cp) / с = S<Cp) Vdom.
(48)
Сочетая это уравнение с уравнениями (43) и (38), получаем после инте-
грирования и последующего преобразования
= 2/3 S(Cp) l/pm = 2/3 SCp Г с. (49)
Для точного определения удельных теплоемкостей растворов пользу-
ются методом «двойникового» калориметра Джоуля [42] и Пфаундлера [43],
значительно усовершенстванным в дальнейшем другими исследователями 2 *.
Этот метод основан ва тепловом уравновешивании. Два практически одинако-
вых калориметра, снабженные одинаковыми электрическими нагреватель-
ными элементами, погружают в термостатированную оболочку. Один кало-
риметр содержит воду, другой—исследуемый раствор. Веса воды и раствора
подобраны таким образом, чтобы прохождение тока через соединенные
последовательно нагревательные элементы вызывало одинаковый подъем
температуры в обоих калориметрах. В то же время температуру оболочки
осторожно повышают, так что теплоотдача через стенки калориметра
практически не 'происходит, и таким образом процесс является
адиабатическим.
Для вычисления ср раствора из данных таких опытов необходимо знать
точные значения отношения сопротивлений нагревательных элементов
и водяного числа обеих калориметрических систем. Отношение сопротив-
1 Библиографию по этому вопросу, содержащую более пятидесяти названий,
можно найти в статье Ричардса [36]. Ссылки на более поздние работы приводятся
в тексте данного параграфа.
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
235
лений можно определить очень точно \ однако найти с достаточной сте-
пенью точности значения водяного числа калориметрических систем нелегко.
Это затруднение можно устранить, если один из калориметров заполнять
всегда одним и тем же количеством воды и определять .каждый раз веса
воды и раствора, при которых в другом (рабочем) калориметре происходит
такое же повышение температуры, как и в первом. Если абсолютное зна-
чение удельной теплоемкости должно быть определено с высокой степенью
точности, необходимо пользоваться водой [44], не содержащей воздуха.
Весь процесс обработки экспериментальных данных, включающий некоторые
небольшие поправки, подробно рассмотрен Рендаллом и Россини [45].
Устройство двойникового калориметра позволяет производить определе-
ния удельных теплоемкостей с точностью до 0,01% и более высокой, однако
значения и Ср , которые можно вычислить из этих данных для очень
разбавленных растворов, являются весьма неточными. Применение урав-
нения (41) связано с неизбежным увеличением ошибок, появляющихся при
экспериментальном определении теплоемкости, что легко показать, продиф-
ференцировав это уравнение и представив его в следующей форме:
. Г 1000 . о ,1 8m п
c4=L~^(Cp“Cp)J^ (50)
и
• . Г 1000 о , I 8с„ _ .
<51>
Из этих уравнений видно, что срс значительно более чувствительно
к ошибке в определении ср, чем к ошибке в определении т. Гукер
и Шминке [4о] прказали, что член (ср — ср) имеет почти постоянное зна-
чение для большого числа водных растворов и равен приблизительно 50.
Поэтому ошибка в 0,1% при определении т мевяет величину <рс всего на
0,05 кал/град. С другой стороны, выражение в скобках в уравнении (51)
по порядку величины равно 1000/лг, откуда следует, что ошибка всего
в 0,01% при определении ср приводит к ошибке в 10 кал/град в величине
<рс при т = 0,01. Поэтому с помощью существующих в настоящее время
методов практически бесполезно производить измерения теплоёмкости при
концентрациях порядка менее чем 0,01 М.
Один из практических способов, с помощью которого можно устранить
эту трудность, состоит в сочетании Ср^ с соответствующими значениями J2,
полученными из температурного коэффициента L2 или из второй произ-
водной коэффициентов активности по температуре. Так как
^'р2 = ^р2 ^2 (52)
не должно зависеть от т, то можно ограничиться вычислениями, относя-
щимися к области концентраций, для которой обе группы данных являются
наиболее достоверными. В табл. 44 приведены результаты подобных вычи-
слений для некоторых электролитов, для которых получаются постоянные
значения С°2. Как видно из таблицы, точность определения Ср2 для первых
1 Гукер, Айрес и Рубин [416] применили нагревательные элементы с переменными
сопротивлениями, причем отношение сопротивлений этих элементов можно было точно
измерить во время протекания опыта. Это усовершенствование имеет важное значение
для регулировки системы, благодаря улучшению которой достигается одинаковое
повышение температуры в обоих калориметрах.
236
ГЛАВА VIII
Таблица 44
Значения С® , вычисленные по уравнению (52) при 25э
т на [1,2] КС1 [3,4] КОН [5,2] NaOH [6,7]
0,2 -29,50 -28,30 —
0,25 — — -30,3 -28,08
0,5 . -29,15 -28,75 -30,2 -27,74
1,0 -29,25 -29,25 -30,62 -28,37
1,5 -29,17 — 28,78 -30,85 -27,52
2,0 -29,75 -30,05 -31,36 -26,24
2,5 — -28,45 —. -26,30
Среднее -29,4 -28,8 -30,7 -27,4
трех электролитов составляет примерно 0,5 кал, а для гидроокиси натрия —
около 1 кал. Следует отметить, что при попытках [47] теоретически опре-
делить Ср2 для электролитов на основании данных по электрострикции [48]
растворителя были получены правильные по порядку величины значе-
ния Ср2, однако при этом не нашли своего отражения наблюдаемые на
опыте специфические различия между данными для разных электролитов.
Следует указать на тот экспериментальный факт, что Ср2 и многие
другие парциальные молярные величины, характеризующие сильные элек-
тролиты, линейно зависят от с1/2 и от т1^ в интервале концентраций
(0,2 — ЗМ), значительно выходящих за пределы той области концентраций,
в которой применимы предельные законы. Благодаря этому можно выра-
зить экспериментальные результаты через параметры уравнения
Cv=C^Scm^. (53)
Экспериментальные значения коэффициентов наклона SCp могут значительно
отличаться от теоретических §(Ср). Для электролитов, представленных в
табл. 45, это уравнение в пределах ошибок опыта правильно отражает опыт-
ные значения Ср2 в области концентраций от т = 0,2 до т = 3. Так как
параметры этого уравнения вычисляются из данных, относящихся к не
очень разбавленным растворам, то численные значения коэффициента накло-
на и отрезка, отсекаемого соответствующей кривой на оси ординат, могут
отличаться от истинных значений Ср2 теоретического предельного коэффи-
циента наклона. Для отрезка, отсекаемого на оси ординат, эта разница
обычно невелика, как видно из сравнения значений С®2, приведенных
в табл. 44 и 45. С другой стороны, экспериментальные значения коэффи-
циентов наклона Sc для тех же 1, 1-валентных электролитов сильно отли-
чаются как друг от друга, так и от теоретического предельного коэффи-
циента наклона S(c >, который при 25° составляет приблизительно 13,36.
Эти отличия не противоречат теории, так как из определений J2 из
дЬ21дТ и дЧиуч /дЗ"2 видно, что SCp сильно меняется с концентрацией
при значениях с, меныпих чем с = 0,2, и приближается к предсказываемой
теорией предельной величине лишь при очень больших разбавлениях. Это
убедительно показано вычислениями Юнга и Махина [49] и иллюстри-
руется рис. 85 и 92.
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
237
Опубликовано лишь несколько работ, содержащих точные данные
калориметрического исследования теплоемкости электролитов с более высо-
ким типом валентности. Эти данные в общем не подчиняются простому
линейному соотношению, которое выражается уравнением (53). В табл. 46
приведены усредненные значения Ср , а также величины отрезков, отсека-
емых соответствующими кривыми на оси ординат, и кажущиеся предель-
ные коэффициенты наклона, полученные из экспериментальных данных
для очень разбавленных растворов. Данные для хлористого бария взяты
из двух разных источников; эти данные хорошо описываются уравне-
нием (53).
Таблица 45
Параметры интерполяционного уравнения ср2=Ср/6’ср У т.
Применимо при 25° в интервале концентраций от 0,2 до 3 М
Электролит!) Sp и p — 0 cp2 Электролит!) So ' p — 0 CP2
НС1 [1] 7,50 -29,20 NaNO 2> 30,0 -12,6
LiCl [1] 7,88 -15,63
NaCl2) 21,6 -23,8 KNO 0 3 28,65 -17,8
КС12) 16,8 -29,0 NH4NO3 [3] 12,1 -2,5
NaBr2) . 20,4 -24,3 LiOH [4] 18,80 -19,98
KBr2) 16,2 -29,5
NaJ2) 24,9 -25,0 NaOH [4] 26,24 — 26,59
KJ2) 20,4 -30,2 KOH [1] 19,88 -32,10
1) Такая же таблицаТдля этих и некоторых других электролитов при 18, 21,5 и 25° приво
дится в работах Россини [2а] и Рендалла и Россини [26].
2) Пересчитано Россини [2а] с помощью данных Рендалла п Россини [26]. См. также [2в].
Таблица 46
Ср2 для высоковалентных электролитов при 25°
m NasSOi [1] K2SO4 [1] BaCU [2]
0 (-50) (-60,6) (-74,36)
0,1 -22,6 -41,0 -57,5
0,2 -8,5 -31,8 -50,5
0,35 5,8 -21,4 -42,9
0,5 15,8 -13,4 -36,7
1,0 39,0 -— -21,1
1,5 . 66,0 — —
Коэффициент
наклона 76 62 54
Отчетливые различия между значениями SCp (табл. 45) для ряда
электролитов с одинаковым типом валентности видны из рис. 55, взятого
из работы Гукера и Рубина [50]. Эти авторы вычислили разность между
изопиестической и изохорической кажущимися молярными теплоемкостями
с помощью уравнения
YCp
[72
±-VT~^-v0T,
2 Pi)
(54)
238
ГЛАВА VIII
в котором аир представляют собой коэффициенты расширяемости и сжи-
маемости. Величины с нулевым индексом относятся к чистому раствори-
телю. Данные Гукера и Рубина приведены в табл. 47. Предельные зна-
чения были получены с помощью уравнения
ИДй] • <55>
где ф"е и — предельные значения кажущейся молярной расширяемости
и кажущейся молярной сжимаемости. Для с—»0 Гукер и Рубин вывели
уравнение
д'?Ср _ а0Т Г £ д^Е_ «о ' ^К_ 1
д У с д У~с '?о L д Vс ?о д Ус 1 ' '
Таблица 47
Значения ?Ср —при 25°
кг Раство- репное вещество 0 0 ,25 0,50 0,75 1 ,00 1,25 1 50
НС1 2,90 2,91 2,71 2,85 3,01 3,10 3,23
LiCl ........ 2,94 2,74 2,70 2,57 2,49 2,41 2,31
NaCl 8,60 8,33 8,14 8,11 8,25 8,48 8,71
КС1 . 7,82 7,32 7,16 6,98 6,89 6,82 6,72
LiOH 5,36 4,92 4,71 4,44 4,23 3,99 3,69
NaOH 11,36 10,66 10,59 10,63 10,78 10,98 11,12
Таблица 48
/ cv су\
Наблюдаемые значения ( — —— ) при 25°
<дУс дусУс^0
HOI LiCI NaCl KCI LiOH NaOH
—0,081 —0,72 —1,94 —2,04 —1,82 —3,27
с помощью которого можно вычислить разность между предельными коэф-
фициентами наклонов кривых для изопиестической и изохорической
систем. Как видно из табл. 48, значения этих разностей для различных
электролитов сильно отличаются друг от друга. Судя по абсолютным вели-
чинам и знакам этих разностей, специфический характер значений предель-
ных коэффициентов наклона кривых для изохорической системы выражен
еще более отчетливо, чем в случае изобарической системы. Однако в об-
ласти концентраций, применявшихся в этих исследованиях, более отчетливо
выражается специфический характер коэффициентов наклона для изопие-
стической системы. Наиболее отчетливо различие между обеими системами
проявляется в том, что <fc всегда имеет более отрицательное значение,
чем <рСр, и что разброс отдельных значений ус значительно больше, чем
в случае срСр. Обе эти особенности ясно видны при сравнении рис. 55 и 56.
Представляет интерес рассмотрение поведения растворов типичных
неэлектролитов путем их сопоставления с растворами электролитов. С этой
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
239
целью на рис. 57 приводится график <рСр для сахарозы, взятый из работы
Гукера и Эйреса [51], который отражает наиболее точную серию измере-
ний, произведенных в широком интервале концентраций. Предельный
коэффициент наклона кривой, изображающей зависимость <рСр от Ус, неви-
димому, равен нулю. Это соответствует теории, согласно которой член,
Рис. 55. Кажущиеся молярные
теплоемкости водных растворов при
постоянном давлении и 25° (изопие-
стические кривые).
Рис. 56. Кажущиеся молярные
теплоемкости водных растворов при
постоянном объеме и 25° (изохоры).
содержащий у с, выражает взаимодействие ионов. График зависимости срс
от с имеет в пределах ошибок опыта линейный характер вплоть до с = 14
Эти закономерности в основном сохраняются, если выбрать в качестве
независимой переменной вместо с мо-
лярную долю или моляльность. Однако
при этом знак кривизны меняется на
обратный, а ее абсолютная величина
возрастает примерно на 50%. Для удоб-
ства вычисления CPi и Ср результаты
были выражены в виде функции от т
следующим уравнением:
?ср = 145,87 + 1,950m — 0,1182m2 (57)
для 20° и
= 151,20 + 1,325m — 0,0704m2 (58)
Рис. 57. Зависимость кажущейся мо-
лярной теплоемкости сахарозы при 25.
от с/2 (верхняя кривая) и от Ус (ниж-
няя кривая).
для 25°. Коэффициент линейного члена мал и уменьшается с ростом тем-
пературы так быстро, что примерно при 40° <рСр для разбавленных раство-
ров почти перестает зависеть от концентрации. В этом отношении сахароза
ведет себя, как идеальное растворенное вещество. Этот вопрос довольно.
1 На подобных же кривых для растворов мочевины не обнаруживается такого,
ясного различия между зависимостями от с или от У с [52].
40
ГЛАВА VIII
подробно разобран Гукером и Эйресом и в особенности Бенневитцем
и Кратцем [53].
Предельные коэффициенты наклона (<9<?Ср / dm)c 0 были вычислены
с помощью уравнений (57) и (58) на основании экспериментальных данных,
полученных для концентраций, превышающих 0,04 М. Тем не менее зна-
чение коэффициента наклона при 25° весьма близко к значению, вычис-
ленному из температурного коэффициента теплот разведения при более
низких концентрациях. Согласно уравнениям (28) и (29), для срв при 20
и 30°
(59)
дт дТ \ дт /10 '
Гукер, Пикард и Планк [5] считают этот способ определения предельного
коэффициента наклона более точным, чем предыдущий; они использовали
полученное таким образом значение предельного коэффициента наклона
для пересчета других параметров уравнения (58), найденных из данных
по удельным теплоемкостям. В окончательной форме их уравнение имеет
следующий вид:
?Ср = 151,50 4- 1,130m - 0,0466m2. . (60)
С помощью этого уравнения экспериментальные значения удельных тепло-
емкостей могут быть выражены в среднем с точностью в 0,005%.
§ 5. Парциальные молярные объемы, кажущиеся молярные объемы
и плотности растворов электролитов
Парциальные молярные объемы и кажущиеся молярные объемы опре-
деляются из значений плотностей растворов. Кажущийся молярный объем
равен в общем случае
или
1000 . j ,. /С9\
= + (62)
если и2 равно с, т. е. числу молей растворенного вещества в 1000 с.и3
раствора. Аналогичным образом
1000 ., 7, , м, /eQ4
^v'~^dd0 (rfo-c?) + -F, (63)
если га2 равно т, т. е. числу молей растворенного вещества в 1000 г рас-
творителя.
Мэзон1 [54а] нашел, что в случае разбавленных растворов ли-
нейно зависит от ]4 с и что эта простая зависимость часто бывает справе-
длива и для концентрированных растворов. Таким образом, если
^v^^v + SvVc (64)
в интервале концентраций, для которого известны плотности растворов,
то последние можно выразить уравнением
, > । (-1^2 — «о?у) с (Syd^c1-
d = d. +----------------Шбб— ’ <65>
1 См. также [546].
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
241
которое получается из уравнений (63) и (64). Это уравнение было выведено
Рутом [55]. Согласно теории, коэффициент ^ydo/lOOO должен быть оди-
наковым для растворов всех сильных электролитов определенного типа
валентности в данном растворителе, а коэффициент при члене, содержа-
щем с в первой степени, должен быть аддитивным свойством ионов.
Однако экспериментальные значения Sy в пределах группы сходных
электролитов весьма различны для растворов разных солей и лишь
по порядку величины равны значению теоретического коэффициента
наклона [уравнение (91) гл. III]. По данным Рута, оба эмпирических
коэффициента аддитивны.
Вирт [56] определил кажущиеся (и парциальные) молярные объемы
хлористого, бромистого и сернокислого калия в растворах хлористого
натрия и нашел, что уравнение (64) описывает экспериментальные резуль-
таты в случае смесей 1,1-валентных солей вплоть до концентрации 1 н.,
если с представляет собой общую концентрацию растворенного вещества.
Опытные данные, полученные для сернокислого калия, также можно опи-
сать с помощью этого уравнения, если заменить с1/з на Г1/2. Это согла-
суется с теоретическим уравнением (91) гл. III, поскольку в качестве пере-
менной взято Г1/3.'
Обычные ошибки опыта, с которыми связано определение концентрации,
не оказывают серьезного влияния на <ьу, но эта величина очень чувстви-
тельна к неточностям при экспериментальном определении плотностей очень
разбавленных растворов. Дифференцируя уравнение (62) при постоянном d
и используя уравнение (65), получаем выражение
'о J С
(66)
справедливое для разбавленных растворов. Величина, заключенная в скобки,
не зависит от с и лежит между М2 и г/2М2. При постоянном с соответ-
ствующее уравнение имеет вид
так что ошибка в 0,001% при определении d вызывает ошибку в 1 см3
в при с = 0,01.
Так как уравнение (64) является линейным, то удобнее всего вычис-
лять парциальные молярные объемы, пользуясь концентрациями с, выра-
женными в молях на 1000 см3 раствора, причем получаются следующие
уравнения [57, 59]:
_ / 1000—ccpv \ dvv
У2 = ФУТ --------~— с1/2—(68)]
Tv ( 2000 + с /з<9?у/<9 Ус J дУс
II
у = / 2000 7? \
1 \2000 + c8/35?v/5 /ё у ’
которые можно вывести из более знакомых уравнений:
(69)
У 2 — фу +
modify
20 У т
V^VI-
М, ,,
2000 дут
(70)
(71)
1 Если в выражении, заключенном в скобки, пренебречь членами, содержащими
концентрацию, то при с=1 для большинства 1,1- и 2,1-валентных электролитов обу-
словленная этим ошибка составляет менее 0,1 смя.
16 Г. Харнед, Б. Оуэн
242
ГЛАВА VIII
с помощью соотношения
(72)
А-z? Г1—С?У I .
т — «о [ 1000 j
Уравнение (72) получается из уравнения (62).
В табл. 49 содержатся значения параметров Sv и ф®,, полученные
различными исследователями на основании данных для плотности при 25°.
Для всех щелочных галогенидов, за исключением фтористого калия, эти
значения были определены Скоттом [60] по данным Бакстера и Уоллеса [61].
Гефкен [57] сочетал эти данные со своими собственными и с данными,
взятыми из некоторых других источников [62 — 65]. Значения, полученные
Скоттом и Гефкеном, почти не отличаются друг от друга. Мы пользуемся
там, где это возможно, значениями Скотта, так как с их помощью вычис-
лены некоторые производные величины, которые будут приведены ниже
(табл. 50). Оба автора составили таблицы значений Sv и ср®, для 0, 25,
50 и 70°, причем Гефкен включил также некоторые значения для 35 и 45°.
Для 25° имеются также данные других исследователей [66].
Таблица 491)
Значения параметров уравнения (64) для водных растворов при 25°
Электролит ?^(=v®) Sy Электролит 4< = V2) Sy
LiCl [1] 17,00 1,488 HCI [2] 18,20 0,83
(17,06) [2] (18,07) [7]
LiBr [1] 24,08 1,159 LiOH [9] —6,0 3,00
LiJ [1] 35,50 0,841 NaOH [9] —6,7 4.18
NaCl [1] 16,40 2,153 KOH [9] 2,9 4,35
(16,61) [4] NH4C1 [10] .... 35,98 1,45 2)
NaBr [1] 23,51 1,760 NH4NO3 [8] . . . . 47,56 0,97
(23,48) [3] (47,24) [3]
NaJ [1] 35,10 1,346 AgNO3 [12] .... 28,01 2,61 2)
KF [2] 6,60 3,35 KN03 [5] 38,18 2,30
KCI [1] 26,52 2,327 K2CrO4 [12] .... 37,13 11,70
(26,81) [3] K2SO4 [6] 32,28 18,11
KBr [1] 33,73 1,939 Na2SO4 [3] 11,47 12,16
KJ [1] 45,36 1,556 . (11,52) [3]
RbCl [1] 31,87 2,219 Na2CO3 [3] —6,70 11,30
RbBr [1] 38,71 2,038 (-6,74) [3]
RbJ [1] 50,31 1,607 CaCl2 [9] 18,25 5,99
CsCl [1] 39,15 2,172 SrCl2 [4] 17,94 9,902)
CsBr [1] 46,19 1,901 BaCla [9] 23,60 4,83
?CsJ [И • 57,74 1,579 LaCl3 [11] 16,02 11,87
1) В тех случаях, когда для приведены два значения, в скобках содержится наиболее точ-
ное значение величины <ру' вычисленное из данных для разбавленных растворов. Эти значения
не были использованы для вычисления Sy, и поэтому их не Следует применять для определения
с помощью уравнения (64) прп больших концентрациях.
2) Вычислено для разбавленных растворов, с < 0,5.
Из табл. 49 видно, что разность между Sv и ср®, для любой пары
1.1-валентных солей с общим ионом фактически не зависит от природы
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ 243
этого общего иона. Такая аддитивность ср®. сохраняется, хотя и с меньшей
точностью, для солей с более высоким типом валентности. Скотт [60]
показал, что среднее отклонение от аддитивности, полученное для всех
возможных сочетаний щелочных галогенидов, обладающих общим ионом,
составляет около 0,07 см3. Из данных Гефкена получается примерно то же
значение среднего отклонения, однако соответствующее значение,. получен-
ное из тех же данных Ла-Мером и Гронволлом [67], примерно в 2 раза
больше. Впоследствии некоторые исследователи пользовались вместо урав-
нения (64) степенной функцией, содержащей т в третьей степени. Такая
более сложная обработка, невидимому, приводит к преувеличенным зна-
чениям отклонений. Во всяком случае, аддитивность значений ср®, подтвер-
ждается с точностью, которая представляется удивительной, если учесть,
что все экстраполяции производятся для относительно концентрированных
растворов (с >0,25). Этот вопрос будет рассмотрен в дальнейшем в связи
с приблизительной аддитивностью Sy.
Точное значение теоретического предельного коэффициента наклона
неизвестно из-за некоторой неопределенности экспериментального значе-
ния dD/dP, которое входит в уравнение (87) гл. III. Между эксперимен-
тальными значениями коэффициентов наклона Sy для разных электроли-
тов, найденными из данных для концентрации выше 0,25 н., наблюдаются
значительные расхождения. Таким образом, эти значения непригодны для
экспериментальной проверки предельного закона, для которой необхо-
димы данные прецизионных измерений плотностей очень разбавленных
растворов. Для того чтобы избежать увеличения экспериментальной ошибки,
связанной с применением уравнения (67), Гефкен, Бекман и Круис (68)
разработали весьма точный дифференциальный метод поплавка для опреде-
ления относительных плотностей. Этот метод является усовершенствованием
способа Лэмба и Ли [69] и других исследователей [70, 71], в котором
потеря в весе поплавка, снабженного железным сердечником и погружен-
ного в раствор, осторожно уравновешивается с помощью соленоида, в поле
которого втягивается поплавок. Дифференциальный метод заключается
в применении двух поплавков в одном и том же термостате, благодаря
чему ошибки, обусловленные колебаниями температуры, уменьшаются с по-
мощью одновременного изменения плотности раствора и чистого раствори-
теля. Следует отметить, что современные очень точные пикнометрические
методы позволяют получать надежные значения <ру для разбавленных
растворов *. Гефкен и Прайс [73] собрали и проанализировали наиболее
точные данные, взятые из различных источников, и установили, что в слу-
чае хлоридов калия и натрия и бромида натрия коэффициенты наклона
в очень разбавленных растворах стремятся к одному и тому же предель-
ному значению. Это значение согласуется с теорией и равняется 1,9 [74].
Эти исследователи показали с помощью графического метода, что для
очень разбавленных растворов разность (<ру — 1,9с1/2) линейно зависит
от с. На рис. 58 изображены графики зависимости величины (сру— 2/35(у)с’/2)
от концентрации для нескольких солей. Из этого рисунка видно, что дан-
ные, полученные с помощью дифференциального метода поплавка, прекрасно
согласуются с результатами, полученными посредством остроумного дила-
тометрического метода, примененного Круисом [75], а также Гефкеном,
Круисом и Солана [76]. Для солей, представленных на рис. 58, опытные
данные удовлетворительно согласуются с теорией, чего нельзя сказать
1 Для определения изотопного состава воды было разработано несколько очень чув-
ствительных методов определения малых различий в плотностях. Насколько нам известно,
эти методы еще не применялись для определения сру при больших разбавлениях.
См. [72];
16*
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
245"
не только на гидролиз, но и на ассоциацию ионов. Гефкен и
Прайс, а также другие авторы [77] показали, что для слабых кислот Уу.
можно количественно выразить через
станты диссоциации.
На рис. 59 изображена графиче-
ски зависимость от Ус для хлори-
стого стронция; ясно виден S-образ-
ный характер кривой, имеющий место
при переходе от предельного закона,
справедливого при больших разбавле-
ниях, к уравнению Мэзона (64), которое
отвечает более концентрированным рас-
творам. Заметные отклонения от уравне-
ния Мэзона появляются, невидимому,
при столь малых концентрациях, что
вопрос о наличии данных для крайних
разбавлений не имеет большого значе-
ния. Более удовлетворительные способы
экстраполяции, в которых используется
параметр а и член, содержащий с в пер-
вой степени, разбираются в приложе-
нии Б и иллюстрируются рис. 169.
До настоящего времени не было
предложено удовлетворительного объ-
яснения справедливости уравнения (64),
однако имеется ряд интересных сооб-
ражений, относящихся к этому вопро-
су. Мэзон (54а) предположил, что если
бы графики зависимости ср от ейа
оставались линейными для пересыщен-
известные для этих кислот кон-
Видны отклонения от теоретического пре-
дельного закона (показан прямой линией)
при умеренных разведениях. Белые круж-
ки—дилатометрический метод; черные-
кружки—метод дифференциальных поплав-
ков.
ных растворов, то при значении с, равном концентрации электролита в чистой
кристаллической соли, достигалось бы гипотетическое максимальное значе-
ние сру. Обозначая величины, соответствующие этому максимуму, с помощью
индекса «макс.», мы получаем
?^макс. = ?” +‘S’v /Смаке.,
(73)
где 1000/емакс. представляет собой молярный объем V2 чистой кристалли-
ческой соли. График зависимости <pv от V2 для галоидных солей ще-
умакс.
лочных металлов состоит из двух прямых линий, одна из которых соот-
ветствует солям лития и цезия, а другая — остальным солям. Величины
отрезков, отсекаемых обеими линиями на оси ординат, составляют при-
мерно 6 см3. Коэффициент наклона прямой для лития и цезия равен еди-
нице, а для других солей он равен 0,86 (^ [/3/4), что указывает на воз-
можность чисто геометрического истолкования объемных соотношений
в растворах. Скотт [78] указал, что уравнение (64) справедливо для солей
лития в ограниченной области концентрации и что если для определения
сру и Sy взять только данные для наибольших концентраций, то^это^при-
вело бы к более низким значениям <р . для солей лития. При этом
значения для лития оказались бы на графике ближе к соответ-
умакс.
ствующим значениям для других щелочных галогенидов, имеющих кристал-
лическую структуру поваренной соли (гранецентрированный куб), чем
к значениям для солей цезия, которые обладают решеткой объемноцентри-
'244
ГЛАВА VIII
Р и’с. 58. Определение кажущегося
молярного объема при бесконечном
разведении.
Проверка теоретического предельного
уравнения для водных растворов при
25°; с* выражено в эквивалентах на литр.
Белые кружки—дилатометрический ме-
черные кружки—метод дифферен-
и_Циальиых поплавков. Ср. рис. 169.
о полученных теми же авторами аналогичных данных для сернокислого
натрия, углекислого натрия и азотнокислого аммония. График зависимости
(<р7—1,9с1/2) от с для азотнокислого аммония1 искривлен в области раз-
бавленных растворов, а аналогичные графики для углекислого натрия
(с поправкой па гидролиз) и сернокислого натрия приобретают линейный
характер в области больших разбавлений только в том случае, если пре-
дельным коэффициентам наклона2 приданы различные значения, соответ-
ственно равные 11,3 и 12,2. Теоретические коэффициенты наклона для
1,2-валентных электролитов должны рав-
няться значениям коэффициентов для
1,1-валентных солей, умноженным на
(З)3/’2, т. е. 9,9, — эта величина и использо-
вана на рис. 58 для хлористого стронция.
На основании изложенного материала
можно сказать, что результаты, получен-
ные дилатометрическим методом и методом
поплавка, подтверждают теорию между-
цонного притяжения для типичных силь-
ных электролитов. В то же время следует
отметить, что для наиболее разбавленных
растворов гидролизующихся солей обна-
руживаются значительные расхождения
между опытными и теоретическими дан-
ными. В случае углекислого натрия, у ко-
торого способность к гидролизу выражена
весьма отчетливо, значения <fv, вычислен-
ные из данных Лэмба и Ли [69] по
определению плотностей, проходят через
минимум при концентрации около 0,01 н.
Кривая, изображающая зависимость
cpv от У"с, приближается к оси ординат
при нулевой концентрации с отрицатель-
ным коэффициентом наклона. Лэмб и Ли
приписали этот эффект влиянию гидроли-
за, а Гефкеи и Прайс [73] рассчитали
численную величину эффекта для каждой
из концентраций, применявшихся в этих
исследованиях. При наименьших концен-
трациях поправка на гидролиз составля-
ет около 1,7 см3', при введении этой
поправки не только устраняется мини-
мум,"но и достигается приближенное согласие опытных данных с теорией,
причем величина расхождения между опытными и теоретическими данными
лежится пределах ошибки при вычислении поправок на гидролиз. Весьма
возможно, что сравнительно небольшие отклонения от теорий, отмеченные
в случае азотнокислого аммония и сернокислого натрия, могут быть
устранены тем же путем 3. В случае солей с более высоким типом валент-
ности, например в случае сернокислого цинка, следует учитывать поправки
1 Гукер и Рубин [416] нашли, что полученным ими данным для этой соли при
несколько более высоких концентрациях соответствует коэффициент наклона, равный
всего 0,966.
/ 2 Указанные здесь коэффициенты наклона соответствуют нашему уравнению (64),
в котором представляет собой кажущийся молярный объем, а с выражено в моль/л.
3 Гидролиз с участием катиона способствует уменьшению в противоположность
влиянию гидролиза (Na2CO3) с участием аниона.
24G
ГЛАВА VIII
рованного куба. Кроме того, если вычислить гипотетические величины V2
для солей цезия, исходя из решетки типа поваренной соли, то данные
для этих солей попадают на кривую, соответствующую данным для солей
натрия, калия и рубидия.
Скотт делает, далее, следующий важный шаг. Он возражает против
того, чтобы чистое кристаллическое твердое вещество рассматривалось
в качестве гипотетического верхнего предела концентрации раствора,
утверждая, что если в понятие этого предельного раствора должны быть
включены свойства твердой'соли, то следует ввести представление о «кри-
тическом объеме разрушения» твердого вещества V*. Скотт предложил
метод определения величины V*х. Для наших целей достаточно будет
отметить, что силы сцепления достигают максимума при расширении кри-
сталла от его нормального равновесного объема до У*, а при дальнейшем
расширении эти силы уменьшаются. Следовательно, при достижении объема
V* происходит переход от твердой кристаллической структуры к более
подвижному жидкому состоянию, и можно предположить, что V* соответ-
ствует гипотетической максимальной концентрации, достигаемой в (пересы-
щенном) растворе данной соли.
В табл. 50 приведены значения V*, полученные Скоттом для галоид-
ных солей щелочных металлов при 25° и соответствующие значения
Ф *=фо +5у|/1000/7*, .(74)
Таблица 50
Значения величин, характеризующих критическое состояние разрушения
Соль V* (25») <Py (25°) V* = ?y/V*
0° 25 = -35“ 45’ 50“ 70“
LiCJ 32,4 25,3 0,78
LiBr 39,1 30,0 0,77
LiJ 51,2 39,2 0,77
NaCl 41,6 26,9 0,68 0,65 0,64 0,65 0,64 0,64 2
NaBr 49,0 31,5 0,66 0,64 0,64 0,64 0,64 0,65
NaJ 61,1 40,6 0,66
KF 37,2 24,0 0,65
KC1 56,6 36,3 0,65 0,64 0,64 0,64 0,64 0,64 2
KBr 64,7 41,3 0,65 0,64 '0,64 0,64 0,64
KJ 78,0 51,0 0,65
RbCl 63,8 40,7 0,65 0,64 0,64
RbBr 72,7 46,3 0.64 0,64 0,64
RbJ 86,7 55,9 0,64
CsCl1) 73,4 47,2 0,64
CsBr1) 82,6 52,8 0,64
CsJ1) • . 97,5 62,7 0,64
CsCl 62,4 48,0 0,77
CsBr 70,4 53,4 0,76
CsJ 83,3 63,2 0,76
1) Гипотетическая структура с решеткой типа поваренной соли,
е) 85°.
1 См. [79].
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ 247
вычисленные с помощью уравнения (64) и параметров, приведенных
в табл. 49. В этой таблице содержатся также значения У* для фтористого
калия при 25° и значения функции
_ ^1/1000/^+<4
Т---у* у* \ '
при других температурах. Эта функция вычислена с помощью значений
Sy и ®о, взятых из работ Скотта [78], Гефкена [57] и Пэске [80], причем
предполагается, что для всех солей
[1 + 1,2 • 10~4(г-25)]. (76)
Влияние изменений V* с температурой на величины ср* и ф* настолько
мало, что нет необходимости знать точное значение коэффициента расши-
рения.
Из уравнения (75) можно получить следующее выражение:
Sy = (ф*У* -<(4) ]/У*/1000 (77)
для величины, которая входит в уравнение зависимости фу от с%. Это вы-
ражение является необычным, поскольку в нем не отражено влияние ион-
ной атмосферы в обычном смысле. Члены ф* и V* относятся к настолько
плотно упакованной системе ионов, что их распределение, повидимому,
подобно распределению ионов в кристаллической решетке, a fy соответст-
вует исчезновению ионной атмосферы при бесконечном удалении ионов
друг от друга. Два следствия из этого эмпирического уравнения представ-
ляют значительный интерес. Во-первых, из табл. 49 и 50 следует, что как
®у, так и ф* являются аддитивными величинами. Поэтому для солей
с одинаковой кристаллической структурой (имеющих одинаковые значе-
ния ф*) из уравнения (77) следует, что б’ур^ООО/У* должно быть адди-
тивной величиной, если У* также подчиняется условию аддитивности
(табл. 50, второй столбец). Однако к отмеченному Рутом и другими иссле-
дователями свойству приблизительной аддитивности Sy следует относиться
с осторожностью, так как в уравнение (77) входит величина ф*, которая
может значительно меняться при переходе от одной кристаллической
структуры к другой. В этом случае величина Sy не является аддитивной.
Второе следствие уравнения (77) связано с тем, что ф* и У* почти не за-
висят от температуры в пределах значительного интервала температур,
поэтому соотношение
^1/2^ Г781
дТ — \.1000> дТ ' °'
представляет собой очень хорошее приближение.
На рис. 60 изображен график зависимости <р* от У* при 25°. Стрелки
указывают на значения, получающиеся для солей лития с помощью при-
ближенных величин Sv и фу, вычисленных только из данных для наиболее
высоких концентраций1. В случае солей цезия стрелки указывают на
точки, для которых У* вычислено, исходя из структуры каменной соли.
Из рис. 60 видно, что ф* (коэффициент наклона прямой) является функ-
цией пространственного расположения ионов в решетке, причем это под-
тверждается тем, что ф* почти совершенно не зависит от температуры.
Этот вывод имеет важное значение, так как если зависимость между Sv
и фу, выражаемая уравнением (75), определяется только геометрическими
1 Согласно некоторым данным, при переходе от малых концентраций к большим
величинам .Sy претерпевает резкие изменения. См. [81].
248
ГЛАВА VIII
условиями критического состояния разрушения, то это указывает на то,
что геометрические условия этого состояния сохраняются, по крайней мере
статистически, во всей области, дл^которой справедливо уравнение (64).
Если это имеет место, то для всех концентраций, за исключением очень
малых, представление о ионной атмосфере можно заменить представлением
о статистической структуре пространственной решетки* 1. Такая точка зрения
Р и с. 60. Графики зависимости от V* для водных
растворов галогенидов щелочных металлов при 25°.
заслуживает более детального исследования, поскольку рентгенографи-
ческие данные также указывают на связь между Sv и скоростью изменения
структуры жидкости, служащей растворителем, при изменении концентрации
ионов [82].
С помощью точных измерений плотности разбавленных растворов моче-
вины [83] и сахарозы 2 было показано, что в случае разбавленных растворов
неэлектролитов линейный характер имеет скорее зависимость от с, чем
от с1/2. Рис. 61, взятый из работы Гукера, Гейджа и Мозера [83], сви-
детельствует о справедливости этого заключения.
х Д. И. Менделеев (Избранные сочинения, т. IV, Л., 1937, стр. 375) установил при-
менимость к солевым растворам уравнения, описывающего изменение плотности раствора
d- с изменением его состава:
где dv—плотность растворителя, п—количество молей растворенного вещества на 100 молей
воды, А и В—эмпирические константы.
Рассматривая решение уравнения для п=оо, т. е. для концентрации чистой соли,
он получил следующее выражение для плотности чистой соли (й2), находящейся в рас-
творе:
1
d=z 1 + 7'
Выведенные соотношения оправдываются на всем диапазоне концентраций с точностью
в ±0,0002 (при 20°). Плотность кристаллического хлористого натрия получается отсюда
равной 1,9855, что дает молярный объем 29,4. В хорошем согласиис этим находится «кри-
тическая» величина, получаемая Скоттом путем экстраполяции по У с, а’именно 26,9
(табл. 50). То же имеет место для ряда других солей. Таким образом, Менделееву первому
принадлежит как сама идея о том, что свойства раствора тесно связаны со свойствами
чистого растворенного вещества, так и количественное выражение этой связи. (Прим, ред.)
2 Приведено у Редлпха и Клингера [846].
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
249-
До сих пор мы уделяли больше внимания <ру, чем V2, поскольку
первая величина получается более непосредственно из данных по^плотностям
и более просто выражается с помощью эмпирических уравнений^типа (64).
Величина V2 не менее важна, однако она будет рассмотрена менее но дроб-
но, так как по своим свойствам она весьма близка к величине <ру.
В очень разбавленных растворах V2 линейно зависит от с1/2 при тех
условиях, при которых для обнаруживается такая же зависимость. При
Рис. 61. Кажущийся молярный объем водных рас-
творов мочевины при 25°.
этом коэффициент наклона dV2jdc112 в полтора раза больше соответству-
ющего коэффициента наклона dyv/dc112, что может быть показано путем диф-
ференцирования уравнения (68). Следовательно, если одна из этих величин
изменяется с концентрацией согласно теоретическому предельному закону,
то и другая величина должна подчиняться этому закону. При умеренных
концентрациях S-образный характер кривой, имеющий место для ©у (см.
рис. 59), в случае V2 несколько усиливается в соответствии с требова-
ниями уравнения (68), а в концентрированных растворах, в которых зави-
симость <ру от с1/2 для многих солей снова становится линейной, на графике
для V2 обнаруживается небольшая кривизна, которая обычно становится
резко выраженной при высоких концентрациях.
Значения <ру, приведенные в табл. 49, равны соответствующим значе-
ниям V°. Для многих практических целей можно находить V2 при средних
концентрациях, умножая последний член уравнения (64) на 3/2 и пользуясь
величиной Ау (= дсру/дс1/2), приведенной в той же таблице.
Зависимости V2 от температуры и давления подробно рассматривают-
ся в § 6 и 7, посвященных расширяемости и сжимаемости, однако неко-
торые интересные особенности этих зависимостей связаны с излагаемыми
вопросами. В ряде исследований [85, 57, 60, 80, 86] имеются указания на
то, что значение ©у для водных растворов электролитов достигает макси-
мума между 60 и 70°. Этот максимум можно наблюдать также на графиках
для <ру, причем с увеличением концентрации температура, соответствующая
точке максимума, обычно немного возрастает, а кривизна графика вблизи
этой точки значительно уменьшается. Поведение величины V2 совершенно
аналогично. Максимальные значения и V® приблизительно аддитивны.
Влияние давления на и V2 представляет особенный интерес, по-
скольку с возрастанием давления увеличиваются кажущийся молярный
объем и парциальный молярный объем. Знак и порядок величины
dV$/dP можно предсказать [87, 88] на основании простой электростати-
ческой теории, которая приводит к уравнению (109) гл. III. Адамс [89}
250
ГЛАВА VIII
показал, что этот любопытный эффект наблюдается для растворов хлори-
стого натрия и сернокислого калия вплоть до давлений в 10 000 атм, но
становится менее резко выраженным при высоких давлениях и высоких
концентрациях. На рис. 62 изображена зависимость парциальных молярных
объемов от давления для этих солей, а также для азотнокислого аммо-
ния [90]. На данном рисунке пунктирные прямые представляют молярные
объемы чистых кристаллических солей, причем к этим прямым при высо-
ких концентрациях и давлениях приближаются кривые для парциальных
молярных объемов. Увеличение парциального молярного объема с ростом
давления, вероятно, обусловлено сжатием растворителя, которое связано
Р и с. 62. Влияние давления на пар-
циальные молярные объемы солей в вод-
ных растворах при 25°.
Молярные объемы чистых твердых солей
представлены пунктирными линиями. Цифры
в правой части графика обозначают концен-
трацию, выраженную в весовых процентах.
с присутствием растворенного вещест-
ва, если процесс растворения сопро-
вождается уменьшением объема1. Из
уравнения (61) видно, что если с уве-
личением давления У? уменьшается
быстрее, чем V, то <ру может при этом
увеличиваться. Это наблюдается в
случае разбавленных водных раство-
ров электролитов при низких давле-
ниях, так как при этих условиях
сжимаемость воды быстро уменьшает-
ся с увеличением давления. При вы-
соких давлениях, когда сжимаемость
очень мало уменьшается при увели-
чении давления, а также в случае
концентрированных растворов, для
которых величина Рв приближается2
к Р, величина меньше меняется
при изменении Р и может даже ме-
няться в обратном направлении. Кро-
ме того, при этих необычных усло-
виях высоких давлений или высоких
концентраций сжимаемость чистой
соли становится важным фактором,
характеризующим сжимаемость рас-
твора. Поэтому тот факт, что величина
•yv и ее изменение с давлением приближаются к соответствующим молярным
величинам для чистого растворенного вещества при этих условиях, нельзя
рассматривать как достаточное условйе идеальности системы [91]3.
1 В работе А. Г. Пасынского [ЖФХ, 11, 606 (1938)] сравнение экспериментальных
величин сжимаемостей воды и растворов электролитов ионами различной величины и ва-
лентности было использовано для вычисления чисел сольватации. О сжимаемости раство-
рителя под влиянием ионов см. также: А. В. Николаев, Изв. АН СССР, ОХН, № 5,
431 (1945). (Прим, ред.)
2 Определение эффективного давления Ре приведено в этой главе.
3 Применение измерений парциальных молярных объемов и теплоемкостей для целей
физико-химического анализа, а также для установления границ применимости существу-
ющих представлений о разбавленных растворах электролитов см. в следующих работах:
Н. К. Воскресенская, Г. Н. Янковс кая, Изв. АН СССР, ОХН, № 1, 3
<(1945); Я. И. Герасимов, Вестник МГУ, № 7, 81 (1948); А. Ф. Капустин-
ский, О. Я. Самойлов, Изв. АН СССР, ОХН, № 5, 472 (1946); А. Ф. К а п у-
•с тинский, И. П. Дэзидерьева, Trans. Far. Soc., 14, № 285, 69 (1946);
А. Ф. Капустинский, И. И. Липилина, ДАН СССР, 62, № 4, 485 (1948);
А. Ф. К а п у с т и н с к и й, В. Р. К л о к м а и, Изв. АН СССР, ОХН, № 4, 259
•(1943); А. Ф. Капустинский, Н. П. К а п у с ти н с кая, там же, № 6, 581
s(1946). (Прим, ред.)
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ 251
§ 6. Парциальная молярная расширяемость, кажущаяся молярная
расширяемость и коэффициент термического расширения
растворов электролитов
Парциальные молярные расширяемости компонентов бинарного рас-
твора определяются выражениями
НМ <та>
и
(Ш- <8°>
Кажущаяся молярная расширяемость растворенного вещества равна
^=(М- <81>
Подставляя в это уравнение значение <?у из (61), получаем
П1\ дТ )р аУ-а^У* 82\
’•Е пч ’ '
где
<88>
является коэффициентом термического расширения раствора и
является коэффициентом термического расширения чистого растворителя.
Кажущуюся молярную расширяемость нетрудно вычислить из плотностей
и их температурных коэффициентов с помощью уравнений
1000 . , , V . м2
®Е (йо“ аао) +rfoao —
= “(а —ао)+аО®у (85)
и
1000 ., , , . м2
— (ао“ —“ао)+“а =
т в mdciQ ' и ' 1 а
1000 / \ । /оп\
=^(“-«о)+а?у> (86>
получаемых путем сочетания уравнений (83) и (84) с уравнением (62) или
(63). Практически необходимые для этих вычислений данные по плотностям
обычно используются для определения <ру, а зная <ру, нетрудно вычислить
<fE по уравнению (81). Гукер [92] построил графики значений <ру, вычис-
ленных из данных по плотностям [57, 61] для хлоридов натрия и калия.
"Значения определялись графически как коэффициенты наклона кривых,
изображающих зависимость <су от Т. На рис. 63 изображена зависимость
этих значений от с для трех различных температур. За исключением
данных, относящихся к хлористому натрию при 0°, точки хорошо укла-
дываются на прямые линии. Таким образом, для интервала концентраций,
применявшихся в исследованиях, <рЕ равно
?е = ?Ое + ^1/'^ ’(87)
252
ГЛАВА VIII
Вследствие значительных различий между величинами SE для солей одного
и того же типа валентности значения <р“, полученные при использовании
этого уравнения для выражения опытных данных, могут сильно отличаться
от истинных предельных парциальных молярных расширяемостей.
В случае разбавленных растворов уравнение (87) получается путем
дифференцирования уравнения (64). Таким образом, уравнение
дфу f dS у aS1 у
'~?Е = \~дТ Г.
(88)
можно использовать в пределах применимости уравнения (64). .Далее,.
Гукер [92] указал, что для большинства 1,1-валентных электролитов заме-
на а на % в этом уравнении почти не влияет на величину -ьЕ вплоть до
Рис. 63. Зависимость кажущей-
ся молярной расширяемости вод-
ных растворов от ]/с при раз-
личных температурах.
Черные кружки—NaCl, белые круж-
ки—КС1.
весьма высоких концентраций, равных не-
скольким единицам моляльности. Следова-
тельно, уравнение
(89)
может служить хорошим приближением. Для
того чтобы в случае более концентриро-
ванных растворов получить линейную за-
висимость, нужно перенести член, содержа-
щий а и зависящий от концентрации, в ле-
вую часть уравнения (89) и построить гра-
фик зависимости [®Е 1/2а6’ус1/2] от с1^.
В тех немногих случаях, для которых име-
ются опытные данные, определение SE пу-
тем подстановки приближенного значения
из (78) в уравнение (89) приводит к
значениям Se> заниженным на 0,001 — 0,002
единицы.
При сочетании уравнений (87) и (64)
со второй формой уравнения (85) получается
выражение
аналогичное уравнению для плотности (65). Значения коэффициентов этого
уравнения, заключенных в скобки, нетрудно определить из коэффициента
наклона кривой, выражающей зависимость (а — а0)/с от с1/2 и отрезка, от-
секаемого этой кривой на оси ординат; сро и SE можно найти из известных
значений®^ и 6V. Значения %у и Sy для ряда электролитов приведены
в табл. 49. К этим значениям Гукер [92] добавил другие, вычисленные из
плотностей, взятых из «International Critical Tables», а также из данных
Гибсона [93] для сернокислого натрия. Значения, полученные Гукером
для коэффициентов уравнений (87) и (90) при 25°, приводятся в табл. 51.
Гукер вычислил также эти значения для хлоридов лития, натрия и калия
при 0 и 50°. График зависимости кажущейся молярной расширяемости
этих электролитов от с изображен на рис. 64. Как видно из этого
рисунка, при 25° положительно, а ®Е уменьшается при увеличении
концентрации. При более низких температурах значение становится
более положительным, а его уменьшение с концентрацией выражено еще
более отчетливо. При более высоких температурах наблюдаются обратные
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
253
Таблица 51
Численные значения коэффициентов и а при 25°
и давлении в 1 атм
Электролит eE=?E+SEc1/3 З/21) a = a0+Ac + Bc *
ioVE !02Se 104A 104B
НС1 3,4 -0,02 0,29 -0,004
LIC1 2,5 -0,65 0,21 -0,069
LiOH2) 4,5 -1,8 0,47 -0,19
NaCl 9,30 -2,10 0,89 -0,216
KCI 8,50 -2,20 0,78 -0,226
NaOII 11,7 -3,5 1,19 -0,36
Na2SO4 22,19 -9,33 2,19 -0,966
1) а0 = 2,55 10-4.
?) Значения дия L1OH следует считать приближенными.
явления. Так, Гукер обнаружил, что при 50° для хлористого лития от-
рицательно, a Se положительно. В случае концентраций, при которых
вычислялось Se для электролитов с одинаковым типом валентности, не
наблюдается приближения их коэффициентов наклона к одинаковому
юбщему значению.
Ё2 и Е} можно выразить через и
д? /дтЧ* путем дифференцирования урав-
нений (70) и (71). Если представляется бо-
лее удобным пользоваться для этой цели
вместо моляльностей концентрациями с, то
получаются следующие уравнения:
Е _1 С * 91 92 * * * * * * * 100° — \ 1/2 dtfE
+ 2000 + с3/2<9?у/<?/Г s
(91)
и
/_________1_______ \ сз/2
J1 1 do \ 2000 + c3lzd?v/d]/c ) d/c
(92)
Оба эти уравнения справедливы для широ-
кого интервала концентраций; Гукер [92]
придал уравнению (91) следующую форму:
Р и с. 64. Кажущиеся молярные
расширяемости водных растворов
при 25°.
Z?2
3000 — c<f° ч <?с₽
------ ---—ci/2 —2L , (93)
2000 + се У дУс
•которая является справедливой только в той области концентраций, в кото-
рой и <ру линейно зависят от с1/2_ Следовательно, если линейно
зависит от с1/2, то зависимость от с1/2 не’может быть линейной. В слу-
чае тех электролитов, для которых имеются опытные данные, отклоне-
ния Ег от линейной зависимости составляют лишь 3 — 4% вплоть до кон-
центрации с = 9, и поэтому трудно решить, которая из двух величии
254
ГЛАВА VIII
(<рЕ или Е2) строго линейно зависит от с1/2. Во всяком случае, графики
зависимости Е2 от (3/2) с1/2 настолько хорошо совпадают с кривыми, пред-
ставленными на рис. 64, что линейный характер этой зависимости не
нуждается в дополнительных доказательствах.
Величины a, <fE, Е2 и Е± для водных растворов мочевины при 27,5°
были найдены [94] из данных по плотностям, относящихся к 25 и 30°.
Так как плотности при обеих температурах выражаются уравнением
d = d0 + Агс + А2с3 + А3с3 -р . . ., (94)
то, согласно уравнению (83), выражение для а должно иметь такую же
форму. Зависимость между коэффициентами аналогичных уравнений для d
и а была выведена Гукером и Мозером1. Выражения, полученные этими
исследователями для коэффициента термического расширения и для кажу-
щегося молярного объема мочевины при 27,5°, можно записать в оконча-
тельном виде следующим образом:
а • 10е = 281,2-р 52,0с —4,71 с2 (95)
и
?у = 44,385 +0,1203с. (96)
Таким образом, согласно уравнению (85),
?Е= 0,0645-0,00468с. (97)
Если принять эту линейную зависимость <рЕ от с (вместо с1/2), то из этих
уравнений получаются выражения
Ж = 0,0645 -0,00936 с + 0,00208 с2 (98)
и
= 0,005085 + 0,0000846 с2 -10"8 с4. (99)
Как и следовало ожидать, в случае растворов неэлектролитов в выра-
жениях для Е2, <рЕ и у отсутствуют члены, содержащие с1/2, а в выраже-
ниях для £i, а и d нет членов, содержащих с3/2. Тот факт, что числен-
ные значения <?Е и Е2 для мочевины приблизительно такие же, как и для
сильных 1,1-валентных электролитов, до настоящего времени не получил
объяснения и представляет собой, видимо, случайное совпадение. Такое
отклонение поведения мочевины от идеального является весьма странным,
так как зависимость для <?v [уравнение (96)] является почти идеальной.
Молярный объем твердой мочевины равен 45,19 см3 при 25°, a V2 при
той же температуре отличается от него не более чем на 2% вплоть до
концентрации с = 10.
§ 7. Парциальная молярная сжимаемость, кажущаяся молярная
сжимаемость и коэффициент сжимаемости растворов электролитов
Парциальные молярные сжимаемости компонентов бинарного раствора
равны
(100)
1 В уравнении 6 из этой статьи множитель перед членом, содержащим с3, должен
быть равен единице.
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
255
и
(101}
Кажущаяся молярная сжимаемость определяется выражением
(102}
__ P?v)
( дР /т ’
которое с помощью уравнения (61) можно представить в виде
/ЗКД _ /"ЗУ? \ _
_ Up/г п\др}т +
п2 п2
(103}
Коэффициенты сжимаемости для раствора и для чистого растворителя равны,
соответственно
^жгхяохж-^х <w4>
и
= (105}
7? < дР )т d,j \дР )т •
Практически кажущуюся молярную сжимаемость можно вычислять
из данных по сжимаемости и плотности с уравнений: 1000 , -j □ гп \ । п ?К cd0 d0 - помощью одного из TOGO ,0 а . , п с (Р Ро) + Ро?у следующих (106}
и
1000 /л о лй \ । и Мг mdd0^ О + ? d 1000 ,й й \ , о (107}
а также с помощью уравнения (102).
Как и в случае можно ожидать, что зависимость ‘<рк от концен-
трации имеет простой вид
= + <108>
так как при дифференцировании уравнения (64) получается выражение
<109>
Далее, поскольку в уравнении (109), не внося значительной ошибки, можно
заменить р на ро, то
^=-[(^ + ^1 • <110>
Для 1,1-валентных электролитов это условие выполняется при концен-
трациях меньше 3 и., при которых разница между значениями ук, вычислен-
ными по уравнениям (108) и (109), составляет примерно 1%. Линейную
зависимость от }/"с можно сохранить вплоть до самых высоких концен-
трации, если преобразовать уравнение (109) следующим образом:
256
ГЛАВА VIII
использоваться в качестве зависимой переменной не ©к, а членом, заклю-
ченным в скобки. Скотт [95, 81] пользуется почти всегда этим уравнением
как для вычислений, так и для теоретических целей; это уравнение,
несомненно, заслуживает предпочтение перед уравнением (108), если только
считать правило Мэзона вполне справедливым. Для составления таблиц
при современном уровне наших сведений можно пользоваться более про-
стым уравнением (108). Ниже будет показано, что в случае некоторых
электролитов предпочтительно совершенно не пользоваться концентрациями,
•обозначаемыми с, а следует употреблять моляльности или весовые доли.
Для области концентраций, в которой ук и линейно зависят от с1/2,
путем сочетания уравнений (108), (110) и (64) с уравнением (106) полу-
чается выражение
р = % + Ac + Bcsi\ (112)
где
1ОООЛ = ?ос-ро?оу (ИЗ)
и
1000 В = SK-%SV. ‘rv (1.14)
Если вместо уравнений (108) и (110) воспользоваться уравнением (109), то в
правой части уравнения (114) появляется дополнительный член 1/2 <5% (% — р).
-Значения р, вычисленные для 3 н. растворов 1,1-валентных электролитов
•с учетом этого члена, отклоняются от прежних значений всего на 0,5%.
В табл. 52 приведены параметры для вычисления и р с помощью
уравнений (108) и (112). Значения А и В вычислены Гукером и Руби-
ном [50], которые из данных Лэнмэна и Мэйра [96] по сжимаемости, отно-
сящихся к давлению 100 — 300 бар, определили коэффициенты сжимаемо-
-сти при давлении в 1 бар. Для этих расчетов они воспользовались рас-
сматриваемым ниже точным методом Гибсона [97]. и 8К были вычислены
Таблица 52
Параметры уравнений (108) п (112) при 25°
и давлении в 1 атм1)
Электролит -1O42) SK • io4 A • 106*" В • 10s
НС1 - 8,3 3,0 -1,66 0,26
1.1С1 — 42,0 9,1 -4,97 0,84
NaCl -51,6 11,4 -5,91 1,04
КС1 -45,2 ' 12,4 -5,73 1,13
LiOH -77,9 17,0 -7,52 1,56
NaOH -89,0 20,9 -8,59 1,90
1) р0 = 45,5 • 10'®.
Е) Дополнительные значения %® см. в таблицах, составленных
Оуэном и Бринкли [1].
с помощью уравнений (ИЗ) и (114) из значений А и В, приведенных
в той же таблице. Необходимые для вычислений значения ©о. и Sv были
взяты из табл. 49. Все значения о®. и Sr для солей и оснований, пред-
ставленных в табл. 52, примерно на 16 — 23% выше соответствующих зна-
чений, полученных Гукером [98] до того, как он воспользовался данными
о сжимаемости при 200 барах, приведенными к 1 атм. Значения п SK
для соляной кислоты, полученные авторами этой книги, на 26 и 30% вьпге
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ 257
значений, полученных Гукером. Зависимость <рк от с1/2 при высоких
концентрациях изображена на рис. 65. Обсуждение результатов, полу-
ченных для более разбавленных растворов, приводится в конце этого
параграфа (табл. 56).
Задача вычисления коэффициентов сжимаемости, отнесенных к 1 атм,
из данных непосредственных измерений зависимости объема от давления
для растворов является весьма слож-
ной. Зависимость р от давления такова,
что р наиболее быстро уменьшается при
низких давлениях, т. е. в таких усло-
виях, когда труднее всего получить
весьма точные экспериментальные дан-
ные. При обычных измерениях опре-
деляют уменьшение объема при увели-
чении давления в области от 200 до
1000 атм. Если начальное давление
равно Ро (обычно Р . - 1 атм), то при
измерении находят объемное сжатие к0,
определяемое для чистой жидкости вы-
ражением 1
V(P)_v(po)
ъ — Ч)
Р и с. 65. Кажущиеся молярные сжи-
маемости водных растворов 1,1-валент-
ных электролитов при 25° и 1 атм.
Это выражение, взятое без нулевых
индексов, представляет собой опреде-
ление объемного сжатия раствора к.
Если произвести большое количество
точных измерений к0 при разных да-
влениях, то можно выразить зависи-
мость к0 от давления с помощью степенного ряда [в качестве независимой
переменной взят прирост давления (Р — Ро)]
к0 щ (Р - Ро) + а2 (Р - Роу + а3 (Р - Р0)з + . . . (116)
и вычислить с помощью обычных методов параметры этого выраже-
ния [99, 100]. Далее, из уравнения (116) следует, что
(»т = Й1 + 2й2 “ Р») + Зяз ’ • • (117)
при любом давлении. Истинная сжимаемость р0 при атмосферном давлении
(Р = Р0=1) равна аг. Получение многочисленных опытных данных, необ-
ходимых для того, чтобы этот простой эмпирический способ вычисления
мог дать точные значения к0, является весьма затруднительным. Описы-
ваемые в следующих параграфах уравнения, возможно, менее удобны для
вычислений, но зато с их помощью можно из одного экспериментального зна-
чения к., очень точно определять 30 для значительного интервала да влепий.
В 1888 г. Тэйт [101] обнаружил для случая воды простую зависи-
мость Р от V, которую можно выразить уравнением
r(P)„(P)_ fdvn\(P) 0,43430
РО у0----{~др)Т ^-в + р-’ <118)
или в интегральной форме
. MPj)=-[4P)-«(oPo)]==cig(-|^), (119)
1 Начиная с этого места и до конца главы, мы будем пользоваться удельными
объемами и т. д. вместо соответствующих молярных величин, и таким образом наше
изложение не будет существенно отличаться от изложения этих вопросов Гибсоном.
17 Г. Харнед, Б. Оуэн
258
ГЛАВА VIII
где С и В — положительные постоянные. Гибсон [97, 102, 103а]1 подробно
исследовал уравнение Тэйта и показал, что оно хорошо описывает экспе-
риментальные результаты Адамса [89а] для воды при 25° вплоть до давле-
ний в 10 000 бар, а также данные Бриджмэна [104] для многочисленных
нелетучих жидкостей при различных температурах в том же интервале
давлений. С этим уравнением согласуются также данные Бриджмэна
для 50 летучих жидкостей в области давлений от 4000 до 12 000 бар
и данные для некоторых твердых веществ.
Некоторые из самых последних2 значений, полученных Гибсоном для В
и C/v0, приведены в табл. 53. Отметим, что С/у0 мало зависит от состава
жидкости и не зависит от температуры [105]. Гибсон и Кинкейд [103а]3
определили физический смысл этих параметров. Член 0,4343С можно
рассматривать как сжатие, вызываемое изменением давления В -\-Р,
а В представляет собой разность между давлением расширения, обуслов-
ленным тепловой энергией, и внутренним (кохезионным) давлением, обу-
словленным притяжением между молекулами, не зависящим от температуры.
Поэтому В, как видно из табл. 53, уменьшается с температурой и увели-
чивается при возрастании между молекулярных сил.
Табл ица 53
Параметры уравнений (118) и (119)
В, бары
Вещество С 2)
25° 45° 65° 85°
Вода [4] 1) 0,3150 2996 3081 3052,2 2939,0
Метиловый спирт [1] . . 0,2208 764 — — —
Этиленгликоль . . . 0,21763 2544 2363 2186 2011
Бензол [2] 0,21591 970 829 701 —
Хлорбензол [3] .... 0,21591 1249,1 1097,8 960,9 835,0
Бромбензо л [3] .... 0,21591 1404,4 1247,3 1103,3 972,0
Анилин [3] 0,21591 1865,2 1678,8 1504,5 1341,6
Нитробензол [3] . . . . Четырехлористый угле- 0,21591 2006,6 1798,3 1605,6 1429,4
род1) 0,21290 867,0 737,7 622,1 —
1) Частное сообщение Р. Гибсона.
2) Отметим, что при выбранных обозначениях величиной, не зависящей от температуры,
вместо С оказывается
^0
Из данных, приведенных в табл- 54, видно, насколько уравнение (118),
в которое входят константы, выведенные из данных измерений сжатия при
высоких давлениях, отражает свойства жидкостей, находящихся под атмо-
сферным давлением. Значения ро при давлении в 1 бар вычислены
с помощью констант, взятых из табл. 53, и сопоставлены с результатами,
полученными из измерений скорости звука в жидкостях при атмосферном
давлении [уравнения (128) и (129)]. Значение ро Для воды (45,7 10“®),
1 См. также [103 6].
2 Встречающиеся в более ранних работах Гибсона величины В и С следует заменять
на В + 1 и 0,4343 С для того, чтобы эти величины имели то значение, которое приписы-
вается им в настоящее время.
3 См. также работы Кинкейда и Эйринга и Хадлстона [1036], в которых рассмотрены
уравнения состояния, подобные уравнению (118).
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
269
Таблица 54
Значения р0 • 106 при давлении в 1 бар,
вычисленные по уравнению (118), и значения той
же величины, полученные из измерений
скорости звука
Жидкость t Ро ю6
По урав- нению Из скорости звука
Бензол 10 86,5 86,4 [1]
» 20 93,1 93,7 [1]
» 30 100,4 101,2 [1]
» 40 108,6 108,6 [1]
» 50 117,8 118,3 [1]
Метиловый спирт . . 25 125,3 125,6 Г1]
Вода 25 45,7 45,2 [2] 1)
45,9 [3]2)
1) Высокие частоты.
2) Низкие частоты.
вычисленное по уравнению (118), только на 1/3% выше среднего из значе-
ний р0, вычисленных с помощью измерений скорости звука. В последующих
вычислениях будет применяться полученная Гибсоном [97] несколько^мень-
шая величина, равная 45,5 • 10~8-
Гибсон [97, 102] применил уравнение Тэйта к растворам, введя в’пего
только один дополнительный параметр. Это важное обобщение уравнения
Тэйта основано на гипотезе Таммана [106], который предположил, что
в присутствии ионизированного растворенного вещества вода в водном
растворе ведет себя так, как будто на нее, кроме атмосферного давления,
действует постоянное эффективное давление Ре. Согласно Тамману, умень-
шение объема и уменьшение сжимаемости при растворении электролита
обусловлено сжатием воды, а не изменением свойств растворенного веще-
ства (рассматриваемого как жидкость).
В соответствии с этим предположением объем 1 г раствора, содержа-
щего г растворителя и г растворенного вещества, можно представить
в виде
«<Р) = М1₽) + (120)
В этом -выражении <Ь2 представляет собой удельный объем чистого раство-
ренного вещества (как жидкости), а Фх — кажущийся удельный объем раство-
рителя в растворе1. Далее, прибавление диссоциирующего на ионы раство-
ренного вещества (в виде жидкости) к 1 г чистой воды вызывает уменьшение
общего объема системы, равное — Ф^- Выраженное с помощью уравне-
ния Тэйта (119), это изменение объема равняется
(121)
при любом давлении Р. Это полезное соотношение мы будем называть
уравнением Тэйта — Гибсона.
1 Для умеренных концентраций и давлений, когда х2'52 значительно меньше, чем
ж1ф1, нет необходимости в точном вычислении ip2- Физический смысл величины ip2 будет
рассмотрен ниже.
17*
260 1 ГЛАВА VIII
Исключая ф[Р) из предыдущих уравнений, получаем
- = Х1С 1g (В + Р + Ре) - х^’ - х. [о(о₽) + с 1g (В + В)]. (122)
Согласно уравнению (119), последний член в правой части этого уравнения
исчезает, если изменение давления происходит при постоянном составе;
следовательно,
fcy(JPo)=— ——(123)
\-° ~г от* е/
Путем дифференцирования уравнения (122) по давлению при постоянных
температуре и составе получаем выражение
р(Г)0(Г) = °в’^Ср-е - х2 , (124)
характеризующее сжимаемость раствора при любом давлении Р. Для случая
умеренных давлений и концентраций можно пренебречь членом? содержа-
щим ф2- Так как В и С характеризуют только растворитель, то Ре является
единственным параметром, зависящим от свойств раствора. Ре можно
вычислить из данных одного опыта в любом удобном для этой цели интер-
вале давлений. Ре непосредственно зависит от молярного объема растворен-
ного вещества. Из определения = (dV/dnJp, т, па, пропорции | х2 =
= п1М1\п2М2 и уравнения (120) следует, что
(125)
С/Лг j
Сочетая это уравнение с основным уравнением
кР^Х^+Хг^Р (126)
и уравнениями (120) и (121), Гибсон [107] получил выражения
-<Р) ,(₽) 0,4343^(7 д (В + Ре) м27.
“’Ь ^В+Р' + р—д— { ’
И
?~(Щ М ^р'>— — 434>ЗГ д(В + Ре) И 27а)
-^2^2 -~вТр7+р дт (1^'а)
Из последнего уравнения видно, что величина парциального молярного
объема растворенного вещества в значительной степени определяется двумя
множителями. Первый из них — д (В + Р^/дт, т. е. скорость изменения
общего внутреннего давления с концентрацией, зависит от свойств раство-
рителя и растворенного вещества. Второй — С/(В Ре-\- Р),— связан с сжима-
емостью растворителя, находящегося под давлением Р-\-Ре, и не зависит
от свойств растворенного вещества.
При вычислении значений р, соответствующих параметрам, приведен-
ным в табл. 52, приращение давления, равное 200 бар, было отнесено
к исходному давлению Рй = 100 бар вместо атмосферного. В этом случае
аргумент логарифмического члена уравнения (123) составляет (В+Ре-\-
+ 300)/(В + Ре + 100), и этот член отражает сжатие чистого жидкого рас-
творенного вещества при изменении давления от 100 до 300 бар. Гибсон [108]
исследовал зависимость Ре от концентрации для большого числа электро-
литов в интервале давлений от 1 до 1000 бар. Некоторые из полученных
им результатов [109] представлены на рис. 66. Так кай член Ре увеличи-
вается по мере увеличения сил. притяжения между ионами и молекулами
воды, обладающими индуцированными или остаточными зарядами, то можно
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
261
Рис. 66. Эффективное давление
(в барах) водных растворов солей
при 25°.
ожидать, что Ре будет увеличиваться с возрастанием плотности зарядов
ионов. Это предположение подтверждается, за немногими исключениями,
представленными на рисунке кривыми, из которых видно, что для ряда
солей с общим ионом значение Ре является наибольшим для тех солей,
у которых второй ион (не являющийся общим) имеет меньший размер, т. е.
обладает большей плотностью заряда. Однако следует отметить, что
в случае ионов самых малых размеров, Li*, Ве++ и Mg++, это правило не
выполняется. Бернал и Фаулер [110] показали, что поляризация, произво-
димая маленькими ионами с высокой плотностью заряда, увеличивает силы,
способствующие образованию открытой структуры воды, при которой
каждая молекула воды окружена четырьмя
другими молекулами, расположенными в
вершинах тетраэдра. Это обстоятельство в
некоторой степени противодействует сжатию
растворителя под влиянием ионов и приво-
дит к ненормально низким значениям Ре.
Более подробное описание этого процесса
принадлежит Гибсону [109], который пред-
полагает также, что специфические отличия
между значениями Ре для подобных ионов
в значительной степени объясняются влия-
нием объема ионов, которое приводит к уве-
личению среднего расстояния между моле-
кулами воды. При этом уменьшается та
часть общего внутреннего давления раствора
В-}-Ре, которая обусловлена взаимодействием
молекул воды. Этот объемный эффект дол-
жен увеличиваться по мере увеличения
размеров ионов. Гибсон считает, что для
учета этого эффекта нужно сравнивать зна-
чения общих внутренних давлений раство-
ров, деленные на квадрат концентрации во-
ды, а не значения эффективных давлений.
Таким образом, если при построении кривых, изображенных на рис. 66,
откладывать по оси ординат вместо Ре значения функции (В + P^/Cw, то
эти кривые в значительной степени утрачивают свой специфический ха-
рактер и результаты для всех 1,1-валентных солей, за исключением данных
для лития, хорошо укладываются на одну кривую. Этот факт убедительно
свидетельствует о том, что влияние объемов ионов имеет существенное
значение.
Роль растворителя отчетливо проявляется в том, что аномальные
свойства солей лития в отношении величины Ре, которые наблюдаются
в случае водных растворов, не имеют места в случае растворов гликоля,
а кривые, изображающие зависимость Ре от т для разбавленных растворов
бромидов натрия и лития, а также иодидов натрия и кадмия, совпадают
в метиловом спирте и сильно расходятся в воде и гликоле [91].
Другое эмпирическое соотношение для зависимости Ре от состава рас-
твора имеет вид
Pe — Lc1C2+i, (1276)
где Lui — специфические величины для данного электролита. Постоян-
ная i настолько мала, что в большинстве случаев ею можно пренебречь.
Она введена в уравнение для того, чтобы отразить тот экспериментальный
факт, что кривые, изображающие зависимость Ре от с^, не проходят
точно через начало координат для всех солей. Согласно этому уравнению,
262
ГЛАВА VIII
Ре можно считать мерой отклонения объемных соотношений для растворов
от идеального случая, поскольку было показано \ что относительные откло-
нения от простого закона смесей для сжимаемостей ряда растворов также
прямо пропорциональны произведению сгс2. Из уравнения (1276) следует,
что Р,. обратно пропорционально квадрату объема. В этом проявляется
аналогия между Ре и внутренним давлением в газах, которое соответствует
члену а/»2 в уравнении Ван-дер-Ваальса. Поскольку Рр зависит от объема,
то эта величина является также функцией давления. Это обстоятельство
может оказаться весьма важным при изучении жидкостей, у которых вели-
чины сжимаемости сравнительно велики. При условиях, в которых приме-
няют уравнения (123) и (124), Рр рассматривается в качестве постоянной
величины для данной системы при постоянном составе и температуре.
Такое упрощение оправдано тем, что эти уравнения описывают данные
по сжимаемости во всей изученной области давлений и позволяют про-
водить экстраполяцию до атмосферного давления. В табл. 55 приведены
некоторые значения сжимаемости, вычисленные для давления в 1 бар
с помощью уравнения (124) из величин Ре, полученных из данных по
сжимаемости при 1000 бар, а также значения, полученные из данных
по скорости звука [111] в растворах при 1 атм. Совпадение результатов
не оставляет желать ничего лучшего.
Таблица 55
Значения сжимаемостей, определенные двумя независимыми методами при 25°
и давлении в 1 бар[1 ]
р 10е
100x2, % вес. Из данных о сжатии при давлении 1000 бар Из данных о скорости звука при давлении 1 бар
КС1 КВг KJ NaCl KOI КВг KJ NaCl
6 41,8 43,2 44,3 40,6 41,7 43,5 44,3 40,4
10 ' 39,5 41,7 37,6 39,4 41,9 37,5
16 36,3 41,7 33,2 36,2 41,7 33,4
20 34,3 38,1 30,7 34,2 38,4 31,0
24 32,4 28,4 32,4 28,8
30 34,6 37,8 34,8 38,0
40 31,1 31,4
45 33,4 33,7
Заканчивая разбор уравнения Тэйта и его обобщенной формы, относя-
щейся к растворам электролитов, следует вкратце остановиться на вопросе
об истинном объеме растворенного вещества. Уравнение (122) можно
использовать для вычисления ф2 при 1 атм из значений Ре, найденных
путем определения сжатия, сопровождающего процесс растворения. Значе-
ния ф2> полученные этим способом [97, 102], практически не зависят от
концентрации и равны удельному объему чистого кристаллического раство-
ренного вещества или немного (на 5 — 30%) его превышают. Если исполь-
зовать уравнение (127а) для определения ф2 из величин Ре и V2, то оказы-
вается, что ф2 несколько уменьшается с концентрацией. Однако если
допустить, что сжимаемость растворенного вещества в растворе в 3 раза
больше, чем в твердом состоянии, то окажется, что ф2 совершенно пе
Концентрации воды (д и электролита сг выражены в молях на литр раствора.
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ 263
зависит от концентрации [107]. Таким образом, удельный объем чистого
растворенного вещества, или, точнее, чистого расплавленного растворен-
ного вещества, невидимому, близок к «истинному» объему растворенного
вещества в растворе. Тот же вывод получается при рассмотрении крити-
ческого объема разрушения V* (§ 5), однако следует отметить, что V*
значительно больше, чем вычисленные величины М2Ь2. Точный характер
зависимости между этими двумя величинами нельзя считать выясненным.
Рассмотренные выше экспериментальные данные относятся к сравнитель-
но концентрированным растворам (с >0,5). Прямые измерения сжимаемости
в случае разбавленных растворов мало пригодны, поэтому для точного
определения значений сжимаемости при достаточно больших разбавлениях
пользуются другими способами. Представляет интерес сравнение этих опыт-
ных данных с результатами, вычисленными на основании теории между-
ионного притяжения. Так же как и в случае других вторых производных
уравнения Дебая — Гюккеля, J2 и Е2, не следует ожидать точного совпа-
дения или К2 с величинами, вычисленными с помощью теоретического
предельного закона в рассматриваемой области концентраций. Однако
интересно выяснить, стремятся ли экспериментальные кривые при наиболь-
ших достигнутых разбавлениях приблизиться к теоретическим или по крайней
мере группируются ли они определенным образом в соответствии с типом
валентности. Напомним, что экспериментальные значения коэффициента
наклона $к, приведенные в табл. 52, для некоторых 1,1-валентных элек-
тролитов по своей величине в 7 раз отличаются друг от друга.
Наиболее Точный метод определения сжимаемости при атмосферном
давлении и малых концентрациях основан на измерении скорости звука
в растворе. Адиабатическая сжимаемость Ps (в бар-1) связана со скоростью
звука и (ван сек-1) в среде с плотностью d уравнением
106
₽s = Sr (128)
Изотермическую сжимаемость р можно вычислить из Ps по известной термо-
динамической формуле
Р = PsCp/Cp = Ps + 0,02391а2Т/с/, (129)
где ср и с„ — теплоемкости среды, а а — ее коэффициент термического рас-
ширения. В водных растворах при 25° разность р и ps составляет всего
около 1%, но под влиянием этой разности значение ак для 1,1-валентных
электролитов может изменяться на 10%. В табл. 54 и 55 эта разность
учтена и приведены значения изотермических коэффициентов. При дальней-
шем изложении будут использованы значения Ps- Небольшие поправки
в случае разбавленных растворов не влияют на выводы, которые можно
сделать на основании этих данных, и к тому же величины, необходимые
для внесения этих поправок, для некоторых растворов неизвестны с до-
статочной степенью точности. В исследованиях, относящихся к измерениям
скорости звука, Ps обычно выражается в абсолютных единицах. Мы выра-
зили эти величины в обратных барах.
Экспериментальная методика непрерывно совершенствовалась, от срав-
нительно грубых опытов Кундта [112] до применения акустического интер-
ферометра Хаббардом и Лумисом[113а] % с помощью которого была достигнута
абсолютная точность в 0,06%. В более поздних исследованиях [114—122]
было использовано важное открытие Дебая и Сирса [123] и Лукаса и Би-
куорда [124], показавших, что при прохождении ультразвуковых волн через
жидкость возникает периодическая неоднородность, которая проявляет себя
См. также [1136].
264 •
ГЛАВА VIII
подобно оптической решетке; в этих исследованиях была достигнута отно-
сительная точность, равная нескольким тысячным процента. Салаи [115]
впервые применил эту методику и получил данные, согласно которым влия-
ние валентности согласуется с закономерностью, установленной теорети-
ческим путем; однако на основании этих результатов Салаи нельзя делать,
окончательных выводов, поскольку полученные им значения (ро — ₽)/?о
относятся только к одной концентрации — 0,1н. Из уравнений (112)—-(114)
видно, что (Р0-^Р) состоит из двух членов, первый из которых —
(Ро ~?к) Ю~3с — не содержит фактора валентности. Следовательно, для
двух солей с различными типами валентности отношение величин (р0 —Р)
не должно обязательно равняться отношению соответствующих факторов
валентности (Svizt)3/2- Влияние валентности было правильно учтено Гуке-
ром [98], а также Фалькенгагеном и Бахемом [125, 122]. Измерения Ба-
хема охватывают область концентраций от 0,5 до 0,03н. и относятся к
значительному числу различных солей. Значения <рх, вычисленные Бахе-
мом непосредственно из значений Ps, линейно зависят от ]/ с, однако коэф-
фициенты наклона, приведенные в табл. 56, согласуются с'теорией не лучше,
чем коэффициенты наклона, полученные Гукером [126, 98, 50] из данных
для более высоких концентраций (табл. 52). Данные, полученные для галоид-
ных солей щелочных и щелочноземельных металлов, в основном согласуются
с теоретическими значениями при всех концентрациях, применявшихся
в этих исследованиях. Однако в случае других электролитов при наиболь-
ших разбавлениях наблюдаются значительные расхождения. Как было
указано, такое же явление наблюдается для более высоких концентраций
при исследованиях кажущихся молярных объемов (см. § 5). Так, например,
для значений углекислого натрия удается добиться согласия с теорией
только с помощью данных, полученных для очень низких концентраций,
а также путем введения поправок на гидролиз.
Таблица 56
Сравнение экспериментальных значений
коэффициентов наклона 8 к
с теоретическими [1]
Электролит sk 104 (2/s)S(.K) • i°4 3)
NaCl 9,0 6,3
КВг 8,2 6,3
ВаС12 26,8 32,6
СаС12 [2]i) 26,0 32,6
Na2CO3 48,8 32,6
MgS04 35,2 50,3
K3Fe (CN)e 58,0 92,4
K4Fe (CN). 100,8 199,4
CaaFe (CN), [2p) 139,0 Около 260
1) При 30°.
2) При 0°.
3) 2+ z-l> Р=2.
Для сжимаемости растворов некоторых неэлектролитов, повидимому,
справедлива та же зависимость от концентрации, что и в случае раство-
ров электролитов. По данным Пермана и Урри [128] для мочевины и саха-
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
265
розы Гукер.[127] построил линейные графики зависимости от ]Лс. Зна-
чения <?к, полученные по данным Бахема [129] для сахарозы, точнее соот-
ветствуют линейной зависимости ср к от с, чем от ]/с; однако две точки для
наименьших концентраций не укладываются ни на одну из этих прямых.
Гибсон нашел, что кажущаяся молярная сжимаемость резорцина [130]
в пределах ошибки опыта линейно зависит от ]/с. Для случая смесей ме-
тилового спирта с водой это соотношение явно неприменим!) к метиловому
спирту, но очень хорошо выполняется в случае воды [130]. Для растворов
частично диссоциированных электролитов получаются противоречивые
результаты. Кажущаяся сжимаемость водных растворов уксусной кислоты
[102] линейно зависит от с1/2 вплоть до концентрации кислоты, равной 40%,
а соответствующие значения для-растворов йодистого кадмия [91] в гликоле
и метаноле линейно зависят от с вплоть до концентрации соли порядка 50%.
Как видно из материала, изложенного в данном и в двух предыдущих
параграфах, с помощью теории междуионного притяжения в ее современ-
ной форме нельзя удовлетворительно объяснить те зависимости для моляр-
ных объемов, расширяемостей, сжимаемостей и т. д., которые наблюдаются
в случае водных растворов в пределах экспериментально изученного интер-
вала концентраций. Экспериментальные определения парциальных моляр-
ных объемов, выполненные с помощью усовершенствованной прецизионной
методики, подтверждают теорию Дебая — Гюккеля при больших разбавле-
ниях. Однако для теоретического истолкования правила Мэзона и подоб-
ных ему линейных соотношений для расширяемости и сжимаемости, неви-
димому, необходимы более глубокие познания относительно структуры
жидкостей, а также, возможно, более совершенная теория распределения
ионов, чем та, которая основана на представлении о ионной атмосфере.
ЛИТЕРАТУРА
1. A. L. R о b inson, J. Am. Chem. Soc., 54, 1311 (1932).
2. W. N e r n s t, W. Orthmann, Sitzber. preuss. Akad. Wiss., 51 (1926); Z. phy-
sik. Chem., 135, 199 (1928).
3. a) E. L a n g e, A. L. Robinson, Chem. Rev., 9, 89 (1931); б) T. W. R i-
c h a r d s, F. T. G ucker, Jr., J. Am. Chem. Soc., 47, 1876 (1925).
4. E. L a n g e, J. M о n h e i m, Z. physik. Chem., A149, 51 (1930).
5 F. T. G u c ke r, Jr., H. G. P i c k a r d, R. W. Pl a n c k, J. Am. Chem. Soc.,
61, 459 (1939).
6. E. Lange, H. S t r e e c k, Z. physik. Chem., A157, 1 (1931).
7. E. Lange, J. M о n h e i m, Z. physik. Chem., A150, 349 (1939).
8. G. В. В о n i n о, V. V a g 1 i o, Nuovo Cimento, 5, 4 (1928).
9. E. Lange, H. S t r e e c k, Naturwiss., 19, 359 (1931).
10. E. A. G u 1 b r a n s e n, A. L. R о b i n s о n, J. Am. Chem. Soc., 56, 2637 (1934).
11. T. F. Yo ung, O. G. Vogel, J. Am. Chem. Soc.,- 54, 3030 (1932).
12. T. F. Y о u n g, V. L. G г о e n i e r, J. Am. Chem. Soc., 58, 187 (1936).
13. T. F. Y о u n g, P. S e 1 i g m a n n, J. Am. Chem. Soc., 60, 2379 (1938).
14. E. Lange, J. Messner, Z. Elektrochem., 33, 431 (1927).
15. E. L a n g e, A. E i c h 1 e r, Z. physik. Chem., 129, 285 (1927).
16. a) F. R. В icho wsky, F. D. Rossini, Thermochemistry of Chemical Sub-
stances, Reinhold Publishing Corp., N. Y., 1936; б) T. F. Young, Science, 85,
48 (1937).
17. а) Г. H. Льюис, M. P e и д а л л, Химическая термодинамика, ОНТИ—Хим-
теорет, Л., 1936; б) W. Schottky, Thermodynamik, Julius Springer, Berlin,
1929.
18. E. Lange, F. Durr, Z. physik. Chem., 121, 361 (1926).
19. A. L. R о b i n so n, W. E. W a 1 1 a c e, J. Am. Chem. Soc., 63, 1582 (1941).
20. E. D о e h 1 e m a n n, E. L a n g e, Z. physik. Chem., A170, 391 (1934).
21. E. L a n g e, A. L. Robinson, J. Am. Chem. Soc., 52, 4218 (1930).
22. J. Wyman, Jr., Phys. Rev., 35, 623 (1930).
23. J. W у m a n, Jr., J. Am. Chem. Soc., 55, 4116 (1933).
24. G. A k e r 1 6 f, J. Am. Chem. Soc., 54, 4125 (1932).
266
ГЛАВА VIII
9
25. L. К о с к.е 1, Ann. Physik, 77, 417 (1925).
26. F. Т. G и с к е г, Jr., Н. G. Р i с к а г d, J. Am. Chem. Soc., 62, 1464 (1940).
27. F. M. Naude, Z. physik. Chem., 135, 209 (1928).
28. A. W. Porter, Trans. Faraday Soc., 13, 123 (1917).
29. F. fl. P r a t t, J. Franklin Inst., 185, 663 (1918).
30. R. В. V a 1 1 e n d e r, E. P. P e r m a n, Trans. Faraday Soc., 27, 124 (1931).
31. T. W. Richards,. L. P. H a 1 1, J. Am. Chem. Soc, 51, 731 (1929).
32. F. D. Rossini, Bur. Standards J. Research, 6, 847 y931).
33. T. W. R i c h a r d s, A. W. Rowe, J. Am. Chem. Soc., 44, 684 (1922).
34. L. J. G i 1 1 e s p i e, R. H. Lambert, J. A. Gibson, Jr., J. Am. Chem.
Soc., 52, 3806 (1930).
35. F. D. Rossini, Bur. Standards J. Research, 6, 791 (1931); 7, 47 (1931).
36. E. L a n ge, J. Monheim, A. L. Robinson, J. Am. Chem. Sgc., 55, 4733
(1933).
37. H. S. H a r n ed, W. J. H a m e r, J. Am. Chem. Soc., 57, 27 (1935).
38. H. S. H a r n e d, R. W. Ehlers, J. Am. Chem. Soc., 55, 2179 (1933).
39. H. S. H a r n e d, J. С. H e с к e r, J. Am. Chem. Soc., 55, 4836 (1933).
40. R. H. La m b e r t, L. J. Gillespie,!. Am. Chem. Soc., 53, 2632 (1931).
41. a) F. T. G u с к e r, J. Phys. Chem., 38, 312 (1934); 6) F. T. G u с к e r, Jr.,
F. D. A у r e s, T. R. R u b i n, J. Am. Chem. Soc., 58, 2118 (1936).
42. J. P. Joule, Mem. Manchester Lit. Phil. Soc., 2, 559 (1845).
43. L. P f a u n d 1 e r, Sitzb. Akad. Wiss. Wien, 59, 2145 (1869).
44. R. J e s s e 1, Proc. Phys. Soc., 46, 759 (1934).
45. M. R a n d a 1 I, F. D. Rossini, J. Am. Chem. Soc., 51, 323 (1929).
46. F. T. Gucker, К. II. S c h m i n к e, J. Am. Chem. Soc., 54, 1358 (1932).
47. F. Zwicky, Physik Z., 26, 664 (1925); 27, 271 (1926); Proc. Nat. Acad. Sci., 12,
86 (1926); H. M. Ev jen, F. Zwicky, Phys. Rev., 33, 860 (1929).
48. T. J. Webb, J. Am. Chem. Soc., 48, 2589 (1926).
49. T. F. Y о u n g, J. S. M a c h i n, J. Am. Chem. Soc., 58, 2254 (1936).
50. F. T. G u с к e r, T. R. Rubin, J. Am. Chem. Soc., 57, 78 (1935).
51. F. T. G u с к e r, F. D. A у r e s, J. Am. Chem. Soc., 59, 447 (1937).
52. F. T. G u с к e r, Jr., F. D. A у r e s, J. Am. Chem. Soc., 59, 2152 (1937);
С. M. Whit e, там же, 58, 1620 (1936).
53. К. Bennewitz, L. Kratz, Physik. Z., 37, 496 (1936).
54. a) D. О. M a s s о n, Phil. Mag. [7], 8, 218 (1929); 6) A. F. Scott, J. Phys. Chem.,
35, 2315 (1931); W. G e f f с к e n, Z. physik. Chem., A155, 1 (1931); 0. R e d 1 i c h,
P. Rosenfeld, Z. Elektrochem., 37, 705 (1931); Z. physik. Chem., A155, 65
(1931).
55. W. C. Root, J. Am. Chem. Soc., 55, 850 (1933).
56. H. E. Wirt h, J. Am. Chem. Soc., 59, 2549 (1937); 62, 1128 (1940).
57. W. G e f f с к e n, Z. physik. Chem., A155, 1 (1931).
58. F. T. G u с к e r, Jr., J. Phys. Chem., 38, 311 (1934); J. Am. Chem. Soc., 60, 2582
(1936).
59. R. E. Gibson, J. Phys. Chem., 38, 320 (1934).
60. A. F. Scot t, J. Phys. Chem., 35, 2315 (1931).
61. G. P. В a x t e г, С. C. Wallace,!. Am. Chem. Soc., 38, 70 (1916).
62. H. H ii t t i g, Z. Elektrochem., 34, 14 (1928).
63. H. К 6 h n e r, Z. physik. Chem., Bl, 427 (1928).
64. P. H 6 1 e m a n n, H. К 6 h n e r, Z. physik. Chem., B13, 335 (1931).
65. Z. S h i b a t a, P. H б 1 e m a n n, Z. physik. Chem., B13, 347 (1931).
•66. a) L. G. Longsworth, J. Am. Chem. Soc., 57, 1185 (1935); б) В. B. Owe n,
S. R. Brinkley, Jr., Chem. 29, 461 (1941); в) K. F a j a n s, 0. Johnson,
J. Am. Chem. Soc., 64, 668 (1942).
67. V. K. L a M e r, T, H. G г о n w a 1 1, J. Phys. Chem., 31, 393 (1927).
68. W. G e f f с к e n, С. В e с к m a n n, A. К r u i s, Z. physik. Chem., B20, 398 (1933).
69. A. B. Lamb, R. E. L e e, J. Am. Chem. Soc., 35, 1666 (1913).
70. F. К о h 1 r a u s c h, W. H a 1 1 w a c h, Wied. Ann., 53, 14 (1894).
71. N. R e g g i a n i, Rend. Reale Accad. dei Lincei [4], 6, 99 (1890).
72. P. С. V 1 n c e n t, Proc. Phys. Soc., 45, 833 (1933); H. E. W i r t h, T. G. T h о m p-
s о n, C. L. U t t e r b a c k, J. Am. Chem. Soc., 57, 400 (1935); 0. E. F r i v о 1 d,
Physik. Z., 21, 529 (1920).
73. W. Geffcken, D. Price, Z. physik. Chem., B26, 81 (1934).
74. 0. R e d 1 i c h, J. Phys. Chem., 44, 619 (1940).
75. A. К r u i s, Z. physik. Chem., B34, 1 (1936).
76. W. Geffcken, A. К r u i s, L. Solana, Z. physik Chem., B35, 317 (1937).
77. R. С. H о a t h e r, C. F. G о о d e v e, Trans. Faraday Soc., 30, 630 (1934);
I. M. К 1 о t z, C. F. Eckert, J. Am. Chem. Soc., 64, 1878 (1942).
78. A. F. S с о 11, J. Phys. Chem., 35, 3379 (1931).
ТЕРМОХИМИЧЕСКИЕ И ПАРЦИАЛЬНЫЕ МОЛЯРНЫЕ ВЕЛИЧИНЫ
267
79, А. Ф. Иоффе, Физика кристаллов, Госиздат, М.—Л,, 1929.
80. В. Pesc'e, Gazz. chim. ital., 66, 99 (1936).
81. A. F. S с о t t, G. L. Bridge r, J. Phys. Chem., 39, 1031 (1935).
82. G. W. Stewart,!. Chem. Phys., 7, 869 (1939).
83. F. T. Gucker, F. W. G a,g e, С. E. Mose r, J. Am. Chem. Soc., 60, 2582
(1938); О. К. С к a p p e, С. Г. Д ем ид ен ко, А, И. Бродский, Acta
Physicochim. URSS, 6, 297 (1937).
84. a) F. Plato, J, D о m k e, H. Harting, Wiss. Abn. Normaleichungskommis-
sion, 2 Hefte, Berlin, Springer, 1900; б) O. R e d 1 i c h, H. Klinger, Monats-
hefte fiir Chemie, 65, 137 (1934).
85. W. R. В о u s f i e 1 d, T. M. Lowry, Trans. Faraday Soc., 6, 15 (1910).
86. 0. R e d 1 i c h, H. Klinger, Monatsh., 65, 137 (1934).
87. W. G e f f c k e n, Z. phys. Chem., A167, 240 (1933).
88. И. P. Кричевский, Acta Physicochem. URSS, 8, 181 (1938); ЖФХ, 11, 305(1938).
89. a) D. H. Adam s, J.. Am. Chem. Soc., 53, 3769 (1931); б) там же, 54, 2229 (1932).
90. L. H. A d a m s, R. E. G i b s о n, J. Am. Chem. Soc., 54, 4520 (1932).
91. R. E. Gibson, J. Am. Chem. Soc., 59, 1521 (1937).
92. F. T. G u c k e r, J. Am. Chem. Soc., 56, 1017 (1934).
93. R. E. Gibson, J. Phys. Chem., 31, 496 (1927); 38, 319 (1934).
94. F. T. G u c k e г, С. E. Mose r, J. Am. Chem. Soc., 61, 1558 (1939).
95. A. F. S с о t t, R. W. W i 1 s о n, J. Phys. Chem., 38, 951 (1934).
96. E. H. L a n m a n, B. J. Mair, J. Am. Chem. Soc., 56, 390 (1934).
97. R. E. G i b s о n, J. Am. Chem. Soc., 56, 4 (1934).
98. F. T. G u c k e r, Chem. Rev., 13, 111 (1933).
99. A. L. M о e s v.e 1 d, Z. physik, Chem., 105, 442, 450 (1923).
100. T. W. R i c h a r d s, W. N. Stull, Carnegie Inst. Pub., 7 (1903); 76 (1907).
101. P. G. Tait, Report on some of the physical properties of fresh-water and of sea-
water, 1888. From the «Physics and Chemistry» of the voyage of H. M. S. Challenger,
Vol. II, Part IV. S. P. LXI.
102. R. E. Gibson, J. Am. Chem. Soc., 57, 284 (1935).
103. a) R. E. Gibson, J. F. Kincaid, J. Am. Chem. Soc., 60, 511 (1938);
6) J. F. Kincaid, H. E у r i n g, J. Chem. Phys., 5, 587 (1937); L. J. H u d-
1 e s t о n, Trans. Faraday Soc., 33, 98 (1937).
104. P. W. Bridgman, Proc. Am. Acad. Arts Sci., 66, 185 (1931); 67, 1 (1932); 68, 1
105. H. Carl, Z. physik. Chem., 101, 238 (1922).
106. G. T a m m a n n, Uber die Beziehungen zwischen den inneren Kraften und Einge-
schaften der Losungen, Voss, Leipzig, 1907, p. 36.
107. R. E. Gibson, Am. J. Sci. [5], A35, 49 (1938).
108. R. E. Gibson, J. Am. Chem. Soc., 57, 290 (1935).
109. R. E. Gibson, Sci. Monthly, 46, 103 (1938).
110. J. D. Bernal, R. H. F о w 1 e r, J. Chem. Phys., 1, 540 (1933).
111. E. B. F г e у e r, J. Am. Chem. Soc., 53, 1313 (1931).
112. A. К u n d t, D. Lehmann, Pogg. Ann., 127, 497 (1866).
113. a) J. С. H u b b a r d, A. L. Loomis, Nature, 120, 189 (1927); Phil Mag. [7], 5
1177 (1928); Phys. Rev., 31, 158 (1928); б) E. B. F г e у e r, J. C. Hubbard,
D. H. Andrews, J. Am. Chem. Soc., 51, 759 (1929); E. B. F г e у e г, там же,
53, 1313 (1931); E. К 1 e i n, W. D. Hershberger, Phys. Rev., 37, 760 (1931).
114. L. Bergmann, Physik. Z., 34, 761 (1933).
115. A. S z a 1 a y, Physik. Z., 35, 639 (1934).
116. R. Wyss, Helv. Phys. Acta, 7, 406 (1934).
117. С. В a c h e m, E. H i e d e m a n n, Z. Physik, 89, 502 (1934); 91, 418 (1934); 94,
68 (1935).
118. E. H i e d e m a n n, N. S i e f e n, Z. Physik., 91, 413 (1934).
119, E. H i e d e m a n n, H. R. A s b a c h, К. H. H о e s c h, Z. Physik., 90, 322 (1934).
120. С. В a c h e m, E. H i e d e m a n n, H. R. A s b a c h, Z. Physik, 87, 734 (1934);
88, 395 (1934).
121. H. Falkenhagen, С. В a c h e m, Z. Elektrochem., 41, 570 (1935).
122. С. В a c h e m, Z. Physik, 101, 541 (1936).
123. P. D e b y e, F. W. Sear s, Proc. Nat. Acad., 18, 409 (1932); P. Debye, Phy-
sik. Z., 33, 849 (1932).
124. R. L u c a s, P. В i q u a r d, J. Phys. Radium, 3, 464 (1932).
125. H. Falkenhagen, С. В a c h e m, Z. Elektrochem., 41, 570 (1935).
126. F. T. G u c k e r, Jr., Am. Chem. Soc., 55, 2709 (1933).
127. F. T. G u c k e r, Chem. Rev., 13, 111 (1933).
128. E. P. P e r m a n, W. D. U r r y, Proc. Roy. Soc., A126, 44 (1929).
129. С. В a c h e m, Z. Physik, 101, 541 (1936).
130. R. E. G i b s о n, J. Am. Chem. Soc., 57, 1551 (1935).
268
ГЛАВА VIII
ЛИТЕРАТУРА К ТАБЛИЦАМ
К табл. 43
1. Е. A. G u lb ransen, A. L. R о b i n s о n, J. Am. Chem. Soc., 56, 2637 (1934;.
2. Н. Н а т т е г s с h т i d, A. L. Robinson, J. Am. Chem. Soc., 54, 3120 (1932).
3. E. Lange, A. L. Robinson, Z. physik. Chem., A148; 97 (1930).
4. E. L a n g e, P. A. L ei gh t on, Z. Elektrochem., 34, 566 (1928).
5. E. L a n g e, J. Monheim, Z. physik. Chem., A150, 349 (1930).
6. M. A n d a u e г, E. L a n g e, Z. physik. Chem., A165, 89 (1933).
7. E. Lange, H. S t г e e c k, Z. physik. Chem., A157, 1 (1931).
К табл. 44
1. H. S. H a r n e d, R. W. Ehlers, J. Am. Chem. Soc., 55, 2179 (1933).
2. E. T. G ,u с к e r, Jr., К. H. S c h m i n к e, J. Am. Chem. Soc., 54, 1358 (1932).
3. H. S. H a r n e d, M. А. С о о k, J. Am. Chem. Soc., 59, 1290 (1937).
4. M. Randall, F. D. R о s s i n i, J. Am. Chem. Soc., 51, 323 (1929).
5. H. S. H arned, M. A. Coo k, J. Am. Chem. Soc., 59, 496 (1937).
6. H. S. H a r n e d, J. С. H e с к e r, J. Am. Chem. Soc., 55, 4838 (1933).
7. F. T. G u с к e r, Jr., К. H. Schminke, J. Am. Chem. Soc., 55, 1013 (1933).
К табл. 45
1. F. T. G u с к e r, Jr., К. H. S c h m i n к e, J. Am. Chem. Soc., 54, 1358 (1932).
2. a) F. D. Rossini, Bur. Standards J. Research, 7, 47 (1931); 6) R a n d a 1 1,
F. D. Rossini, J. Am. Chem. Soc., 51, 323 (1929); в) С. M. Whit e, J. Phys.
Chem., 44, 494 (1940); С. B. Hess, там же, 45, 755 (1941).
3. F. T. G u с к e r, Jr., F. D. А у r e s, T. R. R u b i n, J. Am. Chem. Soc., 58, 2118
(1936).
4. F. T. G ц с к e r, Jr., К. H. S c h m i n к e, J. Am. Chem. Soc., 55, 1013 (1933).
К табл. 46
1. M. R a n d a 1 1, F. D. R о s s i n i, J. Am. Chem. Soc., 51, 323 (1929).
. 2. С. M. White, J. Am. Chem. Soc., 58, 1615 (1936).
К табл. 49
•1. A. F. Scott, J. Phys. Chem., 35, 2315 (1931).
2. W. G e f f с к e n, Z. physik. Chem., A155, 1 (1931); Naturwiss., 19, 321 (1931).
3, W. G e f f с к e n, D. P r i c e, Z. physik. Chem., B26, 81 (1934).
4. А. К r u i s, Z. physik. Chem., B34, 1 (1936).
5. R. E. G i b s о n, J. F. Kincaid, J. Am. Chem. Soc., 59, 25 (1937).
6. H. E. Wirth, J. Am..Chem. Soc., 59, 2549 (1937).
7. H. E. Wirth, J. Am. Chem. Soc., 62, 1128 (1940).
8. F. T. G u с к e r, Jr., T. R. R u b i n, J. Am. Chem. Soc., 58, 2118 (1936).
9. F. T. G u с к e r, Jr., Chem. Rev., 13, 111 (1933).
10. G. J о n e s, S. K. Talley, J. Am. Chem. Soc., 55, 624 (1933).
11. G. J о n e s, R. E. S t a u f f e r, J. Am. Chem. Soc., 62, 335 (1940).
12. G. J о n e s, J. H. С о 1 v i n, J. Am. Chem. Soc., 62, 338 (1940).
К табл. 52
1. В. В. Owen, S. R. Brinkley, Chem. Rev., 29, 461 (1941).
К табл. 53
1. R. E. G i b s о n, J. Am. Chem. Soc., 59, 1525 (1937).
2. R. E. G i b s о n, J. G. К i n c a i d, J. Am. Chem. Soc., 60, 511 (1938).
3. R. E. G i b s о n, О. H. Loeffler, J. Am. Chem. Soc., 61, 2517 (1939).
4. R. E. Gibson, Am. J. Sci. [5], A35, 49 (1938).
К табл. 54
1. E. В. F г e у e r, J. С. H u b b a r d, D. H. Andrews, J. Am. Chem. Soc., 51,
759 (1929).
2. J. С. H u b b a r d, A. L. Loomis, Phil. Mag. [7], 5, 1177 (1928).
3. L. G. Pooler, Phys. Rev., 35, 831 (1930).
К табл. 55
1. R. E. G i b s о n, J. Am. Chem. Soc., 57, 284 (1935), Table IV, p. 291.
К табл. 56
1. С. В a chem, Z. Physik., 101, 541 (1936).
2. F. T. G u с к e r, Chem. Rev. 13, 111 (1933).
Глава IX
ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ АКТИВНОСТИ И ОСМОТИЧЕСКИХ
КОЭФФИЦИЕНТОВ ИЗ ДАННЫХ ПО ТЕМПЕРАТУРАМ ЗАМЕРЗАНИЯ,
ТЕМПЕРАТУРАМ КИПЕНИЯ И ДАВЛЕНИЮ ПАРА
В предыдущей главе были рассмотрены все парциальные молярные
величины, за исключением ДУ и ДА1. Изменения парциальной молярной
свободной энергии или активности растворенных электролитов можно иссле-
довать прямым путем с помощью измерений электродвижущих сил соот-
ветствующих элементов или вычислять по уравнению Гиббса — Дюгема [(22)
гл. I] на основании наблюдаемых на опыте изменений свободной энергии
или активности растворителя. Настоящая глава посвящена наиболее важ-
ным методам определения изменений свободной энергии растворителя и свя-
занных с ней величин — коэффициентов активности и осмотических коэффи-
циентов. Метод электродвижущих сил будет рассмотрен в гл. X.
| 1. Введение
Если растворенное вещество нелетуче, то активность растворителя
можно определить по данным измерений понижения давления его паров, вы-
званного присутствием растворенного вещества. Преимущество этого метода
состоит в том, что он не ограничен какой-либо определенной температурой,
однако с его помощью нельзя достичь той точности, которая достигается
при использовании методов, основанных на измерении температуры замерзания
или температуры кипения, если не соблюдать специальных мер предосторож-
ности. Точные определения цонижения давления пара можно осуществить с
помощью чувствительного дифференциального манометра [1, 2, 3] или из дан-
ных по определению количества пара растворителя, необходимого для на-
сыщения данного объема инертного газа [4]. Статический (манометри-
ческий) метод был разработан главным образом Фрэзером и Ловлейсом [5].
Динамический' метод (насыщение воздуха) в экспериментальном отношении
проще статического; он применялся в большом числе исследований,
среди которых следует отметить работы Беркли [6], Уэшборна [7] и Пирса [8].
Методика определения понижения температуры замерзания достигла боль-
шого совершенства-. Первым исследователем, отказавшимся от разработанного
ранее Раулем [9] и Бекманом [10] метода переохлаждения, был, повиди-
мому, Ролов [11], а Гаусрат [12] и Осака [13] ввели в употребление термо-
пары. В дальнейшем измерение понижения температуры замерзания стало точ-
ным методом благодаря непрерывному совершенствованию методики измерений
и потенциометрического устройства. Ряд существенных усовершенствований
внесли в эту методику Уайт [14], Адамс [15], Рендалл. [16], Харкинс [17]
и Скэтчард [18]. Эти авторы рассмотрели различные источники экспери-
ментальных ошибок и определили величины этих ошибок [19]. Некоторые
менее значительные ошибки, связанные с вычислением коэффициентов
1 Величиной ДУ пользуются очень редко, поэтому мы не рассматриваем ее подробно.
Зависимость Д/»' от температуры почти всегда выражают через ДН, а не через ДД-.
270
ГЛАВА IX
активности из экспериментальных данных, будут обсуждены в этой
главе.
Точное определение повышения температуры кипения является значитель-
но более затруднительным, чем измерение понижения температуры замерза-
ния, вследствие явления перегрева и необходимости тщательного наблюдения
за величиной давления. Кроме того, молярное повышение температуры кипения
меньше, чем соответствующее понижение температуры замерзания, а поэтому
ошибки при измерениях температуры кипения вызывают большую ошибку
при вычислении термодинамических величин, чем ошибки в измерениях
температуры замерзания. Котрель [20], а также Уэшборн и Рид [21] поло-
жили начало успешной разработке метода устранения наиболее серьезного
экспериментального затруднения — явления перегрева, —а Смит [22] достиг
в этом направлении наибольших успехов. Нет необходимости приводить
здесь подробный обзор многочисленных методических усовершенствований
последнего периода, поскольку они подробно освещены в одной из совре-
менных монографий [23] и по своему характеру аналогичны усовершенство-
ваниям методики определения температуры замерзания.
§ 2. Общие уравнения для вычисления активности растворителя
и осмотического коэффициента из данных по понижению температуры
замерзания
Обозначим через а' активность твердого растворителя при темпера-
туре Т', соответствующей температуре замерзания раствора, содержащего т.
молей растворенного вещества на килограмм растворителя; пусть при этих
условиях активность жидкого растворителя равна а®', а активности рас-
творителя и растворенного вещества в растворе равны соответственно а' и
а2- Увеличение Т*7! при изотермическом переходе одного моля чистого
растворителя из жидкого состояния в твердое равно RT'1п а'/а®', а при
переходе одного моля растворителя из состояния чистой жидкости в со-
стояние раствора составляет RT'In а'/а®’ [уравнения (29) и (30) гл. I].
Примем состояние чистого растворителя за стандартное, относительно ко-
торого определяются все активности, т. е. будем считать а®' равным еди-
нице. Таким образом1,
< = «'1 » (О
И
И) =1- (2)
Применяя уравнение (38) гл. I к переходу одного моля чистого раствори-
теля из жидкого состояния в твердое, можно найти зависимость между с/
(и следовательно а[) и понижением температуры замерзания
(3>
где T(s — температура замерзания чистого растворителя. При таком переходе
АН равняется разности между относительными молярными теплосодержаниями
твердого и чистого жидкого растворителей. Так как чистый жидкий рас-
1 Так как по условиям измерения необходимо лишь наличие равновесия между твер-
дым растворителем и каким-либо раствором при Т’, то отсутствие равновесия между твер-
дым и чистым жидким растворителями при всех температурах, кроме не имеет значе-
ния. Состояние чистого растворителя следует выбрать в качестве стандартного для того,
чтобы обычные условия, выраженные уравнениями (1) и (2), соблюдались при всех тем-
пературах.
ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ АКТИВНОСТИ
271
творитель находится в стандартном состоянии, то его относительное моляр-
ное теплосодержание равно нулю. Поэтому
L' Ь'М
dlna'——.—s—dT' =------?----, (4)
s 7?Z 2 7?(Z0 —Э)2 v 7
где L's — относительное молекулярное теплосодержание твердого вещества
при Т'. Хотя температура в этом уравнении является переменной, мы со-
храним индекс «штрих» для напоминания о том, что вычисления произво-
дятся для температуры замерзания раствора. Прежде чем интегрировать
это уравнение, следует выразить L' в виде функции от Т' или 4) .
Если молярные’ теплоемкости твердого и чистого жидкого раствори-
теля выразить в форме степенных рядов с переменной &, то получаем
СО
^-^=2 (5>
1
где Ап — разность между коэффициентами п-х членов обоих рядов. Ао равно
ACpj при 7’0. Применяя уравнение (40) гл. I, получаем
Т' Т' 9 оо
*\dLs — (CPs-C°Pi)dT= - An-i^db, (6)
То То 0 1
откуда1
СО
L;=Ls(0)-2|A-i8n. (7)
1
Подставляя это значение L' в уравнение (4), заменяя а' на а' и разлагая
в ряд (То —I))-2, получаем следующее выражение:
оо оо
где
1-п s=n
(7п==ТГп-^-2(“У^-1 (9>
ьз(0) , V ns >
s= 1
И
1 Первая попытка придать общий вид уравнению (4) (заменив аг на Nt) путем соче-
тания его с выражением, аналогичным уравнению (7), принадлежит, невидимому, Ван-
Лаару [24а], который выразил зависимость Ls от N2 в форме степенного ряда и определил
численные коэффициенты этого ряда из данных по точкам замерзания. В дальнейшем, раз-
вивая этот метод, Ван-Лаар [246] выразил L, через коэффициенты уравнения состояния
Ван-дер-Ваальса для бинарной смеси [24в]. Уэшборн разложил Ls в степенной ряд по
температуре и получил выражение, аналогичное уравнению (7), но подстановка в уравнение
(4) (при замене аг на Л\) приводит к выражению, содержащему логарифмический член,
и поэтому указанное выражение нельзя непосредственно сравнить с приведенным выше.
При дальнейшем «общем интегрировании» Уэшборн, сохраняя логарифмическую форму,
к сожалению, отбрасывает все коэффициенты после второго в уравнении (7). При срав-
нении уравнений Уэшборна с приведенными выше следует обратить внимание на опе-
чатки [24 г]. Обозначения ДСр и L необходимо изменить в соответствии с принятыми
в настоящее время.
272
ГЛАВА IX
Можно показать [уравнение (52)], что vk представляет собой понижение темпе-
ратуры замерзания, отнесенное к молю растворенного вещества, при т, стре-
мящемся к нулю. Численные значения1 этой важной величины для неко-
торых растворителей приведены в табл. 57. Для водных растворов четыре
первых коэффициента, вычисленных по уравнению (9), равны соответст-
венно С± = 1, С2 = 5,2 • 10 4, С3 — — 8,5 • 10~в и — 5 • 10~8.
Таблица 57
Криоскопические постоянные различных
растворителей при давлении, равном 1 атм
(г—температура замерзания)
Растворитель t Л1)
Аммиак -15 0,72
Вода 0 1,858
Уксусная кислота 16,6 3,73
Диоксан 11,7 4,63 2)
Бензол 5,4 5,08
Фенол 25,4 6,11
Хлорное олово . . • -33 13,6
Четыреххлористый углерод . -24 29,6
Камфора . . . : 178,4 37,7 3)
Цикло гексанол 23,2 41,64)
1) Вычислено по уравнению (10).
£) Определено из данных по температурам замерза-
ния [I].
з) Определено из данных по температурам замерза-
ния [2].
СР- [3].
В интегральной форме уравнение (8) имеет вид
СО
In а\ = 100(^ 2 (11)
1
и с помощью уравнения (69) гл. I получается следующее выражение для
осмотического коэффициента при температуре замерзания раствора:
СО
<)2>
1
Для разбавленных водных растворов сулгма первых двух членов равна
- In а[ - 9,696 • 10“3& + 5,1 • 10-6&2 (13)
и
= —1— (1 + 0,54 . 10-3&). (14)
• 1,858 чт ' ' ' >
1 Необходимые для вычислений значения Та и Ls^y взяты из «International Critical
Tables» [25].
ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ АКТИВНОСТИ
273
§ 3. Общие уравнения для вычисления активности
растворителя и осмотического коэффициента из данных
по повышению температуры кипения
При температуре кипения Т' раствора, содержащего тп молей раство-
ренного вещества на килограмм растворителя, активность парообразного
растворителя равна а'„, а активность растворителя в растворе равна a't.
Если принять состояние чистого жидкого растворителя за стандартное
для обеих фаз, то будет равным а{, если давление равно давлению
пара чистого растворителя р0 при То. Поэтому, согласно уравнению (38)
гл. I, активность растворителя, выраженная через теплоту его испарения L'v,
будет определяться уравнением
L'v dT’ Ц </0
d In а[ = — RT,2 = — R(To + q}2 > (15)
а повышение температуры кипения будет равно
(16)
Если выразить в виде степенного ряда
СО
(17)
1
разность между молярными теплоемкостями пара и чистого растворителя
при постоянном давлении1 р0, то, согласно уравнению (40) гл. I, полу-
чаем выражение
СО
L'v = Lv (о) + 3 (18)
1
После подстановки этого выражения в уравнение (15) и разложения в ряд
(T'o-J-O)-2 получается следующее уравнение:
оо ОО
d 1па( = - 3 de = E nC^n~l dQ’
° i i
где
«- (- т-.)-"+s C2^) A!-< < -
' s -1
И
, MrRTl
K “I- 1000Lr(0) •
В табл. 58 приведены значения молярного повышения температуры кипе^-
ния к* для воды и ряда обычных органических растворителей по отношению
к их нормальным температурам кипения. Значения к* и первых трех коэффи-
циентов С* для воды при различных давлениях приведены в табл. 59.
(19)
(20)
(21)
1 Джонсоном [26а] подробно описано вычисление зависимости Lv от температуры
при постоянном давлении р0. Эти вычисления производились им с помощью данных,
полученных при непрерывно меняющемся давлении насыщения. Полученные данные при-
ведены в статье Джонсона и Смита [266].
18 Г. Харнед, Б. Оуэн
274
ГЛАВА IX
Таблица 58
Эбулиоскопические постоянные различных растворителей при давлении, равном
1 атм 1). J—температура кипения
Растворитель t Л* Растворитель t Л*
Аммиак -33,4 0,349 Сероуглерод 46,3 2,41
Вода 100,0 0,513 Бензол 80,2 2,628
Метиловый спирт . . . 64,7 0,862 Циклогексанол .... 116,1 2,78
Этиловый » ... 78,3 1,214 Уксусная кислота . . 118,3 3,15
Двуокись серы .... -10,1 1,45
1) Вычислено с помощью уравнения (21) по данным из «International Critical Tables» [1].
Таблица 59
Эбулиоскопические постоянные воды при различных давлениях1)
t Ро Л* Ж с 1 5 ж 10 С2 108С* ю10с* 4
60 149,4 0,39108 1 -347,900 1072,13 - 325,2
70 233,7 0,41931 1 -339,201 992,13 -284,1
80 355,2 0,44905 1 -331,029 915,88 — 245,9
. 90 525,8 0,48007 1 -323,132 822,64 -198,2
100 760,0 0,51276 . 1 -316,017 784,24 -184,4
1) Вычислено с помощью численных значений констант, применявшихся в работе Смита [1].
Т=273,1 + 1; Л=8,3126 международного джоуля.
Интегрируя уравнение (19) и применяя уравнение (69) гл. I, получаем
СО
(22)
1
и
оо
(23)
1
§ 4. Общие уравнения для вычисления активности растворителя
и осмотического коэффициента из данных по измерению
давления пара и из изопиестических данных
При сочетании уравнения (10) гл. I с выражением для активности
чистого газа при постоянной температуре RT d In а = dF получается выра-
жение, характеризующее изменение активности паров воды с давлением:
< дР )т~ RT •
(24)
ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ АКТИВНОСТИ 275
В случае равновесия общее давление Р равно давлению паров воды р.
Молярный объем можно выразить в следующем виде:
TZ RT
V = -р- — а, (25)
где а является в общем случае функцией Р и Т, а если Р мало, то ос
можно считать зависящим лишь от Т. Сочетая написанные выше урав-
нения и пользуясь пропорциональностью между активностью растворителя
в газовой фазе и в находящемся в равновесии с нею растворе, получаем
Й1па1 = й1п7> — -^pdP. (26)
Интегрирование в пределах от р до р0 (давление пара чистого раствори-
теля) приводит к выражению
р
In й! = In р/р0 (27)
ро
Таким образом, осмотический коэффициент [уравнение (69) гл. I] равен
р
1000 , р 1000 с ]П оо.
С₽ —-----In И •—г,- р \ a dP. (28)
т чтМ± р0 чтМуКТ J v '
ро
При практических вычислениях пренебрегают членами, содержащими а,
так как они меньше экспериментальных ошибок при измерениях давле-
ния пара при комнатных температурах1.
Если известна зависимость осмотического коэффициента или актив-
ности растворителя от концентрации для некоторых стандартных растворов,
то ее нетрудно найти и для других растворов с помощью метода изопиести-
ческого сравнения [27]. Пробы стандартного и исследуемого растворов
взвешивают в серебряных чашках и приводят в хороший тепловой кон-
такт в вакуумном эксикаторе. После того как установится равновесие,
давление пара растворителя становится одинаковым для всех растворов.
После этого равновесные концентрации определяются путем вторичного
взвешивания чашек с растворами. Активность и осмотический коэффициент
исследуемого раствора с концентрацией т, если их выразить через .соот-
ветствующие свойства стандартного раствора с концентрацией mR, описы-
ваются следующими уравнениями:
«1 (№) = «! Н(тд) (29)
и
= (30)
Значения для важнейших стандартных растворов приведены в табл. 60.
В качестве стандартных растворов обычно пользуются растворами хлори-
стого калия или хлористого натрия.
1 Обычно пренебрегают также тем, что значения и <р, вычисленные из прямых
манометрических измерений, определяются при давлении р, которое зависит от состана
раствора. Однако численная величина VJRT такова, что, как видно из уравнения (44)
гл. I, поправка, необходимая для приведения значений аг и к постоянному давлению р0
или к 1 атм, гораздо меньше ошибки опыта.
18*
276
ГЛАВА
Таблица 60
Осмотические коэффициенты стандартных растворов при 25°
т NaCl1) КС11) H2SO.i2) Сахароза8)
0,1 0,9319 0,9257 0,6800 1,0073
0,2 0,9246 0,9129 0,6680 1,0151
0,3 0,9212 0,9062 0,6672 1,0234
0,5 0,9217 0,9002 0,6748 1,0410
0,7 0,9265 0,8979 0,6888 1,0596
1,0 0,9378 0,8994 0,7195 1,0897
1,5 0,9594 0,9056 0,7823 1,1415
2,0 0,9862 0,9148 0,8488 1,1929
2,5 • 1,0170 0,9274 0,9185 1,2436
3,0 1,0498 0,9409 0,9960 1,2923
3,5 1,0847 0,9552 1,0725 1,3379
4,0 1,1219 0,9698 1,1523 1,3805
4,5 0,9854 1,2298 1,4186
5,0 0,99584) 1,3041 1,4541
5,5 1,3766 1,4881
6,0 1,4485 1,5216
7,0 1,5817 ,
8,0 1,6998
3) Эти значения были предоставлены авторам Р. А. Робинзоном. Они были согласованы
с наилучшими результатами определения коэффициентов активности хлоридов и бромидов натрия
и калия с помощью метода электродвижущих сил и по давлению пара (десять независимых изме-
рений), а также с результатами последних определений иаопиестических отношений. Значения
осмотических коэффициентов даны с точностью до четвертого знака после запятой, чтобы можно
было с их помощью провести плавную кривую. Последняя значащая цифра не имеет физическог
значения.
2) Эти значения вычислены из данных, приведенных во втором и третьем столбцах, и изопие-
стических отношений, найденных Скэтчардом, Гамером и Вудом [1], Шеффером, Дженисом и Фер-
гюсоном (2] и Робинзоном [3]. Для концентраций, превышающих 3 М, значения ? вычислены
с помощью данных, приведенных в статье Шэнкмана и Гордона [4].
з) Эти значения вычислены с помощью данных третьего столбца и изопиестических отно-
шений, приведенных в статьях Скэтчарда, Гамера и Вуда [1] и Робинзона и Синклера [5]; исполь-
зованы также неопубликованные данные Робинзона.
1) т=4,81.
§ 5. Вычисление коэффициента активности растворенного
вещества с помощью осмотического коэффициента
или активности растворителя
Это вычисление, которое связано с интегрированием уравневия (45)
гл. I, выполняется с помощью различных аналитических и графических
методов. Мы рассмотрим типичные методы, применяя их к данным, взятым
из нескольких источников. Путем преобразования уравнений (70) и (71)
гл. I получаются следующие основные соотношения:
d In у± = — In — d Inm (31)
И
6?InY±= - ±-d[m(l— <?)], (32)
которые должны быть проинтегрированы. Так как значения In и можно
выразить эмпирически в виде функций от т, то они могут быть исклю-
чены из этих уравнений. К сожалению, абсолютная точность, с которой
ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ АКТИВНОСТИ
277
по этому способу определяется 1пу±, в значительной мере зависит
от вида этих функций, если нижним пределом интегрирования является
ш = 0. С помощью уравнения Дебая — Гюккеля можно выбрать подходящие
функции, посредством которых можно выразить свойства растворов в про-
межутке концентраций от бесконечного разведения до наименьших кон-
центраций, применяющихся в исследовании.
Сочетание уравнения (10) гл. V с уравнением (57) гл. I приводит
к следующему выражению:
<33>
Так как это уравнение применимо только к разбавленным растворам, то
можно пренебречь последним членом. Поэтому, если принять х = А,угс
и к’ = 2,303 то
бИпу±=—к' (1 + ж)~2 dx. (34)
Так как т пропорционально х2 при большом разбавлении, то при под-
становке d In у± из этого уравнения в уравнение (32) получается следующее
выражение:
X X
, к’ С dx к' Г , dx
1 — ср = — \ т -ту--—= —5- \ X2 77——Г5- . (35)
т т J + хг J (1-|-ж)2 ' '
о о
После интегрирования и преобразования получаем
1-»-т{1[1+1-2|"(1 + >)-177]}^‘т. (36)
Функцию а, которая определяется этим уравнением, можно выразить сле-
дующим образом:
ОО со
°-2 = 2 (37)
П=1 ' П=1
если х < 1. Подставляя значение к' в (36), получаем
1 — ср = 0,7676 5(/)в/с. (38)
Любое соотношение, которое используется для выражения зависимости
экспериментальных значений 1 —ср от состава раствора, должно переходить
в это уравнение при больших разбавлениях. Численные значения S(f> и А'
приведены в табл. 7, а значения а —в табл. 61.
Так как приведенное выше уравнение можно применять только в слу-
чае разбавленных растворов, для которых с = у' md0, то можно ввести
следующие обозначения:
(f) ]/~d0, (39)
Am A' j/ d0 (40)
и
co
°m— 2 /7~2 — (41)
n=l
Таким образом,
1 - ср = 0,7676 (42)
Й78
Г Л А В А IX
Это выражение в пределе переходит в уравнение (38), а при средних
разведениях отличается от него на малую величину, которой можно прене-
бречь.
Таблица 61* 1)
Функция о2)
A' Vc 9 А' У с 9 А' Ус G
0,00 1i0000 0,40 0,5986 0,80 0,4036
0,05 0,9293 0,45 0,5668 0,85 0,3863
0,10 0,8662 0,50 0,5377 0,90 0,3703
0,15 0,8097 0,55 0,5108 0,95 0,3552
0,20 0,7588 0,60 0,4860 1,00 . 0,3411
0,25 0,7129 0,65 0,4631 1,05 0,3279
0,30 0,6712 0,70 0,4418 1,10 0,3154
0,35 0,6332 0,75 0,4220 1,15 0,3037
1) При пользовании моляльными концентрациями (т), А'Ус на заменяются
соответственно на Ат и ат-
2) См. табл, 174.
Смит [28] вычислил ср' для растворов хлористого натрия при темпе-
ратуре кипения, используя выражение
1 — ф' = 0,7676 Sm if) 0,5 Вт, (42а)
Наличие члена 0,5 Вт дает возможность применять это уравнение в случае
высоких концентраций [ср. с уравнением (74) гл. III]. Численные значения
Рис. 67. Функция отклонения
3<р' для растворов хлористого на-
трия в воде при различных темпе-
ратурах. Диаметр кружка равен
0,0005.
Ат и В можно выбрать так, чтобы откло-
нение
Sep' = —(1 — ф')+ 0,7676 Sm(/)cmVn'1— 0,5Вт
(43)
было невелико при любой концентрации.
Из сопоставления уравнений (43) и (42) вид-
но, что при бесконечном разбавлении Sep'
должно обращаться в нуль. Сочетая урав-
нения (43) и (42) и интегрируя, получаем
1пу± =
2,303 Sm(/) /т
А-Вт +-
7П
+ $ 77 dm + 8®Z'
6
Два члена, содержащие Sep', определяются
графически. Из рис. 67, который содер-
жит значения Sep', найденные Смитом для
растворов хлористого натрия (2% = 60, 70,
80, 90 и 100°), видно, что изменение величи-
ны обусловленное введением этих чле-
нов, не превышает 2 — 3%.
Следует отметить, что величина, обозначенная через у'±, не является
истинным коэффициентом активности, потому что уравнение (32) было
1 -|- -4m "jC
ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ АКТИВНОСТИ
279
использовано при неизотермических условиях. Из этой величины можно
получить истинный коэффициент активности, если до интегрирования урав-
нения (32) привести все значения ср' к некоторой стандартной температуре Т".
Пользуясь интегральной формой уравнения (38) гл. I
lg а'\ = lg а' + X (45)
и соотношением
,г , 2,303 X
где
X = -Д_____________ (47)
\ 2,303Вт12 ’ ' >
Т'
получаем выражение
х
lgY" = lgY'-4^ (48)
о
применяемое Льюисом и Рендаллом [29а] *.
Вычисление коэффициентов активности из уравнений (44) и (48) является
в значительной степени аналитическим, потому что члены, содержащие Sep'
и dX, весьма мало влияют на окончательный результат. Скэтчард [30]
предложил формальный, чисто аналитический метод вычисления коэффи-
циента активности и осмотического коэффициента, однако обычно поль-
зуются графическим способом. Интегрирование уравнения (32) приводит
К следующему выражению для коэффициента активности:
m
In у± = — (1 — ср) — 2 (49)
о 4 m у
если dm заменить на 2m1/a dm1^. Кривую, выражающую зависимость
(1 — ср)/тт/2 от т1/2, можно использовать для определения обоих членов,
содержащих ср. Значение (1 —ср)/»!1/2 в области разбавленных растворов
настолько чувствительно к экспериментальным ошибкам, что для проведе-
ния кривой от 0,1 М до бесконечного разведения следует применить урав-
нение Дебая—Гюккеля. Для разбавленных растворов из уравнения (42) полу-
чаем выражение
(1 - ср)/!»1/2 = 0,7676Sm (/)am. (50)
При использовании этого уравнения для получения полной кривой, выра-
жающей зависимость (1 — ср)/»!1/2 от w!1/2, необходимо знать А'т для того,
чтобы вычислить ат. Так как а обычно неизвестно, то путем подбора находят
такое значение А'т, при котором уравнение (50) удовлетворяет эксперимен-
тальным данным при самых низких концентрациях. Методика вычисления А'т
иллюстрируется ниже в связи с вычислением /-функции [уравнение (51)].
Уравнения (49) и (50) обычно используют при вычислении коэффициентов
активности по осмотическим коэффициентам, полученным из данных по
давлению пара или из результатов изопиестических измерений. В табл. 146
и 151 приведены значения у±, полученные с помощью таких вычислений.
Для определения коэффициентов активности из данных по понижению
точек замерзания производят графическое интегрирование уравнения Гиббса—
1 См. также [29в].
280
ГЛАВА IX
Дюгема. При этом интегрировании обычно используют так называемую
переменную1 Льюиса и Рендалла [296]
/ = 1---(51)
' у/пА '
Сочетая уравнения (8), (31) и (48), получаем
й 1g У1== 0,4343-Д- У nCX~l-dlgm-^ —. (52)
1
Так как при дифференцировании уравнения (51) получается выражение
-^(i-fidlnm-dj, (53)
а коэффициент С\ равен единице, то уравнение (52) может быть написано
в следующем виде:
,, „ А /о,о ,• о 1 , 0,4343с/0 ст „п-1 1000 АУ ri,
dlgY± = - 0,4343 rfj-jcHgm + _-—2пС„» 54)
2
откуда
= — 0,4343/ —0,8686 Г +
Я со X
Щ4343 С (55)
vA J т п vMt J w х '
0 2 0
Калориметрические данные, необходимые для вычисления Сп, обычно отсут-
ствуют (за исключением Сх и С2). Для воды можно получить значение Cs,
если воспользоваться данными точных измерений температуры замерзания
при средних концентрациях 2. В большинстве случаев всеми членами выше С2
пренебрегают, и уравнение (55) принимает приближенную, но более про-
стую форму:
lgY± = -0,4343/-0,8686 f +
У -
» x
+ 0.8686G (1 - /) db - . (56)
о 1 о
В случае разбавленных растворов, когда можно оставить в правой
части уравнения (54) только два первых члена, нетрудно исключить d lg у±
с помощью уравнения (34). Тогда, прибегнув к способу, уже употребляв-
шемуся при выводе уравнений (38) и (42), можно получить следующие
уравнения:
/ = 0,7676 S(/)c]/c, (57)
/ = 0,7676 £т(/)ат]/щ. (58)
1 Величину /* = 1 — ^1'imK* можно аналогичным образом использовать для вычис-
ления коэффициентов активности из данных по температурам кипения.
2 Юнг [31] показал, что влияние различных членов на коэффициент активности
5,2 М хлористого натрия составляет примерно 0,58% для члена С3, 0,06% для С4 и 0,006%
для С8. По мере разбавления раствора влияние этих членов на коэффициент активности
быстро уменьшается.
ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ АКТИВНОСТИ
•281
Очевидно,, что / = 1 — ср в том интервале концентраций, для которого выве-
дены эти уравнения.
Применение этих уравнений и выбор подходящего значения А'т, необхо-
димого для вычисления ат, иллюстрируется рис. 68. На этом рисунке
изображена зависимость экспериментальных [32] значений j/m1^ от т1^ для
водных растворов хлористого калия. Сплошную кривую, соответствую-
щую этим значениям, можно построить
с большой точностью вплоть до концен-
траций, для которых щ1/2=0,3, и с до-
статочной степенью точности — до = 0,2.
По экспериментальным значениям в про-
межутке между т1/2 = 0,2 и бесконечным
разбавлением нельзя определить форму
кривой на этом участке. Эта часть кри-
вой, изображенная пунктирной линией,
была получена с помощью уравнения (58)для
значения Ат, соответствующего а = 3,6А.
°
При выборе подходящей величины а или Ат
экспериментальное значение Цтп№, опреде-
ляемое из графика для т1/2, приблизитель-
но равного 0,2, может быть использовано
для вычисления Лт^1/2, а следовательно,
и А'т. Это вычисление производится с по-
мощью уравнений (41) и (58) путем ряда
последовательных приближений. Для вод-
ных растворов Спенсер [33] составил та-
блицу теоретических значений jlm1^ и
соответствующих различным значениям
О о
а (от 1 до 8 А), Спенсер пользовался вы-
ражением
Пунктирные кривые 1, 2 и 1 отвечают
о
уравнению (59) соответственно для а=2,
о
8,6 и 7 А. Пунктирная линия з отвечает
о о
уравнению (58) при а=3,6 А.
/ = 0,3738 ст]/т- 0,1640 (г2/а)3103[Х3(я;)-6У3 (х)]-
- 0,0780 (z2/e)5 Ю5 [Хв (ж) - 10У6 (ж)], (59)
в которое входят члены, соответствующие членам высших порядков урав-
нения (42) гл. III для симметричных электролитов. Три пунктирные кри-
вые на рис. 68 построены по данным Спенсера, вычисленным для а, равного
соответственно 2, 3,6 и 7А. Из рисунка видно, какое влияние оказывают
члены высших порядков в случае водных растворов хлористого калия,
так как кривые, построенные с помощью уравнений (59) и (58), явно не
совпадают друг с другом. Из рисунка также следует, что если бы для
экстраполяции экспериментальной кривой было применено уравнение (59),
то значение а следовало бы выбрать большим, чем 3,6. Расхождение между
значениями а, получающимися из этих двух уравнений, равное примерно
О
0,1 А, несущественно, так как оба уравнения применяются при столь высо-
О
ких концентрациях (иг1/2 = 0,2), что параметр а приходится подбирать таким
образом, чтобы компенсировать эффекты, обусловленные приравниванием
с к величине dom и отбрасыванием двух членов в уравнении (55). В связи с
этим экстраполяция сплошной кривой на рис. 68 является эмпирической для
всех значений ]Zт, кроме теоретической величины, соответствующей точке
пересечения кривой с осью ординат. Недавно Скэтчард [18] придал этим
182
ГЛАВА IX
расчетам еще более эмпирический характер, применив для случая 1,1-ва-
лентных электролитов следующее выражение:
7
0,3738m1'2
1 + Вт1'1
Ь Ст,
(60)
•которое при бесконечном разведении переходит в уравнение (58), но не
согласуется с теорией Дебая—Гюккеля при более высоких концентрациях.
Достоинство этого выражения состоит в том, что с его помощью можно
получать приближенные значения температуры замерзания растворов с концен-
трациями вплоть до \ М. Если при расчетах наряду с этим выражением
—(0,3738 m1/2)/(l + 1,7m1/2) + 0,0149m.
Сплошную кривую использовал Скэтчард для получения
усредненных значений У из экспериментальных данных;
пунктирную линию применял Спенсер, использовавший
более ранние экспериментальные данные.
^пользуются графиком отклонений 8/ от значений, вычисленных по урав-
нению (60) на основании экспериментальных данных, то этот эмпирический
способ должен давать результаты, весьма близкие к теоретическим. Этот
способ имеет определенные преимущества. Обычно выбирают такие значения
эмпирических постоянных В и С, чтобы отклонения
0,3738m1/2
1 +7Ут1''2
Ст
(61)
не превышали 0,01 — 0,02. Поэтому построение кривой, выражающей зави-
симость отклонений 8/ от т или от т1!2, является весьма чувствительным
и удобным способом получения усредненных данных и определения значе-
ний j, необходимых для вычисления интегралов в уравнениях (55) или (56).
На рис. 69 кружками представлены данные Скэтчарда и Прентиса [32]
для хлористого калия, причем, согласно этим авторам, зависимость 8/
от т1/2 изображается сплошной линией. На этом рисунке для сравнения
приведена также пунктирная кривая, построенная по значениям j, полу-
ченным Спенсером [33] из более ранних данных. Расхождения между
обеими кривыми не превышают +0,001 от величины /, что является вполне
удовлетворительным, если учесть, что данные, использованные для построе-
ния кривых, взяты из различных источников.
Такого рода графики были использованы для вычисления значений
— 1g для большого числа 1,1-валентных электролитов из многочисленных
-опытных данных Скэтчарда и Прентиса. Эти данные приведены в табл. 123
и будут рассмотрены в гл. XII.
ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ АКТИВНОСТИ
283
ЛИТЕРАТУРА
1. С. D i е t е г i с i, Ann. Physik [3], 50, 47 (1893); 62, 616 (1897); 67, 859 (1899).
2. R ay 1 eigh, Z. physik. Chem., 37, 713 (1901); Trans. Roy. Soc., 196, 205 (1901).
3. A. Smits, Z. physik. Chem., 39, 386 (1906).
4. J. Walker, Z. physik. Chem., 2, 602 (1888).
5. J. C. W. Frazer, B. F. Lovelace, Z. physik. Chem., 89, 155 (1914); J. Am.
Chem. Soc., 36, 2439 (1914); J. C. W. Frazer, B. F. Lovelace, E. Mil-
ler, там же, 38, 515 (1916); J. C. W. Frazer, B. F. Lovelace, T. H. R o-
gers, там же, 42, 1793 (1920); J. C. W. Frazer, B. F. Lovelace,
V. B. S e a s e, там же, 43, 102 (1921); J. C. W. F r a z e r, B. F. Lovela-
ce, W. H. В a h 1 к e, там же, 45, 2930 (1923).
6. В e г к e 1 e у, E. G. J. Hartley, С. V. Burton, Phil. Trans. Roy. Soc.,
A209, 177 (1909); A218, 295 (1919).
7. E. W. Washburn, E. О. H e u s e, J. Am. Chem. Soc., 37, 309 (1915).
8. J. N. Pearce, R. D. Snow, J. Phys. Chem., 31, 231 (1927); J. N. Pearce,
A. F. Nelson, J. Am. Chem. Soc., 54, 3544 (1932); 55, 3075 (1933); J. N. Pear-
ce, L. E. В 1 а с к m a n, там же, 57, 24 (1935); J. N. Pearce, H. C. Eck-
strom, там же, 59, 2689 (1937); J. Phys. Chem., 41, 563 (1936).
9. F. M. R а о u 1 t, Ann. chim. phys..[5], 20, 217 (1880).
10. E. Beckmann, Z. physik. Chem., 2, 638 (1888); 7, 323 (1891).
11. M. R о 1 о f f, Z. physik. Chem., 18, 572 (1895).
12. H. H a u s r a t h, Ann. Physik. [4], 9, 522 (1902).
13. Y. О s a к a, Z. physik. Chem., 41, 560 (1902).
14. W. P. White, J. Am. Chem. Soc., 36, 1859, 1876, 2011, 2292, 2313 (1914).
15. L. H. Adams, J. Am. Chem. Soc., 37, 481 (1915).
16. M. R and all, A. P. V a n s e 1 о w, J. Am. Chem. Soc., 46, 2418 (1924).
17. R. E. Hall, W. D. Harkins, J. Am. Chem. Soc., 38, 2658 (1916).
18. G. S c a t c h a r d, P. T. Jones, S. S. Prentiss, J. Am. Chem. Soc., 54,
2676 (1932).
19. C. Robertson, V. K. L a M e r, J. Phys. Chem., 35, 1953 (1931).
20. F. G. Cottrell, J. Am. Chem. Soc., 41, 721 (1919).
21. E. W. Washburn, J. W. Read,. J. Am. Chem. Soc., 41, 729 (1919).
22. R. P. Smith, J. Am. Chem. Soc., 61, 497 (1939).
23. W. Swietoslawski, Ebulliometry, Chemical Publishing Co., N. Y., 1937.
24. a) J. J. van Laar, Kon. Akad. Wetten. Amsterdam, Verslog Wis.-en Natuur..
11, 478 (1902); б) там же, И, 576 (1903); в) E. W. W a s h b u г n, J. Am. Chem
Soc., 32,653 (1910); г) там же, p. 1636.
25.. International Critical Tables, Vol. V, McGraw-Hill Book Co., N. Y., 1931, p. 130.
26. a) G. C. J о h n s о n, Диссертация, Yale University,, 1940; 6) G. C. Johnson,
R. P. Smith, J. Am. Chem. Soc., 63, 1351 (1941).
27. D. A. S i n с 1 a i r, 1 J. Phys. Chem., 37, 495 (1933); R. A. Robinson, D. A.
Sinclair, J. Am. Chem. Soc., 56, 1830 (1934).
28. R. P. Smith, J. Am. Chem. Soc., 61, 500 (1939).
29. а) Г. H. Льюис, M. P e н д а л л, Химическая термодинамика, ОНТИ—Хим-
теорет, Л., 1936, стр. 272: б) там же, стр. 269; в) М. R е n d а 1 1, L. Е. Y о u n g, J.
Am. Chem. Soc., 50, 989 (1928).
30. G. Scatchard, Chem. Rev., 8, 321 (1931); G. S c a t c h a r d, S. S. Pren-
tiss, J. Am. Chem. Soc., 56, 1486 (1934).
31. T. F. Young, Chem. Rev., 13, 103 (1933).
32. G. S c a t c h a r d, S. S. Prentiss, J. Am. Chem. Soc., 55, 4355 (1933).
33. H. M. Spence r, J. Am. Chem. Soc., 54, 4490 (1932).
ЛИТЕРАТУРА К ТАБЛИЦАМ
К табл. 57
1. С. A. Kraus, R. А. V i n g е е, J. Am. Chem. Soc., 56, 511 (1934).
2. М. F г a n d s е n, Bur. Standards, J. Research, 7, 477 (1931).
3. Е. Schreiner, О. Е. F г i v о 1 d, Z. physik. Chem., 124, 1 (1926).
R табл. 58
1. International Critical Tables Vol. V, McGraw-Hill Book Co., N. Y., 1931, p. 135.
284 ' Г Л А В А IX
К табл. 59
1. R. Р. Smith, J. Am, Chem. Soc., 61, 500 (1939).
К табл. 60
1. G. S с a t с h а г d, W. J. Hamer, S. E. Wood, J. Am. Chem. Soc., 60, 3061
(1938).
2. H. Scheffer, A. A. Janis, J. B. Ferguson, Canadian J. Research,
817, 338 (1939).
3. R. A. Robinson, Trans. Faraday Soc., 35, 1229 (1939).
4. S. S h a n к m a n, A. R. Gordon, J. Am. Chem. Soc., 61, 2370 (1939).
5. R. A. Robinson, D. A. Sinclair, J. Am. Chem. Soc., 56, 1830 (1934).
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
В начале настоящей главы излагаются основные принципы метода
электродвижущих сил, описываются условные обозначения для гальвани-
ческих элементов, а также условия, касающиеся знаков электродвижущей
силы и стандартных электродных потенциалов. Затем излагается термоди-
намика гальванических элементов с жидкостными соединениями и без
жидкостных соединений, причем это изложение связывается с результатами
исследований растворов. Далее подробно рассматриваются гипотетический
потенциал жидкостного соединения, понятие об электрическом потенциале
на границе раздела фаз, проблема индивидуальных химических потенциа-
лов и активностей ионов. В конце главы обсуждается вопрос о тех огра-
ничениях, которые возникают при использовании элементов с жидкостными
соединениями из-за наличия диффузионных потенциалов, а также описы-
вается удобный способ устранения последних.
§ 1. Общие термодинамические свойства элементов. Определение
стандартного электродного потенциала
Ранее было показано, что при постоянных давлении, температуре
и составе изменение свободной энергии, соответствующее обратимой электри-
ческой работе, определяется уравнением (23) гл. I
(^)р, т, п = (ф'— ф") </е.
Условия, соответствующие этому уравнению, осуществляются в гальвани-
ческом элементе, состоящем из двух обратимых электродов, соединенных
раствором соответствующим образом подобранных электролитов. Разность
(ф' — ф") представляет собой разность потенциалов между двумя проводни-
ками из одного и того же металла, соединенными с электродами, de — пере-
несенный заряд, который соответствует изменению свободной энергии, рав-
ному dF. Это уравнение справедливо только для обратимых процессов,
т. е. при строгом соблюдении следующих условий:
1) если через элемент не проходит ток, в нем не происходит никаких
реакций;
2) всякий процесс, происходящий при прохождении тока, можно изме-
нить на обратный путем изменения направления тока.
Кроме того, элемент представляет ценность для термодинамических
исследований лишь в том случае, если известен общий результат всех хими-
ческих изменений, которые в нем происходят. Если все эти условия выпол-
нены, мы можем измерить обратимую электрическую работу (ф' — ф") de
и, следовательно, изменение свободной энергии dF, соответствующее дан-
ной реакции. Если электродвижущая сила элемента ±Е точно уравнове-
шена внешней электродвижущей силой, так что в элементе не происходит
286
ГЛАВА X
заряжения или разряжения, то ±Е представляет собой электродвижущую
силу элемента в условиях, когда вся система находится в равновесии.
Если представить себе, что элемент разряжается против этой внешней
электродвижущей силы до тех пор, пока не протечет количество электри-
чества de, то процесс в элементе произойдет обратимо и произведенная
обратимая электрическая работа будет равна ± Е de. Разность потенциалов
(Ф' — Ф") равняется + Е или — Ев зависимости от условного способа обо-
значения, принятого для знака электродвижущей силы. Таким образом,
уравнение (23) гл. I приобретает следующий вид:
(dF)p, т,п — ±Ede. (1)
Так как при рассмотрении химических реакций нас интересуют изменения
количеств веществ, выраженные в целых числах молей, то, согласно закону
Фарадея, эти изменения сопровождаются прохождением одного фарадея
электричества F или целого кратного числа фарадеев NF; поскольку, со-
гласно уравнению (1), dF пропорционально de, то при прохождении через
элемент NF кулон
(-W,T,n = NFE. (2>
В данном уравнении принято условное обозначение, предложенное Льюисом
и Рендаллом [1], согласно которому положительное значение Е соответ-
ствует уменьшению свободной энергии.
Полное изменение свободной энергии любой химической реакции, выра-
женное через активности исходных веществ и продуктов реакции', дается
уравнением (77) гл. I. Если обозначить через П'а и Па произведения
активностей конечных и исходных веществ, а через П'у, Пу, П'т и Пт.
соответствующие произведения коэффициентов активности и концентраций,
то уравнение (77) гл. I можно написать в виде
AF - AF0 = ДТ in = ДТ In П'т , (3)
На Пу Пт ’ v р
где АД— изменение свободной энергии в общем случае, а ДТ°— изменение-
свободной энергии в том случае, когда активности всех исходных веществ
и продуктов реакции равны единице.
Если свободная энергия реакции измеряется с помощью гальваниче-
ского элемента, то из уравнений (2) и (3) можно получить следующее
уравнение:
Е = Е°-^1п-^ = Е°-^1п^^, (4>
где
Е°= — AT’/NF; (4а >
через. Е° обозначается стандартная электродвижущая сила элемента. Оче-
видно, что Е = Е°, когда активности всех веществ, участвующих в реакции,,
равны единице. Так как состав твердых компонентов (или чистой жидкой
фазы) элемента, как правило, не зависит от концентрации раствора электро-
лита, то обычно принимают активности всех таких компонентов равными еди-
нице и считают, что эти компоненты находятся в стандартном состоянии при
всех температурах при давлении в 1 атм1. Стандартное состояние для
компонентов жидких растворов обычно выбирается так, что т = а, т. е.
у = 1, для каждого растворенного вещества при бесконечном разбавлении.
Так как в газовых смесях парциальные давления пропорциональны кон-
1 Поскольку молярные объемы твердых (и жидких) веществ малы, то можно считать,,
что активности этих веществ равны единице при любых давлениях порядка одной атмо-
сферы [уравнение (44) гл. I]. В случае газов давление должно быть указано.
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
287
центрациям и законы идеальных газов можно применять для практических
целей при малых изменениях давления в области около 1 атм, то актив-
ности газообразных компонентов можно считать равными их парциальным
давлениям. Таким образом, газ находится в стандартном состоянии при
любой температуре, если'его парциальное давление равно 1 атмх. На осно-
вании этого выбора стандартных состояний и после преобразования урав-
нения (4) к виду
„ , RT , П'т RT , П'у
Е + пртгЫ-я— = Е°— In-jT-1- (5)
1 NF Пт NF Пу ' '
очевидно, что Е° численно равно сумме членов левой части уравнения при
бесконечном разведении раствора электролита, так как при этом величина
члена, содержащего у, обращается в нуль. Различные способы определения
важной величины
Гп . RT , П'т "1 гл . .
Е -г хч?- In =Е° (5а)
L NF Пт J(m->0) ' '
будут рассмотрены в § 3 и в следующих главах.
§ 2. Условные способы изображения элементов и электродов;
знаки электродвижущих сил
Так как большинство американских химиков при описании реакций
у электродов и в элементах, а также при определении знаков электродви-
жущих сил придерживаются системы обозначений, предложенной Льюисом
и Рендаллом [1], то эта система принята и в данной книге. В случае новых,
более сложных элементов, недавно описанных в литературе, эти условные
способы изображений элементов несколько видоизменены и дополнены. Все
такого рода дополнительные условности, имеющие ограниченную примени-
мость, оговорены в тех случаях, когда это является необходимым. Ниже
перечислены условные обозначения общего характера.
а) Контакт между отдельными фазами (электродом и раствором) обычно
обозначается вертикальной линией, но при изображении отдельных элек-
тродов2 между фазами ставится дефис. Так, Pt-Ag-AgCl | MG1 (т) пред-
ставляет собой электрод серебро-хлористое серебро в контакте с раствором
хлорида.
б) физическая природа фазы, если она не вполне очевидна, обозна-
чается буквами (тв.), (ж.) и (г.) соответственно для твердой, жидкой и газо-
образной фаз. Обозначения (т), (с) и т. д. выражают концентрации
и относятся к жидким растворам. Твердые растворы редко применяются
в качестве электродов, за исключением применения ртути в качество
одного из компонентов; в этих случаях обычно присутствуют два насы-
щенных твердых раствора, и такая система носит название «двухфазной
амальгамы». Так, Pt-Pb-Hg (2 фазы)-РЬЗО4 (т) | представляет собой
электрод, составленный из насыщенных растворов ртути в свинце и свинца
в ртути.
в) Парциальное давление газа обозначается или буквенно через р, или
числом, например 1 атм и т. д. Если давление не указано, то предпола-
гается, что парциальное давление равно 1 атм.
1 Мы всегда будем пользоваться таким определением стандартных состояний, исклю--
чая некоторые специальные случаи, которые будут особо оговорены.
2 При изображении этих электродов Pt- обычно опускают; так, например, принято
писать Ag-AgCl | MCI (m), Pb-Hg (2 фазы)-РЬЗО41 и т. д. Кроме того, амальгамные
электроды часто обозначают M-Hg (насыщ.) и M^Hg для того, чтобы отличить двух-
и однофазные амальгамы.
288
ГЛАВА X
г) Концентрация растворенного вещества обычно выражается в молях
на килограмм растворителя и обозначается через т. Если имеется несколь-
ко растворенных веществ, то их концентрации обозначают через (т^), (т2)
и т. д., а иногда через Шксь тн+ и т. д. Совместно растворенные вещества
отделяются запятыми, указывающими, что эти' вещества присутствуют
в той же самой фазе.
д) Если нет специальных обозначений, то растворителем является вода.
При использовании смесей растворителей их состав обычно выражается
в весовых процентах, обозначаемых X и Y. Если одним из компонентов
смеси является вода, тогда через X обозначают неводный компонент.
е) Положительный знак над электродом элемента показывает, что при
самопроизвольном разряде элемента в его стандартном состоянии отрица-
тельное электричество переходит от электрода к электролиту. Положитель-
ный знак часто будет опускаться, так как, если записывать обозначение
элемента в общем виде, не указывая численной величины Е° и значений
концентраций, то электродам такого элемента нельзя приписать определен-
ных знаков.
ж) Электродвижущая сила Е считается положительной, если при
самопроизвольном разряде элемента отрицательное электричество течет
внутри элемента справа налево.
Большинство из перечисленных условий можно проиллюстрировать
на примере следующего элемента:
Н2(1 ашж)|НА(ш0), NaA(m1), NaCl(m2), СН3ОН(Х), Н2О (У) | AgCl-Ag+,
который содержит хлористый натрий, слабую кислоту НА и ее натриевую
соль. Растворителем служит раствор метилового спирта (Х%) в воде (У%).
Электрод серебро-хлорид серебра считается положительным. Таким образом,
согласно уравнению ^4),
Е = Е° — In ан+ пс1--
Электродвижущая сила этого элемента была бы равна Е°, если бы мы
могли осуществить условие ав aCj = 1. Как мы увидим далее, Е° представ-
ляет собой также электродвижущую силу элемента
Н2(1 ашж)|НС1(ш), СН3ОН(А), Н2О (У) | AgCl-Ag
при той же температуре, если анас1 — 1.
з) При изображении отдельных электродов электролит располагается
с правой стороны. Следовательно, в случае водородного электрода
Н2(/> атм) | Н+ (шн+)
или в более простой форме
Н2(/>)|Н+ (иг)
электродвижущая сила будет положительна, если отрицательное электри-
чество протекает через электрод справа налево. На этом электроде проис-
ходит реакция
1н2(/>)^П+(т) + 0, (6)
где символом 0 обозначен один фарадей отрицательного электричества.
Если всегда записывать электродные реакции так, чтобы 0 оказывался
в той части уравнения, где находятся продукты реакции, то общее урав-
нение для потенциала те отдельного электрода будет иметь следующий вид:
n RT 1 П'а /п\
==К- NF 1П~ПТ’ (7)
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
289
и для потенциала рассмотренного выше водородного электрода получаем
уравнение
0 RT, ан+
к = я°_ Ь , (8)
Рц
так как N = 1.
и) Стандартный (или нормальный) электродный потенциал -° представ-
ляет собой потенциал данного электрода при активностях всех веществ,
участвующих в электродной реакции, равных единице. Так как потен-
циалы отдельных электродов измерить невозможно, то обычно определяют
их значения по отношению к ^ = 0.
к) При соединении любых двух отдельных электродов в гальваниче-
ский элемент электродвижущая сила элемента равняется алгебраической
разности между потенциалами этих электродов. Значение электродного
потенциала правого электрода вычитается из значения потенциала левого
электрода.
Так, если соединить электрод
Ag-AgCl | С1~(пг)
с водородным электродом, то можно написать
Н2 (/>) |НС1 (пг) | AgCl-Ag. I
Реакции на электродах будут
1н2(р) = П+(щ) + е (9)
Ag + Cl"(m) = AgCl (тв.)-|-Q. (10)
Соответствующие потенциалы
„ ^н(р) = 0—(И)
и
^AgCl = KAgCl-р- 1п—- . (12)
. г аС1
Реакция, протекающая в этом элементе, и его электродвижущая сила
равны соответственно
А Н2 (р) + AgCl'(TB.) -> Ag + HCl(m) (13)
и
„ о RT, ан аш ц/х
«Н(р) — ^AgCl — U — KAgCl-р- 1П '
ИЛИ
Ei(p) = ES-^ln^. (15)
При обычных условиях отрицательное электричество самопроизвольно проте-
кает через этот элемент справа налево и поэтому Ет и Ен положительны.
Если схематически изобразить элемент, составленный из тех же электро-
дов, в обратной последовательности
Ag-AgCl|HCl(m)|H2(p), II
то протекающая в элементе реакция и его электродвижущая сила должны
быть записаны так:
НС1 (m) + Ag —» AgCl (тв.)+ у Н2 (р) , (16)
19 г. Харнед, В. Оуэн
290
ГЛАВА X
И
V ____ро , RT7 j аН аС1 /» 7V
Ьц (Р) — Ьп +-jj-In 1/2 • (1*)
В этом случае ЕП(Р) = — Е1(р) и Еп = — EJ. Если мы ассоциируем эле-
ктродвижущую силу и ее знак с определенной реакцией, происходящей
в элементе, то, конечно, не имеет существенного значения, изобразим ли
мы элемент по схеме I или по схеме II.
Следует отметить, что природу химической реакции, соответствующей
данному электроду или элементу, нельзя определить только на основании
измерений электродвижущей силы. Реакции, протекающие в гальвани-
ческом элементе, какими бы простыми и очевидными они ни казались,
следует рассматривать как гипотетические до тех пор, пока не будет
показано, что термодинамические величины &F, &Н, койстанты равновесия
и т. д., вычисленные на основании измерений электродвижущей силы,
совпадают с соответствующими величинами, вычисленными другими спосо-
бами.
Для того чтобы избежать необходимости записывать парциальные
давления газов, участвующих в протекающей в элементе реакции, обычно
пересчитывают значения электродвижущих сил, измеренные при парциаль-
ном давлении р, на значения, соответствующие парциальному давлению,
равному в точности 1 атм, и записывают последние как «наблюдаемые»
электродвижущие силы. Если преобразовать уравнение (15) для элемента!
к виду
Е«-^1панас1=[Е(р)-^1пр]=Е, (18)
то нетрудно заметить, что выражение в скобках равно электродвижущей
силе элемента при р = 1. Эта электродвижущая сила обозначается через Е.
Таблица 62
_ RT, Т 760' \ ,
пирометрическая поправка —= ш ( —---- ) для разбавленных
\ * Pw /
водных растворов (вольт-105)
\Темпера- \ тура, \ °C Р 0 5 10 15 20 25 30 35 40 45 50 55 60
780 -24 -21 -17 -12 - 4 7 20 40 64 98 140 195 269
770 -8 -6 -1 4 12 24 38 58 82 115 160 217 291
760 7 10 15 21 29 41 56 76 101 136 181 239 314
750 23 26 31 38 46 59 74 95 121 156 202 261 338
740 39 43 48 55 64 76 92 113 141 176 223
Табл. 62 содержит величины поправки
НТ, НТ, 760
2F 2р р_
(19)
Pw
для водных растворов. Эти величины, которые иногда обозначают
через Е(бар.), следует прибавлять к Е(р), чтобы получить Е. В этом уравне-
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТ.ОВ 291
нии полное барометрическое давление Р и упругость пара чистой воды рю
выражены в миллиметрах. Упругость пара воды над разбавленным рас-
твором незначительно отличается от упругости пара чистой воды, и это
различие не оказывает заметного влияния на величину Е, так как изме-
нению pw на 0,5 мм отвечает изменение значений в табл. 62 примерно
на 0,01 мв при комнатной температуре. Линейная интерполяция дает удо-
влетворительные результаты. Однако для концентрированных растворов
необходимо применять уравнение (19) для каждой концентрации и вводить
соответствующие значения pw.
Отметим еще следующее обстоятельство: поскольку для элементов,
рассматриваемых в данном параграфе, характерно отсутствие границы
раздела между растворами одного и того же электролита при различных
концентрациях или между двумя растворами разных электролитов, то их
называют «элементами без жидкостного соединения». Однако нельзя
считатр, что в таких элементах совершенно отсутствуют градиенты кон-
центрации, так как, например, хлористое серебро обладает некоторой
растворимостью. Если весь раствор насыщен хлористым серебром, то
серебро будет осаждаться на водородном электроде и изменять величину
его потенциала; если же раствор не насыщен хлористым серебром, то воз-
никает некоторый градиент концентраций ионов серебра и хлора и в ре-
зультате появляется некоторый, хотя и незначительный, диффузионный
потенциал. Это обстоятельство имеет место для всех элементов, составлен-
ных из электродов такого типа. Очевидно, чем меньше растворимость дан-
ной соли, тем точнее реальный элемент соответствует идеальному элементу
без жидкостного соединения.
§ 3. Экстраполяция данных по электродвижущим силам
и вычисление стандартного молярного электродного потенциала
В общем случае электродвижущая сила элемента определяется уравне-
нием (4). Для нас представляет особый интерес одна специальная форма
этого уравнения. Во-первых, мы будем рассматривать электродвижущую
силу Е при таких условиях, когда все газы, участвующие в реакциях,
протекающих в элементе, имеют давление, равное 1 атм [уравнение (18)].
Во-вторых, будет рассмотрен только тот тип реакций, при которых обра-
зуется электролит с концентрацией т [уравнение (13)]. В этом случае
уравнение (4) принимает следующий вид:
Е = Е° — ^lnYXV-'m+m-' ,
или
Е = Е° —^lnY±m± (20)
в соответствии с уравнениями (31) и (49) гл. I. После преобразования этого
уравнения получаем
(E+^lnm±) = E«-^lnY±. (21)
Величина в скобках должна равняться Е° при т = 0, так как Y± = 1
при бесконечном разведении. Таким образом, задача определения Е° путем
экстраполяции сводится к определению левой части этого уравнения при
т = 0- Так как уравнение (20) гл. III, не содержащее логарифмического
члена в правой части,
lgY±=-^(/)/r (22)
19*
292
ГЛАВА X
позволяет исключить у± из уравнения (21), то Е° может быть вычислено
либо на основании одного точного измерения при очень большом разбавле-
нии, либо из результатов ряда измерений при различных концентрациях.
В последнем случае можно изобразить графически левую часть уравнения (21)
как функцию от j/г и экстраполировать до концентрации с — 0, так что
полученная кривая будет асимптотически приближаться к прямой с теоре-
тическим предельным коэффициентом наклона. Таким графическим опре-
делением пользовались Льюис и Рендалл [2] до появления теории Дебая
и Гюккеля. Лыоис и Рендалл предположили, что левая часть уравнения (21)
линейно зависит от \/~т при бесконечном разведении, и использовали для
экстраполяции такую прямую с произвольным коэффициентом наклона,
которая соответствовала опытным данным для очень разбавленных растворов.
Этот способ экстраполяции не вполне удовлетворителен, так как при самых
низких концентрациях наблюдается искривление прямой и приходится при-
давать слишком большое значение результатам измерений для наиболее
разбавленных растворов, когда получаемые данные менее надежны.
Простой практический прием экстраполяции [3], при которой получается
хорошее соответствие с опытными данными при умеренных концентрациях,
состоит в применении уравнения (22) с дополнительным членом:
lgY±= + (23)
Путем подстановки значения lg у± из этого выражения в уравнение (21)
и преобразования полученного выражения получаем
(Е+wln = Е° - W В'т- (24)
Согласно этому уравнению график зависимости величины, заключенной
в скобки, от т должен представлять собой прямую, отсекающую на оси
ординат отрезок, равный Е°. Многочисленные примеры экстраполяции такого
рода приведены в гл. XI —XV.
Мы не ставили перед собой задачу составить полную таблицу стандарт-
ных потенциалов и приводим лишь те значения Е°, которые имеют не-
посредственное отношение к рассматриваемым вопросам. Таблицы 1 стандарт-
ных свободных энергий ионов и энтропий нами не приводятся вовсе.
§ 4. Определение коэффициентов активности путем экстраполяции
данных по электродвижущим силам концентрационных элементов
без жидкостного соединения
Имеется другой метод экстраполяции, который является менее важным,
чем вышеописанный, но который оказался полезным для определения коэф-
фициентов активности. Для экспериментальных целей иногда представляется
удобным сочетать два элемента ранее описанного типа. В качестве примера
можно привести амальгамный элемент
Ma:Hg|MX(7n1)|AgX-Ag, (25)
где М — щелочный металл и MX —галогенид щелочного металла. Реакция,
происходящая в этом элементе, может быть записана следующим образом:
М (M^Hg) 4-AgX = Ag-(-.MX(7n1). (26)
рассмотрим другой элемент этого типа, в котором концентрация электролита
равна т2:
MJIg|MX(m2)|AgX-Ag. (27)
1 Табличные данные имеются, например, у Латимера [4аЦ см. [46].
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
293
В этом элементе протекает следующая реакция:
М (М Jig) + AgX = Ag + MX (m2) . (28)
Если из этих двух элементов образовать гальваническую цепь
Ag-AgX | MX (ш2) | MxIIg | MX (mi) | AgX-Ag, (29)
то реакция, протекающая в этой цепи, может быть получена путем вычи-
тания уравнения (28) из уравнения (26) и общий результат этой реакции,
который состоит в переносе MX из раствора с концентрацией т2 в раствор
с концентрацией mit можно выразить равенством
MX.(m2) = MX (ин). (30)
Согласно уравнению (30) гл. I, для переноса одного моля получаем следу-
ющее выражение1: _
А
— NFE = dF = F1 - F2 = RT In Ъ™! . (31)
F2
Это уравнение можно получить также, применяя уравнение (4) к эле-
ментам (25) и (27) и вычитая одно из полученных выражений из другого.
Так как отношение т1/т2 известно, то путем измерения электродвижущей
силы подобной гальванической цепи можно определить отношение коэффи-
циентов активности. При определении у обычно придают постоянное
значение — стандартную концентрацию (0,1Л/)— и определяют отношение
у/уо,1, г^е у — коэффициент активности при любой другой концентрации т.
Затем производят экстраполяцию с помощью уравнения Дебая и Гюккеля,
дополненного Гюккелем путем прибавления линейного члена Вс к уравне-
нию (34) гл. III. Таким образом, учитывая уравнение (57) гл. I, можно
написать соотношение
lgY= — Вс —lg (1+ 0,01802vm), (32)
где А и В — эмпирические постоянные при данной температуре. Как было
показано, это уравнение весьма точно выражает опытные данные, получен-
ные при определении активности от низких концентраций вплоть до 1Л/,
а иногда до 2М, и поэтому оно удобно для экстраполяции данных, получен-
ных при высоких концентрациях [5]. Постоянные А и В могут быть опре-
делены различными методами, простейшим из которых является совместное
решение двух уравнений для отношения коэффициентов активности. Так,
согласно уравнению (32),
0 __ S>(/) 1^12 S>(/) 1^11 . , /1 + 0,01802^m2\ /qqx
lg г - ~ ГТлТК+l+Al/r; в (С2С1) ~ lg ki+0,01802^J • <33>
Если написать отношение Y2/T1 для двух разных значений с2 и сх при посто-
янном Yj, то получатся два уравнения, с помощью которых можно опре-
делить А и В. После подстановки этих значений А и В в уравнение (32)
можно вычислять величину у для любой заданной концентрации.
Если имеются надежные опытные данные в области концентраций
от 0,1 М до 1 М, то у можно определить путем экстраполяции с точностью
порядка ± 0,002, что соответствует ошибке в ± 0,1 мв. Поскольку уравне-
ние (32) не выражает с достаточной степенью точности опытные данные
1 В этом параграфе через 7 обозначается средний коэффициент активности у±,
а индексы 1 и 2 относятся к двум различным концентрациям одного и того же элек-
тролита.
294
ГЛАВА X
при концентрациях, значительно превышающих 1 М, то это приводит иногда
к таким ошибкам при экстраполяции, которые значительно превышают
приведенную выше величину ошибки.
§ 5. Вычисление относительного парциального молярного теплосодержания
и относительной парциальной молярной теплоемкости электролитов
из данных по электродвижущим силам
Согласно уравнениям (73) гл. III и (20),
Ч<Э1П7±_ £2 _
дТ RT2 ’
а по уравнению (20)
(Е— Е°) NF . ,
^~=-InY±-Inm.
Поэтому при постоянном составе
<31ny±_ NF д (Е —Е°) _ Г2
дт ~ -,в. дт т ~ nRt2
(34)
L2 = NF^A(^). (35)
Последнее выражение представляет собой уравнение Гиббса — Гельмгольца
и аналогично уравнению (16) гл. I. После дифференцирования оно при-
нимает более простой вид:
Z2 = — NF (Е — Е°) + NF71 . (36)
Эти уравнения показывают, что точность определения L2 из данных по
электродвижущим силам зависит от точности, с которой может быть измерен
температурный коэффициент электродвижущей силы.
Относительная парциальная молярная теплоемкость может быть вычис-
лена из этих соотношений с помощью уравнения (40) гл. I, т. е.
После подстановки значения L2 из уравнения (36) и дифференцирования
получаем
J2 = NF71—^~Е°\ (38)
Далее нужно определить вид функции, которую следует использовать
для вычисления зависимости (Е — Е°) от температуры. Хотя известно
что J2 незначительно меняется с температурой, допустим в первом при-
ближении, что эта зависимость является линейной:
J2 = J2(0) + c'"(71-710), (39)
где /2(о> относится к некоторой стандартной температуре То, а с"постоян-
ная. Интегрируя уравнение (37) между пределами То и Т, получаем
т
^2 = Г/2(0)+ J-2.dT. (40)
\ То -
После подстановки в это уравнение значения J2 из (39) и интегрирования
получаем уравнение
Z2 —a"-f-6"71-|-c"712,
(41)
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
295
где
а" = -^'2(о? — J2 (О)Т'о + с’"Т q/2,
b" = J2W-c"'T0, (42)
с" = с'"/2.
Подставляя величину L2 из (41) в уравнение (35) и интегрируя, получаем
(Е —Е°) = ~(-а” + 1Т + Ь'"ПпТ-^ с"Т2)= (43)
= (а' - а') + (У - b'o) Т + (с’-с'о) Т* + (<*' - ^) Т In Т, (44)
где I — константа интегрирования.
Опытным путем было найдено, что в ограниченной области изменения
температуры (от 0 до 50°) зависимость Е и Е° от температуры для многих
элементов может быть выражена с точностью до ± 0,05 мв следующими
квадратными уравнениями:
Е = а + ЬТ + сТ2, (45)
Е° = а0+Ь0Т + с0Т2, (46)
которые не содержат логарифмического члена.
Если подставить значение (Е —Е°) из (44) в уравнение (36), то полу-
чается
Z2 = - NF [(д' - а') - (d' -d')T- (с’ - с{,) 72]. (47)
Если же использовать для этой цели квадратные уравнения (45) и (46), то
Z2 = — NF [(а — а0) — (с— с0) 72]. - (48)
Соответствующие уравнения для J2 имеют вид
72 = NF[(d'-d') + 2(c'-c')T], (49)
и
72 = 2NF(c-c0)T\ (50)
В случае концентрационных элементов без жидкостного соединения
[уравнение (29)] можно вычислять только относительные значения Ь2 и J2 по
отношению к соответствующим значениям при некоторой стандартной кон-
центрации. Если электродвижущая сила концентрационного элемента
выражается квадратным уравнением
Em — Ед = а + ЪТ сТ~,
то
Нцт) — 7^2 (й) == 7^2 (т) — 7.2 (й) = —NF (а — С?2), (51)
где Ег (т) — относительное парциальное молярное теплосодержание при кон-
центрации т, а Е2(й) — эта же величина при стандартной концентрации. От-
носительная парциальная молярная теплоемкость выражается уравнением
Т'Рг (ш) Ср2 (й) = J2 (m) J2 (R) = 2NFc7\ (52)
Так как константы а, Ъ, с и т. д. обычно вычисляются с помощью
метода наименьших квадратов, то для упрощения вычислений можно ис-
пользовать более простые квадратные уравнения. Если точность измерений
электродвижущих сил не превышает + 0,05 .мв, а температура меняется
в пределах до 40°, то вряд ли есть необходимость в использовании члена
Т In Т или членов с более высокими степенями Т.
296’
ГЛАВА X
§ 6. Концентрационный элемент с жидкостным соединением
Как показал Тейлор [6], уравнение для электродвижущей силы кон-
центрационного элемента, в котором происходит перенос растворенного
вещества через границу раздела жидкостного соединения, можно получить
в общем виде следующим образом. Изобразим этот элемент с помощью
такой схемы:
электрод (1) | т1; щ2..mi, .. ., ms | т\, т'2, . . ., m's | электрод (1),
(53)
где тх, ..., ms и т[....m's—моляльности ионов по обе стороны от гра-
ницы раздела двух растворов, причем электрод (1) обратим по отношению
к иону (1). Нечетные индексы относятся к катионам, четные—к анионам.
Если при обратимом разряде элемента через него протекает один фарадей
электричества, то при этом из одного раствора в другой переходит один
эквивалент ионов (I). Кроме того, ионы всех видов, находящиеся в растворе,
переходят через границу раздела в количествах, определяемых их концен-
трациями и числами переноса. Поэтому если разности концентраций по обе
стороны от границы раздела бесконечно малы, то
F dEj (T) = dF1 — T1dF1+ T2dF2— T3dFa + ... = (54)
= dF± - 2 dFi = (55)
i
^dFx-FdEj. (56)
Ei (t;—электродвижущая сила элемента, которая может быть измерена
непосредственно, Ej обычно называют диффузионным потенциалом. Что
касается «гипотетических» парциальных свободных энергий ионов в правой
части уравнения (55), то необходимо вспомнить положения гл. I, в которой
подчеркивалось, что термодинамика позволяет находить парциальные сво-
бодные энергии, активности и т. д. только для молекулярных компонентов,
в том смысле, как их определяет Гиббс, и не дает возможности вычислять
эти величины для отдельных видов ионов.
Приведенное выше выражение для </Е4 весьма удобно для получе-
ния зависимости между электродвижущими силами концентрационных эле-
ментов с жидкостными соединениями и без них. Допустим, что мы поме-
стили электрод, обратимый по отношению к ионам (2), между двумя рас-
творами элемента (53) и получили таким образом цепь без жидкостного
соединения:
электрод (1) |щ1; ..., ms | электрод (2)| т[, ..., т'е\ электрод (1). (57)
Электродвижущая сила этой цепи связана с переносом ионов (1) и (2)
из одного раствора в другой. Поэтому если концентрации двух раство-
ров отличаются друг от друга на бесконечно малую величину, то электро-
движущая сила с?Е12 выражается уравнением
F с/Е12 = dF± + dF2, или dF2 = —dF± + F dE12. (58)
Аналогичным образом, если вместо электрода (2) поместить электрод (3),
обратимый по отношению к ионам (3), то суммарная электродвижущая
сила йЕ13 такой цепи будет
F</Е13 = dFi — dF3, или dF3 — dFi — FdE13. (59)
Подобные уравнения могут быть получены и для других видов ионов.
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
297
При подстановке этих значений dF3, dF3 и т. д. в уравнение (54)
получаем
FrZEj (Т) = d Fy - Тх dFy -T2dF\ + T2F с?Е12 - Т3 dFy + Т3 F с?Е13± ... (60)
или
F dEt (Т) = dFy - (Ту + Т2 + . . . + 7\) dFу + Т2 F dE12 + Т3 F <Ж13 + .. . (61)
и, следовательно,
(ZEj (Т) = Т3 <Ж12 + Т3 <Ж13 + . . ., (62)'
так как сумма чисел переноса всех ионов равна единице. Такую же фор-
мулу можно получить для случая, когда переносимый ион представляет
собой анион; для этого необходимо соответствующим образом переставить-
индексы катионов и анионов. Так, если в элементе (53) электроды обра-
тимы по отношению к ионам (2), то
сЖз (Т) = Ту </Е21 -|- Т3 </Е23 (63)
Результат, который выражается уравнениями (62) и (63), является
весьма существенным; действительно, в этих уравнениях отсутствуют пар-
циальные свободные энергии ионов dFt, и электродвижущие силы элемен-
тов выражены через величины Е21, Е23 и т. д., соответствующие суммам
парциальных свободных энергий положительных и отрицательных ионов,
причем эти сум^ы представляют собой молярные свободные энергии элек-
тролитов.
Как было показано, с помощью уравнения (54) нельзя определять,
парциальные свободные энергии ионов и, следовательно, измерения электро-
движущих сил элементов с переносом нельзя использовать для получе-
ния данных о парциальных свободных энергиях и активностях ионов.
Точное термодинамическое соотношение, выражаемое уравнениями (62)
и (63), представляет большую ценность, так как оно дает возможность
определять числа переноса из данных по электродвижущим силам элемен-
тов с жидкостным соединением и без него [7]. Предположим, что мы имеем
такого рода элементы, содержащие один электролит. Если электроды этих
элементов обратимы по отношению к аниону, то, согласно уравнению (63),
сЖ2 (Т) = Ту </Е21, (64)
где Ту—число переноса катиона. Из этого уравнения видно, что если известна
функциональная зависимость Е2 (т> от Е21, то можно вычислить Ту. Если эта
зависимость неизвестна, то можно определить Ту графически из наклона
кривой, изображающей изменение Е2(Т) с изменением E2i. Этот метод опре-
деления чисел переноса был разработан Мак-Иннесом и Паркером [8], а также
Мак-Иннесом и Битти [9, 10], которые получили с его помощью весьма точ-
ные результаты. После того как появилось предельное уравнение Онзагера
для чисел переноса, Джонс и Дол [11] и другие исследователи [12] такжо
использовали этот метод, но полученные ими результаты были значительнее
менее точными, чем данные, полученные с помощью метода движущейся
границы (гл. IV).
Браун -и Мак-Иннес [13] использовали весьма точные данные по числам
переноса, полученные по методу движущейся границы, для определения
коэффициентов активности галогенидов путем измерения электродвижущих
сил элементов с переносом. Они применяли элемент
Ag-AgCl | NaCl (сх) | NaCl (с2) | AgCl-Ag. (65)
298
ГЛАВА X
Интегрируя уравнение (64), можно найти электродвижущую силу этого
элемента:
и II
E2(T) = 7’Na^E21=-^^rNadln2/±C. (66)
1 1
Если TNa известно для различных концентраций, то у± можно определять
с помощью этого уравнения с большой точностью. Подробности вычисления
будут рассмотрены в гл. ХП, § 1.
Хотя изложенные выше сведения об элементах с диффузионным по-
тенциалом и достаточны для наших целей, они далеко не исчерпывают
этого сложного и глубокого вопроса. В последнее время в этой области
было выполнено несколько исследований ГерМансом [14] и Кёнигом [15],
которые расширили теорию, исследовав влияние силы тяжести, электри-
ческого и магнитного полей.
ГИПОТЕТИЧЕСКИЙ потенциал ЖИДКОСТНОГО СОЕДИНЕНИЯ
(ДИФФУЗИОННЫЙ ПОТЕНЦИАЛ)
Выше было показано, что путем измерения электродвижущих сил
элементов с жидкостным соединением можно определять парциальные
молярные свободные энергии или линейные комбинации «гипотетических»
свободных энергий ионов. Этот термодинамический результат имеет очень
существенное значение для изучения диффузионного потенциала. Из урав-
нений (55) и (64) получаем соотношение
S S
ЕТ2б/Е12 = Еб/Е1(Т) = «/Л-2 Ti dFt = dF1 - RT 2 (67)
i i
С помощью элемента с жидкостным соединением можно измерить только
величину F cZEj (Г), где «ZEj (ту—разность потенциалов между двумя провод-
никами из одного и того же металла, присоединенными к электродам.
dFi представляет собой совершенно произвольную величину, которую нельзя
'S
вычислить никаким термодинамическим методом. Поэтому величина 2 Тt dFt
i
также является условной, а следовательно, и диффузионный потенциал,'
который выражается вторым членом в правой части приведенных выше
уравнений, тоже является совершенно условным. Если условно приписать
одному из видов ионов в растворе некоторую определенную величину сво-
бодной энергии, тогда свободные энергии других ионов можно вычислить,
используя известные значения молярных свободных энергий. Таким обра-
зом, мы свободны в выборе такого условия, лишь бы оно было полезно
для наших целей. Так, например, было предложено принять [16] FK = Fci
и определять свободные энергии других ионов с помощью этого условного
стандартного значения. Следует отметить, что, согласно уравнению (67),
можно с равным успехом использовать любое другое условие, например
FK = nF(-A, где п — любое конечное положительное число. Харнед [166]
доказал справедливость этого положения с помощью численного метода,
а Тейлор [6]—посредством специально разработанного аналитического спо-
соба. Поскольку это положение является справедливым, нет оснований
предпочитать ионы калия и хлора в качестве стандартных ионов ионам
любого другого электролита.
Указанные трудности нельзя преодолеть путем использования выражений
типа уравнений Плавка и Гендерсона для диффузионных потенциалов.
Это нетрудно показать, написав уравнение для гипотетического диффузи-
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
299
онного потенциала Ej в интегральной форме
3 II S II
-У 2 5 2 5 <68)
if 11
где пределы интегрирования соответствуют двум растворам (I и П), образую-
щим жидкостное соединение. Формулы Планка [17] и Гендерсона [18а, б]
для Ej были получены путем вычисления последнего члена в правой части
этого уравнения. Если даже предположить, что это вычисление выполнено
правильно, то таким путем можно найти лишь часть величины диффузионного
потенциала, так как остается неопределенной та часть, которая включает
гипотетические1 коэффициенты активности ионов. Это положение имеет место
для любых конечных концентраций ионов, причем это затруднение исчезает
лишь в том случае, если растворы по обе стороны от границы раздела фаз
имеют одинаковый состав.
Применяя солевые мостики, содержащие концентрированные растворы
электролитов (насыщенный раствор КС1), можно снизить численную вели-
чину второго члена в правой части уравнения (68), однако и в этом слу-
чае остается совершенно произвольный первый член.
ПРЕДСТАВЛЕНИЕ О РАЗНОСТИ ПОТЕНЦИАЛОВ НА ГРАНИЦЕ РАЗДЕЛА ФАЗ
Сделанный Тейлором вывод о том, что измерение электродвижущих
сил элементов с жидкостным соединением и без него не дает никаких све-
дений относительно свободных энергий ионов, был развит Гуггенгеймом [19].
Гуггенгейм изучал вопрос о разности электрических потенциалов между
двумя точками, находящимися в различных средах, и пришел к выводу,
что эта величина является совершенно произвольной1 и не может быть
определена через величины, подлежащие физическому измерению. Гугген-
гейм проанализировал различие между этим электростатическим потенци-
алом и обычным потенциалом, который определяется в электростатике.
Электростатика основана «на математической теории воображаемой эле-
ктрической жидкости, равновесие и движение которой полностью опре-
деляются электрическим полем. Подобного рода «электричество» фактически
не существует; в действительности существуют только электроны и ионы,
и эти частицы существенно отличаются от гипотетической электрической
жидкости тем, что они все время движутся по отношению друг к другу;
их равновесие является термодинамическим, а не статическим». Условия
термодинамического равновесия этих систем при постоянных температуре
и давлении можно найти с помощью уравнения
dF = 2 2 W ~ 'К) ^е'
1 1
Напомним, что в случае обратимого элемента (ф'— ф") представляет собой
разность потенциалов между двумя металлическими проводниками, при-
соединенными к электродам; (ф' — ф") de—электрическая работа; рг—хими-
ческие потенциалы компонентов в данной фазе. Выразим ту часть работы,
которая соответствует данной фазе, в виде суммы 2 <?е/ dnj, где ф—потен-
1
циал в месте расположения (иона или электрона) с зарядом е,-, а сумми-
1 На стр. 298—301 авторы допускают ту же ошибку, которая была отмечена нами
выше, см. примечание редактора на стр. 24. (Прим, ред.)
300
ГЛАВА X
рование производится по всем таким частицам, присутствующим в данной
фазе. Потенциал ф представляет собой совершенно условную величину.
Р о
(ф'— ф")йе равно сумме фе,с?п>. Таким образом, уравнение (24) гл. I
„ 1 1
для одной фазы имеет вид
S ff
dFp = 2 Hi dnt+ 2 $e! dnb (69>
i i
где первое суммирование производится по всем нейтральным и заряженным
частицам (s), а второе суммирование—по всем заряженным частицам (а).
Если мы рассматриваем процесс, в котором участвуют только заряженные-
частицы, то можно разделить химический потенциал молекулярного ком-
понента рг- на условные химические потенциалы заряженных частиц р/
и написать
о о
dFe = 2 Н/ dnj + 2 ^ei dnjt
i i
или
а
dFe = 2 (н/ + Фе/) dnj. (70)
1
Если ввести величину
Н/^Р/ + фе/, (71)
то получается уравнение
а
dFe = 2 Vi dnj, (72)
1
аналогичное уравнению
с
dF=^ pfdn-L,
i
откуда видно, что в электрохимической системе р,- играет ту же роль,
что и р,- в молекулярной системе. Для многофазной системы, которая
находится в равновесии при постоянных давлении, температуре и заряде,
из уравнения (18) гл. I получается условие равенства значений химиче-
ского потенциала данного компонента во всех фазах, выражаемое уравне-
нием (12) гл. 1. Аналогичным образом при равновесии в электрохимических
системах должно соблюдаться условие равенства электрохимических потен-
циалов р7- (Гуггенгейм и Бренстед) заряженных частиц данного вида во всех
фазах при условии, если эти частицы содержатся во всех фазах. Таким
образом,
— f -'/ — р
Р1=Р1='. .. pj
j (73)
— / -// — о
На = На = • ’ • НА
где через р[, р!-.. р? и т. д. обозначены электрохимические потенциалы
— Г —» —Р
частиц первого рода, через рг, ре • • • Иг—соответствующие значения для
частиц второго рода и т. д. во всех р фазах1. Как было показано выше,
1 Изложенная выше трактовка электрохимического потенциала по существу совпа-
дает с той, которая дается Бренстедом, обозначающим эту величину через X [20].
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
301
для ионов определенного вида величины парциальных молярных свободных
энергий, т. е. р,, являются совершенно условными; следовательно, фе7-
также не имеет определенного термодинамического значения.
На основании приведенных выше соотношений можно, так же как
й раньше, сделать вывод о том, что, несмотря на произвольный характер
величины ру, некоторые линейные комбинации ру не являются произволь-
ными. Так, рассмотрим электролит, диссоциирующий на два иона в одной
<фазе. Тогда, как показал Гуггенгейм,
р. = р - 4- е,- ф,
(7^)
Нс — Нс + ек Ф>
откуда после исключения ф получаем соотношение
р/ Pfc _ Р/ Р'Тс /75ч
еу ек е; ек ’ ( '
так что выражение определяется величинами, которые поддаются
измерению. Как показал Гуггенгейм, нетрудно доказать, что явления диффу-
зии, распределения между двумя фазами, мембранного равновесия, а также
процессы в элементах с жидкостным соединением и без него и скорости
реакций могут быть полностью описаны с помощью электрохимических
потенциалов и что химические потенциалы '-ч и члены фе4 никогда не встре-
чаются отдельно. Это показывает, что эти величины нельзя определить ни
из каких известных нам опытных данных. Гуггенгейм утверждает даже,
что «разность электрических потенциалов между двумя точками, находя-
щимися в различных средах, невозможно измерить».
Эти соображения очень интересны в связи с имеющей вековую давность
проблемой о месте возникновения электрического потенциала в гальвани-
ческом элементе1, например в элементе Даниэля:
Си | Zn | ZnSO4 (m) | CuSO4 (m) | Си.
Общую измеряемую электродвижущую силу можно рассматривать как сумму
разностей потенциалов на границах Си | Zn, Zn | Zn SO4 (m), Zn SO4 (m)|CuSO4 (m)
и CuSO4 (m) | Си. Поскольку выше было показано, что потенциалы в точках,
расположенных внутри разных фаз, не могут быть ни вычислены, ни измерены
никаким из известных методов, то в настоящее время мы не имеем возмож-
ности вычислять значения скачков потенциала на любой отдельной границе
раздела фаз.
Выводы, вытекающие из этого анализа, были предсказаны Гиббсом
{21], который писал: «При рассмотрении электрического потенциала в
электролите и в особенности разности потенциалов между электролитом и
электродом приходится иметь дело с такими величинами, которые не под-
даются физическим измерениям, в то время как разность потенциалов между
проводниками из одного и того же металла, присоединенными к электродам,
представляет собой величину, которую мы можем измерить и измеряем».
1 Вопрос о месте и механизме возникновения электрического потенциала в гальва-
ническом элементе разрабатывался А. Н. Фрумкиным и его школой. См. А. Н. Фрум-
кин, Статья в сборнике «Электрические и электрохимические свойства металлов»,
Госхимиздат, Л., 1928, стр. 116; А. Фрумкин и А. Городецкая, Z. physik.
Chem., 136, 215, 451 (1928); М. И. Темкин, Изв. ОХН АН СССР, № 3, 235 (1946).
Механизм возникновения электродвижущей силы в свете представлений о сольва-
тации ионов впервые был предложен Н. А. Изгарышевым, на работу которого мы
ссылались выше. См. примечание редактора на стр. 23. (Прим, ред.)
302
ГЛАВА X
§ 7. Использование элементов с жидкостными соединениями.
Определение pH. Устранение диффузионных потенциалов
Существуют два типа жидкостных соединений, с которыми обычно'
приходится иметь дело при электрохимических исследованиях: гомоионные
соединения, образующиеся при соприкосновении растворов, различающихся
только концентрацией ионов, и гетероионные соединения, возникающие
при соприкосновении растворов, содержащих различные ионы в одинаковых
или разных концентрациях. Как было показано в § 6, в случае гомоион-
ных соединений диффузионные потенциалы представляют собой величины,
поддающиеся точному термодинамическому определению [уравнение (66)],
и поэтому результаты, полученные с помощью элементов, содержащих
такие соединения, можно использовать для термодинамических расчетов
(гл. XI, § 9, и гл. XII, § 1). В настоящем параграфе будет рассмотрено
применение элементов с гетероионными жидкостными соединениями. Из-
мерение электродвижущих сил этих элементов находит широкое примене-
ние при различных способах определения pH и может при соблюдении
некоторых особых условий давать результаты, имеющие термодинамический
смысл. Эти условия носят экспериментальный характер и применяются для
устранения диффузионных потенциалов, а не для вычисления их величин.
Сначала мы рассмотрим современные методы1 измерения pH и ту
неопределенность, которая вносится в определение этой величины из-за
наличия жидкостных соединений. Затем будет показано, что диффузионные
потенциалы некоторых элементов или комбинаций нескольких элементов
можно уменьшить до очень незначительных величин, применяя в месте
соединения почти тождественные растворы. Такие элементы можно условно
рассматривать как обычные элементы без жидкостного соединения2. На-
конец, будет рассмотрен предельный случай, при котором полное тождество
двух соприкасающихся растворов достигается только путем экстраполяций,,
основанных на ряде измерений. Применимость величин, полученных ’ с по-
мощью такой экстраполяции, с точки зрения термодинамики не вызывает
сомнений, однако практическая возможность выполнения такой экстрапо-
ляции должна быть определена экспериментальным путем3.
Величину pH раствора первоначально определяли [23] как отрицатель-
ный десятичный логарифм концентрации водородных ионов, однако в даль-
нейшем pH стали отождествлять с отрицательным десятичным логарифмом
активности водородных ионов4. Применяя уравнение
рН=—lgaH+, (76)
необходимо иметь в виду, что оно носит совершенно формальный характер,
так как сама величина ан+ не поддается определению (§ 6). Окончательная
форма выражения для pH зависит от метода его определения. Рассмотрим
в качестве типичного примера следующий элемент:
Н21 (исследуемый раствор) | КС1насьПц. | (электрод сравнения). I
1 Подробное изложение более ранних методов см. в книгах Кларка [22а], Мисло-
вицера [226] и Бриттона [22в].
2 См. последний абзац § 2, стр. 291.
3 В связи с излагаемым здесь материалом, интересно обратить внимание чита-
телей на работу Н. А. Измайлова, устанавливающую зависимость коэффициентов ак-
тивности от диэлектрической постоянной и основности среды (ЖФХ, 23, № 5, 1949).
(Прим, ред.)
4 Зёренсен и Линдерштрём-Ланг [24] предложили применять обозначение раН
в тех случаях, когда используется активность водородных ионов, однако мы будем,
во всех случаях применять общепринятое обозначение pH.
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
303-
В качестве электрода сравнения обычно применяют насыщенный или
децинормальный каломельный электрод1. Для специальных целей водородный
газовый электрод замевяют вторичными стандартными электродами: хингид-
ронным, сурьмяным (Sb-Sb2O3) или стеклянным. В любом случае pH опре-
деляется уравнением
Е-Е°рн Е—ЕрН
Р 2,3 ЛГ/F к
(77}
где Е —измеряемая электродвижущая сила элемента I. Так как ЕрН —
постоянная величина, зависящая только от температуры и давления, а также
от природы электродов, то уравнение (77) полностью определяет практически
удобную величину pH, в отношении которой не возникает никаких со-
мнений, если только не пытаться интерпретировать ее согласно уравнению
(76). Численное значение этой величины pH зависит, конечно, от природы
жидкостного соединения и от того, как оно образуется. Это значение pH
нельзя отождествлять с величиной —lgaH+, которая определяется уравне-
нием (76), если не учитывать зависимость ан+ от свойств жидкостного
соединения. Для того чтобы избавиться от этого затруднения при интер-
претации pH и ан+, не устраняя самого жидкостного соединения2, были
сделаны попытки внести поправки на величины диффузионных потенциалов
к значениям Е или Е£н. Ни одна из этих поправок не является вполне
законной с точки зрения термодинамики, и исправление всех эксперимен-
тальных значений Е, например с помощью уравнения Гендерсона [27а]3,
всегда является трудоемким и обычно неосуществимо при практической
аналитической работе.
Недавно были предложены новые значения ЕрН для различных типов
каломельных электродов, причем они, повидимому, включают большую
часть диффузионного потенциала. Эти значения могут быть весьма удобны
и полезны для определения констант диссоциации и других констант рав-
новесия, где не требуется очень высокой точности. Эти значения ЕрН были
определены путем замены исследуемых растворов в элементе I буферными
растворами кислот, для которых константы диссоциации были точно опре-
делены с помощью кондуктометрического метода или из данных по электро-
движущим силам элементов без жидкостного соединения, Определение ве-
личин ЕрН можно проиллюстрировать на примере буферного раствора, со-
держащего слабую кислоту НА и ее натриевую соль. Если рАд предста-
вляет собой отрицательный логарифм термодинамической константы диссоци-
ации этой кислоты4, то, согласно уравнению (76) и уравнению (14) гл. VII,
получается выражение
pH = РАа + lg + lg . (78)
еНА ^НА
1 Этот выбор сделан произвольно и главным образом по соображениям большего
удобства. Хичкок [25а] и Редлих и Клингер [256] проанализировали возможные преиму-
щества применения электродов сравнения, обратимых по отношению к одновалентному
катиону.
2 Определение pH с помощью величин, характеризующих элементы без жидкостных
соединений, предложили Хичкок [25а], Гамер и Акри [26а], Гуггенгейм [266], а также
другие исследователи.
3 Ср. [276].
4 Нет необходимости учитывать различие в значениях КА, полученных при исполь-
зовании моляльных и молярных концентраций, так как Ерн изменяется при этом, как
правило, не больше чем на 0,1 мв.
304
ГЛАВА X
Мак-Иннес, Белчер и Шидловский [28] заменили 1рг/д_/?/НА на—V"V
и преобразовали это уравнение следующим образом:
lg-^ = ptfA-5/~p. (79)
к СНА
Эти исследователи подбирали величину ЕрН таким образом, чтобы график
зависимости членов левой части уравнения (79) от ]/р был линейным в
разбавленных растворах (р-^-от 0,001 до 0,01) и чтобы отрезок, отсекаемый
этой прямой на оси ординат, равнялся рТГА. Два таких графика изобра-
жены на рис. 70. Величины S, определяемые из наклона прямых, на
10—25% больше, чем значения коэффициентов наклона соответствую-
щих уравнению Дебая и Гюккеля для одновалентного иона.
Рис. 70. Определение Ерн при 25°
по уравнению (79). Величина отноше-
ния сА-/еНА непостоянна.
Рис. 71 . Определение Е°н при 25° по
уравнению (78). Величина отношения сА-/сНА
равна единице.
Хичкок и Тейлор [29] заменили 1g (г/А-/.Унл) в уравнении (78) на
—+ и преобразовали это уравнение следующим образом:
Е°р'не?Е-ЛрЙГА-/с 1g—+/« S(/)]/p = E°pH + W (80)
еНА
Ерн равно отрезку, который отсекает на оси ординат прямая, выража-
ющая зависимость членов левой части уравнения от р. В этой работе при-
менялись значения р, лежащие в пределах от 0,01 'до 0,1. Как видно из
рис. 71, графики имеют некоторую кривизну.
В табл. 63 приведены значения Ерн при 25°, полученные с помощью
обоих описанных методов. В этих работах применялся водородный электрод
в различных буферных растворах, а электродом сравнения служил насыщен-
ный каломельный электрод1.
Различия между значениями Ерц, приведенными в табл. 63, обуслов-
лены, вероятно, тем, что диффузионные потенциалы, возникающие при
1 Результаты Мак-Иннеса, Белчера и Шидловского были фактически получены
•с помощью 0,1 н. каломельного электрода, однако эти величины были затем пересчитаны
на обычный электрод сравнения путем вычитания вычисленного этими авторами значения
разности потенциалов (0,09112) между насыщенным и 0,1 н. электродами.
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
305
Таблица 63
Ерн при 25° для элемента:
Н2 | кислота (mJ, соль (mJ | КС1 (насыщ.), HgCl-Hg
Кислота Соль rv) mi EpH
СН3СО3Н СН3СО2К 1 0,2442 [1]
СН3СО2Н CH3CO2Na 4 0,2441 [1]
СН3СО2Н ' CH3CO2Na 1 0,2441[1]
СН3СО2Н CH3CO2Na 0,2440 [1]
КН2РО4 Na2HPO4 1 0,2442 [1]
нво2 NaBO2 1 0,2440 [1]
НС1 CH2OHCO2Li 2 0,2442 [1]
НС1 — — 0,2450 [1]
НС1 KCI 9 0,2445 [1]
НС1 NaCl 9 0,2441 [1]
СН3СО,Н CH3CO2Na 1 0,2446 [2]
С1СН2СО2Н ClCH2CO2Na 1 0,2445 [2]
1) Значения являются приближенными,
mi
соединении различных испытуемых растворов с насыщенным раствором хло-
ристого калия, несколько отличаются друг от друга. Первые семь значений,
определенные для буферных растворов, отклоняются не более чем на 0,1 мв
от их среднего значения (0,2441). Для соляной кислоты с добавкой хло-
ристого натрия получается то же значение. В случае чистых растворов со-
ляной кислоты Ерн значительно больше. Наблюдается заметная разница
между величинами ЕрН, найденными для одного и того же буферного рас-
твора уксусной кислоты с помощью двух различных методов. Это расхож-
дение может быть в некоторой степени обусловлено небольшими различиями
между насыщенными каломельными электродами, которые применялись в
этих двух сериях измерений.
Для того чтобы избежать необходимости приготовлять стандартные
электроды согласно точным предписаниям, рекомендуется в ходе работы
определять Ерн для каждой применяемой электродной системы. Для этой
цели следует пользоваться стандартными растворами, приведенными в табл. 64.
Значения pH этих растворов были вычислены из величин ЕрП, которые содер-
жатся в табл. 63, причем эти значения pH представляют собой новую шкалу
стандартных величин, с помощью которых можно определять термодинамиче-
ские константы диссоциации более точно, чем на основании других данных по
измерениям значений pH. Для определения значения ЕрН любой заданной
электродной системы желательно пользоваться стандартным раствором, зна-
чение pH которого близко к pH изучаемого раствора.
Если стандартный и исследуемый растворы имеют одинаковую ионную
силу и очень близки по составу1, то их диффузионные потенциалы по от-
ношению к системе электрода сравнения будут равны и их можно не при-
нимать во внимание для большинства практических целей. Этот экспери-
ментальный метод устранения влияния жидкостного соединения применялся
Харнедом и Робинзоном [30], Гюнтельбергом и Шиодтом [31], Ларсоном и
1 Преобладающие электролиты (присутствующие в более высокой концентрации)
в обоих растворах одинаковы и концентрации их равны.
20 г. Харнед, Б. Оуэн
ЗС6
ГЛАВА X
Таблица 64
Значения pH некоторых стандартных растворов, применяемых для определения
Е°н по уравнению (77)
—Температура, °C состав раствора1), МОЛЬ/’л 25 [1,2] 25 [3] 38 [2] 38 [3]
0,1 НС1 1,085 1,082
0,1 КН3(С2О4)2-2Н2О (тетроксалат) . 1,480 1,495
0,01 HCI-f-0,09 NaCl 2,058
0,01 HCl+0,09 КС1 . . 2,078 2,075
0,03 KHC4H4Oe (тартрат) 3,567
0,ГКН2С6Н5О, (цитрат) 3,719
0,05 КНС3Н4О4 (фталат) 4,008 4,000 4,025 4,015
0,1 НС2Н3О2+0,1 NaC2H3O2 (ацетат) 4,618 4,640 4,655 4,635
0,01 НС2Н3О2+0,01 NaC2H,O2 . . . 4,714 4,700 4,710
0,025 КН2РО4+0,025 Na2HPO4 . . . 6,857 6,835
0,05 Na2B4O7 9,180 9,070 -
1) Состав растворов, употреблявшихся в работах Хичкока и Тейлора [1,2], определялся при
температурах от 21 до 23°.
Аделлом [32], и его можно проиллюстрировать на примере следующих эле-
ментов:
Н2
НА (т4)
NaCl (m —тин)
КС1 (насыщ.), HgCl-Hg,
Н2
НС1 (т0)
NaCl (m — m0)
КС1 (насыщ, ),|HgCl-Hg.
II
III
Так как из величины электродвижущей силы элемента II можно вычислить
с помощью уравнений (76) и (77) приближенное значение тин в растворе
слабой кислоты НА, то можно подобрать концентрации электролитов в эле-
менте III таким образом, чтобы ионная сила обоих растворов была равна1
т. При соединении этих элементов получается цепь
Н2
НА (т^ НС1 (т0)
NaCl (т—тн) NaCl (т— т0)
IV
в электродвижущую силу которой практически не входит диффузионный
потенциал, если т0 мало, а т — относительно большая величина.
Если предположить, что соляная кислота полностью диссоциирована,
то электродвижущая сила элемента IV является мерой концентрации во-
дородных ионов в растворе слабой кислоты. Таким образом,
EIV=-*Ig^, (81).
1 Практически т0 можно сохранять постоянным и равным примерно 0,01 М, по-
скольку при такой концентрации уже соблюдается условие тн ~ т0. Элемент III может
давать ошибочные результаты при т0 < 0,001, однако это является лишь эксперимен-
тальным, а нэ теоретическим ограничением применимости данного метода»
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
307
так как коэффициенты активности водородных ионов в двух растворах
вследствие симметрии элемента сокращаются. Значения
О
Ага = —— -------(82).
ТОНА т1~~ тН ' '
для растворов солей, найденные этим методом [32], хорошо совпадают со
значениями К^, полученными из данных по электродвижущим силам эле-
ментов без жидкостного соединения [33]. Соответствующие термодинами-
ческие константы диссоциации
^а = ^а(р->0) (83)
также согласуются со значениями, найденными другими методами (табл. 112).
Практически устранение диффузионного потенциала, которое дости-
гается в элементе IV путем уменьшения величин т1 и т0, можно сделать
абсолютно полным, если найти предельную величину некоторой функции
от Е и от концентраций при стремлении значений тх и т0 к нулю как
к своему пределу1 *. Экспериментальное определение этой предельной вели-
чины заключается в измерении электродвижущих сил ряда элементов,
содержащих растворы переменного состава, но с постоянной ионной силой,
что достигается добавлением электролита, который не участвует в элек-
тродных реакциях [35]. При экстраполяции до нулевых концентраций тех
ионов, которые имеются лишь в одном из соприкасающихся растворов,
диффузионный потенциал исчезает. Условия экстраполяции были проана-
лизированы Оуэном и Бринкли [34в]. Влияние инертного электролита
исключается путем последующей экстраполяции до нулевой ионной силы.
Данный метод можно проиллюстрировать на примере следующего элемента:
КС1(ж)т AgNO3(x)m
Ag-AgCl т, ' . KNO3(w) ч AgCl-Ag. V
й 6 KNO3(1 — x)m M ’ KNO3(1— x)m 6 6
Общая ионвая сила каждого раствора равна т, однако некоторая доля этой
величины (ж) обусловлена присутствием ионов хлора в левом растворе
и ионов серебра в правом растворе. Электродвижущая сила элемента опре-
деляется уравнением
Е = Л lg aAg/aAg ± Ej, (84)
где Ej — сумма неизвестных диффузионных потенциалов, к — 2,303 RT/F,
«Ag и aAg — активности ионов серебра в правом и левом растворах. Для
исключения a.\g можно применить уравнение произведения растворимости
^AgCI = °Ag flci, (85)
после чего уравнение (84) принимает вид
Е — A: 1g akg act = — A: 1g AAgci i Ej. (86)
Вводя концентрации xm и коэффициенты активности ионов, получаем
Е —2А 1g жт= — A: lg AAgCI-J-A: lg YCIYAg ± Ej. (87)
Отметим, что два последних члена этого уравнения не могут быть незави-
симо определены с помощью одной лишь термодинамики.
1 Этот способ был предложен в работе Лингарта [34а], его применили Попов
и Кунц [346].
20*
308
ГЛАВА X
Рассмотрение уравнения (87) с учетом симметрии электрохимических
систем, которым это уравнение соответствует, показывает, что при постоян-
ном т и переменном х при экстраполяции до ж = 0 получается уравнение
(E-2A:lgx/n)a;=o= — Mg2srAgci + (*lgYciYAg)^0’ (88)
поскольку при этих условиях Ej равно нулю. Так как при ж = 0 можно
отбросить знак «штрих» у , то последний член поддается термодинами-
ческой обработке и может быть выражен в виде функции от т, так же
как для любого сильного 1,1-электролита. Таким образом, к lg YclYAg можно
заменить на —2kS(^y md0, и уравнение (88) принимает вид
(Е — 2к lg хт -j- mdo')x=o — к lg T^Agci- (89)
На рис. 72 изображено определение члена, заключенного в скобки, путем
экстраполяции при нескольких значениях величины т [35]. Определение
Рис. 72. Устранение Ej при посто-
янной общей моляльности (указана
справа) при 25°.
Рис. 73. Определение значения
— A1g^Agci ПРИ 25°'
величины —/с lg AAgci путем последующей экстраполяции полученных таким
образом значений отрезков оси ординат (— к IgAAgci) до zn = O показано
на рис. 73. Оба применяемых для экстраполяции графика являются линейны-
ми1 в пределах ошибки опыта, равной ±0,1 мв, которая соответствует ради-
усам кружков на рисунках. Аналогичные линейные зависимости были
получены при использовании результатов, полученных с помощью
щих элементов [36, 37]:
следую-
AgAc (ж) т
Н21 НАс (zn), NaAc (zn) | НАс (zn), _ ж) т
Ag-AgCl
КС1 (ж) т
KNO3 (1 — ж) zn
КВг (ж) zn
KNO3 (1 — ж) zn
Ag
VI
AgBr-Ag.
VII
и
Простота функции, изображенной графически на рис. 72, имеет существен-
ное’значение, так как ее форма и возможность ее экстраполяции представ-
1 Линейный характер зависимости, представленной на рис. 72, повидимому, может
быть объяснен на основании уравнения (13) гл. XIV и высокой симметрии обеих систем
электролитов. Линейный характер графиков рис. 73 является следствием, уравнений
(23) и (24).
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ЭЛЕМЕНТОВ
ляют собой основное допущение, не обоснованное термодинамически,
на котором основан этот метод устранения диффузионных потенциалов.
Надежность экстраполяции, с помощью которой исключаются диффу-
зионные потенциалы, может быть проверена путем сравнения вычисленных
термодинамических величин с величинами, полученными другими независи-
мыми методами. В тех случаях, когда подобное сопоставление .возможно,
получается вполне удовлетворительное совпадение [35, 36]. Разность между
величинами стандартных потенциалов электродов серебро-хлористое серебро
и серебро-бромистое серебро, полученная из электродвижущей силы
элемента VII, в точности совпадает с разностью между соответствующими
величинами, найденными непосредственно из электродвижущих сил элемен-
тов без жидкостного соединения. Было также обнаружено, что результаты,
полученные с помощью элементов V и VI, превосходно совпадают друг
с другом, хотя коэффициенты наклона экстраполируемых прямых для этих
двух систем различаются по знаку и по порядку величины. Стандартный
электродный потенциал серебра можно вычислить по уравнению
~Ag = lg ^AgCl + ^Ag-AgCl , (90)
используя значения A?Agci, полученные с помощью элемента V. В случае
элемента VI «Ag получается непосредственно. В табл. 65 сопоставлены
Таблица 65
Стандартный электродный потенциал серебра,
полученный из данных по электродвижущим
силам элементов V и VI
t Элемент V Элемент VI Уравнение (91)
5 -0,8185,, -0,8187 -0,81855
15 -0,8087 -0,8090 -0,8089!
25 -0,7990 -0,7992 —0,79910
35 -0,7891 -0,7892 —0,78915
45 -0,7788 —0,77915 —0,7790e
найденные этими двумя методами значения стандартных электродных потен-
циалов. Полученные результаты хорошо выражаются квадратным уравнением
= - 0,7991 + 0,000988 (t - 25) + 7 • 10"7 (Z - 25)2. (91)
С помощью температурного коэффициента, найденного из уравнения, можно
вычислить величину энтропии иона серебра при 25°, которая прекрасно
совпадает со значениями, полученными современными калориметрическими
методами [46, 38].
ЛИТЕРАТУРА
‘1. Г. Н. Льюис, М. Р е н д а л л, Химическая термодинамика, ОНТИ—Химтео-
рет, Л., 1936.
2. G. N. Lewis, М. Randall, J. Am. Chem. Soc., 43, 1112 (1921).
3. D. I. Hitchcock, J. Am. Chem. Soc., 50, 2076 (1928); N. В jerrum,
A. U n m a c k, Det. Kg. Danske Videnskab. Math. fys. Medd., 9, 1 (1929).
4. a) W. M. Latimer, The Oxidation States of the Elements and Their Potentials in
Aqueous Solutions, Prentice-Hall, N. Y., 1938; 6) W. M. L a t i m e r, K. S. Pl l-
zer, W. V. Smith, J. Am. Chem. Soc., 60, 1829 (1938).
310
I' Л А В A X
5. E. H ii с к e 1, Physik. Z., 26, 93 (1925); G. S c a t c h а г d, J. Am. Chem. Soc.,
47, 2098 (1925); H. S. H а г n e d, там же, 48, 326 (1926); 51, 416 (1929); H. S.
Hamed, G. Akerlof, Physik. Z., 27, 411 (1926).
6. P. B. Taylor, J. Phys. Chem., 31, 1478 (1927).
7. H. Helmholtz, Wied. Ann., 3, 201 (1878); Ber., 7, 27 (1882); Ges. Abhl., 1, 840,
2 979
8. D. A. M a с I n n e s, K. Parker, J. Am. Chem. Soc., 37, 1445 (1915).
9. D. A. M a с I n n e s, J. A. Beattie, J. Am. Chem. Soc., 42, 1117 (1920).
10. W. W. Lucasse, J. Am. Chem. Soc., 47, 743 (1925).
11. G. Jones, M. Dole, J. Am. Chem. Soc., 51, 1073 (1929).
12. W. С. H a m e r, J. Am. Chem. Soc., 57, 662 (1935); H. S. H a r n e d, E. C. D re-
b у, там же, 61, 3113 (1939).
13. A. S. Brown, D. A. M a с I n n e s, J. Am. Chem. Soc., 57, 1356 (1935).
14. J. J. Hermans. Rec. trav. chim., 56, 635, 658 (1937); 58, 99, 2419 (1939).
15. F. О. К о e n i g, J. Phys. Chem., 44, 101 (1940); F. O. Koenig, S. W. Grin-
nell, там же, 44, 463 (1940); 46, 980 (1942).
16. a) D. A. M a с I n n e s, J. Am. Chem. Soc., 41, 1086 (1919); H. S. H a r n e d, там же,
42, 1808 (1920); 6) J. Phys. Chem., 30, 433 (1926).
17. M. P 1 a n c k, Wied. Ann., 39, 161 (1890); 40, 561 (1890).
18. a) P. Henderson, Z. physik. Chem., 59, 118 (1907); 63, 525 (1908); 6) J. J. H e r-
m a n s, Rec. trav. chim., 57, 1373 (1938).
19. E. A. G u g g e n h e i m, J. Phys. Chem., 33, 842 (1929); 34, 1540 (1930).
20. J. N. В r 6 n s t e d, Z. physik. Chem., 143, 301 (1928).
21. The Collected Works of J. Willard Gibbs, Vol. I, Longmans Green Co., N. Y., 1928,
p. 429.
22. a) W. M. Clark, The Determination of Hydrogen Ions, Williams and. Wilkins Co.,
Baltimore, 1928; б) E. Мисловицер, Определение концентрации, водород-
ных ионов в жидкостях, НХТИ, Л., 1930; в) X. Т. С. Бриттон, Водородные
ионы. Определение и значение их в теоретической и прикладной химии, Хим-
теорет, Л., 1936.
23. S. Р. L. S о г ens en, Compt. Rend. Lab. Carlsberg, 8, 1 (1909).
24. S. P. L. S a r e n s e n, K. L i n d e r s t r 0 m-L a n g, Compt. Rend. Lab. Carls-
berg, 15, 40 (1924).
25. a) D. I. Hitchcock,!. Am. Chem. Soc., 58, 855 (1936); б) O. R e d 1 i c h,
H. К 1 i n g e г, там же, 61, 2983 (1939).
26. a) W. J. Hamer, Trans. Electrochem. Soc., 72, 45 (1937); W. J. H a m e r, S. F.
Acree, Bur. Standards J. Research, 23, 647 (1939); б) E. A. Guggenhei m,
J. Phys. Chem., 34, 1758 (1930).
27. a) P. Henderson, Z. physik. Chem., 59, 118 (1907); 63,325, (1908); 6) N.. В j e r-
r u m, A. U n m a c k, K. Danske Videnskab. Selskab. Math-fys. Medd., 9, 1
(1929); E. A. G u g g e n h e i m, T. D. Schindler, J. Phys. Chem., 38,
533 (1934).
28. D. A. Maclnnes, D. Belcher, T. S h e d 1 о v s k y, J. Am. Chem. Soc.,
60, 1094 (1938).
29. D. I. H i t c h с о c k, A. C. Taylor, J. Am. Chem. Soc., 59, 1812 (1937).
30. H. S. H a r n e d, R. A. Robinson, J. Am. Chem. Soc., 50, 3157 (1928).
31. E. Guntelberg, E. Schiodt, Z. physik. Chem., 135, 393 (1928).
32. E. Larsson, B. Adell, Z. physik. Chem., A156, 352 (1931).
33. H. S. Harned, G. M. Murphy, J. Am. Chem. Soc., 53, 8 (1931).
34. a) G. A. L i n h a r t, J. Am. Chem. Soc., 38, 2356 (1916); 6) S. P о p о f f, A. H.
Kunz, там же, 51, 382 (1929); в) В. В. Owen, S. R. Brinkley, J. Am.
Chem. Soc., 64, 2171 (1942).
35. В. В. 0 w e n, J. Am. Chem. Soc., 60, 2229 (1938).
36. В. В. 0 w e n, S. R. Brinkley,!. Am. Chem. Soc., 60, 2233 (1938).
37. В. В. 0 w e n, E. J. King, J. Am. Chem. Soc., 63, 1711 (1941).
38. K. S. Pitzer, W. V. S m i t h, J. Am. Chem. Soc., 59, 2633 (1937).
ЛИТЕРАТУРА К ТАБЛИЦАМ
К табл. 63
1. D. I. Н i t с h с о с к, А. С. Taylor, J. Am. Chem. Soc., 59, 1812 (1934).
2. D. A. M a с I n n e s, D. Belcher, T. S h e d 1 о v s к y, J. Am. Chem. Soc.,
60, 1094 (1938).
К табл. 64
1. D. I. Hitchcock, A. C. Taylor, J. Am. Chem. Soc., 59, 1812 (1937).
2. D. I. H i t c h с о с к, A. C. Taylor, J. Am. Chem. Soc., 60, 2710 (1938).
3. D. A. M a с I n n e s, D. Belcher, T. S h e d 1 о v s к y, J. Am. Chem. Soc.,
60, 1094 (1938).
Глава XI
СОЛЯНАЯ КИСЛОТА
Свойства соляной кислоты в водных и неводных растворах, а также
в смешанных водно-неводных растворителях были исследованы более
подробно, чем свойства любого другого электролита, и они могут служить
иллюстрацией основных свойств ионных растворов для того случая, когда
отсутствуют затруднения, связанные с наличием ионов с зарядом больше
единицы. В начале данной главы будет рассмотрен вопрос об определении'
степени диссоциации этой кислоты в средах с различной диэлектрической
постоянной на основании данных об электропроводности. Затем будут по-
дробно описаны свойства соляной кислотц на основании данных об электро-
движущей силе элемента
Н2 | НС1 (т) | AgCl-Ag.
Полученные величины будут сопоставлены с соответствующими величинами,
вычисленными из данных о температурах замерзания, теплотах разбавления
и теплоемкости. Таким путем будет получено исчерпывающее представле-
ние о зависимости свойств определенного электролита от концентрации,
температуры и диэлектрической постоянной. Все полученные результаты
будут сопоставлены с выводами теории междуионного притяжения.
§ 1. Электропроводность соляной кислоты в смесях диоксан —вода
На основании изучения данных по электропроводности соляной кислоты
в водном растворе (гл. VI, § 2) было показано, что соляная кислота
является типичным сильным электролитом. Этот вывод был основан на том,
что наблюдаемая эквивалентная электропроводность А в случае разбавлен-
ных растворов больше, чем вычисленная по уравнению Онзагера [уравне-
ние (3) гл. VI].
А = А° — (а*А°4-р* ]/с),
причем в пределе опытные данные приближаются к значению, вычислен-
ному по этому уравнению. Поведение соляной кислоты в средах с низкой
диэлектрической постоянной хорошо изучено в работе Оуэна и Уотерса [1],
которые измеряли электропроводность соляной кислоты в смесях диок-
сан—вода, содержавших 20, 45, 70 и 82% диоксана при температурах от 15
до 45°. При 25° диэлектрические постоянные этих растворов, согласно
данным Акерлёфа и Шота [2], раввы соответственно: 60,79; 38,48; 17,69
и 9,53. На рис. 74 изображена зависимость эквивалентной электропровод-
ности Д от с1/2. Экспериментальные значения представлены на графике
в виде кружков, а прямые линии соответствуют теоретической зависимости
по уравнению Онзагера. Результаты, полученные в случае смесей, содержа-
щих 20% диоксана, очень похожи на результаты, найденные для водных
растворов, поскольку экспериментальная кривая лежит выше теоретиче-
ской прямой (рис. 16). Даже для наиболее разбавленного раствора (0,0004 н.)
опытное значение несколько превышает теоретическое. В смесях, содержащих
45% диоксана (7)~40), очевидно, происходит ассоциация ионов. Экспери-
ментальная кривая приближается к теоретической снизу, причем расхождение
312
ГЛАВА XI
между обеими кривыми очень незначительно. В случае смесей, содержащих
70% диоксана, наблюдается значительная ассоциация ионов. В смесях, со-
держащих 82% диоксана, соляная кислота ведет себя как типичный слабый
электролит.
Для определения предельной электропроводности в смесях, содержащих
20 и 45% диоксана, применялась экстраполяция с помощью следующей
Рис. 74. Эквивалентная электропровод-
ность соляной кислоты в смесях диок-
сан—вода при 25°.
Прямые линии отвечают теоретическому пре-
дельному закону. X—весовой процент диоксана.
функции [уравнение (12) гл. VI]1:
до, А + ?* /ё
1 — а* с
В случае смесей, содержащих 70
и 82% диоксана, был применен ме-
тод Фуосса и Крауса [3], усовершен-
ствованный фуоссом [4]. Этот метод
основан на использовании уравне-
ний (7) — (13) гл. VII.
Значения констант диссоциации,
полученные Оуэном и Уотерсом,
приведены в табл. 66. Оуэн и Уотерс
не смогли вычислить точные значе-
ния констант диссоциации для сме-
сей, содержащих 20 и 45% диокса-
на, однако они считают, что в слу-
чае последней из этих смесей К
близко к единице.
Общий характер полученных
результатов находится в соответ-
ствии с теорией ассоциации ионов
Бьеррума. Применяя эту теорию
[уравнение (57) гл. III], Оуэн и
Уотерс вычислили значения сумм
ионных радиусов в ангстремах, так-
же приведенные в табл. 66. Величины, полученные для Смесей, содер-
жащих 82% диоксана, следует считать более надежными, поскольку
Таблица 66
Константы диссоциации соляной кислоты в смесях дпоксан—вода ')
t,°C 70% диоксана 82% диоксана
От К • 102 О а Do К 104 й2)
15 18,72 11 9,1 10,01 2,5 6,6
25 17,69 8 7,9 9,53 2,0 6,3
35 16,72 7 7,7 9,06 1,6 6,2
45 15,80 6 7,4 8,62 1,1 5,8
1) Величины параметров о.»
в таблице 124.
-) Сумма ионных радиусов.
и Р* п необходимые для расчетов величины вязкости можно найти
значения константы диссоциации К для этих смесей несколько более до-
стоверны, чем для смесей с меньшим количеством диоксана.
1 Величины а* и % для смесей диоксан—вода, содержащих 20 и 45% диоксана,
приведены в табл. 124.
СОЛЯНАЯ КИСЛОТА
313
(1)
§ 2. Стандартный потенциал элемента Н21 НС1 (т) | AgCl-Ag в случае
водных растворов
Реакция, происходящая в элементе
Н2|НС1 (тп)| AgCl-Ag,
может быть выражена уравнением
уН2 + AgCl = Ag + НС1 (m).
(2)
Так как при этой реакции образуется один моль хлористоводородной
кислоты, то электродвижущая сила элемента определяется уравнением (21)
гл. X. Таким образом, для 1,1-элек-
тролита ^г50| !
Е2А: lg m — Е° — 2А: 1g у±, (3)
где к = 2,3026 RT/Y. Для экстрапо-
ляции воспользуемся уравнением (24)
гл. X:
Е 4- 2к 1g т —- 2к |/ Г =
= Е° — 2кВ'т. (4)
На рис. 75 левая часть этого
уравнения представлена графически
как функция от т. Как видно из
рисунка, при низких концентрациях
опытные данные ложатся на прямые
линии, и поэтому экстраполяция мо-
жет быть выполнена с большой точ-
ностью. Так как электродный по-
тенциал водородного электрода при т
давлении в 1 атм условно принят Рис. 75. Определение Е° по уравнению (4);,
равным нулю при всех температурах, интервалы между каждыми двумя со-
то найденная таким путем величина седними линиями составляют 5°.
Е°, взятая с обратным знаком, пред-
ставляет собой стандартный потенциал электрода серебро-хлористое серебро.
Определению величины этого стандартного электродного потенциала
было посвящено немало исследований. Ниже будут рассмотрены лишь те
данные, которые нам представляются наиболее точными. Для определе-
ния Е° применялись электроды различных типов. Лингарт [5] использовал
в качестве электрода платиновую проволоку, опущенную в смесь осадка
хлористого серебра с серебром, приготовленным путем электролиза раство-
ра азотнокислого серебра при большой плотности тока. Кармоди [6] при-
менял платиновую сетку, на которой серебро осаждалось электролитически,
а хлористое серебро образовывалось на этом электроде путем электролиза
соляной кислоты1. Нойес и Эллис [8], Робертс [9], Харнед и Элерс [10]
применяли электроды, приготовленные путем нагревания окиси серебра
на платиновой спирали до 500°. Полученное таким образом серебро покры-
валось хлористым серебром путем электролиза в соляной кислоте2.
1 Аналогичный электрод был применен Скэтчардом [7].
2 Несколько отличные типы этого электрода описаны Гюнтельбергом [11а],,
Харнедом [116], Мак-Иннесом и Битти [Ив] и Брауном [Иг].
314
ГЛАВА XI
Величины Е° обычно определяют графическими методами. Прентис
и Скэтчард [12] вычислили Е° с помощью метода наименьших квадратов
на основании • опытных данных, полученных вышеуказанными авторами.
В табл. 67 приведены результаты, полученные при 25°.
Несмотря на различия в методике и в типах электродов, величины
•стандартных потенциалов, найденные разными авторами, превосходно сов-
падают друг с другом. Кроме того, величины, найденные графическим
путем, прекрасно согласуются с величинами, вычисленными по методу
наименьших квадратов.
Таблица 67
Стандартный потенциал электрода
серебро-хлористое серебро при 25°
Исследователь Графический метод Метод наименьших квадратов ’)
Кармоди -0,2223 х) -0,22222
» -0,2222 2) —.
Лингарт -0,2226 3) -0,22253
» -0,2224 4) —
Робертс -0,22240 5) -0,22241
» — -0,22237
Харнед и Элерс .... -0,22239 6) -0,22244
Мак-Иннес — 0,2225 8) —
Вычислено: 1)—Кармоди [1]; 2)—Спенсером [12];
з>—Скэтчардом [3]; 4)—Хичкоком [4J; 5)~Робертсом [5];
6)—Харнедом и Элерсом [6]; 7)—Прентисом и Скэтчар-
дом [7]; 8)—Мак-Иннесом [8].
Таблица 68
Стандартный потенциал электрода серебро-хлористое серебро
в интервале температур от 0 до 60°
г,° с -’’Ag-AgCl =Е° Отклонения, Мв 1) t,° С “^Ag-AgCr Отклонен ия, мв 1)
0 0,23634 -0,02 35 0,21563 -0,02
5 0,23392 -0,01 40 0,21200 0,00
10 0,23126 + 0,04 45 0,20821 +0,04
15 0,22847 4-0,04 50 0,20437 -0,01
20 0,22551 ’ + 0,04 55 0,20035 -0,02
25 0,22239 0,00 60 0,19620 0,00
30 0,21912 -0,04
1) Отрицательные отклонения означают, что вычисленная величина меньше, чем
наблюдаемая.
Необходимо отметить, что величины стандартных потенциалов довольно
сильно зависят от выбора численных значений постоянной к и предельного
коэффициента наклона уравнения Дебая и Гюккеля; Харнед и Райт [13]
вычислили Е° из данных Харнеда и Элерса следующим образом: а) пола-
СОЛЯНАЯ КИСЛОТА
315
гая к=- 0,000198447 и 7 = £ + 2^3 ,1 в соответствии с данными «Internatio-
nal Critical Tables»; б) применяя данные Бёрджа [14], согласно которым
к ~ 0,00019848 7 и 7 = t-\- 273,18. В случае «а» Е° при 25° оказалось рав-
ным 0,22239а, а в случае «б» — 0,22223в.
Применяя надежную методику, Харнед и Элерс определили стандарт-
ные потенциалы электрода серебро-хлористое серебро при температурах
от 0 до 60° с интервалами в 5°. Эти авторы применяли уравнение (4)
и определяли Е° графическим методом. В табл. 68 представлены значения
стандартных потенциалов, полученные Харнедом и Элерсом, а также откло-
нения наблюдаемых значений от вычисленных по методу наименьших квад-
ратов с помощью уравнения
АйС1 = Е° = 0,22239 -645,52 1(Г° (« -25)-
- 3,284 • IO'6 * * (t - 25)2 + 9,948 • IO"9 (Z - 25)3. (5)
При последующих вычислениях будут использованы эти значения, так как
они охватывают широкий интервал температур и получены с помощью
такой же методики, как и большинство рассматриваемых ниже эксперимен-
тальных данных. Отклонения вычисленных значений стандартных потен-
циалов от наблюдаемых меньше, чем суммарная ошибка при измерении
и экстраполяции, которая составляет ± 0,05 мв.
§ 3. Стандартный потенциал элемента Н21 HCI (т), £ (X), Н2О (К) | AgCl-Ag
для смесей вода — органический растворитель
Электродвижущие силы этих элементов в случае смесей вода — мети-
ловый спирт, содержащих 10 и 20% метилового спирта, были определены
Харнедом и Томасом [15]. Нонгебель и Хартли [16] и Вулкок и Хартли [17]
исследовали эти элементы для случаев растворов в чистых метиловом
и этиловом спиртах. Аналогичные измерения в чистом этиловом спирте
при 25° были произведены Даннером [18], а в смесях, содержащих 50
и 99,9% этилового спирта, Харнедом и Флейшером [19]. Лукасе [20]
измерял электродвижущую силу этого элемента в случае смесей глицерин —
вода, содержащих 1 и 5 молярных процентов глицерина, а Скэтчард [21]—
в растворах сахарозы. Наиболее полные измерения такого рода были
выполнены для смесей диоксан — вода *. В этих исследованиях электро-
движущие силы элементов были определены в области температур 0 — 50°
с интервалом в 5° при концентрациях соляной кислоты от 0,001 М до самых
высоких практически достижимых концентраций в смесях диоксан — вода,
содержащих 20, 45, 70 и 82% диоксана, что соответствует диэлектрическим
постоянным растворителя, равным примерно 60, 40, 20 и 10. Диэлектри-
ческие постоянные этих смесей были определены Акерлефом и Шотом [23],
а упругости пара — Говорка, Шефером и Дрейсбахом [24].
При уменьшении диэлектрической постоянной растворителя при экстра-
поляции должны быть учтены члены более высокого порядка в уравнениях
теории междуионного притяжения (42) и (43) гл. III. Для того чтобы
проиллюстрировать влияние этих членов и в то же время продемонстри-
1 Харнед и Моррисон [22а]; Харнед и Кэлмон [226] измеряли плотности смесей:
Харнед [22в] выполнил экстраполяции для 20 и 45% смесей; Харнед и Донелсон [22г]
исследовали свойства (20 и 45%); Харнед и Кэлмон [22д] выполнили экстраполя-
ции (70%); Харнед, Донелсон и Кэлмон [22е] исследовали свойства (70%); Харнед,
Уолкер и Кэлмон [22ж] выполнили экстраполяции (82%); Харнед и Уолкер [22з]
исследовали свойства (82%): Харнед, Моррисон, Уолкер, Донелсон и Кэлмон [22и]
цали общий обзор и критические замечания.
316
ГЛАВА XI
ровать самый метод экстраполяции для смесей, содержащих 20, 45 и 70%,
диоксана, воспользуемся функцией Е0', определяемой уравнением
2Л 1/ с
Е° esE + 2/clgm-—lg(H-0,002Mxym)=E° + /(m). (6)
1 -|- "I/ е
В этом уравнении Е — экспериментальное значение электродвижущей силы
элемента при концентрации т, Е° — стандартный потенциал элемента,. Л' —
параметр, включающий кажущийся
гл. Ill], Mxy — средний молекулярный
Рис. 76. Экстраполяция по уравнению (6)
для смесей диоксан—вода (при 25°).
Справа указаны весовые проценты дионсана.
диаметр ионов а [уравнение (35)
вес растворителя [уравнение (60)
гл. I]. Сначала Е(Г было вычи-
слено с применением приближен-
ного значения а. Затем с помощью
функций, приведенных в табл. 8,
была вычислена составляющая элек-
тродного потенциала, обуслов-
ленная влиянием дополнительных
членов Гронвола, Ла-Мера, Сэнд-
веда, соответствующая этому зна-
чению а. Эти величины, умно-
женные на 2к, обозначены через
Едоп,. Затем величины Е° и
Е°'-Едоп. изображались графи-
чески как функции от т, и в тех
случаях, когда разность Е° — Едоп.
не была постоянной при низких
концентрациях (т < 0,02 М), при-
нималось другое значение а, и
так до тех пор, пока не достигалось
постоянство разности Е° —Едоп .
Значения с получались с помощью табл. 126.
На рис. 76 хорошо видна экстраполяция и влияние дополнительных
членов. На этом рисунке изображена зависимость величин Е° и Е° —Едоп,
от моляльности т при 25° для растворителей, содержащих 20, 45 и 70%
диоксана. Диэлектрические постоянные этих смесей равны соответственно
60,79; 38,48 и 17,69. Как видно из рис. 76, на графиках для Е° имеются
характерные для этой функции искривления вблизи оси ординат. Степень
этих искривлений увеличивается с уменьшением диэлектрической постоян-
ной среды. При использовании уравнений, содержащих дополнительные
члены, этот эффект исчезает, как видно из графиков функции Е° — Едоп ,
которые при значениях а, равных соответственно 5,0; 5,4 и 5,6, и при
концентрациях меньше 0,02 М представляют собой прямые линии с нулевыми
коэффициентами наклона [/(да) — 0]. Значение составляющей, связанной
с влиянием дополнительных членов, для случая смесей с 20% дноксана
настолько мало, что экстраполяция без их учета приводит практически
к тому же значению Е° (отклонение составляет -^0,1 мв). Приведенные
выше значения а хорошо совпадают со значением 5,6, полученным Шид-
ловским и Мак-Иннесом [25] путем измерения электродвижущих сил эле-
ментов с жидкостным соединением для случая водных растворов, и со значе-
нием 5,3, полученным из данных Харнеда и Элерса с помощью измерений
электродвижущих сил элементов без жидкостного соединения с той же
СОЛЯНАЯ КИСЛОТА
317
средой в качестве растворителя. Как видно из табл. 69, функция Е0' — Едоп.
сохраняет постоянное значение при изменении концентраций от 0,002 до
0,02М.
Результаты определения стандартного потенциала в случае смесей
с таким низким значением диэлектрической постоянной, как это имеет
место для смеси, содержащей 82% диоксана, не являются надежными. Это
обусловливается несколькими причинами. Во-первых, теория Дебая и Гюк-
келя в форме расширенного уравнения Гронвола, Ла-Мера и Сэндведа
неприменима в случае растворов с диэлектрической постоянной, равной 10
и менее, поскольку второй член полного уравнения при этом весьма велик.
Так, при концентрации кислоты порядка 0,001 М этот член больше, чем
первый член. Во-вторых, в настоящее время еще нет достоверных определе-
ний электродвижущих сил при концентрациях кислоты, меньших чем
0,001 М. Для эмпирической экстраполяции необходимо знать электро-
движущие силы элементов с концентрацией растворов порядка 0,00005 М
или еще ниже. Ввиду наличия этих затруднений ниже применяется метод,
в котором используется закон действующих масс, а также константы дис-
социации, полученные из данных по электропроводности. Этот метод
не является вполне удовлетворительным, однако он не хуже любого дру-
гого метода, имеющегося в нашем распоряжении в настоящее время.
Таблица 69
Значения экстраполяционной функции Е0' —Е°оп
о ° 5,0 5,4 5,6
m -Х = 20% Х=45% 1 = 70%
0,000 (0,20303) (0,16358) ' (0,06395)
0,001 — — (0,0634) !)
0,002 — — 0,06400
0,003 .0,20298 0,16357 0,06393
0,005 0,20304 0,16358 0,06389
0,007 0,20303 0,16359 0,06391
0,01 0,20305 0,16361 0,06399
0,02 0,20305 0,16360 0,06400
1) При концентрациях ниже 0,002 М точность результатов
значительно уменьшается, нан видно из приведенной величины.
Этот вопрос был подробно рассмотрен Харнедом и Кэлмоном [1].
2) Значения S(/)> использованные в этих вычислениях, при-
ведены в табл. 125, а плотности растворов—в табл. 126.
Согласно уравнению (6), функция Е0'—Е° определяется выражением:
Е0' — Е° = Е — Е° 4- 2# 1g пг — = / (т), (7)
если пренебречь членом, содержащим МХу. Если разность Е0'— Е° может
быть вычислена с помощью какого-либо теоретически правильного способа
при т = 0, тогда с помощью известных значений Е(Г можно найти Е°. Для
того чтобы использовать известные значения константы диссоциации К,
можно выразить электродвижущую силу элемента уравнением
Е — Е° = — 2к 1g атуа, (8)
318
ГЛАВА XI
где а —степень диссоциации, — коэффициент активности кислоты как
сильного электролита при концентрации ионов, равной ат. Сочетая это
уравнение с уравнением (7), получаем искомую функцию
Еа~Е°'-Е°= - 2/с lg aYa - / (m). (9)
1 ~Г 21 у с
Таким .образом, задача сводится к вычислению а и у . Выражая а через
значение термодинамической константы диссоциации [уравнение (19)
гл. VII], получаем
1 К , /ТР
а~~2-----2~±1/ + • (Ю)
2 L V ~^т Го.т]
Применяя теоретическое уравнение с дополнительными членами, можно
вычислить уа и затем начертить график зависимости у от ]/ т . Приве-
Рис. 77. Экстраполяция по уравнениям (9)
и (10) для 82-процентпых смесей диоксан—вода
при 25°.
денное выше уравнение было
затем решено арифметическим
приближенным вычислением1 с
подстановкой значений К, най-
денных из данных по измере-
ниям электропроводности (табл.
66). Экстраполяционные кривые
при 25° показаны на рис. 77,
на котором Ео< и разность.
Е0' — Еа изображены в зависи-
мости от т. При ?п=0 обе эти
функции равны Е°. При экстра-
поляции функции Дебая и Гюк-
ЕО'
можно получить ОШИ-
бочный результат, так как гра-
фик этой функции является
почти прямолинейным, и если
не учитывать наличие ассоциа-
ции ионов, то эту функцию
следовало бы экстраполировать,,
как показано верхней пунктир-
ной линией. Эта экстраполяция приводит к значению Е°, которое примерна
на 10 мв выше истинной величины. Нижней пунктирной линией обозначен
предполагаемый ход истинной кривой. Однако до сих пор отсутствуют до-
стоверные результаты для таких низких концентраций кислоты (мень-
ше 0,001 Л7).
График Е0'— Еа не является Достаточно удобным для экстраполяции,
так как он имеет значительный коэффициент наклона, из-за чего сни-
жается точность экстраполяции. Средний ионный диаметр был принят
равным 6 А. в соответствии с данными Оуэна и Уотерса (табл. 66). Сле-
дует отметить, что это значение близко к тому, которое было найдено для
среднего расстояния сближения ионов в воде и в смесях диоксан — вода,
содержащих 20, 45 и 70% диоксана, из данных по измерениям электро-
движущих сил элементов.
Значения стандартных потенциалов при 25° для некоторых из этих
сред и для концентраций, выраженных в единицах т, с и N, приведены
1 Так как этот расчет является приближенным, то различия между значениями Кг
которые получаются при выражении концентрации в единицах с и т, не принимаются
во внимание.
СОЛЯНАЯ КИСЛОТА 319-
в табл. 70 и отмечены звездочками в соответствии с уравнениями (13)
и (14). Согласно уравнению (54) гл. I, три величины Е„, Ес* и En свя-
заны уравнениями (при 25°)
Е°‘= Е^ +0,1183 lg d0, (И)
E^*=Em -0,1183 lg (1000/>XY). (12)
Таблица 70
Стандартные потенциалы элементов Н2|НС1(пг), растворитель (N2), Н2О (NjJlAgCl-Ag
при 25°. Ni, N2—молярные доли
Растворитель jV2 Do m 0 * «А
Вода . . 0 78,54 0,22239 0,22151 . 0,01602
Метиловый спирт—вода* 1) . . 0,0588 74,0 0,21538 0,21431 0,01124
» » » . . . 0,1233 69,2 0,20881 0,20692 0,00710
» » » . . . 1,00 31,5 -0,0101
Этиловый спирт—вода .... 0,0417 72,8 0,21442 0,21340 0,01123
» » » .... 0,0891 67,0 0,20736 0,20561 0,00763
» » .... 1,000 24,3 -0,0740
Глицерин —вода 0,01 77 0,2146 0,2201 0,0153
» 0,05 72 0,2082 0,2106 0,0107
Изопропиловый спирт—вода . 0,0323 71,4 0,21363 0,21266 0,01095
Диоксан— -вода1) 0,0487 60,8 0,20303 0,20375 0,00554
» » 0,1433 38,5 0,16352 0,16513 -0,02002
» » ...... 0,3231 17,7 0,06395 0,06584 -0,10049
» » 0,4823 9,5 -0,0413 -0,0396 -0,19339
1) Стандартные потенциалы при других
температурах можно найти в табл.
127.
На рис. 78 представлена зависимость Е™ от 1/Z) для сред с большой
диэлектрической постоянной (Z)~ 80 — 60). Начальная точка кривых
в левой стороне рисунка отвечает значению Е™ для чистой воды. Ни одна
из этих кривых не является прямолинейной, и, кроме того, кривые отли-
чаются друг от друга. Графики зависимости величин Ec°’hEjv от ^i/D
имеют аналогичный характер.
Явление переноса кислоты из воды в смешанный растворитель, содер-
жащий воду, может быть выражено количественно следующим образом.
Электродвижущую силу соответствующих элементов при 25° можно выра-
зить с помощью основных уравнений [уравнение (3)]:
Е = Е„ — 0,059151g 7нннгс1 — 0,059151g унуС1, (13)
Е = Em — 0,05915 lg7HHW.1 - 0,05915 lg(14)
где Е„ — стандартный потенциал элемента, [содержащего водный раствор,
YhYci — коэффициент активности в любом из этих растворов, отнесенный
к коэффициенту активности бесконечно разбавленного водного раствора,
принятому за единицу, —стандартный потенциал элемента, содержащего
320
ГЛАВА XI
любую смесь, отнесенный к коэффициенту активности при бесконеч-
ном разбавлении в том же растворителе, принятому за едивицу. Из этих
уравнений получаем выражение
Em —Em — 0,05915 lg (15)
7Н(С1
Верхний индекс в виде звездочки представляет собой специальное обозна-
чение, применяемое при рассмотрении переноса электролита из одной среды
Рис. 78. Зависимость EJ^ от 1/Z) для
г.ред с высокой диэлектрической посто-
янной.
О вода; ? глицерин—вода; С) метиловый спирт—
вода; ф этиловый’спирт—вода; 4- изопропило-
вый спирт—вода; □ диоксан—вода. Пунктирные
линии соответствуют уравнению (18).
в другую. Такое обозначение исполь-
зуется в данном параграфе, а также
в гл. XV, § 7.
Применяя термодинамические урав-
нения к реакции Н+ т СГ -|- Н2О
^±Н3О+ + СГ, которая является основ-
ной реакцией в случае растворов
Рис. 79. Зависимость функции —
—0,05915 1g Ni от 1/D.
О вода; ? глицерин—вода; О метиловый спирт—
вода; ф этиловый спирт—вода; + изопропиловый
спирт—вода; О диоксап—вода. Пунктирные линии
соответствуют уравнению (18).
с большим содержанием воды, из уравнений (13) и (14) можно получить
следующее выражение:
Em - (Em - 0,059151g аш) = 0,05915 1g , (16)
7н3о7с1
где aw — активность воды в любой смеси. Аналогичным образом получаем
уравнение
БЛ- (Е^-0,05915 lg aw) = 0,05915 Ig-^30^1- • (17)
'НзО'С!
Активность чистой воды всегда принимается за единицу. Данные по пар-
циальной упругости пара указывают на то, что в случае растворов, содер-
жащих много воды и небольшое количество органического растворителя,
можно с некоторым приближением заменить аш через молярную долю
воды А’,. При этом допущении получается графическая зависимость вели-
чины Едг* —0,059151g от 1/D, показанная - на рис. 79. В отличие от
данных, представленных на рис. 78, точки, полученные для всех раствори-
СОЛЯНАЯ КИСЛОТА
321
телей, довольно точно ложатся на одну и ту же кривую. Если этот
интересный результат будет подтвержден дальнейшими эксперименталь-
ными исследованиями, он может оказаться весьма важным для обработки
такого рода опытных данных.
Перенос кислоты, соответствующий уравнениям (15), (16) и (17), опре-
делен для раствора с активностью и коэффициентом активности, равными
единице (гипотетический идеальный раствор с моляльностью, равной еди-
нице), т. е. без учета влияния ионных атмосфер. Электростатическая
теория этого влияния была рассмотрена в гл. III, § 10, причем было пока-
зано, что она приводит к уравнению Борна [уравнение (109) гл. III].
Если в этом уравнении заменить ln/±(!) соответствующим выражением
для электродвижущей силы, тогда
для 1,1-электролита при 25° полу-
чается уравнение
w0 р0*
0,05915 —
Z). — D-> v, 1
“‘210'10!~hr2v <18>
Кривые, соответствующие этому урав-
нению, показаны пунктирными ли-
ниями на рис. 78 и 79. Нижние
пунктирные кривые на обоих рисун-
ках отвечают уравнению (18) при
2 1/b} = 1,2, что примерно соответ-
ствует величинам, которые получа-
ются из данных о кристаллографи-
ческих радиусах. Верхние пунктир-
изменении D в широких пределах.
1—метиловый спирт—вода; 2—этиловый спирт—
вода; 3—диоксаи—вода. Пунктирная прямая
отвечает уравнению (18).
ные кривые отвечают этому уравне-
нию при 21 1/^7 — 0,8, причем оэто значение получается, если принять сред-
ний ионный диаметр равным 5 А. и считать радиусы обоих ионов одинаковыми.
Хотя эти приближенные теоретически кривые заметно отличаются от опыт-
ных, все же несомненно, что теоретические вычисления приводят к вели-
чинам йравильного порядка. Рассмотренное явление слишком, сложно, что-
бы его можно было полностью объяснить с помощью такой простой элек-
тростатической теории.
На рис. 80 показан характер зависимости Em от 1/7) для более ши-
рокого интервала изменения диэлектрической постоянной. Кривая 2 пред-
ставляет зависимость Em от 1/7) для смесей вода — этиловый спирт, состав
которых меняется в пределах от чистой воды до чистого спирта. Проме-
жуточные значения Em, не приведенные в табл. 70, получены из данных
Харнеда и Флейшера [19]. Кривая 1 для смесей метилового спирта с во-
дой ввиду недостаточного количества опытных данных имеет приближен-
ный характер. Кривая 3 изображает зависимость Em от 1/7) для смесей
диоксан — вода. Конечная точка этой кривой отвечает раствору с диэлек-
трической постоянной 9,6, еще весьма далекой от диэлектрической постоян-
ной чистого диоксана, для которого 7) = 2,1. Прямая пунктирная линия
соответствует значениям, вычисленным с помощью уравнения Борна.
Следует отметить, что это уравнение хорошо отражает характер измене-
ния величины Em в случае смесей, содержащих от 100 до 2% воды.
Очень быстрое уменьшение величины Em с ростом 1/7), который про-
исходит по мере уменьшения . количества воды в смеси, связано с тем,
что вместо иона оксония Н3О+ появляется ион (растворитель) Н+ согласно
21 Г. Харнед. Б Оуэн
322
ГЛАВА XI
реакции Н30+ + (растворитель) ?Н20 + (растворитель) Н+. В случае сме-
сей Диоксан — вода нет опытных данных для достаточно высокой концен-
трации диоксана, необходимой для возникновения этого эффекта. Вопрос
о влиянии среды будет подробно рассмотрен в гл. XV.
§ 4. Коэффициент активности соляной кислоты в водных растворах
В настоящем параграфе будет подробно рассмотрен вопрос о коэффи-
циенте активности и других термодинамических свойствах соляной кисло-
ты, определенных с помощью экспериментальных данных по электродви-
жущим силам. При этом будет показано, в какой степени применима тео-
рия междуионного притяжения и каковы дополнительные параметры, необ-
ходимые для отражения опытных данных.
Наиболее подробные исследования элемента I были проведены Хар-
недом и Элерсом [10, 26], которые определили э. д. с. этого элемента
в области температур 0 — 60° с интервалами в 5° при концентрациях
от 0,004 до 4 М. Точность полученных ими результатов была порядка
± 0,05.ме. Результаты измерений электродвижущих сил в указанной
области температур выражаются уравнением
Е = Е25 + a (i — 25) + 6 (^ —- 25)а4-с (^ — 25)3. (19)
Константы этого уравнения приведены в статье Харнеда и Элерса [26а].
Данные для концентраций от 0,0001 М до 0,002 М включительно были
получены с помощью графиков зависимости левой части уравнения (4)
от т, применявшихся для определения Е°.
После преобразования уравнения (3) получаем
, , (Е—Е°)
lgY±= -lgzn-v-- (20)
Для вычисления значений у±, приведенных в табл. 128, были использо-
ваны значения Е, полученные по уравнению (19), и значения Е° из табл. 68.
Значения, заключенные в скобки, были получены экстраполяцией данных
по электродвижущим силам; эти значения отвечают концентрациям, кото-
рые лежат за пределами области исследованных концентраций. Величины
для промежуточных температур могут быть получевы интерполяцией.
Ошибка в ± 0,05 мв в определении электродвижущей силы соответствует
ошибке в величине коэффициента активности порядка ± 0,001. Значения
для 20 и 25° очень хорошо согласуются с результатами, полученными
из данных по измерению электродвижущих сил Гюнтельбергом [27],
а также Рендаллом и Юнгом [28]. Акерлёф и Тир [29] распространили
измерения электродвижущей силы элемента I на область концентраций
кислоты от 3 до 16 М. Полученные этими исследователями величины 1g
в интервале температур от 0 до 50° приведены в табл. 162.
Подтверждением правильности этих результатов может служить их
сопоставление с величинами коэффициентов активности при температурах
замерзания растворов у±, вычисленными Рендаллом и Юнгом из данных по
определению температур замерзания Рендалла и Ванзелова [30]. Как видно
из табл. 71, значения коэффициентов активности, вычисленные с помощью
данных по электродвижущим силам элементов и по температурам замерзания,
хорошо совпадают друг с другом. Это совпадение подтверждает правиль-
ность нашего предположения о природе реакции, протекающей в элементе.
СОЛЯНАЯ КИСЛОТА
323
Таблица 71
Экспериментальные и вычисленные средние коэффициенты активности (э. д. с.—
по методу измерения электродвижущих сил; т. замерз.—по данным
для понижения температуры замерзания)
°C 0 251) 60
э. д. с. т. замерз. вычисл. 3. д. с. вычисл. э. д. с. вычисл.
0,001 0,9668 0,9661 0,9667 0,9656 0,9654 0,9632 0,9628
0,002 0,9541 0,9534 0,9541 0,9521 0,9524 0,9491 0,9489
0,005 0,9303 0,9300 0,9311 0,9285 0,9285 0,9235 0,9235
0,01 0,9065 0,9064 0,9080 0,9048 0,9045 0,8987 0,8975
0,02 0,8774 0,8772 0,8798 0,8755 0,8751 0,8666 0,8660
0,05 0,8346 0,8341 0,8366 0,8304 0,8299 0,8168 0,8170
0,1 0,8027 0,8021 0,8032 0,7964 0,7946 0,7813 0,7780
0,2 0,7756 0,7754 0,7749 0,7667 0,7650 0,7437 0,7437
0,5 0,7761 0,774 0,7753 0,7571 0,7563 0,7237 0,7234
1,0 0,8419 0,840 0,8415 0,8090 0,8091 0,7541 0,7567
1,5 0,9452 — - 0,9465 0,8962 0,8960 0,8178 0,8196
2,0 1,078 — 1,081 1,0090 1,0085 0,9072 0,9059
3,0 1,452 — 1,454 1,316 1,313 — —
4,0 2,006 — 2,005 — — — —
1) Значения при 25° и концентрации ниже 0,1 М были получены Шидловским и Мак-Инн&-
сом [1] из данных для элементов с жидкостным соединением.
§ 5. Вычисление коэффициента активности соляной кислоты в водном
растворе с помощью обобщенной теории Дебая и Гюккеля
Из уравнений (57) гл. I, (34) и (42) гл. III можно получить следу-
ющее выражение для 1,1-электролита:
lg Y± = - Д + Д°п’ — !&(! + 0,01802vm), (21).
1 + А ус
где Доп,—общая составляющая, обусловленная дополнительными членами.
Это уравнение выражает общее влияние кулоновских сил на коэффициент
активности полностью диссоциированного электролита, если диэлектриче-
ская постоянная среды, состоящей из кислоты и воды, остается постоян-
ной. Коэффициент активности реального электролита приближается к
соответствующей теоретической величине по мере приближения концентра-
ции раствора к нулю. Для того чтобы отразить поведение реального
электролита, необходимо ввести в уравнение (21) линейный член относи-
тельно с [уравнение (32) гл. X] и члены степенного ряда с переменной с.
Таким образом, получаем
lgY±=- £т£^ + Доп. + Вс+ У Лпс"/2 — lg(1+ 0,01802vm), (22)
где В и Ап — постоянные для данной температуры и давления. Экспери-
ментальные значения коэффициентов активности сильных электролитов
для растворов с концентрациями вплоть до 1 М весьма точно выражаются
уравнением, не содержащим членов в степени выше единицы по отноше-
21*
'324
ГЛАВА XI
нию к концентрации1. Параметры А' и В можно определить путем со-
вместного решения двух уравнений, подобно тому как это было описано
в гл. X, § 4 [уравнение (33) гл. X]. В более ранних исследованиях для
определения параметров А' я. В Гюккель [31], Харнед [32], а также Хар-
нед и Акерлёф [33] пользовались данными для концентраций 0,1—3 М
и уравнением, включающим только линейный член относительно с.
Найденные этими авторами значения параметра А', а следовательно
[уравнение (35) гл. III] и величины среднего расстояния сближения
ионов а несколько занижены. Более точные значения а могут быть най-
дены, если константы уравнения определить из экспериментальных
данных, полученных для пределов концентраций от 0,01 до 1 М. Это
определение является очень точным, так как, например, изменение
величины а от 4,2 до 4,3 вызывает такое изменение первого члена в пра-
вой части уравнения (21), которое соответствует изменению на одну еди-
ницу в третьем десятичном знаке коэффициента активности при концен-
трации раствора 0,1 М.
Харнед и Элерс нашли а по уравнению непосредственно из экспери-
ментальных данных для области температур от 0 до 60° с интервалами
через каждые 5°. Все полученные ими значения а лежат в пределах 4,3 ±0,2,
а девять значений из тринадцати (при температурах 10 — 50°) лежат в пре-
делах 4,33 ± 0,03. Этот результат весьма важен, так так он показывает,
что если величина среднего расстояния сближения ионов и меняется при
изменении температуры, то это изменение очень незначительно. Следует
Отметить, что найденное значение а значительно больше, чем сумма ион-
ных радиусов, определенных из кристаллографических данных. Для раз-
бавленных растворов этот результат не является неожиданным, так как
ионы гидратированы. Шидловский и Мак-Иннес [25] получили из данных
по измерениям электродвижущих сил элементов с жидкостным соедине-
нием значение а = 4,6, применяя уравнение (22), содержащее только пер-
вый, третий и пятый члены. Подставляя в уравнение (22) D'c2 вместо
члена 2 А„си/2, Харнед и Элерс получили уравнение, которое очень хорошо
выражает опытные данные в интервале температур 0 — 60° и для области
концентраций от 0,005 до 2 и даже до 4 М. Для своих вычислений эти
авторы использовали значения и х/<±2, приведенные в табл. 7, и зна-
чение й = 4,3. Оказалось, что постоянные В и D' линейно зависят от
температуры согласно уравнениям
В = 0,1390 — 0,0003.92г, (23)
Z)'= 0,0070 — 0,000033г. (24)
В табл. 71 сопоставлены коэффициенты активности, найденные экспери-
ментальным путем и вычисленные по уравнению (23) при 0, 25 и 60°.
В табл. 72 приведены уравнение и постоянные ау и Ьг, с помощью
которых может быть вычислено значение с из данного значения т. Эта
.таблица содержит также значения параметров А', В и D' и значения
коэффициентов наклона в области температур 0 — 60° с интерва-
лами в 5°. Таким образом, имеются все данные, необходимые для при-
1 Скэтчард и Прентис использовали степенной ряд вида:
§(Л /?+ Вс + Сс312 + Dc*+ • • •
Таблица 72
Данные, необходимые для вычисления коэффициента активности соляной кислоты по уравнению (22)
lg‘( . =-------------------доп. _|_ 2?с jr)'c2 _ 1g (1 0,03604 т),
1 + А'ус
с ,
где — = а, — ь,т
т
Применимы для концентраций от 0 до 4 М (0—25°) и от 0 до 2 М (30—60°)
Коэффици- енты урав- нения t,c
0 5 10 15 20 25 30 35 40 45 50 55 60
&(/) 0,487 0,490 0,494 0,498 0,502 0,506 0,512 0,517 0,522 0,528 0,534 0,541 0,547
А’ 1,395 1,398 1,402 1,405 1,409 1,414 1,418 1,423 1,428 1,433 1,439 1,445 1,450
В 0,1390 0,1371 0,1351 0,1332 0,1312 0,1292 0,1272 0,1253 0,1233 0,1214 0,1194 0,1175 0,1155
D' 0,0070 0,0068 0,0067 0,0065 0,0063 0,0061 0,0060 0,0058 0,0057 0,0055 0,0054 0,0052 0,0050
«1 0,9998 1,0000 0,9995 0,9990 0,9982 0,9972 0,9958 0,9941 0,9922 0,9901 0,9879 0,9855 0,9832
&!- 10s 1,707 1,742 1,760 1,782 1,805 1,817 1,822 1,825 1,825 1,815 1,815 1,805 1,805
Значения члена Доп.
С тп 0 25 , 60 t, с т 0 25 60
0,001 -0,00009 . -0,00010 -0,00011 0,2 -0,00067 -0,00072 -0,00085
0,002 -0,00015 -0,00018 -0,00019 0,5 -0,00045 -0,00049 • -0,-00057
0,005 -0,00027 -0,00030 -0,00036 1,0 -0,00027 -0,00028 -0,00032
0,01 -0,00041 -0,00045 -0,00053 1,5 -0,00017 -0,00019 -0,00020
0,02 — 0,00055 -0,00061 -0,00072 2,0 -0,00012 -0,00013 -0,00015
0,05 -0,00071 -0,00077 -0,00091 3,0 -0,00007 . 0,00007 -0,00008
0,1 —0,00075 — 0,00080 — 0,00094 4,0 — 0,00004 -0,00005 -0,00006
326
ГЛАВА XI
менения уравнения (22). Совпадение вычисленных и экспериментальных
значений у±, приведенных в табл. 71, является превосходным, так как,
за немногими исключениями, максимальное расхождение между этими зна-
чениями не превышает ± 0,001 в единицах коэффициента активности, что
соответствует ошибке в ± 0,05 мв. В случае низких температур (0 и 5°
при концентрациях 0,01—0,1 М) совпадение вычисленных и опытных
значений несколько хуже, чем для более высоких температур. Прини-
мая во внимание хорошее совпадение этих экспериментальных значений
с результатами, полученными из данных о температурах замерзания растворов,
нельзя объяснить это расхождение между вычисленными и опытными зна-
чениями у± при более низких температурах только за счет эксперимен-
тальных ошибок.
На основании общего анализа уравнения (22) можно описать поведе-
ние сильно диссоциированных электролитов. Для водных растворов 1,1-элек-
Р и с. 81. Влияние величины различных
членов уравнения (21) при 25°(а = 4).
галоидных соединений)
тролитов составляющая, обусловлен-
ная дополнительными членами, весь-
ма мала и, как правило, не превы-
шает одной единицы в третьем деся-
тичном знаке коэффициента актив-
ности, поэтому в данном случае нет
необходимости принимать ее во вни-
мание. Для типичного полностью
диссоциированного 1,1-электролита
величина среднего расстояния сбли-
жения ионов а должна быть близка
к 4. Если принять для ряда элек-
тролитов значение а = 4, то пер-
вый член в правой части уравнения
(22) будет одинаков для всех элек-
тролитов. С другой стороны, значе-
ние В может меняться в широ-
ких пределах (от 0,01 до 0,2 для
и может характеризовать специфические особен-
ности электролитов в более концентрированных растворах. Влияние величины
В хорошо иллюстрируется рис. 81, на котором изображены кривые для
1gу±, отвечающие предельному закону [уравнение (20) гл. III], первому
члену правой части уравнения (22), а также сумме этого члена и ли-
нейного члена Вс. Как видно из рисунка, чем больше В, тем более
отчетливым является минимум, характерный для многих сильных элек-
тролитов.
Представляет интерес оценить порядок величины отдельных членов
уравнения (22) для случая реального электролита, например для соляной
кислоты. Для очень разбавленных растворов члены, содержащие с в пер-
вой и более высоких степенях, очень малы по сравнению с членом Дебая
и Гюккеля, однако их влияние проявляется даже при концентрациях 0,001 М.
Так, например, при концентрации в 0,002 М и при 0° первый, второй и
сумма остальных членов равны соответственно —0,02050; —0,00015 и
+ 0,00028. Таким образом, члены Вс+... вдвое превышают величину
члена Доп. При концентрации ОДМ эти члены равны соответственно
— 0,10680; —0,00075 и +0,01394, в то время как при концентрации
4 + получаются значения —0,25485; —0,00004 и +0,6153. Таким обра-
зом, при концентрации 0,1 М линейный член относительно с примерно
в 15 раз превышает величину составляющей, обусловленной дополнитель-
ными членами.
СОЛЯНАЯ КИСЛОТА
327
V7n
Рис. 82. Средний коэффициент активности
соляной кислоты для смесей диоксан—вода.
X—весовой процент дионсана. Кружнами обозначены
результаты для 25°.
§ 6. Коэффициент активности соляной кислоты в средах,
содержащих органические растворители
Характер зависимости коэффициента активности от концентрации
кислоты, температуры и диэлектрической постоянной (или от состава рас-
творителя) можно хорошо проиллюстрировать на примере обширных иссле-
дований смесей диоксан—вода (см. данные табл. 129). На рис. 82 пред-
ставлена зависимость всех приведенных в этой таблице значений 1g
при 25° и некоторых значений при 0 и 50° от т для смесей, содержа-
щих 0, 20, 45, 70 и 82% дио-
ксана. Прямые линии соответ-
ствуют предельному закону тео-
рии междуионного притяжения
при 25°. Значения предельных
коэффициентов наклона
приведены в табл. 125.
Наиболее важный вывод,
который можно сделать на осно-
вании рассмотрения этих ре-
зультатов, состоит в том, что
в случае разбавленных рас-
творов наблюдаемые величины
в основном совпадают с теоре-
тическими, Предельный коэф-
фициент наклона S для
растворов, содержащих 82% дио-
ксана (Z) = 9,53; §(/) = 11,98),
в 23 раза больше, чем для рас-
творов в чистой воде (Z) = 78,54;
= 0,506), и следовательно,
член с, обусловленный
влиянием кулоновских сил, в
первом случае очень велик.
Таким образом, согласно теории, при изменении концентрации кислоты от 0
до 0,1 н., у± меняется в 10 раз сильнее в случае смеси с 82% диокса-
на, чем в случае чистой воды. Если не происходит ассоциации ионов,
то получающиеся графики лежат выше прямых с предельными коэф-
фициентами наклона, что согласуется с теорией, учитывающей среднее
расстояние сближения ионов. В случае сред, содержащих 0, 20 и 45%
диоксана, экспериментальные результаты подчиняются этому требованию
теории, и кривые для всех концентраций лежат выше прямых с предельным
наклоном. По мере уменьшения диэлектрической постоянной среды графики
в области разбавленных растворов приближаются к теоретическим прямым,
и, наконец, как видно на примере растворов, содержащих 70% диоксана,
при концентрациях кислоты ниже 0,002 М экспериментальная кривая
совпадает с теоретической. В случае смесей, содержащих 82% диоксана,
кривая lg y± может лежать даже ниже прямой, отвечающей предельному
коэффициенту наклона, о чем свидетельствуют данные для растнорон с кон-,
центрациями кислоты 0,001 и 0,0015 М. Это явление аналогично тому,
которое наблюдается в случае зависимости электропроводности от
(рис. 74). При всех концентрациях и температура!х коэффициенты активности
уменьшаются с увеличением температуры и, следовательно, относитель-
ное парциальное молярное теплосодержание L2 всегда положительно.
В области более концентрированных растворов на всех кривых, пред-
ставленных на рис. 82, имеется минимум. Это показывает, что суммарный
328
ГЛАВА XI
эффект ослабления кулоновских сил вследствие изменения размеров ионов
.и роста концентрации раствора, вызывающего увеличение активности кис-
лоты, очень значителен для смесей с 82% диоксана. Так, например, со-
гласно предельному закону, при концентрации 0,5 М 1g у± должен рав-
няться — 8,569, в то время как опытное значение равно —1,2173. Если
принять а =6, то lgY±=—1,697, т. е. при этом расхождение между
теоретическими и наблюдаемыми величинами значительно сокращается.
Коэффициент активности соляной кислоты был определен для некото-
рых водных смесей метилового, этилового и изопропилового спиртов и
глицерина с водой, а также для растворов в чистых метиловом и этило-
вом спиртах (табл. 130). Характер зависимости коэффициента активности
от концентрации для указанных сред не отличается какими-либо особенно-
стями и аналогичен тому, который наблюдается в случае водных растворов.
В области концентраций от 0 до 1 М коэффициенты активности можно
вычислять по уравнению
lgт± = -1 ^(^С- + B'm-lg(l + 0,002 МХут). (25)
В случае растворов с высокой диэлектрической постоянной (Z) > 60) точность
определения у± составляет ±0,002, если принять а = 4,3. В случае вод-
ных смесей, содержащих 10 и 20% метилового спирта, 10 и 20% этилового
спирта и 10% изопропилового спирта, значения параметра В', необходимые
для вычислений, равны соответственно 0,1315; 0,1293; 0,1345; 0,1376 и
0,1373. Для растворов в чистых метиловом и этиловом спиртах необходимо
принимать другие значения параметров а и В' [34, 20].
§ 7. Относительное пардиальное молярное теплосодержание
соляной кислоты. Численные методы определения Ь2
из данных по электродвижущим силам
Результаты определения различных термодинамических величин из
данных по электродвижущим силам плохо воспроизводимы и очень чувст-
вительны к незначительным изменениям в условиях опыта. Так, если для
1,1-электролита температурный коэффициент электродвижущей силы най-
ден с точностью ± 0,001 Mejzpad, то ошибка в определении Ь2 составляет
± 7 кал, однако даже из наилучших экспериментальных данных не удает-
ся получать столь точных результатов. Если удается довести точность
определения Ь2 до ±30 кал, то этот результат можно рассматривать как
вполне удовлетворительный. Поскольку относительная точность наилучших
калориметрических определений Ь.2 составляет ±10 кал, то их следует
считать более точными, чем значения, получаемые из данвых по электро-
движущим силам.
При обычном способе определения Ь2 из данных по электродвижущим
силам необходимо большое число экспериментальных точек при различных
температурах. Полученные результаты изображают графически как функ-
ции от концентрации в виде плавной кривой и находят значения электро-
движущей силы для некоторых целочисленных значений концентраций.
Константы соответствующим образом подобранной функции, например типа
уравнения (45) гл. X, находят из этих значений для каждой концентрации
с помощью метода наименьших квадратов или графически с использованием
разностей первого порядка [35]. В случае соляной кислоты Харнед и
Элерс, использовав уравнение (45) гл. X, вычисляли L2 по уравнению (48)
СОЛЯНАЯ КИСЛОТА
329
гл. X. Эти же авторы определяли В2, дифференцируя уравнение Дебая
и Гюккеля (22) аналогично тому, как это делалось при выводе уравнения
(77) гл. III. При этом получается следующее уравнение:
Т = _ 2,3481 • Г дА А дс_ 'I
2— 1+А /2? (1+Л/2^)2Р3/2 1С 2 дТ}
-4,6052.7?^ [(В + 2^С)^ + С(^ + с5г)] . (26)
Табл. 73 содержит значения L2 для 10, 25, 40 и 60°, полученные
двумя методами. В табл. 74 приведены уравнение и параметры, с помощью
которых можно вычислять L2 для всех температур от 0 до 60°. С помощью
значений, полученных первым методом, была построена плавная кривая,
выражающая зависимость этих значений от.т1/2. Для всех температур
от 10 до 60° расхождение между результатами, полученными с помощью
двух методов, не превышает 40 кал, при 0° это расхождение несколько
больше. Второй метод вычисления £2 более сложен, но дает более надежные
Таблица 73
Относительное парциальное молярное теплосодержание
соляной кислоты. (1)—вычислено по уравнению (48) гл. X;
(2)—вычислено по уравнению (26)
t> °C
10 25 40 60
m
0,001 (1) 16 22 27 39
0,005(1) 18 37 60 90
(2) 34 45 59 79
0,01 (1) 19 46 75 117
(2) 48 63 81 108
0,02 (1) ' 27 64 101 155
(2) 64 85 109 145
0,05 (1) 74 120- 169 238
(2) 99 130 165 219
0,1 (1) 101 160 221 300
(2) 135 175 222 295
0,2 (1) 172 238 307 414
(2) 189 242 306 395
0,5 (1) 319 402 488 614
(2) 319 391 483 ’ 608
1,0 (1) 502 626 748 944
(2) 503 611 728 900
1,5 (1) 676 840 1000 1230
(2) 698 831 975 1184
2,0 (1) 866 1040 1236 1506
(2) 900 1059 1230 1470
3,0 (1) 1264 1510 — ——
(2) 1302 1507 —— —
4,0 (1) 1696 1982 —
(2) 1735 1991 — —
330
ГЛАВА XI
Таблица 741)
Константы уравнения La = Бь (0°) + at + ₽f2.
Применимы в интервале температур от 0 до 60° 2)
m Ь2<0 ) О.
0,005 28 0,70 0,003
0,01 39 1,00 0,003
0,02 52 1,30 0,004
0,05 82 1,85 0,006
0,1 ИЗ 2,50 0,008
0,2 159 3,20 0,009
0,5 272 4,70 0,011
1,0 427 6,80 0,015
1,5 615 8,20 0,019
2,0 791 10,00 ► 0,023
3,0 1175 12,45 0,031
4,0 1604 14,70 0,040
1) Анерлёф и Тир [1] вычислили L-z в интервале тем-
ператур от 0 до 50° и для концентраций от 3 до 16 М из
данных по электродвижущим силам.
2) Значения L.2, полученные из данных по электро-
движущим силам, хорошо совпадают с последними кало-
риметрическими результатами Стуртеванта [2]. Максималь-
ное отклонение составляет 31 кал при концентрации 3 М.
результаты. Член dcjdT нетрудно найти из данных по плотностям
растворов; члены dB/дТ и дБ'/дТ известны из уравнений (23) и (24); как
было показано выше, можно.считать, что а не зависит от температуры.
Представляет интерес оценить, каков удельный вес первого, второго
и последнего членов в уравнении (26), определяющем L2. Первый член,
которому на рис. 83 соответствует кривая 1, всегда положителен и со-
ставляет большую часть L2 в разбавленных растворах. Второй член
в случае растворов соляной кислоты отрицателен, так как сдА/дТ> АдсрлБТ.
Сумме первого и второго членов отвечает на рисунке кривая 7 + 2. Так
как в случае растворов соляной кислоты члены dB/дТ и дБ’/дТ отрица-
тельны, то третий член положителен и составляет большую часть
Т2 в концентрированных растворах. Кривая 7 + 2 + 3 представляет всю
правую часть уравнения (26).
Дальнейший анализ зависимости параметров В и Б' от температуры
весьма полезен для выяснения общей закономерности поведения L2 при
изменении концентрации для различных электролитов в концентрирован-
ных растворах. В случае растворов соляной кислоты члены dB/дТ и дБ'/дТ
отрицательны, а третий член уравнения (26) положителен. В некоторых
случаях эти коэффициенты положительны, и тогда третий член уравнения
(26) отрицателен. На рис. 83 кривая 7 + 2 — 3 соответствует электролиту,
для которого коэффициенты dB/дТ и дА'/дТ равны по величине, но про-
тивоположны по знаку коэффициентам для соляной кислоты, причем для
этого электролита а — 4,3. Кривые 7 + 2 + 3 и 7 + 2 — 3 отвечают двум
предельным случаям, так как для большинства 1,1-электролитов значе-
ния L2 лежат между этими кривыми. В качестве примера на рисунке
приведен график зависимости Ь2 от ]/ с для водного раствора гидрата
окиси натрия. Так лак в этом случае величина а приблизительно равна
СОЛЯНАЯ КИСЛОТА
331
3,5, то в области разбавленных растворов эта кривая расположена несколь-
ко выше, чем соответствующая кривая для разбавленного раствора соляной
кислоты. Кривая для NaOH проходит через максимум при т1/2 = 0,4, и
это свидетельствует о том, что коэффициент дВ/дТ положителен. Затем
эта кривая проходит через минимум при т1/2 = 1,2, что указывает на
\Гтп
Рис. 84. Относительное парциальное молярное тепло-
содержание соляной кислоты в смесях диоксан—вода
при 25°.
Прямые линии отвечают предельному закону. X—весовой
процент дионсана.
отрицательную величину члена dD'/дТ. Таков общий характер зависи-
мости от концентрации для водных растворов 1,1-электролитов.
Характер зависимости относительного парциального молярного теп-
лосодержания от диэлектрической постоянной иллюстрируют кривые,
приведенные на рис. 84. На этом рисунке представлена зависимость Т2 от
Утп для смесей диоксан —вода при 25°. Прямые линии, проведенные через
332
ГЛАВА XI
начало координат, отвечают теоретическому предельному уравнению
L2 = 5(H) У"с . Значения 5(н), вычисленные по уравнению (78) гл. III,
приведены в табл. 131. Из рисунка видно, что в случае разбавленных
растворов экспериментальные кривые располагаются тем выше теоретиче-
ских, чем больше содержание диоксана в смеси. Все кривые имеют одина-
ковый характер. После быстрого подъема в области разбавленных растворов
наклоны кривых в дальнейшем уменьшаются, однако затем начинают
снова возрастать в области более концентрированных растворов.
Величина ошибки, связанной главным образом с трудностями, которые
встречаются при экстраполяции, возрастает с увеличением концентрации
диоксана. Для смесей, содержащих 82% диоксана, ошибка в определении
L2 может достигать нескольких сотен калорий. Если относить ошибку
при данной концентрации к величине соответствующей концентрации, то
точность определения L2 является более высокой, достигая величины по-
рядка 100 кал. Уравнения для вычисления L2 для этих смесей приведены
в табл. 132.
§ 8. Относительная парциальная молярная теплоемкость
соляной кислоты
Относительная парциальная молярная теплоемкость определяется урав-
нением (50) гл. X:
J2 = 46120 (с-с0) Т, (27)
где с и с0 —константы уравнений (45) и (46) гл. X. Так как для опреде-
ления этой величины необходимы коэффициенты, которые получаются
путем двукратного дифференцирования исходных данных по электродви-
жущим силам, то этот метод не может давать очень точных результатов.
Таблица 75
Относительная парциальная молярная теплоемкость соляной
и бромистоводородной кислот
m (с - Со). 10е Ja (25°)
НС1 НВг НС1 1) НС1 3) НС1 2)
0,001 0,03 0,04 0,3 0,3
0,005 0,08 0,10 0,9 — 1,1
0,01 0,11 0,15 1,2 — 1,5
0,02 0,15 0,20 1,5 — 2,1
0,05 0,22 0,26 2,1 — 3,1
0,1 0,28 • 0,31 2,9 2,4 3,8
0,2 0,34 0,35 3,7 3,4 4,8
0,5 0,43 — 5,3 5,3 6,0
1,0 0,58 — 7,5 7,5 8,1
1,5 0,74 — 9,1 9,2 10,1
2,0 0,87 — 11,2 10,6 12,0
3,0 1,14 — • 14,1 13,0 15,8
4,0 1,4 —. 16,7 15,0 19,1
1) Уравнение (26).
2) Уравнение (27).
з) Калориметрические данные. Гукер и Шминке [1].
СОЛЯНАЯ КИСЛОТА
333
С другой стороны, интересно сравнить полученные этим методом значения
с результатами, найденными из калориметрических данных. 13 табл. 75
приведены значения (с — с0), из которых можно вычислить J2 для интер-
вала температур 0 — 60°. Харнед и Элерс получили J2 из значений L2,
вычисленных по уравнению (26). Табл. 75 содержит результаты, полу-
ченные при 25°. _
Гукер и Шминке [36] определили J2 для концентрированных растворов
при 25° с помощью калориметрических измерений. Их данные могут быть
выражены уравнением J2(25°) = 7,5 wi1-2; значения, вычисленные по этому
уравнению, также включены в таблицу. Совпадение этих значений с вели-
чинами, полученными из данных по измерениям электродвижущих сил,
является очень хорошим, несмотря на
те трудности, которые встречаются при
определении этих величин.
Зависимость J2 (при 25°) от )/'т
для водных растворов и для смесей
диоксан —вода изображена на рис. 85.
Прямые линии отвечают предельному
уравнению (17) гл. V для относи-
тельной парциальной молярной тепло-
емкости J2(25°)= Scpl^c, выве-
денному на основании теории между-
ионного притяжения. Опытные и тео-
ретические значения J2 для данной кон-
центрации кислоты отличаются всего
лишь на несколько калорий (± 2 кал).
В случае смесей, содержащих 82%
диоксана, точность данных, получаю-
щихся путем экстраполяции (пунктир-
ная линия), невелика; ошибка может
достигать значительной величины
(~ 10 кал). Эти результаты хорошо
иллюстрируют быстрое возрастание
величины J2 в средах с низкой ди-
электрической постоянной.
§ 9. Числа переноса соляной кислоты
в воде и в смесях диоксан —вода при
температурах 0 — 50°
В гл. VI, § 6, в связи с предель-
ным уравнением теории междуионного
притяжения было рассмотрено опре-
Р и с. 85. Относительная парциальная
молярная теплоемкость соляной кислоты
в смесях диоксан—вода при 25°.
Прямые линии отвечают предельному зако-
ну. X—весовой процент диоксана.
деление чисел переноса некоторых
электролитов методом движущейся границы. В гл. X, § 6, было показано,
что число переноса электролита можно определить из данных по измерению
электродвижущих сил элементов с жидкостным соединением или без
жидкостного соединения по уравнению (64) гл. X. Этот метод был при-
менен Харнедом и Дреби [37] в работе по определению числа переноса
катиона хлористоводородной кислоты в водном растворе и в смесях диоксан —
вода при температурах 0—50°. Полученные результаты представляют собой
.исчерпывающие сведения о зависимости чисел переноса электролита от
концентрации, температуры и состава растворителя.
334
ГЛАВА XI
Опытные данные были получены путем измерения электродвижущих
сил Ет и Й элементов
Ag-AgGl | HCI (m), диоксан (X), Н2О (У) | НС1 (щд), диоксан (X),
Н2О(У) | AgCl-Ag
и
Ag-AgCl | HCI (m), диоксан (X), Н2О (К) | Н21 HCI (mR), диоксан (X),
Н2О (У) | AgCl-Ag
с жидкостным соединением и без него. Электродвижущие силы элементов
с жидкостным соединением измерялись Харнедом и Дреби непосредственно,
а электродвижущие силы элементов без жидкостного соединения эти авто-
ры вычисляли из данных, полученных путем измерения электродвижущих
сил элементов
Н21 HCI (т), диоксан (X), Н2О (У) | AgCl-Ag,
описанных в § 3. Через HCI (mR) обозначена постоянная стандартная кон-
центрация.
Согласно уравнению (64) гл. X, число переноса катиона Т+ равно
ТЧ^сЖт/с/Е. (28)
При любой заданной концентрации раствора Т+ представляет собой коэф-
фициент наклона кривой, получаемой при графическом изображении зави-
симости электродвижущей силы Ет элемента с жидкостным соединением
от электродвижущей силы Е элемента без жидкостного соединения. Хорошо
известно, что этот метод, который связан с определением коэффициента
наклона, по своей точности уступает методу движущейся границы, однако
он является надежным и простым и поэтому удобным для обширных
исследований.
На практике коэффициент наклона кривой для зависимости Е от Ег
определяют путем выражения этих величин с помощью эмпирических
функций с последующим дифференцированием [38] или чисто графическим
методом. Гамер [39] и Харнед и Дреби [37] нашли, что описанный ниже
графический метод заслуживает предпочтения перед всеми остальными
испробованными ими графическими и аналитическими методами.
• Так как величины Ет и Е меняются в широких пределах, то при не-
посредственном графическом изображении их зависимости утрачиваются
значащие цифры, необходимые для точного определения коэффициента
наклона. Харнед и Дреби применили метод, посредством которого значе-
ния Е определялись для равных интервалов изменения Ет без потери
значащих цифр. Кроме ^ого, при этом методе можно определять наклон
аналитическим путем и, следовательно, получать более точные результаты.
Функции отклонения, определяемые соотношениями
X = F.~r2klgm/ mR, XT^5ET-[-2klgm/mR, • (29)
дают значения Хт и X, соответствующие величинам Еу и Е. Значения Хт
и X изменяются в очень малых пределах, так что их можно очень точно
представить графически без потери значащих цифр. Харнед и Дреби опреде-
ляли путем графической интерполяции значения Хт при тех концентрациях,
для которых были известны величины X. Затем на основании этих данных
значение разности X — Ху они изображали графически в виде функции от X.
Как видно из уравнений (29), разность X —Хт равна разности Е —Ет.
Из полученного графика находили значения X — Хт для величин Хт, соот-
ветствующих значениям Ет, выбранным таким образом, что интервалы
между ними равнялись 0,(Д в. Эти значения.Ет находили из графика зави-
симости Ет от Хт- Суммируя соответствующие значения X — Хт и Ет,.
получали искомые значения Е, отвечающие последовательности величин Е^
СОЛЯНАЯ КИСЛОТА
335
с интервалами между отдельными величинами в 0,01 в. Полученные таким
образом данные применялись для аналитического определения коэффици-
ента наклона кривых с помощью специальной функции, предложенной
Рутледжом [40]. Эта функция имеет вид
— / (Е) = ^2^ (С'_2Е_2 + C_iE_i С0Е0 + С2Е2), (30)
где б/Е/б/Еу — коэффициент наклона при значении зависимой переменной,
равном Е; Л —величина выбранного интервала изменения независимой пере-
менной; Е 2, Е_х и т. д.—последовательные значения Е; С_2, и т. д.—
постоянные. Имеются 3 группы констант, причем выбор той или иной
из них зависит от того, при каком значении Е определяется наклон
Величины суммарных ошибок равны диаметрам кружков, X—ве-
совой процент дионсана. Касательные, проведенные н начальной
части каждой кривой, отвечают теоретическому предельному
закону.
(Е_х, Ео или Ех). За исключением крайних значений Е, коэффициенты
наклона определялись как среднее из результатов, получаемых по формуле
Рутледжа, взятой для трех возможных значений Е (E_x, Ео п Ej).
Полученные таким образом величины Т+ изображались графически
в виде функции от Ег и из этого графика получались сглаженные вели-
чины 7’+ при определенных концентрациях кислоты. Из этих данных опре-
делялось предельное значение числа переноса по методу Лонгсворта [41],
описанному в гл. VI, § 7. Этот метод состоит в графическом изображении
функции
?п, _Г+А' + 1/2^*у7
+ Д' + 5* /с
где
Д'= А0 — (а*А° + Р*) ]/с (32)
(31)
336
ГЛАВА XI
Рис. 87. Число
кислоты в смесях
переноса катиона соляной
диоксан—вода при 15, 25
и 35°.
Прямые линии отвечают теоретическому предель-
ному закону. X—весовой процент диоксапа.
в зависимости от концентрации кислоты с и экстраполяции этой функции
до нулевой концентрации. Полученные Харнедом и Дреби графики пред-
ставляли собой прямые линии при концентрациях 0,01— 0,1 н.1 и были
вполне пригодны для экстраполяции. Значения Т+ при концентрациях
меньше 0,01 н. были менее надежны, так как определение величины коэф-
фициента наклона кривой при
очень малых концентрациях за-
труднительно.
Как показывает анализ функ-
ции Рутледжа, ошибка в 0,01 мв
в определении значений Е или Ет
вызывает ошибку в 0,001 в зна-
чении Т+. Из величин средних
отклонений наблюдаемых электро-
движущих сил или из средних от-
клонений опытных значений от
плавных кривых можно вычислить
среднюю ошибку в определении Т+.
На основании рис. 86 можно со-
ставить представление о величи-
нах ошибок. На этом рисунке
представлены кривые для чисел
переноса катиона соляной кислоты
при 25° в чистой воде и в смесях
диоксан—вода, содержащих 20, 45
и 70% диоксана. Суммарные ошиб-
ки в определении чисел переноса
соответствуют диаметрам кружков.
Для воды и для трех смесей ди-
оксан—вода эти ошибки равны соот-
ветственно 0,002, 0,005, 0,004 и
0,008.
Табл. 133 содержит оконча-
тельные сглаженные значения Т+
и Т+ для указанных растворите-
лей при температурах 0 — 50° че-
рез каждые 5°. Графики этих зна- '
чений для 15, 25 и 35° приведены
на рис. 87. Прямые линии соответ-
ствуют числам переноса, вычислен-
ным с помощью предельного уравнения Онзагера
П = S(T) Vс = /т . (33)
Значения <э(т) приведены в нижней части табл. 133. Полученные резуль-
таты согласуются с теорией, поскольку по мере уменьшения концентрации
электролита все кривые приближаются к теоретическим прямым.
Как видно из рис. 86, наилучшие результаты были получены для рас-
творов кислоты в чистой воде. Согласно приведенным в табл. 76 данным,
ошибка при определении Т+ не превышает одну или две единицы в третьем
десятичном знаке. В этой таблице значения числа переноса, полученные
1 В случае раствора с 20% диоксана этот график является прямолинейным в интер-
вале концентраций от 0,03 до 0,1 н.; при концентрациях ниже 0,03 н. получаются оши-
бочные результаты.
СОЛЯНАЯ КИСЛОТА
337
Лонгсвортом с помощью более точного метода движущейся границы, сопо-
ставлены с величинами, полученными Харнедом и Дреби из данных по
электродвижущим силам. В этом наиболее благоприятном случае совпадение
является вполне хорошим и отклонения по порядку величины не превыша-
ют величины средней ошибки исходных данных по электродвижущим силам-
Харнед и Дреби измерили, кроме того, электродвижущие силы эле-
ментов с жидкостным соединением для смесей диоксан—вода, содержащих
82% диоксана. При этом были получены менее точнее результаты, чем для
смесей с более низким содержанием диоксана, особенно в тех случаях,
когда концентрация кислоты была меньше 0,05 М. Поскольку других дан-
ных по числам переноса в средах со столь низкими диэлектрическими
постоянными {D (25°) = 9,57) не имеется, то эти результаты представляют
известную ценность и вклйчены в табл. 133.
Таблица 76
Значения Тпри 25°, найденные из данных пн электродвижущим силам
и методом движущейся границы
c Метод 0,005 0,01 0,02 0,05 0 ,100
Движ. границы ...... Э. д. с 0,8239 (0,824) 0,8251 (0,825) 0,8266 (0,827) 0,8292 (0,830) 0/8314 (0,830)
ЛИТЕРАТУРА
1. В. В. О w е n, G. W. Waters, J. Am. Chem. Soc., 60, 2371 (1938).
2. G. A k e r 1 6 f, 0. A. Short, J. Am. Chem. Soc., 58, 1241 (1936).
3. R. M. F u о s s, C. A. Kraus, J. Am. Chem. Soc., 55, 476 (1933).
4. R. M. F u о s s, J. Am. Chem. Soc., 57, 488 (1935).
5. G. A. Linhart, J. Am. Chem. Soc., 41, 1175 (1919).
6. W. R. C ar m od y, J. Am. Chem. Soc., 51, 2905 (1929).
7. G. S c a t c h a r d, J. Am. Chem. Soc., 47, 641 (1925).
8. A. A. N о у e s, J. H. Ellis, J. Am. Chem. Soc., 39, 2532 (1917);
9. E. J. Roberts, J. Am. Chem. Soc., 52, 3877 (1930).
10. H. S. Harned, R. W. Ehlers, J. Am. Chem. Soc., 54, 1350 (1932).
11. a) E. Giintelberg, Z. physik. Chem., 123, 199 (1926); б) H. S. H a r n e d,
J. Am. Chem. Soc., 51, 416 (1929); в) D. A. M a с I n n e s, J. A. Beattie,
там же, 42, 1117 (1920); г) A. S. В г о w n, там же, 56, 646 (1934).
12. S. S. Р г е n t i s s, G. S c a t c h a r d, Chem. Rev., 13, 139 (1933).
13. H. S. Harned, D.D. Wright, J. Am. Chem. Soc., 55, 4849 (1933).
14. R. T. Birge, Phys. Rev. Supplement, 1, 1 (1929).
15. H. S. Harn ed, H. C. Thomas, J. Am. Chem. Soc., 57, 1666 (1935); 58, 761
(1936).
16. G. N о n h e b e 1, H. Hartley, Phil. Mag. (6), 50, 729 (1925).
17. J. W. W о о 1 с о c k, H. Hartley, Phil. Mag. (7), 5, 1133 (1928).
18. P. S. Danner, J. Am.' Chem. Soc., 44, 2832 (1922).
19. H. S. Harned, M. H. F 1 e у s h e r, J. Am. Chem. Soc., 47, 82 (1925).
20. W. W. L u c a s s e, Z. physik. Chem., 121, 254 (1926).
21. G. S c a t c h a rd, J. Am. Chem. Soc., 48, 2026 (1926).
22. a) H. S. Harned, J. 0. Morrison, Am. J. Sci., 33, 161 (1937); J. Am. Chem.
Soc., 58, 1908 (1936); б) H. S. H a r n e d, С. С a 1 m о n, там же, 60, 334 (1938);
в) H. S. H a r n e d, там же, 60, 336 (1938); г) H. S. Harned, J. G. D о n el-
son, там же, 60, 339 (1938); 60, 2128 (1938); д) H. S. Harned, С. С a 1 m о n,
60, 2130 (1938); e) H. S. Harned, J. G. D о nelso n, С. С a 1 m о n, там же,
60,2133 (1938); ж) H. S. Harned, F. Walker, C. Calm о n, там же, 61,
44 (1939); з) H. S. H a r n e d, F. Walker, там же, 61, 48 (1939); и) H. S. Har-
ned, I. О. Morrison, F. Walker, J. G. Donelson, C. Calmon
61, 49 (1938).
22 Г. Харнед, Б. Оуэн
338
ГЛАВА XI
23. G. А к er 1 of, О. A. Short, J. Am. Chem. Soc., 58, 1241 (1936).
24. F. Hovorka, R. A. Schaefer, D. Dreisbach, J. Am. Chem. Soc., 58,
2264 (1936).
25. T. S h e d 1 о v s к y, D. A. Maclnnes, J. Am. Chem. Soc., 58, 1970 (1936).
26. a) H. S. H a r n e d, R. W. E hl e r s, J. Am. Chem. Soc., 55, 652 (1933), table IV;
б) там же, 55, 2179 (1933).
27. E. Giin t elber g, Z. physik. Chem., 123, 199 (1926).
28. M. Randall, L. E. Young, J. Am. Chem. Soc., 50, 989 (1928).
29. G. A к er 1 of, J. W. T e a r e, J. Am. Chem. Soc., 59, 1855 (1937).
30. M. Randall, А. Ц. Vanselow, J. Am. Chem. Soc., 46, 2418 (1924).
31. E. H u с к e 1, Physik. Z., 26, 93 (1925).
32. H. S. Hamed, J. Am. Chem. Soc.,'48, 326 (1926).
33. H. S. Hamed, G. А к e r 1 о f, Physik. Z., 27, 411 (1926).
34. G. S c a t c h a r d, J. Am. Chem. Soc., 47, 2098 (1925).
35. H. S. H a r n e d, L. F. Nim s, J. Am. Chem. Soc., 54, 423 (1932).
36. F. T. G u с к e r, Jr., К. H. Schminke, J. Am. Chem. Soc., 54, 1358 (1932).
37. H. S. H a r n e d, E. C. D r e b y, J. Am. Chem. Soc., 61, 3113 (1939).
38. D. A. Maclnnes, J. A. Beattie, J. Am. Chem. Soc., 42, 1117 (1920).
39. W. J. H a m e r, J. Am. Chem. Soc., 57, 662 (1935).
40. G. Rutledge, J. Math. Phys. M. I. T., 8, 1 (1929); Phys. Rev., 40, 262 (1932).
41. L. G. Longs wo r th, J. Am. Chem. Soc., 54, 2741 (1932).
ЛИТЕРАТУРА К ТАБЛИЦАМ
R табл. 67
1. W. R. С a r m о d y, J. Am. Chem. Soc., 51, 2905 (1929).
2. H. M. Spencer, J. Am. Chem. Soc., 54, 3647 (1933). "
3. G. S c a t c h a r d, J. Am. Chem. Soc., 47, 641 (1925).
4. D. Hitchcock, J. Am. Chem. Soc., 50, 2076 (1928).
5. E, J. Roberts, J. Am. Chem. Soc., 52, 3877 (1930).
6. H. S. H a r n e d, R. W. Ehlers, J. Am. Chem. Soc., 54, 1350 (1932).
7. S. S. Prentise, G. Scatchard, Chem. Rev., 13, 139 (1933).
8. D. A. Maclnnes, The Principles of Electrochemistry, Reinhold Publishing Corp.,
N. Y., 1939, p. 187.
К табл. 69
1. H. S. H a r n e d, С. С a 1 m о n, J. Am. Chem. Soc., 60, 2130 (1938).
К табл. 71
1. T. S h e d 1 о v s к у, D. A. Maclnnes, J. Am. Chem. Soc., 58, 1970 (1936).
R табл. 74
1. G. A к e r 1 6 f, J. W. T e a r e, J. Am. Chem. Soc., 59, 1855 (1937).
2. J. M. ,S turte van t, J. Am. Chem. Soc., 62, 584 (1940).
R табл. 75
1. F. T. G u с к e r, Jr., К. H. S c h m i n к e, J. Am. Chem; Soc., 54, 1358 (1932).
Глава XII1
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
В гл. IX были описаны наиболее существенные работы, посвященные
определению понижения точек замерзания растворов 1,1-электролитов,
а в гл. VIII были рассмотрены наиболее важные исследования по опре-
делению различных термохимических свойств этих растворов. Данная глава
посвящена более широкому обсуждению результатов, полученных путем
определения термодинамических свойств этих растворов.
Важность систематического изучения коэффициентов активности элек-
тролитов была впервые отмечена Льюисом [1]. Применяя для экстраполя-
ции эмпирическую функцию, содержащую две постоянные, Льюис и Лин-
гарт [2], а также Льюис и Рендалл [3] сделали первую попытку вычислить
коэффициенты активности из имевшихся опытных данных. Аналогичная
попытка была предпринята Харнедом [4, 9а], который воспользовался для вы-
числения у эмпирическим уравнением с тремя эмпирическими постоян-
ными. Эти исследования были сделаны до того, как стал известен предель-
ный закон теории Дебая и Гюккеля; при экстраполяции были допущены
большие ошибки и коэффициенты активности были вычислены с точностью,
не превышающей ±0,01.
После того как стал применяться предельный закон теории Дебая
и Гюккеля, были быстро достигнуты существенные успехи в смысле повы-
шения точности экстраполяции. Гюккель [5], Скэтчард [6], Харнед [7],
Рендалл и Юнг [8], применяя предельный закон и обобщенные формы
уравнений теории Дебая и Гюккеля, смогли значительно увеличить точность
экстраполяции. Общий обзор полученных до 1929 г. результатов был еде-,
лан Харнедом [96].
Ниже будет приведен обзор всех известных экспериментальных данных,
касающихся растворов 1,1-электролитов. Далее будут изложены термо-
химические данные с точки зрения их связи с коэффициентами активности..
В первую очередь мы остановимся на свойствах растворов хлоридов натрия
и калия, поскольку они были исследованы особенно подробно как при 25°,
так и при других температурах. Коэффициенты активности этих электро-
литов можно затем использовать в качестве стандартных для определения
1 Харнед и Оуэн, как в этой главе, так и в последующих, ничего не говорят
об энтропии и теплоемкости иона в растворе. Между тем, советскими учеными эти:
вопросы разрабатывались [А. Ф. Капустинский, Изв. АН СССР, ОХН, № 2,
99 (1944); В. А. Киреев, Acta physicochimica URSS, 21, As 3, 555 (1946); E. H. Г a-
п о н, ЖФХ, 21, вып. 9, 1058 (1947); А. Ф. Капустинский, ДАН, 30, As 9,
792, 795 (1941); ЖФХ, 12, As 3—4, 186 (1942); ЖФХ, 15, вып. 9, 1055 (1941); Н. К. Во с-
кресенская, Г. Н. Янковская, Изв. АН СССР, ОХН, As 1, 3 (1945);
А. ф. Капустинский, К. Б. Яцими р с Кий, ЖФХ, 22, вып. 10, 1271
(1948)]. В связи с этим см. также исследования Мищенко по энергиям сольватации ионов
]К. П. Мищенко, Э. Ланге, 7. physik. Chem., 148, 161 (1930); 149 (1930);
К. П. Мищенко, ЖФХ, 10, вып. 7, 865 (1936)]. См. также реферат: А. М. Рубин-
штейн. Вестник АН СССР, As 3, 93 (1948). (Прим, ред.)
22*
340
ГЛАВА XII
коэффициентов активности других электролитов с помощью метода опре-
деления упругости пара в изопиестических условиях (§ 3). j;5 —8 содержат
обзор коэффициентов активности 1,1-электролитов в связи с теорией Дебая
и Гюккеля, а также с ее видоизменениями и обобщениями для случая
концентрированных растворов; § 10 посвящен «солевым эффектам», которые
наблюдаются при действии электролитов на нейтральные молекулы; § 11
содержит сведения о поверхностном натяжении ионных растворов. Оба эти
явления будут рассмотрены в связи с теориями, изложенными в гл.. III,
§ 10 и И.
§ 1. Термодинамические свойства растворов хлористого натрия,
определяемые путем измерения электродвижущих сил, а также
из калориметрических данных
а) Коэффициент активности в разбавленных растворах при 25°.
Имеются многочисленные исследования, посвященные определению электро-
движущих сил элементов без жидкостного соединения, содержащих 1,1-га-
логениды и гидраты окисей [10]. Было обнаружено, что электроды из про-
точных амальгам очень удобны для работы с растворами с концентрацией
выше 0,05 М. При более низких концентрациях эти электроды дают оши-
бочные результаты.
Для того чтобы преодолеть это затруднение, Браун и Мак-Иннес [11а]1
очень тщательно измерили электродвижущую силу элемента
Ag-AgCl [ NaCl (ej | NaCl (с2) [ AgCl-Ag.
Как было показано [уравнение (66) гл. X], электродвижущая сила этого
элемента определяется уравнением
и
2RT Ст,, ...
Е =----р- \ Т+ d In а± , (1)
I
где пределы интегрирования соответствуют концентрациям растворов у элек-
трода I и электрода II. Поскольку числа переноса были очень точно опре-
делены Мак-Иннесом и Лонгсвортом (гл. VI), а электродвижущую силу
такого рода элементов можно измерить при низких концентрациях раство-
ров, то может быть осуществлена надежная экстраполяция, дающая точные
значения коэффициентов активности.
В дифференциальной форме для 25° уравнение (1) имеет вид
dE= —0,1183 (c?lgc + cZIg2/±). (2)
Вместо того чтобы выражать Т+ как функцию от с и затем интегрировать,
можно использовать простой метод, предложенный Лонгсвортом. Значение
числа переноса при любой концентрации определяется уравнением
Т+^7’в+Д71+ (3)
где Тц — число переноса катиона при некоторой определенной концентра-
ции, выбранной в качестве стандартной. В данном случае в качестве стан-
дартной принята концентрация 0,1 н. После подстановки этого значения
Т+ в уравнение (2), преобразования и интегрирования получаем
-Alg2/±slg>±-lg2/B =
CR VR
1 Аналогичные измерения в интервале 15—45° были сделаны Янцем и Гордоном '
[116].
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭлЕКТРОЛИТОВ
341
Первые два члена в правой части этого уравнения можно непосредственно
вычислить из экспериментальных данных, третий член можно найти путем
графического интегрирования, а последний член, величина которого
сравнительно мала, получается с помощью графического интегрирования
с использованием предварительных значений Alg?/±, вычисленных для
первых трех членов. Эти предварительные значения A 1g у± затем испра-
вляют и вычисления повторяют снова до тех пор, пока не, будет достигнута
необходимая точность. Так как у± равно единице при бесконечном разба-
влении, то можно написать
1g у± = С" — A 1g , (5)
где С' — постоянная, равная Ig2/R. Подставляя вместо 1g у ± значение 1g /±
из уравнения (10) гл. V, получаем
A lg 2/± - S(/) \fc = С + А’ (С -Mgy±)\/c. (6)
При графическом изображении левой части этого уравнения как функции
от (С — Algy±) К с можно путем нескольких приближений найти значения
постоянных С и А'. Браун и Мак-Иннес получили таким образом значе-
ния — 0,1081 и —0,1088 соответственно для 1g у± и 1g при концентра-
ции 0,1 М. Вычисленные этими авторами величины у± и у± представлены
в 'табл. 77. Последний столбец содержит величины у±, вычисленные с по-
мощью уравнения (32) гл. X.
Таблица 77
Коэффициент активности хлористого натрия
т у±- У|_ (ВЫЧИСЛ.) 1)
0,005 0,9283 0,9283 0,9281
0,007 0,9171 0,9171 0,9169
0,01 0,9034 0,9032 0,9034
0,02 0,8726 0,8724 0,8724
0,03 0,8513 0,8509 0,8515
0,04 0,8354 0,8348 0,8354
0,05 0,8221 0,8215 0,8224
0,06 0,8119 0,8111 0,8115
0,08 0,7940 0,7927 0,7938
0,1 0,7796 0,7784 0,7796
1) Вычислено по уравнению (32) гл. X; а = 4; В =
= 0,047.
б) Относительное парциальное молярное теплосодержание хлористого
натрия в водных растворах. В табл. 122 мы приводим значения La
при 25° для водных растворов хлористого натрия. Эти данные были допол-
нены Гульбрансеном и Робинзоном [12], которые выполнили калориметри-
ческие измерения при 10, 15, 20 и 25°. Их данные приводятся в табл. 78.
в) Коэффициент активности хлористого натрия в 0,1 М растворе
при температурах 0 — 100°. Пользуясь известной величиной у± = 0,7784
в 0,1Л/ растворе при 25° и значениями Z2 ПРИ этой концентрации,
342
ГЛАВА XII
Относительное парциг хлористого натрия и* Таблица 78 1льное молярное теплосодержание I данных о теплотах разбавлениях) \молъ J
°C m 10 15 20 25
0,0001 0,0005 0,001 0,005 0,01 0,05 0,1 0,2 0,404 0,816 1) Эти значения были л чины при 25° несколько отл 3,0 8,3 11,5 25,6 34,8 43,2 30,2 - 20 -130 олучены Гу ичаются от 4,7 12,0 16,5 35,8 46,0 65,0 57,7 9,4 -89,9 льбрансено данных, пр 5,3 13,1 17,9 38,1 51,0 76,3 76,6 54,2 - 19,4 -169,7. м и Робинз иведенных 6,3 14,8 20,1 42,2 55,8 93,7 99,6 85,0 30,0 — 123,3 оном. Вели- в табл. 122.
приведенными в табл. 78, можно вычислять у0 При других температурах
с помощью уравнения (73) гл. III
2д1п 701 _ £2 (01)
дТ RT2 ’
Зависимость £2(0,1) от температуры имеет следующий вид:
£2(0>1)=—1277 + 4,62 7. (7)
После подстановки значения £2(0,1) в уравнение (73) гл. III, интегриро-
вания и вычисления постоянной интегрирования с помощью значения
70 1 =0,7784 при 25° получаем
- Y0.1 = +1,16251g Т- 3,2358. (8)
Уравнение (8) должно давать точное значение у0 4 для широкого интервала
температур при условии, что относительная парциальная теплоемкость (рав-
ная 4,62) остается постоянной или меняется очень незначительно с изме-
нением температуры. Вычисленные таким образом для различных темпе-
ратур значения у0 1 приведены в табл. 79.
Харнед и Нимс [Юж] измерили электродвижущую силу элемента без
жидкостного соединения:
Ag-AgCl | NaCl (щ) | NaxHg I NaCl (0,1) | AgCl-Ag.
Измерения производились через каждые 5° в интервале от 0 до 40°. Так
как элементы с проточной амальгамой дают ненадежные результаты при
концентрациях ниже 0,05 М, то для экстраполяции приходится использо-
вать результаты, полученные при более высоких концентрациях. Для этой
цели можно использовать функции Дебая и Гюккеля [уравнение (32) или
(33) гл. X]. Как показывает опыт, с помощью этого метода можно осуще-
ствить надежную экстраполяцию, применяя растворы с концентрациями
0,1 — \ М. Если применять эти уравнения для более широкого интер-
вала концентраций (0,1 —ЗА/), то получаются несколько заниженные вели-
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
343
чины а и у±. В третьем столбце табл. 79 приведены результаты, полу-
ченные Харнедом и Куком [13]. Значение среднего расстояния сближения
ионов оказалось равным 4,0 ± 0,1 А при всех температурах. Совпадение
этих значений у0 { с величинами, приведенными во втором столбце таблицы,
является удовлетворительным.
Сочетая уравнение (33) гл. X с уравнениями для определения осмо-
тического коэффициента из данных о повышении температур кипения рас-
творов, Смит [14] получил значения у0 р приведенные в последнем столбце
табл. 79. В своих расчетах Смит также принял среднее расстояние сбли-
жения ионов равным 4,0. Данные Смита прекрасно совпадают с резуль-
татами, полученными по уравнению (8).
Таблица 79
Значения То, 1 для хлористого натрия, определен-
ные тремя независимыми методами
t, ‘С Метод определения
по уравне- нию (8) [1] по уравне- нию (33) гл. X по понижению температур кипения
0 0,7809 0,781
5 0,7809 0,782 —
10 0,7807 0,782 ЯШ»
15 0,7802 0,7815 —
20 0,7793 0,781 —
25 0,7784 0,779 —
30 0,7773 0,776 —
35 0,7757 0,774 —
40 0,7743 0,773 —
50 0,770 — —
60 0,766 - 0,766
70 0,761 — 0,762
80 0,756 — 0,757
90 0,751 — 0,752
100 0,745 — 0,746
В табл. 134 приведены простые уравнения и соответствующие константц
для растворов 1,1-электролитов, позволяющие вычислять с из известных
значений т. Эти уравнения будут полезны для некоторых теоретических
расчетов.
г) Осмотический коэффициент и коэффициент активности хлори-
стого натрия при температурах 0—100°. На основании измерений электро-
движущих сил [10 ж] и температур кипения [15, 16] различными авторами
были вычислены коэффициент активности и осмотический коэффициент хло-
ристого натрия в интервале температур 0 — 100° для концентраций 0,1 — 4 М.
На рис. 88 и 89 представлена зависимость коэффициентов активности
от температуры. Аналогичные кривые для осмотических коэффициентов
изображены на рис. 90. Данные для температур 0 — 40° получены по изме-
рению электродвижущих сил, а для температур 60—100° — по определению
температур кипения. Как видно из рисунков, в обоих случаях наблюдаются
небольшие ошибки. При 35 и 40° значения у , вычисленные на основании
Рис. 88. Зависимость среднего коэффициента ак-
тивности хлористого натрия от температуры для
растворов с концентрациями 0,1; 0,2; 0,5 и 1 М.
О вычислено по уравнению (8); Q вычислено на основа-
нии измерений электродвижущих сил; • вычислено из
данных для температур кипения. Диаметры кружков рав-
ны 0,002. Кривые смещены одна относительно другой.
Рис. 89. Зависимость среднего коэффи-
циента активности хлористого натрия от
температуры при концентрациях раство-
ров 1,5—4 М.
От 0 до 40° вычислено на основании измерений
электродвижущих сил; от 60 до 100°—из дан-
ных для температур кипения. Диаметры круж-
ков'равны 0,002. Кривые смещены одна относи-
тельно другой.
Рис. 90. Осмотические коэффициен-
ты растворов хлористого натрия для
концентрации 0,1—3,5 М.
От 0 до 40° вычислено на основании измере-
ний электродвижущих сил; от 60 до 100°—из
данных для температур кипения. Диаметры
кружков равны 0,002.
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
345
измерения электродвижущих сил, являются в некоторых случаях зани-
женными, в то время как при 60 и 70° значения, полученные на основа-
нии измерения температур кипения, оказываются иногда слишком большими.
Это наиболее заметно для результатов, полученных при 60° для концен-
трированных растворов (1,5 — 4 М). Эти замечания относятся также к осмо-
тическим коэффициентам. Значения у приведены в табл. 135. В интер-
вале температур 35 — 70° эти данные были несколько округлены в соот-
ветствии с изображенными на рисунках кривыми, чтобы можно было
воспроизвести эти кривые. Диаметры кружков на рисунках равны 0,002
единицы у , что соответствует приблизительно 0,15 мв при измерении
электродвижущей силы и 0,0003° при определении температуры кипения.
При 40° максимальное отклонение экспериментальных данных от кривых
составляет 0,004, т. е. примерно 0,3 мв.
д) Относительные парциальные молярные теплосодержание и тепло-
емкость хлористого натрия. Наилучшие определения относительного
Рис. 91. водных растворов хлористого натрия.
О вычислено па основании измерений электродвижущих сил и темпе-
ратур кипения (диаметры кружков равны 50 кал)-, • калориметрические
данные (диаметры кружков равны 30 кал).
парциального молярного теплосодержания на основании измерений электро-
движущих сил и температур кипения были сделаны Смитом и Гиртле [16].
Уравнение (73) гл. III можно написать в следующем виде:
dlnfj. L2
d(i/T) ==~2R '
346
ГЛАВА XII
Отсюда видно, что В2 можно определить из величины наклона касатель-
ных к кривым, изображающим зависимость 1g у± от у. При 0° резуль-
таты практически совпадают с результатами Харнеда и Кука, полученными
путем измерений электродвижущих сил. При 100° получаются менее досто-
верные результаты, так как наклон конечного участка кривой не может
быть точно определен. На рис. 91 изображены результаты Смита и Гиртле
•совместно с калориметрическими данными Робинзона [17], Гульбрансена
•и Робинзона [12], а также Россини [18], приведенными в табл. 136.
На рисунке белыми кружками обозначены значения, полученные путем изме-
рения электродвижущих сил и температур кипения, а черными кружками —
значения, найденные калориметрическими методами. Пунктирная кривая
нанесена по данным Россини, относящимся к 18°. При температурах
10 — 25° наблюдается прекрасное совпадение значений, полученных с помощью
этих совершенно независимых друг от друга методов. При более высоких
температурах калориметрические данные отсутствуют. Хотя данные для
высоких температур являются приближенными, все же с их помощью можно
получить общее представление о поведении В2 в большом интервале темпе-
ратур.
Значения относительной парциальной молярной теплоемкости 72, полу-
ченные Смитом и Гиртле путем измерений температурного коэффициента L2,
приведены в табл. 80 вместе с результатами калориметрических определений.
Таблица 80
Относительная парциальная молярная теплоемкость хлористого натрия
t, °C 0 25 60 25 25
т X. J2 1} Л 2) Л8)
0,05 3,3 3,5 4,8
0,1 — . 5,4 — 5,0 6,8
0,2 — 7,0 — 7,1 9,6
0,5 10 10,0 10 11,2 15,3
. 1,0 20 16,0 12 15,6 21,6
1,5 25 20,0 15 — 26,4
2,0 33 24,0 19 — 31,0
2,5 35 32,0 21- — 34,2
3,0 45 35,0 21 — 37,2
3,5 51 38,0 22 — 40,5
4,0 53 41,0 24 — 43,2
1) По измерениям температур кипения и электродвижущих сил.
г) Получены Гульбрансеном и Робинзоном из значений Ь2, определенных кало-
риметрическим путем.
а) Получены Робинзоном из данных по измерениям теплоемкостей, произве-
денным Рендаллом и Россини [1].
Вплоть до концентрации, равной 1 М, результаты, полученные на основа-
нии измерений электродвижущих сил элементов и температур кипения, совпа-
дают с данными Гульбрансена и Робинзона; при более высоких температурах
эти результаты приближаются к данным Россини [18а].
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
347
§ 2 Термодинамические свойства водных растворов бромистоводородной
кислоты, хлористого калия, бромистого натрия и гидратов окисей натрия
и калия
а) Стандартный потенциал элемента Н21 HBr (т) | AgBr-Ag. После
ранних исследований Льюиса и Сторча [19], а также Харнеда и Джемса [20]
по определению электродвижущей силы элементов, составленных из водо-
родного электрода, соединенного с электродом серебро-бромистое серебро
при 25°, было выполнено много работ [21] по определению электродвижу-
щих сил этого элемента в широком интервале изменения температуры,
причем электролитом служили либо растворы чистой кислоты [22],
либо растворы кислоты, содержащие бромид [23, 24]. Применялись два
типа электродов серебро-бромистое серебро. Первый из них изготовлялся
путем нагревания при 650° смеси, содержащей 90% окиси серебра и 10%
бромата серебра, электроды второго типа приготовлялись путем нагревания
окиси серебра на платиновой спирали при 450° и последующего электро-
лиза в бромистоводородной кислоте или в растворе бромистой соли для обра-
зования бромистого серебра. Харнед, Кэстон и Донельсон показали, что
в отсутствие кислорода эти два типа электродов дают совершенно одина-
ковые результаты.
Не так давно стандартный потенциал электрода серебро-бромистое
серебро был определен с помощью трех независимых способов:
1. Е° приведенного выше элемента, содержащего только бромистоводо-
родную кислоту, была определена методом графической эктраполяции,
описанным в гл. XI, § 2.
2. Харнед и Донельсон определили Е° аналогичным методом путем
измерения электродвижущей силы элементов
Н2|НВг(0,01), LiBr (ш) | AgBr-Ag.
3. Оуэн [25] показал, что в буферных растворах можно непосредственно
сравнивать электродвижущие силы элементов с электродами серебро-бро-
мистое серебро и серебро-хлористое серебро. Рассмотрим элементы
Н21НВО2 (т), NaBO2 (т), NaBr (ш) | AgBr-Ag,
Н21НВО2 (т), NaBO2 (m), NaCl (m) | AgCl-Ag.
Электродвижущие силы этих элементов ЕцВг и Енсь измеренные Оуэном
и Ферингом [26], определяются уравнениями
Еш т?0 RT,
нвг Енвг р- In У^ДвД^н^вг’
Т7Ш Т?0 RT ,
Енс1 = Encl - -p- in YHYcl •
После исключения из этих уравнений Ynmii с помощью термодинамического
уравнения для константы равновесия борной кислоты
__ 7н7во2 'гентвоа
А 7нво3 TOHBO2
и преобразования получаем
Енвг — / (y) — Енвг + -р- In К а. + -р- In т
и
Ецс1 — / (y) = Ehci + у In К а + -р- In т.
348
ГЛАВА XII
Эти уравнения можно применять в отдельности для вычисления КА, а затем
Енвг! можно также исключить КА при условии, если все три электролита
имеют одинаковую концентрацию. Таким образом, вычитая второе уравне-
ние из первого, получаем
НВг = -^НС! “Г ЬНВг — ^нсь
Зная стандартный потенциал —EgC1 электрода серебро-хлористое серебро
и измеряя Енвг и Ена при какой-либо низкой концентрации раствора,
нетрудно вычислить Ецвг- В связи с этим см. рис. 148. Значения Е°, опре-
деленные с помощью этих методов, приведены в табл. 81. Значения Ена,
Таблица 81
Значения стандартного потенциала электрода
серебро-бромистое серебро, определенные
с помощью трех методов
— тсО —Ео
Ag-AgBr НВг
Метод °C 1 2 3
0 0,0813 0,08165 ——
5 0,0796 0,07991 0,07986
10 0,0777 0,07801 0,07800
15 0,0756 0,07593 0,07596
20 0,0734 0,07377 0,07374
25 1) 0,0711 0,07127 0,07134 1)
30 0,0685 0,06872 0,06876
35 0,0658 0,06602 0,06600
40 0,0629 0,06300 0,06306
45 0,0600 0,05995 —
50 0,0569 0,05666 —
1) Джонс и Бекштрём [1] нашли, что стандартный
потенциал этого электрода при 25° равен 0,0712.
необходимые для вычислений по уравнению (9), были получены с помощью
уравнения (5) гл. XI. Совпадение между результатами, приведенными
в последних двух столбцах таблицы, является превосходным, в то время
как данные второго столбца несколько занижены. Уравнение
- ^Ag-AgBr = Ецвг = 0,07131 - 4,99 • НИ (t - 25) - 3,45 • 10~6 (г - 25)2 (10)
отражает результаты, приведенные в столбцах 2 и 3, с точностью до
нескольких сотых милливольта.
б) Термодинамические свойства бромистоводородной кислоты, опре-
деляемые путем измерения электродвижущих сил. Коэффициент актив-
ности, относительные парциальные молярные теплосодержание и тепло-
емкость бромистоводородной кислоты были определены Харнедом, Кэстоном
и Донелсоном [22] в растворах с концентрациями до 1 М при температу-
рах 0 — 60°. Значения у± в этих пределах концентраций и температур,
а также значения L2 и J2 при 25° даны в табл. 137. Поскольку свойства
этой кислоты во всех отношениях очень схожи со свойствами соляной
кислоты, то нет необходимости останавливаться на них более подробно.
ВОДНЫЕ РАСТВОРЫ «СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
349
в) Термодинамические свойства водных растворов хлористого калия.
Свойства водных растворов хлористого калия изучались многими спосо-
бами. Значения электродвижущей силы элементов
Ag-AgCl | KCI (m) | KJIg | KCI (m0) | AgCl-Ag
измерялись при 25° Мак-Иннесом и Паркером [10а], а также Харнедом [Юг];
при 0° —Смитом [27], в интервале 0 — 40° — Харнедом и Куком [28]. Шид-
ловский и Мак-Иннес [29] исследовали элемент
Ag-AgCl | КС1 (сх) | KCI (c2) | AgCl-Ag
при 25°.
Спенсер [30] вычислил коэффициент активности при 0° из данных
о температурах замерзания; Скэтчард и Прентис [31] проделали аналогичный
расчет на основании своих собственных результатов. Сэкстон и Смит [32]
вычислили у± для 100° по данным для температур кипения. Упругость пара
растворов хлористого калия при 20° определили Ловлейс, Фрэзер и Сиз [33].
Для сравнения результатов измерений этими методами может служить
табл. 82. Совпадение значений, полученных различными методами,
Таблица 82
Значения коэффициента активности хлористого калия, определенные различными
методами (э. д. с.—по методу измерения электродвижущих сил; т. замерз.—по
данным для понижения температуры замерзания; упр. пара—по данным для пони-
жения упругости пара)
\ t, °C т 0 20 25
э. д. с.1) Э. Д. С.2) т. замерз.8) т. замерз.4) э. д. с.1) упр. пара 5) э. д. с. 1) э. д. с.в)
0,005 (0,929) 0,929 0,929 (0,927) (0,9275)
0,01 (0,904) 0,903 0,904 (0,901) 0,902
0,05 (0,819) 0,818 0,819 (0,816) (0,815) 0,817
0,1 0,768 0,770 0,771 (0,770) 0,770 0,769 ' 0,770
0,2 0,717 0,717 0,719 0,717 0,718 0,721 0,719 0,719
0,3 0,683 0,688 0,688
0,5 0,642 0,642 0,645 0,644 0,651 0,651 0,651 0,652
0,7 0,613 0,615 0,618 0,627 0,628
1,0 0,588 0,589 0,590 0,588 0,604 (0,604) 0,606 0,607
1,5 0,563 0,562 0,561 0,582 0,585
2,0 0,547 0,548 0,573 0,573 0,576 0,578
2,5 0,540 0,541 0,568 0,572
3,0 0,539 0,540 0,567 0,569 0,573 0,574
3,5 0,540 0,542 0,571 0,574 0,576
4,0 0,574 0,572 0,582 0,581
1) Харнед и Кук [1], элементы без жидкостных соединений.
2) Смит [2], элементы без жидкостных соединений.
з) Спенсер [3], понижение температуры замерзания.
4) скэтчард и Прентис [4], понижение температуры замерзания.
5) Ловелэк, Фрезер и Сиз [5], понижение упругости пара.
6) Шидловский и Мак-Иннес [6], элементы с жидкостным соединением.
является очень хорошим. Более полная сводка коэффициентов активности
для большего интервала температур приведена в табл. 138.
350 ГЛАВА .XII
В табл. 139 приведены константы уравнения, с помощью которого
можно вычислить значения С2 и J2 при температурах 0 — 40°. Эти данные
будут рассмотрены ниже в этом же параграфе.
г) Термодинамические свойства водных растворов бромистого натрия,
определенные путем измерений электродвижущих сил. Для определения
коэффициента активности,, относительных парциальных молярных тепло-
содержания и теплоемкости Харнед и Кроуфорд [Юк] использовали эле-
мент с электродами: проточная амальгама и серебро-бромистое серебро.
Полученные ими коэффициенты активности приведены в табл. 140. Урав-
нения и соответствующие константы, с помощью которых можно вычислять
Ь2 и /2 в интервале 0 — 40°, приведены в табл. 141.
д) Коэффициенты активности гидроокисей щелочных металлов
при 25°. Реакция, протекающая в элементе
Н21 МОН (т2) | MJIg ] МОН (тД | Н2,
где M = Li, Na и т. д., может быть представлена уравнением
МОН (т2) + Н2О (тиД = МОН (тД + Н2О (w2),
. причем электродвижущая сила этого элемента равна
E = 2A:lg^- + £lg^i) , (11)
7i™i aw(m2)
где к = 2,3026 RT/Y, aW(mi) и aw (m2) — активности воды в двух исследу-
емых растворах.
Кнобелем [34], Харнедом [10в], Харнедом и Свинделсом [35], Харне-
дом и Щуплом [10е] на основании измерений электродвижущих сил этих
элементов были вычислены коэффициенты активности гидроокисей калия,
натрия, лития и цезия при 25°. Позднее Харнед и Хеккер [10з] исследовали
элементы, содержащие гидроокись натрия, а Харнед и Кук [36] — элементы,
содержащие гидроокись калия в интервале температур 0 — 35°. Харнед пока-
зал, каким образом может быть вычислена поправка на перенос воды из дан-
ных по электродвижущим силам элементов с помощью уравнения для у±.
Харнед и Хеккер, а также Харнед и Кук применяли аналогичные графические
методы. Согласно уравнению (45) гл. I, активности растворителя и раство-
ренного вещества связаны уравнением
с?1паю = — ^с?1па2. (12)
Так как d In а2 = 'id In ут, то для водного раствора 1,1-электролита имеем
d In aw = — d In ym (13)
и, следовательно,
-Idlna^^ + ^iZlny. (14)
После интегрирования в пределах от до т2 искомый логарифм отноше-
ния активностей определяется уравнением
а ‘т
1 w<rnR'> __ т — тЕ . 1 С ji _J±_ /151
2 lg aw(m1 “2,303.55,51 + 55,51 J mdlS
mR
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
351
Пусть ун и mR представляют собой некоторые стандартные значения коэф-
фициента активности и моляльности. Харнед и Хеккер применяли в каче-
стве стандартного раствор с концентрацией 0,05 М. Искомая величина
левой части этого уравнения может быть вычислена путем арифметического
приближения. В первом приближении можно отбросить второй член в пра-
вой части этого уравнения. Далее вычисляют значения левой части уравне-
ния для различных концентраций и эти значения подставляют совместно
с величинами Е и т в уравнение (11). Затем вычисляют по уравнению (11)
предварительные значения 1g у± / ун и используют их для определения вто-
рого члена уравнения (15) путем графического интегрирования. Таким
образом получают новое значение левой части уравнения (15), которое
можно применить для получения более точных значений отношения коэф-
фициентов активности. Эту операцию повторяют до тех пор, пока не будут
получены значения, удовлетворяющие уравнению1. Повторяя эту опера-
цию 3 или 4 раза, можно получить значения, удовлетворяющие уравне-
нию с точностью до 0,01 мв, что гораздо меньше экспериментальной
ошибки. Таким образом получают окончательную величину отношения
Y±/YB- Применяя соответствующим образом уравнение теории Дебая
и Гюккеля [уравнение (32) гл. X], можно экстраполировать эти результаты
и определить значение ун при стандартной концентрации, а следовательно,,
и при любой другой концентрации.
В табл. 83 приведены значения у± при 25°, полученные этим методом.
Экстраполяция может быть выполнена удовлетворительно для всех раство-
ров гидроокисей, кроме растворов гидроокиси лития, для которых полу-
чаются сравнительно неточные результаты.
Таблица 83
Коэффициенты активности гидроокисей щелочных
металлов при 25°
т L1OH [1] NaOH 1) КОН [3] CsOH [4]
0,05 (0,803) (0,818) (0,824) (0,831)
0,1 0,760 0,766 0,798 0,802
0,2 0,702 0,727 0,757 0,761
0,5 0,616 0,693 0,728 0,748
1,0 0,554 0,679 0,756 0,780
1,5 0,528 0,683 0,814 —
2,0 0,513 0,698 0,888 —
2,5 0,501 0,729 0,974 —
3,0 0,494 0,774 1,081 —
3,5 0,487 0,826 1,215 —
4,0 0,481 0,888 1,352 —
1) Харнед и Хеккер [2а]-от 0,05 до 2М, Акерлёф и Киджелс [261-
otJ-2 до 4М.
е) Термодинамические свойства водных растворов гидроокисей натрия
и калия, определенные путем измерений электродвижущих сил. Коэф-
фициенты активности, относительные парциальные молярные теплосодер-
1 Акерлёф и Киджелс [Юл] применили аналитический метод, дающий возможность
избежать последовательных приближений, но требующий представления Е в виде функ-
ции от т с помощью метода наименьших квадратов.
352
ГЛАВА XII
жание и теплоемкость гидроокиси натрия в водных растворах были опре-
делены Харнедом и Хеккером [Юз] вплоть до концентраций 4 М для тем-
ператур 0 — 35°. Акерлёф и Киджелс [Юл] выполнили весьма обширное
исследование этих растворов в интервале концентраций 0,1 —17 М для
температур 0—70°. Свойства растворов гидроокиси калия при температу-
рах 0 — 40° и вплоть до концентраций 4 М были изучены Харнедом
и Куком [36]. Коэффициенты активности этих гидроокисей приведены
в табл. 142 и 143. В табл. 144 и 145 приведены константы уравнений,
с помощью которых можно вычислить /,2 и Л.
ж) Сравнение величин относительных парциальных молярных тепло-
содержания и теплоемкости, определенных калориметрическим и электро-
химическим способами. На рис. 92 изображена зависимость £2 от т1/2
Рис. 92. Относительные парциальные Рис. 93. Относительные парциальные
молярные теплосодержания некоторых молярные теплоемкости некоторых 1,1-элек-
1,1-электролитов при 18°. тролитов при 25°.
Сплошные линии—вычислено на основании из- Сплошные линии—вычислено на основании из-
мерений электродвижущих сил; пунктирные мерений электродвижущих сил; пунктирные
линии—калориметрические данные. П. 3,—пря- линии—калориметрические данные; П. 3.—пря-
мая, отвечающая теоретическому предельному мая, отвечающая теоретическому предельному
. закону. закону.
при 18° для соляной кислоты, хлористого калия и гидроокисей калия
и натрия, причем значения Л2 вычислены из данных о теплотах разбавле-
ния и по измерениям электродвижущих сил. Сплошные линии изображают
данные, полученные методом электродвижущих сил, пунктирные — калори-
метрические данные. Максимальное расхождение между результатами, полу-
ченными этими двумя методами, составляет около 60 кал. Это совпадение
можно считать хорошим, так как методика эксперимента с элементами
с проточной амальгамой в качестве электрода, которые применялись
во всех трех случаях, весьма сложна. Прямая линия соответствует пре-
дельному закону теории междуионного притяжения. Аналогичным образом
значение L2 для растворов бромистого натрия, определенное путем изме-
рения электродвижущих сил, хорошо совпадает со значением Л2, получен-
ным из калориметрических определений Гаммершмида и Робинзона [37],
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
353
если учесть, что расхождение в 25 кал обусловлено трудностью экстрапо-
ляции.
На рис. 93 изображена зависимость J2 от пг1/2 для хлорида, гидроокиси
калия и для соляной кислоты, полученная с помощью этих методов.
Пунктирными линиямишредставлены результаты, полученные калориметри-
ческим методом, сплошными линиями—данные, полученные путем измерений
электродвижущих сил соответствующих элементов. Совпадение этих резуль-
татов можно считать превосходным, так как максимальное расхождение
между ними составляет около 2 кал. Значения для гидроокиси натрия,
полученные теми же методами (на рисунке не показаны), также хорошо
совпадают друг с другом.
Описанные результаты свидетельствуют о том, что метод электродви-
жущих сил может применяться для определения зависимости величин Л2
и /2 от ™1/2 для большого числа электролитов. Следует помнить, что
с помощью метода электродвижущих сил непосредственно определяется
(F2 — Fl) и у Когда этот метод применяется для определения L2 и
то исходные данные подвергаются при этом весьма строгому испы-
танию, так как в ходе этих вычислений находят первую и вторую про-
изводные от экспериментальных данных. Поэтому калориметрические опре-
деления, как правило, должны давать более точные результаты за исклю-
чением тех случаев, когда электродвижущие силы могут быть измерены
с особенно выбокой точностью. Хорошее совпадение результатов, получен-
ных двумя методами, продемонстрированное на рис. 92 и 93, свидетель-
ствует о том, что наши представления о механизме происходящих в эле-
ментах реакций очень близки к истине.
§ 3. Вычисление коэффициентов активности 1,1-электролитов из данных
по изопиестическим измерениям давления пара
а) Коэффициенты активности при 25°. Как было показано, метод
определения коэффициентов активности путем измерения электродвижущих
сил дает хорошие результаты, однако он применим лишь в тех случаях,
когда электролит диссоциирует на ионы, для которых можно найти обра-
тимый электрод. В случае концентрированных растворов применим метод
определения коэффициентов активности по измерениям упругости пара,
однако абсолютные значения упругости пара до сих пор были точно опре-
делены лишь для небольшого числа растворов электролитов. Значитель-
ного успеха в этой области добились Робинзон и Синклер [38], а также
Робинзон [39]. Эти авторы значительно усовершенствовали метод изопие-
стического измерения упругости пара по Боусфильду [40] (гл. IX, § 4).
Если растворам двух различных солей дать возможность прийти в равно-
весие с одной и той же парообразной фазой, а затем произвести их анализ,
то при условии, что активность и концентрация одной из солей известны,
можно вычислить коэффициент активности другой соли. Пусть mR, ун, т
и —моляльности и коэффициенты активности двух солей, находящихся
в указанных выше изопиестических условиях, и ап— активность стандарт-
ной соли. Так как активности растворителя в двух растворах одинаковы,
то, согласно уравнению (45) гл. I, мы имеем
d In aR = — d In aw = d In a2 (16)
или
й1пу± = сИпув + <Лп^ + 0^-1)сг1п7нтв. (17)
23 Г. Харнед, Б. Оуэн
354
ГЛАВА XII
Так как
с?1пГвтв = 2у=?>
то после интегрирования мы получим
1пу± = 1п-1в+1п^ + 2^(^-1)^. (18)
Отсюда можно вычислить у±, если известны ул и aR. Отношение концен-
траций изопиестических растворов mR/m можно изобразить графически как
функцию от т и таким образом определить второй член правой части
уравнения для всех концентраций заданного интервала. Третий член
в правой части уравнения можно определить графически с помощью кри-
вой, изображающей зависимость (mR/m — 1)/ aR от У aR. Поскольку aR
равно нулю при т = 0, то эта кривая проходит через начало координат.
Проблема приготовления подходящего стандартного электролита
остается пока еще не разрешенной. Робинзон и Синклер применяли в каче-
стве стандартного электролита растворы хлористого калия, Скэтчард,
Гамер и Вуд [41] — растворы хлористого натрия. Последние авторы,
в отличие от первых, придавали большее значение измерениям давления
пара, чем измерениям электродвижущих сил; их стандартные величины
заметно отличаются от соответствующих величин, полученных Робинзоном
(см. табл. 60). В табл. 146 приведены полученные Робинзоном значения у
при 25° для всех щелочных галогенидов, нитратов, ацетатов, п-толуолсуль-
фонатов и таллиевых солей. При этих определениях в качестве стандарт-
ных растворов принимались растворы хлористого калия или натрия. Зна-
чения у которые Робинзон применял для этих солей, практически совпа-
дают со значениями у приведенными в табл. 135 и 138.
Результаты определений коэффициентов активности с помощью метода
изопиестических измерений давления пара весьма точны и хорошо совпа-
дают с данными, полученными путем измерений электродвижущих сил.
Таблица 84
Отношения средних коэффициентов активности при 25°, определенных методами
изопиестической упругости пара и электродвижущих сил
т VNaClО I’KCl TNaBr I’KCl ТКВг ткш iCsCl I’KCl
упр. пара э. д. с. упр. пара э. д. с. упр. пара э. д. с. [3] упр. пара э. д. с. [4;
о,1 1,012 1,013 1,016 1,017 1,003 1,004 0,982 0,984
0,2 1,024 1,020 1,031 1,029 1,006 1,004 0,967 0,966
0,3 1,033 1,032 1,044 1,044 1,007 1,006 — —
0,5 1,049 1,046 1,069 1,068 1,011 1,013 0,929 0,931
0,7 1,067 1,068 1,097 1,094 1,018 1,018 -0,915 0,916
1,0 1,088 1,084 1,136 1,132 1,020 1,019 0,898 0,898
1,5 1,126 1,122 1,203 1,202 1,027 1,026 0,879 0,879
2,0 1,167 1,165 1,273 1,274 1,037 1,032 0,861 0,859
2,5 1,210 1,210 1,346 1,352 1,042 1,039 0,848 0,843
3,0 1,257 1,261 1,426 (1,447) 1,047 1,043 0,838 0,829
3,5 1,307 1,312 1,512 (1,530) 1,052 1,053 —
4,0 1 1,361 1,368 1,612 1,613 1,057 1,061 —
1) Аналогичное сравнение при 15 и 40° сделали Робинзон [1] и Олиник и Гордон [2].
ВОДНЫЕ РАСТВОРЫ .СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ 355
Это подтверждается данными табл. 84, в которой приведены значения
отношений коэффициентов активности различных солей к активности
хлористого калия, причем эти отношения определены с помощью двух
упомянутых методов. В интервале концентраций 0,1 — 3 М наблюдается
прекрасное совпадение. Расхождения значений для более концентрирован-
ных растворов могут быть обусловлены изменениями растворимости
у электродов. Однако возможно, что это объяснение и неправильно,
поскольку опыты при высоких концентрациях не были столь же тщатель-
ными, как опыты с использованием элементов с проточными амальгамными
электродами в интервале концентраций от 0,1 до 3 21/. С электродами
из амальгам лития результаты получаются менее воспроизводимые, чем
в случае амальгам других щелочных металлов. Поэтому для растворов
хлористого и бромистого лития совпадение между величинами, получен-
ными изопиестическим и электрохимическим методами, хуже, чем в дру-
гих случаях1.
§ 4. Вычисление коэффициента активности при различных температурах
и давлениях из данных, относящихся к 25° и атмосферному давлению.
В табл. 122, 45 и 46 приведены значения L2 и J2. Если пренебречь
незначительным изменением /2 с температурой, то приближенное уравне-
'ние для L2 будет иметь следующий вид:
L2 = L2(tR) + J2(tS){T — Tr), (19)
где TR—некоторая стандартная температура. Это уравнение можно при-
менять для вычисления коэффициентов активности при различных темпе-
ратурах из известных величин этих коэффициентов для данной темпера-
туры. Таким образом, согласно уравнению (73) гл. III,
/ a in 7± \ _ i э in /± \ _
\ дт }р,т\ дТ /р,т~ vRT2 '
Если объединить это уравнение с уравнением (19) и проинтегрировать,
то получаем
1 £2(ТД)—_ Л(Тя) J т J.
в‘± v2,3037?7’ vfl (AV)
Если при данной температуре TR известно, можно вычислить константу
интегрирования I (см- § 1 данной главы). Если стандартная температура
равна 25°, то значение TR = 298,1 можно подставить в приведенное выше
уравнение и выразить константу интегрирования как функцию от 1g у
при этой температуре. Таким образом,
lg Y± = 1g Y± (25°) + f L2 (25°) - J2 (25°), (21)
причем
_ 298,1—71__ .„2^
y 2,303 R 298,1 T ’ '
Z = 298,l^--^lg0^). (23)
Значения переменных величин у и z приведены в табл. 2 для интервала
температур 0 — 100°.
1 Другие подобные сравнения см. в статье Робинзона и Харнеда [42].
23*
356
ГЛАВА ХН
Согласно уравнению (86) гл. Ш, зависимость коэффициента активно-
сти от давления имеет вид
/ д In /, \ / д In 7 , \ V.. —
z± — r 2 Г 2 (9/, \
\ ЭР )т, m \ дР /т, m vRT ’ '
Табл. 49 содержит величины V2 и Sy при 25°; зная эти величины, можно
вычислить V2 при этой температуре с помощью приближенного уравнения
V2 = Vl + ^/2)Svi/c (25)
или с помощью более точного соотношения
Т;+(з,у
i+^yc^/aooo 4
которое получается из уравнения (68) гл. VIII. Для других температур
значения V2 и V2 можно вычислить по уравнениям (80), (87) и (91) гл. VIII
и с помощью данных, приведенных в табл. 51.
Для того чтобы выразить V2 — V2 как функцию давления, прежде чем
интегрировать уравнение (24), допустим, что значения парциальной молярной
сжимаемости при 1 атм К2 и Кй2 постоянны в узких пределах изменения
давления; пользуясь уравнением (101) гл. VIII, можем написать
72-^ = (Й2-^)р==1-(Р-1)(^2-^)р=1. (27)
Согласно уравнению (24), получаем
, . (Р-1) (П-(Р-1)2(^-Л)р=1 ,9Й.
lgy± (lgT±)p-l I 2,303^7?7’ 2 - 2,303vRT ~
Из этого уравнения можно вычислять значения коэффициентов активности
при умеренных давлениях. Для более высоких давлений, а также в тех
случаях, когда требуется большая точность, необходимо принимать во вни-
мание изменение К2 — К2 в зависимости от давления. Вплоть до концен-
трации 1 М значение разности К2 — К2 хорошо выражается уравнением
К2-К°2 = (3/2)Як}Гс, (29)
причем значения для некоторых солей можно найти в табл. 52.
Робинзон и Харнед [42] применили для вычисления коэффициентов
активности хлористого натрия в области температур 0 —100° уравнение (20)
в следующем виде:
lgY±= -ф-BlgT + I. (20а)
Из термодинамических данных, приведенных в § 1, и из значений у при 25°
были получены величины А, В и I, приведенные в табл. 85. Применимость
этого уравнения ограничена, поскольку имеется мало надежных данных
о тепловых эффектах при высоких концентрациях. В табл. 86 с целью
сравнения- коэффициентов активности, вычисленных по этому уравнению, со
значениями, приведенными в табл. 135, представлены отклонения опытных
значений от вычисленных. Далее будет показано, что это уравнение дает
достаточно точные результаты для температур 0—100°, хотя, как и следо-
вало ожидать, при высоких концентрациях наблюдаются несколько боль-
шие расхождения.
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
357
После исключения (V2 — У|) и (К2 —Кг) из уравнения (28) с помощью
уравнений (25) и (29) получаей для 1,1-электролитов при 25° следующее
соотношение:
lg Y± = (lg Y±)p=i +0,888 -IO-5(3/2) Sv (P-1) Vc-
-0,444-IO-5 (3/2) SK(P-1)2 j/c. (30).
В табл. 87 показано влияние давлений в 1, 100 и 1000 атм на коэф-
фициенты активности хлористоводородной кислоты для четырех концентра-
ций.кислоты, а также влияние этих же давлений на коэффициенты актив-
ности хлоридов натрия и калия и гидроокиси натрия при концентрации,
равной единице. При 100 атм влияние члена, в который давление входит
во второй степени, незначительно, но при 1000 атм его следует учитывать.
Таблица 85
Данные для вычисления коэффициента активно-
сти хлористого натрия при температурах
от 0 до 100°
Ig^I-^-BlgT
т I А В
о,1 3,5083 152,06 1,2557
0,2 5,0010 221,15 1,7755
0,3 6,1564 275,49 2,1748
0,5 7,9970 364,47 2,8051
0.7 9,5163 440,34 3,3199
1,0 11,4326 535,45 3,9679
1,5 14,0912 668,38 4,8619
2,0 16,3294 779,34 5,6128
Таблица 86
Сравнение наблюдаемых коэффициентов активности хлористого
натрия с коэффициентами активности, вычисленными
по уравнению (20а)
Онабл.— 7 выч. )’
t, °C т 0,1 0,2 , 0.5 1,0 1,5 2,0 i
0 +1 -2 -2 -1 — 2 -2
•10 +1 -1 -2 0 -1 0
20 0 — 2 — 2 -2 -3 -1
30 0 — 2 -2 -3 -4 -1
40 0 — 2 — 2 — 4 — 4 -4
60 0 -1 0 0 -2 -2
80 +1 + 2 + 3 + 1 + 1 0
100 +2 -4-3 + 4- + 1 +4 +2
358
ГЛАВА XII
' Таблица 87
Влияние давления на коэффициенты активности электролитов при 25°
II Cl КС1 NaCl NaOH
р Концентрация, с
0,1 0,5 • 1.0 2,0 1 1 1
1 0,797 0,757 0,807 0,991 0,605 0,6545 0,6775
100 0,7975 0,759 0,809 0,995 . 0,609 0,659 0,686
1000!) 0,803 0,771 0,828 1,0275
10002) 0,802 0,768 0,829 1,021 0,637 0,687 0,745
(3/2) Sv 1,25 3,49 3,23 6,27
(3/2) Sk 10* 4,5 • 18,6 17,1 31,3
1) В уравнении (30) опущен член, содержащий (Р — 1)2.
2) Уравнение (30) использовано полностью.
В двух последних строчках таблицы приведены значения (3/2)5у и (3/2)Sg
по данным табл. 49 и 52.
Коэффициент активности хлористоводородной кислоты мало меняется
при изменении давления. Даже в 2 М растворе изменение давления на
1000 атм изменяет коэффициент активности у± всего лишь на 3%. В случае
других электролитов влияние давления несколько больше. Наибольшее
изменение (10%) было обнаружено в случае коэффициента активности
гидроокиси натрия.
§ 5. Общий обзор коэффициентов активности 1,1-электролитов
в связи с теорией Дебая и Гюккеля
а) Средние расстояния сближения ионов а. Нами приводились много-
численные примеры, иллюстрирующие совпадение наблюдаемых результатов
с расчетными данными, соответствующими предельным законам теории
междуионного притяжения. Кроме того, уравнения, вытекающие из этой
теории, часто применялись нами для экстраполяции. Однако предельные
законы справедливы только в случае бесконечно разбавленных растворов
п, строго говоря, неприменимы к реальным ионным растворам. Причины
отклонений от предельных законов представляют значительный интерес,
так же как и различные теоретические толкования свойств электролитов
при умеренных и высоких концентрациях растворов.
Дебай и Гюккель понимали, что при вычислении кулоновских сил
должны быть учтены конечные размеры ионов, и ввели параметр а, опре-
деляемый как «среднее расстояние сближения ионов». Таким образом, область
применимости предельного закона для коэффициентов активности расши-
ряется и вводятся уравнения (34) и (35), согласно которым
ig/± =---------.. — • (31)
1 + 35,51 а (ОТГ 12 V Г
Позднее Гюккель [43а] дополнил эту теорию предположением о линейной
зависимости диэлектрической постоянной среды от концентрации ионов.
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
359
Введя эту-зависимость, Гюккель получил уравнение, аналогичное по форме
уравнению (32) гл. X:
S’/о /Г
1g /± = - -------t/ z_ + Вс. (32)
1 + 35,57 a (DT)-1!2 /Г
Это уравнение часто применялось нами для экстраполяции данных, получен-
ных путем измерений электродвижущих сил. Согласно теории Гюккеля,
член Вс учитывает влияние изменения диэлектрической постоянной при
изменении концентрации соли. Понижение диэлектрической постоянной при
добавлении ионов обусловливает «высаливание ионов» и приводит к увели-
чению коэффициента активности. Это действие соответствует появлению
сил отталкивания между ионами и противоположно по знаку влиянию
междуионного притяжения, которое выражается первым членом в правой
части уравнения (32). Наоборот, если при увеличении концентрации соли
диэлектрическая постоянная возрастает, то ионы «всаливаются», член Вс
является отрицательным и коэффициент активности уменьшается. Эти
явления соответствуют солевым эффектам, которые, согласно теории Дебая
и Мак-Олея (гл. III, § 10), прямо пропорциональны сумме обратных радиусов
ионов 2 Vfy- Хотя факт изменения диэлектрической постоянной при увели-
чении концентрации ионов и не подлежит сомнению, однако есть все
основания считать, что это влияние не является единственным существен-
ным фактором в случае концентрированных растворов. Поэтому данное
уравнение следует рассматривать в основном как эмпирическое.
О
При определении параметров а и В. из экспериментальных данных
оказалось, что они различны для разных электролитов. Полученные таким
образом значения а нельзя считать совершенно точными, хотя по порядку
величины эти значения и являются правильными. В табл. 88 приведены
О
значения а для некоторых электролитов, вычисленные с помощью различных
уравнений и для разных интервалов концентраций растворов. Во втором
столбце даны значения а [44], вычисленные по уравнению (31) без линейного
°
члена. В следующих трех столбцах содержатся значения а, полученные
Таблица 88
О
Среднее расстояние сближения ионов а
Электролиты Уравнение
(31) (32) (33) (48)
0,005-0,1 М 0,1-1 м 0,1-3 м 0,1-4 М 0,1-3 М
НС1 5,6 4,6 4,3 3,6 4,3
КС1 4,1 4,1 3,6 3,4 3,95 3,2
NaCl 4,4 4,4 4,0 3,6 4,2 3,7
+
путем применения уравнения (32) к экспериментальным данным для раз-
личных интервалов концентраций. В шестом столбце приведены значе-
О
ния а, вычисленные по уравнению
1g /± ----------------------- + Вс + D'c2, (33)
1 + 35,57 а (2)Т)—1/2 /Г
360
ГЛАВА XII
в которое входит добавочный член, содержащий с2. Это уравнение применялось
(гл. XI, § 5) для вычисления коэффициента активности НС1. Последний столбец
содержит значения, вычисленные Ван-Риссельберге и Эйзенбергом [45] по
уравнению (48), которое по форме аналогично уравнению (33). Как видно,
различные методы вычисления а не дают одинаковых результатов, но по-
лученные значения всегда имеют правильный порядок величины и обычно
несколько превосходят соответствующие параметры кристаллических реше-
ток; кроме того, значения а для электролитов HCI, NaCl и КС1 изменяются
в одном и том же порядке: «нс1 > «Naci > «ксь каким бы путем эти зна-
чения ни были получены.
В указанных случаях простое уравнение без линейного члена и с по-
стоянным значением а хорошо согласуется с экспериментальными данными
в широком интервале концентраций (0,005 — 0,1 М), однако такое согласие
наблюдается не всегда. Гронволл, Ла-Мер и Сэндвед [46] показали, что
для азотнокислого калия значение а, вычисленное по этому уравнению,
изменяется с концентрацией раствора и что постоянное значение а получается
лишь в том случае, если воспользоваться обобщенной теорией этих авторов.
Таблица 89
Параметры уравнений (32) и (33) при 25°
Электролиты Уравнение (32), приме- нимо до 1 M Уравнение (33), применимо до 4 М (Г+ + Г—)
О а В О а В D' максим, откл.
HJ 5,0 0,197 5,5 0,1725 0,0128 0,007
НВг 4,4 0,165
НС1 4,3 0,133 4,3 0,1292 0,00615 0,003
LiJ 5,05 0,165 5,0 0,155 0,0113 0,015 2,77
LiBr 4,3 0,130 4,3 0,126 0,0099 0,002 2,56
Li Cl 4,25 0,121 4,25 0,111 0,0070 0,002 2,41
NaJ 4,2 0,100 4,2 0,090 0,0058 0,008 3,13
NaBr 4,1 0,0687 4,2 0,0590 0,0064 0,002 2,91
NaCl 4,0 0,0521 4,2 0,0410 0,0053 0,001 2,76
KJ 3,94 0,0462 3,95 0,0440 0,0016 0,002 3,50
KBr 3,84 0,0282 3,85 0,0247 0,0035 0,001 3,28
KCI 3,8 0,0202 3,85 0,0187 0,0034 0,001 3,14
RbCl 3,6 0,010 3,2 0,0235 0,0023 0,003 3,29
RbBr 3,55 0,010 3,2 0,0193 0,0021 0,005 3,43
RbJ 3,5 0,0085 3,2 0,0162 0,0031 0,004 3,65
CsCl 3,0 0 2,5 0,0229 0,0024 0,006 3,46
CsBr 2,93 0 2,5 0,0162 0,0033 0,008 3,61
CsJ 2,87 0 2,5 0,0140 0 0,006 3,82
NaOH 3,24 0,0460
KOH 3,7 0,1294
В табл. 89 приведены величины параметров а и В уравнения (32)
и величины параметров а, В и D' уравнения (33) для 1,1-галогенидов,
галоидоводородных кислот и гидроокисей калия и натрия при 25°. В § 1
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
361
было подчеркнуто, что с помощью уравнения (32) могут быть получены
хорошие результаты при экстраполяции лишь в том случае, если экспери-
ментальные данные соответствуют концентрациям, не превышающим 1 М.
Коэффициенты активности в растворах с концентрацией 0,1 М, вычислен-
ные с применением параметров табл. 89, тождественны с коэффициентами
активности, приведенными в табл. 146. В интервале концентраций 0,1 — 1 М
наблюдаемые коэффициенты активности и вычисленные по уравнению (32)
хорошо совпадают друг с другом (с точностью ± 0,002 единицы у±) за
исключением трех случаев. Для йодистого лития наблюдается расхождение
между опытными и теоретическими значениями, для бромистого лития
и хлористого цезия экспери-
ментальные и расчетные дан-
ные совпадают в указанных
выше пределах точности вплоть
до концентраций 0,7 М.
Средние столбцы табл. 89
содержат величины парамет-
ров а, В и D' уравнения (33).
В предпоследнем столбце при-
ведены максимальные отклоне-
ния наблюдаемых результатов
(табл. 146) от значений, вы- _2
численных по этому уравнению,
которое справедливо вплоть до
концентраций 4 М. Как видно
из таблицы, совпадение опыт-
ных и расчетных значений почти Р и
во всех случаях является хоро-
шим. Особенно хорошее совпа-
дение наблюдается в тех случаях, когда результаты являются наиболее
надежными. Наиболее серьезные расхождения были обнаружены для рас-
творов иодистых лития и натрия, которые требуют дальнейшего эксперимен-
тального изучения [42].
Последний столбец табл. 89 содержит сумму кристаллографических
1 о
радиусов солей. Порядок величины а является во всех случаях правильным.
О
Для большинства солей а больше, чем сумма кристаллографических ради-
усов, причем это можно объяснить влиянием гидратации ионов. Для солей
цезия а меньше, чем сумма (г,-|-г_). Так как имеются некоторые сомнения
в правильности определения абсолютной величины а, то было бы прежде-
временным делать на основании этого результата какие-либо выводы.
С точки зрения теории ассоциации ионов Бьеррума (гл. III, § 7) следует,
что все эти электролиты являются сильными, так как в большинстве случаев
значения а превосходят q (равное 3,5), а это указывает на очень малую
вероятность ассоциации ионов. Образование ионных пар может иметь место
в некоторой степени в случае солей цезия, но даже в этих растворах оно
является маловероятным. о
Как видно из табл. 89, значения а и В, вычисленные по уравнению (32),
изменяются для разных электролитов в одинаковой последовательности,
и возможно, что между ними может быть установлена некоторая зависимость.
Если это так, то все результаты вплоть до концентраций 1 М могут быть
выражены уравнением с одним параметром и им должно соответствовать
О
одно семейство кривых. На рис. 94 изображена зависимость 1g В от 1g с.
362
ГЛАВА XII
Как видно, в тех пределах, в которых а и В могут быть определены,
наблюдается линейная зависимость между логарифмами этих величин.
Уравнение этой прямой имеет следующий вид:
1g В = 14 1g а -9,75. (34)
О
Следовательно, В пропорционально а в четырнадцатой степени, и если мы
О
пожелаем вычислить В из значений а, то нужно учитывать, что при такой
О
зависимости В будет заметно меняться даже при малом изменении а. Об-
ратная операция — вычисление а из значений В — вполне возможна. Точная
О
численная величина показателя степени а не имеет для нас существенного
значения. Эта величина свидетельствует только о том, что силы отталкивания
О
между ионами зависят от величины а, взятой в определенной степени, и что
двух уравнений, как, например (32) и (34), достаточно для того, чтобы
получить одно семейство кривых для галоидных солей щелочных металлов
и для галоидоводородных кислот в интервале концентраций 0,1 — 1 М1.
Любая зависимость между а и В, полученная для интервала 0,1— 4 М
с помощью уравнения (33), осложняется наличием третьего члена D'c2 * * * *.
Хотя постоянная D', невидимому, не связана с В, все же сомнительно,
чтобы.для такого уравнения с тремя параметрами удалось найти какое-
нибудь количественное соотношение между а и В эмпирическим путем.
Тем не менее тенденция а и В к одновременному возрастанию еще сохра-
няется. В § 8 мы вернемся к анализу соотношения, аналогичного по фор-
ме уравнению (33), причем это соотношение имеет под собой некоторую
теоретическую основу.
Если изобразить величину В в виде функции от 211/7"», т0 П0ЛУча'
ются отдельные кривые для хлористых, бромистых и иодистых солей.
Хотя этот результат и не подтверждает уравнение Дебая и Мак-Олея
[уравнение (106) гл. III], он свидетельствует о том, что коэффициенты
активности в концентрированных растворах могут в значительной степени
определяться величинами ионных радиусов. К более подробному рассмо-
трению этой возможности мы вернемся в § 7.
б) Специфические особенности 1,1-электролитов. На рис. 95 изобра-
?кена зависимость у от ]/т для хлористых, бромистых и иодистых солей
щелочных металлов вплоть до высоких концентраций. Расхождение кри-
вых оказывается весьма большим, причем при увеличении концентраций
наблюдаются резко выраженные индивидуальные различия, обусловленные
1 Стонхилл и Берри [47] выразили сомнение в том, что любое соотношение типа
уравнения (34) сохранится, если изменится от 0,5065 до 0,5103.. Приведенный ими
расчет является ошибочным, так как избранный ими интервал концентраций 0,01—0,10 М
непригоден для определения В, поскольку величина линейного члена меньше, чем ошибка
опыта при 0,01 М, и того же порядка, что ошибка опыта при 0,10 М. Если бы эти авторы
выбрали интервал концентраций 0,1—1,0 М, как это было сделано Харнедом и Оуэном,
О
то вышеуказанное изменение величины должно было бы привести к изменению а
на 0,138 и В на —0,0034, вместо 0,63 и —0,0528, как указывается в их примере. При рас-
четах такого рода, варьируя значения для ряда 1,1-галогенидов, Харнед и Оуэн
О
всегда находили, что В является весьма плавной, быстро возрастающей функцией от а.
Любое, выражающее эту функцию уравнение будет представлять результаты в виде
единственного семейства кривых.
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
363
большими различиями в значениях величины В. Так как все кривые
лежат выше кривой, соответствующей предельному закону, то нет основа-
ний допускать наличие ассоциации ионов. В расположении кривых имеется
определенная закономерность за исключением одного случая. Значения
коэффициентов активности для катионов располагаются в следующей по-
следовательности: литий, натрий, калий, рубидий, цезий. Для анионов
соблюдается последовательность: иодид, бромид и хлорид для солей лития,
натрия, калия и обратная последовательность для солей рубидия и цезия.
При добавлении менее точных результатов для фтористых солей, для кото-
рых последовательность для катионов оказывается обратной, эти законо-
мерности осложняются.
На рис. 96 представлен наиболее яркий пример обратной последова-
тельности для катионов, наблюдающейся в случае гидроокисей, когда
кривые для разных катионов сильно отличаются друг от друга. Наиболь-
шим коэффициентом активности обладает гидроокись цезия, наименьшим —
гидроокись лития. Эта последовательность прямо противоположна той,
которая наблюдается для хлоридов, бромидов и иодидов. Такой эффект
можно частично объяснить взаимодействием ионов и образованием ионных
пар. Согласно теории ассоциации ионов Бьеррума (гл. III, § 7), 1,1-элек-
тролиты считаются сильными, если расстояние сближения ионов больше
или равно 3,5 А. Приведенные на рис. 96 значения у согласуются
со средними расстояниями сближения ионов: 3; 3,5; 4 и 4,2 А соответ-
ственно для гидроокисей лития, натрия, калия и цезия. Согласно теории
Бьеррума, все указанные гидроокиси являются сильными электролитами,
хотя в случае гидроокиси лития может происходитх некоторая ассоциация
ионов. Эти результаты согласуются с выводами, сделанными на основании
измерений электропроводности. Примерно такие же результаты наблю-
даются и в случае ацетатов, хотя для ацетатов расхождение кривых
не столь велико.
364
ГЛАВА XII
Мы полагаем, что «нормальная последовательность» для галогенидов
и обратная — для гидроокисей, ацетатов и др. объясняется взаимодействием
ионов с растворителем. Поскольку ионы меныпего размера, например
ионы лития, обладают более сильным полем, они должны сильнее взаимо-
действовать с диполями растворителя. Образование оболочки из молекул
воды вокруг иона должно приводить к увеличению а по сравнению
с суммой кристаллографических радиусов. В случае галогенидов величина В
Рис. 96. Средние коэффициенты активности 1,1-гид-
роокисей при 25°.
[уравнение (32)] меняется в той же последовательности, что и вели-
чина а, и коэффициенты активности должны располагаться в том же
порядке. Поэтому гидратация, которая уменьшается в ряду Li > Na > К >
> Rb > Cs, является причиной той последовательности коэффициентов
активности, которая наблюдается для галогенидов. Однако этот вид взаи-
модействия между ионами и растворителем может приводить также
к «локализованному гидролизу» в результате реакции с анионами, которые
являются акцепторами протонов. Протоны молеку'л воды выталкиваются
из гидратной оболочки катионов и стремятся образовать связь с акцепто-
рами протонов, вроде ионов ацетата или гидроксила. Этот процесс можно
представить в виде схемы
М+4-Н2О М+...ОН'...Н+,
где точками изображены связи, обусловленные силами взаимодействия
между молекулами растворителя и ионами. Взаимодействие с акцептором
протонов можно представить уравнением
М+. . .ОНА . .Н++ A- М+...ОН“. ..Н+...А“,
причем можно считать, что протон колеблется между крайними положе-
ниями от гидроксильной группы до акцептора протонов. Суммируя .обе
реакции, получаем
\ М++Н2О + А’ л±М4...01Г...Н+...А".
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
365
Эта реакция аналогична процессу образования ионных пар
М+ +А" М+.. . + .. .А“
в том смысле, что в обоих случаях происходит уменьшение числа свобод-
ных ионов в растворе. При этом механизме коэффициенты активности
должны быть меньше, чем значения, вычисленные на основании предполо-
жения о полной диссоциации, причем это понижение будет тем больше,
чем меньше радиус катиона. Таким образом, для соединений, содержащих
гидроокиси, ацетаты и т. п., которые способны связывать протоны, после-
довательность коэффициентов активности будет следующая: Gs > Rb > К >
> Na > Li. В некоторых из этих случаев может происходить обычный
гидролиз, однако он не приводит к изменению числа свободных ионов.
§ 6. Постулаты Бренстеда о специфическом взаимодействии ионов
и вычисление осмотических коэффициентов и коэффициентов
активности для низких концентраций по методу Гуггенгейма
Специфические особенности различных электролитов можно подробно
проиллюстрировать на примерах коэффициентов активности, относительных
теплосодержания и теплоемкости. Теория Дебая и Гюккеля не в состоянии
объяснить различия в свойствах разных электролитов. Теория образования
ионных пар Бьеррума, дополненная теорией образования ионных тройников
Фуосса и Крауса (гл. VII), оказалась очень полезной для понимания
свойств ионных растворов в средах с низкой диэлектрической постоянной.
Однако эта теория не вносит почти ничего нового в вопрос о характере
взаимодействия ионов сильных электролитов в средах с высокой диэлек-
трической постоянной.
Существенный вклад в теорию, способствующий выяснению причин
специфического влияния различных ионов на коэффициенты активности
других электролитов, был сделан Бренстедом [48] до появления теории
Дебая и Гюккеля. На основе его исследований растворимости высоковалент-
ных соединений кобальта в растворах различных солей была создана так
называемая «теория специфического взаимодействия ионов». Основной по-
стулат теории Бренстеда сводится к тому, что «в разбавленных растворах
солей с постоянной общей концентрацией различные ионы подвергаются
одинаковому воздействию со стороны ионов того же знака». Специфиче-
ское электростатическое взаимодействие между ионами в разбавленных
растворах возникает лишь тогда, когда ионы противоположного заряда
приближаются друг к другу настолько, что может проявиться это специ-
фическое влияние. Кроме эффекта взаимодействия ионов, Бренстед учиты-
вал также явление «высаливания», т. е. влияние растворителя. Так как
большинство исследований, подтверждающих теорию специфического взаимо-
действия ионов, было выполнено со смесями двух электролитов, то эта
теория будет подробно рассмотрена в гл. XIV, посвященной свойствам
таких смесей.
Гуггенгейм [49] объединил теорию Дебая и Гюккеля с упрощенной
теорией специфического взаимодействия для объяснения свойств 1,1-элек-
тролитов в интервале концентраций 0 — 0,1 М. Он выбрал стандартный
электролит, обладающий свойствами «идеального электролита Дебая и Гюк-
келя», т. е. такой электролит, рациональный коэффициент активности
которого определяется уравнением (34) гл. III
1g/± =
& (/) /£
1+А / г
366
ГЛАВА XII
Этот электролит подчиняется закону Кулона при любых расстояниях между
ионами, и его ионы представляют собой несжимаемые и неполяризуемые
сферы. Для упрощения вычислений допустим далее, что среднее расстоя-
ние сближения ионов равно 3,05 А при 25°; тогда
1g у------.. (35>
6/± 1+ /Г/2 ! К '
а для 1,1-электролита
<зв>
Гуггенгейм добавил к этому уравнению линейный член для учета всех спе-
цифических эффектов, характеризующих данный электролит. Таким обра-
зом он получил очень простое уравнение
<3,)
Член Хс учитывает понижение диэлектрической постоянной (Гюккель),
образование ионных пар (Бьеррум), влияние размера ионов, поляризуемость
и т. д. Гуггенгейм применил это уравнение для вычисления осмотического
коэффициента и коэффициента активности по измерениям температур замер-
зания и электродвижущих сил при концентрациях ниже 0,1 М. Согласие
с данными по температурам замерзания лежало в пределах от ± 0,0002 до
± 0,0005°.
Это уравнение очень удобно для вычисления коэффициентов актив-
ности в разбавленных растворах благодаря его простоте. Поскольку при
данной температуре первый член справа имеет постоянное значение для
всех 1,1-электролитов, то его достаточно вычислить один раз для каждой
концентрации, и, таким образом, вычисление / сводится к определению
линейного члена Хс. В табл. 147 приведены определенные Гуггенгеймом
значения X для большого числа электролитов и / (X = 0) для стандартного
электролита для ряда концентраций. С помощью этой таблицы можно вычис-
лять/± вплоть до концентрации 0,1 М с точностью порядка 0,5%, чего
вполне достаточно для большинства практических целей.
С теоретической точки зрения, это приближение является слишком
упрощенным даже для данного интервала концентраций (< 0,1 М). При-
меняя уравнение (37) для коэффициентов активности двух электролитов
при одинаковой концентрации, получаем соотношение
1g 4= te?- = -7 с = с’ (38)
/2 72
где (Xj — Х2) и, следовательно, В12 не являются функциями с. Из теории
Дебая и Гюккеля такой результат может получиться лишь при условии,
что среднее расстояние сближения ионов для всех электролитов одинаково.
Конечно, подобный вывод был бы весьма неожиданным. Согласно теории
Дебая и Гюккеля, из уравнения (34) гл. III должно получиться следующее
соотношение:
(39)
Это уравнение не является линейным относительно с, а, напротив, из него
следует, что В12 меняется с изменением концентрации. Зависимость этой ве-
личины от концентрации для двух 1,1-электролитов со значениями а, рав-
ными соответственно 4,0 и 3,5, изображена графически на рис. 97, где
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
367
1 X
— lg(/i//3) представлено в виде функции от с, причем из рисунка видно,
что эта величина действительно зависит от с.
Браун и Мак-Иннес, а также Шидловский и Мак-Иннес получили
значения а, равные 5,6; 4,1 и 4,4 соответственно для хлористоводородной
кислоты, хлористых калия и натрия в том случае, когда они подставили
свои точные экспериментальные данные в уравнение Дебая и Гюккеля
(табл. 88); когда же они воспользовались этим выражением с линейным
членом [уравнение (32)], то получили значения 4,6; 3,7 и 4,0. Их опыт-
ные данные лежали в преде-
лах концентраций 0,005 —
0,1 М. Эти результаты не
согласуются с упрощенным
уравнением Гуггенгейма.
Разность (Хнс1—ХКс1), вычи-
сленная из эксперименталь-
ных значений 1g (уЫс1 / уКс1)>
меняется от 0,24 до 0,15.
В случае 1g (у Hcl/YNaC1) раз-
ность (Хнс1 — XNaC1) меняется
от 0,16 до 0,11 при измене-
нии концентрации от 0,005
до 0,1 М. На рис. 97 круж-
ками обозначены значения
разности (lNaC1-lKcl), полу-
Рис. 97. Теоретическая и экспериментальная за-
1
висимости — (lg(/l//2) от с.
Кривая отвечает теоретическим данным; кружки—экс-
периментальным данным; индексом 1 обозначен NaCl,
индексом 2 — КС1.
ченные из этих результатов. Хотя вычисленные значения а различаются
только на 0,3, все же видно, что величина разности (Хх — ),2) уменьшается
с увеличением концентрации.
Эти вычисления могут быть проверены только с помощью очень точ-
ных опытов. Мы полагаем, что результаты указанных исследований являются
достаточно точными и свидетельствуют о том, что метод Гуггенгейма
является приближенным и представляет ценность для упрощенных практи-
ческих вычислений х.
§ 7. Теория концентрированных растворов галогенидов '
щелочных металлов по Скэтчарду
В § 5 мы останавливались на обобщении первоначальной теории
Дебая и Гюккеля путем введения члена, учитывающего «высаливание»,
возникающее вследствие изменения диэлектрической постоянной среды при
увеличении концентрации ионов в растворе. Мы указывали также, что
хотя этот фактор и должен учитываться в теории концентрированных рас-
творов, однако он недостаточен для объяснения свойств этих растворов.
Действительно, согласно уравнению Гюккеля, получается, что в растворах
соляной кислоты при концентрации около 4 М диэлектрическая постоянная
должна равняться нулю и иметь отрицательное значение при более высо-
ких концентрациях [7, 50].
Наиболее полное теоретическое исследование концентрированных рас-
творов сильных электролитов, в особенности галогенидов щелочных метал-
1 Дополнительные доказательства этого вывода приведены в статье Робинзона
и Харнеда [42].
368
глава XII
лов, было выполнено Скэтчардом [51а]1. Ионы типа благородных газов
можно рассматривать как простейший случай, наиболее удобный для чис-
ленной обработки. Уравнение Скэтчарда для коэффициента активности
имеет следующий вид:
i„Y±=. _ i„(1 + ,, Л</JV0) + [-^ +
ЧгУл У) +-у (тг+
(40)
Дополнительные члены Гронволла, Ла-Мера и Сэндведа в это уравнение
не включены. В этом уравнении через 7VS обозначено число молей элек-
тролита, через N0 — число молей-растворителя, через V4 и У2 — молярные
объемы ионов и через Fs — молярный объем электролита. Параметры Ь±
и Ь2 представляют собой эффективные значения радиусов ионов для процесса
«высаливания». Остальные обозначения определяются следующими уравне-
ниями, где а — сумма эффективных ионных радиусов для процессов столк-
новений ионов (среднее расстояние сближения ионов), а,2, а1п и а20 —плот-
ности энергии их взаимного притяжения,70 — молярный объем растворителя:
vs= *1 + v2,
Vs = ViVi + v2F2,
D = D0V0N0/V,
2 _ 8nN'2s.2z1zi'tjlNs _
v' ГббоятяГ- юооягД, ’
X = j—-------Y,
1 -p xa
Y = Г 1 4- v.a — —------2 In (i 4- xa) 1 /x2a3,
L ' - 1 + xa v 1 7 J ’
A = 2v1v2V1V2 (2a 12 — a10 — a2o)/V
He вдаваясь в подробное обсуждение этих сложных выражений, мы
только укажем на значение каждого члена.
Уравнение (40) представляет собой обобщенное уравнение теории Дебая
и Гюккеля. Первый член в правой части — обычное выражение для пере-
хода от рационального коэффициента активности /± к стехиометрическому
коэффициенту у±. Первые два члена в скобках совпадают по существу
с соответствующими членами уравнения Дебая и Гюккеля, выражающими
взаимодействие зарядов, за исключением того, что х определяется не для
неизменного значения диэлектрической постоянной Ло, а для ее перемен-
ного значения D. В то время как Гюккель принимал, что диэлектрическая
постоянная линейно зависит от молярности электролита, Скэтчард восполь-
зовался соотношением Z) = Л0У0У0/У, полученным Вайменом [52] для рас-
творов неэлектролитов. Влияние этого различия на величину х оказывается
незначительным; оно пропорционально числу ионов в единице объема рас-
творителя. Третий член в скобках отражает «высаливающее» действие, т. е.
взаимодействие ионов с молекулами, и соответствует выражению Дебая
1 Представления, на основании которых выведено уравнение (40), изложены Скэт-
чардом в книге Кона и Эдсэла [516].
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
369
и Мак-Олея для этого эффекта. Эти члены аналогичны тем, которые приме-
няются в теории Гюккеля. Последний член вне скобок введен для учета
взаимодействия между молекулами неэлектролита, т. е. отклонения свойств
растворов истинных неэлектролитов от законов идеальных растворов.
Для применения этой теории необходимо знать температуру, молярный
объем и диэлектрическую постоянную растворителя, валентность, молярный
объем раствора, эффективные радиусы всех ионов для процесса «высалива-
ния», эффективные расстояния сближения ионов а и коэффициент взаимо-
действия между молекулами хА. Предполагается, что ионы щелочных гало-
генидов представляют собой сферы. В этом случае Ь = г иа = г14-г2- В ка-
честве радиусов ионов были приняты значения, найденные Паулингом [53]1
из кристаллографических данных. Величины объемов ионов в растворе были
взяты несколько меньшими, чем 4w3/3. Постоянная А принималась одина-
ковой для всех галогенидов. Теория взаимодействия ионов Бренстеда, ко-
торая исключает взаимодействие между ионами одинакового знака на близких
расстояниях, была тоже использована для упрощения некоторых подстано-
вок . Два параметра были определены эмпирически с помощью осмотических
коэффициентов, а именно: отношение объема иона в растворе к истинному
объему иона и коэффициент А. Первый из этих параметров совпал с тео-
ретическим значением, в то время как для второго параметра получилось
лишь приближенное согласие.
Вычисленные на основании этой теории осмотические коэффициенты
щелочных галогенидов в общем хорошо согласуются с данными, получен-
ными Робинзоном и Синклером, а также Робинзоном. Наиболее важный
результат теории Скэтчарда состоит в том, что свойства ионов типа благо-
родных газов характеризуются их валентностями и размерами, которые
определяются из кристаллографических данных. Как последовательность,
так и величины результатов, полученных для иодидов, бромидов и хло-
ридов, хорошо согласуются с теоретическими данными. Кроме того, эта
теория позволила предсказать обратную последовательность для влияния
катионов в случае фторидов на основании данных о температурах замерзания.
Исследования Скэтчарда представляют собой наиболее успешную по-
пытку теоретического объяснения свойств концентрированных растворов
путем дополнения теории Дебая и Гюккеля. Скэтчард рассматривает от-
дельно взаимодействие между ионами, между ионами и молекулами, а
также между молекулами и обнаруживает зависимость между некоторыми
термодинамическими свойствами электролитов и ионными радиусами, по-
лученными из кристаллографических данных.
§ 8. Теория концентрированных растворов электролитов,
учитывающая поправку Ван-дер-Ваальса на собственный объем
растворенных частиц
Как было показано (§5), коэффициенты активности многих 1,1-электро-
литрв для концентраций 0 — 4 М можно вычислять с большой точностью
пэ уравнению
-|/ р
---------—------------f-5c-|-Z)'c2. мп
1 + 35,57а(/)Г)"1/2 /Г k 7
Числитель первого члена в правой части уравнения учитывает действие
кулоновских сил между ионами, принятыми за точечные заряды. Знамена-
1 См. табл. 6.
24 г. Харнед, Б. Оуэи
370
ГЛАВА XII
тель учитывает эффект, обусловленный конечными размерами ионов и огра-
ничивающий действие кулоновских сил. Члены Вс и Z>'c1 2 положительны
и противоположны по знаку первому члену, они учитывают влияние сил
отталкивания, которые действуют на близких расстояниях и обусловли-
вают сильные эффекты в очень концентрированных растворах.
. На основании анализа теории концентрированных растворов Онзагер
[54] высказал предположение, что эти члены, учитывающие влияние сил
отталкивания, можно выразить в виде обычной поправки на собственный
объем молекул согласно Ван-дер-Ваальсу.
' <42>
где а — «среднее расстояние сближения ионов», V — общий объем. Это рав-
ноценно допущению, что силы отталкивания быстро уменьшаются с уве-
личением расстояния и что характер этих сил определяется наличием
«резкого изменения величины потенциальной энергии на некотором рассто-
янии (г = <?)». Так как параметр а в члене, выражающем поправку на
объем [уравнение (42)], тот же самый, что и в первом члене в правой
части уравнения (41), то эту теорию можно рассматривать как попытку
[упомянутую в § 5] выразить термодинамические свойства электролитов с
помощью уравнения с одним параметром.
Эта теория была разработана более подробно Ван-РиссельбЬрге и Эйзен-
бергом [55а]1 . Проблема подбора соответствующей функции, которая выра-
жает исключение из общего объема некоторых частей пространства,
в случае молекул двух видов 2VX и N2 с диаметрами dA и d2, является очень
трудной. Наиболее успешное решение этой проблемы принадлежит
Урселлу [56а] 2, который развил теорию для общего случая любого числа
молекул и получил полное решение для частного случая двух видов мо-
лекул. Его результаты можно значительно упростить, допуская, что
d1 — d2 = d12 — d+=d_ = d± — a
и что
N1 = N+ = y+N; N2 = N_ = v-N,
v+ + v_ —v,
Ni = (v+ + v_)N = vN,
где через d+, d_ и d± обозначены диаметры катиона, аниона и средний
ионный диаметр электролита. Отметим, что d± равно среднему расстоянию
сближения ионов а и представляет собой не средний диаметр, получаемый
из кристаллографических данных, а некоторый параметр. Для бинарного
электролита выражение для исключаемого объема (In У по Ван-Риссель-
берге) имеет простой вид
1g — 2 у v 64 V V ’ ^4^
где V — объем и ц = утса3. В результате учета исключаемого объема общая
работоспособность раствора меняется на величину А^:
Л* = ^Др?4-У0Др§= -kTlnY-^NikT [1^ ^ + й(Ф)2у2] • <44>
1 Поправку к теории см. [556].
2 См. [566].
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
371
Если, принимая во внимание малую сжимаемость ионных растворов, заме-
нить Лад на Fw, то ван-дер-ваальсовская составляющая коэффициента актив-
ности In/* может быть получена дифференцированием по TV; при Постоян-
ных Р, Т и No. Таким образом,
, ,* _ Ди _ 1 dFw _
7± кТ кТ dNj
Ni , . 15/jVA2 Г 1 /А А3 , 10/ТУА3 ,1 dV ,/сч
V У + )v2 L 2 ) V J dNt ’
Подобным же образом коэффициент активности растворителя выражается
уравнением
. dF* av г 1 /ту,-у . ю/туа3 ,1 av ,,,,х
— ln ft — dV dN0 ~ L 2 С v ) v + 64 С V J v J 5TV0 ' (4b)
Выражая а в ангстремах, переходя к молярным концентрациям и пренебре-
гая последним членом в скобках, получаем из уравнения (45) следующее
выражение для бинарных электролитов:
lg fl = 2,2063 • 10-3 а3с + 2,6269 • 10~6 авс2. (47)
Это теоретическое уравнение соответствует членам Вс -f-D'c2 в уравнении (41).
Вряд ли можно ожидать, чтобы это упрощенное представление было
достаточным для точного истолкования экспериментальных результатов.
В случае соляной кислоты было обнаружено, что экспериментальные
результаты при 25° можно выразить с точностью до ±0,001 в еди-
ницах у, если а, В и D' равны соответственно 4,3; 0,1292 и 0,00615.
Подставляя в теоретическое уравнение (47) значение а = 4,3, получаем
В = 0,1754 и D' = 0,01661. Таким образом, поправка на объем по аналогии
с уравнением Ван-дер-Ваальса более чем достаточна для учета составля-
ющей, обусловленной всеми силами отталкивания между ионами. В коли-
чественном отношении полученный результат является неудовлетворитель-
ным. Аналогичные результаты были получены для других 1,1-электролитов.
Было обнаружено (см. табл. 89), что для выражения экспериментальных
результатов вплоть до концентрации для бромистоводородной кислоты,
хлористых натрия и калия и бромистого натрия надо принять значения а
равными соответственно 4,4; 4,1; 4,0 и 3,8, а значения В равными 0,165;
0,059; 0,041 и 0,019. Теоретические значения В равны соответственно 0,1879;
0,1521; 0,1412 и 0,1259; таким образом, теоретические значения оказыва-
ются сильно завышенными. Это дает возможность предположить, что одно-
временный учет как поправки на собственный объем, так и ассоциации
ионов, которая, по всей вероятности, имеет место в растворах с высокими
концентрациями, должен дать более точное приближение к реальным
условиям.
В § 5 было упомянуто о том, что можно получить уравнение с одним
параметром для вычисления коэффициентов активности 1,1-электролитов
при данной температуре. Ван-Риссельберге и Эйзенберг показали, что
эмпирическое уравнение
lg у--------0±059]Л— 1 Ю31 . 10-3аЗс +8,7564 • 10“7 а6с2,
i + <48>
+ 3,042 v
похожее по форме на уравнение (41), неплохо решает эту задачу в интер-
вале концентраций 0,1 —4 М. Применение уравнения (48) к 1,1-галогенидам
о
дает значения а, которые не очень отличаются от значении, полученных по
24*
372
ГЛАВА XII
уравнению (32) для интервала концентраций 0,05 — 3 М [57]. Эти значения
а ниже, чем вычисленные на основании самых лучших опытных данных
для концентраций раствора 0,005—1 М (см. табл. 89); при использовании
этих значений а для вычисления у , при концентрации 0,1 М получаются
несколько заниженные величины коэффициента активности. Тем не менее,
учитывая сложность поставленной задачи, следует считать, что уравнение
с одним параметром дает очень хорошее приближение.
§ 9. Связь термодинамических свойств ионов с их размерами
и структурой
В результате обширных исследований по определению температур за-
мерзания, выполненных Скэтчардом и Прентисом (гл. IX), и изопиести-
ческих измерений упругости пара, проведенных Робинзоном, были полу-
чены достаточно подробные сведения о термодинамических коэффициентах
многих 1,1-электролитов, позволяющие произвести их примерную классифи-
кацию с точки зрения структуры ионов1. Для удобства можно принять
следующую схему:
Группа 1. Катионы типа благородных газов со следующими анио-
нами:
1) анионы типа благородных газов;
2) анионы, являющиеся акцепторами протонов:
а) малые анионы F-, ОН~ и т. д., »
б) несимметричные анионы НСОО-, СН3СОО- и т. д.;
3) многоатомные анионы типа NOjf, СЮз", СЮ^" и т. д.
Группа 2. Многоатомные катионы Н3О+, NH* и ионы аммония,
в которых атомы водорода замещены органическими радикалами.
Первый класс первой группы был рассмотрен в предыдущих параграфах,
причем оказалось, что индивидуальные свойства очень сильно зависят. от
размера ионов. Взаимодействия специфического характера, которые вызы-
вают обращение последовательности коэффициентов активности для катио-
нов в присутствии ионов фтора и гидроксила, согласно Скэтчарду и Прен-
тису, также обусловлены очень малыми размерами этих ионов. Хотя точ-
ные экспериментальные данные для фторидов почти совершенно отсутствуют,
все же можно полагать, что ион фтора является акцептором протонов, так
как осмотический коэффициент 1 М раствора фтористоводородной кислоты
почти такой же, как осмотический коэффициент муравьиной кислоты при
той же концентрации. Если это допущецие правильно, то механизм взаи-
модействия катионов с растворителем (водой), рассмотренный в § 5, может
иметь существенное значение. В анионе большого размера, вроде ацетат-
иона, заряд расположен, невидимому, на значительном расстоянии от центра
иона. С этим обстоятельством может быть связана способность этих анио-
нов соединяться с протонами и явление обращения последовательности
коэффициентов активности для различных катионов.
Свойства солей с многоатомными анионами, которые не являются акцеп-
торами протонов (соответствующие кислоты — сильные электролиты), объ-
яснить труднее. На рис. 98 изображены, кривые для коэффициентов актив-
ности нитратов щелочных металлов наряду с соответствующими кривыми
для хлоридов, причем такие же кривые получаются для хлоратов и пер-
1 Аналогичные попытки были сделаны Харнедом [9в] в то время, когда имелось
меньше экспериментальных данных. Настоящее изложение воспроизводит обзор, со-
ставленный Скэтчардом и Прентисом [58].
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
373
хлоратов. Хотя анион азотной кислоты представляет собой сложный ион,
порядок расположения кривых относительно катионов такой же, как для
хлоридов, однако разброс кривых для нитратов несколько больше, чем для
хлоридов. Скэтчард и Прентис указали, что поскольку последовательность
по катионам является обратной по сравнению со случаем ионов ацетата,
то эти эффекты нельзя объяснить несимметричным распределением заряда
в ионе. Кроме того, касаясь вопроса о влиянии дипольных моментов, Скэт-
чард и Прентис отмечают: «Тот факт, что поведение ионов галогенидов
отличается от поведения хлорат-ионов, которые должны обладать диполем,
Рис. 98. Средние коэффициенты активности 1,1-хлори-
дов и нитратов при 25°.
не больше, чем от поведения нитратов и перхлоратов, не имеющих дипо-
лей, если только структура ионов в растворе не очень сильно отличается
от их структуры в кристаллическом состоянии, позволяет считать, что
дипольный момент не является важным фактором».
В настоящее время нет достаточного количества экспериментальных
данных о свойствах многоатомных катионов для того, чтобы объяснить их
поведение. Ион гидроксония в растворах сильных кислот ведет себя почти
так же, как ион лития, хотя их размеры, вероятно, весьма различны.
Симметричный ион аммония ведет себя в концентрированных растворах
так же, как меньшие по размеру ионы натрия и калия. Скэтчард и Прен-
тис [59] нашли, что галоидные соли аммония и нитраты обнаруживают
специфические особенности в разбавленных растворах (<0,04 М). Все они
обнаруживают эффект, аналогичный эффекту ассоциации ионов, который
исчезает при более высоких концентрациях в результате действия факторов,
способствующих увеличению коэффициента активности. Ни у одного из
других 1,1-электролитов это специфическое взаимодействие ионов не наблю-
далось. Удовлетворительного объяснения этого явления еще не найдено.
Температуры замерзания растворов алкилзамещенных фторидов и хло-
ридов аммония были точно определены Эбертом и Ланге [60], а также
Ланге [61]. Полученные ими значения осмотических коэффициентов этих
374
ГЛАВА XII
сильных электролитов изображены графически на рис. 99. Пунктирная
линия соответствует предельному закону Дебая и Гюккеля. Все хлориды
являются сильными электролитами, в
киламмониевых оснований при температуре
-замерзания.
I-N(C4H9)4 Cl; 2-N (С3Н,)4 Cl; 3-N (CHS)4 С1;
*-N(CH3)4 F; 5-N (СНз)з (С8Нц) F; 6-N(C3H7)4 F;
7—К(СаНэ)4 F; П. 3.—теоретический предельный
закон.
то время как ионы фтора, невиди-
мому, образуют ионные пары с
этими многоатомными катионами,
так как все соответствующие кри-
вые лежат ниже прямой предель-
ного закона.
На примере поведения хлори-
стого таллия видно, что ассоциа-
ция ионов может иметь место в
случае одноатомного иона со слож-
ной электронной структурой. Кау-
персвейт, Ла-Мер и Барксдел [62]
измерили электродвижущую силу
элемента
Т1 (тв.) | Т1С1 (т) | AgCl-Ag
в температурном интервале 0 — 50е.
Они обнаружили, что их резуль-
таты соответствуют обобщенной
теории Гронвола, Ла-Мера и Сэнд-
веда, если принять а равным 0,93.
Эти авторы показали также, что
если принять для а более вероят-
ное значение 3,0 и дополнить
Сэндведа законом действующих масс, то
функцию Гронвола, Ла-Мера и
можно получить столь же хоро-
шее совпадение, считая константу равновесия равной 0,31. Этот вывод сов-
падает с результатом, полученным Онзагером [63] на основании анализа
отклонений данных по электропроводности от его предельного уравнения.
§ 10. Коэффициенты активности нейтральных молекул в водных
растворах солей
Коэффициент активности нейтральной молекулы в растворе соли может
быть проще всего вычислен из данных по растворимости или на основании
измерений распределения. Пусть имеется газ, находящийся в равновесии со
своим водным раствором согласно реакции типа О2 (газ) = О2 (водн.). Констан-
та равновесия при постоянных Р и Т равна
к = а2 (води.) = „
«2 (Г.) р ’ ' '
если давление газа р настолько мало, что его можно приравнять величине
летучести. Через и /(8) обозначены молярная доля и коэффициент
активности газа в растворе соли. Примем, что в чистой воде /(s) =/(0) = 1
и Y(S) = m(0)M(s)- Тогда /(S)A(S) = Nm, и
55,5 -j~
^s) = A^ = Y(s)55,5 + zn(0) • (50)
Точно такое же соотношение получается для коэффициента активности
жидкого или твердого вещества в растворе соли при условии, что в насы-
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
375
щенком растворе в чистой воде коэффициент активности принят равным
единице.
Подробный обзор экспериментальных данных, полученных до 1927 г.,
был составлен Рендаллом и Файлеем [64]. Этот материал, а также более
поздние исследования по определению коэффициентов активности гелия,
аргона и более сложных молекул, вроде уксусноэтилового эфира1 и диаце-
тонового спирта, будут использованы для иллюстрации сложной природы
наблюдаемых явлений.
Многими исследователями [66] были предложены эмпирические уравне-
ния для определения действия соли на нейтральные молекулы. Из этих
уравнений мы упомянем лишь уравнение Сеченова
lg^ = *A (51)
где 5° и 5 — растворимости нейтральных молекул соответственно в чистом
растворителе и в растворе соли, с — концентрация в молях на литр и ks—
константа. Сравнение этого уравнения с уравнением (50) показывает, что
соотношения
lg/(S) = W, (52)
1gY(s) = /c™!i> (53)
где [х —ионная сила, могут быть полезны для выражения опытных резуль-
татов. Рендалл и Файлей показали, что такое линейное соотношение
представляет собой первое приближение и не может являться совершенно
точным. В то же время эти уравнения хорошо соответствуют эксперимен-
тальным данным, и мы будем их применять в качестве основы для обсу-
ждения результатов.
В табл. 148 приведены коэффициенты «высаливания» кт [уравне-
ние (53)] для гелия" и аргона [67], водорода, кислорода, закиси азота,
двуокиси углерода, иода и ацетилена [64а,б] в растворах многих солей.
Кроме того, таблица содержит подобные же значения для следующих жидко-
стей: уксусноэтилового эфира [65в,г], диацетонового спирта [656], фенилтио-
мочевины [68,666] и фенола [69]. О характере наблюдаемых явлений можно
судить по рис. 100 и 101, на которых представлена зависимость lg у( от
ионной силы для двух газов и двух жидких веществ в растворах многих
солей. Подобие всех изображенных кривых заметно с первого взгляда.
Порядок величины эффекта высаливания одинаков во всех случаях; после-
довательность разных солей, расположенных по силе их «солевого действия»,
также приблизительно одинакова. Отмечаются лишь небольшие отклоне-
ния в этой последовательности расположения солей. Следует отметить,
что даже для столь различных молекул, как закись азота и диацетоновый
спирт, наблюдается аналогия как в отношении порядка величины эффектов,
так и в смысле последовательности расположения солей по степени их
действия.
В гл. III, § 10, было выведено уравнение Дебая и Мак-Олея для
солевых эффектов при действии электролитов на нейтральные молекулы:
1 3
х Акерлёф исследовал Не, Аг [65а], диацетоновый спирт[65б]: Глесстон и Паунд[65в],
Глесстон, Даймонд и Джонс [65г] исследовали уксусноэтиловый эфир.
376
ГЛАВА XII
Как видно из вывода этого уравнения, оно является приближенным соотно-
шением, применимым к разбавленным растворам, и не может давать точных
количественных результатов при высоких концентрациях. Однако представ-
ляет интерес провести качественное сравнение результатов, полученных с
помощью этой теории, с экспериментальными^данными.
Вспомним, что Р является характерной для неэлектролита постоянной
величиной, которая определяется соотношением D = D0 — fin, где п — число
Р ис. 100. Различные солевые эффекты.
Верхний рисунок—N2O; нижний рису-
нок—С2Н4.
Рис. 101. Различные солевые эф-
фекты.
Верхний рисунок—диацетоновый спирт;
нижний рисунок—уксусноэтиловый эфир
молекул неэлектролита, D — диэлектрическая постоянная смеси неэлектро-
лита и растворителя (воды), Do — диэлектрическая постоянная чистого
растворителя, м,—число ионов /-го вида с радиусом bj. Суммирование
производится по всем присутствующим ионам. Коэффициент р положителен
в том случае, когда неэлектролит имеет более низкую диэлектрическую
постоянную, чем чистый растворитель. Так как вода имеет высокую ди-
электрическую постоянную, то для большинства нейтральных веществ /(s)
должно возрастать в водных растворах при увеличении концентрации соли.
Данные, представленные на рис. 100 и 101, подтверждают это предполо-
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
377
жение. Кроме того, согласно теории, lg/(S) должен линейно зависеть от
ионной концентрации, что также согласуется с экспериментальными резуль-
татами, так как Г = 2р.. Наконец, величины солевых эффектов должны
меняться в той же последовательности,
радиусов. Если использовать кристалло-
графические радиусы (табл. 6), эта зако-
номерность не соблюдается и наблюдается
много исключений.
Солевые эффекты, наблюдаемые при
действии хлористых натрия и бария на не-
которые нейтральные молекулы, показа-
ны на1 рис. 102. В растворах хлористого
бария расхождение прямых значительно
меньше, чем в растворах хлористого нат-
рия. Это указывает на то, что для до-
стижения согласия с опытом константу р
следует изменять при переходе от одной
соли к другой.
Все эти положения можно резюми-
ровать следующим образом. Уравнение
Дебая и Мак-Олея совпадает по форме
с эмпирическим уравнением (53) и при-
водит к результатам, имеющим правиль-
ный порядок величины [70]. Это показы-
вает, что влияние электростатических сил
играет важную роль в явлении «высали-
вания». Значение величин ионных радиу-
сов неясно, и сделанное на основании
теории предположение о последователь-
ности величин солевого эффекта для раз-
ных солей не совпадает с наблюдаемыми
результатами. Согласно теории, в водных
растворах всегда должно наблюдаться вы-
саливание, тогда как в действительности
наблюдаются многочисленные примеры об-
ратного эффекта—«всаливания» (этилаце-
тат в растворе йодистого цезия). Эти рас-
хождения могут быть частично устранены,
если ограничить исследования разбавлен-
ными растворами как солей, так и ней-
тральных молекул.
а) Коэффициент активности недиссо-
циированной части некоторых слабых
кислот в растворах солей. В табл. 149
приводятся коэффициенты высаливания кт
что и суммы обратных ионных
Рис. 102. Солевые эффекты для
различных веществ при действии хло-
ристых бария и натрия.
Верхний рисунок—хлористый Оарий:
1—уксусноэтиловый эфир; 2—фенилтио-
мочевина; 3—закись азота; 4—аргон,
фенол; 5—этилен; 6—двуокись кисло-
рода. Нижний рисунок—хлористый на-
трий: 1—фенол; 2—уксусноэтиловый
эфир; 3—фенилтиомочевина; диацетоно-
вый спирт; 4—кислород; 5—закись азота;
в—водород, этилен; 7—аргон.
бензойной, о-толуоловой, о-нитробензой-
ной, салициловой, уксусной и хлоруксусной кислот, вычисленные Рендал-
лом и Файлеем [64а]. Значения коэффициента для первых четырех кислот
были вычислены на основании данных по определению растворимости
Рердама [71] и Гофмана и Лангебека [72], а для уксусной кислоты эти
значения были вычислены по данным Сегдена [73], Мак-Бэна и Кама [74].
Сегден определял распределение уксусной кислоты между амиловым спир-
том и водой; Мак-Бэн и Кам определяли величину, пропорциональную пар-
циальному давлению кислоты над раствором, деленному на концентрацию
378
ГЛАВА XII
недиссоциированных молекул' кислоты в растворе. Рендалл и Файлей изу-
чали распределение моно- и дихлоруксусных кислот между дибутиловым
эфиром и водными растворами солей.
Зависимость величин 1g у для уксусной кислоты от ионной силы
(рис. 103) имеет тот же вид, что и для других неэлектролитов (см. рис. 100,
101 и 102). Наблюдаются лишь
незначительные отклонения в по-
следовательности расположения
кривых, а также явления всали-
вания в случае растворов уксусно-
кислого натрия и азотнокислого
калия.
б) Распределение ацетона и
синильной кислоты между водны-
ми растворами солей и бензолом.
Теория солевых эффектов Дебая.
Гроссом [75] было проведено тща-
тельное исследование влияния
солей на коэффициенты активно-
сти ацетона и синильной кислоты
путем определения распределения
этих веществ между водой или
водными растворами солей и бен-
золом. Эти измерения особенно
удобны для проверки теории «со-
левых эффектов« Дебая (гл. III,
§ 11), так как ацетон понижает,
а синильная кислота повышает
диэлектрическую постоянную
воды.
На основании опытных значений коэффициентов распределения между
бензолом и чистой водой и данных относительно температур замерзания
растворов в бензоле, в сочетании с калориметрическими данными, была
вычислена зависимость коэффициентов активности в бензоле от концентра-
ции. Из этих данных могут быть вычислены коэффициенты активности
изучаемых соединений в растворах солей. Гросс составил таблицы функций
с~ Vo/s/Vo/o’ т \то Л' \т0 /о ’
(55)
где индексы s и 0 указывают на наличие или отсутствие электролитов,
а индекс В относится к растворам в бензоле. В опытах по изучению
распределения, проведенных с разбавленными растворами солей, [cB]ss^ [св]о-
Следовательно, в этих условиях значения отношений (55) приближались
к отношениям коэффициентов активности в растворах солей к коэффициен-
там активности в чистой воде. При более высоких концентрациях солей
можно вносить небольшую поправку на изменение коэффициента активно-
сти при изменении концентрации высаливающегося вещества в бензоль-
ном слое.
Было обнаружено, что явление высаливания зависит от природы
неэлектролита, но практически не зависит от его концентрации и темпе-
ратуры. Приближенно экспериментальные результаты могут быть представ-
лены с помощью следующих уравнений:
у- = 1 — т~ = 1 - Ьтс, (56)
/с /т
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
379
в которых через с обозначена концентрация соли1. На рис. 104 представлены
некоторые результаты, полученные Гроссом. Сплошными и пунктирными
линиями обозначены солевые эффекты соответственно для ацетона и синиль-
ной кислоты. Для азотнокислых цезия и калия, хлористого натрия
и сернокислых магния и лантана
наблюдается любопытное . обращение
солевого эффекта. В этих растворах
ацетон высаливается, а синильная ки-
слота всаливается. В качестве исклю-
чения из этого правила можно отметить
поведение ацетона и синильной кисло-
ты в растворах азотнокислого лития.
Аналогичное обращение солевого эф-
фекта было обнаружено в случае рас-
творов хлористых лития и калия, сер-
нокислого калия и азотнокислого на-
трия. В растворах хлористого магния
это явлейие не наблюдается.
Все эти результаты в основном со-
гласуются с теорией солевых эффектов
Дебая. В гл. III, § 10, было выведено
уравнение (139)
Zi — 1 _ У 7 с
/-1 1000 Jr
Входящие в него функции опреде-
ляемые уравнениями (140) — (142) гл. III,
зависят от ионных радиусов и от ве-
личины/?/, которая находится по урав-
нению (134) гл. III
Рис. 104. Высаливание и всалива-
ние синильной кислоты и ацетона.
Черные кружки—ацетон; белые кружки—
синильная кислота.
* ф210001-1
Постоянная | а | определяется уравнением (133) гл. III
_(А-А) v
Dr V'
где Dr и D2 — диэлектрические постоянные компонентов' 1 и 2, Vr—молярный
объем компонента 1. Компонент 2 высаливается или всаливается. В случае
описанных выше систем компонентом 1 является вода, компонентом 2—ацетон
или синильная кислота.
Так как функции J j являются постоянными величинами, зависящими
от значений | а | и R}, и поскольку /° — коэффициент активности компо-
нента 2. для чистой воды—принимается равным единице, то уравнение (139)
гл. III формально эквивалентно уравнению (56). Следовательно, теория
правильно предсказывает наблюдаемую на опыте линейную зависимость
1//с от концентрации. Далее, согласно теории, синильная кислота должна
всаливаться, так как ее диэлектрическая постоянная больше, чем у чистой
1 Это выражение похоже на уравнение (53), так как
W=lg 7(4)яе 1—1/7(S).
380
ГЛАВА XII
воды, в то время как в случае ацетона должно происходить высаливание.
За исключением опытов с азотнокислым литием и с хлористым магнием,
в остальных случаях все эти положения теории подтверждаются опытными
данными.
На основании измерений диэлектрических постоянных растворов ацетона
и синильной кислоты Гросс получил соответственно следующие уравнения:
D = DX (1-0,0367 с) (57)
и
D = DX (1+0,0039 с). (58)
Таким образом, согласно уравнениям (135) и (137) гл. III, величина | а |
равна 0,0367 для растворов ацетона и 0,0039 для растворов синильной
кислоты. Согласно уравнению (134) гл. Ill, Rj (ацетон) = 2,03 • 10-8 см
и У?, (HCN) = 1,2 • 10~8 см. Для солевых эффектов в разбавленных растворах
солей с помощью этих значений получаются уравнения
1// = 1-^Эг, (59)
а
1//==1+^Г (60)
а
соответственно для ацетона и синильной кислоты, причем 1/а=Е1/а/.
Нельзя ожидать, чтобы эти уравнения давали правильные количест-
венные результаты, так как они представляют собой предельные соотно-
шения, применимые лишь к очень разбавленным растворам, и, кроме того,
как показывают аномальные результаты, полученные в случае растворов
азотнокислого лития и хлористого магния, солевые эффекты, вероятно,
осложняются химическими эффектами, которые нельзя свести к чисто
электростатическим взаимодействиям. Теоретические расчеты дают пра-
вильный порядок величин, так как они приводят для многих солей к зна-
чениям а, равным 0,5 — 2. Для сернокислых лантана и магния наблюдается
очень большой эффект, который примерно совпадает с теоретическими
расчетами.
Явление высаливания воды из водно-диоксановых смесей хлоридами
калия, натрия и лития было исследовано Скэтчардом и Бенедиктом [76]
путем измерения точек замерзания. Применялись содержащие соли смеси
воды и диоксана в отношении 1 : 2 и 2 : 1, при концентрациях солей 0,01—2.
Из значений осмотических коэффициентов были вычислены коэффициенты
высаливания (коэффициенты взаимодействия молекул с электролитом).
При использовании кристаллографических радиусов (табл. 6) было полу-
чено хорошее совпадение экспериментальных данных с расчетами по теории
высаливания Дебая, в то время как теория Дебая и Мак-Олея давала
менее удовлетворительные результаты.
§ 11. Поверхностное натяжение разбавленных
растворов электролитов
Увеличение поверхностного натяжения при добавлении электролитов
впервые наблюдал Гейдвейлер [77]. Швенкер [78] измерял поверхностное
натяжение растворов некоторых 1,1-электролитов при концентрациях
0,015 — 0,2 н. и получил аналогичные результаты.
Теоретическое объяснение влияния междуионного притяжения на поверх-
ностное натяжение, согласно Вагнеру, а также Онзагеру и Сэмарасу, было
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
381
дано выше (гл. III, § И), причем было получено выражение
с
О»
1,012с, 1,467
(61)
которое учитывает влияние прибавления 1,1-электролита на относительное
поверхностное натяжение воды при 25°. Отметим прежде всего, что, со-
гласно теории, отношение а/а0 должно возрастать при увеличении концен-
трации, хотя это предусматриваемое теорией изменение должно быть не-
значительным. Так, например, для воды отношение а/а0 равно 1; 1,00004;
1,0003 и 1,0016 для значений с, равных соответственно 0; 0,001; 0,01 и 0,1.
Онзагер и Сэмарас показали, что их теория дает значения, которые сов-
падают по порядку величины с данны-
ми Гейдвейлера и Швенкера. Совпаде-
ние было не очень точным, но это
расхождение можно, невидимому, объ-
яснить ошибками опыта, так как ре-
зультаты Гейдвейлера несколько отли-
чались от данных Швенкера.
Применяя кварцевый капилляри-
метр, Джонс и Рей [79] с очень боль-
шой точностью измеряли относитель-
ный подъем раствора в капилляре.
Из результатов этих измерений Джонс
и Рей получили величину (а/а0)', кото-
рую мы будем называть «кажущимся
относительным поверхностным натяже-
нием». Воспроизводимость определений
была исключительно хорошей. Наиболь-
шее отклонение для любого раствора
не превышало 0,005%, а среднее откло-
нение составляло 0,002%.
Некоторые из результатов, полу-
ченных Джонсом и Реем для очень
разбавленных растворов хлористого ка-
лия, азотнокислого цезия и сернокис-
лого калия, показаны на рис. 105 в
форме зависимости кажущегося относи-
тельного поверхностного натяжения от
концентрации растворов. Во всех слу-
Р и с. 105. Кажущееся относитель-
ное поверхностное натяжение раство-
ров азотнокислых калия и цезия и
сернокислого калия при 25°.
Приведены также теоретические кривые длн
относительного поверхностного натяжения
согласно уравнению (61).
чаях верхняя кривая изображает теоретические результаты, рассчитанные
по уравнению (61). В области самых разбавленных растворов наблюдается
быстрое уменьшение (а/а0)' Д° тех пор, пока не достигается отчетливый
минимум. После этого значения (а/а0)' начинают возрастать, причем наклон
кривой приблизительно соответствует теоретическому. Если отношение
(а/а0)' представляет собой лишь относительное поверхностное натяжение,
то теория в условиях очень разбавленных растворов, очевидно, не оправ-
дывается. Джонс и Рей показали, что это быстрое уменьшение значе-
ний (а/а0)' наблюдается в случае ионных растворов, однако оно не было
обнаружено в водных растворах сахарозы.
Лангмюр [80а]1 указал, что в случае очень разбавленных растворов
-потенциал кварца обусловливает образование смачивающего слоя в тон-
1 Дол [806] сделал попытку истолковать это явление как следствие одного только
поверхностного натяжения.
382
ГЛАВА XII
ких кварцевых капиллярах. Этот слой является настолько толстым, что
он уменьшает диаметр капилляра в такой степени, что это влияет на точ-
ность измерения поверхностного натяжения. Таким образом, эффект,
который наблюдали Джонс и Рей, не является только мерой относительного
поверхностного натяжения, а обусловлен двумя явлениями: изменением,
поверхностного натяжения и влиянием С-потенциала. Онзагер1 подробно
изучил явление смачивания поверхностей растворами электролитов с теоре-
тической точки зрения и развил количественную теорию влияния С-потен-
циала на подъем раствора в капилляре. Его теория позволила в значи-
тельной мере объяснить расхождение теоретических и экспериментальных
данных, показанное на рис. 105, быстрое уменьшение величины отноше-
ния (а/а0)' в разбавленных растворах и появление минимумов на кривых.
Онзагер не учитывал возможного влияния квантовых сил. Однако ему
удалось весьма точно вычислить С-потенциал кварца на основании изме-
рений поверхностного натяжения Джонса и Рея.
Все описанные выше факты свидетельствуют о том, что теория влияния
междуионного притяжения на поверхностное натяжение, развитая Вагнером,
Онзагером и Сэмарасом, является в первом приближении правильной.
§ 12. Критический обзор
Как видно из приведенного в этой главе обзора свойств 1,1-электро-
литов, в систематизации имеющихся данных встречается ряд затруднений.
Изучение экспериментального материала показывает, что существуют
расхождения между результатами различных исследователей. Так, например,
значения /0 4 (табл. 147), полученные Гуггенгеймом для некоторых электро-
литов, значительно отличаются от значений, найденных Скэтчардом и Прен-
тисом (табл. 123), которые использовали те же опытные данные, полученные
путем определения температур замерзания. Аналитический метод экстрапо-
ляции Гуггенгейма почти во всех случаях дает более низкие значения у ,
чем графический метод, который применяли Скэтчард и Прентис. Данные
Робинзона (табл. 146), полученные с помощью метода определения изопие-
стической упругости пара, для многих электролитов ниже, чем данные
Гуггенгейма. Для случая хлористых калия и натрия измерения электро-
движущих сил элементов с жидкостным соединением, выполненные Брауном
и Мак-Иннесом, а также Шидловским и Мак-Иннесом, имели существенное
значение для определения величины /0 j при 25°, и полученные ими дан-
ные явились хорошим подтверждением совпадения результатов, найден-
ных для этих электролитов различными методами.
До сих пор единственным электролитом, который был исследован
в интервале' температур 0—100°, является хлористый натрий, но, к сожа-
лению, методы исследования этого электролита в случае разбавленных
растворов (< 0,1 М) были неточными. Даже для более концентрированных
растворов и при 25° еще остаются сомнения в точности определения у .
Результаты (табл. 135), полученные путем сочетания данных из измерений
электродвижущих сил, температур кипения и теплот разбавления, не вполне
согласуются со значениями, принятыми Скэтчардом, Гамером и Вудом [41]
в качестве стандартных и полученными ими путем изопиестических изме-
рений упругости пара. ,
Как было показано, создание теории концентрированных растворов
электролитов, учитывающей индивидуальные особенности различных ионов,
является весьма затруднительным, и до сих пор отсутствует точная колй-
1 Онзагер, частное сообщение;
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
383
чественная трактовка влияния различных сложных факторов. В § 5 было
показано, что определение среднего расстояния сближения ионов а на осно-
вании экспериментельных данных представляется сомнительным, а это
равносильно утверждению, что до настоящего времени мы не можем точно
истолковать причины наблюдаемых отклонений от предельного закона.
Теория Скэтчарда, которая учитывает влияние взаимодействия между
ионами, между ионами и молекулами, а также между молекулами, имеет
существенное значение, так как она показывает, что свойства простейших
ионов «типа благородных газов» могут быть истолкованы как функции
ионных радиусов. Однако сомнительно, чтобы линейная комбинация членов,
учитывающих эти эффекты, с членом, входящим в предельное уравнение
Дебая и Гюккеля [уравнение (40)], могла дать больше, чем удовлетвори-
тельное качественное объяснение и классификацию специфических свойств
ИОНОВ'.
Онзагер [54] дал подробный критический разбор и обобщение резуль-
татов теории концентрированных растворов. В гл. II и III было показано,
что возможность расчета термодинамических свойств зависит от предполо-
жения «о линейном наложении ионных атмосфер», которое приводит к про-
порциональности между зарядом и потенциалом. Онзагер показал, что по-
скольку уравнение Пуассона—Больцмана является приближенным, то нельзя
приписывать ионным атмосферам аддитивные свойства в тех случаях,
когда следует учитывать высшие (дополнительные) члены уравнения
Пуассона—Больцмана.
Влияние взаимодействия ионов с растворителем на термодинамические
свойства растворов электролитов еще не выяснено в полной мере.
Бьеррум [81] и Гюккель [436] показали, что можно ожидать увеличения
коэффициента активности в случае концентрированных растворов вследствие
гидратации ионов. В приложении Б, § 7, будет более подробно рассмот-
рена современная теория, представляющая собой дальнейшее развитие
теории Бьеррума.
На основании теоретических соображений о структуре воды и о взаи-
модействии ионов с водой Бернал и Фаулер [82а]1 развили теорию таких
свойств, как кажущиеся молярные объемы и теплоты гидратации ионов.
Они показали, что в жидкой воде молекулы стремятся соединиться
в группы, имеющие форму тетраэдров, в углах которых расположены
атомы кислорода. Эта структура аналогична открытой тетраэдрической
структуре льда [83]. В случае кристаллического состояния молекулы
воды группируются вокруг ионов, и число молекул воды, находящихся
в таком ориентированном состоянии, определяется пространственными усло-
виями, которые зависят от относительных объемов молекул воды и ионов.
В случае водных растворов степень гидратации иона зависит от числа
молекул воды, которые могут расположиться вокруг иона, а также от силы
поля у поверхности этого иона.
Применяя эти соображения к вопросу о парциальных молярных
объемах, Бернал и Фаулер показали, что можно расположить одновалент-
ные катионы в следующей последовательности по степени их гидратации2: •
Li+ > Na+ >К+ > Rb+, Cs+. Поскольку ионы Rb+ и Cs+ имеют большие
• радиусы, сила поля у поверхности этих ионов слишком мала, чтобы
притягивать обладающие диполями молекулы воды, а поэтому гидрата-
ция этих ионов незначительна и ею можно пренебречь. Этот вывод теории
согласуется с тем фактом, что объем водного раствора йодистого цезия
1 См. также [82б[.
2 См. также работу А. Г. Пасынского (Acta physicochimica URSS, 8-
(1938)]. (Прим, ред.)
384
ГЛАВА XII
значительно превышает сумму объемов чистой соли и жидкой воды при
той же температуре. За исключением иона F-, все одновалентные анионы
обладают большим размером и не гидратированы. Многовалентные ионы
подвергаются гидратации, причем, согласно теории, их можно располо-
жить в следующем порядке по степени гидратации: Mg++ > Са++ >
>Sr++>Ba++. Применение изложенных теоретических соображений
можно найти в приложении Б, § 6 и 71.
ЛИТЕРАТУРА
1. G. N. Lewis, J. Am. Chem. Soc., 34, 1631 (1912).
2. G.N. Lewis, G. A. Linhart, J. Am. Chem. Soc., 41, 1951 (1919).
3. G. N. Lewis, M. Randall, J. Am. Chem. Soc., 43, 1112 (1921);
Г. H. Льюис, M. P e н д а л л, Химическая термодинамика, ОНТИ—Хим-
теорет. Л., 1936.
4. И. S. И а г n е d, J. Am. Chem. Soc., 42, 1808 (1920); 44, 252 (1922);
5. E. H ii c k e 1, Physik. Z., 26, 93 (1925).
6.. G. Scatchard, J. Am. Chem. Soc., 47, 648 (1925); 47, 2098 (1925).
7. H. S. H a r n e d, J. Am. Chem. Soc., 48, 326 (1926).
8. M. Randall, L. E. Young, J. Am. Chem. Soc., 50, 989 (1928).
9. X. С. Тей ло p, Физическая химия, ОНТИ—Химтеорет, Л., 1935, гл. XII:
• а) -стр. 777 и сл.; б) табл. IX, стр. 754; в) стр. 801 и сл.
10. a) D. A. Maclnnes, К. Parker, J. Am. Chem. Soc., 37, 1445 (1915);
6) D. A. Maclnnes, J. A. Beattie, там же, 42, 1117 (1920); M. К n о h e 1,
там же, 45, 70 (1923); И. S. Harned, F. Е. Swindells,; там>же, 48, T26~
(1926); в) И. S. И а г n ed, там же, 47, 676 (1925); г) 51, AW0929); д)’Й.’§. Har-
ned, S. М. Douglas, там же, 48, 3095 (1926); е) Н. S. Harned,
О. Е. S с h и р р, там же, 52, 3886 (1930); ж) Н. S. Harn ed, L. F. N i m s, там же, 54,
. 423 (1932); з) H. S. Harned, J.'C. Hecker, там же, 55, 4838 (1933);
и) Н. S. Harned, М. A. Cook, там же, 59, 497 (1937); к) Н. S. Harned,
С. С. Crawford, там же, 59, 1903 (1937); л) G. A. k е г 1 б f, G. К е geles,
там же, 62, 620 (1940).
11. a) A. S. Brown, D. A. Maclnnes, J. Am. Chem. Soc., 57, 1356 (1935);
6) G. J. J a n z, A. R. Gordon, там же, 65, 218 (1943).
12. E. A.- G ul bransen, A. L. Robinson,!. Am. Chem. Soc., 56, 2637 (1934).
13. H. S. Harned, M. А. С о c k, J. Am. Chem. Soc., 61, 495 (1939).
. R. P. Smith, J. Am. Chem. Soc., 61, 500 (1939).
. R..P. Smith, .1. Ami Chem. Soc., 61, 497 (1939).
16. R. P. Smith, D. S. Hirtle, J. Am. Chem. Soc., 61, 1123 (1939).
17. A. L. R о b i n s о n, J. Am. Chem. Soc., 54, 1311 (1932).
18. a) F. D. R о s s 1 n i, Bur. Standards J. Research, 7, 47 (1931); б) там же, 6, 791 (1931).
19. G. N. Lewis, H. Store h, J. Am. Chem. Soc., 35, 2544 (1917).
20. H. S. Harned, G. W. James, J. Phys. Chem., 30, 1060 (1926).
21. A. S. К e s t о n, J. Am. Chem. Soc., 57, 1671 (1935).
22. H. S. H a r n e d, A. S. К e s t о n, J. G. D о n e I s о n, J. Am. Chem. Soc., 58,
989 (1936).
23. H. S. H a r n о d, W. J. Hamer, J. Am. Chem. Soc., 55, 4496 (1933).
24. H. S. Harned, J. G. D о n e 1 s о n, J. Am. Chem. Soc., 59, 1280 (1937).
25. В. B. Owen, J. Am. Chem. Soc., 57, 1526 (1935).
26. В. В. 0 w e n, L. F о e r i n g, J. Am. Chem. Soc., 58, 1575 (1936).
27. R. P. Smith, J. Am. Chem. Soc., 55, 3279 (1933).
28. H. S. H a r n e d, M. A. Cook, J. Am. Chem. Soc., 59, 1290 (1937).
29. T. S h e d 1 о v s к y, D. A. Maclnnes, J. Am. Chem. Soc., 59, 503 (1937).
30. H. M. Spencer, J. Am. Chem. Soc,., 54, 4490 (1932).
31. G. Scatchard, S. S. Prentiss, J. Am. Chem. Soc., 55, 4355 (1933).
32. B. Saxton, R. P. Smit h, J. Am. Chem. Soc., 44, 2826 (1932).
33. B. F. Lovelace, J. C. W. Frazer, V. B. Sea’se, J. Am. Chem. Soc., 43,
102 (1921).
34. M. Knobel, J. Am. Chem. Soc., 45, 70 (1923).
35. H. S. Harned, F. E. Swindells, J. Am. Chem. Soc., 48, 126 (1926).
1 Оригинальная трактовка явления гидратации была дана О. Я. Самойло-
вым [ДАН, 77, 641 (1951)]. Он рассматривает гидратацию не как связывание
ионом определенного числа молекул воды, а как воздействие ионов на трансляционное
движение молекул воды. (Прим, ред.)
ВОДНЫЕ РАСТВОРЫ СИЛЬНЫХ 1,1-ЭЛЕКТРОЛИТОВ
385
36. Н. S. Н а г n е d, М. A. Cook, J. Am. Chem. Soc., 59, 496 (1937).
37. A. L: Robinson, J. Am. Chem. Soc., 60, 1265 (1938); H. Hammerschmid,
A. L. Robinson, там же, 54, 3120 (1932).
38. D. A. Sinclair, J. Phys. Chem., 37, 495 (1933).
39. R. A. Robinson, D. A. Sinclair, J. Am. Chem. Soc., 56, 1830 (1934);
R. A. Robinson, там же, 57, 1161 (1935); 57, 1165 (1935); 59, 84 (1937); Trans.
Faraday Soc., 35, 1217 (1939).
40. W. R. В oust i eld, E, Bousfield, Proc. Roy. Soc., A103, 429 (1923).
41. G. S c a t c h a r d, W. J. Hamer, S. E. W о о d, J. Am. Chem. Soc., 60, 3061
(1938).
42. R. A. Robinson, H. S. H a r n e d, Chem. Rev., 28, 419 (1941).
43. a) E. H u с к с 1, Physik. Z., 26, 93 (1925); б) там же, p. 92.
44. A. S. Brown, D. A. M a с I n n e s, J. Am. Chem. Soc., 57, 1356 (1935);
T. Shedlovsky, D. A. Maclnnes, там же, 59, 503 (1937).
45. P. V an Ryss e Iberghe, S. Eisenberg, J. Am. Chem. Soc., 61, 3030 (1939).
46. T. H. Gronwall, V. K. L a M e r, K. S a n d v e d, Physik. Z., 29, 358 (1928).
47. H. I. Stonehill, M. A. Berry, J. Am. Chem. Soc., 64, 2724 (1942).
48. J. N. В r 6 n s t e d, J. Am. Chem. Soc., 42, 761 (1920); 44, 877 (1922); 44, 938 (1922);
45, 2898 (1923).
49. E. A. Guggenheim, Rep. of Scandinavian Science Congress, Copenhagen 1929,
p. 296; Phil. Mag. (7), 19, 588 (1935); (7), 22, 322 (1936); E. A. Guggenheim,
L. A. Wiseman, там же (7), 25, 45 (1938).
50. E. G ii n t e 1 b e r g, Z. Physik. Chem., 123, 199 (1926).
51. a) G. Scatchard, Physik. Z., 33, 22 (1932); Chem. Rev., 19, 309 (1936);
б) E. J. Cohn, J. T. Edsall, Proteins, Amino Acids and Peptides, Rein-
hold Publishing Corporation, N. V., 1943, Chapter 3, Thermodynamics and Electro-
static Theory. .
52. J. W у m a n, Jr., J. Am. Chem. Soc., 58, 1482 (1936)
53. L. Pauling, J. Am. Chem. Soc., 50, 1036 (1928).
54. L. 0 n s a g e r, Chem. Rev., 13, 73 (1933). ф
55. a) P. Van R у s s e 1 b e r g h e, S. Eisenberg, J. Am. Chem. Soc., 61,
3030 (1939); 6) 62, 451 (1940).
56. a) H. D. U r s e 1 1, Proc. Camb. Phil. Soc., 23, 685 (1927); б) P. Фаулер, Ста-
тистическая термодинамика, Издатинлит, М., 1949.
57. Н. S. Harned, G. A k е г 1 о f, Physik. Z., 27, 411 (1926).
58. G. S с a t с h а г d, S. S. Prentiss, J. Am. Chem. Soc., 56, 807 (1934).
59. G. S c a t c h a r d, S. S. Prentiss, J. Am. Chem. Soc., 54, 2696 (1932).
60. L. Ebert, J. Lange, Z. physik. Chem., A139, 584 (1928).
61. J. Lange, Z. physik. Chem., A168, 147 (1934).
62. LA. C owp er thwai te, V. K. LaMer, J. Barksdale, J. Am. Chem.
Soc., 56, 544 (1934).
63. L. 0 n s a g e r, Physik. Z„ 28, 277 (1927).
64. a) M. Randall, C. F. F a i 1 e y, Chem. Rev., 4, 271 (1927); 6) 4, 285 (1927);
в) 4, 291 (1927).
65. a) G. Ake r 1 of, J. Am. Chem. Soc., 57, 1198 (1935); 6) 51, 984 (1929); в) S.- G 1 a s-
stone, A. Pound, J. Chem. Soc., 127, 2660 (1925), r) S. G 1 a s s t о n e,
D. W. D i m о n d, E. C. Jones, там же, 128, 2935 (1926).
66. а) И. M. С e ч e н о в, Ann. chim. phys. (6), 25, 226 (1892); V. G о г d о n, Z. physik.
Chem., 18, 8 (1895); 6)V. Rothmund, там же, 33, 401 (1900).
67. G. Akerlof, J. Am. Chem. Soc., 57, 1196 (1935).
68. W. В i 1 t z, Z. physik. Chem., 43, 41 (1903); V. Rothmund, 33, 401 (1900).
69. W. Herz, F. H i e b e n t h a 1, Z. anorg. Chem., 177, 363 (1929).
70. G. S c a t c h a r d, Trans. Far. Soc., 23, 454 (1927).
71. H. N. K. R б r d a m, диссертация, Copenhagen, 1926.
72. F. Hoffmann, K. L a n g e b e c k, Z. physik. Chem., 51, 385 (1905).
73. J. N. S ugd en; J. Chem. Soc., 128, 174 (1926).
74. J. W. M с В a i n-, J. Kam, J. Chem. Soc., 115, 1332 (1919).
75. P. Gross, K. Schwarz, Sitz. Akad. Wiss. Wien, 139, 179 (1930); P. Gross,
M. I s e г, там же, 139, 221 (1930).
76. G. Scatchard, M. A. Benedict, J. Am. Chem. Soc., 58, 837 (1936).
77. A. H e у d w e i 1 1 e r, Ann. Physik. (4), 33, 145 (1910); Physik. Z., '3, 329 (1902).
78. G. S c h w e n к e r, Ann. Physik., 11, 525 (1931).
79. G. Jones, W. A. Ray, J. Am. Chem. Soc., 59, 187 (1937); 63, 288 (1941).
80. a) I. Langmuir, Science, 88, 430 (1937); J. Chem. Phys., 6, 873 (1938); б) M. D о 1 e,
J. Am. Chem. Soc., 60, 904 (1938).
81. N. В j e r r u m, Z. anorg. Chem., 109, 275 (1920).
82. a) J. D. В e r n a 1, R. H. Fowler,!. Chem. Phys., 1, 515 (1913); б) T. J. W e b b,
J. Am. Chem. Soc., 57, 2680 (1935).
83, L. Pauling, J. Am. Chem. Soc., 57, 2680 (1935).
2г
5 Г. Харнед, Б. Оуэн
386
ГЛАВА XII
ЛИТЕРАТУРА К ТАБЛИЦАМ
К табл. 79
1. H.S. Н агп ed, J, Franklin Inst., 225, 623 (1928); Е. R. Smith, J. К. Taylor,
Bur. Standards J. Res., 25, 731 (1940).
К табл. 80
1. M. Randall, F. D. Rossini, J. Am. Chem. Soc., 51, 323 (1929).
К табл. 81
1. G. Jones, S. Baeckstrom, J. Am. Chem. Soc., 56, 1524 (1934).
К табл. 82
1. H. S. H a r n e d, M. А. С о о k, J. Am. Chem. Soc., 59, 1290 (1937); H.S. Harned,
там же, 41, 416 (1929).
2. R. P. Smith, J. Am. Chem. Soc., 55, 3279 (1933).
3. H. M. Spencer, J. Am. Chem. Soc., 54, 4490 (1932).
4. G. Scatchard, S. S. Prentiss, J. Am. Chem. Soc., 55, 4355 (1933).
5.. B. F. Lovelace, J. C. W. Frazer, V. B. S e a s e, J. Am. Chem. Soc., 43,
102 (1921).
6. T. S h e d 1 о v s к y, D. A. Maclnnes, J. Am. Chem. Soc., 59, 503 (1937).
К табл. 83
1. H. S. H a r n e d, F. E. S w i n d e 11 s, J. Am. Chem. Soc., 48, 126 (1926).
2. a) H. S. Harned, J. C. Hecker, J. Am. Chem. Soc., 55, 4838(1933);
6) G. А к e r 1 d f, G. К e g e l e s, там же, 62, 620 (1940).
3. H. S. Harned, M. A. Coo k, J. Am. Chem. Soc., 59, 496 (1937).
4. H. S. Harned, О. E. S c h u p p, J. Am. Chem. Soc., 52, 3886 (1930).
К табл. 84
1. R. A. Robinson, Trans. Faraday Soc., 35, 1222 (1939).
2. P. О 1 у n у k, A. R. Gordon, J. Am. Chem. Soc., 65, 224 (1943).
3. H. S. Harned, J. Am. Chem. Soc., 51, 416 (1929).
4. H. S. Harned, 0. E. S c h u p p, J. Am. Chem. Soc., 52, 3886 (1930).
Глава XIII
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
. Присутствие в растворе ионов более высокой валентности приводит
к дальнейшему усложнению теории термодинамических свойств растворов,
даже в случае растворителей с большой диэлектрической постоянной.
Из теоретических соображений, приведенных в гл. III, § 6, 7, явствует,
что, даже если учитывать только кулоновские силы, следует ожидать
отклонений от теории Дебая и Гюккеля в ее первоначальной форме.
Члены высших порядков полного решения уравнения Пуассона—Больцмана
являются функциями валентности ионов. Как показали Гронвол, Ла-Мер
и Сэндвед, для симметричных электролитов удельный вес второго и третьего
членов правой части уравнения (42) гл. III в общей величине 1п/± прак-
тически ничтожен для водных растворов 1,1-электролитов, однако он очень
сильно зависит от величины z, так как множители перед функциями,
заключенными в скобки, содержат z6 и z10. Аналогичные результаты были
получены также для членов Высших порядков уравнения (44) гл. III для
электролитов с несимметричным типом валентности. Точный учет влияния
членов высших порядков является весьма затруднительным, так как он
может до некоторой степени осложняться из-за наличия других эффектов,
например тех, которые учитываются членами ряда 2 Ап сп>2 [уравнение
п>2
(22) гл. XI]. Как было показано, эти дополнительные члены необходимы
для вычисления коэффициентов активности 1,1-электролитов.
Можно внести некоторую ясность в вопрос о многовалентных электро-
литах, если изучить его с точки зрения теории ассоциации ионов Бьеррума
(гл. III, § 7). Было показано, что если b (расстояние наибольшего сближения
положительных ионов с отрицательными) больше, чем q = s21 Zj z2 \/2DkT,
то вероятность ассоциации ионов ничтожна. Для водных растворов 1,1-элек-
тролитов при 25° <?= 3,5 А. Следовательно, для 2,1-, 3,1-, 2,2-, 3,2- и 3,3-
электролитов q равно соответственно 7; 10,5; 14; 21 и 31,5 А. Так как эти
величины значительно больше, чем а для многих высоковалентных элек-
тролитов, то, согласно теории Бьеррума, можно ожидать наличия значи-
тельной ассоциации ионов. Судя по значениям электропроводности, хло-
ристый кальций и другие подобные ему электролиты являются сильными
в разбавленных растворах, а в случае электролитов типа сернокислых
цинка и магния имеет место ассоциация.
Другими электролитами, которые, несомненно, диссоциируют непол-
ностью, являются растворы галогенидов кадмия. Возможно, что в этих
случаях в растворе имеется равновесие следующего вида:
МС12 С1 4-МС1+ ?± М++4-2С1 ,
причем процесс диссоциации определяется не только кулоновскими силами,
но и силами более сложной природы, вроде тех, которые были рассмотрены
25*
388
ГЛАВА XIII
в гл. III, § 7. В этих случаях значения, вычисленные с учетом закона
действующих масс, совпадают с экспериментальными результатами.
Из многочисленного, но разрозненного материала, относящегося к этой
области, будет рассмотрена только ограниченная часть, на примере которой
будут проиллюстрированы наиболее совершенные методы обработки резуль-
татов. В первую очередь будут рассмотрены примеры 2,1-, 3,1-, 2,2-
и 3,2-электролитов, а затем свойства растворов более слабых электролитов,
причем особое внимание будет уделено растворам галогенидов кадмия.
Наконец, будут описаны более подробно свойства водных растворов серной
кислоты.
§ 1. Термодинамика растворов галогенидов магния, кальция,
стронция и бария
Лукасе [1] измерил при 25° электродвижущие силы следующих эле-
ментов, содержащих хлориды кальция, стронция и бария:
Ag-AgCl | МС12 (т) | MxHg | МС12 (тв) | AgCl-Ag.
Скэтчард и Тефт [2] также применили этот элемент для исследования
раствбров хлористого кальция при 25°. Типпетс и Ньютон [3] исследовали
с помощью этого элемента растворы хлористого бария в интервале тем-
ператур 0 — 45°.
На основании тех же соображений, которые были использованы при
выводе уравнения (31) гл. X, можно получить следующее выражение для
электродвижущей силы этого элемента: ' '
2F 1Л
Как видно, из этого уравнения можно вычислить отношение у± к
yR — коэффициент активности при стандартной концентрации. Эти
были получены путем экстраполяции в соответствии с теорией
(1)
Yr, где
данные
Харне-
дом [4], Скэтчардом и Тефтом [2], Джонсом и Долом [5], Типпетсом
и Ньютоном, которые применяли для этого уравнение (33) гл, X или
аналогичное ему соотношение. Дополнительные члены не учитывались
ни в одной из этих работ. Из проведенных до настоящего времени опытов
явствует, что амальгамные электроды, содержащие барий и стронций,
являются воспроизводимыми, а электроды из амальгамы кальция чрез-
вычайно невоспроизводимы, что приводит к ошибочным результатам. Тип-
петс и Ньютон показали, что для амальгамных бариевых электродов сред-
няя величина ошибки при измерении электродвижущих сил составляет
-£ 0,2 мв, в то время как в опытах Скэтчарда и Тефта та же величина
для кальциевых электродов составляла Д: 0,4 мв. Лукасе, а также Скэт-
чард и Тефт применяли проточные амальгамные электроды. Типпетс
и Ньютон пользовались покоящимися электродами, покрытыми слоем масла.
Результаты, полученные для растворов с концентрациями солей меньше
0,01 М, в литературе не приводятся, так как надежность опытных данных
уменьшается с понижением концентрации растворов. В табл. 90 приведены
значения. у± при 25° для растворов хлористых бария и стронция, получен-
ные путем измерений электродвижущих сил элементов с амальгамными
электродами. В табл. 150 содержатся значения коэффициентов активности
хлористого бария при температурах 0 — 45°, полученные Типпетсом и Нью-
тоном.
Как видно из совпадения величин коэффициентов активности при 25°,
определенных Типпетсом и Ньютоном (табл. 90, третий столбец), с величинами,
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
389
Таблица 90
Коэффициенты активности у± хлоридов щелочноземельных металлов при 25°
тп ВаС12 SrC12 С"аС12
упр. пара [3] э. д. с. [1] Э. Д. С. 1) упр. пара [3] э. д. с. 1) упр. пара [3]
0,01 — 0,723 0,723 0,729 —
0,05 — 0,559 0,554 — 0,571 —
0,1 (0,492) 0,492 0,495 (0,514) 0,512 (0,531)
0,2 0,438 ' 0,436 0,439 0,463 0,465 0,482
0,3 0,411 0,411 — 0,440 — 0,462
0,4 0,398 — — 0,430 — 0,456
0,5 0,390 0,390 0,395 0,425 0,427 0,457
0,6 0,386 — — 0,426 — 0,462
0,7 0,384 0,384 — 0,430 — 0,469
0,8 0,385 — — 0,436 — 0,479
6,9 0,388 — — 0,444 — 0,493
1,0 0,392 0,389 0,395 0,455 0,449 0,509
1,1 0,397 — — 0,467 — 0,528
1,2 0,402 — — 0,480 — 0,550
1,3 0,409 — — 0,494 — 0,573
1,4 0,416 — — 0,510 — 0,599
1,5 0,423 0,425 0,425 0,527 0,526 0,626
1,6 0,431 — — 0,546 .—. 0,657
1,7 0,440 — . — 0,566 — 0,690
1,8 0,450 — — 0,587 — 0,726
1,9 — — — 0,611 .— 0,764
2,0 — — — 0,636 0,638 0,807
2,1 — — — 0,664 — 0,853
2,2 — — — 0,694 .—. 0,901
2,5 — — — — 0,772 —
3,0 — — — 1,003 —
1) Харнед [2а]—на основании данных Лукасса [26].
вычисленными на основании данных Лукасса (четвертый столбец), резуль-
таты измерений электродвижущих сил бариевых амальгамных элементов
хорошо воспроизводятся. На основании измерений электродвижущих сил
при 25° Типпетс и Ньютон [6] вычислили также коэффициенты активности
воды в растворах хлористого бария и сравнили эти величины с данными,
полученными непосредственно из измерений упругости пара динамическим
методом. Совпадение результатов полностью подтвердило принципиальную
применимость обоих методов при данной температуре.
Относительное парциальное молярное теплосодержание £3 хлористого
бария было вычислено Типпетсом и Ньютоном на основании измерений
электродвижущих сил, причем их результаты хорошо согласуются с дан-
ными, полученными из калориметрических определений Ричардсом и Долом
[7] при .25°. Наряду с этим данные Типпетса и Ньютона при других тем-
пературах недостаточно надежны; это относится в особенности к экстре-
мальным температурам в условиях их опытов, т. е. к температурам 0
и 45°, так как значения относительной парциальной молярной теплоем-
кости J2, вычисленные на основании этих данных, сильно отличаются
390
ГЛАВА ХШ
от результатов калориметрических измерений. При 25° и концентрации
0,1 М Типпетс и Ньютон получили значение /2, равное 61, в то время
как, согласно калориметрическим измерениям Уайта [8], значение /2 рав-
няется 17,1. Это расхождение связано, невидимому, главным образом
с трудностями, встречающимися при экстраполяции данных, полученных
путем определений электродвижущих сил, а не с большими ошибками
в исходных измерениях.
Осмотические коэффициенты и коэффициенты активности хлоридов
марганца, кобальта, никеля, меди, магния, кальция, стронция и бария,
а также бромида и иодида магния были определены Робинзоном [9] путем
изопиестических измерений упругости пара для концентраций растворов
от 0,1 до 1,6, а в некоторых опытах—до 2 Л/. Стандартным раствором для
всех этих изопиестических измерений служил раствор хлористого натрия (см.
стр. 276 и 354). Значения у при 25° приведены в табл. 151. Для хлористых
бария и стронция, как видно из данных, приведенных в табл. 90, резуль-
таты изопиестических измерений совпадают с результатами, получен-
ными путем измерений электродвижущих сил. Кроме того, значения у+ ,
определенные при помощи кальциевого амальгамного элемента, сильно
отличаются от соответствующих значений, полученных путем измерений
упругости пара.
§ 2. Термодинамика водных растворов сульфатов лития,
натрия и калия
Для определения коэффициентов активности сульфатов лития, натрия
и калия при 25° Акерлёф [10] применял элементы типа
Hg-Hg2SO41M2SO4 (m) | MxHg | M2SO4 (mn) | Hg2SO4-Hg.
В своих более подробных исследованиях по термодинамике водных раство-
ров сернокислого натрия Харнед и Хеккер [11] измеряли электродвижущие
силы элементов
Pb-PbJlg-PbSO, | Na2SO4 (m) | NaJIg | Na2SO4 (0,05) | PbSO4-PbJIg-Pb.
Для этой соли имеются точные данные Рендалла и Скотта [12] о темпера-
турах замерзания, Ланге и Стрика[13]о теплотах разбавления и Рендалла
и Россини [14] о теплоемкости.
Так как с амальгамами щелочных металлов нельзя получить точных
результатов в разбавленных растворах (иг < 0,05 М), то экстраполяция
на нулевую моляльность с помощью уравнения (33) гл. X ненадежна.
В табл. 91 приведены значения у , полученные путем измерений электро-
движущих сил элементов с амальгамными электродами. В этой таблице
стандартные значения, полученные путем экстраполяции с помощью урав-
нения (33) гл. X, заключены в скобки. В третьем столбце таблицы содер-
жатся значения у^_ из данных о температурах замерзания, вычисленные
Рендаллом и Скоттом с помощью предельного уравнения Дебая и Гюккеля.
Полученное ими значение 0,537 при концентрации раствора 0,05 М незна-
чительно отличается от величины 0,532, полученной Харнедом и Хеккером
путем измерения электро движущих сил. Это различие объясняется в основ-
ном теми трудностями, которые встречаются при экстраполяции. Значения
при 25°, определенные путем изопиестического измерения упругости пара,
хорошо совпадают с величинами, полученными путем измерения электро-
движущих сил.
Средние коэффициенты активности сульфатов натрия, лития и калия
Таблица 91
Na2SO4 L12SO4 ' K2SO4 NagSaOe
т Температура, С
ОМ 02) 151) 251) 25«) 253) 351) 251) 25«) 251) 254) 251)
0,01 — (0,719) — — — — — —. — — —.
0,05 (0,532) 0,537 (0,530) (0,529) — (0,529) (0,523) — — — — —
0,075 0,482 — 0,482 0,480 — 0,480 0,475 — — — — —
0,1 0,446 0,449 0,447 0,445 (0,445) 0,445 0,440 (0,468) (0,468) (0,441) (0,441) 0,455
0,2 0,359 0,374 0,364 0,365 0,365 0,366 0,363 0,398 0,398 0,361 0,361 0,382
0,3 0,310 — 0,319 0,322 0,320 0,322 0,321 — 0,362 0,313 0,317 0,340
0,5 0,250 — 0,263 0,268 0,267 0,271 0,270 0,323 .0,322 0,265 0,264 0,292
0,7 — — 0,230 0,234 0,234 0,237 0,238 — 0,299 — 0,233 0,263
1,0 — — — 0,204 0,202 0,204 0,208 0,276 0,281 — — 0,236
1,2 — — — 0,189 0,187 0,187 0,194 — — — —
1,3 — — — 0,182 0,182 0,180 0,187 — — — — —
1,5 — — — 0,171 — — 0,272 0,269 — — 0,212
2,0 — — — — 0,153 — — 0,267 0,269 — — 0,200
1) По методу электродвижущих сил.
2) по данным для температур замерзания].
г) Вычислено по уравнению (33) гл. X. Параметры А=0,809; В=-0,0П; а=3,63.
4) По изопиестическим измерениям упругости пара [ 1 ].
392
ГЛАВА XIII
Харнед и Хеккер вычислили значения L2 и J2 для интервала темпе-
ратур 0 — 35° вплоть до концентрации 2 М на основании своих данных
относительно электродвижущих сил. Полученные ими результаты пред-
ставляют интерес для общей характеристики зависимости этих величин
от концентрации электролита и температуры, однако их точность весьма
невелика. Точное определение значений L2 и J2 с помощью калориметри-
ческих методов было осуществлено Уоллесом и Робинзоном [15] для темпе-
ратур 15, 20 и 25° в пределах от очень разбавленных растворов до 0,4 М.
Их данные можно представить следующими эмпирическими уравнениями:
15°; Г2 = 2650 т1/2-11382 т-ф 7080 т3/2, (2)
20°; Ь2 = 3100 пг112- 12164 т + 8340 т3/2, . . (3)
25°; Е2 = 3644 т1/2 -15468 т +22475 m3/2-15108 m2, (4)
25°; 72 = 84m1/2, (5)
25°; C’P2 = 84mf/2-50. (6)
§ 3. Водные растворы галогенидов цинка, кадмия и свинца
Свойства растворов этих солей были исследованы при помощи элемен-
тов типа
MJHg (одно- или двухфазн.) | МХ2 (m) | AgX-Ag.
Согласно уравнению (21) гл. X, электродвижущая сила такого элемента
равняется
Е = Е° - 4 In Y±m = Е° - 4 In Y±m (4)1/3. (7)
Z Г 1 ± ± АГ
После подстановки значения коэффициента активности из предельного закона
Дебая и Гюккеля с дополнительным линейным членом получаем следу-
ющую разновидность уравнения (24) гл. X:
Е°'= Е+wln т (4)1/3 -S? 2’303 = Е° - ¥ Вт- (8)
При 25° это выражение приобретает вид
Е0' =Е + 0,08873 lgm + 0,01783 — 0,155/m = E°-/(m), (9)
если для упрощения принять т — с. Зависимость Е0' — Е° от концентрации
для некоторых галогенидов кадмия, свинца и цинка показана на рис. 106.
Эти кривые аналогичны кривым Скэтчарда и Тефта [16]; применявшийся
этими авторами метод обработки экспериментальных результатов, получен-
ных для этих растворов, будет изложен ниже. Измерения электродвижущих
сил в случае хлористого кадмия были произведены Хоршем [17], Харне-
дом и Фицджеральдом [18], в случае хлористого свинца — Кармоди [19],
для хлористого цинка — Скэтчардом и Тефтом. Кривые для йодистого
цинка [20] и бромистого [21] и йодистого [22] кадмия построены на осно-
вании данных Бэйтса и Восбурга [23]. Следует отметить, что на кривых
для хлористого и йодистого цинка отсутствует максимум, в то время как
в случае хлористого, бромистого и йодистого кадмия на кривых появляется
максимум, величина которого возрастает при переходе от хлорида к иодиду.
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
393
Существуют два способа обработки этих экспериментальных данных.
Первый из них заключается в применении обобщенной теории Дебая и Гюк-
келя. Ла-Мер, Гронвол и Гриф воспользовались этим Методом и получили
следующие значения среднего расстояния сближения ° ионов соответственно
для хлоридов цинка, свинца и кадмия: 3,8; 1,75 и 1 А. Позднее Бэйтс [21]
использовал уравнение Дебая и Гюккеля с дополнительными членами для
экстраполяции своих данных по электродвижущим силам элемента, содер-
жащего раствор бромистого кадмия. Lgy± вычислялся по уравнениям (41)
и (43) гл. III и по данным для
значений а из табл. 8. Путем
подстановки этих значений в
уравнение (7) были вычислены
предварительные значения Е°
для концентраций 0,0005 —
0,01 М. Полученные значения
Е° представлены графически как
функции от Ут на рис. 107.
Наилучшие результаты получа-
ются, если а_ принять равным
0,6. ПЛтоянство значений при-
ближенной величины Е° сохра-
няется только для концентраций
ниже 0,001 М• Поскольку тео-
рия применима только при та-
ких низких концентрациях и
значение а = 0,6 вряд ли мо-
жет иметь . реальный физиче-
ский смысл, то, вероятно, сле-
дует учитывать влияние дру-
гих факторов, кроме тех, ко-
торые определяются простыми
кулоновскими силами между-
ионного притяжения. Второй
метод, описанный Скэтчардом
и Тефтом [16] и усовершенство-
галогенидов свинца, цинка и кадмия при 25°.
ванный Харнедом и Фицджеральдом [18], основан на предположении о том,
что имеет место неполная диссоциация согласно реакциям следующего типа;
МХ2->МХ+4-Х“
и
МХ+ХМ++ + Х“ или МХ+ + Х-ХМ++4-2Х“.
К этим реакциям применяется закон действующих масс. В случае раство-
ров хлористого и бромистого кадмия, повидимому, как МХ2, так и MX ‘
диссоциируют неполностью, однако мы будем предполагать, что на первой
ступени диссоциация происходит нацело. В случае растворов йодистого кад-
мия это предположение является неправильным. Пусть а —степень дис-
социации МХ+, а К — константа диссоциации этого равновесия; если при-
нять, что Ya(X) = Ya(MX)> т0 “ определяется уравнением
-2—J + V ( ! + —) 4-4--- —
ТО7а(М)/ Г Ч mA(M)/ ТОК(М)
(Ю)
394
ГЛАВА XIII
где уа(М) — истинный коэффициент активности иона М++, равный ам/атп, при-
чем а.т— истинная концентрация ионов. Это означает, что величина у М)
должна быть того же порядка, что и значения, полученные для сильных
электролитов, например для хлористого бария. Поэтому можно вычислить
величину у м для разбавленных растворов по уравнению Дебая и Гюк-
келя:
(И)
Мы приняли для величины а значение 5, т. е. значение того же порядка,
какое было получено для хлористого
0,428
0,426
0,422
0,420
0,424
Е°
0,1
Е°
се-
0,4/8
О 0,02 0,04 0,06 0,08
\/т
Рис. 107. Приближенные значения
для элемента амальгама кадмия-бромид
ребра при 25°, вычисленные по уравнению
(43) гл. III.
бария. При таком значении а влия-
ние дополнительных членов теорети-
ческого уравнения настолько мало,
что при наших вычислениях этими
членами можно пренебречь. Отме-
тим, что 2р' = где rn'i — ис-
тинная ионная концентрация, в от-
личие от стехиометрической * Вели-
чины а, соответствующие данным
значениям К и т, можно получить
следующим образом. Задается неко-
торое исходное значение а для дан-
ных К и
зуют для
НИЮ
т. Это значение
вычисления р' по
исполь-
уравне-
р' = (2а 4-1) т.
(12)
Затем находят уа из уравнения (11).
После этого вычисляют новое
значение а по уравнению (10), ис-
пользуя эту величину уа(М). Полу-
ченное новое значение а вновь при-
и найденное значение р' подстав-
для определения уа(М . Эту процедуру повто-
не будет достигнуто постоянство значений а с
Ya(M) И Р
меняют для вычисления
ляют в уравнение (И)
ряют до тех пор, пока
точностью до 0,1%.
В уравнении (7) фигурируют стехиометрический коэффициент актив-
ности и моляльность. Величину Е можно выразить также с помощью урав-
нения
Е = Е° — 2р In а (1 + а)2 ш3у®,
(13)
где уа — «истинный» средний коэффициент активности, а(а4-1)2ш3—произ-
ведение истинных концентраций. Согласно определению Е0' по уравне-
нию (8), имеем
Е°'-Е° = Е-Е°In4ш3-1^2,303 . (14)
1 Такое обозначение применяется в гл. XV.
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
395
Путем сочетания этого выражения с уравнением (13) получаем
Е0'-Е0 = |У1п4-|^1па(14-а)3-
“4¥ln Ya-12,303 &(/) . (15)
С помощью значений а, вычисленных вышеописанным способом для
различных К, и значений у0, которые принимаются равными значениям
Рис. 109. Экстраполяционные функции
для 0, 25 и 40° (шкала в мв, диаметры
кружков равны 1 же).
Кривые без кружков—зависимость Е0'—Е° от
Ут—рассчитаны по уравнению (15). Белыми
кружками обозначены значения Е0', соответ-
ствующие уравнению (8); черными кружками—
значения Е° [уравнение (16)]. Константы дис-
социации равны 0,01, 0,011 и 0,013 соответ-
ственно при 40, 25 и 0°.
Рис. 108. Зависимость Е0' —Е° от Ут
согласно уравнению (15) для различных
значений К.
той же величины, найденным при из-
мерениях с сильными 2,1-электроли-
тами вроде хлористого бария, мож-
но определить фактор отклонения
Е0' —Е°. Результаты таких вычислений изображены графически на рис. 108
для значений К от 0,005 до бесконечности. При сравнении рис. 108
и 107 можно заметить большое сходство, которое указывает на то, что
эта теория объясняет поведение хлоридов бария, цинка, свинца и кадмия
в разбавленных растворах, если принять для константы диссоциации реак-
ции MX+^tM++4-X- соответственно следующие значения: со; 0,1; 0,04
и 0,01.
Пример более тщательного применения этого метода можно найти
в работе Харнеда и Фицджеральда [18], которые подробно исследовали
водные растворы хлористого кадмия. На рис. 109 представлена получен-
ная этим методом зависимость фактора отклонения Е0' —Е° от У т при
температурах 0, 25 и 40°. Результаты хорошо согласуются со значениями
Е0', вычисленными из экспериментальных данных по уравнению (9). Зна-
чения применявшихся при расчете констант диссоциации, которые лучше
всего согласуются с опытными данными, приводятся в подписи к рис. 109
1 Эти результаты приблизительно согласуются со значением 0,010, вычисленным
из данных по электропроводности Риджеллато и Дэвисом [24].
396
ГЛАВА XIII
Путем преобразования уравнения (13) можно выразить Е° следующим
уравнением:
' Е° = Е + In а (1 + а)1 2 т^. (16)
Точки, необходимые для экстраполяции Е°, были получены путем прибав-
ления к экспериментальным значениям Е второго члена правой части
уравнения (16). Успешный результат экстраполяции показывает, что пу-
тем простого сочетания закона действующих масс и предельного закона
Дебая и Гюккеля можно разработать хороший метод экстраполяции.
Бэйтс [21] тщательно исследовал растворы бромистого кадмия с по-
мощью элемента
Cd-CdaHg | CdBr2 (m) | AgBr-Ag
и также определил стандартный потенциал для температур 0 — 40°. На
основании этого значения, а также значений стандартных потенциалов
электродов серебро-хлористое серебро и серебро-бромистое серебро [урав-
нения (5) гл. XI и (10) гл. XII] можно вычислить стандартный потен-
циал электрода Cd-Cd^Hg | Cd++(а == 1). В табл. 152 приведены стандарт-
ные потенциалы, полученные на основании этих вычислений. В послед-
них двух столбцах значения, полученные Харнедом и Фицджеральдом,
-сравниваются с данными Бэйтса. При 25, 30, 35 и 40° наблюдается очень
хорошее совпадение, однако при более низких температурах появ-
ляется некоторое расхождение результатов, возрастающее с понижением
температуры. Это различие пока еще не поддается объяснению.
Бэйтс и Восбург измеряли электродвижущие силы элементов
Cd-CdJIg | CdJ2 (да) | Hg2J2-Hg,
содержавших в одном случае только растворы йодистого кадмия, в дру-
гом—растворы сульфата и иодида кадмия и в третьем — растворы йоди-
дов кадмия и калия. Таким путем был получен стандартный потенциал
электрода ртуть-иодид одновалентной ртути [23], определен коэффициент
активности иодида кадмия [25], а также были исследованы многочислен-
ные равновесные системы, содержащие эти растворы [26], при 25°].
Бэйтс [22] определил термодинамические величины у , Z2 и J2 с помощью
элемента
Cd-Cd^Hg | CdJ2 (да) | AgJ-Ag
в интервале'температур 5 — 40°.
Растворы хлорида [16, 28а]2, бромида [29] и иодида цинка [20] были
исследованы с помощью элементов
Zn-ZnxHg | ZnX2 (да) | AgX-Ag.
Работа Робинзона и Стокса по изучению растворов хлористого цинка
и работа Бэйтса по исследованию растворов йодистого цинка охватывают
интервал температур 5 — 40°, в то время как Партон и Митчел изучали
растворы бромистого цинка только при 25°. Стандартные потенциалы этих
элементов, а также двухфазных электродов с амальгамой цинка приво-
дятся в табл. 153. Совпадение значений стандартных потенциалов послед-
него электрода, полученных различными авторами, является очень хоро-
1 Более старые исследования галогенидов кадмия см. [27].
2 Числа переноса из данных для элементов с жидкостным соединением приведены
в работе Харриса и Партона [286].
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
397
шим. С помощью стандартных потенциалов элементов и их электродви-
жущих сил по уравнению (7) вычислены значения f±, приведенные в табл. 154.
Относительное парциальное молярное теплосодержание L2 и относи-
тельная парциальная молярная теплоемкость /2 хлорида и иодида цинка,
а также хлорида, бромида и иодида кадмия были вычислены с помощью
уравнений (48) и (50) гл. X на основании данных, полученных путем
измерений электродвижущих сил. Для хлорида и иодида цинка эти вели-
чины можно вычислить на основании данных, приведенных в табл. 155.
Робинзон и Уоллес [30] определили Л2 и /2 вплоть до концентра-
ции 0,2 М из данных по теплотам разбавления растворов. Их результаты,
приведенные в табл. 156, сильно отличаются от величин, вычисленных
по данным для электродвижущих сил, и являются, несомненно, более
точными. Критический разбор методов, применяемых для вычисления зна-
чений L2 и J2 из данных об электродвижущих силах, может внести яс-
ность в этот вопрос.
§ 4. Общие соображения относительно водных растворов
1,2- и 2,1-электролитов
Зависимость коэффициентов активности 1,2- и 2,1-галогенидов и суль-
фатов, а также гидроокиси бария [31] при 25° от т показана на
рис. 110. Верхняя группа кривых от хлористого бария до йодистого маг-
ния включительно отвечает сильным электролитам. Для этих солей вели-
Р и с. 110. Средние коэффициенты активности 1,2- и
2,1-электролитов при 25°.
чина а (среднее расстояние сближения ионов) лежит приблизительно
в пределах от 4А для хлористого бария’ до 6Аодля иодистых магния и цин-
ка. Эти значения меньше, чем величина </=7А, соответствующая теории
ассоциации Бьеррума для 2,1- и 1,2-электролитов, и, следовательно,
в этом случае можно ожидать, что в некоторой степени будет происхо-
дить ассоциация ионов. В любом из этих растворов могут присутствовать
ионы типа [М++Х~] или [МХ]+, причем в концентрированных растворах
хлористого бария их количество может быть значительным. Необходимо
отметить высокие коэффициенты активности йодистого цинка и хлористого
398 ГЛАВА XIII
кобальта. Средняя группа солей, включающая сульфаты щелочных метал-
лов и Гидроокись бария, для которой значение а равно 3 — 4, обнаружи-
вает большую склонность к ассоциации с образованием ионов типа [MSO4]-'
и [Ва (ОН)]+. Хлористый свинец и неполностью диссоциированные галоге-
ниды кадмия составляют третью группу. Обрабатывая данные по электро-
движущим силам с помощью метода, описанного в предыдущем параграфе,
Харнед и Фицджеральд получили для константы диссоциации [CdCl]+
при 25° значение 0,011. Таким же путем Бэйтс [21], а также Бэйтс и Вос-
бург [26] получили значения 0,006 и 0,004 для констант диссоциа-
ции [CdBr]+ и [CdJ]+. На основании результатов Кармоди [32] для эле-
ментов с хлористым свинцом можно определить константу диссоциации
[РЬС1]+, которая имеет величину порядка 0,03.
Эти наблюдения находятся в соответствии с интерпретацией данных
по электропроводности, принадлежащей Онзагеру, а также Риджеллато
и Дэвису (гл. VI, § 3). Эти исследователи обнаружили, что группа солей
от хлористого бария до хлористого магния состоит из очень сильных элек-
тролитов. Вычисленные константы диссоциации [LiSO4]_, [NaSO4]~ и [KSO4]_
равны соответственно 0,23; 0,20 и 0,15 и располагаются в той же после-
довательности, что и коэффициенты активности. Ионы [CaNO3]+, [SrNO3]+
и [BaNO3]+ имеют константы диссоциации того же порядка. Наконец, зна-
чения 0,03 и 0,01 для констант диссоциации [РЬС1]+ и [CdCl]+ хорошо
совпадают со значениями, вычисленными из термодинамических экспери-
ментальных данных. в
Кривые для коэффициентов активности бромида и хлорида цинка
обнаруживают аномальное поведение. В разбавленных растворах обе соли
ведут себя как сильные электролиты; на' основании измерений электро-
движущих сил в этом случае получаются значения а, равные приблизи-
тельно .5—6. При концентрации раствора, превышающей 0,3 М, кривые
для коэффициентов активности этих солей начинают снижаться, пересе-
кая кривые для многих других электролитов. Причина такого поведения
коэффициентов активности этих солей не ясна.
§ 5. Термодинамика водных растворов сернокислого цинка
Брэй [33]'измерял электродвижущие силы элемента
Zn-ZnJIg | ZnSO4 (m) | PbSO4-PbJIg-Pb
при 25° в широком интервале концентраций растворов. Позднее Кауперс-
вейт ’и Ла-Мер [34] исследовали этот элемент при нцзких концентра- -
циях (0,0005 — 0,01 М) в интервале температур 0 — 50°. Относительное
парциальное теплосодержание £2 для низких концентраций определили '
Ланге, Монгейм и Робинзон [35]. Результаты этих исследований могут
служить для иллюстрации некоторых важных свойств 2,2-электролитов.
В дальнейшем, мы будем пользоваться величиной Е0', определяемой
для данного элемента уравнением
EO' = E + ^lnm = E«-^lnY±. (17)
На рис. 111 представлена зависимость Е0' от j/m, полученная на основа- ,
нни данных Кауперсвейта и Ла-Мера. Прямые линии соответствуют пре-
дельному уравнению теории Дебая и Гюккеля. Заметное отклонение от j
предельного закона, характеризующееся выпуклостью кривых, согласуется j
с предсказанием дополненной Гронволом, Ла-Мером и Сэндведом теории j
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
399
Дебая. Повидимому, этот случай представляет собой пример наиболее
строгой, проверки обобщенной теории водных растворов.
Применяя уравнение (42) гл. III, Кауперсвейт и Ла-Мер нашли зна-
чение а, с помощью которого получалось строгое согласие теоретических
результатов с их экспериментальными данными, поскольку вычисленная
ими величина Е° оказалась постоянной во всем интервале концентра-
ций 0,0005 — 0,0171/. Более того, величина а оказалась постоянной и рав-
ной 3,64 для всех температур между 0 и 50°. Хотя при учете добавочных
эффектов, пропорциональных концентрации, численные результаты могут
несколько измениться, указанные концентрации слишком низки для того,
чтобы влияние этих добавочных эффектов по своей величине могло быть
сравнимым с влиянием, обусловленным дополнительными членами.
Прямые линии соответствуют предельному закону.
Величины £2, полученные из этих измерений электродвижущих
сил [36], хорошо согласуются с результами калориметрических определе-
ний Ланге, Монгейма и Робинзона при концентрациях 0,001 — 0,01 М,
как видно из табл. 92. Последние калориметрические значения были
уменьшены нами на 25 кал в соответствии с нашей методикой экстрапо-
ляции, описанной в гл. VIII, § 2.
Брэй [37] определял коэффициенты активности для концентрирован-
ных растворов по методу электродвижущих сил, а Робинзон и Джонс [38] —
Таблица 92
Относительное парциальное молярное теплосодержание сернокислого
цинка при 25° по данным определений электродвижущих сил
и по калориметрическим измерениям
т 0 ,0005 0,001 0,002 0,005 0,01
L2, э. д. с (198) 385 480 674 772
L2, калор 227 327 463 665 765
400
ГЛАВА XIII
по методу изопиестического измерения упругости пара. В табл. 93 приве-
дены эти значения для 25°. Величина у± = 0,387 для концентрации рас-
твора, равной 0,01 М, была взята из работы Кауперсвейта и Ла-Мера
и использована для пересчета данных Брэя. Для получения результатов,
основанных на данных об упругости пара, в качестве исходной величины
было взято значение у± —0,150, также найденное из данных по электро-
движущим силам при концентрации раствора 0,1 М. Во всем интервале
концентраций, в котором измерения производились обоими методами,
наблюдается достаточно хорошее совпадение.
Для 2,2-электролитов # = 14 А. Так как Кауперсвейт и Ла-Мер полу-
чили для раствора сернокислого цинка значение я = 3,6, то можно пред-
полагать, что эта соль диссоциирована неполностью. Принимая это зна-
чение а, получаем по уравнению Бьеррума [(57) гл. Ill], что К = 0,003.
Оуэн и Гэрри [39] измерили электропроводность растворов сульфатов
цинка и меди в воде при 25° и получили из этих данных значения кон-
стант диссоциации, равные соответственно 0,0049 и 0,0043. Согласно дан-
ным Дэвиса [40а], величина К для обеих указанных солей равна 0,0045
при 18°.
Таблица 93
Средний коэффициент активности -1± сульфата цинка в водных растворах при 25°
Метод измерения Э. д. с. У пр. пара Метод изме рения т Э. д. с. Упр. пара
0,0005 ,0,780 — 0,2 0,104 0,104
0,001 0,700 — 0,5 0,0634 0,0626
0,002 0,608 — 1,0 0,0438 0,0434
0,005 0,477 — 1,5 0,0372 0,0378
0,01 0,387 — 2,0 0,0354 0,0350
0,02 0,298 — 2,5 0,0368 0,0360
0,05 0,202 — 3,0 0,0409 0,0397
0,1 0,150 (0,150) 3,5 0,0478 0,0467
§ 6. Коэффициенты активности сульфатов двухвалентных металлов
в водных растворах
Хотя термодинамические свойства водных растворов сульфатов кад-
мия, меди, магния, марганца и никеля изучены не так подробно, как
термодинамические свойства сульфата цинка, все же имеется достаточно
данных, свидетельствующих о том, что поведение коэффициентов актив-
ности этих солей представляет значительный интерес. Во-первых, из данных
о температурах замерзания разбавленных растворов явствует, что коэффи-
циенты активности всех этих электролитов имеют почти одинаковые вели-
чины. Так, например, для концентрации раствора, равной 0,1 М, различ-
ные исследователи получили для коэффициентов активности сульфатов меди,
кадмия, магния и цинка соответственно значения 0,149; 0,150; 0,166
и 0,160. Далее, Ла-Мер и Паркс [406] получили путем измерения электро-
движущих сил следующие значения у для сернокислого кадмия: 0,699;
0,476 и 0,383 при концентрациях растворов, равных соответственно 0,001;
0,005 и 0,01 М. Эти значения почти совершенно точно совпадают с дан-
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
401
ними для сернокислого цинка, приведенными в табл. 93. Ошибки непо-
средственных измерений, а также методов экстраполяции имеют, вероятно,
тот же порядок величины, что и разброс значений у±, полученных издан-
ных о температурах замерзания. Различия между отдельными двухва-
лентными сульфатами оказываются во всех случаях действительно очень
незначительными.
Таблица 94
Средние коэффициенты активности сульфатов двухвалентных металлов при 25°
т ZnSO4 CdSO4 MnSO4 MgSO4 CuSO4 N1SO4
о,1 (0,150) (0,150) (0.150) (0,150) (0,150) (0,150)
0,2 0,104 0,102 0,1056 0,1077 0,1043 0,1049
0,3 0,0831 0,0815 0,0850 0,0877 0,0834 0,0841
0,4 0,0708 0,0692 0,0728 0,0720 0,0708 0,0713
0,5 0,0626 0,0609 0,0643 0,0678 0,0624 0,0628
0,7 0,0520. 0,0501 0,0532 0,0574 0,0515 0,0516
1,0 0,0434 0,0411 0,0441 0,0488 — 0,0426
1,5 0,0368 0,0342 0,0373 0,0430 0,0360
2,0 0,0350 0,0318 0,0351 0,0419 — 0,0343
2,5 0,0360 0,0315 0,0353 0,0441 — 0,0357
3,0 0,0397 0,0327 0,0375 0,0495 — —
3,5 0,0467 0,0351 0,0416 — — —
4,0 — —. 0,0478 — — —
4,25 — 0,0518 —. — —
Этот вывод подтверждается также результатами изопиестических измере-
ний упругости пара, произведенных Робинзоном и Джонсом [38] в рас-
творах с высокой концентрацией (0,1 — 0,4 М). Так, если в качестве примера
принять, что для всех этих сульфатов при концентрации 0,1 М и при
25° у± = 0,150, то для других концентраций получаются значения, при-
веденные в табл. 94. Анализ этих значений показывает, что даже в кон-
центрированных растворах индивидуальные различия между отдельными
2,2-электролитами проявляются в значительно меньшей степени, чем в случае
1,1- и 2,1-электролитов.
§ 7. 1,3-, 3,1-, 3,2-, 1,4-Электролиты
Систематические данные для этих электролитов несимметричного типа
отсутствуют. Только в одном случае (хлористый лантан) имеются данные
для достаточно разбавленных растворов, которые могут быть использованы
для экстраполяции [41]. На основании результатов измерения температур
замерзания были вычислены коэффициенты активности нитрата лантана [42],
феррицианида калия [43], кобальтициацида калия [44] и сульфата лан-
тана [45]. Кроме того, были сделаны изопиестические измерения упругости
пара растворов хлорида лантана [46 — 48], ферроцианида калия [46], суль-
фата алюминия [46] и некоторых 3,1-хлоридов редкоземельных метал-
лов [48]. Хаттокс и Де-Фриз [49] исследовали водные растворы сернокислого
индия при температурах 0 — 35° при помощи элемента ...
In | In2r(SO4)3 (тп) | Hg2SO4-Hg.
Путем измерения электродвижущей силы элемента
Ag-AgCl |, LaCI3 (сх)’| LaCl3 (C2®’AgCl-Ag
26 г. Харнед, ДЗ.'Оуэн
402
ГЛАВА XIII
и пользуясь значениями чисел переноса, Шидловский и Мак-Иннес [41]
определили коэффициенты активности хлористого лантана в пределах кон-
центраций 0,0006 — 0,03333 М с помощью метода, описанного в гл. XII, § 1.
В результате допущенной при вычислениях ошибки был сделан не-
правильный вывод о том, что разбавленные водные растворы хлористого
лантана не подчиняются теории. Мы произвели вычисления заново и пока-
зали, что в первом приближении опытные данные могут быть описаны
формулой Дебая и Гюккеля
. 3,7446j/c /j1CV
1g У л. —------2—7= , (18)
14-4,75 Ус '
которая дает вполне приемлемое значение для среднего расстояния сближе-
ния ионов (5,9 А.). Значения теоретических функций в этом уравнении
были вычислены на основании значений универсальных констант, при-
веденных в табл. 168. Средняя величина разности между вычисленными
и опытными результатами составляет 0,0015 единицы
Таблица 95
Средние коэффициенты активности хлорида лантана,
ферроцианида калия, сульфатов 'алюминия и индия при 25°
т ,ЬаС13 K4Fe(CN)e [1] Al2(SO4)3 [1] In2(SO4)3 [2] И
0,001 0,788 —
0,005 0,637 — — —
0,01 0,559 — —. 0,142
0,02 0,484 — — 0,095
0,03 0,442 — — 0,071
0,05 0,392 (0,189) — 0,054
0,1 0,336 .0,138 (0,0350) 0,035
0,2 0,293 0,107 0,0223 0,022
0,3 0,281 0,088 0,0174 0,017
0,4 0,280 0,076 0,0151 0,015
0,5 0,285 0,067 0,0115 —
0,7 0,305 0,055 0,0133 —
1,0 0,366 0,048 0,0176 —
1,2 0,426 — — —-
1,4 0,503 — . —. —
1,5 0,554 — —
1) Результаты получены для 0, 15 и 30°. Значения Ь2 оценены.
Судя по характеру отклонений опытных данных от теоретических, величину
этой разности можно уменьшить посредством использования дополнительных
членов теории Дебая и Гюккеля1. Поскольку при низких концентрациях
значения мало отличаются от у±, то это уравнение было использовано
для вычислений при концентрации 0,05 М, в результате чего было полу-
чено значение у±, равное 0,392.
1 Шидловский сообщил авторам, что он пересчитал свои результаты заново. Путем
применения формулы Дебая и Гюккеля с добавочным членом Dc\gc ему удалось
снизить среднюю величину расхождения между теоретическими и опытными данными
до 0,0002.
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ 403
Робинзон [46] и Мэзон [48] произвели изопиестические измерения
упругости пара растворов хлористого лантана при высоких концентрациях
(0,05 — 2 М). Коэффициенты активности, полученные с помощью этих
измерений, были приведены в соответствие с величинами, полученными
для низких концентраций [47]; они представлены в табл. 95. Мэзон исполь-
зовал этот метод также для изучения концентрированных растворов хлоридов
алюминия, скандия, иттрия, церия, празеодимия и неодимия. Полученные
им данные приводятся в табл. 157.
1,4- и 3,2-электролиты представлены в табл. 95 ферроцианидом калия,
сульфатом алюминия [46] и сульфатом индия [49]. В этих случаях экстра-
поляция не вполне надежна, но полученные результаты имеют правильный
порядок величин и могут служить в качестве иллюстрации поведения
электролитов этого типа валентности. Для дальнейшего развития теории
растворов этих электролитов необходимы подробные экспериментальные
данные для очень разбавленных растворов.
§ 8. Определение константы диссоциации HSO7
из данных об электродвижущих силах
Последующие параграфы будут в основном посвящены рассмотрению
коэффициентов активности, относительных парциальных молярных тепло-
содержания и теплоемкости серной кислоты в водных растворах. Эти вели-
чины вычисляются из опытных данных, получаемых путем измерения
электродвижущих сил элементов
Н21H2SO4 (т) j PbSO4-PbO2-Pt,
Н21H2SO4 (m) | Hg2SO4-Hg.
Так как серная кислота является частично слабым электролитом, то при
определении стандартных потенциалов этих элементов возникают затрудне-
ния, для устранения которых нужно знать величину константы диссоциации
дантй04‘(н‘(304
А 2Д = "--------- •
mHS04lHS04
Эта величина была определена Гамером [50] с помощью элемента
Н21 NaHSO4 (m1),Na2SO4 (m2),NaCl (m3) | AgCl-Ag,
а также Юнгом, Клотцом и Синглетерри *, которые применяли спектрофото-
метрический метод. В табл. 96 приведены значения, полученные с помощью
обоих методов при различных температурах. Значения, относящиеся к 25°,
находятся в хорошем согласии с величиной 0,0118 (при концентрации,
выраженной в единицах моляльности), вычисленной Шериллом и Нойесом
[52] из данных по электропроводности и числам переноса, которые были
получены Нойесом и Стюартом [53]. Последние авторы внесли в свои резуль-
таты поправки к значениям подвижностей и коэффициентов активности
согласно теории междуионного притяжения. Отметим, что эта величина
0,0118 гораздо ниже значений 0,03, 0,02 и 0,017, полученных ранее другими
исследователями [54], не вводившими всех необходимых поправок,
Вычисление К%А из результатов измерений электродвижущих сил за-
труднительно и связано со сложными арифметическими приближенными
вычислениями [50]. В гл. XV будет показано, что точное определение
констант диссоциации, порядок величины которых меньше 10~3, может быть
осуществлено путем измерений электродвижущих сил элементов без
1 Частное сообщение профессора Т. Ф. Юнга, основанное на диссертациях Клотца
и Синглетерри [51].
26*
404
ГЛАВА ХШ
жидкостных соединений. В случае констант диссоциации, превышающих 10~3,
получить точные результаты значительно труднее. Так как К^а для HSO7
равно приблизительно 10~2, то приведенные данные являются приближен-
ными .
Таблица 96
Константа диссоциации HSOj
i, °C t, ’С К2А
метод э. д. с. [1] спектрофото- метрический метод 1) метод э. д. с. [1] спектрофото- метрический метод 1).
0 0,0148 35 0,0105 0,0075
5 0,0143 0,0180 40 0,0097 —
10 0,0139 — • 45 0,0089 0,0056
15 0,0134 0,0136 50 0,0079 —
20 0,0127 .— 55 0,0070 0,0041
25 0,0120 0,0101 60 0,0060 —
30 0,0113 —
1) Смотри сноску на стр. 403.
Значения констант диссоциации, определенные оптическим методом,
сильнее зависят от температуры, чем результаты, полученные путем из-
мерений электродвижущих сил, и приводят к значению теплоты диссоциа-
ции — АН® = 5200 ± 100 кал, совпадающему со значением 5200 кал, найден-
ным Питцером [55], тогда как значение — полученное путем измерения
электродвижущих сил, оказалось равным 2200 кал. Для устранения этого
расхождения необходимо уточнение методов вычисления К2а из данных
об электродвижущих силах.
§ 9. Стандартный потенциал элемента [56]
H2|H2SO4(m)|PbSO4-PbO2-Pt
а) Определение стандартного потенциала Е° эмпирическим методом.
Если химическую реакцию, происходящую в изучаемом элементе, изобразить
уравнением
Н2 + РЬО2 (тв.) + H2SO4 (m) = PbSO4 (тв.) + 2Н2О,
электродвижущая сила этого элемента определяется выражением
Е = Е° + (RT/2Y) In m^mS04 + (RT/2¥) In y|ys04 - (7?T/2F) In a2w, (20)
где E — измеряемая электродвижущая сила элемента, Е° — стандартный
потенциал. Активность воды в растворе серной кислоты с моляльностью т
обозначена через аш.
Сначала мы используем эмпирический метод экстраполяции [57а].
Уравнение (21) можно преобразовать следующим образом:
E-(/?7,/2F)lnm^mSo4 = E0 + (^/2F)lnYHYso4-(/?7,/2F)lnal (21) *
Если левую часть этого уравнения изобразить графически как функцию
случае очень разбавленных растворов в значения электродвижущих сил вноси-
лась поправка, учитывающая растворимость PbSO4 [56].
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
405
от квадратного корня из моляльности, то в случае разбавленных растворов
должна получаться прямая линия. При нулевой концентрации эта функция
равна Е°, так как в этих условиях последние два члена в правой части
уравнения по определению равны нулю.
Для проверки надежности этого способа экстраполяции второй член
правой части уравнения (21) был вычислен при помощи коэффициентов
активности при 0°, найденных. Рендаллом и Скоттом [12] путем определения
точек замерзания. Очевидно, что разность между этим членом и левой
частью того же уравнения (21) равна значению Е°, если пренебречь очень
малым членом, содержащим aw. Данные, полученные на основании измере-
ния электродвижущих сил, располагаются параллельно этой кривой, как
показали опыты Рендалла и Юнга
[58], относящиеся к другим элек-
тролитам. На рис. 112 изображе-
на экстраполяция левой части
уравнения (21), представленной
в виде функции от для
температур 0, 25, 45 и 60° и для
концентраций растворов 0,0005 —
0,01 М. Нижняя кривая получена
на основании данных о температу-
рах замерзания. Для более отчет-
ливого сравнения с данными, по-
лученными путем измерений элек-
тродвижущих сил, из функции, вы-
численной по температурам замер-
зания, вычиталось 0,002 в. Кривая,
полученная по температурам за-
мерзания, имеет небольшую выпу-
клость при концентрациях ниже
0,002 М, но это может быть обу-
словлено некоторой неопределен-
ностью, допущенной Рендаллом
ции j/m1^ в случае разбавленных
Рис. 112. Зависимость левой части уравне-
ния (21) от )1^2.
и Скоттом при экстраполяции функ-
растворов. Значения, отвечающие точкам,
вычисленным из данных для электродвижущих сил, недостаточно точны
для того, чтобы по ним можно было проверить наличие этой выпуклости,
и поэтому для всех остальных температур были проведены прямые линии.
Значения стандартных потенциалов, полученные этим путем, приведены
во втором столбце табл. 97. После обработки этих значений по методу
наименьших квадратов с целью представить их в форме квадратного урав-
нения было получено следующее выражение:
Е°=1,67694 + 342,17 • 10-6^ + 83,11 • ICW2. (22)
Вычисленные по этому уравнению значения Е° отличаются от опытных
значений не больше чем на 0,1 мв.
б) Вычисление стандартного потенциала Е° с помощью константы
диссоциации IISO?. Применяя описанный выше метод экстраполяции, мы
пользовались стехиометрическими концентрациями. Но, как известно, серная
кислота представляет собой умеренно сильный электролит и вторая стадия
диссоциации протекает не до конца. Это обстоятельство подтверждается
рис. 112, из которого видно, что наклоны экспериментальных кривых
значительно больше, чем наклоны прямых, соответствующих предельному
закону Дебая и Гюккеля для полностью диссоциированного электролита.
406
ГЛАВА XIII
Преимуществом описанного выше метода экстраполяции является его
простота, однако он обладает двумя серьезными недостатками. Во-первых,-
если по оси абсцисс нанесено т1^, то расстояние от последней эксперимен-
тальной точки до нулевой концентрации оказывается слишком большим.
Таблица 97
Стандартные потенциалы элемента
H2|H3SO4(m)|PbSO4-PbO2-Pt
t, °C Использованное уравнение Д (мв)
(21) (25)
0 1,67694 1,67694 0
5 1,67870 1,67846 0,24
10 1,68045 1,67998 0,47
15 1,68228 1,68159 0,69
20 1,68411 1,68322 0,89
25 1,68597 1,68488 1,09
30 1,68800 1,68671 1,29
35 1,68991 1,68847 1,45
40 1,69196 1,69036 1,60
45 1,69396 1,69231 1,65
50 1,69616 1,69436 1,80
55 1,69831 1,69649 1,82
60 1,70044 1,69861 1,83
Во-вторых, для электролитов, диссоциирующих неполностью, экстраполяция
до нулевой концентрации производится с помощью прямой линии, не соот-
ветствующей предельному закону Дебая и Гюккеля. Ниже будет описан
другой метод, который в отличие от предыдущего дает результаты, согласу-
ющиеся с предельным законом.
Если значение истинной концентрации ионов водорода, определяемое
уравнением
^2А = (^hWso4YhYso4) / (Whso4Yhso4), (23)
подставить в уравнение (21), то получается
Е = Е° +(ЙТ/2F) 1п7ПН7Пнзо4 + (ЙГ/2Е) 1hyhYhso4 +
+ (Z?T/2F) ЩК2А/аЪ). (24)
При выводе этого уравнения делается несколько предположений. Во-первых,
допускается, что серная кислота полностью диссоциирует на ионы Н+ и
HSOi". Во-вторых, считается, что т представляет собой истинную, а не
стехиометрическую концентрацию присутствующих веществ. Последнее
дает возможность воспользоваться предельным законом Дебая и Гюккеля
для коэффициентов активности. После подстановки значения коэффи-
циента активности из выражения для этого закона, содержащего доба-
вочный член Рр.', в третий член правой части уравнения (24) получаем
Е0' = Е — (Z?T/2F) In mHWHso4 + 2,303 (RT/ 2F) S(/) 2/ - (RT/ 2F) In K2A =
= E°-(/?77 2F)lnat + ^'. (25)
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
407
Так как при очень низких концентрациях все величины в левой части
уравнения можно либо определить экспериментально, либо вычислить, то
экстраполяция левой части уравнения, выраженной в виде функции от р/,
дает значение Е°. Ионная сила р' вычисляется из истинных концентраций
ионов.
Концентрации каждого из ионов [Н+, HSO7 и SOT] вычисляют по
уравнению (23), а логарифм отношения коэффициентов активности находят
с помощью предельного закона Дебая и Гюккеля. В уравнении (23)
Мн = ® + ®и- Wso4 = mH + *S
и mnsQi — m — ^н, где т —
стехиометрическая концен-
трация, И1н — концентрация
иона сульфата или иона во-
дорода, образующихся при
диссоциации иона HSO7, А —
добавочная моляльность суль-
фат-ионов, которые образу-
ются в результате незначи-
тельного растворения суль-
фата свинца.. Ионная сила
оказывается равной (т +
+ 2тн + 45). 3 начения
и ионной силы получают с
помощью метода последова-
тельных приближений. При
этих вычислениях применяют
экспериментальные значения
А2а, приведенные в табл. 96.
В случае растворов со зна-
чительной ионной силой ве-
личины концентраций, полу-
ченные этим методом, явля-
ются приблизительными, однако в случае разбавленных растворов они стре-
мятся к истинным значениям и обеспечивают правильную экстраполяцию.
Результаты такого рода экстраполяции показаны на рис. 113, на котором
левая часть уравнения (25), т. е. значение Е0', изображена графически
в виде функции от р.'. Окончательные результаты экстраполяции представ-
лены в третьем столбце табл. 97. Эти значения могут быть вычислены по
уравнению
Е° = 1,67699 + 0,000285г 4-1,2467 . 10-в£2, (26)
полученному методом наименьших квадратов. Четвертый столбец таблицы
содержит величины разности между значениями третьего столбца и значе-
ниями, полученными первым методом экстраполяции. Изображенные на
рис. 113 линии имеют при 0° небольшую кривизну, но при более высоких
температурах они представляют собой прямые, что дает возможность срав-
нительно просто осуществлять экстраполяцию.
Теперь мы имеем возможность сравнить два метода экстраполяции.
На рис. 112 проведены прямые через точки, отвечающие величинам Е°, по-
лученным вторым методом экстраполяции, причем эти прямые соответствуют
предельному закону (П. 3.) Дебая и Гюккеля. Прежде всего следует
отметить, что при 0° наклон кривых, экстраполированных по методу
Льюиса и Рендалла, почти точно совпадает с наклоном, соответствующим
предельному закону. Следовательно, при этой температуре оба метода
408
ГЛАВА ХШ
экстраполяции дают одинаковые значения Е°. При более высоких темпера-
турах, особенно нри 60°, наклон кривых, полученных согласно Льюису и
Рендаллу, больше теоретического наклона. Очевидно, что если учитывать
требования теории, то истинные значения Е° должны быть меньше соот-
ветствующих значений, получаемых первым методом. Такое согласование
с теорией представлено на рис. 112 пунктирными линиями, направленными
к величинам Е°, полученным вторым методом. Где-то в области концентра-
ций ниже 0,005 М экстраполяционные кривые должны иметь изгиб и на-
клоны экспериментальных кривых должны приближаться к наклонам,
соответствующим предельному закону. Хотя наклоны представленных на
рис. 112 экспериментальных кривых сильно отличаются от наклонов кри-
вых, отвечающих предельному закону, мы полагаем, что это обусловлено
тем простым фактом, что до сих пор не было получено надежных экспе-
риментальных данных для изучаемых элементов при столь больших раз-
бавлениях. Поскольку наличие у опытных кривых правильного теорети-
ческого наклона является необходимым, то несомненно, что величины Е°,
полученные вторым методом экстраполяции, более точны.
/
§ 10. Определение активности воды в водных растворах серной
кислоты путем измерений электродвижущих сил
и упругости пара
Как было указано в предыдущем параграфе, электродвижущая сила
элемента с электродом сульфат свинца-двуокись свинца определяется
уравнением (20). В этом элементе протекает следующая реакция:
Н2 + РЬО2 + H2SO4 (m) = PbSO4 + 2Н2О.
Для вычисления aw с помощью измерений электродвижущих сил и урав-
нения (20) прежде всего определяется предварительное значение коэффи-
циента активности у', которое находят, пренебрегая членом, содержащим
aw. Из этого значения у' вычисляют предварительное значение aw путем
графического интегрирования общего термодинамического уравнения (45)
гл. I, которое после подстановки величины упругости пара принимает вид
— In aw = In ~ (у -р— игсИпу^). (27)
р 55,5 х. m J 1 у ' '
0
Эти предварительные значения aw подставляют затем в уравнение (20)
и вычисляют новые значения у'. Этот процесс повторяют до тех пор, пока
величины aw не будут удовлетворять уравнению (20). Такое вычисление
наиболее удобно производить для концентраций ниже 0,1 М. При концен-
трациях, превышающих 0,1 М, применяют более простой метод.
Электродвижущие силы элемента
Н21 H2SO4 (m) | Hg2SO4-Hg
определяются уравнением1
тг, RT , 2 RT, , ,оо,
Ei = Ei —-2p-lnmHmso4 —2p-lnyHyS01, (28)
где EJ — моляльный электродный потенциал элемента. Путем сочетания
1 Нижний индекс I введен для обозначения элемента, содержащего сульфат
одновалентной ртути, в отличие от элемента, содержащего двуокись свинца. Этот
индекс употребляется только в данном параграфе.
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
409
уравнений (20) и (28) получаем следующую зависимость:
E + Ei-E0-E?=(29)
из которой нетрудно вычислить aw. Подставляя значения Е° из табл. 97,
Таблица 98
Активность воды в растворах серной кислоты
Метод Вычисл. У пр. пара [1] Вычисл.
Температура, ° с
щ(Н-2ЙО4) 0 25 25 40 60
0,0005 0,99998 0,99998 0,99998 0,99998
0,01 0,99959 0,99960 — 0,99961 0,99962
0,05 0,99809 0,99819 — 0,99822 0,99823
0,1 0,99620 0,9964 .— 0,9964 0,9964
0,5 0,9817 0,9821 0,9821 ' 0,9822 0,9823
1,0 0,9613 0,9620 (0,9620) 0,9624 0,9630
1,5 0,9374 0,9391 0,9389 0,9402 . 0,9415
2 0,9105 0,9136 0,9129 0,9155 0,9180
3 0,8438 0,8506 0,8514 0,8548 0,8602
4 0,7650 0,7775 0,7795 0,7850 0,7950
5 0,6801 0,6980 0,7030 0,7086 0,7229
6 0,5968 0,6200 0,6252 0,6288 0,6505
7 0,5184 0,5453 0,5497 0,5608 0,5815
данным измерений электродвижущих сил и упругости пара.
• Гроллмен и ФрэзерТ(упр. пара); 9 Коллинз (упр.Спара); ОХарнед
и Гамер (э. д. с.).
410
ГЛАВА ХШ
значения Е и EJ, определенные Гамером [56], а также Харнедом и Гаме-
ром [59], и значения Е| из табл. 100, можно вычислить aw. Получен-
ные таким путем величины aw для нескольких температур приведены в
табл. 98.
Как видно из рис. 114, эти данные очень хорошо совпадают с величи-
нами, которые были определены непосредственно из измерений упругости
пара Гроллменом и Фрэзером [60] для разбавленных растворов, и с вели-
чинами Коллинза [61] для более высоких концентраций (0,5 — 4 М). С другой
стороны, данные Шэнкмана и Гордона [62], полученные из измерений
упругости пара, не особенно хорошо совпадают с результатами, найденными
путем измерений электродвижущих сил в области концентраций 5—8 М и
12—17,5 М. Как видно из данных четвертого столбца табл. 98, вплоть до
концентрации 4 М совпадение величин является хорошим, в то время как
при концентрациях 5, 6 и 7 М расхождения между значениями aw, получен-
ными двумя разными методами, составляют около 1%.
§ 11. Коэффициент активности серной кислоты
После исключения последнего члена в правой части уравнения (20)
и преобразования получаем соотношение
Е = Е°-^1пту±41/з, (30)
которое может применяться для вычисления у± на основании данных для
электродвижущей силы элемента с электродами двуокись свинца-сульфат
свинца-платина или же сульфат закиси ртути-ртуть. Элементы с элек-
тродами сульфат закиси ртути-ртуть дают плохие результаты при концен-
трациях ниже 0,05 М, но начиная с этой концентрации и до 17,5 М полу-
чаются хорошо воспроизводимые результаты. В табл. 99 приведены значе-
ния у±, вычисленные на основании данных, полученных разными методами
при нескольких температурах. Второй столбец содержит значения, получен-
ные Рендаллом и Скоттом [12] по измерениям температур замерзания. Как
видно из таблицы, эти значения прекрасно совпадают с результатами, полу-
ченными методом электродвижущих сил. Значения, вычисленные Харнедом
и Гамером для 25°, гораздо ниже, чем ’ результаты, полученные Льюисом
и Рендаллом [576]. Это связано в основном с тем обстоятельством, что ука-
занные авторы применяли при своих расчетах явно заниженные величины
относительного парциального молярного теплосодержания. Приведенные в
таблице результаты на 2,4% выше значений, полученных Баумштарком [63],
а также Шериллом и Нойесом [52].
В табл. 99 приведены также значения у± Для 25°, вычисленные из
данных по упругости пара Шэнкмана и Гордона на основании обычного
стандартного значения коэффициента активности при концентрации 1 М.
Совпадение этих данных с результатами, полученными путем измерения
электродвижущих сил для концентраций 1—4 и 9— 11 Л/ включительно,
является удовлетворительным, в то время как для концентраций 5 — 8 и
12— 16 М наблюдается расхождение результатов. Последний столбец таблицы
содержит значения у±, полученные изопиестическим методом измерения
упругости пара, причем в качестве стандартного раствора применялся рас-
твор хлористого натрия. Хорошее совпадение с результатами, полученными
методом электродвижущих сил, наблюдается в интервале концентраций
0,1-4 М.
В табл. 158 приводятся значения коэффициента активности для тем-
ператур 0—60°, определенные через каждые 5° путем измерения электро-
движущих сил.
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
411
Таблица 99
Средний коэффициент активности серной кислоты в водных растворах
Метод Т'. замерз. [1] э. Д. С. [2] Э. д. с. [2] Упр. пара [3] Упр. пара 1)
Температура С
' 25
0,0005 0,912 0,908 0,885
0,001 0,876 0,873 0,830 — —
0,002 0,825 0,825 0,757 — —
0,005 0,734 0,734 0,639 — —
0,01 0,648 0,649 0,544 — —
0,02 0,553 0,554 0,453 — —
0,05 0,424 0,^26 0,340 — —
0,1 0,341 0,341 0,265 — (0,265)
0,2 0,272 0,271 0,209 — 0,207
0,5 — 0,202 0,154 — 1,154
1,0 — 0,173 0,130 (0,130) 0,131
1,5 — 0,167 0,124 0,124 0,125
2 — 0,170 0,124 0,125 0,126
3 — 0,201 0,141 0,141 0,142
4 — 0,254 0,171 0,168 0,172
5 — 0,330 0,212 0,206 —
6 — 0,427 0,264 0,254 —
7 .— 0,546 0,326 0,315 —
8 — 0,686 0,397 0,385 —
9 — 0,843 0,470 0,466 —
10 — 1,012 0,553 0,557 —-—
И — 1,212 0,643 0,643 —
12 — 1,431 0,743 0,763 —
13 — 1,676 0,830 0,850 —
14 — 1,958 0,969 1,009 .—
15 — 2,271 1,093 1,123 —
16 — 2,612 1,235 1,270 —
17 — 3,015 1,387 — —
17,5 — 3,217 1,473 — —
1) Робинзон [4а]. Из изопиестических отношений т„ вп /т Скэтчард, Гамер
xi2bU4 JNaOl
и Вуд [46],
§ 12. Стандартный потенциал элемента
Н2 |H2SO4 (иг) I Hg2SO4-Hg
Так как измерения электродвижущих сил элементов этого типа в случае
разбавленных растворов недостаточно точны для выполнения надежной экстра-
поляции, то для вычисления стандартного электродного потенциала были
использованы значения у±, полученные путем измерения электродвижущих
сил элементов с электродами сульфат свинца-двуокись свинца. Значения
у± при концентрациях 0,05; 0,07 и 0,1 М, а также величины Е подставля-
лись в уравнение (30), после чего можно было вычислять Е° для каждой
концентрации. Средние значения, полученные из трех определений, приве-
дены в табл. 100. Полученные результаты можно выразить с точностью
412
ГЛАВА XIII
до 4z 0,05 мв следующим квадратным уравнением:
Е° = 0,63495 — 781,44 • 10-4-426,89 • Ю’8?2. (31)
Константы этого уравнения были вычислены с помощью метода наимень-
ших квадратов.
Таблица 100
Стандартный потенциал элемента
Н2 I H2SO4 (m) I Hg2SO4-Hg
i, °C Е0 t, °C Ео
0 0,63495 35 0,60701
5 0,63097 40 0,60305
10 0,62704 45 0,59900
15 0,62307 50 0,59487
20 0,61930 55 0,59051
25 0,61515 60 0,58659
30 0,61107 — —
Значение E°, полученное при 25°, оказалось на 6 мв ниже, чем
значение, полученное Льюисом и Рендаллом [57в] на основании данных
Рендалла и Кэшмана [64]. Это значение Е° оказалось также ниже величины,
вычисленной Бродским [65]. Несомненно, что приведенное в табл. 100 зна-
чение является более точным, так как коэффициенты активности кислоты,
вычисленные по электродвижущим силам элементов с электродами, содер-
жащими сульфат ртути, совпадают с величинами, полученными из электро-
движущих сил элементов с электродами, содержащими двуокись свинца,
в интервале концентраций 0,05 — 7 М. Значения этих коэффициентов актив-
ности при 0° хорошо согласуются с величинами, полученными на основа-
нии данных Рендалла и Скотта о температурах замерзания.
§ 13. Относительные парциальные молярные теплосодержание
и теплоемкость серной кислоты в водном растворе
Харнед и Гамер вычислили значения для области температур 0 — 60°
с помощью данных об электродвижущих силах. Полученные ими резуль-
таты выражаются уравнением
Z/2 = Z/2 (0°) 4* 4* Р?2, (32)
где а и Р — постоянные, L2 (0°) — значение L2 при 0°. Значения констант
этого уравнения приведены в табл. 159.
Ланге, Монгейм и Робинзон [66] определили L2 калориметрическим
методом в пределах концентраций 0,0001 — 0,05 М. На рис. 115 представ-
лена графически зависимость полученных ими значений L2 (черные кружки)
и величин Z/2 — Z/2 (0,05), вычисленных путем измерения электродвижущих
сил (белые кружки) от иг1/®. Совпадение данных можно считать прекрасным,
так как в большинстве случаев расхождение не превышает 20 кал.
На этом рисунке показаны два приема экстраполяции. Прямая линия
соответствует эмпирической экстраполяции Ланге, Монгейма и Робинзона,
которая согласуется с эмпирической экстраполяцией значений электродви-
жущих сил [уравнение (21); рис. 112]. Кривая линия отвечает соответству-
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
413
ющей требованиям теории экстраполяции, которая согласуется со значе-
ниями стандартных потенциалов, полученными с применением предельного
закона [уравнение (25); рис. ИЗ]. Из рисунка видно, что полученная по
первому способу кривая приближается к теоретической только в очень раз-
бавленных растворах (тп < 0,0001 М}. Расхождение между результатами,
полученными двумя методами экстраполяции, составляет около 460 кал
при 25°. Мы полагаем, что экстраполяция, соответствующая требованиям
теории, является более точной, хотя для доказательства этого утвержде-
ния необходимы данные по измерениям электродвижущих сил или теплот
разбавления при достаточно низких концентрациях, которые н настоящее
время отсутствуют.
Для дальнейшего сравнения значения L2 при 25° были представлены гра-
фически в] виде функции от вг^адля всех концентраций на рис. 116. Белыми
Рис. 115. Зависимость L2 — £2 (0,05) от m1^2 для раз-
бавленных водных растворов серной кислоты при 25°.
Белые кружки—э. д. с.; черные кружки—калориметрия.
кружками обозначены результаты, полученные путем измерений электродви-
жущих сил, черными кружками — результаты Ланге, Монгейма и Робинзона,
а кружками с вертикальной черточкой — величины, вычисленные Бренстедом
из данных по измерениям электродвижущих сил. Поскольку у Бренстеда от-
сутствуют данные для достаточно разбавленных растворов, необходимые для
экстраполяции, то ко всем его результатам пришлось прибавить по 2530 вал.
Общий характер результатов Бренстеда и других авторов [67, 57г] оди-
наков, хотя максимальное расхождение между этими результатами состав-
ляет около 300 кал. Пунктирная линия соответствует значениям, получен-
ным на основании калориметрических данных Крейга и Вайнела [68]. Эти
авторы применяли экспериментальный метод экстраполяции, поэтому отвеча-
ющая их данным кривая лежит примерно на 400 кал выше кривой, полу-
ченной с помощью метода электродвижущих сил. Если учесть различие
в методе экстраполяции, то совпадение можно считать вполне удовлетво-
рительным.
Относительная парциальная молярная теплоемкость, которая получается
путем дифференцирования уравнения [32], выражается соотношением
72 = а + 2р«. (33)
Крейг и Вайнел вычислили J2 при 25° из калориметрических данных [69].
Значения J2 при 25°, полученные из данных для электродвижущих сил
414
ГЛАВА XIII
и из калориметрических определений, представлены в табл. 159 и изо-
бражены графически на рис. 117. Обе серии результатов характеризуются
весьма быстрым ростом с увеличением т1^ в области разбавленных
Рис. 116. Зависимость L2 от т1/® Яля водных растворов сер-
ной кислоты при 25°*
О э. д. с.; • калориметрия; 9 э. д. с. (Бренстед). Прямая линия
вблизи начала координат соответствует предельному закону. Пунктир-
ная линия отвечает значениям, вычисленным Крейгом и Вайнелом из
калориметрических данных.
Рис. 117. Относительная парциаль-
ная молярная теплоемкость серной ки-
слоты в водных растворах при 25°.
Белые кружки—э. д. с.; черные кружки—
калориметрия.
Рис. 118. Зависимость числа переноса
катиона серной кислоты в водных растворах
от т1!2.
Прямые линии соответствуют предельному закону..
растворов, после чего значения уменьшаются, проходят через минимум
и затем снова увеличиваются в очень концентрированных растворах.
Наблюдается большое расхождение между результатами, полученными
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ 415
разными методами, причем вероятность ошибочности обеих серий результатов
весьма велика. Калориметрические значения были получены из старых,
недостаточно точных экспериментальных данных. Нельзя также ожидать
большой точности от результатов, представляющих собой вторую произ-
водную от значений, полученных путем измерения электродвижущих сил.
§ 14. Число переноса катиона в водных растворах
серной кислоты
Гамер [70] определил число переноса катиона серной кислоты с по-
мощью данных, полученных из измерений электродвижущих сил элементов
Hg-Hg2SO41H2SO4 (иг) | Н21H2SO4 (пгд) Hg2SO4-Hg; Е,
причем значения Е были получены на основании результатов, рассмот-
ренных в предыдущих параграфах, а также путем измерения электродви-
жущих сил элементов
Hg-Hg2SO41H2SO4 (m) | H2SO4 (mR) | Hg2SO4-Hg; ET.
Для определения числа переноса катиона серной кислоты Гамер исполь-
зовал метод вычисления Т+ с помощью уравнения (64) гл. X, т. е.
7\ — cZFt/cZE, который совершенно аналогичен методу, описанному в гл. XI,
§ 9. Наклоны кривых зависимости Е от Ет для различных концентраций
кислоты были определены с помощью производной функции Рутледжа
[уравнение (30) гл. XI]. Значения Т+ для концентраций 0,05—17 М и для
интервала температур 0 — 60° приведены в табл. 160. Эта таблица содер-
жит также значения предельных чисел переноса (вычисленные из данных
по электропроводности, а также из других данных о числах переноса)
и величины предельных коэффициентов наклона ]/ d0 уравнения
Онзагера
T+ = T0++S^VdoVm. (34)
Зависимость Т+ от пг’/г представлена на рис. 118. Согласно теоретиче-
ским предельным функциям, изображенным в виде прямых линий, значе-
ния Т+ должны возрастать с увеличением концентрации. Необходимые для
проверки этого теоретического положения экспериментальные результаты
для достаточно разбавленных растворов отсутствуют, а имеющиеся данные
непригодны для экстраполяций. Все кривые имеют тенденцию к достиже-
нию предела при уменьшении концентрации кислоты, и можно предполо-
жить, что в более разбавленных растворах они будут приближаться
к теоретическим значениям. Теоретические прямые проведены через зна-
чения предельных чисел переноса, полученные из других источников [70].
ЛИТЕРАТУРА
1. W. W. L u с a s s е, J. Am. Chem. Soc., 47, 743 (1925).
2. G. Scatchard, R. F. Tefft, J. Am. Chem. Soc., 52, 2265 (1930).
3. E. A. Tippetts, R. F. Newton, J. Am. Chem. Soc., 56, 1675 (1934).
4. H. S. H arned, J. Am. Chem. Soc., 48, 326 (1926).
5. G. Jones, M. Dole, J. Am. Chem. Soc., 51, 1084 (1929).
6. R. F. Newton, E. A. Tippetts, J. Am. Chem. Soc., 58, 280 (1936);
M. F. Bechtold, R. F. Newton, там же, 62, 1390 (1940).
7. T. W. Richards, M. Dole, J. Am. Chem. Soc., 51, 794 (1931).
8. С. M. White, J. Am. Chem. Soc., 58, 1620 (1936).
9. R. A. Robinson, R. H. Stokes, Trans. Faraday Soc., 36, 733 (1940);
R. A. Robinson, там же, 36, 735 (1940); R. A. Robinson, R. H. Sto-
kes, там же, 36, 1137 (1940).
416
ГЛАВА XIII
10. G. А к е г 1 б f, J. Am.' Chem. Soc., 48, 1160 (1926); H. S. Harned, G. A ker-
lof, Phys. Z., 27, 411 (1926).
11. H. S. Harned, J. C. Hecker, J. Am. Chem. Soc., 56, 650 (1934).
12. M. Randall, G. N. S с о t t, J. Am. Chem. Soc., 49, 647 (1927)).
13. E. Lange, H. S t r e e c k, Z. physik. Chem., A157, 1 (1931).
14. M. Randall, F. D. Rossini, J. Am. Chem. Soc., 51, 323 (1929).
15. W. E. Wallace, A. L. Robinson, J. Am. Chem. Soc., 63, 958 (1941).
16. G. S c a t c h a r d, R. F. Tefft, J. Am. Chem. Soc., 52, 2272 (1930).
17. W. G. Horsch, J. Am. Chem. Soc., 41, 1787 (1919).
18. H. S. H a r n e d, M. E. Fitzgerald, J. Am. Chem. Soc., 58, 2624 (1936).
19. W. R. C a r m о d y, J. Am. Chem. Soc., 51, 2905 (1929).
20. R. G. Bates, J. Am, Chem. Soc., 60, 2983 (1938).
21. R. G. В a t e s, J. Am. Chem. Soc., 61, 308 (1939).
22. R. G. В a t e s, J. Am. Chem. Soc., 63, 399 (1941).
23. R. G. Bates, W. C. Vosburgh, J. Am. Chem. Soc., 59, 1188 (1937).
24. R i g h e 11 a t о, C. W. Davies, Trans. Faraday Soc., 26, 592 (1930).
25. R. G. Bates, W. C. Vosburgh, J. Am. Chem. Soc., 59, 1583 (1937).
26. R. G. В a t e s, W. C. Vosburgh, J. Am. Chem. Soc., 60, 136 (1938).
27. W. G. Horsch, J. Am. Chem. Soc., 41, 1787 (1919); F. H. G e t m a n, J. Phys.
Chem., 35, 588 (1931); W. W. L u c a s s e, J. Am. Chem. Soc., 51, 2597 (1929).
28. a) R. A. Robinson, R. H. Stokes, Trans. Faraday Soc., 36, 740 (1940);
б) A. C. Harris, H. N. Parton, там же, 36, 1139 (1940).
29. H. N. P a r t о n, J. W. Mitchell, Trans. Faraday Soc., 35, 758 (1939);
R. H. Stokes J. M. Stokes, там же, 41, 488 (1945).
30. A. L. Robinson, W. E. Wallace, Chem. Rev., 30, 195 (1942).
31. H. S. Harned, С. M. Mason, J. Am. Chem. Soc., 54, 1439 (1932).
32. W. R. Carmody, J. Am. Chem. Soc., 51, 2905 (1929).
33. U. B. Bray, J. Am. Chem. Soc., 49, 2372 (1927).
34. I. A. Cowperthwaite, V. K. LaMer, J. Am. Chem. Soc., 53, 4333
(1931).
35. E. Lange, J. Monheim, A. L. Robinson, J. Am. Chem. Soc., 55, 4733
(1933).
36. H. S. Harned, J. Am. Chem. Soc., 58, 360 (1937).
37. U. B. Bray, J. Am. Soc., 49, 2372 (1927).
38. R. A. Robinson, R. S. Jones, J. Am. Chem. Soc., 58, 959 (1936).
39. В. B. Owen, R. W. Gurry, J. Am. Chem. Soc., 60, 3074 (1938).
40. a) C. W. Davies, Trans. Faraday Soc., 23, 351 (1927); б) V. K. L a M e r,
W. G. Parks, J. Am. Chem. Soc., 53, 2040 (1931); 55, 4343 (1933).
41. T. S h e d 1 о v s к y, D. A. Maclnnes, J. Am. Chem. Soc., 61, 200 (1939).
42. R. E. Hall, W. D. Harkins, J. Am. Chem. Soc., 38, 2658 (1916).
43. G. T. Bedford, Proc. Roy. Soc., A83, 454 (1909).
44. C. Robertson, V. K. L a M e r, J. Phys. Chem., 35, 1953 (1931).
45. F. H о v о г к a, W. H. R о d e b u s h, J. Am. Chem. Soc., 47, 1614 (1925);
W. H. Rodebush, там же, 48, 709 (1926).
46. R. A. Robinson, J. Am. Chem. Soc., 59, 84 (1937).
47. R. A. R о b i n s о n, Trans. Faraday Soc., 35, 1229 (1939).
48. С. M. Mason, J. Am. Chem. Soc., 60, 1638 (1938).
49. E. M. H a t t о x, T. DeVries, J. Am. Chem. Soc., 58, 2126 (1936).
50. W. J. Hamer, J. Am. Chem. Soc., 56, 866 (1934).
51. I. M. Klotz, C. R. S i n g 1 e t e г г у, диссертация, University of Chicago (1940),
52. M. S. Sherrill, A. A. Noyes, J. Am. Chem. Soc., 48, 1861 (1926).
53. A. A. Noyes, M. A. Stewart, J. Am. Chem. Soc., 32, 1133 (1910).
54. R. S. Livingston, J. Am. Chem. Soc., 48, 45 (1926); C. Drucker, Z. phy-
sik. Chem., 96, 381 (1920); I. M. К о 1 t h о f f, Rec. trav. chim., 43, 207 (1924).
55. K. S. Pitzer, J. Am. Chem. Soc., 59, 2365 (1937).
56. W. J. Hamer, J. Am. Chem. Soc., 57, 9 (1935).
57. а) Г. H. Л ь и с, M. Р е н д а л л, Химическая термодинамика, ОНТИ—Хим-
теорет, Л., 1935; б) стр. 354, 356; в) стр. 407; г) стр. 95.
58. М. Randall, L. Е. Young, J. Am. Chem. Soc., 50, 989 (1928).
59. H. S. Harned, W. J. Hamer, J. Am. Chem. Soc., 57, 27 (1935).
60. A. G г о 1 1 m a n, J. C. W. Frazer, J. Am. Chem. Soc., 47, 712 (1925).
61. E. M. Collins, J. Phys. Chem., 37, 1191 (1933).
62. S. S h a n k m a n, A. R. Gordon, J. Am. Chem. Soc., 61, 2370 (1939).
63. G. В a u m s t a r k, диссертация, Catholic University of America, Baltimore, Md.,
1933.
64. M. Randall, О. E. Cushman, J. Am. Chem. Soc., 40, 393 (1918).
65. А. И. Бродский, Z'. Electrochem., 35, 833 (1929).
66. E. Lange, J. Monheim, A. L. Robinson, J. Am. Chem. Soc., 55, 4733
(1938).
ВОДНЫЕ РАСТВОРЫ МНОГОВАЛЕНТНЫХ ЭЛЕКТРОЛИТОВ
417
67. J. N. Brons ted, Z. physik. Chem., 68, 693 (1910).
68. D. N. Craig, G. W. Vinal, Вцг. Standards, J. Research, 24, 475 (1940).
69. E. Биро н, ЖРФХО, 31, 201 (1899); S. S о с о 1 i k, Z. physik. Chem., 158,
305 (1932); F. В о d e, Z. Angew. Chem., 2, 244 (1899); S. S а с о 1 i k, Gar-
nier, Bull. Soc. Chim., 27, 8 (1920); M. D. Taylor, диссертация, University
of California, 1931.
70. W. J. H a m e r, J. Am. Chem. Soc., 57, 662 (1935).
ЛИТЕРАТУРА К ТАБЛИЦАМ
К табл. 90
1. Е. A. Tippets, R. F. Newton, J. Am. Chem. Soc., 56, 1675 (1934).
2. a) H. S. Harn e d, J. Am. Chem. Soc., 48, 326 (1926); 6) W. W. L u c a s s e,
J. Am. Chem. Soc., 47, 743 (1925).
3. R. A. Robinson, R. H. Stokes, Trans. Faraday Soc., 36, 735 (1940);
R табл. 91
1. R. A. Robinson, J. M. Wilson, R. H. Stokes, J. Am. Chem; Soc., 63,
1011 (1941).
R табл. 95
1. R. A. Robinson, J. Am. Chem. Soc., 59, 84 (1937).
2. E. M. Hattox, T. DeVries, J. Am. Chem. Soc., 58, 2126 (1936).
R табл. 96
1. W. J. Hamer, J. Am. Chem. Soc., 56, 866 (1934).
R табл. 98
1. S. S h a‘n к m a n, A. R. Gordon, J. Am. Chem. Soc., 61, 2370 (1939).
R табл. 99
1. M. Randall, C. N. Scott, J. Am. Chem. Soc., 49, 647 (1927).
2. H. S. Harned, W. J. Hamer, J. Am. Chem. Soc., 57, 27 (1935).
3. S. S h a n к m a n, A. R. Gordon, J. Am. Chem. Soc., 61, 2370 (1939).
4. a) R. A. Robinson, Trans. Faraday Soc., 35, 1229 (1939); 6) G. Scatchard,
W. J. Hamer, S. E. Wood, J. Am. Chem. Soc., 60, 3061 (1938).
27 Г. Харнед, Б. Оуэн
Глава XIV
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
Определение такого термодинамического свойства, как коэффициент
активности одного электролита в растворе другого электролита, сыграло
важную роль в создании теории междуионного притяжения и в расшире-
нии наших познаний о ионных равновесиях. Два экспериментальных метода
имеют в этой области наиболее существенное значение. Первый метод
состоит в определении растворимости солей в солевых растворах, второй —
в определении коэффициента активности одного электролита в присутствии
другого путем измерения электродвижущих сил.
Исследование растворимости солей в солевых растворах было начато
Нойесом [1] и продолжено Брэем [2] и Гаркинсом [3]. Полученные этими
авторами результаты были использованы Льюисом и Рендаллом [4] в ка-
честве иллюстрации плодотворности разработанного ими представления
о ионной силе, а также были использованы Нойесом [5] в его анализе
теории Дебая и Гюккеля.
Растворимость солей, применявшихся в этих первых исследованиях,
была несколько велика для точной проверки справедливости уравнений
теории междуионного притяжения. Этот недостаток отсутствовал в рабо-
тах Бренстеда [6] и его сотрудников, которые определили растворимость
многих высоковалентных соединений кобальта, обладающих очень малой
растворимостью (~ 0,00005 М) в растворах различных солей. Эти исследо-
вания впервые с особой отчетливостью продемонстрировали влияние валент-
ности ионов на величину коэффициента активности, обнаружили ряд
сложных специфических влияний одних электролитов на свойства других
электролитов и привели Бренстеда к созданию теории специфического
взаимодействия ионов.
Элементы без жидкостного соединения, содержащие смесь электроли-
тов, были впервые применены Харнедом [7], который исследовал влияние
растворов хлористого калия различной концентрации на коэффициент актив-
ности -0,1 М раствора соляной кислоты. Лумис, Эссекс, Мичэм [8] и'Чоу
Минг [9] также исследовали такого рода элементы для того, чтобы изме-
рить коэффициент активности соляной кислоты в растворах хлористого
калия при общей ионной силе, равной 0,1 М. Один из выводов, сделанный
Харнедом на основании этих измерений, состоял в том, что коэффициент
активности, а также относительное парциальное молярное теплосодержание
данного сильного электролита в растворе другого электролита являются
прежде всего функцией общей концентрации электролита или, как пока-
зали Льюис и Рендалл, общей ионной силы. Этот вывод находится в соот-
ветствии с основными уравнениями теории междуионного притяжения,
поскольку в эти уравнения всегда входит Г1/2 —функция концентраций
всех ионов и их валентностей. После этих работ и возникновения теории
междуионного притяжения были выполнены весьма обширные исследова-
ния электродвижущих сил элементов со смесями электролитов. Результаты
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
419
этих исследований являются основой для точного изучения ионных равно-
весий слабых электролитов, в том числе ионов гидроксония в солевых
растворах (гл. XV).
§ 1. Измерения растворимости и теория
междуионного притяжения
Выражения для значений коэффициентов активности сильных электро-
литов в присутствии солей различных типов и концентраций можно
легко вывести на основании определений растворимости. В присутствии
твердой фазы вещества, насыщающего раствор, необходимо, чтобы выпол-
нялось соотношение
^(O)Y(o> = = К+ ) (^-) Q Y? ), (1)
где ^(о) и V' — стехиометрические произведения растворимости соответ-
ственно для воды и для раствора, в котором присутствует другое раство-
ренное вещество, а у(0) и у± — соответствующие средние коэффициенты ак-
тивности. Поэтому в общем случае
1
V ==lgY(o)~1gY±- (2)
Если второе растворенное вещество является неэлектролитом или не имеет
общего иона с изучаемым электролитом, то левую часть этого уравнения
можно заменить через 1g (6’/6’(0)), где S и 5(0) — стехиометрические раство-
римости в растворе соли и в чистой воде. Рассмотрим некоторые резуль-
таты измерений растворимости, которые являются особенно важными для
теории междуионного притяжения.
Вскоре после появления теории междуионного притяжения Дебая
и Гюккеля данные о растворимости стали широко использовать для про-
верки справедливости этой теории. Брецстед и Ла-Мер [10] определили
растворимость комплексных кобальтамминов 1,1-, 2,1- и 3,1-валент-
ных типов. Значительно позже была измерена растворимость аналогичных
соединений типа 2,2 и 3,3 [11]. Эти данные подтвердили, что фактор
валентности в теоретическом уравнении имеет правильное значение, и
показали, что пропорциональность значения 1g у± квадратному корню из
ионной силы соблюдается весьма точно. В некоторых случаях [12] вычи-
сленные на основании этих опытов данные согласуются с численными
значениями теоретического коэффициента наклона. Позднее Ла-Мер и дру-
гие исследователи наблюдали резкое изменение величины наклона при
достижении концентрации растворенного вещества, соответствующей насы-
щенному раствору в воде. Бакстер [13] подтвердил справедливость теоре-
тических данных при 75° путем измерения растворимости йодата серебра
в растворах солей. Измерения растворимости в органических растворителях
с низкой диэлектрической постоянной, как правило, дают результаты, кото-
рые согласуются с теоретическими данными лишь качественно.
Даже в тех случаях, когда осложнения, связанные с ассоциацией
ионов, сведены к минимуму, как, например, в очень разбавленных вод-
ных растворах, коэффициенты активности обнаруживают весьма специфи-
ческие особенности при некоторых комбинациях электролитов со сложным
типом валентности. Это явление было впервые исследовано Бренстедом
и Петерсеном [14] и позднее Ла-Мером [15]. На рис. 119 показаны ре-
зультаты, полученные Ла-Мером и Мэзоном путем определения раствори-
мости 1,3-валентной соли (лютеодинитродиаммино-оксало-кобальтиата) ввод-
ных растворах различных солей при 25°. Как видно из рисунка, в случае
азотнокислого калия и хлористого бария получаются кривые, которые
27*
420
ГЛАВА XIV
удовлетворительно согласуются с теоретической предельной прямой, в то
время как для других солей получаются кривые с характерным «проги-
бом». Наиболее резкие изменения величины наклона наблюдаются для
систем, у которых ион высшей валентности насыщающего раствор вещества
противоположен по знаку иону с высшей валентностью второго растворен-
ного вещества [14]. Ясно выраженный «прогиб» типичен для 3,1- и 1,2-
валентных систем, вроде йодата лантана в растворе сернокислого калия
[16], йодата церия в растворе сернокислого калия [17] и 3,1-валентных
электролитов типа кобальтамминов в растворе сернокислого калия [15, 18].
Рис. 119. Влияние различных солей на растворимость
[Со (NH3),[ [Со (NH3)2 (NO2)2 С2О4]3 в воде при 25°.
• KN03; О ВаС12; Q MgSO4; © K2SO4; Q K3Fe(CN)e; © K4Fe(CN)6.
• Пунктирная линия изображает предельный. закон.
Если в таких системах заменить сернокислый калий на сульфат с сим-
метричным типом валентности, вроде сернокислого магния, то «прогиб» на
кривых заметно уменьшается. Если вместо сернокислого калия взять хло-
ристый магний,, то «прогиб» исчезает совершенно. Когда в растворе отсут-
ствуют ионы с валентностью, превышающей 2, то «прогиб» незначителен,
а иногда вообще не обнаруживается. Так, например, в случае системы
иодат кальция — сульфат калия «прогиб» не наблюдается [19], хотя в дру-
гих 2,1- и 1,2-валентных системах [20] он, несомненно, существует.
Если ионы высшей валентности насыщающей и дополнительной солей
имеют одинаковые знаки, то «прогиб» обычно отсутствует [21, 15, 19].
Наиболее резкое исключение из этого правила наблюдается в случае
3,3- и 2,2-валентных систем, изученных Ла-Мером, Кингом и Мэзоном [22].
Некоторые из описанных результатов можно было бы качественно
объяснить неполной диссоциацией. Ла-Мер обнаружил хорошее соответ-
ствие между отдельными результатами своих опытов и теорией специфи-
ческого взаимодействия ионов Бренстеда. Однако выразить эту связь в виде
количественных соотношений не удалось. Кривые, изображенные на рис. 120
и 121, дают некоторое представление о сложности этих взаимодействий
между ионами даже при концентрациях, соответствующих значениям р
порядка 0,01, и при комбинациях электролитов с такими простыми типами
валентности, как 1,1 и 1,2. Как было показано (гл. XII и XIII), урав-
нение Дебая и Гюккеля в его обобщенной форме может точно отражать
«прогибы» на кривых для коэффициентов активности индивидуальных
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
421
1-KNOS; 2-K2SO4; S-La(NOs)s; 4-La2(SO4)s; 5-MgSO4. .Значения ординат
для разных солей смещены на 0,02 для того, чтобы отдельные кривые не
сливались друг с другом.
электролитов, однако в случае смеси сс
согласуется с опытом, если сохранять
физический смысл. Параметр «о
насыщающей соли в общем слу-
чае отличается от параметра
второй соли, и поэтому вычислить
или хотя бы надежно определить
параметр а для смеси как функ-
цию от ао и as не удается [23].
Вследствие этого нельзя приписы-
вать особое' значение тому обсто-
ятельству, что иногда удается най-
ти такую постоянную величину а,
при которой получается приблизи-
тельное согласие опытных данных
с теоретическими. Более правиль-
ным было бы следующее примене-
ние теории: допуская, что она
справедлива для любых заданных
отношений концентраций, вычи-
слить такие значения а, которые
давали бы результаты, точно со-
гласующиеся с опытными вели-
чинами растворимости в этих сме-
той в различных соотношениях оно не
для & такие значения, которые’имеют
Рис. 121. Влияние различных солей на раство-
римость [Со (NHs)5NCS] [NOs]2 в воде при 25°.
1—KCNS; 2—NaCNS; 3—Ba(CNS)s. Пунктирная
линия соответствует предельному закону.
сях. Таким путем можно изучать зависимость величины а. от ионной силы.
Такое вычисление производят следующим образом: вычисляют 1g у(0
с помощью обобщенного уравнения Дебая и Гюккеля при некотором про-
422
ГЛАВА XIV
извольном значении ад, например равном 1. Полученное значение 1gу(0)
используют вместе с данными по растворимости для вычисления значения
1g у± согласно уравнению (2). Затем обобщенное уравнение решают путем
последовательных приближений относительно величин а, соответствующих
каждому значению 1g у±. После этого все вычисления повторяют для зна-
чений а0, равных 2, 3, 4 и т. д. Партингтон и Стонхилл [23] широко поль-
зовались вычислениями такого рода при обработке своих данных для
2,1-валентных кобальтамминов. Эти авторы нашли такое значение а0 для
данной насыщающей соли, при котором получаются минимальные колебания
величины а в присутствии данной дополнительной соли. Эти значения а0
Таблица 101
Средние значения а (обобщенное уравнение)
Дополнительная соль Насыщающая соль
М (SCN)n rM [Co(NHs)5] <NOs>2 (% — 3,0) [Co(NHs)J 12 (a =4,0) 0
NaSCN 0,96 2,85 4,86
KSCN 1,33 3,03 4,07
Ba (SCN)3 1,35 2,84 5,11
La (SCN)s 1,15 3,88 5,34
и средние значения соответствующих величин а, полученные для смесей,
приведены в табл. 101. Во втором столбце приведены радиусы катионов
дополнительных солей, полученные из кристаллографических данных.
Хотя ион тиоцианата входит" в состав всех дополнительных солей, после-
довательность изменения величины а для различных солей не совпадает
с соответствующей последовательностью величин кристаллографических
радиусов и, кроме того, изменяется в зависимости от природы насыща-
ющей соли. Таким образом, среднее значение величины а является
функцией обеих солей и не характеризует ни одну из них в отдель-
ности. Этот вывод относится не только к системам типа кобальтаамминотио-
цианатов, поскольку аналогичные результаты были получены Хлупеком,
Данеш и Данешовой также для систем, состоящих из йодата церия или
йодата кальция в присутствии различных солей, хотя в этих случаях
колебания величины а не были столь велики.
Крокфорд и Томас [24] дали определение величины а для смесей,
основанное на учете частоты столкновений. Их выражение
_ а1^1с1 4- а2^2с2 /О\
~' k^l + k-iCl '
содержит кинетические константы кг и к2, для вычисления которых необхо-
димо знать величины ионных радиусов в растворах. Если кг, а1 и к2, а-2
характеризуют отдельные соли, то, согласно этому уравнению, послёдова-
„ о
тельность значении а, которая получается для данного ряда дополнительных
электролитов, не должна зависеть от природы насыщающей соли. Этот
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
423
вывод противоречит результатам, приведенным в табл. 101. Таким же
недостатком страдает и более простое уравнение
° 4- ^2^2
<?1 + с2
предложенное Мак-Дугалом [25]. Таким образом, не существует такого
уравнения, с помощью которого можно было бы определить а в виде функ-
ции параметров, характерных для отдельных солей. Возможно, что урав-
нение (3) отвечало бы этому требованию, если бы имелись данные для
вычисления значений к± и к2 как свойств смеси.
Параметр а уравнения Дебая и Гюккеля (35) гл. III связан с индиви-
дуальными величинами а7- посредством следующего уравнения:
р
1 + ха - Zi 1 + ха,
4 •'
В случае очень разбавленных растворов, когда (1 J-xa)-1 можно заменить
через (1—ха), это уравнение можно записать в таком виде:
« = —
р
,2 о
'з aj
Ни одно из этих уравнений не дает представления о физическом смысле а,
потому что не существует точного определения величины а/.
§ 2. Определение коэффициентов активности соляной, бромистоводород-
ной и серной кислот и гидроокисей щелочных металлов в растворах
солей путем измерений электродвижущих сил
Очень важным преимуществом метода электродвижущих сил, приме-
няемого для измерения коэффициентов активности одного электролита
в присутствии другого, является возможность изменять концентрации обоих
электролитов в широких пределах. Метод растворимости имеет тот недоста-
ток, что концентрация одной из солей, а именно концентрация насыщающей
соли, является фиксированной. Метод электродвижущих сил можно при-
менять как для определения коэффициентов активности одного электролита
с постоянной концентрацией в растворе другого электролита с переменной
концентрацией, так и для измерения коэффициентов активности данного
электролита при меняющейся концентрации обоих электролитов в растворе
с постоянной общей моляльностью.
Электродвижущие силы элементов
H2|HX(nh), MXn (m2) | Ag X-Ag,
Н21 H2SO4 (mJ, M2SO4 (m2) | Hg2SO4-Hg
определяются соответственно следующими уравнениями:
Е = Е°— 2,3рЗДГ- 1g рнухт1 (nm2 + mi), (7)
E = E0-^§^1gYHYso4(2mi)2(mi + m2)- (8)
Таким образом, если известны значения их стандартных потенциалов, то
Можно легко определить величины унх и YH2SO4 для растворов галогенидов
и сульфатов.
424
ГЛАВА XIV
В гл. XI, § 2, и в гл. ХШ, § 12, было доказано, что стандартные
потенциалы элементов этого типа могут быть вычислены из данных по
измерению электродвижущих сил этих элементов в условиях, когда они
содержат растворы только чистых кислот. Хотя элементы, не содержащие
дополнительных солей, более удобны
Рис. 122. Экстраполяция Е0' при 25° по
уравнению (9).
для этой цели, Е можно успешно
определять и путем измерения элек-
тродвижущих сил элементов,. содер-
жащих соли. Преобразуя уравне-
ние (7) и используя предельное урав-
нение Дебая и Гюккеля, мы полу-
чаем видоизмененную форму урав-
нения (4) гл. XI, удобную для на-
шей цели:
Е° н=Е+ 2^-02-Г lgm1(«m2 + m1) —
-«^± = е“ + /(р), (9)
где Е = Е при р = 0. На рис. 122
представлена графически зависимость
Е от р для элементов, содержащих хлориды лития, натрия и калия.'
Так как эти прямые практически сливаются в одну, то на । рисунке £они
несколько раздвинуты. Как видно из рисунка, все эти прямые дают при
Ут
Рис. 123. Средние коэффициенты активности
соляной и бромистоводородной кислот в растворах
щелочных галогенидов при 25°.
экстраполяции почти одно и то же значение Е , причем это значение сов-
падает с тем, которое было получено раньше при изучении элемента,
О'
содержащего только кислоту. Зависимость Е от р для этого элемента
изображена пунктирной линией. Элемент, который был использован для
изучения термодинамических свойств гидроокисей (гл. XII, § 2),
Н21 МОН (ту), IК IМ0Н Н*
I I1"! |
можно применять и для определения коэффициентов активности гидро-
окисей в растворах галогенидов и сульфатов. Электродвижущая сила этого
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
425
v 2,303ЯТ ,
Е =-----г---1g
элемента в растворе, галогенидов выражается уравнением
7он аП2О(тД) т1(т1 + т2)
Чм(тд) "(0Н(тд) аНгО
На рис. 123— 125 представлены полученные результаты. Кривые изобра-
жают зависимость коэффициентов активности кислот и оснований при низких
Рис. 124. Средние коэффициенты активности гидрооки-
сей щелочных металлов в растворах соответствующих га-
логенидов при 25°.
Рис. 125. Средние коэффициенты активности кислот и
гидроокисей в растворах солей при 25°.
концентрациях (0,1 — 0,01 М и ниже) от концентраций солей, указанных
в скобках1. В основном эти кривые по своей форме подобны тем, которые
1 Библиографию к данным, использованным для этих рисунков, см. [76, 26].
426
ГЛАВА XIV
наблюдаются для индивидуальных электролитов. На кривых имеется f
характерный минимум, и во многих случаях наблюдается очень крутой . ‘
подъем при переходе к концентрированным растворам. !
Специфические особенности коэффициентов активности можно обобщить ' ?
в основном в форме следующих двух положений: , г.
1. При данной ионной силе и данной концентрации кислоты коэффициент =
активности сильной кислоты в присутствии соли определенного типа валент- V
ности тем больше, чем больше коэффициент активности этой соли в чистом
растворителе. Таким образом, как видно из рис. 123,
VhcKLICI) ^HCUNaC!) > YhCI(KCI) > YllCl(CsCl)
и из рис. 95
7biCl > ^NaCI > YkcI > YcsCI'
Таким же образом из рис. 125 явствует, что
^H2SO4(L12SO4) > ^H2SO4(Na2SO4) > Yh2SO4(K2SO4) ’
а из рис. 110, что
YLi2SO4 > ^Na2SO4 > 7k2SO4’
2. Как видно из рис. 124, для сильных оснований в растворах гало-
генидов обнаруживается противоположная закономерность:
YmOH(CsCI) > YjlOH(KCl) > YmoH(KBt) > YMOH(NaCl)
> YMOH(NaBr) > YmOH(KJ) > YMOH(NaJ) ’
t. e. коэффициенты активности оснований в присутствии солей распола-
гаются в обратной последовательности по сравнению с коэффициентами
активности индивидуальных солей в воде. Далее, из рис. 125 явствует,
что
YmOH(K2S04) > ТмОНДОаг8О4Ц > YmOH(L12SO4) ’
что также подтверждает это правило. Взаимодействие ионов в растворах,
содержащих гидроокиси и кислоты, представляет значительный интерес,
и к этому вопросу мы еще вернемся в дальнейшем. Не так давно были
вычислены коэффициенты активности соляной и бромистоводородной кислот
в растворах, содержащих соответственно хлориды лития, натрия, калия
и бария, а также бромиды лития, натрия и калия для широкого интер-
вала температур (0—50°). Точность этих вычислений составляла ±0,001.
Как будет показано в дальнейшем, эти результаты имеют существенное
значение для определения диссоциации воды и слабых электролитов в рас-
творах солей (гл. XV, § 2 и 8). Поэтому эти данные приведены вместе
с библиографией в табл. 161.
§ 3. Относительное парциальное молярное теплосодержание 0,01 М
соляной и бромистоводородной кислот в растворах галогенидов
По данным для электродвижущих сил элементов, рассмотренных
в предыдущем параграфе, можно вычислить парциальное молярное тепло-
содержание L2 — L2 (0,01) кислоты относительно его величины при 0,01 М,
применив для этого уравнение (73) гл. III или подходящую форму
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
427
уравнения Гиббса—Гельмгольца, На рис. 126 представлены значения £2 этих
кислот в некоторых растворах соответствующих галогенидов, а также
величины L2 Для соляной кислоты в чистой воде. Следует отметить сходство
этих кривых как по форме, так
и по абсолютным значениям ве-
личин. Действительно, значения
относительного парциального мо-
лярного теплосодержания кислот
в растворах хлорида и бромида
лития очень близки к значениям
£2 для соляной кислоты в чистой
воде.
§ 4. Изменение коэффициента
активности одного электролита
в присутствии другого при посто-
янной общей ионной силе
Большое число измерений
электродвижущих сил было вы-
полнено с элементами типа
Н2 | НХ (тД, MXn (mi2) | Ag X-Ag,
у которых поддерживалась посто-
янная ионная сила раствора р.
На основании этих измерений
были определены коэффициенты
Рис. 126. Относительное парциальное мо-
лярное теплосодержание галоидоводородных
кислот в растворах некоторых галогенидов.
активности галоидоводорОдных кислот в
смесях и найдены весьма про-
стые
эмпирические закономерности как в случае разбавленных, так и в
случае концентрированных растворов.
Гюнтельберг [27] произвел исключительно точные измерения электро-
движущих сил указанных элементов, содержащих хлориды лития, натрия,
калия и цезия при общей концентрации 0,1 М при 20 и 2§°. В связи с тем,
что Гюнтельберг обнаружил в этой работе ошибку, обусловленную присут-
ствием следов иона брома в растворах хлористых солей, соответствующие
старые * исследования были им повторены, за исключением измерения
электродвижущих сил элементов, содержащих хлористый цезий. В этой
работе применялись два типа электродов серебро-хлорид серебра, потенциал
которых отличался на постоянную величину 0,185 мв. Один из электро-
дов, дававший большую электродвижущую силу, был приготовлен из серебра,
полученного путем осаждения из раствора азотнокислого серебра при дей-
ствии сернокислого закисного железа. Второй электрод был получен путем
электролитического осаждения серебра из раствора азотнокислого серебра.
Элемент с электродом первого типа имел при концентрации соляной
кислоты, равной 0,1 М, электродвижущую силу 0,35316 при 20° и 0,35233
при 25°. Харнед и Элерс [28] получили при этих же температурах соответ-
ствующие значения 0,35322 и 0,35239, применяя электроды, приготовленные
путем электрического осаждения хлористого серебра на серебре, получен-
ном термическим разложением окиси серебра. Воспроизводимость элемен-
тов Гюнтельберга была порядка ± 0,02 мв, средние значения определялись
с точностью ± 0,01 мв. 0,
На основании уравнения для этого элемента можно определить Е
с помощью выражения
Е0,=Е + ^’1птнтс1 = Е0-^1пг±, (И)
428
ГЛАВА XIV
где Е—электродвижущая сила элемента при постоянной общей моляльности
и переменных концентрациях кислоты и соли. Если тМс1 = 0,1ж, гДе х
меняется в пределах от 0 до 1, то результаты можно выразить простым
уравнением
„О' „О' ' ,
Е =Е(х^0) + Лх, °(12)
где к—изотермическая постоянная. В табл. 102 приведены значения Е(х=0)
и к, которые, согласно Гюнтельбергу, удовлетворяют его опытным данным,
полученным при работе с электродами, приготовленными из химически
осажденного серебра. Прекрасное совпадение между опытными данными
и результатами, вычисленными по уравнению (12), видно из данных, при-
веденных в нижней части табл. 102. Значения к при 25° для тех же солей,
полученные в более ранней работе, составляли 0,00008, 0,00049 и 0,00098.
Таблица 102
Константы уравнения (12) и проверка линейного характера
изменений Е
Соль 1=20 Э (=25°
L1C1 En'=0,23683o+0,00014 x EO'=0,234015+0,00011 x
NaCl E°'=0,23683o + 0,00054 x E°'=0,234015+0,00051 x
КС1 E°'=0,23683o+0,00096 x E0' =0,234015+0,00093 x
CsCl E°'=O,23683o+0,00170a:1) ,
(=25°
Соль X
E (ВЫЧИСЛ.) E (набл.)
0 0,234015 O,234O2o
LiCl 0,5 0,23407о 0,234070
0,9 0,234114 O,23411o
NaCl 0,5 O,23427o O,23427o
0,9 0,234474 0,234456
КС1 0,5 O,23448o 0,23448B
0,9 0,234852 0,234852
!) Значение 0,00170 взято из ранней статьи Гюнтельберга. 2RT/F
- 0,4343=0,116324 и 0,118310 соответственно при 20 и 25°.
Применяя аналогичные элементы с каломельными электродами, Харнед [29]
получил менее точные результаты, из которых он нашел следующие зна-
чения к при 25° для растворов хлоридов лития, натрия и калия: 0,0002,
0,0005 и 0,0009 при величине ошибки порядка 0,0001.
Так как Е°—постоянная величина при данной температуре, то из урав-
о'
нения (11) очевидно, что наблюдаемая линейная зависимость Е от 1g у
влечет за собой как необходимое следствие линейную зависимость 1g у
от концентраций кислоты или соли при постоянной общей моляльности.
Это простое эмпирическое правило соблюдается во многих случаях не только
для разбавленных растворов.
На рис. 127 изображена зависимость у соляной кислоты от логарифма
концентрации кислоты для растворов некоторых хлоридов при общей
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
'429
моляльности, равной 1 и 3 М. Следует отметить, что при уменьшении
концентрации кислоты у стремится к постоянной величине. На рис. 128
1g у соляной кислоты представлен в виде функции от концентрации
кислоты, причем эта зависимость является прямолинейной. Хоукинс [30]
показал, что для смесей соляной кислоты с 1,1-галогенидами при
постоянной общей моляльности эта линейная зависимость сохраняется
вплоть до значения, равного 6 М. Бэйтс и Армстон [31], Мардок
и Бартон [32] также получили линейную зависимость для соляной кислоты
Рис. 128. Линейная зависимость lgf±
соляной кислоты от тнс1 для растворов
1,1-хлоридов при постоянной общей мо-
ляльности.
Верхние три кривые—при концентрации ЗМ,
нижние три кривые—при концентрации 1 М.
Верхняя кривая каждой серии отвечает раство-
ру хлорида лития, средняя кривая (черные
кружки)—раствору хлорида натрия, нижняя
кривая—раствору хлорида калия.
Рис. 127. Зависимость среднего коэффи-
циента активности соляной кислоты в рас-
творах хлоридов от логарифма моляль-
ности кислоты при общей моляльности,
равной 1 и 3 М.
Верхние три кривые—при концентрации ЗМ;
нижние три кривые—при концентрации 1М.
Верхняя кривая каждой серии отвечает раство-
ру хлорида лития, средняя кривая (черные
кружки)—раствору хлорида натрия, нижняя
кривая—раствору хлорида калия.
в присутствии перхлоратов калия и натрия и хлорной кислоты. Точность
их опытов была порядка ±0,1 мв- Очень важно отметить, что эти резуль-
таты можно экстраполировать на нулевую концентрацию кислоты, что
позволяет определить значение 1g у± кислоты при нулевой концентрации
в чистом растворе соли.
На рис. 129 представлена зависимость 1g у^ соляной кислоты в чистом
водном растворе (кривая) и в растворах хлористого цезия [266] при посто-
янной общей концентрации (прямые линии) от mHCi. Прямые линии про-
ведены через соответствующие точки при одинаковой общей моляльности.
Все эти линии при более высоких концентрациях почти точно параллельны.
Такое поведение характерно для соляной кислоты в растворах галогенидов.
Аналогичная линейная зависимость часто наблюдается и в случае рас-
творов, содержащих электролиты с более высокой валентностью. Это под-
тверждается данными, представленными на рис. 130, где изображена
зависимость коэффициента активности соляной кислоты в растворах хло-
ристого алюминия [33] при постоянной общей ионной силе от моляльности
(ионной силы) кислоты. Аналогичные результаты были получены для коэф-
фициентов активности соляной кислоты в растворах хлорида церия [34],
хлоридов бария и лантана [35] и дитцоната натрия [36]. Результаты опре-
деления растворимости сульфата серебра [37] в смесях солей также при-
430'
ГЛАВА XIV
водят к линейной зависимости 1g у насыщающей соли от ее ионной силы
при постоянной общей ионной силе. Необходимо отметить, что эта линей-
ная зависимость наблюдается лишь тогда, когда по оси абсцисс отложены
значения ионной силы изучаемой соли. Так, 1g у± сульфата серебра в раство-
рах смесей сульфатов магния и кадмия, магния и лития, а также сульфатов
алюминия и цинка изменяется линейно с изменением ионной силы сульфата
серебра.
Линейную зависимость 1g у каждого из двух электролитов в смеси
от ионной силы можно представить следующими уравнениями:
IgYi = lg Y(0)i + “12^1 ^SYko)-01^’ (13)
lgY2 = 1gY(0)2 + a21!12 = 1gY2(0)-a21!1l> (14)
где индексы 1 и 2 относятся соответственно к электролитам 1 и 2; средние
коэффициенты активности электролитов 1 и 2 в смесях любого состава
Рис. 129. Зависимость lg 7h7ci от гонс1
для растворов соляная кислота—хло-
ристый цезий.
Прямые линии соответствуют постоянной
общей моляльности.
Рис. 130. Зависимость 1gсоляной ки-
слоты от концентрации кислоты в растворах
соляная кислота—хлористый алюминий при
постоянной общей ионной силе, равной еди-
нице.
при постоянной общей моляльности обозначены через уt и у2; у(0) представляет
собой коэффициент активности электролита 1 при его нулевой концентра-
ции в присутствии электролита 2 при данной ионной силе р; у(0)2-—коэф-
фициент активности электролита 2 при его нулевой концентрации в присут-
ствии электролита 1 при том же значении ионной силы р; у1(0) и у2(0)^*
коэффициенты активности электролитов 1 и 2 в их чистых растворах.
Постоянные а12 и a2i представляют собой коэффициенты наклона кривых,
изображенных на рисунках. Этими удобными обозначениями мы будем
пользоваться при последующих теоретических выводах.
Интересное и важное подтверждение этого явления, типичного для
концентрированных растворов смесей электролитов, было обнаружено в работе
Акерлёфа, Тира и Тарка [38]. На рис. 131 логарифм коэффициента актив-
ности соляной кислоты в растворах хлористого натрия при общей ионной
силе 1 М и при различных температурах представлен графически в виде
функции от моляльности кислоты. Шесть растворителей представляют
собой 10-, 20-, 30-, 40-, 50- и 60-процентные (по весу) смеси метилового
спирта и воды. В пределах точности i 0,1 мв все линии представляют
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
431
собой прямые. Кроме того, для данной температуры наклоны этих прямых
( — а12) не зависят от концентрации спирта, что является неожиданным
результатом. Таким образом, ai2 является функцией температуры.
Линейную зависимость, выражаемую уравнением (13), нельзя рас-
сматривать как универсальный закон. Известен ряд случаев, когда для
достижения соответствия с экспериментальными данными необходимо при-
менять уравнения с более высокими степенями р2 и Это было показано
тнш
Рис. 131. Зависимость lg (7(0)/f±) для соля-
ной кислоты от моляльности кислоты в раство-
рах хлористого натрия при общей моляльности,
равной единице.
Коэффициент активности в чистых растворах кисло-
ты равен X—весовой процент метилового спирта.
Диаметры кружков равны 0,002.
Харнедом и Харрисом [39],
а также Харнедом и Ку-
ком [40], которые измеряли
коэффициенты активности ги-
дроокисей натрия и калия в
mHCl mNaOK
Рис. 132. Зависимость 1g -(^гидро-
окиси натрия от моляльности гидро-
окиси в смесях гидроокись натрия—
хлористый натрий.
Верхние кривые соответствуют общей
концентрации,- равной 5 М, нижние кри-
вые—3 М.
растворах соответствующих хлоридов при помощи следующих элементов:
Н21 МОН (mi), MCI (m2) | Mx Hg | МОН (zzzr) | Н2. *
На рис. 132 изображена зависимость 1g у± гидрата окиси натрия в раство-
рах хлористого натрия с общей моляльностью, равной 3 и 5, от концентра-
ции этого основания. Отклонения полученных кривых от прямолинейности
ясно видны.
Все эти результаты показывают, что линейная зависимость 1g у±
от ионной силы, выражаемая уравнениями (13) и (14), представляет собой
первое приближение, которое может быть использовано при обработке
опытных данных для многих подобных смесей. Конечно, благодаря этому
обстоятельству сложный вопрос о свойствах смесей электролитов может
быть значительно упрощен.
В тех случаях, когда имеет место неполная диссоциация электролитов,,
можно ожидать отклонений от указанной эмпирической зависимости. Так„
432 г Л'А В А XIV
например, Гюнтельберг [276] измерил электродвижущую силу элемента
Pb^HglPbClaOnj), MCI (m2) | AgCl-Ag
при р = 0,1. Он обнаружил, что вместо линейной зависимости, согласно
О'
уравнению (12), величина Е может быть выражена уравнением
Е0/ = Е®х=0)-аж + б//2, (15)
где а и b—константы. Степень диссоциации хлористого свинца в растворах
галогенидов была приближенно оценена.
§ 5. Термодинамическая теория смесей при постоянной
общей моляльности1 *
Для того чтобы изложить вопрос в наиболее простой форме, мы огра-
ничимся рассмотрением растворов смесей двух 1,1-электролитов, обозна-
ченных черев 1 и 2, при постоянной общей моляльности. Кроме того, мы
будем касаться лишь таких смесей, для которых логарифм коэффициента
активности каждого электролита можно считать зависящим линейно от
моляльной концентрации этого электролита. Согласно уравнениям (13)
и (14), 1g у для электролитов 1 и 2 выражается следующими линейными
соотношениями:
1gYi=1gY(O)i + ai2mi=1gYi(O)-'ai2-™20 (16)
lg y!= lg Y(0)2 + а21 mZ = lgY2(0)- a21 mr <17)
Зависимость между a12 и а21 и другими термодинамическими величи-
нами может быть получена с помощью уравнения Гиббса—Дюгема [урав-
нение (45) гл. 1]:
A7! d In ar + d In a2 + Nw d In aw — 0,
где ATj, A2, Nw и Hj, a2, aw —соответственно молярные доли и активности
электролитов 1 и 2 и воды. После подстановки коэффициентов активности
и моляльностей это уравнение приобретает вид
2т1 d lg Yi + 2m2 d lg y2 = — 55,51 d lg aw . (18)
Согласно уравнениям (16) и (17), d lg yj = a12 dm^, rflgy2 = a21rfm2. и, следо-
вательно, уравнение (18) переходит в следующее:
2а12 тг dm1 -[- 2а21 т2 dm2 = — 55,51 d lg aw . (19)
Положим m = m1-]-m2 и ml = mx, где x может принимать любое значение
между 0 и 1. Тогда т2 = (1— х)т. Подставляя эти значения в последнее
уравнение, получаем
2 (а12а21) ж — 2а21 с/ж — — d lg aWj = , (20)
так как дифференцирование уравнения (69) гл. I при постоянных т и Т
дает следующее соотношение:
л„ 2 m. с?р
й ig “ 55,51 (2,303) ‘
1 Последующее изложение почти точно соответствует статье Харнеда [41]. Сообра-
жения, изложенные в работе Гюнтельберга [276], также включены в данный параграф.
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
433
После интегрирования левой части уравнения (20) получаем
X
(«12 + «21) я2 — 2a3iZ = — 1g «» =
о
= _ 55>51 lg aw(x)^_
8%(0)
X
2 С 2
= 2,303m J d<?== 2,303т (21)
0
Это уравнение представляет собой основу для многих последующих выводов.
Если а12 или а21 известно (из опыта), то a2i (или соответственно а12)
можно вычислить с помощью осмотических коэффициентов и коэффициен-
тов активности следующим простым способом. Подставляя значения верх-
него предела (ж = 1) в уравнение (21), получаем
«21 = «12 4 = + 2,303m ^2 (0) ~“?1 (0))’ (22)
где aW(i), aW(2), ?i(0), ?2(0) — активности воды и осмотические коэффициенты
для чистых бинарных растворов электролитов 1 и 2 соответственно.
В гл. I, § 9, было показано, что осмотический коэффициент связан
с коэффициентом активности растворенного вещества соотношением
? = 1 4-1 тс/1ну±.
1
После подстановки этого выражения для ср в уравнение (22) получаем
t,2 Т2(°) т1(0)
«81 = «12+—aQ "icZ ln Y2(0) ~ \ ™*1ПГ1(О)) =
1 1
тп
+ <23>
Уравнения (22) и (23) связывают значения коэффициентов наклона а12 и а21
с осмотическими коэффициентами и коэффициентами активности растворов
чистых электролитов при общей концентрации т. Эти уравнения справедливы
в той мере, в какой соблюдается линейная зависимость логарифмов коэф-
фициентов активности растворенных веществ при постоянной общей моляль-
ности, выражаемая уравнениями (16) и (17). Выводы, которые можно
сделать из этих соотношений, зависят от применяемых для этой цели
термодинамических методов.
а) В общем случае, как видно из уравнения (21), линейная зави-
симость lg y± от х при постоянной общей моляльности приводит к квадра-
тичной зависимости осмотического коэффициента от х, исключая тот случай,
когда «12 =—а21- В последнем случае, очевидно,
(?® — ?о) = 2,303ma21£, (24)
и, следовательно, зависимость от х имеет линейный характер. Для
общего случая, без всяких дополнительных ограничений, результаты пред-
ставлены на рис. 133,а. В этом случае а12 =# — a2i> lgY(o>i Х(0)2> и Т
является квадратичной функцией от х.
28 г. Харнед, Б. Оуэн
434
ГЛАВА XIV
б) Если принять следующее дополнительное условие:
= (25)
72(0).
где В12=#/(т), т. е. зависимость, которая, согласно Акерлёфу и Тома-
су [37], приблизительно соблюдается для высоких концентраций, то
а—общий случай; б—Акерлёф; е—ТЗренстед; г—Гуггенгейм.
получаются результаты, изображенные на рис. 133,6. Подставляя это
выражение в уравнение (23) и интегрируя, получаем
<*12-<*21 = ^12 (26)
Далее, из этого уравнения и уравнений (16) и (17) находим, что
(ai2 — a2i)т — 1g (27)
_____________ 72(0)
1 На этом рисунке авторами допущена ошибка: вместо lgT(Qj2 в0 всех случаях
стоит lg7o<2>- (Прим- рад.)
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ , 435
и
lg Y(0) i = 1g Y(o)2' (28)
Эта зависимость представлена на рис. 133,6.
в) Теория специфического взаимодействия ионов Бренстеда (гл. XII, § 6)
вводит целый ряд дополнительных ограничений. Для того чтобы ознакомиться
с применением этой теории, лучше всего рассмотреть конкретный случай,
например смесь соляной кислоты и хлористого калия. Для значений
логарифма коэффициента активности соляной кислоты и хлористого калия
соответственно в растворах хлористого калия и соляной кислоты можно
написать следующие уравнения:
ln Y нсцкс!+ни) ~ 1“ Y h(H)Yckci)Y h(K)Yckk)Y h(ci)Yci(h)Y(h+k)Y(cd’ (29)
ln Ykckhci+kci) = lnYK(H)Yci(ci)YK(K)Yci(K)YK(ci)Yci(H)Y(K+H)Y(ci)’ (3®)
где Yh(H)» Yci(K) и t- д’ —коэффициенты специфического взаимодействия,
a Y(H+K) и у(С1) — коэффициенты высаливания. Теорию Бренстеда можно
выразить в форме следующих положений:
1. Ионы различного знака действуют друг на друга по-разному, в то
время как ионы одинакового знака оказывают друг на друга одинаковое
влияние. . 5
2. Каждый.ион обладает специфическим «эффектом высаливания» по
отношению ко всем остальным ионам, находящимся в растворе.
Рассмотрим изменение величины x = mnci/m при постоянной моляль-
ности т. Согласно положению 1, Ущщ — Yh(K)~ Yci(ci) = YK(K) = C0Bsfc*
Далее, при постоянной концентрации хлорида коэффициенты взаимодействия
хлор-иона с катионами Yh(ci> и Yk(ci) сохраняют постоянные значения при
изменении состава. Дифференцирование уравнений (29) и (30) приводит
к выражениям
ln Yhci(hci+kci) ln Yci(K)Yci(H) + In Y(h+k>Y(ci)’ (31)
ln Ykckhci+kci) = ln Yci(K)Yckh) + ln Y(k+h)Y(ci> (32)
для любого данного значения х между 0 и 1. Поэтому
ln Yhci(hci+kci) = 1ПТкскнс1+кс1)ф (33)
Согласно нашим предыдущим выводам, это уравнение эквивалентно утвер-
ждению, что а12 == —а21. Этот результат вытекает из теории Бренстеда
о специфическом взаимодействии ионов.
Рассмотрим теперь предельные значения 1пунсцкс1) и ,1пуксцнс1), гДе
индексы указывают, что кислота и соль находятся при нулевой концентрации
соответственно в растворах соли и кислоты. В этом предельном случае
уравнения (29) и (30) принимают вид
lnYHci(Kci) = lnYH(K)YH(ci)Yci(K)Yci(ci)YcKci)’ (3^)
1“ Ykckhci) = 1“ Yk(H)Yk(ci)Yckh)Yci(ci)Y(hci)’ (33)
где через Y(kci> и Y<hci) обозначены коэффициенты высаливания электролитов,
т. е. соответственно Y(K)Y(ci) и Y(H)Y(c1)- 9тих уравнений видно, что если
использовать принятую ранее зависимость 7Н(К) = Ycuci)= Yk(h> и пРеДп°-
ложить, что Yh(ci) = Yci(H) и Yk(ci) = Yci(K)» то получается соотношение
ln Yhci(kci) ln Ykchhci) = 2 In Y(kci)““ 31n Y(HC1). (36}
28*
436
/
a
ГЛАВА XIV
Гюнтельберг [27а] проанализировал физическую сущность и достоверность
этого предположения. Оказалось, что оно зависит от основного допущения
о линейном наложении ионных атмосфер, согласно которому средняя сила,
действующая на ион, определяется потенциалом атмосферы другого иона
[уравнение (17) гл. II].
Как видно из уравнения (36), согласно теории Бренстеда не требуется,
чтобы выполнялось соотношение 1пуНС1(КС1) = 1пуКС1(НС1), которое было полу-
чено Акерлёфом и выражается уравнением (28). Другой результат теории
Бренстеда нетрудно получить путем интегрирования уравнения (33) в пре-
делах от х = О до х~ 1. В результате интегрирования получается выражение
Yhci(kci) + Ykci(hci) = Ykci + 7hci- №7)
Результаты Бренстеда представлены на рис. 133, в, причем а12=—а21,
<f изменяется линейно с изменением состава согласно уравнению (22)
и Y(0)i 7(0)2-
г) Если пренебречь коэффициентами высаливания Бренстеда в уравне-
нии (36), то из уравнения (37) сразу получается
I*1 Ihckkci) = 7кскнс1) “ уОп7нс1 + ^п7кс1)’ • (^)
Согласно принятым нами обозначениям, это означает, что а12 = — а21
и 1g у j — lgy(0)2. Этот случай представлен на рис. 133, г, причем дан-
ное условие вытекает из теории специфического взаимодействия ионов
в интерпретации Гуггенгейма. Уравнение Гуггенгейма (37) гл. XII [42]
приводит к такому выражению для логарифма отношения коэффициентов
активности двух электролитов, которое аналогично соотношению Акерлёфа
и Томаса (25), за исключением того, чтб в нем принята молярная концен-
трация с вместо моляльной т, а также используется рациональный коэф-
фициент активности. Таким образом, согласно уравнению (38) гл. XII,
'2(0)
где В — постоянная. Отсюда получается результат, выраженный уравнени-
ем (28), т. е. lgY(0)1 = lgY(0)2.
§ 6. Общий обзор экспериментальных исследований
смесей 1,1-галогенидов
Прежде чем перейти к обсуждению теорий и закономерностей, выведен-
ных в предыдущем параграфе, мы остановимся на обширных эксперименталь-
ных исследованиях растворов соляной кислоты в присутствии хлоридов.
На основании измерений, упомянутых в § 2 и 4, было вычислено значе-
ние lg Yi кислоты в растворах галогенидов, и, следовательно, из этих дан-
ных можно определить характеристические наклоны а12 для различных
концентраций и температур. По этим данным и по коэффициентам активности
или осмотическим коэффициентам кислот и солей в чистой воде можно
вычислить a2i с помощью уравнений (22) или (23) [43]. Так как а12 вычис-
ляется только по двум экспериментальным величинам, т. е. по данным,
относящимся к раствору чистой кислоты и к 0,01 М раствору кислоты
в присутствии соли при той же общей моляльности, то нельзя рассчитывать
на получение очень точных результатов. Далее, ошибка в вычислении а12
пропорциональна 1/т, и, следовательно, определение этой величины при
концентрациях ниже 0,5 М не отличается большой точностью; поэтому
величины а12 при концентрациях ниже 0,5 М не принимались во внимание.
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
437
Ошибки в измерении электродвижущих сил составляют ±0,1, ±0,2
и ±0,2 мв при концентрациях, равных соответственно 0,5, 1 и 3 М, и при-
водят к ошибкам в определении величины а12, равным соответственно
±0,0034, ±0,0034 и ± 0,0011. Так как использованные данные получены
из многих источников, то трудно оценить все ошибки, но, судя по постоян-
ству результатов, вероятно, достигалась точность порядка ± 0,001. Опре-
деления а21 менее надежны и обладают точностью порядка ± 0,002 лишь
в наиболее благоприятных случаях. В табл. 103 приведены значения а12
Таблица ЮЗ1)
Значения а12 и а21 для смесей 1,1-хлорид—соляная кислота при 25°
т L1C1[2] NaCl [3] КС1 [4] CsCl [5]
“12 — “-21 <*12 — “21 «12 — “21 “-12 — “21
0,1 0,0013 [1] 0,043 [1] — 0,077 [1] 0,143 [2] ,—
0,5 0,006 0,011 0,037 0,057 0,062 0,074 0,105 0,070
1,0 0,005 0,012 0,032 0,058 0,056 0,072 0,100 0,060
1,5 0,005 0,011 — 0,058 0,055 0,069 0,099 0,053
2,0 0,005 0,012 0,031 0,058 0,057 '0,064 0,099 0,046
3,0 0,004 0,013 0,031 0,058 0,062 0,054 0,098 0,041
4,0 [6] -0,0025 — 0,030 — 0,066 0,050 — —
5,0 [6] — — 0,030 — 0,072 — — —
6,0 [6] -0,0086 — 0,029 — — — — . —
!) Значения 1g т± для соляной кислоты и для галогенидов взяты соответственно из табл. 128
и 146.
, Таблица 104
Значения а12 и а21 для смесей соляная кислота—хлорид натрия между 10 и 40°
X. т t, °C 0,1 0,3 0,5 1 2 3
а 12
10 — 0,044 0,040 0,038 0,037 0,037
20 0,046 [1] 0,040 0,036 0,034 0,033 0,033
25 0,043 [1] 0,038 0,034 0,032 0,031 0,031
30 — 0,035 0,032 0,030 0,029 0,029
40 — 0,030 0,028 0,026 0,025 0,025
- а21
10 — 0,068 0,070 0,069 0,066 0,064
20 — 0,060 0,062 0,062 0,060 0,064
25 — 0,057 0,059 0,058 0,058 0,059
30 — 0,054 0,055 0,055 0,055 0,057
• 40 — 0,047 0,047 0,048 0,049 —
438
ГЛАВА XIV
и —а21, а также источники экспериментальных данных, на основании которых
они были вычислены. О влиянии температуры можно судить по табл. 104,
в которой приведены значения тех же величин, найденные для смесей хлорид
натрия — соляная кислота. Более поздние исследования Акерлёфа, Тира
и’Тарка [38] подтверждают, что величина ошибки в определении а21 при
этих измерениях не превышает ±0,001; эти авторы получили значения а12
для концентрации 1 М и для температур 0,— 40°, совпадающие с величинами,
приведенными в таблице, в пределах указанной точности.
Характер зависимости а12 и а21 от концентрации иллюстрируют
рис. 134 и 135. По мере увеличения концентрации в некоторых случаях
(GsGl, NaBr, NaCl) а12 приближается к постоянному значению (рис. 134).
Рис. 134. Зависимость параметра а12
от моляльности.
Рис. 135. Зависимость параметров
а12 и —а21 от моляльности для рас-
творов хлоридов цезия и натрия.
На основании этой закономерности Акерлёф и Томас сформулировали правило,
согласно которому величины а12, так же как и а21, не являются функциями
концентрации в концентрированных растворах. Однако поскольку имеется
ряд случаев, когда это правило не выполняется (например, КВг, KG1
и т. д.), то его следует рассматривать как первое приближение. При
более низких концентрациях (меньше 0,5 М) правило Акерлёфа и Томаса,
очевидно, неприменимо для всех этих смесей.
На рис. 135 изображены значения я12 и а21 для хлоридов цезия и натрия.
Параметр а12 не равен —а21, хотя в случае хлористого натрия при уменьшении
концентрации раствора величина а12, повидимому, приближается к —а21.
Если рассматривать эти результаты с точки зрения теоретических поло-
жений, развитых в предыдущем параграфе, то нетрудно убедиться, что они
являются подтверждением закономерностей, которые были отнесены нами
к общему случаю (рис. 133,а). Это видно на примере рис.' 136, где значе-
ния а12 — а21 и В12, определяемые уравнением (25)
12(0)
изображены в зависимости от т. Как видно, В12 является функцией т
в разбавленных растворах, и, следовательно, согласно теории, lg Y0<i) не
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
439
равен lg Y0(2), так же как а12 —a2i не равно В12, за исключением случаев
бромида и хлорида лития, для которых это равенство, может быть, и не
является вполне точным, но все же соблюдается в пределах ошибок опыта.
При увеличении концентрации значения В12 и а12 — «21 приближаются друг
к другу. Этот факт указывает на удовлетворительную точность приближен-
ного эмпирического правила, согласно которому в очень концентрированных
растворах Н12#=/(т), а также на то, что при этом осуществляются условия,
соответствующие рис. 133,6.
Непосредственное доказательство того, что* осмотический коэффициент
не зависит линейно от концентрации в случае смесей хлорид лития — хлорид
калия при общей моляльности 1, 2 и 3 М, было получено Оуэном и Куком [44а].
Рис. 136. Зависимость В12 и а12 — а21 от моляльности
для растворов 1,1-галогенидов.
B12; О —(»12-*21).
Применяя метод изопиестического измерения упругости пара, эти авторы
непосредственно определили величину изменения осмотического коэффициента
и смогли проверить уравнение (21), которое можно представить в следующем
виде:
(<рж —<р0)= — 2,303а21^ж +1,151 (а12 + а21)тж2. (39)
В этом уравнении концентрация хлорида лития обозначена через тх, а хло-
рида калия — через т(1 — ж), причем т — общая моляльность. Выражая
осмотические коэффициенты через упругости пара, можно преобразовать это
уравнение следующим образом:
= - 0,036а21 + 0,018 (а12 ± а21) х. (40)
Если изображенная графически зависимость левой части этого уравнения
от х представляет собой прямую линию, то можно утверждать, что осмоти-
ческий коэффициент является квадратичной функцией х. Эта зависимость
изображена на рис. 137. Диаметры кружков соответствуют ошибке в ±0,1%
от значений т, что равняется величине ошибки измерений. Как видно из
рисунка, во всех, случаях в пределах этой ошибки зависимость имеет прямо-
линейный характер, что указывает на хорошее совпадение с уравнениями (39)
и (40).
Так как в концентрированных растворах (0,5 М и больше) я12 =5^ — а21,
то теория специфического взаимодействия . ионов в своей простейшей
форме (рис. 133, в или г) подтверждается не вполне точно. Этот факт иллю-
440
ГЛАВА XIV
стрируется кривыми рис. 138, на котором сумма а12 + а21 изображена как
функция от т для смесей соляной кислоты с хлоридами. Поскольку сумма
«12+ «21 не равна нулю, то условия, отвечающие теории специфического
взаимодействия, не выполняются.'' Конечно, это и не удивительно для рас-
творов с такими концентрациями.
Хотя при концентрации раствора порядка 0,1 'М вычисления стано-
вятся весьма чувствительными по отношению к ошибкам, все же оказы-
вается, что при этой концентрации получается правильное расположение
Рис. 137. Формальная проверка 'ура- Рис. 138. Зависимость ai2 + a2i от т.
внений (39) и (40). Радиусы кружков соответствуют величинам
Цифрами обозначена общая моляльность для ошибок.
каждой серии опытов. Ординаты серии опы-
тов для ЗМ смещены вниз на 0,0001 во из-
бежание слияния прямых.
кривых. Так, например, если принять для —lg Yo 1 солян°й кислоты, хло-
ристого калия, хлористого натрия и хлористого цезия (табл. 128 и 146)
значения соответственно 0,0980; 0,1135; 0,1088 и 0,1238, а для а12 — значе-
ния согласно табл. 103, то получаются величины «12 +«21, равные +0,028,
— 0,001 и —0,020 соответственно для растворов, содержащих хлориды цезия,
калия и натрия. При этих расчетах небольшая разность между а12 — а21 и В12
не принималась во внимание. Как видно из рис. 138, в этом случае полу-
чается такое же расположение кривых, как и для более высоких концен-
траций. Величина суммы а12 + а21 явно приближается к нулю с уменьше-
нием концентрации, но для концентрации 0,1 М это предельное значение
не достигается, хотя в случае смесей хлорида лития и калия кривые почти
достигают нуля.
Оуэн и Кук {446] определили 1g у± бромистоводородной кислоты в рас-
творах соляной кислоты путем измерения электродвижущих сил элементов
Н21 НВг (m^HCl (m2) | AgBr-Ag
при значении гпу + т2, равном 0,5, и температурах 0 — 45°. Было обна-
ружено, что 1g уНВг линейно зависит от моляльности бромистоводородной
кислоты. Были найдены значения наклона а12 и вычислены наклоны а21
для переменных концентраций соляной кислоты в этих смесях. Значения
наклона а21 оказались почти совпадающими по величине, но противополож-
ными по знаку по сравнению со значениями а12. Сумма а12 + а21 имеет не-
большую величину при всех температурах.
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
441
Изложенные выше соображения свидетельствуют о том, что теория
Бренстеда о специфическом взаимодействии ионов в ее первоначальном
виде, изложенная в предыдущем параграфе, является в некоторых отноше-
ниях слишком упрощенной. Этот вывод основан на тщательном, анализе
данных о температурах замерзания смесей нитрата калия, хлорида калия,
нитрата лития и хлорида лития, сделанном Скэтчардом и Прентисом [45].
Применяя свое полное уравнение [уравнение (40) гл. XII] которое учиты-
вает изменение диэлектрической постоянной среды при изменении концен-
трации соли, явление высаливания и взаимодействие между молекулами
в смесях, Скэтчард показал на основании этих исследований, что теория
специфического взаимодействия ионов справедлива для концентраций выше
0,1 М и, возможно, для концентраций порядка 1 М.
§ 7. Дальнейший анализ теории специфического
взаимодействия ионов
Хотя приведенные в предыдущем параграфе данные свидетельствуют
о чрезвычайно сложной природе смесей электролитов и трудности точной
проверки теории специфического взаимодействия ионов, Бренстед и Гюн-
тельберг получили много данных, подтверждающих справедливость основ-
ных положений этой теории. Ниже будет рассмотрено несколько примеров,
полученных на основании весьма точных экспериментальных данных.
Одним из непосредственных следствий теории Бренстеда является сле-
дующее равенство:
IglTici(Hci) lAgci(HGi) Ig IRCI(HCI) lg Тнсцнср t д (4!)
ItICI(MCI) lAgCl(MCl) iRCl(MCl) IhcI(MGI)
Путем измерения растворимости мало растворимой соли [CO(NOa) (CNS) •
• (NH3)4] Cl в растворах хлоридов Гюнтельберг [27а] получил возможность
сравнить данные по растворимости со своими результатами измерений
электродвижущих сил элементов, содержащих соляную кислоту в тех же
растворах галогенидов. Из двух последних соотношений [см. уравнение (41)]
получаем выражение
"2" [1S ^rckmci) 1S ‘Srci(hci)] = 1S Yhci(hci) ]S Yhci(mci) == ai2/w- (42)
В табл. 105 приведены данные, полученные при 20°; значение левой части
этого уравнения, деленное на т, содержится в третьем столбце, а значе-
ния а12, полученные на основании измерений электродвижущих сил, при-
водятся в последнем столбце. Эти данные, которые были получены совер-
шенно различными методами, хорошо совпадают друг с другом.
Таблица 105
Проверка уравнения (42) (т=0,1)
Электро- лит SRC1 ’2mIleSRC1(MC1>-lgSRC1 (HC1)J a12
НС1 0,01216
LiCl 0,01219 0,006 0,012
NaCl 0,01238 0,039 0,046
КС1 0,01264 0,084 0,081
CsCl 0,01301 0,147 0,146
442
ГЛАВА XIV
Другим прекрасным примером, доказывающим справедливость зависи-
мости, выраженной уравнением (41), являются результаты измерений раство-
римости комплексных солей с общим анионом [Co(NH3)2 (NO2)4]—. Согласно
теории, г, т. е. отношение растворимостей солей данного типа валентности
в растворах нитратов калия и натрия, должно быть постоянным. Резуль-
таты, приведенные в табл. 106 [66], подтверждают правильность этого тео-
ретического положения. Далее, на основании анализа теории Гуггенгейм [42]
показал, что <
= (W = - - ('ях,)*'1. («)
где г о — постоянная, которая удовлетворяет этой зависимости, если ее при-
нять равной 1,064. Из этого уравнения получаются значения г, равные
Таблица 106
Отношение растворимости
тетранитродиамминокобальТиатов в 0,1М KNO3
к их растворимости в 0,1 М NaNO3 при 20°
Катионы r=sRXn(KNO3)/sRXn(NaNO3)
Со (NH3)4 (С2О4) + со (nh3)4co; Ag + n(ch3); Со (NH3)eCl++ . Со (NH3)SNO*+ Со (NH2C2H4NH2)3++ + Со (NH3)5H2O+ + + 1,034 1,032 1,034 1,033 1,041 1,041 1,045 1,047
1,032, 1,042 и 1,047 для одно-, двух- и трехвалентных катионов. Эти дан-
ные хорошо согласуются с результатами, приведенными в табл. 106.
§ 8. Смеси оснований с хлоридами
Как отмечалось в § 4 (рис. 132), Харнед и Харрис показали, что для
смесей некоторых оснований с хлоридами не имеет места линейная зави-
симость логарифма коэффициента активности основания от концентрации
этого основания при постоянной общей моляльности. Измеряя электродви-
жущие силы элементов
Н21 МОН (т^.МХ (ш2) | MxHg | МОН (тв) | Н2
и
Ag-AgX | МОН (mi),MX (m2) | MJlg | MX (mH) | AgX-Ag,
Харнед и Кук [40] определили одновременно коэффициенты активности
гидроокисей й хлоридов в смесях. Это первое исследование, в котором
были непосредственно определены коэффициенты активности обоих компо-
нентов (электролитов) в смеси. Можно перечислить всего несколько эле-
ментов, пригодных для такого рода определений.
Характер полученных этими авторами данных виден из рис. 139. Две кри-
вые, расположенные в верхней части рисунка, соответствуют смесям гидро-
окиси калия и хлорида калия, в то время как две нижние кривые отвечают
смесям гидроокиси натрия и хлорида натрия; во всех случаях концентрация
равна 1 М. Самая верхняя кривая (белые кружки) представляет зависимость
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
443
логарифма коэффициента активности гидроокиси калия в растворах хлорида
от концентрации гидроокиси. Следующая, расположенная ниже первой, кри-
вая (черные кружки) изображает логарифм коэффициента активности хлорида
в растворе гидроокиси. Значения логарифма коэффициента активности гидро-
окиси lg Yj и логарифма коэффициента активности хлорида lgy2(0) в
чистой воде обозначены стрелками с левой и с правой стороны графика. Ана-
< логичным образом две нижние кривые изображают логарифмы коэффициентов
активности системы гидроокись натрия — хлорид
обозначены значения логарифма коэффициента
активности гидроокиси натрия в растворе хло-
рида натрия, а черными кружками — значения
коэффициента активности хлорида натрия в
той же смеси.
Как видно, зависимость 1g ух и 1g уг со-
ответственно от т2 и тх не является линей-
ной. Харнед и Кук выразили эту зависимость
с помощью следующих квадратных уравнений
с точностью, лежащей в пределах ошибок опыта:
lg Yi = lg Yi со, - «12^2 - ₽i2^2,
lg Y2 = lg Y2 (0) “21^1 — ?21"*1 •
После подстановки этих выражений в
нение (19) и интегрирования получаем
чину изменения осмотического коэффициента:
У(ац=0) У(аа=а:)
натрия. Белыми кружками
19 У ко)
1,85
1,80
(44)
урав-
вели-
1д у ко)
I? Уг«»
19 Уг(о)
0
Р ис. 139.
1g 72 от т2
ной общей
Две верхние npi
смесям КОН—КС
0,5 1
т,—-
— тг О
Зависимости 1g 71 и
и т1 при постоян-
моляльнОсти 1 М.
ривые отвечают
_ ___ _____ —С1; две нижние
кривые—смесям NaOH—МаСЬДиа-
метры кружков равны 0,5 мв.
1,80
2,303 m
— ---2 (“12 + а21) Х1 — т (Р12 — P21) xi +
о
+ "з’7?г^12— ?21)ж1, (45)
где т = игх + т2 - имеет постоянное значение и
х1 = т1/т.
Смеси гидроокиси натрия с хлористым натрием имеют одну особенность:
соответствующие кривые для коэффициентов активности имеют противопо-
ложные наклоны, откуда следует, что а12 и а2Х имеют одинаковый знак.
Это явление не наблюдается для большинства других систем, хотя имеются
сведения, что смеси гидроокиси лития с хлористым литием ведут себя так же.
Харнед и Кук вычислили по уравнению (45) разность между осмоти-
ческими коэффициентами для чистых гидроокисей и для чистых хлоридов,
т. е. (ср(а;1=1) —<р(Ж1=0)). Они сравнили эти разности со значениями, вычис-
ленными на основании измерений электродвижущих сил путем графического
интегрирования уравнения (23), и с соответствующими величинами, получен-
ными по данным о температурах замерзания. Вычисленные этими совер-
шенно независимыми способами результаты совпадают друг с другом’ с удо-
влетворительной точностью.
§ 9. Экстраполяция коэффициентов активности до значений, соответ-
ствующих насыщенным растворам. Вычисление растворимости хорошо
растворимых солей в растворах электролитов
Сведения о термодинамических свойствах электролитов и их смесей
можно использовать для вычисления растворимости хорошо растворимых
солей в солёвЫх растворах. Для этих расчетов' необходим метод экстрапо-
444
ГЛАВА XIV
ляции, с помощью которого можно определять коэффициент активности
насыщающего вещества при больших значениях общей ионной силы рас-
твора смеси солей. Для этой цели Акерлёф и Томас [37] и Акерлёф [46]
применили приближенное эмпирическое уравнение (25), которое можно
написать в следующей форме1:
= (46)
<н (0)
Индекс (0) служит2 для условного обозначения того, что это уравнение приме-
нимо только к простым бинарным растворам индивидуальных электролитов 1
и R при постоянной ионной силе р. В качестве стандартного электролита R
обычно применяется соляная кислота, так как ее коэффициент активности
известен в очень широких пределах концентраций. Акерлёф и Тир [47] опреде-
лили эту величину в интервале температур 0—50° для концентраций 3—16 М.
Их данные приводятся в табл. 162. Изотермическую константу Вщ можно
определить из известных значений и уВ(0) в простых ненасыщенных
растворах или по данным о растворимости с помощью описанного ниже метода.
Произведение растворимости3 электролита, который диссоциирует на два
вида ионов, можно записать следующим образом:
eyj(O) = (^++^-)(O) = (v++v^)m('o), (47)
если в качестве растворителя применяется чистая вода, или:
аР = (mX+ml“), (48)
если растворителем служит раствор, содержащий другие электролиты. Если
эти другие электролиты не имеют общих ионов с насыщающей солью, то аР
также равно (v'l+vl-)mv.
В насыщенных растворах электролита 1 произведение активности ионов
этого электролита при данной температуре является постоянным, и, следо-
вательно,
lg Ki = lg (о) + lg Yi (0) = lg + lg Yi • (49)
Поскольку член 1g (0) в этом уравнении относится к насыщенному рас-
твору электролита 1 в чистой воде, то этот член дается непосредственно
уравнением (46)4. Обозначая ионную силу этого раствора через рч <о)»
получаем
^-Ig-^^-^-lg^iCoj + lgYRfoj + ^Bi pi (0) • (50)
1 В некоторых слвгчаях необходимо включать небольшой постоянный член, так как
кривая для (0) / 7н (о)> экстраполированная от высоких концентраций до ц=0, не
всегда проходит через начало координат. Если на графике обнаруживается резкая
кривизна при высоких концентрациях, к уравнению (46) необходимо добавить член,
содержащий ц2.
2 В случае растворов смесей электролитов индекс (0) опускается.
3 Вопрос об ограничениях и о применимости правила произведения растворимости
был подробно рассмотрен А. Ф. Капустинским [ЖПХ, 16, вып. 1—2, 50 (1943).
См. также А. Ф. Капустинский, ДАН, 28, № 2, 149 (1940); А. И. Брод-
ский, Ж. М. Ш е р ш е в е р, Укр. хим. журн., 2, 289 (1926); Я. И. Ф и а л к о в,
Н. В. А к с е л ь р у д, Укр. хим. журн., 16, вып. 2, 283 (1950); К. Б. Я ц и м и р-
с к и й, ЖОХ, 20, 12, 2133 (1950); Э. Б. Ш т е р н и н а, Изв. Сект., ФХА АН СССР,
19, 34 (1950)]. (Прим, ред.)
4 В .этом уравнении ц представляет собой некоторое экспериментальное значение
ионной силы, которое, лежит в пределах применимости уравнения. Применение уравне-
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
445
Член IgYi в уравнении (49) относится к насыщенному раствору электро-
лита 1 в присутствии одного или нескольких других электролитов при
общей ионной силе Для упрощения мы ограничимся случаем раствора
электролита 1, содержащего только один добавочный электролит, обозна-
чаемый индексом 2, так что рт = Р1 + Р2- Вычисление lg Yi состоит из двух
операций. Предполагаем, что уравнение (46) справедливо для пересыщен-
ных растворов 1 в чистой воде, так что его можно использовать для вычи-
сления lg Yj (0 при ионной силе ру. При этой ионной силе применяем урав-
нение (16) для того, чтобы выразить IgYi через lgY1(0) и Ра- Тогда
IgYi^lgYKO)-»^- (51)
Путем сочетания этого уравнения с уравнением (46) при общей ионной
силе, равной рт, получаем следующее равенство* 1:
IgYi = lgYn(T) + ^B1!J'T — “12^2- (52)
Используя это равенство, можно придать уравнению (49) следующий вид:
1 1
— 1g-^1 = — lg (0) + lg Yb (0) + Вм H (0) =
= — lg + lg YH (Tj + Sri pT — a12p2 • (53)
Это уравнение содержит три независимые постоянные— Вр а12 и ^що)
(или Kj), которые необходимо определить для вычисления значения
а затем растворимости электролита 1 в присутствии другой соли 2. Суще-
ствует целый ряд способов вычисления этих постоянных из опытных данных.
Особый интерес представляют следующие три способа:
1. Если Bri и а12 известны на основании измерений в разбавленных
растворах, а получено путем определения растворимости индивидуаль-
ного вещества в чистой воде, то из уравнения (53)2 * можно найти раство-
римость электролита 1 в растворах, содержащих второе растворенное
вещество при любом значении ионной сиды р2. Ценность этого способа
состоит в том, что он не требует измерения растворимости данного элек-
тролита в смеси.
2. Если известен коэффициент активности электролита 1 в чистой
воде в интервале Достаточно высоких концентраций, необходимых для
вычисления BRi, то ^цо) й а12 могут быть определены с помощью одного
опыта по измерению растворимости в чистом растворителе и одного опре-
деления растворимости в присутствии второго растворенного вещества.
Эти значения коэффициентов активности обычно бывают известны.
3. Если BRi не известно, то значения постоянных можно определить
на основании трех измерений растворимости в присутствии второго раство-
ния (46) при (Xj (0) обычно основано на экстраполяции, так как значения коэффициентов
активности хорошо растворимых солей редко измеряют вплоть до насыщенных растворов.
1 Повидимому, для концентрированных растворов применимость уравнения (51)
носит менее общий характер, чем применимость уравнения (46). К уравнениям (51),
(53) часто приходится добавлять член р12ц|. Мы воздержались от представления этих
соотношений в наиболее общей форме, поскольку введение всякого дополнительного
эмпирического параметра делает менее ясным физический смысл остальных пара-
метров.
2 Решение этого уравнения получается с помощью небольшого числа последова-
тельных приближений, поскольку при этом расчете вначале |хг не известно.
446
ГЛАВА XIV
ренного вещества. Этот результат заслуживает внимания, так как, зная
BRi, можно по уравнению (46) определить коэффициент активности насы-
щающего вещества в чистой воде из данных о его растворимости в смесях
солей при высокой ионной силе. К сожалению, полученные этим способом
коэффициенты активности обычно очень неточны, так как форма этого
уравнения такова, что она способствует увеличению ошибок, обусловлен-
ных неточностями эксперимента и приближенным характером уравне-
ния (53).
Следует отметить, что условием применимости уравнения (53) к вычи-
слению растворимости является независимость BRl и я12 от общей ионной
силы рт- В § 6 было показано, что для некоторых электролитов при боль-
ших концентрациях это условие в отношении Вщ соблюдается очень
хорошо (рис. 136). Можно ожидать, что та часть вычислений, которая
зависит от этой величины, будет вполне точной для многих электролитов.
Однако если предположить, что а12 тоже не является функцией от кон-
центрации, то это может в некоторых случаях привести к значительным
ошибкам. Так, например, из табл. 103 и 104, а также из рис. 134 и 135
явствует, что значения а12 и а21 в случае смесей соляной кислоты с хло-
ристым натрием, при высоких концентрациях очень мало меняются с изме-
нением т, однако в случае смесей соляной кислоты с хлористым калием
те же величины сильно, зависят от концентрации. Эти факты согласуются
с результатами вычислений Акерлёфа1, определявшего растворимость хло-
ридов натрия и калия в растворах соляной кислоты. Акерлёф нашел, что
уравнения
4 lg »*Naci = 0,7782 + 0,0875 рт — 41g рт — IgfR (Т) —
— 0,0530тнс1 + 0,0005тпнс1 (54)
и
у 1g Wei ~ 0,4621 4- 0,118 р-т — 1g р-т — 1g уК(Т) —
— 0,0512тНС1 + 0,00167пгнс1 (55)
хорошо согласуются со значениями растворимости т\аа и ткс1 при 25°. Эти
уравнения аналогичны уравнению (53), за исключением дополнительных
членов тинсь Первые члены в правой части этих уравнений представляют
собой значения J/2lgKlt а —0,0875 и —0,118 — соответственно значения 5Н1
этих систем. Последние два члена в каждом из этих уравнений заменяют
последний член в уравнении (53). Это показывает, что параметр а12 изме-
няется с концентрацией и что это изменение является более сильным
в случае системы с хлоридом калия, чем для системы с хлоридом натрия.
Этот результат находится в соответствии с данными табл. 103, согласно
которым значение2 (— а21) уменьшается быстрее для растворов хлорида
калия, чем для растворов хлорида натрия. Значения этих величин, полу-
ченные из данных о растворимости и из данных для более разбавленных
растворов, совпадают лишь приблизительно; до настоящего времени не уда-
лось добиться точного совпадения данных, полученных из этих двух-
источников.
Акерлёф [466] применил уравнение (53) в обобщенной форме к некото-
рым трех- и четырехкомпонентным системам, а также к пятикомпонентной
системе NaCl-KCl-MgCl2-MgSO4-H2O.
1 Акерлёф, частное сообщение.
2 Отметим, что а21 в табл. 103 соответствует а12 в уравнении (53).
СМЕСИ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
447
ЛИТЕРАТУРА
1. A. A. N о у е s, Z. physik. Chem., 6, 241 (1890); A. A. Noyes, W. С. В г а.у,
J. Am. Chem. Soc., 30, 1643 (1908).
2. W. С. Bray, W. J. Winninghof, J. Am. Chem. Soc., 33, 1663 (1911).
3. W..D. Harkins, W. J. Winninghof, J. Am. Chem. Soc., 33, 1827 (1911);
W. D. Harkins, H. M. Paine, там же, 41, 1155 (1919); W. D. Harkins.
W. T. Pearce, там же, 38, 2679 (1916).
4. G. N. Lewis, M. Randall, J. Am. Chem. Soc., 43, 1112 (1921).
5. A. A. N о у e s, J. Am. Soc., 46, 1098 (1924).
6. a) J. N. В г о n s t e d, J. Am. Chem. Soc., 42, 761 (1920); 6) 44, 877 (1922); в) 44,
938 (1922); 45, 2898 (1923); J. N. Brons ted, A. Petersen, там же, 43,
2265 (1921).
7. a) H. S. H a r n e d, J. Am. Chem. Soc., 38, 1986 (1916); 6) 42, 1808 (1920).
8. N. E. Loomis, J. L. Essex, M. R. Meacham, J. Am. Chem. Soc., 39,
1133 (1917).
9. M i n g Chow, J. Am. Chem. Soc., 42, 497 (1920).
10. J. N. В г о n s t e d, V. K. L a M e r, J. Am. Chem. Soc., 46, 555 (1924).
11. J. N. В r ousted, N. J. Brumbaugh, Am. Chem. Soc., 48, 2015 (1926).
12. V. K. L a M e г, С. V. King, C. F. Mason, J. Ain. Chem. Soc., 49, 363 (1927).
13. W. P. Baxter, J. Am. Chem. Soc., 48, 615 (1926).
14. J. N. В г о n s t e d, A. Petersen, J. Am. Chem. Soc., 43, 2265 (1921).
15. V. K. L a M e r, C. F. Mason, J. Am. Chem. Soc., 49, 410 (1927).
16. V. K. La'Mer, F. H. Goldman, J. Am. Chem. Soc., 51, 2632 (1929).
17. J. В. C h 1 о u p e к, V. Z. DaneS, B. A. D aneSo va, Coll. Czech., 5, 21
(1933).
18. V. K. L a M e r, R. G. Cook, J. Am. Chem. Soc., 51, 2622 (1920).
19. J. В. C h 1 о u p e к, V. Z. Dane J, B. A. DaneSova, Coll. Czech., 6, 116
(1934).
20. E. W. N e u m a n, J. Am. Chem. Soc., 55, 879 (1933); V. K. L a M e r, F. H. Gold-
man, там же, 51, 2632 (1929).
21. L. О ’ N e i 1 1, J. R. Partington, Trans. Faraday Soc., 30, 1134 (1934).
22. V. K. L a M e г, С. V. К i n g, C. F. Mason, J. Am. Chem. Soc., 49,
363 (1927).
23. J.R. Partington, H. J. Stonehill, Phil. Mag. (7), 22, 857 (1936).
24. H. D. С г о с к f о r d, H. C. Thomas, J. Am. Chem. Soc., 55, 568 (1933).
25. F. H. MacDougall, Thermodynamics and Chemistry, John Wiley and Sons,
N. Y., 1926, p. 279.
26. a) H. S. H a r n e d, N. J. Brumbaugh, J. Am. Chem. Soc., 44, 2729 (1922);
H. S. H a r n e d, F. E. Swindells, там же, 48, 126 (1926); H. S. Harned,
G. M. James, J. Phys. Chem., 30, 1060 (1926); G. А к e г 1 6 f, J. Am. Chem.
Soc., 48, 1160 (1926); M. Randall, С. T. Langford, там же, 49, 1445
(1927); M. Randall, G. F. Breckenridge, там же, 49, 1435 (1927);
H. S. Harned, G. Akerlof, Physik. Z., 27, 411 (1926); б) H. S. H a r n e d,
О. E. S c h u p p, J. Am. Chem. Soc., 52, 3892 (1930).
27. a) E. G ii n t e 1 b e r g, Z. physik. Chem., 123, 199 (1926); 6) Studier over Elektro-
lyt-Activiteter, G. E. C. Gads Forlag-Kobenhavn, 1938.
28. H. S. Harned, R. W. Ehlers, J. Am. Chem. Soc., 55,2179 (1933).
29. H. S. Harned, J. Am. Chem. Soc., 48, 326 (1926).
30. J. E. Hawkins, J. Am. Chem. Soc., 54, 4480 (1932).
31. S. R. Bates, J. W. U r m s t о n, J. Am. Chem. Soc., 55, 4068 (1933).
32. P. G. Murdock, R. C. Barton, J. Am. Chem. Soc., 55, 4074 (1933).
33. H. S. H a r n e d, С. M. Mason, J. Am. Chem. Soc., 53, 3377 (1932).
34. С. M. Mason, D. B. Kellam, J. Phys. Chem., 38, 689 (1934)..
35. M. . R a n d a 1 1, G. F. Breckenridge, J. Am. Chem. Soc., 49,
1435 (1927).
36. P. G. Murdock, R. C. Barton, J. Am. Chem. Soc., 55, 4074 (1933).
37. G. Akerlof, H. C. Thomas, J. Am. Chem. Soc., 56, 593 (1934).
38. G. А к e r 1 6 f, J. W. T e a r e, H. E. T u r c k, J. Am. Chem. Soc., 59, 4916
(1937).
39. H. S. Harned, J. M. Harris, J. Am. Chem. Soc., 50, 2633 (1928).
40. H. S. Harned, M. A. Cook, J. Am. Chem. Soc., 59, 1890 (1937).
41. H. S. H a r n e d, J. Am. Chem. Soc., 51, 1865 (1935).
42. E. A. Guggenheim, Phil. Mag. (7), 19, 588 (1935).
43. H. S. Harned, J. Am. Chem. Soc., 37, 1865 (1935).
44. а) В. В. 0 w e n, T. F. Cooke, J. Am. Chem. Soc., 59, 2273 (1937); 6) p. 2277.
45. G. S c a t c h a r d, S. S. Prentiss, J. Am. Chem. Soc., 56, 2320 (1924); G. Scat-
chard, Chem. Rev., 19, 309 (1936).
448
ГЛАВА XIV
46. a) G. А к е г 1 б f, J. Am. Chem. Soc., 56, 1439 (1934); 6) J. Phys. Chem., 41, 1053
(1937); G. Akerlof, H. E. T u r.c k, J. Am. Chem. Soc., 56, 1875 (1934);
H. S. Harned, J. Franklin Institute, 225, 623 (1938).
47. G. Akerlof, J. W. T e a r e, J. Am. Chem. Soc., 59, 1855 (1937).
ЛИТЕРАТУРА К ТАБЛИЦАМ
К табл. 103 ,
1. Е. Giintelberg, Z. physik. Chem., 123, 199 (1926).
2. Н. S. Harned, H. R. С о p s о n, J. Am. Chem. Soc., 55, 2206 (1933).
3; H. S. Harned, J. Am. Chem. Soc., 57, 1865 (1935).
4. H. S. Harned, W. J. Hamer, J. Am. Chem. Soc., 55, 2194 (1933).
5. H. S. Harned, О. E. S c h u p p, J. Am. Chem. Soc., 52, 3892 (1930).
6. J. E. Hawkins, J. Am. Chem. Soc., 54, 4480 (1932).
К табл. 104
1. E. G ii n t e 1 b e r g, Z. physik. Chem., 123, 199 (1926).
Глава XV
ДИССОЦИАЦИЯ И ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА
СЛАБЫХ ЭЛЕКТРОЛИТОВ1 2
В гл. VII были рассмотрены новейшие методы вычисления констант
диссоциации сильных электролитов в средах с низкой диэлектрической
постоянной и слабых электролитов в водных растворах из данных
по электропроводности. Константы диссоциации, полученные из этих дан-
ных, в большинстве случаев определены точно только для 25°, и поэтому
их нельзя использовать для вычисления теплот диссоциации и других
термодинамических величин, связанных с-реакциями диссоциации.
Почти все точные данные о зависимости констант диссоциации от
температуры были получены путем измерения электродвижущих сил эле-
ментов без жидкостных соединений, содержащих слабые электролиты.
Кроме того, все имеющиеся в настоящее время точные значения констант
диссоциации для растворов в смесях воды с неводными растворителями в ши-
роком интервале температур были получены также путем измерения электро-
движущих сил элементов без жидкостных соединений. В настоящей главе
будут рассмотрены основы этого метода и его применение к определению
констант диссоциации воды, слабых кислот и- амфолитов в воде и в водных
растворах солей, а также в смесях воды с органическими растворителями.
Будут описаны методы определения зависимости констант диссоциации
от температуры и рассмотрены таблицы термодинамических величин, свя-
занных с реакциями диссоциации.
Вообще можно использовать два типа элементов без жидкостного со-
единения. Элементы первого типа, содержащие буферные растворы, особенно
удобны для определения констант диссоциации в случае чистых раствори-
телей, смешанных растворителей и растворов солей. С помощью элементов
второго типа, содержащих небуферные растворы, можно получать дополни-
тельные данные о влиянии среды. Метод электродвижущих сил во многих
случаях заслуживает предпочтения по сравнению с методом определения
цонстант диссоциации из данных по электропроводности.
Для изучения слабых электролитов иногда применяются элементы
с неизвестными жидкостными соединениями, но поскольку эти элементы,
как правило, не пригодны для получения точных термодинамических
результатов, то они будут учитываться в настоящем обзоре лишь как второ-
степенный источник, дополняющий данные, полученные при работе с эле-
ментами без жидкостного соединенияа.
С помощью разработанного в последнее время совершенно объективного
фотоэлектрического метода были произведены точные определения кон-
стант диссоциации, а также степени диссоциации в растворах солей. Мы
1 Некоторые части данной главы совпадают по содержанию с обзором Харнеда
и Оуэна [1].
2 О применении элементов с жидкостным соединением см. гл. X, § 7.
29 г. Харнед, Б. Оуэн
450
ГЛАВА XV
рассмотрим некоторые результаты, полученные с помощью этого метода,
и сравним их с данными, полученными путем измерения электродвижущих
сил.
§ 1. Константа диссоциации воды
* Поскольку вода является наиболее важным слабым электролитом,
термодинамике процесса ее диссоциации было уделено большое внимание.
Первые точные определения константы диссоциации воды были выполнены
с помощью метода электропроводности [2] и путем измерения электродви-
жущих сил элементов с жидкостными соединениями. Если принять во вни-
мание большие экспериментальные трудности, с которыми пришлось столк-
нуться в процессе этих исследований, то следует считать, что значения
/£^(0,59 • 10-м при 18° и 1,04 • 10“14 при л25°), полученные из данных
по измерениям электропроводности, прекрасно совпадают со значениями
(0,58 • 10-14 при 18° и 1,008 • 10~J1 при 25°), полученными в последнее
время методом измерения электродвижущих сил. Часто для определения
К,,: применялись элементы с жидкостными соединениями
Н21 NaOH (m) | NaCl (m) [HGl (m) | H2,
аналогичные тем, которые использовал Аррениус [За]
Применяя уравнение Льюиса и Сарджента [4] для диффузионных
потенциалов и подставляя в это уравнение средние активности ионов,
Льюис, Брайтон и Себастьян [5] получили для величины Kw при 25°
значение 1,012 -10'11, хорошо совпадающее со значением 1,008-10-14, най-
денным путем измерений электродвижущих сил элементов без жидкостных
соединений.
Первым применением элементов без жидкостных соединений для изуче-
ния диссоциации воды было определение ионного произведения /Пцигон
и определение функции коэффициента активности ун у0Н/аН20 в растворах
солей при 25° [6]. Было показано, что последняя величина стремится
к значению,, соответствующему предельному закону для коэффициентов
активности в разбавленных растворах солей [7]. Для расчетов было исполь-
зовано значение константы диссоциации, полученное из других данных.
При последующих исследованиях составлялись различные_комбинации из эле-
ментов без жидкостных соединений:
Н21 НХ (иг) | AgX-Ag, I
MX (m2) |AgX-Ag, II
H2|HX(/W1), MX(m2) | AgX-Ag, III
которые служили как для определения константы диссоциации, так и для
определения »4imon 11 7н1'он/ан->о в растворах солей.
Электродвижущие силы этих элементов можно выразить уравнениями
E = E°-^lnaHax (1)
II
Е = Е°-^1пуыухтытх, (2)
где индексом X обозначены. хлорид, бромид или иодид. Значения, придава-
емые величинам у , ух, тп и тх, находят, как правило, с помощью термо-
динамических соображений. Рассмотрим элемент I, содержащий только
1 Сводка ранних определений Kw имеется в статье Бинса и Окса [36].
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
451
га лоидо во дородную кислоту. Все изложенные в гл. XI данные свидетель-
ствуют о том, что галоидоводородные кислоты — очень сильные электролиты.
Предположим, что галоидоводородные кислоты полностью диссоциированы
при любом значении ионной силы раствора и, следовательно, т.ц представ-
ляет собой сумму моляльных концентраций протонов и сольватированных
протонов, а тих — сумму концентраций иона галоида и сольватированного
иона галоида. Любые отклонения от этого предположения потребуют испра-
вления величин mHm0H и УнУон/ан2о в РаствоРах солей, но не будут
оказывать влияния на определение константы диссоциации Kw. Далее,
поскольку тн и тх — истинные стехио-
метрические моляльности, то у ух пред-
ставляет собой произведение коэффици-
ентов активности полностью диссоцииро-
ванного электролита, подчиняющегося
предельному закону теории междуион-
ного притяжения.
Эти рассуждения полностью прило-
жимы к элементу
Н21 МОН (тД, MX (п?2) | AgX-Ag, II
который применяется для определения
константы диссоциации воды. Предпо-
лагают, что сильное основание МОН
(обычно гидроокись щелочного металла)
и MX полностью диссоциированы, так
что цгОн равно сумме моляльностей ио-
нов гидроксила и гидратированных
ионов гидроксила. Электродвижущая
сила этого элемента определяется урав-
нением (2). В этом случае концентрация
водородных ион®в определяется из урав-
нения для диссоциации воды
к _ ТнТон
Aw = ——----7Инт0Н (о)
ан2о
Рис. 140. Определение величины
(—0,05915 lg'£'„) в растворах различ-
ных солей при 25°.
и выражается как функция независимой переменной »гон- Так как индексы Н
и ОН отвечают также и гидратированным ионам, то показатель степени х
либо равен единице, либо больше единицы, в зависимости от степени гидра-
тации. Есть данные, свидетельствующие о том, что гидратация протонов
в водных растворах является полной, однако ввиду отсутствия точных дан-
ных о гидратации ионов мы принимаем условно, что х = 1. Это допущение
не вводит ошибки в последующие термодинамические выводы, но им не сле-
дует пренебрегать при объяснении механизма диссоциации. Таким образом,
если принять х=1, то, исключая ти путем сочетания уравнений (2) и (3),
получаем следующее выражение:
(En-E")F тх _ Тон _
2.303 RT +lg™0H g7x«H2o g W‘
(4)
Так как член lg Y0II / Yx «На0 Для разбавленных растворов пропорционален
ионной силе, то при графическом изображении левой части уравнения как
функции ет р. можно путем прямолинейной экстраполяции до бесконечного
разбавления получить значение —1g Кх11. На рис. 140 показаны шесть
примеров независимых экстраполяций такого типа. Если опыт проводится
29*
452
ГЛАВА XV
только для определения Kw, то может оказаться удобным сохранить отноше-
ние тпх/^он постоянным [8] при ряде разбавлений. Однако в тех опытах,
результаты которых представлены на рис. 140 \ тх меняется, в то время
как тОн сохраняет постоянное значение, равное 0,01, и, таким образом,
эти данные могут быть использованы также для последующих вычислений
других величин. Как видно из рисунка, во всем интервале изменения р
получаются прямые линии, причем все они пересекаются в одной точке.
Воспроизводимость значений электродвижущих сил составляет примерно
0,05 мв, точность определения величины К,,: путем экстраполяции полу-
чается порядка 0,2% при 25°.
Существует также другая комбинация элементов, которую можно исполь-
зовать для определения константы диссоциации воды без предварительного
вычисления стандарнюго потенциала Е°. Соединяя элемент типа II с эле-
ментом типа III
Н21 НХ (mJ, MX (m2) | AgX-Ag, III
получаем выражение
(Ец —Ещ) F _ (ун тп тх)п1 _ (ту тх)1П
2,303 ДТ1 * Cil fx m£l'”х(m£lmxiji
Второе равенство основано на допущении, что значения коэффициента актив-
ности кислоты в кислотно-солевых растворах и в растворах гидроокись —
соль одинаковы. Это допущение хорошо соблюдается в данных условиях,
поскольку концентрации кислоты и гидроокиси составляют всего 0,01 М.
В следующем параграфе будет рассмотрен метод, с помощью которого можно
получить данные для концентраций кислоты ft гидроокиси, равных нулю,
путем ряда экстраполяций при постоянной ионной силе, однако, как
правило, поправка не превышает 0,03 мв при тх = 0,01, и ею можно пре-
небречь. Таким образом, значение функции YhYoh^ilo’ найденное путем
Измерений электродвижущих сил элементов II и III, можно приравнять ее
зйачению в чистом растворе соли MX при ионной силе, соответствующей
сумме mx-|-m2. Имея в виду это условие, можно путем исключения (тп)п
из уравнений (5) и (3) получить выражение
(Ец —EHl)F , (теНтах)ш , ТНТОН , /ох
2,303 ЯГ lg(™X/™oH)ii lg ан2о g^W’
гДе Yh Тон/ан2о Дан0 без индекса II. Согласно теории Дебая— Гюккеля,
эта величина равна
, ТнТон 3 *>629 • 10<> г-
Сочетая это соотношение с уравнением (6), получаем выражение
_ la К' — (En~Eiii)F _, (тентх)ш ,
г.зознг 8 (znx/m0H)n
, 3,629 • 10е , ,/ . /о,
+ (Д^/2 |/р = - 1g + 7 (и) • (8)
Так как все члены левой части данного уравнения определяются экспери-
ментально, то, изображая левую часть уравнения графически в виде
функции от р, получаем при экстраполяции на бесконечное разбавление
1 Харнед и Шупп [9] исследовали систему CsOH—CsCl; Харнед и Гамер [10]—
систему КОН—КС1; Харнед и Копсон [11] — систему LiOH—LiCl; Харнед и Маннвейлер
[12] — систему NaOH—NaCl; Харнед и Донельсон [13] —систему LiOH— LiBr; Харнед
и Гири [14] —систему Ва (ОН)2 - ВаС12.
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
453
величину —1g А^. Несколько полученных таким путем кривых показано
на рис. 141; как видно, наклоны этих кривых отличаются от наклонов
соответствующих кривых на рис. 140. Эта разница является существенным
преимуществом для определения точки пересечения кривых с осью орди-
нат [15]. Важнейшим достоинством метода экстраполяции, изображенного
на рис. 141, является его независимость от значений стандартных электрод-
ных потенциалов. Как видно из рис. 141, этот метод экстраполяции дает тоже
значение величины Kw, что и метод, представленный рис. 140.
Рис. 141. Определение Kw при 25° на основании
уравнения (8).
В табл. 107 приведены значения Kw, полученные независимо в упо-
мянутых выше исследованиях. Наиболее сомнительные в смысле их точ-
ности результаты взяты в скобки и не были использованы при вычислении
средних величин, приведенных в последнем столбце.
Таблица 107
Константа диссоциации воды (Р=1 атм)
Kw Ю“
t, °C КС1 [1] NaCl [2] ВаСЦ [3] ЫВг [4] Среднее значение
0 0,11421) 0,1134 (0,1125) (0,1132) 0,1139
5 (0,1860) 0,1850 (0,1834) 0,1842 0,1846
10 (0,293) 0,2919 (0,2890) 0,2921 0,2920
15 (0,452) 0,4505 0,4506 0,4504 0,4505
20 0,681 0,6806 0,6815 0,6806 0,6809
25 1,008 1,007 1,009 1,007 1,008
30 1,471 1,470 1,466 1,467 1,469
35 2,008 2,091 2,090 (2,068) 2,089
40 2,916 2,914 2,920 — 2,919
45 4,016 4,017 4,023 — 4,018
50 5,476 5,482 5,465 — 5,474
55 7,297 — .— — 7,297
60 9,614 — — — 9,614
1) Пересчитано.
454
ГЛАВА XV
§ 2. Функция коэффициента активности уцуон/«н2о и ионное
произведение тицтон в случае водных растворов солей
При первых определениях [6, 9] величины унУон/ан2о наряду с иссле-
дованием элементов, описанных в предыдущем параграфе, возникла
необходимость в измерениях электродвижущих сил элементов следующих
типов:
Н21 МОН (гаД MX (m2) j Мж Hg | МОН (m3) | Н2 IV
и
Ag-AgX | MX (m2) | Мж Hg | MX (mJ | AgX-Ag. V
Этот метод основан на определении значений трех величин, унух,
УмУон/ангО и умух, в растворах чистой соли MX при постоянной концен-
трации. С помощью этих величин можно получить затем значение
Унуои/«н2о в растворе соли для этой частной концентрации. Таким образом,
(УнУх) (УмУон/«н2о)/(УмУх) = УнУон/ян2о- (9)
Значение умУх определяется с помощью элемента V методом, описан-
ным в гл. 10, § 4. Элементы 1,и III, или элемент III и значение Е°,
полученное из электродвижущей силы элемента I, можно использовать
для определения унУх в смеси НХ и MX, взятых в различных соотношениях,
при заданной постоянной общей моляльности. Затем можно на основании
линейной зависимости IgynYx от тнх, которая была рассмотрена в гл. XIV,
§ 4, получить значение уцух в чистом растворе соли путем экстраполяции
на нулевую концентрацию кислоты. Применение этого же метода для
обработки результатов, полученных при измерении электродвижущих сил
элемента IV, дает значение умуон/«н2о для растворов солей при нулевой
концентрации гидроокиси.
Описанный метод является точным и термодинамически правиль-
ным, но трудоемким, так как он связан с применением неудобных в ра-
боте электродов с проточными амальгамами. При использовании элементов
типа II и III этот недостаток устраняется. Поскольку значение Ки,
известно, то для непосредственного определения унУон/«н2о можво записать
уравнение (6) следующим образом:
(Бц —Бщ) F _ (raHrax)iii . , 7i-i7qh
2,303 ДУ ‘^х/Мп “ 8 «н2о
(Ю)
Ионное произведение 7интОн находят путем деления Кш на унуон/ян2о
согласно уравнению (3). Этот метод был применен для определения дис-
социации воды в водных растворах хлоридов и бромидов натрия, калия
и лития, а также хлоридов цезия, стронция и бария в интервале концен-
траций 0 — 3 М и для температур от 0 до 50°, а в некоторых случаях до 60°.
Ни в коем случае нельзя забывать, что при этих вычислениях прини-
малось, что все сильные электролиты (НХ, МОН и MX), которые содер-
жатся в этих элементах, полностью диссоциированы при всех концентра-
циях. Если это предположение несправедливо, то значение тнтон не совпадает
точно с произведением истинных моляльностей этих ионов в солевых
растворах. Конечно, полученные до сих пор результаты могут быть исправ-
лены в дальнейшем с учетом любых отклонений от полной диссоциации,
которые будут обнаружены. Однако эти поправки не влияют на значения
дантон и Kw при нулевой концентрации соли, так как указанные откло-
нения исчезают при экстраполяции.
Влияние температуры и концентрации на величину тн ( = нгнтОн)
в случае растворов хлористого. [10] калия изображено на рис. 142,
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
455
из которого видно, что диссоциация достигает максимума при концен-
трации, равной примерно 0,7 М, и что этот максимум становится более
четким при более высоких температурах.
На рис. 143 представлена зависимость величин унТон/«н2о и mHm0H
от ]/"р. для ряда растворов солей при 25°. Полная сводка результатов,
ристого калия в интервале температур 0—60° через
каждые 5°.
полученных для различных температур, приведена в табл. 163. Эти дан-
ные были частично обобщены Харнедом и Куком [16] с помощью урав-
нения Дебая и Гюккеля
4 -2^>^.+ви + (у/г, (И)
ё «изо 1 ’
в котором
£(/> = 1,814 . W/IDTyiz, (12)
(13)
Значения D приведены в табл. 3. Для удобства вычислений и составления
таблиц было принято, что эмпирические параметры В и С линейно зависят
от температуры:
= + (14)
С = с0 + с^, (15)
а параметр 8 с температурой не меняется. Значения констант даны в табл.108.
Несмотря на произвольный выбор обычных величин а для растворов
456
ГЛАВА XV
хлоридов и бромидов и допущение о простой зависимости параметров от
температуры, с помощью приведенных в табл. 108 констант можно выра-
жать экспериментальные результаты с точностью 0,5—И% для всех концен-
траций вплоть до 3 М и для любой температуры в интервале 0—40°.
Исключение составляют растворы хлористого лития, для [которых при
Р и с. 143. Произведение коэффициентов активности
ионов и ионное произведение воды для растворов неко-
торых галогенидов при 25°.
температурах ниже 10° ошибки превышают 1%. В случае растворов хло-
ристых натрия и калия это уравнение удовлетворительно отражает резуль-
таты вплоть до 50°. Величины среднего отклонения ДСредн. наблюдаемых
значений ^унуон/«н2о от значений, вычисленных по уравнению (11), приве-
дены в последнем столбце табл. 108. Если принять во внимание все пара-
метры, то эти отклонения не будут превышать экспериментальных ошибок.
Таблица 108
Константы уравнений (13)—(15)
Соль О а Ьо bj ю4 Со С1 • 10* дсредн.
КС1 3,6 0,266 5,20 -0,0350 -4,88 0,0027
NaCl 3,6 0,198 2,00 -0,0085 -2,0 0,0032
UC1 3,6 0,039 2,00 +0,0325 -4,0 0,0025
КВг 4,2 0,205 12,75 -0,016 -9,0 0,0022
NaBr 4,2 0,157 6,75 +0,010 -6,57 0,0035
LiBr 4,2 0,000 0,000 + 0,0475 -2,20 0,0041
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
457
Последовательность расположения кривых на рис. 143 заслуживает
некоторого внимания. При данной концентрации диссоциация воды в рас-
творе данной соли тем больше, чем меньше радиусы ионов этой соли
в кристаллическом состоянии. Это явление можно качественно объяснить
тем, что молекулы воды сильнее поляризуются при непосредственном
влиянии ионов меньшего размера с более высокой плотностью заряда.
В настоящее время нет достаточно надежных данных, на основании
которых можно было бы определить точную функциональную зависимость
диссоциации растворителя от ионных радиусов, но закономерный характер
этой зависимости и наличие упомянутой обратной пропорциональности
явствует из рис. 144. Это явление проиллюстрировали Харнед и Маннвей-
лер [12], произвольно прини-
мавшие в качестве абсцисс ве-
личины 1/а+4-1/а-). Мы
принимали в качестве независи-
мой переменной величину ^1/а?,
так как средняя плотность заря-
да ионов прямо пропорциональна
этой величине. Значения ионных
радиусов были взяты из табл. 6.
Хотя было бы преждевре-
менным делать какие-либо коли-
чественные заключения на осно-
вании рис. 144, но характер ура-
внения
lg Woh = 1g _= А _ в У 4,
rtII2O m£ImOH ^ai
(16)
которое отвечает прямым линиям,
проходящим через данные точки,
позволяет предположить, что на
Рис. 144. Зависимость ‘ГцУоп^НаО для Рас"
творов солей от ионных радиусов соли-
1 — 2,ОНИ; 2—11/; 3 — 0,5/И; 1-0,11.1/
(4 = 25°).
основании такого рода результатов
можно разбить эффект действия соли на две различные части. Влияние
междуионных сил можно представить членом А, в то время как влияние поляри-
зации молекул растворителя может быть учтено только членом который
пропорционален средней плотности зарядов. Правильность такого разделе-
ния подтверждается тем фактом, что параметр В все время увеличивается
с ростом концентрации, в то время как параметр А вначале уменьшается,
достигает минимального значения при концентрации около 0,4 71/, а затем
возрастает при увеличении концентрации в соответствии с общеизвестным
поведением коэффициента активности сильного 1,1-электролита.
§ 3. Влияние температуры и давления на диссоциацию
воды. Термохимические величины [17]
Ввиду отсутствия точного теоретического уравнения для выражения
константы диссоциации как функции от температуры, а также в связи с тем,
что значения констант диссоциации известны точно только для сравнительно
узкого интервала температур (0 — 60°), трудно выбрать то или иное эмпи-
рическое уравнение. Обычный метод состоит в том, что зависимость Д2/?
от Т представляют в виде степенного ряда
ДЯ9 = Л-В/’-С1Г2-|- ...,
ДС^ = - В-2СТ+ .
(17)
(18)
458
Г Л* А В A XV
После подстановки значения ДЯ? в уравнение (86) гл. I и интегрирова-
ния получаем
1п^!а==--А--|1п7’-^7’ + .»4- ... (19)
Знаки перед членами выбраны таким образом, чтобы константы, получен-
ные из экспериментальных данных, были положительными. В случае ре-
зультатов, лежащих в узком интервале температур, никогда не приходится
применять члены со степенями выше У2 в уравнении (17). Определение
констант уравнения (19) из экспериментальных данных с помощью метода
наименьших квадратов представляет собой очень трудоемкий процесс,
и поэтому желательно иметь более простые выражения. Одним из допу-
стимых упрощений является предположение о независимости ДС?. от Т, т. е.
о линейной зависимости ДУГ? от температуры. При этом получаются
следующие уравнения:
Д//? = Л-ВГ, (20)
ДС°.= -В, (21)
1пУГи,= -А_^1пГ + А (22)
Другое уравнение для InTf, которое является непосредственным след-
ствием свойств электродвижущих сил, требует некоторых разъяснений.
Было обнаружено, что для изучаемого интервала температур эксперимен-
тальные значения электродвижущих сил можно в пределах ошибки опыта
(+0,05 мв) выразить при помощи квадратного уравнения относительно Т.
Йз уравнения (4) явствует, что если Еп и Е° выражаются квадратным
уравнением относительно Т, то 1п7Гю можно представить следующим урав-
нением:
+ A (23)
и, следовательно,
Д#? = Л-СУ2, (24)
ДС?.= -2СГ. • (25)
Конечно, нельзя считать, что ДСр. совершенно не зависит от Т или
что эта величина является линейной функцией от Т, как это следует
из уравнений (21) и (25). Однако далее будет показано, что оба уравне-
ния (22) и (23) можно применять для вычисления Kw в интервале темпе-
ратур 0 — 60° с одинаковой степенью точности. Этот факт свидетельствует
о том, что данные вычисления нельзя использовать для выяснения харак-
тера зависимости ДС?. от Т.
Константы уравнений (19), (22) и (23) были найдены с помощью
метода наименьших квадратов из значений Kw, приведенных в табл. 107.
Основанные на этих данных уравнения для —lg Kw с численными значе-
ниями параметров, а также соответствующие численные уравнения для
определения ДУ?, Д/Z?, ДС?. и ДД1?, полученные из уравнения (19), при-
ведены в табл. 109. Данные, получаемые с помощью трех уравнений,
прекрасно совпадают друг с другом. Так, средние величины отклонения
расчетных данных от опытных составляют для этих трех уравнений соот-
ветственно всего 0,00045, 0,0005 и 0,0005 (в единицах —lgKw), а макси-
мальное отклонение составляет 0,0014. Среднее отклонение соответствует
ошибке в 0,03 мв, а максимальное — в 0,08 мв, что не превышает воспроизво-
димости результатов опытов по определению электродвижущих сил. Такой
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
459 ‘
результат показывает, что на основании этих данных нельзя решить,
является ли величина ДСр. постоянной, или найти характер ее зависи-
мости от температуры.
Таблица 109
Численные коэффициенты для вычисления (— 1g Ка) по уравнениям (19), (22) и (23)
и для вычисления ДяР, ДЯ?, ДС®{ и Д5® по уравнениям, полученным из уравнения (19)
1. Уравнение (19) и т. д.
1g К№= — _24^’39 + 35,3944 — 0,008530т —11,8261 lg Т (19а)
(среднее отклонение наблюдаемых результатов от вычисленных составляет
0,00045 в единицах IgK. Максимальное отклонение равно 0,0014).
&F® = 23981,15 + 54,1047 Т lg Т — 161,9308 Т + 0,039025 Г2.
ДЯ® = 23984,15 —23,497 Т —0,039025 Т2. (17а)
ДС°_=— 23,497 — 0,07805 Т. (18а)
Д5» = — 54,1047 lg Т— 0,07805 Т + 138,4338.
2. Уравнение (22).
6013 79
lg ---23,6521 lg Г+ 64,7013 (22а)
(среднее отклонение составляет 0,0005. Максимальное отклонение равно 0,0014).
3. Уравнение (23).
1g — 4479,99 + 6,0875 —0,017060 Т (23а)
(среднее отклонение составляет 0,0005. Максимальное отклонение равно 0,0014).
Максимальное расхождение между значениями вычисленными
по этим трем уравнениям, составляет 40 кал при 0°. При 25° полученные
из уравнений (18), (21) и (25) значения ДСр. составляют соответственно
— 46,7, —47,0 и —46,5. Однако при более высоких и более низких темпера-
турах наблюдаются значительные расхождения. Так, например, при 0° три
уравнения дают значения —44,8, —47,0 и —42,6 кал. Поскольку все
уравнения одинаково хорошо согласуются с экспериментальными данными,
то нельзя сказать, какой из этих результатов ближе к истинному значению.
На рис. 145 представлена зависимость величин (— igKw), вычисленных
по уравнениям (19а), (22а) и (23а), от температуры. Все кривые имеют
максимум при температуре около 550э. Данные о значении Kw при таких
высоких температурах получены Нойесом и Като [18], а также Сосмэном [19]
на основании исследования гидролиза ацетата аммония. Эти данные, пока-
занные на рис. 145 кружками, определенно указывают на то, что макси-
мальное значение Kw действительно достигается при температуре, совпа-
дающей со значением Т, рассчитанным по уравнениям (19а), (22а) и (23а).
Эти результаты могут быть использованы также для вычисления &Нг —
теплоты диссоциации воды в растворах солей. В растноре с концентрацией m
теплота диссоциации воды равна
= Ян+ + 77 оп- - ЯНзо (26)
или в иной форме
Д77, = Д77° + LH++-^он-^що- (27)
Таким образом, Д_/7\ можно разделить на две части. Первая часть Д#<
не зависит от концентрации соли и связана с температурным коэффициентом
460
ГЛАВА XV
In К по уравнению (86) гл. I. Вторая часть теплоты диссоциации воды
(£н+ + АОн---^н2о) зависит от концентрации и может быть вычислена по
уравнению (38) гл. I из температурного коэффициента функции In (упТон/«н»о).
Таким образом,
д уу._RT2 & ^н^он/янгр) (28)
Сравнение этого уравнения с уравнением (6) показывает, что АН, можно
определить непосредственно из данных об электродвижущих силах с помощью
Рис. 145. Зависимость константы дис-
социации воды от температуры.
Кружки соответствуют экспериментальным
данным. 1—уравнение (19а); 2—уравне-
ние (22а); 3—уравнение (23а).
Рис. 146. Полная теплота диссоциации воды
в растворах некоторых галогенидов при 20°.
Аналогичным образом остальные
величины определяются уравнениями
а(Еп-Е1и)
дТ
ACPi = FT
ПЕп~Еш)
дТ2
(30)
(31)
AAj = F
На основании вычисления теплот диссоциации воды в растворах различ-
ных солей было установлено [20, 12, 21], что зависимость АН° от р1/2 имеет
линейный характер для всех концентраций' с точностью до эксперименталь-
ных ошибок, составляющих 30 — 80 кал. Согласно теории, такая зависи-
мость должна соблюдаться для суммы LH+ 4- L0H---Анщпри большом разбав-
лении. Поскольку эта закономерность, вопреки ожиданиям, наблюдается
и при более высоких концентрациях, то этим можно воспользоваться для
определения значения Д#? путем экстраполяции AHi до бесконечного раз-
бавления. На рис. 146 изображена зависимость Д//{ от т1/2 для растворов
пяти различных солей при 20°. Если данные для растворов отдельных солей
экстраполировать независимо друг от друга, все точки пересечения прямых
с осью ординат располагаются с разбросом приблизительно в ± 30 кал
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
461
по отношению к значению Д/7?, равному 13 710 кал-, при других темпера-
турах наблюдается примерно такая же точность экстраполяции. Для большей
ясности на рисунке не нанесены опытные данные, а представлены лишь
прямые линии с полученными экспериментальным путем значениями коэф-
фициента наклона. Теоретический предельный наклон показан на рис. 146
пунктирной линией.
Из сравнения данных рис. 146 с соответствующими калориметрическими
определениями, представленными на рис. 54, явствует, что точки пересе-
чения с осью ординат (отвечающие значению кН®), полученные двумя неза-
висимыми методами, отличаются всего на 40 кал, что лежит в пределах
ошибок опыта обоих методов. В гл. VIII, § 3, было показано, что (— &Htj
в данном солевом растворе нельзя отождествлять с ^Нп — непосредственно
измеренной теплотой нейтрализации, так как процесс нейтрализации
сопровождается разбавлением соли. Далее, хотя Д//* представляет собой
теплоту нейтрализации с учетом поправки на теплоту разведения соли, эта
величина не совпадает с ( — если только величины теплосодержания
ионов не являются аддитивными при всех концентрациях. Поэтому мы воз-
держиваемся от сравнения ( — с Д/7* при различных концентрациях,
ограничиваясь сопоставлением их предельных значений при бесконечном
разбавлении 2.
Согласно уравнению (85) гл. I, влияние давления на диссоциацию воды
выражается уравнением
= f32.
< дР )т 2,3 RT ’ '
где
ДУ?=Иш + Уоп--<о. (33)
Величина Вцао равна молярному объему чиетой воды или 18,07 см3 при 25°
и давлении, равном 1 атм. Парциальный молярный объем диссоциированной
воды можно вычислить, составляя линейную комбинацию величин У| для
сильных кислот, оснований и солей, так как при бесконечном разбавлении
ионные объемы являются аддитивными. Например, можно написать
Ен+ + Уон-7-- V° (ис1) + Уг (кон) — V2(kci)-
Точно так же можно найти -йГон- путем сочетания величин К% для
сильных кислот, оснований и солей, причем ТГн2о равно 18,02 ро/с?0- Если
допустить, что не зависит от Р, то
Ё1(щ = У30-^°(Р-1) (34)
по уравнению (101) гл. VIII. Здесь У|(Р) представляет собой парциальный
молярный объем при любом давлении Р, а У0 и К<} — соответствующие
величины при Р—1. Путем сочетания предыдущих трех уравнений полу-
чается приближенное соотношение
щ СР-1)3
g#w(P=1) 2,3 ЯГ 4,6 ЯГ ’
справедливое в случае низких давлений.
Весьма удобно, что нет необходимости пренебрегать изменением Д7Г°
с давлением, поскольку, как было показано [22], пока величина К° велика
1 Следует отметить, что при сравнении (— Д/Д-) и обнаруживается хорошее
совпадение этих величин друг с другом £20, 12, 21].
462
ГЛАВА XV
по сравнению с молярной сжимаемостью чистого растворенного вещества,
предельный парциальный молярный объем растворенного вещества при любом
давлении может быть найден с помощью уравнения
V°2 (Р) = v°2 - к°2 (Р -1). (36)
Это уравнение отличается от уравнения (34) множителем (В 1)/(В + Р),
который учитывает зависимость от давления в значительных пределах.
Сочетая это уравнение с уравнениями (32) и (33), получаем выражение
lg (J^) = _ ^=1L + + IL Г(Р — 1) — (В + 1) In (A±L-) 1 ,'
W (Р — 1) ' 1x4 L £> -j- 1 у J
(37)
которое можно применять в интервале давлений в несколько тысяч атмосфер.
В табл. 110 приведены средние значения ДУ? и ДУГ?, а также резуль-
таты, полученные при подстановке этих значений, в уравнение (37). Кон-
станта диссоциации возрастает с увеличением давления, причем ее отно-
сительное увеличение тем больше, чем ниже температура. При темпера-
турах, близких к нулю, К„: увеличивается в 3 раза при изменении давле-
ния от 1 до 1000 бар. Положительный температурный коэффициент ДУ°
(и ДУ° для некоторых других реакций диссоциации) указывает на то, что
при достаточно высоких температурах ДУ? может сделаться положитель-
ным, что приведет к уменьшению значения Kw при повышении давления.
Влияние давления на ЬН® и Д5? при 25° показано в двух последних
столбцах табл. 110. Эти термохимические величины, так же как и вели
чина ДС?., с увеличением давления становятся более отрицательными.
Таблица 110 [1]
Влияние давления на диссоциацию чистой воды
t, °C Р(бар-106) 5 15 25 35 45 । ДИ?(25°> AS?(25c)
КЯ/КЮ(Р„1)
1 1 1 1 1 1 13 500 -18,7
200 1,246 1,225 1,202 1,180 1,163 13 200 -19,4
400 1,543 1,490 . 1,435 1,384 1,345 12 900 -20,1
600 1,896 1,800 1,703 1,612 1,545 12 600 -20,8
800 2,317 2,163 2,009 1,868 1,766 12 200 -21,8
1000 2,816 2,585 2,338 2,154 2,009 И 800 -22,8
ДР? -26,1 -24,9 -23,4 -21,8 -20,6 —
ДйГ? г — — - 0,00522!) — — —
1) При всех температурах применялось значение определенное при 25°.
2) Один бар=О,9869-10-а нормальных атмосфер.
К сожалению, имеющиеся экспериментальные данные недостаточно
точны для получения точных сведений относительно влияния давления
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
463
на температуру, соответствующую максимальному значению Kw или, в более
общем случае, К, для слабых электролитов. Краткий обзор этого вопроса
и влияния давления на реакции диссоциации в растворах солей сделан
Оуэном и Бринкли [22].
§ 4. Определение констант диссоциации слабых кислот и оснований
Константы диссоциации слабых кислот могут быть вычислены из
экспериментальных значений электродвижущих сил элементов без жидко-
стных соединений, содержащих небуферные или буферные растворы. Эле-
мент с небуферным раствором
H2|HR(W1),MX(7n2)|AgX-Ag VI
был впервые применен для определения функций диссоциации / (тх — щн)
и функции коэффициента активности YHYAc/YHAc в солевых растворах [23]
и позднее для определения константы диссоциации К а [24]. Из этих экспе-
риментальных данных для уксусной кислоты было получено значение Ка,
равное 1,75 • 10~5 при 25°. Дальнейшие определения [25] показали, что
величина КА мало изменяется в интервале 20 — 30°. Эти результаты согла-
суются с найденными в последнее время более точными значениями, полу-
ченными с помощью элементов, содержащих буферные растворы. Обстоя-
тельный анализ результатов, полученных при использовании элементов,
содержащих небуферные растворы, будет приведен в § 7, где будет пока-
зано, что с помощью этих элементов можно определять коэффициенты
активности и функции диссоциации слабых кислот в растворах солей,
а также константы диссоциации. Далее будет тщательно изучено влияние
среды на свойства веществ, входящих в состав этих элементов.
Наиболее непосредственное электрометрическое определение' константы
диссоциации слабой кислоты может быть осуществлено с применением
элемента следующего типа [26]:
Н21 HR (mJ, MR (mJ, MCI (mJ | AgCl-Ag. VII
Такие элементы могут служить только для определения констант дис-
социации, но для этой цели они очень удобны. Буферное действий смесей
растворов используется для того, чтобы точно и просто измерять малые
концентрации водородных ионов при относительно низких значениях ион-
ной силы. Экстраполяция полученных данных производится на корот-
кие промежутки и является практически прямолинейной. Концентрация
слабой кислоты HR и концентрации двух солей MR и MCI подбираются
приблизительно равными; в качестве катиона М обычно служит натрий
или калий. Применение электрода серебро-хлористое,] серебро имеет ряд'
практических преимуществ, однако в тех случаях, когда этот электрод
неприменим, можно использовать и другие электроды.
Электродвижущая сила этого элемента определяется по уравнению (2),
из которого YhW'T[ можно исключить с помощью выражения для константы
диссоциации слабой кислоты:
Yh Yr
У hr
mIl TOR
mIIR
В результате получается следующее уравнение:
(EVII-E°)F mclmHR . YcHhr
”Z303 HF“ + lg ~lg ^~lg—
(38)
(39)
464
ГЛАВА XV
Так как mcj = m3, mHR = mx— т-п и mR = m2-|-mH, т0 члены левой части
этого уравнения известны, если тн очень мало по сравнению с тх и т2.
Если значение тн настолько велико, что им нельзя пренебречь, то его
можно оценить следующим образом [26— 28]. Сначала вычисляют предва-
рительное значение КА по уравнению (39) при помощи предельного закона
для коэффициентов активности и при допущении mx/m2. Если
это значение меньше, чем 10“4, то тн с достаточной точностью можно
представить выражением тн — К^т^т^. В случае необходимости можно
найти более точное значение тн, используя более точную величину КА,
которая определяется с помощью метода экстраполяции, описанного ниже.
Число таких последовательных приближений тем больше, чем больше вели-
чина КА. Константы диссоциации порядка 10*4 и больше также можно
определять этим методом, но при относительно больших значениях ин либо
приходится прибегать в весьма большому числу последовательных прибли-
жений [29]1, либо совсем отказываться от’применения уравнения (39).
В последнем случае пользуются вычислением «кажущихся» концентраций
водородных ионов [24] непосредственно из значений электродвижущих сил
(§ 7) и с помощью, уравнения (2). Преобразуя это уравнение и выражая
коэффициенты активности при помощи предельного закона, получаем сле-
дующую зависимость:
(EVTT — Е°) F __
- 1gтц = -^-303 Д7, - + 1gтС1 -2§(/)у pd0. (40)
Ионная сила равна приблизительно
+ + ' (41)
Некоторая неточность, связанная с применением предельного закона, кото-
рый не учитывает влияния среды, состоящей из неионизированных молекул
слабого электролита, исключает возможность определений истинного зна-
чения тц этим методом, но различие между тн и тй быстро уменьшается,
с разбавлением и в пределе исчезает. Таким образом, пользуясь уравне-
нием (38) и предельным законом, можно вычислить кажущуюся константу
ионизации АД, соответствующую тУ Следовательно,
--- т'„ 1т, + ml,)
ig^= -2s(/)Kк+ig; (42i
lg К а можно получить путем экстраполяции [30] величин lg k'A.
Если К а < 10_ 9, как, например, в случае борной кислоты, то при
больших разбавлениях следует учитывать гидролиз аниона кислоты R.
Так как тн в растворах таких слабых кислот очень мало, можно считать
mHR = mx -ф тон 11 тп = m-z — тон и оценить значение тОн по константе
гидролиза Kh аниона кислоты. Достаточно точное значение величины тон
можно получить из соотношения тон —7^/;m2/mi, а соответствующее значе-
ние величины Kh получается из отношения Kw и грубо приближенной'
величины КА-
В уравнении (39) последний член правой стороны содержит логарифм
отношения двух коэффициентов активности ионов одинакового типа валент-
ности и логарифм коэффициента активности нейтральной молекулы. Так
как обе эти величины незначительно изменяются с ионной силой при
большом разбавлении, то, изображая графически зависимость левой части
уравнения (39) от р, можно определить — 1g АД. На рис. 147 изображена
1 См. гл. XIII, § 8.
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
465
экстраполяция, с помощью которой была вычислена константа диссоциации
уксусной кислоты [26, 31] при 25°.
Склонностью бора к образованию комплексных многоосновных ионов
даже в сравнительно разбавленных растворах объясняется особый вид
графика экстраполяционной функции в случае борной кислоты. Определе-
ние константы первой ступени диссоциации борной кислоты (Н3ВО3) при 25°
Р и с. 147. Определение константы диссоциации уксусной
кислоты по уравнению (39) при 25°.
Диаметры кружков равны 0,02 мв.
Р и с. 148. Определение константы диссоциации борной ки-
слоты по уравнению (39) при 25°.
Отношение т^^^/т^^принято равным: 1 (верхняя кривая), 1,18
(средняя кривая) и 2,69 (нижняя кривая).
показано на рис. 148. Изменение отношения mi/m2 оказывает значительно
более сильное влияние на расположение и форму кривых, чем это наблю-
далось в случае уксусной кислоты. Верхние кривые на рис. 148 становятся
горизонтальными при р 0,01, что устраняет необходимость в экстраполя-
ции ниже этой концентрации. Далее, уравнение (39) можно преобразовать
таким образом, что может быть определено Е°, если известна величина Ка.
30 Г. Харнед, Б. Оуен
466
ГЛАВА XV
Эти обстоятельства, а также устойчивость иодидов в щелочных растворах
позволяют значительно легче определить стандартный потенциал электрода
серебро-иодистое серебро [32] в буферных растворах борной кислоты, чем
по измерениям электродвижущей силы элементов, содержащих разбавлен-
ные растворы иодистоводородной кислоты. Оказалось, что применение буфер-
ных растворов имеет значительные преимущества также при определении
других стандартных потенциалов [33, 34] (см. гл. XII, § 2).
Для многоосновных кислот экстраполяционная функция усложняется
вследствие наличия ступенчатой диссоциации и присутствия многовалент-
ных ионов, но это практически не имеет значения в тех случаях, когда
отношения констант нескольких процессов диссоциации превышают 10®
или 104. Для иллюстрации хода вычислений [27] ниже будет определена
константа второй ступени диссоциации фосфорной кислоты. Для определе-
ния этой константы применялся следующий элемент:
H2|MH2PO4(m1),M2HPO4(m2), MCI (m3) | AgCl-Ag, VIII
электродвижущая сила которого определяется уравнением (2). Константа
второй ступени диссоциации фосфорной кислоты равняется
К2А = 7н7нРО* тн-тнР^ . (43)
ЧН2РО4 даН2РО4
Путем исключения у тн из уравнений (2) и (43) получается уравнение
04)
т "гнро4 7нро4
Последний член справа содержит в числителе коэффициенты активности
двух одновалентных ионов, а в знаменателе — коэффициент активности двух-
валентного иона. Применяя к этому члену уравнение Дебая и Гюккеля,
можно при большом разбавлении заменить его выражением —2§(/) [/”
в результате чего уравнению (44) можно придать следующий вид:
<45>
Определение —lgJ^2A с помощью уравнения (45) показано на рис. 149 [27].
В фосфатных буферных растворах порядок величины тн составляет 10”’,
т. е. тн настолько мало по сравнению с величинами т1 й т2, что оно
не принималось во внимание при вычислении значений тн2ро4 и тлро4- Как
будет показано, в общем случае можно не учитывать как тн, так и тон,
если т4/т2^1 и значение К лежит между 10-5 и 10~9.
Электрометрическое определение констант диссоциации слабых основа-
ний в принципе аналогично описанному выше . способу, за исключением
необходимости применять электроды, заменяющие электрод серебро-хлористое
серебро в случае аммиачных и других основных растворов, в которых хло-
ристое серебро хорошо растворимо. Было показано, что электрод из амаль-
гамы натрия [35] дает результаты, совпадающие с результатами, получен-
ными при работе с электродом серебро-хлористое серебро в случае таких
систем, для которых могут быть использованы оба электрода. Было пред-
ложено также применять электрод из амальгамы таллия [36]. Электрод
серебро-иодистое серебро также пригоден для этой цели, и мы используем
его для иллюстрации метода. Электродвижущая сила элемента
H2 |BOH(m1),BJ(m2)| AgJ-Ag IX
может быть выражена уравнением (2), если заменить ион хлора на ион
иода. Произведение YHmH можно исключить путем совместного решения
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
<467
уравнения (2) и выражений
TZ 7ц 7он „
л w —- — :— ТПц Шон (46),
аН2О
и
7 в 7он тв тон
7вон твон
(47)
После подстановки — ]/ud0 ± Pt1 вместо 1g ув /?вон получается: сле-
дующая экстраполяционная функция:
(ETv — Е°) F mBmT ---
2ЖДТ +1g^ + 1g^"j-g(/)/ K = lg^B±^- (48)
’ вон
На основании некоторых предварительных измерений [37] в растворах,
содержащих аммиак и иодистый аммоний при 25°, было найдено, что зави-
симость членов левой части уравнения (48) от ц является линейной вплоть
Нижняя кривая—соли натрия, mi=m2. Верхняя кривая—однозамещен-
ный фосфат натрия и двузамещенный фосфат калия, 4mi=3m2.
до концентраций порядка р^0,1. Полученное путем экстраполяции зна-
чение Кв равно 1,75 • 10-5, что по порядку величины соответствует ожидае-
мому значению. Более надежные значения величин Кв получаются на осно-
вании данных относительно основной ионизации аминокислот; эти данные
будут рассмотрены в следующем параграфе.
Общеизвестные методы определения констант диссоциации с помощью
визуальной колориметрии, как прямые, когда слабый электролит или его
ионы окрашены, так и косвенные, связанные с применением цветных инди-
каторов и стандартных буферных растворов, значительно уступают в точ-
ности современным кондуктометрическим и электрометрическим методам.
Однако если заменить визуальное сравнение интенсивности окраски доступ-
ной в настоящее время объективной фотоэлектрической методикой [38], то
тем самым будет устранен наиболее важный источник ошибок этого способа.
С помощью «фотоэлектрической колориметрии» может быть достигнута очень
большая точность. Фотоэлектрический метод был впервые применен для
точного определения константы диссоциации Гальбаном и Эбертом [39] при
30*
468
ГЛАВА XV
изучении пикриновой кислоты. Аппаратура и методика исследования описаны
Гальбаном и Зидентопфом [40]. Принцип этого метода очень прост и состоит
в применении закона Ламберта и Бэра в интегральной форме
= (48а)
Здесь /0 и I -— интенсивность света до и после прохождения через слой
раствора толщиной х сантиметров, %— константа, зависящая от природы
раствора, от температуры и длины волны света. Мы будем придерживаться
обозначения Гальбана и называть g коэффициентом поглощения; величина
gc = (1/ж) 1g (70/7) называется поглощением.
Гальбан [41] исследовал условия, при которых применимо уравне-
ние (48а), и показал, что эти условия выполнялись в-его опытах. Он сделал
предположение, что поглощение света анионом кислоты не зависит от при-
роды присутствующего катиона. Это допущение создает некоторую неопре-
деленность, однако замена одной добавляемой нейтральной соли на другую
очень мало влияет на величину поглощения в разбавленных растворах,
а концентрация водородных ионов настолько мала, что специфическим влия-
нием этих ионов можно пренебречь.
Гальбан и Кортюм [416] определяли степень диссоциации а-динитро-
фенола (1, 2, 4) путем сравнения величин поглощения растворов этой кислоты
с величинами, поглощения растворов ее щелочных солей при той же ионной
силе. Так как при данной стехиометрической концентрации с поглощение
в растворе кислоты — gca, а в растворе соли —gc, то степень диссоциации а
равна отношению этих величин. В случае небуферного раствора кислоты
имеет место следующее выражение для константы диссоциации слабой
кислоты HR:
& __ Ун Уд. сн cr _ У-д у-r, со?
А Уин снн Унн а
Если ввести обозначение kA~ca.2/(i — а) и выразить коэффициенты актив-
ности с помощью предельного закона, то это уравнение принимает вид
lg#A = - 2S(/)]/^0 + 1g кА. . (50)
Экспериментальные значения 1g кА для а-динитрофенола, растворенного
в чистой .воде и в различных растворах солей [416, 42], представлены гра-
фически в виде функции от ]/ рс?0 в верхней части рис. 150. Прямая линия
проведена так, что ее наклон имеет теоретическое значение 1,013. В нижней
части рис. 150 изображена зависимость величины 1g кА—1,013 от
концентрации недиссоциированной кислоты сц, а почти горизонтальная
прямая линия, проведенная через эти точки, соответствует значению
1g КА = 5,9103. Хотя данные для чистых растворов слабой кислоты (верхняя
кривая) могут быть более точно представлены прямой, имеющей наклон
примерно на 20% выше теоретического, результаты для растворов, содер-
жащих добавочные соли, удовлетворительно выражаются прямой с теорети-
ческим значением наклона вплоть до р = 0,0004 и выше. Принимая во вни-
мание выводы, сделанные при анализе данных, изображенных на рис. 33
и 34, следует ожидать некоторого нарушения непрерывности при р 0,0002,
где данные для растворов чистой кислоты совпадают с данными для кислоты
в растворах солей, так как концентрация недиссоциированных молекул
изменяется непрерывно ниже этой точки, а выше ее практически является
постоянной. На рис. 33 влияние нейтральных молекул среды заметно уже
при у- — 0,0002 и увеличивается так быстро, что данные при больших кон-
центрациях нельзя использовать для экстраполяции, если это влияние ранее
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
469
не было учтено, как это сделано на рис. 34. Однако в случае рис. 150
условия совсем иные. Влияние среды практически постоянно, начиная с
[1 = 0,0002 и выше, и, таким образом, результаты, относящиеся к области
графика, соответствующей более концентрированным растворам, можно
использовать для более надежной проверки предельного закона, чем резуль-
таты для более разбавленных растворов. Поэтому можно использовать дан-
ные вплоть до р = 0,0004 для определения предельного коэффициента
наклона и считать, что эти опытные данные удовлетворительно соответ-
ствуют теоретическому коэффициенту наклона в пределах ошибок опыта.
Р и с. 150. Определение константы диссоциации
а-динитрофенола в воде при 25°.
Белые кружки—чистая слабая кислота; черные кружки—
слабая кислота в присутствии нейтральных солей.
Среднее отклонение точек от кривых составляет около 0,25%, и лишь для
пяти или шести точек отклонение достигает 0,5%. Эти отклонения имеют
тот же порядок величины, что и отклонения, встречающиеся при электро-
метрических определениях констант диссоциации с помощью элементов без
жидкостных соединений (среднее отклонение 0,05 мв, максимальное — 0,1 ли)1.
Однако следует отметить, что колориметрический метод часто применяется
при таких концентрациях, которые лежат за пределами, доступными методу
исследования с помощью гальванических элементов.
§ 5. Определение констант диссоциации амфолитов
Определение константы диссоциации наиболее важного амфолита — воды
путем измерения электродвижущей силы элементов без жидкостных соеди-
нений было рассмотрено в § 1. Все другие амфолиты, исследованные
этим же методом, представляют собой алифатические аминокислоты. Как
было показано ранее [43 — 46], электронейтральные молекулы этих соеди-
нений в растворе являются главным образом диполярными ионами (амфионы),
несущими положительный и отрицательный заряды. Обозначая такие моле-
кулы через Z±, можно изобразить кислотную и основную диссоциации
простых аминокислот с помощью следующих схем:
ZH+^±Z± + H+ . (51)
1 Гальбан и Кортюм [416] при оценке точности электрометрического метода не вво-
дят поправку на влияние имеющихся в растворе примесей, тогда как при оценке точности
разработанного ими фотоэлектрического метода они вносят соответствующие поправки.
470
ГЛАВА XV
(52)
(53)
(54)
И
ZOH-5± Z± + ОН~.
Соответствующие константы диссоциации равны [44, 35]
& — wz wh .
А Izh WZH
и
ТГ 7z 7 ОН WZWOH
л в =--- '--- •
Tzoh WZOH
Для определения кислотной диссоциации наиболее удобны элементы,
содержащие примерно равные концентрации аминокислоты и ее соляно-
кислого соединения. Электродвижущая сила элемента
Н21Z* (т^), HZC1 (m2) | Ag Cl-Ag X
определяется уравнением (2); после исключения Унтн с помощью уравне-
ния (53) получаем выражение
^+lg^H+lgbi._feb. (55)
Последний член в левой части этого уравнения можно заменить выраже-
нием — 2§(/)|/<>.с/0 ± Pfi, а неизвестный линейный член перенести в пра-
вую часть уравнения, чтобы получить экстраполяционную функцию
(Е2ZH - 2gw= (56)
"Ч
очень похожую на соответствующую функцию, применявшуюся в случае
фосфорной кислоты. Так как mci = m2, twz = 7^i + wh и ®zh = тг — тн, т0
член, в который входят концентрации, можно вычислить путем последова-
тельных приближений, исходя из предварительного значения К л, которое
получается, если не учитывать значения тн. В случае аминокислот
для достижения удовлетворительной точности требуется большое число
последовательных приближений, поскольку для этих кислот значения К а
имеют порядок величины 10~3 —10'2. Можно воспользоваться более про-
стым способом — представить уравнение (2) в следующем виде:
- ig = (E2^Jr)F + !g ™ci + lg YhYci, (57)
и вычислить mH с помощью приближенного выражения
lg YhYci 2 §(/)}/ ?d0.
Рис. 151 иллюстрирует применение уравнения (56) для вычисления
К а (//-аланина [47]. Нижняя кривая представляет собой графическую зави-
симость членов левой стороны уравнения (56) от у. для каждой данной
температуры. Верхние, почти горизонтальные кривые соответствуют дру-
гой функции, получаемой при подстановке в уравнение (56) вместо члена
— 2§(/д/щ70 известных значений lg YhYci для чистых водных растворов при
той же ионной силе и температуре. Так как обе экстраполяционные функ-
ции должны давать одно и то же значение величины —1g КА, то разли-
чие в их наклонах весьма удобно практически, поскольку оно способствует
точному определению искомой точки пересечения с осью ординат.
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
471
Константы основной диссоциации амфолитов можно определять с по-
мощью элементов типа
Н21Z* (mx), MZOH (m2), MCI (m3) | AgCl-Ag,
XI
в которых берутся равные концентрации компонентов. MZOH представляет
собой подходящую калиевую или натриевую соль амфолита, а MCI —соот-
ветствующий хлорид. Строго гово-
ря, обозначение MZOH соответ-
ствует гидратированной форме со-
ли. Явление гидратации влияет
на механизм диссоциации, но не
отражается на численном значе-
нии Кв-
Исключая унтн и уОнтон
при помощи уравнений (2) и (54)
и выражения для константы дис-
социации воды, получаем
(Eyt— E°)F тг,т7
2>ZRT g mZ0H + lg =
= Ig gB-lg10172^ . (58)
7zoh
ной диссоциации dZ-аланина по уравнению (56)
при 25°.
При построении верхней кривой —
было заменено экспериментальными значениями
lg VjjYc1 для чистых растворов хлористого [.на-
трия (табл. 160).
Последний член справа содержит
только величины, относящиеся к
нейтральным молекулам, а также
отношение двух аналогичных коэф-
фициентов активности ионов. По-
этому этот член является линейной функцией р при большом разбавлении.
Рис. 152 иллюстрирует применение этой экстраполяционной функции для
определения константы основной диссоциации глицина [48].
Рис. 152. Определение константы основной диссоциа-
ции глицина при 25 (нижняя кривая) и 35° (верхняя
кривая) по уравнению (58).
После того как Ig^B вычислен, величина YC1YZ«H2O / YZ0H в растворе
соли может быть определена с помощью уравнения (58), так как левую
часть уравнения можно найти из экспериментальных данных. Аналогич-
ным образом можно определить YclYZH/yz для раствора солей с помощью
уравнения (55).
472
ГЛАВА XV
Робертс и Кирквуд [49] использовали для определения коэффициен-
тов активности хлористого калия и глицина у3 в водных растворах,
содержащих оба эти вещества в моляльных концентрациях т2 и т3,
элементы типа: Ag-AgCl | КС1 (тге2) | КС1 (ти2), глицин (тге3) | AgCl-Ag.
Путем измерения электродвижущей силы этого элемента можно опреде-
лить / у±, где у±— коэффициент активности хлористого калия в отсутст-
вие глицина. Из уравнения
V J Т, Р, m2 \ / Т, Р, т3 ’ ' '
которое вытекает из условий интегрирования уравнения (11) гл. I, можно
вычислить у3. Значения этих величин при 25°, по данным Робертса и
Кирквуда, могут быть представлены эмпирическими уравнениями
1g = — 0,08942т3 4- 0,1226«?.//2//>?3 + 0,01074тз — 0,0628/w2m3 (60)
У±
и
lgy3 = - 0,1789 m2-0,06278m| +0,1635m2/2. (61)
§ 6. Вычисление констант диссоциации и термодинамических функций
Д#?, ДА? и ACapi
Константы диссоциации, полученные с помощью описанных в предыду-
щих разделах методов, приведены в табл. 164 — 166. Табл. 164 содержит
Р и с. 153. Зависимость логарифма константы
диссоциации от молярной доли диоксана (J =25э)*.
IIP—пропионовая кислота; НАс—уксусная кислота;
HF—муравьиная кислота.
константы диссоциации, найден-
ные для водных растворов при
различных температурах. В
табл. 165 приведены константы
диссоциации воды, муравьиной,
уксусной и пропионовой кислот
и глицина в смесях диоксан —
вода для температур 0 — 50°
с интервалами через 5°. Табл. 166
содержит кислотные и основные
константы диссоциации некото-
рых аминокислот в водных рас-
творах между 1 и 50°. Во всех
случаях при соответствующих
таблицах имеются ссылки на
первоисточники.
Константа диссоциации силь-
но зависит от диэлектрической
постоянной среды. В случае али-
фатических кислот и воды эта
зависимость выражается в том,
что К уменьшается в 10s раз при
изменении D от 80 (чистая во-
да) до 10 (82% диоксана). Так
как определения были проведены
только для пяти смесей дибксана
и воды, служивших в качестве
растворителей (табл. 165), то эти результаты нельзя точно интерполировать
с помощью одного графика. Харнед и Фаллон [50] предложили метод интер-
1 На рис. 153 авторы допустили ошибку в знаках при \%К (ордината). (Прим: ред.)
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
473
полиции, основанный на том факте, что 1g К почти линейно зависит от
молярной доли воды или диоксана. Этот метод показан на рис. 153, на
котором значение [1g К — 1g ТГ(дг2=о)] при 25° представлено в виде функции
от молярной доли диоксана TV2- Максимальное отклонение эксперименталь-
ных величин для воды, уксусной и пропионовой кислот от линейной зави-
симости составляет около 0,05 или около 1% от 1g К. Графическое изобра-
жение этих отклонений от простой линейной зависимости можно исполь-
зовать для точной интерполяции. Соответствующая кривая для муравьиной
кислоты отличается большей кривизной.
Так же как й при исследовании диссоциации воды (§ 3), было пред-
ложено несколько эмпирических уравнений для выражения зависимости
констант диссоциации слабых электролитов от температуры и для вычисления
термохимических функций. При обобщении экспериментальных результатов,
касающихся поведения ДСр;, встречаются те же затруднения, как и в слу-
чае воды. Данные табл. 111 хорошо иллюстрируют это обстоятельство.
Таблица 111
Константа диссоциации муравьиной кислоты [1]
А=[ 1£1^а1выч. I- 1gпабл.
t, °C -fg КА Д-104
(1) (2) (3) (4) (5)
0 3,7857 — 5 — 5 -6 -22 -32
5 3,7719 +6 -|-5 + 7 -{-6 — 5
10 3,7625 +5 +4 + 6 + 14 +3
15 3,7572 '-7 -8 - 5 + 6 -4
20 3,7533 -2 -2 + 6 + 2
25 3,7515 +5 +6 + 4 + 11 +5
30 3,7525 + 13 + 15 + 10 + 11 -И
35 3,7577 . 0 -|-3 -3 -6 -4
40 3,7655 -15 - 13 -18 -25 -18
45 3,7734 -И -9 -13 -21 -8
50 3,7825 +3 -|-5 0 -5 + 15
55 3,7940 +11 + 11 + 11 + 13 +39
60 3,8094 _ 9 -6 + 1 + 12 +49
Д (общее) 85 83 86 158 195
Д (среднее) 6,5 6,4 6,6 12 15
1)18Кд= 30,9965-^Ц,— — 0,00758417—10,4000 IgT, (62)
2) 1g Ад-56,7186-^^1-20,8000 IgT, (63)
3) IS Хд--134у’85 +5,2744-0,0151682 Т, (64)
4) 1g Кд=-8,0502+0,028803Т-4,826-Ю-5Т2, (65)
5) 1g КА = -8,1868 + 0,02978Т-5-10_»Т2.. (66)
Во втором столбце этой таблицы приводятся экспериментальные значения
— Ig^T.i для муравьиной кислоты. В других столбцах таблицы содержатся
отклонения этих значений от величин, вычисленных с помощью пяти урав-
нений, приведенных в нижней части таблицы. Константы уравнений были
474
ГЛАВА XV
получены с помощью метода наименьших квадратов. Как видно, первые три
уравнения по своей форме аналогичны уравнениям (19), (22) и (23), кото-
рые применялись для вычисления константы диссоциации воды (табл. 109).
Вычисленные и экспериментальные значения 1g К отличаются в среднем
только на 0,0007 для всех трех уравнений. Из этих данных можно сделать
тот же вывод, что и в случае диссоциации воды, а именно: на основании
этих результатов нельзя найти зависимость ДС£ от температуры. Выше
было показано, что имеются данные, свидетельствующие о том, что константа
диссоциации воды достигает максимального значения вблизи температуры 250°,
т. е. далеко от того интервала температур (0 — 60°), где значение константы
известно точно. Соответствующий максимум в случае муравьиной и других
алифатических кислот достигается почти при комнатной температуре. Было
обнаружено, что вблизи максимума для вычисления IgA" также можно при-
менять квадратное уравнение относительно Т. Это иллюстрируется величи-
нами отклонений, приведенными в последних двух столбцах табл. 111.
Первый из этих расчетов был сделан с помощью уравнения (65), все три
константы которого были получены методом наименьших квадратов. Послед-
ний столбец содержит отклонения опытных величин от вычисленных по
уравнению (66), в. котором коэффициент при Т2 был принят равным 5-10~6,-
а другие константы были найдены с помощью метода наименьших квадра-
тов. Совпадение результатов не является таким же хорошим, как при
использовании первых трех уравнений, поскольку средние отклонения
(соответственно 0,0012 и 0,0015 в единицах IgA) в этом случае примерно
вдвое больше. Таким образом, для получения более точных результатов
уравнения (62), (63) и (64), несомненно^ более пригодны, чем квадратные
уравнения.
Константы диссоциации многих слабых электролитов (табл. 164) имеют
максимум в интервале 0 — 60°. Харнед и Эмбри [51] сделали предположе-
ние, что это является свойством всех слабых электролитов. Эти авторы
показали также, что уравнение
lg A —1g Ав= —р(£ —0)2 (67)
в первом приближении выражает результаты достаточно точно (± 0,002
в единицах IgA) при условии, если оно применяется вблизи температуры 6,
при которой константа диссоциации имеет максимальное значение А'д. Та-
ким образом, константа диссоциации может быть выражена с помощью двух
эмпирических констант 6 и Aj и универсальной константы р, которая ока-
палась равной 5,- 10'5. После раскрытия скобок и преобразования уравне-
ние (67) приобретает вид
1g К = (lg Ае -p02) + 2p0f- pt2. (68)
Это уравнение идентично уравнению (66); возможность его применения в слу-
чае муравьиной кислоты иллюстрируется данными последнего столбца
табл. 111. Хотя это уравнение и не выражает наиболее точные опытные
данные так же хорошо, как другие рассмотренные выше уравнения, однако
оно дает достаточно хорошее приближение и его удобно применять, поскольку
нет необходимости прибегать к методу наименьших квадратов. Так, после
преобразования получается выражение
lgA + ^2 = (lgAe-p62) + 2^. (69)
Левая часть этого уравнения определяется из экспериментальных данных
и затем представляется графически в виде функции от t. При этом должна
получаться прямая линия с наклоном 2р0, отсекающая на оси ординат при
Z = 0 отрезок, равный (1g Ае — рО2). Так как р известно, то Ао и 0
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
475
можно определить. Соответствующие кривые приведены на рис. 154, на
котором сумма pt2 и мантиссы IgA" для муравьиной, уксусной, пропионо-
вой и хлоруксусной кислот представлена в виде функции от t. При тем-
пературах 0 — 45° точки ложатся на прямые линии в пределах точности
определения констант диссоциации. При температурах 50 — 60° наблюда-
ются отклонения [52].
Все эти уравнения в свое время применялись для обработки опытных
данных. Систематические вычисления термодинамических величин по кон-
стантам диссоциации, приведенным в таб
Харнедом и Оуэном [1] с помощью урав-
нения (67), а также Эвереттом и Вин-Джон-
сом [53] с помощью уравнения (22).
Последнее уравнение выражает экспери-
ментальные данные более точно, чем квад-
ратное уравнение, и поэтому оно пред-
почтительнее для вычисления термодина-
мических величин.
Харнед и Оуэн применили для этой
цели уравнение (23) в следующем виде
Рис. 154. Графическое изображе-
ние зависимости левой части урав-
нения (69) от температуры для уксус-
ной, пропионовой, муравьиной и
'хлоруксусной кислот.
[54] 1;
-4p--C*T + D*, (70)
откуда 1
&.Fi — A' — D'T-\-C'T2, (71)
kHot = A'-СТ2, (72)
&C°Pt= -2СТ, (73)
kS°i = D' -2СТ, (74)
^=/5*. (75)
lgAe = Z)*-2j/c*4^ (76)
В этих уравнениях А', С и D' в 2,3026 R
раза больше, чем, соответственно, А*, С*
и D*. Выбор этого метода вычисления основан на том факте, что все экспе-
.риментальные значения электродвижущих сил можно выразить с опреде-
ленной точностью во всем интервале температур с помощью квадратного
уравнения относительно Т. Возможно, что для получения более точных
результатов или при исследованиях в области температур, выходящих за
пределы 60°, квадратное уравнение для Е может оказаться непригодным,
однако до сих пор не встретилось необходимости в применении более слож-
ных уравнений. В качестве уравнения, типичного для тех уравнений, кото-
рые применяются для определения К с помощью экстраполяции, можно
рассмотреть уравнение (39), которое при р = 0 принимает вид
(EVH~Е°) F I , mClmHR
2,303RT ' mR
-(1g КА^=й.
(77
Так как E° и Еуц являются квадратичными функциями от Т, а член,
содержащий моляльности, не зависит от температуры, то отсюда получаются
уравнения (70) и (71) для IgA и ДА?. Легко получаются также уравне-
ния (72) — (76) для других термодинамических величин.
1 Большинство приведенных далее вычислений выполнено Р. А. Робинзоно .
476
ГЛАВА XV
Константы А*, С*, D*, А', С и D' уравнений (70) и (74) приведены
в табл. 167. В этой таблице содержатся, кроме того, значения темпера-
тур Т’д, соответствующих максимуму констант диссоциации, а также значе-
ния (—lgKs). В табл. 112 приведены значения ДЯ-, ДС£. и ДД”? при 25°,
вычисленные с помощью уравнений (70) —(74). Эти результаты хорошо
совпадают с данными, полученными Эвереттом и Вин-Джонсом, которые при-
меняли уравнения (20) — (22). Наиболее значительное расхождение с дан-
ными этих авторов имеет место в случае определения ДСр. для воды, когда
Эверетт и Вин-Джонс получили значение — 51 кал вместо приведенного
в таблице значения —46,5 кал.
Таблица 112
AF°, ДИ®, ДС° . п A.S? при 25°, кал
Электролиты ДЯ? 1 АС₽г AS?
Кислоты
Вода . . . ' 19089 13519 -46,5 -18,7
Муравьиная 5117 -23 -41,7 -17,6
Уксусная . 6486 -92 -36,5 -22,1
Пропионовая 6647 -163 -38,3 -22,8
Масляная 6574 -693 -36,4 -24,4
Хлоруксусная 3901 -1158 -40,0 -17,0
Молочная 5267 -99 -40,7 -18,0-
Гликолевая 5225 175 -38,8 -16,9
Серная (2) 2721 - 5200 -50,0 -26,5
Щавелевая (2) 5846 -1577 -57,8' -24,9
Малоновая (2) 7767 -1157 -60,0 -29,9
Фосфорная (1) 2896- -1773 ' -50,7' -15,7
» (2) 9829 822 -45,1 -30,2 ,
Борная 12596 3328 -45,0 -31,1
Аминокислоты
Глицин А 3205 1156 -32,2 -6,9
» В 5753 2713 -21,9 -10,2
fZZ-Аланин А 3191 3203 714 773 -34,2 -37,3 -8,3 -8,2
» В 5627 5631 2787 2539 -44,8 -29,9 -9,5 -10,4
dl-a-Амино-н-масляная А . . . 3123 298 -34,1 -9,5
» В . . . 5681 2824 -29,9 -9,6
cZZ-a-Амино-н-валерьяновая А . . 3161 487 -34,2 -9,0
» В . . 5714 2685 -38,7 -10,1
tZZ-Норлсйцин А 3183 528 -33,1 -8,9
» В 4314 2593 -32,3 -5,8
t/Z-a-Аминоиаомасляная А . . . 3215 . 492 -38,0 -9,1
» В . . . 5170 1988 -31,5 -10,7
Валин А 3118 59 -37,8 -10,3
» В 5835 2872 -32,7 -9,9
cZZ-Лсйцин А 3176 382 -36,8 -9,4
» В 5796 2705 -32,5 ' -10,4
cZZ-Изолейцин А 3163 264 -38,1 -9,7
» В 5786 2772 -28,6 -10,1
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
477
Продолжение табл. 112
Вода и кислоты в смесях диоксан—вода и метиловый спирт—вода
Состав смеси X ДН° 0 АСр.
Вода в .ЛГ-процентной смеси ди-
оксан—вода 20 19940 13515 -50,4 -21,5
То же 45 21470 13190 -54,0 -27,8
» » Муравьиная кислота в АГ-процент- 70 24353 12662 -49,4 -39,2
ной смеси диоксан—вода . . . 20 5701 359 -43,4 -17,92
То же 45 6944 1067 -48,1 -19,72
» » 70 9569 1474 -46,2 -27,16
» » Уксусная кислота в АГ-процентной 82 12002 2509 -109,1 -31,84
смеси диоксан—вода 20 7217 -52 —44,0 -24,4
То же 45 8602 -442 -51,1 -30,3
» » 70 11348 -610 -51,7 -40,1
» » Пропионовая кислота в АГ-процент- 82 13827 -1338 -124,5 -50,8
ной смеси диоксан—вода . . . 20 7455 -48 -42,0 -25,5
То же 45 8937 -206 -46,8 -30,7
» » 70 11746 -201 -47,6 -40,1
» » Уксусная кислота в АГ-процентной 82 14203 -1064 -120,1 -51,2
смеси метиловый спирт — вода 10 6690 30 -43,3 -22,3
То же Глицин КА в АГ-процентной смеси 20 6929 168 — 47,1 -22,7
диоксан—вода 0 3205 1156 -32,2 -6,9
То же 20 3?86 1183 -34,1 -8,1
» » 45 4233 993 -32,4 -10,9
» » - Глицин Кв в АГ-процентной смеси 70 5412 829 -30,9 -15,4
диоксан—вода 0 5753 2713 -21,9 -10,2
То же 20 6428 2869 -27,4 -11,9
» » . . .’ 45 7511 3342 — 19,5 -14,0
» 70 8968 3763 —- -17,5
Наиболее точные результаты были получены для алифатических кислот
и воды в водных растворах и в смесях диоксан —вода с более высокой
диэлектрической постоянной (7) > 30). В смесях с высоким содержанием
диоксана определение Е° (гл. XI, § 3) ненадежно, и ошибка при опреде-
лении константы диссоциации возрастает по мере уменьшения диэлектри-
ческой постоянной. Для смесей, содержащих 70% диоксана, результаты
еще вполне надежны, но в случае смесей с содержанием диоксана 82%
опытные данные являются уже приближенными. ,
Между значениями констант для (ZZ-аланина, полученными двумя неза-
висимыми способами, наблюдается расхождение, которое достигает 1 — 2%,
как видно из табл. 164 и 166Г Если требуется абсолютное значение вели-
чип АДили Кв, то заслуживают предпочтения данные, приведенные в табл. 164,
так как величины, имеющиеся в табл. 166, были получены на основании
весьма скудного количества экспериментальных данных и без полного уда-
ления воздуха из элемента. С другой стороны, термодинамические вели-
478
ГЛАВА XV
чины, полученные с помощью данных табл. 166, могут быть более точными,
так как в этом случае измерения производились в более широком темпе-
ратурном интервале.
Количество работ, посвященных определению констант диссоциации
с помощью элементов с жидкостным соединением или по кривым электро-
метрического титрования, весьма велико [55]. В табл. ИЗ приводятся
некоторые значения, полученные при использовании элементов с жидкост-
ным соединением и принадлежащие к числу наилучших результатов, полу-
ченных при помощи современной экспериментальной техники и методов:
экстраполяции, основанных на теории междуионного притяжения. Соответ-
ствующие величины, полученные при использовании элементов без жидкост-
ных соединений, были интерполированы до значений при 18°. Согласно данным
табл. ИЗ, результаты, полученные двумя методами, отличаются в среднем
на 1,5%, причем это совпадение следует считать исключительно хорошим,
так как очень часто встречаются расхождения в 10 раз, что является,
вероятно, скорее следствием неопределенности, связанной с внесением
поправок на диффузионный потенциал, чем результатом значительных
экспериментальных ошибок. Это обстоятельство подчеркивает преимущество
применения элементов без жидкостных соединений или использования таких
комбинаций элементов (гл. X, § 7), в которых исключается влияние диф-
фузионного потенциала.
Таблица ИЗ
Сравнение констант диссоциации, полученных по данным
об электродвижущих силах элементов с жидкостными и без жидкостных
соединений, а также по данным об электропроводности
Кислота \ . °C 18° 25°
ПО 0. Д. с. [1] элементов с жидкост- ными соедине- ниями ПО э. д. с. элементов без жидкост- ных соедине- ний ПО э. д. с. элементов без жидкост- ных соедине- ний по данным об электро- проводности
Муравьиная . . 1,79 • 10~4 1,76 • 10-4 1,772 • 10-‘ 1,830 • 10-«
Уксусная . . . . . . . 1,73 • Ю"5 1,75 • 10-6 1,754 • 10-6 1,758 • 10-»
Пропионовая . . • . . 1,32 • 10"5 1,34 • IO"5 1,336 • ю-6 1,347 • 10-6
и-Масляная . . . . . . 1,53 • 10-5 1,56 • IO'5 1,515 • IO-5 1,512 • 10-6
Хлоруксусная 1,49 • 10~3 1,44 • 10-3 1,379 IO'3 1,400 • 10-6
а-Молочная . . . . . . 1,38 • 10-'1 1,37 • IO-4 1,374 • 10-4 1,391 • 10-4
Гликолевая . . .... 1,46 • 10-4 1,455 • IO-5 1,475 • IO-*
В табл. ИЗ в двух последних столбцах сравниваются константы дис-
социации при 25°, определенные путем измерения электродвижущих сил
элементов без жидкостных соединений и константы диссоциации, излу-
ченные из данных по электропроводности (табл. 40). Для уксусной и мас-
ляной кислот наблюдается хорошее согласие, тогда как для муравьиной
кислоты1 имеется сильное расхождение между результатами.
Точность данных по тепловым эффектам оценить очень трудно. Все те-
трудности, которые встречаются при определении таких величин, как Ь2
и J2 для сильных электролитов (гл. XI —XIII) путем дифференцирования,
1 Это расхождение между результатами в случае муравьиной кислоты вызывает
недоумение, так как при обоих определениях применялась одна и та же проба кислоты.
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
479
имеют место также при определении ДЯ? и ДСр. реакций диссоциации.
В наиболее благоприятных' случаях (вода и растворы алифатических кислот
в воде) ДЯ? при 25° определяется с точностью до 50 кал. В случае наименее
надежных результатов (кислотные и основные константы аминокислот)
ошибки могут достигать 250 кал. Так, для кислотной диссоциации гли-
цина Стуртевант [56] получил с помощью калориметрических измерений
значение 906 кал, что на 250 кал меньше величины, приведенной в табл. 111.
Значения ДСр. для интервала температур 15 — 35° могут быть, в лучшем
случае, определены с точностью в ± 1 кал. В наиболее неблагоприятных
случаях имеют место ошибки порядка ±7 кал и выше. Более подробный
анализ этих ошибок можно найти в работах Вальде [57], Питцера [58],
Эверетта и Вин-Джонса [59], а также Харнеда и Оуэна [1].
Общий обзор термодинамики равновесных процессов диссоциации и раз-
личных теорий, предлагавшихся для объяснения зависимости констант дис
социации от температуры, будет дан в § 10 после теоретического анализа
результатов, полученных с помощью элементов с небуферными растворами
и с жидкостными соединениями, и после рассмотрения влияния среды
и солевых эффектов (§ 7 и 8).
' § 7. Влияние среды. Элемент с небуферным раствором
без жидкостного соединения
В гл. XI, § 3, было рассмотрено влияние изменения состава смесей,,
состоящих из воды и органического растворителя, на стандартный потен-
циал элемента
Н21 НС1 (т), растворитель (Х),вода (У) | AgCl-Ag.
Там же было показано, что с помощью уравнения Борна (109) гл. III
можно правильно оценить порядок величины этого эффекта изменения
потенциала, однако оно непригодно для точных вычислений. Рассмотрим
вопрос о влиянии среды на свойства сильных и слабых электролитов в более
общем виде.
При рассмотрении влияния растворителя (среды) на величины коэф-
фициента активности и константы диссоциации ионизирующего растворен-
ного вещества следует различать первичный и вторичный эффекты среды.
В случае раствора электролита, в котором присутствуют нейтральные
(неводные) молекулы, суммарный эффект среды определяется как логарифм*
отношения коэффициента активности электролита в присутствии нейтраль-
ных молекул к коэффициенту активности в чистой воде при той же кон-
центрации электролита. Оба коэффициента активности в этом отношении
отнесены к единице, как к коэффициенту активности при бесконечном раз-
бавлении в чистой воде. Первичным эффектом среды обозначают тот предел,
к которому приближается суммарный эффект, когда концентрация электро-
лита стремится к нулю. Вторичный эффект среды всегда определяется как
разность между суммарным и первичным эффектами.
Для того чтобы при пользовании этими обозначениями не возникали
недоразумения, необходимо точно установить стандартные состояния, при.
которых коэффициенты активности принимаются равными единице. В данном
параграфе все коэффициенты активности в любых средах считаются рав-
ными единице при бесконечном разбавлении в чистой воде. В том случае,
если коэффициент активности записан с индексом в виде звездочки (у*), то
это значит, что он принят за единицу при бесконечном разбавлении в той
среде, в которой данное вещество растворено. Если этой средой является
чистая вода, применяется верхний индекс нуль (у0). Нижний индекс нуль
480
ГЛАВА XV
показывает, что концентрация электролита равна нулю. Таким образом,
для всех концентраций можно написать
lgY==lgYo + lgY*, (78)
или, прибавляя — 1g у0 к обеим сторонам уравнения (78), получаем
lgi = lgYo + lgJ • (79)
I I
Совокупность трех членов этого уравнения представляет собой зависимость,
связывающую суммарный первичный и вторичный эффекты среды [60].
Первичный эффект среды учитывается уравнениями (108) и (109) гл. III.
Значения —1g Ад для смесей вода — органический растворитель, при-
веденные в табл. 165, были получены при помощи элементов
H2|HR(m1), NaR (m2), NaCl (m3), растворитель (X), H2O (У) | AgCl-Ag
согласно уравнению (39). Существует два простых равноценных способа
выражения электродвижущей силы таких элементов, а именно:
F 2ЕЗдР~ ~ ]g - lg YnYci (80)
И
F(E — E0*) , i * * /on
- 2.3ДГ = - mHmci - YSYer (81)
В первом выражении —E° представляет собой стандартный потенциал элек-
трода серебро-хлористое серебро в чистой воде. Во втором выражении
— Е°* представляет собой стандартный потенциал в смешанном растворителе.
Путем сочетания этих двух уравнений и сравнения результата с уравне-
нием (78) можно показать, что первичный эффект растворителя по отно-
шению к ионам водорода и хлора выражается непосредственно, через зна-
чения двух стандартных потенциалов. Таким образом,
bY ^liglHlcL=F(E°-E0*)
ё ionol 2 ё 7*7*! 4,6 ЯГ
(82)
Первичный эффект органического растворителя по отношению к коэф-
фициенту активности и константе диссоциации уксусной кислоты можно
определить путем экстраполяции данных, полученных при работе с выше-
описанными элементами, согласно уравнению (39). Применяя уравнение (81),
мы получаем величину Ад, которая является функцией среды, так как у*
всегда равно единице при бесконечном разбавлении. Напишем выражения
Г2 =2Н1П
lA Thr
и к
mHR ’
(83)
термодинамическая константа диссоциации для чистой воды Ад связана
со свойствами кислоты в смешанных растворителях следукпцими уравне-
ниями:
К л = YAfeA = Yo2AYl2feA = YM- (84)
Следовательно,
<85>
В табл. 114 приводятся значения Е0*, полученные для смесей метило-
вый спирт —вода и диоксан — вода, и соответствующие величины IgYonci
и lgYoA. Следует отметить, что влияние уксусной кислоты как растворителя
на соляную кислоту больше, чем соответствующее влияние метилового
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
481
спирта и диоксана, и что влияние двух последних растворителей на уксус-
ную кислоту сильнее, чем на соляную кислоту. Влияние молекул уксусной
кислоты, как среды, на диссоциированную уксусную кислоту пока еще не
определено экспериментально.
Таблица 114
Первичные эффекты среды при 25° для смесей органический растворитель—вода
X—весовой % органического растворителя
Растворитель X 0 *1) Е , lg Y0HCl lgY0A 2)
муравьи- ная кислота уксусная кислота пропио- новая кислота
Вода . . . 0 0,22239 0,0 0,0 0,0 0,0
Метиловый спирт .... 10 0,21535 0,0595 — 0,0741 —
» » .... 20 0,20881 0,1148 —. 0,1614 —
Диоксан . 20 0,20303 0,1636 0,2143 0,2678 0,2961
ъ 45 0,16352 0,4976 0,6200 0,7755 .0,8392
» . . . 70 0,06395 1,3392 1,6318 1,7823 1,869
» 82 -0,0415 2,230 2,52 2,69 2,77
Уксусная кислота .... 10 0,21050 0,1005 — — .—
» .... 20 0,19682 0,2161 — — .—
» » .... 30 0,18091 0,3506 — — —
» .... 40 0,16211 0,5095 — — —
» .... 50 0,13945 0,7010 — — —
» » • . . . 60' 0,11150 0,9373 — — —
1) Табл. 70; уксусная кислота [1].
2) По данным табл. 165.
Если применить для экстраполяции уравнение (80) вместо уравне-
ния (81), то возникает такое же положение, как и в случае элементов,
содержащих небуферные растворы слабых кислот. Уравнение (80) можно
записать в следующем виде:
+ lg = - 1g КЛ + 2 1g 7д - 2 lg YlIcl. (86)
Экстраполяция членов левой части уравнения к нулевому значению ионной
силы дает величину (— 1g КА-\- 2 lg Y04— 21g ?0ПС1), так что, зная К а, можно
найти разность между первичными эффектами растворителя по отношению
к слабой кислоте и к соляной кислоте. Таким образом, 1g уод нельзя опре-
делить с помощью вышеописанного элемента, если неизвестны либо значе-
ние 1g у0НС1, либо Е°* [уравнение (82)].
Рассмотрим теперь элемент, содержащий цебуферный раствор, типа
Н2| HR (щх), NaCl (щ2)| AgCl-Ag,
впервые примененный для определения констант диссоциации [24, 25]
и коэффициентов активности [23] слабых электролитов в растворах солей.
В таких элементах концентрация слабой кислоты поддерживается постоян-
ной, равной некоторой заданной величине тих, в то время как концентрация
соли иг2 изменяется. Если преобразовать выражение для электродвижущей
31г. Харнед, Б. Оуан
482
ГЛАВА XV
силы этих элементов и прибавить к обеим сторонам уравнения величину
21gYHCi’ получаем соотношение
-^J^- + lg^3 + 21gr^CI= -lgmH-21g-^-. (87)
Правая часть этого уравнения учитывает суммарный эффект среды — слабой
кислоты по отношению к ионам хлора и водорода. Эта величина обычно
бывает неизвестна, и поэтому целесообразно ввести кажущуюся концентра-
цию водородных ионов Шн, определяемую выражением
lg ®ii = lg + 2 lg ,
TJhci
(88)
которая может быть вычислена непосредственно из значений членов левой
части уравнения (87). Соответствующий кажущийся коэффициент активности
слабой кислоты у' выражается через термодинамическую константу диссо-
циации с помощью уравнения
тг 7h7r тн
Аа =
(89)
----------
7Hr wi-wh
аналогичного зависимости
К 2h7r_ ^2Н =
7hr W1-WH а’
(90)
которая связывает истинный коэффициент активности с истинной концен-
трацией иона водорода. В обоих выражениях все коэффициенты активности
приняты равными единице при бесконечном разбавлении в чистой воде.
Из уравнений (88) — (90) получаем выражение
Ig.Yi = lg Ya - 2 lg + i lg , (91)
которое показывает, что lg у'А определяется через истинные коэффициенты
активности и действительно должен выражать свойства истинного коэффи-
циента активности в том случае, когда содержащий тА1 член мал. Если кон-
центрацию слабой кислоты поддерживать постоянной, меняя в то же время
ионную силу путем изменения концентрации соли т2, можно получить ряд
значений тй и к'А для (тх — тн)-моляльного раствора недиссоциирующего
слабого электролита в воде. Поскольку Иц гораздо меньше, чем т1; то
состав растворителя является почти совершенно постоянным и первичный
эффект среды, а также, в первом приближении, суммарный эффект среды
можно считать не зависящими от концентрации соли. Поэтому с помощью
экстраполяции целесообразно определять значение коА:
*0A = Lim[*A]. (92)
н-* о
Логарифм соответствующей величины
ТоА = ЫтпЮ (93)
связан с первичным эффектом среды, так как, согласно уравнению (91),
он должен быть равен
lg Yoa — 2 lg Yohci’
если зависящие от концентрации члены при экстраполяции исчезают.
Экстраполяция 1g к'А произведится с помощью уравнений
lg Ка. — 2 р' d0 = 1g к'0А ± 2Рр' (94)
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
483
или
1 + А' У у.' d0
(95)
в которых вместо р.'( = т2 + тн) применяется «•( = m2 + тн) как очень хоро-
шее приближение. При использовании численной величины &(/), относя-
щейся к чистой воде, не учитывается вторичной эффект среды — слабой кис-
лоты, но это допущение является неизбежным, так как влияние молекул HR
Рис. 155. Определение значения к0А для
уксусной кислоты при 25° при различных
значениях т^.
Значения mi, считая сверху вниз, равны: 10,2;
5,41; 1,0 0,52; 0,2 и 0,1. Пунктирная линия
отвечает гипотетической предельной кривой
при условии, что mi=0.
Р и с. 156. Определение для ук-
сусной кислоты при 25°.
значениях о' необходимо применять уравнение (95).
Характер величины у'А таков, что она должна обращаться в единицу
при т1 = 0, поскольку в этих условиях растворителем является чистая вода-
Согласно уравнению (89), экстраполяция к'йА до = 0 должна давать зна-
чение К\. Эта экстраполяция показана на рис. 156. Тот факт, что эта;
экстраполяция в пределах экспериментальных ошибок оказывается линей-
ной, не имеет теоретического значения, так как в настоящее время невозь
можно оценить влияние некоторых приближенных допущений, сделанных;
при вычислениях. Известно, что аналогичная зависимость 1g у0НС1 от кон-
центрации не является линейной [60], а характер поведения lgy0A не-
известен Т
1 В статье Харнеда и Оуэна [24] третье равенство в уравнении (17) неправильно^
так как в нем не учитывается влияние среды—уксусной кислоты—на истинное значе-
ние Третья фраза на стр. 5081 указанной статьи неправильна, поскольку в ней.
величина относится к «данному растворителю», а не к чистой воде. Эти ошибки были
исправлены в другой статье Харнеда и Оуэна [1], которой мы и придерживаемся
в данном изложении.
31*
484
ГЛАВА XV
§ 8. Коэффициенты активности слабых кислот
в растворах солей
Как было показано в § 2, функция 1g (ун уон / аН2о) для воды в рас-
творах солей может быть получена из данных по электродвижущим силам
соответствующих элементов без жидкостных соединений. Метод электродви-
жущих сил применим также для определения аналогичных функций для
других слабых электролитов. Подобные результаты могут быть получены
также с помощью оптического метода, описанного в § 4. В настоящем пара-
графе будет проведено вычисление функции lg ун yR / yHR для уксусной
кислоты из данных по электродвижущим силам и для а-динитрофенола
по результатам оптических измерений. Сначала будет рассмотрен метод
электродвижущих сил, так как он основан на результатах, описанных
только что в § 7.
На основании почти параллельного расположения кривых, изображен-
ных на рис. '155, для низких концентраций можно сделать вывод о том,
что пунктирная линия, проведенная через точку пересечения кривой с осью
ординат (1g Кд) будет отражать свойства бесконечно разбавленной уксусной
кислоты в растворах хлористого натрия. В таких растворах эффект среды
(слабой кислоты) равен нулю. Это обстоятельство свидетельствует о наличии
очень важного ограничения по отношению к тем результатам, которые
могут быть получены при работе с элементами без жидкостных соединений,
содержащими небуферные растворы. Если не учитывать специальных термо-
динамических данных, эти элементы дают значение ' величины кА или
/ (mi — тн) в растворах солей при нулевой концентрации слабой кис-
лоты (tw1 = O). В растворе, содержащем конечное количество слабого элек-
тролита, с помощью таких элементов нельзя определить величину кА.
С другой стороны, этот метод показывает путь решения данной задачи,
так как он указывает, что для этого необходимо знать влияние среды —
слабой кислоты —или зависимость кА от тх. Когда будут выполнены даль-
нейшие исследования в этой области и установлены общие законы влияния
изменения состава растворителя, тогда, можно будет определить значение
ти в кислотно-солевых растворах различного состава, а также важную
величину лгн в буферных растворах.
Если эффект среды (слабой кислоты) равен нулю (тг — 0), то можно
отбросить индексы-штрихи в уравнениях (92) и (93). Если учесть уравне-
ние (88), то видно, что наклон пунктирной линии можно считать равным 23
в уравнении типа
2 lg Ya = lg КА - 1g кА = ~ + 2{3р. (96)
1 + А' у
Это уравнение можно использовать для вычисления константы диссоциации
и коэффициента активности слабой кислоты в растворах солей.
В табл. 115 приведены значения кА при 25° для растворов некоторых
солей, полученные Харнедом и Хиккеем [61] \ Эти значения представлены
графически на рис. 157, из которого видно, что полученные кривые анало-
гичны кривым, изображающим зависимость mHw0H от р.1/2 для растворов
галоидных солей (рис. 143). В случае концентрированных растворов вели-
чина солевого эффекта имеет тот же порядок, как и в случае воды, и согла-
суется с первыми, менее точными результатами Харнеда и Робинзона [23],
которые получили тот же порядок величины этого эффекта и для слабой
1 Диссоциация молочной кислоты в растворах хлористого натрия в интервале
0—37,5° была определена Хиккеем [62]. •
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
485
Таблица 115
Константа диссоциации уксусной кислоты при бесконечном
разбавлении в растворах солей при 25°
кА 105
И ЫС1 NaCl KOI Back
0 1,754 1,754 1,754 1,754
0,02 2,290 2,292 2,302 2,292
0,03 2,401 2,401 2,415 2,404
0,06 2,630 2,622 2,650 ' 2,635
0,11 2,874 2,850 2,891 2,885
0,21 3,167 3,101 3,151 3,190
0,51 3,546 3,315 3,340 3,609
1,01 3,670 3,158 3,071 3,799
2,01 3,432 2,475 2,182 3,680
3,01 — 1,824 — —
гидроокиси —монометиламина. В случае растворов галогенидов щелочных
металлов наибольшая диссоциация имеет место в растворах, содержащих
катионы с наименьшим радиусом, и поэтому замечания, сделанные в § 2
Рис. 157. Константа диссоциации уксусной ки-
слоты при бесконечном разбавлении в растворах
хлоридов при 25°.
относительно специфического влияния солей на диссоциацию воды, приме-
нимы также к слабым кислотам и гидроокисям. В общем случае диссоциа-
ция слабых электролитов в концентрированных растворах в значительной
степени зависит от силы электрического поля ионов.
Харнед и Хиккей определили этим методом кА для уксусной кислоты
в растворах хлористого натрия при температурах 0 — 40°. Они нашли, что
зависимость константы диссоциации от температуры можно выразить слег
дующим соотношением:
(97>
lg кА = 1g - 5 • 10-® (t - &)2,
486
ГЛАВА XV
похожим на уравнение (67). Это приводит к интересному выводу о том,
что 1g линейно зависит от температуры. Действительно, путем сочетания
уравнений (96) и (97) с уравнением (67) получается
1g Тд = (1/2), 1g (Л»/А:») + 2,5 • 10-®(&2-62)_5 . 10~6 (& - 6) t. (98)
Величины IgA:» и & приведены в табл. 116. Средние отклонения опытных
значений. 1g кА от величин, вычисленных по уравнению (98), обозначены
через Дсредн. •
Таблица 116
Параметры уравнения (97) для уксусной кислоты
при бесконечном разбавлении в растворах
хлористого натрия
И -lg fc» лсредн.
0,00 4,7544 22,6 0,0009
0,02 4,6388 25,1 0,0007
.0,03 4,6189 25,6 0,0007
0,06 4,5798 26,4 0,0007
0,11 4,5432 27,6 0,0009
0,21 4,5061 29,2 0,0011
0,51 4,4752 32,5 0,0009
1,01 4,4957 35,7 0,0012
2,01 4,5875 45,0 0,0029
3,01 4,7132 50,9 0,0038
На рис. 158 изображено поведение коэффициентов активности несколь-
ких слабых электролитов в растворах хлористого натрия. Для сравнения
на рисунке приводятся коэффициенты активности сильного электролита (соля-
ной кислоты) в растворах той же соли. Результаты для а-динитрофенола
приводятся по данным Гальбана и Кортюма [416]. Хотя соответствующие
этой кислоте точки в отличие от точек, отвечающих другим слабым электро-
литам, расположены выше кривой для соляной кислоты, это различие не-
значительно. Близкое сходство в поведении слабых кислот 1, 1-валентного
типа в растворах солей имеет весьма общий характер. Функции коэффи-
циента активности для 24 слабых кислот, изученных Ларсоном и Аделлом [63]
с помощью элементов с жидкостными соединениями, почти совершенно точно
располагаются между верхним и нижним пределами, соответствующими
точкам, полученным для а-динитрофенола и воды на рис. 158. Исходя из
этих данных, можно сказать, что коэффициенты активности нормальных жир-
ных кислот практически не отличаются друг от друга. Введение метильной,
фенильной или гидроксильной группы в анион кислоты не оказывает замет-
ного влияния, если только введенные радикалы не займут а-положения
по отношению к карбоксильной группе. Подробный анализ влияния строе-
ния кислот был сделан Ларсоном и Аделлом, а также Гюнтельбергом
и Шиодтом [64].
Наличие члена Yhr в Функйии коэффициента активности слабых
Кислот имеет существенное значение для определения последовательности
расположения различных солей по вызываемым ими солевым эффектам при
высокой ионной силе (рис. 103). Для а-динитрофенола в растворах хлоридов
натрия и калия кривые пересекаются при концентрации, равной примерно
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
487-
2 М, и таким образом при более высоких концентрациях указанная после-
довательность оказывается обратной по сравнению с той, которая наблюдается
при низких концентрациях. Это явление представляется весьма необычным,
трофенола).
так как в разбавленвых растворах последовательность расположения солей
по степени их влияния на растворы 7-динитрофенола является такой же, как
и в случае относительно мало деформируемых галоидоводородных кислот.
Q—NaCl; КС1; ф—ВаС12. Сплошные кривые отвечают коэф-
фициентам активности соляной кислоты в растворах тех же
солей.
Кроме того, Гальбан, Кортюм и Зейлер [42] показали путем определения
растворимости, что для этой кислоты yHR очень сильно меняется при пере-
ходе от одной соли к другой. Полученная ими зависимость величин lgyHR
от Г для нескольких растворов солей изображена графически на рис. 159.
Различие в величине солевых эффектов хлоридов натрия и калия по отно-
шению к yHR настолько велико, что если эти опытные значения исполь-
488
ГЛАВА XV
зовать для исключения yIIR из функции yHyR/ yHR, для которой наблю-
дается обращение последовательности расположения солей по величине их
солевых эффектов при концентрации, равной примерно 2 М, то оказывается,
что уже не будет иметь места обращение этой последовательности солевых
эффектов для функции уа yR ни при какой концентрации. На рис. 160
показана зависимость 1g уп yR от р. для а-динитрофенола в растворах хло-
ридов натрия, калия и бария, а для сравнения изображена эта зависимость
также для 1gун ус1 в тех же растворах. Как видно из рисунка, последова-
тельность расположения кривых одинакова для обеих кислот, причем кри-
вые для слабой кислоты расположены выше, чем соответствующие кривые
для соляной кислоты. Описанные любопытные аномалии еще не получили
объяснения.
Элементы без жидкостных соединений были использованы Батчелдером
и Шмидтом [65] для определения диссоциации аланина, аспарагиновой кис-
лоты, аргинина и орнитина в растворах хлоридов калия, натрия и бария.
§ 9. Гидролиз анионов органических кислот в растворах солей
С помощью функций коэффициентов активности YHYR/YnR и уыуОн/ ан»о
можно исследовать влияние солей на гидролиз аниона R" (при бесконечном
разбавлении HR). Для реакции гидролиза
R- + Н2О HR -И ОН"
имеет место следующая зависимость:
К — Kw — ^он WHR WOH , 7w !;w 2 »
>O1R WR ~ -tAkA*='lh h
(99)
Так как четыре величины yw, kw, ya и кА в отдельности известны для не-
которых растворов солей, то для этих растворов можно вычислить YZl и ки-
В табл. 117 приведены полученные Харнедом и Хиккеем значения kh для
гидролиза ацетатного иона в растворах хлорида натрия при температурах
0 — 40° и в растворах хлоридов калия, лития и бария при 25°. При данной
ионной силе и при 25° в случае хлорида бария наблюдается наибольший
Таблица 117
Функция гидролиза kh — kw/kA для иона ацетата при бесконечном разбавлении
в растворах хлоридов
kh • 1010
0 10 20 25 30 40 25 25 25
NaCl КС! L1C1 Ваш 2
0,0 0,684 0,688 3,88 5,74 8,40 17,11 5,74 5,74 5,74
0,02 0,697 1,706 3,92 5,79 8,45 17,29 5,77 5,81 5,83
0,03 0,704 1,701 3,92 5,79 8,45 17,29 5,74 5,83 5,85
0,06 0,706 1,714 3,93 5,79 8,44 17,31 5,69 5,93 5,96
0,11 0,708 1,720 3,95 5,82 8,50 17,39 5,65 6,08 6,05
0,21 0,719 1,741 3,98 5,84 8,54 17,36 5,61 6,27 6,29
0,51 0,738 1,770 4,03 5,91 8,57 17,44 5,65 6,53 6,81
1,01 0,761 1,820 4,09 5,96 8,65 17,68 5,57 6,87 7,43
2,01 0,801 1,864 4,11 5,93 8,54 17,48 5,85 7,13 8,28
3,01 0,818 1,858 4,02 5,74 8,27 17,05 — — 9,02
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
48».
Р и с. 161. Зависимость 1g 7л от цпри
гидролизе иона ацетата при бесконечном-
разбавлении в растворах хлористого нат-
рия при 25°.
солевой эффект. Для других солей значения солевого эффекта уменьшаются
при переходе от хлористого лития к хлористому натрию и затем к хлори-
стому калию.
Зависимость функции коэффициента активности lg от ]/ р для раствора
хлористого натрия при 25° представлена графически на рис. 161, причем
значения этой функции изображены в виде кружков, радиус которых соот-
ветствует точности порядка 0,1% от величины ул. Этот рисунок иллюстри-
рует два интересных обстоятельства. Общее изменение yh с концентрацией
соли очень мало и составляет менее
реакции гидролиза сохраняется и при
температурах 30 и 40°, но не наблю-
дается при более низких температу-
рах. Второе любопытное обстоятель-
ство касается той формы, которую
кривая приобретает в области раз-
бавленных растворов. Так как, со-
гласно уравнению (99), yft равно
Y пас Yoh /ан2о Где ’ то кРивая> изобра-
жающая зависимость lgyh от
должна приближаться к точке пере-
сечения с осью ординат при 1g у = 0
с наклоном,' равным нулю. Из
рисунка видно, что это условие не
соблюдается даже при самых низких
значениях концентраций, равных 0,02 71/. Чтобы показать, насколько боль-
шие разбавления могут потребоваться для того, чтобы обнаружить опреде-
ленные признаки перехода кривой в горизонтальное положение, на рисунке
проведена кривая, соответствующая отношению двух гипотетических одно-
валентных ионов, подчиняющихся уравнению Дебая и Гюккеля. Величины а
были приняты равными 4,0 и 4,5, а линейный член 0,005р был подобран
таким образом, чтобы обеспечивалось приближенное совпадение с данными
при высоких значениях ионной силы. Численные величины этих параметров
настолько произвольны, что им нельзя придавать серьезного значения, но
они показывают достаточное совпадение экспериментальных данных с теорией.
Если учесть разность значений а, равную 0,5 А, то обращает на себя вни-
мание малая величина эффекта, который был измерен экспериментально-
§ 10. Теоретические соображения
Данные о константах диссоциации слабых электролитов, приведенные
в предыдущих ыараграфах, были предметом многочисленных дискуссий
[51, 58, 59, 66, 1, 54]. Однако ни в одной из этих дискуссий не было полу-
чено точных и исчерпывающих ответов на многочисленные возникавшие
вопросы. При наличии столь сложного положения дальнейший анализ
экспериментальных данных, в их связи с важнейшими переменными вели-
чинами и с учетом имеющихся теоретических предположений, приведет
к интересным выводам и постановке новых задач.
На рис. 162 представлена зависимость — IgA, соответствующего кон-
центрации, выраженной в моляльности, от 1 / D при 25° для воды,
муравьиной, уксусной и пропионовой кислот в смесях диоксан — вода. Как
указал Харнед [67], эти графики не являются прямолинейными, и поэтому
уравнение Борна (109) гл. III неприменимо. Если изобразить такую же за-
висимость для логарифма константы диссоциации, соответствувлцего концент-
490
ГЛАВА XV
рации, выраженной в молярности или в молярных долях, то получаются
аналогичные результаты. Это отклонение от значений, соответствующих
уравнению Борна, не является неожиданным, поскольку подобный результат
наблюдался также для стандартных потенциалов элемента водород-хлори-
стое серебро (гл. XI, § 3). Такие же результаты получаются и при всех
остальных температурах в интервале 0 — 50°.
Гэрни [66а] выдвинул теорию для объяснения зависимости констант
диссоциации от температуры, причем Боген [666] придал этой теории форму,
более удобную для обработки экспериментальных результатов. Прежде всего
рассмотрим два типа реакций диссоциации:
RNHa+ + H2O rnh2 + h3o+, г
RGOOH + H2O RGOO' + H3O+. II'
При реакциях типа Г происходит переход протона, но не возникают новые
заряженные частицы.. Эти реакции являются изоэлектрическими. Реакции
типа II' сопровождаются как пе-
реходом протона, так и возник-
новением новых заряженных ча-
стиц. При реакциях третьего типа
Рис. 162. Зависимость констант диссо-
циации от диэлектрической постоянной для
смесей диоксан—вода.
HP—пропионовая кислота; НАс—уксусная кис-
лота; HF—муравьиная кислота.
(Ц=со)
и электрическую Д7*’°(эл.) составляющие,
ДF0 = ДF(°d=ж) + ДF° (эл.),
н2ро; + н2о нро;-+н3о+
пр
происходит образование новой за-
ряженной частицы и затрачивается
добавочная работа на отделение
положительно заряженного протона
от отрицательного иона.
Согласно уравнению Борна,
электрическая составляющая изме-
нения стандартной свободной энер-
гии процесса II' дается следующим
равенством:
(«о»
Если полную свободную энергию
AF0 разделить на химическую
тогда
(101)
(D.„,+<(± + ^) (102)
Д7?° = — RT\nK = —TJTlnA"
или
-lntf= - 1^=00)+^^ Гу-+-г) • (103)
После подстановки в уравнение (86) гл. I
51ПЛГ _ ДН° ^In^D-oo) А#(Р = о°) (104)
~дт ~~ R-Т- И дТ ~ RT2 К '
и дифференцирования получаем
+ С [ А (1 + Т **?) ] , (105)
Диссоциация слабых электролитов
491
где
с=^(±+±У
(106)
Рис. 163. Графическое изображение
. „о 1 (, , TdDX
зависимости от l + pgp J’
согласно уравнению (105), для воды
и уксусной кислоты.
Уравнение (105) применялось Богеном для реакций типа 1Г и ПГ.
Справедливость уравнения (105) основана на двух факторах. Во-пер-
вых, на предположении о том, что уравнение Борна является точным в
интервале температур 0 — 60°, причем в случае водных растворов диэлек-
трическая постоянная изменяется в этом интервале от 88 до 67. Второе
допущение касается зависимости ДЯ°О=^ от температуры. Из результатов
измерений Эверетта и Вин-Джонса [68], относящихся к диссоциации иона
аммония, и данных Педерсена [69], опре-
делявшего константы диссоциации анили-
на и иона о-хлоранилина, следует, что
для этих изоэлектрических реакций дис-
социации, соответствующих типу Г, 1g К
зависит от температуры, согласно урав-
нению
\%К = А-^, (107)
где А и В — постоянные. Если это спра-
ведливо, то Д77® является постоянным и
ACpt = 0. Однако при более поздних
исследованиях [536] было установлено,
что диссоциация ионов моно-, ди- и триме-
тиламмония сопровождается изменением
теплоемкости соответственно на 7, 20 и
41 кал. Вслед за Гэрни Боген предпо-
ложил, что для изоэлектрических реак-
ций ДЯ{°о=0о) не зависит от Т. Несмотря
на сомнительность этого предположения
и неточность уравнения Борна, Богену
удалось получить некоторые результаты,
которые приводят к правильному порядку
величины ионного радиуса. Как будет
видно из некоторых дальнейших вычислений, весь этот вопрос настолько
сложен, что он не может быть решен с помощью таких упрощенных допу-
щений.
Согласно уравнению (105), при графическом изображении зависимости
Д7/° от —у ^1 + Т должна получаться прямая линия с наклоном С
и значением ординаты в точке пересечения с осью ординат, равным Д7/(о=га).
Такая зависимость показана на рис. 163 для воды и уксусной кислоты
в водных растворах. Значения Д/Z0 были вычислены по уравнению (68)
с применением параметров, приведенных в табл. 167. Графики представ-
ляют собой почти прямые линии, но каждый из них обладает заметной
кривизной, причем эти отклонения от прямолинейности превышают ошибки
эксперимента. Аналогичные результаты были получены для воды, муравь-
иной, уксусной и пропионовой кислот в смесях диоксан—вода. С помощью
значений С, найденных приближенно по наклонам кривых, были получены
величины среднего радиуса г, который определяется уравнением
..............WA»
2 1,1
(107а)
492
ГЛАВА XV
Эти величины приводятся в табл. 118. Подобные же результаты были
получены для этих кислот, а также Для других кислот в водных раство-
рах Богеном; который нашел, что значения г лежат в пределах 0,65 — 0,85.
Хотя значения г имеют правильный порядок величины, однако на
основании этих значений нельзя сделать каких-либо достоверных выводов.
Таблица 118
Значения средних ионных радиусов [1], вычисленные
с помощью уравнения (106)
Растворитель—смеси воды с диоксаном; X—% диоксана;
О
значения г (Л) вычислены по уравнению (107)
X н2о НСООН СНзСООН С2Н5СООН
0 0,61 0,68 0,77 0,74
20 0,86 1,00 0,99 1,04
45 1,51 1,70 1,60 1,75
70 3,62 3,88 3,47 3,76
82 — 2,38 2,09 2,17
В Случае водной среды эти значения слишком малы, а в случае 70-про-
центных смесей диоксан —вода они слишком велики. Изменения этих
значений при переходе от одного растворителя к другому очень трудно
объяснить.
Следует отметить, что путем сочетания уравнений (103) и (105) можно
получить уравнение
lg# = , (108)
где А, В и С — постоянные. Это уравнение, которое отличается по форме от
эмпирических уравнений, рассмотренных в § 3 и 6, очень хорошо отра-
жает опытные данные1.
Неприменимость упрощенной электростатической теории в случае водных
растворов становится очевидной при рассмотрении теплоемкостей реакций.
На основании изучения значений ДС£„ собранных в табл. 112, можно про-
извести следующую приближенную классификацию соответственно различ-
ным типам реакций:
НА + Н3О А~4-Н3О+,
+nh3rco2h +.h2o^+nh3rco;+н3о+,
nh2rco; + н2о +nh3rco; + он-,
— (36 —>41),
^Ср. = —36 (среднее),
ДС°. —32 (среднее).
Теплоемкости процессов кислотной диссоциации амфолитов изменяются
от —32 до —38, в то время как теплоемкости процессов основной диссо-
циации этих же амфолитов изменяются от —29 до —39, если отбросить
аномальное значение —22 кал для основной диссоциации глицина. Необ-
1 На основе изложенной выше теории Ла-Мер и Бресчиа [70] получили выражение
для эмпирической константы р в уравнении (67) Харнеда и Эмбри. Маге, Ри и Эйринг
[52] дополнили представления Гэрни и Богена, более детально рассмотрев механизм
процесса диссоциации и влияния температуры и изменения растворителя на константы
диссоциации.
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
493
ходимо отметить, что хотя эти реакции принадлежат к совершенно различ-
ным типам, все же ДСр; приблизительно одинаково для всех этих процес-
сов. Эти результаты можно выразить в более точной форме, если сравнить
их с какой-либо обычной стандартной реакцией. В качестве такой реакции
можно выбрать следующую:
Н2О + Н2О^Н3О+ + ОН-, ДС°.^-47,
хотя и другие реакции, например диссоциация уксусной кислоты, также
могут служить в качестве стандартных. Вычитая по очереди каждую из
трех приведенных выше реакций из реакции диссоциации воды и рассматри-
вая полученные реакции вместе с первой из исходных, получаем:
1) HA + H2OZ±A- + H3O+, ДС°г^-(36-^41),
2) А- + Н2О^НА + ОН-,
3) +nh3rco;+h2o^+nh3rco2h+oh-,
4) +NH3RCO2 + H2O^NH2RCO2- + H3O+,
ДС°. (6-» И),
ДС°.^-11,
ДС°.^-16.
Изоэлектрическая реакция 2 сопровождается значительным изменением
ДСр., что противоречит предположению о независимости от темпе-
ратуры, а также результату, выраженному урав-
нением (107). На рис. 164 изображена графиче-
ски зависимость ДС'р. от числа углеродных ато-
мов первых четырех кислот алифатического ряда.
Как видно, при переходе к более сложным
кислотам происходит явное уменьшение —ДС^.
Это не имело бы места, если бы для изоэлек-
трических реакций ДСр. равнялось нулю. Вычи-
тая уравнение 3 из уравнения 4, получаем дру-
гую изоэлектрическую реакцию:
5) +NH3RCO2H + О1Г NH2RCO2 + Н3О+,
ДС°.^-5,
для которой ДС«г действительно мало. Принимая
во внимание возможные ошибки опыта, можно счи-
тать, что эта величина близка к нулевому значению.
Из приведенных данных вытекают два общих
вывода: 1) для изоэлектрических реакций вели-
чина — ДСр. значительно меньше, чем для других
реакций; это показывает, что данная величина в
Рис. 164. Зависимость
теплоемкостей реакций дис-
социации от числа атомов
углерода в алифатических
кислотах.
Пунктирные кружки—данные
Эверетта и Вин-Джопса, урав-
нение (22). Сплошные круж-
ки—табл. 164, уравнение (70);
диаметры кружков равны 1 кат.
значительной степени обусловлена кулоновскими силами; 2) из этих данных
ясно также, что существенное значение имеет не только электрический тип,
но и химический тип реакций. Этот фактор в значительной степени услож-
няет все явление.
Ввиду непригодности уравнения Борна для интерпретации равновесия
диссоциации и термодинамических свойств ионов были предприняты попытки
определить эти свойства, исходя из ориентации молекул воды вокруг ионов.
Основываясь на данных о структуре жидкой воды и ориентации ее молекул
вокруг ионов, Бернал и Фаулер [71] вычислили теплоты растворения ионов
в воде. Эли и Эванс [72] дополнили эту теорию вычислениями энтропии
ионов, сделанными на основании исследований Латимера [73]. Эверетт и
Коульсон [66в] сделали попытку применить этот способ для вычисления
494
ГЛАВА XV
ионных теплоемкостей и для оценки изменений теплоемкости при процессе
диссоциации слабой кислоты. После суммирования эффекта ориентации
молекул воды, находящихся в непосредственной близости от ионов, и
электростатического эффекта Борна, учитывающего влияние молекул, на-
ходящихся вне тетраэдрической сольватной оболочки молекул воды, они
получили такие значения — ДСр., которые были меньше опытных значений
даже в том случае, когда ионные радиусы были приняты равными 1 А.
Они предположили, что это несоответствие можно объяснить другими при-
чинами, главным образом наличием «свободного объема» молекул воды1.
§ 11. Дополнительные наблюдения
Более поздние исследования, посвященные изучению зависимости кон-
• стант диссоциации от среды и температуры, показали, что необходима боль-
шая осторожность при попытках создать теорию, устанавливающую зави-
симость констант диссоциации от строения слабых электролитов. Вин-Джонс
Рис. 165. Зависимость логарифмов от-
ношений констант диссоциации от диэлек-
трической постоянной для смесей диок-
сан— вода при 25°.
HF—муравьиная кислота; НАс—уксусная
кислота; HP—пропионовая кислота.
Рис. 166. Зависимость констант диссо-
циации уксусной и масляной кислот от
температуры.
[75] показал, что отношение констант диссоциации двух слабых электро-
литов представляет собой функцию диэлектрической постоянной среды.
Согласно его данным (полученным для сред с высокой диэлектрической
постоянной), логарифм этого отношения линейно зависит от 1/7). Получен-
ные за последнее время более полные экспериментальные данные не под-
тверждают наличия этой линейной зависимости. На рис. 165 изображена
графически зависимость логарифма отношения констант диссоциации муравь-
иной и уксусной кислот (верхняя кривая), а также уксусной и пропионо-
вой кислот (нижняя кривая) от 1/7). При уменьшении 1/7) кривизна этих
линий возрастает, и их экстраполяция до 0 (7) = оо) без использования
какой-либо точной теоретической функции не может быть осуществлена.
1 Обширные исследования относительной силы кислот, растворенных в бутиловом
спирте, были выполнены Уотеном и Хамметом [74а] с помощью электрометрического
метода, а также Мэзоном и Килпатриком [746], применившими фотоэлектрический коло-
риметр. Эти исследования были дополнены путем изучения других растворителей Мин-
ником и Килпатриком [74в], а также Эллиотом и Килпатриком [74г].
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
495
Из рис. 166, на котором изображена зависимость—1g АД от температуры
для уксусной и масляной кислот, видно, что отношение констант, диссоци-
ации двух электролитов сильно меняется с температурой. Действительно,
если продолжить кривые до температур ниже 0°, то они пересекаются при
— 15°. Выше этой температуры уксусная кислота диссоциирована сильнее,
чем масляная, в то время как ниже этой температуры масляная кислота
оказывается более сильной.
Величину константы диссоциации обычно рассматривают как меру силы
электролита, а отношение констант диссоциации двух электролитов — как
меру их относительной силы. Приведенные выше опытные данные показы-
вают, что относительная сила зависит от характера среды и от температуры
и вовсе не является постоянной величиной. Длн правильного понимания
кислотного и основного равновесий необходимо учитывать влияние этих и
других переменных, как, например, давления и концентрации соли.
ЛИТЕРАТУРА
Owen, Chem. Rev., 25, 31 (1939).
Heyd wei Iler, Z. physik. Chem.,
". К о h 1 r a u s c h, Z. physik.
1. H. S. Harned, В. B.
2. F. К о h 1 r a u s c h, A.
Ann. Physik., 53, 209 (1894); “F.
(1903); A. H eyd weiller, Ann. Physik. [4], 28, 503 (1905).
3. S. Arrheni us, Z. physik. Chem., 11, 808 (1893); б) H. T. Beans,
k e s, J. Am. Chem. Soc., 42, 2116 (1920).
G. N. Lewis, I "
G. N. Lewi s, T. B.
39, 2245 (1917).
H. S. Harned,
F. E. Swindells,
J. Phys. Chem., 30, 1060 (1926).
H. S. Harned, Trans. Faraday Soc., 23, 462 (1927).
E. J. Roberts, ’ ' ~ '
H. S. Harned.
H. S.' ”
H. S.
H. S.
H. S.
II. S.
H. S.
H. S.
H. S.
A. A.
R.
H. S.
H.
B. B.
H. S.
H. S.
H. S.
H. S.
L. F.
B. B.
W. J.
L. F.
H. S.
B. B.
burgh, там же, 62, 1817 (1940).
В. В. " ’ "
R. G.
H. S.
E. J.
B. B.
E.
H. H a i b a n, L. Ebert, Z. physik. Chem., 112, 359 (1924).
4.
5.
6.
7.
8.
9.
10.
И.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
В.
S.
14, 317 (1894)-,
Chem., 42, 193
Е. Т. О а-
L. W. Sargent, J. Am. Chem. Soc., 31, 363 (1909).
m ” Brighton, R. L. Sebastian, J. Am.
J. Am. Chem. Soc., 47, 930 (1925); H. S.
Chem. Soc.,
Harned,
там же, 48, 126 (1926); H. S. Harned, G. M. James,
33.
34.
35:
36.
37.
38.
39.
J. Am. Chem. Soc., 52, 3877 (.1930).
О. E. S c h u p p, J. Am. Chem. Soc., 52, 3892 (1930).
W. J. Hamer, J. Am. Chem. Soc., 55, 2194 (1933).
H. R. С о p s о n, J. Am. Chem. Soc., 55, 2206 (1933).
G. E. M a n n ' ’ ’
J.G. Donels
C. G. Geary,
H. R. С о p s о
M. А. С о о k,
R. A. Robinson, Trans. Faraday Soc., 36, 973 (1940).
H a r n e d,
H a r n e d,
H a r n e d,
a r n e d,
a r n e d,
a r n e d,
a r n e d,
a r n e d,
Noyes, Y. Kato, Publications Carnegie Institution, 63, 153 (1907).
S о s m a n, Publications Carnegie Institution, 63, 193 (1907).
Harned, W. J. Hamer, J. Am. Chem. Soc., 55, 4496 (1933).
Harned, J. Franklin Inst., 225, 623 (1938).
Owen, S. R. Brinkley, Chem. Rev., 29, 461 (1941).
Harned, " ‘ ” T ‘ " """
H a r n e d,
Harned,
H a r n e d,
N i m s, J.
Owen, J.
Hamer,.
N i m s, J. Am. Chem. Soc., 56, 1110 (1934). ’
Harned, R. W. Ehlers, J. Am. Chem. Soc., 55, 652 (1933).
Owen, J. Am. Chem. Soc., 57, 1526 (1935); R. K. Gould, W.C.Vos-
Н
н
н
н
н
weiler, J. Am. Chem. Soc., 57, 1873 (1935).
on, J. Am. Chem. Soc., 59, 1280 (1937).
J. Am. Chem. Soc., 59, 2032 (1937).
n, J. Am. Chem. Soc., 55, 2206 (1933).
J. Am. Chem. Soc.,.59, 2304 (1937).
R. A. Robinson, J. Am. Chem. Soc., 50, 3157 (1928).
В. B. Owen, J. Am. Chem. Soc., 52, 5079 (1930) .
G. M. Murphy,!. Am. Chem. Soc., 53, 8 (1931).
R. W. Ehlers, J. Am. Chem. Soc., 54, 1350 (1932).
Am. Chem. Soc., 55, 1946 (1933).
. Am. Chem. Soc., 56, 1695 (1934).
J. Am. Chem. Soc., 56, 860 (1934).
Owen, L. F о e r i n g, J. Am. Chem. Soc., 58, 1575 (1936).
Bates, W. C. Vosburgh, J. Am. Chem. Soc., 59, 1188 (1937).
Harned, H. B. Owen, J. Am. Chem. Soc., 52, 5091 (1930).
Roberts, J. Am. Chem. Soc., 56, 878 (1934).
Owen, J. Am. Chem. Soc., 56, 2785 (1934).
Meyer, H. Rosenberg, Vierteljahres Schr. Astron. Ges., 48, 3 (1913).
496
ГЛАВА XV
40. Н. Halban, К, Siedentopf, Z. physik. Chem., 100, 208 (1922); 103, 71
(1922).
41. a) H. Halban, L. Ebert, Z. physik. Chem., 112, 321 (1924); H. Halban,
J. E i s e n b r a n d, там же, 122, 337 (1926); 146, 294 (1930); H. Halban,
G. Kortum, там же, A170, 212 (1934); б) p. 351; в) H. Halban, М. Sei-
ler, там же, А181, 70 (1937); G. К о г t ii m, там же, ВЗЗ, 243 (1936).
42. Н. Halban, G. К о г t ii m, М. Seiler, Z. physik. Chem., A173, 449 (1935).
43. E. Q. Adams, J. Am. Chem. Soc., 38, 1503 (1916).
44. N. В j e r r u m, Z. physik. Chem., 104, 147 (1923).
45. J. T. E d s a 1 1, M. H. Blanchard, J. Am. Chem. Soc., 55, 2337 (1933).
46. J. W у rft a n, Jr., T. L. M с M e e к i n, J. Am. Chem. Soc., 55, 908, 915 (1933).
47. L. F. N i m s, P. K. Smith, J. Biol. Chem., 101, 401 (1933).
48. В. B. Owen, J. Am. Chem. Soc., 56, 24 (1934).
49. R. M. Roberts, J. G. Kirkwood, J.. Am. Chem. Soc., 63, 1373 (1941).
50. H.S. Harned, L. D. Fallon, J. Am. Chem. Soc., 61, 2377 (1939).
51. H.S. Harned, N. D. E m b r e e, J. Am. Chem. Soc., 56, 1050 (1934).
52. J. L. Magee, T. R i, H. E у r i n g, J. Chem. Phys., 9, 419 (1941).
53. a) D. H. Everett, W. F. K. Wynne-Jones, Trans. Faraday Soc., 35, 1380
(1939); 6) 37, 373 (1941); в) J. F. J. D i p p у, H. O. Jenkins, там же, 37, 366
(1941).
54. H.S. Harned, R. A. Robinson, Trans. Faraday Soc., 36, 973 (1940).
55. The International Critical Tables, Vol. VI, McGraw-Hill Book Co., N. Y., 1929,
p. 259—304.
56. J. M. S t-u r t e v a n t, J. Am. Chem. Soc., 62, 1879 (1940).
57. A. W. W a 1 d e, J. Phys. Chem., 39, 477 (1935).
58. K. S. Pitzer, J. Am. Chem. Soc., 59, 2365 (1937).
59. D. H. Everett, W. F. K. W у n n e - J о n e s, Trans. Faraday Soc., 35, 1380
(1939).
60. В. B. Owen, J. Am. Chem. Soc., 54, 1758 (1932).
61. H. S. Harned, F. C. Hickey, J. Am. Chem. Soc., 59, 1284 (1937).
62. F. C. Hickey, J. Am. Chem. Soc., 62, 2916 (1940).
63. E. Larsson, B. Adell, Z. physik. Chem., A157, 342 (1931); A156, 381 (1931).
134. E. Guntelberg, E. SchiOdt, Z. physik. Chem., 135, 393 (1928).
65. A. C. Batchelder, C. L. A. Schmidt, J. Phys. Chem., 44, 880 (1940):
44, 893 (1940).
66. a) R. W. Gurney, J. Chem. Phys. , 6, 499 (1938); б) E. С. В a u g h a n, там же
7, 951 (1939); в) D. H. Everett, C. A. Coulson, Trans. Faraday Soc.,
36, 633 (1940); r) J. F. J. Dipp y, Chem. Rev., 25, 131 (.1940).
67. H.S. H a r n e d, J. Phys. Chem., 43, 275 (1939).
68. D. H. Everett, W. F. K. Wynne-Jones, Proc. Roy. Soc., A169, 190 (1938).
f69. K. Pedersen, K. Danske Vidensk. Selsk. Skr., 14, 9 (1937); 15, 3 (1937).
70. V. K. L a M e r, F. Brescia, J. Am. Chem. Soc., 62, 617 (1940).
71. J. D. Bernal, R. H. Fowler, J. Chem. Phys., 1, 515 (1933).
72. D. D. E 1 e у, M. G. Evan s, Trans. Faraday Soc., 34, 1093 (1938).
73. W. M. Latimer, K. S. Pitzer, С. M. S h a n s к y, J. Chem. Phys., 7, 108
(1939).
74. a) L. A. Wooten, L. P., H a m m e t t, J. Am. Chem. Soc., 57, 2289 (1935);
6) R. B. Mason, M. Kilpatrick, там же, 59, 572 (1937); в) L. J. M i n-
n i с к, M. Kilpatrick, J. Phys. Chem., 43, 259 (1939); r) J. H. Elliott,
M. Kilpatrick, там же, 45, 454 (1941); 45, 466 (1941); 45, 472 (1941); 45, 485
(1941).
75. W. F. К. W у n n e-J ones, Proc. Roy. Soc., 140, 440 (1933).
ЛИТЕРАТУРА К ТАБЛИЦАМ
К табл. 107
1. Н. S. Harned, W. J. Hamer, J. Am. Chem. Soc., 55, 2194 (1933).
2. H. S. Harned, G. E. M a n n w e i 1 e r, J. Am. Chem. Soc., 57, 1875 (1935).
3. H.S. Harned, C. G. Geary, J. Am. Chem. Soc., 59, 2032 (1937).
4. H. S. Harned, J. G. D о n e 1 s о n, J. Am. Chem. Soc., 59, 1280 (1937).
К табл. 110
1. В. В. Owen, S. R. Brinkley, Jr., Chem. Rev., 29, 461 (1941).
ДИССОЦИАЦИЯ СЛАБЫХ ЭЛЕКТРОЛИТОВ
497
К табл. 111
1. Н. S. Harned, R. A. Robinson, Trans. Faraday Soc., 36, 973 (1940).
К табл. 112
1. Е. Larsson, В. Adell, Z. physik. Chem., A156, 352, 381 (1931); A157, 342
К табл. 114
1. В. В. О we n, J. Am. Chem. Soc., 54, 1758 (1932).
К табл. 118
1. H.S. Й arned, Т. R. D е d е 11, J. Am. Chem. Soc., 63, 3308 (1941).
32 г, Харнед, Б. Оуэн
ПРИЛОЖЕНИЕ А
Таблица 119
Эквивалентная электропроводность водных растворов электролитов при 25°
с, Жв.]л Элек- тролиты 0 ft,0005 0,001 0,005 0,01 0,02 0,05 0,1 Ссылки на лите- ратуру
НС1 426,16 422,74 421,36 415,80 412,00 407,24 399,09 391,32 1 3)
LiCl 115,03 113,15 112,40 109,40 107,32 104,65 100,11 95,86 1,2,3
NaCl 126,45 124,50 123,74 120,65 118,51 115,76 111,06 106,74 , 4‘)
КС1 149,86 147,81 146,95 143,55 141,27 138,34 133,37 128,96 V)
NH4C1 149,7 — .— —• 141,28 138,33 133,29 128,75 5
КВг 151,9 — 146,09 . 143,43 140,48 135,68 131,39 61)
Na J 126,94 125,36 124;25 121,25 119,24 116,70 112,79 108,78 8
KJ 150,38 «— — 144,37 142,18 139,45 134,97 131,11 Ч
KNO, 144,96 142,77 141,84 138,48 132,82 132,41 126,31 120,40 1
KHCO3 118,00 116,10 115,34 112,24 110,08 107,22 — — 9
NaO2CCH3 91,0 89,2 88,5 85,72 83,76 81,24 76,92 72,80 10
NaO2C(CH2)2CH3 82,70 81,04 80,31 77,58 75,76 73,39 69,32 65,27 И
NaOH 247,8 245,6 244,7 240,8 238,0 — — — 18
AgNO3 133,36 131,36 130,51 127,20 124,76 121,41 115,24 109,14 1
MgCl2 129,40 125,61 124,11 118,31 114,55 110,04 103,08 97,10 12
CaCl2 135,84 131,93 130,36 124,25 120,36 115,65 108,47 102,46 12
SrCl2 135,80 131,90 130,33 124,24 120,29 115,54 108,25 102,19 12
BaCl2 139,98 135,96 134,34 128,02 123,94 119,09 111,48 105,19 12
Na2SO4 129,9 125,74 124,15 117,15 112,44 106,78 97,75 7 89,98 Ч
CuSO4 133,6 121,6 115,26 94,07 83,12 72,20 59,05 50,58 132)
ZnSO4 132,8 121,4 115,53 95,49 84,91 74,24 61,20 52,64 13 2)
LaCl3 145,8 139,6 137,0 127,5 121,8 115,3 106,2 99,1 6, 14
K3Fe(CN)6 174,5 166,4 163,1 150,7 — — — — 15 “
K4Fe(CN)6 184,5 — 167,24 146,09 134,83 122,82 107,70 97,87 16, 17
1) Неопубликованные данные Шидловокого и Лонгсворта, приведенные в книге Мак-
«Оз [7].
2) В эти значения были внесены поправки, учитывающие присутствие ионов М(ОН)+ и HSO 4.
8) Значения А вплоть до с=12 для каждых 10° в интервале 5—65° получены Оуэном и Сью-
м Г191 [
4) Данные для интервала температур 15—45° приводятся в работе Ганнинга и Гордона [20].
Таблица 120
Числа переноса катионов в водных растворах при 25°, определенные методом движущейся границы
Электролиты
(Т+)
Концентрация, экв/л
Ссылки
на
литературу
НС1 426,17 +0,04507 0,8209 0,8251 0,8266 0,8292 0,8314 0,8337 1
NaC2H3O2 90,99 +0,03336 0,5507 0,5537 0,5550 0,5573 0,5594 0,5610 2
КС2Н3О2 114,40 +0,07467 0,6427 0,6498 0,6523 0,6569 0,6609 — 6
KNO3 144,96 +0,00297 0,5072 0,5084 0,5087 0,5093 0,5103 0,5120 2
NH4C1 149,94 -0,00363 0,4909 0,4907 0,4906 0,4905 0,4907 0,4911 2
КС1 149,86 -0,00376 0,4906 0,4902 0,4901 0,4899 0,4898 0,4894 1
KJ 150,29 -0,00430 0,4892 0,4884 0,4883 0,4882 0,4883 0,4887 2
КВг 151,63 -0,00596 0,4849 0,4833 0,4832 0,4831 0,4833 0,4841 2
AgNO3 133,36 -0,01604 0,4643 0,4648 0,4652 0,4664. 0,4682 — 2)
NaCl 126,43 -0,04910 0,3963 0,3918 0,3902 0,3876 0,3854 0,3821 1
LiCl 115,03 -0,08514 0,3364 0,3289 0,3261 0,3211 0,3168 0,3112 1
CaCl2 135,84 -0,26174 0,4380 0,4264 0,4220 0,4140 0,4060 0,3953 2
Na2SO4 129,9 +0,0632 0,386 0,3848 0,3836 0,3829 0,3828 0,3828 2
K2SO4 153,3 +0,1482 0,479 0,4829 0,4848 0,4870 0,4890 0,4910 4
LaCl3 145,9 -0,5491 0,477 0,4625 0,4576 0,4482 0,4375 0,4233 5
VI необходимо
Если полученные данные
выражать
концентрацию в молях
на литр.
1) При вычислении величины по уравнению (37а) гл.
изобразить графически как функцию от квадратного корня из эквивалентной концентрации, то предельные коэффициенты наклона для последних
четырех солей, приведенных в таблице, будут равны —0,18508; +0,0447; +0,1048 и—0,3170.
2) Мак-Иннес и Кауперсвейт, приведено в статье Мак-Иннеса и Лонгсворта [3].
32*
500
ПРИЛОЖЕНИЕ А
V.
Таблица 121
<pL, кажущееся относительное молярное теплосодержание
Часть I
Разбавленные водные растворы при 25°
У m Элек- \ Тролиты 0,01 0 ,02 0,04 0,06 0,08 0,10 0,15 0,20 0,25 0 ,30 Д 1) ССЫЛКИ на лите- ратуру
НС1 4,8 9,6 19,2 29 38 48 71 93 114 134 0 l8)
LiCl 4,3 8,4 16,6 25 33 40 60 78 96 114 81 2
LiBr 4,2 8,2 16,2 24 32 39 59 76 93 110 7 3,16
NaCl 4,5 8,5 17,0 25 33 40 55 67 77 83 2 4
NaBr 4,2 8,4 16,4 24 31 38 52 62 69 73 2 5,9
NaNO3 4,3 8,5 16,5 23 29 34 43 44 40 31 2) 4,5 7
NaC103 4,2 8,4 17,0 24 30 35 43 49 48 43 2) 1 7
NaBrOg 4,3 8,5 16,7 24 30 34 39 38 34 26 2) 1 7
NaJO3 4,0 7,5 14,0 19 21 21 16 0 -24 -57 2) 2 7
NaOH 4,8 9,5 19,0 28 36 44 62 77 90 99 0 8
KF 4,7 9,2 18,3 27 36 45 66 83 99 113 6 6
KCI 4,5 8,5 16,0 24 31 38 • 54 65 72 77 2 9,10,12
KBr 4,2 8,2 15,6 23 29 36 47 55 61 64 2 5, 9
KNO3 4,1 8,0 15,1 20 23 24 22 12 -6 — 29 3) 4 12
KClOg 4,2 8,2 15,5 21 26 29 28 19 4 -192) 3 13 ’
KC1O4 ' 4,3 8,0 13,0 16 16 14 -3 -28 -59 -109 2) 3 13
RbF 4 8 14 20 26 31 44 57 69 80 4) 4 12
CsCl 4 8 15 21 27 32 43 49 51 51 5) 2,5 16
NH4C1 4,4 8,6 17 25 33 39 53 66 78 88 е) 7 И9)
Li2SO4 24 47 91 135 177 218 307 377 438 488 3 15
Na2SO4 23 44 84 119 150 175 216 237 243 237 2 15
K2SO4 22 42 81 116 146 171 214 238 249 250 2 15
Rb2SO4 21 41 79 114 143 166 200 215 226 219 2 15
Cs2SO4 20 39 71 99 121 139 160 161 152 137 3 15
MgCl2 24 47 89 129 167 202 280 350 415 471 0 14
MgBra 23 45 86 124 161 196 271 334 389 437 0 . 14
Mg(NO3)2 23 44 81 119 155 187 254 304 344 376 5 15
CaCl2 24 47 88 127 165 199 275 340 398 445 1 14
CaBr2 23 44 83 120 154 186 252 308 355 394 1 14
Ca(NO3)2 22 43 79 111 136 158 200 222 230 224 5 15
SrCl2 23 46 86 124 161 195 270 332 381 420 1 14
SrBr2 . 23 44 182 120 152 182 244 293 333 366 1 14
Sr(NOg)2 21 40 172 99 122 140 166 169 159 135 5 15
ПРИЛОЖЕНИЕ А
501
Продолжение табл. 121
лек- тролиты 0,01 0,02 0,04 0,06 0,08 0,10 0,15 0,20 0,25 0,30 Д1) Ссылки НН лите- ратуру
ВаС12 23 46 86 124 160 194 268 329 375 412 2 14
ВаВг2 22 43 81 117 150 180 240 287 325 356 1 14
Ba(NO3)2 19 36 59 70 72 66 20 -46 -128 -223 5 15
MgSO4 44 118 154 369 463 542 685 784 842 -25 15
CaSO4 47 125 283 415 523 611 700’) -25 12
ZnSO4 51 127 273 396 501 581 731 826 896 953 -25 17
CdSO4 70 180 386 554 693 807 1018 1150 1246 1324 -40 17
CuSO4 62 159 345 501 618 713 885 1012 1109 1183 -35 17
1) Приближенное значение прироста при новой экстраполяции.
2) При Ут = 0,3162.
3) При Ут^= 0,35, ?/ = — 55.
4) При Ут — 0,35; 0,40; 0,45 и 0,50; <= 91, 101, 111 и 120, соответственно.
5) При Ут = 0,35 и 0,40; = 49 и 44, соответственно.
в) При Ут = 0,4; 0,5; 0,6; 0,72; 0,88 и 1; fl = 101, 111, 127, 136, 142 и 143, соответственно.
7) При Ут = 0,125.
8) Стуртевант [1а]. Исправленные значения см. [16].
0) Приводятся также данные для хлорида метиламмония.
Часть II
Водные растворы при 25°
Электролиты m HCl LiCl LiBr Nad NaBr NaOH KF KCI KBr
0,1 140 119 117 83 73 102 117 78 64
. 0,2 191 162 156 90 72 117 143 81 70
0,3 229 194 185 84 63 121 155 76 46
0,4 261 220 210 72 53 119 163 63 29
0,5 290 242 232 58 41 114 169 48 9
0,6 317 263 252 42 27 108 174 33 -13
0,7 342 283 271 26 13 101 179 18 -35
0,8 368 300 289 9 - 3 94 183 3 -58
0,9 391 318 306 -7 -20 86 186 -12 -81
1,0 414 334 322 -23 -37 78 190 -26 -104
1,2 460 365 352 -57 -72 61 198 -55 -148
1,5 526 410 394 -105 -124 38 209 -99 -212
1,7 569 438 421 -136 -160 25 215 -128 -251
2,0 633 479 460 -177 -212 8 223 -169 -306
2,5 739 547 524 -224 -294 -10 234 -236 -394
3,0 847 615 588 -304 -364 -17 246 -300 -480
502
ПРИЛОЖЕНИЕ А
Продолжение табл. 121
Электролиты
НС1 LiCl LiBr NaCl NaBr NaOH KF KCI KBr
т
/
3,5 954 2) 683 652 -355 -424 -12 260 -355 -559
4,0 — 758 716 -395 -482 3 279 -405 -631
' 4,5 — 838 780 -427 -533 36 2) 305 -448 -697
5,0 — 921 849 -453 -578 — 334 - 472 4) -760
5,5 — 1007 920 -470 -617 — 368 — -819
6,0 — 1098 994 -4833) -648 — 404 — -839*)
1) Ссылки на литературу указаны в части I этой таблицы.
а) Экстраполировано.
а) Для насыщенного раствора, т=6,12.
4) Для насыщенного раствора, т=4,82.
а) Для насыщенного раствора, т=5,68.
Таблица 122 х)
L2, относительное парциальное молярное теплосодержание
Часть!
Разбавленные водные растворы при 25°
У~ m Электролиты 0,01 0,02 0,04 0,06 0,08 0,10 0,15 0,20 0,25 0,30
HCI 7,2 14,3 28,6 43 57 71 105 136 165 193
LiCl 6,4 12,5 24,7 37 49 60 89 115 141 165
LiBr 6,3 12,2 24,0 35 47 59 86 111 136 159
NaCl 6,5 12,5 24,0 35 46 57 77 92 100 104
NaBr 6,3 12,2 24,2 35 45 54 70 79 83 82
NaNO3 6,0 11,5 22,5 31 40 46 51 40 27 13 2)
NaClO3 . 6,6 12,9 24,4 34 41 47 55 55 45 27 2)
NaBrOg 6,5 12,7 24,2 33 39 43 42 33 22 8 2)
NaJOg 5,8 11,0 19,8 24 24 20 0 -41 -88 —1432)
NaOH 7,1 14,0 28,0 42 54 65 89 104 117 125
KP 7,0 13,8 27,4 41 54 66 94 117 136 151
KCI 6,5 12,5 24,0 36 46 55 71 82 88 91
KBr 6,1 12,1 23,0 33 41 48 62 69 71 68
KNO3 5,7 11,0 20,3 26 29 28 12 -17 -57 -103 3)
КС 10 з 6,7 12,3 21,7 28 32 32 20 -6 -37 -77 2)
KC1O4 6,2 11,3 16,6 17 13 4 -34 -86 -148 -228 2)
NH4C1 6,7 13 25 37 47 56 75 91 105 ' 121 2)
Li2SO4 35 69 135 200 260 317 424 508 578 620
Na2SO4 33 64 122 170 206 229 261 264 241 287
KaSO4 32 62 119 164 200 226 262 272 266 233
ПРИЛОЖЕНИЕ А
503
Продолжение табл. 122х)
У" т Электролиты^. 0,01 0,02 0,04 0,06 0,08 0,10 0,15 0 ,20 0,25 0 ,30
RbaSO4 31 61 115 162 195 216 233 233 225 201
CsaSO4 29 57 102 136 161 176 172 152 124 87
MgCl2 36 70 130 187 240 287 388 484 567 626
MgBra 34 67 -125 181 233 278 372 453 519 564
Mg(NO3)a 32 62 121 175 224 264 339 393 437 465
CaCla 35 68 127 185 237 282 378 463 530 576
CaBr2 34 65 120 172 219 260 340 410 462 498
Ca(NO3)a 31 62 114 152 183 212 245 250 234 191
SrCla 34 66 125 180 232 277 372 443 492 528
SrBr2 33 64 119 170 216 254 324 383 423 452
Sr(NO3)2 30 58 102 137 159 180 185 161 116 . 46
BaCl2 34 66 124 180 231 275 369 435 480 513
BaBra 33 64 118 169 214 251 317 372 412 439
Ba(NO3)2 27 51 75 80 68 37 -65 -195 -352 -528
MgSO4 79 191 380 525 631 715 865 937 951 —
CaSO4 80 208 418 588 712 802 882 4) — — —
ZnSO4 86 202 415 595 698 765 893 989 1064 1120
CdSO4 119 290 567 785 939 1071 1266 1380 1467 1542
CuSO4 106 256 518 711 826 930 1115 1242 1321 1375
1) ссылки на литературу см. в табл. 121.
2) При У~т = 0,3162.-
3) При ]/гТп_= 0,35, 1.2= —149.
4) При Ут = 0,125.
Часть II
Водные растворы при 25°
Электролиты m HC1 LiCl LIBr NaCl NaBr Na OH KF KCl KBr
0,1 202 173 166 102 78 127 155 91 70
0,2 273 234 223 90 58 133 179 72 39
0,3 332 279 267 62 31 122 190 44 1
0,4 383 318 305 28 2 105 197 12 -42
0,5 430 352. 339 -10 -30 89 203 -21 -89
0,6 475 384 371 -48 -66 69 209 -55 -136
0,7 518 313 400 -85 -100 51 215 -85 -180.
0,8 560 440 424 -120 -136 33 220 -115 -224
0,9 604 467 448 -156 -173 13 226 -146 -268
1,0 645 491 471 -188 -206 — 4 231 -176 -309
Продолжение табл. 122
\. Эл ектролиты т \. HCI L1CI ЫВг NaCl NaBr NaOH KF KCl KBr
1,2 728 540 515 -252 -280 -37 240 -233 -390
1,5 853 614 580 -343 -382 -70 254 -316 -507
1,7 934 665 625 -398 — 448 -81 262 -370 -579
2,0 1055 744 696 -466 -538 -86 274 -446 -678
2,5 1269 885 821 -556 -654 -73 291 -558 -823
3,0 1484 1044 960 -626 -751 -24 318 -648 -956
3,5 1690 2) 1205 1097 -671 -832 57 376 -721 -1076
4,0 1375 1237 -688 -897 180 456 -782 -1181
4,5 — 1524 1386 -683 -945 — 546 -828 -1269
5,0 1743 1544 -656 -981 — 643 - 853 *) -1345
5,5 —. 1947 1700 -620 -1001 — 754 — *» -1392
6,0 — 2163 1865 -570 3) -992 — 884 — -1412 5)
1) Ссылки на литературу см. в табл. 121
2) Экстраполировано.
з) Для насыщенного раствора, т=6,12.
Ч) Для насыщенного раствора, т=4,82.
5) Для насыщенного раствора, т=5,68.
Таблица 123 х)
Значения —1g при температурах замерзания водных растворов2)
m Элёктро- литы 0,005 0,01 0,02 0 ,05 0,1 0,2 0,3 0,5 0,7 1,0 Ссылки на лите- ратуру
LiCl 0,0343 0,0463 0,0605|0,0830 0,1017 0,1192 0,1262 0,1281 0,1220 0,1055 4
LiBr 0,0298 0,0400 0,0525 0,0720 0,0877 0,1008 0,1041 0,0996 0,0875 0,0621 4
LiNO3 .... 0,0304 0,0412 0,0543 0,0760 0,0947 0,1125 0,1204 0,1246 0,1220 0,1122 2
LiC103 .... 0,0302 0,0406 0,0537 0,0745 0,0915 0,1070 0,1131 0,1141 0,1080 0,0923 5
LiC104 .... 0,0290 0,0386 0,0506 0,0692 0,0835 0,0940 0,0948 0,0856 0,0697 0,0396 5
LiOsCH .... 0,0311 0,0424 0,0573 0,0826 0,1057 0,1312 0,1465 0,1637 0,1729 0,1802 6
LiO2CCH3 . . 0,0309 0,0421 0,0564 0,0799 0,1000 0,1201 0,1298 0,1373 0,1370 0,1296 6
NaCl 0,0306 0,0416 0,0557 0,0804 0,1039 0,1309 0,1476 0,1690 0,1824 0,1948 4
NaBr 0,0282 0,0377 0,0503 0,0721 0,0928 0,1161 0,1300 0,1465 0,1554 0,1622 4
NaNO3 .... 0,0311 0,0428 0,0584 0,0870 0,1165 0,1543 0,1812 0,2214 0,2525 0,2907 2
NaC103 .... 0,0316 0,0433 0,0588 0,0865 0,1139 0,1482 0,1718 0,2066 0,2330 0,2644 5
NaClO4 ... 0,0321 0,0439 0,0588 0,0857 0,1116 0,1429 0,1640 0,1936 0,2151 0,2393 5
NaO2CH . . . 0,0308 0,0416 0,0557 0,0794 0,1012 0,1262 0,1415 0,1606 0,1720 0,1829 6
NaO2CCH3 . . 0,0306 0,0412 0,0544 0,0754 0,0928 0,1089 0,1160 0,1199 0,1172 0,1074 6
KCl 0,0317 0,0434 0,0587 0,0857 0,1121 0,1430 0,1632 0,1903 0,2093 0,2302 4
KBr 0,0313 0,0428 0,0578 0,0839 0,1091 0,1392 0,1590 0,1855 0,2038 0,2236 4
KNO3 0,0329 0,0461 0,0645 0,1001 0,1391 0,1923 0,2336 0,2979 0,3502 0,4158 2
KC1O3 .... 0,0301 0,0418 0,0583 0,0913 0,1277 0,1771 .— — — 5
KC1O4 .... 0,0347 0,0492 0,0697 —. — — —. .— — — 5
KO2CH .... 0,0302 0,0406 0,0543 0,0774 0,0989 0,1221 0,1356 0,1509 0,1589 0,1648 6
KO2CCH3 . . . 0,0306 0,0411 0,0544 0,0750 0,0915 0,1061 0,1113 0,1107 0,1034 0,0867 6
NH4C1 .... 0,0405 0,0555 0,0732 0,1025 0,1298 0,1616 0,1818 0,2081 0,2249 0,2423 За3)
NH4Br .... 0,0451 0,0605 0,0786 0,1082 0,1351 0,1657 0,1849 0,2097 0,2260 0,2427 За3)
NH4J 0,0375 0,0509 0,0674 0,0947 0,1193 0,1479 0,1659 0,1897 0,2052 0,2218 За3)
NH4NO3 . . . 0,0401 0,0547 0,0736 0,1064 0,1390 0,1806 0,2101 0,2542 0,2884 0,3306 За 3)
(NH4)2SO4 . . 0,1308 0,1749 0,2294 0,3194 0,4023 0,4994 0,5633 0,6514 0,7141 0,7836 За3)
') Для CsCl и иодидов LI, Na, К и Cs значения —1g т+ имеются вплоть До концентрации
0,1 н. См. [1]. ~ т
2) Вычислено по уравнению (55) гл. IX, причем член — ~~ не принимается во внимание.
О
3) Относительно четырехзамещенных хлоридов и иодидов аммония см. [36].
Таблица 124
Диэлектрические постоянные и вязкости смесей диоксан—вода.
Константы уравнений (22) и (23) гл. V
Весовой % диоксана t, °C Do lOO^o а* *
20 15 64,01 1,689 0,3258 35,73
20 25 60,79 1,292 0,3345 47,17
20 35 57,73 1,017 0,3439 60,42
20 45 54,83 0,8243 0,3542 75,29
45 15 40,70 2,453 0,6425 30,85
45 25 38,48 1,837 0,6642 41,66
45 35 36,37 1,430 0,6878 54,14
45 45 34,39 1,142 0,7131 68,62
70 15 18,72 2,483 2,060 44,94
70 25 17,69 1,918 2,131 58,85
70 35 16,72 1,522 2,207 75,03
70 45 15,80 1,232 2,290 93,83
82 15 10,01 2,106 5,268 72,46
82 25 9,53 1,671 5,389 92,02
82 35 9,06 1,356 5', 532 114,39
82 45 8,62 1,117 5,683 140,12
Таблица 125
Предельные коэффициенты наклона для коэффициентов активности смесей
диоксан—вода. Диэлектрические постоянные
X—весовой % диоксана
i, °C X = 20 X = 45
D S(7) D S(7)
0 69,16 0,6989 44,28 1,364
5 67,39 0,7072 43,05 1,385
10 65,68 0,7156 41,86 1,406
15 64,01 0,7245 40,70 1,429
20 62,38 0,7339 39,57 1,453
25 60,79 0,7437 38,48 1,477
30 59,94 0,7540 37,41 1,503
35 57,73 0,7648 36,37 1,530
40 56,26 0,7760 35,37 1,557
45 54,83 0,7877 34,39 1,586
50. 53,43 0,7999 33,43 1,616
x = = 70 X'= = 82
i, CC D S(/)
0 20,37 4,373 — —-
• 5 19,81 4,437 10,52 11,47
10 19,25 4,510 10,27 11,58
15 . 18,72 4,581 10,01 11,72
20 18,20 4,657 9,77 11,85
25 17,69 4,738 9,53 11,98
30 17,20 4,820 9,29 12,16
35 16,72 4,907 9,06 12,30
40 16,26 4,995 8,84 12,46
45 15,80 5,092 8,62 12,64
50 15,37 5,185 — —
1 ' Таблица 126
Плотности чистых растворителей и растворов соляной кислоты в смесях
диоксан—вода. Параметры приведенных ниже уравнений
С
(Значения с можно определить, зная т, с помощью уравнения —=d0—Am с точ-
ностью 0,1%). Д—разность между вычисленными и опытными значениями, выражен-
ная в %. Дг относится к уравнению для d, Д2—к уравнению для , X—весовой %
диоксана [1].
Х = 20
d — d0 + am — Ьта + ет lg m; уравнение применимо до концентрации 3 М.
t, °C do а Ь е Л1 А Дг
0 1,0271 0,0133 0,00032 0,008 0,02 0,02159 0,16
5 1,0245 0,0142 0,00035 0,007 0,02 ' 0,02116 0,12
10 1,0219 0,0149 0,00025 0,004 0,02 0,02077 0,09
15 1,0193 0,0154 0,00005 0,000 0,02 0,02038 0,05
20 1,0167 0,0160 0,00013 — 0,02 0,02013 0,03
25 1,0141 #0,0161 0,00013 — 0,01 0,01989 0,02
30 1,0115 0,0167 0,00019 — 0,02 0,01965 0,01
35 1,0090 0,0168 0,00020 — 0,01 0,01954 0,01
40 1,0063 0,0168 0,00018 — 0,02 0,01933 0,01
45 1,0038 0,0166 0,00015 — 0,02 0,01922 0,02
50 1,0014 0,0164 0,00014 — 0,01 0,01933 0,02
Х = 45
d = rf0+ am — Ьта; уравнение применимо до концентрации 3 М.
i, °C do а ь Д1 А Дг
, 0 1,0484 — — — (0,02267)!) 0,07
5 1,0450 0,0143 0,00005 0,01 0,02223 0,05
10 1,0419 0,0145 0,00008 0,01 0,02209 0,05
15 1,0386 0,0144 0,00002 0,01 0,02171 0,07
20. 1,0353 0,0147 0,00004 0,01 0,02148 0,06
25 1,0319 0,0150 0,00010 0,01 0,02138 0,04
30 1,0282 0,0153 0,00011 0,01 0,02109 0,03
35 1,0246 0,0156 0,00012 0,01 0,02072 0,02
40 1,0210 0,0157 0,00012 0,01 0,02052 0,01
45 1,0175 0,0160 0,00013 0,01 0,02021 0,02
50 1,0139 0,0163 0,00016 0,01 0,01995 0,01
X = 70
d = d0 + am + emlg m; уравнение применимо до концентрации 1,5 М.
1, °C do а е Д1 А Дг
0 1,0619 — — — (0.02559)1) —
5 1,0570 0,0123 0,0085 0,03 0,02489 0,13
10 1,0522 0,0128 0,0080 0,03 0,02420 0,12
1) Экстраполировано.
Продолжение табл. 126
(, °C do а' £ • Л1 А Дз
15 1,0474 0,0135 0,0060 0,02 0,02352 .0,11
20 1,0426 0,0142 0,0040 0,02 0,02275 0,07 •
25 1,0378 0,0150 0,0020 0,02 0,02200 0,05
30 1,0332 0,0155 0,0015 0,01 0,02128 0,05
35 . 1,0285 0,0159 0,0000 0,01 0,02077 0,02
40 1,0239 0,0165 0,01 0,01998 0,02
45 1,0194 0,0167 — 0,01 0,01968 0,02
50 1,0148 0,0169 — 0,01 0,01926 0,02
X = 82
d — d0 + ат; уравнение применимо до концентрации 0,6 М.
£, °C do а Л1 А Д2
5 1,0540 0,0152 0,01 0,0224 0,01
10 1,0488 0,0159 0,02 0,0220 0,02
15 1,0436 0,0165 0,02 0,0212 0,02
20 1,0387 0,0165 0,01 0,0210 0,01
25 1,0338 0,0166 0,01 0,0210 0,01
30 1,0288 0,0165 0,01 0,0206 0,01
35 1,0236 0,0173 0,01 0,0198 ' 0,01
40 1,0183 • 0,0178 0,01 0,0198 0,02
45 1,0130 0,0183 0,03 0,0178 0,05
Таблица 127
Стандартные потенциалы элемента
Н2 j НС1 (т), растворитель (X), Н2О (У) | AgCl-Ag.
X и Y—весовые %
Смеси диоксан—вода
X = 0; Е° = 0,22237 — 639,64 • 10"» (£ — 25) — 3,181 • 10“’ (t — 25)а,
X = 20; Е°*= 0,20303 — 760,5 • 10'«(I — 25) — 3,70 • 10"» (£ — 25)3,
X = 45; Е°*= 0,16352 — 1135 • 10“’ (t — 25) — 3,70 • 'КГ6 (t — 25)3,
X = 70; Е°*= 0,06395 — 1767 • КГ6 (I — 25) - 3,70 • 10"’ (t — 25)2,
X = 82; Е°* = 0,0413 — 2370 • 10"’ (I — 25) — 8,80 • 10'6 (I — 25)2.
Эти соотношения применимы в пределах от 0 до 50э включительно
Смеси метиловый спирт—вода
X. = 10; Е°* = 0,21818 — 555,63 • IO'6 (t — 20) — 4,128 • 10~6 (t — 20)2,
X = 20; Е°* = 0,21151 — 529,10 • КГ6 (t — 20) — 4,706 • ИГ6 (t — 20)а.
Эти соотношения применимы в пределах от 0 до 40° включительно.
Смеси этиловый спирт—вода [I]1)
X = 10; Е°*= 0,21900 — 5,03 • КГ4 (t — 20) — 3,82 • 10~6 (t — 20)2,
X = 20; Е°*= 0,21025 — 4,19 • Кг4 (t — 20) — 6,0 • 10“6 (t — 20)2.
Смеси изопропиловый спирт—вода [2]
ЛГ = 5; Е°*= 0,22110 — 5,7425 • 10~4 (t — 20) — 3,8357 • 10*’ (t — 20)2,
X = 10; Е°*= 0,21666 — 5,3324 • КГ4 (г — 20) — 4,7405 • КГ6 (t — 20)2,
X = 20; Е°*= 0,20905 — 4,9001 • КГ4 (t — 20) — 6,8362 • 10“’ (t — 20)2.
Смеси глицерин—вода [3]
X = 50; Е°*= 0,18392 — 7,45 • Кг4 (£ — 25) — 3 • l(T’(t — 25)2.
Это соотношение применимо в пределах от 0 до 90° включительно.
1) Исправленные данные.
Средний коэффициент активности соляной кислоты в воде г)
Таблица 128
t, °C m 0 5 10 ’ 15 20 25 30 35 40 4 5 50 X 55 60
0,0001 (0,9890) (0,9886) (0,9890) (0,9890) (0,9892) (0,9891) (0,9890) (0,9886) (0,9885) (0,9883) • (0,9879) (0,9879) (0,9879)
0,0002 ,(0,9848) (0,9847) (0,9846) (0,9844) (0,9844) (0,9842) (0,9835) (0,9838) (0,9833) (0,9835) (0,9831) (0,9833) (0,9831)
0,0005 (0,9756) (0,9756) (0,9756) (0,9757) (0,9759) (0,9752) (0,9747) (0,9745) (0,9741) (0,9741) (0,9738) (0,9735) (0,9734)
0,001 (0,9668) (0,9662) (0,9666) (0,9661) (0,9661) (0,9656) (0,9650) (0,9647) (0,9643) (0,9644) (0,9639) (0,9636) (0,9632)
0,002 0,9541 0,9539 0,9544 0,9530 0,9527 0,9521 0,9515 0,9513. 0,9505 0,9504 0,9500 0,9497 0,9491
0,005 0,9303 0,9300 0,9300 0,9297 0,9294 0,9285 0,9275 0,9268 0,9265 0,9261 0,9250 0,9240 0,9235
0,01 0,9065 0,9056 0,9055 0,9055 0,9052 0,9048 0,9034 0,9025 0,9016 0,9008 0,9000 0,8990 0,8987
0,02 0,8774 0,8768 0,8773 0,8770 0,8768 0,8755 0,8741 0,8731 0,8715 0,8704 0,8690 0,8680 0,8666
0,05 0,8346 0,8344 0,8338 0,8329 0,8317 0,8304 0,8285 0,8265 0,8246 0,8232 0,8211 0,8195 0,8168
0,1 0,8027 0,8023 0,8016 0,8000 0,7985 0,7964 0,7940 0,7918 0,7891 0,7872 0,7850 0,7829 0,7813
0,2 0,7756 0,7756 0,7740 0,7717 0,7694 0,7667 0,7630 0,7604 0,7569 0,7538 0,7508 0,7474 0,7437
0,5 0,7761 0,7730 0,7694 0,7658 0,7616 0,7571 0,7526 0,7477 0,7432 0,7387 0,7344 0,7292 0,7237
1,0 0,8419 0,8363 0,8295 0,8229 0,8162 0,8090 0,8018 •0,7942 0,7865 0,7790 0,7697 0,7628 0,7541
1,5 0,9452 0,9365 0,9270 0,9154 0,9065 0,8962 0,8849 0,8740 0,8601 0,8517 0,8404 0,8276 0,8178
2,0 1,078 1,068 1,053 1,039 1,024 1,009 0,9929 0,9755 0,9602 0,9481 0,9327 0,9186 0,9072
3,0 1,452 1,427 1,401 1,373 1,345 1,316 — — — — —. — —
4,0 2,006 1,960 1,911 1,862 1,812 Л, 762 — — — — — —
1) Вычислено из опытных значений электродвижущих сил. Величины для концентраций, меньше 0,002 Af, были получены путем графической экстрапо-
ляции с использованием данных Харнеда и Элерса [1]. Андерсон [2] тщательно изучил элемент Ha | НС1 (m) | AgCl-Ag, содержащий соляную кислоту с кон-
центрациями 0,00002 —0,003М. Были внесены поправки на растворимость хлористого серебра и на другие факторы. Коэффициент активности при 25°
оказался равным 0,9751, 0,9653 И 0,9523, соответственно, для концентраций 0,0005, 0,001 и 0,002 М. Эти величины очень хорошо совпадают со
значениями, приведенными в таблице и полученными с помощью экстраполяции.
ПРИ Л.0 Ж Е Н И Е А . 509'
Таблица 129
Средние коэффициенты активности соляной кислоты в смесях диоксан—вода *)
X — весовой % диоксана
t, °C т 0 10 29 25 30 40 50
X — 20
0,005 0,007 0,902 0,889 0,900 0,886- 0,898 0,883 0,896 0,880 0,895 0,880 0,892 0,876 0,889 0,871
0,01 0,872 0,869 0,865 0,862 0,861 0,857 0,851
0,02 0,835 0,830 0,825 0,821 0,820 0,814 0,808
0,03 0,811 0,805 0,800 0,796 0,795 0,788 0,781
0,05 0,780 0,774 0,768 0,763 0,762 0,755 0,748
0,07 0,759 0,753 0,746 0,740 0,740 0,732 0,725
0,1 0,736 0,729 0,722 0,720 0,716 0,708 0,701
0,2 0,696 0,688 0,681 0,676 0,673 0,665 0,656
0,3 0,682 0,675 0,667 0,661 0,658 0,649 0,639
0,5 . 0,684 0,675 0,666 0,660 0,656 0,646 0,633
0,7 0,649 0,690 0,679 0,672 0,667 0,655 0,641
1,0 0,736 0,725 0,712 0,704 0,698 0,683 0,666
1,5 0,830 0,815 . 0,797 0,786 0,777 0,755 0,732
2,0 0,959 0,938 •0,913 0,898 0,885 0,855 0,823
3,0 1,337 1,293 1,245 1,219 1,195 1,141 1,085
X = 45
0,003 0,849 0,846 0,844 0,842 0,839 0,834 0,828
0,005 0,824 0,817 0,811 0,808 0,803 0,795 0,786
0,007 0,802 0,793 0,786 0,782 0,777 0,767 0,757
0,01 0,776 0,766 0,758 0,753 0,747 0,737 0.725
0,02 0,720 0,707 0,697 0,692 0,686 0,673 (/,660
0,03 0,683 - 0,671 0,661 0,654 0,649 0,635 0,622
0,05 0,637 0,624 0,613 0,607 0,600 0,586 0,573
0,07 0,605 0,593 0,583 0,577 0,570 [0,557 0,545
0,1 0,579 0,566 0,553 0,547 0,540 0,525 0,512
0,2 0,529 0,514 0,503 0,496 0,488 0,474 0,459
0,3 0,511 0,496 0,484 0,476 1,466 0,453 0,438
0,5 0,503 0,487 0,473 0,465 0,456 0,440 0,423
0,7 0,513 0,495 0,480 0,471 0,461 0,443 0,424
1,0 0,547 0,526 0,508 0,497 0,485 0,463 0,442
1,5 0,640 0,612 0,585 0,570 0,555 0,524 0,496
2,0 0,773 0,733 0,695 0,676 0,655 . 0,614 0,575
3,0 1,191 1,112 1,037 1,001 0,962 0,887 0,818
1) Библиографию См. под номером [22]
в описке литературы к гл.
XI.
510
ПРИЛОЖЕНИЕ A
Продолжение табл. 129
*» °с т 0 10 20 25 30 40 50
Х = 70
0,001 0,719 0,713 0,705 0,700 0,696 0,686 0,675
0,0015 0,672 0,665 0,656 0,651 0,647 0,636 0,624
0,002 0,641 0,633 0,623 0,618 0,613 0,601 0,589
0,003 0,589 0,582 0,573 0,568 0,563 0,552 0,540
0,005 0,530 0,521 0,510 0,505 0,499 0,487 0,473
0,007 0,488 0,479 0,468 0,462 0,457 0,444 0,431
0,01 0,446 - 0,436 0,425 • 0,418 0,413 0,401 0,388
0,02 0,369 0,359 0,348 0,342 0,336 0,324 0,312
0,03 0,328 0,318 0,308 0,303 0,297 0,286 0,275
0,05 0,283 0,274 0,264 0,258 0,253 0,243 0,232
0,07 0,259 0,249 0,239 0,234 0,229 0,219 0,208
0,1 0,236 0,226 0,217 0,212 0,207 0,197 0,188
0,2 0,204 0,194 0,185 0,180 0,175 0,165 0,156
0,3 0,193 0,182 0,173 0,168 0,163. 0,154 0,144
0,5 0,191 0,179 0,169, 0,163 0,158 0,147 0,137
0,7 0,200 0,187 0,175 0,168 0,162 0,150 0,139
1,0 0,227 0,211 0,195 0,187 . 0,179 0,165 0,151
1.5 0,303 0,277 0,252 0,240 0,228 0,207 0,187
Д' = 82
°с 15 25 35 45
m 5
0,001 0,4242 0,4129 0,3979 0,3795 0,3592
0,0015 0,3725 0,3627 0,3488 0,3318 0,3129
0,002 0,3369 0,3277 0,3147 0,2990 0,2810
0,003 0,2862 0,2781 0,2682 0,2553 0,2378
0,005 0,2319 ' 0,2267 0,2181 0,2062 0,1916
0,007 0,2019 0,1977 0,1900 0,1791 0,1654
0,01 0,1744 0,1707 0,1629 0,1529 . 0,1412
0,015 0,1472 0,1440 ’ 0,1371 0,1282 0,1176
0,02 0,1311 0,1274 0,1213 0,1131 0,1035
0,03 0,1112 .0,1076 0,1020 0,0946 0,0868
0,05 0,0912 0,0876 0,0826 0,0766 0,0698
0,07 0,0780 0,0756 0,0713 0,0659 0,0569»
0,1 0,0701 0,0675 0,0634* 0,0582 0,0525
0,15 0,0627 0,0597 0,0560 0,0513 0,0460
0,2 0,0589 0,0560 ' 0,0521 0,0476 0,0425
0,3 0,0563 0,0532 0,0490 0,0443 0,0392
0,5 0,0595 0,0554 •0,0504 0;0445 0,0386
ПРИЛОЖЕНИЕ А 511
Таблица 130
jj Средний коэффициент активности соляной кислоты в органических растворителях
и в смесях ВОДЫ с органическими растворителями.
N2 = молярная доля органического растворителя
Метиловый спирт
IV 2 =“ 0,0588 [1] N2 = 0,1233 [1]
i / • т 0° 25° 40° 0° 25° 40°
0,001 0,964 0,962 ' 0,961 0,961 0,959’ 0,957
: о,оо2 0,951 0,948 0,946 0,946 0,943 0,941
0,005 0,926 0,922 0,919 0,919 0,915 0,912
1 0,01 0,901 0,897 0,893 0,893 0,888 0,884
0,02 0,872 0,866 0,861 0,862 0,856 0,850
0,05 0,825 0,819 0,812 0,814 0,806 0,798
’ 0,1 0,790 0,780 0,772 0,771 0,762 0,751
; - 0,2 0,762 0,747 0,736 0,741 0,727 0,715
" 0,5 0,754 0,737 0,718 0,726 0,708 0,693
1,0 0,809 0,783 0,756 0,772 0,747 0,722
• 1. 1,5 0,898 0,861 0,827 0,855 0,814 0,781
’ 2,0 1,020 0,966 0,917 0,965 - 0,911 0,860
; i 1 ЛГ2 = 1 [2]
jt т 25’ т 25° т 25°
! 0,00236 0,826 0,01444 0,658 0,0733 0,470
L 0,002683 0,817 0,01722 0,638 0,0751 0,468
• 0,002980 0,809 0,01986 0,621 0,0947 0/443
0,003161 0,804 0,02363 0,601 0,1155 0,423
Г; 0,00494 ’ 0,766 0,02549 0,592 0,4802 0,325
0,00542 0,758 0,04261 0,532 0,5574 0,322
0,00711 0,732 0,04356 0,530 . — —
0,00986 0,699 0,05312 0,507 — —
Этиловый спирт
N2 = 0,0417 [3] N2 = 0,0891 [3] N2 = 0,5 [4,5] Д’2 = 1 [4,5]
т 25° т 25° т 25’ т 25°
0,00631 0,914 0,00470 0,915 0,005 0,815 0,005 0,728
0,00758 0,907 0,00787 0,894 0,01 0,757 0,01 0,632
0,01088 0,891 0,01015 0,883 0,02 0,676 0,02 0,544
0,01987 0,864 0,01983 0,850 0,05 0,586 0,0249 0,514
0,04210 0,824 0,04150 0,809 0,1 0,521 0,0423 0,445
0,05100 0,814 0,05166 0,794 0,2 0,471 0,05 0,426
0,07085 0,795 0,07123 0,775 0,5 0,432 0,1 0,352
0,0800 0,788 0,0816 0,770 1,0 0,449 0,1242 0,327
0,0885 0,783 0,0924 0,761 1,5 0,510 0,1782 0,300
512
ПРИЛОЖЕНИЕ А
Продолжение табл. 130
N2 = 0,0417 [3] = 0,0891 [3] Лг2 = 0,5 [4,5] N1 = 1 [4,5]
т 25° т 25° т 25° т 25°
0,1091 0,773 0,1042 0,751 2,0 0,582 0,2 0,286
0,1990 0,744 0,3038 0,699 2,5 0,697 0,4437 0,218
0,2999 0,731 0,4751 ’ 0,698 — — 1,0 0,177
0,5050 0,730 0,7309 0,709 — — 1,050 0,168
0,7014 0,743 ' 1,0216 0,741 -х- — 1,481 0,159
0,9946 0,776 1,549 0,819 — — 3,62 0,150
1,499 0,856 2,079 0,930 — — — —
1,994 0,954 — — — — — —
Изопропиловый спирт [3]
= 0,0323
25° т 25 т 25’
0,001862 0,948 0,03558 0,830 0,2990 0,726
0,004019 0,927 0,04855 0,813 0,4451 0,723
0,006356 0,911 0,06685 0,795 0,6993 0,737
0,008616 0,899 0,07947 0,785 0,8863 0,757
0,00892 0,898 0,1119 0,766 1,0 0,770
0,02089 0,858 0,1921 0,740 — —
Глицерин [5]
т Лг2 = 0,01 Лг2 = 0,05
25’ 25°
0,002 0,951 —
0,005 0,924 0,898
‘ 0,01 0,902 0,885
0,02 0,873 0,858
0,05 0,826 0,810
0,1 0,798 0,775
0,2 0,764 0,^44
0,28 0,756 0,738
0,38 0,753 0,738
0,5 0,755 0,737
0,7 0,772 0,760
1,0 , 0,810 0,801
1,5 0,901 0,901
2,0 1,019 1,030
2,5 1,161 1,190
3,0 1,345 1,385
4,0 1,792 1,914
ПРИЛОЖЕНИИ А
513
Таблица 131
Предельные коэффициенты наклона S(l) Для в смесях диокеан—вода
X — весовой % диоксана
t, °C х 0 20 45 70 82
0 433 945 2 480 7 480
10 537 1 147 2 930 8 940 14 630
20 654 1364 3 440 10 570 17 610
25 717 1485 3 720 11470 19 210
30 785 1605 4 010 12 410 20 910
40 931 1872 4 640 14 450 24 520
50 ’ 1090 2173 5 340 16 730 —
Таблица 132
Параметры уравнений Ь2 = “ + ₽ Т2, J2 = и значения L2 и J2 при 25°. Уравнения
применимы в интервале от 0 до 50° 1)
X = весовой % диоксана
m — а La (25») J2 (25»)
Х = 20
0,005 77 0,00208 108 1,2
0,007 77 0,00254 149 1,5
0,01 72 0,00277 169 1,7
0,02 77 0,00346 235 2,1
0,03 87 0,00393 262 2,3
0,05 87 0,00438 302 2,6
0,07 . 96 0,00485 335 2,9
0,1 144 0,00554 348 3,3
0,2 298 0,00808 420 4,8
0,3 435 0,01015 467 6,1
0,5 682 0,01384 548 8,3
0,7 871 0,01684 625 10,0
1,0 1156 0,02123 731 13,8
1,5 1517 0,02723 903 16,2
2,0 1753 а, 03207 1097 19,1
3,0 2013 0,03946 1494 23,5
X = 45
0,003 640 0,00923 180 5,5
0,005 619 0,01061 324 6,3
0,007 630 0,01154 395 6,9
0,01 609 0,01223 478 7,3
0,02 651 0,01408 600 8,4
0,03 669 0,01500 664 8,9
0,05 703 0,01638 753 9,8
0,07 735 0,01730 802 10,2
0,1 ’ 754 0,01823 866 10,9
1) Библиографию см. под номером [22] в списке литературы к гл; XI.
33 Г. Харнед, Б. Оуэн
514
ПРИЛОЖЕНИЕ А
Продолжение табл. 132
m 1 — ft L2 (25°) (25°)
0,2 839 0,02077 ' 1007 12,4
0,3 934 0,02284 1096 13,6
0,5 1179 0,02723 1241 . .16,2
0,7 1430 0,03138 1359 18,7
1,0 1704 0,03646 1536 21,7
1,5 1999 0,04292 1815 25,6
2,0 2169 0,04799 2095 28,6
3,0 2285 0,05561 2657 33,1
2 = 70
0,001 701 0,0129 '446 7,7
0,0015 862 0,0157 533 9,4 -
0,002 994 0,0180 606 10,7
0,003 1121 0,0196 621 11,7 ’
0,005 ' . 1233 0,0228 793 13,6
0,007. 1^6 0,0242 884 14,4
0,01 1270 . 0,0254 987 15,1
0,02 1299 0,0282 1207 16,8
0,03 1348 0,0295 1273 17,6
0,05 1389 0,0316 1419 18,8
0,07 1397 0*0328 1518 19,6
0,1 1413 0,0341 1617 20,3
0,2 1417 0,0371 1880 22,1
0,3 1477 0,0397 2051 23,7
0,5 1558 0,0438 2334 26,1
0,7 1635 0,0475 2586 28,3
1,0 1697 0,0519 2915 30,9
1,5 1700 0,0579 3445 34,5
Т = 82
0,001 5190 0,0752 1493 44,8
0,0015 5710 0,0824 1612 49,1
0,002 6550 0,0930 1714 55,4
0,003 7800 0,1082 1815 64,5
0,005 9240 0,1258 1939 75,0
0,007 10100 0,1364 2021 81,3
0,01 10500 0,1418 2101 84,5
0,015 10860 0,1468 2185 87,5
0,02 11000 0,1488 2223 88,7
0,03 11120 0,1510 2298 90,0
0,05 11240 0,1538 2427 91,7
0,07 11360 0,1560 2503 93,0
0,1 11480 0,1589 2640 94,7
0,15 11630 0,1624 2802 96,8
0,2 11860 0,1666 2945 99,3
0,3 12200 0,1742 3280 103,9
0,5 12830 0,1880 3876 112,1
ПРИЛОЖЕНИЕ'А Й5.
Таблица 135
Числа переноса катиона Т+ соляной кислоты в воде и в смесях диоксан—вода [1]
X - весовой % диоксана
X t, °C m 0 5 10 15 20 25 30 35 40 45 50
Х=0
0,0 — 0,842 0,837 0,831 0,826 0,821 0,816 0,811, 0,806 0,801 0,796
0,005 —. 0,844 0,840 0,834 0,829 0,824 0,819 0,814’ 0,809 0,804 0,799
0,01 — 0,845 0,841 0,835 0,830 0,825 0,821 0,816 0,811 0,806 0,801
0,02 — 0,846 0,842 0,836 0,832 0,827 0,822 0,818 0,813 0,808 0,803
0,05 — 0,848 0,844 0,838 0,834 0,830 0,825 0,821 0,816 0,811 0,806
0,1 — 0,850 0,846 0,840 0,837 0,832 0,828 0,823 0,819 0,814 0,810
0,2 — 0,851 0,847 0,843 0,839 0,835 0,830 0,827 0,823 0,818 0,814
0,5 —, 0,854 0,850 0,846 0,842 0,838 0,834 0,831 0,827 0,822 0,819
1,0 — 0,855 0,852 0,848 0,844 0,841 0,837 0,833 0,829 0,824 0,821
1,5 '— 0,857 0,853 0,849 0,845 0,842 0,839 0,835 0,830 0,825 0,822
2,0 —. 0,857 0,853 0,849 0,846 0,843 0,839 0,835 0,831 0,826 0,822
3,0 — . 0,858 0,854 0,850 0,846 0,843 0,840 0,836 0,832 0,827 0,823
Х=20
0,0 0,856 0,851 0,846 0,841 0,836 0,831 0,825 0,821 0,816 0,810 0,805
0,005 0,861 0,855 0,850 0,845 0,840 0,835 0,829 0,825 0,820 0,814 0,809
0,01 0,862 0,857 0,851 0,846 0,841 0,836 0,831 0,827 0,821 0,816 0,811
0,02 0,865 0,859 0,853 0,848 0,843 0,838 0,833 0,829 0,824 0,818 0,813
0,05 0,867 0,861 0,856 0,851 0,846 0,841 0,837 0,832 0,827 0,822 0,816
0,1 0,868 0,862 0,857 0,852 0,848 0,843 0,839 0,834 0,829 0,823 0,818
0,2 0,869 0,863 0,858 0,853 0,849 0,844 0,840 0,835 0,830 0,825 0,820
0,5 0,867 0,862 0,857 0,852 0,847 0,843 0,838 0,833 0,829 0,823 0,818
1,0 0,864 0,860 0,854 0,849 0,844 0,840 0,836 0,831 0,826 0,821 0,816
1,5 0,862 0,857 0,852 0,847 0,842 0,838 0,834 0,829 0,824 0,819 0,814
2,0 0,860 0,855 0,850 0,845 0,841 0,836 0,832 0,828 0,823 0,818 0,813
3,0 0,856 0,852 0,847 0,842 0,838 0,833 0,829 0,825 0,820 0,816 0,811
Х=45
0,0 0,828 0,824 0,820 0,816 0,811 0,806 0,801 0,796 0,791 0,787 0,783
0,005 0,833 .0,829 . 0,825 0,821 0,816 0,811 0,807 0,801 0,797 0,793 0,788
0,01 0,835’ 0,830 0,827 0,823 0,818 0,813 0,809 0,804 0,799 0,795 0,790
0,02 0,838 0,833 0,829 0,825 0,820 0,816 0,811 0,807 0,802 0,798 0,793
0,05 0,842 0,837 0,833 0,829 0,824 0,820 0,816 0,812 0,807 0,803 0,798
0,1 0,845 0,840 0,836 0,831 0,827 0,823 0,819 0,816 0,811 0,807 0,803
0,2 0,849 0,844 0,840 0,834 0,830 0,826 0,823 0,820 0,816 0,812 0,807
0,5 0,851 0,846 0,842 0,836 0,833 0,829 0,826 0,822 0,819 0,815 0,811
1,0 0,851 0,846 0,841 0,836 0,832 0,828 0,825 0,822 0,819 0,815 0,811
1,5 0,850 0,845 0,840 0,835 0,832 0,828 0,824 0,821 0,818 0,814 0,810
2,0 0,849 0,844 0,839 0,835 0,831 0,827 0,824 0,820 0,817 0,813 0,809
3,0 0,847 0,843 0,838 0,833 0,830 0,825 0,822 0,817 0,814 0,810 0,807
33*
516
ПРИЛОЖЕНИЕ А
0,0 II ' 0,772 0,768 0,764 0,760 0,755 0,750 0,746 0,742 0,738 0,734
0,005 — 0,781 0,778 0,774 0,770 0,766 0,761 0,757 0,753 0,750 0,747
0,01 — 0,783 0,780 0,777 0,773 0,769 0,764 0,760 0,756 0,753 0,751
0,02 — 0,786 0,783 0,779 0,776 0,772 0,767 0,763 0,760 0,757 0,755
0,05 — 0,788 0,785 0,782 0,778 0,774 0,770 0,766 0,763 0,760 0,758
0,1 — 0,789 0,786 0,783 0,780 0,775 0,771 0,768 0,765 0,762 0,759
' 0,2 — 0,789 0,786 0,784 0,780 0,776 0,771 0,768 0,765 0,762 0,759
0,5 — 0,789 0,786 0,783 0,779 0,774 0,770 0,766 0,764 0,760 0,757
1,0 — 0,788 0,785 0,782 0,777 0,772 0,768 0,764 0,762 0,758 0,754
1,5 • — 0,788 0,784 0,781 0,776 0,771 0,766 0,763 0,760 0,757 0,752
АГ=82
t, °C т X. 5 10 15 20 25 30 35 40 46
0,0 0,677 0,675 0,673 0,672 0,670 0,668 0,667 0,665 0,663
0,05 0,742 0,735 .0,730 0,726 0,722 0,717 0,712 0,708 0,702
0,1 0,767 0,764 0,762 0,759 0,756 0,754 0,752 0,750 0,747
0,2 0,755 0,751 0,747 0,744 0,740 0,738 0,735 0,732 0,729
0,3 0,718 0,715 0,711 0,708 0,705 0,702 0,699 0,696 0,693
0,5 0,660 0,657 0,654 0,651 0,648 0,645 0,642 0,639 0,637
Теоретические предельные коэффициенты наклона &(Т)Уз0
уравнения 7’+=§(Г)Ул<10 Ут
X (, °C 0 20 45 79
15 0,04278 0,04906 0,06773 0,1629
25 0,04509 0,05195 0,07186 0,1642
35 0,04714 0,05507 0,07492 0,1664
ПРИЛОЖЕНИИ А
517
Таблица 134
Значения функций дли перехода от моляльных концентраций т к молирным
концентрациям с дли водных растворов 1,1-электролитов
— d0 — Ат + Вт*-, da - 0,9970
т и и
Точность сй± 0,05%
I. t = 25°C. Уравнение применимо до концентрации 4 М1)
Анион Катион Cl Вг J NO3
A в A в A В A в
Li 0,0182 0 0,0247 0,0002 0,0358 0,0009 0,0289 0,0004
Na 0,0183 0 0,0245 0 0,0356 0,0008 0,0288 0
К 0,0284 0,0003 0,0345 0,0005 0,0458 0,0014 0,0392 0,0007
Rb 0,0331 0,0004 0,0395 0,0008 0,0508 0,0016 0,0439 0,0008
Cs 0,0400 0,0008 0,0470 0,0015 0,0580 0,0021 0,0503 0
II. В=0 для всех веществ, за исключением бромистого натрия
t, °C do NaBr [1] HC1 [2] HBr [3] KCl [4] NaCl [5]
A в A A A A
0 0,9999 0,0212 -0,0003 0,0171 0,0228 0,0263 0,0160
5 1,0000 0,0222 -0,0001 0,0174 0,0232 0,0265 0,0166
10 0,9997 0,0231 -0,0000 0,0176 0,0236 0,0266 0,0171
15 0,9991 0,0237 0,0000 0,0178 0,0239 0,0267 0,0176
20 0,9982 0,0244 0,0001 0,01805 0,0241 0,0270 0,0180
25 0,9970 0,0248 0,0002 0,0182 0,0243 0,0272 0,0183
30 0,9957 0,0252 0,0002 0,0182 0,0244 0,0273 0,0186
35 0,9940 0,0255 0,0003 0,01825 0,0245 0,0274 0,0188
40 0,9922 0,0257 0,0004 0,01825 0,0245 0,0276 0,0189’
45 0,9901 — — 0,01815 0,0245 — —
50 0,9879 .— — 0,01815 0,0245 — —
55 0,9855 — — 0,01805 0,0244 — —.
60 0,9832 — —- 0,01805 0,0244
1) Р. А. Робинзон, частное сообщение.
i
Средний коэффициент активности хлористого натрия в интервале 0-100° [4]
Таблица 135
\ t, °C т \ 0 0 1) 5 10 15 20 25 [2] 25 [3] 30 35 40 50 60 70 80 90 100 2)
о,1 0,781 0,781 0,781 0,781 0,780 0,779 0,778 0,778 0,777 0,776 0,774 0,770 0,766 0,762 0,757 0,752 0,746
0,2 0,731 0,731 0,733 0,734 0,734 0,733 0,732 0,732 0,731 0,7295 0,728 (0,725) 0,721 0,717 0,711 0,705 0,698
0,5 0,671 0,673 0,675 0,677 0,678 0,679 0,679 0,678 0,679 (0,679) (0,678) (0,675) (0,671) 0,667 0,660 0,653 0,644
1,0 0,6375 0,635 0,6435 0,649 0,652 0,654 0,656 0,654 0,657 (0,657) 0,657 (0,656) (0,654) 0,648 0,641 0,632 0,622
1,5 0,626 — 0,6355 0,6425 0,648 0,652 0,6555 0,658 0,658 (0,660) (0,661) (0,662) (0,659) (0,655) 0,646 0,638 0,629
2,0 0,630 • — 0,6425 0,652 0,659 0,665 0,670 0,670 0,674 (0,676) (0,678) (0,678) (0,676) 0,672 0,663 0,651 0,641
2,5 0,641 — 0,659 0,667 0,677 0,684 0,691 — 0,695 (0,697) (0,698) (0,699) (0,696) (0,692) 0,685 0,674 0,659
3,0 0,660 — 0,677 0,691 0,702 0,7115 0,719 0,718 0,724 0,7255 (0,728) (0,728) (0,726) (0,721) (0,712) 0,700 0,687
3,5 0,687 — 0,706 0,721 0,735 0,744 0,752 — 0,756 (0,759) 0,761 (0,762) (0,760) (0,758) 0,742 0,730 0,716
4,0 0,717 — 0,7395 0,757 0,772 0,783 0,791 — 0,797 0,800 (0,802) (0,802) (0,799) (0,791) 0,777 0,763 0,746
1) Из данных по определению понижения температуры замерзания [1].
2) Величины от 60 до 100° основаны на данных по повышению температуры кипения [5]. Величины в скобках получены с помощью кривых рис. 88 и 8 .
ПРИЛОЖЕНИЕ А
519
Таблица 136
Относительное парциальное молярное теплосодержание растворов
хлористого натрия- [1]х)
t, °C т X. 0 10 20 25 30 40 60 70 80 90 100
0,5 - 330 - 185 - 90 - 45 15 170 300 400 500 600 700
1,0 - 630 - 440 -250 -180 -100 30 310 430 550 670 760
1,5 - 860 - 680 -430 -330 -260 - 60 240 420 580 720 800
' 2,0 -1120 - 840 -560 -450 -330 -130 280 450 650 820 1020
2,5 -1300 -1070 -710 -560 -380 -140 290 480 700 890 1040
3,0 -1650 -1190 -770 -580 -400 -150 300 500 730 910 1040
3,5 -1800 -1230 -800 -590 -420 -170 310 520 820 960 1120
4,0 -1850 -1380 -850 -620 -440 -150 360 590 890 1100 1300
1) Результаты получены с помощью калориметрических измерений,
жущих Сил и из данных по повышению температуры кипения.
методом
электродви-
Таблица 137
Средний коэффициент активности бромистоводородной кислоты
в водных растворах [1], а также значения L2 н Jz при 25°
(, °C т X. о 5 10 15 20 25 30 35
0,001 0,967 0,967 0,967 0,966 0,966 0,966 0,966 0,965
0,005 0,932 0,932 0,932 0,930 0,930 0,930 0,929 0,928
0,01 0,910 0,910 0,909 0,908 0,907 0,906 0,906 0,905
0,02. 0,883 0,883 0,883 0,882 0,879 0,879 0,879 0,878
0,05 0,843 0,843 0,843 0,842 0,838 0,838 0,837 0,834
0,1 0,812 0,812 0,811 0,808 0,807 0,805 0,804 0,802
0,2 0,793 0,791 0,790 0,787 0,785 0,782 0,780 0,777
0,5 0,806 0,803 0,800 0,797 0,793 0,790 0,784 0,781
1,0 0,900 0,894 0,889 0,888 0,877 0,871 0,864 0,856
t, °с т X. 40 45 50 55 60 1.2 72
0,001 0,964 0,964 0,964 0,963 0,963 22 0,4
0,005 0,928 0,927 0,926 0,924 0,924 47 0,8
0,01 0,904 0,904 0,902 0,900 0,898' 64 1,0
0,02 0,877 0,875 0,873 0,871 0,869 85 1,5
0,05 0,833 0,831 0,830 0,827 0,826 124 2,2
0,1 0,800 0,797 0,795 0,791 0,788 163 2,9
0,2 0,774 0,772 0,769 0,765 0,758 ^22 3,9
0,5 0,776 0,772 0,767 0,764 0,760 338 5,6
1,0 0,850 0,844 0,838 0,831 0,823 500 7,7
520
ПРИЛОЖЕНИЕ А
Таблица 138
Средний коэффициент активности хлористого калия в водных растворах [1]
i, °C т х. 0 5 10 15 20 25 30 35 40
о,1 0,768 0,769 0,769 0,769 0,770 0,769 0,768 0,767 0,765
0,2 0,717 0,718 0,718 0,719 0,718 0,719 0,718 0,717 0,715
0,3 0,683 0,685 0,687 0,687 0,688 0,688 0,687 0,685 0,682
0,5 0,642 0,646 0,648 0,650 0,651 0,651 0,651 0,648 0,646
0,7 0,613 0,619 0,623 0,624 0,627 0,628 0,629 0,627 0,626
1,0 0,588 0,595 0,598 0,601 0,604 0,606 0,604 0,604 0,603
1,5 0,563 0,570 0,576 • 0,579 0,582 0,585 0,585 0,585 0,585
2,0 0,547 0,554 0,562 0,568 0,573 0,576 0,578 0,579 0,578
2,5 0,540’ 0,549 0,556 0,562 0,568 0,572 0,574 0,575 0,575
3,0 0,539 0,549 0,556 0,562 0,567 0,571 0,573 0,574 0,573
3,5 0,540 0,550 0,558 0,565 0,571 0,574 0,577 0,578 0,578
4,0 — — 0,563 0,569 0,574 0,579 0,582 0,584 0,585
Таблица 139
Относительное парциальное молярное теплосодержание
и теплоемкость водных растворов хлористого калия
Z/2 “ (0°) aj «72 = а + 2^J [1]
Уравнения применимы в интервале от 0 до 40°
т Ь2(0°) а
0,05 (34) 2,0 0,014
0,1 - 15 3,5 0,025
0,2 - 80 5,1 0,030
0,3 - 190 7,5 . 0,034
0,5 - 280 9,4 0,037
0,7 - 430 12,0 0,040
1,0 - 570 14,4 0,045
1,5 - 760 18,4 0,049
2,0 - 920 22,2 0,055
2,5 -1000 23,0 0,060
3,0 -1025 25,4 0,066 .
3,5 -1200 27,7 0,072
4,0 -1270 29,6 0,079
ПРИЛОЖЕНИЕ А
521
Таблица 140
Средний коэффициент активности бромистого натрия в водных растворах [I]1)
т . 1, °C 0 5 10 15 20 25 30 35 40
0,1 (0,784) (0,784) (0,784) (0,783) (0,783) (0,782) (С 7 11 . ,779) (0,777)
0,2 0,738 0,739 0,741 0,740 0,741 0,740 0,739 0,737 0,734
0,3 0,713 0,716 0,718 0,720 0,718 0,718 0,717 0,715 0,712
0,5 0,685 0,689 0,693 0,693 0,695 0,695 0,694 0,692 0,689
0,7 0,670 0,675 0,681 0,684 0,683 0,687 0,686 0,685 0,685
1,0 0,659 0,667 0,675 0,680 0,684 0,686 0,687 0,686 0,686
1,5 0,664 0,673 0,686 0,693 0,699 0,703 0,706 0,708 0,707
2,0 0,679 0,693 0,708 0,719 0,727 0,734 0,739 0,741 0,743
2,5 0,708 0,727 0,745 0,738 0,769 0,773 0,784 0,789 0,791
3,0 0,745 0,766 0,787 0,802 0,815 0,826 0,834 0,839 0,842
3,5 0,787 0,811 0,834 0,852 0,866 0,878 0,887 0,893 0,896
4,0 0,832 0,858 0,885 0,905 0,921 0,934 0,945 0,951 0,954
1) Значения повышения температуры кипения для водных растворов бромистого калия были
намерены Джонсоном и Смитом [21 вплоть до концентрации 5 М в интервале температур от 60
до 100°. ,
Таблица 141
Относительное парциальное молярное теплосодержание
и теплоемкость водных растворов бромистого натрия [1]
Z<2 = (0°) 4“ 4“ *^2 — а 4- 2^i
Уравнения применимы в интервале от 0 до 40°
т Ч (0°) а ₽
0,1 - 23 4,6 0,015
0,2 - 140 8,2 0,023
0,3 - 210 11,8 0,027
0,5 - 400 15,5 0,036
0,7 - 580 21,8 0,044
1,0 - 830 23,5 0,049
1,5 -1140 27,4 0,055
2,0 -1390 30,6 0,061
2,5 -1600 33,6 0,066
3,0 -1790 37,0 0,073
3,5 -1940 41,4 0,079
4,0 -2020 43,2 0,086
522
ПРИЛОЖЕНИЕ А
Таблиц а 142
Средний коэффициент активности гидрата окиси натрия в водных растворах1)
т . t, °с 0 5 10 15 20 25 30 35
0,05 0,820 0,821 0,820 0,820 0,819 0,818 0,818 0,816
0,1 0,767 0,768 0,768 0,767 0,766 0,766 0,765 0,764
0,25 0,713 0,715 0,716 0,717 0,714 0,713 0,712 0,712
0,5 0,684 0,688 0,690 0,692 0,693 0,693 0,693 0,694
1,0 0,660 0,668 0,672 0,676 0,678 0,679 0,680 0,678
1,5 0,661 0,669 0,673 0,681 0,682 0,683 0,685 0,683
2,0 0,674 0,682 0,689 0,694 0,696 0,698 0,700 0,698
2,5 0,696 0,708 0,717 0,724 0,727 0,729 0,730 0,726
3,0 0,736 0,751 0,762 0,769 0,774 0,774 0,775 0,772
3,5 0,792 0,801 0,816 0,822 0,825 0,826 0,827 0,822
4,0 0,857 0,874 0,882 0,887 0,889 0,888 0,888 0,882
1) От 0,05 до 1,5 М измерения производились Харнедом и Хепнером [1].
Акерлёфа и КиджелсаЦ2] и табл. 176.
См. также работу
Таблица 143
Средний коэффициент активности гидрата окиси калия в водных растворах [1]
т t,.°c 0 5 10 15 20 25 30 35
0,05 (0,829) (0,828) (0,828) (0,827) (0,825) (0,824) (0,823) (0,822)
0,1 0,795 0,796 0,798 0,798 0,798 0,798 0,796 0,793
0,15 0,778 0,778 0,778 0,777 0,776 0,774 0,773 0,771
0,25 0,758 0,757 0,759 0,758 0,757 0,757 0,753 0,751
0,35 0,738 0,740 0,740 0,739 0,739 0,739 0,736 0,733
0,5 0,737 0,736 0,735 0,734 0,732 0,728 0,725 0,725
0,75 0,742 0,742 0,743 0,743 0,741 0,740 0,740 0,736
1,0 0,755 0,756 0,758 0,757 0,756 0,756 0,755 0,752
1,5 0,809 0,812 0,815 0,815 0,814 0,814 0,812 0,809
2,0 0,889 0,886 0,890 0,890 0,889 0,888 0,884 0,879
2,5 0,974 0,978 1,981 0,982 0,980 0,974 0,972 0,965
3,0 1,088 1,091 1,094 1,093 1,087 1,081 1,072 1,065
3,5 1,219 1,229 1,231 1,229 1,219 1,215 1,199 1,195
4,0 1,391 1,395 1,389 1,381 1,361 1,352 1,334 1,314
ПРИЛОЖЕНИЕ А
523
Таблица 144 [1]
Относительное парциальное молярное тепло-
содержание и теплоемкость водных растворов
гидрата окиси натрияг)
Z/2 = ^2 (0°) + ®
т -La(0°) а
0,05 7 6
0,1 70 9,5
0,25 200 15
0,5 400 20
1,0 680 27
1,5 820 30,5
2,0 940 35
1) Подробные таблицы значений La и Ja в интервале
температур от 0 до 70° и до концентрации Л равной t7 М,
для растворов гидроокиси натрия приведены у Акерлёфа
и Киджелса [2].
Таблица 145
Относительное парциальное молярное теплосодержание
и теплоемкость водных растворов гидрата окиси калия [1]
L2 = L2(0°)+af + fit2; 7a = a + 2₽t
Уравнения применимы в интервале от 0 до 40°
т ь2(0°) а Р
0,05 37 2,4 0,017
0,1 41 3,6 0,029
0,15 30 4,7 0,030
0,25 -1 6,4 0,032
0,35 -35 8, 2 0,034
0,5 -95 10,4 0,036
0,75 -180 13,6 0,040
1,0 -270 16,2 0,043
1,5 -335 20,6 0,050
2,0 -381 24,6 0,057
2,5 -390 28,2 0,063
3,0 -356 31,5 0,070
3,5 -335 34,5 0,078
4,0 -226 37,4 0,081
524
ПРИЛОЖЕНИЕ А
Таблица 146
Средние коэффициенты активности 1,1-эяектролитов при 25°
Часть!
т L1C1 NaCl KCl RbCl CsCl
о,1 0,792 0,778 0,769 0,764 0,755
0,2 0,761 0,734 0,717 0,709 0,693
0,3 0,748 0,710 0,687 0,675 0,653
0,5 0,742 0,682 0,650 0,634 0,604
0,7 0,754 0,668 0,626 0,607 0,573
1,0 . 0,781 0,658 0,605 0,583 0,543
1,5 0,841 0,659 0.585 0,559 0,514
2,0 0,931 0,671 0,575 0,547 0,495
2,5 1,043 0,692 0,572 0,540 0,485
3,0 1,174 0,720 0,573 0,538 0,480"
3,5 0,753 0,576 0,539 0,476
' 4,0 0,792 0,582 0,541 0,474
4,5 — 0,590 0,544 0,474
5,0 — — — 0,547 0,476
т ЫВг NaBr КВг RbBr CsBr
о,1 0,794 0,781 0,771 0,763 0,754
0,2 0,764 0,739 0,721 0,706 0,692
0,3 0,757 0,717 0,692 0,674 0,652
0,5 0,755 0,695 0,657 0,634 0,603
0,7 0,770 0,687 0,637 0,606 0,570
1,0 0,811 0,687 0,617 0,579 0,537
1,5 0,899 0,704 0,601 0,552 0,504
2,0 1,016 0,732 0,596 0,537 0,486
2,5 1,166 0,770 0,596 0,527 0,474
3,0 1,352 0,817 0,600 0,521 0,468
3,5 0,871 0,606 0,518 0,462
4,0 — 0,938 0,615 0,517 0,460
4,5 . — — 0,517 0,459
5,0 — — — 0,518 0,460
т L1J NaJ KJ RbJ CsJ
о,1 0,811 ' 0,788 0,776 0,762 0,753
0,2 0,800 0,752 0,731 0,705 0,691
0,3 0,799 0,737 0,704 0,673 0,651
0,5 0,819 0,726 0,675 0,631 0,599
0,7 0,848 0,729 0,659 0,602 0,566
1,0 0,907 0,739 0,646 0,575 0,532
1,5 1,029 0,772 0,639 0,548 0,495
2,0 1,196 0,824 0,641 0,533 0,470
2,5 1,423 0,889 0,649 0,525 0,450
3,0 1,739 0,967 0,657 0,519 0,434
3,5 1,060 0,667 0,518 —
4,0 — 0,678 0,517 —
4,5 — — 0,692 0,519 —
5,0 — — — 0,520
ПРИЛОЖЕНИЕ А
525
Продолжение табл. 146
т LINOs NaNOs KNOs RbNO3 CsNOs
о,1 0,788 0,758 0,733 0,730 0,729
0,2 0,751 0,702 0,659 0,656 0,651
0,3 0,737 0,664 0,607 0,603 0,598
0,5 0,728 0,615 0,542 0,534 0,526
0,7 0,731 0,583 0,494 0,484 0,475
1,0 0,746 0,548 0,441 0,429 0,419
1,5 0,783 0,509 0,378 0,365 0,354
2,0 0,840 0,481 0,327 0,319 —
2,5 0,903 0,457 0,293 0,284 —
3,0 . 0,973 0,438 0,266 0,256 —.
3,5 1,052 0,423 0,244 0,235 —
4,0 — 0,410 — 0,216 .—
4,5 — 0,398 *— 0,200 —
.5,0 — 0,388 л— — —
5,5 .— 0,380 .— — —
6,0 — 0,373 — — —
т LiAci) NaAc KAc RbAc CsAc
о,1 0,782 0,791 0,796 0,797 0,798
0,2 0,740 0,755 0,767 .0,771 0,773
0,3 0,718 0,741 0,752 0,759 0,763
0,5 0,698 0,740 0,751 0,760 0,765
0,7 0,691 0,741 0,755 0,769 0,777
1,0 0,690 0,757 0,779 0,795 0,802
1,5 0,709 0,799 . 0,839 0,859 0,868
• 2,0 0,734 0,854 0,910 0,940 0,952
2,5 0,769 0,920 0,993 1,034 1,046
3,0 0,807 0,993 1,086 1,139 1,153
3,5 0,847 1,070 1,187 1,255 1,277
4,0 0,893 — — — —
т NaF KF NaCNS KCNS JHJ
о,1 0,764 0,774 0,787 0,769 0,818
0,2 0,708 0,727- 0,750 0,716 0,807
0,3 0,675 0,701 0,731 0,685 0,811
0,5 0,631 0,672 0,715 0,646 0,839
0,7 0,602 0,657 0,710 0,623 0,883
1,0 0,572 0,649 0,712 0,600 0,965
1,5 —. 0,649 0,725 0,574 1,139
2,0 —. 0,663 0,751 0,558 1,367
2,5 — 0,684 0,784 0,548 1,656
3,0 — 0,713 0,820 0,542 2,025
3,5 —. 0,748 0,860 0,537
4,0 0,790 0,911 0,533 —
4,5 —- —. —1 0,531 —
5,0 —г — — 0,529 ——
1) Ас означает радикал уксусной кислоты.
526
ПРИЛОЖЕНИЕ А
Продолжение табл. 146
т CsNO3 LiS'H NaS KS T1NO3 Т1С1О4 Т1АС AgNOa
0,1 0,729 0,773 0,764 0,760 0,701 0,730 0,748 0,731
0,2 0,651 0,729 0,708 0,701 0,605 0,652 0,684 0,654
0,3 0,598 0,698 0,672 0,662 0,544 0,599 0,643 0,603
0,5 0,526 0,664 0,624 0,607 — 0,527 0,588 0,534
0,7 0,475 0,642 0,592 0,562 <— — 0,552 0,483-
1,0 0,419 0,621 0,551 0,509 <— — 0,513 0,428.
1,5 0,354 0,595 0,502 0,438 — — 0,472 0,362
2,0 — 0,574 0,460 0,387 — — 0,444 0,315.
2,5 0,565 0,428 0,349 — — 0,422 0,280
3,0 — 0,563 0,403 0,318 — — 0,405 0,252
3,5 п.. . - 0,566 0,385 0,294 .— — 0,390 0,229
4,0 — 0,573 0,368 — — — 0,377 0,210’
4,5 — 0,584 — — — — 0,365 0,194
5,0 — — — — — 0,354 0,181
5,5 — — — — — 0,345 0,169
6,0 — — — — — 0,336 0,159’
8,0 — — — — — .— —- 0,129
10,0 — — — — — — — 0,110
13,0 — — — — — — — 0,0908
Н S — радикал п-толуолсульфоновой кислоты.
Библиографию, относящуюся к части I, см. в списке литературы к этой таблице под номе-
ром [1].
Часть П1)’2)
m NaClO3 [2] KCIO3 [3j NaBrOa [3] КВгС’з [4] LiClOt [5] NaClOi [5] NaH2PO4 [6] KH2PO4 [6J
0,1 0,772 0,749 0,7582) 0,745 0,812 0,775 0,744 0,731
0,2 0,720 0,681 0,696 0,674 0,794 0,729 0,675 0,653
0,3 0,688 0,635 0,657 0,625 0,792 0,701 0,629 0,602
0,5 0,645 0,568 0,605 0,552 0,808 0,668 0,563 0,529
0,7 0,617 0,518 0,569 0,834 0,648 0,517 0,477
1,0 0,589 0,528 0,887 0,629 0,468 0,421
1,4 0,563 0,489 0,979 0,616 0,420 0,369’
2,0 0,538 0,450 1,158 0,609 0,371
2,5 0,525 0,426 1,350 0,609 0,343
3,0 0,515 1,582 0,611 0,320
3,5 0,508 1,866 0,617 0,305
4,0 2,18 0,626 0,293
5,0 0,649 0,276
6,0 0,677 0,265
1) Данные для очень высоких концентраций см. в табл. 176.
2) Обзор данных по осмотическим коэффициентам и коэффициентам активности см. в работа
Робинзона и Стокса [7].
ПРИЛОЖЕНИЕ А
527
Таблица 147
Константы и значение /± (стандартные) уравнения Гуггенгейма [уравнение (37),
гл. XII] 1g /± = 1g /±(Х = 0) + X с. Приведены также значения у0
Электролит A *0,1 m /±U=o)
НС1 +0,240 0,801 0,001 0,966
LiCl +0,195 0,793 0,005 0,927
NaCl +0,130 0,781 0,01 0,901
KCl +0,072 0,771 0,02 0,867
LiC10s +0,243 0,802 0,03 0,844
NaC10s +0,035 0,764 0,04 0,825
KC1OS -0,143 0,734 0,05 0,810
LiC104 +0,330 0,818 0,06 0,797
NaC104 +0,065 0,770 0,07 0,786
KC1O4 -0,435 — 0,08 0,776
LiNO3 +0,226 0,799 0,09 0,767
NaNOs +0,000 0,758 0,1 0,758
KNO3 -0,206 0,723 — —
CsNO3 +0,00 (0,758) — —
NaJO3 ... -0,35 0,700 — —
KJO3 -0,35 0,700 — —
LiOOCH , +0,122 0,780 — —
NaOOCH +0,148 0,785 — —
KOOCH +0,165 0,787 — —
LiOOC.CH3 +0,183 0,791 — —
NaOOC.CH3 +0,252 0,804 — —
KOOC-CH3 +0,252 0,804 — —
Таблица 148
Коэффициенты высаливания кт (25°) [уравнение (53) гл. XII]
Электролит Hell] Ar[l] H2[2] Oa [2] N2O [2] CO2 12] C2H4 [2] CH3COOC2H5 [3] C5H12O2 [4] ®<йн2’Нб[51 C6H6OH [6]
НС1 — — 0,020 0,022 0,016 . 0,006 — — .— —. —
LiCl -0,015 0,037 0,066 — 0,081 — — 0,088 0,077 — 0,181
NaCl 0,053 0,058 0,094 0,132 0,101 — 0,093 0,166 0,139 0,140 0,220
NaBr — — — — 0,089 — 0,077 0,119 0,109 — —
NaJ — — — — — — — 0,024 0,041 0,029 —
NaNO3 0,050 0,058 0,080 — 0,072 (J2)0,053 0,050 0,074 — 0,019 —
Na2SO4 —. — — — 0,110 (J2)0,100 0,098 — 0,159 0,133 —
KC1 0,055 0,061 0,078 — 0,085 0,059 0,062 0,143 0,118 0,118 0,245
KBr — — — — 0,063 0,045 0,049 0,105 0,090 0,055
KJ — — — — 0,058 0,032 — 0,037 0,034 0,057 —
KNO3 — — 0,061 — 0,047 0,025 0,030 0,060 — 0,013 —
nh4no3 — — — — 0,007 — — 0,027 — -0,057 —
BaCl2 — 0,070 — — 0,073 0,060 0,067 0,080 — 0,074 0,069
SrCl2 — 0,033 — — — — — — — — 0,064
CaCl2 — 0,018 0,065 — 0,064 — 0,058 — — — 0,062
Ca (NO3)2 —7 — — — 0,041 — 0,028 0,022 — — 0,062
MgCl2 — 0,012 — — — — 0,055 — 0,060 — 0,060
MgSO4 — — 0,057 — . 0,070 — 0,064 0,127 0,105 0,080 —
A1C13 — 0,021 0,028 — — — 0,044 — 0,043 0,063
A12(SO4)3 — .— — — 0,045 — 0,043 — 0,076 — —
LiJ -0,038 — — — — — — — — — —
HC1O4 -0,048 -0,022 *— — — — — — — — —
ПРИЛОЖЕНИЕ А
529
Таблица 149
Коэффициенты высаливания кт (25°) [уравнение (53) гл. XII] некоторых слабых кислот
^Электролиты Кислоты Бензой- ная ©-Толу- оловая Нитро- бензой- ная Салици- ловая Уксус- ная Хлорук- сусная
NaCl 0,191 0,232 0,180 1,196 0,066 0,088
КС1 0,152 — — 0,140 0,033 0,026
NaNO3 ...... 0,064 0,089 — -0,078 — —
LiCl ...... — — — — 0,075 —
NaAc — — — — -0,014 —
NaOOC • CH2C1 . — — — — — -0,016
BaCl2 0,100 0,134 0,105 — — 0,056
Ba(NO3)2 . . . . 0,030 0,040 0,004 — — —
KBr — — — — 0,014 0,008
KNO3 ...... 0,025 — — -0,006 -0,020 -0,043
KSCN — — — — 0,010 —
C12H22O11 • • • • — — — — 0,025 —
Таблица 150
' Средний коэффициент активности хлористого бария [1]
t, °C т л. 0 15 25 35 45
0,01 0,725 0,727 0,723 0,720 0,710
0,05 0,555 0,565 0,559 0,554 0,536
0,1 0,483 0,498 0,492 0,492 0,487
0,2 0,422 0,442 0,436 0,436 0,431
0,3 0,394 0,416 0,411 0,411 0,405
0,5 0,371 0,395 0,390 0,390 0,382
0,7 0,365 0,390 0,384 0,384 0,376
1,0 0,377 0,395 0,389 0,389 0,381
1,5 0,410 0,410 0,425 0,417 0,409
34 г. Харнед, Б. Оуэн
Таблица 151
Средние коэффициенты активности 2,1-валентных солей при 25° *)
m Мн Cl2 [31 CoCla [3] NiCia [3] CuCl2 [3] FeCla [6] MgC12 [1] CaC12 [2] 2) 1 - SrC12 [2] BaC12 [2] MgBr2 [1] MgJ2 [1] BaBra [4] я* N 0Э О g о Ca (NOa)2 [4] N 0Э О g о (s
0,1 (0,522) (0,526) (0,526) (0,501) (0,525) (0,565) (0,531) (0,514) (0,492) (0,582) (0,599) (0,513) (0,517) (0,480) (0,513) (0,455)
0,2 0,474 0,482 0,483 0,447 0,480 0,520 0,482 0,463 0,438 0,546 0,577 0,465 0,469 0,421 0,464 0,379
0,3 0,454 0,466 0,468 0,423 0,463 0,507 0,462 0,440 0,411 0,547 0,585 0,446 0,448 0,391 0,443 0,338
0,4 0,446 0,463 0,465 0,409 0,459 0,508 0,456 0,430 0,398 0,560 0,607 0,438 0,437 0,373 0,434 0,311
0,5 0,446 0,465 0,468 0,405 0,460 0,514 0,457 0,425 0,390 0,579 0,637 0,437 0,433 0,360 0,432 0,292
0,6 0,448 0,473 0,476 0,403 0,467 0,527 0,462 0,426 .0,386 0,604 0,676 0,439 0,431 0,351 0,434 0,276
.0,7 0,455 0,483 0,489 0,403 0,475 0,542 0,469 0,430 0,384 0,635 0,723 0,444 0,432 0,344 0,438 0,263
0,8 0,463 0,496 0,504 0,405 0,486 0,563 0,479 0,436 0,385 0,671 0,782 0,452 0,435 0,339 0,445 0,253
0,9 0,474 0,514 0,522 0,408 0,501 0,587 0,493 0,444 0,388 0,714 0,851 0,463 0,438 0,336 0,453 0,244
1,0 0,486- 0,538 0,542 0,411 0,519 0,613 0,509 0,455 0,392 0,764 0,929 0,473 0,443 0,334 0,463 0,236
1,2 0,516 0,578 0,595 0,419 0,558 0,680 0,550 0,480 0,402 0,885 1,112 0,500 0,455 0,332 0,485 0,224
1,4 0,554 0,635 0,660 0,430 0,607 0,764 0,599 0,510 0,416 1,032 1,353 0,534 0,469 0,333 0,513 0,214
1,6 0,596 0,706 0,737 0,442 0,668 0,867 0,657 0,546 0,431 1,214 1,651 0,572 0,487 0,335 0,541 0,207
1,8 0,637 0,785 0,826 0,454 0,739 0,986 0,726 0,587 0,450. 1,440 — 0,616 0,507 0,339 0,577 0,201
2,0 0,682 0,884 0,938 0,466 0,817 1,143 0,807 0,636 — — — 0,666 0,528 0,343 0,614 0,197
1) См. табл. 177.
2) Шидловский и Мак-Иннес [5] получили значения т± для 25° и для концентраций ниже 0,1 М из данных для элементов с жидкостным соединением.
3) Р. А. Робинзон, частное сообщение.
Таблица 152
Стандартные потенциалы элементов:
Cd-CdxHg | CdCla (m) | AgCl-Ag; Е“ (CdCla),
Cd-Cd^Hg | CdBra (m) | AgBr-Ag; Е» (CdBra),
Cd-CdxHg | Cd++(a = 1);
(,'С Е» (CdCla) во (CdBra) те» [1] те» [2]
0 0,5815 0,3452
5 0,5804 0,4250 0,3465 0,3452
10 0,5790 0,4248 0,3477 0,3468
15 0,5776 0,4243 0,3491 0,3483
20 0,5758 0,4236 0,3503 0,3499
25 0,5730 0,4227 0,3515 0,3514
30 0,5716 0,4215 0,3526 0,3528
35 0,5696 0,4201 0,3539 0,3541
40 0,5673 0,4185 0,3553 0,3554
Таблица 153
Стандартные потенциалы элементов:
Zn-ZnxHg | ZnCla (m) | AgCl-Ag; E® (ZnCla),
Zn-Zn^Hg | ZnJa (m) | AgJ-Ag; E® (ZnJa),
Zn-Zn^Hg | Zn++(a=l); *®
1,'С Е0 (ZnCla) [1] E»(ZnJa)[2] «о [2] 75° [i]
• 5 0,6176 0,7646
• 10 0,9962 0,6161 0,7642 0,7639
15 0,9919 0,6145 0,7637 0,7635
20 0,9885 0,6126 0,7632 0,7630
25 0,9848 0,6105 0,7627 0,7625
30 0,9810 0,6083 0,7622 0,7619
35 0,9770 0,6059 0,7617 0,7614
40 0,9728 0,6038 0,7612 0,7608
Zn-Zn^Hg tZnBra (m) I AgBr-Ag; <=25°; Е» (ZnBra) - 0,8339; = 0,7628 [3].
Таблица 154
Средние коэффициенты активности галогенидов цинка и кадмия и хлористого свинца
Часть!. при 25°
тп PbCia [1] ZnCla [2] ZnBra [3] ZnJa [4] CdCl2 [5] CdBra [6] CdJa [7]
0,0005 0,902 — 0,880 0,855
0,001 0,859 — w 1 1 — 0,819 0,787 —
0,002 0,803 — — 0,851 0,743 0,699 —
0,003 — ч 1 1 0,833 — 4 — —
0,005 0,704 0,789 - 0,799 0,623 0,570 0,490
0,007 — — — -•* 0,772 — 0,520 0,441
0,01 0,612 0,731 * 1 0,746 0,524 0,468 0,379
0,02 0,497 0,667 0,685 0,690 0,456 0,370 0,281
0,05 0,628 0,605 0,621 0,304 0,259 0,167
0,1 0,575 0,555 0,578 0,228 0,189 0,108
0,2 0,459 0,517 0,564 0,163 [8] 0,132 0,0685
0,5 — 0,394 0,490 0,624 0,1001 0,0789 0,0382
0,7 0,367 0,485 0,701 0,0825 0,0651 0,0310
1,0 — 0,337 0,492 — 0,0664 0,0533 0,0254
1,5 0,306 (1,49 М) 0,500 — 0,0523 0,0425 —
2,0 — 0,282 (2,83 М) 0,516 — 0,0439 — 0,0183
34*
532
ПРИЛОЖЕНИЕ А
Продолжение табл. 154
Часть II. при различных температурах
X. (, °C 10 20 30 40 5 1Л 30 40
т ZnCla Zula
0,005 0,794 0,791 0,787 0,783 0,808 0,802 0,797 0,793
0,007 — —— —- 0,782 0,775 0,770 0,765
О', 008 — — — 0,772 0,765 0,759 0,754
0,01 0,737 0,733 0,728 0,723 0,757 0,750 0,744 0,738
0,02 0j673 0,669 0,663 0,657 0,701 0,694 0,687 0,680
0,03 . 0,635 0,631 0,625 0,617 0,671 0,664 0,655 0,648
0,05 0,587 0,582 0,575 0,566 0,634 0,627 0,617 0,609
0,07 0,556 0,551 0,543 0,532 0,611 0,604 0,594 0,585
0,1 0,525 0,520 0,510 0,497 0,592 0,585 0,574 0,564
0,2 0,476 0,465 0,452 0,434 0,581 О', 572 0,559 0,546
0,5 0,453 0,439 0,419 0,393 0,650 0,638 0,614 0,593
0,7 0,433 0,409 0,379 0,347 0,740 0,723 0,687 0,65б
0,8 0,415 0,384 0,349 0,313 0,787 0,766 0,724 0,687
1,0 0,394 0,357 0,318 0,280 — — — —
^х t, °C 0 40 5 15 30 40
т х. CdCl2 CdBra
0,0005 0,885 0,872 0,850 0,854 0,855 0,853
0,001 0,834 0,811 0,777 0,784 0,787 0,784
0,002 0,746 0,739 0,688 0,696 0,699 0,696
0,005 0,659 0,607 0,553 0,564 0,571 0,569
0,007 — 0,504 0,514 0,521 0,518
o;qi 0,545 0,505 0,453 0,463 0,468 0,465
0,02 0,444 0,408 0,358 0,366 0,370 0,367
0,03 0,309 0,317 0,320 0,317
0,05 0,318 0,292 0,250 0,257 0,259 0,256
0,07 — 0,215 0,221 0,223 0,221
0,1 0,237 0,218 0,182 0,187 0,189 0,186
0,2 0,127 0,130 0,132 0,129
0,5 0,0753 0,0779 0,0787 0,0772
0,7 —_ 0,0619 0,0643 0,0650 0,0638
1,0 0,0505 0,0526 0,0532 0,0522
1,2 0,0449 0,0467 0,0473 0,0465
1,5 __ 0,0400 0,0418 0,0424 0,0417
- 1,8 — — 0,0359 0,0376 0,0383 0,0376
i> °C 5 15 20 25 30 40
т CdJa [9]
0,002 >0,566 0,596 0,607 0,615 0,622 0,629
0,005 0,445 0,472 0,483 0,492 0,499 0,506
0,007 0,391 0,417 0,428 0,436 0,443 0,440
0,01 0,338 0,364 0,374 0,382 0,389 0,397
0,02 0,243 0,265 0,274 0,281 0,287 0,294
0,05 0,141 0,156 0,162 0,167 0,171 0,177
0,07 0,113 0,126 0,131 0,135 0,139 0,143
0,1 0,0891 0,0989 0,103 0,107 0,110 0,113
0,2 0,0562 0,0625 0,0652 0,0675 0,0694 0,0718
0,5 0,0307 0,0342 . 0,0356 0,0369 0,0379 0,0393
0,7 0,0254 0,0282 0,0294 0,0304 0,0312 0,0323
1,0 0,0210 0,0233 0,0242 0,0250 0,0257 0,0265
1,5 0,0173 0,0191 0,0198 0,0205 0,0210 ' 0,0216
2,0 0,0155 0,0171 0,0177 0.0183 0,0187 0,0192-
ПРИЛОЖЕНИЕ А
533
Таблица 155
Z2 и хлористого и йодистого цинка из данных
по электродвижущим силам.
Константы уравнений Z,2=a+?T’2 и J2=2$T
Уравнения применимы в интервале от 0 до 40° ZnCl2 [1]
т —а Е2(25°) 72(25°)
0,005 295 0,0060 237 4
0,01 718 0,0116 316 7
0,02 1540 0,0219 403 . 13
0,03 2140 0,0295 485 18
0,05 3380 0,0453 644 27
0,07 4400 0,0587 812 35
0,1 5700 0,0752 981 45
0,2 9340 0,1224 1530 73
0,3 12110 0,1639 2450 98
0,5 11760 0,1773 ' 3990 106
0,7 11430 0,1846 4970 110
1,0 10290 0,1846 6110 НО
ZnJ2 [2]
т —Л g. 10 * L2(25°) J2(25°)
0,005 739 106 204 6
0,007 848 125 259 7
0,008 895 134 294 8
0,01 1152 166 324 10
0,02 1719 240 413 15
0,03 2095 291 488 18
0,05 2623 360 576 22
0,07 3009 411 640 25
0,1 3381 461 720 28
0,2 4291 586 917 35
0,5 6852 932 1432 56
0,7 8740 1200 1922 72
0,8 9405 1301 2160 78
Таблица 156
Относительное парциальное молярное теплосодержание растворов
галогенидов кадмия [1]
°C т 15 25 15 25 15 25
CdCl2 CdBr2 CdJa
0,0001 47 67 — 4 6 -287 -157
0,0005 112 157 -74 — 7 -819 -564
0,001 162 232 -134 -23 -1148 -828
0,005 362 634 -343 -108 -2155 -1587
0,01 472 759 -414 -131 -2622 -1941
0,05 702 993 -540 -127 -3700 -2789
ОН 778 1134 -591 -70 -4074 -3108
0,2 860 1323 -639 -9 — —
0,4 — — -686 -5 — —
Таблица 157
Коэффициенты активности хлоридов трехвалентных металлов при 25° [I]1)
m AlCls ScCls | YC1S | LaCls Cecis PrCls NdCls SmCls EuCls
0,05 0,409 0,395 0,392 0,392 0,392 0,390 0,389 0,385 0,389
0,1 0,360 0,342 0,336 0,336 0,331 0,333 0,332 0,331 0,335
0,2 0,308 0,297 0,293 0,292 0,292 0,292 0,291 0,290 0,294
0,3 0,323 0,302 0,288 0,281 0,279 0,278 0,279 0,280 0,283
0,5 0,354 0,319 0,297 0,285 0,282 0,280 0,282 0,284 0,288
0,7 0,415 0,363 0,328 0,305 0,306 0,301 0,308 0,306 0,313
1,0 0,578 0,474 0,412 0,366 0,366 0,362 0,368 0,373 0,383
1,2 0,750 0,582 0,494 0,426 0,423 0,423 0,431 0,442 0,453
1,4 1,002 0,724 0,606 0,503 0,502 0,500 0,514 0,526 0,543
1,6 1,374 0,913 0,750 0,600 0,598 0,597 0,617 0,643 0,666
1,8 1,946 1,165 0,946 0,724 0,732 0,722 0,753 0,795 0,830
2,0 — — 1,216 0,883 0,906 0,883 0,928 0,997 1,076
1) Вычислено заново Р. А. Робинзоном для получения данных, согласующихся с результа-
тами, найденными путем изопиестического определения упругости пара но отношению к новым
стандартным растворам (см. приложение Б, §5), и отнесено нами к значению для LaCls при
0,05 М (см. табл. 95).
Таблица 158
Средний коэффициент активности серной кислоты в водных растворах из данных
по электродвижущим силам [ 1] г)
\ i, °C m x. 0 10 20 25 30 40 50 60
0,0005 0,912 0,901 0,890 0,885 0,880 0,869 0,859 0,848
0,0007 0,896 0,880 0,867 0,857 0,854 0,841 0,828 0,814
0,001 0,876 0,857 0,839 0,830 0,823 0,806 0,790 0,775
0,002 0,825 0,796 0,769 0,757 0,746 0,722 0,701 0,680
0,003 0,788 0,754 ' 0,723 0,709 0,695 0,669 0,645 0,622
0,005 0,734 0,693 0,656 0,639 0,623 0,593 0,566 0,533
0,007 0,691 0,647 0,608 0,591 0,574 0,543 0,515 0,489
0,01 0,649 0,603 0,562 0,544 0,527 0,495 0,467 0,441
0,02 0,554 0,509 0,470 0,453 0,437 0,407 0,380 0,356
0,03 0,495 0,453 0,417 0,401 0,386 0,358 0,333 0,311
0,05 0,426 0,387 0,354 0,340 0,326 0,301 0,279 0,260
0,07 0,383 0,346 0,315 0,301 0,290 0,266 0,246 0,228
0,1 0,341 0,307 0,278 0,265 0,254 0,227 0,214 0,197
0,2 0,271 0,243 0,219 0,209 0,199 0,161 0,166 0,153
0,5 0,202 0,181 0,162 0,154 0,147 0,133 0,122 0,107
1,0 0,173 0,153 0,137 0,130 0,123 0,111 0,101 0,0922
1,5 0,167 0,147 0,131 0,124 0,117 0,106 0,0956 0,0869
2,0 0,170 0,149 0,132 0,124 0,118 0,105 0,0949 0,0859
3,0 0,201 0,173 0,151 0,141 0,132 0,117 0,104 0,0926
4,0 0,254 0,215 0,184 0,171 0,159 0,138 0,121 0,106
5,0 0,330 0,275 0,231 0,212 0,196 0,168 0,145 0,126
6,0 0,427 0,350 0,289 0,264 0,242 0,205 0,174 0,150
7,0 0,546 0,440 0,359 0,326 0,297 0,247 0,208 0,177
8,0 0,686 0,545 0,439 0,397 0,358 0,296 0,246 0,206
9,0 0,843 0,662 0,527 0,470 0,425 0,346 0,285 0,237
10,0 1,012 0,785 0,618 0,553 0,493 0,398 0,325 0,268
11,0 1,212 0,930 0,725 0,643 0,573 0,458 0,370 0,302
12,0 1,431 1,088 0,840 0,742 0,656 0,521 0,418 - 0,339
13,0 1,676 1,261 0,965 0,851 0,750 0,590 0,471 0,379
14,0 1,958 1,458 1,104 0,967 0,850 0,664 0,525 0,420
15,0 2,271 1,671 1,254 1,093 0,957 0,741 0,583 0,462
16,0 2,612 1,907 1,420 1,234 1,076 0,828 0,647 0,511
17,0 3,015 2,176 1,604 1,387 1,204 0,919 0,712 0,559
17,5 3,217 2,316 1,703 1,471 1,275 0,972 0,752 0,589
' !) Справедливость этих значений проверена в пределах концентраций от 0 до 4 М и от 9
до 11 М включительно.
ПРИЛОЖЕНИЕ А
535
Таблица 159
Относительные парциальные молярные теплосодержание и
теплоемкость серной кислоты fl].
Константы уравнения La=La(0°)4-at+J}ta
m L2 (0°) а. g-102 L2 (25°) Ja(25°) (э. д. с.) J2(25°) (калор.)
0,0005 397 7,228 6,748 620 И
0,001 858 7,678 6,706 1092 И —
0,002 1481 7,038 6,664 1699 10 —
0,005 2 503 6,948 6,676 2 719 10 —
0,01 3 244 7,538 6,691 3 474 И 6
0,02 3 729 7,328 6,713 3 954 И 7
0,05 4 192 7,128 6,628 4 411 10 8
0,1 4 672 7,748 6,712 4 908 И 9
0,2 . 4 903 8,008 6,798 5145 11 И
0,5 5 063 8,268 6,828 5 313 12 15
1,0 5 310 10,168 7,118 5 608 14 21
2,0 5 766 11,068 7,658 6 091 15 27
3,0 6 607 11,528 7,488 6 942 15 25
4,0 • 7 464 11,908 7,458 7 809 15 21
6,0 9 059 12,578 7,638 9 421 16 16
8,0 10 399 13,898 7,908 10 795 18 13
10,0 И 474 16,768 8,518 11 946 21 20
12,0 12 434 18,218 8,748 12 944 23 24
14,0 13 402 16,168 8,308 13 858 20 26
16,0 14 320 12,438 7,388 14 677 16 28
17,5 14 961 11,508 7,488 15 296 15 29
Таблица 160
Числа переноса катиона серной кислоты [1] в водных растворах
°C т 0 10 15 25 35 45 60
0,0 (0,840) (0,829) (0,824) (0,813) (0,801) (0,788) (0,761)
0,05 0,839 0,834 0,830 0,819 0,807 0,793 0,770
0,10 0,838 0,834 0,829 0,819 0,807 0,793 0,770
0,20 0,837 0,833 0,829 0,819 0,806 0,792 0,770
0,50 0,834 0,828 0,824 0,815 0,801 0,787 0,764
1,00 0,828 ' 0,822 0,818 0,808 0,793 0,779 0,755
2,00 0,816 0,808 0,803 0,793 0,779 0,763 0,737
3,00 0,803 0,793 0,788 0,776 0,762 0,747 0,720
5,00 0,772 0,762 0,756 0,744 0,730 ' 0,715 0,689
8,00 0,720 0,708 0,702 0,690 0,676 0,663 0,641
10,00 0,682 0,672 0,666 0,655 0,642 0,629 0,610
12,00 0,638 0,629 0,625 0,616 0,605 0,595 0,578
14,00 0,591 0,584 0,580 0,573 0,564 0,556 0,543
17,00 0,512 0,508 0,506 0,502 0,498 0,494 0,488
S(T)V^”do 0,0331 0,0362 0,0370 0,0375 0,0363 0,0331 0,0250
536
ПРИЛОЖЕНИЕ А
Таблица 161
Средние коэффициенты активности соляной и бромистоводородной кислот
в растворах галогенидов. Концентрация кислоты—0,01 М. Концентрация соли—т
Часть I. Соляная кислота
сс 7П 0 5 10 15 20 25 30 35 ’ 40 45 50
Соль = КС1 [1}
0,0 0,906 0,906 0,906 0,905 0,905 0,905 0,904 0,903 0,902 0,901 0,900
0,01 0,876 0,875 0,875 0,874 0,874 0,874 0,873 0,871 0,871 0,869 0,866
0,02 0,856 0,855 0,853 0,853 0,853 0,852 0,851 0,850 0,850 0,847 0,844
0,03 0,841 0,841 0,840 0,839 0,838 0,837 0,836 0,834 0,834 0,831 0,828
0,05 0,819 0,819 0,817 0,817 0,816 0,816 0,814 0,812 0,812 0,809 0,806
0,1 0,786 0,786 0,786 0,784 0,783 0,782 0,780 0,778 0,777 0,773 0,769
0,2 0,753 0,753 0,752 0,750 0,749 0,747 0,746 0,743 0,742 0,739 0,734
0,5 0,712 0,712 0,711 0,709 0,708 0,706 0,704 0,701 0,699 0,695 0,690
1,0 0,731 0,727 0,728 0,725 0,723 0,720 0,716 0,712 0,709 0,702 0,697
1,5 0,758 0,757 0,754 0,750 0,747 0,743 0,738 0,732 0,728 0,722 0,713
2,0 0,801 0,749 0,796 0,791 0,786 0,781 0,773 0,767 0,761 0,753 0,743
3,0 0,893 0,890 0,882 0,875 0,868 0,860 0,851 0,841 0,831 0,820 0,808
3,5 0,939 0,933 0,926 0,917 0,908 0,899 0,888 0,876 0,867 0,853 0,839
Соль = NaCl [2]
0,01 0,877 0,876 0,876 0,875 0,874 0,874 0,873 0,872 0,871 0,870 0,869
0,02 0,856 0,856 0,856 0,855 0,855 0,854 0,853 0,851 0,850 0,849 0,847
0,05 - 0,821 0,821 0,821 0,819 0,819 0,818 0,816 0,814 0,812 0,810 0,807
0,1 0,789 0,789 0,788 0,786 0,785 0,784 0,781 0,779 0,777 0,774 0,771
0,2 0,758 0,758 0,757 0,754 0,753 0,752 0,749 0,747 0,745 0,742 0,739
0,5 0,738 0,737 0,736 0,733 0,732 0,730 0,727 0,724 0,721 0,718 0,715
1,0 0,765 0,764 0,762 0,-759 0,756 0,754 0,750 0,746 0,742 0,738 0,733
2,0 0,898 0,896 0,893 0,888 0,883 0,878 0,871 0,864 0,856 0,847 0,838
3,0 1,103 1,099 1,094 1,086 1,077 1,068 1,056 1,043 1,029 1,014 0,999
Соль=LiCl [3]
0,01 0,02 0,878 0,859 0,878 0,859 0,881 0,861 0,879 0,859 0,877 0,857 . —
0,05 — — — 0,826 0,826 0,827 0,824 0,822 — -— ——
0,1 — 0,798 О', 797 0,796 0,793 0,789 >
0,2 — — 0,769 0,767 0,766 0,762 0,760 — — —
0,5 — 0,762 0,759 0,757 0,753 0,749 — — —
1,0 — — 0,812 0,806 0,801 0,793 0,787 —- —
1,5 — 0,896 0,887 0,879 0,869 0,858 — —
2,0 — —- 1,012 0,999 0,986 0,972 0,958 — —- —
3,0 — — —. 1,334 1,308 1,284 1,257 1,232 — —- —
4,0 — — — 1,791 1,748 1,708 1,665 1,624 — — —
Соль = ВаС1а [4]; ц-ионная сила раствора
°с и 0 5 10 15 20 25 30 35 40 45 50
0,01 0,906 0,906 0,905 0,905 0,905 0,905 0,903 0,903 0,902 0,901 0,900
0,02 0,876 0,876 0,875 0,875 0,875 0,875 0,873 0,872 0,871 0,870 0,869
0,03 0,855 0,855 0,855 0,855 0,855 0,854 0,853 0,852 0,851 0,850 0; 848
0,05 0,829 0,828 0,827 0,827 0,827 0,826 0,825 0,824 0,823 0,821 0,820
0,07 0,808 0,808 0,808 0,808 0,808 0,807 0,805 0,804 0,802 0,801 0,799
0,1 0,788 0,788 0,787 0,787 0,787 0,786 0,784 0,783 0,781 0,779 0,777
0,2 0,747 0,748 0,748 0,747 0,747 0,746 0,744 0,741 0,739 0,736 0,733
0,5 0,710 0,709 0,709 0,708 0,707 0,705 0,702 0,698 0,694 0,690 0,686
0,7 0,704 0,704 0,703 0,702 0,700 0,698 0,694 0,691 0,686 0,682 0,677
1.0 0,707 0,706 0,705 0,704 ’0,702 0,699 0,693 0,690 0,685 0,680 0,675
2,0 0,760 0,758 0,756 0,752 0,748 0,743 0,737 0,731 0,724 0,716 0,708
3,0 0,84 7 0,844 0,841 0,836 0,830 0.823 0,815 0,805 0,796 0,785 0,774
ПРИЛОЖЕНИЕ А
537
Продолжение табл. 161
Часть II. Бромистоводородная кислота
Г, °C т 0 5 10 15 20 25 30 35 40 45 50
Соль = КВг [5]
0,0 0,910 0,910 0,909 0,908 0,907 0,906 0,906 0,905 0,904 0,903 0,902
0,01 0,881 0,880 0,879 0,877 0,876 0,874 0,874 0,873 0,871 0,870 0,868
0,02 0,861 0,860 0,858 0,856 0,855 0,853 0,852 0,852 0,849 0,847 0,846
0,03 0,845 0,844 0,843 0,841 0,834 0,838 0,837 0,835 0,833 0,831 0,829
0,05 0,824 0,824 0,822 0,821 0,820 0,818 0,817 0,814 0,812 0,811 0,810
0,1 0,792 0,792 0,790 0,787 0,785 0,783 0,782 0,778 0,776 0,773 0,771
0,2 0,760 0,759 0,757 0,754 0,752 0,750 0,748 0,744 0,741 0,738 0,736
0,5 0,728 0,728 0,725 0,722 0,722 0,717 0,714 0,710 0,705 0,701 0,697
1,0 0,748 0,745 0,741 0,737 0,732 0,728 0,724 0,718 0,712 0,706 0,700
1,5 0,777 0,774 0,770 0,765 0,760 0,756 0,751 0,743 0,736 0,729 0,722
2,0 0,840 0,836 0,831 0,825 0,818 0,810 0,803 0,794 0,785 0,776 0,767
3,0 0,974 0,967 0,958 0,948 0,937 0,926 0,916 0,903 0,890 0,874 0,864
Соль=МаВг [5]
0,01 0,884 0,883 0,882 0,880 0,879 0,878 0,878 0,878 0,875 0,876 0,874
0,02 0,866 0,866 0,865 0,863 0,861 0,859 0,858 0,858 0,854 0,852 0,850
0,03 0,850 0,850 0,848 0,846 0,844 0,842 0,841 0,841 0,837 0,835 0,833
0,05 0,829 0,828 0,827 0,824 0,822 0,821 0,820 0,818 0,815 0,813 0,811
0,1 0,801 0,799 0,797 0,795 0,793 0,791 0,789 0,788 0,783 0,781 0,779
0,2 0,780 0,778 0,775 0,772 0,769 0,767 0,765 0,765 0,759 0,756 0,753
0,5 0,774 0,772 0,768 0,764 0,761 0,756 0,752 0,747 0,739 0,735 0,730
1,0 ' 0,833 0,827 0,821 0,814 0,808 0,801 0,795 0,788 0,778 0,770 0,762
1,5 0,934 0,926 0,916 0,906 0,895 0,884 0,875 0,865 0,852 0,840 0,828
2,0 1,050 1,038 1,026 1,009 0,995 0,981 0,967 0,954 0,936 0,921 0,905
3,0 1,362 1,337 1,311 1,284 1,258 1,233 1,208 1,184 1,156 1,131 1,106
Соль = ЫВг [6]
0,0 0,911 0,910 0,910 0,909 0,908 0,907 0,905 0,904 0,902 0,901 0,899
0,01 0,885 0,885 0,884 0,883 0,882 0,880 0,878 0,877 0,874 0,872 0,870
0,02 0,866 0,867 0,866 0,865 0,864 0,863 0,861 0,858 0,856 0,853 0,850
'0,03 0,854 0,854 0,853 0,852 0,851 0,849 0,847 0,844 0,841 0,838 0,835
0,05 0,834 0,834 0,834 0,833 0,831 0,829 0,827 0,824 0,821 0,817 0,814
0,07 0,821 0,821 0,821 0,820 0,818 0,816 0,813 0,811 0,807 0,804 0,799
0,1 0,810 0,809 0,807 0,807 0,805 0,802 0,799 0,796 0,793 0,789 0,785
0,2 0,796 0,794 0,792 0,789 0,787 0,783 0,780 0,776 0,772 0,767 0,763
0,3 0,796 0,793 0,791 0,788 0,784 0,780 0,776 0,772 0,768 0,763 0,758
0,4 0,802 0,799 0,796 0,792 0,788 0,784 0,780 0,775 0,770 0,765 0,760
0,6 0,824 0,822 0,818 0,814 0,809 0,805 0,800 0,794 0,789 0,783 0,777
1,0 0,911 0,905 0,898 0,892 0,885 0,878 0,870 0,863 0,856 0,848 0,840
1,5 1,061 1,050 1,039 1,028 1,017 1,006 0,996 0,985 0,974 0,964 0,954
2,0 1,255 1,241 1,227 Г, 213 1,197 1,160 1,168 1,152 1,137 1,121 1,105
3,0 1,775 1,748 1,720 1,694 1,667 1,641 1,615 1,589 1,564 1,538 1,514
538
ПРИЛОЖЕНИЕ А
Продолжение табл. 161
Часть III. Соляная кислота при 25°.
Концентрация кислоты—mf, концентрация соли—т2
ГП2 (zni = 0,01) CsCl [7] m2 (тг=0,05) AICI3 [8] m2 (т1 = 0,01) СеС1з[9] m2 (т1 = 0,01) SrCla [Ю]
0,0 0,905 0,0 0,830 0,0 0,904 0,0 0,904
0,01 0,875 0,005 0,809 0,005 0,839 0,025 0,797
0,03011 0,836 0,075 0,799 0,01 0,805 0,05 0,761
0,07 0,795 0,01 0,789 0,03 0,745 0,075 0,743
0,1 0,773 0,02 0,763 0,05 0,717 0,1 0,731
0,2 0,730 0,03 0,750 0,075 0,699 0,2 0,706
0,4 0,685 0,05 0,728 0,1 0,689 0,3 0,711
0,7 0,656 0,07 0,714 0,165 0,662 0,5 0,739
1,0 0,644 0,1 0,708 0,25 0,655 0,75 0,801
1,34 0,638 0,2 0,716 0,375 0,664 1,0 0,888
1,5 0,639 ' 0,4 0,797 0,5 0,698 1,5 1,121
2,0. 0,641 0,6 0,920 0,75 0,767 2,0 1,460
3,0 0,672 1,0 2,0 1,402 3,96 1,0 0,855 2,5 1,944
Таблица 162 [I]1)
Значения 1g для соляной кислоты в концентрированных водных растворах
X. °C т X. 0 10. 20 25 30 40
3 0,1544 0,1377 0,1205 0,1120 0,1028 0,0841 0,0650
' 4 0,2955 0,2732 0,2503 0,2389 0,2269 0,2025 0,1775
5 0,4427 0,4145 0,3857 0,3714 0,3564 0,3260 0,2951
6 0,5922 0,5581 0,5233 0,5059 0,4879 0,4516 0,4146
7 0,7413 0,7011 0,6603 0,6400 0,6190 0,5765 0,5335
8 0,8877 0,8415 0,7948 0,7714 0,7475 0,6990 0,6500
9 1,0297 0,9776 0,9250 0,8988 0,8718 0,8175 0,7625
10 1,1661 1,1082 1,0498 1,0208 0,9908 0,9306 0,8699
И 1,2962 1,2326 1,1684 1,1366 1,1036 1,0377 0,9712
12 1,4196 1,3503 1,2803 1,2457 1,2097 1,1380 1,0657
13 1,5366 1,4613 1,3855 1,3479 1,3090 1,2314 1,1531
14 1,6476 1,5662 1,4841 ‘ 1,4436 1,4016 1,3178 1,2334
15 1,7536 1,6656 1,5770 1,5333 1,4878 1,3975 1,3065
16 1,8559 1,7608 1,6650 1,6179 1,5687 1,4711 1,3730
Ч В этой работе приведены также подробные таблицы значений для Lj, J2 и т. д.
Таблица 163
Функция активности ионов воды 7н1он/®НзО для растворов солей
КС1 [1]
\t, °C и 0 5 10 15 20 25 *30 35 40 45 50
0,01 0,819 0,818 0,818 0,818 0,817 0,816 0,816 0,812 0,809 0,809 0,808
0,02 0,765 0,762 0,764 0,762 0,759 0,760 0,760 0,754 0,753 0,751 0,750
0,03 0,732 0,729 0,726 0,727 0,726 0,726 0,725 0,720 0,718 0,715 0,713
0,04 0,707 0,709 0,706 0,706 0,704 0,702 0,700 0,696 0,694 0,691 0,687
0,06 0,672 0,672 0,669 0,671 0,669 0,668 0,667 0,662 0,661 0,655 0,655
0,11 0,621 0,621 0,622 0,623 0,619 0,618 0,617 0,612 0,611 0,605 0,602
0,21 0,574 0,576 0,576 0,576 0,572 0,574 0,573 0,567 0,565 0,562 0,558
0,51 0,534 0,536 0,537 0,538 0,535 0,534 0,534 0,527 0,524 0,519 0,514
1,01 0,600 0,596 0,601 0,599 0,595 0,592 0,588 0,580 0,574 0,563 0,558
1,51 0,698 0,689 0,696 0,693 0,677 0,672 0,665 0,652 0,643 0,633 0,620
2,01 0,816 0,815 0,813 0,807 0,795 0,787 0,774 0,761 0,744 0,728 0,710
3,01 1,128 1,126 1,109 1,095 1,075 1,056 1,037 1,006 0,979 0,948 0,925
3,51 1,313 1,301 1,285 1,264 1,234 1,205 1,183 1,147 1,109 1,075 1,043
NaCl [2]
0,02 0,764 0,764 0,763 0,763 0,760 0,759 0,759 0,756 0,752 0,752 0,751
0,03 0,726 0,728 0,732 0,728 0,726 0,725 0,725 0,720 0,717 0,715 0,715
0,06 0,663 0,665 0,666 0,665 0,664 0,664 0,663 0,658 0,653 0,650 0,649
0,11 ' 0,612 0,613 0,613 0,611 0,608 0,607 0,605 0,600 0,595 0,591 0,589
0,21 0,562 0,562 0,559 0,559 0,557 0,556 0,553 0,549 0,544 0,540 0,538
0,51. 0,519 0,519 0,520 0,518 0,516 0,514 0,513 0,508 0,502 0,498 0,497
1,01 0,542 0,543 0,542 0,541 0,537 0,535 0,531 0,524 0,517 0,511 0,507
2,01 0,694 0,698 0,699 0,696 0,691 0,696 0,681 0,667 0,654 0,640 0,629
3,01 0,975. 0,982 0,985 0,982 0,972 0,962 0,998 0,925 0,898 0,878 0,855
LiCl [3]
\t, °C 15 20 25 30 35 °C P- \ 15 20 25 30 35
0,02 0,758 0,758 0,759 0,759 0,758 0,35 0,466 0,464 0,462 0,459 0,456
0,03 0,717 0,718 0,719 0,718 0,717 0,40 0,456 0,454 0,452 0,449 0,447
0,05 0,664 0,663 0,665 0,664 0,662 0,45 0,449 0,446 0,443 0,440 0,440
0,06 0,645 0,646 0,645 0,644 0,642 0,50 0,442 0,439 0,437 0,433 0,431
0,10 0,591 0,590 0,589 0,587 0,584 1,01 0,409 0,405 0,400 0,395 0,390
0,11 0,581 0,580 0,579 0,575 0,572 1,51 0,408 0,402 0,397 0,390 0,383
0,15 0,546 0,544 0,543 0,540 0,536 2,01 0,427 0,418 0,410 0,401 0,393
0,20 0,515 0,513 0,511 0,508 0,505 2,51 0,459 0,449 0,438 0,427 0,416
0,25 0,493 0,491 0,489 0,486 0,483 3,01 0,496 0,483 0,470 0,456 0,442
0,30 0,478 0,476 0,474 0,471 0,468 — — — — — —
KBr [4]
0(< и 0 5 10 15 20 25 30 35 40
0,02 0,778 0,775 0,774 0,771 0,769 0,766 0,764 0,764 0,761
0,03 0,744 0,739 0,739 0,736 0,734 0,731 0,729 0,728 0,725
0,04 0,719 0,715 0,713 0,709 0,710 0,709 0,706 0,704 0,701
0,06 0,685 0,683 0,680 0,678 0,676 0,675 0,672 0,671 0,668
0,11 0,635 0,633 0,628 0,630 0,628 0,624 0,621 0,621 0,617
KBr [4]
Продолжение табл. 163
°C и 0 5 10 15 20 25 30 35 40. .
0,21 0,583 0,583 0,582 0,582 0,581 0,579 0,576 0,574 0,570
0,51 ‘0,540 0,540 0,540 0,540 0,541 0,536 0,532 0,529 0,523
1,01 0,601 0,601- 0,600 0,598 0,595 0,590 0,584 0,578 0,570
1,51 0,677 0,677 0,677 0,672 0,667 0,661 0,651 0,642 0,630
2,01 0,798 0,795 0,795 0,785 0,776 0,764 0,752 0,739 0,721
3,01 1,062 1,053 1,045 1,029 1,009 0,993 0,971 0,947 0,921
NaBr [4]
0,02 0,777 0,773 0,773 0,771 0,769 0,767 0,765 0,765 0,761
0,03 0,745 0,742 0,741 0,737 0,735 0,731 0,728 0,726 0,723
0,04 0,715 0,711 0,711 0,707 0,705 0,703 0,701 0,699 0,695
0,06 0,678 0,675 0,674 0,670 0,668 0,666 0,662 0,660 0,655
0,11 0,624 0,622 0,621 0,618 0,615 0,612 0,608 0,606 0,601
0,21 0,571 0,569 0,570 0,568 0,566 0,565 0,563 0,561 0,558
0,51 0,524 0,525 0,528 0,527 0,526 0,524 0,520 0,518 0,513
1,01 0,552 0,551 0,553 0,551 0,550 0,547 0,542 0,539 0,532
1,51 0,648 0,640 0,638 0,633 0,626 0,618 0,609 0,600 0,592
2,01 0,752 0,746 0,740 0,730 0,718 0,706 0,693 0,679 0,662
3,01 1,044 1,020 1,007 0,983 0,959 0,931 0,905 0,878 0,847
LiBr [5]
0,02 0,778 0,778 0,777 0,773 0,771 0,769 0,770 0,769 0,767
0,03. 0,736 0,736 0,738 0,735 0,733 0,731 0,733 0,731 0,729
0,04 0,707 0,708 0,709 0,706 0,704 0,702 0,704 0,701 0,698
0,06 0,659 0,662 0,663 0,661 0,660 0,658 0,659 0,657 0,653
0,08 0,624 0,628 0,630 0,629 0,628 0,627 0,628 0,625 0,622
0,11 0,590 0,593 0,594 0,593 0,592 0,590 0,591 0,588 0,584
0,21 0,525 0,527 0,528 0,526 0,524 0,522 0,522 0,518 0,513
0,31 0,492 0,493 0,494 0,491 0,489 0,486 0,484 0,481 0,476
0,41 0,469 0,470 0,469 0,467 0,464 0,461 0,460 0,456 0,451
0,61 0,442 0,442 0,441 0,438 0,435 0,432 0,430 0,426 0,421
1,01 0,426 0,426 0,424 0,420 0,416 0,412 0,409 0,404 0,398
1,51 0,442 0,437 0,436 0,430 0,425 0,419 0,415 0,408 0,400
2,01 0,478 0,472 0,469 0,462 0,455 0,449 0,443 0,434 0,424
2,51 0,553 0,549 0,544 0,535 0,527 0,517 0,509 0,498 0,486
ВаС12 [6]
\t, °C и \ 0 5 10 15 20 25 30 35 40 45 50
0,03 0,727 0,728 0,723 0,720 0,716 0,716 0,719 0,716 0,711 0,710 0,713
0,05 0,676 0,677 0,671 0,668 0,665 0,660 0,665 0,663 0,658 0,655 0,658
0,07 0,637 0,638 0,637 0,633 0,629 0,626 0,628 0,624 0,619 0,616 0,617
0,1 0,597 0j598 0,594 0,593 0,589 0,587 0,589 0,584 0,579 0,574 0,574
0,2 0,516 0,517 0,515 0,514 0,511 0,507 0,507 0,502 0,495 0,490 0,488
0,5 0,426 0,423 0,421 0,418 0,416 0,412 0,410 0,404 0,397 0,391 0,385
0,7 0,391 0,394 0,392 0,390 0,386 0,383 0,381 0,375 0,368 0,362 0,358
1,0 0,364 0,367 0,364 0,363 0,361 0,357 0,354 0,350 0,343 0,337 0,333
2,0 0,337 0,341 0,339 0,337 0,334 0,330 0,326 0,320 0,313 0,307 0,301
3,0 0,339 0,344 0,344 0,341 0,337 0,333 0,328 0,320 0,310 0,302 0,292
Были получены также эначения для хлорида цеэия [7], хлорида стронция [8], сульфатов на-
трия, калия и лития [9] при 25°.
*йММй
каМЙйЙМШй
Таблица 164
Опытные значения констант диссоциации в воде. Все значения соответствуют моляльным концентрациям 1)
Кислоты ’) Температура, ° С Ссыл- ки на лите- ратуру
0 5 10 1,5 20 25 30 35 40 45 50 55 60
Муравьиная КА 104 1,638 1,691 1,728 1,749 1,765 1,772 1,768 1,747 1,716 1,685 1,650 1,607 1,551 1
Уксусная КА • 10» 1,657 1,700 1,729 1,745 1,753 1,754 1,750 1,728 1,703 1,670 1,633 1,589 1,542 2
Пропионовая КА 10» 1,274 1,305 1,326 1,336 1,338 1,336 1,326 1,310 1,280 1,257 1,229 1,195 1,160 3
к-Масляная КА • 10» 1,563 1,574 1,576 1,569 1,542 1,515 1,484 1,439 1,395 1,347 1,302 1,252 1,199 4
Хлоруксусная .... КА 10» 1,528 —• 1,488 1,440») — 1,379 1,3084) — 1,230 — — — — 5
Молочная КА • 104 1,287 — 1,361 2) — — 1,374 — 1,336») — — 1,270 — — 6
Гликолевая КА 104 1,334 —— 1,427 2) — — 1,475 — 1,471») — — 1,415 — — 7а
Серная к2А 102 — 1,80 — 1,36 .— 1,01 — 0,75 — 0,56 — 0,41 — 6)
Угольная К2А 1(Р 2,36 2,77 3,24 3,71 4,20 4,69 5,13 5,62 6,03 6,38 6,73 — — 7в
Щавелевая X2jl • 103 5,91 5,82 5,70 5,55 5,40 5,18 4,92 4,67 4,41 4,09 3,83 — — 8а
Малоновая KzA 10» 2,140 2,165 2,152 2,124 2,076 2,014 1,948 1,863 1,768 1,670 1,575 1,469 1,362 8%
Фосфорная • 10» 8,968 — 8,394 2) — — 7,516 — 6,531») — — 5,495 — — 9
А21 • 10» — — — — 6,056 6,226 6,349 6,430 6,471 6,475 6,439 — — 10
Борная Аминокислоты К А 10“ — 3,63 4,17 4,72 5,26 5,79 6,34 6,86 7,38 — 8,32 — — И
Глицин • 10» — — 3,94 — 4,31 4,47 4,50 — 4,81 — — — 12
\кБ 10» — — 4,68 5,12 5,57 6,04 6,52 6,98 7,43 7,87 — — — 12
Аланин (КА 10» — — — — 4,47 4,57 4,66 4,71 4,74 4,76 — — — 13
\кБ 10» — — —— — 6,90 7,47 8,08 8,61 9,10 9,60 — — — 13
1) См. табл. 182.
2) При 12,5°.
з) При 18°.
4) При 32°.
5) При 37,5°.
в) Частное сообщение Т. Ф. Юнга на основании данных, полученных Клотцем и Синглетерри [76].
’) Тартер и Гарретсон [14] определили константы диссоциации сернистой кислоты при 25“ из данных по э. д. с. элементов без жидкостных соединении.
К11 и КгЛ. оказались равными соответственно 1,72 • Ю~2 и 6,24 • 10~з.
Таблица 165
Константы диссоциации в смесях диоксан—вода и метиловый спирт—вода. Все значения соответствуют моляльный концентрациям1)
X- весовой % органического растворителя
X Температура, 'С
0 5' 10 15 20 25 30 35 40 45 50
Вода в смесях диоксан—вода[1]
О2) • Ка • 101* 0,1139 0,1846 0,2920 0,4505 0,6809 1,008 1,469 2,089 2,919 4,018 5,474
20 К„ 10« 0,2702 0,4375 0,6918 1,067 1,622 2,399 3,477 4,922 6,947 9,531 12,87
45 Kw Ю16 0,2114 0,3409 0,5349 0,8188 1,234 1,809 2,594 3,655 5,077 6,914 9,277
70 к„ 1018 0,1789 0,2819 0,4348 0,6511 0,9654 1,395 1,974 2,743 3,779 5,064 6,719
Муравьиная кисл'ота в смесях диоксан—вода [2]
О3) Кд 104 1,638 1,691 1,728 1,749 1,765 1,772 1,768 1,747 1,716 1,685 1,650
20 Кд 10* 6,412 6,548 6,625 6,656 6,651 . 0,605 6,519 6,394 6,243 6,077 5,876
45 Кд 10е 8,702 8,702 8,614 8,488 8,318 8,099 7,834 7,537 7,212 6,867 6,510
70 Кд 10* 11,077 10,876 10,641 10,347 10,005 9,634 9,213 8,778 8,310 7,842 7,359
82 Кд 10’ — 1,883 1,836 1,774 1,690 1,588 1,472 1,339 1,208 1,079 —
Уксусная кислота в смесях диоксан —вода [За]4)
О3) Кд • 105 1,657 1,700 1,729 1,745 1,753 1,754 1,750 1,728 1,703 1,670 1,633
20 Кд 10е 4,75 4,87 4,98 5,05 5,09 5,11 5,08 5,03 4,95 4,86 4,73
45 Кд 10’ 4,78 4,89 4,96 4,96 4,96 4,93 4,86 4,75 4,61 4,44 4,28
70 ^•10» 4,75 4,83 4,89 4,83 4,83 4,78 4,69 4,56 4,42 4,22 4,05
82 Кд Ю11 — 7,41 7,50 7,55 7,55 7,24 6,92 6,61 6,09 5,49 —
1) См. табл. 183.
2) Табл. 107.
3) Табл. 164.
4) Проверено Харнедом [36].
Пропионовая кислота в смесях диоксан—вода [4]
0 К а Ю5 1,274 1,305 1,326 1,336 1,338 1,336 1,326 1,310 1,280 1,257 1,229
20 К а Ю6 3,175 3,267 3,337 3,385 3,412 3,417 3,403 3,370 3,319 3,252 3,172
45 К А • 10’ 2,641 2,713 2,764 2,796 2,808 2,801 2,776 2,734 2,677 2,607 2,526
70 КА 10’ 2,299 2,364 2,410 2,439 2,450 2,444 2,422 2,386 2,336 2,274 2,202
82 К А • 1011 — 3,797 3,917 3,966 3,946 3,860 3,716 3,524 3,293 3,035 —
Уксусная кислота в смесях метиловый спирт—вода [5]
10 К А 105 1,138 — 1,200 .— 1,242 1,247 1,237 — 1,214 — —
20 К А 10е 7,38 — 7,94 — 8,24 8,34 8,30 —- 8,19 — —
Глицин в смесях диоксан—вода1)
20 К а . 103 1,832 1,953 2,067 2,169 2,257 2,352 2,428 2,481 2,533 2,573 2,591
45 К а 104 6,339 6,707 7,034 7,348 7,626 7,847 8,091 8,232 8,350 8,457 8,484
70 К А 105 8,933 9,350 9,759 10,01 10,43 10,84 10,98 11,15 11,26 11,36 11,39
20 КБ • Ю5 1,179 1,317 1,473 1,618 1,781 1,937 2,088 2,241 2,407 2,548 2,690
45 Кв • 10е 1,789 2,024 2,272 2,541 2,840 3,120 3,408 3,707 4,015 4,342 4,680
70 Кв Ю’ 1,47 1,68 1,91 2,13 2,39 2,66 2,94 3,24 3,60 3,96 4,34
1) [ба]—для значений Ка; [66]—для значений Кв.
Константы диссоциации аминокислот [1] в водных растворах
Все значения соответствуют моляльным концентрациям
Таблица 166
Аминокислота Темпёратура, °C
1 12,5 25 37,5 50
dZ-Аланин Ал • 103 3,75 4,14 4,49 4,68 4,66
Кв 105 4,83 6,10 7,40 8,73 9,86
cZZ-a-Амино-н-масляная кислота КА 103 4,63 4,90 5,18 5,15 5,05
К в 105 4,25 5,50 6,81 6,22 9,38
di-a-Амино-н-валерьяновая кислота КА 10» 4,21 4,57 4,81 4,91 4,86
Кв Ю5 4,04 5,18 6,47 7,64 8,63
dl-Нор лейцин КА 103 4,04 4,41 4,62 4,74 4,70
Кв 10* 4,41 5,62 6,87 8,05 9,16
a-Аминоизомасляная кислота КА 10» 3,81 4,17 4,40 4,46 4,41
Кв 10« 1,14 1,38 1,61 1,84 1,99
dl Валин КА 103 4,79 5,05 5,18 5,11 4,90
Кв • 10» 3,24 4,21 5,27 6,28 7,28
rfZ-Лейцин КА 10’ 4,14 4,49 4,70 4,71 4,65
Кв • IO’ 3,57 4,52 5,60 6,71 7,59
dZ-Изолейцин КА 10s 4,32 4,59 4,81 4,82 4,66
Кв Ю« 3,61 4,57 5,77 6,79 7,85
35 Г. Харнед, Г>. Оуэн
Таблица 167 *)
Константы уравнений (70)—(76) гл. XV для вычисления констант диссоциации, AF;, ДН?, ДС° , Д6'? -1» 7Г„ и Та
Ъ' Ъ' *1 О g g.
j*
lg Z = — ^- + D* — C*T A' = 2,3026RA*
= A' —D'T + C'T2 D' 2,3026 RD*
Д#° = A' —CT2 C = 2,3026 RC*
&Cp——2CT
= D' — 2CT
Гв = lg Kt = D* - 2/CM*
Часть I. Водные растворы
Кислоты А* D» С’ А' D' сг гв -1g
Муравьиная 1342,85 • 5,2743 0,015168 6143,59 24,1301 0,069395 297,5 3,7519
Уксусная 1170,48 3,1649 0,013399 5354,99 14,4795 0,061301 295,6 4,7555
Пропионовая 1213,26 3,3860 0,014055 5550,71 15,4911 0,064302 293,8 4,8729
н-Масляная 1033,39 2,6215 0,013334 4727,80 11,9935 0,061004 278,4 4,8026
Хлоруксусная 1049,05 5,0273 0,014654 4799,45 23,0001 0,067043 267,6 2,8143
Молочная. . 1304,72 4,9639 0,014926 5969,15 22,7100 0,068887 295,7 3,8620
Гликолевая 1303,26 4,7845 0,014236 5962,47 21,8893 0,065130 302,6 3,8302
Угольная (2) 2902,39 6,4980 0,02379 13278,55 29,7286 0,10884 349,3 10,121
Щавелевая 1539,31 7,1966 0,021200 7042,40 32,9247 0,096991 269,5 4,2285
Малоновая 1703,31 6,5810 0,022014 7792,71 30,1083 0,100715 278,2 5,6659
*) См. табл. 184.
Продолжение табл. 167
Кислоты А* о» А' • D' С - 1g А»
Фосфорная (1) 1264,51 7,6601 0,018590 5785,18 35,0452 0,085050 260,8 2,0368
» (2) 1648,88 3,2542 0,016534 7543,69 14,8880 0,075644 315,8 7,1885
Борная 2193,55 3,0395 0,016499 10035,58 13,9058 0,075484 364,6 8,9923
Аминокислоты Глицин А 1300,53 5,5277 0,011792 5949,98 25,2894 0,053949 332,1 2,3045
» - В 1307,30 2,5629 0,008038 5980,95 11,7254 0,036773 403,3 3,9202
rfZ-Аланин А (1383,06) (6,3639) (0,013661) (6327,55) (29,1151) (0,062502) (318,2) (2,3297)
1271,17 5,6650 0,012548 5815,65 25,9176 0,057408 318,3 2,3226
» В . . . . 2069,10 7,7123 0,01643 9466,22 35,2841 0,075159 354,9 3,9481
(1529,44) (4,2704) (0,010966) (6997,25) (19,5373) (0,050168) (373,5) (3,9201)
<7£-а-Амино-н-масляная А 1174,74 5,3735 0,012487 5374,48 24,5840 0,057129 299,8 2,2866
» В 1591,68 4,4429 0,010965 7282,0 20,3264 0,05017 381,0 3,9126
dl-a-Амино-н-валерьяновая А . . . 1222,02 5,5238 0,012553 5590,79 25,2716 0,05743 312,0 2,3094
» В . . . 1852,42 6,2544 0,014191 8474,9 28,6141 0,064923 361,3 3,9999
dZ-Норлейцин А 1193,30 5,2850 0,012130 5459,40 24,179 0,055495 313,7 2,3242
» В . 1619,10 5,7984 0,011842 7407,45 26,5279 0,054178 369,8 1,9591
Л-а-Амино-изомасляная А .... 1344,95 6,3053 0,013924 6153,20 28,8470 0,063703 310,8 2,3497
» В . . . . 1460,04 4,5471 0,011540 6679,41 20,8032 0,052796 355,7 3,6624
dZ-Валин А 1245,31 6,0251 0,013868 5697,34 27,5650 0,063447 299,6 2,2863
» В 1694,53 4,9842 0,012004 7752,54 22,8029 0,054919 375,7 4,0360
dZ-Лейцин А 1283,60 6,0027 0,013505 5872,52 27,4626 0,061786 308,3 2,3244
» В 1651,61 4,8479 0,011933 7556,18 22,1793 0,054594 372,0 4,0310
rfZ-Нзолсйцин А 1298,09 6,1967 0,013959 5938,81 28,3502 0,063863 205,0 2,3169
» В 1537,47 4,0396 0,010482 7033,99 18,4813 0,047956 383,0 3,9893
<М*
Продолжен е табл. 167
Часть II. Вода и кислоты в смесях диоксан—вода и метиловый спирт—вода
X-—весовой % органического растворителя
X А» D* С* А' О' С т<> -1g к0
Вода в смесях диоксан—вода
0 4470,99 6,0875 0,01706 20454,96 27,8506 0,078050 511,9 11,3796
20 4596,78 6,3108 0,0184867 21030,45 28,8722 0,084577 498,7 12,1260
45 4641,08 5,7231 0,019784 21233,13 26,1834 0,090513 484,3 13,4414
70 4377,47 2,2289 0,018116 20027,10 10,1973 0,082882 491,6 15,5815
Муравьиная кислота в смесях диоксан—вода
35*
20 1339,04 5,0628 0,015983 6126,17 23,1626 0,072917 289,8 4,1766
45 1333,79 4,6393 0,017634 6102,16 21,2248 0,080676 275,0 5,0602
70 1181,65 1,9920 0,016922 5406,09 9,1135 0,077419 264,2 6,9513
82 3006,85 13,2110 0,040005 13755,07 60,4408 0,18302 274,1 8,7232
Уксусная кислота в смесях диоксан—вода
20 1423,45 4,2934 0,016136 6512,34 19,6425 0,073822 297,0 5,2917
45 1568,31 4,5387 0,018736 7175,08 20,7650 0,085718 289,3 6,3027
70. 1549,12 2,5194 0,018933 7087,29 11,5264 0,086619 286,0 8,3119
82 3763,40 16,0925 0,045641 17217,71 73,6238 0,208809 287,2 10,1194
Продолжение табл. 167
X А* D* С* А' D' С те -1g к8
Пропионовая кислота в смесях диоксан—вода
20 1356,57 3,67038 0,015384 6206,67 16,7930 0,070385 297,0 5,4662
45 1480,12 3,52870 0,017163 6771,94 16,1447 0,078525 293,7 6,5516
70 1508,10 1,65390 0,017466 6879,96 7,5670 0,079914 293,8 8,6112
82 3680,11 15,05723 0,044030 '16837,48 68,8908 0,20145 289,1 10,4015
Уксусная кислота в смесях метиловый спирт— вода
10 1417,19 4,5806 0,015874 6483,70 20,9564 0,072624 298,8 4,9055
20 1572,21 5,3447 0,017279 7192,92 24,4522 0,079052 301,7 5,0796
КА глицина в смесях диоксан—вода
0 1300,53 5,5277 0,011792 5949,98 25,2894 0,053949 332,1 2,3043
. 20 1368,94 5,6875 0,612493 6263,16 26,0216 0,057161 331,0 ' 2,5806
45 1273,49 4,7113 0,011887 5826,47 21,5553 0,054386 327,3 3,0702
70 1187,30 3,3894 0,011322 5432,12 15,5073 0,051801 323,8 3,9435
глицина в смесях диоксан—вода
0 1307,30 2,5629 0,008038 5980,95 11,7254 0,036673 403,3 3,9203
20 1519,89 3,3800 0,010046 6953,78 15,4642 0,045964 389,0 4,4350
45 1364,90 1,1999 0,007139 6244,69 5,4898 0,032663 437,2 5,0433
70 895,27 -3,328 0,000818 4096,02 -15,227 0,003743 — —
ПРИЛОЖЕНИЕ А
549
ЛИТЕРАТУРА К ТАБЛИЦАМ ПРИЛОЖЕНИЯ А
К табл. 119
1. Т. Shedlovsky, J. Am. Chem. Soc., 54, 1411 (1932).
2. D. A. Maclnnes, T. Shedlovsky, L. G. Longsworth, J. Am.
Chem. Soc., 54, 2758 (1932).
3. K. A. Krieger, M. Kilpatrick, J. Am. Chem. Soc., 59, 1878 (1937).
4. T. Shedlovsky, A. S. Brown, D. A. Maclnnes, Trans. Electrochem.
Soc., 66, 165 (1934).
5. L. G. Longsworth, J. Am. Chem. Soc., 57, 1185 (1935).
6. G. Jones, C. F. Bickford, J. Am. Chem. Soc., 56, 602 (1934).
7. D. A. M a с I n n e s, Principles of Electrochemistry, Reinhold Publishing Corp., N. Y.
8. P. A. L a s s e 1 1 e, J. G. Aston, J. Am; Chem. Soc., 55, 3067 (1933).
9. T. Shedlovsky, D. A. Maclnnes, .1. Am. Chem. Soc., 57, 1705 (1935).
10. D. A. Maclnnes, T. S h e d 1 о v s к y, J. Am. Chem. Soc., 54, 1429 (1932).
11. D. Belcher, J. Am. Chem. Soc., 60, 2744 (1938).
12. T. Shedlovsky, A. S. Brown, J. Am. Chem. Soc., 56, 1066 (1934).
13. В. B. Owen, R. W. Gurry, J. Am. Chem. Soc., 60, 3074 (1938).
14. L. G. Longsworth, D. A. Maclnnes, J. Am. Chem. Soc., 60, 3070 (1938). .
15. G. S. H a r t 1 e y, G. W. Donaldson, Trans. Faraday Soc., 33, 457 (1937).
16. G. Jones, F. C. J e 1 e n, J. Am. Chem. Soc., 58, 2561 (1936).
17. E. Swift, J r,, J. Am. Chem. Soc., 60, 728 (1938).
18. V. S i v e r t z, R. E. R e i t m e i e г, И. V. Tartar, J. Am. Chem. Soc., 62,
1379 (1940).
19. В. B. Owen, F. H. S w e e t о n, J. Am. Chem. Soc., 63, 2811 (1941).
20. И. E. Gunning, A. R. Garden, J. Chem. Phys., 10, 126 (1942).
К табл. 120
1. L. G. Longsworth, J. Am. Chem. Soc., 54, 2741 (1932).
2. L. G. Longsworth, J. Am. Chem. Soc., 57, 1185 (1935).
3. D. A. Maclnnes, L. G. Longsworth, Chem. Rev., 11, 171 (1932).
4. G. S. Hartley, G. W. Donaldson, Trans. Faraday Soc., 33, 457 (1937).
5. L. G. Longsworth, D. A. Maclnnes, J. Am. Chem. Soc., 60, 3070 (1938).
6. D. J. L e R о y, A. R. Gordon, J. Chem. Phys., 6, 398 (1938).
К табл. 121
1. a) J. M. Sturtevant, J. Am. Chem. Soc., 62, 584 (1940); 6) 62, 3265 (1940).
2. E. Lange, F. D ii r r, Z. physik. Chem., 121, 361 (1926).
3. E. Lange, E. Schwartz, Z. physik. Chem., 133, 129 (1928).
4. A. L. Robinson, J. Am. Chem. Soc., 54, 1311 (1932).
5. H. Hammerschmid, A. L. Robinson, J. Am. Chem. Soc., 54,’ .3120
(1932).
6. E. Lange, A. Eichler, Z. physik. Chem., 129, 285 (1937).
7. E. Lange, A. L. Robinson, Z. physik. Chem., A148, 97 (1930).
8. J. M. Sturtevant, J. Am. Chem. Soc., 62, 2276 (1940).
9. J. W ii s t, E. Lange, Z. physik. Chem., 116, 161 (1925).
10. E. Lange, P. A. Leighton, Z. Elektrochem., 34, 566 (1928).
И. H. S t r e e c k, Z. physik. Chem., A169, 103 (1934).
12. E. Lange, J. Monheim, Z. physik. Chem., A150, 349 (1930).
13. M. A n d a u e r, E. Lange, Z. physik. Chem., A165, 89 (1933).
14. E. Lange, H. S tr eeck, Z. physik. Chem., A152, 1 (1931).
15. E. Lange, H. S t r e e c k, Z. physik. Chem., A157, 1 (1931).
16. E. Lange, J. Messner, Z. Elektrochem., 33, 431 (1927).
17. E. Lange, J. Monheim, A. L. Robinson, J. Am. Chem. Soc., 55, 4733
(1933).
К табл. 123
1. J. Lange, Z. physik. Chem., A177, 193 (1936).
2. G. Scatchard, P. T. Jones, S. S. Prentiss, J. Am. Chem. Soc., 54,
2690 (1932).
3. a) G. S c a t c h a r d, S. S. Prentiss, J. Am. Chem. Soc., 54, 2696 (1932);
6) J. Lange, Z. physik. Chem., A168, 147 (1934).
4. G. Scatchard, S. S. Prentiss, J. Am. Chem. Soc., 55, 4355'(1933).
5. S c a t c h a r d, S. S. Prentiss, P. T. Jones, J. Am. Chem. Soc., 56, 805
(1934).
550 ПРИЛОЖЕНИЕ А
6. G. Sea t ch ard, S. S. Prentiss,!. Am. Chem. Soc., 56, 807 (1934).
К табл. 126
1. H. S. H a r n e d, С. С a 1 m о n, J. Am. Chem. Soc., 60, 334 (1938).
R табл. 127
1. A. P a t t e г s о n, W. A. F e 1 s i n g, J. Am. Chem. Soc., 64, 1478 (1942).
2. ' R. L. Moore, W. A. F e 1 s i n g, J. Am. Chem. Soc., 69, 1076 (1947).
3. H. S. Harned, F. H. M. N e s t 1 e r, J. Am. Chem. Soc., 68, 665 (1946).
R табл. 128
1. H. S. Harned, R. W. Ehlers, J. Am. Chem. Soc., 55, 2179 (1933).
2. N. J. Anderson, диссертация, University of Chicago (1934).
R табл. 130
1. H. S. Harned, H. C. Thomas, J. Am. Chem. Soc., 58, 761 (1936).
2. G. Nonh eb el, H. Hartley, Phil. Mag. [6], 50, 298, 729 (1923).
3. H. S. Harned, С. С a 1 m о n, J. Am. Chem. Soc., 61, 1491 (1939).
4. H. S. Harned, M. H. F 1 e у s h e r, J. Am. Chem. Soc., 47, 82 (1925).
5. W. W. Lucasse, X. physik. Chem., 121, 254 (1926).
R табл. 133
1. H. S. Harned, E. C. D r e b y, J. Am. Chem. Soc., 61, 3113 (1939).
R табл. 134
1. H. S. Harned, С. C. Crawford, J. Am. Chem. Soc.^59, 1903 (1937).
2. H. S. Harned, R. W. Ehlers, J. Am. Chem. Soc., 59, 2179 (1933).
3. H. S. Harned, A. S. К e s t о n, J. G. D о n e 1 s о n, J. Am. Chem. Soc., 58,
989 (1936).
4. H. S. Harned, M. A. Cook, J. Am. Chem. Soc., 59, 1290 (1937).
5. H. S. Harned, M. A. Cook, J. Am. Chem. Soc., 61, 495 (1939).
R табл. 135
1. G. Scatchard, S. S. Prentiss, J. Am. Chem. Soc., 55, 4855 (1933).
2. H. S. H a r n e d, L. F. N i m s, J. Am. Chem. Soc., 54, 423 (1932).
3. H. S. H a r n e d, J. Am. Chem. Soc., 51, 416 (1929).
4. R. A. Robinson, Trans. Faraday Soc., 35, 1222 (1939).
5. R. P. Smith, D. S. II i r t 1 e, J. Am. Chem. Soc., 61, 1123 (1939).
R табл. 136
l.R. P. Smith, D. S. H i r t 1 e, J. Am. Chem. Soc., 61, 1123 (1939).
R табл. 137
1. H. S. H a r n e d, A. S. К e s t о n, J. G. D о n e 1 s о n, J. Am. Chem. Soc., 58,
989 (1936).
R табл. 138
1. H. S. H a r n e d, M. A. Cook, J. Am. Chem. Soc., 59, 1290 (1937).
R табл. 139
1. H. S. Harned, M. A. Cook, J. Am. Chem. Soc., 59, 1290 (1937).
R cmp. 140
1. H. S. Harned, С. C. Crawf or d, J. Am. Chem. Soc., 59, 1903 (1937).
2. G. C. Johnson, R. P. Smith, J. Am. Chem. Soc., 63, 1351 (1941).
R cmp. 141
1. H. S. Harned, С. C. Crawford, J. Am. Chem. Soc., 59, 1903 (1937).
ПРИЛОЖЕНИЕ А
551
R стр. 142
1. Н. S. Harned, J. С. Hecker, J. Am. Chem. Soc., 55, 4838 (1933).
2. G. А к e г 1 6 f, G. К e g e 1 e s, J. Am. Chem. Soc., 62, 620 (1940).
R cmp. 143
1. H. S. Harned, M. A. Cook, J. Am. Chem. Soc., 59, 496 (1937).
R cmp. 144
1. H. S. Harned, J. C. Hecker, J. Am. Chem. Soc., 55, 4838 (1933).
2. G. А к e r 1 6 f, G. К e g e 1 e s, J. Am. Chem. Soc., 62, 620 (1940).
К табл. 146
1. H. S. Harned, M. A. Coo k, J. Am. Chem. Soc., 59, 496 (1937).
R табл. 146
1. R. A. Robinson, D. A. Sinclair, J. Am. Chem. Soc., 56, 1830 (1934);
R. A. Robinson, там же, 57,. 1161 (1935); 57, 1165 (1935); 59, 84 (1937);
R. A. Robinson, Trans. Faraday Soc., 35, 1217 (1939); R. A. Robinson,
J. Am. Chem. Soc., 62, 3131 (1940), 63, 628 (1941); H. S. H a r n e d, R. A. Ro-
binson, Trans. Faraday Soc., 37, 302 (1941); R. A. Robinson, H. S. H a r-
n e d, Chem. Rev., 28, 419 (1941).
2. J. H. Jones, J. Am. Chem. Soc., 65, 1353 (1943).
3. J. H. Jone s, J. Am. Chem. Soc., 66, 1672 (1944).
4. J. H. Jones, J. Am. Chem. Soc., 69, 1066 (1947).
5. J. H. Jones, J. Phys. Chem., 51, 516 (1947).
6. J. M. Stoke s, Trans. Faraday Soc., 45, 612 (1949).
7. R. A. Robinson, R. H. Stokes, Trans. Faraday Soc., 45, 612 (1949).
R табл. 148
1. G. Akerlof, J. Am. Chem. Soc., 57, 1196 (1935).
2. M. Randall, C. F. F a i 1 e y, Chem. Rev., 4, 271 (1927).
3. Glass tone, A. P о u n d, J. Chem. Soc., 127, 2660 (1925); S. G 1 a s s t о n e,
A. Dimond, D. W. Pound, J. Chem. Soc., 128, 2935 (1926).
4. G. Akerlof, J. Am. Chem. Soc., 51, 984 (1929).
5. W. В i 1 t z, Z. physik. Chem., 43, 41 (1903); V. Rothmund, там же, 33, 401
(1900).
6. W. Herz, F. H i e b e n t h a 1, Z. Anorg. Chem., 177, 363 (1929).
R табл. 150
1. E. A. Tippetts, R. F. Newton, J. Am. Chem. Soc., 56, 1675 (1934).
R табл. 151
1. R. A. Robinson, R. H. Stokes, Trans. Faraday Soc., 36, 733 (1940).
2. R. A. Robinson, Trans. Faraday Soc., 36, 735 (1940).
3. R. A. Robinson, R. H. Stokes, Trans. Faraday Soc., 36, 1137 (1940).
4. R. A. Robinson, Trans. Faraday Soc., 36, 1135 (1940).
5. T. Shedlovsky, D. A. Maclnnes, J. Am. Chem. Soc., 59, 503.
6. R. H. Stokes, R. A. Robinson, Trans. Faraday Soc., 37, 419 (1941).
R табл. 152
1. H. S. Harned, M. E. Fitzgerald, J. Am. Chem. Soc., 58, 2624 (1936).
2. R. G. Bates, J. Am. Chem. Soc., 61, 308 (1939).
R табл. 153
i. R. A. Robinson, R. H. Stokes, Trans. Faraday Soc., 36, 740 (1940).
2. R. G. Bates, J. Am. Chem. Soc., 60, 2983 (1938).
3. H. N. Parton, J. W. Mitchell, Trans. Faraday Soc., 35, 758 (1938).
R табл. 154
1. W. R. Carmody, J. Am. Chem. Soc., 51, 2905 (1929).
2. R. A. Robinson, R. H. Stokes, Trans. Faraday Soc., 36, 740 (1940).
3. H. N. Parton, J. W. Mitchell, Trans, Faraday Soc., 35, 758 (1939).
4. R. G. Bates, J. Am. Ctem, Soc., 60, 2983 (1938).
552
ПРИЛОЖЕНИЕ А
5. Н. S. Harned, М. Е. Fitzgerald, J. Am. Chem. Son., 58, 2624 (1936).
6. R. G. Bates, J. Am. Chem. Soc., 61, 308 (1939.)
7. R. G. Bates, W.C. V о s b u r g h, J. Am. Chem. Soc., 59, 1583 (1937).
8. R. A. Robinson, Trans. Faraday Soc., 36, 1135 (1940).
9. R. G. В a t e s, J. Am. Chem. Soc., 63, 399 (1941).
К табл. 155
1. R. A. Robinson, R. H. Stokes, Trans. Faraday Soc., 36, 740 (1940).
2. R. G. Bates, J. Am. Chem. Soc., 60, 2983 (1938).
К табл. 156
1. A. L. Robinson, W. E. W a 1 1 a c e, Chem. Rev., 30, 195 (1942).
К табл. 157
1. С. M. Mason, J. Am. Chem. Soc., 60, 1638 (1938); 63, 220 (1941).
К табл. 158
1. H. S. Harned, W. J. Hamer, J. Am. Chem. Soc., 57, 27 (1935).
К табл. 159
1. H. S. Harned, W. J.
Hamer, J. Am. Chem. Soc., 57, 27 (1935).
К табл. 160
1. W. J. Hamer, J.
Am. Chem. Soc., 57, 662 (1935).
К табл. 161
1. H. S.
2. H. S.
3. H. S.
4. H. S.
5. H. S.
6. H. S.
7. H. S.
8. H. S.
9. С. M.
10. J. Е.
Harned,
Harned,
Harned,
H a r n e d,
Harned,
H a r n e d,
H a r n e d,
H a r n e d,
Al a s о n,
V a n c e, J.
W. J. Hamer, J. Am. Chem. Soc., 55, 2194 (1933).
G. E. Mannweiley, J. Am. Chem. Soc., 57, 1873 (1935).
H. R. С о p s о n, J. Am. Chem. Soc., 55, 2296 (1933).
C. G. Geary, J. Am. Chem. Soc., 59, 2032 (1937).
W. J. Hamer, J. Am. Chem. Soc., 55, 4496 (1933).
J. G. D о n e 1 s о n, J. Am. Chem. Soc., 59, 1280 (1937).
О. E. S c h u p p, Jr. , J. Am. Chem. Soc., 52, 3892 (1930).
С. M. Mason, J. Am. Chem. Soc., 53, 3377 (1931).
D. B. Kellam, J. Phys. Chem., 38, 689 (1934).
Am. Chem. Soc., 55, 2729 (1933).
К табл. 162
1. G. А к e г 1 6 f, J. W. T e a r e, J. Am. Chem. Soc., 59, 1855 (1937),
К табл. 163
1. H. S. Harned, W. J. Hamer, J. Am. Chem. Soc., 55, 2194 (1933).
2. H. S. Harned, G. E. M annweiler, J. Am. Chem. Soc., 57, 1873 (1933).
3. H. S. Harned, H. R. С о p s о n, J. Am. Chem. Soc., 55, 2206 (1933).
4. H. S. Harned, W. G. Hamer, J. Am. Chem. Soc., 55, 4496 (1933).
5. H. S. Harned, J. G. D о n e 1 s о n, J. Am. Chem. Soc., 59, 1280 (1937).
6. H. S. Harned, C. G. Geary, J. Am. Chem. Soc., 59, 2032 (1937),
7. H. S. Harned, О. E. S c h u p p, Jr., J. Am. Chem. Soc., 52, 3892 (1930).
8. J. E. Vance, J. Am. Chem. Soc., 55, 2729 (1933).
9. G. А к e r 1 о f, J. Am. Chem. Soc., 48, 1160 (1926).
К табл. 164
1.
2.
3.
4.
5.
6.
7.
H.
H.
H. s.
H. s.
D. D.
L. F.
a) L. F.
8.
N. D. E m b r e e, J. Am. Chem. Soc., 56, 1042 (1934).
R. W. Ehlers, J. Am. Chem. Soc., 55, 652 (1933).
R. W. Ehlers, J. Am. Chem. Soc., 55, 2379 (1933).
R. 0. Sutherland, J. Am. Chem. Soc., 56, 2039 (1934).
J. Am. Chem. Soc., 56, 314 (1934).
Nims' P.K. Smith, J. BioLChem., 113, 145 (1936).
1 e t e
S. R.
a) H. S.
m e r,
S.
S.
H
H
H
H
a r n e d,
a r n e d,
a r n e d,
ar n ed,
Wright,
N i m s, J. Am. Chem. Soc., 58, 987 (1936); б) I. M. Klotz, C. R. Sin g-
r г у, диссертация, University of Chicago, 1940; в) H.S. Harned,
Scholes, Jr., J. Am. Chem. Soc., 63, 1706 (1941).
H a r n e d, L. D. F a 1 1 о n, J. Am. Chem. Soc., 61, 3111 (1939); 6) W. J. H a-
J. О. В u r t о n, S. F. Acree, Вur.oStandard J. Research, 24, 292 (1940).
ПРИЛОЖЕНИЕ А
553-
9. L. F. N i m s, J. Am. Chem. Soc., 56, 1110 (1934).
10. L. F. N i m s, J. Am. Chem. Soc., 55, 1946 (1933).
11. В. B. Owen, J. Am. Chem. Soc., 56, 1695 (1934); 56, 2785 (1934).
12. В. B. Owen, J. Am. Chem. Soc., 56, 24 (1934),
13. L. F. Nims, P. K. Smith, J. Biol. Chem., 101, 401 (1933).
14. H. V. T а г t а г, H. H. C a r r e t s о n, J. Am. Chem. Soc., 63, 808 (1941).
К табл. 165
1. H. S. H a r n e d, L. D. Fallon, J. Am. Chem. Soc., 61, 2374 (1939).
2. H. S. Harned, R. S. Done, J. Am. Chem. Soc., 63, 2579 (1941),
3. a) H. S. Harned, G. L. К a z a n j i a n, J. Am. Chem. Soc., 58, 1912 (1936);
H. S. Harned, L. D. F a 1 1 о n, там же, 61, 2377 (1939); б) H. S. H ar n ed,
J. Phys. Chem., 43, 275 (1938).
4. H. S. Harned, T. R. D e d e 1 1, J. Am. Chem. Soc., 63, 3308 (1941).
5. H. S. Harned, N. D, E m b r e e, J. Am. Chem. Soc., 57, 1669 (1935).
6. a) H. S. Harned, С. M. Birdsall, J. Am. Chem. Soc., 65, 54 (1943); 6) 65,
1117 (1943).
К табл. 166
1. P. К. Smith, A. C. Taylor, E. R. B. Smith, J. Biol. Chem., 122, 109
(1937).
ПРИЛОЖЕНИЕ Б
§ 1. Диэлектрическая постоянная воды.
Перерасчет теоретических предельных коэффициентов наклона
и функций, в которые входит диэлектрическая
постоянная воды. Исправление табл. 1, 7, 11, 13.
Таблицы теоретических предельных коэффициентов наклона
для парциальных молярных объемов, расширяемостей и сжимаемостей.
Функция g(x)
Во время работы над настоящей монографией при вычислении теоретиче-
ских предельных коэффициентов наклона и других величин, в которые входит
диэлектрическая постоянная воды D, мы базировались на результатах изме-
рений Ваймена [1], выражаемых уравнением (1) гл. V:
1) = 78,54 [1-4,6 10~3 (i - 25) + 8,8 • IO-8 (Z — 25)2].
Когда эта работа была почти закончена, Ваймен и Ингэлс [2] внесли
в результаты, полученные Вайменом, поправку на тепловое расширение
резонатора, который им применялся, и предложили, новое уравнение:
71 = 78,54[1-4,579 • 10~3(Z-25) +11,9-- 10-e(i-25)2 + 28-l(Te(i-25)3], (1)
выражающее исправленные результаты. Значения D, получаемые с помощью
этих двух уравнений, отличаются друг от друга меньше чем на 0,25%
в интервале температур 0 — 50° и не больше чем на 1% при 100°. Это раз-
личие—того же порядка величины, что и расхождения между значениями
Ваймена и значениями Дрэйка, Пирса и Доу [3], которые выражаются
уравнением1 2
D = 78,57 [1 - 4,61 • 10-3 (i - 25) + 15,5 10~8 (i - 25)2]. (2)
Таким образом, ошибка в определении абсолютного значения диэлектри-
ческой постоянной, во всяком случае, не меньше, чем расхождения между
результатами, полученными с помощью приведенных выше уравнений, и
представлялось нецелесообразным производить перерасчет всех значений
предельных коэффициентов наклона. Хотя за последнее время не появились
новые экспериментальные данные, под влиянием которых наша точка зре-
ния относительно неопределенности абсолютных значений D могла бы
измениться, большинство физико-химиков, повидимому, предпочитает поль-
зоваться уравнением (1). Это обстоятельство, а также исправление основных
физических констант N, г, к и др. Бир джем [4]2 побудило нас вычислить
заново все теоретические предельные коэффициенты наклона на основе
уравнения (1) и исправленных значений, приведенных в табл. 168.
При этом изменение тех коэффициентов наклона, в которые входит
только диэлектрическая постоянная, а не ее производные, составляет около
1 Это уравнение применимо в интервале температур 10—604
2 Ср. табл. 1 и табл. 168.
ПРИЛОЖЕНИЕ Б
555
Таблица 168
Основные физические константы [1]
Обозначение Константа Численное значение
S F То R Я к кал15 Заряд электрона Число Фарадея » » » » Объем идеального газа (0° С, норм, атмосфера) Температура таяния льда (абс. шкала) 0° С (1 атм) Газовая постоянная » » Число Авогадро (химическая шкала) Газовая постоянная, рассчитанная на одну молекулу Нормальная атмосфера Литр Малая калория » » 4,8025х- 10“10 абс. эл. ст. ед. 965012 межд. кулон • г-экв~1 230605 «ал15 • в-1 г-экв^1 2,89247 • 1014 абс. эл. ст. ед. • г-жв~1 22414,6 cms • моль~1 273,16°К 8,31436 • 107 эрг • град~1 • моль"1 1,98646г кал15 град~г • моль"1 6,02283 • 1023 моль'1 1,380474 • 10-1в эрг • град'1 1,013246 • 10е дин • см“2 1000,028 с.и3 4,1855 абс. джоуля 4,1847 межд. джоуля
0,5%, и это не оказывает сколько-нибудь существенного влияния на вели-
чины, которые получаются путем экстраполяции — Е°, lg у±, A0, Kw и др.
С другой стороны, те коэффициенты наклона, в которые входит первая или
вторая производная диэлектрической постоянной (§(Н), §(ср)> §(У> и др.), меня-
ются весьма значительно. Таким образом, вычисленные заново значения §(СР)
и §(у) в табл. 169 отличаются от соответствующих значений, приведенных
в гл. VIII, примерно на 30%. Следует отметить, что это значительное
отклонение (30%) является не столько мерой «ошибочности» прежних зна-
чений коэффициентов наклона, сколько показателем большой чувствитель-
ности значений этих коэффициентов наклона к ошибкам в определении
производных D. Новые значения столь же чувствительны к этим ошибкам,
как и старые, и решить вопрос о степени их точности можно лишь путем
тщательных и точных экспериментальных определений зависимости диэлек-
трической постоянной воды от температуры и давления.
В числе теоретических величин, приведенных в табл. 169, встречается
несколько функций, которые были впервые определены лишь в последнее
время. Подробности вычислений содержатся в статье Оуэна и,Бринкли [5];
ниже приведены данные о тех свойствах воды, которые необходимы для
этих вычислений. Область температур, для которых определены значения
а*, Р*, §(Н) и 5<с ), значительно расширена.
Коэффициент расширения воды а определялся с помощью уравнения
Тилтона и Тейлора [6]:
(г — 3,9863)у t + 288,9414%
J 508929,2 + 68,12963) ’ . ''
выражающего опытные данные Шаппюи [7] для области температур 0 — 42°.
Значения а в интервале температур 45 — 85° основаны на данных Уайта [8]
и Смита [9]. Первая и вторая производные D по температуре были полу-
чены из уравнения (1). Значения Р и его производных по температуре и
Таблица 169
Теоретические значения коэффициентов для водных растворов 1,1 -электролитов :)
t, °C S(/) о А'/а S(V) -W(V)l°a S(H) h . S(Cp) S(K) S(E) ft*
0 0,4883 0,3241 3,700 0,1189 509,2 57,45 -0,0222 6,520 0,00165 -0,0050 0,2195 29,61
5 0,4921 0,3249 3,684 0,1186 543,4 ' 58,25 +0,0051 7,123 0,00159 -0,0015 0,2212 35,07
10 0,4960 0,3258 3,685 0,1190 580,4 59,56 0,0275 7,657 0,00155 +0,0016 0,2230 40,77
15 0,5002 0,3267 3,701 0,1199 620,2 61,30 -0,0462 8,164 0,00152 0,0045 0,2249 46,78
20 0,5046 0,3276 3,732 0,1212 662,6 63,40 0,0619 8,674 0,00151 0,0071 0,2268 53,24
25 0,5091 0,3286 3,776 0,1230 707,7 65,86 0,0752 9,194 0,00151 0,0096 0,2289 60,19
30 0,5139 0,3297 3,833 0,1252 755,5 68,64 0,0866 9,731 0,00151 0,0120 0,2311 67,51
35 0,5189 0,3307 3,902 0,1279 806,2 71,70 0,0966 10,300 0,00153 0,0143 0,2333 75,21
40 0,5241 0,3318 3,982 0,1310 860,0 75,07 0,1054 10,922 0,00155 0,0166 0,2356 83,21
45 0,5295 0,3330 4,075 0,1345 917,1 78,78 0,1130 11,606 0,00158- 0,0189 0,2381 91,41
50 0,5351 0,3341 4,180 0,1385 978,1 82,87 0,1196 12,365 0,00162 0,0212 0,2406 99,72
55 • 0,5410 0,3353 4,297 0,1428 1043,2 87,33 0,1255 13,208 0,00167 0,0236 0,2432 108,37
60 0,5470 0,3366 4,427 0,1477 1112,8 92,24 0,1306 14,142 0,00172 0,0260 0,2460 117,41
65 0,5534 0,3379 4,569 0,1530 1187,6 97,63 0,1350 15,185 0,00179 ' 0,0285 0,2488 126,79
' 70 0,5599 0,3392 4,725 0,1589 1268,1 103,46 0,1389 16,354 0,00186 0,0312 0,2517 136,58
75 0,5668 0,3406 4,895 0,1655 1355,2 110,11 0,1422 17,673 0,00195 0,0339 0,2548 146,68
80 0,5739 0,3420 5,079 0,1722 1449,4 117,34 0,1449 19,169 0,00204 0,0368 0,2580 157,03
85 0,5813 0,3435 5,279 0,1797 1551,8 125,35 0,1472 20,851 0,00215 0,0398 0,2614 167,71
90 0,5891 0,3450 5,495 0,1879 — — — — 0,00226 — 0,2649 178,61
95 0,5972 0,3466 5,728 0,1968 — — — — 0,00239 — 0,2685 189,74
100 0,6056 0,3482 5,979 0,2064 — — — — 0,00254 — 0,2723 201,04
18 0,5028 0,3273 3,718 0,1206 645,3 62,54 0,0559 8,469 0,00151 0,0061 0,2261 50,59
Фактор w' w" w’4/2 w'w’v/2 w'^12 w'w"^/2 1 w,'il2 w'v/2 w,'<l2 w’Q w*
1) Для электролитов с более высоким типом валентности следует умножать цифры каждого столбца на фактор валентности, приведенный в последней
строчке каждого столбца.
ПРИЛОЖЕНИЕ Б
557
давлению были найдены с помощью уравнения Тэйта [уравнение (118)
гл. VIII], которое в случае воды имеет следующий вид:
₽ = 7вТр--
Зависимость В от температуры выражается уравнением [10]1
В = 2996,0 + 7,5554 (t - 25) - 0,17814 (i - 25)2 + 0,000608 (t - 25)3. (5)
Производные D по давлению были получены с помощью эмпирического
уравнения Оуэна и Бринкли [11]2
1 / dD\ 0,1754
Т>Ьр)=Ър-
В настоящее время мы не знаем, насколько точно уравнение (6) выра-
жает зависимость диэлектрической постоянной воды от давления при тем-
пературах, отличных от 20°, однако поскольку оно является единственным
уравнением, которым мы располагаем, и, кроме того, оно было тщательно
проверено с применением других жидкостей, мы воспользовались этим
уравнением для определения §<у), §<к> и для всех температур, кото-
рые встречаются в таблице. В § 4 будут рассмотрены экспериментальные
доказательства того, что значения коэффициента наклона 5<у), повидимому,
определены правильно с точностью до двух значащих цифр при комнатной
температуре. Слабая зависимость от температуры и изменение знака
S(E) при низких температурах также подтверждаются опытными данными
[12, 13], однако изученные в этих исследованиях концентрации слишком
высоки, чтобы можно было проверить правильность численных значений этих
теоретических коэффициентов наклона.
Таблица 170
Функция g(x). Дополнительные значения к табл. 17
X 9 (х) X В (х) X а (х)
2,6 0,11721 3,2 0,10367 3,8 0,09272
2,8 0,11236 3,4 0,09977 4,0 0,08952
3,0 0,10786 3,6 0,09612 4,2 0,08656
§ 2. Электропроводность и числа переноса.
Дополнения к табл. 26, 27 и 29
Проверяя справедливость уравнения (4) гл. VI, Ланге [14] воспользо-
вался значением теоретического предельного коэффициента наклона 5(Д) и
показал, что уравнение
Д = Д°-—5(Д) |/ с + Вс (7)
хорошо выражает опытные данные для 560 сильных электролитов в различ-
ных растворителях при концентрациях вплоть до 0,05 М, а в некоторых
случаях и до 0,10 М. Он обнаружил также, что параметр В может быть
выражен следующим уравнением:
в=3-^-С[42’--1]а». (8)
1 В и Р выражены в барах.
2 Вопрос о вычислении множителя 0,1754 разобран в статье Оуэна и Бринкли [51.
558
ПРИЛОЖЕНИЕ Б
Таблица 171
Сопоставление вычисленных значений Л° и ZJ с экспериментальными значениями ’),
из которых они вычислены
Электро- лит Обозна- чение величины Температура, сС Ссылки на лите- ратуру
5 J 15 25 35 45 55
НС1 Д° 297,53 362,16 426,20 489,11 550,34 609,38 2)
297,57 362,07 426,24 489,15 550,30 609,47 3)
426,16 [3]
0,8403 0,8304 0,8209 0,8115 0,8021 0,7926 2)
0,842 0,831 0,821 0,811 0,801 fill
0,8210 [121
LiCl де 70,28 91,62 114,99 140,17 166,94 195,15 2)
70,30 91,60 114,99 140,18 166,92 195,14 [Я
115,03 [31
114,95 [5J
т" 0,3240 0,3296 0,3360 0,3422 0,3476 0,3523 2)
0,3368 [12]
NaCl А» 77,81 101,14 126,50 153,74 182,69 213,23 2)
77,84 126,49 213,28 [41
101,18 126,45 153,75 182,65 [6]
126,45 [7]
грО 0,3894 0,3927 0,3964 0,4002 0,4039 0,4072 2)
0,3929 0,3962 0,4002 0,4039 [131
0,3963 [12]
KCl Л» 94,23 121,03 149,85 180,41 212,43 245,67 2)
94,26 149,88 245,69 [81
121,07 149,85 180,42 212,41 [6[
149,86 [7]
грО 0,4959 0,4925 0,4905 0,4889 0,4873 0,4855 2)
0,4928 0,4905 0,4889 0,4872 [14]
0,4906 [12]
КВг А» 95,99 122,81 151,67 182,23 214,17 247,15 2)
96,00 151,68 247,15 [8]
122,81 151,64 182,24 214,17 [61
151,63 [91
гр 0 0,4868 0,4854 0,4846 0,4840 0,4834 0,4826 2)
0,4847 [151
KJ А» 95,32 121,83 150,35 180,56 212,13 244,74 2)
95,32 121,83 150,34 180,60 212,13 244,73 [8]
150,38 4)
грО 0,4902 0,4893 0,4889 0,4885 0,4880 0,4873 2)
0,4887 [16]
1) Все величины отнесены к стандартным значениям Джонса и Брэдшоу [1].
2) Данные, приведенные в соответствующих строках, рассчитаны с помощью уравнения (9)
и значений параметров, проведенных в табл. 172.
3) Данные, взятые нз статьи Оуэна и сьютова [2], снова экстраполированы с помощью урав-
нения (11) гл. VI.
4) Л. Г. Лонгсворт, частное сообщение, приведенное в статье Ганнинга и Гордона [10].
ПРИЛОЖЕНИЕ Б
559
Коэффициент С отражает специфические особенности различных растворите-
лей и равен 0,19 в случае воды.
В течение последнего десятилетия были тщательно измерены электро-
проводности и числа переноса многих солей и соляной кислоты в интервале
температур 40 — 60°. Результаты этих измерений настолько многочисленны,,
что мы не будем их приводить для различных температур и концентраций
и ограничимся лишь значениями при бесконечном разведении (табл. 171).
Соответствующие значения для концентраций вплоть до 0,01 — 0,02 н. можно-
найти в оригинальных статьях, ссылки на которые приведены в таблице.
На основании этих предельных значений, а также соответствующих значе-
ний при 25° для тех же электролитов и йодистого натрия, приведенных
в табл. 119 и 120, можно составить весьма точную сводку «предпочтитель-
ных» значений предельных подвижностей семи простых ионов для интерва-
ла температур 5 — 55°. Эти значения и их зависимость от температуры
могут быть выражены с помощью кубического уравнения
Х° = Х(°25’) + a (Z - 25) + b (£ - 25)2 + с (t - 25)3, (9}
параметры которого приведены в табл. 172.
Значения А° и Т+, вычисленные с помощью уравнения (9) и парамет-
ров из табл. 172, сопоставлены в табл. 171 с опытными значениями.
Разность между вычисленными и опытными значениями А° составляет
в среднем меньше 0,02%, и лишь в случае бромистого калия при 25°"
Таблица 172
Параметры уравнения (9), выражающего зависимость
предельных подвижностей ионов от температуры
в интервале 5—55°
Иои :0 x(25°) a b- io2 c • io4
Н* 349,85 4,81595 -1,03125 -0,7670
Li* 38,64 0,88986 +0,44075 -0,2042
Na* 50,15 1,09160 0,47150 -0,1150
К* 73,50 1,43262 0,40563 -0,3183
Cl' 76,35 1,54037 0,46500 -0,1285
Br~ 78,17 1,54370 0,44700 -0,2300
J- 76,85 1,50893 0,43750 -0,2170
(см. табл. 119) превышает 0,04%. Поэтому при составлении сводки значе-
ний подвижностей величина Д° для этой соли, равная 151,9, была отбро-
шена. Если учесть, что экспериментальные значения Т+ для соляной
кислоты (ссылка [11] в табл. 171) выражены с точностью лишь до трех
значащих цифр, совпадение между вычисленными и опытными значениями
можно считать удовлетворительным. Следует отметить, что значение 0,4837
(ссылка [16] в табл. 171) для бромистого калия было заменено значением
0,4847 (ссылка [15] в табл. 171) для того, чтобы получить наилучшее сов-
падение между значениями подвижностей.
При изучении подвижностей ионов был обнаружен весьма интересный
результат, который заключается в изменении относительной последователь-
ности величин предельных подвижностей ионов хлора и иода при увеличе-
нии температуры. Так, при 5° Хщ равно 47,51 и меньше которое равно
48,60. При 55° Ащ равно 126,40, и уже больше X®, равного 125,47.
560
ПРИЛОЖЕН И ЕВ
Фуосс и Шидловский [15] недавно обнаружили, что при определении
константы диссоциации с помощью той или иной из предложенных ими
функций1 F (Z) и S (Z) получаются различные значения К. Если обозна-
чить эти значения через К(ру и K(S)> то можно вычислить разность их
•обратных величин с помощью уравнения
(Ю)
Для большинства слабых электролитов (К < 10'5) эта разность очень мала,
однако в случае умеренно сильных электролитов (К > 10~3 *) она может ока-
заться заметной. Так, например, в случае сернокислого цинка путем
экстраполяции, аналогичной приведенной на рис. 27, получаются значе-
ния K(S) = 0,0049 и К(ру — 0,0052, которые отличаются друг от друга
на 6%.
§ 3. Коэффициенты диффузии хлористого калия при концентрациях
О—0,5.1/ и хлористого кальция при концентрациях 0,002—0,005 М
в водных растворах
С помощью усовершенствованного в последнее время метода определе-
ния дифференциальных коэффициентов диффузии электролитов путем изме-
рения электропроводности [16, 17а]2 были впервые получены результаты,
точность которых достаточно велика, чтобы их можно было использовать
для проверки теории Онзагера и Фуосса [18]. Во втором столбце табл. 173
приведены экспериментальные значения коэффициента диффузии хлористого
калия при 25° для концентраций, выраженных в молярностях и приведен-
ных в первом столбце этой таблицы.
Если использовать значения констант, приведенных в табл. 168, то
вместо численных' значений 16,632; 1,074; 22,00 и 9,18 в уравнениях (57)
и (58) гл. VI получаются соответственно значения 16,629; 1,0748; 22,148
и 9,304. После подстановки этих значений, а также других величин, при-
веденных в нижней части таблицы, уравнения (57), (58), (60) и (61) гл. VI
приобретают следующий вид:
Я == 4,958 • 1013 (1 + , (И)
. • 1020 —40,253 — 18,96С? (1,249/7), (12)
(1+с^)=1-------------+0,0490с-сф(</), (13)
\ 9с } (1 +1,249 /с )2 Y V v ’
, . 0,00959 с —0,00348 cs/2 ....
C’i> (d) = —-------------------Гт- . (14)
’ х ' 0,99707 + 0,00959 с—0,00232cs/2 v '
Все эти уравнения были использованы для вычисления значений, при-
веденных в третьем столбце таблицы. Функция ср (1,249/с) определялась
путем интерполяции данных, приведенных в табл. 14.
Опытные и теоретические значения хорошо совпадают друг с другом
во всем интервале концентраций. В случае значений, отвечающих концен-
1 См. гл. VII, стр. 185—189.
2 См. также статью Харнеда [176], в которой помещен подробный обзор работ, содер-
жащих количественное рассмотрение вопроса о диффузии в растворах электролитов.
В статье Лонгеворта [17в] приводится обзор экспериментальных методов. Обе указанные
статьи содержат подробную библиографию по данному, вопросу.
ПРИЛОЖЕНИЕ Б
561
Таблица 173
Экспериментальные и вычисленные значения
дифференциального коэффициента диффузии
хлористого калия в водном растворе при 25°
С XV 1(Р X Ю5 * *
Эксперим. Вычисл. 1)
0,00000 — (1,9958) (1,996)
0,00125 1,961 1,960 1,997
0,00194 1,954 1,953 1,997
0,00325 1,943 1,943 1,996
0,00585 1,931 1,929 1,998
0,00704 1,924 1,924 1,996
0,00980 1,918 1,915 1,999
0,01261 1,908 1,907 1,997
0,02654 1,879 1,883 1,992
0,03992 1,877 1,870 2,003
0,04620 1,872 1,866 2,002
0,05450 1,860 1,861 1,995-
0,06074 1,856 1,858 1,994
0,1298 1,838 1,840 1,994
. 0,3323 1,842 1,839 1,999
0,5276 1,852 1,853 1,995
Среднее 1,997
1) Численные значения величин, применявшихся для теоре-
тических вычислений согласно уравнениям (11)—(14) : Т=273,16;
I) = 78,54; т,0 == 8,949 10~3; Л° = 73,52; л“ = 76,34; а = 3,8;
2В = 0,0213; §(^= 0,5092; d (25°) = 0,99707 + 0,04811с—
- 0,00232 С8/®-
трациям меньше 0,05 н., правильность теоретических значений не подлежит
сомнению, однако совпадение теоретических и опытных данных при более
высоких, концентрациях является неожиданным. Возможно, что это совпаде-
ние обусловлено удачным выбором соли и температуры опыта, поскольку
при 25° относительная вязкость водного раствора хлористого калия меняется
меньше чем на 0,3% при изменении концентраций от 0 до 0,5 н.
Эти результаты представляют собой первые экспериментальные данные,
которые весьма точно подтверждают справедливость предельного уравнения
Нернста для коэффициента диффузии электролита при бесконечном раз-
ведении. Этот вывод наглядно подтверждается значениями приведенными
в последнем столбце таблицы; эти значения получены путем сложения
теоретических разностей (Vo— -^теор. с опытными значениями 3 оп. Прекрас-
ное совпадение 2>'й с предельным значением в особенности при концен-
трациях меньше 0,01 н., служит убедительным доказательством справедли-
вости предельного уравнения Нернста для этого 1,1-валентного электролита.
Приведенные на рис. 167 кривые, соответствующие 20, 25 и 30°,
иллюстрируют теоретические вычисления. Кривая 1 отвечает предельному
уравнению (62) гл. VI, которое после вычисления при 25° с помощью
уравнения (63) гл. VI приобретает вид
3 • 108= 1,9958-1,170J/7. (15)
36 г. харнед, Б. Оуэн
562
ПРИЛОЖЕНИЕ В
Кривая 3 соответствует результатам, которые получаются при отбрасывании
электрофоретической составляющей, причем этой кривой отвечает уравнение
.[10® = 1,9958 + . (16)
Кривая 2 соответствует уравнениям (11) —(14), и, следовательно, разность
между кривыми 2 и 3 характеризует влияние электрофоретической состав-
ляющей. Результаты, взятые из табл. 173, изображены кружками, и, судя
по расположению этих кружков, для правильного выражения эксперимен-
Рис. 167. Коэффициент диффузии хлористого
калия в воде.
Q—кондуктометрический метод; х—метод ячейки
с диафрагмой; Д—данные Коэна и Бруинса; □—дан-
ные Ламма.
тальных данных следует учиты-
вать электрофоретическую состав-
ляющую. Крестиками обозначены
значения, вычисленные Гордо-
ном [19] из результатов выпол-
ненных им измерений с помощью
ячейки с диафрагмой, а также
из данных Мак-Бэна и Доусо-
на [20], а также Хартли и Ран-
никса [21]. Результаты этих из-
мерений [176] были использо-
ваны Гордоном, который исполь-
зовал для калибровки ячейки
результаты, полученные кондук-
тометрическим методом для кон-
центраций ниже 0,01 н. (табл.
173). При низких концентрациях
совпадение результатов, получен-
ных обоими методами, является
хорошим, однако при более вы-
соких концентрациях результа-
ты, которые дает метод ячейки
с диафрагмой, несколько ниже
результатов, полученных методом
электропроводности. Данные Коэ-
на и Бруинса [22], полученные
по методу анализа слоев, а также
данные Ламма [23], полученные
по его методу шкалы, также изо-
бражены на рис. 167. Поскольку принципы описанных методов определения
коэффициентов диффузии весьма различны, можно считать совпадение
результатов, полученных различными методами, удовлетворительным.
Хорошее совпадение этих экспериментальных данных с теоретическими
значениями не может служить доказательством применимости теории к элек-
тролитам всех типов. На рис. 168 представлены результаты Харнеда и
Леви [24], которые определяли коэффициент диффузии хлористого кальция
в водных растворах при низких концентрациях при 25°. Как видно из.
рисунка, уже при концентрациях, близких к 0,001 М, наблюдаются значи-
тельные отклонения опытных значений от теоретических. Этот результат
можно сравнить с аналогичным отклонением от теории, наблюдающемся в слу-
чае числа переноса иона кальция, которое было продемонстрировано на рис. 17.
Описанные выше результаты имеют существенное значение, поскольку
они наводят на мысль, что теория Онзагера и Фуосса, которая столь
успешно описывает зависимость коэффициента диффузии хлористого калия
от концентрации, является либо неправильной, либо недостаточно точной,
в случае электролитов с более высоким типом валентности.
ПРИЛОЖЕНИЕ Б
563
§ 4. Экстраполяция кажущихся молярных объемов и теплот разведения.
Дополнение к табл. 61
В гл. VIII было указано на ошибки, которые допускаются при экстра-
поляции кривых, выражающих зависимость кажущихся молярных величин
от с1/2, а также было показано, что если использовать параметр а, но не
вводить член, включающий с в первой степени (стр. 222, 223), получаются
неудовлетворительные результа-
ты. С другой стороны, если ис-
пользовать член, содержащий с,
но не учитывать параметр а, по-
видимому, удается добиться при-
емлемых результатов при экстра-
поляции кажущихся молярных
объемов. Как было показано
(стр. 244,245) в случае хлори-
стого калия, бромистого натрия
и хлористого стронция графики
зависимости -------^-S^c1/2 J
от с являются почти прямоли-
нейными, если принять 2/3
равным 1,9 для 1,1-валентных
солей, и 1,9 (3)3/2, т. е. 9,9,—
для хлористого стронция. К со-
жалению, при использовании
этих численных значений, пре-
дельногб коэффициента наклона
не удается получить удовлетво-
рительных результатов в слу-
чае других сильных электро-
Р и с. 168. Коэффициент диффузии хлористого
кальция в воде при 25°.
Верхняя кривая—теоретическая; Q—кондуктометри-
ческий метод; •—метод ячейки с диафрагмой.
литов таких же типов валент-
ности, и согласно нашим вы-
числениям, результаты которых
приведены в табл. 169, теорети-
ческое значение 2/3 при 25°
должно составлять примерно
Оуэн и Бринкли [5] недавно
2,5, а не 1,9.
показали, что при использовании пара-
метра а и дополнительного члена, содержащего с, при линейной экстрапо-
ляции 1 получаются значения, которые совпадают с теоретическими значе-
ниями §>(у) и §(Н). Как было показано, уравнения для и <ря, соответ-
ствующие2 уравнениям (87) и (77) гл. III для V2 и L2, имеют следующий
вид:
—S(V)C1/2 +y^(V)C
(17)
1 В функциях, которые используются для экстраполяции, не были учтены допол-
нительные члены (стр. 52—54), и эти функции применялись только к <рг и Чн для 1,1-элек-
тролитов. К электролитам более высокого валентного типа эти функции могут быть при-
менены лишь после тщательного выяснения влияния дополнительных членов. Кроме того,
неизвестные значения производных д In a/дТ и <? In а/дР приняты равными нулю.
2 При переходе от переменной Г к более удобной переменной с мы заменили
через
а также К с через УС Г и т. д.
(V) в (V) А
36*
.564
ПРИЛОЖЕНИЕ Б
И
= 4 2(н)с1/2 + т ^(Н) с‘
В этих уравнениях влияние параметра а учитывается функциями
о Z 1 <?lnZ> InX/dlnD IX-1 ,,Q4
~дР—т ₽а) — т <19>
и
( 1 SdlnD . 1\ . 1 l^dlnD ,1,1 /9П\
2(*)ЧгТ^Сп^ + г) + тазК-^+г +TaJ • (2°)
Используя уравнения (1) и (4) —(6), получаем отсюда для всех температур
выражения
= 0,3513о (21)
И
= (22>
где значения h при различных температурах приведены в табл. 169. Функ-
ция а, определяемая уравнением (36) гл. IX, приведена в табл. 61. Более
подробные значения а, необходимые для описываемого ниже способа экстра-
поляции, содержатся в табл. 174. Теоретические коэффициенты наклона
5(У) и §(Н) приведены в табл. 169.
ф О данные Круиса; + данные Геффкена и Прайса.
В окончательной форме, удобной для экстраполяции, можно записать
уравнения (17) и (18) в следующем виде:
[?у — }%)2(юс1/2] =¥y + -^--K(V)C (23)
и
[сРн-сРн-4 S(H)S(H)C1/2J =='40j-^;j + -|x(H)c. (24)
В последнем уравнении у'н — кажущееся молярное теплосодержание при
некоторой заданной стандартной концентрации.
На рис. 169 и 170 представлена зависимость левых частей этих уравне-
ний от с. Оба графика являются прямолинейными в пределах точности
экспериментальных данных. При составлении графика, изображенного на
рис. 169, использованы значения для хлористого калия при 25°, полу-
ПРИЛОЖЕНИЕ Б
565
Таблица 174х)
Функция с
° = i [4Н-ж- (44-я:)'1 -2 in (1+х)]
X а X а X в X а
0,00 1,000000 0,50 0,537675 1,00 0,341117 2,00 0,176041
0,01 0,985178 0,51 0,532127 1,02 0,335718 2,05 0,171274
0,02 0,970704 0,52 0,526668 1,04 0,330451 2,10 0,166704
0,03 0,956568 0,53 0,521298 1,06 0,325313 2,15 0,162321
0,04 0,942758 0,54 0,516014 1,08 0,320299 2,20 0,158114
0,05 0,929263 0,55 0,510813 1,10 0,315405 2,25 0,154074
0,06 0,916074 0,56 0,505695 1,12 0,310627 2,30 0,150191
0,07 0,903182 0,57 0,500657 1,14 0,305962 2,35 0,146458
0,08 0,890577 0,58 0,495699 1,16 0,301405 2,40 0,142867
0,09 0,878250 0,59 0,490817 1,18 0,296953 2,45 0,139410
0,10 0,866193 0,60 0,486010 1,20 0,292604 2,50 0,136082
0,11 0,854399 0,61 0,481278 1,22 0,288353 2,55 - 0,132875
0,12 0,842859 0,62 0,476617 •1,24 0,284198 2,60 0,129783
0,13 0,831565 0,63 0,472028 1,26 0,280136 2,65 0,126801
0,14 0,820510 0,64 0,467507 1,28 0,276164 2,70 0,123924
0,15 0,809687 0,65 0,463055 1,30 0,272279 2,75 0,121147
0,16 0,799090 0,66 0,458669 1,32 0,268479 2,80 0,118464
0,17 0,788712 0,67 0,454348 1,34 0,264761 2,85 0,115872
0,18 ’ 0,778547 0,68 0,450091 1,36 0,261123 2,90 0,113367
0,19 0,768589 0,69 0,445896 1,38 0,257562 2,95 0,110945
0,20 0,758832 0,70 0,441763 1,40 0,254077 3,00 0,108601
0,_21 0,749271 0,71 0,437689 1,42 0,250665 3,05 0,106333
0,22 0,739900 0,72 0,433675 1,44 0,247323 3,10 0,104138 •
0,23 0,730714 0,73 0,429718 1,46 0,244051 ' 3,15 0,102011
0,24 0,721707 0,74 0,425817 1,48 0,240846 3,20 0,099951
0,25 0,712876 0,75 0,421972 1,50 0,237705 3,25 0,097955
0,26 0,704216 0,76 0,418182 1,52 0,234628 3,30 0,096019
0,27 0,695721 0,77 0,414444 1,54 0,231613 3,35 0,094142
0,28 0,687388 0,78 0,410759 1,56 0,228658 3,40 0,092320
0,29 0,679212 0,79 0,407125 1,58 0,225761 3,45 0,090553
0,30 0,671189 0,80 0,403542 1,60 0,222921 3,50 0,088837
0,31 0,663316 0,81 0,400008 1,62 0,220136 3,55 0,087170
0,32 0,655588 0,82 0,396522 1,64 0,217405 3,60 0,085551
0^33 0,648002 0,83 0,393083 1,66 0,214727 3,65 0,083979
0,34 0,640554 0,84 0,389692 1,68 0,212099 3,70 0,082450
0,35 0,633241 0,85 0,386346 1,70 0,209521 3,75 0,080963
0,36 0,626060 0,86 0,383045 1,72 0,206992 3,80 0,079518
0,37 0,619007 0,87 0,379788 1,74 0,204510 3,85 0,078112
0^38 0,612078 0,88 0,376574 1,76 0,202073 3,90 0,076743
0,39 0,605272 0,89 0,373403 1,78 0,199682 3,95 0,075412
0,40 0,598585 0,90 0,370273 1,80 0,197335 4,00 0,074115
0,41 0,592014 0,91 0,367185 1,82 0,195030 4,05 0,072853
0,42 0,585557 0,92 0,364137 1,84 0,192766 4,10 0,071623
олз 0,579210 0,93 0,361128 1,86 0,190543 4,15 0,070425
0,44 0,572971 0,94 0,358158 1,88 0,188360 4,20 0,069257
0,45 0,566838 0,95 0,355226 1,90 0,186216 4,25 0,068119
0,46 0,560809 0,96 . 0,352332 1,92 0,184109 4,30 0,067009
О'47 0,554880 0,97 0,349475 1,94 0,182039 4,35 0,065927
0,48 0,549049 0,98 0,346654 1,96 0,180005 4,40 0,064871
О', 49 0,543315 0,99 0,343868 1,98 0,178006 4,45 0,063842
0,50 0,537675 1,00 0,341117 2,00 0,176041 4,50 0,062837
1) Указанная таблица заимствована авторами у С. Р. Бринкли и Р. Ф. Бринкли.
566
ПРИЛОЖЕНИЕ Б
ченные Круисом [25, 26] *, а также Геффкеном и Прайсом [27], а в случае
графика на рис. 170 —значения теплот разведения растворов хлористого
натрия при 25°, измеренные Робинзоном [28]. Значения параметра а, рав-
ного 3,8 для хлористого калия и 4,0 для хлористого натрия, взяты из табл. 89.
Следует отметить, что они были получены независимо от коэффициентов
активности, выражаемых уравнением (32) гл. XII.
На основе коэффициентов наклона прямых, изображенных на рис. 169
и 170, и отрезков, отсекаемых этими прямыми на оси ординат, можно полу-
чить следующие уравнения:
<py = 26,742 + 2,517S(V)cV2 + 0,85c; а = 3,8 (25)
для хлористого калия при 25° и
?ff-?or = 471,8SfH)c‘/2 + 217c; а = 4,0 (26)
для хлористого натрия при 25°. Далее, для обработки этих результатов
можно использовать то обстоятельство, что константы 0,85 и 217 в уравне-
ниях (25) и (26) представляют собой коэффициенты при с в уравнениях (23)
Рис. 170. Относительное кажущееся молярное теплосодержание
хлористого натрия в воде при 25°.
О—данные Робинзона.
и (24). Эти коэффициенты в 2 раза меньше соответствующих коэффициентов
перед последним членом в уравнениях
&(У)с1/2
V2-V°2
1 + А'с1!2
(1+Л'с1/2)2
+ ^(У)С,
(27)
е „ ri/2 W/rr-.c
Т __ s>(H)c , -----------СЮ----- „
Ь2— 1+Л,с1/2 -Г(1 + Л'с1/2)2 + Л<Н> ’
(28)
которые можно получить путем подстановки выражений S(V) = i^(V) (2jv/zf)1/2’
^(v; = Ж’(У) S и K(V) = ^(V) S B уравнение (87) гл. Ill и 5(Н) =
= ^>(H)(Sv/zb1/2> WZ(H) = 5F(ff) ijvyZ; и K(H) в уравнение (77)
гл. III. Поскольку значения теоретических констант 1У(У) и W(H) содержатся
в табл. 169 наряду с предельными коэффициентами наклона, то с помощью
этих уравнений нетрудно вычислить относительные парциальные молярные
величины V2— К” и L2.
1 Приводимые значения получены с помощью интерполяции.
ПРИЛОЖЕНИЕ Б
567
§ 5. Коэффициенты активности. Новые и исправленные данные для
1,1-, 2,1- и 1,2-электролитов
Точные значения коэффициентов активности хлористого натрия, хлори-
стого калия, бромистого калия и хлористого кальция в разбавленных водных
растворах для температур 15 — 45° были получены Гордоном и его сотруд-
никами из данных по числам переноса и электродвижущим силам элементов
с жидкостным соединением с помощью метода Брауна и Мак-Иннеса, опи-
санного в гл. XII, § 1. Полученные результаты можно выразить с помощью
уравнения
lg Y± = — + Вт- (29)
1 + А' у т
Значения параметров Л' и В, а также ссылки на литературные источ-
ники приведены в табл. 175. Посредством уравнения (29) получаются зна-
чения 1g у± при 0,1 М для трех 1,1-электролитов, которые очень хорошо
совпадают с соответствующими значениями, найденными с помощью элемен-
тов без жидкостного соединения и приведенными в таблицах в гл. XII,
Таблица 175
Параметры уравнения (29) для вычисления коэффициентов
активности; применимы в интервале концентрации
от 0 до 0,1 М
, Параметры Температура, 0 С
15 25 35 45
О
Хлористый натрий; а = 4,12 [1а]1)
0,4966 0,5049 0,5141 0,5254
Г>А' 1,343 1,350 1,357 1,364
В 0,022 0,031 0,034 0,033
О Хлористый калий; а = 3,97 [2а]2)
А’ 1,300 1,307 1,314 1,321
В -0,0140 — 0,0075 — 0,0055 —0,004а
О Хлористый кальций; а = 4,575 А [За] 3)
1,7321 1,7615 1,7925
1 А' 2,588 2,600 2,613
В 0,198 0,203 0,185
Бромистый калий [4а]4)
t = 25°; а = 4,ЗА S(/)/<V= 0,5049; А' = 1,420;
______________ В= - 0,014
1) Значения чисел переноса взяты из статьи Олгуда и Гордона [16].
2) Значения чисел переноса взяты из статьи Олгуда, Ле-Роя и Гор-
дона [26J.
з) Значения чисел переноса взяты из статьи Кенена, Мак-Леода
и Гордона [36]. _
4) Значения чисел переноса взяты из статьи Кейена и Гордона [46],
568
ПРИЛОЖЕНИЕ Б
и в приложении А. Коэффициент активности хлористого кальция при кон-
центрации 0,1 М равен 0,518, т. е. значительно отличается от значе-
ния 0,531, приведенного в табл. 151, однако согласуется с исправленными
значениями, приведенными в табл. 176. С помощью этого метода Батлер
и Гордон [29] впервые произвели точное определение коэффициента актив-
ности соли в разбавленных смесях органического растворителя с водой. Они
определили у хлористого натрия в смеси, содержащей 50 молярных про-
центов метилового спирта.
Правильность некоторых значений средних коэффициентов активности
для более высоких концентраций была независимым путем подтверждена
Стоксом [30] с помощью метода, при котором раствор соли при 25° приво-
дится в равновесие с водой, находящейся при'более низкой температуре,
через газовую фазу. Путем точного измерения разности температур находят
температуру воды и определяют ее активность из данных по стандартным
значениям, давлений пара, приведенным в «International Critical Tables».
Результаты, полученные для концентрированных растворов хлоридов натрия
и кальция, а также для гидрата окиси натрия, свидетельствуют о том, что
этот метод является весьма точным.
После 1943 г. Робинзон и Стокс исправили свои старые результаты
и значительно расширили свою работу по изопиестическим измерениям давле-
ния пара растворов 2,1- и 1,2-электролитов. Они получили также резуль-
таты для ряда электролитов при очень высоких концентрациях, на основа-
нии которых они предприняли попытку вычислить коэффициенты активно-
сти этих электролитов, используя в качестве стандартных растворы серной
кислоты и хлористого кальция. В табл. 176 приведены средние коэффи-
циенты активности этих электролитов, а также ряда 1,1-электролитов,
коэффициенты активности которых были определены при высоких концен-
трациях. Из сопоставления с табл. 146 видно, что эти новые значения
коэффициентов активности мало отличаются от прежних в области концентра-
ций, не превышающих 2 М, однако при 4 М это расхождение достигает
примерно 1%.
Табл. 177 содержит новые значения коэффициентов активности 2,1- и 1,2-
электролитов, которые могут служить в качестве дополнительных данных
к табл. 151. В случае хлоридов магния и стронция, а также бромистого
и йодистого магния новые значения коэффициентов активности сильно отли-
чаются от старых, и поэтому они были также включены в таблицу.
В табл. 177 содержатся также коэффициенты активности хлористого, бро-
мистого и йодистого цинка, которые значительно отличаются от соответ-
ствующих значений, приведенных в табл. 154. Все остальные результаты,
приведенные в таблице, относятся к электролитам, для которых эти значе-
ния раньше не были известны.
§ 6. Обзор новых работ по теории электролитов
В обзоре, помещенном в конце гл. XII, было указано, что трудно
ожидать быстрых успехов в области дальнейшего выяснения природы и коли-
чественной трактовки сложных взаимодействий, имеющих место в растворах
электролитов. Новые представления в вопросе об отклонениях от предель-
ного закона Дебая и Гюккеля были развиты Франком [31а] Ч Эти пред-
ставления основаны на модели, предложенной в свое время Дебаем и Пау-
лингом [32] для доказательства независимости предельного закона для
химического потенциала и для коэффициента активности от изменений
1 Вопрос о взаимодействии растворенных веществ с водой получил дальнейшее
.развитие в работах Франка и Эванса [316].
Таблица 176
Средние коэффициенты активности некоторых 1,1-электролитов, серной кислоты, хлористого кальция
и азотнокислого кальция при 25° и высоких концентрациях1)
т NaCl [2] NaOH [3]з) LiCl [4] LiBr [5] LiNOs [6] H2SO4 [7] CaCl2 [8] НСЮ4 [9] Ca (NO3)2 [10]
о,1 0,778 0,776 0,790 0,796 0,788 0,2655 0,518 0,803 0,485
0,2 0,735 0,727 0,757 0,766 0,752 0,2090 0,472 0,778 0,426
0,5 0,681 0,693 0,739 0,753 0,726 0,1557 0,448 0,769 0,363
1,0 0,657 0,679 0,774 0,803 0,743 0,1316 0,500 0,823 0,336
1,5 0,656 0,683 0,838 0,896 0,783 — — 0,923 0,336
2,0 0,668 (0,700) 0,921 1,015 0,835 0,1276 0,792 1,055 0,345
3,0 0,714 0,774 1,156 1,341 0,966 0,1422 1,483 1,448 0,380
4,0 0,783 0,890 1,510 1,897 1,125 0,1700' 2,934 2,08 0,435
5,0 0,874 1,060 2,02 2,74 1,310 0,2081 5,89 3,11 0,507
6,0 0,986 1,280 2,72 3,92 1,515 0,2567 11,11 - 4,76 0,592
7,0 — 1,578 3,71 5,76 1,723 0,3166 18,28 7,44 0,690
8,0 — 1,979 5,10 8,61 1,952 0,386 26,02 11,83 0,801
9,0 — 2,51 6,96 12,92 2,19 0,467 34,20 19,11 0,935
10,0 — 3,18 9,.40 19,92 2,44 0,559 43,0 30,9 1,065
11,0 — 4,04 12,55 31,0 2,69 0,661 '— 50,1 1,184
12,0 — 5,11 16,41 46,3 2,95 0,770 — 80,8 1,311
14,0 — 7,91 26,2 104,7 — 1,017 — 205,0 1,538
16,0 — 11,38 37,9 198,0 — 1,300 — 500,0 1,724
18,0 — 15,15 49,9 331,0 — 1,608 — — 1,917
20,0 — 19,0 62,4 485,0 — 1,940 — — 2,008
22,0 — 22,7 — — — 2,300 — — —
24,0 — 26,1 — — — — — —
26,0 — 29,0 — — — — — — —
29,0 — 33,2 . — — — — — — —
1) Подробные таблицы осмотических коэффициентов и коэффициентов активности приведены в статье Робинзона и Стокса [1].
2) Отнесено к значению 0,700, соответствующему концентрации, равной 2 М.
Таблица 1771)
Средние коэффициенты активности 2,1- и 1,2-электролитов и нитрата тория при 25°
т MgC12 MgBr2 MgJ2 СаВгг Ca J2 STC12 SrBr2 SrJ2 BaJ2 Mg (NO3)2 Sr(NO3)2 Ba (NO3)2 Co (NO3)2
0,1 0,529 0,550 0,580 0,532 0,560 0,511 0,526 0,553 0,542 0,523 0,478 0,428 0,518
0,2 0,489 0,519 0,558 0,492 0,531 0,462 0,483 0,520 0,509 0,481 0,410 0,342 0,471
0,5 0,481 0,545 0,614 0,491 0,561 0,430 0,467 0,536 0,523 0,470 0,329 — 0,445
0,7 0,506 0,599 0,698 0,522 0,614 0,434 0,484 0,578 0,562 0,489 0,302 — 0,455
1,0 0,570 0,723 0,892 0,597 0,741 0,461 0,535 0,680 0,649 0,537 0,275 — 0,490
1,4 0,709 0,975 1,291 0,747 0,992 0,524 0,643 0,885 0,814 0,632 0,253 — 0,563
2,0 1,053 1,614 2,43 1,121 1,640 0,670 0,906 1,407 1,221 0,842 0,232 — 0,726
3,0 2,32 4,26 7,93 2,54 — 1,126 — — — — — — 1,182
4,0 5,54 12,2 29,0 6,28 — 1,977 — — — — — — 1,972
т ZnC12 ZnBr2 ZnJ2 Zn(ClO4)2 Zn(NO3)2 Mg(C104)2 Na2Fu2) Na2Ma3) Rb2SO4 CS2SO4 K2CrO4 Na2CrO4 Th(NO3)4
о,1 0,515 0,547 0,581 0,581 0,531 0,590 0,465 0,430 0,451 0,456 0,456 0,464 0,279
0,2 0,462 0,510 0,559 0,564 0,489 0,578 0,402 0,354 0,374 0,382 0,382 0,394 0,225
0,5 0,394 0,511 0,610 0,629 0,473 0,647 0,335 0,272 0,279 0,291 0,292 0,307 0,189
0,7 0,369 0,528 0,683 0,720 0,489 0,739 0,323 0,250 0,249 0,262 0,263 0,280 0,191
1,0 0,339 0,552 0,800 0,929 0,535 0,946 0,319 0,231 0,219 0,235 0,235 0,253 0,207
1,4 0,309 0,567 0,928 1,386 0,625 1,385 0,323 0,219 0,196 0,214 0,214 0,233 0,246
2,0 0,289 0,572 1,028 2,74 0,817 2,65 0,343 0,213 — — 0,196 0,222 0,326
3,0 0,287 0,598 1,123 9,99 1,363 9,19 — 0,220 — — 0,190 0,236 0,486
4,0 0,307 0,664 1,259 38,8 2,31 — — — — — — 0,285 0,647
1) Стокс [1]. В этой статье указаны все работы, в которых впервые были получены приводимые данные.
2) Na2Fu—фумарат натрия.
з) Na2Ma—малеат натрия; Робинзон и Смит [2].
ПРИЛОЖЕНИЕ Б
571
диэлектрической постоянной в непосредственной близости от центрального
иона.
Согласно модели Дебая и Паулинга, центральный ион представляет
собой сферу с радиусом г = а и с зарядом ze, расположенным в центре этого
иона. Предполагается, что в области внутри иона, а также внутри сферы
с радиусом г = R > а диэлектрическая постоянная непрерывно меняется
с радиусом, в то время как вне этой области, т. е. для значений г от г — R
jip г = оо, диэлектрическая постоянная сохраняет неизменное значение.
На основании этой модели и соответствующих граничных условий выводится
сложное уравнение для потенциала иона, которое после разложения в ряд
по степеням концентрации сводится к сумме, состоящей из члена, соответ-
ствующего предельному закону, и членов, содержащих первую и более
высокие степени концентрации.
Франк выводит уравнение Дебая и Паулинга иным способом и получает
выражение не для потенциала, а для свободной энергии иона, окруженного
ионной атмосферой. Затем он анализирует выводы, которые следуют из этой
теории для области умеренных концентраций, если приписать диэлектриче-
ской постоянной ряд различных значений внутри сферы с радиусом, рав-
ным R, и принять неизменное значение макроскопической диэлектрической
постоянной воды (78,54 при 25°) вне этой сферы. На основе своих вычисле-
ний Франк пришел к заключению, что если диэлектрическая постоянная
внутри сферы с радиусом R не превосходит 25 и если ионы могут прибли-
жаться друг к другу на расстояние, равное сумме кристаллографических
радиусов (г+4-г_), то, согласно этой теории, должны получаться очень
большие отрицательные отклонения от предельного уравнения Дебая и Гюк-
келя, что не соответствует экспериментальным результатам. Эти отклонения
тем менвше, чем меньше разность между R и а. Если ионы гидратированы
и если эти гидратированные ионы представляют собой непроницаемые сферы,
тогда внутри этого слоя молекул воды достигается диэлектрическое насы-
щение и R — a. При этом допущении, а также с учетом уменьшения числа
молекул растворителя из-за гидратации получаются положительные отклоне-
ния от экспериментальных данных. Если же допустить, что ионы гидрати-
рованы и в то же время они могут проникать сквозь гидратные оболочки
противоположно заряженных ионов, тогда результаты теории могут быть
приведены в соответствие с опытными данными. Эта модель, учитывающая
возможность ассоциации ионов в результате их проникновения сквозь гид-
ратную оболочку, будет более подробно рассмотрена в следующем параграфе.
В гл. XII, § 5а, мы упоминали о представлении Гюккеля [33], согласно
которому линейный член Вс в уравнении (32) гл. XII отражает понижение
макроскопической диэлектрической постоянной растворителя вследствие при-
бавления соли. Этот вопрос был рассмотрен Хастедом, Ритсоном и Колли [34]
с точки зрения произведенных ими измерений диэлектрической постоянной
и углов потерь в концентрированных растворах электролитов при частотах,
соответствующих длинам волн в 10,3 и 1,25 см. К сожалению, их Экспери-
ментальная методика не позволяет определять эти величины для разбавлен-
ных растворов. Согласно данным указанных авторов, полученные ими резуль-
таты в интервале концентраций 0,5 — 2М можно выразить уравнением
Z» = Z»0-(8+ + 8_)C, (30)
где D — статическая диэлектрическая постоянная, Do — диэлектрическая
постоянная воды, 8+ и 8_ — положительные константы, характеризующие
соответственно катионы и анионы. При концентрациях выше 2 М линейная
зависимость D от с уже не соблюдается и полученные результаты свиде-
тельствуют о том, что выше этой концентрации сумма (3+ + 8_) уменьшается
с ростом концентрации. Из своих данных Хастед, Ритсон и Колли вычислили
572
ПРИЛОЖЕНИЕ Б
минимальные величины гидратации на основе допущения о том, что ионы
притягивают молекулы воды с образованием внутренних гидратных оболочек,
в результате чего эти молекулы не оказывают влияния на величину диэлек-
трической постоянной. Если обозначить через п минимальное число молекул
воды во внутреннем слое, а через — молекулярный вес воды, тогда
jD = £’o-2SLc = £’o-(3+ + 8-)c- (31)
Вне этой области происходит сравнительно менее значительное понижение
диэлектрической' постоянной, обусловленное притяжением молекул воды
к ионам. В случае хлористого натрия, для которого (8+ + 8_) = 11, авторы
на основании теоретических соображений относительно вращения молекул
воды, притянутых к ионам, предполагают, что 8+— 8 и В_ = 3. Далее они
допускайт, что в случае хлористого натрия уменьшение диэлектрической
постоянной, обусловленное внешним слоем молекул воды, составляет % от соот-
ветствующего уменьшения, обусловленного внутренним слоем. Из этих дан-
ных они получили следующее выражение для числа гидратации катиона п+
хлористого натрия:
n+==(o+-2)^-. (32)
Кроме того, предполагается, что это уменьшение S+ на 2 единицы имеет
место для всех катионов, так что уравнение (32) имеет общий характер.
В табл. 178 приведены некоторые значения о+, о_ и п+, вычисленные
с помощью этого уравнения.
Таблица 178
Постоянные й+, й_ и некоторые минимальные значения гидратных чисел
дли положительных ионов
Обозначение величин н+ Li + Na + к+ Rb + Mg+ + Ва++ La +++ ci-
в+, 5- 17 11 8 8 7 24 22 35 3
п+ 10,5 6 4 4 3,5 15,5 14 23
Ритсон и Хастед [35] развили теорию диэлектрического насыщения
вблизи ионов на основе теории диэлектрических свойств жидкостей Онза-
гера [36], а также на основе усовершенствования теории Кирквуда [37].
Полученные ими результаты качественно согласуются с опытными значе-
ниями понижения диэлектрической постоянной. Кроме того, они подвергли
анализу теории концентрированных растворов и рассмотрели влияние сле-
дующих факторов: поправки на ван-дер-ваальсовский ко-объем, ассоциации
ионов по Бьерруму, изменения диэлектрической постоянной и изменения
структуры растворителя в присутствии растворенного вещества. Ритсон
и Хастед пришли к выводу, что необходимо целеустремленное теоретическое
рассмотрение проблемы взаимодействия ионов с растворителем.
§ 7. Гидратация ионов, коэффициенты активности и осмотические
коэффициенты
В гл. XII, § 7 и 8, были подробно рассмотрены некоторые теории, пред-
ложенные для объяснения зависимости коэффициентов активности электроли-
тов в концентрированных растворах от концентрации. Две из этих теорий,
представляющие собой дальнейшее развитие теории Дебая — Гюккеля, а именно
теории Гюккеля и Скэтчарда, в значительной степени сводятся к определе-
ПРИЛОЖЕНИЕ В
573
нию зависимости коэффициента активности от взаимодействий между ионами,
между ионами и растворителем, а также между молекулами при изменении
диэлектрической постоянной, обусловленном изменениями концентрации
электролита. Другая теория, развитая Ван-Риссельбергом, была основана
на определении влияния ван-дер-ваальсовского ко-объема. В конце гл. XII
была упомянута теория гидратации ионов, предложенная Бьеррумом [38]
еще до появления теории Дебая и Гюккеля. Ниже мы рассмотрим теорию
Стокса и Робинзона [39], которая представляет собой видоизменение и даль-
нейшее усовершенствование теории Бьеррума.
Для вывода уравнений этой теории мы не будем пользоваться методом
Стокса и Робинзона, которые применяли уравнение Гиббса—Дюгема, а восполь-
зуемся выводом Харнеда1, основанным непосредственно на законе действую-
щих масс. Допустим, что равновесия, существующие в водном • растворе
электролита, можно изобразить следующими реакциями и уравнениями:
1) СА^С+ + А~, .
2) С+ + ЛН2О^С+-АН2О,
3) а~+/н2о^а--/н2о,
X] d^-d— / й2,
K2 = uh/d+a*, (33)
К2 = а//а_а{0,
где Xj, К2 и Х3 —константы закона действующих масс, а+, а_ — активности
негидратированных ионов; ал, а}- — активности гидратированных ионов;
aw — активность воды. Перемножая три выражения закона действующих
масс, получаем
КгК2К3 =
х2х3
ahaj
(34)
где п = (& + /). Если принять условие aw = l при бесконечном разведении
электролита, тогда Х2Х3 = 1и, следовательно,
и^и— — aha; /aw, (35)
После введения коэффициентов активности и моляльностей и логарифми-
рования получаем выражение
1g lg Т'±щ' - ~ lg dw.
(36)
активности
В этом выражении у представляет собой средний коэффициент
электролита, т — его стехиометрическую моляльность, — коэффициент
активности гидратированных ионов, т' — их моляльность. Если допустить,
что электролит полностью диссоциировин при всех концентрициях, т' можно
выразить следующим уравнением:
, 55,51m т
55,51—пт 1—0,018nm ’
(37)
При приближении концентрации электролита к нулю, согласно закону
действующих масс, должна достигаться полная гидратация ионов. Следова-
тельно, можно допустить, что коэффициент активности гидратированных
ионов выражается уравнением Дебая и Гюккеля [уравнение (10) гл. V],
т. е.
lgY±= — . ~ — lg (1 + 0,018wz').
1 1 + А у с
(38)
1 См. в книге Тэйлора [40].
ПРИЛОЖЕНИЕ В
574
Если подставить в уравнение (36) это значение ч'±, исключить т' с помо-
щью уравнения (37) и выразить А' с через уравнения (И) и (12) гл. V,
получаем соотношение х
1g Т± =-------S(/) lg - lg [1 - 0,018 (n- v) иг]. (39)
1 + 0,3286a /Г/2
Это соотношение представляет собой «усовершенствованное» уравнение Бьер-
рума, применяемое Стоксом и Робинзоном, с тем отличием, что они заме-
няют с и Г/2 на т и р.
Это уравнение содержит два параметра (а и п), которые можно варьиро-
вать, и с его помощью можно выражать коэффициент активности с точно-
стью, которая не уступает точности уравнения (32) гл. XII, однако в более
широком интервале концентраций. Это уравнение труднее использовать для
практических целей, чем уравнения (32) и (33) гл. XII, так как для этого
необходимо вычислить aw из экспериментальных значений коэффициентов
активности. В табл. 179 приведены некоторые значения а и п, взятые из
более подробной таблицы Стокса и Робинзона.
В гл. XII, § 5 и 8, мы отметили, что можно вывести уравнение с од-
ним параметром для вычисления коэффициентов активности в значительном
интервале концентраций. С этой целью Стокс и Робинзон используют соот-
ношение
«={Ц(3°п + Р+)]1/3 + г--Д) , (40)
с помощью которого можно исключить а из уравнения (39). Таким образом,
в уравнении (39)' оетается лишь один параметр п. Соотношение (40) осно-
вано на представлениях Бернала и Фаулера [41] о кажущихся молярных
объемах ионов и воды. Делается допущение, что большие анионы С1_, Вт-,
J- гораздо меньше гидратированы, чем имеющие меньшие размеры кати-
оны Li+, Na+ и т. д., так как г_ равно кристаллографическому радиусу
аниона. Кроме того, предполагается, что эффективный объем молекулы воды
составляет 30 А3, и если обозначить через У+ кажущийся ионный объем
f3 11/3
(30/г 4- Р+) J равно эффективному «идеаль-
ному» радиусу гидратированного катиона. Для определения Стокс
и Робинзон вычисляли кажущиеся молярные объемы некоторых электроли-
тов при концентрации 1 М из данных по плотностям растворов. Получен-
ные ими результаты можно выразить с помощью эмпирического уравне-
ния Ккаж. = 6,47 (т-^Я_), где г+ и г_ — кристаллографические радиусы
(табл. 6). Если объем каждого аниона составляет 6,47 r‘i_ А3, тогда объем
катиона будет К+= Укаж. — 6,47v_r^/v+, где v_/v+ —число анионов, связан-
ных с одним катионом. Из этого соотношения вычисляют У+, а гх нахо-
дят из У+. При этом оказывается, что (гг + г_) превышает значения дополу-
чаемые с помощью уравнения (39) с двумя параметрами, на 0,7А для
галогенидов щелочных металлов и на 1,ЗА для галогенидов щелочнозе-
мельных металлов. Что касается физического смысла величины, обозначенной
через Д в уравнении (40), то делается допущение, что она характеризует
проникновение анионов в гидратную оболочку катиона [42] или деформацию
ионов в электрическом поле.
В четвертом столбце табл. 179 приведены значения п, с помощью ко-
торых получается наилучшее совпадение вычисленных значений с опытными
данными. В пятом столбце содержатся значения а, вычисленные с помощью
ПРИЛОЖЕНИЕ Б
575
Таблица 179
Константы уравнения (39), а также совместных уравнений (39)—(40)
Электролит Уравнение (39) Уравнения (39)—(40)
п О а п а R
НС1 8,0 4,47 7,3 4,84 0,1—1
L1C1 7,1 4,32 6,5 4,66 0,1—2
NaCl 3,5 3,97 3,5 3,97 0,1—5
КС1 1,9 3,63 1,9 3,63 0,1—4
RbCl 1,2 3,49 1,25 3,47 0,1—2
MgCl2 13,7 5,02 13,9 4,99 0,1—1
СаС12 12,0 4,73 11,9 4,75 0,1—1,8
SrCl2 10,7 4,61 10,8 4,60 0,1—1,8
ВаС12 7,7 4,45 8,4 4,29 0,1—1
уравнения (40), причем, как видно из таблицы, эти значения прекрасно
совпадают с величинами в третьем столбце. Расхождение между вычислен-
ными таким образом коэффициентами активности и опытными значе-
ниями не превышает ± 0,0015 единиц у± для интервала концентраций,
указанного в последнем столбце, причем в случае хлоридов кальция и
стронция сверх ожиданий также получается очень хорошее совпадение вы-
численных' данных с опытными.
Некоторые критические замечания относительно этой теории были вы-
сказаны Стоксом и Робинзоном, которые указали, что параметр п следует
рассматривать не как число молекул воды в слое, окружающем ион, а как
«число, которое должно учитывать средний эффект влияния всех тех взаимо-
действий между ионами и растворителем, которые велики по сравнению
с кТ». Согласно более ранним теориям гидратации, гидратные числа на-
столько велики, что при высоких концентрациях (например, ~ 7 М для
соляной кислоты) все молекулы воды связаны с ионами и воды, как рас-
творителя, совсем не остается. Таким образом, уравнение (37) следует
усовершенствовать с тем, чтобы с увеличением концентрации электролита
значение п уменьшалось, причем для объяснения этого явления надо до-
пустить, что происходит проникновение ионов в гидратные оболочки других
ионов. В этих условиях смысл понятия «объем свободного растворителя»
становится весьма неопределенным, и поэтому применимость этой теории
ограничена промежуточной областью концентраций (вплоть до 1 — 2 Д/),
в которой проникновение оказывает меньшее влияние на величину тп'.
Для количественного объяснения зависимости между концентрацией
раствора и активностью воды в очень концентрированных растворах Стокс
и Робинзон выдвинули следующую гипотезу. Они обнаружили, что азотно-
кислый кальций, который образует насыщенный раствор с концентра-
цией 8,4 М при 25°, легко дает пересыщенные растворы и при дальней-
шем повышении концентрации образует полутвердые гели. Переход между
этими состояниями осуществляется непрерывно, и давление пара является
непрерывной функцией концентрации. На основании этих данных Стокс
и Робинзон считают, что в очень концентрированных растворах этого и дру-
гих электролитов имеются ионы с частично заполненными гидратными обо-
лочками, ионы с мономолекулярными гидратными оболочками, а также
ионы с двумя или большим числом более связанных слоев. На основании
аналогии этой модели с картиной полимолекулярной адсорбции делается
предположение о применимости в данном случае соответствующим образом
576
ПРИЛОЖЕНИЕ В
видоизмененной изотермы адсорбции Брунауэра, Эммета и Теллера [43]
или ее усовершенствованной формы, предложенной Андерсоном [44]. По-
лучающееся при этом уравнение приводит к результатам, которые хорошо
совпадают с данными, найденными путем измерения давления пара раство-
ров ряда электролитов в интервале концентраций 4 — 22 М,
§ 8. Данные о диссоциации сильных электролитов, полученные
путем изучения рамановских спектров
Вопрос о диссоциации умеренно сильных и очень сильных электроли-
тов был рассмотрен в обстоятельном обзоре Редлиха [45] \ который уделил
значительное внимание тем несомненным доказательствам присутствия
недиссоциированных молекул, которые могут быть получены на основании
изучения рамановских спектров. Наличие вибрационных спектров представ-
ляет собой тот критерий, с помощью которого можно надежно отличать
молекулы от взаимодействующих между собой ионов. Редлих и Розен-
фельд [46] полагают, что истинную концентрацию ионов в данном растворе
можно определить путем сравнения спектра этого раствора со спектром
раствора полностью диссоциированного электролита, содержащего тот же
ион. Как известно, интенсивность рамановских спектров нитратов щелочных
металлов пропорциональна их концентрациям [47, 48], что является дока-
зательством полной диссоциации этих солей. Для определения концентра-
ции нитрат-ионов. ас в растворе азотной кислоты со стехиометрической кон-
центрацией с приготовляют такой раствор азотнокислого натрия, у которого
интенсивность рамановских спектров равна интенсивности соответствующих
спектров, наблюдаемых в случае раствора азотной кислоты. Поскольку
стандартней раствор нитрата с концентрацией cR полностью диссоциирован.
Cr равно ас. В табл. 180 приведены значения а, найденные таким способом;
из этих значений была вычислена термодинамическая константа диссоциа-
ции, которая оказалась равной 21,4.
Таблица 180
Степень диссоциации азотной кислоты при температуре, близкой к 25°
С о,1 1 2 3 4 6 8 10 12 14
а 0,997 0,978 0,95 0,90 0,85 0,72 0,56 0,42 0,28 0,16
Юнг и Блатц [49] использовали имеющиеся данные по рамановским
спектрам [50] для выяснения характера тех сложных равновесий, которые
имеют место в растворах серной кислоты при различных концентрациях.
Интенсивность полос спектра, отвечающих ионам ЭОГ- и НЗОГ, можно срав-
нить с интенсивностью полос для растворов (NH4)2SO4 и KHSO4, в то время
как полоса спектра, отвечающая H2SO4, идентифицируется на основании
максимального значения ее интенсивности, которое наблюдается в случае
раствора, содержащего по 50 молярных процентов Н2О и SO3. Рис. 171
хорошо иллюстрирует результаты, полученные зтим методом анализа. На
этом рисунке изображена зависимость выраженных в процентах относительных
количеств HHSO4, HSO7 и SO^- в растворе серной кислоты от с. На осно-
вании данных о значениях степени диссоциации можно дать количественную
трактовку таких свойств, как теплоты разведения, поверхностное натяжение
[51] и кажущиеся молярные объемы.
1 В этой статье содержится подробная библиография.
ПРИЛОЖЕНИЕ В
577
<-------
Рамановские спектры растворов йодноватой кислоты [52], трихлоруксус-
ной кислоты [53] и хлорной кислоты [486] свидетельствуют о том, что эти
кислоты неполностью диссоциированы. Изучение рамановских спектров может
Р и с. 171. Относительные количества серной кислоты, присутствующие
в виде не диссоциированной кислоты, бисульфат-иона и сульфат-иона, вы-
раженные через величины вертикальных отрезков между смежными ли-
лиями.
дать ряд новых важных результатов при условии, если будут усовершен-
ствованы методы определения относительной интенсивности спектров.
§ 9. Диссоциация некоторых умеренно сильных электролитов
В табл. 181 приведены взятые из обзора Редлиха [46] значения кон-
стант диссоциации некоторых умеренно сильных электролитов, найденные
различными методами. Эта таблица может служить в качестве дополнения
к табл. 22. Мы не приводим ссылок на литературные источники этих дан-
ных, поскольку эти ссылки имеются в обзоре Редлиха.
Таблица 181
Константы диссоциации некоторых умеренно сильных
электролитов
Электролит к Электролит к
HJO3 0,17 РЬВг+ 0,07
QP13COOH 0,22—0,23 Pbj+ 0,035
NOa OaN^ ^OH 0,16 H2SO3 0,012—0,017
no2 CHC1.2COOH 0,14 HSeO4~ 0,01
H4P2O7 0,14 ^>СООН 0,006
РЬС1+ 0,06—0,1 no2
37 г. Харнед, Б. Оуэн
578 ПРИЛОЖЕНИЕ Б
§ 10. Константы диссоциации и диссоциация слабых кислот
в растворах солей
В табл. 182—184 приведены точные значения констант диссоциации !
слабых электролитов в воде, в водных растворах солей, а также в смесях
воды с органическими растворителями; эти значения были получены в последние
годы с помощью цепей без жидкостных соединений посредством методики,
описанной в гл. XV. В табл. 184 содержатся параметры уравнений
(70) —(76) гл. XV, которые, как нами было показано, удобны для вычис-
ления стандартных термодинамических функций, соответствующих реакциям
диссоциаций1.
Первая ступень диссоциации угольной кислоты
Н2О + СО2 Н2СО3 Н++ нсо3-
быЛа тщательно изучена Харнедом и Дэвисом [55], а также Харнедом
и Боннером [56], которые измеряли электродвижущую силу элемента
Pt-H21 NaJICO3 (m(), NaCl(m2), СО2 (m3) | AgCl-Ag.
Общее уравнение для электродвижущей силы этого элемента имеет следу-
ющий вид: f
Е = Е° - In mH^ci YhYci — Ес, (41)
где Ес=—2Z772F In РН2. В этом уравнении фигурирует член Ес, так как
при измерении применялись смеси водорода и газообразной двуокиси угле-
рода, в которых содержание двуокиси углерода изменялось от 18 до 80%.
Константу диссоциации можно выразить следующим уравнением:
дг анансо3 1н7нсозтонтонсоз ’УнТнсоз д,
1А аН2ОаСО2 аН201С02тС02 ан2о7со2 1А
В качестве стандартного состояния было выбрано условие Yh==Yhco3 =
= yC02 = «н2о — 1 для чистой воды, и, следовательно, АДд является изотер-
мической константой для всех концентраций соли. Исключая тн из урав-
нений (41) и (42), получаем выражение
F
(Е + Ес — Е°) 2 3026ЯГ + Шс°2 + mHC0 =
’ НСОз
= (43)
, *Н 1НСО3
которое определяет экстраполяционную функцию — IgA'. Поскольку лога-
рифмический член, содержащий коэффициенты активности, равен нулю
для чистой воды, то —lg Kia можно получить путем экстраполяции правой
части этого уравнения на нулевую концентрацию для всех растворенных
веществ. Для получения точных результатов приходится прибегать к двум
экстраполяциям — сначала на нулевую ионную силу, а затем на нулевую
концентрацию двуокиси углерода. Моляльную концентрацию двуокиси
углерода можно вычислить с помощью закона Генри
тсо2 =
1 Дженкинс [54] снова исследовал эмпирические выражения [уравнения (22) ц (23) ;
гл. XV], служащие для определения изменения константы диссоциации с температурой,
и предложил более сложное выражение, содержащее четыре эмпирические константы.
Из сравнения значений вычисленных с помощью этого уравнения с эксперименталь-
ными значениями видно, что это уравнение не превосходит по степени точности более
простое уравнение. При этих вычислениях снова проявилась трудность определения тер-
модинамических функций с помощью дифференцирования эмпирических выражений для
свободной энергии (см. стр. 478, 479).
Таблица 182
Экспериментальные значения констант диссоциации слабых кислот в воде и в водных растворах солей. Все значения
соответствуют моляльным концентрациям
Кислота Растворитель Температура, °C Ссылки на лите- ратуру
0 5 10 15 20 25 30 35 40 45 50
Угольная Вода 10’ 2,64 3,04 3,44 3,81 4,16 4,45 4,71 4,90 5,04 5,13 5,19 1
0,1 М NaCl 4,84 5,16 5,84 6,49 7,11 7,66 8,14 8,54 8,85 9,06 9,16 2
0,2 М NaCl . 10’ 5,15 5,93 6,70 7,44 8,14 8,78 9,33 9,79 10,15 10,39 10,51 2
0,5 М NaCl . к14 • 10’ 6,23 •7,14 8,04 8,90 9,70 10,41 11,03 11,53 11,92 12,19 12,32 2
0,7 М NaCl 1А ’ 10’ 6,62 7,56 8,47 9,34 10,15 10,81 10,51 12,03 12,45 12,75 12,93 2
1,0 М NaCl &1А Ю’ 7,02 7,98 8,92 9,80 10,63 11,37 12,02 12,58 13,03 13,37 13,58 2
Фосфорная Вода аг2А • 10» 4,85 5,24 5,57 5,89 6,12 6,34 6,46 6,53 6,58 6,59 6,55 3
Фенолсульфоновая » К^А Ю10 4,45 5,20 6,03 6,92 7,85 8,85 9,89 10,94 12,00 13,09 14,16 4
Г лицин » А1Д . 10’ — 3,82 3,99 4,17 4,32 4,46 4,57 4,66 4,73 4,77 4,79 5 .
Бензойная » *!А • 105 — — — — 6,24 6,29 6,27 6,24 — — ч
Лимонная » /г1А • IO* 6,03 6,31 6,69 6,92 7,21 7,45 7,66 7,78 7,96 7,99 8,04 6
» аг2А . 105 1,45 1,54 1,60 1,65 1,70 1,73 1,76 1,77 1,78 1,76 1,75 6
» к ЗА 10’ 4,05 4,11 4,14 4,13 4,09 4,02 3,99 3,78 3,69 3,45 3,28 6
1) X. Н. Партон и А. В; Джонс, частное сообщение.
*!£
580
ПРИЛОЖЕНИЕ Б
Константы диссоциации слабых кислот в смесях органический
X—весовой %
X Темпе-
0 5 10 15
Пропионовая кислота в смесях
0[1] КА • Ю5 1,274 1,305 . 1,326 1,336
10 ' КА • 105 0,881 ' — 0,885 —
20 КА . 105 0,603 — 0,608 —
Пропионовая кислота в смесях
, 10 КА 105 0,870 — 0,900 —
20 ХА . 105 0,673 — 0,755 —
Пропионовая кислота в смесях
5 КА 105 1,015 1,038 1,052 1,061
10 КА 105 0,780 0,803 0,815 0,823
20 КА 105 0,431 • 0,448 0,459 0,466
н-Масляная кислота в смесях
5 КА 105 1,190 1,197 1,189 1,177
10 КА 105 0,922 0,929 0,925 0,916
20 КА 105 0,466 0,475 0,476 0,473
Уксусная кислота в смесях
X Темпе-
0 55 60 10 65 15 70
50 КА • 10» 4,78 5,07 4,96 4,95 5,10 4,81 5,22 4,65
ПРИЛОЖЕНИЕ Б
581
Таблица 183
растворитель—вода. Все значения соответствуют моляльным концентрациям
органического растворителя
ратура, ' С
20 25 30 35 40 45
метиловый спирт—вода [2]
1,338. 1,336 1,326 1,310 1,280 1,257 1,229
0,9.17 0,947 0,909 —. 0,838 — —
0,586 0,578 0,553 — 0,525 — —
этиловый спирт—вода [2]
• 0,900 0,900 0,906 — 0,902
. 0,771 0,781 0,788 — 0,780 — —
изопропиловый спирт—вода [3]
1,060 1,053 1,044 1,032 1,012
0,824 0,820 0,812 0,800 0,785 — —
0,466 0,466 0,462 0,454 ' 0,444 — —
изопропиловый спирт—вода [4]
1,157 1,133 1,104 1,077 1,040 —
0,907 0,888 0,864 0,839 0,813 — —
0,465 0,456 0,446 0,432 0,417 — —
глицерин—вода [5] (0—90°)
ратура, 1 с
20 75 25 80 30 85 35 90 4 0 45 50
5,32 4,47 5,35 4,31 5,38 4,14 5,37 3,93 5,33 5,27 5,18
* ‘ Константы уравнений (70)—(76) гл. XV для констант диссоциации, AF® , ДН® Табл ДСО^-lg^ и — 2 ]/С*А* и ц а 184
mHmAc Л = TirUc = 1нАс Часть I дные раство] -к 'п Uc IhAc Л* г0=^; ig k, = d* А' = 2,3026 RA* £>' = 2,3026 RD*
1 1 1.1 II С) ' II II II * V ° а тНАс л* D'T’ + C'T’2 С'Т2 2С'Т Во
С' = 2,3 )Ы 026 RC*
Кислота А* ь* с* А1 Dr С . Ti -1g К0
Фосфорная *2А 1979,5 5,3541 0,019840 9055,0 24,491 0,090751 317 7,1795
Фсиолсульфоповая . .• %2А 1961,2 1,1436 0,012139 8970,8 5,231 0,055525 402 8,3928
Бензойная ^1А 1590,2 5,394 0,01765 7319,1 29,247 0,080733 301 4,2014
Лимонная %АА 1255,6 4,5635 0,011673 5143,3 20,874 0,053394 328 3,094
» 2А 1585,2 5,4460 0,016399 7250,9 24,911 0,074012 311 5,751
» К?,Л 1814,9 6,3664 0,022389 8301,6 29,121 0,102410 285 6,383
Часть II
Угольная кислота (-ЙГ1А), (к1А) в водных растворах NaCl[lj
mNaCl А* D* С* mNaCl А* D* С*
0,0 3404,71 14,8435 0,032786 0,5 3158,38 13,5235 0,029892
0,1 3266,11 14,0256 0,030811 0,7 2955,23 12,2139 0,027721
0,2 3228,50 13,8363 0,030401 1,0 2786,18 11,1187 0,025885
1: ,, ч Л >
,1 :ц .1. ill, л
Hi-1wi'g
mNaCl А* D* С* mNaCl А* £>* С*
Борная кислота в водных растворах NaCl [2]
0,0 2291,9 3,6865 0,017560 0,725 1695,2 0,0750 0,010799
0,02 2221,1 3,2837 0,016595 1,25 1496,2 -1,1962 0,008662
0,07 2136,4 2,7805 0,015590 2,00 1264,9 -2,7439 0,006053
0,36 1881,8 1,2322 0,012782 3,00 984,7 -4,7340 0,002626
Глицин (Ка) в водных растворах NaCl [3]
0,0 1259,9 5,3980 0,011817 1,25 1277,1 5,2559 0,011412
0,1 1278,0 5,5030 0,011974 2,00 1213,1 4,6187 0,010234
0,3 1292,4 5,5727 0,012067 3,00 1026,6 3,0809 0,007529
0,725 1297,4 5,5234 0,011931
Пропионовая кислота в смесях изопропиловый спирт —вода
X 0 1213,26 3,3860 0,014055 X 10 1403,858 4,5143 0,016406
5 1191,0865 3,1909 0,013995 20 1771,125 6,7087 0,020457
н-Масляная кислота в смесях изопропиловый спирт—вода
mNaCl А* D* С* mNaCl А* £)♦ С*
0 1033,39 2,6215 0,013334 10 z 1217,523 3,6903 0,015625
5 1048,516 2,6719 0,013753 20 1459,369 5,0187 0,018325
Уксусная кислота в смесях глицерин—вода (0—90°)
50 | 1321,4256 I 3,4148 I 0,014268
584
ПРИЛОЖЕНИЕ Б
Используя газовые смеси с самыми различными концентрациями двуокиси
углерода, Харнед и Дэвис показали, что присутствие двуокиси углерода
в растворе не оказывает значительного влияния на — IgJf'.'B связи с этим
обстоятельством отпадает необходимость в применении двойной экстрапо-
ляции и значительно упрощается экспериментальная методика.
Значение константы диссоциации к1А для растворов солей можно
получить, исключая Kia из уравнений (42) и (43):
- Igi.A = (Е - Е« + Ес) + lg -^1 + lgVc]. (45)
HCU3
Все величины в правой части этого уравнения известны, поскольку
|/ IhYcp т- е. коэффициент активности соляной кислоты при нулевой кон-
центрации в растворах хлористого натрия (табл. 161), был определен.
Моляльность двуокиси углерода можно вычислить по уравнению
тСО2 = со2> (^6)
если воспользоваться константами закона Генри для растворов солей, кото-
рые были определены Харнедом и Дэвисом. Опытные значения KiA и kiA
для. растворов хлористого натрия приведены в табл. 182, а константы урав-
нения (70) гл. XV. для вычисления температурной зависимости этих величин
содержатся в табл. 184.
Логарифмируя уравнение (42) и подставляя вместо lgYHYHC0 выражение
Дебая — Гюккеля с дополнительным линейным членом, получаем
-lg Aia = -lg^iA--|^7^+-5V-lgYC02«n20. (47)
Было’ показано, что для растворов хлористого натрия в интервале концен-
траций 0 — 1 М
IgYco^te/r^0^ (48)
(.о;
где а — изотермическая постоянная. Кроме того, приближенное уравнение
— lgaH2o = ?™ (49)
достаточно точно выражает опытные данные, и, следовательно, уравнению (47)
можно придать следующий простой вид:
- lg/c1A= -Ig/GA-+ ж (50)
1 + V н
если принять А' равным единице при всех температурах. Ниже приведены
уравнения для величин, необходимых для вычисления ktA в интервале
температур 0 — 50° и для концентраций 0—1 М:
lgKlA= — 3404,71/7’+14,8435 — 0,03278674 (51)
= 0,48834-0,75545 • Ю’8/4-0,1743 • 10~5г24-0,11665 • 10*7/3, (52)1
В = о,066 + 1,92 • 10-3/ —0,0176 • 10-V2. (53)
Харнед и Боннер показали, что эти уравнения выражают опытные данные
для — Ig/ciA со средним отклонением, которое составляет 0,001 в интервале
температур 0 — 10°, 0,001 — 0,002 для температур 15 — 30° и 0,002 — 0,004
для температур 35 — 50°. Максимальное отклонение равно 0,0063 при 50°
для 1 М раствора хлористого натрия.
1 См. [57].
ПРИЛОЖЕНИЕ Б
585-
Значения а в уравнении (48), с помощью которых можно вычислять
'gYcoj для всех изученных температур и концентраций, выражается уравнением
а = 0,1190 - 0,833 • 10~37 + 0,666 • 10-5£2. (54>
Концентрация двуокиси углерода выражается уравнением
}gmco2 — ]gS0PCG2 — ар., (55)
причем lg Sо можно вычислить с помощью выражения
— 1g 3*0 =—2385,73/Т н-14,0184— 0,0152647’. (56)
На основании результатов этих исследований можно вычислять точные
значения многих важных величин, характеризующих водные растворы дву-
окиси углерода. Кроме стандартных термодинамических величин AF°, А 77? ,
ЛСр. и Д5“ были определены величины KlA, klA, 7нГнсо3/7со2ан2о, 7н1'нсо3,
усо2 и mco3 для растворов солей. Следует, однако, отметить, что с помощью
этого метода нельзя получить никаких сведений относительно диссоциации
кислоты Н2СО3, а только данные относительно реакции Н2О-|-СО2, т. е. для
суммы реакций
Н2О + СО2 7- Н2СО_,
и .
н2со3 н+ + нсо;.
Посредством кинетических калориметрических измерений Роутон [58]
определил величины теплосодержания этих реакций, а также константу
скорости второй реакции. Он использовал для этой цели метод быстрого
смешения раствора бикарбоната натрия с соляной кислотой. Эта смесь про-
текает с известной скоростью через трубку, в которой помещены термопары,
расположенные на некотором расстоянии друг от друга. Результаты изме-
рений температуры совершенно определенно указывают на то, что при взаи-
модействии с сильной кислотой реакция Н+ -ф НСОГ —» Н2СО3 протекает
практически "мгновенно, в то время как константа скорости к0 более медлен-
ной реакции Н2СО3 —> Н2О-фСО2 поддается измерению. Путем независимых
определений [59] скорости образования двуокиси углерода' в буферных
смесях бикарбоната с какодилатом (7*п = 5,5— 7) была найдена константа
скорости кг быстрой реакции Н+ + НСО-Г —>Н2О-|-СО2. Из величины отно-
шения Йо/#! была определена константа диссоциации Н2СО3, которая оказа-
лась равной примерно 2 • 10~4.
Оуэн и Кинг [60] определили диссоциацию борной кислоты в растворах
хлористого натрия с помощью элементов без жидкостных соединений,, содер-
жащих буферные растворы. Особенность этой работы состояла _ в таком
выборе стандартного состояния, при котором коэффициенты активности при-
нимались равными единице для любого раствора хлористого натрия с кон-
центрацией т. Таким образом, все растворы солей рассматривались в ка-
честве отдельных растворителей. Способ математической обработки опытных
данных аналогичен тому, который применяется в случае смосей воды
с органическими растворителями, и был описан в гл. XV, § 7. Хотя этот
метод имеет некоторые формальные и логические преимущества, он не дает
таких результатов, которые нельзя было бы получить с помощью старых
методов, применение которых было рассмотрено в этом параграфе на примере
изучения равновесия в растворе угольной кислоты.
Плодотворность применения термодинамического метода к определению
констант диссоциации, а также к изучению диссоциации слабых кислот
в данном растворителе и в растворах солей с использованием результатов
измерений электродвижущих сил элементов без жидкостных соединений
588
ПРИЛОЖЕНИЕ Б
можно проиллюстрировать на примере работы Бэйтса [61], который приме-
нил этот метод к изучению слабых кислот, у которых различные ступени
диссоциации перекрываются. Он показал, как можно использовать этот
.метод для определения произведения КГК2 для двухосновных кислот, а также
произведений КУК2 и К2КЛ для трехосновных кислот. Бэйтс и Пинчинг [62]
применили этот метод к изучению трех констант диссоциации лимонной
кислоты. Полученные ими результаты приведены в табл. 182, а константы
уравнений для вычисления термодинамических функций содержатся
в табл. 184. Найденные этими авторами значения — lgK2A и — Ig-STan для
18, 25 и 37° хорошо совпадают с результатами, которые получили Бьеррум
и Унмак [63] с помощью элементов с жидкостными соединениями, однако
значения —lgKtA, полученные этими двумя методами,, отличаются при-
мерно па' 0,06.
§ 11. Переход от простых ионов к сложным аггрегатам
и полимерным электролитам
Изложенный в настоящей монографии материал сознательно ограничен
вопросами, относящимися к теоретическому и экспериментальному изучению
свойств растворов, которые содержат ионы, обладающие сравнительно
простым строением. Поскольку за последнее время было выполнено большое
число исследований, посвященных электролитам, состоящим из ионов
е длинными цепочками и из многовалентных ионов, а также протеинам, мы
-сочли необходимым вкратце остановиться на результатах, которые иллюстри-
руют изменение свойств типичного ряда растворов солей при изменении
структуры ионов в направлении ее усложнения.
На рис. 172 изображены кривые зависимости средних коэффициентов
активности от для натриевых солей первых десяти нормальных жир-
ных кислот [64]. Кривые для коэффициентов активности первых четырех
членов ряда аналогичны соответствующим кривым для простых сильных
1,1-электролитов в водных растворах, представленных на рис. 95, 96 и 98.
Только в случае натриевой соли валериановой кислоты впервые обнаружи-
вается отклонение от нормального поведения, поскольку кривая для коэф-
фициента активности этой соли пересекает соответствующую кривую для
натриевой соли масляной кислоты. Начиная с капроновокислого натрия,
это отличие от поведения простых 1,1-электролитов становится очень замет-
ным и возрастает еще больше с увеличением числа атомов углерода в ионе.
-Эти результаты указывают на образование мицелл во всех растворах, начи-
ная с раствора соли, содержащей шесть атомов углерода. Особенно сильное
отклонение от обычного поведения наблюдается в случае солей, содержащих
девять и десять атомов углерода.
Наличие резкого перехода от простых ионов к мицеллам очень хорошо
•подтверждается изменением характера зависимости эквивалентной электро-
проводности от концентрации. Этот эффект изображен на рис. 173. Харак-
терные изломы на кривых зависимости электропроводности от у'с для выс-
ших членов ряда солянокислых алкиламинов [65, 66] указывают на обра-
зование мицелл. Эти изломы являются очень резкими в случае солянокис-
лых октадециламина и гексадециламина и делаются более плавными
по мере уменьшения числа атомов углерода в катионе. Кривая для соля-
.нокислого октиламина уже является типичной для нормального сильного
1,1-электролита. Концентрация, при которой происходит этот переход от
поведения типичных электролитов к поведению, характерному для мицелл,
очень сильно зависит от структуры растворенного вещества иг от природы
^растворителя. Поведение такого рода электролитов в смешанных растворителях
ПРИЛОЖЕНИЕ Б
58Т
Р пр. 172. Средние коэффициенты активности натриевых солей однооснов-
ных жирных кислот в воде при 25э.
1—муравьинокислый натрий; 2—уксуснокислый натрий; 3—пропионовокислый нат-
рий; 4—маслянокислый натрий; 5—валерьяновокислый натрий; 6—капроновокислый
натрий; 7—гептиловокислый натрий; 8 — каприловокнелый натрий; 9—пеларгоново-
кислый натрий; 10—каириновокислый натрий; II. 3.—теоретический предельный
закон.
Рис. 173. Эквивалентные электропроводности хлористоводо-
родных алкиламинов в воде при 25°.
8—октил-; 10—децил-; 12—додецил-, 14—тетрадецил-; 16—гексадецил-
п 18—октадецилампн.
и в присутствии более простых посторонних электролитов было подробно
исследовано1 Краусом, Тартаром, Ролстоном и их сотрудниками. Интересное
теоретическое объяснение описанных явлений было предложено Дебаем [68].
1 Типичные исследования такого рода описаны в работах, указанных под рубри-
кой [67а]. Обзор некоторых из этих работ содержится в статье Григера [676].
588
ПРИЛОЖЕНИЕ Б
В одной из последних работ Фуосса [69] рассмотрено применение
описанных методов исследования к области, которая не рассматривается
в настоящей монографии, а именно к растворам ионов больших размеров
и весьма сложного состава, близких по своим свойствам к протеинам.
Фуосс и его сотрудники исследовали свойства растворов электролитов
с очень длинными цепочками (образующихся путем полимеризации), обла-
дающих известной структурой и большим числом зарядов. При изучении
электропроводности, вязкости и осмотического давления растворов этих
веществ был обнаружен целый ряд новых интересных явлений [70, 71].
ЛИТЕРАТУРА К ПРИЛОЖЕНИЮ Б
1. J. Wy m an, J г., Phys. Rev., 35, 623 (1930).
2. J. W у m a n, Jr., E. N. I n g a 1 1 s, J. Am. Chem. Soc., 60, 1182 (1938).
3. F. H. Drake, G. W. Pierce, M. T. Do w, Phys. Rev., 35, 813 (1930.).
4. В. T. В i r g e, Rev. Mod. Phys., 13, 233 (1941).
5. В. B. Owen, S. R. Brinkley, J r., Ann. N. Y. Acad.. Set, 51, 753 (1949).
6. L. W. Tilton, J. K. Taylor, I. Research Nat. Bur. Stand., 18, 205 (1937).
7. P. Ch appuis, Travaux et Memoires du Bureau International des Poids et Mesurcs,
13, D40 (1907).
8. .1. R. White, диссертация, Yale University, 1944.
9. J. S. Smith, диссертация, Yale University, 1943.
10. ' В. B. Owen, J. Chem. Ed., 21, 59 (1944).
11. В. B. Owen, S. R. Brinkley, Jr., Phys. Rev. (2), 64, 32 (1943).
12. F. T. Gucker, .1 r., J. Am. Chem. Soc., 55, 2709 (1943).
13. R._ E. G i b s о n, O. N. Loeffler, J. Am. Chem. Soc.., 63, 443 (1941).
14. J. Lange, Z. phys. Chem., A188, 284 (1941).
15. R. M. F u о s s, T. S h e d 1 о v s k y, J. Am. Chem. Soc., 71, 1496 (1949).
16. • H. S. H а г n e d, D. M. French, "Ann. N. Y. Acad. Sci., 46, 267 (1945).
17. a) H. S. Harned, R. L. Nuttall, J. Am. Chem. Soc., 69, 737 (1947); 71,
. 1460 (1949); Ann. N. Y. Acad. Sci., 51, 781 (1949); б) H. S. H a r n e d, Chem. Rev.,
40, 461 (1947); в) G. L о n g s w о r t h, Ann. N. Y. Acad. Sci., 46, 211 (1945).
18. L. О n s a g e r, R. M. F u о s s, J. Phys. Chem., 36, 2689 (1932).
19. A. R. Gordo n, J. Phys. Chem., 5, 522 (1937); Ann. N. Y., Acad. Sci., 46, 285 (1945).
20. J. W. M с В a i n, C. R. D a w s о n, Proc. Roy. Soc., A118, 32 (1935).
21. G. S. Hartley, D. F. R u n n i с 1 e s, Proc. Roy. Soc., A168, 401 (1938).
22. E. C oh e n, H. R. Bruins, Z. Physik. Chem., 103, 337 (1923).
23. O. Lamm, Nova Acta Reg. Sci. Upsala, IV, 10 (6), (1937).
24. H. S. Harned, A. L. Levy, J. Am. Chem. Soc., 71, 2781 (1949).
25. A. К r u i s, Z. physik. Chem., B34, 1 (1936).
26. A. К r u i s, Z. physik. Chem., B34, 13 (1936).
27. W. G e I f c k e n, D.' Price, Z. physik. Chem., B26, 81 (1934).
28. A. L. Robinson, J. Am. Chem. Soc., 54, 1311 (1932).
29. J, P. Butler, A. R. Gordon, J. Am. Chem. Soc., 70, 2276 (1948).
30. R. H. Stokes, J. Am. Chem. Soc., 69, 1291 (1947).
31. a) H. S. Frank, J. Am. Chem. Soc., 63, 1789 (1941); б) H. S. Frank, .1. Chem.
Phys., 13, 479, 493 (1945); H. S. Frank, M. W. Evans, там же, 13, 507
(1945).
32. P. Debye, L. Paulin g, J. Am. Chem. Soc., 47, 2129 (1925).
33. E. H ii c k e 1, Physik, Z„ 26, 93 (1925).
34. J. B. Hasted, D. M. R i t s о n, С. H. Collie, J. Chem. Phys., 16, 1 (1948).
35. D. M. R i t s о n, J. B. Hasted, J. Chem. Phys., 16, 11 (1948).
36. L. О n s a g e r, J. Am. Chem. Soc., 58, 1486 (1936).
37. .1. G. К i r k w о о d, J. Chem. Phys., 4, 592 (1936).
38. N. В j e r r u m, Z. anorg. Chem., 109, 275 (1920).
39. R. H. S t о k e s, R. A. R о b i n s о n, J. Am. Chem. Soc., 70, 1870 (1948);
R. A. Robinson, R. H. Stokes, Ann. N. Y. Acad. Sci., 51, 593 (1949).
40. X. С. T e й и о p, Физическая химия, т. I, ОНТИ, Химтеорет, Л., 1935, р. 776—778.
41. J. D. В е г п а 1, R. Н. Fowler, J. Chem. Phys., 1, 515 (1933).
42. Н. S. Fran k, J. Am. Chem. Soc., 63, 1789 (1941).
43. S. В r u n a u e r, P. H. Emmett, E. Teller, J. Am. Chem. Soc., 60, 309
(1938).
44. R. B. A n d e r s о n, J. Am. Chem. Soc., 68, 686 (1946).
45. O. R e d 1 i c h, Chem. Rev., 39, 333 (1946).
46. O. Redlich, P. Rosenfeld, Stizber. Acad. Wiss. Wien, Math.-Naturw.
Klasse (Abt. II, b) 145, 87 (1936).
П Р И Л О ЖЬЕ Н И Е Б
589
47. N. R. R а о, Indian, J. Phys., 15, 185 (1941).
48. а) О. R е d 1 i с h, J. В i g е 1 е i s о n, J. Am. Chem. Soc., 65, 1883 (1943);
б) O. R ed 1 i c h, E. K. Holt, J. В i g e 1 e i s e n, там же, 66, 13 (1944).
49. T. F. Y о u n g, L. А. В 1 a t z, Chem. Rev., 44, 93 (1949).
50. R. M. Bell, M. A. Jepessen, J. Chem. Phys., 2, 711 (1934); 3, 245 (1935);
J. C h e d i n, Ann. Chim. (11), 8, 243 (1937); H. G e r d i n g, W. J. N i j v e 1 d,
Rec. trav. chim., 59, 1206 (1940); H. Nisi, Japan, J. Phys., 5, 119 (1929);
N. G. Pa i, Phil. Mag., 20, 616 (1935); N. R. R a o, Proc. Roy. Soc., A144, 159
(1934); N. R. R a o, Indian, J. Phys. 14, 143 (1940).
51. T. F. Young, S. R. Grinstead, Ann. N. Y. Acad. Sci., 51, 765 (1949).
52’ N. R. Rao, Indian, J. Phys., 16, 71 (1942).
53. N. R. Rao, Indian J. Phys., 17, 332 (1943).
54. H. O. Jenkins, Trans. Faraday Soc., 40, 19 (1944); 41, 138 (1945).
55. H. S. Harned, R. Davis, J r., J. Am. Chem. Soc., 65, 2030 (1943).
56. H.S. Harned, F. T. Bonner, J. Am. Chem. Soc., 67, 1026 (1945).
57. G. Scatchard, J. Am. Chem. Soc., 65, 1249 (1943).
58. F. J. W. Roughton, J. Am. Chem. Soc., 63, 2930 (1941).
59. R. Brinkman, R. M a r g a r i a, F. J. W. R о u g h t о n, Phil. Trans.
Roy. Soc., A332, 65 (1933).
60. В. B. Owen, E. J. King, J. Am. Chem. Soc., 65, 1612 (1943).
61. Bates R. G., J. Am. Chem. Soc., 70, 1579 (1948).
62. R. G. Bates, G. D. Pinching, J. Am. Chem. Soc., 71, 1274 (1949).
63. N. В j e r r u m, A. U n m a c k, Kgl. Danske, Videnskab, Selskab. Malh.-fys.,
Medd. 9, No 1 (1929).
64. E. R. Smith, R. A. Robinson, Trans. Faraday Soc., 38, 70 (1942).
65. A. W. Ralston, C. W. H о e r r, J. Am. Chem. Soc., 64, 772 (1942).
66. A. W. Ralston, C. W. H о e r r, E. J. Hoffman, J. Am. Chem. Soc.,
64,. 97 (1942).
67. a) P. E. Grieger, C. A. Kraus, J. Am. Chem. Soc., 70, 3803 (1948); A. B.
S с о t t, H. V. Tartar, E. C. L i n g a f e 1 t e г, там же, 65; 698 (1943);
A. W. К a 1 s t о n, D. N. E g g e n b e r g e г, там же, 70, 2918 (1948);
б) P. F. Grieger, Ann. N. Y. Acad. Sci., 51, 827 (1949).
68. P.- Debye, Ann. N. Y. Acad. Sci., 51, 575 (1949).
69. R. M. F u о s s, Science, 108, 545 (1948).
70. R. M. F u о s s, U. P. Strauss, Ann. N. Y. Acad. Sci., 51, 836 (1949).
71. U. P. Strauss, R. M. F u о s s, J. Polymer Sci., 4, 457 (1949).
ЛИТЕ PA ТУРА
К ТАБЛИЦАМ ПРИЛОЖЕНИЯ Б
К табл. 168
1. R. Т. Birge, Rev. Mod. Phys., 13, 233 (1941).
К табл. Ill
1. G. Jones, В. C. Bradshaw, J. Am. Chem. Soc., 55, 1780 (1933).
2. В. B. Owen, F. H. Sweeten, J. Am. Chem. Soc., 63, 2811 (1941).
3. T. Shedlovsky, J. Am. Chem. Soc., 54, 1411 (1932).
4. F. W. Tober, диссертация, Yale University, 1948.
5. K. A. Krieger, M. Kilpatrick, Jr., J. Am. Chem. Soc., 59, 1878 (1937).
6. G. C. Benson, A. R. Gordon, J. Chem. Phys., 13, 473 (1945).
7. T. S h e d 1 о v s к y, A. S. Brown, D. A. Maclnnes, Trans. Electrochem.
Soc., 66, 165 (1934).
8. H. Z e 1 d e s, диссертация, Yale University, 1947.
9. G. Jones, C. F. Bickford, J. Am. Chem. Soc., 56, 602 (1934).
10. H. E. Gunning, A. R. Gordon, J. Chem. Phys., 10, 126 (1942).
11. H. S. Harned, E. C. D r e b y, J. Am. Chem. Soc., 61, 3113 (1939).
12. L. G. Longsworth, J. Am. Chem. Soc., 54, 2741 (1932).
13. R. W. Allgood, A. R. Gordon, J. Chem. Phys., 10, 124 (1942).
14. R. W. Allgood, I. D. L e R о y, A. R. Gordon, J. Chem. Phys., 8, 418
(1940).
15. A. G. Keenan, A. R. Gordon, J. Chem. Phys., 11, 172 (1943).
46. L. G. Longsworth, J. Am. Chem. Soc., 57, 1185 (1935).
К табл. 175
1. a) G. J. J a n z, A. R. G о r d о n, J. Am. Chem. Soc., 65, 218 (1943); 6) R. W. A 1-
1 g о о d, A. R. Gordon, J. Chem. Phys., 10, 124 (1942).
590
ПРИЛОЖЕНИЕ Б
2. a) W. J. Hornibrook, G. J. J anz, A. R. Gordon, J. Am. Chem. Soc.,
64, 513 (1942); 6) R. W. Allgood, D. J. L e R о y, A. R.' Gord on, J. Chem.
Phys., 8, 418 (1940).
3. a) H. G. M c L e о d, A. R. Gordon, J. Am. Chem. Soc., 68, 58 (1946);
6) A. G. Keenan, H. G. McLeod, A. R. Gordon,!. Chem. Phys.,
13, 468 (1945).
4. a) E. A. M c W i 1 1 i a m , A. R. Gordon, J. Am. Chem. Soc., 65, 984 (1943);
6) A. G. Keenan, A. R. Gordon, J. Chem. Phys., 11, 172 (1943).
К табл. 176
1. R. A. Robinson, R. H. Stoke s, Trans. Faraday Soc., 45, 612 (1949).
2. R. II. Stokes, B. J. Levien, J. Am. Chem. Soc., 68, 333 (1946).
3, R. H. Stoke s, J. Am. Chem. Soc., 67, 1689 (1945).
4. R. A. R o b i n s о n, Trans. Faraday Soc., 41, 756 (1945).
5. R, A. R'obinso n, H. J. M с С о a c h, J. Am. Chem. Soc., 69, 2244 (1947).
6. R. A.. R о b i n s о n, J. Am. Chem. Soc., 68, 2402 (1946).
7. R. H. Stokes, Trans. Faraday Soc., 44, 295 (1948).
8. R. H. Stoke s, Trans. Faraday Soc., 41, 637 (1945).
9. R. A. R obinson, О. J. Baker, Trans. Roy. Soc., N. Z., 76, 250 (1946).
10. R. H. Stokes, R; A. Robinson, J. Am. Chem. Soc., 70, 1871 (1948).
К табл. 177
1. R. H. Stoke s, Trans. Faraday Soc., 44, 295 (1948).
2. R. A. R obins’on, P. K. Smith, E. R. B. Smith, Trans. Faraday Soc., 38„
63 (1942).
К табл. 182
1. H. S. Harned, R. Davis, J r., J. Am. Chem. Soc., 65, 2030 (1943).
2. H. B. Harned, F. T. Bonner, J. Am. Chem. Soc., 67, 1026 (1945).
3. R. G. Bates, S. F. Acree, J. Res. Nat., Bur. Standards, 30, 129 (1943).
4. R. G. Bates, G. L. Siegel, S. F. Acree, J. Res. Nat. Bur. Standards, 31, 205«
(1943),
5. E. J. К i n g, J. Am. Chem. Soc., 67, 2178 (1945).
6. R. G. Bates, G. D. Pinching, J. Am. Chem. Soc., 71, 1274 (1949).
К табл. 183
1. H. S. Harned, R. W. Ehlers, J. Am. Chem. Soc., 55, 2379 (1933).
2. A. Patterson, W. A. F e 1 s i n g, J. Am. Chem. Soc., 64, 1480 (1942).
3. R. L. M о о r e, W. A. F e 1 s i n g, J. Am. Chem. Soc., 69, 2420 (1947).
4. W. A. Felsing, M. May, J. Am. Chem. Soc., 70, 2904 (1948).
5. H. S. Harned, F. H. M. N e s t 1 e r, J. Am. Chem. Soc., 68, 966 (1946).
К табл. 184
1. H. S. Harned, F. T. Bonner, J. Am. Chem. Soc., 67, 1026 (1945).
2. В. B. Owen, E. J. К i n g, J. Am. Chem. Soc., 65, 1612 (1943).
3. E. J. Kin g, J. Am. Chem. Soc., 67, 2178 (1945).
ИМЕННОЙ У К А 3 Л Т Е JI Ь
Адамс (Е. Q. Adams) 496
Адамс (L. Н. Adams) 249, 258. 267, 269,
283
Аделл (В. Adell) 306, 310, 486, 496, 497
Аизер (М. Iser) 379, 385
Айрес (F. D. Ayres) 235, 239, 240, 266, 268
Акерлёф (G. С. Akerlof) 73, 119, 137, 228,
265, 310, 311, 315, 322, 324, 330, 337,
338*, 351, 352, 375, 384, 385, 386, 390,
416, 430, 432, 436, 438, 444, 446, 447,
522, 523, 551—553
Акри (S. F. Acree) 302, 310, 552, 590
Аксельрод Н. В. 444
Анваер Б. 226
Андауэр (М. Andauer) 268, 549
Андерсон (N. J. Anderson) 508, 550
Андерсон (R. В. Anderson) 576, 588
Ансон (М. L. Anson) 180
Армстон (J. W, Urmston) 429, 447
Аррениус (S. Arrhenius) 179, 180, 201, 215,
450, 495
Асбах (Н. R. Asbach) 267
Асмус (Е. Asmus) Д81
Астин (А. V. Astin) 137, 177
Астон (J. G. Aston) 181, 549
Бакстер (G. Р. Baxter) 242, 266
Бакстер (W. Р, Baxter) 419, 447
Балке (W. Н. Bahlke) 283
Барак (М. Barak) 178
Барксдел (J. Barksdale) 374, 385
Бартон (R. С. Barton) 429, 447
Батлер (J. Р. Butler) 568, 588
Батлер (J. А. V. Butler) 65, 73
Батсон (F. М. Batson) 214
Батчельдер (А. С. Batchelder) 488, 496
Баумштарк (G. Baumstark) 410, 416
Бахем (С. Bach'em) 264, 265, 267, 268
Бекман (С. Beckmann) .243, 266
Бекман (Е. Beckmann) 283
Бекштрём (S. Baeckstroip) 348, 386
Бельчер (D. Belcher) 181, 216, 304, 310,
549 .
Бенедикт (М. A. Benedict) 380, 385 .
Бенневптц (К. Bennewitz) 178, 240, 266
Бенсон (G. С. Benson) .589
Бергман (L. Bergmann) 267
Беркели (Berkeley) . 283 • . - -
Бернал (J. D. Bernal) 261, 267, 383, 385,
493, 496, 574, 588 '
Берри-(М. А. Вену) 362, 385
Бехтольд (М. F. Bechtold) 415
Бигельэйзен. (J. Bigeleisen) .589
Бикуорд (Р. Biquard) 263, 267
Бикфорд^С. F. Bickford) 178, 181, 549, 589*
Бильтц (W. Biltz) 385, 551
Бингхэм (Е. С. Bingham)- 179
Бинс (Н. Т. Beans) 450, 495
Бирдж (В. Т. Birge) 96, 116, 118, 137, 315г
337, 554, 588, 589
Бирдсэлл (С. М. Birdsall) 553
Бирон Е. (Biron Е.) 417
Битти (J. A. Beattie) 297, 310, 313, 337г
338, 384
Биховский (F. R. Bichowsky) 226, 265-
Бирр (Е. R. Birr) 215
Бланчерд (М. Н. Blanchard) 496
Блатц (L. A. Blatz) 576, 589
Блэкман (L. Е. Blackman) 283
Блюментрит (М. Blumentritt) 34, '45, 116,
209, 215
Боген (Е. С. Baughan) 490—492, 496
Боде (F. Bode) 417
Боллингер (D. М. Bollinger) 139, 177
Боллингер (G. М. Bollinger) 139, 178
Бонино (G. В. Bonino) 222, 265
Боннер (F. Т. Bonner) 578, 589, 590
Борн (М. Born) 65, 73, 214, 321, 490, 49$
Боусфильд (Е. Bousfield) 385
Боусфильд (W. R. Bousfield) 267, 353, 38&
Брайтон (Т. В. Brighton) 450, 495
Брамбог (N. J. Brumbaugh) 447
Браун (A. S. Brown) 181, 310, 340, 341,
367, 382, 384, 385, 549, 567, 589
Брекенридж (G. F. Breckenridge) 447
Брен дель (В. Brendel) 2-15
Бренстед (J. N. Bronsted) 32, 177, 300, 310,
365, 369, 385, 413, 414, 417, 418—420,.
432, 435,.441, 447
Бресчиа (F. Brescia) 492, 496
Бриджер (G. L. Bridger)' 267
Бриджмэн (Р. W. Bridgeman) 258, 267
Бринкли (R. F. Brinkley) 565
Бринкли (8. R. Brinkley) 266, 268, 307,.
310, 463, 495, 496, 555, 557, 563, 585, 588
Бринкман (R. Brinkman) 589
Бриско (Н. Т. Briscoe) 173, 180
Бриттон (Н. Т. S. Britton) 302, 310
Брицке Э. В. 226 .
Бродский А. И. 4, 23, £0, 61, 167, 267,
412, 416, 444 .. -
Брокман (F. G. Brockman) J81, 216
Брунауэр (S. Brunauer) 576, 588
Бруине (Н. R. Bruins) 562, 588
Брэдшоу (В, С. Bredshaw) 140, 141, 152,
160, 169, 178, 179, 180, 558, 589
592
ИМЕННОЙ УКАЗАТЕЛЬ
Брэй (U. В. Bray) 398, 399, 400, 416
Брэй (W. С. Bray) 146, 156, 178, 180, 185,
214 418 447
Брюлль (W. ВгйП) 181
Булок (Е. Bullock) 178
Буш (G. Busch) 178, 214
Бьен (G. S. Bien) 197, 214, 215
Бьеррум (N. Bjerrum) 29, 30, 32,-33, 45, 54,
55, 57—59, 61, 73, НО, 125, 137, 182,
189, 198, 309, 310, 312, 361, 383, 385,
398, 496, 572, 573, 574, 586, 588, 589
Бэдфорд (G. Т. Bedford) 416
Бэйтс (В. G. Bates) 392, 393, 396, 397, 416,
495, 551, 552, 586, 589, 590
Бэйтс (S. В. Bates) 429, 447
Бэкер (L. Е. Baker) 183, 214
Бэкер (О. J. Baker) 590
Бэлл (В. М. Bell) 589 ,
Бэнкс (W. Н. -Banks) 178
Бэр (Beers) 468
Бэртон (С. V. Burton) 283
Бэртон (J. О. Burton) 552
Ваглио (V. Vaglio) 222, 265
Вагнер (С. Wagner) 70, 73, 178, 380, 382
Вагнер (К. W. Wagner) 139, 177
Ваймен (J. Wyman) 119, 137, 228, 229, 265,
368, 385, 496
Вайнел (G. W. Vinal) 413, 414, 417
Валледдер (В. В. Vallender) 229, 266
Вальде (A. W. Walde) 479, 496
Вальден (Р. Walden) 142, 155, 178, 184,
189, 192, 194, 214
Ваизелов (А. Р. Vanselow) 283, 322, 338
Ван-Лаар (J. J. van-Laar) 33, 45, 271,
283
Ван-Риссельберге (Р. van-Rysselberghe)
176, 180, 360, 370, 371, 385, 573
Вазастьерна (J. A. 'Wasastierna) 137
Вернон (Е. L. Vernon) 116, 171, 179
Веселовский Б. 226
Виземан (L. A. Wiseman) 385
Вильсон (J. М. Wilson) 417
Вильсон (В. W. Wilson) 267
Вильсон (W. S. Wilson) 34, 45, 101, 105,
116, 135
Вильямс (G. V. Williams) 179
Вин (М. Wien) 116, 205, 206, 208, 210, 215
Винджи (В. A. Vingee) 214, 283
Вин-Джонс (W. F. К. Wynne-Jones)
177, 475, 476, 479, 491, 494, 496
Виноград (J. В. Vinograd) 177, 180
Винцент (Р. С. Vincent) 266
Вирт (Н. Е. Wirth) 241, 266, 268
Вольфенден (J.' Н. Wolfenden) 168, 172,
179, 181
Восбург (W. С. Vosburgh) 392, 396, 398,
416, 495, 552
Воскресенская Н. К. 250, 339
Вуд (О. Wood) 229
Вуд (S. Е. Wood) 276, 284, 354, 385, 411, 417
Вулкок (J. W. Woolcock) 315, 337
Вустер (С. В. Wooster) 214
Вэнс (J. Е. Vance) 552
Вюст (J. Wtist) 501
Гальбан (Н. Halban) 467, 468, 469, 486,
487, 495, 496
Гамер (W. J. Hamer) 231, 266, 276, 284,
303, 310, 384, 385, 403, 409, 410, 411
415—417, 448, 452, 495, 496, 552
Гаммершмид (Н. Hammerschmid) 268, 352,
385, 549 '
Ганнинг (Н. Е. Gunning) 141, 169, 180,
181, 498, 549, 558, 589
Гапон Е. Н. 339
Гарнье (Garnier) 417
Гарретсон (Н. Н. Garretson) 541, 553
Гартинг (Н. Harting) 267
Гатчек (Е. Hatschek) 179
Гаусрат (Н. Hausrath) 269, 283
Геддес (J. A. Geddes) 215
Гейдвейлер (A. Heydweiller) 380, 381, 385,
495
Гейдж (F. W. Gage) 248, 267
• Гельмгольц (Н. Helmholtz) 310
Гемант (A. Gemant) 208, 211, 215
Гендерсон (Р. Henderson) 298, 299, 303,
310
Герасимов Я. И. 250
Гердинг (Н. Gerding) 589
Германе (J. J. Hermans) 93, 116, 175, 180,
183, 214, 298, 310
Герсбергер (W. D. Hersberger) 267
Герц (Р. Hertz) 33, 45, 156, 178
Герц (W. Herz) 385, 551
Гесс (С. В. Hess) 268
Гетман (F. Н. Getman) 416
Гефкен (W. Geffcken) 242, 243, 245, 247,
266—268, 564, 566, 588
Гиббс (J. W. Gibbs) 19, 20, 22—24, 32, 296,
301, 310
Гибенталь (F. Hiebenthal) 385, 551
Гибсон (J. A. Gibson) 231, 266
Гибсон (R. Е. Gibson) 252, 256, 257—261,
265, 267, 268, 588
Гильберт (D. Hilbert) 83, 116
Гири (С. G. Geary) 452, 495, 496, 552
Гиртле (D. S. Hirtle) 343, 346, 384, 550
Гитторф (W. Hittorf) 160, 179
Глесстон (S. Glasstone) 375, 385, 551
Говорка (F, Govorka) 315, 338, 416
Гольдман (F. Н. Goldman) 447
Гольдшмидт (V. М. Goldschmidt) 121, 137
Горбачев С. В. 4
Гордон (А. В. Gordon) 141, 169, 176, 179,
180, 181, 284, 340, 354, 384, 386, 410,
416, 417, 549, 559, 562, 567, 568, 588, 590
Гордон (V. Gordon) 385
Горенбейн Е. Я. 167
Горин (М. Н. Gorin) 157, 158, 165, 178
Городецкая А. В. 301
Гофман (Е. J. Hoffmann) 589
Гофман (F. Hoffmann) 377, 385
Гош (Y. С. Ghosh) 33, 45, 156, 178
Гренье (W. L. Groenier) 223, 224, 265
Григер (Р. Е. Grieger) 588, 589
Гриннелл (Grinnell S. W.) 310
Гринстед (S. R. Grinstaed) 589
Гриф (L. J. Greiff) 53, 72, 137, 393
Гроллмэн (A. Grollman) 409, 410, 416
Гронволл (Т. Н. Gr on wall) 48, 53, 54, 72,
137, 243, 266, 316, 317, 360, 368, 385,
387, 393, 398
Гросс (Р. Gross) 63, 69, 73, 378, 379, 380,
385
Грюнайзен (Е. Griineisen) 168, 179
ИМЕННОЙ УКАЗАТЕЛЬ
593
Гуггенгейм (Е. A. Guggenheim) 32, 299—
301, 303, 310, 365, 366, 367, 382, 385,
432, 436, 442
Гудив (С. F. Goodeve) 266
Гукер (F. Т. Gucker) 62, 73, 220, 221, 229,
230, 235, 237—240, 244, 248, 251—254,
256, 257, 264, 265—267, 268, 332, 338, 588
Гульбрансен (Е. A. Gulbransen) 223, 224,
265, 268, 341, 342, 346, 384
Гульд(R. К. Gould) 495
Гарни (R. W. Gurney) 490 , 491, 492 , 496
Гэрри (R. W. Gurry) 180, 181, 400, 416, 549
Гэтти (О: Gatty) 73
Гюккель (Е. Hiickel) 33, 34, 45, 54, 74, 85,
142,- 178, 293, 310, 324, 338, 339, 358,
359, 368, 383—385, 571, 572, 588
Гюнтельберг (Е. Guntelberg) 48, 50, 72,
305, 310, 313, 322, 337, 338, 385, 427,
428, 432, 436, 441, 447, 448, 486, 496
Дайк (Р. Н. Dike) 177
Даймонд (D.. W. Dimond) 375, 385, 551
Данеш (V, Z. Danes) 422, 447
Данешойа (В. A. DaneSova) 422, 447
Даннер (Р. S. Danner) 315, 337
Даркен (L. S. Darken) 181, 215, 216
Даусон (С. R. Davson) 175, 180, 562, 588
Дебай (Р. Debye) 33, 34, 38, 45, 48, 54, 63,
66, 68, 73, 74, 85, 97, 116, 142, 178, 206,
215, 263, 571, 587, 588, 590
Деделл (Т. R. Dedell) 497, 553
Дейбнер (A. Deubner) 206, 215
Демиденко С. Г. 267
Денисон (R. R. Denison) 158, 178
Де-Фриз (De Vries) 401, 416, 417
Джемс (G. М. James) 347, 384, 447, 495
Джемс (W. A. James) 180
Дженис (A. A, Janis) 276, 284
Дженкинс (Н. О. Jenkins)'496, 578, 589
Джеппесен (М. A. Jeppesen) 589
Джессел (R. Jessel) 266
Джиллеспай (L. J. Gillespie) 231, 232, 266
Джозефе (R. С. Josephs) 139, 177
Джой (W. Е. Joy) 168, 172, 179
Джонс (A. Jones) 579
Джонс (Е. С. Jones) 375, 385
Джонс (G. Jones) 79, 116, 139—141, 155,
156, 160, 165, 168, 169, 172, 177—181,
268, 297, 310, 348, 381, 382, 385, 386,
388, 415, 549, 558, 589 .
Джонс (J. Н. Jones) 551
Джонс (Р. Т. Jones) 180, 283, 549
Джонс (R. S. Jones) 399, 401, 416
Джонс (Т. Т. Jones) 137
Джонсон (Е. G. Johnson) 214
Джонсон (G. G. Johnson) 273, 283
Джонсон (О. Johnson) 266
Джоуль (J. Р. Joule) 266
Джэксон (R. G. Jackson) 179
Диппи (J. F. Dippy) 496
Диссельхорст (Н. Diesselhorst) 178
Дитеричи (С. Dieterici) 283
Дол (М. Dole) 45, 79, 116, 155, 156, 160,
165, 168, 171, 178—181, 214, 297, 310,
381, 385, 388, 389, 415
Долеман (Е. Doehlemann) 228, 265
Домке (J. Domke) 267
Дон (R. S. Done) 553
Дональдсон (G.W. Donaldson) 179, 181, 549
38 г. Харнед, Б. Оуэн
Донелсон (J. G. Donelson) 315, 337, 347,
348, 384, 452, 495, 496, 550, 552
Дорошевский А. 191
Доу (М. Т. Dow) 119, 588
Дреби (Е. С. Dreby) 310,333, 334, 338, 550,
589
Дрейк (F. Н. Drake) 119, 137, 554, 588
Дрейсбах (D. Dreisbach) 315, 337
Друкер (С. Drucker) 416
Дрью (Е. Drew) 179
Дуглас (S. М. Douglas) 384
Думанский A. R. 191
Дэвис (С. W. Davies) 149, 152, 156, 177,
178, 179, 181, 188, 215, 395, 398, 400,
416
Дэвис (R. М. Davis) 137, 578, 589, 590
Дэзидерьева И. П. 250
Дэлиен (F. Е. Dalian) 173, 180
Дюрр (F. Durr) 228, 265, 549
Елен (F. С. Jelen) 181, 549
Зак (Н. Sack) 205, 206, 215
Зейлер (М. Seiler) 487, 496
Зелигман (Р. Seligmann) 224, 225, 265
Зелинский Н. Д. 191
Зёренсен (S. Р. L. Sorensen) 302 , 310
Зигель (G. L. Siegel) 590
Зидентопф (К. Siedentopf) 468, 496
Зифен (N. Siefen) 267
Изгарышев Н. А. 4, 23, 301
Измайлов Н. А. 50, 302
Ингэлс (Е. N. Ingalls) 117, 137, 554, 558
Иоос (G. Joos) 34, 45, 116, 209, 215
Иоффе А. Ф. 247, 267
Каблуков И. А. 4, 167, 191
Казаньян (G. L. Kazanjian) 553
Какраварти (A. S. Chakravarti) 172, 180
Кам (J. Кат) 377, 385
Канеко (S. Kaneko) 152, 178
Капустинская Н. П. 250
Капустинский А. Ф. 4, 57, 121, 226, 250,
339, 444
Карапетьянц М. X. 4
Карл (И. Carl) 267
Кармоди (W. R. Carmody) 313, 314, 337,
338, 392, 398, 416, 551
Карпентер (Р. G. Carpenter) 175, 180
Като (Y. Kato) 459, 495
Кауперсвейт (J. A. Cowperthwaite) 73,
179, 374, 385, 398, 399, 400, 416, 499
Кёнер (Н. Kohner) 266
Кёниг (F. О. Koenig) 298, 310,
Кенен (A. G. Keenan) 567, 590
Кибрик (A, Kibrick) 149, 180
Киджельс (G. Kegeles) 351, 352, 384, 386,
522 551 553
Кильпатрик (М. Kilpatrik) 145, 178, 216,
493, 496, 549, 589 , .
Кильпатрик (М. L. Kilpatrik) 181
Кинг (С. V. King) 420, 447
Кинг (Е. J. King) 310, 589, 590
Кинкэид (J. F. Kincaid) 258, 267, 268
Киреев Н. А, 339
Кирквуд (J. G, Kirkwood) 33, 472, 496,
572, 588
КистяКовский В. А. 4
594
ИМЕННОЙ УКАЗАТЕЛЬ
Клак (В. W. Clack) 175, 180
Кларк (W. М. Clark) 45, 303, 310
Клейн (Е. Klein) 267
Клингер (Н. Klinger) 248, 267, 302, 310
Клокман В. Р. 250
Клотц (J. М. Klotz) 266, 403, 416, 541, 552
Кнобель (М. Knobel) 350, 384
Кокель (L. Kockel) 228, 266
Кокс (N. L. Сох) 214, 216
Кокс (W. М. Сох) 179, 181
Колли (В. Collie) 179
Колли (С. Н. Collie) 571, 572, 588
Колли (С. R. Collie) 179
Коллинз (Е. М. Collins) 409, 410, 416
Кольнин (J. Н. Colvin) 181, 268
Кольрауш (F. Kohlrausch) 140, 141, 155,
159, 178, 179, 202, 215, 266, 495
Кольтгоф (J. М. Kolthoff) 416
Кон (Е. J. Cohn) 368, 385
Копсон (Н. R. Copson) 452, 495, 552
Кортюм (G. Kortum) 468, 469 , 486 , 487,
496
Котрель (F. G. Cottrell) 270, 283
Коульсон (С. A. Coulson) 493, 496
Коэн (Е. Cohen) 562, 588
Крапивин С. 191
Кратц (L. Kratz) 240, 266
Краус (С. A. Kraus) 56—60, 73, 125, 137,
156, 177, 178, 185, 186, 189—192, 194,
195, 197—199, 214, 215, 283, 312, 337,
365, 587, 589
Краус (Р. В. Kraus) 214, 216
Кригер (К. A. Krieger) 145, 178, 549, 589
Кричевский И. Р. 267
Крокфорд (Н. D. Crockford) 422, 447
Кроуфорд (С. С. Crawford) 350, 384, 550
Круис (A. Kruis) 243, 266, 268, 564, 566,
588
Крэйг (D. N. Craig) 413, 414, 417
Крэмер (Н. A. Kramers) 33
Кук (М. A. Cook) 175, 180, 268, 342, 346,
349, 350, 352, 384, 385, 431, 442, 443,
447, 455, 495, 550, 551
Кук (R. G. Cook) 447
Кук (Т. F. Cooke} 439, 440, 447
Кундт (A. Kundt) 263, 267
Кунц (А. Н. Kunz) 307, 310
Курант (R. Courant) 83, 116
Кэди (Н. Р. Cady) 158, 179
Кэллем (D. В. Kellam) 447, 552 •
Кэлмон (С. Calmon) 315, 337, 338, 550
Кэннан (R. К. Саппап) 149, 180
Кэстон (A. S. Keston) 347, 348, 384, 550
Кэшман (О. Е. Cushman) 412, 416
Кюхлер (К. Kiichler) 178
Ламберт 468
Ла-Мер (V. К. La Мег) 48, 53, 54, 72, 73,
137, 1,83, 214, 243, 267, 283, 316, 317,
360, 368, 374, 385, 387, 393, 398, 399,
400, 416, 419, 420, 447, 492, 496
Ламперт (R. Н. Lampert) 231, 232, 266
Ланге (Е. Lange) 178, 220—222, 227, 228,
231, 265, 266, 339, 390, 398, 399, 412, 413,
' 410, 549
Ланге (J. Lange) 178, 373, 385, 549, 557,
588
Лангебек (К. Langebeck) 377, 385
Лангер (Т. W. Langer) 181, 215, 216
Лангмюир (J> Langmuir) 381, 385
Лангфорд (С. Т. Langford) 447
Ланде (A. Lande) 137
Ландольт (Landolt—Bomstein) 180
Ланженен (Р. Langevin) 77, 116, 213
Ларсон (Е. Larsson) 305, 310, 486, 496,
497
Ларсон (W, D. Larson) 215
Лассель (Р. A. Lasselle) 181, 549
Латимер (W. М. Latimer) 292, 309, 310,
493, 496
Лауренс (V. D. Laurence) 172, 179, 181
Лени (A. L. Levy) 562, 588
Лениен (В. J. Levien) 590
Лейгтон (Р. A. Leighton) 268, 549
Леман (D. Lehmann) 267
Лемке Н. 182
Лемм (О. Lemm) 562, 588
Ле-Рой (D. J. Le Roy) 158, 178, 180, 549,
567, 589, 590
Леффлер (О. N. Loeffler) 268, 588
Ли (N. С. Li) 181
Ли (R. Е. Lee) 243, 244, 266
Ливингстон (R. S. Livingston) 403, 416
Лингарт (G. A. Linhart) 307, 309, 310, 313,
337, 384
Лингафельтер (Е. С. Lingafelter) 589
Линдерштрём-Ланг (К. Linderstr0m-Lang)
302, 310
Липилина И. И, 250
Лиу (Т. Н. Liu) 180
Ловлейс (В. F. Lovelace) 269, 283, 349,
384, 386
Лодж (О. Lodge) 158, 178
Ломоносов М. В. 4
Лонгснорт (L. G. Longsworth) 158—160,
164—167, 178, 179, 181, 214, 266, 335,
338, 340, 498, 499, 549, 558; 560, 588,
589
Лоренц (R. Lorenz) 154, 156, 178
Лоури (Т. М. Lowry) 267
Лукас (R. Lucas) 263, 267
Лукасе (W. W. Lucasse) 310, 315, 337, 388,
415, 416, 417
Лумис (A. L. Loomis) 263, 267, 268
Лумис (N. Е. Loomis) 417, 447
Льюис (G. N. Lewis) 24, 32, 179, 227, 265,
279, 280, 283, 286, 287, 292, 309, 338,
347, 384, 407, 408, 410, 412, 416, 418,
447, 450, 495
Лэмб (А. В. Lamb) 243, 244, 266
Лэнман (Е. Н. Lanman) 256, 267
Лэттэй (R. Т. Lattey) 155, 178, 214
Людер (W. F. Luder) 139, 177, 214
Маге (J. L. Magee) 492, 496
Майкельс (F. Michels) 210
Мак-Вэн (J. W. McBain) 158, 173, 176—
178, 180, 377, 385, 562, 588
Мак-Вильям (Е. A. Me William) 590
Мак-Дугалл (F. MacDougall) 423, 447
Мак-Иннес (D. A. Mclnnes) 45, 158, 160,
166, 167, 178, 179, 181, 202—204,214—
216, 297, 304, 310, 311, 314, 316, 323,
324/ 337, 338, 340, 341, 349, 367, 382,
384—386, 402, 416, 498, 499, 530, 549,
551 567 589
Мак-Интош (L. R. McIntosh) 214, 215
Мак-Коч (Н. J. McCoach) 590
именной указатель
595
Мак-Леод (Н. G. McLeod) 567, 590
Мак-Микин (Т. L. McMeckin) 496
Мак-Олэй (J. McAulay) 63, 73
Максвелл (С. Max-wall) 75, 115
Мальш (J. Malsch) 206, 215
Маннвейлер (G. Е. Mannweiler) 452, 457,
495, 496, 552
Маргариа (R. Margaria) 589
Мардок (Р. G. Murdock) 429, 447
Мартин (A. R. Martin) 156, 178
Мартин (A. W. Martin) 216
Мартин (W. Е. Martin) 177
Махин (J.' S. Machin) 236, 266
Мейер (Н. F. Meier) 169, 181, 215, 216
Мейер (Е. Meyer) 495
Менделеев Д. И. 4, 218
Мертон (Т. R. Merton) 179
Месснер (J. Messner) 265, 549
Мид (D. J. Mead) 211—216
Миллер (Е. Miller) 269
Миллер (W. L. Miller) 159, 179
Мильнер (R. Milner) 33, 45
Минник (L. J. Minnick) 493, 496
Мисловицер (Е. Mislowitzer) 302, 310
Миттелыптедт (О. Mittelstaedt) 215
Митчел (J. W. Mitchell) 396, 416, 551
Михаэль (W. Michael) 178
Михельс (F. Michels) 215
Мичэм (М. R. Meacham) 418, 447
Миценко К. П. 339
Мозер (С. Е. Moser) 248, 254, 267
Монгейм (J. Monheim) 221, 231, 265, 266,
268, 398, 399, 412, 413, 416, 549
Монэй (R, W. Money) 180
Моррисон (J. О. Morrison) 315, 337
Моэсвельд (A. L. Т. Moesveld) 267
Муалье (J. С. Moilliet) 179
Мур (R. L. Moore) 550, 590
Мэзон (С. F. Mason) 419, 420, 447
Мэзон (С. М. Mason) 403, 416, 552
Мэзон (М. Mason) 552
Мэзон (R. В. Mason) 494, 496
Мэй (М. Мау) 590
Мэйр (В. J. Mair) 256, 267
Мэррэй-Раст (D. М. Murray-Rust) 178
Мэрфи (G. М. Murphy) 310, 495
Мэссон (D. О. Masson) 158, 178, 240, 245,
265, 266
Мюллер (Н. Miiller) 48, 72
Нейман (Е. W. Neuman) 447
Нельсон (A. F. Nelson) 283
Нернст (W. Nemst) 45, 95, 116, 158, 174,
177, 178, 180, 220, 265
Нестлер (F, Н. М. Nestler) 590
Нивельд (W. J. Nijveld) 589
Николаев А. В. 250
Нильсон (R. F. Nielson) 177
Нимс (L. F. Nims) 338, 342, 384, 495, 496,
550, 552, 553
Ниси (Н. Nisi) 589
Ноде (F. М. Naude) 229, 266
Нойес (A. A. Noyes) 33, 45, 179, 202, 215,
216, 313, 337, 403, 410, 416, 418, 447,
459, 495
Нонгебель (G, Nonhebel) 315, 317, 550
Нортроп (J. Н. Northrup) 173, 176, 180
Нуттал (R. L. Nuttal) 588
Ньютон (R. F. Newton) 388, 389, 390, 415,
417, 551
Оакс (Е. Т. Oakes) 450 , 495
Олгуд (R. W. Allgood) 177, 180, 567, 589,
590
Олиник (Р. Olynyk) 354, 386
О’Нейль (L. O’Neill) 447
Онзагер (L. Onsager) 33, 34, 45, 48, 70—74,
77, 80, 83—85, 87, 90, 92—94, 97, 98,
99, 101, 105, 110—114, 116, 129, 133,
135, 136, 137, 142, 145, 146, 147, 149,
150, 166, 173, 175, 176, 178, 180, 189,
207, 210, 211, 212, 215, 297, 370, 374,
380, 382, 383, 385, 398, 560, 572, 588
Ортман (W. Orthmann) 265
Осака (Y. Osaka) 269, 283
Освальд (Р. Osswald) 178
Оствальд (W, Ostwald) Г78, 201, 215
Оуэн (В. В. Owen) 178, 179, 180, 181, 215,
266, 268, 307, 310, 311, 312, 318, 337,
347, 362, 384, 400, 416, 439, 440, 447,
449, 463, 475, 479, 483, 495—498, 549,
553, 555, 557, 558, 563, 585, 588, 589, 590
Паранджпэ (G. R. Paranjpe) 172, 180
Паркер (Е. W. Parker) 140, 141, 178
Паркер (Н. С. Рагкег) 140, 141, 178
Паркер (К. Parker) 297, 310, 349, 384
Паркс (W. G. Parks) 400, 416
Партингтон (J, R. Partington) 160, 422, 447
Партон (Н. N. Parton) 396, 416, 551, 579
Паскаль (Р. Pascal) 417
Пасынский А. Г. 250, 383
Паттерсон (A, Patterson) 550, 590
Паулинг (L. Pauling) 120, 137, 369, 385,
571, 588
Паунд (A. Pound) 375, 385, 551
Педерсен (К, Pedersen) 491, 496
Перман (Е. Р. Perman) 229, 264, 266, 267
Петерсен (A. Petersen) 419, 447
Пикар (Н. G. Pickard) 221, 229, 230, 240,
265
Пинчин.г (G. D. Pinching) 586, 589, 590
Пирс (J. N. Pearce) 269, 283
Пирс (W. Т. Pearce) 447
Пирс (G. W. Pierce) 119, 137, 554, 588
Писаржевский Л. В. 182, 191
Питцер (К. S. Pitzer) 309, 310, 404, 416,
479, 496
Планк (М. Planck) 214, 298, 299, 310
Планк (R. W. Planck) 221, 229, 230, 240,
265, 266
Плато (F. Plato) 267
Плесков В. А. 23, 167
Плотников В. А. 167, 191
Поллиссар (М. j. Polissar) 116
Попов (S. Popoff) 307, 310
Портер (A. W. Porter) 156, 178, 229, 266
Прайс (D. Price) 243, 244, 245, 266, 268,'564,
566, 588
Прасад (В. Prasade) 172, 180
Прасад (F. Prasade) 180
Пратт (F. R. Pratt) 229, 266
Прендергаст (М. J. Prendergast) 140, 178
Прентис (S. S. Prentiss) 180, 282, 283, 314,
324, 337, 349, 372, 373, 382, 384, 385,
386, 441, 448, 549, 550
Пуазейль (J. L. Poiseuille) 168, 179
38*
596
ИМЕННОЙ УКАЗАТЕЛЬ
Пулер (L. G. Pooler) 268
Пфаундлер (L. Pfaundler) 266
Пэйдж (L. Page) 77, 116
Пэйн (Н. М. Paine) 447
Цэске (В. Peace) 247, 267
Райт (С. Р. Wright) 178
Райт (D. 'D. Wright) 314, 337, 552
Ралстон (A. W. Ralston) 589
Ранад (J. D. Ranade) 172, 180
Ранниклс (D. F. Runnicles) 562, 588
Рао (N. R. Rao) 589
Рауль (F. М. Raoult) 269, 283
Реджиани (N. Reggiani) 266
Редлих (О. Redlich) 248, 266, 267, 303, 310,
576, 577, 588, 589
Рей (W. A. Ray) 381, 382, 385
Рейтмейер (R. Е.-Reitmeier) 181, 216, 549
Ренгольм (J. Е. Renholm) 145, 178
Рендалл (М. Randall) 24, 32, 227, 235, 265,
266, 268, 269, 279, 280, 283, 286, 287,
292, 309, 322, 338, 339, 375, 377, 378,
384, 385, 386, 390, 405, 407,408, 410,
412, 416—418, 447, 551
Рердам (Н. N. К. Rordam) 377, 385
Ри (Т. Ri) 492, 496
Рид (J. W. Read) 270, 283
Риджелато (Е. С. Righellato) 178, 179, 180,
395, 398, 416
Рикхоф (Н. Rieckhoff) 215
Ритсон (D. М. Ritson) 571, 572, 588
Ричардс (Т. W. Richards) 220, 231, 234,
265, 266, 267, 388, 415
Робертс (Е. J. Roberts) 313, 314, 337, 338
Робертс (В. М. Roberts) 472, 495, 496
Робертсон' (С. Robertson) 283, 416
Робинсон (A. L. Robinson) 219, 220, 223,
224, 231, 265, 266, 268, 341, 342, 346,
352—353, 354, 355, 367-, 369, 382, 384—
386, 388, 392, 397, 399, 412, 413, 416,
549, 566, 588
Робинсон (R. A. Robinson) 181, 265, 275,
276, 283, 284, 305, 310, 390, 396, 399,
401, 403, 411, 415, 416, 417, 457, 475,
495, 496, 497, 517, 526, 530, 534, 551, 552,
568, 570, 573, 574, 575, 580, 589, 590
Родебуш (W. Н. Rodebush) 416
Роджерс (Т. Н. Rogers) 269
Розенберг (Н. Rosenberg) 495
Розенфельд (Р. Rosenfeld) 266, 576, 588
Ролов (М. Roloff) 269, 283
Ролстон (A. W. Ralston) 587, 589
Ротмунд (V. Rothmund) 385
Ротрок (D. A. Rothrock) 215
Россини (F. D. Rossini) 226, 231, 235, 265,
266, 268, 346, 384, 386, 390, 416
Роу (A. W. Rowe) 231, 266
Роутон (F. J. W. Roughton) 585, 589
Рубин (Т. R. Rubin) 235, 237, 238, 244,
256, 266, 268
Рубинштейн А. М. 339
Рут (W. С. Root) 241, 247, 266
Рутледж (G. Rutledge) 335, 338, 415
Рэлей (Rayleigh) 283
Сарджент (L. W. Sargent) 450, 495
Саханов (A. Sachanov) 185, 214
Светославский (W. Swietoslawsky) 221, 283
Свинделс (F. Е. Swindells) 350, 384, 386,
447, 495
Свифт (Е. Swift) 214, 549
Себастьян (R. L. Sebastian) 450, 495
Сегден (J. N. Sugden) 377, 385
Семенченко В. К. 4, 55, 57
Сеченов И. М. 375, 385
Сзалай (A. Szalay) 267
Сивертц (V. Szvertz) 181, 216, 549
Сиз (V. В. Sease) 283, 349, 384, 386
Синглетерри (С. R. Singleterry) 403, 416,
541, 552
Синклер (D. A. Sinclair) 283, 284, 353,
354, 369, 385, 551
Сирс (F. W. Sears) 263, 267
Скарре О. К. 267
Сколе (S. R. Scholes) 552
Скотт (А. В. Scott) 589
Скотт (A. F. Scott) 242, 245—247, 256, 266,
267, 268
Скотт (G. N. Scott) 390, 405, 410, 416, 417
Скэтчард (G. Scatchard) 73, 180, 260, 276,
281, 282, 283, 284, 310, 313—315, 324,
337—339, 349, 354, 368, 369, 372, 373,
380, 382, 384—386, 388, 392, 411, 415,
416, 417, 441, 448, 519, 550, 572, 589
Смит (Е. R. Smith) 158, 179
Смит (Е. R. В. Smith) 553, 570, 589, 590
Смит (J. S. Smith) 555, 588
Смит (Р. К. Smith) 496, 552, 553, 570, 590
Смит (R. Р. Smith) 270, 273, 283, 284, 343,
346, 349, 384, 521, 550
Смит (W. V. Smith) 309, 310
Смите (A. Smiths) 283
Сноу (R. D. Snow) 283
Соколик А. 417
Соколик С. 417
Солана (L. Solana) 243, 266
СоСмэн (R. В. Sosman) 459, 495
Спенсер (Н. М. Spencer) 281, 283, 314, 338,
349, 384, 386
Стил (В. D. Steele) 158, 178
Стокс (G. G. Stokes) 86, 182, 184
Стокс (R. Н. Stokes) 396, 415, 416, 417, 526,
551, 552, 568, 573—575, 588, 590
Стонхилл (Н. I. Stonehill) 362, 385
Стонхилл (Н. J. Stonehill) 422, 447
Сторч (Н. Storch) 347, 384
Стрик (Н. Streeck) 221, 265, 268, 390, 416,
549
Стуртевант (J. М. Sturtevant) 330, 338, 479,
496, 502, 549
Стэлл (W. N. Stall) 267
Стюарт (G. W. Stewart) 267
Стюарт (М. A. Stewart) 403, 416
Сьютон (F. Н. Sweeton) 181, 498, 549, 558,
589
Сэзерлэнд (R. О. Sutherland) 552
Сэзерленд (W. Sutherland) 33, 45
Сэкстон (В. Saxton) 169, 181, 215, 216, 349,
384
Сэмарас (N. N. I. Samaras) 70—73, 129,
137, 380, 382
Сэмис (С. S. Samis) 162, 163, 179
Сэндвед (К. Sandved) 48, 53, 54, 72, 137,
316, 317, 360, 368, 374, 385, 386, 398
Тамман (G. Tamman) 259, 267
Тарк (Н. Е. Turck) 430, 438, 448
ИМЕННОЙ УКАЗАТЕЛЬ
597
Тартар (Н. V. Tarter) 181, 216, 541, 549,
553, 587, 589
Теллер (Е. Teller) 576, 588
Темкин М. И. 301
Тефт (R. F. Tefft) 388, 392, 393, 415, 416
Тильтон (L. W. Tilton) 555, 588
Типпетс (Е. A. Tippets) 388, 389, 390, 415,
417, 551
Тир (J. W. Теаге) 322, 330, 338, 430, 438,
447, 448, 552
Тобер (F. W. Tober) 589
Толлей (S. К. Talley) 179, 181, 268
Толлерт (Н. Tollert) 179
Тольман (R. С. Tolman) 45
Томас (Н. С. Thomas) 315, 337,422,432,
436, 438, 444, 447, 550
Томпсон (Т. G. Thompson) 266
Томсицек (W. J. Tomsicek) 215
Томсон (J. J. Tomson) 73
Топп (N. Е. Торр) 149, 180
Трахтенберг Ф. 167
Тэйлор (А. С. Taylor) 304, 306, 310, 553
Тэйлор (Н. S. Taylor) 32, 45, 160, 178, 179,
202, 215, 384,- 573, 588
.Тэйлор (J. К. Taylor) 555, 588
.Тэйлор (М. D. Taylor) 417
Тэйлор (Р. В. Taylor) 296, 298, 299, 310
Тэйт (Р. G. Tait) 257, 259, 267
Уайс (R. Wyss) 267
Уайт (С. М. White) 266, 268, 390, 415
Уайт (J. R. White) 555, 588
Уайт. (W. Р. White) 269, 283
Уиннингоф (W. J. Winninghof) 447
Уиттэкер (Е. Т. Whitaker) 116
Уитхэм (W. С. D. Whetham) 158, 178
Улих (Н. Ulich) 178, 187, 215
Улих (Н. Н. Uhlig) 214
Унмак (A. Unmack) 178, 309, 310, 586, 589
Уолкер (F. Walker) 315, 337
Уолкер (J. Walker) 283
Уоллес (С. С. Wallace) 266
Уоллес (W. Е. Wallace) 242, 265, 392, 397,
416
Уотен (L. A. Wooten) 494, 496
Уотерс (G. W. Waters) 216, 311, 312, 318,
337
Уотсон (С. N. Watson) 116
Урри (W. D. Urry) 264, 267
Урселл (Н. D. Ursell) 370, 385
Усанович М. И. 167
Уттербак (С. L. Utterback) 266
Уэбб (Т. Y. Webb) 266
Уэшборн (Е. W. Washburn) 178, 179, 269,
270, 271, 283
Файлей (С. F. Failey) 375, 377, 378, 385, 551
Фаллон (L. D. Fallon) 472, 496, 552, 553
Фалькенгаген (Н. Falkenhagen) 34, 35,
45, 74, 77, 79, 83, 97, 98, 101, 116, 133,
137, 171, 179, 180, 206, 208, 210, 215,
264, 267
Фаулер (D. L. Fowler) 33, 214
Фаулер (R. Н. Fowler) 261, 267, 383, 385,
493, 496, 574, 588
Фаянс (К. Fajans) 266
Фельсинг (W. A. Felsing) 550, 590
Фергюсон (A. Ferguson) 156, 178
Фергюсон (J. В. Ferguson) 276, 284
Феринг (L. Foering) 347, 384, 495
Фиалков Я. А. 167, 444
Фицджеральд (М. Е. Fitzgerald) 392, 393,
395, 396, 398, 416, 551, 552
Фишер (W. Fischer) 101, 116
Флейшер (Н. Fleischer) 109, 116, 210,-215
Флейшер (М. Н. Fleyscher) 315, 321, 337,.
550
Фогель (Vogel) 156, 178
Фогель (О. Q. Vogel) 229, 265
Фолк (К. G. Falk) 179
Форнуолт (Н. J. Fornwalt) 179, 181
Франдсен (М. Frandsen) 283
Франк (Н, 8. Frank) 568, 571, 588
Франклин (Е. С. Franklin) 179
Фрейер (Е. В. Freyer) 267, 268
Фрёлих (F. Frolich) 109, 116, 210, 215
Френч (D. М. French) 588
Фривольд (О. Е. Frivold) 266, 283
Фридман (L. Friedman) 175, 180
Фрумкин А. Н. 301
Фрэзер (J. С. W. Frazer) 269, 283, 349, 384,
386, 409, 410, 416
Фуосс (R. М. Fuoss) 34, 45, 55, 60, 73, 77,
80, 83, 84, 85, 87, 92, 97, 98, 116, 125,
137, 145, 146, 149, 150, 173, 175—178,
180, 186, 187, 189, 190—192, 194—199,
211—216, 312, 337, 365, 560, 587, 588,
599
Хаббард (J. С. Hubbard) 263, 267, 268
Хадлстон (L. Y. Hudleston) 258, 267
Хаммет (L. Р. Hammet) 494, 496
Ханг (Т. С. Huang) 179
Хант (F. L. Hunt) 146, 180
Харкинс (W. D. Harkins) 416, 417, 447
Харнед (Н. S. Hamed) 26, 61, 73, 137, 175,
180, 231, 266, 268, 290, 305, 310, SIS-
SI?,' 321, 322, 324, 328, 337—339, 342,
343, 346, 347, 348, 349, 350—352, 355,
362, 367, 372, 388, 389, 390, 392, 393,
395, 396, 398, 409, 410, 415—418, 427,
428, 431, 432, 457, 472, 474, 475, 479,
483—485, 489, 495—497, 522, 530, 542,
551, 552, 553, 560, 562, 578, 580, 589,
590
Харрис (А. С. Harris) 396, 416
Харрис (J. М. Harris) 431, 442, 447
Хартли (Е. G. J. Hartley) 283
Хартли (G. S. Hartley) 160, 179, 181, 549,
562, 588
Хартли (Н. Hartley) 178, 315, 337, 550
Хастед (J, В. Hasted) .571, 588
Хаттокс (Е. М. Hattox) 401, 416, 417
Хейсе (Е. О. Heuse) 283
Хеккер (J. С. Hecker) 61, 73, 137, 266, 268,
350, 351, 352, 384, 386, 390, 392, 416,.
522 551
Хелвах (W. Hallwach) 266
Хидеман (Е. Hiedemann) 267
Хиккей (F. С. Hickey) 484, 485, 496
Хичкок (D. J. Hitchcock) 303, 304, 306,
309, 310, 314, 338
Хлупек (J. В. Chloupek) 422, 447
Ходаков Ю. В. 57, 121
Холборн (L. Holborn) 178
Холемаи (Р. Holemann) 266
Холл (L. Р. Hall) 231, 266
Холл (R. Е. Hall) 283, 416
5.98
ИМЕННОЙ УКАЗАТЕЛЬ
Холлингшид (Е. A. Hollingshead) 180
Холт (Е. К. Holt) 589
Холфельдер.(Ь. Р. Hohlfelder) 181
Хорнибрук (W. J. Hornibrook) 520
Хорш (W. G. Horsch) 322, 416
Хотер (R. С. Hoather) 266
Хоукинс (J. Е, Hawkins) 429, 447, 448
Хоуэс (W. W. Hawes) 214
Хоэш (К. Н. Hoesch) 267
Христиан (S. М. Christian) 139, 177, 181
Худ (G. R. Hood) 181
Хупер (G, S. Hooper) 215
Хир (С. W. Ноегг) 589
Хюттиг (Н. Hutting) 266
Цан (Н. Zahn) 206, 215
Цвики (F. Zwicky) 266
Цедлес (Н. Zedles) 589
Цирклер (J. Zirkler) 180
Чедин (J. Chedin) 589
Ченцова Л. Г, 226
Чоу Минг (Chow Mijng) ’418, 447
Шамовский Л. 226
Шанский (С. М. Shansky) 496
Шаппюи (Р. Chappui) 555, 588
Шварц (Е. Schwartz) 549
Шварц (К. Schwarz) 73, 378, 385
Швенкер (G. Schwenker) 380, 381, 385
Шерилл (М. S. Sherill) 202 , 215, 403, 410,
416
Шершевер Ж. М, 444
Шефер (R. A. Schaefer) 315, 338
Шеффер (Н. Scheffer) 276, 284
Шибата (Z. Shibata) 266
Шидловский (Т.’Shedlovsky) 139, 149, 153,
154, 158, 164, 166, 177, 178, 179, 180,
181, 187, 203, 204, 215, 216, 304, 310,
316, 323, 324, 338, 349, 367, 382, 384,
385, 386, 402, 416, 498, 530, 549, 551,
560, 588, 589
Шиле (J, Schiele) 116, 211, 215
Шиндлер (Т. D. Schindler) 303
Шиодт (Е. Schiodt) 305, 310, 486, 496
Шмельцер (С. Schmelzer) 116, 208, 209,
215
Шмидт (С. L. A. Schmidt) 488, 496
Шмик (Н. Schmick) 214
Шминке (К. Н. Schminke) 266, 268, 332,
333, 338
Шот (О. A. Short) 137, 311, 315, 337, 338
Шоттке (W. Schottky) 227, 265
Шпрунг (A. Sprung) 179
Шрейнер (Е. Schreiner) 283
Штауффер (R. Е. Stauffer) 179, 181, 268
Штернина Э. Б. 444
Штраус (U. Р. Strauss) 589
Шупп (О. Е. Schupp) 350, 384, 386, 447,
448, 450, 495, 552
Шэнкман (S. Shankman) 284, 410, 416,
417
Эберт (L. Ebert) 373, 385, 467, 468, 495
Эванс (М. G. Evans) 493, 496, 568, 588
Эверетт (D. Н. Everett) 475, 476, 479, 491,
493, 496
Эвьен (Н. М. Evjen) 266
Эггенбергер (D. N. Eggenberger) 589
Эгю (В. Hague) 139, 177
Эдселл (J. Т. Edsall) 368, 385, 496
Эйзенберг (S. Eisenberg) 360, 370,^371, 385
Эйзенбранд (J. Eisenbrand) 466
Эйкен (A. Euken) 45
Эйринг(Н. Eyring) 258, 267, 492, 496
Эйхлер (A. Eichler) 265, 549
Эккерт (С. F. Eckert) 266
Экштром (Н. С. Eckstrom) 116, 208, 209,
210 215 283
Элерс (R. W. Ehlers) 61, 73, 266, 313, 314,
316, 322, 324, 328, 333, 337, 338, 427,
447, 495, 550, 552, 590
Эли (D. D. Eley) 493, 496
Эллиот (J. Н. Elliott) 494/496
Эллиот (М. A. Elliott) 214, ‘216
Эллис (J. Н. Ellis) 313, 337
Эмбри (N. D. Embree) 474, 492, 496, 552,
553
Эмде (F. Emde) 73, 137
Эммет (Р. Н. Emmett) 576, 588
Энгель (G. Angel) 214
Эндрьюс (D. Н. Andrews) 267, 268
Эндрьюс (D. Н. Andrews) 177
Эппельбэй (М. R. Appelbay) 179
Эпштейн (Р. S. Epstein) 32
Эршлер Б. В. 23
Эссекс (J. L. Essex) 418, 447
Юнг (L. Е. Young) 280, 283, 322, 338, 339,
384, 405, 416
Юнг (Т. F. Young) 223, 224, 225, 229, 236,
265, 266, 403, 541, 576, 589
Янке (Е. Jahnke) 73, 137
Янковская Г. Н. 250, 339
Янц (G. J. Janz) 340, 384, 589, 590
Яцимирский К. Б. 121, 339, 444
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адсорбционный потенциал, определение
70—71
Азот, закись, см. Закись азота
Азотная кислота, степень диссоциации,
таблица 576
Активность воды в растворе серной кис-
лоты, определение 408—410
— выражение через моляльность 27
--------молярность 27
— гидратированных ионов 573
— зависимость от состава 26
---------- —• температуры и давления 25
— иоиов, средняя, определение 24
— компонента системы 25
— коэффициент, см. Коэффициент актив-
ности
— негидратированных ионов 573
— раствора, уравнение 275
— растворителя, вычисление из данных
для температур замерзания, уравнение
270—272
-------------------кипения, уравне-
ние 273—274 .
:-------по измерениям давления пара,
уравнение 274—276
-----------изопиестическим данным, урав-
нение 274—276
— свойства 27
— уравнение 24
— электролита 23—24
Й1-Аланин, изменение термодинамических
функций при кислотной диссоциации
476
--------------основной диссоциации 476
— константа кислотной диссоциации 541,
544
-----------максимальная 546
----------- определение 471
----основной диссоциации 541, 544
----------- максимальная 546
Алюминий сернокислый, высаливание ней-
тральных молекул 376
----коэффициент активности средний 402
— хлористый, высаливание нейтральных
молекул 376
----коэффициент активности 534
Амальгамные элементы, см. Элементы
амальгамные
dZ-a-Амино-и-ваперьяиовая кислота, из-
менение термодинамических функций
при кислотной диссоциации 476
----------------основной диссоциации
476
«ZZ-a-Амино-н-вацерьяновая кислота, кон-
станта кислотной диссоциации 544
-------------максимальная 546
------- основной диссоциации 544
-------------максимальная 546 ' .
dl-a-Аминоизомасляная кислота, измене-
ние термодинамических функций при
кислотной диссоциации 476
---------------- основной диссоциации
476
-----константа кислотной диссоциации
544
------------- максимальная 546
-------основной диссоциации 544
-------------- максимальная 546
dl-a-Амино-н-масляная кислота, изменение
термодинамических функций при кислот-
ной диссоциации 476
-----—----------основной диссоциации
476
— — константа кислотной диссоциации
544
------------- максимальная 546
-------— основной диссоциации 544
------------- максимальная 546
Аммоний азотнокислый, высаливание ней-
тральных молекул 376
-----кажущийся молярный объем 242
-----коэффициент активности, средний
при температурах замерзания водных
растворов 504
-----парциальный молярный объем, влия-
ние давления 250
— бромистый, коэффициент активности,
средний при температурах замерзания
водных растворов 504
— иодистый, коэффициент активности,
средний при температурах замерзания
водных растворов 504
— сернокислый, коэффициент активности,
средний при температурах замерзания
водных растворов 504
— хлористый, кажущееся относительное
молярное теплосодержание 500
-----кажущийся молярный объем 242
-----коэффициент активности, средний
при температурах замерзания водных
растворов 504
— — относительное парциальное моляр-
ное теплосодержание 502
----- числа переноса 499
----- эквивалентная электропроводность
498
600
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Амфолиты, константы диссоциации 548
-------- определение 469—472
— коэффициенты активности 470—471
Аргон, коэффициент высаливания 528
— различные солевые эффекты 377
Ассоциация ионов, теория Бьеррума 54—57
Атмосфера иона, мера относительной не-
симметричности 85
-----уравнение, теория Дебая 38—42
— ионная, см. Ионная атмосфера
Ацетат ион, гидролиз в растворах хлори-
стых солей, таблица 448—489
Ацетилен, коэффициенты высаливания 528
Ацетон, высаливание и всаливание раз-
личными солями 379
Балансное детекторное устройство 139
Барий азотнокислый, константа диссоциа-
ции иона 149
-----кажущееся относительное молярное
теплосодержание 501
-----коэффициент активности средний 570
— — относительное парциальное моляр-
ное теплосодержание 503
— бромистый, кажущееся относительное
молярное теплосодержание 501
-----коэффициент активности средний
397, 530
-----относительное парциальное моляр-
ное теплосодержание 503
— гидрат окиси, константа Диссоциации
иона по данным электропроводности
— железистосинеродистый, электропро-
водность, влияние силы поля 209
— железосинеродистый, электропровод-
ность', влияние силы поля 209
— иодистый, коэффициент активности сред-
ний 570 ,
— хлористый, влияние на коэффициент
• активности соляной кислоты 425—426
-----высаливание нейтральных молекул
376, 377
-----кажущаяся молярная сжимаемость
264
-----кажущееся относительное молярное
теплосодержание 501
-----кажущийся мольный объем 242
-----коэффициент активности средний
389, 397, 529—530
-----молярная электропроводность, таб-
лица 157
-----относительная вязкость, предельный
коэффициент наклона 170
-----относительное парциальное моляр-
ное теплосодержание 503
----- парциальная молярная теплоем-
кость 237
----- эквивалентная электропроводность
498
----- эффективное давление 261
Барометрическая поправка водных раство-
ров, таблица 290
Бензойная кислота, константа диссоциации,
определение методом электропровод-
ности 205
--------кислотной диссоциации 579
--------------максимальная 582
-----коэффициенты высаливания 529
Бензол, коэффициент сжимаемости 259
Борная кислота, изменение термодинами-
ческих функций при диссоциации 476
-----константа диссоциации 465, 541
-----в растворах хлористого нат-
рия 583
----------- максимальная 546
-----pH 0,05н. раствора 306
-----применение для определения стан-
дартных потенциалов элементов 347
Бромистоводородная кислота, коэффициент
активности, средний в растворах бро-
мистых солей 537
-------------------галоидных солей ще-
лочных металлов 424
-----—-------------солей 423—426
--------------- — водных растворов, таб-
лица 519
-----относительная парциальная моляр-
ная теплоемкость, таблицы 332, 519
-----относительное парциальное моляр-
ное теплосодержание в растворах бро-
мистых солей 426—427
— —------------таблица 519
-----среднее расстояние сближения ионов
360
-----стандартный потенциал элемента,
содержащего бромистоводородную кис-
лоту 348
-----термодинамические свойства раство-
ров 347—348
mpem-бутиловый спирт—вода, диэлектриче-
ские постоянные смесей 120
Буферные растворы, значения pH, таблица
306
-----применение для определения кон-
стант диссоциации 463—469
--------при определении стандартных
потенциалов 347
-----стандартные потенциалы, таблица
305
Валентность, фактор, значение для коэф- '
фициентов активности 121—123
----------- электропроводности 131
dZ-Валин, изменение термодинамических
функций при кислотной диссоциации 476
-------------— основной диссоциации 476
— константа кислотной диссоциации 544
-------------максимальная 546
— — основной диссоциации 544
—-- ----- — максимальная 546
Вероятность нахождения иона в поле дру-
гого иона, см. Теория образования ион-
ных пар
Вода, активность в водных растворах сер-
ной кислоты, определение 408—410
-------------------по данным измере-
ний электродвижущих сил и упругости
пара 409
—-------растворах серной кислоты, таб-
лица 409
— диссоциация в растворах солей 454—457
--------------зависимость от ионного
радиуса соли 457
-----------хлористого калия 455
-----влияние давления, таблица 462
-----температуры и давления 457—462
— диэлектрическая постоянная 119
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
601
Вода, изменение термодинамических функ-
ций при диссоциации 476
— константа диссоциации в растворах
различных солей 451
--------смесях диоксан—вода 542, 547
--------зависимость от температуры 449,
460
----- — определение экстраполяцией 453
-------- таблица 453
— коэффициент сжимаемости 258—259
— молярное повышение температуры ки-
пения 274
-----понижение температуры замерзания
272
— средний ионный радиус, определение из
температурной зависимости 492
— суммарная теплота диссоциации в раст-
ворах галоидных солей 460
— теплота диссоциации, определение 232
— функции активности ионов в растворах
солей, таблица 539—540
Водород, коэффициенты высаливания 528
— различные солевые эффекты 377
Возмущение ионной атмосферы, сила 74
Время запаздывания 74—76
-----по Ланжевену, определение 77, 115,
213 . v
-----процесс образования ионной группы
76—77
— релаксации ионной атмосферы, см. Ион-
ная атмосфера
Всаливание 63—65
— коэффициент 435, 436
— коэффициенты 528, 529
— синильной кислоты и ацетона 379
— уксусной кислоты 377, 378
Вторичные эффекты, см. Среда
Высаливание 63—65, 367, 368, 376
— синильной кислоты и ацетона 379
Высаливающее действие ионов на ней-
тральные молекулы 376—380
—• — определение 368
Вязкость в неводных растворителях, пре-
дельные коэффициенты, таблица 173
— относительная, предельная зависимость
171
-----предельные коэффициенты наклона
170—171
— параметры уравнений, таблица 172
— предельные теоретические коэффициен-
ты наклона, таблица 172
— предельный закон 129—130
— смесей диоксан—вода, таблица 505
— теория Фалькенгагена 77
— уравнение 78
— электролитов, зависимость от концен-
трации 168—173
— электростатическая составляющая 83—
84
Газовая постоянная 117, 555
-----рассчитанная на одну молекулу 555
Газы, константа равновесия, уравнение
374
— коэффициенты активности 375
— растворимость в растворах солей 374—
377
Гальванический элемент, Обратимый 22
Гальванометр Пашена 221
Гексадециламин, эквивалентная электро-
проводность, 587
Гелий, коэффициенты высаливания 528
Гидратация ионов 361—364, 572—576
Гидратированные ионы, коэффициент ак-
тивности, выражение 573
Гидратированный ион, эффективный иде-
альный радиус, выражение 574
Гидратное число иона, уравнение 572
Гидратные числа положительных ионов,
таблица 572
Гидраты окисей щелочных металлов, коэф-
фициенты активности в растворах солей
423—426
Гидролиз анионов органических кислот в
растворах солей 488—489
— локализованный 364
— реакция, влияние солей, уравнение 488
Гликолевая кислота, изменение термоди-
намических функций при диссоциации
476
-----константа диссоциации 478, 541
-----------максимальная 545
Глицерин—вода, диэлектрические постоян-
ные смесей 120
-------константа диссоциации уксусной
кислоты 580—581, 583
-------- коэффициент активности соляной
кислоты, таблица 512
— ----стандартные потенциалы, таблица
'507
Глицин, изменение термодинамических
функций при кислотной диссоциации
476
------------основной диссоциации 476
— константа кислотной диссоциации 541,
579
-----------в растворах хлористого нат-
рия 583
-------------смесях диоксан—вода 543,
548
—----------максимальная 546
-----основной диссоциации 471, 541
-----------в смесях диоксан—вода 543—
548
-----------максимальная 546
Грамм-калория 117
Давление внутреннее (кохезионное) 258
— эффективное, см. Эффективное давле-
ние
Двуокись углерода, коэффициенты выса-
ливания 528
Деформация, относительная скорость 78
— стационарная 78
Дециламин, эквивалентная электропро-
водность 587
Диацетоновый спирт, коэффициенты выса-
ливания 528
-----различные солевые эффекты 376—377
а-Динитрофенол, константа диссоциации,
определение 469
— коэффициенты активности ионов и не-
диссоциированных молекул в растворах
солей 487
Диоксан—вода, вязкость смесей, таблица
505
-------диэлектрические постоянные,
таблица 505
602
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Диоксан—вода, константы диссоциации
алифатических кислот, глицина и воды
542—543, 547—548
--------коэффициент активности соля-
ной кислоты, таблица 509—510
--------относительная парциальная мо-
лярная теплоемкость, таблица 513—
514 .
--------относительное парциальное моляр-
ное теплосодержание, таблица 513
--------плотности растворов соляной
кислоты, таблица 506—507
--------— чистых растворителей, таб-
лица 506—507
-----.— предельные коэффициенты на-
клона для коэффициентов активности,
таблица 505
--------стандартные потенциалы, табли-
ца 507
--------числа переноса соляной кислоты,
таблица 515—516
Диссоциация, влияние силы поля 136, 137
— воды в растворах хлористого калия 455
-----влияние температуры и давления
457—463
— и скорость реакции 77
—кислотная и основная, уравнение 468
— константа, см. Константа диссоциации
— сильных электролитов 576—577
— слабых кислот в растворах солей 578—
586
— — электролитов, теория 109—116
— степень 77
— угольной кислоты, первая ступень 578
— умеренно сильных электролитов 577
— чистой воды, влияние давления 462
— электролитов, изменение термодинами-
ческих функций 476-—477
Диффузионный потенциал. 296—298, 299,
302—309
— — устранение его при постоянной об-
щей моляльности 308
Диффузия, дифференциальный коэффи-
циент 174
— интегральный коэффициент 173
— коэффициент, см. Коэффициент диффу-
зии
— простого электролита, теория 94
— теория 132
-----значения функций, таблица 132
-----ионных сил 88—89
— электролитов 173—174
-----теория 93—96
Дихлоруксусная кислота, константа дис-
социации 577
Диэлектрическая постоянная 118—120
-----в переменном поле, уравнение 100—
101
-----влияние на образование ионных пар
189—190.
----- воды 554—557
-------- таблица 119
-----зависимость от концентрации ионов,
уравнение 358—359
-----------частоты, теория 97—109
-----макроскопическая 65
-----предельные коэффициенты наклона
и функции 554—557
-----связь с частотой тока 205—208
Диэлектрические постоянные, влияние ча-
стоты тока, таблица 134
----смесей вода—органический раство-
ритель, таблица 120
----смесей диоксан—вода, таблица 505
-------------параметры уравнения 120
Додециламин, эквивалентная электропро-
водность 587
Европий хлористый, коэффициент актив-
ности 534
Емкость сосуда для определения электро-
проводнбсти 139
Железо хлористое, коэффициент активности
Закись азота, коэффициенты высаливания
528
----различные солевые эффекты 376—377
Закон Генри 578
— действующих масс 31
— диффузии электролитов 95
— Кольрауша 91, 166
— Кулона 55
— Ламберта и Бэра 468
— разведения Оствальда 185
— распределения Максвелла—Больцмана
87
— Стокса 86—89, 182
— Фарадея 286
— Фика 93
Идеальный газ, нормальный объем 117
dZ-Изолейцин, изменение термодинамиче-
ских функций при кислотной диссо-
циации 476
-------------основной диссоциации 476
— константа кислотной диссоциации 544
---------- максимальная 546
----основной диссоциации 544
----------максимальная 546
Изопропиловый спирт—вода диэлектриче-
ские постоянные смесей 120
----------константы диссоциации про-
пионовой и н-масляной кислот 580—
581, 583
----------коэффициент активности соля-
ной кислоты средний, таблица 512
----------стандартные потенциалы, таб-
лица 507
Импульсивные методы определения элек-
тропроводности 208
Индий сернокислый, коэффициент актив-
ности средний 402
Интерполяция зависимости констант дис-
социации алифатических кислот от
диэлектрической постоянной среды 472—
473
Иодистоводородная кислота, коэффициент
активности средний 525
----среднее расстояние сближения ионов
360
Йодноватая кислота, константа диссоциа-
ции 577
Ион водорода,изменение электропровод-
ности в смесях соляная кислота—ка-
лий хлористый 145
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
603
Ион калия, изменение электропроводности
в смесях соляная кислота—калий хло-
ристый 145
— коэффициент трения, см. Коэффициент
трения иона
— потенциал, см. Потенциал иона
— центральный, связь диэлектрической
постоянной с радиусом иона 571
Иональная концентрация, определение 47
------ связь с ионной силой, уравнение 50
Ионная атмосфера 33—36, 74
------в отсутствие внешних полей, урав-
нение 38—42
-----время релаксации 45, 74—77,
98—99
-----возмущение 74, 107
----- деформация 78
-----поведение в возмущенных состоя-
ниях 44
------ потенциал 42
— — термодинамические функции, урав-
нения 121
-----уравнения движения 38
—-------для возмущенного состояния 42
— группа, время запаздывания процесса
образования 77
— электропроводность ирна хлора, зави-
симость от концентрации 168
Ионное произведение воды в случае водных
растворов солей 454—457
-----выраженное через коэффициенты ак-
тивности 454
-----определение 450
Ионные атмосферы, свойства 33
— взаимодействия 368
-----специфические 365
-----квадруполи и электропроводность
191—198 '
-----теория образования 58—60
— пары, длинные, определение 55
-----константы диссоциации 189—191
-----короткие, определение 55
-----образование 185—188
-----размер, влияние на ассоциацию 198—
201
--------------электропроводность 198—
201
— поля 103—107
— радиусы из данных кристаллографии,
таблица 120—121
-----------электропроводности, таблица
158
-----констант диссоциации 490
-----—- теплот диссоциации 490—491
-----средние, определение из темпера-
турной зависимости, таблица 492
— тройники и электропроводность 191—198
-----константы диссоциаций 191—195,
200
-----размер, влияние на ассоциацию 198—
201
--------влияние на электропроводность
58—60
-----теория образования 58—60
Ионный параметр 123
-----определение 51
— радиус средний, уравнение 491
Ионы, влияние размеров и строения на
ассоциацию 198—200
Ионы, влияние размеров а строения на
электропроводность 198—200
— диполярные, определение 469
— и молекулы, взаимодействие 368
— определение силы переноса 85
— полимерных электролитов 586
— простые 586
— с мицеллярной структурой 586
— степень гидратации, см. Гидратация
. ионов
— хлора, зависимость ионной электро-
проводности от концентрации 168
Иттрий хлористый, коэффициент актив-
ности 534
Кадмий, амальгамный электрод, стандарт-
ный потенциал 391, 531
— азотнокислый, коэффициент активно-
сти средний 530
— бромистый, данные по электродвижу-
щим силам элемента, зависимость от
концентрации 393
— — коэффициент активности средний
397, 531—532
------ относительное парциальное моляр-
ное теплосодержание 533
-----электродвижущая сила, зависимость
концентрации 393
— иодистый, коэффициент активности
средний 397, 531—532
-----относительное парциальное моляр-
ное теплосодержание 533
— малоново кислый, константа диссоциа-
ции 149
— сернокислый, кажущееся относитель-
ное молярное теплосодержание 501
-----константа диссоциации 149
-----коэффициент активности средний 401
-----относительное парциальное моляр-
ное теплосодержание 503
— хлористый, коэффициент активности
средний 397, *531—532
-----относительное парциальное моляр-
ное теплосодержание 533
----- электродвижущая сила, зависимость
от концентрации 393
Кажущиеся молярные объемы электроли-
тов 240—248
Кажущийся молярный объем неэлектро-
литов, влияние давления 249—250
--------параметры уравнения 242
--------электролита, параметры уравне-
ния, таблица 242^
Калий азотнокислый, высаливание ней-
тральных молекул 376, 378, 379
-----кажущееся относительное молярное
теплосодержание 500
-----кажущийся молярный объем 242
-----константа диссоциации 149
--------- — таблица 148
-----коэффициент активности средний
373, 525
-------------при температурах замерза-
ния водных растворов 504
-------- диффузии 177
-----относительное кажущееся молярное
теплосодержание 222
--------парциальное молярное теплосо-
держание 502
604
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Калий азотнокислый, поверхностное натя-
жение разбавленных растворов 381
----- числа переноса 161, 499
----- эквивалентная электропроводность
498
— бромистый, высаливание нейтральных
молекул 376, 378
-----кажущаяся молярная сжимаемость
284
-----Кажущееся относительное молярное
теплосодержание 500—502
-----кажущийся молярный объем 242
-----коэффициент активности средний
363, 524
—---------- — влияние бромистоводород-
ной кислоты и гидрата окиси калия
424—426
------------ — при температурах за-
мерзания водных растворов 504
-----критическое состояние разрушения,
величина 246—248
-----относительное кажущееся молярное
теплосодержание 222
--------парциальное молярное тепло-
содержание 502—504
----- парциальная молярная теплоем-
кость 237
-----среднее расстояние сближения ионов
360
----- числа переноса 499, 558
- — — эквивалентная электропроводность
498, 558
— — эффективное давление 261
— бромноватокислый, коэффициент актив-
ности средний 526
— гидрат окиси, кажущийся молярный
объем 242
—-------коэффициент активности 351
--------------в растворе хлористого ка-
лия 443
-----—--------средний 364, 522
-----------------в растворах галоид-
ных солей калия и сернокислого'калия
425—426
--------относительная парциальная мо-
лярная теплоемкость 352, 523
--------относительное парциальное мо-
лярное теплосодержание 352, 523
-------- парциальная молярная тепло-
емкость 236
--------среднее расстояние сближения
ионов 360
-----— термодинамические свойства рас-
творов 350—352
— железистосинеродистый, кажущаяся
молярная сжимаемость 264
— — эквивалентная электропроводность
498
— железосинеродистый, кажущаяся мо-
лярная сжимаемость 264
-----коэффициент активности средний
402
----- эквивалентная электропроводность
*— иодистый, высаливание нейтральных
молекул 378
----кажущийся молярный объем 242
-----коэффициент активности средний
363, 524
Калий иодистый, коэффициент активности
средний, влияние гидрата окиси калия
425—426
-----критическое состояние разрушения,
величина 246—248
-----парциальная молярная теплоем-
кость 237
— — произведение электропроводности на
вязкость 184
-----среднее расстояние сближения ионов
360
— — числа переноса 499, 558
•— — эквивалентная электропроводность
498, 558
— муравьинокислый, коэффициент актив-
ности, средний при температурах замер-
зания водных растворов 504
— роданистый, коэффициент активности
средний 525
— сернокислый, кажущееся относитель-
ное молярное теплосодержание 500
— — кажущийся молярный объем 242
------ коэффициент активности средний
391, 397
-------------влияние гидрата окиси ка-
лия и серной кислоты 425—426
-----относительное парциальное моляр-
ное теплосодержание 502
-----парциальная молярная теплоемкость
237
-----парциальный молярный объем,
влияние давления 250
-----поверхностное натяжение разбавлен-
ных растворов 381
-----числа переноса 499-**
— п-толуолсульфоновокислый, коэффици-
ент активности средний 526
— углекислый, эквивалентная электро-
проводность 498
— уксуснокислый, коэффициент активно-
сти средний 525
-------------при температурах замерза-
ния водных растворов 504
----- числа переноса 499
— фосфорнокислый, коэффициент актив-
ности средний 526
— фтористый, кажущееся относительное
молярное теплосодержание 500—502
-----кажущийся молярный объем 242
-----коэффициент активности средний
525
-----критическое состояние разрушения,
величина 246—248
-----относительное кажущееся молярное
теплосодержание 222
-----парциальное молярное теплосодер-
жание 502—504
— хлористый, высаливание нейтральных
молекул 376, 378
-----кажущаяся молярная расширяе-
мость 252—253
— —-------сжимаемость 256—257
-----кажущееся относительное молярное
Теплосодержание 500—502
-----кажущиеся молярные теплоемкости
при постоянном объеме и постоянном
давлении 238—239
-----кажущийся молярный объем 242,
244
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 605
Калий хлористый, коэффициент активности
в растворе гвдрата окиси калия 443
------------ влияние давления 358
-------------средний 363, 373, 524
ч ----------- влияние соляной кислоты
и гидрата окиси калия 424—426
-----при температурах замер-
зания водных растворов 504
-----'--таблица 520
-------- таблица 349
---------------- — диффузии 177
--------критическое состояние разрушения,
величина 246—248
--------осмотический коэффициент 276
--------относительная вязкость, предельный
коэффициент наклойа 170
-------- парциальная молярная теплоем-
кость 352
------------------ Таблица 520
----относительное кажущееся молярное
теплосодержание 222
— — — —---------в смешанных раство-
рителях 229
—-------парциальное молярное теплосо-
держание 352, 502—504
-—--------------таблица 520
---- парциальная молярная теплоем-
кость 236—237
----понижение температуры замерзания,
функции 282
----среднее расстояние сближения ионов
359—360
----термодинамические свойства раство-
ров 349—350
----удельная электропроводность стан-
дартных растворов, таблица 141
----------числа переноса 101, 163, 499, 558
----- эквивалентная электропроводность
498, 558
----экстраполяция по данным темпе-
ратур замерзания 281—282
----электропроводность предельная,
ионная 137
— — эффективное' давление 261
— хлорноватокислый, кажущееся отно-
сительное молярное теплосодержание
222, 500
----коэффициент активности средний
526
--------— — при температурах замерза-
ния водных растворов 504 ,
—-------парциальное молярное теплосо-
держание 502
— хлорнокислый, кажущееся относитель-
ное молярное теплосодержание 222, 500
-----константа диссоциации 149
------коэффициент активности, средний
при температурах замерзания водных
растворов 504
— хлорнокислый, относительное парциаль-
ное молярное теплосодержание 502
— хромовокислый, кажущийся молярный
объем 242
----коэффициент активности средний
530, 570
Калориметр двойной Джоуля—Пфаундлера
221, 234
Кальций азотнокислый, высаливание ней-
тральных молекул 376
Кальций азотнокислый, кажущееся отно-
сительное молярное теплосодержание
500
-----коэффициент активности средний
350, 397, 569
-----относительное парцйальное моляр-
ное теплосодержание 503
— бромистый, кажущееся относительное
молярное теплосодержание 500
-----коэффициент активности средний
570
-----относительное парциальное моляр-
ное теплосодержание 503
— железосинеродистый, кажущаяся мо-
лярная сжимаемость 264
— иодистый, коэффициент активности
средний 570
----- эффективное давление 261
— сернокислый, кажущееся относитель-
ное молярное теплосодержание 221,
501
-----константа диссоциации 149
-----относительное парциальное моляр-
ное теплосодержание 503
— хлористый, влияние на коэффициент
активности соляной кислоты 425—426
-----высаливание нейтральных моле-
кул 376
-----кажущаяся молярная сжимаемость
254
-----кажущееся относительное молярное
теплосодержание 500
— — кажущийся молярный объем 242
-----коэффициент активности средний
389, 397, 530, 569
-----относительное парциальное моляр-
ное теплосодержание 503
— — числа переноса 162, 499
----- эквивалентная электропроводность
498
-----электропроводность иона хлора 168
Кислород, различные солевые эффекты
377
— коэффициенты высаливания 528
Кобальт азотнокислый, коэффициент актив-
ности средний 570
— сернокислый, константа диссоциации
149
— хлористый, коэффициент активности
средний 397, 530
Кобальтаммины, растворимость в воде,
влияние солей 419—422
Компонент, определение 21, 24, 25, 29
Константа диссоциации 77
— — ассоциированной ионной пары 56
-----борной кислоты 465
— — в сильном поле 137
-----воды 450—453
--------определение методом измерения
электродвижущих сил 450 , 467
— — — — — — электропроводности
450
-------------экстраполяции 453
-------- таблица 453
-----зависимость от температуры 473—
476, 485—486, 494
-----и коэффициент активности слабой
кислоты в растворах солей, уравне-
ние 484
606 ПРЕДМЕТНЫЙ указатель
Константа диссоциации иона HSO~, опре-
деление 403—404 .
----------- таблица 404
-----ионного тройника 59, 126
-----кажущаяся, уравнение 464
-----кислотой глицина в растворах хло-
ристого натрия, таблица 583
--------уравнение 470
-----определение кондуктометрическим
методом 303
----- основной, уравнение 470
------ слабой кислоты, электрометриче-
ское определение 463
-----слабых электролитов в растворах
солей, уравнение 584
-----уксусной кислоты’203, 204, 465, 541
-----------в растворах хлористых солей,
таблица 485
----- фосфорной кислоты, вторая сту-
пень, уравнение 466—467
-----чистой воды, связь со свойствами
кислоты в смешанном растворителе,
уравнение 480
— .равновесия образования ионной пары
56 i
-----процесса диссоциации ионной пары
189
-----химической реакции 30—32
----------- уравнение 31
— скорости диссоциации 77
----- рекомбинации 77
Константы диссоциации алифатических
кислот, таблица 478
----- аминокислот, таблица 544
-----воды, зависимость от температуры
460
--------и кислот в смесях диоксан—вода
и метиловый спирт—вода, таблицы
542—543, 547—548
-----вычисление 185—188
— — зависимость их отношений от диэлек-
трической постоянной 494
--------от температуры 494
-----и термодинамические функции
472—479
-----кислот.и глицина при температурах
максимума диссоциации, константы
уравнений, таблица 582—583
--------при температурах максимума дис-
социации, константы уравнений, табли-
ца 545-—548
--------таблица 541
-----конечные 146—149
-----определение из данных по электро-
движущим силам 303
--------методом визуальной калоримет-
рии 467
--------— фотоэлектрической калори-
метрии 467
— — слабых кислот в воде и в растворах
солей, таблица 579
--------------растворах солей 578—586
--------------смесях органический рас-
творитель—вода 580—581
—----------и оснований, определение
463—469
-----солей и комплексных ионов, табли-
ца 149
Константы диссоциации угольной и бор-
ной кислот в растворах хлористого
натрия, таблица 582—583
-----умеренно сильных электролитов,
таблица 577
— предельного коэффициента наклона для
электропроводности, таблица 131
— уравнений для констант диссоциации
. 545—548, 582—583
--------произведения коэффициентов ак-
тивности ионов, таблица 456
------ электропроводности 505
— уравнения для стандартного электрод-
ного потенциала элемента 428
----для электродвижущей силы, табли-
ца 428
-----коэффициента активностй 527, 575
— физические, основные, таблицы 117,
555
Концентрационные цепи, уравнение 23
Концентрация водородных ионов 302
--------кажущаяся, уравнение 482
-------- определение 451
Коэффициент активности 24, 28, 94, 343,
365—367, 427, 433, 567, 572—576
-----в разбавленных растворах 340—345
-----вандерваальсовская составляющая,
уравнение 371
-----вычисление из данных по числам
переноса 567 . •
-----------—— — электродвижущим си-
лам элементов 567
--------по методу Гуггенгейма 365—367
-----зависимость от диэлектрической по-
стоянной и основности воды 302
-----------концентрации 367
--------------предельный закон 48—51
-----------среднего расстояния сближе-
ния ионов, высаливание 361—362
-----иона, предельный закон Дебая—
Гюккеля 49—50
--------уравнение 51—52
-----истинный, связь с концентрацией
иона водорода 482
-------- средний 394
-----моляльный 50
-----обобщенная теория Дебая 323—325
--------------параметры и уравнения,
таблица 325
-----общая составляющая 323
-----определение по предельному зако-
ну 186
--------путем экстраполяции 292—293
-----практический 27
-----при различных температурах и да-
влениях 355—358
-----растворенного вещества, вычисле-
ние по активности растворителя 276—
282
-----------------осмотическому коэффи-
циенту растворителя 276—282
------------------------- функция о,
таблица 278
-----растворителя, уравнение 371
--------рациональный, уравнение 29
-----рациональный, 27, 29, 68
-----связь с осмотическим коэффициен-
том и коэффициентом наклона, уравне-
ние 433
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 607
Коэффициент активности слабой кислоты
кажущийся, уравнение 482
-----слабых кислот в растворах солей 377
-----средний моляльный, уравнение 354
--------серной кислоты, таблица 534
-----— соляной кислоты в воде, табли-
ца 508
-----------------смешанных растворите-
лях, таблица 511—512
--------стехиометрический 394
--------хлористого натрия, таблица 341
-----теория концентрированных раство-
ров Скэтчарда 367—369
-----уравнение Скэтчарда 368
-----функция для водных растворов
солей 454—457
-----экстраполяция 443—446
-----электролита в смеси, зависимость
от ионной силы, уравнение 430
--------изменение в присутствии другого
электролита 427—432
--------константы уравнений, таблица
575
-------- средний, уравнение 573
------- электролитов, определение по пони-
жению температуры замерзания, урав-
нение 279—280
— всаливания 435, 436, 528, 529
— высаливания, определение 380
-----таблицы 528, 529
— вязкости, определение 78
— диффузии бесконечно разбавленного
раствора, значение 174
-----дифференциальный, определение ме-
тодом электропроводности 560
--------хлористого калия, таблица 561
-----хлористого калия 562
-----:--кальция 563.
-----теоретический и экспериментальный
177
-----уравнение 76, 93—96
— наклона 236, 237, 244, 247, 249,252, 335
— осмотический, см. Осмотический коэф-
фициент
— расширения воды, уравнение 555
— расширяемости 217, 251
— сжимаемости растворов, параметры ура-
внений, таблицы 257, 258
----- таблица 259
-----уравнение 255
----- электролитов 254—265
— специфического взаимодействия ионов
435—436
— термодинамического расширения рас-
творов электролитов 251—254
— трения нона 38
Коэффициенты активности 292, 324, 328,
353, 358, 365, 374, 389, 400, 484, 534,
567—568, 572—575 _
-----гидратов окисей щелочных метал-
лов, таблица 351
-----нонрв воды, произведение для рас-
творов некоторых галоидных солей 456
-----------—. зависимость от ионных
радиусов солей 457
—> — нейтральных молекул в водных рас-
творах солей 374—378
.----1,1-валентных электролитов, тео-
рия Дебая—Гюккеля 358—365
Коэффициенты активности, параметры
уравнения, таблица 567
-----предельные коэффициенты наклона
смесей диоксан—вода, таблица 505
-----различных электролитов 567—568
-----растворов смеси двух электролитов,
различные ннтерпретацнн свойств
433—434
-----сернокислых солей двухвалентных
металлов 400—401
-----слабых кислот в растворах солей
484—489
-----средние 323, 363, 364, 373, 391,
397, 401, 402, 419, 421, 424—425,
509, 524—526, 536—538, 569—570, 587
--------азотнокислого тория, таблица 570
--------гидратов окисей щелочных метал-
лов-----в растворах соответствующих
солей 423—426
--------2,1-валентных электролитов,
таблица 529—532, 570
--------кислот в растворах солей, табли-
цы 536—538
-----------и гидратов окисей в раство-
рах солей 425—426
--------натриевых солей одноосновных
жирных кислот 587
--------некоторых солей, таблица 402
—-------1,2-валентных электролитов, та-
блица 570
--------ряда электролитов, таблица 569
--------серной кислоты в растворах
солей 423—426
--------сернокислых солей 2-валентиых
металлов, таблица 401
-------------натрия, лития и калия,
таблица 391
--------сильных электролитов 1,1-валент-
ных, таблица 508—527
-------------- определение по методу
Гуггенгейма, таблица 527
-------------— отношение, таблица 354
------ — смесей сильных электролитов, за-
висимость от концентрации кислоты или
соли при постоянной ионной силе 429—432
--------соляной н бромистоводородной
кислот в растворах солей 423—426
----------- кислоты в концентрирован-
ных водных растворах, таблица 538
-----------------смесях диоксан—вода,
таблица 509—510
--------хлористых солей щелочноземель-
ных металлов, таблица 389
-------- электролитов при температурах
замерзания, таблица 504
-----хлористых солей трехвалентных ме-
таллов, таблица 354
-----электролитов, в растворах хлори-
стого натрия 486—487
--------влияние давления, таблица 358
--------по изопиестическим измерениям,
таблица 353—355
— высаливания газов и жидкостей в рас-
творах электролитов, таблица 528
-----слабых кислот, таблица 529
— наклона 213, 236, 243, 244, 264, 335, 433
-----теоретические для парциальных
молярных объемов 1,1-валентных элек-
тролитов, таблица 556
608
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Коэффициенты наклона теоретические для
расширяемости 1,1-валентных электро-
литов, таблица 556
— сжимаемости 256
Криоскопические постоянные растворите-
лей, таблица 272
Кристаллографические радиусы 321, 361,
571
-----солей, сумма 360—361
Критический объем расширения твердого
вещества 2-46
а-Кротоновая кислота, константа диссо-
циации, определение методом электро-
проводности 205
Лантан хлористый, кажущийся молярный
объем 242
-----коэффициент активности 534
----------- средний 402
-----относительная вязкость, предельный
коэффициент наклона 170
----- числа переноса 162
----- электропроводность иона хлора 168
t/Z-Лейцин, изменение термодинамических
функций при кислотной диссоциации
476
-------------основной диссоциации 476
константа кислотной диссоциации 544
--------— максимальная 546 *
-----основной диссоциации 544
----------- максимальная 546
Лимонная кислота, константа диссоциа-
ции I, II, III ступени 579
-----—максимальная I, II, III ступени
579
Литий азотнокислый, коэффициент актив-
ности средний 373,' 525, 569
-------------при температурах замер-
зания водных растворов 504
— бромистый, кажущееся относительное
молярное теплосодержание 500—502
-----кажущийся молярный объем 242
-----коэффициент активности средний
363, 524, 569
-------------влияние бромистоводород-
ной кислоты 424—426
-------------при температурах замерза-
ния водных растворов 504
-----критическое состояние разрушения,
значение 246—248
-----относительное парциальное моляр-
ное теплосодержание 502—504
-----среднее расстояние сближения ионов
360
-------- эффективное давление 261
— гидрат окиси, кажущаяся молярная
сжимаемость 256—257
--------кажущийся молярный объем
242
--------коэффициент активности 351
------------- средний 364
----------------в растворах солей ли-
тия 425—426
--------разности между кажущимися мо-
лярными изопиестическими и изохо-
ристическими теплоемкостями 238—239
— железистосинеродистый, влияние силы
поля на электропроводность 209
Литий иодистый, кажущийся молярный
объем 242
-----коэффициент активности средний
363, 524
-----критическое состояние разрушения,
значение 246—248
-----среднее расстояние сближения ионов
360
— муравьинокислый, коэффициент актив-
ности, средний при температурах замер-
зания водных растворов 504
— сернокислый, кажущееся относитель-
ное молярное теплосодержание 500
-----коэффициент активности средний
391, 397
-------------- влияние гидрата окиси
лития и серной кислоты 425-—426
-----относительное парциальное моляр-
ное теплосодержание 502
— п-толу о л сульфоновокислый, коэффи-
циент активности средний 526
— уксуснокислый, коэффициент активно-
сти средний 525
--------------при температурах замерза-
ния водных растворов 504
— хлористый, высаливание нейтральных
молекул 376, 378
-----кажущаяся молярная расширяе-
мость 253
-----------сжимаемость 256—257
-----кажущееся относительное молярное
теплосодержание 500—502
-----кажущийся молярный объем 242
-----коэффициент активности средний
363, 373, 524, 569
-------------- влияние соляной кислоты
и гидрата окиси лития 424—426
--------------при температурах замер-
зания водных растворов 504
— — критическое состояние разрушения,
значение 246—248
-----относительное парциальное моляр-
ное теплосодержание 502—504
----- парциальная молярная теплоем-
кость 237
-----разность между кажущимися' моляр-
ными изопиестическими и изохориче-
скими теплоемкостями 238—239
-----среднее расстояние сближения ио-
нов 360
-----числа переноса 499, 558
—- — эквивалентная электропроводность
498, 558
— хлорноватокислый, коэффициент актив-
ности, средний при температурах замер-
зания водных растворов 504
— хлорнокислый, коэффициент активно-
сти средний 526
--------------при температурах замерза-
ния водных растворов 504
Магний азотнокислый, кажущееся относи-
тельное молярное теплосодержание 500
-----коэффициент активности средний
570
-----относительное парциальное моляр-
, ное теплосодержание 503
— бромистый, кажущееся относительное
молярное теплосодержание 500
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 609
---------------- - ।----------------——
Магний бромистый, коэффициент активно-
сти средний 397, 530, 570
-----относительное парциальное моляр-
ное теплосодержание 503
— иодистый, коэффициент активности
средний 397, 530, 570
— сернокислый, высаливание нейтраль-
ных молекул 376, 379
-----диэлектрическая постоянная, влия-
ние частоты тока 207 •
-----кажущаяся молярная сжимаемость
254 ,
-----кажущееся относительное молярное
теплосодержание 221, 501
-----константа диссоциации 149
-----коэффициент активности средний
401
-----относительное парциальное моляр-
ное теплосодержание 503
-----электропроводность, влияние силы
поля 209, 210
-----------частоты тока 206—207
----- эффективное давление 261
— хлористый, высаливание нейтральных
молекул 376 '
—----кажущееся относительное молярное
теплосодержание 500
-----коэффициент активности средний
397, 530
-----относительное парциальное моляр-
ное теплосодержание 503
— — эквивалентная электропроводность
498
-— хлорнокислый, коэффициент активности
средний 570
Малоновая кислота, изменение термодина-
мических функций при диссоциации
по второй ступери 476
-----константа диссоциации по второй
ступени 541
-----------максимальная 545
Марганец сернокислый, коэффициент ак-
тивности 401
-----относительная вязкость, предельный
коэффициент наклона 471
— хлористый, коэффициент активности
средний 397, 530
н-Масляная кислота, изменение термодина-
мических функций при диссоциации 476
-----константа диссоциации 478, 541
-----в смесях изопропиловый
спирт—вода 580—581
-----------зависимость от температуры
494
----------- максимальная 545
--------------в смесях изопропиловый
спирт—вода 583
-----------определение методом элект-
ропроводности 205
Математические функции 117—137
Медь азотнокислая, коэффициент актив-
ности средний 530
— малоновокислая, константа диссоциа-
ции 149
— сернокислая, кажущееся относительное
молярное теплосодержание 501
-----константа диссоциации 149
-----коэффициент активности средний
39 г. Харнед. В. Оуэн
Медь сернокислая, относительная вяз-
кость, предельный коэффициент на-
клона 171
-----относительное парциальное моляр-
ное теплосодержание 503
----- эквивалентная электропроводность
498
— хлористая, коэффициент активности
средний 397, 530
Междуионное притяжение, теория 33
Метиловый спирт, коэффициент сжимае-
мости 259
Метиловый спирт—вода, смеси, константа
диссоциации пропионовой кислоты
580—581
----------------уксусной кислоты 543
---------------- максимальная
548
-----------средний коэффициент актив-
ности соляной кислоты 511
Метод адиабатической тепловой компен-
сации 217
—: Дебая 66
— изопиестического сравнения 275
— электрического изображения Вагнера 70
Мицеллы 586
Модель центрального иона Дебая и Пау-
линга 571 I
Молекулы, взаимодействие, теория кон-
центрированных растворов 369
Молочная кислота, константа диссоциа-
ции, определение методом электропро-
водности 205
а-Молочная кислота, изменение термоди-
намических функций при диссоциации
476
-----константа диссоциации 478, 541
----------- максимальная 545
Моляльность ионов средняя 27
— определение 27
— стехиометрическая 28
Молярность ионов средняя 27
— определение 27
— стехиометрическая 28
Молярные объемы кажущиеся и теплота
разведения 563—566
Молярный объем хлористого калия кажу-
щийся 564
Моноэтиламмоний, гидроокись, константа
диссоциации, определение методом
электропроводности 205
Мочевина, кажущийся молярный объем 249
Муравьиная кислота, изменение термоди-
намических функций при диссоциации 476
-----константа диссоциации 478, 541
-----в смесях диоксан—вода 542,
547
-----------зависимость от диэлектриче-
ской постоянной 490, 494
----------------молярной доли диок-
сана 472—473
----------------температуры 474—475
----------максимальная 545
-----------определение методом элект-
ропроводности 205
-------- — таблица 473
-----коэффициент активности, влияние
среды 481
-----средний ионный радиус 492
610
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Напряжение сдвига, составляющая 78—81
Напряженность действующего на ион поля,
уравнение 99
Натрий азотнокислый, высаливание нейт-
ральных молекул 376
-----кажущееся относительное молярное
теплосодержание 500
-----коэффициент активности средний
373, 525
--------------при температурах замер-
зания водных растворов 504
-----относительное парциальное моляр-
ное теплосодержание 502
— бромистый, влияние на коэффициент
активности бромистоводородной кисло-
ты и гидрата окиси, натрия 425—426
-----высаливание нейтральных молекул
376
-----кажущееся относительное молярное
теплосодержание 500—502
-----кажущийся молярный объем 242, 244
----- коэффициент активности средний
363, 524
--------------при температурах замерза-
ния водных растворов 504
—-------------' таблица 521
-----критическое состояние разрушения,
величина 246—248
— — относительная парциальная моляр-
ная теплоемкость 521
-----относительное парциальное моляр-
ное теплосодержание 502—504, 521
-----парциальная молярная теплоемкость
237 .х
— — среднее расстояние сближения ионов
360
-----; термодинамические свойства рас-
творов 350
----- эффективное давление 261
— бромноватокислый, кажущееся относи-
тельное молярное теплосодержание 500
-----коэффициент активности, средний
526
—---относительное парциальное моляр-
ное теплосодержание. 502
— валерьяновокислый, коэффициент ак-
тивности средний 587
— гвайяколат, константа диссоциации
и предельная электропроводность 187
— гептиловокислый, коэффициент актив-
ности средний 587
— гидрат окиси, кажущаяся молярная
расширяемость 253
--------------сжимаемость 256—257
--------кажущееся относительное моляр-
ное теплосодержание 500—502
--------кажущиеся молярные теплоемко-
сти, изопиестические и изохорические
238—239
--------кажущийся молярный объем 242
--------коэффициент активности 351
—-------------в растворе хлористого нат-
рия 443
-------------- влияние давления 358
---------------средний 364, 522, 569
----------------- в растворах солей нат-
рия 425—426
--------относительная парциальная мо-
лярная теплоемкость 523
Натрий, гидрат окиси, относительное пар- <
циальное молярное теплосодержание
352, 502—504, 523 J
--------парциальная молярная теплоем-
кость 236 ’ . J
--------среднее расстояние сближения «
ионов 360 ,
--------термодинамические свойства рас- ?
Дворов 350—352
-------- эквивалентная электропровод- i
ность 498
--------эффективное давление 261
— иодистый, влияние на коэффициент ак- ’
тивности гидрата окиси калия 425—426 ’
-----высаливание нейтральных молекул
-----кажущийся молярный объем 242 ;
•-----коэффициент активности средний
363, 524 ]
-----критическое состояние разрушения, J
величина 246—248 -- |
-----парциальная молярная теплоемкость i
237 \
-----произведение электропроводности на
вязкость 184 j
-----среднее расстояние сближения ионов '
360
----- эквивалентная электропроводность
498
— —эффективное давление водных рас-
творов 261
— йодноватокислый, кажущееся относи-
тельное молярное теплосодержание 500
-----относительное парциальное моляр-
ное теплосодержание 502
— каприловокислый, коэффициент актив-
ности средний 587
— каприновокислый, коэффициент актив-
ности средний 587
— капроновокислый, коэффициент актив-
ности средний 587 i
— малеиновокислый, коэффициент актив-
ности средний 570
— маслянокислый, коэффициент актив-
ности средний 587
— муравьинокислый, коэффициент актив-
ности средний 504, 587
— пеларгоновокислый, коэффициент ак-
тивности средний 587
— пропионовокислый, коэффициент актив-
ности средний 587
----- эквивалентная электропроводность i
498 ;
— роданистый, коэффициент активности ;
средний 525
— серноватистокислый, коэффициент ак-
тивности средний 391, 397 >
— сернокислый, влияние на коэффициент ,
активности гидрата окиси натрия и сер-
ной кислоты 425—426
-----высаливание нейтральных молекул
376
-----кажущаяся молярная, расширяв- >
мость 253
-----кажущееся относительное молярное
теплосодержание 500
-----кажущийся молярный объем 242
-----коэффициент активности средний ,
391, 397 -1
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
61 f
Натрий сернокислый, относительное пар-
циальное молярное теплосодержание
502
-----парциальная молярная теплоемкость
237
----- числа переноса 499
----- эквивалентная электропроводность
----- эффективное давление 261
— л-толуол сульфоновокислый, коэффици-
ент активности 526
— углекислый, кажущаяся молярная сжит
маемость 264
-----кажущийся молярный объем 242
— уксуснокислый, коэффициент активно-
сти средний 525, 587
--------------при температурах замер-
зания водных растворов 504
-—: — числа переноса 499
----- эквивалентная электропроводимость
498
— фосфорнокислый, коэффициент актив-
ности средний 526
— фтористый, коэффициент активности
средний 525
— фумаровокислый, коэффициент актив-
ности средний 570
— хлористокислый, коэффициент актив-
ности средний 526
— хлористый, влияние на коэффициент
активности соляной кислоты и гидрата
окиси натрия 424—426
-----высаливание нейтральных молекул
376—379
-----кажущаяся молярная расширяе-
мость 252—253
-----------сжимаемость 256—257, 264
-----кажущееся относительное молярное
теплосодержание 500—502
>---кажущиеся молярные теплоемкости
водных растворов, изопиестические и
изохорические 237—239
-----кажущийся молярный объем 242
-----константы разведения, таблица 223
-----коэффициент активности в растворе
гидрата окиси натрия 443
-----------влияние давления 358
----------- средний 363, 373, 524, 534,
569
--------------вычисление при темпера-
турах от 0 до 100° 357
'----'-------зависимость от температу-
ры 344
--------------при- температурах замер-
зания водных растворов 504
— ------------таблица 518
-----------таблица 341, 343
—-------диффузии 176—177
'----коэффициенты активности теорети-
ческие и наблюдаемые 357
-----критическое состояние разрушения,
величина 246—248
-----осмотические коэффициенты раство-
ров разных концентраций 278, 343—345
-----значение функции а 278
-----относительная вязкость, предельный
коэффициент наклона 170
-------- парциальная молярная теплоем-
кость, таблица 345—346
Натрий хлористый, относительное пар-
циальное молярное теплосодержание
345—346, 502—504
— --------------таблица 342, 599
-----парциальная молярная теплоемкость
237
--------------при постоянном давлении
237
-----парциальный молярный объем, влия-
ние давления 250
-----среднее расстояние сближения ионов
359—360
-----теплота разведения, таблица, 219—220
-----термодинамические свойства раство-
ров 340—346
-----числа переноса 161, 163, 499, 558
----- эквивалентная электропроводность
498, 558
— — электропроводность иона хлора 168
— — эффективное давление 261
— хлорноватокислый, коэффициент-актив-
ности средний 526
--------------при температурах замерза-
ния водных растворов 504
-----кажущееся относительное молярное
теплосодержание 500
-----относительное парциальное моляр-
ное тейлосодержание 502
— хлорнокислый, коэффициент активно-
сти, средний при температурах замер-
зания водных растворов 504
— хромовокислый, коэффициент активно-
сти средний 570
Никель сернокислый, константа диссоциа-
ции 149
-----коэффициент активности средний
401
— хлористый, коэффициент активности
средний 397, 530
о-Нитробензойная кислота, константа дис- .
социации 577
-----коэффициенты высаливания 529
dZ-Норлейцин, изменение термодинамиче-
ских функций при кислотной диссоциа-
ции 476
-------------- основной диссоциации 476
— константа кислотной диссоциации 544
— t-------максимальная 546
— — основной диссоциации 544
-----------максимальная 546
Нормальная атмосфера 117, 555
Обобщенный момент 57
Обращение солевого эффекта, см. Солевой
эффект, обращение 1
Объем идеального газа 555
— растворенных частиц, поправка Ван-
дер-Ваальса 369—372
Объемная сила 80, 86
Одно-двухвалентные электролиты, кажу-
щееся относительное молярное тепло-
содержание, предельные коэффициенты
наклона 225
Одно-одновалентные электролиты, кажу-
щееся относительное молярное теплосо-
держание, предельные коэффициенты
наклона 224
Октиламии, эквивалентная электропровод-
ность 587
39*
612
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Октодепиланилин, эквивалентная электро-
проводность 587
Осмотические коэффициенты 276, 365, 374,
439, 572—575
-----стандартных растворов, таблица 276
-----хлористых и фтористых четырех-
замещенных алкиламмониевых основа-
ний 374
Осмотический коэффициент 51, 52, 270—
272, 273, 274, 275—282, 343, 423
-----вычисление из данных для темпера-
тур замерзания, уравнение 270—272
------------------- кипения, уравнение
273—274
--------------по измерениям давления
пара, уравнение 274—276,
-----------изопиестических данных, урав-
нение 274—276
--------по методу Гуггенгейма 365—367
;----зависимость от моляльности, про-
верка уравнения 439—440
-----практический, уравнение 29
----- растворителя 29
— рациональный 29
-----связь с коэффициентом активности
30, 433
------ уравнение 51—52
Параметр ионнрй, см. Ионный параметр
Параметры уравнений для коэффициента
активности, таблица 360
-----сжимаемости 257—259
— уравнения вязкости 172
-----диэлектрической постоянной, табли-
ца 120
-----зависимости предельных подвижно-
стей ионов от температуры 559
-----константы кислотной диссоциации,
таблица 486
-----коэффициента активности 567
-----относительного кажущегося моляр-
ного теплосодержания, таблица 227
-----• — парциального молярного тепло-
содержания 513—514
-----относительной парциальной моляр-
ной теплоемкости 513—514
-----парциального молярного объема 242
-----теплоемкости при постоянном давле-
нии 237
----- электропроводности разбавленных
растворов 151, 152
Парциальные молярные величины 21—22
-----объемы солей, влияние давления
249—250
-------- электролитов 240—243
—----------определение из значений
плотностей раствора 240
--------— связь с концентрацией, урав-
нение 241
— относительные молярные теплосодер-
жание и теплоемкость, коэффициенты
наклона в водных растворах, таблица
128
Парциальный молярный объем компонента,
уравнение 26
--------растворенного вещества, опреде-
ление 260
Парциальный молярный объем электроли-
та, уравнение 26, 62
— относительный молярный объем, пре-
дельное уравнение 62
pH, определение 302—306
----постоянной уравнения, таблица 305
— некоторых стандартных растворов, таб-
лица 306
— уравнение 303, см. также Концентра-
ция водородных ионов
Первичные эффекты для смесей органи-
ческий растворитель—вода, таблица 481
Первичный эффект, см. Среда
Пикрат тетраэтиламмоний, электропровод-
ность, влияние диэлектрической постоян-
ной 143—144
Пикриновая кислота, константа диссо-
циации 577
Плотности растворов соляной кислоты
в смесях диоксан—вода, таблица 506—
507
Поверхностное натяжение разбавленных
растворов электролитов 380—382
------ растворов электролитов, предель-
ный закон 70—72
----электролитов, значения функций,
таблица 129
-------- уравнение 32
--------уравнения 128—129
Поверхностный слой 70
Подвижности иоиов предельные, зависи-
мость от температуры, параметры урав-
нения, таблица 559
Подвижность ионов, зависимость от темпе-
ратуры 559
----определение 90
---- предельная, определение 90
---- уравнение 174
Полимерные электролиты 586
Поляризация электродов 139
Постоянная сосуда, определение 140
Постоянные Дебая и Гюккеля для водных
. растворов, таблица 122
Потенциал адсорбционный, см. Адсорб-
ционный потенциал
— асимметрии колеблющегося иона 99
— атмосферы иона, уравнение 99
— величина, определение 87
— возмущающей силы 74—75
Потенциал диффузионный, см. Диффузион-
ный потенциал
— иона и его атмосферы, уравнение, теория •
Дебая 38—42, 51
----электрический 38
— стандартный, см. Стандартный потен-
циал элементов
— термодинамический, см. Термодинами-
. ческий потенциал
— химический, см. Химический потенциал
— электродный, см. Стандартный электрод-
ный потенциал
— электрокинетический, см. Электрокине-
тический потенциал
— электростатический, см. Электростати-
ческий потенциал
Потенциалы асимметрии, значения 99
— и силы в переменном поле, уравнения
97—99
— условия симметрии 103—104
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
613
Правило Вальдена, произведение электро-
проводности на вязкость 182—184
----------------влияние температуры,
таблица 184
— Пнсаржевского 182
— произведения растворимости, ограниче-
ния 444
— эмпирическое Оствальда—Вальдена—
Бредига 142
Празеодим хлористый, коэффициент актив-
ности 534
Предельное значение числа переноса, метод
Лонгсворта 335
— уравнение для коэффициента диффузии
электролита 561
--------относительного парциального мо-
лярного объема электролита 62
----------------теплосодержания 60—
61
--------относительной парциальной мо-
лярной теплоемкости 60—61
------ — расширяемости электролита 62
--------сжимаемости электролита 62—63
--------чисел переноса 160—162
Предельные коэффициенты наклона 49,
128,,170—171, 172, 173 , 212 , 222, 224,
225, 237, 505, 516, 554—560
—------для парциальных относительных
молярных величин 127, 128
Предельный закон для вязкости 129—130
— — Дебая и Гюккеля 64
----для зависимости коэффициента ак-
тивности от концентрации 48—51
--------коэффициента активности 186
--------поверхностного напряжения эле-
ктролитов 70—72
-------- электропроводности электроли-
тов 89—93
— коэффициент наклона 49, 50, 63, 122,
130, 131, 222, 236, 243, 557, 563
--------уравнение для коэффициента ак-
тивности сильных электролитов
221—225
Притяжение междунонное, см. Теория
междуионного притяжения
Произведение электропроводности на вяз-
кость некоторых ионов см. также Пра-
вило Вальдена 185
Пропионовая кислота в смесях дноксан—•
вода, константа диссоциации 543
—-----------------— максимальная 548
---------------средний ионный радиус 492
-----------------изопропиловый спирт—вода,
константа диссоциации 580—581
----------------------максимальная 583
--------- — метиловый спирт—вода, кон-
станта диссоциации 580—581
----------этиловый спирт—вода, кон-
станта диссоциации 580—581
----— изменение термодинамических функ-
ций при диссоциации 476
—• — константа диссоциации 470, 541
—---------влияние среды 481
----------- зависимость от диэлектриче-
ской постоянной 49С, 494
----------------молярной доли днокса-
на 472—473
----------------температуры 474—475
----------максимальная 545
н-Пропноновая кислота, константа диссо-
циации, определение методом электро-
, проводностн 205
Пространственная решетка, статистическая
структура 248
Протоны молекулы воды, связь с акцепто-
рами протонов, схема процесса 364
Пространственная решетка, статистическая
структура 248
Процесс заряжения, уравнение 47—48
Работоспособность, метод вычисления 47
— уравнение 20
Радиусы ионные, см. Ионные радиусы
— кристаллографические, см. Кристалло-
графические радиусы
Разность потенциалов 22
-----на границе раздела фаз 299—301
----------------- уравнение 299—300
----- уравнение 286
Рамановские спектры, применение для
исследования диссоциации 576
Раствор демальный, определение 140
Растворимость и теория междуионного
притяжения, измерения 419—423
--------специфического ионного взаимо-
действия 441—442
— нейтральных веществ и электролитов,
изменение при действии солей 63—70
— проверка уравнения для ряда электро-
литов, таблица, 441
— стехиометрическое произведение 419
— тетраннтродиаммннокобальтиатов в рас-
творах азотнокислых калия и натрия,
отношение, таблица 442
— хорошо растворимых солей в растворах
электролитов, вычисление 442—446
Растворы электролитов концентрирован-
ные 240, 243, 353, 367, 369—372
-----разбавленные 29, 150—152, 213, 221,
231, 237, 240, 242, 280, 341—343, 365,
419, 445, 500—501
Расширяемость, кажущаяся молярная
251—254
— парциальная молярная 251—254
— относительная парциальная малярная,
предельное уравнение 62
Реакции изоэлектрические 490
Рекомбинация ионов, скорость 77
Рубидий азотнокислый, коэффициент ак-
тивности средний 373, 525
— бромистый, кажущийся молярный объем
242
-----коэффициент активности средний
363, 524’
— — критическое состояние разрушения,
величина 246, 248
-----среднее расстояние сближения ноиов
360
— ноднстый, кажущийся молярный объем
242
-----коэффициент активности средний
363, 524
-----критическое состояние разрушения,
величина 246, 248
-----среднее расстояние сближения ионов
360
— сернокислый, кажущееся относительное
молярное теплосодержание 500
614
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Рубидий сернокислый, коэффициент актив-
ности средний 570
-----относительное парциальное моляр-
ное теплосодержание 503
— уксуснокислый, коэффициент активно-
сти средний 525
— хлористый, кажущийся молярный объем
242
-----коэффициент активности средний
363, 373, 524
-----критическое состояние разрушения,
величина 246 , 248
—: — среднее расстояние сближения ионов
360
— фтористый, кажущееся относительное
молярное теплосодержание 500
Салициловая кислота, коэффициенты вы-
саливания 529
Самарий хлористый, коэффициент актив-
ности в водных растворах 534
Сахароза—вода, днэлектрическне постоян-
ные смесей 120
— кажущаяся молярная теплоемкость 239
— осмотические коэффициенты стандарт-
ных растворов 276
— относительная вязкость, предельный
коэффициент наклона 171
— относительное кажущееся молярное те-
плосодержание 230
Свинец бромистый, константа диссоциа-
ции иона 577
— иоднстый, ион, константа диссоциации
577
— хлористый Ё°' —Ё°, зависимость от кон-
центрации 393
-----ион, константа диссоциации 577
-----коэффициент активности средний
397 531
Свободная энергия 20, 22, 31, 277, 286
— — и константа равновесия 31
-----изменение, выраженное через актив-
ности, уравнение 286
-----общее увеличение 22
-----•- парциальная молярная 22, 24, 25, 27
-----------связь с активностью, ура-
внение 25
-----------------осмотическим коэффи-
циентом, уравнение 29
------------ уравнения 25, 27
-----связь с гальваническим элементом,
уравнение 286
— —--------- электрической работой 277
—, —г*Уравнение 20
.Свойцдва колмиативные, определение 26
Селеновая кислота, ион, константа диссо-
. цнйцни 577
Серебро азотнокислое, кажущийся стан-
дартный молярный объем 242
-----константа диссоциации 149
-----коэффициент активности средний 526
-----числа переноса в водных растворах
499
----- эквивалентная электропроводность
в водных растворах 498
— хлористое, коэффициенты активности,
средние в водных растворах солей 421
-----уравнение пронзведення растворимо-
сти 307
Серная кислота, изменение термодинамиче-
ских функций при диссоциации по вто-
рой ступени 476
---константа диссоциации по второй
ступени 541
-------------------- определение мето-
дом электропроводности 205
-----коэффициент активности 410—411
-----------средний в водных растворах,
таблицы 411, 534
--------------в растворах сернокислых
солей 425—426
--------------------солен, определение
. методом электродвижущих сил 423—426
--------------при высоких концентра-
циях 569
-----осмотические коэффициенты стан-
дартных растворов 276
-----относительная парциальная моляр-
ная теплоемкость 412—415
-----:--------—в водных растворах 414
-------------------------таблица 535
-----относительное парциальное молярное
теплосодержание 412—415
-----------------в водных растворах
413—414
------------------------- таблица 535
-----числа переноса катиона в водных
растворах 415
-----------.----------зависимость ot '
моляльности 414
---------------------- таблица 535
Сернистая кислота, константа диссоциа-
ции 577
Сжимаемость адиабатическая, уравнение
263
— изотермическая, уравнение 263
— кажущаяся молярная 254—265
--------коэффициенты наклона, таблица
264
---------параметры уравнений, таблица’
256
-------- уравнение 255
— коэффициент, см. Коэффициент сжимае-
мости
— метод определения по измерениям ско-
рости звука 263
— определения по скорости звука, табли-
цы 259, 262
— относительная парциальная молярная,
предельное уравнение 62
— парциальная молярная 254—265
-------- уравнение 254—255
— растворителя под влиянием ионов 250
— ультразвуковые волны 263—264
Сила объемная 80
Силы переноса ионов 85
Сильные электролиты 101—109, 182, 208—
210, 339—383, 418—446, 563, 576—577
-----влияние сильных попей, теория 101—
109
-----эффект Вина 208—210
Символ Кронекера 93
Синильная кислота, всалнвание и высали-
вание различными солями 379
-----распределение между водными рас-
творами солен и бензолом 378—380
Скандий хлористый, коэффициент актив-
ности 534
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Скорости реакций диссоциации молекул
и Ионов 77
Скорость деформации сдвига 78—81
— ионов 109
— перецоса ионов 84—85
— реакции диссоциации, уравнение 77
-----рекомбинации, уравнение 77
— химической реакции 76
Слабые электролиты 109—115, 182, 201—
205, 210—213, 449—495, 560
— — влияние сильных полей 109—115
-----диссоциация 201—205
—»— константы диссоциации по методу
’ электропроводности, таблица 205
-----эффект Вина\10—213
Смеси ацетон—вода 120
— бутиловый спирт, трет.—вода 120
глицерин—вода 120, 319, 320, 507,
512, 580—581, 583
— двух электролитов, различная интер-
. претацня свойств 432—433
— дноксан—вода 120, 312, 316, 318, 319,
320, 321, 327, 331, 333, 335,' 336, 481,
490, 505, 506, 507, 509, 513—514,
515—516, 542—543, 547—548
-----предельные коэффициенты наклона
для коэффициентов активности 505
— изопропиловый спирт—вода 120, 319,
320, 507, 512, 580—581, 583
— маннит—вода 120
— метиловый спирт—вода 120, 167, 319,
320, 321, 481, 507, 511, 543, 548, 580—
581
— 1,1-Валентных гаплоидных электроли-
тов 436—441
— оснований с хлоридами 442—443
— н-пропиловый спирт—вода 120
— сахароза—вода 120
— сильных электролитов 418—446
— уксусная кислота—вода 481
— 1,1-хлорид—соляная кислота яри 25°,
характеристические коэффициенты на-
клона, таблица 437
— хлористый натрий—вода 580—581
--------соляная кислота 146
-------------характеристические накло-
ны, таблица 437
— электролитов, термодинамическая тео-
рия 432—436
— этиленгликоль—вода 120
— этиловый спирт—вода 120 , 319 , 320,
321, 507, 511, 580—581
Солевое действие 375
Солевой эффект, обращение для ацетона
и синильной кислоты 379
Солевые эффекты в растворах уксусной кис-
лоты 378
-----для различных веществ 376—378
-----теория Дебая 378—380
-----уравнения 380
Сольватация, числа 250
Соляная кислота, высаливание нейтраль-
ных молекул 376
-----вычисление коэффициента активно-
сти 323—326
-----кажущаяся молярная расширяе-
мость 253
——--------- сжимаемость водных раство-
ров 256—257
Соляная кислота, кажущаяся молярнай
теплоемкость при постоянном давле-
нии 239
-------------------объеме 239
-----кажущееся. относительное молярное
теплосодержание водных растворов
500—502
-----кажущиеся молярные теплоемкости,
изопиестические и изохорические 237
----------------;-----разность , табли-
ца 238
----------— — — — — уравнение 237
-----кажущийся стандартный молярный
объем 242
-----константа диссоциации, влияние
среды 481
-----константы диссоциации в смесях
диоксан—вода 312
-----коэффициент активности в водных
растворах 322—323
—------:------растворах галоидных со-
лей 425—426
----------------------щелочных метал-
лов 424
----------------солей, определение ме-
тодом электродвижущих сил 423—
426
----------------хлористых солей 423—
426
--------------смесях диоксан—вода 327
----------влияние давления 358
—---------вычисление, таблица 325
------------ средний в водных растворах,
таблица 508
----------------концентрированных вод-
ных растворах 538
----------------различных растворите-
лях, таблица 51Г
----------------растворах хлористых
солей 536, 538
----------------смесях дноксан—веда
509—510
-------------------органический рас-
творитель—вода, таблица 511—512
----------------средах, содержащих ор-
ганический растворитель 327—328
----------------органических раствори-
телях, таблица 511—512
-----коэффициенты активности средние,
экспериментальные и вычисленные, таб-
лица 323
-----относительная парциальная моляр-
ная теплоемкость 333—352
-----------------в смесях диоксан—вода
333
----------------таблица 332
-----относительное парциальное моляр-
ное теплосодержание 328—332, 352
-----в растворах галогени-
дов 426—427
-------------------смесях диоксан—во-
да 331
----------------водных растворов 502,
504
----------------зависимость от концен-
трации 331
----------------константы уравнения,
таблица 330
----------:-----таблица 329
616
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Соляная кислота, парциальная молярная
теплоемкость, вычисление 236—237
-----pH стандартных растворов 306
-----плотности в смесях диоксан—вода,
таблица 506—507
-----средние расстояния сближения ионов
в водных растворах 359—360
----- числа переноса 333—337, 558
-----------в водных растворах при 25°
161,499
-----------катиона в воде и в смесях диок-
сан—вода, таблица 515—516
----- ----------- смесях диоксан—вода
335—336
----- эквивалентная электропроводность
558
--------— в водных растворах 498
----- электропроводность в смесях диок-
сан—вода 311—312
Сосуд для определения электропроводности
139—140
Специфическое взаимодействие ионов, тео-
рия 365—367, 435—436
Среда, влияние на свойства электролитов
479—484
--------стандартные потенциалы 320—321
— вторичный эффект, определение 479
— зависимость между суммарным, первич-
ным и вторичным эффектами, уравнение
480
— первичные эффекты для смесей органи-
ческий растворитель—вода 481
— первичный эффект органического рас-
творителя для ряда кислот, таблица 481
— связь между суммарным, первичным и
вторичным эффектами 479
— суммарный эффект, определение 479
Среднее расстояние наибольшего сближе-
ния ионов 146
-----сближения ионов 359, 370, 422
-----------в растворах некоторых элек-
тролитов, таблица 359
--------— — случае смеси солей 422
-----------и коэффициент активности
358—362
-----------электролитов, параметры урав-
нений, таблица 359—361
Средние расстояния сближения ионов
358—362
Средний ионный диаметр электролита,
определение 370
— коэффициент активности 419
— Стехиометрический моляльный коэффи-
циент активности 27, 49
Средняя активность ионов 24
— моляльность ионов 27
— молярность ионов 24
Стандартное состояние 19, 21, 25, 27, 286
-----для гальванических элементов
286—287
Стандартные потенциалы элемента
H2|H2SO4(m)|PbSO4-PbO2-Pt, таблица
406 ,
--------Н2|НС1 (ли), S (X), Н2О (Y)|AgCl-Ag
в смесях растворителей, таблица 507
— растворы 138—141, 275—276, 305—306
— электроды 302
Стандартный потенциал элементов типа
Н2|НХ (mJ, MX„(m2)|AgX-Ag, уравне-
ние 423
Стандартный потенциал элементов типа
H2|llX(zn1),MXn(zn2)|AgX-Ag, вычисле-
ние коэффициентов активности раство-
ров галогенидов путем экстраполяции
-----------MxHg|MX2 (m)|AgX-Ag, урав-
нение 392
-----элемента 112|НВг (ли) jAgBr-Ag 347-г-
--------H2|H2SO4(zn) IPbSO4-PbO2-Pt 404
—408
-----------вычисление с помощью кон-
станты диссоциации HSOj 405—408
------------определение методом экстра-
поляции 404—405
—----------определение с помощью кон-
станты диссоциации, зависимость от
ионной силы, экстраполяция 407
----------- уравнение 404
--------H2|H2SO4(m), Hg2SO4-Hg 411—
412
—----------таблица 412
--------Н2|НС1 (m)|AgCl-Ag 313
-----------в смешанных растворителях,
таблица 319
-----------зависимость от диэлектриче-
ской постоянной среды 320—321
--------H2|HCl(m),S (X), Н2О (У)| AgCl-Ag
для смесей вода—органический раство-
ритель 315—322
-----—1----значения экстраполяционной
,функции, Е0'—Е°Доп., таблица 317
-----------методы экстраполяции экспе-
риментальных данных 316—318
--------типа Н2|НВ (mJ, NaCl (m2)|
AgCl-Ag 481—482
--------CdxHg |CdBr2(m)| AgBr-Ag, вы-
числение по уравнению 394
-----— Cd-Cdz Hg |CdBr2 (m)\ AgBr-Ag,
таблица 531
-----Cd-CdxHg | CdCl2 (m)| AgCl-Ag,
таблица 531
--------Zn-Zn^Hg |ZnJ2 (m) | AgJ-Ag, таб-
лица 531
—-------Zn-Zn^Hg | ZnCl2(m) | AgCl-Ag таб-
лица 531
-----электрода, серебро-бромистое се-
ребро, таблица 348
--------серебро-хлористое серебро, таб-
лица 314
— электродный молярный потенциал,
определение с помощью экстраполяции
291—292
-----потенциал 285—287
-------- кадмия 531
-------- определение 289
--------серебра из данных по электродви-
жущим силам элементов, таблица 309
-------- цинка 531
Степень диссоциации азотной кислоты,
таблица 576
— — влияние силы поля, уравнение 115,
137
Стехиометрическая моляльность электро-
лита 28, 573
— молярность электролита 28
Стронций азотнокислый, кажущееся отно-
сительное молярное теплосодержание
в водных растворах 500
ПРЕДМЕТНЫЙ указатель
617
Стронций азотнокислый, коэффициент
активности средний 570
-----относительное парциальное молярное
теплосодержание в водных растворах
503 *
— бромистый, кажущееся относительное
молярное теплосодержание в водных
растворах 500
-----коэффициент активности средний
570
-----относительное парциальное молярное
теплосодержание в водных растворах
503
• — иодистый, коэффициент активности
средний 570
— хлористый, влияние на коэффициент
активности соляной кислоты 425—426
-----кажущееся относительное молярное
теплосодержание в водных растворах
500
• ,----кажущийся стандартный молярный
объем 242, 244, 245
-----коэффициент активности средний 389,
397, 570
----------средний в водных растворах
530
-----относительное парциальное молярное
теплосодержание в водных растворах
503
----- эквивалентная электропроводность
в водных растворах 498
Таллий азотнокислый, константа диссоциа-
ции 149
-----коэффициент активности средний 526
— уксуснокислый, коэффициент активно-
сти средний 526
— хлористый, константа диссоциации 149
— хлорнокислый, коэффициент активности
средний 526
Температура абсолютная, функции, табли-
ца 118
Теория ассоциации ионов Бьеррума 54—58,
125—126
-------, функции 125—126
-------, функция IgQ(b), таблица 126
— Бренстеда 365—367, см. также Ионное
взаимодействие
— влияние частоты тока на электропро-
водность 132—135
— Дебая 38—42, 65—70
-----Гюккеля 53—54, 323—326, 358—365
------- основное уравнение 53
-----и Мак-Олэя 63—65
-----уравнение для атмосферы иона,
см. Атмосфера иона
— диффузии 132
— концентрированных растворов Скэт-
чарда 367—369
-------электролитов с учетом поправки
Ван-дер-Ваальса 366^—372
— междуионного притяжения 156, 189—
190 , 419—423
— образования ионных пар 59
-------тройников, значения функций
таблица 127
— и квадруполей 58—60
— растворов, гидратная Менделеева 4
физическая 4
Теория растворов химическая 4
— солевых эффектов Дебая 378—380
— специфического взаимодействия ионов
435—436, 441—442
-----------уравнение 435—436
— Фуосса и Крауса 58—60, см. также Ион-
ные тройники и квадруполи
— электролитов 568—572
Теплоемкости, кажущиеся молярные, изо-
пиестические и изохорические, разность,
таблица 238
-------------------- уравнение 237
Теплоемкость иона в растворе 339
— кажущаяся молярная, изопиестическая
и изохорическая 237, 239
— молярная чистого растворителя 232
— относительная молярная 26
-----парциальная молярная 294—295,
332—333, 345—346
-----------вычисление из данных „ню
электродвижущим силам 294—295*
-----------для смесей диоксан—вода,
таблица 513—514
-----------параметры уравнения для
смесей диоксан—вода, таблица 513—514
-----------соляной и бромистоводород-
ной кислот, таблица 332
-----------сравнение калориметриче-
ских и электрометрических способов
определения 352—353
-----------предельное уравнение 60—61
----------- уравнение 413
— парциальная молярная 26, 232
--------для высоковалентных электроли-
тов, таблица 237
--------некоторых электролитов, вычис-
ление, таблицы 236—237
--------параметры уравнения, таблица
237
--------при постоянном давлении 232
--------таблицы 236, 237
— раствора общая 232
----- удельная 233
— растворенного вещества, кажущаяся
молярная, уравнение 233
— реакций диссоциации, зависимость от
числа углеродных атомов 493
— увеличение при реакции диссоциации
491
— чистого растворителя, удельная 233
— электролитов, относительная парциаль-
ная молярная 352
--------------таблица 519—523, 533, 535
Теплосодержание и теплоемкость, относи-
тельные парциальные молярные серной
кислоты в водном растворе 412—415
-----------------хлористого натрия
345—346
— изменение при реакциях диссоциации
491
— кажущееся молярное 564
— относительное кажущееся молярное
217—230
-----------для ряда электролитов, таб-
лица 500—501
--------параметры уравнения, табли-
ца 227
-----парциальное молярное 217—230 (
294—295, 328—33 2
618
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Теплосодержание, относительное парциаль-
ное молярное, вычисление из данных
по электродвижущим силам 294—295,
328—339
-----галоидоводородных кислот в
растворах галоидных солей 426—427
-----------для ряда электролитов, таб-
лицы 502, 503
-----------значения и параметры уравне-
ния, таблица 513—514
—--------— предельное уравнение 60—61
-----------предельные коэффициенты на-
клона для смесей диоксан—вода, таб-
лица 513—514
-----------сравнение калориметрических
и электрометрических способов опре-
деления 352—353
— парциальное молярное 25
— раствора общее, определение 218
— растворенного вещества, относительное
кажущееся молярное 218
— растворителя, парциальное молярное 218
— твердого растворенного вещества, отно-
сительное парциальное 227
— хлористого натрия, кажущееся относи-
тельное молярное 566
— уравнение 20, 25
— электролитов, относительное парциаль-
ное молярное 352
--------------- таблицы 519—523, 533, 535
Теплота диссоциации 230—232
-----воды, суммарная в растворах неко-
торых галоидных солей 460
---------уравнение 459—460
— нейтрализации 230—232
-----при постоянной концентрации 232
— разведения 217—230
— растворения парциальная 227
-----суммарная 227
Термические константы неорганических
веществ 226
Термодинамическая теория смесей при по-
стоянной общей моляльности 432—436
Термодинамические свойства водных рас-
творов сернокислого цинка 398—399
-----------сернокислых солей лития, на-
трия и калия 390—392
-----ионов, связь с размерами и структу-
рой 372—374
-----растворов галоидных солей магния,
кальция, стронция и бария 388—390
— функции воды и кислот в смесях: диок-
сан—вода и метиловый спирт—вода,
таблица 477
-----ионных атмосфер, см. Ионная атмо-
сфера
Термодинамический множитель 174
• — потенциал, определение 44
Термохимические величины 457—463
н-Тетрабутиламмониевая соль окситрифе-
нилборной кислоты, характеристиче-
ские величины для эллипсоидальной мо-
дели диполя 196
Тетрабутиламмоний азотнокислый, кон-
станты электропроводности, таблица 197
-----электропроводность, влияние тем-
пературы 198
— бромистый, электропроводность, влия-
ние силы поля и частоты 212—213
Тетрабутиламмоний, пикрат, коэффициент
активности, определение из эффекта
Вина 212
------ электропроводность', влияние силы
поля' 212
—----характеристические величины для
эллипсоидальной модели диполя 196
— фтористый, осмотический коэффициент
374
— хлористый, осмотический коэффициент
н-Тетрабутиламмоний, пикрат, константы
диссоциации в различных растворите-
лях 200
н-Тетрабутиламмоний, уксуснокислый, ха-
рактеристические величины для эллип-
соидальной модели диполя 196
Тетрадециламин, эквивалентная электро-
проводность 587
Тетраизоамил аммоний азотнокислый, кон-
станта диссоциации, влияние диэлектри-
ческой постоянной 190—191, 195
-----константы диссоциации в смесях
диоксан—вода 190, 194
— пикрат, характеристические величины
для эллипсоидальной модели диполя
196
— растворы солей в бензоле, крнстанты
диссоциации, таблица 199
Тетраметиламмоний фтористый, осмотиче-
ский коэффициент 374
— хлористый, осмотический коэффициент
374
-----произведение электропроводности на
вязкость 184
Тетрапропиламмоний фтористый, осмоти-
ческий коэффициент 374
— хлористый, осмотический коэффициент
374
Тетраэтиламмоний, пикрат, произведение
электропроводности на вязкость 184
-----электропроводность, влияние ди-
электрической постоянной 144
о-Толуиловая кислота, коэффициенты вы-
саливания 529
Торий азотнокислый, коэффициент сред-
ний 570
н-Трибутиламмоний, пикрат, характери-
стические величины для эллипсоидаль-
ной модели диполя 196
Триизоамиламмоний, пикрат, характери-
стические величины для эллипсоидаль-
ной модели диполя 196
Трихлоруксусная кислота, константа дис-
социации 577
Углекислота, константа диссоциации по
второй ступени 541
-----------максимальная 545
----------- первой ступени, определение
методом электропроводности 205
-----------------в воде и в растворах со-
лей 579
--------------------растворах хлористо-
го натрия 582
Удельная электропроводность 138—139
-----смесей соляная кислота—хлористый
натрий, влияние добавок электролитов,
таблица 146
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
619
Удельная электропроводность стандарт-
ных растворов хлористого калия, таб-
лица 141
----- уравнение 139
Удельное сопротивление раствора 139
Уксусная кислота в растворах солей, кон-
станта диссоциации, зависимость от
температуры 485
-----изменение термодинамических функ-
ций при диссоциации 476
-----константа диссоциации 203, 204, 465,
541
-----------в растворах хлористого нат-
рия, таблица 486
----------------хлористых солей 484—
485
-------------смесях глицерин—вода
580—581
---------------- диоксан—вода 547
-------------- — метиловый спирт— во-
да 543
-----------влияние среды 481, 483
—----------зависимость от диэлектриче-
ской постоянной 490, 494
----------------молярной доли диок-
сана 472—473
----------------температуры 474—475,
491, 494
-----------максимальная 545
•—-----------в смесях диоксан—вода
547
—-------7----------метиловый спирт—
вода 548
-----------определение по методу экстра-
поляции 483
------------------- электропроводности
205, 478
-----------по данным для электродвижу-
щих сил 478
----- коэффициенты высаливания 529
— — различные солевые эффекты 378
-----средний ионный радиус, определе-
ние из температурной зависимости 492
-----электропроводность, влияние силы
поля 211
Уксусноэтиловый эфир, коэффициенты вы-
саливания 528
-----различные солевые эффекты 376—377
Упругость пара, вычисление активности
растворителя 274—275
--------коэффициентов активности 353—
355, 390, 401—403, 411, 524—526, 529
--------осмотического коэффициента
274—276
-----значение коэффициента активности
хлористого калия 349
-----метод изопиестического сравнения
275, 353—355, 400 '
-----определение активности воды в вод-
ных растворах серной кислоты 408—410
Уравнение Батлера 65
— Бьеррума 574
— Бант-Гоффа 32
— Гиббса 32
-----Гиббса—Гельмгольца 294
-----Дюгема 84, 280 , 573
— Дебая и Гюккеля, члены высших поряд-
ков, таблица 124
— Дебая и Мак-Олэя 64—65
Уравнение Дебая и Мак-Олэя для соле-
вых эффектов 375—376
— для зависимости электропроводности
от температуры 196
----коэффициента активности 52
----— диффузии 93
----осмотического коэффициента 52—53
----потенциала атмосферы вокруг иона
99
— Лоренца 156
— Менделеева для плотности солевых рас-
творов 248
— непрерывности 82
— Нернста предельное 561
— Онзелера 90
— Пуассона 39, 43
— Сеченова 375
— 4 Тильтона и Тейлора 555
— Тэйта 257—258, 557
----Гибсона 259
— электропроводности 157
Уравнения движения 38
— для чисел переноса 163—166
— Пуассона для бинарных систем 102—103
Фенол, коэффициенты высаливания 528
— различные солевые эффекты 377
Фенолсульфоновая кислота, константа дис-
социации по второй ступени 579
------------------- максимальная 582
Фенолтиомочевина, коэффициенты высали-
вания 528
— различные солевые эффекты 377
Физико-химический анализ 250
Физические константы, значения 117—
137
----основные, таблицы 117, 555
Формула Дебая и Гюккеля 402
— разведения Оствадьда 156
Фосфорная кислота, изменение термодина-
мических функций при диссоциации по
второй ступени 476
---------------------- первой ступени
476
----константа диссоциации 577
----------по второй ступени 546, 579
------------------- максимальная 582
------------- первой ступени 541
----------;--------определение мето-
дом электропроводности 205
Фотоэлектрический метод определения кон-
стант диссоциации 467—469
Фториды четырехзамещенных алкиламмо-
ниевых оснований, осмотические коэф-
фициенты 374
Функции абсолютной температуры, таблица
118
— Гиббса 20
— диэлектрической постоянной воды 119
— для вычисления поверхностного натя-
жения электролитов 129
— теории Бьеррума 126
— перехода от моляльиых концентраций
к молярным, таблица 517
— теории ассоциации ионов Бьеррума
125—126
----диффузии 132
----образования ионных тройников
126—127
620
ПРЕДМЕТНЫЙ указатель
Функции термодинамические, электроста-
тических свойств ионных атмосфер,
вычисление 121
— уравнения для коэффициента активно-
сти, таблицы 278, 565
-----теории эффекта Вина 107—108,
135—136, 557
Функция активности 23
----- ионов воды, таблица 539—540
— гидролиза для иона ацетата, таблица 488
— коэффициента активности для растворов
солей 454
----------------константы уравнений,
таблица 456
:— распределения, определение 34—36
-----скорость изменения во времени 37, 42
— экстраполяционная для электродвижу-
щих сил 317, 395
Характеристические величины для эллип-
соидальной модели диполя, таблица 196
— коэффициенты 117—137
----- наклона 118
-----— значения 117—137
— параметры для галоидных солей щелоч-
ных металлов 438—439
-------соляной кислоты с хлористыми со-
лями 440
Характеристическое расстояние между ио-
• нами 440
Химический потенциал 19—25
-----вычисление электростатической со-
ставляющей 48
-----методы вычисления 47—48
------ процесс заражения 48
-----составляющая неэлектролита, опре-
деление 64—65
-----уравнение 20, 300
л-Хлорбензойная кислота, константа дис-
социации, определение методом элек-
тропроводности 205, 478
о-Хлорбензойная кислота, константа дис-
социации, определение методом электро-
проводности 205
п-Хлорбензойная кислота, костанта дис-
социации,’ определение методом элект-
ропроводности 205
Хлориды четырехзамещенных алкиламмо-
ниевых оснований, осмотические коэф-
фициенты 374
Хлорная кислота, коэффициент активности
средний 569
Хлоруксусная кислота, изменение термоди-
намических функций при диссоциации 476
-----констайта диссоциации 541
-----зависимость от температуры
474—475
----------максимальная 545
—---------определение методом электро-
проводности 205
• -по данным для электродвижу-
щих сил 478
•----коэффициенты высаливания 529
-----электропроводность, влияние силы
ноля 211
Цезий азотнокислый, коэффициент актив-
ности средний 373, 525, 526
•----относительная вязкость, предельный
коэффициент наклона 170
Цезий азотнокислый, поверхностное на-
Ц^тяжение разбавленных растворов 381
— бромистый, кажущийся молярный объем
-----коэффициент активности средний
363, 524
-----критическое состояние разрушения,
величина 246—248
-----среднее расстояние сближения ионов
360
-----эффективное давление 261
— гидрат окиси, коэффициент активности
-------------- средний 364
—---------------в растворе хлористого
цезия 425—426
— иодистый, кажущийся молярный объем
—----коэффициент активности средний
363, 524
-----критическое состояние разрушения,
величина 246—248
— •— среднее расстояние сближения ионов
360
— сернокислый, кажущееся относитель-
ное молярное теплосодержание 500
--коэффициент активности средний 570
-----относительное парциальное молярное
теплосодержание 503
— уксуснокислый, коэффициент активно-
сти средний 525
— хлористый, влияние на коэффициент
активности соляной кислоты и гидрата
окиси цезия 425—426
— •— кажущееся относительное молярное
теплосодержание 500
-----кажущийся молярный объем 242
----- коэффициент активности средний
363, 373, 524
-----критическое состояние разрушения,
величина 246—248
-----среднее расстояние сближения ионов
360
Церий хлористый, коэффициент активно-
сти 534
Циануксусная кислота, константа диссо-
циации, определение методом электро-
проводности 205
Цинк азотнокислый, коэффициент актив-
ности средний 570
— бромистый, коэффициент активности
средний 397, 531, 570
— иодистый, коэффициент активности
средний 397, 531—532, 570
-----относительная парциальная моляр-
ная теплоемкость 533
-----относительное парциальное моляр-
ное теплосодержание 533
-----электродвижущая сила, зависимость
от концентрации 393
— малоновокислый, константа диссоциа-
ции 149
— сернокислый, кажущееся относительное
молярное теплосодержание 501
— •— константа диссоциации 149
----------- и предельная электропровод-
ность 188
-----коэффициент активности средний
400—401
ПРЕДМЕТНЫЙ указатель
621
Цинк сернокислый, относительное пар-
циальное молярное теплосодержание
399, 503
----- эквивалентная электропроводность
498
— — электродвижущая сила, зависимость
от концентрации 399
— стандартный потенциал 531
— хлористый, коэффициент активности
средний 397, 531—532, 570
— — относительная парциальная моляр-
ная теплоемкость 533
-----относительное парциальное моляр-
ное теплосодержание 533
— — электродвижущая сила, зависимость
от концентрации 393
— хлорнокислый, коэффициент активности
средний 570
Частота переменного тока 97—99
— тока, влияние на эффект Вина 101—102
Числа переноса, значения, таблица 337
-----катиона серной кислоты в водных
растворах, таблица 535
— — — соляной кислоты в воде и смесям
диоксан—вода 333—337
--------— таблица 515—516
-------------предельные коэффициенты
наклона, таблица 516
-----катионов, 1,1-валентных электроли-
тов 161
--------электролитов несимметрического
типа 162
— — определение методом Гитторфа
158—160
—----------движущейся границы 158—160
-------- — уравновешенной границы 160
— — — — экстраполяции 165
-----предельное уравнение 160—163
— — предельные для электролитов, опре-
деленные методом движущейся грани-
цы, таблица 499
— — при умеренных разбавлениях, урав-
нения 163—166
— — проверка уравнения 164
-----хлористого калия и хлористого нат-
рия, таблица 163
— сольватации, вычисление 250
Число Авогадро 117, 555
— переноса 91
— значение при любой концентрации,
уравнение 340
— — катиона в водных растворах серной
кислоты 415
— —------------------зависимость от
концентрации 414
— — — соляной кислоты в смесях диок-
сан—вода 335, 336
— — определение по электродвижущим
силам элемента 297, 333—334
— — предельное значение 335
— Фарадея 117, 555
Щавелевая кислота, изменение термодина-
мических функций при диссоциации при
второй ступени 476
— — константа диссоциации максималь-
ная 545
— — — — по второй ступени 541
Щавелевая кислота, константа диссоциа-
ции максимальная первой ступени,
определение методом электропроводно-
сти 205
Щелочные металлы, гидраты окисей, ко-
эффициенты активности средние в рас-
творах солей 423—426
Эбулиоскопические постоянные раствори-
телей, таблица 274
Эквивалентная электропроводность в силь-
ном поле 137
-----водных растворов солей, влияние
валентности 143
— — общее уравнение 144
----- определение 138—140
-----сравнение теоретических и опытных
результатов 142
—• —• уравнение 186
---электролитов, таблица 498
Эквивалентные электропроводности для
хлористоводородных алкиламинов 587
Экранирование поля иона атмосферой 111
Экстраполяционная функция для электро-
движущих сил, таблица 317
Экстраполяционные функции для электро-
движущих сил 395
Экстраполяция зависимости константы дис-
социации от концентрации для слабых
электролитов 203—204
-----электропроводности сильных элек-
тролитов от концентрации 150—154
— определение кажущихся молярных
объемов 563—566
— — констант диссоциации 451—453
---— — амфолитов 470—472
— — — — многоосновных кислот 465—
467
— — слабых кислот с учетом влия-
ния среды 482—484
— — константы диссоциации из данных
измерений электропроводности 186—188
— коэффициента активности 351, 390—
391, 443—446
— — по данным для температур за-
мерзания 279—282
— ------------— электродвижущих
сил 292—294
— — относительного кажущегося моляр-
ного теплосодержания 222—228
— — стандартного молярного электродного
потенциала 291—292
— — электродного потенциала 313,
315—316, 318, 405—408, 424
— • — теплот разведения 219—220 , 563—
566
— — — нейтрализации для растворов с
постоянной концентрацией 232
------ электродвижущих сил 396
Электрическая работа обратимого процес-
са, определение 20
— — разряжения ионов 63
Электрод водородный 288
Электродвижущая сила гальванического
элемента 289
— —-------константы уравнения, табли-
ца 428
---зависимость от концентрации, экстра-
поляционные функции 395
622 ПРЕДМЕТНЫЙ указатель
Электродвижущая сила элемента, зави-
симость от ионной силы, экстраполя-
ция 451
--------стандартная, определение 313
Электродный потенциал 23
-----равновесный, зависимость от раство-
рителя 23
--------механизм возникновения 23
-----серебра, стандартный, уравнение
302
Электроды 287—291, 302—303, 313—315,
347—348, 466, 531, см. также Стандарт-
ные электродные потенциалы
— амальгамные 350, 388—390, 390—392,
393—397
-----содержащие амальгамы щелочнозе-
мельных металлов 388—390
-----------щелочных металлов 390—392
'— ----кадмиевые амальгамы 392—397
--------цинковые амальгамы 392—393,
395—397
— поляризация 139
— способы изображения 287—291
— сраинения 303
-----для определения pH 302
— стандартные 302
. — электрическая емкость 139
Электрокинетический потенциал 80
Электролиты, диффузия, теория Онзагера
93—97
— 1,1-валентные, функции для перехода
от моляльных концентраций к моляр-
ным, таблица 517
— сильные, см. Сильные электролиты
— слабые, см. Слабые электролиты
Электрон, заряд 117, 555
Электропроводности предельные, ионные
в водных растворах, таблица 169
Электропроводность в переменном поле,
уравнение 100—101
— влияние сильного поля 208—214
-----сильных полей, функции, таблица
557
-----частоты, теория 132—135
--------тока 97—100
— водных растворов, значения постоянных
для предельного коэффициента наклона,
таблица 131
— зависимость от концентрации 153—154
--------частоты, теория 97—109
-г закон квадратного корня Кольрауша
155
— и числа переноса 557—560
----------- электролитов, таблица 558
— измерение с помощью мостика 138, 139
— иона хлора растворов хлористого нат-
рия и лития в смесях метиловый спирт—
вода, таблица 167
— молярная, влияние частоты, таблица 133
----- уравнение 100—101
— неводных растворов 167
— пикрата тетраэтиламмония, влияние ди-
электрической постоянной 144
— предельная ионная 166—168
--------влияние концентрации 166—168
— предельный закон 89—93
----- параметры, таблица 505
— разбавленных растворов, минимум
191—194 ' ‘
Электропроводность разбавленных раство-
ров, параметры уравнения, таблица 152
-------разность теоретических и опыт-
ных значений 151—152
-------уравнение, определение парамет-
ров 150—151
— растворов сернокислого магния, влия-
ние частоты 206—207
— связь с частотой тока 205—208
— стандартные растворы 138—141
— теория Онзагера 84—86
-----ионных сил 88—89
— удельная, см. Удельная электропровод-
ность
— уравнение 109
-----Онзагера 140—146
-----теории эффекта Вина, значения
функций, таблицы 135—136
— уравнения 155—158
— эквивалентная, см. Эквивалентная элек-
тропроводность
— электролитов, влияние сильных полей
135—137
-----при слабых электрических полях
89—93
-------низких частотах 89—93
Электростатическая составляющая вязко-
сти, уравнение 83
Электростатические силы 80
Электростатический потенциал, уравнение
Эпектррфоретический эффект 44, 85—88,
106—107
'------влияние силы ионного поля 106—
109
Электрохимия неводных растворов 167, 191
Элемент амальгамный 292
— без жидкостного соединения с небуфер-
ным раствором 479—484
— гальванический, обратимый 22
— Даниэля 301
— концентрационный без жидкостного со-
единения 295
--------------определение коэффициен-
тов активности 292—294
-----Нортропа—Мак-Бэна 173
-----с жидкостным соединением 296—298
--------------электродвижущая сила,
. уравнение 296
— стандартная электродвижущая сила,
определение 286
Элементы 22, 288—291, 292—294, 296—
298, 301—303, 307, 313, 315—319, 347,
350, 388, 390, 392, 394, 398, .403—404,
406, 408—409, 411—412, 415, 423—424,
427, 431—432, 440, 442, 450—452, 454,
463, 466, 470—472, 478, 479—481, 507,
531, см. также Стандартные потен-
циалы элементов
— без жидкостного соединения 291
— гальванические 288—290 , 301, 308—
309
— общие термодинамические свойства
285—287
— с жидкостным соединением, определе-
ние pH 301—309
— способы изображения 287—291
Энергии сольватации ионов 339
Энергия в стандартном состоянии 19
ПРЕДМЕТНЫЙ указатель
623
Энергия иона в растворе 339
— поверхностная 32
— полный дифференциал 19
— свободная, см. Свободная энергия
— сольватации ионов 23
— фазы 19
Энтропия 19, 20
— и теплоемкость иона в растворе 339
Этилен, различные солевые эффекты 376—
377
Этилен гликоль—вода, диэлектрические по-
стоянные смесей 120
Этиловый спирт—вода, диэлектрические
постоянные смесей 120
-----------константа диссоциации про-
пионовой кислоты 580—581
-----------коэффициент активности со-
ляной кислоты, таблица 511
---1-------стандартные потенциалы, та-
блица 507
Эффект Вина 45, 209—210, 212—213
— — влияние частоты тока 212
----:---------и концентрации растворен-
ного вещества 213
----для сильных электролитов 209—
210
--------слабых электролитов 210—214
----значения функций 101—108
— высаливания 435
— Грюнайзена 168
— солевой 65
— среды 204
----определение 204
— электрофоретический, см. Электрофоре-
тический эффект
Эффективное давление 259
----уравнение 261
— расстояние между ионами 110
Эффекты солевые, уравнения 68—69
СОДЕРЖАНИЕ
Предисловие ....................•....................(.................. 3
Указатель обозначений................................................... 5
Глава I. Общее термодинамическое введение................t............. 19
§ 1. Энергия, энтропия и химические потенциалы Гиббса ....... 19
§ 2. Теплосодержание, работоспособность (максимальная работа изотерми-
ческого процесса) и свободная энергия............................ 20
§ 3. Системы при постоянных составе и заряде.................• 21
§ 4. Системы при постоянных давлении, температуре и заряде. Парциаль-
ные молярные величины .......................................... 21
§ 5. Системы при постоянных температуре и давлении. Обратимый галь-
ванический элемент..................................... .... 22
§ 6. Функция активности. Активность электролита.................. 23
§ 7. Зависимость активности от температуры и давления ........... 25
§ 8. Зависимость активности от состава при постоянных температуре и дав-
лении. Определение коэффициентов активности ..................... 26
§ 9. Коэффициент активности и осмотический коэффициент растворителя. 29
§ 10. Константа равновесия химической реакции..................... 30
§ И. Поверхностное натяжение. Уравнение адсорбции Гиббса.......... 32
Глава II. Общие основы теории междуионного притяжения и свойства ионных
атмосфер.............................................................. 33
Ионная атмосфера.............................................. 34
а) Функции распределения.................................. 34
б) Скорость изменения функции распределения во времени ... 37
в) Уравнения движения..................................... 38
г) Основные уравнения для потенциалов иона и его атмосферы
в отсутствие внешних полей. Теория Дебая................. 38
д) Общие уравнения для ионных атмосфер в возмущенном
состоянии................................................. 42
е) Общий обзор поведения ионных атмосфер в возмущенных
состояниях................................................ 44
Глава III. Теория термодинамических свойств растворов электролитов ... 46
§ 1. Свойства 1/х ........................... 46
§ 2. Методы вычисления работоспособности и химического потенциала 47
§ 3. Процесс заряжения и вычисления электростатической составляющей
химического потенциала..................................... 48
§ 4. Предельный закон для зависимости коэффициента активности от кон-
центрации ...................................................... 48
§ 5. Влияние кажущихся диаметров ионов. Уравнения для коэффи-
циента активности и осмотического коэффициента................... 51
§ 6. Развитие теории Дебая и Гюккеля с учетом членов высших поряд-
ков (Гронвол, Ла-Мер и Сэндвед).................................. 53
§ 7А . Теория ассоциации ионов Бьеррума.......................... 54
§ 7Б . Теория образования ионных тройников и квадруполей по Фуоссу и
Краусу ........................................................ 58
§ 8. Предельные уравнения для относительного парциального моляр-
ного теплосодержания и относительной парциальной молярной
теплоемкости .................................................. "60
§ 9. Предельные уравнения для относительного парциального молярного
объема, расширяемости и сжимаемости электролитов................. 62
СОДЕРЖАНИЕ
625
§ 10. Увеличение или уменьшение растворимости нейтральных веществ
и электролитов при добавлении соли к их растворам....................... 63
§ 11. Теоретический предельный закон для поверхностного натяжения рас-
творов электролитов.................................................. 70
Глава IV. Теория необратимых процессов в растворах электролитов.............. 74
§ 1. Время запаздывания.......................................... 74
§ 2. Вязкость. Теория Фалькенгагена............................... 77
§ 3. Электропроводность. Теория Онзагера......................... 84^
А. Общие соображения и предварительный обзор теории............... 84
Б. Очерк общей теории электропроводности согласно Онзагеру ... 86
§ 4. Диффузия электролитов. Теория Онзагера ... ............... 93
§ 5. Зависимость электропроводности и диэлектрической постоянной силь-
ных электролитов от частоты. Теория Дебая и - Фалькенгагена . . 97
§ 6. Теория влияния сильных полей на свойства сильных электролитов.
Эффект Вина........................................................... 101
§ 7. Влияние сильных полей на диссоциацию слабых электролитов. Теория
Онзагера.............................................................. 109
Глава V. Значения физических констант, характеристических коэффициентов
наклона и математических функций........................................... 117
§ 1. Основные физические константы..................................117
§ 2. Уравнения, выведенные в гл. III, для вычисления термодинами-
ческих функций из электростатических свойств ионных атмосфер , 121
§ 3. Уравнения, выведенные в гл. IV, для необратимых процессов ... 129
Глава VI. Экспериментальное исследование необратимых процессов в растворах
сильных электролитов. Электропроводность, числа переноса, вяз-
кость и диффузия........................................................... 138
§ 1. Эквивалентная электропроводность А. Стандартные растворы . . . 138
§ 2. Уравнение электропроводности, выведенное Онзагером. Сравнение
с опытом............................................................... 140 V
§ 3. Расширение области применимости уравнения Онзагера. Вычи-
сление Д°.......................................................... . . 146
§ 4. Другие уравнения электропроводности. Критический обзор......... 155\4
§ 5. Определение чисел переноса по методу движущейся границы. Основ-
ные уравнения ... •..................... . . . •.................... 158
§ 6. Предельное уравнение Онзагера для чисел переноса. Сравнение с экс-
периментальными данными............................................... 160
§ 7. Уравнения для чисел переноса при умеренных разбавлениях. Вы-
числение т”.......................................................... 163
§ 8. Предельная ионная электропроводность. Закон Кольрауша............ 166 V
§ 9. Вязкость растворов сильных электролитов и ее зависимость от кон-
центрации ............................................................ 168
§ 10. Диффузия электролитов..............................•.......... 173
Глава VII. Кулоновские силы и ассоциация ионов. Слабые электролиты. Влия-
ние частоты и силы поля..................................................... 182
§ 1. Закон Стокса и правило Вальдена................................. 182
§ 2- Образование ионных пар. Вычисление констант диссоциации . . 185
§ 3. Проверка теории междуионного притяжения Бьеррума. Влияние ди-
электрической постоянной на образование ионных пар.................... 189
§ 4. Ассоциация с образованием сложных аггрегатов. Ионные тройники,
квадруполи и минимумы электропроводности.............................. 191
§ 5. Влияние размеров и строения ионов на электропроводность и ассо-
циацию .................................................. ......... 198
§ 6. Диссоциация слабых электролитов............................. • 201
§ 7. Экспериментальное исследование влияния частоты переменного тока
на электропроводность и диэлектрическую постоянную...................- 205
§ 8. Влияние сильного электрического поля на электропроводность раство-
ров электролитов...................................................... 208
ГлаваУШ. Термохимические величины, парциальные молярные объемы и коэф-
фициенты расширяемости и сжимаемости............................. 217
§ 1. -Введение.................................................... 217
§ 2. Относительное кажущееся молярное теплосодержание, теплота разве-
дения и относительное парциальное молярное теплосодержание ... 217
40 г. Харнед, Б. Оуэн
626
СОДЕРЖАНИЕ
§ 3. Теплота нейтрализации и теплота диссоциации.................... 230
§ 4. Парциальная молярная теплоемкость при постоянном давлении . . . 232
§ 5. Парциальные молярные объемы, кажущиеся молярные объемы и плот-
ности растворов Электролитов ...................................... 240
§ 6. Парциальная молярная расширяемость, кажущаяся молярная рас-
ширяемость и коэффициент термического расширения растворов элек-
тролитов .................................................... .... 251
§ 7. Парциальная молярная сжимаемость, кажущаяся молярная сжи-
маемость и коэффициент сжимаемости растворов электролитов. . . 254
Глава IX. Вычисление коэффициентов активности и осмотических коэффициен-
тов из данных по температурам замерзания, температурам кипения
и давлению пара .......................................................... 269
§ 1. Введение....................................................... 269
§ 2. Общие уравнения для вычисления активности растворителя и осмоти-
ческого коэффициента из данных по понижению температуры замер-
зания ............................................................ 270
§ 3. Общие уравнения для вычисления активности растворителя и.осмоти-
ческого коэффициента из данных по повышению температуры кипения 273
§ 4. Общие уравнения для вычисления активности растворителя и осмо-
тического коэффициента из данных по измерению давления пара
й из изопиестических данных........................................ 274
§ 5. Вычисление коэффициента активности растворенного вещества с по-
мощью осмотического коэффициента или активности растворителя . . 276
Глава X. Термодинамика гальванических элементов........................ • 285
§ 1. Общие термодинамические свойства элементов. Определение стандарт-
ного электродного потенциала........................................ 285
§ 2. Условные способы изображения элементов и электродов; знаки электро-
движущих сил........................................... ' 287
§ 3. Экстраполяция данных по электродвижущим силам и вычисление
стандартного молярного электродного Потенциала...................... 291
§ 4. Определение коэффициентов активности путем экстраполяции данных
по электродвижущим, силам концентрационных элемейтов без
жидкостного соединения.............................................. 292
§ 5. Вычисление относительного парциального молярного теплосодержа-
ния и относительной парциальной молярной теплоемкости электро-
литов из данных по электродвижущим силам............................ 294
§ 6. Концентрационный элемент с’ жидкостным соединением ....... 296
Гипотетический потенциал жидкостного соединения (диффузионный
потенциал)..................................................... 298
Представление о разности потенциалов на границе раздела фаз . . . 299
§ 7. Использование элементов с жидкостными соединениями. Определе-
ние pH. Устранение диффузионных потенциалов......................... 302
Глава XI. Соляная кислота.................................................. ЗЦ
§ 1. Электропроводность соляной кислоты в смесях диоксан—вода . . 311
§ 2. Стандартный потенциал элемента Н2 | HCI(m) I AgCl-Ag в случае
водных растворов.........................'. . . .................... 313
§ 3. Стандартный потенциал элемента Н2 ) НС1 (т), S (X), Н2О (У) | AgCl-Ag
для смесей вода—органический растворитель........................... 315
§ 4. Коэффициент активности соляной кислоты в водных растворах ... 322
§ 5. Вычисление коэффициента активности соляной кислоты в водном
растворе с помощью обобщенной теории Дебая и Гюккеля................ 323
§ 6. Коэффициент активности соляной кислоты в средах, содержащих
органические растворители........................................... 327
§ 7. Относительное парциальное молярное теплосодержание соляной кис-
лоты. Численные методы определения L2 из данных по электро-
движущим силам................................................. 328
§ 8. Относительная парциальная молярная теплоемкость соляной кис-
лоты ...........................'.................................. 332
§ 9. Числа переноса соляной кислоты в воде, и в смесях диоксан—вода
при температурах 0—50°...................’.......................... 333
Глава XII. Водные растворы сильных .1,1-злектролитов...................... 339
§ 1. Термодинамические свойства растворов хлористого натрия, определяе-
мые путем измерения электродвижущих сил, а также из калори-
метрических данных ................................................. 340
СОДЕРЖАНИЕ
627
§ 2. Термодинамические свойства водных растворов бромистоводородной
1 кислоты, хлористого Калия, бромистого натрия и гидратов окисей
натрия и калия .............................................. '347
§ 3. Вычисление коэффициентов активности 1,1-электролИтов из данных ,
по изопиестическим измерениям давления пара........................ 353
§ 4. Вычисление коэффициента активности при различных температурах и
давлениях из данных, относящихся к 25° и атмосферному давлению 355
§ 5. Общий обзор коэффициентов активности 1,1-электролитов в связи
с теорией Дебая и Гюккеля.......................................... 358 -
§ 6. Постулаты Бренстеда о специфическом взаимодействии ионов и вы-
числение осмотических коэффициентов и коэффициентов активности
для низких концентраций по, методу Гуггенгейма..................... 365
§ 7, Теория концентрированных растворов галогенидов щелочных метал-
лов по Скэтчарду.................................................. 367
§ 8. Теория концентрированных растворов электролитов, учитывающая
поправку Ван-дер-Ваальса на собственный объем растворенных частиц 369
§ 9. Связь термодинамических свойств ионов с их размерами и структурой 372
§ 10. Коэффициенты активности нейтральных молекул в водных растворах
солей.............................................................. 374
§ 11. Поверхностное натяжение разбавленных растворов электролитов . . 380
§ 12. Критический обзор...................................................... 382
Глава XIII. Водные растворы многовалентных электролитов....................... 387
§ 1. Термодинамика растворов галогенидов магния, кальция, стронция
и бария............................................................ 388
§ 2. Термодинамика водных растворов сульфатов лития, натрия и калия 390
§ 3. Водные растворы галогенидов цинка, кадмия и свинца..................... 392
§ 4. Общие соображения относительно водных растворов 1,2-и 2,1-элект-
ролитов .......................................................... 397-
§ 5. Термодинамика водных растворов сернокислого цинка...................... 398
§ 6. Коэффициенты активности сульфатов двухвалентных металлов в вод-
ных растворах ..................................................... 400
§ 7. 1,3-, 3,1-, 3,2-, 1,4-электролиты................................... 401
§ 8. Определение константы диссоциации HSO- из данных об электро-
движущих силах..................................................... 403
§ 9. Стандартный потенциал элемента Н2 | H2SO4 (m) | PbSO4-PbO2-Pt . . . 404
§ 10. Определение активности воды в водных растворах серной кислоты
путем измерений электродвижущих сил и упругости пара............... 408
§ 11. Коэффициент активности серной кислоты................................. 410
§ 12. Стандартный потенциал элемента Н21 H2SO4 (т) | Hg2SO4-Hg.. 411
§ 13. Относительные парциальные молярные теплосодержание и тепло-
емкость серной кислоты в водном растворе............................ 412
§ 14. Число переноса катиона в водных растворах серной кислоты . . . 415
Глава XIV. Смеси сильных электролитов............................................. 418
§ 1. Измерения растворимости и теория междуионного притяжения . . . 419
§ 2. Определение коэффициентов активности соляной, бромистоводородной
и серной кислот и гидроокисей щелочных металлов в растворах
. солей путем измерений электродвижущих сил................................. 423
§ 3. Относительное парциальное молярное теплосодержание 0,01 М со-
ляной и бромистоводородной кислот в растворах галогенидов .... 426
§ 4. Изменение коэффициента активности одного электролита в присут-
ствии другого при постоянной общей ионной силе...................... 427
§ 5. Термодинамическая теория смесей при постоянной общей моляль-
ности ......................................................... • . 432
§ 6. Общий обзор экспериментальных исследований смесей 1,1-галбгенидов 436
§ 7. Дальнейший анализ теории специфического взаимодействия ионов . . 441
§ 8. Смеси оснований с хлоридами ,.................................; 442
§ 9. Экстраполяция коэффициентов активности до значений, соответст-
вующих насыщенным растворам. Вычисление растворимости хорошо
растворимых солей в растворах электролитов.......................... 443
Глава XV. Диссоциация и термодинамические свойства слабых электролитов 449
§ 1. Константа диссоциации воды........................................... 450
§ 2. Функция коэффициента активности 7н7он/аНаО и ионное произве-
дение в случае водных растворов солей ................. 454
§ 3. Влияние температуры и давления на диссоциацию воды. Термохи-
мические величины................................................ 457
628
СОДЕРЖАНИЕ
§ 4. Определение констант диссоциации слабых кислот и оснований . . . 463
§ 5. Определение констант диссоциации амфолитов............... 469
§ 6. Вычисление констант диссоциации и термодинамических функций
, ДЯ°, AS? и ДС“ .......................................... 472
гг
§ 7. Влияние среды. Элемент с небуферным раствором без жидкостного
соединения................-................................ • 479
§ 8. Коэффициенты активности слабых кислот в растворах солей .... 484
§ 9. Гидролиз анионов органических кислот в растворах солей.• 488
§ 10. Теоретические соображения................................ 489
§ 11. Дополнительные наблюдения................................ 494
П р и ложениеА.................................................... 498
Приложение Б........................................................ 554
§ 1. Диэлектрическая постоянная воды. Перерасчет теоретических предель-
ных коэффициентов наклона и функций, в которые входит диэлектри-
ческая постоянная воды. Исправление табл. 1, 7, 11, 13. Таблицы
теоретических предельных коэффициентов наклона для парциальных
молярных объемов, расширяемостей и сжимаемостей. Функция g (х) 554
§ 2. Электропроводность и числа переноса. Дополнения к табл.26, 27, 29 557*
§ 3. Коэффициенты диффузии хлористого калия при концентрациях
0—0,5 М и хлористого кальция при концентрациях 0,002—0,005 М
в водных растворах........................................ 560
§ 4. Экстраполяция кажущихся молярных объемов и теплот разведения.
Дополнение к табл. 61......................................... 563
§ 5. Коэффициенты активности. Новые и исправленные данные для 1,1-,
2,1-электролитов.............................................. 567
§ 6. Обзор новых работ по теории электролитов.................. J68 ь
§ 7. Гидратация ионов, коэффициенты активности и осмотические коэф-
фициенты .................................................... 572
§ 8. Данные о диссоциации сильных электролитов, полученные путем
изучения рамановских спектров............................ • 576
§ 9. Диссоциация некоторых умеренно сильных электролитов....... 577
§ 10. Константы диссоциации и диссоциация слабых кислот в растворах
солей......................................................... 578
§ И. Переход от простых ионов к сложным аггрегатам и полимерным
электролитам.................................................. 586
Именной указатель................................................... 591
Предметный указатель................................................ 599
Редактор Я. Б. Арон
Художник М. Г. Ровенский Технический редактор А. Н. Никифорова
Сдано в производство 17/1 1952 г. Подписано к печати 2/VI 1952 г. А 04610. Бумага 70х1081/1в=
= 19,6 бум. л. 53,8 печ. л. Уч.-издат. л. 56,9. Изд. № 3/561 Цена 41 р. 85 к.
(по прейскуранту 1952 г.) Зак. 23
16-я типография Глдвполиграфиздата ppp Совете Министров СССР, Москва, Трехпрудный пер., 9.
21
ОПЕЧАТКИ
Стра- ница Строка Напечатано Следует читать
79 уравне- „ dvx r.0 dvx
ние (30) lo Qy
132 18 св. и (53) гл. VI и (58) гл. VI
204 18 св. а = 4 a = 4
222 25 св. а 0 a
233 уравне- ние (40) J = n±J। -р ^2*^ J == f^J! 4“ n%J2 ’ : n2?J
258 табл. 53, С c
графа вто- «0 v0
рая
371 уравне- ние (46) . dF* dV = 1П/{=^^ =
г i /'iViV2 -Шv+ [W-+
64 < И ) J oW0 ’ 10/IViY 1 dV + 64 < V ) v J C'NO-
441 20 сн. [CO (NO2) (CNS) • [Co (NO2) (CNS) •
461 14 сн. to Лц+—Л-ОН- Л'11+ + A OH-
461 уравне- ние (34) ^(P) = F!bW-l) ^{P)=7°--^(P-1)
461 9 сн. a V° и Ko a Fj и ~K?2
461 4 сн. ±K° ±K°-
461 3 сн. K° K°2
514 табл. 132, графа пятая J2 (25°) J2 (25°)
Зак. 23.