/





























































































































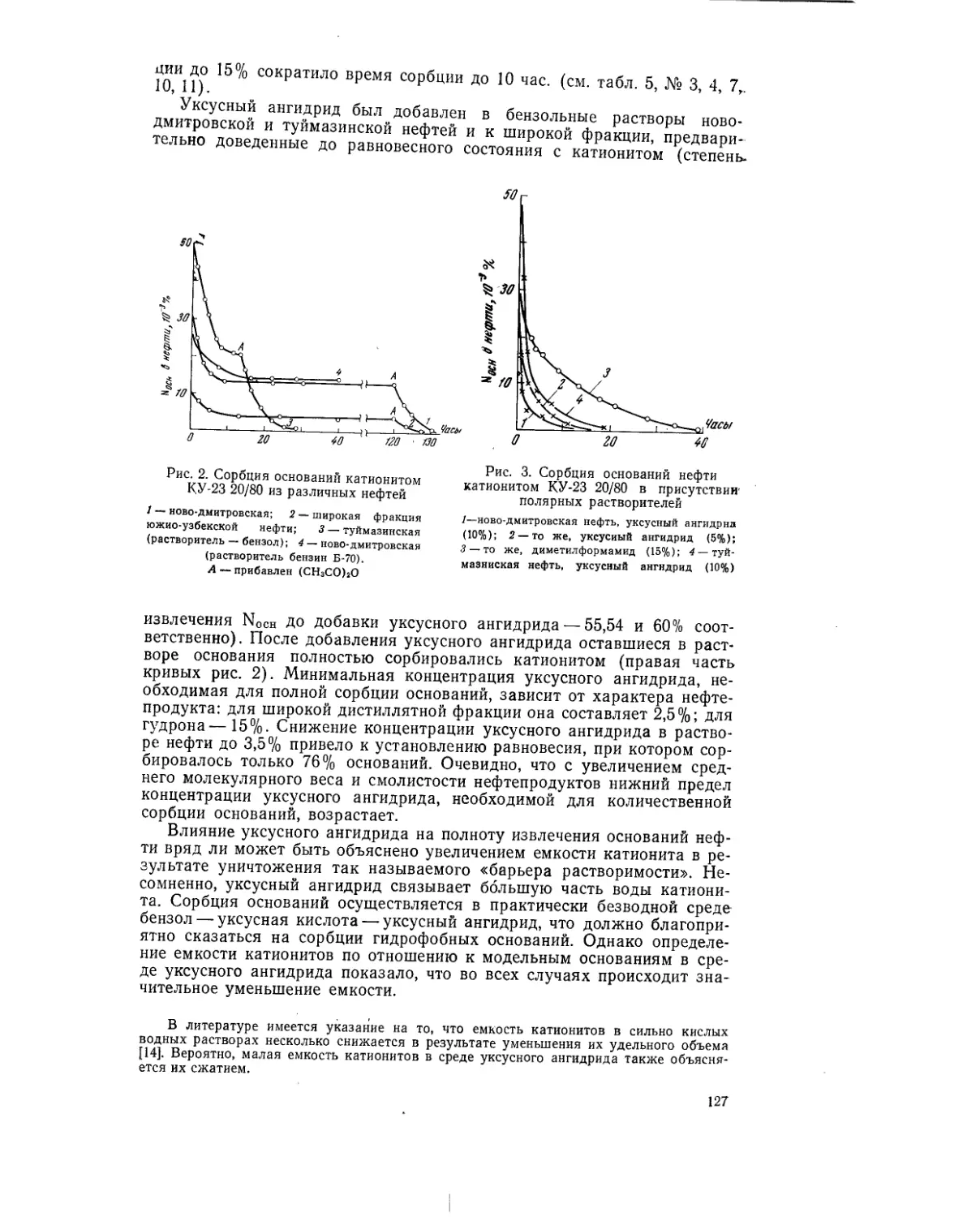
























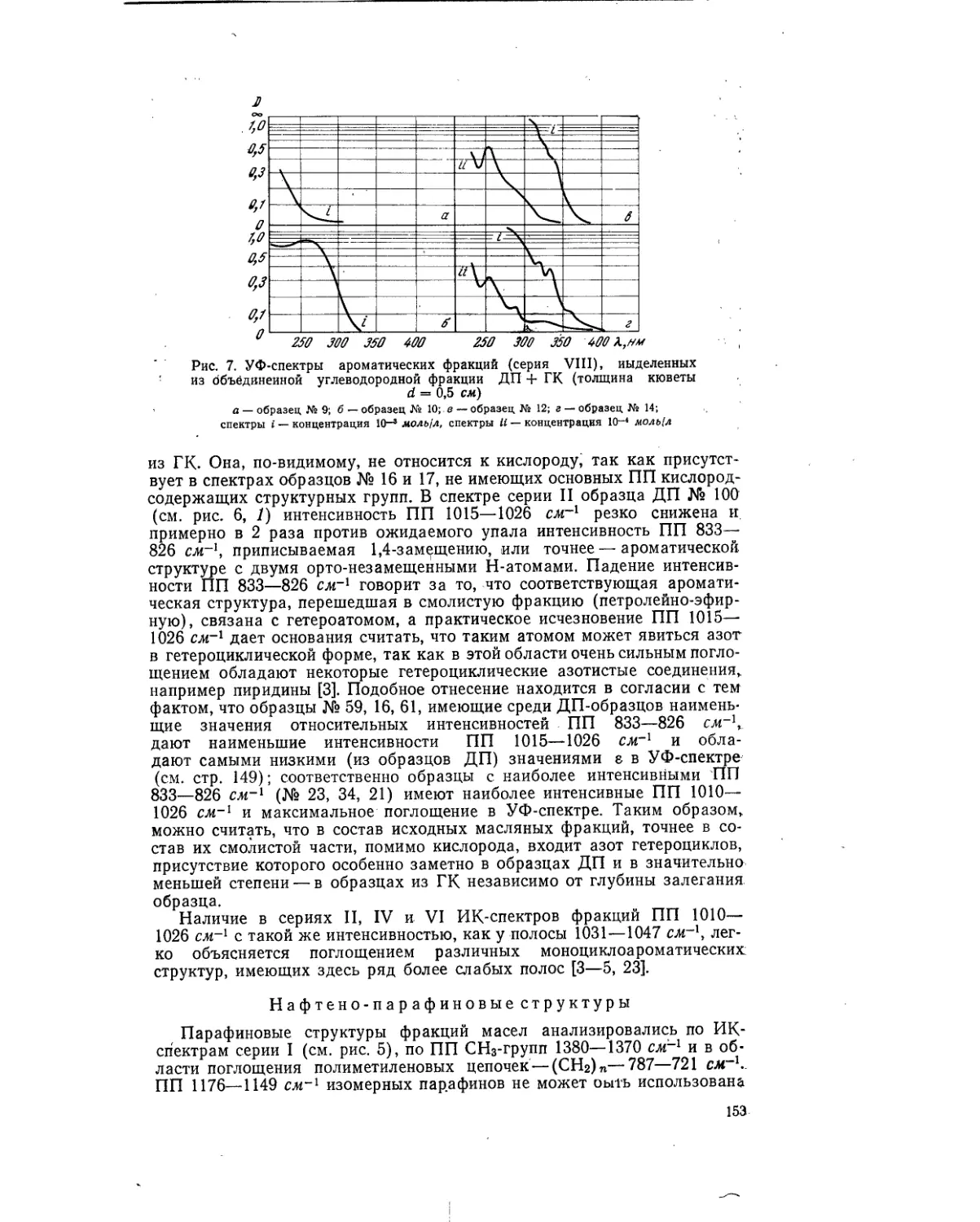






























































Текст
АКАДЕМИЯ НАУК СССР
ИНСТИТУТ НЕФТЕХИМИЧЕСКОГО СИНТЕЗА им. А. В. ТОПЧИЕВА
МЕТОДЫ АНАЛИЗА
ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ НЕФТИ,
ИХ СМЕСЕЙ
И ПРОИЗВОДНЫХ
Сборник 2
ЭЛЕМЕНТНЫЙ И ГРУППОВОЙ АНАЛИЗ;
ХИМИЧЕСКИЕ, ХРОМАТОГРАФИЧЕСКИЕ
И СПЕКТРАЛЬНЫЕ МЕТОДЫ
ИЗДАТЕЛЬСТВО «НАУКА»
Москва- -1969
УДК 547.912 : 543.8
В сборнике дано подробное описание оригинальных и
усовершенствованных аналитических методов, подвергнутых
тщательной экспериментальной проверке: метод анализа ин-
дивидуального состава бензинов путем газо-жидкостной ка-
пиллярной хроматографии, компонентный микроанализ неф-
тей и битумов, групповой микрохроматографический, анализ
средних и высших фракций нефти. Описываются методы груп-
пового выделения сульфидов в виде сульфоксидов из фрак-
ций нефти, разделение и характеристика смесей сульфидов ц
их производных аналитической и препаративной тонкослой-
ной хроматографией в сочетании с газо-жидкостной хрома-
тографией и анализом стереомоделей изомеров. Разработана
аппаратура и метод полуавтоматического экспресс-анализд
иа серу и галоген. Приводится методика определения азота,
углерода и водорода с газохроматографическим окончанием
анализа, а также метод количественного извлечения азотис-
тых оснований из нефти и их получение в виде концентра-
тов. Сборник содержит данные по применению спектроскопии
(ИК-, К.РС- и УФ-) к исследованию структурно-группового со-
става масел и к изучению насыщенных, непредельных и арома-
тических сульфидов и их смесей.
Рекомендуемые методики несомненно будут полезны широ-
кому кругу работников научно-исследовательских институтов,
контрольных и исследовательских лабораторий промышленности
органического синтеза и нефтехимической промышленности и
разнообразных отраслей применения синтетических и природ-
ных органических веществ.
Сборник может быть использован в качестве ценного ме-.
тодического пособия преподавателями и студентами высше^
школы и техникумов соответствующего профиля.
Ответственный редактор
доктор химических наук Г. Д. ГАЛЬПЕРН
2-5-3
343-69(1)
ОТ РЕДАКТОРА
За девять лет, прошедших со времени выпуска в свет первого сбор-
ника настоящей серии, существенно выросла техническая оснащенность
отечественных лабораторий исследовательских и производственных
предприятий и учреждений, занимающихся изучением состава, свойств
и реакций смесей природных органических соединений (в частности,
нефтей) и их производных, а также изысканием путей их рационального
использования в народном хозяйстве.
Максимальное значение в этой связи приобрела разработка совре-
менных методов разделения многокомпонентных смесей как исходных
веществ, так и продуктов их превращения, в том числе множества
смесей, получаемых в результате нефтехимического синтеза и развития
многочисленных направлений синтетической органической химии и
смежных с ней областей.
Настоящий сборник посвящен преимущественно последним достиже-
ниям в области химических и физико-химических методов разделения
сложных смесей органических соединений и выделению отдельных
компонентов и групп компонентов из таких смесей.
Успешное развитие работ, посвященных разделению многокомпо-
нентных смесей, теснейшим образом связано с успехами в области ана-
литического контроля за процессами разделения. Последнему также
уделено необходимое внимание в настоящем сборнике.
Особое внимание обращено на хроматографические методы, отлича-
ющиеся высокой эффективностью и позволяющие работать со смесями
разного качественного состава в широком диапазоне масс, от микро-
количеств до препаративных. Разнообразие современных методов хрома-
тографии позволяет успешно использовать их как в многокаскадных
схемах разделения и препаративного выделения веществ, так и для
аналитического контроля.
Необходимо, однако, подчеркнуть, что совершенствование хромато-
графических способов разделения не только не отменяет, но, наоборот,
требует параллельного развития химических методов, используемых
либо в одной из стадий многокаскадных схем ‘разделения смесей, либо
для индикации хода разделения, либо для синтеза эталонов, необходи-
мых при калибровке хроматограмм' так же как и в спектральных
методах анализа.
Сочетанию спектральных методов анализа с методами разделения
также уделено внимание в настоящем сборнике.
Современное развитие капиллярной газо-жидкостной хроматографии
с пламенно-ионизационным детектированием позволяет нам рекомен-
довать методику экспресс-определения индивидуального состава таких
сложных углеводородных смесей, как бензины прямой гонки. Наиболее
трудная задача индикации множества пиков на получаемых хромато-
3
граммах успешно решена в результате применейия химического способа
«равновесной изомеризации» простейших углеводородов в эталонные
смеси определенного, строго постоянного состава.
Если анализ бензинов решается практически по однокаскадной схеме
(мы не учитываем здесь стадию предварительной, сравнительно грубой
ректификации), то для разделения высших фракций нефти необходимы
схемы многокаскадного разделения.
Для предварительного разделения тяжелых частей нефти и битумов
вместо применяемых до недавнего времени громоздких вариантов ком-
понентного анализа по Маркусону и Васильеву предложена изящная
методика микроанализа.
Приводится методика элюентной колоночной микрохроматографии
(выполняемой в режиме линейной хроматографии) фракций масел, га-
зойлей, средних топливных фракций и других смесей углеводородов, вы-
кипающих от 150° С и выше, которая в сочетании с систематическим ана-
лизом элюируемых фракций микрорефрактометрическим и спектральным
способами (в инфракрасной и ультрафиолетовой областях) отличается
высокой информативностью в отношении группового состава исследуе-
мых смесей.
Из работ в области гетероатомных соединений нефти в настоящем
сборнике детально рассмотрены методы препаративного выделения
сульфидов из средних фракций нефти путем химической модификации
их в сульфоксиды и экстрактивного или хроматографического извлече-
ния последних из оксидатов. Эти процессы контролируются методами
потенциометрии и тонкослойной хроматографии.
Детально рассмотрено сочетание аналитической и препаративной
тонкослойной хроматографии сульфидов и их производных с газо-
жидкостной хроматографией. Особое внимание при этом было уделено
разделению смесей структурных и стерических изомеров и контролю
таких химических реакций, как дегидратация, гидрирование, превраще-
ние сульфоксидов в непредельные сульфиды.
Систематические исследования в области синтеза и разделения
смесей органических соединений серы и их производных потребовали
дальнейшего усовершенствования пиролитического лампового метода
элементного анализа веществ на содержание серы и галогенов, реко-
мендованного нами в сборнике 1. Детальное описание вновь созданной
полуавтоматической установки позволяет химикам-аналитикам и синте-
тикам широко использовать рекомендуемую методику экспресс-анализа
для контроля состава применяемых и получаемых веществ на своем
рабочем месте без помощи специальной аналитической лаборатории.
Среди органических соединений серы, особенно широко представлен-
ных в средних фракциях нефти, важнейшими являются насыщенные
сульфиды разного строения с примесью разнообразных арилсульфидов.
От насыщенных сульфидов возможен переход к гораздо более реакцион-
носпособным непредельным сульфидам. Для структурной характеристи-
ки перечисленных групп сульфидов существенное значение имеют
спектральные методы анализа. Им посвящена глава по исследованию
молекулярных и электронных спектров большого числа индивидуальных
сульфидов, моделирующих компоненты нефти и их производные.
Второй группой гетероатомных соединений нефти, рассматриваемых
в настоящем сборнике, являются азотистые основания. Малая концен-
трация органических соединений азота в нефтях прежде всего требует
дальнейшего усовершенствования методов элементного анализа. Необ-
ходимое повышение точности и чувствительности определения N, С и Н
достигнуто сочетанием пиролитического метода разложения анализиру-
емых веществ с газохроматографическим окончанием анализа, позволя-
ющим работать с малыми количествами вещества. Описание рекоменду-
4
емой методики ацидометрического функционального анализа азотистых
соединений нефти дано нами в предыдущем сборнике.
Наиболее трудной и весьма актуальной задачей, стоящей перед хи-
мией нефти, является количественное извлечение суммы азотистых ос-
нований из сырых нефтей и нефтяных остатков. Она была впервые ус-
пешно решена нами с помощью ионообменных смол для сорбции и до-
полнительных химико-экстракционных приемов для десорбции оснований
нефти, поглощенных специальными крупнопористыми катионитами. Опи-
сание рекомендуемой методики, дающей азотистые концентраты, сто-
кратно обогащенные по N, и примеры, иллюстрирующие ее применение,
будут способствовать накоплению сведений о природе этой группы весь-
ма малоисследованных компонентов нефти.
В сборнике дано подробное описание сравнительно простой переделки
стандартного рефрактометра, которая превращает его в очень удобный
прибор для микроанализа рефрактометрическими и дисперсиометриче-
скими методами, в частности теми, описание которых приведено в сбор-
нике.
Методики, рассмотренные в настоящем сборнике, либо непосред-
ственно, либо с небольшими модификациями могут быть успешно ис-
пользованы для решения широкого круга химико-аналитических задач,
возникающих при изучении разнообразных вопросов синтеза, выделения,
разделения и характеристики органических веществ, получаемых разны-
ми путями, в особенности на базе нефти и нефтепродуктов.
В заключение просим наших читателей присылать свои замечания
по опубликованным методикам с целью их дальнейшего усовершенство-
вания, а также пожелания относительно содержания последующих вы-
пусков сборника в Институт нефтехимического синтеза им. А. В. Топчи-
ева АН СССР (Москва В-71, Ленинский проспект, 29, Лаборатория
гетероатомных соединений нефти).
Г. Д. Гальперн
АНАЛИЗ ПРЯМОГОННЫХ БЕНЗИНОВ
МЕТОДОМ ГАЗО-ЖИДКОСТНОЙ ХРОМАТОГРАФИИ
С ПРИМЕНЕНИЕМ КАПИЛЛЯРНЫХ КОЛОНОК
Э. К. Брянская, 3. К. Оленина, Ал. А. Петров
Предлагаемая методика анализа позволяет определить индивидуаль-
ный состав бензиновых фракций нефтей и конденсатов, выкипающих до
150° С. Эти фракции представляют собой сложные смеси углеводоро-
дов различного строения. До 125° С выкипает 70 компонентов, в интер-
вале 125—150° С выкипает 130 компонентов. Анализ исследуемых
фракций проводят на хроматографе с ионизационным детектором и
капиллярной колонкой (неподвижная фаза—сквалан) с использованием
эталонных углеводородов.
Синтез индивидуальных эталонных углеводородов доступен не каж-
дой лаборатории, поэтому в основу методики положено применение
«вторичных эталонов» — калибровочных смесей и их хроматограмм [1].
Состав смесей был первоначально расшифрован с помощью индивиду-
альных углеводородов, синтезированных в лаборатории геохимии нефти
ИГиРГИ.
При идентификации пиков на хроматограмме следует руководство-
ваться порядком выхода углеводородов, который неизменен для данной
неподвижной фазы и температуры термостатирования колонки.
Идентификация углеводородов по удерживаемым объемам в усло-
виях капиллярной хроматографии невозможна для рассматриваемых
сложных смесей, так как эти характеристики не сохраняют постоянства
даже при одинаковых условиях хроматографирования.
Для более полной расшифровки состава фракций рекомендуется
хроматографировать каждую фракцию при двух температурах. При
этом селективность неподвижной фазы и порядок выхода углеводородов
изменяются, что позволяет расшифровать состав углеводородных смесей,
выходящих при одной из температур в виде одного пика,
Таким путем состав фракции н. к.—125° С удается расшифровать
практически нацело. Из-за чрезвычайной сложности фракций 125—150° С
их качественный состав нельзя считать расшифрованным на 100%.
Описание подготовки к анализу фракций 125—150° С приведены ниже.
1. АНАЛИЗ БЕНЗИНОВОЙ ФРАКЦИИ
н.к.— 125° С
1. Подготовка к анализу фракций н.к. —125° С
На ректификационной колонке эффективностью 20—50 теоретических
тарелок из исследуемой нефти или конденсата (100—200 мл) отгоняют
фракции I — н. к.— 110°С и II — НО—125° С и определяют их выход на
сырье.
Во избежание потерь фракции хранят в холодильнике.
7
2. Приготовление калибровочной смеси алканов
Изомеризацией легкодоступных углеводородов (н-октан, изооктан,
я-нонан и др.) можно получить смесь, содержащую все изомеры исход-
ного углеводорода, а также все алканы состава С4—С7 и С9—Сю, так
как наряду с изомеризацией идет реакция деструктивного алкилирова-
ния.
Изомеризация протекает по ступенчатому механизму, поэтому
изомеризат нормального парафина обогащен малоразветвленными
изомерами, а в изомеризате изооктана преобладают ди- и тризамещен-
ные изомеры. ,
Для получения калибровочной смеси в склянку вводят 5—10 мл
углеводорода, добавляют кусочек свежего бромистого алюминия (10%
по весу) и оставляют на 20—24 часа при комнатной температуре. Для
изомеризации изооктана достаточно 6—8 час. Для прекращения реакции
следует слить верхний бесцветный слой изомеризата, нейтрализовать
его раствором КОН и отделить углеводородный слой. От более высоко-
кипящих углеводородов освобождаются перегонкой.
3. Хроматографирование фракции н.к. — 125е С
Хроматографирование бензиновой фракции проводят на капиллярной
колонке длиной 50—100 м и внутренним диаметром 0,2—0,3 мм. Для
нанесения неподвижной фазы приготовляют раствор сквалана в серном
эфире (1:4 по объему). 3 мл раствора пропускают через колонку под
давлением 1,5—2 атн, эффективность такой колонки составляет пример-
но 1000 теоретических тарелок на 1 погонный метр.
Для каждой фракции снимают хроматограммы при двух температу-
рах: для фракции н? к.— 110° С при 30 и 80° С (давление газа-носителя
на входе в колонку соответственно меняется от 1,2 до 0,6 ати)., для
фракции 110—125°С при 50 и 80°С (давление 1,2 и 0,8 ати).
Так как изменение температуры влияет на порядок выхода компо-
нентов, необходимо как можно точнее придерживаться указанных
температур.
4. Качественная расишфровка хроматограмм
Качественную расшифровку хроматограмм следует начинать с иден-
тификации пиков парафиновых углеводородов, для чего необходимо
снять хроматограммы калибровочных смесей в тех же условиях. В ос-
новном парафиновые углеводороды выходят в порядке возрастания
температур кипения. На рис. 1 и 2 приведены хроматограммы изомери-
затов н-октана и изооктана, а в табл. 1 указан порядок выхода угле-
водородов. Этими хроматограммами можно пользоваться как калибро-
вочными. В табл. 2 даны относительные времена удерживания алканов
состава С6—С8, которые можно использовать для расшифровки состава
калибровочных смесей.
Сравнивая хроматограммы бензиновых фракций и изомеризатов,
снятые в одинаковых условиях, идентифицируют пики парафиновых
углеводородов. Для упрощения расшифровки в пробу бензинов можно
добавить смесь реперов: н-гексан, н-гептан, н-октан. Возросшие на хро-
матограммах пики относятся к этим углеводородам. «Привязать» к
реперам разветвленные алканы уже не составляет труда.
Для идентификации циклопарафиновых и ароматических углеводо-
родов (бензол, толуол) во фракциях н. к.— 110°С и ПО—125°С необхо-
димо пользоваться хроматограммами, приведенными на рис. 3, 4, и
табл. 3, в которой дан порядок выхода углеводородов, найденных во
фракциях н. к.— 110°С и НО—125°С. Хроматограммы были получены
8
Таблица 1
Углеводороды *, найденные
в изомеризатах алканов,
н порядок их выхода
Углеводо- № . Углеводо-
п.п род п.п род
1 uao-Cj
2 h-Cj
3 мао-Сз
9
10
11
12
13
14
15
16
17
н-Се
22 ДМП
24 ДМП
223 ТМБ
33 ДМП
2 МГ
23 ДМП
3 МГ
3 ЭП
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
224 ТМП
н-С?
22 ДМГ
25 ДМГ
24 ДМГ
223 ТМП
33 ДМГ
234 ТМП
233 ТМП
23 ДМГ
2 МГп
4 МГп
34 ДМГ
3 МГп
3 ЭГ
33 h-Cs
Рис. 1. Хроматограмма изомеризата
я-октана. Сквалан, 50° С
* Расшифровку названий см.
в табл. 3.
№
Отнесение пиков см. в табл. I
добавлением в калибровочную смесь алканов индивидуальных нафте-
новых и ароматических углеводородов (пики показаны пунктиром).
С повышением температуры хроматографирования наблюдается
увеличение времени удерживания циклопарафиновых углеводородов
относительно сетки алканов. Это может служить дополнительным
качественным критерием принадлежности пика к тому или иному классу
углеводородов.
5. Расчет хроматограмм
Количественная расшифровка хроматограмм проводится по методу
внутренней нормализации с измерением высоты пиков h и расстояния
максимума пика от ввода пробы I.
Процентное содержание каждого компонента, считая на исследуемую
фракцию, определяется отношением /Хй/2/Х/г.
Ниже приводится примерная форма записи при расчете каждой
хроматограммы:
Ха пика Компоненты /, мм h, мм IX Л, мм2 Выход на фракцию % на нефть
Если при одной температуре пики на хроматограмме представляют,
собой дуплеты или триплеты, то при другой температуре углеводороды
выходят в виде простых пиков или в других комбинациях.
»
Таблица 2
Рис. 2. Хроматограмма изомеризата
изооктана. Сквалан, 50° С
Отнесение пиков см. в табя. 1
Относительное время удерживания
алканов Се—С8
(время удерживания н-Ce принято за 1;
неподвижная фаза — сквалан)
Углеводород • Т.кип., <>С *♦ Относительные вре- мена удерживания при температуре
30° С I 50° С | 80» С
22 ДМБ 49,741 0,47 0,53 0,55
23 ДМБ 57,988 0,65 0,75 0,76
2 МП 60,272 0,70 0,75 0,76
3 МП 63,282 0,82 0,86 0,89
н-Cf, 68,740 1,00 1,00 1,00
22 ДМП 79,198 1,25 1,25 1,27
24 ДМП 80,50 1,39 2,31 1,29
223 ТМБ 80,883 1,50 1,46 1,45
33 ДМП 86,064 1,80 1,7Т 1,66
2 МГ 90,052 2,09 1,90 1,85
23 ДМП 89,784 2,18 2,10 2,00
3 МГ 91,851 2,30 2,70 2,04
3 ЭП 93,475 2,57 2,31 2,24
224 ТМП 99,238 2,64 2,40 2,34
н-С? 98,428 3,18 2,67 2,50
2233-тетраМБ 106,30 — —
22 ДМГ 106,840 3,62 3,20 2,97
25 ДМГ 109,103 4,13 3,50 3,17
24 ДМГ 109,429 4,20 3,64 3,29
223 ТМП 109,841 4,43 3,82 3,52
33 ДМГ 111,968 4,45 4,05 3,71
234 ТМП 113,467 4,95 4,45 3,90
233 ТМП 114,760 5,22 4,82 4,33
23 ДМГ 115,607 5,42 4,82 4,22
2 МЗЭП 115,650 5,42 4,82 4,22
2 МГп 117,646 5,73 5,00 4,33
4 МГп 117,709 5,92 5,18 4,34
34 ДМГ 117,725 5,97 5,32 4,40
3 МГп 118,925 6,16 5,40 4,40
3 ЭГ 118,534 6,44 5,62 4,79
h-Cs 125,665 8,17 7,15 5,82
• Расшифровку названий см. в табл. 3.
** См. «Физико-химические свойства ин-
дивидуальных углеводородов». М., Гостоп-
техиздат, 1960.
Например, во фракции н. к.— 110° С метилциклопентан при 30° С
выходит вместе с 2,2-диметилпентаном, а при 80° С — с 2,4-диметилпен-
таном; при 50° С н-октан выходит вместе с транс-1,2-диметилциклогек-
саном, а при 80° С они делятся. Поэтому для каждой фракции состав
рассчитывают по хроматограммам при двух температурах и определяют
средний процент углеводорода на исследуемую фракцию.
Зная выход фракции на сырье, можно пересчитать процентное содер-
жание компонента или суммы компонентов на нефть (конденсат). Ввиду
того что при ректификации часть компонентов одной фракции попадает
в смежные, общее содержание каждого компонента в нефти (конденса-
10
1
zz
I
II
to
! к
not
mS
30
/" Л'; I
Яремя
Рис. 3. Хроматограмма углеводородных смесей, выкипающих
в интервале 30—110° С
а — сквалан, 30° С; б — сквалан, 80° С. Отнесение пиков см. в табл. 3
Рис. 4. Хроматограмма углево-
дородных смесей, выкипающих
в интервале ПО—125° С
а — сквалан, Б0° С; б — сквалан,
80° С, Отнесение пиков см. в табл. 3
Таблица 3
Порядок выхода углеводородов *, выкипающих до 125° С
(в стандартных условиях при температурах 30, 50 и 80° С)
Фракция н. К. — но °C Фракция 111-125° С
пика 30° с пика 80° С пика 50° С пика 80° С
g g g
1 изо-Cs 1 изо-С5 Г 22 ДМГ 1 22 ДМГ
2 н-Са 2 н-С5 1 [ 12 ДМЦП ц 2 12 ДМЦП ц
3 22 ДМБ 3 22 ДМБ 2 ИЗ ТМЦП Г113 ТМЦП
4 ЦПентан С ЦПентан 3 мцг 3 [25 ДМГ
5 23 ДМБ 4 23 ДМБ 4 25 ДМГ Г МЦГ
6 2 МП 1г мп 5 24 ДМГ 4 [ 24 ДМГ
7 3 МП 5 3 мп 6 ЭЦП Г ЭЦП
Q н-Се 6 Н-С<5 7 223 ТМП 5 [223 ТМП
г 22 ДМП 7 22 ДМП 8 124 ТМЦА тт Г124 ТМЦП тт
9 (мцп 8 Г 24 ДМП 9 33 ДМГ О [ 33 ДМГ
10 24 ДМП [МЦП 10 Толуол 7 Толуол
И Бензол 9 223 ТМБ И 123 ТМЦП тт 8 123 ТМЦП тт-
223 ТМБ 10 Бензол 12 234 ТМП 9 234 ТМП
12 г 33 ДМП 11 33 ДМП ] 23 ДМГ 10 Г 23 ДМГ
13 [ЦГексан 12 ЦГ ексан 13 23 МЭП [23 МЭП
14 2 МГ 13 2 МГ 1 233 ТМП И 2 МГп
г 23 ДМП 14 23 ДМП 14 112 ТМЦП 233 ТМП
15 [И ДМЦП 15 Г И ДМЦП 15 2 МГп 12 4 МГп
16 3 МГ 1з МГ 16 4 МГп 13 112 ТМЦП
17 13 ДМЦП Ц 16 Г13 ДМЦП Ц 17 34 ДМГ 34 ДМГ
18 13 ДМЦП т [3 ЭП ( 3 МГп 14 3 МГп
19-., 12 ДМЦП т 17 13 ДМЦП т 18 1з эг 3 ЭГ
J3 ЭП 18 12 ДМЦП т ( 124 ТМЦП ЦТ 15 124 ТМЦП цц
20 'н-С, 19 Н-С7 19 11133-тетраМЦП 16 f 124 ТМЦП цт
21,, 12 ДМЦП ц 20 22 ДМГ 20 123 ТМЦП тц [ 1133-тетраМЦП
22 ДМГ 21 12 ДМЦП ц f 13 ДМЦГ ц 17 123 ТМЦП тц
22 1 113 ТМЦП 99 ( ИЗ ТМЦП 21 [ 14 ДМЦГ т 13 ДМЦГ ц
(мцг 125 ДМГ г И ДМЦГ 18 14 ДМЦГ т
23 25 ДМГ 9а (мцг 22 (13 МЭЦП ц 11 ДМЦГ
24 ЭЦП 124 ДМГ г 13 МЭЦП т 13 МЭЦП ц
25 24 ДМГ 24 (ЭЦП 23 1 12 МЭЦП т 19 [ 13 МЭЦП т
26 223 ТМП 1223 ТМП 24 111 МЭЦП [ 12 МЭЦП т
27 124 ТМЦП тт 25 Г124 ТМЦП тт 20 н-Сз
28 Толуол [33 ДМГ 25 < 1234-тетраМЦП 21 И МЭЦП
29 33 ДМГ 26 Толуол 112 ДМЦГ т 22 1234-тетраМЦП ттт
30 123 ТМЦП тт 27 123 ТМЦП тт ( 13 ДМЦГ т 23 12 ДМЦГ т
26 114 ДМЦГ ц 24 Г13 ДМЦГ т [ 14 ДМЦГ ц
* Шифр названий углеводородов: н-С5— н-Св —- нормальные парафины, М — метил, ДМ —
диметил, ТМ — трнметил, Э — этнл, Пр— пропил, Б — бутан, П—пентан, Г — гексан, Гп — геп-
тан, О —октан, т —транс, ц—цис, Ц —цикло. Цифры перед шифром показывают положение
заместителей (числа всюду однозначные, запятые между цифрами опущены).
те) находят сложением его долей (на исходное сырье) во фракции I, II
и фракции 125—150° С.
В таблицах состава бензиновых фракций содержание некоторых,
компонентов дается в сумме, так как из-за сходства структуры и близо-
сти температур кипения некоторые углевбдороды на сквалане не
делятся.
12
Рис. 5. Хроматограмма бензиновой фракции н.к.— 110° С из грозненской нефти
а — сквалан, 30° С; б — сквалан, 80° С. Отнесение пиков см. в табл. 3
Например, во фракции ПО—125° С не делятся цис-1,3- и транс-1,4-,
-я также транс-1,3- и цис-1,4-диметилциклогексан, транс-1,2- и транс-1,3-
диметилциклопентан.
В качестве примера на рис. 5 и 6 приведены хроматограммы бензи-
новых фракций, выделенных из грозненской нефти.
И. АНАЛИЗ БЕНЗИНОВОЙ ФРАКЦИИ 125—150° С
Исследование бензиновых фракций, выкипающих до 125° С [1—3],
показало, что в нефтях присутствуют в больших концентрациях термо-
динамически устойчивые изомеры и что количественное распределение
углеводородов подчинено следующим закономерностям:
1. Среди алканов преобладают нормальные и малоразветвленные
(моно- и дизамещенные изомеры), трехзамещенные и изомеры с чет-
вертичным атомом углерода находятся в незначительных количествах.
Распределение изомерных алканов с одинаковым количеством замести-
телей соответствует равновесным концентрациям.
2. Геометрические изомеры алкилцикланов находятся в равновесных
•соотношениях, т. е. преобладают наиболее термодинамически устойчи-
вые легкокипящие изомеры.
Эти закономерности и экспериментальные данные по равновесным
соотношениям пространственных изомеров нафтеновых углеводородов
13
Рис. 6. Хроматограммы бензиновой фракции 110—125° С из грозненской нефти
а — сквалан, 50° С; б — сквалан, 80° С. Отнесение пиков см. в табл. 3
состава С9 были использованы при исследовании состава бензиновой
фракции 125—150° С.
В этом температурном интервале кипят почти все углеводороды,
состава С9, всего около 130 (35 алканов, 21 алкилциклогексанов, 55 ал-
килциклопентанов, 20 бицикланов и алкилбицикланов). Все они могуг
присутствовать в нефтях или конденсатах. Благодаря близости физиче-
ских свойств многих из них идентификация их затруднительна, и опре-
деление полного компонентного состава фракции 125—150° С практиче-
ски недоступно.
Предлагаемая методика ограничивается задачей определения таких,
углеводородов, которые находятся в нефтях или конденсатах в относи-
тельно больших количествах, сумма которых составляет 90—95% всей
фракции.
Методика анализа фракций 125—150° С отличается от приведенной,
выше для фракций н. к.— 125° С тем, что из фракции 125—150° С необ-
ходимо предварительно выделить ароматические углеводороды, так
как при снятии хроматограмм их пики настолько перекрывают пики-
парафиновых и нафтеновых углеводородов, что количественная расшиф-
ровка становится невозможной.
Отогнанную на ректификационной колонне фракцию 125—150° С
хроматографией на силикагеле разделяют на ароматическую и парафи-
но-нафтеновую части, которые в дальнейшем порознь анализируют на»
капиллярном хроматографе.
Ароматические углеводороды при температуре 80° С хорошо делятся
на сквалане и выходят в порядке повышения температуры кипения
(рис. 7). Для определения процента ароматического углеводорода на
фракцию необходимо определить суммарное содержание ароматических
14
3
мб
Рис. 7. Хроматограмма
ароматических углеводородов,
содержавшихся в бензиновой,
, фракций 125—150° С.
Сквалан, 80° С
1 — этилбензол; 2 — п-ксилол;
3 — л-ксилол; 4 — о-ксилод
Рис. 8. Хроматограммы изомеризата м-вднаца
а — сквалан, 80“ С; б — сквалан, 106° С.
7 — н-октан; 2 — 2,3,5-трнметилгексан; 3 — 2,2-диметилгептаи; 4 — 2,4-диметилгептаи; 5 — 2,4-метил-,
этилгексан; 6 — 2,6-диметнлгептан; 7 — 2,5-диметилгептан; 8 — 3,5-диметилгептан; 9 — 3,3-диметилгеп-
тан; 19 — 2,3-диметнлгептан; 11—4-этилгептан; 12 — 3,4-диметилгептан; 13— 4-метнлоктан; 14 — Я-ме-
тилоктаи; 15 —< 3-этилгептаи; 16— 3-метилоктан; 17—19 — алканы состава., Сю; 39.— н-нонаи
углеводородов во фракции; зная выход фракции, можем рассчитать,
процент углеводорода на нефть (конденсат).
Хроматограммы парафино-нафтеновой части снимают при темпера-
туре 80 и 106° С, в этих же условиях снимают и хроматограммы изоме-
ризата «-нонана, так как предварительно необходимо идентифицировать
алканы состава Сэ. Для определения области алканов Сэ на хромато-
грамме изомеризата в него добавляют в качестве реперов «-октан и
н-нонан. Пользуясь предлагаемой в методике хроматограммой (рис. 8),
и относительными временами удерживания алканов, состава Сэ (табл.
15 .
4), можно идентифицировать пики алканов на хроматограмме изомери-
зата.
В парафино-нафтеновую часть бензиновой фракции добавляют
реперы, снимают хроматограмму и сравнением полученной хромато-
граммы с хроматограммой изомеризата, полученной при той же темпе-
ратуре, определяют пики алканов.
Для идентификации пиков алкилцикланов используют порядок вы-
хода углеводородов при температурах 80 и 106° С, приведенных в табл.
5 и 6 (рис. 9).
Рис. 9. Хроматограммы парафино-нафтеновых углеводородов состава Св,
содержащихся во фракции 125—150° С
.а —сквалан, 80° С (отнесение пиков см. в табл. 5); б — сквалан, 106° С (отнесение пиков
см. в табл. 6)
Следует помнить, что состав фракции 125—150° С различен для
-нефтей разной природы: просты по составу бензиновые фракции кон-
денсатов и парафинистых нефтей, более сложны фракции нафтеновых
нефтей. Однако независимо от характера нефтей в хроматограммах
«наблюдается много общего, поэтому в качестве эталонной достаточно
иметь одну расшифрованную хроматограмму.
Таблица 4
Относительные времена удерживания алкаиов состава С8, присутствующих
во фракции 125—150° С (сквалан)
Углеводород Температура колонки, °C Углеводо род Температура колонки, °C
80 106 80 106
н-Октан 50,7 62,2 3,3-Диметилгептан 65,3 74,8
2,3,5-Триметилгексан 55,4 2,3-Диметилгептан 73,3 80,4
2,2-Диметилгептан 56,4 | 67,2 4-Этилгептан 75,2
2,4-Димети лгептан 58,2 68,5 4-Метилоктан 77,0 J-82,5
2,4-Метилэтилгексан 59,6 70,0 2-Метилоктан 78,0 83,2
2,6-Диметилгептан 60,6 70,0 З-Этилгептан 79,3 84,6
2,5-Диметилгептан 62,4 72,0 З-Метилоктан 81,2 86,0
3,5-Диметил гептан 63,4 72,7 н-Нонан* 100,0 юо'о
* Время удерживания н-нонана принято за 100.
16
Таблица 5
Порядок выхода парафино-нафтеновых углеводородов, содержащихся
во фракции 125—150° С (сквалан, температура 80°)
Ki пика Углеводород * К пика Углеводород
1 н-Октан 21 4ЭГп + 1234-тетраМЦП ццт
2 1234-тетраМЦП ттт 22 34 ДМГп
3 12 ДМЦГ т 23 4МО 4- 1234-тетраМЦП цтц
4 (13 т + 14 ц) ДМЦГ 24 2 МО
5 6 235 ТМГ 22 ДМГп-{-1124-тетраМЦП тц+ 1134-тетраМЦП ц + цзоПрЦП 25 ЗЭГп 4- 135 ТМЦГ цт 4- 124 ТМЦГ тц 4- бицикло [3,3,0]- октан
7 24 ДМГп 26 ЗМО-|-1,3 МизоПрЦП ц 4- т
8 9 10 24 МЭГ 26 ДМГп 11 ДМЗЭЦП 27 28 Не идентифицирован 14 ДМ2ЭЦП цц 4- 14 ДМ2ЭЦП цт 4- 13 ДМ2ЭЦП тц 4- бицикло- [13,2,1]-октан
И 25 ДМГп 29 12 ДМЗЭЦП тц
12 13 35 ДМГп 4- ПрЦП + 1234-тет- раМЦП тцт 12 ДМЦГ ц 30 12 ДМЗЭЦП цт 4- 124 ТМЦГ тт 4- 4-третБЦП-|-1,2 ДМ4ЭЦП цц-|- 4- 12 ДМ4ЭЦП цт
14 1234-тетраМЦП цтт -}- 33 ДМГп 31 12 МПрЦП т
15 16 ЭЦГ 14 ДМ2ЭЦП тт 32 123 ТМЦГ тт 4- 13 МПрЦП ц 4- 4- 12 ДМ1ЭЦП т
17 ИЗ ТМЦГ + 14 ДМ2ЭЦП тц 4- + 13ДМ1ЭЦП + 12ДМ4ЭЦП тт 33 34 13 МПрЦП т4- 124 ТМЦГ цц 124 ТМЦГ цт 4-112 ТМЦГ
18 135 ТМЦГ цц 4-114 ТМЦГ 4- + 13 ДМ2ЭЦП тт 35 12 ДЭЦП т 4- изоБЦП -|- 1135- тетраМЦГ
19 12 ДМЗЭЦП тт 36 13 МЭЦГ ц 4-13 ДЭЦП ц 4- т
20 23 ДМГп 37 14 МЭЦГ т 4- н-нонан
* Расшифровку названий см. в табл. 3.
Таблица 6
Порядок выхода парафино-нафтеновых углеводородов, содержащихся
во фракции 125—150°С (сквалан, температура 106°С)
JMe пика Углеводород * № пика Углеводород
1 н-Октан 16 2МО
2 1234-тетраМЦП ттт 17 ЗЭГп
3 (12ц 4- 13т 4- 14ц) ДМЦГ 4- 4- 235 ТМГ 4- 22 ДМГп 18 ЗМО 4- 135 ТМЦГ ЦТ 4- 1234- тетраМЦП ццт
4 24 ДМГ п4-И34-тетраМЦН ц4~т 19 124 ТМЦГ тц
5 1134-тетраМЦП ц4-изоПрЦП 20 13 МизоПрЦП ц 4- т 4- бицикло-
6 26 ДМГп 4- 24 МЭЦП [3,3,0]октан
7 25 ДМГп 21 14 ДМ2ЭЦП цт -|- цц -}-
8 35 ДМГп 4-И ДМЗЭЦП 4-13 ДМ2ЭЦП тц
9 33 ДМГп 4- ПрЦП 4- 1234- 22 12 ДМЗЭЦП тц
тетраМЦП тцт 23 12 ДМЗЭЦП цт 4- 12ДМ4ЭЦП цц 4-
10 12 ДМЦГ ц 4-1234-тетраМЦП 4- 12 ДМ4ЭЦП тц 4- mpem-БЦП
ЦТТ 24 124 ТМЦГ тт 4- 12 МПрЦП т
11 ЭЦГ 25 13 МПрЦП ц -}- 12 ДМ1ЭЦП
12 14 ДМ2ЭЦП тт 4- 14 ДМ2ЭЦП тц 26 13 МПрЦП т 4- 123 ТМЦГ тт
13 (ИЗ 4- 114 4- 135 цц) ТМЦГ 4- 4- 12ДМ4ЭЦП тт 4- 13ДМ1ЭЦП цт 4- 13 ДМ2ЭЦП тт 27 124 ТМЦГ цц 4- 124 ТМЦГ цт 4- -|- 12 ДЭЦП т 4- изоБЦП 4- 4- 112 ТМЦГ 4-н-Нонан
14 23 ДМГп 4- 12ДМЗЭЦП тт 4- 28 13 МЭЦГ ц 4- 13 ДЭЦП цт
15 4- 4ЭГп 34 ДМГп 4- 4МО 4- 1234- тетраМЦП цтц 29 14 МЭЦГ т
* Расшифровку названий см. в табл. 3.
2 Методы анализа
17
Таблица 7
Рис. 10. Хроматограмма группы нафтеновых
углеводородов состава Сэ
а — сквалан, 80° С. Отнесение пиков: 1 — 1,4-диметил-
2-этилциклопентан тт; 2— 1,4-диметил-2-этилцикло-
пентан тц + 1,1,3-триметилциклогексан + 1,2-днметил-
4-9тилциклопентаи + 1,3-диметнл-1-этилциклопентан;
3 — 1,1,4-трнметнлциклогексан + 1,3,5-трнметилцик-
логексан + 1,3-диметил-2-этилцнклопентан тт; 4 — 1,2-
диметил-3-этилциклопентан тт.
б — дибутнлтетрахлорфталат, 105° С. Отиесенне пиков:
1 — 1,4-диметил-2-этнлциклопентан тт + 1,4-диметил-
2-этнлциклопентан тц; 2 — 1,2-днметил-4-этнлцикло-
пентаи тт; 3—1,3-диметил-1-этилциклопентан + 1,3-
днметил-2-этилциклопентан тт; 4 — 1,1,3-триметилцик-
логексаи + 1,2-диметил-З-этилциклопентая тт; 5 —
1,1,4-трнметилциклогексан; 6 — 1,3,5-трнметилцикло-
гексан
Равновесный состав смеси некото-
рых нафтеновых углеводородов С»
при температуре 600 ° К
Углеводород Стереоизомеры Содержа- ние, %
транс, транс 40,6
14 ДМ2ЭЦП транс, цис цис, транс 45,7 6,6
цис, цис 7,1
транс, транс 70,7
12 ДМЗЭЦП транс, цис цис, транс 9,7 16,6
цис, цис 3,0
транс, транс 71,4
13 ДМ2ЭЦП транс, цис 25,6
цис, цис 3,0
13 МПрЦП цис транс 47,0 43,0
124 ТМЦГ транс, цис транс, транс цис, цис '73,4 9,8 8,4
цис, транс 8,4
135 ТМЦГ цис, цис цис, транс 73,6 26,4
Количественная расшифровка хроматограммы в области пиков
между этйлциклогексаном и 2,3-диметилгептаном (рис. 10, а) весьма
затруднительна, поскольку в двух пиках содержится семь нафтеновых
углеводородов. Эти углеводороды хорошо делятся на дибутилтетрахлор-
фталате при 105°С (рис. 10, б), но использовать указанную фазу для
анализа бензиновой фракции конденсатов и ряда нефтей нельзя, так
как монозамещенные изомеры нонана, присутствующие в большом
количестве, в этом случае полностью закрывают пики циклопентановых
углеводородов. Поэтому приходится ограничиться определением суммар-
ного содержания указанных углеводородов.
С приемлемой для ряда лабораторий точностью содержание некото-
рых неразделяющихся компонентов бензиновой фракции можно подсчи-
тать, исходя из равновесных соотношений их пространственных изомеров
[4—6]. Например, изомеры 1,2,3,4-тетраметилциклопентана в равновес-
ной при 600° К смеси находятся в следующем соотношении:
ттт .... 53,7%
тцт. . . . 12,0%
ттц .... 24,8%
ццт.... 5,8%
цтц.... 3,4%
ццц ... 0,3%
Таблица 8
Состав бензиновой фракции и. к. — 125° С грозненской парафинистой нефти
Углеводороды Содержание, вес.% на фракцию Углеводороды Содержание, вес. % на фракцию Углеводороды Содержание, вес. % на фракцию
Парафиновые 24 ДМГ 0,63 123 ТМЦП тт 0,95
н-С5 з;зэ 23 ДМГ 1 0,32 123 ТМЦП тц 0,31.
изо-Сз 2,79 23 МЭП J 11 МЭЦП 0Д6
н-С6 8,15 34 ДМГ 0,17 13 МЭЦП ц 0,50
2МП 4,17 33 ДМГ 0,06 13 МЭЦП т 1 1,0»
ЗМП 2,96 22 ДМГ 0,06 12 МЭЦП т J
23 ДМБ 0,50 223 ТМП 0,02 1234-тетра МЦП ттт 0,22
22 ДМБ 0,04 233 ТМП Следы Цгексан 3,52
н-С? 10,60 234 ТМП 0,15 МЦГ 11,0
2МГ ЗМГ 2,06 4,11 Нафтеновые 13 ДМЦГ ц 14 ДМЦГ т 1,74 1,03
3 эп 0,38 Цпентан 0,53 12 ДМЦГ т 1,26
24 ДМП 0,44 МЦП 4,23 13 ДМЦГ т | 14 ДМЦГ ц J
23 ДМП 1,17 ЭЦП 0,81 0,44
22 ДМП 0,01 И ДМЦП 1,32
33 ДМП 223 ТМП 0,006 0,003 13 ДМЦП ц 13 ДМЦП т 2,23 1,96 Ароматические
н-Cg 6,42 12 ДМЦП т 3,74 Бензол 0,22
2 МГп 3,67 12 ДМЦП ц 0,43 Толуцл 1,92
3 МГп 2,22 113 ТМЦП 1,60
4 МГп 0,81 112 ТМЦП 0,28
3 ЭГ 0,09 124 ТМЦП тт 1,38
25 ДМГ 0,61 124 ТМЦП цт 0,17
Таблица 9
Состав парафино-нафтеновой части фракции 125—150° С грозненской нефти
Углеводород Содержа- ние, % на фракцию Углеводород Содержа- ние, % на фракцию
1234-тетраМЦП ттт 1,16 4 ЭГп Следы
12 ДМЦГ т 4,1 34 ДМГп 3,77
(13 т + 1,4 ц) ДМЦГ 1,62 4 МО 3,39
22 ДМГп 0,85 2 МО 5,21
1124-тетраМЦП цт 0,29 3 ЭГп 0,75
24 ДМГп 1,52 3 МО 4,93
Изопропил ЦП Следы 14 ДМ2ЭЦП цц, цт + 1,72
26 ДМГп 5,11 +13 ДМ2ЭЦП тц
НДМЗЭЦП 0,58 124 ТМЦГ тц 3,10
25 ДМГп 2 0 12 Д МЗЭЦП цт + 12 ДМ4ЭЦП тц 0,58
35 ДМГп 1,02 и цц + mpem-БЦП + + 124 ТМЦГ тт
1234-тетраМЦП тцт 0,26 123 ТМЦГ + 13 МПрЦП ц ’ 0,86
33 ДМГп 1,23 13 МПрЦП т + 124 ТМЦГ цц 0,40
ПрЦП 0,15 124 ТМЦГ цт + 112 ТМЦГ * 0,54
12 ДМЦГ ц 1,20 12 ДЭЦП т + изоБЦП + 0,88
ЭЦГ 9,44 + 1135-тетраМЦГ * 1
14 ДМ2ЭЦП тт 1,38 13 МЭЦГ ц + 13 ДЭЦП ц + т 3,28
113 ТМЦГ + 14 ДМ2ЭЦП тц + 7,88 14 МЭЦГ т 2 07
4- 12ДМ4ЭЦПтт + 13ДМ1ЭЦП ’ Бицикло [3,3,0] октан + Ц62
114+135) ТМЦГ + + 13 ДМ2ЭЦП тт ° 2,86 + 13 МизоПрЦП * н-Нонан 16,59
12 ДМЗЭЦП тт 1,95
23 ДМГп 6,11
* Первым указан преобладающий компонент.
2
1»
Зная процент на фракцию 1234-тетраМЦП ттт, который выходит на
хроматограмме отдельным пиком, можем определить содержание ос-
тальных его изомеров, выходящих в сумме с углеводородами. Присут-
ствие в нефтях изомеров цтц и ццц из-за их термодинамической неустой-
чивости маловероятно, и практически ими можно пренебречь. Приведен-
ные в табл. 7 данные по равновесным соотношениям некоторых
нафтеновых углеводородов состава С9 [4, 6] могут быть использованы
при определении компонентного состава бензиновой фракции 125—
150° С.
Здесь принята номенклатура стереоизомеров, использованная Петро-
вым и сотр. в работе [7]. Взаимное положение заместителей отсчитыва-
ется попарно от каждого предыдущего с каждым последующим, начиная
с первого, в порядке нумерации углеродных атомов.
Рассчитав хроматограммы при двух температурах, определяют
процент углеводорода на парафино-нафтеновую часть, а затем — на
фракцию и сырье. Для количественных подсчетов рекомендуется
в основном пользоваться данными хроматограммы, снятой при 80° С,
так как на хроматограмме, снятой при 106° С, деление некоторых компо-
нентов менее четкое.
В табл. 8 и 9 в качестве примера представлены данные по составу
грозненского бензина.
ВЫВОДЫ
1. Разработан метод анализа индивидуального состава прямогонных
бензинов, основанный на применении капиллярной газо-жидкостной хро-
матографии и вторичных калибровочных эталонов, получаемых равно-
весной изомеризацией легкодоступных углеводородов в присутствии
бромистого алюминия.
2. Метод иллюстрирован примерами анализа фракций, выкипающих
до 150° С.
Институт геологии и разработки горючих ископаемых
МНДП СССР и АН СССР
ЛИТЕРАТУРА
1. Э. К. Брянская, В. А. Захаренко, Ал. А. Петров. Нефтехимия, 6, 784 (1966).
2. Э. К. Брянская, В. А. Захаренко, Ал. А. Петров. Там же, стр. 904.
3. 3. К. Оленина, А. А. Петров. Нефтехимия, 7, 323 (1967).
4. О. А. Арефьев, В. А. Захаренко, Ал. А. Петров. Нефтехимия, 6, 506 (1966).
5. О. А. Арефьев, В. А. Захаренко, Ал. А. Петров. Нефтехимия, 7, 842 (1967).
6. Е. В. Всеволожская, О. Е. Морозова, Ал. А. Петров. Нефтехимия, 4, 142 (1964).
7. С. С. Берман, В. А. Захаренко, Ал. А. Петров. Там же, стр. 850.
КОМПОНЕНТНЫЙ АНАЛИЗ ТЯЖЕЛОЙ ЧАСТИ НЕФТЕЙ
И БИТУМОВ В МАЛЫХ НАВЕСКАХ
Д. К. Жестков
Множество классов соединений, входящих в состав тяжелой части
нефтей и битумов, не позволяет осуществлять их четкое разделение по
одноступенчатой схеме анализа. В практике исследования нефтей и
битумов в качестве первой предварительной ступени разделения иссле-
дуемую навеску обычно делят на группы компонентов, число которых
может быть различно в зависимости от поставленных задач.
При упрощенном компонентом анализе нефти и битумы разделяют
на группы соединений, более или менее резко различающихся по моле-
кулярным массам: а) масла, б) смолы бензольные, в) смолы спирто-
бензольные, г) «асфальтены». Такой упрощенный компонентный анализ
по широко распространенной технике его проведения [1—3] требует не
менее 1,0—0,5 г вещества.
СХЕМА КОМПОНЕНТНОГО АНАЛИЗА ТЯЖЕЛЫХ
НЕФТЕПРОДУКТОВ И БИТУМОВ
Компонентный анализ предусматривает выделение масел, смол бен-
зольных, смол спирто-бензольных и фракции «асфальтенов», включаю-
щих, кроме собственно асфальтенов, также асфальтогеновые кислоты и
их ангидриды (см. схему).
В случае необходимости, применив ряд других растворителей, можно
разделить смолы и «асфальтены» на более узкие фракции, отличающиеся
преимущественно полярностью. Выделение одноименных компонентов
рекомендуемым методом проводят в условиях, близких к принятым
в обычных битуминологических анализах [3, 4]. Это позволяет получать
результаты, сопоставимые с прежними битуминологическими исследова-
ниями. Получаемые фракции могут быть подвергнуты дальнейшему
изучению современными микрометодами.
РЕКОМЕНДУЕМАЯ МЕТОДИКА
Рекомендуемая методика позволяет разделить тяжелую часть нефти
и тяжелые нефтепродукты (с температурой начала кипения выше
250°С), а также битумы на фракции масел, смол бензольных, смол
спирто-бензольных и асфальтенов при исходных навесках 50—150 мг.
Ниже мы остановимся на детальном описании рекомендуемой техни-
ки эксперимента и оценки получаемых результатов.
21
СХЕМА КОМПОНЕНТНОГО АНАЛИЗА
ТЯЖЕЛЫЕ ФРАКЦИИ НЕФТИ,
ОСТАТОЧНЫЕ НЕФТЕПРОДУКТЫ
ИЛИ БИТУМЫ
I
I
Осаждение из раствора
петролейного эфира
(или нормального бензина)
а. Выделение и определение содержания асфальтенов
Осаждение асфальтенов рекомендуется проводить выбранным оса-
дителем (петролейным эфиром или нормальным бензином) в микро-
отстойнике (рис. 1 и 2, а), который для принятых навесок 50—150 мг
имеет диаметр широкой части 40—50 мм, длину 60—70 мм, вес без
пробок 10—12 г. В доведенный до постоянного веса отстойник с помощью
малого шпателя вносят образец нефти или битума (рис. 1, а), вес кото-
рого определяют по разности (вес отстойника с образцом минус вес
пустого отстойника). Если вещества так мало, что перенос его шпателем
в отстойник невозможен, то образец растворяют в нескольких каплях
бензола, раствор вносят в отстойник пипеткой и затем, удаляют бензол,
подавая в отстойник через капилляр струю азота (рис. 1, б). Таким
образом удается переносить 25—30л«г вещества. Твердые или имеющие
густую консистенцию битумы растворяют в отстойнике в малом количе-
стве бензола (0,1—0,2 мл). Растворение навески осуществляется быстро
при легком покачивании и вращении отстойника в горизонтальном
положении.
После растворения навески в отстойник из микробюретки со специ-
ально оттянутым носиком (рис. 2, а) вводят выбранный осадитель
(петролейный эфир, нормальный бензин и т. п.) в 40-кратном количестве
по отношению к весу образца или его раствора в бензоле. Отстойник
закрывают пробками, шлифы которых обязательно надо смочить дистил-
лированной водой, что препятствует обычному «выползанию» масел из
отстойника. Наполненный закрытый отстойник осторожно вращают
в горизонтальном положении для хорошего перемешивания всей смеси
22
Рис. 1. Способ осаждения асфальтенов
а — внесение шпателем навески в отстойник для осаждения асфальтенов; б — уда-
ление бензола нз битума в отстойнике для осаждения асфальтенов; в — переме-
шивание исследуемой навески с осадителем; г — осаждение асфальтенов в поло-
жении многочасового отстоя в отстойнике
(рис. 1, в) и ставят с мокрыми ватками на шлифах (рис. 1, г) в темноту.
По прошествии 12—20 час. пробки открывают, отстойник ставят в верти-
кальное положение над воронкой с пористой пластиной № 4, все масла
и смолы смывают тем же осадителем (петролейным эфиром или нор-
мальным бензином) в микроэкстрактор на силикагель (рис. 2, б).
Полноту отмывки масел и смол с отстойника и пористой воронки
проверяют по исчезновению люминисцентного возбуждения под ультра-
фиолетовым светом. Количество осадителя, идущего на промывку,
составляет 25—50 мл. При выходе асфальтенов примерно 10 мг крат-
ность веса асфальтенов к весу осадителя составляет 1:2500—1: 5000,
что обусловливает хорошую отмывку асфальтенов от масел и смол.
В тех случаях, в которых исследуемая навеска содержит очень много
твердых углеводородов, рекомендуется повторное переосаждение ас-
фальтенов.
Отмытые асфальтены после незначительного подсушивания смывают
горячим бензолом (или хлороформом) со стенок отстойника и пористой
торонки во взвешенный стаканчик (рис. 2, в). После выпаривания
растворителя на водяной бане в токе азота стаканчик с асфальтенами
доводят до постоянного веса и по разности весов определяется выход
асфальтенов.
б. Выделение масел и фракций смол
При отмывании асфальтенов петролейным эфиром (или другим
ъыбранным растворителем) раствор масел и смол фильтруют под сла-
бым разрежением через воронку со стеклянной пористой пластинкой
в микроэкстрактор с силикагелем (см. рис. 2, б) (с силикагеля необхо-
димо заранее снять теплоту смачивания). Разделение масел и смол
рекомендуется вести на силикагеле марки АСК крупностью помола
23
Рис. 2. Способ выделения
масел и смол
а — введение отмеренного количе-
ства осадителя в отстойник для
осаждения асфальтенов; б — ми*
кроэкстракционный прибор в рабо-
чем положении при отмывании ас-
фальтенов от масел и смол: /—от-
стойник для осаждения асфальте-
нов, 2 — вороика с пористой стек-
лянной пластинкой № 4, S — корко-
вая пробка, 4—мнкроэкстрактор,
5 — силикагель, 6 — вата; в — пере
нос асфальтенов во взвешенный
стаканчик; г — микроэкстракцнои-
ный прибор в рабочем положении
при экстракции масел н смол
30—150 меш и активностью 6,0—7,0 мл по бензолу [5]. Навеска силика-
геля зависит от его активности и подбирается экспериментально. Для
этого новую партию силикагеля активируют в сушильном, шкафу при
150—160° С в течение 5—6 час., заряжают несколько микроэкстракторов
разным количеством силикагеля и вносят в каждый микроэкстрактор
одинаковую навеску нефти или битума. При экстракции масел выбран-
ным растворителем наблюдается опускание нижней границы окрашенных
смол. При слишком большой навеске силикагеля масла вымываются
очень медленно и остается большой слой не окрашенного смолами си-
ликагеля. При малой навеске силикагеля окрашенная смолами нижняя
граница доходит до самого низа силикагелевого слоя в микроэкстрак-
торе, масла вымываются слишком быстро и получаются сильно окра-
шенными. Результаты такого анализа завышены по маслам, занижены
по смолам и, как правило, плохо воспроизводимы. При правильно
подобранной навеске силикагеля граница окрашенных смол при экстрак-
ции масел останавливается на 20—25 мм выше нижней ватки, на которой
24
лежит силикагель. В этом случае анализ проходит быстро без потери*
точности его результатов.
После полной отмывки асфальтенов микроэкстрактор отсоединяют
от вакуум-линии, отвод для вакуум-линии закрывают экстрагированной*
корковой пробочкой, вместо воронки ставят обратный холодильник,,
в колбочку наливают свежую порцию петролейного эфира (или другого*
выбранного растворителя) и включают нагрев
(рис. 2, г). Растворитель экстрагирует с силика-
геля масла.
Конец экстракции масел определяют по ин-
тенсивности свечения под ультрафиолетовым све-
том— после 2 час. экстракции свежим раствори-
телем интенсивность свечения растворителя в
колбочке не должна превышать 2—3 баллов [6].
При соблюдении этого условия расхожде-
ния между параллельными анализами не превы-
шают ±1,0%. Если за конец экстракции прини-
мается момент, когда очередная порция свежего
растворителя в колбочке после 2 час. экстракции
остается бесцветной, то расхождение между дву-
мя параллельными опытами достигает 2—4%.
После окончания экстракции масел масля-
ные экстракты сливают, растворитель отгоняют,
а концентрированную масляную фракцию смыва-
ют тем же растворителем в приемник для сушки
масел и доводят в нем до постоянного веса.
Удаление следов растворителя из малых коли-
честв масляной фракции в бюксах или колбоч-
ках сопровождается нежелательным явлением
«выползания» масел и частичной их потерей, а
также некоторой дифференциацией масел по.
стенкам сосуда. В последнем случае при взятии
части масел для дальнейших исследований часто
получаются большие расхождения в результатах
анализа параллельных проб.
Сушку масляных фракций удается осущест-
вить быстро (почти без потерь и без дифферен-
циации навески с помощью простого прибора, предложенного М. Н. Ла-
рионовым (рис. 3). Прибор состоит из тонкостенного приемника 2
(диаметр широкой части 20 мм, общая высота 150 мм, вес не более
10 г), подшипника 1 из корковой пробки на штативной лапке, капилля-
ра для подачи азота, 5, резиновой трубки 3 и электромотора 4 мощ-
ностью 40—60 вт со скоростью вращения 800—1000 об/мин.
В приемник вносят 5—10 мл раствора масляной фракции и вращают'
его с помощью электромотора в вертикальном положении. Внутрь прием-
ника подается ток азота по капилляру 5. Стенки приемника можно подо-
греть маленькой электропечкой или сильной электрической лампой до-
температуры 40—60° С. Раствор масла под действием центробежной
силы распределяется тонким слоем по стенкам приемника и, омываемый
током азота, быстро отдает все летучие фракции. После отстоя в верти-
кальном положении масла, находящиеся на стенках приемника, стекают
в нижний отросток, откуда легко берутся длинным капилляром или
тонким шпателем для дальнейших исследований.
После окончания экстракции масел приступают к экстракции смол
бензолом, а затем спирто-бензольной смесью. Конец экстракции опреде-
ляется, как описано выше. Одноименные экстракты сливают вместе,
Рис. 3. Прибор для
быстрой сушки малых
количеств масел
I — подшипник из корковой?
пробки; 2 — приемник; 3 —
резиновая трубка; 4 — элек-
тромотор 800—1000 об/мии;
5 — капилляр подачи азота
25-
^растворитель отгоняют, а фракции смол доводят до постоянного веса
*в тарированных бюксах в токе азота при нагревании до 40—60° С. По
выходам фракций вычисляют их весовой процент на нефть или битум.
ЭКСПЕРИМЕНТАЛЬНАЯ ПРОВЕРКА СОПОСТАВИМОСТИ
РЕЗУЛЬТАТОВ АНАЛИЗА ТЯЖЕЛОЙ ЧАСТИ НЕФТИ
И БИТУМОВ МИКРО- И МАКРОМЕТОДАМИ
Для проверки описанного метода была взята серия образцов нефтей
и битумов месторождений о. Сахалина. Перед анализом нефти и выде-
ленные из пород битумы доводились до постоянного веса на приборе
для сушки малых количеств (см. рис. 3).
Сравнительные результаты определения компонентного состава сахалинских
нефтей и битумов (в вес. %)
Образец Масла Смолы Асфальтены Потери Метод анализа
бензоль- ные спирто- бензоль- ны е
Битум из асфальтового 52,6 12,7 14,9 21,1 +1,3 1 Метод малых навесок
•озера в районе г. Охи 52,4 12,7 14,8 21,2 +1,1 !
52,6 12,3 14,3 21,3 +0,4 Макрометод
Нефть месторождения 76,5 IT, 3 3,9 1,8 —0,5 Метод малых навесок
Юха 75,0 16,2 4,5 1,9 —2,4 Макрометод
Нефть месторождения 95,1 2,5 0,9 0,1 -1,4 1 Метод малых навесок
Нутово 94,9 2,6 1,1 0,1 -1,3 J
94,7 2,3 1,2 0,3 -1,5 Макрометод
‘ Нефть естественного вы- хода пос. Минеральное 78,0 79,8 14,3 14,2 3,2 3,1 1,8 1,8 —2,7 1 —1,1 J Метод малых навесок
79,5 14,5 з,о 2,2 -0,8 Макрометод
Нефть месторождения 76,8 16,5 3,2 1,8 -1,7 1 Метод малых навесок
Уйглекуты 77,0 16,3 3,0 1,7 -2,0 J
78,5 15,9 3,1 2,0 —0,5 Макрометод
Битум из канавы место- 16,0 5,8 21,1 54,6 —2,5 1 Метод малых навесок
>рождения Уйглекуты, 16,2 6 0 22,0 54,6 -1,2 I
•образец I 16,0 6,3 22,5 54,7 -0,5 Макрометод
То же, образец’!!! — 5.2 30,1 39,1 +1,5 } Метод малых навесок
25,6 5,2 31,3 39,4
25,8 5,0 30,0 39,6 +0,4 Макрометод
То же, образец V 57,3 8,4 22,0 10,0 —2,3 1 Метод малых навесок
57,2 8,5 22,6 9,5 —2,2 J
57,0 9,1 22,5 10,6 -0,8 Макрометод
"Нефть месторождения 90,5 — 1.1 Метод малых навесок
Некрасовка 90,4 1,0 5,0 0,9 -2,7 Г
89,0 2,0 4,0 1,4 -3,6 Макрометод
Битум из пород пос. Ми- 79,3 12,0 4,5 2,0 -2,2 I Метод малых навесок
„неральное 79,1 11,7 4,9 2,1 -2,2 J
79,2 12,1 5,0 2,1 -1,6 Макрометод
'При меча н-и е. Перед анализом нефти доводились до постоянного веса в * тех же усло-
виях, что и битумы.
26
Сравнительные результаты анализов приведены в таблице, из которой
видно, что метод малых навесок дает результаты, совпадающие с ана-
логичными определениями макрометодами.
ВЫВОДЫ
Разработан метод определения и простейшая аппаратура для компо-
нентного анализа тяжелой части нефти, тяжелых нефтепродуктов и
битумов в навесках от 50 до 150 мг, включающий а) выделение и
весовое определение асфальтенов; б) выделение и весовое определение
масляной, бензольной и спирто-бензольной фракций.
Описанная методика дает результаты, практически совпадающие
с результатами аналогичных определений, выполненных макрометодом.
Институт нефтехимического синтеза им. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. В. А. Успенский, О. А. Радченко. Описание основных методов битуминологического
исследования при обработке материалов опорного бурения. М, Гостоптехиздат,
1955.
2. В. А. Успенский, А. П. Шишкова, Н. А. Антоновская. Исследование серии битуми-
нозных образований, связанных определенными генетическими отношениями. Труды
ВНИГРИ, геохимический сборник, т. 1. Л., Гостоптехиздат, 1949.
3. И. Н. Петрова, И. П. Карпова, Н. Ф. Касаткина. «Геохимическое изучение органи-
ческого вещества девона». Сб. «Об условиях образования нефти» (под редакцией
3. Л. Маймин). Л., Гостоптехиздат, 1955.
4. Руководство по анализу битумов и рассеянного органического вещества горных по-
' род (под редакцией Успенского В. А., Радионовой К. Ф., Горской А. И. и Шишко-
вой А. П.). Л., «Недра», 1966.
5. Б. А. Казанский, Е. А. Михайлова. Адсорбционный хроматографический метод раз-
деления углеводородов. Рефераты докладов на совещании по применению хрома-
тографического метода М. С. Цвета в химическом анализе. М., Изд-во АН СССР,
1953.
6. В. Н. Флоровская. Люминисцентно-битумииологический метод изучения и поисков
нефтяных месторождений. М., Гостоптехиздат, 1954.
МИКРОХРОМАТОГРАФИЧЕСКИЙ АНАЛИЗ
УГЛЕВОДОРОДНЫХ СМЕСЕЙ
Д. К. Шестков, Г. Д. Гальперн
I. ЛИТЕРАТУРНЫЙ ОБЗОР у
При исследовании состава нефтей или продуктов их переработки
прежде всего необходимо упростить состав сложной смеси, пользуясь
сочетанием различных методов разделения молекул, как по массам, так
и по строению. Разделение молекул по массам проводится главным
образом с помощью различных типов перегонок. Разделение же по типу
молекул проводится многими методами: селективной экстракцией,,
кристаллизацией, клатрацией, рядом абсорбционных способов и, нако-
нец, высокоэффективными методами современной газо-жидкостной и
тонкослойной хроматографии.
Адсорбционные методы за последние 20—30 лет превратились,
в мощное средство разделения и анализа нефти и нефтепродуктов.
Однако не всегда изучение объекта необходимо и возможно доводить
до стадии определения индивидуального состава. В большинстве совре-
менных работ различные способы ректификации и абсорбционные
способы разделения предшествуют идентификации индивидуальных
составляющих с помощью газо-жидкостной хроматографии. Часто-
определение группового состава является конечной целью исследования.
Хроматографические методы в этом отношении широко используются
как самостоятельные приемы разделения, так и.в качестве предваритель-
ных каскадов в многокаскадных схемах аналитической и препаративной
дифференциации смесей сложного состава.
Первые опыты хроматографического отделения ароматических углеводородов от
парафино-нафтеновой части в бензино-дигроиновой фракции осуществили Дей [1, 2],
Энглер [3], Гурвич [4, 5], Тарасов [6]. Позднее Россини, Майер и Форциати [7, 8],
а также Беликовский, Павлова, Гофман и др. [9] своими исследованиями подтверди-
ли перспективность этого метода применительно' к легким и средним фракциям неф-
ти. Хроматография на силикагеле вошла существенной составной частью в предложен-
ный Ландсбергом, Казанским и сотр. [10] метод определения индивидуального углево-
дородного состава бензинов прямой гонки. Впоследствии многие исследователи стали
широко применять хроматографический метод для разделения легких, средних и тя-
желых фракций нефти и для разделения крекинг-продуктов [11—13]. Аллибон [14]
впервые осуществил хроматографическое разделение масляных фракций на различных
сорбентах при большом разведении масла петролейным эфиром. Вслед за ним многие
авторы сообщили о преимуществе метода хроматографии перед другими методами
разделения [15], о выделении чистых ароматических углеводородов [16, 17], об отделе-
нии нормальных парафиновых углеводородов изостроения от нафтеновых [18], о вы-
делении чистых нафтеновых углеводородов [19] и о выделении ряда индивидуальных
нормальных парафиновых углеводородов от C2i до Сзо [20, 21].
28
Много работ посвящено изучению адсорбционной способности угле-
водородов различных групп [22—24]. При разделении на силикагеле
искусственных смесей и исследовании нефтяных фракций установлен
ряд общих закономерностей по сорбируемости углеводородов в зависи-
мости от их химического строения, которые могут быть коротко сформу-
лированы следующим образом:
1. Ароматические углеводороды обладают значительно большей
сорбируемостью, чем метановые и нафтеновые.
2. Нафтеновые углеводороды сорбируются сильнее метановых, хотя
разница эта невелика.
3. Разветвленные метановые углеводороды сорбируются несколько
сильнее, чем углеводороды нормального строения. Некоторые авторы в
этом вопросе [8] приходят к выводу, что метановые углеводороды нор-
мального строения удерживаются силикагелем более прочно, чем мета-
новые углеводороды изостроения.
4. Увеличение количества ароматических колец значительно повы-
шает сорбируемость углеводородов: нарастание количества двойных
связей в молекуле углеводорода способствует росту сорбируемости
последнего.
5. Рост числа алифатических цепей и увеличение их длины во
всех случаях уменьшают сорбируемость ароматических углеводородов.
При хроматографическом разделении на флоридине [25], алюмосили-
кате или угле [26], окиси алюминия [27, 28] и других сорбентах законо-
мерности распределения углеводородов разных групп несколько варьи-
руют.
Для исследования углеводородных смесей необходимо получить их
в чистом виде, т. е. отделить от неуглеводородных компонентов — смол,
сернистых, азотистых и кислородных соединений. Многими исследова-
телями [10, 29—33] показано, что хроматографическим разделением на
силикагеле бензино-лигроиновых фракций нефти сравнительно легко
•отделить углеводородную часть от неуглеводородных примесей.
В масляных фракциях часть неуглеводородных компонентов с ярко
выраженной полярностью легко отделяется в хроматографической ко-
лонке вместе со смолами. Но в высококипящих фракциях нефтей при-
сутствуют сравнительно низкомолекулярные неуглеводородные компо-
ненты в виде азотистых, кислородных и сернистых соединений (суль-
фиды, тиофены, азотистые гетероциклы и др.). Эти соединения, по-види-
мому, присутствуют в нефтях частью в виде более или менее сложных
«ассоциатов» (Рубинштейн, Крейн и др.), сорбируемых нефтяными смо-
лами и полициклическими ароматическими углеводородами. Вероятно,
причина этого явления заключается в слабых взаимодействиях типа до-
норно-акцепторных с участием гетероатомов азота и серы.
Указанные «ассоциаты» частично разрушаются при хроматографии
на силикагеле, что приводит к выделению соответствующих гетероатом-
ных соединений совместно с углеводородами. При этом для однотипных
хроматографических фракций отмечалось несоответствие молекуляр-
ных масс гетероатомных компонентов молекулярным массам углеводо-
родов. Например, часть органических соединений серы выделяется сов-
местно с полициклическими ароматическими углеводородами с суще-
ственно большими молекулярными массами. Другая часть органических
соединений серы, по-видимому, более насыщенных, выделяется до аро-
матических углеводородов. Это создает особые трудности при хромато-
графии сернистых нефтепродуктов и объясняет противоречивость соот-
ветствующих литературных данных.
При хроматографическом разделении на силикагеле масляных фрак-
ций одним исследователям удавалось получить метано-нафтеновую
часть без сернистых соединений [34—36], а у других [37—39] и в мета-
29
но-нафтеновой части элементарный анализ показывал присутствие се-
ры. Но все исследователи утверждают, что с ароматическими углево-
дородами всегда десорбируется некоторое количество сернистых соеди-
нений и тем большее, чем выше цикличность выделенных ароматиче-
ских фракций.
Вероятно, при применении адсорбентов кислого характера, напри-
мер силикагеля, невозможно освободиться от неуглеводородных (в ос-
новном сернистых) примесей. При применении адсорбентов основного
характера намечается возможность получения чистых ароматических
углеводородов. Так, Шнейдер [40], исследуя нефти методом хроматогра-
фии на окиси алюминия (десорбенты — петролейный эфир и смесь трех-
хлористого этилена со спиртом), получил чистые ароматические угле-
водороды. Автор утверждает, что неуглеводородные примеси масляных,
фракций кислого характера обладают сильными адсорбционными свой-
ствами по отношению к основным адсорбентам (окиси алюминия) и по-
этому адсорбируются в первую очередь, не поддаются десорбции непо-
лярными десорбентами и сравнительно легко отделяются от углеводо-
родной части. Эти исследования подтверждены также и другими
авторами. Однако возможность отделения при помощи адсорбционной
хроматографии ароматических углеводородов от производных ряда тио-
фена, имеющих аналогичное строение, вызывает сомнение.
В каждом отдельном случае исследователи в зависимости от постав-
ленных задач подбирают условия опыта и методику исследования хро-
матографических фракций. В практике исследовательских работ очень
часто желательно вести исследования с малыми количествами вещества
либо из-за недостатка вещества, либо в целях экономии времени. В на-
стоящее время описаны методы хроматографического разделения не-
больших количеств углеводородных смесей [41—44]. Однако исходные
навески в описанных методах или относительно велики (от 500 до 1000
мг), или результаты анализа не дают той степени детальности и точно-
сти, которую можно получить, применяя ниже рассматриваемый вари-
ант микрометода.
И. СХЕМА ГРУППОВОГО АНАЛИЗА СМЕСЕЙ УГЛЕВОДОРОДОВ
(СРЕДНИЕ И ВЫСШИЕ ФРАКЦИИ НЕФТИ)
Предполагаемая схема группового микроанализа позволяет разде-
лить методом элюэнтной хроматографии смеси углеводородов на алка-
но-нафтеновую часть, ароматические углеводороды и фракцию, в кото-
рой в основном концентрируются гетероатомные соединения (кислород-
ные, сернистые и азотистые).
Метод применим к навескам исследуемой смеси от 10 до 100 мг. Та-
кие навески дают возможность брать большие весовые соотношения ве-
щество— адсорбент (равные 1:100 и более), значительно сохранять
время анализа и получать четкое деление. Навеску 10 мг можно разде-
лить на 20 микрофракций по 0,2—1,0 мг, а применение рефрактометра
для работы с малыми количествами (описание которого дается ниже)
позволяет в таких микрофракциях определять показатель преломления
и относительную дисперсию. В процессе анализа фиксируется объем'
растворителя, пошедшего на десорбцию каждой микрофракции (или
время выхода каждой микрофракции при постоянной скорости прохож-
дения десорбента).
По этим трем показателям все отобранные в одном опыте и взвешен-
ные микрофракции разбиваются сначала на две группы:.
1. Фракции, выделяемые в первой стадии элюирования, в которых
показатель преломления последовательно увеличивается, относятся к
углеводородным с примесью гетеро атомных соединений.'
30
2. Фракции, выделяемые во второй стадии элюирования, в которых,
показатель преломления последовательно уменьшается, относятся к ге-
тероатомным соединениям с примесью ароматических углеводородов..
А. Отнесение микрофракций к группам углеводородов
Эффективность определения группового состава в очень большой-
степени зависит от точности определения границ раздела групп углево-
дородов. Поэтому в схеме предусмотрено определение границы между
алкано-нафтеновыми и ароматическими углеводородами по двум ха-
рактеристикам: по значениям относительной дисперсии и по значениям;
объема растворителя, пошедшего на десорбцию микрофракций (без
учета значений показателя преломления).
К алкано-нафтеновым углеводородам относятся те микрофракции,
которые выходят из микрохроматографической колонки до резкого-
подъема значений объема растворителя, пошедшего на вымывание
микрофракции, и имеют значение относительной дисперсии меньше
19,0—19,5 единиц.
К ароматическим углеводородам относятся те микрофракции, кото-
рые выходят из микрохроматографической колонки после резкого подъ-
СХЕМА ГРУППОВОГО АНАЛИЗА МАСЛЯНОЙ ФРАКЦИИ НЕФТИ И БИТУМОВ
МАСЛЯНАЯ ФРАКЦИЯ
(масла + петролейноэфирные смолы)
i
i
Микрох ром атогр афическое
разделение на 15—20 микрофракций
4 1 I J- I I J- I J- J j
□ □□□ □ □ □□□□□□'
I 1 I J I
Определение по каждой микрофракцнн:
1) веса и выхода
2) объема растворителя, пошедшего на
десо рбцию
3) показателя преломления
4) относительной дисперсии
I 1 I I I
31
ема значений объема растворителя, пошедшего на вымывание микро-
*фракций, и имеют значение относительной дисперсии больше 19,0—19,Ъ
единиц.
Б. Отнесение микрофракций к подгруппам углеводородов
Излагаемая ниже методика применима к анализу сложной смеси
достаточно большого числа индивидуальных углеводородов (к широким
фракциям).
По данным рекомендуемого микрохроматографического анализа
возможно ориентировочно оценить строение углеводородов, входящих в
состав исследуемой смеси с началом кипения не ниже 250°С (см. схе-
му на стр. 31).
В табл. 1 приведены значения показателя преломления, по которым,
с учетом значений относительной дисперсии и объемов растворителя,
пошедшего на вымывание каждой микрофракции, можно отнести угле-
водородную часть исследуемой навески к следующим подгруппам уг-
леводородов:
1) алканы нормального или слаборазветвленного строения (если
.известно, что в исследуемой смеси отсутствуют заметные количества
твердых нафтеновых);
2) алканы изостроения;
3) нафтены моноциклические;
4) нафтены бициклические;
5) нафтены три- и полициклические;
6) ароматические моноциклические;
7) ароматические бициклические;
.8) ароматические три- и полициклические.
Таблица!
Пределы показателя преломления для ориентировочной оценки
углеводородов, входящих в состав нефтяных фракций с н.к.250° С и выше *
Подгруппа углеводородов т-г 20** Пределы пр
Алканы нормального или слабо- разветвленного строения Алканы изостроения Нафтены моноциклические Нафтены бициклические Все фракции, застывающие выше 4-20° (если в смеси нет заметных ко- личеств твердых нафтенов) От 1,45 до 1,47 От 1,47 до 1,48 От 1,48 до 1,49
Нафтены три- и полициклические От 1,49до значения «д, в области
которого происходит резкий подъем значений относительной дисперсии и объемов растворителя, пошедшего на вымывание микрофракции
Ароматические моноциклические От значении п^, в области которо-
Ароматические бициклические Ароматические три- и полицикли- ческие го происходит резкий подъем значе- ний относительной дисперсии и объе- мов растворителя, пошедшего на вымывание микрофракции, до 1,53 От 1,53 до 1,55 От 1,55 и выше
I.
* Для фракций с температурой выкипания 150—250° С пределы показателя пре-
ломления должны быть дополнительно уточнены.
** С учетом значений относительной дисперсии и объемов растворителя, пошед-
шего на вымывание каждой микрофракцин.
:32
III. РЕКОМЕНДУЕМАЯ МЕТОДИКА
Область, применения предлагаемого метода — разделение углеводо-
родных смесей с н.к. 150° С и выше в навесках от 10 до 100 мг в тече-
ние 1—2 рабочих дней.
Для разделения фракций с н.к. 250° С и выше возможно примене-
ние той же аппаратуры, но с существенно упрощенным вращающимся
столиком для отбора микрофракций. Подобрав соответствующие адсор-
бенты и десорбаты, микрометодом можно эффективно делить и неугле-
водородные смеси. Относительная быстрота эксперимента и малый рас-
ход исследуемого вещества позволяют рекомендовать предлагаемый
метод для подыскания условий разделения элюэнтной хроматографией
различных смесей.
Рекомендуемая техника микрохроматографического разделения вы-
годно отличается от ранее предложенных тем, что позволяет проводить
фракционировку на заранее заданное количество микрофракций, ори-
ентировочный вес которых определяется экспериментатором заранее
или в процессе работы. Последнее обстоятельство дает возможность
четко определять границы разделяющихся веществ.
Прежде чем перейти к обсуждению и обоснованию метода в целом,
дадим полное описание аппаратуры и экспериментальной техники.
А. Аппаратура, реактивы, манипуляции
Аппаратура для ми крохромато граф ич еског о
разделения фракций с началом кипения 150°С и выше
1. Прибор для микрохроматографии с микроректификационным
голиком (рис. 1).
Стеклянная колонка 5 внутренним диаметром 3—4 мм и длиной, в
зависимости от величины исходной навески, от 15 до 75 см (на нижнем
конце хороший шлиф). Кран 3 имеет риски для тонкой регулировки по-,
дачи десорбента. Азот перед подачей его в капилляры, предназначенные
для высушивания 6 и досушивания 7, очищают, пропуская его через
хромовую смесь, силикагель, прокаленный хлористый кальций и слой
ваты. Ток азота регулируют винтовыми зажимами. Размеры сосуда для
десорбента 1 и напорной трубки 2 не лимитированы.
На вращающемся приемном столике из органического стекла (пред-
ставлен отдельно на рис. 2) собраны микроректификационные ячейки с
горячей 7, нейтральной 6 и холодной 5 зонами. Внизу горячей зоны яче-
ек помещены одинаковые нагревательные спирали 8 из нихромовой
проволоки диаметром 0,4—0,5 мм и длиной 70—100 мм, на которые по-
дают регулируемый ток силой от 0 до 10 а при напряжении до 20 в
(проволоки нихромовых спиралей могут нагреваться до темно-красного
каления). В холодных зонах микроректификационных ячеек располо-
жены специальные каналы 14 для подвода сухого холодного газа с тем-
п^атурой примерно —25° С. Для его выхода из холодной зоны преду-
смотрены специальные отверстия 3 в закрывающихся заслонках. В ка-
честве охлаждающего газа используют СО2 из баллона с редуктором,
осушенный прокаленным хлористым кальцием или ангидроном. Высу-
шенный газ для охлаждения пропускают через две спиральные мед-
ные трубки 13 (см. рис. 1) внутренним диаметром 8 мм, длиной спи-
ральной части около 100 см. Обе спирали опущены в сосуд Дьюара с
сухим льдом. Одновременную подачу сухого холодного газа к охлаж-
даемой зоне двух ректификационных ячеек осуществляют через теп-
лоизолированные концы тех же самых медных трубок. Теплоизоляция
удобно осуществлена обклеиванием концов медных трубок полосами’
3 Методы анализа 33
пенопласта 14. Подача охлаждающего газа сразу в две ректифика-
ционные ячейки осуществлена с целью синхронизации удаления раст-
ворителя из отбираемой фракции и досушивания уже отобранной.
Такой прием позволяет осуществить непрерывный процесс элюирова-
ния продукта в стационарном режиме.
2. Микроприемники (20—25
штук) представляют собой запаян-
ные с одного конца отрезка тонко-
стенных трубочек с надетыми на них
кольцами из тефлона (рис. 3). Дно
микроприемников оттянуто в виде
небольшого конуса.
J--7
10
Рис. 1. Прибор для микрохроматографии
фракций с н.к. 150° С и выше
1 — сосуд для десорбента; 2 — напорная
трубка; 3 — трехходовой кран; / — шлиф; 5 —
микрохроматографическая колонка; 6 — капил-
ляр высушивания; 7 — капилляр досушивания;
8 — общая трубка для подачи сухого азота;
9 — микроректификационный столик; 10 — про-
вода низкого напряжения; 11 — понижающий
трансформатор с выключателями; 12 — трубки
подачи сухого углекислого газа; 13 — медиые
спиральные трубки; 14 — кожух из пенопласта;
15 — крышка сосуда Дьюара; 16 — штатив
Рис. 2. Вращающийся микроректифика-
ционнын столик для приема фракций
с н.к. 150° С и выше
1 — ножка; 2 — отверстие крепежного винта;
3 — отверстие в заслонке ректификационной
ячейки; 4— заслонка; 5 — холодная зона ячей-
ки; 6 — нейтральная зона ячейки; 7 — горячая
зона ячейки; 8 — нагревательная спираль;
9—‘11 — токопровода; 12 — микроприемник; 13 —
тефлоновое кольцо; 14— отверстие канала для
подвода холодного сухого углекислого газа
в холодную зону ячейки; 15 — запорный крю-
чок ячейки
3. Подставка для микроприемников (рис. 4) из любого листового
металла. Гнезда в подставке оцифрованы. Перед взвешиванием микро-
приемников подставку заземляют на 5—10 мин. для снятия электроста-
тического заряда.
4. Промывалки с грушами (3 штуки) (рис. 5).
5. Капилляры для внесения навесок.
6. Металлическая спица с зазубринкой на одном конце для внесе-
ния и извлечения ватного тампончика из микрохроматографической ко-
лонки. ; 3 1И
34
Рис 3. Микроприемники
для хроматографических
фракций с кип. 150° С
и выше
/ — микроприемннк; 2 — теф-
лоновое кольцо
Рис. 4. Подставка для
микроприемников
Рис. 5. Промывалка с грушей
Рис. 6. Прибор для микрохро-
матографии фракций с н. к.
250° С и выше
/—штатив; 2 — колонка; 3— на-
порная трубка; 4 — сосуд для де-
сорбеита; 5 — вращающийся столик
для микроприемников; 6 — капил-
ляры подачи азота; 7 — лупа;
8 — край регулировки подачи де-
сорбента; 9 — микропечь
Рис. 7. Вращающийся столик для приема
фракций с н. к. 250° С и выше
1— капилляр для подачи десорбата; 2— капилляр
для высушивания; 3 — капилляр для досушивания;
4 — микропечь; 5 — штатив; 6 — вращающийся сто-
лик для микроприемников; 7 — чистый запасной
мнкроприемиик; 8 — микроприемник, в который от-
бирается фракция; 9—мнкроприемиик, в котором
досушивается фракция; 19 — лупа
3
35
7. Пинцеты с пластмассовыми кончиками.
8. Баллон с сжатым азотом.
9. Баллон с сжатой углекислотой.
10. Редуктор газовый (2 штуки, для СОг иЫг).
И. Герметичная емкость на 2—5 л с двумя трубками для сушки уг-
лекислого газа под давлением примерно 0,5 атм.
13. Медные трубки длиной 1,5 м, внутренним диаметром 8 мм (2 шту-
ки). Медные трубки после предварительного отжига свивают в спирали
по размеру сосуда Дьюара, а выходные концы их длиной примерно
35 см теплоизолируют (см. рис. 1).
14. Микровесы с точностью взвешивания 0,00002 г.
15. ЛАТР-1 (2 штуки, присоединяют друг к другу последовательно)
для тонкой регулировки температуры нагревательных спиралей 8 (см.
рис. 2).
Упрощенная аппаратура
для микрохроматографического разделения
фракций с началом кипения 250° С и выше
1. Прибор для микрохроматографии (рис. 6) с вращающимся при-
емным столиком 5. Стеклянная колонка 2 внутренним диаметром 3—4 мм
и длиной в зависимости от величины навески от 15 до 75 см\ в нижнем
конце колонки хороший шлиф. Кран 8 имеет риски для более тонкой ре-
гулировки подачи десорбента. Азот перед подачей его в капилляры 6
очищают, пропуская через хромовую смесь, силикагель, прокален-
ный хлористый кальций и вату. Ток азота регулируют винтовыми зажи-
мами (на рисунке не показаны). Микропечь 9 с регулируемым нагревом
от 40 до 150° С. Размеры сосуда для десорбента 4 и напорной трубки 3
произвольны. Вращающийся приемный столик отдельно представлен на
рис. 7.
2. Микроприемники (20—25 штук) (рис. 8); размеры даны ориенти-
ровочно. Нижние концы микроприемников оттянуты на конус. Принятая
форма микроприемников — эвапораторов предотвращает «выползание»
углеводородов с началом кипения выше 250°С при отдуве растворителя.
Рис. 9. Подставка
с микроприемниками
для фракций, кипящих при
250° и выше
Рис. 8. Микроприемник
для отбора фракций
с н. к. 250° С и выше
3. Подставка для микроприемников (рис. 9) сделана из любого ли-
стового металла. Гнезда в подставке отмечены цифрами. Перед взвеши-
ванием приемников подставку для снятия электростатического заряда
заземляют на 5—10 мин.
4. Промывалка с грушами (3 штуки) (см. рис. 5).
5. Капилляры для внесения навесок.
6. Металлическая спица с зазубринкой на конце для внесения и из-
влечения ватных тампончиков из микрохроматографической колонки.
7. Пинцеты с пластмассовыми кончиками.
8. Микровесы с точностью взвешивания до 0,00002 г.
9. Редуктор газовый (для N2 или СО2).
10. Баллон с сжатым азотом или углекислым газом.
11. Микропечь 9 (см. рис. 6) и ЛАТР-2 для регулировки темпера-
туры.
Реактивы
1. Силикагель марки МСК, крупность помола 0,07—0,15 мм, актив-
ность по бензолу [45] не менее 10—12 мл. Чистота силикагеля проверя-
ется в холостом опыте. Через микрохроматографическую колонку с 1 г
силикагеля пропускают 10 мл спирто-бензольной смеси (1:1). Привес
приемника, после высушивания растворителя, не должен быть более
0,0002 г. Если привес приемника больше, то экстрагируют силикагель в
аппарате Сокслета спирто-бензольной смесью, повторяют экстракцию
петролейным эфиром, испаряют с силикагеля растворитель и кипятят
при постоянном перемешивании в многократно сменяемой дистиллиро-
ванной воде. После высушивания до воздушно-сухого состояния сили-
кагель, активируют нагреванием в сушильном шкафу в течение 5 час.
при температуре 180—230° С. В процессе очистки и при хранении сили-
кагель нужно предохранять от сорбции из воздуха различных веществ,
которые при последующем испытании на чистоту силикагеля вымывают-
ся растворителями и дают недопустимый привес.
2. Вата, проэкстрагированная спирто-бензольной смесью (1:1).
3. Пентан или петролейный эфир с пределами выкипания 30—50° С.
При анализе фракций, кипящих выше 250° С, допустимо применение
петролейного эфира с к. к. 90° С. Петролейный эфир предварительно
очищается моногидратом H2SO4 с последующей нейтрализацией, про-
мывкой и перегонкой.
4. Бензол криоскопический.
5. Спирто-бензольная смесь (1:1).
Все применяемые растворители не должны люминисцировать в ульт-
рофиолетовом свете, и после испарения 10 мл привес приемников, в ко-
торых проводилось испарение, не должен превышать 0,00002 г.
Сборка и разборка прибора
1. На штативе 16 (см. рис. 1) укрепляют емкость для десорбента
1 с напорной трубкой 2 и трехходовым краном 3.
2. Вставляют чистую сухую микрохроматографическую колонку 5 с
изогнутым капиллярным кончиком 1 (см. рис. 7).
3. Под капиллярный кончик микрохроматографической колонки под-
водят вращающийся отборный столик с вставленными микроприемни-
ками (см. рис. 7) так, чтобы конец капилляра находился в центре микро-
приемника и не доходил до его дна на 10—15 мм.
4. Укрепляют капилляр отдува 2 (см. рис. 7) азотом параллельно
концу капилляра микрохроматографической колонки, но на 3—4 мм
выше.
Параллельная установка двух длинных капилляров представляет
известные трудности. Для облегчения такой установки подводят воз-
можно ближе капилляр микрохроматографической колонки к капилля-
ру отдува' азотом, складывают их вместе и обматывают мягкой тонкой
проволочкой так, чтобы они плотно и без перекосов были прижаты друг
к другу (рис. 10, а, б). Естественно, в стекле возникнут напряжения, ко-
торые снимают прогреванием капилляров выше проволочки мягким пла-
менем спички (рис. 10, в). После остывания и удаления проволочки ка-
пилляры сохраняют правильное расположение (рис. 10, г).
5. Укрепляют второй капилляр подачи азота (капилляр досушива-
ния) в центре второго микроприемника. Опусканием, вращением и по-
37
следующим подыманием столи-
ка убеждаются в правильном
положении всех трех капилля-
ров при смене микроприем-
ников.
6. При анализе «легких»
фракций (с н. к. 150° С и вы-
ше) следует пользоваться от-
борным столиком с микрорек-
б тификационными ячейками
Рис. 10. Параллельная установка капилляров
(см. рис. 2), который может
быть использован без охлаж-
дающей системы при микро-
хроматографическом разделе-
нии более высоко кипящих
смесей. Разделение фракций,
кипящих выше 250° С, удобнее
проводить на упрощенном вра-
щающемся отборном столике
(см. рис. 7) с микропечкой 4.
При сборке аппаратуры
для работы с «легкими» фрак-
циями после установки капил-
ляров подводят и укрепляют
на отдельном штативе две параллельные медные трубки охлаждающей
системы 13, 14 (см. рис. 1), помещенные в сосуд Дьюара, и устанавлива-
ют их так, чтобы они без перекосов входили в соответствующие отвер-
стия 14 (см. рис. 2) отборного столика. При опускании, вращении и по-
следующем подымании столика трубки охлаждающей системы должны
входить в соответствующие отверстия всех других микроректификаци-
онных ячеек. После внесения в сосуд Дьюара твердой углекислоты и
соединения резиновыми шлангами медых трубок с осушающей емкостью
и редуктором на баллоне сжатого газа охлаждающая система готова к
работе.
7. Наливают в сосуд для десорбента 1 (см. рис. 1) пентан или очи-
щенный петролейный эфир и заполняют им всю напорную трубку до
шлифа микроколонки.
8. Снимают микроколонку, вносят в нее маленький комочек экстра-
гированной ваты и проталкивают его металлической спицей к капилляр-
ному концу. Ватка должна прочно держаться в микроколонке, но не
создавать большого сопротивления прохождению растворителей. Пере-
вернув микроколонку вверх шлифом, заполняют ее заданным количест-
вом силикагеля при непрерывном постукивании по колонке полуваку-
умной резиновой трубкой для равномерного уплотнения адсорбента.
Вторым ватным тампоном из экстрагированной ваты закрепляют сили-
кагель в колонке.
9. Перед внесением в микрохроматографическую колонку навески
следует снять с силикагеля теплоту смачивания. Для этого в перевер-
нутую вверх шлифом микрохроматографическую колонку маленькими
порциями заливают петролейный эфир до тех пор, пока весь силика-
гель будет смочен.
Разборка прибора после анализа
1. Повернув трехходовой кран 3 (см. рис. 1), выливают раствори-
тель из сосудов 1 и напорной трубки 2.
2. Вынимают пробку трехходового крана.
3. Снимают микрохроматографическую колонку и ее шлиф присое-
38
диняют к шлангу водоструйного насоса. Отсасывают растворитель и
высушивают силикагель под разрежением, после чего вынимают ват-
ные тампоны, а силикагель легко высыпается из микроколонки. Тре-
тий ватный тампон вытаскивают зазубренным концом металлической
спицы.
Если в процессе анализа или при разборке прибора сломался капил-
лярный кончик микрохроматографической колонки, то его легко можно
сделать собственными силами. Для этого в небольшом остром пламе-
ни стеклодувной горелки оттягивают длинный толстостенный капилляр
Рис. 11. Изгибание капиллярного кончика микроколонки
и формируют его кончик. Закрепляют микроколонку в штатив и нагре-
вают на маленьком мягком пламени капилляр у самой микроколонки.
Под собственным весом капилляр сгибается под нужным углом. Закреп-
ляя микроколонку в разных положениях и нагревая капилляр мягким
пламенем (можно даже от спички), можно легко получить желаемый
изгиб капиллярного кончика микроколонки (рис. 11).
Б. Процесс работы при разделении фракции
Взятие навески
Оптимальная величина исходной навески исследуемого вещества
равна 25—35 мг. Силикагеля рекомендуется брать соответственно 2,5—
3,5 г.
Навеску вещества берут на микроаналитических весах с точностью
до 0,00002 г по разности.
Жидкие и легкоподвижные продукты берут пипеткой с длинным ка-
пиллярным концом.
Вязкие и парафинистые продукты лучше предварительно подогреть
на водяной бане и взять пипеткой с толстым капиллярным кончиком.
Подогретое вещество в пипетке перед взвешиванием надо выдержать
около весов 10—15 мин. до полного выравнивания температуры, взве-
сить, подогреть уже всю пипетку и внести вещество в микроколонку.
Повторное взвешивание пипетки с остатком вещества также следует
проводить только после выдерживания у весов.
Тугоплавкий продукт, который взять пипеткой не удается, следует
брать маленьким шпательком.
Во всех случаях взятия навески надо тщательно следить за тем, что-
бы вещество было внесено в глубину микроколонки, перевернутой вверх
шлифом, желательно прямо на ватный тампон.
Избегать размазывания навески по стенкам микроколонки! (Если
часть навески попала на наружную сторону шлифа микроколонки, то
опыт испорчен.)
После внесения навески смывают возможные следы вещества со сте-
нок микроколонки 2—3 каплями петролейного эфира из промывалки
39
(см. рис. 5) с очень тонко оттянутым носиком. Раствору дают впитать-
ся в силикагель й повторяют смывание 2—3 раза. Когда весь раствор
впитается, на ватный тампон вносят 0,5 мл свежего силикагеля и за-
крывают вторым ватным тампоном. Последняя операция необходима
для предотвращения возможного проникновения части навески в раст-
воритель, когда микроколонка будет поставлена на место.
Установление скорости прохождения растворителя
через микрохроматогр афическую колонку
Микрохроматографическую колонку с внесенной в нее навеской ис-
следуемого вещества перевертывают вниз шлифом и ставят (см. рис. 1)
так, чтобы изогнутый капиллярный кончик находился в центре микро-
приемника на высоте 10—15 мм от дна, а капилляр отдува азотом был
параллелен и располагался выше на 3—4 мм (см. рис. 7).
После закрепления микроколонки постепенно открывают трехходо-
вой кран и добиваются прохождения десорбента через колонку со ско-
ростью 0,10—0,05 мл/мин, что визуально соответствует 5—6 каплям чи-
стого петролейного эфира при тонко оттянутом кончике.
В самом начале десорбции из изогнутого капиллярного кончика мик-
роколонки поступает вытесняемый воздух, поэтому трудно судить о
скорости поступления десорбента в колонку. В этом случае рекомен-
дуется наполнить запасной микроприемник петролейным эфиром на '/з
высоты, опустить в него носик микроколонки и наблюдать выделение
пузырьков воздуха из капиллярного кончика. По скорости выделения
пузырьков можно оценить скорость поступления растворителя в микро-
колонку.
Время прохождения фронта углеводородов через всю высоту мик-
рсколонки может быть рекомендовано порядка 25—40 мин. Более быст-
рое прохождение фронта углеводородов отрицательно сказывается на
четкости разделения, особенно в первых микрофракциях.
После вытеснения воздуха из микроколонки через изогнутый капил-
ляр поступает чистый растворитель. Опусканием, поворотом и после-
дующим подъемом вращающегося отборного столика (см. рис. 2) подво-
дят чистый тарированный микроприемник к концу изогнутого капил-
ляра микроколонки и окончательно устанавливают трехходовым кра-
ном рекомендованную скорость десорбента (0,10—0,05 мл {мин) или не-
много меньшую.
Трехходовой кран до конца вымывания всей метано-нафтеновой
фракции и части ароматических углеводородов больше регулировать
нельзя!
Десорбция и улавливание метано-нафтеновых
углеводородов
а) Улавливание фракций с т. кип. 150° С и выше
При анализе «легких» фракций углеводородов (с началом кипения
не ниже 150°) необходимо провести микроректификационную отгонку
растворителя, не потеряв при этом выделенных микрофракций. Для
этого пускают ток охлаждающего газа через установленные медные
трубки 8 (см. рис. 1) и одновременно включают микропечки 8 горячей
зоны (см. рис. 2) работающей ректификационной ячейки. Нагрев и ох-
лаждение регулируют так, чтобы слабый ток азота из капилляра высу-
шивания 6 (см. рис. 1) способствовал удалению всех паров растворите-
ля, а микрофракция, выделяясь в холодной зоне, стекала вниз по стен-
кам микроприемника.
Поступление следующей капли элюата в микроприемник должно
произойти после удаления паров растворителя предыдущей капли.
40
При правильно подобранном режиме, через 15—20 мин. непрерывно-
го элюирования наступает момент, когда одна капля элюата высохла
в микроприемнике и не оставила (как все предыдущие) никаких сле-
дов, а высыхание следующей капли дало на стенках, главным образом
в холодной зоне, заметную пленку или кольцо углеводородов, которые
не испаряются до падения следующей капли. Поступление первых пор-
ций десорбированных углеводородов очень четко фиксирует момент
начала выделения микрофракций, а непрерывное удаление растворите-
ля позволяет приближенно, на глаз, определять количество накопив-
шихся углеводородов в микроприемнике с точностью 0,5—1,0 мг.
Последнее обстоятельство дает возможность разделить навеску ис-
следуемого вещества на заранее заданное количество микрофракций
десорбата, ориентировочный вес которых может быть задан экспери-
ментатору или установлен им самим в процессе анализа.
После отбора первой микрофракции рекомендуется смыть следы уг-
леводородов с капилляров одной каплей петролейного эфира из промы-
валки (см. рис. 5) и поворотом отборного столика (см. рис. 2) быстро
подставить под носик микроколонки микроприемник следующей ректи-
фикационной ячейки, одновременно зафиксировав время смены микро-
приемников.
Пока отбирается вторая микрофракция, первый микроприемник до-
сушивается. Для досушивания «легких» фракций опять применяют ме-
тод микроректификационной отгонки. При включенной микропечке 8
(см. рис. 2) пускают ток охлаждающего газа и ток азота в капилляр
досушивания 7 (см. рис. 1) такой интенсивности, чтобы пары раствори-
теля были удалены из микроприемника, а выделенные углеводороды
остались.
«Легкие» микрофракции при стоянии могут «выползти» из микро-
приемников, если в них остался растворитель. Поэтому микрофракции
надо по возможности быстрее досушивать. В связи с этим рекоменду-
ется в один микроприемник отбирать не более 4—5 мг, так как из боль-
ших микрофракций труднее отогнать следы растворителя.
б) Улавливание фракций с температурой кипения , 250° С и выше
При микрохроматографическом разделении «тяжелых» смесей угле-
водородов (фракций с температурой кипения 250° и выше) охлаждение
микроприемников применять не следует.
Для удаления растворителя из микроприемника пускают в капилляр
отдува слабый ток чистого сухого азота, а в случае очень большого ох-
лаждения микроприемника, когда он покрывается росой или даже ине-
ем, включают нагрев микропечки.
При анализе «тяжелых» фракций скорость поступления элюата из
микроколонки, ток азота из капилляра досушивания и нагрев микро-
печки регулируют так, чтобы капля десорбата, упавшая на дно микро-
приемника, под действием струи азота растекалась бы без брызг по
стенкам и высыхала, не оставляя никаких видимых следов. После вы-
сыхания первой капли, через 1—2 сек. должна упасть вторая капля
и т. д.
Как и в предыдущем случае, если скорость подачи растворителя вы-
держана правильно, то через 15—25 мин. непрерывного наблюдения
экспериментатор сможет уловить момент, когда одна капля высохла в
микроприемнике и не оставила (как и все предыдущие) никаких сле-
дов, а высыхание следующей капли дало на стенках ясно заметную
пленку или кольцо углеводородов, которые не испаряются до падения
следующей капли. Такой прием позволяет очень четко определить нача-
ло поступления первых порций десорбированных углеводородов и за-
фиксировать время начала выделения микрофракций.
Непрерывное удаление растворителя из микроприемника с одно-
временным медленным поступлением в него десорбата позволяет опре-
41
делить на глаз количество накопившихся в микроприемнике углеводо-
родов с точностью 0,2—0,5 мг.
Последнее обстоятельство дает возможность разделить навеску
исследуемого вещества на заранее заданное количество микрофракций,
ориентировочный вес которых также может быть заранее задан экспе-
риментатору или установлен им самим в процессе анализа.
После отбора первой микрофракции рекомендуется смыть следы
углеводородов с капилляров одной каплей петролейного эфира из про-
мывалки (см. рис. 5) и поворотом столика (см. рис. 7) быстро подста-
вить под носик микроколонки микроприемник следующей ячейки, одно-
временно зафиксировав время смены микроприемников. Пока отбирает-
ся вторая микрофракция, в первый микроприемник по капилляру
досушивания 3 (см. рис. 7) подают ток азота.
При разделении «тяжелых» углеводородных смесей ток азота из
капилляра досушивания должен быть такой, чтобы обеспечить распре-
деление всей микрофракции тонким слоем (без брызг) по стенкам
микроприемника для интенсификации удаления растворителя. Микро-
фракции, в которых остался растворитель, надо возможно быстрее
досушивать, в противном случае углеводороды с растворителем неизбеж-
но «выползут» по стенкам микроприемника.
В связи с этим рекомендуется отбирать в один микроприемник не
более 3—4 мг, так как большие микрофракции трудно быстро высушить.
в) Окончание десорбции метано-нафтеновых углеводородов
Концентрация углеводородов в элюате за время хроматографирова-
ния обычно не остается постоянной. При микрохроматографировании
минеральных масел первые порции элюата содержат относительно
много углеводородов. Поэтому при отборе в первые 3—4 микроприем-
ника испарение двух-трех капель элюата дает 0,2—0,7 мг углеводо-
родов. Затем концентрация углеводородов в фильтрате постепенно
падает, и соответственно количество петролейного эфира, которое необ-
ходимо испарить, чтобы набрать 0,5—1,0 мг микрофракции, возрастает.
При десорбции последних порций метано-нафтеновых углеводородов
накапливание 0,2—0,3 мг микрофракции занимает более 15 мин. Затем
наступает период, когда поступление углеводородов с элюатом практи-
чески прекращается на довольно большой промежуток времени — 20—
50 мин. Это явление перерыва в поступлении углеводородов хорошо
заметно и четко фиксируется при регистрации времени смены микро-
приемников. После некоторого промежутка времени углеводороды опять
начинают поступать с элюэнтом, но гораздо медленнее, чем в первый
период. После перерыва отбирают еще 2—3 микрофракции по 0,2—
0,4 мг, на этом десорбцию метано-нафтеновых (и самых первых арома-
тических) фракций заканчивают.
Выделение фракции ароматических углеводородов
и гетероатомных соединений
Оставшиеся в микрохроматографической колонке углеводороды как
в случае анализа «легких», так и в случае анализа «тяжелых» фракций
могут быть десорбированы петролейным эфиром, но дальнейший про-
цесс вымывания будет происходить крайне медленно, что связано с очень
большой затратой времени. Для ускорения процесса элюирования не-
обходимо заменить петролейный эфир на бензол. Для этого через трех-
ходовой кран нужно слить петролейный эфир из сосуда для элюэнта и
напорной трубки и заполнить их бензолом.
Скорость прохождения бензола через микрохроматографическую
колонку регулируют так, чтобы скорость отгона растворителя на от-
борном столике (при работе с «легкими» фракциями) или отдув его
азотом (при работе с «тяжелыми» смесями) соответствовала поступле-
42
нию новых порций элюэнта и можно было бы следить за количеством
накапливающейся микрофракции. Естественно, что отобранные микро-
фракции должны быть досушены так же, как при отборе метано-нафте-
новой части анализируемой смеси. Регистрация времени смены микро-
приемников уже не является обязательной, и на этой стадии анализа
вполне допустимо многократное регулирование скорости потока бензола
вплоть до полного его прекращения.
Бензол, проходя через силикагель, десорбирует часть ароматических
углеводородов, которые вместе с растворителем попадают в очередной
микроприемник. Удаляя растворитель, наблюдают накапливание микро-
фракции и меняют приемники поворотом столика. С течением времени
интенсивность выделения микрофракций бензолом сильно уменьшается,
тогда таким же приемом заменяют бензол на спирто-бензольную
смесь (1 : 1).
Уже первые порции спирто-бензольной смеси десорбируют все остав-
шиеся ароматические углеводороды и большую часть гетероатомных
соединений. Это заметно по движению вдоль микрохроматографической
колонки (снизу вверх) окрашенного кольца. Когда это кольцо подойдет
к капиллярному кончику микрохроматографической колонки, следует
почаще менять микроприемники, чтобы впоследствии точнее уловить
границу, где кончаются ароматические углеводороды (с примесью гете-
роатомных соединений) и начинаются гетероатомные соединения (с при-
месью ароматических углеводородов).
Опыт считают законченным тогда, когда после вымывания спирто-
бензольной смесью интенсивно окрашенных гетероатомных соединений
в очередном микроприемнике испарение 2—5 мл элюэнта не дает замет-
ных на глаз количеств микрофракции.
В. Табличное и графическое оформление результатов анализа
(на примере микрохроматографического разделения масляной фракции
из современных морских осадков Берингова моря)
Для более наглядного пояснения процесса анализа, порядка измеря-
емых величин, форм записей и расчетов, а также графического оформле-
ния результатов целесообразно привести конкретный числовой пример
микрохроматографического анализа. В качестве такого конкретного
примера дается полное описание хода всего анализа масляной фракции,
выделенной из современных морских осадков Берингова моря. Образцы
выделены О. К. Бордовским из донных проб, отобранных во время
экспедиции 1950—1951 гг. на «Витязе».
Перед анализом подготовлен прибор (см. рис. 6) с чистой микрохро-
матографической колонкой 2, три промывалки (см. рис. 5) с петролейным
эфиром, бензолом, спирто-бензольной смесью (1 : 1), силикагель, экстра-
гированная вата, пипетка с тонко оттянутым кончиком для взятия навес-
ки, набор тарированных микроприемников на подставке (см. рис. 9) и
два листа бумаги, разграфленные по форме табл. 2 и 3. Графа 3 табл. 2
заполнена результатами взвешивания при тарировании микроприем-
ников.
Большое число выделяемых микрофракций, каждая из которых
впоследствии характеризуется множеством цифровых показателей, за-
ставляет во избежание путаницы вести регистрацию получаемых данных
по заранее составленным табличным формам. Характер эксперименталь-
ной работы с микроколичествами при большом числе маленьких микро-
приемников не позволяет применять громоздкие лабораторные журна-
лы для записей. Поэтому для микрохроматографического анализа
рекомендуется пользоваться небольшими табличными формами (см.
табл. 2 и 3) на отдельных листках, которые дл^/удобства можно
43
Таблица 2
Анализ масляной фракции, выделенной из современных осадков Берингова моря
№ гнезда 1 № на микро- приемнике Вес микроприемников, мг Выход микро- фракции Время смены микроприемни- ка, мин. Объем раствори- теля, мл
до опыта после опыта мг нарастаю- щим ито- гом
мг вес. %
1 2 3 4 5 6 7 8 9
Растворитель — петролейный эфир
1 20 Р —33, Ш + 1, НТ —0 Р —45, Ш—1, НТ —1 1,19 1,19 2,2 10 0,50
2 36 10, Р—17, Ш + 2, НТО 10, Р —41, Ш + 4, НТ —2 2,44 3,63 6,6 14 0,70
3 9 10, Р—46, Ш + 2, НТО 10, Р —85, Ш 0, НТ + 1 3,87 7,56 13,7 17 0,85
4 37 100, Р —46, Ш —3, НТ + 1 100, Р —100, Ш + 4, НТ + 1 5,47 13,03 23,6 20 1,00
5 15 100,5, Р—11, Ш + 3, НТ + 1 100, 5, Р —90, Ш-5, НТ 0 7,73 20,76 37,6 24 1,20
6 34 100,5, Р—12, Ш + 1, НТ —1 100, 5, Р —84, Ш—2, НТ + 1 7,19 27,95 50,6 28 1,40
7 26 100,50, 5, Р —30, Ш+2, НТ —1 100, 50, 5, Р —93, Ш —5, НТ + 1 6,21 34,16 61,8 31 1,55
8 35 100,50, 10, Р—4, Ш + 5, НТ 0 100, 50, 10, 5, Р-59, Ш + 1, НТ 0 10,46 44,62 80,7 42 2,10
9 33 100,50, 20, Р — 5, Ш — 1, НТ 0 100, 50, 20, Р — 24, Ш — 2, НТ 0 1,89 46,51 84,1 60 3,00
10 13 200,10, Р — 10, Ш + 2, НТ—1 200, 10, Р —15, Ш—1, НТ 0 Растворитель — бензол 0,46 46,97 85,0 105 5,25
11 2 200,10, Р — 12, Ш + 5, НТ 0 200, 10, Р —12, Ш + 4, НТ —2 0,01 — •— 200 10,00
12 1 200,50, Р —47, Ш —3, НТ 0 200, 50, Р—58, Ш 0, НТ 0 1,13 48,10 87,0 — —
13 17 200,50, 5, Р —12, Ш —3, НТ 0 200, 50, 5, Р — 38, Ш+ 1, НТ 0 2,64 50,74 91,8 — —
14 И 200,50, 10, Р — 46, Ш + 4, НТ 0 200, 50, 10, Р — 57, Ш + 4, НТ 2 1,12 51,86 93,8 — *-*
15 5 200,200, Р — 6, Ш + 4, НТ 0 200, 200, Р —15, Ш+1, НТ 0 0,87 52,73 95,4 — —
16 8 200,200, Р—13, Ш + 3, НТ 0 200, 200, Р —19, Ш +1, НТ + 1 0,57 53,30 96,4 — —-
17 19 500,Р —18, Ш —3, НТ 0 500,Р — 24,Ш+4,НТ + 2 0,65 53,95 97,6 — —‘
18 50 500,Р—27, Ш + 2, НТ 0 500, Р — 27, Ш + 4, НТ + 1 0,01 — — — —
Примечание. Р — показание рейтера, Ш — показание шкалы, НТ — показание нулевой точ-
ки.
Расчет результатов анализа;
Взято масляной фракции . . . 55,29 мг
Взято силикагеля.............. 5,5 г
вещество
Весовое соотношение ———
силикагель
1
100
Скорость пропус-
кания петролей-
ного эфира . . 0,05 мг/мин
Выход масляной
фракции .... 53,95 мг (97,6 вес.%)
Потери.............1,34 мг (2,4 вес.%)
44
прикрепить на жесткий картон соответствующего размера. Когда анализ
закончен, эти заполненные формы вместе с графиком сброшюровы-
ваются в специальный лабораторный журнал.
В перевернутую вверх шлифом микрохроматографическую колонку
при равномерном уплотнении засыпали 5,5 г силикагеля и закрыли его
ватным тампончиком. После снятия теплоты активации на верхнюю
ватку пипеткой наносили 55,29 мг масляной фракции. Вес масляной
фракции определили по разности веса пипетки с веществом и веса пи-
петки с остатками вещества, о чем внизу табл. 2 сделали соответствую-
щую запись и произвели расчет весового соотношения вещество—сили-
кагель (1:100). После впитывания всей навески в колонку засыпали
небольшое количество свежего силикагеля и закрепили его ватным там-
пончиком. Сосуд для десорбента 4 (см. рис. 6) и напорную трубку 3 до
шлифа заполнили петролейный эфиром. Микрохроматографическую ко-
лонку 2 перевернули вниз шлифом и поставили на место (см. рис. 6).
Краном 8 отрегулировали скорость прохождения петролейного эфира
примерно 0,05 мл/мин и записали эту скорость внизу табл. 2. Пустили
ток азота в капилляр высушивания 6 и включили нагрев микропечки 9.
Десорбент по каплям поступал на дно микроприемника и спокойно, без
вскипания, испарялся и отдувался азотом. Следующая капля десорбента
падала уже в сухой микроприемник. После появления первых признаков
поступления углеводородов в микроприемник гнезда 1 (номер на микро-
приемнике 20) включили секундомер, по которому отмечали в графе 8
табл. 2 время смены этого и следующих приемников. После смены деся-
ти микроприемников отбор фракции в микроприемник гнезда И (номер
на микроприемнике 2) продолжался 95 мин., а количество углеводородов
в этом микроприемнике, определенное на глаз, оставалось ничтожно
малым. Поэтому в этом опыте после смены микроприемника гнезда 11
петролейный эфир был заменен на бензол, а после микроприемника гнез-
да 15 (номер на микроприемнике 5) —на спирто-бензол. После замены
микроприемника гнезда 11 (после смены петролейного эфира на
бензол) секундомер за ненадобностью был остановлен, дальнейшая
регистрация времени смены микроприемников не производилась. После
остановки секундомера немного подрегулировали кран 8 (рис. 6),
увеличили нагрев микропечки 9 и прибавили поток высушивающего газа
для лучшего испарения бензола. В микроприемник гнезда 17 (номер на
микроприемнике 19) десорбат собирался и высушивался более 20 мин.
Видимой на глаз микрофракции очень мало, поэтому опыт закон-
чили на отборе микроприемника гнезда 18 (номер на микроприемнике
50).
После опыта досушенные микроприемники взвесили, результат взве-
шивания в виде перечисления гирь, положения рейтера, показания
шкалы и нулевой точки записали в графу 4 табл. 2, а вычисленный по
разности «до опыта» и «после опыта» вес полученных микрофракций
внесли в графу 5. В графе 6 вес ^выделенных микрофракций в милли-
граммах внесен нарастающим итогом сверху вниз, а в графе 7 — в весо-
вых процентах. Время смены микроприемников, умноженное на скорость
прохождения петролейного эфира через микрохроматографическую ко-
лонку (~0,05 мл/мин), перевели в объем растворителя (графа 9), по-
шедшего на десорбцию каждой микрофракции (нарастающим итогом).
Внизу табл. 2 подсчитали весовой процент десорбированной части
навески, потери в миллиграммах и в весовых процентах.
Далее в табл. 3 заполнили графы 1 и 2. Из каждого микроприемника
тонко оттянутым капиллярным кончиком пипетки отбирали часть микро-
фракции и переносили ее на заранее отмеченное место линзы рефракто-
метра, переделанного для работы с малыми количествами вещества.
Часть выделенных микрофракций оказалась застывшей, измерение nD
и Z на рефрактометре проводили при повышенной температуре. Резуль-
45
Таблица 3
Анализ масляной фракции, выделенной из современных морских осадков Берингова моря
№ гнезда Ne на при- емнике Показатель прелом- ления без поправки „t nD Показ атель преломления с поправкой „20 nD Определение величины Z Относительная дисперсия 20 WFC
+ Z — Z ^ср
1 2 3 4 5 6 7 8
1 20 1,4695 (37,5° С) 1,4771 41,2—41,5—41,2—41,5—41,6 41,8—41,9—41,7—41,8—41,8 41,6 17,0
2 36 1,4704 (36,5° С) 1,4779 41,3-41,5—41,4—41,3—41,4 41,7—41,9—41,8—41,8—41,8 41.6 17,0
3 9 1,4420 (36,0° С) 1,4595 41,3—41,4—41,5—41,6—41,8 41,8—41,8—41,7—41,7—41,8 41,6 17,0
4 37 1,4580 (35,0° С) 1,4640 41,3—41,5—41,4—41,4—41,4 41,9—41,8—41,7—41,7—41,9 41,6 17,0
5 15 1,4580 (35,0° С) 1,4640 41,4—41,6—41,5—41,4—41,4 41,9—41,7—41,8—41,8—41,8 41,6 17,0
6 34 1,4632 (31,0° С) 1,4676 41,4—41,3—41,2—41,4—41,4 42,0-41,8—41,9—41,6—41,7 41,6 1740
7 26 1,4734 (21,0° С) 0,4738 41,5—41,4—41,4—41,3—41,4 41,8—41,8—41,8—41,7—41,9 41,6 17,0
8 35 1,4890 4 1,4890 41,0—41,5—41,6—41,5—41,4 41,6—41,9—41,9—41,8—41.8 41,8 17,0
9 33 1,5070 1,5070 41,4—41,5—41,4—41,3—41,4 42,0—41,9—41,5—41,8—41,8 41,8 17,0
10 13 1,4950 1.4950 39.5—39.8—39.5—39.6—39.6 40.8—40.0—40.4—40.4 40,0 21,0
11 2 —» — —
12 1 1,5000 1,5000 39,4—39,8—39,6—39,5—39,7 40,2—40,8—40,2—40,4—40,4 40,0 21,1
13 17 1,5391 ] (20,0° С) 1,5000 37,5—35,8—35,5—35,4—35,6 36,4—36,5—36,4—36,3—36,4 36,0 25,0
14 11 1,5490 1,5490 34,6—34,4—34,8—34,6 —34,6 35,4—35,8—35,4—35,0—35,4 35,0 25,0
15 5 1,5482 1,5482
16 8 1,5410 1,5410
17 19 1,5242 1,5242
18 50 —* —*
таты измерения nJ записывали в графу 3 табл. 3, причем у значений
nJ в скобках ставили температуру линз рефрактометра. Пересчитан-
ные при помощи табл. 4 значения nD20 записали в табл. 3, графу 4.
Таблица 4
Температурные поправки показателя преломления для смесей алкано-
нафтеновых углеводородов [59]
Пределы выкипа- ния фракции, °C Поправка показателя преломления на 1° С Пределы выкипания фракции, °C Поправка показателя преломления на 1° С
150-200 0,00042 350-400 0,00038
200—250 0,00040 400—450 0,00036
250—300 0,00039 450—500 0,00035
300—350 0,00039 500—550 0,00035
Определение числа Z (отсчет по лимбу компенсатора) проводили 5 раз
вправо от нуля, 5 раз влево (табл. 3, графы 5 и 6), выведенное среднее
значение Zcp внесли в графу 7. Величина Z мало зависит от температу-
ры определения, поэтому никаких температурных поправок вносить не
надо. Значения Zcp определяли только в микрофракциях, десорбирован-
ных петролейным эфиром и бензолом, так как микрофракции, десорби-
рованные спирто-бензолом, были окрашены и Z в них определяется с
большой ошибкой.
Из табл. 3 по величинам nD20 (графа 4) и Zop (графа 7) при помощи
графика определили значения относительной дисперсии ® каждой
микрофракции, десорбированной петролейным эфиром и бензолом. Най-
денные значения со внесли в графу 8, табл. 3 (график со см. на стр. 204).
Графическое оформление результатов анализа проводилось согласно
рис. 12. Для построения кривой nJ0— весовой процент взяты данные из
табл. 2 графы 7 и табл. 3 графы 4; для кривой со — весовой процент —
данные из табл. 2 графы 7 и табл. 3 графы 8; для кривой v — весовой
процент — данные из табл. 2 графы 7 и 8.
Расчет группового состава
По одновременному подъему кривых со — весовой процент и v —
весовой процент (см. рис. 12) определен микроприемник, в который
десорбировалась последняя метано-нафтеновая фракция. Это оказался
микроприемник в гнезде 9 (номер на приемнике 33). По снижению кри-
вой nD20 — весовой процент установили, что последние ароматические
углеводороды были уловлены в микроприемник гнезда 15 (номер на
приемнике 5). Следовательно, по данным рис. 1 и табл. 2 (графа 7)
устанавливаем, что в анализируемой масляной фракции определены
следующие группы соединений (в вес. %):
Метано-нафтеновые углеводороды......... 84,1
Ароматические углеводороды.............. 11,3
Гетероатомные соединения................. 2,1
Потери................................... 2.5
Всего. . . . 100,0
Ориентировочная оценка подгрупп углеводородов
На графике рис. 12 проводим пунктирные горизонтальные линии по
пределам показателя преломления, взятым из табл. 1. Поскольку
в исследуемом образце совершенно неизвестно наличие или отсутствие
47
Рис. 12. Десорбционные кривые микрохроматографического
разделения масел современных морских осадков
I — метановые; II — нафтеновые моноциклические; III — нафтеновые би-
циклические; IV ароматические моноциклические; V — ароматические
бициклические; VI — гетеросоедднеиия
твердых нафтеновых углеводородов, все фракции, имеющие nD20 1,47
или ниже, относим к метановым углеводородам без разбивки их на
нормальные и изостроения.
Далее на основании остальных пределов «о20 ориентировочно опреде-
ляем содержание (в вес. %) моноциклических, бициклических, три- и
полициклических нафтенов, в случае необходимости применяя графиче-
скую интерполяцию. Границей конца три- и полициклических нафтенов
можно считать микроприемник в гнезде 9 (номер на приемнике 33)
(см. рис. 12).
Область ароматических углеводородов также разбивается на под-
группы по значениям nD20 (см. табл. 1).
Результат такой ориентировочной оценки масел из современных
морских осадков Берингова моря с проверкой их спектральными мето-
дами приведен в табл. 14 и 15 (спектральное исследование микрофрак-
ций проведено в нашей лаборатории Б. А. Смирновым, см. стр.131—157).
IV. ОБОСНОВАНИЕ МЕТОДА
А. Обоснование способа интерпретации результатов
микрохроматографического анализа для определения группового
состава
Рекомендуемая методика микрохроматографического анализа дает возможность
из небольшой навески получить 15—20 микрофракций, в которых показатель прелом-
ления обычно сначала последовательно увеличивается до какой-то определенной вели-
чины, а затем начинает уменьшаться. Во многих методах анализа [46, 47], использую-
щих хроматографическое деление на силикагеле, принимается, что фракции, имеющие
показатель преломления ниже 1,49, относятся к метано-нафтеновым углеводородам.
Это не совсем правильно, так как в настоящее время известно, что полициклические
нафтеновые углеводороды, присутствующие, например, в высококипящих фракциях
эмбенских и бакинских нефтей, содержащих примесь сернистых соединений, имеют
48
значение выше 1,49 (около 1,4931; 1,4962; 1,4970). Фракция нафтеновых угле-
водородов дистиллята ильской иефти, выкипающего в пределах 420—470° С, имеет
значения от 1,4970 до 1,5041. На этом основании некоторые авторы [48] считают,
что изменение показателя преломления при хроматографии не может быть однознач-
ным критерием для установления границы между нафтеновыми и ароматическими
углеводородами.
В качестве критерия для установления границы между метано-нафтеновыми и аро-
матическими углеводородами может быть принято не условное значение показателя
преломления, а величина относительной дисперсии. Значение относительной диспер-
t — пс
сии ®fcd —5--------- Ю3, как указывает Иоффе [49], для легких аро-
—1
матических углеводородов колеблется в пределах 29—33 единиц. Легкие метановые и
нафтеновые углеводороды имеют значение относительной дисперсии, примерно одина-
ковое и равное 17,5 единиц. Для тяжелых нафтеновых углеводородов, выделенных из
высококипящих фракций различных нефтей, значения относительной дисперсии ко-
леблются в пределах 18—19 единиц, как видно из табл. 2, 3 и 5. Из этих же таблиц
видно, что все ароматические углеводороды имеют значения относительной дисперсии
значительно больше 20 единиц.
При большом весовом соотношении вещество — силикагель (1:100 и более) по-
падание сернистых соединений в метано-нафтеновую часть маловероятно. При хрома-
тографическом разделении ряд сернистых соединений десорбируется вместе с арома-
тическими углеводородами (часто в сопоставимых концентрациях). Однако величины
относительной дисперсии сернистых соединений (табл. 6) того же порядка, что и аро-
матических углеводородов. Поэтому сернистые соединения, десорбирующиеся вместе
с ароматическими углеводородами, не могут внести сильных искажений в порядок из-
меряемых величин относительной дисперсии и повлиять на результаты определения
ненасыщенных углеводородов.
Правильность определения границы между метано-нафтеновыми и ароматически-
ми углеводородами по скачку значений относительной дисперсии и четкость этой
границы можно хорошо проконтролировать другим показателем — «выходной кривой»,
выражающей зависимость количества эллюированного вещества от объема прошедше-
го растворителя [50, 51]. В микрохроматографическом разделении объем десорбента,
пошедшего на вымывание каждой микрофракции, резко меняется при переходе от
метано-нафтеновых к ароматическим углеводородам. Поэтому все микрофракции, по-
лученные до резкого подъема «выходной кривой», построенной по времени выхода или
по объему десорбента, относились к метано-нафтеновым углеводородам. В описывае-
мом методе мнкрохроматографического разделения масла при активности силикагеля
12—14 мл по бензолу [45] и весовом соотношении вещество — силигакель, равном
1 : 100, удавалось четко отделить петролейным эфиром насыщенные углеводороды от
ненасыщенных, так как после вытеснения насыщенных углеводородов из колонки не-
которое время (в отдельных опытах до 2 час.) вытекал чистый растворитель [52].
Установить резкую границу между ароматическими углеводородами и гетеросое-
динениями нефти не представляется возможным, так как эти классы веществ при хро-
матографии на силикагеле четко не разделяются. Многочисленными работами [53, 54]
установлено, что понижение значений показателя преломления в конце хроматографи-
ческого разделения масел обусловлено выходом части гетеросоединений, не содержа-
щих существенных количеств ароматических углеводородов. Поэтому начало прогрес-
сивного уменьшения значений показателя преломления в конце десорбции можно при-
нять за условную (но хорошо воспроизводимую) границу конца вымывания основной
части суммы ароматических углеводородов и тиофенов и появления первых десорби-
рованных фракций, содержащих преимущественно гетеросоединения неароматической
природы.
На основании изложенного выше можно произвести следующую
обработку экспериментальных данных для малосернистых нефтепродук-
тов:
1. По полученным цифровым данным составляют график изменения
физико-химических свойств последовательно выделяемых микрофракций
масла, взятого на исследование.
2. По значению показателя преломления микрофракций определяют
границу конца вымывания углеводородов с примесью слабополярных
гетероатомных соединений. и начала выхода полярных гетероатомных
соединений, возможно с примесью высркоцикличных ароматических угле-
водородов. По определенной границе вычисляют выходы суммы углево-
дородов (и слабополярных соединений серы) и отдельно — суммы по-
д . Методы анализа 49
Таблица 5
Дисперсиометрическая характеристика тяжелых нефтяных углеводородов
по литературным данным
Фракция ,20 “FCD Яр — Яс „20 nD Данные о составе и возмож ном строении (вычислено)
Фракции, выделенные при адсорбционном анализе дистиллята жирновской нефти,
выкипающего в пределах 420—500° С [48]
Фракция: № 1 17,7 97,2 1,4785 0,8615 4iH24-2,11
№ 2 17,8 97,9 1,4745 0,8638 . —
№ 3 17,8 98,0 1,4751 0,8647 СпН2л-2,31
№ 4 17,9 98,5 1,4758 0,8668 —
№ 5 18,1 99,1 1,4771 0,8705 4iH24-2,81
№ 6 18,2 99,7 1,4790 0,8734 4»Н24-2,77
№ 7 18,4 101,2 1,4828 0,8789 4гН24-2,73
№ 8 19,2 105,8 1,4881 0,8882 —
№ 9 20,9 115,5 1,4936 0,8953 —
№ 10 21,8 120,5 1,5049 0,9186 4гН24-9,47
Нафтеновые углеводороды, выделенные из высококипящих фракций различных нефтей [48]
Фракция:
из остаточного сырья 18,2 1,4747 0,8688 Производные декалина и
сернистых нефтей ’ дициклопентила
то же 18,2 ^100 1,4751 0,8624 То же
из эмбенской смоли- 18,2 1,4786 0,8709 Смесь ди- и трицикло-
стой нефти гексильных нафтенов
из китайской нефти 18,2 1,4819 0,8778 То же
из катанглиской неф- 18,3 1,4866 0,8895 Смесь конденсированных
ти три- и тетрациклических
нафтенов
из эмбенской смоли- 18,4 1,4980 0,9030 Тетра- и пентацикличе-
стой нефти <_ 1QQ ские нафтены
из дистиллятов сер- 18,1 1,4714 0,8558 Смесь моно- и дицикло-
нистых нефтей пентильных нафтенов
из катанглиской 18,4 1,4888 0,8988 Производные трицикло-
нефти гексильных нафтенов
из ильской нефти 18,5 1,4991 0,9266 Производные три-и тетра-
цикло гексильных нафте-
нов
Нафтеновые углеводороды, полученные при гидрировании смол [55]
Фракция: № 1 18,8 102 1,5016 0,9228 4гН24-6,57 (« — "42)
№ 2 18,4 99,8 1,4988 0,9130 СпН2я-6,04 (п = 33)
Полициклические нафтеновые углеводороды, полученные
при гидрировании высокомолекулярных ароматических углеводородов [55]
Фракция: из парафинистых сернистых нефтей: ароматические уг- леводороды 30,7 173,2 1,5493 0,9734 4^24-15,36 (« — 35,07)
нафтеновые угле- водороды 17,9 97,7 1,4915 0,9030 4^24-5,52 (« = 36,8)
S0
Таблица 5 (окончание)
Фракция ,20 FCD •Rp — Rq „20 nD 420 Данные о составе н возмож- ном строении (вычислено)
ароматические угле- водороды 29,2 164,0 1,5410 0,9631 СпН2л-1б,и (« = 43,42)
нафтеновые углево- дороды Фракции из малопа- рафииистых и мало- сернистых нефтей: 17,9 97,7 1,4918 0,9029 Спн2«-б,12 (« = 41,22)
ароматические угле- водороды 33,8 191,0 1,5599 0,9902 ^пН2л-17,92 (« = 36,4)
нафтеновые углево- дороды 18,0 97,7 1,4979 0,9192 -6,6 (« = 34,3)
ароматические уг- леводороды 30,8 172,0 1,5439 0,9751 ^п^2п~24,3 (п ~ 65,0)
нафтеновые углево- дороды 18,2 99,0 1,5017 0,9228 ^n^2n-i0,3 (« = 59,1)
Фракции десорбционного анализа деасфальтированного и депарафинированного
концентрата ромашкинской нефти [56]
Фракции: 17,6 17,8 97,0 98,0 1,4741 1,4760 0,8605 0,8656 СпН2л-2,04 СпН2л-2,69
№ № 1 4
№ 7 18,0 99,0 1,4779 0,8695 СпН2л-2,85
№ 10 18,3 100,6 1,4850 0,8837 СпН2л-3,63
№ 11 19,4 106,7 1,4908 0,8911 СпН2л-6,24
№ 18 22,0 122,1 1,5087 0,9096 4iH2n-10,34
№ 15 23,8 132,3 1,5139 0,9258 CnH2n-12,76
Полициклические нафтеновые углеводороды, полученные гидрированием
ароматических углеводородов (смешанного строения) [19]
Фракции: № la 47,5 281 1,628 1,061
№ 16 18,6 100 1,505 0,940
№ Па 35,2 201 1,573 1,004
№ Пб 18,5 100 1,502 0,918
№ Ша 22,4 124 1,508 0,918
Шб 18,3 100 1,485 0,889
Одно нафтеновое коль-
цо -f- тРи ароматических
кольца (конденсирован-
ных по типу фенантрена)
Четыре нафтеновых коль-
ца (получены гидрирова-
нием 1а)
Два нафтеновых коль-
ца + два ароматических
кольца (конденсирован-
ных по типу нафталина)
Четыре нафтеновых коль-
ца (получены гидрирова-
нием Па)
Два циклопарафиновых
кольца -]- одно аромати-
ческое кольцо
Три циклопарафиновых
кольца (получены гидри-
рованием 1Па)
Примечание.
“FCD = («“-!)• 10 ’
Относительная дисперсия вычислялась по данным работы [57]:
4* 51
лярных гетероатомных соединений в весовых процентах ко всей навеске
масляной фракции.
3. По значению относительной дисперсий и одновременно по резкому
повышению объема десорбента, требующегося для вымывания 1 мг
вещества, устанавливают границу разделения метано-нафтеновых и
ароматических углеводородов независимо от значения показателя пре-
ломления. Эта граница позволяет вычислить процент каждой микро-
фракции по отношению к сумме углеводородов.
Таблица 6
Величины относительной дисперсии и показатели преломления
некоторых малополярных сераоргаиических веществ
Вещество Т. кип., °с ,20 “fcd „20 nD
н-Амилмеркаптан 126,5 21,0 1,4469
Тиациклопентан 120,9 22,6 1,5016
Тиациклогексан 141,8 22,2 1,5068
Диэтилдисульфид 152,6 26.1 1,5073
2-Метилтиофен 112,5 ’ 31,7 1,5204
3-Метилтиофен 115,4 31,2 1,5204
Для сернистых и высокосернистых нефтепродуктов необходимы дополнительные
исследования. Механическое перенесение показателей, рекомендуемых для малосер-
нистых продуктов, на продукты с повышенным содержанием органических соедине-
ний серы недопустимо.
Б. .Экспериментальная проверка метода на искусственных смесях
индивидуальных углеводородов
Для проверки описанного метода определения группового состава были взяты
две серии индивидуальных углеводородов.
Первая'серия составлена из углеводородов, выкипающих в пределах 150—250° С
(табл. 7), вторая серия — из углеводородов с пределами кипения выше 250° С (табл.
8). Ввиду отсутствия чистых и достаточно сложных высококипящих метановых угле-
Таблица 7
Индивидуальные углеводороды, использованные для составления смесей, кипящих выше
150° С
Углеводород Т. кип., °C и20 nD ,л 20 WFCD
брутто- формула структурная формула
С10Н22 СНз-(СН2)8-СНз 174 1,4120 17,2
СцНзо СН3-СН2-СН—(СН2)4-СН—СН2—СНз С2Н5 С2Н5 232 1,4438 17,5
СцН22 С2Нп-(СН2)9-СНз 204 1,4428 18,0
С9Н12 С3Н5-СН2-СН2-СНз СНз 158 1,4919 29,0
СцН1в СНз—<^=^—CHZ СНз СНз 199 1,4996 30,0
52
водородов, взята фракция некрасовской нефти (о. Сахалин), из которой сульфирова-
нием и двукратным хроматографированием на силикагеле была выделена узкая фрак-
ция предельных углеводородов, условно названная нами «нефтяные метановые» (см.
табл. 8).
Таблица 8
Индивидуальные углеводороды, использованные для составления серии смесей,
кипящих выше 250° С
Углеводород Определено Вычислено
брутто- формула структурная формула “FCD е % ‘э н, % мол. вес j С, % % ‘н и 0> п Ч о 2
С17Н34 СвНц—CH—СН2—СН (СН3)2 18,0 1,4576 85,68 14,25 241 85,71 14,29 238
СМ2-С5Нц
С27Н52 (СвНц-СН2—СН2)2—CH—С10Н21 18,0 1,4750 86,18 13,80 374 86,17 13,83 376
СазНав (CeHu-CH2-CH2)sCH-CH2-CHs 18,0 1,4893 86,63 13,27 328 86,75 13,25 332
Ани
С17Н28 С3Н5—С (CHg)2—С8Н17 24,5 1,4858 87,80 12,05 230 87,98 12,07 232
С27Н40 (СвН5—СНг-СНЖ—СН —С10Н21 Более 25,0 1,5148 88,99 10,98 357 89,01 10,99 364
Из первой серии относительно низкокипящих углеводородов составлены две сме-
си, состав которых дан в табл. 9.
Эти относительно иизкокипящие смеси были разделены на микрохроматографиче-
ской колонке с применением охлаждения (см. рис. 1), а результаты анализа сведены в
табл. 9.
Из второй серии высококипящнх углеводородов были составлены три смеси
(табл. 10), но эти высококипящие смеси анализировались на упрощенной аппаратуре
для мнкрохроматографического разделения (см. рис. 6), результаты которого сведены
в табл. 12. ,
Из табл. И и 12 видно, что ошибка в определении весового процентного содержа-
ния метано-нафтеновой части не превышает +1,5 абс. %.
Результаты определений группового состава, полученные микрохроматографичес-
ким методом, проверялись хроматографическим разделением и последующим анали-
зом больших навесок масляной фракции нефти месторождения Некрасовка (о. Саха-
лин). Пятидесятиградусные фракции этой нефти были разделены микрохроматографи-
ческим методом из навесок 30—60 мг и методом хроматографии навесок 3—6 г. При
этих параллельных определениях тщательно соблюдались как одинаковые условия раз-
деления (силикагель и растворители из одной и той же партии), так и одинаковые ус-
ловия определения физико-химических показателей отбираемых фракций. Расчет груп-
пового состава был проведен для микрохроматографического разделения рекомендуе-
мым методом, а для макрохроматографического разделения расчет группового соста-
ва проводился тремя способами:
1) по показателю преломления, все фракции, имевшие больше 1,49 единиц, отно-
сились к ароматическим;
2) по петролейному эфиру, все фракции, вымытые петролейным эфиром, относи-
лись к метано-нафтеновым;
3) по удельной дисперсии, все фракции, имевшие удельную дисперсию больше
100 единиц, относились к ароматическим.
Данные определения группового состава разными методами сведены в табл. 13.
Из таблицы видно, что микрохроматографический метод определения группового со-
става дает результаты, ближе всего совпадающие с аналогичными определениями в
микромасштабе, расчитанными по величинам удельной дисперсии. Необходимо отме-
тить, что микрометод дает более точные результаты с меньшнм разбросом зна-
чений при параллельных определениях группового состава. Это объясняется
возможностью в процессе микрохроматографии отбирать микрофракции желаемого ве-
са с большим весовым соотношением вещество — силикагель, равным 1 : 100, тогда как
при макрохроматографии обычно применяют весовое соотношение от 1 : 10 до 1 :30.
53
Таблица 9
Состав искусственных-смесей (кипящих выше 150° С), использованных
для проверки микрохроматографического метода
Углеводород Содержание компонентов „20 nD “FCD
брутто- формула структурная формула вес. % нарастаю- щим ито- гом, вес. %
С10Н22 С14Н30 Смесь 1 СН3-(СНг)8-СН3 СН3—СН3—СН-(СН2)4-СН—СН3- СНз 18,3 23,2 18,3 41,5 1,4120 1,4438 17,2 17,5
С11Н22 1 1 С2Н5 С2Н5 CfiHn-(CH2)4-CH3 20,9 62,4 1,4428 18,0
с,н13 Сумма метано-нафтеновых СсН5-СН2-СН2—СНз СНз 17,1 62,4 79,5 1,4245 1,4919 17,5 29,0
CuHie СНз-^^-СН , СНз 20,5 100,0 1,4996 30,0
Сумма ароматических — 37,6 1,4950 «29,5
Вся смесь , —- 100,0 1,4520 ж 22,5
С10Н22 С14Н30 Смесь 2 СН3-(СН2)8—СНз СН з-СНг-СН-(СНг)4—СН—СН2—СНз 9,0 11,4 9,0 20,4 1,4120 1,4438 17,2 17,5
Снн23 С2Н5 С2Н5 С6Нп-(СН2)4-СН3 10,1 30,5 1,4428 18,0.
С9Н12 Сумма метано-нафтеновых С0Н5—СН2-СН2-СН3 СНз 29,3 30,5 59,8 1,4245 1,4919 17,5 ж29,0
CuHie CHa-Q-CH7 СНз ХСН3 40,2 100,0 1,4996 «30,0
Сумма ароматических — 69,5 1,4955 «29,5
Вся смесь — 100,0 1,4750 «26,6
Таблица 10
Состав искусственных смесей (кипящих выше 250° С), использованных
для проверки микрохроматографического метода анализа
Углеводород Содержание компонентов 20 nD “FCD
брутто- формула структурная формула вес. % нарастающим итогом. вес. %
Смесь 1
С17Н34 С27Н52 С25Н43 Нефтяные метановые СН2—СН(СН3)2 СвНцСН"7 СН2—СзНц (СвНц—СН2—СН2)2—CH—С10Н21 (CeHu-CHa-CHsHCH—СН2—СвНц 88,3 19,4 20,0 20,3 39,3 57,7 79,7 100,00 1,4571 1,4576 4,4750 1,4893 о о о о ОО 00 00 00 и » 1?
Вся смесь ... — 100,00 1,4719 -18,0
Смесь 2
Нефтяные метановые 11,0 11,0 1,4571 «18,0
СН2—СН (СН3)2
С17Н34 СвНцСН7" 20,1 31,1 1,4576 —18,0
\н2-С8Ни
(CeHu—СН2—СНг)гСНСюНи 19,2 50,3 1,4750 —18,0
С25Н43 (СвНи-СН2-СН2)2СН—СН2СвНи 24,3 74,6 1,4893 -18,0
С17Н28 С6Н5—С (CHg^CgHu 25,4 100,0 . 1,4858 —24,5
Вся смесь. . . . — 100,0 1,4719 «19,6
Смесь 3
Нефтяные метановые 9,0 9,0 1,4571 — 18,0
СН2—СН(СН3)2
С17Н34 СзНиСН7 16,8 25,8 1,4576 —18,0
СНз-СЛц
С27Н52 (СвНц—С Но—СН2)аСН—CioHsi 16,0 41,8 1,4750 «18,0
С25Н46 (СзНп-СНз—СН2)2СН-СН2СвН11 20,3 62,2 1,4893 -18,0
С17Н28 С8Н5-С(СНз)2-СзН17 21,1 83,3 1,4858 24,5
С27Н40 (С6Н5-СН2-СН2)2СН—С10Н21 16,7 100,0 1,5148 Более 25,0
Вся смесь. . . — 100,0 1,4810 24,0
55
Таблица 11
Результат проверки микрохроматографического метода на смесях индивидуальных углеводородов, кипящих выше 150° С
Навеска смеси, мг ! Состав смеси Взято Получено Расхождение, вес. %
вес. % gQ С Q g£ . 3 вес. % gQ е аЭ»^ 03 по группам в сумме (потери)
Смесь 1
42,35 (Метано-н афтеновые ^Ароматические 62,4 37,6 1,4245 1,4950 «17,5 =29,5 61,4 36,9 1,4245 1,4950 «17,5 >25 —1,0 1 —0,7 J —1,7
qz 79 (Метаио-нафтеновые 62,4 1,4245 «17,5 62,0 1,4240 -17,5 -°’i У 2 0
(Ароматические 37,6 1,4950 «29,5 36,0 1,4948 >25 1,6 J
Смесь 2
30,35 гМетано-нафтеновые (Ароматические 30,5 69,5 1,4245 1,4955 «17,5 «29,5 30,1 69,0 1,4240 1,4952 «17,5 >25 —0,4 —0,5
QQ 07 (Метано-н афтеновые 30,5 1,4245 «17,5 30,1 1,4242 «17,5 —0,4
аУ to/ (Ароматические 69,5 1,4955 «29,5 67,3 1,4950 >25 —2,2
В. Обоснование способа интерпретации результатов
микрохроматографического анализа для ориентировочного
определения подгрупп углеводородов
В литературе [58] имеются указания, подтвержденные большим эксперименталь-
ным материалом, о возможности четкого разеделения масляной части нефтей методом
хроматографии на силикагеле при весовом соотношении вещество — силикагель поряд-
ка 1 :50, причем фракции углеводородов можно делить по показателям преломления.
Ниже приведены пределы показателя преломления подгрупп углеводородов по
литературным данным [58]:
Подгруппа углеводородов Пределы
Парафины нормального и изостроения . . 1,45—1,47
Нафтены моно- и бициклические........... 1,47—1,48
Нафтены три- и полициклические.......... 1,48—1,50
Ароматические с одним ядром............. 1,50—1,53
Ароматические с двумя ядрами............ 1,53—1,55
Полициклические ароматические........... 1,55 и более
В микрохроматографическом анализе при весовом соотношении вещество — сили-
кагель, равном 1 : 100, и при наличии десорбционных кривых по показателю преломле-
ния такой прием вполне приложим для ориентировочной оценки цикличности углево-
дородов в относительно широких нефтяных фракциях.
В рекомендуемом анализе оценка подгрупп углеводородов может быть проведена
по данным табл. 1 с большей степенью вероятности потому, что граница разделения
метано-нафтеновых и ароматических углеводородов устанавливается очень четко (по
значению относительной дисперсии и одновременно по «выходной кривой»), В резуль-
тате четкого деления и анализа последних десорбируемых насыщенных углеводородов
в ряде случаев было установлено, что три- и полициклические нафтеновые углеводоро-
ды имеют Пд более 1,50. Это подтверждают и многочисленные литературные данные
(см. табл. 5).
Ориентировочное отнесение хроматографически выделяемых подгрупп углеводоро-
дов по условным пределам показателя преломления возможно потому, что число инди-
видуальных углеводородов в естественных маслах (или в других достаточно сложных
рмесях) очень велико, а десорбция углеводородов с силикагеля идет в первом приб-
56
Таблица 12
Результаты проверки микрохроматографического метода иа смесях индивидуальных углеводородов, кипящих выше 250° С
Навеска смесн Состав смеси Взято Получено Расхождение, вес. %
вес. % л20 "о „20 “FCD вес. % л20 nD „20 WFCD ПО группам в сумме (потери)
с чесь 1
25,03 (Метановые 38,3 61,7 1,4571 1,4789 < —18,0 —18,0 'месь 2 101,8* 1,46 (в среднем) ж18,0 4-1,8 4-1,3
1 Нафтеновые
38,52 /Метано-нафтеновые 74,5 22,6 1,460 (в среднем) 1,4858 -18,0 24,5 —0,1) —2,8/ —2,9
74,6 25,4 1,4719 1,4858 —18,0 24,5
1Моноциклические
Смесь 3
Метано-нафтеновые 62,2 1,4571 «18,0 63,1 1,46 (в среднем) ж18,0 4-0,94'
41,37 Моноциклические ароматические . . 21,1 1,4858 24,5 20,7 1,4858 24,5 —0,4 —1,4
Бициклические ароматические .... 16,7 1,5148 25,8 14,8 1,5148 >25,0 —2,0 ,
Метано-нафтеновые' 62,2 1,4571 «18,0 68,0 1,46 (в среднем) «18,0 4-0,9
36,92 Моноциклические ароматические . . . 21,1 1,4858 24,5 19,6 1,4858 24,5 —1,6 —2,8
Бициклические ароматические .... 16,7 1,5148 25,8 14,7 1,5148 >25,0 —2,0 .
Смесь не разделилась.
Т’а'б ли ц а 13
Сравнительные результаты определения группового состава фракций нефти месторождения Некрасовка (о. Сахалин)
Темпера- тура отбора фракции, °C Выход фракции, вес. % на фракцию 300-500° С Определено методом микрохроматографии Определено методом макрохроматографии
навеска, мг ! групповой состав, вес. % на фракцию навеска, мг групповой состав рассчитан по 20 вес. % на фракцию* групповой состав рассчитан по петролейному эфиру, вес. % на фракцию2* групповой состав рассчитан по удельной дисперсии, % на фракцию 8*
метано- нафтеновые аромати- ческие потери метаио- нафтеновые аромати- ческие потери метано- нафтеиовые аромати- ческие потери метано- нафтеновые аромати- ческие поте и
300—350 25,07 40,12 66,0 32,1 1,9 5236 62,5 35,6 1,9 66,3 31,8 1,9 64,1 34,0 1,9
37,26 67,5 30,9 1,8 4470 64,2 31,8 4,0 69,2 26,8 4,0 67,1 28,9 4,0
л4* пшах 1,5 1,2 0,3 — 1,7 3,8 2,1 2,9 5,0 2,1 3,0 5,1 2,1
350—400 28,84 39,91 67,6 30,5 2,1 6003 62,0 32,8 5,2 72,5 22,3 5,2 67,4 27,4 5,2
60,58 66,8 31,5 1,7 4310 59,0 37,7 3,3 69,1 27,3 3,3 64,1 32,6 3,3
Атах 0,8 1,2 0,4 — 3,0 4,9 1,9 3,1 5,0 1,9 3,3 5,2 1,9
400—450 18,90 34,56 62,1 37,9 — 5023 61,0 37,2 1,8 62,0 36,2 1,8 62,9 35,3 1,8
33,50 62,6 35,9 1,5 3795 57,5 38,8 3,7 67,2 29,1 3,7 59,2 37,1 3,7
^тах 0,5 2,0 1,5 — 3,5 1,6 1,9 5,2 7,1 1,9 3,7 1,8 1,9
450—500 13,77 50,67 55,5 44,5 — 4210 54,7 42,4 2,9 56,5 40,6 2,9 55,1 42,0 2,9
9Q £5 ЛЗ 9 Л 7 3210 51 1 Л 5 5 3.4 60.7 35.9 3.4 52.8 43.8 3.4
Атах 0,‘б 1,3 6,7 — 3,6 3,1 0,5 4,2 4,7 0,5 2,3 1,8 0,5
500—550 14,13 46,95 51,9 46,7 0,4 3279 42,1 54,4 3,5 54,0 42,5 3,5 50,5 46,0 3,0
42,97 52,6 47,4 — 3715 37,5 59,4 3,1 60,4 36,5 3,1 49,3 47,6 3,1
Атах 0,7 0,7 0,4 — 4,6 5,0 0,4 6,4 6,0 - 0,4- 1,2 1,6 0,4
300—550 100,00 42,12 63,5 35,0 1,5 5160 60,2 37,2 2,6 66,2 31.2 2,6 62,7 34,7 2,6
Вычислено по труп- 62,5 35,8 1,7 — 57,4 40,7 1,9 65,8 31,4 2,8 61,2 35,7 3,1
повому составу дне- .... ... .. . .
тиллятов и ИХ ВЫХО-
ду для фракции 300—550® С
* Все фракции с пд более 1,49 относились к ароматическим.
-* Все фракции, вымытые петролейным эфиром, относились к метаио-иафтеновым.
’• Все фракции, имевшие удельную дисперсию более 100 единиц, относились к ароматическим.
<• Dnrvnwnpwwp upwnv ПЯПЯЛЛРЛЬНЫМИ ОПЫТЭМ (МаКСИМЗЛЬНОС).
лижении в порядке возрастающего отношения С: Н и как следствие — в порядке воз-
растающих значений ng*. Необходимо отметить, что из индивидуальных углеводоро-
дов можно подобрать такие, не очень сложные, искусственные смеси, при хромато-
графии которых более цикличные углеводороды будут выходить раньше менее циклич-
ных. Но такие отдельные, в общем немногочисленные отклонения от общего правила
не могут в сложных смесях резко нарушить общую закономерность.
Фиксируя выход насыщенных микрофракций, застывающих выше +25°, можно
ориентировочно определить количество твердых углеводородов, относящихся к пара-
финам, если заранее известно, что в смеси нет заметных количеств твердых нафте-
новых.
Таблица 14
Результаты микрохроматографического разделения масляной фракции
современных морских осадков Берингова моря
(навеска масла 55 лег, весовоэ соотношение вещество — силикагель 1 :100)
№ микро- фракции мг Зыход вес. % „20 nD гч20 “fcd Объем (У) десорбента нарастающим итогом
Метано-нафтеновые углеводороды
1 3,4 6,2 1,4775 17,0 0,70
2 3,8 6,9 1,4495 17,0 0,85
3 5,4 9,8 1,4640 17,0 1,00
4 7,7 14,0 1,4640 17,0 1,20
5 7,1 12,9 1,4670 17,0 1,40
6 6,2 11,3 1,4733 17,0 1,50
7 10,4 18,9 1,4890 17,0 2,10
8 1,8 з,з 1,5070 17,0 3,00
Итого . . 45,8 83,3
Ароматические углеводороды
9 0,4 0,7 1,4950 21,0 5,25
10 1,0 1,8 1,5000 21,0
11 2,6 4,7 1,5391 25,0
12 1,1 2,0 1,5490 25,0
13 0,8 1,4 1,5482 25,0 —
Итого . . 5,9 10,6 1 1
Петролейноэфирные смолы (включающие гетеросоединения)
14 0,5 1,о 1,5410 Ьк,
15 0,6 1,1 1,5242 1— —
Потери * 3,3 6,1 —
Итого . . 55,0 100,0
* «Потери», т. е. ие вымытые из колонки вещества,- причислены к петролейио-
эфириым смолам.
59
Все сказанное в отношении предельных углеводородов, десорбирующихся послед-
ними, целиком относится и к первым фракциям ненасыщенной части. Начало вымыва-
ния ненасыщенных углеводородов хорошо фиксируется и служит отправной точкой для
определения моно-, би-, три- и полициклических подгрупп ароматических углеводоро-
дов. К сожалению, в практической работе с естественными маслами почти все микро-
фракции ненасыщенной части (кроме первых трех-четырех очень маленьких по весу)
получаются слабо окрашенными. В окрашенных фракциях нельзя точно определить
величину относительной дисперсии, а следовательно, использовать ее для оценки цик-
личности этих микрофракций. Поэтому рекомендуется и в ненасыщенной части иссле-
дуемого масла оценивать количество подгрупп углеводородов по условным пределам
показателя преломления (см. табл. 1).
Таблица 15
Сопоставление результатов микрохроматографического анализа и спектроскопических
исследований масла современных морских осадков Берингова моря
№ мвкро- фрак- ции Выход, вес. % Грубоориентировочная оценка класса углеводородов по микрохроматографии Оценка структурно-группового состава по результатам ИК- и УФ-спектроскопии
1 6,2)
2 3 4 6,91 9,8 14,0' Метановые Слабозамещенные парафиновые То же и появление нефтеновых струк-
5 12,9 Метановые, возможны следы наф-' тур Слабозамещенные метановые цепи со
6 11,3 Метановые + моно- и бицикличес- кие нафтеновые все возрастающим количеством нафте- новых колец
7 8 18,9 3,3 Нафтеновые бициклические; воз- , можны следы моноциклических нафтеновых Нафтеновые три- и полицикличес- кие Следы олефиновых структур
Ито го Метановые 53 % Нафтеновые . . . . 30 % (по графику рис. 12.) Общее количество нафтеновых струк- тур — не менее 2/з от суммы выде- ленных фракций
9 0,7 Ароматические моноциклические Наличие олефиновых структур при отсутствии (меньше 1 %) ароматических
10 1,8 Ароматические моноциклические Ароматические полизамещенные мо- ноциклы
11 4,7 Ароматические моноциклические с примесью бициклических Ароматические моноциклы с примесью бициклов с одним гидрированным кольцом: ароматические углеводороды с двумя бензольными кольцами, сое- диненные мостиками
12 2,0 Ароматические бициклические Бициклические структуры, в том чис- ле производные нафталина и гидриро- ванные трициклические конденсиро- ванные ароматические системы
13 1,4 Ароматические бициклические Бициклические структуры типа наф- талина с примесью нелинейно конден- сированных соединений типа фенан- трена, хризена и т. п.
14 1,0 Ароматические неизвестного стро- ении с примесью гетеросоединений Бициклические структуры с заметным количеством поликонденснрованных систем типа фенантрена, хризена н т. п.
15 1,1 Гетеросоединення Различные гетеросоединения
Г. Экспериментальная проверка результатов
микрохроматографического анализа другими методами
Проверка чистоты разделения и предложенных приемов интерпретации результа-
тов микрохроматографического анализа была проведена методами инфракрасной (ИК)
и ультрафиолетовой (УФ) спектроскопии. В качестве объекта для такой проверки были
взяты микрофракции масел из современных морских осадков Берингова моря.
Сведения о выделении этих микрофракций с подробными цифровыми данными
приведены на стр. 131.
Результаты разделения масел современных морских осадков сведены в табл. 14,
а данные по спектральным исследованиям микрофракций сведены в табл. 15 и сопо-
ставлены с результатами ориентировочной оценки весовых процентов подгрупп углево-
дородов по рекомендуемым (см. табл. 1) пределам показателя преломления.
Из табл. 15 видно, что определение группового состава и оценка строения углево-
дородов масляной фракции, проведенная по рекомендуемой методике, находится в хо-
рошем соответствии с данными спектрального анализа.
ВЫВОДЫ
1. Разработана конструкция микрохроматографической колонки и
установлен режим элюэнтной хроматографии с одновременным отгоном
растворителя, дающий возможность проводить групповой адсорбцион-
ный анализ углеводородных смесей с температурой кипения от 150° С и
выше с навесками 10—50 мг. Разработанный метод адсорбционного
микроанализа проверен на смесях индивидуальных углеводородов, неф-
тяных фракциях и на маслах из современных морских осадков.
2. При адсорбционном элюэнтном методе микроанализа углеводо-
родных смесей (если разделение на метано-нафтеновую и ароматиче-
скую группы углеводородов проведено по скачку относительной диспер-
сии и «выходной кривой») групповой состав определяется с точностью
±1,0 вес. % для метано-нафтеновой и ±2,0 вес. % для ароматической
части исследуемой смеси.
3. Уточнены пределы показателей преломления микрофракций для
ориентировочного определения степени цикличности нафтеновых и аро-
матических углеводородов, а также для оценки количества полярных
гетероатомных соединений.
Институт нефтехимического синтеза им. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. D. Т. Day. Prac. Am. Phil. Soc., 36, 112 (1897).
2. D. Т. Day. Ind. Tech. Petroleum Rev., Supplement to issue of August, 25, 10 (1900).
3. C, Engler, E. Albrecht. Z. angew. Chem., 1901, 389.
4. L. Gurwitsch. Koll. Z„ 11, 17 (1912).
5. L. Gurwitsch. Petroleum (Berlin), 8, 65 (1912).
6. Б. Тарасов. Нефт. хоз., № 10, 47 (1926).
7. E. D. Rossini, В. I. Mair, A. F. Forziati, A. R. Glasgow, С. B. Willingham. Oil Gas
J., 41, 106 (1942); Petroleum Refiner, 21, 73 (1942); Proc. Am. Petroleum Inst., 23,
7 (1942).
8. В. I. Mair, A. L. Gaboriault, F, D. Rossini. Ind. Eng. Chem., 39, 1072 (1947),
9. А. С. Беликовский, С. H. Павлова, П. С. Гофман, 3. В. Дриацкая. Нефт. хоз., № 9,
30 (1947).
10. П. Ландсберг, Б. А. Казанский, П. И. Бажулин и др. Определение индивидуаль-
ного углеводородного состава бензинов прямой гонки комбинированным методом.
М., Изд-во АН СССР, 1959.
11. С. Р. Сергиенко, А. А. Михновская. Труды Комиссии по аналитической химии АН
СССР, т. VI. М„ Изд-во АН СССР, 1955, стр. 162—170.
12. А. В. Топчиев, И. А. Мусаев. Химия и технология топлив и масел, № 12, 1 (1956).
13. А. В. Топчиев, И. А. Мусаев, Г. Д. Гальперн. ДАН СССР, 115, 740 (1957).
14. В. С. Allibone. J. of the Institute of Petroleum, 27, 94, 209 (1941).
15. E. E. Handael. C. A., 35, 7166 (1941).
16. D. Swern. C. A., 38, 3570 (1944).
17. P. I. Wilson. C. A., 40, 3888 (1946).
61
18. В. Фукс. Труды IV Международного нефтяного конгресса, т. 6. М., Изд-во АН
СССР, 1956, стр. 185.
19. Ф. Д. Россини, Б. Д. Мэйр, А. Д. Стрейф. Углеводороды нефти. М., Гостоптехиздат,
1957.
20. С. Р. Сергиенко, Е. В. Лебедев. Изв. АН ТуркмССР, № 5, 16 (1959).
21. Е. В. Лебедев. Кандидатская диссертация. М., 1959.
22. В. I. Mair. A. F. Forziati. J. of Reserch of the Nation. Bur. Stand., 32, 165 (1944).
23. В. I. Mair, J. Res. Nat. Bur. Stand., 34, 435 (1945).
24. G. Dinneen, C. Bailley, J. Smith, I. Ball. Analyt. Chem., 19, 992 (1947).
25. G. W. Nederbragt, J. J. Jong. Rec. trav. chim., 65, 831 (1946).
26. С. M. Брещенко. Химия и технология топлив и масел, № 3, 53 (1959).
27. С. Р. Сергиенко, Л. И. Квитковский, Ю. Т. Гордаш, А. А. Петров. ДАН СССР, 128,
769 (1959).
28. Л. И. Квитковский. Кандидатская диссертация. М., 1959.
29. Б. А. Казанский, А. Ф. Плате. Труды Всесоюзного совещания по химии и перера-
ботке нефти. М., Изд-во АН СССР, 1953, стр. 21.
30. Б. А. Казанский, Г. С. Ландсберг. Изв. АН СССР, ОХН, 1951, 100.
31. Е. А. Михайлова, Б. А. Казанский. Труды Всесоюзного совещания по хроматогра-
фии. М., Изд-во АН СССР, 1952, стр. 155.
32. F. D. Rossini. Analyt. Chem., 20, ПО (1948).
33. Af. F. Bell. Analyt. Chem., 22, 1005 (1950).
34. Af. С. Бугуславская, А. С. Беликовский. Нефт. хоз., № 3, 52 (1947).
35. Af. R. Lipkin. Analyt. Chem., 20, 130 (1948).
36. G. N. Beaven. J. of the Institute of Petroleum, 36, 89 (1950).
37. R. Van-Nes, N. I. Waterman. J. of the Institute of Petroleum 38, 998 (1952).
38. С. P. Сергиенко, А. А. Махновская. ДАН СССР, 91, 103 (1953).
39. И. Абрамсон. Химия и химическая технология, № 8, 85 (1954).
40. К W. Schneider. Erdol und Kohlei № 2, 87 (1949).
41, Б. Д. Луфт, Е. С. Шер. Зав. лаб., № 7, 784 (1955).
42. В. В. Веселов. Труды Вост. Сиб. филиала АН СССР, серия хим., вып. 3, 55 (1955).
43. И. Spira. Erdol und Kohle, 13, № 1, 27 (1960).
44. Руководство по анализу битумов и рассеянного органического вещества горных по-
род. Л., «Недра», 1966.
45. Б. А. Казанский, Е. А. Михайлова. Рефераты докладов иа совещании по примене-
нию хроматографического метода М. С. Цвета в химическом анализе. М., Изд-во
АН СССР, 1953.
46. С. И. Павлова, 3. В. Дриацкая, П. С. Гофман. Методы исследования нефтей и неф-
тепродуктов. М., Гостоптехиздат, 1955.
47. Труды Всесоюзного совещания по хроматографии. М., Изд-во АН СССР, 1952.
48. Л. Г. Жердева, В. А. Потанина, Б. Б. Кроль. Химия и технология топлив, № 2,
13 (1956).
49. Б. В. Иоффе. ЖОХ, 16, 1121 (1946).
50. И. А. Фукс. Усп. химии, 18, 206 (1949).
51. Af. М. Синявин. Там же, стр. 186.
52. R. G. Clerc, С. R. Kincannon, Т. Р. Weir. Analyt. Chem., 7, 864 (1950).
53. И. В. Калечиц и др. Труды Вост. Сиб. филиала АН СССР, серия хим., вып. 3,
19 (1955).
54. И. В. Калечиц, Н. И. Павлова. Рефераты докладов на совещании по применению
хроматографического метода М. С. Цвета в химическом анализе. М., Изд-во АН
СССР, 1953, стр. 20.
55. Л. Г. Жердева, Ф. П. Сидляренок, И. И. Велизарьева. Химия и технология топлив,
№ 1, 17 (1956). ’
56. Л. Г. Жердева. Химия и технология топлив, № 7, 13 (1956).
57. Б. В. Иоффе. Руководство по рефрактометрии для химиков. Изд. Ленинградского
ун-та, 1956.
58. А. Бестужев, Д. Баржан. IV Международный нефтяной конгресс, т. VI, 1956,
стр. 211.
59. Химический состав нефти и нефтепродуктов. ГрозНИИ, 1955, стр. 133.
ПОЛУАВТОМАТИЧЕСКАЯ УСТАНОВКА
ДЛЯ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ СЕРЫ
И ГАЛОГЕНОВ (ХЛОРА, БРОМА, ИОДА)
В ОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ И НЕФТЕПРОДУКТАХ
Н. П. Волынский
Ранее было показано [1—4], что пиролитический ламповый метод
(«метод двойного сожжения») позволяет легко и достаточно точно опре-
делять количественное содержание серы и галогенов (хлора, брома,
иода) во всевозможных органических соединениях и нефтепродуктах.
Метод позволяет также проводить одновременное определение серы и
галогена (хлора или брома) из одной навески. На одно определение
серы или галогенов (хлора или брома) требуется около 30—50 мин.
В настоящем сообщении дается описание сконструированной нами
полуавтоматической установки для количественного экспресс-анализа
органических соединений и нефтепродуктов на содержание серы и гало-
генов.
I. Описание установки
В основу работы установки положен пиролитический ламповый
метод, заключающийся в том, что навеску испаряют и пиролизуют
в кварцевом стаканчике, открытый конец которого введен в дожигающее
пламя микрогорелки. Продукты испарения и пиролиза полностью сгора-
ют в дожигающем пламени, а образующиеся при этом окислы серы,
галогены или галогеноводородные кислоты поглощаются раствором
в абсорбере, через который при сожжении просасывается воздух. Оста-
ющийся в стаканчике кокс дожигают, не прекращая просасывание воз-
духа через абсорбер, путем введения в стаканчик небольшого количества
воздуха через кварцевый капилляр. В качестве поглотительного раство-
ра при определении серы, хлора или брома (а также при одновременном
определении серы и галогена) удобнее всего применять примерно 10 мл
0,05 N раствора соды в смеси с 3 мл 6%-ной перекиси водорода.
Свободные галогены (хлор, бром) восстанавливаются перекисью
водорода в щелочной среде до ионов хлора или брома:
На12 + Н2О2 + Na2CO3H 2NaHal + Н2О + О2 + СО2.
Рациональность и универсальность пиролитического лампового мето-
да для сожжения навески подтверждена многолетним опытом. Оконча-
ние анализа на S, С1, Вг проводят ацидиметрическим титрованием; на
все определение, включая приведение прибора в исходное положение,
заполнение абсорбера поглотительными растворами, сожжение и титро-
вание, требуется не более 5 мин. Навеску берут на аналитических весах
63
Рис. 1. Общий вид полуавтоматической установки
для определения содержания серы, хлора или брома
в органических соединениях и нефтепродуктах
(АДВ-200) примерно за 3 мин. Вся установка собирается на одном
лабораторном штативе Бунзена (рис. 1) так, чтобы обеспечить наиболь-
шее удобство для работы и исключить лишние движения аналитика.
Если в прежних конструктивных оформлениях пиролитического лам-
пового метода приходилось затрачивать много времени и внимания для
64
подведения к неподвижному абсорберу бюреток с поглотительными и
титрующими растворами или для смывания раствора в сосуд для титро-
вания, то в данном случае, наоборот, к неподвижным бюреткам подво-
дится вращающийся вокруг вертикальной оси абсорбер, в котором и
производится титрование. Абсорбер и ламповое стекло показаны на
рис. 2. Устройство для поворачивания абсорбера вокруг вертикальной
оси видно на рис. 3, а также на рис. 4. Оно является главной конструк-
тивной особенностью данной полуавтоматической установки.
Абсорбер (см. рис. 3) постоянно укреплен на специальном съемном
держателе 1 с фетровой прокладкой, цилиндрическая ручка которого
легко надевается на стержень 2, ввинченный в штатив, и вращается
вокруг вертикальной оси. Своим нижним торцом ручка упирается
в бобышку 3, укрепляемую на стержне на желаемой высоте. В верхнем
торце бобышки высверлены три лунки, в которые заходит примерно на
1 мм фиксатор 4 — короткий пруток диаметром 3 мм, припаянный к
ручке держателя. Лунки высверлены таким образом, что фиксируют
абсорбер в положениях «наполнение», «сожжение» или «титрование».
Фиксированные положения абсорбера («поворот до щелчка») создают
удобства для работы и значительно обегчают труд аналитика.
Для предотвращения случайных поломок бюреток и абсорбера при
невнимательной работе на бобышке имеется колпачок 5 с прорезью
в торце, устанавливаемый таким образом, что снятие абсорбера со
стержня возможно лишь после отведения абсорбера от бюреток- Во всех
других положениях абсорбера снятию его вверх будет препятствовать
фиксатор, упирающийся верхним концом в торец колпачка с внутренней
стороны. При свободном положении держателя абсорбера между верх-
ним концом фиксатора и торцом колпачка с внутренней стороны должен
оставаться зазор приблизительно в 2 мм. Дополнительная защита но-
сиков бюреток достигается с помощью ограждающего кольца
(рис. 5).
К держателю кольца припаяно устройство для подведения к носикам
бюреток воронки, соединенной шлангом с колбой для сбрасываемых
растворов. Вместо диоксановой горелки, работа с которой представляет
известное неудобство, в данной установке дожигающее пламя создается
специальной газовой микрогорелкой, конструкция которой показана на
рис. 6, а также на рис. 4.
Основание горелки 1 (см. рис. 6) выточено из стандартной горелки
Теклю. Выходное отверстие в ниппеле 2 запаяно третником; в нем
обыкновенной иглой сделано маленькое отверстие, через которое посту-
пает нужное для оптимальной величины пламени («*40 мм) количество
газа. В связи с этим отпадает необходимость каждый раз заново уста-
навливать нужную величину пламени регулировочным винтом 3 горелки,
который работает, следовательно, в двух положениях: «газ открыт» и
«газ закрыт».
В корпусе горелки 4 сделано несколько отверстий диаметром 2 мм,
через которые поступает воздух, благодаря чему пламя перестает быть
коптящим и приобретает устойчивость, не срывается потоком воздуха,
просасываемым через ламповое стекло и абсорбер. На корпусе горелки
на резьбе устанавливается и закрепляется контргайкой так называемая
«корона» 5, выполняющая три функции: предотвращение случайного
отклонения пламени от лампового стекла, фиксирование положения
стаканчика относительно пламени и фиксирование положения кварце-
вого капилляра относительно стаканчика. Капилляр вводят в стаканчик
через воронку 6, припаянную латунью к короне. Высоту горелки регули-
руют в определенных пределах резьбовым соединением корпуса с ниппе-
лем и ниппеля с основанием горелки. Установленное положение фикси-
руется контргайками на ниппеле. При этом расстояние от верхней
5 Методы анализа
65
^Янутр "'^ &
~70
Рис. 2. Детали из стекла и
кварца
а — абсорбер; б — ламповое стекло;
в — туманоуловитель; г — кварце-
вый стаканчик с колпачком;
д — кварцевый капилляр; е — счет-
чик пузырьков воздуха
Рис. 3. Устройство для
поворачивания абсорбера
вокруг вертикальной оси
1 — держатель; 2 — стержень;
3 — бобышка; 4 — фиксатор (при-
паять третником); 5 — колпачок
Рис. 4. Взаимное расположение держателя абсорбера, микрогорелки
и крана управления иа штативе
Рис. 5. Ограждающее и сбросное
устройство для бюреток
1 — воронка; 2 — сбросный патрубок; 3 — пре-
дохранительное устройство для бюреток;
4 — ограничитель перемещения воронки
Рис. 6. Газовая микрогорелка
(из нержавеющей стали)
1 — основание горелки; 2 — ниппель; 3 — регу-
лировочный винт; 4 — корпус горелки; 5 —ко-
рона; 6 — воронка
5
кромки короны до нижней кромки зонтика лампового стекла должно
составлять 5 мм. Перемещение горелки (при установке) в горизонталь-
ной плоскости обеспечивается специальной скамьей и скользящей по
ней во всех направлениях плоской скобой, к которой припаяно латунью
основание горелки (см. рис. 4). После установки скоба фиксируется
двумя винтами. Горелка всегда готова к работе и действует безот-
казно.
Быстрота сожжения навески и малая величина дожигающего пламе-
ни гарантируют отсутствие влияния каких-либо примесей в газе на
результаты определения. Последнее периодически контролируется хо-
лостыми опытами (примерно раз в неделю, а также после наполнения
емкостей установки вновь приготовленными растворами). •
Просасывание воздуха через абсорбер при сожжении, а равно и по-
дача воздуха в абсорбер для перемешивания во время титрования
осуществляется в нашем случае с помощью .переоборудованного насоса
Комовского, приводимого в движение электродвигателем. Скорость
просасывания воздуха в обоих направлениях регулируют краном управ-
ления, показанным на рис. 7 и рис. 4. Остаточное давление в вакуумной
части крана составляет примерно 160 мм рт. ст., давление (избыточное)
в нагнетательной части — около 300 мм рт. ст. Конструкция крана пред-
усматривает сравнительно небольшой угол поворота пробки крана (при-
мерно 50°) от режима сожжения до режима титрования (т. е. на угол
около 25° по часовой стрелке и 25° против часовой стрелки от нейтраль-
ного положения). Это создает большое удобство для аналитика, осо-
бенно при массовых анализах. Взаимное расположение держателя аб-
сорбера, микрогорелки и крана управления на плоскости штатива
хорошо видно на рис. 4. Необходимые для работы растворы соды и
Рис. 7. Кран управления
Рис. 8. Устройство для крепления
бутылей с растворами соды, кислоты
и воды
68
кислоты, а также дистиллированная вода помещаются в трех бутылях
(емкостью по 500 мл), укрепленных хомутиками в самой верхней части
штатива (см. рис. 1 и 8). Посредством стеклянных трубок раствор соды
подается в дозатор раствора соды, раствор кислоты — в бюретку для
титрования, а дистиллированная вода — в промывалку.
Дозатор раствора соды (рис. 9 и 10) позволяет поворотом пробки
крана в течение нескольких секунд подать в абсорбер определенное и
постоянное количество раствора, а именно около 10 мл— 0,05 N раство-
ра ЫагСОз в дистиллированной воде (см. также рис. 10). Носик дозатора
располагается на таком уровне относительно крана, что при вытекании
отмеряемого дозатором количества раствора мениск остающейся в систе-
ме жидкости устанавливается в самой узкой нижней части дозатора.
Это обеспечивает точность работы дозатора. При наладке нового при-
бора важно подобрать и всегда применять такую концентрацию раствора
соды, при которой на титрование его в холостом опыте требовалось бы
от 9,0 до 9,9 мл 0,05 N раствора H2SO4. Устройство дозатора перекиси
водорода показано на рис. 11.
Рис. 9. Дозатор
раствора соды
Во избежание быстрого разложения перекиси водорода дозатор по-
крашен черной краской. Поворотом пробки крана в абсорбер подается
около 3,5 мл 6%-ного раствора перекиси водорода. Количества переки-
си водорода, находящегося в дозаторе (100 мл), хватает примерно на
30 определений.
Устройство бюретки для титрования изображено на рис. 12. Конструк-
ция бюретки гарантирует точность автоматической установки нуля. При
этом от аналитика не требуется особого внимания и затраты активного
времени. Носик бюретки оттянут таким образом, что истечение 10 мл
раствора происходит за 50 сек. Применяемый 0,05 N раствор кислоты
удобно приготовлять из фиксанала серной кислоты. Конструкция промы-
валки приведена на рис. 13. Благодаря наличию ролика шланг, подаю-
щий воду, всегда находится под некоторым натяжением, при отпускании
шланга он втягивается в кожух. На конце нержавеющей трубки, соеди-
ненной с резиновым шлангом, имеется пять отверстий диаметром 0,7 мм
(четыре отверстия в радиальном направлении), что позволяет тщательно
смывать изнутри верхние части абсорбера минимальным количеством
воды.
69
Рис. 10. Нижняя часть полуавтоматической установ-
ки для определения серы, хлора и брома в органиче-
ских соединениях
Рис. 11. Дозатор раствора
перекиси водорода
II. ПОРЯДОК ПРОВЕДЕНИЯ АНАЛИЗА
Определение содержания серы или галогенов в органических соеди-
нениях производят следующим образом. Отводят сбросное устройство
от бюреток и поворачивают абсорбер против часовой стрелки до щелчка
«наполнение». Поворотом пробки крана содового дозатора наполняют
дозатор до появления фонтанчика в верхней части, после чего поворачи-
вают пробку в противоположном направлении. При этом отмеренное
дозатором количество раствора сливается в абсорбер через его широкую
горловину. Не ожидая окончания сливания содового раствора, поворо-
том пробки крана дозатора перекиси водорода заполняют сифонное
устройство до начала сливания перекиси водорода в абсорбер, после
чего перекрывают подачу перекиси водорода. Далее левой рукой откры-
вают расположенный позади бюретки кран и наполняют бюретку раство-
ром серной кислоты до появления фонтанчика в верхней части. Избыток
растворов при наполнении дозатора и бюретки сливается через шланги
в колбу для сбросных растворов. По окончании сливания раствора соды
из дозатора в абсорбер поворачивают абсорбер по часовой стрелке до
70
Рис. 12. Бюретка для титрования
раствором серной кислоты
1
Рис. 13. Промывалка
/ — трубка из нержавеющей стали; 2 — стеклян-
ная бусинка; 3 — стеклянная трубка; 4 — кожух;
5 — шланг; б — ролик
щелчка «сожжение», вставляют в широкую горловину туманоуловитель
(на пробке № 23, рис. 2) и надевают ламповое стекло так, чтобы зонтик
находился левее микрогорелки на —10 мм. На приведение аппарата
в положение готовности к сожжению навески требуется всего около
36 сек.
Стаканчик с навеской вставляют в шарнирно-укрепленный держатель
(рис. 14). Зажигают микрогорелку, включают воздуходувку, поворотом
крана управления по часовой стрелке устанавливают необходимую
скорость просасывания воздуха через абсорбер, после чего подводят
ламповое стекло к микрогорелке. При этом верхняя часть пламени при-
обретает иглообразную форму. За счет просасываемого воздуха погло-
тительный раствор увеличивается в объеме, столб пенящейся жидкости
заполняет абсорбер примерно до середины шарообразного расширения.
Это обеспечивает полное поглощение продуктов сгорания навески. Да-
лее снимают колпачок со стаканчика и быстро вводят открытый конец
стаканчика через прорезь в короне в пламя микрогорелки. Осторожно
нагревают стаканчик коротким шипящим пламенем горелки Теклю,
стараясь лучше испарить навеску, чем пиролизовать ее в стаканчике. Это
уменьшает количество остающегося в стаканчике кокса, на выжигание
которого требуется время.
При сожжении навески дожигающее пламя может значительно уве-
личиваться в объеме и приобретать окраску или светиться. В конце
сожжения сильно нагревают стаканчик и при этом вводят в него через
воронку в короне кварцевый капилляр, по которому подается воздух.
Момент введения капилляра показан на рис. 4. На том же рисунке вид-
на укрепленная под скамьей микрогорелки толстостенная пробирка,
предназначенная для хранения капилляра. Маленькое отверстие в
донышке пробирки служит для сбрасывания давления и предотвраще-
71
Рис. 14. Шарнирное устройство для крепления кварцевого
стаканчика
ния выброса пробки с капилляром из пробирки. Подача воздуха в ка-
пилляр осуществляется той же воздуходувкой, которая обслуживает
прибор; количество воздуха, подаваемого через кварцевый капилляр,
устанавливают раз и навсегда с помощью кусочка толстостенного стек-
лянного капилляра с очень малым отверстием. Стеклянный капилляр,
размер которого подбирают опытным путем, помещают в разрезе
шланга между промывалкой с водой, служащей счетчиком пузырьков,
и кварцевым капилляром. Скорость подачи воздуха для дожигания кок-
са не должна быть слишком высокой во избежание отклонения пламени
или слишком низкой, что увеличивало бы время, требуемое для полного
сгорания кокса. Обычно для дожигания кокса требуется около 30—
60 сек.
Сожжение веществ с большим содержанием серы или галогенов
следует проводить с такой скоростью, которая бы обеспечила полное
поглощение продуктов сгорания в абсорбере. Образование тумана в
правой части абсорбера (над поглотительным раствором) может приво-
дить к несколько заниженным результатам. На сожжение навески,
включая дожигание кокса, требуется в большинстве случаев от 2 до
4 мин. По окончании сожжения гасят пламя микрогорелки, снимают
ламповое стекло и вешают его на специально предусмотренный для это-
го держатель (см. рис. 10) с правой стороны прибора; вынимают пипет-
ку с индикатором из бутылочки, укрепленной на держателе для лампо-
вого стекла, и вводят одну каплю раствора метилоранжа в узкую откры-
тую горловину абсорбера. При анализе соединений с высоким содержа-
нием серы или галогенов следует после этого смыть узкую горловину
небольшим количеством воды из промывалки. Затем поворачивают аб-
сорбер по часовой стрелке до щелчка «титрование» и поворотом крана
управления против часовой стрелки устанавливают продавливание
72
воздуха через абсорбер. При этом жидкость передавливается в узкую
часть абсорбера и хорошо перемешивается воздухом. Затем открывают
полностью кран бюретки для титрования и следят за изменением ок-
раски раствора.
При появлении розовой окраски всего раствора прекращают при-
бавление кислоты; не изменяя положение абсорбера, вводят трубочку
промывалки сверху в узкую горловину абсорбера и смывают ее неболь-
шим количеством воды. Далее открывают пробку туманоуловителя и
также смывают его небольшим количеством воды, закрывают тумано-
уловитель, снимают его с абсорбера, смывают верхнюю часть абсорбера
и ставят туманоуловитель на место. После этого переключают краном
направление подачи воздуха через абсорбер (т. е. ставят кран в поло-
. жение сожжение»), затем тут же вновь переключают на режим продав-
ливания воздуха через абсорбер. За счет запаса щелочного раствора в
пористой пластинке слегка перетитрованная вначале система переводит-
ся в несколько недотитровэнную. Прибавлением нескольких капель кис-
лоты добиваются отчетливого перехода окраски от желтой до розова-
той. Благодаря малому разбавлению переход окраски виден достаточно
хорошо даже при электрическом освещении. Примененная нами систе-
ма титрования позволяет вести титрование в очень быстром режиме
(полное открытие крана бюретки) и в то же время не требует от анали-
тика постоянного напряжения для того, чтобы не перетитровать и во-
время остановить прибавление кислоты. Всего на титрование уходит
около 1 мин. По окончании титрования пробку крана управления ста-
вят в нейтральное положение, выключают воздуходувку, снимают тума-
ноуловитель, снимают держатель вместе с абсорбером, выливают со-
держимое и, не споласкивая абсорбер, ставят его обратно. Прибор го-
тов для заполнения его растворами для следующего анализа.
Специально наливать воду на пластинку туманоуловителя не обя-
зательно, для работы достаточно количество воды, сохранившееся в
пластинке от предыдущего смывания. Иногда полезно ввести в тумано-
уловитель каплю раствора метилоранжа. Красное окрашивание при
сожжении укажет на проскок определяемых элементов. При аккурат-
ной работе вес стаканчика после сожжения образца (если вещество
сгорает полностью, без оставления золы) не меняется; поэтому во мно-
гих случаях при массовых анализах взвешивание пустого стаканчика
можно производить лишь для периодического контроля.
Содержание серы, хлора или брома вычисляется по формулам:
o/Q— (а —5) М-1603
Р
у С1 ~ М-3546
%Вг= («т^-7992
где а — количество (в мл) кислоты, пошедшее на титрование поглоти-
тельного раствора в холостом опыте; b — количество (в мл) кислоты,
пошедшее на титрование поглотительного раствора после сожжения
навески; N — нормальность кислоты, применяемой для титрования;
Р — величина навески (в мг).
При титровании 0,05 N раствором кислоты формулы приобретают
следующий вид:
0/С_ . (а-6)-80,15
/0 о р ,
оу gp (д Ь) *399,6
73
Таблица 1
Результаты определения содержания серы в дибеизилсульфок-
сиде, CmHuOS (А), 4-меркаптогептеие-1, C7HUS (Б) и 2,4-ди-
меркаптогептане, CjHieSa (В)
Навес- ка , мг Время сож- жения + время до- жигания кокса, мин. а — Ъ, мл Содержание серы, % Абсолют- ная ошиб- ка опреде- ления А,% Относи- тельная ошибка оп- ределения А-100
найдено вычис- лено
0/ Q ’ /и /о отеор.
24,8 4 с 4,28 оединение 13,831 А —0,08 —0,57
19,3 3 3,29 13,661 13,91 —0,25 —1,80
20,0 4 3,54 14,191 +0,28 +2,02
16,9 3 2,94 13,94' +0,03 +0,22
17,5 1 3 Соединение I 5,30 1 24,271 Б —0,35 —1,42
10,6 3 3,30 24,951 24,62 +0,33 +1,34
14,6 1 3 1 4,54 1 24.92J +0,30 +1,22
13,2 1 3 Соединение 1 6,42 I 38,931 В | 39,03 I —0,05 I —0,13
10,9 1 3 I 5,24 1 38,53/ | —0,50 —1,23
Таблица 2
Результаты определения содержания хлора в 5-5,5-трихлорпента-
ноле, C5H9CI3O (А) и 4-хлоргептене-1, C7H13CI (Б)
Навес- ка, мг Время сожже- ния Ц- вре- мя дожи- гания кок- са, мин. а — Ъ, мл Содержание хл'ора % Абсолют- ная ошиб- ка опреде- ления А, % Относи- тельная ошибка определе- ни я А-100
найдено вычис- лено
%С1теор.
17,7 I 2
50,7 I 3
Соединена А
8,13 7,02 5,34 5,36 7,22 55,23 55,81 56,02 55,90 54,94J 55,55 —0,32 +0,26 +0,47 +0,35 —0,61 —0,57 +0,47 +0,85 +0,63 —1,1
Соединение Б
2,70 7,66 27,041 26.75J 26,74 +0,30 +0,01 +1,12 +0,04
Приведенные в табл. 1—3 данные по анализу соединений с извест-
ным содержанием серы, хлора и брома вновь подтверждают надеж-
ность пиролитического лампового метода, в частности, в предлагаемом
новом оформлении, которое позволяет вести анализ с максимальной от-
носительной ошибкой в пределах ± 2% при средней ошибке 0,7%. Оп-
ределение содержания серы в нефтепродуктах ничем не отличается от
определения содержания серы в органических соединениях, за исклю-
чением лишь необходимости брать большую навеску. Так, при содержа-
нии серы около 0,5% следует сжигать примерно 500 мг нефтепродукта.
При анализе бензинов, содержащих менее 0,1% серы, следует вместо
74
газовой микрогорелки укрепить лампу, применяемую для ускоренного
лампового метода определения серы в нефтепродуктах [5]. При опреде-
лении содержания серы и хлора или серы и брома из одной навески
сожжение проводят обычным образом, определяют условную кислот-
ность, а затем количественно переносят содержимое абсорбера в сосуд
Таблица 3
Результаты определения содержания брома в п-дибромбензоле,
СбЩВга (А), 2-бром-4-оксйгептаие, C7Hi5BrO (Б) и2,4-дибром-
гептане, C7Hi4Br2 (В)
Навес- ка, МВ Время сожжения, + время дожит ания кокса, мин. а — Ь, мл Содержание бро- ма, % Абсолют- ная ошиб- ка опреде- ления Д, % Относи- тельная ошибка определе- ния А-МО
найдено вычис- лено
% ВгтеОр.
Соедине ние А
34 ,2
30,7
29,8
24,5
24,4
17,9
34,7
25,3
3
3
3
3
3
2
2
66,95%
67,03
68,12
68,50
67,14
67,64
66,79
67,60^
67,75
—0,80
—0,72
+0,37
+0,75
—0,61
—0,11
—0,96
—0,15
—1,18
—1,06
+0,55
+1,10
—0,90
—0,16
—1,42
—0,25
Соединение Б
31,0 | 3 | 3,18 | 40,99 | 40,96| 4-0,03 | +0,07
Соединение В
I 4,65 I 61,941 I д, 0,1 0,00 I 0,00
| 4,03 | 61,94/ j 0,00 | 0,00
для потенциометрического определения галоген-ионов по данным рабо-
ты [3]. Прибор не предназначен для непосредственного определения
иода. В случае необходимости проведения анализа на содержание иода
в абсорбер следует поместить ~15 мл дистиллированной воды и приба-
вить две капли гидразингидрата. По окончании сожжения содержимое
переливают в сосуд для меркуриметрического титрования J~ [3].
ВЫВОДЫ
Предложена полуавтоматическая установка для количественного
экспресс-анализа органических соединений состава CHS, CHOS,
CHONS на содержание серы и соединений состава CHHal, CHOHal,
CHONHal на содержание галогенов (хлора или брома).
Институт нефтехимического синтеза им. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. Н. П. Волынский, И. К. Чудакова. Зав. лаб., № 12, 1435 (1955).
2. Н. П. Волынский, Г. Д. Гальперн, И. К. Чудакова. Зав. лаб., № 1, 27 (1957). ,
3. И. К. Чудакова, Г. Д. Гальперн, Н. П. Волынский. Методы анализа органических
соединений нефти, их смесей и производных, сб. 1. М., «Наука», 1960, стр. 107.
4. И. К. Чудакова, Г. Д. Гальперн, И. П. Волынский. Там же, стр. 21.
5. Н. П. Волынский. Зав. лаб., № 5, 536 (1955).
75
ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ СЕРЫ И ЕЕ ПРИМЕНЕНИЕ
К ИССЛЕДОВАНИЮ СУЛЬФИДОВ (ТИПА ВСТРЕЧАЮЩИХСЯ
В НЕФТЯХ) И ИХ ПРОИЗВОДНЫХ*
Н. Караулова
I. Введение
Общие положения, методика работы и многочисленные применения
метода тонкослойной хроматографии (ТСХ) изложены в двух моногра-
фиях [1, 2]. В монографии [2] приведены примеры ТСХ соединений раз-
личных классов, содержащих атом серы в молекуле (главным образом
биологически активных): сульфолипидов, витамина Bi, биотина, суль-
фонамидов, пенициллинов, фенилтиогидантоинов и серусодержащих
инсектицидов. В монографии [1] указаны условия разделения производ-
ных тиофена (как полярных, так и неполярных).
За время, прошедшее после написания названных выше монографий, появились
новые исследования, касающиеся ТСХ сераорганических соединений. Полученные при
этом данные могут оказаться полезными при изучении сераорганических соединений
нефти; краткий обзор их уместно привести.
Петшик и Штегер [3] с помощью ТСХ обнаружили примеси диэтилднтиофосфор-
иой кислоты (ДДТФ), образующиеся при ее синтезе [4]. ДДТФ наносилась на AI2O3,
закрепленную крахмалом. Элюэнтом служила смесь «-гептан—ацетон (10:1 по объ-
ему), проявителем — раствор HjJOe в НСЮ4, содержащий V2O5. При этом ДДТФ оста-
валась на старте, примеси—(ЕЮ)2 PSSEt и (ЕЮ)2 PSH — двигались; в этих же
условиях двигался диэтилсульфид.
Хавирж, Хромый и Вржестл [5] предложили использовать для чувствительного из-
бирательного проявления сераорганических соединений при ТСХ коммерческий препа-
рат флуоресцеин-1,3,6,8-тетрамеркуртриацетата (ТМФ). Вещества, содержащие
группы — SH и — C(S)NH2 гасят желто-зеленую флуоресценцию ТМФ. Сульфиды и
полисульфиды гасят флуоресценцию ТМФ только после восстановления натрием
в жидком аммиаке. Проявление с ТМФ не менее, чем на порядок чувствительнее, не-
жели открытие при помощи нитропруссида.
Тиоллактоны и соответствующие меркаптокислоты
(^СООН
SH
разделяли [6] на закрепленном слое силикагеля. Тиоллактоны легко размыкаются при
действии щелочей, образующаяся при этом SH-группа обнаруживается действием 2%-
ного нитропруссида натрия в 75%-ном спирте. Лучше всего шло деление с элюэнтом
диизопропиловый эфир — изооктан 3 : 2.
* Экспериментальная часть работы выполнялась при участии В. Ш. Шайхразие-
вой, В. Д. Никитиной, Т. С. Бобруйской, И. В. Черепановой, Ф. В. Козловой.
76
Чистота у-тиапиронов
нов)
(в частности присутствие примесей соответствующих пиро-
О S S
S О S S
необходимая для полярографических исследований [7], устанавливалась с по-
мощью ТСХ (адсорбент — незакрепленный слой А12О3 третьей активности, температура
19° С, элюэнт —эфир, проявитель 0,1 Лг КМпО4, подкисленный фосфорной кислотой).
ТСХ в комбинации со спектроскопией применена Майером, Росмусом и Фабианом
£8] для открытия, разделения и идентификации 1,2-дитиол-З-тионов, 1,2-дитиол-З-онов,
их анилов, а также производных 1,2-тиазолин-5-тионов и ксантотеновой кислоты. При
исследовании сераорганических соединений нефти интересен прием, использованный
авторами: ртутные комплексы сернистых соединений растворяют в ацетоне и наносят
на пластинку со слоем силикагеля; стартовое пятно обрабатывают сероводородом, вы-
делившееся сераорганическое соединение хроматографируют.
Принцлер и сотр. (9} изучали тонкослойную (нисходящую) хроматографию алкил-
и арилтиолов, диалкил-, алкиларил- и диарилсульфидов на закрепленном слое окиси
алюминия. При адсорбционной хроматографии элюэнтом служил петролейный эфир,
проявителем — бромтимоловый синий. Авторы пришли к заключению, что при разде-
лении в этих условиях смеси алкил- или арилтиола с соответствующим сульфидом
различия Rf 102 составляют всего лишь 1—2 единицы (и не более 4), т, е. что адсорб-
ционная хроматография для решения таких задач недостаточно эффективна. Кроме
того, при адсорбционной хроматографии воспроизводимость Р; вследствие колебаний
влажности воздуха невысока. Авторы добились хорошей воспроизводимости (S(Rf) =
= 0,013) при распределительной хроматографии с «обращенной фазой»; адсорбент
влажная А12О3, пропитанная на 70% цетаном. Колебания атмосферной влажности при
этом уже не оказывали влияния. Этот метод позволил достичь удовлетворительного
разделения внутри каждого класса соединений. Для движения тиолов применялся
элюэнт — 96%-ная СН3СООН— ацетонитрил 1:3, проявитель — нитропруссид натрия.
В этих условиях были разделены амил-, гексил-, октил-, децил-, и тетр а децилмер капта-
ны. Арилтиолы элюировали смесью метанол — хлороформ — вода 5:15:1; разделение
нечеткое, о- и я-тиокрезолы разделить не удалось. Сульфиды — ди-н-пропил, ди-н-бу-
тил-, ди-я-октил- и ди-н-додецил- и асимметричные — алкилоктил- (алкил — Ci—С3,
Св, Се, Сю, С12) и метилдодецил- были разделены элюэнтом хлороформ — метанол 1 :3
(проявитель — КМпО4) • В каждой группе соединений адсорбируемость только для не-
которых соединений подчинялась уравнению Мартина (см. [2]).
Эртель и Хорнер (10] рекомендуют в качестве универсальных проявителей (в том
числе для насыщенных углеводородов) смесь раствора бихромата натрия или перман-
ганата калия и концентрированной серной кислоты. С помощью сернокислого раство-
ра перманганата была проявлена смесь фенилбензилсульфида, сульфоксида и сульфона,
разделенная ТСХ на силикагеле; элюэнт — хлороформ; К/ равно соответственно 0,95;
0,08 и 0,27.
Эту же последовательность хода Р/ в ряду сульфиды > сульфоны > сульфокси-
ды установили Фишбейн и Фаукерс, изучившие ТСХ 11 сульфоксидов, 20 сульфонов и
14 сульфидов [11]. Разделение осуществлено с помощью ТСХ на закрепленном слое
силикагеля. В качестве элюэнтов применяли смесь толуол — этилацетат 1 : 1 или же
2,5%-ный раствор ацетона в бензоле. Разделены диалкилсульфиды (алкил —Сз, “зо-
С3, С4, мзо-С4, зтор-Сд, Сз, изо-Сз, Се) и тиофан; диарилсульфиды (арил — СзНз,
С6Н5СН2, п-С1СеН4, n-NH2C6H4), диалкилсульфоксиды (алкил — Ci, С3, С4, мзо-
С4, Сз, Сз) и тиофансульфоксид; диарилсульфоксиды (арил — СзНз, СзНзСН2, я-СН3С3Н4,
п-С1СзН4); диалкилсульфоны (алкил — Ci — Сз) и дивинилсульфон; диарилсульфоны
(арил — СбН3, я-СН3С3Н4, я-С1С6Н4, я-РС3Н4, n-NH2C3Ht, jm-NH2C3H4); смешанные
ароматические сульфоны — я-хлорфенилметил-, я-хлорфенил-2,4,5-трихлорфенил-,
я-хлорфенилфенил- я-фтор-ж-нитрофенил-. Для замещенных арилсульфонов после-
довательность хода Rf такая: я-хлорфенил > я-фторфенил > я-аминофенил; для
изомерных аминофенилсульфонов — м-аминофенил > я-аминофенил. Для изученных
сульфоксидов, сульфонов и сульфидов характерно линейное увеличение Р/ с ростом
длины цепи.
При хроматографировании на бумаге цистеин окисляется в цистин, декарбоксили-
руется и затем претерпевает ряд более сложных превращений. Разделительная ТСХ
была применена для деления цистеина и продуктов его окисления на силикагеле, за-
крепленном гипсом [12]. Элюэнтами служили смеси: бутанол — уксусная кислота — во-
да (12:3:5 по объему); бутанол — пиридин — вода (1:1:1 по объему); фенол —
вода (4:1 — вес : объем).
77
ТСХ на пластинках с незакрепленным слоем AI2O3 была использована [13] для ка-
чественного анализа смеси а-замещенных винилсульфонов и цис- и транс- (3-замещен-
ных винилсульфонов (элюэнт — смесь петролейного эфира и эфира в отношениях от
1:1 до 1:2, проявитель — иод). Этим же методом авторы контролировали окисление
замещенных винилсульфидов н гидрирование замещенных винилсульфонов [14].
На X научной сессии по химии сераорганическнх соединений Прилежаева, Снегоц-
кий и Сюндюкова [15] сообщили о применении ТСХ для контроля окисления Р-оксисуль-
фндов до сульфонов. Методом ТСХ на окисн алюминия с применением элюэнта гек-
сан—эфир были разделены смеси RSCH2CH2OH, RSOCH2CH2OH, RSO2CH2CHOH и
РБОгСН^СНз. Для гомологов внутри каждого класса компонентов этой смеси R/ прак-
тически одинаковы. Авторами работы [15] установлено, что предел примеси винилокс-
этнлеульфида в соответствующем сульфоне, который можно обнаружить, пользуясь ТСХ,
составляет 0,2—0,3%. Там же Дронов с сотр. [16] описал разделение смеси сульфолана
и его галондопронзводных на окисн алюминия методом ТСХ; для каждого компонента
был определен порог чувствительности.
В нашей лаборатории за последние годы (1963—1966) тонкослой-
ная хроматография сераорганическнх соединений нашла широкое при-
менении при синтезе и исследовании реакций модельных сульфидов
(типа встречающихся в нефтях), а также при изучении превращений
сульфидов средних фракций нефти. Были найдены условия ТСХ для
представителей различных классов сераорганическнх соединений: ди-
алкилсульфидов, замещенных винилсульфидов, циклоалкилтиолов,
циклоалкилсульфидов, циклоалкилдисульфидов [17]; диалкилсульфокси-
дов, циклоалкилсульфоксидов, тиабициклансульфоксидов, арилалкил-
сульфоксидов, диарилсульфоксидов, арилалкенилсульфоксидов, суль-
фоксидов полуароматических тиабицикланов [18]; ароматических и по-
луаромэтических тиабицикланов [19]; тиабицикланолов [20]; тиабицик-
ланонов, тиабицикланов [21] и некоторых других (табл. 1). Большая
часть изученных сераорганическнх соединений по молекулярным весам
соответствовала сераорганическим соединениям средних фракций неф-
ти и их производным. Условия движения для перечисленных в табл. 1
соединений [кроме (C6Hi3)2S, (C6Hi3)2SO, (C6H5CH2)2SO, тиофансуль-
фоксида и диэтилдитиофорной кислоты] найдены впервые.
С помощью ТСХ контролировались скорость и глубина превраще-
ний в таких реакциях, как окисление сульфидов в сульфоксиды; терми-
ческая перегруппировка арилалкенилсульфидов; дегидратация тиаби-
цикланолов; восстановление дисульфидов в тиолы; присоединение ди-
этилдитиофосфорной кислоты к ненасыщенным сульфидам, полученным
из насыщенных сульфидов ромашкинского и южно-узбекского кероси-
на и др.
ТСХ успешно применялась для контроля разделения смесей сераор-
ганических соединений фракционированной перегонкой, микрохрома-
тографией и обычной колоночной хроматографией. С помощью ТСХ
исследовалась устойчивость сераорганическнх соединений при газо-
жидкостной хроматографии и по отношению к адсорбентам, применяв-.
мым при колоночной хроматографии.
Впервые применительно к сераорганическим соединениям была раз-
работана препаративная тонкослойная хроматография (ПТСХ) для
различных классов сераорганическнх соединений: сульфидов ненасы-
щенных [20] и насыщенных [21, 22]; тиабицикланолов [20]; кетосульфи-
дов, сульфоксидов [22]. Сочетанием методов ПТСХ (при контроле
с помощью ТСХ) и газо-жидкостной хроматографии (ГЖХ) удалось осу-
ществить препаративное разделение смесей стереоизомерных тиабицик-
ланолов, тиабицикланов и тиабициклансульфоксидов и дать количест-
венную оценку стереоизомерного состава смесей, образующихся при
нестереоспецифическом синтезе.
78
Брутто-фор-
мула
CjHgOS
C4H11O2PS2
CgHitjOaS
CeHj2S
CgHgOS
CgHuO^PSg
CgHisSg
CgHgS
c9h8s
gCHioS
CgHioS
CgHioS
Таблица 1
Условия аналитической ТСХ сераорганических соединений
Мол. вес Название, структурная формула Адсорбент Элюэнт
104 Тиофансульфоксид J 1 !L В ж 0,08
186 0 Диэтилдитиофосфорная кислота (С2Н5О)2Р^ \sh А Б 0,028
146 Ацетокситиофан ососн3 SH А г 0,149; 0,39
106 Цикло гексилтио л | | Б и 0,56
152 2-Тиаиндансульфоксид || [ 1 II s В Ж 0,08
266 Тиофаиилдиэтилдитиофосфат S 1 V>C2H,)2 А Г 0,55
172 Димер дигидротиофеиа А Г 0; 0,62
148 У\ 2-Метилтиаинден || || /XsZ\h3 СНз А А 0,44
148 - Ои А А 0,62
150 Аллилфенилсульфид C6H5SCH2CH=CH2 Б И 0,36 0,35
150 2-Метил-1-тиаиндан 1 || | S/\s/\cH3 А А
150 У\/\ 1-Тиахроман | || \/\/ S Б И 0,32
79
Таблица 1 (продолжение)
Брутто-фор- мула Мол. вес. Название, структурная формула Адсорбент Элюэнт
CsHwOS CsHhOS C9H14OS QHjsS QHxeOS CjoHiqS 166 168 170 156 172 142 z\/\ 1-ТиахромансульфоксидГ || | v¥ II О Изопропилфенилсульфоксид (CH8)2CH-SO-CeH5 2-Метил-1-тиабицикло-(4,3,0)-нонанон- -4 (смесь стереоизомеров) О II 2-Метил-1-тиабицикло-(4,3,0)-нонан S сн8 2-Метил-1-тиабицикло- (4,3,0)-нонан- сульфоксид (смесь стереоизомеров) S сн8 6-Метил-1-тиадигидронафталин в в в в в Б Ж ж Е В Ж И 0,23 0,41 0,25; 0,42 0,40; 0,51 0,3; 0,54; 0,61; 0,68 0,57; 0,68
C10H12S 164 2,5-Диметил-1-тиаиндан Н8С-/\ । V\s/\ 5 сн3 Б или В И 0,42
C10H12S C10H12OS 164 180 6-Метил-1-тиахроман Н8С— о Аллилбензилсульфоксид cgh5ch2sch2ch=ch2 II О Б или В В И Ж 0,35 0,19
Таблица I (продолжение)
Брутто-фор- мула Мол. вес Название, структурная формула Адсорбент Элюэнт
CwH^OS 180 6-Метил-1-тиахромансульфоксид 10 II 0 В ж 0,19
CioblieOS 184 2-Метил-1-тиадекалон-4 (смесь стерео- изомеров) О ui S СНз в Е 0,23; 0,45
CioHijS 170 2-Метил-1-тиадекалин (смесь стерео- изомеров) | 1 \/\А „ S СНз в В 0,36; 0,47
CioHigOS 186 2-Метил-1-тиадекалиисульфоксид (смесь стереоизомеров) | 1 J \:н3 11 в Ж 0,36; 0,50
CioHigS 186 0 2,4-Диметил-1-тиабицикло-(4,3,0)-но- наиол-4 (смесь стереоизомеров) Н3С ОН S СНз Б д 0,26; 0,39 0,52; 0,68
C10H20S 172 цис-2,8-Диметил-5-тианонен-3, изо- СзНн—S—СН==СНСН(СН3)а А Б 0,56
CioH'oS 172 транс-2,8-Диметил-5-тианоиеи-3 изо- СзНц—S—СН=СНСН(СНз)з А Б 0,56
Ci«H22S 174 Диизоамилсульфид [смесь структурных изомеров, главным образом (СНз)2СН(СН2)25(СН2)2СН(СН3)2 и (СН3)2СН(СН2)25СН(СНз)СН(СНз)2 с примесью (СН3)2СНСН(СНз)5СН(СН3)СН(СНз)2] А Б 0,3
CioH22OS 190 Диизоамилсульфоксид (смесь таких же структурных изомеров, как в случае диизоамилсульфида) 1 в в Ж 3 0,38 0,61
6 Методы анализа
81
Таблица 1 (продолжение)
БруттО-фор-
мула
Мол.
вес
Название, структурная формула
Адсорбент Элюэнт
CnHigOS
CuH2oS
СцНгоОЗ
СцНгдОБ
Ci2H10OS
C12H12S
C12H14O2S
C12H22OS
C12H22OS
198
184
200
200
202
198
222
214
214
2,6,8-Триметил-1-тиабицикло- (4,3,0)-
нонанон-4 (смесь стереоизомеров)
О
НзС
S СНз
НзС
2,6,8-Триметил-1 -тиабицикло-(4,3,0)-
нонан (смесь стереоизомеров)
S СНз
НзС
2,6,8-Триметил-1-тиабицикло- (4,3,0)-
нонансульфоксид (смесь стереоизоме-
ров) ________/\
В
В
Е
В
НзС
В
Ж
0,40; 0,50;
0,65
0,63; 0,75
0,50; 0,72
СНз
S
НзС ||
О
2,4-Диметил-1-тиадекалол-4 (смесь
стереоизомеров) Н3С ОН
НзС
Б
Д
0,13; 0,2®
S СНз
Дифенилсульфоксид СвН5—SO—С6Н5
Дициклогексилсульфид СвНц—S—СвНц
Ацетокси- 6-метил-1 -тиатетралин
НзС
В
в
А
Б
Ж
3
В
И
0,48
0,63
0,14
0,0Ф
S ОСОСНз
Дициклогексилсульфоксид
СвНц—SO—СвНц
2,4,6,8-Тетраметил-1-тиабицикло-(4,3,
0)-нонанол-4 (смесь стереоизомеров)
Н8С ОН
В
В
Б
Ж
Ж
д
0,37
0,39
0,20; 0,40
HSC
S СНз
НзС
82
Таблица 1 (о кончание)
Брутто-фор- мула Мол. вес Название, структурная формула Адсорбент Элюэнт Rf
C12H22S2 230 Дициклогексилдисульфид СвНц—S2—СвНц A Б в и 0,53 0,48
CitHgeS 202 Ди-я-гексилсульфид CeHi8—S— CeHi8 A А 0,21
C12H26OS 218 Ди-я-гексил сульфоксид СвН1з—SO—СвН18 В В A Ж 3 А 0,39 0,59 0
CuHuOS 230 Дибензилсульфоксид CeH5CH2-SO-CH2CeH5 В Ж 0,20
C11H31O2S3P 358 Диэтилдитиофосфат 2,8-диметил-5- тианонена-3 мзо-СзН115СНСН2СН(СН8)2 S—Р< A Б 0,16
CieHiaOS 252 З-Нафтилфенилсульфоксид О II z\/x/s\z\ II 1 1II \/\z \/ В В Ж 3 0,49 0,67
QoHMOS 302 Ди-р-нафтилсульфоксид О II z\/\/s\z\/\ 1II 1 1 III \/\z \/\z В Ж 0,48
C20H42OS 330 Ди-н-децилсульфоксид C10H21 SO—СюН; 1 в в ж 3 0,53 0,72
II. АНАЛИТИЧЕСКАЯ ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ (ТСХ)
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ СЕРЫ
1. Методика работы
Применена восходящая ТСХ на незакрепленном слое сорбента (ме-
тод описан в монографии [1]), толщина слоя сорбента 0,5 мм\ исполь-
зована адсорбционно-элюэнтная хроматография. Абсолютные значения
Rf, полученные с помощью этого метода (см. табл. 1), не обладали, как
отмечено ран,ее (см. [9]), высокой воспроизводимостью, однако при раз-
делении смесей сохранялась постоянная разность Rf, при работе с под-
ходящим в каждом случае свидетелем получались вполне удовлетвори-
тельные результаты.
Адсорбенты: А. Силикагель марки «АСК», крупности 0,063—
0,16 мм1 2 * * * * * В., освобожденный от феррогеля, активность 6 (по бензолу).
Б. Окись алюминия коммерческая, марки «для хроматографии», от-
сеянная от пыли, активность III (по Брокману).
В. То же, что в Б, но активность II.
Элюэнты (все отношения — объемные): А. 1,16% диизопропило
вого эфира в изооктане.
Б. 1,18% диизопропилового эфира в изооктане.
В. 1,96% » » »
Г. 20% » » »
6* 83
' Д. Изооктан — этиловый эфир 3 : 2.
Е. 10% изооктана в диизопропиловом эфире.
Ж. Ацетон — четыреххлористый углерод 1:4.
3. Хлороформ.
И. Изооктан.
Проявитель: пары иода.
Использовались пластинки 13X18 см и 4X18 см для одномерной
и 13 X 13 см для двухмерной ТСХ. Расстояние от старта до фрон-
та ГО—12 см. Индивидуальное вещество наносилось обычно в виде 1%-
ного раствора в гексане (только сульфоксиды — в виде растворов в
хлороформе). Смеси веществ наносились в соответственно большей
концентрации. Время проявления хроматограмм от нескольких минут
до получаса. Полученные данные см. в табл. 1.
2. Общие рекомендации
Силикагель (адсорбент А) можно рекомендовать для ТСХ сульфидов,
в том числе ацетоксисульфидов, и для разделения тиаинданов и тиаинде-
нов. Для разделения стереоизомеров тиабицикланов и их производных
адсорбент А непригоден. Однако не исключено, что можно будет достичь
разделения стереизомеров, используя более мелкие и узкие (монодис-
персные) фракции силикагеля. Адсорбент А обладает тем достоинством,
что движение и испарение элюэнта после ТСХ на нем происходит зна-
чительно быстрее, чем на адсорбентах Б и В.
Окись алюминия (адсорбенты Б и В) пригодна для ТСХ сульфидов,
дисульфидов и сульфоксидов. На этих адсорбентах возможно разделе-
ние стереоизомерных тиабицикланов, тиабицикланонов, тиабициклано-
лов и тиабициклансульфоксидов. Применение Б и В не оптимизировано —
выбор определялся тем, что было в наличии. Улучшения разделения,
так же как и в предыдущем случае, можно ожидать при увеличении мо-
нодисперсности адсорбента.
Элюэнты, как изооктан, так и растворы диизопропилового эфира
в изооктане А—В рекомендуются для движения и разделения сульфи-
дов. Постепенное увеличение в этом ряду доли полярного компонента
позволяет варьировать значение Rf и выбрать оптимальное для каждой
конкретной задачи.
Элюэнт Г (20% диизопропилового эфира, 80% изооктана) исполь-
зовался для разделения ацетокситиофана, димера дигидротиофена и
тиоф анилдиэтилдитиофосф ата.
Элюэнты Д (изооктан — этиловый эфир) иЕ (изооктан в диизопропи-
ловом эфире), в которых полярный компонент преобладает, целесооб-
разно использовать для разделения стереоизомеров полярных соедине-
ний— тиабицикланолов и тиабицикланонов.
Элюэнты Ж (ацетон — четыреххлористый углерод) иЗ (хлороформ)
подобраны для ТСХ сульфоксидов. Элюэнт Ж рекомендуется также
для разделения стереоизомеров сульфоксидов, образующихся за счет
различного пространственного положения сульфоксидной группы. При
работе с элюэнтом 3 рекомендуется трехкратное хроматографирование.
3. Примеры
1. Контроль окисления сульфидов в сульфоксиды
Для движения сульфоксидов применяли адсорбент В, элюэнты Ж
и 3. Чувствительность метода — 2,5—10 мкг (в зависимости от приро-
ды сульфоксида), что на порядок выше, чем при хроматографировании
на бумаге. При хроматографировании смеси сульфоксида и соответству-
84
ющего сульфида, в условиях дви-
жения сульфоксидов, сульфид
четко отделяется от сульфоксида
и уходит на линию фронта. В ус-
ловиях движения сульфидов (см.
табл. 1) сульфоксид остается на
старте. Это позволяет контроли-
ровать часто применяемое при
исследовании сульфидов нефти и
при синтезе соответствующих мо-
дельных соединений окисление
сульфидов в сульфоксиды и полу-
чать качественное представление
о составе продуктов реакции. Так,
чистый стереоизомер 2-метил-
1-тиадекалина (транс-транс-, см.
[22]) окисляют перекисью водоро-
да в уксусной кислоте. Пробы ре-
акционной смеси периодически
анализируют с помощью ТСХ в
условиях движения сульфидов
(адсорбент В, элюэнт В); через
15 мин. проба показывает, что
сульфида нет и, следовательно,
Рис. 1. Строение тиабициклансульфок-
сидов (модели Стюарта — Бриглеба).
a — гра«с-цис-гра«с-2-метил-1-тиадекалинсуль-
окисление окончено.
Окисление сульфидов пере-
кисью водорода идет нестереоспе-
цифично; при этом следует ожи-
ДЭТЬ уДВОеНИЯ числа стереоизоме- фоксид; б — транс~транс-транс~2 -метил-1-тиа
ров за счет двух ВОЗМОЖНЫХ Ори- декалиисульфоксид
ентаций сульфоксидной группы.
Из транс-транс-2-метил-1-тиаде-
калина могут образоваться транс-цис-транс и транс-транс-транс-сулъдр-
оксиды (правила номенклатуры те же, что и для замещенных тиабици-
кланов [21], причем SO-группа считается старшим заместителем) (рис. 1).
ТСХ 2-метил-1-тиадекалинсульфоксида в условиях движения сульф-
оксидов обнаруживает два пятна с Rf 0,36 и 0,50. О выделении этих
стереоизомеров препаративной тонкослойной хроматографией см. ниже
(стр. 91).
2. Контроль дегидратации карбинолов
Дегидратация карбинолов может быть одной из стадий .при синте-
зах модельных замещенных моно- и битиацикланов (см., в частности,
работы [20—23]). Так, например, 2,4-диметил-1-тиадекалол-4 и 2,4-димб-
тил-1-тиабицикло-(4, 3, 0)-нонанол-4 (оба карбинола в виде смеси стере-
оизомеров) дегидратируют нагреванием в присутствии серной кислоты
соответственно в 2,4-диметил-1-тиаокталин и 2,4-диметил-1-тиабицикло-
нонен. Время, необходимое для дегидратации, определяют периодиче-
ски, анализируя пробы реакционной смеси с помощью ТСХ в условиях
движения карбинолов (адсорбент Б, элюэнт Д); при этом образующие-
ся тиабициклены уходят на линию фронта.
Из данных ТСХ (рис. 2) следует, что 2,4-диметил-1-тиабицикло-(4,
3, 0)-нонанол-4 дегидратируется быстрее 2,4-диметил-1-тиадекалола-4.
Для обоих карбинолов скорость дегидратации отдельных стереоизоме-
ров различна.
85
3. Исследование устойчивости органических
соединений серы
Органические соединения серы могут изменяться при длительном
соприкосновении с адсорбентами, например в условиях колоночной хро-
матографии. Так, Лаба и Грачева [24] наблюдали стереоспецифическое
отщепление сульфиновых кислот от 1,2-дисульфонов при выдержива-
нии на пластинках с окисью алюминия в течение 6 час.
Мы нашли, что при микроэлюэнтной колоночной хроматографии сме-
си £{ас-гранс-изомеров 2-метил-Гтиадекалона-4 (требующей не менее
10 час.) происходит изомеризация с образованием транс-транс-изомера;
при этом часть продукта осмоляется.
При работе с сульфидами и их производными целесообразно прове-
рить устойчивость органического соединения серы в предполагаемых
условиях хроматографии. Одним из методов, позволяющих во многих
случаях получить на это ответ, является двухмерная ТСХ. На несколь-
ко стеклянных пластинок размером 13X13 см наносят слои адсорбента
толщиной 0,5 мм. На один из углов каждой пластинки одновременно
впитывают исследуемый образец органического соединения серы и так-
же одновременно элюируют все пластины (в условиях движения образ-
ца). После этого одну из пластин тотчас же подвергают элюированию
в направлении, перпендикулярном первому. Остальные пластины перед
повторным (двухмерным) хроматографированием выдерживают в те-
чение различных промежутков времени. Если изменений вещества за
время контакта не произошло, то, после проявления хроматограммы,
пятна находятся на диагонали; при изменениях образца пятна сходят
с диагонали (часто появляются стартовые пятна).
Рис. 2. Дегидратация
тиабицикланолов
(контроль методом ТСХ)
а — 2,4-диметил-1-тиадекалол-4;
б — 2,4-диметил-1-тиабицикло-
(4, 3, 0)-нонанол-4.
Цифрами обозначено время ре-
Рис. 3. 2-Метил-1-тиадекалин после выдерживания
на окиси алюминия; контроль изменений методом
двумерной ТСХ
а — в течение 8 час.; б —в течение 24 час.
акции (часы)
В качестве иллюстраций может служить поведение смеси стереоизо-
меров 2-метил-1-тиадекалина на нейтральной окиси алюминия (рис. 3).
За 8 час. сульфид не изменяется — пятна находятся вблизи диагонали,
за 24 часа заметны четкие изменения образца, часть его остается на
старте.
Органические соединения серы могут претерпевать изменения и при
газо-жидкостной хроматографии. В тех случаях, когда нет аппаратуры
для препаративной ГЖХ, можно моделировать условия ГЖХ и исследо-
вать продукт с помощью ТСХ. Две капли исследуемого образца тща-
86
дельно перемешивают с несколькими граммами фазы ГЖХ и выдержи-
вают в атмосфере газа-носителя при температуре и в течение проме-
жутка времени, равных температуре и времени выхода образца при
ГЖХ. Фазу экстрагируют этиловым эфиром, полученный экстракт ана-
лизируют методом ТСХ. Отсутствие изменений тонкослойной хромато-
граммы, конечно, не является строгим доказательством того, что обра-
зец при ГЖХ не изменился, изменения же в ТС-хромэтограмме свиде-
тельствуют о превращениях образца при ГЖХ.
Рис. 4. Превращение стерео-
изомеров 2-метил-1-тиабицикло-
(4, 3, 0)-нонанона-4 при нагре-
вании на фазе ГЖХ, контроль
по ТСХ
А А' 6 6'
Так, например, при нагревании в условиях ГЖХ двух различных
стереоизомеров 2-метил-1-тиабицикло-(4, 3, 0)-нонанона-4 по меньшей
мере один из стереоизомеров изменяется, изомеризуясь в другой
(рис. 4).
4. Разделение смесей сульфидов различного
строения
а) Разделение предельных и непредельных сульфидов сходного
'Строения. При исследовании сульфидов средних фракций нефти в на-
шей лаборатории используется реакция Пуммерера [25]. С помощью
.этой реакции насыщенные сульфиды превращают в ненасыщенные.
RSCHjj R' ±^RSCHaR'____________ RSCH=GHR.
II (CH3CO)20,t°
О (реакция Пуммерера)
При этом может происходить частичное окислительно-восстановитель-
ное диспропорционирование сульфоксида с образованием исходного суль-
фида. Так, например, в результате нагревания диизоамилсульфоксида
в среде уксусного ангидрида после ректификации продукта реакции бы-
ла получена смесь ^ыс-траяс-изомеров 2,8-диметил-5-тианонена-3, со-
держащая около половины диизоамилсульфида. Такую смесь путем га-
зо-жидкостной хроматографии вследствие близости времен удержива-
ния разделить не удалось. Однако в условиях ТСХ (адсорбентА, элюэнт
Б) наблюдалось два четких пятна (рис. 5). Одно пятно с 7?^=О,3 соот-
ветствовало диизоамилсульфиду, другое cR/=0,5 — смеси цис-транс-пзо-
меров 2,8-диметил-5-тианонена-3. Данные ТСХ послужили основанием
для выбора условий колоночной элюэнтной хроматографии, с помощью
которой был отделен диизоамилсульфид. После его удаления оставшая-
ся смесь цис-транс-изомеров была четко разделена с помощью препа-
ративной ГЖХ на цис- и транс-2,8-диметил-5-тианонены-3. Таким об-
разом, целесообразным оказалось сочетание методов ГЖХ и ТСХ.
87
При взаимодействии упомянутой выше смеси диизоамилсульфида и
2,8-диметил-5-тианонена-3 с диэтилдитиофосфорной кислотой в реакцию
вступает только непредельный сульфид. Полученный продукт и неиз-
мененный диизоамилсульфид разделяют перегонкой, ТСХ (адсорбент
Е, элюэнт Б) позволяет контролировать ход разделения и чистоту полу-
ченных соединений (рис. 5).
Рис. 5. Тонкослойная хромато-
графия смеси диизоамилсульфи-
да и цис- и транс-2,8-диметил-5-
тианоненов-3 (Л); диэтилдитио-
фосфорной кислоты (Б); диизо-
О амилсульфида (В); диэтилди-
тиофосфата 2,8-диметил-5-тиа-
нонена-3 (Г)
б) Разделение смесей стереоизомеров тиабицикланов. 2-Метил-1 -тиа-
декалин получают восстановлением 2-метил-1-тиадекалона [21]. Пос-
ледний синтезируют по [21] или [26] циклизацией продукта присоедине-
ния сероводорода к пропенилциклогексенилкетону. Как для тиакето-
на, так соответственно и для продукта его восстановления возможно об-
разование четырех пространственных изомеров (рис. 6).
Из стереохимических соображений следует ожидать большую ве-
роятность образования трех из них (номенклатуру см. в работах [19],
Рис. 6. Строение тиадекалинов (модели Стюарта — Бриглеба)
а — транс-цис-2-ыетил-1 -тиадекалин; б — цис-траис-2-метнл-1-тиадекалин; в — траис-транс-2-метнл-
1-тиадекалнн; г — цис-цис-2-метил-1 -тиадекалин
88
[21]), а именно: транс-2-;иетал-г{ыс-1-тиадекалина, цис-2-метил-транс-1*
тиадекалина и транс-2-метил-транс-1-тиадекалина.
Действительно, по данным ГЖХ можно обнаружить четыре ожидае-.
мых стереоизомера, два из которых резко преобладают (рис. 7, пики а, б).
Методом ТСХ (адсорбент В, элюэнт В) удается разделить такую смесь нд
Рис. 7. Анализ смеси
стереоизомеров
2-метил-1 -тиадекалина
I — ГЖХ; II — ТСХ той же снеси и
фракций после ПТСХ
два компонента ч(см. рис. 7, II): с Rf 0,37 и j0,49. Один из них (Rf 0,37>
представляет собой, по-видимому, транс-транс-изомер. (Такой транс-^
транс-изомер с Rf 0,37 был синтезирован нами независимо восстановле-.
нием транс-транс-2-метил-1-тиадекалона.)
Упомянутую смесь стереоизомеров 2-метилтиадекалина с помощью»
препаративной ГЖХ разделить не удалось. Данные ТСХ послужили ос-
нованием для разработки методики ПТСХ, позволившей достичь раз-,
деления (см. ниже).
III. ПРЕПАРАТИВНАЯ ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ
(ПТСХ) ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ СЕРЫ
1. Методика работы
Адсорбенты: А. Нейтральная окись алюминия активность II.
Б. Нейтральная окись алюминия, активность III.
Элюэнты: А. Эфир — изооктан 1:5 (соотношения объемные).
Б. Изооктан.
В. Ацетон — четыреххлористый углерод 1:4.
Г. Изооктан — эфир 3 :2.
Проявитель: Раствор 0,5 г перманганата калия в 25 мл концентриро-.
ванной серной кислоты. При проявлении в зонах нахождения адсорби-.
рованных веществ фиолетовая окраска адсорбента изменяется на свет-
ло-желтую или бурую.
На стеклянную пластину размером 21X30 см наносят, разравни-
вая стальным катком с соответствующим зазором, слой адсорбента,
толщиной 2 мм. По всей длине короткой стороны пластины намечают,,
отступя от края на 1,5—2 см, линию старта в виде небольшого углубле-
ния. На эту линию равномерно наносят пипеткой раствор хроматогра-
фируемой смеси. Дают растворителю улетучиться, после чего пласти-
ну помещают в камеру с элюэнтом и производят двукратное элюирова-
ние (в одном и том же направлении). После испарения элюэнта хрома-
тограмму проявляют, смачивая оба края вдоль длинных сторон пласти-
89.
ны проявителем (проявитель наносят с помощью пипетки с тонко
оттянутым кончиком).
После проявления краев намечают зоны разделения веществ поперек
-пластины и собирают их, всасывая адсорбент с помощью фильтра с по-
ристой пластинкой № 2. Адсорбент на фильтре исчерпывающе вымыва-
ют эфиром (полноту элюирования и ход разделения контролируют ме-
тодом ТСХ в условиях движения соответствующих соединений). Если
«при однократной ПТСХ получают промежуточные фракции, то их хро-
матографируют многократно до тех пор, пока не будет достигнуто чет-
кое деление.
2. Примеры
1. Разделение сульфидов
Адсорбент —А, элюэнт — Б. Загрузка — раствор 0,1—0,15 г суль-
>фида в 2 мл изооктана. Время хроматографирования 3—4 часа.
а) Разделение стереизомеров 2-метил-1-тиадекалина. Смесь стерео-
изомеров, «л20 1,5132 (см .рис. 7, /) методом ПТСХ разделена на две
“фракции, А и Б (см. рис. 7, //), общий выход после ПТСХ составляет
96% от загрузки. Фракция A (nD20 1,5160) составляет 44,3% от получен-
ного продукта, фракция Б (nD20 1,5138) соответственно — 55,7%. По
данным ГЖХ, фракция А содержит на один изомер менее, нежели ис-
-ходная смесь (см. рис. 7, I и 8).
Рис. 8. Газо-жидкостная
хроматография фракции А
после ПТСХ
2-метил-1-тиадекалина
Такую смесь уже значительно легче делить препаративной ГЖХ;
’четкое разделение пиков дает возможность рассчитать соотношение изо-
меров. Фракция Б представляет собой, по-видимому, один стереоизо-
мер, отличный от транс-транс-2-метил-тиадекалина, о чем свидетельст-
вует ГЖХ искусственных смесей т/?ояс-транс-2-метил-1-тиадекалина с
фракциями А и Б (рис. 9).
Таблица 2
Стереоизомерный состав смеси 2-метил-1-тиадекалинов
Пики по рис. 8, 9 Относительные удерживаемые объемы (стан- дарт С„Н>4) Состав фракций, ПТСХ, % Содержание стереоизомеров, %
А (45 %) Б (55 %) во фракциях ПТСХ В ИСХОДНОЙ смеси
А 1 Б
а 1,29 46,5 5 21 3 24
б 1,36 0 95 0 52 52
в 1,57 40,0 Следы 18 Следы 18
г 1,69 13,5 0 5,0 0 6
•80
Состав фракций ПТСХ А и Б рассчитывают так: взвешивают пики
ГЖХ и, принимая, что каждый пик соответствует одному стереоизоме-
ру, экстраполируют, учитывая выходы фракций Л и Б, до состава исход-
ной смеси (табл. 2) (условия ГЖК см. [22]). Аналогично выполнено [20]
деление смесей стереоизомеров 2-метил-1-тиабицикло-(4, 3, 0)-нонанов
и 2,6,8-триметил-1-тиабицикло-(4,3,0)-нонанов (см. табл. 1).
б) Отделение 2,4-диметил-1-тиадекалина от 2,4-диметил-1-тиаокта-
.линов. 2,4-Диметил-1-тиаокталины (смесь стереоизомеров и структур-
ных изомеров с различным положением двойной связи) гидрируют над
Рис. 9. Газо-жидкостная хроматография ГЖХ Рис. 10. Контроль ПТСХ
•искусственной смесн транс-гранс-2-метил-1-тиадекалина 2-метил-1-тнадекалинсульф-
1 — с фракцией А (1 : 1); II -с фракцией Б ОКСИДЭ методом АТСХ
Pd/C при 50° и 70 атм в 2,4-диметил-1-тиадекалин, причем гидрирова-
ние полностью не идет. Продукт гидрирования делят с помощью ПТСХ
на две фракции. Из каждых 0,100 г получают 0,013 г (13%) фракции
-с nD20 1,5290, состоящей, по данным ГЖХ и ИК спектроскопии, главным
образом из ненасыщенных сульфидов, и 0,075 г (75%) фракции с Hd29
1,5214, представляющей собой, по данным ГЖХ и ИК спектроскопии,
2,4-диметил-1 -тиадекалйн. Аналогичным образом очищают 2,4-диметил-
1-тиабицикло-(4, 3, 0)-нонан от примеси 2,4-диметил-1-тиабицикло-(4,3,
•0)-ноненов [20].
2. Разделение стереоизомеров сульфоксидов
Адсорбент — Б; элюэнт — В; загрузка — раствор 0,14—0,15 г суль-
фоксида в 2 мл изооктана; время хроматографирования 5—6 час. Смесь
«стереоизомерных 2-метил-1-тиадекалинсульфоксидов с nD20 1,4986 и
Rf 0,36 и 0,50 (см. выше) была разделена на две фракции — А и Б
(рис. 10), общий выход равен 87% от загрузки. Фракция A (nD20 1,4721 и
R/ 0,38) составляет 70,8%, а фракция Б (Rf 0,57, т. пл. 74,5—76°С после
•перекристаллизации из изооктана) соответственно 29,8% от загрузки.
По аналогии с данными по замещенным тиациклогексансульфокси-
дам можно фракции А приписать транс-конфигурацию, а фракции Б —
цис-конфигурацию (см. рис. 1). Аналогично [22] делят смеси стереоизо-
мерных 2-метил- и 2,6,8-триметил-1-тиабицикло-(4,3,0)-нонансульфо-
.ксидов.
91
Рис. 11. Строение тиадекалолов
(модели Стюарта — Бриглеба)
а — транс-транс-транс-2,4-диметил-
1-тиадекалол-4; б — цис-транс-транс-
2,4-диметил-1-тиа дека лол-4
3. Разделение стереоизо-
меров тиабицикланонов
Адсорбент — Б; элюэнт — А; за-
грузка— раствор 0,1—0,2 г кето-
сульфида в 10 объемах изооктана..
Однократное элюирование требует'
около 1 часа, испарение элюэнта —
0,5 часа. Общее время хроматогра-
фирования 3—4 часа.
Смесь стереоизомерных 2-метил-
1-тиабицикло- (4,3,0) -нонанонов, nD27
1,5242 с Rf 0,30 и 0,43 (см. табл. 1)
разделена на две фракции—А и Б-
с общим выходом 81,5% от загруз-
ки. Фракция А (nD2a 1,5278, Rf 0,30)
составляла 45%, а фракция Б (nD20‘
1,5170, Rf 0,43)—55% от загрузки.
В условиях ГЖХ [22] фракция Б не
изменяется, но фракция А частично*
изомеризуется в Б. Фракция Б вы-
ходит при ГЖХ в виде индивидуаль-
ного пика, а фракция А — в виде
двух примерно равных пиков, один-
из которых Б. Можно предполагать,
что второй пик во фракции А обна-
руживается как результат измене-
ний при ГЖХ стереоизомера, да-
ющего первый пик, т. е. что фрак-
ции А и Б, полученные путемз
ПТСХ, представляют собой индиви-
дуальные стереоизомеры. 4 * * *
4. Разделение стереоизомеров тиабицикл анолов
Адсорбент — Б; элюэнт — Г. Применяют трехкратное элюирование;,
загрузка — раствор 0,1—0,2 г карбинола в изооктане. Выходы 85—95%..
6
Рис. 12.
а —ТСХ тиабициклановов 1Н,
б — контроль
Анализ тиабицикланолов
IV, IV и полученных из них карбинолов I, la, II, V;.
ПТСХ карбинолов с помощью АТСХ
62
При получении тиабицикланолов по реакции Гриньяра из соответст-
вующих тиабицикланонов для каждого стереоизомера возможно обра-
зование двух карбинолов с цис- или транс-положением ОН-группы (по
отношению к ангулярному атому водорода, ближайшему к атому серы)
(рис. II).
Так, при синтезе 2,4-диметил-1-тиадекалола-4 (I) и 2,4-диметил-1-ти-
абицикло-(4,3,0)-нонанола-4 (II) соответственно из 2-метил-1-тиадекало-
на-4 (III) и 2-метил-1-тиабицикло-(4,3,0)-нонанона-4 (IV), по данным
ТСХ, наблюдается удвоение числа пятен; синтез 2,4,6,8-тетраметил-1-тиа-
<бицикло-(4,3,0)-нонанола-4 (V) из 2,6,8-триметил-1-тиабицикло-(4,3,0)-
нонанона-4 (VI), вероятно, протекает стереоспецифично (рис. 12).
Один из стереоизомеров 2,4-диметил-1-тиадекалола-4 [вероятно
транс-транс-транс-изомер (la)] вследствие различия в температурах
плавления выделяется в виде кристаллов уже в процессе синтеза. Ма-
точный раствор (16) после отделения твердого 2,4-диметил-1-тиадекало-
ла-4 (1а), а также 2,4-диметил-1-тиабицикло-(4,3,0)-нононолы-4 (II) и
*2,4,6,8-тетраметил-1-тиабицикло-(4,3,0)-нонанолы-4 (V) методом ПТСХ
делят на фракции, обнаруживающиеся в виде одного пятна (см. рис. 12
>и табл. 3).
Таблица 3
Разделение тиабицикланолов методом ПТСХ
Фракции ПТСХ
А Б В Г
Т. пл.* 0,16 15 1,5271 0,30
38-40° С
(44—45° С)
1,5233 0,26 16 1,8181 0,39
Отсут-
ствует
Чб 1,5280 0,13 85
0,26
II 1,5218 0,26 23
0,39
0.52
0,68
V 1,5028 0,20 47
0,40
1,5088 0,20 53 1,5072 0,40
Отсут-
ствует
1,5220 0,52 7 1,5255 0,68
• После перекристаллизации из изооктана.
Можно предполагать, что фракции, полученные после ПТСХ карби-
нолов, представляют собой индивидуальные стереоизомеры.
выводы
Рассмотрены литературные данные по ТСХ сераорганических соеди-
нений за 1962—1965 гг.
Впервые приведены условия ТСХ ряда сераорганических соединений.
Приведены примеры применения ТСХ при синтезе и превращениях
сераорганических соединений.
Предложена методика препаративной тонкослойной хроматографии
(ПТСХ) различных классов сераорганических соединений: тиабицикла-
«онов, тиабицикланолов, тиабицикланов и тиабициклансульфоксидов.
93
Показано, что методика ПТСХ сераорганических соединений пригод-
на для полного или частичного разделения смесей стереоизомеров.
Институт нефтехимического синтеза им. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. А. А. Ахрем, А. И. Кузнецова. Тонкослойная хроматография. М., «Наука», 1964.
2. Хроматография в тонких слоях (под ред. Э. Шталя). М., «Мир», 1965.
3. Н. Petschik, Е. Steger. J. Chromat., 9, 307 (1962).
4. М. И. Кабачник, Т. А. Мастрюкова. Изв. АН СССР, ОХН, 1953, 121.
5 Л Havir, V. Chromy. Vrestal. Chem. Listy, 59, 431 (1965).
6 F. Korte, J. Vogel. J. Chromat., 9, 381 (1962).
7. C. Parkanyi, R. Zahradnik. Collect. Czech. Chem. Commun., 27, 1355 (1962).
8. R. Mayer, P. Rasmus, J. Fabian. J. Chromat., 15, 153 (1964).
9. H. W. Prinzler, D. Pape, M. Teppke. J. Chromat., 19, 375 (1965).
10. H. Ertel, L. Horner. J. Chromat., 7, 268 (1962).
11. L. Fischbein, J. Faukers. J. Chromat., 22, 323 (1966).
12. G. J. Banker, B. L. Tonge. J. Chromat., 12, 53 (1963).
13. £ П. Грачева, В. И. Лаба, Н. К. Ку львовская, М. Ф. Шостаковский. ЖОХ, 33,
2493 (1963).
14. Е. П. Грачева, В. И. Лаба, М. Ф. Шостаковский. ЖОХ, 33, 2501 (1963).
15. Е. Н. Прилежаева, В. И. Снегоцкий, В. X. Сюндюкова. X Научная сессия по химии
сераорганических соединений нефти и нефтепродуктов (тезисы докладов). Уфа,
1966, стр. 61.
16. В. И. Дронов, В. А. Снегоцкая, Л. И. Коленченко, Д. П. Ворончихина. Там же,
стр. 61.
17. Е. Н. Караулова, Г. Д. Гальперн, В. Д. Никитина, И. В. Черепанова, Д. К- Жестков,.
Л. Р. Барыкина, Г. Ю. Пек, Ф. В. Козлова. Нефтехимия (в печати). .
18. Е. Н. Караулова, Т. С. Бобруйская, Г. Д. Гальперн. ЖАХ, 21, 893 (1966).
19. Е. Н. Караулова, Г. Д. Гальперн, В. Д. Никитина, И. В. Черепанова. Нефтехимия»
7, 774 (1967).
20. Е. Н. Караулова, Г. Д. Гальперн, В. Ш. Шайхразиева, И. В. Черепанова, Л. Р. Ба+
рыкина. Нефтехимия, 8, 102 (1968).
21. Е. Н. Караулова, В. Ш. Шайхразиева, Г. Д. Гальперн. Химия гетероциклических
соединений, № 1, 51 (1967).
22. Е. Н. Караулова, Г. Д. Гальперн, В. Ш. Шайхразиева, И. В. Черепанова. Нефтехи-
мия, 7, 812 (1967).
23. R. Onesta, G. Castelfranchi. Gazz. chim. ital., 89, 141, 1127 (1959).
24. В. И. Лаба, Е. П. Грачева. ДАН СССР, 155, 1357 (1964).
25. Е. Н. Караулова, Г. Д. Гальперн, В. Д. Никитина. IX Научная сессия по химии се-
раоргаиических соединений нефти и нефтепродуктов (тезисы докладов). Уфа, 1965*.
стр. 47.
26. И. Н. Назаров, А. И. Кузнецова, И. А. Гурвич. ЖОХ, 20, 2150 (1950).
МЕТОДЫ ПОЛУЧЕНИЯ СУЛЬФОКСИДОВ
ИЗ СРЕДНИХ ФРАКЦИЙ СЕРНИСТЫХ НЕФТЕЙ
(ГРУППОВОЕ ВЫДЕЛЕНИЕ СУЛЬФИДОВ В ВИДЕ
СУЛЬФОКСИДОВ)
Е. Н. Караулова, Т. А. Бардина, F. Д. Гальперн
В 1960 г. нашей лабораторией была опубликована методика коли-
чественного выделения сульфидов из сернисто-ароматических концентра-
тов керосиновых фракций нефти [1]. Суть ее заключалась в селективном,
окислении сульфидов до сульфоксидов перекисью водорода в присут-
ствии уксусной кислоты в качестве растворителя и в последующем хро-
матографическом выделении образующихся сульфоксидов. В этом спосо-
бе отправным пунктом исследования была общеизвестная аналитическая
методика окисления сульфидов перекисью водорода в среде ледяной
уксусной кислоты.
Неясность роли уксусной кислоты и деталей механизма реакции,
а также сложность и сравнительно высокая стоимость этой методики за-
ставили нас рассматривать ее как предварительную. В последующие-
годы мы продолжали поиск более совершенных вариантов окислитель-
ных методов выделения сульфидов в виде сульфоксидов [2].
В итоге исследований мы предлагаем несколько методов окисления
сульфидов и нефтяных дистиллятов и выделения сульфоксидов из окис-
ленных смесей. Часть методов может быть успешно применена на опыт-
ных и полупромышленных установках, некоторые методы пригодны-
главным образом для лабораторного использования.
1. ВЫДЕЛЕНИЕ СУЛЬФОКСИДОВ ИЗ ОКИСЛЕННЫХ ДИСТИЛЛЯТОВ
А. Хроматографический метод (элюэнтно-вытеснительный)
Метод основан на значительно большей адсорбируемости сульфокси-
дов по сравнению с ароматическими углеводородами и неокисленными
сернистыми соединениями. Он применим к растворам сульфоксидов в не-
полярных жидкостях (а также в нефтяных дистиллятах) любой кон-
центрации, в том числе к весьма разбавленным. Рекомендуемая методи-
ка включает предварительное хроматографирование пробной загрузки.
В результате однократного хроматографирования растворов сульфокси-
дов в неполярных растворителях концентрация сульфоксидов возрастает
не менее чем в 20 раз. Способ, подробно описанный ниже, дает возмож-
ность получить рафинат, полностью свободный от сульфоксидов. Хрома-
тографический метод наиболее целесообразно использовать при таких
лабораторных работах, как выделение сульфоксидов из окисленных
нефтяных дистиллятов с малым содержанием сульфоксидной серы (на-
9F
пример, менее 0,3%). Хроматографический метод [3] может быть успешно
применен для очистки индивидуальных сульфоксидов от примеси нео-
кисленных сульфидов, для очистки сульфидов (полученных восстановле-
нием сульфоксидов) от примеси невосстановленных сульфоксидов, а так
же во всех других случаях, где необходимо количественное разделение
«сульфидов и сульфоксидов.
Аппаратура
Стеклянные хроматографические колонки разных размеров.
Реактивы
1. Технический силикагель марки «АСК» активностью не менее 8—
10 по о-оксихинолину (определение активности см. ниже).
2. Бензол.
3. Этиловый спирт — ректификат.
4. Петролейный эфир с к. к. не выше 60° С, не содержащий аромати-
ческих углеводородов (формолитовая проба отрицательная!).
Методика работы
а) Определение емкости силикагеля пд о-оксихинолину. В колонку
'(внутренний диаметр 10 мм) помещают 10,0 г силикагеля и пропуска-
ют 10%-ный раствор о-оксихинолина в бензоле. Отмечают объем вытек-
шего бензола в тот момент, когда окрашивание столба силикагеля до-
стигает низа колонки.
Емкость силикагеля * = 10/9 X число мл бензола.
б) Хроматографирование пробной загрузки. Колонку (внутренний
диаметр 10 мм) наполняют 8 г силикагеля. Силикагель смачивают пет-
ролейным эфиром. Затем в колонку вводят такое количество окисленно-
го дистиллята, которое соответствует содержанию 0,04 г сульфоксидной
серы (оно отвечает отношению силикагель — сульфоксидная сера, рав-
ному 200: 1).
После впитывания загрузки колонку промывают бензолом. Вытека-
ющий бензол проверяют на отсутствие сульфоксидов, отбирая по 10 мл
элюата, из которого испаряют бензол; в остатке проводят качественную
пробу на сульфоксиды по методике, приведенной в работе [4]. Если ре-
акция положительна, то повторяют хроматографирование пробы, увели-
чив относительное количество силикагеля. Если силикагеля достаточно,
то продолжают промывание бензолом до объема десорбата, равного
300 мл. Последние 10 мл элюата вновь проверяют на сульфоксиды. При
отсутствии сульфоксидов в пробе можно считать, что сульфоксиды иссле-
дуемой окисленной фракции (при данном относительном количестве си-
ликагеля) бензолом не вымываются. В тех случаях, в которых бензол
может вымыть сульфоксиды, при хроматографировании основной массы
окисленной фракции не следует брать бензол в избытке. Хроматографи-
рование пробы требует не более одного рабочего дня.
в) Хроматографирование основной массы окисленного продукта.
Колонку наполняют таким же силикагелем, какой был взят для хро-
матографирования пробы. Относительное количество силикагеля опре-
деляют на основании хроматографирования пробы. Силикагель смачи-
вают петролейным эфиром, вводят окисленную фракцию и промывают
колонку бензолом до полного элюирования неокисленных компонентов.
Если при хроматографировании пробы оказалось, что избыток бен-
зола может вытеснить сульфоксиды, то следует более точно определить
* Емкость силикагеля, определенная этим методом, представляет собой удельную
адсорбцию, умноженную на 100 (см. [3]).
96
начало вытекания чистого бензола. Для этого перед концом вымывания
несульфоксидных компонентов отбирают пробы элюата (по 50 мл) и
следят за уменьшением остатка после удаления бензола. После вымы-
вания неокисленных компонентов все сульфоксиды вытесняют спир-
том. Из десорбата отгоняют спирт до постоянного веса остатка.
Пример 1
Широкая фракция южно-узбекской нефти, т. кип. 200—400° С, содержавшая около
1,5% сульфидной серы, окислена гетерогенно-эмульсионным методом с добавкой малых
количеств серной кислоты (см. ниже, стр. 103). Получен углеводородный слой с содер-
жанием сульфоксидной серы (Sso)-1,1%. Ход хроматографического выделения суль-
фоксидов см. табл. 1; количество силикагеля 200 г/г Sso. Состав продукта:
С14.1 Н27,9 SO1 ,38-
Пример 2
«Бензольный элюат», полученный при выделении сульфоксидов из окисленной ши-
рокой фракции южно-узбекской нефти, как описано в примере 1, окислиют в растворе
уксусного ангидрида и метилэтилкетона (см. ниже, стр. 101). После обработки получа-
ют 433 г окисленного продукта с содержанием Sso 0,38%. Хроматографированием вы-
деляют 16,7 г сульфоксидов состава С[5 H27,s SOi,4> содержащих 80бщ 12,1 %, Sso 9,4%.
Всего учтено по общей сере 96,1%, по сульфоксидной сере 99,1%.
Б. Кислотно-экстракционные методы извлечения сульфоксидов
Рекомендуемые способы основаны на использовании основных
свойств сульфоксидов и их способности образовывать труднораствори-
мые и слабодиссоциированные соли.
Выделение сульфоксидов через перхлораты
Перхлораты сульфоксидов практически нерастворимы как в легких
насыщенных углеводородах, так и в углеводородной части исходных ке-
росинов. При действии одного (или более) эквивалента 42 %-ной хлор-
ной кислоты на раствор сульфоксидов в керосине перхлораты сульфо-
ксидов количественно выделяются в отдельную жидкую фазу, распола-
гающуюся между углеводородным и водяным слоями [5]. Раствором
соды или щелочи сульфоксиды выделяются из перхлоратов количест-
венно:
R2S+HO-C1O4- + NaOH R2SO + NaC!O4 + Н2О.
Выделяющиеся перхлораты сульфоксидов могут захватывать до 10%
(от веса перхлоратов) углеводородных примесей. Для получения более
чистых сульфоксидов перхлораты перед нейтрализацией отмывают пет-
ролейным эфиром. Это позволяет получать перхлораты такой же чисто-
ты, как при хроматографировании, описанном выше. Метод выделения
сульфоксидов через перхлораты менее трудоемок и более быстр, нежели
хроматографический. Однако область применения перхлоратного метода
ограничена несколько более высоким нижним пределом по содержанию
сульфоксидной серы в растворах — не менее 0,3% (это соответствует
3—4% сульфоксидов в окисленном дистилляте).
Перхлоратный метод применим для очистки сульфоксидов от приме-
сей углеводородов и неокисленных сульфидов.
В присутствии хлорной кислоты возможно осуществление одностадий-
ного гетерогенно-эмульсионного окисления и выделения сульфоксидов
(см. ниже, стр. 100). Перхлоратный метод выделения сульфоксидов
можно применять на крупных лабораторных и опытных полупромыш-
ленных установках.
Стабильность перхлоратов, по-видимому, аналогична наблюдавшей-
ся стабильности перхлоратов многочисленных азотистых оснований, низ-
шие представители которых взрывоопасны, а высшие сравнительно
7 Методы анализа ду
42) Зак. 2300
устойчивы, выдерживают трение, удар и нагрев до 100° и более. Специ-
альные испытания влажных перхлоратов сульфоксидов, выделенных из
окисленной широкой фракции южно-узбекской нефти (см. ниже) на тре-
ние и удар, показали, что они не взрывчаты. Рекомендуется влажные
перхлораты сульфоксидов нейтрализовать сразу по получении, превра-
щая их в сульфоксиды. Категорически недопустимы сушка и хранение
перхлоратов сульфоксидов без специального исследования их стабиль-
ности и безопасности в отношении спонтанной взрываемости при терми-
ческих и механических воздействиях.
Реактивы
1. Хлорная кислота 42%-ная.
2. Петролейный эфир, т. кип. 40—60° (или гексан, или гептан).
3. Сода (или едкий натр).
4. Диэтиловый эфир (или бензол, или хлороформ).
5. Прокаленный сульфат магния (или натрия).
Методика работы
Раствор сульфоксида в гептане или окисленный нефтяной дистил-
лят встряхивают 10—15 мин. в делительной воронке с 42%-ной хлорной
кислотой, взятой из расчета 1,5 экв. на 1 экв. сульфоксидной серы. При
отстаивании перхлораты сульфоксидов образуют отдельную фазу —
средний слой, находящийся между углеводородным и водным слоями.
Перхлораты сульфоксидов (средний слой) отделяют и • многократно
промывают петролейным эфиром (гексаном или гептаном) до тех пор,
пока после испарения 30 мл промывной жидкости не перестанет оста-
ваться маслянистый остаток. Отмытые перхлораты подщелачивают
20%-ным раствором едкого натра или соды, выделившиеся в виде вяз-
кой массы сульфоксиды растворяют в эфире и экстрагируют водный
слой тем же растворителем. Вместо эфира можно воспользоваться бен-
золом или хлороформом. Экстракт соединяют с раствором сульфоксидов,
отмывают минимальным количеством воды до pH 7 в промывных водах
и сушат прокаленным сульфатом магния. Растворитель отгоняют под ко-
нец при остаточном давлении 2—4 мм до прекращения уменьшения веса
остатка (взвешивают с точностью до 0,1 г).
Пример 3
135,5 г окисленной широкой фракции южно-узбекской нефти, содержащей Sso
1,2%, Бсульфид 0,4%, обрабатывают 20 г 42%-ной НС1О4. Выделяют сульфоксиды со-
става Ci3,iH24,28SO. Баланс экстракции см. табл. 1.
Выделение сульфоксидов через сульфаты — «сернокислотный метод»
(предварительная методика)
Нами рекомендуется методика извлечения сульфоксидов непосред-
ственно из окисленных нефтяных дистиллятов * однократной экстракци-
ей 62%-ной серной кислотой, взятой в количестве 6 экв. на 1 экв. сульф-
оксидной серы [6]. Сульфаты сульфоксидов в отличие от перхлоратов
не образуют отдельной фазы, но переходят в воднокислотный слой. По-
тери при сернокислотной экстракции несколько выше, нежели при пер-
хлоратной, но дешевизна и безопасность этого метода позволяют реко-
мендовать его для использования на промышленных установках. Мето-
Извлечение сульфоксидов из пентанового раствора окисленного сернистоарома-
тического концентрата нефти Уоссон (Техас) многократной экстракцией 80%-ной сер-
ной кислотой описано Томсоном и сотр. [7]. F
98
дика принята как предварительная, так как возможны уточнения коли-
чества кислоты в зависимости от природы и количества сернистых со-
единений в различных дистиллятах.
Реактивы
1. Серная кислота 60—62%-ная.
2. Петролейный эфир, т. кип. 40—60° С (или гексан или гептан).
3. Едкий натр (20%-ный раствор).
4. Диэтиловый эфир.
5. Прокаленный сульфат натрия (или магния).
Методика работы
Раствор сульфоксида в гептане или окисленный нефтяной дистиллят
встряхивают 10—15 мин. в делительной воронке с 62%-ной серной кис-
лотой, взятой из расчета 6 экв. на 1 экв. сульфоксидной серы. После
расслаивания нижний слой (раствор сульфатов сульфоксидов в разбав-
ленной серной кислоте) отделяют, отмывают петролейным эфиром (гек-
саном или гептаном), как описано при экстракции хлорной кислотой,
нейтрализуют раствором едкого натра. Сульфоксиды при этом всплыва-
ют, их извлекают этиловым эфиром. Эфирный экстракт отмывают малы-
ми порциями воды до pH 7 в промывных водах и сушат. Остаток после
отгонки растворителя доводят до постоянного веса, как описано ранее.
Пример 4 ;
324,0 г окисленной широкой фракции южно-узбекской нефти, содержащей Sso
1,1% и ЗсульфидО,35%, обрабатывают 55 г 62%-ной серной кислоты. Баланс экст-
ракции см. табл. 1.
И. ОКИСЛЕНИЕ СУЛЬФИДОВ НЕФТЯНЫХ дистиллятов
В ранее разработанной методике окисления сульфидов в сернистоаро-
матических концентратах [1] был использован 27—30 %-ный водный
раствор перекиси водорода. Тот же окислитель использован в новых ме-
тодах окисления дистиллятов, не требующих предварительного получе-
ния концентратов [2, 5, 6]. Необходимое количество перекиси водорода
отвечает стехиометрическому уравнению
R2S + Н2О2 —R2SO + Н2О.
Ориентировочно содержание сульфидной серы в исходном сырье оп-
ределяется потенциометрическим титрованием образца йодатом калия
[8].
Недавно было установлено [9], что некоторые индивидуальные тиа-
бицикланы при определении [8] сульфидной серы дают значительно за-
вышенные значения. Однако эти же сульфиды при окислении теоретиче-
ским количеством перекиси водорода образуют эквивалентные количест-
ва сульфоксидов. Не исключена возможность того, что иодатометриче-
ское определение сульфидной серы в некоторых видах исходного сырья,
предназначенного для окисления, дает лишь приближенное представ-
ление о необходимом эквиваленте Н2Ог.
Опыт работы .с нефтяными дистиллятами показал, что баланс окис-
ления по сульфидной сере, определенной иодатометрически, можно со-
ставить только весьма ориентировочно. Балансы по общей и особенно по
сульфоксидной сере могут быть даны с гораздо большей точностью, так
как применяемые здесь аналитические методы менее чувствительны к
различиям в строении окисляемых сульфидов.
Во избежание большого избытка перекиси водорода (против необ-
ходимого по теории для окисления сульфидов до сульфоксидов) можно
7* 99
Таблица 1
Выделение суммы сульфоксидов из окисленной нефтяной фракции
Продукт Количество Зсульфид, % SSO’ %
г % во фрак- ции к исход- ной во фрак- ции к исход- ной
Хроматографический метод (пример 1)
Исходная окисленная широкая фракция 29,8 100 0,4 100 1,1 100
Элюат бензольный 26,2 88 0,4 97 0 0
Десорбат спиртовый 2,5 8 0 0 12,7 95
Сумма ... 28,7 96 97 95
Экстракция хлорнс й кислотой (пример 3)
Исходная окисленная широкая фракция 135,5 100 0,4 100 1,2 100
Рафинат Сульфоксиды, выделенные из 119,1 88 0,4 93 0 0
перхлоратов 12,3 10 0 0 12,5 91
Продукт, вымытый петролейным эфиром из перхлоратов .... 3,5 2 0 0 1,0 2
Сумма . . . 134,9 100 93 93
Экстракция серной кислотой (пример 4)
Исходная окисленная широкая фракция 324,0 100 0,35 100 1,1 100
Рафинат Сульфоксиды, выделенные из 295,0 91 0,35 90 0,05 5
сернокислотного раствора . . . 25,3 8 0 0 12,7 90,3
Продукт, вымытый петролей- ным эфиром из сернокислотно-
ГО слоя 3,7 1 0 0 0,8 0,7
Сумма . . . 324,0 100 90 96
рекомендовать проведение предварительных опытов окисления малых
навесок. Варьируя количество взятой перекиси водорода вблизи экви-
валента по отношению к сульфидной сере в окисляемой фракции,
следует подобрать то количество окислителя, которое приводит к мак-
симальному содержанию сульфоксидной серы *. Максимум по сульфо-
ксидам отвечает минимуму по неокисленным сульфидам и минимуму
побочно образующихся продуктов высших степеней окисления, в пер-
вую очередь сульфонов.
Рекомендуемый нами новый способ превращения сульфидов нефтя-
ных фракций в сульфоксиды заключается в гетерогенно-эмульсионном
окислении в присутствии добавок сильных кислот в каталитических или
же в эквивалентных сульфидной сере количествах; при этом окисляется
* Следует напомнить, что определять сульфоксидную серу в окисленной фракции
можно только после раскисления Н2О2 [2].
100
50—75% всех сульфидов, присутствующих в нефтяном дистилляте; при
наличии в нефтяных фракциях сульфидов, не окисляющихся методами
гетерогенно-эмульсионного окисления, рекомендуется повторное окисле-
ние фракций, освобожденных от сульфоксидов, но уже в гомогенной
среде — в смеси уксусного ангидрида и метилэтилкетона. Последний ме-
тод применим также в случае трудноокисляющихся индивидуальных
ароматических сульфидов.
А. Гетерогенно-эмульсионное окисление
Окисление с добавкой эквивалентных количеств хлорной кислоты
(«перхлоратно-эмульсионное» окисление)
Окисление сульфидов нефтяных фракций перекисью водорода в при-
сутствии эквивалентных количеств хлорной кислоты протекает очень
быстро [5]. Механизм происходящих при этом процессов детально не
изучен, однако было высказано предположение, что в апротонных сре-
дах хлорная кислота сначала образует комплекс с сульфидом, который
далее окисляется перекисью водорода. В результате окисления образу-
ется слабодиссоциированный перхлорат сульфоксида, который выводит-
ся из сферы реакции:
r2s + нсю4 -» r2s+h • сю;,
r2s+h.cio; + h2o2^r2s+ho.cio; + н2о.
Не исключено, что окисление идет в тройном комплексе сульфид —
перекись водорода — хлорная кислота (с переносом заряда):
н2о2 + нсю4 ->н3о; • сю;,
+ _
Н8О2-С1О4 + R2S-> | _ SR2 R2SOH.C1O4 4- н2о ±222 О = SR2 + КС1О4 + Н2О.
tio4'z
Окисление сульфидов и выделение сульфоксидов в виде перхлора-
тов происходит практически в одну стадию. Перхлораты сульфоксидов
после отделения легко превращаются в соответствующие сульфоксиды
(или их гидраты) простой обработкой водными щелочами. Этот способ
весьма удобен для препаративных работ, а также может быть применен
на крупных лабораторных и опытных установках. Метод позволяет реа-
лизовать получение сульфоксидов удовлетворительной степени чистоты
в таких количествах, которые необходимы для всестороннего испытания
их свойств и поиска областей применения.
Метод проверен на широках прямогонных фракциях: сборной южно-
узбекской нефти (температура отбора 200—400°С), арланской нефти
(температура отбора 150—325° С) и на дизельном топливе из нефтей
Татарии (т. кип. 200—365° С); содержание сульфидной серы в этих ши-
роких фракциях найдено в пределах 0,5—1,5%.
Аппаратура
1. Реактор для окисления пробных загрузок — цилиндрический сосуд
диаметром 35 мм, высотой 150 мм. Для перемешивания применяют стек-
лянную турбомешалку — «колокольчик Витта» (суммарная площадь
верхних отверстий должна быть не менее площади нижнего засасываю-
щего отверстия). Скорость вращения мешалки не менее 1500 об/мин.
2. Реактор для окисления основной массы окисляемого дистиллята
представляет собой цилиндрический сосуд из молибденового стекла с
101
полусферическим дном диаметром НО мм и высотой 350 мм. На реактор
накладывается крышка из плексигласа с отверстиями для мешалки,
термометра и капельной воронки; крышка укрепляется лабораторным
зажимом на том же штативе, что и реактор. Для перемешивания при-
меняют стеклянную турбомешалку такого же устройства, как описано
выше.
Реактивы
1. Перекись водорода 27—30%-ная; содержание ее должно быть
точно определено одним из обычных аналитических методов.
2. Хлорная кислота 57%-ная.
3. Эмульгатор (например мерзолят натрия).
4. Петролейный эфир (т. кип. 40—60° С) или гексан, или гептан.
5. Сода (или едкий натр), 20%-ный раствор.
6. Диэтиловый эфир.
7. Сульфат магния, прокаленный (или сульфат натрия).
Методика работы
К навеске дистиллята (0,3—0,5 кг) при интенсивном перемешивании
прибавляют за 5—6 мин. смесь, состоящую из перекиси водорода, хлор-
ной кислоты и эмульгатора (в окисляющей смеси концентрация хлорной
кислоты 40—42%). На 1 экв. сульфидной серы берут 1 экв. перекиси во-
дорода, I—1,5 экв. хлорной кислоты и 0,5% от общего веса эмульгатора
реакционной смеси. В процессе окисления реакционную смесь охлажда-
ют (водой с температурой 10—12° С) так, чтобы температура в реакторе
не превышала 25—30° С. Перемешивают полчаса, после чего дают смеси
отстояться не менее 3 час. Перхлораты сульфоксидов образуют средний
слой между углеводородным и водным слоями. Перхлораты отделяют,
затем отмывают и разлагают щелочами, как описано выше.
/ Пример 1
К 350,6 г широкой фракции сборной южно-узбекской нефти (т. кип 200—400° С,
50бщ2,4%, 8сульфид ,1,5%) отдельными порциями всего за 6 мин. прибавляют смесь
19,2 г 30%-ной перекиси водорода, 0,4 г мерзолята натрия и 50,7 а 57%-ной хлорной
кислоты. Реактор охлаждают водой, поддерживая температуру в реакционной смеси 25—
30° С. Через 12 мин. раскисляется 90% Н2Ог (контроль йодометрическим методом в от-
дельных пробах, взятых из реакционной смеси). Перемешивание продолжают около
0,5 часа (до полного раскисления перекиси водорода). Отчетливое образование трех
слоев наблюдается уже через 5—15 мин. после остановки мешалки. Для обеспечения
полного расслоения смеси дают отстояться 2—3 часа, при этом верхний слой стано-
вится прозрачным.
Средний слой — перхлораты сульфоксидов (окрашен) — отделяют и обрабатыва-
ют, как описано ранее. Получают 29,9 г сульфоксидов состава Cis.s^.sSOi.a (препа-
рат гигроскопичен); S общ 13,4%, Sso 11,5% и 313,5 г рафината, в котором S ощб
1.3%, SОуЛьфИд 0,2%, S s0 отсутствует.
Всего окислено 86,7% сульфидов, находившихся в широкой фракции (считается
по содержанию сульфидной серы).
Пример 2
К 419,6 г фракции арланской нефти (т. кип. 150—325° С, 80бщ 1,6%, 8Сульфид
1,0%) отдельными порциями за 6 мин. прибавляют смесь 13 г 30%-ной перекиси водо-
рода, 32,5 г хлорной кислоты (57%-ной) и 0,5 а мерзолята натрия. Реактор охлажда-
ют водой, поддерживая температуру реакции 25—30° С. Перемешивание продолжают
0,5 часа (до полного раскисления перекиси водорода). Контроль за раскислением пере-
киси водорода проводят, как в примере 1. Затем смеси дают расслоиться. Образуется
три слоя.
Средний слой — перхлораты сульфоксидов — отделяют и обрабатывают, как опи-
сано ранее (см. пример 1). Получают 20,5 г окисленного продукта состава
Си.вНгмЗОьг (препарат гигроскопичен) с 12,5%, Ss0 10,3% и 390 а рафината,
в котором 80бщ 0,95%, 8супьфид 0,2%, Ss0 отсутствует.
Всего окислено 70% сульфидов, находившихся в широкой фракции (считается по
содержанию сульфидной серы).
102
"Окисление с каталитическими добавками серной кислоты
Навеску нефтяного дистиллята окисляют смесью перекиси водорода,
серной кислоты и эмульгатора. Количество серной кислоты составляет
0,1—0,2 экв. по отношению к сульфидной сере (0,2—0,4% к общему весу
реакционной смеси) (6]. Окисление этим способом идет значительно
медленнее, нежели перхлоратным, однако простота, безопасность и де-
шевизна кислоты делают такой вариант метода перспективным для про-
мышленного применения. Сульфоксиды после окончания окисления вы-
деляют одним из трех перечисленных выше способов. Проводить окис-
ление, совмещая его с сернокислотной экстракцией в одну стадию, как
описано в опыте с хлорной кислотой, нецелесообразно. Окисление в при-
сутствии значительных количеств серной кислоты (60 %-ной) сопровож-
далось сильным осмолением продукта.
Аппаратура та же, что и в предыдущем способе.
Реактивы
1. Перекись водорода — та же, что и в предыдущем способе.
2. Серная кислота 96—98%-ная.
3. Эмульгатор (например мерзолят натрия).
4. Реактивы, необходимые для выделения сульфоксидов, перечислены
выше при описании соответствующих методов.
Методика работы
Приготовление окислителя. Перекись водорода охлаждают до 0—5“
и при 10° С отдельными порциями прибавляют концентрированную
серную кислоту, после чего в этот раствор вводят эмульгатор (перекись
водорода берут из расчета 1—1,2 экв. к сульфидной сере, серной кисло-
ты 0,2—0,4%, эмульгатора 0,5% к суммарному весу реакционной смеси).
К навеске окисляемого дистиллята при интенсивном перемешивании при
20—25° С постепенно прибавляют окислитель. Процесс контролируют,
как при перхлоратном окислении. По окончании реакции смесь перено-
сят в делительную воронку и выдерживают до расслоения. Воднокис-
лотный слой отделяют. Из органического слоя выделяют сульфоксиды
либо хроматографированием, либо кислотно-экстракционным способом,
Пример 3
436 г широкой фракции южно-узбекской нефти (т. кип. 200—400° С, 8СуЛьфид
1,5%) окисляют смесью, состоящей из 28,4 г 30%-ной Н2О2, 2 г 98%-ной H2SO4 и 1,14 г
мерзолята натрия. Время окисления 2 часа. Углеводородный слой содержит Ssq 1,1%,
Зсульфид 0,4%; окислилось 75% сульфидов. Хроматографирование части углеводо-
родного слоя см. ранее в табл. 1 (пример 1).
Б. Окисление сульфидов до сульфоксидов в гомогенной среде
Окисление сульфидов до сульфоксидов в среде уксусного ангидрида
и смесей его с метилэтилкетоном основано на экспериментально уста-
новленном факте повышения глубины и скорости окисления трудноокис-
ляемых сульфидов в этих средах [2, 10].
Наблюдаемый эффект, вероятно, может быть объяснен тремя фак-
торами:
1. Повышением основности образующихся сульфоксидов.
2. Наличием примеси уксусной (и, вероятно, надуксусной) кислоты,
способствующей протонизации исходного сульфида и затем взаимодей-
ствующей с образующимися сульфоксидами.
3. Малой степенью диссоциации в растворе уксусного ангидрида.
103
возникающих ацетатов сульфоксидов, благодаря чему они выводятся из
сферы реакции подобно тому, как это наблюдается для перхлоратов.
Применение в качестве растворителя уксусного ангидрида (или ук-
сусного ангидрида с добавкой метилэтилкетона) при окислении суль-
фидов перекисью водорода дает возможность получить с удовлетвори-
тельными выходами сульфоксиды из сульфидов, не окисляющихся в
других условиях [1]. Метод применим как к индивидуальным сульфидам
(в частности ароматическим), так и к «трудноокисляемым» сульфидам
нефтяных дистиллятов.
Метод проверен на «трудноокисляемых» сульфидах широкой фрак-
ции южно-узбекской нефти, «трудноокисляемых» сульфидах широкой
фракции «арланской нефти» и на 2-фенилнафтилсульфиде.
Метод пригоден для получения сульфоксидов из нефтяных дистил-
лятов в количествах, достаточных для лабораторных исследований.
Аппаратура
Трехгорлые колбы разных размеров, хроматографические колонки.
Реактивы
1. Перекись водорода 27—30%-ная; содержание ее должно быть
точно определено одним из обычных аналитических методов.
2. Уксусный ангидрид.
3. Метилэтилкетон.
4. Этиловый эфир (при работе с индивидуальными сульфидами).
5. Бензол.
6. Спирт.
7. Петролейный эфир, т. кип. <С60° С, формолитовая проба отрица-
тельна.
8. Хлорная кислота 42%-ная.
9. Технический силикагель марки «АСК» активностью не менее 8—10
по о-оксихинолину.
10. Сульфат магния прокаленный.
Методика работы
Сырьем могут служить индивидуальные сульфиды или дистилляты
сернистых нефтей. В последнем случае предварительно определяют в
исходном сырье содержание сульфидной серы.
1. Окисление
Навеску окисляемого продукта смешивают с уксусным ангидридом
(или с уксусным ангидридом и метилэтилкетоном). Количества раство-
рителей определяются условиями гомогенизации смеси в каждом отдель-
ном случае, причем уксусного ангидрида берут не менее 25 молей на
1 г-атом сульфидной серы. При перемешивании прибавляют перекись
водорода, взятую в количестве, рассчитанном на окисление сульфидной
серы до сульфоксидной (моль на моль, без избытка). После прибавления
окислителя реакционную смесь выдерживают до тех пор, пока в пробе
смеси не перестанет обнаруживаться перекись водорода (обычно 40—
48 час.).
2. Обработка реакционной смеси
Растворители отгоняют в вакууме при остаточном давлении 2 мм
рт. ст. под конец при слабом нагревании смеси; температура в бане не
должна превышать 30—45°С*. Остаток после отгонки растворителя
* При повышении температуры может иметь место взаимодействие сульфоксидов
с уксусным ангидридом (реакция Пуммерера).
104
(при работе с индивидуальными сульфидами) растворяют в эфире, ра-
створ отмывают 10%-ным раствором соды от следов уксусной кислоты
и сушат прокаленным сульфатом магния. Растворитель отгоняют, оста-
ток, в случае твердых сульфоксидов, перекристаллизовывают из подхо-
дящего растворителя. При работе с нефтяными дистиллятами или с
жидкими сульфоксидами продукт после отгонки растворителей промы-
вают 10%-ным раствором соды и сушат прокаленным сульфатом маг-
ния, после чего хроматографируют, как описано ранее (при малой кон-
центрации сульфоксидов, недостаточной для кислотной экстракции).
Пример 4
0,55 г 2-нафтилфенилсульфида окисляют, растворив в 21,4 мл уксусного ангидрида.
Получают масло, кристаллизующееся при растирании; выход сульфоксида (по содер-
жанию Sso) равен 56% • Кристаллы отделяют и перекристаллизовывают из гептана.
Вес 0,21 г, т. пл., 2-нафтилфенилсульфоксида 71° С. Найдено, %: С 76,1; Н 5,1;
CieHijSO. Вычислено, %: С 76,1; Н 4,8.
Пример 5
Широкую фракцию сборной южно-узбекской нефти (т. кип. 200—400° С, 50бщ 2,4%,
5Сульфид 1,5%) окисляют методом гетерогенно-эмульсионного окисления [10]. Обра-
зующиеся при этом сульфоксиды отделяют хроматографированием и получают часть
широкой фракции, так называемый «бензольный элюат» (см. табл. 1, пример 1), со-
держащий «трудноокисляемые» сульфиды (сульфидной серы 0,4%).
0,5 кг этого продукта окисляют в растворе, в смеси 159 г уксусного ангидрида
и 206 г метилэтилкетона. После обработки получают 433 г окисленного продукта, со-
держание SS0 0,38%, что соответствует окислению 70% «трудноокисляемых» суль-
фидов. Хроматографирование см. пример 2.
Пример 6
Широкую фракцию арланской нефти (т. кип. 150—280°, 50бщ 1,6%, 5сульфВД
1,0%) подвергают «перхлоратно-эмульсионному» окислению, как описано на стр. 100
настоящего сборника, и образующиеся при этом сульфоксиды отделяют (см. там же);
получают рафинат, содержащий «трудноокисляемые» сульфиды (сульфидной серы
0,2%). 217 г этого рафината окисляют в смеси 100 мл уксусного ангидрида и 250 мл
метилэтилкетона. После отгоики основной массы растворителей получают 287 г окис-
ленного продукта с содержанием Ss0 0,12%, что соответствует окислению 70% «труд-
ноокисляемых» сульфидов. Сульфоксиды выделяют хроматографированием (табл. 2)»
Таблица 2
Хроматографическое выделение сульфоксидов из окисленного «рафината»
фракции арланской нефти
Фракция Вес Sso> %
г % во фракции к исходной
Исходная окисленная фракция Беизольиый элюат Спиртовый десорбат 239,1 234,1 2,2 100,0 98,0 1,0 0,12 0 10,9 100,0 0 85,5
Сумма 236,3 99,0 85,5
Состав продукта: Cib.kHso^SOi^i
10F>
выводы
1. Разработана методика хроматографического выделения сульфо-
ксидов из окисленных нефтяных фракций, включающая предваритель-
ное хроматографирование пробной загрузки. Рекомендуемая методика
хроматографического выделения применима также к разбавленным ра-
створам сульфоксидов в неполярных жидкостях; полярные растворите-
ли—уксусный ангидрид и метилзтилкетон предварительно отгоняют.
2. Для хроматографического отделения сульфоксидов рекомендован
технический силикагель марки АСК; для определения его емкости в ка-
честве стандартного вещества рекомендован о-оксихинолин.
3. Разработана методика выделения сульфоксидов в виде перхлора-
тов из окисленных нефтяных фракций при содержании в них сульфо-
ксидной серы не менее 0,3%.
4. Предложен метод экстракции сульфоксидов из окисленных нефтя-
ных дистиллятов разбавленной (60 %-ной) серной кислотой.
5. Предложен способ гетерогенно-эмульсионного селективного окис-
ления сульфидов в дистиллятах сернистых нефтей действием перекиси
водорода в присутствии эквивалентного количества хлорной кислоты,
при котором образующиеся сульфоксиды выделяются в виде перхлора-
тов в одну стадию.
6. Предложен способ гетерогенно-эмульсионного селективного окис-
ления сульфидов в дистиллятах сернистых нефтей действием перекиси
водорода в присутствии каталитических количеств серной кислоты.
7. Предложен метод селективного окисления «трудноокисляемых»
сульфидов до сульфоксидов перекисью водорода в уксусном ангидриде
или смеси его с метилэтилкетоном.
Институт нефтехимического синтеза им. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. Е. Н. Караулова, Г. Д. Гальперн. Сб. «Методы анализа органических соединений
нефти, их смесей и производных», сб. 1. М., Изд-во АН СССР, 1960, стр. 101.
2 Е. Н. Караулова, Г. Д. Гальперн, Т. А. Бардина, Т. С. Бобруйская. Нефтехимия,
6, 480 (1966).
3. Е. Н. Караулова, Г. Д. Гальперн, Л. Д. Аристова, Т. А. Бардина, В. М. Коршунова.
Нефтехимия, 5, 755 (1965).
4. Е. Н. Караулова, Г. Д. Гальперн, ЖОХ, 29, 3033 (1959),
5. Е. Н. Караулова, Г. Д. Гальперн, Т. А. Бардина. ДАН СССР, 173, 104 (1967); Авт.
свид. 186454 (1966); Бюлл. изобр., № 19, 29 (1966).
6. Е. Н. Караулова, Г. Д. Гальперн, Т. А. Бардина, А. С. Харитонов. Авт. свид. 206579
(1967); Бюлл. изобр., № 1, 37 (1968).
7. С. J. Thompson, Н. I. Coleman, R. F. Hopkins, Н. Т. Rail. J. Chem. Eng. Data, 9,
293 (1964).
8. Г. Д. Гальперн, Г. П. Гирина, В. Г. Лукьяница. Сб. «Методы анализа органических
соединений нефти, их смесей и производных», сб. 1. М., Изд-во АН СССР, 1960,
стр. 58.
9. Е. Н. Караулова, В. Ш. Шайхразиева, Г. Д. Гальперн. Химия гетероциклических
соединений, № 1, 51 (1967).
10. Е. Н. Караулова, Г. Д. Гальперн, Т. А. Бардина, Т. С. Бобруйская. Авт. свид.
175958 (1964); Бюлл. изобр., № 21, 18 (1965).
ЭЛЕМЕНТНЫЙ АНАЛИЗ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
< ГАЗОХРОМАТОГРАФИЧЕСКИМ ОПРЕДЕЛЕНИЕМ ПРОДУКТОВ
РАЗЛОЖЕНИЯ.
I. МЕТОД ОПРЕДЕЛЕНИЯ АЗОТА
А. В. Непряхина, И. К. Чудакова, Г. А. Новикова,
Г. Г. Радикова, О. II. Доманина
I. ВВЕДЕНИЕ
Элементный органический анализ обычно связан с окислительным
или восстановительным разложением органического вещества с целью
превращения элементов, входящих в его состав, в простые продукты,
удобные для последующего количественного определения.,
В последние годы выявилось, что для целей конечного определения
продуктов разложения чрезвычайно перспективным является использо-
вание газовой хроматографии.
В литературе по газовой хроматографии (1,2] описаны условия разделения смесей
низкокипящих газов, таких, как СО2, О2, Н2, N2, СН4, С2Н2 и др., т. е. газов, которые
могут являться конечными продуктами разложения органического вещества при при-
менении рдзных методов «минерализации». Газовая хроматография открывает воз-
можность одновременного определения нескольких элементов (из одной навески) без
значительного снижения точности определения и с заметным сокращением длитель-
ности анализа. С помощью хроматографии можно также корректировать условия про-
ведения количественного разложения органических веществ.
Важно найти способы полного разложения и превращения продуктов разложения
в удобную для хроматографирования форму. Этого можно достичь, проводя разложе-
ние в небольшом замкнутом объеме, что позволяет осуществить последующее еди-
новременное введение продуктов превращения анализируемой пробы на хроматографи-
ческую колонку.
Создание соответствующих аналитических приборов позволяет проводить количе-
ственный элементный анализ с достаточной быстротой и точностью. За рубежом ряд
фирм уже выпускает так называемые CHN-анализаторы [3—6].
Метод газовой хроматографии позволяет проводить определение со-
держания отдельных компонентов смеси при малых концентрациях, что
представляет существенный интерес для определения микроконцентра-
ций элементов, например азота в нефтях и нефтепродуктах. Определение
азота в подобных веществах производят по методу Дюма или Кьельда-
ля, а также по их многочисленным модификациям [7]. Трудности и ошиб-
ки анализа в этом случае связаны с необходимостью разложения боль-
ших навесок вещества, что обусловлено и применяемыми методами
окончания анализа. В литературе имеется описание ряда методов опре-
деления азота с использованием газовой хроматографии.
В 1961 г. Рейтсема и Олфин (8] предложили метод быстрого определения азота в
органических веществах, основанный на сочетании сожжения и газовой хроматогра-
107
фии. Из приведенной ими схемы видно, что метод разработан для анализа жидких ве-
ществ. Авторы придают большое значение конструкции камеры сожжения. Была
применена металлическая трубка (длина 20 мм. диаметр 6 мм), запол-
ненная плотным пучком длинных тонких медных проволочек, которые предварительно»
протравливали водным раствором нитрата железа, а затем прокаливали иа воздухе.
При высокой эффективности сожжения перепад давления в такой колонке очень мал.
Заметив, что оптимальная температура сожжения зависит от типа анализируемого-
соединения, авторы установили опытным путем, что для полного сожжения органи-
ческих соединений вполне достаточно 700° С. Сожжение производилось очень быстро-
(весь анализ длится 2 мин.) в токе гелия, проходящего со скоростью 160—189 см* [мин.
Азот, содержащийся в органических соединениях, при сожжении давал пик, иденти-
фицированный как двуокись азота. Для разделения применялись колонки, заполнен-
ные силикагелем. Температура колонки 50—100° С. Детектором служил катарометр.
Процент азота определяли сравнением полученных данных с результатом анализа
чистых азотсодержащих соединений, проводимого в аналогичных условиях. Объем:
пробы составлял 0,05—0,001 мл. Авторы отмечают, что такой отбор невзвешенной
пробы может являться источником ошибки анализа.
Парсонс, Пеннигтон и Уолкер [9] для определения азота использовали сочетание-
быстрого сожжения с газохроматографическим окоичаиием анализа. Точность анали-
за 0,1 абс. %. Навески (1—10 мг) органических и неорганических соединений (бро-
мацетанилид, никотиновая кислота, тиомочевина, нитрат аммония и др.) смешивали
с 0,8 г перманганата серебра, добавляли немного окиси меди и сжигали в кварцевой
трубке, помещенной в высокочастотную индукционную печь, в атмосфере гелия. Про-
дукты сгорания током гелия (скорость 13 мл/мин) пропускали через конвертор, за-
полненный на 5% окисью меди и на 95% восстановленной медью. В конверторе осу-
ществлялось конечное окисление продуктов сгорания и восстановление окислов азота
до элементарного азота. Образующаяся вода и углекислота поглощались ангидроном
и аскаритом.
Терентьев, Туркельтауб, Бондаревская и Домочкина ([10] предложили метод хро-
матографического определения азота с использованием восстановительного способа
разложения. Навеску органического вещества (5—10 мг) в платиновой или кварцевой
лодочке разлагали в замкнутом объеме кварцевой трубки, содержащей слой «никели-
рованной» сажи, в атмосфере гелия при давлении 20 мм рт. ст. Температура разложе-
ния 900° С. При анализе веществ состава С, Н, О, N на хроматограмме отмечаются-
два пика: СО и N2. Авторы указывают, что в некоторых случаях в продуктах разло-
жения в незначительных количествах появляется метан (например при анализе аце-
танилида). Разделение проводили на колонке, заполненной молекулярными ситами 5А.
Расчет азота проводили по площадям пиков хроматограмм, сравнивая их с калибро-
вочными кривыми, которые строили по площадям пиков, полученным при разложении,
различных навесок мочевины. Точность определения азота 0,4%.
Пахомова и Чумаченко [11] предложили метод определения азота, в котором сис-
тема разложения и хроматограф связаны при помощи мембранного переключателя:
потока гелия, позволяющего герметично закрывать реакционную систему в момент
разложения вещества. Разложение осуществлялось в кварцевой пробирке (длина-
160 мм, диаметр 11 мм), закрывающейся металлической пробкой, имеющей два капил-
ляра для ввода газа-носителя и вывода продуктов сгорания. Авторы рекомендуют
применять в качестве окислителя органических соединений (в атмосфере гелия) окись
никеля, нагретую до 900—1000° С. При этом образуются только СОг, Н2О и N2- Для
разделения применялась колонка, заполненная активированным углем марки БАУ-2.
Скорость гелия 30 мл/мин. Содержание азота вычислялось по калибровочной кривой,
построенной на основании измерения высот пиков при тридцати определениях карба-
зола. Среднее отклонение при определении азота составляло 0,2—0,3%.
Предложен также ряд методов определения азота совместно с углеродом и водо-
родом [12—14].
Анализ опубликованных работ и опыт нашей работы показывает,,
что использование газовой хроматографии действительно открывает
большие возможности для определения азота в элементном анализе.
Используя принцип пиролитического разложения навески органического-
вещества, преимущество которого убедительно показано работами Кор-
шун и Климовой [15, 16], мы разработали соответствующее приспособле-
ние с автоматическим обогревом и нашли условия сожжения органиче-
ских веществ, удовлетворяющие требованиям как количественного окис-
ления навески анализируемого вещества до простых веществ, так и их.
хроматографирования.
В качестве окислителя была выбрана окись меди в виде порошка и
крупинок размером 0,25—0,5 мм, широко используемая в органическом
анализе.
108
В последние годы появились работы по исследованию активности
юкислов металлов, применяемых в качестве окислительных катализато-
ров в элементном анализе. В качестве окисляемого вещества брали ме-
тан. Вечержа и Синек [17] показали, что в токе азота (скорость
15 мл/мин) окись-закись кобальта окисляет метан при температуре
750°С примерно на 60%, а окись меди в тех же условиях не работает.
Каинц и Хорватиш [18] также на примере окисления метана, но в токе
двуокиси углерода, показали, что окислы металлов располагаются по их
активности в следующем порядке:
MnO2>CuO, Co3O4>Fe2O3>NiO.
В нашей лаборатории были проведены опыты [19] по пиролитиче-
скому. разложению ряда органических веществ (ацетанилид, нитробен-
зол, производные гидразина, полиакрилонитрил, термообработанные по-
лиакрилонитрилы и др.) в замкнутом объеме, в среде гелия с
использованием в качестве окислителя окиси меди. Установлено, что в
этих условиях при соотношении навеска — окись меди примерно
1:10 000 происходит полное разложение органического вещества до
СО2, Н2О, N2 при температуре 710—830° С.
В настоящей работе рекомендуется метод определения азота в орга-
нических веществах, основанный на пиролитическом окислении навески
анализируемого вещества до образования конечных продуктов N2,
СО2, Н2О. По окончании разложения образовавшиеся продукты вытес-
няются током гелия в хроматографическую колонку, заполненную сили-
кагелем. После элюирования из колонки азот регистрируется катаромет-
ром и по площади пика определяется его количество.
Полнота превращения в конечные продукты обеспечивается 1) про-
ведением разложения в замкнутом объеме в статических условиях;
2) увеличением поверхности и эффективности окиси меди за счет ее
размельчения; 3) проведением постепенного, равномерного разложения
и окисления анализируемого вещества путем поддержания оптимально-
го температурного режима всего объема окислителя в период сожжения.
Установлено, что при соблюдении указанных условий сожжения нет
необходимости в применении восстановленной меди, функцию которой
выполняют сами продукты разложения органического вещества.
II. РЕКОМЕНДУЕМАЯ МЕТОДИКА
Аппаратура и реактивы
1. Хроматограф ХЛ-4.
2. Трубка для сожжения.
3. Краны запирающие.
4. Нагреватель.
5. Стаканчик кварцевый.
6. Силикагель МСМ.
7. Окись меди порошкообразная и в проволоке.
8. Газ-носитель — гелий.
9. Микровесы (Бунге).
Схема установки приведена на рис. 1.
1. Для хроматографического измерения продуктов сожжения приме-
няется хроматограф ХЛ-4, в котором кран дозатор с дозировочным объ-
емом заменен на три запирающих игольчатых крана 9 и трубку для
сожжения 10 (рис. 1). Колонка 8 (длина 1 м) заполнена силикагелем
МСМ. Температура термостатирования колонки +70° С.
2. Трубка для сожжения 10 изготовляется из нержавеющей стали *
* Ранее [19] было дано описание аппаратуры с кварцевой трубкой.
109
марки 1 X 18 НТ-9, с отводом для подачи газа-носителя. Длина трубки
160 мм, внутренний диаметр 10 мм. Трубка герметично закрывается
металлической пробкой с тефлоновым уплотнением. Для предотвраще-
ния перегрева пробки трубка в этом месте имеет утолщение и ребрис-
тую поверхность (воздушный холодильник) длиною 36 мм. Трубка ук-
реплена на металлической подставке, на которой монтируется также-
электронагреватель.
Рис. 1. Схема установки
для определения азота
1 — баллон с гелием; 2 — редуктор;
3 — кран тонкой регулировки с об-
разцовым манометром иа 6 кгс]см*\
4 — капилляр; 5 — осушитель газа;
6 — дроссель газа; 7 — детектор;
8 —колонка; 9—запирающие краны;.
10 — трубка для сожжения; // — са-
мописец Э1ИП-09
3. Три запирающих двухходовых игольчатых крана 9 обеспечивают
надежное отключение от трубки потока гелия на период разложения на-
вески. Соединительные трубки возможно меньшей длины — металличе-
ские (нержавеющая сталь), внутренний диаметр 1 мм.
Рис. 2. Кривые нагревания
и охлаждения трехсекцион-
ной электропечи
4. Нагреватель представляет трехсекционную открытую (без тепло-
изоляции) электропечь, которая изготовлена из нихромовой проволоки
диаметром 2 мм. Длина проволоки одной секции равна 970 мм. Элек-
тронагреватель обладает малой инерционностью, что видно из рис. 2.
Трубка для сожжения вставляется внутрь спиралей электронагревате-
ля. Секции включаются последовательно через заданные интервалы
посредством реле времени типа ВС 10-64. Реле времени обеспечивает
продолжительность работы каждой секции от 1 до 30 мин.
Трубки для сожжения, запирающие краны и электронагреватель,,
были сконструированы и изготовлены в СКВ ИНХС АН СССР.
5. Кварцевые стаканчики для навесок длиной 90 мм и внутренним
диаметром 6—7 мм.
6. Силикагель марки МСМ (фракция 0,25—0,5 мм), применяемый в
хроматографии.
7. Окись меди проволочную измельчали до фракции 0,25—1 мм. Эту
110
фракцию и порошкообразную окись меди прокаливали в муфеле в те-
чение 2 час. при Температуре около 700° С. Хроматографическую чис-
тоту окиси меди проверяли нагреванием ее в кварцевом стаканчике в
условиях анализа. Отсутствие пиков показывает, что окислитель не име-
ет примесей.
Анализ
Навеску анализируемого вещества (1—5 мг) на дне кварцевого ста-
канчика * для лучшего контакта с окислителем засыпают порошкообраз-
ной окисью меди (0,5 см3) , а затем — проволочной окисью меди почти
до краев стаканчика (3 см3). Для того чтобы окислитель не рассыпал-
ся, стаканчик закрывают «пробкой» из медной проволоки (спираль).
Рис. 3. Схема расположения
стаканчика в трубке
для сожжения
1 — 700—710°; II — 730—750°;
• /// — 730—750° С
Стаканчик с навеской и окислителем вносят в трубку для сожже-
ния в токе гелия. Трубку продувают гелием до полного вытеснения воз-
духа, что фиксируется на диаграммной ленте хроматографа (на первой
чувствительности). Гелий подается со скоростью 15—20 мл/мин. Затем
трубку для сожжения отключают от потока гелия и включают автомати-
ческое сожжение. Сначала включают секцию I электронагревателя, че-
рез 5 мин.— секцию // и еще через 5 мин.— секцию III. Таким образом,
секция I работает все 30 мин., секция // — 25 мин. и секция /// — 20 мин.
Все сожжение продолжается 30 мин. Расположение стаканчика с навес-
кой в трубке для сожжения показано на рис. 3. В течение первых
10 мин., когда включены секции I и II электронагревателя, производят
подготовку окислителя и одновременно происходит предварительный
пиролиз вещества. В остальные 20 мин., после автоматического включе-
ния секции /// происходит полное завершение процесса разложения и
окисления. Режим сожжения рассчитан на разложение трудносжигае-
мых соединений. Однако для особо термоустойчивых производных по-
лиакрилнитрилов температура сожжения повышается до 800—830° С.
После автоматического выключения электронагревателя поток гелия
направляется через трубку. Образовавшиеся продукты окисления вы-
дуваются гелием в хроматографическую колонку, где и разделяются.
В зависимости от концентрации азота и величины навески запись пика
азота производится на 1,2 или 4 чувствительности.
Расчет
Ввиду отсутствия контрольных и регистрирующих приборов ВЫСОКОЙ
точности, которые поддерживали бы длительное время постоянство ус-
ловий анализа, предварительная калибровка прибора не производи-
лась. Ежедневно определяется так называемый титр (Г), под которым
понимается количество азота в у на 1 мм2 площади пика, полученного от
сожжения навески стандартного вещества в условиях анализа.
7 _ АР Г т i
iuo-sp.L J ’
* Для взятия навесок жидких веществ применяли следующие способы: для лету;
чих веществ — закрытый капилляр, для высококипящих —открытый Капилляр, для
пастообразных и маслянистых (нелетучих) тг лодочку. Взятые таким образом навески
помещали на дно стаканчика и засыпали окисью.меди. .
Ш
где А — теоретическое содержание азота в стандартном веществе
(в %), Р — навеска стандартного вещества (в у); Лр — площадь пика
азота, полученная при сожжении стандартного вещества (в л«ж2).
Площадь пика определяется по формуле
Sp = НаК,
где Н — высота пика (в мм);. а — ширина пика на половине высоты
(в мм) (включая толщину левой стороны пика); К-—чувствительность.
Измерения ширины пиков производятся лупой с окулярмикрометром.
Расчет содержания азота в анализируемом веществе производится
цо формуле
7\S -100
%N= —
* V
где Т — титр азота по стандартному веществу (в у/мм2); Sx — площадь
пика азота анализируемого вещества (в л«Л42); Рх — навеска анализиру-
емого вещества (в у).
Пример расчета титра азота по ацетанилиду и определения процент-
ного содержания азота в тиомочевине дан в табл. 1. Для тиомочевины
% NTeopeT. = 36,81.
Таблица 1
Расчет титра и опргделение азота
Вещество
T-Sv-loo
%N =-----б-----
Ацетани-
лид
Тиомо-
чевина
2,779 51,0
1,050 75,0
2,62
2,40
534,4
720,0
10,37-2779
100-534,4
= 0,539
0,539-720,0-100
1050
= 36,84
Величина титра при сожжении различных стандартных веществ в
течение дня постоянна. Например, титр азота по ацетанилиду равен
0,500 у [мм2, по динитробензолу — 0,501 у [мм2.
III. РЕЗУЛЬТАТЫ АНАЛИЗОВ
С помощью описанного метода быд проведен анализ ряда веществ
различного состава и строения, включая полимеры, а также нефтепро-
дуктов. Результаты представлены в табл. 2.
Как видно из табл. 2, точность определения азота в веществах раз-
личного строения, включая полимеры, и нефтепродуктах составляет
±0,1 —0,2%.
ВЫВОДЫ
Разработан метод определения азота в органических веществах раз-
личного состава и строения с газохроматографическим окончанием ана-
лиза. Для получения количественных результатов используется предва-
рительное пиролитическое разложение анализируемого вещества и
окисление продуктов разложения в присутствии окиси меди. Продол-
жительность анализа 50 мин. Точность определения азота ±0,1—0,2%.
112
Таблица 2
Определение азота в веществах различного состава и строения
Вещество Содержание азота, %
вычислено найдено
СН3СО—NH—\* o2n no2 * \У\/ и h2ncsnh2 * CioH8.CeH2(NOs)2OH * он O2N 1 no2* \zV 1 no2 10,37 16,67 36,81 11,76 18,34 12,38 18,66 7,82 6,82 10,22 10,32 10,24 10,28 16,78 16,66 16,58 16,68 36,74 36,72 37,02 11,70 11,75 11,74 18,41 18,35 18,34 18,50 12,44 12,26 18,45 18,30 7,81 7,70 6,66 7,30 10,50 10,18
\nh/=0 OH O2N 1 no2 \У\/ и 1 no2 rrn V\z\z N y\/4 NHs o: COOH
* Эти соединения использовались позже в качестве стандартов при определении
титра.
8 Методы анализа
113
Таблица 2 (окончание)
Содержание азота, %
Вещество вычислено найдено
/ \n-ch2—/ \ 7,32 7,46
\—/j, \=/ 7,48
О
(C12H25)3N-»O 2,60 2,64 2,65
СНз сн=сн2
^Si"7 СН2 6,44 6,63 6,59
% j/ СН—СН2—N
\н2
Полиакрилонитрил 26,40 26,45 26,40 26,36
Полимер 4,73 4,71 4,79
Тример CggH^SiNs Концентраты азотистых оснований, выделенных 6,83 6,70 6,70
из нефти, состава CHNSO № 1 2,38** 2,59 2,46
№ 2 4,33 ** 4,46 4,56
№ 3 1,56** 1,61 1,50
** Содержание азота определено методом потенциометрического титрования в не-
водной среде в лаборатории гетероатомных соединений нефти.
Институт нефтехимического синтеза им. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. А. А. Жуховицкий, Н. М. Туркельтауб. Газовая хроматография. М., Гостоптехиздат,
1962.
2. И. И. Лулова, Л. И. Пигузова, А. И. Тарасов, А. К. Федосова. Химия и технология
топлив и масел, № 8, 59 (1961).
3. W. Walisch. Chem. Вег., 94, 2314 (1961).
4. Н. Weitkamp, F. Korte. Chemie Ing. Techn., 35, 429 (1963).
5. H. 'Weitkamp, К- Maynz, F. Korte. Z. analyt. Chem., 205, 81 (1964).
6. Yanagimoto. CHN Corder Model MT-1.
7. H. H. Безингер, Г. Д. Гальперн, Т. И. Овечкина. Методы анализа органических со-
единений нефти, их смесей и производных, сб. 1. М., Изд-во АН СССР, 1960,
стр. 132.
8. R. Н. Reitsema, N. L. Allphin. Analyt. Chem., 33, 355 (1961).
9. M. A. Parsons, S. N. Pennington, J. M. Walker. Analyt. Chem., 35, 842 (1963).
10. А. П. Терентьев, H. M. Туркельтауб, E. А. Бондаревская, Л. А. Домочкина. ДАН
СССР, 148, 1316 (1963).
11. И. Е. Пахомова, М. Н. Чумаченко. Изв. АН СССР, серия хим., 1965, 1138.
12. F. Nightingale, I. М. Walker. Analyt. Chem., 34, 1435 (1962).
13. А. А. Москвина, Л. В. Кузнецова, С. А. Добычин, М. И. Розова. ЖАХ, вып. 6,
749 (1964).
14. М. Н. Чумаченко, И. Е. Пахомова. ДАН СССР, 170, 125 (1966).
15 М. О. Коршун, В. А. Климова. ЖАХ, № 6, 274 (1947).
16. М. О. Коршун. ЖАХ, № 7, 96 (1952).
17. М. Veiefa, L. Synek. Mikrochim. Acta, № 2, 208 (1960).
18 G. Kainz, H. Horwatitsch. Mikrochim. Acta, № 1—2, 7 (1962).
19. А. В. Непряхина, И. К. Чудакова, Г. А. Новикова, Г. Г. Лукина. ЖАХ, № 6,
909 (1967).
ЭЛЕМЕНТНЫЙ АНАЛИЗ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
С ГАЗОХРОМАТОГРАФИЧЕСКИМ ОПРЕДЕЛЕНИЕМ ПРОДУКТОВ
РАЗЛОЖЕНИЯ.
II. МЕТОД ОДНОВРЕМЕННОГО ОПРЕДЕЛЕНИЯ
УГЛЕРОДА, ВОДОРОДА, АЗОТА
И. К. Чудакова, А. В. Непряхина, О. Н. Дома ни па,
Г. А. Новикова, Г. Г. Радикова
I. ВВЕДЕНИЕ
Выше было рассмотрено [1] применение газовой хроматографии к
элементному анализу для определения азота.
Для одновременного определения углерода и водорода или углерода,
водорода и азота методом газовой хроматографии в литературе приве-
дено несколько вариантов анализа. Общая для всех методов схема ана-
лиза включает химическое превращение пробы анализируемого вещест-
ва в простые продукты с последующим хроматографическим разделе-
нием образовавшейся смеси и количественной регистрацией каждого
компонента смеси с помощью катарометра.
Основные затруднения в применении газовой хроматографии для
анализа заключаются в необходимости единовременной подачи продук-
тов окисления на хроматографическую колонку и в необходимости раз-
деления смеси, состоящей из газов и паров воды.
Различные варианты газохроматографических методов элементного
анализа отличаются применяемыми способами преодоления указан-
ных трудностей.
Дазуолт и Брандт [2] предложили способ определения углерода и водорода, при
котором пробу (2—6 мг) окисляют на окиси, меди при температуре 750° С в потоке
кислорода. Продукты окисления (двуокись углерода и вода) проходят через реактор
с карбидом кальция, где пары воды превращаются в ацетилен. Ацетилен и двуокись
углерода конденсируют в ловушке, охлаждаемой жидким азотом. Давление в ловуш-
ке поддерживают на уровне 110 мм рт. ст во избежание конденсации кислорода. Пос-
ле сожжения пробы сконденсированные продукты размораживают и в потоке гелия
вводят в хроматографическую колонку с силикагелем. В качестве детектора был при-
менен катарометр. Продолжительность анализа 20 мин. Ошибка определения по уг.
лероду 0,5 абс. %; по водороду — 0,1 абс. %.
Сандберг и Мареш [3] модифицировали метод (2], исключив применение кислоро-
да. Навеска вещества сжигается в токе гелия при температуре 750° С в присутствии
окиси меди. Продолжительность сожжения не менее 26 мни. Исключение кислорода
упрощает аппаратуру и устраняет возможность возникновения взрывоопасной смеси
Oj—С2Н2.
Фогель и Кватроне [4] сжигали органические вещества (8—11 мг) в стационарных
условиях, в наполненной кислородом бомбе. Для анализа часть продуктов сожжения
направляли из бомбы в специальное газоотборное устройство хроматографа. Пары
воды и двуокись углерода анализировали на хроматографической колонке с доде-
цилфталатом на диатомитовой земле; температура колонки 104° С, газ-носитель —
8*, 115
кислород. Продолжительность анализа 17 мин. Точность определения по углероду
±0,24%, по водороду ±0,1%.
Лускина, Сявцилло, Терентьев и Туркельтауб [5] проводили сожжение органиче-
ского вещества в присутствий окиси меди в замкнутой трубке (кварц) при остаточном
давлении 10 мм рт. ст. Сожжение продолжалось 10 мин. при температуре 700° С. Про-
дукты сожжения вытесняли током гелин на хроматографическую колонку. Средние
отклонения при анализе органических веществ состава СНО составляли для углеро-
да 0,4 абс. %, для водорода 0,16 абс. %.
Вечержа [6, 7] предложил метод автоматического определения углерода, водоро-
да, азота, в котором хроматографическая колонка заменена химическими поглотите-
лями. В качестве окислителя использована закись-окись кобальта при температуре
600—700° С. Для определения каждого элемента применялся соответствующий газ-носи-
тель. При определении углерода сожжение проводят в токе кислорода. Образующуюся
воду и окислы азота поглощают ангидроном и двуокисью марганца, а двуокись угле-
рода определяют по теплопроводности. При определении водорода вещество сжигают
в токе азота; воду восстанавливают железными стружками до водорода; после погло-
щения двуокиси углерода водород фиксируют катарометром. При определении азота
вещество сжигают в токе СО2, элементарный азот определяют также по теплопроводно-
сти. Точность определения углерода ±0,46%, водорода ±0,16%, азота ±0,27%.
Найтингел и Уолкер [8] разработали метод одновременного определения углеро-
да, водорода и азота быстрым сожжением (в течение 30 сек.) анализируемой пробы
с помощью индукционной печи. В качестве окислителей использованы перманганат
серебра и окись меди. Быстрое сожжение пробы с катализатором в потоке гелия по-
зволяет непосредственно без предварительного концентрирования разделять простые
продукты окисления в хроматографической колонке. Навеску анализируемого веще-
ства, смешанного с окислителем, сжигали в угольном тигле, футерованном кварцем.
Продукты окисления проходили через реактор, заполненный на */з окисью меди и на
г/з металлической медью дли завершения окисления и восстановления окислов азота.
Далее газовый поток проходил через реактор с карбидом кальция, где вода превра-
щалась в ацетилен. Карбид кальция в реакторе заменяли новым перед каждым ана-
лизом. Смесь простых продуктов (азот, двуокись углерода, ацетилен) разделяли на
хроматографической колонке с молекулярными ситами 5А. Среднее отклонение при
определении углерода ±0,52%, водорода ±0,22%, азота ±0,58%.
Общая продолжительность анализа 1 час 45 мин.
Москвина, Кузнецова, Добычин и Розова [9] сжигали пробу анализируемого сое-
динения, смешанную с окисью меди, в атмосфере гелия при температуре 650° С. Сож-
жение продолжалось 12 мин. при остаточном давлении 5—10 мм рт. ст. Азот, двуокись
углерода и воду разделяли на колонке, заполненной пористым тефлоном, пропитан-
ным триэтаноламином при температуре 97—98° С. Продолжительность анализа 25—
30 мин. Средняя квадратичная ошибка для углерода 0,74%, для водорода 0,22%, для
азота 0,46%.
В предложенном Чумаченко и Пахомовой [10] методе одновременного опре-
деления углерода, водорода и азота с применением газовой хроматографии окисление
вещества осуществляют окисью никеля при температуре 900—950° С в атмосфере
гелия в герметично закрывающейся реакционной пробирке. Продолжительность
сожжения 1—2 мин. Образовавшаяся вода превращается в ацетилен над карбидом
кальция. Полученные азот, двуокись углерода и ацетилен разделяют на колонке, за-
полненной активированным углем при температуре 120° С, скорость газа-носителя
170 мл!мин. Точность определения ±0,2%.
В настоящее время ряд зарубежных фирм выпускают так называемые CHN-ана-
лизаторы [11—14]. Однако, как указывается в работе [15], до сих пор они не полу-
чили широкого распространения главным образом из-за их дороговизны.
Накопленный нами опыт работы по определению азота в органи-
ческих соединениях с газохроматографическим окончанием анализа
[1, 16] позволил перейти к разработке метода одновременного определе-
ния углерода, водорода и азота. При этом мы применили способ раз-
ложения и количественного окисления анализируемых веществ (на-
вески 1—5 мг) окисью меди в замкнутом объеме в среде' гелия при
температуре 710—830° С, как и при определении азота, описание кото-
рого дано выше [1].
Образующаяся вода подвергается электролизу. Продукт электроли-
за — кислород определяется (так же как азот и двуокись углерода) хро-
матографически. Измерение кислорода производится после азота и дву-
окиси углерода.
116
Применение электролиза в элементном анализе описано в литерату-
ре [17—19].'Однако во всех случаях определение количества воды осно-
вано на кулонометрическом измерении.
II. РЕКОМЕНДУЕМАЯ МЕТОДИКА (ПРЕДВАРИТЕЛЬНАЯ)
Аппаратура и реактивы
1. Хроматограф ХЛ-4.
2. Металлическая трубка для сожжения.
3. Краны запирающие (два комплекта из трех кранов).
4. Электронагреватель.
5. Стаканчик кварцевый.
6. Электролитическая ячейка.
7. Силикагель МСМ.
8. Окись меди порошкообразная и в проволоке, ч.д.а.
9. Газ-носитель — гелий. Узел осушки газа-носителя.
10. Микровесы (Бунге).
Трубка для сожжения, все газовые коммуникации и узлы установки
выполнены из нержавеющей стали марки 1Х18НТ-9, так как включение
электролитической ячейки требует особо надежной герметичности уста-
новки [20]. Схема установки приведена на рис. 1.
Рис. 1. Схема установки для определения С, Н и N
1 — баллон с гелием; 3 — редуктор; 3 — край тонкой регулировки с образцовым мано-
метром на 6 кгс/см2; 4 — узел осушки газа-носителя с кранами; 5 — детектор; 6 — ко-
лонка; 7 — запирающие краны; в —трубка для сожжения; 9 — электролитическая
ячейка; 10 — самописец ЭПП-09
Изменения в газовой схеме хроматографа ХЛ-4, трубка для сожже-
ния, электронагреватель, а также подготовка реактивов подробно опи-
саны в предыдущей статье [1].
Установка содержит два комплекта из трех запирающих игольчатых
кранов 7 (см. рис. 1). Три крана одного комплекта обслуживают труб-
ку для сожжения, а три крана другого — электролитическую ячейку. Эти
краны.обеспечивают надежное отключение потока гелия от трубки для
сожжения на период разложения навески и от электролитической ячей-
ки на период разложения образующейся-воды.
В предварительных опытах была использована электролитическая
ячейка [9] из влагомера типа АВЭ-1 ВНИИГаза [21]. Принцип действия
ячейки состоит в извлечении влаги из анализируемого потока газов
пленкой фосфорного ангидрида и последующем разложении воды на
117
водород и кислород путем электролиза. В настоящее время проходит
испытание ячейка, специально разработанная для элементного анализа
и изготовленная в СКВ ИНХС АН СССР.
Узел осушки газа-носителя гелия 4 состоит из змеевика, охлаж-
даемого в сосуде Дьюара твердой углекислотой до температуры — 80° С.
Узел осушки снабжен тремя игольчатыми кранами для продувки гелия
через систему или в атмосферу.
Предварительно метод одновременного определения С, Н, N испы-
тывался на макетной установке, состоящей из отдельных узлов, сконст-
руированных и изготовленных в СКВ ИНХС АН СССР в сочетании с
хроматографом ХЛ-4 и электролитической ячейкой от влагомера типа
АВЭ-1, позволившей предложить единый прибор для определения С,
Н, N [22].
Анализ
Сожжение навески анализируемого вещества проводят в режиме,
описанном ранее [1].
После автоматического выключения электронагревателя продукты
сожжения продувают гелием через электролитическую ячейку, где воду,
Рис. 2. Хроматограмма,
полученная при анализе
ацетанилида
образовавшуюся при сожжении, поглощает фосфорный ангидрид, а азот
и двуокись углерода подают на колонку. На время прохождения про-
дуктов сожжения через ячейку (3 мин.) электрический ток отключают.
После этого ячейку закрывают кранами, включают электрический ток
и проводят электролиз воды. Одновременно производят разделение азо-
та и двуокиси углерода на хроматографической колонке. В зависимости
от концентрации азота и величины навески регистрацию пика азота
производят на 1, 2 или 4 чувствительности. Пик углекислоты обычно ре-
гистрируют на 8 чувствительности. К тому времени, когда окончена ре-
гистрация двуокиси углерода, заканчивается электролиз в электролити-
ческой ячейке (контролируется по микроамперметру). Переключая
краны ячейки, выдувают гелием образовавшийся кислород в хрома-
тограф и регистрируют на диаграмме. Хроматограммы азота, двуокиси
углерода и кислорода представлены на рис. 2.
Расчет
Ежедневно определяют титр (Г), под которым понимается количест-
во элемента в у на 1 л/л2 площади пика каждого элемента, полученной
от сожжения навески стандартного вещества в условиях анализа.
Т = АР/100-Sp,
118
где А — теоретическое содержание элемента в стандартном веществе
(в %); Р— навеска стандартного вещества (в у)! Sp — площадь пика
элемента, полученная при сожжении стандартного вещества.
Площадь пика определяется по формуле
SP = НаК,
где Н — высота пика (в мм); а — ширина пика на полувысоте (включая
толщину левой стороны пика); К — чувствительность.
Измерения производятся десятикратной лупой с окуляр-микромет-
ром.
Расчет процентного содержания элементов (С, Н, N) в анализиру-
емом веществе производится по формуле
%Э,= TSxA00/Px,
где Т — титр элемента по стандартному веществу (в у/мм2); Sx — пло-
щадь пика элемента, полученная при сожжении анализируемого ве-
щества (в мм2); Рх — навеска анализируемого вещества (в у).
Примеры расчета приведены в табл. 1.
Таблица 1
Пример расчета титра углерода, водорода и процентного содержания углерода,
водорода в сахарозе
(% Стеорет =42,10; % Нтеорет = 6,43)
S
с
«Л
о „
5 *
К к
rs -юо
% Э = ^-£—
х
1,058
1,058
1,400
1,400
59,4 7,0 8
Н 130,0 2,6 2
С 78,2 7,0 8
Н 158,5 2,8 2
3324
6760
4379
8876
т _42,10-1058
с 100-3324 — 0,134
Т„ = 6’43-1058 _ 0 100
н .100.6760
о%С^О,134;43;9-1ОО=41,9О
QZH — 0,Ю0-8876-100_д
z 1400
Величина титра при сожжении различных стандартных веществ
в течение дня постоянна. Например, титр углерода по сахарозе равен
0,134 у] мм2, по динитробензолу — 0,134 у/мм2.
III. РЕЗУЛЬТАТЫ АНАЛИЗОВ
Предварительные опыты по одновременному определению углерода,
водорода, азота в органических веществах описанным выше методом
проводились на веществах состава CHNO.
В табл. 2 приведены результаты анализа некоторых веществ.
Из табл. 2 видно, что метод,дает точность определения азота ±0,2;
углерода ±0,4; водорода ±0,1%'абс.
Работа продолжается совместно с СКВ ИНХС АН СССР по созда-
нию прибора и расширению применимости метода для органических
соединений различного состава и строения.
119
Таблица 2
Определение С, Н, N
Вещество Содержание N, % Содержание С, % Содержание Н, %
вычислено найдено . вычислено найдено вычислено найдено
Сахароза С12Н220ц — 42,10 41,90 42,30 6,43 6,34 6,39
Т-Фенил-х-л-ксилилмасляная кислота — — 80,56 80,76 7,51 7,52
Ацетанилид QHsNHCOOCHs 10,37 10,21 10,39 71,10 70,99 70,93 6,67 6,58 6,79
Динитробензол CoH4(N02)2 16,67 16,50 16,75 42,86 43,29 43,24 2,38 2,23 2,14
ВЫВОДЫ
Дана предварительная методика одновременного определения С, Н,
N, по которой навеска анализируемого вещества подвергается пироли-
тическому разложению в замкнутом объеме (в присутствии окиси меди)
при температуре 710—830° С; вода подвергается электролизу. Образу-
ющиеся СО2, N2 и О2 определяются с помощью газовой хроматографии.
Продолжительность анализа 50—60 мин.
Институт нефтехимического синтеза нм. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. А. В. Непряхина, И. К. Чудакова, Г. А. Новикова, Г. Г. Радикова, О. Н. Доманина.
Настоящий сборник, стр. 107.
2. A. A. Duswalt, W. W. Brandt. Analyt. Chem., 32, 210, 1960.
3. О. E. Sundberg, C. Marech. Ibid., p. 212.
4. A. M. Vogel, I. I. Quattrone. Ibid., p. 1754.
5. Б. M. Л у скина, С. Я. Сявцилло, А. П. Терентьев, Н. М. Туркельтауб. ДАН СССР,
141, 869 (1961).
6. М. Vegeta. Collect. Czech. Chem. Comm., 28, 2278, 2298 (1961).
7. M. Veiefa. Acta chim. Hungar., 26, 511 (1961).
8. C. F. Nightingale, I. M. Walker. Analyt. Chem., 34, 1435 (1962).
9. А. А. Москвина, JI. В. Кузнецова, С. А. Добычин, M. И. Розова. ЖАХ, № 6,
749 (1964).
10. M. H. Чумаченко, И. E. Пахомова. ДАН СССР, 170, 125 (1966).
11. W. Walisch. Chem. Вег., 94, 2314 (1961).
12. H. Weitkamp, F. Korte. Chemie Ing. Techn., 35, 429 (1963).
13. H. Weitkamp, Д. Maynz, F. Korte. Z. analyt. Chem., 205, 81 (1964).
14. Yanagimoto. С. H. N. Corder, Model МТ-1 (реклама).
15. Chem. Eng. News, 43, № 33, 41 (1965).
16. А. В. Непряхина, И. К. Чудакова, Г. А. Новикова, Г. Г. Лукина. ЖАХ, № 6 (1967).
17. Г. Ф. Анисимова, В. А. Климова. ЖАХ, 18, 412 (1963).
18. F. Salzer. Z. analyt. Chem., 205, 66 (1964).
19. F. Salzer. Microchem. J., 10, 27 (1966).
20. С. Г. Поляк, А. В. Непряхина, Л. H. Татаринова, Г. В. Виноградов. Зав. лаб., № 5,
572 (1966).
21. А. К- Карпов, П. А. Фроловский, Н. Р. Шорохов, 3. С. Филатова. Газ. пром., 4,
37 (1962).
22. А. В. Непряхина, И. К. Чудакова, А. А. Константинов, С. А. Крашенинников. Авт.
свид. 191877 (1966); Бюлл. изобр., №4, 129 (1967).
ПОЛУЧЕНИЕ КОНЦЕНТРАТОВ
АЗОТИСТЫХ ОСНОВАНИЙ НЕФТИ
Н. Н. Беаингер, Г. Д. Гальперн, В. Н. Даричева
Изучению состава азотистых соединений нефти в настоящее время
уделяется значительное внимание. Это связано с развитием методов
каталитической переработки нефтяных дистиллятов и с вредным влия-
нием азотистых соединений на эксплуатационные свойства нефтепро-
дуктов. Однако несмотря на успехи, достигнутые в этой области нефте-
химии, природа главной массы азотистых соединений нефти до сих пор
остается не изученной.
Одной из причин этого является трудность их выделения. Методы
выделения так называемых «нейтральных» азотистых соединений отсут-
ствуют вообще. Единственным методом выделения нефтяных оснований
до сих пор являлось извлечение их водными растворами минеральных
кислот. Помимо практических трудностей, связанных с образованием,
устойчивых эмульсий, метод дает возможность получать представитель-
ные концентраты оснований лишь из фракций, выкипающих до 350—
400°С (керосин, лигроин). С увеличением температуры выкипания
фракций, т. е. с увеличением их молекулярного веса, степень извлечения,
оснований резко падает. Из газойля водными растворами кислот удается
извлечь около 40% оснований [1], из асфальтено-смолистой части водно-
спиртовыми растворами серной кислоты было извлечено оснований*
8,5% [2].
Вместе с тем известно, что' азотистые соединения концентрируются
именно в высокомолекулярной части нефти [3—5]. Неполное извлечение
оснований с большим молекулярным весом водными растворами кислот
вероятнее всего может быть объяснено гидрофобностью как самих,
оснований, так и образующихся солей, обусловленной значительным
преобладанием углеводородной части в молекуле (свыше 20 атомов
углерода на 1 атом азота). Недостатки метода извлечения оснований,
водными растворами кислот в значительной степени могут быть преодо-
лены при использовании катионитов. В этом случае извлечение основа-
ний можно проводить из любой неводной среды. При этом исчезает
«барьер растворимости» и одновременно исключается возможность гид-
ролиза образующихся солей. Известно, что применение неводных сред
позволило разработать весьма чувствительные методы количественного
ацидиметрического титрования нефтяных оснований. В неводных средах
происходит сдвиг равновесия реакции в сторону образования соли
кислота + основание соль
даже для очень слабых оснований с р/(н2о9—14.
121
В последние годы описано несколько случаев использования катионитов для сорб-
ции нефтяных оснований. Мандей и Иве [6] в работе, посвященной вопросу о коли-
чественном удалении кислот, фенолов и оснований из товарных нефтепродуктов, изу-
чили зависимость емкости ряда сульфокислотных катионитов от степени поперечной
сшивки и содержания воды. Наиболее важным фактором оказалось содержание воды
в смоле. Путем частичной замены воды на метанол авторам удалось значительно ус-
корить сорбцию оснований на смолах и провести извлечение оснований из керосина,
газолина и газойля с использованием 65—94% емкости смолы. Однако из легких сма-
зочных масел и газойля основания полностью не извлекались. Подобные же резуль-
таты были получены Снидером и Бюллем [7] при извлечении оснований из топлив ка-
тионитом Даулайт С-10 с целью их количественного определения методом УФ-спект-
:р оскопи и.
Задачей настоящего исследования явилась разработка метода полу-
чения представительных концентратов оснований из высокомолекуляр-
ных нефтяных фракций и сырых нефтей с помощью сульфокислотных
катионитов.
Работа состоит из двух частей: 1) изучение сорбции индивидуальных
оснований и 2) подбор оптимальных условий для сорбции и десорбции
оснований нефти.
I. СОРБЦИЯ ИНДИВИДУАЛЬНЫХ ОСНОВАНИИ
Существенным фактором, ответственным за неполную сорбцию
нефтяных оснований, наряду с содержанием воды является величина
•сорбируемых молекул. Заполнение активных центров в зерне ионооб-
менной смолы связано с диффузией сорбата вглубь зерна. При изучении
•сорбции больших молекул органических оснований наблюдали замед-
ленную диффузию их вглубь частицы ионита [8—10]. В связи с этим
нами была исследована зависимость емкости ряда сульфокислотных
катионитов с различной степенью сшивки и разного способа приготовле-
ния от величины сорбирующихся молекул. Характеристика катионитов
приведена в табл. 1. Были испытаны Н-формы катионитов Дауэкс 50,
КУ-1, КУ-2 и серия крупнопористых катионитов КУ-23, применение ко-
торых представлялось нам особенно перспективным.
Сорбатами служили гетероциклические индивидуальные ароматиче-
ские основания различного молекулярного веса и структуры (табл. 2),
моделирующие в некоторой мере типы соединений, находимых в нефтях.
Главная масса азотистых соединений нефти представлена веществами
сложного строения, включающими, кроме ароматического гетероцикли-
ческого ядра, алкильные, нафтеновые и ароматические радикалы.
Ряд модельных соединений подобран так, что в него включены ос-
нования с частично или полностью экранированным атомом азота.
Сорбция таких соединений должна протекать наиболее трудно. Сорб-
цию проводили из бензольных растворов.
В табл. 3 приведены результаты, полученные при определении общей
емкости катионитов по иону натрия и а-пиколину и относительные
емкости би-, три- и тетрациклических оснований. Измерения были вы-
полнены в обычно принятых статических условиях [11].
Емкость по а-пиколину (ЕПик) практически для всех катионитов
хорошо совпадала с емкостью по натрию в водных растворах (ENa).
Исключение составили катиониты КУ-1 и КУ-23 4/50, показавшие низ-
кую емкость по отношению ко всем испытанным сорбатам. Степень
использования емкости катионитов бициклическими основаниями —
хинолин (Ехин) и 2-фенилпиридин (Ефп) — сохраняется высокой (90—
100%) у катионитов КУ-23 с содержанием ДВБ 4 и 12%, и Дауэкс 50 с
содержанием ДВБ 2,4 и 8%. С увеличением содержания ДВБ сорбция
бициклических оснований заметно падает. Степень использования емко-
сти составляет 30—80% от Ешп;. Во всех случаях 2-фенилпиридин адсор-
122
Таблица 1
Характеристика катионитов
К атионит Функцио- нальные группы Содержа- ние Н2О в воздуш- но-сухом образце, % Насыпной вес, мл/г Матрица
Дауэкс 50x2 SO3H 22 0,7 Сополимер стирола и
Дауэкс 50x4 SO3H 23 0,7 дивинилбензола
Дауэкс 50x8 SO3H 21 —-
Дауэкс 50X12 SO3H 16
КУ-2 SO3H 17 0,7
SO3H, он Конденсационная смола
КУ-1 И 0,7 из п-фенолсульфокислоты и формальдегида
КУ-23 4/20* SO3H 37 0,8 Сополимер стирола и ди-
КУ-23 4/50 SO3H 21 0,6 винилбензола
КУ-23 4/100 SO3H 47 0,5
КУ-23 12/20 SO3H 25 0,7
КУ-23 12/50 SO3H 27 0,7
КУ-23 12/80 SO3H 29 0,8
КУ-23 20/50 SO3H 38 —
КУ-23 20/80 SO3H 40 0,5
КУ-23 25/50 SO3H 37 0,7
* Числитель указывает процент дивинилбензола (ДВБ); знаменатель — количество
растворителя, взятого при полимеризации (в %).
Таблица 2
Характеристика индивидуальных азотистых оснований,
использованных в качестве модельных соединений
Основание Мол. вес Т. кип., (плавле- ния), °C р^ню
Пиридин 79,05 116 8,8
а-Пиколин 93,06 129 8,0
Хинолин 129,06 238 9,2
2-Фенилпиридин 155,2 270 8,7
2-Фенилхинолин 205,1 >300 9,35
2-Фенил-5,6-бензохинолин . . 255,3 (188) —
Примечание. Степень чистоты всех соединений по содержанию
азота 100%, за исключением 2-фенилпиридина, чистота которого равна
98,2%.
бируется хуже, чем хинолин. По отношению к три- и тетрациклическим
-основаниям заметной емкостью обладали лишь катиониты КУ-23 20/50
и 20/80, хотя и у них использование емкости составляло лишь 16—30%.
У остальных катионитов емкость по фенилхинолину (£фХ) и фенилбен-
зохинолину (£фбх) равна нулю.
Столь резкое падение сорбционной способности катионитов при пере-
ходе от моно- и бициклических оснований к три- и тетрациклическим
123
Таблица 3
Емкость катионитов по основаниям различного молекулярного веса
(растворитель — бензол)
Основание Na Псн N 11 । \/\^ N II L/=\ \? N Г1 L—ч А ч УР) N
Мол. вес 23 93,1 129,1 155,3 205,1 255,3
Катноннт Емкость сухого катионита, мг-экв/г Относительная емкость, % от емкости по а-пиколину
Дауэкс 5,1 5,2 90 90 0
50X2
Дауэкс 5,7 5,2 97 90 0 0
50x4
Дауэкс 5,5 4,4 102 90 5 0
50X8
Дауэкс 50X12 5,5 3,9 62 70 0 0
КУ-1 5,1 1,9 5 3 0 0
КУ-2 4,9 См.» 4,3 52 4 0 0
КУ-23 5,1 102 77 2 0
4/20
КУ-23 1,6 100 100 0 0
4/50
КУ-23 ь—1 4,9 100 94 4 а
4/100
КУ-23 __ 5,2 100 51 5 0
12/20
КУ-23 4,9 100 25 0 0
12/50
КУ-23 12/80 — 5,1 93 37 4 0
КУ-23 5,4 60 40 30 26
20/50
КУ-23 20/80 — 5,9 75 66 20 16
КУ-23 25/50 5,7 80 75 9 6
* Efta для катионитов КУ-23 лежит в пределах .5,0—5,6 мг-экв.
(Ж
может быть связано как с уменьшением растворимости этих соединений
в воде, что затрудняет диффузию их на границе органический раствори-
тель— вода, так и с малыми размерами пор в зерне ионитов.
Из всех испытанных образцов катиониты КУ-23 20/50 и 20/80 отли-
чаются наиболее высоким содержанием воды (см. табл. 1). Их повышен-
ная емкость по отношению к 2-фенилхинолину и 2-фенил-5,6-бензо'
хинолину, по-видимому, связана преимущественно с наличием достаточно-
крупных каналов, по которым более крупные молекулы оснований
могут проникать внутрь зерен. Были сняты кривые сорбции 2-фенил-
пиридина и 2-фенил-5,6-бензохинолина (см. рис. 1). Найдено, что емкость
по 2-фенил-5,6-бензохинолину для катионитов КУ-23 20/50 и 20/80
124
практически остается постоянной в течение 96 час. и не поднимается
выше 1,4 и 0,8 мг-экв N соответственно. Максимальное значение емкости
достигается уже через 20 час. Величина Е$п для катионитов Дауэкс 2,
КУ-23 4/20, 4/100 и 12/80, измеренная через 24 часа, не соответствовала
их общей емкости (так называемая «кажущаяся емкость» [8]). Через 72
часа была достигнута емкость, хорошо совпадавшая со значением Ддик
Рис. 1. Сорбция 2-фенилпиридина из бензольного раствора
различными катионитами
1 - КУ-23 4/100; 2 — Дауэкс 50X2; 3 - КУ-23 4/20; 4 — КУ-23 12/80;
5 — КУ-23 12/50
для этих же катионитов («истинная емкость»). Для катионита КУ-23
12/50 заметного увеличения не наблюдалось даже после 96-часового
контакта катионита с раствором.
Таким образом установлено, что в серии испытанных катионитов
удовлетворительной сорбционной способностью по отношению к три- и
тетрациклическим ароматическим основаниям обладают только крупно-
пористые катиониты с содержанием дивинилбензола 20%. Эти катиони-
ты обладают также удовлетворительной емкостью по отношению к моно-
и бициклическим основаниям при сорбции из бензола.
И. СОРБЦИЯ ОСНОВАНИЙ НЕФТИ
Исходя из изложенного выше, в качестве сорбентов для нефтяных
оснований были выбраны крупнопористые катиониты марки КУ-23 20/50
и 20/80. Извлечение оснований было проведено из туймазинской нефти,
ново-дмитровской нефти (Грозненский бассейн), гудрона и широкой
•фракции товарной южно-узбекской нефти. Краткая характеристика
нефтепродуктов приведена в табл. 4. Сорбцию проводили в статических
условиях при перемешивании. Контроль за ходом сорбции оснований
•осуществляли потенциометрическим титрованием аликвотных объемов
в среде уксусной кислоты или уксусного ангидрида. Титрант — 0,1 N
раствор хлорной кислоты в уксусной кислоте.
Предварительными опытами было найдено, что емкость катионитов
КУ-23 X 20 по отношению к нефтяным основаниям составляет в среднем
0,3—0,4 мг-экв N на 1 г сухого катионита. Исходя из этой емкости,
-брали 100%-ный избыток катионита по отношению к N0Ch- Были сняты
125
Таблица 4
Характеристика нефтей и нефтепродуктов, использованных для получения
концентратов азотистых оснований
Нефтепродукт Nqch» % $обЩ’ % [Примечание
Нефть сборная, тунмазинская 0,048 1,89 Парафинистая
Нефть ново-дмнтровская (Грозный) Нефть южно-узбекская 0,029 0,11 »
широкая газойлевая фракция . . 0,010 2,40 Температура отбора! 200—400° С
гудрон 0,12 5,02 Остаток выше 550° С
Таблица 5
Сорбция оснований нз нефти
№ п. п. Нефтепродукт (раствор в бензоле) Количе- ство (СНаСО)2О, % от об- щего объема Время сорбции, часы N0CH- %х10~3 на нефть Извлечено ^осн* % от исходного
ДО сорбции после сорбции
1 Ново-дмитровская нефть 10 29 13 55
2 То же — 120 29 12 59’
3 » » 5 14 29 Нет 100-
4 » » 3,5 13 29 7 76
5 Ново-дмитровская нефть в бен- — 16 29 14 52
золе
6 Туймазинская нефть — 11 48 22 54
7 То же 5 16 48 Нет 100
8 Широкая фракция южно-уз- бекской нефти — 13 10 4 60
9 То же — 96 10 4 60-
10 » » 2,5 12 10 Нет 100
11 Гудрон южно-узбекской нефти 15 23 120 » 100-
кривые сорбции оснований из туймазинской и ново-дмитровской нефтей
и из широкой фракции. Нефть предварительно разбавляли бензолом или
бензином в отношении 1—4; сорбцию из широкой фракции вели без
растворителя. В этих условиях процесс сорбции описывается левой
частью кривых 1, 2 и 3 и кривой 4 рис. 2. Степень извлечения оснований
составляла 55—60% (табл. 5, № 1, 2, 5, 6, 8, 9). Прибавление в раствор
свежих порций катионита, а также увеличение времени контакта катив'
нита с раствором до 120 час. не привели к дальнейшему извлечению ос-
нований.
Ранее при исследовании концентратов нефтяных оснований нами
было установлено, что лучшей средой для отделения оснований от сопут-
ствующих им нейтральных азотистых соединений и сульфидов с помощью
ионообменных смол является уксусный ангидрид [12, 13]. Поэтому было
проверено влияние уксусного ангидрида на процесс извлечения основа-
ний из нефти. Оказалось, что в присутствии уксусного ангидрида сорб-
ция оснований проходит количественно (рис. 3). Уксусный ангидрид
влияет не только на полноту извлечения оснований, но значительно уве-
личивает скорость сорбции. Из раствора ново-дмитровской нефти,
содержащей 5 объемн. % уксусного ангидрида, количественное извлече-
ние оснований было достигнуто через 14 час. Увеличение его концентра-
126
f™!?)0 15% сократило вРемя сорбции до 10 час. (см. табл. 5, № 3, 4, 7,.
Уксусный ангидрид был добавлен в бензольные растворы ново-
дмитровской и туймазинской нефтей и к широкой фракции, предвари-
тельно доведенные до равновесного состояния с катионитом (степень.
1 — ново-дмитровская; 2— широкая фракция
южио-узбекской нефти; 3 — туймазинская
(растворитель — бензол); 4 — ново-дмитровская
(растворитель бензин Б-70).
А— прибавлен (CHgCOhO
1—ново-дмитровская нефть, уксусный ангидрид
(10%); 2 —то же, уксусный ангидрид (5%);
3 — то же, диметилформамид (15%); 4 — туй-
мазниская нефть, уксусный ангидрид (10%)
извлечения N0CH до добавки уксусного ангидрида — 55,54 и 60% соот-
ветственно). После добавления уксусного ангидрида оставшиеся в раст-
воре основания полностью сорбировались катионитом (правая часть
кривых рис. 2). Минимальная концентрация уксусного ангидрида, не-
обходимая для полной сорбции оснований, зависит от характера нефте-
продукта: для широкой дистиллятной фракции она составляет 2,5%; для
гудрона—15%. Снижение концентрации уксусного ангидрида в раство-
ре нефти до 3,5% привело к установлению равновесия, при котором сор-
бировалось только 76% оснований. Очевидно, что с увеличением сред-
него молекулярного веса и смолистости нефтепродуктов нижний предел
концентрации уксусного ангидрида, необходимой для количественной
сорбции оснований, возрастает.
Влияние уксусного ангидрида на полноту извлечения оснований неф-
ти вряд ли может быть объяснено увеличением емкости катионита в ре-
зультате уничтожения так называемого «барьера растворимости». Не-
сомненно, уксусный ангидрид связывает большую часть воды катиони-
та. Сорбция оснований осуществляется в практически безводной среде
бензол — уксусная кислота — уксусный ангидрид, что должно благопри-
ятно сказаться на сорбции гидрофобных оснований. Однако определе-
ние емкости катионитов по отношению к модельным основаниям в сре-
де уксусного ангидрида показало, что во всех случаях происходит зна-
чительное уменьшение емкости.
В литературе имеется указание на то, что емкость катионитов в сильно кислых
водных растворах несколько снижается в результате уменьшения их удельного объема
[14]. Вероятно, малая емкость катионитов в среде уксусного ангидрида также объясня-
ется их сжатием.
127
Тем не менее, несмотря на уменьшение общей емкости катионитов,
количество нефтяных оснований, сорбирующихся в присутствии уксус-
ного ангидрида, увеличивается почти вдвое.
Возможно, что влияние уксусного ангидрида заключается в разруше-
нии ассоциатов гетероатомных соединений нефти и высвобождении из
них оснований. Уже неоднократно отмечалось, что при выделении неф-
тяных оснований в виде концентратов в последние попадает значитель-
ное количество сернистых и кислородных соединений. Извлечение этих
соединений, идущее симбатно с извлечением оснований, нельзя объяс-
нить повышенной растворимостью их в водных или водно-спиртовых
растворах минеральных кислот. Более детальное изучение состава кон-
центратов показало, что разделение, например, азотистых соединений,
обладающих свойствами оснований и сульфидов, не удается ни метода-
ми адсорбционной хроматографии [15], ни ионообменной [12]. Вместе с
тем разделение легко прошло после окисления сульфидов до сульфок-
сидов. Остаточные сернистые соединения (вероятно тиофеновой приро-
ды) и кислородные соединения отделить от азотистых оснований до сих
пор не удалось. Эти наблюдения в сочетании с результатами, получен-
ными при сорбции оснований на катионитах, приводят нас к заключе-
нию, что большая часть гетероатомных компонентов нефти существует
в виде сложных, весьма устойчивых ассоциатов, образованных как за
счет р-электронов атомов серы, кислорода и азота, так и за счет л-свя-
зей конденсированной ароматической части молекул.
На ассоциацию сульфидов средних и высших фракций нефти с кон-
денсированными ароматическими углеводородами недавно было обра-
щено внимание в работе Крейна и Рубинштейна и сотр. [15]. Гетеро-
циклические азотистые основания нефти полностью представлены
ароматическими системами. По-видимому, их донорно-акцепторное
взаимодействие с сульфидами нефти, представленными преимуществен-
но тиацикланами, осуществляется легче, чем взаимодействие сульфидов
с ароматическими углеводородами. Существованием ассоциатов, ве-
роятно, объясняется приуроченность гетероатомных соединений к смо-
листым компонентам нефти. С увеличением молекулярного веса нефтя-
ных фракций и усложнением структур входящих в них соединений
вероятность образования таких ассоциатов увеличивается. Можно пред-
положить, что в широкой фракции не менее 40% азотистых оснований
находится в виде подобных комплексов. В сырых нефтях в виде круп-
ных ассоциатов, по-видимому, находится не менее 50% азотистых осно-
ваний. Размеры ассоциированных частиц настолько велики, что они не
сорбируются катионитами. Неполная сорбция оснований из газойля и
легкого масла, отмеченная в работах [6, 7], вероятно, также связана
с существованием ассоциатов. В присутствии уксусного ангидрида—
полярного растворителя с кислыми свойствами — происходит разруше-
ние ассоциатов (вероятно частичное), азотистые основания оказываются
свободными, либо связанными в частицы меньшего размера, которые
могут сорбироваться на катионите.
Для проверки этого предположения была проведена сорбция ос-
нований из ново-дмитровской нефти в присутствии другого полярного
растворителя-—диметилформамида. В противоположность уксусному
ангидриду диметилформамид является растворителем со слабовыра-
женными основными свойствами. В присутствии диметилформамида
(15 объемн.%) сорбция оснований также прошла количественно, но
значительно медленнее (см. рис. 3, кривая 4).
Таким образом, из углеводородной среды на катионите К.У-23Х20
сорбировалось около 50% оснований из сырых нефтей и 60%—из ши-
рокой фракции. Количественная сорбция оснований достигалась только
128
после прибавления в раствор полярного растворителя — уксусного ан-
гидрида или диметилформамида.
Не исключено, что близость результатов, получаемых с добавками
разной полярности, частично обусловлена амфотерным характером аро-
матических систем, детально рассмотренных в работах Шатенштей-
на [16].
При применении уксусного ангидрида не следует забывать о возмож-
ности ацилирования гидроксильных и амино-групп в исследуемых сис-
темах. Однако последнее Обстоятельство на данном этапе работы мож-
но не принимать во внимание, так как содержание первичных и вторич-
ных аминов в нефтях ничтожно [17].
Ацетилирование кислородных соединений, попадающих в концен-
трат, также не создает осложнений при исследовании азотистых соеди-
нений нефти.
III. ДЕСОРБЦИЯ НЕФТЯНЫХ ОСНОВАНИЙ
При контакте катионита с нефтью имерт место адсорбция и хемо-
сорбция. В связи с этим десорбцию проводили в две стадии. Физически
связанный сорбат, а также часть нефтепродукта, механически удержи-
ваемого между зернами сорбента, удаляли промыванием катионита по-
лярным растворителем (спирт). Хемосорбированную часть десорбиро-
ровали спиртовым раствором щелочи или аммиака. В дальнейшей ра-
боте аммиаку, по чисто техническим причинам, было отдано предпочте-
ние.
Особое внимание пришлось уделить вымыванию оснований после
проведения химической десорбции. Выделенные из нефтей концентра-
ты представляют собой смолистые вещества с ограниченной раствори-
мостью не только в воде, но и в спирте. Поэтому после десорбции хемо-
сорбированной части катионит приходится многократно и тщательно
Таблица 6
Баланс по десорбции оснований нефти и характеристика полученных концентратов
раст- .о, N, мг Найдено ^ОСН’ Содержание NOch> % S3 • СО си О - о д ГЛ
Нефтепродукт (раствор в бензоле) 5? «о О к я аа 2 О Ьй 2 о й ф £ 2 о м S о СЛ ф ф <у sc S Й S = о р.’О,
cu s ф ф S < Си иэ Си S3 ю Си Е w S3 о S £* •&Q в- я S3 ю 2 Си к о I'g. •&3 S'© S о. S3 о № § CU ф . © ф Ф о4- Ч S’ S3 Z* 'g.S О Е- S °
о ою U и о О ф и м ф 03 < оз ф со м Л) USh eft?
Ново-дмитровская нефть . . . Нет 45,2 2,4 41,7 0,19 3,20 0,40 97,4
То же 5* 39,2 Нет 35,4 Нет 1,90 1,06 90,3
» » 5** 69,1 » 66,3 » 2,65 1,28 95,9
» » 39,4 » 40,2 » 2,82 1,72 102,1
Ново-дмитровская нефть в бен- 49,1 » 49,4 » 2,60 1,27 100,1
зине
Широкая фракция южно-уз- бекской нефти 2,5** 126,9 » 114,8 » 3,81 2,65 32,0
Сборная туймазинская нефть 10 ** 76,0 —• 66,3 — 1,91 1,05 92,5
* Уксусный ангидрид и новая порция .катионита добавлены после сорбции оснований из бен-
зольного раствора и удаления катионита.
** Уксусный ангидрид добавлен сразу.
•** Добавлен диметилформамид вместо уксусного ангидрида.
9 Методы анализа
129
промывать бензолом или серным эфиром, а в некоторых случаях — аце-
тоном, иначе большая часть оснований остается на катионите. Баланс
опытов по выделению азотистых оснований из нефтей в виде концентра-
тов, а также краткая характеристика полученных концентратов приве-
дены в табл. 6.
Следует отметить, что при проведении сорбции в статических усло-
виях физический десорбат практически не содержал оснований.
Содержание азота в полученных концентратах составляет в среднем
2,6—3,2%. В концентрате, выделенном из широкой фракции, содержа-
ние азота приближается к 4%. Концентраты, полученные в присутствии
полярного растворителя, характеризуются довольно высоким содержа-
нием серы. Все концентраты представляют собой черные или темно-ко-
ричневые смолистые вещества с различной степенью подвижности.
IV. РЕКОМЕНДУЕМАЯ МЕТОДИКА
На основании изложенного выше мы рекомендуем следующую мето-
дику выделения нефтяных оснований в виде концентратов (см. схему).
СХЕМА ВЫДЕЛЕНИЯ АЗОТИСТЫХ ОСНОВАНИЙ НЕФТИ
130
А. Сорбция нефтяных оснований
Для упрощения дальнейшего исследования концентратов сорбцию
оснований проводят в две стадии: сначала из бензольного раствора, за-
тем из раствора с добавкой уксусного ангидрида.
Аппаратура
Сорбцию и десорбцию оснований проводят в трехгорлой колбе,
снабженной мешалкой с затвором и обратным холодильником. Третье
горло колбы служит для удаления растворов и пропускания аммиака.
Растворы из колбы отсасывают с помощью пористого фильтра, пред-
ставляющего собой трубку, доходящую до дна колбы.
Ход выделения оснований
Навеску нефти или нефтепродукта, выкипающего выше 400° С, раз-
бавляют бензолом или бензином в отношении 1 :4 по объему *, добавля-
ют катионит из расчета 100 г на 1 кг нефти и раствор энергично переме-
шивают. Через 7—10 час. мешалку останавливают, раствору дают хорошо
осесть и отбирают 25—50 мл раствора для определения N0CH по ранее
описанной методике [18]. Перемешивание продолжают еще 3 часа, за-
тем анализ повторяют. Если уменьшение N0CH не превышает 0,001 —
0,002%, раствор пересасывают в другую такую же колбу. В колбу до-
бавляют свежий катионит из расчета 150 г на 1 кг нефти и приливают
уксусный ангидрид в таком количестве, чтобы концентрация его в раст-
воре составляла 5—10%.
Как и в первом случае, сорбцию ведут при перемешивании. Пробы
на анализ отбирают через 10—14 час. По окончании сорбции раствор
из колбы отсасывают.
В том случае, если концентрация N0CH в растворе, достигнув неко-
TQporo предела, не уменьшается даже при длительном перемешивании,
необходимо сменить катионит и увеличить концентрацию уксусного
ангидрида в растворе.
Б. Десорбция нефтяных оснований
Десорбцию с первой и второй порций катионита проводят раздель-
но. Для удаления остатков нефти и физически связанного сорбата кати-
онит 2—3 раза при перемешивании и нагревании промывают бензолом,
затем спиртом до тех пор, пока отсасываемый растворитель останется
бесцветным или слабоокрашенным.
К отмытому катиониту добавляют этанол (около 500 мл на 100 г
катионита); колбу помещают в ледяную баню и в раствор пропускают
сухой газообразный аммиак до насыщения раствора. Раствор оставляют
на 10—12 час., сначала в ледяной бане, затем при комнатной темпера-
туре. Спиртовый раствор части освободившихся оснований отсасывают.
Остальную часть оснований тщательно извлекают из катионита, при
перемешивании, несколькими порциями кипящего бензола и затем сер-
ным эфиром до получения бесцветных растворов. Спиртовый, бензоль-
ный и эфирный растворы объединяют, растворитель отгоняют. Концен-
траты оснований представляют собой темные смолистые массы.
Выделение оснований из ново-дмитровской нефти
(Noch — 0,029%)
Пример
В колбу загружено 375 мл (316 г) нефти, 1625 мл бензола и 35 г катионита. Пос-
ле перемешивания в течение 10 час. Noch в нефти снизился до 0,013%. Анализ, прове-
денный еще через 4 часа, дал то же значение Noch.
* Из фракций, выкипающих до 400° С, сорбцию оснований ведут без растворителя.
9* 131
Раствор нефти перенесли в другую колбу, добавили в него 1 00 лсл уксусного
ангидрида и 45 г катионита. Перемешивали 9 час. Анализ показал, что основания сор-
бированы катионитом полностью. Десорбция первой (30 г) и второй (45 г) порций
катионита была проведена раздельно.
К 30 г катионита прилили 150 мл этанола, колбу поместили в ледяную баню и при
перемешивании в нее пропускали газообразный аммиак до насыщения (6 час.). Раст-
вор оставили на ночь. Спиртовый раствор отсосали, а в колбу прилили 100—150 мл
бензола. Экстракцию вели при нагревании, перемешивали в течение 15—20 мин. Эту опе-
рацию повторили 3—4 раза, пока бензол не остался бесцветным. Затем катионит также
промыли серным эфиром. В спиртовый раствор переходит лишь незначительное ко-
личество десорбата, главная масса его вымывается бензолом. Так как существенной
разницы в содержании Noch н S в части концентрата, экстрагируемого спиртом и бен-
золом, не было, отгонку растворителей проводят последовательно: сначала отгоняют
спирт, к остатку в колбе добавляют бензольный экстракт и отгоняют бензол и, нако-
нец, добавляют эфириый экстракт и отгоняют эфир. Получено 1,5 г концентрата, со-
держащего Noch 3,20%.
Десорбция оснований со второй порции катионита была проведена по той же ме-
тодике; получено дополнительно 2,0 г концентрата с содержанием Noch 1,88%.
ВЫВОДЫ
Предложен метод получения концентратов азотистых оснований из
сырых нефтей и нефтепродуктов с помощью крупнопористых сульфо-
кислотных катионитов. Показано, что исчерпывающая сорбция оснований
нефти на катионитах проходит только в присутствии полярных раство-
рителей (уксусный ангидрид, диметилформамид). Впервые получены и
охарактеризованы полные концентраты оснований из сырых нефтей.
Институт нефтехимического синтеза им. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. D. М. Jewell, G. К- Hartung. Chem. and Eng. Data, 9, 297 (1964).
2. M. А. Абдурахманов, H. Д. Рябова. Сб. «Химия сера- и азоторганических соедине-
ний, содержащихся в нефтях и нефтепродуктах», т. 3. Уфа, 1960, стр. 207.
3. F. Р. Richter, Р. D. Caesar, S. L. Meisel, R. D. Offenauer. Ind. Eng. Chem., 44,
2601 (1952).
4. В. В. Гецеу. Аз. нефт. хоз., № 8, 32 (1956).
5. Н. L. Lochte, Е. R. Littmann. The petroleum acides and bases. London, 1955.
6. W. A. Monday, A. Eaves. Fifth world Petroleum Congress, section V, Paper 9, 1959,
p. 103.
7. L. R. Snyder, В. E. Buell. Analyt. Chem., 34, 689 (1962).
8. Л. Ф. Яхонтова, E. M. Савицкая, Б. П. Брунс. ДАН СССР, 111, 388 (1956).
9. Г. В. Самсонов, С. Е. Бреслер. Коллоидн. ж., 18, 88 (1956).
10. Л. Ф. Яхонтова, Е. М. Савицкая, Б. П. Брунс. ДАН СССР, 115, 358 (1957).
11. К. М. Салдадзе, А. Б. Пашков, В. С. Титов. Ионообменные высокомолекулярные
соединения. М., Госхимиздат, 1960, стр. 87.
12. И. И. Безингер, М. А. Абдурахманов, Г. Д. Гальперн. Нефтехимии, 1, 149 (1961).
13. И. Н. Безингер, М. А. Абдурахманов, Г. Д. Гальперн. Нефтехимия, 1, 583 (1961).
14. Г. В. Самсонов. Коллоидн. ж., 18, 592 (1956).
15. С. Э. Крейн, И. А. ' Рубинштейн, Е. А. Попова. IX научная сессия по химии
сераорганических соединений нефтей и нефтепродуктов (тезисы докл,). Уфа, 1965,
стр. 59.
16. А. И. Шатенштейн. Изотопный обмен и замещение водорода в органических соеди-
нениях. М., Изд-во АН СССР, 1960.
17. Н. И. Безингер, Г. Д. Гальперн, Т. И. Савостьянова. Сб. «Химия, технология и при-
менение производных пиридина н хинолина». Рига, 1960, стр. 55.
18. П. П. Безингер, Г. Д. Гальперн. Сб. «Методы анализа органических соединений
нефти, их смесей и производных», сб. 1. М., Изд-во АН СССР, 1960, стр. 147.
1
СРАВНИТЕЛЬНОЕ ИЗУЧЕНИЕ СОСТАВА СЛОЖНЫХ СМЕСЕЙ
ОРГАНИЧЕСКИХ ВЕЩЕСТВ КОМБИНИРОВАННЫМ
СПЕКТРАЛЬНЫМ МИКРОМЕТОДОМ АНАЛИЗА
Б. А. Смирнов
При возрастающем количестве методов исследования, дающих воз-
можность все более детально проникнуть в структуру и свойства орга-
нических соединений, особую роль приобретают микрометоды, и в пер-
вую очередь те из них, в которых исследуемое вещество не уничтожается
и может быть использовано для последующего изучения. К их числу
относится спектральный метод, позволяющий получить сведения о при-
сутствии в веществе различных структурных групп*: —(СН2)П>5—
(СН3)2СН—R, а-, цис- и транс-непредельных, ароматических, С = О,
О
—ОН, — с< ; SH и т. д. Полученные таким образом сведения
ЧОН
дают представление о наличии некоторых структурных фрагментов как
в индивидуальных соединениях, так и в разнообразных смесях. Необхо-
димо всегда помнить, что не все структуры могут быть определены
спектральным методом. Так, например, в сложных органических смесях
практически невозможно различить парафиновые цепи—(СН2)П—-по
числу звеньев п (можно лишь указать наличие цепей с и п^4);
нельзя отличить пятичленные от шестичленных нафтеновых колец; не
определяется тетразамещенная двойная связь и т. д. Комбинация спек-
трального метода с микрохроматографическим, описание которого дано
в настоящем сборнике (стр. 28), применительно к нефтям и битумам
позволяет определить количественный состав не только по основным
группам соединений — парафинов, нафтено-парафинов, непредельных,
ароматических и т. д., но и по подгруппам, например, нормальных пара-
финов, изопарафинов и алкилбензолов определенных типов замещения,
нафталиновых и др. Такой комбинированный метод анализа приобретает
особую ценность в ряде задач, где необходимо сравнительное изучение
состава большого набора микроколичеств фракций, например фракций
масел различных нефтей (разных месторождений), разных горизонтов
одного месторождения, ископаемых битумов и т. д. Принципиальные
основы предлагаемого метода имеют большую общность и могут найти
применение при изучении смесей самого различного состава. Однако
необходимо подчеркнуть; что невозможно дать единый стандартный
метод для решения любых аналитических задач данного типа. Метод
решения каждой конкретной задачи определяется рядом факторов:
* Под термином «структурная группа» будем понимать как собственно структур-
ные группы, так и функциональные группы, как это принято в спектроскопии.
133
Зак. 2300
корректностью постановки задачи, предполагаемым составом смеси
(углеводороды, сернистые, азотистые соединения и т. д.) и ее физико-
химическими свойствами (вязкость, сорбируемость, реакционная способ-
ность и т. д.), экспериментальными особенностями метода, такими,
например, как правильный выбор аналитически выгодных спектральных
областей и рядом других.
Для наиболее полного извлечения сведений из результатов анализа
каждая задача должна рассматриваться как самостоятельное научное
исследование. В этом смысле предлагаемый метод является логической
схемой исследования для решения указанного круга задач.
Дальнейшее изложение будет идти с учетом последующего примене-
ния метода только к сложным углеводородным смесям, где индивиду-
альные спектральные характеристики отдельных компонентов полностью
взаимно перекрыты и образуют «фон» спектра, а полосы поглощения
соответствуют присутствию в смеси определенных структурных групп
независимо от характера и последовательности соединения этих групп
в молекулах. В этом смысле указанная схема анализа может быть
разработана для смесей любого состава.
Ниже дано общее описание методики измерений комбинированным
спектральным микрометодом и детально разобран способ ее применения
на примере решения конкретной битуминологической задачи.
1. МЕТОД СПЕКТРАЛЬНОГО СТРУКТУРНО-ГРУППОВОГО АНАЛИЗА
В УЛЬТРАФИОЛЕТОВОЙ И ИНФРАКРАСНОЙ ОБЛАСТЯХ СПЕКТРА
Основными спектральными областями, применяемыми для аналити-
ческих целей, являются УФ- и ИК-области спектра. Ввиду наличия
у каждой из них ряда специфических особенностей дадим их краткое
описание. При этом будет предполагаться, что читатель знаком с мето-
дикой получения спектров поглощения в УФ- и ИК-областях с элемен-
тарными приемами обработки и расшифровки спектрограмм с помощью
таблиц частот и атласов спектров [1—13].
Для удобства напомним выражение закона Ламберта — Бера, иначе
называемого законом Бугера, и связанных с ним обозначений, которыми
мы будем пользоваться в дальнейшем:
Z)* /
/x,v=/o,vc \ In —- = 1п-т- = D* = e*d%,
X, v
где /0,v, Ix,v — соответственно интенсивности монохроматического излу-
чения частоты м и того же излучения, прошедшего через вещество;
Dv* — оптическая плотность; d— толщина образца; % — концентрация
(в моль/л); ev * — коэффициент поглощения вещества при частоте v;
Av — -100% пропускание, выражаемое в процентах.
'о, V
Обычно вместо натурального логарифма In ~L берут десятичный
1X, 'J
log • В этом случае для оптической плотности Dv название сохра-
*Х, V
няется, а константа поглощения ev называется коэффициентом экстинк-
ции. Обе последние величины отличаются от Dv * и ev * постоянным мно-
жителем а — loge = 0,4343:
Dv — a,D*, ev = ае*.
Дадим также объяснение термину «структурно-групповой состав»
(СГС). Под СГС будем понимать содержание каждой СГ в процентах
134
по отношению к общему количеству определяемых данным методом С Г.
Например, СГС гексилбензола содержит 50% гексильных групп и 50%
ароматических колец.
Ультрафиолетовая область
Под ультрафиолетовой (УФ) спектральной областью будем понимать
участок спектра 220—500 нм, т. е. собственно ультрафиолетовую область
спектра 220—400 нм и область видимого света 400—500 нм. В УФ-обла-
сти поглощают структурные, группы с сопряженными С = С-связями.
К ним относятся полиеновые структуры типа каротиноидов и в особен-
ности ароматические структуры [6—13]. Все остальные углеводороды
прозрачны в УФ-области, что делает исследования в этой области
чрезвычайно удобными для определения ароматических структур ввиду
отсутствия маскирующего действия других структурных групп. С другой
стороны, это является и недостатком метода УФ-спектроскопии, ограни-
чивающим его применение единственным классом углеводородов, если
не считать полиеновых, редко встречающихся в горючих ископаемых.
В случае присутствия наряду с ароматическими значительных количеств
полиеновых структур, с чем мы встречаемся, например, в продуктах
термической переработки нефти, метод УФ-спектроскопии часто требует
предварительного разделения смеси на ароматическую и непредельную
фракции.
Наиболее общей характеристикой смеси ароматических углеводоро-
дов является соотношение моноциклических, бициклических (нафтали-
новых) и полициклических конденсированных структур *.
Анализ спектральных данных показывает, что для смесей моноцикли-
ческих соединений максимум поглощения будет лежать в области
255—275 Нм с е~400. Для смеси алкилбензолов эта полоса может иметь
тонкую структуру — не более трех слабых пиков, из которых наиболее
интенсивным может быть либо центральный, либо длинноволновый.
В сложных смесях моноциклических соединений тонкой структуры этой
полосы обычно не наблюдается.
Для бициклических (нафталиновых) соединений максимум поглоще-
ния приходится на область 275—290 нм и 8=5000 н-8000. Так же как и
для фенильных производных, эта полоса поглощения в зависимости от
состава может быть слегка структурирована — порядка 3—4 слабых
полос. Характерной особенностью поглощения нафталиновых являются
два четких пика в области 310—330 нм с s~400-:-600, из которых наи-
большей интенсивностью обладает длинноволновый. По этой области
в основном происходит определение нафталиновых структур.
По характеру поглощения полициклические конденсированные струк-
туры могут быть разбиты на линейноконденсированные — типа антра-
цена, и нелинейноконденсированные — типа фенантрена. Для первых
характерным является интенсивное поглощение в области 240—250 нм
с 8—1 • 10б —ь- 2 • 106, и в особенности четко структурированная широкая
полоса поглощения с 3—5 максимумами в области 310—380 нм при
среднем для полосы значении 8~3000. Для вторых наиболее интенсивное
поглощение приходится на область 250—260 нм при 8 = 40 000-4-60 000;
аналитической областью их определения является интервал 280—310 нм
* Под указанными структурами будем подразумевать таковые вне зависимости от
характера (типа) заместителей, а также степени конденсации с неароматическими
(нафтеновыми) кольцами, так как эти соединения имеют сходные спектры поглоще-
ния в УФ-области. Поэтому, например, к моноциклическим структурам будем отно-
сить, помимо алкилбензолов, такие соединения, как инданы, тетралины и др., в одну
группу с нафталиновыми попадут инденовые, а также антраценовые и фенантреновые
с одним гидрированным кольцом (крайним) и т. д.
135
Рис. 1. Схематические кривые поглощения основных типов ароматических
структур в УФ-области
/ — моноциклические; 2 — бициклические (нафталиновые); 3 —полициклические нели-
иейноконденсироваиные; 4 — полициклические лииейноконденсированные; 5 — типич-
ная кривая полиеновых CHs—(CH—CH)e—CHS; 6— УФ-спектр углеводородной арома-
тической фракции
при е= 10 000-4-20 000 с 2—3 пиками поглощения, из которых в сложных
смесях обычно сохраняется лишь наиболее интенсивный — длинноволно-
вый у 300 нм, и областью 320—360 нм с тремя отчетливыми пиками и
е~500, интенсивность которых возрастает с увеличением длины волны.
К сожалению, эта последняя область частично перекрывается с обла-
стью поглощения линейноконденсированных структур и может быть
использована полностью только в их отсутствии.
Рассмотренные выше кривые поглощения основных типов аромати-
ческих структур схематически представлены на рис. 1, где для удобства
по оси ординат вместо значений е отложены значения логарифма этой
величины. Там же нанесена типичная кривая поглощения полиеновых
структур. Увеличение числа сопряженных С = С-связей сдвигает всю
полосу в длинноволновую область. Отсюда нетрудно получить общий
вид кривых поглощения различных смесей ароматических структур.
Так, для наиболее распространенного типа природных ароматических
смесей (нефти, битумы), в которых содержание моноциклов>бициклов>
>полициклов, общий контур кривой будет иметь вид, показанный на
рис. 1 типичной линией 6. Вид этой кривой свидетельствует об одном из
существеннейших правил в УФ-спектроскопии: из-за большой разницы
значений е характеристических областей поглощения анализируемых
структур для получения наиболее полных данных необходимо записать
УФ-спектры исследуемой фракции при нескольких концентрациях, начи-
ная с максимально возможной. При максимальной концентрации будут
«пробиваться» аналитические полосы, лежащие на слабопоглощающем
длинноволновом крыле спектра; по мере разведения мы сможем после-
довательно наблюдать появление аналитических полос во все более
сильнопоглощающих участках спектральной кривой. Как видно из
136
рис. 1, этот метод не позволяет определить моноциклические структуры
в присутствии полициклических структур из-за отсутствия характеристи-
ческого поглощения моноциклических структур в других областях, кроме
области ~270 нм, где все остальные ароматические структуры обладают
значительно большим поглощением.
Отметим, что наличие в молекулах гетероатомов сильно увеличивает
поглощение в УФ-спектре, не меняя существенно характеоа спектра.
Поэтому правильное определение группового состава углеводородных
смесей возможно только для образцов, в которых примесь гетероатомных
соединений не превышает нескольких процентов.
Благодаря высоким значениям е в УФ-спектре этот метод требует
на анализ очень небольших навесок. Так, для моноциклических структур,
обладающих наименьшим поглощением в УФ-спектре при концентрации
1 мг!мл и Л4-~300, получим кривую поглощения с значением оптической
плотности в максимуме ~ 1,0, что соответствует пропусканию ~10%.
Очевидно, что для определения остальных ароматических структур тре-
буются еще меньшие навески. Так как все нафтено-парафиновые угле-
водороды прозрачны в УФ-свете, то, растворив навеску в таком углево-
дороде и выпарив его после снятия спектра, можно сохранить образец
для дальнейших исследований. Обычно в качестве растворителя
в УФ-спектроскопии применяются спектрально чистые изооктан и этанол.
Инфракрасная область
Под инфракрасной (ИК) областью обычно понимается длинноволно-
вый участок спектра с частотами v, меньшими 10 000 см~\ или длинами
волн X, большими 1 мкн. В отличие от поглощения УФ-области в ИК-об-
ласти поглощают все органические соединения, а также все структурные
группы, из которых эти соединения составлены. С точки зрения приме-
нимости для аналитических целей наиболее разработанной является
область частот от 5000 до 600 с.и-1, или в длинах волн от 2 до 15 мкн.
Полосы поглощения, расположенные в этой области, соответствуют
колебаниям подавляющего большинства структурных групп. По харак-
теру поглощения в этой области могут быть условно выделены следую-
щие интервалы: область 5000—2500 см~\ характеризующая валентные
колебания водородного атома С—Н, О—Н, N—Н и т. д. в различных
структурных группах, и область v<2000 см~\ характеризующая коле-
бания самых разнообразных структурных групп — различного типа
замещенных ароматических структур, олефиновых групп, парафинов
и др. Полосы поглощения в этой последней области обусловлены как
деформационными колебаниями Н-атомов, так и более сложными коле-
баниями, происходящими с изменением межатомных углов и расстояний
всех атомов, входящих в данную структурную группу. Каждая структур-
ная группа характеризуется своим набором полос, число которых, по-
ложение по спектру и интенсивности в большей или меньшей степени
зависят от строения и состава остальной части молекулы. В связи с этим
каждая молекула обладает своим, присущим только ей спектром. Не
существует двух разных веществ (молекул) с одинаковыми спектрами,
так же как не могут два разных спектра принадлежать одному и тому
же веществу (молекуле) [1—5, 9, 10].
Полосы поглощения, характеризующие различные структурные груп-
пы, можно подразделить на три типа. К первому будем относить полосы,
практически сохраняющие интенсивность и общий вид (контур) вне за-
висимости от строения и состава остальной части молекулы; при этом
положение полосы может меняться в небольших пределах, порядка
полуширины полосы. Примером такого типа полос может служить
полоса 1382—1375 см~' для одиночной СН3-группы, полоса 721 —
137
719,5 см-1 для полиметиленовой цепи— (СН2)П>5, полоса 975—965 см-1
для транс-олефиновой конфигурации и т. д. Такие полосы называются
характеристическими. В большинстве своем это интенсивные полосы
и поэтому являются основными в спектральном анализе.
Для полос второго типа как положение, так и интенсивности могут
значительно меняться в зависимости от строения молекулы. Интервал
изменения положения этих полос значительно превосходит их полуши-
рину. По интенсивности эти полосы обычно бывают несколько слабее
полос первого типа, однако в большинстве своем также относятся к
категории сильных. В качестве примера можно привести полосу валент-
ного колебания свободной О—Н-группы с интервалом частот 3700—
3500 см-1, полосу 730—675 см-1 для цпс-олефиновой конфигурации,
полосу 745—705 см-1 для 1,2,3-триалкилбензола и т. п.
Наконец, к третьему типу относятся полосы, положение, интенсив-
ность, а также количество которых могут сильно меняться в зависимо-
сти от строения молекулы. Обычно это полосы средней и малой интен-
сивности. Примером может служить система полос поглощения нафте-
новых структур в области 1000—850 см~г.
Следует различать два вида задач структурно-группового анализа.
К первому виду относятся как задачи определения строения индивиду-
альных соединений, спектр поглощения которых неизвестен (например
впервые синтезированных), так и идентификации соединений, спектры
которых имеются в литературе. Первая из этих задач касается строго
индивидуальных соединений; вторая задача носит название «определения
индивидуального состава» и применяется для смесей ограниченного
компонентного состава (не более 10 соединений). Ко второму виду от-
носятся задачи определения группового состава сложных смесей. Эта
последняя и будет интересовать нас в дальнейшем.
Наиболее просто определяются структурные группы, которые имеют
среди совокупности характеризующих их полос, хотя бы одну характе-
ристическую полосу первого типа. Положение таких полос, а также
примерная интенсивность на одну группу устанавливается на основании
изучения спектров индивидуальных соединений. Поэтому по таким по-
лосам может быть надежно определено не только присутствие данной
структурной группы, но в некоторых случаях и относительное количест-
венное содержание ее в смеси. Полосы поглощения второго типа менее
надежны для анализа. Контур таких полос в сложных смесях не соот-
ветствует их контуру в индивидуальных соединениях, так как является
результатом суммирования этих полос, смещенных друг относительно
друга в зависимости от состава исследуемой смеси. Поэтому их иденти-
фикация в смесях на основании изучения только спектров индивидуаль-
ных соединений может привести к ошибкам, если в этой же спектраль-
ной области присутствуют полосы, относящиеся к другим структурам.
Однако обычно даже в сложных природных смесях компонентный
состав не бывает полностью произвольным, а содержит какие-то пре-
имущественные структуры. В этом' случае интервалы частот для таких
полос оказываются значительно суженными, что приближает их к пер-
вому типу, увеличивая тем самым степень надежности определения
соответствующих им структур.
По полосам поглощения третьего типа анализ в общем случае не-
возможен. В сложных смесях такие полосы взаимно усредняются, давая
равномерный фон поглощения *. В тех случаях, когда полосы этого
* Отсюда следует на первый взгляд парадоксальный вывод, о том, что спектры
сложных смесей внешне проще спектра любого из входящих в него соединений, так как
в спектре индивидуальных веществ есть полосы всех трех типов, а в спектре смеси
только первый и второй типы. Аналогичная картина наблюдается и в случае полиме-
ров, спектр которых внешне проще, чем у мономеров.
138
типа группируются в достаточ-
но узком спектральном интер-
вале, в спектре смеси появля-
ется область поглощения, со
-своими полосами, по которым
может быть проведено качест-
венное определение данной
структурной группы. В этом
случае контур указанной обла-
сти поглощения будет также
специфичен. Однако практиче-
ски он не может быть ни выве-
ден, ни проверен по спектрам
индивудальных соединений.
Его отнесение может быть сде-
лано только на основании изу-
чения спектров сложных сме-
сей, содержащих данную i
структурную группу, а также |
.дополнительных физико-хими- ;
ческих данных. Примером та- i
кого рода может служить об- i
ласть поглощения нафтеновых
структур в смесях (рис. 2).
Для сравнения там же нанесен
спектр изопропилциклопентана
и 1,3-диметилпенталана. Необ-
ходимо подчеркнуть, что ука-
занные полосы поглощения
Рис. 2. Кривая поглощения нафтено-
парафиновых структур в смесях
в области 850—1000 см-1
1 — изопропилциклопентан (модуль 10); 2 —
1,3-днметилпенталаи (модуль 20); 3 — харак-
терное поглощение нафтеновых структур
смеси, несмотря на свою специфичность, не имеют физического смысла,
аналогичного полосам поглощения в спектрах индивидуальных веществ,
а являются лишь своеобразной огибающей — «статистической суммой»
полос в данной области. Аналогичный смысл имеет УФ-максимум по-
глощения смеси в области 240—250 нм (рис. 1).
Как выше отмечалось, в настоящее время существуют подробные
таблицы отнесения групповых частот, определение которых в принципе
возможно по ИК-спектрам. Однако в действительности такая возмож-
ность сильно зависит от группового состава смеси. Опыт работы пока-
зывает, что в сложных углеводородных смесях наиболее надежно могут
быть определены структурные группы и их совокупности, представлен-
ные в табл. 1. Под структурными группами (СГ), обозначенными в таб-
лице как «нафтеновые циклы», следует понимать нафтено-парафиновые
структуры, так как нафтеновые кольца определяются вместе с алкиль-
ными заместителями с длиной цепи не выше С4. Данные этой таблицы
по ароматическим структурам более правильно относить не к типу за-
мещения (число и расположение заместителей в бензольном кольце) ,'
.а к числу и расположению незамещенных Н-атомов, так как поглощение
в этой спектральной области связано с неплоскими колебаниями атомов
водорода в цикле. При таком понимании в указанных интервалах
частот могут поглощать соответствующие гетероциклические соединения.
Например, 2,3- и 2,4-диметилхинолины. содержащие группу из четырех
незамещенных Н-атомов в углеводородном цикле и по одному в гетеро-
цикле, имеют интенсивные полосы в области 755 и 900—850 см~1, соот-
ветствующие орто-замещению для углеводородного кольца и пента-
замещению для гетероцикла [3]. Это относится также к полиядерным
соединениям, для которых показано [14], что присутствие в них одиноч-
ных, двойных, тройных и четверных С—Н-групп вызывает появление
139
в спектрах полос поглощения соответственно в областях 900—860, 860—
800, 810—750 и 770—735 см-1 независимо от места и взаимного располо-
жения групп друг относительно друга. Указанные спектральные интер-
валы не противоречат данным табл. 1.
Данные табл. 1 получены на жидких образцах *. Если среди изучен-
ных образцов имеются твердые фракции, необходимо учесть возможные
изменения в спектре, обусловленные изменением агрегатного состояния.
Так, например, для твердых нафтено-парафиновых углеводородов нефти
с длинными парафиновыми цепями установлена температурная зависи-
мость структуры и интенсивности полосы вблизи 720 см-1, отражающая
фазовые переходы жидкость->-твердое и твердое->-твердое тело [16].
Так как в ИК-области спектра поглощают все вещества, то примене-
ние растворителей становится здесь нежелательным из-за возможности
перекрытия полос растворителя и исследуемого вещества. В ИК-спектре
считаются интенсивными полосы поглощения со значениями 8^10**.
В редких случаях е превосходит 100 — это в основном полосы поглоще-
ния гетероатомных структур, таких, например, как карбонил. Для угле-
водородов максимальное значение е не превосходит 150, что выгодно-
отличает ИК-спектр от УФ-спектра, где разброс значений е достигает
нескольких порядков (~1000 раз). Практически каждая структурная
группа имеет в ИК-спектре по крайней мере одну полосу с е около 40.
Поэтому для анализа здесь достаточно получить спектр при одной,
толщине поглощающего слоя (кюветы) за каковую можно принять-
ЗО^с^бО мкн. Так как размер рабочей части светового пучка стан-
дартных приборов (ИКС-14, UR-10) в плоскости расположения кюветы
в рабочем канале не превосходит 2,0X0,5 см, то при толщине слоя
~40 мкн и плотности образца р-~0,8 г/см? необходима навеска ~3 мг.
При желании рабочий участок можно без ущерба для качества записи,
спектра довести до размера ~1,0X0,3 см. В этом случае навеска веще-
ства должна быть около 1 мг. Для работы с такими количествами нужно
иметь специальные приспособления, так как стандартные кюветы к ука-
занным приборам требуют значительно больших количеств вещества.
~0,5 г. Одним из возможных способов решения проблемы является
использование микроприставки, выпускаемой некоторыми зарубежными'
фирмами, в которых используется конденсор светового пучка с микро-
кюветой. При их использовании необходимо иметь в виду, что такой
конденсор, фокусируя пучок света на малую площадь, сильно ее разо-
гревает— до температур ~100°С. В особенности это касается приборов
с мощными источниками, как, например UR-10. Такое повышение темпе-
ратуры может вызвать различные изменения в образце — испарение
летучих компонентов, осмоление и др. В этом случае необходимо пред-
усмотреть надежную систему охлаждения образца.
Другим способом решения задачи является простое диафрагмиро-
вание рабочего пучка до требуемых размеров так, чтобы в прибор по-
падал только свет, предварительно прошедший через исследуемое ве-
щество. При этом потеря интенсивности будет в основном происходить
из-за диафрагмирования пучка по высоте, так как фактическая ширина
пучка света, попадающего в монохроматор прибора, определяется ши-
* Возможность использования этой таблицы была проверена нами ранее на при-
мере анализа моноциклоароматической фракции (выше 325° С) высокомолекулярной
части абуродесской нефти (Египет). Получено, что основу этой фракции составляет
сумма 1,4-+ 1,2,4-замещенных; значительно меньше 1-+1,3-+1,3,5-замещенных и не
обнаружено 1,2-+ 1,2,3-замещенных. В дальнейшем этот же продукт был исследован
методом окисления перманганатом калия; полученные данные полностью совпали со
спектральными [15].
** Иногда в литературе по ИК-спектроскопии встречаются значения е, рассчитан-
ные для концентраций (в жоль/см3). Соответствующие значения е будут в 100 раз. болы-
ше здесь указанных.
140
Таблица 1
Таблица отнесения спектральных интервалов поглощения к некоторым
углеводородным группам
3.
Структурная группа (тип замещения) Интервал частот, см~* £, моль!Л; ; на группу Поглощающая группа Примечание
Парафины
723,5—721,5 2,1 -сн2- Узкая, четкая полоса. В твердых продуктах расщепляется на две: 726 и 719 см"1
1 1 1 Ш СО (N Е е е и и и т । т 725,5—724 729,5—726 743—734 2,9 3,5 5 1 1 1 ci d с-1 EXE ООО 1 1 1 Полоса, обнаруживается только, если полоса для слабая. В против- ном случае наличие этих групп устанавливается качественно по уширению всей полосы и смещению максимума к большим частотам
СНз—R 1383—1377 18 -СНз Симметричное деформа- ционное колебание С — Н изолированной конечной СН»-группы
(СН3)2СН—R 1389—1381 1372—1368 40 40 (СН3)2СН- (СНзЬСН- Расщепление полосы сим- метричного деформаци- онного колебания С— Н в результате взаимодей- ствия соседних СНд-групп
(CH3)SCH-R 1171—1163 10 (СНз)2СН- Скелетное колебание
R-CH2-CH—сн2—r 1159—1151 7 —СН2-СН—СН2— То же
СНз СНз
Нафтены
Нафтеновые циклы 1000—850 •— — Качественное определение по области поглощения. Содержит три широких максимума, интенсивность которых падает с умень- шением частоты
Олефины
СНг=СН—R (а-олефины) 916—909 1004—991 120 45 Е Е О о II II сМ Е Е и о > Неплоское деформа- | ционное колебание | С — Н при двойной J связи Валентное С = С-колеба-
1645—1650* 35 СН2=СН—
R-C-R' II СЬ12 (винилиденовая структура) 892—887 1661—1639* 140 40 С=СН2 С=СН2 нне Неплоское деформацион- ное =С—Н-колебание Валентное С=С-колеба- ние
R
Хн=сн 974—964 100 СН=СН Неплоское деформацион- ное =С—Н-колебание
X'
{транс-) 1667 * 0 СН=СН Валентное С==С-ко пебание
Я R' 729—675 60 сн=сн Неплоское деформацион- ное =С— Н-колебание
\н=сн/
{цис-) 1661—1631* 10 сн=сн ВалентноеС=С колебание
141
Таблица 1 (окончание)
Структурная группа (тип замещения) Интервал частот, см~1 е, молъ/Л'См на группу Поглощающая группа Примечание
R—СН=С—R' СН3 833—808 40 —сн=с— СНз Неплоское деформацион- ное =С- Н-колебание
R—СН=С—R' СНз 926—908 10 —сн=с— 1 СНз То же
04 1 ел d-g II X о 1 04 1692—1667* 6 Араме —СН=С- СНз ипические Валентное С=С-колебанне
1,2,4- 882—872 2 СвН8 Поглощение связано с
1,3- 882—872 2 С6Н4 ч группой С—Н
1,3,5- 882—872 1 СбНз
1,3,5- 854—840 20 СвНз То же
1,4- 833—826 20 С6Н4 Поглощение связано с
1,2,4- 814—806 10 СсНз группой Ч „ С—н (Lh
1,2,3- 780—770 10 С6Нз Поглощение связано с
1,3- 780—770 10 сен4 группой V с—н 1 ,, с—н с—н
1,2,3- 748—742 5 QHs Поглощение связано е
1- 748—742 10 С6Н5 группой
1,2- 748—742 10 С6Н4 1 о— о." a I 1 : а а
1,3,5- 710—680 5 С6Нз Отнесение неизвестно
1- 710—640 20 С6н5
1,3- 710—680 10 С6Н4
* Частоты обычно дают общий пик в силу недостаточного разрешения NaCl-призмы в это&
области и могут служить структур. лишь для качественного определения суммарного присутствия олефиновых.
риной входной щели (а не входного зрачка), которая в указанных при-
борах не превосходит 1 мм.
Мы дадим здесь описание упрощенного варианта такой кюветы для
ИКС-14, пригодной для малолетучих и достаточно вязких образцов,,
которую легко можно сделать подручными средствами. От стандартной
кюветы для ИКС-14 используется корпус держателя (рис. 3, 1) в окош-
ко которого плотно вставляется диафрагма (рис. 3, 2). Установка щели
диафрагмы в строго вертикальном положении проверяется по ее изоб-
ражению в плоскости входной щели. Вместо стандартной крышки дер-
жателя кюветы изготовляется новая крышка 6 с щелевым окном шири-
ной 0.5 см и салазками 6 (см. рис. 3), в которые плотно входит
диафрагма типа «ласточкин хвост» для ограничения по высоте пучка
света. Соответствующее диафрагмирование пучка света в канале срав-
нения ИКС-14 проводится по области прозрачности образца.
142
Рис. 3. Микрокювета для малолетучих образцов
/ — корпус держателя кюветы; 2 — диафрагма; 3 — прокладки; 4, 5 — КВг-пластинки, 5 — крышка держателя; 7 — диафрагма типа
«ласточкин хвост»; 8—стеклянная лопаточка
Для всех углеводородов, а также для большинства гетероатомных
соединений таковой является область 2200—1800 см~1. Для указанных
соединений в этой области существует лишь фоновое поглощение около
5_7% (Л—95%). Компенсация света в канале сравнения до указан-
ной величины может быть достигнута металлическими сетками с ячей-
кой не менее 1 мм2 (во избежание явления дифракции, меняющего
спектральный состав света), стопой КВг-пластинок, каждая из которых
отражает примерно 8% падающего на нее света независимо от часто-
ты, а также верньером в рабочем канале. Нельзя диафрагмировать свет
в канале сравнения по высоте, это приводит к большим ошибкам в оп-
ределении величины пропускания А.
Собственно кювета состоит из двух КВг-пластинок 4, 5 (см. рис. 3)
и двух прокладок 3 размером примерно 3X0,5 см, определяющих толщи-
ну кюветы. В качестве одной из пластинок можно использовать КВг-
окошко из комплекта окон, прилагаемых к прибору. Другая КВг-пла-
стинка должна быть не более 1 см шириной и не менее 3 см длиной и
может быть изготовлена (выколота) из битых КВг-окошек того же при-
бора. Исследуемое вещество аккуратно наносят стеклянной лопаткой
8 (см. рис. 3) на КВг-пластинку так, чтобы получился слой прямоу-
гольной формы, например 1,5 X 0,3 см (— 1,5 мг) *; на края пластинки
кладут прокладки так, чтобы они не касались образца. В противном
случае часть образца неизбежно затянется капиллярными силами в
зазор между прокладками и окошками. Образец прикрывают другой
(узкой) КВг-пластинкой, слегка придавливают и помещают в кювето-
держатель так, чтобы образец находился строго в центре и по ширине
целиком покрывал щель диафрагмы, которая предварительно выбира-
ется исходя из этих же соображений. Затем надевают крышку и акку-
ратно слегка затягивают гайками. При этом надо следить за равномер-
ностью степени сжатия кюветы, ни в коем случае не допуская
пережимания какой-либо стороны по отношению к другим, во избежа-
ние образования клиновидного слоя и связанного с этим явления «сбе-
гания» образца в более тонкую часть. Это особенно существенно для
маловязких, текучих образцов, что собственно и ограничивает приме-
нение кюветы данной конструкции. Именно по этой причине верхняя
KBr-пластинка делается узкой, шириной примерно 1 см, что ограничи-
вает растекание образца на большую площадь. Максимальное количе-
ство образца, потребное при полном заполнении всего пространства
между прокладками —3X1 см, будет ~ 10 мг.
Практика многолетней работы показала, что не было случая, чтобы
не удавалось снять спектр образца в количестве 4—5 мг (смеси нафте-
но-парафинов С1з—С15), так как при таких количествах даже в случае
«сбегания» образца к одной из сторон (всегда к одной из «длинных»)
образуется достаточно широкая полоска ( — 3 мм и более), которая
легко выводится на щель диафрагмы. После снятия спектра образец
смывают подходящим растворителем, доводя до постоянного веса, пос-
ле чего образец может быть употреблен для дальнейшей работы.
Получаемый таким образом спектральный материал в общем случае
не позволяет произвести прямые оценки абсолютных количеств иссле-
дуемых структур, а годится лишь для качественной оценки относитель-
* Опыт работы на ИКС-14 показывает, что при работе с полосками вещества ме-
нее 1 см длиной илн 0,2 см шириной не удается достаточно корректно скомпенсиро-
вать пучок света в канале сравнения в области поглощения паров воды (2000—
1500 сдг1). На английском приборе фирмы Unicam SP-200, у которого входной зра-
чок — 1,5 X 0,5 см (у ИКС-14 входной зрачок — 2,5 X 0,5 см) работа с полосками ве-
щества — 1,0X0,2 см не вызывает затруднений. Кроме того, в приборе предусмотрена
компенсация линии 100%-ного пропускания до 10% исходного значения 10, что также
облегчает работу. Аналогичная компенсация имеется у прибора UR-20.
144
ного их содержания в данном образце и изменения этого содержания
от образца к образцу. Относительное содержание структурных групп
оценивается по относительным интенсивностям (за вычетом фона) соот-
ветствующих полос поглощения по отношению к полосе 1466 см~\ ин-
тенсивность которой принимается за 10. Эта полоса выбирается в ка-
честве эталонной по той причине, что поглощение здесь обусловлено
С—Н-колебаниями разнообразных углеводородных групп (—СНз—,
—СНз, ^-С—Н и т. д.), и интенсивность этой полосы при данной тол-
щине кюветы должна примерно сохраняться для всех углеводородных
образцов. Более корректно было бы брать значения относительных оп-
тических плотностей. Проверка показывает, что в пределах точности
метода оба способа дают одинаковые результаты.
Комбинированный метод
На основании проведенного здесь краткого обзора аналитические
возможности УФ- и ИК-методоВ в отдельности могут быть сведены к
следующему.
УФ-метод дает сведения о СГС ароматической части образца, игно-
рируя все остальное.
ИК-метод в принципе может дать сведения о содержании в образце
любых СТ; однако перекрытие полос поглощения различных СГ сильно
снижает его возможности,.
Очевидно, что совместное применение обоих методов улучшает ка-
чество анализа, в этом случае анализ СГС ароматической части пред-
варительно выполняется по УФ-спектрам. Знание состава ароматиче-
ской части позволяет в ИК-спектрах отделить полосы поглощения,
соответствующие ароматическим структурам, от полос поглощения дру-
гих СГ, способствуя тем самым их лучшему определению. К сожале-
нию, на практике такое отождествление полос поглощения ароматиче-
ских структур в ИК-спектрах по результатам анализа в УФ-области
спектра не всегда может быть выполнено в силу специфического проис-
хождения поглощения ароматических структур в ИК-области; если в
УФ-спектре характер поглощения обусловлен числом и взаимным рас-
положением ароматических колец, то в ИК-области поглощение опре-
деляется числом и расположением незамещенных Н-атомов ароматиче-
ских колец вне зависимости от степени их конденсации (см. табл. 1).
Значительно лучшие результаты могут быть получены, если иссле-
дуемую смесь подвергнуть препаративному хроматографическому раз-
делению на узкие фракции и их исследовать спектральными методами.
Каждая такая фракция представляет собой смесь веществ определен-
ного типа соединений, спектр которой содержит спектральные призна-
ки СГ только этого типа. Изучение узких фракций и сопоставление со
спектром исходной смеси позволяет уточнить отнесение полос погло-
щения, разобраться в системе -перекрывающихся полос и проконтроли-
ровать собственно метод разделения. Если исследуемая смесь имеется
в достаточном количестве и является единственным образцом, предва-
рительное изучение ее спектров не является обязательным. Основной
анализ ведется по спектрам узких фракций и по данным хроматограм-
мы. Спектры исходной смеси снимаются для контроля, «на всякий слу-
чай». Хуже обстоит дело, когда исследуемая смесь имеется в микроко-
личествах. В этом случае спектр исходной смеси является единствен-
ным способом определения ее состава.
Существует большой круг задач, частично отмеченных выше, в ко-
торых исследованию подвергается набор фракций, имеющихся в_малых
количествах, порядка нескольких миллиграммов. В этом случае может
быть предложен следующий способ их анализа:
1 0 Методы анализа
145
1. Для всех исходных фракций снимают УФ- и ИК-спектры, по ко-
торым проводят предварительное определение их СГС.
2. Образцы сходного СГС объединяют и затем делят на микрохро-
матографической колонке по показателю преломления на ряд узких
фракций (см. стр. 28).
3. Полученные после хроматографии узкие фракции анализируют
спектрально; с помощью этих спектров и хроматограммы уточняют от-
несение полос поглощения в спектрах исходных фракций к соответст-
вующим СГ.
Из полученных таким образом данных могут быть определены!
а) СГС исходных фракций, б) СГС объединенных образцов, в) содер-
жание (в процентах по весу) отдельных групп соединений в объединен-
ных образцах.
Наиболее существенным моментом здесь является объединение об-
разцов по принципу сходства их СГС и последующее разделение. Это
объединение должно проводиться с таким расчетом, чтобы в результа-
те последующего разделения были бы четко выделены интересующие
группы соединений, количества которых составляли бы не менее 1—2
мг. Если к числу интересующих относятся фракции, поглощающие в
УФ-области, то количества их могут быть уменьшены до долей милли-
грамма. В этом случае применение ИК-метода анализа возможно толь-
ко после регистрации УФ-спектров и последующего объединения не-
скольких таких фракций в одну.
Перейдем теперь к разбору конкретной задачи, на примере которой
могут быть наглядно продемонстрированы все особенности метода.
II. ОПЫТ ПРИМЕНЕНИЯ КОМБИНИРОВАННОГО СПЕКТРАЛЬНОГО
МИКРОМЕТОДА АНАЛИЗА К ИЗУЧЕНИЮ СГС ФРАКЦИЙ МАСЕЛ
СОВРЕМЕННЫХ МОРСКИХ ОСАДКОВ
В качестве примера использования изложенного выше метода рас-
смотрим конкретную задачу по изучению СГС масел, выделенных из
битумов донных отложений Берингова моря, Индийского и Тихого оке-
анов и района Антарктики, охватывающих время их формирования до
106 лет.
Целью работы было определение состава масел и выяснение связи
состава: 1) с биогеохимическими особенностями различных областей
Мирового океана, 2) с возрастом вмещающих осадков, 3) с биохимиче-
ским составом морских организмов, 4) с углеводородной частью совре-
менных нефтей. Выяснение этих вопросов имело бы большое значение
для изучения начальных этапов процесса нефтеобразования. Очевидно,
что наиболее объективные данные для решения последних двух вопро-
сов можно получить при изучении объединенных образцов, дающих ус-
редненную картину химического состава.
Исследованы фракции масел двух типов донных отложений — дно-
черпательных проб (ДП), характеризующих начальные этапы процес-
са преобразования осадков, и образцов грунтовых колонок (ГК) более
древнего происхождения. Описание образцов дано в работах [17—20].
Благодаря принятому методу разделения битумов [17, 18] во фракции
масел, помимо углеводородов, попадают так называемые петролейно-
эфирные смолы, считающиеся наименее полярной частью фракции смол.
Наиболее детальному изучению были подвергнуты образцы Берин-
гова моря, на примере которых и был отработан предлагаемый метод.
Изучение фракций масел остальных бассейнов было начато лишь после
завершения анализа масел Берингова моря, что позволило значительно
упростить и сократить их анализ. Метод анализа представлен на сле-
дующей схеме.
146
СХЕМА АНАЛИЗА СОСТАВА ФРАКЦИЙ МАСЕЛ, ВЫДЕЛЕННЫХ ИЗ БИТУМОВ
ДОННЫХ ОТЛОЖЕНИЙ БЕРИНГОВА МОРЯ
ИСХОДНЫЕ ФРАКЦИИ МАСЕЛ:
* УВ — углеводородная.
** Без фракции 15.
Сняты серии
спектров:
ИК-1; УФ-VII
ИК*П
ИК-Ш
ИК-IV
ИК-V
УФ-VIII
ик-vi
Анализ фракций масел Берингова моря
Согласно изложенному выше методу, сначала были сняты УФ- и
ИК-спектры исходных фракций: 8 образцов ДП и 11 образцов, выде-
ленных из ГК. Предварительное изучение спектральных данных пока-
зало, что фракции масел имеют качественно близкий СГС — полосы
поглощения в УФ- и ПК-спектрах группируются в одних и тех же спек-
тральных интервалах. Тем не менее было отмечено определенное отли-
чие спектров образцов ДП от образцов из ГК по полосам поглощения,
отнесенным к ароматическим структурам, о чем ниже будет сказано
подробнее. Для изучения возможных изменений СГС в процессе диаге-
10’ 147
неза осадков интересно было исследовать ДП образцы отдельно от ГК.
Указанные образцы были предварительно объединены в две соответ-
ствующие пробы, каждая из которых была разделена на углеводород-
ную и смолистую части методом, аналогичным применявшемуся для
разделения битумов [17]. Для полученных четырех фракций были сня-
ты УФ- и ИК-спектры. Основной целью настоящей работы было иссле-
дование СГС углеводородной части масел. В каждом из объединенных
образцов ее было около 50 мг. Предварительная оценка концентрации
ароматических углеводородов в этих образцах дала примерно 10%, т. е.
около 5 мг. Учитывая неизбежные потери при разделении и, с другой
стороны, необходимость выделения по крайней мере четырех аромати-
ческих фракций, можно было ожидать получения некоторых фракций,
в основном полициклических, в количествах, явно недостаточных для
анализа. Поэтому было решено объединить обе углеводородные фрак-
ции в одну и эту объединенную фракцию (ДП 4- ГК) разделить микро-
хроматографическим методом на узкие фракции*. Ввиду чрезвычайно
малых количеств ароматических фракций, полученных после разделе-
ния, эти фракции после снятия их УФ-спектров были сведены в объеди-
ненную ароматическую фракцию, количество которой ~ 2,5 мг оказа-
лось уже вполне достаточным для получения ИК-спектра.
В результате были получены спектры поглощения в ИК-области в
интервале 5000—630 см~х, (2—16 мкн) следующих образцов:
I. Всех исходных фракций масел битумов ДП (8 образцов) и об-
разцов из ГК (И образцов).
II. Углеводородной части объединенной фракции масел ДП (обра-
зец 100) и углеводородной части объединенной фракции масел образ-
цов из ГК (образец 101).
III. Смолистой части каждой из этих двух объединенных фракций.
IV. Объединенной углеводородной фракции (ДП + ГК).
V. Нафтено-парафиновых фракций 1—7 (из восьми), выделенных из
фракции ДП + ГК-
VI. Объединенной ароматической части, полученной объединением
ароматических фракций 10—14, выделенных из фракции ДП + ГК.
Спектры поглощения в УФ-области** в интервале 500—220 нм.
VII. Всех исходных фракций масел ДП и ГК.
VIII. Ароматических (фракций 10—15) и двух промежуточных фрак-
ций 8 и 9 (нафтено-парафиновые — ароматические), выделенных из
фракции ДП + ГК.
Ароматические структуры
УФ-спектры исходных фракций масел (серия VII) имеют сходное
строение, аналогичное представленному на рис. 4 с основным максиму-
мом в области 255—265 нм. Если принять средний молекулярный вес
всех фракций Л?'=250 [21], то можно рассчитать коэффициенты молярной
экстинкции е для главного максимума. Результаты расчетов приводятся
ниже.
Приведенные данные показывают, что все исследованные образцы
обладают значительным поглощением, доходящим в ряде случаев до
3—4Х104. Если учесть, что по методу выделения эти фракции должны
содержать в основном нафтено-парафиновые углеводороды, прозрачные
* Автор выражает глубокую благодарность Д. К. Жесткову, выполнившему это
разделение по разработаному им методу (см. его статью в настоящем сборнике).
** УФ-спектры сняты в оптической лаборатории ИНЭОС АН СССР на спектрови-
зоре с монохроматором СФ-4. Автор выражет благодарность сотрудникам этой лабора-
• тории И. Я. Качкуровой и Л. П. Лариной, выполнившим эту работу. ИК-спектры сня-
ты на спектрофотометре ИКС-14 с NaCl-призмой.
148
Фракции масел ДП
№ образца 23 34 21 31 17 61 59 16
8 32800 31700 28700 16200 12700 7700 3700 1800
Фракции масел из ГК
образца 83 40 80 62 41 86 42 39 85 84 64
8 12500 11700 9300 8200 8200 6400 5100 2340 1750 1170 640
в УФ-области, то приходится допустить, что поглощающими должны
быть вещества с ел = 2бо«л< ~ 105. Такие значения е являются максималь-
ными для известных органических веществ. К ним относятся, в частности,
молекулы с сопряженными С = С-связями, с кислородсодержащими заме-
стителями, а также ароматические соединения — одноядерные и некото-
рые конденсированные с гетероатомами и непредельными заместителями.
Отметим, что высшие конденсированные ароматические углеводороды
в соответствии с принятой методикой разделения битумов не должны
присутствовать в исследованных фракциях в больших количествах, а по-
падают во фракции смол и асфальтенов.
Рис. 4. УФ-спектр образца
№ 83 серии VII
1 — концентрация 2,4 • 10-2 модъ/л;
2 — концентрация 4,8 • 10~5 моль!л
Сравнение спектров серий I и VII показывает отсутствие корреляции
между интенсивностью поглощения С = О (основная кислородная полоса
в ИК-спектре) и интенсивностью главного максимума в УФ-спектре.
Таким образом, следует считать, что поглощение в УФ-спектре в основ-
ном обусловлено молекулами, содержащими ароматические ядра —
бензольные, гетероциклические. Количество таких молекул для некото-
рых образцов, по-видимому, составляет не менее */з- На этом основании
последующий анализ СГС ароматической части фракций масел выполнен
в предположении, что основу ее составляют одноядерные с различным
числом и типом заместителей, что подтверждается данными анализа
УФ-спектров серий VII и VIII. В соответствующую группу ароматических
углеводородов, кроме алкилпроизводных бензола, попадают системы с
несколькими изолированными и коньюгированными ароматическими
кольцами (но не конденсированными) и полициклические с одним аро-
матическим и с одним или несколькими полиметиленовыми циклами
(последние могут быть конденсированными). ИК-спектры образцов
149
Рис. 5. ИК-спектры фракций масел серии I
У—ДП образец Кз 61 (модуль 40); 2 — ДП образец № 31 (модуль 30); 3 — образец из ГК № 80
серии I (рис. 5) имеют сходное строение. Соответствующие полосы
группируются в сравнительно узких спектральных интервалах. Интер-
валы, в которых замещенные бензола имеют наиболее интенсивные
полосы, представлены в табл. 1. Сравнительное изучение спектров серий
I—VI показывает, что специфическое поглощение в этих областях
в спектрах серии I действительно вызвано ароматическими структурами.
В исходных образцах ДП 1, 2 (см. рис. 5) основную массу составляет
структура 1,4-замещения (полоса — 830 смт1), которой значительно
больше, чем 1,2,4-замещенных (—810 см-1), присутствующих в этих
образцах в малых количествах, за исключением № 16, где содержание
1,2,4-замещенных структур достаточно велико. Также мало в этих
образцах структур 1,2- + 1,2,3-замещенных ( — 745 см-1) и практически
не обнаруживается 1-+1,3-+1,3,5-замещенных ( — 700 см-1). Образцы
№ 59, 16 и 61 могут быть выделены в особую подгруппу, так как в них
относительное содержание структуры 1,4- ниже, чем в остальных,
и в небольших количествах обнаруживается структура 1,3,5-замещенных
(-850 см--1).
Все исходные образцы, взятые из ГК, характеризуются по сравнению
с ДП повышением относительного содержания 1,2,4-замещенных, а так-
же других структур, в особенности 1,2-+1,2,3-замещенных (см. рис. 5).
По характеру относительного содержания структур 1,2,4- и 1,4-замещен-
ных эти образцы могут быть разбиты на две группы. В одну из них
входят образцы № 39—42, 62, 64, в которых количество 1,2,4-замещен-
150
них примерно равно 1,4-- Другая группа объединяет остальные образцы
№ 80, 83—86, характеризуемые большим относительным содержанием
структур 1,2,4-замещенных. Таким образом, как и в образцах ДП, ос-
новную массу ароматических структур составляет сумма 1,2,4-+1,4-за-
мещенных, далее следует 1,2-+ 1,2,3- и меньше всего 1-+1,3-+1,3,5-за-
мещенных, из которых 1,3,5-замещенные практически нигде не обнару-
живаются.
Что касается ароматических структур с большим числом заместите-
лей, то они по ИК-спектрам не могут быть идентифицированы ввиду
отсутствия достаточно надежных данных.
Определение углеводородных полициклических ароматических соеди-
нений наиболее надежно проводится в УФ-области спектра. При нали-
чии в образце заметных количеств гетеросоединений такое определение
становится невозможным, так как последние имеют на порядок большие
значения е. Изучение этого вопроса по ИК-спектрам, где маскирующе-
го действия гетеросоединений не наблюдается, показало, что спектры
образцов из ГК имеют относительно слабую полосу поглощения (ПП)
в области 1047—1031 см-1, где поглощение приписывается нафталино-
вым структурам [22]. В образцах ДП в этой области имеется более ин-
тенсивная широкая ПП с максимумом 1026—1015 см-1. Изучение ИК-
спектра серии II объединенного образца ДП с удаленной смолистой
частью (образец № 100, рис. 6), где ПП 1026—1015 см-1 резко ослаб-
лена, показывает, что и в дночерпательных образцах имеется ПП в том же
интервале 1047—1031 см~1 с примерно такой же интенсивностью, как в
образцах из колонок. На наличие нафталиновых указывает ИК-спектр
серии VI ароматической части объединенной углеводородной фракции,
где в согласии с данными работы [22] присутствует указанная выше
полоса 1047—1031 см-1 и ПП 744 см-1, по интенсивности превосходящая
соседнюю ПП 722 см-1. Для чисто моноциклоаромэтических фракций
интенсивность ПП 722 см-1 значительно выше, чем 744 см-1 *. В спект-
рах серии I исходных масел ПП 744 см-1 экранируется интенсивной
ПП 722 см-1. Таким образом, можно считать, что в небольших количе-
ствах нафталины есть во всех исследованных фракциях масел.
Данные о строении полиядерных соединений получены на основании
серии VIII УФ-спектров ароматических фракций № 10—15 (рис. 7, б—г).
Эти соединения не имеют характеристических ПП в ИК-области спект-
ра, кроме ПП аналогичных тем, которые имеют одноядерные (см.
табл. 1), что делает невозможным суждение о полиядерных только на
основании серии I ИК-спектров исходных фракций.
Спектр фракций 10 (рис. 7, б) имеет широкий максимум в области
250—265 нм (е<1000) без тонкой структуры, что можно объяснить при-
сутствием во фракции ароматических пол из а мешенных моноциклов.
Спектр фракции 11 имеет более узкий и интенсивный (е~ 1000) глав-
ный максимум — при 255 нм и ряд слабых пиков в области 280—305 нм,
указывающих на появление бициклических соединений с одним гидриро-
ванным кольцом типа производных индана и тетралина; возможно при-
сутствие ароматических углеводородов с двумя бензольными кольцами,
соединенными алкильными и аллильными мостиками, производных сти-
рола, индена, а также полициклических конденсированных систем, со-
держащих одно бензольное кольцо, в частности типа стероидных.
Спектр фракции 12 (см. рис. 7, в) имеет в области X < 315 нм ту же
* Подобное соотношение интенсивностей, по-видимому, объясняется тем, что
ПП 722 см-1 принадлежит только полиметиленовым цепям. Ароматические структуры
не имеют здесь характеристических ПП (см. табл. 1). При одинаковом молекулярном
весе моно- и бициклических фракций процент, приходящийся на парафиновые ради-
калы, в первом случае выше. Отсюда и большая относительная интенсивность ПП
722 см-1.
151
Рис. 6. ИК-спектры объединенных фракций масел с удаленной смолистой
частью
/—образец № 100 (модуль 24); 2 — образец № 101, серия II (модуль 14); 3 — спектр аро-
мэтической части, серия VI
структуру, что и предыдущий образец с более четкой областью поглоще-
ния 270—310 нм. Кроме того, в спектре наблюдаются два плеча в обла-
сти 323—333 нм, что указывает на появление здесь, помимо структур
фракции 11, производных нафталина; вероятно присутствие конденси-
рованных соединений с нафталиновым ядром типа аценафтена, гекса-
гидропирена и т. д., а также производных флюорена, в том числе бенз-
производных.
Спектр фракции 13 аналогичен предыдущему, только вместо плеч
появляются интенсивные пики нафталиновых ядер при 333 и 318 нм и
появляется новый пик при 357 нм. В спектре фракции 14 (см. рис. 7, г}
последние три пика более резки и появляется пик 286 нм. Эти особенно-
сти указывают на появление во фракциях 13 и 14 заметных количеств
производных нелинейно-конденсированных соединений типа фенантрена,
хризена и т. п.
В спектре фракции 15 (хвост хроматографического разделения) все
пики, кроме главного, практически исчезают, что, по всей вероятности,
связано с концентрацией в этой фракции различных гетероатомных со-
единений.
Таким образом, ароматическая часть масел образцов ДП + ГК пред-
ставлена следующими структурами: одиночные ядра; полициклические
конденсированные соединения, содержащие одно бензольное кольцо;
нафталины; полициклические конденсированные соединения, содержа-
щие нафталиновое ядро; ароматические нелинейно-конденсированные
ядра с числом колец не более 4. Не обнаружено признаков существова-
ния линейно-конденсированных структур типа антрацена.
Как выше отмечалось, в ИК-спектрах серии I исходных масел име-
ются две ПП в области 1047—1010 см-1, одна из которых 1'031—1047 см~1
объясняется поглощением нафталинов. Сравнительное изучение этой
серии спектров со спектрами серий II, IV и VI фракций с удаленной
смолистой частью (см. рис. 5, 6) показывает, что вторая ПП 1010—
1026 см-1 значительно более интенсивна в ДП образцах, чем в образцах
152
Рис. 7. УФ-спектры ароматических фракций (серия VIII), иыделенных
из объединенной углеводородной фракции ДП + ГК (толщина кюветы
d = 0,5 см)
а — образец №9; б — образец № 10; в — образец № 12; г — образец Кг 14;
спектры i — концентрация 10“3 мсль1л, спектры U — концентрация 10“4 молъ(л
из ГК. Она, по-видимому, не относится к кислороду, так как присутст-
вует в спектрах образцов № 16 и 17, не имеющих основных ПП кислород-
содержащих структурных групп. В спектре серии II образца ДП № 100
(см. рис. 6, 1) интенсивность ПП 1015—1026 см~1 резко снижена и.
примерно в 2 раза против ожидаемого упала интенсивность ПП 833—
826 см-1, приписываемая 1,4-замещению, или точнее — ароматической,
структуре с двумя орто-незамещенными Н-атомами. Падение интенсив-
ности ПП 833—826 см-1 говорит за то, что соответствующая аромати-
ческая структура, перешедшая в смолистую фракцию (петролейно-эфир-
ную), связана с гетероатомом, а практическое исчезновение ПП 1015—
1026 см-1 дает основания считать, что таким атомом может явиться азот
в гетероциклической форме, так как в этой области очень сильным погло-
щением обладают некоторые гетероциклические азотистые соединения,
например пиридины [3]. Подобное отнесение находится в согласии с тем
фактом, что образцы № 59, 16, 61, имеющие среди ДП-образцов наимень-
щие значения относительных интенсивностей ПП 833—826 см-1,.
дают наименьшие интенсивности ПП 1015—1026 см-1 и обла-
дают самыми низкими (из образцов ДП) значениями е. в УФ-спектре
(см. стр. 149); соответственно образцы с наиболее интенсивными ПП
833—826 см-1 (№ 23, 34, 21) имеют наиболее интенсивные ПП 1010—
1026 см-1 и максимальное поглощение в УФ-спектре. Таким образом,
можно считать, что в состав исходных масляных фракций, точнее в со-
став их смолистой части, помимо кислорода, входит азот гетероциклов,
присутствие которого особенно заметно в образцах ДП и в значительно
меньшей степени — в образцах из ГК независимо от глубины залегания
образца.
Наличие в сериях II, IV и VI ИК-спектров фракций ПП 1010—
1026 см-1 с такой же интенсивностью, как у полосы 1031—1047 см~х, лег-
ко объясняется поглощением различных моноциклоароматических;
структур, имеющих здесь ряд более слабых полос [3—5, 23].
Нафтено-парафинов ы еструктуры
Парафиновые структуры фракций масел анализировались по ИК-
спектрам серии I (см. рис. 5), по ПП СНз-групп 1380—1370 смт1 и в об-
ласти поглощения полиметиленовых цепочек — (СНг)п—787—721 смт1.
ПП 1176—1149 см~х изомерных парафинов не может оыть использована
153
достаточно надежно, так как в этой области имеют сильные ПП кисло-
родсодержащие фрагменты. Во всех образцах наблюдалась относитель-
но высокая интенсивность ПП 1381 см-1, говорящая о значительном ко-
личестве парафиновых структурных групп, величина которых может быть
установлена на основании изучения ПП 787—721 см-1. Анализ этой по-
лосы показал, что во всех исследованных образцах наблюдается более
или менее значительное размытие полосы в сторону коротких длин
волн с небольшими пиками или плечами. Главный максимум располо-
жен в области 723,5—721,5 см-1. Положение коротковолновых пиков,
вырождающихся на некоторых спектрах в плечи (область 769—763 и
749—743 см-1), не дает оснований приписать их полиметиленовым
структурам с п < 5, так как соответствующие им ПП лежат в несколько
других областях спектра (см. табл. 1). Как выше отмечалось, эти ПП
приписаны нами ароматическим структурам, имеющим в этих спект-
ральных интервалах интенсивные ПП, что подтверждается анализом
ПК-спектра серии VI ароматического образца (см. рис. 6, 3).
Отсюда следует, что основную массу парафиновых структур состав-
ляют длинные цепи нормального строения — (СН2)П>5—, а коротко-
цепное ветвление представлено в основном СН3-группами.
Наблюдается корреляция между интенсивностью ПП С = О-группы
1740—1710 см-1, ПП полиметиленовой цепочки ~722 см-1 и ПП СН3-
группы (1381 см-1), образцы с наиболее интенсивной ПП С = О-группы
имеют наиболее интенсивную ПП — (СНг)п>5— и наименее интенсивную
ПП СН3-группы, что видно из табл. 2. Это говорит за то, что кислород
в значительной степени связан с полиметиленовыми цепями. Превраще-
ния кислородсодержащих соединений с выделением кислорода приводят
к увеличению числа СН3-групп. В образцах с большим содержанием
С = О, по-видимому, до половины парафиновых цепей связано с кисло-
родом.
Наблюдаемое в ряде спектров (образцы № 61, 86, 83, 80 и др.) рас-
щепление ПП 721,5 см-1, когда эта полоса достаточно интенсивна и од-
новременно интенсивна ПП С = О-группы, указывает на присутствие в
образце твердых продуктов, содержащих длинные полиметиленовые
Таблица 2
Относительные интенсивности ПП групп С=О
(1740—1710 слГ1), — (СН2)П>В — (~722 слГ1) и —СН3 (1381 слС1)
№ образца с=о Полиметиле- новые группы СН3-груп- пы № образца С=О Полиметиле- новые группы сн8- группы
Дночерпательные пробы Грунтовые колонки
61 10,8 6 5,1 83 9 5,6 4,6
59 10,0 4,1 5,4 86 8 5,4 5,1
34 4,2 2,8 5,8 84 7 4,6 5,5
31 3 2,8 6,5 62* 7 2,4 5
23* 2,8 2 6 64 7 4 5,4
21 2,6 2,9 6,7 85 4 4 6,3
17 — 1,8 6,5 41 4 2,7 6
16 — з,з 7,8 80 3,6 3 6
40 3 2,2 6,2
39 2 2 6,1
42 1,6 3 6,3
* Данные недостаточно точны, в особенности для ПП — (СН2)П — из-за чрезвычайно малых ко-
-личеств образцов (1—3 мг).
•154
Рис. 8. ИК-спектры нафтено-парафиновых фракций (серия V), выделенных
из объединенной углеводородной фракции ДП + ГК
1 — образец № 3; 2 — образец № 7 (модуль 10)
цепи и С = О. Все эти образцы, за исключением № 61, из ГК (5 из И).
В ряде этих образцов твердые частицы видны невооруженным глазом.
Изучение серии V ИК-спектров углеводородных фракций (рис. 8) дает
дополнительные данные о нафтено-парафиновых структурах масел.
Фракции 1—3 (см. рис. 8) имеют идентичные спектры, характерные для
слабо замещенных парафинов. На это указывает малое поглощение в
области 1250—1000 см~\ где обычно проявляются различные типы за-
мещения парафиновой цепи, сильное поглощение на 722 см-1 полимети-
леновой цепи —(СН2)П>5— и отсутствие в области 770 см-1 ПП коротких
цепей с п < 5.
Во фракциях 4—7 появляется характерное поглощение в области
1000—840 см-1 с двумя широкими максимумами на 960 и 850 смг1.
С увеличением номера фракции (соответствует последовательности,
в которой они отбирались) интенсивности этих ПП растут примерно в
3 раза. Одновременно примерно на эту же величину падает интенсив-
ность ПП 722 смг1 без какого-либо заметного изменения интенсивности
остальных полос, в том числе и ПП 1381 см-1 СН3-групп. Лишь во фрак-
ции 7 появляется слабая ПП 738,5 смг1, которая может быть объяснена
появлением структурных групп —(СН2)2— (см. рис. 8).
Наличие в спектрах последних четырех фракций характерной обла-
сти поглощения 1000—850 смг1 с двумя максимумами указывает на при-
сутствие в этих фракциях нафтеновых колец. Подобные полосы в спект-
рах нафтено-парафиновых фракций сложного состава наблюдались на-
ми ранее [24].
Таким образом, нафтено-парафиновая часть фракции масел пред-
ставлена длинноцепочечными алкильными структурами нормального
строения — (СН2)п>5—. Короткоцепное ветвление представлено в ос-
новном СНз-группами.
Найдены нафтеновые структуры. По данным хроматографического
разделения и значений «п20, среди этой части структур присутствуют зна-
чительные количества полициклических нафтенов.
155
Наблюдаемая корреляция между изменением относительных интен-
сивностей полос С = О-, —(СН2) п>5— и СНз-групп говорит за то, что
кислород, попавший во фракции масел, в значительной степени связан
с полиметиленовыми цепями.
Непредельные структуры
Наблюдаемая в ИК-спектрах всех исследованных образцов (см.
рис. 5) характерная узкая ПП в интервале 977—964 см-1 примерно оди-
наковой интенсивности определяет присутствие тушнс-олефиновых струк-
тур R'HC = CHR. Наличие ПП в интервале 890—884 см-1 указывает йа
возможное присутствие винилиденовых структур RZRC = CH2, наимень-
шее количество которых содержится в образцах из ГК (№ 39, 40, 41, 42,
62, 64). В остальных образцах оно примерно одинаково. По ПП в интер-
вале 917—908 см-1 могут быть идентифицированы а-олефиновые струк-
туры RHC = CH2. Наибольшую интенсивность в этой области имеют об-
разцы № 83 и 86 из ГК-
Наиболее интенсивными являются ПП, приписываемые транс-, наи-
меньшие — а-олефиновым структурам, что указывает соответственно на
их максимальное и минимальное присутствие в исследованных образ-
цах. Это подтверждается данными анализа углеводородных фракций,
полученных после хроматографического разделения объединенной угле-
водородной фракции ДП + ГК. В нафтено-парафиновых образцах №4—7,
выделенных из ДП+ГК, наблюдается отчетливый пик транс-оле-
финов на 970 см~\ а также пик на 893 см-1 винилиденовой структуры.
Поглощение а-олефинов в области 920—910 см-1 попадает на крыло
полосы 970—940 см-1 и потому отчетливо не проявляется. Количество
непредельных в этих фракциях невелико, так как даже промежуточная
фракция 8 в концентрации ~10-3 моль!л прозрачна в УФ-области. За-
метная непредельность наблюдается только в следующих фракциях:
фракция 9 имеет незначительное поглощение в УФ-области спектра, уве-
личивающееся от 255 нм в коротковолновую сторону (см. рис. 7, а), что
типично для молекул с несопряженными С = С-связями. В спектре моно-
циклоароматической фракции 10 (см. рис. 7, б) практически отсутствует
минимум на 256—238 нм, характерный для таких углеводородов, что
также говорит о присутствии здесь несопряженных С=С-групп.
Присутствие непредельных структур с внутренней С = С-связью типа
транс-форм подтверждается наличием в УФ-спектрах исходной серии
VII (см. рис. 4) ряда характерных полос в области 555—370 нм, припи-
сываемых обычно цепочкам вида (—СН = СН—)п. Подобные структуры,
по-видимому, связаны со смолистой частью масел, так как в УФ-спект-
рах конечной серии VIII соответствующих им ПП не обнаружено.
Таким образом, несопряженные С = С-группы в отличие от сопря-
женных присутствуют не только в смолистой, но и в углеводородной
части масел как в собственно олефиновых молекулах (образец 9), так и
в качестве заместителей ароматических структур (образец 10), на что
указывает также ИК-спектр серии VI ароматической части объединен-
ной углеводородной фракции (см. рис. 6, 3). Количество олефиновых
структур в углеводородной части не превышает нескольких процентов.
Кислород
Присутствие кислорода в основном в виде С = О-групп установлено
по наличию в ИК-спектрах серии I (см. рис. 5) ПП в области 1740—
1710 см-1, преимущественно в районе 1740 смт1, относящейся в большей
степени к карбонилу жирных кислот и сложных эфиров. Кроме этого,
в спектрах имеются полосы в области 1315—Г300 и 1175—1150 см-1,
которые также могут быть отнесены к жирным кислотам и сложным
156
эфирам, и полоса в области 1125—1100 см~\ указывающая на прису-
ствие в образцах О—Н-групп спиртов различного строения, а также
эфирных групп. Наибольшей достоверностью обладает ПП 1740—
1710 см-1 как наиболее интенсивная из полос кислорода и лежащая в
относительно пустой области спектра, наименьшей — более слабые по-
лосы 1315—1300 и 1175—1150 см~\ лежащие в области интенсивного
/R'
поглощения замещенных парафинов типа R—сн , (СН3)2СН—R
хсн3
и т. д., а также слабая ПП 1125—1100 см-1.
Из образцов ДП наибольшей интенсивностью полосы 1740 см-1 обла-
дают образцы № 59 и 61, а в образцах № 16 и 17 эта ПП отсутствует.
В остальных образцах ее интенсивность примерно одинакова. ПП, ха-
рактеризующие О—Н-группу, примерно одинаковы во всех образцах
ДП, кроме № 16 и 17, где они отсутствуют.
В образцах из ГК ПП 1740 см-1 наиболее интенсивна в образцах
№ 83, 86, 84, 62, 64; ПП 1125—1100 см~1 практически отсутствует в об-
разцах № 39, 40, 41, 42, 84, 85. В остальных образцах она также весьма
слаба.
Таким образом, как в образцах ДП, так и в образцах из ГК кисло-
род представлен в основном С = О и R—СО—О—R'-группами и в значи-
тельно меньшей степени О—Н-группами.
Из результатов проведенного выше анализа можно проследить со-
держание отдельных структур или дать СГС исходных фракций масел
в пределах изученных СГ, а для объединенного образца, помимо СГС,
получить данные о содержании (в процентах по весу) отдельных групп
соединений. Так, например, для СГС объединенной фракции масел
получены следующие результаты. Основную часть углеводородной ча-
сти масел составляют парафиновые цепи —(СН)п>з— нормального
строения ( — 50%); изомерные структуры наблюдаются в очень неболь-
ших количествах (—1%); далее следуют нафтено-парафиновые
(»40%), не менее половины которых поликольчатые.
Найдены непредельные структуры (—1,5%) в порядке убывания:
транс-олефиновые, винилиденовые и а-олефиновые.
Ароматическая часть масел (—10%) в основном представлена одно-
ядерными структурами ( — 70% от ароматической части) с различным
числом и типом заместителей — алкилбензолы, стиролы и т. д. Среди
них могут присутствовать ароматические углеводороды типа терфени-
лов с алкильными и олефиновыми соединительными мостиками, бици-
клические соединения с одним гидрированным кольцом типа производ-
ных индана, тетралина и, в частности, стероидные формы, частично де-
гидрированные. Характерной особенностью ароматической фракции яв-
ляется отсутствие длинноцепочечного —(СН2) п>5-ветвления. Основную
массу одноядерных ароматических углеводородов составляет сумма
1,4-+1,2,4-замещенных. Далее следуют 1,2- 1,2,3- и менее всего 1 + 1,3+
+ 1,3,5-замещенных. Из конденсированных ароматических структур об-
наружены производные нафталина ( — 20%) и нелинейно-конденсиро-
ванные (—10%) с числом колец не более четырех типа фенантрена,
хризена и т. д. Не обнаружены линейно-конденсированные ароматиче-
ские структуры типа антрацена.
Из гетероатомных соединений обнаружены кислород- и азотсодер-
жащие. Первые представлены в основном С = О-группами жирных кис-
лот и сложных эфиров и в значительно меньшей степени—О—Н-груп-
пами спиртов. Вторые, по-видимому, гетероциклические азотистые сое-
динения, связанные с 1,4-замещенным одноядерными ароматическими
структурами. В табл. 3 приведены данные по СГС и составу по группам
соединений этой объединенной фракции масел (ДП + ГК).
157
Таблица 3
Данные по СГС н содержанию различных групп соединений
в объединенной фракции масел Берингова моря (в %)
Группа соединений Масла битумов
по СГС по группам соединений
Нормальные парафины 50 31
Изопарафины — —
Нафтено-парафины 39,5 55
Олефины 1,5 2
Ароматические (сумма) с том числе: моноциклические (преимущественно 9 12
1,4-|-1,2,4-замещенные) 6 8,5
бициклические полициклические нелинейно-кон- 2 2,5
денсированные полициклические линейно-конден- 1 1
сированные . — —
Как было указано в начале этого раздела, помимо образцов Берингова моря, были
изучены фракции масел других бассейнов Мирового океана. Для , изучения их СГС
оказалось достаточным получить ИК- и УФ-спектры только исходных фракций без
последующего объединения и разделения на хроматографической колонке, так как
спектры этих исходных фракций оказались' по своему характеру близкими спектрам
фракций масел Берингова моря — полосы поглощения в УФ- и ИК-спектрах группиро-
вались в тех же спектральных интервалах, незначительно отличаясь относительными
интенсивностями отдельных полос. В силу этого анализ СГС масел указанных бассей-
нов был проведен по упрощенному способу, используя отнесения полос, полученные
на образцах Берингова моря. Проведение анализа по всей программе могло бы дать
сведения о зависимости состава масел от био-геохимических особенностей отдельных
областей океанов. Однако установленные различия в составе исходных фракций не
позволили получить на этом пути отчетливых данных. В основном этот неуспех, по-
видимому, связан с недостаточным количеством поступившего материала. В какой-то
мере это могло быть связано с особенностями принятого метода выделения исходных
фракций. Однако, учитывая, что максимальная разница значений содержа-
ния СГ в исходных фракциях всех исследованных бассейнов лежит в пределах уста-
новленной нами общей последовательности их содержания (см. табл. 3) и что ошиб-
ка в определении состава не превосходит ±10% от величины абсолютного содержания
указанных СГ (групп соединений), результаты усреднения состава по совокупности
разумно подобранных образцов должны дать хотя бы частичные ответы на осталь-
ные вопросы, поставленные перед исследователями. Действительно, на основании по-
лученных данных можно отметить следующее.
Исходя из известных данных по химсоставу нефтей с учетом имеющихся среди
них различий, можно с уверенностью утверждать, что в основных чертах СГС масел
битумов совпадает с СГС углеводородной части обычных нефтей.
Групповой состав масел практически не зависит от возраста и строения осадков,
фациальных условий бассейна, что говорит об однородности как исходного материа-
ла, так и условий его превращения в углеводороды.
Если разбить исследованные образцы фракций масел на следующие группы:
I) дночерпательные образцы Берингова моря, 2) образцы колонок Берингова моря,
дночерпательные образцы Тихого и Индийского океанов и района Антарктики, 3) об-
разцы из колонок океанов, то в этой последовательности можно проследить измене-
ния содержания отдельных ароматических структур, а именно: а) уменьшение содержа-
ния 1,4-замещенных по сравнению с 1,2,4-, если в образцах 1-й группы 1,4- больше,
чем 1,2,4-, то во 2-й группе 1,2,4- больше 1,4-, а в 3-й группе 1,4-замещенные отсутству-
ют, б) увеличение содержания остальных типов замещения 1-+ 1, 2-+ 1,'3- и др.;
в) увеличение содержания бициклических и полициклических структур; г) обнаружива-
ется также заметное уменьшение кислородсодержащих молекул и особенно гетероцик-
158
лических азотистых соединений, которые отчетливо наблюдаются только в образцах
1-й группы *.
Автор надеется, что проведенный подробный разбор конкретного
примера с указанием целей исследования и способа их достижения пред-
лагаемым методом анализа позволит более полно, а главное наглядно
оценить его достоинства и недостатки, а также пути усовершенствова-
ния метода применительно к новым конкретным задачам.
Институт биологической физики Академии наук СССР'
Институт нефтехимического синтеза им. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. С. N. R. Rao. Chemical Appl. of Infrared Spectroscopy Acad. Pr. New York — London,
1963.
2. А. Наканиси. Инфракрасные спектры и строение органических соединений. М.,
«Мир», 1965.
3. Л. Беллами. Инфракрасные спектры сложных молекул. М., ИЛ, 1963.
4. Б. А. Смирнов. Итоги науки. Сб. «Химия нефти и газа». М., Изд-во АН СССР,
1958, стр. 414.
5. Н. A. Szimanski. Infrared Band Handbook. Plenum Press. N. Y., 1963; Suppl. 1,
2—1964; Suppl. 3, 4—1966.
6. L. Lang, J. Szoke, G. Varsany, M. Vizesy. Absorption Spectra in the Ultraviolet and
Visible Region. Vol. 1—5. Budapest, 1959—1965.
7. R. A. Friedel, M. Orchin. Ultraviolet Spectra of Aromatic Compounds. New York —
London, 1951.
8. M. M. К у саков, H. А. Шиманко, M. В. Шишкина. Ультрафиолетовые спектры погло-
щения ароматических углеводородов. М., Изд-во АН СССР, 1963.
9. А. Дункан, В. Горди, Р. Н. Джонс и др. Применение спектроскопии в химии. М.,
ИЛ, 1959.
10. А. А. Бабушкин, П. А. Бажулин, Ф. А. Королев и др. Методы спектрального анали-
за. Изд-во МГУ, 1962.
11. А. Гиллем, Е. Штерн. Электронные спектры поглощения органических соединений.
М„ ИЛ, 1957.
12. Ultraviolet Spectral Data, 1955. Amer. Petrol. Inst. Res. Project 44.
13. A. Eucken, К- H. Hellwege. Lando It—Bornstein Zahlenwerte u. Functionen a. Physik,
Chemie, Aufl. 6, Bd. 1, T. 3. Springer, Berlin, 1951, S. 231.
14. M. P. Groenewege. Spectrochim. Acta, № 5, 579 (1956).
15. А. А. Михновская, A. M Ораби, С. P. Сергиенко. Изв АН ТуркмССР, серия хим.,
№ 3,(1960). •
16. И. Е. Лейфман. Диссертация. М., МГУ — МИНХиГП, 1967.
17. О. К- Бардовский. Диссертация. М., ИГиРГИ АН СССР, 1959.
18. О. К. Бардовский, Б. А. Смирнов. Геохимия, № И, 1009 (1962).
19. О. К. Бардовский. Труды Ин-та океанологии АН СССР, 42, 89 (I960).
20. О. К- Бардовский. Сб. «Среда и процессы нефтеобразования». М., «Наука», 1964;
сб. «Океанологические исследования», № 4. М., Изд-во АН СССР, 1964.
21. В. В. Вебер, А. Е. Горская, Е. А. Глебовская. Битумообразование в четвертичных
осадках и генезис нефти. М., ГТТИ, 1960.
22. С. Р. Сергиенко, М. П. Тетерина, А. А. Михновская. Труды Ин-та нефти, 10,.
153 (1957).
23. Н. L. McMurrey, V. Thornton. Analyt. Chem., 24, 318 (1952).
24. Е. Н. Караулова, Б. А. Смирнов, Г. Д. Гальперн. Нефтехимия, 1, 339 (1961).
25. Б. А. Смирнов. 2-й Международный океанографический конгресс. Тезисы докладов.
М., «Наука», 1966.
26. Б. А. Смирнов. Океанология, 9, 791 (1969).
27. Б. А. Смирнов. Изв. АН СССР, сер. биологич., 1969, 532.
* Более подробно научные результаты работы, в том числе о возможной связи-
СГС масел с биохимическим составом морских организмов, были доложены на II Меж-
дународном океанографическом конгрессе [25—27].
ИССЛЕДОВАНИЕ СПЕКТРОВ СУЛЬФИДОВ*
Л. Р, Барыкина, Г. Ц. Гальперн, А. Н. Бислинекий,
Н. А. Шиманко
В настоящее время интенсивно проводятся синтез и всесторонние ис-
следования органических соединений серы, присутствие которых пред-
полагается или установлено в нефтяных фракциях, а также производных
этих соединений. Наибольший интерес как потенциальный источник
сырья для производства растворителей, экстрагентов, флотореагентов,
биологически активных и других веществ представляют сульфиды раз-
ного строения. Значительное место в исследовании состава и строения
этих веществ занимают спектральные методы, для использования кото-
рых необходим справочный материал в виде структурно-частотных кор-
реляций, получаемый при анализе индивидуальных соединений, модели-
рующих компоненты исследуемых систем.
Для успешного установления строения сульфидов всегда желатель-
но, а часто и необходимо применение совокупности ряда физико-хими-
ческих методов: ИК-, КРС-, ПМР-, УФ- и масс-спектроскопии, а также
газо-жидкостной хроматографии **.
Полученные спектральные данные и доступный литературный мате-
риал позволили выявить ряд новых аналитически ценных структурно-
частотных корреляций, а также на более широком экспериментальном
материале подтвердить правильность ранее выявленных корреляций
или, напротив, обнаружить их ограниченность. Результаты данной ра-
боты помогут облегчить выбор спектрального метода исследования для
решения конкретных структурных задач.
ИК-спектры веществ, взятых в виде жидких пленок, записывали на
спектрометрах ИКС-14 и UR-10. Точность определения значений ча-
стот в максимумах поглощения составляла ±2 см-1. Спектры КРС ре-
гистрировали с помощью спектрографа ИСП-51 фотографическим и фо-
тоэлектрическим способами. Значения частот в этих спектрах определя-
ли с точностью ± 1 см-1 путем компарирования негативов; интенсивно-
сти линий в максимуме /0 и степени деполяризации р определяли из
фотоэлектрических записей спектров. При этом интенсивность линии
v = 770 см-1 метилциклогексана была принята за 100.
УФ-спектры получены на двухлучевом самопишущем спектрофото-
метре EPS-2; в качестве растворителя использован изооктан или
н-гептан.
* Отдельные части данной работы ранее были доложены [1] и опубликованы [2].
** Большинство исследованных нами препаратов было проанализировано И. В. Че-
репановой методом ГЖХ на газо-жидкостном хроматографе ХТ-1 СКВ ИНХС АН
СССР, благодаря чему мы с большой долей вероятности могли судить о степени чи-
стоты исследованных соединений.
160
Спектры ПМР записаны на спектрометре JEOL; в качестве раствори-
теля использован CCU Методы синтеза и свойства изученных сульфи-
дов описаны в работах [3—7].
I. КОЛЕБАТЕЛЬНЫЕ СПЕКТРЫ АЛИФАТИЧЕСКИХ СУЛЬФИДОВ
Сравнение ИК- и КРС-спектров алифатических и некоторых алицик-
лических сульфидов типа RSR' со спектрами углеводородов родствен-
ного строения позволило обнаружить некоторые структурно-частотные
корреляции для сульфидов этого типа.
Для анализа спектральных закономерностей использован как ориги-
нальный, так и обширный литературный материал.
А. Обзор литературы
В литературе [8—11] приводится около тридцати ИК-спектров насы-
щенных сульфидов типа RSR'; спектры КРС семи сульфидов этого типа
даны в работах [12—18].
В ряде работ обсуждался вопрос о влиянии атома серы на частоты
деформационных колебаний СН2- и СНз-групп, присоединенных не-
посредственно к атому серы. Шиманский [19, 20] указывает следующие
значения частот асимметричных и симметричных деформационных
колебаний СНз-групп, присоединенных к атому серы: 1430±20 и 1325—
1300 см-1 соответственно*; он отмечает более низкие значения частот
ножничных колебаний метиленовых групп, расположенных вблизи атома
серы, чем в углеводородах (около 1415 вместо 1470 см-1}.
Шеппард [21] показал, что частоты симметричных деформационных
колебаний метильных групп зависят от химической природы (электро-
отрицательности) гетероатома, присоединенного к СН3-группе, но прак-
тически не зависят от массы и размеров этого атома. Частоты, обуслов-
ленные рассматриваемыми колебаниями, в случае соседства метильной
группы с атомами N, О, F, электроотрицательность которых больше
трех, оказываются более высокими, чем в углеводородах (1380 см-1).
Наибольшая частота, около 1475 см~\ наблюдается для атома фтора.
Соседство с атомом хлора, электроотрицательность которого равна
трем, обусловливает поглощение около 1355 см-1, т. е. несколько ниже,
чем в углеводородах.
Соседство с метильной группой атомов Р, Sb,Br, Se, As, Ge, I, Sn,
а также атома S, электроотрицательность которых меньше трех, вызы-
вает понижение частот симметричных деформационных колебаний
СНз-групп до 1325—1200 см-1.
Частоты асимметричных деформационных колебаний СН3-групп,
присоединенных к разнообразным гетероатомам, могут наблюдаться
в области 1480—1380 см-1, однако встречающееся расщепление полосы
затрудняет надежность ее идентификации.
Скотт и Мак-Кало [22] произвели анализ спектров простейших суль-
фидов, использовав данные работ [17, 18, 22—26], полученные при
расчетах нормальных колебаний простых молекул. Были обнаружены
частоты, характерные для групп CH3S, C2H5S, (CH3)2CHS, (CH3)3CS,
обусловленные маятниковыми колебаниями групп СН3, маятниковыми,
веерными и крутильными колебаниями групп СН2, валентными С—С и
деформационными колебаниями С—С—S, С—S—С. Оказалось, что на
эти частоты лишь незначительно влияет природа второй замещающей
группы, будь то атом водорода или (СНз)3С, что может быть обусловле-
* Были усреднены частоты 16 сульфидов; структураR была весьма разнообразной,
в некоторых случаях содержались атомы N или О
11 Методы анализа
161
но изолирующим влиянием тяжелого атома серы и большей длиной
С—S-связи по сравнению с С—С-связью.
Шеппард [9] эмпирически обнаружил частотно-структурные корреля-
ции между частотами симметричных С—S—С-валентных колебаний
(асимметричные С—S—С-валентные колебания он не рассматривал)
и характером алкильных групп, присоединенных к атому серы. Для
группировки CH3S он указал характерную область поглощения 705—
685 см~1, для RCH2S — 660—630 см-1, для RR'CHS —630—600 см-1-
для RR'R"S — 600—570 см-1.
Однако, сравнивая рассчитанные значения частот симметричных и
антисимметричных С—S—С-валентных колебаний (табл. 1) с эмпири-
чески найденными Шеппардом значениями этих частот, можно видеть,
что структурно-частотные корреляции для С—S—С-валентных колеба-
ний должны быть более сложными, чем полагал Шеппард.
Таблица 1
Частоты С—S—С-валентных колебаний простейших сульфидов (в см *)
Конфор- мация Структурная формула Максимумы поглощения Литература
KPC ик
симм. антисимм. симм. антисимм.
СН3—S—СН3 691 с. 741 ср. 691 с. 741 ср. [24, 27]
С2 QH5-S-С2Н5 639 с. 778 оч. с. 640 ср. 781 ср. [23, 26]
Ci CgHs-S-CaHs 657 с. 764 оч. сл. 660 ср. 764 ср. [23, 26]
Csi, CjHs—S—С2Н5 693 дублет) 693 ср. ^дублет) [23, 26]
с ^транс СНа-З-СгНб 650 723 653 ср. 727 ср. [22, 28]
с ^гош CH3—S—C2H5 650 675 653 ср. 668 сл. [22, 28]
С1 СН3—S-CH (СН3)2 636 723 639 с. 724 с. 125]
Сз CH3-S—CH (CH3)2 610 659 612 ср. — [25]
Из таблицы следует, что для поворотных изомеров С—S—С-валент-
ные колебания могут иметь несовпадающие значения частот; как сим-
метричные, так и антисимметричные С—S—С-валентные колебания
могут вызывать полосы значительной интенсивности. Следовательно,
в спектрах сложных молекул должны присутствовать не одиночные
полосы С—S—С-валентных колебаний, а группы полос, что нами и наб-
людалось.
Б. Обсуждение результатов
а. ИК-спектры
При рассмотрении спектров ряда алкилсульфидов [8, 10, 22] нами
было обнаружено, что частота маятникового колебания СНз-группы,
входящей в изопропильную группировку, отделенную от атома серы
одной или двумя метиленовыми группами, наблюдается в области
1169—1174 см-1. В случае непосредственного присоединения изопропиль-
ной группы к атому серы частота имеет более низкое значение (1149—
1158 сл!"1). Частоты симметричных деформационных колебаний изопро-
пильной (1385—1380 и 1372—1365 см-1, сильные) и трет-бутильной
(1405—1385 см-1, средняя; 1372—1365 см-1, сильная) группировок в уг-
леводородах и сульфидах оказываются аналогичными.
В табл. 2 представлены частоты маятниковых колебаний полимети-
леновых цепочек (СН2)„ ряда сульфидов, а также для сравнения —
аналогичные частоты углеводородов.
162
Таблица 2
Частоты маятниковых колебаний (в слГ1) полиметиленовых
цепочек (CHs)n сульфидов RSR' и углеводородов
п Частоты колебаний Число рас- смотренных сульфидов
для углево- дородов для сульфидов
6 724—722 720—722 5 '
5 724—723 720—722 2
4 726—724 Нет надежных
данных
3 729-726 725-730 3
2 743-734 749—736 5
1,а 776—770 759 1
1,6 785—780 787 —781 6
Можно видеть*, что при п = 6,5,3,2ивслучаеп = 1.,когдаСН2-группа
заключена между группой СН3 и атомом серы, значения частот маятни-
ковых колебаний сульфидов близки соответствующим частотам углево-
дородов. Для п=4 мы не располагали надежными данными. Когда
СН2-группа заключена между группой СН3 и третичным Cj-атомом,
в углеводородах поглощение наблюдается при 776—770 ел-1, когда
СН2-группа заключена между атомом S и третичным С-атомом, погло-
щение наблюдалось при 759 см-1, т. е. при более низком значении часто-
ты (л=1,.а). Когда в углеводороде СН2-груп,па заключена 'Между
СН3-группой и четвертичным углеродным атомом, поглощение наблюда-
ется около 785—780 см-1. Практически в той же области поглощает
метиленовая группа в сульфиде, заключенная между атомом серы и
СНз-группой (n = 1, б).
Проведенный нами анализ спектров девяти алкилсульфидов, в ко-
торых СН3-группа присоединена непосредственно к атому серы [8],
показал, что частоты асимметричного и симметричного деформационных
колебаний СН3-групп наблюдаются в областях 1441—1429 и 1323—
1307 см-1 соответственно.
Полоса ножничных деформационных колебаний группы/СН2) рас-
положенной рядом с атомом серы, наблюдается в большом числе
спектров ([8, 10, 11] и наши данные) в области 1439—1410 см-1. Однако
в спектрах некоторых сульфидов, содержащих группировку/CHaS, не
обнаруживается полосы в указанной области, что может быть связано
с недостаточной дисперсией призмы из хлористого натрия [8, 11].
Из табл. 3 видно, что в углеводородах и сульфидах с монозйМёщен-
ным циклогексановым кольцом наблюдаются полосы при 1350, 1260,
1200 и 890 см-1-, в сульфидах, не содержащих циклогексанового кольца,
например («-Ci0Hai)aS и в дициклогексилсульфоксиде, приводимых
в таблице для сравнения, некоторые из этих полос слабы или отсутству-
ют. В спектрах всех сульфидов с циклогексановым кольцом наблюдается
сильная полоса около 1000 ел-1.
Шиманский [20] указывает, что полоса 1000 см-1 наблюдается в слу-
чае присоединения циклогексанового кольца к Cl, Br, SH, ОС2Н3, NO2,
* К сожалению, число имеющихся данных ограничено, точность определения час-
тот в некоторых случаях недостаточна.
If *63
Таблица 3
Характерные полосы ИК-поглощения монозамещенного циклогексанового кольца
в углеводородах и сульфидах (в слС1)
Т ип колеба- ния * Моноалкил- циклогекса- новые углеводо- роды [20] 0’4) 1 J—S— —С10Н21-Н 1 1 s II 1 XZ“S~\Z\ 1 II ч/ (h-Ci0H2i)2S СМ)
6 (СН2) 1350 с. 1345 с. 1343 с. ср. 1337 ср. 1349 сл. ср. 1345 ср. 1331 ср.
6 (СН2) 1260 с. 1266 с. 1263 с. 1262 с. — 1262 ср. 1252 ср. сл.
6 (СН2) 1200 ср. сл. 1205 с. 1203 с. 1198 с. — —
6 (СН2) 1230 ср. сл. 1185 ср. 1178 ср. сл. 1179 ср. сл. 1172 сл. 1189 ср. 1176 ср.
6 (СН2) 1105 сл. — — — — —
6 (СН2) 1020 ср. 1020 ср. сл. 1028 сл. 1024 с. ср. — 1024 с. ср.
6 (СН2) 950 ср. с. 1000 оч. с. 999 с. 999 с. — 1004 с.
г (СН2) 920 ср. сл. 920 оч. сл. 915 сл. — 920 оч. сл.
г (СН2) 890 оч. с. 890 оч. с. 887 с. 893 ср. 890 сл. 893 ср.
&(СС) 845 оч. с. 860 ср. сл. 825 ср. 859 ср. сл. 821 ср. 866, 858 826, 819 842 оч. сл. 852 ср.
V кольца 800—750 ср. сл. 752 ср. 740 ср. сл. 736 с. 734 ср. с. — 748 ср. сл.
* Приняты следующие обозначения типов колебаний: v — валентные, 8—деформационные,
г — м аятниковые.
Ge и Si, тогда как в моноалкилциклогексанах она смещена к 950 или
1020 см-.1.
Полоса 845 см-1, наблюдаемая в моноалкилциклогексанах, в цикло-
рексилсульфидах претерпевает расщепление на две полосы; в случаях
присоединения циклогексанового кольца к атомам С1, Вг и ОН-группе
она смещается к 810 см-1 [20].
Совокупность характерных ИК-частот позволяет определить наличие
монозамещенного циклогексанового кольца в сульфиде.
Согласно данным работ [11], интенсивная полоса или дублет при
1000 см-1 обнаруживается также в ряде спектров сульфидов общего
строения R (СНз)СН—S—R', что следует иметь в виду при анализе
систем, содержащих циклогексильный радикал.
б. Спектры КРС*
В литературе описан ряд спектров КРС алифатических сульфидов
[12—14, 29], в том числе ди-н-пропилсульфид (I) и ди-н-бутилсульфид
(II), однако данные разных источников довольно значительно различа-
ются между собой. . •
В спектрах сульфидов (I) — (IV)’ типа RSR', где R и R' — нормальные
углеводородные цепочки, обнаружены все основные линии, характерные
для нормальных парафинов [30] (табл. 4): поляризованные линии ва-
лентных полносимметрцчныгх колебаний, цепочки в области 810—
895 см-1 (р = 0,2—0,5); весьма слабые'линии деформационных колеба-
ний цепочки в области 203—506 см-1; линия деформационного колебания
* Таблицы спектров КРС вновь исследованных сульфидов см. ниже.
164
СН2-группы около 1300 см-1, интенсивность которой возрастает с увели-
чением числа СН2-групп; обычно дублетная линия деформационных ко-
лебаний СНз-групп в пределах 1440—1458 см-1.
Таблица 4
Характерные линии углеводородных колебаний сульфидов с нормальными
парафиновыми цепочками
(II) С,—S—С,
(III) С,—S—С7
(IV) C„-S-C2
Интерпретация
(I) Сз-S-C,
V, СЛ)-1
V, см~1
Валентные полно- 844
симметричные ко- 895
лебания цепочки
Деформационные 353
колебания цепочки 378
396
437
Деформационные
колебания групп
СН2
Деформационные
колебания групп
СНз
504
1294
1447
9 0,5
14 0,56
15 } °’4
0 —
10 ш. 0,5
0 —
15 0,81
49 дв. 0,86
842
872
895
337
355
4271
436/
1300
1446
0
20
25
7
7
10
0,50
0,52
822)
840/
867
894
363 1
407 I
506 J
807 6
357 0
432 0
506 3
27 0,64 1305
56 ш. 0,86 1440,
1458
34 0,78 1303
59 0,86 1446
23
53
о Р
V, см~*
В спектрах сульфидов (V) — (VII) типа RSR', где R — изоамильный
радикал, т. е. имеется изолированный третичный атом углерода, присут-
ствуют, помимо слабых линий деформационных колебаний в области
250—350 см-1, характерные для разветвленных углеводородов линии
около 850, 950, 1145 и 1180 см-1, а также линия деформационных коле-
баний СНз-групп (обычно двойная) около 1450 см-1 (табл. 5). В спектре
сульфида (V) линия 1145 см~г, по-видимому, перекрыта соседней широ-
кой интенсивной линией 1120 смт1. Линии около 760 см~х валентных
полносимметричных колебаний цепочки трудно отличить от линий анти-
симметричных валентных колебаний группы С—S—С.
В спектре сульфида (VIII) наблюдаются все линии, характерные для
моноалкилзамещенного циклогексанового кольца [30], линии валентных
симметричных колебаний остова 747—852 см-1, линии деформационных
колебаний остова 243 и 440 см-1, а также линии, характеризующие ко-
лебание шестичленного кольца— 1028, 1159—1178 (полоса), 1265,
1338 см-1.
Интенсивности всех перечисленных линий близки интенсивностям
линий соответствующих углеводородов.
В спектре КРС дициклогексилсульфида вызывают недоумение линии 1002 и
1590 см-1, из которых первая поляризована. Нами были исследованы три различных
образца дициклогексилсульфида, полученные различными методами. Интенсивности
каждой из указанных линий в препаратах заметно различаются, что заставляет счи-
тать их линиями примеси. Действительно, метод ГЖХ обнаруживает в данных препа-
ратах примеси, количество которой составляет в наиболее загрязненном препарате
около 10%. Уф-спектр содержит полосу с максимумом 260 ммк (6 = 700), которая, как и
линии КРС 1002 и 1590 см-1, характерна для ароматических соединений. Между тем
по условиям синтеза присутствие таких соединений исключается. Помимо этого,
в спектрах КРС всех трех препаратов отсутствует линия 3050 см-1, характерная для
фенильного кольца. Таким образом, вопрос о происхождении указанных двух линий
остается пока невыясненным.
165
Таблица 5
Характерные линии углеводорэдных колебаний сульфидов с изопарафиновыми цепочками
Интерпретация (v)L- с -с—s-с—с—L с (VI) | C-C-C-S-C-C-C с (VII) C-C-C-S-C-C=C а
V, СМ-"1 А р Л> р V, СМ-1 4 р
Валентные 791 15
полносиммет- 828 28 0,15 827 3 —— 825 0
ричные коле- 862 0 — 860 0 — 853 0 —
бания углерод- —- — — — — — 871 0 —
ного остова — — — 905 20 ш. 0,58 —
— — — 927 0 —- 930 12 —
960 16 0,82 954 6 955 10 0,8
Перект ыта линр ей 979 . 5 — 984 10 0,8
1120 см 1046 7 0,5 1147 12 0,6
1169 13 0,4 1180 8 • — 1181 0 —
Деформацион- ные колебания групп СНз 1454^дв. 53 0,82 1453 ш. 55 0,86 1451 39 0,86
В спектрах сульфидов (I)—IV) имеется интенсивная (Л> = 154-40)
деполяризованная линия 1030—1070 см-1, интенсивность которой в
спектрах нормальных парафинов обычно меньше (/о=5н-2О). В спект-
рах сульфидов интенсивность этой линии уменьшается при удлинении
углеводородной цепочки.
, • Таким образом, спектры исследованных сульфидов типа RSR', где R
и R' — алкильные радикалы, содержат все основные линии, характер-
ные для соответствующих углеводородов, причем интенсивность боль-
шинства линий имеет тот же порядок величины, что и для углеводоро-
дов.
В спектре сульфида (VII) обнаружена линия 1637 см-1, вызванная
валентным колебанием С = С-связи, и линия 1405 см-1, обусловленная
деформационным колебанием группы СН2, занимающей а-положение
относительно двойной связи [30]. Обе линии в сульфидах имеют более
низкие значения частот, чем в углеводородах соответствующего строе-
ния. Аналогичное явление имеет место в случае бензилаллилсульфида,
для которого наблюдаются частоты 1403 см-1 (48) и 1635 см-1 (135);
для углеводородов соответствующего строения частоты следующие:
1416—1418 см-1 (19—28) и 1642 см-1 (65—100).
В спектрах КРС всех рассмотренных сульфидов, а также в спектрах
ряда других сульфидов с нормальными и изопарафиновыми цепочками
(13, 31] присутствует деполяризованная линия Г417—1427 см-1, частич-
но перекрываемая линией — 1450 см~1. В спектрах насыщенных углево-
дородов такой линии не наблюдается [30].
Все алифатические сульфиды типа RSR' характеризуются интенсив-
ными поляризованными группами линий в области 630—720 см-1, обу-
словленными валентными симметричными колебаниями группы С—S—С.
Линии антисимметричных валентных колебаний группы С—S—С ле-
жат в области 700—800 см-1. Эти линии деполяризованы и обычно менее
интенсивны, чем линии симметричных колебаний. В частности, в спектре
дициклогексилсульфида их интенсивность снижена до /о=О. Венкате-
сваран [13] впервые отметил, что чем сложнее состав сульфида, тем
большее число линий валентных колебаний наблюдается в его спектре.
Вероятно (см. выше), они принадлежат разным стереоизомерам.
166
Линии деформационных колебаний v? группы С—S—С * лежат в об-
ласти 250—350 сл/-1, т. е. в той же области, что и деформационные ко-
лебания углеводородной цепочки. И те и другие в большинстве случаев
весьма слабы и трудно отличимы друг от друга.
Однако в спектрах диметилсульфида и ди-«-пропилсульфида линии
284 ** и 285 сл-1 *** соответственно имеют довольно высокую интен-
сивность.
Спектры КРС вновь исследованных сульфидов
типа RSR**** (Av, см~1).
I. Ди-«-пропйлсульфид СН3(СН2)2—S—(СН2)2СН3 285(20; 0,5),
353(9; 0,4), 378(15; 0,4), 369(0), 437(10 ш; 0,5), 504(0), 536(0), 628(0),
648(26; 0,18), 679(11; 0,6), 710(10), 737(21; 0,63), 752(12), 787(1?; 0,30),
844(9; 0,5), 895(14; 0,56), 1034(40; 0,56), 1095(11 ш; 0,30), (1137—1152—
—1165] (4; 0,3), [1207—1218] (4; 0,8), 1244(7; 0,8), 1294(15; 0,81),
1333(10; 0,56), 1358(6), 1378(9; 0,8), 1421(33; 0,96), 1447 (49 ш. дв;
0,86), 2735(9), 2845(31), 2872(140; 0,1), 2906(240; 0,4), 2133(210; 0,4),
2967(120;0,8).
II. Ди-м-бутилсульфид СН3(СН2)3—S—(СН2)зСНз 284(0, полоса)
335(7), 357(7), [427—436] (10; 0,6), 444(0), 631(10) [649 (25)—659 (22)—
—678.(18)] (0,30) 703(16), 730(20)—750(25; 0,59), 798(16; 0,30), 823(0),
842(0), 872(20; 0,50), 895(25; 0,54), 914(7), 940(0), 976(6), 922(0),
1005(7), 1019(0), 1053(35; 0,67), 1102(23; 0,33), 1138(6ш),[1188— 12Г5](5),
1224(4; 0,5), 1286(0), 1300(27; 0,4), [1338—1342](9), 1379(8), 1423(28;
0,8), 1446 (56; 0,86),2735(1),2856(120; 0,3),2872(160; 0,2),2906(250; 0,3),
2937(190, 0,3), 2963(100; 0,6).
III. Ди-м-гептилсульфид СН3(СН2)6—S—(СН2)6СН3 278(0), 363(4),
[407—506] (6, полоса), 524(0), 562(0), 655(12 ш д; 0,16), 672(7), [707—
734—754—770] (10; 0,53), [822—840] (5), 867(6), 894(9; 0,44), 955(0),
1024(11; 0,45), [1052(5) —1067(17)—1078(20)] (0,74), 1116(17; 0,47),
1171(6; 0,5), 1208(3), 1240(5 ш; 0,5), 1275(0) 1305(34; 0,78), 1338(9),
1371(0), 1417(0), [1440—1458](59; 0,86), 2727(0), [2853(150) —
—2875(150)](0,2), 2900(220; 0,4), 2939(160), 2961 (75; 0,5).
IV. н-Гексилэтилсульфид CH3(CH2)s—S—СН2СН3 230(0), 254(5 ш),
[332—342—357](0), 506(3), 594(5), 632(18) 656(36; 0,14), [669—690]
(23; 0,40), 744(12 д; 0,4), 770(9), 807(6), [846—855] (5), 889(7; 0,2),
910(0), 975(12 ш; 0,61), [1051 (15) —1072(13)] (0,3), 1113(12; 0,5) 1136(0),
1159(0), 1172(6 ш; 0,6), 1214(0), 1246(6), 1260(6), 1274(6), 1303(23;
0,75), 1335(0), 1348(0), 1379(7), 1419(0), 1446(53; 0,86), 2726(6),
2853(100; 0,2), 2868(140; 0,2), 2911 (210; 0,3), 2923(230;0,3),2967(100,0,6).
V. Диизоамилсульфид (СН3)2СН (СН2)2—S—(СН2)СН(СН3)2 206(6)
225(0), 255(0), 273(0), 314(0), 350(0), 385(6), 422(15; 0,26), 451(0),
465(0), 490(0), 658(9 ш; 0, 17), [678—690] (4 ш; дв), 710(0,5), 758(19;
0,27), 828(28; 0,15), 862(0), 922(6; 0,8), 960(16; 0,82), 996(5), 1034(6;
0,37), [1066—1079](5 ш), 1120(21; 0,52), 1169(13; 0,4), 1199(11; 0,3),
1226(6 ш; 0,8), 1278 (12;0,6), 1305(20; 0,6), 1340(26; 0,72), 1358(0),
1385(9), 1422(23; 0,77), 1454(53 дв; 0,82), 2662(6), 2720(10), 2843 (0),
2869(196; 0,2), [2902—2923](189; 0,3), 2961(183; 0,7).
* При отнесении рассмотренных сульфидов к группе симметрии Сгв.
** /0 = 6 (при 15-балльной системе интенсивностей) [31].
*** 10 = 20 (наши измерения).
**** Частоты линий даны в см-1, первое число в скобках представляет интенсив-
ность линии /о, второе — степень ее деполяризации р; д — диффузная линия, ш — широ-
кая, дв — двойная, р — резкая, ф — линия с сильным фоном, п — поляризованная, дп —
деполяризованная; разделение частот черточкой указывает на составной характер
линии.
167
VI. Изоамил-н-пропилсульфид (СН3)2СН(СН2)2—S— (СН2)2СН3
263(0), 278(0), 312(7; 0,2), 349(0 ш), 382(0 ш), [415—425](11), 456(0),
483(11; 0,1), 508(8; 0,4), 550(1), 574(0), 592(2), 617(6), 655(22 ш; 0,37),
689(30 ш; дв; 0,35), 734(12; 0,67), 756(8), [778(28)—791 (15)](0,53), 827
(3), 845(4 ш), 905(20 ш; 0,58), 927(0), 954(6), 979(5), 1003(4), 1033(17;
0,47), 1069(0), 1108(7 ш), 11'46(7; 0,5), 1180(6), 1221(0), 1242(10; 0,3),
1279(0), 1291(15; 0,54), [1321—1332](13; 0,41), 1386(10; 0,56), 1415(16),
1453(55 ш; 0,86), 2659(11), 2733(0), 2872(180; 0,2), 2908(190; 0,3), 2931
(170; 0,3), 2967(160; 0,7).
VII. Изоамилаллилсульфид (CH3)2CH(CH2)2— S— (СН2)2СН = СЬЬ
221(7 шд), 283(0), 293(0), 322(0), [344—353](И), 379(0), 419(26 ш; 0,37)’,
483(13; 0,2), 504(9), 529(0), 552(3), 580(0), 597(18; 0,23), 626(6),
663(23; 0,27), 689(50; 0,30), 747(65; 0,52), 767(0), 786(32; 0,51), 825(0),
853(1), 911(26; 0,81), 930(12), 955(10; 0,8),984(10; 0,8), 1004(7), 1018(3),
1070(7 ш; 0,3), 1110(7 д; 0,8), 1147(12; 0,6), 1165(0), 1184(15); 1201(30;
0,71), 1227(22), 1237(25; 0,64), 1271 (0), 1293(48; 0,37), [1320—1345]( 11),
1364(0), 1383(0), 1405(42; 0,65), 1427(0), [1451 (39) —1465(3)] (0,86),
1503(0), 1534(3), 1554(0), 1611(7), 1637(106; 0,23), 2657(0), 2726(5),
2870(120; 0,2), 2908(170; 0,2), 2935(110; 0,2), 2961(130; 0,8), 2977(120;
0,6), 3011(70; 0,2), 3075(33; 0,7).
VIII. Дициклогексилсульфид С6Нц—S—С6НП 204(0), 226(26; 0,27),
243(22; 0,23), 322(4), 376(13; 0,8), 440(17; 0,40), 471(5), 512(10 д; 0,5),
569(9), 663(0), 695(10; 0,4), 715(4), 747(44 ; 0,23), 784(0), 802(16; 0,30),
820(37; 0,30), 852(22; 0,34), 870(0), 888(0), 912(0), 940(0), 1002(29;
0,27), 1028(60; 0,78), 1049(19; 0,81), 1074(8), 1091(5), 1124(9; 0,7),
[1159—1184] (11; 0,81), 1205(22; 0,68), 1237(0), 1265(47; 0,78), 1294(19;
0,81), 1338(29 ш; 0,71), 1422(0), 1444(78; 0,85), 1590(5), 2656(10),
2849(250; 0,2), 2885(150 ш; 0,4), 2931 (320; 0,4).
Из рассмотрения изложенных выше данных о колебательных спект-
рах алифатических сульфидов следует, что в спектрах КРС наблюдают-
ся характерные группы интенсивных поляризованных линий в области
630—720 см-1 (симметричные валентные колебания С—S—С) и деполя-
ризованных линий в области 700—800 см-1 (антисимметричные валент-
ные колебания С—S—С).
Число их в ИК- и КРС-спектрах для данного сульфида связано с
числом возможных поворотных изомеров.
Для спектров КРС сульфидов с нормальными алифатическими ради-
калами характерна интенсивная деполяризованная линия 1030—
1070 см-1; в спектрах углеводородов соответствующего строения эта ли-
ния значительно менее интенсивна.
Частоты маятниковых колебаний полиметиленовых цепочек (СН2) п
в сульфидах снижаются при увеличении п аналогично тому, как это на-
блюдается для углеводородов.
В ИК-спектрах сульфидов с изопропильной группировкой (СН3)2СН—
маятниковые колебания СН3-групп наблюдаются, как и в углеводородах,
в области 1174—1169 см-1, если изопропильная группа отделена от ато-
ма серы метиленовым мостиком; если изопропильная группа присоеди-
нена к атому серы непосредственно, то частоты этих колебаний наблю-
даются в области 1149—1160 см-1.
Ножничные колебания СН2-групп и антисимметричные деформаци-
онные колебания СН3-групп, присоединенных к атому серы, наблюда-
ются при более низких значениях частот (1440—1410 сл«_1),чем в углево-
дородах, а также в случае тех сульфидов, в которых рассматриваемые
группировки отделены от атома серы метиленовым мостиком. Частоты
симметричных деформационных колебаний СН3-групп, непосредственно
присоединенных к атому серы, лежат в области 1305—1325 см-1, т. е. в-
168
области более низких частот, чем в случаях углеводородов и тех суль-
фидов, в которых СНз-группа не связана с атомом серы.
В спектрах КРС сульфидов наблюдаются в той же области (1417—
1427 см-1) деполяризованные линии, интенсивности которых падают с
увеличением молекулярного веса сульфида; в спектрах парафиновых
углеводородов такие линии отсутствуют.
Частоты дублета 1384, 1365 см~1, обусловленного деформационными
колебаниями изопропильной группы, в сульфидах не зависят от удален-
ности этой группировки от атома серы и совпадают с частотами углево-
дородов. В тех сульфидах, где один или оба радикала являются цикло-
гексановыми кольцами, в ИК-спектрах наблюдаются все частоты, харак-
терные для монозамещенных алкилциклогексанов, за исключением по-
лосы 950 см~\ которая в случае сульфидов смещается к 1000 см-1.
В спектре КРС дициклогексилсульфида наблюдаются все характер-
ные линии монозамещенных циклогексанов.
ВЫВОДЫ
1. Спектр КРС позволяет отличить алифатический сульфид от соот-
ветствующего ему по структуре углеводорода.
2. По спектру КРС можно установить, имеют ли радикалы сульфида
нормальное или изо-строение.
3. Спектры ИК и КРС позволяют обнаружить в сульфиде присутст-
вие монозамещенного циклогексанового кольца.
4. По ИК-спектру сульфида можно определить, присоединена ли изо-
пропильная группировка непосредственно к атому серы или отделена от
него метиленовым мостиком.
II. ИССЛЕДОВАНИЕ КОЛЕБАТЕЛЬНЫХ СПЕКТРОВ НЕПРЕДЕЛЬНЫХ
сульфидов и возможности ИХ ИСПОЛЬЗОВАНИЯ
В СТРУКТУРНО-ГРУППОВОМ АНАЛИЗЕ
Настоящий раздел работы посвящен исследованию частот поглоще-
ния в инфракрасной области, обусловленных валентными колебаниями
связи С = С и неплоскими деформационными колебаниями связи = СН
в группировках —НС = СН— в цис- и транс-форме, —СН = СН2,
^>С=СН2 и/С = СН — непредельных сульфидов. Для сравнения исполь-
зованы средние значения частот поглощения этих группировок в углево-
дородах. Обсуждаются данные для частот линий, соответствующих коле-
баниям двойной связи С = С в спектрах комбинационного рассеяния не-
предельных сульфидов.
А. Исследование спектров
Вопрос о влиянии атома серы на частотные характеристики непре-
дельных соединений до сих пор недостаточно освещен в литературе.
Спектральные данные накапливаются весьма медленно, что связано с
большими трудностями получения необходимых индивидуальных соеди-
нений в достаточно чистом виде.
Тарбел и Мак-Кол [32] впервые правильно отнесли частоту 938 см-1
пропенилфенилсульфида к деформационному колебанию группы—СН =
= СН— в транс-форме, тогда как эта группа в углеводородах поглоща-
ет при 980—960 см-1 [33].
В работе Шорыгина и др. [34] методом КРС исследована область
валентных колебаний связи С = С для шести а-непредельных сульфидов
и обнаружено значительное снижение частоты по сравнению с частота-
ми углеводородов соответствующего строения [35]. Сравнение с соответ-
ствующими частотами а-непредельных эфиров показало, что «группа
169»
SR сильнее, чем OR влияет на частоты С = С и С=С и на оптические
параметры молекул».
В работе Купина и Петрова [36] приводятся ИК- и ПМР-спектры че-
тырех а-непредельных сульфидов с одним незамещенным Н-атомом при
двойной связи. Авторы считают, что «делать какие-либо выводы на ос-
новании ИК- и ПМР-спектров не представляется возможным».
Настоящее исследование показало, что в сульфидах, подобно углево-
дородам, число и место заместителей атомов водорода у двойной связи
•определяет частоту ее валентного колебания и возможно влияет на ин-
тенсивность. Интенсивности полос в настоящей работе не рассматрива-
ются. Для сульфидов с a-положением атома серы относительно двой-
ной связи характерно поглощение в области 1620—1575 С.И-1; для 0-не-
предельных сульфидов поглощение наблюдается практически в той же
спектральной области (1674—1635 см-1), что и у соответствующих алке-
нов (1675—1638 см-1). То же, вероятно, будет справедливо для сульфи-
дов, у которых атом серы более удален от двойной связи (у, бит. д.).
В табл. 6 и 7 приведены частоты валентных колебаний связи С = С
индивидуальных а- и 0-непредельных сульфидов, со ссылкой на литера-
турный источник. Данные сгруппированы по числу и месту заместителей
(тип замещения) при двойной связи. Для легкости сопоставлений тут
же приведены средние значения валентных колебаний связи С = С в уг-
леводородах с аналогичным типом замещения.
Для ряда сульфидов в таблицы включены частоты колебаний двой-
ной связи С = С, полученные методом КРС.
Анализ данных позволяет обнаружить ряд характерных особенно-
стей, полезных в аналитической работе:
1) В непредельных сульфидах частоты колебаний двойной связи
С = С, измеренные методами ИК-поглощения и КР света, совпадают в
пределах точности измерений (структуры № 23, 34, 36).
2) Если атом серы заключен между двойной связью и ароматиче-
ским кольцом (структуры № 6, 18, 21, 22), частота С = С-валентного ко-
лебания наблюдается в тех же областях, что и в сульфидах с атомом
серы, заключенным между двойной связью и алкильным радикалом.
3) Если два атома серы занимают в молекуле положения а и а' по
отношению к двойной связи, то наблюдается аддитивное влияние каж-
дого атома на С = С-валентное колебание, выражающееся в удвоенном
смещении частоты к меньшим значениям (данные КРС для структуры
№ 26) в сравнении со смещением, вызванным одним а-атомом серы
(структуры № 23, 25).
Интересно подробнее остановиться на спектрах поглощения структур
№ 27—30 (в области 1597—1613 см-1), двойная связь в которых заклю-
чена между бензольным кольцом и атомом серы. Авторы [36] считают
невозможным делать какие-либо заключения о структуре молекул на
основании ИК- (и ПМР-) спектров, в частности, потому, что «полоса
двойной связи, сопряженная с фенильным радикалом, налагается на по-
лосу фенильного ядра».
Однако, как впервые отмечено Катаевой и сотр. [37] и подтверждав
ется нашими наблюдениями, сопряжение фенильного кольца с атомом
серы вызывает резкое увеличение интенсивности полосы 1580 см-1 и ис-
чезновение или весьма значительное уменьшение интенсивности полосы
1600 см-1 фенильного кольца. Аналогичное изменение интенсивности
1С = С-валентных колебаний фенильного кольца, сопряженного с двойной
связью, отмечается в работе [33] для углеводородов.
В ИК-спектрах рассматриваемых сульфидов полосы поглощения
1597—1613 см-1 можно считать обусловленными преимущественно ва-
лентными колебаниями С = С-связи, так как вклад полос 1600 см-1 фе-
нильного кольца должен быть мал вследствие сопряжения с атомом
серы.
170
Таблица 6
'Частоты ИК-поглощения и КР-света, вызванные валентными колебаниями связи С=С
при а-положеиии атома серы в непредельных сульфидах
№ соедине- ния Структура Частота полосы поглоще- ния, СМ~1 Смещение, Ду, см—1 Литерату- ра
А. Среднее’значение частот связи С=С группы —НС=СН2 в углеводородах
V. =1643±5 слГ1 (ИКС), 1642 сл-1 (КРС) [33, !5]
1 С-S—С=С 1587 56 [38]
1586* 57 [42]
2 и-С,—S—С=С 1588 55 —
-3 С=С—S—С=С 1588 55 [39]
4 С=С—S-С—С—S—С=С 1577 66 —
5 1580 63
S—с=с
Ph—S—С=С 1600 43 [40]
7 C=C-S—С-С 1586* 57 [41]
8 C=C-~S—Сз-н 1586* 57 [34]
9 С=С—S— 1586* 57 [34]
10 . С=С— S—Сотрет. 1586* 57 [34]
Б. Среднее значение частот связи С=С группы J)C= =СН2 в углеводородах
vB = 1653±5 см 1 [33]
11 С—С—S—С—Сотрет и 1600 53 —
II С
12 н-Сз—S—С—Сотрет н 1590 63 —
II С
13 н-С4—S—С—Сотрет 1595 58 —
14 С—C-—S-’—С—С-г“С—C——S—С—sC 1600 53 [41]
II II
с с
15 С—С—S—С-С—с—с=с 1600 53 [41]
II с
16 с—с—s^c—с—с—с=с—с II 1600 53 [41]
II с
17 с—с—S- О-С—С—С=С—с— с 1600 53 [41]
II с
18 Ph—S—С—Сотрет в смеси с 1602 51 —.
II с
Ph—S—С=С—Сотрет
(транс)
19 С—С—S—С—С 1612* [34]
|| 1609* [42]
С 1606* [42]
19а С—-С—S—Сз-н II
196 ;с С—С—-S >—С4 -н 1607* [42]
II С
Таблица 6 (окончание)
№ соедине- ния Структура Частота полосы поглоще- ния, СМ"1 Смещение, Ду, см—1 Литерату- ра
В. Среднее значение частот связи С=С транс-группы —НС=СН— в углеводородах
vB = 1673+5 (ИКС), 1674 см-' (КРС) [33, 35]
20 изо-Сз—S—С=С—Сз-изо 1614 59 —
21 Ph—S—С=С—С (смесь цис- и транс-изомеров) 1614 59 —
22 Ph—S—С=С—Сотрет (в смеси с 18) 1618 55 1—
23 н-С6—S—С=С—С^-н 1610 63
(смесь цис- и транс-изомеров) 1610* 61 —
Г. Среднее значение частот связи С=С цис-группы —НС—СН— в углеводородах
vr = 1657+5 см-1 (ИКС); 1653 см"1 (КРС) [33, 35], для циклогексена 1616 слС1 [33],
для 3-алкилзамещенных циклогексена 1651—1660 см1 [43]
24 изо-Cs—S—С=С—^Сз-изо 1610 47
25 О 1609* 48 [441
23 н-Cg-—S—С—С—С4-н 1610 47
(смесь цис- и транс-изомеров) 1610* 48 —*
26 С—С—S—С=С—S—С— С 1546* 55 (110 : 2) [341
22 Ph—S—С=С—С (смесь цис- и транс-изомеров) 1614 43 —
Д. Среднее значение частот связи С=С-группы \c=CHR в углеводородах
уц=1670 ±5 см 1 [33], для 1-алкилзамещенных циклогексена 1673 см 1 [43]
27 Ph—С=С—S—С 1 С 1597 73 [36]
28 Ph—С=С—S—С—С 1 С 1613 57 [36]
29 Ph—С=С—S—с—с с-с 1603 67 [36]
30 Ph—С=С—S—Ph 1 С 1613 57 [36]
31 S 1653 17 —г
* Получено методом КРС.
Шиманский [20] указывает, что в результате сопряжения двойной
связи с ароматическим кольцом наблюдается значительное снижение ча-
стоты валентного колебания С = С-связи, но не более чем на 30 см-1.
Можно с большой долей вероятности полагать, что в рассматривае-
мых сульфидах частота валентного колебания связи С = С имеет зани-
женное значение из-за сопряжения с ароматическим кольцом.
172
Таблица 7
Частоты ИК-поглощения и КР-света, вызванные валентными колебаниями связи С=С
при р-положении атома серы в непредельных сульфидах
№ •соеди- нения Структура Частота полосы поглоще- ния, СМ"1 Смещение Av, см~1 Литерату- ра
А. Среднее значение частот связи С=С группы —НС=СН2 в углеводородах
vA = 1643 ±5 смГ1 (ИКС); 1642 см-1 (КРС) {33, 35]
32 С—S—С—с=с 1643 9 [38]
33 С=С-С—S—с—с=с 1637 6 [45]
.34 С—С—С—с—S- с—с=с 1638 5
1 1637*
с
35 Ph—S-С—С=С 1640 3 —.
36 Ph—C-S—С-С=С 1631 12
1635* 7
37 о-СНз—Ph—S—С—C=C 1640 3 —
38 Ph—S—С—C=C 1 1639 4 ]46]
1 c
Б. Среднее значение частот связи С=С-группы ifuc-3-алкилзамещенных циклогексенов
vB =1654+6 см-1 (ИКС) 143]
39 40 Y Y С/Э 00 00 1640 1642 14 12 [45] [45]
В. Среднее значение частот связи С=С группы транс-НС=СН — - в углеводородах
vB = 1673 + 5 см-1 (ИКС); 1674 см-1 (КРС) [33, 35]
41 С—С=С—С—S—С—С=С—С 1674 —1 [45]
42 Ph—S—С—С=С—С 1670 3 [46]
* Получено методом КРС.
В сульфиде № 31, который имеет тот же тип замещения двойной
связи, что и сульфиды № 27—30, но в котором отсутствует фенильное
кольцо, валентная частота наблюдается при 1653 см-1.
В табл. 8 приведены литературные и полученные нами частоты не-
плоских деформационных колебаний Н-атомов при двойной связи в
сульфидах и углеводородах родственного строения. Для каждого суль-
фида указано отклонение Av см-1 частоты деформационного колебания
от среднего значения соответствующей частоты в углеводородах.
Разброс величин этих отклонений внутри групп сульфидов родствен-
ного строения в случае деформационных колебаний больше, чем в слу-
чае валентных колебаний, что согласуется с данными для углеводородов
[33] и объясняется значительным влиянием структуры молекулы на эти
частоты.
Группа — НС = СН2 в а-непредельных сульфидах вызывает погло-
щение в областях 960—948 и 889—855 см-1, алкены поглощают при
995—985 и 915—905 см-1. Следовательно, a-положение серы относи-
тельно двойной связи снижает частоты деформационных колебаний.
В сульфиде № 3 атом серы сопряжен с двумя а- и а'-непредельными
группировками, частоты поглощения которых 6=сн = 960 см~1 и 6=сн2=
= 883 и 874 см-1 имеют, однако, более низкие значения, чем углеводоро-
173
Таблица 8
Частоты ИК-поглощення, вызванного неплоскимн деформационными колебаниями грушг
=СН— и ==СН2 при а- и р-положениях атома серы в непредельных сульфидах
(см-1)
Положение
двойной
связи
№
соединения
Частота
8=с—н>сл1-1
Смещение
относительно
8д, см—1
Частота Смещение
_ , относительно
5=СН2> см 8а<, см—1
А. Средние значения частот колебаний группы - -НС=СН2 в углеводородах
«А = 990 ±5 см -1; бА, =910±5 см-1 [33]
а 1 952 38 855 55
а 3 960 30 883, 874 27,36
а 6 948 42 889 21
3 32 990 0 916 —6
3 33 986 4 916 —6
3 34 988 2 915 —5
3 35 987 3 917 —6
3 36 991 —1 918 —8
3 37 976 -И 917 —7
3 38 987 3 919 —9
Положе- ние двой- ной связи № соедине- ния Частота полосы поглощения, СМ-1 Смещение относительно 8g, см—1 Положе- ние двой- ной связи № соедине- ния Частота полосы поглоще- ния, см—1 Смещение относи- тельно 8 g, см—1
Б. Среднее значение частот колебаний группы \с=СН2 в углеводородах бБ =891 ±6 см-1 В. Среднее значение частот колебаний» группы транс-НС=СН—. в углеводо-
а а а а а а а а Г 11 12 13 14 15 16 17 18 . Область п ifuc-HC=Cf" бг = 800- [33] 860 31 857 34 860 31 847 44 847 44 847 44 846 45 875 26 оглощения группы — углеводородов родах а а а . Ct 3 ₽ Д. Область группы г- 6д= 840— 6в = 970±10 см-1 [33]
20 21 43* 23 41 42 деформ -НС=С -.790 см 941 29 937 33 910 30 (60: 2)' 938 32 961 9 933 37 961 9 934 36 ационных колебаний S углеводородов -1 [33]**
-650 см-1 [33]
Положение двойной связи № соединения Частота полосы поглощения, см—1 Положение двойной связи № соединения Частота полосы поглощения, СМ—1
а 24 732, 725, 703 а 31 776, 920
а 23 750, 725, 702 а 27 *** 915
3 39 759, 735, 723 а 28 *** 910
3 40 762, 738, 723 а 29 *** 900, 930
а 30 *** 920'
П р и'м е ч а н и е. Структуры и ссылки на литературу см. в табл. 6 и 7.
* Структура соединения 43: С—S—С=С—S—С [46],
•• 1-Замещенные циклогексены поглощают при 8д=800 и 8д< — 920 см—1 [43].
*•* Полоса 8д перекрывается областью поглощения фенильной группы.
174
ды. В сульфиде № 1 атом серы сопряжен с одной непредельной группой,,
для которой частоты 6=сн = 952 и б=сн2=855 см-1 имеют более низкие-
значения, чем в сульфиде № 3, что, возможно, объясняется более силь-
ным влиянием атома серы на единственную непредельную группировку.
В сульфиде № 6 S=CH = 948 и б=сн2 = 889 см-1. Не исключено, что на:
частоты этих колебаний оказывает влияние бензольное кольцо, связан-
ное с атомом серы.
В сульфидах p-ряда обнаруживается поглощение в интервалах час-
тот б=сн=976—991 и S=Ch2=916—919 см-1, причем среднее значение
частоты б=сн2 несколько выше соответствующего среднего значения в
углеводородах.
Группа /С = СН2 в а-непредельных сульфидах поглощает в обла-
сти более низких частот 875—846 см-1, чем у алкенов соответствующего,
строения — 885—897 см-1.
Сульфиды № 11—13, весьма близкого строения, поглощают в узком:
интервале 857—860 см-1; сульфиды № 14—17, в структуре которых
имеется тройная или двойная связь в 6-положении к атому серы, погло-
щают при 847 см-1. Сульфид № 18 поглощает при 875 см-1; относительно-
высокая частота поглощения, возможно, обусловлена фенильным заме-
щением у атома серы.
Спектрами сульфидов ₽-ряда с группировкой/ С = СН2 мы не распо-
лагаем.
В сульфидах с транс-группой —НС = СН— при а-положении атома
серы относительно двойной связи деформационные колебания наблюда-
ются при 937—940 см-1; частоты углеводородов в среднем на 30 см-1
выше. '
В бис-сульфиде С—S—С = С—S—С а- и а'-атомы серы вызывают
вдвое большее снижение: деформационной частоты, чем один а-атомсерьг
в прочих сульфидах этого типа; деформационное колебание проявляется
при 910 см-1. Сульфиды № 21 и 23 представляют собой смеси цис- и
транс-изомеров. Возможно, что цис-структуры вызывают поглощение в.
рассматриваемой области, как это нами наблюдалось для структуры
№ 24, но интенсивность его значительно меньше, чем в -спектрах транс-
структур.
Следует отметить появление в спектрах соединений № 20 и 23 группы
полос с максимумом около 815 смт1, которая, возможно, повторится в-
спектрах цис-сульфидов.
Мы располагали только двумя спектрами p-непредельных сульфидов^
с транс-группировкой —НС = СН— (№ 41 и 42), причем степень чистоты
этих соединений неопределенна. Полоса 961 см.-1, очевидно, вызвана не-
плоскими деформационными колебаниями Н-атома при двойной связи и;
не выходит из области частот, характеризующей поглощение в алкенах.
Происхождение полосы 933 см-1 неясно, если ее не относить к примеси
изомера а-ряда.
Прежде чем рассматривать область деформационных колебаний;
сульфидов, содержащих цис-группировку —НС = СН—, остановимся на
обсуждении данных для углеводородов, содержащих эту группировку.
Наканиси [47] указывает область 730—675 см~'- для алкенов и их произ-
водных и 800—650 см-1 (с несколькими полосами равной интенсивности)
для цикленов, причем отнесение этих полос ему неясно. Расчет цис-ди-
замещенных этилена, произведенный Свердловым [48], дает в низкоча-
стотной области полосы 685, 894, 88Г и 972 см-1.
Эйзен [43] для 3-замещенных циклогексенов обнаружил характерное-
поглощение при 721, 697 и 674 см-1.
Число сульфидов цис-ряда, охарактеризованных ИК-спектрами, не-
175.
достаточно для надежного выявления полос, определяемых наличием
рассматриваемой группировки, поэтому мы были вынуждены использо-
вать спектры смесей цис- и транс-изомеров соединений № 21 и 23.
Можно предположить, что для а-непредельных сульфидов ^ас-ряда
характерны полосы при 935, 725, 703 смг\ а для p-непредельных суль-
фидов этого ряда — при 759, 735 и 723 см~1. Не исключено, что некото-
рые из указанных полос вызваны С—S-валентными колебаниями.
Группировка с = СН— в алкенах вызывает поглощение в интер-
вале 840—790 смгх [33], в цикленах наблюдаются две полосы в интерва-
ле 850—790 см~х [47]. В спектрах 1-замещенных циклогексенов были об-
наружены две характерные полосы — при 800 и 920 см —1 [43].
В спектре сульфида № 31 имеются интенсивные полосы 920 и 776
сл/-1, соответствующие, по-видимому, полосам 920 и 800 см~1 углеводо-
родов родственного строения (1-алкилзамещенных циклогексенов). Бо-
лее низкое значение полосы 776 сл/-1 в сульфиде вместо 800 сл/-1 в угле-
водородах обусловлено, очевидно, влиянием а-атома серы на группиров-
ку \с=сн—.
' Таблица 9
Характерные частоты непредельных сульфидов
Углеводород а-Непредельные сульфиды З-Непредельные сульфиды
структура, частота (см—1) структура, частота (см—1) Av/2 структура, частота (см—1) Avl,3
Валентные колебания
rch=ch2 rsch=ch2 rsch£ch=ch2
1643+5 1587 + 10 56 1639+5 4
r2c=ch2 RSCR'=CH2 RSCH2CR'=CH2
1653+5 1598+10 55
RCH=CHR' RSCH=CHR' RSCH2CH=CHR'
(транс) (транс) (транс)
1675+5 1614 + 5 59 1672+5 1
RCH=CHR' RSCH=CHR' RSCH2CH=CHR'
(Цис) (цис) (Цис)
1656 + 5 1610±10 46 1641 (циклич.) 15
Неплоские деформационные колебания
rch=ch2 rsch=ch2 rsch2ch=ch2
990± 5, 953+6, 37 936+5, 4
910+5 874 + 13 36 917 + 2 7
r2c=ch2 RSCR'=CH2 RSCH2CR'=CH2
890 + 6 855±8 36 — —
RCH=CHR' RSCH=CHR' RSCH2CH=CHR'
(транс) (транс) (транс)
970+10 939+2 31 961 + 1 9
В спектрах сульфидов № 27—30 [36] наблюдается поглощение около
915 и 845 сл/-1. Однако интересная для нас область ниже 780 сл/-1, в ко-
торой можно ожидать появление полосы, характерной для а-непредель-
ной группировки, в исследуемых соединениях маскируется поглощением
фенильного кольца.
Полученные результаты в связи с актуальностью задачи применения
метода ИК-спектроскопии к анализу непредельных органических суль-
фидов, несмотря на ограниченность исходных данных, представлялось
176
6
Рис. 1. Структурночастотная
корреляция для С=С алентных
и С—Н неплоских
деформационных колебаний
а- и ;(3-непредельных сульфидов
а — область валентных колебаний;
б — область деформационных коле-
баний
Принятые обозначения: «—» — интер-
валы частот; О — непредельные уг-
леводороды; • — а-непредельные
сульфиды; ® — Р-непредельные
сульфиды
ш=сн2 о о
R2C=CHr
RHC-CHR транс •
Ж7
а
ннс=сн2
r2c=ck2
RHC=CMR цис о
RH£=CHR транс ♦ -<8
R2C=CHR • С
/550 /600 ' /650 /700
V,CM~r
целесообразным подвергнуть простейшему усреднению. Средние данные
для валентных и деформационных частот а- и p-непредельных сульфидов
собраны в табл. 9 и наглядно представлены на рис. 1, демонстрирую-
щем, что ряд частот разных структур взаимно налагается. Это наложе-
ние необходимо учитывать при анализе методом ИК-спектроскопии.
Смеси непредельных сульфидов a-ряда с непредельными углеводоро-
дами аналогичны по ИК-спектрам в области валентных колебаний
С = С-связи смесям сульфидов a-ряда с непредельными сульфидами
p-ряда. Смеси непредельных сульфидов p-ряда с углеводородами практи-
чески неразличимы методом ИК-спектроскопии. Сульфиды у, 6 и после-
дующих рядов (с большим удалением атома S от двойной связи), по-
видимому, как правило, будут аналогичны сульфидам р-ряда.
Аналитические возможности ИК-спектроскопии, вытекающие из рас-
смотренных выше закономерностей, поясняются ниже разбором некото-
рых конкретных примеров.
Б. Примеры применения ИК-спектроскопии к анализу
непредельных сульфидов
Установленные для непредельных сульфидов структурно-частотные
корреляции оказались весьма полезными при решении ряда практиче-
ских задач *.
Пример 1
Методом препаративной газо-жидкостной хроматографии из двухкомпонентной
смеси предполагаемых стереоизомеров 2,8-диметил-5-тианонена-3 были выделены от-
дельные компоненты, каждый нз которых давал один симметричный пнк на хромато-
грамме. На основании ИК-спектров одному из компонентов была приписана транс-
конфигурацня (структура № 20), другому — чис-конфигурация (структура № 24) **.
В ИК-спектре сульфидов № 20 н 24 наблюдались соответственно полосы поглоще-
ния 1613, 1610 и 941, 935 см~'. Полоса 1610 см-1 обладала примерно в 2 раза большей
интенсивностью, чем 1613 см-1, что можно объяснить меньшей симметрией структуры
№ 24 по сравнению с № 20 (см. табл. 6). Это обстоятельство указывает на транскон-
фигурацню структуры № 20 и чис"Конфигурацню структуры № 24. Полоса 941 см-1
примерно в 2 раза интенсивнее полосы 935 cjh-1, что также указывает на транс-конфн-
гурацию структуры № 20 и цис-конфигурацию структуры № 24. Подтверждение этого
* Следует иметь в виду, что некоторые а-непредельные сульфиды при хранении
претерпевают изменения, сопровождающиеся появлением в ИК-спектрах полосы пог-
лощения в области 1720—1750 см-1, интенсивность которой возрастает с увеличением
длительности хранения.
** Спектры ПМР были получены Г. Ю. Пеком в ИХПС АН СССР на спектрометре
JEOL; в качестве растворителя использован СС14.
12 Методы анализа 177
заключения было даио методом ПМР. В спектре ПМР каждого сульфида был обна-
ружен двухпротонный квинтет в области 5,8—6,6 м. д., свидетельствующий о наличии
двойной связи; константа спин-спинового взаимодействия I = 18 гц для сульфида № 20
и I — 10 гц для сульфида № 24, что позволило уверенно приписать цис-конфигурацию
соединению № 24 и транс-конфигурацию — соединению № 20 [49].
Пример 2
Пропеиилфенилсульфид (структура № 21), дававший два пика на газо-жидкостной
хроматограмме, исследовали ИК-методом. Требовалось установить, нет ли в нем при-
меси аллилфенилсульфида. Обнаруженная в спектре полоса 1614 см~х могла быть
вызвана валентными С = С колебаниями группы —НС = СН—, занимающей «-положение
по отношению к атому серы как в цис-, так и в транс-конфигурациях. Сильная полоса
937 см~х также свидетельствует об a-положении этой группировки по отношению к
атому серы. Очевидно, структура с 0-положением атома серы относительно двойной
связи отсутствует в ощутимых количествах, так как нет характерных для нее полос
поглощения. Отсюда следует, что исследованный продукт в основном является смесью
цис- и транс-изомеров пропенилфенилсульфида.
Пример 3
При исследовании гексйлгексенилсульфида (структура № 23) методом газо-жидко-
стной хроматографии обнаружено два пика почти равной интенсивности.
В ИК-спектре полоса 1610 см~х, по-видимому, была вызвана С = С-валентными ко-
лебаниями как цис-, так и транс-форм группировки —НС=СН—, занимающей «-поло-
жение относительно атома серы. Полоса 938 см~х характерна для неплоского деформа-
ционного колебания этой группировки. Сильная полоса 702 см~х, возможно, характери-
зует цис-группировку. Следовательно, данный продукт — смесь цис- и транс-изомеров
гексилгексенил-а-сульфида и не содержит ощутимых примесей непредельных 0-суль-
фидов.
Пример 4
Необходимо определить положение двойной связи в продуктах дегидратации 2,4-ди-
метилтиадеканола-4 (I) [50].
НО СН3
S СНз
(IV)
S СНз
(Ш)
S СНз
(VI)
S СНз
(V)
Теоретически представлялись возможными три направления реакции, приводящей
к образованию у- и (3-иепредельных сульфидов (II), (III) и (IV). Однако уже первая
съемка ИК-спектров в области валентных колебаний показала, что, кроме частот этих
непредельных сульфидов, в смеси продуктов дегидратации отчетливо проявляется ча-
стота 1606 см~х, которую можно было связать с присутствием сульфидов «-ряда.
Метод газо-жидкостной хроматографии также показал, что в продуктах дегидрата-
ции в заметных концентрациях присутствуют не три, а по крайней мере пять соедине-
ний; образование такого числа соединений можно было объяснить, допуская вторич-
ную миграцию двойной связи из (3- в a-положение с образованием сопряженной груп-
пировки— S — С-С<. Последнему типу структур отвечают формулы (V) и (VI). По-
виднмому, рассматриваемые структуры охватывают все возможные изомеры, образую-
щиеся в соизмеримых концентрациях при данном синтезе.
Число этих структур соответствует числу основных пиков ГЖХ. По-видимому, каж-
дый из пиков относится к одному из рассмотренных индивидуальных соединений
(II) — (VI). Количество вещества, имевшегося в нашем распоряжении, было недоста-
точно для выделения каждого из этих соединений в чистом виде.
Рассмотрение структур показывает, что соединение (II) содержит группировку
>С=СНз в у-црложении относительно атома серы; (IV) и (VI) содержат группы
>С=С<, а (III) и (V) —группы >С=СН— в а- и 0-положениях.
Четырехзамещенцая двойная связь, имеющаяся в структурах (IV) и (VI), по-види-
мому, в ИК-области также не обладает характеристическими частотами высокой ин-
тенсивности, как и в соответствующих углеводородах, на что указывалось выше. По-
178
этому при дальнейшем рассмотрении мы не учитывали возможный вклад этих струк-
тур в указанные спектры.
Естественно, что для смеси изомеров ИК-спектр в валентной и деформационной
областях оказался весьма сложным; однако попытка его интерпретации представляла
существенный интерес в связи с отсутствием данных по ИК-спектрам сульфидов
с группировкой >С=СН—.
Для углеводородов типа R2 C = CHR характерна частота 1670 см~1. По-видимому,
наблюдаемая частота 1667 см-1 связана со структурой (III), относящейся к (3-ряду.
Нами показано, что для сульфидов |3-ряда других типов структур не существует час-
тотных различий между углеводородами и сульфидами. С известной степенью прибли-
жения можно принять, что в спектре у-сульфида (II) семициклической двойной связи
(которая аналогична семициклической связи соответствующих углеводородов) отвеча-
ет наблюдаемая частота 1644 см-1. Частоту 1606 см-1, по-видимому, можно связать со
структурой (V), относящейся к a-ряду, для которого наблюдается существенное сниже-
ние частоты по сравнению с сульфидами p-ряда и углеводородами соответствующего
строения.
В ИК-спектре исследуемого продукта наблюдались интенсивные полосы 972,893,886,
850, 823 см-1, некоторые из которых, возможно, обусловлены деформационными коле-
баниями = С—Н-связи.
Неплоские деформационные колебания группы =СН в углеводородах с семицикли-
ческой двойной связью обычно наблюдаются около 890 см-1; предполагая, что частота
этого колебания в у-непредельном сульфиде (II) не должна значительно отличаться от
углеводородной, мы можем приписать деформационному колебанию сульфида (II) час-
тоту 893 или 886 см-1. Вторая из них может быть вызвана пульсационным колебанием
кольца. Частоты 850 и 823 см-1 с некоторой долей вероятности могут быть приписаны
деформационным колебаниям (3-структур (III) и (IV) соответственно.
С большей' достоверностью это отнесение можно будет сделать тогда, когда ока-
жутся доступными ИК-спектры индивидуальных непредельных циклических сульфидов
соответствующего строения.
Исходя из сказанного выше можно полагать, что в исследуемой сложной смеси
присутствуют структуры (II), (III), (IV): не исключено присутствие структур (IV) и
(VI), которые ие могут быть охарактеризованы с помощью ИК-спектров. Вероятно,
что для сульфидов с группировкой ^>С = СН—, относящихся к [3,-ряду, характерны ча-
стоты вблизи 1670 и 850 см-1, а для относящихся к а-ряду — вблизи 1610 см-1.
ВЫВОДЫ
1. Валентные vc=c и деформационные 6=Сн, 6=СНг -колебания а-не-
предельных сульфидов наблюдаются при более низких частотах, чем у
соответствующих углеводородов.
2. Валентные vc=c и деформационные 6-колебания (3-непредельных
сульфидов и углеводородов родственного строения наблюдаются прак-
тически в одинаковых областях.
3. Частоты vc=c -валентных и 6 деформационных колебаний непре-
дельных алкил-сульфидов в основном определяются числом и положе-
нием насыщенных замещающих групп при двойной связи.
4. Частоты vc=c -валентных колебаний непредельных сульфидов, по-
лученные методами ИК-поглощения и КР света, совпадают в пределах
точности измерений.
5. Частоты валентных колебаний цис- и транс-изомеров группы
—НС = СН—для а-непредельных сульфидов совпадают в пределах точ-
ности измерений.
6. Если а- и а’-положения относительно группировки —НС = СН—
занимают атомы серы, снижение частот как валентных, так и деформа-
ционных колебаний вдвое больше, чем в случае присоединения к этой
группировке только одного атома серы.
7. Если один атом серы заключен между двумя непредельными груп-
пировками С = С, снижение частоты валентного колебания этих группи-
ровок практически не отличается от случая, когда атом серы присоеди-
нен только к одной непредельной группировке.
8. Частоты неплоских деформационных 6 = СН-колебаний сульфидов
больше зависят от всей структуры молекулы, чем частоты валентных
колебаний vc=c-
9. Если атом серы заключен между двойной связью и ароматическим
13 Методы анализа 179
кольцом, частота валентного колебания связи С = С наблюдается в той
же области, что и у сульфидов с алкильным радикалом на месте арома-
тического кольца.
10. Если двойная связь С = С заключена между ароматическим
кольцом и атомом серы, частота валентного колебания двойной свя-
зи уменьшается вследствие взаимодействия с каждой из этих группи-
ровок.
11. Колебательные спектры непредельных сульфидов можно эффек-
тивно использовать в структурно-групповом анализе. Весьма полезно'
сочетание методов ИК-, КРС-, ПМР-спектроскопии с газо-жидкостной
хроматографией.
III. ИССЛЕДОВАНИЕ КОЛЕБАТЕЛЬНЫХ СПЕКТРОВ
АРОМАТИЧЕСКИХ СУЛЬФИДОВ
А. Обзор литературы
В ряде работ [8, 11, 12, 37, 51—54] приводятся колебательные спект-
ры или таблицы частот сульфидов типа RSR', где R—ароматическое
кольцо. Структурные формулы сульфидов, спектры которых рассмотре-
ны ниже, и ссылки на соответствующую литературу представлены в
табл. 10.
Шишкина [51], сопоставляя спектры КРС фенилциклогексилсульфи-
да со спектрами дифенилсульфида, фенилметилсульфида и фенилэтил-
сульфида, отметила в них ряд общих частот, близких по величине к со-
ответствующим частотам в спектрах монозамещенных бензолов. Катае-
ва и сотр. [37] в ИК-спектрах сульфидов с группировкой фенил — сера
обнаружили весьма интенсивную полосу около 1585 см-1, тогда как по-
лоса 1605 см—1, обычно весьма интенсивная в спектрах алкилбензолов,
отсутствовала или была очень слаба; подобная инверсия интенсивностей
полос 1605 и 1585 см-1 отмечена Беллами [55] при сопряжении аромати-
ческого кольца с двойной связью С = С.
В работе Кулика, Егорова и др. [56], посвященной исследованию ИК-
спектров и полярных эффектов в пара-производных бензола, содержа-
щих группы SCF3, SOCF3, SO2CF3, интенсивная полоса 1080± 10 см-1
отнесена к колебанию арил—-сера.
Интерпретация колебательных частот в ИК- и КРС-спектрах ме-
тилфенилсульфида и этилфенилсульфида дана в работе [57].
Б. Обсуждение результатов
а. ИК-спектры
Шиманский [20] указывает на ряд ИК-частот моноалкилзамещенно-
го бензольного кольца, нечувствительных к структуре заместителя. Близ-
кие частоты обнаруживаются в полученных нами спектрах ароматиче-
ских сульфидов с монозамещенным бензольным кольцом (табл. 11, А):
695±5 см-1 (оч. с.), 739±6 см-1 (оч. с.), 837±8 см~х (оч. с.);
902+15 см-1 (сл.), 1028+7 см-1 (пер. инт.), 1072 + 8 см-1 (ср.);
1155±5 см-1 (сл.), 1181 ± 10 см~ (сл.), 1235 + 5 см~х (пер. инт.),
1321 + 12 см-1 (сл.), 1443+10 см-1 (с.).
В ИК-спектрах тех сульфидов, в которых атом серы отделен от бен-
зольного кольца метиленовым мостиком, наблюдаются, как и в моно-
алкилбензолах, полосы при 1494 + 5 см~' (оч. с.), 1584 см-1 (сл.), 1606
см -1 (ср.). Первая из этих полос в спектрах сульфидов с атомом серы,
непосредственно присоединенным к бензольному кольцу, имеет несколь-
ко сниженное значение частоты—1479 + 3 см~\ полоса 1585 см-1
имеет весьма значительную интенсивность, а полосы 1605 см-1 не наб-
людается.
180
Таблица 10
Ароматические сульфиды, колебательные спектры которых использованы в данном
сообщении
(Ph — фенил или фенилен)
Формула Литература по спектрам Формула Литература по спектрам
ик крс ик КРС
Ph—S—Ph* Данное [51,52] Ph—CH2S—СН2—СН= Дани ое
сообщение, [И] =СН2** сообщение
Ph—S— СН2—СН=СН— [37]
PhCH2—S—CH2Ph [И] [12,52] -CHg
Ph—S—CHg [8] [51, 52, (смесь цис- и транс-)
54, 57] Ph—S—СН2—СН=СН — [37] —
Ph—S—C2H5 — [51, 54, 57] —СН2—Ph (смесь цис- и транс-)
Ph—S—С3Н7-Я ** Данное сообщение, Данное сообще- я-CHg—Ph—8-СзН7-я** Данное сообщение
[Ю] ние o-CHg-Ph—S-СюНл-я** То же
Ph—:S—CgHi?-# [8] — (вероятна примесь «-изо- мера)
Ph—S—СюН21-н [8] — о-CHg— Ph—S—С3Н7-Я** Данное —
Ph—.S—C6HU (ЦГ) — [51] (с примесью я-изомера) сообще- ние
Ph—CH2—S—CH3 [8] — о-CHg—Ph—S—СН2СН= То же
Ph—CH2—S—Ph Ph—CH2—S—Ph—СН3-я [53] — =СН2** (с примесью я-изомера)
[53] — [36]
Ph—(CH2)2—S—Ph* ** Ph—СН=С(СНд)—S-CHg
Данное сообщение — [36] —
Ph—CH=C(CHg)—S— -c2H5
Ph—(CH2)2—S—CH3 [8] — [36] —
Ph—СН=С(С2Н5)—S—
Ph—S-CH=CH—CH3** (смесь цис- и транс-) Данное сообщение, — -C2H5
[37] Ph—CH=C(CHg)—S—Ph [36] —
Ph—S—СН2—СН= Данное —
=CH2** сообщение,
[И]
* Синтезирован Е. Н. Карауловой (58].
** Синтезированы В. Г. ЛукьяницеЙ и Г. Д. Дергуновой по методикам, приведенным в ра-
боте [4].
*** Синтезирован В. Я. Родионовым.
В табл. И, Б и В представлены полученные нами данные о погло-
щении сульфидов RSR', где R—1,2- или 1,4-дизамещенное ароматиче-
ское кольцо, и для сравнения даны частоты характерного поглощения
1,4- и 1,2-дизамещенных алкилбензолов [59].
В области 700—1450 см-1 сульфиды и алкилбензолы имеют характер-
ные полосы поглощения в аналогичных областях, в области 1475—
1650 см~х поглощение сульфидов зависит от строения групп, связанных с
атомом серы.
1,4-Дизамещенные алкилбензолы имеют сильную полосу поглощения
в области 1521—1510 см-1 и слабую в области 1656—1633 см-1. В ИК-
спектре н-пропил-п-толилсульфида наблюдается сильная полоса при
1496 см-1 и слабые полосы при 1566, 1600 и 1640 см-1.
13*
181
Сравнение частот ароматических углеводородов и сульфидов
Соединение Характерные
Моноалкилбензолы [59] 691+7 751+15 837+10 908±10 сл. ср. сл. 962+6 СЛ. 982+6 1001±1
оч. с. оч. оч. с. оч. сл. пер.инт.с.
А. Сульфиды с монозамещенным
Ph—S—Ph Ph-^S—СНз Ph—S—СзН7-« Ph—S—>CH=CH—CH3 Ph—S—CH2—CH=CH2 Ph—S—CH2—CH=CH— -CHS ph—S—.CH2—CH=CH— —CH2—Ph Ph—CH2—S—СНз Ph—CH2—S—CH2—Ph Ph—CH2—CH2—S—"Ph Ph—CH2—CH2—S—CH3 Ph—CH2—S—CH2—CH = —CH2 700 оч, с. 745 оч. с. 691 оч. с. 739 оч. с. 695 оч. с. 740 оч. с. 700 оч. с. 733 с. 690 оч. с. 733 оч. с. 842 сл. Нет 834 сл. » 831 сл. 898ср.сл. 829 оч. сл. 894 ср. сл. 845 оч.сл. Налож. =СН деф. 970 сл. 967 ср. Нет » > Нет 1005 ср. » 999 оч.сл. » Нет « > Налож. 1010 сл. =СН деф.
Нет Нет
699 оч. с.725 оч. с. 699 оч. с. Нет 690 оч. с. 745 оч. с. 700 оч. с. 740 оч. с. 700 оч. с. 737 с. 829 оч.сл. 912 ср. 830 сл. 915ср.сл. 845 оч.сл. 920 оч. сл. Нет Нет 829 сл. 894оч.сл. 959 ср. 945 оч. сл. 945 оч.сл. 965 ср. Нет 972 ср. 1002 сл. 990 сл. Нет 975 оч.сл. 1010 сл. 977ср.пл. 1005 ср. сл. Нет Нет
i ,4-Диалкил бензолы [59] n-CH3Ph—S—СзНу-н 860—800 с. 810 оч. с. 1000—1070 сл. (2 полосы) Б. Су 1020 с. ср. льфид с | 1100 с. 1125-1090 сл. 1,4-дизамещенным | 1123 ср.
1,2- Диалкилбензолы [59] e-CH3Ph—S—iCk)H2i-h e-CHsPh^S—CH2—CH= =CH2 e-CH3Ph—S—C3H7-M * Согласно данным рабо щения в областях 1525—1475 зольного скелета. *• Валентн 770—735 с. 748 оч. с. 746 оч. св 748 оч. с, гы [20]? не исключен см~* и 1465—1440 сл- ое колебание C=sC-c 1000—1070 сл. (2 полосы) в. Су 1050 с. 1070 с. 1048 ср. 1069 ср. 1054с.ср. 1072с.ср а возможность поя S обусловленных в вязи вызывает погл льфид с 1098 сл. 1091 сл. 1099 с. ср. вления сг рентными ощение в 1125—1090 сл. 1,2-дизамещенным 1120 оч. сл. 1111 оч. сл. Нет льных полос погло- колебаниями бен- этой области.
1,2-Дизвмещенные алкилбензолы имеют сильную полосу поглощения
в области 1509—1504 см~1 и полосу средней интенсивности в области
1620—1617 см-1. В ИК-спектрах сульфидов сорто-замещенными бензоль-
ным кольцом наблюдается интенсивная полоса около 1590 см~1 и поло-
са средней интенсивности около 1493 см~г; последняя, возможно, обус-
182
Таблица И
с фенильной и фениленовой группами в ИК-спектрах
частоты, сл*”1
1029±5 1072+1 1156±5 1177±6 1240+8 1324+10 1387+6 1451±12 1499+7 1588+9 1606±7
С. ср. ср. ср. СЛ. СЛ. СЛ. с. ОЧ. с. СЛ. ср.
бензольным кольцом
1029 с. 1027 с. 1072 1068 с. ср. 1074 ср. 1085 с. 1091 с. 1160 ср. сл. 1157 сл. 1182 ср. сл. 1185 сл. Нет 1233 сл. 1308 сл. ср. 1316 ср. 1390 ср. сл. 1381 сл. 1443 оч. с. 1441 оч. с. 1482 оч. с. 1479 оч. с. 1583 оч. сг 1585 оч. с. Нет 1600 оч. сл.
1026 ср. с. 1068 ср. 1090 с. 1153 сл. 1179 сл. 1236 ср. с. 1332 сл. Нет 1453 с. 1477 оч. С. 1583 оч. с. Нет
1024 ср. 1070 ср. 1086 с. 1155 оч. сл. 1198 оч. сл. 1235 ср. 1334 ср. 1374 ср. 1436 оч. с. 1476 оч. с. 1586 оч. с. »
1026 ср. с. 1067 ср. 1087 с. 1156 оч. сл. 1178 оч. сл. 1238 ср. с. 1310 оч. сл. 1380 ср. сл. 1438 с. 1480 оч. с. 1585 оч. с. »
д а н н ь X 1585 с. »
данных 1586 с. 1605 ср.
1029 с. 1072 с. Нет 1153 сл. 1182 сл. 1242 с. 1319 ср. 1387 сл. 1451 с. 1493 с. 1587 ср. сл. 1600 с.
1Q20 ср. СЛ. 1070 с. ср. » 1150 ОЧ. СЛ; 1180 оч. сл. 1230 с. ср. 1320 ср. сл. Нет 1450 с. ср. 1495 с. 1587 ср. 1600 с. ср.
1031 ср. СЛ. 1076 ср. 1096 ср. 1160 оч. сл. 1185 оч. сл. 1230 сл. 1320 сл. > 1440 с. ср. 1400 с. ср. 1482 с. ср. 1500 ср. 1585 с. ср. 1605 ср. сл.
1035 ср. СЛ. 1080 ср. сл. Нет 1147 ср. сл. 1180 сл. 1233 ср. 1320 сл. ср. » 1440 с. 1500 с. 1588 ср. сл. 1605 ср.
1028 ср. 1072 с. ср. > 1155 сл. 1170 сл. 1236 сл. пл. Нет 1380 сл. 1450 с. 1492 оч. с. 1584 ср. сл. 1602 ср.
1175—1225 сл. См. • 1521— 1510 — 1656-1633 сл.
бензольным кольцом
1215 ср. 1460 оч. с. 1498 оч. с. 1566 сл. 1600 сл. | 1640 сл.
1175—1225 с л. См.* 1509— 1504 с. 1620—1617 ср.
бензольным кольцом
tl230 оч. сл. 1470 ОЧ. с. 1495 ср. 1575 ср. сл0 (пл.) 1588 оч. с. Нет
1223 ср. 1475 оч. с. 1492 ср. 1575 ср. сл, (пл.) 1591 ср. См.**
1208 сл. ср. 1474 оч. с. 1500 ср. 1577 ср. сл, (пл.) 1592 с. ср. Нет
ловлена присутствием примеси пара-изомеров в исследованных препа-
ратах.
В ИК-спектрах всех ароматических сульфидов, в которых фениль-
ное кольцо непосредственно присоединено к атому серы, наблюдается
полоса 110—1085 см~1, сильная в случае моно- и ди-пара-замещенного
бензольного кольца, слабая —для орто-замещенного бензольного коль-
ца. Авторы работы [56] приписывают эту полосу колебаниям группипов-
ки фенил — сера.
183
- В ИК-спектрах сульфидов с монозамещенным бензольным кольцом
имеется одна или две полосы в области 460—520 еле-1 (моноалкилза-
мещенные бензолы поглощают в области 460—575 см-1 [60]).
Полоса около 500 см-1 в сульфиде п-СН3—С6Н4—S—С3Н7, возможно,
характеризует 1,4-замещенное бензольное кольцо (1,4-дизамещенные
бензолы поглощают в области 460—550 см~' [60]).
Наблюдаемые нами полосы ИК-поглощения ди-орто-замещенных
сульфидов № 20, 21, 22 (см. табл. 6) вблизи 438, 500 (слабо разрешен-
ный дублет) и 560 см-1, возможно, связаны с колебаниями 1,2-дизаме-
щенного бензольного кольца (1,2-диалкилбензолы имеют полосы по-
глощения около 450, 500 и 580 см"1 [60]).
Все сульфиды, имеющие винильную группировку, поглощают в обла-
стях 555—565 и 590—596 см-1 (в алкенах винильная группировка вы-
зывает поглощение около 555 и 630 см-1). В спектрах насыщенных
сульфидов и ароматических сульфидов без винильной группировки эти
области обычно прозрачны [60].
б. Спектры КРС*
Исследование спектров КРС сульфидов (I) — (IV) отчетливо указы-
вает на общее различие, существующее между спектром сульфида (II),
в котором фенильное кольцо не связано непосредственно с S-атомом,
и спектрами сульфидов (I), (III), (IV), где такая связь имеется: в
спектре сульфида (II) с монозамещенным бензольным кольцом с боль-
шой точностью сохраняются частоты всех характерных линий моноза-
мещенных бензолов (табл. 12) [61], тогда как в спектрах сульфидов
(I), (III), (IV) имеется ряд существенных отличий от спектров углево-
дородов, аналогичных по структуре [52, 62—66].
Эти отличия заключаются в следующем: 1) частоты некоторых ха-
рактерных линий по сравнению с соответствующими углеводородами за-
нижены на 6—30 см-1 (см. табл. 12, Б и В);
2) появляются некоторые интенсивные поляризованные линии, отсут-
ствующие в спектрах аналогичных углеводородов: в спектре сульфида
(III) (орто-замещение)—линия 801 см-1 (см. табл. 12, Б), в спектрах
сульфидов (I), (III), (IV) — (моно-, диорто- и пара-замещение) — линия
1092—1096 слН (см. табл.12, А, Б и В).
Из сопоставления, сделанного Кольраушем [15, 67] для ряда моно-
замещенных бензолов и толуолов с различными элементами и группа-
ми элементов, присоединенными к фенильному кольцу, следует, что в
спектрах гетероатомных монозамещенных бензолов и пара-замещенных
толуолов линия ~ 1090 см-1 аналогична линии ~1200 см-1 соответ-
ствующих ароматических углеводородов, причем частота ее убывает по
мере возрастания относительной массы радикала, как это видно для
следующего ряда монозамещенных бензолов: F— 1218 см-1; SH— 1097—
1118 см~1; С1—1083 см-1; NO2—1074 см-1; Вг—1071 см~1; J—
1060 см-1. Для пара-замещенных толуолов: F—1213 , см-1; С1—
1090 см-1; Вг— 1069 см-1; J— 1054 см-1.
Сопоставление спектра сульфида (I) с монозамещенным фенильным
кольцом со спектрами сульфидов такого же типа из работы [51] показы-
вает, что в спектре (I) присутствуют линии, отмеченные в этой работе и
характерные по своим частотам для монозамещенных бензолов: 614,
1000, 1026, 1157, 1182, 1586 см-1.
Характерные для ароматических углеводородов линии ~1580 и
— 1600 см-1 в спектрах сульфидов (I), (III), (IV), т. е. в случае сосед-
ства серы с фенильным кольцом, отличаются двумя интересными особен-
* Таблицы спектроа КРС вновь исследованных ароматических сульфидов см.
ниже.
Таблица 12
Сравнение характерных частот (в слГ1) ароматических углеводородов и сульфидов
с фенильной и фениленовой группами в спектрах КРС
Углеводороды Ph—R Сульфид (II) Ph—С—-S—С—С=С Разность частот, см-1 Сульфид (Г) Ph—S—С—С—С Разность частот, см—1
622—624 (37—60; 0,73—0,90) 771—818 (23—250; 0,1—0,3) 1002—1005 (230—500; 0,07—0,17) 1029—1033 (73—125; 0,10—0,24) 1156—1158 (25—40; 0,54—0,86) 1179—1192 (47—54; 0,30—0,77) 1203—>1212 (54—110; 0,1—0,21) 1583—1586 (21—44; 0,62—0,82) 1604—1606 (69—110; 0,59—0,93) 618 (47; 0,81) 810 (37; 0,30) 1002 (310; 0,13) 1029 (58; 0,12) 1160 (37; 0,90) 1181 1(~3) крыло линии] 1200 (94; 0,33) 1583 (37; 0,88) 1604 (150; 0,81) 4—6 1 —Г 614 (42; 0,6) 1000 (375; 0,17) 1026 (135; 0,18) 1157 (42; 0,70) 1182 (19; 0,3) 1092 (140; 0,29) 1586 (220; 0,72) 8—10 2—5 3—7 Г1"" —»
Б В
Углеводороды o-R-Ph-R' Сульфид (III) Ph—СН,-о S—C,0H2i-« Разность частот, СМ—1 Углеводороды n-R—Ph—R' Сульфид (IV) Ph—СНа-л S—СзН,-« Разность частот, см-1
586 (35) 1220 (150) 1382 (6—65) 1580 (21—40) 1600—1609 555 (42) 801 (82) 1093 (47) 1202 (87) 1376 (26) 1571 (37) 1591 (126) 31 18 6 9 9—18 642 (50) 780—310 (250) 1204 (100) 1380 (10—80) 1577 (1—12) 1612—1620 636 (83) 797 (129) 1096 (460) 1210 (57) 1378 (60) 1571 (0) 1601 (300) 6 6 11—19
костями: 1) в спектрах сульфидов (!!!) и (IV) с дизамещенным аро-
матическим кольцом эти линии, как указывалось выше, занижены по
частоте; 2) в спектре сульфида (!) с монозамещенным бензольным коль-
цом, сопряженным с атомом S, линия — 1600 см-1 отсутствует, а линия
~ 1580 см-1, сохраняя частоту и степень деполяризации, характерные
для моноалкилзамещенных бензолов (1583—1586 см-1; 0,62—0,82), имеет
интенсивность (/о = 22О) гораздо большую, чем у углеводородов. Такое
же явление наблюдается [51] у четырех других сульфидов подобного же
типа.
Принимая во внимание тот факт, что соседняя линия ~ 1600 см-1
в спектрах исследованных нами сульфидов (!!!) и (IV) с дизамещенным
бензольным кольцом занижена по частоте по сравнению с углеводоро-
дами на 9—19 см-1, можно предположить, что в спектре сульфида (!)
высокая интенсивность линии 1586 см-1 объясняется наложением на нее
заниженной по частоте линии ~ 1600 см-1. Степени деполяризации обе-
их рассматриваемых линий близки между собой (0,6—0,9).
185
Валентные колебания С—S—С в рассматриваемых ароматических
сульфидах, как и в случае алифатических сульфидов, представлены
двумя группами линий: 643—686 см-1 (симметричные колебания) и 720—
792 см-1 (антисимметричные колебания). Линии первой группы поляри-
зованы, второй — деполяризованы. У сульфидов с бензольным кольцом,,
присоединенным к S-атому [(I), (III), (IV)], основные линии каждой
группы значительно менее интенсивны (7о=17—29), чем у сульфида (II)
(/о=57—75) с метиленовым мостиком между атомом серы и бензольным
кольцом.
В спектрах сульфидов, содержащих аллильную группировку, таких
как (II), изоамилаллилсульфид (VII)* и диаллилсульфид [68], имеется
интенсивная поляризованная линия 590 см-1, относимая Шеппардом [9J
также к С—S—С-валентному колебанию.
Ниже приведены полные данные по спектрам КРС вновь исследован-
ных ароматических сульфидов (Av, сл~1).
1. Фенил-н-пропилсульфид Ph—S(CH2)2—СН3
170(0), 202(0), 230(0) 279(2 ш), 410(0), 493(0), 590(1), 614(42; 0,6,),
647(17 ш), 700(35; 0,5); [720—739] (21), [760—774] (10), 788(12 ш),
828(10), 872(0), 897(19; 0,1), 942(25; 0,3), 978(0), 1О0О(375; 0,17),
1026(135; 0,18), 1049(0), 1092(140; 0,29), 1109(4), 1130(1), 1157(42; 0,70),
1182(19; 0,3), 1209(4), 1240(8), 1273(8), [1287—1300](12), 1325(0),
1341(12), 1355(0), 1368(10), [1388—1392](0), [1418—1424] (17), 1442(29),
1482(21), 1508(0), 1564(0), 1586(220; 0,72), 1617(0), 1652(0?), 2660(26),
2870(150; 0,1), 2921(135 ш; 0,2), 2965(54; 0,5), 3004(12), 3058(210),
3157(31).
II. Бензилаллилсульфид Ph—СН2—S—СН2—СН = СН2
[201—208—222] (25; 0,67), 248(10), 293(7 ш), 320(0), 347(12 ш), 411(30;
0,47), 440(0), 450(0), 472(22; 0,30), 520(0), 540(0), 564(15; 0,8),
594(28; 0,26), 618(47; 0,81), 652(0), 686(75; 0,24), 706(57; 0,25),
725(0), 744(63; 0,52), 769(57; 0,76), 792(0), [807(37)—816(30)] (0,32),
836(0), 847(3), 902(8), 919(12; 0,61), 936(10), 945(22; 0,25), 964(0),
973(0), 1002(310; 0,13), 1029(58; 0,12), 1073(0), 1100(1), 1123(0),
1160(37; 0,90), 1181(0), 1200(94; 0,33), 1222(0), 1245(97; 0,38),
1294(50; 0,32), 1327(0), 1349(0), 1403(48; 0,53), 1424(30; 0,7), 1450(7),
1496(7), 1583(37; 0,88), 1604(150; 0,81), 1635(135; 0,24), 2909(150; 0,2),
2949(70; 0,6), 2977(60; 0,2), 3009(100; 0,2), 3053(200; 0,3), 3074(70; 0,6),
3160(4), 3192(6).
III. Децил-о-толилсульфид о-СН3—Ph—S—СщНщ-н
259(15), 440(0), 555(42; 0,3), 575(0), 675(29; 0,3), [703(0)—730(0)—
744(13)], 801(82; 0,24), 833(0), 955(0), 996(21; 0,23), 1050(118; 0,23),
1072(0), 1093(47; 0,38), 1128(16; 0,3), 1160(21; 0,5), 1202(87; 0,26),
1236(0), 1303(34; 0,8), 1376(26; 0,6), [1436—1452] (45; 0,73), 1571(37),
1591(126; 0,84), 2737(0), 2849(110; 0,2), 2883(0), [2899(140; 0,4) —
2925(150; 0,3)—2969(45; 0,4)], 2989 (0), 3015(10), 3059(55; 0,5).
IV. Пропил-п-толилсульфид п-СН3—Ph—S—С3Н?-н
261(17 шд), 307(26 шд; 0,4), 338(0), 507(0), 636(83; 0,73), 656(20; п),
698(0), [725—744] (17), 797(129; 0,14), 820(0), 901(11), 937(0), 957(0),
1037(54; 0,35) **, 1078(0), 1095(460; 0,28), 1182(46; 0,48); 1210(57; 0,20),
1231(0), 1292(11), 1310(11), 1378(69; 0,53), 1453(29 шд; 0,82), 1494(29;
0,82), 1516(0), 1571(0), 1601(300; 0,73), 2736(12), 2870(100; 0,2),
2920(230; 0,2), 2949(70), 2973(70; 0,6), 2997(0), 3035(70), 3049 ***.
* См. стр. 166.
** Возможно, здесь совмещено «повторение» линии 1095 см-1 от возбуждающей
HgX=4348 А с основной линией 1037 см~‘.
*** Интенсивность этой линии не могла быть измерена вследствие наложения ли-
нии Hg X = 5025 А.
186
выводы
1. В ИК- и КРС-спектрах ароматических сульфидов имеются группы
линий, характеризующие бензольное кольцо определенного типа заме-
щения, близкие по частотам к линиям, характерным для углеводородов
соответствующего строения, что позволяет обнаружить с помощью
спектра присутствие бензольного кольца в сульфиде.
2. Спектры КРС позволяют отличить ароматический сульфид от
углеводорода соответствующего ему строения.
3. КРС- и ИК-спектры позволяют определить, присоединен ли атом
серы непосредственно к бензольному кольцу или отделен от него мети-
леновым мостиком (сульфиды с моно-, ди-орто- и ди-пара-замещенным
бензольным кольцом).
4. ИК- и КРС-спектры позволяют обнаружить в сульфиде строения
RSR', где R— ароматическое кольцо, группировку С = С, входящую
в состав R'.
IV. ЭЛЕКТРОННЫЕ СПЕКТРЫ СУЛЬФИДОВ
Электронные спектры насыщенных сульфидов характеризуются по-
глощением в далекой ультрафиолетовой области. Интересно было выяс-
нить аналитические возможности спектроскопии в ближней ультрафио-
летовой области для установления строения а- и p-непредельных и аро-
матических сульфидов.
Имеется значительное количество работ, в которых приводятся дан-
ные об электронных спектрах сульфидов. Интерес представляют обзо-
ры Прайса и Оае [69], Джаффе и Орчина [70], Пассерини [71].
Нами был проведен анализ как вновь полученного, так и литератур-
ного материала об ультрафиолетовых спектрах поглощения ряда упомя-
нутых сульфидов.
А. Непредельные сульфиды
Мы наблюдали (рис. 2, кривые 1, 2) большую интенсивность и длин-
новолновое смещение в спектрах p-непредельных сульфидов (в которых
атом серы и связь С = С разделены СНг-группой) относительно Спектров
алифатических сульфидов [72], что указывает на наличие некоторого
взаимодействия между ненасыщенной системой и атомом серы. Это под-
тверждается литературными данными табл. 13: в спектрах р-непредель-
ных сульфидов указаны перегиб в области 237—250 нм (1g е = 2,30—3,20)
и коротковолновый максимум в области 202—215 нм (1g е = 3,38—3,42).
Взаимодействие атома серы с двойной связью значительно больше у
а-непредельных сульфидов; в их спектрах полностью отсутствует адди-
тивность поглощения. Действительно, спектры 3, 4 (см. рис. 2) отли-
чаются от спектров 1, 2 по контуру: коротковолновые полосы в них
претерпевают значительный батохромный сдвиг, сместившись из об-
ласти ниже 220 нм для (/, 2) в область 220—234 нм-, при этом интен-
сивность поглощения значительно возрастает. Это подтверждается,
например, спектрами соединений № 9 и 23, 5 и 20 (см. табл. 13), струк-
туры которых попарно различаются лишь положением двойной связи
(а- или р-): для сульфидов с а-положением двойной связи обнаружи-
вается батохромный сдвиг и увеличение интенсивности коротковолновой
полосы.
Данные табл. 13 позволяют заключить, что на спектры непредельных
сульфидов строение алкильной цепи оказывает второстепенное влияние;
в основном характер спектра зависит от близости атома серы к двойной
связи С = С. В работе [34], посвященной изучению структуры и спектров
ряда алкилвинилсульфидов, наиболее вероятным считается существо-
вание а-иепредельных сульфидов в плоской S-транс и в почти плоской
S-цис-форме.
187
Рис. 2. УФ-спектры поглощения непредельных (а) и ароматических (б)
сульфидов
I — (СН3)2 СН(СН2)2 — S — СН2СН = СН2; 2 — СН2 = СНСН2 — S — СН2 — ;
^-CH3(CH2)5—S — CH = CH(CH2)3CH3 (модуль 0,4); 4— | I II „„ (модуль 0,2);
\ /к ✓ ” ЬН3
A H21C„S-/X
Д - - S - (СН2)2 - СН3; 6 - НзС ;
Л —S—(модуль 0,8)
ч/ ч/
7 н-Н7С3 S (модуль 0,4);
Ч/-СНз
В случае а-непредельных структур переход от молекул с ацикли-
ческой сульфидной группировкой к молекуле с тиациклогексановым
кольцом не вызывает существенных изменений в спектре последней
(см. рис. 2, кривая 4), что подтверждается литературными данными
(см. табл. 13); однако переход к молекуле тиациклопентена вызывает
красное смещение.
В спектрах . р-тиациклопентена и р-тиациклогексена (см. № 25 и
23, табл. 13) коротковолновые максимумы смещены в синюю сторону,
причем у последнего смещение больше. Авторы работы [73] полагают,
что в циклических сульфидах при передаче взаимодействия между ато-
мом серы и двойной связью существенную роль играют конформацион-
ные факторы.
Наличие двух двойных связей, попарно занимающих а- или 0-поло-
жение относительно атома серы (№ 13—16, 18, табл. 13), приводит к
большему красному смещению, чем в однонепредельных сульфидах, что
свидетельствует о способности атома серы к передаче взаимодействия
на обе двойные связи.
В работе [73] было произведено сопоставление УФ-спектров непре-
дельных сульфидов винильного и аллильного типов и их физико-химиче-
188
Таблица 13
УФ-спектры непредельных сульфидов
№ п. п. Структура X, нм * Iff е Растворитель Лите- ратура
а-ряд
1 С=С—S—С 230 4,2 Этиловый спирт [70]
240 4,0
2 С=С—S—С—с 234,1 2,87 н-Гексан [74]
250,0 2,47
228 3,84 н-Гептан [34]
240 3,60
3 С=С—S—Сз-м 228,0 3,73 » [34]
1240,0} 3,63
4 С=С—S—Ct-H 228,0 3,82 » [34]
[240,0] 3,70
229,0 3,84 Циклогексан [73]
[240J 3,74
228 3,81 Метиловый спирт [73]
[239—240] 3,72
5 % 1 СЛ 1 о II о 1 ? о 232 3,80 Циклогексан [73]
[248] 3,65
229 3,78 Метиловый спирт [73]
[248] 3,64
6 н-Св—S—С=С—С4-м 230 3,26 н-Гептан
[250] 3,15
7 С=С—S—С-С 223' 3,81 » [34]
[ С [245] 3,60
8 С=С—S—Сотрет 234,0 3,59 » [34]
249,0 3,63
9 226 3,72 Циклогексан [73]
|| 251 3,41
226 ' 3,66 Метиловый спирт [73]
249 3,30
10 228 3,87 Изооктан
СП [253] 3,42
1 1 с \/\5/ С
11 227 3,87 Циклогексан [73]
j/4ji I [260] 3,25
1 I 1 225 3,85 Метиловый спирт [73]
\s/\Z 256 3,25
12 . : 237 — Циклогексан [73]
1 1 263 __
236 — Метиловый спирт [73]
262 —
13 с=с—s—с=с 242 3,93 н-Гексан [74]
259 3,87
255,0 3,75 Этиловый спирт [40]
275,0 3,7
* Скобки указывают на перегиб в полосе поглощения.
189
Таблица 13 (окончание)
№ п. п. Структура X, нм * 1g е Растворитель Лите- ратура
14 15 16 0 аал 1 1 \As/\/ с 1 /\/\/\ fill 236—-238 278 234 276 238 3,72 3,38 2,82 3,79 м-Гексан [75] [76] [77]
17 18 с—С—S—с II (Ч«с) С—C—S—с С=С—C-S-C—с=с (см.*) 254,0 [285,0] 3-ряд 224 221 220 4,01 3,48 3,35 3,33 3,22 «-Гептан Циклогексан Этиловый спирт Метиловый спирт [34] [73] 178] 73]
19 н-С4—S—С—С—С 218 [238] 210—215 [238] 3,25 2,77 3,24 Циклогексан Метиловый спирт [73] [73]
20 Н-С4—rS —С—тС=С—с 215 [237] [214] [237] 3,39 2,60 3,38 2,64 Циклогексан Метиловый спирт [73] [73]
21 22 23 wao-Cs—S—С—С=С ** Ph—С—S—С-;С=С* п [235] [240 [260] [266] 202 [250] 206 [255] 3,20 3,15 2,65 2,43 3,42 2,30 3,30 н-Гептаи Изооктан Циклогексан Метиловый спирт [73] [73]
24 25 Н-С4—S —/ [237] 208 [230—233] 211 [232] 2,63 Циклогексан » Метиловый спирт [73] [73] [73]
26 Н-С4—S—| [238] 2,43 Циклогексан [73]
Эти спектры не регистрировались в области ниже 220 нм.
190
ских характеристик (теплоты сожжения и гидрогенизации, молекуляр-
ные рефракции и дипольные моменты, длины С—S-связей). Результаты
сопоставления свидетельствуют об очень малой степени взаимодействия
р-электронов атома серы и двойной связи в основном состоянии, поэто-
му смещение максимумов в УФ-спектрах непредельных сульфидов авто-
ры приписывают преимущественно взаимодействию в возбужденном
состоянии.
Однако экспериментальные данные и расчеты Попова и Кагана [42]
по спектрам поглощения непредельных сульфидов в ИК-области согла-
суются с допущением взаимодействия в основном состоянии. Для окон-
чательного решения этого вопроса необходимы дальнейшие исследо-
вания.
Из изложенного выше следует, что для определения положения двой-
ной связи (а- или 0-) относительно атома серы в индивидуальном суль-
фиде может быть успешно привлечен метод УФ-спектроскопии. С той же
целью могут использоваться колебательные спектры, однако обсужда-
емый метод незаменим при определении наличия и положения тетраза-
мещенной двойной связи, когда ИК- и ПМР-методы неприменимы (на-
пример, соединение № 11, табл. 13).
Б. Ароматические сульфиды
На рис. 2, б представлены электронные спектры изученных нами аро-
матических сульфидов. В спектре аллилбензилсульфида (см. рис. 2,
кривая 2) проявляется бензоидная полоса типа В2и с максимумами 262
и 268 нм, налагающаяся на полосу поглощения 0-непредельного сульфи-
да. Это указывает на ограниченность взаимодействия неподеленной пары
электронов атома серы с полностью конъюгированной л-электронной
системой бензольного кольца.
Литературные данные [78—81] по УФ-поглощению ароматических
сульфидов свидетельствуют об исчезновении тонкой структуры бензоль-
ной полосы В2и, проявляющейся около 260 нм, и увеличении ее интен-
сивности в случае непосредственного присоединения атома серы к бен-
зольному кольцу; если же сера и бензольное кольцо разделены метиле-
новыми группами, тонкая структура этой полосы проявляется.
Приведенные на рис. 2,6 спектры ароматических сульфидов 5—7
с фенильным кольцом, присоединенным к атому серы, имеют интенсив-
ную сложную полосу поглощения с максимумом в области 250—260 нм.
Наличие этой полосы ряд авторов [70] связывает с наложением полосы
бензоидного перехода типа В2и и полосы, обусловленной сопряжением
серы и л-электронной системы бензола. Установлено, что в алкилфенил-
сульфидах очень существенны спектральные эффекты, обусловленные
изгибом плоскости молекулы за счет особенностей строения алкильных
заместителей и их положения в бензольном кольце [79]. Стерические
препятствия сопряжению неподеленной пары электронов атома серы и
л-электронов бензольного кольца, возникающие за счет орто-заместите-
ля, можно обнаружить в молекуле децил-о-толилсульфида, что проявля-
ется в заметном гипсохромном сдвиге ее максимума поглощения 6 отно-
сительно максимума поглощения в спектре пропилпаратолилсульфида 7.
Интересно сопоставить спектры поглощения дифенилсульфида 8 и ди-
фенилоксида [82]. Характер спектра 8 указывает на наличие значитель-
ного сопряжения вдоль всей молекулы, что следует из существенного
отличия этого спектра от спектра тиоанизола или фенилпропилсульфи-
да 5. С другой стороны, спектр дифенилоксида подобен спектру анизола.
Отсюда можно заключить, что в отличие от молекулы дифенилоксида,
являющейся некопланарной (фенильные кольца в ней расположены во
взаимно перпендикулярных плоскостях [82], молекуле дифенилсульфида
191
можно приписать, по-видимому, значительно более плоское строение,
что подтверждает выводы работы [83].
На основании спектральных данных было сделано заключение [34],
что степень нарушения копланарности в молекуле винилфенилсульфида
значительно меньше, чем в молекуле винилфенилового эфира, спектр
которого подобен спектру анизола. С другой стороны, наблюдается су-
щественное различие между спектрами винилфенилсульфида и тиоани-
зола [70]. Эти особенности можно, по-видимому, объяснить различием в-
электронном строении атомов серы и кислорода [84].
В. Тиаииданы
Электронные спектры поглощения алкилзамещенных тиаинданов
(рис. 3), отличаясь от спектров инданов [85], близких спектрам алкил-
бензолов, по своему характеру подобны спектрам алкиларилсульфидов.
Спектры алкилтиаинданов характеризуются на порядок более высо-
ким коэффициентом экстинкции в главном максимуме, расположенном
при 250 нм, чем спектры углеводородов близкого строения. Небольшой
перегиб в спектрах алкилтиаинданов наблюдается при 260 нм-, в ин-
тервале 285—310 нм имеется вторая полоса малой интенсивности с мак-
симумом при 290 нм и перегибом при 295 нм (для замещенных в гетеро-
цикле) или соответственно 297 и 305 нм (для 2,5-замещенного).
•9»*^ гЛ*? 3900 34-00 v,m'’
S,S\----—,----------р——q--------р—г-------
тиаинданов
СНз (модуль 1,0)
7_сн3 (модуль
220 240 260 280 ЗОО 2., нм
Переход от ациклического сульфида к гетероциклическому связан с
более четким проявлением тонкой структуры полос, однако отличным от
того, которое наблюдается в изоциклических аналогах инданового ряда.
Как и следовало ожидать, положение и строение алкильного заме-
стителя в пятичленном кольце весьма незначительно сказывается на
192
характере электронных спектров тиаинданов, влияя в некоторой мере на
их интенсивность. Так, заметно воспроизводится уменьшение интенсив-
ности обеих полос при a-положении заместителя относительно атома
серы (см. рис. 3, спектры 2, 3) по сравнению с соединениями, имеющими
заместители в p-положении. Наличие второго a-заместителя, находяще-
гося в ароматическом кольце, уже более существенно сказывается на
спектре замещенного тиаиндана. Так,
обе полосы поглощения в спектре
2,7-диметилтиаиндана (рис. 4)* замет-
но смещены в коротковолновую сторо-
ну относительно спектра 2,5-диметил-
тиаиндана (рис. 3, спектр 6), совпадая
по положению со спектрами монозаме-
щенных тиаинданов. Это может указы-
вать на наличие взаимодействия пери-
метильной группы с атомом серы, при-
водящего к снижению взаимодействия
неподеленной пары электронов атома
серы с л-электронной системой арома-
тического кольца. Судя по спектру
2,5-диметилтиаиндана, в котором на-
блюдается заметное смещение обеих
полос в длинноволновую сторону, от-
носительно спектров остальных соеди- Рис. 4. УФ-спектр поглощения
нений, можно ожидать, что между 2,7-диметилтиаиндана
строением спектров поглощения в бли-
жней ультрафиолетовой области и числом и положением алкильных за-
местителей в ароматическом кольце алкилтиаинданов существуют
аналитически важные корреляции.
Данное рассмотрение электронных спектров позволяет сделать ряд
заключений, которые могут быть полезны при исследовании структуры
сульфидов.
Выводы
1. С помощью УФ-спектроскопии представляется возможным опре-
делять положение (а- или р-), занимаемое атомом серы относительно
двойной связи.
2. По положению максимума интенсивной полосы в области 228—
238 нм возможно в ряде случаев отличать сульфиды с одиночной а-не-
предельной группировкой относительно атомов серы от сульфидов с дву-
мя а-непредельными группировками (в ациклических сульфидах и шести-
членных гетероциклах).
3. Исчезновение тонкой структуры и увеличение интенсивности
(lge~4) полосы ~260 нм в УФ-спектре ароматического сульфида
может служить указанием на непосредственное присоединении атома
серы к бензольному кольцу, а появление тонкой структуры и уменьше-
ние интенсивности этой полосы более чем на порядок — на наличие меж-
ду кольцом и серой углеводородного мостика.
В работе принимали участие лаборанты Ф. В. Козлова, М. С. Лен-
товская, Л. Я- Виноградова, Е. Н. Кондрашкова (ИНХС) и Е. И. Рыба-
кова (МХТИ).
* Численные значения интенсивностей полос не были получены из-за недостаточ-
ного количества образца.
J93-
Выражаем благодарность химикам-синтетикам Н. П. Волынскому,
Е. Н. Карауловой, В. Г. Лукьянице (ИНХС АН СССР), Т. А. Даниловой
(МГУ) за предоставление индивидуальных сернистых соединений и
В. И. Лабе, Н. П. Петуховой (ИОХ АН СССР), В. Я. Родионову (МХТИ)
за предоставление ИК-спектров индивидуальных сульфидов.
Институт нефтехимического синтеза им. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. Одиннадцатая научная сессия по химии сераорганических соединений нефтей и
нефтепродуктов (Тезисы докладов). Уфа, 1968, стр. 59.
2 Г. Д. Гальперн, Л. Р. Барыкина, Е. Н. Караулова. Изв. АН СССР, серия хим.,
1967, 2123.
ЗИН Тиц-Скворцова, Р. Д. Левина, А. И. Леонова, Е. А. Карасева. ЖОХ, 21,
242 (1951).
4. И. И Тиц-Скворцова, Р. Д. Левина, А. И. Леонова, Т. А. Данилова. ЖОХ, 22,
135 (1952).
5. Н. П. Волынский, Г. Д. Гальперн, В. В. Смольянинов. Нефтехимия, 1, 473 (1961).
6. Е. Н. Караулова, А. С. Некрасов. Ж. прикл. химии, 28, 10 (1955).
7. Е. Н. Караулова, Г. Д. Гальперн, В. Д. Никитина, Л. Р. Барыкина и др. Нефтехи-
мия (в печати).
8. Catalog of Infrared Spectral Data. Amer. Petrol. Inst. Res. Project 44.
9. N. Sheppard. Trans. Faraday. Soc., 46, 429 (1950).
10. P. Д. Оболенцев, Ю. И. Котов, E. H. Челов. Сб. «Химия сераорганических соедине-
ний, содержащихся в нефтях и нефтепродуктах», т. 4. М., Гостоптехиздат, 1961,
стр. 39.
И. Г. Ф. Большаков, Е. А. Глебовская, 3. Г. Каплан. Инфракрасные спектры и рентге-
нограммы гетероорганических соединений. Л., «Химия», 1967.
12. V. Thatte, A. Ganesan. Phil. Mag., 15, 51 (1933); Nature, 127, 306 (1931).
13. S. Venkatesvaran. Indian J. Phys. 6, 51 (1931).
14. Raman Spectral Data. Amer. Petrol. Inst. Res. Project 44.
15. К. Кольрауш. Спектры комбинационного рассеяния. М., ИЛ, 1952.
16. Н. IF. Thompson. Trans. Faraday. Soc., 37, 38 (1941).
17. M. Hayashi, T. Shimanuochi, S. Mizushima. J. Chem. Phys., 26, 608 (1957).
18. D. Scott, H. Finke. J. Am. Chem. Soc., 74, 4656 (1952).
19. H. A. Szimanski. Infrared Band Handbook (append. 1), 476 (1963).
20. H. A. Szimanski. Interpreted Infrared Spectra, vol. 1. N. Y, Plenum Press, 1964.
21. N. Sheppard. Trans Faraday Soc., 51, 1465 (1955).
22. D. W. Scott, I. P. Me Cullough. J. Am. Chem. Soc., 80, 3555 (1958).
23. M. Hayashi. J. Chem. Soc. Japan, Pure, Chem. Soc. (Nipon Kagaki Zassi), 77, 1804
(1956).
24. P. Д. Оболенцев, Ю. И. Котов, E. H. Челов. Сб. «Химия сера- и азоторганических
соединений, содержащихся в нефтях и нефтепродуктах», т. 3. Уфа, 1960, стр. 105.
25. I. Me Cullough, Н. Finke, et al. J. Am. Chem. Soc., 77, 6119 (1955).
26. D. Scott, H. Finke et al. J. Am. Chem. Soc., 74, 4656 (1952).
27. I. Me Cullough, W. Hubburd et al. J. Am. Chem. Soc., 79, 561 (1957).
28. D. W. Scott, H. L. Finke et al. J. Am. Chem. Soc., 73, 261 (1951).
29. Б. В. Айвазов, С. M. Петров, В. P. Хайруллина, В. T. Дпрынцева. Физико-химиче-
ские константы сераорганических соединений. М., «Химия», 1964.
30. М. М. Сущинский. Труды ФИАН, 12, 54 (1960).
31. I. R. Alikins, Р. I. Hendra. Spectrachim. Acta, 22, 2075 (1966).
32. D. S. Tarbell, M. A. McCal. J. Am. Chem. Soc., 74, 48 (1952).
33. C. N. R. Rao. Chemical applications of infrared spectroscopy. New York — London,
1963.
34. П. П. Шорыгин, M. Ф. Шостаковский, E. H. Прилежаева, T. H. Шурина, Л. Г. Сто-
ляров, А. П. Гения. Изв. АН СССР, ОХН, 1961, 1571.
35. И. Брандмюллер, Г. Мозер. Введение в спектроскопию комбинационного рассеяния
света. М., «Мир», 1964.
36. Б. С. Купин, А. А. Петров. ЖОрХ, 3, 975 (1967).
37. Л. М. Катаева, Е. Г. Катаев, Г. А. Чмутова, Т. Г. Маннафов. Материалы XV сове-
щания по спектроскопии, т. 1. М., ВИНИТИ АН СССР, 1964, стр. 318.
38. С. Price, R. Gillis. J. Am. Chem. Soc, 75, 4750 (1953).
39. K. Georgieff, A. Dupre. Canad. J. Chem, 37, 1104 (1959).
40. C. Price, H. Morita. J. Am. Chem. Soc, 75, 4747 (1953).
41. А. А. Петров, M. П. Форест. ЖОХ, 2, 1178 (1966).
42. E. M. Попов, Г. И. Каган. Оптика и спектроскопия, XI (6), 730 (1961).
43. Ю. Эйзен, С. Ранг и др. Изв. АН ЭстССР, 16(2), 101 (1967).
44. П. А. Акишин, Н. Г. Рамбиди и др. Вестник МГУ, 3, 77 (1954).
194
45. С. Соре, D. Morrison, L. Field. J. Am. Chem. Soc., 72, 59 (1950).
46. W. E. Parham, R. F. Matter. J. Am. Chem. Soc., 81, 3386 (1959).
47. К. Наканиси. Инфракрасные спектры и строение органических соединений. М.,
«Мир», 1965.
48. А. А. Бабушкин, П. А. Бажулин и др. Методы спектрального анализа. М., Изд-во
МГУ, 1962.
49. Н. Бхакка, Д. Уильямс. Применение ЯМР в органической химии. М., «Мир», 1964.
50. Е. Н. Караулова, Г. Д. Гальперн, В. Ш. Шайхразиева, И. В. Черепанова, Л. Р. Ба-
рыкина. Нефтехимия, 8(1), 101 (1968).
51. М. В. Шишкина. ЖФХ, 27 (12), 1877 (1953).
52. Landolt-Bornstein. Zahlenwerten und Funktionen aus Physik, Chemie, Astronomie,
Geophysik und Technik, 6te Aufl., Springer, Verlag, Bd. 1, T. 2, 1951.
53. N. Lozach, G. Gwilowzo. Bull. Soc. Chim. France., 1957, 222.
54. L. Kahovec, A. W. Reitz. Monatshefte fur chemie, 69, N 5/6, 363 (1936).
55. Л. Беллами. Инфракрасные спектры сложных молекул. М., ИЛ, 1963.
56. В. Ф. Кулик, Ю. П. Егоров и др. Ж. структури. хим., 4 (4), 541 (1963).
57. Л Н. S. Green. Spectrochim. acta, 18, 39 (1962).
58. Е. Н. Караулова, Т. А. Бардина, Г. Д. Гальперн, Т. С. Бобруйская. Нефтехимия,
6, 480 (1966).
59. Н. Me Murrey, V. Thornton. Analyt. Chem., 24(2), 318 (1952).
60. F. Bentley, K- Wolfarth. Spectrochim. Acta, 15(3), 165 (1959).
61. Г. С. Ландсберг, П. А. Бажулин, M. M. Сущинский. Основные параметры спектров
комбинационного рассеяния углеводородов. М., Изд-во АН СССР, 1956.
62. Б. А. Казанский, Г. С. Ландсберг и др. Изв. АН СССР, серия физич., 18, 704 (1954).
63. Г. С. Ландсберг, Б. А. Казанский, П. А. Бажулин и др. Определение индивидуаль-
ного углеводородного состава бензинов прямой гонки комбинированным методом.
М„ Изд-во АН СССР, 1959.
64. Н. Fromherz, Н. Bueren. Angew. Chem., А, 59, № 5/6, 142 (1947).
65. В. Т. Алексанян, X. Е. Стерин. Изв. АН СССР, ОХН, 1961, 1437.
66. Е. Т. Трещдва, Ю. Н. Панченко, Н. И. Васильева и др. Оптика и спектроскопия, 8,
371 (1960).
67. К. W. F. Kohlrausch, A. Pongratz. Monatshefte fur Chemie, 63, № 5/6, 427 (1933).
68. К. Kohlrausch, W. Stockmair. Z. phys. Chem., 32, 292 (1935).
69. Ch. Price, S. Oae. Sulfur bonding. Ronald Press, N. Y., 1962.
70. H. Jaffe, M. Orchin. Theory and Applications of Ultraviolet Spectroscopy. Pergamon
Press, 1963.
71. R. S. Passerini. Ultraviolet absorbtion spectra of organic sulfur compounds. Pergamon
Press, Organic sulfur compounds. Ed. by N. Kharasch, vol. 1, 1961, p. 57.
72. P. Д. Оболенцев, H. С. Любопытова. Сб. «Химия сераорганических соединений, со-
держащихся в нефтях и нефтепродуктах», 1, Уфа, Баш. ФАН СССР, 1958, стр. 105;
т. 2, М., Гостоптехиздат, 1959, стр. 31; т. 7, М.—Л., «Химия», 1964, стр. 281.
73. М. Prochazka, М. Palececk. Collection, 32(9), 3049 (1967).
74. J. Басе. J. Phys. Chem., 64(10); 1573 (1960).
75. Л Str at in g et al. Angew. Chem., 74(13), 465 (1962).
76. T. И. Купина. Диссертация. Саратов, 1968.
77. С. К- Клименко. Диссертация. Саратов, 1968.
78. Е. Fehnel, М. Carmach. J. Am. Chem. Soc., 71, 84, 2889 (1949).
79. J. Degani, A. Mangini, A. Trombetti, C. Zauli. Spectrochim. Acta, 23A, 1351 (1967).
80. E. Campaige, S. Osborn. J. Org. Chem., 22(5), 561 (1957).
81. П. П. Шорыгин, Л. С. Егорова. ДАН СССР, 117, 856 (1957).
82. M. М. Ку саков, Н. А. Шиманко, М. В. Шишкина, М. Г. Руденко, И. Г. Турянчик.
Ж. прикл. спектроскопии, 8, 302 (1968).
83. A. Cerniani et al. Boll. Sceint. della facolta di chimica ind. di Bologna, 12, Ns 3,
114 (1954).
84. Я. К. Сыркин, M. E. Дяткина. Химическая связь и строение молекул. М.—Л.,
ГНТИХЛ, 1946.
85. М. М. Ку саков, Н. А. Шиманко, М. В. Шишкина. Ультрафиолетовые спектры погло-
щения ароматических углеводородов. М., Изд-во АН СССР, 1963.
РЕФРАКТОМЕТР ДЛЯ РАБОТЫ
С МАЛЫМИ КОЛИЧЕСТВАМИ ВЕЩЕСТВА
Д. R. Жестков
Стандартный рефрактометр типа Аббе требует для определения отно-
сительной дисперсии и показателя преломления не менее 10 мг вещества.
Описанные методы микрорефрактометрии [1, 2] с применением папирос-
ной бумаги не всегда дают четкую границу светотени и обладают рядом
других недостатков. Между тем на обычном рефрактометре марки РЛУ
или ИРФ-22 после предлагаемой ниже переделки можно определять
значения показателя преломления и относительной дисперсии малолету-
чих веществ, которые имеются в количествах 0,2—0,3 мг.
I. ПЕРЕДЕЛКА СТАНДАРТНОГО РЕФРАКТОМЕТРА ИФР-22
Переделка рефрактометра заключается в уменьшении зазора между
гипотенузными гранями измерительной и осветительной призм.
Ниже дается описание переделки рефрактометра марки ИФР-22.
Прежде чем приступить к переделке выделенного для этой цели
рефрактометра, необходимо лекальным угольником первого класса точ-
ности установить отсутствие зазора между металлической оправой и
плоскостью измерительной призмы в двух взаимно перпендикулярных
направлениях. Если видимого на просвет зазора нет, то выбранный
экземпляр рефрактометра может быть переделан без особой сложности.
Если обнаружен зазор между измерительной призмой и ее оправой, то
необходимо вынуть измерительную призму из оправы и вклеить ее зано-
во без зазора с плоскостью оправы, что рекомендуется делать только
в специальных оптических мастерских, так как всякое изменение поло-
жения измерительной призмы в оправе или ее шлифовка потребует
в дальнейшем сложной юстировки.
Верхняя призмй рефрактометра, осветительная, несет более простую
оптическую функцию и величина ее углов не влияет на показания при-
бора [1]. Поэтому можно разобрать крепление и снять осветительную
призму с прибора.
Снятую осветительную призму шлифуют до почти полного отсутствия
зазора между плоскостью самой призмы и ее металлической оправы.
Шлифовку осветительной призмы производят вручную на ровной пло-
скости толстого зеркального стекла корундовым порошком № 120. На
ровный стол кладут стекло, наносят на него немного шлифовального
порошка с водой и начинают шлифовку без нажима, плавными движе-
ниями, поворачивая призму вокруг вертикальной оси. При шлифовании
призма должна все время описывать фигуру, напоминающую цифру 8
196
(рис. 1). Через каждые 10—15 мин. тщательно смывают весь шлифо-
вальный порошок со стекла и призмы, призму вытирают досуха и у
призмы проверяют зазор на просвет при движении лекального угольни-
ка в двух взаимно перпендикулярных направлениях (рис. 2). Если за-
зор велик, то повторяют шлифование с новой порцией корунда. Необхо-
димо добиться равномерного зазора по всей плоскости осветительной
призмы. Величина зазора определяется визуально. Зазор считается до-
статочным тогда, когда в щели между стеклом и лекальным угольни-
ком появятся 2—3 дифракционные полосы. При большем зазоре коли-
чество вещества, необходимое для измерения, превысит 0,1—0,3 мг; при
меньшем зазоре на границе светотени рефрактометра появятся много-
численные дифракционные, полосы, затрудняющие точные отсчеты вели-
чин показателя преломления.
Рис. 1. Шлифование осветительной призмы
После шлифования осветительной призмы высота ее оправы несколь-
ко уменьшится и прежнее крепление уже оказывается непригодным. По-
этому для установки сошлифованной осветительной призмы необходимо
переделать ее крепление. Для этого старые осевые отверстия крепежной
петли, через которые проходила ось, рассверливают до диаметра 6 мм.
Вытачивают из латуни три вкладыша 6—8 внешним диаметром 6 мм с
эксцентричным каналом диаметром 3 мм (рис. 3). Два крайних вклады-
ша 6 и 8 необходимо сделать с головками в виде гаек. Смещение центра
канала во вкладышах от их геометрической оси достаточно сделать в
1 мм. Вкладыши вставляют в гнезда, приготовленные для них в петле,
и через асимметричный канал пропускают калиброванную стальную ось
диаметром 3 мм. Далее прикрепляют переделанную петлю к измеритель-
ной и осветительной призмам. Чтобы при установке не повредить изме-
рительную призму, нужно на нее положить кусочек тонкой, мягкой
фольги или папиросной бумаги. Открывая и закрывая осветительную
призму и подпиливая выступ верхней части петли (см. рис. 3), устра-
няют возможность его зацепления за нижнюю часть петли.
Окончательная установка осветительной призмы производится с на-
несением примерно 0,5—0,7 мг какой-либо нелетучей жидкости в центр
плоскости измерительной призмы. Закрыв осветительную призму, вра-
щают головки вкладышей 6, 8 (см. рис. 3) до тех пор, пока вещество
при легком нажиме на осветительную призму будет равномерно распре-
деляться по максимальной площади, не уходя при этом в сторону от
центра. Распределение вещества хорошо видно через выходное окно
измерительной призмы, если во входное окно осветительной призмы на-
править пучок сильного света. После окончательной установки освети-
тельной призмы латунные вкладыши петли закрепляются специальны-
ми винтами 9.
14 Методы анализа
197
Рис. 2. Проверка зазора между плос-
костью призмы и ее металлической
оправы лекальным угольником в двух
взаимно перпендикулярных направле-
ниях (а) и (б)
Рис. 3. Переделанная крепежная петля
рефрактометра ИФР-22
1 — нижняя часть петли; 2 — верхняя часть петли;
3, 4 — крепежные болты; 5 — стальная ось диамет-
ром 3 мм; 6, 8 — вкладыши с эксцентричным ка-
налом и головкой; 7 — вкладыш с эксцентричные
каналом; 9 — крепежные винты вкладышей
На переделанном приборе опытным путем находят то место на приз-
мах, при нанесении на которое 0,1—0,3 мг вещества через окуляр реф-
рактометра наблюдается достаточно хорошая граница светотеневого*
поля. Это место можно отметить легкими штрихами на металлической
оправе призмы. После переделки показания рефрактометра необходимо*
проверить по известным веществам и проградуировать для отсчета ве-
личин относительной дисперсии.
Необходимо отметить, что рефрактометр с малым зазором между
призмами требует особенно бережного обращения. Пыль из воздуха или
твердые частицы в исследуемом веществе при закрывании призм могут
испортить полировку измерительной призмы и прибор выйдет из строя.
II. ПРОВЕРКА И ГРАДУИРОВКА РЕФРАКТОМЕТРА
Рефрактометры, обычно применяемые в химических лабораториях, и
способы работы с ними подробно рассмотрены в общем руководстве
Иоффе по рефрактометрическим методам в химии [3].
Рефрактометр ИФР-22 представляет один из современных вариан-
тов рефрактометра Аббе с компенсатором дисперсии в виде двух призм
Амичи. Отсчет по шкале показателей преломления и Z позволяют полу-
чать значения nDl и по разности (пр* — «с4) —среднюю дисперсию. Для
получения надежных значений необходима проверка и градуировка при-
бора. Для этого может быть использована методика, рекомендованная
Иоффе для анализа бензинов [4]. Ниже дано описание этого метод,, гра-
дуировки рефрактометра применительно к средним и тяжелым фракци-
ям нефти, которую мы использовали в своей работе.
Проверку рефрактометра начинают с определения правильности из-
готовления компенсатора дисперсии данного экземпляра прибора. В ли-
тературе [4—6] имеются указания, что ошибки в измерении дисперсии
могут достигать нескольких десятков процентов. Для проверки поме-
щают каплю воды между призмами и освещают призменный блок бе-
лым светом. Вращая' маховик компенсатора, добиваются обесцвечива-
ния граничной линии, наводят на нее визирный крест трубы и произво-
198
дят отсчет по шкале tirf. Затем, продолжая вращать маховичок, уста-
навливают второе положение компенсатора, при котором граница свето-
тени резка и бесцветна, вновь устанавливают визирный крест на свето-
теневую границу и делают отсчет п^1. Оба отсчета nDf должны совпасть
с точностью 0,0001. Большие расхождения указывают на то, что компен-
сатор данного экземпляра прибора имеет «пирамидальную ошибку». -
Для ее устранения необходимо все определения показателя преломле-
ния производить при обоих положениях компенсатора и брать среднее
значение п^.
Желтая линия D спектра не должна отклоняться призмами Амичи,
но если компенсатор имеет «призматическую ошибку», то такое откло-
нение наблюдается. Для обнаружения призматической ошибки компен-
сатора рефрактометр в затемненной комнате освещают монохромати-
ческим желтым натриевым светом. Затем определяют tirf воды при не-
скольких положениях компенсатора. Если призматической ошибки нет,
то при любом положении компенсатора отсчет по шкале пв* будет од-
ним и тем же. Если нет возможности сменить рефрактометр на лучший,
то в случае обнаружения призматической ошибки необходимо снять
кривую поправок и в дальнейших измерениях вносить поправки в зна-
чения по* [1]. На показания nDl рефрактометр юстируется по нескольким
значениям показателя преломления. Лучше если значения показателя
преломления эталонов охватывают возможно больший участок шкалы,
по которым можно составить таблицу или график поправок.
Для определения относительной дисперсии на рефрактометре ИФР-22
определяют угол поворота лимба компенсатора, при котором граничная
линия светотени бесцветна. Необходимые для вычисления дисперсии
значения коэффициентов даются в прилагаемых к прибору дисперсион-
ных таблицах. Фирменный номер таблицы должен совпадать с двух-
значным числом в правом нижнем углу шкалы прибора. Необходи-
мо иметь в виду, что эти таблицы являются серийными, а отдельные
приборы серии могут дать значительные отклонения при их использова-
нии. Юстировка компенсатора дисперсии практически неосуществима..
Поэтому может быть рекомендован способ экспериментального построе-
ния номограммы, по которой легко и быстро можно определять значе-
ния (о с точностью порядка 0,5 единиц, что вполне достаточно для
микрохроматографического анализа углеводородных смесей.
III. ГРАДУИРОВКА РЕФРАКТОМЕТРА
При построении номограммы определения относительной дисперсии
следует иметь в виду, что, кроме описанных поправок на значения п,
следует учитывать главные возможные субъективные ошибки экспери-
ментатора и ошибки, связанные с природой применяемых источников
света.
Субъективные ошибки связаны с особенностями цветового зрения
и утомляемостью глаза у различных людей. (Измерять дисперсию с по-
мощью компенсатора рефрактометра могут только лица с нормальным
цветовым зрением.) Граничная линия полного внутреннего отражения
не бывает совершенно бесцветной. Между светлым и темным полями
наблюдается слабый цветовой оттенок. Разные наблюдатели по-разно-
му оценивают окраску этой полоски. Обычно экспериментатор хоро-
шо воспроизводит свою установку компенсатора, а бесцветную, По
мнению других наблюдателей, линию считает красной или синей. Рас-
хождения между результатами измерений разных наблюдателей в оди-
наковых условиях систематичны и могут достигать 0,9 единиц Относи-
тельной 'дисперсии.
14-
19»
Другим источником существенной субъективной ошибки может явить-
ся быстрая утомляемость цветного зрения у экспериментатора. Для
уменьшения случайных ошибок в измерении угла (Z) поворота компен-
сатора, обесцвечивание границы светотени надо повторить 10 раз (5 раз
влево от нуля, 5 раз вправо от нуля) и брать среднее значение Z. Каж-
дый раз надо тщательно устанавливать компенсатор, желательно 5 раз
Рис. 4. Графическое определение Z как функции nD20
при со = const для метаново-нафтеновых углеводородов
подходить с красного и 5 раз с синего конца спектра. При определениях
цветовое зрение наблюдателя утомляется, что приводит к неверной ха-
рактеристике цвета. Рекомендуется после очередной установки грани-
цы на бесцветность отвести глаз от зрительной трубы и посмотреть 5—
10 сек. на дневной свет или какой-нибудь белый предмет, затем повто-
рить оценку окраски границы светотени. При таком определении Z
ошибка в средних значениях, связанная с неточностью установки ком-
пенсатора, будет сведена до минимума.
Ошибки, вызываемые применением дневного или искусственного све-
та от ламп накаливания, вызываются различиями спектрального соста-
ва излучения этих источников. При дневном свете, более богатом сини-
ми лучами, обесцвечивание линии достигается при более «синем» поло-
жении компенсатора по сравнению с его установкой при электрическом
освещении. Результаты измерения дисперсии при прочих равных усло-
виях при дневном свете получаются завышенными примерно на 0,4—
0,6 единиц по сравнению с таким же определением при электрическом
свете. Поэтому градуировку прибора и работу на нем желательно про-
водить с постоянным источником света. Очень удобны малогабаритные
осветители для микроскопов, например типа ОИ-19.
При калибровке рефрактометр по относительной дисперсии (и) исхо-
.200
дят из уравнения, справедливого для рефрактометров типа Аббе, дан-
ного в форме
k>fcd = 20^ + —0Д cos 3Z,
nD~ 1 nD — 1
где cofcd* — относительная дисперсия; — показатель преломления;
Z — число делений шкалы лимба компенсатора, соответствующего пол-
ной ахроматизации пограничной линии светотени; А и В — характерные
для данного прибора функции.
В координатах cos 3 Z, nJ этому уравнению соответствует не-
которая поверхность. Проекции сечений этой поверхности плоскостями
«л* = const на плоскость п.в* = 0 являются прямые линии. Калибровка
рефрактометра сводится к определению положения ряда таких прямых
и завершается построением номограммы — Z —
Приступая к градуировке рефрактометра, приготавливают по не-
скольку грамм фракций метано-нафтеновых углеводородов, выкипаю-
щих в пределах 5—10° С, с различными nD20. Необходимо, чтобы эти уз-
кие фракции не содержали ароматических и непредельных углеводоро-
дов, а также гетероатомных соединений. Для этого фракции после раз-
гонки очищают серной кислотой или пропускают через 20—30-сантимет-
ровый слой силикагеля. Желательно, чтобы приготовленные метано-наф-
теновые фракции охватывали интервал кипения от 80—90 до 200—
220° С. Относительная дисперсия всех таких фракций будет одинакова
и примерно равна 17,5. Каждую фракцию поочередно наносят на приз-
мы рефрактометра и устанавливают Z лимба компенсатора на правую
и на левую от нуля сторону, учитывая все ранее сделанные замечания.
Выводят среднее значение Z для каждой фракции и одновременно
фиксируют значения nD20. Все измерения проводят при термостатирован-
ном (20 + 0,2° С) призменном блоке. Далее наносят на график Z —
(рис. 4) полученные данные, графически выравнивают их и экстраполи-
руют в область значений «о20 = 1,50—1,60. Для более уверенной экстра-
поляции надо нанести несколько точек значений Z в области ио20 = 1,50—
1,60. Необходимые для этого метано-нафтеновые фракции можно полу-
201
чить из тяжелых минеральных масел после их вакуумной разгонки на
выкипающие в пределах 5—10° С фракции с последующим пропускание
ем узкой фракции через колонку с активированным силикагелем. Пер-
вые порции фильтрата будут иметь со» 17,5 при больших значенияхnD20.
В отобранных порциях фильтрата определяют среднее значение Z и nL20.
Если полученные значения Z и nD20, нанесенные на график рис. 4, ля-
гут около экстраполированной линии со, то полученные образцы пригод-
ны для дальнейшей градуировки.
Рис. 6. Номограмма nD —«в— Z
Из части измеренных и нанесенных на график метано-нафтеновых
фракций приготовляют серию смесей с точно известным одинаковым со-
держанием выбранного ароматического углеводорода. Вычисляют зна-
чение относительной дисперсии этой серии смесей по формуле.
Р (шар шмет. нафт.) ।
®смеси — IQQ С^мет.нафт.,
где со'смеси — относительная дисперсия полученной смеси: р — концент-
рация ароматического углеводорода в процентах по весу; шар — относи-
тельная дисперсия ароматического углеводорода, взятого для приготов-
ления смеси; соМет. нафт-—средняя относительная дисперсия метановых и
нафтеновых углеводородов, равная 17,5.
Сравнительные результаты определения показателя преломлении
и относительной дисперсии (при 20° С)
Вещество На обычном рефрактометре Аббе На переделанном рефрактометре Аббе
показатель преломления относительная дисперсия показатель преломления относительная дисперсия
3,8-Диэтилдекаи 1,4438 17,2 1,4438 17,2
5-Бутилдекан 1,4292 18,0 1,4290 17,9
4-Бутилнонан 1,4222 18,0 1,4224 17,9
Нефтяная фракция 1,4778 22,2 1,4776 22,1
То же 1,4850 23,6 1,4850 22,5
Толуол 1,4950 32,3 1,4952 Больше 25
202
Составив ряд смесей с одинаковой относительной дисперсией, опреде-
ляют на рефрактометре nD20 и среднее значение Z для каждой смеси.
Наносят на рис. 4 эти значения, графически сглаживают, экстраполиру-
ют обе полученные прямые через равные промежутки значений nD20. Из
верхних и нижних интерполяционных точек проводят ряд горизонталь-
ных прямых и получают серии значений Za и Zb.
Далее на подсобном графике (рис. 5) в координатах cos 3Z— со от-
мечают серии точек айв. Соответствующие точки соединяют прямыми
линиями, интерполируют значения со, nL26 и получают все исходные дан-
ные для построения номограммы. На графике cos 3 Z — со параллельно
оси cos 3Z целесообразно построить ось значений Z. После этого, счи-
тывая величины со, наносят на большой график рис. 6 с координатами
Z — nD20 значения со, графически сглаживая кривые относительной дис-
персии. Один из переделанных таким образом рефрактометров после
проверки и градуировки дал показания, сведенные в таблицу (стр. 204).
Институт нефтехимического синтеза им. А. В. Топчиева
Академии наук СССР
ЛИТЕРАТУРА
1. Б. В. Иоффе. Руководство по рефрактометрии для химиков. Изд-во Ленинградского
ун-та, 1956.
2. И. И. Алиханов. Зав. лаб., 8, 964 (1955).
3. Б. В. Иоффе. Рефрактометрические методы химии. Л., ГНТИХЛ, 1960.
4. Б. В. Иоффе. ЖОХ, 16, 1121 (1946).
5. L. Dodd. Rev. Scient. Instruments, 2, 466 (1931); J. Apt. Soc. Amer., 22, 477 (1932).
6. Б. В. Иоффе. Зав. лаб., 14, 1003 (1948).
СОДЕРЖАН И Е
От редактора................................................................ 5
Анализ прямогонных бензинов методом газо-жидкостной хроматографии с при-,
менением капиллярных колонок. Э. К. Брянская, 3. К. Оленина, Ал. А. Петров 7
I. Анализ бензиновой фракции и. к.— 125° С...........................
II. Анализ бензиновой фракции 125—150° С............................... 13
Литература................................................................ 20
Компонентный анализ тяжелой части нефтей и битумов в малых навесках.
Д. К. Жестков.............................................................. 21
I. Схема компонентного анализа тяжелых нефтепродуктов н битумов 21
II. Рекомендуемая методика............................................ 21
III. Экспериментальная проверка сопоставимости результатов анализа тя-
желой части нефти и битумов микро- и макрометодами......................... 26
Литература................................................................. 27
Микрохроматографический анализ углеводородных смесей. Д. К. Жестков,
Г. Д. Гальперн ............................................................ 28
I. Литературный обзор................................................. 28
II. Схема группового анализа смесей углеводородов (средние н высшие
фракции нефти) ....................................................... 30
А. Отнесение мнкрофракций к группам углеводородов................. 31
Б. Отнесение мнкрофракций к подгруппам углеводородов............... 32
III. Рекомендуемая методика........................................... 33
А. Аппаратура, реактивы, манипуляции............................... 33
Б. Процесс работы при разделении фракций........................... 39
В. Табличное н графическое оформление результатов анализа (на при-
мере микрохроматографического разделения масляной фракции из сов-
ременных морских осадков Берингова моря) .......................... 43
IV. Обоснование метода............................................... 48
А. Обоснование способа интерпретации результатов мнкрохроматогра-
фического анализа для определения группового состава .............. 48
Б. Экспериментальная проверка метода на искусственных смесях ин-
дивидуальных углеводородов ........................................ 52
В. Обоснование способа интерпретации результатов мнкрохроматогра-
фического анализа для ориентировочного определения подгрупп углево-
дородов .......................................................... 56-
Г. Экспериментальная проверка результатов микрохроматографического
анализа другими методами........................................... 61
Литература............................................................ . 61
205.
Полуавтоматическая установка для количественного определения серы и гало-
генов (хлора, брома, иода) в органических соединениях и нефтепродуктах.
Н. П. Волынский........................................................... 63
I. Описание установки................................................. 63
II. Порядок проведения анализа......................................... 70
Литература.................................................................. 75
Тонкослойная хроматография органических соединений серы и ее применение
к исследованию сульфидов (типа встречающихся в нефтях) и их производных
Е. Н. Караулова............................................................. 76
I. Введение.....................................................- • • 76
II. Аналитическая тонкослойная хроматография (ПТСХ) органических соеди-
нений серы............................................................. 83
III. Препаративная тонкослойная хроматография (ПТСХ) органических сое-
динений серы........................................................... 89
Литература................................................................. 94
Методы получения сульфоксидов из средних фракций сернистых нефтей (груп-
повое выделение сульфидов в виде сульфоксидов). Е. Н. Караулова,
Т. А. Бардина, Г. Д. Гальперн............................................... 95
1. Выделение сульфоксидов из окисленных дистиллятов......................... 95
А. Хроматографический метод (элюэнтно-вытеснительный)............... 95
Б. Кислотно-экстракционные методы извлечения сульфоксидов .... 97
II. Окисление сульфидов нефтяных дистиллятов................................ 99
А. Гетерогенно-эмульсионное окисление...............................101
Б. Окисление сульфидов до сульфоксидов в гомогенной среде .... 103
Литература............................................................... Ю6
Элементный анализ органических соединений с газохроматографическим опре-
делением продуктов разложения I. Метод определения азота. А. В. Непряхина,
И. К- Чудакова, Г. А. Новикова, Г. Г. Радикова, О. Н. Доманина 107
I. Введение...........................................................107
II. Рекомендуемая методика ............................................109
III. Результаты анализов................................................112
Литература..................................................................114
Элементный анализ органических соединений с газохроматографическим опреде-
лением продуктов разложения. II. Метод одновременного определения углерода,
водорода, азота. И. К. Чудакова, А. В. Непряхина, О. Н. Доманина. Г. А . Нови-
кова Г. Г.Радикова 115
I. Введение...........................................................115
II. Рекомендуемая методика (предварительная).......................... 117
III. Результаты анализов................................................119
Литература..................................................................120
Получение концентратов азотистых оснований нефти. Н. Н. Безингер, Г. Д. Галь-
перн, В. Н. Каричева 121
I. Сорбция индивидуальных оснований...................................122
II. Сорбция оснований нефти............................................125
III. Десорбция нефтяных оснований...................................... 129
IV. Рекомендуемая методика..............................................130
А. Сорбция нефтяных оснований.......................................131
Б. Десорбция нефтяных оснований.................................... 131
Литература......................................,...........................132
Сравнительное изучение состава сложных смесей органических веществ комби-
нированным спектральным микрометодом анализа. Б. А. Смирнов 133
I. Метод спектрального структурно-группового анализа в ультрафиолетовой
и инфракрасной областях спектра....................................... 134
206
II. Опыт применения комбинированного спектрального микрометода анали-
за к изучению СГС фракций масел современных морских осадков . . 146
Литература..................................................................159
Исследование спектров сульфидов. Л. Р. Барыкина, Г. Д. Гальперн, .4. Н. Кас-
линский, Н. А. Шиманко 160
1. Колебательные спектры алифатических сульфидов.......................161
А. Обзор литературы................................................161
Б. Обсуждение результатов..........................................162
II. Исследование колебательных спектров непредельных сульфидов и воз-
можности их использования в структурно-групповом анализе .... 169
А. Исследование спектров...........................................169
Б. Примеры применения ИК-спектроскопии к анализу непредельных
сульфидов..........................................................177
III. Исследование колебательных спектров ароматических сульфидов . . . 180
А. Обзор литературы................................................180
Б. Обсуждение результатов..........................................180
IV. Электронные спектры сульфидов..................................... 187
А. Непредельные сульфиды...........................................187
Б. Ароматические сульфиды..........................................191
В. Тиаинданы.......................................................192
Литература..................................................................194
Рефрактометр для работы с малыми количествами вещества. Д. К. Жестков 196
I. Переделка стандартного рефрактометра ИФР-22.......................196
II. Проверка и градуировка рефрактометра................. . . . . 198
III. Градуировка рефрактометра........................................ 199
Литература.............................................................. 203
УДК 553.981.8.665, 612.2 — 404
Анализ прямогонных бензинов методом газо-жидкостной хроматографии с применением
капиллярных колонок. Брянская Э. К., Оленина 3. К., Петров А. А. «Ме-
тоды анализа органических соединений нефти, их смесей и производных», сб. 2. М„ «Наука»,
1969, стр. 7—20.
Разработан метод исследования углеводородного состава фракции н. к.— 150° С пря-
могонных бензинов. В основу метода положен порядок выхода углеводородов при данной
температуре на данной фазе. В качестве неподвижной фазы применялся сквалан. Найдены
относительные времена удерживания алканов состава Св — Св. Приведен компонентный
состав бензиновой фракции н. к.— 150° С грозненской нефти. Для расшифровки состава бен-
зиновых фракций предложен метод получения смеси индивидуальных углеводородов путем
изомеризации в присутствии бромистого алюминия любого парафинового углеводорода,
абл. 8. Иллюстраций 19. Библ. 8 назв.
УДК 5.43.063.665.451.05
Компонентный анализ тяжелой части нефтей и битумов в малых навесках. Жестков Д. К.
«Методы анализа органических соединений нефти, их смесей и производных», сб. 2. М., «Нау-
ка», 1969, стр. 20—27.
Описываемый метод предусматривает разделение тяжелой части нефти тяжелых нефте-
продуктов или битумов на фракции масел, смол бензольных, смол спнрто-бензольных и ас-
фальтенов при исходных навесках 50—150 мг. В случае необходимости путем применения
ряда других растворителей смолы и асфальтены разделяют иа более узкие фракции. В мето-
дике даио детальное описание простых приборов (стекло) и рекомендуемой техники экспери-
мента.
Осаждение асфальтенов проводится в специальном отстойнике при 40-кратном разбав-
лении исследуемой навески петролейным эфиром. Перенос смол и масел иа силикагель в мик-
роэкстрактор, а также перенос асфальтенов во взвешенный стаканчик осуществляется быстро
и удобно с минимальными потерями. Полнота отмывки асфальтенов от масел и смол контро-
лируется по люминесцентному свечению (лампа ПРК). Разделение масел и смол проводится
в микроэкстракторах последовательной экстракцией с силикагеля (марки АСК» активностью
6—7 мл по бензолу) петролейным эфиром, бензолом н спирто-бензолом. Извлечение фракций
заканчивали, когда свежий растворитель в колбочке после 2 час. экстракции имел интенсив-
ность свечения, при облучении ультрафиолетом, не более 2—3 баллов.
Рекомендуемая методика дает результаты, практически совпадающие с результатами
аналогичных определений, выполненных макрометодами.
Таблиц 6. Иллюстраций 3. Библ. 6 назв.
УДК 547.21; 543.544.063.
Микрохроматографический анализ углеводородных смесей. Жестков Д. К., Галь-
перн Г. Д. «Методы анализа органических соединений нефти, их смесей н производных»,
сб. 2. «Наука», 1969, стр. 28—62.
Дано описание микрохроматографической колонки с приспособлениями и методики элю-
энтной микрохроматографии(в режиме линейной хроматографии) с одновременным отгоном
растворителя, позволяющих проводить групповой адсорбционный анализ углеводородных
смесей с т. кнп. от 150° С и выше в навесках 10—50 мг. Метод проверен на смесях индивиду-
альных углеводородов, нефтяных фракциях и на маслах из современных морских осадков.
Установлено, что точность метода ± 1,0 вес. % для метанонафтеиовой и ± 2,0 вес. %
для ароматической части исследуемой смеси (индикация — по скачку относительной диспер-
20
сии и «выходной кривой»). Приведены пределы пр микрофракций для ориентировочного
определения степени цикличности нафтеновых и ароматических углеводородов, а также для
оценки количества полярных гетероатомных соединений. Рекомендуемая техника разделения
выгодно отличается от ранее предложенных тем, что позволяет проводить фракционировку
иа заданное количество микрофракций, ориентировочный вес которых определяется экспе-
риментатором заранее или в процессе работы, что позволяет четко определять границы раз-
деляющихся веществ.
Метод, при подборе соответствующих адсорбентов и десорбентов, может быть рекомен-
дован для использования в химических лабораториях, занимающихся получением и иссле-
дованием органических веществ разного состава и строения.
Таблиц 16. Иллюстраций 12. Библ. 58 назв.
УДК 543.845.0634-543.848.063: 665.521
Полуавтоматическая установка для количественного определения серы и галогенов (хлора,
брома? иода) в органических соединениях и нефтепродуктах. Волынский Н. П. «Ме-
тоды анализа органических соединений нефти, их смесей и производных», сб. 2. М., «Наука»,
1969, стр. 63—75.
Дано детальное описание установки для экспресс-аиализа. Сожжение навески прово-
дят пиролитическим ламповым методом, заключающемся в том, что навеску испаряют и (или)
пиролизуют в кварцевом стаканчике, открытый конец которого введен в дожигающее пламя,
создаваемое газовой микрогорелкой. Продукты пиролиза полностью сгорают в дожигающем
пламени, а образующиеся при этом окнслы серы, галогены галогеноводороды поглощаются
в абсорбере, через который во время сожжения просасывают воздух. Поглотительным рас-
твором служит смесь 10 мл 0,05 N Na2CO3 н 3—4 мл ~ 6%-ной Н2О2, которые вводятся в
абсорбер полуавтоматическими дозаторами. Окончание анализа на S, С1, Вг проводят ацидо-
метрическим титрованием (по метилоранжу) в абсорбере. На все определение уходит около
5 мни. Установка собрана иа одном штативе Бунзена таким образом, что обеспечивает удоб-
ство работы н исключает лишние движения аналитика; абсорбер укреплен на съемном дер-
жателе, поворачивающемся вокруг вертикальной оси, что позволяет устанавливать его в три
фиксированные положения: наполнение, сожжение, титрование. В случае определения иода
в абсорбер помещают 15 мл дистиллированной воды н 2 капли гндразингидрата. По оконча-
нии сожжения переносят раствор в сосуд для меркуриметрического определения.
Таблиц 3. Иллюстраций 14. Библ. 5 назв.
2G9
УДК 543.544 : 45 : 547,29 : 665.41.094.3.
Тонкослойная хроматография органических соединений серы,' ее применение к исследованию
сульфидов (типа встречающихся в нефтях) н нх производных. Караулова Е. Н. «Ме-
тоды анализа органических соединений нефти, их смесей и производных», сб. 2. М., «Наука»,
1969, стр. 76—94.
Рассмотрены литературные данные по тонкослойной хроматографии (ТСХ) органиче-
ских соединений серы (СС) за 1962—1965 гг. Найдены условия ТСХ различных классов СС,
прн этом использованы элюэнты с постепенно возрастающей полярностью; приведены при-
меры применения ТСХ при синтезе и превращениях СС. Для сульфоксидов определен ниж-
инй предел чувствительности метода ТСХ, равный 2,5—10 мкг. Разработана методика пре-
паративной ТСХ (ПТСХ) для тиабицикланонов, тиабицикланолов, тиабицикланов и тиаби-
циклансульфоксидов. С помощью ПТСХ удается осуществить полное нли частичное разде-
ление стереоизомеров перечисленных выше классов СС. Сочетанием методов ТСХ, ПТСХ и
ГЖХ можно количественно оценить состав смесей стереоизомеров, образующихся при
нестереоспецифичном синтезе; в качестве примера приводится исследование состава смеси
стереоизомерных 2-метил-1-тиадекалинов.
Таблиц 3. Иллюстраций 12. Библ. 26 назв.
УДК 665.51 575.(4) : 665.547.932.943.546.11—39
Методы получения сульфоксидов нз средних фракций сернистых нефтей (групповое выделен
ние сульфидов в виде сульфоксидов). Караулова Е. Н., Б а р д и и а Т. А., Галь-
перн Г. Д. «Методы анализа органических соединений нефти, их смесей и производных»,
сб. 2. М., «Наука», 1969, стр. 95—106.
Предложены методы окисления сульфидов в нефтяных дистиллятах (СНД) до суль-
фоксидов (СО) и методы выделения СО из полученных оксидатов. Гетерогенно-эмульсионное
окисление СНД осуществляют действием Н2О2 либо в присутствии эквивалентного количе-
ства НС1О4 (при этом СО выделяются в виде перхлоратов в одну стадию), либо в присутст-
вии каталитических количеств H2SO4 ; в последнем случае СО извлекают 60%-ной H2SO4.
Предложен метод селективного окисления «трудноокисляемых» СНД (не окисляющихся
первыми двумя методами) до СО действием Н2О2 в (МеСО)аО (I) или в смеси (I) с MeCOEt.
Рекомендована методика хроматографического выделения СО, включающая предварительное
хроматографирование пробной загрузки; адсорбент — технический силикагель «АСК», ем-
кость его определяют по о-оксихинолину, при Sso в окисленных дистиллятах 0,3%-ной
СО выделяют в виде перхлоратов действием 42%-ной НС1О4; сульфиды регенерируют из пер-
хлоратов подщелачиванием.
Таблиц 2. Библ. 7 назв.
УДК 543.544 : 543.80
Элементный анализ органических соединений с газохроматографнческнм определением про-
дуктов разложения 1. Метод определения азота. Непряхина А. В,, Чудакова
И. К», Новикова Г. А., Радикова Г. Г., Доманина О. Н. «Методы ана-
лиза органических соединений нефти, нх смесей и производных», сб. 2. М.» «Наука», 1969,
стр. 107—114.
Предлагается метод определения азота в азотсодержащих органических соединениях
с газохроматографическим окончанием анализа. Навеска анализируемого вещества (1 — 5 ла),
помещенная в кварцевый стаканчик длиной 90 мм, диаметром 6—7мм , засыпается порошкооб-
разной и зерненной окисью меди. Стаканчик с навеской и окислителем помещаются в трубку
для сожжения (ТС) из нержавеющей стали длиной 160 мм, внутренним диаметром 10 мм.
Окисление анализируемого вещества до Ns, СО2 и Н2О осуществляется за счет кислорода
окиси меди в замкнутом объеме в среде гелия при температуре 750—850° при использовании
предварительного пиролитического разложения навески анализируемого вещества. Процесс
окисления полностью завершается внутри стаканчика. Дополнительные зоны окисления от-
сутствуют. Необходимость восстановленной меди отпадает, так как функции ее выполняют
продукты разложения органического вещества. Нагрев ТС производится с помощью трех-
секционного электронагревателя. Секции включаются последовательно автоматически через
заданные интервалы посредством реле времени.
Продолжительность сожжения 30 мин. Образующийся свободный азот идентифици-
руется газохроматографнческнм методом. Хроматографическая колонка длиной 1 м, диамет-
ром 6 мм заполнена силикагелем МСМ. Температура колонки 70° С. Детектор по теплопро-
водности. Скорость гелня 20 — 30 мл! мин.
Предложенным методом проведено определение азота в ряде органических соединений
с точностью ± 0,3 абс.%
Таблиц 1. Иллюстраций 3. Библ. 19 назв.
УДК 543.544 : 543.80
Элементный анализ органических соединений с газохроматографическим определением продук-
тов разложения И. Метод одновременного определения углерода, водорода и азота. Непр я-
хииа А. В., Чудакова Й. К., Доманина О. Н., Новикова Г. А.,
Радикова Г. Г. «Методы анализа органических соединений нефти, их смесей и произ-
водных», -сб. 2. М., «Наука», 1969, стр. 115—120.
Предлагается метод одновременного определения С, Н, N в органических соединениях
с газохроматографическим окончанием анализа. Навеска анализируемого вещества (1 — 5 мг)
подвергается сожжению так же, как при определении азота (см. предыдущий реферат).
Продолжительность сожжения 30 мии. Продукты окисления вытесняются гелием и
проходят-через электролитическую ячейку, где происходит накопление и последующее элек-
тролитическое разложение воды. Водопоглощающий слой — Р2О5. Продукты пиролиза и
электролиза (N2, СО2 , О2) идентифицируются газохроматографическим методом. Разделение
компонентов смеси газов происходит иа колонке длиной 1 м, диаметром 6 мм, которая запол-
нена силикагелем МСМ. Температура колонки 70°. Детектор по теплопроводности. Скорость
гелия 20 — 30 мл/мин.
Предложенным методом проведен анализ ряда органических соединений с точностью
± 0,3 абс. % для углерода, и азота и ± 0,1 — 0,2 абс. % для водорода.
Таблиц 1. Иллюстраций 2. Библ. 22 назв.
210
УДК 665.547.96 4; 545.544.6
Получение концентратов азотистых оснований нефти. Безингер Н. Н., Га ль п ер в
Г. Д., Каричева В. Н. «Методы анализа органических соединений, нх смесей и произ-
водных», сб. 2. М., «Наука», 1969, стр. 121—132.
Разработан метод выделения азотистых оснований из сырых нефтей и нефтепродуктов
в виде концентратов. Метод основан на сорбции оснований крупнопористыми сульфокислот-
нымн катноинтамн КУ-23 н последующей десорбции их аммиаком в спиртовом растворе.
Показано, что количественная сорбция оснований достигается только в присутствии поляр-
ных растворителей (уксусный ангидрид, днметилформамид). Полученные концентраты, помимо
азота, содержат значительное количество кислородных и сернистых соединений. Высказано
предположение, что неполная сорбция оснований из углеводородных сред связана с суще-
ствованием ассоциатов гетероатомных соединений нефти, образованных как за счет р-элек-
тронов атомов N, S и О, так и за счет я-связей конденсированных ароматических систем.
Таблиц 6. Иллюстраций 3. Библ. 18 назв.
УДК 539.194-541.57.
Сравнительное изучение состава сложных смесей органических веществ комбинированным
спектральным микрометодом анализа. Смирнов Б. А. «Методы анализа органических
соединений нефти, нх смесей н производных», сб. 2. М., «Наука», 1969, стр. 133—159.
Рассмотрен метод спектрального (в УФ-н ИК-области спектра) структурно-группового-
(СГ) анализа сложных углеводородных смесей. Описаны приспособления для применения
указанного метода к мнкроколичествам вещества. Для получения наилучших результатов
СГ анализа исследуемый образец предварительно подвергают препаративному хроматографи-
ческому разделению. Даиа подробная схема такого комбинированного спектрально-хромато-
графического микрометода, являющегося особенно ценным в задачах, где необходимо срав-
нительное изучение состава большого набора микроколичеств фракций. Обсуждены условия,
выполнение которых необходимо для наиболее полного извлечения сведений из результатов-
анализа.
В качестве примера использования метода рассмотрена конкретная задача по изучению
СГ состава масел, выделенных из битумов донных отложений различных районов Мирового
океана. Подробно рассмотрены цели исследования и способы их наилучшего достижения.
Принципиальные основы предлагаемого метода имеют большую общность н могут най-
ти применение при изучении смесей самого различного состава.
Табл. 4. Иллюстраций 8. Библ. 25 назв.
УДК 539.19 4-541.57
Исследование спектров сульфидов. Барыкина Л. Р., Гальперн Г. Д., Кис-
лннскнй А. Н., Шиманко Н. А. «Методы анализа органических соединений неф-
ти, их смесей н производных» сб. 2. М.» «Наука», 1969, стр. 160—195.
Рассмотрены особенности ИК-и КРС-спектров насыщенных непредельных н ароматиче-
ских сульфидов по сравнению со спектрами углеводородов родственного строения, а также-
УФ-спектры сульфидов. Выявлены структурно-частотные корреляции, которые рекомендо-
ваны для использования прн решении ряда аналитических задач. Даны примеры структур-
ного анализа некоторых сульфидов и нх смесей.
Таблиц 13. Иллюстраций 5. Библ. 85 назв.
УДК 535.322.4; 543.063
Рефрактометр для работы с малыми количествами вещества. Жестков Д. К. «Методы
анализа органических соединений нефти, их смесей и производных», сб. 2. М.» «Наука»
1969, стр. 196—203.
Описан способ переделки стандартного рефрактометра ИФР-22 с целью определения
показателя преломления я величин относительной днсперснн малолетучнх веществ в коли-
чествах 0,2—0,3 мг с обычной точностью.
Согласно предлагаемому способу, не трогая измерительную призму, снимают освети-
тельную призму н вручную сошлнфовывают ее до равномерного зазора между плоскостью
призмы и ее металлической оправой. Величина зазора определяется визуально до появления
2—3 дифракционных полос. Сошлифованную призму ставят на место прн помощи переделан-
ной крепежной петлн.
Проверку показаний переделанного рефрактометра и его калибровку проводят по уг-
леводородным фракциям и чистым ароматическим углеводородам. Калибровка заканчивается*
построением номограммы.
Таблиц 1. Иллюстраций 6. Бнбл. 6 назв.
Методы анализа органических соединений
нефти, их смесей и производных, сб. 2
Утверждено к печати
Институтом нефтехимического синтеза
им. А. В. Топчиева
Академии наук СССР
Редактор Л. С. Поваров
Технический редактор А. П. Ефимова
Сдано в набор 23/V 1969 г.
Т-15352. Подписано к печати 18/XI 1969 г.
Формат 70X108’/i6. Бумага типогр. № 2.
Издательство «Наука»
Печ. л. 13,25. Усл.-печ. л. 18,55. Уч.-изд. л. 17,8.
Тираж 2200 экз. Тип. 2300
Цена 1 р. 19 к.
Москва, К-62, Подсосенский пер., 21
2-я типография издательства «Наука»
Москва, Г-99, ШубиискнЙ пер., 10